/
























































































































































































































































































































































Author: Роговин З.А.
Tags: искусственные волокна химия неорганическая химия легкая промышленность издательство химия химические волокна
Year: 1974




Text
3. А. Роговин
основы
ХИМИИ И ТЕХНОЛОГИИ
ХИМИЧЕСКИХ ВОЛОКОН
ТОМ II
ПРОИЗВОДСТВО
синтетических волокон
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ»
1974
3. А. Роговин
основы
ХИМИИ И ТЕХНОЛОГИИ
ХИМИЧЕСКИХ ВОЛОКОН
ИЗДАНИЕ ЧЕТВЕРТОЕ,
ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Допущено Министерством высшего
и среднего специального образования СССР
в качестве учебного пособия
для студентов высших технических учебных заведений
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ»
1974
вП752
УДК 677.4Э4.6/.7 @75.8)
Р59
Роговин 3. А.
Основы химии и технологии химических волокон. Т. II.
Изд. 4-е, перераб. и доп. М., «Химия», 1974.
344 с, 10 табл., 52 рис., список литературы 471 ссылок.
В двух томах книги изложены основы химии и технологии
химических волокон (искусственных и синтетических).
В первом томе освещаются общие принципы и методы
получения химических волокон всех видов, а также основы
химии и технологии производства искусственных волокон.
Во втором томе изложены основы химии и технологии
синтетических волокон.
Книга предназначается в качестве учебного пособия для
студентов вузов. Она может быть полезна широкому кругу
научных н инжеиерно-техиических работников
исследовательских институтов промышленности химических волокон и
других отраслей народного хозяйства, произнодящих исходное
сырье и перерабатывающих" искусственные и синтетические
волокна.
31412-073 ,„ _. 6П752
050@1)-74
© Издательство «Химия», 1974 г.
СОДЕРЖАНИЕ
Глава 1. Общие сведения о производстве синтетических волокон ...... 9
1.1. Возникновение и развитие производства синтетических волокон . . 9
1.2. Перспективы развития производства синтетических волокон ... 12
1.3. Классификация синтетических волокон и особенности их
производства '4
1.3.1. Классификация синтетических волокон 14
1.3.2. Особенности производства синтетических волокон 16
Глава 2. Производство полиамидных волокон 18
2.1. Синтез поликапроамида 21
2.1.1. Получение капролактама 21
2.1.2. Свойства капролактама 26
2.1.3. Полимеризация капролактама 28
2.1.4. Свойства поликапроамида 46
2.2. Синтез полигексаметиленадипамнда 47
2.2.1. Получение мономеров 48
2.2.2. Получение анида (полигексаметиленадипамида) 50
2.3. Синтез полиэнантоамида 52
2.3.1. Получение аминоэнантовой кислоты 53
2.3.2. Получение полиамида эиаит 54
2.4. Синтез полиамидов иайлон 6, 10 и ундекан (найлон II) .... 55
2.5. Синтез полиамида до декан (иайлон 12) 56
2.6. Синтез полиамидов из пяти- и шестичленных лактамов 58
2.7. Синтез поли^-амидов полимеризацией четырехчленных лактамов . 60
2.8. Свойства полиамидов капрон и анид 61
2.9. Формование полиамидных волокон 63
2.9.1. Основные параметры процесса формования . 69
2.9.2. Получение волокна капрон непрерывным методом 72
2.10. Последующая обработка нити 74
2.11. Особенности производства полиамидной кордной нити 81
2.12. Особенности производства полиамидного штапельного волокна . . 83
2.13. Особенности производства полиамидного моноволокна 85
2.14. Использование отходов 86
2.15. Свойства полиамидных волокон 88
2.16. Модифицированные полиамидные волокна 99
6 Содержание
2.16.1. Методы физической модификации полиамидных волокон . . 99
2 16 2. Методы химической модификации полиамидов и волокон . 100
Литература
Глава 3. Производство полиэфирных волокон 117
3.1. Производство полиэтилентерефталатных волокон 118
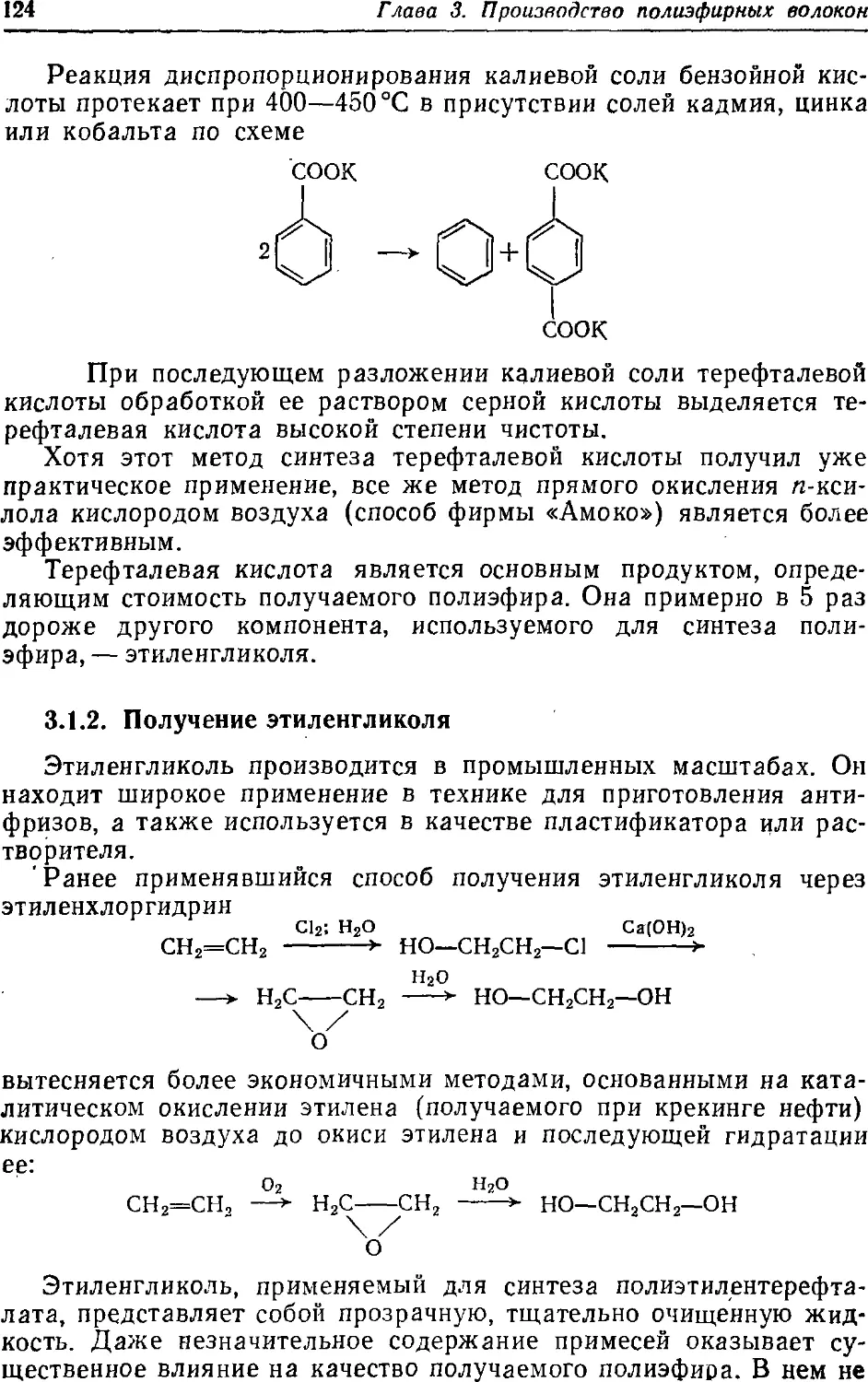
3.1.1. Получение терефталевой кислоты 119
3.1.2. Получение этиленгликоля 124
3.1.3. Синтез диэтиленгликольтерефталата 125
3.1.4. Синтез полиэтилентерефталата 131
3.1.5. Свойства полиэтилентерефталата 140
3.1.6. Формование полиэтилентерефталатного волокна 142
3.1.7. Последующая обработка полиэтилентерефталатного волокна 146
3.1.8. Особенности производства кордиой нити 149
3.1.9. Особенности производства штапельного волокна 151
3.1.10. Особенности производства моноволокна 152
3.1.11. Использование отходов 153
3.1.12. Свойства полиэтилентерефталатного волокна и области его
применения ' 154
3.2. Модифицированные полиэфирные волокна 161
3.2.1. Улучшение накрашиваемости волокон 161
3.2.2. Получение извитых и объемных волокон . 166
3.2.3. Уменьшение склонности волокна к пиллингу 168
Литература 169
Глава 4. Производство полиуретановых волокон 171
Литература 175
Глава 5. Полиформальдегидное волокно 176
Литература 178
Глава 6. Производство полиакрилонитрильиых волокон 179
6.1. Получение акрилонитрила 180
6.2. Синтез полимеров и сополимеров акрилонитрила 182
6.2.1. Суспензионная полимеризация 185
6.2.2. Полимеризация в растворе 185
6.3. Свойства полиакрилонитрила 193
6.4. Получение полиакрилонитрильного волокна 194
6.4.1. Формование полиакрилоиитрильной нити 198
6.4.2. Формование полиакрилонитрильного штапельного волокна . 200
6.4.3. Регенерация растворителя 203
6.4.4. Последующая обработка полиакрилонитрильного волокна . 205
64.5. Свойства полиакрилонитрильного волокна 208
Содержание
6.5. Модифицированные полиакрилонитрильные волокна 211
6.5.1. Волокна из сополимеров акрилонитрила 211
6.5.1.1. Волокна из сополимеров с низким содержанием второго
мономера 211
6.5.1.2. Волокна из сополимеров с высоким содержанием второго
мономера 221
6.5.2. Волокна из привитых сополимеров акрилонитрила .... 223
6.5.3. Волокна из смесей полиакрилонитрила с другими
полимерами 224
Литература 226
Глава 7. Производство поливинилхлоридных волокон 229
7.1. Поливинилхлоридные волокна 230
7.2. Перхлорвиниловые волокна 235
7.3. Волокна из сополимеров винилхлорида 244
Литература 247
Глава 8. Производство поливинилспиртовых волокон 248
8.1. Получение поливинилового спирта 248
8.2. Приготовление прядильного раствора 252
8.3. Формование волокна 253
8.4. Последующая обработка волокна 255
8.5. Свойства и области применения поливинилспиртового волокна . 263
8.6. Модифицированные поливинилспиртовые волокна 266
Литература 268
Глава 9. Производство полиолефиновых волокон 270
9.1. Полипропиленовые волокна 271
9.1.1. Синтез полипропилена 272
9.1.2. Свойства полипропилена 274
9.1.3. Получение полипропиленового волокна 281
9.1.4. Свойства полипропиленового волокна 284
9.1.5. Химическая модификация полипропиленовых волокон . . . 288
9.2. Полиэтиленовые волокна '. 292
9.2.1. Получение и свойства полиэтилена 292
9.2.2. Получение полиэтиленового волокна ¦ 293
Литература 295
Глава 10. Производство волокон из фторсодержащих полимеров ..... 297
10.1. Получение волокна тефлон 297
10.2. Получение волокна фторлон 300
Литература 302
8 Содержание
Глава Л. Термостойкие и жаростойкие волокна 303
11.1. Получение термостойких волокон 304
11.1.1. Волокна из ароматических полиамидов и полиэфиров . . . 306
11.1.2. Волокна из полигетероциклических полимеров 310
11.1.3. Волокна из лестничных полимеров 319
11.2. Получение жаростойких волокон 320
11.2.1. Исходное сырье 321
11.2.2. Получение углеродного волокна из полиакрилонитрильиого
волокна 321
11.2.3. Получение углеродного волокна из вискозной кордной нити 325
11.2.4. Свойства и области применения углеродных волокон . . . 327
Литература 329
Глава 12. Сверхпрочные и высокомодульные волокна 331
12.1. Характеристика новых типов жесткоцепных волокиообразующих
полимеров .332
12.2. Особенности процесса формования волокна 335
Литература 339
Предметный указатель 340
Глава 1
ОБЩИЕ СВЕДЕНИЯ О ПРОИЗВОДСТВЕ
СИНТЕТИЧЕСКИХ ВОЛОКОН
Промышленное производство синтетических волокон, начатое
30—35 лет назад, является принципиально новым этапом в
развитии производства химических волокон.
В результате разработки эффективных методов получения
синтетических волокон значительно расширилась сырьевая база
промышленности химических волокон и, главное, создались реальные
условия для массового производства волокон с новыми свойствами,
которыми не обладают натуральные и искусственные волокна.
Этим и объясняется бурное развитие производства волокон из
синтетических полимеров.
Динамика развития мирового производства синтетических
волокон характеризуется следующими цифрами:
Годы 1940 1950 1955 1960 1966 1970 1972
Объем производства, тыс. т . . . . 2,5 77 254 710 2483 4924 6862
Как видно из этих данных, за 32 года мировое производство
синтетических волокон увеличилось с 2,5 почти до 7000 тыс. т.
За последние 10 "лет выпуск синтетических волокон возрос
более чем на 5800 тыс. т. В 1968 г. мировое производство этих
волокон впервые превысило производство искусственных волокон.
В Советском Союзе производство синтетических волокон
развивается быстрыми темпами. Выработка этих волокон в 1975 г.
достигнет 440 тыс. т.
Число отдельных видов синтетических волокон,
вырабатываемых в промышленности, непрерывно увеличивается,
совершенствуются методы получения этих волокон и расширяются области их
применения. Если в 1952 г. было известно 10—12 видов
синтетических волокон, то в 1970 г. на опытных установках и в
производственных условиях вырабатывалось уже свыше 30 видов этих волокон.
Организация промышленного производства синтетических
волокон явилась результатом длительных исследований больших
коллективов ученых, конструкторов и технологов многих стран.
1.1. ВОЗНИКНОВЕНИЕ И РАЗВИТИЕ ПРОИЗВОДСТВА
СИНТЕТИЧЕСКИХ ВОЛОКОН
Впервые производство синтетических волокон было начато
в середине 30-х годов текущего столетия. Для получения волокон
были использованы некоторые типы карбоцепных синтетических
10 Глава 1. Общие сведения о производстве синтетических волокон
полимеров, в частности сополимер винилхлорида и винилацетата
(для волокна виньон), поливиниловый спирт, хлорированный по-
ливинилхлорид (для волокна ПІД). Волокно формовали из
растворов полимеров сухим или мокрым способом. Получение этих
волокон не было связано с каким-либо принципиальным изменением
технологического процесса производства искусственных волокон,
в частности ацетатного. По ряду причин производство указанных
волокон значительного развития не получило.
В 1938—1939 гг. после длительных исследований начинается
промышленное производство полиамидного волокна найлон. Для
производства этого волокна, обладающего рядом новых ценных
свойств, потребовалось разработать принципиально новый метод
формования — не из растворов, а из расплава. Волокно найлон
нашло широкое применение для выработки предметов народного
потребления и многих технических изделий. Производство его
получило большое развитие сначала в С1Ш$, а затем и в других
странах.
В конце 40-х годов начинается промышленное производство
нового типа синтетического волокна — полиэфирного волокна
терилен, которое в последние годы развивалось в различных странах
очень высокими темпами.
После того как в 1942—1944 гг. в результате длительных
исследований был найден доступный растворитель для полиакрилонит-
рила, появилась возможность организовать промышленное
производство нового высококачественного синтетического волокна — по-
лиакрилонитрильного, которое в настоящее время является одним
из основных типов многотоннажных синтетических волокон.
В последние годы разработаны методы получения новых
синтетических волокон из различных виниловых полимеров, волокон из
стереорегулярных полимеров, в первую очередь из полипропилена,
и высокоэластичных волокон на. основе полиуретанов. Нашли
промышленное применение способы получения синтетических волокон,
обладающих специфическими свойствами.
К таким волокнам в первую очередь следует отнести
термостойкие и жаростойкие, а также сверхпрочные, высокомодульные
волокна.
В ближайшие годы, по-видимому, будут применяться в
производственных условиях принципиально новые способы формования
карбоцепных волокон из гелей, латексов и суспензий.
Основные этапы развития производства синтетических волокон
приведены в табл. 1.1.
Объем мирового производства синтетических волокон в 1972 г.
составил (в тыс. т) :
Полиамидные .
Полиэфирные .
. 2424
. 2508
Полиакрилонп-
трнльные . . .
Другие виды . .
Итого . . . .
1270
660
6862
1.1. Возникновение и развитие производства синтетических волокон
11
Таблица 1.1. Основные этапы развития производства
синтетических волокон
Годы
1932—1934
1932—1938
1934—1936
1938-1940
1940-1946
1942—1950
1942-1943
1954-1956
1954—1956
1955-1958
1960-1962
1962
1965-1970
1971 — 1973
Волокно
Виньон
Найлон 6,6
пц
Перлон
Терилен
Куралон
Полиакрилони-
трильное
Тефлон
Фторлон
Полно лефиновое
НТ-1 (Аш-Тэ-1)
номекс
Ликра (лайкра),
спандекс
—
—
Исходный полимер
и характеристика этапа развития
синтетических волокон
Сополимер винилхлорида и
винилацетата
Продукт поликонденсации гек-
саметилендиамина и адипи-
новой кислоты
Синтезирован новый класс
высокомолекулярных
соединений — полиамидов и
разработан новый метод
формования волокон — из расплава
Хлорированный поливинил-
АЛОрИД
Поликапроамид. Получен
новый класс полиамидов
Полиэтилентерефталат. Для
производства волокон
использован новый класс ге-
тероцепных соединений —
полиэфиров
Поливиниловый спирт
Полиакрилонитрил. Найден
новый технически
приемлемый растворитель для по-
лиакрилонитрила
Политетрафторэтилен.
Предложен новый метод
формования волокна
Получен новый вид волокна
из растворимых в ацетоне
фторсодержащих
полимеров
Линейный полиэтилен и сте-
реорегулярный пропилен.
Предложен новый метод
синтеза полимеров
Полиамид. Получен новый
тип термостойкого волокна
Сополимеры макродиизоциа-
натов и полиэфиров.
Организовано производство
высокоэластичных волокон
Различные типы термостойких
и жаростойких волокон
Сверхпрочное гетероцепное
синтетическое волокно,
значительно превышающее по
прочности все природные и
химические волокна
Страна
США
США
Германия
Германия
Англия
Япония
Германия,
США
США
СССР
США, Италия
США
США, Японии
СССР, США,
Япония
СССР. США
12 Глава 1. Общие сведения о производстве синтетических волокон
Суммарная выработка трех важнейших видов синтетических
волокон — полиамидных, полиэфирных и полиакрилонитрильных —
превысила 90% общего производства всех синтетических волокон.
1.2. ПЕРСПЕКТИВЫ РАЗВИТИЯ ПРОИЗВОДСТВА
СИНТЕТИЧЕСКИХ ВОЛОКОН
Кроме указанных основных типов синтетических волокон в
будущем значительное развитие, по-видимому, получат полиолефи-
новые, поливинилспиртовые и поливинилхлоридные волокна. Для
производства волокон, обладающих специфически ценными
свойствами,— термо- и жаростойких, хемостойких, биологически
активных, ионообменных и полупроводниковых — все в большем
количестве будут использоваться новые классы полимеров.
Наиболее крупным техническим достижением последних лет
является получение сверхпрочных волокон, разрывная прочность
которых в 2—3 раза'превышает прочность всех известных
природных и химических волокон (см. гл. 12). Широкое использование
этих волокон, обладающих, как правило, не только очень высокой
прочностью, но и термостойкостью, для производства корда,
армирующих материалов и других изделий должно резко повысить
эффективность их применения в народном хозяйстве.
Технический прогресс в производстве синтетических волокон
развивается главным образом в следующих направлениях:
разрабатываются новые способы получения мономеров из более
дешевых и доступных видов сырья для производства известных
типов волокнообразующих полимеров;
подвергаются дальнейшей рационализации существующие
методы синтеза мономеров (наиболее значительные результаты . в
этом направлении в последние годы были достигнуты в области
синтеза капролактама и акрилонитрила) ;
разрабатываются и уже осуществлены в производственных
условиях методы получения различных синтетических волокон, резко
уменьшающих капиталовложения и повышающих
производительность труда;
внедряются в производство непрерывные процессы синтеза гете-
роцепных полимеров (в первую очередь полиамидов и полиэфиров)
и формования из них волокон;
при синтезе карбоцепных полимеров осуществляется
полимеризация мономеров в растворах и формование волокна (например,
полиакрилонитрильного) из этих растворов;
начинают применять новые, эффективные методы формования
волокон из высококонцентрированных гелей (поливинилхлорид-
ных и поливинилспиртовых), суспензий и латексов, а также из
полимеров, находящихся в термопластичном состоянии, при высоких
давлениях.
1.2. Перспективы развития производства синтетических волокон
13
Таблица 1.2. Основные методы модификации свойств
синтетических волокон
Метод модификации
Результаты
Волокна, для которых метол
может быть использован
Синтез сополимера
Введение боковых групп
в элементарное звено
макромолекулы
полимера
Синтез привитых и блок-
сополимеров или, что в
большинстве случаев
более целесообразно,
прививка к макромолекулам
готовых волокон
Образование поперечных
химических связей
между макромолекулами
Введение новых
функциональных групп
Улучшение структуры
волокон (изменение
соотношения между
кристаллической и аморфной
фракциями) путем
вытягивания, термообработки и
других воздействий
Введение небольших
добавок термо- и светостаби-
лизаторов при синтезе
полимера
Увеличение молекулярного
веса полимера
Формование нолокон из
смеси полимеров (из
раствора или из
расплава)
Изготовление изделий из
смесей синтетических и
природных или
искусственных волокон
Нарушение
регулярности строения
полимера. Улучшение
растворимости полимера
и повышение
эластичности и накрашивае-
мости волокна,
снижение температуры
плавления и
размягчения
Снижение степени
кристалличности,
замедление процесса
кристаллизации волокна
Повышение
растворимости полимера.
Изменение окрашивае-
мости, гидрофильно-
сти, термостойкости
и других свойств
волокна
Уменьшение текучести
и растворимости,
повышение
термостойкости волокна
Повышение гидрофиль-
ности, улучшение ок-
рашиваемости н
светостойкости волокна
Изменение прочности,
текучести, , модуля,
термостойкости и
эластичности волокна
Повышение термо- и
светостойкости
волокна
Увеличение разрывной
и усталостной
прочности волокна
Изменение хемостойко-
сти, гидрофильности,
эластичности и
других свойств волокна
Изменение
гидрофильности изделий, их
эластичности и
устойчивости к деформациям
Карбоцепные (например,
полиакрилонитриль-
ные) и гетероцепные
Полиэфирные
Карбоцепиые (полиак-
рилонитрильные и по-
ливинилспиртовые) и
гетероцепные
Карбоцепные (полиоле-
финовые, поливииил-
спиртовые) и
гетероцепные
Полиамидные н поли-
акрилоиитрильные
Большинство карбо- и
гетероцепных
Гетероцепные
(полиамидные) и
карбоцепные
Полиамидные и
полиэфирные
Карбоцепные и
гетероцепные
Карбо- и гетероцепиые
14 Глава t. Общие сведения о производстве синтетических волокон
Продолжаются работы по изысканию новых методов
модификации свойств полимеров и получаемых из них волокон, в частности
для повышения термо-и хемостойкости, гидрофильное™, а также
по улучшению окрашиваемое™, изменению степени
кристалличности и эластичности волокон и т. д. Для решения этих задач,
часто определяющих масштабы производства отдельных видов
синтетических волокон и их конкурентоспособность с другими
волокнами, могут быть использованы все известные методы
модификации свойств полимерных материалов, а также некоторые
дополнительные способы, применимые только для изменения
свойств волокон и получаемых из них изделий.
Основные методы, которые могут быть использованы для
модификации свойств синтетических волокон, приведены в табл. 1.2.
Вопрос о возможности и целесообразности применения этих
методов более детально рассмотрен в последующих главах.
1.3. КЛАССИФИКАЦИЯ СИНТЕТИЧЕСКИХ ВОЛОКОН
И ОСОБЕННОСТИ ИХ ПРОИЗВОДСТВА
1.3.1. Классификация синтетических волокон
В зависимости от особенностей химического строения
макромолекул синтетические волокна могут быть подразделены на гете-
роцепные и карбоцепные.
Гетероцепные волокна. К этому классу относятся волокна,
макромолекулы которых кроме атомов углерода содержат в основной
цепи атомы кислорода, азота или других элементов. Гетероцепные
волокна подразделяют на полиамидные, полиэфирные и полиуре-
тановые.
Полиамидные волокна. В эту группу включены все виды
волокон, полученных из различных полиамидов. Такими волокнами
являются поликапроамидные, полигексаметиленадипамидные,
полиэнантоамидные, полиундеканамидные и др.
Поликапроамидные волокна формуют из поликапроамида —
полимера, синтезированного из капролактама. Эти волокна
выпускаются в разных странах под различными названиями, например
«капрон», «дедерон», «силон», «найлон 6».
Полигексаметиленадипамидные волокна (анид, найлон 6,6 и
др.) вырабатывают из полимера, полученного поликонденсацией
гексаметилендиамина и адипиновой кислоты.
Полиэнантоамидные волокна (найлон 7) формуют из полиэнан-
тоамида — полимера, полученного поликонденсацией ш-аминоэнан-
товой кислоты.
Полиундеканамидные волокна (ундекан, найлон 11, киана),
вырабатываемые из полиундеканамида — полиамида,
синтезированного из ш-аминоундекановой кислоты.
1.3. Классификация и особенности производства синтетических волокон 15
Полиэфирные волокна. Эта группа волокон включает
волокна, вырабатываемые из различных продуктов
поликонденсации терефталевой кислоты или ее эфиров с гликолями (волокно
лавсан, терилен, дакрон и др.).
Полиуретановые волокна. К полиуретановым относят
высокоэластичные волокна из полимеров, полученных
полимеризацией диизоцианатов с полиэфирами. Эти волокна известны под
общим названием волокна «спандекс».
Гетероцепные волокна — основной класс синтетических
волокон, получивший наиболее широкое распространение. В
промышленных масштабах вырабатываются в основном два вида гетеро-
цепных волокон — полиамидные и полиэфирные — и в небольших
количествах высокоэластичное полиуретановое волокно.
Наибольшее распространение в предыдущие годы получили полиамидные
волокна. Это объяснялось присущими им ценными свойствами,
широкой сырьевой базой для их производства и в значительной
степени тем, что методы получения исходных материалов* а также
процессы формования и последующей обработки разработаны для
полиамидных волокон раньше и более детально, чем для других
гетероцепных волокон.
В последние годы наиболее быстрыми темпами развивается
производство полиэфирных волокон. Масштабы производства этих
волокон в отдельных странах уже превысили объем производства
полиамидных волокон, и эта тенденция в ближайшие годы будет,
по-видимому, все более усиливаться. Резкое увеличение
производства полиэфирных волокон объясняется значительной
рационализацией технологического процесса получения полимеров и волокон
из них, а главным образом — специфически ценными свойствами
полиэфирных волокон, определившими целесообразность их
применения для изготовления широкого ассортимента товаров народного
потребления и изделий технического назначения.
Разработаны также методы получения синтетических волокон
из других гетероцепных полимеров, в частности поликарбонатов
и полиформальдегида.
Карбоцепные волокна. К этому классу синтетических волокон
относят волокна, макромолекулы которых содержат в основной
цепи только атомы углерода.
Вырабатываемые карбоцепные волокна подразделяются на по-
лиакрилонитрильные, поливинилхлоридные, поливинилспиртовые,
полиолефиновые и фторсодержащие.
Полиакрилонитрильные волокна (нитрон, орлон и др.)
получают из полимеров и сополимеров нитрила акриловой кислоты.
Поливинилхлоридные волокна вырабатывают из полимеров и
сополимеров винилхлорида (волокна типа ровиль) и винилиденхло-
рида (волокно совиден, саран и др.), а также из хлорированного
поливинилхлорида (волокно хлорин).
16 Глава 1. Общие сведения о производстве синтетических волокон
Поливинилспиртовые, полиолефиновые и фторсодержащие
волокна получают соответственно из поливинилового спирта (волокно
винол, куралон), полиолефинов (полиэтиленовые и
полипропиленовые волокна) и фторсодержащих полимеров (волокно тефлон,
фторлон).
Особую группу синтетических волокон, получающих в послед«
ние годы все более широкое применение, составляют
термостойкие, жаростойкие и сверхпрочные волокна, вырабатываемые из
различных полимеров.
1.3.2. Особенности производства
синтетических волокон
Технологический процесс получения синтетических волокон
существенно отличается от процесса производства основных видов
искусственных волокон. В то же время отдельные стадии процесса
получения гетероцепных волокон значительно отличаются от
таковых при производстве большинства карбоцепных волокон.
Производство синтетических волокон характеризуется
следующими особенностями:
при формовании волокон любым способом (из раствора,
расплава илн из размягченного полимера) полимер химически не
изменяется; получаемое волокно (кроме волокон из поливинилового
спирта и некоторых термостойких волокон) по химическому
составу идентично с исходным полимером;
для повышения комплекса механических свойств синтетические
волокна значительно вытягивают (на 400—2000%); вытягивание
волокна проводится при формовании на прядильной машине или,
преимущественно, в процессе последующей обработки при
нормальной или повышенной температуре;
синтетические волокна (кроме волокна тефлон)
термопластичны, поэтому в большинстве случаев они подвергаются
терморелаксации (термофиксации), в результате чего повышается
равномерность структуры волокна, увеличивается его удлинение и
значительно снижается усадка при повышенных температурах,
в частности в горячей воде;
варьируя условия формования, вытягивания и термофиксации,
в широких пределах изменяют механические свойства получаемых
волокон.
Основные различия в методах производства гетеро- и
карбоцепных волокон сводятся к следующему:
так как волокнообразующие гетероцепные полимеры плавятся
без разложения, волокна, как правило, формуют из расплава
(кроме полиуретановых и большинства термостойких волокон,
температура плавления которых выше температуры разложения);
карбоцепные волокна формуют в основном из растворов или
1.3. Классификация и особенности производства синтетических волокон 17
из высококонцентрированных гелей*, и только волокна из поли-
олефинов получают из расплава;
скорость формования гетероцепных волокон из расплава и
соответственно производительность одного прядильного места
значительно выше, чем при получении карбоцепных волокон такой же
толщины из растворов;
гетероцепные комплексные нити вытягивают при нормальной
температуре (полиамидные нити) или при несколько повышенных
температурах (полиэфирные нити) в процессе крутки на крутильно-
вытяжных машинах, а при производстве штапельного волокна —
между вытягивающими вальцами в отдельных секциях прядильно-
вытяжного агрегата; карбоцепные волокна, как правило,
вытягивают при температурах выше 100°С.
Основные особенности производства гетеро- и карбоцепных
синтетических волокон могут быть суммированы следующим
образом:
Гетероцепные волокна Карбоцепные волокна
Исходное сырье Фенол, бензол, акрило- Этилен, ацетилен, метан,
нитрил, ацетилен, ци- пропилен
клогексан, га-кс нлол
Метод получения по- Поликонденсация, пре- Цепная радикальная
полимера вращение циклов в ли- лнмеризация (кроме
йй ф)
Растворители,
применяемые при
получении прядильного
ТІЯРТПППЯ
Udt» i oKJyd
Метод формования
Скорость
формования, м/мин
мокрый способ
сухой способ
из расплава
Вытягивание
степень, %
условия
неиныи полимер
Не применяются *
Из расплавл *
—
600—4000
300—400
При нормальной или по-
сиитеза полиолефинов)
Ацетон, вода, диметил-
формамид
Из раствора, гелей .н
расплава
20—60
100—300
500-800
200—2000
При повышенной темпе-
р р
вышенной температуре ратуре на прядильной
на крутильно-вытяж- машине или иа агре-
ной машине или между гате для производства
вальцами на агрегате штапельного волокна
для производства
штапельного волокна
• Кроме термостойких волокон.
* По-видимому, в дальнейшем при подаче высоковязкого полимера,
находящегося в термопластичном состоянии, к прядильному насосику, а затем
к фильере под высоким давлением 100—200 кгс/см2 A07 —2-Ю7 Па) и более
могут быть получены волокна из расплава или термопластичного состонния
большинства карбоцепиых полимеров,
Глава 2
ПРОИЗВОДСТВО ПОЛИАМИДНЫХ ВОЛОКОН
Полиамидными называются синтетические волокна, получаемые
из полимеров, у которых отдельные звенья макромолекулы
соединены между собой амидными группами —N—С—
I II
H О
HN(CH2)^COHN(CH2)^COHN
или
Соотношение между числом метиленовых и амидных групп
в макромолекулах разных полиамидов существенно различается.
Как уже отмечалось выше, полиамидные волокна — это первый
класс синтетических волокон, производство которых получило
широкое промышленное развитие.
Разработка методов получения синтетических гетероцепных полимеров,
пригодных для формования волокон, была начата коллективом исследователей
во главе с Карозерсом в 1929 г. в Вильмингтоне (США).
В 1934 г. Карозерс синтезировал полиамид из 9-аминононановой кислоты и
впервые в мире получил из него волокно. В 1935 г. также в лабораторных
условиях было сформовано волокно из полиамида, образующегося в результате
поликонденсации гексаметилендиамина и адипиновой кислоты, получившее
впоследствии название «найлон 6,6» *. Потребовалось еще три года для разработки
приемлемого в технологическом и аппаратурном отношении метода получения
полимера и волокна найлон 6,6 в производственных условиях. В 1938 г. было
начато строительство первого завода по производству волокна найлон 6,6 в Си-
форде (США), который в 1939 г. был введен в эксплуатацию. В последующие
годы производство полиамидного волокна типа найлон 6,6 получило широкое
развитие как в США, так и в других странах (Англии, Италии, Японии,
Франции).
* В ряде стран (США, Англия) все виды синтетических волокнообразую-
щих полиамидов обозначаются термином «найлон». Химический состав
полиамида характеризуется числом (пишется после слова «найлон»), равным числу
атомов углерода в молекуле мономера. Например, найлон 6 обозначает, что
полиамид синтезирован из є-аминокапроновой кислоты NH2(CH2bCOOH или из
ее лактама HNfCHibCO; найлон 11—из со-аминоундекановой кислоты
NH2(CH2)ioCOOH. Два числа указывают на то, что полиамид получен из
диамина и дикарбоновой кислоты, причем первое число обозначает число атомов
углерода в молекуле диамина, второе — число атомов углерода в дикарбоновой
кислоте. Например, найлон 6,6 получают из гексаметилендиамина ЭДНЫСНгЭеМНг
и адипиновой кислоты НООС(СН2),|СООН; найлон 6,10 — из
гексаметилендиамина и себациновой кислоты НООС(СНг)бСООН и т. д.
В большинстве случаев полиамидным волокнам присваиваются различные
торговые названия.
2.1. Синтез поликапроамида 19
В 1938 г. в Германии Шлак впервые получил полиамид нагреванием
капролактама с хлористоводородной солью аминокапроновой кислоты, несмотря на
то, что в работе Карозерса и Берхета [7], опубликованной в 1930 г.,
указывалось: «полимеризация капролактама невозможна ни в присутствии, ни в
отсутствие катализаторов». В 1939 г. на заводе полиамидного волокна в Лихтенберге
(Берлин) было начато опытное производство полиамидного волокна перлон из
поликапроамида. Независимо от исследований немецких химиков, которые до
1945 г. не были известны, работы по получению полиамида из капролактама и
формованию волокна из получаемого полимера (поликапроамида) проводились
в Советском Союзе (И. Л. Кнунянц, 3. А. Роговин, Ю. А. Рымашевская,
Э. В. Хаит) и в Чехословакии (О. Вихтерле). В 1945—1950 гг. в Советском
Союзе были проведены систематические исследования основных закономерностей
синтеза полиамидов, получаемых как путем полимеризации капролактама
(И. Л. Кнунянц, 3. А. Роговин, Ю. А. Рымашевская, Э. В. Хаит, А. А. Стрепи-
хеев), так и методом поликонденсации (В. В. Коршак и сотр.).
В настоящее время производство полиамидных волокон
является многотоннажным и непрерывно развивается. Полиамидные
волокна используются во все больших количествах как для
изготовления различных изделий народного потребления, так и для
производства разнообразных материалов для технических целей.
Рост мирового производства полиамидных волокон
характеризуется следующими данными:
Годы 1940 1945 1950 1955 1960 1966 1970 1972
Объем производства,
тыс. т 2.5 20 62 175 392 1214 1660 2424
Как видно из этих цифр, производство полиамидных волокон
развивалось весьма интенсивно. В 1972 г. выпуск полиамидных
волокон увеличился по сравнению с выпуском в 1960 г. почти
в 6,2 раза. '
В последние годы темпы развития производства полиамидных
волокон несколько снизились, однако согласно прогнозам {2]
ожидается, что к 1980 г. мировое производство полиамидных волокон
достигнет 4 млн. т. Увеличилось число видов полиамидных
волокон, вырабатываемых как в промышленных масштабах, так и
особенно в опытных производствах.
В промышленных масштабах выпускаются следующие
полиамидные волокна: капрон (СССР), дедерон (ГДР), перлон (ФРГ),
силон (ЧССР), найлон 6 (США), анид (СССР), найлон 6,6 (США,
Англия, Италия), найлон 6,10 (США), найлон 11 (США).
В небольшом масштабе вырабатываются волокна найлон 7,
найлон 12 (ФРГ, Япония) из продукта полимеризации додекалак-
тама, получаемого из нефтехимического сырья, поли- ?-амидные
(Япония), формуемые мокрым способом из растворов поли-р-ами-
дов.
Особое место среди полиамидных волокон занимают так
называемые термостойкие полиамидные волокна, получаемые из
термостойких полимеров с температурой плавления выше 3Q0°C.
Методы синтеза этих полиамидов (преимущественно путем
20 Глава 2. Производство полиамидных волокон
поликонденсации ароматических дикарбоновых кислот и
диаминов), и особенно формование из них волокон, существенно
отличаются от методов получения обычных полиамидных волокон.
Условия получения и свойства термостойких полиамидных
волокон рассматриваются в гл. 11.
Наиболее широкое распространение получили волокна типа
капрон и найлон 6,6, объем производства которых составляет
свыше 95% общего количества вырабатываемых полиамидных
волокон.
Среди новых заводов полиамидных волокон все большее
значение приобретают заводы, вырабатывающие волокно из поликап-
роамида.
Даже в США, где был разработан и впервые освоен в
промышленном масштабе метод получения волокна найлон 6,6, в
последние годы введены в эксплуатацию заводы, вырабатывающие
волокно типа найлон 6. Это свидетельствует о наметившейся
тенденции к расширению производства волокна из поликапроамида.
Если в общем мировом производстве полиамидных волокон на
долю волокон типа найлон 6 в 1950 г. приходилось менее 30%, то
в 1970 г. удельный вес этих волокон превысил 45%- В США
волокна типа найлон 6 до 1953 г. вообще не производились, а
в 1970 г. их выработка достигла 37% общего выпуска
полиамидных волокон.
В 1967 г. полиамидные волокна производились в 45 странах
на 207 заводах [3]. В последующие годы число заводов по
производству полиамидных волокон продолжало увеличиваться.
Все полиамидные волокна, кроме термостойких и волокон из
?-полиамидов, формуются из расплава. Технологический процесс
производства полиамидных волокон включает следующие три
основные стадии: 1) синтез полимера; 2) формование волокна и 3)
вытягивание и последующую обработку.
В зависимости от химического строения и свойств полиамидов
число и последовательность операций на каждой стадии
технологического процесса могут несколько изменяться.
Примерные схемы технологического процесса получения
текстильных нитей капрон и анид при периодическом способе
производства приведены в табл. 2.1.
Как правило, полиамиды синтезируются на заводах
синтетических волокон, а не на химических заводах, где вырабатываются
соответствующие мономеры.
Полиамидные волокна могут быть получены и по непрерывной
схеме. При реализации этого технически прогрессивного метода
значительно упрощается технологический процесс и его
аппаратурное оформление (см. разд. 2.9.2.).
К настоящему времени имеется уже значительное число
разработанных методов синтеза полиамидов, которые подробно
описаны в периодической литературе и монографиях [4, с. 45—99;
2.1. Синтез поликапроамида
21
Таблица 2.1. Примерные схемы технологического процесса
получения текстильных нитей капрон и анид при периодическом
способе производства
Капрон
Анид
Получение полиамида
1. Плавление кайролактама
2. Полимеризация капролактама
1. Приготовление раствора соли АГ
2. Поликондеисация соли АГ
3. Получение ленты или жилки
4. Дробление ленты (получение крошки)
5. Экстракция низкомолекулярных
фракций из крошки
6. Отжим крошки
7. Сушка крошки
5. Подсушивание крошки
Формование волокна из расплава
Вытягивание и отделка нитн
1. Вытягивание и одновременная крутка
2. Окончательная крутка
3. Обработка горячей водой для
удаления низкомолекулярных фракций и
фиксации волокна
4. Отжим
5. Сушка
6. Перемотка
3. Прогрев волокна (термофиксация)
4. Сушка
5. Перемотка
Примечание. Не все нз приведенных операций необходимы. Например, процесс
окончательной крутки при производстве текстильной нити может быть исключен,
необходимая крутка достигается в процессе кручении с вытягиванием.
5, с. 267—284]. Ниже приводятся основные данные об условиях
получения важнейших типов полиамидов, а также краткое
описание новых методов синтеза, представляющих практический ин
терес.
2.1. СИНТЕЗ ПОЛИКАПРОАМИДА
Исходным материалом для синтеза поликапроамида (капрона)
является лактам є-аминокапроновой кислоты — капролактам.
Доступность исходного сырья, простота методов производства
капролактама, высокий выход продукта определяют большие
масштабы производства этого мономера, а тем самым поликапроамида
и капронового волокна.
2.1.1. Получение капролактама
Сырьем для получения капролактама могут служить фенол,
бензол или циклогексан, а также анилин и толуол. Капролактам
может быть синтезирован также из ацетилена через динитрил
22 Глава 2. Производство полиамидных волокон
адипиновой кислоты или из фурфурола через тетрагидрофуран
и адиподинитрил [6]. Эти методы не получили пока
промышленного применения и поэтому в данной книге не рассматриваются.
Наиболее освоено в производственных условиях получение кап-
ролактама из фенола и бензола. Однако использование других
исходных веществ имеет большое значение для дальнейшего,
более широкого развития производства поликапроамидных волокон.
Капролактам из фенола. Гидрированием фенол превращают в
циклогексанол:
ОН
Гидрирование фенола осуществляется в присутствии
катализатора (никеля) при давлении 15—20 кгс/см2 A,5- 10б —2- 10б Па)
и температуре 135—150°С; выход циклогексанола достигает 95%
от теоретического.
Полученный циклогексанол подвергают дегидрированию В
результате образуется кетон — циклогексанон:
„СН2
хснон
ч /СНг
сн2
Дегидрирование проводят при 400—450 °С и нормальном
давлении на цинк-железном катализаторе (90: 10). В качестве
катализаторов применяют также сплав цинка и хрома (при 360—380 °С)
или сплавы магния (при 240—300°С). Конверсия циклогексанона
за один проход составляет 70—80%- Полученную смесь
циклогексанона и циклогексанола разделяют дистилляцией. Выход
циклогексанона достигает 92—94% от теоретического:
В результате взаимодействия с гидроксиламином
циклогексанон превращается в оксим циклогексанона (циклогексаноноксим):
JCH2 СН2
Н2Сг ХСО NH2QH H2C/^C=
2\ /
СН2 СН2
Оксимирование производят сернокислым гидроксиламином *
* Сернокислый гидроксиламин получают взаимодействием нитрита аммония
с раствором бисульфита аммония и сернистым ангидридом. На первой стадии
реакции образуется дисульфонат гидроксиламина по схеме
NH4NO2 + NH4HSO3 + SO2 —* N(OH)(SO3NH,J
который при повышенной температуре гидролизуется, образуя сульфат
гидроксиламина (HONHhSC>
2.1. Синтез поликапроамида 23
при 20 °С. Выделяющаяся при реакции серная кислота
нейтрализуется аммиаком, вследствие чего температура реакционной массы
повышается до 90 °С. Реакция протекает без катализаторов, выход
оксима циклогексанона составляет 90—93% от теоретического.
При действии на оксим циклогексанона концентрированной
серной кислотой (96—98%-ной) или, что лучше, олеумом
(содержащим 18—25% SO3) происходит перегруппировка (изомеризация)
оксима циклогексанона в лактам є-аминокапроновой кислоты (изо-
оксим циклогексанона) :
CH2-CO
Г \
NH
Изомеризация протекает очень быстро. Эта реакция сильно
экзотермична. Чтобы температура не поднималась выше 80—85 °С,
реакционную массу необходимо охлаждать. В производственных
условиях процесс изомеризации осуществляется непрерывно.
По окончании изомеризации серная кислота, находящаяся
в реакционной смеси, нейтрализуется водным раствором аммиака
или едкого натра при охлаждении. Реакционная масса при этом
разделяется на два слоя: нижний слой — насыщенный раствор
солей, и верхний слой, имеющий примерно следующий состав: 63%
лактама, 33% воды, 4% солей. Лактам, находящийся в верхнем
слое, сливается, а затем очищается от примесей повторной
ректификацией под вакуумом.
При работе по описанной схеме из 1 кг фенола получается
0,9—0,95 кг капролактама.
В последнее время предложен новый способ получения
капролактама из циклогексанона, минуя стадию оксимирования и
последующей перегруппировки [7]. На первой стадии циклогексанон
окисляют перкислотами, в частности надуксусной кислотой, при
30—40 °С. При этом образуется лактон є-оксикапроновой кислоты
(капролактон):
СН2 снзсг СН2—СО
Н2С^ ХСО ^оон Н2Сг \
і І *- I Ю + СНзСООН
СН2 СН2—СН2
На второй стадии капролактон амидируется аммиаком в
присутствии катализатора (никеля, рения или палладия) на угле при
175—220 °С и давлении 10—35 кгс/см2 A • 106 —3,5-106 Па) с
образованием капролактама:
СН2—СО СН2—СО
2| ^О -^- 2| ^NH+H,0
Н2СЛч / Н2С\ /
24
Глава 2. Производство полиамидных волокон
Капролактам из циклогексана или бензола. Метод получения
капролактама из фенола имеет ряд недостатков: высокая
стоимость фенола, многостадийность процесса, большой расход
неорганических реагентов и др. Эти. недостатки в значительной степени
устраняются при применении других методов, основанных иа
использовании для синтеза капролактама циклогексана,
выделенного из нефтяного сырья или полученного гидрированием бензола.
Циклогексан различными способами перерабатывается в циклогек-
саноноксим и затем в капролактам. К таким методам относятся {8]:
1) окисление циклогексана кислородом воздуха
.^н2
Н2
N
Н2С4. /CH2
сн2
H2i
сн2
сн2
N
Н2
-н2
сн2
NOH H:
NH2OH
>-
\
н2с4 /
СНо СН;
NH
2) нитрование циклогексана
СН2
СН2
сн2
N:=noh
2с N:
Н2СХ. yQiW}
HN(CH2MCO
12V-' %>П2 HNO3
>-
jH2 ch2 сн2
3) фотохимическое нйтрозирование циклогексана
:-Н2 ^
NOCl + HCi + Av НгС^ N:=NOH • HCI ,
> і і —> hn(ch2Mco
2 vch2 2 !
Первый метод — окисление циклогексана кислородом
воздуха — приводит к получению смеси циклогексанола и циклогексанона в
соотношении 1:1. Жидкофазное окисление циклогексана осуществляется при 120—
130 °С и давлении 15—20 кгс/см2 A,5 • 106 — 2 • 106 Па) в присутствии
катализатора— нафтената кобальта @,02—0,05% от массы циклогексана) или стеарата
марганца. Превращение циклогексанона за одну ступень составляет около 5—10%,
поэтому окисление проводится, как правило, в многоступенчатом реакторе.
Суммарный выход циклогексанола и циклогексанона составляет 65—75%.
Второй метод — нитрование циклогексана — дает возможность
получить наряду с нитроциклогексаном адипиновую кислоту @,4 т на 1 т капро-
2.1. Синтез поликапроамида 25
лактама). Суммарный выход нитроциклогексана и адипиновой кислоты
составляет около 80% от массы циклогексана.
Нитрование производится 95%-ной HNO3 при 120—125 °С и давлении
3—5 кгс/см2 C • 105—5 • 105 Па).
Полученный нитроциклогексан восстанавливают в циклогексаноноксим
водородом в присутствии катализатора (Zn, Сг) при 140—160 °С под давлением
100—120 кгс/см2 A07—1,2 • 107 Па). Выход составляет 55—60%. Значительно
более высокий выход циклогексаноноксима (85—90% от теоретического)
достигается, если процесс восстановления нитроциклогексана происходит в
присутствии жидкого аммиака и катализатора (металлическая медь) при 80—130 °С и
давлении 150—200 кгс/см2 A,5 • 107—2 • 107 Па).
Однако этот метод может быть экономичным только при условии
одновременного использования образующейся адипиновой кислоты для получения
пластификаторов, синтеза полиамида типа найлон 6,6 или для других целей.
ТреТИЙ МеТОД — фоТОХИМИЧеСКОе НИТрОЗирОВаНИе Ц И KJI О-
гексана [9] — приводит непосредственно к образованию хлоргидрата
циклогексаноноксима, из которого получают оксим и затем капролактам.
В качестве источников света для инициирования реакции применяются
электрические лампы, в частности люминесцентные. Выход циклогексаионоксима
составляет 80—90% , от теоретического. При наличии дешевой электроэнергии
использование этого метода может представить практический интерес.
По этой схеме с 1963 г. работает завод в Японии (фирма «Токус-Кадон»)
производительностью 10 тыс. т капролактама в год. Источник облучения —
ртутно-кварцевые лампы мощностью 10 кВ. Расход электроэнергии на 1 кг
получаемого капролактама составляет около 5 кВт • ч A,8 • 104 кДж). При
применении более мощных источников облучения B0 кВт) расход электроэнергии на 1 кг
продукта снизился до 4,4 кВт • ч A,6 • 104 кДж) [10].
Капролактам из бензола через аиилии. Капролактам можно получить также
из анилина. Анилин гидрируют над никелем в присутствии паров воды при
200—250 °С. В результате этой реакции и последующего отщепления аммиака
при действии воды получается смесь продуктов примерно следующего состава
(в %):
Циклогексанол 60—65
Анилин 20
Циклогексиламин 5
Примеси 5—10
Циклогексанол отделяют дистилляцией. Побочные продукты подвергают
повторной обработке для получения капролактама по указанной выше схеме.
Эффективность этого способа может быть значительно повышена прямым
окислением циклогексяламина в циклогексаноноксим перекисью водорода в
присутствии катализаторов (соли урановой, молибденовой или вольфрамовой кислот):
?Н2 ?Н2
НгО'NjLHNH, н2О2 Н2Г
ХСН2 г 2 Gib
Выход оксима циклогексанона составляет 94—95%. Основным недостатком
этого способа при реализации его в промышленности является сравнительно
высокая стоимость перекиси водорода.
При получении капролактама из бензола или циклогексана описанными выше
методами расширяется сырьевая база, снижается себестоимость капролактама и
получаемого из него волокна. Благодаря этому повышается
конкурентоспособность поликапроамидного волокна с другими видами синтетических, и в
частности полиамидных волокон.
Капролактам из толуола. Этот метод, разработанный и реализованный в
промышленном масштабе фи мой «Снна-Вискоза» (Италия), представляет
26 Глава 2. Производство полиамидных волокон
существенный интерес в отношении дополнительного расширения сырьевой базы
для получения капролактаме.
Толуол окисляют до бензойной кислоты кислородом воздуха в присутствии
солей кобальта при 130—140°С и давлении 4—5 кгс/см2 D- 105—5 • 105 Па).
Гидрированием бензойной кислоты при 120—130 °С в присутствии палладия, иа-
иесеиного иа уголь, получают циклогексанкарбоиовую (гексагидробензойную)
кислоту:
СНз о2 Г^^СООН н2 — -С00Н
При взаимодействии циклогексанкарбоновой кислоты с нитрозилсерной
кислотой происходит декарбоксилирование и в результате последующих обработок
образуется капролактам по схеме
С00Н O=N-OSO3H
Выход капролактама составляет 85% от теоретического.
Удельный вес различных методов получения капролактама
в мировом производстве характеризуется следующими данными
(в%):
Исходное сырье 1967 г. 1970 г.
Фенол 37,4 31,4
Циклогексан 55,0 62,1
в том числе
окисление 52,2 58,6
фотохимическое нитрознрование . . 2,8 3,5
Толуол . . . . 1.7 1,1
Другие виды сырья 5,9 5,3
Как видно из приведенных данных, основным видом сырья для
производства капролактама является циклогексан,
перерабатываемый по описанной выше окислительной схеме.
Представляют интерес опубликованные данные о себестоимости
капролактама, получаемого из различных видов сырья. Если
принять себестоимость капролактама, полученного из фенола, за 100%,
то себестоимость капролактама из анилина составляет 95%, из
толуола — 70%, из циклогексана по окислительной схеме — 65% и
по схеме фотохимического нитрозирования — 53%. Эти данные
показывают, что наиболее экономичными являются методы получения
капролактама из циклогексана.
2.1.2. Свойства капролактама
Капролактам плавится при 69—71 °С, кипит при нормальном
давлении при 258 °С. Характерной особенностью его является
растворимость почти во всех растворителях как полярных, так и ле-
2.1. Синтез поликапроамида 27
полярных. Капролактам растворяется в воде и в бензоле, в ацетоне
и в петролейном эфире, в растворах кислот и солей.
Капролактам, применяемый для производства волокна, должен
удовлетворять ряду требований; основным из них является
отсутствие примесей.
При анализе капролактама определяют его температуру
плавления, растворимость в воде (проба должна полностью
растворяться в воде, образуя прозрачный раствор) и наличие примесей,
обладающих восстановительной способностью.
Капролактам, применяемый для производства синтетического
волокна, должен отвечать следующим требованиям:
Перманганатное число, с, не менее 2000
Содержание летучих оснований, мл 0,1 н.
раствора H2SO4 на 100 г капролактама,
не более 3
Температура кристаллизации, °С, не ниже . . 68,5
Влажность, %, не более 0,2
Прозрачность 25%-ного водного раствора,
см, не менее 100
Механические примеси, %, не более 0,0001
Перманганатное число характеризует содержание в
капролактаме примесей, обладающих восстановительной способностью *.
Существенное влияние на перманганатное число оказывают
непредельные соединения. Например, после гидрирования
капролактама в течение 1 ч при 70—75 °С в присутствии никелевого
катализатора содержание этих примесей снижается в 2—3 раза.
Соответственно резко увеличивается перманганатное число (с 120 до
30 000 с), а окраска 50%-ного водного раствора лактама
уменьшается в 5—10 раз [11].
Большое значение имеет также содержание в капролактаме
анилина и нитробензола. Например, добавление 0,001% анилина от
массы капролактама снижает перманганатное число с 8000 до
400 с, одновременно увеличивается количество летучих оснований
[12]. Добавление такого же количества нитробензола резко
ухудшает прозрачность водных растворов капролактама.
Следовательно, эти примеси должны быть полностью удалены при
получении капролактама.
Влияние количества и характера примесей на процесс
полимеризации и свойства получаемого полимера еще недостаточно
изучено. Дальнейшие исследования в этом направлении
представляют большой интерес.
* Методы анализа капролактама — см. Контроль производства химических
волокон. Справочное пособие. Под ред. А. Б. Пакшвера и А А Конкина M
«Химия», 1967. 607 с.
28 ' Глава 2. Производства полиамидных волокон
2.1.3. Полимеризация капролактама
Полимеризация капролактама — циклического соединения —
является наиболее характерным примером нового типа реакции
синтеза полимеров — превращения циклов в линейные полимеры.
Эта реакция была детально исследована советскими учеными A3,
14, 15].
Реакция превращения циклов в линейные полимеры подробно
рассматривается в курсе химии высокомолекулярных соединений
[16, с. 182—204, 17]. Поэтому мы ограничимся только кратким
изложением основных положений, знание которых необходимо для
понимания механизма реакции и установления параметров
технологического процесса получения поликапролактама.
Основные закономерности процесса получения полимеров путем
полимеризации циклов существенно отличаются .от
закономерностей реакции цепной полимеризации и поликонденсации
линейных мономеров. В отличие от других методов при полимеризации
гетероциклов химические связи в молекуле мономера вновь не
образуются и не исчезают, а имеет место лишь превращение
химических внутримолекулярных циклических связей в линейные, т. е.
происходит раскрытие кольца. Возможность превращения
гетероциклов в линейные полимеры определяется термодинамической
устойчивостью и напряженностью циклов. Последняя изменяется
в зависимости от ряда факторов, основными из которых являются
число звеньев и характер функциональных групп в цикле. Чем
больше напряженность цикла, тем меньше его термодинамическая
устойчивость и тем легче и полнее цикл превращается в линейный полимер.
Условия получения полимера определяются в большинстве
случаев числом звеньев в цикле {18]. Четырехчленные и пятичленные
лактамы, гетероциклы которых не напряжены, не полимеризуются
при использовании воды в качестве инициатора.
Процесс полимеризации таких лактамов протекает в
присутствии более активных катализаторов, чем те, которые используются
при полимеризации семичленных лактамов (см. разд. 2.6), и
осуществляется по механизму анионной полимеризации.
Гетероциклы с восемью и более членами в цикле при
соответствующих условиях проведения процесса полностью превращаются
в полимеры. Семичленные циклы, к которым относится
капролактам, также полимеризуются, ^однако процесс полимеризации идет
не до конца, и в полученном полимере всегда содержится
некоторое количество мономера. Это усложняет отдельные стадии
технологического процесса получения поликапроамида и капронового
волокна *.
* Эти закономерности относятся в основном к гетероциклам, содержащим
метиленовые группы. При введении в молекулу циклического соединения
функциональных групп (ОН, ОСНз и др.) может значительно измениться
устойчивость цикла и способность его к полимеризации.
2.1. Синтез паликапраамида 29
При одном и том же числе атомов в цикле возможность
полимеризации мономера и количественное соотношение полимера и
мономера в полученном продукте в значительной степени зависят
от наличия и характера боковых групп в цикле. Эти группы
понижают напряженность цикла, изменяют энтропию системы и тем
самым уменьшают способность мономера к полимеризации.
Например [19], при введении боковых метильных групп в молекулу кап-
ролактама (С-метилкапролактам) скорость полимеризации
понижается и одновременно увеличивается содержание мономера в
полученном полиамиде. Необходимо, однако, отметить, что для легко
полимеризующихся циклов (8—13 звеньев в цикле) наличие
заместителей в цикле не оказывает существенного влияния на
скорость их полимеризации B0].
Механизм превращения капролактама в полимер не вполне
выяснен. Различают:
гидролитическую полимеризацию (при использовании в
качестве активатора воды) ;
катионную полимеризацию, осуществляемую в присутствии
безводных кислот как катализаторов;
анионную полимеризацию в присутствии щелочей и щелочных
металлов.
Гидролитическая полимеризация. При гидролитической
полимеризации капролактама, по мнению многих исследователей
A3, 21], наиболее вероятно, что процесс протекает по типу
ступенчатой полимеризации. В начальной стадии реакции в результате
взаимодействия мономера с водой (активатор) образуется амино-
капроновая кислота:
/NH н2о
(СН,)/ [ > HOOC(CH2MNH2
которая реагирует с капролактамом, и получается димер:
HOOC(CH,MNH2 + (CH2V I —-»HOOC(CH2MNH—OC(CH2MNH2
Затем молекула димера реагирует с молекулой капролактама
с образованием тримера и т. д. вплоть до получения продукта со
степенью полимеризации, определяемой условиями проведения
реакции.
Наиболее медленной стадией процесса превращения
капролактама в полимер является первая стадия — образование аминокап-
роновой кислоты при взаимодействии капролактама с активатором.
Поэтому полиамид образуется гораздо быстрее при применении
в качестве исходного продукта не капролактама, а аминокапроно-
вой кислоты.
ЗО Глава 2. Производство полиамидных волокон
Возможна, однако, и другая схема реакции. В начальной
стадии процесса также образуется аминокапроновая кислота:
ОС—(СН2M—NH + Н2О —> HOOC(CH2MNH2
молекулы которой реагируют между собой по
поликонденсационному механизму с образованием димера:
HOOC(CH2MNH2+HOOC(CH2MNH2 —> HOOC(CH2MNH—CO(CH2MNH2 + Н2О
Выделяющаяся вода снова реагирует с капролактамом с
образованием аминокапроновой кислоты, которая вступает в реакцию
конденсации с димером, и т. д. вплоть до образования полимера.
Эта схема, не изменяющая общего представления о характере
реакции, менее вероятна и не подтверждается экспериментальными
данными.
Катионная полимеризация. При катионной полимеризации
капролактама, осуществляемой в безводной среде, реакция также
протекает ступенчато. В результате при полном'отсутствии воды
получается, по-видимому, промежуточный продукт — гидрохлорид
N-аминокапроилкапролактама:
/NH неї /NCO(CH2NNH3 СГ
2(CH2M( І » (СН2)/ |
хсо хсо
который реагирует с капролактамом, раскрывая цикл и
обусловливая полимеризацию по схеме [22]
ЛЧССКСНгЬЙНз СГ NH
( | + п(СН2M( |
ЧСО
Л}СО(СН2M[МНСО(СН2)В]ПЙН3 СГ
—> (сн2M( |
чсо
Катализаторами катионной полимеризации капролактама
являются минеральные кислоты. Однако большинство кислот не
может быть использовано, так как при высоких температурах они
окисляют или разлагают мономер или полимер (азотная и серная
кислота). Кроме того, при высоких температурах резко возрастает
летучесть некоторых кислот (например, хлористоводородной).
Практический интерес может представлять только фосфорная
кислота. В присутствии небольших количеств этой кислоты @,2—
0,5%) капролактам полимеризуетея достаточно быстро при
нормальном давлении. Например, в присутствии 0,5% Н3РО4 (в % от
массы лактама) реакция полимеризации капролактама
заканчивается в течение 2—3 ч [23]. Энергия активации этой реакции
46 ккал/моль [24].
Основным преимуществом полимеризации капролактама в
присутствии фосфорной кислоты является протекание процесса при
2.1. Синтез поликап/юамида 31
нормальном давлении в течение непродолжительного времени.
Выяснение возможности и целесообразности промышленного
использования этого катализатора представляет значительный интерес.
Полимеризация капролактама в присутствии фосфорной
кислоты может протекать по различным механизмам. По данным
Гелей и сотр. [23], детально исследовавших этот процесс,
полимеризация капролактама в присутствии этой кислоты протекает по
гидролитическому механизму. При 250 °С ортофосфорная кислота
переходит в пирофосфорную по схеме
2Н3РО4 —> Н4Р2О7 + Н2О
и выделяющаяся вода является инициатором полимеризации
капролактама.
Однако возможно, что в присутствии Н3РО4, благодаря более
интенсивному окислению капролактама кислородом, находящимся
в небольших количествах в реакционной среде, и образованию пе-
рекисных групп, синтез полиамида может частично протекать и по
радикальному механизму. Этот вывод подтверждается, по мнению
венгерских исследователей, тем, что типичные ингибиторы
радикальных процессов (пирогаллол или гидрохинон), введенные в
реакционную смесь, затрудняют полимеризацию капролактама в
присутствии Н3РО4. При гидролитической полимеризации этот эффект не
должен иметь места.
Наличие фосфорной кислоты, взаимодействующей с конечными
аминогруппами макромолекул полиамида, стабилизирует
молекулярный вес полиамида при последующем его плавлении, подобно
тому, как это имеет место при введении небольших количеств
карбоновых кислот.
Анионная полимеризация. При анионной полимеризации
капролактама наиболее широко применяемыми катализаторами
являются Na, NaOH, NaHCO3, Na2CO3, обычно в присутствии сока-
тализаторов.
Этот метод полимеризации лактамов, который в некоторых
случаях, как указано ниже, является единственно приемлемым при
полимеризации пяти- и шестичленных циклов (см. разд. 2.6),
представляет значительный интерес.
Анионная полимеризация капролактама осуществляется в
присутствии металлического натрия, щелочных металлов или
щелочных солей карбоновых кислот по механизму нуклеофильной атаки
анионом лактама атома углерода карбонильной группы лактама,
являющейся наиболее электрофильной группой в полимеризую-
щейся системе. Для получения воспроизводимых результатов при
щелочной полимеризации капролактама, так же как и других
лактамов, необходимо связать незначительное количество воды,
находящейся в реакционной среде. Для этого в реакционную смесь
добавляют небольшое количество алкиламинов, например ацил-
лактамов [25]. Эти реагенты, являющиеся сокатализаторами в
32 Глава 2. Производство полиамидных волокон
процессе анионной полимеризации, получили широкое применение
при щелочной полимеризации капролактама [26]. Количество сока-
тализатора в 1,5—2 раза превышает содержание Na-капролактама.
Механизм действия катализаторов основан на образовании
с капролактамом или его анионом производного, содержащего
полярный заместитель у атома азота в амидной группе
капролактама.
Образующиеся в незначительных количествах производные
капролактама принимают участие в начале роста цепи при условии
присутствия сильного основания и большого избытка безводного
свободного л актам а.
В качестве сокатализаторов можно применять также эфиры
карбоновых и минеральных кислот, амиды и спирты. Наиболее
активными сокатализаторами являются ацилкапролактам и, в
частности, бензоилкапролактам и ацетилкапролактам {27]:
ОССНз
ОС—(СН2M—N
Анионная полимеризация капролактама протекает ступенчато
при взаимодействии аниона капролактама с конечными имидными
группами растущей цепи. Детальней механизм и условия
осуществления этой интересной реакции рассмотрены в специальных
обзорных статьях [26, 28].
Параметры процесса полимеризации. Реакция полимеризации
является равновесной и обратимой:
. n[HN—(СН2M-СО] ч=* ¦¦—HN(CH2NCOHN(CH2MCO—¦
Чем выше температура реакции и ниже концентрация мономера
в реакционной смеси (при полимеризации в растворе), тем выше
содержание мономера в продукте реакции при достижении
равновесия.
Присутствие кислорода и других окислителей вызывает при
повышенной температуре побочные процессы окисления и
разложения полимера, но не влияет на скорость превращения циклов в
полимер. Скорость полимеризации капролактама в присутствии
воздуха и в среде инертного газа одинакова.
Основными параметрами процесса полимеризации
капролактама являются его продолжительность, температура, количество
и природа активатора (катализатора) и стабилизатора, характер
среды.
Продолжительность процесса. Влияние
продолжительности полимеризации на выход и свойства полимера
схематически показано на рис. 2.1. С увеличением продолжительности по-
з
2.1. Синтез поликапроамида 33
лимеризации возрастает выход полимера (до достижения
равновесия) и одновременно повышается его молекулярный вес*.
Время, необходимое для полимеризации капролактама, тем
меньше, чем выше температура и чем больше количество
активатора. Большое влияние на скорость процесса оказывает характер
активатора.
Характерной особенностью реакции полимеризации
капролактама является наличие начального индукционного периода, в
течение которого происходит
постепенный гидролиз капролактама [14].
При температуре полимеризации
230—260 °С индукционный период
при применении воды в качестве
активатора составляет 30—40 мин.
Реакция полимеризации
капролактама по окончании индукционного
периода протекает аутокаталитиче- ¦. ам/ктмшшь „оттритии.
ски. это объясняется, по-видимому, ґ
тем, что функциональные группы Рис. 2.1. Влияние продолжитель-
аминокислоты (NH2 и СООН), так ности полимеризации капролак-
же как и вода, ускоряют раскрытие тама 1;а ВЫХ°Д и св°йсгва
полицикла капролактама [29]. Макси- ,_выход поЛИИемрТ :,_молекулярвый
МаЛЬНЭЯ СКОРОСТЬ реакции ДОСТИГает- вес полимера; 3 — содержание мономера
СЯ При Превращении В ПОЛИМер 40— В Я°™капроамИде.
42% мономера.
Температура. Температура полимеризации определяет
количественное соотношение полимера и мономера, а также
низкомолекулярных фракций в получаемом полиамиде. Чем выше
температура, тем больше мономера содержится в поликапроамиде после
достижения равновесия:
Температура полимеризации, °С ... 200 230 260 290
Содержание мономера в полиамиде *,% 1—1,5 3—4 6—7 9—10
* Кроме капролактама в состав экстрагируемой низкомолекуляриой
фракции входит и некоторое Количество олигомеров—циклических ди-, три- и
тетрамеров [30] (около 30—35% от массы экстрагированного капролактама).
Олигомеры в определенных условиях могут полимеризоваться так же, как и
капролактам.
С повышением температуры усиливается деструкция полимера
в результате действия воды, имеющейся в реакционной смеси, и
соответственно снижается молекулярный вес полимера. Чем выше
температура полимеризации, тем' тщательнее должен очищаться
азот, применяемый в качестве среды при полимеризации, от
* Повышение молекулярного веса в процессе полимеризации имеет место
только при гидролитической полимеризации. При полимеризации капролактама
по анионному механизму (в частности, в присутствии металлического натрия)
молекулярный вес в начальной стадии процесса достигает максимума и затем
закономерно понижается (см. разд. 2,1.3).
2 Зак. 234
34
Глава 2. Производство полиамидных волокон
кислорода. Это необходимо для устранения окисления и
разложения полимера. Влияние температуры на скорость процесса и
свойства получаемого полимера схематически показано на рис. 2.2.
При повышении температуры реакции на 20 °С скорость
реакции увеличивается в 2 раза. Температура реакции должна
выдерживаться с точностью ± 1 °С.
Полимеризация капролактама осуществляется при 250—260 °С.
Дальнейшее повышение температуры нецелесообразно, так как при
этом повышается содержание
мономера в получаемом полиамиде
и увеличивается возможность
окисления полимера. Снижение
температуры при
гидролитической полимеризации
ограничивается температурой плавления
поликапролактама. '
Поликонденсация
аминокислот или низкомолекулярных
полиаминокислот может быть
осуществлена при более низких
температурах, чем температура плав-
( 520 °С)
Температура полимеризации
Рис. 2.2. Влияние температуры поли-
рур, рур
меризации на скорость процесса и ления ПОЛимера (на 5—20 °С)
свойства полимера: [31] Энергия активации реакции
/—выход полимера; 2— молекулярный вес; l J " - л.
3—продолжительность реакции; 4—содер- ПОЛИКОНДЄНСаЦИИ В ТВерДОИ фазе
жаиие мономера в поликапроамиде. составляет 90—180 ККЭЛ/МОЛЬ
C78—756 кДж/моль) и
значительно превышает энергию активации той же реакции,
протекающей в жидкой фазе [38—45 ккал/моль A60—189 кДж/моль)].
Этот метод может представить известный практический интерес
при использовании его для, дополнительного нагрева поликапро-
амидной крошки (например, при 200 °С) с целью уменьшить
содержание в ней низкомолекулярных фракций. В этом случае
отпадает необходимость экстракции мономера и последующей
сушки крошки.
' Активаторы или катализаторы. Характер применяемых
активаторов или катализаторов существенно зависит от метода
полимеризации капролактама. При гидролитической полимеризации,
наиболее широко применяемой для синтеза волокнообразующих
полиамидов, активатором служит вода. При ионной
полимеризации капролактама в качестве катализаторов используются, как
уже указывалось, некоторые минеральные кислоты, щелочи,
щелочные металлы и другие реагенты.
Основной недостаток применения воды как активатора —
необходимость проведения полимеризации' капролактама под
давлением, что усложняет аппаратурное оформление процесса. При
увеличении количества воды ускоряется полимеризация и снижается
молекулярный вес полимера (возрастает интенсивность гидролиза
2.1. Синтез поликапроамида
35
полиамида). Содержание мономера в поликапроамиде при этом
практически не изменяется (рис. 2.3).
Индукционный период и общая продолжительность
гидролитической полимеризации уменьшаются при добавлении небольших
количеств кислоты. Это действие кислот объясняют [32]
ускорением гидролиза капролактама (т. е. образования е-аминокапроно-
it
Количестдо воды
Рис. 2.3. Влияние количества
активатора (воды) на скорость
полимеризации и свойства получаемого
полиамида:
I — продолжительность реакции; 2 —
содержание мономера в поликапроамиде; 3—
молекулярный вес полимера.
Продолжительность
полимеризации
Рис. 2.4. Изменение степени
полимеризации полиамида в процессе
полимеризации:
/—в присутствии воды; 2—в присутствии
металлического натрия или Ыа2СОз, К2СО3,
NaHCO3, NaOH.
вой кислоты) в присутствии ионов гидроксония, образующихся
при диссоциации добавленной кислоты:
АсН + Н2О
Ас" + Н3О+
Очень эффективными активаторами полимеризации
капролактама являются аминокислоты. В присутствии этих соединений
полимеризация капролактама протекает без индукционного периода,
благодаря чему значительно ускоряется процесс. Под действием
аминокислот капролактам полимеризуется при нормальном
давлении. Существенное влияние на скорость полимеризации
оказывает состав аминокислоты.
В качестве активаторов реакции ступенчатой полимеризации
капролактама могут быть использованы также амины (бензил-
амин, гексаметилендиамин) и карбоновые кислоты [33].
Как уже указывалось выше,- эффективными катализаторами
реакции анионной полимеризации капролактама являются
металлический натрий, едкий натр или щелочные соли) В присутствии
небольшого количества этих реагентов @,2—0,5%)
продолжительность процесса полимеризации сокращается с 6—8 ч до 5—15 мин.
При применении в качестве катализатора щелочной соли кислого
эфира щавелевой кислоты [34—36], которая при 230—260 °С де-
карбоксилируется, полимеризация капролактама заканчивается в
течение 30—60 с«
Глава 2. Производство полиамидных волокон
500
І 400
300
zoo
wo
і г з ч
Быстрая полимеризация капролактама в присутствии щелочей
может быть осуществлена только в безводной среде. Содержание
мономера и низкомолекулярных фракций в поликапроамиде,
получаемом этим методом, такое же, как в полиамиде, синтезируемом
обычным методом.
Одной из основных особенностей процесса быстрой
полимеризации капролактама в присутствии щелочей является характер
изменения степени полимеризации получаемого полиамида в
процессе полимеризации (рис. 2.4).
Данные [37] о влиянии
количества катализатора (NaOH) и
продолжительности полимеризации на
молекулярный вес (определенный
по значению относительной
вязкости) полиамида приведены на
рис. 2.5.
Как видно из рис. 2.5, при
полимеризации в присутствии Na или
щелочных соединений в начальной
стадии процесса (через 10—15 мин
после начала полимеризации)
образуется высокомолекулярный
полиамид, степень полимеризации
которого в 3—4 раза превышает
степень полимеризации полиамида,
получаемого при синтезе
капролактама в присутствии воды (при той
же температуре). При дальнейшем
увеличении продолжительности
полимеризации начинается
постепенное снижение молекулярного веса
этого полиамида. Примерно через
6 ч выдерживания полиамида при
260 °С среднее значение степени
полимеризации полимера
снижается в 4 раза [38]. Поэтому при
синтезе методом быстрой
полимеризации поликапроамида, используемого для формования волокна,
необходимо после завершения процесса полимеризации
выдерживать расплавленный полимер несколько часов для получения
сравнительно однородного по молекулярному весу продукта.
Такое увеличение продолжительности процесса значительно
снижает эффективность метода быстрой полимеризации
капролактама.
Необходимо, однако, отметить, что указанный недостаток
может быть устранен при добавлении к реакционной смеси ацетил-
капролактама [39]. Например, если при щелочной полимеризации
о
50 WO 150
Продолжительность
полимеризации пик
Рис. 2.5. Влиииие
продолжительности полимеризации и количества
катализатора (NaOH) на
молекулярный вес поликапроамида
(температура полимеризации 260 °С):
/, 2, 3, 4—количество NaOH
соответственно 2, 1, 0,5 и '/s мол. %.
2.1. Синтез поликапроамида 37
добавить 0,005 моль ацетилкапролактама, то вязкость расплава
в течение длительного выдерживания не изменяется *.
Полимеризация, катализируемая натриевой солью капролак-
тама, имеет индукционный период. При проведении этой реакции
в присутствии N-алкиламида (например, ацетилкапролактама)
индукционного периода нет, и поэтому скорость реакции
полимеризации в этом случае значительно выше, чем в отсутствие амида.
Метод быстрой полимеризации капролактама пока еще
недостаточно разработан. Дальнейшие исследования в этой области
должны определить технико-экономическую целесообразность
использования этого метода в производственных условиях, в
частности при осуществлении непрерывного процесса полимеризации
капролактама и формования волокна.
Регуляторы молекулярного веса. В процессе
формования волокна при плавлении поликапроамида, содержащего на
концах макромолекул активные функциональные группы, может
произойти дополнительная поликонденсация и соответственно
повышение молекулярного веса полимера и вязкости расплава. Во
избежание этого одну из концевых групп полимера — карбоксильную
или аминогруппу — блокируют. Для блокирования аминогрупп
обычно добавляют к полимеру карбоновые кислоты, которые
образуют замещенные амиды:
HOOC(CH2MHN[CO(CH2MNH]„CO(CH2MNH2 + RCOOH —».
—». HOOC(CH2MHN[CO(CH2MNH]„CO(CH2MNHCOR + Н2О
Для блокирования карбоксильных групп применяются амины
или NaOH. В результате блокирования одной из активных
функциональных групп дальнейший процесс поликонденсации
становится невозможным и молекулярный вес и соответственно вязкость
расплава полимера при его последующем нагревании и плавлении
не изменяются.
Реагенты, применяемые для блокирования функциональных
групп полиамида, называются регуляторами. Обычно блокируют
частично аминогруппы, добавляя в начальной стадии
полимеризации небольшие количества уксусной, адипиновой, салициловой или
других органических кислот; при этом образуются
соответствующие замещенные амиды. Однако лучше использовать нелетучие
кислоты, концентрация которых в системе на различных стадиях
технологического процесса не изменяется, например стеариновую
или бензойную.
В качестве регуляторов могут быть использованы также и
соединения, блокирующие обе концевые реакционноспособные группы
* Интересно отметить, что охлажденный расплав капролактама, в котором
содержится 0,005 моль NaOH или металлического натрия, при нормальной
температуре не теряет каталитической активности в течение длительного времени
(до 7 месяцев) [40].
38 Глава 2. Производство полиамидных волокон
(карбоксильные и аминогруппы), например амиды кислот или
соли карбоновых кислот и первичных аминон [41].
Введение этих добавок дает возможность регулировать
молекулярный вес поликапроамида. Чем больше количество регулятора,
тем ниже молекулярный вес получаемого полимера.
А. Н. Быков и сотр. [42] использовали в качестве стабилизаторов цветные
аминоантрахиноиы, в частности а- и ?-аминоантрахиноны и 1,5-диаминоантрахи-
нон, в количестве 0,2—1 % от массы мономера. Полимеризацию капролактаме
они проводили в обычных условиях. Применение цветных регуляторов
обусловливает получение окрашенных полиамидов — розового, красного и оранжевого
цвета, которые нормально перерабатываются в волокно. Поскольку цветные
регуляторы, как и регуляторы других типов, химически связаны с концевыми
группами макромолекулы полимера, окраска получаемых волокон вполне стойка
к различным воздействиям.
Среда. Полимеризация капролактама всегда проводится в
отсутствие растворителей. При полимеризации капролактама в
растворе уменьшается выход полимера и снижается его
молекулярный вес вследствие уменьшения межмолекулярного
взаимодействия активных функциональных групп.
Полимеризация капролактама осуществляется в атмосфере
инертного газа, обычно азота, тщательно очищенного от
кислорода. Присутствие даже небольших количеств кислорода в
реакторе не допускается по следующим причинам:
при высоких температурах полимеризации капролактама
кислород частично окисляет получаемый полимер, что приводит к
изменению цвета (потемнению) полиамида и формуемого из него
волокна;
в результате частичного окисления полиамида и
незначительного его разложения может образоваться полимер сетчатой
структуры, который не плавится, засоряет отверстия фильер и
затрудняет последующее вытягивание волокна.
Поэтому необходимо, чтобы в азоте, применяемом для
полимеризации, содержалось меньше 0,005% кислорода. Чем выше
температура, при которой происходит полимеризация капролактама и
формование волокна, тем ниже предельно допустимое содержание
кислорода в азоте.
Азот, получаемый при разделении воздуха методом глубокого
охлаждения и содержащий до 1 % кислорода, подвергают
дополнительной очистке.
Очистка, в результате которой содержание кислорода
снижается до 0,005%, заключается в пропускании азота над медными
стружками или медной спиралью, нагретыми до 450—500 °С. При
этой температуре кислород связывается медью. Когда основное
количество меди переходит в окись, ее регенерируют пропусканием
водорода, и она может быть снова использована для связывания
кислорода. Такой способ очистки азста имеет, однако, ряд
недостатков, к которым, в частности, относится большой расход элек-
2.1. Синтез поликапроамида 39
троэнергии и необходимость установки электропечей для очистки
азота и регенерации меди.
Известен более простой и рациональный способ очистки азота,
который заключается в добавлении к азоту водорода (на ОД—
0,2% больше, чем содержание кислорода в азоте). Азотоводород-
ную смесь пропускают над палладиевым катализатором при 20—
25 °С. Образующуюся воду отделяют пропусканием азота через
осушительное устройство. При этом методе очистки азота
значительно уменьшается расход электроэнергии и упрощается
аппаратурное оформление процесса [43].
Аппаратура. При аппаратурном оформлении процесса
полимеризации капролактаме должна быть учтена необходимость
точного регулирования температуры в пределах ± 1 °С и проведения
реакции в атмосфере инертного газа.
Капролактам полимеризуют в аппаратах периодического или
непрерывного действия.
При осуществлении процесса в аппарате периодического
действия реакцию проводят в автоклавах под давлением. Иногда
автоклавы рассчитаны одновременно и на работу под вакуумом для
отсоса паров воды и лактама в конце процесса. Однако
необходимость работы под вакуумом и под давлением усложняет
конструкцию и обслуживание аппарата. Для упрощения конструкции
аппарата в большинстве случаев мешалку внутри автоклава не
устанавливают, и перемешивание реакционной массы происходит
только в результате кипения воды.
Аппарат для полимеризации обогревается электричеством,
жидкими высокотемпературными теплоносителями или их парами,
циркулирующими в его рубашке.
Электричеством трудно обеспечить равномерность обогрева, тая
как реакционная масса у стенок аппарата нагревается сильнее, чем
это требуется. Поэтому электрический способ обогрева в
промышленной практике пока не используется. Однако вполне возможно,
что при дальнейшем усовершенствовании этот метод обогрева
получит практическое применение.
Из высокотемпературных органических теплоносителей (ВОТ)
наибольшее применение получили так называемая динильная смесь
(иначе — даутерма, динил) и смесь ароматизированных нефтяных
масел (теплоноситель АМТ-300). Для обогрева может быть
использован также пар высокого давления.
Динилом называется эвтектическая смесь, содержащая 26,5%
дифенила С6Н5—С6Н5 и 73,5% дифенилового эфира (С6Н5JО,
кипящая при 258 °С. В качестве теплоносителя можно применять и
чистый дифенил, температура кипения которого та же, что и ди-
нильной смеси. Однако динил плавится при более низкой
температуре A2,3°С), чем дифенил G0°С). Это основное преимущество
динила по сравнению с дифенилом, который п и обычной
40 Глава 2. Производство полиамидных волокон
температуре застывает в трубах, что затрудняет разогрев
в'начале производственного процесса.
Преимуществом применения динильной смеси является
возможность обогрева аппарата парами теплоносителя при нормальном
давлении или даже под вакуумом (при температуре ниже 258°С),
недостатком — относительная сложность получения дифенила и ди-
фенилового эфира, а также специфический запах и некоторая
токсичность паров смеси.
При работе с динилом вся система, по которой циркулирует
жидкая смесь или ее пары, должна быть тщательно
герметизирована во избежание частичного улетучивания динила, что приводит
к потерям теплоносителя и к ухудшению условий труда.
На ряде заводов химического волокна (капронового и других
гетероцепных волокон, формуемых из расплава полимера)
применяется теплоноситель АМТ-300. Этот теплоноситель застывает при
—30 °С, кипит при 300—320 °С. Преимущество АТМ-300 перед
динилом — значительно меньшая токсичность, недостаток — окис-
ляемость на воздухе и невозможность (согласно требованиям
техники безопасности) нагрева теплоносителя выше 280 °С.
При использовании водяного пара высокого давления для
обогрева автоклава полностью устраняется возможность загрязнения
воздуха токсичными веществами, но требуется применять трубы
и арматуру, рассчитанные на работу под давлением выше 50—
60 кгс/см2 F-Ю6 Па). Этот недостаток не имеет в настоящее
время большого значения, если учесть, что использование пара
высокого давления широко применяется в различных отраслях
промышленности. Необходимо также отметить, что водяной пар
имеет в 3—4 раза более высокую энтальпию (теплосодержание) и
обладает более высоким коэффициентом теплоотдачи, чем пары
динила, поэтому паровой обогрев аппаратов значительно
экономичнее; кроме того, уменьшается продолжительность нагрева
аппарата перед началом полимеризации.
Периодический процесс полимеризации. Капролактам,
применяемый для полимеризации, предварительно расплавляют при 85—
90°С в специальном аппарате — расплавителе. В этом аппарате,
снабженном рубашкой и мешалкой, проводится перемешивание
капролактама с активатором и стабилизатором.-Затем реакционная
смесь отфильтровывается под давлением азота через ткань на
обогреваемом керамическом или стеклянном фильтре и подается в
автоклавы или более совершенные аппараты — так называемые
трубы НП (непрерывной полимеризации), преимущественно
применяемые в настоящее время.
Для равномерного распределения двуокиси титана в полимере
при получении матированного волокна требуемые количества ТіОг
добавляют в виде водной суспензии одновременно с
капролактамом в автоклав через 2—3 ч после начала полимеризации
2.1. Синтез поликапроа.шда 41
По окончании полимеризации расплавленная масса под
давлением 1,5—2 кгс/см2 A,5- 105 —2- 105 Па) выдавливается в
течение 1—1,5 ч из аппарата, а иногда пропускается через песочный
фильтр для очистки от механических примесей и вытекает через
широкую щель в виде ленты или жилки. Ленту пропускают обычно
через ванну с водой для охлаждения.
Измельчение ленты. Полимер, полученный в виде ленты или
жилки, как правило, непосредственно не используется для
формования волокна. Для проведения последующих операций, в
частности экстракции лактама из полиамида и сушки, необходимо
значительно увеличить поверхность полимера. Для этого выходящая из
ванны с водой лента подается на рубильный станок и измельчается,
а полученная крошка пневматическим транспортом направляется
на следующую операцию.
Большое значение имеет однородность крошки по величине.
Мелкая крошка будет просыпаться на плавильной решетке
прядильной машины, крупная крошка — медленно плавиться. При
нормальном измельчении крошка имеет следующие размеры:
ширина— 7—8 мм, длина —8—10 мм, толщина — 2—3 мм.
Экстракция лактама из поликапроамида. Как уже указывалось,
процесс полимеризации капролактама протекает не до конца. В
получаемом полиамиде содержится в зависимости от температуры
полимеризации большее или меньшее количество мономера. При
температуре полимеризации 260 °С содержание мономера и
низкомолекулярных водорастворимых фракций составляет 10—12% от
массы полиамида.
Олигомеры, входящие в состав водорастворимой фракции,
могут быть как циклическими, так и линейными (олигомеры амино-
капроновой кислоты). Олигомеры составляют 25—35% от общего
количества водорастворимых фракций, причем основное количество
олигомеров — циклические соединения. Соотношение циклических
димеров, тримеров и тетрамеров в поликапроамиде, полученном
при проведении реакции полимеризации при 250—260 °С, равно
5:2:7 {44].
Циклический димер плавится при 248 °С, а циклический тример
при 261 °С. Линейные олигомеры плавятся при более низких
температурах: димер при 197°С, а тетрамер при 206°С. Наличие
лактама, кипящего при 258 °С, ухудшает условия формования волокна,
так как его пары, выходящие из фильеры вместе с расплавленным
полимером, могут обрывать формующиеся волокна. Поэтому в тех
случаях, когда полимеризация капролактама и формование
волокна из поликапроамида проводятся раздельно, мономер и
низкомолекулярные водорастворимые фракции удаляют из крошки
экстракцией горячей водой. Экстракция проводится путем ряда
последовательных обработок крошки при 95—100 °С. После первой
обработки содержание водорастворимых фракций в полиамиде
снижается с 10—12 до 4—5%, после второй обработки — до 2—3%
42 Глава 2. Производство полиамидных волокон
и после третьей — до 0,5—1%. Для экстракции применяется
метод противоточной промывки.
В последнее время на ряде предприятий экстракция лактама и
других низкомолекулярных водорастворимых фракций из поли-
капроамида, а также сушка крошки после экстракции
производится в аппаратах непрерывного действия. Естественно, что при
таком аппаратурном оформлении процесса продолжительность
этих операций сокращается и одновременно уменьшаются
затраты труда.
Водный раствор, полученный после экстракции, направляется.
на вакуум-выпарку. Это необходимо не только для регенерации
лактама (в результате регенерации лактама удельный расход
мономера уменьшается на 6—8%), но и по санитарным требованиям.
Метод регенерации лактама из промывной воды путем
выпаривания недостаточно экономичен (для регенерации 1 кг лактама
необходимо испарить свыше 20 кг воды), однако более
рациональные методы пока не разработаны.
Сушка поликапроамида. По окончании экстракции крошка
иногда отжимается в центрифуге и с влажностью 10—15%
поступает на сушку. Содержание влаги в высушенном продукте не
должно превышать 0,05%, в противном случае выделяющиеся при
формовании пары воды вызывают повышенный обрыв волокон.
Наиболее затруднительно удаление последних 1—1,5% влаги, и
именно этим определяется продолжительность сушки. Обычно
сушка полиамида производится в барабанных вакуумных
сушилках 18—24 ч при ПО—125 °С и остаточном давлении 5—6 мм рт. ст.
F70—800 Па).
Для устранения частичного окисления крошки сушку
желательно проводить при температуре не выше 80 °С, а для ускорения
процесса целесообразно применять более глубокий вакуум. После
сушки крошку следует выгружать в токе азота.
Продолжительность отдельных стадий технологического
процесса синтеза поликапроамида составляет (в ч) :
Растворение лактама и подготовка его к полимеризации . 3,5—4
Полимеризация капролактаме и формование ленты или
жилки 15—24
Экстракция низкомолекулярных соединений и отжим
крошки 15—20
Сушка крошки 18—24
Общая продолжительность технологического процесса
получения поликапроамида периодическим методом достигает 50—60 ч.
Потери лактама на отдельных стадиях технологического
процесса получения полиамида и подготовки его к формованию не
должны превышать 2%.
Непрерывный процесс полимеризации.Непрерывная
полимеризация капролактама может быть осуществлена не только при
быстром проведении процесса в присутствии металлического натрия
2.1. Синтез полдкапроамида 43
или щелочей, но и при более медленном его проведении в
присутствии активаторов.
В СССР этот способ ранее использовался при получении
штапельного волокна из поликапроамида, а в настоящее время он
является основным и при выработке комплексной нити.
Процесс непрерывной полимеризации проводят при
нормальном давлении, что упрощает его аппаратурное оформление. В
качестве активаторов применяют не только воду, но и реагенты,
отщепляющие при высокой температуре воду, которая в момент
выделения разрывает цикл капролактама и тем самым обеспечивает
начало процесса полимеризации. Такими активаторами являются
аминокислоты или соли дикарбоновой кислоты и диамина, которые
при температуре 260—270 °С вступают в реакцию
поликонденсации и выделяют воду. Когда применяются эти реагенты, в процессе
полимеризации образуется не поликапроамид, а сополимер
капролактама с 3—5% (от массы капролактама) соли АГ — соль ади-
пиновой кислоты и гексаметилендиамина (см. разд. 2.2.2.). Ввиду
незначительного содержания соли АГ в полиамиде получаемый
сополимер заметно не отличается по свойствам от
поликапроамида.
Продолжительность процесса полимеризации составляет 12—
24 ч. В большинстве случаев полимеризация капролактама
производится в вертикальной или U-образной трубе [45, с. 94],
[46, с. 73—84].
Внутренняя поверхность стальной трубы для полимеризации
покрыта эмалью или специальной сталью. Труба обогревается ди-
нилом или паром высокого давления. Высота ее 10—12 м, диаметр
0,25—0,5 м. Чтобы исключить смешение мономера и других
низкомолекулярных продуктов, находящихся в верхней части трубы,
с полимером, образующимся в нижней части, в ней установлены
перфорированные алюминиевые диски. Эти диски расположены на
расстоянии 25—50 см друг от друга. Через отверстия в дисках и
протекает расплавленная масса. Одна из схем трубы для
непрерывной полимеризации приведена на рис. 2.6.
Процесс полимеризации в трубе проводится при 260—275 °С.
В большинстве случаев температура в отдельных ее секциях
различается: максимальная температура B60—275 °С)
поддерживается в средней секции, а в верхней и нижней секциях
температура несколько ниже B50°С).
Производительность трубы НП при высоте 12 м и
продолжительности полимеризации 18—24 ч составляет 5—6 т/сут. Загрузка
лактама в трубу и регулирование температуры осуществляются
автоматически [45, 46].
Вытекающий из трубы расплавленный поликапроамид
направляется по трубопроводу на прядильную машину. Если
полимеризация и формование волокна разделены, то полимер выпускается
в виде ленты или жилки, дальнейшая обработка которых
44
Глава 2. Производство полиамидных волокон
осуществляется так же, как и при периодическом методе
получения поликапроамида.
Аппарат оригинальной конструкции для непрерывной
полимеризации предложен советскими исследователями [47]. В этом аппа-
2
і Уробень
лакглама
/ісекция
Рис. 2.6. Схема трубы для непрерывной полимеризации капролактама:
/—крышка; 2—смотровые стекла; 3 — штуцер для уровнемера; 4—наружная труба; 5 —
внутренняя труба; 6— паровая рубашка; 7—карманы для электронагревательных пакетов;
8—разгрузочный штуцер; S—соединительный патрубок; 10—стержень с перфорированными
дисками; // — поворотная регулировочная заслонка.
рате (рис. 2.7) достигается равномерная скорость течения
расплава по всему сечению и объему. В корпусе аппарата находятся
2.1. Синтез поликапроамида
45
закрепленные на его днище коаксиально расположенные цилиндры
(один в другом), между которым-и смонтированы не доходящие до
дна аппарата и выступающие над цилиндрами перегородки. Для
снижения скорости движения средних, наиболее быстро
перемещающихся слоев расплава, в пространстве между цилиндрами и
перегородками установлены
насадки в форме змеевиков.
Капролактам, непрерывно
поступающий в аппарат сверху
вниз, вместе с необходимыми
количествами инициатора^-(воды) и
регулятора (бензойной кислоты)
непрерывно стекает в первую
секцию (цилиндр) аппарата, где
и происходит первая стадия
процесса полимеризации и
образуется низкомолекулярный
полимер. Расплавленный полимер че-
рез перегородку поступает во
вторую секцию аппарата, затем
в третью, в которой завершается
процесс полимеризации и
получается поликапроамид
требуемого молекулярного веса. Общая
продолжительность пребывания
полимера в этом аппарате
несколько меньше, чем в трубе НП,
и при температуре реакции
260 °С составляет 18—22 ч.
Основными преимуществами
аппарата этого типа по
сравнению с трубой НП являются:
компактность, благодаря че-
Рис. 2.7. Аппарат для непрерывной
полимеризации капролактама
конструкции ВНИИСВ:
/—корпус аппарата; 2—крышка аппарата;
3—цилиндрические перегородки, ие
доходящие до днища 6; 4— цилиндры (секции
аппарата, закрепленные на диище 6); 5—иа-
садки в виде змеевиков; 7—штуцеры для
подачи капролактама в аппарат.
му уменьшается
производственная площадь, необходимая для
установки аппарата;
снижение расхода тепла на обогрев аппарата вследствие
значительно меньшей теплоотдачи цилиндров (секций аппарата),
установленных коаксиально по отношению друг к другу;
более однородное молекулярно-весовое распределение
получаемого полимера, так как вея масса лактама и полиамида находится
в аппарате примерно одинаковое время; в результате
сформованное волокно обладает более высоким комплексом
физико-механических свойств.
Эти аппараты для непрерывной Полимеризации капролактама
успешно работают на ряде заводов капронового волокна и,
по-видимому, в ближайшие годы найдут широкое промышленное применение.
46 Глава 2. Производство полиамидных волокон
2.1.4. Свойства поликапроамида
Основными показателями, характеризующими свойства
поликапроамида и пригодность его для формования волокон, являются
молекулярный вес, содержание низкомолекулярных фракций,
влажность и вязкость расплава полимера.
Молекулярный вес. Для формования волокна применяется
полимер с молекулярным весом 16 000—22 000 (степень
полимеризации 150—200). Несмотря на сравнительно небольшой
молекулярный вес, поликапроамид и получаемые из него изделия обладают
высокой механической прочностью, что объясняется наличием
большого числа водородных связей между макромолекулами полимера.
Молекулярный вес полиамида в производственных условиях
определяется обычно по вязкости 0,5%-ного раствора полимера
в крезоле или 1%-ного раствора в 45%-ной или 84%-ной серной
кислоте. При степени полимеризации 200 удельная вязкость 0,5%-
ного раствора поликапролактама в крезоле равна 0,5, а 1%-ного
раствора того же полимера в 84%-ной H2SO4— 1,2.
Содержание низкомолекулярных фракции. Содержание
водорастворимых низкомолекулярных фракций определяют путем
экстракции их горячей водой (кипячением в течение 15 ч.)
Содержание капролактама в полиамиде можно определить экстракцией
капролактама бензолом или эфиром, так как димер и тример
в этих растворителях не растворяются.
Влажность. Определение крайне незначительных количеств
влаги, содержащейся в полиамиде, связано с большими
трудностями. Обычно применяемые методы длительного высушивания до
постоянной массы в большинстве случаев недостаточно точны для
этой цели. Поэтому небольшие количества влаги целесообразно
определять нитридным методом, исходя из взаимодействия
полиамида с нитридом магния при повышенной температуре B75 °С) :
Mg3N2 + 6H2O —> 3Mg(OHJ + 2NH3
Выделяющийся при этом аммиак поглощается 0,1 н. раствором
соляной кислоты. Избыток кислоты оттитровывают.
Определить содержание воды в полиамиде можно также и иодо-
метрически с применением для титрования реактива Фишера.
Реакция протекает по схеме
І2 + SO2 + 2Н2О —> 2HI + H2SO4
Исследуемый образец поликапроамида растворяют в смеси
фенола и абсолютированного спирта и затем титруют реактивом
Фишера. Этим методом содержание влаги можно определить с
точностью до тысячных долей процента. Получаются вполне
воспроизводимые результаты, хотя они и несколько ниже (на 0,01 —
0,02%), чем определенные нитридным методом.
Вязкость расплава. Одной из основных технологических
характеристик полиамидов, в частности поликапроамида, является
2.1. Синтез поликапроамида 47
вязкость расплава, поступающего в прядильный насосик, а затем
в фильеру.
Важным фактором, определяющим вязкость расплава, является
молекулярный вес полимера. Например, при увеличении
молекулярного веса с 17 000 до 30 000 вязкость расплава повышается в 5—
6 раз. В результате давление, требуемое для подачи расплава к
фильере, возрастает с 2 B-Ю5 Па) до 22 кгс/см2 B2-105 Па) [48].
Вязкость расплава зависит не только от молекулярного веса
полимера, но и от содержания различных примесей, в частности
капролактама (рис. 2.8) и воды.
Чем больше этих веществ находит-
ся в полиамиде, тем меньше при
прочих равных условиях вязкость
расплава.
В присутствии 0,1% воды или
0,2—0,3% капролактама (от массы
полиамида) вязкость расплавлен-
ного полимера снижается в
2—Зраза. Возможность значительно пони- Содержание кагіролактапа.
зить вязкость расплава и тем
самым температуру формования, не из- Рис. 2.8. Влияние содержания
меняя молекулярный вес полимера, капролактама на вязкость рас-
путем введения различных добавок плава поликапроамида.
представляет определенный интерес.
Аналогичный эффект достигается при добавлении
пластификаторов, хорошо растворимых в капролактаме, например арилсуль-
фонамидов общей формулы ArSO2NHR. Добавление 5—10%
пластификатора от массы капролактама перед полимеризацией
снижает вязкость расплава полимера одного и того же молекулярного
веса в 2—3 раза [49]. Эффект добавления пластификатора тем
больше, чем выше молекулярный вес полиамида.
Большое влияние на вязкость расплава различных полиамидов
одного и того же молекулярного веса оказывает химический состав
полиамида, определяющий в значительной степени гибкость цепи.
Вязкость расплава одного и того же полимера существенно
зависит от температуры. Например, изменение вязкости расплава
поликапроамида с молекулярным весом 17 000 с повышением
температуры характеризуется следующими данными [50]:
Температура, °С.... 230 240 250 260 270
Вязкость расплава, П 7500 6500 5500 48O0 2700
2.2. СИНТЕЗ ПОЛИГЕКСАМЕТИЛЕНАДИПАМИДА
Как уже указывалось, исходными - продуктами для
получения полигексаметиленадипамида (анид, найлон 6,6) являются
адипиновая кислота НООС(СНгLСООН и гексаметилендиамцн
48 Глава 2. Производство полиамидных волокон
H2N(CH2NNH2. Для производства полиамидов этого типа могут
быть применены также другие дикарбоновые кислоты, например пи-
мелиновая НООС(СН2MСООН и себациновая НООС(СН2)8СООН,
и другие диамины, например тетраметилендиамин H2N(CH2LNH2.
Изменяя химический состав исходных мономеров, в частности
число метиленовых групп в молекулах диамина и дикарбоновой
кислоты, можно изменять температуру плавления полиамида и
получаемого из него волокна.
2.2.1. Получение мономеров
Сырьевая база для получения мономеров, используемых для
синтеза полиамидов типа" анид, (найлон 6,6) шире, чем для
производства капролактама. Кроме фенола, бензола и циклогексана,
которые могут быть использованы для получения как
капролактама, так и адипиновой кислоты и гексаметилендиамина, для
синтеза мономеров, применяемых в производстве полиамида анид,
в качестве исходных веществ могут служить фурфурол и ацетилен,
бутадиен, а также акрилонитрил.
Получение адипиновой кислоты. Эту кислоту получают из
фенола по следующей схеме:
ОН'
СНОН
н2 н2с/ хсн2 HNO
—^ У I 3 > НООС(СН2LСООН
При гидрировании фенола образуется циклогексанол, который
окисляют 65—68%-ной азотной кислотой при 45—60°С (без
катализатора или на медно-ванадиевом катализаторе) в адипиновую
кислоту.
Адипиновую кислоту можно получить и из циклогексана,
который, так же как при производстве капролактама, окисляют при
120—140°С кислородом воздуха до циклогексанола. Затем
циклогексанол окисляют азотной кислотой до адипиновой кислоты.
Азотной кислотой можно окислить непосредственно
циклогексан, однако выход продукта реакции в этом случае незначителен
[51] и образуется большое количество побочных продуктов, что
делает этот способ экономически неэффективным.
Адипиновую кислоту целесообразно получать из циклогексана
методом двухстадийного окисления. На первой стадии циклогексан
окисляют кислородом воздуха при 140—145 °С под давлением
10 кгс/см2 A0-105 Па) в присутствии нафтената кобальта как
катализатора. В результате образуется смесь циклогексанола и
циклогексанона. На второй стадии указанную смесь окисляют
кислородом воздуха или 54—56%-ной азотной кислотой при 55—85 °С
в присутствии катализаторов — меди и метаванадата аммония. Вы-
2.2. Синтез полигексаметиленадипамида 49
ход адипиновой кислоты составляет 90—93% от теоретического.
В промышленности этот метод нашел наиболее широкое
применение.
Адипиновая кислота может быть получена также из бензола
через нитробензол, анилин и циклогексанол; переработка цикло-
гексанола в адипиновую кислоту производится по обычной схеме.
Получение гексаметилендиамина. Адипиновая кислота является
также исходным веществом для получения второго компонента,
необходимого для синтеза полигексаметиленадипамида, —
гексаметилендиамина.
Гексаметилендиамин из адипиновой кислоты получают по
следующей схеме. При пропускании паров адипиновой кислоты и
аммиака над катализатором при 320—330 °С образуется динитрил
адипиновой кислоты:
НООС(СН2LСООН + 2NH3 ~^+ H2NOC(CH2LCONH2 -^-> NC(CH2LCN
В качестве катализатора обычно применяют фосфат бора или
продукт взаимодействия алюминия и вольфрамовой кислоты или
соли кобальта.
Промежуточным продуктом реакции является диамид
адипиновой кислоты, который при высокой температуре над катализатором
дегидратируется.
При гидрировании динитрила адипиновой кислоты на
катализаторах (никель или кобальт) при температуре 160 °С и давлении
20 кгс/см2 B-Ю6 Па) получается гексаметилендиамин:
NC(CH2LCN —* H2N(CH2NNH2
Гидрирование целесообразно проводить в присутствии
безводного аммиака, который тормозит побочные реакции. Выход
гексаметилендиамина составляет 85—95%.
Для производства гексаметилендиамина могут быть использованы также
ацетилен и формальдегид.
Гексаметилендиамин из ацетилена получают' по следующей схеме. При
взаимодействии ацетилена с формальдегидом образуется 1,4-бутиндиол:
СН=СН + 2СН2О —> НО—Н2С—С=С—СН2—ОН
При гидрировании 1,4-бутиндиола в присутствии никелевого катализатора
получают 1,4-бутендиол:
но—н2с—с=с—сн2—он —+ но—н2с—нс=сн—сн2—он
В присутствии CuCI2 1,4-бутендиол реагирует с HCN и образуется дициан-
бутен:
НО-Н2С—НС=СН—СН2—ОН + 2HCN —у
—у NC—Н2С—НС=СН-СН2—CN + 2Н2О
Дицианбутен восстанавливают до гексаметилендиамина:
NCHjCHC=CHCH3CN —> H3N(CH,NNH,
50 Глава 2. Производство полиамидных волокон
Дицианбутен, а следовательно, и гексаметилендиамин можно получить
также по следующей схеме. Бутадиен хлорируют при 65—75 °С с образованием
1,4-дихлорбутадиеиа:
' С12
СН2=СН—СН=СН2 >- С1СН2СН=СНСН2С1
При взаимодействии дихлорбутадиеиа с NaCN при 90 °С получается ди-
цианбутеи:
С1СН2СН=СНСН2С1 + 2NaCN —>- NCCH2CH=CHCH2CN + 2NaCl
Гидрированием дицианбутена получают гексаметилеидиамин. По
имеющимся данным, по этой схеме производится гексаметилеидиамии в количестве
20 тыс. т/год на одном из заводов фирмы «Дюпои».
Перспективным методом получения исходного сырья для
производства соли АГ является предложенный советскими
исследователями [52] синтез динитрила адипиновой кислоты гидродимериза-
цией акрилонитрила. Если учесть, что акрилонитрил может быть
получен не только из ацетилена, но и при взаимодействии
пропилена и аммиака (см. разд. 6.1), то очевидно, насколько
использование в качестве исходного продукта акрилонитрила вместо
бензола или фенола расширяет сырьевую базу для производства соли
АГ, а соответственно и волокна анид (найлона 6,6).
Димеризацию и частичное восстановление акрилонитрила
производят в электролизере непрерывного действия в присутствии
гидротропных солей в ячейке с ртутным катодом и платиновым
анодом. Реакция протекает по схеме
2Н
2СН2=СН—CN > NC—(СН2L—CN
Образующийся при электролизе адиподинитрил непрерывно
экстрагируется из продуктов реакции органическим растворителем,
а не вступивший в реакцию акрилонитрил возвращается в реактор.
Выход целевого продукта реакции почти количественный.
Побочные продукты реакции — пропионитрил и небольшое количество
высших олигомеров.
Полученный динитрил адипиновой кислоты может быть
использован для получения гексаметилендиамина и адипиновой кислоты.
Предполагается, что этот метод синтеза позволяет снизить
стоимость адиподинитрила на 25—30% {53].
2.2.2. Получение анида (гексаметиленадипамида)
Как уже указывалось выше, при поликонденсации адипиновой
кислоты с гексаметилендиамином выделяется вода и образуется
полиамид — полигексаметиленадипамид:
Н2С—СН2—СООН H2N—СН2—СН2—СН2
| + | =ё=>:Н2О +
Н2С—СН2—СООН H2N—СН2—СН2—СН2
+ CO-(CHjL-CONH-(CH?)e-NHCO-(CH,L-CONH-(CH,)e-NH--.
2.2. Синтез полигексаметиленадипамида
Полученный полимер широко используется для производства
волокна типа анид (найлон 6,6).
Основные закономерности, характеризующие этот метод
синтеза полимеров, рассматриваются в курсе химии
высокомолекулярных соединений и изложены в специальных монографиях.
Ниже приводятся некоторые данные, которые необходимо
учитывать при разработке параметров технологического процесса
получения анида.
Реакция поликонденсации является обратимой. Чем полнее
удаляется выделяющаяся в процессе реакции вода, тем выше
молекулярный вес получаемого полимера. Следовательно, тщательное
удаление воды из сферы реакции — одно из основных условий
получения высокомолекулярного полиамида.
Так же как и при полимеризации циклов, увеличение
продолжительности реакции приводит к повышению молекулярного веса
получаемого полимера.
Активаторы реакции образования полиамидов типа анид
методом линейной поликонденсации пока неизвестны. Поэтому
основным методом регулирования скорости реакции является изменение
температуры.
Одним из главных факторов, влияющих на молекулярный вес
получаемого полимера, является соотношение мономеров {54, с.
81—85]. Максимальный молекулярный вес полиамида достигается
при эквимолярном соотношении обоих компонентов. Избыток
одного из компонентов приводит в результате аминолиза (действия
избытка диамина) или ацидолиза (действия избытка дикарбоно-
вой кислоты) при высокой температуре к частичной деструкции
полимера и к снижению его молекулярного веса. Чем больше
избыток одного из компонентов, тем ниже при прочих равных условиях
молекулярный вес полимера. Поэтому исходным веществом для
поликонденсации служит не смесь адипиновой кислоты и гекса-
метилендиамина, а их средняя соль, так называемая
H2N—(CH2N—NH2 • НООС—(СН2L—СООН
соль АГ, которая отличается по растворимости от исходных
мономеров и поэтому может быть очищена от избытков мономеров
перекристаллизацией из различных растворителей {в частности, из
метилового спирта).
В отличие от полимеризации капролактама, при которой в
получаемом продукте устанавливается определенное равновесие
между полимером и мономером, реакция синтеза
полигексаметиленадипамида идет практически до конца. При достижении
равновесия получается полиамид, содержащий менее 1 %
низкомолекулярных соединений. Этот факт объясняется тем, что циклизация
соли АГ, содержащей в молекуле 14 звеньев, крайне затруднена и
практически не имеет места.
52 Глава 2. Производство полиамидных волокон
Синтез полиамида протекает в две стадии. Вначале получают
соль АГ смешением растворов адипиновой кислоты и гексамети-
лендиамина в метаноле. Соль АГ выпадает в осадок, который
очищается перекристаллизацией и сушится. Затем проводится
поликонденсация соли АГ в атмосфере азота в автоклаве,
обогреваемом динилом. Выгрузка расплавленного полиамида из автоклава,
его фильтрация, формование ленты и ее измельчение
осуществляются так же, как при получении поликапроамида.
Отличия синтеза анида от процесса получения поликапроамида
заключаются в следующем.
1. Полиамид типа анид (найлон 6,6) имеет более высокую
температуру плавления B55°С), чем капрон B15°С). Поэтому
поликонденсация соли АГ протекает при более высокой температуре
B75—280°С). Продолжительность процесса 10—16 ч.
2. Высокомолекулярный продукт получается, только если
полностью удалена вода, выделяющаяся при поликонденсации. Для
этого на последней стадии поликонденсации в аппарате создается
вакуум.
3. Экстракция мономера из измельченного полиамида, а
следовательно, и регенерация мономера из промывных вод
исключается из технологического процесса, так как при
поликонденсации соли АГ реакция практически идет до конца.
Автоклав, в котором получают анид, должен быть рассчитан
на работу под давлением 12—14 кгс/см2 A2-105—14-105 Па) на
первой стадии процесса поликонденсации и под вакуумом (для
возможно более полного удаления выделяющейся воды) — на
второй стадии.
Принципиально возможно осуществить непрерывный процесс
поликонденсации полиамида типа анид и формования из него
волокна. Такая схема технологического процесса применяется на
ряде зарубежных предприятий, вырабатывающих волокно
найлон 6,6.
Процесс поликонденсации можно осуществить и в твердой фазе
при 180—188°С [55] (температура плавления соли АГ 193—195°С).
При проведении реакции в твердой фазе можно использовать
катализатор, в частности борную кислоту [56], в то время как при
поликонденсации в расплаве каталитического действия эти реагенты
не оказывают. Поликонденсация в твердой фазе представляет пока
только теоретический интерес.
2.3. СИНТЕЗ ПОЛИЭНАНТОАМИДА
Исходным продуктом для получения полиэнантоамида (энанта)
является ю-аминоэнантовая кислота NH2(CH2NCOOH.
Применение для производства полиамида аминокислоты, а не ее лактама
дает возможность значительно ускорить синтез энанта по
сравнению с получением капрона.
2.3. Синтез полиэнантоамида 53
2.3.1. Получение аминоэнантовой кислоты
о-Аминоэнантовую кислоту можно получить двумя методами:
из этилена и четыреххлористого углерода и из циклогексанона
через є-капролактон.
Получение ю-аминоэнантовой кислоты из этилена и ССЦ по
методу, разработанному А. Н. Несмеяновым и Р. X. Фрейдлиной,
основано на использовании реакции теломеризации, при которой
цепная полимеризация в присутствии большого количества
регулятора обрывается (останавливается) на стадии получения
низкомолекулярных продуктов [57, 58]. В качестве регулятора обычно
применяется ССЦ.
При взаимодействии этилена и ССЦ в присутствии
инициатора — азодинитрила изомасляной кислоты или других
инициаторов, распадающихся с образованием свободных радикалов,
получается смесь тетрахлоралканов:
пСН2=СН2 + СС14 —>- Cl(CH2CH2)nCClj
Реакция проводится при давлении 100—150 кгс/см2 A00 • 105—
150- 103 Па) и температуре 90—100 °С. Значение п, т. е. состав
тетрахлоралканов, изменяется в зависимости от соотношения ССЦ
и этилена в реакционной смеси *. Чем больше этилена приходится
на 1 моль ССЦ, тем выше молекулярный вес получаемых
тетрахлоралканов.
Следовательно, при реакции теломеризации всегда получается
не индивидуальный продукт, а смесь продуктов. Тетрахлоралканы
разделяют разгонкой в вакууме.
Для производства аминоэнантовой кислоты используется
1,1,1,7-тетрахлоргептан, получаемый в указанных условиях
проведения процесса в количестве 30% от суммы тетрахлоралканов.
Из 1,1,1,7-тетрахлоргептана при нагревании с
концентрированной серной кислотой при 90—100 °С получается ю-хлорэнантовая
кислота:
С1(СН2NСС13 —> С1(СН2NСООН
Обработкой ю-хлорэнантовой кислоты 25%-ным водным
раствором NH4OH при 100 °С под давлением 5—6 кгс/см2 E- 105—
6 • 105 Па) получают ю-аминоэнантовую кислоту:
С1(СН2NСООН —> H2N—(СН2N—СООН
Очистка этой кислоты от примесей производится с помощью
ионообменных смол.
По аналогичной схеме из 1,1,1,9-тетрахлорнонана
получают ю-аминопеларгоновую кислоту H2N—(CH2)s—СООН, а
* При 4—4,5 моль этилена на 1 моль ССЦ получается смесь
тетрахлоралканов, содержащая около 40% (от суммы всех продуктов реакции) алканов
с л=.2, около 30% ся=3, около 15% с п = 4 и около 10% ся = 5.
54 Глава 2. Производство полиамидных волокон
из 1,1,1.11-тетрахлорундекана — и-аминоундекановую кислоту
H2N— (CH2)io—GOOH.
Возможность рационального и экономичного получения
полиамида и волокна энант зависит от эффективного использования
всех аминокислот, образующихся при теломеризации этилена и
последующей переработке тетрахлоралканов, е-аминовалериановой
H2N(CH2LCOOH, cu-аминоэнантовой, и-аминопеларгоновой и и-
аминоундекановой кислот. Аминопеларгоновая и аминоундекано-
вая кислоты могут быть, так же как и аминоэнантовая кислота,
использованы для синтеза соответствующих полиамидов и
получения из них волокон.
Использование аминовалериановой кислоты, образующейся
в количестве 35—40% от массы смеси всех аминокислот,
получаемых при реакции теломеризации, для синтеза полиамида и
последующего формования из него волокна представляет значительные
трудности. При поликонденсации аминовалериановой кислоты
образуется устойчивый шестичленный цикл — пиперидон, который
в обычных условиях гидролитической полимеризации не полиме-
ризуется. В последнее время получены данные (см. разд. 2.6),
показывающие, что в определенных условиях шестичленный цикл
раскрывается и пиперидон полимеризуется.
Другим недостатком метода синтеза аминоэнантовой кислоты
теломеризацией этилена является большой расход ССЦ при
получении тетрахлоралканов и отсутствие экономичных методов
использования значительного количества НС1, выделяющегося на
последующих стадиях процесса получения указанных аминокислот.
2.3.2. Получение полиамида энант
Полиамид энант получают поликонденсацией и-аминоэнанто-
вой кислоты:
/iH2N(CH2NCOOH —> NH—(СН2N—CONH—(СН2N—СО h H2O
Поликонденсация и-аминоэнантовой кислоты производится при
250—260°С (температура плавления полимера энант 225°С) в тех
же аппаратах, в которых получают полиамиды капрон и анид.
Так как полиаминоэнантовая кислота обладает высокой
термостойкостью, не уступающей термостабильности полиамида капрон,
волокно энант можно получить непрерывным методом. В отличие от
процесса получения поликапроамида при синтезе полиэнантоамида
образуется полимер, который почти не содержит
низкомолекулярных фракций. Содержание в нем водорастворимых фракций не
превышает 1%, что является основным преимуществом энанта
перед полиамидом капрон.
Поликонденсация и-аминоэнантовой кислоты с целью
получения полиамида необходимого молекулярного веса проводится с
добавкой регулятора — адипиновой или уксусной кислоты.
2.4. Синтез полиамидов найлон 6,10 и ундекан (найлон 11) 55
Опубликованы данные о новом методе получения
синтетического волокна (найлон 7) из эфиров м-аминоэнантовой кислоты
[59], которые в свою очередь получают из циклогексанона через
е-капролактон по схеме
^СН2 3 Чюн СН2—(СН2K—СО НС1^
/сн2 *~ сн2 o *~
"сн2
циклогексаион надуксусная е-капролактон
кислота
ROH NaCN
—> С1(СН2M—СООН »¦ С1(СН2M—COOR *~
е-хлоркапроновая эфир е-хлор-
кислота капроновой кислоты
Н2
—> NC—(СН2N—COOR »- H2N—(CH2N—COOR
нитрил эфира эфир ш-амииоэиантовой
эиаитовой кислоты кислоты
где R = СН3, С2Н5 или С3Н7.
Поликонденсация эфиров аминоэнантовой кислоты проводится при 270—
280 "С. Существенным преимуществом этого метода получения волокна из поли-
аминоэнантовой кислоты является образование в результате поликонденсации
только полиамида одного типа и соответственно волокна одного вида.
2.4. СИНТЕЗ ПОЛИАМИДОВ НАЙЛОН 6,10
И УНДЕКАН (НАЙЛОНИ)
Исходными продуктами для получения полиамида найлон 6,10
являются гексаметилендиамин и себациновая кислота, для унде-
кана (найлон 11, рильсан)—м-аминоундекановая кислота. Синтез
гексаметилендиамина описан выше, поэтому здесь приводятся
схемы получения только себациновои и м-аминоундекановой кислот.
Себациновая кислота. Исходным сырьем для синтеза себациновои кислоты
является касторовое масло, т. е. глицериновый эфир рищинолевой кислоты.
Щелочным омылением выделяют рицинолевую кислоту, которая при дальнейшем
нагревании со щелочью при повышенной температуре расщепляется на себаци-
новую кислоту и 2-октанол (каприловый спирт):
СН3(СН2MСНОНСН2СН=СН(СН2OСООН —>
рицииолевая кислота
—>¦ НООС(СН2)8СООН + СН3(СН2MСНОНСН3
Аминоундекановая кислота. Из касторового масла получают также и ами-
ноундекановую кислоту. При 300 °С в вакууме рицинолевая кислота распадается,
образуя ундециленовую кислоту и альдегид энантовой кислоты:
СН3(СН2NСНОНСН2СН=СН(СН2OСООН —>
—» CH2=CH(CH2)SCOOH + Cl
56 Глава 2. Производство полиамидных волокон
которые разделяют отгонкой с водяным паром. Ундециленовую кислоту
обрабатывают НВг в присутствии перекиси бензоила. Полученную при этом бромунде-
кановую кислоту подвергают аммонолизу, в результате чего образуется амино-
ундекановая кислота:
Br(CH2)laCOOH + NH3 —> H2N(CH2I0COOH+HBr
В последнее время опубликованы данные о получении амнноундекановой
кислоты непосредственным аминнрованнем ундецнленовой кислоты, что упрощает
условия ее получения.
Как уже отмечалось выше, аминоундекановую кислоту можно
получить также в качестве одного из компонентов реакции тело-
меризации этилена.
Полиамид найлон 6,10. Этот полиамид получают
поликонденсацией себациновой кислоты и гексаметилендиамина:
СН2—СН2—СН2—СН2—СООН HjN— СН2—СН2—СН2
I + I —¦
СН2—СН2—СН2—СН2—СООН H2N—СН2—СН2—СН2
—> СО—(СН2)8—CONH—(СН2N—NH + Н2О
Полиамид ундекан (найлон 11, рильсан). Полиамиды этого типа
синтезируют путем поликонденсации ?-аминоундекановой кислоты:
H2N(CH2),0COOH —> NH(CH2I0CONH(CH2I0CO .+Н2О
2.5. СИНТЕЗ ПОЛИАМИДА ДОДЕКАН (НАЙЛОН 12)
В последнее время разработан метод получения полиамида ¦
найлон 12 [ CO(CH2)uNHOC(CH2)nNH ], исходя из лак-
тама аминододекановой кислоты (додекалактама).
Производство этого полиамида и волокна из него стало
возможным после того, как Циглером и Вилке [60] был разработан
в 1957 г. метод получения циклододекатриена из бутадиена.
Каталитической тримеризацией бутадиена в присутствии
катализаторов Циглера {TiCl'4 + А1(С2Н5)з] получают двенадцатичлен-
ный цикл— 1,5,9-циклододекатриен:
/ \
СН СН
СН
сн2
\ /
сн2—сн=сн
СН.
Этот процесс осуществляется в растворе бутадиена в неполярном
растворителе при 40—80 °С. Полученную смесь цис- и
транс-изомеров 1,5,9-циклододекатриена без разделения гидрируют с обра-
2.5. Синтез полиамида додекан (найлон 12) 57
зованием циклододекана, который затем фотонитрозированием
перерабатывают в додекалактам:
СН2 СН2 СН2
/ \
сн2 сн2
І І
СН2 СН2 —> ОС—(СН2)„—NH
сн.
\- /2
СН2—СН2—СН2
циклододекан додекалактам
Этот метод синтеза реализован фирмой «Хюльз», которая
организовала опытно-промышленное производство додекалактама
(производительностью 6 тыс. т/год) в ФРГ, а также в Японии.
Додекалактам можно получить и другими способами, широко
применяемыми при синтезе капролактама, исходя из циклогек-
сана. Из этих способов наибольший интерес представляют:
окисление циклододекана до циклододеканона и последующая
переработка кетона в оксим и затем в додекалактам по обычной
схеме;
получение нитроциклодекана нитрованием циклододекана
азотной кислотой при 125 °С {61] и последующей переработкой его в
оксим по схеме, применяемой при получении капролактама из нит-
роциклогексана (см. разд. 2.1.1.).
Додекалактам хорошо полимеризуется обычными методами
гидролитической полимеризации в присутствии воды.
В соответствии с изложенными выше закономерностями
превращения лактамов в линейные полимеры полимеризация тринад-
цатичленного цикла — додекалактама — протекает практически до
конца. Содержание низкомолекулярных фракций в найлоне 12 не
превышает 0,5—0,6%.
Имеются данные, что в ФРГ предполагают к 1975 г. довести
производство найлона 12 до 45 тыс. т/год {62].
Окисляя циклододекан, можно получить декандикарбоновую
кислоту НООС(СН2)юСООН, которую предполагают использовать
для получения найлона 6,12 путем ее поликонденсации с гексаме-
тилендиамином. Основным преимуществом производства найлона 12
перед описанным выше методом получения найлона 11 является
использование нефтехимического, а не пищевого сырья.
Волокно найлон 12, содержащее в элементарном звене
максимальное количество метиленовых групп по сравнению со всеми
другими полиамидными волокнами, вырабатываемыми в
настоящее время, обладает наибольшей гидрофобностью, а также
высокими изоляционными и адгезионными свойствами.
Температура плавления полиамида найлон 12 ниже, чем
других полиамидов, используемых для производства волокон, и
составляет 180 °С [63]. Пониженная температура плавления ограничивает
58 Глава 2. Производство полиамидных волокон
области применения этого волокна, сочетающего высокую
прочность волокон типа найлон 6 и найлон 6,6 с низким водопоглоще-
нием @,85% при относительной влажности воздуха 65%) и малой
плотностью A,01 г/см3).
Волокно найлон 12 обладает высокой морозостойкостью и
может быть использовано при температурах от —50 до —70 °С, в то
время как волокно капрон используется обычно при температурах
не ниже — 30 СС.
Для производства кордной нити найлон 12 непригоден ввиду
пониженной температуры плавления и низкого начального модуля.
Учитывая приведенные выше данные, можно сделать вывод, что
производство волокна 12, основанное на использовании доступного
многотоннажного нефтехимического сырья и обладающего рядом
технически ценных свойств, может, по-видимому, найти в
ближайшие годы широкое применение.
2.6. СИНТЕЗ ПОЛИАМИДОВ ИЗ ПЯТИ-
И ШЕСТИЧЛЕННЫХ ЛАКТАМОВ
Синтезировать полиамиды из пяти- н шестичлеиных лактамов (найлон 4 н
найлон 5) можно только методом анионной полимеризации. При использовании
этого метода пяти- и шестичленные циклы размыкаются даже быстрей, чем семи-
членные циклы [64]. Используя метод анионной полимеризации и применяя
наряду с катализатором также и сокатализатор, удалось осуществить
полимеризацию пятичленного гетероцикла — пирролидона (бутиролактама) :
2 ' *~" •— HN(CH2KCOHN(CH2KCO—
и получить полиамид найлои 4 н волокно из иего.
Полимеризация пирролидона методом анионной полимеризации проводится
при 50—60 С в присутствии калиевой соли пирролидона, используемой в
качестве катализатора. Для ускорения этого процесса, согласно патентам Нея [65],
рекомендуется добавлять хлорангидриды кислот, в частности адипиновой
кислоты или N-ациллактам, т. е. использовать ту же смесь катализаторов, которая
применяется по методу Вихтерле при анионной полимеризации капролактама.
Однако в этих условиях в присутствии 0,001—0,002 моль калиевой солн
пирролидона и 0,001—0,002 моль сокатализатора, в частности N-бензоилпирро-
лидона, на 1 моль лактама процесс протекает очень медленно и реакция
заканчивается через 24—36 ч. В литературе нет данных о возможности
интенсифицировать этот процесс повышением температуры или увеличением количества
катализатора.
Предварительным условием осуществления этой реакции, так же как и
других ионных реакций, является тщательное удаление следов воды и других
соединений, содержащих подвижный водород, из мономера и растворителя
(углеводорода), применяемого при полимеризации пирролидона. Выход полимера
составляет 80—85%. Найлон 4 недостаточно устойчив к действию повышенных
температур, поэтому формование волокна из него проводится не из расплава,
а из раствора в муравьиной кислоте сухим или мокрым способом.
Температура плавления иайлона 4 равна 265 °С, прочность полученного
волокна около 45 гс/текс D50 мН/текс), гигроскопичность несколько выше, чем
у капрона, и составляет 5% при относительной влажности воздуха 65%. Если
Таблица 2.2. Условия получения и некоторые свойства основных типов полиамидов,
используемых для производства синтетических волокон
Условия получения и свойства
Мономер, применяемый для синтеза
полиамида
Исходное сырье для получения
мономера
Метод синтеза полиамида
Основные параметры процесса
синтеза полиамида
продолжительность, ч
температура, °С
активаторы нли катализаторы
Содержание растворимых ннзкомо-
лекуляриых фракций в полиамиде,
% от массы полимера
Свойства полиамида
температура плавлення, °С
степень полимеризации
Капрон
(найлои 6)
Капролактам
Фенол, бензол,
циклогексан
Полимеризация
(превращение
цикла в линейный
полимер)
16—21 *
250-260
Применяются
10-12
215
150-200
Анид
(найлон 6,6)
Соль АГ
Фенол, бензол,
циклогексан, акрило-
нитрнл, ацетилен
Поликонденсация
10-15
275-280
Не применяются
0,5—1
255
80-100**
Энаит
(найлон 7)
Аминоэнантовая
кислота
Этилен и четырех-
хлористый
углерод, фурфурол,
циклогексанон
Поликонденсация
8-Ю
250-260
Не применяются
0,5— 1
225
150-200
Додекан
(иайлон 12)
Додекалактам
Бутадиен
Полимеризация
(превращение
цикла в
линейный полимер)
220-240
Применяются
0,5-1
180
• При использовании щелочного катализатора продолжительность процесса составляет 5—15 мин.
'• Считая на элементарное, звено—CO(CH2LCOHN(CH2NNH~.
60 Глава 2. Производство полиамидных волокон
удастся повысить термостойкость этого полимера, то можно будет формовать
волокно из расплава.
Были получены также сополимеры капролактаме с пирролидоном по
реакции анионной полимеризации. На начальной стадии этой реакции пирролидон
полимеризуется быстрей, чем капролактам, что, по-видимому, объясняется
большей кислотностью пирролидона.
Интересно отметить, что пирролидон вступает в реакцию сополимеризации
с капролактамом в таких условиях, в которых сам пирролидон не
полимеризуется. Сополимеризация является одним из эффективных способов синтеза
трудно полимеризуемых лактамов.
Осуществлена также полимеризация шестичленного лактама (пиперидона) [66]
в присутствии катализатора (КОН + N-ацетилпнперидон) при температуре
45—80 °С:
СН2
I —>¦ NH(CH2LCONH(CH2LCO—¦•
СС
Н2С\ /СО
NH
Температура плавления полипиперидона 259 °С. Максимально достигнутый
выход полимера составляет 76% от теоретического. С повышением температуры
реакции выход полимера и его молекулярный вес снижаются.
Данные, характеризующие условия синтеза и некоторые
свойства основных типов полиамидов, применяемых для производства
синтетических волокон, представлены в табл. 2.2. Плотность всех
полиамидов, приведенных в таблице, одинакова —1,14 г/см3;
регуляторы молекулярного веса применяются во всех случаях.
2.7. СИНТЕЗ ПОЛИ-Э-АМИДОВ ПОЛИМЕРИЗАЦИЕЙ
ЧЕТЫРЕХЧЛЕННЫХ ЛАКТАМОВ
В последнее времн показана возможность [67] получения нового класса
полиамидов полимеризацией четырехчленных лактамов (?-лактамов). Синтез
этих лактамов был разработан Графом [68].
При взаимодействии SO3 с хлорцианом получается хлорангидрид N-карбонил-
сульфоаминовой кислоты (содержащей изоцианатную группу):
SO3 + C1CN —> C1O2SN=CO
который присоединяется к олефинам по месту двойной связи с образованием
ІМ-сульфонилхлорид-Р-лактама:
R
R—С—СН2
N—С=О
I
C1SO2
При последующем отщеплении сульфонилхлоридной группы образуется
свободный ?-лактам [69]:
R
R—С—СН2
HN—С=О
где R = Н, СНз, С2Н3 и т. д.
Раскрытие цикла четырехчленного лактама, т. е. его полимеризация,
происходит только в щелочной среде методом анионной полимеризации, В качестве
2.8. Свойства полиамидов капрон и анид 61
катализатора используются, так же как при анионной полимеризации капролак-
тама, Na-?-лактам в присутствии небольшого количества ациллактама [70].
Напряженные четырехчленные гетероциклы раскрываются значительно
быстрей и легче, чем пяти- и шестичленные циклы. Полимеризация ?-лактамов,
например лактама ?-аланина, растворенного в диметилсульфоксиде, протекает
количественно при комнатной температуре за несколько минут.
Характерной особенностью полимеризации напряженных ?-лактамов является
количественное превращение мономера в полимер. В получаемом полимере
практически не содержится мономера.
При анионной полимеризации ?-лактамов получаются высокомолекулярные
полиамиды, молекулярный вес которых значительно превышает молекулярный
вес обычных полиамидов. Молекулярный вес поли^-амидов составляет
60 000—75 000.
Ввиду того что напряженные четырехчленные циклы раскрываются
значительно легче, чем пяти- и даже семичленные циклы, могут быть с большим
выходом получены поли^-амиды, исходя из С-замещеиных ?-лактамов.
Поли^-аланин получен Бреслоу [71] по другой интересной схеме — методом
анионной полимеризации акриламида в присутствии алкоголята натрия в среде
инертного растворителя (пиридин, хииолии) :
о о о
II I! II
СН2=СН —> СН2=СН—С—NH(CH2—СН2—CNH)„—СН2—СН2—CNH2
CONHj,
Однако этот метод не получил практического применения. Условия получения и
свойства волокна из поли^-амидов приведены ниже (см. разд. 2.16.2).
2.8. СВОЙСТВА ПОЛИАМИДОВ КАПРОН И АНИД
Полиамиды капрон и анид (найлон 6,6) имеют ряд общих
свойств и отличаются только по некоторым показателям.
Полиамиды обоих типов плавятся без разложения, растворяются в
одном и том же ограниченном числе растворителей — феноле,
крезоле, 4—5 н. растворах минеральных кислот, муравьиной кислоте.
Из этих полиамидов получают волокна и пластические массы,
обладающие высокой прочностью и эластичностью.
Молекулярный вес полиамидов в производственных условиях
определяют обычно путем вискозиметрических измерений
растворов в крезоле, в 84%-ной серной кислоте или в 80%-ной
муравьиной кислоте. При выдерживании полиамидов в этих кислотах при
20 °С в течение 50 ч или в течение 10 ч при 50 °С они почти не де-
структируются. Молекулярный вес капрона (растворитель —
крезол) и анида (растворитель — H2SO4), определяемый вискозимет-
рически, рассчитывают соответственно по уравнениям
[i\] = 24 • Ю~4м°-51 и [п] = 11. ю~4м°-51
где [г|] — характеристическая вязкость;
M — молекулярный вес растворенного полимера.
В качестве растворителя для вискозиметрического определения
молекулярного веса полиамида предложен также 70%-ный водный
62 Глава 2. Производство полиамидных волокон
раствор хлоральгидрата [72]. В этом случае степень полимеризации
определяют по уравнению
[г\] = 1,996 • 10М0'76
Молекулярный вес полиамидов оказывает существенное
влияние на проведение отдельных стадий технологического процесса
получения волокна, в частности на одну из важнейших операций —
вытягивание. По данным Кларе [73; 74, с. 165], поликапроамид
с молекулярным весом менее 10 000 не может быть вытянут на
300—400%, что необходимо для получения прочного волокна.
Оптимальное значение молекулярного веса этих полимеров,
используемых для формования волокна,— 15 000—20 000.
Прн получении высокопрочной (более 80 гс/текс) (800 мН/текс),
кордной нити молекулярный вес поликапроамида должен быть
увеличен до 25 000—28 000.
При дальнейшем, повышении молекулярного веса способность
полиамидов к вытягиванию снова ухудшается. По данным Губерта,
поликапроамид с молекулярным весом выше 100 000 уже не
плавится. При повышении молекулярного веса полиамида прочность
получаемого волокна увеличивается, особенно при применении
горячей вытяжки. Это видно из следующих данных [48]:
Молекулярный
вес полиамида
Кратності
вытяжки
Вытягивай
20 000
30 000
4,0
4,4
> Прочность,
гс/текс
не при 20'
42,4
47.4
Вытягивание прн 150
17 000
20 000
30 000
5,0
4,8
4,8
50,0
75,0
78,0
Удлинение,
%
¦с
17,9
16,6
°С
11,8
11,3
11,2
К сожалению, указанные исследователи не приводят данных об
изменении других практически важных показателей волокон,
например усталостной прочности и эластичности, которые при
повышении молекулярного веса полимера также должны значительно
повыситься.
Капрон и найлон 6,6 обладают значительной
полидисперсностью. Это установлено Грилем при исследовании поликапроамида
[75, 76], а также Вилотом [77], который определял
полидисперсность полиамидов методом фракционного осаждения, разделяя
эти препараты на 7—9 фракций. При среднем значении степени
полимеризации полиамида 102 степень полимеризации отдельных
фракций изменялась от 58 до 382.
Полидисперсность полимеров можно определить также методом
турбидиметрического титрования, применяя дли этого 0,05%-ный
раствор полиамида в смеси фенола, воды и этанола в соотношении
2:1:1. Осадитель — 96%-ный этанол 1781.
2.9. Формование полиамидных волокон 63
Полиамиды капрон и анид одинаково легко кристаллизуются.
Основные различия между полиамидами капрон и анид (найлон
6,6) сводятся к следующему.
1. Полиамиды типа капрон не разлагаются и не деструкти-
руются даже при длительном выдерживании при температурах
формования волокна. Полиамиды типа найлон 6,6 в этих условиях
начинают разлагаться с выделением СО2, NH3 и других продуктов.
Эта особенность затрудняет осуществление непрерывного процесса
синтеза полиамида найлон 6,6 и формования из него волокна.
2. Полиамиды капрон и найлон 6,6 обладают различной
растворимостью в концентрированных кислотах. Капрон растворяется
в 4,2 н. НС1 A53 г/л) при 20 °С, а найлон 6,6 — только при
нагревании. Найлон 6,6 растворим в концентрированном растворе СаСЬ
в метаноле. В этом же растворителе A7,5—33,6% СаСЬ в
метаноле) растворим и капрон. Растворение протекает без деструкции,
и молекулярный вес полиамида в этом растворителе не изменяется
в течение нескольких суток.
3. Температура плавления найлона 6,6 на 40°С выше, чем
капрона, что обусловливает различия в температурном режиме
получения этих полиамидов и формования из них волокна.
Стоимость капролактама и соли АГ примерно одинакова, хотя
в некоторых странах соль АГ стоит на 5—10% дороже
капролактама. Необходимо также учесть, что удельный расход соли АГ при
синтезе полиамида на 6—8°/о больше, чем капролактама, что
объясняется выделением воды при поликонден'сации соли АГ.
2.9. ФОРМОВАНИЕ ПОЛИАМИДНЫХ ВОЛОКОН
Полиамидные волокна можно формовать из раствора и из
расплава. Однако формование этих волокон из раствора для обычных
полиамидов, температура плавления которых ниже температуры
разложения, никогда не применяется и, по-видимому, не будет
применяться, так как использование реагентов, в которых растворимы
полиамиды, нецелесообразно по многим соображениям.
Формование волокон из концентрированных растворов полиамидов в
муравьиной кислоте, феноле или крезоле сухим способом трудно
осуществимо (вследствие сравнительно высокой температуры
кипения этих растворителей) и не эффективно по сравнению с более
простым методом формования из расплава. Еще менее экономично
формование волокна из растворов мокрым способом. Однако этот
способ применяется в тех случаях, когда формование из расплава
или из растворов сухим способом невозможно, например при
получении волокна из некоторых типов термостойких полиамидов
(см. разд. 11.1.1.) или поли-р-амидов.
В производственных условиях основным методом получения
обычных полиамидных волокон является формование из расплава.
Принципы формования волокна из расплавов изложены ранее
64
Глава 2. Производство полиамидных волокон
(см. т. I). Ниже приводятся данные, характеризующие условия
формования полиамидных волокон из расплава и аппаратурное
оформление этого процесса *.
При_ получении полиамидного волокна обычным методом
измельченный полимер (крошку) загружают в бункер прядильной
машины, рассчитанный на подачу крошки на одно или несколько
прядильных мест. В бункер загружают 100—150 кг измельченного
полимера (насыпная плотность крошки 0,5—0,6 кг/л). Это
количество обеспечивает формование волокна в течение 2—3 дней. После
Рис. 2.9. Плавильная решетка.
загрузки крошки бункер герметически закрывают и пропускают
азот для удаления следов кислорода. Вместо азота можно
применять водяной пар, что проще и дешевле.
Крошка плавится на плавильной решетке, являющейся важной
деталью прядильной машины, применяемой при формовании
волокна из расплава **. Плавильная решетка (рис. 2.9) представляет
собой змеевик из нержавеющей стали, укрепленный в чаше,
имеющей два патрубка. Через верхний патрубок поступает
измельченная крошка, а через нижний вытекает расплавленный полиамид.
По змеевику пропускают пары динила. Расплавленный полиамид
стекает в приемный конус под решеткой (так называемое
«болото»). В конусе находится 400—500 г расплавленного полиамида,
что необходимо для бесперебойного питания прядильных насоси-
ков. Производительность плавильной решетки при получении
текстильной нити при 260—270 °С составляет обычно 28—56 г
расплавленного полиамида в минуту. ,
В последнее время на ряде заводов проводились опыты по
обогреву прядильных головок не динилом, а электричеством. При этом
методе нагрева отпадает необходимость оборудовать динильные
котельные и прокладывать трубопроводы для циркуляции паров
динила на прядильной машине. Кроме того, улучшаются условия
труда на прядильных машинах — исключается возможность загряз-
* Подробное описание устройства прядильных машин для формования
полиамидных волокон прнводитси в соответствующих руководствах [79].
** При осуществлении метода непрерывного синтеза полиамида и
формовании из него волокна необходимость в бункере и плавильной решетке отпадает.
2.9. Формование полиамидных волокон 65
нения воздуха парами динила. Одновременно значительно
уменьшается расход электроэнергии.
На некоторых заводах используются плавильные решетки на
несколько фильер (например, на 4—8 фильер). При этом
существенно упрощается аппаратурное оформление процесса
формования полиамидных волокон и примерно в 2 раза уменьшается длина
прядильной машины.
Габаритные размеры прядильной машины дополнительно
уменьшаются и повышается производительность прядильного места при
приемке нити из нескольких фильер на одну бобину с получением
на ней нескольких паковок.
В последнее время все более широко применяется метод
плавления крошки в шнеках (экструдерах)—так называемый метод
экструдерного формования. Благодаря непрерывному
перемешиванию в экструдерах, обогреваемых до 260—280 °С электричеством,
крошка плавится значительно быстрей, чем на плавильной решетке
(в течение 1,5—2 мин). Из экструдера расплав без промежуточного
накапливания его в конусе поступает в насосный блок. Один
экструдер обслуживает 8—16 шахт. Продолжительность
пребывания расплава на прядильной машине при этом не превышает
5 мин, и содержание низкомолекулярных фракций в полученной
нити в 1,5—2 раза меньше, чем при формовании на прядильной
машине с использованием плавильных решеток. Поэтому в
большинстве случаев нить, получаемая методом экструдерного
формования, не требует дополнительной промывки для удаления
низкомолекулярных фракций. Преимуществом этого метода является
также возможность переработки полиамида с более высоким
молекулярным весом, что особенно существенно при получении
высокопрочных кордных нитей.
Расплавленная масса, вытекающая из плавильной решетки или
из экструдера, попадает в насосный блок, представляющий собой
стальной цилиндр, в котором расположены два зубчатых
прядильных насосика — напорный и дозирующий.
Прядильный насосик работает при температуре 250—290 °С и
давлении 20—60 кгс/см2 B-Ю6—6-Ю6 Па), в зависимости от
вязкости расплава, поэтому насосики изготовляют из
высококачественной стали, легированной ванадием и молибденом.
Применение в блоке двух прядильных насосиков при формовании
полиамидного волокна объясняется необходимостью обеспечить
равномерную подачу расплава к фильере. Первый насосик подает
несколько больше расплава, чем второй, и поэтому создает
повышенное давление, необходимое для нормальной работы второго
насосика.
Перед поступлением в фильеру (диаметр фильеры 60 мм,
диаметр отверстия 0,2—0,3 мм) расплав фильтруется через кварцевый
песок, находящийся на металлических сетках. Обычно
применяются три слоя кварцевого песка общей толщиной 8—10 мм.
3 Зак. 234
Глава 2. Производство полиамидных волокон
Каждый последующий слой состоит из зерен меньшего размера,
благодаря чему повышается степень очистки расплава.
Вытекающие из фильеры струйки расплавленного полиамида
поступают в шахту прядильной машины (рис. 2.10), где они
обдуваются током холодного
воздуха и, застывая,
превращаются в тонкие волокна.
В последнее время
фильеры, применяемые для
формования полиамидных волокон,
часто покрывают кремнийор-
ганическими полимерами. В
результате уменьшается
прилипание расплава к фильере
и засорение ее отверстий.
Общий вид фильерных
комплектов, применяемых для
формования капроновой
текстильной нити, приведен на
рис. 2.11.
Условия охлаждения
волокна в шахте оказывают
большое влияние на
однородность нити по толщине и на
равномерность ее
окрашивания. Поэтому температура,
влажность и количество
воздуха, подаваемого в шахту,
имеют очень существенное
значение для получения
высококачественного волокна.
Высота шахты 3—5 м. При
_,. скорости формования волокна
Рис. 2.10. Шахта прядильной машины. 600 м/мин и высоте шахты
5 м продолжительность
пребывания нити в шахте 0,5 с. При формовании волокна найлон 6,6
рекомендуют вдувать в шахту сверху вниз водяной пар или
влажный воздух (при относительной влажности не менее 90%). В этих
условиях волокно, выходящее из шахты, сорбирует некоторое
количество влаги и последующее увлажнение его на дисках не
требуется. При формовании волокна капрон подобное увлажнение
в шахте нецелесообразно, так как это волокно, содержащее
капролактам, может частично слипаться. Капроновая нить, выходящая
из шахты прядильной машины, почти не содержит влаги (не более
0,1—0,2%). Непосредственный прием нити с такой низкой
влажностью на бобину нецелесообразен, так как сухая нить сильно
электризуется. Кроме того, при постепенном увлажнении нить удли-
2.9. Формование полиамидных волокон
67
няется и намотка на бобине становится рыхлой. Нить постепенно
сползает с бобины, что делает невозможным последующее
кручение и вытягивание.
Для устранения указанного недостатка капроновую нит^
выходящую из шахты прядильной машины, пропускают над двумя ди-
Рис. 2.11. Фильерные комплекты:
а—для фильеры с числом отверстий от I до 40; б — для фильеры
с числом отверстий от 40 до 80.
сками, вращающимися в ванночках с водной эмульсией замасли-
вателя. Нить, проходя над дисками, несколько увлажняется и
замасливается. Затем нить огибает два прядильных диска (сначала
нижний, потом верхний) и наматывается на бобину (рис. 2.12).
Влажность капроновой нити, содержащей 3,5—4,5%
низкомолекулярных фракций, после подобной обработки составляет 4—5%.
Для замасливания (препарации) применяют обычно водные
эмульсии минерального масла. Иногда в состав препарирующих
композиций вводят вещества, склеивающие волокна и
облегчающие последующую обработку нити. Количество замасливателя
составляет 0,8—1,5% от массы волокна.
Эмульсии, наносимые на нить при выходе ее из шахты
прядильной машины, должны:
значительно снижать обрывность нити при вытягивании и
кручении;
уменьшать электризуемость волокна;
обладать достаточной стойкостью к повышенным температурам;
легко смываться с волокна при водных обработках [80].
Обычно основой замасливателя являются масла, в частности
вазелиновое масло, недостаточно стойкое при повышенных
температурах термической вытяжки; оно постепенно разлагается, что
обусловливает пожелтение нити. Для устранения этого недостатка
вазелиновое масло и другие масла, входящие в препарирующий
состав, были заменены [81, с. 92] полисилоксановой жидкостью,
которая не изменяется при нагреве до 200 °С. В качестве бо*
лее вффективных вмульгаторов вместо обычно применяемых
68
Глава 2. Производство полиамидных волокон
сульфированных препаратов, обладающих пониженной
термостойкостью, предложено использовать оксиэтилированные соединения.
Препарирующий состав, включающий полисилоксановую
жидкость и оксиэтилированные эмульгаторы, обладает достаточно
высокой стойкостью к повышенным температурам и поэтому
применяется на практике.
Рис. 2.12. Приемная часть прядильной машины для полиамидных
волокон.
Равномерного увлажнения нити по ее толщине при
кратковременной обработке (при скорости формования 800 м/мин нить
соприкасается с диском только 0,01—0,08 с) не удается достигнуть.
Поэтому выравнивание влажности происходит при длительном
выдерживании нити на бобине, вследствие чего намотка становится
более рыхлой.
Чтобы замедлить поглощение влаги волокном и тем самым
сохранить требуемую плотность намотки нити на бобине, в
прядильном и крутильном цехах поддерживают пониженную влажность
воздуха. Обычно относительная влажность воздуха в прядильном
цехе при 18—22 °С составляет 40—45%. При формовании волокна
найлон 6,6 [82, с. 137] относительная влажность воздуха в цехе
может достигать 70—72%.
Для уменьшения влажности воздуха прядильного цеха при
формовании капроновой нити его приходится охлаждать, что связано
со значительным расходом холода и электроэнергии. Поэтому
разработка методов равномерного и быстрого увлажнения волокна,
выходящего из шахты прядильной машины, представляет большой
практический интерес.
2.9. Формование полиамидных волокон 69
2-9.1. Основные параметры процесса формования
Основными параметрами процесса формования полиамидных
волокон из расплава являются скорость формования, вязкость
расплава, температура на плавильной решетке и в шахте, толщина
злеі\іентарного волокна и комплексной нити.
Скорость формования. Как уже указывалось, скорость
формования из расплава значительно выше, чем из раствора полимера.
Полиамидная нить формуется со скоростью от 500 до 1200 м/мин.
На некоторых заводах полиамидного волокна скорость
формования повышают до 4000—5000 м/мин. Чем больше толщина
элементарного волокна, тем меньше скорость формования.
В результате высокой скорости приема нити фильерная
вытяжка при формовании волокна из расплава значительно больше,
чем при формовании из раствора. Например, при скорости
формования 800 м/мин фильерная вытяжка составляет 2000—2500%-
Несмотря на такую фильерную вытяжку, на бобину принимается
почти неориентированное волокно. Это объясняется тем, что
вытягивание происходит в основном около фильеры, когда
образующееся из расплава волокно еще не застыло и, следовательно,
устойчивая и необратимая ориентация макромолекул не может
быть достигнута.
Однако при дальнейшем повышении скорости приема нити
ориентация агрегатов макромолекул и соответственно прочность
волокна значительно повышаются. Например, при увеличении
скорости приема нити до 4000—5000 м/мин происходит такая же
ориентация макромолекул, как при формовании волокна со скоростью
800—1000 м/мин и последующем вытягивании его на 350—400%-
Следовательно, при формовании волокна с такой высокой
скоростью необходимость вытягивания его на крутильно-вытяжных
машинах отпадает.
При скорости более 2500 м/мин, когда в процессе формования
происходит ориентация макромолекул волокна, существенное
влияние на физико-механические свойства нити оказывает величина
фильерной вытяжки [83].
Зависимость физико-механических показателей капроновой
нити от фильерной вытяжки * характеризуется следующими
данными:
Диаметр отверстий
фильеры, мм
0,25
0,6
1,0
3,8
Фильерная
вытяжка, %
120
168
480
6100
Прочность,
гс/текс
28,8
35,5
34,8
36,5
Удлинение.
%
120
120
93,5
84,8
* Число отверстий в фильере постоянное; скорость формования составляла
8000 м/мнн.
70 Глава 2. Производство полиамидных волокон
Как видно из приведенных данных, с увеличением фильерной
вытяжки закономерно повышается прочность нити и снижается
удлинение.
При повышении скорости формования на 100 м/мин
максимально достигаемая степень вытягивания на последующих стадиях
технологического процесса уменьшается примерно на 10%,
прочность нити, получаемой на прядильной машине, повышается на
2 гс/текс B0 мН/текс) и удлинение снижается на 2%. Так, если
при скорости приема нити на прядильной машине 600 м/мин
максимальная степень последующего вытягивания волокна составляет
470%, то при приеме той же нити со скоростью 1160 м/мин она
может быть вытянута в тех же условиях всего на 350%.
Следовательно, при изменении скорости формования необходимо учитывать
соответствующее изменение степени последующего вытягивания
нити.
Вязкость расплава. Обычно вязкость расплава на плавильной
решетке составляет 500—1500 П E0—150 Па • с).
Температура на плавильной решетке. Температура, которую
надо поддерживать на плавильной решетке, зависит от вязкости
расплава полиамида при его плавлении. Чем больше вязкость
расплава, тем выше должна быть температура на плавильной
решетке, чтобы обеспечить требуемую вязкость -расплавленной
массы, проходящей через песочные фильтры и отверстия
фильеры.
Чем выше температура, тем больше опасность частичного
разложения полиамида и тем тщательнее должна быть проведена
очистка азота от следов кислорода. Максимально допустимое
содержание кислорода в азоте, непрерывно пропускаемом над
плавильной решеткой, при формовании волокна капрон и анид
составляет 0,005%. При 290—300 °С начинается термическая деструкция
и разложение полиамида. Следовательно, повышение температуры
на плавильной решетке выше 280—290 °С недопустимо.
Поскольку температура плавления полиамида типа найлон 6,6
выше температуры плавления капрона, температура на
плавильной решетке при формовании волокна найлон 6,6 должна быть
выше. Возможность изменения температуры при формовании
волокна найлон 6,6 более ограничена, чем при формовании волокна
капрон. Это существенный недостаток процесса формования
волокна из полиамида найлон 6,6.
Реакция полимеризации капролактама, как уже указывалось,
обратима. Поэтому при нагревании на плавильной решетке поли-
капроамида, из которого в процессе экстракции в основном
удалены мономер и другие низкомолекулярные водорастворимые
продукты, вновь образуется некоторое количество мономера, димера
и тримера. Так как общая продолжительность нахождения поли-
капроамида при температуре 250—260 °С (на плавильной решетке,
в прядильном насосике и фильере) не превышает 20—30 мин, ко-
2.9 Формование полиамидных волокон 71
личество мономера в полиамиде ниже, чем то, которое
соответствует состоянию равновесия при данной температуре.
Содержание мономера в волокне капрон составляет 3—4%, а общее
содержание водорастворимых продуктов — 5—6%- Однако наличие даже
такого количества низкомолекулярных продуктов в волокне в
большинстве случаев нежелательно, так как, выделяясь из волокна, эти
примеси оседают на деталях машин, в результате чего
затрудняется последующая обработка нити. Поэтому волокно капрон
в большинстве случаев подвергают промывке для удаления
низкомолекулярных фракций.
При формовании волокна найлон 6,6 мономер не образуется,
и поэтому лить не промывают.
Температура в шахте. Влияние температуры в шахте на
свойства получаемого волокна исследовано недостаточно. Согласно
опубликованным данным [84], при повышении температуры в шахте
удлинение невытянутого волокна снижается. Например, при
температуре в шахте 20, 120, 175 °С удлинение невытянутой нити
составляет соответственно 405, 285 и 200%.
Если формуемая нить проходит через обогреваемую шахту, то
полностью вытянутое волокно, не требующее последующего
вытягивания, получается при скорости формования 2000 м/мин, а не
4000 м/мин, как это имеет место при движении нити через необо-
греваемую шахту.
В обычных условиях формования значительно повышать
температуру в шахте нецелесообразно. Если сформованное при
нормальной температуре в шахте волокно принимать в ванну,
температура которой 50—60 °С, оно получается аморфным. При
последующем нагреве при температуре выше 180°С это волокно
кристаллизуется. В результате повышается. плотность, значительно
(в 5—6 раз) увеличивается модуль Юнга, снижается
гигроскопичность и количество красителя, сорбируемого волокном.
Толщина элементарного волокна и комплексной нити. Толщина
комплексной нити, получаемой формованием из расплавов, может
изменяться в значительно более широких пределах, чем при
формовании из раствора. Это объясняется тем, что при формовании
из расплава нет необходимости испарять большие количества
растворителей, а также высокой прочностью и эластичностью нити,
вследствие чего создается возможность получения моноволокна
толщиной 300—500 текс и тонкой нити толщиной 3—1,5 текс,
используемой в трикотажной промышленности. Толщина
элементарного волокна составляет 0,4—0,2 текс. При получении нити для
технических целей толщина элементарного волокна может быть
повышена. Необходимо учитывать, что в результате последующего
вытягивания на 350—400% толщина нити уменьшается в 3,5—4
раза. Следовательно, на прядильной машине нить должна быть
в 3,5—4 раза толще готовой нити.
72 Глава 2. Производство полиамидных волокон
2.9.2. Получение волокна капрон
непрерывным методом
При непрерывном процессе полимеризации капролактама и
формования волокна до поступления полимера в фильеру из него
должны быть удалены капролактам и его олигомеры; для этого
расплав подвергают демономеризации. В зависимости от условий
проведения этого процесса, и особенно от его аппаратурного
оформления, на прядильную машину может поступать расплав,
содержащий различное количество низкомолекулярных фракций.
Содержание низкомолекулярных фракций в расплаве может
быть примерно такое же D—4,5%), как в расплаве, поступающем
в фильеру при формовании волокна описанным выше
периодическим методом. Это волокно должно быть дополнительно промыто
для удаления повышенного количества низкомолекулярных
фракций, затрудняющих последующую переработку волокна (см.
разд. 2.10).
В поступающем на прядильную машину расплаве, а
следовательно и в получаемом волокне, может содержаться пониженное
количество капролактама и низкомолекулярных фракций, не
превышающее 2—2,5% от массы полимера, т. е. столько же, сколько
в расплаве, подаваемом в фильеру из экструдера. При таком
содержании низкомолекулярных фракций дополнительной промывки
волокна, как показал длительный опыт работы, не требуется.
Для демономеризации поликапроамида (или, точнее, для
удаления основного количества мономера и части олигомеров)
предложены два метода: отгонка с перегретым паром и отсос под
вакуумом. Каждый из этих методов имеет свои преимущества и не
достатки.
Демономеризация поликапроамида перегретым паром. При
отгонке капролактама перегретым паром методом, разработанным
Киевскими экспериментальными мастерскими совместно с
Киевским комбинатом химических волокон, расплав подается к
прядильному насосику через форсунку, в которой к вытекающим
струйкам полимера подводится сухой перегретый пар при 300°С.
Пар увлекает с собой пары лактама и димера, а расплав подается
насосиком через песочный фильтр в фильеру. Вязкость расплава
полиамида в результате указанной обработки значительно
повышается.
Преимуществом этого метода является отсутствие аппаратуры,
работающей под вакуумом, и необходимости применения вакуум-
насосов. Недостатком — повышенное содержание
низкомолекулярных фракций, остающихся в расплаве, а следовательно, и в
волокне, составляющее 4—4,2 %•
Имеются данные [85], что при очень небольшом содержании
воды в расплаве удается снизить содержание низкомолекулярных
фракций в получаемом волокне до 3,2%.
2.9. Формование полиамидных волокон 73
Капролактам, отгоняемый с паром, конденсируется, образуя
10— 15%-ный водный раствор, который подвергается
упариванию.
Метод демономеризации поликапроамида паром, по-видимому,
целесообразно применять при формовании штапельного волокна,
для которого несколько повышенное содержание
низкомолекулярных фракций не имеет существенного значения.
Демономеризация поликапроамида под вакуумом. Отсос паров
капролактама под вакуумом производится в тонком слое,
стекающем по стенке аппарата, при остаточном давлении 1 мм рт. ст.
A33,3 Па). В расплаве, находившемся под вакуумом 30—40 мин,
остается 2—2,5% водорастворимых фракций. Если остаточное
давление снизить до 0,5 мм рт. ст. F6,6 Па), содержание этих
фракций уменьшается до 1%.
Вследствие низкого содержания водорастворимых фракций
в расплаве можно с помощью вакуумной демономеризации
получить нить (без последующей промывки), содержащую менее 3%
низкомолекулярных фракций, соответствующую по этому
показателю требованиям, предъявляемым к кордной нити. Следовательно,
этот метод демономеризации может быть использован для
получения не только штапельного поликапроамидного волокна, но и
кордной и текстильной нитей, которые могут быть переработаны в
изделия без промывки и сушки.
Как уже указывалось выше, при отгонке под вакуумом
удаляются не только пары капролактама, но и пары димеров, а также
некоторое количество тримеров капролактама. Пары этих олиго-
меров, обладающие более высокой температурой кипения, чем
пары капролактама, осаждаются на трубопроводах конденсатора
отгоняемого капролактама, что осложняет работу установки и
обусловливает необходимость ее периодической остановки для
чистки.
Для устранения указанного недостатка советские исследователи
предложили [86] каталитически разлагать олигомеры до
капролактама. Для этого пары отсасываемых низкомолекулярных фракций,
содержащие 2—10% циклических олигомеров, направляют в
вакуум-аппарат, заполненный гранулированным катализатором.
Разложение олигомеров происходит при 240—280 °С и остаточном
давлении 2—5 мм рт. ст. B66,6—666,5 Па) в течение 0,5—1 мин.
Из вакуум-аппарата пары отогнанного капролактама поступают
на конденсацию. Полученный 98%-ный капролактам направляется
на ректификацию. Введение дополнительного аппарата заметно не
усложняет систему демономеризации поликапроамида.
Эффективность непрерывного метода. Осуществление
непрерывного метода синтеза полиамида и формования из него волокна
даст значительный технико-экономический эффект. Расчеты
показывают, что при производстве кордной нити из поликапроамида
по непрерывной схеме (без промежуточного получения крошки)
74 Глава 2. Производство полиамидных волокон
капитальные затраты снижаются на 13%, эксплуатационные
расходы— на 10% и трудовые затраты — на 17%.
На процесс удаления низкомолекулярных соединений из
капроновой крошки при синтезе поликапроамида периодическим
способом приходится 53,1% капитальных вложений и 51,8% трудовых
затрат в химическом цехе завода капронового волокна {87].
Метод непрерывной полимеризации и формования волокна
капрон применяется в производственных условиях при получении
штапельного волокна и кордной нити. Этот же метод может быть
использован и при получении текстильной нити, при формовании
которой количество расплава, подаваемого в единицу времени на
прядильную машину, значительно меньше. Однако при получении
полиамидной текстильной нити в большинстве случаев пока
используется описанный выше так называемый полунепрерывный
метод (непрерывный процесс полимеризации мономера, дробления
полимера, экстракции и сушки крошки и последующее плавление
ее в экструдере). Так как время пребывания крошки в экструдере
не превышает 5 мин, то и без демономеризации в фильеру
поступает расплав поликапроамида, содержащий только 1,5—2%
низкомолекулярных фракций. В этом случае промывка полученной
текстильной нити также является излишней.
Приведенные выше данные свидетельствуют о перспективности
и целесообразности более широкого применения непрерывного
метода получения капронового волокна, в первую очередь в
производстве кордной нити.
Как уже указывалось, непрерывным методом можно получить
и волокно найлон 6,6, но для этого требуется повысить
термостабильность полиамида.
2.10. ПОСЛЕДУЮЩАЯ ОБРАБОТКА НИТИ
При производстве капроновой текстильной нити отделка ее
(подготовка, к переработке на текстильных предприятиях) ранее
включала следующие операции: 1) предварительное кручение; 2)
вытягивание нити с одновременным кручением; 3) окончательное
кручение; 4) промывку на бобинах с одновременной фиксацией
крутки; 5) сушку на бобинах; 6) перемотку на бобинажных
машинах; 7) сортировку и упаковку.
В настоящее время число отделочных операций, как это будет
показано ниже, заметно уменьшилось.
Условия кручения полиамидной нити существенно отличаются
от кручения нитей из искусственных волокон. Основные отличия
заключаются в том, что процесс кручения совмещен с
вытягиванием нити в 3,5—4 раза. Крутильно-вытяжные машины, на
которых осуществляется вытягивание, по конструкции (наличие
вытяжных приспособлений) несколько отличаются от крутильных ма-
2.10. Последующая обработка нити
75
шин, применяемых для кручения текстильных нитей из
искусственных волокон.
Применявшийся ранее для повышения связности элементарных
волокон в нити процесс предварительного подкручивания нитей на
кольцекрутильных машинах исключен из общей схемы
технологического процесса текстильной обработки нити. Для повышения
связности элементарных волокон нить подшлихтовывают; в
препарирующий состав добавляют немного поливинилового спирта,
который склеивает между собой
элементарные волокна.
Необходимая величина крутки
полиамидной нити может быть
достигнута в одну стадию путем
кручения с одновременным
вытягиванием или (в большинстве случаев)
путем дополнительного кручения
после вытягивания. Так как крутиль-
но-вытяжная машина сложнее по
конструкции, чем обычная
крутильная машина, для повышения ее
производительности требуемую крутку
обычно получают в две стадии. На Рис. 2.13. Изменение свойств по-
первой стадии нить подвергается
одновременно кручению и
вытягиванию, на второй стадии нить
докручивают на обычной крутильной
машине.
Вытягивание полиамидного волокна — одна из важнейших
операций технологического процесса производства. Как уже
указывалось выше, характерной особенностью полиамидных волокон
является их способность вытягиваться при нормальной
температуре на 300—400%. В результате происходит значительное
повышение степени ориентации макромолекул или их агрегатов в
волокне, что приводит к соответствующему изменению механических
свойств. Получить высококачественное полиамидное волокно,
обладающее ценными механическими, а следовательно, и
эксплуатационными свойствами, без вытягивания не представляется
возможным. Изменение свойств полиамидного волокна после вытягивания
показано на рис. 2.13. Как видно из этих данных, при
вытягивании повышается прочность, модуль эластичности и теплостойкость
волокна, а удлинение снижается.
При вытягивании полиамидного волокна степень его
кристалличности (содержание кристаллической фракции)
заметно не изменяется [88]. Теплота растворения в
муравьиной кислоте ориентированного полиамидного волокна лишь
незначительно меньше, чем неориентированного A1,53 кал/моль
вместо 12,64 кал/моль). Плотность полиамидного вытянутого
Степень вытягивания
лиамидного волокна при его
вытягивании:
/ — прочность; 2 — удлинение; 3 — модуль
эластичности; 4—плотность;
5—понижение прочности при 150 °С.
76 Глава 2. Производство полиамидных волокон
свежесформованного волокна составляет 1,13 г/см3, а после
вытягивания на 300% — 1,132—1,138 г/см3. Продольное набухание волокна
в воде после вытягивания уменьшается в 2—2,5 раза, а
гигроскопичность волокна типа капрон снижается с 4,8 до 3,8%.
Данные [89] о влиянии степени вытягивания на свойства
волокна найлон 6,6 приводятся ниже:
Степень Прочность, Удлинение, Гигроскопич-
вытягнвания гс/текс , % ность •*, %
250
345
447
500
538
607* '
2,2
3,1
4,6
5,8
6,6
8,2
160
89
50
36
22
21
4,67
4.52
4,29
3,90
3,75
3,54
* При нагревании.
** При 25 °С и относительной влажности воздуха 72%.
При вытягивании полиамидного волокна выше определенного
оптимума в результате снижения эластического удлинения может
уменьшиться величина эластической работоспособности *,
являющаяся, по Берингеру, одним из основных критериев для оценки
носкости и потребительской ценности изделий. Следовательно, для
получения волокна, обладающего наиболее высокими
эксплуатационными свойствами и соответственно наибольшим значением
эластической работоспособности, необходимо обеспечить не
максимальную, а оптимальную степень вытягивания. По данным этого
исследователя, максимальная эластическая работоспособность
полиамидного волокна, используемого для изготовления изделий
народного потребления, достигается при вытягивании на 330%, т. е. в
4,3 раза.
Оптимальная степень вытягивания, при которой получается
волокно, обладающее наиболее высокими потребительскими
свойствами, зависит от его толщины, а также от условий эксплуатации
изделий. При получении полиамидной кордной нити, которая
должна иметь наиболее высокую прочность, степень вытягивания
увеличивают до 400%. При такой степени вытягивания прочность
нити из полиамида с нормальным молекулярным весом
увеличивается до 65—75 гс/текс F50—750 мН/текс), а из полиамида
повышенного молекулярного веса—до 80—82 гс/текс (800—
820 мН/текс).
Схема вытягивания нити на крутильно-вытяжной машине
показана на рис. 2.14. Предварительно подкрученная невытянутая
нить со шпули / поступает через нитепроводник 2 на питающий
цилиндр 4 и затем в вытяжной механизм, который состоит из диска
7, получающего принудительное вращение, вспомогательного ро-
* Эластическая работоспособность — произведение прочности волокна
[о кгс/мм2 (Н/м2)] на высокоэластическое удлинение (в %).
2.10. Последующая обработка мити
77
лика 6 и неподвижной тормозной агатовой палочки 5.
Вытягивание нити происходит на участке между питающим цилиндром и
диском и начинается непосредственно после прохождения ею
агатовой палочки. Степень вытягивания определяется соотношением
скоростей цилиндра и диска. На вытяжном механизме нить делает
один-два оборота вокруг диска 7 и
вспомогательного ролика 6. Затем
нить проходит через нитепровод-
ник 8, бегунок 9, подвергается
кручению и наматывается на патрон 10. /-
Тормозная агатовая палочка 5
фиксирует то место, где начинается
вытягивание нити. Наличие этой
палочки на крутильно-вытяжной
машине имеет существенное
значение для получения нити с
однородными физико-механическими
свойствами.
Скорость питания на машине
100—300 м/мин, в зависимости от
толщины нити. Число оборотов
веретена в 1 мин 8 000—10 000. Чем
тоньше нить, тем больше скорость
питания. Допустимая степень
вытягивания нити и возможность
осуществления этого процесса без обрыва
волокон зависят от ряда факторов,
в частности от качества полимера и
условий проведения всех
предыдущих стадий технологического
процесса.
На заводе им. К. Маркса изго- Рис. 2.14. Схема вытягивания нити
товлена высокоскоростная крутиль- на крутильно-вытяжной машине:
но-вытяжная машина КВ-150-И4. /-шпуля; 2,
10
питяцио va
ПИТаНИЯ На
МаШИНе
у; нитещюводпики;
3 —фарфоровый валик; 4 —питающий ци-
линдрГ 5_тормозиая агатовая палоч-
6 й 7
линдр 5тормозиая агатовая палоч
СОСТЭВЛЯеТ 550—750 М/МИН, ЧИСЛО ка-6 — вспомогательный ролик; 7 — диск;
г і 9 — бегунок; 10 — патрон для вытянутой
OOOpOTOB Веретен В 1 МИН — ОТ нити.*
5000 до 8000; обычно нить
получает крутку 15—30 витков/м. Схема заправки и прохождения
нити при вытягивании на машине КВ-150-И4 показана на
рис. 2.15.
В отдельных случаях вытягивание полиамидной нити
производится непосредственно на прядильной машине и скорость приемки
нити составляет 3500—4000 м/мин. Этот метод, имеющий свои
преимущества и недостатки по сравнению с вытягиванием на крутйль-
но-вытяжных машинах, используется для вытягивания нитей
толщиной 15—20 текс.
Рис. 2.15. Схема прохождения нити на крутилыю-вытяжнои машине
КВ-150-И4:
/ — поворотный кронштейн; г—натяжное приспособление; 3— ннтеводитель;
4—грузовой валнк; 5—цилиндр питающего аппарата; 6 — свободный ролик; 7— кольцедер-
жатель; 8— направляющая стойка; 9— приспособление для приема заправочного
конца (невытянутогр участка нити).
2.10. Последующая обработка нити 79
Условия вытягивания полиамидной нити зависят от [90]:
качества полиамида, в частности молекулярного веса,
содержания низкомолекулярных фракций и влажности;
скорости формования;
температуры формования;
равномерности нити по толщине;
препарирующего состава;
влажности и температуры воздуха в крутильном цехе, а также
от содержания влаги в свежесформованном волокне;
температуры вытягивания.
Наличие следов кислорода при полимеризации мономера или
формовании волокна может привести к образованию частично
сшитого полимера и тем самым к значительному ухудшению
способности нити к вытягиванию.
При содержании в полиамиде более 0,1% влаги в процессе,
формования элементарных волокон в них могут образоваться
пузырьки, также ухудшающие способность нити к вытягиванию.
Как уже указывалось выше, с увеличением скорости
формования и величины фильерной вытяжки уменьшается максимальная
степень вытягивания нити на крутильно-вытяжной машине.
Чем больше равномерность элементарных волокон по толщине,
тем меньше возможность их обрыва при вытягивании нити.
Большое влияние на условия вытягивания оказывает замасли-
ватель, применяемый на прядильных машинах. При
использовании водных эмульсий в нить вносится повышенное количество
влаги, вследствие чего на ее поверхности высаживаются
низкомолекулярные фракции. Вытягивание в этом случае затрудняется
в результате загрязнения нитепроводящих деталей на крутильно-
вытяжных машинах.
Заметно влияют на вытягивание волокна влажность и
температура воздуха в крутильном цехе. Относительная влажность
воздуха в цехе при кручении капроновой нити, содержащей
капролактам, должна составлять 55—65%, а при кручении нити типа
найлон 6,6 может быть повышена до 72%. Температура в цехе должна
поддерживаться в пределах 17—18 °С. При температуре ниже 16 °С и
выше 28—30 °С способность нити к вытягиванию ухудшается [91].
По вопросу о возможности сравнительно длительное время
выдерживать полиамидную нить перед ее вытягиванием единой точки
зрения пока нет. По одним данным [92], во избежание затруднений
при вытягивании свежесформованное волокно до его поступления
на вытяжную машину можно выдерживать не более 4—6 ч; по
другим данным [93], выдерживание невытянутого полиамидного
волокна в течение 30 сут не ухудшает его способности к вытягиванию.
На крутильно-вытяжной машине нить получает крутку в
среднем 20—30 витков/м. Такая сравнительно высокая крутка не
является необходимой для многих изделий, вырабатываемых из
Полиамидных нитей. Общая тенденция к снижению крутки и тещ
80 Глава 2. Производство полиамидных волокон
самым к значительному уменьшению трудоемкости текстильных
операций, характерных для производства текстильных нитей в прр-
мышленности химических волокон вообще, имеет место и в
производстве полиамидных нитей. Значительное количество текстильной
нити выпускается с круткой 8—15 витков/м, что приводит к
повышению производительности крутильно-вытяжных машин.
На передовых предприятиях для повышения
производительности труда паковки нити, снимаемые с крутильно-вытяжных
машин, увеличены с 300—500 г до 2—3 кг.
Механические свойства полиамидной нити, вытянутой при
нормальной температуре, удовлетворяют требованиям большинства
потребителей. Однако кордная нить должна иметь более ^высокую
прочность и, что особенно существенно, пониженное удлинение, не
превышающее 15—20%. Для получения такой нити вытянутая
полиамидная нить подвергается дополнительному вытягиванию на
100—150% при повышенной температуре A00—150°С). При этом
прочность нити возрастает на 10—20 гс/текс A00—200 мН/текс),
а удлинение снижается до 15—20%. Одновременно заметно
повышается теплостойкость и модуль эластичности нити. Благодаря
этому улучшаются эксплуатационные свойства полиамидного
корда, т. е. уменьшается разнашиваемость шин.
Обе стадии вытягивания высокопрочной кордной нити
осуществляются на одной и той же крутильно-вытяжной машине.
Первая стадия вытягивания (на 350—400%) проводится при 20°С на
пути между бобиной с нитью и первым диском. Дополнительное
вытягивание (на 100—150%) осуществляется между
обогреваемыми электричеством до 165—170 °С вторым и третьим диском.
Вытягивание кордной нити при повышенной температуре можно
провести также над нагретой поверхностью (утюгом) между
вторым и третьим диском, вращающимися с различной скоростью.
Дополнительная крутка необходима также для текстильной
нити, применяемой, например, для изготовления борта чулок. Это
кручение вытянутой нити проводится на этажных крутильных
машинах. Скорость питания на этих машинах 60—90 м/мин; скорость
вращения бобины 800—1000 об/мин. Дополнительная крутка
достигает 100—120 витков/м. Для тех изделий, которые изготовляются
из нитей с высокой круткой A000 витков/м), дополнительное
кручение нити проводится на крутильных фабриках.
Вытянутая полиамидная нить в горячей воде обычно
усаживается на 6—8%. Во избежание этого скрученную нить
целесообразно подвергнуть термообработке горячей (80—100 °С) водой или
насыщенным паром при 100—120 °С в автоклавах. Ввиду сильной
усадки нити бобины должны быть изготовлены из прочного
материала— стали или дюралюминия. Термообработка необходима
также для снятия напряжений вытянутой нити и фиксации крутки.
Для капроновой нити процесс термообработки совмещают
с промывкой горячей водой с целью вымывания водорастворимых
2.11. Особенности производства полиамидной кордной нити 81
низкомолекулярных фракций, образовавшихся в процессе
формования. Капролактам вымывается из нити при промывке ее на
бобине водой при 90—95 °С. Эта операция применяется все реже, так
как нить, содержащую до 2,5—3% низкомолекулярных фракций,
не требуется промывать. Такое содержание низкомолекулярных
фракций характерно для нити, получаемой современными
непрерывными методами (см. разд. 2.9.2), и при использовании метода
экструдерного формования волокна из крошки (см. разд. 2.9.1).
Как показали результаты исследований, проведенных в
последние годы на Энгельсском комбинате химических волокон и иа дру^
гих заводах, производящих капроновую пить, промывка ее для
удаления низкомолекулярных фракций может быть исключена и
при переработке обычной текстильной нити, используемой в
трикотажной и ткацкой промышленности. В этом случае отпадает
необходимость сушки и последующей перемотки нити, вследствие
чего уменьшаются затраты труда в отделочных цехах.
При промывке текстильной нити препарационный состав
смывается. Наличие в промывной воде веществ, примененных для
препарации, затрудняет регенерацию из нее капролактама. После
окончания промывки нить на бобинах обычно снова обрабатывают пре-
парационным составом.
Аппаратурное оформление процесса промывки полиамидного
волокна аналогично аппаратурному оформлению процесса
промывки вискозной текстильной нити на бобинах.
После окончания промывки и препарации в большинстве
случаев через слой нити на бобине 3—5 мин продувают воздух. Затем
бобины отжимают в центрифуге. Содержание влаги в нити- после
отжима снижается с 30 до 10%. Отжатую нить сушат в сушилках
4—6 ч при 90—95 °С. Высушенную нить выдерживают в помещении
с кондиционной влажностью воздуха F5%) До поглощения
стандартного количества влаги C,5—4%). После этого нить
перематывают на бобинажных машинах на шпули, сортируют и
упаковывают. Если препарирующий состав не был нанесен после промывки,
то нить обрабатывают при перемотке. В качестве препарирующих
веществ рекомендуется применять кроме замасливателя
поливинил ацетат, поливиниловый спирт, эфиры полиакриловой кислоты
с небольшими добавками борной кислоты и глицерина или гликоля
для уменьшения электризуемое™ волокна, а также поверхностно-
активные вещества. Количество препарирующих веществ,
наносимых на нить, составляет от 3 до 5% от ее массы.
2.11. ОСОБЕННОСТИ ПРОИЗВОДСТВА ПОЛИАМИДНОЙ
КОРДНОЙ НИТИ
Полиамидные волокна являются основным материалом,
используемым для производства шинного корда. В этой важнейшей
области применения высокопрочной нити полиамидный корд в
последние годы все в большей степени вытесняет вискозный корд и
82 Глава 2. Производство полиамидных волокон
занял почти монопольное положение при изготовлении кордной
ткани для авиационных и грузовых автомобильных шин.
Основными преимуществами полиамидной кордной нити по сравнению
с вискозной являются: более высокая прочность (на 30—40%),
значительно более низкая плотность, что позволяет уменьшить
массу кордной ткани в шинах, и более высокие усталостные
характеристики, обеспечивающие повышенную ходимость шин.
Ходимость шин с каркасом из полиамидного корда на 30—50%
больше, чем с каркасом из вискозного корда, что имеет большое
технико-экономическое значение. Указанные обстоятельства и
определяют большую эффективность использования полиамидной
кордной нити, несмотря на то, что стоимость ее в наших условиях
на 20—25% выше стоимости вискозной кордной нити.
В Советском Союзе в ближайшие годы удельный вес
полиамидного корда в общем производстве этого материала резко возрастет.
Необходимо, однако, отметить, что полиамидная кордная нить
имеет недостатки, которые, по-видимому, будут ограничивать
дальнейшее расширение областей ее применения. Основным
недостатком полиамидных нитей является низкий начальный модуль.
Вследствие этого каркас, а следовательно, и сама шина при
эксплуатации разнашивается, и тем самым повышается ее
сопротивление качению. Другой недостаток —значительное снижение
прочности нити при повышенных температурах, имеющих место при
эксплуатации шин A00—120°С).
Основные особенности производства высокопрочной
полиамидной кордной нити сводятся к следующему.
1. Для формования волокна применяется поликапроамид,
молекулярный вес которого на 20—30% выше молекулярного веса
полимера, используемого для производства текстильной нити и
штапельного волокна. Получение полиамида более высокого молекулярного
веса достигается изменением количества регулятора, вводимого в
реакционную смесь при полимеризации.
2. Экстракция водорастворимых фракций из измельченной
крошки проводится более тщательно; содержание этих фракций
в полиамиде не должно превышать 0,5%.
3. Толщина кордной нити в последнее время на большинства
заводов повышена до 90 и даже 180 текс. Естественно, что при
этом значительно увеличивается производительность каждого
прядильного места. Число отверстий в фильере при формовании
кордной нити толщиной 90 текс равно 140. Скорость формования
кордной нити достигает 550 м/мин.
4. Суммарная степень вытягивания нити составляет 420—450%,
причем при вытягивании на крутильно-вытяжных машинах
относительно тонкой кордной нити весь процесс проводят при нормальной
температуре, а кордную нить толщиной 90 текс и более
вытягивают в две стадии: на 350—400% при нормальной температуре и
дополнительно на 100—150% при повышенной температуре.
2.12. Особенности производства полиамидного штапельного волокна 83
5. Крутка нити на крутильно-вытяжной машине составляет
20 витков/м. После дополнительного кручения на машинах
двойного кручения суммарная крутка составляет 420—450 витков/м для
нити толщиной 90 текс и 320—350 витков/м для нити толщшгой-
180 текс.
6. В результате сильного вытягивания прочность кордной нити
достигает 70—75 гс/текс, а удлинение соответственно снижается до
15—17%. Так же как и для других типов кордной нити, одним из
направлений рационализации производства полиамидной кордной
нити является увеличение ее толщины. Например, при повышении
толщины капроновой кордной нити с 90 до 180 текс число слоев
кордной ткани в каркасе шины может быть уменьшено с 14 до 10.
Согласно подсчетам [94] такое увеличение толщины кордной нити
дает возможность уменьшить на 11 % капитальные затраты на
производство кордной ткани и на 21% сократить затраты труда.
Значительная экономия достигается и в шинной промышленности.
Повышение производительности труда рабочих на прядильных
кордных машинах может быть достигнуто при увеличении массы
паковки нити. Например, в последнее время применением цепных
нитеукладчиков на прядильных машинах удалось увеличить массу
паковки кордной нити до 12 кг.
2.12. ОСОБЕННОСТИ ПРОИЗВОДСТВА ПОЛИАМИДНОГО
ШТАПЕЛЬНОГО ВОЛОКНА
Добавление 15—20% полиамидного штапельного волокна
к хлопку или к шерсти значительно повышает устойчивость
полученных изделий к различным деформациям и резко увеличивает
срок их службы. Поэтому применение штапельного полиамидного
волокна в смеси с другими волокнами для изготовления изделий
технического назначения (например, технических сукон) и товаров
народного потребления имеет большое народнохозяйственное
значение.
Схема производства штапельного волокна несколько отличается
от схемы производства текстильной и кордной нитей. Как правило,
капроновое штапельное волокно всегда получают непрерывным
методом.
¦ Расплав полиамида подается к прядильной головке под
давлением 15—20 кгс/см2 A,5 • 106—2 • 106 Па) по трубопроводу,
обогреваемому динилом или электричеством. В каж'дой шахте
прядильной машины устанавливается несколько фильер D—6), что
обеспечивает значительное уменьшение габаритных размеров
прядильных машин. Число отверстий в фильере составляет 200—300.
Важной задачей является дальнейшее увеличение числа отверстий
в фильере и соответственно повышение производительности
прядильного места.
84
Глава 2. Производство полиамидных волокон
Штапельное полиамидное волокно формуют с меньшей
скоростью D00—500 м/мин), чем текстильную нить. Получаемое
волокно вытягивают в 3—3,5 раза на вальцах в несколько стадий.
При вытягивании толстого жгута полиамидных волокон он
разогревается до 100 °С. После вытягивания прочность штапельного
волокна составляет 37—38 гс/текс C70—380 мН/текс).
и
Рис. 2.16. Схема механизма для непрерывного вытягивания и резки волокна;
1, 2 — диски; 3 —крючки; 4 —полые оси вращения; 5 —тарелки; ff —манжеты; 7 —дисковый
нож.
Советскими исследователями [95] разработан непрерывный процесс
получения штапельного волокна, начиная от полимеризации капролактама и кончая
сушкой вытянутого резаного волокна. Непрерывная полимеризация капролактама
производится в трубах НП. После демономеризации расплавленный полимер
поступает к групповым прядильным головкам на 4 фильеры и затем
индивидуальными зубчатыми иасосиками подается к отдельным фильерам.
Следующая стадия непрерывного процесса — вытягивание жгута толщиной
500—200 текс—производится на машине оригинальной конструкции. Вытягивание
жгута происходит не между роликами или дисками, вращающимися с
различной скоростью, а путем растяжения его коротких участков с помощью крючков.
Схема механизма для непрерывного вытягивания и резки волокна приведена
на рис. 2.16.
Механизм состоит из дисков / и 2, по окружности которых на равном
расстоянии один от другого укреплены крючки 3. Угол между плоскостями дисков
устанавливаетси в зависимости от требуемой степени вытягивания жгута.
Вытягивание волокна производится при скорости формования 500—600 м/мии.
2.13. Особенности производства полиамидного моноволокна 85
Вытянутое и разрезанное волокно вентилятором подается в циклон, а затем
В каскадную башню отделочной машины. После отмывки капролактама и
замасливания волокно поступает на сушку.
Волокно с пониженным удлинением C0—35% вместо 60—70%
для обычного штапельного полиамидного волокна) можно получить
различными методами, в частности дополнительным вытягиванием
при повышенной температуре, как это имеет место при вытягивании
кордной нити, обработкой жгута горячей водой под натяжением и
последующей сушкой его также под натяжением. Однако эти
операции усложняют технологический процесс. Поэтому производство
штапельного волокна с пониженным удлинением целесообразно
только в том случае, если заметно улучшается его переработка
в текстильной промышленности.
Для облегчения переработки полиамидного штапельного
волокна в смеси с другими волокнами необходимо повысить его
сцепляемость и.извитость. Для этого могут быть использованы
следующие методы {96]:
получение волокна из сополимеров;
применение профилированных фильер;
придание извитости волокну путем механической обработки;
непродолжительная обработка химическими реагентами,
вызывающими набухание волокна.
Хорошая извитость может быть придана волокну
непродолжительной B—5 мин при 30—40 °С) обработкой химическими
реагентами, например 23%-ной серной кислотой, 57—63%-ным
раствором хлорида цинка или разбавленным 2—4%-ным фенолом.
Аналогичный эффект достигается и при обработке волокна 60—
7О°/о-ным водным раствором капролактама при 90—100 °С в течение
1—5 мин.
Однако все эти.методы химической обработки требуют
дополнительного расхода реагентов и специальной аппаратуры для их
осуществления. Поэтому наиболее широко используется
механический способ — гофрирование жгута с последующей его резкой.
2.13. ОСОБЕННОСТИ ПРОИЗВОДСТВА
ПОЛИАМИДНОГО МОНОВОЛОКНА
Полиамидное моноволокно толщиной 500—300 текс обладает
достаточно высокой эластичностью. В последнее время тонкое
моноволокно A,6—2,2 текс) нашло сравнительно широкое
применение для изготовления не только лески и щетины, но и чулочных
изделий.
Применение моноволокна вместо нити, состоящей из большого
числа волокон, представляет большой интерес, так как при этом
устраняется наиболее трудоемкий процесс — кручение,
необходимый для обеспечения связности волокон в нити.
Глава 2. Производство полиамидных волокон
Особенно~большой интерес представляет использование
моноволокна соответствующей толщины в качестве кордной нити.
Успешное решение этой задачи значительно упростит и удешевит
производство полиамидного корда.
При производстве моноволокна повышенной толщины процесс
формования всегда осуществляется по непрерывной схеме.
Вытекающий из аппарата расплавленный полиамид подается насосиком
Рис. 2.17. Вытягивание полиамидного моноволокна.
в фильеру, выдавливаемая из нее струйка расплава поступает в
воду и застывает. Образующееся моноволокно принимается на
мотовило или на другую паковку и затем вытягивается в 3,5—4 раза
(рис. 2.17). Вытянутое моноволокно обычно подвергают
термофиксации. Производительность прядильного места при производстве
моноволокна повышенной толщины составляет 100—150 кг/сут.
Прочность моноволокна при диаметре 0,55 мм достигает 27—
40 кгс/мм2 B,7-108—4-Ю8 Н/м2), удлинение 35%.
2.14. ИСПОЛЬЗОВАНИЕ ОТХОДОВ
Одной из особенностей производства полиамидных волокон,
в частности текстильной нити, является повышенное количество
отходов (рвани), в несколько раз превышающее количество отходов,
2.14. Использование отходов 87
получающихся при производстве искусственных волокон. Отходы
образуются в результате формования с большой скоростью и при
кручении, особенно при вытягивании с одновременным кручением
нити, когда возможен обрыв отдельных волокон.
Одна из основных задач рационализации технологического
процесса производства полиамидного волокна — резкое уменьшение
количества отходов на отдельных стадиях процесса путем
повышения стабильности полиамида, улучшения условий формования
волокна и его вытягивания. Однако до тех пор, пока в производстве
полиамидного волокна имеются отходы, особенно важно
рационально использовать их.
Отходы на различных 'стадиях технологического процесса
различаются по свойствам. Отходы, получаемые на прядильной
машине, представляют собой некрученое и невытянутое волокно;
отходы при кручении и вытягивании — крученую и вытянутую нить.
В отличие от большинства видов химических волокон
полиамидные волокна, и особенно капрон, могут быть снова расплавлены
или деполимеризованы до мономера. Эта особенность определяет
основные направления использования отходов волокна из поли-
капроаиида.
Вытянутые нити могут быть использованы после некоторых
дополнительных обработок для получения штапельного волокна. Этот
способ использования отходов наиболее эффективен.
Все остальные волокнистые отходы, получаемые в производстве
капроновых волокон, а также отходы полиамидов, образующиеся
в химическом цехе, могут быть переработаны без разложения
полимера до мономера или с разложением полиамида до мономера.
Отходы полиамида и волокна расплавляют и после фильтрации
используют для получения моноволокна, а иногда и штапельного
волокна. Эти же отходы могут быть переработаны литьем под
давлением в разнообразные изделия (шпули, челноки и т. д.).
Указанные методы использования отходов сравнительно просты,
и в тех случаях, когда они не очень загрязнены и волокно не
окрашено, повторное плавление их целесообразно.
Разложение отходов полиамида и волокна капрон до капролак-
тама осуществляется нагреванием со щелочью при 300—310°С.
В этих условиях равновесие между полимером и мономером
сдвигается в сторону образования мономера. Получаемый капролактам
отгоняется и после многократной дистилляции под вакуумом снова
применяется для полимеризации. При этой обработке
капролактам частично разлагается, в результате чего выход мономера не
превышает 75—85% от массы обрабатываемого полимера. Более
высокий выход мономера достигается при обработке отходов
горячей водой C00 °С) под давлением. При такой обработке выход
мономера составляет свыше 90% от массы полимера.
В последнее время сравнительно широкое применение получил
процесс деполимеризации отходов капронового производства в
88 Глава 2. Производство полиамидных волокон
кислой .среде. Для этого отходы предварительно разрезаются,
размалываются на дезинтеграторе и промываются раствором соды для
удаления препарирующего состава, находящегося в волокне.
Деполимеризация полиамида проводится в две стадии:
частичная деполимеризация водой, содержащей небольшое количество
фосфорной кислоты @,7—1 % от массы полиамида), при 180°С;
для окончательной деполимеризации в аппарат подается
дополнительное количество этой кислоты до образования 10—15%-ного
водного раствора Н3РО4. Эту обработку проводят при 250°С в
течение 3 ч. Продукты реакции очищают на ионитах, обрабатывают
для окисления примесей раствором КМпО4, выделяющиеся
нерастворимые примеси отфильтровывают, а образовавшийся
капролактам направляют на ректификацию. Выход капролактама
составляет 82—85% от теоретического.
Для регенерации мономера могут быть использованы все виды
отходов, в том числе окрашенные и матированные волокна.
Для разложения отходов, получаемых при производстве
волокна найлон 6,6, применяют более сложные методы.
2.15. СВОЙСТВА ПОЛИАМИДНЫХ ВОЛОКОН
Полиамидные волокна обладают комплексом ценных свойств,
определяющих целесообразность, а в ряде случаев необходимость
их широкого использования для изготовления разнообразных
изделий. Остановимся на показателях, характеризующих основные
свойства полиамидных волокон.
Прочность. Полиамидные волокна имеют высокую прочность
при разрыве — 40—50 гс/текс в сухом состоянии. Путем увеличения
степени вытягивания волокна до 400—420% прочность можно
повысить до 70—75 гс/текс. Если "нить подвергнуть дополнительному
вытягиванию при повышенной температуре или повысить
молекулярный вес полиамида, прочность нити может быть доведена до
80—85 гс/текс. Однако такое повышение прочности целесообразно
только при получении кордной нити, строп, канатов и других
аналогичных изделий, при эксплуатации которых высокая разрывная
прочность имеет основное значение.
Уменьшение прочности в мокром состоянии у полиамидного
волокна не превышает 10%.
Эластичность. Полиамидные волокна характеризуются высокими
эластическими свойствами, что определяет значительную величину
обратимых удлинений и устойчивость к многократным деформациям.
Удлинение полиамидных нитей составляет 20—25%. Для
кордной нити требуется более низкое удлинение, не превышающее 15—
18%. Снижение удлинения достигается в ряде случаев
дополнительным вытягиванием волокна. Удлинение волокна в мокром
состоянии на 3—5% выше, чем в сухом.
2.15. Свойства полиамидных волокон 89
Полностью обратимое удлинение составляет 35—40% от общего
удлинения волокна. При нагрузке до 20 кгс/мм2 B-Ю8 Па)
эластическое удлинение волокна капрон составляет 90—95% от
общего удлинения, в то время как у шерсти при таких же нагрузках
оно не превышает 60—65%. а у вискозного штапельного волокна
{97] —30—40%.
Прочность полиамидной нити в узле достигает 95% от
прочности обычной нити (без узелка), у шерсти этот показатель равен
85%.
Устойчивость к многократным деформациям, характеризуемая
в известной степени числом двойных изгибов, выдерживаемых
волокном до разрыва, у полиамидного волокна примерно в 100 раз
больше, чем у вискозного штапельного волокна, и в среднем в 10
раз больше, чем у хлопка [97]. По этому показателю полиамидные
волокна превосходят все природные и химические волокна.
Устойчивость полиамидного волокна к многократным деформациям, так же
как и к ряду других воздействий, значительно изменяется в зависимости от
молекулярного веса и, по-видимому, от химического состава полиамида (числа
метиленовых групп в элементарном звене). Например, при повышении
молекулярного веса полиамидного волокна перлон с 10 000 до 15 000 и затем до
18 000 число двойных изгибов, выдерживаемых волокном до разрыва,
повышается соответственно с 500 до 1000 и до 6000.
Недавно опубликованы интересные данные [98] о том, что окрашенные
пигментными красителями полиамидные волокна обладают в 5—8 раз более
высокой усталостной прочностью, чем неокрашенные волокна. Авторы объясняют
это тем, что частицы пигментов, так же как других наполнителей, выполняя
роль зародышей кристаллизации, обусловливают ускоренную кристаллизацию
полимеров и соответственно уменьшение размеров кристаллитов, в результате
чего улучшаются усталостные свойства волокна.
Истираемость. Полиамидные волокна обладают наиболее
высокой устойчивостью к истиранию, превосходя по этому практически
важному показателю все другие волокна. Если устойчивость
полиамидного волокна к истиранию принять за 100%, то у хлопка
оно составит 10%, у шерсти 5% и у вискозного штапельного
волокна 2% {99]. Такая высокая устойчивость к истиранию и
определяет целесообразность добавления небольших количеств
полиамидного штапельного волокна к другим волокнам, в частности
к шерсти, для значительного повышения устойчивости получаемых
изделий к истиранию.
Гигроскопичность. Полиамидные волокна характеризуются
сравнительно невысокой гигроскопичностью. При относительной
влажности воздуха 65% эти волокна поглощают 3,5—4% влаги.
Плотность. Полиамидные волокна имеют значительно меньшую
плотность, чем искусственные и природные. Плотность волокна
капрон и найлон 6,6 составляет 1,135—1,14 г/см3, волокна унде-
кан— 1,04 г/см3.
Теплостойкость и термостойкость. Полиамидные волокна
обладают недостаточно высокой теплостойкостью. При температуре
140 °С прочность полиамидного волокна обратимо снижается на
90
Глава 2. Производство полиамидных волокон
40—50%. Одновременно заметно повышается удлинение и
снижается модуль эластичности.
Полиамидные волокна, полученные из алифатических
полиамидов, обладают невысокой термостойкостью, что является
существенным недостатком этого класса синтетических волокон.
Сравнительно непродолжительный нагрев приводит к значительному
необратимому снижению прочности полиамидного волокна.
Например, после нагрева 5 ч при 140°С
прочность полиамидного
волокна, определенная при
нормальной температуре, снижается на
40%, а удлинение —на 70%.
При нагреве полиамидного
волокна при 160 °С на воздухе в те-
. чение 1 ч степень полимеризации
макромолекул волокна снижается
в 2 раза. Если это волокно нагре-
_^ ^ вать в среде азота в течение та-
с по по 160 180 zoo кого же времени, степень поли-
Теплературл,°С меризации почти не снижается
Рис. 2.18. Изменение степени поли- Даже ПРИ 180°С (РИС- 218>- ЭтИ
700
80
меризации макромолекул
полиамидного волокна при прогреве 1 ч:
/—на воздухе; 2—в среде азота.
данные подтверждают
предположение о том, что недостаточно
высокая термостойкость
полиамидных волокон объясняется
интенсивным процессом термоокислительной деструкции полимера
при повышенных температурах.
Термостойкость полиамидного волокна можно значительно
повысить {100] при добавлении небольших количеств различных ан-
тиоксидантов или стабилизаторов. В качестве добавок могут быть
использованы органические и неорганические вещества.
К веществам, рекомендуемым для практического использования
в качестве термостабилизаторов, предъявляется ряд требований,
из которых основное значение имеют следующие: 1) высокая
эффективность в процессах термо- и фотохимической деструкции; 2)
стойкость к высоким температурам при синтезе и переработке
полиамидов; 3) хорошая совместимость с полиамидом; 4) низкая
летучесть; 5) малая растворимость в воде; 6) по возможности
отсутствие способности окрашивать полиамидную нить. Кроме того,
термостабилизаторы не должны быть токсичными. Этому
комплексу требований удовлетворяет только сравнительно
незначительное количество стабилизаторов, получивших или получающих в
настоящее время практическое применение.
К наиболее эффективным стабилизаторам относятся
неорганические стабилизаторы, в частности смесь солей меди и KI в
соотношении 1 : 1, добавляемых в количестве 0,1—0,2% от массы
мономера. Однако растворимость этих реагентов в воде, значительно
2.15 Свойства полиамидных волокон
SI
ограничивает возможность их применения, например, для
термостабилизации капроновой кордной нити. При промывке вытянутой
нити горячей водой эти стабилизаторы в большей или меньшей
степени вымываются. Поэтому данную смесь можно применять для
термостабилизации полиамидных волокон типа анид, которые при
производстве не подвергаются водным обработкам, или для
капронового волокна, получаемого в условиях, при которых промывка
100
80
60
40
*Г
N. #
N
О л
j
2
j
1 1 1
150 175
Температура, С
г оо
Рис. 2.20. Влияние стабилизатора
@,5 %) иа термостойкость капронового
корда:
/ — стабилизированный корд; 2—нестабили-
зированиый корд.
•з- о . го \о бо во wo
Продолжительность прогрвЗа, ч
Рис. 2.19. Влияние стабилизатора
@,5%) иа изменение молекулярного
веса полиамида (характеризуемого
значением удельной вязкости) после
нагрева при различных температурах:
/—стабилизированный корд структуры
35.5/4,2; 2— нестабилизированиый корд
^структуры 34,5/4,2.
нити на бобинах является излишней. Необходимо отметить, что
эти соли частично могут вымываться и при пропитке кордной ткани
на шинных заводах.
Из органических стабилизаторов детально исследованы в
нашей стране и уже широко применяются производные /г-фенилен-
диамина [101], в частности И.^-ди-р-нафтил-га-фенилендиамин:
При введении в полиамид 0,4—0,5% этого стабилизатора
прочность волокна после нагрева 2 ч при 200 °С снижается на 15—
20%, в то время как нестабилизированная кордная нить в тех же
условиях теряет 80—85%) начальной прочности.
При введении более 0,5% этого стабилизатора (от массы
волокна) понижается однородность структуры волокон и
соответственно снижается прочность. В результате введения стабилизатора
92 Глава 2. Производство полиамидных волокон
происходит уплотнение как поверхностных, так и внутренних слоев
волокна [102]. Некоторые данные об эффективности действия этого
стабилизатора приведены на рис. 2.19 и 2.20.
При добавлении стабилизатора одновременно резко
увеличивается число двойных изгибов, выдерживаемых волокном.
Например, если нестабилизированный корд выдерживает при 130 °С до
разрушения 27 тыс. циклов, то стабилизированный корд в тех же
условиях обработки выдерживает 340 тыс. циклов.
Если нагревать стабилизированную и нестабилизированную
кордные нити при указанных температурах в атмосфере инертного
газа, прочность и число двойных изгибов почти не изменяются.
Следовательно, стабилизатор повышает устойчивость полиамидных
волокон только к термоокислительным воздействиям.
Однако этот стабилизатор имеет ряд существенных недостатков,
затрудняющих возможность его практического применения.
Основными недостатками являются [103]: 1) изменение цвета
(потемнение) при действии света, что ограничивает использование
стабилизированного волокна; 2) уменьшение прочности связи капроновой
ткани с резиной при хранении шины вследствие миграции
стабилизатора на поверхность, что приводит к снижению ее
эксплуатационных свойств; 3) повышенная токсичность [104].
В качестве стабилизатора применяется также соединение амин-
ного типа следующего состава {105, с. 158J:
-О—С2Н4ОС2Н4—О-
СН3О—f \>—NH HN—f х)—OCH,
Обычно для этой цели используются производные оксидифенил-
амина с различными заместителями в молекуле. Наиболее
эффективными термостабилизаторами этого класса являются 2,2-
бисC-метоксифениламино)феноксидиэтиловый эфир (так
называемый стабилизатор Н-1) и 1,4-бисфеноксидиатиловый эфир [106].
Преимущество этих стабилизаторов перед описанным выше
заключается в том, что они не изменяют адгезии кордной нити
к резине даже при длительном хранении. Однако и эти
стабилизаторы также постепенно, хотя и менее интенсивно, окрашивают
волокна. Поэтому отходы кордной нити и ткани, содержащие
стабилизаторы М,Ы-ди-р-нафтил-я-фенилендиамин или стабилизатор
Н-1, нельзя использовать в текстильной промышленности для
изготовления изделий различного ассортимента [103]. Недостатком
обоих стабилизаторов является также их летучесть в процессе
вытягивания при повышенной температуре и при термообработке
кордной ткани.
К органическим термостабилизаторам относятся также
металлсодержащие гетероциклические соединения, особенно хелатные со-
2,15, Свойства полиамидных волокон 93
единения меди (стабилин 9). Основными преимуществами этих
стабилизаторов перед описанными выше являются [103] невы-
мываемость и нелетучесть, а также возможность получения
волокон, не окрашивающихся даже при длительном хранении.
Адгезия ткани к резине при введении стабилина 9 не
ухудшается. Эффективность его действия такая же, как и других
термостабилизаторов, но усталостная прочность волокон со
стабилином 9 в несколько раз меньше, чем с Ы^-ди-р-нафтил-п-фе-
нилендиамином.
Эффективными стабилизаторами являются аминофенолы,
особенно такие, в молекуле которых группы NH2 находятся в гс-поло-
жении к ОН группам. Отсутствие свободной ОН группы в молекуле
фенолсодержащего стабилизатора резко снижает его
эффективность [107]. '
В качестве термо- и фотостабилизаторов, не окрашивающих
волокно при длительном хранении, могут быть использованы и
другие классы органических веществ, в частности продукт
конденсации формальдегида и замещенных монофенолов. Эти фенолы
имеют следующий состав:
Однако ингибиторы фенольного типа (действие которых, так же
как и аминов, основано на взаимодействии с образующимися
в макромолекуле полиамида радикалами и превращении их в
малоактивные соединения) менее активны, чем ингибиторы,
полученные на основе производных ароматических аминов.
Фосфорорганические соединения, преимущественно продукты
взаимодействия фосфорной кислоты с фенолами, и особенно с бис-
фенолами, значительно повышают стойкость волокон к термо- и
фотоокислительной деструкции. Однако использование этих
ингибиторов, как правило, нецелесообразно. При повышенной
температуре они распадаются с выделением фенолов, которые, так же как
при применении ингибиторов фенольного типа, реагируют с
макромолекулами полиамида, обусловливая их частичное сшивание
в процессе формования. Вытягивать частично сшитые волокна
трудно из-за повышенной обрывности.
Сравнительные испытания указанных выше стабилизаторов
показали, что по комплексу свойств — прочности при нормальной
температуре, термостойкости (сохранении прочности после нагрева
2 ч при 200°С), устойчивости связи с резиной, и в особенности по
устойчивости к многократным деформациям, наилучшими
свойствами обладает кордная нить, содержащая стабилизатор Н-1.
Однако эти данные не являются бесспорными, и для
окончательного вывода необходимо провести дополнительные исследования.
94
Глава 2. Производство полиамидных волокон
Антиоксиданты, или вещества, ингибирующие
термоокислительную деструкцию полимера, протекающую по радикальному
механизму, находят все более широкое промышленное применение.
В большинстве стран основное количество полиамидной кордной
нити выпускается с такими добавками. Этот метод повышения
термостабильности не является специфичным для полиамидных
волокон и может быть использован для всех других химических
волокон.
Стабилизатор можно вводить в полиамид опудриванием крошки
или добавлением его в раеплавленный капролактам при
полимеризации.
В работе [108] показано, что при наличии антиоксидантов
капроновое волокно можно формовать не в токе азота, а на воздухе.
Если это подтвердится при дальнейшей производственной проверке,
процесс формования термостабилизированного волокна значительно
упростится. Очень существенно, что стабилизаторы, являющиеся
одновременно ингибиторами цепных радикальных процессов, ловы-
шают не только термостойкость волокна, но и его усталостную
прочность.
Светостойкость. Недостаточно высокая светостойкость
полиамидного волокна, особенно матированного двуокисью титана,—
существенный недостаток синтетических волокон этого вида.
Пониженная светостойкость полиамидов объясняется их
сравнительно легкой окисляемостью. Это подтверждается тем, что
в среде инертного газа (N2, CO2) скорость фотохимической
деструкции полиамидов очень невелика. Наиболее легко окисляются ме-
тиленовые группы, находящиеся в элементарном звене
макромолекулы рядом с группой NH.
Светостойкость полиамидного волокна можно значительно
повысить введением в волокно небольших добавок солей металлов,
в частности марганца и хрома. Некоторые данные,
характеризующие влияние солей марганца на изменение прочности и удлинения
волокна перлон после облучения, приведены в табл. 2.3.
Таблица 2.3. Влияние солей марганца * на изменение прочности
и удлинения волокна перлон после облучения [109, с. 452]
Добавка
Без добавки (матированное волокно)
Ацетат марганца
Нитрат марганца
Стеарат марганца
Хлорид марганца
Потери после облучения, %
к начальному значенню
прочности
82
20
16,3
6,91
1,84
удлинения
85
5,6
20,5
12,5
0,38
* Соли марганца добавлены в количестве 0,01 моль на 1 моль капролактаме.
2.15 Свойства полиамидных волокон
Уменьшение потери прочности в результате облучения
достигается и при добавлении солей хрома. Например, волокно найлон
6,6, обработанное солями хрома @,02—0,2% в пересчете на
металлический хром), после 4 месяцев облучения на солнечном свету
(весна, лето) потеряло 25% прочности, в то время как
необработанное волокно в тех же условиях потеряло 80% прочности [100].
Соли хрома, в частности ацетат хрома, можно добавлять в
небольших количествах (не более 0,1%) перед полимеризацией капро-
лактама. В таком количестве он не влияет на процесс
полимеризации.
Повышения светостойкости можно достигнуть и введением в
волокно органических добавок, в частности салола, гидрохинона,
резорцина, а также 2-меркаптобензимидазола @,1 — 1% от массы
волокна).
Существенным недостатком большинства светостабилизаторов,
так же как и термостабилизаторов, является окрашивание волокна
и их постепенная миграция на поверхность волокна в процессе
переработки и эксплуатации изделий, что приводит к ухудшению
внешнего вида изделий и снижению эффективности вводимых
добавок.
В большинстве случаев стабилизаторы и антиоксиданты,
значительно тормозящие процессы термоокислительной деструкции,
замедляют и процесс фотохимической деструкции. Это имеет место
при добавлении органических и неорганических соединений и
подтверждает идентичность механизма распада макромолекулы
полиамида при термоокислительной и фотохимической деструкции.
Однородность структуры. Полиамидное волокно, полученное формованием
из расплава, так же как и волокна, полученные формованием нз раствора, имеет
неоднородную структуру. Например, волокно снлон, вырабатываемое методом
непрерывной полимеризации, формования и вытягивания, характеризуется
наличием поверхностного слоя (ориентационной оболочки), который набухает
в 25%-ной серной кислоте значительно менее интенсивно, чем внутренний слой.
Толщина этого поверхностного слоя составляет 2,2 мкм. Температура плавлення
поверхностного слоя выше, чем внутреннего; окрашивается он менее
интенсивно [110].
Значительное понижение степени кристалличности волокон найлон 6 и 6,6 и
соответственно снижение прочности и повышение удлинения имеет место при
обработке их раствором иода в насыщенном водном растворе иодида калня [111].
Данные, характеризующие изменение механических свойств ннтей найлон 6 н
6,6 после их обработки при 20 °С в течение 4 ч 20%-ным раствором нода в
насыщенном водном растворе нодида калия, приводятся ниже:
Условия обработки
Без обработки
Обработка горячей водой . .
Обработка раствором нода,
промывка холодной водой
Обработка раствором иода,
промывка горячей водой
п и 60-70 °С
Прочность,
гс/текс
58
48
27—28
30-32
Удлинениеі
20
30
35-38
50
96 Глава 2. Производство полиамидных волокон
При обработке в указанных условиях нить усаживается на 15—22%. Если
обработку нити производить в тех же условиях раствором KI, не содержащим
иода, то свойства ее заметно не изменяются. Дальнейшее изучение этого
явления, и особенно причин неодинаковых изменений свойств разных видов
полиамидных волокон при обработке их раствором иода в водном растворе иодида
калия, может дать практически важные результаты.
Другие показатели. Полиамидные волокна, так же как и другие
синтетические волокна, обладают стойкостью к действию
микроорганизмов (гниению), но недостаточно стойки в условиях тропиков.
Кроме того, эти волокна нестойки к щелочам и концентрированным
минеральным кислотам, а также к окислителям.
Наряду с высокими потребительскими и эксплуатационными
свойствами полиамидные волокна имеют ряд существенных
недостатков, затрудняющих их переработку и практическое
использование.
Недостатки полиамидных волокон и некоторые способы их
устранения. К основным недостаткам полиамидных волокон
относятся следующие.
Низкий модуль эластичности. Полиамидные волокна
имеют значительно меньший модуль, чем другие химические
волокна. Например, усилие, необходимое для вытягивания
капронового волокна на 1%, в 4—5 раз ниже, чем полиэтилентерефталат-
ного. Из-за низкого модуля затрудняется, как уже указывалось,
использование полиамидного корда в шинах. Начальный модуль
волокна капрон в 2 раза ниже, чем найлона 6,6.
Пониженная сцепляем ость. Чрезмерная гладкость
полиамидных волокон обусловливает пониженную сцепляемость их
с другими волокнами. Например, при смешивании с шерстью
капроновое штапельное волокно в процессе эксплуатации «вылезает»
на поверхность ткани, вследствие чего нарушается структура и
ухудшается внешний вид изделия. Повышенная гладкость волокна
и малая его сцепляемость с другими волокнами вызывает отрыв
отдельных нитей от общей структуры ткани и образование ворса
на ее поверхности. В результате высокой прочности полиамидных
волокон, и особенно их высокой устойчивости к истиранию, эти
нити не обрываются, а скатываются на поверхности ткани в
шарики, что также ухудшает внешний вид изделий (так называемый
«пиллинг эффект»).
Повышенной гладкостью полиамидных нитей объясняется
также и частый спуск петель в чулках и других трикотажных
изделиях из полиамидных волокон, а также неприятный блеск этих
изделий.
Для уменьшения гладкости полиамидных волокон их
обрабатывают кислотами или другими реагентами (в частности, раствором
ZnCl2), однако при этом полностью не устраняется указанный
недостаток.
Наиболее интересным и реальным методом устранения
чрезмерной гладкости волокна является, по-видимому, метод формования
2,15. Свойства полиамидных волокон
97
полиамидного волокна через фильеры с профилированными
(рис. 2.21) отверстиями [112]. Получаемые этим методом волокна
называются профилированными. Поперечные срезы таких волокон
в зависимости от формы отверстий фильеры показаны на рис. 2.22.
Рис. 2.21. Форма отверстий профилированных фильер.
Механические свойства профилированных волокон, за
исключением удлинения, не отличаются от аналогичных свойств волокон,
получаемых при формовании через фильеры с круглыми
отверстиями. Волокна с профилированным сечением обладают
пониженным, блеском и почти не требуют матирования. Это обстоятельство
Рис. 2.22. Срезы профилированных волокон:
а—форма отверстий фильеры; б—форма поперечного среза волокна.
очень важно, если учесть трудность матирования полиамидных
волокон, особенно при их получении непрерывным методом, а также
резкое понижение светостойкости волокон при введении даже
небольших количеств TiO2.
Носкость чулок, полученных из профилированного полиамидного
волокна, благодаря меньшему числу узлов и затяжек,
образующихся в процессе их носки, больше носкости обычных полиамидных
чулок. При изготовлении чулок из профилированного
полиамидного волокна нет необходимости придавать высокую крутку
нити A000—1200 витков/м) во избежание спуска петель [113], а
4 Зак. ?31
Глава 2. Производство полиамидных волокон
достаточна крутка, равная 400—500 витков/м. Одновременно
повышается кроющая способность волокна, что приводит к уменьшению
на 20—23% расхода волокна при изготовлении многих изделий.
Профилированные волокна при сохранении других
механических свойств вытягиваются в меньшей степени, чем волокна
круглого сечения {114]. Это объясняется тем, что они формуются при
значительно большей фильерной вытяжке.
Полые профилированные волокна обладают более высокой
извитостью и устойчивостью к многократным деформациям, а также
меньшей плотностью (на 10—12%), чем обычные волокна.
Получение полых волокон реализовано преимущественно при
производстве штапельного волокна. Эти волокна характеризуются
более высокими теплоизоляционными свойствами, улучшенным
внешним видом, повышенной объемностью и сцепляемостью и
повышенными эксплуатационными свойствами. Прочность этих
волокон не ниже 30 гс/текс при удлинении 55—70%.
Метод получения профилированных волокон представляет
большой практический интерес, так как он дает возможность
существенно улучшить качество * изделий из полиамидных и других
видов синтетических волокон, формуемых из расплава. Изделия из
профилированных, и особенно из полых профилированных
волокон, лишены одного из основных дефектов, присущих изделиям из
полиамидных волокон, — пиллинга.
Плохой гриф. Полиамидные волокна недостаточно упруги на
оіцупь, т. е. имеют плохой гриф.
Недостаточная гигроскопичность. Вследствие
пониженной гигроскопичности полиамидных волокон изделия из них
препятствуют быстрому испарению пота, что ухудшает их
гигиенические свойства [115, 116]. Например, через перлоновые ткани пот
испаряется в 7 раз медленнее, чем через ткани из гидрофильных
волокон [117].
Сравнительная оценка волокон капрон и анид. Существенный
интерес для определения направления дальнейшего развития
производства полиамидных волокон имеет детальное изучение свойств
волокон капрон (найлон 6) и анид (найлон 6;6) и сопоставление
соответствующих показателей..
Опубликованные данные {118] показывают, что теплостойкость
полиамидных волокон не зависит от температуры их плавления.
Например, температура, при которой прочность нити снижается
на 50%, для волокон найлон 6 и найлон 6,6 одна и та же, степень
кристалличности также одинакова. Усталостная прочность и
светостойкость найлона 6 выше, чем найлона 6,6. Однако эти данные,
имеющие большое значение для определения преимуществ и недо-
* Опыт носки чулок, изготовленных из профилированного капронового
волокна, показал, что они лучше обычных чулок: меньше затяжек (в 3—4 раза)
и спусков петель (на 11%),
2.16. Модифицированные полиамидные волокна 99
статков основных типов полиамидных волокон, требуют
дополнительной проверки.
Области применения. Описанные выше свойства полиамидных
волокон определяют области их применения.
Полиамидная текстильная нить наиболее широко используется
для изготовления чулочно-носочных изделий, а также бельевых и
платьевых тканей.
Штапельное полиамидное волокно в смеси с другими волокнами
(хлопок, шерсть, вискозное волокно) широко применяется для
изготовления тканей. Содержание полиамидных волокон в смеси
обычно не превышает 20—25%. Так как другие волокна, входящие
в смесь, обладают высокой гигроскопичностью, пониженное
поглощение влаги полиамидным волокном в этом случае не имеет
существенного значения. Использование полиамидного штапельного
волокна в смеси с другими волокнами очень эффективно и дает
возможность значительно увеличить срок службы изделия. По
данным Берингера {118], при добавлении к вискозному штапельному
волокну 15% капронового штапельного волокна срок носки
мужских носков увеличивается с 87 до 170 дней, а при увеличении
содержания капронового штапельного волокна в смеси до 30% —
до 240 дней. Срок службы ткани из смеси, содержащей 70%
вискозного и 30% полиамидного штапельного волокна, на 35%
больше, чем ткани, изготовленной только из вискозного волокна.
Полиамидная техническая нить применяется при изготовлении
высокопрочной кордной ткани, прочных не гниющих рыболовных
сетей, строп, канатов, фильтровальных тканей, устойчивых к
действию различных химических реагентов-(кроме концентрированных
кислот) и технических тканей (в смеси с другими волокнами).
2.16. МОДИФИЦИРОВАННЫЕ ПОЛИАМИДНЫЕ ВОЛОКНА
Так же как и при направленном изменении свойств других
химических волокон, для модификации полиамидных волокон могут
быть использованы методы физической и химической модификации.
2.16.1. Методы физической модификации
полиамидных волокон
, Из различных методов физической модификации наибольшее
практическое применение получили:
методы получения высокопрочных полиамидных волокон;
методы получения высокообъемных и высокоэластичных нитей.
Высокопрочные полиамидные волокна и нити, так же как и
другие волокна, вырабатываемые из расплава, получаются, как
правило, при изменении условий вытягивания волокна. Основным
методом повышения степени вытягивания, а следовательно, ориентации
и прочности волокна, является, как уже указывалось выше, прове-
100 Глава 2. Производство полиамидных волокон
дение этого процесса при повышенной температуре. Большой
интерес представляет выяснение возможности регулировать на
отдельных стадиях технологического процесса надмолекулярную
структуру полиамидных волокон и степень их кристалличности.
Так же как и для других термопластичных химических волокон,
удельный вес высокообъемных и высокоэластичных полиамидных
нитей по отношению к общему количеству полиамидных нитей
непрерывно увеличивается. Для получения этих волокон
используются ранее описанные методы (см. т. I).
2.16.2. Методы химической модификации
полиамидов и волокон
Описанные выше полиамиды и полиамидные волокна,
получившие практическое применение, характеризуются регулярным
строением макромолекул (закономерным чередованием в них различных
атомных групп), что обусловливает образование большого числа
водородных связей и соответственно низкую растворимость
полимеров, и наличием ограниченного числа функциональных групп
в макромолекуле.
Для изменения в требуемых направлениях основных свойств
полиамидов (растворимости, температуры плавления и др.)
разрабатываются методы их модификации.
Для химической модификации свойств полиамидов и волокон
могут быть использованы следующие методы:
нарушение регулярности строения;
изменение соотношения между метиленовыми и амидными
группами;
введение новых функциональных групп;
введение ароматических ядер и гетероциклов;
синтез привитых сополимеров;
структурирование (сшивание) волокон;
получение химически окрашенных волокон.
Естественно, что волокна, получаемые из модифицированных
полиамидов, будут отличаться по своим свойствам от обычных
полиамидных волокон. Ниже приводятся краткие сведения об
основных методах химической модификации полиамидов и свойствах
волокон на их основе.
Нарушение регулярности строения. Этот метод модификации
сводится к получению сополимеров. Некоторое применение
получили продукты совместной поликонденсации капролактама и соли
АГ, а также капролактама и соли ТГ *.
При совместном нагревании (поликонденсации) капролактама
и соли АГ в тех же условиях, в которых получаются полиамиды
капрон и анид, образуется сополимер следующего строения:
OC(CH2MNHOC(CH2LCONH(CH2NNHOC(CH2MNH
¦ Соль ТГ — соль терефталевой кислоты и гексаметилендиамииа.
2.16. модифицированные полиамидные волокна
101
В отличие от полиамидов капр.он и анид продукт сополиконден-
сации не имеет регулярного строения, так как чередование
четырех, пяти и шести метиленовых групп, находящихся в
элементарном звене между амидными группами, в макромолекуле может быть
различно в зависимости от соотношения исходных компонентов.
Нарушение регулярности строения полиамида приводит к
уменьшению образования водородных связей между
макромолекулами и соответственно к повышению растворимости полиамидов
и снижению температуры их плавления. Кроме того, смешанные
полиамиды кристаллизуются
значительно трудней, чем полиамиды ре- |
гулярного строения.
Резкое повышение растворимости
полиамидов, получаемых сополикон-
денсацией, по сравнению с
растворимостью полиамидов капрон и анид
является одним из немногих
примеров в физико-химии полимеров,
когда различная растворимость
определяется не различиями в
молекулярном весе и химическом составе,
а только нарушением регулярности
строения макромолекул полимера.
Влияние соотношения мономеров
на свойства получаемого полиамида
показано на рис. 2.23 и 2.24.
Как видно из рис. 2.24, температура плавления сополимеров
значительно ниже, чем полиамидов капрон и найлон 6,6. Например,
продукт, полученный при совместной поликонденсации капролак-
тама E0 мол. %) и соли АГ E0 мол. %), плавится при 160—
165 °С. Одновременно значительно повышается растворимость
полиамидов.
Полиамиды, синтезированные из смеси мономеров, содержащей
от 25 до 70% капролактама, полностью растворяются в смеси
метанола или этанола и воды (90:10) при повышенных
температурах D0—50 °С). При этом образуются не только разбавленные,
но и концентрированные растворы. Эти модифицированные
полиамиды применяются для формования пленок из раствора и в
качестве лаков или клеев. Для получения волокон они пока не
используются.
Сополиамиды обладают также и повышенной
гигроскопичностью. Если, например, при 100%-ной относительной влажности
воздуха полиамиды типа найлон 6,6 сорбируют 7% воды, то сопо-
лиамид, содержащий 40% гексаметиленадипамида и 60%
капролактама, поглощает в тех же условиях 14% воды.
Полиамиды нерегулярной структуры могут быть получены не
только совместной поликонденсацией различных мономеров, но и
15 50 15
Соль АГ, %
Рис. 2.23. Влияние соотношения
мономеров на свойства
модифицированного полиамида:
/—растворимость в смеси метанола или
этанола и воды (9D : 10); 2 —
интегральная теплота растворения.
102
Глава 2. Производство полиамидных волокон
совместным плавлением полиамидов капрон и анид при 280—285°С
в течение 8—20 ч. В этих условиях происходит перераспределение
связей (переамидирование) между макромолекулами и получается
полиамид, аналогичный по ряду свойств сополимеру соли АГ и
капролактама. В отличие от полиамидов регулярной структуры,
мутных и непрозрачных, полиамиды нерегулярной структуры
обладают высокой прозрачностью.
Полиамидные волокна нерегулярного строения значительно
сильнее усаживаются, при повышенной температуре, чем волокна
Z6Q г
°* Z1Q
I
І 200
110
700
75 50 25
Капролактам, %
Рис. 2.24. Зависимость
температуры плавления
модифицированного полиамида от., соотношения
мономеров.
15 50 75
Соль fir, %
100
капрон и найлон 6,6. Для уменьшения усадки волокно необходимо
подвергать термофиксации. Например, если усадка
необработанных волокон в кипящей воде составляет 13%, то после нагрева при
100°С она снижается до 5%, а после нагрева при 130°С усадка
дополнительно снижается на 2%, т. е. до 3% {119]. Эти волокна
могут быть использованы в качестве одного из компонентов смеси
волокон при производстве объемных высокоэластичных нитей. '
Интересный тип модифицированного гетероцепного
синтетического волокна получен советскими исследователями [120] при
взаимодействии диэтиленгликольтерефталата с капролактамом при
температурах, используемых для синтеза полимеров из этих
мономеров. С изменением соотношения амидных и эфирных звеньев
в молекуле полученного полиамидоэфира закономерно изменяются
свойства волокна, сформованного из этого сополимера.
С увеличением содержания в смешанном полимере звеньев
сложного эфира (до 20% от массы сополимера) снижается
прочность и гигроскопичность волокна; одновременно повышается его
модуль и термостойкость. По-видимому, такое модифицированное
волокно, соединяющее ряд ценных свойств полиамидных и
полиэфирных волокон, может представить практический интерес.
Волокно, полученное из сополимера бициклического лактама,
в частности лактама 4-аминоциклогексанкарбоновой кислоты и
капролактама {121], обладает такой же теплостойкостью, как и
2.16. Модифицированные полиамидные волокна 103
капроновое волокно, но значительно более высокой
термостойкостью.
Модифицированное полиамидное волокно было получено также
методом привитой сополнмеризации. Реакцию сополимеризада«
инициировали перекисными и гидроперекисными группами,
которые предварительно вводили в волокно при использовании редокс-
системы Fe3+—Н2О2. Таким путем была осуществлена прививка
к капроновому волокну полимеров акрилонитрила, стирола, метил-
метакрилата и акриловой кислоты. Количество привитого полимера
составляло 'до 100—140% от массы полиамидного волокна [122].
Свойства полученных привитых сополиамидов, характер и
количество полимера, которое наиболее целесообразно использовать
для прививки, пока еще детально не исследованы. Работы в этом
направлении могут дать интересные и практически ценные
результаты для направленного изменения свойств модифицированных
полиамидных волокон.
Изменение соотношения между метиленовими и амидными
группами. Этот метод модификации полиамидных волокон можно
наиболее просто осуществить при получении полиамидов типа
найлон 6,6, применяя для поликонденсации различные дикарбоновые
кислоты и диамины {123].
Изменение соотношения между числом метиленовых и амидных
групп не влияет на растворимость полиамида (при сохранении
регулярной структуры), но значительно влияет на его температуру
плавления. Как правило, с увеличением числа неполярных
метиленовых групп, расположенных в макромолекуле полиамида между
полярными амидными группами, температура плавления полимера
снижается {124].
Полиамиды с нечетным числом метиленовых групп в
элементарном звене плавятся при более низкой температуре, чем
полиамиды с четным числом этих групп. Например, температура
плавления полиамида энант, в элементарном звене которого находится
6 метиленовых групп, на 10 °С выше, чем поликапроамида,
содержащего 5 таких групп в элементарном звене макромолекулы. Эта
особенность, которую необходимо учитывать при регулировании
свойств получаемых полиамидов, объясняется, по-видимому, боль-'
шей трудностью образования водородных связей между
макромолекулами с нечетным числом метиленовых групп в элементарном
звене.
С увеличением числа метиленовых групп в элементарном звене
увеличивается гибкость молекул и соответственно повышается
степень кристалличности полимера {125]. Одновременно возрастает
теплота плавления полиамида. Например, теплота плавления
полиамидов капрон, энант и ундекан составляет соответственно 12,5,
15,7 и 17,5 кал/г.
Благодаря повышению плотности упаковки при увеличении
числа метиленовых групп в элементарном звене макромолекулы
104
Глава 2. Производство полиамидных волокон
возрастает модуль эластичности, резко увеличивается число
двойных изгибов, выдерживаемых волокном, и уменьшается плотность
волокна. Это подтверждается следующими данными [126]:
Волокно Капрон
Число метиленовых групп в
элементарном звене 5
Плотность, г/см3 1,14
Прочность, гс/текс 44
Удлинение, % 28
Модуль (при 3%-ном растяжении),
кгс/мм3 (МН/м3) 166
A 660)
Число двойных изгибов при
напряжении 12 кгс/мм2 A20 МН/м2). . 2 800
Число циклов до разрушения
(устойчивость к истиранию) ...... 2300
Потеря прочности в мокром
состоянии, % 9
Гидрофильность (при 65%-ной от-
восительной влажности
воздуха), % 3,8
Энант
6
1,10
40
28
280
B800)
4 000
2 200
13
Пелартон
8
1,08
38-40
24.
420
D 200)
40 000
4 000
2,0
Ундекан
10
1,04
36-44
27
700
G 000)
56 000
6 000
0
2,2
1,4
0,6
Кроме того, с увеличением плотности упаковки повышается
стойкость полиамида к гидролизу (например, ундекан значительно
более стоек к гидролизу, чем
капрон) и ухудшается растворимость
в муравьиной кислоте [127].
Зависимость основных свойств
полиамидов от числа
метиленовых групп в элементарном звене
макромолекулы схематически
показана на рис. 2.25.
Частичная замена
метиленовых групп на атом кислорода или
серы приводит к снижению
температуры плавления полиамида [128].
Число петаленоВых групп
В макронолщле
В последнее время получены
волокна из поли^-амидов [129],
синтезированных полимеризацией ?-лактамов (см.
разд. 2.7). Этот класс полиамидов
плавится при высокой температуре
(например, поли-Р-валин при 296 °С, поли-
?-аминомасляная кислота при 340°С),
поэтому формовать волокно из
расплава этих полимеров не удается. Из поли-
?-амидов волокно формуют мокрым
способом из 15—30%-ных растворов полимера в муравьиной или серной кислоте
или в смеси роданистого кальция и метанола. Осадительной ванной служат
¦одяые растворы солей. Свежесформованиое волокно вытягивали в 7 раз;
прочность его 18—27 гс/текс, удлинение 30—35%. Гигроскопичность волокна такая
же, как капрона.
Рис. 2.25. Влияние числа метиЛено-
вых групп в элементарном звене
макромолекулы на изменение свойств
полиамидов:
/ — температура плавления; 2—степень
кристалличности; 3—плотность; 4—
стойкость к гидролизу; 5—модуль эластичности.
2.16. Модифицированные полиамидные волокна 105
Из-за значительно более сложного процесса формования, необходимости
получать волокна из растворов, а не из расплава, сравнительно низких
механических свойств волокон и отсутствия каких-либо преимуществ перед обычными
полиамидными волокнами волокна из поли^-амидов неперспективны.
Введение новых функциональных групп. Как уже указывалось,
одним из методов модификации является введение в
макромолекулу полиамида новых функциональных групп.
Введение метильной группы. При частичном
замещении водорода в группе NH (N-замещенные полиамиды) или
в группе СН2 (С-замещенные полиамиды) группой СН3
значительно уменьшается интенсивность межмолекулярного
взаимодействия и соответственно снижается температура плавления
полиамида [130, с. 134]. Так, при замене водорода метильной группой
в 25% групп NH (от их общего числа в молекуле) температура
плавления полиамида снижается с 255 до 200 °С [131], а при замене
водорода в 50% групп NH — до 60°С. При замене водорода
метильной группой во всех группах NH и, следовательно, при
практически полном устранении возможности образования водородных
связей между макромолекулами вместо твердого, прочного,
высокоплавкого полиамида получается жидкость, затвердевающая при
очень низкой температуре (—75°С).
Данные об изменении температуры плавления и удлинения
полиамидного волокна найлон 6,6 при замещении водорода в
различном числе групп NH группой СНз приводятся ниже:
Число групп СНз. % Температура Удлинение
от общего числа групп плавления, °С волокна, %
0
25
50
70
100
255
200
60
—50
—75
20
150
1400
__
__
По-видимому, при применении для производства волокон N-за-
мещенных полиамидов невысокой степени замещения удается
дополнительно повысить эластичность волокон, т. е. получить каучу-
коподобные волокна.
Уменьшение интенсивности межмолекулярного взаимодействия
и понижение температуры плавления имеет место и при получении
С-замещенных полиамидов. В этом случае на температуру
плавления влияет не только число метальных групп в макромолекуле, но
и положение их в элементарном звене [132]:
Строение элементарного звена Температура
CO(CH2LCOHN(CH2NNH плавления,
COCH2CHCH2CH2COHN(CH2NNH 216
СН3
. СО(СВД4СОНЫСН2СН2СНСН2СН2СН2Ш 189
106 Глава 2. Производство полиамидных волокон
Введение полярных групп. При введении в молекулу
полиамида полярных групп, в частности гидроксильных, значительно
улучшаются некоторые практически важные свойства полиамида и.
получаемых из него волокон (гигроскопичность и накрашивае-
мость). Гидрофильные группы наиболее просто могут быть введены
путем замещения водорода в группе NH. Существенный интерес
представляют N-замещенные полиамиды, полученные при
взаимодействии полиамида с эпоксисоединениями, в частности с окисью
этилена [133].
Продукт, полученный при взаимодействии полиамида типа
найлон 6,6 с окисью этилена, носит название «гидроксиэтилнайлон».
Эта реакция, представляющая собой один из вариантов реакции
синтеза разветвленных или привитых полимеров, протекает по
схеме
\ . \
NH N—СН2СН2О(СН2СН2О)„Н
| + (п+ 1)С2Н4О —*> '
с=о
Обычно получают разветвленный сополимер, в котором с окисью
этилена прореагировало от 15 до 40% общего количества групп
NH, находящихся в макромолекуле полиамида'.
Разветвленные полиамиды существенно отличаются по
свойствам от N-замещенных полиамидов, описанных выше. Они
обладают повышенной гибкостью и гидрофильностью, характерной для
полиэтиленоксида. Температура плавления гидроксиэтилнайлона
невысокой степени замещения незначительно отличается от
температуры плавления найлона 6,6. Например, при введении 15% окиси
этилена (от массы полиамида) температура плавления полиамида
типа найлон снижается до 235—240 °С. Продукт такой степени
замещения растворим в тех же растворителях, что и исходный
полиамид. Гидрофильность полиамида и проницаемость его для
водяных паров при введении оксиэтильных групп значительно
повышаются. Так, при введении 32% окиси этилена (от массы
полиамида) проницаемость водяных паров через пленку
модифицированного полиамида повышается в 5—6 раз.
Синтез гидроксиэтилнайлона сравнительно сложен, и волокно
из модифицированного полиамида этого типа целесообразно
получать, по-видимому, только в особых случаях.
Интересный тип полиамида, модифицированного введением
новых функциональных групп, был получен советскими
исследователями [134] при использовании вместо дикарбоновой кислоты
калиевой соли 5-сульфоизофталевой кислоты. Этот мономер в виде соли
с гексаметилендиамином вводили в реакционную смесь при
полимеризации капролактама в количестве 5—6% (от массы капро-
дактама). Механические свойства волокна не изменялись.
2.16. Модифицированные полиамидные волокна 107
Наличие кислотной группы в макромолекуле полиамида
значительно улучшает его окрашиваемость, а также позволяет получить
антимикробное волокно (соли с Ag или Си), устойчивое к
многократным стиркам.
Некоторого повышения гидрофильности полиамидов или
полученных из них волокон можно достигнуть обработкой их
формальдегидом. Эта реакция может протекать по двум схемам.
По первой схеме образуется метилольное производное
полиамида:
\ \
с=о с=о
| +СН2О —> |
NH N—СН2ОН
/ /
В результате введения гидроксильных групп повышается гидро-
фильность полиамида.
По второй схеме образуются химические связи между
макромолекулами полиамида вследствие взаимодействия формальдегида
с двумя макромолекулами полимера:
\
c=o
2 1
NH
/
i-ch2o -
c=o o=c
-+ 1 1 +H20
N—CH2—N
/ \
Детальней эта реакция рассматривается ниже. Согласно
данным японских исследователей [135], с увеличением числа вводимых
оксиметильных групп повышается прочность волокна (при
увеличении массы волокна на 20%), удлинение, устойчивость к истиранию
и удару, а также количество сорбируемой влаги.
Максимальное количество оксиметильных групп было введено
при обработке волокна в течение 3 ч раствором формальдегида
в метаноле в присутствии Н3РО4 (катализатор) при 60°С и pH 0,9.
Это волокно, названное «пластифицированным» волокном найлон 6,
вырабатывается в Японии в опытном масштабе.
Существенный интерес представляет новый тип полиамидного
волокна, полученный из полиамида, синтезированного
взаимодействием бис- (л-аминоциклогексил) -метана и дикарбоновой
алифатической кислоты, содержащей свыше 8 атомов углерода, по схеме
H2N—U у—CH2—P у—NH2 + НООС(СН2)„СООН —>
_> NH <f^ CH2—f^ NHCO(CH2)„CO 1- H20
где п — 8— 12.
Сведения о получении такого волокна, названного «киана»,
появились впервые в 1968 и 1969 г. [1361.
108 Глава 2. Производство полиамидных волокон
Основная особенность волокна киана заключается в том, что
по грифу и по внешнему виду оно существенно отличается от
других полиамидных волокон и приближается к натуральному шелку.
Кроме того, благодаря наличию в элементарном звене
макромолекулы полиамида гидрированных ароматических группировок, это
волокно обладает более высоким модулем, чем другие полиамидные
волокна, и по данному показателю приближается к полиэфирному
волокну.
Плотность волокна киана 1,03 г/см3, прочность — около
30 гс/текс, удлинение 30%, гигроскопичность ниже, чем волокон
найлон б и 6,6 B,5% при относительной влажности воздуха 65%).
Формование волокна производится из расплава. По-видимому, если
будут разработаны доступные методы получения мономеров,
волокно киана найдет более широкое промышленное применение.
Структурирование (сшивание) полиамидных волокон. В
последнее время проведено большое количество исследований по
обработке полиамидных волокон би- и полифункциональными
соединениями для образования поперечных химических связей между
макромолекулами или их агрегатами и повышения тем самым их
термостойкости, усталостной прочности и других ценных свойств. Для
получения сшитых полиамидных волокон применялись различные
бифункциональные соединения. Рассмотрим некоторые способы
сшивания.
Обработка волокна 5%-ным раствором хлористой серы
в ксилоле при 60 °С [137]. Реакция протекает по схеме
CO(CH2MNHCO(CH2MNH
+ S2C12 —>
CO(CH2MNHCO(CH2MNH '
СО(СН2) 5NCO(.CH2MNH
I
—> s + неї
CO(CH2MNCO(CH2MNH
Выделяющийся НС1 связЕївается органическими основаниями.
В результате указанной обработки значительно повышается термостойкость
волокна. Например, после иагрева при 150 °С в течение 24 ч исходное капроновое
волокно теряет 55% начальной прочности, а сшитое волокно, содержащее 3,7%
связанной серы, теряет всего 5,2% прочности. Прочность сшитого волокна,
содержащего 6,3% связанной серы, при таком нагреве не только не уменьшается,
но даже увеличивается на 14,4%. При нагреве этих волокон при 200 °С в
течение 1 ч необработанное волокно полностью теряет прочность, а волокно,
содержащее 3,7% серы, теряет всего 23,8% начальной прочности [138]. Несколько
повышается и теплостойкость волокна. Например, волокно капрон при 150 °С
теряет 60% прочности, а сшитое волокно — 40%. Одновременно увеличивается
модуль волокна и снижается его текучесть *.
* Извитость штапельного волокна можно значительно повысить, если его
обработать раствором полухлористой серы. Образующийся пзвиток обладает
высокой устойчивостью.
2.16. Модифицированные полиамидные волокна 109
Однако этот метод сшивания волокон не может быть рекомендован для
практического применения. Основные недостатки — необходимость проведения
реакции в органическом растворителе в присутствии основания и значительное
(почти на 30%) снижение прочности и удлинения волокна, что, по-видимому,
объясняется деструкцией волокна при действии НС1, выделяющегося в процессе
реакции.
Обработка волокна 5%-ным раствором цианурхлорнда
в ксилоле прн 90 °С [139]. Цианурхлорид содержит три атома хлора,
различающихся между собою реакционноспособностыо. При взаимодействии с
капроновым волокном в указанных выше условиях цианурхлорид реагирует как
моно- или бифункциональное соединение. Результаты, достигаемые прн
обработке капронового волокна этим реагентом, примерно те же, что и при
обработке раствором хлорида серы, и приведенные выше недостатки характерны
также и для данного метода сшивания.
Обработка капронового волокна раствором дихлор-
ангидрнда днкарбоновой кислоты в органическом растворителе
в присутствии органического основания для связывания выделяющегося НС1 [137].
Реакция протекает по схеме
NH NH NH
IN I I
(СН2M . (СН2M (СН2M
СО СО СО
2| II
NH + С1СО(СН2)ЛСОС1 —у N С(СН2)ХС—N + НС1
І І II II I
(СН2M (СН2M О О (СН2M
СО ' СО СО
Реакция проводится также в безводном органическом растворителе, что
является существенным недостатком этого метода.
Обработка волокна растворами диизоцианатов, в
частности т о л у и л е н-2,4-д и и з о ц и а н а т о м. При обработке этим реагентом,
так же как н при обработке другими бифункциональными соединениями,
начальная температура текучести волокна повышается с 50 до 130—150 °С,
одновременно повышается термостойкость волокна.
В отличие от других сшивающих реагентов, которые взаимодействуют, как
правило, только с одним типом функциональных групп макромолекулы
полиамидов, диизоцианат может реагировать с концевыми карбоксильными и
аминогруппами, а также с иминогруппами, что соответственно повышает скорость
реакции [140].
Количество вступившего в реакцию диизоцианата существенно зависит от
степени ориентации волокон. Чем больше степень ориентации, тем меньше
диизоцианата вступает во взаимодействие с капроновым волокном. Это имеет место
и прн обработке другими бифункциональными соединениями.
Обработка волокна диизоцианатом проводилась в токе аргона в растворе
абсолютированного ксилола при 140 °С в течение 0,3—12 ч [141], что, естественно,
делает неприемлемым этот метод для использования в производстве.
Дополнительным недостатком описанного метода сшивания полиамидного
волокна является токсичность диизоцианатов. Для устранения этого недостатка
были использованы в качестве сшивающего реагента блокированные днизо-
по
Глава 2. Производство полиамидных волокон
Ц60
І
І «О
І
Г
О
1
цианаты, обладающие значительно меньшей токсичностью. Такие диизоцианаты
'представляют собой продукт присоединения фенола или о-хлорфенола к
диизоцианату. В процессе взаимодействия блокированного диизоцианата с
полиамидом эти соединения отщепляются и реакция диизоцианата с полиамидом
протекает по обычной схеме.
При сшивании диизоцианатами одновременно с повышением термостойкости
значительно повышается и светостойкость полиамидного волокна.
Обработка волокна формальдегидом или его
производными. Принцип этого метода изложен выше. Существенным его преимуществом
является возможность проведения реакции сшивания в
отсутствие органических растворителей. Обработать
полиамидное волокно формальдегидом можно двумя
способами.
Обработка готового волокна формальдегидом в
присутствии борной кислоты (катализатор). Обработка
проводится при 135—140°С в течение 30—120 мин водным
раствором формальдегида под давлением.
Обработка крошки полиамида сополимером
формальдегида и диоксана. При плавлении крошки
сополимер ^медленно разлагается (при 220—240 °С) и
выделяющийся формальдегид равномерно сшивает волокно
в процессе формования [142].
Для завершения процесса сшивания сформованное
волокно после вытягивания дополнительно нагревают
при 175 °С в течение 30—40 мин. Этот способ
сшивания проще. Кроме того, реакция протекает более
равномерно, чем при первом варианте. В результате
получается волокно, обладающее более высокой
прочностью при нормальной и повышенных
температурах.
Температура нулевой прочности волокна, сшитого
формальдегидом, повышается на 70—150 °С по
сравнению с температурой нулевой прочности
необработанного полиамидного волокна. Одновременно повышается
стойкость не только к термическим, но и к
термоокислительным обработкам, что, по-видимому,
объясняется частичным замещением (при сшивании) атома
водорода в группе NH [143]. Значительно повышается
также устойчивость волокна к многократным
деформациям при повышенных температурах, т. е.
увеличивает«! усталостная прочность. Этот показатель
(прочность в % от исходной) имеет большое практическое значение; его определяют
после 100 циклов растяжения [144].
Повышение усталостной прочности в значительной степени зависит от
степени вытягивания волокна (рис. 2.26). При вытягивании волокна более чем на
400% этот эффект заметно уменьшается. Однако наряду с повышением
усталостной прочности в результате сшивания капронового волокна формальдегидом
существенно снижаются некоторые другие показатели. Например, прочность и
удлинение волокна при этом уменьшаются на 10—40% [145].
Плотность сшитого волокна увеличивается в зависимости от числа сшивок
с 1,146 до 1,163 г/см3.
Обработка волокна семикарбазидом. Существенный интерес
представляют опубликованные в последнее время данные об одновременном
заметном повышении термо- и теплостойкости волокна в результате обработки его
10—40%-ным водным раствором семикарбазида NH2NHCONH2. Обработку
вытянутого волокна этим реагентом проводили 5—20 ч при 80—100 °С. Затем
обработанное волокно подвергали термообработке при 130 °С в токе азота
6 ч [146]. В результате этой обработки прочность волокна заметно не изменяется,
во в 1,5—2 раза уменьшается потеря прочности после нагрева при 200 °С в тече-
200 300 Ш 500
Степень бытягибшшя1 %
Рис. 2.26.
Усталостная прочность
капроновых волокон в
зависимости от степени
вытягивания
исходного волокна:
/—исходное волокно;
2—сшитое B,2 мол. %
волокно.
Литература 111
ниє 2 ч и резко повышается теплостойкость волокна. Механизм этой реакции
недостаточно выяснен; по-видимому, и в этом случае имеет место сшивание
макромолекул, однако сшиваются другие функциональные группы, что и
приводит к такому существенному повышению термостойкости волокна.
Суммируя приведенные данные, можно сделать вывод, что
приемлемый для производства метод сшивки полиамидного волокна
пока не разработан.
Получение химически окрашенных полиамидных волокон. Метод
модификации свойств полиамидных волокон, разработанный
преимущественно советскими исследователями, можно осуществить
в различных вариантах. Наибольший интерес представляют
следующие варианты.
Синтез поликапроамида, содержащего на конце
макромолекулы ароматические аминогруппы {147].
После диазотирования этих групп и сочетания их с различными
азосоставляющими получаются волокна, окрашенные в
зависимости от строения амина или фенола, применяемого в качестве
азосоставляющего, в различные цвета: темно-синий, фиолетовый,
темно-коричневый, серый {148].
Недостатком этого варианта является необходимость
дополнительных обработок сформованного волокна, преимуществом —
возможность получения широкой гаммы цветов.
Добавление к капролактаму цветных аминов
антрахинонового ряда, играющих роль стабилизаторов при
полимеризации капролактама [149]. Эти амины реагируют с
концевыми карбоксильными группами макромолекулы поликапроамида,
входят в его состав и обусловливают образование химически
окрашенного полимера, а затем и волокна. Этот тип окрашенного
полимера имеет следующее строение:
NHCO(CH2N—NH-
Добавление к капролактаму активных (процио-
новых) красителей, содержащих активный атом хлора (хлор-
триазиновую группировку), который реагирует с концевыми
аминогруппами макромолекулы полимера. При синтезе полимера в
результате взаимодействия этих красителей с полимером образуется
химически окрашенный полиамид [150]. Естественно, что
добавляемый краситель должен обладать достаточно высокой
термостойкостью и не разлагаться при температуре 260—280 °С, при которой
происходит полимеризация капролактама.
112 Глава 2. Производство полиамидных волокон
ЛИТЕРАТУРА
1. Са rot h er s W., Berchet G., J. Am. Soc, 1930, v. 52, p. 5289—5291.
2. Chem. Ind., 1970, v. 22, N 3, p. 123—127.
3. S n і d e r О. E., Richardson В. І. In: Encyclopedia of Polymer Science
and Technologie. V. 10. New York — London, Interscience Publisher, D.
Wiley, 1969, p. 347.
4. К л яре Г. Химия и технология полиамидных волокон. Пер. с немецк. Под
ред. Д. И. Тумаркина и 3. А. Роговина. М., Гизлегпром, 1956. 247 с.
5. Хоппф Н., Мюллер Е., Венгер Ф. Полиамиды. Пер. с немецк. Под
ред. А. Б. Пакшвера. М., Госхимиздат, 1958. 452 с; Волокна из
синтетических полимеров. Пер. с англ. Под ред. Р. Хилла. М., Издатинлит, 1957.
• 452 с.
6. Артемьев А. А., Генкина Е. В. В кн.: Современные методы синтеза
мономеров для гётероцепных волокнообразующих полимеров. М., ВИНИТИ,
1961, с. 41—56.
7. Chem. Age, 1965, v. 93, N 23, p. 18—19.
8. Бабкин Б. А., В є п д є л ь ш т є й н Е. Г., Генкина Е. В., Хим. волокна,
1961, № 4, с. 3—6.
9. Артемьев А. А., Стрельцова А. А., Генкина Е. В. и др., Хим.
наука и пром., 1968, т. 3, № 5, с. 629—632.
10. Артемьев А. А., Вен д е льштей н Е. Г., ЖВХО им. Д. И.
Менделеева, 1966, т. 11, с. 612—616.
11. Иоселиани Э. Г., Ругинский В. Р., Чичагов В. Н. и др., Хим.
волокна, 1969, № 5, с. 5—7.
12. Никулина Л. Е., Королева В. Р., Хим. волокна, 1968, № 6, с. 16—18.
13. Кнунянц И. Л., Роговин Э. А., Р ы м а ш е в с к а я Ю. А., Хаит Э. В.,
ЖОХ, 1947, т. 17, с. 987; 1326-1329. -
14. С к у р а т о в С. М., Стрепихеев А. А., Воеводский Н. В. и др.,
ДАН СССР, 1954, т. 95, № 4, с. 829—831; т. 95, № 5, с. 1017—1020.
15. Скуратов С. М., Стрепихеев А. А., Канарская Е. Н., Коллоид,
ж., 1952, т. 14, № 3, с. 187—200.
16. Стрепихеев А. А., Деревицкая В. А., Слонимский Г. Л.
Основы химии высокомолекулярных соединений, М., «Химия», 1966. 514 с.
17. Во лохи на В. А., ЖВХО им. Д. И. Менделеева, 1962, т. 7, № 2, с. 140—
146.
18. С а г о t h е г s W., J. Am. Chem. Soc, 1929, v. 51, p. 2548—2550.
19. Роговин 3. A., Стрепихеев А. А., Прокофьева А. С, ЖОХ, 1947,
т. 17, № 7, с 1321 — 1325.
20. В о л о x и h a А. В., Хим. волокна, 1966, № 4, с 3—8.
21. Скуратов С. М., Стрепихеев А. А., Воеводский В. В. и др.,
ДАН СССР, 1952, т. 86, № 6, с. 1155-1156.
22. Reinisch G., Ja gar W., Faserforsch, u. Textiltechn., 1961, Bd. 12,
S. 448—452.
23. Geley F., Szafner A, Holly Z. e. a., J. Polymer Sei., 1961, v. 52,
p. 245—249.
24. Geley F., Szafner A., J. Polymer Sei., 1962, v. 53, p. 455—457.
25. Hall H. K., J. Am. Chem. Soc, 1958, v. 80, p. 6404—6406; S с h amp e-
tie G., Sekiguschi H., Compt. Rend., 1958, v. 24, p. 108—112.
26. S t e h І і с e k I., S e b e n d a I., W і с h t e r 1 e O., Coll. Czechoslov. Chem.
Commun., 1964, v. 29, p. 1236—1240.
27. S tec G., Gechill G., Europen. Polymer J., 1970 v 6, p. 233—239.
28. S e k y r u m u X., К а с a k y H-., «Успехи химии», 1969, 38, с. 2075; S t e h 1 i-
cek Г., Se ben da I., Wichterle О., Coll. Czechoslov. Chem Commun.,
1964, v. 29, p. 1236—1240.
29. Скуратов С. M., Воеводин 'В. В., Стрепихеев А. А и др.,
«Ученые записки МГУ», 1953, вып. 164, с. 103—110.
30. Fritzsche E., Ко го se J., Faserforsch, u. Textiltechn., 1959, Bd 10,
S. 248—252.
Литература 113
31. В о л о х и и а А. В., Кудрявцев Г. И., Хим. волокна, 1959, № 5, с. 13—16.
32. Скуратов С. М, Воеводский В. В., Стрепихеев А. А. и др.,
ДАН СССР, 1954, т. 95, № 4, с. 591—594.
33. Ma g ига T., J. Polymer Sei., 1958, v. 31, p. 383—386.
34. Han ford W., Jo и ce R, J. Polymer Sei., 1948, N 3, p. 167.
35. Wichterle О., Faserforsch, и. Textiltechn., 1955, Bd. 6, S. 237—240.
36. Sebenda L, Kralicek 1., Faserforsch, u. Textiltechn., 1955, Bd. 6,
S. 653—657.
37. S cha a f I., Faserforsch, u. Textiltechn., 1959, Bd. 10, S. 328.
38. Griel W., Faserforsch, u. Textiltechn., 1956, Bd. 7, S. 207—210.
39. Griel W., S с ha a f 1, Makromol. Chern., 1959, Bd. 32, S. 177—180.
40. H am an A., Faserforsch, u. Textiltechn., 1958, Bd. 9, S. 351—354.
41. Каторжное H. Д., Воителев Ю. А., Просяник Ю. В., Хим.
волокна, 1964, № 6, с. 23—26.
42. Б ы к о в А. Н., Ермолаева Е. А., Кириллова Т. М. и др., Хим.
волокна, 1962, № 4, с. 4—8.
43. Туманов Н., Хим. волокна, 1960, № б, с. 54—56.
44. К а то р ж н о в Н. Д., Хим. волокна, 1966, № 1, с. 3—6.
45. Б р а в е р м а н П. Ф., Ч а ч х и а н и А. Б., Оборудование и механизация
производства химических волокон. М., «Машиностроение», 1967. 324 с.
46. К ля ре Г. Химия и технология полиамидных волокон. Пер. с немецк. Под
ред. Д. И. Тумаркина и 3. А. Роговина. М., Гизлегпром, 1956. 247 с.
47. Пат. ФРГ 193507; индийск. пат. 122366; франц. пат. 2024705.
48. Полуденная В. М., Волкова Н. М., Архангельский Д. Н., Хим.
волокна, 1969, № 5, с. 6—10.
49. Полуденная В. М., Волкова Н. С, Архангельский Д. Н. и др.,
Хим. волокна, 1969, № 2, с. 10—12.
50. Волкова Н. С, Дорожкин А. С, Семенова И. С. и др.. Хим.
волокна, 1966, № 2, с. 40—42.
51. Весельчакова Т. В, Преображенский В. А., Гольдман А. М.
и др.. Хим. пром., 1971, № 3, с. 3—5; № 4, с. 243—246.
52. Кнунянц И. Л., Вязанкнн К. С, Изв. АН СССР, ОХН, 1957, № 2,
с. 238—240.
53. Chem. Ind., 1966, v. 15, N 12, p. 790—791.
54. Ko p ш а к В. В., Ф р у_н зе Т. М., ДАН СССР, 1955, т. 103, № 3, с. 551—554;
№ 5, с. 643—646; Синтетические гетероцепные полиамиды. М., Изд-во АН
СССР, 1962. 523 с.
55. Хрипко в Е. Г., Харитонов В. П., Кудрявцев Г. И., Хим. волокна,
1970, № 6, с. 63—66.
56. Волохина А. В., Кудрявцев Г. И., Высокомол. соед., 1959, т. 1,
с. 1724—1727.
57. Фрейдлина Р. X., Васильева Е. И., Хим. наука и пром., 1957, т. 2,
с. 2—8.
58.0вакнмян Г. Б., Б е с п р оз в а н н ы й М. А., Беэр А. А., Хим. наука
и пром., 1957, т. 2, с. 12—16.
59. Horn G., Fr е иге В., Veneyard H. e. a., Angew. Chem., 1962, Bd. 74,
S. 531—536; Хим. и технол. полимеров, 1961, № И, с. 68—72.
60. W і I ke G., Angew. Chem., 1957, Bd. 69, S. 397—402.
61 Chem. Ind. Techn., 1964 v. 36, N 9, p. 960—962.
62. Text. Rundschau, 1967, Bd. 20, N 12, S. 207—208.
63. Griel W., Busthem S., «Kunststoffe Rundschau» 1970, № 1 S. 2—6.
64. Ne y O., Angew. Chem., 1962, Bd. 74, S. 523—530.
65. Barnes I., Nammy F., Ney I., Пат. США 2806841 A957)- 2739959
A956).
66. Ioda N., Mijaks A., J. Polymer Sei., 1960, v. 43, N 14, p. 118—122,
67. В e s t і a n H., Angew. Chem., 1968, Bd. 80, S. 179—185.
68. Graf R., Angew. Chem., 1968, Bd. 80, S. 111—124.
69. Graf R., Ann., 1963, Bd. 661, S. 111—124,
114 Глава 2. Производство полиамидных волокон
70. Graf R., Lohaus G., Borner R. e. a., Angew. Chem., 1962, Bd. 74,
S. 523-529.
71. Breslow D. S., Hulse G., Hetlack A., J. Am. Chem. Soc, 1957, v. 79,
p. 3760—3763.
72. Bus лак W., G ere y F., Faserforsch. u.Textiltechn, 1954, Bd. 5, S. 490—494.
73. Klare H., Faserforsch, u. Techtiltechn., 1955, Bd. 6, S. 219—225.
74. Кляре Г. Химия и технология полиамидных волокон. Пер. с немецк. Под
ред. Д. И. Тумаркина и 3. А. Роговина. М., Гизлегпром., 1956. 247 с.
75. Gordienko A., Griel W., Sieb er H., Faserforsch, u. Textiltechn., 1955,
Bd. 6, S. 105—110.
76. Griel W., Faserforsch.. u. Textiltechn., 1955, Bd. 6, S. 260—263.
77. Wiloth F., Makromol. Chem., 1954, Bd. 14, S. 156—159.
78. Бардина Г. M., Сперанский А. А., Муравьев В. К. и др., Хим.
волокна, 1968, № 2, с. 26—28.
79. Б р а в е р м а н П. Ф., Ч а ч х и а н и А. Б. Оборудование и механизация
производства химических волокон. М., «Машиностроение», 1967. 324 с;
Фншман К. Е., X р у з н и Н. А. Производство волокна капрон. М.,
«Химия», 1967. 247 с.
80. Я б л о ч ни к Н. С, Б а ш и н а В. С, К о в т у н е н к о В. Т. н др.. Хим.
волокна, 1968, № 4, с. 25—27.
81. Чернецкин Е. К., Салтыкова 3. П., H и ко нова Е. А. и др.
В кн.: Гетероцепные волокна. М., «Химия», 1967, с. 92.
82. Кляре Г. Химия и технология полиамидных волокон. Пер. с немецк. Под
ред. Д. И. Тумаркина и 3. А. Роговина. М., Гизлегпром, 1956. 247 с.
83. К у Д р я в ц е в Г. И., Б е р н а ц к а я Е. Э., Ш е в е л к н и Л. Ф., Текст,
пром., 1958, № 2, с. 15—17.
84. Griel W., Ver s a um er H., Faserforsch, u. Textiltechn., 1958, Bd. 9,
S. 226—229.
85. Кончинская Л. M., Ф и ш м а н К. Е., Хнм. волокна, 1970, № 2,
с. 69—71.
86. Харитонов В. Н., Чуравьев В. К., Высоцкий В. И. и др., англ.
пат. 1217052 A968); итал. пат. 836370 A968); франц. пат. 1588464 A968);
пат. США 3607102 A963).
87. Евсюкова В. С, Бакуменко Т. Л., Харитонов В. М., Хим.
волокна, 1966, № 4, с. 60—63.
88. Михайлов Н. В., Файнберг Э. 3., Коллонд. ж., 1956, т. 18, № 1,
с. 41—43; № 2, с. 200—203.
89. Bez О., Schul 1er Е., Faserforsch, u. Textiltechn., 1955, Bd. 6, S. 152—154.
90. Qu і g I. В., Text. Res. J., 1950, v. 23 p. 280—284.
91. Klare H., Faserforsch, u. Textiltechn., 1955, Bd. 6, S. 219—225.
92. Grobe V., Versaume г Н., Faserforsch, u. Textiltechn., 1963, Bd. 7,
S. 288—291.
93. Носов М. П., Болдина Л. И., Хим. волокна, 1965, № 3, с. 15—17.
94. Евсюкова В. С, Бакуменко Т. Л., Хим. волокна, 1966, № 5,
с. 60—63.
95. Каторжное Н. Д., Прокофьева А. С, Купинский Р. В. и др.,
Хнм. волокна, 1959, № 8, с. 11—15.
96. Fritz sehe E., W е с k 1 є і n A., Faserforsch. u. Textiltechn. 1959 Bd 10,
S. 475—479.
97. Wreit I., Text. Rundschau, 1952, Bd. 7, S. 323—326.
98. Пекарский M. Ш., Пакшвер А. Б., Беленький Л И. Хим
волокна, 1970, № 2, с. 74—77.
99. Mullard R„ Thornton P., J. Text. Praxis, 1952, v. 43, p. 413—416.
100. Sommers Y., Man-Made Text., 1955, v. 32, p. 58—61.
101. Михайлов H. В., Токарева Л. Г., Ковалева М. В. Высокомол.
соед., I960, т. 2, № 4, с. 581—585; То к а р е в а Л. Г., M и х а й л о в H В.,
ЖВХО им. Д. И. Менделєєви, 1966, т. 11, с. 243—249.
102. Горбачева В. О., Красова И. И., Токарева Л. Г. и др., Хим.
волокна, 1964, № 3, с, 19—23,
Литература 115
103. Смирнов Л. H., Харитонов В. М., Антипина Г. И. и др. Синтез
и исследование эффективности химикатов для полимерных материалов.
Вып. 4. Изд-во «Тамбовская правда», 1970, с. 152.
104 Данилевский С Л., Брейтман А. Я., ЖВХО им. Д. И. Менделеева,
1966, т. И, № 3, с. 303—304.
105. К у т ь и н а Л. В., Г а в р и л о в С. Г., В е с е л о в а М. А. и др. Синтез н
исследование эффективности химикатов для полимерных материалов.
Вып. 3. Изд-во «Тамбовская правда», 1962, с. 158.
106. О л ей ник В. Г., Кутьи на Л. В., Рудеико Л. Г. и др., Хим.
волокна, 1970, № 3, с. 6—10.
107. Плешаков М. Г., Смирнова Г. П., Меркурьева Е. В., Хим.
волокна, 1967, № 1, с. 26—28.
108. Токарева Л. Г., Михайлов Н. В., Потемкина 3. П. и др., Хим.
волокна, 1961, № 3, с. 15—18.
109. Хопфф Г., Мюллер А., Венгер Ф. Полиамиды. Пер. с немецк. Под
ред. А. Б. Пакшвера. М., Госхимиздат, 1957. 452 с.
ПО. Schwert a s sek К., Faserforsch, u. Textiltechn., 1954, Bd. 5, S. 433—436.
111. Ford I., Warmer aer H., J. Text. Inst., 1960, v. 51, p. 80—82.
112. Bohr in ger H., В oll and F., Faserforsch, u. Textiltechn., 1955, Bd. 6,
S. 199—203.
113. Bohr in ger H., Melliand Textilber., 1955, Bd. 36, S. 677—680.
114. Bohr in ger H., Bo Hand F., Faserforsch, u. Textiltechn., 1958, Bd. 9,
S. 405.—406.
115. Mecheels O., Melliand Textilber., 1955, Bd. 36, S. 1129—1132.
116. Nesby G., Text. Res. J., 1959, v. 32, p. 763—767.
117. Hohen see H., Faserforsch, u. Textiltechn., 1954, Bd. 5, S. 299—302.
118. В or in ger H., Bo Hand T., Faserforsch, u. Textiltechn., 1956, Bd. 7,
S. 345—349.
119. Lu de wig H., Faserforsch, u. Textiltechn., 1955, Bd. 6, S. 277—280.
120. О п p и u. 3. Г., Лазуткина Т. П., Ш а б л ы г и н М. В. и др., Хим.
волокна, 1966, № 4, с. 23—26.
121. Волохина А. В., Мурашкина СИ., Харитонова А. С. и др..
Хим. волокна, 1970, № 3, с. 8—11.
122. Юркевич В. В., Конкин А. А., Хим. волокна, 1969, № 6, с. 15—18.
123. Коршак В. В., Фрунзе Т. М., Матвеева Н. Г., «Успехи химии»,
1956, т. 25, с. 419—422.
124. Коршак В. В., Фрунзе Т. М., ДАН СССР, 1955, т. 103, № 4 с. 623—
627; № 5, с. 643—646; Изв. АН СССР, ОХН, 1955, № 3 с. 551—554; № 4.
с. 756—759; № 5, с. 934—938; 1956, № 1, с. 98—101.
125. Михайлов Н. В., Шейн Т. Н., Горбачева В. О. и др., Высокомол.
соед., 1959, т. 1, с. 185—189; Михайлов Н. В., Ф айн бер г Э. 3.,
Высокомол. соед., 1959, т. 1, с. 201—203.
126. Горбачева В. О., Шейн Т. И., Красова И. И. и др., Хим. волокна,
1966, № 3, с. 24—27.
127. Muller A., Pfluger R., «Kunststoffe», 1961, Bd. 50, S. 203—205.
128. H і 11 R, W a 1 k e г G., J. Polymer Sei., 1948, v. 3, p. 609.
129. BestianH., Angew. Chem., 1968, Bd. 80, S. 312—317.
130. Волокна из синтетических полимеров. Пер. с англ. Под ред. Р. Хилла. М.,
Издатиилит, 1957. 505 с.
131. Charch W., Seh і ver s I., Text. Res. J., 1954, v. 29, p. 536—537.
132. Hill R., J. Soc. Dyers Colour., 1952, v. 68, N 5, p. 158—161.
133. Haas H., Cohen I., Ogbesly A. e. a., J. Polymer Sei., 1955 v 15,
p. 427—429.
134. Мандросова Ф. M., Богданов M. H., Кравченко Т. В и др.,
Хим. волокна, 1968, № 5, с. 14—18. '
135. Takischita T., M a cd a S., Sasaoka M., Chem. of High. Polymers,
Japan, 1969, v. 26, p. 216—220; Takischita T., Macda S., Naka-
gawa A., Chem. of High. Polymers, Japan, 1969, v. 26, p. 221—229.
136. Am. Dyestuff Rep., 1969, v. 58, N 25, p. 21, 35.
116 Глава 2. Производство полиамидных волокон
137. Смольникова Л. Г., Конкин А. А., Хим. волокна, 1966, № 1, с. 2—5.
138. Смольникова Л. Г., Конкин А. А., Хим. волокна, 1966, № 3, с. 21—24.
139. Смольникова Л. Г., Конкин А. А., Хим. волокна, 1965, № 3, с. 2—4.
140. Perry Е., Savory I., Polymer Preprints, 1967, v. 8, № 1, p. 729, 730.
141. Бежуашвнли Т. Б., Конкин А. А., Хим. волокна, 1971, № 2, с. 9—11.
142. Семенова А. С, Васильева-Соколова Е. А., Кудрявцев Г. И.
и др.. Хим. волокна, 1971, № 1, с. 73—76.
143. Сперанский А. А., Богданова С. А., Ложкин Б. В. и др., Хим.
волокна, 1968, № 5, с. 11.
144. Семенова А. С, Васильева-Соколова Е. А., Михайлов Н. В.
и др., Хим. волокна, 1970, № 5, с. 24—27.
145. Васильева-Соколова Е. А., Семенова А. С, Роговина А. А.,
¦ Хим. волокна, 1970, № 3, с. 66—68.
146. Кутьина Л. В., Жигоцкий А. Г., Соловьева Л. С. и др., Хим.
волокна, 1969, № 1, с. 53—56.
147. Богданов М. Н., Лещинер А. У., Плишкевич Л. А., Высоко^ол.
соед., 1966, т. 8, с. 903—905; 1423—1426.
148. Богданов M. H., Лещинер А. У., Пляшкевич Л. А., Хим. волокна,
1967, № 2, с. 8—10.
149. Быков А. Н., Ермолаева Е. А., Кириллова Т. М. и др.. Хим.
волокна, 1962, № 4, с. 9—11.
150. Быков А. Н., Ермолаева Е. А., Кириллова Т. М. и др., Хим.
волокна, 1966, № 4, с. 26—28.
Глава 3
ПРОИЗВОДСТВО ПОЛИЭФИРНЫХ ВОЛОКОН
Возможность получения линейных полиэфиров —
высокомолекулярных веществ, у которых отдельные звенья макромолекулы
соединены между собой сложноэфирными группами —С—О—, и фор-
О
мования из них волокна была впервые показана Карозерсом и Ар-
вином {1] в 1929 г. Однако полиэфиры, синтезированные ими путем
поликонденсации алифатических дикарбоновых кислот и гликолеи,
имели температуру плавления ниже 100 °С и поэтому не
представляли существенного интереса для производства синтетических
волокон.
Исследования в области синтеза полиэфиров, пригодных для
производства высококачественных синтетических волокон, были
продолжены английскими химиками Уинфилдом и Диксоном.
В 1941 г. они получили высокоплавкие полиэфиры, образующие
прочные нити, используя в качестве исходного компонента для
поликонденсации с этиленгликолем не алифатическую, а
ароматическую дикарбоновую кислоту — терефталевую кислоту,
карбоксильные группы которой находятся в «-положении.
Впервые волокно из полиэфира терефталевой кислоты и этилен'-
гликоля было получено Уинфилдом и Диксоном в 1944 г. на
небольшой опытной установке. В 1949 г. в Англии была введена
в эксплуатацию первая производственная установка по синтезу
полиэфиров и получению из них волокон. С 1950 г. начинается
выработка полиэфирного волокна в промышленных масштабах
в Англии, Канаде и в США. В последующие годы производство
полиэфирных волокон из полиэтилентерефталата значительно
увеличилось.
Доступность исходного сырья, ценные свойства полиэфирного
волокна, превосходящего по отдельным показателям другие
синтетические волокна, обусловили широкое развитие производства
этих волокон для изготовления технических изделий и предметов
народного потребления.
В настоящее время волокна из полиэтилентерефталата
вырабатываются в значительных количествах в Англии и Канаде
(терилен), в США (дакрон), в СССР (лавсан), ФРГ, Италии, ГДР и
ряде других стран.
Как указывалось выше, из всех синтетических волокон
производство полиэфирных волокон развивается наиболее быстрыми
118 Глава 3. Производство полиэфирных волокон
темпами. Например, если в 1960 г. мировое производство
полиэфирных волокон составило 123 тыс. т, то в 1967 г. оно увеличилось
до 753 тыс. т, в 1969 г. — до 1350 тыс. т и в 1972 г. превысило
2500 тыс. т. Удельный вес полиэфирных волокон среди других
типов синтетических волокон увеличился с 16,2% в 1960 г. до 39,5%
в 1972 г. В 1972 г. полиэфирные волокна заняли первое место среди
синтетических волокон. Согласно опубликованным в литературе
прогнозам [2], объем производства этого волокна к 1980 г.
достигнет 4,6 млн. т.
В Советском Союзе промышленное производство полиэфирного
волокна лавсан начато в 1960 г. на Курском комбинате
химического волокна. В последующие годы производство этого волокна
непрерывно увеличивалось, особенно после ввода в эксплуатацию
в 1970 г. крупного комбината полиэфирного волокна в г.
Могилеве. К 1975 г. производство полиэфирных волокон у нас в стране
превысит 90 тыс. т/год
Необходимо отметить, что полиэтилентерефталат не является едянственным
видом полиэфиров, который может быть использован для получения прочного
волокна. Не говоря уже о возможностя яспользованяя других ароматяческих
дикарбоновых кислот, содержащих карбоксяльные группы в га-положения, и
других бифункциональных алифатических гликолей, принципиально кристаллические
волокнообразующие полиэфиры могут быть синтезированы путем
поликонденсации ароматическях диолов, содержащях гидроксильные группы в га-положении,
с алифатической дикарбоновой кислотой, например адипиновой *. Синтез
подобных полиэфиров и сопоставление свойств волокон, получаемых из таких
полимеров и из полиэтилентерефталата, представляет большой интерес.
Практическое значение для производства волокон могут иметь также
полиэфиры, полученные из мономеров, образующихся при химической переработке
лигнина, а также поликарбонаты — линейные полиэфиры, синтезируемые путем
поликонденсации, например, дифенилолпропана с фосгеном:
СН3
л—^
+ СОС12 —>
Волокно из поликарбонатов производится в небольшом объеме в США под
названием «лексел».
3.1. ПРОИЗВОДСТВО ПОЛИЭТИЛЕНТЕРЕФТАЛАТНЫХ ВОЛОКОН
Полиэтилентерефталатными называются волокна, полученные
из полиэфира, образующегося путем поликонденсации терефтале-
вой кислоты и этиленгликоля (лавсан, терилен, дакрон).
* Полиэфиры такого строения синтезированы В. В. Коршаком и С. В. Ви-
иоградовой [3].
3.1. Производство полиэтилентерефталатных волокон 119
Исходным сырьем для получения полиэтилентерефталата,
используемого для производства синтетического волокна, является
терефталевая кислота и этиленгликоль.
Применение в качестве исходного компонента для процесса ге-
терополиконденсации дикарбоновой ароматической кислоты,
имеющей симметричное строение, — обязательное условие получения
высокоплавкого полимера, способного образовывать прочные нити.
Данные о влиянии положения карбоксильных групп в молекуле
ароматической дикарбоновой кислоты на свойства получаемых
полиэфиров при поликонденсации с этиленгликолем {4] приводятся
ниже:
"карток Способность ^ZltllllT Способность
Кислота сильных полимера полнмепа к волокиообразо-
групп к «Рнсталлизации полимера, ваиию .
Терефталевая . . 1;4 Высокая 265 Очень высокая
Изофталевая . . 1,3 Не кристалли- 102—107 Обладает прядо-
зуется мостыв^
Фталевая .... 1,2 То же Низкая Не обладает пря-
домостью
Как видно из этих данных, при использовании ароматических
дикарбоновых кислот, карбоксильные группы которых находятся
в положении 1, 3 и 1, 2, образуются низкоплавкие некристаллизую-
щиеся полимеры, из которых не могут быть получены прочные
волокна *.
Если этиленгликоль, используемый в качестве одного из
компонентов в реакции поликонденсации, является продуктом
многотоннажного производства химической промышленности, то терефтале-
вую кислоту начали получать в больших масштабах только в связи
с организацией производства полиэтилентерефталатного волокна.
3.1.1. Получение терефталевой кислоты
Для получения терефталевой кислоты могут быть использованы
разные виды химического сырья. Наибольший интерес
представляет получение терефталевой кислоты из я-диалкилзамещенных
бензола (в частности, из «-ксилола) и из толуола {7].
Терефталевая кислота из я-ксилола. Последний получают
вместе с другими изомерами (о- и ж-кеилолом) преимущественно из
нефти. При каталитическом риформинге нафтеновых соединений
происходит их ароматизация и получается смесь, содержащая
различные количества изомеров ксилолов.
Содержание п-, м- и о-изомеров в смеси ксилолов,
выделяющихся из каменноугольной смолы, составляет примерно 20, 50 и
30% соответственно.
* Дополнительные сведения о влиянии симметричного положения карб-
рксильных групп в молекуле дикарбоновой кислоты иа способность получаемого
полиэфира к кристаллизации и к образованию прочных волокон имеются в
работах Ватцера [5], а также в специальных монографиях [6, с. 129—14?],
120 Глава 3. Производство полиэфирных волокон
Соотношение в смеси этих изомеров, получаемых из нефти
методом гидроформинга, составляет: 21% /г-ксилола, 46% о-ксилола,
18% м- ксилол а. В состав смеси входит также 15% этил бензол а.
Отделить /г-ксилол от других изомеров, в частности от лг-кси-
лола, методом разгонки не удается ввиду незначительной разницы
в температурах кипения этих изомеров. Температура кипения «-
ксилола 138 °С, а ж-ксилола 144 °С. Поэтому изомеры разделяют
фракционной кристаллизацией при низких температурах (—70°С).
Таким путем может быть выделено около 70% /г-ксилола от общего
количества его в смеси изомеров. Содержание я-ксилола в
конечном продукте достигает 97—98%.
Остальные изомеры ксилола могут быть использованы для
других целей, в частности для получения фталевого ангидрида,
применяемого в производстве красителей, а также модифицированных
полиэфиров, в молекуле которых наряду с терефталевой кислотой
содержится некоторое количество звеньев изофталевой кислоты.
Кроме того, смесь о- и лг-изомеров можно подвергнуть
изомеризации для их частичного превращения в «-ксилол. Изомеризация
осуществляется методом пиролиза при высокой температуре G00 °С)
или в Присутствии различных катализаторов при 350—700 °С. В
результате изомеризации образуется смесь ксилолов, содержащая
18—20% /г-ксилола.
Терефталевую кислоту можно получить окислением «-ксилола
в одну или в две стадии.
Одностадийный процесс. /г-Ксилол окисляют
кислородом воздуха при 160—170 °С в присутствии катализатора — 0,3%
Со(ОНJ:
н3с—/?—сн, -^ ноос—/~~Ч—соон
Однако выход терефталевой кислоты при этом методе составляет
всего 10%, что делает его непригодным для практического
использования.
В последние годы разработан новый вариант одностадийного
окисления /г-ксилола кислородом воздуха; он нашел практическое
применение как один из наиболее экономичных и перспективных
методов. Терефталевая кислота этим методом в наиболее
значительных количествах производится фирмой «Амоко» (США), и его
часто называют способом Амоко. Сообщается [8, с. 14] о том, что
свыше 50% мирового производства терефталевой кислоты
вырабатывается этим методом.
Окисление /г-ксилола осуществляется по радикальному
механизму кислородом воздуха в растворе «-ксилола в уксусной
кислоте B00—400% от массы /г-ксилола) при 350 °С под давлением
14—15 кгс/см2 A,4 • 106—1,5 • 106 Па) в течение 30—60 мин. В
качестве катализатора применяется бром A—2% от массы
уксусной кислоты), являющейся инициатором образования свободных
3.1. Производство полиэтилентерефталатных волокон 121
радикалов. Обычно применяется смесь катализаторов (свободный
бром и бромид калия или тетрабромацетилен, а также соли
кобальта или марганца).
В результате реакции, проведенной в указанных выше условиях,
получается терефталевая кислота высокой степени чистоты,
которая после дополнительной очистки может быть использована для
синтеза диэтиленгликольтерефталата.
Известен также метод получения терефталевой кдслоты
окислением n-ксилола азотной кислотой (при 150 °С) в присутствии
солей ртути:
сн3 соон
4NO + 4Н2О
СН3 СООН
Однако этот метод, предложенный еще в 1943 г., является менее
экономичным и не может конкурировать с методами
каталитического окисления n-ксилола кислородом воздуха. Достаточно
указать, что для окисления 1 т n-ксилола требуется 3,5 т 100%-ной
азотной кислоты.
Двухстадийный процесс. Окисление n-ксилола в две
стадии в ряде случаев более экономично, чем в одну. На первой
стадии получается n-толуиловая кислота, которую затем окисляют
в терефталевую кислоту:
°2 Л N\ О2
н, с—С У-сн3 —* н3с—f ^>—соон
—>- НООС—\/—СООН
n-Толуиловую кислоту можно получить окислением я-ксилола
кислородом воздуха при 250 °С и 60 кгс/см2 F • 106 Па) в
присутствии уксуснокислых или нафтеновых солей кобальта, марганца
или свинца. n-Толуиловую кислоту окисляют 30—35%-ной НЫОз
при 100 °С. Выход терефталевой кислоты, получаемой этим
методом, составляет 60—75% от массы исходного п-ксилола.
Диметиловый эфир терефталевой кислоты —
диметилтерефталат (ДМТ). До последнего времени для синтеза
полиэфира применялась не терефталевая кислота, а ее
диметиловый эфир. Это объясняется тем, что чистый продукт, пригодный
для синтеза полимера требуемого молекулярного веса, проще
получить, используя для процесса поликонденсации не терефталевую
кислоту, а ее диметиловый эфир.
Диметилтерефталат, используемый непосредственно для синтеза
полиэтилентерефталата, может быть получен из n-ксилола через
n-толуиловую кислоту.
122 Глава 3. Производство полиэфирных волокон
В результате окисления л-ксилола кислородом воздуха при
температуре выше 100 °С в присутствии нафтената или олеата
кобальта получается л-толуиловая кислота, которую этерифицируют
метиловым спиртом при 175—180 °С и давлении 25—28 кгс/см2
B,5-Ю6—2,8-106 Па). Образующийся при этом метиловый эфир
л-толуиловой кислоты (метил-л-толуилат) окисляют кислородом
воздуха в тех же условиях, но при более высокой температуре
A50—200°С) до монометилового эфира терефталевой кислоты.
При этерификации этого эфира получается диметиловый эфир
терефталевой кислоты:
сн3он
ООН —¦>
монометиловый эфир
терефталевой кислоты
—> Н3СООС—f%—СООСНз
диметиловый эфир
терефталевой кислоты
Полученный диметилтерефталат дистилляцией или другими
методами * очищают от примесей (метилового спирта, метилового
эфира n-толуиловой кислоты).
Диметилтерефталат, применяемый для производства полиэтилентерефталата,
должен удовлетворять следующим основным требованиям:
Температура плавления, °С 140,6
Содержание альдегидов, %, менее 0,05
Содержание метил-л-толуилата, %, менее 0,05
Зольность, %, менее 0,01
Коэффициент омыления 576—580
Содержание воды, %, менее '. . 0,02
Кислотное число, мг КОН/г, не более 0,05
Наиболее вредными примесями являются альдегиды и соединения железа.
В присутствии этих веществ получается сильно окрашенный полимер. Наличие
небольших примесей монофункциональных кислот или спиртов приводит к
снижению молекулярного веса образующегося полиэфира в результате
блокирования концевых групп растущих цепей. По данным Б. В. Петухова [9, с. 14], при
содержании в реакционной смеси всего 0,05% метилового эфира л-толуиловой
кислоты реакция роста макромолекулы полимера прекращается. Поэтому чистота
диметилтерефталата имеет важнейшее значение для синтеза
высококачественного волокиообразующего полимера.
* Диметилтерефталат можно отделить от других эфиров
перекристаллизацией из бензола, в котором он не растворяется.
3.1. Производство полиэтилентерефталатных волокон 123
При получении в качестве конечного продукта окисления не диметилового
эфира, а терефталевой кислоты необходимо очистить ее от примесей л-толуило-
вой и нзофталевой (получаемой окислением jn-ксилола) кислот.
Расход га-ксилола составляет 0,7—0,9 кг на 1 кг диметилового эфира
терефталевой кислоты.
Терефталевая кислота из толуола. Из толуола терефталевую
кислоту можно получить различными способами, из которых
наибольший интерес представляет синтез этой кислоты из л-толуило-
вого альдегида или из бензойной кислоты. Синтез терефталевой
кислоты из п-толуилового альдегида осуществляется по следующей
схеме:
// \ СО // ^ ;?> HNO3 // ^
н3с—? y —>¦ н3с—г р—с^ >- ноос—Р у—соон
- n-Толуиловый альдегид получают обработкой толуола окисью
углерода при 40 °С и давлении 50 кгс/см2 E • 10б Па) в
присутствии хлорида алюминия, терефталевую кислоту — окислением
этого альдегида 30%-ной HNO3 при 200°С и давлении 30 кгс/см2
C-Ю6 Па).
Основной недостаток этого способа — большой расход хлорида
алюминия, достигающий 1 кг А1С13 на 1 кг получаемой
терефталевой кислоты. Выход n-толуилового альдегида 82% от массы
превращенного толуола.
Из n-толуилового альдегида через хлорангидрид трихлор-п-то-
луиловой кислоты может быть получен и диметилтерефталат:
CI2 // \ CH3OH
»¦ CI3C—<f ^—COCI >•
\H \=/
н3соос—?\—соосн
Хлорангидрид трихлор-п-толуиловой кислоты получают
хлорированием n-толуилового альдегида в паровой фазе при 240°С под
действием ультрафиолетовых лучей. Диметилтерефталат
образуется при обработке хлорангидрида трихлор-п-толуиловой
кислоты метанолом в присутствии серной кислоты.
Терефталевую кислоту можно получить также через хлорметил-
толуол по методу, разработанному в ГДР [10].
Метод использования в качестве исходного продукта толуола
путем окисления его до бензойной кислоты и последующего дис-
пропорционирования ее калиевой соли с образованием калиевой
соли терефталевой кислоты получил в последнее десятилетие
промышленное применение в Японии.
124 Глава 3. Производство полиэфирных волокон
Реакция диспропорционирования калиевой соли бензойной
кислоты протекает при 400—450 °С в присутствии солей кадмия, цинка
или кобальта по схеме
COOK COOK
COOK
При последующем разложении калиевой соли терефталевой
кислоты обработкой ее раствором серной кислоты выделяется те-
рефталевая кислота высокой степени чистоты.
Хотя этот метод синтеза терефталевой кислоты получил уже
практическое применение, все же метод прямого окисления л-кси-
лола кислородом воздуха (способ фирмы «Амоко») является более
эффективным.
Терефталевая кислота является основным продуктом,
определяющим стоимость получаемого полиэфира. Она примерно в 5 раз
дороже другого компонента, используемого для синтеза
полиэфира, — этиленгликоля.
3.1.2. Получение этиленгликоля
Этиленгликоль производится в промышленных масштабах. Он
находит широкое применение в технике для приготовления
антифризов, а также используется в качестве пластификатора или
растворителя.
'Ранее применявшийся способ получения этиленгликоля через
этиленхлоргидрин
С12; Н2О Са(ОНJ
СН2=СН2 >- НО—СН2СН2—Cl >-
Н2О
но—сн,сн,—он
вытесняется более экономичными методами, основанными на
каталитическом окислении этилена (получаемого при крекинге нефти)
кислородом воздуха до окиси этилена и последующей гидратации
ее:
о2 н2о
СН2=СН, —>¦ Н2С СН2 v НО—СН2СН2—ОН
О
Этиленгликоль, применяемый для синтеза полиэтилентерефта-
лата, представляет собой прозрачную, тщательно очищенную
жидкость. Даже незначительное содержание примесей оказывает
существенное влияние на качество получаемого полиэфира. В нем не
3.1. Производство полиэтилентерефталатных волокон 125
должно быть хлорсодержащих веществ и высококипящих
одноатомных спиртов, которые могут блокировать растущие цепи
полимера при поликонденсации.
Особенно вредно наличие примесей ди- и полигликолей,
которые снижают температуру плавления получаемого полимера и его
термостабильность, а также альдегидов, уменьшающих
термостабильность полимера (см. разд. 3.1.12).
Содержание дигликоля (димера этиленгликоля) в этиленгли-
коле не должно превышать 0,2%.
3.1.3. Синтез диэтиленгликольтерефталата
Исходным продуктом для синтеза полиэтилентерефталата
является дигликолевый эфир терефталевой кислоты — диэтиленгли-
кольтерефталат (ДЭГТ). Этот своеобразный бифункциональный
мономер, который используется для синтеза линейного полиэфира
по реакции поликонденсации, может быть получен различными
методами, из которых наибольшее практическое значение имеют два
основных метода:
синтез диэтиленгликольтерефталата переэтерификацией
гликолем диметилового эфира терефталевой кислоты;
непосредственная этерификация терефталевой кислоты этилен-
гликолем.
Практическое значение может представить также
разрабатываемый в последнее время метод получения
диэтиленгликольтерефталата взаимодействием терефталевой кислоты с окисью
этилена (оксиэтилирование терефталевой кислоты).
Рассмотрим вкратце преимущества и недостатки этих методов.
Синтез диэтиленгликольтерефталата переэтерификацией диме«
тилтерефталата. Реакция переэтерификации осуществляется по
схеме
СООСНз СОО(СН2JОН
+ 2НО—(СН2J—ОН —> Г J +2СН3ОН
СООСНз СОО(СН2JОН
Основным преимуществом этого метода, как отмечалось выше,
до последнего времени являлась возможность получения чистого
продукта. Путем перегонки ДМТ очищали от примесей диметило-
вых эфиров других фталевых кислот, что обеспечивало получение
дигликолевого эфира терефталевой кислоты, содержащего
минимальные количества других эфиров.
Переэтерификация диметилтерефталата проводится в
присутствии избытка гликоля. Эта реакция должна быть доведена до
конца, так как при наличии в реакционной смеси небольшого
количества диметилового или метилгликолевого эфира терефталевой
126
Глава 3. Производство полиэфирных волокон
кислоты при последующей реакции поликонденсации снижается
молекулярный вес образующегося полиэфира. Процесс
переэтерификации проводится 4—6 ч в присутствии небольших количеств
@,05—0,1%) катализаторов— PbO, CaO, MgO, a также солей
низших жирных кислот с двухвалентными металлами, например аце-
гата цинка @,01—0,02% от массы диметилтерефталата),
ацетатов марганца, кобальта и свинца, бората свинца и т. п.
Теоретическим оеъем Выделяющегося метанола.
І
1
Продолжительность процесса., ч
Рис. 3.1. Влияние химического состава катализатора на скорость реакции
переэтерификации диметилтерефталата:
/—Cd(CH3COOJ; 2—Zn(CH3COOJ; 3-РЬ(СН3СООJ-ЗН2О; 4-Со(СН3СООJ-4Н2О:
S— Ва(СН3СООJ-ЗН2О; в—Mg(CH3COOJ• 4Н2О; 7—Ba[Sn(OCH2CH2OH)el; S—H2SO4; 9—л-то-
луолсульфокислота.
Скорость реакции переэтерификации зависит от количества
добавленного катализатора, и особенно от его химического состава.
Катализатор не должен вызывать побочных процессов,
приводящих к разложению полиэфира при последующей его переработке
при повышенных температурах.
Данные [11] о влиянии состава добавленного катализатора на
скорость реакции переэтерификации диметилтерефталата
приведены на рис. З.1.- Эффективность действия катализатора
характеризовалась количеством метанола, выделяющегося при
взаимодействий диметилтерефталата с этиленгликолем.
Как видно из рис. 3.1, наиболее эффективными катализаторами
реакции переэтерификации являются уксуснокислые соли кадмия,
свинца и цинка. Уксуснокислые соли кобальта, бария и магния
действуют менее активно. Наименее эффективные катализаторы —
кислоты, в частности серная кислота и л-толуолсульфокислота.
Каталитическим действием при реакции переэтерификации
обладают также терефталевая и адипиновая кислоты, в присутствии
3.1. Производство полиэтилентерефтаЛатных волокон 127
которых скорость переэтерификации повышается в 2—3 раза [12].
Однако эти кислоты являются значительно менее активными
катализаторами, чем ацетат цинка.
Существенное влияние на скорость процесса переэтерификации
оказывает характер применяемого эфира терефталевой кислоты.
Например, при замене диметилового эфира терефталевой кислоты
на диэтиловый эфир той же кислоты процесс переэтерификации
значительно замедляется [10].
Энергия активации реакции переэтерификации диметилтерефта-
лата составляет 9,5—10,5 кал/моль D0—44 Дж/моль).
Выделяющийся при переэтерификации метиловый спирт отгоняется и
конденсируется.
Начальная температура реакции переэтерификации ДМТ
должна быть ниже температуры кипения гликоля и составляет
обычно 180 °С. К концу процесса температуру постепенно
повышают до 205 ± 5 °С. Это объясняется необходимостью отгонки не
вступившего в реакцию избытка гликоля. В результате повышения
температуры при реакции переэтерификации образуется не только
диэтиленгликольтерефталат, но и значительное количество
низкомолекулярных полиэфиров (со степенью полимеризации 2—4).
Смесь дигликолевого эфира терефталевой кислоты и
низкомолекулярных полиэфиров подвергают фильтрации для отделения
примесей и нерастворенного катализатора, а затем ее используют для
синтеза полиэфира.
Недостатком описанного метода получения дигликольэтиленте-
рефталата является необходимость предварительного синтеза ди-
метилтерефталата. Использование метанола при переэтерификации
делает этот процесс взрыво- и огнеопасным, поэтому значительно
повышаются требования техники безопасности, что удорожает
строительство цеха и осложняет его эксплуатацию.
Этерификация терефталевой кислоты. Указанные недостатки
описанного выше метода устраняются при синтезе дигликолевого
эфира терефталевой кислоты взаимодействием терефталевой
кислоты и этилен гликоля. Этот процесс был впервые осуществлен
в 1963 г. [13].
Предварительным условием реализации этого перспективного
метода является получение чистой 99,99%-ной терефталевой
кислоты, содержащей не более 0,01% примесей. Одним из основных
достижений в производстве полиэфирных волокон, реализованном
в промышленном масштабе, является разработка в последние годы
метода получения терефталевой кислоты такой чистоты.
Терефталевая кислота высокой степени чистоты, получаемая
многократной перекристаллизацией аммиачной соли терефталевой
кислоты из насыщенных водных растворов при повышенной темпе*
ратуре или экстракцией под давлением, вырабатывается в
производственных условиях.
128 Глава 3. Производство полиэфирных волокон
Синтез дигликолевого эфира терефталевой кислоты
взаимодействуем этой кислоты с этиленгликолем протекает по схеме
СООН СОО(СН2JОН
\\ + 2НО—(СН2J—ОН
СООН
Реакция начинается в гетерогенной и заканчивается в
гомогенной среде, так как образующийся эфир терефталевой кислоты
растворим в избытке этиленгликоля. Этерификация проводится в
присутствии избытка этиленгликоля B,5—3 моль на 1 моль
терефталевой кислоты) при высокой температуре A90—200°С). Применения
катализатора, как правило, не требуется, так как реакция этерифи-
кации, катализируемая обычно кислыми реагентами, в данном
случае ускоряется автокаталитически карбоксильными группами
терефталевой кислоты. Действие карбоксильных групп дополнительно
усиливается благодаря их частичной диссоциации в присутствии
следов воды, выделяющихся при реакции этерификации *.
Добавление небольших количеств минеральных кислот (серной
кислоты или сульфокислоты), являющихся катализаторами этой
реакции, дает возможность повысить скорость этерификации
терефталевой кислоты в 20—30 раз [14]. Однако существенным
недостатком этих реагентов, определившим нецелесообразность их
применения, является возможность частичной дегидратации
этиленгликоля при высоких температурах и образования диэтиленгликоля.
Поэтому при необходимости повысить скорость этерификации
терефталевой кислоты применяют менее активные катализаторы, не
обладающие указанными выше недостатками, например сурьмяную
кислоту или тетрабутоксититан. В присутствии этих реагентов
скорость этерификации повышается в 3—4 раза.
Для дополнительного повышения скорости этерификации
терефталевой кислоты реакцию проводят под давлением 3—12 кгс/см2
C-Ю5—12-105 Па) при 250—260°С в течение 1,5—2ч**. При
таких температурах катализатор не требуется.
Синтез диэтиленгликольтерефталата этерификацией
терефталевой кислоты находит все более широкое применение. Например,
в США в 1968 г. этим методом вырабатывалось 25% от общего
* При реакции переэтерификации ДМТ благодаря блокированию групп
СООН такое автокаталитическое ускорение реакции, естественно, не имеет
места.
** Переэтерификацию ДМТ при таких высоких температурах проводить
нецелесообразно, так как давление в аппарате повышается до 50—60 кгс/см3
E-10е—6-10е Па).
3.1. Производство полиэтилентерефтамтных волокон 129
количества ДЭГТ [15], а в 1971 г. количество ДЭГТ, получаемого
этим методом, намечалось увеличить до 60%.
По сравнению с переэтерификацией ДМТ метод получения
ДЭГТ прямой этерификацией терефталевой кислоты имеет
следующие преимущества.
1. Значительно уменьшается вредность и взрывоопасность
производства (в результате устранения метанола из
производственного цикла).
2. Снижается стоимость получаемого полиэфира. В настоящее
время стоимость терефталевой кислоты высокой степени чистоты
не превышает стоимости ДМТ. Так как молекулярный вес
терефталевой кислоты составляет 166, а ДМТ—194, расход последнего
на получение 1 т ДЭГТ примерно на 100—150 кг превышает
расход терефталевой кислоты. Применение в качестве исходного
продукта терефталевой кислоты обусловливает существенное
снижение стоимости ДЭГТ и полиэфирного волокна. Предварительные
подсчеты [16] показали, что себестоимость полиэфирного волокна
при использовании терефталевой кислоты на 5,5% ниже, чем при
применении для синтеза полиэфира ДМТ.
3. Скорость этерификации терефталевой кислоты этиленглико-
лем, по данным советских исследователей, примерно в 2 раза выше
скорости переэтерификации им эфира терефталевой кислоты.
4. Количество этиленгликоля, вводимого в реакционный аппарат
при этерификации терефталевой кислоты, значительно меньше, чем
при переэтерификации ДМТ. Например, если при
переэтерификации вводится до 2,5 моль этиленгликоля на 1 моль ДМТ, то при
этерификации терефталевой кислоты благодаря проведению
реакции при более высокой температуре достаточно ввести в
реакционную смесь не более 2 моль этиленгликоля на 1 моль
терефталевой кислоты.
Соответственно уменьшаются потери при ректификации и,
следовательно, снижается удельный расход этиленгликоля.
Наряду с указанными преимуществами синтез ДЭГТ прямой
этерификацией терефталевой кислоты имеет на данной стадии
разработки этого метода ряд существенных недостатков.
При синтезе ДЭГТ прямой этерификацией терефталевой
кислоты наряду с эфиром терефталевой кислоты образуется
небольшое количество молекул этого эфира, содержащих диэтиленглико-
левые, а возможно, и триэтиленгликолевые группировки
следующего состава:
COO[(CH2JOj„H
СОО[(СН2JО]„Н
где я == 2 или 3.
5 Зак. 231
130 Глава 3. Производство полиэфирных волокон
Наличие ди- и триэтиленгликолевых групп, которые образуются,
по-видимому, в результате побочной реакции поликонденсации эти-
ленгликоля, приводит к нарушению регулярности строения
макромолекул полиэфира и соответственно к ухудшению ряда ценных
свойств получаемого полимера и изготовляемых из него волокон.
Снижается температура плавления полиэфира и повышается
ползучесть (пластическое удлинение) получаемых волокон. Например,
при наличии в молекуле полиэфира 3—4% ди- и
триэтиленгликолевых групп (от общего числа этиленгликолевых групп) температура
плавления полимера снижается на 5—7 °С, а пластическое
удлинение повышается на 2—4%. Последнее особенно нежелательно при
производстве полиэфирных волокон для технических изделий, в
частности для получения кордной нити. Поэтому при производстве
корда ДЭГТ, полученный методом прямой этерификации терефта-
левой кислоты, пока используется в сравнительно ограниченных
масштабах.
В полиэфире, полученном поликонденсацией ДЭГТ,
синтезированного прямой этерификацией терефталевой кислоты, содержится
2—3% ди- и тригликолевых групп от общего числа
этиленгликолевых групп вместо 0,5—0,8%,обычно находящихся в молекуле
полиэфира, синтезированного на основе ДМТ.
Вода, выделяющаяся при этерификации терефталевой кислоты,
уносит с собой 1—2% этиленгликоля (от общего количества
отгоняемой воды). Задача очистки этой воды после конденсации ее
паров и регенерации из нее этиленгликоля пока не решена.
Поскольку суспензия терефталевой кислоты в этиленгликоле,
используемая для этерификации, обладает отчетливо выраженными
тиксотропными свойствами, усложняется технологический процесс
получения ДЭГТ и его аппаратурное оформление.
При применении для . синтеза полиэфира в качестве
исходного продукта терефталевой кислоты усиливается коррозия
аппаратуры.
Несмотря на перечисленные недостатки, преимущества метода
получения ДЭГТ прямой этерификацией терефталевой кислоты
настолько значительны, что он, по-видимому, найдет в ближайшие
годы еще более широкое промышленное применение.
Получение диэтиленгликольтерефталата из терефталевой
кислоты и окиси этилена. Большой интерес представляет синтез ДЭГТ
обработкой терефталевой кислоты не этиленгликолем, а окисью
этилена по схеме
СООН СОО(СН2JОН
СООН СОО(СН2JОН
3.1. Производство полиэтилентерефталатных волокон 131
Этот способ имеет следующие принципиальные преимущества:
отпадает необходимость синтеза этиленгликоля, получаемого,
как известно, гидратацией окиси этилена;
не выделяются побочные продукты (в частности, вода) при эте-
рификации, что существенно упрощает аппаратурное оформление
процесса;
этерификация терефталевой кислоты окисью этилена протекает
значительно быстрее, чем этиленгликолем, так как окись этилена
является более активным алкилирующим реагентом;
этерификация протекает при более низкой температуре A20—
150 °С).
Основным недостатком этого метода получения ДЭГТ является
возможность синтеза эфира терефталевой кислоты не с
этиленгликолем, а с полигликолем, т. е. эфира, содержащего полигликолевые
группы. Это объясняется тем, что окись этилена легко реагирует не
только с группами СООН, но и с группами ОН (как, например, при
оксиэтилировании целлюлозы). Для устранения этого недостатка,
затрудняющего практическое использование метода, имеющего ряд
значительных технико-экономических преимуществ, необходимо
подобрать соответствующий катализатор и условия реакции, при
которых исключается возможность образования полигликолей.
По литературным данным [17], этот метод получил уже
практическое применение в Японии (фирма «Тейджин»).
3.1.4. Синтез полиэтилентерефталата
Синтез полиэтилентерефталата происходит по своеобразной
реакции линейной гомополиконденсации дигликолевого эфира
терефталевой кислоты по схеме
НОСН2СН2ООС—/ \—COOCHjCHjOH :*=t
:<=> ОС—/ \—СООСН2СН2ООС—/ \—COOCHjCH2O f-
+ НО(СН2JОН
По-видимому, начальной стадией этой реакции является алкого-
лиз сложноэфирной связи при действии групп ОН мономера с
образованием свободной терефталевой кислоты и отщеплением
этиленгликоля. Эта реакция является равновесной и обратимой, и для
того, чтобы сдвинуть ее в сторону образования полимера, в
молекуле которого между звеньями имеются связи того же типа, что и
в молекуле мономера, необходимо непрерывно отводить
выделяющийся этиленгликоль из сферы реакции. Чем полней удаляется
этиленгликоль (при повышенной температуре под вакуумом), тем
больше вероятность образования полиэфира с высоким
молекулярным весом. Следовательно, при синтезе полиэфира, так же как и
132 Глава 3. Производство полиэфирных волокон
других полимеров, получаемых по реакции поликонденсации,
молекулярный вес полимера и степень завершенности реакции
определяются полнотой удаления низкомолекулярного соединения
(в данном случае этиленгликоля), выделяющегося при реакции
поликонденсации.
Известным подтверждением этого предположения является
установленный В. А. Мягковым и Л. П. Репиной [18] факт
необходимости присутствия в реакционной смеси при синтезе полиэтилен-
терефталата небольших количеств воды, обусловливающей
возможность частичного омыления дигликолевого эфира терефталевой
кислоты в начальной стадии процесса и образования
карбоксильных групп, необходимых для начала реакции поликонденсации.
При реакции поликонденсации получается некоторое количество
побочных продуктов: до 2,5% циклического димера
СОО(СН2J—О(СН2JОСО
:оо(сн2J—О(сн2Jосо
и до 1,5% олигомеров циклического строения [19] со степенью
полимеризации, равной 3 и 4.
Одновременно с основной реакцией поликонденсации при
высоких температурах протекают побочные реакции, из которых
наибольшее значение имеют:
дегидратация этиленгликолевой группы, находящейся на конце
-макромолекулы, в результате чего в молекуле полиэфира
образуется некоторое количество двойных связей по схеме
—осн2сн2он ——>¦ —осн=сн2
¦—Н2О
образование полигликолей, которые также входят в молекулу
получаемого полиэфира.
Реакция поликонденсации дигликолевого эфира терефталевой
кислоты ускоряется при добавлении катализаторов, как правило,
тех же веществ, что и при реакции переэтерификации диметилте-
рефталата. Наиболее активным катализатором является бензоаг
железа [11]. Однако применение железа или его солей
нецелесообразно, поскольку в этом случае, так же как и при прибавлении
солей большинства других металлов, получаются окрашенные
полимеры. Наиболее широко в качестве катализатора используются
Sb2O3 или аморфный Со203 (кристаллическая модификация Со2О3
не растворяется в гликоле).
Ниже приводятся данные [II], характеризующие влияние
различных катализаторов на скорость процесса поликонденсации
дигликолевого эфира терефталевой кислоты:
3.1. Производство полиэтилентерефталатных волокон 133
Удельная вязкость
і, 0,5%-ного раствора
Катализатор полиэфира через 2,5 ч
после начала реакции
Fe(C6H5COJ 0,70
ZnCI2 0,60
Co(CH3COO)j-4H2O 0,57
ZnSO4 0,56
PbO 0,44
AICl, 0,38
Без катализатора 0.21
Скорость реакции определяли по значению удельной вязкости
получаемого полимера, которая тем больше, чем выше скорость
реакции. Катализаторы вводили в количестве 5 • 10~* моль на
1 моль дигликолевого эфира терефталевой кислоты.
Однако увеличение скорости реакции поликонденсации не
является единственным и даже основным критерием при выборе типа
катализатора для получения полиэфира в производственных
условиях. Необходимо учитывать также влияние катализатора на
интенсивность побочных реакций термической деструкции
получаемого полимера, дегидратации и поликонденсации этиленгликоля,
цвет и полидисперсность получаемого полимера. Например,
максимальная термодеструкция полимера происходит в присутствии
ацетата цинка, а ацетат кадмия оказывает значительно меньше
влияния на этот процесс [20].
До настоящего времени не удалось найти такие катализаторы,
которые ускоряли бы только процесс поликонденсации или пере-
этерификации, не ускоряя одновременно нежелательные побочные
реакции дегидратации этиленгликоля или образования полигли-
колей.
Катализатор оказывает некоторое влияние также и на
температуру плавления получаемого полиэфира. Например, температура
плавления полиэфиров, полученных в присутствии ацетата цинка,
на 5—6 °С ниже, чем полиэфиров, синтезированных с применением
в качестве катализатора ацетата свинца или кобальта. Это
объясняется [2,1] различным влиянием катализаторов на побочную
реакцию поликонденсации этиленгликоля и включением в
макромолекулу полиэфира некоторого количества ди- и триэтиленгликоля,
снижающих температуру плавления получаемого полимера.
Катализатор оказывает также влияние на полидисперсность
получаемого полиэфира. По данным Б. В. Петухова, при
использовании наиболее активных катализаторов (например, ацетата
цинка) получаются полиэфиры, обладающие большей
полидисперсностью. В присутствии менее активных катализаторов (например,
ацетата кобальта, и особенно ацетата кальция) образуются
полимеры, значительно более однородные по молекулярному весу.
134 Глава 3. Производство полиэфирных волокон
Количество катализатора, добавляемого при проведении
реакции переэтерификации и поликонденсации, составляет 0,02—0,04%
от массы диметилтерефталата.
Судя по опубликованным данным, наиболее приемлемым
катализатором является смесь, состоящая из 0,02% (от массы
диметилтерефталата) ацетата кальция и 0,015% трехокиси сурьмы, а
также смесь ацетата марганца со Sb2O3.
Обязательным условием получения высокомолекулярных
полиэфиров является полная отгонка низкомолекулярного компонента,
выделяющегося в процессе поликонденсации, — этиленгликоля.
Поликонденсация дигликолевого эфира терефталевой кислоты
осуществляется при 270—280°С и высоком вакууме (остаточное давление
менее 1 мм рт. ст.), так как только в этих условиях удается
практически полностью удалить этиленгликоль и получить полиэфир
достаточно высокого молекулярного веса B0 000 — 30 000).
С увеличением продолжительности поликонденсации
понижается полидисперсность получаемого полиэфира, уменьшается
содержание наиболее низкомолекулярных и высокомолекулярных
фракций. Такое изменение полидисперсности в зависимости от
продолжительности процесса характерно для всех гетероцепных
полимеров, получаемых по реакции поликонденсации.
Стабилизаторы для блокирования концевых групп
макромолекул при поликонденсации в большинстве случаев пока не
применяются, хотя блокирование этих групп (в первую очередь гидрок-
сильных) обусловливает, как это будет показано ниже (см. разд.
3.1.12), повышение термостойкости полиэфиров и получаемых из
них изделий.
Дальнейшее повышение температуры поликонденсации
нецелесообразно, так как при 300 °С начинается разложение
образующегося полиэфира. Для получения однородного по молекулярному
весу полимера реакционная масса подвергается непрерывному
перемешиванию. Перемешивание способствует также более
полному удалению гликоля, выделяющегося в процессе
поликонденсации.
По сравнению с аппаратурой для синтеза полиамидов
аппаратурное оформление технологического процесса получения полиэти-
лентерефталата несколько сложнее, так как поликонденсация
проводится при высокой температуре в аппарате с мешалкой, который
должен быть герметичным при глубоко]» вакууме.
Периодический процесс получения полиэтилентерефталата. По-
лиэтилентерефталат получают периодическим или непрерывным
способом, используя в качестве исходного продукта ДМТ или
ДЭГТ.
При синтезе полиэфира из ДМТ и этнленгликоля основными
технологическими операциями являются [22]:
1) расплавление днметилового эфира терефталевой кислоты или
растворение его в этиленгликоле;
3.1. Производство полиэтилентерефталатных во.гокон
135
2) переэтерификация диметилового эфира терефталевой
кислоты— получение дигликолевого эфира терефталевой кислоты и
низкомолекулярных полиэфиров;
3) фильтрация реакционной смеси;
4) поликонденсация;
5) получение твердого высокомолекулярного полиэфира в виде
ленты, нити или блока;
Рис. 3.2. Схема установки для синтеза полиэтилентерефталата ііериодическим
способом:
/ — растворитель диметилтерефталата; 2—дозирующие насосы; 3—подогреватель этилеи-
гликоля; 4—автоклав для переэтерификации диметилтерефталата; 5—конденсатор мета»
нола; 6— сборник метанола; 7—фильтр расплава; 8—автоклав для поликонденсации
дигликолевого эфира терефталевой кислоты; 9—конденсатор этиленгликоля; ГО—вентиль с
автоматическим устройством; // —вакуумные насосы; 12 — водяная ванна;/3 —рубильный станок!
14—бак для смешения крошки; 75—сушилка; IS—бачок для сбора конденсата.
6) дробление полиэфира;
7) сушка крошки.
Схема установки для получения полиэтилентерефталата
периодическим способом показана на рис. 3.2.
При непрерывном процессе поликонденсации и формования
волокна расплавленный полиэфир поступает непосредственно на
прядильную машину и операции 5—7 исключаются.
Расплавление или растворение ДМТ. Диметилтере-
фталат расплавляют в аппарате, который представляет собой котел
из нержавеющей стали, снабженный плавильной решеткой и
рубашкой (температура плавления диметилтерефталата 140°С).
Аппарат обогревается паром под давлением 5,5—8 кгс/см2 E,5 • 105—
8- 105 Па) при 160—175°С. Расплав диметилтерефталата из рас-
плавителя насосом перекачивается в автоклав для переэтерифика-
ции, куда одновременно подается горячий этиленгликоль,
содержащий катализатор. '-
136 Глава 3. Производство полиэфирных волокон
По другой схеме диметилтерефталат растворяют в горячем эти-
ленгликоле. Эта операция производится в растворителе с
электрообогревом. Раствор диметилтерефталата должен быть сразу же
передан в автоклав для переэтерификации, иначе в аппарате
начнет выделяться метанол. При работе по этой схеме катализатор
@,05—0,1% от массы диметилтерефталата) вводят
непосредственно в автоклав для переэтерификации при энергичном
перемешивании реакционной смеси.
Переэтерификация ДМ Т. Начальная температура
процесса переэтерификации составляет 160—180 °С, конечная
достигает 240—250 °С.
В зависимости от скорости подъема и максимальной
температуры реакции продолжительность процесса изменяется от 5 до
9 ч. Ввиду сравнительно высокой температуры реакции аппарат
для переэтерификации диметилтерефталата целесообразно
обогревать ВОТ, в частности динилом или АМТ-300 (см. с. 39). В качестве
теплоносителя может быть использован также насыщенный
водяной пар при давлении 60—75 кгс/см2 F • 106—7,5 • 106 Па). В
начальной стадии процесса переэтерификации отгоняется метанол, а
затем при повышении температуры до 240—250 °С — избыток эти-
ленгликоля.
Поликонденсация. Образовавшийся в результате
переэтерификации диметилтерефталата дигликолевый эфир терефталевой
кислоты подается в аппарат для поликонденсации. В большинстве,
случаев масса продукта при поликонденсации меньше, чем при
переэтерификации, и не превышает (при периодическом способе)
1,5—2,5 т. Поскольку переэтерификация диметилтерефталата
производится в аппарате большой емкости, продукт переэтерификации
из него подается в несколько аппаратов для поликоиденсации.
Продолжительность процесса поликонденсации составляет 3—
8 ч, начальная температура реакции равна 240—260°С, конечная
достигает 280—290 °С. Остаточное давление в аппарате при отгонке
этиленгликоля, выделяющегося при реакции поликонденсации, не
превышает 0,8—2 мм рт. ст. A07—267 Па). Для блокирования
концевых групп ОН в макромолекуле полимера и повышения
термостабильности получаемого полиэфира в конце процесса
добавляют (на ряде заводов) небольшое количество ортофосфорной
кислоты или ее эфиров @,04—0,05% от массы ДМТ).
Контроль процесса поликонденсации, и в частности
молекулярного веса получаемого полиэфира, заключается в определении
вязкости расплава. Чем больше молекулярный вес расплавленного
полиэтилентерефталата, тем выше при одной и той же температуре
вязкость расплава и тем больше при прочих равных условиях
сопротивление вращению мешалки в аппарате, т. е. тем больше
потребляемая мощность при перемешивании. Следовательно,
определяя изменение силы тока при вращении мешалки, можно, не
беря пробы из аппарата, с достаточной для практических целей
3.1. Производство полиэтилентерефталатных волокон 137
точностью установить степень завершенности реакции и
молекулярный вес получаемого полиэфира. Этот метод контроля может
быть использован и при синтезе других гетероцепных полимеров.
Дальнейшая обработка полученного полиэфира не отличается
от аналогичной обработки полиамида анид.
Получение твердого полимера. При периодическом
методе производства твердый полиэфир получают сухим или
мокрым способом.
При сухом способе расплавленный полиэфир через широкую
щель принимается на барабан или разливается в небольшие
изложницы. Расплавленный полимер затвердевает на поверхности
барабана или в изложницах разливочной машины (без
охлаждения) и затем направляется на дробление. Однако и при этом
способе барабан всегда обливают водой и затем ленты поступают для
охлаждения в ванну с водой. Поэтому данный способ можно только
условно называть сухим.
Так как содержание влаги в полиэфире не превышает 0,05%,
он не нуждается в сушке. Полученный после дробления
измельченный полимер (крошка) проходит через перфорированную сетку,
задерживающую частицы, размер которых больше определенной
величины (обычно 8—10 мм). Крошка, получаемая из нескольких
партий полиэфира, смешивается во вращающемся барабане и
подается в бункер прядильной машины.
При мокром способе расплавленный полимер, вытекающий из
аппарата для поликонденсации, попадает на движущуюся
стальную ленту в виде ленты или пучка толстых нитей (жилки) и затем
поступает в бассейн с водой. Лента или жилка разрезаются на
мелкие кусочки. Так как содержание влаги в полиэфире при
получении его этим способом превышает допустимый предел, крошка
сушится в вакуум-сушилке при 120—190 °С. Содержание влаги
в ней после сушки составляет 0,04—0,05%.
Непрерывный процесс получения полиэтилентерефталата.
Так же как и при производстве других химических волокон, одним
из основных направлений технического прогресса в производстве
полиэфирных волокон является осуществление непрерывного
процесса синтеза полиэфира или, что более целесообразно, создание
непрерывного процесса получения полимера и переработки
расплава полимера в волокно.
Основные трудности при создании непрерывного процесса
синтеза полиэтилентерефталата обусловлены недостаточной
термостойкостью этого гетероцепного полимера. При высокой
температуре имеют место разрыв сложноэфирной связи между звеньями
макромолекул, окисление метиленовых групп с образованием
двойной связи в макромолекуле полимера и другие побочные
процессы.
Для уменьшения возможности протекания, этих процессов и
обеспечения получения высококачественного полимера основные
138 Глава 3. Производство полиэфирных волокон
стации синтеза полиэфира необходимо проводить в короткий срок
и при возможно более низких температурах. Для этого процесс
целесообразно проводить в нескольких реакторах, в каждом из
которых реакция протекает при оптимальных для данной стадии
температуре и давлении.
Указанный принцип-положен в основу непрерывного метода
получения полиэтилентерефталата, разработанного фирмой «Виккерс-
Циммер» [23], который находит в последние годы все более
широкое применение.
Непрерывный процесс синтеза полиэфира разработан и
реализован в двух вариантах. По первому варианту полиэтилентерефта-
лат получают из ДМТ и этиленгликоля, по второму варианту
исходным продуктом является терефталевая кислота.
Непрерывный процесс поликонденсации проводится
последовательно в двух или трех реакторах. В первом реакторе, в котором
осуществляется этерификация терефталевой кислоты и
предварительная поликонденсация (получаются олигомеры со степенью
полимеризации 4—6), образуется низковязкая масса. Температура
реакции 270°С, остаточное давление 30 мм рт. ст. D000 Па). В
качестве катализатора рекомендуют смесь трехокиси и триацетата
сурьмы, которые проявляют активность только при 270—280 °С.
В последнем (обычно третьем) реакторе поликонденсация
завершается. Вязкость расплава образовавшегося сравнительно
высокомолекулярного полиэфира повышается до нескольких тысяч пуаз.
Для отгонки этиленгликоля, выделяющегося при поликонденсации,
остаточное давление в этом аппарате поддерживают ниже 1 мм рт. ст.
A33,3 Па). Вытекающий из аппарата расплав насосами или
шнеком подается по трубопроводу на прядильную машину.
Общая продолжительность синтеза полиэфира из терефталевой
кислоты и этиленгликоля непрерывным способом составляет 7 ч.
При использовании в качестве исходного продукта ДМТ
продолжительность процесса увеличивается до 9 ч, что, однако,
значительно меньше общей продолжительности синтеза полиэфира
периодическим способом.
При непрерывном процессе получения полиэфира из
терефталевой кислоты и этиленгликоля и формования волокна [24] паста,
состоящая из этих продуктов, поступает в смеситель и затем через
теплообменник в аппарат для этерификации, в котором
одновременно происходит предварительная поликонденсация. Реакция
проводится в тонком слое при 270 °С, продолжительность реакции
менее 30 мин. Вторая стадия процесса осуществляется в двух
многокамерных реакторах. В первом реакторе остаточное давление
1 мм рт. ст. A33,3 Па), температура 280°С, во втором создается
более глубокий вакуум. Реакция протекает в тонком слое,
продолжительность ее 6 ч. При соблюдении заданных параметров
процесса (продолжительности, температуры и др.) получается
полиэфир требуемого молекулярного веса.
3.1. Производство полиэтилентерефталатных волокон
139
Каждый из реакторов снабжен мешалкой и обогревается дини-
лом. Предложено [25] последнюю стадию процесса
поликонденсации проводить в шнеке, который обеспечивает хорошее
перемешивание вязкого расплава.
Расплав полиэфира шнеком подается на прядильную машину.
От момента выхода расплавленного полиэфира из реактора до
поступления его на прядильную машину, как правило, проходит не
Гликоль
і Метанол
Краситель.
Рис. 3.3. Схема получения полиэтилентерефталата из ДМТ и этиленгликоля
непрерывным способом:
/ —расплавптель; 2 —сборник для этиленглнколя; 3 — аппарат для переэтерификацин
4— первый аппарат для поликонденсации; 5 — второй аппарат для поликонденсации; S—пря
дильная машина; 7—смеситель.
более 8—10 мин. При необходимости в расплав вводят
матирующие вещества, красители или другие добавки. В получаемом
непрерывным способом полиэфире не содержится влаги, которая
остается даже в высушенной крошке, получаемой периодическим
способом. Благодаря отсутствию гидролитической деструкции
полиэфира при синтезе его непрерывным способом получается
полимер более высокого молекулярного веса.
Схема непрерывного процесса получения ДЭГТ из ДМТ и этиленгликоля
приведена иа рис. 3.3. Расплавленный ДМТ из расплавителя / непрерывно
поступает в аппарат 3 для переэтерификации, представляющий собой каскад
колони, в которые из сборника 2 также непрерывно подается этиленгликоль,
содержащий катализатор. Переэтерификация производится при нормальном давлении.
Для отгонки избытка гликоля температура повышается постепенно от 170
до 290 °С.
Метанол, выделяющийся в токе инертного газа при переэтерификации,
содержащий некоторое количество ДМТ и этилеигликоля, отгоняется из колонны
в начале процесса. Последние два компонента отделяются от метанола и после
ректификации возвращаются в производство.
Образовавшийся диэтнленгликольтерефталат поступает в аппарат для
предварительной конденсации, в котором процесс проводится при остаточном
140 Глава 3. Производство полиэфирных волокон
давлении 30 мм рт. ст. D000 Па). В этом аппарате завершается отгонка избытка
этиленгликоля. Остальные стадии процесса проводятся в таких же аппаратах,
в которых осуществляется синтез полиэфира из терефталевой кислоты и этилен-
гликоля.
Полученный полиэфир подается по трубам непосредственно на прядильную
машину. В отдельных случаях полимер выпускается в виде ленты и затем
подвергается такой же обработке, как и при получении полиэтилентерефталата
периодическим способом.
Непрерывный процесс синтеза полиэтилентерефталата и
формования из него волокна имеет следующие преимущества:
исключаются операции получения крошки, ее тщательной сушки
и транспортировки;
устраняется необходимость повторного плавления полиэфира на
прядильной машине, благодаря чему значительно упрощается
конструкция и уменьшается высота этой машины;
облегчается автоматизация процесса;
в каждом аппарате проводится только одна операция, поэтому
в нем можно поддерживать постоянные вакуум и температуру;
получается более однородный продукт;
значительно уменьшаются капиталовложения и
производственные площади.
Недостатком непрерывного процесса по сравнению с
периодическим является значительно более ограниченная возможность
изменения ассортимента волокон. Поэтому в ряде случаев наряду
с поточной линией для получения полимера непрерывным способом
создают поточные линии для синтеза полиэфира периодическим
способом, например полиэфира, окрашенного в массе, глубокома-
тированного B—2,5% ТЮ2), и т. п. Мошность каждой из
непрерывных поточных линий достигает 20 000 т/год, в то время как
мощность одной поточной линии для получения полиэфира
периодическим способом не превышает 2000—3000 т/год.
3.1.5. Свойства полиэтилеитерефталата
Полиэтилентерефталат, применяемый для формования волокна,
имеет обычно молекулярный вес 20 000—30 000 (степень
полимеризации 85—120). Минимальный молекулярный вес полимера, при
котором еще возможно получение волокон, составляет 12 000—
13 000 (степень полимеризации 65—70).
Качество полиэтилентерефталата, применяемого для получения
волокна, характеризуется в основном молекулярным весом и
степенью полидисперсности, а также температурой его плавления
(характеризующей чистоту продукта) и степенью окраски.
Определение молекулярного веса. Полиэтилентерефталат растворяется при
повышенных температурах в феноле, крезоле, тетрахлорэтане и нитробензоле.
Определение молекулярного веса на основании вискозиметрических измерений
проводят в смеси фенола и тетрахлорэтана A : 1) или фенола п ацетилеитетра-
хлорида A : 1). Молекулярный вес рекомендуется вычислять по уравнению
3.1. Производство полиэтилентерефталатных волокон
141
где [ri] — характеристическая вязкость;
M — молекулярный вес.
Предложены также методы определения молекулярного веса полиэфира на
основании результатов вискозиметрических определений значений [г\] в феноле
при 50 °С. В этом случае молекулярный вес вычисляют по уравнению:
h]=5,517-10-W-707
При проведении вискозиметрических определений в о-хлорфеноле при
25 °С [26] молекулярный вес находят по уравнению
[т)]=6,56-10~4М0'73
Характеристическая вязкость полиэтилентерефталата, предназначенного для
получения штапельного волокна и текстильной нити, равна 0,55—0,70. Для
производства кордной нити требуется полимер более высокого молекулярного веса,
и [ту| для такого полимера должна быть увеличена до 0,90.
Предложен также метод определения среднечислового молекулярного веса
полиэфира, исходя из содержания концевых групп в его макромолекулах [27].
Концевые группы СООН определяют титрованием раствора полиэфира в ани-
лине 0,05 M раствором КОН, а концевые группы ОН — ацетилированием
полиэфира, растворенного в нитробензоле, бромацетилбромидом. Естественно, что
этот метод может быть использован только для определения молекулярного веса
нестабилизированного полиэфира.
Определение полидисперсности. Путем сопоставления результатов
определения молекулярного веса полиэфира вискозиметрическим методом и по
содержанию концевых групп можно приближенно определить полидисперсность
исследуемых препаратов.
Полидисперсность полиэфира, применяемого для формования волокна, можно
определить также методами фракционирования. Например, Уберейтер и Готце [28]
определяли полидисперсиость фракционным осаждением полиэтилентерефталата
лигроином из его растворов в л-крезоле *. Этими же исследователями
предложен другой метод фракционирования путем растворения полиэфира в диметил-
формамиде при 150 °С и последующем постепенном снижении температуры
раствора. Данным методом препарат полиэфира был ими разделен на 5—6 фракций.
Предложен также метод фракционного осаждения полиэтилентерефталата
бензином из 1°/о-ного раствора полиэфира в смеси фенола и хлорбензола A : 1).
Полимер в этой смеси растворяли при 100 °С.
Исследования показали, что полиэтилентерефталат
неоднороден по молекулярному весу. Например, при среднем молекулярном
весе этого полимера 15 000 молекулярный вес отдельных фракций,
Таблица 3.1. Данные, характеризующие зависимость между вязкостью раствора
н расплава полиэтилентерефталата
Удельная вязкость
0.5К-НОГО раствора
полиэтилентерефталата
в трикрезоле при 20 °С
Вязкость гасплава (в П) при температуре
270 °С
280 °С
290 °С
0,23
0,25
0,30
0,36
850
2800
4300
500
1200
2І50
420
950
1200
Полиэфир плавнтси, но не обладает текучестью
* Одни из немногих растворителей, в котором этот полиэфир растворяется
при нормальной температуре.
142
Глава 3. Производство полиэфирных волокон
определенный по содержанию концевых групп, изменялся от 5000
до 17 000 [28, 29].
При этих исследованиях применялся метод фракционного
осаждения «-ксилолом из 1%-ных растворов полиэфира в смеси
фенола и тетрахлорэтана A : 1). При
средней степени полимеризации 121
фракционированием было выделено 17
фракций, степень полимеризации
которых изменялась от 48 до 513.
С увеличением молекулярного веса
и соответственно удельной вязкости
раствора полиэфира повышается и
вязкость расплава. Данные [30, с. 36],
характеризующие зависимость между
вязкостью раствора и расплава
полиэфира, приведены в табл. 3.1.
Данные, приведенные в табл. 3.1,
показывают, что при сравнительно
незначительном увеличении
молекулярного веса (удельной вязкости раствора)
полиэфира резко увеличивается
вязкость расплава.
Интенсивность снижения вязкости
расплава полиэтилентерефталата при
высоких температурах (в интервале
270—300 °С) тем больше, чем выше
молекулярный вес полимера (рис. 3.4)
[31].
270
180 290
Tenmpamt/pa., °С
300
Рис. 3.4. Зависимость вязкости
расплава
полиэтилентерефталата от температуры и
молекулярного веса полимера:
7 — 32 000; 2 — 29 000; 3 — 28 000;
4 — 26 000; 5 — 25 000; 5—24 000;
7—23 000; Я—16 500; 9 —13 500;
10— 11500.
3.1.6. Формование полиэтилентере-
фталатного волокна
Формование полиэфирного волокна
из расплава осуществляется по такой
же технологической схеме и на тех же
прядильных машинах, что и
формование полиамидных волокон.
Характерное отличие в аппаратурном оформлении процесса
формования полиэфирных волокон от полиамидных заключается
в отсутствии в большинстве случаев на прядильных машинах
плавильных решеток. Расплав подают к прядильному насосику при
помощи шнеков (экструдеров), в которых одновременно
происходит плавление полимера. Часто вертикально расположенные шнеки
подают крошку на перфорированные алюминиевые или серебряные
плиты, обогреваемые электричеством. На этих плитах и
производится плавление измельченного полимера.
3.1. Производство полиэтилентерефталатных волокон 143
Непрерывный процесс синтеза полимера и формования из него
волокна нашел практическое применение при формовании
штапельного волокна и технической нити, в частности кордной нити.
Специфическое преимущество этого способа при производстве
кордной нити заключается в том, что таким путем можно получить
полиэфирную нить с более высоким молекулярным весом (на 10—
15%). Последнее обстоятельство объясняется тем, что в данном
случае в полиэфире нет влаги и поэтому в процессе формования
молекулярный вес полимера не снижается.
Вязкость расплава полиэтилентерефталата, используемого для
формования волокна, составляет 2000—2500 П, т. е. примерно в
2 раза выше, чем у расплава поликапроамида. Поэтому при
формовании полиэфирного волокна применяют фильеры, как правило, с
значительно большим диаметром отверстий. При получении волокна
из полиэтилентерефталата диаметр отверстий фильеры составляет
0,5 мм вместо 0,25 мм при формовании полиамидных волокон.
Из различных видов полиэфирных волокон основное значение
имеет штапельное волокно. Объем производства штапельных
волокон составляет около 75% общего количества вырабатываемых
полиэфирных волокон. В последние годы начинает увеличиваться
производство полиэфирной кордной нити, а также текстурирован-
ной текстильной нити.
При получении штапельного волокна, когда необходимо
подавать к фильере значительно больше расплава, чем при формовании
текстильной нити, рекомендуется расплавлять полиэфир в экстру-
дере, который обогревается электрообогревателями по зонам.
Выдавливаемый экструдером расплав подается к прядильным насоси-
кам для нескольких прядильных мест. При этой схеме подачи
расплава отпадает необходимость устанавливать плавильные решетки
над каждым прядильным местом; кроме того, не ограничивается
количество полимера, подаваемого в прядильный насосик.
Соответственно может быть увеличено число отверстий в фильере и
значительно повышена производительность каждого прядильного места.
Такую схему подачи полимера к прядильному насосику (при
периодическом способе производства) целесообразно использовать и
при формовании штапельного волокна из расплавов других
синтетических полимеров (полиамидов, полиолефинов).
Термическая деструкция и разложение полиэтилентерефталата
с выделением СО2 начинается при 290—300 °С. Формовать волокно
при Температурах ниже 270—275 °С нельзя, поскольку температура
плавления полиэфира составляет 258—260 °С. Поэтому интервал
между температурой формования полиэфирного волокна и
температурой его разложения не превышает 15—20 °С, что
обусловливает необходимость очень точно выдерживать температуру в
процессе формования и затрудняет проведение этого процесса.
Интенсивность деструкции нестабилнзированного
полиэтилентерефталата характеризуется снижением вязкости его растворов
144
Глава 3. Производство полиэфирных волокон
Із
1>}
\
•
гео°с
¦ 280°С
о ч- 8 к 16 го гч
•> Продолжительность ^прогрева, ч
Рис. 3.5. Интенсивность термической
деструкции лолиэтилентерефталата
в зависимости от продолжительности
нагрева.
в смеси фенола с тетрахлорэтаном (рис. 3.5). Из рис. 3.5 видно,
что если при длительном выдерживании при 260°С молекулярный
вес полимера (определяемого вискозиметрически) заметно не
снижается, то при 280 °С вязкость раствора значительно
снижается.
Снижение молекулярного веса происходит тем интенсивней, чем
выше молекулярный вес полиэфира. Например, при формовании
волокон из полиэфиров с M =
= 22 000 и M = 30 000 их
молекулярные веса в результате
повторного плавления снижаются
соответственно на 4000 и 10000 [32].
Пребывание расплава полиэфира
в плавильной головке должно
быть непродолжительным,
поскольку полиэтилентерефталат
недостаточно термостабилен.
Необходимость уменьшения
продолжительности пребывания
расплавленного полиэфира на прядильной
машине объясняется также и тем,
что активные функциональные
группы макромолекул полиэфира
(в отличие от полиамидов) в процессе поликонденсации обычно
не блокируются. Вследствие этого возможно их дополнительное
взаимодействие и соответствующее изменение (в большинстве
случаев снижение) молекулярного веса и вязкости расплава при
повторном плавлении полиэфира в процессе формования.
Недостаточная термостабильность расплава полиэтилентерефталата и
изменение его вязкости в процессе формования — существенный
недостаток полимера, для устранения которого необходимо
блокировать концевые группы макромолекулы в процессе
поликонденсации.
Как уже указывалось выше, разработаны методы блокирования
концевых групп ОН макромолекулы полиэтилентерефталата эте-
рификацией их фосфорной кислотой в процессе поликонденсации
при 280°С в присутствии катализатора @,003% ацетата цинка)
[33]. Во избежание дополнительной деструкции сложноэфирных
связей в макромолекуле полиэфира количество фосфорной кислоты
не должно превышать 0,1% от массы диметилтерефталата, и
вводить ее желательно не в начале, а в конце реакции
поликонденсации.
Расплавленный полиэтилентерефталат более стоек к действию
кислорода воздуха, чем полиамиды. Поэтому требования в
отношении допустимого содержания кислорода в азоте, в среде
которого происходит формование полиэфирного волокна, менее жесткие,
чем при формовании полиамидного волокна. Однако и в этом
3.1. Производство полиэтилентерефталатных волокон 145
случае содержание кислорода в азоте не должно превышать
0,01%.
Так же как и при формовании полиамидных волокон, при
производстве полиэфирных волокон могут применяться
профилированные фильеры.
В отличие от полиамидных волокон, которые частично
кристаллизуются уже в прядильной шахте, полиэфирная нить при быстром
охлаждении в шахте не кристаллизуется и может продолжительное
время оставаться в аморфном (застеклованном) состоянии.
Поэтому при формовании нити из полиэтилентерефталата получается
волокно с аморфной структурой. Возможность кристаллизации, а
следовательно, и последующего вытягивания полиэфирного волокна
сильно зависит от температуры в шахте. Чем выше температура
в шахте, т. е. чем вероятнее частичная кристаллизация полимера
в процессе формования волокна, тем меньше максимально
возможное последующее вытягивание. Если, например, при температуре
в шахте 30 °С максимальная степень вытягивания волокна
составляет 900%, то при 60 и 90 °С она снижается соответственно до 600
и 500% [30].
Существенное влияние на максимально возможную степень
последующего вытягивания волокна оказывает скорость формования
и фильерная вытяжка. Повышение скорости формования на
100 м/мин (в интервале 460—1150 м/мин) снижает максимально
возможную степень последующего вытягивания на 10% [34].
Аналогично, хотя и в меньшей степени, влияет повышение фильерной
вытяжки.
Так как гидрофильность полиэфиров значительно ниже, чем
полиамидов, то свежесформованное полиэтилентерефталатное
волокно, выходящее из шахты прядильной машины, поглощает мало
влаги. Поэтому удлинение нити на бобине в результате увлажнения
и сползание ее с бобины (что характерно для полиамидных
волокон и обусловливает необходимость тщательного
кондиционирования воздуха в прядильных и отделочных цехах заводов
полиамидных волокон) при получении полиэфирного волокна почти не имеют
места.
Скорость формования полиэфирного волокна составляет 400—
1500 м/мин. Предложено формовать эти волокна со скоростью
до 6000 м/мин. В этом случае вытягивание волокна в основном
происходит в процессе' его формования на прядильной машине.
Однако эти предложения пока практического применения не
получили, так как при повышении скорости формования выше
определенного предела B000—2500 м/мин) дополнительного повышения
прочности волокна не наблюдается, а удлинение остается
аномально высоким. Поэтому целесообразно совмещать процессы
формования и вытягивания. При этом выходящая из шахты нить
принимается на диск и затем поступает на второй диск, вращающийся
с более высокой скоростью, в 3—4 раза превышающей скорость
і
148 Глава 3. Производство полиэфирных волокон
первого диска. Скорость приемки вытянутого волокна в этом
случае достигает 4000 м/мин.
Чем больше скорость формования, тем больше фильерная
вытяжка и тем меньше максимально возможная степень
последующего вытягивания волокна. Зависимость максимальной ориента-
ционной вытяжки от фильерной вытяжки для полиэфирных
волокон выявляется значительно более
отчетливо, чем для полиамидных.
Увеличение фильерной вытяжки в
2 раза уменьшает максимальную степень
последующей ориентационной вытяжки
на 20% [32]. Частичная ориентация
волокна, происходящая на прядильной
машине при повышении скорости
формования, обусловливает повышение
прочности получаемого вытянутого волокна
(рис. 3.6).
"ідо 500 юоо При формовании штапельного волок-
скороапь (рорпоШя.фин на из полиэтилентерефталата число от-
Рис. 3.6. Зависимость проч- ВеРСТИЙ в Фильере составляет 300-500,
ности волокна лавсан от а Диаметр отверстия фильеры —0,5—
скорости формования 0,6 мм. Такое число отверстий не
является предельным.
В последнее время число отверстий в фильере при формовании
штапельного волокна увеличено до 600—1000, что, естественно,
обеспечивает значительное повышение производительности каждого
прядильного места. Скорость формования штапельного волокна
составляет 600—1200 м/мин.
3.1.7. Последующая обработка
полиэтилентерефталатного волокна
Обработка полиэфирных волокон после формования почти не
отличается от аналогичной обработки полиамидных волокон типа
анид (за исключением условий вытягивания). Так как в
сформованном полиэфирном волокне содержится незначительное
количество низкомолекулярных продуктов, специальной водной обработки
не требуется.
Вытягивание полиэфирного волокна. В отличие от
полиамидных полиэфирные волокна всегда вытягивают при повышенных
температурах.
При вытягивании комплексной нити обогрев ее производится
в две стадии: сначала на верхнем диске при 80—90 °С и затем
при прохождении нити над металлической пластиной (утюгом),
обогреваемой электричеством.
Обычно нить при вытягивании нагревают на
крутильно-вытяжной машине. Для этого перед поступлением на вытяжной ролик
нить пропускают над нагретой поверхностью.
3.1. Производство полиэтилентерефталатных волокон
147
Необходимо отметить, что температура стеклования полиэфиров
не является постоянной [32], а уменьшается с увеличением
молекулярного веса. Например, температура стеклования полиэфиров
с молекулярным весом 11500, 16 000 и 29 000 составляет
соответственно 65, 59 и 51 °С.
60
50
,40
JO
го
10
\
V
\
>
—
-\
є о
40
I? 7,4 7,8 1,1 2,6 3,0 3,4 3,8
Кратность бытя гадания
Рис. 3.7. Влияние температуры
вытягивания полиэфирного волокна на
величину усадки в кипящей воде.
зо
40 60 SO WO
¦Температура^
1Z0
Рис. 3.8. Зависимость прочности
полиэфирного волокна различного
молекулярного веса от температуры
вытягивания:
/ — молекулярный вес 17 500; 2 — 20 000;
3 — 24 000; 4 — 25 000 (кратность
вытягивания 5).
Степень вытягивания полиэфирной нити и условия проведения
этого процесса оказывают существенное влияние на последующую
усадку волокна при повышенных температурах [35]. Если
невытянутое полиэфирное волокно усаживается при кипячении в воде на
55%, то волокно, вытянутое на 350%, усаживается в кипящей воде
всего на 13%. Чем больше степень вытягивания полиэфирных
волокон, тем меньше последующая их усадка в кипящей воде.
При одной и той же степени вытягивания усадка нити в воде
при 100 °С зависит от температуры, при которой она вытягивалась.
Чем выше температура вытягивания (до определенного предела),
тем меньше при прочих равных условиях усадка нити в кипящей
воде.
Зависимость усадки полиэфирной нити от температуры
вытягивания показана на рис. 3.7. Приведенные на рис. 3.7 данные
показывают, что при вытягивании нити на 400% при 80°С усадка
волокна в кипящей воде составляет 8%.
.В зависимости от температуры и натяжения при вытягивании
полиэфирное волокно может быть аморфным или частично
закристаллизовавшимся [36]. При вытягивании волокна при температуре
выше 100—120 °С вследствие преобладания процесса тепловой
дезориентации над процессом ориентации заметного повышения
прочности не происходит. Максимальное повышение прочности
волокна достигается при 80—90 °С, причем полученный эффект, как
148 , Глава 3. Производство полиэфирных волокон
это видно из рис. 3.8, в значительной степени зависит от
молекулярного веса полиэфира [37].
Чем больше молекулярный вес полимера, тем выше
температура, при которой достигается (при одной и той же степени
вытягивания) максимальная прочность волокна.
Способность полиэфирного волокна к вытягиванию в
значительной степени зависит от продолжительности его выдерживания до
этой операции. Например, если свежесформованное полиэфирное
волокно с молекулярным весом 17 500 можно вытянуть на 500%,
то после хранения его в течение 24 ч и 5 сут максимально
возможная степень вытягивания снижается соответственно до 460 и 400%.
При хранении этого же волокна в течение 10 дней оно практически
полностью теряет способность к вытягиванию [38]. Однако эти
данные не являются бесспорными.
Время выдерживания невытянутого волокна, в течение которого
оно сохраняет способность к вытягиванию, в значительной степени
зависит от молекулярного веса волокна, определяющего скорость
кристаллизации полимера. Чем выше молекулярный вес полимера,
тем медленней происходит кристаллизация и тем дольше волокно
сохраняет способность к вытягиванию. Например, при
молекулярном весе полиэфирного волокна 22 000 оно сохраняет способность
после выдерживания 10 сут вытягиваться при повышенных
температурах на 450—480% [38].
В результате вытягивания полиэфирного волокна значительно
повышается не только прочность, но и начальный модуль. Для
полиэфирного волокна этот показатель при вытягивании изменяется
в большей степени^ чем для полиамидных, что объясняется
большей жесткостью макромолекул этого полимера. Например, при
вытягивании нити на 400% модуль эластичности повышается в 4 раза
по сравнению с модулем того же волокна, вытянутого на
100—150%.
Вытянутая и скрученная полиэфирная нить, как и полиамидная,
подвергается термофиксации — обработке в автоклаве при 105—
125 °С в течение 20—40 мин в среде насыщенного пара или при
140—160 °С на воздухе.
До и после обработки нити паром автоклав вакуумируют. Это
необходимо для быстрого и равномерного прогревания нити на
паковке и для подсушивания нити после обработки паром.
Термофиксация нити всегда производится на эластичных паковках.
После завершения термофиксации и выдерживания на бобинах
3—4 ч нить перематывается на выходные паковки.
Во время термообработки, особенно при высоких
температурах, несколько повышается плотность нити — с 1,37 до 1,38 г/см3
(в результате дополнительной кристаллизации полимера) и
уменьшается поглощение красителя и сорбция воды.
148 , Глава 3. Производство полиэфирных волокон
это видно из рис. 3.8, в значительной степени зависит от
молекулярного веса полиэфира [37].
Чем больше молекулярный вес полимера, тем выше
температура, при которой достигается (при одной и той же степени
вытягивания) максимальная прочность волокна.
Способность полиэфирного волокна к вытягиванию в
значительной степени зависит от продолжительности его выдерживания до
этой операции. Например, если свежесформованное полиэфирное
волокно с молекулярным весом 17 500 можно вытянуть на 500%,
то после хранения его в течение 24 ч и 5 сут максимально
возможная степень вытягивания снижается соответственно до 460 и 400%.
При хранении этого же волокна в течение 10 дней оно практически
полностью теряет способность к вытягиванию [38]. Однако эти
данные не являются бесспорными.
Время выдерживания невытянутого волокна, в течение которого
оно сохраняет способность к вытягиванию, в значительной степени
зависит от молекулярного веса волокна, определяющего скорость
кристаллизации полимера. Чем выше молекулярный вес полимера,
тем медленней происходит кристаллизация и тем дольше волокно
сохраняет способность к вытягиванию. Например, при
молекулярном весе полиэфирного волокна 22 000 оно сохраняет способность
после выдерживания 10 сут вытягиваться при повышенных
температурах на 450—480% [38].
В результате вытягивания полиэфирного волокна значительно
повышается не только прочность, но и начальный модуль. Для
полиэфирного волокна этот показатель при вытягивании изменяется
в большей степени^ чем для полиамидных, что объясняется
большей жесткостью макромолекул этого полимера. Например, при
вытягивании нити на 400% модуль эластичности повышается в 4 раза
по сравнению с модулем того же волокна, вытянутого на
100—150%.
Вытянутая и скрученная полиэфирная нить, как и полиамидная,
подвергается термофиксации — обработке в автоклаве при 105—
125 °С в течение 20—40 мин в среде насыщенного пара или при
140—160 °С на воздухе.
До и после обработки нити паром автоклав вакуумируют. Это
необходимо для быстрого и равномерного прогревания нити на
паковке и для подсушивания нити после обработки паром.
Термофиксация нити всегда производится на эластичных паковках.
После завершения термофиксации и выдерживания на бобинах
3—4 ч нить перематывается на выходные паковки.
Во время термообработки, особенно при высоких
температурах, несколько повышается плотность нити — с 1,37 до 1,38 г/см3
(в результате дополнительной кристаллизации полимера) и
уменьшается поглощение красителя и сорбция воды.
150 Глава 3. Производство полиэфирных волокон
Значительное повышение адгезии полиэфирной кордной нитн
к резине достигается пропиткой кордной ткани или нити
блокированными изоцианатами — растворами диизоцианатов в толуоле или
в дихлорэтане или водной суспензией диизоцианатов, концевые
группы которых блокированы, например, фенолами.
Пропитку нити дисперсией изоцианатов можно осуществить
непосредственно на прядильной машине, вводя блокированные
изоцианаты в эмульсию, применяемую в качестве препарирующего
состава. В этом случае отпадает необходимость пропитки кордной
ткани на шинных заводах.
Блокированные изоцианаты имеют существенный недостаток —
при нагреве нити или ткани для образования свободных
изоцианатов выделяются фенолы, в результате чего значительно
ухудшаются условия работы в цехе. Приводятся данные [40],
указывающие на возможность повышения адгезии полиэфирных нитей
к резине при использовании эпоксидных смол, которые также
можно вводить в препарирующий состав. Однако адгезия к резине
нити, обработанной эпоксидными смолами, недостаточна.
Преимущества и недостатки полиэфирной кордной нити по
сравнению с полиамидной полностью еще не выяснены.
К основным преимуществам полиэфирной кордной нити по
сравнению с полиамидной относятся:
меньшая разнашиваемость автомобильных шин;
меньшие потери прочности ткани в процессе эксплуатации
шины * вследствие более высокой термостойкости полиэфирного
корда;
значительное превосходство полиэфирных кордных тканей по
устойчивости к удару, что повышает эксплуатационные свойства
шин (ударная прочность полиэфирной кордной нити примерно
в 4 раза выше этого показателя для полиамидной нити и в 20 раз
выше, чем для вискозной [41], поэтому из полиэфирной кордной
нити изготовляют тормозные парашюты для скоростных
самолетов) ;
высокая усталостная прочность при повышенных температурах
A20—140 °С).
Недостатками полиэфирной кордной нити по сравнению с
полиамидной являются:
более низкая прочность связи полиэфирной нити с резиной;
сравнительно быстрая дополнительная кристаллизация
вытянутой кордной нити в процессе эксплуатации шины, приводящая
к снижению ее эластичности и повышению хрупкости.
Устранение дополнительной кристаллизации вытянутой нити
является одним из основных условий широкого промышленного
* При применении для изготовления пневматических шин полиамидного
корда, содержащего термостабнлнзаторы, полиэфирный и полиамидный корды
по этому показателю становятся почти равноценными.
3.1. Производство полиэтилентерефталатных волокон 151
применения полиэфирного корда. Для замедления этого процесса
предложен ряд методов, из которых наибольший интерес
представляет повышение молекулярного веса полиэфира с 15 000—18 000
до 25 000-—30 000, так как с увеличением длины макромолекул
затрудняется кристаллизация полимера.
Интересным направлением, наметившимся в последние годы,
является использование смеси полиэфирной кордной нити с
полиамидной или вискозной; при применении смешанных кордных
нитей улучшаются эксплуатационные свойства кордной ткани.
Как указывалось выше, полиэфирный корд нашел уже
промышленное применение, и масштабы производства высокопрочной
кордной нити непрерывно увеличиваются.
3.1.9. Особенности производства штапельного
волокна
При получении штапельного полиэфирного волокна отдельные
невытянутые нити соединяются в общий жгут. Эта операция
в большинстве случаев производится непосредственно после
формования.
Обычно пучок волокон (жгутик), выходящий из прядильной
машины, укладывается в тазы. В каждом тазу помещается по
100-^250 кг волокна. Иногда загрузка таза увеличивается до
500 кг. Тазы с волокном B0—30) устанавливают в 2—3 ряда, и
жгутики из всех тазов собираются вместе, образуя общий жгут
(ленту), который поступает на штапельный агрегат.
На этом агрегате проводятся следующие операции:
вытягивание, гофрирование, термообработка, обработка антистатиком и
упаковка волокна в кипы.
Жгут вытягивают на вытяжных станах в две стадии. На первой
стадии его вытягивают на 400—420%, а на второй — на 20—60%.
Жгут проходит в закрытой камере над нагретой поверхностью или
омывается током горячего воздуха при 140—170 °С. Температура
вытягивания на второй стадии процесса несколько ниже (ПО—
120 °С).
При получении высокоусадочного штапельного волокна первая
стадия процесса вытягивания производится без предварительного
нагрева волокна. Этот процесс называется холодным
вытягиванием.
После вытягивания жгут подвергается механическому
гофрированию для придания извитости и повышения тем самым сцепляе-
мости волокна при последующей текстильной переработке.
Для устранения усадки при последующей обработке в горячей
воде и выравнивания напряжений в вытянутом волокне жгут после
гофрирования обрабатывают горячим воздухом или паром при
130—155 °С. Термообработка производится без натяжения в
условиях, обеспечивающих возможность некоторой усадки волокна.
152 Глава 3: Производство полиэфирных волокон
Усадка термофиксированного штапельного волокна при
последующей обработке в горячей воде не превышает 1,5—3%. Обработка
горячим воздухом производится в секции проходного аппарата
в течение 0,5—1 мин, и затем жгут после обработки эмульсией
антистатического препарата поступает на резку.
Резка жгута производится на машине, аналогичной по
конструкции машинам, применяемым для этого в производстве других
видов химических волокон.
Для снижения электризуемости гидрофобного полиэфирного
волокна и облегчения переработки его обрабатывают поверхностно-
активными веществами (например, продуктом конденсации высших
жирных спиртов с окисью этилена). Количество антистатического
препарата, наносимого'на волокно, составляет 0,3—0,5% от его
массы.
Иногда последовательность операций при отделке штапельного
волокна меняется — вытянутый жгут после гофрирования и
обработки антистатиком поступает в сушилку, В первых зонах сушилки
при 105—110°С происходит сушка жгута, а в последующих при
135—160°С — фиксация извитости. Выходящий из сушилки жгут
поступает на резку.
Разрезанное на штапелъки различной длины, в зависимости от
условий последующей переработки, волокно подается
пневматическим транспортом в пресс для упаковки. В большинстве случаев
волокно отправляют потребителям в виде жгутов, которые
разрезаются на мелкие отрезки на текстильных фабриках.
Параметры технологического процесса получения полиэфирного
штапельного волокна контролируются и регулируются
автоматически.
3.1.10. Особенности производства моноволокна
В последние годы полиэфирное моноволокно находит все более
широкое применение. Этот тип волокна имеет ряд преимуществ
перед полиамидным моноволокном, что и определяет
целесообразность его использования в различных отраслях народного
хозяйства.
Основным отличием полиэфирных моноволокон и вообще
полиэфирных волокон от полиамидных является более высокий
начальный модуль и соответственно пониженная текучесть нити, а также
значительно более низкая (в 6—8 раз) сорбция влаги, что
определяет целесообразность применения полиэфирных волокон в тех
случаях, когда при эксплуатации изделий проводят мокрые обработки.
Наибольшее народнохозяйственное значение имеет применение
полиэфирных моноволокон для следующих целей:
изготовление сеток бумагоделательных машин (для замены
латуни и бронзы);
3.1. Производство полиэтилентерефталаткых волокон 1ГЗ
производство фильтровальных сеток для фильтрации пульпы
(вместо нержавеющей стали);
изготовление для мельничных (элеваторных) машин щеток,
которые работают при 100—120 °С; для этих изделий используется
дополнительное преимущество полиэфирных волокон перед
полиамидными— их более высокая термостойкость;
изготовление струн для теннисных ракеток, скрипок,
роялей и т. п.
Полиэфирное моноволокно производится по той же
технологической схеме, что и полиамидное моноволокно, т. е.
преимущественно непрерывным методом. Расплавленный полиэфир через
отверстия фильеры диаметром 1,5—3 мм поступает в водную ванну
при 50—60 °С. Чем выше температура ванны, тем больше усадка
невытянутого волокна и тем выше прочность вытянутого
моноволокна [42]. В этой ванне при скорости формования 20—40 м/мин
полимер застывает и образуются нити. Так как фильера обычно
имеет 10—15 отверстий, то при формовании получается пучок
моноволокон, которые вытягиваются на 400—450%.
Вытягивание моноволокна происходит в горячей воде при 95—
99°С. Вытянутое волокно в большинстве случаев для
кристаллизации и снятия напряжений подвергается термообработке под
натяжением или в мотках при 180—240°С. В результате такой
обработки получается волокно, почти не усаживающееся при
последующем нагреве (усадка около 3%), что обеспечивает стабильность
формы получаемых изделий.
Для изготовления моноволокна, используемого в изделиях,
подвергающихся при эксплуатации динамическим нагрузкам,
необходимо применять полимер повышенного молекулярного веса.
3.1.11. И спользование отходов
Наиболее эффективным способом утилизации отходов
полиэфира и волокна является гидролитическая деструкция, в
результате которой образуется терефталевая кислота или диметилтере-
фталат. Для этого отходы полиэтилентерефталатного волокна
подвергаются гидролизу в воде или в растворе щелочи или алкоголизу
при высоких температурах. Полное омыление отходов в результате
их гидролиза в воде при давлении 15 кгс/см2 A,5 • 106 Па)
достигается через 5 ч, при давлении 20 кгс/см2 B-Ю6 Па)—через 1ч.
Выделяющаяся терефталевая кислота выпадает в осадок,
отфильтровывается, промывается и направляется на этерификацию [43].
Гидролиз отходов полиэтилентерефталата в 5—7%-ном
растворе NaOH производится в течение 1—2 ч при более низкой
температуре и более низком давлении, а именно 9—10 кгс/см2
@,9 • 106—1-Ю6 Па). Однако этот метод неэкономичен из-за
большого расхода едкого натра, реагирующего с терефталевой
кислотой с образованием натриевой соли.
154 Глава 3. Производство полиэфирных волокон
Наиболее экономичным методом является разложение отходов
метанолом. При обработке полиэфира метанолом в течение 3—
6 ч при 280 °С и давлении 27 кгс/см2 B,7- 10s Па) образуется
диметилтерефталат (80% от теоретического), который после
перегонки (без дополнительной обработки) может быть использован
для синтеза полиэтилентерефталата. Известным недостатком этого
метода является необходимость применения при разложении
отходов более высокого давления, чем при гидролизе.
Температура при метанолизе полиэтилентерефталата может
быть снижена до 150 °С, давление — до 15 кгс/см2, если процесс
проводится в присутствии катализаторов [44]. В качестве
катализаторов можно использовать соли свинца, цинка или марганца,
т. е. в основном те же вещества, которые являются катализаторами
реакции поликонденсации.
Алкоголиз может быть осуществлен путем обработки полиэфира
Этиленгликолем. Этим методом в качестве продукта реакции
получается непосредственно диэтиленгликольтерефталат, который
после очистки можно использовать для получения
полиэтилентерефталата.
Волокнистые отходы не всегда целесообразно подвергать
гидролитической деструкции для регенерации исходных продуктов.
Эти отходы могут найти эффективное применение в качестве
наполнителей.
3.1.12. Свойства полиэтилентерефталатного
волокна и области его применения
Из большого числа разнообразных полиэфиров,
синтезированных различными исследователями, для получения высокопрочного
и высокоплавкого волокна применяется только полиэфир,
образующийся при поликонденсации этиленгликоля и терефталевой
кислоты. Специфически ценные свойства полиэтилентерефталатного
волокна объясняются составом и строением полимера. В частности,
большое влияние на свойства этого полиэфира оказывает:
большая жесткость цепи благодаря наличию симметричных
гс,п'-фениленовых групп в макромолекуле;
высокая полярность сложноэфирных групп в макромолекуле
полимера;
наличие водородных связей между сложноэфирными группами
(или группами СО) и водородом в бензольном кольце, которые
способствуют тому, что макромолекулы сильно вытянуты и
обладают высокой степенью асимметрии.
Волокно, полученное из такого полимера, обладает
способностью быстро кристаллизоваться *. Кристаллизация в вытянутом
* Кристаллический полиэтилентерефталат в отличие от аморфного
непрозрачен [45].
S.I. Производство полиэтилентерефталатных волокон 155
(ориентированном) состоянии обусловливает высокие механические
свойства волокна.
Волокно из полиэтилентерефталата характеризуется
следующими показателями.
Прочность и удлинение. По этому показателю полиэтилентере-
фталатное волокно почти не уступает полиамидному. Прочность
обычной полиэфирной нити составляет 40—50 гс/текс @,4—
0,5 Н/текс), высокопрочной кордной нити — 60—80 гс/текс @,6—
0,8 Н/текс). В мокром состоянии прочность нити (волокна) не
изменяется.
Дальнейшее увеличение прочности нити может быть достигнуто
при повышении молекулярного веса полиэфира до 30 000 и более,
что связано с необходимостью перерабатывать более вязкие
расплавы. Прочность полиэфирной нити можно увеличить повышением
степени ее вытягивания при использовании для формования так
называемых изоморфных полиэфиров (см. разд. 3.2).
Удлинение обычной полиэтилентерефталатной нити составляет
20—25%, а высокопрочной—10—12%.
Эластичность. Полиэфирное волокно высокоэластично. При
вытягивании до 5—6% удлинение полностью обратимо. Этим и
объясняется высокая устойчивость полиэфирных волокон и
получаемых из них изделий к сминанию, превышающая устойчивость к сми-
нанию всех других волокон.
При нормальной влажности воздуха изделия из полиэфирного
волокна обладают в 2—3 раза более высокой устойчивостью
к сминанию, чем шерстяные изделия. При повышенной влажности
(например, при относительной влажности воздуха 90%) сминае-
мость изделий из полиэфирного волокна также значительно меньше,
чем шерстяных изделий.
Модуль полиэфирной нити в 3—5 раз выше, чем полиамидной,
и в 2 раза больше, чем хлопковой и вискозной. Это свойство,
являющееся одним из важных преимуществ полиэфирной нити, имеет
большое значение при определении областей ее применения (в
частности, при производстве кордной ткани).
Плотность. Плотность полиэфирного волокна A,38 г/см3)
выше, чем полиамидного, и существенно изменяется в зависимости
от степени его кристаллизации. Например, плотность аморфного
полиэфирного волокна 1,33 г/см3, а полностью
кристаллизованного— 1,47 г/см3. Зная плотность полиэфирных волокон,
полученных в различных условиях, можно приблизительно определить их
среднюю степень кристалличности. Плотность полых волокон
снижается до 0,6—1,2 г/см3.
Гигроскопичность. Полиэфирное волокно негигроскопично; это
является ценным свойством при использовании его в качестве
электроизоляционного материала и существенным недостатком
при крашении и отделке в производстве предметов народного
156 Глава 3. Производство полиэфирных волокон
потребления. Водопоглощение полиэфирного волокна при
относительной влажности воздуха 65% составляет 0,4%.
Термостойкость. По стойкости к повышенным температурам
полиэфирное волокно превосходит все природные и большинство
химических волокон (кроме термостойких волокон) . Например,
после нагрева 1000 ч при 150 °С полиэфирное волокно теряет не
более 50% прочности, в то время как все другие волокна
полностью разрушаются после нагрева при этой температуре в
течение 200—300 ч.
Термостойкость полиэфирного волокна дополнительно
повышается при блокировании концевых групп ОН макромолекул
полиэфира в процессе поликонденсации и введении в состав полимера
термостабилизаторов. Например, прочность нестабилизованного
полиэфирного волокна после нагрева 2 ч при 200 °С снижается на
40%, а при нагреве в этих же условиях волокна из полиэфира,
у которого концевые группы ОН этерифицированы фосфорной
кислотой, потеря прочности волокна не превышает 23%.
Одновременно достигается стабильность цвета волокна при нагреве —
волокно не желтеет.
Однако термическая деструкция полиэфиров обусловливается
не только влиянием концевых групп ОН. Наибольшее влияние на
снижение термостойкости оказывают альдегидные группы,
находящиеся в незначительном количестве в молекуле окисленного эти-
ленгликоля, или образующиеся в результате окисления концевых
групп ОН и деструкции полимера. Такое же влияние оказывают
оставшиеся в полимере катализаторы, которые инициируют распад
альдегидных и перекисных групп и обусловливают распад
макромолекулы полиэфира при высоких температурах по радикальному
механизму [46].
Удаление альдегидных групп из макромолекулы полиэфира
восстановлением (обработкой раствором боргидрида натрия) или
окислением хлоритом натрия значительно повышает
термостойкость полимера. Следовательно, для эффективной
термостабилизации полиэфиров необходимо кроме блокирования концевых
групп ОН ингибировать реакцию окисления, протекающую по
цепному механизму, связать металлы, применявшиеся в качестве
катализаторов при реакциях переэтерификации или поликонденсации
и оставшиеся в полимере, а также связать или разрушить
альдегидные группы, находящиеся в молекуле полиэфира. Эти задачи
в основном могут быть решены при использовании в качестве
стабилизатора фосфорной кислоты. Последняя не только блокирует
концевые группы ОН путем их этерификации, но, по-видимому, и
связывает альдегидные группы по реакции Арбузова и реагирует
с остатками катализатора, образуя соответствующие соли фосфор1
ной кислоты или комплексные соединения.
Эффективными термостабилизаторами являются также
фосфористая кислота, добавляемая в таких же количествах, как и фос-
3.1. Производство полиэтилентерефталатных волокон
157
форная кислота @,01—0,02% от массы ДМТ), и трифенилфосфит,
который наиболее интенсивно замедляет скорость окисления эти-
ленгликоля и образующегося полимера. Поэтому полиэфир,
полученный в присутствии трифенилфосфита, содержит значительно
меньше альдегидных групп, а волокно из такого полимера желтеет
при формовании в минимальной степени.
Дополнительным преимуществом трифенилфосфита по
сравнению с фосфорной кислотой является более высокая температура
0,35 г
260 270 Z80 2.90
letinepamypa, С
300
Рис. 3.10. Влииние количества и характера фосфитов на изменение удельной
вязкости полиэтилентерефталата прн плавлении:
/—без фосфита; 2—0,025?« три-п-гргг-бутилфенилфосфита (от массы ДМТ); 3—0,025?« п-до-
децилфеиилфосфита; 4—0,25% трифеиилфосфита; 5—0,25% три-я-гргг-бутилфеаилфосфита.
кипения и большая термическая стойкость. Кроме того, алкилфе-
нолы, и в частности трифенилфосфат, являются ингибиторами
реакций цепного распада. Поэтому целесообразно в качестве
термостабилизаторов применять смесь фосфитов и алкилфенолов.
Эфиры фосфористой кислоты добавляют (в количестве 0,01 —
0,25% от массы ДМТ) в виде 15%-ного раствора фосфита в эти-
ленгликоле. Небольшие добавки эфиров фосфористых кислот
замедляют деструкцию (уменьшение молекулярного веса) полиэфира
при его плавлении, особенно при 280—290 °С (рис. 3.10) [47].
Трифенилфосфит, являясь трехфункциональным соединением,
имеет существенный недостаток: при блокировании концевых групп
ОН может создавать трехмерную сетку между макромолекулами и
тем самым затруднять вытягивание сформованного волокна.
Эфиры фосфористой кислоты, применяемые в качестве
стабилизаторов, химически реагируют по реакции переэтерификации
с полиэтилентерефталатом, образуя смешанные полиэфиры.
158 Глава 3. Производство полиэфирных волокон
В результате снижается температура перехода полимера в вязко-
текучее состояние.
Эффективными термостабилизаторами полиэфиров, согласно
полученным в последнее время данным [48], являются смеси терпен-
фенолов, например 2,6-диизоборнилфенола с три-л-трет-бутил-фе-
нилфосфитом. Количество вводимого стабилизатора составляет
0,5—1,0- Ю-3 моль/моль ДМТ.
Стабилизированное полиэфирное волокно обладает значительно
более высокой усталостной прочностью, чем волокно, не
содержащее стабилизаторов [49]. Усталостная прочность, характеризуемая
числом двойных изгибов, выдерживаемых волокнами до разрыва,
дополнительно повышается в результате терморелаксации
волокна.
Имеются стабилизаторы, которые наряду с увеличением
термической устойчивости полиэфира повышают его молекулярный вес
[50]. К таким стабилизаторам относится, например, дифениловый
эфир угольной кислоты С6НбО—СО—ОСвНб. Увеличение
молекулярного веса можно объяснить этерификациеи концевых групп ОН
макромолекулы полиэфира бифункциональным соединением, а
повышение термостабильности — связыванием свободных радикалов,
появляющихся при термораспаде макромолекулы полиэфира по
радикальному механизму, феноксильными радикалами,
образующимися при распаде дифенилового эфира.
Стойкость к низким температурам. Полиэфирные волокна
обладают также высокой стойкостью к низким температурам. Пленка
из полиэтилентерефталата остается эластичной при —50 °С и не
становится хруякой даже при —60 °С [51, с. 331].
Истираемость. По устойчивости к истиранию полиэфирное
волокно лучше гидратцеллюлозных и синтетических карбоцепных
волокон, но хуже полиамидных. Устойчивость к истиранию
полиэфирных волокон в сухом состоянии в 4—4,5 раза, а в мокром —
в 2—2,5 раза меньше, чем полиамидных.
Светостойкость. Полиэфирные волокна обладают значительно
более высокой светостойкостью, чем большинство природных и
химических волокон [46]. По этому показателю полиэфирное
волокно уступает только полиакрилонитрильному.
Несмотря на сравнительно высокую светостойкость
полиэфирного волокна, этот показатель может быть дополнительно повышен
путем добавления в процессе поликонденсации или перед
формованием волокна небольших количеств светостабилизаторов. К таким
добавкам, значительно повышающим стойкость пленок из
полиэтилентерефталата к действию облучения, относится салол и диэти-
ловый эфир 2,5-диокситерефталевой кислоты, а также производные
бензотриазола.
Хемостойкость. Полиэфирное волокно обладает высокой
стойкостью к-кислотам и окислителям, значительно превышающей
стойкость к этим реагентам полиамидных волокон. Однако поли-
3.1. Производство полизтилентерефталатных волокон
159
эфирное волокно недостаточно стойко к некоторым минеральным
кислотам (азотной и серной), и особенно к щелочам (рис. 3.11).
Сохранение формы. Характерной особенностью изделий
народного потребления, изготовленных из полиэфирных волокон или из.
wo
80
60
40
10
-
-
\\
\
1 1 1
w
^70°C
1 .
0
00
80
SO
40
20
w го зо 40
Концентрация
- \ \ о
If с 7°а°
Г 1 I I
50 60
H2SO
\
\
\
ko с
I Т
70
10 Zu J0 W 50 60 70
Концентрация НК[Оз,%
О 5 10 15 го 15 30 35
Концентрация HCI,%
Э5°С
0 10 го 30 40 50 60 70 80
Концентрация
О 1 4 6 8 10 1Z Ш 1В
Концентрация
2 4- ' В 8 10 іг П
Концентрация
Рис. 3.11. Стойкость полиэфирных волокон к неорганическим кислотам и
щелочам (продолжительность обработки 72 ч).
их смесей с другими волокнами, является устойчивость формы
изделия, в частности плиссе. После стирки внешний вид изделия не
ухудшается. По способности к сохранению приданной в процессе
переработки формы полиэфирные волокна значительно
превосходят шерсть, не говоря уже о гидратцеллюлозных волокнах. Шер-
степодобный вид полиэфирного волокна и получаемых из него
изделий, теплота на ощупь, а также большая объемность этого
волокна обусловливают целесообразность использования его
в смеси с шерстью, хлопком и с другими волокнами для
изготовления разноообразных изделий. Содержание полиэфирного волокна
в смеси может быть различным, и даже превышать 50%.
160 Глава 3. Производство полиэфирных волокон
Изделия из полиэфирных волокон легко стираются и быстро
высыхают. Усадка изделий из полиэтилентерефталатного волокна,
вытянутого при повышенной температуре, сравнительно
незначительна и может быть дополнительно уменьшена после термической
обработки (термофиксации). В результате термофиксации
повышаете» также стабильность формы изделий и уменьшается их
сминаемость [52]. Поэтому термообработка готовых "тканей или
трикотажных изделий, независимо от термообработки волокна,
является обязательным условием получения несминаемых
текстильных изделий.
Другие свойства. Так же как и другие синтетические волокна,
полиэфирное волокно обладает высокой стойкостью к действию
бактерий и микроорганизмов. Это волокно не горит, а только
плавится.
Существенный недостаток волокна из полиэтилентерефталата^—
плрхая окрашиваемость, что объясняется высокой
кристалличностью и отсутствием в макромолекуле полиэфира реакционно-
способных функциональных групп. Например, скорость диффузии
одного и того же красителя в полиэфирное волокно примерно
в 500 раз меньше, чем в ацетатное или полиамидное волокно. Для
улучшения окрашиваемости полиэфирного волокна разработан ряд
методов: крашение волокна в массе, нарушение регулярной
структуры полимера при получении смешанных полиэфиров, а также
проведение процесса крашения при высокой температуре A80—
225 °С) и повышенном давлении.
Полиэфирные волокна можно окрашивать также и при
нормальном давлении (при 95—98°С), если в красильную ванну
вводить так называемые переносчики * — вещества, вызывающие
разрыхление структуры волокна (например, 2,2'-диоксидифенил,
салициловая и бензойная кислоты, фенол, л-крезол, ?-нафтиламин,
бензиловий спирт и др.).
Однако практическое использование указанных веществ
ограничено токсичностью большинства из них, а главное, трудностью
очистки сточных вод, содержащих большее или меньшее
количество этих реагентов.
В последние годы разработаны и реализованы в
производственных условиях новые методы крашения изделий из полиэфирных
волокон, обеспечивающие возможность интенсивного и быстрого их
окрашивания.
Области применения. Рассмотренные свойства полиэфирных
волокон определяют области их применения. Благодаря высокой
устойчивости к сминанию и способности к сохранению формы,
хорошему внешнему виду и другим свойствам полиэфирное волокно
в чистом виде или в смеси с другими волокнами целесообразно
• Характеристика' переносчиков приводится в специальных руководствах
(см. Фурне Ф. Синтетические волокна, М., «Химия», 1970, с. 472).
3.2. Модифицированные полиэфирные волокна ^ 161
- _
применять для широкого ассортимента товаров народного
потребления— одежной и костюмной тканей, верхнего трикотажа и т. д.
Вследствие относительно невысокой устойчивости к истиранию (по
сравнению с полиамидными волокнами) использование этого
волокна для производства чулочно-носочных изделий
нецелесообразно. Благодаря высокой светостойкости полиэфирное волокно
может быть использовано для изготовления заназесен, парусов
и других изделий, для которых этот показатель имеет большое
значение.
Высокий модуль наряду с большой прочностью обусловливает
целесообразность применения полиэфирного волокна для
производства кордной ткани, транспортерных лент, приводных ремней,
пожарных рукавов и других аналогичных изделий.
Высокие диэлектрические свойства и низкая гигроскопичность
определяют перспективность использования полиэфирного волокна
в качестве электроизоляционного материала.
В последние годы области применения полиэфирных волокон
непрерывно увеличиваются.. Эти волокна получили широкое
применение для изготовления высокообъемных тканей и трикотажных
изделий, хирургических нитей и искусственного меха и т. п. Кроме
того, полиэфирное волокно используется для электроизоляции
очень тонких проводов.
3.2. МОДИФИЦИРОВАННЫЕ ПОЛИЭФИРНЫЕ ВОЛОКНА
Свойства полиэфирных волокон можно изменять в широких
пределах путем модификации структуры и химического состава
этих волокон. Благодаря отсутствию в макромолекуле полиэтилен-
терефталата реакцпонноспособных групп описанные выше методы
химической модификации, в частности методы превращения реак-
ционноспособных функциональных групп, не могут быть
использованы для направленного изменения свойств полиэфирных волокон.
К основным задачам, которые могут быть решены путем
модификации свойств полиэфирных волокон, следует отнести:
улучшение накрашиваемости;
повышение комплекса механических свойств волокон;
получение извитых и объемных волокон;
уменьшение склонности волокна к пиллингу.
3.2.1. Улучшение накрашиваемости волокон
Выше уже указывалось, что наряду со специфическими
ценными свойствами полиэфирное волокно имеет ряд недостатков
(трудность окрашивания, недостаточная устойчивость к
истиранию), затрудняющих его переработку и практическое
использование. Для устранения этих недостатков необходимо понизить
кристалличность получаемого полиэфирного волокна.
G Зак >-,!
162 ' Глава 3. Производство полиэфирных волокон
Понижение кристалличности полиэтилентерефталата без
заметного ухудшения ценных свойств этого полимера и получаемых
из него волокон может быть достигнуто уменьшением регулярности
его строения. Для этого могут быть использованы следующие
методы:
синтез смешанных полиэфиров путем применения в процессе
поликонденсации кроме терефталевой кислоты и этиленгликоля
других дикарбоновых кислот и гликолей;
получение блок-сополимеров полиэтилентерефталата с другими
полимерами.
Получение смешанных полиэфиров. Возможность получения
смешанных полиэфиров детально исследована Грилем и
Гофмейстером [53].
При частичной замене при поликонденсации этиленгликоля бу-
тандиолом или 1,2-проїіандиолом НОСН2—СНОН значительно по-
СНз
нижается кристалличность волокна, но одновременно сильно
уменьшается прочность и температура его плавления. Поэтому
данный метод снижения регулярности структуры полиэфира для
практических целей непригоден.
При исследовании возможности применения в процессе поли-
конденсации наряду с терефталевой кислотой небольшого
количества другой дикарбО'НОвой кислоты было установлено [53], что для
получения сополиэфира с достаточно удовлетворительными
механическими свойствами целесообразно применять себациновую
кислоту. При применении дикарбоновых кислот с более низким
молекулярным весом заметно понижается термостойкость'полимера.
Полиэфир, полученный совместной поликонденсацией
этиленгликоля, терефталевой кислоты и небольшого количества себаци-
новой кислоты (до 20% от массы сополиэфира), обладает рядом
технически ценных свойств. Несмотря на более низкую
температуру плавления B20°С), такой полиэфир более стоек, чем поли-
этилентерефталат, к повышенным температурам, применяемым при
формовании волокна. При добавлении 10% себациновой кислоты
[53] прочность получаемого волокна не снижается, но
увеличивается его удлинение (с 17 до 28%). Волокно из такого
сополиэфира обладает значительно более высокой устойчивостью к
изгибу, чем волокно из полиэтилентерефталата. Если, например, поли-
этилентерефталатное волокно, вытянутое в 4,8 раза, выдерживает
400 изгибов, то волокно, получаемое из сополиэфира указанного
состава, выдерживает свыше 10000 изгибов. Накрашиваемость
волокна из этого сополиэфира примерно на 50% выше, чем волокна
из полиэтилентерефталата. Светостойкость полиэфирных волокон
обоих видов одинакова.
Сополиэфиры, содержащие в макромолекуле даже небольшие
количества алифатических дикарбоновых кислот, обладают более
3.2. Модифицированные полиэфирные волокна 163
низкой температурой кристаллизации. Например, температура
кристаллизации сополиэфира, в состав которого входит 15%
алифатической дикарбоновой кислоты (пимелиновой, азелаиновой или
себациновой), снижается на 40 °С [54]. Одновременно'уменьшается
степень кристалличности полиэфира и формуемого из него
волокна.
Учитывая высокую термостойкость, устойчивость к
многократным деформациям и повышенную накрашиваемость, можно
предположить, что волокно из сополиэфира терефталевой и
себациновой кислот и этиленгликоля (при соотношении терефталевой и
себациновой кислот 9:1), несмотря на более низкую температуру
плавления, найдет практическое применение.
Интересный метод получения волокон из сополиэфиров был
разработан советскими исследователями [55]. Смешанный полиэфир
синтезировали путем поликонденсации полиэтиленгликоля с
терефталевой кислотой, к которой добавлено небольшое количество
E—10% от массы терефталевой кислоты) периоксиэтилового эфира
n-оксибензойной кислоты НООС—<f /—OCHoCHjOCHa- Из этого
сополиэфира обычными методами формовали волокно, получившее
название «оксон».
Как и другие волокна, полученные из смешанных полиэфиров,
волокно оксон отличается от лавсана более низкой температурой
плавления и большей усадкой в кипящей воде. Усадка волокна
тем больше, чем больше количество метилового эфира /г-этокси-
бензойной кислоты в макромолекуле сополиэфира и чем сильнее
нарушена регулярность его строения. Благодаря меньшей
регулярности строения волокна оксон окрашивается значительно легче,
чем лавсан. Равномерная и интенсивная окраска изделий из
волокна оксон может быть достигнута без применения переносчиков.
Существенный интерес представляет метод, основанный на
введении в молекулу сополиэфира небольшого числа A—2%)
звеньев другой ароматической кислоты, содержащей реакционно-
способные группы, например сульфоизофталевой кислоты [56]
(в виде калиевой соли).
Другим видом сополиэфирного волокна является кодель,
волокно, которое производилось ранее в США из сополимера,
образующегося в результате совместной поликонденсации
терефталевой кислоты, этиленгликоля и ароматического гликоля. Этот
смешанный полиэфир обладает высокой термостойкостью и
повышенной температурой плавления B90—295°С).
Смешанный полиэфир можно получить также при совместной
поликонденсации терефталевой и изофталевой кислот с этиленгли-
колем. Этот тип сополиэфйра нашел в ряде стран применение для
производства модифицированных полиэфирных волокон.
Волокна из сополиэфиров, обладающие пониженной
регулярностью строения и меньшей кристалличностью, целесообразно
в*
164 Глава 3. Производство полиэфирных волокон
использовать, по-видимому, только для производства изделий
народного потребления, для которых высокая прочность и
термостойкость не требуются, а большее значение имеет легкость
накрашивания, повышенная эластичность и усадка в горячей воде для
получения определенных текстильных эффектов.
В промышленном масштабе в США выпускаются два типа
волокон из сополиэфиров с изофталевой кислотой (волокно викрон);
или с сульфоизофталевой кислотой (волокно дакрон 64).
Практический интерес представляет разработанный Б. В. Пету-
ховым и С. М. Кондрашовой [57] метод синтеза так называемых
изоморфных полиэфиров. Основная цель такого синтеза —
повышение гибкости макромолекулы. Поэтому при синтезе
изоморфных полиэфиров небольшое число звеньев терефталевой
кислоты заменяют другими дикарбоновыми кислотами, молекулы
которых при одинаковой длине с молекулами терефталевой кислоты
обладают меньшей жесткостью.
В качестве таких кислот были использованы адипиновая или
гексагидротерефталевая (циклогексан-1,4-дикарбоновая) кислота
(точнее, диметиловые эфиры этих кислот). Так как размеры
молекул терефталевой кислоты и этих кислот одинаковы, регулярность
строения сополиэфира не изменяется. В результате увеличения
гибкости макромолекул степень зсристалличности и плотность
сополимера не только не понижаются, а, наоборот, увеличиваются.
Одновременно повышается возможная степень вытягивания
волокна и соответственно возрастает прочность (при одном и том же
удлинении) или удлинение (при одинаковой прочности волокна).
Оптимальное количество второй дикарбоновсй кислоты,
вводимой в состав сополиэфира, составляет 2—4% от массы
терефталевой кислоты. Если вместо адипиновой кислоты взять эквимоляр-
ное количество, например, себациновой кислоты, молекула которой
больше, чем молекула терефталевой кислоты, то дополнительного
упрочнения получаемого волокна не происходит. Синтез
изоморфных полиэфиров (а также и других синтетических полимеров)
является одним из наиболее эффективных методов внутренней
пластификации получаемых волокон.
Волокно из изоморфного сополиэфира, содержащего 4,2% ди-
метилового эфира адипиновой кислоты (т. е. одно звено на 20—30
звеньев диметилтерефталата), можно вытянуть на 600—620%, т. е.
примерно на 100—150% больше, чем волокно из полиэтилентере-
фталата [58]. Соответственно повышается прочность вытянутого
волокна. Аналогичные результаты получаются при формовании
олокна из сополиэфира, в молекулу которого введено 3—4% диме-
гилового эфира не адипиновой, а гексагидротерефталевой
кислоты [59].
Благодаря большей кристалличности текучесть изоморфных
волокон под нагрузкой меньше, чем волокон из полиэтилентере-
фталата. По этой же причине и модуль при повышенных темпера-
3.2. Модифицированные полиэфирные волокна 165
турах A20—160 °С) у этого типа сополиэфирных волокон
примерно в 2 раза выше. Кроме того, в результате увеличения гибкости
макромолекул эти волокна обладают значительно большей
устойчивостью к многократным деформациям (двойным изгибам).
Метод химической модификации, основанный на синтезе
привитых сополимеров, для полиэфиров и волокон из них является
малоперспективным. Реакция привитой сополимеризации для этого
класса гетероцепных полимеров может быть осуществлена только
при облучении их лучами высоких энергий, что затрудняет
применение этого метода.
Получение блок-сополимеров. Вторым направлением
модификации свойств полиэфирных волокон является синтез
блок-сополимеров полиэтиленоксида H (ОСН2СН2)ПОН с молекулярным весом
1350—2800 и полиэтилентерефталата [60]. Этот блок-сополимер
имеет следующее строение:
[—OCHjCHaOOC—f~\—СО—1 -O-I-
L \ / Im
Волокно, полученное формованием из расплава
блок-сополимера, содержащего 7—10% полиэтиленгликоля, имеет такую же
прочность, как и волокно из полиэтилентерефталата, но в 3 раза
более высокую гигроскопичность и накрашиваемость и в 10 раз
более высокую устойчивость к изгибу. Однако по светостойкости
волокно из блок-сополимера указанного состава значительно^
уступает полиэтилентерефталатному. Если волокно из
полиэтилентерефталата, подвергнутое действию ультрафиолетовых лучей в
течение 20 ч, теряет 9% прочности, то потеря прочности волокна из
блок-сополимера достигает 61,3%. Этот существенный недостаток
волокна из блок-сополимера ограничивает возможность его
практического применения.
Предложен новый метод синтеза блок-сополимеров
полиэтилентерефталата [61], который заключается в том, что в качестве
второго компонента применяется полиэфир, полученный при
поликонденсации этиленгликоля с ароматическими (фталевой и изофта-
левой) или жирными дикарбоновыми кислотами. Такой
блок-сополимер синтезируют по реакции обмена путем совместного
плавления полиэтилентерефталата с одним из полиэфиров указанного
состава A0—20% от массы полиэтилентерефталата) в вакууме
в течение 3 ч *.
Волокно из этого блок-сополимера имеет такую же прочность,
как полиэтилентерефталатное, но значительно лучшую (в 2—
* При увеличении продолжительности обработки в результате
дополнительно протекающей реакции переэтерификации получается ¦ не блок-сополимер, а
смешанный сополимер.
166 Глава 3. Производство полиэфирных волокон
3 раза) накрашиваемость. Данные, характеризующие другие
свойства этого волокна (эластичность, термостойкость), в
литературе не приводятся.
3.2.2. Получение извитых и объемных волокон
Получение извитых и объемных полиэфирных волокон, так же
как и других термопластичных волокон, основано на использовании
двух или нескольких типов полиэфиров, отличающихся
температурой плавления или молекулярным весом, а следовательно, и
степенью усадки волокон и изделий при повышенной температуре
(в частности, в горячей воде).
Практически применяются три способа решения этой задачи:
получение бикомпонентных волокон;
использование для изготовления волокон сополиэфиров,
отличающихся от полиэтилентерефталата температурой плавления и
усадкой при повышенных температурах;
применение полиэфирного волокна, подвергнутого холодному
вытягиванию.
Получение бикомпонентных волокон. При производстве
бикомпонентных волокон расплав полиэфира подается к фильере двумя
насосиками. Один из насосиков подает расплав
полиэтилентерефталата, а второй — расплав полимера, обладающего на 20—30 °С
более низкой температурой плавления, чем полиэтйлентерефталат.
В. качестве таких полимеров могут быть использованы:
сополиэфиры, в частности сополимер, полученный
поликонденсацией этиленгликоля с диметилтерефталатом и небольшим
количеством (8—10% от массы ДМТ) диметилизофталата;
полиамиды типа капрон;
полиэтйлентерефталат более низкого молекулярного веса,
волокно из которого обладает соответственно большей усадкой.
Соотношение этих полимеров и полиэтилентерефталата может
быть различным, но в большинстве случаев оно составляет 1 : 1.
Формование и вытягивание волокна, полученного из смеси
полимеров указанного состава, производится по обычной схеме. При
нагреве нити или готовых изделий при 140—160 °С волокно
приобретает извитость в результате различной усадки составляющих
его компонентов. Этот способ применяют в большинстве случаев
при изготовлении трикотажных изделий.
Получение волокон из сополиэфиров. При использовании
второго способа получения извитых и объемных нитей волокна из
полиэтилентерефталата и сополиэфиров формуют раздельно. В
качестве исходного волокнообразующего полимера для получения
высокоусадочного волокна применяют обычно сополиэфир,
содержащий 5—10% изофталевой или 10% адипиновой кислоты.
Волокно с высокой усадкой формуют из сополиэфира,
синтезированного по реакции поликонденсации в присутствии 8—10 мол. %
3.2. Модифицированные полиэфирные волокна 167
калиевой соли этиленгликольсульфонатизофталата [62, с. 26].
Получение сополиэфира приводит к некоторому снижению прочности
получаемого волокна. Это объясняется нарушением регулярности
структуры полимера и снижением его температуры плавления^
Например, введение в макромолекулу 8 мол. % звеньев этиленгли-
кольизофталата снижает температуру плавления с 255 до 241 °С-
Если в молекуле полиэфира будут звенья, в состав которых входит
сульфогруппа, то одновременно значительно улучшается окраска
волокна дисперсными красителями и, кроме того, волокно
приобретает способность окрашиваться основными красителями.
Принципиально высокоусадочное волокно можно получить из
смешанных полиэфиров при использовании в процессе
поликонденсации не смеси различных дикарбоновых кислот, а различных
диолов (добавлением небольших количеств пропиленгликоля и
других гликолей). Однако этот способ получения смешанных
полиэфиров для производства волокна не нашел пока практического
применения.
Полученное из сополиэфиров волокно после вытягивания и
последующей обработки смешивают перед изготовлением пряжи
с обычным волокном лавсан, а пряжу или готовое изделие
подвергают термообработке горячей водой ,в процессе крашения. В
результате различной усадки двух типов полиэфирных волокон
получается извитое (объемное) изделие *.
Получение извитого волокна холодным вытягиванием. Поли-
этилентерефталатное волокно, обладающее повышенной усадкой
при нагреве, получают методом холодного вытягивания. Сущность
метода заключается в том, что полиэфирное волокно, содержащее
незначительное количество влаги (являющейся в известной
степени пластификатором волокна и снижающей температуру его
стеклования на 45—50°С), начинают вытягивать при нормальной!
температуре. В этом случае применяются значительно большие
усилия, чем при вытягивании волокна в условиях повышенных
температур (выше температуры стеклования полимера).
Поскольку при вытягивании волокно разогревается,
заключительная часть процесса протекает фактически при температурах,
близких к температуре стеклования волокна. Однако степень
кристалличности такого волокна ниже, а напряжение в волокне выше,
чем у волокна того же химического состава, подвергнутого
вытягиванию в обычных условиях после предварительного нагрева до
температуры, превышающей температуру его стеклования.
Следовательно, сущность метода холодного вытягивания заключается
в том, что волокно вытягивают без предварительного нагрева.
* В последнее время объемное полиэфирное волокно начинают получать
смешением штапельного волокна лавсан, подвергнутого вытягиванию в
различных условиях. Этот метод используется, в частности, на Курском комбинате
химического волокна.
168 Глава 3. Производство полиэфирных волокон
Вытянутое волокно обладает, благодаря меньшей степени
кристалличности, значительно более высокой усадкой при повышенных
температурах, чем обычное волокно лавсан. При нагреве до 100°С
оно усаживается на 50%.
Требования к усадке модифицированных полиэфирных волокон
различны в зависимости от их назначения. Усадочные нити,
применяемые для получения тканей, сравнительно мало усаживаются
в кипящей воде (на 3—5%), а основная усадка (на 25—40%)
происходит при термообработке изделий на воздухе при 200 °С.
Высокоусадочные полиэфирные волокна, используемые для
изготовления трикотажа, должны в основном усаживаться в кипящей воде, а
дополнительная усадка при нагреве на воздухе не должна
превышать 3—5%. Такая разница в свойствах и условиях обработки
этих волокон и получаемых из них изделий определяется
химическим составом получаемого сополиэфира [63]. Как правило,
высокоусадочными называются такие полиэфирные волокна, усадка
которых при тепловых обработках (в кипящей воде, на воздухе при
180—200°С) составляет 30—50%.
Содержание высокоусадочных волокон в пряже при получении
высокоусадочных изделий составляет 20—30% от общего
количества полиэфирного волокна.
3.2.3. Уменьшение склонности волокна к пиллингу
Уменьшения пиллинга, который, так же как и у полиамидных
волокон, значительно ухудшает внешний вид изделий, можно
достигнуть различными способами. Наибольшее применение получили
два способа: уменьшение прочности волокна и увеличение крутки
пряжи.
Уменьшение прочности волокна. Снизить прочность волокна
можно путем уменьшения молекулярного веса исходного полимера.
Чем меньше прочность и выше хрупкость волокна, тем легче
обрываются отдельные волокна, образующие шарики и комочки на го-'
товых изделиях. Поэтому при получении волокна,
предназначенного для изготовления трикотажных изделий, при эксплуатации
которых разрывная прочность не имеет существенного значения,
молекулярный вес полиэфира снижают до 15 000. Этот способ
борьбы с пиллингом является одним из немногих примеров в
производстве химических волокон, когда целью проводимых
обработок является снижение прочности волокон и их устойчивости к
истиранию.
Повышение крутки пряжи. Чем выше крутка, тем меньше
возможность обрыва отдельных волокон, образующих шарики или
комочки. Однако повышение крутки связано с увеличением числа
крутильных машин и затрат труда, поэтому на практике данный
способ применяется редко.
Литература 169
ЛИТЕРАТУРА
1 С a rot hers W., Arvin I., J. Chem. Soc, 1929, v. 51, p. 2560—2562.
2. Chem. a. Ind., 1970, v. 22, N 3, p. 123—126.
3 Коршак В. В., Виноградова С. В., Высокомол. соед., 1959, т. I,
с. 834—837.
4 Hill R., J. Soc. Dyers Colour., 1952, v. 68, p. 158—162.
5. В atze r H., Angew. Chem., 1952, Bd. 67, S. 552—556.
6. Коршак В. В., Виноградова С. В. В кн.: Гетероцепные полиэфиры.
М., Изд-во АН СССР, 1958. 403 с.
7. Артемьев А., X аилов В., Хим. наука и пром., 1956, т. 1, № 1,
с. 22—27.
8. С к р и п к и н а И. А., Т ю л ю к а н о в а Ю. П. Производство полиэфирных
волокон в СССР и за рубежом. М., НИИТЭХИМ, 1969. 45 с.
9. Петухов Б. В. Полиэфирные волокна. М, Госхимиздат, 1960. 87 с.
10. Griel W, Faserforsch, u. Textutechn., 1953, Bd. 4, S. 465—469.
11 Griel W„ Schnock G., Faserforsch, u. Textutechn., 1957, Bd. 8, S. 408—
411.
12. Му ромова Р. С Шарапова И. А., Хим. волокна 1963, № 1, с. 19—22.
13. Chem. -Ztg.. 1963, Bd. 87, S. 497, 498.
14. Репина Л. П., Крем ер Е. Б., Айзенштейн Э. М, Хим. волокна,
1969. № 6, с. 9—12.
15. Chem. Ind., 1969, N 8, p. 537, 538; Chem. Age, 1969, v. 99, N 2, p. 621—624.
16. Зуева В. H., Евсюков В. С., Айзенштейн Э. М. и др., Хим.
волокна, 1969, № 2, с. 59—60.
17. Chem. Eng. News, 1967, v. 74, N 9, p. 102—104.
18. Мягков В. А., Репина Л. П., Хим. волокна, 1963. № 3, с. 25—27.
19. Goodman I., An gew. Chem., 1962. Bd. 74, S. 606—609.
20. Петухов Б.. В., Терехова Г. М., Хим. волокна. 1961, № 5, с. 24—26.
21. Петухов Б. В. Докторская диссертация. ВНИИСВ, Калинин, 1962.
22. Lud ewig H. «Polyestern», Akademie Verlag DDR. 1965 S. 65—121
23. Schellen H„ «Chemiefasern», 1965, N 12. S. 923—925.
24. Hardung H., H є і m о N., Ind. Eng. Chem., 1969, v. 62, N 3, p. 13.
25. Scheller H„ «Chemiefasern», 1965. N 12, S. 926.
26. Marschall I„ Todd A., Trans. Faraday Soc, 1953, v. 49, p. 67—70.
27. Матвеева С. П., Мягков В. А. Хим. волокна, 1959, № 5 с 18—20
28. Uber r ei te r К., Gotze F., Makromol. Chem.. 1955, Bd. 29 S. 61—66.
29. Gerke К., Rein і seh G., J. Polymer Sei., 1969, v. A7, p. 1571—1574.
30. Петухов Б. В., Полиэфирные волокна. AV, Госхимиздат, 1960. 87 с.
31. Михайлов Н. В., Горбачева В. О., Айзенштейн Э М. и др. Хим.
волокна, 1964, № 5, с. 22—25.
32. А йзен штей н Э. М., Петухов Б. В., Хим. волокна, 1964 № 4,
с. 24—27.
33. Терехова Г. М., Петухов Б. В., Хим. волокна, 1960 № 2 е 8—11.
34. Ludewig H., 1. с, S. 155.
35. Lud ewig H., Raman H., Both W. e. a., Faserforsch, u. Textutechn.,
1960, Bd. 2. S. 15—19.
36. Ward I, Text. Res. J., 1961, v. 31, p. 650, 651.
37. Геллер Б. Э., Петухов Б. В., Высоцкая 3. П. и др Хнм волокна,
1967, № 4, с. 66—70.
38. Айз ен штейн Э. М., Петухов Б. В., Хим. волокна 1964 № 6,
с. 18—20.
39. Александрийский С, Кошевой Л., Брянцев В Хим волокна
1969, № 1, с. 76—79.
40. В eke г I.. «Chemiefasern», 1966, N 4, S. 276—278.
41. Кремер Е. Б., Айзенштейн Э. И., Петухов Б. В Хим волокна,
1967, № 4, с. 51—54.
42. Геллер Б. Э., Петухов Б. В., Глаз ко веки и Ю В Хим волокна
1968, № 1, с. 13—15.
170 Глава 3. Производство полиэфирных волокон
43 Сыч Л. С, Козлов В. И., Петухов Б. В. и др., Хим. волокна, 1959,
№ 6, с. 16—19.
44. Александру Л., Даскалу Л., Кристеску И., Хим. волокна, 1962,
№ 3, с. 25—28.
45 Горбачева В. О., Михайлов Н. В., Коллоид, ж., 1958, т. 20, № 1,
с. 38—41.
46. К ов а рек а я Б. М., ЖВХО им. Д. И. Менделеева, 1966, т. 11, с. 42—46.
47. Терехова Г. М., Михайлов Н. В., Токарева Л. Г., Хим. волокна,
1964, № 4, с, 33—3.6.
48. Михайлов Н. В., Терехова Г. М., Токарева Л. Г., Хим. волокна,
1970, № 4, с. 15—18.
49. M и х а й л о в Н. В., Токарева Л. Г., Горбачева В. О. и др., Хим.
волокна, 1970, № 5, с. 9—12.
50. Васильев П., Кремер Е. Е., Айзенштейн Э. М., Хим. волокна, 1970,
№ 2, с. 15-18.
51. Кор ш а к В. В., В ин огр а д о в а С. В. В кн.: Гетероцепиые полиэфиры.
М., Изд-во АН СССР, 1958. 403 с.
52. Whin fie ld T. R., Z. ges. Textilind., 1954, N 13, S. 817—819.
53. Griehl W., Hoffmeiset R., Faserforsch, u. Textiltechn., 1955, Bd 6
S. 504-509.
54. Farkai R., Boder G., Faserforsch, u. Textiltechn, 1957, Bd. 8, S. 114—118.
55. Богданов M. И., Петухов Б. В., К о н д р а ш о в а С. М., Хим. волокна,
1959, № 6, с. 21-24.
56. Репина Л. П., Пакшвер А. Б., Хим. волокна, 1966, № 5, с. 26—29.
57. Петухов Б. В., Кондрашова С. М., Высокомол. соед. 1961, т. 3,
с. 657—660.
58. П е т у х о в Б. В., К о н д р а ш о в а С. М., Хим. волокна, 1962, № 1, с. 55—58.
59. П е т у х о в Б. В., К о н д р а ш о в а С. М., Хим. волокна, 1962 № 4 с 10—13
60. Golem an I., J. Polymer Sei., 1954, v. 14, p. 15—18.
61. Kresse P., Faserforsch, u. Textiltechn., 1961, Bd. 12, S. 353—357.
62. P e n и h а Л. П., Пакшвер А. Б., Р о з е н г а у з С, С. и др. В кн.;
Гетероцепные волокна. М., «Химия», 1967', с. 26—30.
63. Timm D., Melliand Textilber., 1970, Bd. 51, S. 177—180,
Глава 4
ПРОИЗВОДСТВО ПОЛИУРЕТАНОВЫХ ВОЛОКОН
Полиуретанами называют высокомолекулярные соединения,
макромолекулы которых содержат уретановую группу —NH—С—О—.
О
Линейные полиуретаны относят к классу кристаллических во-
локнообразующих гетероцепных полимеров. Наличие
дополнительного атома кислорода в уретановой группе по сравнению с амид-
ной повышает гибкость цепи, что обусловливает более низкую
температуру плавления этих полимеров, чем у полиамидов.
Полиуретаны обычно получают взаимодействием диизоциана-
тов с гликолями. Например, при взаимодействии бутандиола-1,4
и гексаметилендиизоцианата получается полиуретан (так
называемый перлон U)
n[HO(CH2LOH] +n[OCN(CH2NNCO] —¦>
—»- O(CH2LOCONH(CH2NNHCO
который используется для получения моноволокна, щетины и
различных пластмасс.
Для переработки в волокна полиуретаны типа перлон U менее
пригодны, чем полиамиды, так как они быстро кристаллизуются,
и поэтому получаемые нити плохо вытягиваются.
Высокоэластичные волокна. Разработаны методы производства
на основе полиуретанов новых видов синтетических волокон,
получивших благодаря высокой эластичности название
«высокоэластичных» (спандекс-волокна) [1]. Производство этих волокон
начато в 1966 г. Мировое производство их в 1973 г. превысило
15 тыс. т.
Высокоэластичные волокна обладают рядом специфических
ценных свойств. Например, при прочности 6—8 гс/текс F0—
80 мН/текс) удлинение таких волокон достигает 600—800%.
Таким удлинением обладают только каучукоподобные материалы.
Высокоэластичные волокна позволяют расширить ассортимент
изделий народного потребления, вырабатываемых из химических
волокон.
Для производства высокоэластнчных волокон используются
в основном высокоэластичные полиуретаны, синтезированные по
методу, предложенному Байером, для получения каучукоподобного
172 Глава 4. Производство полиуретановых волокон
полимера — вулколана [2]. Технологический процесс производства
этого волокна состоит из нескольких стадий. Основными стадиями
являются следующие:
синтез волокнообразующего полимера;
получение прядильного раствора и подготовка его к
формованию;
формование волокна и регенерация растворителя;
отделка волокна (при формовании мокрым способом).
Для придания высокой эластичности полимеру в
макромолекулы его необходимо ввести гибкие блоки, не содержащие
полярных групп. В качестве таких блоков используются сравнительно
низкомолекулярные простые эфиры (например, полиэтиленоксид
или сополимер этиленгликоля с пропиленгликолем) или сложные
алифатические полиэфиры (например, продукт поликонденсации
этиленгликоля и адипиновой кислоты). Молекулярный вес этих
полиэфиров 2000—3000. На концах молекул содержатся реакцион-
носпособные группы ОН.
Низкомолекулярные полиэфиры реагируют с диизоцианатами,
которые добавляют в количестве, превышающем 1 моль на 1 моль
полиэфира. В результате получаются так называемые макроди-
изоцианаты, содержащие на конце молекулы изоцианатные группы
_N=C = O:
2HOR'OH + 3OCNRNCO —у
—¦* OCNHRNOCOR'OCONHRNHOCOR'OCONHRNCO
где HOR'OH — молекула полиэфира.
При указанном выше соотношении компонентов получается
диизоцианат с молекулярным весом около 5000.
Благодаря наличию на конце макромолекулы высокореакцион-
носпособных изоцианатных групп создается возможность
получения высокомолекулярных полимеров при последующем
взаимодействии макродиизоцианатов с соединениями, содержащими
подвижный атом водорода, например с диаминами. Эта реакция протекает
очень быстро при комнатной или несколько повышенной
температуре по схеме
—> CONHRNHCONH NH
где OCN • • • -NCO — молекула макродиизоцианата.
При эквимолярном соотношении макродиизоцианата и диамина
получается высокомолекулярный полимер (в зависимости от
условий проведения реакций), в котором полиуретановые блоки
соединены между собой теми же группами, что и в молекуле
полимочевины /—NH—С—NH—>
h
Если при этой реакции применяется избыток
макродиизоцианата, то образуется сшитый полиме , так как избыточное количе-
Волокно вирен 173
ство (по сравнению с эквимолярным) молекул макродиизоцианата
реагирует с водородом, входящим в группу NH.
Следовательно, гетероцепной полимер, используемый для
получения высокоэластичных волокон, представляет собой
блок-сополимер, макромолекулы которого не содержат полярных групп.
Изменяя характер исходных мономеров, используемых при
синтезе полиэфиров, состав диизоцианатов и диаминов, а также
соотношение отдельных компонентов, можно значительно изменять
свойства полимеров и получаемых из них волокон.
Высокоэластичные волокна (лайкра, вирен) в сравнительно
небольших масштабах вырабатываются в различных странах.
Некоторые данные о составе и условиях получения этих волокон
приводятся ниже.
Полиуретановые волокна можно формовать из олигомеров
(макродиизоцианатов) или из растворов полимеров в апротонных
растворителях (диметилформамиде, диметилсульфоксиде, диметил-
ацетамиде). Путем формования волокна из олигомеров получают
в США волокно вирен.
Волокно вирен. Исходный полиэфир — продукт
поликонденсации этиленгликоля и небольшого количества пропиленгликоля-1,3
с адипиновой кислотой. Молекулярный вес полиэфира 2000. В
качестве диизоцианата используется дифенилдиизоцианат.
Получаемый макродиизоцианат представляет собой вязкую жидкость,
которая непосредственно используется для получения волокна. Эта
жидкость выдавливается через отверстия фильеры в осадительную
ванну — водный раствор диамина. Так как диамин реагирует
с изоцианатными группами по приведенной выше схеме очень
быстро, то на поверхности формующихся волокон образуется
прочная оболочка; образующаяся нить принимается на бобину.
Свежесформованная нить обрабатывается кислотами для
нейтрализации остатков непрореагировавшего диамина. Обычно для
такой обработки применяется 1 —10%-ный раствор органической
кислоты. Для завершения процесса формования и отверждения
полужидкой внутренней части волокна нить обрабатывается водой
под давлением. При этом также образуется полимочевина и
отщепляется СОг. Реакция протекает по схеме
OCN—R—NCO + Н2О + OCN—R—NCO —>
—> OCNH—R—NHCONH—R—NH \- СО2
Следовательно, при производстве волокна вирен используется
новый принцип получения волокна, при котором синтезвысоко-
молекулярного полимера из низкомолекулярных
олигомеров происходит в процессе формования.
Существенным недостатком этого способа является
значительное ухудшение условий труда, определяемое наличием в
прядильной ванне низкомолекулярных алифатических диаминов,
обладающих высоким давлением паров.
174 Глава 4. Производство полиуретановьсх волокон
Полиуретановое волокно можно получить также из растворов
полимера. При растворении полимера в раствор добавляют
небольшое количество матирующего вещества (ТіОг), антиоксиданта
и стабилизатора, препятствующего пожелтению волокна при его
эксплуатации. По этому способу предварительно получают
высокомолекулярный полиуретан путем взаимодействия макродиизоциа-
ната с диамином (обычно для этой цели применяют этилендиамин).
Пол'ученный полимер растворяют в одном из указанных выше
апротонных растворителей и из полученного прядильного раствора,
содержащего 15—30% полимера, формуют волокно сухим или
мокрым способом.
Формование полиуретановой нити сухим способом по
сравнению с мокрым способом имеет ряд преимуществ. Затруднением
при формовании этих волокон сухим способом является
необходимость создания высоких температур в шахте A50—170 °С) для
быстрого испарения высококипящего апротонного растворителя при
одновременном увеличении высоты шахты до 8—9 м. Скорость
формования полиуретановой нити сухим способом составляет 500—
700 м/мин. Методом сухого формования получают полиуретановые
нити в. США (волокно лайкра), в ФРГ и в Японии.
-Полиуретановые волокна производятся в США, Японии и
Бельгии также и мокрым способом формования в ванне, содержащей
водный раствор растворителя. Содержание воды в осадительной
ванне, определяющее ее коагулирующую способность, зависит от
олщины элементарного волокна, скорости формования и
температуры ванны.
Обычно формование мокрым способом осуществляется при
скорости 40—80 м/мин и температуре ванны 90—95 °С. Сформованное
волокно промывается, сушится и принимается на паковку.
Исследуется возможность формования полиуретанового волокна
из расплава. Разработка этого перспективного и экономичного
метода получения высокоэластичного волокна находится пока в
начальной стадии.
Высокоэластичные полиуретановые нити имеют ряд
существенных преимуществ Чіеред резиновыми нитями. Основными
преимуществами являются следующие [3]:
более высокая прочность (в 2—3 раза);
более высокая эластичность (в 2—3 раза);
пониженная плотность A,2 г/см3 вместо 1,4 г/см3);
более высокая устойчивость к истиранию;
значительно более высокая устойчивость к многократным
деформациям (в 10—20 раз).
Существенным недостатком полиуретановых волокон является
их сравнительно невысокая термостойкость: при нагревании до
150 °С они желтеют, так как начинается термическая деструкция.
Разработка методов термостабилизации этих волокон имеет боль-
Литература 175
шое значение для расширения областей применения изделий из
них и повышения их эксплуатационной ценности.
Высокоэластичные волокна в чистом виде или в смеси с
другими волокнами используют для изготовления спортивной одежды,
корсажных, хирургических, ортопедических и других изделий.
Содержание полиуретанового волокна в изделиях, получаемых из
смеси волокон, составляет 10—15%. Как правило, полиуретановую
нить используют в качестве стержня, вокруг которого навивают
нити из других волокон. Из таких комбинированных нитей
получают несминаемые эластичные материалы.
ЛИТЕРАТУРА
1. Rince H., «Chimia», 1962, Bd. 16, № 4, S. 93—98; Angew. Chem., 1962,
Bd. 16, S. 621—626.
2. Bayer P., Angew. Chem., 1946, Bd. 59, S. 257—268.
3. Кошевой Л. Г., Галиулина Ф. К-, Айзенштейн Э. М., Хим.
волокна, 1967, № 2, с. 6—10.
Глава 5
ПОЛИФОРМАЛЬДЕГИДНОЕ ВОЛОКНО
Доступность исходного сырья и высокие механические свойства
полиформальдегида, а также возможность формования волокна
из расплава определили целесообразность использования
полиформальдегида для производства синтетических волокон. Такие
работы были проведены преимущественно в Советском Союзе на
кафедре химических волокон Киевского института легкой
промышленности под руководством А. В. Юдина, а также в Германской
Демократической Республике и в других странах.
Полиформальдегид, применяемый в производстве пластических
масс, может быть использован и для формования волокон. Его
получают методом катионной или анионной полимеризации *. Для
производства волокна может быть использован гомополимер
формальдегида или его сополимер с небольшим количеством второго
мономера. Исходным сырьем для синтеза полимера является
формальдегид или его тример — триоксиметилен (триоксан)
СНО
хСН2—Оч
О JCH2. Температура плавления триоксана 62 °С, тем-
^СН2—<У
пература кипения 115 °С. По сравнению с формальдегидом
триоксан имеет следующие преимущества [1]: он более хемостоек, что
позволяет проще осуществить очистку полимера, и его легче
транспортировать.
Гомополиформальдегид СН2О—СНг—О—СН2—О
обладает низкой термостабильностью и начинает интенсивно
разлагаться при 120°С. Естественно, что использовать такой полимер
для производства волокна нельзя. Начальную температуру
разложения полиформальдегида можно резко повысить (до 250 °С)
блокированием концевых групп ОН, в частности ацетилированием
или путем синтеза сополимера формальдегида, содержащего
небольшое количество второго компонента — окиси этилена или ди-
оксалана СН2—СН2, который располагается преимущественно на
О О
\/
СН9
* Детальней о синтезе полиформальдегида — см. Е п и к о л о п я н Н. С,
В о л ь ф с о п С. А. Химия и технология полиформальдегида. М., «Химия», 1968.
279 с
Полиформальдегидное волокно 177
концах макромолекул. Этот метод получения термостабильного
полимера получил практическое применение. Молекулярный вес
полиформальдегида, используемого для получения волокна,
40 000—50 000.
Технологический процесс получения полиформальдегидного
волокна включает три основные стадии: формование, вытягивание и
термообработку.
-Формование волокна. Полиформальдегидное волокно формуют
из расплава стабилизированного полимера при 185—220 °С
(температура плавления полимера ~160°С). Вязкость расплава при
этих температурах значительно выше вязкости расплавов
полиамидов и приближается к вязкости расплавленных полиэфиров.
Поэтому подача расплава к прядильному насосику осуществляется
так же, как при формовании полиэфирного волокна, — при
помощи шнека. Скорость формования полиформальдегидного волокна
из расплава, так же как и при получении других гетероцепных
волокон, достигает 800—1000 м/мин, фильерная вытяжка—1000—
1500% [2].
Вытягивание волокна. Полиформальдегидное волокно
вытягивают при нормальной температуре в одну или в две стадии на
200—400%, а затем дополнительно на 500—800% при повышенной
температуре A40—150°С). Это волокно легко кристаллизуется,
поэтому при длительном выдерживании оно теряет способность к
вытягиванию на холоду. Однако при температурах, близких к
температуре плавления полиформальдегида, максимально возможная
степень вытягивания волокна не уменьшается.
Термообработка. Вытянутое волокно подвергается
термообработке при 130—150 °С в свободном состоянии или под натяжением.
Свойства. Вытянутое и термообработанное
полиформальдегидное волокно характеризуется следующими показателями.
Прочность волокна в зависимости от степени его вытягивания
составляет 50—100 гс/текс, удлинение — 8—16%- Следовательно,
полиформальдегидное волокно, полученное вытягиванием свеже-
сформованного волокна на 800—1000%, является одним из
наиболее прочных среди обычных типов синтетических волокон.
Волокно обладает высокой гидрофобностью; при относительной
влажности воздуха 65% оно сорбирует всего 0,2% влаги. Поэтому
в мокром состоянии прочность и удлинение волокна заметно не
изменяются.
Плотность волокна A,38—1,42 г/см3) превышает плотность
большинства других синтетических волокон.
Начальный модуль волокна в 1,5—2 раза выше модуля
полиамидного волокна. Эластические свойства полиформальдегидного
волокна невысоки.
Светостойкость волокна — высокая; она приближается к
светостойкости полиакрилонитрильного волокна [3].
178 Глава 5. Полиформальдегидное волокно
Теплостойкость волокна превышает теплостойкость
полипропиленового, но заметно уступает по этому показателю полиамидному
волокну [4].
Термостойкость полиформальдегидного волокна также
невысока. При нагревании при температурах выше 125 °С прочность
волокна резко снижается в результате термоокислительиой
деструкции. Интенсивность этого процесса, по-видимому, может быть
значительно уменьшена при добавлении термостабилизаторов.
Волокно стойко к органическим растворителям, но
недостаточно стойко к минеральным кислотам и щелочам, под действием
которых постепенно разрываются ацетальные связи между
элементарными звеньями макромолекул.
Области применения полиформальдегидных волокон; пока не
определены. Это волокно не обладает какими-либо
специфическими ценными свойствами. Пониженная температура плавления
волокна и соответственно более узкий температурный интервал, в
котором могут быть использованы изделия из него, а также
повышенная плотность и низкая гигроскопичность ограничивают
области использования полиформальдегидного волокна (по сравнению с
другими синтетическими гетероцепными волокнами). По-видимому,
в некоторых случаях полиформальдегидное волокно, учитывая его
высокую прочность, может заменить полиамидное и
полипропиленовое волокна. Целесообразность этого будет в основном
определяться экономикой — стоимостью, полимера и получаемого из него
волокна. Дальнейшие исследования в этом направлении, а также
проведение технико-экономических исследований, и особенно
сравнение свойств разных типов гетероцепных синтетических
волокон и поведения их в эксплуатации, должны внести ясность в
вопрос о масштабах производства и ассортименте изделий, для
изготовления которых целесообразно использовать полиформальдегид-
ные волокна.
ЛИТЕРАТУРА
1. Егоров Б. А., Грж им а л овский Л. С, Юдин А. В., Хим. волокна,
1969, № 4, с. 35—38.
2. Гойхман А. Ш., Носов М. П., Бычков М. И. и др., Хим. волокна,
1968, № 4, с. 69—72.
3. Егоров Б. А., Юдин А. В., Герасимова Л. С. и др., Хим. волокна,
1969, № 1, с. 55—57.
4. Dietrich К., R ei ni sch G., Versaume r H. е, a., Faserforsch, u. Textil-
techn., 1971, Bd. 22. S. 95—98,
Глава 6
ПРОИЗВОДСТВО ПОЛИАКРИЛОНИТРИЛЬНЫХ
волокон
Среди карбоцепных волокон наиболее широкое промышленное
применение нашли синтетические волокна из полимеров и
сополимеров нитрила акриловой кислоты (акрилонитрила).
Нитрил акриловой кислоты уже в течение сравнительно
длительного времени вырабатывается в промышленном масштабе для
получения синтетического бутадиен-нитрильного каучука (продукт
совместной полимеризации бутадиена и акрилонитрила).
Использование полиакрилонитрила для производства волокна
затруднялось из-за неплавкости и невозможности получения
концентрированных прядильных растворов этого полимера. Так же
как и большинство других карбоцепных полимеров, полиакрило-
нитрил не плавится без разложения. Поэтому нить, состоящую из
большого числа волокон, можно получать пока только
формованием из растворов полимера. Из-за нерастворимости
полиакрилонитрила в обычных растворителях длительное время существовало
мнение о том, что этот полимер обладает сетчатой .структурой и
использование его для производства волокна вообще невозможно.
Это предположение оказалось ошибочным.
В 1932 г. Рейн [1] установил, что полиакрилонитрил растворим в
концентрированных растворах некоторых неорганических- солей (ZnCU, роданиды Na и
Са) и в четвертичных аммониевых основаниях. Однако эти растворители в то
время ие получили практического применения.
В 1942 г. Рейн в Германии и почти одновременно Хаутц в США [2]
предложили новый органический растворитель для получения прядильных
концентрированных растворов полиакрилонитрила. Таким растворителем, который ранее не
вырабатывался в значительных количествах, оказался диметилформамид,
получивший в настоящее время широкое применение в производстве полиакрило-
нитрильного волокна.
Образцы полиакрилонитрильного волокна были получены в
1943 г. Потребовалось около 5 лет для того, чтобы в 1948 г. было
организовано опытное производство этого волокна. С 1950 г.
начинается промышленное производство полиакрилонитрильного
волокна в США и почти одновременно — в ГДР и в ФРГ.
В настоящее время полиакрилонитрильные волокна
вырабатываются во многих странах. Волокна из полиакрилонитрила или в
большинстве случаев из сополимеров акрилонитрила выпускаются
под фирменными названиями: «волкрилон», «предана» (ГДР),
«орлон», «акрилан», «креслан», «зефран» (США), ' «дралон»,
180 Глава 6. Производство полиакралонитрильных волокон
«редон» (ФРГ), «нитрон» (СССР), «куртел» (Англия), «крилор»
(Франция) )и т. д.
Динамика мирового производства полиакрилонитрильных
волокон * (в тыс. т) характеризуется следующими цифрами [3]:
Годы
1962
1964
1966
1968
1970
1972
Текстильная
нить
2,3
2,3
3,6
4,6
5,2
Штапельное
волокно
297,5
454,3
727,9
997,4
1264
Всего
45
299,8
456,6
731,5
1002,0
1269,2
Как видно из этих данных, производство
полиакрилонитрильных волокон за последние 8 лет увеличилось более чем в 4 раза
и в 1972 г. составило свыше 20% общей выработки всех
синтетических волокон. Выпуск полиакрилонитрильных волокон растет
быстрее, чем выпуск других карбоцепных волокон.
В отличие от других синтетических волокон полиакрилонитриль-
ные волокна вырабатываются в основном в Виде штапельного
волокна. Объем производства нитей, используемых преимущественно
для специальных целей, в частности для получения жаростойких
волокон (см. разд. 11.2.2), не превышает 0,5% общего производства
полиакрилонитрильных волокон.
В Советском Союзе волокно из полиакрилонитрила (точнее, из
сополимеров акрилонитрила) выпускается под названием «нитрон»
с 1963 г. В девятой пятилетке производство полиакрилонитрильного
волокна резко увеличится и в 1975 г. будет в более чем 9 раз
превышать объем производства этого волокна в 1970 г.
6.1. ПОЛУЧЕНИЕ АКРИЛОНИТРИЛА
Синтез нитрила акриловой кислоты, являющегося исходным
мономером для получения полиакрилонитрила, может быть
осуществлен различными методами. Наибольший практический
интерес представляют следующие методы получения акрилонитрила:
из окиси этилена, ацетальдегида, ацетилена и особенно из
пропилена.
Акрилонитрил из окиси этилена. Синтез акрилонитрила из
окиси этилена протекает в две стадии.
На первой стадии при взаимодействии окиси этилена с
синильной кислотой получается зтиленциангидрин:
н2о
HjC CH2 + HCN *- НО—СН2—CHj—CN
ч у * ' катализатор
О
* В мировой статистике к полиакрилонитрильным волокнам отнесены также
и волокна из сополимеров акрилошприла, содержащие небольшое количество
(Ь—15%) второго мономера.
б.!. Получение акрилонитрила 181
На второй стадии в результате дегидратации этиленциангид-
рина при 180—200 °С в присутствии окислов металла (А12О3) или
солей щелочных металлов образуется акрилонитрил:
НО—СН2—СН2—CN »- СН2=СН
—Н2О і
CN
Однако этот метод является наименее экономичным. На 1 т
акрилонитрила расходуется 1,05—1,4 т окиси этилена и 0,62 т
синильной кислоты [4].
Акрилонитрил из ацетальдегида. По этому методу вначале
получают а-оксипропионитрил (нитрил молочной кислоты)
взаимодействием (при 50—60 °С) уксусного альдегида (ацетальдегида) и
синильной кислоты:
н А,
Затем сс-оксипропионнитрил дегидратируют при 650°С в
присутствии катализатора (85%-ной Н3РО4):
Этот метод может представить определенный интерес при
получении уксусного альдегида прямым окислением этилена. На 1 т
акрилонитрила расходуется 0,93 т ацетальдегида и 0,56 т
синильной кислоты [4].
Акрилонитрил из ацетилена. Синтез акрилонитрила из
ацетилена, получивший широкое промышленное применение, основан на
непосредственном присоединении HCN к С2Н2:
^CH + HCN —> СН2=СН
CN
Эта реакция осуществляется в жидкой фазе в присутствии
катализатора (СиС12 и хлориды Na, К или NH4 в соотношении 1 :0,3)
или в газовой фазе при 450—700 °С под давлением 1—4 кгс/см2
A • 105 Па—4 • 105 Па) в присутствии окисных катализаторов
(например, ZnO).
Выход акрилонитрила по отношению к израсходованному
ацетилену и синильной кислоте составляет 80 и 85%. На 1 т
акрилонитрила расходуется 0,64 т ацетилена и 0,58 т HCN. При наличии
дешевого ацетилена, в частности при синтезе его из природного
газа, этот метод получения акрилонитрила может найти широкое
применение в промышленности.
182 Глава 6. Производство полиакрилонитрильных волокон
Акрилонитрил из пропилена. Получение акрилонитрила из
пропилена основано на окислении пропилена и аммиака кислородом
воздуха (так называемый процесс окислительного аммонолиза):
СН2=СН
Реакция осуществляется при 450—475 °С под давлением
3 • 105 Па при стехиометрическом соотношении компонентов.
Смесь пропилена, аммиака, воздуха и водяных паров
пропускается через кипящий слой катализатора — смесь окислов
металлов переменной валентности, нанесенных на пористый носитель
(обычно силикагель).
В качестве исходных продуктов для изготовления
.катализатора используются соли.висмуту (молибдат или вольфрамат
висмута), олова, рутения и других металлов.
Этот метод синтеза акрилонитрила, реализованный на ряде
предприятий в Советском Союзе и за рубежом, является в
настоящее время наиболее перспективным и экономичным. Например,
в Японии, начиная с 1967 г., акрилонитрил производится только из
пропилена и аммиака. Стоимость этого мономера соответственно
снизилась в 2 раза [5].
Основные преимущества данного способа заключаются в том,
что синильная кислота и окись этилена или ацетальдегид
заменяются более доступными пропиленом и аммиаком. На 1 т
акрилонитрила расходуется 0,9 т пропилена (в пересчете на 100%-ный),
0,4 т аммиака и 3500 м3 воздуха [4].
Стоимость акрилонитрила, полученного из пропилена, и
соответственно полиакрилонитрила значительно снижается и тем самым
повышается конкурентоспособность полиакрилонитрильного
волокна с.другими синтетическими волокнами.
6.2. СИНТЕЗ ПОЛИМЕРОВ И СОПОЛИМЕРОВ
АКРИЛОНИТРИЛА
Основным волокнообразующим полимером, используемым для
получения полиакрилонитрильных волокон, является не полиакри-
лонитрил, а его сополимеры, содержащие небольшие количества
E—10%) второго мономера или двух других мономеров. В
промышленности волокно из чистого полиакрилонитрила почти не
вырабатывают ввиду его низкой эластичности, плохой окрашивае-
мости и недостаточной устойчивости к истиранию. В качестве
винильных мономеров, используемых в виде добавок при синтезе
волокнообразующих сополимеров, в многочисленных патентах и
статьях предложено большое количество различных соединений
(см. разд. 6.5.1), которые в большинстве случаев не используются
по технологическим или экономическим соображениям.
6.2. Синтез полимеров и сополимеров акрилонитрила 183
Наибольшее практическое применение для синтеза сополимеров
получили мономеры, образующие с акрилонитрилом сополимеры
нерегулярного строения. Нарушение регулярности повышает
гибкость цепи сополимера, вследствие чего увеличивается эластичность
получаемых волокон. Из таких мономеров, добавляемых в
количестве 6—12% от массы акрилонитрила, наиболее широко
используются метилакрилат, метилметакрилат и винилацетат *.
Целесообразность использования того или иного мономера
определяется условиями проведения процесса полимеризации и
требованиями к свойствам волокон. Например, при сополимеризации
мономеров в растворе роданистых солей целесообразно
использовать метилакрилат (94% акрилонитрила и 6% метилакрилата),
скорость полимеризации которого такая же, .как и у
акрилонитрила, что облегчает проведение процесса демономеризации
полимера. С экономической точки зрений рекомендуют применять
винилацетат, стоимость которого ниже стоимости других
мономеров, используемых в качестве второго компонента при
полимеризации. Добавление второго компонента, как правило, снижает
скорость полимеризации акрилонитрила, причем снижение скорости
пропорционально количеству добавляемого второго мономера.
Введение в реакционную смесь винилацетата обусловливает снижение
скорости полимеризации в большей степени, чем добавление экви-
молярных количеств метилакрилата.
Для синтеза сополимеров акрилонитрила применяются также
мономеры, содержащие реакционноспособные группы,
преимущественно кислотные, введение которых в макромолекулу сополимера
улучшает окрашиваемость волокна и повышает его гидрофиль-
ность. В качестве таких мономеров (добавляемых в количестве
0,5—2% от массы акрилонитрила) обычно используются итаконо-
вая и акриловая кислоты, а в последнее время и различные суль-
фонаты (аллилсульфонат, металлилсульфонат, сульфированный
стирол). Наиболее высокой константой скорости полимеризации
характеризуется металлилсульфонат.
Широкое применение получили также тройные сополимеры
акрилонитрила, содержащие наряду с акрилонитрилом 8—10% (от
массы акрилонитрила) других мономеров. При получении волокон,
обладающих повышенной усадочностью, количество этих
мономеров в сополимере повышается.
При синтезе волокнообразующего полимера или сополимера
акрилонитрила пока используется только метод радикальной цепной
полимеризации.
Принципиально полимеризацию акрилонитрила можно осуществить и по
реакции ионной полимеризации, например в присутствии ионов лития, которые
с диметилформамидом образуют комплексные соединения. Ионная полимериза-
* Более детально о составе модифицированных полиакрилонитрилышх
волокон и об их свойствах см, ниже, оазд. 6.5.1.
184 Глава 6. Производство по.шакрилонитрильных волокон
ция акрилонитрнла протекает значительно быстрей, чем реакция радикальной
полимеризации. Продолжительность процесса не превышает 40—60 с, причем
за это время достигается 100%-ная конверсия мономера. Однако, несмотря на
указанные преимущества, метод ионной полимеризации акрилонитрила не
получил практического применения по следующим причинам:
при проведении этой реакции в растворе требуется растворитель очень
высокой степени чистоты (например, 99,99%-ный диметилформамид);
в реакционной среде не должно быть даже следов воды;
катализатор для реакции ионной полимеризации значительно дороже
инициатора для реакции радикальной полимеризации.
Полимеризация акрилонитрила методом радикальной
полимеризации может быть осуществлена в суспензии или в растворе
с образованием концентрированного раствора полимера,
используемого непосредственно для формования волокна. Оба способа
получения полиакрил.онитрила широко применяются на
практике.
Каждый из указанных способов имеет свои преимущества и
недостатки. Основным и очень существенным преимуществом метода
полимеризации в растворе является значительное упрощение
технологии получения полимера и формования из него волокна, а
также возможность осуществления непрерывного процесса синтеза
полимера и формования из него волокна.
Отпадает ряд операций как при получении полимера
(высаживание полимера из суспензии, промывка его и сушка), так и при
использовании его для производства волокна (растворение
полимера при получении прядильного раствора).
Однако метод . суспензионной полимеризации акрилонитрила
имеет пока некоторые специфические преимущества,
определяющие целесообразность использования его в ряде случаев. К таким
преимуществам относятся:
возможность регулирования состава получаемого сополимера
в более широких пределах, чем при синтезе сополимера
акрилонитрила в растворе, что имеет особое значение при производстве
так называемых усадочных волокон, применяемых для выработки
различных изделий народного потребления;
более простая регенерация осадительной ванны, так как
различные примеси и побочные продукты, образующиеся в процессе
полимеризации, остаются в водном растворе, из которого
высаживается полимер;
высушенный полимер не содержит акрилонитрила, поэтому
в процессе формования волокна нет вредных выделений, как это
имеет место при формовании волокна из прядильного раствора,
полученного в процессе полимеризации акрилонитрила в растворе.
Если указанные недостатки метода полимеризации
акрилонитрила в растворе удастся устранить, он получит еще более широкое
применение.
6.2. Синтез полимеров и сополимеров акрилонитрила 185
6.2.1. Суспензионная полимеризация
Этот способ является в настоящее время основным при синтезе
различных карбоцепных полимеров. Характерным отличием
акрилонитрила от большинства других мономеров, используемых для
синтеза волокнообразующих карбоцепных полимеров, является
растворимость, хотя и ограниченная, в воде. При 20 °С в воде
растворяется 7% акрилонитрила. Поэтому при суспензионной
полимеризации процесс начинается в водном растворе акрилонитрила,
образующиеся олигомеры выпадают из водного раствора и
дальнейшая полимеризация происходит уже в гетерогенной среде.
При полимеризации в суспензии используют водорастворимые
инициаторы или соответствующие
окислительно-восстановительные системы, в частности персульфат аммония и метабисульфит
натрия.
Суспензионная полимеризация и сополимеризация
акрилонитрила проводится в следующих условиях: продолжительность
процесса 20—120 мин, температура 30—50 °С, содержание
акрилонитрила в смеси 10—20%. Количество инициатора изменяется в
зависимости от заданного молекулярного веса полимера. Конверсия
мономера составляет 80—85%.
6.2.2. Полимеризация в растворе
Этот способ полимеризации нашел пока практическое
применение только при синтезе волокнообразующего полимера или
сополимера акрилонитрила. Сущность его заключается в том, что
полимеризация акрилонитрила проводится в растворителе, в котором
растворяется мономер и образующийся полимер. Полученный
концентрированный раствор полимера используется непосредственно
для формования волокна.
Характерной особенностью процесса полимеризации
акрилонитрила в растворе является сравнительно низкая степень конверсии
мономера, не превышающая 50—70%. Не вступивший в реакцию
мономер отгоняется под вакуумом из образовавшегося
концентрированного раствора полимера (процесс демономеризации) и снова
используется для полимеризации. Дальнейшее повышение
конверсии мономера не рекомендуется во избежание образования
разветвленного полимера и получения продукта, обладающего
повышенной полидисперсностью. Надежных экспериментальных данных
о влиянии степени конверсии мономера на строение получаемого
полимера и свойства волокна пока нет.
Демономеризация осуществляется в тонком слое при
небольшом вакууме (остаточное давление 40—70 мм рт. ст.) при 50—60 °С.
Полностью удалить мономер из получаемого раствора в
производственных условиях не удается, поэтому прядильный раствор,
поступающий на прядильную машину, содержит 0,2—0,3% мономера.
186 Глава 6. Производство полиакрилониїрильних волокон
Из большого числа веществ, в которых растворяется поли-
акрилонитрил, практическое применение нашли органические
растворители— диметилформамид, диметилацетамид, диметилсульфок-
сид и этиленкарбонат, а также концентрированные водные растворы
некоторых неорганических солей — роданида натрия или хлорида
цинка.
Особое место среди растворителей
полиакрилонитрила^используемых для получения прядильных растворов, занимает азотная
кислота.
Ниже схематически излагаются условия синтеза
полиакрилонитрила или его сополимеров в растворах в различных
растворителях и преимущества и недостатки каждого из этих методов.
Полимеризация акрилонитрила в диметилформамиде.
Диметилформамид является одним из наиболее доступных и широко
используемых растворителей полимера и сополимеров
акрилонитрила, содержащих небольшое количество второго мономера. Этот
растворитель используется также и при приготовлении прядильных
растворов из гомополимера и сополимеров акрилонитрила,
полученных методом суспензионной полимеризации.
Диметилф.ормамид получается из метанола, окиси углерода и
аммиака. При взаимодействии метанола с аммиаком при 420—
450^С под давлением 50 кгс/см2 E • 106 Па) в присутствии окиси
алюминия и каолина в качестве одного из основных продуктов
реакции образуется диметиламин:
2CH3OH + NH3 —> NH(CH3J + 2Н2О
В результате реакции между метанолом и окисью углерода при
повышенной температуре в присутствии катализатора (алкоголята
метилового спирта) получается метиловый эфир муравьиной
кислоты:
СНзОН + СО —> НСООСНз
При взаимодействии диметиламина и метилового эфира
муравьиной кислоты образуется диметилформамид:
NH(CH3J + НСООСНз —> HCON(CH3J + СН3ОН
Вместо метилового эфира муравьиной кислоты для синтеза
диметилформамида можно использовать и концентрированную
(84%-ную) муравьиную кислоту:
НСООН + NH(CH3J —> HCON(CH3J + Н2О
Преимуществом полимеризации акрилонитрила в растворе
диметилформамида является возможность получения эквиконцентри-
рованных растворов полимера значительно более низкой вязкости,
чем при использовании других растворителей. Поэтому при
переработке эквивязких растворов полимера в диметилформамиде
можно применять значительно более концентрированные растворы,
чем п и использовании прядильных растворов полиакрилонитрила
6.2. Синтез полимеров и сополимеров акрилонитрим 187
или сополимеров акрилонитрила в других растворителях.
Например, при формовании мокрым способом концентрация полимера
в растворе диметилформамида составляет 20—25% (вместо 11 —
13% для эквивязких растворов в растворах солей), вязкость
раствора 400—500 П D0—50 Па • с) при 25 °С. В результате
повышения температуры прядильного раствора до 40 °С при подаче его на
прядильную машину вязкость раствора снижается до 100—150 П
A0—15 Па -с). При формовании волокна сухим способом
концентрацию полимера в растворе повышают до 30—32%.
Дополнительным преимуществом применения
диметилформамида является более простая регенерация его из осадительной ванны
(см. разд. 6.4.3).
Однако использование диметилформамида в качестве
растворителя имеет ряд существенных недостатков, основным из которых
является токсичность. Допустимая концентрация паров
диметилформамида в воздухе не должна превышать 0,03 мг/м* а в
водостоках 10 г/м3. При ректификации диметилформамида при
температуре выше 100 °С он частично разлагается с выделением
муравьиной кислоты (вызывает повышенную коррозию
ректификационных колонн) и диметиламина (токсичный продукт).
Основным недостатком диметилформамида при использовании
его в качестве среды при полимеризации является малая скорость
полимеризации акрилонитрила. Как правило, продолжительность
полимеризации или сополимеризации акрилонитрила в диметил-
формамиде при конверсии мономера 70% составляет 16—24 ч, что
в 8—10 раз превышает продолжительность полимеризации в
большинстве других растворителей. Такая малая скорость
полимеризации объясняется, по-видимому, наличием в диметилформамиде
примесей аминов, которые обрывают растущую цепь и таким образом
замедляют или даже ингибируют процесс радикальной
полимеризации. Учитывая указанные недостатки, полимеризацию
акрилонитрила в среде диметилформамида применяют сравнительно
редко. При получении прядильного раствора из полимера,
синтезированного методом суспензионной полимеризации, этот
растворитель используется достаточно широко.
Полимеризация акрилонитрила в диметилацетамиде. Диметил-
ацетамид получается по такой же схеме, как и диметилформамид,
но вместо муравьиной кислоты применяется уксусная кислота:
(CH3JNH + СН3СООН —> CH3CON(CH3J + Н2О
Диметилацетамид по сравнению с диметилформамидом менее
токсичен и агрессивен, меньше разрушает ректификационные
колонны. Недостатком диметилацетамида является более высокая
вязкость эквиконцентрированных растворов, что обусловливает
необходимость понижения концентрации полимера в растворе на
1—2% и увеличения объема отработанной осадительной ванны,
поступающей на регенерацию. Кроме того, у диметилацетамида
188 Глава 6. Производство помшкрилонитрильных волокон
более высокая температура кипения, чем у диметилформамида
A65 вместо 151 °С). Диметилацетамид образует с водой азеотроп,
устойчивый до 100 °С. Поэтому для полного отделения воды от
диметилацетамида его подвергают повторной ректификации при
температурах выше 100 °С.
Данных о сравнительных скоростях полимеризации и сополиме-
ризации акрилонитрила в диметилформамиде и в диметилацетамиде
в литературе не имеется.
Полимеризация акрилонитрила в диметилсульфоксиде. Диме-
тилсульфоксид является одним из сравнительно доступных
растворителей полиакрилонитрила. Опыты по формованию полиакрило-
нитрильного волокна из растворов в диметилсульфоксиде были
проведены в СССР [6] и Японии. В последнее время этот
растворитель получил применение при производстве полиакрилонитриль-
ного штапельного волокна в Японии.
Диметилсульфоксид может быть получен окислением диметил-
сульфида, или, что значительно более эффективно,
взаимодействием метанола с сероводородом по схеме
2CH3OH + H2S —*¦ (CH3JSO + H2O
Преимуществом диметилсульфоксида при использовании его
в качестве растворителя при синтезе полимера в растворе является
высокая скорость полимеризации акрилонитрила.
Продолжительность процесса при конверсии мономера 70% и
температуре полимеризации 70 °С не превышает 1,5—2 ч.
Наличие кислорода в реакционной среде при. полимеризации
акрилонитрила в растворе диметилсульфоксида, особенно при
температурах ниже 60 °С, сильно замедляет и даже ингибирует
процесс. Поэтому полимеризацию акрилонитрила в среде
диметилсульфоксида проводят в атмосфере инертного газа. Используемый
в качестве растворителя диметилсульфоксид должен содержать
минимальное количество воды и диметилсульфида. При наличии в
диметилсульфоксиде 5% воды прядильный раствор желатинирует
уже на третий день [7].
Другое преимущество диметилсульфоксида заключается в том,
что он не вызывает коррозии аппаратуры при формовании волокна
и при регенер-ации отработанной осадительной ванны.
Обычно принято считать, что в отличие от диметилформамида
диметилсульфоксид не токсичен. Однако имеются данные о
частичном разложении диметилсульфоксида при формовании волокна.
При этом выделяются небольшие количества диметилсульфида,
обладающего неприятным запахом и повышенной токсичностью.
Недостатком диметилсульфоксида при использовании его в
качестве среды при полимеризации акрилонитрила является
повышенное количество акрилонитрила (до 3% от массы
диметилсульфоксида), поступающего вместе с растворителем в виде азеотропа
6.2. Синтез полимеров и сополимеров акрилонитрила 189
в прядильный раствор и затем в осадительную ванну. Это
количество акрилонитрила выделяется при формовании волокна и
регенерации осадительной ванны, что резко повышает вредность
работы в прядильном цехе. Необходима дополнительная тщательная
проверка этих данных, так как если они подтвердятся, то
использование диметилсульфоксида в качестве среды при полимеризации
или растворителя при получении прядильного раствора до
устранения этого серьезного недостатка будет сильно затруднено.
Дополнительным недостатком диметилсульфоксида является
более высокая вязкость эквиконцентрированных растворов полиакри-
лонитрила в этом растворителе, значительно превышающая
вязкость растворов в диметилформамиде, что обусловливает
необходимость снижения концентрации полимера в прядильном растворе.
Полимеризация акрилонитрила в этиленкарбонате. Этиленкар-
бонат получают при взаимодействии этиленгликоля с дихлорангид-
ридом угольной кислоты (фосгеном) по схеме
СН2ОН
] +СОС12 —> (СН2ОJСО + 2НС1
СН2ОН
В последнее время разработан метод получения
этиленкарбоната взаимодействием окиси этилена и окиси углерода, что будет
способствовать снижению стоимости этого растворителя.
Этиленкарбонат, являющийся хорошим растворителем для
полимеров и сополимеров акрилонитрила [8], применяется для
производства полиакрилонитрильного волокна в Румынии.
Основными преимуществами этиленкарбоната являются низкая
токсичность, отсутствие коррозии аппаратуры и высокая скорость
полимеризации A,5—2 ч при конверсии 60—70% акрилонитрила).
К недостаткам этиленкарбоната следует отнести высокую
температуру плавления C6°С), что обусловливает необходимость
обогрева всех аппаратов и трубопроводов, по которым поступает
этот растворитель, а также более высокая вязкость прядильных
растворов и сравнительно легкий гидролиз растворителя при
повышенных температурах, в частности при ректификации.
Полимеризация акрилонитрила в концентрированных растворах
роданистых солей. Полимеры и сополимеры акрилонитрила
растворяются в концентрированных водных растворах роданистых
солей, в частности в 51—52%-ном растворе роданида натрия.
В качестве инициатора при полимеризации и сополимеризации
акрилонитрила используют динитрилизомасляной.кислоты в
количестве 0,5—1% от массы мономера. Изменяя количество этого
реагента, можно регулировать молекулярный вес образующегося
полимера. В качестве регулятора используются также небольшие
добавки изопропилового спирта.
Предельная концентрация мономера или смеси мономеров
в реакционной среде ограничивается их растворимостью в
190 Глава 6. Производство полиакрилонитрияьных волокон
концентрированных водных растворах роданидов и составляет 20—¦
25% от массы раствора.
Полимеризация акрилонитрила в растворе роданида натрия и
последующее формование волокна из получаемых
концентрированных растворов полимера осуществлены по непрерывной схеме
в производственных условиях в СССР, Англии, Польше и
Югославии. Основными преимуществами этого метода полимеризации
и сополимеризации акрилонитрила в растворе являются высокая
скорость процесса A,5—2 ч при конверсии мономера 50—70%) и
низкая токсичность вследствие малой летучести. Однако, как уже
указывалось выше, в прядильном растворе после демономеризации
остается 0,2—0,3% от массы раствора акрилонитрила, который,
выделяясь на прядильной машине, обусловливает повышенную
вредность условий труда и необходимость капсуляции прядильной
машины. Кроме того, как показывает опыт работы, растворы по-
лиакрилонитрила в - роданистых солях вызывают заболевание
кожи — дерматит. Для однозначного выяснения вопроса о
вредности работы при использовании прядильных растворов в этом
растворителе необходимы специальные исследования.
Растворы роданистых солей при применении их в качестве
растворителей гомополимера и сополимеров акрилонитрила имеют ряд
существенных недостатков по сравнению с другими
растворителями. Основными недостатками являются:
необходимость изготовления всех аппаратов, трубопроводов и
выпарных установок нз специальных сталей, содержащих не менее
3% молибдена и титана, что повышает стоимость оборудования;
высокая вязкость эквиконцентрированных растворов,
значительно превышающая вязкость раствора полиакрилонитрила в
других растворителях, что вынуждает применять для формования
волокна прядильные растворы, содержащие всего 12—13% полимера;
сложность процесса регенерации отработанной осадительной
ванны (см. разд. 6.4.3).
Вследствие указанных недостатков водные растворы роданистых
солей не нашли практического применения в качестве растворителя
полимера, получаемого методом суспензионной полимеризации, и
используются только при полимеризации или сополимеризации
акрилонитрила в растворе.
Скорость полимеризации акрилонитрила в растворе роданистых
солей или других неорганических солей в значительной степени
зависит от содержания в растворе ионов трехвалентного железа,
которые действуют как ингибитор и агент передачи цепи, снижая
молекулярный вес получаемого полимера [9]. Поэтому применение
растворов роданистых солей, содержащих минимальное количество
железа, является предварительным условием высокой скорости
полимеризации и получения полимера заданного молекулярного веса.
Существенное влияние на скорость полимеризации оказывает
количество органических и минеральных примесей, образующихся
6.2. Синтез полимеров и сополимеров акрилонитрила 191
в качестве побочных продуктов при полимеризации. Эти примеси
переходят при формовании волокна в осадительную ванну и затем
остаются при регенерации в растворе роданида натрия, который
повторно используется в качестве растворителя при полимеризации
акрилонитрила. Состав этих примесей и влияние каждого из
компонентов на скорость полимеризации и свойства полимера
достаточно не изучены. На основании эмпирических данных принимают,
что содержание этих примесей не должно превышать 3% от массы
роданидов.
Полимеризация акрилонитрила в концентрированных растворах
хлорида цинка. Впервые данные об условиях полимеризации
акрилонитрила в концентрированных F0%-ных) водных растворах
хлорида цинка или смеси солей цинка и кальция C6,8% ZnCl2 и
19,4% СаС12) были опубликованы в 1955 г. Хуньяром и Гребе [10].
Раствор для полимеризации содержал 10—16% акрилонитрила и
84—90% водного раствора солей. Во избежание образования
разветвленного полимера они добавляли регулятор (эфиры тиоглико-
левой кислоты). В этих условиях был синтезирован полимер с
молекулярным весом 70000—120 000, который был использован для
формования волокна.
Водные растворы солей цинка и кальция при применении их
в качестве растворителя имеют те же преимущества и недостатки,
что и растворы роданида натрия. Дополнительным недостатком
применения этих солей является большая вредность ионов цинка в
водоемах и необходимость более тщательной очистки сточных вод.
Полимеризация акрилонитрила в азотной кислоте.
Концентрированная E5—68%-ная) азотная кислота используется в качестве
растворителя в Японии при производстве полиакрилонитрильного
волокна кашмилон.
Основным преимуществом азотной кислоты при использовании
ее в качестве растворителя полиакрилонитрила является низкая
стоимость и простота регенерации, к недостаткам азотной кислоты
следует отнести ее токсичность, из-за которой ухудшаются
условия труда, а также агрессивность, обусловливающую
необходимость изготовления аппаратуры из специальных сортов стали или
титана. Получаемые концентрированные растворы полимера или
сополимера акрилонитрила в азотной кислоте недостаточно
стабильны, и их можно хранить и перерабатывать при температуре
не выше 3—5 °С. При более высоких температурах начинается
постепенное окисление полимера, что приводит к получению
волокна, которое окрашивается неравномерно. Естественно, что
переработка прядильных растворов при пониженных температурах
связана с большим расходом холода. Кроме того, это приводит
к значительному повышению вязкости растворов и понижению
концентрации полимера в них.
Регенерация отработанной осадительной ванны при этом методе
получения полимера сводится к отгонке разбавленного водного
192
Глава 6. Производство полиакрилонитрильных волокон
раствора B0%-ного) азотной кислоты. Концентрирование такой
кислоты представляет большие трудности, поэтому целесообразно
использовать ее для производства азотных удобрений.
Комбинирование заводов по производству поли-
акрилонитрильного волокна с заводом
азотных удобрений является
характерной особенностью рассматриваемого
способа производства полиакрилони-
трильного волокна.
Суммируя приведенные данные,
можно сделать вывод, что пока нет
исчерпывающих данных, достаточных
для окончательной оценки технико-
экономической эффективности
применения отдельных растворителей для
проведения полимеризации и сополи-
меризации акрилонитрила в растворе
Как будет показано ниже, свойства
волокон, получаемых из растворов
полимеров и сополимеров акрилонитрила в
различных растворителях, примерно
одинаковы. Использование тех или
иных растворителей кроме патентных
соображений, которые играют
существенную роль при определении
направления технического прогресса в
капиталистических странах, зависит от
сырьевой базы, необходимой для
производства растворителя, стоимости
растворителя, возможности изготовления
аппаратуры из коррозионно-стойкой
стали, условий регенерации
растворителя, требований к чистоте сточных вод и ряда дополнительных
требований. Технико-экономические сравнения экономичности
использования различных типов растворителей с учетом приведенных
выше преимуществ и недостатков отдельных способов до
настоящего времени не проведены. Поэтому пока можно сделать только
следующие предварительные выводы. Если стоимостьдиметилформ-
амида принять за 100%, то перспективная стоимость других
растворителей составит [11, с. 265—270]:
Диметилсульфоксид 115
Роданид натрия E0%-ный раствор) ... 58
Этиленкарбопат 38
Азотная кислота F5%-ная) 5,2
Наиболее агрессивными растворителями являются водный рас-
тво роданида натрия и азотная кислота.
Продолжительность, ч
Рис. 6.1. Влияние характера
растворителя на скорость
полимеризации акрилонитрила
в растворе и на молекулярный
вес полученного полимера:
сплошная линия — выход (в %);
пунктирная — удельная
вязкость [8]:
/ — роданид натрия; 2 —диметилфор-
ыамид; 3 —этиленкарбонат;
4—диметилсульфоксид.
6.3. Свойства полиакрилониТри.Ш
193
Растворы полимеров или сополимеров акрилонитрила в диме-
тилформамиде, диметилацетамиде, диметилсульфоксиде, в водных
растворах роданистых солей можно перерабатывать при
нормальной температуре, в этиленкарбонате — при 40—50 °С, а в азотной
кислоте — при 3—5 °С.
Наименее токсичны диметилсульфоксид и этиленкарбонат,
наиболее токсична азотная кислота (пары).
Наивысшая скорость полимеризации или сополимеризации
акрилонитрила достигается в 51%-ном растворе роданида натрия.
Наиболее медленно эти процессы протекают в диметилформамиде
(рис. 6.1). При одних и тех же условиях полимеризации полимер
наиболее высокого молекулярного веса получается при проведении
реакции в растворе роданистых солей.
Минимальная вязкость эквиконцентрированных растворов
полимера характерна для диметилформамида, максимальная — для
68%-ной азотной кислоты. Соответственно максимальная
концентрация полимера в прядильном растворе, которая может быть
достигнута при переработке эквивязких растворов полимера, будет
в растворах в диметилформамиде, а минимальная — в азотной
кислоте.
6.3. СВОЙСТВА ПОЛИАКРИЛОНИТРИЛА
Оптимальный молекулярный вес полиакрилонитрила,
используемого для получения волокна, составляет 40 000—60 000 [12]. При
молекулярном весе полимера ниже 10 000 волокно не формуется, а
при молекулярном весе выше 70 000 уменьшается прочность
получаемого волокна [13] вследствие необходимости понижения
концентрации полиакрилонитрила в растворе. Однако эти данные не
являются бесспорными, так как
советские исследователи [14] показали,
что при увеличении молекулярного
веса полиакрилонитрила с 100000 до
600000 прочность получаемого
волокна повышается, особенно при его
значительном вытягивании (рис. 6.2).
Одновременно возрастает число
двойных изгибов, выдерживаемых
волокном.
При определении молекулярного веса
полиакрилонитрила в растворах диметил-
формамида вискозиметрическим методом
рекомендуется пользоваться уравнением
где [т|] — характеристическая вязкость;
M — молекулярный вес;
a = 0,72—0,75; '
К = 2,5—4.
/ Z 3 4 s „
Молекулярный, бес Цхщ~5
Рис. 6.2. Зависимость прочности
волокна при различной степени его
вытягивания от молекулярного
веса:
/, 2, 3 — степень вытягивания
соответственно 525, 730 U 1090».
7 Зак. 234
194 Глава б. Производство полиакрилонитрильных волокон
Полиакрилонитрил, так же как и другие синтетические полимеры, является
полидисперсным продуктом. Из 2%-ных растворов полиакрилонитрила в диметил-
формамиде фракционным осаждением гептаном было выделено 9 фракций
полимера, отличающихся между собой по молекулярному весу (от 17 000 до
135 000) [2]. Аналогичные результаты были получены и в других работах.
Советскими исследователями разработан метод определения полидисперс-
иости фракционным осаждением полиакрилоиитрила скипидаром из его
1—2%-ных растворов в диметилформамиде. Этим методом им удалось разделить
исследуемый препарат полиакрилоиитрила на 9—11 фракций, причем по
молекулярному весу крайние фракции отличались между собой более чем в 9 раз
[15, с. 43].
Полидисперсиость полиакрилоиитрила, в частности содержание в нем низко-
молекулярных фракций, оказывает существенное влияние на способность волокна
к вытягиванию. Чем больше в полимере фракций с молекулярным весом ниже
10000, тем меньше предельная степень вытягивания и меньше прочность
получаемого волокна. С увеличением содержания в полимере иизкомолекуляриых и
высокомолекулярных фракций повышается неоднородность прядильного раствора,
что приводит к повышениому засореиию отверстий фильер [16].
Как уже указывалось, полиакрилонитрил растворяется в
ограниченном числе растворителей. Он растворим в
концентрированной серной кислоте, в некоторых неорганических солях и в
приведенных выше органических соединениях.
В концентрированных растворах E0—60%-ных) перхлоратов
(Na, Ca, Ba, Mg, Al) и роданидов (Na, К, NH4, Ca), a также в
растворе ZnCb или смеси ZnCb с другими солями (NaCl, СаСЬ)
полиакрилонитрил растворяется при повышенных температурах
неограниченно, образуя вязкие концентрированные растворы [17].
В работе Хаутца [2] описано более 30 органических веществ,
растворяющих полиакрилонитрил, и приведены интересные данные
о влиянии функциональных групп в молекуле этих соединений на
их растворяющую способность. Все органические соединения,
растворяющие полиакрилонитрил, представляют собой
сильнополярные жидкости (например, диметилсульфон, диметилсульфоксид и
диметилформамид).
Интересно отметить, что ни формамид, ни диэтилформамид
почти не растворяют полиакрилонитрил [2], и в этом отношении
диметилзамещенные формамида обладают особыми свойствами.
Полиакрилонитрил растворяется также в капролактаме, м- и га-
нитрофеноле (в о-нитрофеноле не растворяется); в о-, м- и га-фе-
нилендиамине, бутиролактоне, 2-пирролидоне и в ряде других
растворителей [18].
6.4. ПОЛУЧЕНИЕ ПОЛИАКРИЛОНИТРИЛЬНОГО ВОЛОКНА
Приготовление прядильного раствора. Технологическая схема
получения прядильного раствора и подготовки его к формованию
полиакрилонитрильного волокна (рис. 6.3) принципиально не
отличается от аналогичных схем, применяемых при формовании
искусственных и других синтетических волокон из растворов.
6.4. Получение полиакрилонитрильного волокна
195
При получении прядильного раствора- полиакрилонитрила
необходимо учитывать следующие особенности.
Для приготовления прядильного раствора полиакрилонитрила,
синтезированного методом суспензионной полимеризации, в каче;
стве растворителя используют диметилформамид. Содержание воды
в диметилформамиде не должно превышать 1%. При увеличении
Рис. 6.3. Схема процесса приготовления прядильного раствора
полиакрилонитрила:
1 — мерннк диметилформамида; 2 —бункер-дозатор полиакрилонитрила; 3 — смеситель;
4 — подогреватель; 5—дорастворитель; 6,9—фильтр-прессы для первой и второй
фильтрации соответственно; 7—бак; 8—эвакуатор непрерывного действии (для удалении воздуха).
количества воды растворяющая способность диметилформамида
понижается и соответственно увеличивается минимальная
температура, необходимая для полного растворения
полиакрилонитрила [19]:
Содержание воды
в
диметилформамиде, вес. % 0 1 2 3 4
Минимальная
температура
растворения
полиакрилонитрила,
°С -10 10-20 50-60 60-70 80-90
Кроме того, при наличии воды в диметилформамиде снижается
стабильность получаемых прядильных растворов. Чем больше
содержится воды в прядильном растворе, тем быстрее повышается
его вязкость во времени. Чем выше молекулярный вес полимера
]96 Глава б. Производство полиакрилонитрильных волокон
и чем выше концентрация полимера в растворе, тем ниже должно
быть предельное содержание воды в растворе [20], в противном
случае резко повышается вязкость раствора, что приводит к его
застудневанию.
Значительное изменение растворимости полиакрилонитрила и
температуры, необходимой для его растворения, а также
стабильности этих растворов во времени при сравнительно небольшом
изменении содержания воды в диметилформамиде необходимо
учитывать при установлении параметров процесса растворения
полиакрилонитрила. Поэтому для получения стандартных и
воспроизводимых результатов при приготовлении прядильных растворов,
если используется полимер, полученный методом суспензионной
полимеризации, необходимо применять диметилформамид с
постоянным содержанием влаги и тщательно высушенный полиакри-
лонитрил. Содержание влаги в полимере не должно
превышать 0,5%.
Скорость самопроизвольного повышения вязкости
концентрированного раствора полиакрилонитрила в диметилформамиде зависит
от полидисперсности полимера и температуры, при которой
производится растворение. Чем меньше полидисперсность
полиакрилонитрила, тем ниже скорость желатинирования раствора [21]. При
температуре растворения 80—115 °С образуются растворы, более
стабильные, т. е. обладающие меньшей скоростью
желатинирования, чем растворы, полученные при температурах растворения
ниже 70 и выше 120 °С. Причины этого явления пока не выяснены.
Растворение полиакрилонитр.ила (точнее, его набухание) в
диметилформамиде начинается при нормальной температуре, но
затем в большинстве случаев температуру растворения повышают до
80—90 °С. При этой температуре процесс длится 2—5 ч. В
последнее время все более широкое распространение получает метод
непрерывного растворения в шнеках. Продолжительность процесса
при этом сокращается до 10—30 мин.
Состав и свойства прядильного раствора (концентрация
полимера в растворе и вязкость) зависят от метода формования
волокна. Так же как и при получении всех других химических
волокон, прядильный раствор, применяемый для формования полиак-
рилонитрильного волокна сухим способом, обладает значительно
более высокой вязкостью, и соответственно концентрация полимера
в растворе выше, чем при формовании мокрым способом. При
формовании полиакрилонитрильного волокна мокрым способом
вязкость прядильного раствора составляет 300—500 с, а при
формовании сухим способом — 600—1000 с (при молекулярном весе
полимера 40 000—60 000). Концентрация полиакрилонитрила в
прядильном растворе при формовании мокрым способом достигает
18—20%, а при сухом способе —30—32%.
Так как в большинстве случаев выпускается матированное по-
лиакрилонитрильное волокно, в прядильный раствор добавляют
0,73
1,76
1,99
3,70
1,64
11
65
127
245
585
6.4. Получение полиакрилонитрильного волокна 197
0,7—1 % ТіО2 (от массы полимера). При крашении в массе в
раствор вводят также пигменты в виде пасты.
Как уже указывалось выше, вязкость эквиконцентрированных
растворов одного и того же полимера или сополимера акрилониг-
рила в значительной степени зависит от характера применяемого
растворителя. Влияние природы растворителя на вязкость 10%-ных
растворов сополимера акрилонитрила и метилакрилата
характеризуется следующими данными [22]:
Вязкость Вязкость
растворителя, 10%-ного раствора
сП сополимера, П
Диметилформамид
Диметилсульфоксид
Этиленкарбонат (87%-ный,
температура плавления 23 °С)
Роданид натрия E1%-ный водный
раствор)
Азотная кислота F5%-ная) ....
Как видно из этих данных, вязкость эквиконцентрированных
растворов сополимера в концентрированной азотной кислоте
примерно в 50 раз выше, чем в диметилформамиде. Необходимо
отметить, что вязкость растворов полимера в водных растворах эти-
ленкарбоната и HNO3 значительно снижается при изменении
содержания воды в этих растворителях. Например, при повышении
концентрации HNO3 в водном растворе с 59 до 67% вязкость экви-
концентрированного прядильного раствора снижается с 400 до
150 с, а при повышении концентрации этиленкарбоната в водном
растворе с 82 до 87% вязкость снижается в 2—2,5 раза. При
дальнейшем повышении концентрации HNO3 или этиленкарбоната
в водном растворе вязкость прядильных растворов снова
повышается.
Вследствие применения прядильных растворов повышенной
вязкости увеличивается и давление при фильтрации. При формовании
волокна мокрым способом фильтрация производится под
давлением 10—20 кгс/см2 (МО6 Па —2-Ю6 Па).
Для ускорения удаления воздуха из прядильного раствора обез-
воздушивание его можно проводить под вакуумом при повышенных
температурах D5—50°С).
Прядильный раствор после однократной фильтрации при
повышенной температуре G0—75 °С) и обезвоздушивания подается на
прядильную машину.
Формование волокна. Полиакрилонитрильное волокно, так же
как и другие термопластичные волокна, можно формовать из
полимера, находящегося в размягченном состоянии (без применения
растворителей или с добавкой небольшого количества
пластификатора) или из концентрированных растворов полимера.
198 Глава 6. Производство полиакрилонитрильных волокон
При формовании волокна из размягченного полимера отпадает
необходимость в применении растворителей и их регенерации, а
также в приготовлении прядильных растворов и подготовке их
к формованию. По-видимому, для осуществления этого
перспективного способа необходимо использовать сополимеры акрилонит-
рила, содержащие в макромолекуле повышенное количество
второго гибкоцепного мономера. Так как при применении такого
полимера для формования волокна может значительно ухудшиться
ряд ценных свойств волокна, в частности температура плавления,
целесообразно осуществить сшивание получаемого волокна, что
должно повысить его теплостойкость и эластичность. Разработка
метода формования полиакрилонитрильного волокна без
применения растворителей представляет значительный интерес.
В настоящее .время полиакрилонитрильные волокна различных
типов получают только формованием из растворов сухим или
мокрым способом. Формование волокна сухим способом производится
преимущественно при получении нити и при использовании в
качестве растворителей органических веществ с температурой
кипения 150—160 °С. Естественно, что растворы полиакрилонитрила
в неорганических солях не могут быть использованы для
формования волокна сухим способом. Из различных органических
растворителей практическое применение для формования нити сухим
способом получил диметилформамид. Сухой способ формования
нити из растворов полимера в диметилформамиде реализован
в производственных условиях в ФРГ, США и Японии.
Температура кипения диметилформамида значительно выше,
чем большинства растворителей, используемых при формовании
других химических волокон сухим способом. Это обстоятельство
обусловливает необходимость изменения основных параметров
процесса формования по сравнению, например, с формованием
ацетатной нити.
6.4.1. Формование полиакрилонитрильной нити
Для формования полиакрилонитрильной нити сухим способом
применяются машины такой же конструкции, как и для
формования ацетатного волокна. На прядильную машину поступает
прядильный раствор, нагретый до 100—120 °С [23]. Естественно, что
испарение в шахте высококипящего растворителя обусловливает
необходимость формования волокна при значительно более
высокой температуре, чем при формовании волокон из ацетоновых
растворов. Поэтому шахта прядильной машины высотой 4—& м
нагревается до 300—400 °С. Снизу в шахту подается воздух при
температуре 100°С; проходя через шахту, нагретую до такой высокой
температуры, он нагревается до 200 °С.
Другим характерным отличием формования
полиакрилонитрильной нити сухим способом является высокая концентрация
6.4. Получение полиакрилонитрильного волокна 199
паров растворителя в шахте D00—500 г/м3), превышающая
верхний предел взрывоопасности. Отсасываемая из шахты прядильной
машины газовоздушная смесь охлаждается, основное количество
паров диметилформамида конденсируется, а оставшийся в
газовоздушной смеси после ее охлаждения растворитель улавливается
водой. Разбавленный водный раствор диметилформамида
поступает на ректификацию или в ряде случаев сжигается.
Необходимость почти полного испарения высококипящего
растворителя из формующейся нити и сравнительно медленная
диффузия растворителя из нити при высокой концентрации паров
растворителя в шахте обусловливают низкие скорости формования
даже при сухом способе получения этого волокна. Выходящая из
шахты нить принимается на галету или на бобину со скоростью
50—100 м/мин. Эта нить содержит 3—5% диметилформамида.
Вытягивание нити в 6—7 раз в большинстве случаев производится
на прядильной машине между двумя дисками, нагреваемыми до
130—140 °С. Скорость приемки вытянутого волокна 250—350 м/мин.
Вытянутое волокно подвергается промывке на бобине для
удаления остаточного количества диметилформамида и релаксации нити
и затем сушится.
Полиакрилонитрильная нить может быть получена и мокрым
способом из растворов полиакрилонитрила в диметилформамиде.
Сведений о формовании этой нити из растворов полимера в других
растворителях в литературе нет. Для формования нити
применяются фильеры с 50—500 отверстиями. Толщина вытянутой нити
5—20 текс. Характерной особенностью процесса формования нити
мокрым способом является низкая скорость приемки невытянутой
нити, не превышающая 5—7 м/мин. Свежесформованная нить
вытягивается в б—7 раз, промывается в две стадии, сушится,
замасливается и поступает на кручение. Скорость приемки вытянутой
нити 40—50 м/мин, прочность нити 30—35 гс/текс C00—
350 мН/текс).
Для изготовления ряда технических изделий требуются
высокопрочные нити E0—55 гс/текс). При получении такой нити
молекулярный вес исходного полимера или сополимера повышают с 60 000
до 120 000. Соответственно концентрацию полимера в растворе при
переработке эквивязких прядильных растворов снижают до 10—
12%. Кроме того, вытянутую нить дополнительно вытягивают при
160 °С на 50—70%.
Высокопрочная нить обладает повышенной хрупкостью и
пониженным удлинением. После терморелаксации нити при 200 °С и ее
усадки на 8—10% удлинение нити повышается до 10%.
Интересным и перспективным способом формования полиакри-
лонитрильной нити является так называемый сухо-мокрый
способ, разработанный советскими исследователями [24].
Выдавливаемые из фильеры струйки прядильного вязкого раствора
проходят путь в 3—5 см на воздухе и затем поступают в осадительную
200
Глава 6. Производство полиакрилонитрильных волокон
ванну. Скорость формования такой нити, частично скоагулиро-
вавшей на воздухе, повышается в 2—3 раза и составляет 120—
150 м/мин.
Схема формования волокна через воздушную прослойку сухо-
мокрым способом приведена на рис. 6.4.
Как указывалось выше, производство полиакрилонитрильной
нити составляет крайне незначительную часть общего количества
вырабатываемого полиакрилонитриль-
ного волокна @,4—0,5%). Такая нить
используется только для изготовления
1 технических изделий.
6.4.2. Формование полиакрилонит-
рильного штапельного волокна
Характерным отличием метода
формования полиакрилонитрильного
штапельного волокна от формования
других штапельных волокон мокрым
способом является большое число
растворителей, которые используются или
могут быть использованы для
приготовления концентрированных
прядильных растворов полимеров или
сополимеров акрилонитрила. При
полимеризации или сополимеризации
акрилонитрила в растворе в
производственных условиях может
использоваться несколько растворителей. При получении прядильных
растворов методом растворения полиакрилонитрила, синтезированного
методом эмульсионной полимеризации, количество используемых
растворителей дополнительно увеличивается. Естественно, что
основные параметры процесса формования, в частности состав оса-
дительной ванны, температура и скорость формования, должны
быть соответственно изменены.
Рассмотрим основные закономерности процесса формования
полиакрилонитрильного штапельного волокна мокрым способом.
Осадителями при формовании волокна из растворов
полиакрилонитрила в органических растворителях служат органические
жидкости, в которых полиакрилонитрил не растворяется и не
набухает, или водные растворы, содержащие большее или меньшее
количество растворителя.
Для высаживания полиакрилонитрила из прядильного раствора
предложены различные жидкости, в частности глицерин. В ГДР для
этой цели применяется гексантриол СН3СНОНСН2СНОНСНОНСНз,
который является отходом производства синтетического каучука.
Температура осадительной ванны при использовании многоатом-
Рис. 6.4. Схема формования
волокна через воздушную
прослойку:
/ — фильера; 2—направляющее
устройство; 3 — осадительная ванна;
4 — приемное устройство.
6.4. Получение полиакрилонитрильного волокна 201
ных спиртов составляет 90—100 °С. Такая высокая температура
ванны усложняет процесс формования и ухудшает условия труда.
Основное преимущество глицериновой осадительной ванны по
сравнению с водной заключается в том, что волокно можно
формовать с более высокой скоростью.
В качестве основного компонента осадительной ванны
предложено применять высшие спирты и жирные кислоты, а также
неполярные жидкости, например керосин, четыреххлористый углерод'
и т. д. Имеются сведения, что наиболее прочное волокно
получается при применении в качестве осадительной ванны высших
спиртов (Сю—Си) или высших жирных кислот (С]7—Сгі).
Однако использование летучих и легко воспламеняющихся
жидкостей, таких, как керосин, естественно, нецелесообразно и не
может быть рекомендовано для практического применения.
Основным преимуществом формования волокна в органических
ваннах до последнего времени являлась возможность проведения
процесса в мягких условиях. Однако применение органических
осадительных ванн в производственных условиях нецелесообразно,
так как этот метод значительно менее экономичен, чем формование
в водных ваннах. Изменяя состав водной ванны, в последнее время
удалось сформовать волокно, не уступающее по эластичности
волокну, получаемому в органических ваннах.
При формовании в водных ваннах существенное, а в ряде
случаев решающее влияние на свойства получаемого волокна
оказывает концентрация растворителя в осадительной ванне. Чем выше
концентрация растворителя в ванне, тем больше равномерность
структуры и выше эластичность волокна. Например, минимальная
концентрация диметилформамида в осадительной ванне, при
которой получается волокно со сравнительно удовлетворительными
эластическими свойствами, составляет 30—40%. Применение ванны,
содержащей меньшее количество диметилформамида,
нецелесообразно, так как при этом значительно ускоряется процесс коагуляции
и получается более хрупкое волокно. Наоборот, при увеличении
концентрации диметилформамида в ванне до 60—70% в результате
замедления процесса коагуляции повышается эластичность
волокна, и в частности уменьшается потеря прочности в петле.
Одновременно возрастает плотность и уменьшается пористость волокна.
Влияние концентрации диметилформамида в осадительной
ванне на свойства волокна нитрон характеризуется следующими
данными [25]:
Содержание днметил- Ппочнпеть Уплине Число
формамида в осади- "^текс шЛ % Двойных
тельной ванне, % гс/текс ние, % изгибов
15-20
30-35
40-45
50—55
60-63
23,6
27,4
27,2
29,4
27,8
22,0
24,0
23,0
22,0
26,0
6740
9640
13470
1 4725
14430
202 Глава 6. Производство полиакрилонитрильных волокон
Эти данные показывают, что при увеличении концентрации ди-
метилформамида в осадительной ванне несколько повышается
прочность и удлинение и значительно возрастает эластичность
волокна, характеризуемая числом двойных изгибов, выдерживаемых
волокном до разрушения.
Наиболее мягкие условия коагуляции достигаются при
формовании волокна в 70—80%-ном водном растворе диметилфэрмамида.
При такой концентрации и даже в 55—60%-ном водном растворе
вся вода связывается диметилформамидом в результате
образования гидратов C—4 моль воды на 1 моль диметилформамида).
Следовательно, в этих условиях формование волокна происходит
фактически в органической ванне.
Предельная концентрация растворителя в осадительной ванне
различна в зависимости от характера применяемого растворителя
и изменяется от 10—12% (при формовании волокна из растворов
полимера в роданистых солях) до 70—80% (при формовании
волокна из растворов полимера в дйметилформамиде). Вопрос об
оптимальной и максимально допустимой концентрации
растворителя в водной ванне при формовании волокна из растворов поли-
акрилонитрила в других растворителях достаточно полно не
исследован.
Формование полиакрилонитрильного волокна мокрым способом
производится, как правило, с отрицательной фильерной вытяжкой
при низких скоростях формования. Обычно скорость приемки нити,
выходящей из осадительной ванны, составляет 5—8 м/мин. Однако
благодаря тому, что свежесформованная нить вытягивается в 5—
7 раз, скорость приема сформованного и вытянутого штапельного
волокна составляет 35—40 м/мин.
Несмотря на низкую скорость формования, количество волокна,
получаемого в единицу времени с одной фильеры, в несколько раз
больше, чем при формовании любого другого вида штапельного
волокна. Это объясняется тем, что при формовании
полиакрилонитрильного штапельного волокна применяются фильеры с
30 000—40 000 отверстиями. Однако даже такое количество
отверстий в фильере, значительно превышающее число отверстий в
фильерах, используемых при формовании других штапельных
волокон мокрым способом, не является предельным. В
производственных и в опытно-промышленных условиях применяются уже
фильеры с 150 000 и более отверстиями. Учитывая трудность заправки и
обслуживания прядильного места, а также возможность
увеличения засоряемости фильер с таким большим числом отверстий,
следует дополнительно проверить целесообразность их применения.
По-видимому, на данном этапе оптимальное число отверстий
находится в пределах 40 000—60 000.
Как правило, формование волокна в водных осадительных
ваннах в отличие от формования в органических ваннах
проводится при пониженных температурах. Например, волокно из прядиль-
6.4. Получение полиакрилонитрильного волокна 203
ных растворов полиакрилонитрила в диметилформамиде формуют
при 10—12 °С. При этом получается волокно с более плотной
структурой, содержащее меньшее количество пор и обладающее высокой
устойчивостью к многократным деформациям. При 22—24 °С и
более высокой температуре прядильной ванны эластические
свойства получаемого волокна ухудшаются. Формование волокна при
температуре осадительной ванны ниже 10 °С экономически
нецелесообразно. Кроме того, при таких температурах ухудшаются
условия труда. При получении волокна из растворов полимера в
роданистых солях температура осадительной ванны составляет 10—
12 °С, а при формовании волокна из растворов полимера в азотной
кислоте должна быть снижена до 0—5 °С, что является
дополнительным недостатком этого способа. Длина пути нити в ванне
составляет 0,25—0,5 м.
Форма среза волокна и плотность его структуры при
формовании волокна из диметилформамидных растворов зависит от pH
осадительной ванны. При снижении pH ванны с 3 до 1 (путем
добавки в ванну небольших количеств щавелевой кислоты)
набухание свежесформованного волокна уменьшается в 1,5—2 раза [26].
Одновременно снижается скорость сорбции и количество
поглощаемого волокном красителя. Следовательно, чем ниже pH
прядильной ванны, тем больше плотность структуры полученного
волокна. При понижении pH прядильной ванны изменяется форма
среза — вместо круглого среза волокно имеет бобовидный срез.
Плотность волокна зависит также и от характера применяемого
растворителя. Волокна, сформованные из растворов полимера в
диметилформамиде, обладают меньшей плотностью и большей
пористостью, чем волокна, сформованные из растворов полимеров в
диметилсульфоксиде [27].
6.4.3. Регенерация растворителя
Обязательной стадией технологического процесса получения
полиакрилонитрильного штапельного волокна является
регенерация растворителя из осадительной ванны и из промывных вод.
Условия регенерации различны в зависимости от характера
применяемого растворителя. Если в качестве растворителя при
полимеризации акрилонитрила в растворе или при получении
прядильного раствора растворением полиакрилонитрила, синтезированного
методом суспензионной полимеризации, используются органические
растворители (диметилформамид, диметилацетамид, диметилсульф-
оксид) с температурой кипения 150—180°С, то регенерация
производится испарением воды из отработанной осадительной ванны и
последующей перегонкой органического растворителя.
Все минеральные и нелетучие органические примеси,
находящиеся в осадительной ванне, при указанном методе регенерации
204 Глава 6. Производство полиакрилонитрильных волокон
остаются в кубовом остатке и регенерированный растворитель не
содержит этих примесей.
Для уменьшения гидролиза диметилформамида и диметил-
ацетамида'их регенерация обычно проводится под вакуумом при
90—100 °С.
При полной регенерации органических растворителей из
отработанной осадительной ванны и промывных вод (при промывке
противотоком) удельный расход растворителя не должен
превышать 0,1—0,15 кг на 1 кг волокна.
Регенерация роданистых солей или других неорганических
солей (ZnCl2, СаС12) из отработанных осадительных ванн
значительно сложнее, чем органических растворителей (особенно при
использовании этих солей при полимеризации акрилонитрила в
растворе). Получить концентрированный раствор солей, пригодный
для повторного использования в качестве растворителя при
полимеризации акрилонитрила методом отгонки растворителей, не
представляется возможным. Для очистки этих растворителей от
примесей используют следующий метод регенерации:
выпариванием избытка воды из отработанной осадительной
ванны в двухкорпусном вакуум-выпарном аппарате повышают
концентрацию роданидов в растворе с 11 до 52%;
для полного удаления солей железа, попадающих в прядильный
раствор- и в осадительную ванну (в результате постепенной
коррозии аппаратуры), концентрированный раствор роданида
пропускают через ионитовые фильтры, улавливающие ионы только
поливалентных, а не одновалентных металлов.
Однако методом ионного обмена нельзя очистить раствор
роданида от органических примесей, образующихся в растворе при
полимеризации акрилонитрила. Состав этих примесей пока не
исследован, так же как не изучено влияние их на скорость
полимеризации и состав получаемого полимера. Обычно принимается (без
достаточного обоснования), что содержание органических
примесей в растворе полиакрилонитрила не должно превышать 3% от
массы полимера.
Для получения после регенерации раствора роданида,
содержащего не более 3% органических примесей, необходимо 15—20% от
общего количества регенерированного роданида полностью
очистить от органических примесей. Для этого используют метод
кристаллизации роданидов и метод экстракции.
Кристаллизация роданидов из
концентрированного раствора. Отбирают 15—20% от общего объема
регенерированного 52%-ного водного раствора роданида, нагревают его
до 60°С и затем мгновенно испаряют в вакууме. В результате
такого испарения происходит дополнительное испарение воды и
роданиды выпадают в виде кристаллов, которые растворяют в
концентрированном водном растворе роданистых солей. Маточный
раство , содержащий еще значительное количество роданистых
6.4. Получение полиакрилонитрильного волокна 205
солей, направляют на ионообменники для улавливания этих солей.
Оставшийся водный раствор, содержащий только органические
примеси, поступает совместно со сточными водами на
биологическую очистку.
Метод экстракции. Примерно такое же количество
регенерированного раствора роданида A5—20%) направляют на
экстракцию. Этот раствор обрабатывается концентрированной серной
кислотой. В результате обменной реакции образуется HCNS,
которая экстрагируется из раствора изопропиловым спиртом.
Оставшийся водный раствор после нейтрализации серной кислоты
поступает на биологическую очистку для разложения органических
примесей и затем сливается в водоемы. К раствору HCNS в изо-
пропиловом спирте добавляют водный раствор NaOH.
Образующийся NaCNS переходит в водный раствор, который добавляется
к основному количеству регенерированного роданида, а изопропи-
ловый спирт снова используется для экстракции.
6.4.4. Последующая обработка
полиакрилонитрильного волокна
Свежесформованное полиакрилонитрильное волокно
подвергается вытягиванию, промывке, замасливанию, сушке,
термообработке и гофрированию. Все эти операции при выработке
штапельного волокна проводятся, как правило, в жгуте, который в случае
необходимости подвергают резке после сушки.
Вытягивание. Полиакрилонитрильные волокна вытягивают,
так же как и другие карбоцепные волокна, между двумя или
несколькими парами вытяжных дисков или между вытяжными
вальцами при сравнительно невысоких скоростях. Характерной
особенностью процесса вытягивания полиакрилонитрильных волокон
является значительно более высокая степень вытягивания, чем при
получении даже высокопрочных гетероцепных синтетических
волокон. Средняя степень вытягивания полиакрилонитрильных
волокон составляет 600—800%. Вытягивание волокна, осуществляемое
при повышенных температурах, может быть проведено в одну или
две стадии в различных вариантах.
Вытягивание в одну стадию при 90—100 °С проводится в
горячей воде, содержащей небольшое количество растворителя.
При вытягивании жгута в две стадии первая стадия
вытягивания (на 50—100%) предварительно промытого или непромытого
волокна осуществляется при нормальной температуре. Затем
частично вытянутый жгут тщательно промывается в 2—3 ваннах.
Промывные воды, содержащие 4—2,5% растворителя, испсльзуют-
ся для добавления в осадительную ванну. На второй стадии жгут
вытягивается (на 400—500%) над нагретой поверхностью прн
160—180 °С.
206 Глава б. Производство полиакрилонитрильных волокон
В некоторых случаях обе стадии процесса вытягивания
проводятся непосредственно одна за другой, а затем вытянутое волокно
промывается.
Максимально возможная степень вытягивания полиакрилонит-
рильного волокна зависит от температуры. Например, при 100, 140
и 15у8°С в пластификационной ванне волокно можно вытянуть
соответственно на 675, 1220 и 2040% [28]. В результате
вытягивания значительно уплотняется структура волокна, что выявляется,
в частности, в заметном замедлении скорости диффузии красителя
в волокно. Например, коэффициент диффузии красителя
(кислотного синего) в волокно при его вытягивании на 400% понижается
с 1,6-10~7 (для свежесформованного волокна) до 3,4-10~8, а при
дополнительном вытягивании еще на 150%—до 8,6-10~9. После
сушки этого волокна при 120 °С коэффициент диффузии красителя"
дополнительно уменьшается в 100 раз [29].
Замасливание. Вытянутое, и промытое волокно обрабатывается
водным раствором, содержащим замасливатель и антистатик, и
затем поступает на сушку. Чтобы волокно не пожелтело, его сушат
при температуре не выше 80 °С. Обычно при сушке волокно
усаживается на 20—25%. При сушке резко уменьшается внутренняя
поверхность волокна — с 200 м2/г для несушеного волокна до 0,5—
1 м2/г для высушенного волокна. Соответственно снижается
скорость диффузии различных веществ в высушенное волокно.
Поэтому большинство обработок, например поверхностное крашение,
химические превращения реакционноспособных функциональных
групп в макромолекуле полимера или сополимера акрилонитрила,
рекомендуется проводить в процессе отделки несушеного волокна.
Термообработка. Условия, в которых производится
термообработка (терморелаксация) волокна, оказывают существенное
влияние на улучшение комплекса свойств полиакрилонитрильного
волокна. Этот процесс детально изучен советскими исследователями
C0, 31].
Влияние условий термообработки на изменение прочности и
удлинение полиакрилонитрильной нити характеризуется
следующими данными [32]:
Прочность, Удлинение»
гс/текс %
Без релаксации 41,8 10,5
Нагрев в течение 1 ч при 150 °С
иа жесткой паковке 45,5 9,7
в мотках 45,8 14,8
Эти цифры показывают, что в результате нагрева
полиакрилонитрильной нити в течение 1 ч при 150 °С на жесткой паковке
повышается прочность, а если процесс проводить в условиях
свободной усадки нити (нить в мотках), то увеличивается и прочность, и
удлинение нити.
В результате терморелаксации и обусловленной этой
обработкой увеличения интенсивности межмолекулярного взаимодействия
6.4. Получение полиакрилонитрильного волокна 207
значительно повышается термостойкость волокна. Например, если
температура, при которой начинается усадка для вытянутого не-
прогретого полиакрилонитрильного волокна составляет 75 °С, то
для терморелаксированного волокна она достигает 125 °С.
Одновременно на 15—50% повышается устойчивость к истиранию
(особенно при нагреве под натяжением) и к многократным
деформациям, а также стойкость к ультрафиолетовому облучению [30].
В результате термообработки дополнительно снимаются
внутренние напряжения, исчезают поры и получается волокно более
однородной структуры, обладающее более высокой усталостной
прочностью. Если проводить термообработку волокна в свободном
состоянии, в условиях, при которых может происходить
дополнительное усаживание (на 2—5%), то после этой обработки
получается безусадочное волокно.
После прогрева нити в свободном состоянии заметно
повышается также один из наиболее существенных показателей,
характеризующих эластические свойства волокна, — относительная прочность
в петле. Относительная прочность нити в петле после нагрева при
200°С увеличивается с 37 до 60%.
На относительную прочность нити в петле влияет не только
температура нагрева, но и характер среды, в которой производится
термообработка. Например [30], после нагрева при 150 °С в
атмосфере насыщенного водяного пара относительная прочность нити
в петле достигает очень большой величины — 90,1%, а после
нагрева на воздухе — только 58,5%- Однако в последнем случае
устойчивость нити к истиранию выше, чем при нагреве в атмосфере
пара.
Термообработка полиакрилонитрильного штапельного волокна
производится в автоклавах при 130 °С в течение 30 мин или в
сушилках при 150°С в течение Юс.
Гофрирование. Эта операция необходима для повышения
извитости и тем самым увеличения сцепляемости волокна при
последующем изготовлении пряжи. В настоящее время, как правило,
метод гофрирования полиакрилонитрильного волокна механической
обработкой не применяется, так как при этом, заметно снижается
его прочность.
Гофрирование осуществляется обычно методом перевода
волокна в пластическое (размягченное) состояние. Для этого жгут
пропускают в течение 0,5—0,8 с между плитами, которые обогреваются
инфракрасными лучами или паром до 200 °С. Нагретый жгут
поступает без натяжения в камеру, в которой он укладывается в виде
петель при одновременном быстром охлаждении. В результате
жгут сминается и образуется устойчивая извитость волокна.
Гофрированный жгут в случае необходимости подвергается
резке.
208
Глава 6. Производство по.шакрилонитрильных волокон
6.4.5. Свойства полиакрилонитрильного волокна
Полиакрилонитрильное волокно обладает достаточно высокой
прочностью. После вытягивания свежесформованной нити на 800—
1200% прочность ее составляет 35—40 гс/текс. Как указывалось
выше, для изготовления технических изделий применяют нить
прочностью 50—60 гс/текс. Для штапельного волокна (в ряде случаев
и для нити), используемого для изготовления изделий народного
потребления, такая высокая прочность не требуется. Поэтому жгут
вытягивают только на 400—600%, что обеспечивает получение
волокна с прочностью 22—25 гс/текс. Менее вытянутое волокно
окрашивается интенсивней, чем сильно вытянутое.
В мокром состоянии прочность полиакрилонитрильного волокна
благодаря низкой гигроскопичности почти не уменьшается.
Прочность мокрого волокна составляет 95—98% от прочности в сухом
состоянии.
Полиакрилонитрильное волокно менее эластично, чем
полиамидное, но превосходит по этому показателю ацетатные и
вискозные .волокна. Удлинение сухого волокна 22—35%. В мокром
состоянии удлинение волокна не изменяется.
Начальный модуль полиакрилонитрильной нити несколько
ниже,' чем полиэфирной, но значительно (в 2—3 раза) выше, чем
полиамидной.
При относительной влажности воздуха 65 и 100%
полиакрилонитрильное волокно сорбирует соответственно 0,9—1 и 6% влаги.
Плотность волокна 1,16—1,18 г/см3.
Полиакрилонитрильное волокно обладает сравнительно
высокой термостойкостью. Его можно использовать непродолжительное
время при 180 и даже 200 °С. При длительном (в течение
нескольких недель) выдерживании этого волокна при 120—130°С его
прочность, определяемая при нормальной температуре, ле
изменяется.
Данные [2] об изменении прочности и удлинения
полиакрилонитрильного волокна после сравнительно длительного нагрева при
200 °С на воздухе и в атмосфере азота приведены в табл. 6.1.
Таблица 6.1. Изменение прочности и удлинения полиакрилонитрильного
волокна после нагрева при 200 °С
Волокно
Исходное (непрогретое)
Нагретое
в азоте
на воздухе
Прочность,
сухого
30,6
26,0
17,5
гс/текс
мокрого
30,5
25,6
16,2
Удлинение
сухого
15,7
12,9
7,3
мокрого
17,0
13,7
9,2
6.4. Получение полиакрилонитрильного волокна 209
Из табл. 6.1 видно, что после нагрева при 200 °С в условиях,
исключающих возможность одновременной окислительной
деструкции полиакрилонитрила (нагрев в атмосфере азота), деструкция
волокна и соответственно снижение его прочности незначительны.
По термостойкости полиакрилонитрильное волокно превосходит
почти все карбоцепные волокна и не уступает полиэфирному.
Однако при нагреве или обработке при температурах вцше
120—150 °С происходит постепенное пожелтение волокна,
сопровождаемое выделением небольших количеств HCN. Интенсивность
этих процессов значительно увеличивается в присутствии
кислорода, а в среде инертного газа или в вакууме HCN не выделяется
[33]. Отщепление HCN происходит в основном по радикальному
механизму. Добавление реагентов, распадающихся с образованием
радикалов (например, перекиси бензоила), увеличивает количество
выделяющейся HCN, a добавка реагентов, связывающих
образующиеся радикалы, уменьшает выделение HCN.
Интенсивность пожелтения волокна при повышенных
температурах (при прочих равных условиях) зависит от количества
карбоксильных групп в макромолекуле полимера. Чем больше групп
СООН содержится в макромолекуле, тем интенсивнее
окрашивается волокно. Степень пожелтения волокна при нагреве в
присутствии кислорода значительно больше, чем в среде инертного газа,
и повышается с увеличением числа «аномальных» (кетоиминных)
групп в макромолекуле полимера [34]. По теплостойкости поли-
акрилонитрильные и полиамидные волокна равноценны. Например,
при 100 и 150°С эти волокна теряют соответственно 20—23 и 50%
прочности. Если определять прочность непосредственно при
повышенной температуре, то при 200°С они сохраняют всего 10% от
прочности при нормальной температуре.
Теплостойкость можно значительно повысить обработкой
полиакрилонитрильного волокна полифункциональными соединениями,
обеспечивающими сшивание макромолекул полимера (см.
разд. 6.5.1).
В отношении стойкости к повышенным температурам
полиакрилонитрильное волокно обладает специфическими особенностями,
резко отличающими его от большинства природных и других
химических волокон. После длительного нагрева
полиакрилонитрильного волокна (или тканей) при 200—300 °С постепенно изменяется
его цвет (делается черным и блестящим) и оно становится
нерастворимым. Одновременно значительно уменьшается прочность
волокна и резко повышается его термостойкость. При нагревании
в пламени бунзеновской горелки при 600—800°С предварительно
термообработанное в указанных условиях волокно (так
называемое черное полиакрилонитрильное волокно) не разрушается и
сохраняет после прокаливания прочность и эластичность,
естественно, значительно меньшие, чем исходного волокна. Такое
термообработанное модифицированное полиакрилонитрильное волокно
210 Глава б. Производство полиакрилонитрильных волокон
является одним из наиболее термостойких волокон органического
происхождения (см. разд. 11.2.2) и может быть использовано при
очень высоких температурах B000—3000°С).
Полиакрилонитрильное волокно обладает очень высокой
стойкостью к свету и атмосферным воздействиям, превышающим
аналогичные показатели почти всех природных и химических волокон,
кроме волокна фторлон. После комбинированного воздействия
света и погоды в течение года полиамидные, ацетатные и вискозные
волокна, а также натуральный шелк полностью теряют прочность;
прочность хлопкового волокна снижается на 95%, а полиакрило-
нитрильного — всего на 20%.
Устойчивость полиакрилонитрильного волокна к истиранию в
5—10 раз ниже, чем полиамидного и полиэфирного. Чем больше
прочность и соответственно степень вытягивания волокна (сверх
определенного оптимального значения), тем меньше его
устойчивость к истиранию. Устойчивость полиакрилонитрильного волокна
к истиранию можно увеличить, если процесс формования проводить
в более мягких условиях.
Преимуществом полиакрилонитрильного волокна является
хороший и теплый гриф, шерстеподобный вид, очень высокая
объемная эластичность, низкая теплопроводность, приближающаяся к
теплопроводности шерсти, а также легкость очистки. Эти
показатели определяют целесообразность использования
полиакрилонитрильного волокна для изготовления изделий народного
потребления определенного ассортимента.
Изделия из полиакрилонитрильного волокна стойки к
окислителям, но недостаточно стойки к концентрированным щелочам и
серной кислоте, особенно при повышенной температуре. Эти
реагенты омыляют нитрильные группы, что сопровождается
деструкцией макромолекул. Следовательно, по хемостойкости полиакрило-
нитрильные волокна значительно уступают другим карбоцепным
волокнам. Например, при воздействии 5—20%-ного раствора
NaOH в течение 8—20 ч полиакрилонитрильное волокно полностью
разрушается.
К недостаткам полиакрилонитрильного волокна можно отнести
также его горючесть, повышенную хрупкость (пониженная
прочность нити в петле) и легкую электризуемость [35].
Однако отмеченные недостатки могут быть в большей или
меньшей степени устранены изменением условий формования или
последующей обработкой полиакрилонитрильного волокна. О
методах повышения эластичности волокна указано ниже (см. разд. 6.5).
Улучшение окрашиваемости достигается при получении волокна не
из полиакрилонитрила, а из сополимеров акрилонитрнла с
небольшим количеством другого мономера, содержащего активные
функциональные группы (см. разд. 6.5.1.1), изменением структуры
волокна или применением новых красителей.
6.5. Модифицированные полиакрилонитрильные волокна . 211
Полиакрилонитрильное волокно в чистом виде или в смеси с
шерстью или другими волокнами широко используется для
изготовления верхнего трикотажа и различных тканей. При
изготовлении многих трикотажных изделий полиакрилонитрильные волокна
имеют преимущество перед всеми другими синтетическими
волокнами.
Полиакрилонитрильные волокна целесообразно применять для
изготовления изделий, подвергающихся длительному воздействию
света и погоды, — это гардины, изделия, используемые в тропиках,
а также рыболовные сети и снасти и др. Вследствие низкой
устойчивости к истиранию полиакрилонитрильное волокно
нецелесообразно применять для изготовления чулочно-носочных изделий.
Высокоусадочные полиакрилонитрильные волокна используют
для изготовления объемной пряжи. Для получения таких волокон,
обладающих большей усадкой (в горячей воде или при
повышенных температурах), чем обычное полиакрилонитрильное волокно,
используют сополимеры акрилонитрила, содержащие 10—22%
второго мономера. Для того чтобы повысить усадку волокна в
горячей воде, термообработку его после вытягивания не производят.
Такое волокно усаживается в горячей воде на 25—30%, в то время
как волокно, полученное из сополимера обычного состава,
усаживается в тех же условиях на 4—5%.
6.5. МОДИФИЦИРОВАННЫЕ
ПОЛИАКРИЛОНИТРИЛЬНЫЕ ВОЛОКНА
Свойства полиакрилонитрильных волокон можно изменять в
широких пределах, используя различные методы их модификации.
Из модифицированных волокон наибольший интерес представляют:
волокна из сополимеров акрилонитрила;
волокна из привитых сополимеров полиакрилонитрила;
волокна из смесей полиакрилонитрила с другими полимерами.
6.5.1. Волокна из сополимеров акрилонитрила
В настоящее время полиакрилонитрильные волокна получают
не из полимеров, а из сополимеров акрилонитрила. В зависимости
от состава сополимеры акрилонитрила могут быть условно
подразделены на две группы: 1) с низким содержанием второго
мономера; 2) с высоким содержанием второго мономера.
Количество второго компонента, входящего в состав
сополимеров первой и второй группы, обычно составляет соответственно
5—10 и 40—60%.
6.5.1.1. Волокна из сополимеров
с низким содержанием второго мономера
Сополимеры, содержащие более 80—85-% акрилонитрила, и
формуемые из них волокна по основным показателям
(растворимости, механическим свойствам, термостойкости) заметно не
212 Глава 6. Производство полиакрилонитрильных волокон
отличаются от полиакрилонитрила. Хотя эти сополимеры
растворяются в тех же растворителях, что и полиакрилонитрил, но
минимальная температура, при которой происходит их растворение
(например, в диметилформамиде), ниже, чем для
полиакрилонитрила. Этот факт дополнительно свидетельствует о том, что при
нарушении регулярности строения полиакрилонитрила улучшается
его растворимость.
Так как сополимеры акрилонитрила даже при небольшом
содержании второго мономера имеют менее регулярное строение, чем
полиакрилонитрил, получаемые из них волокна обладают более
высокими эластичностью и усадочностью, чем полиакрилонитриль-
ное волокно.
Химический состав второго мономера, добавляемого в
небольших количествах в процессе сополимеризации акрилонитрила,
определяется свойствами волокон, которые должны быть
получены.
Число винильных мономеров, предложенных в качестве сомоно-
меров акрилонитрила, очень велико, однако по многим причинам
применение нашли только некоторые из них.
Мономеры, используемые в качестве небольших добавок при
синтезе волокнообразующих сополимеров акрилонитрила, можно
условно подразделить на четыре группы: 1) улучшающие накра-
шиваемость; 2) повышающие гидрофильность; 3) увеличивающие
эластичность и 4) повышающие реакционную способность
сополимера и получаемого из него волокна.
Мономеры, улучшающие накрашиваемость волокон. Эти
мономеры могут быть в свою очередь подразделены на следующие
классы.
1. Мономеры, которые содержат функциональные группы
основного характера. Наличие таких групп в макромолекуле полимера
позволяет окрашивать получаемые волокна кислотными
красителями. К этому классу относятся 2-винилпиридин, 2-метил-5-винил-
пиридин, N-винилимидазол, акриламид и другие мономеры
аналогичного состава.
Советскими исследователями изучены условия получения
сополимера акрилонитрила с 2-метил-5-винилпиридином и формование
из него волокна [36].
Эквиконцентрированный раствор этого сополимера
акрилонитрила в 50—55%-ном водном растворе роданида натрия обладает
в 3—4 раза более низкой вязкостью, чем сополимер акрилонитрила
с сс-винилпиридином, что, по-видимому, объясняется меньшей
степенью асимметрии макромолекул сополимера акрилонитрила с
2-метил-5-винилпиридином. Волокно можно формовать из 10—
16%-ных растворов этого сополимера в растворе роданида натрия
(осадительная ванна содержит 8—10% водного раствора роданида
натрия) и из растворов в диметилформамиде (осадительная
ванна— водный раствор диметилформамида) [37].
6.5. Модифицированные полиакрилонитрильные волокна 213
2. Мономеры с функциональными группами кислотного
характера. Эти мономеры придают волокну способность окрашиваться
основными красителями. К этому классу мономеров относятся
акриловая и метакриловая кислоты, хлоракриловая кислота, итако-
новая кислота и ряд других мономеров, содержащих
карбоксильные группы.
В последнее время синтезирован также сополимер акрилонит-
рила с производным стильбендисульфокислоты
C6H5—NHCONH ^ у—СН=СН—^ J NHCONH—С6Н5
\sO3Na NaO3s/
Содержание этого мономера в сополимере составляет 3,3% [38].
Были синтезированы сополимеры акрилонитрила с N-аллиламино-
карбоновыми кислотами с использованием при реакции сополиме-
ризации N-аллилглицина или Ы-аллил-р-аланина [39], а также
сополимер акрилонитрила с N-аллил и Ы-металлил-Ы-метиламино-
сульфонатом [40]. Практическое применение получили и
сополимеры акрилонитрила с винилсульфонатом.
3. Мономеры, содержащие функциональные группы, способные
при последующем взаимодействии с красителями обеспечить
химическое окрашивание волокна и очень высокую прочность окраски.
К таким мономерам относятся, например, и-аминостирол, акролеин
и метакролеин. Например, сополимер акрилонитрила с и-амино-
стиролом содержит ароматическую аминогруппу. Эта аминогруппа
подвергалась диазотированию, а затем проводилось сочетание
полученного диазосоединения с различными азосоставляющими с
образованием полимерного красителя [41].
Ароматическая аминогруппа может быть введена в молекулу
сополимера также путем синтеза сополимера акрилонитрила с
небольшим количеством 4-винилсульфонил-2-аминоанизола [42]. Этот
сополимер имеет следующее строение:
СН2—СН—СН2—СН— • •
CN
SO,
NH,
Для получения интенсивно окрашенного волокна путем
последующего азосочетания достаточно ввести в молекулу сополимера
акрилонитрила 2—3% этого мономера.
Химически окрашенное волокно можно получить также
путем синтеза сополимера акрилонитрила с винилсульфоновыми
214 Глава 6. Производство полиакрилонитрильных волокон
красителями или с водорастворимыми азокрасителями,
содержащими винильную группу и способными к реакции полимеризации
или сополимеризации [43]. Например, при синтезе сополимера,
окрашенного в красный цвет, в качестве красителя применяют
производные гамма-кислоты следующего состава:
ОН
H,C=HCOCHN
SO3H
При этом образуется интенсивно-окрашенный низкомолекулярный
сополимер акрилонитрила. Чем ниже молекулярный вес
сополимера, тем больше красителя вводится в молекулу сополимера.
Изменяя химический состав винилсульфонового красителя, можно
получить сополимеры акрилонитрила, содержащие 0,3—2,5%
красителя и окрашенные в красный, желтый, синий и фиолетовый
цвета. Такой сополимер добавляют в количестве 6—8% к волокно-
образующему полимеру или сополимеру акрилонитрила [44]. При
формовании получается окрашенное волокно, устойчивое к
различным обработкам.
Мономеры, повышающие гидрофильность волокна. Эти
мономеры должны содержать карбоксильные, гидроксильные или
аминогруппы, которые, как уже указывалось выше, одновременно
улучшают окрашиваемость сополимера.
Такие группы содержит сополимер акрилонитрила с винил-
капролактамом [45]
—СН2—СН—СН2—СН—
CN N—(СН2M—СО
Из этих сополимеров было получено волокно, при последующей
обработке которого 1%-ным водным раствором КОН частично
омыляются лактамные циклы и в макромолекуле полимера
появляются аминогруппы и карбоксильные группы (звенья винилами-
нокапроновой кислоты).
Эти реакционные группы обеспечивают также возможность
сшивания полимера, т. е. образования поперечных химических
связей между макромолекулами в результате их взаимодействия в
определенных условиях с бифункциональными соединениями.
Получены волокна из сополимеров акрилонитрила с небольшим
количеством A0—20%) акриловой или метакриловой кислоты [46]. Эти
волокна обладают повышенной гидрофильностью (при
относительной влажности воздуха 65% они сорбируют 3—5% паров воды) и
хорошо окрашиваются основными красителями. Наличие
карбоксильных групп в элементарном звене макромолекулы полимера
дает возможность осуществить сшивание этих волокон действием
б 5. Модифицированные полиакрилонитрильные волокна
215
поливалентных металлов или других бифункциональных
соединений и тем самым дополнительно повысить теплостойкость волокна.
Некоторые данные [47], иллюстрирующие влияние сшивания на
изменение теплостойкости волокон, приведены в табл. 6.2.
Нитрон
А-20*
Капрон
Лавсан
Вискозное
Таблица 6.2. Влияние
сшиваии,я на теплостойкость
полиакрилопитрильного волокна
В л к
Сшитое волокно А-20 **
Температура (в °С), при
которой
волокно сохраняет
№% прочности
164
171
194
222
- 278
288
прочности
122
134
180
206
240
266
* Волокно, полученное из сополимера акрилоннтрила и акриловой кислоты.
•* Волокно А-20, дополнительно обработанное раствором уксуснокислого кальция. При
этом образуются солевые мостики между макромолекулами.
Приведенные в табл. 6.2 данные показывают, что сшитые
волокна из сополимера акрилонитрила, содержащего карбоксильные
группы, значительно превосходят по теплостойкости не только
нитрон, но и гетероиепные синтетические волокна капрон и лавсан
и не уступают по этому показателю одному из наиболее
теплостойких химических волокон — вискозному.
Волокно, обладающее повышенной гидрофильностью и окраши-
ваемостью и способное к последующему сшиванию путем
термической обработки, можно получить также синтезом сополимера
акрилонитрила с небольшим количеством метилолакриламида [48].
Сшивание полиакрилонитрильных волокон возможно
различными способами. Кроме описанного выше метода наибольший
интерес представляют следующие методы.
1. Обработка гексаметилендиамином волокна, полученного из
сополимера акрилонитрила, в макромолекуле которого содержится
небольшое количество глицидилметакрилата [49]. В результате
такой обработки значительно уменьшается усадка волокна при
повышенных температурах. Например, если исходное необработанное
волокно усаживается на 3% при 110 °С, то волокно, содержащее
после указанной обработки 17,5% гексаметилендиамина,
усаживается в такой же степени только после нагревания до 210 °С.
2. Обработка волокна 30—35%-ным водным раствором гидра-
зингидрата с последующим прогревом его в среде азота при
120—160 °С в течение 1—4 ч [50]. После такой обработки,
приводящей к образованию химических связей между макромолекулами,
теплостойкость волокна значительно повышается. Например, если
216 v , Глава 6. Производство полиакрилонитрильных волокон
модифицированное волокно сохраняет при 200°С всего 6%
начальной прочности, то волокно, подвергнутое обработке гидразингидра-
том в указанных условиях, сохраняет при этой температуре 50—
72% начальной прочности.
3. Обработка полиакрилонитрильного волокна в кислой среде
этиленгликолем. В этих условиях нитрильные группы частично
омыляются до карбоксильных, которые реагируют с
этиленгликолем с образованием химических межмолекулярных связей [51].
Волокно, обработанное этиленгликолем, нагревают при 115—150°С.
Характерной особенностью этого метода сшивания является
пониженная электризуемость модифицированного волокна [52].
4. Обработка волокна семикарбазидом, тиосемикарбазидом,
гидроксиламином или сульфидом аммония. В последнем случае в
сополимере появляются тиоамидные группы, которые в результате
последующего взаимодействия также образуют химические связи
между макромолекулами.
Все указанные методы сшивания полиакрилонитрильного
волокна, приводящие к образованию химических связей между
макромолекулами, изменяют его свойства в одном и том же
направлении — значительно повышают теплостойкость, устойчивость
к многократным деформациям при повышенных температурах и
в ряде случаев увеличивают термостойкость волокна. Наибольший
эффект в отношении повышения теплостойкости достигнут при
обработке волокна тиосемикарбазидом. Эта реакция, по-видимому,
протекает по схеме
S
СН2—СН |-H2N—NH—С—NH2 —> СН2—СН
CN C=NH S
I II
NH— NH—С—NH2
Волокно, обработанное этим реагентом, даже при 400°С
сохраняет около 40% начальной прочности.
Сшитое волокно обладает повышенной стойкостью к
химическим реагентам, в частности к минеральным кислотам и щелочам.
Из всех рассмотренных методов повышения теплостойкости и
улучшения других показателей волокна при повышенных
температурах наиболее экономичной и доступной является обработка его
водным раствором сульфида аммония или гидросульфида аммония
NH4HS для введения в макромолекулу небольшого числа тиоамид-
ных групп. Реакция протекает по схеме
/NH4JS
СН2—СН—СН2—СН > СН2—СН—СН2—СН
II II
CN CN CN C=S
1
NHL
—
10
20
85—90
80—82
65—72
6.5. Модифицированные полиакрилонитрильные волокна 217
Мономеры, повышающие эластичность волокон. К числу таких
мономеров относятся эфиры акриловой кислоты, в частности ме-
тилакрилат, а также винилацетат. Однако при введении в состав
сополимера сравнительно небольших количеств винилацетата!
значительно понижается термостойкость волокна и увеличивается его
усадка в горячей воде. Одновременно снижается температура
стеклования волокна, что видно из следующих данных [53]:
„ Содержание Температура стекло-
полимер винилацетата. % вания волокна, °С
Полиакрилонитрил
Сополимер акрилонитрила . .
То же
Волокно из сополимера акрилонитрила и винилацетата,
обладающего повышенной усадочностью при повышенных
температурах, целесообразно использовать для изготовления искусственного
меха.
Мономеры, повышающие реакционную способность сополимера.
К таким мономерам относятся вещества, содержащие
функциональные группы, способные к взаимодействию с различными
соединениями и обеспечивающие возможность последующих
химических превращений сополимера акрилонитрила и получаемых из
него волокон. Из этих мономеров значительный интерес
представляет дикетен
I I
о—с=о
При сополимеризации дикетена с акрилонитрилом получается
сополимер [54], содержащий ?-лактоновую группировку:
сн2=сн СН7С\ ¦•-сн.сн-сн.-с-...
1 + о; ;сн2 —> (JN o( ;сн2
CN \ / N /
С С
о
і
Содержание дикетена в сополимере составляет 5—10%.
Наличие ?-лактоновой группировки дает возможность при последующем
взаимодействии с различными реагентами (например, аммиаком,
гидроксиламином, гексаметилендиамином, тиогликолевой кислотой,
диэтаноламином, гидразином) ввести в макромолекулу сополимера
различные функциональные группы.
Схема возможных полимераналогичных превращений сополимера
акрилонитрила и дикетена приводится ниже.
Как видно из этой схемы, в макромолекулу сополимера акрилонитрила и
дикетена могут быть путем последующих превращений введены аминогруппы,
гидроксильные, карбоксильные и амидные группы. При взаимодействии с
¦218 Глава 6. Производство полиакрилонитрильных волокон
бифункциональными соединениями, например с гексаметилендиамином образуются
химические связи между макромолекулами что приводит к значительному
повышению термостойкости получаемых волокон. Например, температура иулечой
прочности таких волокон составляла 320 вместо 140 С для обычного полиакрило-
нитрильного волокна [55]. Модифицированное волокно окрашивается кислотными
и активными красителями, причем благодаря наличию в нем ?-лактоиовых гр\п-
пировок возможно образование химических связей с красителями, в молекуле
которых содержатся аминогруппы.
Схема возможных полимераналогичных превращений сополимера
акрилонитрила и дикетена
ОН . ОН
I І
—Є—CH.CHCH,— —CHjCHOHj—С
II II
СН2 CN CN СН2
с=о с=о
NH,
NHNH2
ОН
*—СН2СНСН2—С—.» •
CN СН2
с=о
I
NHOH
H2N—NH2
ОН CN
С—СН2СНСН2
СН2 н.\(СН2сн2онJ
| -s
С=О
N(CH2CH2OHJ
NH3
H2N—ОН
L
HS—CH2—СООН
с
А
ОН
СНгСНСНг—С
CN СН2
С=О
Ан
н2№сн2N>;н2> (СН2N
NH
U
CN CH2 CN
СНгСНСН,—С—СН2СН-
ОН
J
S—СН2—СООН
—с—сн2снсн2—
I I
СН2 CN
соон
он
•¦—СН2СНСН2—С—
сн2
1
соон
6.5. Модифицированные полиакрилонитрильные волокна 219
Определенный интерес представляет также синтезированный
японскими исследователями сополимер акрилонитрила,
содержащий 12—15% глицидилметакрилата [56]:
CHS
—сн2—сн—сн2—сн2—с—
CN ОС—ОСН2—НС СН2
V
Наличие высокореакционноспособной эпоксигруппы создает
возможность проведения многих превращений и введения в
макромолекулу сополимера различных функциональных групп. Например,
при взаимодействии этого сополимера с диэтаноламином
получается модифицированный сополимер акрилонитрила, содержащий
гидроксильные группы и обладающий повышенной гидрофиль-
ностью:
CH3 сдон
СН2—СН—СН2—С + HN —>
CN ОС—ОСН2—НС СН3 С2Н4ОН
о
сн3
_ ..-сн^сн-снД-... - н
N ОС—ОСН2— СН—СН3—N
он Чслон
Волокно, полученное из этого модифицированного сополимера
акрилонитрила, поглощает в 2—3 раза больше влаги, чем волокно
из сополимера акрилонитрила с глицидилметакрилатом, и легко
окрашивается кислотными и прямыми красителями. Наличие в
волокне эпоксигруппы дает возможность получать химически
окрашенное волокно, а также сшитое волокно.
Ввести в молекулу новые функциональные группы и нарушить
регулярность строения полимера можно также, частично
осуществляя полимераналогичные превращения полиакрилонитрила. Нит-
рильная группа достаточно реакционноспособна и при действии
тех или иных реагентов может превращаться в различные
функциональные группы. Например, при гидразидировании полиакри-лони-
трильного волокна обработкой его спиртовым раствором
гидразина или гидразинсульфата получено модифицированное волокно,
220 Глава б. Производство полиакрилонитрилькых волокон
макромолекулы которого имеют следующее строение [57]:
NH2—NH2
—сн2—сн—сн2—сн— *- —сн2—сн—сн2—сн—•¦
CN CN NH2NH—C=NH CN
—> • • —Сг^^-СН—СН2—СН— • •
NH2NH—C=O CN
Введение в макромолекулу полиакрилонитрильного волокна
гидразидной группы СО—NH—NH2 и других групп основного
характера придает волокну способность окрашиваться кислотными и
прямыми красителями.
Интересные результаты были получены при формовании
волокна из растворов модифицированного полиакрилонитрила,
содержащего 7% тиоамидных групп. Эти группы вводили в
макромолекулу полиакрилонитрила при обработке его растворами сульф-
гидрата аммония. При формовании волокна из растворов этого
полимера в осадительной ванне, содержащей 50—60% диметил-
формамида, было получено волокно [58], несколько уступающее
полиакрилонитрильному волокну по прочности и по устойчивости
к истиранию, но значительно превосходящее его по устойчивости
к двойным изгибам (в 15 раз) и по относительной прочности в
петле.
Частичная замена нитрильных групп на тиоамидные
значительно улучшает свойства волокон, получаемых на основе сополимеров
акрилонитрила. Например, при частичной замене нитрильных
групп на тиоамидные в макромолекуле волокна санив из
сополимера акрилонитрила и винилиденхлорида (см. разд. 6.5.1.2) резко
повышается температура текучести этого волокна (с 50—70 до
200—250 °С) и его теплостойкость. При 140 °С такие
модифицированные волокна сохраняют 65—75% начальной прочности, в то
время как исходное волокно санив сохраняет всего 5—10%
прочности. Начальный модуль при повышенных температурах
увеличивается в несколько раз. Одновременно заметно повышается
устойчивость волокна к многократным деформациям и к истиранию.
Необходимость использования для формования волокна
значительного числа сополимеров акрилонитрила, заметно не
отличающихся по свойствам и являющихся в основном
взаимозаменяемыми, не является бесспорной, особенно если учесть, что пока не
могут быть достаточно четко сформулированы сравнительные
преимущества и недостатки каждого сополимера и полученных из него
волокон. Разработка преимущественно в США методов получения
целого ряда сополимеров акрилонитрила может быть объяснена
спецификой развития техники в условиях господства
капиталистических монополий; она, по-видимому, была осуществлена главным
образом с целью обойти патенты конкурирующих фирм.
6.5. Модифицированные полиакрилонитрильные волокна 221
Для того чтобы сделать достаточно обоснованный вывод о
наиболее рациональном составе сополимеров акрилонитрила этого
типа, необходимо провести дополнительные исследования.
6.5.1.2. Волокна из сополимеров
с высоким содержанием второго мономера
При получении сополимера акрилонитрила, содержащего 40—
60% второго мономера, значительно нарушается регулярность
строения макромолекул, что приводит к изменению ряда основных
свойств полимера, в частности растворимости. В отличие от поли-
акрилонитрила и сополимеров акрилонитрила, содержащих 85—
90% акрилонитрила, сополимеры с большим количеством второго
мономера растворяются в более доступных растворителях,
например в ацетоне. Благодаря этому значительно упрощается и
удешевляется стоимость производства сополимерного волокна по
сравнению со стоимостью производства полиакрилонитрильного
волокна, хотя при этом несколько снижается теплостойкость этого
волокна.
Из сополимеров акрилонитрила, содержащих значительное
количество второго компонента, применение для производства
волокна получил сополимер акрилонитрила и винилхлорида.
Известный интерес может представить и сополимер акрилонитрила и ви-
нилиденхлорида.
Волокно из сополимера акрилонитрила и винилхлорида.
Опытное производство этого волокна было начато в 1947—1949 гг. Нити,
получаемые из этого сополимера в производственных условиях, в
США называются «виньон Н», а штапельное волокно — «дайнел».
Синтез сополимера акрилонитрила и винилхлорида затрудняется
различной скоростью полимеризации этих мономеров. Винилхло-
рид полимеризуется значительно медленнее, чем акрилонитрил.
Поэтому процесс эмульсионной полимеризации указанных
мономеров проводится в особых условиях. Вначале к винилхлориду
добавляют небольшое количество нитрила акриловой кислоты, а
затем через определенные промежутки времени вводят небольшими
порциями остальное количество акрилонитрила. Иногда
полимеризацию проводят в одну стадию. Но в этом случае в реакционной
смеси содержится значительно больше винилхлорида, чем должно
быть в получаемом сополимере [59].
При содержании от 20 до 60% акрилонитрила сополимер
акрилонитрила и винилхлорида растворяется в ацетоне. Чем больше
содержание акрилонитрила в сополимере, тем выше термостойкость,
светостойкость и прочность получаемого волокна. Поэтому при
производстве волокна из растворов сополимера в ацетоне необходимо
стремиться к тому, чтобы сополимер содержал максимально
возможное количество акрилонитрила,
222 Глава б. Производство полиакрилонитрильных волокон
Волокно из сополимера акрилонитрила и винилхлорида можно
формовать сухим и мокрым способами. В прядильный раствор
добавляют небольшое количество стабилизатора @,5—0,25% дибу-
тиллаурината олова), повышающего светостойкость получаемого
волокна. Так как сформованное волокно в дальнейшем
подвергается значительному вытягиванию (в 8—10 раз), на прядильной
машине получается волокно значительно большей толщины C,33—
2,2 текс), чем это требуется.
Благодаря наличию в макромолекуле волокна виньон H
значительного числа звеньев винилхлорида гигроскопичность этого
волокна еще меньше, чем полиакрилонитрильного. При
относительной влажности воздуха 65% количество влаги, поглощаемой этим
волокном, составляет менее 0,5%. Плотность волокна виньон H
выше, чем полиакрилонитрильного, и составляет 1,28—1,32 г/см3.
Волокна типа виньон H и дайнел используются как для
изготовления изделий технического назначения (спецодежда,
фильтрующие материалы, диафрагмы и т. д.), так и для производства
товаров народного потребления. Ассортимент изделий,
вырабатываемых из этих волокон, в основном совпадает с ассортиментом
изделий из полиакрилонитрильного волокна. В последнее время
волокно типа дайнел получило широкое применение для
производства искусственного меха, используемого, в частности, для
изготовления дамских шуб.
Волокна виньон H и дайнел имеют в технико-экономическом
отношении два существенных преимущества перед полиакрило-
нитрильным волокном. Они вырабатываются из более доступного
и более дешевого сырья, так как примерно около 50% общего
количества акрилонитрила заменяется винилхлоридом, а вместо ди-
метилформамида применяется ацетон. Второе преимущество
заключается в том, что волокна виньон H и дайнел формуют из
растворов полимера в ацетоне, а не из растворов в диметилформами-
де, в результате чего упрощается технологический процесс
получения волокна и регенерация растворителей. Однако это волокно
обладает значительно меньшей термостойкостью, чем полиакрило-
нитрильное.
Волокно из сополимера акрилонитрила и винилиденхлорида.
Вместо винилхлорида при получении ацетонорастворимых
сополимеров акрилонитрила может быть применен винялиденхлорид:
СН,=СН + СН2=СС12 —> СН2~СН—СН,—CCI
І І І
CN CN Cl
При содержании в сополимере 40—60% акрилонитрила он
полностью растворяется в ацетоне.
Применение винилиденхлорида вместо винилхлорида имеет ряд
преимуществ. Скорость полимеризации акрилонитрила и винил-
6.5. Модифицированные полиакрилонитрильные волокна 223
иденхлорида примерно одинакова, и поэтому синтез сополимера
проще, так как в реактор одновременно вводят оба мономера в
тех соотношениях, в которых они должны находиться в
макромолекуле получаемого сополимера. Полимеры и соответственно
сополимеры винилиденхлорида обладают более высокой
термостойкостью, чем сополимеры винилхлорида. Поэтому рабочая
температура, при которой может быть использовано волокно из
сополимера акрилонитрила и винилиденхлорида, на 10—20 °С выше, чем
для волокна виньон Н.
Способ получения волокна из сополимера винилиденхлорида
(при содержании в сополимере 40% акрилонитрила), так
называемого волокна санив, был разработан 3. А. Роговиньш и
3. А. Зазулиной [60]. Волокно санив формовали мокрым способом
в осадительной ванне, содержавшей 3—5% ацетона.
Сформованное и высушенное волокно дополнительно вытягивали на 300—
800%. Большое влияние на свойства волокна санив, как и на
свойства других волокон, оказывает^молекулярный вес исходного
полимера. Оптимальный молекулярный вес сополимера санив,
используемого для получения волокна, составляет 150 000—250 000.
Учитывая доступность винилиденхлорида и указанные выше
технологические преимущества, а также несколько более высокую
термостойкость волокна санив по сравнению с волокном виньон Н,
можно сделать вывод, что если в результате дальнейших
исследований будет установлена целесообразность производства карбо-
цепных волокон из сополимеров акрилонитрила, содержащих
значительное количество второго компонента, то этот вариант
получения ацетонорастворимого сополимера акрилонитрила может
представить практический интерес.
6.5.2. Волокна из привитых сополимеров акрилонитрила
Первая попытка получения разветвленных привитых
сополимеров акрилонитрила и использования их для формования волокна
была сделана Хуньяром и Рейхертом [18]. Они синтезировали
привитые сополимеры полиакрилонитрила с винилацетатом или с
акриловой кислотой и сравнивали волокна, получаемые из этих
сополимеров, с волокнами из сополимеров того же состава,
синтезированных обычными методами эмульсионной полимеризации.
Содержание акрилонитрила в привитых сополимерах
составляло в большинстве случаев 90—95%. В сополимере
поливинилового спирта с акрилонитрилом содержание полиакрилонитрила
составляло от 70 до 95%.
Для прививки в качестве исходного продукта используют
сополимеры акрилонитрила, содержащие небольшое количество реак-
ционноспособных групп (например, сополимер акрилонитрила,
содержащий небольшое количество 4-р-сульфониланизола) [61].
Следовательно, в молекулы сополимера было введено некоторое коли-
224 Глава 6. Производство полиакрилонитрильных волокон
чество ароматических аминогрупп, которые в присутствии солей
ванадия образуют окислительно-восстановительную систему,
позволяющую привить мономер по радикальному механизму. В этих
условиях гомополимер не образуется.
Используя указанную систему, к полиакрилонитрилу
прививали 30—45% полиакриловой кислоты или других полимеров
(полиметил- или полибутилакрилаты). Прививка указанных
полимеров к полиакрилонитрильному волокну увеличивает устойчивость
его к истиранию в 2—5 раз. В качестве исходного продукта для
осуществления процесса прививки были использованы также
сополимеры акрилонитрила с небольшим количеством метакролеина
[62].
Привитые сополимеры акрилонитрила можно получить не
только прививкой различных полимеров к молекуле полиакрилонитри-
ла, но и прививкой значительных количеств полиакрилонитрила к
другому полимеру, в частности к природному полимеру. Так была
осуществлена прививка полиакрилоиитрила к карбоксилметилцел-
люлозе и получен привитой сополимер (содержит 10% карбокси-
метилцеллюлозы и 90% полиакрилонитрила), растворимый в ди-
метилформамиде [63]. При добавлении такого привитого
сополимера к полиакрилонитрилу B5—35% от массы
полиакрилонитрила) и последующем формовании волокна из раствора этой смеси
было получено волокно, гигроскопичность которого (благодаря
наличию небольших количеств гидрофильной карбоксиметилцел-
люлозы) в 10—12 раз превышала гигроскопичность обычного поли-
акрилонитрильного волокна. Это волокно обладало повышенной
устойчивостью к истиранию и к многократным деформациям.
Прививка мономеров к полиакрилонитрилу может быть
осуществлена к исходному полимеру или к готовому волокну.
Вязкость прядильных растворов из привитых сополимеров
акрилонитрила, имеющих разветвленную структуру, значительно
больше вязкости растворов полиакрилонитрила, что затрудняет
их переработку. Этого можно избежать при прививке тех же
полимеров к готовому волокну. Однако в этом случае привитые цепи не
принимают участия в формировании структуры полимера, поэтому
полученные модифицированные волокна обладают более низким
комплексом механических свойств, чем волокна, сформованные из
привитых сополимеров акрилонитрила.
6.5.3. Волокна из смесей полиакрилонитрила
с другими полимерами
Точных данных о производстве полиакрилонитрильных волокон
из смесей полимеров в литературе нет. Имеется только указание
о том, что волокно акрилан получают из раствора двух линейных
сополимеров акрилонитрила. Первый из них — сополимер акрило-
иитрила (95%) и винилацетата E%), второй — сополимер акри-
6.5. Модифицированные полиакрилонитрильные волокна 225
лонитрила E0%) и винилпиридина E0%). Используя в качестве
второго компонента при формовании полиакрилонитрильного
волокна полимеры или сополимеры различного состава, можно в
сравнительно широких пределах изменять свойства получаемых
волокон.
^ Для повышения устойчивости волокна к истиранию и улучше*
нию его эластических свойств к полиакрилонитрилу добавляют
небольшие количества гибкоцепного полимера. Например, при
формовании волокна из смеси полиакрилонитрила и полиметил-
акрилата (в смеси содержалось от 5 до 20% полиметилакрилата с
вязкостью 100000 П A05 Па-с) было получено волокно,
обладающее в 2—3 раза более высокой устойчивостью и к двойным
изгибам, чем волокно, полученное из сополимера акрилонитрила [64].
Смеси полимеров указанного состава в разбавленных растворах
расслаиваются, и только в концентрированных (вязких) прядильных
растворах расслаивание значительно замедляется. Такие растворы
даже при выдерживании их в течение 3—5 дней не расслаиваются.
Хорошие результаты достигаются при формовании волокон из
смесей полимеров или сополимеров акрилонитрила с полидиенами
(с различными каучуками). Например, волокно, полученное из
15%-ного раствора полиакрилонитрила и бутадиен-нитрильного
каучука (при содержании в смеси от 3 до 20% каучука) в диме-
тилформамиде, обладает в 6 раз более высокой устойчивостью
к истиранию и в 15—20 раз большей устойчивостью к
многократным деформациям, чем обычное полиакрклонитрильное волокно
[65]. Наличие в полидиене небольшого количества двойных связей
дает возможность «сшить» полученное волокно и таким образом
повысить его теплостойкость.
Интересные результаты были получены при формовании
волокна из растворов полиакрилонитрила и привитого сополимера
акрилонитрила (8—30%) и ацетата целлюлозы в диметилформ-
амиде [66]. Привитой сополимер содержал 67% полиакрилонитрила
и 33% ацетата целлюлозы; степень полимеризации привитых
цепей полиакрилонитрила 2300. Введение в раствор небольших
количеств привитых сополимеров повысило совместимость
полиакрилонитрила и ацетата целлюлозы в растворе. Сформованное из
растворов в диметилформамиде волокно превосходило (на 30—50%)
по прочности ацетатное волокно, обладало более высокой
устойчивостью к истиранию и выдерживало в 2—4 раза больше двойных
изгибов.
Волокно, полученное из смеси, содержащей 20% привитого
сополимера, имело в 5 раз меньше пор, чем обычное полиакрильное
волокно, причем объем макропор уменьшился в 25 раз. Авторы
работы [66] считают, что уменьшение пористости является основной
причиной повышения комплекса свойств волокна'. Этот фактор до
последнего времени вообще не учитывался при исследовании
структуры и свойств получаемого волокна, _ ¦ - •
8 Зак 234
226 Глава 6. Производство полиакрилонитрильных волокон
Волокна из смеси полимеров и сополимеров акрилонитрила,
значительно отличающихся между собой по температурам
размягчения и по термомеханическим свойствам и соответственно по
степени усадки при повышенных температурах (в частности, при
обработке горячей водой), могут быть использованы для выработки
объемной пряжи.
В заключение необходимо отметить, что существенное
преимущество модификации полиакрилонитрильных волокон
формованием их из смеси полимеров заключается в отсутствии
необходимости синтезировать новые сополимеры акрилонитрила, так как
изменение свойств получаемого волокна в требуемом направлении
достигается в результате изменения количества добавляемых
полимеров, вырабатываемых в промышленных масштабах. В тех
случаях, когда вводимый в состав смеси полимер не совмещается
с гомополимером или сополимером акрилонитрила в
концентрированных растворах, добавляют небольшое количество C—5% от
массы смеси) привитого сополимера акрилонитрила с мономером,
образующим второй полимер, который вводят в состав смеси.
> ЛИТЕРАТУРА
1. Rein I., Z. ges. Textilind., 1954, Bd. 56, N 13, S. 812—815.
2. HautzR., Text. Res. J., 1950, v. 20, N 11, p. 786—790.
3. Промышленность хим. волокон, 1974, № 1, с. 9.
4. Антонов В. Н., Хим. волокна, 1961, № 3, с. 3—6.
5. Полякова 3. П., Хим. пром. за рубежом, 1969, № 12, с. 48—50.
6. Б о г о м о л о в Б. Д., Д о р о х и н а И. С, Клименков В. С, Хим.
волокна, 1962, № 2, с. 14—16.
7. Клименков В. С, Дорохииа И. С, Буреикова В. В. и др., Хим.
волокна, 1966, № 2, с. 18—21.
8. H a m G., Ind. Eng. Chem., 1954, v. 46, p. 391—395.
9. GrizoA., PetorD, OsberH, Chem.-Ztg., 1967, Bd. 68, S. 299—302.
10. -H un jar A., Grobe V., Faserforsch, u. Tcxtiltechn., 1955, Bd. 6, S. 548—552.
11. Циперман В. Л., Е всю ков M. Д., Пакшвер Э. А. и др., В кн.:
Карбоцепные волокна. Под ред. А. Б. Илкшвера. М., «Химия», 1966, с. 265—
270.
12. Koch P., «Fibres», 1955, v. 16, N 9, p. 314—318.
13. Hunjar A., Faserforsch, u. Textiltecha, 1955, Bd. 6, S. 30—34.
14. Кама лов С, Короткое А. А., Красулина В. Н. и др., Хим.
волокна, 1966, № 6, с. 9—12.
15. П а к ш в е р А. Б., Геллер Б. Э. Химия и технология производства
волокна нитрон. М., Госхимиздат, 1960. 143 с.
16. Бе дер H. M., Пакшвер А. Б., Хим. волокна, 1963, № 5, с 9—11
17. Hunjar A., Grobe V., Faserforsch, u. Textiltechn., 1965, Bd. 16, S 496—
498.
18. Hunjar A., Reichert H., Faserforsch, u. Textiltechn., 1956, Bd. 7,
S. 165—169.
19. Hunjar A., Moller W., Faserforsch, u. Textiltechn., 1955, Bd. 6, S. 442—
446.
20. Пакшвер Э. А., Геллер Б. Э., Виноградов Г В, Хим. волокна,
1959, № 2, с. 21—29.
21. lost К-, «Reyon Zellwolle», 1959, Bd. 9, N 1, S. 29—32.
Литература 227
22. Циперман В. Л., Пакт в ер А. Б., Пакт в ер Э. А. В кн.: Кацбо-
цепные волокна. М., «Химия», 1966, с. 158—166.
23. Scherman I., Text. World, 1947, v. 97, p. 101—104; 205—208.
24 Юннцкий В. П., Пакшвер Э. А., Соколова А. П., Хим. волокна,
1970, № 5, с. 11—14.
25 Гришина Т. Я. Мичурина Г. А., Пакшвер Э. А., Хим. волокна,
1959, № 4, с. 3—7.
26. Grobe V., В ies ekc R., Faserforsch, u. Textiltechn., 1969, Bd. 20, S. 30—33.
27. Зверев M. П., Бо рик А. Г., Кострова К. А., Хим. волокна, 1968,
№ 6, с 48—51.
28. Котина Е В., К л имен ко в В. С. «Научно-исследовательские труды
ВНИИВ», 1958, вып. 4, с. 58—62.
29. Геллер Б. Э., Пакшвер А. Б., Хим. волокна, 1959, № 6, с. 16—19.
30. Аш М. А., Геллер Б. Э., Хим. волокна, 1961, № 1, с. 30—34.
31. Котина В. Е., Бунарева 3. С, Косова Р. М., Хим. волокна, 1960,
№ 4, с. 10—12.
32. Котина В. Е., К л и м е н к о в В. С, Демина Н. В. и др., Хим. волокна,
1959, № 1, с. 30—34.
33. Ma k s chin W., Ulbricht I., Faserforscb. u. Textiltechn., 1969, Bd. 20,
с 321 325
34. Ulbricht I, Makschin W., Faserlorsch. u. Textiltechn., 1969, Bd. 20,
S. 170—173.
35. U 1 brich H. M., Melliand Textilber., 1956, Bd. 37, S. 185—189.
36. Ж а р к о в a M. А., Р а с с о л о в а Э. А., Кудрявцев Г. И. и др., Хим.
волокна, 1963, № 2, с. 8—11.
37. Дюрнбаум В. С, Клименков В. С, .Хим. волокна, 1963, № 4, с. 8—11.
38. Бур люк 3. И. Бедер H. M., Крупцов Б. К. В кн.: Карбоцепные
волокна. М., «Химия», 1966, с. 44—47.
39. Reichert H., Grobe V., Faserforsch, u. Textiltechn., 1969, Bd. 20,
S. 317—320.
40. Grobe V., Reichert H., Faserforsch, u. Textiltechn., 1969, Bd. 20,
S. 131—134.
41. Кудрявцев Г. И., Васильева-Соколова Е. А. и др., ЖПХ, 1959,
т. 32, № 11, с. 2594—2597.
42. Пеньков а М. П., Коикин А. А., Хим. волокна, 1965, № 3, с. 12—15.
43. Архипцев В. М., Быков А. Н., Хим. волокна, 1967, № 3, с. 19—22.
44. Пайкачев Ю. С, Быков А. Н., Силантьева В. Г. и др., Хим
волокна, 1970, № 4, с. 18—20.
45. Кудрявцев Г. И., Ра ее олова Э. А., Романова Г. А и др. Хим.
волокна, 1966, № 3, с. 12—16.
46. Васильев Ю. В., Р о г о в и н 3. А., Хим. волокна, 1961, № 6, с. 13—19.
47. Васильев Ю. В., Роговий 3. А., Хим. волокна, 1961, № 4, с. 42—46.
48. К а м а л о в Я. С., Голицин Б. Э., Ростовский Е Н. и др., Хим.
волокна, 1967, № 2, с. 16—18.
49. Кама лов Я. С, Гладкий А. Ф., Иванов H. H. и др., Хим волокна,
1967, № 3, с. 21—23.
50. Р о м а н о в а Т. А., Жаркова М. А., Кудрявцев Г. И. и др., Хим.
волокна, 1968, № 5, с. 23—26.
51. Fester W., Melliand Textilber., 1969, Bd. 45, S. 1869—1372.
52. Я сни ко в Л. Я., Пакшвер Э. А., Хим. волокна, 1968, № 4, с. 54—56.
53. Дорохииа И., Клименков В., Абкин А., Хим. волокна 1962 № 2,
с. 5—8.
54. Габриелян Г. А., Роговин 3. А., Хим. волокна, 1963, № 5, с. 2—5.
53. Габриелян Г. А., Станченко Г. И. .Роговий З А Хим волокна
1965, № 1, с. 13—16. ...
56. Iwakura I., Kurosaki T., Nakabayasche N., Makromol Chem.,
1961, v. 44—46, p. 570—572.
57. Кудрявцев Г. И., M a т я ш Т. А., Жаркова M. A. и др., Хцм, волокна,
1961, № 4, с, 13—15.
228 Глава 6. Производство полиакрилонитрильных волокон
58. Шал аби С. Э., Габриелян Г. А., Конкин А. А., Хим. волокна, 1970,
№ 6, с. 68—71.
59. Koch P., «Fibres», 1955, v. 16, p. 174—177.
60. Роговин 3. А., З азу лин а 3. А., Текст, пром., 1955, № 2, с. 18—22.
61. Пеньков а М. П., С к о р о б о г а т о в а Л. Ф., Конкин А. А., Хим.
волокна, 1968, № 5, с. 18—21.
62 Эль-Гарф С, Станченко Г. И., Конкин А. А. и др., Хим. волокна,
1968, № 5, с. 5—8.
63. M и г р а н я н Т., П е н ь к о в а М. П., Лившиц Р. М. и др., Хим. волокна,
1970, № 2, с. 25—29.
64. Кр а со вс к а я С. Б., Зазулнна 3. А., Конкин А. А., Хим. волокна,
1970, № 2, с. 19—22.
65. П е т р о с »н В. А., Габриелян Г. А., Роговин 3. А., Хим. волокна,
1971, № 1, с. 20—23.
66. 3 а в ь я л о в а 3. А., Петрова Н. П., П а к ш в е р Э. А., Хнм. волокна,
1968, № 5, с. 20—23.
Глава 7
ПРОИЗВОДСТВО ПОЛИВИНИЛХЛОРИДНЫХ
волокон
К поливинилхлоридным волокнам относятся волокна,
полученные из полимеров и сополимеров винилхлорида — поливинилхло-
ридные (ровиль, термовиль), волокна из хлорированного поли-
винилхлорида (хлорин, ПЦ), из сополимеров винилиденхлорида
(совиден, саран) и др. Эти волокна представляют определенный
интерес благодаря ряду ценных свойств (высокой хемостойкости,
малой теплопроводности и др.) и, главным образом, доступности и
дешевизне сырья. Исходным сырьем для производства
винилхлорида являются ацетилен и этилен.
Из ацетилена винилхлорид получают путем непосредственного
взаимодействия СгН2 с хлористым водородом:
CH^CH+HCl —> СН2=СНС!
в жидкой или паровой фазе.
Наиболее часто применяется парофазное гидрохлорирование
ацетилена, которое проводится при 120—180°С над
катализатором— активированным углем, пропитанным раствором сулемы.
Получаемый продукт содержит свыше 99,9% винилхлорида.
Из этилена винилхлорид получается в две стадии. При
взаимодействии этилена с хлором образуется дихлорэтан:
СН2=СН2 + С12 —> СН2С1—СН2С1
Путем термического дегидрохлорирования, осуществляемого
при 500—600°С и давлении 10 кгс/см2 A-Ю6 Па) на
активированном угле в присутствии катализатора, получается винилхлорид:
СН2С1—СН2С1 —у СН2=СНС1 + НС1
Этот способ может быть экономичным при условии
рационального применения выделяющегося при реакции НС1. Последний
можно использовать для взаимодействия с ацетиленом и
образования винилхлорида по приведенной выше схеме. Однако
осуществление этого способа, при котором одновременно используются
этилен и ацетилен, представляет определенные трудности. Поэтому
более приемлемым является способ окислительного
гидрохлорирования этилена с образованием дихлорэтана:
СН2=СН2 + 2НС1 + [О] —>¦ CjH4Cl2 + Н2О
Реакция осуществляется в газовой фазе при 250—300 °С.
Катализаторами служат металлы переменной валентности Си, Fe, Cr.
230 Глава 7. Производство поливинилхлоридных волокон
Дихлорэтан подвергается дегидрохлорированию по приведенной
выше схеме, а выделяющийся НС1 используется в качестве гидро-
хлорирующего агента для синтеза дихлорэтана по реакции
окислительного гидрохлорирования.
Перспективным и экономичным методом синтеза винилхлорида
является метод прямого хлорирования этилена [1]. Для этого
применяется смесь этилена и хлора в соотношении 5: 1. Температура
реакции 400—500°С. Так как в данном случае отпадает
необходимость дегидрохлорирования дихлорэтана, естественно, что
указанный метод получения винилхлорида должен быть более
экономичным.
. 7.1. ПОЛИВИНИЛХЛОРИДНЫЕ ВОЛОКНА
В промышленности поливинилхлорид получают эмульсионной
полимеризацией хлористого винила:
СНа=СНС1 —>- СН2—СНС1—СН2—СНС1
В состав реакционной смеси входит вода, эмульгатор
(ализариновое или триэтаноловое мыло, некаль, натриевая соль изобутил-
нафталинсульфокислоты и др.), инициатор (перекись бензоила, па-
рофор N—динитрил азодиизомасляной кислоты, персульфат
калия, перекись водорода), стабилизатор (поливиниловый спирт) и
регулятор (фосфорная кислота и ее соли).
Принципиально полимеризацию винилхлорида можно
проводить в растворе (лаковым методом) аналогично тому, как это
имеет место при синтезе других карбоцепных волокнообразующих
полимеров (например, полиакрилонитрила). Полимеризация
винилхлорида в растворителе, например в тетрагидрофуране или в
диметилформамиде, и использование образовавшегося
концентрированного раствора непосредственно для формования волокна
представляют определенный практический интерес.
Поливинилхлорид, обладающий сравнительно высокой
степенью стереорегулярности, удалось получить методом радикальной
полимеризации при низких температурах [2]. Так же как и для
других полимеров, в частности полидиенов, получаемых методом
радикальной полимеризации, стереорегулярность синтезируемого
поливинилхлорида тем выше, чем ниже температура
полимеризации. Температура стеклования так называемого
низкотемпературного поливинилхлорида равна 100°С вместо 80—82 °С для
полимера, получаемого обычным методом суспензионной
полимеризации при повышенных температурах. Благодаря более высокой
степени стереорегулярности низкотемпературный поливинилхлорид
имеет более высокую плотность, чем обычный поливинилхлорид
A,442 вместо 1,408 г/см3). Усадка волокна, получаемого из
низкотемпературного поливинилхлорида, при повышении температуры
7.1. Поливинилхлоридные волокна 231
значительно меньше (в 3—4 раза), чем волокна из обычного
суспензионного поливинилхлорида.
Однако использовать низкотемпературный поливинилхлорид для
формования волокна трудно, так как концентрированные растворы
этого полимера в диметилформамиде малоустойчивы даже при
повышенных температурах и быстро желатинируются.
Стереорегулярный поливинилхлорид синтезирован путем
полимеризации хлористого винила в присутствии триалкилалюминия
и ТіС13. Этот полимер значительно отличается по свойствам от
поливинилхлорида, получаемого методом радикальной
полимеризации. Например, стереорегулярный поливинилхлорид плавится
при 130—150 °С и разлагается при 180—190 °С [3]. Если эти данные
правильны, то появляемся принципиальная возможность
формования поливинилхлоридного волокна из расплава. Стереорегулярный
поливинилхлорид не растворяется ни в одном из известных
растворителей. Поэтому возможность формования волокна из растворов
Данного полимера пока исключается.
Поливинилхлорид получил широкое применение во многих
отраслях промышленности, где он перерабатывается различными
методами. Применение этого наиболее доступного полимера,
обладающего рядом ценных свойств, для производства синтетических
волокон затрудняется его ограниченной растворимостью в
доступных растворителях. Поэтому, несмотря на заманчивость
предложения о формовании волокон из поливинилхлорида, высказанного
еще в 1913 г. Клатте (Германия) [4], способ в течение длительного
времени не находил применения в промышленности.
Впервые волокно из поливинилхлорида было получено в 1930 г.
Хубертом (Германия) мокрым способом формования из растворов
полимера в циклогексаноне в осадительную ванну, содержащую
30% уксусной кислоты [4]. Однако этот способ оказался
экономически неприемлемым и не получил практического применения.
В дальнейшем метод производства волокна из
поливинилхлорида разрабатывался во Франции, где в 1941 г. Гребе был
предложен новый способ получения этого волокна.
Начиная с 1951 г. поливинилхлоридное волокно производится
в промышленном масштабе под названием «ровиль». Волокно
ровиль формуют сухим способом из растворов поливинилхлорида в
смеси ацетона и сероуглерода (при соотношении этих
растворителей 1:1). При этих соотношениях ацетон и CS2 образуют
азеотропную смесь, кипящую при 37 °С. В Японии в качестве
растворителя поливинилхлорида используется смесь ацетона и бензола в
соотношении 1 : 1.
В 1953 г. волокно из поливинилхлорида начали вырабатывать
в производственных условиях в ФРГ, а также в Италии (волокно
мовиль). В Японии поливинилхлоридное волокно выпускается под
названием «тевирон». В СССР это волокно вырабатывается пока
в опытно-промышленном масштабе.
232 Глава 7. Производство поливинилхлоридных волокон
Технологическая схема производства поливинилхлоридного
волокна принципиально не отличается от схемы получения других
карбоцепных волокон. Поливинилхлорид, используемый для
формования волокна, имеет сравнительно высокий молекулярный вес.
Степень полимеризации этого полимера составляет от 1000 до
2500, что соответствует молекулярному весу от 60 000 до 150 000 [5].
В зависимости от применяемого метода формования волокна
для получения прядильных растворов в промышленности
используются растворители двух типов: 1) смесь ацетона и сероуглерода
(или бензола) — при сухом способе формования; 2) тетрагидрофу-
ран — при мокром.
Тетрагидрофуран стоит значительно дороже, чем смесь ацетона
и сероуглерода. Кроме того, он также токсичен и действует
раздражающе на кожу человека.
В качестве растворителя поливинилхлорида при формовании
волокна (особенно штапельного) мокрым способом советскими
исследователями предложен диметилформамид [6]. Этот
растворитель, широко используемый при производстве полиакрилонитриль-
ного волокна, имеет ряд существенных технико-экономических
преимуществ перед взрывоопасными смесями растворителей,
применяемых для формования волокна сухим способом (смесь ацетона
с сероуглеродом или бензолом) и тетрагидрофураном. Основными
преимуществами диметилформамида как растворителя являются
более низкая вязкость получаемых прядильных растворов [7] и
меньшая токсичность. Промышленное производство
поливинилхлоридного волокна этим способом намечается осуществить в СССР
в ближайшие годы. Необходимо учитывать, что
концентрированные растворы поливинилхлорида в диметилформамиде образуются
при повышенных температурах F0—70°С). При понижении
температуры и длительном хранении этих растворов образуются гели.
Желатинирование прядильных растворов происходит тем быстрей,
чем ниже температура, выше концентрация полимера в растворе
и чем выше молекулярный вес поливинилхлорида [8].
Температура растворения поливинилхлорида в
диметилформамиде определяет в значительной степени структуру образующегося
прядильного раствора, а следовательно, и свойства получаемого
волокна. Например, при повышении температуры растворения с 80
до ПО °С прочность волокна повышается с 25 до 31 гс/текс; число
двойных изгибов увеличивается в 2 раза. Дальнейшее повышение
температуры растворения до 125 °С приводит к ухудшению свойств
волокна [9]. Наличие в прядильном растворе до 15% воды
значительно (в 5—6 раз) уменьшает число двойных изгибов,
выдерживаемых волокном до разрушения, по сравнению с аналогичным
показателем для волокна, сформованного из прядильного раствора,
содержащего менее 0,5% воды.
Существенное влияние на свойства волокон оказывает
концентрация полимера в прядильном растворе. При повышении концен-
7.1. Поливинилхлоридные волокна 233
трации поливинилхлорида в растворе с 19,6 до 27,5% и мокром
способе формования повышается прочность волокна с 18,4 до
27,0 гс/текс A84—270 мН/текс) и число двойных изгибов (в 3—
3,5 раза). Одновременно снижается удлинение волокна. При
снижении концентрации полимера в растворе до 16,5% волокна
склеиваются между собой в момент формования [10].
Принципиально волокно можно получить из поливинилхлорида,
находящегося в термопластичном состоянии (в присутствии
небольших количеств растворителя или пластификаторов). Для этой
цели необходимо применять хорошо стабилизированный поливи-
нилхлорид, который не разлагается при непродолжительном
нагревании до 150—200 °С.
Сухим способом формования из поливинилхлорида
вырабатывается нить и штапельное волокно во Франции, ФРГ, Италии и
Японии. Формование волокна осуществляется на машинах,
применяемых для получения ацетатного волокна.
При формовании поливинилхлоридного волокна сухим
способом вместо прядильных растворов применяются
высококонцентрированные гели, которые подаются на прядильную машину при
высоком давлении 50—100 кгс/см2 E-Ю6—107 Па). В этом
заключается основное отличие процесса формования сухим способом
поливинилхлоридного волокна от формования этим способом
других видов карбоцепных волокон.
Сформованное поливинилхлоридное волокно обладает
невысокой прочностью. В результате вытягивания при повышенной
температуре на 200—800% и последующей термообработки
механические свойства его значительно улучшаются.
Нить и штапельное волокно, полученные сухим способом
формования, после вытягивания подвергают термообработке сначала
в кипящей воде под натяжением, а затем в условиях,
обеспечивающих возможность некоторой усадки волокна. В результате этой
обработки температура размягчения и начальная температура
усадки повышаются на 25—30 °С. Оптимальная температура
термообработки 120 °С. Волокно, обработанное 10—20 мин под
натяжением в указанных условиях, не усаживается в воде при 80—
100 °С [11].
Штапельное волокно целесообразно получать мокрым способом
формования, особенно при использовании в качестве растворителя
диметилформамида. Формование волокна из растворов полимера
в этом растворителе производится в осадительной ванне,
содержащей 80—85% диметилформамида и 15—20% воды. Волокна,
полученные в ваннах, содержащих 30—50% воды, вследствие жестких
условий высаживания полимера не способны вытягиваться и
обладают низкой прочностью [12].
Штапельное волокно, полученное мокрым способом, после
вытягивания заметно не отличается по свойствам от волокна,
сформованного сухим способом. Особенностью волокна, полученного
234 ,. Глава 7. Производство поливинилхлоридных волокон
мокрым способом формования является более низкое удлинение
A6-18%).
Специфическими преимуществами поливинилхлоридного
волокна являются:
высокая хемостойкость, превышающая стойкость большинства
природных и химических волокон;
негорючесть и невоспламеняемость;
очень низкая теплопроводность;
высокие электроизоляционные свойства;
высокая устойчивость к истиранию [13];
высокая светостойкость, приближающаяся к светостойкости по-
лиакрилонитрильного волокна.
Светостойкость поливинилхлоридных волокон зависит от
содержания низкомолекулярных фракций и железа. Чем больше
содержится этих фракций и чем выше концентрация железа в волокне
(более 0,005%), тем ниже светостойкость волокна. Волокно,
полученное из диметилформамидных растворов поливинилхлорида,
менее стойко к фотохимической деструкции, чем волокно,
сформованное из растворов полимера в тетрагидрофуране или в цикло-
гексаноне [14]. Для выяснения причин этого явления необходимо
провести дополнительные исследования.
Наиболее эффективным светостабилизатором
поливинилхлорида, повышающим его стойкость к фотохимической деструкции,
является окись дибутилолова.
Наряду с перечисленными преимуществами поливинилхлорид-
ные волокна имеют существенные недостатки. Волокно начинает
усаживаться при 70—75 °С, что, естественно, исключает
возможность кипячения или крашения изделий из поливинилхлоридного
волокна при повышенной температуре. Этот недостаток в
известной степени устраняется при производстве волокна из полимера,
обладающего более высокой степенью стереорегулярности, а
также при более высокой степени вытягивания волокна.
Необходимо отметить, что усадка волокна при повышенных
температурах для некоторых изделий, например искусственного меха
с подпушком, войлока и др., не только не недостаток, но в
известной степени преимущество этого волокна.
Поливинилхлоридное волокно плохо окрашивается из-за того,
что оно не набухает в воде и трудно перерабатывается вследствие
легкой электризуемости. Волокно обладает также низкой
стойкостью к у-лучам. После такой обработки даже при пониженных
температурах сильно снижается его прочность и удлинение [15].
Поливинилхлоридное волокно используется для производства
разнообразных изделий технического назначения, для которых
указанные выше преимущества имеют существенное значение. Это
волокно в чистом виде или в смеси с другими волокнами начинают
применять и для изготовления некоторых изделий народного
потребления, которые не подвергаются кипячению в воде (например,
1.2. Йерхлорваниловые волокна 235
купальные костюмы, пуловеры, дамские шубки, ковры, гардины
и т. д.) [16]. При добавлении 30—50% волокна термовиль к шерсти
заметно повышается устойчивость тканей к истиранию и
одновременно улучшаются их теплоизоляционные свойства. Прочность
таких тканей по сравнению с чистошерстяными заметно не
изменяется [17]. Благодаря наличию трибоэлектрического эффекта
белье из поливинилхлоридного волокна, так же как и из ряда
других гидрофобных синтетических волокон, в частности из хлорина
(хлорированного поливинилхлорида), начали применять для
лечения радикулита.
Низкая стоимость исходного полимера и сравнительная
простота технологического процесса привели к тому, что поливинил-
хлоридное волокно стало одним из наиболее дешевых и
доступных видов синтетических карбоцепных волокон. Если удастся
преодолеть затруднения, связанные с применением смеси
растворителей, в состав которой входит сероуглерод, и реализовать метод
формования штапельного волокна из растворов поливинилхлорида
в диметилформамиде, то этот вид карбоцепных волокон получит
широкое промышленное применение.
7.2. ПЕРХЛОРВИНИЛОВЫЕ ВОЛОКНА
Несмотря на то что полнвинилхлорид является одним из
наиболее доступных карбоцепных полимеров, растворимость его
только в ограниченном числе растворителей и связанные с этим
трудности в процессе формования обусловили многочисленные попытки
получить волокна из полимеров, обладающих в основном теми же
свойствами, что и поливинилхлорид, но в отличие от него
растворимых в доступных растворителях.
Одним из способов модификации поливинилхлорида для
улучшения его растворимости является дополнительное хлорирование
этого полимера. Из хлорированного поливинилхлорида —
перхлорвинила — получают модифицированное волокно хлорин.
С увеличением до определенного предела содержания хлора в
макромолекуле поливинилхлорида и, следовательно, с понижением
регулярности строения макромолекул уменьшается интенсивность
межмолекулярного взаимодействия и соответственно изменяется
комплекс свойств полимера аналогично тому, как это имеет место
при получении сополимеров нерегулярного строения. С
повышением содержания хлора до 63—65% увеличивается растворимость
полимера в доступных растворителях, в частности в ацетоне,
снижается температура размягчения и уменьшается вязкость экви-
концентрированных растворов полимера [18, с. 28].
При дальнейшем увеличении количества хлора растворимость
хлорированного поливинилхлорида в ацетоне снижается;
продукты, содержащие 72—73% хлора, уже не растворяются в
ацетоне.
236 Глава 7. Производство поливинилхлоридных волокон
На растворимость перхлорвинила в ацетоне влияет не только
содержание хлора, но и равномерность его распределения в
макромолекулах полимера. Многочисленные попытки хлорирования по-
ливинилхлорида в водной суспензии, в которой поливинилхлорид
не набухает, не привели к положительным результатам.
Получающийся при этом продукт, содержащий 64—65% хлора, не
растворяется в ацетоне, что объясняется неравномерностью
хлорирования в таких условиях. Сравнительно равномерно хлор
распределяется в молекуле полимера только при хлорировании в растворе.
Поэтому дополнительное хлорирование поливинилхлорида для
получения полимера, растворимого в ацетоне, всегда проводят в
растворе. В качестве растворителей применяют вещества, полностью
растворяющие поливинилхлорид и продукты его хлорирования и
практически не взаимодействующие с хлором. Наиболее
приемлемыми растворителями являются тетрахлорэтан и хлорбензол.
Хлорирование проводится в течение 24—40 ч при 80—120°С. Для
удаления НС1, выделяющегося в процессе реакции, через раствор
продувают азот. При хлорировании поливинилхлорида в тетра-
хлорэтане (ГДР) по окончании реакции раствор охлаждают до
температуры от —10 до —20 °С и затем высаживают полимер из
раствора, добавляя метанол. Высаженный полимер
отфильтровывают и сушат во вращающемся барабане под вакуумом в течение
24 ч при 70 °С.
Продолжительность хлорирования поливинилхлорида в
хлорбензоле 7—9 ч. По окончании хлорирования перхлорвинил
высаживают из раствора горячей водой. Хлорбензол отгоняется с
водой в виде азеотропной смеси, кипящей при 92 °С. Перхлорвинил,
высаженный из раствора органическим разбавителем, а не водой
содержит меньше низкомолекулярных фракций полимера и других
примесей, растворимых в органических растворителях.
Существенным недостатком этого способа является образование в качестве
побочного продукта полихлорбензолов, обладающих неприятным
запахом. Эти вещества прочно удерживаются полимером, не
удаляются в процессе получения волокна и его отделки и, оставаясь
в волокне, обусловливают неприятный запах волокна и
получаемых из него изделий.
Механизм реакций, протекающих при дополнительном
хлорировании поливинилхлорида, не вполне выяснен. По-видимому, в
первую очередь(хлор замещает водород в группе СН2,
находящейся в а-положенйи к группе СНС1. Одна из возможных схем
строения перхлорвинила:
—сн2—сна—сна—сна—
Неограниченная растворимость дополнительно хлорированного
поливинилхлорида в ацетоне и в других доступных растворителях
обусловила целесообразность использования этого полимера
вместо поливинилхлорида для производства синтетических волокон."
7.2. Перхлорвиниловые волокна ¦ 237
Однако этот метод не получил и, по-видимому, не получит в
дальнейшем широкого распространения. Это объясняется, в частности,
тем, что перхлорвинил примерно в 2 раза дороже поливинилхло-
рида, и поэтому себестоимость волокна хлорин на 30—60% выше
поливинилхлоридного [7] при примерно одинаковых свойствах
указанных волокон.
Волокно из перхлорвинила, содержащего 63—65% хлора,
вырабатывается в сравнительно небольших количествах в ГДР
(волокно ПЦ) и в Советском Союзе (волокно хлорин).
Перхлорвинил производится обычно на заводах,
вырабатывающих поливинилхлорид. На завод синтетического волокна
поступает уже высушенный хлорированный поливинилхлорид. Этот
полимер используется для производства нити и штапельного
волокна. К перхлорвинилу, предназначенному для производства волокна,
предъявляются следующие требования:
Содержание хлора, % 63,0—65,0
Зольность, % 0,03—0,08
Содержание железа, %, не более 0,001
Удельная вязкость 0,2%-ного раствора
полимера в циклогексаноне 0,2
Степень полимеризации 900—1000
Небольшие количества железа резко понижают растворимость
перхлорвинила и стабильность получаемых растворов. Наличие
даже 0,005% железа в растворе перхлорвинила может привести
к значительному повышению вязкости концентрированных
растворов, а при длительном выдерживании — к образованию
нерастворимого геля. Механизм действия таких небольших добавок железа
не выяснен.
При повышении степени полимеризации полимера с 420 до
1150 прочность волокна, получаемого в одних и тех же условиях,
увеличивается в 1,8 раза, а число двойных изгибов,
выдерживаемых волокном, более чем в 13 раз [19]. Увеличение содержания
низкомолекулярных фракций в перхлорвиниле (так же как и в
других полимерах) приводит к значительному снижению
прочности и удлинения, а также к уменьшению числа двойных изгибов,
выдерживаемых волокном до разрушения.
Технологический процесс производства волокна хлорин
достаточно прост и не отличается от процесса получения других карбо-
цепных волокон.
Для получения прядильного раствора перхлорвинил растворяют
при нормальной температуре в сухом ацетоне. При наличии
небольшого количества воды @,8—1%) растворимость полимера
снижается. Поэтому тщательное высушивание полимера и
обезвоживание ацетона является необходимым условием для получения
концентрированного прядильного раствора. Для формования
волокна применяют 25—34%-ный раствор перхлорвинила в ацетоне.
238
Глава 7. Производство поливинилхлоридных волокон
Вода
¦5
і
Раствор подготавливают к формованию так же, как и при
получении ацетатного волокна.
Полученный концентрированный прядильный раствор
подвергается трехкратной фильтрации, отстаивается для удаления
пузырьков воздуха и поступает на
прядильную машину. Формование
волокна хлорин может
осуществляться мокрым.л сухим
способами. Соответственно
различается состав прядильного
раствора. Для формования волокна
мокрым способом применяют
раствор с вязкостью 50—100 с, а
концентрация полимера в растворе
составляет 25—30%. При сухом
способе формования вязкость
раствора повышают до 300—500 с,
а концентрацию полимера в
растворе—до 32—34%.
Волокно хлорин получают
мокрым способом формования. В
качестве осадительной ванны
применяется разбавленный раствор
ацетона в воде. Основное
количество ацетона из осадительной
ванны регенерируется
ректификацией отработанной
водно-ацетоновой смеси. Чем выше
концентрация ацетона в отработанной
ванне, тем меньше объем ванны,
поступающей на ректификацию, и
тем меньше расход пара при
ректификации. Однако при
увеличении концентрации г жетона
возрастает количество р.э.'. "зорителя,
уносимого волокном, и повышаются его потери в результате
испарения с поверхности ванны.
Максимально допустимое содержание ацетона в осадительной
ванне различно при формовании текстильной нити и штапельного
волокна. При формовании нити концентрация ацетона в ванне
составляет 3,5—4%. При формовании штапельного волокна
содержание ацетона в осадительной ванне может быть увеличено до
8—10%. Во избежание значительного испарения ацетона из ванны
формование волокна хлорин проводится при 20°С.
Аппаратурное оформление процесса формования волокна
хлорин мокрым способом имеет существенные особенности. Путь нити
в ванне достигает 250 см, поэтому формование проводится не в го-
вода
Яря$ш?ьный
' раствор
Рис. 7.1. Схема формования нити
хлорин мокрым способом:
/—червяк; 2—гидрозатвор; 3—заправочный
кран; 4—напорная труба; 5—форсунка;
6 —фильера; 7 —корпус шахты; в—трубка;
9, 10 — соответственно верхний и нижний
прядильные диски; // — замасливающая
шайба; /2—приемные бобины.
7.2. Перхлорвиниловые волокна 239
ризонтальной ванне, а в вертикально установленной отдельно для
каждого прядильного места стеклянной трубке высотой 2—2,5 м
(рис. 7.1). При повышении температуры формования свыше 28—
30 °С снижается прочность и удлинение волокна.
Выходящая из трубки через воронку нить принимается на
верхний прядильный диск 9, затем вытягивается между верхним и
нижним прядильными дисками на 130—150%, замасливается и
принимается на бобину*
При обеспечении требуемой длины пути нити в ванне нить хлорин
может быть получена и на прядильных машинах других типов,
применяемых для формования химических волокон мокрым способом.
Волокно на бобине содержит 4% ацетона и 30% воды (от
массы сухого волокна). Бобина с текстильной нитью выдерживается
в цехе 24 ч для удаления ацетона (остается 0,2% ацетона от
массы волокна) и затем сушится 30—40 ч при постепенно
повышающейся в различных зонах сушилки температуре (от 35 до
60°С). В процессе сушки происходит частичная терморелаксация
волокна. Очевидно, что для удаления легколетучего растворителя
такой длительной сушки не требуется, и продолжительность этой
операции может быть значительно сокращена.
Высушенная нить подвергается кручению с одновременным
замасливанием и затем перематывается на шпули крестовой
намотки. Для препарации нити применяются гидрофильные вещества,
нанесение которых на нить уменьшает ее электризуемость
(глицерин, поливиниловый спирт и другие вещества, содержащие группы
ОНиСООН).
Вытекающая из трубки отработанная осадительная ванна,
содержащая 4—10% ацетона, поступает на ректификацию или
используется после разбавления для промывки свежесформованного
штапельного волокна (жгута), выходящего из ванны.
Скорость формования нити хлорин мокрым способом не
превышает 30—40 м/мин.
Волокно хлорин можно дополнительно вытягивать на 300—
700%. Однако перхлорвиниловая нить, вырабатываемая в
Советском Союзе и в ГДР, не подвергается дополнительному
вытягиванию при повышенных температурах, как это имеет место при
производстве других карбоцепных волокон. Вследствие этого физико-
механические показатели ее значительно ниже, чем для нитей из
других карбоцепных полимеров.
Штапельное волокно хлорин также получают мокрым способом.
Основные отличия этого процесса от формования комплексной нити
заключаются в следующем:
концентрация ацетона в отработанной осадительной ванне
повышается до 8—10%;
выходящий из осадительной ванны жгут обрабатывают 1 —
2%-ным водным раствором ацетона для удаления основного
количества растворителя, удержанного волокном;
240
Глава 7. Производство поливинилхлоридных волокон
температура осадительной ванны (вода) 13—15 °С; при
повышении температуры коагуляция полимера из прядильного раствора
происходит неравномерно.
Рнс. 7.2. Прядильная машина для формования
штапельного волокна хлорин мокрым способом.
Для формования штапельного волокна применяют фильеры с
2000—3000 отверстиями. Однако такое число отверстий не
является оптимальным. Учитывая опыт формования карбоцепных
штапельных волокон других видов, число отверстий в фильере
целесообразно увеличить минимум до 10 000—15 000. Скорость формо-
рания штапельного волокна составляет 15—20 м/мин.
7.2. Перхлорвиниловые волокна
241
Машина для формования штапельного волокна хлорин и ГШ
мокрым способом показана на рис. 7.2. По конструкции машины
для формования штапельного волокна хлорин существенно
отличаются от машин, применяемых для формования мокрым способом
других химических волокон. На машине имеется 6 радиально
расположенных секций. Из каждой секции выходит жгут, содержащий
// Ю
т
Рис. 7.3. Схема последующей обработки хлоринового штапельного волокна:
/ — секция прядильной машины; 2 — общий жгут; 3, 4, 5, 6— промывные ванны; 7 —ванна для
замасливания волокна; S — триовальцы; 9— компенсатор натяжения; 10— гофрировочиая
машина; Л —приемный ящик; 12— питающие валики; 13 — узлоуловитель; 14—резательная
машина; 15—сушилка; 16— разрыхлитель; 17— пневмотранспортер; IS—пресс.
30 000—32 000 волокон. Жгуты отдельных секций соединяются в
общий жгут, состоящий из 200 000 волокон. При такой конструкции
прядильной машины длина пути нити в ванне для волокон,
выходящих из каждой фильеры, одинакова. Каждая секция капсулиро-
вана во избежание испарения ацетона и имеет местный отсос. На
каждую фильеру подается 40 л/ч прядильной воды, для того чтобы
концентрация ацетона в воде, вытекающей из.машины, составляла
9—10%. Использование этой машины для формования
штапельного волокна других видов в ряде случаев может представить
практический интерес.
Общий жгут 2 (рис. 7.3) передается в первую промывную
ванну 3 и поступает на первые триовальцы 8. В промывных
ваннах 3—6 волокно отмывается от ацетона. В ванне 7 жгут
обрабатывается замасливающей эмульсией и вальцами через рифленые
алюминиевые цилиндры и компенсатор натяжения 9 подается на
гофрировочную машину 10. После резки волокно сушится,
разрыхляется и упаковывается.
242 Глава 7. Производство поливинилхлоридных ^ "»со«
В промывных ваннах вода циркулирует по принципу
про;.г;потока. Вытекающая из первой промывной ванны вода содержит
0,3% ацетона и используется в качестве осадительной ванны.
Четвертая (последняя) промывка проводится теплой водой E0°С) для
удаления следов ацетона.
В первой зоне сушилки температура составляет 35—40 °С, во
второй зоне, куда поступает волокно, из которого полностью
удален ацетон, температура повышается до 60—70°С.
По сравнению с мокрым способом формования комплексной
нити хлорин сухой способ имеет ряд преимуществ, в частности
значительно повышается скорость формования (в 5—8 раз) и
отпадает необходимость сушки нити. Однако сухой способ формования
нити хлорин, обладающей такими же показателями, как
получаемая мокрым способом, пока не разработан.
Волокно хлорин, полученное мокрым способом, характеризуется
следующими показателями.
Прочность волокна хлорин в сухом состоянии составляет
13—15 гс/текс, удлинение — 30—40%. При вытягивании свеже-
сформованного волокна прочность его может быть увеличена до
25—30 гс/текс.
Хлориновое волокно гидрофобно и при нормальной влажности
воздуха поглощает не более 0,1—0,15% влаги, поэтому в мокром
состоянии его прочность и удлинение не изменяются.
Волокно хлорин обладает высокой стойкостью к большинству
химических реагентов (в частности, разбавленных и
концентрированных кислот и щелочей), а также к действию микроорганизмов.
По хемостойкости волокно хлорин, так же хаки поливинилхлорид-
ное, превосходит все химические волокна (кроме волокон из фтор-
полимеров).
При 90—100 °С хлорин начинает деформироваться, что
свидетельствует о его недостаточной термостойкости. При вытягивании
волокна на 300—400% температура начала деформации
повышается на 20—30 °С.
К фотохимическим воздействиям хлориновое волокно обладает
недостаточной стойкостью. При облучении солнечным светом и
атмосферных воздействиях прочность и удлинение волокна
постепенно понижаются. Одновременно изменяется химический состав
полимера — отщепляется некоторое количество НС1, и в
макромолекуле полимера появляются двойные связи. Для повышения
светостойкости необходимо по возможности удалить из исходного
полимера низкомолекулярные фракции и ввести в волокно
небольшие количества @,5—1% от массы волокна) стабилизаторов.
Последние повышают светостойкость волокна в 2—4 раза.
Гигроскопичность и термостойкость волокна хлорин могут быть
повышены путем синтеза привитых сополимеров. Прививка к
перхлорвинилу может быть осуществлена преимущественно
радиационным методом. При прививке к волокну хлорин 3% полиметакри-
7.2. Перхлорвиниловые волокна 243
ловой кислоты [20] гигроскопичность волокна повышается до 3—
3,5%. С увеличением количества привитой полиметакриловой
кислоты количество влаги, сорбируемой волокном, значительно
увеличивается. При прививке к хлорину полиакрилонитрила
увеличивается термостойкость модифицированного волокна. Например,
если после нагрева в течение 24 ч при 100 °С прочность волокна
хлорин снижается с 18 гс/текс A80 мН/текс)) до 11 гс/текс
A10 мН/текс), то у модифицированного волокна, содержащего 20—
25% привитого полиакрилонитрила, прочность не снижается [21].
Детально свойства волокна хлорин (в частности, упругое и
высокоэластическое удлинение,, устойчивость к многократным
деформациям, усталостная прочность, интенсивность старения и т. д.)
не исследованы.
Описанные выше свойства волокна хлорин определяют области
его применения. Это волокно наиболее целесообразно использовать
в первую очередь для изготовления изделий, от которых требуется
высокая стойкость к различным агрессивным химическим
реагентам (фильтровальные ткани, спецодежда, прокладки и т. д.). Эти
изделия можно использовать при сравнительно невысоких
температурах (не выше 60—75°С). Из волокна хлорин можно
изготовлять негниющие рыболовные сети и снасти. После фильтрации
вискозы через хлориновые ткани они выдерживают более 70
стирок, в то время как хлопчатобумажные ткани выдерживают только
10 стирок [22]. Волокно хлорин применяют также для изготовления
медицинского белья.
Модифицированное хлориновое волокно. Советскими
исследователями предложен метод получения модифицированного волокна
из перхлорвинила — ацетохлорина [23]. Это волокно получают из
ацетоновых растворов смеси полимеров, содержащей 80—85%
хлорина и 15—20% ацетата целлюлозы. При введении сравнительно
небольших количеств ацетата целлюлозы заметно повышается
термостойкость волокна.
В отличие от волокна хлорин, которое начинает усаживаться
при 80—90 °С, ацетохлорин не деформируется при нагреве до 100—
ПО °С. Так же как и хлорин, волокно ацетохлорин является
негорючим материалом и поэтому может быть использовано для
изготовления изделий, для которых это свойство имеет важное
значение.
Существенным затруднением при получении волокна
ацетохлорин является плохая совместимость растворов указанных
полимеров в ацетоне. Через несколько дней прядильный раствор
расслаивается, что усложняет получение равномерного и однородного по
составу волокна. Чтобы устранить расслаивание растворов,
смешивать их целесообразно непосредственно перед формованием
(например, при помощи насосика).
Модифицированное волокно хлорин можно получить из смеси
хлорина и нитрата целлюлозы (волокно винит он) [24]. Добавление
244 Глава 7. Производство поливинилхлоридных волокон
сравнительно небольшого количества A0—15% от массы хлорина)
нитрата целлюлозы значительно повышает температуру
размягчения волокна (на 60—80°С).
Растворы хлорина и нитрата целлюлозы в ацетоне
термодинамически несовместимы, однако они не расслаиваются в течение
нескольких дней, что обеспечивает возможность их переработки в
волокно обычным методом. Волокно формуют мокрым способом
в 6—8%-ном водном растворе ацетона (осадительная ванна).
7.3. ВОЛОКНА ИЗ СОПОЛИМЕРОВ ВИНИЛХЛОРИДА
Практическое применение для производства волокон получили
сополимеры винилхлорида с винилацетатом, акрилонитрилом и
винилиденхлоридом.
Первые два сополимера при определенном соотношении
мономеров растворяются в ацетоне, образуя вязкий концентрированный
раствор. Сополимер винилхлорида и винилиденхлорида не
растворяется ни в ацетоне, ни в других органических доступных
растворителях.
Волокно из сополимера винилхлорида и винилацетата.
Производство волокна из сополимеров винилхлорида и винилацетата
было начато в 1939 г. В 1945—1948 гг. производство этого волокна
достигло 2 тыс. т/год. При появлении новых карбоцепных волокон,
значительно превосходящих виньон по ряду показателей,
дальнейшее увеличение производства этого волокна прекратилось, а в
настоящее время это волокно в производственных условиях не
вырабатывается.
Свойства волокна из сополимера винилхлорида и винилацетата
мало отличаются от свойств других волокон, получаемых из
полимеров и сополимеров винилхлорида.
Волокно из сополимера винилхлорида и винилиденхлорида.
Сополимер винилхлорида и винилиденхлорида получается при
совместной полимеризации этих мономеров:
СН2=СНС1 + СН2=СС12 —> СН2—СНС1—СН2—СС12
Винилиденхлорид (хлористый винилиден) получается дегидрохло-
рированием трихлорэтана спиртовыми или водными растворами
щелочей.
В производственных условиях эта обработка производится
известковым молоком:
СН2С1—СНС12 + Са(ОНJ —> СН2=СС12 + СаС12 + Н2О
Этот процесс обычно проводят при температуре около 100°С. Трн-
хлорэтан получается хлорированием этилена. Следовательно,
сырьевая база для получения винилхлорида и винилиденхлорида
одна и та же.
Основным преимуществом поливинилиденхлорида или продукта
совместной полиме изации винилиденхло ида с винилхло идом по
7.3. Волокна из сополимеров винилхлорида 245
сравнению с поливинилхлоридом является более высокая
термостойкость, а также большая текучесть при повышенной
температуре, что облегчает формование из него волокна.
Производство волокон из сополимера винилиденхлорида было
начато в 1939 г. Эти волокна носят название «саран».
Поливинилиденхлорид, а также сополимер винилхлорида и
винилиденхлорида при различном содержании винилхлорида в
сополимере (сополимер, применяемый для производства волокна,
содержит от 10 до 60% винилхлорида) не растворяются в органических
растворителях. Растворители для этих полимеров (в которых
можно было бы получить вязкие концентрированные растворы) пока
не найдены, поэтому волокно получают из полимера, находящегося
в термопластичном состоянии (экструзией). Из сополимера
винилхлорида и винилиденхлорида, так же как из поливинилиденхло-
рида, формуют пока только моноволокно. Принципиально процесс
формования указанных волокон можно осуществить на прядильной
машине, применяемой для формования волокон из расплава,
подавая вязкую массу к прядильному насосику при помощи шнека
аналогично тому, как это имеет место при формовании полиэфирного
и полипропиленового волокон. По-видимому, таким путем удастся
получить текстильную нить.
Минимальный диаметр моноволокна саран, получаемого из
размягченного полимера, 0,05 мм, толщина 12—ЗО текс.
Технологический процесс получения волокна саран и
аппаратурное оформление этого процесса существенно отличаются от
таковых для других карбоцепных волокон.
Полимеры и сополимеры винилиденхлорида легко
кристаллизуются. Так как вытягивать закристаллизованное волокно очень
трудно, а в ряде случаев вообще невозможно, то свежесформован-
ное волокно сначала охлаждают (так называемый процесс
закалки). Этим достигается значительное замедление процесса
кристаллизации и сохранение волокна в течение более или менее
длительного времени в неравновесном (переохлажденном) аморфном
состоянии. Например, кристаллизация свежесформованного
волокна, полученного из сополимера, содержащего 90%
винилиденхлорида и 10% винилхлорида, при 100°С начинается через несколько
секунд, при 20 °С — через несколько минут, а при 0°С — через 24 ч.
По-видимому, закалка волокна перед его вытягиванием имеет
значение при получении волокон не только из полимеров и
сополимеров винилиденхлорида, но и из кристаллических полимеров
других типов. Проведение соответствующих исследований представляет
большой интерес.
Технологический процесс получения волокна типа саран
включает следующие операции: формование, закалка, вытягивание при
нормальной температуре и перемотка.
Формование волокна из размягченного сополимера
осуществляется на машинах, аналогичных по конструкции и принципу
246 Глава 7. Производство поливинилхлоридных волокон
работы экструдерам, применяемым в промышленности пластических
масс для переработки термопластичных материалов. Сущность
метода заключается в выдавливании материала, переходящего при
повышенной температуре в пластическое состояние, через
профилированное отверстие — мундштук или отверстия фильеры при
давлении 150—250 кгс/см2 A5-Ю6 Па —25-106 Па). Такое
давление создается обычно при помощи шнека, подающего нагретую
вязкую массу к фильере. Равномерность получаемого волокна по
толщине при таком способе формования значительно ниже, чем
при формовании обычными способами с использованием
прядильных насосиков. Перед подачей в экструдер измельченный полимер
в большинстве случаев подвергают предварительному вальцеванию
для равномерного перемешивания со стабилизатором, который
обычно прибавляют к полимеру. Стабилизаторы связывают
хлористый водород, выделяющийся при частичном разложении
полимера при повышенных температурах, и таким образом
препятствуют дальнейшему автокаталитическому протеканию реакции.
После вальцевания масса поступает в экструдер, обогреваемый
до 130—170 °С паром или жидким ВОТ. Для уменьшения
термического разложения полимера продолжительность пребывания его
в экструдере сводят к минимуму, уменьшая объем массы,
находящейся в машине. Расстояние от шнека до фильеры 50—60 см. Так
как некоторые металлы, особенно железо, медь и цинк,
каталитически ускоряют термический распад поливинилиденхлорида, то
детали машины, соприкасающиеся с полимером, не должны
содержать этих металлов или могут иметь специальные покрытия.
При формовании волокна типа саран применяют фильеры не
только с одним отверстием, как это обычно имеет место при
производстве моноволокна, но и с 15—18 отверстиями. Это дает
возможность значительно повысить производительность экструдера.
.. Свежесформованное волокно пропускается через охлаждающую
ванну @—10°С), где оно подвергается закалке.
После закалки охлажденное волокно вытягивают при 10—20 °С.
Вытянутое волокно наматывается на бобину (в случае
необходимости сушится) и направляется на переработку. Формование,
закалка, вытягивание и перемотка проводятся на одном агрегате.
Волокно типа саран имеет очень низкую гигроскопичность (при
относительной влажности воздуха 65% сорбирует менее 0,1%
влаги) и более высокую термостойкость, чем поливинилхлоридное
волокно. При кипячении в воде волокно саран не усаживается. Этот
вид синтетического волокна обладает такой же хемостойкостью,
как и другие волокна, получаемые на основе поливинилхлорида.
Области применения волокна саран ограничены вследствие того,
что пока удается получить только моноволокно. В настоящее время
волокно из сополимера винилхлорида и винилиденхлорида
используется для изготовления негорючих декоративных и обивочных
тканей.
Литература 247
ЛИТЕРАТУРА
1. Антонов В. П., Хим. волокна, 1961, № 3, с. 3—5.
2. 3 а в ь я л о в В. Д., Глазковский Ю. В., Зубов Л. Н. и др., Хим.
волокна, 1969, № 2, с. 29—32.
3. Э т л и с В., Минскер К-, Рыл ов Е., Высокомол. соед., 1959, т. 1,
с. 1403—1406.
4. Rein Н., Z. ges. Textilind., 1954, Bd. 57, N 13, S. 812—815.
5. Koch R., Mod. Text. Mag., 1955, v. 36, N 8, p. 45—47.
6. Фихман В. Д., Зубов Л. Н., Пакшвер Э. А. авт. свид. 134871 A960);
Бюлл. изобр., 1961, № 1.
7. Бакунеико Т. Л., Фихман В. Д., Хим. волокна, 1963, № 5, с. 69—72.
8. 3 у б о в Л. Н., Пакшвер А. Б. В кн.: Карбоцепные волокна М.
«Химия», 1966, с. 124—127.
9. Зубов Л. Н., Пакшвер А. Б., Фихман В. Д. и др., Хим. волокна,
1968, № 1, с. 26—28.
10. Фихман В. Д., Аш М. А., Пакшвер А. Б., Хим. волокна, 1966, № 1,
с. 11—14.
11. Фихман В. Д., Алексеева В. М., Пакшвер А. Б., Хим. волокна,
1965, № 2, с. 12—15.
12. Фихман В. Д., Аш М. А., Воробьева Е. В., Хим. волокна, 1965, № 1,
с. 28—30.
13. Hochstaidter L, Text. Res. J., 1958, v. 28, p. 78—82.
14 В а и м а н Э. Я., Пакшвер А. Б., Фихман В. Д., В кн.: Карбоцепные
волокна. М., «Химия», 1966, с. 251—254.
15. Карпов В. Л., Юркевич В. Г., Кавалеров а Л. М. и др. Хим.
волокна, 1966, № 6, с. 18—21.
16. Urlich H. M., Melliand Textilber., 1956, Bd. 37, S. 185—188.
17. Albert I., «Rayon Zellwolle», 1955, Bd. 5, N 12, S. 842—845.
18. Геллер Б. Э. Химия и технология хлоринового волокна. М., Гизлегпром.,
1958. 123 с.
19. Геллер Б. Э., Текст.пром., 1956, № 11, с. 24—25; ЖОХ, 1951 т 24 с 1058—
1061.
20. Уем а нов X. У., M у с а е в У. Н., Турсунов Д. и др., Хим юлокна,
1967, № 4, с. 73^-75.
21. Турсунов Д., Мусаев У., Таллиев Р. и др., Хим. волокна 1968
№ 5, с. 74—76.
22. В и р є з у б А. И., Г и н з б е р г М. А., Андреева Л. Э. и др , Хим
волокна, 1969, № 6, с. 67—70.
23. Михайлов Н., У х ан о в а 3., К а ретин а Г., Хим. волокна 1959 № 3
с. 18—20. ' ' " '
24. M и х а и л о в Н. В., Покровская Н. В., Хим. волокна, 1969, № 5,
с. 13—16.
Глава 8
ПРОИЗВОДСТВО ПОЛИВИНИЛСПИРТОВЫХ
волокон
Волокно из поливинилового спирта — поливинилспиртовое —
обладает специфическими свойствами, отличающими его от всех
других синтетических волокон. Этот вид волокна является
единственным гидрофильным синтетическим волокном,
вырабатываемым в производственных масштабах. В зависимости от метода
последующей (после формования) обработки гигроскопичность
поливинилспиртового волокна изменяется в широких пределах (по
этому показателю оно не уступает хлопку).
Производство водорастворимого поливинилспиртового волокна
было начато в Германии в 1934 г. Однако растворимое в воде
волокно, естественно, могло найти только ограниченное применение.
Потребовалось еще 10—12 лет для разработки экономичного
метода получения волокна из этого полимера, нерастворимого в воде
и обладающего необходимым комплексом механических свойств.
Производство нерастворимого в воде волокна из поливинилового
спирта было начато в 1948—1950 гг. в Японии. До 1957—1958 гг.
Япония была единственной страной, в которой это волокно
вырабатывалось в промышленном масштабе (под названиями «кура-
лон» и «винилон»). В 1959 г. объем производства этих волокон в
Японии составил около 17 тыс. т. В настоящее время
поливинилспиртовое волокно вырабатывается в США (винал), в КНДР (ви-
нолон) и в ряде других стран. Мировое производство волокон из
поливинилового спирта * в 1970 г. превысило 100 тыс. т.
8.1. ПОЛУЧЕНИЕ ПОЛИВИНИЛОВОГО СПИРТА
В отличие от других карбоцепных полимеров поливиниловый
спирт не может быть получен путем полимеризации
соответствующего мономера (винилового спирта). Это объясняется тем, что
виниловый спирт в свободном состоянии не существует, так как
в момент образования сразу изомеризуется и получается уксусный
альдегид:
/он ^°
СН2=С —*¦ СН3—С
* Под названием «волокна из поливинилового спирта» мировая статистика
учитывает волокна не только из поливинилового спирта, но и из сополимеров,
содержащих не менее 85 мол.% звеньев поливинилового спирта или его
производных (например, эфнров).
8.1. Получение поливинилового спирта 249
Поэтому поливиниловый спирт получают не из мономера, а
омылением полимера сложного эфира винилового спирта, который
является вполне стабильным. Из сложных эфиров винилового спирта
наиболее доступен винилацетат, получаемый присоединением
уксусной кислоты к ацетилену в присутствии катализаторов (ацетат
цинка на активированном угле) :
СН3—СООН + CbfeCH —> СН3—СО—О-СН=СН2
который и используется в качестве исходного мономера для
синтеза поливинилацетата, а затем и поливинилового спирта. Для этой
цели могут применяться и другие эфиры винилового спирта, в
частности винилформиат. Однако эти мономеры не получили
практического применения.
Следовательно, получение поливинилового спирта складывается
из двух процессов: синтеза поливинилацетата полимеризацией ви-
нилацетата
СН2=СН —> СН2СН—СН2—СН
« I II
ОСОСНз ОСОСНз ОСОСНз
и омыления полученного поливинилацетата растворами кислот или
щелочей
СН,—СН—СН2—СН—•• —> СН2—СН—СН2—СН
I I II
ОСОСНз ОСОСНз ОН ОН
Полимеризация винилацетата обычно осуществляется по
радикальному механизму с использованием в качестве инициаторов
перекисных соединений.
Процесс полимеризации может проводиться в эмульсии или в
растворе. При полимеризации винилацетата в растворе (обычно
в растворе метанола) последующее омыление поливинилацетата
также проводится в растворе, что значительно ускоряет процесс и
повышает однородность продукта омыления.
Для устранения возможности образования сильно
разветвленного полимера при полимеризации винилацетата в растворе
процесс необходимо прекращать при конверсии 60—65% мономера.
Синтез поливинилацетата, а следовательно, и поливинилового
спирта с минимальной степенью разветвленности макромолекул
является предварительным условием получения высокопрочного
поливинилспиртового волокна, которое может быть использовано
для различных целей бе*з дополнительного ацеталирования.
Принципиально возможно получение стереорегулярного
поливинилацетата, а следовательно, и поливинилового спирта. Одним
из наиболее реальных методов повышения стереорегулярности
является понижение температуры полимеризации. Так же как и при
синтезе других карбоцепных полимеров, при снижении температуры
реакции радикальной полимеризации количество разветвлений в
макромолекуле получаемого полимера уменьшается.
250 Глава 8. Производство поливинилспиртовых волокон
Поливинилацетат, полученный методом радикальной
полимеризации при пониженных температурах (около 0°С), образует при
последующем омылении поливиниловый спирт высокой степени
стереорегулярности. Из этого полимера получаются волокна,
обладающие такой же прочностью, как волокна, сформованные в тех же
условиях из растворов поливинилового спирта, синтезированного
при повышенной температуре F0°С). Однако уменьшение числа
разветвлений в молекуле поливинилового спирта значительно
увеличивает водостойкость волокна и температуру его размягчения и
снижает его растворимость. Зависимость растворимости поливинил-
спиртового волокна от температуры полимеризации исходного
мономера характеризуется следующими данными:
Температура полимеризации Степень вытягивания Растворимость волокна
внннлацетата, °С волокна, % в кипящей воде, %
—30 400 0,5
10 400 17,5
30 400 100
60 430 100
В Японии получены препараты синдиотактических полимеров
винилового спирта, которые были разделены на 2 фракции,
отличающиеся по фазовому состоянию и по плотности. Плотность син-
диотактической модификации поливинилового спирта составляет
1,348 г/см3, а аморфной — 1,265 г/см3 [1]. Плотность полимера,
используемого для формования волокон (содержит 65%
кристаллической фракции), достигает 1,320 г/см3.
Омыление поливинилацетата обычно осуществляется в растворе
метанола спиртовым раствором щелочи при 30 °С в течение 10—
15 мин. Количество NaOH, применяемого при омылении,
составляет 0,1—0,3 моль на одно элементарное звено макромолекулы.
В этих условиях происходит не только омыление, но и алкоголиз
поливинилацетата. Это подтверждается тем, что при омылении
расходуется всего 10% NaOH от теоретически необходимого для
омыления всех ацетильных групп и связывания выделяющейся при
этом уксусной кислоты [2].
При омылении около 70% ацетильных групп получается
частично омыленный поливинилацетат, растворимый в воде. Для
производства прочных волокон требуется поливиниловый спирт,
содержащий менее 0,2% ацетильных групп. При наличии
повышенного количества таких групп, так же как и небольшого числа
разветвлений в макромолекуле поливинилового спирта, затрудняется
кристаллизация полимера и ориентация макромолекул при
вытягивании, а тем самым и получение волокна с высокими
механическими свойствами.
Гидроксильные группы в молекулах поливинилового спирта,
используемого для получения волокна, находятся в положении 1,3.
Однако в макромолекулах имеется небольшое количество гликоле-
8.1. Получение поливинилового спирта 251
вых групп (до 2% от общего числа групп ОН в макромолекуле),
в которых гидроксильные группы находятся в положении 1,2.
Количество гликолевых групп в макромолекуле поливинилового
спирта зависит от температуры полимеризации и от характера ис
ходного мономера, используемого для полимеризации. Например,
если в макромолекуле поливинилового спирта, получаемого
омылением поливинилацетата, полимеризация которого
осуществлялась при 40 °С, содержится 1,58 мол.% гликолевых групп, то при
повышении температуры полимеризации до 130 °С количество этих
групп в молекуле увеличивается до 2,75 мол.% [3].
Поливиниловый спирт, полученный омылением поливинилфор-
миата, содержит меньше гликолевых групп, чем омыленный поли-
винилацетат.
Гликолевые группы уменьшают стойкость поливинилового
спирта к окислительным воздействиям и снижают степень его
кристалличности [4].
Полученный после омыления поливиниловый спирт в некоторых
случаях промывается метанолом для удаления избытка щелочи и
ацетата натрия, образовавшегося после омыления.
Требования к поливиниловому спирту, используемому для
получения волокна, в литературе не приводятся. Степень
полимеризации такого полимера обычно составляет 1200—1600. По данным
Сакурада, при увеличении степени полимеризации поливинилового
спирта выше оптимума (более 1400—1800) прочность волокна не
только не возрастает, но даже снижается. Например, прочность
волокна, сформованного из полимера со степенью полимеризации
1400—1600, 2400 и 3000, составляет соответственно 90, 80 и
70 гс/текс. Однако эти данные требуют дополнительной проверки.
При определении степени полимеризации поливинилового
спирта вискозиметрическим методом (растворитель — вода) можно
воспользоваться следующим уравнением:
[П] =^8,87- 10-4М0'62
где [ті] — характеристическая вязкость;
M — молекулярный вес.
Поливиниловый спирт является полидисперсным продуктом.
Фракционирование его может быть осуществлено различными
способами, в частности методом фракционного осаждения из водных
растворов полимера при добавлении н-пропилового спирта [5]. Было
выделено 12 фракций с молекулярным весом от 26 000 до 20 000.
Методы определения полидисперсности поливинилового спирта,
и в частности содержания низкомолекулярных фракций, пока не
унифицированы, так же как не исследован вопрос о влиянии
различных факторов на растворимость поливинилового спирта и
вязкость концентрированных растворов. Например, при наличии в
составе полимера небольшого количества A0—15%) низкомолеку-
2Г>2 Глава 8. Производство поливинилспиртовых волокон
лярных фракций со степенью полимеризации около 270 заметно
снижается прочность волокна и особенно уменьшается число
двойных изгибов, выдерживаемых волокном до разрушения.
Технология получения поливинилспиртового волокна несколько
отличается от процесса производства других карбоцепных волокон.
Основными операциями являются:
приготовление прядильного раствора и подготовка его к
формованию;
формование волокна;
вытягивание, промывка и сушка волокна;
дополнительное вытягивание при повышенных температурах;
ацеталирование для образования поперечных химических
связей между макромолекулами;
промывка, мыловка и сушка.
8.2. ПРИГОТОВЛЕНИЕ ПРЯДИЛЬНОГО РАСТВОРА
Поливиниловый спирт, поступающий для получения волокна,
промывается холодной водой для удаления ацетата натрия (если
он не был предварительно промыт метанолом). Максимально
допустимое содержание этой соли в полимере 0,2—0,3%-
Основным растворителем поливинилового спирта является вода.
Поливиниловый спирт растворяется также в диметилформамиде,
в водных растворах органических оснований и в фенолах. Однако
очевидно, что применение этих растворителей для приготовления
прядильных растворов нецелесообразно.
Прядильный раствор поливинилового спирта для формования
волокна сухим способом содержит 40—45% поливинилового
спирта. При формовании волокна мокрым способом применяется 14—
16%-ный раствор поливинилового спирта в воде.
Концентрированные растворы поливинилового спирта в воде при нормальной
температуре недостаточно стабильны. Только при увеличении
температуры раствора до 50—60 °С стойкость повышается и вязкость
раствора заметно не изменяется в течение нескольких дней.
Поэтому все операции по получению прядильного раствора и его
подготовке к формованию, как правило, проводятся при повышенных
температурах (80—95°С).
Сравнительно незначительное повышение концентрации
полимера обусловливает значительное повышение вязкости раствора.
При повышении концентрации полимера в прядильном растворе на
2% вязкость раствора увеличивается примерно в 2 раза. Например,
при повышении концентрации поливинилового спирта в растворе с
12 до 16% вязкость раствора увеличивается с 500 до 2000 П [6].
При возрастании концентрации полимера в растворе до 20%
вязкость раствора увеличивается до 6500 П.
Прядильный раствор поливинилового спирта, так же как и
концентрированные растворы других волокнообразующих полимеров,
8.3. Формование волокна .. . 253
сильно структурирован, и поэтому при повышении температуры
вязкость его резко снижается. Например, при увеличений
температуры с 20 до 50 °С вязкость раствора уменьшается в 3—3,5 ра-
за [7].
Условия подготовки прядильного раствора поливинилового
спирта к формованию такие же, как и при получении других
химических волокон. Для повышения однородности прядильного
раствора, улучшения фильтруемое™ и уменьшения засорения фильер
рекомендуется [8] добавлять в прядильный раствор небольшое
количество @,3% от массы раствора) поверхностно-активных
веществ, аналогичных по составу добавляемым в вискозу, в частности
оксиэтилированные амины или алкилфенолы (ОП-10).
8.3. ФОРМОВАНИЕ ВОЛОКНА
Волокно из растворов поливинилового спирта можно получать
сухим или мокрым способом формования. При использовании для
формования волокна поливинилового спирта стереорегулярной
структуры, образующего прядильные растворы очень высокой
вязкости, применяют метод формования нити из полимера,
содержащего небольшие количества растворителя и находящегося в
размягченном состоянии (так называемый метод полурасплава) [1].
Этот метод используется в Японии для получения комплексной
нити. Он может быть использован для формования и других карбо-
цепных волокон. Высоковязкий раствор (размягченный полимер)
подается шнеком при повышенной температуре к прядильному
насосику, а затем к фильере. Выдавливаемые через отверстия
фильеры струйки термопластичного полимера (в результате
испарения растворителя в шахте прядильной машины) затвердевают
и превращаются в тонкие волокна, которые образуют нить.
Полученная нить принимается на бобину. , ¦
Изучение условий формования волокна этим способом было
проведено советскими исследователями [9]. Для формования
использовались препараты поливинилового спирта со степенью
полимеризации 1300—1700, пластифицированные водой или спиртом
A00—150% от массы полимера). При добавлении такого
количества воды температура течения поливинилового спирта резко
снижается (с,220—230 до 80—95°С). Пластифицированный полимер
перед формованием гранулируют. Температура в шахте
изменяется в зависимости от характера применяемого пластифицирующего
реагента в пределах 130—200°С. Свежесформованное волокно,
содержащее 10—30% воды, подсушивается и затем подвергается
вытягиванию при повышенной температуре, так же как«и обычное
поливинилепиртовое волокно, полученное из растворов.
Сухой способ формования поливинилспиртовой нити из водных
растворов не получил еще широкого применения, хотя
преимущества его при формовании нити очевидны.
254 Глава 8. Производство поливинилспиртовых волокон
Основным способом получения поливинилспиртового волокна
является пока мокрый способ формования, занявший в
производстве штапельного волокна почти монопольное положение.
При формовании поливинилспиртового волокна, так же как и
других синтетических волокон, происходят только
физико-химические, а не химические процессы. Поэтому формование производится
однованным способом.
В качестве осадительной ванны применяются органические
жидкости или концентрированные водные растворы минеральных
солей. Из органических жидкостей, смешивающихся с водой,
наибольшей коагулирующей способностью по отношению к
поливиниловому спирту обладает ацетон, затем циклогексанон и этиловый
спирт [6].
Формование волокна в органической ванне происходит более
мягко, чем в растворах солей. Коагулирующая способность
указанных органических соединений в 3—10 раз меньше, чем водных
растворов обычно применяемых электролитов [10].
При формовании в ванне, содержащей органический осадитель,
волокно набухает значительно меньше, чем в водных растворах
солей, в результате чего оно имеет более однородную и плотную
структуру. Этот способ может, по-видимому, представлять интерес
при получении водорастворимых волокон, приобретающих в
последнее время определенное значение.
В настоящее время при формовании поливинилспиртового
волокна мокрым способом в качестве осадительной ванны
используются преимущественно растворы солей.
Наибольшей, коагулирующей способностью среди растворов
сернокислых солей (при концентрации в ванне 350—400 г/л)
обладает сульфат натрия и несколько меньшей — сульфат аммония.
При уменьшении концентрации Na2SC>4 в растворе ниже 350 г/л
механические свойства получаемых волокон, в первую очередь
прочность и усталостные свойства, ухудшаются.
Коагулирующая способность отдельных анионов солей при
формовании поливинилспиртового волокна убывает в следующей
последовательности [11]:
SO?" > СОз2" > СНзСОО" > СГ > Ж)з~
Скорость формования поливинилспиртового штапельного
волокна 7—10 м/мин, длина пути нити в ванне 150—220 см.
Оптимальная температура формования 40—50 °С [12].
Формование волокна при повышенной температуре
обусловливается не только стремлением увеличить скорость коагуляции
прядильного раствора, но и, главным образом, необходимостью
устранить высаживание солей на деталях прядильных машин.
Формование волокна мокрым способом проводится, как
правило, с отрицательной фильерной вытяжкой (—40%) —(—80%).
8.4. Последующая обработка волокна 255
Чем меньше филье^ная вытяжка (т. е. чем больше величина
отрицательной вытяжки), тем больше последующая термопластифика-
ционная вытяжка и тем выше прочность получаемого волокна [13].
Например, если формование проводили с фильерной вытяжкой
—71,5%, то максимально возможная последующая термопласти-
фикационная вытяжка составляла —1470%, а прочность
получаемого волокна равнялась 70 гс/текс. При фильерной вытяжке
—53Д% максимальная последующая вытяжка составляла 880%,
а прочность волокна равнялась 54,8 гс/текс. При дальнейшем
изменении фильерной вытяжки до —37,4% максимальная величина
последующей вытяжки снижалась до 65%.
Свежесформованное волокно на прядильной машине
подвергается пластификационной вытяжке на 200—300%. Вытягивание
волокна проводится в пластификационной ванне, содержащей 300—
400 г/л Na2SO4, при 70—80 °С в одну или в две стадии.
Волокно, получаемое при формовании в водно-солевых ваннах,
неоднородно по структуре. Срез этого волокна характеризуется
наличием ядра и оболочки.
8.4. ПОСЛЕДУЮЩАЯ ОБРАБОТКА ВОЛОКНА
Свежесформованное поливинилспиртовое волокно сорбирует
значительное количество сульфата натрия (до 80% от массы
волокна) [14], поэтому после формования нить подвергают промывке.
Если для вискозного волокна эта операция не представляет
никаких затруднений, то отмывка солей из растворимого в воде поли-
винилспиртового волокна производится в особых условиях.
Волокно отмывают под натяжением 2—4%-ным раствором сульфата
натрия при пониженной температуре C—5 °С) во избежание
частичного растворения волокна.
Водорастворимое поливинилспиртовое волокно начинают
применять при изготовлении бумаги для производства фильтров,
используемых для очистки масел в двигателях внутреннего сгорания
в автомобильной, тракторной и авиационной промышленности. При
этом значительно повышается эффективность фильтров:
повышается скорость фильтрации и на 20—25% увеличивается срок
эксплуатации двигателей [15].
На сушку поступает волокно, содержащее 0,2—2% сульфатов
(от массы волокна). Обычно волокно сушится под натяжением при
75—90°С. Высушенное волокно растворимо в воде при температуре
выше 60 °С.
Вытягивание волокна. Несмотря на указанную выше
возможность использования водорастворимых волокон, широко
применяться могут только волокна, нерастворимые в воде, незначительно
набухающие и ограниченно усаживающиеся в горячей воде. Это
может быть достигнуто главным образом в результате увеличения
межмолекулярного взаимодействия, которое достигается при
256 Глава 8. Производство поливинилрпиртовых волокон
повышении степени ориентации макромолекул или их агрегатов
или при образовании поперечных химических связей между ними
(сшивание).
Степень ориентации макромолекул поливинилспиртового
волокна, так же как и других волокон, может быть увеличена
вытягиванием при нормальной и особенно при повышенной температуре.
Большое влияние на максимально возможную степень
вытягивания и соответственно на прочность получаемого волокна
оказывает содержание ацетильных групп. С увеличением числа этих
групп в молекуле поливинилового спирта уменьшается
максимальная степень вытягивания и снижается прочность волокна [16]:
Содержание ацетильных "^ГвышенныГ* Прочно",ьмД?™ Гс/текс
гРупп> % температурах, % (мн/текс)
0,21 290 75,0G50)
0,39 260 73,1 G30)
1,70 240 66,9 F69)
С увеличением содержания ацетильных групп в волокне
снижается температура начала усадки волокна в воде и температура
его размягчения, что объясняется уменьшением регулярности
строения макромолекул и степени кристалличности волокна.
Кристалличность волокна возрастает с повышением степени его
вытягивания, особенно при наличии последующего процесса
терморелаксации. Изменение степени кристалличности
поливинилспиртового волокна на отдельных стадиях производства
характеризуется следующими данными [6]:
Кристалличность,
%
Исходный полимер 25
Свежесформованное волокно
невытянутое 30
вытянутое на 300% ....... 40
вытянутое на 500% 50
Вытянутое и терморелаксированное
волокно 70—75
С увеличением степени вытягивания волокон с 300 до 500%
плотность волокна повышается с 1,30 до 1,32 г/см3 [17].
Дополнительное вытягивание волокна производится при 220—
240 °С (так называемое термическое вытягивание) по обычной для
карбоцепных волокон схеме. Штапельное поливинилспиртовое
волокно дополнительно вытягивают на 30—100%, высокопрочное —
на 350—500%. В результате значительно снижается набухание и
усадка волокна в воде.
Чем больше степень пластификационного вытягивания на
прядильной машине, тем меньше максимально возможная степень
последующего вытягивания при повышенной температуре и тем
8.4. Последующая обработка волокна 257
меньше прочность волокна. При одинаковой степени термического
вытягивания волокна, невытянутого на прядильной машине,
прочность волокна выше, а удлинение ниже, чем при пластификацион-
ном вытягивании:
Степень Прочность Удлинение
вытягивания, волокна, гс/текс волокна.
% (мН/текс) %
Пластификационная
вытяжка 225 35—45 15—25
C50-450)
Термическая вытяжка . . 225 55—65 5—10
E50-650)
Термическое вытягивание осуществляется при 220—240 °С в
среде горячего воздуха. Увеличение продолжительности процесса
с 5 до 15 с повышает степень ориентации волокна и его
водостойкость [18].
Терморелаксация (термофиксация) волокна. Вытянутое
волокно подвергается дополнительной кратковременной термообработке
при 220—230 °С. Влияние этой обработки на изменение свойств
волокна выявляется для поливинилстшртового волокна в
значительно большей, степени, чем для других карбоцепных волокон.
В результате терморелаксации вытянутых поливинилспиртовых
волокон, осуществляемой в свободном состоянии, снижается
прочность и начальный модуль и одновременно значительно
повышается удлинение и эластические свойства волокна.
Условия проведения процесса терморелаксации оказывают
существенное влияние не только на усадку несшитого волокна в
горячей воде, но и на усадку сшитого волокна (после его ацетали-
рования). При повышении температуры выше 200 °С увеличивается
плотность волокна, показатель двойного лучепреломления и
температура усадки волокна [19]. Например, плотность волокна,
нагретого при 200 и 230 °С, составляет соответственно 1,304 и
1,310 г/см3. Повышение плотности волокна в результате
термообработки характеризуется также значительным уменьшением
количества иода, сорбируемого волокном [20].
Температура и продолжительность нагрева оказывают также
большое влияние на степень кристалличности волокна. Это видно
из следующих данных:
Температура нагрева, °С 80
¦ Продолжительность, с 3600
Степень кристалличности волокна *, °/0 50
120
300
49
160
100
53
200
100
64
225
100
68
* Степень кристалличности исходного волокна 30%.
В ряде случаев в результате термообработки волокно желтеет.
Это изменение цвета волокна является результатом окислитель-
9 Зі] к 234
258
Глава 8. Производство поливинилспиртовых волокон
ного действия кислорода воздуха в присутствии щелочи. Наличие
небольшого количества NaOH на волокне объясняется гидролизом
при повышенных температурах ацетата натрия, частично
адсорбированного волокном. Для устранения этого явления перед
термообработкой волокна необходимо полностью удалить ацетат
натрия.
Нагрев при температуре выше 200 °С заметно не влияет на
усадку поливинилспиртового волокна в горячей воде (табл. 8.1).
Таблица 8.1. Влияние температуры нагрева на изменение усадки
поливинилспиртового волокна и воде
Температура
нагрева. °С
130
160
180
200
210
Усадка
'65 °С
34,4
6,69
2,11
0,99
0,42
волокна (% от первоначальной
в воде при
85 °С
64,2
50,4
36,2
3,4
1.4
длины)
95 °С
70,5
65.9
61,1
8,8
2,29
Сшивание. Образование поперечных химических связей между
макромолекулами необходимо для получения нерастворимого
волокна, дающего минимальную усадку в кипящей воде. Как уже
указывалось выше, волокно, полученное из стереорегулярного
полимера, или высокопрочное волокно, сформованное из растворов
обычного полимера, обладающее повышенной степенью
кристалличности, не требует сшивания.
Химические связи между макромолекулами поливинилового
спирта Могут быть образованы при взаимодействии различных
бифункциональных соединений с реакционноспособными гидроксиль-
ными группами этого полимера. Для осуществления этой реакции,
так же как и для сшивания целлюлозы, могут быть использованы
альдегиды и диальдегиды, диэпоксисоединения, диизоцианаты и
другие ди- и полифункциональные соединения. Единственное
отличие сшивания целлюлозы от сшивания поливинилового спирта
заключается в том, что при обработке целлюлозы этими
соединениями могут реагировать первичные и вторичные группы ОН, а в
молекуле поливинилового спирта — только вторичные.
Наиболее широкое применение получил метод сшивания,
заключающийся в обработке вытянутого термообработанного
поливинилспиртового волокна альдегидами, в частности
формальдегидом.
При взаимодействии формальдегида с поливиниловым спиртом
образуются ацетальные связи между отдельными звеньями внутри
СН2—СН—СН2—СН
8.4. Последующая обработка волокна ' 259
макромолекул и между ними:
сн2о
СН,—СН—СН2—СН ——» СН2—СН—СН2—СН ЬН2О
I і II
ОН ОН О—СН2—О
• • —СН,—СН—СН2—СН—'• •
II II
он он о о
^ \;н2 + н2о
он он о о
II II
СН2—СН—СН2—СН СН2—СН—СН2—СН
Процесс ацеталирования должен проводиться в таких условиях,
чтобы взаимодействие альдегида с поливиниловым спиртом
протекало по возможности межмолекулярно, а не внутримолекулярно.
Это дает возможность уменьшить расход формальдегида. Однако
пока нет достаточно надежных методов раздельного определения
числа внутри- и межмолекулярных ацетальных связей, и поэтому
не удается изучить влияние различных факторов на соотношение
скоростей реакции внутримолекулярного и межмолекулярного
ацеталирования поливинилового спирта.
Обработка волокна обычно производится раствором
следующего состава (в %) *:
Серная кислота . . 20 Формальдегид . . 2—5
Сульфат натрия . . 25 Вода 50
Серная кислота является катализатором реакции
ацеталирования поливинилового спирта. Сульфат натрия вводится в состав
ацеталирующей ванны для снижения набухаемости волокна и
уменьшения возможности их склеивания. Модуль ацеталирующей ванны
достигает 40. Температура обработки составляет 60—70 °С,
продолжительность обработки — 25—30 мин. В этих условиях с
формальдегидом реагирует 30—40% от общего количества групп ОН.
Ацеталирование происходит преимущественно в аморфных
участках волокна. Чем выше суммарное число прореагировавших групп
ОН при внутри- и межмолекулярном ацеталировании, тем выше
теплостойкость волокна, характеризуемая величиной усадки в
кипящей воде.
Влияние степени ацеталирования на изменение теплостойкости
(после обработки в кипящей воде) поливинилспиртового волокна
* Этот состав ацеталирующей ванны не является оптимальным. В частности,
недостаточно обосновано столь высокое содержание серной кислоты в растворе.
9*
260 Глава 8. Производство поливинилспиртовых волокон
видно из следующих данных:
Степень ацеталировання. Усадка,
% от общего числа % от исходной
групп ОН длины
До 30 Очень большая
30—35 12
35—40 4
40—45 2-3
45—50 0,2
Повышение степени ацеталирования более 30—40% от общего
числа групп ОН нецелесообразно, так как в результате
значительного увеличения числа химических связей между
макромолекулами возрастает жесткость и хрупкость волокна и соответственно
ухудшаются его эксплуатационные свойства.
Существенное влияние на свойства пояивинилспиртового
волокна при одной и той же степени ацеталирования оказывает его
структура и степень кристалличности. Например, если
формальдегидом обрабатывается невытянутое и не подвергнутое
термообработке волокно, обладающее рыхлой структурой, то при
нормальной степени ацеталирования C0—40% замещенных групп ОН от
общего числа таких групп) получается волокно, сильно
усаживающееся в кипящей воде. Данные [6], характеризующие зависимость
усадки сшитого волокна в кипящей воде от степени его
кристалличности, приводятся ниже:
Кристал- Степень ацеталирования, Усадка волокна после кипяче-
личность % от общего числа ния в воде в течение 30 мин,
волокна, % групп ОН % от первоначальной длины
82,1
77,0
71,4
0-2,5
Набухание поливинилспиртового волокна в воде также зависит
от его структуры. С повышением степени кристалличности
набухание волокна в воде значительно уменьшается, что подтверждается
следующими данными [21]:
Степень кристалличности волокна, % 20 30 40
Набухание волокна в воде, % 500 200 100
Следовательно, нерастворимое и не усаживающееся в горячей
воде поливинилспиртовое волокно получается только при ацетали-
ровании волокна определенной структуры и степени
кристалличности. Поэтому выбор условий проведения процесса сшивания и
установление оптимальной степени ацеталирования волокна
зависят от предварительной обработки волокна. Чем выше
кристалличность волокна, тем ниже степень ацеталирования, требуемая для
получения нерастворимого безусадочного волокна.
После обработки формальдегидом волокно тщательно
промывается, обрабатывается замасливающими препаратами и сушится.
Несмотря на значительный эффект, достигаемый в результате
30
35
40
65-70
63,5
53,2
44,9
37,2
84. Последующая обработка волокна 261
обработки поливинилспиртового волокна формальдегидом,
доступность и низкую стоимость формальдегида, применение этого
реагента в производственных условиях нежелательно из-за его
вредности (раздражающего действия на слизистую оболочку глаз).
Этим обстоятельством в основном объясняется проведение
большого количества исследований, посвященных выяснению
возможности замены формальдегида другими «сшивающими» реагентами.
В Японии в качестве ацеталирующего реагента в промышленном
масштабе применяется бензальдегид.
Поливинилспиртовое волокно, обработанное бензальдегидом,
обладает более высокой эластичностью, чем ацеталированное
формальдегидом.
Условия ацеталирования гіоливинилспиртового волокна
бензальдегидом изучались советскими исследователями [22]. Перед
ацеталированием волокно подвергалось термообработке при 220 °С
п течение 6—13 мин для повышения стойкости волокна к ацетали-
рующей смеси. Волокно ацеталировали 2—2,5%-ной водной
эмульсией бензальдегида. В качестве катализатора реакции
ацеталирования применялась серная кислота. Оптимальная концентрация
кислоты в ацеталирующей смеси была 10—12%. Для снижения
набухания волокна при ацеталировании к эмульсии добавляют
15—20% Na2SO4 (от массы эмульсии). При проведении реакции
в течение 50—60 мин при 60—70 °С получается волокно,
усаживающееся в кипящей воде не более чем на 5—6%-
Из других реагентов, предложенных для ацеталирования
поливинилспиртового волокна, известный интерес представляют диаль-
дегиды. При действии этих реагентов повышается вероятность
образования межмолекулярных, а не внутримолекулярных ацеталь-
ных связей и соответственно увеличивается эффективность
обработки. Для этого могут быть применены алифатические диальде-
гиды, содержащие различное число метиленовых групп в молекуле
(глиоксаль, диальдегид адипиновой кислоты), и ароматические
диальдегиды (диальдегид фталевой или изофталевой кислоты).
Характер применяемого диальдегида оказывает определенное
влияние на свойства ацеталированного волокна, в частности на его
эластичность [23]. Сшивание предлагается производить, например,
обработкой поливинилспиртового волокна раствором, содержащим
3% диальдегида терефталевой или изофталевой кислоты, 20%
H2SO4, 38% СН3ОН, 39% Н2О [24]. Однако применение раствора
ацеталирующего реагента в органическом растворителе
нецелесообразно.
Советскими исследователями предложен метод ацеталирования
поливинилспиртового волокна водным раствором диальдегида ма-
леиновой кислоты. Обработка волокна производится раствором,
содержащим 1,5—2% диальдегида малеиновой кислоты, 7% H2SO4)
20% Na2SO4 и 71% воды, в течение 1,5 ч при постепенном
повышении температуры от 20 до 70 °С. Снижение усадки волокна и
262 Глава 8. Производство поливинилспиртовых волокон
получение волокна, нерастворимого в кипящей воде, достигается
при степени ацеталирования 15—17%, т. е. в 2—2,5 раза более
низкой, чем при обработке волокна формальдегидом.
В результате взаимодействия диальдегида малеиновой кислоты
с поливиниловым спиртом получается сшитое волокно:
о—СН—СНо—СН—СН?—СН—Сгі2—СН— • •
І І І І
ОН О—СН—О ОН
ІІ
СН .
ОН О СН О ОН
[III
СН2—СН—СН2—СН—СН2—СН—СН2—СН
Благодаря наличию двойных связей между макромолекулами
создается возможность осуществления последующей модификации
сшитого волокна, в частности прививки различных карбоцепных
полимеров.
Нерастворимость и отсутствие усадки волокна в кипящей воде
достигаются и при обработке поливинилспиртового волокна диэпо-
ксисоединениями, и в частности диглицидиловым эфиром.
Интересный способ ацеталирования диальдегидами и
получения тем самым химически окрашенного волокна предложен
советскими исследователями [25]. В качестве ацеталирующего реагента
применяются производные дисперсных (антрахиноновых) кр-асите-
лей. Окраска волокна не изменяется ни при кипячении, ни при
экстракции органическими растворителями.
Определенный интерес представляет способ получения
сшитого волокна без применения низкомолекулярных ацеталирующих
реагентов, основанный на периодатном методе окисления (йодной
кислотой) поливинилового спирта. По данным Сакурада, этот
способ заключается в следующем. Препарат поливинилового спирта
обрабатывают йодной кислотой. В результате такой обрабо,тки
происходит окисление гликолевых групп и образование
альдегидных групп. Раствор этого модифицированного полимера (так
называемого диальдегида поливинилового спирта) добавляют в
небольших количествах к раствору обычного поливинилового спирта.
Из смеси двух полимеров формуют волокно по обычной схеме.
При последующем прогреве этого волокна диальдегид
поливинилового спирта взаимодействует с поливиниловым спиртом с
образованием сшитого полимера (образование так называемых сенд-
вич-полимеров). Целесообразно продолжить исследования в этом
направлении, так как при использовании указанного способа
получения нерастворимого поливинилспиртового волокна устраняются
основные недостатки обычных методов ацеталирования, в
частности формилирования волокна.
S.S. Свойства и применение поливинилспиртового волокна 263
8.5. СВОЙСТВА И ОБЛАСТИ ПРИМЕНЕНИЯ
ПОЛИВИНИЛСПИРТОВОГО ВОЛОКНА
Прочность волокна винол зависит от условий получения и на^
значения этого волокна. У штапельного волокна, используемого в
смеси с другими волокнами или в чистом виде для изготовления
изделий народного потребления, прочность в сухом состоянии
составляет 30—40 гс/текс C00—400 мН/текс). Прочность волокон,
предназначенных для изготовления технических изделий,
повышают до 40—60 гс/текс D00—600 мН/текс) увеличением степени
вытягивания. У высокопрочного волокна прочность достигает 60—
65 гс/текс, а при необходимости может быть доведена до 80 гс/текс.
В опытном масштабе получены поливинилспиртовые волокна с
прочностью 90—100 гс/текс.
Советскими исследователями была показана возможность [26]
получения нерастворимого в горячей воде высокопрочного поли-
винилспиртсвого волокна (80—110 гс/текс, удлинение 8—10%),
сформованного из обычного поливинилового спирта. Это достигнуто
так называемым термопластичным вытягиванием волокна в узком
интервале повышенных температур, близких к температуре
кристаллизации поливинилспиртового волокна. Дополнительному
вытягиванию на 150—275% подвергалось обычное поливинилспирто-
вое волокно с прочностью 45 гс/текс [27].
Возможность получения высокопрочного нерастворимого в
горячей воде поливинилспиртового волокна из обычного полимера
(не стереорегулярного) без ацеталирования представляет большой
научный и практический интерес. Однако, так же как и некоторые
другие виды сверхпрочных волокон (например, фортизан), это
волокно обладает значительно меньшей устойчивостью к
истиранию и к многократным деформациям, чем обычное поливинилспир-
товое волокно.
Наличие гидрофильных групп в макромолекуле полимера
обусловливает уменьшение прочности волокна в мокром состоянии.
Интенсивность снижения прочности в мокром состоянии зависит
от числа поперечных химических связей между макромолекулами,
а также, по-видимому, от природы реагента, применяемого для
сшивания. У обычного волокна (сшитого формальдегидом)
прочность в мокром состоянии снижается на 15—25%> а удлинение
на 5—8%.
Удлинение волокна изменяется в широких пределах: от 30—35%
для штапельного волокна, используемого в смеси с шерстью, до
8—10% для высокопрочной текстильной нити.
Гигроскопичность, т. е. количество влаги, поглощаемой
волокном, в котором в результате ацеталирования блокировано 30—:
35% от общего числа групп ОН, составляет 4,5—5% при
относительной влажности воздуха 65%, а при 100%-ной относительной
264 Глава 8. Производство поливиниЛспиртовых волокон
влажности—12%. По гигроскопичности поливинилспиртовое
волокно приближается к хлопку.
Плотность волокна изменяется в зависимости от структуры
исходного полимера (степени его стереорегулярности и разветвлен-
ности, ориентации и кристалличности). Плотность обычного
волокна составляет 1,30—1,31 г/см3.
Начальный модуль поливинилспиртового волокна в 2—3 раза
выше, чем полиамидного, и в 1,5 раза больше, чем полиэфирного
волокна. Модуль высокопрочного волокна дополнительно
повышается в 2—3 раза.
Устойчивость к истиранию поливинилспиртового волокна также
выше, чем у большинства химических волокон; по этому показателю
оно несколько уступает только полиамидным волокнам.
Эластические свойства волокна из поливинилового спирта ниже,
чем у большинства карбоцепных, и особенно гетероцепных
синтетических волокон, но значительно выше, чем у природных и
искусственных целлюлозных волокон.
Теплостойкость ацеталированного поливинилспиртового волокна
выше, чем у большинства карбоцепных синтетических, волокон.
Температура размягчения и начало разложения таких волокон
составляет 220°С, благодаря чему изделия из этого волокна можно
гладить (с известной предосторожностью). По-видимому, так же
как для большинства других химических волокон, термостойкость
этого волокна можно повысить введением добавок (если удастся
устранить возможность дегидратации волокна при повышенных
температурах).
Этот вывод подтверждается данными, полученными советскими
исследователями [28]. Для устранения или снижения интенсивности
термоокислительного распада поливинилспиртового волокна при
температурах выше 180—200°С в волокно вводилось небольшое
количество термостабилизаторов, значительно замедляющих
термоокислительный распад других синтетических полимеров, в
частности полиамидов (см. разд. 2.15). Наиболее эффективными
термостабилизаторами для поливинилспиртового волокна оказались
фосфорсодержащие соединения — трифенилфосфит и другие эфи-
ры фосфористой кислоты.
Прочность поливинилспиртового волокна при повышении
температуры снижается в меньшей степени, чем для большинства
синтетических волокон. Это объясняется наличием поперечных
химических связей между макромолекулами.
Светостойкость этого волокна значительно выше, чем
целлюлозных волокон. Например, после ультрафиолетового облучения
в течение 30 ч прочность вискозного и хлопкового волокон
снижалась на 53—55%, а поливинилспиртового волокна — на
22% [29].
Окрашиваемость волокна облегчается наличием в
макромолекуле поливинилового спирта гидроксильных групп. Это волокно,
3.5. Свойства и применение поливинилспиртового волокна 265
так же как и целлюлозные волокна, окрашивается прямыми
красителями и некоторыми красителями для ацетатного волокна.
Хемостойкость —один из важных показателей,
характеризующих возможность применения изделий из данного волокна в
некоторых отраслях промышленности. Как и другие карбоцепные
волокна, поливинилспиртовое волокно стойко к кислотам и
щелочам, а также не разрушается микроорганизмами. Однако при
действии концентрированных кислот омыляются ацетальные связи,
что приводит к повышению растворимости волокна. Поэтому
поливинилспиртовое волокно растворимо, особенно при температуре
50 °С и более высокой, в концентрированных минеральных
кислотах.
Из приведенных данных видно, что поливинилспиртовое волокно
по ряду свойств приближается к упрочненным гидратцеллюлозным
волокнам и обладает даже некоторыми преимуществами перед
ними. Основными преимуществами этого волокна по сравнению с
вискозными являются:
более высокая стойкость к кислотам и щелочам;
более низкая плотность;
более высокие эластичность и прочность (для высокопрочного
волокна);
устойчивость к действию микроорганизмов.
Однако по сравнению с вискозным волокном
поливинилспиртовое волокно имеет ряд недостатков: более узкая сырьевая база,
сложность получения поливинилового спирта, трудность промывки
водорастворимого волокна, а также необходимость применения
длительной и вредной для здоровья операции — обработки
формальдегидом. Устранение этой операции, как уже указывалось, будет
иметь большое значение для дальнейшего развития производства
поливинилспиртового волокна.
Области применения. Гидрофильное поливинилспиртовое
волокно, в особенности штапельное, может успешно использоваться
в смеси с хлопковым и вискозными волокнами или с шерстью
для изготовления разнообразных изделий народного
потребления. Упрочненные и высокопрочные нити можно применять для
изготовления рыболовных сетей и снастей, канатов,
фильтровальных материалов.
В последнее время высокопрочные и высокомодульные поли-
винилспиртовые волокна, обладающие минимальной ползучестью,
начинают использовать при производстве армированных изделий.
Эти волокна обладают наиболее высоким модулем из всех обычных
типов химических волокон. Получаемые на их основе
армированные пластики имеют существенное преимущество перед
стеклопластиками по плотности, устойчивости к деформациям при кручении
и изгибах. Кроме того, эти волокна обладают высокой адгезией к
различным смолам (эпоксидным, фенольным, полиэфирным), пре-
росходящей адгезию стеклянных волокон к этим смолам [30].
266 Глава 8. Производство поливинилспартовых волокон*
,., , j.
Использование этих волокон в качестве армирующих материалов,
по-видимому, в дальнейшем будет непрерывно расширяться.
В литературе нет данных о возможности применения
высокопрочного поливинилспиртового волокна для производства шинного
корда, хотя использование для этой цели высокопрочного (80—
90 гс/текс), высокомодульного эластичного волокна пониженной
плотности (по сравнению с вискозным волокном) является
перспективным.
Поливинилспиртовые волокна находят применение также при
производстве бумаги [31]. В отличие от других синтетических
волокон поливинилспиртовые волокна обладают большей гидрофиль-
ностью, лучшей смачиваемостью и способностью диспергироваться
в воде, что обеспечивает целесообразность применения их при
получении бумаги мокрым способом. Количество добавляемого
поливинилспиртового волокна составляет 10—20% от массы целлюлозы.
Введение в состав композиции термопластичного волокна облегчает
склеивание целлюлозных волокон в бумаге. Полученная из смеси
целлюлозных и поливинилспиртовых волокон бумага и картон
используются для фильтрации воздуха, моторных топлив, масел,
особенно для отделения их от небольших количеств воды.
8.6. МОДИФИЦИРОВАННЫЕ ПОЛИВИНИЛСПИРТОВЫЕ
ВОЛОКНА
Большие возможности имеются и для модификации свойств
поливинилспиртового волокна аналогично модификации свойств
целлюлозных волокон, содержащих те же реакционноспособные гидр-
оксильные группы.
Для химической модификации этих волокон могут быть
использованы различные методы, из которых наиболее
перспективным является синтез привитых сополимеров и осуществление по-
лимераналогичных превращений реакционноспособных групп в
макромолекуле полимера.
Свойства получаемых волокон могут быть модифицированы
также формованием волокна из растворов смеси поливинилового
спирта с другими водо- или щелочерастворимыми полимерами,
например с казеином.
Систематические исследования в области модификации
поливинилспиртовых волокон проведены советскими исследователями,
и результаты этих работ суммированы в монографии [32, с. 223].
Для прививки виниловых полимеров к поливиниловому спирту
в молекулу этого полимера в процессе ацеталирования вводили
небольшое количество карбоксильных групп (в результате
обработки малеиновой кислотой) или альдегидных групп (при ацета-
лировании малеиновым, терефталевым или другим диальдегидом)
[33]. При последующей обработке волокна, содержащего неболь-
a.6. Модифицированные поливинилспиртовые волокна 267
шое количество альдегидных групп, разбавленным раствором
перекиси водорода эти группы переводили в гидроперекисные. При
распаде последних образуются макрорадикалы, к которым
прививались различные мономеры (акриловая кислота, 2-метил-5-винйл-
пиридин, акрилонитрил) [34].
Для прививки к поливинилспиртовому волокну был применен
также и другой метод, использованный ранее для прививки к
целлюлозному волокну, — метод предварительного введения в
макромолекулу ароматической аминогруппы и последующего ее диазоти-
рования [35]. При прививке этим методом метилвинилпиридина
были получены анионообменные волокна.
Значительный интерес представляет метод частичной или
полной дегидратации поливинилового спирта. Сущность этого метода
заключается в следующем. Поливинилспиртовое волокно
обрабатывают дегидратирующими реагентами (NaHSO4, бензолсульфо-
кислота) в присутствии катализаторов в вакууме или в атмосфере
азота в среде органических растворителей (толуол, СС14) при
75—200°С в течение 8—20 мин [36]. При полной дегидратации
поливинилового спирта образуется волокно с .системой сопряженных
двойных связей —СН=СН—СН=СН, обладающее
полупроводниковыми свойствами [37]. Такое волокно характеризуется высокой
термостойкостью и может быть использовано в качестве
ионообменного волокна, образующего комплексные соединения с
солями серебра, меди и других металлов [38].
Дегидратированное при 200 °С поливинилспиртовое волокно
сохраняет 50% начальной прочности, а при 300°С —25—30%
начальной прочности [39]. После нагрева такого волокна на воздухе
при 200 °С в течение 50 ч оно сохраняет 85—95% прочности
исходного дегидратированного волокна (прочность определяли при
нормальной температуре).
Дегидратированное поливинилспиртовое волокно способно
вступать в реакцию диенового синтеза, в результате которого
образуются термостойкие волокна, не разлагающиеся при нагреве до
400 °С.
Указанными методами модификации А. И. Меос и Л. А. Вольф
получили поливинилспиртовые волокна, обладающие теми же
свойствами, что и модифицированные гидратцеллюлозные
волокна — негорючестью и повышенной светостойкостью. Они получили
также бактерицидные волокна присоединением к
модифицированному поливинилспиртовому волокну небольших количеств нитро-
фурилакролеина (волокно летилан) [40]. Эти волокна обладают не
только антимикробным, но и антигрибковым действием.
Получены также различные ионообменные поливинилспиртовые
волокна, например слабые катиониты (прививкой акриловой или
метакриловой кислоты) и сильные катиониты, содержащие суль-
фогруппы. Сульфогруппы вводили методом ацеталирования ди-
альде'гидами и последующей обработкой раствором бисульфата
268 Глава 8. Производство поливинилспиртовых волокон
калия или сульфированием дегидратированного волокна [41].
Такие же ионообменные волокна получены при взаимодействии
поливинилспиртового волокна с водорастворимыми сульфопроиз-
водными цианурхлорида [42].
Приведенные данные показывают, что модифицированные
волокна (кроме дегидратированных, содержащих систему
сопряженных двойных связей), обладающие примерно одинаковыми
свойствами, могут быть получены химическими превращениями
реакционноспособной группы в двух гидроксилсодержащих
полимерах — целлюлозе и поливиниловом спирте. Учитывая более
высокую стоимость и меньшую доступность поливинилспиртовых
волокон по сравнению с гидратцеллюлозными, в частности с вискозным
волокном, можно сделать вывод, что модифицированные поливи-
нилспиртовые волокна целесообразно использовать только в тех
случаях, когда по тем или иным причинам не могут быть
применены модифицированные вискозные волокна. Это относится в
первую очередь к ионообменным волокнам, используемым в кислых
ередах, в которых макромолекулы целлюлозы постепенно гидро-
лизуются и деструктируются.
ЛИТЕРАТУРА
1. Wells D., Morgan N.. Text. Res. J., 1960, v. 30, p. 668—671.
2. S a k u г a d a I., Koll. Z., 1954, Bd. 139, N 3, S. 155—159.
3. Сакур ада И., Хим. и технол. полимеров, 1964, т. 8, № 10, с. 80—107.
4. Перепелкин К. Е., Бородина О. О., Шешков Н. К., Хим. волокна,
1962, № 4, с. 17—19.
б. Ефимова С. Г., Быркииа Л. Ф., Панков С. П., Хим. волокна, 1966,
№ 4, с. 17—20.
6. Aleksandru L., Grohanu Т., Markt an E. e. a., Faserforsch, u.
Textiltechn., 1960, Bd. 11, S. 213—218.
7. Перепелкин К. E., Константинова Г. В., Хим. волокна, 1961, № 6.
с. 9—11.
8. Начиикин О. И., Шурьева Г. Г., Константинова Н. Г. и др.,
Хим. волокна, 1965, № 6, с. 26—28.
9. Перепелкин К. Е., Рабинович В. И., Хим. волокна, 1964 № 3,
с. 11—14.
10. Начин кин О. И., Перепелкин К. Е., Хим. волокна 1963 № 2,
с. 12—14.
11. Начин кин О. И., Перепелкин К. Е., Хим. волокна 1963 № 1
с. 13-15.
12. П ер епе л ки н К. E., H а чин кин О. И., Хим. волокна 1964 № 2,
с. 19—21.
13. Рабинович В. И., Перепелкин К. Е., Хим. волокна 1965 № 2,
с. 18.
14. Me ос А. И., Вольф Л. А., Еерещак Л. П., Хим волокна 1961 № 5,
с. 21—23.
15. Смирнов В. С, Шапиро Е. С, Пром. хим. волокон, 1969, № 8, с. 5—7.
16. Перепелкин'К. Е., К а д и и с к и й Б. Б., Бородина О. О. и др.. Хим.
волокна, 1967, № 1, с. 20—23.
17. Афанасьева Г. Н., Вольф Л. А., Калинина Т Н. и др Хим
волокна, 1967, № 5, с. 6—8.
Литература
269
с. 15—17.
др., Хим.
: Карбоцепные
волокна, 1960,
Я-, Козлова М. В., Фокин Е. П., Хим. волокна,
А., Афанасьева Г. Н., Хим. волокна, 1963,
A., Me ос А. И. и др., Хим. волокна,
18. Л е н и н к о в О. С., У т е в с к и й Л. Е., П е р е п е л к и н К. Е. и др., Хим.
волокна, 1969, № 6, с. 45—47.
19. У те веки й Л. Е., Яновская Н. Б., Хим. волокна, 1966, №
20. П е р е п е л к и н К. Е., У т е в с к и й Л. Е., Орлова Н. И.
волокна, 1964, № 5, с. 17—19.
21. M о t h о у m а Т., «Kunststoffe», 1960, Bd. 50, S. 3—6.
22. M е ос А. И., Шиманский П. Е., Вольф Л. А. В кн
волокна. М., «Химия», 1966, с. 210—214.
23. Dorret В., Text. Manufactur, 1956, v. 82, p. 636; 984—986.
24. M e о с А. И., Вольф Л. А., Гиллер С. А. и др,, Хим.
№ 4, с. 19—21.
25. Ко л он т а р о в И.
1966, № 4, с. 15—17.
26. Me ос А. И. Вольф Л.
№ 3, с. 18-20.
27. А ф а н а с ь е в а Г. Н., Вольф Л
1963, № 5, с. 16—18.
Токарева Л. Г., Ж а н д а р е в а 3. А., Михайлов Н. В., Хим. волокна,
1966, № 5, с. 12—14.
П е р е п е л к и н К. Е., П е р е п е л к и н а М. Д., Текст, пром., 1963, № 6,
с. 20-21.
Перепел кии К. Е., Ленников О. С, Утевский Л. Е. и др., Хим.
волокна, 1969, № 5, с. 69—71.
31. Дергачев В. С, Иванов С. Н., Перепел кин К. Е. «Научные труды
ЛТА», 1971, № 143, с. 73—76.
32. Me ос А. И., Вольф Л. А. Химические волокна со специальными
свойствами. М., «Химия», 1971. 223 с.
33. Максимов А. М., Вольф Л. А., Меос А. И. и др., Хим. волокна,
1969, № 5, с. 14—17.
34. Бабкина И. Ю., Гордеев Ю. М., Китае в К. Н., Хим. волокна, 1970,
№ 3, с. 13—16.
35. Сергеева Л. М., Роговин 3. А., Хим. волокна, 1967, № 1, с. 27—29.
36. Вольф Л. А., Меос А. И., Кириленко Ю
Д. И. Менделеева, 1966, т. 11, с. 654—658.
37. Кириленко Ю. К., Меос А. И., В
с. 209—212.
38. Вольф Л. А., Меос А. И., Удобин 3. А., Хим. волокна
1820
28.
29.
30.
к,
Ю. К. и др.,
Вольф Л. А., ЖПХ,
ф
с. 18—20.
39. Вольф Л. А., Кириленко Ю. К., Хим. волокна, 1969,
40. Вольф Л. А., Меос А. И., Кот едкий В. В. и др., Х
N° 6, с. 16—19.
41. Вольф Л. А., Кириленко Ю. К., Котецкий
Д. И. Менделеева, 1966, т. 11, с. 631—635.
42. Сергеева Л. С, Вирник А. Д., Роговин 3. А., Хим
№ 2, с. 25—28.
№
ЖВХО им.
1965 т 38
1968 № 5,
3, с. 15—17.
Хим. волокна 1963,
В. В. ЖВХО им.
волокна 1967,
Глава 9
ПРОИЗВОДСТВО ПОЛИОЛЕФИНОВЫХ
волокон
Из синтетических полимеров, используемых для производства
волокон, одними из наиболее доступных являются полиолефияы.
Однако еще 15—20 лет назад получение волокон из полимеров
этого класса, в макромолекуле которых не содержится полярных
групп, считалось нецелесообразным вследствие низкой прочности
и теплостойкости вырабатываемых из них изделий. Формование
волокон с ценным комплексом свойств стало возможным1 лишь
после того, как были разработаны методы синтеза полиэтилена
строго линейной структуры и особенно стереорегулярных (изотак-
тических) полимеров из а-олефинов (пропилена, бутилена и др.).
При использовании таких полимеров удалось резко улучшить
свойства получаемых материалов (возрастает интенсивность
межмолекулярного взаимодействия и соответственно повышается весь
комплекс физико-механических свойств полимера) и тем самым
создать необходимые условия использования полиолефинов для
производства волокна.
К стереорегулярным относятся природные гетероцепные
полимеры (например, целлюлоза) и синтетические карбоцепные
высокомолекулярные соединения, полученные цепной полимеризацией
в особых условиях. Синтез стереорегулярных карбоцепных
полимеров осуществляется преимущественно по ионному механизму *
с применением гетерогенных катализаторов, предложенных Цигле-
ром и Натта [1, с. 100—104]. Полученные в этих условиях полимеры
а-олефинов имеют стереорегулярную структуру и содержат в эле*
ментарном звене асимметричные атомы углерода одной и той же
пространственной конфигурации.
По сравнению с высокомолекулярными соединениями
нерегулярной структуры стереорегулярные полимеры имеют более
вытянутые макромолекулы в конденсированном состоянии,
повышенную плотность, более низкую растворимость, высокие
физико-механические свойства (в частности, более высокую прочность и
температуру плавления или размягчения).
Для стереорегулярных поли-а-олефинов характерны: типичная
рентгенограмма кристаллических полимеров, явление двойного лу-
* Установлено, что в определенных условиях стереорегулярные полимеры
могут быть синтезированы также и методом радикальной полимеризации.
9.1. Полипропиленовые волокна 271
чепреломления, возможность ориентации и наличие
высокоэластического удлинения.
Естественно, что этот метод резкого улучшения свойств одного
из наиболее доступных полимеров должен был найти применение
и в производстве синтетических волокон, так как он позволяет
значительно расширить число мономеров и полимеров, которые
могут быть использованы для получения высококачественных
волокон.
Первое сообщение [2] о получении волокон из стереорегулярных
поли-а-олефинов появилось в 1956 г. Начиная с 60-х годов
текущего столетия, производство волокон из полиолефинов строго
линейного или стереорегулярного строения приобретает
промышленное значение. В 1963 г. мировое производство полиолефиновых
волокон превысило 40 тыс. т, в 1965 г. составило 160 тыс. т, а в
1970 г. — 230 тыс. т.
Оба мономера (этилен и пропилен), используемые для синтеза
полиэтилена и полипропилена, получаются при пиролизе и
крекинге нефти. Выход этилена и пропилена при пиролизе нефти
составляет соответственно 8—10 и 4—5% (от массы сырой нефти),
а при крекинге 1,5—3%. В отличие от этилена, являющегося
исходным сырьем при синтезе разнообразных продуктов (этанола,
уксусной кислоты и др.) и различных винильных мономеров (винил-
хлорида, винилиденхлорида, винилацетата), пропилен не находит
пока столь широкого, квалифицированного применения.
Использование этого доступного сырья для синтеза дешевых
волокнообразующих полимеров представляет большой
практический интерес. Этим обстоятельством, а также некоторыми
специфическими преимуществами полипропиленового волокна
объясняется тот факт, что производство волокна из полипропилена
получило наиболее широкое развитие. Выработка этого волокна в
1970 г. составила более 85% общего количества получаемых
полиолефиновых волокон.
Методы получения различных стереорегулярных полиолефинов
и линейного полиэтилена (так называемого полиэтилена низкого
давления) и технологический процесс переработки их в волокно
примерно идентичны. Поэтому ниже вкратце излагается
технология наиболее массового типа полиолефиновых волокон —
полипропиленового волокна — с указанием особенностей производства и
свойств других полиолефиновых волокон, в частности
полиэтиленового волокна.
9.1. ПОЛИПРОПИЛЕНОВЫЕ ВОЛОКНА
Синтез полипропилена, являющегося исходным материалом для
получения не только волокон, но и пластических масс, как
правило, производится на химических заводах. На заводы
синтетического волокна обычно поступает готовый высушенный полимер, в,
большинстве случаев — гранулированный.
272 Глава 9. Производство помюлефиновых волокон
9.1.1. Синтез полипропилена
Полипропилен может быть получен периодическим и
непрерывным способами.
При периодическом способе полимеризация пропилена
проводится без растворителя (полимеризация в массе) или в
присутствии реагента, растворяющего мономер. В последнем случае
уменьшается количество мономера, загружаемого в реакционный
аппарат, но значительно облегчается отвод тепла. В качестве
растворителя применяются гептан и бензин «галоша» или, что
значительно лучше, пропан, а также пропан-пропиленовая фракция.
Полимеризация пропилена протекает по ионному механизму в
присутствии катализатора — смеси триалкилалюминия (обычно
триэтилалюминия) с трех- или четыреххлористым титаном, взятых
в различных соотношениях. Триэтилалюминий может быть
заменен триизобутилалюминием, благодаря чему заметно уменьшается
опасность воспламенения катализатора.
Соотношение между компонентами, входящими в состав смеси
катализаторов, существенно влияет на содержание
кристаллической и аморфной фракций в получаемом полимере. Количество
катализатора, применяемого для полимеризации пропилена,
составляет обычно 0,3—0,6% массы мономера. Выход полимера
достигает 90—95% общего количества мономера, загруженного в
аппарат.
По завершении реакции давление в аппарате снижают, а
растворитель отгоняют. Образующийся полимер нерастворим в
применяемом растворителе и выпадает из раствора в виде белого
порошка, который после разложения катализатора спиртом
промывают водой, отжимают и сушат.
Свойства получаемого полимера зависят от продолжительности
и температуры полимеризации, концентрации мономера в
реакционной смеси и состава катализатора (в частности, от
соотношения триэтилалюминия и треххлористого титана).
Продолжительность полимеризации. Чем больше количество
катализатора и выше концентрация мономера в реакционной смеси
(т. е. чем выше давление в аппарате), тем меньше должна быть
продолжительность полимеризации. Обычно продолжительность
полимеризации (при конверсии до 90% мономера) составляет 4—6 ч.
С увеличением продолжительности процесса молекулярный вес
получаемого стереорегулярного полимера и соотношение
кристаллических и аморфных фракций в нем не изменяются.
Температура полимеризации. С повышением температуры
полимеризации закономерно снижается молекулярный вес полимера.
Одновременно повышается скорость реакции полимеризации.
Данные о влиянии температуры полимеризации на изменение
соотношения кристаллической и аморфной фракций в полимере
в литературе не приводятся.
9.1. Полипропиленовые волокна 273
Концентрация мономера. Чем выше концентрация мономера,
тем больше скорость полимеризации и тем выше молекулярный
вес получаемого полимера [2].
Количество катализатора. Расход катализатора, применяемого
при полимеризации пропилена, зависит от его активности.
Активность катализатора характеризуется количеством пропилена (в
молях), вступившего в реакцию полимеризации, которое приходится
на 1 моль катализатора в 1 ч при давлении 1 кгс/см2 A05 Па).
Активность катализатора зависит от соотношения компонентов
каталитической смеси. Например, при полимеризации пропилена
на каталитической системе триизобутилалюминий и ТіС14
максимальная скорость полимеризации достигается при соотношении
этих компонентов, равном 1,5—2,5. При увеличении этого
соотношения скорость полимеризации понижается.
Естественно, что с увеличением количества катализатора
скорость реакции повышается. Однако возможность значительного
повышения скорости реакции лимитируется необходимостью
отвода большого количества тепла, выделяющегося в процессе
реакции. Кроме того, с увеличением концентрации катализатора в
реакционной смеси закономерно понижается молекулярный вес
получаемого полимера.
Соотношение компонентов каталитической системы определяет
также структуру и свойства получаемого полимера. От характера
галогенида титана, применяемого в качестве одного из
компонентов каталитической системы, зависит в основном соотношение
между кристаллической и аморфной фракциями в получаемом
полипропилене. Это относится также и к другим стереорегулярным
полимерам, получаемым ионной полимеризацией в присутствии
гетерогенного катализатора. Если, например, при полимеризации в
одних и тех же условиях и использовании в качестве компонента
каталитической системы треххлористого титана содержание
кристаллической фракции в получаемом полимере составляет 85—95%,
то при замене этого реагента эквимольным количеством четырех-
хлористого титана содержание кристаллической фракции снижается
до 60—65% *.
Содержание кристаллических фракций в высокомолекулярном
стереорегулярном полипропилене, используемом для производства
синтетических волокон, достигает 85—95%.
Существенное влияние на изменение молекулярного веса
полимеров, получаемых по реакции стереорегулярной полимеризации,
оказывают добавки триэтиламина, производных тиомочевины, и
особенно водорода [4]. Молекулярный вес полиолефина снижается
пропорционально корню квадратному из концентрации водорода в
реакционной смеси.
* Более детально влияние химического строения соединений, входяіцих
в состав комплексных катализаторов, на выход кристаллического (стереорегу-
лярного) полиолефина рассмотрено в работе [3, с. 20—48].
274 Глава 9. Производство полиолефиновых волокон
9.1.2. Свойства полипропилена
Для характеристики свойств полипропилена, используемого для
получения волокон, так же как и других волокнообразующих
полимеров, основное значение имеют:
структура полимера, и в частности соотношение
кристаллической и аморфной фракций;
молекулярный вес полимера;
полидисперсность полимера;
стойкость к термоокислительным и фотохимическим
воздействиям;
текучесть полимера.
Методы исследования полипропилена и определения его
основных показателей не унифицированы и детально не разработаны.
Опубликованные по этому вопросу данные кратко суммированы
ниже.
Структура полимера. Стереорегулярные полимеры в отличне от
других природных и синтетических высокомолекулярных
соединений могут быть разделены путем фракционного растворения на
аморфную и кристаллическую фракции. Аморфные фракции даже
высокого молекулярного веса благодаря нерегулярному строению
макромолекул и соответственно значительно меньшему
межмолекулярному взаимодействию растворяются при нормальной
температуре в различных неполярных растворителях, в частности в
эфире, толуоле, гептане. Стереорегулярная кристаллическая фракция
полипропилена достаточно высокого молекулярного веса не
растворяется при нормальной температуре ни в одном из известных
растворителей. Только при 80—100 °С эта фракция растворяется
в некоторых неполярных растворителях (уайт-спирите, толуоле,
хлорированных углеводородах).
Резкое различие в растворимости аморфной и кристаллической
фракций полипропилена используется для их разделения и
определения соотношения этих фракций в полимере, применяемом для
формования волокна. Однако в неполярных растворителях при
нормальной температуре кроме аморфных фракций растворяются
также и низкомолекулярные стереорегулярные фракции
полипропилена [5]. Поэтому результаты определения содержания
аморфных фракций путем фракционного растворения, особенно
полипропилена, подвергнутого термической или термоокислительной
деструкции, могут оказаться в ряде случаев завышенными.
Однозначных данных о влиянии содержания аморфной
фракции в полипропилене на условия формования и свойства
получаемого волокна пока нет. Как правило, с увеличением содержания
аморфной фракции повышается текучесть расплава
полипропилена и пластичность получаемого волокна. Одновременно
снижается прочность волокна и появляется липкость, что усложняет
его последующую текстильную обработку.
9.1. Полипропиленовые волокна 275
Молекулярный вес аморфной фракции полипропилена может
в ряде случаев превышать молекулярный вес кристаллической
фракции. Например, молекулярный вес аморфной фракции
полипропилена, полученного полимеризацией пропилена в присутствии
триизобутилалюминия и четыреххлористого углерода, составляет
200 000, а кристаллической — 100 000—160 000. Температура
плавления аморфной и кристаллической фракций при переходе в вяз-
котекучее состояние составляет соответственно 75—80 и 160—
170 °С.
Соотношение кристаллической и аморфной фракций можно
регулировать, изменяя условия получения полимера или частично
удаляя аморфную фракцию из полипропилена. Возможность
регулировать соотношение этих фракций является характерной
особенностью стереорегулярных полимеров, и в частности
полипропилена, используемого для формования волокна.
Молекулярный вес полипропилена определяют в основном вис-
козиметрическим методом. Для этого препараты стереорегуляр-
ного полимера растворяют при 80—100 °С в тетралине или
декалине. Расчет средневесового молекулярного веса производится по
формуле [6]
[т]] = 1,04 - 10М0'8
где [т)] — характеристическая вязкость раствора полипропилена в декалине
при 135 °С.
Для определения молекулярного веса можно воспользоваться
и осмометрическим методом. Так как растворы стереорегулярного-
полипропилена стабильны только при повышенной температуре,
то определение осмотического давления следует проводить также
при повышенной температуре [7].
Волокно обычно получают из стереорегулярного
полипропилена с молекулярным весом 80 000—250 000 (степень
полимеризации 1900—5900). С повышением молекулярного веса затрудняется
переработка полипропилена из расплава, но одновременно
увеличивается прочность получаемого волокна (при увеличении
молекулярного веса до известного предела). Зависимость
механических свойств волокна при формовании его из раствора от
молекулярного веса полипропилена характеризуется следующими
данными [8]:
Молекулярный вес Прочность, гс/текс Удлинение, %
200 000 62,7 26
118000 56 27
78 000 51,0 27
Полидисперсность. При характеристике полипропилена обычно
ограничиваются определением содержания аморфной и
кристаллической фракций. Однако определение полидисперсности для
полипропилена, используемого при получении волокон, не менее
существенно, чем для других волокнообразующих полимеров.
276
Глава 9. Производство полиолефиновых волокон
І 20
10
О 50 WO 150 ZOO
Продолжительность , ч
Рис. 9.1. Стойкость полипропилена (J)
и полиэтилена B) к
термоокислительной деструкции при 150 °С.
Возможность проведения этого анализа обычно применяемыми
методами фракционного растворения или осаждения затрудняется
различной растворимостью кристаллической и аморфной фракций.
Поэтому при изучении полидисперсности полипропилена, а также
других стереорегулярных полимеров необходимо раздельно
определять полидисперсность аморфной и кристаллической фракций.
Для этого предложен следующий
метод [9].
Полидисперсность аморфной
фракции определяют
фракционным осаждением полипропилена
ацетоном из 0,5%-ного раствора
его в толуоле. Таким путем
аморфная фракция стереорегу-
лярного полипропилена была
разделена на 6 фракций. Молекуляр-
150 ные веса 1-й и 5-й фракций
различались более чем в 12 раз.
Кристаллическая фракция
подвергалась фракционному
растворению в уайт-спирите путем
обработки полипропилена этим
растворителем (при модуле 100) в течение 4 ч при различных
температурах, постепенно повышающихся от 20 до 100 °С. При
повышении температуры обработки значения молекулярного веса
фракций, переходящих в раствор, соответственно увеличивались. Этим
методом образец кристаллического полипропилена был разделен
на 5 фракций, причем молекулярный вес крайних фракций
отличался более чем в 10 раз.
Кристаллическая фракция полипропилена растворяется
значительно хуже, чем аморфная фракция препарата, обладающего в
5—6 раз более высоким молекулярным весом. Этот факт является
дополнительной иллюстрацией большого влияния структуры
полимера на его растворимость.
Несмотря на трудоемкость описанного метода анализа,
определять полидисперсность целесообразно для того, чтобы иметь
более детальную характеристику полипропилена, используемого для
формования волокна.
Известен и другой метод фракционирования полипропилена.
В качестве бинарной смеси для фракционирования была
использована смесь декалина и ацетофенона [10]. Фракционирование
проводили на колонке при различных температурах (в интервале
НО—170 °С).
Стойкость к термоокислительным и фотохимическим
воздействиям. Полипропилен, так же как и другие полиолефины,
сравнительно стоек к повышенным температурам при отсутствии
одновременно протекающих окислительных процессов. Например, при
9.1. Полипропиленовые волокна 277
нагреве стереорегулярного полипропилена в глубоком вакууме или
в атмосфере инертного газа (азота) в течение 3 ч при 150 °С
удельная вязкость его растворов заметно не изменяется. В отсутствие
кислорода [11] полиолефины обладают достаточной стойкостью
даже при 300 °С. Однако полипропилен, в элементарном звене
которого содержится третичный атом углерода и подвижный атом
водорода (метильной группы), значительно менее стоек к действию
кислорода при повышенных температурах, чем другие
полиолефины, в частности полиэтилен. Даже после непродолжительного
нагрева полипропилена на воздухе при 15Q°C его молекулярный
вес уменьшается более чем в 30 раз и соответственно резко
снижается прочность волокна.
Данные о стойкости полипропилена и полиэтилена к
термоокислительной деструкции, характеризуемой количеством
поглощенного кислорода, приведены на рис. 9.1. Как видно из рис. 9.1,
скорость термоокислительной деструкции полипропилена значительно
больше, чем полиэтилена.
В одних и тех же условиях нагрева аморфная фракция
полипропилена деструктируется-интенсивнее, чем кристаллическая.
Интенсивность термоокислительной деструкции полипропилена резко
уменьшается при добавлении небольших количеств антиоксиданта.
Данные о влиянии некоторых антиоксидантов на изменение
свойств полипропилена после нагрева на воздухе в течение 3 ч
при 150°С приводятся ниже:
Антнокситнт Растворимость Удельная вязкость
Антиоксидант в толуоле 0,5%-ного раствора
Без добавок 65 0,05
Дикрезилпропан 19,5 1,10
Три-грег-бутилфенол .... 16,5 1,02
о, о'-Диоксидифенил 18,7 1,12
Антиоксиданты добавляли в количестве 0,5% от массы
полимера. Влияние различных антиоксидантов на изменение
характеристической вязкости полипропилена [12] после прогрева при 200 °С
показано на рис. 9.2.
При добавлении неозона Д или дифениламина в количестве
0,5% вязкость полипропилена после нагрева в течение 30 мин при
200 °С почти не изменяется, в то время как вязкость исходного
(не содержащего антиоксиданты) полимера после нагрева в тех
же условиях в течение 5 мин снижается более чем в 4 раза. Опыты
показали, что для ингибирования термоокислительной деструкции
достаточно добавить 0,1—0,25% антиоксиданта. Увеличение
количества антиоксиданта не дает существенных результатов.
Влияние химического состава стабилизаторов на эффективность
их действия при термоокислительной деструкции полипропилена
изучено советскими исследователями. Согласно полученным
данным, наиболее эффективны смеси серосодержащих органических
соединений и производных фенолов, причем при использовании
278
Глава 9. Производство полиолефиновых волокон
этих смесей наблюдается отчетливо выраженный синергетический
эффект [13]. В качестве серосодержащих соединений рекомендуется
применять тиокислоты (тиоуксусную, хлортиоуксусную) [14], тио-
мочевину, и особенно ее производные, содержащие фенольные или
Продолжительность прогреба, мин
Рис. 9.2. Влияние различных антноксидантов (прн добавлении их в
количестве 0,5%) на характеристическую вязкость растворов полипропилена:
/ — неозон Д; 2—нонол; 3—дифениламин; 4—три-грег-бутилфенол; 5—дикреэнлпро-
пан; ff—пирогаллол; 7—меркаптобензимидазол; 8—без антиоксидантов.
нафтильные радикалы, например дифенилтиомочевину или динаф-
тилтиомочевину [15]:
¦HN-
г_г—мн—/ \
-С-
II
S
. На рис. 9.3 приведены данные, иллюстрирующие
комбинированное действие двух стабилизаторов (каждый из них
сравнительно малоэффективен) при термоокислительной деструкции
полипропилена [16]. Как видно из рис. 9.3, каждый из примененных
стабилизаторов (додецилсульфид и 2,6-ди-г/?ег-октил-4-метилфе-
нол) мало йнгибируют реакцию окисления полипропилена, в то
время как смеси этих реагентов в значительно меньших мольных
количествах оказывают сильное ингибирующее действие, заметно
увеличивая индукционный период этой реакции.
Для предотвращения или замедления процесса термоокисли:
тельной деструкции полипропилена кроме антиоксидантов могут
быть применены и другие типы стабилизаторов, механизм дей-,,
ствия которых основан на взаимодействии с макрорадикалами, об-
9.1. Полипропиленовые волокна
279
I
гоо
700
О 0,0Ь 0,08 0,12 0,16
Концентрация стаіиліипторцг
моль/кг
Рис. 9.3. Зависимость периода
индукции термоокислительной
деструкции полипропилена от природы
стабилизатора:
/ — додецил сульфид; 2 — 2і6-ди-грег-октил-
4-метилфенол; 3 —смйсь додецилсульфида
и 2,6-ди-грег-окгил 4-метилфенола.
разующимися в начальной стадии цепного распада
макромолекулы, или на разрушении образовавшихся перекисных групп.
Полипропилен недостаточно стоек и к фотохимическим воздей--
ствиям. Однако и этот практически важный показатель может
быть значительно улучшен
добавлением небольших количеств
фотостабилизаторов. Такими
стабилизаторами являются
преимущественно пигменты, поглощающие
или отражающие
ультрафиолетовые лучи с длиной волны 300—
400 нм, а также сажа. На
защитное действие этих добавок
существенное влияние оказывает
их цвет.
В результате добавления
небольших количеств пигментов [17]
потеря прочности волокна после
облучения значительно
уменьшается. Если волокно, не
содержащее стабилизаторов, после об"
лучения в течение 250 ч в
определенных условиях полностью
теряет прочность, то при добавлении некоторых пигментов (в
частности, голубого и желтого цвета) после облучения в течение 1000 ч
прочность волокна снижается всего на 20—25%.
Одним из наиболее эффективных светостабилизаторов, действие
которого основано на сорбции ультрафиолетовых лучей, является
2-окси-4-алкоксибензофенон [18], а также га-октилфенилсалицилат
и триоксибензофенон. Стойкость волокна к термоокислительной
деструкции при добавлении этого класса светостабилизаторов не
повышается. Поэтому целесообразно добавлять смесь
стабилизаторов— антиоксидантов и веществ, сорбирующих
ультрафиолетовые лучи.
Текучесть. Одним из важных показателей, определяющих
условия формования волокна из расплава, является текучесть
полимера. При исследовании реологических свойств полипропилена
необходимо учитывать ряд специфических особенностей, присущих
этому классу стереорегулярных полимеров.
Вязкость расплава полипропилена значительно выше вязкости
расплава гетероцепных полимеров, используемых для формования
волокна из расплава. Например, вязкость расплава
полипропилена, поступающего на формование волокна, в 30—40 раз выше
вязкости расплава капрона или лавсана [8]. Это обстоятельство
обусловливает необходимость изменения аппаратурного
оформления процесса формования волокна из расплава полипро--
пилена.
280
Глава 9. Производство полиолефиновых волокон
Кристаллическая и аморфная фракции полипропилена резко
различаются по текучести, а также по характеру
термомеханических кривых (рис. 9.4).
Изменение молекулярного веса полимера не влияет на общий
вид термомеханической кривой, но оказывает существенное
влияние на изменение начальной температуры плавления материала
3Z0
180
240
г-,
1
и
и
|
II
[
1
по
во
о го чо во во юо по но wo wo zoo
Tennepamypa, °С
Ряс. 9.4. Термомеханические кривые кристаллической и аморфной фракций
полипропилена:
/—исходный полимер. [Г|] = 1,35; 2 —кристаллическая фракция исходного полимера, [Г|] = 1,45,
3—фракция, растворимая в толуоле после прогрева при 150 °С в течение 5 ч. [r|J = 0,01;
4—фракция, растворимая в толуоле после прогрева при 180 °С в течение 12 ч, [ті] = 0,03.
[19]. Например, при уменьшении молекулярного веса
кристаллической фракции в 5 раз начальная температура плавления
полипропилена снижается примерно на 50 °С. Следовательно, изменяя
молекулярный вес полимера и соотношение кристаллической и
аморфной фракций в нем, можно в широких пределах изменять
текучесть полипропилена. При возрастании температуры с 200 до
260 °С вязкость расплава полипропилена уменьшается в 2—4 раза.
При добавлении небольших количеств эффективных антиоксидан-
тов можно использовать эту зависимость для повышения текучести
расплава полипропилена.
Текучесть полипропилена можно повысить также при
добавлении пластификаторов. Например, при введении небольших
количеств E—10% от массы полимера) аморфной фракции
полипропилена или каучукоподобного полимера, хорошо совмещающегося
с полипропиленом (например, полиизобутилена), текучесть
расплава значительно повышается и, таким образом, облегчается
формование волокна [20].
9.1. Полипропиленовые волокна 281
Реологические исследования расплавов полипропилена
проведены недостаточно полно. Не изучена, например, возможность
значительного снижения вязкости расплава полипропилена
добавлением небольших количеств низкомолекулярных веществ
аналогично тому, как это имеет место при получении полиамидных волокон.
Этот вопрос имеет большое значение для направленного
регулирования свойств расплава полипропилена при формовании из него
волокна.
9.1.3. Получение полипропиленового волокна
Технологический процесс производства полипропиленового
волокна принципиально не отличается от получения других
синтетических волокон, формуемых из расплава.
Обычно технологический процесс производства
полипропиленового волокна включает следующие основные операции: 1)
формование; 2) вытягивание; 3) терморелаксацию; 4) крутку и
перемотку (при получении текстильной нити).
Формование волокна. Полипропиленовое волокно можно
получить формованием из раствора или из расплава полимера.
Как уже указывалось выше, стереорегулярный полипропилен
растворяется при повышенных температурах A00—160 °С) в
различных растворителях, в частности в тетралине, декалине, высоко-
кипящей A20—180 °С) фракции керосина и в других неполярных
растворителях. Используя эти растворители, можно получить
концентрированные вязкие растворы, содержащие 25—30% полимера,
из которых можно формовать волокно сухим или мокрым
способом. Естественно, что для испарения этих растворителей в шахту
прядильной машины должен подаваться воздух или инертный газ,
нагретый до 150—200 °С.
Единственным преимуществом способа формования волокна из
растворов полимера является возможность использовать
полипропилен более высокого молекулярного веса и получать волокна с
более высокими механическими свойствами. По-видимому, в
отдельных случаях для получения тонкого полипропиленового
волокна повышенной прочности этот способ может представить
известный интерес.
Если имеется возможность получения волокна из того же
полимера формованием из расплава, то нецелесообразно применять
способ формования из растворов, так как он менее экономичен.
Формование из расплава является основным способом получения
волокна из полипропилена и других широко применяемых поли-
олефинов.
Для формования волокна из расплава полипропилена
используются прядильные машины, подобные по конструкции машинам,
применяемым при производстве полиамидных и полиэфирных
282 Глава 9. Производство полиолефиновых волокон
волокон. Однако прядильные машины, применяемые для
формования полипропиленового волокна, имеют некоторые отличия.
Основное отличие прядильных машин для формования волокна
из полипропилена состоит в том, что вследствие более высокой
вязкости расплава полимера, составляющей от 1000 до 3000 П,
осуществить его равномерную подачу к прядильному насосику
самотеком, как это имеет место, например, при формовании
полиамидного волокна, не представляется возможным. Поэтому
высоковязкий расплав подается к прядильному насосику при помощи
обогреваемого шнека, имеющего обычно 3—4 зоны обогрева.
Температура расплава, поступающего в насосик, составляет 230—240 °С,
а в выходном отверстии шнека и в фильере — соответственно 260
я 240 °С. За время прохождения полипропилена через шнек
гранулы полипропилена расплавляются, и расплавленный
полипропилен под давлением 20—30 кгс/см2 подается к прядильному
насосику B-Ю6 Па —3-Ю6 Па).
При применении такого шнека продолжительность пребывания
расплава в зоне высоких температур снижается со 100 до
10—15 мин. В результате интенсивность термоокислительной
деструкции полипропилена уменьшается.
Температура в шнеке на 80—100 °С выше, чем в расплавопро-
воде или на фильере, а температура, при которой происходит
формование волокна, на 80—120 °С превышает температуру плавления
полипропилена. При недостаточно тщательной стабилизации
полипропилена при формовании происходит термоокислительная"
деструкция полимера, причем интенсивность этого процесса тем больше,
чем выше молекулярный вес исходного полимера.
Конструкция шнека прядильной машины, в частности
отношение длины к диаметру, число зон обогрева и температура в этих
зонах, степень сжатия расплавленного полимера при прохождении
через шнек оказывают существенное влияние на процесс
формования. Влияние этих факторов на условия формования и свойства
получаемых волокон детально рассмотрены в работе [3, с. 278].
Для обеспечения равномерного формования и устранения
обрыва волокон (в процессе формования) и дополнительной
деструкции полимера на прядильной машине необходимо предотвратить
попадание воздуха в расплав. Поэтому порошкообразный материал,
содержащий обычно некоторое количество воздуха, непригоден для
формования волокна. Полипропилен применяется в виде плотных
гранул; он содержит небольшое количество @,2—0,5%) антиокси-
данта, а в случае необходимости — и пигментного красителя.
Скорость формования волокна из расплава достигает 500 м/мин,
высота шахты 4—6 м. Формованием из расплава полипропилена
можно получить моноволокно и комплексную нить, а также
штапельное волокно.
Формование волокна из высоковязкого расплава
полипропилена производится через фильеры с большим диаметром отверстий
9.1. Полипропиленовые волокна 283
@,5—0,65 мм), чем при получении полиамидных волокон. Поэтому,
как правило, полипропиленовое волокно формуют с большой филь-
ерной вытяжкой.
Для упрочнения сформованное полипропиленовое волокно
подвергается вытягиванию. »
Полипропиленовую нить обычно вытягивают на 400—800°/о
при 100—140 °С. Максимально возможная степень вытягивания и
свойства получаемого волокна в значительной степени зависят от
условий вытягивания, и особенно от температуры. Для получения
высокопрочного полипропиленового волокна его следует
дополнительно вытягивать при температурах, близких к температуре
плавления полимера A70—180°С), и под натяжением для устранения
возможности необратимого течения волокна. При таких
температурах увеличивается максимально возможная степень вытягивания
(с 530% при 120 °С до 770% при 170—180 °С) и значительно
повышается прочность волокна — с 66,4 до 84 кгс/мм2 F64—
840 Мн/м2). Одновременно снижается удлинение нити (с 45 до
33%) и повышается плотность волокна (с 0,9127 до 0,9265 г/см3)
[21].
При одной и той же температуре максимально возможная
степень вытягивания волокна зависит от структуры
полипропилена [22]. Полимеры с менее упорядоченной и менее совершенной
структурой легче деформируются. Для волокон из такого
полипропилена могут быть достигнуты более высокие степени
вытягивания и ориентации, а следовательно, и более высокие
физико-механические свойства, чем для волокон из полимера, обладающего
более совершенной структурой.
Структуру волокна можно регулировать различными методами,
в частности изменением условий охлаждения свежесформованного
волокна перед вытягиванием. При быстром охлаждении волокна
образуется менее упорядоченная структура волокон, а при
медленном охлаждении — более устойчивая и упорядоченная структу-
ра [23].
Степень вытягивания полипропиленового волокна различна в
зависимости от его назначения. Например, от полипропиленового
волокна, используемого в смеси с шерстью для изготовления
изделий народного потребления, требуется более высокое удлинение и
меньшая прочность. Поэтому такое волокно вытягивают на 400—
500%- При получении высокопрочного волокна для изготовления
канатов степень вытягивания повышают до 700—800%.
Так же как и другие карбоцепные волокна, вытянутое
полипропиленовое волокно подвергается термообработке, т. е. прогреву
при повышенной температуре. Волокна, применяемые для
изготовления изделий народного потребления, целесообразно прогревать
без натяжения. После нагрева волокна при 100°С в течение 30 мин
прочность волокна не изменяется, удлинение повышается
незначительно (с 17,9 до 23%), а число двойных изгибов, выдерживаемых
284 Глава 9. Производство полиолефиновых волокон
волокном до разрыва, увеличивается почти в 5 раз [24].
Одновременно значительно уменьшается усадка волокна в горячей воде,
что имеет существенное значение для изделий, подвергаемых
стирке.
Непродолжительный нагрев полипропиленового моноволокна
A0 мин при 120—140 °С) повышает его устойчивость к истиранию
в 4—5 раз. При более продолжительном нагревании устойчивость
к истиранию понижается, по-видимому, в результате
дополнительной кристаллизации и образования трещин в волокне [25].
Так же как и при переработке других гибких полимеров, из
расплава полипропилена и полиэтилена могут быть получены не
только комплексная нить и штапельное волокно, но и моноволокно
(по той же технологической схеме, что и моноволокно из
полиамидов и полиэфиров). Образующееся при застывании струек
расплава моноволокно или пучок моноволокон направляется в ванну с
водой, где оно охлаждается и затем подвергается вытягиванию в
8—10 раз при повышенной температуре A05—130°С). -
Вытянутые волокна подвергаются термообработке в натянутом
состоянии в токе горячего воздуха или перегретого пара.
Прочность вытянутого моноволокна составляет 40—50 гс/текс
D00—500 мН/текс), удлинение — 15—25%.
9.1.4. Свойства полипропиленового волокна
Полипропиленовое волокно обладает комплексом ценных
эксплуатационных свойств. Ниже рассматриваются показатели,
характеризующие основные свойства волокна.
Прочность полипропиленового волокна в сухом и мокром
состоянии достаточно высока. Например, прочность волокна,
используемого для изготовления изделий народного потребления,
составляет 35—40 гс/текс, а для изделий технического назначения — 60—
80 гс/текс. В мокром состоянии прочность полипропиленового
волокна не изменяется. Прочность волокна, сформованного и
вытянутого в одних и тех же условиях, понижается с увеличением
содержания аморфных или низкомолекулярных кристаллических
фракций в исходном полимере.
Удлинение волокна в сухом и мокром состоянии одинаковое и
составляет 30—40%; удлинение высокопрочного волокна—12—
15%.
Эластичность полипропиленового волокна достаточно высокая.
При вытягивании волокна на 5 и 10% эластическое удлинение
составляет соответственно 98 и 95% от общего удлинения.
Следовательно, по величине эластического удлинения это волокно почти
не уступает полиамидным и превосходит большинство
синтетических во око .
9.1. Полипропиленовые волокна 285
Гигроскопичность полипропиленового волокна, так же как и
других волокон из стереорегулярных полиолефинов, практически
равна нулю. Это значительно затрудняет окраску
полипропиленовых волокон и возможность их использования в изделиях
народного потребления. Окрашивание этих волокон пока производится,
как правило, в массе (введением пигментов в полимер перед
формованием). Вследствие очень низкой гигроскопичности
полипропиленовое волокно используется для изготовления изделий народного
потребления только в смесях с другими волокнами, в частности с
шерстью.
Крайне низкая гигроскопичность полипропиленового волокна
обусловливает значительную электризуемость этих волокон, что
затрудняет их переработку и ограничивает использование в
изделиях народного потребления. При отделке для уменьшения элек-
тризуемости на волокно наносят небольшое количество
поверхностно-активных веществ, которые резко уменьшают электрическое
сопротивление волокон. Например, при относительной влажности
воздуха 60 ±1%, температуре 18 °С и количестве наносимого
поверхностно-активного вещества 1 % от массы волокна удельное
электрическое сопротивление полипропиленового волокна
составило (в Ом-см) [26]:
Необработанное волокно . . . Более 1013
Волокно, обработанное
авиролем 1,1-10s
полиэтиленгликолем ... 7,2 • 105
лаурилпиридинийсульфатом 5,3 • 105
При пониженной влажности воздуха влияние
поверхностно-активных веществ выявляется в меньшей степени.
Поверхностно-активные вещества смываются с волокна при первой же водной
обработке (стирке изделий). Поэтому обеспечить устойчивое
снижение электризуемости волокон или полученных из них изделий при
использовании этого класса соединений не представляется
возможным.
Для придания полиолефиновым волокнам устойчивых
антистатических свойств, сохраняющихся после многократных стирок,
существенный интерес представляет метод, предложенный 3. Г.
Серебряковой и сотр..[27]. При формовании волокна к полипропилену
добавляют небольшое количество поли-2-метил-5-винилпиридина
D—9% от массы полипропилена), который затем алкилируют
обработкой волокна йодистым метилом. Образующаяся четвертичная
соль полиметилвинилпиридина обладает высокой гидрофильно-
стью, и поэтому сильно снижает электризуемость волокна.
Например, удельное электрическое сопротивление волокна, содержащего
5—6% четвертичной соли поли-2-метил-5-винилпиридина,
снижалось с 5-Ю13 (для исходного волокна) до 109—108 Ом-см. Хотя
четвертичная соль этого полимера растворима в воде, но будучи
введена в волокно, в процессе его формования она инклюдируется
286 Глава 9. Производство полиолефиновых волокон
в волокне и не удаляется из него при водных обработках. Поэтому
достигнутое таким образом резкое снижение электрического
сопротивления волокна сохраняется и после двадцати стирок. Хотя ал-
килирование полиметилвинилпиридина йодистым алкилом
неэкономично, но этот принцип снижения электризуемости гидрофобных
волокон при дальнейшем его упрощении и удешевлении может
практически применяться.
Плотность полипропиленового волокна является наиболее
низкой @,91 г/см3) среди всех природных и химических волокон. Это
волокно, так же как и полиэтиленовое, не тонет в воде.
Естественно, что такая низкая плотность" является существенным
преимуществом полипропиленового волокна при использовании его для
изготовления ряда изделий технического назначения.
Хемостойкость полипропиленового волокна является также
одним из его преимуществ. Это волокно обладает высокой
стойкостью к действию кислот (например, азотной и серной) и
щелочей различных концентраций, не уступая по этому важному
показателю такому хемостойкому волокну, как хлорин. Так же как и
другие синтетические волокна, полипропиленовое- волокно
устойчиво к действию микроорганизмов.
- Устойчивость к истиранию полипропиленового волокна ниже,
чем полиэтиленового; по этому показателю полипропиленовое
волокно значительно уступает полиамидным волокнам. Методы
повышения устойчивости полипропиленовых волокон к истиранию пока
не разработаны.
Термо- и теплостойкость полипропиленового волокна
недостаточно высокая, что является одним из основных его недостатков.
Термостойкость волокна из полипропилена может быть
значительно повышена введением антиоксидантов. Например, волокно, не
содержащее антиоксидантов, полностью разрушается после
нагрева в течение 8 ч при 140 °С [12]. При содержании в волокне 0,5%
антиоксидантов (неозона Д) прочность и удлинение волокна не
изменяются даже после нагрева при той же температуре в течение
25 ч.
При разработке методов улучшения теплостойкости
полипропиленового волокна, т. е. уменьшении потери прочности
непосредственно при повышенных температурах, встречаются значительные
трудности. Полипропиленовое волокно размягчается при 140 °С и
плавится при 160—165 °С. По этой причине полипропиленовое
волокно пока нельзя использовать для изготовления корда и
резинотканевых изделий, так как процесс вулканизации резины в
промышленности производится обычно при 150—160 °С.
Разработка методов повышения температуры плавления
полипропиленового волокна на 40—50 °С или получения неплавкого
волокна и соответственно волокон с высоким начальным модулем
создаст принципиальную возможность использования
высокопрочного полипропиленового волокна для выработки корда. Учитывая
9.1. Полипропиленовые волокна 287
низкую плотность этих волокон, высокую эластичность,
однотипность химического строения полиолефиновых волокон и каучука
и, следовательно, достаточно высокую адгезию между этими
материалами, можно предположить, что при успешном решении
этой задачи полипропиленовое волокно сможет стать одним из
перспективных химических волокон для производства кордной
ткани.
При 100 °С полипропиленовое волокно обратимо теряет свыше
40% прочности, а при 120 °С при приложении очень небольших
усилий начинается течение волокна. Поэтому использование
изделий лз полипропиленового волокна при температурах выше 100 °С
затруднено, а в ряде случаев не представляется возможным.
Температура так называемой нулевой прочности этого волокна
составляет 160—170 °С.
Теплостойкость волокна, по-видимому, можно повысить путем
образования небольшого числа поперечных химических связей
между макромолекулами полипропилена, т. е. сшиванием
полипропилена, что должно привести к резкому снижению текучести
полимера и вырабатываемых из него изделий.
Образование поперечных химических связей между
макромолекулами полимера, не содержащими активных реакционноспособ-
ных групп, может быть осуществлено различными методами.
Наибольший интерес представляют следующие методы:
получение сополимера пропилена с небольшим количеством
диена (в частности, изопрена) и последующей вулканизацией
изделий из этого сополимера аналогично вулканизации бутилкау-
чука;
облучение волокон лучами высоких энергий, в частности у-пу-
чами.
Синтез сополимеров пропилена с небольшим количеством
изопрена (от 1 до 10% от массы пропилена) был осуществлен
советскими исследователями [28]. Однако попытка провести
вулканизацию этих сополимеров различными методами не привела пока к
положительным результатам. Текучесть полимеров после этой
обработки не изменилась.
Попытки образовать химические связи между макромолекулами
полипропилена, действуя у-излучением F0Со), также не привели
к положительным результатам [29]. Образование химических связей
между макромолекулами происходит при дозе излучения, равной
125—150 Мрад. Образование таких связей характеризуется, в
частности, значительным уменьшением усадки волокна в кипящей воде.
Однако одновременно происходит интенсивная деструкция волокна,
в результате которой прочность его снижается в 2—2,5 раза при
одновременном значительном снижении удлинения. Естественно,
что такой метод не может быть рекомендован для практического
использования.
288 Глава 9. Производство полиолефиновых волокон
9.1.5. Химическая модификация
полипропиленовых волокон
Свойства полипропиленовых волокон можно улучшить и
изменить в требуемом направлении путем химической модификации.
Для этого могут быть использованы те же методы, которые
применяются для других химических волокон. Наибольший интерес
представляют следующие методы:
получение волокон из сополимеров пропилена с другими олефи-
нами;
использование смесей полимеров для формования волокон;
синтез блок- (алломеров) и привитых сополимеров
полипропилена.
Сополимеры пропилена и этилена, содержащие 30—40%
звеньев пропилена, приобретают свойства каучуков. Такой сополимер
(без двойных связей) находит все большее применение в
резиновой промышленности как один из новых типов синтетического
каучука со специфически ценными свойствами. Вследствие нарушения
регулярности строения исходного волокнообразующего полимера
получается волокно, обладающее более низкими прочностью и
температурой усадки, более высоким удлинением, чем волокна из
полиолефинов регулярной структуры [30]. Возможно, что в
будущем этим методом можно будет вырабатывать дешевые
высокоэластичные волокна.
Советскими исследователями проведены опыты по формованию
волокна из смеси стереорегулярного полипропилена с
кристаллическим полистиролом [31]. Полистирол добавляли для снижения
текучести волокна. Так как эти полимеры не совмещаются, получать
из указанной смеси волокно с высокие комплексом механических
свойств не представляется возможным.
Такие же результаты получались при формовании волокна из
смеси полипропилена и поли-2-винилпиридина G—12% от массы
полипропилена), но наличие в волокне пиридиновых групп
значительно улучшило его накрашиваемость кислотными и дисперсными
красителями [32]. Улучшение накрашиваемости волокна
достигалось также разрыхлением структуры при добавлении к
полипропилену перед формованием 2—10% сополимера акрилонитрила со
стиролом. Волокно, сформованное из этой смеси, сорбировало в
4—4,5 раза больше красителя, чем волокно, полученное из
полипропилена регулярной структуры [33]. Однако и в этом случае
механические свойства волокна снижаются.
Не нашел применения также метод модификации свойств
полиолефиновых волокон синтезом блок-сополимеров. Однако в
последние годы разработан метод получения так называемых
алломеров — полимеров, макромолекулы которых содержат большее или
меньшее число блоков нескольких мономеров. Этот тип
блок-сополимеров, синтезируемый то'лько методом анионной полимеризации,
9.1. Полипропиленовые волокна 289
путем образования в качестве промежуточных соединений так
называемых «живых» полимеров [34, с. 99—102], обладает такой же
степенью кристалличности, как гомополимеры олефинов
регулярного строения. Поэтому применение алломеров дает возможность
производить материалы, которые существенно отличаются по
свойствам от аналогичных материалов, получаемых из смесей тех же
полимеров или из статистических сополимеров. Например, алло-
меры полипропилена и полиэтилена обладают лучшими
свойствами, чем обычные сополимеры этилена и пропилена, а также гомо-
полипропилен [35, с. 196—220]. Использование этого класса
модифицированных полполефинов для производства волокон может
привести к получению практически ценных результатов.
Наибольшее число исследований по химической модификации
полиолефиновых волокон посвящено синтезу различных привитых
сополимеров, в частности привитых сополимеров полипропилена.
Получены привитые сополимеры полиолефинов с большинством
полимеризующихся винильных мономеров. Так как привитые
сополимеры полиолефинов с этими мономерами не растворяются и
не плавятся, перерабатывать их в волокна обычными методами не
удается. Поэтому прививка производится к волокну или к получен-
номучиз него изделиям. Из-за отсутствия в макромолекуле
полиолефинов реакционноспособных функциональных групп
ограничивается число методов, которые могут быть использованы для
образования макрорадикалов в молекуле полимеров, что является
необходимым для прививки различных винильных полимеров
методами радикальной полимеризации.
Образовать макрорадикалы в молекуле полиолефинов можно,
как правило, физическими методами, а для полипропиленового
волокна, содержащего в элементарном звене макромолекулы легко-
окисляемую метильную группу, также и химическими методами.
Из физических методов инициирования радикальной реакции
привитой сополимеризации наибольший интерес представляют
радиационные методы (лучи высоких энергий), а также образование
макрорадикалов в результате механохимическои деструкции,
которая в большей или меньшей степени всегда имеет место при
формовании волокон из расплава.
Для уменьшения количества образующегося при прививке го-
мополимера целесообразно предварительно облучать полиолефи-
новое волокно. В результате облучения в волокне образуются пе-
рекисные и гидроперекисные группы (при облучении
полипропилена преимущественно гидроперекисные, а при облучении
полиэтилена — перекисные группы), распадающиеся затем с образованием
макрорадикалов. При последующей обработке облученных
волокон раствором или водной эмульсией мономера в присутствии
восстановителя (в частности, FeSO4) происходит только процесс
прививки; образования гомополимера, как правило, не наблюдается
[36]. Используя этот метод предварительного облучения полиоле-
10 Зак. 234
290 Глава 9. Производство полиолефиновых волокон
финового волокна (прививка на так называемом постэффекте),
осуществили прививку к полипропиленовому волокну 20—25% (от
массы волокна) полиакриловой кислоты, поли-2-метил-5-винилпи-
ридина и полиакрилонитрила.
Прививка этих полимеров приводит к снижению прочности и
начального модуля волокна (при одновременном повышении его
удлинения). Образование боковых групп в результате введения
даже жестких полимеров (например, полиакрилонитрила) не
повышает термостойкости и теплостойкости полиолефинового
волокна [37]. Следовательно, для улучшения этих практически ценных
свойств волокна методы прививки не являются достаточно
эффективными. Однако в результате прививки полимеров, содержащих
реакционноспособные полярные функциональные группы
(полиакриловая кислота и полиметилвинилпиридин), значительно
повышается гигроскопичность волокна и улучшается накрашиваемость.
Например, при прививке к полиэтиленовому волокну 20% (от массы
волокна) полиакриловой кислоты, гигроскопичность его
повышается в 10—15 раз и приближается к гигроскопичности хлопка [38].
Такое резкое изменение этого важного показателя имеет большое
значение и создает предпосылки для дальнейшего расширения
областей применения этих волокон.
Привитые сополимеры полиолефиновых волокон с
полиакриловой кислотой и с полиметилвинилпиридином обладают высокими
ионообменными свойствами. Учитывая высокую хемостойкость
полиолефиновых волокон и их стойкость к агрессивным веществам,
можно предположить, что эти модифицированные полиолефиновые
воло'кна могут представить значительный интерес в качестве
ионообменных волокнистых материалов с высокоразвитой
поверхностью, особенно в условиях, в которых более доступные
ионообменные волокна (целлюлозные) не могут быть применены (например,
при улавливании ценных и редких металлов из сильнокислых
растворов).
Отметим, что при прививке^ к полиэтиленовому волокну 30—
40% полиметилвинилпиридина устойчивость его к истиранию
повышается в 8—10 раз [39].
Образование макрорадикалов может быть осуществлено также
непосредственно в процессе формования волокна (без
предварительной обработки полиолефинов). В присутствии даже
минимальных количеств кислорода воздуха при высоких температурах, при
которых происходит формование волокон, в макромолекуле
полимеров образуются перекисные и гидроперекисные группы, при
распаде которых появляются макрорадикалы. Специфическое
затруднение при использовании этого метода прививки (так же как и
других способов образования макрорадикалов на полиолефиновых
волокнах) заключается в том, что в полиолефиновых волокнах
всегда содержатся термо- и светостабилизаторы. Механизм
действия этих добавок основан главным образом на ингибированин
9.1. Полипропиленовые волокна 291
процессов деструкции полимеров, протекающих, как правило, по
цепному радикальному механизму. Эти стабилизаторы могут
реагировать и с макрорадикалами, обусловливающими начало реакции
прививки, инактивировать их и таким образом препятствовать
этой реакции.
Опыты показали, что в присутствии стабилизаторов количество
прививаемого полимера значительно уменьшается. Например, если
при нагреве волокна из нестабилизированного полиэтилена
низкого давления при 300 °С в течение 1 ч'образуется до 0,02% пере-
кисных групп, достаточных для последующей прививки 20—25%
полимера (полиакриловой кислоты, полиметилакрилата), то при
такой обработке стабилизированного волокна образуется только
0,005—0,008% перекисных групп и количество привитого полимера
составляет 5—8%от массы волокна [40].
При введении в полиолефиновое волокно эффективных
стабилизаторов количество прививаемого полимера дополнительно
уменьшается, а в ряде случаев прививка вообще не происходит.
Это обстоятельство значительно затрудняет использование метода
прививки по реакции радикальной полимеризации для
модификации свойств термостабилизированных полиолефиновых волокон.
Наличие в молекуле полипропилена легко окисляющегося
третичного атома углерода создает возможность введения в
макромолекулу перекисных групп и при обработке в более мягких
условиях, например при ультрафиолетовом облучении при 50 °С [41]
или при предварительном озонировании полипропиленовых
волокон [42]. Используя эти методы прививки, прививали 30—50%
полиакрилонитрила и 20—30% полигликольметилакрилата [43],
введение которого в молекулу привитого полимера, так же как и
введение полиакриловой кислоты, значительно повышает
гигроскопичность и улучшает окрашиваемость волокна.
Суммируя, можно сделать вывод, что привитой сополимериза-
цией можно значительно улучшить некоторые практически важные
свойства полиолефиновых волокон (накрашиваемость,
гигроскопичность, светостойкость) и получить модифицированные волокна,
которые могут быть . эффективно использованы, например, в
качестве ионообменных. Однако специфические затруднения,
возникающие при прививке к стабилизированным полиолефиновым
волокнам, значительно ограничивают возможность широкого
использования этого метода модификации.
Описанные свойства полипропиленового волокна определяют
области его применения. Высокая прочность, эластичность и
стойкость к действию микроорганизмов, а также низкая плотность
полипропиленового, волокна определяют целесообразность
использования этого волокна в первую очередь для изготовления изделий
технического назначения, в частности рыболовных снастей и сетей,
канатов, плавучих средств и т. д. Как уже отмечалось, большой
интерес представляет использование полипропиленовых волокон
10*
292 Глава 9. Производство полиолефиновы'х волокон
в промышленности резиновых технических изделий, и особенно для
изготовления шинного корда. Однако это станет возможным
только после того, как будет повышена теплостойкость
полипропиленового волокна.
Все более широкое применение находят окрашенные в массе
полипропиленовые волокна для изготовления ковровых изделий,
несмотря на то что требования к устойчивости этих изделий к
истиранию не всегда могут быть выполнены.'
До последнего времени считали нецелесообразным
использовать полипропиленовые волокна для изготовления изделий
народного потребления из-за того, что эти волокна практически не
поглощают влагу. В настоящее время в различных странах
разработан ассортимент тканей из смеси полипропиленовых и
гидрофильных волокон (шерсти, хлопка, вискозного волокна). Из
полипропиленового волокна можно изготовлять плательные ткани,
одеяла и другие изделия.
Синтезировано сравнительно большое количество полиолефи-
нов стереорегулярного строения с более высокой температурой
плавления, чем у полипропилена, например полиметилпентен-1
(полипропилэтилен) и поли-З-метилбутен-1 с температурой
плавления соответственно 205 и 250 °С. Однако эти полимеры не
получили пока применения для производства синтетических волокон
из-за более высокой стоимости по сравнению с полипропиленом
или вследствие трудности их переработки. Поэтому наряду с
полипропиленом практический интерес для формования волокон
представляет пока только полиэтилен.
9.2. ПОЛИЭТИЛЕНОВЫЕ ВОЛОКНА
Производство полиэтиленового волокна, так же как и
полипропиленового, стало возможным только после разработки методов
синтеза полиэтилена линейного строения на стереоспецифических
катализаторах при низком давлении. В результате ввода в
эксплуатацию ряда заводов по производству полиэтилена низкого
давления в нашей стране созданы реальные предпосылки для
организации многотоннажного производства полиэтиленового волокна.
9.2.1. Получение и свойства полиэтилена
Полиэтилен линейного строения, применяемый для формования
волокна, так же как и для производства других изделий, может
быть получен полимеризацией этилена при низком давлении —
1—6 кгс/см2 A05—6-Ю5 Па) на металлоорганических
катализаторах Циглера или при среднем давлении — 35—70 кгс/см2
C,5-106—7-Ю6 Па).
Полиэтилен низкого и среднего давления практически не имеет
разветвлений в макромолекулах. Он стоек к воде и водным па-
9.2. Полиэтиленовые волокна 293
рам. При нормальной температуре полиэтилен обладает высокой
стойкостью к концентрированным минеральным кислотам (серной,
соляной, азотной и др.) и щелочам, а также ко многим
растворителям.
Плотность полиэтилена низкого давления 0,94—0,96 г/см3,
среднего давления — 0,96 г/см3, т. е. несколько выше, чем у
полипропилена, но значительно ниже, чем у других природных и
синтетических волокнообразующих полимеров. *
9.2.2. Получение полиэтиленового волокна
До последнего времени из полиэтилена вырабатывали в
основном моноволокно. Получение текстильной нити может быть
осуществлено по технологической схеме, применяемой при производстве
полипропиленового волокна.
Характерным отличием, которое необходимо учитывать при
разработке технологических параметров процесса получения
полиэтиленового волокна, является более высокая вязкость расплава
полиэтилена по сравнению с вязкостью расплава полипропилена
при примерно одинаковом молекулярном весе этих полимеров [44].
Поэтому формование полиэтиленового волокна из расплава
проводится при более высоких температурах C50—370°С), чем
формование полипропиленового волокна. Эта температура близка к
температуре термического разложения полиэтилена.
Из полиэтилена низкого давления, макромолекулы которого
имеют линейное строение, получается волокно, обладающее
достаточно высокими физико-механическими свойствами [45]. Волокно
из гранулированного и стабилизированного полиэтилена формуют
на такой же прядильной машине, как и волокно из
полипропилена, но при несколько измененных параметрах процесса.
Для повышения прочности полиэтиленовое волокно также
подвергается значительному вытягиванию (на 1000—1500%). Ввиду
низкой температуры стеклования полиэтилена (—21 °С) волокно
можно вытягивать при температурах выше температуры
стеклования полимера, т. е. от 20 до 120 °С. Чем выше температура
вытягивания, тем больше максимальная степень вытягивания и тем
меньше усадка вытянутого волокна. Например, при повышении
температуры вытягивания с 20 до ПО °С максимально возможная степень
вытягивания увеличивается с 170 до 1060% [46]. Длительное
выдерживание невытянутого полиэтиленового волокна (до 320 дней) не
уменьшает его способности к вытягиванию. При одинаковой
степени вытягивания волокна существенное влияние на его' прочность
оказывает температура вытягивания или последующего нагрева
волокна.
Комплекс механических свойств вытянутого волокна зависит от
молекулярного веса'полиэтилена:
O94
Глава 9. Производство полиолефиновых волокон
Кратность
вытягивания
6
7
6
7
Прочность,
гс/текс (мН/текс)
Молекулярный вес 52 000
22,0 B20)
22,2 B22)
Молекулярный вес 71 000
29,8 B98)
37,5C75)
Число двойных
изгибов
50
25
330
250
Начальный модуль,
кгс/мм2
(МН/и1)
350 C500)
370 C700)
420 D200)
450 D500)
Как видно из приведенных данных, с повышением
молекулярного веса полиэтилена с 52 000 до 71 000 при одинаковой степени
вытягивания закономерно повышается прочность волокна, число
двойных изгибов, выдерживаемых волокном до разрыва и, что
весьма существенно, начальный модуль [47].
Из полиэтилена высокого давления, макромолекулы которого
имеют разветвленную форму, получается волокно, обладающее
низкими механическими свойствами. Прочность такого волокна
не превышает-9—13 гс/текс. Естественно, что использование
такого волокна нецелесообразно.
Сравнительные данные о свойствах волокон, полученных из
стереорегулярного полипропилена, линейного и разветвленного
полиэтилена приводятся ниже:
Плотность,
Г/СИ»
гс/текс
(мН/текс)
ние, %
Температура
при размягчения
I0Q°C, % (без нагрузки), °С
Волокно из
стереорегулярного
полипропилена 0,90—0,91 50—65
Волокно из
полиэтилена
высокого
давления
разветилен-
ного, с
низким
молекулярным
весом . . .
разветвленного, со
средним
молекулярным
весом . . .
E00-650)
15-25
10
149-151
0,92
0,92
9—13 45-50
(90-130)
50-60
40-50
18-21 25-30
A80-210)
низкого
давления,
линейного строения 0,93-0,96 60-70 10—20 5-Ю
F00-700)
85-86
Ю4—ПО
127—132
Эти данные показывают, что полипропиленовое волокно имеет
более высокую температуру размягчения и несколько более
низкую плотность, чем волокно, полученное из линейного полиэтилена
Литература 295
(низкого давления). По механическим свойствам эти два вида
полиолефиновых волокон примерно равноценны.
Известным преимуществом полиэтиленового волокна перед
пропиленовым является его более высокая стойкость к
фотохимическим и термоокислительным воздействиям. Однако при наличии
соответствующих стабилизаторов полипропиленовое волокно и по
этим показателям не уступает полиэтиленовому.
Значительно более высокая стойкость полиэтиленового волокна
к термоокислительным воздействиям определяет и более высокую
термостойкость полиэтиленового волокна. Например, после
нагрева при 100 °С прочность полиэтиленового волокна,
определяемая при нормальной температуре, заметно не изменяется, в то
время как полипропиленовое волокно после нагрева при 80 °С
теряет 12—20% прочности [48]. Благодаря более высокой стойкости
полиэтиленового волокна к радиационным и окислительным
воздействиям сшивание этого волокна, а следовательно, и
существенное повышение его теплостойкости и улучшение других ценных
свойств можно осуществить без заметной деструкции и снижения
прочности. Сшивание (структурирование) полиэтиленового
волокна можно, по-видимому, производить аналогично
структурированию полиэтиленовых пленок путем радиационного облучения- в
определенных условиях. Температура размягчения таких пленок
(так называемого ирратена) повышается почти на 100 °С (т. е. до
200 °С), что, естественно, значительно расширяет области их
применения.
ЛИТЕРАТУРА
1. Стрепихеев А. А., Деревицкая В. А., Слонимский Г. Л. Основы
химии высокомолекулярных соединений. М., «Химия», 1966. 514 с.
2. H ear le L., Skinner Silk. Rayon., 1956, v. 30, p. 114—118.
3. К о и к и н А. А., Зверев М. А. Полиолефиновые волокна. М., «Химия»,
1966. 278 с.
4. Натта Д., Хим. и технол. полимеров, 1960, № 7—8, с. 112—120.
5. Роговин 3. А., Дружинина Т. В., Хим. волокна, 1959, № 4,
с. 302-305.
6. A n g Е., M a r k H., Makromol. Chem., 1959, Bd. 29, S. 679—682.
7. Гильман И. С, Роговин 3. А., Высокомол. соед., 1959, т. 1, с. 619—622.
8. Клименков В. С, Зверев М. П., Груздев В. А. и др., Хим.
волокна, 1958, № 4, с. 19—22.
9. Роговин 3. А., Дружинина Т. В., Изв. вузов. «Химия и химическая
технология», 1958, № 1, с. 139—142.
10. Ф и л ь б е р т Д. В., M о л ь к о в а Г. Н., П а к ш в е р А. Б., Хим. волокна,
1965, № 5, с. 6—8.
11. Ma dor sky S., J. Polymer Sei., 1952, v. 9, p. 133—136.
12. Груздев В. А., Клименков В. С, С е р к о в а Л. А. и др., Хим
волокна, 1960, № 6, с. 19—21.
13. П л е ш а к о в М. Г., Тихонова Т. И., Грибова Т. А и др Хим
волокна, 1966, № 2, с. 14—16.
14. Плешаков М. Г., Тихонова Т. И., Вольштейн Е M и др Хим
волокна, 1966, № 4, с. 12—15.
15. Плешаков М. Г., Тихонова Т. И., Хим. волокна, 1968' № 4, с, 76—78.
296 Глава 9. Производство полиолефпновых волокон
16. Шляпников Ю. A., M я л л е р В. Б., H е ф м а н М. Б. и др., Высокомол.
соед., 1963, т. 5, с. 702—705.
17. Er lieh V., Text. Res. J., 1959, v. 29, p. 679—681.
18. Попов А. Г., Токарева Л. Г., Михайлов H. В., Хим. волокна, 1966,
№ 6, с. 26—29.
19. Роговия 3. А., Коварская Б. М, Изв. вузов. «Химия и химическая
технология», 1958, № 2, с. 361—363.
20. Нечаева С. А., Роговин 3. А., Хим. волокна, 1960, № 1, с. 10—13.
21. Половихина Л. А., Зверев М. П., Хим. волокна, 1969, № 5, с. 12—14.
22. Дячик И., Ямбрях М., Ковач Я., Хим. волокна, 1964, № 4, с. 2—4.
23. Sobul H., Tobata V., J. Polymer Sei., 1959, v. 39, p. 427—430.
24. Нечаева С. А., Роговин 3. А., Хим. волокна, 1960, № 1, с. 10—12.
25. Исаева В. И., Фильберт Д. В., Васильев Ю. В. и др., Хим.
волокна, 1968, № 6, с. 25—28.
26. Агафонова Л. Л., Лущейкин Г. А., Серебрякова 3. Г., Хим.
волокна, 1968, № 2, с. 51—53.
27. А г а ф о н о в а Л. Л., Лущейкин Г. А., Серебрякова 3. Г., Хим.
волокна, 1968, № 3, с. 16—18.
28. Волкова Н. С, X у т о р е в а Г. В., Крендель Б. А. и др., Высокомол.
соед., 1959, т. 1, с. 1759—1762.
29. Нечаева С. А., Малинский Ю. М., Роговин 3. А Хим. волокна,
1966, № 3, с. 7—10.
30. Дружинина Т. В., Андриченко Ю. Д., Конкин А. А. и др., Хим.
волокна, 1963, № 3, с. 15—18.
31. Зверев М. П., Бычков Р. А., Костина Т. Ф. и др Хим. волокна,
1964, № 3, с. 15—17.
32. Бычков Р. А., Зверев М. П., Хим. волокна, 1966, № 2, с. 14—16.
33. Фяльберт Д. В., Муравьев В. П., Пакшвер А. В. В кн.:
цепные волокна. М., «Химия», 1966, с. 181 —189.
34. Деревицкая В. А., Стрепихеев А. А., Слонимский Г. Л. Химяя
высокомолекулярных соединений. М., «Химия», 1966. 514 с.
35. К о н к я н А. А., Зверев М. П. Полиолефиновые волокна. М. «Химяя»,
1968. 278 с.
36. У Ж у н-ж у й, Стасюк X. А., Кочергинская Л. А. и др., Хим.
волокна, 1963, № 5, с. 12—16.
37. У Жун-жуй, Конкин А. А., Роговин 3. А., Хим. волокна, 1964,
№ 6, с. 14—16.
38. Дружинина Т. В., Конкин А. А. В кн.: Карбоцепные волокна. М.,
«Химяя», 1966, с. 97—101.
39. Д р у ж и и я я а Т. В., Конкин А. А., Хим. волокна, 1968, № 6, с. 46—48.
40. Костров Ю. А., Соловская Н. Л., Хим. волокна, 1966, № 2, с. 25—27.
41. Костров Ю. А., Конкин А. А., Хим. волокна, 1966, № 6, с. 22—25.
42. Манясек 3., Мичко M., Павлинец И. и др., Хим' волокна, 1964,
№ 4, с. 7—10.
43. Павлянец И., Л аз ар М., Манясек 3., Хим. волокна 1964 № 4,
с. 7—10.
44. Дружяняна Т. В., Конкнн А. А., Виноградов Ю А Хйм
волокна, 1962, № 1, с. 25—28.
45. Дружинина Т. В., Андриченко Ю. Д., Конкин А А я др. Хим.
волокна, 1962, № 2, с. 17—19.
46. Дружинина Т. В., Андриченко Ю. Д., Конкнн А А. Хим. волокна,
1967, № 1, с. 67—69.
47. Аверьянов А. А., Фильберт Д. В., Конкин А А, Хим волокна,
1968, № 1, с. 29—31..
48. Ткаченко А., Чернявская Г., Шрубович В и до Хим волокна,
1970, № 2, с. 30—32.
Глава 10
ПРОИЗВОДСТВО ВОЛОКОН
ИЗ ФТОРСОДЕРЖАЩИХ ПОЛИМЕРОВ
В промышленности пластических масс широко применяются
полимеры, получаемые из фторсодержащих а-олефинов —
фторопласты. Среди этих полимеров наибольшее распространение
получил политетрафторэтилен, известный под названием «фторопласт»
(СССР) или «тефлон» (США).
Отличительная особенность политетрафторэтилена — высокая
хемостойкость и термостойкость. По химической стойкости фтор-
содержащие полимеры превосходят все известные природные и
синтетические высокомолекулярные соединения. Естественно, что
использование этих полимеров для производства синтетических
волокон, обладающих специфически ценными свойствами,
представляет большой интерес.
В опытно-производственных или в опытных условиях
вырабатывается волокно тефлон из политетрафторэтилена и волокно фтор-
лон из фторсодержащих сополимеров, растворяющихся в ацетоне.
10.1. ПОЛУЧЕНИЕ ВОЛОКНА ТЕФЛОН*
Исходный полимер для формования волокна тефлон —
политетрафторэтилен — синтезируют путем полимеризации тетрафтор-
этилена. Этот мономер в промышленности получают из фтористого
водорода и хлороформа по следующей схеме:
HF+СНСІз —> CHFjCl
Образующийся дифторхлорметан подвергается пиролизу при
700 °С;
CHF2C1 —> CF2=CF2 -f HCl
Полимеризация тетрафторэтилена проводится в присутствии
перекисей (например, перекиси водорода) при 70—80 °С и
давлении 40—100 кгс/см2 D-Ю6—10-Ю6 Па). Получаемый полимер
имеет линейную структуру:
НаОг
CF2=CF2 *- CF2—CF2—CF2—CF2
* Более детально о технологии получения этого волокна — см. Сигал М. Б.,
Козиорова Т. И. Синтетические волокна из дисперсий полимеров. М.,
«Химия», 1972, 145 с.
Варшавский В. Я. В кн.: Карбоцепные синтетические волокна. М.,
«Химия», 1974, с. 445—491.
1/,10 Зек. 234
298 Глава 10. Производство волокон из фторсодержащих полимеров
Политетрафторэтилен не растворяется ни в одном из известных
растворителей, а также не может быть расплавлен или размягчен
без разложения. Следовательно, ни один из методов, обычно
используемых при формовании волокон (из раствора, из расплава и
даже продавливанием размягченного полимера), не может быть
применен для получения волокна тефлон. При производстве этого
волокна используется принципиально новый способ формования —
из суспензии полимера, образующейся в процессе эмульсионной
полимеризации *.
Технологический процесс получения волокна тефлон обычно
включает следующие стадии.
В суспензию полимера, полученную при полимеризации тетра-
фторэтилена, добавляют заранее приготовленный раствор
водорастворимого полимера (загустителя) и эту смесь направляют на
прядильную машину для формования волокна.
К добавляемому в качестве загустителя полимеру
предъявляются определенные требования. Этот полимер должен:
обладать хорошей способностью к волокнообразованию;
хорошо растворяться в среде, используемой для образования
суспензии фторсодержащего полимера;
полностью разлагаться с образованием газообразных
продуктов при последующем процессе спекания.
Наиболее приемлемыми загустителями, удовлетворяющими
указанным требованиям, являются поливиниловый спирт или
ксантогенат целлюлозы. При растворении этих загустителей в воде,
являющейся средой при суспензионной полимеризации тетрафтор-
этилена, образуется вязкий прядильный раствор, содержащий в
качестве наполнителя водную суспензию политетрафторэтилена.
Содержание политетрафторэтилена в суспензии составляет 35—
40%.
Для повышения стабильности суспензии и загустителя в состав
полимера рекомендуется вводить в качестве стабилизатора
небольшое количество поверхностно-активного вещества.
Волокно тефлон получают методом мокрого или сухого
формования. Сформованное волокно обладает, как правило, низкими
физико-механическими показателями (прочность около 0,8 гс/текс).
Следовательно, волокно, полученное из суспензии, нуждается в
дополнительных обработках. Важнейшими из них являются спекание
и вытягивание.
Спекание заключается в быстром (в течение неско. ьких
секунд) нагревании волокна при температуре около 385 °С. При этой
температуре загуститель разлагается, продукты деструкции —
газообразные вещества — удаляются, а частицы полимера
сплавляются и образуется монолитное волокно более высокой прочно-
* Этот метод начинают применять и при формовании волокна из эмульсии
волокнообразующих полимеров других типо'в.
10.1. Получение волокна тефлон 299
сти. В отличие от относительно крупных кусочков
политетрафторэтилена, которые плавятся настолько медленно, что процесс
разложения начинается еще до того, как полимер расплавится,
высокодисперсные частицы этого полимера сплавляются до начала
разложения полимера.
Для дополнительного упрочнения волокно тефлон вытягивают
при 200—250 °С на 300—500%. Если температуру вытягивания
повысить до 330 °С, то максимально возможная степень вытягивания
увеличивается до 700—800% [1]. По аналогичной схеме получается
не только текстильная нить, но и моноволокно.
В Советском Союзе разработан метод получения волокна
полифен из водной дисперсии политетрафторэтилена формованием в
водной солевой ванне [2].
Волокно тефлон (полифен) характеризуется следующими
показателями.
Плотность волокна составляет 2,2 г/см3, т. е. значительно выше,
чем у всех природных и других химических волокон. Высокая
плотность является недостатком фторсодержащих волокон.
Прочность волокна сравнительно невелика— 12—16 гс/текс при
удлинении 13—15%. Однако ввиду значительно большей
плотности при сопоставлении показателей волокна тефлон с
показателями других волокон меньшей плотности (например, с
полиамидными) пользоваться прочностью, выраженной в гс/текс, не
следует, так как это приводит к ошибочным выводам. Например
прочность в гс/текс (мН/текс) волокна тефлон в 3—4 раза ниже,
чем полиамидного. Если же сопоставить показатели, выраженные
в кгс/мм2 (мН/м2), то прочность волокна тефлон C5 кгс/мм2)
будет всего в 2 раза ниже, чем прочность полиамидных волокон.
Следовательно, в таких случаях целесообразно характеризовать
волокно показателем прочности в кгс/мм2. Вытягиванием волокна при
370°С на 800—900% прочность можно повысить [1]до20—25 гс/текс,
что, естественно, расширяет области его применения. Одновременно
с увеличением прочности повышается и теплостойкость волокна,
характеризуемая, в частности, величиной усадки при повышенных
температурах.
Гигроскопичность волокна тефлон ничтожна. При
относительной влажности воздуха 65% это волокно поглощает меньше 0,01 %
влаги. Следовательно, прочность и удлинение волокна в мокром
состоянии не изменяются. Тефлон является самым гидрофобным
волокном из всех химических волокон.
Термостойкость волокна тефлон превышает аналогичный
показатель почти всех природных и большинства химических волокон.
Это волокно выдерживает нагрев до 250 °С без разложения и бфз
необратимого изменения прочности (при последующем
определении этого показателя при нормальной температуре). Например,
после нагрева волокна тефлон в течение 8 дней при 260 °С и
последующего (после охлаждения) испытания его при нормальной
300 Глава 10. Производство волокон из фторсодержаЩих полимеров
температуре прочность волокна снижается не более чем на 25—
30%. Усадка ткани при выдерживании ее при 260 °С составляет
20—25%. В течение небольшого промежутка времени (нескольких
минут) волокно тефлон может быть использовано при 315 °С.
Теплостойкость волокна тефлои, так же как и других
синтетических волокон, невысока. При повышенных температурах не
только значительно понижается прочность, но в отличие от
термопластичных синтетических волокон одновременно снижается
удлинение волокна:
Температура испытании и иагрева, °С ...... 20 150 200
Прочность, гс/текс 14,5 3,6 2,7
Удлинение, % 3 7,8 7
Хемостоикость волокна тефлон исключительно высокая.
Волокно и получаемые из него изделия стойки к разнообразным
агрессивным реагентам при нормальной и при повышенной
температуре. Например, при кипячении в концентрированной серной или
азотной кислоте, в царской водке или в 50%-ном растворе NaOH
при 100 °С механические свойства этого волокна не изменяются.
Коэффициент трения волокна тефлон — наиболее низкий среди
всех типов химических волокон.
Модуль эластичности волокна тефлои сравнительно
невысокий — 144 гс/текс.
Указанные свойства и определяют области применения волокна
тефлон. Учитывая ограниченную сырьевую базу и значительно
более высокую стоимость политетрафторэтилена по сравнению с
другими синтетическими волокнообразующими полимерами, можно
сделать вывод, что использование этого волокна целесообразно в
тех случаях, когда ткани из других синтетических волокон не
могут быть применены (например, в очень агрессивных средах, в
частности в концентрированной азотной кислоте или
концентрированном растворе перекиси водорода).
10.2. ПОЛУЧЕНИЕ ВОЛОКНА ФТОРЛОН
Метод получения волокна фторлон из фторсодержащих
полимеров разработан в Советском Союзе 3. А. Роговииым и 3. А. За-
зулиной [3—5]. В отличие от политетрафторэтилена некоторые
фторсодержащие сополимеры растворимы в доступных
растворителях, в частности в ацетоне. Это обстоятельство упрощает
технологический процесс производства волокна фторлон и дает
возможность значительно повысить его прочность по сравнению с
прочностью волокна тефлон.
Так же как и другие полимеры, фторлои является полндисперс-
ным продуктом. Фракционирование этого полимера проводили
методом фракционного осаждения изооктаном из ацетоновых
растворов [6]. Средневесовой молекулярный вес фторлона, используемого
для производства волокна, составляет 70 000—100 000.
10.2. Получение волокна фторлон 101
Волокно фторлон из ацетоновых растворов можно формовать
сухим и мокрым способами (осадительная ванна — 4%-ный
водный раствор ацетона). В отличие от большинства карбоцепных
волокон волокно фторлон можно вытягивать при нормальной
температуре на 300—400%- Дополнительное вытягивание волокна
производится при 140—150°С. Суммарно волокно фторлон можно
вытянуть на 1600—2000%.
При последующей терморелаксации (в условиях свободной
усадки) при 120—130 °С в течение 1 ч удлинение волокна фторлон,
так же как и других термопластичных волокон, повышается с 8—
10% до 12—20% без снижения прочности.
Волокно фторлон выпускается обычно в виде нити,
состоящей из очень тонких волокон. Толщина вытянутого волокна 0,05—
0,11 текс, т. е. это волокно значительно тоньше других волокон,
вырабатываемых в производственных условиях.
Волокно фторлон характеризуется следующими показателями.
Плотность волокна фторлон составляет 2,13 г/см3.
Прочность волокна фторлон, подвергнутого вытягиванию на
800—1500%, достигает 100—120 кгс/мм2, что превышает
прочность любого другого химического волокна и почти всех природных
волокон (кроме рами и льна). Однако такая высокая прочность
волокна (и, следовательно, пониженное удлинение), достигаемая
при усложнении технологического процесса (дополнительное
вытягивание волокна при повышенных температурах), не всегда
является необходимой. В ряде случаев целесообразно
применять изделия из волокон, вытянутых только при нормальной
температуре на 300—400%. Прочность такого волокна составляет 50—
60 кгс/мм2, что вполне обеспечивает высокие эксплуатационные
свойства изделий из него.
Удлинение волокна в зависимости от степени вытягивания нити
и наличия процесса терморелаксации составляет от 8 до 40%. По
эластичности волокно фторлон незначительно уступает только
полиамидному.
Гигроскопичность волокна при относительной влажности
воздуха 65% составляет всего лишь 0,04%. Прочность и удлинение
волокна в мокром состоянии не изменяются.
Светостойкость волокна фторлон высокая. По этому
показателю волокно фторлон превосходит многие химические волокна и
даже полиакрилонитрильное волокно.
Теплостойкость и термостойкость волокна фторлон, полученного
из полимера нерегулярной структуры, более низкие, чем волокна
тефлон. С увеличением степени вытягивания термостойкость
волокна повышается. Однако и для высокопрочного волокна фторлон
максимальная температура, при которой могут применяться
изделия из этого волокна, не превышает 120—130 °С. По этому
показателю волокно фторлон значительно уступает волокну из
политетрафторэтилена.
302 Глава 10. Производство волокон из фторсодержащих полимеров
Хемостойкость волокна фторлон к действию агрессивных
реагентов такая же высокая, как и волокна тефлон. Это волокно
стойко к концентрированной азотной кислоте при нормальной и
при повышенной температуре.
Приведенные замечания о сравнительно ограниченных
сырьевых ресурсах производства волокна тефлон относятся и к
производству волокна фторлон.
Волокно фторлон целесообразно применять для изготовления
фильтровальных тканей, спецодежды, ниток, прокладок и других
аналогичных изделий, подвергаемых при эксплуатации
воздействию агрессивных веществ, когда природные и другие
химические волокна не могут быть использованы. Основным
преимуществом фторлона перед волокном тефлон является значительно
более высокая прочность и простота технологического процесса его
получения, недостатком — более низкая теплостойкость.
ЛИТЕРАТУРА
1. M оте лье 3. H., Сигал M. Б., В а рш а иски й В. Я., Хим. волокна, 1971,
№ 1, с. 53—55.
2. Сигал М. Б., К о з и о р о в а Т. Н., Хим. волокна, 1961, № 3, с. 37—40.
3. З а з у л и н а 3. А., Яковлев И. И., Роговин 3. А.,
«Научно-исследовательские труды МТИ», 1956, т. 18, с. 71—75.
4. Роговин 3. А., Заэулина 3. А., Марцинковская P. H Текст,
пром., 1957, № 5, с. 6—8.
5. Роговин 3. А., З азу лин а 3. А., Изв. вузов. «Химия и химическая
технология», 1958, № 1, с. 137—140.
6. Гильман И. С, Лебедушкина С. Б., Хим. волокна, 1963, № 6,
с. 28—30.
Глава 11
ТЕРМОСТОЙКИЕ И ЖАРОСТОЙКИЕ ВОЛОКНА
Термостойкие и жаростойкие волокна .приобретают большое
значение в связи с все возрастающими потребностями народного
хозяйства в теплоизоляционных и конструкционных материалах,
способных длительное время работать при повышенных и высоких
температурах. Волокна, так же как и другие полимерные
материалы, используемые при повышенной температуре, разделяются на
две группы: термостойкие и жаростойкие.
Термостойкими называются волокна, которые могут быть
использованы в течение десятковій сотен часов при температурах
250—400 °С в качестве изоляционных и конструкционных
материалов. Для этого термостойкие волокна должны обладать высокими
прочностью и эластичностью, а также высоким начальным
модулем. Требования в отношении рабочих температур, при которых
могут и должны использоваться термостойкие волокна, не
являются постоянными. Основной тенденцией в этой области
является непрерывное повышение рабочих температур. Если еще 5—
10 лет тому назад получение волокон, которые можно использовать
в течение длительного времени при 200—300 °С, являлось одним из
наиболее существенных достижений в производстве синтетических
волокон, то в настоящее время требования к рабочим
температурам повышены минимум на 100 °С. По-видимому, эта тенденция
сохранится и в ближайшие годы.
Жаростойкими называются волокна, которые могут быть
использованы в качестве теплоизоляционных и конструкционных
материалов при 2000—3000 °С. Десять лет тому назад создание, а тем
более промышленное производство таких органических волокон
казалось вообще невозможным, а в 1970 г. мировое производство
жаростойких волокон (по ориентировочным подсчетам) уже
превысило 1000—1500 т.
Организация производства термо- и жаростойких волокон
является одним из наиболее важных достижений в промышленности
химических волокон за последнее десятилетие. Естественно, что эти
волокна не являются волокнами массового потребления, поэтому
они вырабатываются в ограниченных масштабах. Однако это не
уменьшает их значения, так как без преувеличения можно
утверждать, что без таких волокон дальнейшее развитие техники в
некоторых важных отраслях народного хозяйства было бы невозможно.
304 Глава 11. Термостойкие и жаростойкие волокна
Термо- и жаростойкие волокна могут быть разделены на три
группы (неравноценных по значению и перспективности): волокна
на основе неорганических, элементоорганических и органических
полимеров.
Для производства неорганических волокон используются SiO2,
карбид кремния, производные борной кислоты, силикаты
алюминия и окислы различных металлов. Этот класс термостойких,
точнее, жаростойких волокон, обладает наиболее высокой стойкостью
к высоким температурам. Однако низкие эластические свойства
этих волокон, сложность получения и высокая стоимость
ограничивают масштабы производства и области их применения.
Рассмотрение условий получения и свойств неорганических волокон выходит
за рамки данной книги. Этим волокнам посвящены специальные
статьи [1].
Из элементоорганических полимеров для производства
термостойких волокон представляют интерес кремнийорганические и
фторорганические полимеры.
Кремнийорганические полимеры широко применяются для
изготовления термо- и жаростойких эластомеров (например, каучу-
ков и лаков). Однако из-за отсутствия достаточного количества
полярных групп в молекулах мономеров и нерегулярного строения
этих полимеров получить термостойкие волокна с требуемым
комплексом свойств (высокими прочностью и начальным модулем)
пока не удается. Вполне вероятно, что в ближайшие годы будут
разработаны методы получения таких волокон.
Волокна регулярного строения из фторсодержащих полимеров
(например, из политетрафторэтилена) могут быть использованы
при температурах эксплуатации до 280°С. Однако из-за
специфических условий формования (см. разд. 10.1) получение
высокопрочных волокон из этого полимера крайне затруднено.
Для получения термо- и жаростойких волокон используются в
основном органические полимеры определенного строения.
Принципы и условия получения термо- и жаростойких химических
волокон существенно различаются между собой, поэтому
целесообразно рассматривать их раздельно.
11.1. ПОЛУЧЕНИЕ ТЕРМОСТОЙКИХ ВОЛОКОН
Термостойкие волокна, используемые в различных областях
техники, должны одновременно обладать и высокой
теплостойкостью [2], т. е. в незначительной степени снижать, хотя и
обратимо, комплекс механических свойств, в первую очередь прочность
при высоких температурах.
Теплостойкие волокна можно получить химической
модификацией известных многотоннажных синтетических волокон.
Образование химических связей между макромолекулами или их
агрегатами (сшивание) путем обработки вытянутых полиамлдных и по-
11.1. Получение термостойких волокон 305
лиакрилонитрильных волокон различными полифункциональными
соединениями значительно повышает их теплостойкость, но не
улучшает термостойкости волокна.
Наиболее перспективным является использование для
получения термостойких волокон специального класса термостойких во-
локнообразующих полимеров, химическое строение которых
определяет их термическую стойкость и температурный интервал
эксплуатации изделий.
Для создания термостойких полимеров необходимо обеспечить
интенсивное взаимодействие между макромолекулами, высокую
степень кристалличности полимера и наличие объемных
заместителей в цепи, затрудняющих свободное вращение отдельных цепей
или сегментов, но не препятствующих их плотной упаковке
(наличие жестких цепей) [3]. Исходные полимеры должны быть
достаточно высокого молекулярного веса, так как от этого зависят
способность волокон к вытягиванию и их механические свойства.
Полимеры должны растворяться в некоторых растворителях. Для
выполнения этих требований необходимо, чтобы макромолекулы
волокнообразующего полимера содержали ароматические или
циклические звенья, обусловливающие жесткость цепи, высокий
начальный модуль, тепло- и термостойкость волокон.
Как правило, такие волокна не могут быть получены из карбо-
цепных полимеров. Для этой цели могут быть использованы только
некоторые гетероцепные полимеры, в макромолекуле которых
содержатся указанные выше группы. Следовательно, термостойкие
волокнообразующие полимеры могут быть получены не по
реакции полимеризации, а по реакции поликонденсации, в ряде случаев
с последующей циклизацией полученного материала. ¦
Технологические методы получения термостойких гетероцепных
волокон существенно отличаются от методов производства
многотоннажных синтетических гетероцепных волокон (полиамидных и
полиэфирных). Если волокна будут использоваться при 250°С и
более высокой температуре, то, естественно, температура
плавления исходных полимеров' и получаемых из них волокон должна
быть выше, чем температура их эксплуатации. Получить такие
волокна формованием из расплава нельзя, так как температура
плавления этих полимеров, как правило, превышает температуру их
разложения. Поэтому термостойкие органические волокна можно
формовать только из растворов полимеров. Термостойкие волокно-
образующие полимеры растворяются в ограниченном числе
органических апротонных растворителей, обладающих высокой
растворяющей способностью по отношению к различным классам
полимеров.
Указанные растворители, к которым относится диметилформ-
амид, диметилацетамид, N-метилпирролидон и диметилсульфоксид
(используемые при производстве волокон из полимеров и
сополимеров акрилонитркла), получают все более широкое применение
306 Глава 11. Термостойкие и жаростойкие волокна
при формовании волокон из термостойких полимеров. Улучшить
растворяющую способность этого класса растворителей можно, как
это впервые показано при получении волокон из ароматических
полиамидов, путем добавления к апротонным растворителям
небольших количеств солей (LiCl, СаС12, MgCb). Такие бинарные
смеси (например, 90—95% диметилформамида и 5—10% LiCl)
являются новым классом растворителей, которые используются для
получения концентрированных прядильных растворов,
применяемых для формования термостойких волокон. Детальное изучение
механизма действия добавленных солей и возможности
использовать эти смеси при формовании карбоцепных и других гетероцеп-
ных волокон представляет большой интерес.
В качестве растворителя при получении термостойких волокон
может быть использована и концентрированная серная кислота,
которая является наиболее дешевым веществом. Однако
использование ее связано с большими трудностями, так как ухудшаются
условия-труда. Если эти трудности будут преодолены, серная
кислота найдет широкое применение.
Последующие процессы вытягивания и упрочнения
сформованных термостойких волокон производятся по обычной схеме, при
повышенных температурах на различных стадиях технологического
процесса.
Для получения термостойких волокон используются
ароматические полиамиды и полиэфиры, гетероциклические и лестничные
полимеры.
11.1.1. Волокна из ароматических полиамидов
и полиэфиров
Как известно, замена алифатических звеньев на ароматические
в макромолекулах указанных гетероцепных волокнообразующих
полимеров значительно повышает их температуру плавления и
термостойкость. Температура плавления ароматических полимеров
значительно выше их температуры разложения, поэтому синтез
обычными методами поликонденсации в расплаве проводить
нельзя. Ароматические полимеры можно получить следующими тремя
способами:
поликонденсацией на поверхности раздела фаз ароматических
диаминов, растворенных в воде, и дихлорангидридов ароматических
дикарбоновых кислот, растворенных в неполярных растворителях,
не смешивающихся с водой [4]; реакция проводится при низких
температурах (от —20 до 0°С) для уменьшения возможности
разложения дихлорангидрида дикарбоновой кислоты, находящейся на
поверхности водной фазы;
поликонденсацией при пониженной температуре в среде, в
которой растворимы исходные мономеры и синтезированный полимер;
//./. Получение термостойких волокон
307
вместо дикарбоновой кислоты применяют более реакционноспособ-
ное соединение — дихлорангидрид кислоты в присутствии реагента,
связывающего НС1, выделяющийся при реакции;
поликонденсацией мономеров в эмульсии, например в системе
тетрагидрофуран — вода [5].
В качестве исходных продуктов для синтеза термостойких
полимеров могут быть использованы различные ароматические
диамины и ароматические дикарбоновые кислоты. Наибольшее
практическое применение получили продукты поликонденсации фени-
лендиамина и фталевых кислот. При одинаковом составе исходных
продуктов положение реакционноспособной группы в молекуле
мономера оказывает существенное, а в ряде случаев решающее
влияние на тепло- и термостойкость получаемых полимеров и их
растворимость. Наиболее высокой термостойкостью обладает
полиамид, синтезированный методом поликонденсации п-фениленди-
амина и терефталевой кислоты (температура разложения 500—
600°С) [6]. Но полученный полимер плохо растворяется, поэтому
переработка его в волокно сопряжена со значительными
трудностями. Из этого класса термостойких волокнообразующих
полимеров наиболее широко применяется продукт взаимодействия л«-фе-
нилендиамина и изофталевой кислоты. Из этого полимера в США
вырабатывается волокно номекс.
Температура плавления полиамида, синтезированного методом
поликонденсации .w-фенилендиамина и дихлорангидрида
изофталевой кислоты, составляет 410 °С, а полиамид, полученный
взаимодействием того же диамина с терефталевой кислотой, не плавится
при нагревании до 500 °С [7]. Реакция поликонденсации .и-фенилен-
диамина и дихлорангидрида изофталевой кислоты протекает по
схеме
H2N C1OC
СОС1 —*¦
\—/ -неї
СО—
Эти ароматические полиамиды растворяются в различных апро-
тонных растворителях, например в диметилформамиде, причем
растворимость значительно улучшается при добавлении небольших
количеств E—8% от массы раствора) хлоридов магния или
кальция, а особенно хлорида лития. Такие бинарные смеси
растворителей впервые были использованы при формовании термостойкого
волокна из приведенного полиамида.
308 Глава il. Термостойкие и жаростойкие волокна
Полиамид, из которого вырабатывается волокно типа номекс,
растворяется и в других органических растворителях, например
в диметилсульфоксиде, капролактаме, N-метилпирролидоне, но
на практике применяется только бинарная смесь, состоящая из
диметилформамида и хлоридов магния, кальция или лития
E-8%).
Для формования волокна мокрым способом применяется 15—
25%-ный раствор полимера. Чем больше LiCl содержится в смеси,
тем выше стабильность раствора и тем больше может быть
количество воды в смеси растворителей, необходимое для получения
устойчивых растворов. Например, при содержании в прядильном
растворе 2—3% LiCl максимально допустимое количество воды
составляет 1% (от массы раствора), а при повышении
концентрации LiCl в растворе до 6—8% содержание воды может быть
увеличено до 4%.
Введение в состав прядильного раствора небольших количеств
LiCl обусловливает также понижение вязкости эквиконцентриро-
ваиных растворов полимера. Минимальная вязкость достигается
при наличии в растворе 5% LiCl. Формование волокна номекс из
раствора полимера в диметилформамиде или в диметилацетамиде
производится при низких скоростях E—8 м/мин) в осадительной
ванне — водном растворе того же растворителя, содержащего 30—
50% воды [3].
Температура осадительной ванны 50—70 °С. Вытягивание
волокна номекс осуществляется обычно в две стадии. Для фиксации
структуры вытянутого волокна и его дополнительной
кристаллизации производится термообработка при 360 °С.
Волокно номекс обладает высокими механическими
свойствами [7]: прочность 46—55 гс/текс D60—550 мН/текс); удлинение
20—40%; начальный модуль ' 1300—1500 кгс/мм2 О3'103—
15-Ю3 МН/м2). Плотность волокна 1,35 г/см3.
Характерной особенностью волокна номекс, так же как и
других термостойких волокон, не содержащих гидрофильных
функциональных групп, является сравнительно высокая гигроскопичность,
составляющая 5% при относительной влажности воздуха 65%.
Это объясняется, по-видимому, пористой структурой волокна.
Изделия из волокна номекс можно в течение длительного
времени эксплуатировать при 250°С. При температуре 280°С волокно
сохраняет 50% начальной прочности, следовательно, оно обладает
высокой теплостойкостью. Температура нулевой прочности этого
волокна 370°С. После нагрева волокна номекс при 175°С в
течение 2000 ч прочность волокна снижается на 20—25% (8] (после
охлаждения).
Так же как и другие ароматические полиамидные волокна,
волокно номекс характеризуется высокой радиационной стойкостью.
Вытянутое волокно растворяется только в концентрированной
серной кислоте, диметилформамиде и диметилацетамиде [91.
//./. Получение термостойких волокон 309
К классу термостойких полиамидов относятся также
ароматические полисульфонамиды, получаемые взаимодействием 4,4'-ди-
аминодифенилсульфона и дихлорангидридов фталевых кислот, в
частности изофталевой кислоты [10]. Реакция протекает по схеме
СОСІ
-неї
HN
Наличие группы SO2 между фенильными ядрами повышает
гибкость полимера и его растворимость и соответственно облегчает
условия переработки и улучшает эластические свойства
получаемых волокон.
Реакция поликонденсации при синтезе полисульфонамидов, так
же как и при получении полиамида типа номекс, производится на
поверхности раздела фаз или в растворе. При проведении реакции
поликонденсации в среде, в которой растворяются мономеры и
получаемый полимер (например, в диметилацетамиде), образуется
концентрированный прядильный раствор, который после
фильтрации поступает на прядильную машину. Сформованное волокно
подвергается вытягиванию в две стадии (пластификационная вытяжка
при 20 °С на 200—250% и дополнительное вытягивание при 300—
350 °С на 30—60%).
При замене изофталевой кислоты терефталевой получается
полимер с более высокой степенью кристалличности и
соответственно с более высокой термостойкостью и температурой плавления.
Из такого полимера в Советском Союзе [3] получено волокно суль-
фон, по ряду показателей превосходящее волокно номекс. В
частности, волокно сульфон обладает более высокой термо- и
теплостойкостью. Например, при 300—310 °С волокно сульфон сохраняет
50% начальной прочности; после нагрева в течение 100 ч на
воздухе при 300°С оно необратимо теряет только 10—15% прочности.
В течение непродолжительного времени волокно сульфон можно
использовать и при 400°С. При этой температуре оно сохраняет
20—25% начальной прочности.
Для производства термостойких волокон ароматические
полиэфиры применяются значительно реже ароматических полиамидов
или полисульфонамидов. Это объясняется в основном тем, что
ароматические полиэфиры имеют более низкую температуру
плавления или разложения, чем ароматические полиамиды. Лишь
немногие полиэфиры плавятся выше температуры 350—380 °С. Из
ароматических полиэфиров для получения волокна иногда
применяются полиарилаты — продукты поликонденсации бис-фенолое
310
Глава U. Термостойкие и жаростойкие волокна
и дихлорангидридов ароматических дикарбоновых кислот
(например, фенолфталеина и изо- или терефталевой кислоты). Реакция
протекает по схеме [11, с. 132—174]
ОН
С1ОС
юсі + н
—ос-
/А.
-соо
-неї
Систематические исследования в области синтеза различных
полиарилатов проводятся советскими исследователями [12].
Имеются сведения, что волокна из полиарилатов выпускает фирма
«Дюпон» [13]. Волокно формуют сухим способом из растворов полимера
в циклогексаноне.
11.1.2. Волокна из полигетероциклических
полимеров
Известно, что наиболее термостойкими синтетическими гетеро-
цепиыми полимерами являются полимеры, содержащие
ароматические звенья и систему сопряженных циклов. Подобные полимеры
применяются для производства термостойких пластмасс,
конструкционных и изоляционных материалов.
При получении волокон полигетероциклические полимеры в
большинстве случаев синтезируют в две стадии.
На первой стадии по реакции поликонденсации образуется ге-
тероцепной полимер линейной структуры, растворимый в тех же
апротонных растворителях, в которых растворяются описанные
выше волокнообразующие ароматические полиамиды. Получаемое
из этого полимера волокно является только полупродуктом, так
как обладает низкой термостойкостью.
//./. Получение термостойких волокон 311
На второй стадии в результате термической или химической
обработки волокна происходят дальнейшие превращения полимера
(преимущественно дегидратация) и в макромолекулах образуются
гетероциклы (процесс циклизации). Осуществить эти превращения
в исходном полимере, предназначенном для получения волокон,
в большинстве случаев не представляется возможным, так как
гетероциклические полимеры не плавятся и не растворяются в
известных растворителях. Чем больше степень циклизации линейных
полимеров, осуществляемая на сформованном, а в ряде случаев на
уже вытянутом волокне, тем выше термостойкость волокна.
Разработаны также методы синтеза в одну стадию некоторых
полимеров циклической структуры (в частности, полиимидов и пир-
ронов), которые растворяются в некоторых растворителях.
Поэтому волокно можно в ряде случаев формовать из растворов уже
циклизованного полимера.
Дегидратация может быть достигнута действием химических
реагентов (химическая циклизация) или повышенных температур
(термическая циклизация). Каждый из этих способов имеет свои
преимущества и недостатки, однако в большинстве случаев метод
термической циклизации, не требующей расхода химических
реагентов, является более экономичным. Для того чтобы процесс
дегидратации стал возможным, исходный полимер и сформованное
из него волокно должно содержать реакционноспособные группы,
которые в результате указанных обработок могут отщепляться с
выделением молекул воды или других низкомолекулярных
соединений и образованием циклов. Для получения таких полимеров
необходимо также, чтобы один из мономеров был тетрафункцио-
нальным соединением и содержал различные функциональные
группы, отличающиеся между собой по реакционной способности.
Реакцию поликонденсации в этом случае проводят в таких
условиях, при которых в тетрафункциональном соединении способен
реагировать только один тип функциональных групп и,
следовательно, реакция поликонденсации протекает как обычная реакция
линейной поликонденсации бифункциональных соединений.
Для синтеза полигетероциклических полимеров используются
различные классы би- и тетрафункциональных мономеров.
.Соответственно образуются различные по строению полимеры и
волокна на их основе. Большинство этих термостойких волокон не
вышло (кроме полипиромеллитимидного волокна) из стадии опытной
и опытно-промышленной проверки, хотя, бесспорно, этот класс
термостойких волокон найдет в дальнейшем более широкое
применение. Из волокон, получаемых на основе гетероциклических
полимеров, наибольший интерес представляют, полибензоксазольные,
полиоксадиазольные, полипиромеллитимидные и полибензимид-
азольные волокна.
Различные представители этого класса термостойких
полимеров отличаются в основном по строению и характеру
312
Глава 11. Термостойкие и жаростойкие волокна
тетрафункционального соединения, используемого для синтеза
линейного гетероцепного полимера. Технологическая схема получения
полимера, подготовки его к формованию, процесс формования,
последующего вытягивания и циклизации для всех этих волокон
примерно одинаковы. Рассмотрим вкратце методы получения
указанных типов термостойких волокон.
Полибензоксазольные волокна. Волокнообразующий полимер
синтезируется по реакции поликонденсации тетрафункционального
соединения — бис-о-оксидиаминов с дихлорангидридом изо- или те-
рефталевой кислоты или 4,4'-дифенилоксикарбоновой кислоты [14].
Реакция протекает по схеме
НО
ОН
+ гаСЮС—R'—СОСІ
НО,
ОН
-неї ш/ \NH—CO—R'—СО
Обычно этот класс гетероциклических полимеров получают
взаимодействием 3,3'-диоксибензидина и дихлорангидрида изофтале-
вой кислоты:
H,N-
ш
Путем дегидратации полибензоксамида получается
гетероциклический полимер — полибензоксазол:
Как видно из этой схемы, поликонденсация проводится в таких
условиях, при которых из двух типов функциональных групп,
входящих в молекулу 3,3'-диоксибензидина, в реакцию вступают
только более реакционноспособные группы NH2. В образующемся
линейном полимере содержатся реакционноспособные группы ОН.
11.1. Получение термостойких волокон 313
При поликонденсации в растворе получается
концентрированный раствор полимера в апротонном растворителе
(преимущественно в диметилацетамиде). Формование полибензоксазольного
волокна, так же как и других полигетероциклических
термостойких волокон, проводится мокрым способом во избежание
преждевременной частичной циклизации, которая может иметь место при
формовании волокна сухим способом при повышенных
температурах.
Сформованное полибензоксазольное волокно подвергается
вытягиванию обычно также в две стадии. Затем при повышенной
температуре происходит циклизация, или, точнее, циклодегидратация
волокна. Этот процесс начинается еще при вытягивании волокна
при повышенной температуре. Циклизация может проводиться в
вакууме или при нормальном давлении в атмосфере инертного
газа. Химическая циклизация этого волокна пока не была
осуществлена. Реакция термической дегидратации без одновременного
разложения образующегося полимера циклической структуры
начинается с заметной скоростью при 200 °С и завершается при
300—400 °С [15].
Циклизация волокна протекает полней и в течение более
короткого времени, если нагреванию подвергается невытянутое или
незначительно вытянутое волокно [16]. Реакция циклизации в
ориентированных полибензоксазольных волокнах, так же как и в
других полигетероциклических волокнах, протекает в 10—15 раз
медленней, чем в неориентированных волокнах [17]. Поэтому
приведенная схема получения полигетероциклических волокон путем
циклизации уже вытянутых при нормальной и при повышенной
температуре волокон не является универсальной, и в ряде случаев
целесообразно осуществлять циклизацию частично вытянутого
волокна. Если нагрев волокон для циклизации проводить под
натяжением, то в этих условиях происходит их дополнительное
упрочнение.
Полибензоксазольные волокна рекомендуется вытягивать на
100—300% при комнатной температуре и дополнительно на 15—
50% при высокой температуре D90—500 °С) [18].
Полибензоксазольное волокно является одним из наиболее
термостойких гетероцепных волокон. После нагрева в течение 48 ч при
300 °С прочность волокна снижается всего на 23%, а после
нагрева при 400°С в течение 3,5 ч оно сохраняет более 55% исходной
прочности. Это волокно обладает высокой стойкостью к
ультрафиолетовому излучению, к действию концентрированных
растворов щелочей и серной кислоты, но нестойко к сильным
окислителям. Прочность волокна изменяется в зависимости от
молекулярного веса исходного линейного полимера и степени вытягивания
в пределах 22—50 гс/текс B20—500 мН/текс) при удлинении
18—10%. Даже циклизованное волокно, не содержащее полярных
групп, поглощает в стандартных условиях 12% влаги [19], что
11 Зак. 234
314 Глава 11. Термостойкие и жаростойкие волокна
может быть объяснено только дефектами структуры этого волокна.
Этот тип полигетероциклических волокон, впервые полученный
в СССР, по-видимому, не сможет в ближайшие годы получить
практического применения из-за того, что один из исходных
продуктов, используемых для синтеза полимера, — 3,3'-диоксибенз-
идин — является канцерогенным продуктом. Производство этого
волокна станет возможным только в том случае, если удастся найти
неканцерогенные тетрафункциональные соединения.
Полиоксадиазольные волокна. Линейные оксадиазольные
полимеры— полупродукты производства полиоксадиазольных волокон
получаются взаимодействием дихлорангидридов изо- или терефта-
левой кислоты с дигидразидами бифункциональных ароматических
кислот, являющихся тетрафункциональными соединениями, так как
каждая гидразидная группа содержит по две реакционноспособные
группы. Полигидразиды линейной структуры получаются из этих
мономеров по следующей схеме:
О О
ХҐ'
II
C— NHNH2
Так нее-как и при получении других гетероцепных полимеров,
реакция поликонденсации проводится при пониженных
температурах в диметилацетамиде или в N-метилпирролидоне в присутствии
небольшого количества LiCl, который образуется при
взаимодействии введенного в раствор LiOH с выделяющимся при реакции
НС1.
Циклизация этих типов волокон, осуществляемая в основном
термическим методом, — длительной обработкой волокна в течение
50—100 ч при 200—300 °С [20], происходит по схеме
NH—NH _н2о
11.1. Получение термостойких волокон 315
Этот класс полигетероциклических волокон также является
одним из наиболее термостойких. После нагрева этих волокон на
воздухе при 300°С в течение 700 ч они теряют только 50%
начальной прочности [21].
Разработан также одностадийный синтез этого полимера в
растворе олеума при повышенных температурах. Этот способ имеет
ряд существенных технико-экономических преимуществ, так как
вместо дихлорангидридов кислот используются соответствующие
дикарбоновые кислоты, а вместо дигидразидов — гидразинсульфат.
Реакция протекает по схеме
+ 2nNH2NH2 • H2SO4 —>
Полипиромеллитимидные волокна. Исходным сырьем для
синтеза этого важного класса термостойких волокон, получивших
практическое применение [22], служит пиромеллитовая кислота —
1,2,4,5-бензолтетракарбоновая кислота, точней, диангидрид этой
кислоты, обладающий более высокой реакционной способностью в
реакции поликонденсации, чем сама кислота, а также
ароматический диамин. Для синтеза полиимидов [23] используют
ароматические диамины, в частности 4,4'-диаминодифенилоксид или диами-
нодифенилсульфид, т. е. диамины типа [24]
H,N-
Пиромеллитовую кислоту синтезируют каталитическим
окислением дурола — 1,2,4,5-тетраметилбензола (получаемого из нефти)
по схеме
НООС. ^ /СООН
носк
Полученная пиромеллитовая кислота обрабатывается большим
избытком раствора уксусного ангидрида в уксусной кислоте. В
результате обменной реакции образуется диангидрид пиромеллито-
вой кислоты.
Синтез полипиромеллитимида осуществляется в две стадии. На
первой стадии реакции, проводимой в растворе в диметилацетамиде
И*
316
Глава П. Термостойкие и жаростойкие волокна
при 15 °С, образуется полиамидокислота по схеме
О О
NH,
нооо
соон
Этот линейный полимер со степенью полимеризации 1000 (М =
¦=400 000) [25] растворяется в полярных растворителях (в
частности, в амидных) и в спиртах. Волокно можно формовать сухим и
мокрым способами. Полученное волокно вытягивают на 100—200%
при повышенных температурах и затем подвергают циклизации.
Превращение полиамидокислот в полиимид протекает по схеме.
Химическая циклизация заключается в обработке волокна
дегидратирующими реагентами, в частности раствором уксусного
ангидрида в пиридине, связывающем уксусную кислоту,
образующуюся при гидратации уксусного ангидрида.
Преимущество этого метода — высокая прочность получаемых
волокон, недостаток — сложность и более высокая стоимость
процесса, обусловливаемая расходом химических реагентов и
необходимостью их последующей регенерации. Термическая циклизация
значительно проще и сводится к нагреву волокна при 150—250 °С.
Имеются данные, указывающие на то, что полиимидное
волокно, циклизоващюе химическим методом, имеет аморфную
структуру, а при термической циклизации получается значительно более
упорядоченная структура [26]. Одним из основных требований яв-
//./. Получение термостойких волокон 317
ляется максимально полная циклизация, так как наличие в
макромолекуле полиимида нециклизованных звеньев снижает
термостойкость волокон. Этот показатель тем ниже, чем больше число
нециклизованных звеньев в макромолекуле.
Полиимидное волокно является одним из наиболее
термостойких; его можно использовать в течение длительного времени при
300—350 °С. После нагрева при 300 °С в течение 100 ч это волокно
сохраняет 60—64% от начальной прочности, определяемой при
нормальной температуре, а при 400 °С волокно сохраняет около 40%
начальной прочности [27]. Температура нулевой прочности поли-
имидного волокна, носящего в СССР название «аримид» [28], выше
700 °С. Начальная прочность этого волокна 45—50 гс/текс D50—
500 мН/текс) при удлинении б—8%. Волокно не плавится и не
горит, не усаживается в горячей воде при 95°С [29]. При
температуре 200—250 °С срок эксплуатации полиимидного волокна
достигает 1000—10 000 ч, а при 350 °С на воздухе или при 400 °С в
инертной среде составляет 1—2 ч [25].
Полиимидные волокна, так же как и другие
полигетероциклические волокна, устойчивы к ядерному излучению. Например, при
дозе 1000 Мрад эти волокна теряют не более 20% исходной
прочности, в то время как обычные полиамидные волокна полностью
разрушаются при значительно меньших дозах C00 Мрад). Поли-
акрилонитрильные волокна при дозе 500 Мрад теряют более 70%
начальной прочности {27]. Полиимидные волокна сравнительно
быстро разрушаются в растворах щелочей и перегретом водяном
паре. Этот тип полигетероциклических волокон является одним из
наиболее перспективных для изготовления тканей и других
материалов, используемых при температурах до 350°С.
Полибензимидазольные волокна. Полимеры, используемые для
получения термостойких волокон этого класса, синтезируются по
тому же принципу, что и полиимиды. Основное отличие синтеза по-
либензимидазолов от синтеза полиимидов заключается в том, что
в качестве тетрафункционального соединения используется не тет-
ракарбоновая кислота, а тетрамин, в частности 3,3'-диаминобенз-
идин, который вступает в реакцию поликонденсации с дикарбоно-
вой кислотой (обычно с дифениловым эфиром изофталевой
кислоты). При одностадийном способе синтеза и циклизации полимера
реакция может быть представлена схемой
HjN
HN
—c' T 1
N
NH
fY4c
ЛЛл
N
318
Глава 11. Термостойкие и жаростойкие волокна
Поликонденсация, как правило, проводится в растворе в
присутствии реагента, который растворяет исходный мономер и
образующийся полимер.
Если при синтезе полимер не был циклизован, то сформованное
волокно подвергается термической обработке (до или после
вытягивания).
Свойства полибензимидазольных волокон заметно не
отличаются от свойств полиимидных волокон.
Кроме указанных типов термостойких полигетероциклических
волокон, полученных из гомополимеров с регулярной структурой,
термостойкое волокно получают также из ароматических
сополимеров, с регулярной структурой. Советскими исследователями [30]
синтезированы термостойкие сополимеры, содержащие в цепи
имидные и 1,3,4-оксадиазольные циклы, и сформованы волокна на
их основе. Сополимер получают поликонденсацией в растворе гидр-
азида /г-аминобензойной кислоты, диангидрида пиромеллитовой
кислоты и дихлорангидрида терефталевой кислоты по схеме
HN—
НООО
Сополимер растворяется и циклизуется в апротонных
растворителях; циклизация протекает на сформованном и вытянутом
волокне.
Чем больше в полученном сополимере оксидиазольных циклов,
тем больше его стойкость при повышенных температурах.
Например, волокно, циклизация которого проводилась в отсутствие
катализатора, ускоряющего образование оксидиазольных циклов,
после нагрева на воздухе в течение 100 ч при 300 °С сохраняло
20—30% начальной прочности. Если циклизация проводилась в
присутствии катализатора и, следовательно, количество
оксидиазольных циклов в молекуле сополимера значительно больше, то
после нагрева в тех же условиях волокно сохраняет 78—84%
начальной прочности [31].
11.1. Получение термостойких волокон
319
11.1.3. Волокна из лестничных полимеров
Из лестничных полимеров (так называемых пирронов)
получают изделия (в частности, волокна), используемые при
температурах, не превышающих 500°С. Исходными продуктами для
синтеза лесгничных полимеров служат ароматические тетрамины и
тетракарбоновые кислоты (в частности, диангидрид пиромеллито-
вой кислоты).
Так же как и при получении полиимидов, синтез может быть
одно- или двухстадийный. На первой стадии реакции,
осуществляемой в растворе полифосфорной кислоты, образуется полиамино-
амидокарбоновая кислота. В отличие от полиамидокислоты,
синтезируемой на первой стадии процесса получения полиимидов, этот
полимер не растворяется в апротонных растворителях и
растворяется только в полифосфорной или концентрированной (98%-ной)
серной кислоте. Первая стадия синтеза полимера протекает по
схеме
О О
¦NH,
NH—•
ноос
В получаемом продукте сохраняются два типа реакционноспо-
собных групп, обеспечивающих возможность дальнейших
превращений, в частности циклизации при повышенной температуре
A50—300 °С):
В полифосфорной кислоте реакция получения циклизованных
полимеров может быть осуществлена в одну стадию. Так как пир-
роны не растворяются в обычных апротонных растворителях,
используемых для формования других термостойких волокон, то
формование пирронового волокна производится из растворов полимера
820 Глава 11. Термостойкие и жаростойкие волокна
в 98%-ной H2SO4 [31] или в олеуме. Необходимость проведения
формования из растворов полимера в концентрированной серной
кислоте мокрым способом с использованием в качестве осадитель-
ной ванны разбавленного раствора H2SO4 является пока одной из
трудностей при формовании пирронового волокна. Циклизация
производится путем нагрева вытянутого пирронового волокна при
высоких температурах. Это волокно обладает высокой
термостойкостью и может быть использовано в течение длительного
времени при 300—350 °С.
При 500 °С волокно сохраняет свыше 50% начальной прочности.
После вытягивания (при температуре выше 300 °С) прочность
пирронового волокна составляет около 40 гс/текс D00 мН/текс) при
удлинении 4%. Так же как и другие термостойкие волокна, оно
обладает высоким начальным модулем.
Как видно из приведенных выше данных, ни один из описанных
типов термостойких органических волокон, содержащих
ароматические звенья или гетероциклы, не может быть использован
длительное время при температурах выше 400 °С. Для создания
волокон, обладающих достаточно высокой эластичностью, которые в
соответствии с требованиями ряда отраслей новой техники должны
длительное время использоваться при 400—450°С, необходимы
дальнейшие систематические исследования.
11.2. ПОЛУЧЕНИЕ ЖАРОСТОЙКИХ ВОЛОКОН*
Как указывалось выше, в связи с развитием техники возникла
Потребность в создании жаростойких волокнистых материалов,
предназначенных для эксплуатации при температурах до 2000—
2500 °С. Несмотря на необычайную научно-техническую сложность,
эта проблема за последнее десятилетие успешно решена. К тому
времени, когда появилась необходимость в жаростойких волокнах,
было известно большое число жаростойких материалов, к которым
относятся углерод, карбиды, нитриды, металлы и их сплавы,
окислы металлов и др. Превращение этих материалов в волокна
представляло собой новую и сложную задачу, так как обычные методы
формования химических волокон из расплавов и растворов
полимеров в большинстве случаев оказывались непригодными. В
настоящее время разработано большое число способов получения
жаростойких волокон. Наибольшие успехи достигнуты в
производстве жаростойких углеродных и борных волокон. В данном
разделе рассматриваются принципы получения и свойства только
углеродных волокон. Краткие сведения о других жаростойких
волокнах приводятся в монографиях [32].
* Этот раздел напнсан А. А. Конкиным.
11.2. Получение жаростойких волокон 321
11.2.1. Исходное сырье
Исходными продуктами для получения углеродных волокон
являются химические и природные волокна, а также некоторые
олигомеры с высоким содержанием углерода (новолаки, резолы,
нефтяной и каменноугольный пеки), лигнин и др. Эти вещества
независимо от их химической природы вначале превращают в
волокна, которые затем перерабатывают в углеродные волокна.
Таким образом, во всех случаях исходным материалом служат
волокна, ибо только такая форма материала позволяет получить
углеродные волокна.
Сущность метода состоит в том, что органические волокна в
определенных, строго контролируемых условиях подвергаются
термодеструкции. В результате этой обработки отщепляются иные, кроме
углерода, атомы (в виде разнообразных соединений), частично
выделяются углеродсодержащие соединения и осуществляется
переход от органических к углеродным волокнам. В процессе
термораспада полимера протекают две группы реакций: деструкция,
приводящая к образованию различных газообразных и смолоподобных
соединений и рекомбинация, в результате которой образуются
полимерные продукты, обогащенные углеродом.
Технологический процесс получения углеродного волокна
складывается из трех основных стадий: подготовки волокна,
карбонизации (при 900—1500°С) и графитации A500—2800°С).
Волокна, предназначенные для переработки в углеродные
материалы, должны удовлетворять следующим требованиям: 1) не
плавиться при карбонизации; 2) давать высокий выход
углеродного волокна; 3) получаемое углеродное волокно должно обладать
высокими физико-механическими свойствами.
Для получения углеродных волокон были исследованы почти
все типы химических и природных волокон, но по ряду причин
практическое применение нашли только полиакрилонитрильное и
вискозное (кордное) волокна. В Японии разработаны процессы
получения углеродных волокон из пеков [33] и лигнина [34].
Углеродные волокнистые материалы выпускаются в виде нитей, тканей,
лент, жгутов, ваты и т. п.
11.2.2. Получение углеродного волокна
из лолиакрилонитрильного волокна
Исследования в области получения углеродных волокон из по-
лиакрилонитрильных волокон (ПАН-волокна) были начаты в
СССР [35], а затем в Японии и Англии. В Англии организовано
опытно-промышленное производство этого типа высокопрочного
высокомодульного углеродного волокна. Конечным товарным
продуктом может быть волокно окисленное, карбонизованное или гра-
фитированнос,
322 Глава II. Термостойкие и жаростойкие волокна
Окисление ПАН-волокна кислородом воздуха сопровождается
большим числом реакций, основными из которых являются
циклизация полиакрилонитрила (ПАН) с образованием лестничного
полимера и присоединением кислорода, благодаря чему в процессе
карбонизации облегчается дегидрирование полимера.
По-видимому, наиболее достоверной является схема циклизации ПАН [36],
согласно которой в зависимости от глубины протекания реакции
образуются нафтиридиновые циклы (I), межмолекулярные связи
между этими звеньями'макромолекул (II) и гетероциклы (III):
сн ^сн "сн "сн
"СН
N
CN CN
сн, сн,| сн^І сн,
сн ^сн
CH2 CH2 CH;
III
На химические превращения ПАН в процессе окисления влияют
многие факторы. Введение кислорода и других окислителей
ускоряет циклизацию. Новые связи при циклизации образуются в
результате миграции пары электронов группы CN. Водород при
?-углероде приобретает кислый характер. Следовательно,
эффективная атака на атом азота и присутствие оснований ускоряют
циклизацию. Поэтому щелочи, амины, амиды щелочных металлов,
многие нуклеофильные реагенты являются катализаторами
циклизации [37]. В процессе превращения ПАН выделяется NH3, HCN,
Н2О и другие продукты. В полимере обнаружены парамагнитные
центры [38], поэтому многие исследователи считают, что циклизация
ПАН протекает по цепному механизму. В литературе приводятся
П.2. Получение жаростойких волокон
323
7,4
о
широкие диапазоны режимов окисления ПАН-волокна [39]:
продолжительность 1—44 ч, температура 200—300 °С. При осуществлении
процесса получения высокопрочного волокна, разработанного а
Англии, рекомендуется температура циклизации 220 °С,
продолжительность— 24 ч.
Окисленное ПАН-волокно, названное из-за окраски «черный
ПАН», представляет собой технически ценный продукт. К его
специфическим свойствам относится негорючесть. Оно может
применяться в качестве фильтровальных материалов, для изготовления
огнестойких пластиков, листового огнестойкого материала и т. д.
Материал выдерживает длительную
эксплуатацию при 200 °С.
Большое влияние на свойства
получаемых волокон оказывает ориентацион-
ное вытягивание в процессе окислений"
[40]. При окислении в свободном
состоянии волокно претерпевает большую
усадку, сопровождающуюся дезориентацией
структуры. Вытягивание или даже
окисление в условиях, предотвращающих
усадку, приводит к значительному
увеличению прочности и модуля Юнга
углеродного и графитового волокон (рис.
11.1). Только благодаря этому
технологическому приему удалось получить
высококачественное углеродное волокно.
При карбонизации ПАН-волокна
происходят глубокие химические и
структурные превращения и осуществляется
переход к углеродному волокну, а также
изменяются его физико-механические свойства. В процессе
карбонизации уменьшается масса волокна, особенно интенсивно при
280—420 °С; кроме того, волокно дает усадку. Остаток постепенно
обогащается углеродом, но в продукте реакции при нагреве до
1000 °С сохраняется большое количество (8—10 вес.%) азота,
который, по-видимому, входит в состав гетероциклов. На глубоких
стадиях термораспада и образовании углерода существенную роль
играют гемолитические реакции, и особенно реакции
рекомбинации; это подтверждается изменением концентрации парамагнитных
центров, которые возникают при температуре нагрева около 300 °С;
максимальное количество парамагнитных центров наблюдается при
600 °С, а при более высоких температурах количество их
постепенно уменьшается.
Структура исходного волокна полностью исчезает при 300°С.
Образование ароматических циклов происходит на начальной
стадии термообработки (выше 300°С), чему способствует лестничная
структура окисленного ПАН. При температурах нагрева около
Ю 0 го ЧЧ
Изменение длины
Подокна при окислении, %
Рис. 11.1. Влияние
натяжения при окислении
ПАН-волокна на модуль Юнга
углеродного и графитового
волокна:
I, 2 —температура карбонизации
соответственно 1000 и 1500 °С;
3— температура графитизации
2500 °С.
324
Глава //. Термостойкие и жаростойкие волокна
800 °С и более высокой температуре происходит агрегация
ароматических плоскостей с образованием графитоподобных структур:
Углеродные волокна относятся к неграфитирующимся формам
углерода. Физическая структура получаемых волокон очень
сложна и мало изучена. В них содержатся аморфные сетчатые
структуры различных гибридных форм углерода и графитоподобные
500 г
шо то то woo гооо ггоо гчоо
Температура, °С
Рис. 11.2. Влияние высокотемпературной обработки иа
механические свойства волокна.
структурные элементы. В процессе карбонизации сохраняется
фибриллярная структура волокна [41]. В этом заключается
существенное отличие углеродных волокон от других жаростойких волокон.
В результате деструкции полимеров прочность волокна вначале
снижается, достигая минимального значения при нагреве до 250—
300 °С, что соответствует максимальному уменьшению массы
полимера. При дальнейшем повышении температуры нагрева
прочность начинает возрастать. Аналогично изменяется модуль Юнга.
Улучшение механических свойств совпадает с началом процесса
ароматизации и образованием двухмерных структур углерода.
В процессе карбонизации происходит также резкое уменьшение
11.2. Получение жаростойких волокон 325
электрического сопротивления волокна и оно приобретает
полупроводниковые свойства. Графитированное волокно обладает
металлической проводимостью.
По литературным, данным, конечная температура
карбонизации находится в пределах 900—1500 °С, продолжительность —
0,5—4 ч. Отмечается, что при медленном нагреве получается
волокно более высокого качества [40—42]. Согласно ряду патентов,
проведение карбонизации на жестком каркасе позволяет получить
углеродное волокно с прочностью до 300 кгс/мм2 C-Ю3 МН/м2)
и модулем Юнга 25-103—30-103 кгс/мм2 B5-104—30-104 МН/м2).
При графитации содержание углерода в волокне повышается
до 99%. Наиболее существенную роль играют физические
процессы, сопровождающиеся изменением гибридных форм углерода
и увеличением размеров кристаллитов. Как видно из рис. 11.2,
прочность волокна при повышении температуры нагрева до 1500—1600 °С
возрастает, затем начинает уменьшаться, а начальный модуль
волокна непрерывно возрастает. Графитация проводится в среде
аргона в течение нескольких минут.
11.2.3. Получение углеродного волокна
из вискозной кордной нити
Технологический процесс получения углеродного волокна из
вискозной кордной нити состоит из трех стадий: подготовки сырья,
карбонизации и графитации. Важнейшими являются две
последние стадии.
Подготовка тканей (волокна) сводится к удалению
органических примесей (замаеливателя) обработкой органическими
растворителями 143] или поверхностно-активными веществами.
Большое значение придается сушке, так как наличие влаги приводит
к получению хрупкого волокна. Сушка проводится в жестких
условиях [44] (температура 100—120°С, продолжительность 15 ч).
В процессе карбонизации происходят сложные химические и
физические превращения целлюлозы, на скорость и характер
которых влияют различные факторы: природа катализатора, темпера-
турно-временные режимы, характер среды, свойства исходного
материала и т. д.
Термодеструкция целлюлозы протекает в узкой области
температур B50—350°С) и сопровождается интенсивным уменьшением
массы волокна и его усадкой. В результате термодеструкции
целлюлозы образуются летучие соединения (преимущественно НгО,
СО2, СО), а также левоглюкозан, смолы и углеродный остаток.
В работе [45] идентифицировано 37 соединений, среди которых
найдены альдегиды, кетоны, кислоты и др. К основным типам
реакций относится дегидратация'и деполимеризация целлюлозы,
более глубокий распад промежуточных продуктов и образование
углеродного полимера. Большое влияние оказывают катализаторы,
326 Глава И. Термостойкие и жаростойкие волокна
в качестве которых применяются антипирены, кислоты и основания
Льюиса, соли металлов переменной валентности и др. [46].
Действие катализаторов (антипиренов) * проявляется в следующем:
увеличивается выход углерода и уменьшается выход смол;
разложение целлюлозы начинается при более низких температурах;
суммарная скорость деструкции повышается; в присутствии
катализаторов на первой стадии термораспада более интенсивно
протекает дегидратация целлюлозы и таким образом
подавляются реакции, приводящие к
образованию смол. Эффективное
действие катализаторов
проявляется при их небольшом
содержать^ yi нии (I—2% от массы целлюлозы).
« ' уь* При повышении температуры
* ' г -^ • элементарный состав пека
упрощается, но его строение из-за
многообразия переходных форм
ї:0'7г углерода усложняется.
^ ^ ^—^ ^—^ Аморфизация начинается при
Степень бытягибания,% 240—270 °С, при 250—350 °С ДО-
стигается полная аморфизация
целлюлозы, а фаза двухмерной
Юнга волокна; температура графити- упорядоченности углерода появ-
зации 2780 —2870 °С, продолжитель- ЛЯЄТСЯ при 300—400 °С [47].
ность 0,20 — 0,27 с. В процессе карбонизации
изменяются свойства волокна.
Качественно наблюдается такая же картина, как и при
получении углеродного волокна из ПАН-волокна. Прочность и начальный
модуль вначале уменьшаются (до 300°С), а затем увеличиваются;
электропроводность и теплопроводность резко возрастают,
коэффициент линейного расширения уменьшается, теплоемкость мало
изменяется.
Карбонизация проводится в инертной среде, которой могут
служить азот, продукты распада целлюлозы, природный газ и т. п. [48].
Карбонизация проводится очень медленно, особенно на первых
стадиях процесса, характеризующихся высокой скоростью
реакций. При нагреве до 300—400 °С подъем температуры
рекомендуется проводить со скоростью 5—10°С/ч, а при дальнейшем
нагреве— 60—100°С/ч [49]. Общая продолжительность карбонизации
составляет 200—250 ч [50].
Графитация осуществляется также в инертной среде при очень
быстром подъеме температуры. Максимальные механические
свойства достигаются при продолжительности графитации 0,1 с [51].
При графитации совершенствуется структура углерода [52] и
соответственно увеличивается прочность и начальный модуль волокна.
Эти показатели непрерывно возрастают с увеличением
температуры графитации [51].
11.2. Получение жаростойких волокон 327
Вытягивание также оказывает существенное влияние на
свойства углеродного волокна. При применении в качестве исходного
сырья вискозной кордной нити вытягивание следует осуществлять
на стадии карбонизации и графитации. В результате вытягивания
повышается прочность и начальный модуль углеродного и графи-
тированного волокон (рис. 11.3). Благодаря вытягиванию удалось
получить углеродное волокно с исключительно высокими
механическими свойствами: прочность 200—300 кгс/мм2 B-Ю3—
3-Ю3 МН/м2), модуль Юнга 20-103—50-103 кгс/мм2 B0-104—
50-104 МН/м2).
11.2.4. Свойства и области применения
углеродных волокон
Углеродные волокна обладают уникальными механическими
свойствами. В зависимости от назначения изготовляются карбони-
зованные или графитированные материалы, каждый из которых
является технически ценным продуктом. Углеродные волокнистые
материалы применяются как таковые или, чаще, в виде
армированных пластиков, изготовленных на их основе.
Прочность углеродных волокнистых материалов изменяется в
пределах от 30 до 300 кгс/мм2 C00—3000 МН/м2), а модуль
Юнга —от 5000 до 40 000 кгс/мм2 E-104—40-104). Особого
внимания заслуживают высокопрочные, высокомодульные волокна.
Вследствие низкой плотности углерода A,6—2 г/см3) эти волокна
по удельным значениям прочности и модуля (отношение этих
величин к плотности) превосходят все жаростойкие волокна,
металлы и сплавы.
Из-за сетчатой структуры и большой концентрации я-сопряже-
ний углеродные волокна обладают исключительно высокой термо-
и хемостойкостью. При повышении температуры механические
свойства не уменьшаются, а, наоборот, возрастают:
Температура, °С 20 540 1100 1650
Прочность ширины полоски,
кгс/см 0,9-2,9 1,1—3,2 1,4-4,3 1,8-6
Среди всех материалов только углеродные волокна обладают
такими специфическими свойствами.
Углеродные волокна стойки к органическим растворителям,
щелочам и кислотам, но недостаточно стойки к действию
окислителей, в том числе к кислороду воздуха. На воздухе предельная
температура эксплуатации углеродных волокон составляет 350—
400 °С. Однако стойкость волокон к окислителям можно
значительно повысить нанесением на их поверхность тонкого защитного
слоя карбидов (SiC) или нитридов (BN). Благодаря высокой
термостойкости углеродные материалы широко применяются в
качестве теплозащитных и теплоизоляционных средств,
328
Глава 11. Термостойкие и жаростойкие волокна
Изменяя параметры процесса, можно получать волокна с
различными электрофизическими свойствами (удельное
сопротивление 0,002—106 Ом-см), благодаря чему они применяются для
изготовления разнообразных по назначению
электронагревательных элементов (для костюмов, отопления помещений, термопар
и др.).
Углеродные волокна можно получать с очень высокой активной
поверхностью. Из волокна саран получено углеродное волокно с
активной поверхностью 700—1000 м2 [53]. Активация водяным
паром позволяет значительно повысить активную поверхность
углеродного волокна, полученного на основе вискозной кордной нити.
Такие волокна являются прекрасными сорбентами. При нанесении
на волокно катализаторов получаются каталитические системы с
очень развитой поверхностью.
Высокопрочные высокомодульные волокна используются для
изготовления конструкционных пластиков. Связующим служат
эпоксидные или полиэфирные смолы, силовым каркасом углеродное
волокно. Для сравнения приводим показатели эпоксиуглепластиков
и других материалов:
Плотность,
г/см3
Сплав алюминия
HV30WP ... 2,7
Сплав титана
ДТД5173 ... 4,5
Эпоксистеклопла-
стик (S —
стекло) 2,12
Эпоксиуглепла-
стик
высокопрочное волокно . 1,6
высокомодульное
волокно . . 1,7
Модуль Юнга удельная Удельный
кгс/"мм2(МН/м2) Я-|° • прочность, модуль
кгс/мм (яім/м ) кгс/мм2(МН/м2) аїр Я-10-3/р
Прочность,
34,5 C45)
93,1(931)
192,5A925)
131,0A310)
56,0 E60)
6,9 F9)
11A10)
6,9 F9)
17,2A72)
20,4 B04)
12,8
26,0
91,0
82,2
33,0
2,55
2,45
3,2
10,8
12,0
Как видно из этих данных, по удельной прочности эпоксиугле-
пластики уступают только эпоксистеклопластикам, а по удельному
модулю превосходят все другие конструкционные материалы.
Конструкционные пластики на основе углеродного волокна
используются в самолетостроении и вертолетостроении, а в
перспективе они найдут применение в судостроении, химическом и
текстильном машиностроении. Исключительно большое значение
имеют углепластики, полученные из обычного волокна (тканей);
они применяются в качестве теплозащитных материалов.
. По термостойкости органические связующие уступают
углеродному волокну. Поэтому в последние годы проводятся интенсивные
Литература - - - . 329
исследования по получению композиций, в которых в качестве
связующего применяются керамика, металлы или сплавы металлов.
Для углеродных волокон открывается еще одна перспективная
область применения. Введение углеродных волокон в полимеры
значительно (не менее чем в 10 раз) повышает их
износоустойчивость, что может значительно увеличить срок службы изделий из
резины и пластмасс.
К недостаткам углеродных волокон относится высокая
стоимость, что отчасти объясняется небольшими объемами их
производства, а также низкое удлинение (менее 1%), ограничивающее его
применение. По-видимому, при увеличении масштабов производства
стоимость углеродных волокон значительно снизится.
ЛИТЕРАТУРА
1. Strahl G. К., «Chemiefasern», 1963, N 6, S. 426—429; Тверская Л.,
Хим. пром. за рубежом, 1966, № 10, с. 44—48.
2. К у д р я в ц е в Г. И., В о л о х и н а А. В., ЖВХО им. Д. И. Менделеева,
1966, т. 11, с. 665—671.
3. Кудрявцев Г. И., Щетиинн А. М., Хим. волокна, 1968, № 6, с. 2—5.
4. Preston I., J. Polymer Sei., 1966, v. 7, p. 601—604.
5. Соколов Л. Б., Высокомол. соед., 1965, т. 7, с. 601—604; Соколов Л. Б.
Поликоиденсационный метод синтеза полимеров. М., «Химия», 1966. 322 с.
6. Den е-Her t В., Moore В., W г е g h t W., J. Polymer Sei., 1964, v. 132.
p. 363—367.
7. Престон И., Смит Р., Стемон Г. Новое в производстве химических
волокон. Пер. с англ. Под ред. 3. А. Роговина и С. П. Папкова. М., «Мир»,
1968. 227 с.
8. Le R е g G., Me С и п е К-, Text. Res. J., 1962, v. 32, p. 762—766.
9. Хим. волокна, 1968, № 6, с. 72, 73.
10. Rognei G., Noel I., Compt. Rend., 1967, № 1, p. 63—66 № 2, p. 178—
181.
11. Коршак В. В., Виноградова С. В. Термостойкие полимеры. М.,
«Наука», 1964. 411 с.
12. Коршак В. В., Виноградова С. В. Полиарилаты. М., Изд-во АН
СССР, 1964. 69 с.
13. Morgan P., Skott H., J. Polymer Sei., 1969, A, v. 21, p. 2437—2439.
14. Якубович В. С, Мясников Г. В., Браз Г. И. и др., ДАН СССР,
1964, т. 159, с. 630—634; К u b о t а Т., J. Polymer Sei., 1964, v. 2, p. 655—658.
15. Кард аш И. Е., Ардашников А. Я., Якубович В. С. и др.,
Высокомол. соед., 1967, А, т. 9, с. 1914—1918.
16. Spain R., Pi с klese mer L., Text. Res. J., 1966, v. 36, p. 619—622;
Fraser A., Fitzgereld W., J. Polymer Sei., 1967, A, v. 19, p. 95—98.
17. Fraser A., S era s о ki X., J. Polymer Sei., 1966, v. 44, p. 1649—1652.
18. Аидричеико Ю. Д., Конкин" А. А., Хим. волокна, 1968, № 5, с. 25—28.
19. Ан д р ичен ко Ю. Д. Кандидатская диссертация. МТИ, 1970.
20. F г a s е г A., F і t z e g e r є 1 d W., J. Polymer Sei., 1967, С, v. 19, p. 95—98
21. Fraser A., Reed T., J. Polymer Sei., 1967, С, v. 19, p. 89—92.
22. I г vin R., Sweeny W., J. Polymer Sei., 1867, С, v. 19, p. 41—44.
23. Bower G., Frost L., J. Polymer Sei., 1969, A, v. 1, p. 3135—3138.
24. Vol lmert В., «Kunststoffe», 1966, Bd. 56, S. 680—682.
25. Рудаков А. П., Бессонов M. И., К о т т о и M. M. и др., Хим волокна,
1966, JN? 5, с. 20-22,. г •¦¦.¦»
330 Глава 11. Термостойкие и жаростойкие волокна
26. П р о х о р о в О. В., К а р ж а в и н Л. Н., Флоринскии Ф. С, Хим.
волокна, 1970, № 5, с. 73—76.
27. Оприц 3. Г., Кудрявцев Г. И., К а ржа вин Л. Н. и др., Хим.
волокна, 1970, № 3, с. 61—64.
28. Кудрявцев Г. И., Балаклейцева Л. Ф., Хим. волокна, 1970, № 5,
с. 42—45.
29. Хим. волокна, 1969, № 5, с. 66.
30. Токарев А. В., Чикурина Л. В., Кудрявцев Г. И. и др., Хим.
волокна, 1970, № 5, с. 20—23.
31. Gloon W., Appl. Polymer Sei., 1967, N 6, p. 151—154; «Polymer», 1966,
v. 7. p. 819; Da van s P., Marvel G., J. Polymer Sei., 1965, A, v. 3,
p. 3549—3553; Boll V., Pezdiztz W., J. Polymer Sei., 1965, В, v. З,
p. 974—978.
32. Каролл-Порчинский ІД. Материалы будущего. Пер. с англ. Под ред.
Н. В. Михайлова. М., «Химия», 1966. 238 с. Современные композиционные
материалы. Пер. с англ. Под ред. Л. Браутмана и Р. Крока. М., «Мир», 1970.
672 с.
33. Rubb. Plast. Age, 1969, v. 50, N 5, p. 328—330; Oma ni S., «Carbon», 1965,
v. 3, p. 31—34; О m a n і S. e. a., «Carbon», 1966, v. 4, p. 425—429;
Orna ni S. e. a., Bull. Chem. Soc. Japan, 1970, v. 43, N 10, p. 3291—3294.
34. Яп. пат. 1111299; Nikawa Sumio, Chem. Econ. a. Eng. Rev., 1970, v. 2,
N 3, p. 43, 44.
35. Котииа В. Е., Конкнн А. А., Косова P. M. Авт. свид. 134869 A960);
Бюлл. изобр. и товари, знаков, 1966, № 8.
36. G r a s s і g N., Novel N., J. Polymer Sei., I960, v. 48, p. 79—82; Gras-
si g N.. Neill T., J. Polymer Sei., 1959, v. 39, p. 211—214; Grass і g N,
Nay N., Neile T., J. Polymer Sei., 1958, v. 31, p. 205—208; Grassig N.,
N a y N., J. Polymer Sei., 1962, v. 56, p. 189—192.
37. McGartney I., J. Res. Nat. Bur. Standards, 1953, v. 525, p. 123—127; Over-
berger С G., Juki H., Trakawa W., J. Polymer Sei., 1960, v. 25,
p. 127—131.
38. Гейдрих M. A., Давыдов Б. А., Изв. АН СССР, Сер. хим., 1955, № 4,
с. 636—639.
39. Франц. пат. 1541287; 1556024; 1501286; 1438803.
40. Watt W., Jon son V., «Polymer Preprints», 1968, v. 9, p. 1245, 1246.
41. К а р г и и В. А., Литвинов И. А., Высокомол. соед., 1965, т. 7, с. 226—228.
42. Франц. пат. 14308803; пат. США 3255696.
43. Пат. США 3179605; 3290489.
44. Франц. пат. 1406529; пат. США 3116975.
45. Schwenker R., Beck L., J. Polymer Sei., 1963, N 2, p. 331—334.
46. Voheer О., S perk E., Ber. Deutsch. Keram. Gesellschaft (Bonn), 1966,
Bd. B43, N 3, S. 199—202; франц. пат. 1462564; 1524502; пат. США 3235329;
3305315; англ. пат. 179324; 1033009; пат. ФРГ 1242551; Schmidt D.,
Jokes W., Chem. Eng. Progr., 1962, v. 58, N 10, p. 42.
47. Ф и а л ко в A. C. h др., «Химия тбердого топлива», 1968, № 3, с. 116—119.
48. Котина В. Е., Конкин А. А., Горбачева В. С. и др., Хим волокна,
1969, № 6, с. 1—4.
49. Англ. пат. 894458; пат. США 3116975.
50. Франц. пат. 1406529; Bull, de l'Institut textile de France 1969, N 145,
p. 863—865.
51. Gibson N.. Langlois G., «Polymer Preprints», 1968, v. 9, p. 1376—1379.
52. Racon R., Schalamon W., «Polymer Preprints», 1968, v. 9, p. 286.
53. Boucher A., Cooper R., Erevatt D., «Carbon», 1970, v. 8, p. 597—601.
Глава 12
СВЕРХПРОЧНЫЕ И ВЫСОКОМОДУЛЬНЫЕ
ВОЛОКНА*
При создании самолетов со сверхзвуковыми скоростями и
космических летательных аппаратов возникла потребность в
конструкционных материалах особо высокой прочности. Обычно
используемые в авиационной промышленности материалы (в частности,
сталь) хотя и имеют относительно высокую прочность (до
80 кгс/мм2), но обладают очень большой плотностью (до 7,8 гс/с:«3).
Это приводит к утяжелению конструкций, поскольку летательные
аппараты испытывают значительные перегрузки из-за высоких
ускорений при взлете и торможении. Обычные полимерные
материалы в виде литых монолитных конструкций, обладающие
невысокой плотностью (не превышающей 1,5 г/см3), характеризуются
незначительной прочностью, достигающей максимум 10—
15 кгс/мм2 A50МН/М2).
Именно поэтому было обращено особое внимание на получение
новых типов волокон, которые благодаря резко выраженной
анизотропии механических свойств способны выдерживать большие
нагрузки, прилагаемые в направлении оси волокон. Высокопрочные
кордные искусственные и синтетические волокна обладают
прочностью до 50—90 кгс/мм2. При сравнительно невысокой плотности
(максимальная плотность целлюлозных волокон 1,50—1,52 г/см3)
удельная прочность таких волокон, т. е. прочность, отнесенная к
массе, значительно превосходит прочность стали лучших сортов.
Однако и эти показатели не удовлетворяют все возрастающим
требованиям ряда отраслей промышленности. Кроме того, серьезным
недостатком указанных типов химических волокон являются
относительно низкие модули упругости и высокая деформируемость.
Этими обстоятельствами объясняются предпринимаемые в
последние годы в СССР и за рубежом попытки получения новых
типов химических волокон, обладающих более высокими прочностью
и начальным модулем. Такие волокна обладают еще одним
существенным преимуществом — при их использовании в качестве
армирующих (упрочняющих) материалов достигается также большая
устойчивость к распространению случайных трещин и дефектов в
конструкции. В неармированных пластиках возникшая трещина
быстро распространяется на все сечение детали, а в материалах,
* Эта глава написана С. П. Папковым и Г. И. Кудрявцевым.
332 Глава 12. Сверхпрочные и высокопрочные волокна
армированных волокнами, трещины обрываются на границе
пластик — волокно.
Теоретические расчеты показывают (см. т. I, разд. 5.1), что в
идеальном случае прочность химических волокон могла бы
достигнуть 500—600 кгс/мм2 E000—6000 Мн/м2).
12.1. ХАРАКТЕРИСТИКА НОВЫХ ЖЕСТКОЦЕПНЫХ
ВОЛОКНООБРАЗУЮЩИХ ПОЛИМЕРОВ
Поиски новых полимеров для получения высокопрочных
волокон привели к неожиданному открытию класса полимеров, для
которых равновесному состоянию отвечает не беспорядочное, а
упорядоченное взаимное расположение макромолекул. Эти полимеры
построены из очень жестких макромолекул. Если макромолекула
полимера очень жесткая (т. е. если свободное вращение
элементарных звеньев резко ограничено), то беспорядочное расположение
цепей оказывается энергетически невыгодным и равновесному
состоянию отвечает уже более или менее строгий порядок во
взаимном расположении макромолекул.
К числу таких жесткоцепных полимеров относятся полимеры,
цепи которых состоят из бензольных колец, а между звеньями нет
промежуточных атомов или групп, обеспечивающих ограниченно
свободное вращение вокруг ординарных химических связей. В
результате взаимодействия между бензольными кольцами соседних
звеньев, находящихся близко друг к другу, создается большой
энергетический барьер для свободного вращения и макромолекула
в растворе приобретает уже не форму статистического клубка, как
это имеет место для гибкоцепных полимеров, а форму жесткого
стержня.
Примерами подобных жесткоцепных полимеров могут служить
поли-га-бензамид
• •—СО—/ \—NH—••
элементарным звеном в котором является остаток га-ам,инобензой-
ной кислоты, а также поли-га-фенилентерефталамид
СО—(f\—CONH
получаемый сополиконденсацией и-фенилендиамина и терефтале-
вой кислоты.
Близкое расположение соседних бензольных колец в этих
полимерах устраняет гибкость цепи. Синтезированы также и другие
полимеры, сходные по строению с указанными, которые также
обладают жесткими цепями. В качестве примера можно указать на
12.1. Характеристика жесткоцепных волокнообразующих полимеров 333
сополимер 4,4'-диаминобензанилида, аминобензгидразида и тере-
фталевой кислоты и другие аналогичные по строению
ароматические полиамидогидразиды.
Упорядочение жестких стержнеобразных макромолекул
заключается в том, что они образуют группы взаимно ориентированных
параллельно расположенных цепей, которые размещаются подобно
спичкам в коробке. Однако при таком упорядочении жесткие
макромолекулы отличаются от истинных кристаллов тем, что в этом
случае нет строгого трехмерного порядка. Но и двухмерное
упорядочение макромолекул или их агрегатов придает этим полимерам
особые свойства, промежуточные между свойствами истинных
кристаллов и жидкостей, для которых типично отсутствие дальнего
порядка. Такие системы с частично упорядоченным
расположением молекул известны давно и для некоторых низкомолекулярных
веществ с асимметричным строением молекул. Они обычно
называются, «жидкими кристаллами», что формально достаточно точно
характеризует их структуру. Жидкие кристаллы обладают
двойным лучепреломлением.
Оказалось, что и жесткоцепные полимеры обладают такими же
свойствами, т. е. способны переходить в жидкокристаллическое
состояние. Если приготовить серию растворов жесткоцепного
полимера с постепенно повышающейся концентрацией полимера, то
до определенной концентрации стержнеобразное строение
макромолекул не мешает свободному размещению их в растворе. Такой
раствор ведет себя как обычный раствор полимера. Но при
критической концентрации, когда макромолекулы уже не могут
свободно размещаться в заданном объеме, наступает взаимное
упорядочение полимерных цепей и образование взаимно ориентированных
агрегатов макромолекул. При этом резко изменяются свойства
раствора. Он становится мутным из-за возникновения поверхностей
раздела между агрегатами макромолекул и растворителем и
рассеяния света на этих поверхностях; появляется двойное
лучепреломление, а в некоторых случаях резко снижается вязкость, что
объясняется упорядочением в расположении макромолекул
растворенного вещества.
Изменение вязкости раствора поли-я-бензамида в диметилацет-
амиде, содержащего хлорид лития, при изменении концентрации
полимера в растворе показано на рис. 12.1 [1]. Как видно из этого
рисунка, при достижении критической концентрации, которая в
данном случае равна 5%, вязкость раствора резко снижается. При
более высокой концентрации полимера в растворе проявляются и
отмеченные новые оптические свойства, которые не наблюдаются
в растворах с концентрацией полимера ниже критической.
Из растворов, обладающих свойствами жидких кристаллов,
получают волокна, резко отличающиеся по механическим
показателям от волокон, сформованных из растворов обычных
волокнообразующих полимеров. Взаимная упорядоченность макро-
334
Глава 12. Сверхпрочные и высокопрочные волокна
молекул в растворах сохраняется и в волокне. Поскольку
равновесному состоянию отвечает двухмерный порядок во взаимном
расположении макромолекул, такое волокно достаточно
подвергнуть небольшой вытяжке, чтобы расположить упорядоченные
агрегаты макромолекул вдоль оси волокна и таким образом устойчиво
ориентировать полимер. Для этого достаточна вытяжка, не
превышающая 100%, в то время как даже при 8—10-кратном
вытягивании волокна из гибкоцепных полимеров- такая высокая
ориентация не достигается. Соответственно и прочность волокон из таких
жесткоцепных полимеров оказывается
S0Br очень высокой и достигает 250 кгс/мм2
B500 МН/м2).
Сложность получения подобных
волокон заключается в том, что
указанные жесткоцепные полимеры плохо
растворяются в обычных растворителях. Они
растворимы только в концентрированной
кислоте и апротонных растворителях ти-
j па диметилацетамида с добавкой хло-
5 700
% 600
I 500
^400
|Г зоо
| 200
І то
Рис. 12.1. Изменение
вязкости в зависимости от
концентрации поли-га-бензамида
в растворе.
0 і г з ч 5 .в 7 8 ридов лития или кальция. Волокно фор-
Кощентрация,/0 ЩЮТ из 10—15%-ных растворов
полимера в осадительную ванну,
содержащую разбавленные водой растворители
или другие водные растворы.
Сформованное волокно вытягивают на 100—
150%. Последующая термическая
обработка при повышенных температурах приводит к замене
несовершенной кристаллической структуры полимера более совершенной,
в результате чего дополнительно значительно повышается
начальный модуль волокна и снижается удлинение.
Прочность новых волокон, известных под названиями «терлон»
[2] (СССР), «файбр Би» [З], ПРД-49 [4] (фирма «Дюпон», США*),
«волокно Х-500» [5] (фирма «МонЬанто», США), достигает 150—
250 кгс/мм2 A500—2500 МН/м2), удлинение 1,5—4% и модуль
упругости (растяжения) 500—13000 кгс/мм2 E-Ю3—
130-Ю3 МН/м2).
Появилось также сообщение [6] о разработке в СССР нового
сверхпрочного волокна вниивлон Н.
Основное назначение сверхпрочных волокон — армирование
пластических масс (фенолоформальдегидных и эпоксидных смол)
и получение сверхпрочного шинного корда.
Сравнительные данные о механических свойствах волокна
файбр Би и других волокон, используемых для изготовления
шинного корда, приводятся ниже [7]:
* Фирма Дюпон в настоящее время дала этим волокнам новые названия —
«кевлар» и «кевлар 49»; см. Chem, Age, 1973, № 2919, p. 11.
12.2. Особенности процесса формования волокна
335
Волокно Стальная Стрклмюллкип
файбр Би проволока стекловолокно
Прочность, гс/текс (мН/текс) . . . 18A80) 3,4C4) 9,8(98)
Удлинение, % 4 1,7 4,8
Модуль, гс/текс (мН/текс) 3200C,2- 103) 1800 A8-Ю3) 2400B4-103)
Прочность в петле, гс/текс (мН/текс) 72G20) 145A450) 33C30)
Вискозный Полиамидный Полиэфирный
корд корд корд
Прочность, гс/текс (мН/текс) ... 3,9 C9) 8,7 (87) 7,2 G2)
Удлинение, % 15 21 15
Модуль, гс/текс (мН/текс) 450D,5.103) 300 C-Ю3) 600 F-Ю3)
Прочность в петле, гс/текс (мН/текс) 16A60) 40D00) 29B90)
Масштабы производства новых волокон пока невелики, но, судя
по положительным результатам их применения, можно ожидать
в ближайшие годы увеличения объема производства до десятков
тысяч тонн в год.
Получение этих волокон следует рассматривать как первый,
но очень важный этап в производстве сверхпрочных волокон.
В процессе научных и технологических поисков будут, вероятно,
достигнуты новые значительные успехи. В частности, кроме очень
высокой ориентации макромолекул полимера в волокне
существенное значение для повышения прочности волокон имеет
ликвидация макро- и микродефектов структуры. Устранение дефектности
волокон является сложной, но технически разрешимой задачей, и
поэтому можно ожидать в дальнейшем приближения прочности
новых волокон к теоретической [8].
12.2. ОСОБЕННОСТИ ПРОЦЕССА ФОРМОВАНИЯ
ВОЛОКНА
Описанные в предыдущем разделе необычные свойства жестко-
цепных полимеров, и особенно их склонность к самоупорядочению,
определяют особый характер процессов получения сверхпрочных
нитей [9].
Независимо от природы полимера эти волокна формуют из
растворов мокрым или сухо-мокрым методом.
Основные технологические операции при использовании обоих
методов формования одни и те же: получение, фильтрация и обез-
воздушивание прядильного раствора; формование, промывка,
сушка и термовытяжка волокна; крутка и перемотка на выходную
паковку при получении нити.
При получении сверхпрочных волокон отсутствует обычная для
технологического процесса получения синтетических волокон
стадия ориентационного вытягивания. Это и отличает условия
получения волокон из жесткоцепных полимеров, так как исключается
необходимость дополнительной ориентации макромолекул.
Мокрый метод формования. Приготовление прядильного
раствора сводится к растворению порошкообразного, тщательно
336 Глава 12. Сверхпрочные и высокопрочные волокна
высушенного полимера или к приданию требуемых свойств
сиропам, которые получают при поликонденсации мономеров в
растворе. Так как жесткоцепные полимеры плохо растворяются даже
в таких растворителях, как серная, метилсерная, хлорсульфоновая
и другие минеральные кислоты, а также в дим етил ацетамиде и ме-
тилпирролидоне, важнейшим условием получения гомогенного
раствора является минимальное содержание (не более 0,05%) воды
в системе полимер — растворитель. Для этого полимер тщательно
подсушивают в вакуум-сушилках. Органические растворители
предварительно обезвоживаются до аналитически неопределяемого
содержания влаги.
Концентрация прядильного раствора должна соответствовать
критической точке перехода системы в анизотропное состояние,
так как при этом достигается минимальная вязкость
системы [10].
Если полимер в процессе его синтеза не выпадает из раствора,
то образующийся концентрированный раствор может быть
использован для формования волокна. Для этого концентрацию полимера
в растворе доводят до заданной величины, затем раствор
усредняют смешением с растворами других партий полимера, в ряде
случаев удаляют из него хлористый водород (когда полимер
получают поликонденсацией диамина и дихлорангидрида дикарбоновой
кислоты).
Хлористый водород связывают при нейтрализации сиропа
гидроокисью лития, кальция или аммония. При отфильтровывании
выпадающего осадка — хлоридов кальция или аммония —
возникают затруднения из-за высокой вязкости прядильных растворов.
Поэтому для нейтрализации предпочитают применять гидроокись
лития, соли которого хорошо растворяются в органических
растворителях.
Вследствие высокой вязкости прядильных растворов, доходящей
в некоторых случаях до 800—1000 П, фильтровать их приходится
под давлением, а обезвоздушивать — новым методом [11]. Как
известно, процесс обезвоздушивания включает две стадии:
выделение растворенного воздуха (т. е. рост объема дисперсной
фазы) ;
выделение пузырьков воздуха из жидкости и разрушение пены.
Наиболее эффективной является дегазация системы под
вакуумом в кипящем тонком слое. Однако этот метод непригоден для
обезвоздушивания растворов жесткоцепных полимеров из-за
трудности удаления всех пузырьков из очень вязкой жидкости. Кроме
того, иногда длительная дегазация под вакуумом приводит к
образованию на поверхности раствора полимерной пленки. Поэтому
метод дегазации растворов жесткоцепных полимеров основан на
ускорении первой стадии процесса созданием условий для
максимального роста объема дисперсной фазы и коалесценции газовых
пузырьков, выделяющихся из жидкости. Для этого раствор попе-
12.2. Особенности процесса формования волокна 337
ременно выдерживают под давлением и вакуумом, а в некоторых
случаях предварительно насыщают инертным газом.
Второй период дегазации сводится к удалению только тех
пузырьков, диаметр которых превышает критический; после их
удаления прядильный раствор становится ненасыщенным по
отношению к пузырькам воздуха.
Мокрое формование волокон из жесткоцепных полимеров,
проводится в условиях, максимально способствующих самоориентации
макромолекул. Для этого в качестве осадительной ванны
применяют мягкие коагулирующие реагенты, а формование проводят
при отрицательных или нулевых фильерных вытяжках.
Выше указывалось, что для достижения требуемого
ориентированного состояния формуемых волокон, достаточно подвергнуть
их небольшой вытяжке для того, чтобы расположить
упорядоченные агрегаты вдоль оси волокна. Это достигается непосредственно
на прядильной машине при пластификационной вытяжке свеже-
скоагулированного полимерного геля. Волокно, снимаемое с
прядильной машины, имеет достаточно высокую прочность [до
100 гс/текс A00 омН/текс)].
Высокие модуль и прочность при удлинении волокна более
5—6% обусловливают пригодность свежесформованных нитей для
производства кордной ткани и резиновых технических изделий.
После тщательной промывки и сушки волокон, что обычно
проводится на прядильной машине, их подвергают термообработке при
температурах от 500 до 600°С.
Благодаря термообработке в волокне образуются водородные
связи, происходит кристаллизация и фиксация структуры. В
результате этих процессов резко повышаются прочность и начальный
модуль волокна, а удлинение становится менее 5%. При
термообработке можно волокно дополнительно вытянуть. Однако это не
приводит к существенным изменениям свойств волокна, и в ряде
случаев термическая вытяжка даже уменьшает прочность
получаемых нитей (табл. 12.1) [10].
Сухо-мокрый метод формования принципиально отличается от
описанного выше мокрого метода [12]. Предварительным условием
использования этого метода является применение
высококонцентрированных A8—25%-ных) прядильных растворов. При такой
концентрации полимера растворы при нормальной температуре
не текут. Для того чтобы перевести такой раствор в вязкотекучее
состояние, его нагревают до максимально допустимой
температуры. Например, растворы жесткоцепных полимеров в серной
кислоте подогревают до 80—85°С.
При более высоких температурах происходит быстрая
деструкция полимера. Формование нитей производится с использованием
прядильных головок, аналогичных применяемым при формовании
нитей из расплава. Выходящие из фильеры жидкие струйки
нагретого вязкого раствора проходят короткое воздушное пространство
338
Глава 12. Сверхпрочные и высокопрочные волокна
Таблица 12.1. Свойства волокон нз сополимеров л, л'-диамннобензанилиднна
с терефталонлдихлорндом (I) и п,п'-бис-(хлоркарбоннл)-дифеннлом (II)*
Состав
сополимера
1:11
1:0
0,75 0,25
0,5:0,5
0,25:0,75
0,1**
Условия
термообработки
кратность
вытяжки
1.16
1,08—1,00
1,07—1,01
1,07
1,08
темпера-
597
600—580
600—550
600
600
Свойства
кристалличность
Низкая
Следы
*
Низкая
необработанного
волокна
прочность,
гс/текс
(мН/текс)
57,6
E76)
44,1
D4!)
79,2
G92)
86,4
(864)
50,4
E04)
удлинение,
!!,0
12,0
8,0
9,6
9,0
Свойства волокна после
термообработки
кристалличность
Высокая
Средняя
Высокая
прочность,
гс/текс
(мН/текс)
90,9
(909)
127,8
A278)
118,8
A188)
163,8
A638)
118,8
A188)
удлинение.
1,2
2.4
2,1
2.5
2.4
* Химическая структура сополимеров:
NH
NH С 7 NHCO С V NHCC
'• Концентрация полимера в растворе 11 вес. %.
СО (II)
и поступают в водную ванну. Расстояние между плоскостью
фильеры и зеркалом жидкой ванны составляет от 1 до 2 см.
В качестве осадительной ванны обычно применяют воду,
содержащую небольшое количество растворителя или органические
соединения слабого коагулирующего действия (спирты, этиленгли-
коль и др.).
Получение волокна с высокими прочностными свойствами
зависит от следующих основных факторов:
температуры осадительной ванны, которая должна быть как
можно более низкой;
фильерной вытяжки, величина которой обеспечивает
устойчивое формование;
полноты отмывки нити от растворителя.
Особая роль перечисленных факторов объясняется,
по-видимому, необходимостью создать наиболее благоприятные условия
для ориентации элементов структуры и одновременно для
наиболее полного устранения релаксационных процессов, приводящих
к понижению степени ориентации. С этой точки зрения
применение пластификационной вытяжки нецелесообразно. Свежесформо-
Литература 339
ванное волокно после сушки приобретает механические свойства,
характерные для данного типа волокна. Последующая
термообработка не увеличивает прочности и проводится только для
повышения начального модуля волокна.
ЛИТЕРАТУРА
1. Калмыкова В. Д., Кудрявцева Г. И., Папко в С. П., Воло-
хина А. В., Иов лева М. М., Милькова Л. П., Куличихин В. Г.,
Бандур ян С. И., Высокомол. соед., 1971, т. 13Б, с. 707—709.
2. Хим. волокна, 1972, № 6, с. 20.
3. Rubb. World, 1972, v. 116, N 4, p. 47, 48.
4. «The Engineer», 1972, p. 60—63.
5. Chem. Eng. News, 1972, v. 50, № 16, p. 33, 34.
6. Хим. волокна, 1971, № 1, с. 76.
7. Rubb. World, 1972, v. 166, N 1, p. 56—58.
8. П а п к о в С. П., Хим. волокна, 1973, № 1, с. 3—5.
9. Кудрявцев Г. И., ЖВХО им. Д. И. Менделеева, 1972, № 6, с. 625—629.
10. Франц. пат. 2010353 A970).
11. Матвеев В. С, Журавлев Л. В., Кудрявцев Г. И., Хим. волокна,
1971, № 4, с. 6—8; Матвеев В. С, Журавлев Л. В.,
Кудрявцев Г. И., Пере пел кин К. Е., Хим. волокна, 1971, № 6, с. 16—18.
12. Пат. США 138029 A971); 172515 A971).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адипиновая кислота 47, 48
Азотная кислота 191, 192, 197
Акрилан 179
Акрилонитрил 180 ел.
Активаторы полимеризации 34, 35, 43
Амииокапроковая кислота, 23, 29
Аминоундекановая кислота 18, 55, 56
Аминофенолы 93
Аминоэиаитоваи кислота, синтез 53,
54, 56
АМТ-300 39
Анид 14, 19, 47, 48, 98, 99
технологическая схема получения
21
Анионная полимеризация 31, 32
Антиоксиданти 277 ел.
Аппарат для непрерывной
полимеризации капролактаме 45
Ацеталироваиие 258 ел.
Ацетальдегид 181
Ацетилен 181
Ацетохлорин 243
Беизол 24
Бикомпонентные иолокна 166
Винал 248
Винилацетат 183, 244
Вииилиденхлорид 222, 223, 244 ел.
Вииилхлорид 221, 222, 244 ел.
Вииитрон 243, 244
Винол 16, 263
Винолон 248
Вииьон 11
В ирей 173, 174
Вниивлон 334
Волокна
из фторсодержащих полимеров
297 ел.
полиакрилонитрильные 179 ел.
полиамидные 18 ел.
поливинилепиртовые 248 ел.
поливинилхлоридные 229 ел.
полиолефиновые 270 ел.
полиуретаиовые 171 ел.
полиформальдегидные 176 ел.
полиэфирные 117 ел.
сверхпрочные и высокомодульные
331 ел.
Волокна
термостойкие и жаростойкие
303 ел.
Волькрилон 179
Высокопрочные полиамидные
волокна 99
Высокотемпературные органические
теплоносители (ВОТ) 39, 40,
64
Высокоэластичные волокна 11, 171 ел.
Вытягивание 205, 206, 255 ел.
Вязкость расплава 46, 47
Гексаметилен 55
Гексаметилендиамин 47 ел.
Гетероцепные волокна 14, 15, 17
Гигроскопичность 89, 165, 263, 285,
286, 299 ел.
Гидролитическая полимеризация 29,
30
Гидрофильиость 214 ел.
Гидрохинон 31
Гофрирование 207
Дакрон 117
Дедерон 14, 19
Дигликольтерефталат 130, 131
Диизоциаиаты 150
Диметилацетамид 187, 188
Диметилсульфоксид 188, 189, 192, 197
Диметилтерефталат 121, 122, 135, 136,
139
Диметилформамид 186, 187, 197
Динил 39, 40, 64
Диэтиленгликольтерефталат 125 ел.
Додекан, синтез 56 ел.
Доделактам 56, 57
Дополнительная круГка 80
Дралон 179
Жаростойкие волокна
исходное сырье 321
определение 303
получение из вискозной иити 325,
326
полиакрилонитрильного
волокна 32J ел.
свойства н области применения
327 ел.
Предметный указатель
841
Замасливание 67, 206
Зефран 179
Извитость волокна 85, 108
Ингибиторы полимеризации 31
Инициаторы полимеризации 31
Ионообменные волокна 267, 268
Истираемость 89, 158
Капролактам
свойства 26, 27
синтез 21 ел.
полимеризация 28 ел.
Капролактон 23
Капрон 14, 19, 20, 98, 99
получение непрерывным методом
72 ел.
текстильная обработка 74 ел.
технологическая схема получения
21
Карбоцепные волокна 15 ел.
Катализаторы полимеризации 30, 32,
34 ел., 126 сл„ 133, 273
Катионная полимеризация 30, 31
Киана 14, 107
Классификация синтетических
волокон 14 ел.
Креслан 179
„Ксилон 180
Крутильно-вытяжная машина КВ-150-
И4 77, 78
п-Ксилол 119 ел.
Куралон 11, 16,248
Куртел 180
Лавсан 117
?-Лактамы 104
Лестничные полимеры 319, 320
Ликра 11
Модификация свойств синтетических
волокон 13
Модифицированные волокна
полиакрилонитрильные 211 ел
223 ел.
полиамидные 99 ел.
поливииилепиртовые 266 ел.
полиэфирные 161 ел., 288 ел.
Модуль эластичности 96, 264, 300
Метилакрилат 183
Метилметакрилат 183
Механизм для непрерывного
вытягивания и резки волокна 84
Найлои
6 14, 18 ел., 59 сл„ 107
6,6 11, 14, 18 ел., 47, 48, 59 ел., 76
Найлон
6,10 19, 55, 56
7 14, 19, 55, 59
11 19, 55, 56
12 19, 56 ел.
Накрашиваемость волокон 212 ел.,
264, 265
Нитрон 15, 180
Номекс 11, 308
Однородность структуры 95
Окнсь этилена 130, 131, 180, 181
Орлон 15, 170
Отрицательная фильерная вытяжка
254, 255
Перлон 11, 19
Перманганатиое число 27
Перхлорвинил 235, 237
Перхлорвиниловые волокна 235 ел.
Пиллинг 96, 168
Пирогаллол 31
Пирроны см. Лестничные полимеры
Плавильная решетка 64
Пластификатор 47
Пластификационная вытяжка 257
Плотность 89, 155, 264, 286, 299, 301
Полиакрилонитрил 193, 194
Полиакрилонитрильные волокна 11,
15, 179 ел.
модифицированные 211
накрашиваемость 212 ел.
получение 194 ел.
— акрилонитрила 180 ел.
— приготовление прядильного
раствора 194 ел.
— углеродного волокна 321 ел.
— формование текстильной нити
198 ел.
— штапельного волокна 200 ел.
последующая обработка волокна
205 ел.
свойства 208 ел.
синтез полимеров и сополимеров
акрилонитрила 182 ел.
Полиамидные волокна 14, 18 ел.
кордные нити 81 ел.
марки 19
модифицированные 99 ел.
моноволокно 85, 86
области применения 99
свойства 88 ел.
термостойкие 19
формование 63 ел.
— основные параметры 69 ел.
штапельное 83 ел.
Поли-6-амиды 60, 61
342
Предметный указатель
Полиамиды из пяти- и шестичленных
лактамов 58
Поли-л-бензамид 332
Полнбензимидазольные волокна 317,
318
Полибензоксазольные волокна 312 ел.
Поливиниловый спнрт 248 ел.
Поливинилспиртовые волокна 16,
248 ел.
ацеталированиё 258 ел.
марки 248
модифицированные 266 ел.
области применения 265, 266
получение поливинилового спирта
248 ел.
приготовление прядильного
раствора 252, 253
свойства 263, 264
формование и последующая
обработка волокна 253 ел.
Полнвинилхлорид 230 ел.
Поливинилхлоридные волокна 15,
229 ел.
волокна из сополимеров вииилхло-
рида 244 ел.
перхлорвиииловые волокна 235 ел.
Полигексаметиленадипамид 47 ел.
Полнгексаметиленадипамидные
волокна 14
Полидисперсность 62, 63, 141, 275, 276
Поликапроамнд
демономеризация перегретым
паром 72, 73
— под вакуумом 73
деполимеризация 87, 88
свойства 46 ел.
синтез 21 ел.
— аппаратура 39 ел.
— параметры полимеризации
32 ел.
сушка 42
Полимеризация в растворе 185 ел.
растворители 192, 193
Полиоксадиазольные волокна 314,
315
Полиолефиновые волокна 11, 16
полипропиленовые 271 ел.
полиэтиленовые 292 ел.
Полипиромеллитимидные волокна
315 ел.
Полипропилен
свойства 275 ел.
синтез 272 ел.
Полипропиленовые волокна
свойства 284 ел.
— полипропилена 275 ел.
синтез полипропилена 272 ел.
формование 281 ел.
химическая модификация 288 ел.
Политетрафторэтилен 297
Полиундеканамидные волокна 14
Полиуретаиовые волокна 15, 171 ел.
Поли-л-феиилентерефталамид 332
Полиформальдегидное волокно 176 ел.
Полиэнантоамид 52 ел.
Полиэнантоамидиые волокна 14
Полиэтилен 292, 293, 294
Полиэтиленовые волокна
получение волокна 293 ел.
— и свойства полиэтилена 292,
293
Полиэтилентерефталат
гидролиз отходов 153
свойства 14
синтез 131 ел.
— непрерывный процесс 137 ел.
— периодический процесс 134 ел.
Полиэфирные волокна 15
извитые и объемные 166 ел.
модифицированные 161 ел.
области применения 160, 161
определение 118
особенности производства кордной
нити 149 ел.
моноволокна 152, 153
— — штапельного волокна 151,
152
получение терефталевой кислоты
119 ел.
¦— этиленгликоля 124, 125
свойства 154 ел.
синтез диэтилеигликольтерефтала-
та 125 ел.
формование и последующая
обработка волокна 146 ел.
Предана 179
Пропилен 182
Профилированные фильеры 85, 97,
145
Прочность 88, 155, 263, 284, 299, 301
Прядильная шахта 66
ПЦ (волокно) 11, 229, 237
Реакционная способность 217 ел.
Регуляторы молекулярного веса 37,
38
Редои 180
Рильсан 55, 56
Ровиль 15, 229, 231
Роданистые соли 189, 190, 192, 197
Саран 15, 229
Сверхпрочные и высокомодульные
волокна 331 ел.
жесткоценные волокиообразующие
полимеры 332 ел.
формование 335 ел.
Предметный указатель
343
Светостабилизаторы 94, 95
Светостойкость 94, 95, 158, 210, 264,
301
Себацииовая кислота 55
Семикарбазид ПО
Силон 14, 19
Совиден 15, 229
Соль АГ 43, 50, 51, 100
Сополиэфиры 166, 177
Спаидекс-волокиа см. Высокоэластич-
иые волокна
Стабилин 9 93
Степень кристалличности 75, 260
Стереорегулярность 230, 231
Стойкость
к низким температурам 158
— термоокислительным и фотохи-
¦ мическим воздействиям 276 ел.
Структурирование 108 ел.
Сульфон 309
Суспензионная полимеризация 185
Сухо-мокрып способ формования 199
200, 337 ел.
Сцепляемость 96
Сшивание 215, 216, 258 ел.
Тевирон 231
Текучесть 279 ел.
Теплоемкость 264
Теплостойкость 89 ел., 209, 301
Терефталевая кислота 117, 119 130,
131
этерификация 127 ел.
Терилен 11, 117
Терлон 334
Термическая вытяжка 257
Термовиль 229
Термообработка нити 80, 81, 206, 207
Терморелаксация (термофиксация)
257, 258
Термостабилизаторы 90 ел., 156, 157
Термостойкие волокна
яз ароматических полиамидов и
полиэфиров 306 ел.
— лестничных полимеров 319, 320
— полигетероциклических
полимеров 310 ел.
определение 303
Термостойкость 89 ел., 156 ел. 208,
209, 301
Тефлон 11, 16, 297 ел.
Толуилеи-2,4-диизоцианат 109
Толуол 25, 26, 123 ел.
Триоксиметилен 176
Труба для непрерывной
полимеризации капролактама 44
Углеродные волокна 321 ел.
получение 321 ел.
свойства и области применения
327 ел.
Удлинение 155, 263, 284
Уидекан 14
Усталостная прочность волокна 110
Устойчивость к истиранию 264, 286,
287
Файбр Би (волокно) 334, 335
Фенол 22, 26, 48, 49
Физическая модификация волокон 99,
100
Фильерная вытяжка 69
Фильерные комплекты 67
Формальдегид ПО
Фосфорная кислота 31
Фторлон 11, 16, 300 ел.
Фторопласт 297
Фторсодержащие волокна 16, 297 ел.
тефлон 297 ел.
фторлои 300 ел.
Х-500 (волокно) 334
Хемостойкость 158, 159, 265, 286, 300,
302
Химическая модификация волокон
100 ел.
введение новых функциональных
групп 105 ел.
нарушение регулярности строения
100 ел.
получение химически окрашенных
волокон 111
структурирование 108 ел.
Хлорид цинка 191
Хлорин 15, 229, 237
модифицированное волокно 243,
244
формование мокрым способом 238,
239
штапельное волокно 239 ел.
Циаиурхлорид 109
Циклогексан 24 ел.
Экструдеры 65
Эластическая работоспособность 76
Эластические свойства 264
Эластичность 88, 89, г55, 217, 284
Эмульгаторы 67
Этиленгликоль 124, 125
Этиленкарбонат 189, 192, 197
ЗАХАР АЛЕКСАНДРОВИЧ РОГОВИН
ОСНОВЫ ХИМИИ И ТЕХНОЛОГИИ
химических волокон
Редактор | Гершман Б. г. \
Технический редактор Коган В. В.
Художник Петров Г. А.
Корректоры raepufluna Л. В.. Ваничкова Н. А,
Т. 19049 Сдано в наб. 21/VI 1974 г. Подп. в печ. 14/XI 1974 г.
Формат бумаги 60x90'/»- Бумага тип. № 2. Усл. печ. л. 21.5.
Уч.-изД. л. 24,53. Тираж 8500 экз. За к. 234. Изд. № 734.
Цена і р. 02 к.
Издательство сХимая». 107076, Москва, Стромынка, 13
Ордена Трудового Красного Знамени
Ленинградская типография № 2 имени Евгении Соколовой
Союзполиграфпрома при Государственном комитете
Совета Министров СССР по делам издательств,
полиграфии и книжной торговли.
198052, Ленинград, Л-52, Измайловский проспект, 39