/

















































































































































































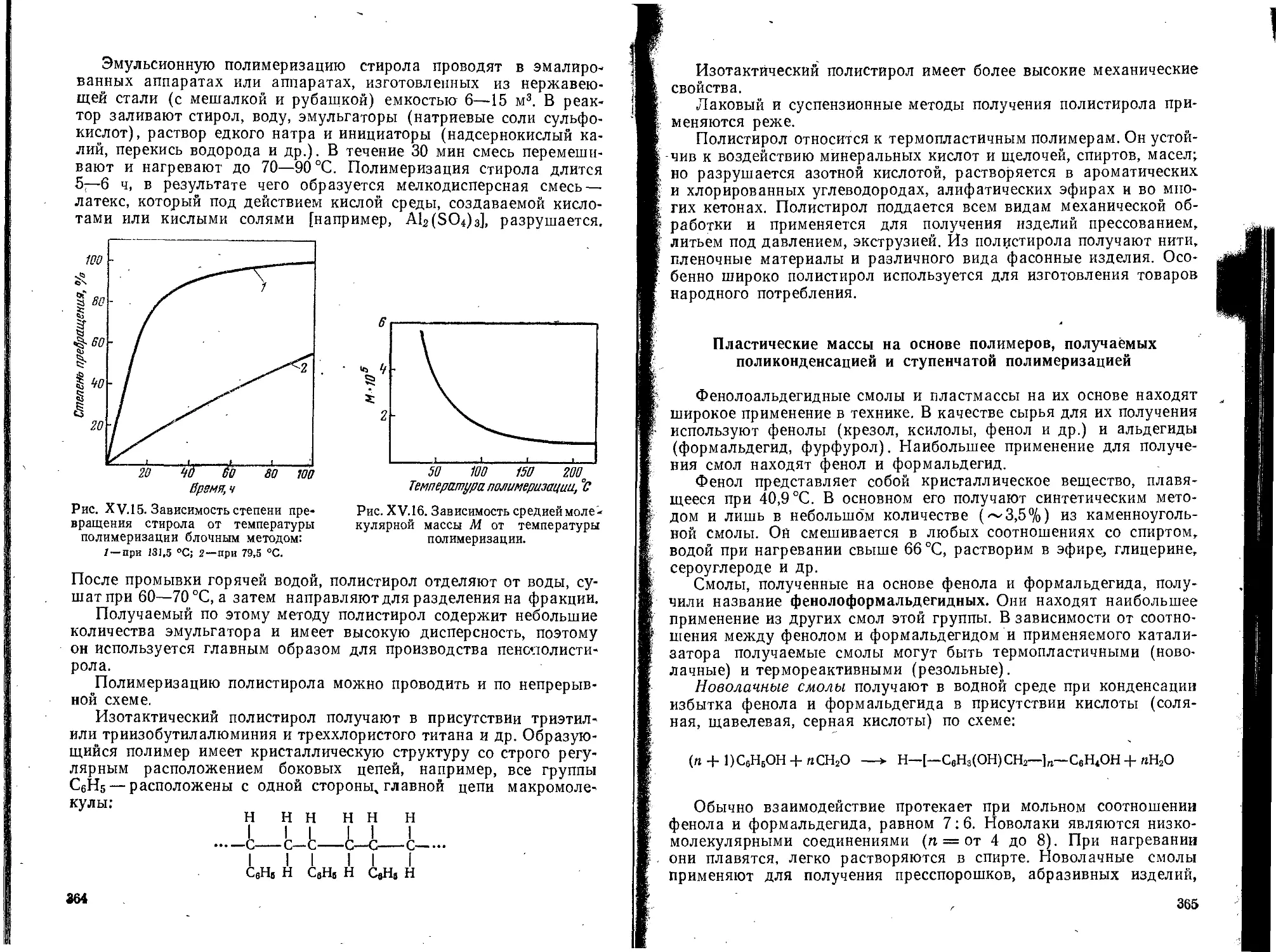






















Text
6 П7
0,28
УДК 66.01(075.8)
Общая химическая технология. Под редакцией
проф. Амелина А. Г. М., «Химия», 1977.
400 стр., 202 pffc„ 24 табл., 45 библиографических ссылок.
Книга является учебным пособием по курсу «Общая химическая
технология» для студентов хнмико-технологических специальностей
вузов. В ией изложены общие закономерности химической технологии,
основы теории, расчета и подход к выбору химических реакторов,
рассмотрены гетерогенные н каталитические процессы и их аппаратурное
оформление. Приведены методы организации химико-технологических
Процессов, даны сведения о химическом сырье, воде и источниках энер-
гии, Описаны производства важнейших химических продуктов — серной
и азотной кислот, аммиака, продуктов основного органического синтеза
и высокомолекулярных соединений.
Книга будет полезна инженерно-техническим работникам химиче-
ской и нефтехимической промышленности.
31401-31
°050(01)-77
31-77
6П7
Авторы: АМЕЛИН А. Г., МАЛАХОВ А. И., ЗУБОВА И. Е„
ЗАЙЦЕВ В. Н.
Рецензенты: проф. ТЕРПИЛОВСКИИ Н. Н. (кафедра ОХТ Казанского
химико-техиологического института) и проф. КСЕНЗЕНКО В. И. (ка-
федра ОХТ Московского института тонкой химической технологии
им. М. В. Ломоносова).
31401-31
050(01)-77
31-77
@ Издательство «Химия», 1977 г.
СОДЕРЖАНИЕ
Предисловие .........................................................
Условные обозначения ............................................9
Введение ............................................................. И
Содержание химической технологии.................о..............12
Значение химической промышленности в народном хозяйстве .... 15
Роль курса «Общая химическая технология» в подготовке инженера-
химика-технолога ...............................................17
ЧАСТЬ ПЕРВАЯ. ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ ХИМИКО-ТЕХНОЛОГИЧЕСКОГО
ПРОЦЕССА.................................. .... ...................19
Глава I. Общие сведения..............................................19
1. Химико-техиологический процесс н его содержание...................20
2. Основные технологические понятия н определения....................20
3. Материальный и энергетический балансы.................... .... 26
4. Качество продукции............................................... 31
5. Экономическая эффективность химического производства..............33
Примеры ........................................................37
Глава II. Физико-Химические закономерности в химической технологии . . 40
1. Классификация химических реакций..................................40
2. Факторы, влияющие на состояние равновесия...........•.............42
Сдвиг равновесия под влиянием температуры.......................44
Сдвиг равновесия под влиянием давления..........................47
Сдвиг равновесия под влиянием концентрации реагирующих веществ . 48
3. Кинетика химико-технологических процессов.........................50
Понятие о мнкро- и макрокинетике................................50
Влияние различных факторов иа скорость химических процессов, про-
текающих на микроуровне.........................................52
Примеры .......................................................63
Глава III. Типы химико-технологических процессов . . ............. . 66
1. Гомогенные процессы...............................................66
Скорость гомогенных процессов...........;.......................68
2. Гетерогенные процессы........................................... 69
Скорость гетерогенных процессов............................... 70
Коэффициент скорости процесса...................................70
Поверхность контакта фаз....................................... 76
' Движущая сила процесса.........................................77
1* 3
3. Математическое моделирование — основной метод расчета химических про-
цессов ........................................................
Моделирование процессов в системе газ — твердое и жидкость — твер-
дое .....................................................
Моделирование процессов в системе газ — жидкость и жидкость —
жидкость .................................................
Примеры ..................................................
78|
81
89
93
Глава IV. Каталитические процессы....................................... 94
1. Общие закономерности каталитических реакций...........................95
2. Гетерогенный катализ..................................................99
3. Кинетика гетерогенно-каталитических реакций........................... 101
4. Свойства и приготовление твердых катализаторов.......................105
ЧАСТЬ ВТОРАЯ. ХИМИЧЕСКИЕ РЕАКТОРЫ ,................................106
Глава V. Общие положения......................................... 106
• Исходные данные для расчета реакторов............................106
Уравнение материального баланса реактора .................... 107
Классификация реакторов ...... -..............................109
Глава VI. Реакторы с различными режимами движения среды.............109
1. Реакторы периодические............................................109
Реактор идеального смешения периодический ... -г.................ПО
2. Реакторы непрерывного действия....................................113
Реактор идеального вытеснения................................. 114
Реактор идеального смешения непрерывный . . ....................118
Каскад реакторов............................................... 120
3. Реакторы полунепрерывные..........................................125
4. Сравнение реакторов различных типов...............................125
5. Истинное время пребывания.........................................133
6. Динамическая характеристика реактора.............................134
Примеры ...................................................... 137
Глава VII. Реакторы с различным тепловым режимом....................140
1. Классификация реакторов с различным тепловым режимом...........141
2. Уравнение теплового баланса реактора.............................142
3. Политропический режим............................................144
4. Адиабатический режим .......................................... 147
5. Изотермический режим.............................................148
6. Условия поддержания устойчивого режима работы реактора...........149
7. Параметрическая чувствительность............................ . . 155
8. Выбор типа реактора с учетом теплового режима....................157
9. Создание оптимального теплового режима в реакторах...............159
Примеры ..................................................... 162
Глава VIII. Устройство реакторов.................................. .
1. Реакторы для гомогенных процессов....................................165
2. Реакторы для гетерогенных некаталитических процессов................166
Реакторы для проведения реакций в системах г—т и ж—т .... 167
Реакторы для проведения реакций в системах г—ж и ж—ж . . . .169
3. Реакторы для гетерогенно-каталитических процессов
Теплообмен в реакторах .............................
ЧАСТЬ ТРЕТЬЯ. НЕКОТОРЫЕ ТЕХНОЛОГИЧЕСКИЕ ПРОЦЕССЫ ХИМИЧЕСКИХ
ПРОИЗВОДСТВ .................................................
171
176
181
Глава IX. Сырье, вода, энергия............................................ 182
1. Сырье . . . ......................................................
Классификация сырья............................................
Сырьевые ресурсы...............................................
Рациональное и комплексное использование сырья ................
Обогащение сырья...............................................
2. Вода ............................................................
Виды и качество потребляемой воды..............................
Очистка воды...................................................
3. Энергия ....................Ь....................................
Виды энергии.................................v.................
Источники энергии................................-.............
Рациональное использование энергии ............................
4. Обезвреживание сточных вод и отходящих газов.....................
182
183
184
186
189
193
194
196
199
200
201
204
206
Глава X. Организация химико-технологического процесса...................209
1. Химическая, принципиальная и технологическая схемы..................210
2. Выбор параметров процесса...........................................219
3. Подбор аппаратуры.................................................. 222
4. Выбор материалов для изготовления аппаратуры........................226
5. Выбор контролируемых и регулируемых параметров......................227
6. Проектирование химико-технологического процесса.....................233
Глава XI. Производство серной кислоты.................................. 235
1. Общие сведения ............................................... . 235
2. Методы получения серной кислоты...................................238
3. Производство серной кислоты контактным методом из флотационного
колчедана .............................................................238
Химическая и принципиальная схемы производства............. 238
Физико-химические основы и технологические схемы отдельных стадий
производства ................................................... 239
Технико-экономические показатели..................................251
4. Производство серной кислоты контактным методом из серы......252
5. Усовершенствование производства серной кислоты............. 954
Примеры ..................................................255
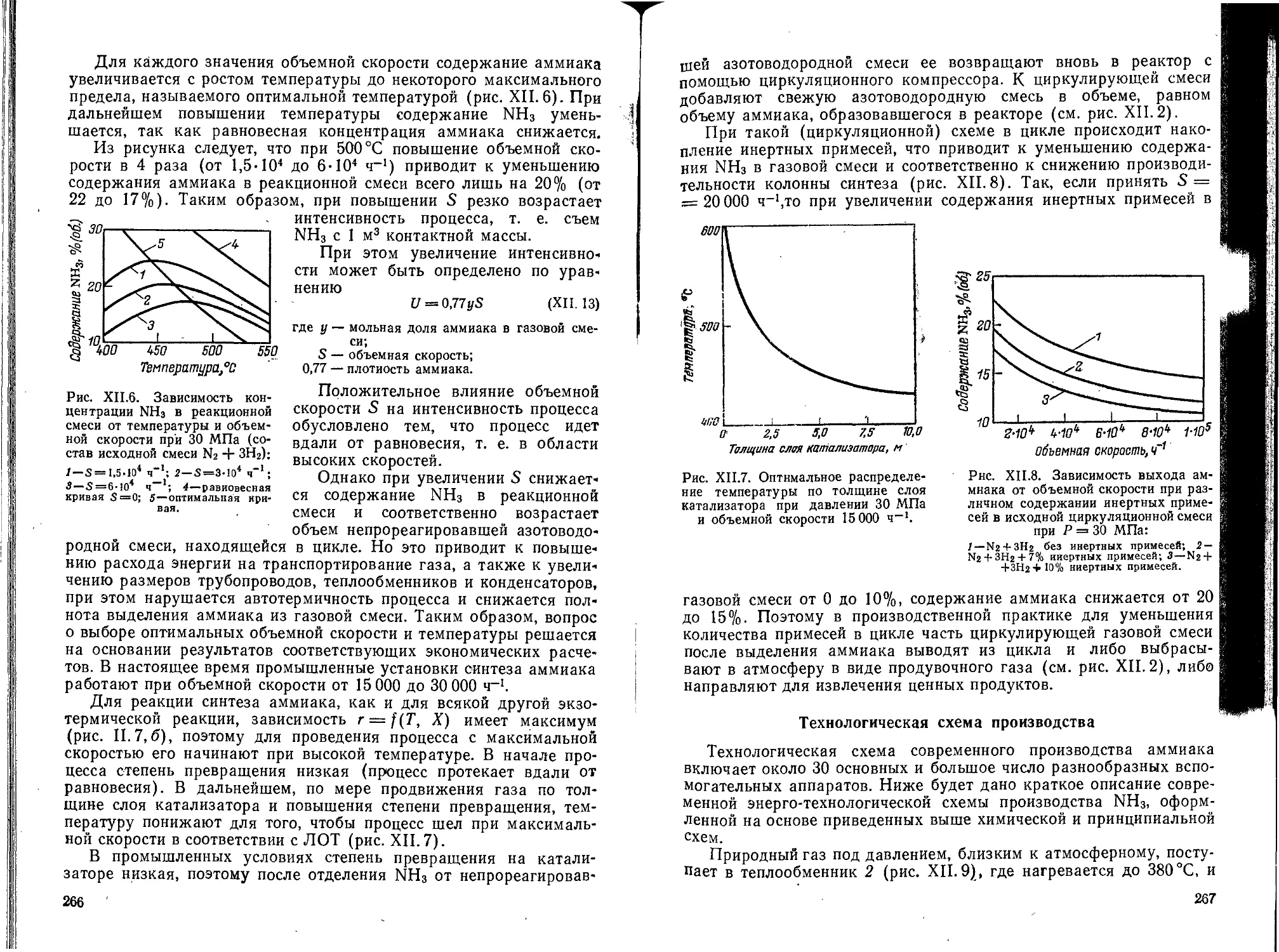
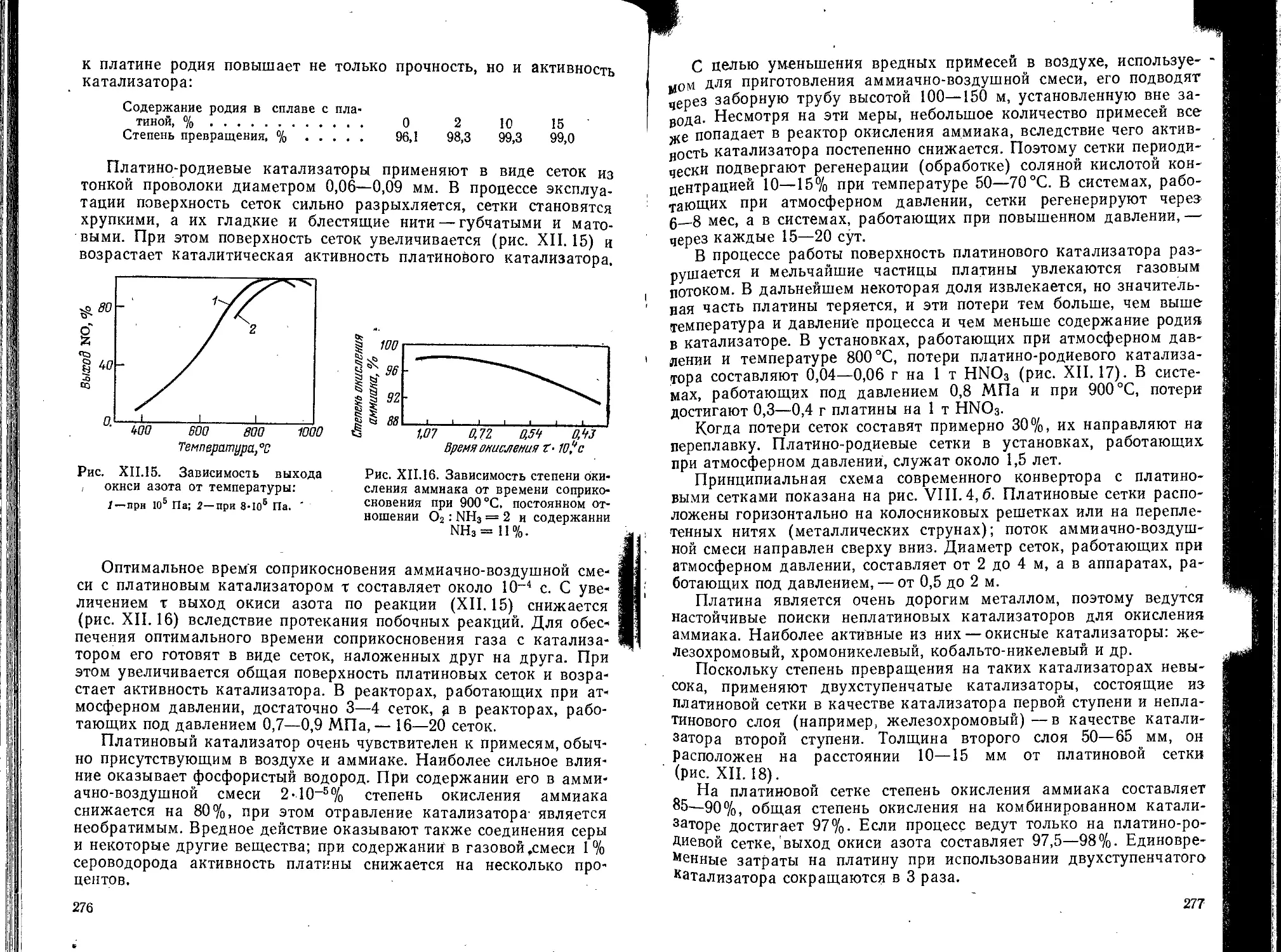
Глава XII. Производство аммиака и азотной кислоты............. 256
1. Производство аммиака......................................... . . 256
Химическая и принципиальная схемы производства.................258
Физико-химические основы производства..........................260
Технологическая схема производства . . <.......................267
Технико-экономические показатели ..............................271
2. Производство азотной кислоты......................................271
Химическая и принципиальная схемы производства разбавленной
азотной кислоты из аммиака.....................................273
Физико-химические основы производства..........................274
Технологическая схема производства разбавленной азотной кислоты . 281
Концентрирование азотной кислоты...............................284
Прямой синтез концентрированной азотной кислоты из окислов азота . 286
Технико-экономические показатели.........................' . . 288
Примеры .......................................................288
5
Глава ХШ. Химическая переработка топлива....................... 290
1. Переработка твердого топлива..................................293
Коксование ............................................... 293
Продукты коксования и их использование......................294
Коксовые печн ............................................. 295
Переработка коксового газа..................................297
Полукоксование .............................................299
Газификация твердого топлива................................300
2. Переработка нефти.............................................303
Методы переработки нефти н нефтепродуктов...................305
Физические методы...........................................307
Химические методы...........................................310
Очистка нефтепродуктов......................................317
3. Переработка природных газов...................................318
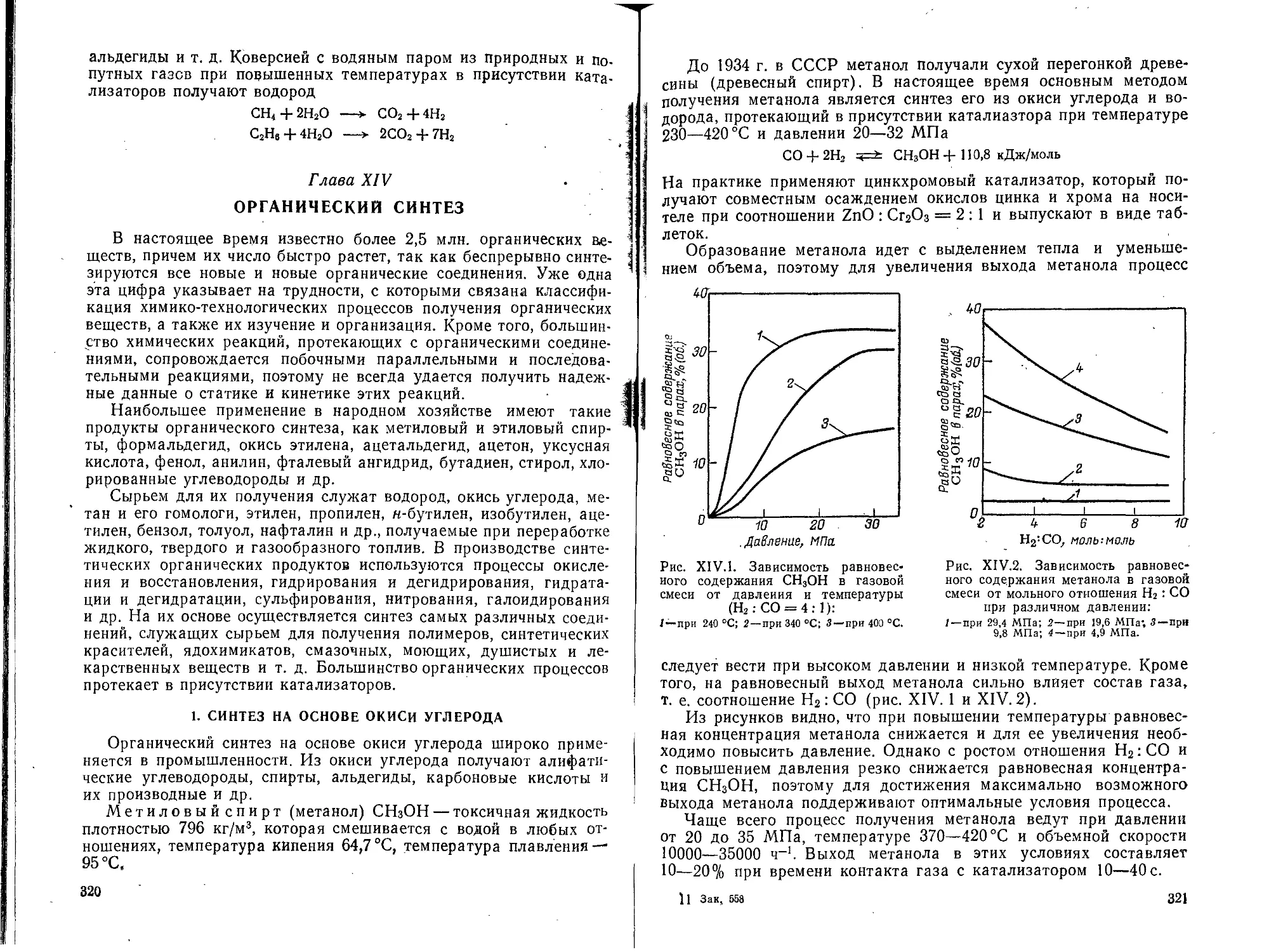
Глава XIV. Органический синтез . .............................. 320
1. Синтез на основе окиси углерода...............................320
2. Синтез на основе предельных углеводородов.....................324
3. Синтез на основе непредельных углеводородов...................326
4. Синтез на основе ацетилена....................................330
5. Нитробензол ................................................. 333
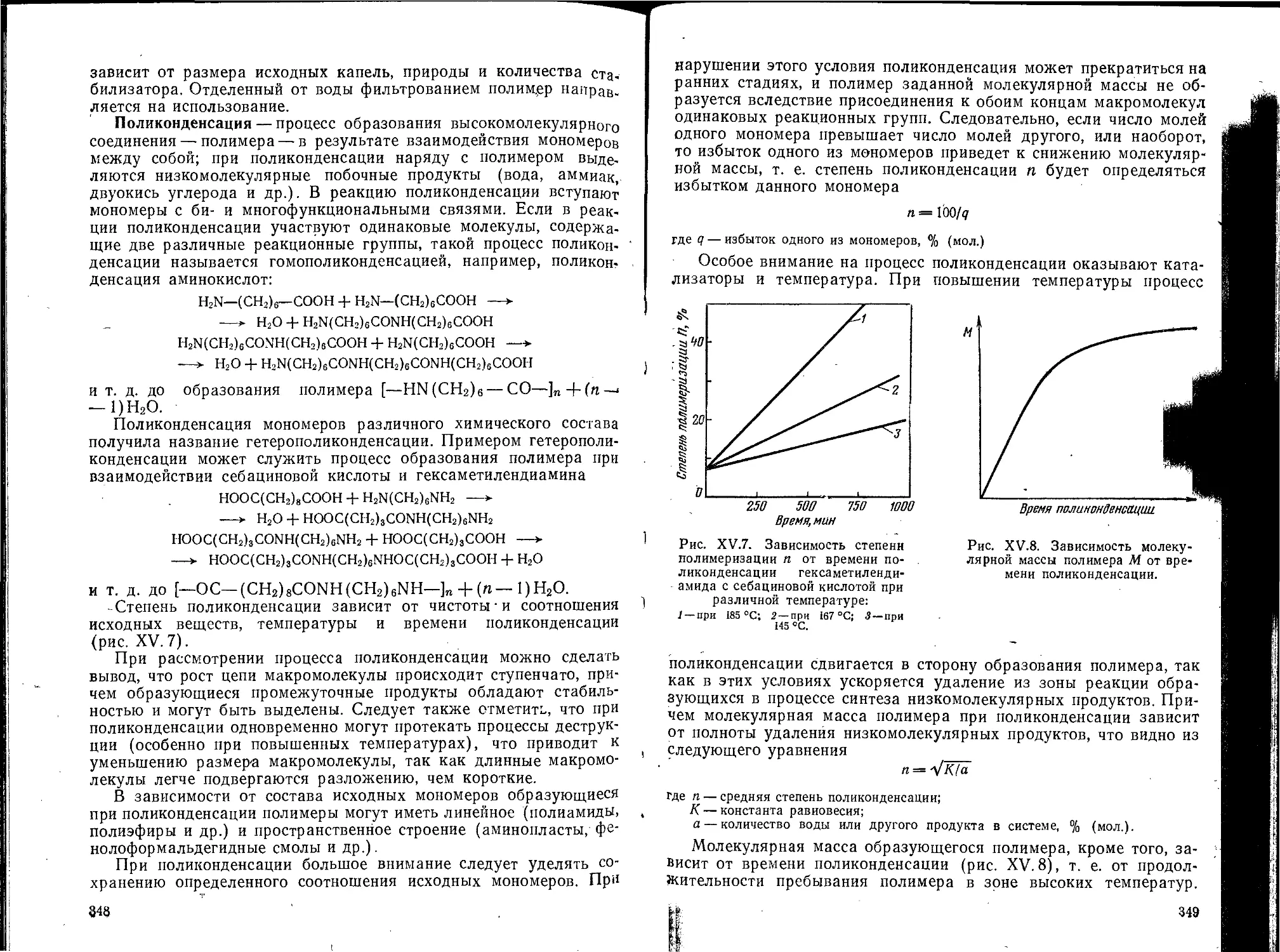
Глава XV. Высокомолекулярные соединения..........................335
1. Введение .....................................................335
Классификация высокомолекулярных соединений.................337
Синтез высокомолекулярных соединений........................338
Свойства высокомолекулярных соединений......................351
2. Производство пластических масс............................... 354
Пластические массы на основе полимеров, получаемых цепной поли-
меризацией .................................................357
Пластические массы на основе полимеров, получаемых поликонденса-
цией и ступенчатой полимеризацией...........................365
3. Производство каучука и резины.................................374
Сырье и материалы резинового производства...................374
Изготовление резиновых изделий..............................382
Глава XVI. Заключение............................................385
1. Плазмохнмические процессы.....................................386
2. Радиационно-химические процессы ..............................389
3. Биохимические процессы........................................390
Литература ......................... 393
Предметный указатель.............................................395
ПРЕДИСЛОВИЕ
/' ° -
Настоящий учебник составлен в соответствии с новой програм-
мой * и состоит из трех частей:
1) основные закономерности химико-технологических процес-
сов;
2) химические реакторы;
3) типовые технологические процессы химических производств.
В первой и второй частях курса излагаются теоретические ос-
новы химической технологии, а в третьей приведены примеры
оформления наиболее типичных и хорошо изученных химико-тех-
нологических процессов.
В настоящее время химическая технология характеризуется
переходом от описательной к точной науке, поэтому, наряду с
изложением общих физико-химических закономерностей, большое
внимание в учебнике уделяется приемам установления связей
между различными параметрами химико-технологического процес-
са. Это дает возможность использовать математическое моделиро-
вание и электронно-вычислительную технику для установления
оптимальных значений параметров процесса, обеспечивающих мак-
симальную экономическую эффективность химического процесса.
При этом учебник построен так, что учащийся знакомится не
только с отдельными физическими и химическими операциями, со-
ставляющими химический процесс, но и с общими принципами
оформления всего химико-технологического процесса в целом. Та-
ким образом студент становится подготовленным для изучения
последующих дисциплин, предусмотренных учебным планом и
отражающих некоторые особенности химического производства
(моделирование и оптимизация, экономика и организация произ-
водства, охрана труда и др.).
После курса «Общая химическая технология» студент в даль-
нейшем закрепляет и расширяет свои знания и совершенствуется
как инженер, но первые и наиболее запоминающиеся сведения о
химико-технологическом процессе студент получает при изучении
* Программа курса «Общая химическая технология» для химико-технологи-
ческих высших учебных заведений, химических факультетов политехнических ву-
зов и факультетов химического машиностроения утверждена Учебно-методнче-
скнм управлением по высшему образованию 9 апреля 1975 года.
«Общей химической технологии»; в этот период он «рождается»
как инженер-химик-технолог, поэтому роль курса в подготовке
инженера-химика-технолога весьма существенная.
Введение и главы I—III, IX—XII и XVI написаны А. Г. Амели-
ным; 1лава IV—А. Г. Амелиным и И. Е. Зубовой; главы V—VIII—
А. Г. Амелиным, И. Е. Зубовой и В. Н. Зайцевым; глава XIII —
А. Г. Амелиным и А. И. Малаховым, главы XIV и XV—А. И. Ма-
лаховым.
Авторы выражают глубокую благодарность академику
Н. М. Жаворонкову и чл.-корр. АН СССР В. В. Кафарову за
ценные советы, а также благодарят рецензентов проф. В. И. Ксен-
зенко и к. т. н. Н. Н. Терпиловского за большой труд по рецензи-
рованию рукописи и ценные замечания.
УСЛОВНЫЕ ОБОЗНАЧЕНИЯ
А, В, ... — реагирующие вещества
j?. 5,... — продукты реакции
a Ь.....г, S, ... — коэффициенты стехиометрических уравнений при вещест-
вах А, В, R, S ...
В — скорость потока вещества, кмоль • ч'1
Сд — концентрация вещества А, кмоль • м~3
Ср — теплоемкость смеси, кДж • кмоль'1 • К'1
Ср — теплоемкость смеси, отнесенная к 1 моль основного компо-
нента смеси (обычно вещества А), кДж • кмоль'1 • К'1
D — коэффициент диффузии (молекулярной и турбулентной),
М2 • Ч'1
d — диаметр, м
Е — энергия активации, кДж
F — поверхность соприкосновения, м2
И — высота общая, м
h — высота, расстояние между полками и т. д., м
Д// — теплота реакции, кДж
К — константа равновесия; коэффициент теплопередачи,
кДж • м'2 • ч-1 • К'1
k — константа скорости реакции, (м3 - кмоль'1) • ч*1
L — длина реактора, м
I — длина, м
пг — число реакторов в каскаде; коэффициент
JNА — количество вещества А, кмоль
п — порядок реакции, коэффициент
Р — общее давление, Па
рА — парциальное давление вещества А в газе, Па
Q — количество тепла, кДж; мощность установки, т • год'1
R — газовая постоянная; радиус частицы, м
Гд. гв, .... rR, rs, ... — скорость реакции, относенная к единице объема
жидкой фазы, кмоль • м~3 • ч'1
г — скорость процесса (единицы измерения изменяются в за-
висимости от характера процесса)
5 — объемная скорость, ч-1; энтропия, Дж-моль-^К-1; се-
бестоимость, руб • т'1 ,
Т — абсолютная температура, К
t — температура, °C
V — объем, м3
о — объемная скорость потока, м3 • ч~>
Vr — объем реактора, м3
w — скорость потока, м • ч'1
X — степень превращения, доли
х, у. z— координаты
а — коэффициент теплоотдачи, кДж • м'1 • ч'1 • К”1
9
Р — коэффициент массоотдачи, кмоль • м"2 • ч*1 • К'1
Д — движущая сила процесса (ДС, Др, ДГ)
б — толщина, м
р — плотность, кг • М"3
в — относительное изменение объема системы в ходе реакции
Л — коэффициент теплопроводности (турбулентная и молеку
лярная), кДж • м~2 • г'1 • К*1
т — время, ч, с
<р — селективность
Ф — выход продукта
Верхние индексы:
* — равновесное значение
" — различные значения параметров
Нижние индексы:
г — газ
ж — жидкость
т — твердое
р — значение на границе раздела фаз
О — начальное состояние системы, или условие на входе
к — конечное состояние системы
ВВЕДЕНИЕ
Наука, изучающая способы и процессы переработки сырья в
предметы потребления и средства производства, носит название
технологии. Под понятием «способы и процессы переработки»
понимают ряд последовательных операций, проводимых с сырьем
в различных машинах и аппаратах с целью получения из него
заданного продукта, непосредственно используемого человеком
или же в свою очередь служащего сырьем для получения других
продуктов или средств производства.
Человеческую деятельность можно разбить на две области.
Одна из них направлена на духовное воспитание человека, а вто-
рая — на удовлетворение его материальных потребностей. С раз-
витием материальной базы социалистического общества человек
все больше и больше внимания будет уделять духовной деятельно-
сти, однако в современных условиях основная часть времени еще
затрачивается людьми на удовлетворение своих материальных
потребностей. Поскольку целью технологии является производство
предметов потребления, то отсюда вытекает важный вывод о том,
что в настоящее время технология является одной из основных
научных дисциплин.
Для современной технологии характерны две области. Одна
состоит в том, что технология изучает массовое производство,
масштабы которого достигают огромных размеров; для этой цели
строятся крупные высокопроизводительные установки и производ-
ственные комплексы.
Следует отметить, что такие важнейшие дисциплины, как мате-
матика, физика, теоретическая химия и некоторые другие, разви-
ваются отвлеченно без тесной связи с технологией. Эти дисципли-
ны устанавливали общие закономерности явлений, а технология,
используя эти закономерности, разрабатывала оптимальные
условия оформления и ведения того или иного технологического
процесса. Однако характерная направленность современной
науки — это углубляющийся и расширяющийся процесс органиче-
ского ее срастания с промышленностью и техникой. Поэтому мно-
гие важнейшие технологические процессы сейчас разрабатываются
совместно учеными и инженерами самых разнообразных специ-
альностей: технологами, физиками, математиками, химиками и др.
11
При этом наиболее отчетливо проявляется ведущая роль техно-
лога: он должен квалифицированно сформулировать задачу, а
также успешно использовать полученные учеными результаты
для оформления технологического процесса.
Технология делится на механическую и химическую.
Механическая технология изучает процессы, при кото-
рых изменяется форма, внешний вид или физические свойства
материалов, а химическая технология рассматривает
процессы изменения химического состава и свойств материалов.
Исходя из этого, можно дать такое определение: химическая тех-
нология — наука о способах и процессах химической переработки
сырья.
Указанное деление в значительной степени условно, так как
при механической переработке материала часто меняются и его
химические свойства. Так, например, некоторые металлы долгое
время могут находиться на воздухе без заметного изменения. Од-
нако, если их подвергнуть физическому воздействию и тонко из-
мельчить, полученные металлические порошки самопроизвольно
загораются на воздухе. Это объясняется тем, что с измельчением
вещества увеличивается его поверхностная энергия, а следователь-
но, повышается химическая активность вещества.
Современное химическое производство включает большое число
разнообразных физических и химических операций, которые тесно
связаны между собой, поэтому химическая технология изучает
совокупность физических и химических процессов и пути их осу-
ществления в промышленном масштабе.
Содержание химической технологии
Еще во времена глубокой древности человек использовал
приемы химической технологии для переработки природного сырья
в продукты и предметы потребления. На протяжении многих веков
химические производства представляли собой разрозненные руч-
ные ремесла, технологические особенности которых базировались
преимущественно на опытных и случайных результатах.
Химическая промышленность стала оформляться в XIX веке,
когда бурный рост промышленности вызвал большой спрос на
химическую продукцию. Этому также способствовало открытие
выдающимися учеными важнейших закономерностей в области
физики и химии; В начальный период оформления химической
технологии большой вклад в ее развитие внесли русские ученые
М. В. Ломоносов, А. М. Бутлеров, Д. И. Менделеев и многие
другие.
Дальнейшее развитие химической промышленности характери-
зуется более глубоким использованием законов математики, фи-
зики и химии и других точных наук для совершенствования хими-
ко-технологических процессов. И в этот период всемирное призна-
ние в области химии и химической технологии получили труды
12
советских ученых М. А. Ильинского, И. А. Каблукова, А. Е. Фа-
ворского и многих других.
В середине нашего столетия были сделаны первые реальные
шаги для того, чтобы с помощью точных математических методов
вывести законы, объясняющие течение отдельных стадий химиче-
ского процесса.
Глубокое проникновение математики в химическую технологию
особенно интенсивно происходит в последние годы, чему способст-
вует бурное развитие вычислительной техники и кибернетики.
В результате этого химическая технология становится точной нау-
кой; именно это является ее характерной чертой в настоящий
период.
Число веществ, используемых человеком в своей практической
деятельности, очень велико и беспрерывно возрастает, поскольку
ежедневно открываются и синтезируются все новые вещества.
В настоящее время насчитывается около 3 млн. веществ; около
300 тыс. неорганических и более 2,5 млн. органических, каждое из
них отличается от другого своими свойствами. Многие из этих
веществ получаются в результате химической переработки, поэто-
му число технологических процессов весьмц велико. Отсюда сле-
дует, что химическая технология чрезвычайно многообразна и
сложна.
Химические производства можно разделить на две группы; _»а
производства неорганических и органических веществ.
Промышленность неорганических веществ включает;
1) производства основных химических веществ (кислоты, щелочи,
соли, удобрения и др.);
2) производство тонких неорганических продуктов (реактивы, ред-
кие элементы, полупроводники, фармацевтические препараты
и др.);
3) электрохимические производства (хлор, щелочи, кислород, во-
дород и др.);
4) металлургия (черная, цветная, металлургия благородных и
редких металлов и др.);
5) производство силикатов (стекло, цемент, керамика и др.);
6) производство минеральных красок и пигментов
Промышленность органических веществ включает:
1) основной (тяжелый) органический синтез (спирты, кислоты,
эфиры, переработка СН4, СО, Н2, С2Н4 и др.);
2) производство полупродуктов и красителей;
3) тонкий органический синтез (фармацевтические препараты,
кино-фотореактивы и др.);
4) производство высокомолекулярных веществ (пластические мас-
сы, искусственные и синтетические волокна, каучук и др.);
5) переработка горючих материалов (нефти, угля, сланцев и др.);
6) производство пищевых продуктов (сахар, жиры и др.).
С технологической точки зрения такое деление условно, так как
процессы получения некоторых неорганических и органических
13
веществ имеют много общего. Так, реакция получения аммиа-
ка — неорганического продукта, и реакция получения метилового
спирта (метанола) — органического продукта, очень сходны. Обе
реакции
N2 4- ЗН2 -< J- 2NH3 + Qt
СО + 2Н2 CH3OH+Q2
проходят с выделением тепла и уменьшением объема. Сходны и
условия синтеза: аммиак получают при температуре около 500°C
и давлении 30 МПа; синтез метилового спирта проводят при тем-
пературе около 250 °C и давлении 25 МПа.
Из приведенных данных видно, что химическая технология
устанавливает закономерности и изучает процессы не только ос-
новной химической промышленности, но и многих других важней-
ших отраслей техники, так как в основе практически любого
производства лежат процессы, связанные с химическим взаимодей-
ствием. Действительно, теперь уже трудно указать отрасль
промышленности, в которой не нашли бы применения методы и
средства химической технологии. Без использования этих методов
не были бы возможны успехи в современной атомно-ядерной
технике, радиотехнике, электронике, космонавтике и во многих
других областях техники.
Трудности, возникающие в этом направлении, обусловлены, с
одной стороны, обилием химико-технологических процессов, а, с
другой — недостаточной их изученностью. Вследствие этого для
большинства химических процессов не установлена зависимость
между отдельными параметрами и, следовательно, не могут быть
-составлены математические описания. Последнее исключает воз-
можность математического моделирования, являющегося эффек-
тивным средством установления оптимальных условий интенсифи-
кации процесса. Чтобы установить связь между параметрами
технологического процесса, необходимо провести всесторонние тео-
ретические и экспериментальные исследования, опытные работы, а
также анализ действующих предприятий с целью глубокого изу-
чения каждого химико-технологического процесса.
Основной задачей современной химической технологии стано-
вится не описание химических процессов и аппаратов, а установ-
ление точных данных, выражаемых в математической форме, о
зависимости как отдельных стадий, так и всего процесса в целом
от различных факторов, т. е. математическое описание химико-
технологического процесса.
В настоящее время, в век научно-технического прогресса, хими-
ческая технология бурно развивается и совершенствуется, исполь-
зуя новейшие достижения в самых разнообразных областях науки
и техники. Особый интерес представляют плазменно-химические
процессы, при которых под действием высокой температуры исход-
ные вещества превращаются в ионизированные газы и реагируют
•с образованием различных продуктов. Перспективы развития хи-
14
мической технологии на основе плазмохимических процессов весь-
ма значительны, так как в высокоионизированной плазме могут
протекать процессы, невозможные в других условиях.
Новые широкие возможности открываются перед химической
технологией в результате применения ультразвука, при котором
происходит механическое воздействие упругих колебаний на обра-
батываемую среду. Этот метод используется для диспергирования
твердых и жидких веществ, коагуляции аэрозолей и эмульсий,
обезвоживания, уменьшения кристаллообразования на стенках
сосудов, снятия пересыщения, интенсификации некоторых гетеро-
генных процессов и т. д.
Большое применение в химической промышленности находят
фотохимические, фотокаталитические и радиационно-химические
процессы. Последним принадлежит особенно большое будущее,
так как эти процессы протекают при воздействии ионизирующих
излучений высоких энергий. При этом создается мощное воздей-
ствие на вещество, разрушаются связи между атомами и обра-
зуются свободные радикалы либо валентно-ненасыщенные атомы.
При массовом производстве химических продуктов исключи-
тельно важное значение приобретает повышение эффективности
использования сырья и энергии, интенсификация процессов и раз-
работка новых технологических схем, а также снижение содержа-
ния вредных примесей в сточных водах и отходящих газах путем
совершенствования технологических процессов. Большое практи-
ческое значение имеет разработка энерготехнологических процес-
сов, при которых тепло химических реакций используется для
получения энергии, потребляемой в самом процессе либо выдавае-
мой на сторону в виде электроэнергии или энергетического пара.
Значение химической промышленности
в народном хозяйстве
Химическая промышленность является одной из основных от-
раслей народного хозяйства. Ассортимент выпускаемой ею продук-
ции чрезвычайно велик и разнообразен: в настоящее время отече-
ственная химическая промышленность выпускает около 50 000 на-
именований продукции. Важнейшими химическими продуктами
являются удобрения, ядохимикаты, кислоты и щелочи, лаки и
краски, синтетические волокна, пластические массы, синтетический
каучук, многочисленные производные углеводородов и т. д. Огром-
ное значение для народного хозяйства имеет химическая перера-
ботка каменного угля, нефти, природного газа, дерева и другого
углеродсодержащего сырья. Велико значение химической промыш-
ленности в области производства атомной энергии, оборонной
промышленности, освоения космоса и др.
XVIII съезд Коммунистической партии Советского Союза, про-
ходивший в 1939 г., объявил третью пятилетку пятилеткой химии.
В дальнейшем ЦК КПСС и Совет Министров СССР приняли ряд
15
важнейших постановлений, в которых подчеркивалось важное зна-
чение химической промышленности и предусматривались ускорен-
ные темпы ее развития. В Программе Коммунистической партии
Советского Союза записано: «Одна из крупнейших задач — все-
мерное развитие химической промышленности, полное использова-
ние во всех отраслях народного хозяйства достижений современ-
ной химии, в огромной степени расширяющей возможности роста
народного богатства, выпуска новых, более совершенных и деше-
вых средств производства и предметов народного потребления» *.
И далее в Программе указывается, что к числу важнейших меро-
приятий, которые будут в дальнейшем предметом особой заботы
партии, относится... «—исследование химических процессов, разра-
ботка новых, наиболее совершенных технологических методов, соз-
дание высококачественных и дешевых искусственных и синтетиче-
ских материалов для всех отраслей народного хозяйства» **.
В решениях XXV съезда КПСС записано «Продолжить в широ-
ких масштабах техническое перевооружение химической промыш-
ленности, внедрение агрегатов большой единичной мощности, не-
прерывных одностадийных технологических процессов с макси-
мальным использованием энергии химических реакций»***. При
этом предусмотрено увеличение прбизводства химической промыш-
ленности на 60—65% и повышение производительности труда на
59—61%. Темпы роста химической промышленности выше, чем
темпы роста многих других отраслей промышленности (табл. 1).
Таблица 1. Увеличение производства промышленной продукции
. (в %)
Пятилетки Вся промышленность Химическая промышленность
1966—70 гг. 50 70
1971-75 гг. 44 70
1976—80 гг. 35-39 • 62,5
По объему производства важнейших продуктов тяжелой про-
мышленности СССР опережает США и занимает первое место
в мире. Однако по ряду продуктов химической промышленности
СССР занимает только второе место, хотя существующий разрыв у
быстро сокращается, так как темпы роста продукции химиче-
ской промышленности в СССР более высокие, чем в США
(табл. 2).
• Программа Коммунистической партии Советского Союза, Госполитиздат,
1972, стр. 70.
•* Там же, стр. 126.
*** Основные иаправлеиия развития народного хозяйства СССР на 1976—
1980 годы, Политиздат, М., 1976, стр. 30.
16
Таблица 2 Производство некоторых видов продукции в СССР и США
(Население, млн. чел.: СССР—253; США — 212)
ч СССР в 1975 г., млн. т СССР к США, % СССР в J980 г„ млн, т Среднегодовой прирост с 1951 по 1975 гг., %
СССР США
Нефть и газ. коид 491 119 620-640 10,8 1.8
Уголь 701 120 790—810 3,8 0,6
Сталь 141 130 160—170 6,8 0,8
Минеральные удобрения 90 131 143 12,2 5,9
Серная кислота 18,6 68 — 9,1 3,5
Химические волокна 0,96 32 1,45-1,5 15,8 6,5
Цемент 122 188 143—146 10,4 2,1
Роль курса «Общая химическая технология»'’
в подготовке инженера-химика-технолога
Все многообразие процессов" химической технологии можно
свести к пяти основным группам процессов;
1) гидродинамические;
2) тепловые;
3)- диффузионные (или массообменные);
4) механические;
5) химические.
К первой группе процессов относится перемещение жидкостей
я газов по трубопроводам и аппаратам, перемешивание в жидких
Рис. 1. Схема подготовки инженеров-химиков-техиологов.
17
средах, а также обработка неоднородных жидких и газовых си-
стем; скорость этих процессов определяется законами механики
и гидромеханики.
Группу тепловых процессов составляют процессы нагревания,
охлаждения, конденсации, выпаривания, теплообмена и другие,
скорость которых определяется законами теплопередачи.
Группа диффузионных процессов связана с переносом вещества
из одной фазы в другую.
К механическим относятся процессы дробления, измельчения,
грохочения, транспортирования твердых материалов, гранулирова-
ния и др.
Наиболее важную и многообразную группу составляют химиче-
ские процессы, связанные с изменением химического состава и
свойств вещества, скорость протекания которых определяется за-
конами химической кинетики,
Первые четыре группы процессов изучаются в курсе «Процессы
и аппараты», пятая группа процессов изучается в курсе «Общая
химическая технология».
Одновременно с изучением физико-химических закономерностей
химико-технологического процесса, а также теории и методов-
расчета реакторов в курс „Общей химической технологии1* вклю-
чены методы определения оптимальных параметров технологиче-
ского режима и основные вопросы, связанные с организацией
химико-технологического процесса. Таким образом, в процессе
изучения курса студент впервые знакомится не только с отдель-
ными аппаратами или операциями, но и содержанием и оформле-
нием всего химико-технологического процесса в целом (см. рис. 1).
После этого студент подготовлен для изучения ряда последующих
дисциплин, предусмотренных учебным планом. Завершает свою
подготовку студент на профилирующих кафедрах (спецкафедрах),
где ощ изучает достаточно подробно особенности какой-либо узкой
специальности.
ЧАСТЬ ПЕРВАЯ
ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ
ХИМИКО-ТЕХНОЛОГИЧЕСКОГО ПРОЦЕССА
Глава I
ОБЩИЕ СВЕДЕНИЯ
Изучение химико-технологического процесса ведут с двух по-
зиций: создание самого процесса, т. е. разработка процесса и
сооружение цеха (установки) для получения данного продукта,
и эксплуатация производства.
В настоящее время развитие производства идет в направлении
внедрения автоматических методов контроля и регулирования тех-
нологическими процессами... «со все большим переходом к цехам
и предприятиям-автоматам»...*. В этом случае число инженеров-
химиков, занятых вопросами эксплуатации, будет невелико. Роль
персонала, обслуживающего такие автоматизированные производ-
ства, будет сведена главным образом к наблюдению за исправ-
ностью технологического оборудования, а также приборов автома-
тического контроля и регулирования, а сам технологический про-
цесс будет регулироваться соответствующими автоматами. Боль-
шинство же инженеров-химиков будет связано с разработкой
технологического процесса и его оформления.
Процесс создания химического производства складывается из
следующих операций:
1) разработка химико-технологического процесса;
2) составление проектной документации;
3) построение цеха, т. е. сооружение здания, монтаж оборудо-
вания, размещение аппаратов и приборов автоматического контро-
ля и регулирования.
В курсе «Общая химическая технология» изучаются вопросы,
посвященные главным образом решению первых двух задач, ко-
торые наиболее тесно связаны с сущностью химического производ-
ства. Но прежде следует ознакомиться с основными элементами
химико-технологического процесса.
* Программа Коммунистической партии Советского Союза, Госполитиздат,
1961, стр. 71.
19
1. ХИМИКО-ТЕХНОЛОГИЧЕСКИЙ ПРОЦЕСС
И ЕГО СОДЕРЖАНИЕ
Химико-технологический процесс (ХТП) включает ряд физиче-
ских, физико-химических и химических процессов (операций) и (
складывается из трех основных стадий. На рис. I. 1 изображена
принципиальная схема процесса получения продуктов по реакции:;
A—+R
А—>S
или А'
В первой стадии протекают только физические процессы, поэ-
тому исходный реагент А химически неизмененным переходит во
вторую стадию, где происходит химическая реакция и образуются
продукты и S. Однако обычно реакция не идет до конца и часть
Рис. 1.1. Принципиальная схема про-
стейшего химнко-технологического
процесса:
7 —подготовка сырья; 2—химические пре-
вращения; 3—выделение целевого продукта.
реагента А остается без измене-
ния. В третьей стадии химиче- ’
ских превращений нет, здесь
происходит разделение продук-
тов: выделяются целевой про-
дукт R, побочный продукт S и
оставшийся исходный реагент А,
который может быть возвращен
в начало процесса.
Таким образом, из трех основ-
ных стадий ХТП первая и третья
стадии — подготовка сырья и выделение целевых продуктов —
в большинстве случаев относятся к физическим или физико-хими-
ческим процессам; вторая стадия—к химическим.
Процессы, протекающие при проведении физических операций, :
рассматриваются в курсе «Процессы и аппараты». В курсе «Об-
щая химическая технология» изучают теоретические основы хими-
ческих процессов, протекающие в реакторах, а также способы
оформления и организации химико-технологических процессов.
2. ОСНОВНЫЕ ТЕХНОЛОГИЧЕСКИЕ ПОНЯТИЯ
И ОПРЕДЕЛЕНИЯ
Показателем, характеризующим эффективность работы машин,
аппаратов, цехов и заводов в целом, служит производительность.
Производительность — это количество выработанного про-
дукта или переработанного сырья в единицу времени
П = В/т (1.1)
где П — производительность;
В — количество продукта;
т — время.
Производительность измеряется в килограммах в час (кг-ч-1),
в тоннах в сутки (т-сут-1) или в кубических метрах в сутки
20
(м3-сут-1) и т. д. Максимально возможная производительность,
называется мощностью.
Для сравнения работы аппаратов и установок различного
устройства и размеров, в которых протекают одни и те же хими-
ческие процессы, используется понятие интенсивность.
Интенсивность — это производительность, отнесенная к.
какой-либо величине, характеризующей размеры аппарата, — к его-
объему или сечению; она, например, может быть выражена в виде
уравнения
.где Vr—объем аппарата (реактора).
Интенсивность может измеряться количеством продукта, полу-
чаемого в течение единицы времени с единицы объема аппарата,
например (кг-ч-1-м~3), или с единицы сечения аппарата
(т • сут-1 • м-2) и т. д.
Расход сырья, воды, энергии и различных реагентов, отнесен-
ный к единице целевого продукта, называют расходным коэф-
фициентом
₽=Q/B (1-3)
где Q — расход сырья, реагента и др.
Расходные коэффициенты выражают в тоннах на тонну (т.-т-1),.
кубических метрах на тонну (м3-т-1), киловатт-часах на тонну
(кВт-ч-т-1) и т. п.
Глубина протекания реакции, от которой зависит степень ис-
пользования сырья и другие показатели химико-технологического
процесса, характеризуется степенью превращения и выходом про-
дукта, а для сложных реакций, кроме того, селективностью.
Степень превращения — это отношение количества ре-
агента, вступившего в реакцию, к его исходному количеству.
Например, для простой необратимой реакции типа А -> R сте-
пень превращения выражается уравнением
где Хд—степень превращения реагента А;
NА 0’ А — количество исходного реагента А в начале и конце процесса.
Степень превращения выражают в долях или в %, в последнем
случае
N, п-Х.
= Л^° Л-Ю0 (1.5)
Из уравнения (1.4) следует, что
na-na.^~xa) ' <L6>
21
Если реакция протекает без изменения объема, то
v о ~са < _ СА
СА.О СА.О
та,еСА0, Сд—концентрация исходного реагента А в начале и в конце про-
цесса.
Из уравнения (1.7) находим, что
сл = сл.о(1-^)
(1.7)
(1-8)
В тех случаях, когда объем реакционной смеси изменяется в
течение процесса, в функциональную зависимость C=f(XA) вво-
дят относительное изменение объема системы, выражаемое урав-
нением
V —V V
хл=1 хд-о
ухл=о Гхл=о
(1-9)
где бд — относительное изменение объема системы;
объем реакционной смесн при ХА = 0 и ХА = 1.
В частном случае — при линейном изменении объема реакцион-
ной смеси во времени — зависимость V от степени превращения
можно записать
V = V0(l+eAXA) (1.Ю)
С учетом уравнений (1.6) и (1.10) текущая концентрация ис-
ходного реагента может быть выражена в виде
С ,*А ^.оО-Хд)
А V ^о(1+еЛ)
Но Na, о/Vo = С а, о, поэтому
1 ~ХЛ
с = с — А а’°1+*аха (1.11)
или
с. п — С .
X — Л. 0 А А СА.0 + СА*А (1.12)
Значение отношения VxA-i/Vхл=о Для реакции
’ аА + 6В+ ... =r/? + sS+ ... (1.13)
«определяют по уравнению
(I. 14)
где § — доля стехиометрической смеси исходных реагентов в реакционной
смеси.
Для стехиометрической смеси (3=1.
Выход продукта — это отношение количества полученно-
го целевого продукта к его количеству, которое должно быть
22
получено по стехиометрическому уравнению. Для необратимой
реакции A -+R имеем
Л nr
Ф„ =----—
R Nn
R, max
(1.15)
где ®R ~ выход целевого продукта;
Nr — количество продукта R в конце процесса;
Nr max — максимально возможное количество продукта R.
Однако в данном случае Nn, max = NA, 0, a NR = NA, с — NAt
поэтому для необратимых процессов
ф - na.0-»a_x>
R~ ^.max “ *A,Q A
Для обратимых реакций важным понятием является равно-
весная степень превращения; для реакции А R она
описывается уравнением
(I. 16)
ЛЛ~ V
• Д. о
где X*—равновесная степень превращения;
Ид — количество исходного реагента А в состоянии равновесия.
Для обратимых реакций Nr,max = поэтому
Л Nr
ф/? = —<
R К*
Ut г»
где Nr — количество продукта R в состоянии равновесия.
Но Nr = Na. о — Na, a Nr = Na, оХа, поэтому из уравнения (I. 18)
следует, что для обратимой реакции (I. 15)
ф Nr NA,0~NA __ ХА
R Nr Na 0Х'а Хд
(I- 17)
(1.18>
(I-19)
Уравнения (I. 14) и (I. 19) справедливы также по отношению к
простым необратимым и обратимым реакциям, протекающим с из-
менением объема реакционной смеси. Например, для реакций
аА ЬВ Ц- ... ——► rR sS .
” аЛ + 6В+ ... rR + sS + ...
1Имеем
фд= = Nr = a ^А'° = NA.o-Na = „ ^R.max JL/у Na,q A a A.o
.< ф - Nr ___t(Na.Q-Na') NA.a-NA XA
"I R Л * ъ 1 >< *i
23
Селективностью называется отношение количества целе|
того продукта к общему количеству получаемых продуктов. Селек^
тивность характеризует процессы, в которых протекают сложные
параллельные и последовательные реакции с получением несколь-
ких продуктов, что часто встречается на практике. Например, если
в процессе протекают параллельные реакции
и целевым является продукт R, то селективность будет выражаться
в виде
nr
(1-20)
п О
.«•де <рд — селективность;
AfD> — количество продукта R и S.
Поскольку для рассматриваемой параллельной реакции NR-}-Ns=a
—Na, о — Na, то
Фд---д? _ N 21)
;УЛ,0 А
Чтобы установить связь между X, Ф и <р,
NA NА
часть уравнения (1.15) на » тогда с
(1.4) и (1.21) получим
ф - Na.0~na nr
R NА, 0 N А, 0 NA, Q~ NА
умножим правую
учетом уравнений
= ХЛФЛ (I 22)
Полученную зависимость Ф = {(Х, ср) можно записать в таком
виде:
Ф == X — для простой необратимой реакции (Д -> R)
Ф — Х/Х* — для простой обратимой реакции (Д R)
(
ф = Х<р — для сложной реакции I А
\ >S
В производственных условиях с целью уменьшения расходных
коэффициентов сырья стремятся иметь возможно более высокие
.'Значения X, <р и Ф.
Скорость химической реакции определяется количе-
ством прореагировавшего исходного вещества или количеством
полученного целевого продукта в единицу времени в единице
объема системы. Например, скорость реакции А -*• R выражается
уравнением
1 dN, 1 dN„
~rA V'~dx rx==V" dr (I
тде Гд, rR — скорости реакции по отношению к исходному реагенту А и по от-
ношению к целевому продукту /?;
V — объем системы;
х — время.
24
В уравнении (I. 23) изменение числа молей прореагировавшего-
исходного реагента А отнесено к единице объема системы. Это из-
менение можно отнести к различным величинам и, таким образом,
получить следующие уравнения
ГА = 1 dNA dx (I.24>
1 dN.
rA = “77 A dx (1.25)
Iff A ~ i m dNA dx (1.26)
на которой происходит реакция;
фаз,
где F — поверхность соприкосновения
р'г— объем реактора;
т — масса твердого вещества.
Выбор уравнения для определения скорости реакции обуслов-
лен удобством его использования; иногда более приемлемым
может оказаться уравнение, отличающееся от приведенных выше.
Если объем системы не изменяется в течение реакции, тогда из-
уравнения (I. 23) следует
dNA
_______v_
A dx
ИЛИ
dCA
dx
(1.27>
г - dC А
A dx
Скорость реакции зависит от концентрации реагирующих ве-
ществ и температуры. Скорость реакции можно выразить
в виде
(1.28>
dC.
-fA — ~lT-kCA
где k — константа скорости реакции.
Подставив в уравнение (I. 29) значения Са из уравнения (I. 8)
находим для реакции первого порядка
d (с л. о О — *л)] , г ,< у ,
---------о С1 “ ха)
(1-29>
или
dXA
-ir = kV~XA)
Для реакции второго порядка 2А -> 7? имеем
dC.
dx л
или после подстановки значения СА из уравнения (I. 8) находим
dXA
(1.30>
(131)
(132)
25
41
Скорость химико-технологического процесса является функцией
скоростей физических и химических процессов и в общем виде
может быть выражена уравнением
r==f (ГфР Гф2.Гфп- ГХ1- Гх2' • • • Гх«) <L 33>
где г — скорость химико-технологического процесса; - й
гф1’ ГФ2’ гфл-~ СКОРОСТИ физических процессов; "г
г.,,.... — скорости химических процессов. «
X1 Хл ЛП
3. МАТЕРИАЛЬНЫЙ И ЭНЕРГЕТИЧЕСКИЙ БАЛАНСЫ
При разработке химико-технологических процессов проводятся
разнообразные расчеты для количественной оценки протекающих
•операций, а также для определения оптимальных значений пара-
метров технологического процесса. Во всех случаях при расчетах
учитываются законы гидродинамики, тепло- и массопередачи и
химической кинетики, поэтому расчеты материальных потоков
обычно сочетаются с энергетическими расчетами, для этого со-1
ставляют материальный и энергетический балансы.
Материальный б ал аир— это вещественное выражение
закона сохранения массы вещества, согласно которому во всякой
замкнутой системе масса веществ, вступивших во взаимодействие, I
равна массе веществ, образовавшихся в результате этого взаимо-
действия, т. е. приход вещества 2Вприх равен его расходу SBpaCx. '
Таким образом, уравнение материального баланса можно предста-
вить в виде
прих ~ у ^расх (1.34)
Для периодических процессов материальный баланс составля-
ют в расчете на одну операцию, для непрерывных процессов — за
единицу времени.
Материальный баланс составляют по уравнению основной
суммарной реакции с учетом параллельных и побочных реакций.
Он может быть составлен для всех веществ, участвующих в про-
цессе, или только для одного какого-либо вещества. Обычно учи-
тываются не все протекающие реакции и получаемые побочные
продукты, а лишь те, которые имеют существенное значение, т. е.
материальный баланс носит приближенный характер.
Материальный баланс составляют для процесса в целом или
для отдельных его стадий. При этом учет массы веществ произво-
дится отдельно для твердой, жидкой и газовой фаз, поэтому в
общем виде материальные балансы выражаются обычно в виде
уравнения
Вт + 5ж + 5г = 5т + 5ж + Д 35)
где Вт, Вж, Вг — массы твердых, жидких и газообразных веществ, поступаю-
щих в производство или на данную операцию в единицу вре-
мени;
Вт, Вж, Вг — массы получаемых продуктов.
26
Иногда при проведении практических расчетов могут не при-
умяться во внимание отдельные фазы (твердая, жидкая или
газообразная) либо в одной какой-либо фазе учитывается сущест-
вование нескольких разных веществ, поэтому уравнение (1.35)
соответственно упрощается или усложняется. При проектировании
обычно задаются массой целевого продукта; массу сырья и массу
| побочных продуктов определяют по уравнению материального ба-
ланса.
Примером составления уравнении материального баланса
отдельных аппаратов могут служить материальные балансы реак-
торов (см. гл. VI). Для того чтобы ознакомиться с составлением
материального баланса химического процесса, в результате кото-
рого образуется несколько продуктов, в качестве примера соста-
3 вим материальный баланс печи кипящего слоя КС-450 для обжига
I флотационного колчедана.
I Флотационный колчедан содержит минерал пирит FeSj, при обжиге которого
i образуется сернистый ангидрид SO2, служащий сырьем для получения серной
( кислоты.
Суммарная реакция обжига колчедана описывается уравнением
4FeS2 + ПО2 = 2Fe2O3 + 8SO2 + 3416 кДж (а)
В процессе обжига небольшое количество образующегося сернистого ангидрида
окисляется до серного ангидрида
| SO2 ^/2^2 ч—SO3 -|- 96 кДж (бу
Так как поступающий в процесс флотационный колчедан и кислород, пода-
ваемый с воздухом, содержат влагу, при составлении баланса в статье «Приход»
помимо колчедана и воздуха необходимо учитывать влагу колчедана и пары
воды из воздуха.
В статье «Расход» необходимо учитывать массу Fe2Os (огарка) и массу ос-
новных компонентов, входящих в состав образующейся газовой смеси.
С учетом приведенных данных общее уравнение материального баланса в
рассматриваемом случае запишется в виде
Вкол + ввл. кол + 5 воз + 5вл. воз = 5ог + 5SO2 + 5SO3 + SO2 + 5N2 + SH2O (B)
где ВКОл, Ввл. кол- 5воз- Ввл. воз — соответственно масса сжигаемого колчедана
(сухого), воды в колчедане, воздуха (сухого),
воды в воздухе, кг • ч-1;
Вог, BSOi, ^sos> 50г> Bn2- ®н2Осоответственно масса образующегося огарка,
SO2, SO3, О2, N2 и Н2О, кг • ч-’.
Условия расчета-.
Производительность печи КС-450 в пересчете на
серную кислоту (100% H2SO4), кг-ч-1 . . . .
Степень Использования серы в колчедане . . .
Содержание, %
серы в сухом колчедане . . . : ..........
влаги в колчедане .......................
серы в огарке...........................
SO2 в сухом обжиговом газе .............
SO3 в сухом обжиговом газе..............
Относительная влажность воздуха, % . . . . ;
BH2so4 — 20 833
Р = 0,885
С,, = 41
^-ВЛ = 6
СОг — 1
6’sO2 = I4,3
cso3 ~ 0,1
Ф = 50
27
Расчет:
Количество серы, колчедана, требуемое для обеспечения производительности печи;
МsBHlSo, 32,06 • 20 833 • 1000
*5= ‘W='
Масса сухого колчедана:
Bs• 100
Вкол = ~
Gs
98,08 • 885
7695-100
41
7695 кг • ч-1
18 768 кг • ч_
Масса влаги в колчедане:
ВВкол^-вл
вл. кол
18 768 - 6 (<пп
100-Свл 100-6 1198 к1"4
Массу огарка определяем из формулы:
(160 - cs) Й 160-41
Bor = k |Г,А. = -q6Q~—f • •18 768 = 0,748 • 18 768 = 14 038 кг - ч->
160 — cs (ог)
Масса серы в огарке:
5S (or)
Bpr^-S (or)
100
14 038-1
“TOO------140КГ.Ч-»
Потери серы с огарком:
&S (ог) ‘ *°°
BS
140-100
~7695 = 1’82%
Масса выгоревшей серы:
Bs - Bs (ог) = 7695 - 140 = 7555 кг • ч~’
Общий объем SO2 + SO3:
(Bs-Bs(or))22,4 7555-22,4
V SOj+SOj “
Afs
32,06
5279 м3 • ч—1
Здесь и далее в расчете объем газов приведен к нормальным условиям. Объем
'SOat
v _ VSOH-SO,CSO2 5279-14,5 *
VsO1 cso + cso “ 14,5 + 0,1 -5243 м 4
Объем SO3:
Vsos= ^soj+so, ^soa “ 5279 — 5243 =36 м3 • ч-1
Концентрация кислорода в обжиговом газе определяется по формуле:
Г — „-Fm- п(т~ !)1г Гт. ПК Л (/Л —0,5) 1л
СОг~П { JOO J CsO= “ L + °’5-------100--J CsO2
где л —содержание кислорода в воздухе (л = 21%);
т — стехиометрическое отношение числа молекул кислорода к числу моле-
кул двуокиси серы (т = 1,375):
= 21 - 1,375 - —~ 14,5 - 1,375 + 0,5 21 °’5) 0,1 -2,06%
28
Объем
Объем
сухого обжигового газа:
Учп • 100 5243 100
Иг =—2^--------=----—-— =36 159 м3 ч-1
i/SO2 14’5
кислорода в обжиговом газе:
VTCn 36 159-2057
Vn = =----------= 744 м3 • ч-1
°* 100 100 М
азота в обжиговом газе:
Объем
у == Иг - (rSo2 + Ks0. + ЕОг) = 36 159 - (5243 + 36 + 744) = 30 136 м3• ч->
Объем сухого воздуха/поступающего на обжиг колчедана (воздух содержит
79% N2):
VN • 109 30 136-100
Vbo3 = -7^-------------79-------38 147 м3 • ч~1
Объем паров влаги в воздухе (при 20 °C и относительной влажности воздуха
<р = 50%, давление паров воды в нем равно Рн2о = ^’^ мм Рт- ст-);
^возРн2О _ 38 147-8,77 _ з
VH2O - 7б0 _ рНг0 7Q0 - 8,77 445 М ‘ 4
Общий объем паров воды в обжиговом газе:
• ВВл-22,4 1198-22,4
инго—44/7 18
-wh,o
Полученные при расчете данные приведены в табл. I. 1.
4- 445 = 1936 м3 • ч -1
Таблица 1.1. Материальный баланс печи КС-450
Приход Количество Расход Количество
кг-ч 1 м3.ч-> кг-ч 1 м3*ч 1
Колчедан Влага колчедана Сухой воздух Влага с воздухом 18 768 1 198 49 400 358 38147 445 Огарок Обжиговый газ SO2 SO3 о2 n2 Н2О 14 038 15 337 129 1 063 37 600 1 556 5 243 36 744 30 136 1 936
Всего . . . 69 724 , 38 592 Всего . . . 69 724 38 095
В основу энергетического баланса положен закон
сохранения энергии, согласно которому количество энергии, вве-
денной в процесс, равно количеству выделяющейся энергии, т. е.
приход энергии равен ее расходу.
29
Химико-технологические процессы связаны с затратой различ-
ных видов энергии — тепловой, механической, электрической.
Поскольку в этих процессах тепловая энергия имеет наибольшее
значение, для них обычно составляют тепловой баланс. В этом
случае закон сохранения энергии формулируется так: приход
тепла в данной производственной операции SQnpi,x должен быть
равен расходу тепла в той же операции SQpacx, т. е.
X Сприх — расх (1.36)
Тепловой баланс составляют по данным материального баланса
с учетом тепловых эффектов химических реакций и физических
превращений, протекающих в аппарате, а также с учетом подвода
или отвода тепла. В общем виде тепловой баланс выражают урав-
нением:
QT + Q,;K + Qr+Q(1, + Q,.> + Qn=Q; + Q; + Q; + Q;, + Q; + Q,n (1.37)
где QT, Q», Qr — тепло, вносимое с поступающими в аппарат твердыми, жид-
кими и газообразными материалами;
QT, Qx, Qr — тепло, уносимое выходящими материалами;
<2ф, (?ф — тепло, выделяемое и поглощенное при физических процессах;
Qp, Qp — тепло экзо- и эндотермических реакций;
Q , Qn — тепло, подводимое в аппарат извне и выводимое из аппарата.
Некоторые из приведенных статей прихода (или расхода) мо-
гут отсутствовать, тогда уравнение (1.37) соответственно упро-
щается.
Для рассмотренного ранее процесса обжига колчедана в печи КС-450 общее
уравнение теплового баланса (1.37) запишется в виде:
Скол 4" Сал. кол + Своз 4" Сал. аоз 4- Ср — Сог 4- Сг 4- Сф 4- Сп (г)
где Скол* Свл. кол, Своз* Саз. воз ~ количество тепла, вносимое в печь сухим воз-
духом и влагой воздуха, кДж • г1;
Ср — количество тепла, выделяющегося по реакции
(а), кДж • ч-1;
Сог, Сг — количество тепла, выносимое из печи огарком
и обжиговым газом, кДж ч*1;
Сф — количество тепла, расходуемое на испарение
воды, содержащейся в колчедане, и иа испа-
рение воды с целью получения пара, кДж •
• »г1;
Сп — потеря тепла в окружающую среду, кДж ч-1.
В уравнении (г) не учитывается тепло образования серного ангидрида по
реакции (б), так как оио мало.
Результаты расчета теплового баланса процесса обжига флотационного кол-
чедана показывают, что в статье «Приход» около 91% тепла составляет тепло
реакции (табл. 1.2).
Основное количество выделяющегося тепла уносится образующимся обжи-
говым газом (45,3%) и расходуется для получения пара (45,5%).
Пар является побочным продуктом, его стоимость высокая, поэтому его
получение и использование в процессе либо па стороне имеет большое практиче-
ское значение для снижения себестоимости основного (целевого) продукта.
30
Таблица 1.2. Тепловой баланс печи КС-450
~~ Приход Количество РАСХОД Количество
10 3кДж-ч 1 % 10~3кДж-ч 1 %
Тепло сухого чедана Тепло влаги чедана Тепло сухого духа Тепло влаги духа Тепло горения чедана кол- кол- воз- воз- кол- 204 101 993 13,4 100 656 0,20 0,10 0,97 0,01 98,72 Тепло огарка Тепло обжигового газа Тепло на получе- ние пара в ки- пящем слое Тепловые потери 8 380 46 128 46 437 1 022 8,2 45,3 45,5 1,0
Всего ... 101 954 100 Всего ... 101 967 100
Потери в окружающую среду малы (около 1%), однако и они непрерывно
•снижаются, поскольку единичная мощность как производственных агрегатов, так
и печей КС увеличивается.
4. КАЧЕСТВО ПРОДУКЦИИ
В большинстве случаев качество химических продуктов опре-
деляется их чистотой или содержанием в них основного вещества.
Производство высокочистых или концентрированных продуктов
важно не только с точки зрения качества конечного продукта,
идущего на народное потребление, но и по многим другим причи-
нам. Например, применение концентрированных продуктов повы-
шает интенсивность процессов, в которых они используются в
качестве сырья. Это приобретает особое значение для химической
промышленности, продукция которой представляет собой глав-
ным образом сырье или средство производства, а не предмет
непосредственного потребления.
Повышение концентрации полезного вещества в многотоннаж-
ных химических продуктах имеет большое практическое значение
также с точки зрения экономии на транспортных перевозках, что
может быть показано на примере перевозки фосфорных удобре-
ний.
Государственными планами развития нашего народного хозяйства предусма-
тривается увеличить производство, минеральных удобрений к концу 1980 г. до
ст Т в ГОд- Из этого количества примерно 30% приходится на производ-
во фосфорных удобрений, что составляет около 45 млн. т в пересчете на про-
УдобРение (суперфосфат), содержащее 18,7% Р2О5; присутствующий в этих
орениях балласт (81,3%) удорожает перевозку удобрений. При увеличении
леп^СКа конЦентРиРованных удобрений, в частности двойного суперфосфата, со-
Ржащего 45—56% Р2О5 (в среднем 50% Р2О5), получится экономия средств,
31
которую нетрудно определить. (В СССР фосфорные удобрения перевозятся в
среднем на расстояние 1000 км и стоимость перевозки удобрений железнодорож-
ным транспортом на такое расстояние составляет 3 руб. за 1 т). Если концентра-
ция Р2О5 в удобрениях возрастает с 18,7 до-50%, то экономия составит
Э = (4,5 107 — 1"~ 3 = 84 600 000 руб.
\ OU /
Необходимо также учитывать, что экономия от повышения доли концентри-
рованных удобрений возрастет за счет перевозки удобрений от железнодорож-
ной станции, где разгружаются удобрения, до места их применения, а также
при внесении удобрений в почву.
Особенно большие требования к чистоте химических продуктов
предъявляются при производстве химических реактивов и особо-
чистых веществ, применяемых в некоторых технологических про-
цессах. Так, например, содержание окислов азота и хлора в реак-
тивной серной кислоте должно быть не более 1 • 10~4%, а содержа-
ние мышьяка— не более 3-10~6%. Большое значение имеет содер-
жание примесей в продуктах, применяемых в качестве полупро-
водниковых материалов и в ядерной технике.
Качество каждого химического продукта, т. е. его состав и
свойства, должны удовлетворять требованиям, изложенным в
государственных стандартах (ГОСТ), где указываются основные
показатели, характеризующие продукт или предмет.
По мере совершенствования химико-технологических процес-
сов, а также по требованию потребителей химических продуктов
стандарты систематически пересматриваются. При этом прово-
дится большая исследовательская работа по оценке возможностей
промышленности, вырабатывающей тот или иной продукт, а также
по установлению обоснованности выдвигаемых потребителями
требований. В результате такой работы составляется новый стан-
дарт, в котором предусматриваются более качественные показа-
тели продукции (все или некоторые из них).
Десятая пятилетка названа пятилеткой эффективности и ка-
чества. При этом проблема качества понимается очень широко.
«Она охватывает все стороны хозяйственной деятельности» ...
«Вот почему на повышение качества продукции должны быть
нацелены весь механизм планирования и управления, вся система
материального и морального поощрения, усилия инженеров и
конструкторов, мастерство рабочих» *.
Таким образом, вопросам повышения качества продукции в
ближайшие годы будет уделяться особое внимание, и, следова-
тельно, должна быть проделана большая работа по пересмотру
существующих стандартов.
* Л. И. Брежнев, Отчет Центрального Комитета КПСС и очередные задачи ‘
партии в области внутренней и внешней политики, Политиздат, Москва, 1976 г.> |
стр. 53. ' J
32
5. ЭКОНОМИЧЕСКАЯ ЭФФЕКТИВНОСТЬ
ХИМИЧЕСКОГО ПРОИЗВОДСТВА
Экономическая эффективность является самым важным показа-
телем, характеризующим совершенство химико-технологического
процесса. Она зависит, во-первых, от мощности установки, на
которой вырабатывается химический продукт, и, во-вторых, от
того, насколько полно использованы новейшие достижения науки
и техники при разработке и оформлении данного технологического
процесса.
Экономическая эффективность характеризуется тремя основны-
ми показателями: капитальные затраты, себестоимость продукции
и производительность труда. Все эти показатели тесно связаны
между собой и должны рассматриваться в комплексе.
Капитальные затраты — это сумма всех затрат, произ-
веденных при строительстве данного цеха или предприятия в це-
лом. Естественно, что во всех случаях следует стремиться к тому,
чтобы эти затраты были минимальными. Однако применение
новой, более совершенной аппаратуры, коррозионно-устойчивых
материалов, автоматических методов контроля и регулирования
процесса связано с увеличением материальных затрат, но одно-
временно приводит к снижению эксплуатационных затрат и себе-
стоимости продукции. Поэтому в каждом отдельном случае при
разработке проекта все эти вопросы решаются с учетом конкрет-
ных условий.
Удельные капитальные затраты — более наглядный показатель,
чем капитальные затраты; они получаются от деления общей
стоимости установки (цеха) на ее годовую мощность
P = KIQ (1.38)
где Р — удельные капитальные затраты, руб • т-1 • год-1;
К. — капитальные затраты, руб;
Q — мощность установки, т • год*1.
Например, стоимость одной контактной сернокислотной систе-
мы мощностью Q = 360 тыс. т серной кислоты в год составляет
/( = 17 600 тыс. руб. Следовательно, удельные капитальные затра-
ты равны
D 17 600 000 ,оо , , .
Р = X/Q = 36000 = 48,9 руб т-> год-1
С увеличением единичной мощности установки удельные капи-
тальные затраты снижаются. Эмпирическое уравнение для выра-
жения зависимости удельных капитальных затрат от единичной
Мощности установки, т. е. одного производственного агрегата,
имеет такой вид
? = aQ-0’4 (1.39)
Где а коэффициент, зависящий от характера химического производства.
2 Зак. 558 \ оо
В качестве примера определим снижение удельных капиталь-
ных затрат при увеличении единичной мощности установки в два
раза, т. е. при изменении мощности от Qi до Q2 = 2Qi. Для этого
подставим в уравнение (1.39) принятые значения и определим
отношение
Р2 aQ70A 2Q~°’i / 1 \0’4'
— = 4 ----— = -) =0,76
?! aQj~0’4 С-0’4 \2j
ИЛИ
Р2 = 0,76?,
Таким образом, при увеличении мощности установки вдвое
удельные капитальные затраты, а соответственно и общие капи-
тальные затраты снижаются на 24%.
Значение коэффициента а, входящего в уравнение. (1.39), на-
ходят для каждого продукта с использованием практических
данных.
Например, в производстве серной кислоты при мощности установки Q =
= 360 000 т • год'1 удельные капитальные затраты составляют Р = 48,9 руб •
• т-1 • год'1. Подставив эти данные в уравнение (1.39), получим 48,9 =
= а(360 000)-0"4, откуда а = 8,2-103.
Следовательно, уравнение для расчета удельных капитальных затрат для
сернокислотных установок будет иметь вид
? = 8,2 • 1O3Q~0,4 (1.40)
Аналогично устанавливают зависимость Р = f(Q) и для других химических
производств.
Полной себестоимостью называется денежное выражение
затрат данного предприятия на изготовление и сбыт единицы про-
дукции. Затраты предприятия, непосредственно связанные с про-
изводством продукции, называются фабрично-заводской себестои-
мостью, которая слагается из следующих статей:
1) сырье, полуфабрикаты и основные материалы, непосред-
ственно участвующие в химических реакциях производства;
2) топливо й энергия на технологические цели;
3) заработная плата основных производственных рабочих;
4) амортизация — отчисления Ам на возмещение 'износа ос-
новных производственных фондов, зданий, сооружений, оборудо-
вания и др. (приближенно Ам = A/10Q = Р/10);
5) цеховые расходы, включающие затраты на содержание и
текущий ремонт основных производственных фондов (в том числе
и зарплату вспомогательных и ремонтных рабочих), а также за-
траты на содержание административно-управленческого персонала
цеха, охрану труда и технику безопасности;
6) общезаводские расходы.
Из себестоимости основного продукта обычно вычитается стои-
мость побочных продуктов,, полученных из того же сырья.
34
Учет себестоимости ведут по специальной форме (табл. 1.3),
в которой отражаются все элементы себестоимости. Форма являет-
ся одинаковой для всех химических производств.
Таблица 1.3. Элементы себестоимости 1 т фосфорной кислоты
(неупаренной, в пересчете на 100% Н3РО4)
Статьи расхода Количество Стоимость, руб Доля себестоимости, %
Апатитовый концентрат (100% Р2О5), т Серная кислота, т Электроэнергия, кВт ч Пар, МДж Вода, м3 Воздух, тыс • м3 Зарплата с начислениями Амортизационные отчисления Цеховые расходы Общезаводские расходы 1,04 2,42 133 0,84 37 0,7 51,16 40,92 1,81 0,96 0,24 1,56 1.13 5,7 6,04 2,44 82 4,2 1,0 5,1 5,4 2,2
Заводская себестоимость Внепроизводственные расходы 111,96 2,10 100,0
Полная себестоимость 114,06
Соотношение затрат по различным статьям себестоимости
сильно изменяется для различных химических производств. Важ-
нейшей статьей в большинстве случаев являются затраты на
сырье; в среднем по химической промышленности они составляют
60—70% себестоимости. Топливо и энергия в среднем составляют
около 10% себестоимости, однако в электрохимических и электро-
термических производствах электроэнергия представляет одну из
главных статей расхода. Так, например, в производстве элемен-
тарного фосфора на электроэнергию приходится 40% себестои-
мости.
Заработная плата основных рабочих в химической промышлен-
ности невелика ввиду высокой степени механизации и автоматиза-
ции производственных процессов, она составляет в среднем лишь
около 4% себестоимости. Однако в ряде химических производств
зарплата превышает 20% себестоимости. Отчисления на амортиза-
цию составляют обычно 10—15% себестоимости; для фосфорной
кислоты они значительно меньше,'гак как применяемое при этом
оборудование сравнительно простое и дешевое.
В состав себестоимости включены амортизационные отчисле-
ния (что отражает влияние капитальных затрат) и все виды
заработной платы (что отражает производительность труда), по-
этому в первом приближении можно принять, что экономическая
2* 35
эффективность химического производства характеризуется себе-
стоимостью продукции.
Зависимость между себестоимостью и единичной мощностью
производственного агрегата приближенно выражается уравнением
S = mQn (1.41)
где S — себестоимость продукта, руб • т-1;
Q — мощность цеха (установки), т год-1;
т, п — коэффициенты (п — —0,2-4-0,3).
Из уравнения (1.41) следует, что при увеличении мощности
установки вдвое от Qi до Q2 — 2Qi и, принимая п = 0,2, себестои-
мость составит
S, nQ2-0’2 (2QI)~0’2 / Q, \0’2 ( 1 \0'2
—-------П2 =(—Ч =(- =0,87
S, nQ"0’2 Q"0,2 \ 2Q] ) \ 2 /
т. е. себестоимость снизится на 13%.
Производительность труда это количество продукции,
вырабатываемой рабочим в единицу времени, или количество ра-
бочего времени, затрачиваемого на выработку единицы продукции.
Так же как и удельные капитальные затраты и себестоимость
продукции, производительность труда зависит главным образом
от техники производства и мощности установки. С увеличением
единичной мощности установки вдвое производительность труда
для многих химических производств возрастает на 60—80%.
Такое соотношение обусловлено тем, что при внедрении меха-
низации и автоматического контроля и регулирования процесса
нет существенной разницы в обслуживании большого или малого
производственного агрегата, так как наблюдение за протеканием
процесса ведется с помощью установленных приборов. С увеличе-
нием размеров аппаратов затраты труда возрастают лишь при
ремонте этих аппаратов, а также при их пуске и остановке.
Производительность труда является одним из важнейших по-
казателей, характеризующих совершенство химико-технологиче-
ского процесса, .значение этого показателя ярко отражено в мате-
риалах XXV съезда КПСС: «Для того чтобы успешно решать
многообразные экономические и социальные задачи, стоящие перед
страной, нет другого пути, кроме быстрого роста производитель-
ности труда, резкого повышения эффективности всего обществен-
ного производства» *. Для этого надо «...полагаться не на привле-
чение дополнительной рабочей силы, а только на повышение про-
изводительности труда. Резкое сокращение доли ручного труда,
комплексная механизация и автоматизация производства стано-
вятся непременным условием экономического роста **.
Из приведенных данных видно, что с увеличением единичной
мощности установки все показатели, отражающие экономическую
* Л. И. Брежнев, Отчет Центрального Комитета КПСС и очередные задачи
партии в области внутренней и внешней политики, Политиздат, М., 1976, стр. 52.
** Там же, стр. 52.
36
эффективность производства, улучшаются: снижаются удельные
капитальные затраты и себестоимость продукции и повышается
производительность труда. Однако возможности при использова-
нии этого мощного фактора для увеличения экономической эффек-
тивности производства ограничены, так как для выбранного места
строительства мощность нового производства не может быть про-
извольно увеличена. Она имеет строго определенное, оптимальное
значение, зависящее от многих факторов, которые не могут про-
извольно изменяться: от потребности в вырабатываемом продукте
в районе строительства предприятия, от возможности изготовле-
ния аппаратуры большой мощности, от стоимости перевозки сырья
и получаемого продукта и др. В связи с этим к разработке проекта
строительства нового химического производства приступают лишь
после составления технико-экономического доклада (ТЭД), где
соответствующими расчетами устанавливаются оптимальная мощ-
ность предприятия и некоторые другие показатели, закладывае-
мые в проект.
Примеры
Пример 1.1. Определить степень превращения SO2 по реакции
2SO2 + О2 <-Е- 2SO3 ' (а)
или
аА + ЬВ rR (б)
где а — 2; b — 1; г = 2.
Условия:
Состав реакционной смеси в начале процесса,
% (об.)
сернистого ангидрида......................CSO2 0=Сд_ о=7,5
кислорода................................... СО2 = 10,3
азота..................................... CN = 82,2
Содержание сернистого ангидрида в реакционной
смеси в конце процесса, % (об.)...................б’дО=.Сз = 2,5
Решение:
Вначале по уравнению (1.9) определим коэффициент изменения объема, а
затем по уравнению (I. 14) найдем степень превращения
Ел-^-1/^=0-!
Так как в исходной реакционной смеси содержатся SO2 и воздух (доля кис-
лорода в котором представляет собой избыток по отношению к стехиометрии для
реакции окисления SO2, а кроме того, в ней содержится балластный азот), расчет
отношения V Xa_1/Vx 0 ведут по уравнению (1.14)
В соответствии с уравнением реакции (а) и'с учетом условий примера доля
^метрической смеси исходных реагентов в общей реакционной смеси соста-
-|-С. п— 7,5+ 7,5-4-
л'° л’° а _ 2 = л 1125
100 - 100 -б.1125
с
₽—:
37
Следовательно
*%=1/^л=о - 1 - 0,1125 (1 - = 0,9625
Подставив полученные значения в уравнения (1.9) и (1.12), находим
VX 1
е . --- 1 = 0,9625 - 1 = - 0,0375
С, . - С, 7,5 - 2,5
Ха ~ СА,'о + САеА ~ 7,5+ 2,5 (-0,0375) = °’675
или Хл = 67,5%.
Пример I. 2. Рассчитать степень превращения и выход аммиака по реакции
N2 + ЗН2 2NH3 + Q (а)
или
аЛ + 6В rR (б)
где а = 1; b = 3; г == 2. ? '
Условия-.
Известно, что после некоторого промежутка времени в конечной газовой сме-
си стало (Vb,^=30 моль и /Vn,T=20 моль; процесс ведут при / = 500 °C и
Р = 30 МПа. В этих условиях при состоянии равновесия в газовой смеси содер-
жится С^=26,4% NH3.
Решение.
Чтобы определить степень превращения Хв по уравнению (1.5), необходимо
прежде рассчитать количество водорода в газовой смеси в начале процесса NB, оЗ
оно может быть определено из следующего соотношения
^,0=^it + D = 8° + D ’ (в)
где D — количество водорода, вступившего в реакцию с образованием NHs.
Поскольку на 1 моль ЫНз расходуется b/г моль Н2 [уравнение (б)], а общее ко-
личество аммиака в конечной газовой смеси Vr,t = 20 моль, то D= Nв,хЫг—1
==.20-3/2 = 30 моль. Следовательно, NB, о = 80 + 30 = ПО моль.
Подставив полученные данные в уравнение (1.5), находим
Увп+Ув 110-80
Хд’т== ^.о ~^°0’273
или Хв = = 27,3%.
Чтобы определить выход продукта Фй по уравнению (I. 18)', необходимо
вначале определить количество аммиака в конечной газовой смеси, соответствую-
щее состоянию равновесия (Nк), исходя из того, что содержание аммиака в этой
смеси составляет Сд = 26,4%.
Искомое значение находим по уравнению
n*r
. = 0,264
ЛГд + ЛГсм
где М*м — количество азотоводородной смеси в условиях равновесия (свобод-
ной), моль.
Но Мсм можно определить также из соотношения
(г)
Ь<м = ^см, 0 = ^,0-^
где JVcm, о—количество исходной азотоводородной смеси (до синтеза), моль.
38
Подставив значения коэффициентов и NB, о = НО моль, находим
Nr "ПГ- + С = ’ ’О -Цр- = 146,67 (д)
Из уравнений (г) и (д) находим
или Nr = 30,63 моль.
Подставив полученные величины в уравнение (I. 18), находим
Л’ 20,0
фл> = тЛ = "57?7я' = °’653
Я N^ 30,63
или Фв = 65,3%.
Из приведенных данных видно, что для рассмотренных условий разница ме-
жду Хв и Фв значительная: Хв = 27,3%, а Фв = 65,3%.
Выход продукта в рассматриваемом примере можно также определить по
уравнению
= (ЫЭ)
Для этого необходимо кроме Хв рассчитать также равновесную степень пре-
вращения Хв по уравнению
* b
М* Nr —
* NB _ К Г
ЛВ~ ft М
в, о в, о
где N*B — количество водорода, которое взаимодействует с азотом до образова-
ния аммиака в состоянии равновесия, моль.
Подставив коэффициенты и найденные значения N % и NB, 0, находим
X* = 30,3 ' У2 = о 4177
В по
С учетом полученных значений Хв и Х*в выход будет равен
X 0 273
Фв == —=-----------= 0,653, или 65,3%
R Хв 0,4177
Пример I. 3. Определить удельные капитальные затраты (УКЗ) и их сниже-
ние при увеличении мощности установки, а также вывести уравнение функцио-
нальной зависимости УКЗ от мощности.
Условия:
Общая стоимость установки по производству фосфорной кислоты мощностью
(одной технологической линии) Qi = 150 тыс. т-год"1 (в пересчете на 100%
Н5РО4) составляет К = 4,885 млн. руб. 1
Мощность установки возрастает до Qs = 250 тыс. т-год-1.
Решение.
1. Подставив значения К и Q в уравнение (1.38), находим'
К _ 4 885 000
Q 150 000
—'32 руб. 60 коп.
39
2. Зная величину Р, по уравнению (J. 39) получаем
32,6== а • 15OOOO-0,4
откуда а = 3,84 • 103. Следовательно, подставив а в уравнение (1.39), находим
Р = 3,84 • 103Q-°’4 (а>.
3. По уравнению (а) определяем снижение удельных капитальных затрат
Р2 3,84-1O3Q“0’4 /Q]V0’4 ( 150 А"°’4
Рх ~ 3,84- 103Qj-0’4 ~ V Q2) ~ \250 7 “ ’
или Рг = 0,81 Pi-
' Следовательно, снижение капитальных затрат составляет 19%.
Глава II
ФИЗИКО-ХИМИЧЕСКИЕ ЗАКОНОМЕРНОСТИ
В ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
Основное внимание в главе уделено закономерностям химиче-
ских процессов (взаимодействий), поскольку они относятся к
области химической технологии и обычно являются наиболее от-
ветственным элементом химико-технологического процесса.
Взаимная связь закономерностей химико-технологического про-
цесса очень сложная и выражается в виде системы уравнений. По-
скольку совместное решение этих уравнений практически невоз-
можно, приходится рассматривать отдельные стороны процесса и
их взаимное влияние. Прежде всего необходимо установить осо-
бенности протекания основной химической реакции, определяющей
выход целевого продукта, с какой полнотой и какой скоростью
протекает эта реакция.
В связи с этим рассмотрим с точки зрения химической техно-
логии некоторые вопросы, относящиеся к особенностям химических
реакций.
1. КЛАССИФИКАЦИЯ ХИМИЧЕСКИХ РЕАКЦИЙ
Существует ряд признаков, по которым можно классифициро-
вать химические реакции. Так, например, реакции делятся на про-
стые и сложные. При протекании простой реакции типа
А —► R (И. 1>
ИЛИ
Д-Р в —> R (11.2)
достаточно одного стехиометрического уравнения для ее описания.
К сложным относятся реакции, состоящие из двух или более
взаимосвязанных между собой простых реакций: например парал-
лельные реакции:
40
* последовательные реакции
j А ->R-> N (II. 4)
- Для описания сложных реакций необходимо несколько стехио-
* метрических уравнений.
' Химические реакции подразделяются на обратимые и необра-
3 -тимые. В действительности все реакции обратимы, так как при
'' соответствующих условиях любая реакция может протекать с за-
i метной скоростью как в прямом, так и в обратном направлении.
’• Однако в рассматриваемых условиях многие химические реакции
протекают в одном из направлений с ничтожно малой, практиче-
ски нулевой скоростью, поэтому такие реакции условно считают
необратимыми. Например, реакция поглощения СО2 известковым
> молоком
й СО2 + Са(ОН)2 —> СаСО3 + Н2О
! практически необратима, так как образующаяся соль СаСО3 мало
растворима в воде и выпадает в осадок.
С точки зрения кинетики химические реакции классифицируют-
• ся по молекулярности реакции либо по порядку реакции.
Молекулярность реакции характеризуется числом молекул, при
одновременном взаимодействии которых происходит элементар-
‘ ный акт химического взаимодействия. По этому признаку химиче-
ские реакции можно разделить на моно-, би- и тримолекулярные.
Можно говорить и о более высокой молекулярности, но, в дей-
? ствительности, одновременное столкновение даже трех молекул
‘ уже является маловероятным. В тех случаях, когда уравнение
реакции указывает, что в реакции принимает участие большее
число молекул, сумма стехиометрических коэффициентов а и Ь в
» уравнении
аА + ЬВ -> rR + sS ± Q (II. 5)
«у
больше трех. Процесс протекает практически сложнее и идет че-
рез две или большее число стадий. В каждой из этих реакций
взаимодействие осуществляется при столкновении двух и редко
? трех молекул.
Порядок реакции п равен сумме показателей степени у концен-
траций в уравнении, выражающем зависимость скорости реакции
от концентрации реагирующих веществ. Так, например, для реак-
ции (II. 5), рассматриваемой в качестве модельной, скорость пря-
мой реакции выражается уравнением
| г = kCaACbB (II. 6)
। Исходя из определения, порядок этой реакции равен п — а -ф- Ь.
?; По порядку реакции разделяются на реакции первого, второго и
; третьего порядка. Известны реакции, порядок которых выражает-
s «я дробным числом и нулем (реакции нулевого порядка). Порядок
§ Каталитических реакций почти всегда ниже, чем их молекуляр-
I ЙОсть- Например, для реакции (II. 5) теоретическое уравнение
скорости прямой реакции выражается уравнением (II. 6), а фак*
тические показатели степени для реакции, протекающей в при-
сутствии. катализатора, будут ниже
r = kCa;cb’
где а' < а, Ь' <. Ь.
Следовательно, порядок реакции п= (а'’-j-b') < (а+ 6).
Если один из реагентов находится в большом избытке, то его
концентрация изменяется незначительно в течение процесса и
может быть принята постоянной. В этом случае уравнение ско- :-
роста реакции упрощается и порядок реакции снижается. Напри- S
мер, если исходный реагент А будет находиться в большом из- |
бытке, уравнение скорости прямой реакции (II. 5) запишется
в виде
r = kCaACbB = k'CbB
где k' = kCaA,
и, следовательно, общий порядок реакции уменьшается.
Порядок реакции п может отличаться от молекулярности реак-
ции т. - |
В производственной практике повышение скорости реакции I
часто достигается путем создания избытка дешевого компонента.
В химической технологии распространена классификация по
фазовому признаку; по которому различают гомогенные реакции,
когда реагирующие вещества находятся в одной какой-либо фазе,
и гетерогенные реакции, когда реагирующие вещества находятся
в разных фазах. По этому же признаку подразделяются и реак-
торы, в которых их осуществляют. 1.
2. ФАКТОРЫ, ВЛИЯЮЩИЕ НА СОСТОЯНИЕ РАВНОВЕСИЯ t
I
При разработке технологического процесса прежде всего не-»*
обходимо установить, какие реакции будут протекать при перера-^
ботке выбранного сырья и насколько глубоко проходит основная
реакция, обеспечивающая получение заданного продукта, т. е. ка-1
ков максимальный выход целевого продукта. *
Определяя изменение изобарно-изотермического потенциала |
(энергии Гиббса) для реакций, которые протекают в рассматри-|
ваемом процессе, и сравнивая их, устанавливают термодинами- f
чески наиболее вероятную реакцию. |
Изменение изобарно-изотермического потенциала ДО либо рас- ’
считывают, либо определяют на основе экспериментальных дан-
ных. Для расчета ДО используют |
AG = ^(«iAG1-)fe-^(«iAGi)H (II. 7)!
где п{ — число молей компонента i, участвующего в реакции; I
AGf — изменение изобарно-изотермического потенциала компонента I. |
42
Из уравнения (II. 7) видно, что величина ДО представляет
собой разность между изменениями изобарно-изотермических по-
тенциалов при образовании из простых веществ конечных продук-
тов и исходных реагентов-. Для реакции (II. 5) из уравнения (II. 7)
следует
AG = (г ЛСобр. д + S ДСобр.я) - (« Д(гобр. А + В AGo6p, в) (II. 8)
При определении ДО на основе экспериментальных данных
обычно исходят из теплового эффекта реакции (найденного опыт-
ным путем). Для этого пользуются уравнением
= — Т bS (II. 9)
где AG— тепловой эффект реакции;
AS — изменение энтропии при реакции.
Значение ДД находят по разности между энтропией конечных
продуктов и исходных веществ
Д5 = £(пА)к-Е(«А)н (И. 10)
Для реакции (II. 5) уравнение (11.10) примет вид
AS = (rSR + sSs) - (aSA + bSB) (11.11)
Значения ДОобр и S находят по справочным таблицам.
Вероятность протекания химической реакции определяется ве-
личиной ДО. Существует следующая зависимость этой величины:
AG >0 — наиболее вероятное течение реакции слева направо,
AG < 0 — наиболее вероятное течение реакции справа налево,
AG = 0 — существует равновесие.
Исходя из уравнения (II.9), ориентировочное представление
о значении ДО и, следовательно, о вероятности той или иной
реакции, протекающей при невысокой температуре, можно полу-
чить на основе теплового эффекта реакции: чем более экзотер-
мична реакция при данной температуре, тем вероятнее реакция.
Представление о том, насколько глубоко может протекать
химическая реакция, можно получить на основании данных о рав-
новесии этой реакции. Условия достижения равновесия имеют
большое практическое значение, поскольку ими определяется пол-
нота использования сырья.
В состоянии равновесия, к которому стремятся все химические
реакции, скорости прямой и обратной реакций становятся одина-
ковыми, а соотношение концентраций компонентов в рассматри-
ваемой системе остается неизменным. Так, например, условия
равновесия реакции (II. 5) можно записать так:
^c“c£ = ^css
где и kt — константы скоростей прямой и обратной реакций.
43
Отсюда следует
где Кс — константа равновесия.
Уравнение (II.12) справедливо только для реакции в равно-
весных условиях.
В зависимости от того, в каких единицах выражаются концен-
трации реагирующих компонентов, константа равновесия прини-
мает разное численное значение.
При взаимодействии газов константу равновесия обычно вы-
ражают через парциальные давления реагирующих газовых ком-
понентов. Например, для реакции (II. 5) имеем
= (11.13)
РдРа
где A/V — изменение числа молей газов в результате реакции.
В рассматриваемом случае
ДАТ = (г + S) _ (а + Z>) (Ц.14)
В общем случае
Ay = (v; + v'+ ... +v')-(v! + v2+ ... +v„) (11.15)
где Vi Va, — коэффициенты при реагентах, вступающих в реакцию;
v2,..., v'— коэффициенты при продуктах реакции.
С целью наиболее полного использования сырья желательно
вести технологический процесс в таких условиях, чтобы получить
наиболее высокую степень превращения исходного вещества. По-
этому большой практический интерес представляет установление
влияния различных факторов на равновесие системы (т. е. на
равновесную степень превращения X*), а также разработка прие-
мов, позволяющих сдвинуть равновесие в сторону образования
целевых продуктов.
Основными параметрами, влияющими на равновесие химиче-
ских реакций, являются температура, давление и концентрация
реагирующих веществ. Эти параметры обычно используются на
практике для сдвига равновесия в желаемую сторону, т. е. для
регулирования равновесной степени превращения.
Сдвиг равновесия под влиянием температуры
В соответствии с принципом Ле-Шателье, если на систему,
находящуюся в равновесии, воздействовать извне путем измене-
ния какого-либо условия, определяющего положение равновесия,
то в этой системе усилится такой процесс, течение которого ослаб-
ляет влияние произведенного воздействия. При этом положение;
44
авновесия сместится в соответствующем направлении. Например,
если протекает обратимая экзотермическая реакция окисления
сернистого ангидрида
S02 + >/202 SO3 + Q
то за счет выделяющегося тепла повышается температура реак-
ционной смеси, равновесие сдвигается влево и равновесная кон-
центрация целевого продукта (SO3) уменьшается. Для того что-
бы этого не произошло, необходимо отводить тепло. Для обрати-
мой эндотермической реакции, протекающей с поглощением тепла,
например, при конверсии метана водяным паром
СН4 + Н2О ^=> CO + 3H2-Q '
когда происходит охлаждение системы, необходимо, наоборот,
подводить тепло для того, чтобы сдвинуть равновесие в сторону
получения продуктов реакции.
Обычно устанавливают функциональную зависимость X* от
константы равновесия [X* = f(Kp)]. Вид этой зависимости опре-
деляется типом химической реакции. Например, для простой обра-
тимой реакции АR зависимость Х* = /(КР) может быть полу-
чена Следующим путем. Константа равновесия этой реакции вы-
ражается уравнением
Xp=PrIpa Ю
где рл, р*я — парциальное давление соответственно реагентов А и В в состоянии
равновесия.
Определим значения р*А и р^ и подставим их в уравнение (а).
Для рассматриваемой реакции по уравнению (I. 17) имеем
у* _ NА, 0~№д _ Рл, 0 —Рд _ РД
Ла~ N D Р
А, О РА.О
откуда
P*R~PXA (б)
где Р — общее давление.
Но
РА - 1 у*
-р-— 1 — лА
или
. Ра = Р^-Ха) ' (В)
Подставив в (а) полученные значения для р*А и р*, находим:
„ РХА _ ХА
" р^-ХА~) 1-Гл
откуда
* КР 1
(IU6>
Кр
45
Для реакции А 2/? аналогична получаем
^=V-4?T^ (1,'17)
Влияние температуры на равновесие, т. е. функциональная за-
висимость K$ = f{T}, отражается уравнением изобары Вант-Гоф-
фа, которое имеет вид
d In Яр Д/Z
ЙТ = ~»Т2 (I 18)
После интегрирования этого уравнения в пределах изменения
температуры от 7\ до Т2 и при условии, что ЛЯ не зависит от ве-
личины Т, получаем
Рис. II. 1. Зависимость рав-
новесной степени превраще-
ния X* от температуры Т:
1 — для экзотермической реак-
ции; 2—для эндотермической ре-
акции.
Условия применения этого уравнения
ограничены небольшим интервалом тем-
ператур, поскольку в действительности
ДЛГ зависит от Т.
Для определения константы равнове-
сия при значительном изменении темпе-
ратуры применяют эмпирическое уравне-
ние типа
!^р=2де+(;|1^' + ^ + а/-|-в,
(II. 20)
В практических расчетах часто пользуются приближенным
уравнением
Wr + B==T + S ~ (IL21)
где
(,L22)
В справочной литературе имеются значения константы равно-
весия или коэффициенты в приведенных уравнениях для многих
реакций. Если же эти сведения отсутствуют, значения констант
равновесия находят экспериментально. Для этого опытным путем
для каждой рассматриваемой реакции в состоянии равновесия
определяют концентрации исходных реагентов и полученных про-
дуктов при нескольких температурах. Подставив эти данные в
уравнение (II. 13), находят константу Лр. Зная константу равно-
весия, определяют коэффициенты в уравнениях (11.20) и (11.21).
Зная зависимости
Г = f (Яр) (а)
И
ЯР = Ф(П (б)
46
еТпудио установить зависимость Х* = Ф(Т). Для этого необхо-
димо в уравнение (а) подставить значение Кр из уравнения (б)
Г = Г^р) = П<р(Г)] = Ф(Г) (в)
Эта зависимость является одной из самых главных в химиче-
ской технологии, поскольку величина X* характеризует макси-
мально возможное извлечение целевых продуктов из перерабаты-
ваемого сырья, а температура оказывает сильное влияние на ско-
рость химических реакций.
Р Вид функциональной зависимости Х* — Ф(Т) для экзотерми-
ческих и эндотермических реакций неодинаков: для экзотермиче-
ских реакций X* уменьшается при повышении температуры, а для
эндотермических реакций — возрастает (рис. II. 1). Такой харак-
тер зависимости вытекает из уравнений (II. 16) и (II. 17).
Действительно, из уравнения (11.21) следует, что для экзо-
термической реакции значение Кр уменьшается при повышении
температуры, следовательно, член 1/Кр увеличивается, а X* —
уменьшается. Для эндотермической реакции, наоборот, при повы-
шении температуры значение Кр возрастает, поэтому член 1/Кр
в уравнении (II. 16) уменьшается, а X* увеличивается.
Сдвиг равновесия под влиянием давления
Давление является также важным фактором, с помощью кото-
рого в производственных условиях регулируют равновесия хими-
ческих процессов и, следовательно, изменяют максимально воз-
можную степень использования сырья.
Качественное влияние изменения давления на равновесие
химических реакций можно установить также на основе принципа
Ле-Шателье. Для этого рассмотрим три реакции, в которых общий
объем газов в процессе реакции изменяется в различной степени:
в первой — объем газов уменьшается, во второй — увеличивается,
а в третьей — остается без изменения
-|- ЗН2 » 2NH3 * (а)
СН4 С + 2Н2 (б)
СО + Н2ОГ =?=> Н2 + СО2 (в)
Поскольку реакция (а) протекает с уменьшением объема и,
следовательно, с уменьшением давления, то, исходя из принципа
Ле-Шателье, для смещения равновесия слева направо необходимо
повышать давление. На практике так и поступают — процесс ведут
под давлением 32,0 МПа (320 кгс/см2).
Для реакции (б), протекающей с увеличением объема, для
смещения равновесия слева направо необходимо, наоборот, пони-
жать давление. На равновесие реакции (в) изменение давления
Не влияет, так как объем системы в процессе реакции не изме-
няется.
47
Сдвиг равновесия под влиянием концентрации
реагирующих веществ
Концентрация реагирующих веществ оказывает сильное влия-
ние на выход целевых продуктов, что видно из уравнения для
определения /Ср любой реакции. Например, для реакции
CO + HSO =?=> СО2 + Н2 (а)
РсоРщо
При увеличении концентрации исходных реагентов, т. е. при
увеличении знаменателя, соответственно возрастает числитель,
т. е. .повышаются концентрации продуктов реакции (поскольку Кр
для данной температуры величина постоянная). С другой стороны,
при уменьшении концентрации продуктов реакции, т. е. при умень-
шении числителя, соответственно снижаются концентрации исход-
ных веществ, т. е. увеличивается степень их переработки. Оба эти
фактора используются на практике для сдвига равновесия в сто-
рону получения целевого продукта, и, следовательно, для увели-
чения его выхода.
Рассмотрим влияние каждого из этих факторов.
Для снижения концентрации продуктов реакции их выводят из
системы частично или полностью по мере накопления. В резуль-
тате вывода равновесие реакции сдвигается в сторону образова-
ния целевого продукта не только за счет уменьшения концентра-
ции продуктов реакции в системе, но и вследствие одновременного
повышения концентрации исходных веществ в реакционной смеси
(общий объем реакционной смеси уменьшается при выводе из нее
продуктов реакции).
Наиболее простым способом вывода продуктов из. зоны реак-
ции является их связывание химическим путем с другими, вводи-
мыми извне веществами. В качестве примера рассмотрим процесс
конверсии окиси углерода водяным паром [см. уравнение (а)].
Соотношение между концентрациями веществ и константой равно-
весия в данном случае выражается уравнением (б).
На практике равновесие этого процесса сдвигается в сторону
образования целевого продукта (водорода) введением негашеной
извести СаО, которая взаимодействует с СО2 с образованием
СаСОз и выводит ее из реакции. При этом снижается рС02; по-
скольку Кр при данной температуре величина постоянная, возра-
стает концентрация Н2.
Чаще используют способ, заключающийся в переводе всех или
отдельных продуктов реакции в другое фазовое состояние и вы-
воде их из системы. Этот процесс обычно осуществляют по цир-
куляционной (циклической) схеме (рис. II. 2).
Смесь исходных реагентов А и В поступает в реактор 1, в ко-
тором происходит химическое взаимодействие между реагентами.
48
На выходе из реактора в газовой смеси содержится продукт реак-
ции и непрореагировавшие исходные реагенты А и В. Газовая
месь поступает в конденсатор 2, где она охлаждается. При этом
Рис. II.2. Циркуляционная схе-
ма химико-технологического
процесса с выводом продукта
реакции:
/ — реактор: 2— конденсатор.
Рис. П.З. Схема химико-технологического
процесса с выводом продукта реакции R:
1-3 реакторы; 4, 5—конденсаторы.
Рис. II.4. Зависимость
равновесного давления
водорода рц2 от пар-
циального давления паров
воды рН20 для реакции
со+н2о^н2+ СО2.
продукт реакции R конденсируется и выводится из системы, а ис-
ходные реагенты А и В возвращаются в процесс. В качестве при-
мера осуществления такой схемы можно привести процесс синтеза
аммиака из N2 и Н2 на катализаторе:
N2 4- ЗН2 2NH3 + Q
При проведении реакции с образованием
двух продуктов, например по модельной
реакции (П.5)
А + В +=> R + S
процесс ведут по схеме, изображенной на
рис. П.З. Схема проста и не требует особых
пояснений.
создают избыток па-
Увеличение выхода продукта реакции за
счет сдвига равновесия в результате повы-
шения концентрации исходных реагентов
также часто осуществляют на практике;
обычно повышают концентрацию наиболее
дешевого компонента. Так, при получении
водорода конверсией СО водяным паром
ров воды, поскольку водяной пар относительно дешев. При уве-
личении рНз0 соответственно возрастают значения рс02 и рН2.
Однако по мере повышения рНг0 эффект уменьшается, что можно
проиллюстрировать рис. II. 4. Как видно из рисунка,
поэтому при значительном повышении рН20 величина р^2 возра-
стает в меньшей степени. Кроме того, значительное повышение
dP»2o
49 ’
рНз0 неэкономично, так как при этом увеличивается объем аппа-
ратуры, возрастает расход электроэнергии и процесс перестает
быть автотермичным (объем газов сильно возрастает, а количество
выделяющегося тепла увеличивается незначительно).
Исходя из изложенного выше, можно сделать следующие вы-
воды:
при повышении температуры равновесие смещается в сторону
получения
1) целевых продуктов (->) для реакций А R— Q
2) исходных реагентов (-<-) для реакций А R + Q
при повышении давления равновесие смещается в сторону по-
лучения
1) целевых продуктов (->) при A'V <z 0 (при уменьшении V)
2) исходных продуктов (-<-) при AN > 0 (при увеличении V)
3) не изменяется (+±Q при AN = 0 (при V = const)
при повышении концентрации исходных регентов равновесие
смещается в сторону образования целевых продуктов (->).
3. КИНЕТИКА ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
Описанные выше термодинамические данные, приводящие к
увеличению выхода продуктов реакции, часто находятся в проти-
воречии с кинетикой процесса, поэтому для установления опти-
' мальных технологических условий необходимо одновременно учи-
тывать как термодинамические, так и кинетические факторы. Так
например, с точки зрения термодинамики экзотермический про-
цесс синтеза аммиака или окисления сернистого ангидрида на
катализаторе желательно вести при низкой температуре, так как
равновесная степень превращения увеличивается при понижении
температуры. Однако скорость реакции, наоборот, при уменьше-
нии температуры снижается, поэтому на практике выбирают такую
оптимальную температуру Tout, при которой обеспечивается макси-
мальный выход продукта в единицу времени, т. е. максимальная
интенсивность.
Понятие о микро- и макрокинетике
Процессы, протекающие в жидкостях и в газах, обычно рас-
сматриваются с двух точек зрения^ процесс взаимодействия от-
дельных молекул — на микроуровне и процесс взаимодействия
агрегатов молекул — на макроуровне.
В системе на микроуровне жидкость представляет собой сво-
бодные индивидуальные молекулы, движущиеся в различных
направлениях, сталкивающиеся и смешивающиеся со всеми дру-
гими молекулами данной жидкости; в системе на макроуровне
жидкость представлет собой совокупность большого числа неболь-
ших глобул, т. е. групп молекул, как бы заключенных в оболочку.
Предполагается, что эта внешняя оболочка химически.инертна и
инСтВенное назначение ее состоит в том, чтобы сохранить условно
СрИнятую индивидуальность каждой глобулы.
n'J весьма условно потоки жидкости в микро- и макросостоянии
показаны на рис. II. 5. Особенности микро- и макросостояния
moJkh° также проследить при смешении жидкостей: чем выше
вязкость смешивающихся жидкостей, тем ярче проявляются в.ней
признаки макросостояния. В качестве примера представим себе
сосуд с мешалкой, в кото-ом весьма интенсивно перемешивается
Рис. II.5. Поток идеализированных жидкостей в микро- и макроса-
стояниях:
1 — жидкость в мнкросостоянии; 2—жидкость н макросостояннн.
жидкость с малой вязкостью (вода). В эту жидкость вливают дру-
гую жидкость тоже малой вязкости (этиловый спирт). Эти жид-
кости хорошо перемешиваются и смесь быстро становится одно-
родной; условно можно принять, что смешение происходит на
микроуровне.
Если в тот же сосуд вместо этилового спирта вливать очень
вязкую жидкость (например, глицерин), а интенсивность переме-
шивания снизить (уменьшить частоту вращения мешалки), глице-
рин разобьется на отдельные капли (глобулы) и жидкость не
скоро станет однородной. Условно можно принять, что смешение
происходит на макроуровне.
В микросостоянии жидкость не обладает никакими свойствами,
обусловленными присутствием глобул молекул и, наоборот, свой-
ства жидкости, находящейся в макросостоянии, в значительной
степени определяются наличием глобул. Реальная смесь в той или
иной степени проявляет промежуточные свойства, которые зависят
от свойств жидкости и от системы, в которой происходит сме-
шение.
Все вопросы, изучаемые в курсе физической химии, — химиче-
ское равновесие, кинетика химических реакций и др. — рассматри-
ваются на микроуровне, т. е. на уровне отдельных молекул. Такой
подход позволяет проанализировать влияние различных факторов
в идеализированных условиях.
В промышленных условиях при оформлении подавляющего
большинства химических процессов необходимо учитывать многие
сопутствующие физические процессы, связанные с макросостоя-
пием системы и накладывающиеся на основной химический про-
цесс. Важнейшими из них являются; а) диффузия исходных
51
реагентов в зону реакции и продуктов реакции из зоны реакции,
б) выделение и распределение тепла.
На оба эти процесса сильное влияние оказывают аэрогидро-
динамические условия (т. е. характер движения газа или жид-
кости), поскольку от них зависят конвективный перенос тепла и
диффузия вещества.
Таким образом, при изучении реального химико-технологиче-
ского процесса необходимо учитывать влияние диффузии, тепло-
передачи и конвекции, т. е. процесс следует рассматривать на
макроуровне.
Влияние различных факторов на скорость
химических процессов, протекающих на микроуровне
Цель всякого химического процесса состоит в том, чтобы по-
лучить целевой продукт из имеющегося сырья с возможно более
низкой себестоимостью. Для этого необходимо обеспечить мини-
мальные расходные коэффиценты электроэнергии, воды, сжатого
воздуха и прежде всего сырья, так как в большинстве случаев
сырье является наиболее крупной статьей в себестоимости про-
дукции.
Для снижения расходного коэффициента сырья технологиче-
ский процесс необходимо вести так, чтобы степень превращения
X, выход целевого продукта Ф и селективность <р (для сложных
реакций) были возможно более высокими и достигались в воз-
можно более короткое время, т. е. скорость процесса должна быть
возможно более высокой. Это объясняется тем, что скорость ха-
рактеризует интенсивность процесса, а интенсивность является
одним из основных показателей, определяющих экономичность
химического производства.
Скорость химической реакции выражается уравнением
'-зт-‘с" W'-23»
где п — порядок реакции.
Но константа скорости реакции k выражается уравнением Арре-
ниуса
__Е
k = koe~*r (11.24)
где k0 — предэкслоненциальный множитель;
Е — энергия активации.
Подставив значение k из уравнения (11.24) в уравнение (II. 23),
находим
Е
г = koe RT Сп (11.25)
Из уравнения (II.25) следует, что скорость реакции зависит
от температуры и от концентрации реагирующих компонентов.
52
ттля реакции в газовой фазе скорость г зависит от давления, а для
каталитических процессов —и от активности катализатора. Таким
образом, в общем случае г = f(C, Т,Р, Кат).
Рассмотрим способы увеличения скорости реакции г для про*
пессов, протекающих на микроуровне. При этом во всех случаях
будем устанавливать условия ведения процесса, при которых до-
стигается наиболее высокое значение г в течение всего рассматри-
ваемою промежутка времени.
Температура оказывает сильное влияние на скорость хи-
мической реакции, так как в уравнении Аррениуса температура
входит в‘показатель степени, однако это влияние неодинаково для
различных типов реакции.
Скорость простой необратимой реакции
_>/? ± Q описывается уравнением (11.25),
из которого следует, что при С = const и
увеличении температуры Т скорость реак-
ции возрастает по экспоненциальному за-
кону (рис. II. 6).
Зависимость скорости простой обра-
тимой экзотермической реакции типа
+ Q от температуры выражается
k2
более сложным уравнением, так как в этом
случае общая скорость реакции зависит
от разности между скоростями прямой и
обратной реакций
г = Г] — г2 = (11.26)
Рис. П.6. Зависимость-
скорости реакции г от
температуры Т для про-
стой необратимой реак-
ции (Л -> R ± Q).
где r2, г2; г — скорость соответственно прямой, обратной и суммарной реакций;
615 k2 — константы скорости прямой и обратных реакций.
Для анализа уравнения (II. 26) подставим в него
Сд^оО-Яд)’ *2 = *>/*с> с« = сд.о^д
и получим
^ = ^д.о(1-^д)-7~Сд.о^
или
г = ^Сд.ог ет[!-Хд (!+-£)] (11.27)'
Из уравнения (II.27) следует, что для некоторой постоянной
с повышением температуры суммарная скорость реакции с
одной стороны должна возрастать за счет увеличения е , а с
Другой стороны снижаться, так как для экзотермических реакций
константа равновесия уменьшается при повышении температуры
и> следовательно, возрастает член 1/Ас и уменьшается мно-
житель в квадратных скобках. В связи с этим при повышении
53
температуры скорость реакции вначале увеличивается (от г = О,
когда начинается реакция), достигает максимального значения,
а затем снижается. Таким образом, зависимость r = f(T) прохо-
дит через максимум (рис. II. 7).
Из уравнения (11.27) следует также, что при увеличении ХА
(при прочих равных условиях) суммарная скорость реакции сни-
жается. Поэтому для случая, когда Х2 > Xi кривая зависимости
Г2 = ф(Л расположится ниже кривой, соответствующей X,
(рис. II. 7), а кривая зависимости г3 = ф(7') (когда Х3 > Х%
> Х{) расположится еще ниже, и т. д. Кривая АВ, соединяющая
максимумы полученных таким образом кривых, является линией
оптимальных температур (ЛОТ).
Для простой обратимой эндотермической реакции типа А
R— Q характер зависимости r = f(T) также может быть уста-
новлен на основе уравнения (11.27). При повышении температуры
____________________________________________________________Е
скорость реакции возрастает за счет увеличения как члена е RT
так и значения Кс. При увеличении ХА (при прочих равных усло-
виях) общая скорость реакции снижается (рис. II. 8).
Установим зависимость XA = f(T) для тех же реакций. Эта
зависимость является очень важной, поскольку степень превра-
щения характеризует относительное количество переработанного
исходного реагента (см. гл. I).
Для простой необратимой реакции зависимость
ХА = f(T) выражается S-обратной кривой (рис. II. 9). Такая за-
висимость объясняется тем, что при низкой температуре скорость
процесса мала, поэтому за промежуток времени т в реакцию
вступает лишь незначительная часть исходного реагента А, а по-
тому невелик подъем кривой XA = f(T) (рис. II. 9). По мере
повышения температуры скорость реакции увеличивается экспо-
ненциально в соответствии с уравнением (11.25), что приводит к
резкому подъему кривой. Но с увеличением ХА концентрация ис-
ходного реагента уменьшается и соответственно снижается рост
скорости реакции, поэтому функциональная зависимость ХА =
~ f (Т) асимптотически приближается к единице.
Для простой обратимой экзотермической реакции типа А
R + Q зависимость XA — f(T) при некотором времени т вна-
чале возрастает, достигает максимального значения, а затем
снижается, поскольку процесс ограничен равновесной степенью
превращения X* (рис. II. 10). Кривая, соответствующая т2 >
> Ti, расположится выше кривой п, а кривая т3 > т2 расположит-
ся еще выше и т. д. (рис. II. 10). Кривая АВ, соединяющая мак-
симумы, отражает зависимость X = f(T) и является линией
оптимальной температуры (ЛОТ). Ее иногда называют линией
оптимальной температурной последовательности. Последняя по-
казывает, что для простой обратимой экзотермической реакции
существует не какая-то оптимальная температура, а температур-
ная последовательность, обеспечивающая максимальную скорость
54
обратимой экзотермической реакции:
’ (Х( < Х2 < Х3 < X.)
Рис. II.8. Зависимость скорости г
Рис. II.9. Зависимости степени пре-
вращения X от температуры Т для
простой обратимой реакции А -> R±Q..
Рис. 11.10. Зависимость степени превращения X от температуры Т для простой'
обратимой экзотермической реакции А R + Q (Т[ < т2 < т3 < т4 < т5)
Рис. 11.11. Зависимость сте-
пени превращения X от тем-
пературы Т для простой
обратимой эндотермической
реакции A J* R — Q (X*,
Хф — степень превращения
•соответственно равновесная
и фактическая).
'процесса. Чтобы создать такие оптимальные условия, процесс
следует вначале вести при высокой температуре, когда скорост
Га велика и из-за низкого значения ХА не может быть достигнута
высокая Х*А. Для увеличения Х"А необходимо снизить температуру
(повышать Ха) и вести процесс по ЛОТ.
Для простой обратимой эндотермической реакции типа I
Д++А1— Q вопрос о влиянии температуры решается однозначно |
и с точки зрения термодинамики (т. е. с точки зрения условий рав- |
новесия),ис точки зрения кинетики про- |
цесса, так как при повышении темпера- |
туры увеличиваются и равновесная X* и
фактическая степень превращения Х$
(рис. II. 11). Однако и в этом случае су-
ществует оптимальняа температура Топ,.,
при которой достигаются наиболее вы-
годные условия для ведения технологи-
ческого процесса, так как при повыше-
нии температуры уменьшается наклон
dX , т—г
кривой, т. е. производная 0- По-
этому невыгодно сильно увеличивать
температуру,поскольку значение Ха воз-
растает незначительно. Однако в неко-
торых случаях повышение температуры
оказывает и отрицательное влияние на
химико-технологический процесс.
Чтобы составить общее представле-
ние о зависимости X = f(T) для всех ти-
пов рассмотренных реакций, проанали-
зируем рис. II. 12.
Из рис. II. 12 видно, что при малых значениях X характер
кривых X = f(T) во всех случаях одинаков, при повышении тем-
пературы величина X резко возрастает. Такая зависимость объяс-
няется тем, что с повышением температуры увеличивается ско-
рость реакции, а влияние термодинамических факторов (т. е.
обратной реакции) незначительно. Из рис. II. 12 также следует,
что для достижения степени превращения Xi температура должна
быть тем выше, чем меньше продолжительность процесса.
С повышением X влияние термодинамических факторов увели-
чивается. Степень этого влияния зависит от типа реакции, что и
определяет характер кривых, приведенных на рис. II. 12. Для
необратимых реакций кривые зависимости XA = f(T) асимптоти-
чески приближаются к единице. Для обратимых реакций кривые
зависимости XA = f(T) ограничиваются кривой равновесной сте-
пени превращения X*, которая, как мы уже знаем, для экзотерми-
ческой реакции с повышением температуры уменьшается, а для
эндотермических реакций возрастает.
Из приведенных данных следует, что температура оказывает
.56
только положительное, но и отрицательное влияние на показа-
тели ХТП. Отрицательное влияние температуры состоит в сле-
дующем:
а) увеличение потерь целевого продукта вследствие его испа^
рения и образования побочных продуктов;
б) снижение прочности и химической стойкости материалов;
в) уменьшение величины X* в экзотермических реакциях;
г) возможное снижение селективности сложных реакций.
Рис. 11.12. Зависимость
степени превращения X
от температуры Т при
различном времени про-
цесса (Т, > Т2 > Тз).
Поскольку селективность является важным показателем ХТП,
установим зависимость ф = f(T’) для параллельных реакций типа
ku Ei.n,
-------Л
А —
-------+ S
кц Ег> Лг
В этом случае селективность по целевому продукту R выражается
Уравнением (1.20), из которого следует
m = NR = rR = rp/rs = f (LS.'X
Ng + Ns 4- rs rg/rs + 1 \ rs )
(II. 28)
Подставив в это уравнение значения rR и rs из уравнений (II. 23)
й (П. 25), находим
Е,
k'oe ^тС^~Пг
Е,
k''e~ &
/ А/ \
= Н —~е & с^-*1 ) (II. 29)
\fe0 /
57
Phc. 11.13. Зависимость кон-
станты скорости реакции k от
температуры 1/Г.
Все величины, входящие в уравнение (11.29), постоянны, за
исключением температуры, поэтому при изменении температуры
изменяется также и селективность, и в зависимости от значения
разности Е2 — Ei влияние температуры может быть либо положи-
тельным, либо отрицательным. Поскольку при низкой темпера-
туре снижается скорость процесса, то и в данном случае суще-
ствует некоторая наиболее выгодная оптимальная температура
Tout как с точки зрения термодинамики (равновесная), так и с
точки зрения кинетики, при которой достигается требуемая селек-
тивность и, в конечном итоге, наиболь-
шая экономичность процесса.
Аналогичная закономерность на-
блюдается и в последовательных ре-
акциях. Поэтому для того, чтобы в
производственных условиях обеспе-
чить наиболее высокую эффективность
процесса, его необходимо проводить в
области температурного оптимума.
Таким образом, температура явля-
ется мощным фактором интенсифика-
ции химического процесса, но возмож-
ности для использования этого факто-
ра ограничены возможностью отрица-
тельного влияния на процесс.
В каждом конкретном случае долж-
на быть установлена экономически
рациональная температура; она всегда несколько ниже ГОпт, най-
денной теоретическим путем, поскольку при теоретическом расчете
не все факторы учитываются.
Значения энергии активации Е и предэкспоненциального множителя й0 в
уравнении Аррениуса (11.24), необходимого для проведения практических расче-
тов, обычно находят по значениям константы скорости реакции, найденным экс-
периментально при двух температурах. В качестве примера представим уравне-
ние Аррениуса в виде
. . , , Е 0,4£ 1 , .
lgfe = igfeo- —ige = lgfe0--yy (a)
Так как k0, Е и R величины постоянные, то зависимость, выражаемая урав-
нением (а), является прямолинейной (рис. II. 13). При этом
tga = -^y - ' (б)
При очень высокой температуре можно принять
Тогда
k = ka (г)
Приведенные Данные позволяют по двум известным значениям k (при раз-
личной температуре) определить
а) константу скорости при любой температуре (см. рис. II. 13);
68
• б) энергию активации Е [уравнение (б)] по графику на рнс. II. 13 и угол
наклона <х,
в) преДэкспонени-иальнь1И множитель ka (продлив прямую до пересечения с
осью ординат, см. рис. II. 13).
Концентрация реагирующих веществ оказывает влияние на
кОрость реакции всех типов (кроме реакций нулевого порядка):
о увеличением концентрации исходных реагентов скорость реакции
возрастает. Так например, суммарная скорость модельной хими-
ческой реакции (II. 5) равна разности между скоростями прямой
и обратной реакций
r = r’l-r2 = klCaACbB-k2CrRCss (11.30)-
Из этого уравнения следует, что, чем больше значения Са и
Св, тем выше г, а также, что с течением времени суммарная ско-
рость реакции г снижается, так как концентрации исходных ре-
агентов (Са, Св) уменьшаются, а концентрации продуктов реакции
(CR, Cs) увеличиваются. Поэтому для того, чтобы судить об из-
менении скорости реакции, строят кривые зависимости концентра-
ций реагентов, участвующих в реакции, от времени, т. е. устанав-
ливают зависимость С = f(x).
Рассмотрим эту зависимость для различных типов реакций.
Скорость необратимой реакции наиболее простого - типа
k
А—>R выражается уравнением (11.23) *
dCA
— г,=----~^ = kC,
A dX &
После интегрирования этого уравнения в пределах изменения г
от 0 до т, а концентрации С а от начального значения С а, о до Са»
и, принимая, что k не зависит от т, находим
J сА }
СА.О А О
откуда
= или СА = СА^ = -Ц^ (11.31)
ьл, о е
Из уравнения (11.31) следует, что по мере течения реакции,.
т- е. по мере увеличения т, концентрация исходного вещества Са
снижается по экспоненциальному закону, соответственно умень-
шается и скорость реакции (рис. II. 14,а). Из рис. II. 14, а также
видно, что для того, чтобы довести реакцию до конца, необходимо-
большое время процесса т, так как Са асимптотически прибли-
жается к нулю. На практике реакцию не доводят до конца, ее оста-
навливают через некоторый промежуток времени, когда С а > О
и Движущая сила процесса достаточно велика; такое время можно-
назвать оптимальным тОпТ. В результате того, что реакция прохо-
дит не полностью, в конце процесса получают смесь продуктов А
59
i
и /?. Следовательно, процесс,следует организовать таким образом
чтобы после стадии химического превращения была стадия раз’
деления, а также так, чтобы после выделения продукта 7? исход,
ный реагент А вновь был использован в реакции, т. е. процесс
необходимо оформить по циклической схеме.
Для простых обратимых реакций зависимость C — f(x) более
сложная. Например, для простой обратимой реакции типа [
процесс останавливается в тот момент, когда концентрация исход.
ного реагента Са достигнет значения концентрации этого реагента £
в состоянии равновесия С а (рис. II. 14, б). Поскольку вблизи^
равновесия скорость взаимодействия очень мала, процесс пре-
кращают по достижении некоторого оптимального времени Топт,
при котором разность С а—С а (движущая сила процесса) еще
значительная и, следовательно, скорость процесса достаточно
высока.
Скорость реакции в этом случае выражается уравнениями
— гл = k^CA — k2CR (а)
rR ~ k2CR ~ klCA
Из сложных реакций рассмотрим только необратимые парал-
лельные и последовательные реакции.
Для необратимых параллельных реакций типа
Л- R : / '
А —
S
изменение концентрации исходного реагента и целевого продукта
R во времени показано на рис. 11.15 скорости реакций могут
60
ь выражены такими уравнениями
-^ = ^A + ^ = ^(fe1 + fe2)
rR = klCA
rS = k2CA
(в)
(Г)
(Д)
Наиболее простая схема двух необратимых последовательных
еакиий может быть представлена в виде
А—►/?—> N
А
•Я
•S
х
Рис. 11.15. Изменение концен-
трации компонентов С парал-
лельных необратимых реакций
А во времени.
с течением времени значение Са снижается, a Cr вначале уве-
личивается, достигает некоторого максимального значения, а за-
’теМ убывает вследствие его разложения и образования продукта
При этом концентрация промежу-
точного продукта R в некоторый мо- f
мент времени зависит от соотношения
скоростей каждой из последователь-
ных реакций.
Различают три случая:
ki~>k2 fei<fe2 К « k2
На рис. II. 16 показано изменение
концентрации A, R и N во времени
для каждого из этих случаев. Из ри-
сунка видно, что если k\ /г2, то при
прочих равных условиях, при полном
расходе продукта А образуется смесь,
содержащая продукты R и N. Если
ki С k2, то промежуточный продукт
R по мере его образования интенсив-
но расходуется, поэтому с самого на-
чала реакции происходит накопление вещества N. Если « k2,
то кривые зависимости CR = f(x) и CN = f(x) занимают проме-
жуточное положение между кривыми, соответствующими k\ k2
и £1 < k2.
Скорость рассмотренных последовательных реакций выражает-
ся такими уравнениями
~ГА~ klCA .
dC»
TR ~ ~dx~ ~ ~ k2CR
rS = k2CR •
Максимальная концентрация промежуточного продукта Cr до-
стигается при условии dCR/dx = 0.
Давление также оказывает большое влияние на скорость хи-
мических процессов, особенно в тех случаях, когда процессы
пРотекают в газовой фазе или же при взаимодействии газов с
61
жидкостями и твердыми веществами. Это объясняется тем, чТо
при повышении давления уменьшается объем газовой фазы ц
соответственно увеличивается концентрация реагирующих веществ
Общую скорость модельной реакции (II. 5), если она необр^
тима или обратима, но протекает вдали от равновесия, можно
выразить в виде уравнения
г = - ГА = kpaApbB (а)
где рл, рв — парциальные давления исходных реагентов.
Но парциальное давление каждого реагента пропорционально
общему давления Р: рл = IP и рв = тР (где /, т— постоянные),
Рис. 11.16. Изменение
концентрации компонен-
тов С последовательной
необратимой реакции
А —> R N
во времени.
Подставив эти значения в уравнение (а), находим
r = klaPambPb = k'Pa+b = k'P" (11.32)
где k' = klamb;
п — порядок реакции (п = а + Ь).
Таким образом, скорость реакции пропорциональна давлению
в степени, равной порядку реакции. Следовательно, изменение
давления наиболее сильно влияет на реакции высокого порядка
(рис. II. 17).
Однако повышение давления имеет предел, поскольку dXjdP
—> О (рис. II. 18); кроме того, увеличение давления приводит к воз-
растанию расхода электроэнергии, изменению свойств газа и жид-
кости (в частности к повышению их вязкости) необходимости при-
более прочных конструкционных материалов и т. д.
в производственных условиях следует учитывать все
и устанавливать так называемое оптимальное давление
менения
Поэтому
ФаКТ при котором достигается наиболее высокая экономическая
АЛективность технологического процесса.
Э<В некоторых случаях повышение давления дает возможность
вести процесс при температуре, превышающей температуру кипе-
ля растворов при атмосферном давлении (что также способ-
ствует увеличению скорости процесса). Примером этому может
служить процесс варки целлюлозы, проводимый при температуре
Рис. 11.17. Зависимость скорости
реакции г от давления Р для га-
зовых реакций различного по-
рядка.
Рис. 11.18. Зависимость степени
превращении X от давления Рдля
простой необратимой реакции
A-+R.
140—150 °C и давлении 0,6—0,8 МПа. В этих условиях значи-
тельно возрастает скорость процесса и предупреждается кипение
раствора.
В твердофазных процессах при сверхвысоких давлениях проис-
ходит перестройка электронных оболочек атомов и деформация
молекул, что может оказывать существенное влияние на скорость
протекающих процессов. ~
Катализаторы оказывают исключительно большое влияние на
скорость химических реакций (см. гл. IV), снижая энергию акти-
вации, которая входит в показатель степени уравнения Аррениуса
,(П.24). В результате этого скорость реакции в присутствии ката-
лизатора иногда увеличивается в миллионы раз. Обычно катали-
затор выбирают с таким расчетом, чтобы он обладал селективным
Действием, т. е. ускорял процесс получения целевого продукта.
Примеры
„ Пример ПЛ. Установить функциональную зависимость X* — f (Кр) для реак-
По ур°ц^чении окиси углерода восстановлением двуокиси углерода, протекающей
СОа -f- С < 2СО , (а)
62
63
Решение.
Учитывая, что концентрация углерода величина постоянная (твердая фаза)
константа равновесия этой реакции выражается уравнением 1
VO/4 (б)
* *
где рсо, рСОг — парциальные давления СО и СО2 в газовой смеси в состоянии
равновесия.
Условия расчета:
Количество СО2 в начале процесса Nco3, о = 1 моль.
Количество СО2, вступившее в реакцию (при достижении равновесия), М =
.= у моль.
Количество СО2, оставшееся в газовой смеси при равновесии
^СОг = ~ У
Из каждой прореагировавшей молекулы СО2 образуются две молекулы СО. По-
скольку прореагировало у моль СО2, количество СО в газовой смеси при равно-
весии равно:
QQ = 2y
Парциальные давления СО и СО2:
= ^СО р = 2У р = 2У р
рС° Л^соЛ^СО Ц~у)+2у 1+у
_ _ г_. Х~У г,
РС°2 ^СО2 + ^СО (1~у)+2у^ 1+У
Подставив полученные значения рсо и рСОз в уравнение (б) для Кр, находим
1 к Рёо_Г 2У р]2Г 1+у 1 W р
₽ = ^”LT+7^J L(f-pj>J = T=^T?
откуда . ________
Р = Л/' 4Р + Кр
Равновесную степень превращения определяем из уравнения (I. 15)
у* ^со,, о ~ -^СО; _ 1 ~ U ~ У) _
Х = Ncn о 1 У
1>U2> U
Подставив значения у из уравнения (б), находим
По уравнению (г) можно найти значение X* при любой температуре, поскольку
величину Кр легко рассчитать по теоретическим формулам или определить экс-
периментально.
Пример II. 2. Доказать, что для простой обратимой экзотермической реакции
функциональная зависимость г — ЦТ) имеет максимум и, что по мере увеличе-
ния значения X положение максимума смещается в сторону низких температур-
Решение.
Скорость простой обратимой экзотермической реакции
А +=+
выражается уравнением .
64
При
что Кс
постоянных значениях С а и Св, т. е. при некоторой постоянной величине X,
записать Св/Сл = Р, или Св = рСл. Подставив. Св в уравнение (а) и
= ki/ki, или ki — k2Ke, получаем
Г = WA ~ k2CR = k2^A ~ k2$C А = k2CА “ ₽>
(б)
или
_ Ег
= CAk^e ^(^с-Р)
CAk
-4г(*с-₽)
еЛГ
(в)
При увеличении температуры Т первый
При увеличении температуры Т первый сомножитель в уравнении (в)
пастает, а второй сомножитель уменьшается (так как снижается константа
новесия экзотермической реакции). Из этого же уравнения следует, что
е2
у = 0 и г = 0, так как е При некотором значении Т (когда Кс —
__й = 0) получим, что г = 0. Отсюда следует, что г = f(T) имеет экстремум;
дальнейший анализ показывает, что эта функция имеет .максимум.
Из уравнения (в) следует, что чем выше значение р (т. е. чем больше
Св/Са или X), тем при более низкой температуре величина г будет достигать
максимального значения, поскольку при достижении низкой температуры (при
большем значении Кс) разность (Кс — Р) будет равна 0.
Пример II. 3. Определить выход продуктов R и S параллельных реакций
А —
ksEi
воз-
рав-
при
при абсолютных температурах Т = 700, 800 и 900 К, если известно, что раз-
ность между энергиями активации этих реакций Ез— Ев = £2—£1 = 2400 Дж*
моль"1, а R = 8,3 Дж • моль-1 К-1-
Решение.
Подставив значения Ев, Ев, Т и R в уравнение (11.29), находим
при
700 К
/ k'Q е,-е2
ЧРтоо = f I —77- е
'*0
h' 24000 \ / f/
КГ 1 = И _°_ е8,3-700 1 = И __ . 62
/ \ kQ / \ kQ
при
при
800 К
900 К
/ k'r, 24000 \ / k'
<P8oo = f(-^e8’3-800 ) = f (4--37
/ \йй
/ k' 24000 \
<Peoo = f М-е8’3-903 =f
\*о /
Из полученных данных следует
= f . 62^ : f 25 1 = 2,5
Фээо ' '
Таким образом, при повышении температуры процесса от 700 до 900 К отиоси-
ельный выход целевого продукта R снижается в 2,5 раза.
3 Зак, 558
k'
^•25
«0 -
65
Глава III
ТИПЫ ХИМИКО-ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
И СПОСОБЫ ИХ ИНТЕНСИФИКАЦИИ
Ранее указывалось, что на практике чаще всего используется
классификация химических реакций по фазовому признаку, соот-
ветственно различают гомогенные и гетерогенные реакции. По
этому же признаку наиболее часто подразделяют химические про-
цессы и реакторы, в которых эти процессы осуществляются.
В гомогенных системах все реагирующие вещества находятся
водной какой-либо фазе: газовой (г), жидкой (ж) или твердой (т).
В гетерогенных системах реагирующие вещества находятся в раз-
ных фазах: газ — жидкость (г — ж), газ — твердое (г — т),
жидкость — твердое (ж— т), две несмешивающиеся жидкости
(ж — ж) и две твердые фазы (т — т). Наиболее часто в промыш-
ленных процессах встречаются системы г — ж, г — т и ж — т. Ино-
гда в промышленных процессах участвуют три или четыре фазы,
например г — ж — т, г —ж — ж, г — ж — т — т.
Обычно за отдельные фазы принимают только основные компо-
ненты и не учитывают наличие малых количеств примесей. Так,
например, в системах ж — ж и ж — т часто содержится газовая
фаза, поскольку процессы проводятся в присутствии воздуха или
других газов, или же в присутствии паров, так как жидкие компо-
ненты частично испаряются. Но газовую фазу учитывают только
в том случае, если она оказывает существенное влияние на про-
цесс.
Некоторые процессы начинаются в гомогенной среде, а затем
в результате появления новой фазы система переходит в гетероген-
ную. Например, при получении полистирола к жидкому стиролу
добавляют перекись бензоила и нагревают, при этом происходит
полимеризация стирола с образованием новой фазы — твердого по-
листирола.
Скорость реакции в гомогенных системах более высокая, чем
в гетерогенных, так как в первом случае реакции протекают на
уровне отдельных молекул (так называемый микроуровень). По-
этому в практических условиях обычно стремятся перевести гете-
рогенный процесс в гомогенный (путем плавления или растворе-
ния твердых реагирующих веществ, абсорбции или конденсации
газов).
1. ГОМОГЕННЫЕ ПРОЦЕССЫ
Строго гомогенные процессы, т. е. процессы, протекающие в од-
ной фазе, встречаются в промышленности сравнительно редко, так
как любое вещество содержит следы различных примесей, находя-
щихся в другой фазе. Например, в 1 мл чистого горного воздух3
содержится около 1000 взвешенных частиц, а в 1 мл дистиллиро-
ванной воды до 20000 частиц. Поскольку следы инородных при-
есей часто активно влияют на ход процесса как катализаторы
иди ингибиторы, большинство процессов лишь условно можно от-
нести к гомогенным.
Число таких условно гомогенных процессов велико в техноло-
ги и неорганических, и органических веществ. Например, окисле-
ние сероводорода и паров серы кислородом воздуха в производ-
стве серной кислоты
2H2S -}- ЗО2 яв 2SO2 -}- 2Н2О -р Q
S -f- О2 « SO2 -}- Q
протекает в гомогенной газовой фазе, несмотря на наличие в воз-
духе большого числа твердых частиц. К числу гомогенных процес-
сов относят также окисление окиси азота до двуокиси азота кисло-
родом воздуха в производстве азотной кислоты
2NO -f- Оа к - 2NOj -f- Q
и многие другие.
Особенно многочисленны и разнообразны гомогенные процессы
в газовой фазе, осуществляемые в технологии органических ве-
ществ. Примером этому может служить сжигание всевозможных
видов газообразного топлива и, в частности, природного газа. Про-
цесс сжигания различного жидкого топлива также в большинстве
случаев является гомогенным процессом, так как всякое жидкое
топливо предварительно испаряется, а образовавшиеся пары затем
окисляются кислородом воздуха.
В технологии органических веществ сущность многих гомоген-
ных процессов в газовой фазе состоит в том, что газообразные
исходные вещества или пары, полученные испарением жидкости,
обрабатываются тем или иным газообразным компонентом: хло-
ром, сернистым ангидридом, окислами азота и др., при этом обыч-
но протекают параллельные и последовательные реакции.
Например, при термическом воздействии хлора на метан при
250—400 °C получают ряд соединений:
СН4 + С12 —► НС1 + CHjCl (хлористый метил)
СН3С1 + С12 —► НС1 + CHjCls (хлористый метилен)
t СН2С12 + С12 .—► НС1 + СНС18 (хлороформ)
СНС13 + С12 —► НС1 + СС14 (четыреххлористый углерод}
Из большого числа процессов, идущих в жидкой фазе, к гомо-
генным можно отнести процессы нейтрализации водных растворов
Кислот водными растворами .щелочей. Например, при взаимодей-
ствии аммиачной воды и серной кислоты в коксохимическом про-
изводстве получают сульфат аммония
2NH4OH + Н,8О4 — (NH4)2SO4 + 2Н2О
К гомогенным реакциям относятся также некоторые обменные
Реакции, проходящие в растйбрах
KCl + NaNO3 NaCl-|-KNO3
66
3»
67
В жидкой фазе получают простые и смешанные эфиры из спир.
тов, например,, при разложении этилсульфата метиловым спиртом
C2H5OSO2OH + СН3ОН C2H5OCH3 + H2so4
В гомогенной среде осуществляют такой важный процесс, как
получение адипиновой кислоты
ЗСбНиОН + 8HNO3 —> ЗС6НюО4 + 7Н2О + 8NO
и многие другие.
Скорость гомогенных процессов
Гомогенные процессы в большинстве случаев протекают в ки-
нетической области, т. е. общая скорость процесса определяется
скоростью химической реакции и подчиняется закономерностям,
установленным для процессов, протекающих на микроуровне. Ско-
рости гомогенных процессов, в основе которых лежат простые не-
обратимые реакции первого и второго порядков (Л->7? и 2Л ->/?),
выражаются уравнениями
ах/
= -х.\
ах,
1Т“=^.о(’-*А)2
из которых следует, что для повышения скорости необходимо уве-
личивать значения k (путем повышения температуры или приме-
нения катализатора) и СА,о- Так как на практике в большинстве
случаев гомогенные реакции являются сложными (параллельными
или последовательными), то в зависимости от значения констант
скоростей этих реакций концентрация продуктов реакций будет
изменяться во времени различно (см. рис. 11.16). При этом, чем
выше порядок реакции, тем большее влияние оказывает концен-
трация исходного реагента на скорость- соответствующей реакции.
Например, при наличии двух параллельных реакций первого и
второго порядков увеличение С а, о в два раза изменяет соотно-
шение скоростей этих реакций в 4 раза.
После интегрирования приведенных выше уравнений в преде-
лах изменения ХА от 0 до ХА находим
_______ха ха: о_
n'2 ^.oO-W-^o)
Если ХА, о = 0, то
2,3 , 1
Tn = l = —lgT=x~
т - *А.О
П=2
(а)
(б)
(в)
(г)
68
' Полученные уравнения (а) — (г) позволяют определить время
т необходимое для достижения заданной конечной степени пре-
вращения Ха- Из этих уравнений следует, что константа скорости
реакции первого порядка измеряется в ч-1, а реакции второго по-
рядка — в м3 • кмоль-1 • Ч'1.
г Гомогенный процесс на макроуровне протекает в том случае,
когда на химическую реакцию накладываются другие физические
или физико-химические процессы. Например, при взаимодействии
двух жидких исходных реагентов или их растворов скорость гомо-
генного процесса зависит от условий и скорости перемешивания
жидкостей; если химическое взаимодействие протекает при подо-
греве, то скорость гомогенного процесса будет зависеть также от
способа подвода тепла.
2. ГЕТЕРОГЕННЫЕ ПРОЦЕССЫ
Большинство химико-технологических процессов относятся к ге-
терогенным; при этом огромное разнообразие гетерогенных про-
цессов затрудняет их классификацию.
Механизм гетерогенных процессов сложнее гомогенных, так
как реагирующие вещества находятся в разных фазах и подвод их
к поверхности раздела фаз, где
происходит химическое взаимо-
действие, а также массообмен
между фазами осуществляются в
результате молекулярной и кон-
вективной диффузии, которые на-
кладываются на основной хими-
ческий процесс. Усложнение вно-
сят также процессы теплообмена
и Процессы, обусловленные осо-
бенностями гидродинамики пото-
ка. Только с учетом всех факто-
ров, влияющих на технологиче-
ский процесс, можно установить
условия, обеспечивающие макси-
мальную его интенсивность, и уп-
Рис. III. 1. Схема горения угля:
/—уголь; 2—зола;- 3—пограничный слой
газа.
равлять этим процессом.
Примером гетерогенного процесса может служить процесс го-
рения угля, который складывается из пяти стадий (рис. III. 1):
внешняя диффузия Ог (через пограничный газовый слой);
внутренняя диффузия О2 (через слой золы);
химическая реакция;
внутренняя диффузия СОг (через слой золы);
внешняя диффузия СОг (через пограничный слой газа).
Горение угля сравнительно простой пример; на практике обыч-
но протекают более сложные процессы. Однако в любом гетеро-
генном химическом процессе можно выделить три основных одно-
временно протекающих процесса;
69
I) диффузия реагентов к границе раздела фаз;
2) химическая реакция;
3) диффузия продуктов реакции из зоны реакции.
Скорость гетерогенных процессов
Для установления оптимальных параметров гетерогенных про.
цессов и их аппаратурного оформления и проектирования необхо.
димО прежде всего изучить статику (т. е. равновесие) и кинетику
(т. е. скорость) этих процессов.
Равновесие в гетерогенных системах зависит от температуры,
давления и концентрации как исходных реагентов, так и продуй
тов реакции, скорость же взаимодействия реагентов, находящихся
в разных фазах, зависит не только от скорости химической реак.
ции, но и от многих других факторов (как любой процесс, проте»
кающий на макроуровне). Поэтому в общем виде скорость гетеро*
генного процесса выражается следующим уравнением;
r = KF&C (III. 1)
где г — скорость гетерогенного процесса;
К. — коэффициент скорости процесса;
F — поверхность контента фаз;
ДС — движущая сила процесса.
Рассмотрим влияние различных факторов на каждый из пара*
метрор, входящих в уравнение (111.1), а также способы увеличе-
ния общей скорости процесса.
Коэффициент скорости процесса
Коэффициент включает в себя многие факторы, влияющие на
скорость гетерогенного процесса. В большинстве практических слу-
чаев влияние этих факторов неодинаково. Так, например, химиче-
ский процесс обычно состоит из нескольких стадий, а его общая
скорость определяется скоростью наиболее медленной (лимити-
рующей) стадии. Поэтому для интенсификации процесса необхо-
димо прежде всего определить, какая из стадий является наиболее
медленной, и ускорить ее. Такими наиболее медленными стадиями,
каждая из которых может тормозить весь процесс, являются
1) химическая реакция;
2) диффузия;
3) одновременно химическая реакция и диффузия.
В первом случае, скорость диффузии велика по сравнению со=
скоростью химической реакции; тогда говорят, что процесс проте-
кает в кинетической области. Во втором случае, скорость химиче-
ской реакции значительно больше скорости диффузии — процесс
протекает в диффузионной области (во внешне- или внутридиффу*
знойной). В третьем случае, скорости отдельных стадий соизме-
римы, тогда говорят, что процесс протекает в переходной (сме-
шанной) области.
70
После установления лимитирующей стадии процесса прини-
мают меры, обеспечивающие повышение скорости этой стадии. Так,
если процесс протекает в кинетической области, создают условия,
ускоряющие химическую реакцию; если процесс протекает в диф-
фузионной области, то ускоряют процесс диффузии; если же
процесс протекает в переходной области, то создают условия, по-
вЫшающие и скорость химической реакции, и скорость диффузии.
Для установления лимитирующей стадии процесса существует не-
сколько приемов. Рассмотрим наиболее важные из них.
Ранее было показано, что температура оказывает сильное влия-
ние на скорость химических реакций [см. уравнение (11.24)]. Так
при увеличении температуры на 10 °C скорость химической реак-
ции в некоторых случаях возрастает в 2—3 раза.
Скорость диффузии газов зависит от температуры в значи-
тельно меньшей степени, приближенно эта зависимость выражает-
ся уравнением:
D = аТ2 (III. 2)
где D — коэффициент диффузии;
а — постоянный коэффициент.
Из уравнения (III. 2) следует, что при повышении температуры
на Ю°C скорость диффузии увеличивается всего на 3—5%. Это
разное влияние температуры используют
для определения лимитирующей стадии
процесса.
На рис. III. 2 кривая 1 отражает тем-
пературную зависимость скорости диф-
фузии исходного реагента в зону реакции
(гф = /(Т)] в соответствии с уравнением
(III. 2). Кривая 2 отражает функцио-
нальную зависимость скорости химиче-
ской реакции от температуры [гх. р =
= Ф(7’)] в соответствии с уравнением
Аррениуса. Общая скорость отражена на
рисунке двумя отрезками кривых 3 и 3'.
Кривая 3 берет свое начало при темпе-
ратуре Тн, когда процесс химического
взаимодействия начинает протекать с
заметной скоростью. Затем наблюдается
.резкий подъем кривой (см. рис. III. 2).
В точке А, где пересекаются кривые 1
и 2, скорость химической реакции и скорость диффузии равны Гф—
*= Гх.Р = г. В дальнейшем величина г равна Гф.
Общая скорость процесса не может превышать самую низкую
составляющую ее скорость Гф г гх, р.
Кривая 4 построена по опытным данным.
Лимитирующая стадия гетерогенного процесса может быть уста-
новлена опытным путем. Так, если опыт показывает, что повыше-
ние температуры оказывает сильное влияние на скорость процесса,
Рис. III. 2. Зависимость ско-
рости диффузии, химической
реакции и общей скорости
процесса от температуры:
/—скорость диффузии; 3—ско-
рость химической реакции; 3—об-
щая скорость процесса теорети-
ческая; 4—общая скорость про-
цесса, установленная экспери-
ментально.
71
то процесс протекает в кинетической области (рис. III. 3,
область Z). Если при дальнейшем повышении температуры ее влия-
ние на скорость общего процесса уменьшается, значит процесс
перешел в переходную область (рис. III. 3, область II). Если же
далее при повышении температуры общая скорость процесса прак-
тически не изменяется, значит процесс протекает в диффузионной
области (рис. III. 3, область III).
Изменение скорости потока также может быть использовано
для определения лимитирующей стадии процесса, поскольку она’
оказывает существенное влияние на скорость внешней диффузии.
роста процесса г от температуры Г:
/ — кинетическая область; //—переходная
область; III—диффузионная область.
Рис. Ш.4. Зависимость общей ско-
рости процесса г от скорости пото-
ка w:
I—диффузионная область; //—переходная
область; III—кинетическая область.
Так, на кривой зависимости общей скорости процесса от скорости
потока г — f(w) можно выделить три области: диффузионную, пе-
реходную и кинетическую. В. диффузионной области наблюдается
значительное влияние скорости потока (рис. III. 4, область Z); в пе-
реходной области влияние скорости потока невелико (область II)
и в кинетической области скорость потока не влияет на скорость
процесса (область ///).
Влияние скорости потока на общую скорость процесса можно
проследить также на графике,отражающем зависимость г = f(T),
если нанести кривые этой зависимости для различных скоростей
потока (рис. III.5). Ход кривых r = f(w) подтверждает тот факт,
что область III является диффузионной (с увеличением w скорость
процесса возрастает), и указывает на способ дальнейшего повы-
шения общей скорости процесса в рассматриваемом случае.
Влияние внутренней диффузии определяют проведением опы-
тов с зернами различного размера. Опыты проводят при такой тем-
пературе и такой скорости потока, когда дальнейшее их увеличение
не влияет на скорость процесса. Результаты опытов могут быть
показаны в виде графической зависимости г — f(l/R), где R— ра-
диус частицы (рис. III.6). Из рис. III. 6 видно, что при 1/R < l//?i
размер частиц оказывает влияние на общую скорость процесса,
следовательно, эта область является внутридиффузионной
72
Чтобы получить представление о взаимном влиянии скорости
внещней диффузии (гф) и скорости химической реакции (гх.р) на
обшую скорость гетерогенного процесса, рассмотрим процесс хи-
мического взаимодействия между газом и твердым реагентом,
Рис. III. 5. Зависимость общей ско-
рости процесса г от температуры Т
и скорости потока w (ац < а>2 <
/—кинетическая область; // — переходная
область; ///—диффузионная область.
Рис. III. 6. Зависимость общей ско-
рости процесса г от радиуса частиц
твердого реагента /?.
когда необратимая реакция первого порядка протекает с образо-
ванием газообразного продукта. Последний диффундирует через
пограничный слой газа и попадает в основ-
ной его поток. Реакция протекает по урав-
нению
Рис. III. 7. Схема взаимо-
действия газообразного
реагента А с твердым
реагентом В:
газ; 2—твердый реагент;
3—’пограничный слой газа.
А (г) + В (т) -> R (г) (III. 3)
.(индекс г относится к газу, индекс т отно-
сится к твердому реагенту).
Реакция протекает на плоской поверх-
ности твердого реагента, к которой из тур-
булентного потока газа диффундирует ис-
ходный реагент А через прилегающий к
поверхности пограничный слой газа толщи-
ной б. При этом изменение концентрации
по толщине слоя происходит що линейному
закону (рис. III. 7).
В данном случае процесс включает диф-
фузионный (физический) перенос газооб-
разного реагента, скорость которого Гф, и
химическую реакцию, скорость которой гх. р.
Скорость диффузионного переноса реагента, отнесенная к еди-
йиЦе поверхности, выражается уравнением
r.= — D
Ф
АС
6
(а)
73
где D — коэффициент диффузии;
в — толщина пограничного слоя газа;
АС — движущая сила процесса, равная
ДС = С4,г-С4,т (б>
СЛ(Г и ^A,t~ концентрация реагента А в объеме газа к у твердой поверхности
(в зоне реакции).
Подставив значение АС из уравнения (б) в уравнение (а), на-
ходим:
ГФ= д' г ~ СА, т) = ~ ₽г (СЛ, г — СА, т) W
где fr — коэффициент скорости массоотдачи в газовой фазе, отнесенный к еди-
нице поверхности:
₽г=4 <iv-4>
Скорость химического взаимодействия для рассматриваемой
реакции выражается уравнением:
гх, р ^т^А, т
где kt — константа скорости реакции.
Для установившегося процесса
гф = гх.р (Д>
Следовательно
гф= гх. р = 0г (С.4, г С.4, т) = ^т^А, т
откуда
Из уравнения видно, что Са, т = /(Мт), т. е. Са, т зависит и
от скорости диффузионного переноса, и от скорости химической
реакции.
Подставив в уравнение (е) значения СА,? из уравнения
(III. 5), получаем
гф в гх. р “ “ ktCA, pr + fcT СА, Т = - 1/Лт + 1/рг СА. г <Ш- 6>
Так как концентрацию реагента А в зоне реакции СА,г опреде-
лить невозможно и, следовательно, нельзя определить движущую
силу процесса АС — СА, г—Са, т, отнесем общий коэффициент
скорости процесса К к концентрации исходного реагента в основ-
ном газовом потоке С а, г, т. е. на границе пограничного слоя газа.
При этом получим
-К = -^₽-------(IIL7>
GA. г GA, г
Из уравнения (III. 6) находим:
К= 1Дт + 1/₽г . (П1,8>
74
(III. 9)
№ + ₽r
Из приведенных данных следует, что для реакции первого по-
рядка, когда концентрация исходного реагента изменяется по тол-
щине пограничного слоя по линейному закону, обратная величина
коэффициента скорости процесса является суммой химического и
диффузионного сопротивлений.
Анализируя уравнение (III.9), можно установить влияние от-'
дельных параметров на величину К. Если k? рг, из уравнения
(III. 9) следует
1/К==1/рг, или К = Рг
т. е. общая скорость процесса определяется скоростью диффузии
(процесс протекает в диффузионной области).
Для повышения скорости такого процесса необходимо увели-
чить коэффициент рг- Обычно это осуществляют путем уменьше-
ния толщины пограничного слоя газа [б в уравнении (III. 4)]. Для
этого увеличивают скорость газового потока или усиливают его
турбулизацию. Влияние упомянутых факторов на величину б
можно проследить на примере уравнения движения жидкой фазы
в трубе:
*=64,2r?^
где d — диаметр трубы;
Re —критерий Рейнольдса;
п wd
v
w — скорость потока;
v—кинематическая вязкость.
Из уравнения видно, что б уменьшается почти прямопропор- .
ционально скорости потока (пропорционально к>7/8).
Когда kT < Рг, из уравнения (III. 9) следует, что К « ki- Сле-
довательно, общая скорость процесса определяется скоростью хи-
мической реакции (процесс протекает в кинетической области).
Для увеличения kT и общей скорости процесса необходимо повы-
шать температуру (величина kT экспоненциально возрастает при
повышении температуры [уравнение (11.24)]) или применять ката-
лизатор, снижающий энергию активации. При протекании сложных
реакций применяют катализатор селективного действия.
В общем случае, когда протекает не одна, а несколько реак-
ций, зависимость коэффициента скорости процесса выражается бо-
лее сложными уравнениями; в общем виде эту зависимость можно
выразить так:
для кинетической области
Ккин f (^1> ^поб) ' (III. 10)
kt, ka, kaaQ — константы скоростей прямой, обратной и побочных реакций.
75
цля диффузионной области
Кдиф = ф(°Рo'it d;,...) (III. 11)
где Pi, D2 — коэффициенты диффузии исходных газовых веществ в зону реак-
ции;
Рр Р^ — коэффициенты диффузии продуктов реакции из зоны реакции;
для переходной области ’
*пер = Ф(*1> k2> *поб> Dx, D2,..., D{, Р',...) (III. 12)
Из приведенных данных видно, что коэффициент скорости про-
цесса К зависит от многих показателей, поэтому способы его уве-
личения устанавливают с учетом особенностей каждого химико-
технологического процесса.
Поверхность контакта фаз
Поверхность контакта фаз может быть увеличена главным об-
разом за счет соответствующего аппаратурного оформления цро-
цесса, т. е. путем применения различных по устройству реакторов,
Рис. III. 8. Реакторы для осуществления химических процессов
г — ж:
а—башня с насадкой; б—полая башня; /—башня; 5—холодильник; 3—насос.
Например, в системе г — ж процесс часто проводят в башнях
с насадкой или в полых башнях. В первом случае насадка оро-
шается жидкостью, которая, стекая вниз, смачивает насадку
(рис. ПГ. 8). Газ проходит через насадку и соприкасается с жид-
костью, смачивающей насадку. Поверхность жидкости, т. е. по-
верхность контакта фаз (Г); тем больше, чем больший объем на-
садки приходится на единицу объема пропускаемого газа, чем
меньше размер насадки и чем более развита поверхность этой на-
садки.
76
Поверхность 1 м3 различных по размерам и устройству насадок
приведена в справочной литературе. Так, например, суммарная
поверхность правильно уложенных керамических колец размером
са у 50X5 мм составляет 100 м2 • м*3, а колец размером 100 X
у 100 X Ю —60 м2 • м“3-
В полых башнях поверхностью соприкосновения фаз служит
поверхность капель, образующихся при разбрызгивании жидкости
в башне с помощью различных устройств (рис. III. 8, б). Общая
поверхность (поверхность контакта фаз F) определяется по фор-
муле: .
где F — поверхность, приходящаяся на 1 кг разбрызгиваемой жидкости;
р — плотность жидкости, г/см3;
R — радиус капель, см.
Так, если плотность жидкости р = 1 г/см3, из уравнения (III. 13)
следует:
Л, см........ 1,0 0,1 0,01 0,001
F, м2 ........ 0,3 3 30 300
В некоторых случаях в системах г — ж процесс осуществляется
в барботажных и пенных аппаратах, в которых газ в виде отдель-
ных пузырьков барботирует через слой жидкости. При этом по-
верхностью контакта служит внутренняя поверхность пузырьков:
чем меньше размер пузырьков газа и чем выше слой жидкости,
тем больше поверхность F.
Для системы г—т увеличение поверхности соприкосновения
фаз достигается измельчением твердой фазы. Газообразное веще-
ство приводят в соприкосновение с измельченным исходным веще-
ством самыми разнообразными способами, например, твердые ча-
стицы вещества располагают на полках реактора, а поток газа
Движется над полками. В других случаях тонко измельченное
твердое исходное вещество распыляют в'потоке газообразного ис-
ходного вещества в полом объеме; таким образом сжигают пыле-
видное топливо в топках паровых котлов.
В реакторах с псевдоожиженным (кипящим) слоем поверх-
ность соприкосновения фаз тем больше, чем мельче частицы зер-
нистого материала и чем выше слой этого материала.
Движущая сила процесса
Движущая сила процесса выражается уравнением
ЛС = Сп.ф-С3.р (III. 14)
Где Сп. ф — концентрация исходного вещества в передающей фазе;
С3. р — концентрация исходного вещества в той же фазе в зоне реакции.
77
Из уравнения (III. 14) следует, что существуют два способа
повышения АС: за счет увеличения Сп. <j>. и снижения С3. р.
Движущая сила процесса возрастает, если увеличить концен,
трацию исходных реагентов в передающей фазе (Сп. ф.) или пову.
сить давление, а также если выводить продукты реакции из сферу
взаимодействия.
Увеличение давления является эффективным способом пову.
шения ДС, особенно для системы г — ж, так как в этом случае
ДС = - р*
где рг — парциальное давление исходного газообразного реагента в газовой фазе’
р* — равновесное давление исходного реагента у поверхности жидкости.
При повышении давления процесса рг увеличивается, а р*
остается постоянным, следовательно, при увеличении давления
растет также ДС.
Вывод продуктов из сферы реакции также является эффектив-
ным способом увеличения движущей силы и широко используется
Рис. Ш.9. Изменение движу-
щей силы процесса ДС во вре-
мени т.
в самых разнообразных производст-
венных процессах. При выводе про-
дуктов реакции из зоны реакции
уменьшается объем реакционной сме-
си, а Сг {или рт) соответственно уве-
личивается.
Рост ДС за счет снижения р* на
практике осуществляется редко. Для
систем г — ж это достигается путем
уменьшения температуры^ так как од-
новременно понижается равновесное
давление вещества над жидкостью
(р*). Однако практические возможно-
сти для использования этого приема
ограничены, поскольку при понижении
температуры сильно уменьшается константа скорости реакции
(уравнение Аррениуса) и общая скорость процесса.
По мере протекания реакции концентрация Стуменьшается, со-
ответственно снижается движущая сила ДС, а также уменьшается
общая скорость процесса, поэтому на практике в целях поддержа-
ния высокой скорости процесса его ведут при достаточно большом
значении ДС, ограничивая время пребывания реакционной смеси
в реакторе некоторым оптимальным значением тОпт (рис. III. 9).
3. МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ - ОСНОВНОЙ МЕТОД
РАСЧЕТА ХИМИЧЕСКИХ ПРОЦЕССОВ
В промышленности встречаются самые разнообразные гетеро-
генные процессы; особенно широкое распространение получили ге-
терогенные процессы в системах г — гиг — ж, на их основе оформ-
лены многочисленные крупнотоннажные химические производства.
78
р качестве примера можно привести следующие процессы!
Общая схема Пример
аА (г) + В (т) -> /? (т) 0,50г + Zn —> ZnO
Л {г) + 6В (т)-> г/? (г) Ог + 2С —► 2СО
aX(r) + 6B(T)->rJ?(r) + sS(T) 1102 + 4FeS2 —► 8SO2 + 2Рег0>
А (ж) + В (т) -> R (г) + S (т) НгО + СаСг —► С2Н2 + Са(ОН)2
А (г) + В (ж) -> В (ж) SO3 + H2O —> H2SO4
Моделирование — это метод исследования, при котором свой-
ства объекта изучаются не на самом объекте, а на его модели,
в которой специально создаются такие же либо аналогичные ре-
альному процессу условия. При этом исходят из того, что, если
существуют две системы, которые ведут себя аналогичным обра-
зом, то более удобную для исследования систему называют м о-
делью, или аналогом.
Различают два вида моделирования: физическое и математиче-
ское.
При физическом моделировании процессы в объекте
и в модели не отличаются по физической природе, но сами систе-
мы различаются, например, размерами.
В этом случае опытные данные, описывающие такие системы,
можно представить в форме безразмерных комплексов, составлен-
ных комбинацией различных физических величин-и линейных раз-
меров. Такая форма описания позволяет распространить найден-
ные зависимости на группу подобных друг другу явлений, харак-
теризующихся постоянством определяющих безразмерных комп-
лексов, или критериев подобия (Рейнольдса, Фруда, Архимеда,
Пекле, Прандтля, Нуссельта и др.). Поэтому физическое модели-
рование сводится к воспроизведению постоянства определяющих
критериев подобия в модели и в объекте.
При математическом моделировании процесс изу-
чается на математической модели, представляющей собой описа-
ние на языке математики отдельных сторон процесса. Это описа-
ние объединяет опытные факты и устанавливает взаимную связь
между параметрами процесса, связь выражается в виде математи-
ческих уравнений.
Исследование математической модели обычно проводят на элек-
тронно-вычислительной машине (ЭВМ), обладающей большими
возможностями в отношении проведения вычислительных опера-
ций. Во многих случаях на основе результатов, полученных при
проведении исследований на ЭВМ, представляется возможным ве-
сти проектирование химических процессов без проведения иссле-
дований на опытной установке, но при условии достаточно полного
изучения кинетики процесса в лабораторных условиях.
Каждая из указанных моделей может быть использована для
нахождения оптимальных условий при оформлении как отдельных
Физических и химических операций, так и всего химического
79
производства в целом. Математическое моделирование имеет ряд
существенных достоинств по сравнению с физическим моделирова-
нием, так как оно не связано с воспроизведением реального про.
цесса и может быть использовано практически для описания
любых процессов: физических, химических, биологических и др.
Это позволяет при относительно небольших материальных затра-
тах исследовать различные варианты технологического и аппарат
турного оформления процесса, изучить его основные особенности
и найти оптимальное решение.
Математическое моделирование включает три этапа:
1) составление математической модели;
2) решение математической модели (составление алгоритма);
3) установление адекватности модели изучаемому объекту.
Под составлением математической модели (математического
описания процесса) подразумевается составление формул (урав-
нений), графиков, таблиц, количественно описывающих зависимо-
сти между различными параметрами процесса. К таким парамет-
рам относятся скорость химических реакций, концентрация ре-
агентов, температура и расход вещества, геометрические размеры
аппаратов и т. д.
Второй этап заключается в создании алгоритма, т. е. разра-
ботке методов решения математического описания для нахождения
численных значений определяемых параметров.
На третьем этапе моделирования должно быть показано, что
принятая модель с достаточной точностью описывает моделируе-
мый объект.
Наиболее сложным и трудоемким этапом математического мо-
делирования является первый, так как для составления математи-
ческого описания должны быть известны зависимости между мно-
гочисленными параметрами технологического процесса, выражен-
ные в математической форме. Эти зависимости устанавлйвают на
основе всесторонних исследований отдельных аппаратов, узлов и
всего химико-технологического процесса в целом. При этом в боль-
шинстве случаев получают системы громоздких нелинейных урав-
нений высшего порядка, включающих большое число неизвестных.
Исследование такой системы связано с огромными трудностями,
поэтому в каждом отдельном случае с целью упрощения матема-
тического описания устанавливают степень влияния отдельных
параметров на экономическую эффективность процесса и, по воз-
можности, исключают из системы уравнений те параметры, кото-
рые оказывают незначительное влияние на общую эффективность
производства. Правда, при этом несколько снижается точность по-
лучаемых результатов, однако уменьшается число вычислительных
операций. Ведущая роль на этом этапе (при составлении и упро-
щении математического описания) принадлежит инженеру-химику,
который, хорошо зная процесс, должен составить математическое
описание, представляющее разумный компромисс между желае-
мой точностью и сложностью получаемых выражений.
80
Все физические и химические операции, составляющие техно-
логическую схему производства, должны быть оформлены так,
чтобы обеспечивалась возможно более высокая экономическая
эффективность всего производства в целом. При этом для каждой
физической и химической операции должны быть составлены ма-
тематические описания всех протекающих в них процессов. На
оСНовании полученных данных составляют математическую модель
процесса в целом, позволяющую с помощью современных вычи-
слительных средств найти оптимальные значения параметров, за-
кладываемых в проект и обеспечивающих минимальную себестои-
мость получаемого продукта.
В настоящее время нахождение оптимальных значений пара-
метров, основанное на математическом моделировании, для подав-
ляющего большинства химических процессов невыполнимо ввиду
отсутствия достаточно полного математического описания отдель-
ных аппаратов, узлов и всего производства в целом. Поэтому вы-
бор оптимальных значений параметров на основе математического
моделирования производят только для наиболее важных физиче-
ских и химических операций; остальные же операции оформляются
пока на основе обычных инженерных расчетов с выбором значе-
ний параметров по результатам лабораторных и опытных работ,
анализа показателей работы действующих производств и инже-
нерной интуиции. Достаточно полные математические описания
составлены в настоящее время только для некоторых наиболее
простых и хорошо изученных технологических процессов.
Моделирование процессов в системах газ — твердое |
и жидкость — твердое
Модель взаимодействия должна достаточно точно отражать
действительную картину процесса, а описывающие ее уравнения
должны быть простыми и удобными для использования в техноло-
гических расчетах.
Для реакций твердых частиц с газом или жидкостью возмож-
ны две идеализированные модели взаимодействия: квазигомоген-
ная модель и модель частицы с невзаимодействующим ядром.
Квазигомогенная, или условно гомогенная, модель предпола-
гает, что окружающий твердую частицу газ проникает внутрь нее.
и реакция протекает во всем объеме частицы. Следовательно, весь
Реагент постепенно превращается в продукт реакции (рис. III. 10).
Дак, из рис. III. 10 видно, что концентрация твердого реагента
о незначительно изменяется по сечению частицы (по радиусу /?)
и> следовательно, скорость реакции и.степень превращения также
изменяются незначительно во всем объеме.
Модель частицы с невзаимодействующим ядром основана на
том, что реакция начинается на внешней поверхности частицы и
°на реакции постепенно перемещается внутрь частицы, иными
Сл°вами, происходит фронтальное продвижение зоны реакции
81
внутрь частицы. За фронтом продвижения зоны реакции остаются
продукт реакции и инертная часть твердого реагента, обычно на--
зываемая золой. Следовательно, по мере протекания реакции раз-
мер ядра частицы твердого реагента уменьшается; при этом кон-
центрация исходного реагента в золе равна нулю, а в ядре частицы
она сохраняет первоначальное значение и постоянна по сечению
частицы (рис. III. 11).
Поскольку на практике часто встречается вторая модель, она
и будет принята нами в дальнейшем для математического описа-
Ro О Ro Я Ro О Ro Я
Рис. ШЛО. Квазигомогенная модель
процесса (До — радиус частицы;
Ст — концентрация твердого ре-
агента);
а — низкая степень превращения; б—высо
кая степень превращения
Рис. Ш.11. Модель процесса с не-
взаимодействующим ядром:
а—низкая степень превращения; б—высо-
кая степень превращения; 1 — ядро; 2—зо-
ла; 3—зона реакции.
ния модели процесса. Уравнения модели выводятся применительно
к системе г — т, но полученные результаты справедливы также и
ддя системы ж — т.
Модель частицы с невзаимодействующим ядром. Рассмотрим в качестве при-
мера процесс горения частицы угля; он складывается из пяти.отдельных стадий
(стр. 69). В реальных условиях некоторые из перечисленных стадий могут от-
сутствовать. Если, например, в результате реакции газообразные продукты не
образуются, будут отсутствовать внутренняя и внешняя диффузии продуктов
реакции (стадии 4 и 5). Однако в любом случае весь процесс представляет со-
бой ряд последовательных сопротивлений, тормозящих реакцию. Причем, сопро-
тивление, возникающее на отдельных стадиях, может быть различным, поэтому
для вывода расчетных уравнений и установления способов интенсификации про-
цесса в целом следует прежде всего определить лимитирующую стадию, которая
в наибольшей степени определяет скорость всего процесса. Таким образом, что-
бы провести анализ реального технологического процесса, необходимо прежде
всего определить, протекает ли процесс в области внешней или внутренней диф-
фузии, в кинетической области или же он протекает в переходной области.
Для вывода уравнений взаимодействия в системе г—т рассмотрим необрати-
мую реакцию, в которой газообразный реагент А (г) вступает в химическое вза-
имодействие с твердой частицей (сферической формы) с образованием твердого
продукта.
Л(г)+В(т)-Ж(т)
(III. 18)
82
Исходя из модели частицы с невзаимодействующим ядром, рассмотрим три
Учая взаимодействия, когда процесс лимитируется внешней диффузией, вну-
пенней диффузией либо скоростью химической реакции.
тРе задача состоит в том, чтобы установить влияние различных факторов на ин-
рнсивиость процесса. В данном случае интенсивность процесса характеризуется
пеменем т, в течение которого сферическая частица твердого реагента В(т) пер-
оначального радиуса Ro прореагирует настолько, что ее радиус станет равным
р или же прореагирует полностью, когда R = 0.
Л’ Таким образом, необходимо установить функциональную
оазличных факторов.
F Первый случай взаимодействия — процесс
лимитируется внешней диффузией (рис. III. 12)
При этом протекает быстрая химическая реак-
ция, поэтому концентрация газообразного ре-
агента А (г) у поверхности ядра твердой ча-
стицы очень мала и ее можно принять равной
,нуЛК> (Са,г = 0). Поскольку скорость вну-
тренней диффузии велика, можно принять
С = 0. Таким образом, снижение СЛ про-
исходит по толщине пограничного слоя от С А г
до СЛЛо = О (см. рис. III. 12).
В данном случае, как и в рассмотренном
ранее аналогичном процессе взаимодействия
газа с твердой плоской поверхностью (стр. 73),
процесс включает два акта: диффузионный
перенос исходного реагента А через погранич-
ный слой газа (физический процесс), скорость
которого Гф, и химическую реакцию, скорость
которой равна Гх. р.
Для установившегося процесса, т. е. для
стационарных условий, скорости этих процес-
сов равны между собой, поэтому можем напи-
сать
зависимость т от
Рнс. III.12. Изменение концен-
трации реагента А в ходе реак-
ции, лимитируемой внешней
диффузией:
1—ядро; 2—зола; 3—пограничный
слой газа.
Определим каждую скорость и прирав
няем их.
Скорость диффузии через пограничный слой газа можно выразить в виде
Уравнения
гф = ₽гАС (б)
где рг — коэффициент скорости массоотдачи по газу, т. е. диффузней;
ДС—движущая сила процесса.
Так как скорости химической реакции и внутренней диффузии велики и
Сл>л = 0 н СЛ(Л, = 0, то „
!^С = СА> г — CARi = Сл, R — 0 = СЛ, г (в)
^ = PA.r (III. 16)
Скорость рассматриваемой химической реакции, т. е. скорость уменьшения
количества реагентов А и В, отнесенная к единице поверхности контакта фаз,
одинакова для каждого из этих реагентов и выражается уравнениями
1 dlV. 1 dRR
гх.р = —= (г)
где к*
Я г — поверхность контакта фаз;
A’ Nв — количества исходных реагентов А н В.
83
Для сферической частицы
F = 4я*о (д)
Поэтому уравнение (г) запишется в таком виде
1 dN
Гх-!> = ~ (е)
4л7?0 dx 1
Подставив полученные значения для гх. р и г$ в уравнение (а), находим
1 dN.
РгСАг = -
4Л-<\0 иХ
ИЛИ
dNA = - 4лЛоРгСл> rdr (III. 17)
Количества прореагировавших реагентов А и В определяют из уравнений:
dNA ~ dNB = PBdV = PBd (4/3 л 7?3) (III. 18)
или
dNA — 4npBR2dR
где V — объем частицы;
р„ — плотность частицы.
Приравняв правые части уравнений (III. 17) и (III. 18), находим (
— 4л^ргСл r dx = 4лрBR2 dR
откуда
R2 dR
Рг^А rR0
। пределах изменения т от 0 до т и R от Ro до
В
( Я2 dR = -о Pg (/?03 - R3)
i 3₽гСдЛ2^0 7
dx — —
Интегрируя это уравнение в
R, получим
Рд
₽/* А г^О
т, е.
_ РдЯр
з₽/\
з
о
(Ш. 19)
При полном превращении реагентов В радиус ядра равен О (R = 0), по-
этому, подставив это значение в уравнение (III. 19), находим:
_ Рд^о -
" 3?гСА г
где тп — время полного превращения частицы.
Разделив пЗчленно уравнение (III. 19) на (III. 20), получаем:
*п \rJ
(III. 20)
(III. 21)
В ряде случаев удобно определять время контакта через степень превраше-
ния Хв, которая в данном примере может быть выражена следующим образом
X Vb' ~ Vb’ r 1 _ Vb’ ‘ (з>
в vb, к, Vb. к,
где Хв — степень превращения реагента В;
Vb VB, д —начальный и конечный объем частицы.
84
Pio объем частицы радиусом Ко и R можно выразить в виде
^В, 4/з ЛЛд И ^В1^===4/зЛЛ
Тогда, подставив эти объемы в уравнение (ж), получим
Хв = 1
D
1?о /
(III. 22)
подставив Хв в уравнения (III. 19) и (III. 21), находим
Рд^О У
3₽гС4,Г Л
г
^п=*В
(III. 23)
(III. 24)
Как видно из уравнения (III. 24), зависимость Хв от г/тп является примо*
линейной (рис. Ш. 13, кривая /).
Таким образом, из уравнений (III. 19) и (III. 20) следует, что для интенси-
фикации процесса, протекающего во внешнедиффузионной области (либо для
уменьшения времени, затрачиваемого на его проведение), необходимо:
Рис. 111,13. Зависимость степени пре-
вращения Хв от г/гп:
1—лимитирующая стадия — внешняя диффу-
зня; 2—лимитирующая стадия —внутренняя
Диффузия; 3—лимитирующая стадия—хи-
мическая реакция.
Рис. III.14. Изменение концентрации?
реагента А в ходе реакции, которая
лимитируется внутренней диффузией;
/ — ядро; 2— зола; 3— пограничный слой
газа.
а)в уменьшить размеры твердых частиц, поскольку Ro входит в числитель
правой части уравнения (III. 20) и с уменьшением его значения снижается и т;
б) увеличить коэффициент скорости массоотдачи по газу, применяя переме-
вание или увеличивая скорость газового потока;
в) увеличить концентрацию реагента А в газовой фазе Слт-
“торой случай — процесс лимитируется внутренней диффузией (рис. III. 14).
pear В этом слУчае можно записать г$ = гх. р (где Гф — скорость диффузии-
пита через слой золы, т. е. скорость внутренней диффузии).
Скорость внутренней диффузии можно выразить с помощью уравнения Фика;
dC.
85
а скорость химической реакции выражается ранее полученным уравнением (м
стр. 84, где вместо Яо следует подставить Я 1 '
1 dRA 1 dNA
rx'pS=~T'~dX~ = ~‘^iRr'~dx~
где D — коэффициент диффузии реагента А через слой золы.
Приравняв значение гх. р и г$, получим
1 dN. dC. dN. dR
---7"nf • = D , или — = 4nD dCa
. 4лЯ2 dx dR dx R2 Л
dN A
Примем вначале, что - --- - не зависит от R. Тогда
или
dNA С dR Г
— \ = \ dG.
dx J Я2 J А
Я» Сд, г
dN. Г I I 1
~dX~ [~ Т + ЯП = 45lD аСЛ'
dNA Г I I
dx [Яо Т]“4яВСЛ,г
(III. 26)
dN А
Допущение, что ~ не зависит от R, не соблюдается в реальных усло-
виях. Размер ядра изменяется во времени, поэтому по мере увеличения толщины
слоя золы скорость диффузии газа по толщине этого слоя снижается, соответ-
dNA „
ствеино изменяется и - Для получения уравнения, соответствующего реаль-
ным условиям, подставим в уравнение (III. 26) значение dNд из уравнения
(III. 18). В результате получаем
4лрЯ2<1ЯГ1 11^ , [>BRdR f \ 1\
dx Lflo я]- П д>г DC„r (я0 я)
Д,г
После интегрирования уравнения получаем
R 1
рв 1 (
^.4*7
А О
Рд
ПС.
Л.г
Я»
(III. 27)
или
i2
на
(III. 28)
Чтобы определить полное время превращения твердой частицы, подставим в
уравнение (III. 28) значение R = О
РвЪ
тп
(III. 22)
^СЛ.г
se
g лй Разделить почленио уравнение (III. 28) на уравнение (III. 29), тогда полу-
чим
(III. 30)
— = 1-3
тп
Чтобы выразить т/тп = f (Хл), определим значение Я/Яо из уравнения (III. 22)
(III. 31)
£-0-V-
я подставив его в уравнение (III. 30), находим
J~3(1-V’ + 2(1-XB)
(III. 32)
ТП
Полученная зависимость отражена на рис. III. 13, из которого
видно, что если процесс лимитируется внутренней диффузией, то его
ная скорость выше скорости процесса, протекаю-
щего в области внешней диффузии.
Из уравнений (III. 28) и (III. 29) следует,
что интенсивность процесса, протекающего во вну-
тридиффузионной области, сильно зависит от сте-
пени измельчения, поскольку радиус частицы вхо-
дит в числитель во второй степени. Для интенси-
фикации процесса необходимо также создавать
условия, ускоряющие диффузию газа через слон
золы (в частности, повышать ее пористость), и
увеличивать концентрацию реагента А в газовой
фазе (Сд,г).
Третий случай взаимодействия — процесс ли-
митируется химической реакцией (протекает в
кинетической области). Так как скорости процес-
сов внешней и внутренней диффузии велики и не
лимитируют общий процесс, то концентрация ре-
агента А по толщине пограничного слон и по тол-
щине слоя золы практически не изменяется и
может быть принята постоянной, равной Cat-
В зоне реакции (у поверхности ядра частицы)
она скачкообразно снижается до нуля
(рис. III. 15).
Таким образом, скорости переноса исходного
реагента А в результате и внешней и внутренней
Диффузии настолько велнки, что общая скорость
процесса целиком определяется скоростью хими-
ческой реакции. Если эту скорость отнести
^единице поверхности контакта фаз, то ее можно выразить в виде уравне-
rx.p~ k(^A,r •
ГДе ы~~ констапта скорости химической реакции.
Но скорость убыли исходного реагента А в результате химической реакции
Жет быть также выражена в виде
1 dNA 1 dNA
Гх‘р р' я 4лЯ2 ’ dx
Ваи„ПРиРавняв полученные значения для гх. р и сделав необходимые преобразо-
няя> находим
dNA
- dx~ kC^-AnR*
(кривая 2)
относитель-
Яд Я OR Яо я
Рис. III.15. Изменение кон-
центрации реагента А в хо-
де реакции, протекающей
в кинетической области:
/— ядро; 2—зола; 3—погранич*
ный слой газа.
(III. 33)
87-
Но
dNA = tetpBW dR
dT=s~^7dR
или после интегрирования
.поэтому
>0
При R = 0 уравнение примет вид
, _ РвЛО
п~ kC -.
А, г
Разделив почленно уравнение (III. 34) на уравнение (III. 35), а также учитыва?
уравнение (III. 31), находим
(Ш.36,
Зависимость, выражаемая уравнением (III. 36), представлена иа рис. III, 13
(кривая 3), из которого следует, что, если процесс протекает в кинетической об-
ласти, то степень превращения находится примерно в такой же зависимости от
т/tn, что и при протекании процесса во внутридиффузионной области.
Из уравнений (Ш. 34) и (III. 35) видно, что для интенсификации процесса,
протекающего в кинетической области, необходимо увеличивать константу скоро-
сти реакции k (путем повышении температуры и применении катализатора),
уменьшать размер частиц (Ro входит в уравнения,в первой степени) и повышать
концентрацию реагента А в газовой фазе СА,Г.
В табл. III. 1 приведены уравнения для определения времени,
в течение которого первоначальный радиус частицы /?0 станет
равным нулю или равным R для трех рассмотренных выше слу-
чаев.
Таблица III. 1. Время взаимодействия газа
с твердой частицей сферической формы
(П1.35.
Конечный радиус равен
нулю (R=0)
Конечный радиус равен К
Полученные кривые зависимости Хв = /(т/тп) позволяют опре-
иТь область, в которой протекает процесс. Для этого проводят
Де> опытов, в которых определяют Хв при различных значениях
сеР затем строят кривую Ав = /(т/тп) и сравнивают ее с теоре-
^ческими кривыми. Если опытная кривая совпадает с кривой 1
тИ рис. III-13, процесс протекает в области внешней диффузии;
опытная кривая совпадает с кривой 2 или 3, это указывает
е т0> что процесс протекает в области внутренней диффузии или
в кинетической области.
При изменении условий гетерогенный химический процесс
может переходить из одной области в другую. Это можно
проиллюстрировать графической зависимостью общей скорости
процесса от температуры r — f(T)
(рис. Ш. 16).
При низкой температуре общая
скорость процесса определяется ско-
ростью .химической реакции, посколь-
ку она мала. При Tj прямые 1 и 2 пе-
ресекаются в точке А, при этом Гф —
= Гх.р. При дальнейшем повышении Т
общая скорость процесса ограничива-
ется значением гВц. диф (2), так как
последняя увеличивается в этих усло-
виях в меньшей степени, чем ~
(Ввн. диф О?'2) .
В точке В, где пересекаются
МЫе 2 И 3, Гвн. диф = Гвнут. диф.
дальнейшем повышении Т общая
рость процесса снижается, так
она изменяется ----------“
чем Гвн. диф. (Пвнут.дИф = ЬТ.
Таким образом подтверждается сделанный ранее вывод, что во-
всех случаях общая скорость процесса не может быть выше скоро-
сти составляющих ее отдельных этапов.
1\. р
пря-
При
ско-
как
в меньшей степени,
Область внешней диффузии
_ — Рв^°
1 -звсд.
= а
т = а
'о ) J “ аЛВ
Рв^о
6^Л,Г “₽
Область внутренней диффузии
в{ф_3(1_хв)2'з + 2(1_Лв)-|
Кинетическая область
Рд^о
kC. г ~Y
Л, г
т3 =
общей,
темпе-
Рис. III.16. Зависимость
скорости процесса г от
ратуры Т:
1~rx.-pmfV)- ^вн. диф
3"'гвиутр. диф. = ф 4 опытная-
кривая.
Моделирование процессов в системах газ — жидкость
и жидкость — жидкость
Закономерности, отражающие процессы, протекающие в си-
стемах г—ж и ж—ж, аналогичны, поэтому ниже будут рассмотре-
ИЬ1 только процессы в системах г—ж.
При соприкосновении газа, содержащего реагент А, с жид-
костью, содержащей растворенный реагент В, газообразный1
Реагент абсорбируется жидкостью и растворяется в ней. Рассмот-
рим условия, при которых реагенты вступают во взаимодействие
в соответствии с уравнением
А (г) + В (ж) -> R (ж)
(III. 37)
8»
88
В зависимости от свойств реагентов и жидкости профили кон-
центраций реагентов в пограничных слоях газа и жидкости могут
быть различными.
Рассмотрим условия, когда взаимодействие между реагентами
происходит в пограничном слое жидкости. В этом случае общее
уравнение скорости процесса такое же, что и для рассмотренной
ранее системы г—т, и должно отражать две стадии взаимодействия
реагентов — массообмен реагентов между фазами и собственно
химическую реакцию. Соотношение скоростей этих стадий может
изменяться в широких пределах.
В одном, предельном, случае проте-
кает настолько быстрая химическая
реакция, что скорость диффузии ве-
ществ целиком лимитирует общую
скорость процесса. В другом случае
химическая реакция протекает на-
столько медленно, что газообразный
реагент абсорбируется жидкостью и
достаточно равномерно распреде-
ляется в ней, при этом общая ско-
Рис. III.17. Распределение кон-
цёитрации реагентов реакции
Лг + Вж -> в пограничных
слоях газа и жидкости:
/«—поверхность раздела фаз; ZZ —погра-
ничный слой газа; ZZZ—пограничный
слой жидкости; ZV — турбулентный по-
ток газа; И—турбулентный поток жид-
кости; V7 —граница зоны реакции;
2—толщина пограничного слоя жидко-
сти; zr—толщина слоя жидкости, в ко-
тором протекает реакция.
рость процесса целиком определяет"
ся скоростью химической реакции.
Встречающиеся на практике процес-
сы в системах г—ж в большинстве
случаев занимают промежуточное
положение между двумя этими пре-
дельными случаями и, следователь-
но, общая скорость таких процессов
зависит как. от скорости переноса
массы реагентов между фазами, так
и от скорости химического взаимодействия.
Если реакция протекает с большой скоростью и осуществляется
в области пограничного слоя жидкости, процесс проходит через
ряд стадий (рис. III. 17), основными из них будут следующие:
1) диффузия реагента А из турбулентного потока газа через
пограничный слой газа II к поверхности раздела фаз I;
2) диффузия реагента А от поверхности раздела фаз I в объем
пограничного слоя жидкости III.
- 3) диффузия активного компонента В из турбулентного потока
жидкости через пограничный слой жидкости III (в зону реакции VI)',
4) химическая реакция в пограничном слое жидкости;
5) диффузия продукта реакции R из пограничного слоя жидко-
сти III в турбулентный поток жидкости V.
Общая скорость реакции, отнесенная к единице поверхности
раздела фаз, в рассматриваемом случае выражается уравнением
1 dNA 1 dNB
~ rA=° ~ rB=X~ Т' d-t F" dx ^А,т(РА.т Ра.м)^
“ ₽Л.ж (сл.г “ °) “ ₽в,ж (св.ж - °) (HI. 38)
90
где Рл.г — коэффициент скорости массоотдачи реагента А в газовой фазе;
й , ™ — коэффициент скорости массоотдачи реагентов А и В в жидкой
₽А* В’ фазе.
Глубина зоны жидкой фазы, в которой протекает реакция
/см, рис. III. 17), зависит от скорости химической реакции, кон-
центрации реагентов А и В, а также от коэффициентов диффузии
реагентов А и В в жидкости. Чем выше скорость реакции и выше
концентрация реагента В, тем меньше зона реакции.
При очень большой скорости (мгновенной реакции) и высокой
концентрации реагента В процесс завершается у поверхности раз-
дела фаз. В этом случае сопротивление пограничного слоя газа
становится определяющим для процесса в целом, — процесс про-
текает во внешнедиффузионной области. Соответственно уравне-
ние (III. 38) упрощается
~гА-----гв=РгрЛ1Г (III. 39)
Для интенсификации такого процесса необходимо увеличивать
парциальное давление реагента А в газовой фазе (Ра, г) и повы-
шать коэффициент скорости массоотдачи по газу ((Зг), что дости-
гается увеличением скорости газового потока и его турбулизацией. .
Влияние скорости химической реакции на скорость процесса
можно установить на основе уравнения скорости Диффузии ре-
агентов в пограничном слое жидкости, которое имеет вид
д3Сп
~дх DB дгг (Ш. 40)
где DB — коэффициент диффузии реагента В в жидкой фазе;
гв — скорость реакции в жидкой фазе.
Для реагента А можно записать аналогичное уравнение.
Левая часть уравнения (III. 40) отражает общую скорость из-
менения концентрации реагентов В в любом элементарном объеме
жидкой фазы. Первый член правой части уравнения характеризует
скорость молекулярной диффузии реагента В в пограничном слое
жидкости к поверхности контакта фаз, а второй член (гв)—ско-
рость химической реакции, т. е. изменение концентрации реагента
В в растворе в результате химической реакции.
Для стационарного состояния, т. е. для установившегося про-
цесса, концентрация реагента В во времени в любой точке реак-
тора постоянна, поэтому дСв]<к = 0. Следовательно, в рассматри-
ваемом случае процесс протекает в жидкой фазе в соответствии
с уравнением
d3CB
DB~d? гв
из которого следует, что чем выше скорость реакции, тем с боль»
*ией скоростью протекает процесс диффузии реагента В в жидкой
Фазе. Другими словами, с повышением скорости химической реак-
ции возрастает градиент концентрации реагента В в жидкой фазе,
81
(III. 41)
а это соответственно приводит к увеличению движущей силы
процесса и к повышению скорости абсорбции реагента А жидкой
фазой.
По мере уменьшения скорости химической реакции могут суще-
ствовать различные режимы. Например, если скорость химической
реакции и скорость диффузии в газовой фазе соизмеримы, процесс
протекает в смешанной (переходной) области (рис. III. 18). Если
реакции охватывает весь пограничный
слой жидкости III и попадает в тур-
булентный поток жидкости К
В этом случае скорость процесса за-
висит как от скорости диффузии в
жидкой фазе, так и от скорости хи-
мической реакции. Для повышения
скорости такого процесса необходи-
мо увеличивать скорость диффузии
реагента А в пограничном слое газа
к поверхности раздела фаз, а также
.увеличивать скорость химической
реакции.
Общее представление о влиянии
различных факторов на скорость
процесса в системе г—ж можно по-
скорость реакции мала, зона
Рис. Ш.18. Распределение кон-
центрации реагентов реакции
Лг + Вж-> Яж в пограничных
-слоях газа и жидкости при малой
скорости химической реакции (обо-
значения). лучить на основе уравнения скоро-
сти абсорбции.
В отсутствие химической реакции скорость абсорбции реаген-
та А из газовой фазы жидкостью, отнесенная к единице поверхно-
сти раздела фаз, выражается уравнением:
ГФ (III. 42)
где
лРг = Лл,г-Рл
^ж~ С А ~ ^А,ж
Ра г» Ра ~ парциальное давление исходного реагента А в газовой фазе и дав-
ление у поверхности раздела фаз;
Сд, ж — концентрация реагента А в жидкости у поверхности раздела фаз и
на границе пограничного слоя жидкости.
Если абсорбируемый реагент А вступает в химическую реакцию
с реагентом В, растворенным в жидкости, то уравнения для расче-
та скорости абсорбции усложняются. При этом влияние химиче-
ской реакции на скорость абсорбции можно учесть введением
поправок в уравнение физической абсорбции (III. 42) либо в ко-
эффициент ж, либо в ДСЖ.
В первом случае необходимо вместо рА,ж подставить р^ ж,
тогда уравнение для расчета скорости абсорбции по жидкой фазе
Примет вид
. -^.0=Рл.жДСж (Ш.43)
S2
где ₽д ж — коэффициент массоотдачи в жидкой фазе при протекании в ней хи-
! мической реакции, отнесенный к движущей-силе при физической аб-
сорбции (ДСЩ).
Отношение X — называют коэффициентом ускорения.
РЛ (ж)
Коэффициент X показывает, во сколько раз химическая реакция,-
сопровождающая абсорбцию, увеличивает количество абсорбиро-
। ванного газа по сравнению с чистой физической абсорбцией.
; Во втором случае влияние химической реакции на скорость
абсорбции можно учесть путем увеличения движущей силы аб-
сорбции, при этом вместо ДСЖ необходимо подставить ДСЖ + б.
Тогда уравнение для скорости абсорбции примет вид
j -'х.Р = ₽4,ж(ДСЖ + д)
где б — увеличение движущей силы в жидкой фазе при наличии в ней химиче-
ской реакции.
J Чтобы иметь представление о том, насколько повышается ско-
рость абсорбции при наличии химической реакции, ниже приведе-
но сравнение скоростей абсорбции СО2 водой и водными раствора-
ми NaOH и КОН в башне с насадкой:
Коэффициент
массопередачи
Вода ............................... 1
Водные растворы:
NaOH (3 н., 15% Na в виде NajCOs) . 45
КОН(2н., 15% К в виде К2СОз) ... 75
Примеры
Пример III. 1. Сравнить увеличение скорости прямой реакции окисления сер-
t чистого ангидрида на ванадиевом катализаторе с увеличением скорости диффу-
’ зии газа при повышении температуры от Л == 693 К до Т2 = 703 К-
Реакция протекает по уравнению
2SO3 + O2^2SO3 . (а)
2А + В =ё=* 27? (б)
Условия. х,
Энергии активации реакции (а) на ванадиевом катализаторе составляет
Е — 268 кДж • моль-1. Зависимость скорости диффузии газов от температуры
выражается уравнением
D2/Dl = (Тз/Т^ (в)
где £>ь D2— коэффициенты диффузии газов соответственно при температурах
I Г] и Т2; п — коэффициент, равный 1,5—2,5; в расчете принимаем п = 2.
Решение.
в . Скорость прямой реакции (б) при температурах и Т2 будет составлять
_ Е
rl = klCA = kQe ЯТ'СА
_ Е
г = k С — k е С
Г2 R2CA R0e
93
откуда
Е
Г1 _ в
kae Ы
Подставив в полученное уравнение Е =• 268 000 Дж • моль-1 и R = 8,3 Дж г
г моль-1 • К-1, находим
11 = е 8,3 \ 093-7Й J = е0,662 = 1 94
Г1
или г2 = 1,94 гр
Таким образом, при увеличении температуры на 10 К скорость реакции воз-
растает на 94%, т. е. почти в два раза.
Подставив значения Т\, Тг и п в уравнение (в), находим
или
£>1 = 1,026£>1
т. е. увеличение составляет 2,6%.
Глава IV
КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ
Каталитические процессы в настоящее время составляют
основу химической технологии, причем область их применения
расширяется: около 90% новых производств, освоенных за послед-
ние годы химической промышленностью, основаны на взаимодейст-
вии, протекающем в присутствии катализаторов.
Под катализом понимают изменение скорости химических
реакций под воздействием веществ — катализаторов, которые, уча-
ствуя в процессе, остаются после его окончания химически неиз-
менными. Катализ называется положительным, если катализатор
ускоряет реакции, и отрицательным, если скорость реакции под
воздействием катализатора снижается.
Применение катализаторов облегчает практическое осуществле-
ние многих химических реакций; скорость некоторых из них уве-
личивается в тысячи и даже миллионы раз. Очень многие промыш-
ленные процессы удалось осуществить только благодаря примене-
нию катализаторов.
К числу каталитических процессов относятся важнейшие
крупнотоннажные производства, например такие, как получение
водорода, аммиака, серной и азотной кислот и многих других
важнейших химических продуктов. Особенно велико и разнообраз-
но применение катализа в технологии органических веществ и в
производстве высокомолекулярных соединений.
Отрицательный катализ применяется значительно реже: ката-
лизаторы, замедляющие скорость процесса, называют также инги-
биторами.
04
Катализаторами могут быть вещества, находящиеся в любом
дз трех агрегатных состояний. К твердым катализаторам можно
отнести металлы и их оксиды, например Fe при синтезе аммиака,
pt при окислении аммиака, V2O5 при окислении SO2, AI2O3 при
крекинге нефтепродуктов и др.
Жидкими катализаторами служат обычно кислоты и основания,
например, H2SO4 и Н3РО4 применяются при алкилировании арома-
тических углеводородов, при изомеризации «-бутилена в изобути-
лен и др.
Примером газообразных катализаторов может служить BF3 в
процессах полимеразиции некоторых углеводородов.
Каталитические процессы можно разделить на две группы:
гомогенные и гетерогенные. В гомогенно-каталитических реакциях
реагирующие вещества и катализатор составляют одну фазу, а в
гетерогенно-каталитических реакциях — разные фазы. В особую
группу выделены микрогетерогенные и ферментативные каталити-
ческие процессы. Микрогетерогенный катализ происходит в жид-
кой фазе с участием коллоидных частиц металлов в качестве
катализаторов. Ферментативный катализ наблюдается в биологи-
ческих системах с участием сложных комплексов (часто белковой
природы), называемых ферментами.
Учение о катализе представляет очень интересную и увлека-
тельную область знаний, имеющую исключительно большое прак-
тическое значение. В настоящее время в мире проводятся очень
широкие и глубокие исследования в области изучения каталитиче-
ских процессов; этим заняты десятки тысяч ученых.
1. ОБЩИЕ ЗАКОНОМЕРНОСТИ КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
Каталитические реакции подчиняются общим законам химии
и термодинамики, но имеют при этом свои особенности, так как
в них всегда участвует один дополнительный компонент — катали-
затор. Действие катализаторов принципиально отличается от дей-
ствия других факторов, способствующих интенсификации химиче-
ских реакций, например, температуры, давления, радиационного
воздействия и др. Повышение температуры может ускорять
реакцию вследствие увеличения энергетического уровня реагирую-
щих молекул, т. е. их активации за счет вводимого извне тепла.
При этом изменяется внутренняя энергия системы и смещается
положение равновесия.
Катализатор же не влияет ни на равновесие химической реак-
ции, ни на все другие термодинамические характеристики реак-
ций. Изменяя в равной степени скорость прямой и обратной реак-
ций, катализатор способствует повышению скорости достижения
равновесия при данных условиях.
Теория каталитических процессов относится к числу сложных
и недостаточно полно изученных областей современной физической
Химии. В настоящее время еще нет общей теории, позволяющей
95
предвидеть каталитическое действие различных веществ на ту или
иную химическую реакцию. Существует несколько теорий, объяс-
няющих механизм действия катализаторов, из которых наиболее
распространенной теорией, служащей основой современных пред-
ставлений о катализе, является теория промежуточных соединений.
Согласно этой теории, медленную реакцию между исходными
веществами можно заменить двумя или несколькими более быст-
рыми реакциями' с участием катализатора, который образует с
исходными веществами промежуточные непрочные соединения.
Ускоряющее действие катализатора состоит в понижении энергии
активации реакций образующихся промежуточных соединений, что
оказывает очень сильное влияние на скорость реакции, поскольку
в уравнение Аррениуса '
Е
k = koe
энергия активации Е входит в показатель степени.
При практическом применении большое значение имеет техно-
логическая характеристика промышленных катализаторов (ак-
тивность, температура зажигания, производительность, селектив-
ность, отравляемость, прочность и др.).
Наиболее важной характеристикой катализаторов является их
активность, т. е. мера ускоряющего действия катализатора по
отношению к данной реакции. Активность определяется уравне-
нием
— -77“ & ~~ ЛЕ
А = ^- = ?°е = е = е~^ (IV. 1)
kDe
где А — активность катализатора;
k, kK — константы скорости реакции без катализатора и в присутствии катали-
затора;
ЕЕ —Снижение энергии активации под действием катализатора:
АЕ = Е — ЕХ (IV. 2)
Е, Ек — энергия активации реакции без катализатора н в присутствии катали-
затора.
Ускоряющее действие катализатора можно наглядно проследить' на при-
мере окисления сернистого ангидрида
2SO2 Ч- О2 =f=t 2SO3 + Q
которая в отсутствие катализатора протекает крайне медленно (прн 420 °C, или
693 К энергия активации составляет приблизительно 420 000 Дж/моль). Прн про-
ведении этой реакции на ванадиевом катализаторе (V2Os) — Ек — 268 000 Дж •
моль"1 К"1- Подставив эти величины в уравнение (IV. 1) н учитывая, что
7? = 8,3 Дж • моль"1 К"1, получаем:
ДЕ 420 000 - 268 000
д = ^. = е^==е 8,3-693 =^6’4=4 = 3.1011
к
т. е. скорость реакции возрастает в сотни миллиардов раз.
96
'В' Температурой зажигания катализатора называют ми-
! «нимальную температуру реагирующей смеси, при которой процесс
I Иначинает протекать с достаточной для практических целей ско-
ростью. Чем активнее катализатор, тем ниже температура зажига-
ния, что особенно важно при проведении экзотермических обрати-
Рнс. IV.1. Зависимость сте-
пени превращения X экзо-
термической обратимой реак-
ции от температуры Т для
катализаторов различной
активности прн А{ < А2.
мых реакций, так как при этом соответственно повышается степень
превращения. Для реакции А R + Q указанное положение отра-
жено на рис. IV. 1.
При работе ца катализаторе Ai с температурой зажигания Д, i
(кривая /) максимальная степень превращения составляет Xj. При
работе на более активном катализато-
ре Аг с более низкой температурой за-
жигания Т3:2 (кривая 2) максимальная
степень превращения более высокая и
соответственно будет равна X?.
При понижении температуры зажи-
гания облегчается оформление катали-
тического процесса, так как расширяет-
ся рабочий интервал температур (между
Т3 и максимальной температурой),
упрощается конструкция реактора,
уменьшается предварительный подогрев
реагентов и становится устойчивее тех-
нологический режим.
Отравление катализато-
ра — это частичная или полная потеря
его активности в результате действия
посторонних примесей — контактных
ядов. Отравление может быть обрати-
мым и необратимым. При обратимом отравлении примеси снижают
активность катализатора временно, пока они присутствуют в зоне
катализа; по удалении ядов катализатор восстанавливает свою
прежнюю активность. При необратимом отравлении активность
катализатора не восстанавливается и после удаления контактных
ядов из зоны реакции.
Активность твердого катализатора может снижаться также
вследствие уменьшения активной поверхности катализатора под
воздействием, например, высоких температур, при осаждении на
поверхности катализатора продуктов реакции или пыли, механи-
ческого разрушения катализатора и по многим другим при-
чинам.
Важной особенностью катализаторов является их избира-
тельность (селективность) по отношению к определен-
ным реакциям. В сложных реакциях (параллельных и последова-
тельных), где термодинамически возможно образование несколь-
ких продуктов, катализатор позволяет ускорить только одну
Целевую реакцию; естественно, что это имеет большое практическое
значение.
4 Зак. 558
' 67
Для сложной реакции типа
селективность выражается уравнением (11.28):
Из этого уравнения видно, что при некоторой заданной темпе-
ратуре Т путем подбора соответствующего катализатора можно
изменять разность Е2— Е\ и, таким образом, создавать возможно-
сти для получения только или главным образом целевого продукта.
Примером избирательности катализатора может служить реак-
ция окисления NH3 до NO на платиновом катализаторе
4NH3 + 5О2 = 4NO + 6Н2О + 986 кДж
протекающая полностью за десятитысячные доли секунды, т. е.
практически мгновенно. Одновременно с указанной реакцией про-
текают две параллельные реакции, при которых образуются N2O
и N2:
4NH3 + 4О2 = 2N2O + 6Н2О +1105 кДж
4NH3 + ЗО2 = 2N2 + 6Н2О + 1269 кДж
Эти реакции термодинамически более выгодны, поскольку они
сопровождаются выделением большого количества тепла, однако
практически они не протекают, поскольку в присутствии платино-
вого катализатора их скорость неизмеримо мала.
Особенно сильно селективность проявляется в сложных органи-
ческих реакциях. Так, например, этиловый спирт в зависимости от
типа катализатора и условий проведения процесса катализа мо-
жет превращаться в следующие продукты: этилен СН2=СН2,
диэтиловый эфир СН3—СН2—О—СН2—СН3, ацетон СН3СОСН3,
бутадиен СН2=СН—СН = СНг, ацетальдегид СН3СНО и другие
продукты. Следовательно, применяя соответствующий катализатор,
из одного и того же сырья можно получить различные целевые
продукты.
В присутствии катализатора обычно снижается порядок реак-
ции, причем, чем активнее катализатор, тем этот эффект сказы-
вается сильнее, т. е. тем ниже порядок реакции. В связи с этим,
кинетика каталитических реакций обычно описывается уравне-
ниями, найденными эмпирически, и формальный порядок таких
каталитических реакций будет выражаться как в виде целого, так
и дробного числа.
Влияние катализатора на порядок реакции можно проследить на примере
реакции окисления SO2
2SO2 + О2 х 2SO3 + Q
98
скорость которой без катализатора может быть выражена в виде следующего
уравнения:
dCer\ п
~ rso2=°rso,= '^1' = kicso2co1 (IV. 5)
где CsOj’ ^so,’ Со2 ~ текущие концентрации. Таким образом
п = 2 + 1 =3.
порядок реакции
При проведении этой реакции на ванадиевом катализаторе ее скорость вы-
ражается уравнением Борескова:
dCso, , ( cso2 ~ cso2 'ч°’8
~rSO2-----dr 2
cso, 7co2
(IV. 4)
где С so — равновесная концентрация SOj. В этом случае п = 0,8+ 1 = 1,8.
Для наиболее активных платиновых катализаторов справедливо уравнение
Тейлора и Ленера:
-rso =—=
s°2 dr
CSOs — cso2
:3
VCSO3
(IV. 5)
Из этого уравнения следует, что п = 1.
Таким образом в рассмотренном примере
при проведении ее без катализатора до 1,8 и
порядок реакпии снижается с 3
1 в присутствии катализатора.
В гомогенно-каталитических реакциях скорость процесса зави-
сит от концентрации не только реагирующих веществ, но и катали-
затора.
Основным недостатком гомогенного катализа является труд-
ность выделения катализатора из конечной продукционной смеси,
в результате чего часть катализатора теряется, а целевой продукт
загрязняется. Однако в последнее время ведутся обширные иссле-
дования в области высокоактивных катализаторов гомогенного
катализа, которые, присутствуя в малых дозах, вызывают цепные
реакции. Поскольку количество вводимого катализатора невелико,
после реакции он не извлекается из реакционной смеси, а остается
в целевом продукте, не снижая качества получаемого целевого
продукта.
Большой интерес к гомогенному катализу объясняется главным
образом тем, что при подборе соответствующих катализаторов ин-
тенсивность гомогенных процессов очень высока. Это объясняется
тем, что гомогенные реакции протекают на микроуровне (на уров-
не отдельных молекул), когда вероятность столкновения молекул
реагирующих веществ с молекулами катализатора весьма значи-
тельная.
2. ГЕТЕРОГЕННЫЙ КАТАЛИЗ
Большинство известных промышленных каталитических реак-
ций — это реакции между газообразными реагентами на твердых
катализаторах. Изменение реакционного пути происходит в этом
случае благодаря образованию промежуточных непрочных про-
дуктов взаимодействия реагирующих веществ с катализатором.
4* 99
В общем случае процесс гетерогенного катализа на твердых
пористых катализаторах складывается из нескольких элементар-
ных стадий (рис. IV. 2):
1) внешняя диффузия реагирующих веществ из ядра потока
к поверхности зерен катализатора (через пограничный слой газа);
2) внутренняя диффузия реагентов в порах зерна катализатора;
3) активированная адсорбция веществ на поверхности ката-
лизатора с образованием поверхностных непрочных химических
Рис. IV.2. Элементарные стадии
гетерогенного катализа:
1—турбулентный поток газа; 2—-погра-
ничный слой газа; 3— наружная поверх-
ность катализатора; 4—поры катализа-
тора; 5 — внутренняя поверхность пор;
I— внешняя диффузия; II— внутренняя
диффузия; III — активированная адсорб-
ция; /V—перегруппировка атомов ла
поверхности; V—десорбция продукта;
V/ — виутреиияи диффузия продукта;
V// —внешняя диффузия продукта.
и увеличением их пористости
соединении — активированных ком-
плексов;
4) перегруппировка атомов с об-
разованием поверхностных ком-
плексов продукт — катализатор;
5) десорбция продукта с поверх-
ности;
6) внутренняя диффузия продук-
та в порах зерна катализатора;
7) внешняя диффузия продукта
реакции от поверхности зерна ката-
лизатора в ядро потока (через по-
граничный слой газа).
В отличие от некаталитического
гетерогенного процесса, протекаю-
щего в системе г—т, в данном слу-
чае появляются дополнительные
промежуточные стадии, в частности,
активированная адсорбция молекул
исходных веществ на поверхности
катализатора (стадия III). При
этом желательно, чтобы твердые ка-
тализаторы имели большую легко
доступную поверхность, что дости-
гается уменьшением размера зерен
В ряде случаев внутренняя поверх-
ность таких катализаторов достигает десятков и даже сотен квад-
ратных метров на 1 см3 катализатора.
При наличии пористого катализатора реакция протекает как
ца внешней, так и на внутренней поверхности гранул катализатора.
Часто внутренняя поверхность в тысячи раз превышает внешнюю
поверхность, в этом случае влияние последней на процесс невелико.
Чтобы судить о роли каждой из этих поверхностей, примем, например, вну-
треннюю поверхность катализатора равной 10 м2-см-3, илн 107 м2-м3 (в дей-
ствительности она бывает более 100 м2 • см*3), и определим их соотношение для
(ранулы (зерна) катализатора диаметром d = 0,005 м (5 мм):
1 о7 — nd3
Гвн 107р _ 6 па 107 - 0,005 _
Fa = nd2 nd2 = 6
где v — объем гранулы, м3.
100
Из соотношения видно, что внутренняя поверхность значительно превышает
наружную поверхность. Следовательно, в данном случае в практических рас-
четах наружную поверхность можно не учитывать.
Так как при гетерогенном катализе процесс протекает главным
образом на внутренней поверхности катализатора, то для описа-
ния большинства каталитических процессов более подходит квази-
гомогенная модель, а не модель частицы с невзаимодействующим
ядром, которая была использована при рассмотрении некаталити-
ческих гетерогенных процессов г — т (стр. 82).
В настоящее время установлено, что не вся поверхность ката-
лизатора однородна, поэтому катализ осуществляется только на
так называемых активных центрах.
3. КИНЕТИКА ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКЦИЙ
В общем случае, суммарное уравнение скорости всего процесса
гетерогенного катализа должно включать описание каждой из
его стадий. Но точно так же, как и при протекании гетерогенного
некаталитического процесса, не все его стадии оказывают равное
влияние на скорость катализа. В большинстве случаев одна из
стадий является наиболее медленной, лимитирующей процесс; она
и определяет его скорость, поэтому для изыскания путей интен-
сификации такого процесса важно прежде всего установить лими-
тирующую стадию.
Из анализа гетерогенных некаталитических процессов известно,
что если наиболее медленной стадией, лимитирующей общую ско-
рость, является диффузионный перенос газообразного вещества
через пограничный слой газа, т. е., если процесс протекает во
внешнедиффузионной области, эффективным средством его уско-
рения служит увеличение скорости газового потока. На этом осно-
ван наиболее часто применяемый экспериментальный метод опре-
деления влияния диффузии на скорость каталитического процесса.
Для этой цели проводят серию опытов по определению скоро-
сти каталитической реакции при различной скорости потока реак-
ционной смеси, но при постоянном отношении объема катализатора
к объему смеси VK/Vr = const, или ~ const (здесь VK—
объем катализатора; Уг — объем реакционной газовой смеси; f —
площадь сечения контактной трубки; w — линейная скорость га-
зового потока).
При увеличении w (что достигается уменьшением f) скорость
реакции — rA = dXA[dr- будет возрастать только до тех пор, пока
процесс протекает во внешнедиффузионной области (рис. IV. 3).
На участке кривой от w = 0 до w = Wi скорость потока оказывает
влияние на скорость реакции и, следовательно, процесс протекает
во внешнедиффузионной области.
Влияние внутренней диффузии исследуют путем проведения
серии опытов при скорости потока w > Wj (в области, где .внеш-
няя диффузия уже не оказывает влияния на общую скорость
101
процесса). Опыты проводят на зернах катализатора различного
размера, результаты опытов выражают в виде графической зави-
симости г = <р • На графике ' эта зависимость имеет такой
же характер, что и для некаталитических процессов в системе г—г
(рис. IV. 4).
Активное действие катализатора обусловлено прежде всего
предварительной адсорбцией реагирующих веществ на поверхно-
сти катализатора (стадии III—V на рис. IV. 2), что оказывает
большое влияние на скорость гетерогенного катализа.
Адсорбция является самопроизвольным процессом, поэтому она
сопровождается убылью энергии системы и связана с выделением
Рис. IV.3. Влияние внешней
диффузии (скорости потока w)
на скорость каталитического
процесса г.
Рис. IV.4. Влияние внутренней
диффузии (радиуса зерен катали-
затора /?) на скорость каталити-
ческого процесса г.
тепла. Существует два вида адсорбции — физическая и химическая
(последняя называется также активированной адсорбцией, или
хемосорбцией). Процесс катализа связан с хемосорбцией, но хемо-
сорбция может быть обратимой и необратимой. Естественно, что
в процессе катализа хемосорбция должна быть обратимой, так как
активные центры должны непрерывно возобновлять свою функцио-
нальную деятельность по отношению к реагентам. Необратимая
адсорбция вызывает отравление катализатора.
С целью установления функциональной зависимости скорости
гетерогенного катализа от различных факторов проводят всесто-
ронние исследования, что позволяет определить влияние различных
показателей на скорость адсорбции и дёсорбции реагентов поверх-
ностью катализатора,' а также на скорость протекания других ста-
дий, определяющих каталитический процесс в целом. Полученные
при этом данные используют для составления теоретических урав-
нений, позволяющих установить общие закономерности в идеаль-
ных условиях.
Для практических целей обычно применяют эмпирические урав-
нения, которые получают путем тщательного изучения влияния
различных факторов на скорость конкретного каталитического
процесса в условиях, близких к производственным. При этом идеа-
102
лизированные модели служат отправной точкой для наиболее эф-
фективного анализа экспериментальных данных.
Одним из эмпирических уравнений является уравнение (IV. 4).
Это уравнение справедливо для сравнительно узкой области изме-
нения концентрации сернистого ангидрида и кислорода (оно при-
менимо для газов, получаемых при обжиге в воздухе флотацион-
ного колчедана). Сейчас предложено свыше 20 эмпирических урав-
нений для этой же реакции.
Нахождение кинетических уравнений и определение оптималь-
ных параметров является главной целью научных исследований в
области каталитических процессов, так как эти данные исполь-
зуются затем для расчета каталитических реакторов.
Температура оказывает весьма существенное влияние на ката-
литические процессы, так как при повышении температуры увели-
чивается константа скорости реакции [уравнение (11.23)] и одно-
временно изменяется константа равновесия [уравнение (11.20)].
Для процессов, проходящих в кинетической области, повышение
температуры всегда способствует приближению процесса к состоя-
нию равновесия. Но, как известно, для обратимых реакций равно-
весная степень превращения X* при повышении Т уменьшается
для экзотермических реакций и увеличивается для эндотермиче-
ских реакций. Поэтому закономерности, отражающие суммарную
скорость реакции и действительную степень превращения для экзо-
термических и эндотермических реакций, совершенно различны.
При этом наблюдается такая же функциональная зависимость
X = f(T), как и для некаталитических процессов (см. рис. II.9
и II. 10).
Время контакта (время соприкосновения) реагирующих ве-
ществ с катализатором также является важной технологической
характеристикой каталитического процесса, так как оно опреде-
ляет его интенсивность. При расчете реакторов время контакта
определяют по уравнению
т=1/к/1/ (IV. 6)
где т — время контакта;
VK—объем катализатора;
V — объем реакционной смеси, проходящей через катализатор в единицу
времени.
Величина, обратная времени контакта, называется объемной
скоростью и выражается уравнением
S = 1/t (IV. 7)
где S — объемная скорость (объем реакционной смеси, проходящей через еди-
ницу объема катализатора в единицу времени), м3 (газа) • м-3 (ката-
лиз.) • с-1 = с-1.
При увеличении объемной скорости обычно снижается степень
превращения, однако при этом возрастает интенсивность работы
аппарата, т. е. увеличивается количество целевого продукта, полу-
чаемого с единицы объема катализатора в единицу времени. Это
объясняется тем, что при увеличении скорости потока реагирую-
103
щая система в большей мере удалена от равновесия, процесс
протекает в области высоких скоростей за счет большой движущей
силы АС = Рт— Рт.
В качестве примера приведем данные о влиянии объемной
скорости на интенсивность каталитического процесса синтеза ам-
миака N2 + ЗН2 2NH3, протекающего при Т = 450 К:
S, с-1 % NH3 в газовой смеси Интенсивность катализатора, кг/(мЗ-ч)
0(равновесие) 45
10 000 25 1950
30 000 18 4280
50 000 16 5640
Из приведенных данных следует, что при увеличении скорости газа в 5 раз
содержание NH3 в конечной смеси снижается только в 1,5 раза, а интенсивность
процесса возрастает в 3 раза.
Интенсивность катализатора выражают в виде уравнения:
G = pzS (IV. 8)
где G—производительность катализатора, кг-ч^-м-3;
р — плотность реагента при нормальных условиях, кг • м~3 (для-аммиака
р = 0,7708);
z — мольная доля целевого продукта в газовой смеси;
S — объемная скорость, ч-*.
Из уравнения (IV. 8) видно, что при увеличении объемной ско-
рости производительность катализатора возрастает. Однако воз-
можности для увеличения S ограничены, так как при этом степень
превращения ХА уменьшается (снижается концентрация целевого
продукта, что затрудняет выделение его из реакционной смеси),
возрастает расход энергии и нарушается автотермичность экзо-
термической реакции вследствие относительного увеличения объ-
ема реакционной смеси.
Если проводить экзотермическую реакцию с различным време-
нем соприкосновения реакционной смеси с катализатором т, полу-
чают серию кривых на осях координат Т—ХА с максимальным
значением Хтах (см. рис. II. 8), что позволяет установить опти-
мальную температурную последовательность, обеспечивающую
наиболее эффективное ведение процесса. Его следует начинать
при высокой температуре, когда скорость процесса велика и, сле-
довательно, достигается высокая интенсивность, а затем вести про-
цесс при снижении температуры и увеличении времени соприкос-
новения реагирующих масс, при этом возрастает значение ХА
(стр. 56).
Применение давления является одним из способов повышения
степени превращения при промышленном осуществлении обрати-
мых каталитических реакций, проходящих с уменьшением объема.
Давление становится решающим фактором, когда активность ка-
тализатора и величина X* низкие.
104
4. СВОЙСТВА И ПРИГОТОВЛЕНИЕ ТВЕРДЫХ
КАТАЛИЗАТОРОВ
Промышленные твердые катализаторы обычно не являются
индивидуальными веществами. Они представляют собой, за ред-
ким исключением, сложную смесь, называемую контактной мас-
сой. В контактной массе одни вещества являются собственно ка-
тализаторами, другие — носителями, а третьи служат активато-
рами.
Носители — термостойкие, инертные, пористые вещества, на
которые осаждением или другими способами наносят катализатор.
Применение носителей улучшает свойства катализаторов и уде-
шевляет их. В качестве носителей применяют пемзу, асбест, сили-
кагель и другие пористые вещества.
Активаторы или промоторы — вещества, повышающие актив-
ность основного катализатора, например, окисли щелочных ме-
таллов увеличивают активность железных катализаторов в синтезе
аммиака и ванадиевых катализаторов при окислении двуокиси
серы.
Механизм действия активатора может быть самым различным:
они могут образовывать химические соединения, твердые растворы,
могут изменять электрофизические свойства поверхности и т. д.
Активаторы могут также увеличивать активность катализатора,
развивая и стабилизируя его поверхность. Последние называются
структурными.
Качество катализаторов характеризуют следующими основ-
ными показателями:
активностью;
избирательностью действия;
устойчивостью к ядам и термостойкостью;
механической прочностью;
доступностью и дешевизной;
теплопроводностью, которая должна быть возможно более вы-
сокой.
Катализатор обычно готовят в виде зерен, таблеток, гранул.
Иногда катализаторы применяют в виде тончайших сеток, изго-
товленных из металлов или сплавов.
Наиболее часто катализаторы изготавливают следующим об-
разом:
1) осаждением гидроокисей или карбонатов (из растворов
их солей) на носителе с последующим формованием и прокали-
ванием;
2) совместным прессованием всех компонентов катализатора
с вяжущим веществом;
3) сплавлением нескольких веществ;
4) пропиткой пористого носителя раствором, содержащим ка-
тализатор и активатор.
ЧАСТЬ ВТОРАЯ
ХИМИЧЕСКИЕ РЕАКТОРЫ
Глава И
ОБЩИЕ ПОЛОЖЕНИЯ
Химический реактор — это основной аппарат любого химиче-
ского процесса; от его устройства и показателей работы в значи-
тельной степени зависит экономическая эффективность всего хи-
мического производства.
Теория химических реакторов — это раздел общей химической
технологии, в котором изучаются вопросы промышленного оформ-
ления химических реакций. Задача этого раздела в конечном счете
состоит в разработке методов расчета реакторов с получением
данных, необходимых для их проектирования. Расчет реакторов —
это первая ступень в создании химического производства.
Исходные данные для расчета реакторов
При выборе конструкции и определении размеров любого
реактора необходимо принимать во внимание различные факторы
и, прежде всего, располагать данными о скорости протекающих
химических реакций, а также о скорости массо- и теплопередачи.
Обычно задаются производительностью установки и степенью пре-
вращения, а концентрацию реагентов, температурный режим и
другие показатели технологического процесса рассчитывают на
основе опытных и теоретических данных.
Наиболее важным показателем, отражающим совершенство хи-
мического реактора, является интенсивность протекающего в нем
процесса. Но интенсивность тем выше, чем меньше время, затра-
чиваемое на получение единицы заданного продукта, поэтому глав-
ной задачей при изучении химических процессов, протекающих в
реакторах любого типа, является установление функциональной
зависимости времени пребывания реагентов в реакторе от различ-
ных факторов. Эту зависимость можно выразить в виде уравнения
т = /(Х, С, г) (V. 1)
где т — время пребывания реагентов в реакторе;
X — степень превращения исходного реагента;
С—начальная концентрация исходного реагента;
г — скорость химической реакции.
106
Уравнение, связывающее четыре указанных параметра, яв-
ляется математическим описанием модели реактора; в дальнейшем
будем его называть уравнением реактора (иногда его называют
характеристическим уравнением).
Уравнение материального баланса реактора
Основанием для получения уравнения реактора любого типа
является материальный баланс, составленный по одному из ком-
понентов реакционной смеси. Составим такой баланс по исходному
реагенту А при проведении простой необратимой реакции
В общем виде уравнение материального баланса записывается
так
ВА (пр) = ВА (расх) . <v- 2)
где ВА (пр) — количество реагента А, поступающего в единицу времени в тот
реакционный объем, для которого составляется баланс;
В а (расх) — количество реагента 4, расходуемого в единицу времени в реак-
ционном объеме.
Учитывая, что поступивший в реактор реагент А расходуется
в трех направлениях, можно записать:
ВА (расх) ~ ВА (к. р) ВА (ст) + ВА (пак)
где Вд (х р) — количество реагента А, вступающее в реакционном объеме в хи-
мическую реакцию в единицу времени;
Вд (ст) — сток реагента А, т. е. количество реагента А, выходящее из реак-
ционного объема в единицу времени;
ВЛ(нак)—накопление реагента А, т. е. количество реагента А, остающееся
в реакционном объеме в неизмененном виде в единицу времени.
С учетом уравнения (V.3) уравнение (V. 2) записывается в
таком виде
ВА (пр) = ВА (х. р) ВА (ст) + ВА (пак)
Разность между ВА(Пр) и ВА(Ст) представляет собой количество
реагента А, переносимое конвективным потоком ВА(КОНв)
ВА (конв) ВА (пр) ВА (ст) 5)
Принимая это во внимание, уравнение (V. 4) можно записать
ВА (пак) ВА (конв) ВА (х. р)
В каждом конкретном случае уравнение материального баланса
принимает различную форму. Баланс может быть составлен для
единицы объема реакционной массы, для бесконечно малого (эле-
ментарного) объема, а также для реактора в целом. При этом
можно рассчитывать материальные потоки, проходящие через
объем за единицу времени, либо относить эти потоки к 1 молю
исходного реагента или продукта.
Так, в общем случае, когда концентрация реагента непостоянна
в различных точках реактора или непостоянна во времени, мате-
риальный баланс составляют в дифференциальной форме для эле-
107
ментарного объема реактора. При этом исходят из уравнения кон*
вективного массообмена (см. А. Г. Касаткин. Основные процессы
и аппараты химической технологии. М., «Химия» 1971, стр. 414),
в которое вводят дополнительный член гА, учитывающий протека-
ние химической реакции. Тогда это уравнение примет вид
дСА
дх
дСА
w*~dT
дС. дС.
у ду dz
(д2С, д2С. д2С.\
+ D '-гт-+ —-г-+—г/) + (V.7>
\ дх2 ду2 dz2 J А ' '
где СА — концентрация реагента А в реакционной смеси;
х, у, z — пространственные координаты;
Wy, wz— составляющие'скорости потока;
D — коэффициент молекулярной и конвективной диффузии;
гА — скорость химической реакции.
Левая часть уравнения (V.7) характеризует общее изменение
концентрации исходного вещества во времени в элементарном
объеме, для которого составляется материальный баланс. Это на*
копление вещества А. Этому члену соответствует величина Вуцнак>
в уравнении баланса (V.6).
Первая группа членов правой части уравнения (V. 7) — произ*
ведения составляющих скорости потока вдоль осей координат
на градиенты концентраций — отражает изменение концентрации
реагента А вследствие переноса его реакционной массой в направ*
лении, совпадающем с направлением общего потока.
Вторая группа членов правой части уравнения (V. 7)—произ-
ведение D на сумму вторых производных от концентрации по х,
у, z — отражает изменение концентрации реагента А в элементар*
ном объеме в результате переноса его путем диффузии.
Указанные две группы правой части уравнения характеризуют
суммарный перенос вещества в движущейся среде путем конвек-
ции и диффузии, в уравнении (V. 6) им соответствует величина
Луцкопп) (такой суммарный перенос вещества называют конвек-
тивным массообменом, или конвективной диффузией).
И, наконец, член гА показывает изменение концентрации ре-
агента А в элементарном объеме за счет химической реакции.
Ему в уравнении (V. 6) соответствует величина ВА(Х. р).
В зависимости от типа реактора и режима его работы диффе-
ренциальное уравнение материального баланса (V. 7) может быть
преобразовано, что облегчает его решение.
В том случае, когда параметры процесса постоянны во всем
объеме реактора и во времени, нет необходимости составлять
баланс в дифференциальной форме. Баланс составляют в конеч-
ных величинах, взяв разность значений параметров на входе в.
реактор и на выходе из него.
Все процессы, протекающие в химических реакторах, подраз-
деляют на стационарные (установившиеся) и нестационарные
(неустановившиеся). К. первым относят процессы, при которых в
108
системе или в рассматриваемом элементарном объеме реакцион-
ной смеси параметры процесса (например, концентрация ре-
агента А, температура и т. д.) не изменяются во времени, поэтому
в реакторах отсутствует накопление вещества (или тепла) и произ-
водная от параметра по времени равна нулю. Так, при стационар-
ном режиме для реагента А можно записать
<ЭС,
гд(иак) = -аГ- = ° (V-8)
При нестационарных режимах параметры непостоянны во вре-
мени и всегда происходит накопление вещества (тепла). В этих
условиях уравнение примет вид:
<ЭС.
ВА(нак)“-^0 (V-9)
Классификация реакторов
При классификации реакторов принимают во внимание сле-
дующие основные признаки:
1) характер операции, протекающей в реакторе;
2) режим движения реакционной среды;
3) тепловой режим;
4) фазовое состояние реагентов.
По первому признаку реакторы делят на периодические,
непрерывные и полунепрерывные. Реакторы непрерыв-
ные, т. е. с непрерывной подачей реагентов, в свою очередь под-
разделяются по характеру движения реакционной среды (т. е. по
гидродинамической обстановке в реакторе) на реакторы
идеального вытеснения и реакторы идеального
смешения.
Обычно изучение реакторов слагается из двух этапов: вначале
в изотермических условиях, когда температура реакционной смеси
постоянна, а затем с учетом изменения температуры реакционной
смеси.
Глава VI
РЕАКТОРЫ С РАЗЛИЧНЫМИ РЕЖИМАМИ
ДВИЖЕНИЯ СРЕДЫ
Уравнение реактора любого типа получают из уравнения ма-
териального баланса, составленного для данного типа реактора
по одному из реагентов рассматриваемой реакции.
Ниже приведен вывод уравнений для различных типов реакто-
ров при условии, что влияние температуры на скорость реакции
не учитывается.
1. РЕАКТОРЫ ПЕРИОДИЧЕСКИЕ
Реакторы периодические характеризуются единовременной за-'
грузкой реагентов. При этом процесс складывается из трех стадий:
загрузки сырья, его обработки (химическое превращение) и
109
Рис. VI.1. Реактор идеаль-
ного смешения периодиче-
ский.
Ni л — начальное
выгрузки готового продукта. После проведения этих стадий они по-
вторяются вновь, т. е. работа реактора осуществляется циклически.
Продолжительность одного цикла, проводимого в периодиче-
ском реакторе, определяется по уравнению:
Тп = т + тисп (VI. 1) «
где тп — полное время цикла;
т—рабочее время (затрачиваемое на проведение химической реакции);
Т(ВСП) ~ вспомогательное время (затрачиваемое на загрузку реагентов и вы-
грузку продукта).
Реактор идеального смешения периодический
Этот реактор, называемый сокращенно РИС-П, представляет
собой аппарат с мешалкой, в который периодически загружают '5
исходные реагенты (рис. VI. 1). В таком
реакторе создается весьма интенсивное
перемешивание, поэтому в любой момент
времени концентрация реагентов одина- J
кова во всем объеме реактора и изме- J
няется лишь во времени, по мере проте- j
кания химической реакции. Такое пере- j
мешивание можно считать идеальным, 1
отсюда и название реактора.
Изменение концентрации исходного «
реагента А во времени и в объеме ре-
актора показано на рис. VI. 2. Обозначе- -<
ния, приведенные а рис. VI. 1 и VI. 2,
имеют следующие значения:
количество исходного реагента А в реак- J
ционной смеси (при загрузке в реактор);
Сл, о —начальная концентрация реагента А в реакционной j
смеси; 1
Ха, о — начальная степень превращения реагента А; j
NA, ^а> ХА — то же, в конце процесса; »
т — время; Л
^ — пространственная координата (координата места).
Периодические химические процессы по своей природе всегда
являются нестационарными (неустановившимися), так как в ходе
химической реакции изменяются параметры процесса во времени
(например, концентрация веществ, участвующих в реакции, т. е.
происходит накопление продуктов реакции).
Для расчета периодического реактора надо знать его уравне-
ние, позволяющее определить рабочее время т, необходимое для
достижения заданной степени превращения ХА при известной на*
чальной концентрации вещества СА,о и известной кинетике про*
цесса, т. е. при известной скорости химической реакции гА.
Основой для получения уравнения реактора является диффе*
ренциальное уравнение (V. 7). Это уравнение может быть преобра*
О
^А,ОгСА,0’^,0
110
зовано; исходя из того, что в РИС-П вследствие интенсивного
перемешивания все параметры одинаковы во всем объеме реак-
тора в любой момент времени (см. рис. VI. 2). В этом случае про-
Рис. VI.2. Распределение концентрации реагента в периодическом
реакторе идеального смешения:
а—по времени; б—по месту (по объему).
изводная любого порядка от концентрации по осям х, у, z равна
нулю. Поэтому можно записать
вС. вС. дС.
wx-^- — wy- — а>г——4- = о (VI.2)
дх у ду dz '
(д2С. д2С, д2С,\
+ + =0 (VI.3)
\ дх2 ду2 dz2 ) ’
С учетом полученных значений уравнение (V. 7) упрощается и
может быть записано не в частных производных, а в виде обыкно-
венного дифференциального уравнения
Если рассматриваемая выше реакция протекает без изменения
объема реакционной смеси, текущая концентрация исходного ве-
щества выражается уравнением (1.5). Подставив это значение
й уравнение (VI. 4), получим . -
*Кл.о(1-*л)] ,.г
dx А
или
dX,
. (VI-5)
Знак минус перед величиной гА указывает на то, что в процессе
происходит убыль исходного реагента.
Ill
Интегрируя уравнений (VI.5) в пределах изменения времени
от 0 до т и степени превращения от 0 до ХА, получим уравнение
РИС-П
Рассмотрим некоторые частные случаи решения этого уравне-
ния. Скорость простой необратимой реакции n-го порядка выра-
жается уравнением (11.28)
Подставив полученное значение для (—гА) в уравнение (VI.6),
получим •
ХА
С dX.
I ---d.
С учетом уравнения (1.5) находим
ХА ХА
f dXA _ 1 f dXA
Х = Са,°) ^до(1-*а)п==
Для необратимой реакции нулевого порядка, когда
уравнения (VI.8) получаем
______1 СЛ dXA СА,0ХА
*С°а7о</ О- *а)° k
Для необратимой-реакции первого порядка, когда п — 1, анало-
гично из уравнения (VI.8) находим
Хл ХА
__1 { dXA .4 -Ч'-ХА) 1
(VI. 7)
(VI. 8)
п = О, из
(VI. 9)
или
1 ,
Т = —• 1п
k
1
'~ХА
(VI. 10)
До сих пор интегрирование производилось в пределах изменения
ХА от 0 до ХА. Если начальная степень превращения не равна 0,
тогда во всех случаях конечные результаты изменяются. Так, на-
пример, для реакции нулевого порядка из уравнения (VI. 8) имеем
ХА
т - 1 f dXA _ С А.
""° *^М0(1_^)0 k
112
Соответственно для реакции первого порядка
ХА
1 Г dXA 1 I 1Х„
или
1
:п=1=Т1п
1-*Л.О
1-^А
В тех случаях, когда в РИС-П проводится реакция, порядок
которой отличается от 0 и 1 (0 #= n #= 1) интегрирование уравне-
ния (VI.6) связано с трудностями, поэтому определение рабочего
времени производят методом графического интегрирования. Для
этого, взяв за основу уравнение
(VI. 6), строят графическую зависи-
мость — = f (Xд) и вычисляют
ГА
площадь под кривой между началь-
ным и конечным значениями степе-
ни превращения (рис. VI. 3):
*А
*~СА,о\ -^TdXA-CA.0S (VI’n)
О А
где:
ха
Рис. VI. 3. Графический расчет
реактора идеального смешения
периодического.
Уравнение (VI. 6) является ма-
тематическим описанием модели
РИС-П. Исходя из этого уравнения
представляется возможность определить размеры реактора, а так-
же исследовать эту модель с точки зрения нахождения оптималь-
ных значений всех входящих в него параметров.
Реакторы периодического действия просты по конструкции,
требуют небольшого вспомогательного оборудования, поэтому они
особенно удобны для проведения опытных работ по изучению хи-
мической кинетики. В промышленности же они обычно исполь-
зуются в малотоннажных производствах и для переработки отно-
сительно дорогостоящих химических продуктов. Большинство же
промышленных процессов оформляется с использованием реакто-
ров непрерывного действия.
2. РЕАКТОРЫ НЕПРЕРЫВНОГО ДЕЙСТВИЯ
В реакторах непрерывного действия (их иногда называют про-
точными реакторами) питание реагентами и отвод продуктов
Реакции осуществляется непрерывно.
113
Если в периодическом реакторе можно непосредственно, по
часам, измерить продолжительность реакции, поскольку показа-
тели процесса меняются во времени, то в реакторе непрерывного
действия этого сделать нельзя, так как при установившемся ре-
жиме в этих реакторах параметры не меняются со временем.
В связи с этим для непрерывных реакторов применяют понятие
условного времени пребывания реагентов в системе (вре-
мени контакта), которое определяется по уравнению:
T = jzr/7O (VI. 13)
1Де т — условное время пребывания;
Vr — объем реактора;
Va—объем реакционной смеси, поступающей в реактор в единицу времени
(объемный расход реагентов).
Реакторы идеального вытеснения
Реактор идеального вытеснения (РИВ) представляет собой
трубчатый аппарат, в котором отношение длины трубы L к ее
диаметру d достаточно велико. В реактор непрерывно подаются
Рис, VI. 4. Реактор идеального вы-
теснения и изменения концентра-
ции реагента Сд и степени пре-
вращения XА по длине реактора.
исходные реагенты, которые пре-
вращаются в продукты реакции по
мере перемещения их по длине ре-
актора (рис. VI. 4).
Гидродинамический режим в
РИВ характеризуется тем, что лю-
бая частица потока движется толь-
ко в одном направлении по длине
реактора, обратное (продольное)
перемешивание отсутствует; отсут-
ствует также перемешивание по се-
чению реактора. Предполагается,
что распределение вещества по это-
му сечению равномерное, т. е. зна-
чения параметров реакционной сме-
си одинаковые.
Каждый элемент объема реак-
ционной массы dVr движется по
длине реактора, не смешиваясь с
предыдущими и последующими эле-
ментами объема, и ведет себя как
поршень в цилиндре, вытесняя все, что находится перед ним. По-
этому такой режим движения реагентов называется иногда порш-
невым или режимом полного вытеснения.
Состав каждого элемента объема последовательно изменяется
по длине реактора вследствие протекания химической реакции.
Так, например, концентрация исходного реагента А постепенно
меняется по длине реактора от начального СА. 0 до конечного зна-
чения СА (см. рис. VI. 4). Следствием такого режима движения
514
реакционной смеси является то, что время пребывания каждой ча-
стицы в реакторе одно и то же.
Для составления математического описания РИВ исходят из
дифференциального уравнения материального баланса (V. 7), пре-
образуя его на основе указанных выше особенностей этого ре-
актора.
Поскольку в РИВ реакционная смесь движется только в одном
направлении (по длине /), то для первой группы членов правой
части уравнения (V. 7) можно записать (выбрав за направление
оси х направление движения потока реагентов в реакторе):
дСА л дСА
Wy =0; — Шг = —5— *= 0
и ду dz
дСА дСА
— WX -Г2- = — W ,
х дх д1
(а)
(б)
где w — линейная скорость движения реакционной смеси в реакторе;
I — длина (длина пути, пройденного элементом объема реакционной смеси
в реакторе).
Так как в идеальном реакторе каждый элемент объема реак-
ционной смеси не смешивается ни с предыдущими, ни с после-
дующими объемами, а также отсутствует радиальное перемеши-
вание (нет ни продольной, ни радиальной диффузии, а молекуляр-
ная диффузия мала), то
д2СА , д^А
. дх2 ду2
д2СА
dz2
(в)
С учетом вышесказанного уравнение (V. 7) для реактора иде-
ального вытеснения принимает вид z ’
дСА
дт
дС.
___А
д1
(VI. 14)
w
Это уравнение материального баланса является математиче-
ским описанием потока реагента в реакторе идеального вытесне-
ния при нестационарном режиме (когда параметры процесса не
только меняются по длине реактора, но и непостоянны во вре-
мени). Подобный режим характерен для периодов пуска и
остановки реактора. Член дСА1дх характеризует изменение концен-
трации А во времени для данной точки реактора, т. е. накопление
вещества А в этой точке.
Стационарный режим характеризуется тем, что параметры в
каждой точке реакционного объема не меняются во времени
(дСА/дх = 0). В этом случае уравнение (VI. 14) принимает вид
dC,
= (VI. 15)
Если объем реакционной смеси не меняется в процессе, спра-
ведливо уравнение (1.8), после дифференцирования которого по-
лучаем
dCA-~CA,OdXA (О
115
Но в любой момент времени т имеем
или dl/dx = w (д) dl=wdx (е)
Подставив полученное (VI. 15), находим значение для dCA п dl в уравнение dX. dX==CA.<)—^ ' (VI. 16)
После интегрирования уравнения (VI.16) в пределах измене-
ния степени превращения от 0 до ХА получаем
хА
х=‘Сл,о $
dXA
~ГА
(VI. 17)
Из полученных данных видно, что уравнение для РИВ в об-
щем виде такое же, как и для РИС-П [см. уравнения (VI. 8) —
(VI. 10)], поэтому для РИВ при различных значениях п можно
записать
1
т =-------------г
п—п bz-ra-1
ЙСА,О
ХА
dXA
(VI. 18)
__ С А, 0ХА
п-0~ k
1 , 1
тп=1— k In 1 -ХА
(VI. 19)
(VI. 20)
Для реакций, порядок которых 0=И='п=И= 1, пользуются графи-
ческим методом, который описан применительно к РИС-П
(стр. 113). При этом
ха
‘-Ca.oJ -=77"a=Ca,os (VI.21)
о А
где
ха
S=( —1—dX =-Д— (VI. 22)
o’ ~Га а’°
В уравнениях для РИС-П величина т — время проведения реак-
ции от загрузки исходного реагента до выгрузки продуктов реак-
ции, а в уравнениях для РИВ т — время, в течение которого реак-
ционная смесь проходит через РИВ от входа в реактор до Выхода
из него.
Если в процессе реакции происходит изменение объема реак-
ционной смеси, то в уравнения (VI. 16) и (VI. 17) необходимо
не
подставить значение скорости реакции с учетом изменения объема-
реакционной смеси [в соответствии с уравнением (1.12)]:
/ 1-Хл х"
- rA = kCnA -j-v—$- I (VI. 23)-
A A V + елХл J
Тогда уравнение РИВ запишется в виде
хА
т 1 с 0 + *Л)"
^АО1 J (1-^Г А
(VI. 24)-
В реальном реакторе гидродинамическая обстановка от-
личается от обстановки в идеальном реакторе. Например, в реаль-
ном реакторе вытеснения помимо поршневого движения основного-
потока по длине реактора воз-
можно перемешивание потока в
продольном и радиальном на-
правлениях. Естественно, модель
реактора усложняется. При нали-
чии продольного перемешивания
(по оси х) уравнение реактора
вытеснения может быть получено
из уравнения (V. 7), если принять
те же условия, что и при выводе
уравнения для РИВ — справедли-
вость уравнений (а) и (б). Ис-
ключение составляет лишь тот
факт, что член д2Сл!дх'2 в уравне-
нии (в), характеризующий изме-
Рис. VI. 5. Зависимость отношения
объема реального реактора вытесне-
ния к объему реактора идеального-
вытеснения Рр/РИд от степени пре-
нение концентрации вследствие вращения ХА и от DJwL.
турбулентной диффузии по оси х
(по длине I), в данном случае отличается от нуля. При этом усло-
вии из уравнения (V. 7) следует
<эсл
дх
дСА _1_ П д'СА _1_ Г
W ---— + "г------Д Т Гл
dl L dl2 А
(VI. 25)
где Dl — коэффициент продольного перемешивания.
Такая модель трубчатого реактора называется диффузионной
однопарамегрической моделью, так как ею учитывается один
диффузионный параметр — продольное перемешивание.
Степень отклонения показателей такого реактора от идеального
зависит от трех величин: коэффициента продольного перемешива-
ния (конвективной диффузии) DL, линейной скорости потока w и
Длины реакторов I. Эти величины сведены в безразмерный комплекс
Dl/(wL). Степень отклонения показателей такого реактора от
показателей РИВ зависит от значения этого комплекса и может
быть выражена через соотношение объемов реального Ер и иде-
ального реакторов Еид, необходимых для достижения одинаковой
степени превращения ХА (рис. VI. 5).
117
Если = наблюдается режим идеального вытеснения; в
этом случае Рр/Рид = 1, а зависимость Vp/Уид от ХА изображается
в виде прямой, параллельной оси абсцисс (см. рис. VI. 5). Если
~-> 0, то Ур/Уид > 1; при этом с увеличением ХА отношение
Vp/Уид возрастает.
D
Таким образом, отношение Рр/Рид зависит от комплекса
.и ХА и в общем виде выражается в виде уравнения
fD. \
: Ха) (VL26>
о,
Чем выше т. е. чем больше отклонение гидродинами-
ческого режима в реальном реакторе от режима в идеальном
реакторе, тем необходим больший объем реального реактора, и эта
разница возрастает с увеличением значения ХА. Все эти соотно-
шения необходимо принимать во внимание при расчете реального
реактора.
Для учета не только продольного, но и радиального перемеши-
вания в реакторе следует поступить так же, как и при выводе
однопараметрической модели, однако в этом случае необходимо
сохранить и второй член в уравнении (в), отражающий переме-
шивание в радиальном направлении. При этом уравнение прини-
мает вид
дС. дС. д2С. д2С.
Sr — w~ir + DL-^ + ^R-^- + rA (VI. 27)
►где D& — коэффициент радиального перемешивания;
R — радиус трубы реактора.
Такую модель называют двухпараметрической диффузионной
моделью, поскольку ею учитываются два диффузионных парамет-
ра — продольное и радиальное перемешивание.
Реактор идеального смешения непрерывный
Реактор идеального смешения непрерывный (РИС-Н) представ-
_ляет собой аппарат с мешалкой, в который непрерывно подают
,реагенты, и также непрерывно выводят из него продукты реакции
(рис. VI. 6).
В РИС-Н наблюдается резкое изменение концентрации исход-
ного реагента при входе в реактор в результате мгновенного
•смешения поступающей смеси с реакционной массой, уже находя-
щейся в реакторе, где концентрация исходного реагента значитель-
но ниже, чем концентрация исходного реагента в поступающей
смеси (рис. VI. 7).
;318
Точка, соответствующая входу реагентов в реактор, нанесена
на ось абсцисс правее начала координат, что дает более нагляд-
ное представление об изменении концентрации исходного вещества
при входе реакционной смеси в реактор. Благодаря тому, что в
рИС-Н реакционная смесь мгно-
венно перемешивается, во всем
объеме реактора одинакова концен-
трация исходного реагента и она тем
ниже, чем больше время пребыва-
ния реагентов в реакторе. По этой
же причине по всему объему реак-
тора одинакова и степень превра-
щения и скорость реакции. Таким
образом, для РИС-Н характерным
является отсутствие градиента па-
раметров как во времени, так и в
объеме реактора, поэтому уравне-
О
Рис. VI. 6. Реактор идеального-
смешения непрерывный.
ние материального баланса состав-
ляют сразу для реактора в целом. При этом градиенты параметров-
в дифференциальной форме заменяются разностью значений пара-
метров на входе в реактор и на выходе из него. С учетом этих.
у
Рис. VI. 7. Изменение параметров процесса в РИС-Н:
а—концентрация реагента Сд; б—степень превращения Хд; в—скорость реак-
ции Гд.
особенностей для установившегося режима уравнение (V. 6) мож«-
но записать
гА(х.Р) = ^(конв) (VI. 28).
Но ВАх. р = (~га) vr и 5Л(К0нв)= V (Сл, о — СА), поэтому, подставив
эти значения в уравнение (VI.28), находим
V С. п- С .
(" га) vr “ v (Са. о - САУ, ---
нли
t = (VI. 29)
ГА ГА
119>
Для простой необратимой реакции n-ного порядка с учетом
уравнений (11.28) и (I. 5) уравнение (VI. 29) принимает вид
С. пХ. С. пХ. X.
т = ...4»о_4. ------4i_2_4--==-----—4------- (V1.30)
kCnA kCXQ (1 - XА)п ЛСХо'О-Яа)"
Для реакции нулевого порядка
_ СА,аХА "=° ЙС°л>0 _ СА. 0ХА _. СА, 0 СА (VI. 31)
k k
для реакции первого порядка . СА,0ХА СА. йХА ХА (VI. 32).
kCA kCA,^~Xj Й(!-Хл)
Если объем реакционной смеси изменяется в процессе реакции,
в уравнение (VI. 30) необходимо подставить значение С л из урав*
нения (I. 12), при этом
т СА,.ХА _
kcnA kcn ( х~ха У ^;0‘(1-хл)"
AOv+<^J
(VI. 33> -
*
В реальном реакторе смешения исходная реакцион-
ная смесь перемешивается с реакционной смесью, находящейся в
реакторе, не мгновенно, как в РИС-Н, а постепенно, поэтому кон-
центрация исходного реагента в объеме реактора неодинакова и,
следовательно, зависимость = /(«/) непрямолинейна, как это
показано на рис. VI. 7, а.
Отклонение реального реактора от идеального можно выразить
в виде уравнения
тр/тнд = б>1 (VI. 34)
тде тр, тид — условное время пребывания реакционной смеси для достижения
заданной степени превращения Хл в реальном и идеальном реак-
торах смешения;
д — коэффициент отклонении реального реактора от идеального. ,
Каскад реакторов
Рассмотрим работу нескольких реакторов идеального вытесне-
ния, соединенных последовательно — каскад реакторов идеального
вытеснения (К-РИВ), показанных на рис. VI. 8.
Обозначим через ХА, i, ХА, 2 ••• XAt m_i, ХА,т, степень превра-
щения исходного реагента в первом, втором и т. д. реакторах. Для
т-го реактора в соответствии с уравнением (VI. 17) можно
.записать
.120
Решая совместно уравнение (а) и уравнение ’(VI. 13) для реак-
торов, соединенных последовательно, получаем
Твыт \ Vr ._у Vr.i vr,l + vr,2+ +vr.m_
CA, 0 ^4.0^0 “ ^4,0^0 ^4.0^0
Xr'1 dX. XeA dX. XAr m dX X r’m dX.
= \ -------— + \ -----— + ... + \ -----------— = \ --------—
x _ rA J ~ГA Y J ~ TA у J — ГА
ЛА, 0 ' xl %A, m—1 -^4,0
(6)
Следовательно, tn реакторов идеального вытеснения общим
объемом Vr обеспечивают такую же степень превращения исходного "
реагента, как и один реактор
идеального вытеснения такого же
объема.
ВА,0 ^А,0
ХА,0
Рис. VI. 8. Схема реакторов идеаль-
ного вытеснения, соединенных после-
довательно (НА р XAi 2, ХА, 3 — степе-
ни превращения реагента А после
реакторов).
В единичном реакторе идеаль-
• кого смешения не достигается
высокая степень превращения, так
как концентрация исходных ве-
ществ в нем мгновенно снижает-
ся до конечного значения и весь
процесс протекает при низкой концентрации. Поэтому очень часто
применяют ряд последовательно расположенных РИС-Н — каска
реакторов (К-РИС, рис. VI-9). Концентоания исходного реагент
Ха,к~Ха,з
Рис. VI. 9. Каскад реакторов идеального смешения.
Сд в такой системе снижается до конечного значения не сразу, а
постепенно от реактора к реактору (рис. VI. 10).
В каждом реакторе концентрация исходного реагента в объеме
постоянна и равна концентрации его на выходе из реактора. Изме-
нение концентрации исходного вещества в нем происходит так'же,
как и в РИС-Н, т. е. скачком, при входе реакционной смеси в
121
реактор. Однако рабочая концентрация Са в каскаде поддержи-
вается выше, чем в единичном реакторе смешения, и при увеличе-
нии числа реакторов приближается к значению концентрации в
РИВ.
Расчет каскада реакторов заключается в определении числа
ступеней (числа реакторов) т, необходимых для достижения за-
данной степени превращения ХА- Существуют графический и
аналитический методы расчета каскада реакторов.
Графический метод расчета каскада реакто-
ров прост и позволяет рассчитать К-РИС для реакции любого
Рис. VI. 10. Изменение концентрации
‘реагента А в каскаде реакторов иде-
ального смешения.
Рис VI. И. Графический метод расче-
та каскада РИС.
порядка. В основе расчета лежит уравнение (VI. 29), из которого
для т-го реактора К-РИС [с учетом уравнения (1-8)] следует
^А.0ХА __ СА.0~САА __ СА, т-Х ~ А. т ( «
см- _Гд - -гАА -
где СА }, СА т — концентрации исходного вещества А на входе в т-й реак-
тор и на выходе из него;
тсм — условное время пребывания реагента в камере смешения.
Из уравнения (а) находим
-гл= -2_СЛ (VI 35)
тсм см
Концентрации реагента на входе в (т — 1)-й реактор СА, m-i
и время пребывания тСм в нем величины известные и постоянные,
так как они даются по условию. Таким образом, из уравнения
{VI. 35) следует, что для m-го реактора зависимость —rA = f(CA)
изображается в виде прямой с углом наклона, а, для которого
tga= — 1/тсм (VI.36)
С другой стороны, скорость реакции описывается кинетическим
уравнением—гл = (кривая на рис. VI. 11). Поэтому точка
122
рассчитать
(а>
(б>
(VI. 37>
уравнения
(в)
(г>
(д>
пересечения прямой и кривой характеризует концентрацию исход-
ного реагента в m-ом реакторе.
Таким образом, для проведения расчета К-РИС графическим
методом необходимо вначале построить кривую по кинетическому
уравнению (рис. VI. 11), а затем из точки, лежащей на оси абс-
цисс, для которой СА;т-1 = Сд;0, провести прямую с тангенсом
угла наклона —1/тсм до пересечения с кривой в точке М Опустив
перпендикуляр из точки М на ось абсцисс, получают значение-
концентрации исходного реагента в первом реакторе (см.
рис. VI. 11). Эта же концентрация является исходной для входа
во второй реактор.
Для нахождения концентрации во втором реакторе СА, 2 опера-
цию повторяют, взяв в качестве исходной точку Ca,i- Такие опера-
ции продолжают повторять до тех пор, пока в последнем реакторе
не будет достигнута заданная конечная концентрация СА<К. Так
как время пребывания во всех реакторах обычно принимают оди-
наковым, то постоянен и угол наклона прямых, следовательно, они
параллельны.
Число ступеней и будет числом реакторов в каскаде, необхо-
димым для достижения заданной степени превращения ХА> к.
Аналитический метод расчета К-РИС включает
алгебраический и итерационный методы. Рассмотрим наиболее-
простой алгебраический метод, который позволяет
К-РИС для реакции первого порядка (n = 1).
Уравнение (VI. 35) можно представить в виде
СА, т ~ хтг А = С A, т-1
Для реакции первого порядка
~ГА = kCA,m
Подставив это значение в уравнение (а), находим
СА. т + XmkC А, т~ С A. m(1 + kxm) = СА.п-1
Отсюда
с.
С = Л» т—\
А>т 1 -J- kx
m
и, соответственно, для т — 1 и т = 2 действительны
г == СА' 0
Ж1 l +
с — 1
Л, 2 1 + kx2
или
С 1 с
С — 0 . ______ л. о____
а 2 1 + • 1 + *т2 (1 4-*тг1)(1 + kx2)
Если время пребывания во всех реакторах одинаково, то
С
(1 + kxm)m
(VI.38>
123
Логарифмируя это уравнение, находим
Jg-^-
1g (1 + £тт)
(VI 39)
Полученное уравнение устанавливает зависимость m от отно-
шения С а, о/ С a, m (которое отражает степень переработки реаген-
та), и величины ktm, пропорциональной
Рис. VI. 12. Зависимость сте-
пени превращения Х^ от
объема kx и числа т реак-
торов в каскаде;
1—т = 1(РИС-Н); 2—т=2; 3 —
т=4; 4—т = оо(РИВ).
объему каждого реактора в каскаде.
Чтобы получить зависимость ХА от
m и объема каждого реактора, пред*
ставим уравнение (VI. 38) в виде
С . m 1
_л' т- =________ (е\
Сло (l+*rm)m
и учтем, что из уравнения (1.5) следует
К А. т ~ 1 С А, т/СА, О
Тогда из уравнений (е) и (ж) находим
г -1 1
a Q + (VI.40)
Из данных табл. VI. 1 и рис. VI. 12, полученных из решения
уравнения (VI. 40), видно, что общий объем реакторов в К-РИС
зависит от т и Хх \_kx = f(m, Ха)].
Таблица VI.1. Относительный общий объем реакторов (fet) в К-РИС
т Хл =0,5 Хд=0,9 Хл = 0,98
kx I kxm kx 1 kxm kx feTm-T kxm
1 (РИС-Н) 1 1 9 1 50 1
2 0.82 1,2 4,3 2,1 12,2 4,1
3 0,76 1,3 3,1 2,9 6,6 7,6
При этом с увеличением числа реакторов общий объем каскада
реакторов, необходимый для достижения заданной степени пре-
вращения, уменьшается. Особенно значительно это уменьшение
при высоких степенях превращения.
Для системы, состоящей из последовательно соединенных реак-
торов различного типа, расчет ведут последовательно для каждого
реактора в отдельности и в зависимости от числа и типа
124
последовательно соединенных реакторов получают соответст-
вующие результаты. В качестве примера рассмотрим систему
рис-н—РИВ—РИС-Н (рис. VI. 13).
Рис. VI. 13. Схема последовательно соединенных реакторов идеаль-
ного смешения (1, 3) и идеального вытеснения (2).
На основе уравнений (VI. 28) и (VI. 17) можно записать
3. РЕАКТОРЫ ПОЛУНЕПРЕРЫВНЫЕ
В полунепрерывных реакторах одна из вспомогательных опе-
раций — загрузка реагентов или выгрузка продуктов реакции —
осуществляется периодически, а вторая — непрерывно. Примером
такого реактора может служить доменная печь, в которую непре-
рывно загружают твердую шихту, а готовый продукт (чугун) вы-
пускают периодически. В печи разложения СаСО3 с получением
СаО и СО2, наоборот, шихта (уголь и СаСО3) загружаются перио-
дически, а продукты реакции (СаО и СО2) выводятся непрерывно.
Так же осуществляется процесс в газогенераторах: уголь (шихта)
загружается периодически, а продукт реакции — генераторный
газ — выводится непрерывно.
4. СРАВНЕНИЕ РЕАКТОРОВ РАЗЛИЧНЫХ ТИПОВ
Важнейшими показателями работы реактора, определяющими
Экономичность химического процесса, являются:
1) размер реактора, от которого зависит его интенсивность;
2) избирательность протекающего в нем процесса, т. е. селек-
тивность;
3) выход продукта.
125
При протекании простых необратимых реакций типа А ->
превращение идет в одном направлении, и чем выше степень пре-
вращения, тем больше выход продукта. Поэтому при выборе типа
реактора для таких реакций имеет значение только первый фак-
тор из числа приведенных выше, т. е. размер реактора, необходи-
мый для достижения заданной степени превращения.
Уравнения РИС-П и РИВ [уравнения (VI, 6) и (VI, 16)] оди-
наковые, поэтому время протекания химической реакции, необхо-
димое для достижения заданной степени превращения, в этих
реакторах одно и то же. Но в РИС-П полное время процесса скла-
дывается из вспомогательного времени и рабочего времени [урав-
нение (VI,!)], а в РИВ вспомогательные операции отсутствуют;
Phc.VI. 14. Изменение концентраций исходного реагента СА в РИВ
и РИС-Н.
процесс протекает непрерывно, поэтому интенсивность РИВ выше,
чем РИС-П.
Рассмотрим вначале этот, наиболее простой случай. Определим
теперь время т, необходимое для достижения одинаковой степени
превращения в РИВ и РИС-Н.
В РИВ происходит постепенное изменение концентрации по
длине реактора, а в РИС-Н наблюдается резкое снижение концен-
трации до конечного значения по любой пространственной коорди-
нате (рис. VI. 14).
Из рис. VI. 14 видно, что в РИВ по сравнению с РИС-Н более
высокая средняя концентрация исходного реагента и соответствен-
но выше скорость реакции, так как она пропорциональна вели-
чине СА
— rA = kC’A = kCA
Для необратимых реакций нулевого порядка (и=0) это поло-
жение не влияет на выбор типа реактора, поскольку для таких
реакций—г а = kCA — kC°A = k и, следовательно, скорость про-
цесса и объем реактора не зависят от концентрации реагента.
126
Этот вывод следует также из уравнений (VI. 9) и (VI. 31)
т — т — Са-°Ха
1ВЫТ --------------------- LCM - £
где Твыт> Тем — время, необходимое для достижения степени превращения Хл
в РИВ и РИС-Н.
Для реакций, порядок которых п > 0, тип реактора имеет важ-
ное значение, так как для достижения одинаковой степени превра-
щения в реакторе смешения нужно большее время, чем в реакторе
вытеснения (тсм > тВыг) и, следовательно, интенсивность РИС-Н
ниже.
РИС-Н
Рис. VI. 15. Зависимость степени
превращения ХА от времени пре-
бывания т:
/-тсм (РИС-Н); 2-Гвыт (РИВ)
1
Покажем это на примере простых необратимых реакций первого порядка.
В этом случае, исходя из полученных ранее уравнений, находим для РИВ урав-
нение (VI. 20)
1 , I
Твыт Ш J ______ , ИЛИ ’ In "j
Для РИС-Н по уравнению (VI. 32) находим
_ h ХА
Тем — k (I - хАу или ktcu ~~ 1 - хА
Из приведенных выше уравнений следует
ХА
Тем
Твыт - 1 ~й>1
1 >~ХА
При этом a =f(XA): чем выше Хл, тем больше значение а, т. е. тем боль-
шие неравенство Тем > тВЫт (табл. V1-2 и рис. VI-15).
К Таблица VI.2. Значение kx, необходимое для достижении степени
И превращения ХА (для реакции п = 1)
feTCM
&ТВыт
1- Степень t превращения Значения fet \м п с “° выт
РИВ РИС-Н
0,1 0,105 в,111 1,06
[ 0,5 0.639 1,0 1,44
f 0,9 2,303 9 3,9
127
Для сравнения реакторов при проведении в них реакций любо-
го порядка пользуются графическим методом (рис. VI. 16). Для
этого графически определяют время пребывания в реакторах
вытеснения и смешения из уравнений
Твыт = СА. о J = —"л = сл.о\ыТ (VI.21)
0J А о А
X. ( 1 Л
Тсм = Сл> о = С А, 0 ) ХА = СА, 0ScM (VI. 41)
Из рис. VI. 16 видно, что площадь прямоугольника больше
площади, ограниченной кривой. С другой стороны, из уравнений
(VI. 21) и (VI. 41) следует, что отношение площадей SCM и 5ВЫТ
Рис. VI. 16. Графическое со-
поставление характеристик
РИВ и РИС-Н.
Рис. VI. 17. Графический расчет
последовательных реакторов
(РИС-Н - РИВ - РИС-Н).
равно соотношению между условным временем пребывания реаген-
тов в реакторах смешения и вытеснения (т. е. объемов реакторов),
Т f"* с с т/
выт __ ЬД> О ВЫТ _ , выт _ v г, выт
Тсм С А. О^см ^см Vr, см
(VI. 42)
Для нескольких реакторов различных типов, соединенных
последовательно, графическим методом можно определить как об-
щую, так и промежуточную степень превращения (рис. VI. 17).
При проведении сложных., реакций судить об эффективности
реактора по его размерам недостаточно. В этих случаях реактор
должен обеспечивать также возможно более высокую селектив-
ность процесса.
Рассмотрим зависимость селективности процесса от типа реак-
тора на примере параллельных реакций:
ал'*'-'
128
Селективность выражается уравнением
+ 'r + 's
rR/rS
rR/rS + 1
(VI. 43)
и является функцией отношения гв/rs-
Подставив значения rR и rs в уравнение (VI. 43), находим
<₽л =f (?•) = •-§£) = f(b~С^'~П!) (VL
V S / \я2 и А / 4 2 /
Из уравнения (VI.44) видно, что при достоянной температуре
в каждом конкретном случае (когда известен порядок основной и
побочной реакций) селективность зависит только от концентрации
Са, так как соотношение констант скорости реакций в этих
условиях величина постоянная. Тогда в зависимости от разности
щ — п2 влияние СА на <₽д может быть либо положительным, либо
отрицательным.
Если порядок основной реакции выше порядка побочной реак-
ции, т. е. ni > п2, и, следовательно, «1 — п2 = а, то из уравнения
(VI. 44) следует
(Гр \
7^ =/ fCJ (а)
fS J ^ 2 /
Таким образом, при увеличении концентрации исходного веще-
ства С а селективность возрастает. Следовательно, для достижения
высокой селективности необходимо поддерживать концентрацию
исходного реагента на максимально высоком уровне, т. е. выгодно
применять РИВ или К-РИС, так как в этих реакторах средняя
концентрация реагента Са выше, чем в РИС-Н.
Если порядок основной реакции ниже порядка побочной реак-
ции, т. е. ni < п2 и, следовательно, «1 — п2 = —а, то аналогично
из уравнения (VI. 38) получаем
(—
1*2
\ *2 J
(б)
Из этого уравнения видно, что при увеличении СА селектив-
ность снижается. При этом выгодно применять РИС-Н, так как
концентрация исходного вещества в нем ниже, чем в РИВ (см.
рис. VI. 8). В данном случае изменение величины СА оказывает
на эти параметры (интенсивность реактора и селективность про-
цесса) противоположное действие, при снижении концентрации
интенсивность уменьшается, а селективность возрастает. Какое
из указанных требований целесообразнее удовлетворить, можно
решить после проведения соответствующего технико-экономическо-
го анализа.
Если п\ = п2, а следовательно, и щ — «2 = 0, из уравнения
следует
5 Зак, 558
129
т. е. селективность не зависит от концентрации исходного реагента
и, следовательно, тип реактора не влияет на селективность. В этом
случае для повышения селективности изменяют температуру или
применяют катализатор селективного действия (вследствие чего
изменяется отношение ki/kz).
Полученные результаты о влиянии отношения rR/rs на селек-
тивность для реакций различного порядка представлены в
табл. VI. 3.
Таблица VI.3. Селективность различных реакций
П1 и П2
VR = f (Сд)
Фд = Д(Хд)
Рекомендуе-
мый тип
реактора
Н1 > п2
щ — п2 = а
«1 = П2
nt — n2 = 0
Не зависит
0Т СА И ХА
Любой
От типа реактора может зависеть не только степень превра-
щения исходных реагентов и селективность, но и выход целевого
продукта. Поэтому при выборе реактора рекомендуется учитывать
связь между ХА, <рл и Фд.
Для РИС-Н величины ХА, фн, срн во всем объеме постоянны,
поэтому связь между приведенными параметрами выражается со-
отношением (I. 16)
% =
Для РИВ эти же величины меняются по длине реактора, по-
этому для него применяют выражение
ХА
~ dXA
о
(VI. 45)
Графическое изображение уравнений (I. 16) и (VI. 45) позволяет
установить тип реактора, обеспечивающий максимальный выход
целевого продукта. Действительно, из уравнения (I. 16) следует,
133
ЧТО ВЫХОД Продукта фя, СМ> достигаемый в РИС-Н, представлен
на графике площадью прямоугольника со сторонами, равными ХА
и фя, см (рис. VL 18).
Для РИВ условия другие.
Из уравнения (VI. 45) видно,
что в этом реакторе выход
продукта Фя, выт представлен
на графике площадью под кри-
вой сря = f(XA) между началь-
ным и конечным значениями
степени превращения. С увели-
чением ХА (т. е. при уменьше-
нии СА) наблюдаются два слу-
чая:
1) селективность снижает-
ся, когда П] > п2
VI. 18, б);
2)
стает,
VI. 18, в).
Таким образом, выход про-
дукта зависит от типа реакто-
ра и разности между порядком
основной и побочной реак-
селективность
когда Hi < г
s
ций.
(рис.
возра-
(рис.
St
^я.еыт
п,>п2
а ^?,см
?R
Рлвыт
J п} <пг
ХЛ X
Рис. VI. 18. Выход продукта в за-
висимости от селективности <р и степе-
ни превращения ХА:
а — РИС-Н; б-РИВ (ni > П2); з-РИВ (n i<rta).
Особенно наглядно влияние на величину Фя типа реактора и
значения разности nt—п2, если сравнить графики, соответствую-
щие РИС-Н и РИВ, при П1 > п2 и «1 < п2 (рис. VI. 19).
Рнс. VI- 19. Сравнение РИС-Н и РИВ по выходу продукта:
а—РИС-Н и РИВ при ni>rt2‘> б —РИС-Н и РИВ при п\ <п.2.
Из рис. VI. 19, а видно, что когда щ > п2 и фя уменьшается
с увеличением ХА, площадь под кривой больше прямоугольника,
5*
131
Только для РИВ
132
следовательно, Фй, ВЫт больше Фя, см, поэтому выгодно применять
рИВ. В том случае, когда «1 < «2 и <ря увеличивается при повыше-
нии Ха (рис. VI. 19, б), площадь под кривой меньше площади
прямоугольника, поэтому ФИ( Выт < Фя см и выгоднее применять
РИС-Н.
При осуществлении реакций различных типов в К-РИС необхо-
димо учитывать, что, меняя число реакторов в каскаде, можно
изменять как степень превращения Ха> так и селективность про-
цесса и выход целевого продукта (для сложной реакции). При
этом необходимо учитывать, что при увеличении числа реакторов
в каскаде характер зависимости между степенью превращения,
селективностью и выходом целевого продукта в К-РИС будет при-
ближаться к зависимости, существующей для РИВ.
В табл. VI. 4 обобщены типы и характеристики реакторов при
Т -= const.
5. ИСТИННОЕ ВРЕМЯ ПРЕБЫВАНИЯ
Если объем реакционной смеси не изменяется во времени
r(V = Vo), то для всех типов реакторов истинное время пребывания
реагентов в реакторе ти равно условному времени пребывания т,
рассчитанному по уравнению (VI. 13).
Если V=/=Vo, что имеет существенное значение главным обра-
зом для газообразных реагентов, то для РИВ и РИС-Н истинное
время пребывания отличается от условного времени пребывания
и равенство между ти и т не соблюдается. В частном случае, при
линейном изменении объема реакционной смеси от степени превра-
щения, исходя из уравнений (1.28), (1.7) и (1.9), имеем
;___1 dNA <Ч"л,оО-*л)] NA,bdXA
А V dx = VQ (1 + гАХА) dx = Vo (1 + елХл) dx =
СА.О dXA
(1+елХл)* dx
Интегрируя это уравнение, получаем для РИВ
? dXA
Аналогично для РИС-Н можно получить
СА.ЬХА
(VI. 47)
Подставив в эти уравнения значение — гА из уравнения (VI. 23),
находим для необратимой реакции n-го порядка
для РИВ
____1 ?0+^аГ'
“ kcxQ (1-^)" dA
(VI. 48)
133
для РИС-Н
О + ^лГ1 х
«То'О-^лГ А
(VI. 49>
В РИС-П газовые реакции проводят очень редко, поэтому ре-
жим работы этого реактора, когда V ф Vo, здесь не рассматри-
вается.
Жидкость
Индикатор
Рис. VI. 20. Схема
реактора для опреде-
ления кривых отклика.
6. ДИНАМИЧЕСКАЯ ХАРАКТЕРИСТИКА РЕАКТОРА
Ранее указывалось, что в реальных реакторах заданная сте-
пень превращения достигается при большем времени пребывания
реагентов в реакторе, чем это следует из уравнений, полученных
для идеальных реакторов. В реальном реакторе вытеснения это
обусловлено наличием продольного и попереч-
ного перемешивания, отражаемых коэффи-
циентами Z)L и £>д уравнения (VI. 27), а в ре-
альном реакторе смешения — тем, что не до-
стигается полное (идеальное) перемешивание
реакционной смеси и коэффициент 6 > I
[уравн. (VI. 34)].
Степень отклонения реального реактора
от идеального определяют экспериментально.
Для этого в жидкость или газ, поступаю-
щие в реактор, вводят индикатор, а затем че-
рез некоторые промежутки времени измеряют
концентрацию этого индикатора в жидкости
на выходе из реактора. По полученным дан-
ным строят так называемые кривые отклика.
В результате анализа этих кривых опреде-
ляют не только значения коэффициентов £Д,
Dr и а, но такие важные характеристики, как
минимальное, максимальное и среднее время
пребывания элемента объема жидкости в ре-
акторе, особенности переходного периода в реакторе (режима пе-
рехода из одного установившегося состояния в другое) и др.
Наиболее широкое распространение получили два метода вво-
да индикатора (два вида возмущения): ступенчатый и импульс-
ный.
Ступенчатый метод ввода индикатора чаще при-
меняют на практике; объясним его на примере реактора вытесне-
ния. Сверху реактора в момент времени то по всему поперечному
сечению непрерывно вводят небольшой объем индикатора (напри-
мер, какого-либо красителя) (рис. VI. 20). Затем определяют
изменение во времени концентрации индикатора в жидкости на вы-
ходе из реактора. По полученным данным строят кривую измене-
ния концентрации индикатора на выходе во времени Cm = f(x) —
Отбор проб
индикатора
134
кривую отклика; при ступенчатом вводе индикатора ее называют
f-выходной кривой.
Для РИВ кривая отклика представляет собой такую же сту-
пенчатую прямую, как и для условий входа (рис. VI.21,а и б), по-
скольку в РИВ отсутствует продольное перемешивание. В этом
случае концентрация индикатора на входе жидкости в реактор
мгновенно достигает некоторого постоянного значения по всему
сечению реактора, которое сохраняется на всем пути движения
жидкости. Наличие индикатора в жидкости на выходе ее из реак-
тора обнаруживается через некоторый промежуток времени (зави-
сящий от скорости движения жидкости и высоты реактора) и его
концентрация в дальнейшем не изменяется, так как он беспрерывно
вводится в жидкость на входе ее в реактор.
Рис. VI. 21. Кривые отклика при ступенчатом вводе индикатора:
л—на входе в реактор; б—на выходе из идеального реактора; в—на выходе из
реального реактора при наличии продольного перемешивания.
Для реального реактора вытеснения кривая отклика имеет вид
S-образной кривой (рис. VI.21,в),так как благодаря продольному
перемешиванию, которое всегда наблюдается в реальном реакторе,
часть индикатора выводится из реактора раньше, а часть — позже
основной массы; постоянная концентрация индикатора устанавли-
вается через более продолжительный промежуток времени, чем
в РИВ.
При импульсном методе ввода индикатора в жид-
кость, поступающую в, реактор, по всему сечению реактора вводят
небольшой объем индикатора мгновенно (импульсно), а затем, как
и при ступенчатом методе, в моменты времени ть тг, ... опреде-
ляют концентрацию индикатора на выходе из реактора и строят
кривую отклика, при импульсном вводе индикатора ее называют
С-выходной кривой (рис. VI. 22). Поскольку индикатор вводится
мгновенно, его концентрация на входе имеет бесконечно большое
значение. В РИВ элемент объема с такой концентрацией передви-
гается без изменения по длине реактора от входа к выходу
(рис. VI.22, а и б).
Для реального реактора при наличии продольного перемеши-
вания С-выходная кривая имеет максимум (см. рис. VI. 22, в).
•Этот максимум зависит от значения коэффициентов продольного
я поперечного перемешивания.
Так как в РИС-Н происходит интенсивное перемешивание
Жидкости, то при ступенчатом вводе индикатора его концентрация
135
на выходе постепенно повышается от 0 в момент времени то до не-
которого постоянного значения (рис. VI. 23, а). При импульсном
Рис. VI. 22. Кривые отклика при импульсном вводе индикатора:
а—на входе в реактор; б—на выходе нз РИВ; в—на выходе из реального
реактора при наличии продольного перемешивания.
вводе индикатора его концентрация на выходе снижается от неко*
торого максимального значения в момент времени то до 0 через
некоторый промежуток времени (рис. VI.23,б).
Рис. VI. 23. Кривые отклика в РИС-Н:
а—при ступенчатом вводе индикатора; в—при импульсном вводе индикатора.
Ввод индикатора имитирует возмущение в потоке, таким воз-
мущением может быть изменение температуры, концентрации ве-
щества, скорости движения жидкости и др.
Сравнение кривых отклика идеальных и реальных реакторов
позволяет уточнить математическую модель реактора и, соответ-
ственно, повысить точность расчета,
136
Примеры
Пример VI. 1. Определить объем РИВ для проведения гомогенной реакции
разложения фосфина-.
4РН3 (газ) —>• Р4 (газ) + 6Н2 (газ) (1)
или
4А —> Р + 6S (2)
Условия-.
Давление, Па.............................. 45,1 • 104
Скорость подачи фосфина ВА. 0. кмоль • с-1 . 4,53 • 10-4
Степень превращения
начальная ХА 0........................... О
конечная ХА.......................... 0,85
Температура Т, К........................ 648,9
Константа скорости реакции (—rA=kCA),
с-1........................................2,78 • КГ3
Решение-.
Объем реактора определяем по уравнению
= = (VI. 13)
сл.о
где Vr — объем реактора, м3;
т — время пребывания реагентов в реакторе, с;
Vo — объемная скорость реакционной смеси, м3 • с*1;
ВА 0 — скорость подачи исходного реагента, кмоль-с-1;
СА 0 — концентрация исходного вещества в смеси, кмоль • м*3.
Из уравнения (6.13) следует
Для рассматриваемой реакции 1-го порядка (п=1) характеристическое
уравнение РИВ имеет вид
ХА ХА
г dX. г dX.
* = М <VL17>
О л о л
«ли, подставив значения т из уравнения (а), находим
ХсА X
Vr = BA.0\ (б)
. . . 0 я
В даииом случае объем реагентов находится в прямой зависимости от Хл,
поэтому справедливо уравнение (I. 12)
с =с
А CA..1+SaXa
Подставив значение СА в уравнение (б), находим
0 + *А)
Vr =
ВА,0
kCA,0
dXA
(в)
о
137
в результате деления, находям
1 + 8д
(1+еЛ):(1_Хл)-----вд + тТгЛ
А
Подставив это значение в уравнение (в) и интегрируя, получаем
в.п г 1 1
г'“^1(1 + 8д),пТ^-еЛ] (г>
Д,0 L A J
Значение вл определяем по уравнению (I. 10)
VXA-t “
Относительный объем реакционной смеси рассчитываем на основе уравнения
реакции, из которого следует
7 — 4
^ri=H6 = 7; ^хд-о = 4> поэтому ел—— = 0,75
Начальную концентрацию исходного вещества определяем по уравнению
„ р 45,1-10* nn0R ,
СД. 0 ЦТ 8,042 • 103 • 648,9 0,086 КМоль ’м
Если давление измеряется в Па (Н/м2), то /? = 8,042-103 Н-м-кмолщ'-К-1.
Подставив найденные величины в уравнение (г), получаем объем реак-
тора
у -------4,53 '10 ---(। + 0,75) 1п--!------0,75 • 0,85 = 4,363 м3
2,78 10-3 - 0,086 1 -0,85
Пример VI. 2.
Определить скорость подачи в РИС-Н двух растворов, содержащих реагенты.
А и В. Реагенты взаимодействуют по уравнению
«I
Скорости подачи реагентов одинаковы и должны быть такими, чтобы за
время их пребывания в реакторе прореагировало 75% вещества В.
Условия'.
Объем реактора Vr, м3........................ 0,18
Концентрация, кмоль • м-3
реагента А в первом потоке Сд_ п............. 4,2
реагента В во втором потоке Св п....... 2,4
Константы скорости, м3 • кмоль-1 • с-1
kx.......................................... 0,12
k2 ...................................... 0,05
Степень превращения реагента В Хв....... 0,75
Решение:
Объем потоков определяем по уравнению (VI. 13)
^обш = ^ + ^ = 2Гл = Гг/т (а>
138
(б)
С другой стороны, для РИС-Н время пребывания можно определить по уравне-
«иЮ Г -Г
~ГА
Приравняв правые части уравнений (а) и (б), находим
М-'л)
(в)
ГД"2(САО-Сд)
Для решения уравнения (в) необходимо прежде всего определить (—гд).
Скорости реакции равны, поэтому справедливо равенство
— ГА = — гв = V АСв ~ k2CRCS
(г)
Таким образом, для определения —гд необходимо иайти концентрации исходных
реагентов и продуктов реакции в потоке на выходе из реактора, а также концен-
трации реагентов А и В иа входе в реактор (после смешения потоков):
4,27. 4,2
СА. о = + v~ = — = 2,1 кмоль •м-3
2,47 2,4
Сп п = = 1,2 кмоль • м-3
5,0 7д+гв 2
С А = СА. О — (СВ, 0 — ^в) = Сд, 0 — [СВ, 0 “ СВ. О 0 — “
= С . „ — С пХк — 2,1 — 1,2 0,75 = 1,2 кмоль• м-3
А» v О» v D
CR — C„ п — С„ пХ = 1,2 — 1,2 • 0,75 = 0,3 кмоль • м-3
О Bt и D< и В
CD =С =Сп = 1,2• 0,75 = 0,9 кмоль• м~3
д\> и о Ot U о
Подставив полученные значения в уравнение (г), а затем значения —гд
в уравиеиие (в), находим
— гА = — гв = 0,12 • 1,2 • 0,3 — 0,05 • 0,9 • 0,9 = 2,7 • 10~4 кмоль • м-3 • с-1
v _v _ 0,18-27- 10~4 ,._4 з
VA~VB 2 • (2,1 - 1,2) 2,7 ’ 0 М ‘ С
400
200
Пример VI. 3.
Определить степень превращения реагента А в каскаде, состоящем из двух
реакторов идеального смешения.
Условия:
Скорость подачи реакционной смеси 70,
Л • МИИ-1..........'...................
Объем каждого реактора Vr, л...........
Начальная концентрация реагента СА 0,
моль-л-1 . '...........................
Коистаита скорости реакции А, мин-1....
Порядок реакции п ..............
1,2
1,6
0,76
В процессе реакции объем реакционной смеси не изменяется (ед =0).
Решение:
. Для первого реактора из уравнений (VI. 13), (VI. 19) и (11.23) получим
Т__7Г. t_CA.0XA.l.
T“V п
-rA^kCnA = kCnA^(\-XA)n
А
139.
Из этих уравнений следует
Vr СА 0^,1
Vo kCnA^\-XAtl)n
АСдоО-Лл)’1
После преобразования находим
ХА , VrfeC^-1 200 ...
-----dd = _Г—did— = ___ .16.1,2~°>24 =-0,762
(!-W'7b 400
(а)
Методом последовательных приближений находим Хл, i = 0,47.
Концентрацию реагента в первом реакторе и, следовательно, на входе во
второй реактор, определяем по уравнению (1.9):
СА, 1 = С А, 0 С1 “ ХА, 1) ~ 1,2 “ °’47) = 0,636 кмоль • л-‘
Аналогично тому, что сделано ранее дли первого реактора, я с учетом зна-
чения С а, 1 по уравнению (а) находим степень превращения реагента А во вто-
ром реакторе ХА,г и сравниваем с количеством реагента А на входе во второй
реактор
Хал УгЬС^~' _ 200
(I-4>2)o,76= Vo 400
1,6 - 0,696“0,24 = 0,89
Отсюда методом последовательных приближений находим ХА 2 = 0,515.
Степень превращения реагента А по отношению к его первоначальному ко-
личеству можно определить из соотношения
1 “ ХА, 2 = (1 - ХА, 1) С “ Х'А, 2) = (1 - 0,47) (1 - 0,515) = 0,257
Таким образом, общая степень превращения после двух реакторов составит
Хл, 2 = 0,743.
Глава VII
РЕАКТОРЫ С РАЗЛИЧНЫМ ТЕПЛОВЫМ РЕЖИМОМ
До сих пор при определении условий проведения процессов
в реакторах влияние температуры не учитывалось, т. е. исследова-
лись процессы в изотермических условиях при Т = const. Между
тем в большинстве случаев температура в процессе изменяется и
оказывает существенное влияние на кинетику, статику и селектив-
ность химических процессов. Поэтому в большинстве практиче-
ских случаев в реакторе создают определенный температурный
режим, обеспечивающий наиболее высокую эффективность процес-
са. В зависимости от теплового эффекта протекающих реакций, а
также от оптимального температурного режима, который необхо-
димо поддерживать в реакторе, от реакционной смеси либо отводят
тепло, либо к ней подводят тепло, или же температурный режим
в реакторе сохраняют таким, каким он самопроизвольно устанав-
ливается в соответствии с тепловым эффектом реакции.
В связи с этим выбор теплового режима в реакторах и разра-
ботка методов его поддержания имеют большое практическое зна-
чение.
140
Уравнения, отражающие работу реактора с учетом влияния
температуры, могут быть получены из уравнений материального
баланса, приведенных в гл. VI при условии, что в эти уравнения
вместо постоянного значения константы скорости (что справед-
ливо для изотермических условий) вводят ее значение из уравне-
ния (11.23).
k = k,e'ElRT
Так, например, если это сделать для РИС-П (или РИВ), то с уче-
том уравнения (VI. 7), получим
ХА ХА
г dX± = 1 г dXA
kcnA j (1-хл)"е-адг (а)
Температура реакционной смеси Т является сложной функцией
многих переменных (степени превращения исходного реагента,
теплового эффекта реакции, теплообмена с окружающей средой и
др.), поэтому интегрирование уравнения (а) связано с большими
трудностями. В связи с этим на практике поступают следующим
образом. Для каждого типа реактора составляют уравнения те-
плового баланса, а затем в эти уравнения вводят необходимые
данные из уравнений материального баланса, поскольку тепловой
баланс зависит от количества прореагировавшего исходного реа-
гента, от массы реакционной смеси и других показателей, что от-
ражается уравнениями материального баланса.
1. КЛАССИФИКАЦИЯ РЕАКТОРОВ
С РАЗЛИЧНЫМ ТЕПЛОВЫМ РЕЖИМОМ
Каждый тип реактора может работать в трех режимах: адиа-
батическом, изотермическом и политропическом.
При адиабатическом режиме в реакторе отсутствует теплооб-
мен с окружающей средой и тепло химической реакции полностью
расходуется на изменение температуры реакционной смеси.
При изотермическом режиме путем подвода или отвода тепла
в реакторе поддерживают постоянную температуру в течение всего
процесса.
При политропическом режиме температура в реакторе непо-
стоянна, при этом часть тепла может отводиться от реакционной
смеси или подводиться к ней.
Адиабатический и изотермический режимы представляют собой
предельные идеальные случаи, которые на практике почти не на-
блюдаются. Однако режим многих реакторов в производственных
Условиях приближается к этим крайним моделям, поэтому с до-
статочной для практических целей точностью эти реакторы могут
быть рассчитаны по уравнениям, полученным для адиабатического
а изотермического режимов.
141
2. УРАВНЕНИЕ ТЕПЛОВОГО БАЛАНСА РЕАКТОРА
Основой для расчета реакторов с учетом теплового режима
служит уравнение теплового баланса, составленное обычно на еди-
ницу времени. В общем виде это уравнение может быть записано
так:
Qnp = Qpacx (VII. 1)
Для экзотермической реакции приход и расход тепла можно
выразить в виде
Qnp = Qpear + Qx. р (VII. 2)
Qpacx = Qnax + Qnpon + Qt (VII. 3)
где Qpear — количество тепла, вносимого исходными реагентами;
Qx. р — количество тепла, выделяющегося при химической реакции;
QnaK — количество тепла, накапливающегося в реакторе;
Спрод — количество тепла, уносимого продуктами;
QT — количество тепла, выводимого теплообменом.
Подставив эти значения в уравнение (VII. 1), находим
Qnax= (Qrtpoa Qpear) ~ Qr 4" Qx. р (VII. 4)
Где Qnpoa — Qpear — Qkohb
Здесь Qkohb — обозначает количество тепла, выносимого кон-
вективным потоком, с учетом которого получим
QnaK = Qkohb — Qt Ч~ Qx. р (VII. 5)
Полученное уравнение теплового баланса (VII. 5) может при-
нимать различную форму в зависимости от типа реактора и теп-
лового режима процесса,
В общем случае температура и другие параметры процесса из-
меняются как в объеме реактора, так и во времени, поэтому урав-
нения теплового баланса составляют в дифференциальной форме
(подобно тому, как это было принято при составлении уравнения
материального баланса). Для этой цели используют дифференци-
альное уравнение конвективного теплообмена (А. Г. Касаткин,
Основные процессы и аппараты химической технологии. М., «Хи-
мия», 1971, стр. 294):
„ дГ „с дТ , дТ . дТ \ ,
рСп -z-= — рС„ I wx Ь р wz -г— I -р
Л ’ дт ’ \ дх и ду dz) 1
’ , . ( д2Т д2Т \
+ Л к дх2 + ду2 + dz2 ) ^VIL 6)
где р, Ср — плотность и удельная теплоемкость реакционной смеси;
х, у, z — пространственные координаты;
Щ*, Щу, — составляющие скорости движения потока в направлении осей;
Л — коэффициент теплопроводности реакционной смеси.
Чтобы использовать это уравнение по отношению к простой не-
обратимой реакции A->/? + Q, его составляют по одному из ком-
понентов реакционной смеси (любому) и вводят в него дополни-
тельные члены, учитывающие отвод тепла в результате теплооб-
мена и тепло реакции. Если составить баланс по компоненту А и
142
ввести в уравнение дополнительные члены, то дифференциальное
уравнение конвективного теплообмена (уравнение теплового ба-
ланса) может быть записано в таком виде:
„ дТ - / дТ , дТ , дТ \ ,
рср~д^~~ рСр ёГ + w«~d^ + Wz ) +
/ д2Т д2Т д2Т \
+ Л (-ГТ + 44- + 44-1 -^ЛАТ+гЛН (VII. 7)
\ дх2 ду2 1 dz2 ) УД А ' '
где Гуд ~ удельная поверхность теплообмена (на единицу объема реакционной
смеси);
К — коэффициент теплопередачи;
ДТ = Т — ТхЛ;
Т — температура реакционной смеси;
Тхд — температура хладоагента;
ДЯ — тепловой эффект реакции.
Левая часть уравнения (VII. 7) характеризует скорость накоп-
ления тепла в элементарном объеме, для которого составляется
тепловой баланс. Этому члену соответствует величина в уравне-
нии (VII. 5)
_ дТ
Унак — РСр-^-
Первая группа членов правой части уравнения (VII.7) опре-
деляет скорость конвективного переноса тепла по соответствую-
щим координатам (х, у, z) в элементарном объеме.
Вторая группа членов правой части уравнения (VII. 7) опреде-
ляет скорость отвода тепла в результате молекулярной и конвек-
тивной теплопроводности реакционной среды.
Первая и вторая группы членов правой части уравнения
(VII. 7) соответствуют члену Qkohb в уравнении (VII. 5)
п п f дТ , дТ , дт\ ,
Скова РСр дх +wy ду + wz дг ) +
+ + + (VII. 8)
\ дх2 ду2 dz2 J
Третья и четвертая группы членов правой части уравнения
(VII. 7) характеризуют скорость отвода тепла путем теплообмена
и скорость подвода тепла в результате химической реакции
QT = FWKAT (VII. 9)
Сх.р='-А^ (VII. 10)
Общее решение уравнения (VII. 7) связано с большими труд-
ностями, поэтому в зависимости от характера реакции, теплового
режима и режима движения реакционной среды, т. е. от гидроди-
намической обстановки в реакторе, в уравнение (VII. 7) вводят
соответствующие упрощения. Это позволяет найти решение разно-
образных задач с достаточной для практических целей точностью.
143
Введем такие упрощения для различных реакторов при раз* i.
ных режимах их работы: адиабатическом, изотермическом и поли* I '
тропическом. Поскольку политропический режим представляет со-
бой общий случай, т. е. при составлении теплового баланса такого ' ’ J
реактора учитываются все виды подвода и отвода тепла, начнем J
с него. Из уравнения теплового баланса для политропического ре-
актора легко определить частные случаи:
для адиабатического реактора, когда уравнение (VII. 9) может
быть записано в виде QT = 0; и для изотермического реактора,
когда уравнение (VII. 8) запишется как—Qkohb = 0.
Рассмотрим реакторы в той же последовательности, которая
была принята в главе VI, при этом к каждому названию реактора
Рис. VII. 1. Политропические реакторы идеального смешения
РИС-Н-П с отводом тепла через стенку (а) и отводом тепла с по-
мощью охлаждающих элементов (б).
добавим букву П, т. е. «политропический»: РИС-П-П, РИВ-П,
РИС-Н-П.
Отвод или подвод тепла выполняются различно. Так, например,
для РИС-Н-П эта операция осуществляется через стенку реактора
или с помощью теплообменных элементов, расположенных внутри
реактора (рис. VII. 1).
3. ПОЛИТРОПИЧЕСКИЙ РЕЖИМ
РИС-П-П. В периодическом реакторе QKohb=0. Тогда уравне-
ние (VII. 7) можно представить в виде:
Но для РИС-П по уравнению (VI. 5) имеем
J А, О ^А <*А, О ^А
Л = ___, нлигА = —~sr~
Подставив в уравнение (а) значение для гА и произведя переста-
новки и сокращения, находим
bHdXk = C' dT + ----dx (VII. 11)
gA.O
где c'p — теплоемкость реакционной смеси, отнесенная к единице массы исход-
ного реагента:
рС_
<=-^- (VII. 12)
СА,0
Преобразуем последний член правой части уравнения (VII. 11),
умножив числитель и знаменатель на Vr. В результате получаем
Гуд/СДГ F^Kl^TVr j FK^T
с d’b— с v N
СА,0 СА,0Иг ЛА,0
где F — общая поверхность теплообмена в реакторе.
с учетом F и NA уравнение (VII. 11) запишется в виде
ДЯ dXA = CpdT + ~^-dx (VII. 13)
РИВ-П. В этом реакторе изменение температуры происходит
только в одном направлении— по длине реактора (т. е. по оси х),
при этом
а Г дТ — дТ — о
дх dl ду dz
Изменение температуры за счет молекулярной теплопроводно-
сти мало и его можно не учитывать; поскольку отсутствует пере-
мешивание, нет изменения температуры за счет турбулентной
диффузии. Таким образом
(дЧ д2Т
\ дх2 ду2 дг2 )
С учетом этого общие уравнения для РИВ при установившемся
режиме (когда <5нак = 0) будут следующими:
Qx. р Фконв "Р
глдя = рср®4г +• (а)
Для РИВ по уравнению (VII. 16) имеем
г -
A dx
а
Подставив эти значения в уравнение (а) и произведя переста-
новки и сокращения, находим
144
145
Преобразуем последний илей* этого уравнения, умножив числи-
тель и знаменатель на V и учтя, что dx — dl/w
Fyaf( кТ FyaK кТ V dl F'KkT
~^TTdx== cA^wv =~BZTdl
F V
где F ------— поверхность теплообмена в пересчете на единицу длины реак-
тора;
В0 = СА< 0V — расход реагента.
С учетом сделанных преобразований находим: •
Д/z dXA = CpdT + di (VII. 14>
, °A,0
г
РИС-Н-П. Для этого реактора характерно отсутствие градиента
параметров как во времени, так и в объеме реактора, поэтому
уравнение теплового баланса составляют в целом для реактора.
Тогда градиенты параметров в дифференциальной форме можно
заменить разностью между значениями параметров на входе в ре-
актор и на выходе из него.
При установившемся режиме из уравнения (VII. 5) следует
Qx. р = Qkohb + Qt (VII. 15>
Величины, входящие в это уравнение, для рассматриваемого
случая можно определить следующим образом:
С, „X. AtfV,
С. aX.kHVr
-°vArlv-~ -cA_oxA^v
Qkohb — pQp V (Т — Т0)
QT=^FKkT
(б)
(в)
(г)
где F — общая поверхность теплообмена в реакторе.
Подставив полученные значения в уравнение (VII. 15) и про-
изведя сокращения и перестановки, находим
^ХА = -^-(Т-Т0) + -5—у
СЛ, О СЛ, (Г
или
ДЯ^С^Г-ГоН^^-
(VII. 16>
Исходя из полученных общих уравнений, отражающих поли-
тропический режим работы реакторов, найдем расчетные уравне-
ния для режимов адиабатического и изотермического.
146
4. АДИАБАТИЧЕСКИЙ РЕЖИМ
РИС-П-А представляет собой реактор с мешалкой, стенки кото-
рого изолированы (рис. VII. 2).
Так как при адиабатическом режиме отвод тепла отсутствует,
то последний член правой части уравнения (VII. 13) равен нулю
ЛКДГ ,
Qt = -v-с?т=0 (а)
А. о
Тогда из уравнения (VII. 13) получим
кН dXA = Ср dT
(VII. 17)
Если принять кН и Ср постоянными,
уравнения (VII.17) находим
кНХА = Ср[Т -TQ) (VII.18)
или
Т=Т „ + ^-Х. (VII.19)
О Ср А
Поскольку То величина постоян-
ная,- функциональная зависимость
T = f(XA) является линейной. При
этом наклон прямой к оси абсцисс бу-
дет определяться уравнением
tga = Atf/(?p (VII. 20)
то после, интегрирования
Рис. VII. 2. Адиабатический
реактор идеального смешения
периодический.
При проведении химической реакции до конца, т. е. при ХА= 1»
из уравнения (VII. 19) получаем
Г' = Г0 + -^-=Г0 + Д7ад
СР
(VII- 21)
где Т' — максимально возможная температура для экзотермической реакции в
адиабатическом процессе (при ХА = 1);
ДГад — адиабатическое изменение температуры.
В РИВ-А и РИС-Н-А также отсутствует теплообмен, поэтому из
уравнений (VII. 14) и (VII. 16) получаем
для РИВ-А
kHdXA=CpdT (VII. 22)
для РИС-Н-А
кН ХА~ С'р(Т ~ Г (VII. 23)
Если принять что кН и Ср не зависят от Т, то из уравнения
(VII.22) следует:
дя Хл = С'р (Г-Го) (VII. 24)
Таким образом, для всех типов реакторов при адиабатическом
режиме их работы и при постоянных значениях кН и Ср тепловой
147
баланс выражается в виде одинаковых уравнений (VII. 18),
(VII.23), (VII.24). Это и следовало ожидать, так как при адиаба-
тическом режиме тепло химической реакции расходуется только
на нагревание реакционной смеси.
5. ИЗОТЕРМИЧЕСКИЙ РЕЖИМ
Изотермические реакции проводят на практике только в не-
прерывных реакторах, так как для поддержания постоянной тем-
пературы в реакторе периодического действия отвод тепла должен
изменяться во времени, что в промышленных условиях осуще-
ствить трудно. В связи с этим изотермические реакторы периоди-
ческого действия на практике не применяются и здесь не рассмат-
риваются.
В изотермических реакторах путем подвода или отвода тепла
поддерживается постоянная температура в течение всего процесса.
В тех случаях, когда температура реакционной смеси на входе
в реакторы равна температуре на выходе из него (т. е. Т = То),
для РИВ-И и РИС-Н-И можно записать
СрйГ = 0 н с'р( Г-Го) = О
С учетом этих значений полученные ранее уравнения (VII. 14) и (VII. 16) примут вид для РИВ-И д/zdxA=(vn 25) для РИС-Н-И = (vn.26) А, 0 Ниже приведены полученные уравнения тепловых балансов ре- акторов различных типов:
Реактор Режим работы
политропический адиабатический изотермический
РИС-П РИС-П-П РИС-П-А —
РИВ РИС-Н bHdXA = C'dT+^^dx ‘ А.о РИВ-П &HdXA = С' dT+ F'hK-— dl °Л.о РИС-Н-П до у — С’ IT Т 4- 6ffdXA = CpdT РИВ-А &ffdXA = C'pdT РИС-Н-А ЬН ХА==С'р(Т-Т0) РИВ-И &ffdXA = -^dt ВА,0 РИС-Н-И ЛИ? ДЯ ХА ~ ' в “ ° А. 0
WJ Ад ^р V i О/ 1 Ft
148
е. УСЛОВИЯ ПОДДЕРЖАНИЯ УСТОЙЧИВОГО
РЕЖИМА РАБОТЫ РЕАКТОРА
Решить уравнение теплового баланса — значит найти перемен-
ные параметры, при которых будет соблюдаться равенство между
приходом тепла и его расходом (Qnp = Qpacx) . Однако в произ-
водственных условиях обычно необходимо, чтобы это равенство'
соблюдалось при возможно более высокой степени превращения я
при условии обеспечения устойчивой работы реактора.
Понятие устойчивости формулируется следующим образом: си-
стема считается устойчивой, если после наложения какого-либо
возмущения она самопроизвольно возвращается в прежнее состоя-
ние при снятии этого возмущения.
При неустойчивом состоянии незначительное отклонение како-
го-либо параметра технологического процесса от его первоначаль-
ного значения (температуры, концентрации, давления и др.), что
в производственных условиях всегда может иметь место, приводит
к отклонению от стационарного состояния в реакторе; это откло-
нение увеличивается во времени, а режим реактора не возвра-
щается в исходное состояние и после снятия возмущения.
Проведем анализ устойчивой работы реактора на примере-
РИС-Н-А при проведении в нем реакции A -)- Q. Из уравнения
l(VII. 15) следует, что тепловой баланс этого реактора при стацио-
нарном режиме выражается в виде уравнения
Qx. р = Qkohb (а)1
где
(б>
Для РИС-Н из уравнения (VI. 29) имеем
Ха
тгд _ _ xkQe~EIRT
С А, О С А. О С А, О
са.о(У-ха)
или после преобразования получаем
= — <VIL27>
Подставив это значение в уравнение (б), находим
Qx.p = -^^------ (VII. 28>
Из уравнения (VII. 28) следует, что при повышении темпера-
туры величина. Qx.p стремится к постоянному значению АН, а при
понижении — к нулю.
Полученные зависимости Qx.p = f(T) и Фкоав = ф(7’) показаны
«а рис. VII. 3.
149-
Угол наклона прямой QHOhb = ф(Т’) можно определить по урав-
нению:
, рС в
tga = C'=-F-^- (VII.29)
ga о
Так как количество выделяющегося тепла Qx. р пропорционально
степени превращения ХА, а при установившемся процессе Qx. р =
= Qkohb, то на осях координат рис. VII. 3 показана также величи-
на Ха, но с учетом соответствующего масштаба.
Если 5-образную кривую на рис. VII. 3, а совместить с прямой
на рис. VII. 3, б и поместить их на общий график, то в зависимости
от значения параметров, входящих в уравнения, показанные выше,
. Рис. VII. 3. Диаграммы зависимости прихода и расхода тепла от
температуры для РИС-Н-А:
a~Qx. Р=КТ); б-Ок=Ф(Л.
взаимное расположение кривой и прямой может быть различным.
На рис. VII. 4 показаны некоторые наиболее характерные вари-
анты такого расположения.
Все точки пересечения кривой и прямой, показанные на
рис. VII. 4 (К; L, М), соответствуют состоянию теплового равно-
весия, когда скорость прихода тепла равна скорости расхода
тепла. Однако не все режимы, соответствующие точкам пересече-
ния, равноценны и не все могут быть рекомендованы для про-
мышленных условий.
Режим, показанный на рис. VII. 4, а (кривая и прямая пере-
секаются в точке К), соответствует высокой степени превращения
и обеспечивает устойчивую работу реактора, поэтому такой режим
представляет практический интерес. Устойчивость режима в этом
случае может быть подтверждена следующими примерами.
При снижении температуры от Тк до Л (см. рис. VII. 4, а)
возникает неравенство Qx.p> Qkohb, поэтому после снятия возму’
щения процесс возвращается в прежнее состояние, т. е. в точку К.
(Это объясняется тем, что в результате возмущения приход тепла
становится больше расхода и реакционная смесь нагревается).
При повышении температуры до Т2 возникает неравенство
150
Qkohb, поэтому после снятия возмущения процесс также
возвратится в положение, характеризуемое точкой Л, поскольку
отвод тепла больше прихода, и реакционная смесь охлаждается.
Режим работы реактора, показанный на рис. VII. 4, б (кривая
и прямая пересекаются в точке L), соответствует низкой степени
превращения; следовательно, этот режим практического интереса
не представляет.
При режиме, показанном на рис. VII. 4, в, тепловое равновесие
достигается в трех точках К, М, L, где пересекаются кривая и пря-
мая. Однако все эти режимы не представляют практического
интереса: точка L соответствует низкой степени превращения, а
точка М — неустойчивому режиму (так как после повышения или
понижения температуры процесс не возвращается в точку Л1 после-
151
снятия возмущения). Последнее можно проиллюстрировать приме,
ром. Так, при понижении температуры от Тм до 7\ возникает не-
равенство Qx.p< Qkohb, поэтому после снятия возмущения режим
не восстановится (не возвратится в точку Л4), а перейдет в стацио-
нарное состояние, соответствующее точке L, характеризуемой низ-
кой степенью превращения. При повышении температуры от Тм до
Т2 возникает неравенство Qx. р > Qkohb, поэтому после снятия воз-
мущения процесс не возвратится в состояние, соответствующее точ-
ке М, а перейдет в состояние, соответствующее точке /<. Хотя точ-
ка К на рис. VII. 4, в и соответствует
высокой степени превращения и
устойчивому режиму, однако этот
режим обладает малым запасом
устойчивости. Действительно, сни-
жение температуры от Тк до Л (см.
pHC.VIL4,e) при Qx. р<Qkohb ПРИВО-
ДИТ режим в неустойчивую область,
а затем неизбежно в точку L, для ко-
торой характерна малая величина ХА.
Таким образом, взаимное рас-
положение кривой и прямой, пока-
занное на рис. VII. 4, а, с точкой пе-
ресечения К. соответствует режиму,
при котором достигается высокая
степень превращения и создается
устойчивый режим, следовательно,
этот режим представляет наиболь-
ший практический интерес.
из невыгодного режима в оптималь-
ный, соответствующий точке К, изменяют параметры технологиче-
Рис. VII. 5. Диаграммы Qx р = ( (Г)
и <2к = ф(П Для адиабатического
непрерывного реактора с разным
наклоном прямой (а2 < аД
Чтобы перевести процесс
•ского процесса и изменяют тем самым взаимное расположение пря-
мой и S-образной кривой.
Так, например, если сохранить все условия, соответствующие
рис. VII. 4, б, но увеличить время пребывания реагентов в реак-
торе т, то количество выделяющегося тепла Qx. р возрастет [урав-
нение (VII. 28)]. Соответственно, S-образная кривая сместится
влево в положение, показанное на рис. VII. 4, а, когда кривая и
прямая пересекаются в точке К.
Возможно, не изменяя положения S-образной кривой, переме-
щать линию теплоотвода вправо или влево от ее первоначального
положения либо изменять угол ее наклона. Для перемещения
прямой вправо повышают температуру реакционной смеси То на
входе в реактор. Тогда в соответствии с уравнением (в) прямая
перемещается в положение, показанное на рис. VII. 4, б
пунктиром, при этом прямая и кривая пересекаются в точке К.
Для уменьшения угла наклона прямой увеличивают концентра-
цию реагента А в поступающей в реактор смеси (СА, о). В этом
случае уменьшается tg а [уравнение (VII. 29)].
352
Как видно из рис. VII. 5, при уменьшении угла наклона от at
до а2 точка пересечения кривой и прямой перемещается из точки
д в точку К, соответствующую большому значению ХА и устойчи-
вому режиму.
При проведении обратимых экзотермических реакций условия
осложняются, так как при этом изменяется вид S-образной кривой
{(она достигает максимума, а затем снижается). Это объясняется
тем, что для обратимых экзотермических реакций равновесная сте-
пень превращения Ад уменьшается при увеличении температуры
(рис. VII. 6).
Из рис. VII. 6 видно, что кривая достигает максимального зна-
чения в точке К, а затем снижается, располагаясь вдоль равно-
весной кривой Ха- Поэтому на практике процесс ведут так, чтобы
Рис. VII. 6. Диаграмма X = f (Т)
для обратимой экзотермической
реакции:
1—кривая прихода тепла (QXi р); 2 —
прямая расхода тепла (QK); 5—прямая
расхода тепла прн проведении процесса
в несколько стадий.
термических реакций.
прямая линия (линия теплоотвода) проходила либо через точку К
на кривой 1 (Ха = max), либо через точку Р, когда прямая обра-
зует меньший угол наклона аг- Это обычно достигается повыше-
нием концентрации исходного реагента СА, о- Для увеличения ко-
нечной степеми превращения процесс ведут в несколько стадий;
при этом реакционную смесь после первой стадии охлаждают, вы-
деляют из нее целевой продукт, а аатем реакционную смесь вновь
нагревают и направляют в реактор второй и последующих стадий.
При эндотермических реакциях происходит поглощение тепла
'(—\Н), в этом случае температура реакционной смеси на входе в
реактор выше температуры в реакторе. Тогда QK0Ha = —Ср (Г — То)
по уравнению (VII. 29), tg а становится отрицательным, а угол
наклона будет больше 90°. При таких условиях прямая всегда
будет иметь только одну точку пересечения с S-образной кривой
Крис. VII. 7).
Перемещение точки пересечения в область высокой степени
превращения обычно достигается передвижением прямой вправо.
153.
Для этого повышают температуру реакционной смеси на входе в
реактор от То до Т при постоянном угле наклона ai (см. рис. VII. 7)
или уменьшают угол наклона от at до аг при постоянной темпера-
туре То за счет уменьшения Са,о’- соответственно пересечение кри-
вой и прямой перемещается в точку К или М.
Таким образом, для каждого практического случая существует
оптимальный, наиболее выгодный режим работы реактора, кото-
рый должен .быть установлен с учетом ряда факторов, влияющих
на экономичность процесса. Этот режим рассчитывают путем под-
Рнс. VII. 8. Диаграмма Qx p=f (Т)
и QT = ф (Т) для изотермического
реактора при проведении простой
экзотермической реакции A->/?+Q:
р: 2-^пР«вЛ,о нГхл; 3~
«т п₽н ВА, 0 (,ВА, 0>В'а, о) « Гхл:
<-о"'при Вд.он<'л«л>ГхЛ)-
бора соответствующих значений па-
раметров.
Приведенный графический ме-
тод определения оптимальных усло-
вий работы РИС-Н-А может приме-
няться также для расчета других
режимов работы реактора. Напри-
мер, для РИС-Н-И по уравнениям
(VII. 5) н (VII. 6) имеем
Qx.p Qt
A„v ДКДТ FK
BA,0
Следовательно
(r-^)
(Vi i. зо)
Qr = -~-(T -Т^)
D А. 0
(VII. 31)
Как видно из уравнения
(VII. 27), приход тепла (кНХА) в
уравнении (VII. 30) выражается
S-образной кривой. Правая часть
уравнения на рисунке (VII. 30) характеризует отвод тепла изобра-
жаемый прямой с углом наклона
tga =
FK
ВА. 0
(VII. 32)
Из уравнения (VII. 32) следует, что этот наклон можно менять
путем изменения поверхности теплообмена F, коэффициента тепло-
передачи К и скорости подачи исходного вещества ВА< 0, а переме-
щение прямой вправо достигается повышением температуры хладо-
агента ГхЛ (рис. VII. 8).
Для РИС-Н-П по уравнению (VII. 16) получаем
^X/=Cp(r-ro) + T^(r-^) (VII.33)
А, 0
где приход тепла определяется членом КНХА и может быть изоб-
ражен в виде S-образной кривой, а отвод тепла — в виде прямой.
154
при этом отвод тепла складывается из конвективного тепла и
тепла, передаваемого охлаждающим элементам.
Наклон прямой определяется значениями Ср, F, К, В а, о; Для
параллельного перемещения прямой изменяют температуру реак-
ционной смеси на входе в реактор TQ и температуру хладоагента
Т^(^т==т—
7. ПАРАМЕТРИЧЕСКАЯ ЧУВСТВИТЕЛЬНОСТЬ
При изменении начальных значений параметров процесса со-
ответственно изменяются значения параметров в реакторе и на
выходе из него. При этом расчеты и опыт показывают, что суще-
ствуют такие условия, при которых режим работы реактора стано-
вится чрезвычайно чувствитель-
ным к незначительным измене-
ниям исходных параметров. Ве-
личину, характеризующую изме-
нение какого-либо конечного па-
раметра процесса при изменении
начального значения какого-либо
параметра, называют парамет-
рической чувствитель-
ностью. Этот показатель имеет
большое практическое значение и
должен учитываться при разра-
ботке и оформлении химических
процессов.
Качественно влияние парамет-
рической чувствительности мож-
но установить на примере про-
стой экзотермической реакции
проводимой в РИВ.
На рис. VII. 9 показаны измене-
ния температуры реакционной
смеси и степени превращения по
Рнс. VII. 9. Параметрическая чувстви-
тельность РИВ:
/—температура стеики Тст=270 К; 2 —
80 К: 3-Гст= 290 К; 4-Гст=300 К;
5-Тст = 310 К; 5-Гст = 320 К.
длине реактора, эти данные полу-
чены при нескольких значениях температуры стенки реактора Тс?
и при условии что эта температура поддерживается постоянной по
всей длине реактора.
На рис. VII. 9 видно, что малое снижение температуры стенки
Т'ст (ниже 310 °C) приводит к очень сильному уменьшению темпе-
ратуры реакционной смеси в горячей области реактора, где резко
возрастает степень .превращения (ХА). Следовательно, реактор
обладает высокой параметрической чувствительностью по отноше-
нию к температуре стенки. Подобная чувствительность может
наблюдаться по отношению к другим параметрам (концентрации
реагента, объему реакционной смеси, коэффициенту теплопере-
дачи и др.).
155
Рис. VII. 10. Реактор с непо-
движным слоем катализатора н
внешним теплообменником
Так, в качестве примера рассмотрим параметрическую чувстви-
тельность реактора, в котором на неподвижном слое катализатора
протекает обратимая реакция (рис. VII. 10).
Параметрическая чувствительность по отношению к темпера,
туре реакционной смеси на входе в реактор может быть выраже-
на в виде производной dT2/dT\ и пока-
зывает, как изменяется температура
на выходе из реактора (Т2) при изме-
нении температуры на входе (Т\) на
I К. Этот показатель позволяет уста-
новить граничные условия, при кото-
рых нарушается автотермичность про-
цесса и реактор «затухает».
Количество тепла, затрачиваемого
на нагревание реакционной смеси Qb
и количество тепла, передаваемого
этой же смеси в теплообменнике Q2, можно определить из урав-
нений:
dQt — VCpdTt (а)
dQ2 = KF d (Т2 — Т(б)
еде У — объемный поток реакционной смеси;
Ср — теплоемкость смеси;
д — коэффициент теплопередачи;
F — поверхность теплообмена.
Для того, чтобы в работе реактора создавался запас устойчи-
«ости, должно соблюдаться неравенство
rfQi > dQ2
или откуда VCp dTl >KFd(T2-Ti) d(T2-Ti) VCP dTi < KF (VII. 34)
-или dT2 VCp dTi <! 1 KF (VII. 35)
но из теплового баланса следует
ИЛИ VCp(T2-T3)=*KF(T2-Ti) . (в) VCp T2-Ti KF T2 - T3 (Г)
Подставив полученное соотношение в уравнение (VII. 35), на-
водим
<1-4- Тг ~ Т' <VII 36)
dTl <1 + т2-т3 (V1L3J
На рис. VII. 11 приведены рассчитанные по уравнению (VII. 36) значения
параметрической чувствительности dT2/dTi реактора, изображенного 11а
J56
рис. VII- Ю. при различном времени контакта газовой смеси, содержащей серни-
стый ангидрид (7,5% SO2, 10,5% О2), с ванадиевым катализатором. Пунктирная
линия соответствует правой части неравенства (VII. 36); расчет сделан для опти-
мальных условий процесса при Л = 440 °C, /2 = 585 °C, ta = 453 °C. Для этих
,с"”‘ 1,
+ t2 - ta + 585 - 453 ~ 21
Из рис. VII. 11 видно, что при времени контакта Ti =0,52 с реактор рабо-
тает на границе «затухания», поскольку кривая 1 пересекает пунктирную прямую
при 440 °C; такую же температуру имеет реак-
ционная смесь на входе в реактор (^= 440 °C).
При вдвое большем времени контакта (т2 =
= 1,03 с) кривая 2 пересекает пунктирную пря-
мую при 420 °C. Поскольку ti = 440 °C, запас
устойчивости составляет 440 — 420 — 20 °C.
При тз = 1,55 с запас устойчивости еще
выше (440 — 412 = 28°C, см. рис. VII.11).
Таким образом, чем больше т, т. е. чем боль-
ше загружено катализатора в реактор, тем ниже
температура «затухания» и тем больше запас
устойчивости.
8. ВЫБОР ТИПА РЕАКТОРА
С УЧЕТОМ ТЕПЛОВОГО РЕЖИМА
Важнейшим показателем, определяю-
щим выбор реактора при проведении
простой необратимой реакции, является
скорость процесса, которая должна быть
максимальной. Она зависит от концен-
трации реагента и температуры процес-
са, т. е. г = f(C, Т). Рассмотрим измене-
ние этих параметров в реакторах различ-
ного типа при работе их в адиабатиче-
ском режиме (рис. VII. 12).
Из рис. VII. 12 видно, что при про-
ведении эндотермической реакции более
эффективным является РИВ, так как средние значения Са, Т,
(~га) в нем выше, чем в РИС-Н.
При проведении экзотермической реакции в адиабатическом
режиме возможны два случая, зависящие от степени влияния на
скорость реакции каждого из параметров Са и Т, поскольку с
течением времени, т. е. с увеличением Ха> температура повы-
шается, а значение Са уменьшается.
В первом случае повышение скорости реакции вследствие уве-
личения температуры преобладает над снижением скорости этой
Реакции за счет уменьшения концентрации реагента [см.
рис. VII. 12, кривые 1 на графике — rsblT = f(l) и —гсм = ф(//)].
“ этом случае при проведении процесса в РИВ функциональная
зависимость имеет максимум [см. рис. VII. 12, кривая 1 на графике
"Твыт = f (01-
Рис. VII. 11. Зависимость
параметрической чувстви-
тельности слоя катализатора
Л2/Л[ от начальной темпе-
ратуры ti и времени сопри-
косновения реагента с ката-
лизатором т:
1 — Ti=0,52 с; 2—т2==1,03 с:
3—Тз»1,55 с.
157
Во втором случае снижение скорости процесса в результате
уменьшения концентрации реагента настолько значительное, что
оно не компенсируется повышением скорости процесса вследствие
повышения температуры реакционной смеси за счет тепла реакции.
При этом скорость реакции снижается (см. рис. VII. 12, кривая 2
Рис. VII. 13. Области применения
различных типов реакторов.
Рис. VII. 14. Сочетание РИС-Н и
РИВ, обеспечивающее оптималь-
ные условия процесса.
на графике —гВыт = f(0 и —rCM = <p(z/)]. Такие условия наблю-
даются при низком тепловом эффекте реакции и при протекании
процесса в области высокой степени превращения, когда количе-
ство выделяющегося тепла невелико.
Условия, соответствующие первому случаю, показаны на
рис. VII. 13 и VII. 14. Из рис. VII. 13 видно, что вначале на участ-
158
ке от Ха, о до Ха, i (когда скорость реакции достигает максималь-
ного значения —г a, i) процесс выгодно вести в РИС-Н, так как
в этом реакторе процесс протекает при постоянной конечной ско-
рости реакции — гА, ь которая имеет наибольшее значение. В даль-
нейшем (на участке от ХА, i до ХА, 2) невыгодно вести процесс в
рИС-Н, так как в этой области процесс будет протекать при
самой низкой скорости —г а, г, соответствующей конечным'значе-
ниям параметров (СА, 2 и Т2).
В РИВ скорость процесса в этой области будет плавно изме-
няться от rA,i до гА, 2, и, следовательно, средняя скорость процес-
са будет выше.
При проведении обратимых и сложных реакций возникают до-
полнительные требования при выборе реакторов, такие, как обес-
печение высокой селективности и выхода по целевому продукту.
9. СОЗДАНИЕ ОПТИМАЛЬНОГО ТЕПЛОВОГО РЕЖИМА
В РЕАКТОРАХ
Как указывалось ранее, температура является одним из самых
мощных факторов, влияющих на скорость химических процессов,
поэтому на практике устанавливают оптимальный температурный
режим каждого химического процесса с учетом условий равнове-
сия протекающих реакций, кинетических факторов, селективности
по целевому продукту, термостойкости аппаратуры и т. д. При
работе реактора в политропическом и изотермическом режимах
имеет место теплообмен с окружающей средой, поэтому, регулируя
этот теплообмен, можно приблизить температурный режим реак-
тора к оптимальному.
При адиабатическом режиме работы реактора теплообмен с
окружающей средой отсутствует, поэтому для создания оптималь-
ного температурного режима применяют несколько последова-
тельно соединенных реакторов и после каждого из них преду-
сматривают нагревание реакционной смеси (при эндотермиче-
ских реакциях) или охлаждение смеси (при экзотермических ре-
акциях).
На рис. VII. 15 в качестве примера показаны схемы оформле-
ния эндотермической реакции А -> R — Q в реакторах вытеснения
и смешения, куда поступают- подогретые исходные реагенты. По
мере протекания реакции температура реакционной смеси и кон-
центрация исходного реагента в ней снижаются; соответственно
снижается и скорость процесса. Если процесс осуществляют в не-
скольких последовательно соединенных реакторах, после которых
реакционная смесь дополнительно нагревается (рис. VII. 15,в и г),
скорость процесса более высокая, чем без дополнительного подо-
грева смеси. При такой оформлении процесса на каждой стадии
создается адиабатический режим, а в целом режим приближается
к политропическому и тем в большей степени, чем больше число
стадий.
159
В тех случаях, когда при проведении экзотермических реакций
количество тепла, выделяющегося в результате реакции, доста-
Рнс. VII. 15. Зависимость скорости эндотермических реакции г от степени пре-
вращения X для различных комбинации реакторов вытеснения и смешения:
1—адиабатический процесс; 2—подвод тепла; а—реактор вытеснения; б—каскад реакторов
смешения; б, г —каскад реакторов вытеснения и смешения с промежуточным подогревом.
точно для нагревания исходных реагентов до температуры начала
процесса, создают условия теплообмена, обеспечивающие подогрев
Рнс. VII. 16. Схема адиабатиче-
ского реактора вытеснения, рабо-
тающего в автотермическом ре-
жиме:
/—теплообменник; 2—реактор.
поступающих исходных реагентов
за счет тепла реакции. Если при
этом исключается необходимость
подвода тепла извне, процесс
называют автотермическим (рис.
VII. 16).
Труднее создать определенный
температурный режим при проведе-
нии обратимых экзотермических ре-
акций. В этом случае оптимальной
является не какая-то одна темпера-
тура, а температурная последова-
тельность: процесс необходимо при-
близить к ЛОТ, т. е. начинать при
высокой температуре, а затем, по
мере увеличения X, температуру не-
обходимо снижать.
Существует несколько применяемых на практике приемов, обес-
печивающих достижение оптимального температурного режима.
160
g одном случае процесс проводят в несколько стадий, поддержи-
вая на каждой стадии адиабатический режим и охлаждая реакци-
онную смесь после каждой стадии. Во втором случае тепло
Рис. VII.17. Схемы оформления экзотермических обратимых реакций (А R+Q)'.
а> б—каскады реакторов идеального вытеснения и смешения; в» г—схемы с промежуточным
вводом холодных исходных реагентов; б—трубчатый реактор вытеснения с непрерывным
отводом тепла.
от реакционной смеси отводят непрерывно по мере выделения его
в процессе реакции.
На рис. VII. 17, а показан каскад реакторов вытеснения с про-
межуточным теплообменом между горячей реакционной смесью
и холодными исходными реагентами. Схема построена таким об-
разом, что на подогрев может подаваться регулируемое количе-
ство исходных реагентов. Это позволяет изменять в необходимых
пределах температуру реакционной смеси на входе в каждый
6 Зак, 558 1 61
реактор. В каждом реакторе наблюдается адиабатический разогрев
(линии I—III), а в теплообменниках происходит охлаждение реак-
ционной смеси (на рис. VII. 17, а понижение температуры реак-
ционной смеси показано в виде линий, параллельных оси абсцисс).
На рис. VII. 17, б изображен каскад реакторов смешения и
график зависимости X — f(T), носящей ступенчатый характер, по-
скольку в реакторе смешения изменение температуры происходит
скачкообразно.
Схемы, показанные на рис. VII. 17, в и г, отличаются промежу-
точным вводом холодных реагентов. При этом охлаждение реак-
ционной смеси между ступенями каскада сопровождается не только
изменением температуры, но и изменением ее состава (т. е. сни-
жением концентрации исходного реагента и степени превраще-
ния). Достоинство такой схемы оформления процесса состоит в ее
простоте, обусловленной отсутствием теплообменников. Недоста-
ток схемы заключается в том, что в результате промежуточного
добавления исходных реагентов (для которых ХА = 0) сни-
жается степень превращения с Xt до Хг, X2 до Х2 и т. д.
(рис. VII. 17, в, г). Поэтому для достижения заданной конечной
степени превращения требуется большее общее время пребывания
реагентов в каскаде, чем при работе по схемам, показанным на
рис. VII. 17ча и б.
В схеме на рис. VII. 17, д изображен реактор идеального вытес-
нения с теплообменом по всей длине его реакционной зоны. Хо-
лодные реагенты поступают в межтрубное пространство, по мере
продвижения нагреваются от То до Д и входят в реактор (цент-
ральную трубу). В начале в реакторе температура быстро повы-
шается за счет большой скорости процесса, обусловленной высо-
кой концентрацией исходных реагентов. В этих условиях скорость
выделения тепла превышает скорость отвода тепла. В дальнейшем
по . мере увеличения Хл и уменьшения концентрации исходных
реагентов скорость реакции снижается и соответственно пони-
жается температура реакционной смеси. Этим объясняется слож-
ный характер кривой XA = f(T). Вначале эта кривая распола-
гается вблизи адиабаты (рис. VII. 17,6, пунктирная линия), затем
выходит вправо от ЛОТ, а в конце процесса смещается влево от
ЛОТ за счет интенсивного теплообмена в этой части реактора.
Во всех рассматриваемых случаях фактический температурный
режим приближается к ЛОТ и тем в большей степени, чем боль-
ше число степеней в каскаде (см. рис. VII. 17, а—г). Кроме того,
в схемах на рис. VII. 17, а, б, д обеспечивается автотермичность
процесса.
Примеры
Пример VII. 1. Определить количество тепла, которое необходимо отводить
в РИС-Н при проведении в нем обратимой реакции.
А + В R -|- 18 000 кДж моль-1
с тем, чтобы обеспечить максимальную степень превращения.
162
Условия-.
Температура поступающей реакционной смеси
t0, °C......................................... 15
Теплоемкость реакционной смеси, отнесенная к
молю продукта Ср, кДж • моль"’ • К" .... 400
Экспериментальная зависимость между ХА и t:
t, °C........... 5 15 25 35 40 42 45 55 65
ХА.............. 0,18 0,31 0,46 0,56 0,58 0,60 0,59 0,49 0,30
Решение.
Принимаем, что реактор работает в политропическом режиме (РИС-Н-П).
Тепловой баланс реактора при таком режиме выражается уравнением (VII. 16)
ДДЛЛ = С;(Т-7’О) + -^- .
из которого следует, что количество тепла, которое необходимо отводить от ре-
акционной смеси, составляет
= (а)
А. 0
Для решения уравнения (а) необходимо определить максимальную степень
превращения (ХА, шах) и температуру реакционной смеси, при которой дости-
гается эта степень превращения.
Из приведенных экспериментальных данных следует, что максимальная сте-
пень превращения составляет ХА, max — 0,6' и достигается это значение при
t — 42 °C. Подставив значения всех параметров в уравнение (а), находим
FF = 18 000 • 0,6 — 400 (42 — 15) = 10 800 — 10 800 = 0
ВА,о
Таким образом, для обеспечения оптимальных условий работы реактора теп-
ло отводить в теплообменниках не следует,
Для проведения такого процесса следует применять адиабатический реактор
идеального смешения непрерывный (РИС-Н-А).
Пример VII. 2. Определить количество отводимого или подводимого тепла и
поверхность теплообмена каждого из двух последовательно соединенных РИС-Н
при проведении в них реакции:
А з=>: R + 75 420 кДж-кмоль-1
Условия:
Количество реагента А. подаваемого в реактор ВА 0,
кмоль • с-1............................................0,2
Температура поступающей реакционной смеси 10, °C .... 15
Теплоемкость реакционной смеси Ср, кДж • моль-1 • °C-1 . . 1676
Коэффициент теплопередачи К, кДж • м—2 • ч—1 • °С—1 .... 419
Средняя разность At между t0 и /охл, °C.................... 10
В 1-ом реакторе температура реакционной смеси ti = 49 °C и степень пре-
вращения ХА, 1=0,42; во 2-м реакторе t2 = 42 °C, а Ял, 2 = 0,7.
Решение.
В указанных условиях рассматриваемые реакторы идеального смешения ра-
ботают в политропическом режиме, поэтому для 1-го реактора с учетом задан-
ных конечных условий его работы по уравнению (VII. 16) получаем
a^ = c'p(g-/0) + -^-
u а л
6*
163
ИЛИ
= 75 420 • 0,42 - 400 (49 - 15) = — 25 310 кДж • моль-1
°А,0
Полученное отрицательное значение указывает на необходимость подвода
тепла.
Общее количество подводимого тепла составляет
FK АТ = — 25 310- ВА 0= -25 310.0,2 = — 5062 кДж-мин-1
Необходимая поверхность обмена
„ 5062-60 5062-60 ,
Fl"----= 419~10 =72’5 м
Аналогично для 2-го реактора по уравнению (VII. 16) получаем
FK дг - АН (ХЛ12 - ХА<,) - с'р (12 - 9 =
& А, О
= 75 420 (0,7 - 0,42) - 1676 (49 - 42) = 32 852 кДж • моль-1
Полученное положительное, значение указывает на необходимость отвода
тепла. Количество отводимого тепла равно
FK ДТ = 32 852 • Вл> 0 = 32 852 • 0,2 = 6570 кДж - мин-1
Тогда необходимая поверхность теплообмена составит
„ 6570•60 6570-60 _. ,
f2=-y^-=4T97To- = 94M
Пример VII. 3. Определить температуру нагревания реагента А на входе в
адиабатический проточный реактор смешения РИС-Н при осуществлении экзо-
термической реакции A-+R.
Известна зависимость Ха = f(t):
t. °C........... 20 40 50 60 70 80 90 100
ХА............. 0,02 0,12 0,24 0,37 0,58 0,82 0,92 0,95
Условия:
Тепловой эффект реакции АН, Дж-моль—1 . . . —160
Теплоемкость сред С кДж/(моль • °C).......3,2
Степень превращения ХА ........... 0,93
Отвечают ли полученные результаты устойчивому режиму?
Решение:
Для проточного реактора идеального смешения РИС-Н уравнение теплового
баланса имеет вид:
дмл=с;(т-т0)
Воспользуемся графическим методом решения теплового баланса: по известной
зависимости Ха — ЦТ) строим график. Полученная S-образная кривая выра-
жает также зависимость тепла химической реакции от температуры
в соответствующих координатах Qx. р = АНХа = f (Т). Из графика опреде-
ляем, что степени превращения ХА = 0,93 соответствует температура в реак-
торе 95 °C.
Температуру на входе в реактор можно определить аналитически по приве-
денному выше уравнению:
С'Т - АНХ, 3,2 • 95 - 160 • 0,93
Та = -£-------Д =--------—--------= 52 °C
р
3,2
164
Это же значение можно получить из графика. Для этой цели строим линию
теплоотвода [QT = С'р (Т — Гр)], которая должна пройти через точку #
= 0,93; Т = 95 °C) с углом наклона tg а = Ср. Из полученных результатов
следует, что выбранные параметры процесса обеспечивают устойчивый тепловой
режим.
Глава VIII
УСТРОЙСТВО РЕАКТОРОВ
Применяемые в промышленности реакторы по своему устрой-
ству могут быть самыми разнообразными: простой резервуар и
емкость с мешалкой, и полая или с насадкой колонна, и доменная
печь, и сложный аппарат с катализатором, атомный реактор и
многие другие. Для проведения одного и того же химического про-
цесса может быть предложено много различных конструкций реак-
торов.
В реакторе протекают не только химические, но и физические
процессы, сопровождаемые переходом исходных реагентов из
одной фазы в другую, связанные с подводом или отводом тепла,
а также с перемешиванием реакционной массы разнообразным
способом и т. д.
Разнообразие реакторов и сложность гидродинамической об-
становки в них затрудняет проведение полной классификации.
В зависимости от критерия, положенного в основу классификации,
один и тот же реактор может быть отнесен к различным классам,
поэтому едва ли целесообразна попытка провести полную систе-
матическую классификацию реакторов. Однако, можно выделить
несколько важнейших критериев или признаков, по которым целе-
сообразно производить классификацию реакторов. К таким при-
знакам, определяющим устройство реактора, можно отнести агре-
гатное состояние исходных реагентов и продуктов реакции, т. е.
фазовое состояние реагентов; периодический или непрерывный
характер производства, гидродинамический режим в реакторе, а
также тепловую обстановку в реакторе.
При конструировании реакторов, кроме этих основных факто-
ров, следует также учитывать общие требования, которым должен
Удовлетворять реактор (см. главу V). Необходимо также добавить
требования, непосредственно определяющие конструкцию реак-
тора: в нем должно быть обеспечено определенное время пребы-
вания реагентов в зоне реакции в заданном режиме, создана тре-
буемая поверхность контакта фаз, высокая скорость массоперег
Дачи и определенное давление, а такЖе обеспечен необходимый
теплообмен.
Рассмотрим устройство реакторов различных типов для про-
ведения гомогенных, гетерогенных и гетерогенно-каталитических
Процессов. При этом будут учтены наиболее важные показатели:
гидродинамический режим и тепловая обстановка в реакторе.
165
Как и прежде, все рассуждения будут относиться к условиям
экзотермических реакций, однако полученные результаты спра-
ведливы также и по отношению к проведению эндотермических
процессов.
1. РЕАКТОРЫ ДЛЯ ГОМОГЕННЫХ ПРОЦЕССОВ
Периодические гомогенные процессы проводят в резервуарах,
кубах или автоклавах, в которых реакционную смесь перемеши-
вают каким-либо способом. Примером такого реактора может
систем для проведения гомогенных реакций:
а—кубовый реактор непрерывного действия с перемешиванием; б —реактор полу-
пернодического действия с перемешиванием; в—реактор периодического действия с
перемешиванием; г—каскад кубовых реакторов непрерывного действия; д—то же, с
распределением сырья; е—трубчатый реактор непрерывного действия; ж— то же, с
предварительным смешиванием сырьевых потоков; з—трубчатый реактор непрерывно-
го действия с поперечным распределением сырья; и—охлаждаемый однотрубный реа-
ктор; к—многотрубный реактор с теплообменом.
служить РИС-П. Для непрерывных процессов используются реак-
торы РИВ или РИС-Н, а также комбинации этих реакторов.
К наиболее распространенным непрерывным реакторам для
гомогенных реакций относится охлаждаемый трубчатый реактор
вытеснения (рис. VIII. 1), представляющий собой видоизмененный
вариант рассмотренного ранее РИВ. Реактор изготовлен в виде
166
змеевика, охлаждаемого снаружи. Большая поверхность труб обе-
спечивает интенсивный отвод тепла в тех случаях, когда тепловой
эффект реакции очень велик.
Каскад реакторов смешения, приведенный на рис. VIII. 1,д, от-
личается от рассмотренного ранее К-РИС тем, что реагент В до-
бавляется в аппарат постепенно. Это позволяет поддерживать оп-
тимальный режим процесса.
2. РЕАКТОРЫ ДЛЯ ГЕТЕРОГЕННЫХ
НЕКАТАЛИТИЧЕСКИХ ПРОЦЕССОВ
При создании реакторов для проведения гетерогенных про-
цессов необходимо принимать во внимание несколько факторов,
усложняющих конструкцию. Во-первых, в гетерогенных системах
компоненты находятся в различных фазах, поэтому протекающие
в них процессы связаны с переносом вещества через поверхность
соприкосновения фаз. В этом случае на скорость процесса значи-
тельное влияние оказывают физические факторы: размер и со-
стояние поверхности раздела фаз, диффузия вещества из одной
фазы к поверхности раздела фаз и в объем второй фазы, а также
обратная диффузия образующихся продуктов и т. д. Поэтому
конструкция реактора для гетерогенных процессов должна обес-
печивать наилучшие условия для массопередачи и, кроме того,
создавать возможно большую поверхность соприкосновения фаз.
В ряде случаев в конструкции аппарата должны быть предусмот-
рены обновления поверхности контакта фаз.
В гетерогенной системе, в отличие от гомогенной, каждая фаза
может иметь свой режим движения реагентов; возможны также
различные комбинации режимов. Кроме того, в этой системе могут
существовать различные потоки фаз: прямоток =5, противо-
ток ^.перекрестный ток -у->.
И, наконец, конструкция реактора зависит от того, в каких
фазах находятся реагенты. Так, конструкция реакторов для осу-
ществления процессов в системе г — т отличается от конструкции
реакторов для системы г — ж.
При разработке гетерогенного процесса необходимо учитывать,
что правильный выбор конструкции аппарата позволяет в значи-
тельной степени интенсифицировать процесс, переводя его из од-
ной области протекания в другую. Например, уменьшая интенсив-
ным перемешиванием диффузионное сопротивление, можно пере-
вести процесс из диффузионной области в кинетическую и далее
Уже повышать скорость химической реакции.
Реакторы для проведения реакций
в системах г-т и ж-т
Характеристика промышленных реакторов во многих случаях
Довольно близка по своим показателям к характеристике реакто-
ров с идеальным режимом движения реакционной смеси, поэтому
167
последние могут служить исходной моделью для'расчета и деталь-
ного исследования промышленных реакторов. При этом реакторы
для проведения гетерогенных процессов г — т и ж — т с достаточ-
йой степенью точности можно отнести к одной из двух групп:
реакторы, в которых реакционная смесь находится в условиях,
близких к режиму вытеснения, и реакторы, в которых условия
близки к режиму смешения.
Рис. VIII.2. Принципиальные схемы аппаратов для проведения иеката-
литических реакций между газом и твердым веществом или жидкостью
и твердым веществом:
а—противоточный аппарат, работающий в режиме вытеснения; б —аппарат с парал-
лельным током, работающий в режиме вытеснения; в—аппарат с перекрестным током,
работающий в режиме вытеснения; г— аппарат со смешанной организацией потока,
работающий в режиме вытеснения (с механической мешалкой); ,д — полупернодический
реактор, газ—в режиме вытеснения; е — реактор для превращения твердого веще-
ства в потоке газа, твердая фаза—в режиме смещения, газ —в промежуточном режиме
между Смешением и вытеснением; ж— аппарат с псевдоожиженным слоем, твердая
фаза— в режиме смешения, газ— в промежуточном режиме.
В реакторе, изображенном на рис. VIII. 2, а, создается режим
вытеснения при противотоке реагентов. Примером такого реактора
является доменная печь и печь для обжига СаСОз-
На рис. VIII. 2, б показан реактор, представляющий собой вра-
щающийся барабан. Здесь также создается режим; вытеснения, но
при прямотоке реагентов. Такой реактор применяют для сушки
веществ, чувствительных к перегреву.
Перекрестный ход реагентов и режим вытеснения создается в
топках паровых котлов, где на движущейся колосниковой решетке
(рис. VIH. 2, е) находится твердое топливо, а через него проду-
вается воздух.
В полочной печи для обжига твердого сырья (рис. VIII. 2 г)
организовано смешанное движение потоков, работающих в ре-
жиме вытеснения, но с перемешиванием твердой фазы.
168
к недостаткам реакторов вытеснения относится либо незначи-
тельное перемешивание фаз (рис. VIII.2,а и а), либо полное от-
сутствие обновления поверхности контакта фаз (рис. VIII. 2,5).
Такие реакторы невыгодно применять в тех случаях, когда про-
цесс протекает в области внешней диффузии; рациональнее ис-
пользовать реакторы второй группы, в которых твердый материал
находится в режиме смешения.
Конструктивно реакторы второй группы могут быть оформлены
б двух вариантах: первый — это полая камера, в которой переме-
шивание твердого материала происходит в потоке газа (например,
печи для пылевидного обжига колчедана, рис. VIII. 2, е). Тонкоиз-
мельченный твердый материал вдувается с помощью форсунки в
полый реактор, где и происходит его взаимодействие с потоком
газа (кислород воздуха) прн интенсивном перемешивании.
Второй вариант реакторов — это аппараты с псевдоожи-
женным (так называемым кипящим) слоем твердого материала
(рис. VIII. 2, ж). Псевдоожиженный слой твердых частиц обра-
зуется при продувании газа снизу вверх сквозь слой твердого
зернистого материала с такой скоростью, при которой частицы
как бы взвешиваются, плавают и пульсируют в потоке газа. Од-
нако при поддержании такой скорости потока частицы не должны
покидать пределы взвешенного слоя; создается впечатление, что
материал кипит.
При применении реактора с псевдоожиженным слоем твердый
материал подается на решетку, а снизу поступает газ. Продукт
реакции или зола непрерывно выводятся из реактора. В таких
реакторах твердый зернистый материал находится в режиме вы-
теснения, а характеристика газового потока трудно поддается
определению. Обычно считают, что в этом случае создается про-
межуточный режим движения газа: между режимами смешения и
вытеснения.
Достоинство аппаратов с кипящим слоем состоит в том, что
при высоких скоростях газового потока снижается внешнедиффуз-
ное сопротивление газовой фазы в десятки и сотни раз. Благодаря
этому в газовой фазе значительно возрастает коэффициент массо-
передачи. Таким образом, в тех случаях, когда взаимодействие
газа с твердым веществом протекает во внешнедиффузионной обла-
сти, выгодно применять реакторы этого типа. Необходимо также
отметить благоприятные условия для отвода тепла в таких аппара-
тах: коэффициент теплоотдачи от кипящего слоя к поверхности
составляет 500—1000 кДж-м2-ч_1"К-1. Естественно, что это
обстоятельство имеет большое значение для процессов, протекаю-
щих с выделением тепла.
Реакторы для проведения реакций в системах г — жиж — ж
Такие реакторы чаще всего конструируются по принципу аб-
сорбционных аппаратов обычно непрерывного действия, реже при-
меняются реакторы полупериодические с непрерывным питанием
169’
газом, еще реже периодические реакторы (преимущественно для
систем ж — ж).
Рассмотрим наиболее распространенные конструкции.
Насадочная скрубберная и полая башни, показанные на
рис. VIII. 3, а и г, широко применяются при оформлении самых
разнообразных гетерогенных процессов в системе г — ж. В таких
башнях газообразный реагент движется снизу вверх навстречу
орошающей жидкости. В насадочной башне жидкость смачивает
Рис. VIII.3. Принципиальные схемы аппаратов для проведения реакций
между газом и жидкостью и между двумя труднорастворимыми жид-
костями:
а —насадочная колонна (режим вытеснения); б—барботажная колонна с колпачковыми
тарелками (режим вытеснения); в—барботажная колонна с сетчатыми’ тарелками
(режим вытеснения); г—распределительная колонна (режим вытеснения); б —реактор
идеального смешения (с сепаратором)—одностадийный контакт фаз; е — каскад реак-
торов смешения, противоточная система.
насадку башни, при этом создается большая поверхность сопри-
косновения фаз. В полой башне соприкосновение фаз осуществ-
ляется через поверхность образующихся капель жидкости. Суще-
ственные достоинства этих реакторов состоят в том, что они
имеют несложную конструкцию, обладают малым гидравлическим
сопротивлением, доступны и просты в обслуживании. Недостатки
башен заключаются в том, что они громоздки и малоинтенсивны.
В этих башнях создают как противоток, так и параллель-
ный ток.
Барботажные колонны, показанные на рис. VIII. 3,6 и в, так-
же широко используются в химической и других отраслях про-
мышленности. В этих реакторах газ с большой скоростью прохо-
дит через отверстия в сетчатых тарелках или через колпачки в
колпачковых тарелках и, проходя через слой жидкости, образует
пузырьки. Жидкость перетекает с верхних тарелок на нижние.
170
Такие реакторы конструктивно сложнее башен и обладают
более высоким гидравлическим сопротивлением. Однако за счет
большой скорости газового потока сопротивление внешней диф-
фузии мало. Этим и объясняется их широкое применение при
оформлении технологических схем.
В баке с мешалкой (рис. VIII. 3, с?) обеспечивается интенсив-
ное смешение газообразного и жидкого или двух жидких реаген-
тов и создается большая поверхность соприкосновения фаз. Од-
нако эти аппараты относительно сложны и малопроизводительны,
поэтому их используют для производства малотоннажных и доро-
гих химических продуктов.
В трубе Вентури, используемой для проведения реакций в си-
стеме г — ж, в потоке газообразного реагента разбрызгивается
жидкость. Скорость газа очень велика — 100—150 м-с-1, поэтому в
потоке газа возникают мощные турбулентные пульсации, способ-
ствующие дополнительному дроблению жидкости на очень мелкие
капли. В результате создается большая поверхность соприкосно-
вения фаз и происходит интенсивное перемешивание потоков, при
этом снижается сопротивление внешней диффузии. Достоинство
такого реактора состоит в том, что в нем создается высокоинтен-
сивный процесс; его недостатки заключаются в большом гидрав-
лическом сопротивлении и необходимости установки специальных
аппаратов для выделения увлекаемых газом мельчайших капель
жидкости.
Высокая интенсивность процесса обеспечивается также в пен-
ных аппаратах, в которых через слой жидкости, находящейся на
решетке, пропускают снизу вверх поток газа с такой скоростью,
что создается взвешенный слой подвижной пены в виде движу-
щихся пленок, струй и капель жидкости, тесно перемешивающихся
с пузырьками и струями газа. При снижении скорости газа пен-
ный режим переходит в барботажный, а при увеличении скорости
газа взвешенный слой подвижной пены разрушается и она уносится
в виде потока капель. В пенных аппаратах развивается большая
поверхность соприкосновения между жидкой и газовой фазами,
происходит постоянное обновление этой поверхности, что способ-
ствует высокой интенсивности процесса. Благодаря этому простые
по конструкции пенные аппараты находят все более широкое при-
менение в промышленности в виде однополочных и многополоч-
ных аппаратов.
3. РЕАКТОРЫ ДЛЯ ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ
ПРОЦЕССОВ
В настоящее время насчитываются десятки тысяч гетерогенно-
каталитических процессов, осуществляемых в различных реакто-
рах, которые часто называют контактными аппаратами, или кон-
верторами. По своему устройству эти аппараты весьма разнооб-
разны, они различаются по способу контакта между катализатором
171
и реагирующими веществами, по структуре потоков взаимодей-
ствующих веществ или способам отвода тепла и т. д. Такое
разнообразие контактных аппаратов затрудняет их классифи-
кацию.
Наиболее часто контактные аппараты классифицируют в за-
висимости от состояния катализатора в них. По этому признаку
контактные аппараты делятся на три группы: с неподвижным
слоем катализатора, с псевдоожиженным слоем катализатора и
с движущимся слоем катализатора.
Рассмотрим вначале реакторы с неподвижным слоем катали-
затора. Режим движения газового потока в таких аппаратах при-
ближается к режиму идеального вытеснения. Газообразный ре-
агент поступает в реактор сверху или снизу и проходит через слой
зернистого катализатора, расположенного на решетках или в тру-
бах (рис. VIII. 4).
В контактном аппарате с одним слоем катализатора
(рис. VIII. 4, а) газ, подогретый до температуры зажигания, про-
ходит через слой зернистого катализатора, расположенного на
решетке. Соотношение между объемом катализатора и объемным
расходом газообразного реагента должно быть таково, чтобы
время соприкосновения т было достаточным для достижения за-
данной степени превращения.
Контактный аппарат с катализатором в виде сетки применяет-
ся в тех случаях, когда скорости-реакции очень велика. Например,
окисление аммиака до окиси азота проводят на катализаторе, ко-
торый представляет собой несколько слоев платиновых сеток, рас-
положенных между верхней и нижней коническими крышками
контактного аппарата (рис. VIII. 4, б). Время соприкосновения т
составляет около 10-4 с. .При уменьшении т снижается степень
превращения, а при увеличении т уменьшается выход NO—целе-
вого продукта за счет образования побочных продуктов.
Многополочные аппараты, показанные на рис. VIII. 4, в — д,
применяются для проведения реакций с большим положительным
или отрицательным тепловым эффектом. После каждой стадии
контактирования реакционная газовая смесь охлаждается или
нагревается, в таких условиях процесс будет протекать в темпе-
ратурном режиме, близком к оптимальному, и при высокой сте-
пени превращения.
Аппараты на рис. VIII. 4, в — <9 различаются по способу отвода
тепла. В первых двух аппаратах охлаждающие элементы распо-
ложены внутри аппарата, а в аппарате на рис. VIII .4,6 охлажде-
ние смеси осуществляется в теплообменниках, расположенных вне
аппарата. В эти выносные теплообменники газ направляют после
каждого слоя катализатора.
В контактном аппарате, показанном на рис. VIII. 4, и, тепло-
обменные элементы также расположены внутри аппарата. Они
состоят из двух концентрических труб, погруженных в катализа-
тор. Газ поступает вначале по внутренней трубе, а затем проходит
172
Рис. VIII.4. Принципиальные схемы контактных аппаратов с неподвиж-
ным катализатором:
а—с одним неподвижным слоем катализатора; б—с катализаторной сеткой; в—полон*
ный аппарат с промежуточным охлаждением реагентов посторонним хладоагентом
во внутренних теплообменниках; е—полочный аппарат с промежуточным охлаждением
реагентов холодной исходной газовой смесью, поступающей на катализ; d—полочный
аппарат с промежуточным охлаждением во внешних теплообменниках; е—полочный
аппарат с вводом холодных реагентов между ступенями процесса; ж—трубчатый
аппарат с охлаждением посторонним хладоагентом; з—трубчатый аппарат с охлаж-
дением реагентов холодной исходной газовой смесью; и-—трубчатый аппарат с двой-
ными теплообменными трубами.
173
кольцевое пространство между трубами. При движении газ на-
гревается до температуры зажигания и поступает в слой катали-
затора. Наличие теплообменных элементов в слое катализатора
обеспечивает отвод тепла в течение всего процесса, что позволяет
получить в одном слое катализатора такие же результаты, как и
в аппаратах с несколькими слоями и промежуточным охлажде-
нием.
Особое положение занимают реакторы с псевдоожиженным и
с движущимся катализатором. Эти реакторы начали применяться
в промышленности сравнительно недавно, примерно начиная с
Рис. VIII.5. Принципиальные схемы контактных аппаратов с псевдоожи-
женным слоем катализатора и с движущимся катализатором:
а —аппарат с одним псевдоожиженным слоем катализатора; б —полочный многослой-
ный аппарат с псевдоожиженными слоями катализатора; в—аппарат с движущимся
катализатором; /Л—контактный аппарат; 2 — регенератор; 3—каталнзаторопровод;
5— подъемник.
50-х годов. Однако благодаря специфическим особенностям, рас-
пространение этих реакторов в промышленности происходит
весьма интенсивно.
Реакторы с.псевдоожиженным слоем могут быть однослойными
и многослойными (рис. VIII. 5). В однослойном реакторе
(рис. VIII. 5, а) часть катализатора непрерывно выводится на ре-
генерацию и такое же количество регенерированного катализа-
тора возвращается в реактор. Эти реакторы получили широкое
распространение в органической химии в тех случаях, когда ката-
лизатор быстро снижает свою активность. В большинстве случаев
причиной снижения активности катализатора является образова-
ние кокса на его поверхности.
В многослойном контактном аппарате с псевдоожиженным
слоем (рис. VIII. 5, б) создаются условия, обеспечивающие дости-
жение высокой степени превращения, так как при увеличении
числа слоев катализатора температурный режим приближается
к ЛОТ.
174
’ В реакторе с движущимся катализатором (рис. VIII. 5, в) газ
поступает снизу, а в верхней части реактора происходит распыле-
ние катализатора, который, падая, встречает поток газа. Катали-
затор собирается в нижней части реактора, а затем выводится из
него и подъемником вновь подается в верхнюю часть реактора.
Такие аппараты часто применяются в процессах, где катали-
затор быстро теряет свою активность (например, при каталитиче-
ской переработке нефти). Непрерывное выведение катализатора
из реактора позволяет одновременно вести процесс катализа и
регенерацию катализатора (рис. VIII. 5, а и в). Такая система по-
зволяет постоянно поддерживать высокую активность катализа-
тора в процессе.
В промышленности в большинстве случаев применяют реак-
торы с неподвижным слоем катализатора; на их основе оформлены
многие крупнотоннажные каталитические процессы. Однако реак-
торы с неподвижным слоем катализатора и реакторы с псевдо-
ожиженным слоем имеют свои достоинства и недостатки, поэтому
при выборе типа реактора решающее значение имеют экономиче-
ские соображения. Рассмотрим наиболее существенные особен-
ности этих реакторов.
В неподвижном слое катализатора происходит адиабатичёское
изменение температуры по высоте слоя катализатора. Поэтому,
если протекает эндотермическая реакция, то по мере прохожде-
ния исходных реагентов через слой катализатора и увеличения
степени превращения температура реакционной смеси снижается.
При экзотермической реакции, наоборот, наблюдается нагревание
смеси по толщине слоя, т. е. температура возрастает от входа
газа к его выходу. Подобные тепловые режимы, в большинстве слу-
чаев, далеки от оптимальных условий, так как при эндотермических
реакциях необходимо дополнительное нагревание реакционной сме-
си, а для обратимых экзотермических реакций, наоборот, следует
понижать температуру в ходе процесса (отводить тепло). Поэтому
реакторы с одним неподвижным слоем катализатора редко исполь-
зуются для проведения процессов в адиабатическом режиме; они
применяются лишь тогда, когда реакция протекает с небольшим
тепловым эффектом либо когда скорость реакции мала.
Таким образом, для того чтобы в реакторе с неподвижным
или кипящим слоем катализатора создать оптимальный темпера-
турный режим, необходимо по мере протекания процесса отводить
или подводить тепло, т. е. применять специальные теплообменные
устройства.
Преимуществом реактора с кипящим слоем является также
возможность подачи реагентов при температуре, ниже темпера-
туры зажигания катализатора. При поступлении таких реагентов
в кипящий слой и смешении с более горячей реакционной смесью,
находящейся в реакторе, происходит скачкообразное повышение
их температуры до постоянного и более высокого значения, чем
температура зажигания катализатора.
175
В реакторах с неподвижным слоем нельзя применять мелко-
зернистый катализатор, так как он слеживается и создает боль-
шое гидравлическое сопротивление, в псевдоожиженном слое, на-
против, успешно применяется мелкозернистый катализатор, так
как с уменьшением зерен катализатора возрастает его активность.
Для осуществления химических процессов, протекающих с боль- ?
шой скоростью (но процесс тормозится диффузионным переносом f
вещества), рационально применять реакторы с псевдоожиженным
слоем, так как при этом уменьшается диффузионное сопротивление
и увеличивается общая скорость процесса.
В аппаратах с неподвижным слоем замена катализатора свя-
зана с остановкой производства. В псевдоожиженном слое за-
мена катализатора осуществляется легко, при работающем реак-
торе (без его остановки). Это особенно удобно для процес- ;
сов, в которых требуется частая регенерация катализатора 5
(см. рис. VIII. 5, а и в).
Существенный недостаток реакторов с кипящим слоем состоит ’
в том, что в процессе работы происходит истирание катализатора - |
(около 10% в год) и целевой продукт загрязняется катализатор-
ной пылью. Поэтому в реакторе с кипящим слоем, в отличие от , |
реактора с неподвижным слоем, применяют особо прочный ката- I
лизатор, а также предусматривают специальные устройства для |
выделения из реакционной смеси измельченного катализатора, г
Кроме того, в аппаратах с кипящим слоем катализатора создается
большое гидравлическое сопротивление.
И, наконец, поскольку режим процесса в неподвижном слое
катализатора близок к режиму идеального вытеснения, а режим
в кипящем слое близок к режиму идеального смешения, то для
процесса, протекающего в кинетической области, время пребыва-
ния, необходимое для достижения заданной степени превращения
реагентов, всегда больше в реакторе с кипящим слоем чем в аппа-
рате с неподвижным слоем. В связи с этим общее количество
катализатора в псевдоожиженном слое больше, чем в неподвиж-
ном слое.
Из приведенных данных видно, что для эффективного ведения
каталитического процесса первостепенное значение имеет обеспе-
чение определенного (наиболее выгодного оптимального) темпе-
ратурного режима. Достижение такого режима в практических
условиях зависит в первую очередь от конструкции аппарата и
способа организации в нем теплообмена, поэтому ниже будут
рассмотрены отдельно способы создания определенного темпера-
турного режима в реакторах.
Теплообмен в реакторах
Существуют разнообразные способы, а также различные при-
способления, с помощью которых обеспечивается и поддерживает-
ся оптимальный температурный режим в реакторе. Относительно
176
просто эта задача решается при проведении гомогенных реакций.
В качестве примера может служить политропический реактор
идеального смешения с отводом тепла либо через стенки реактора,
либо с помощью охлаждающих элементов, расположенных внутри
реактора, рассмотренный ранее (см. рис. VIII.2,а и б), а также
реакторы, приведенные на рис. VIII. 6.
Значительно сложней поддерживать оптимальный температур-
ный режим при проведении гетерогенно-каталитических процессов
и особенно обратимой экзотермической реакции. В этом случае
а 5 в
Исходные- г
вещества
Теплоноситель
Исходные 4 Продукты
вещества'^ -^рёаищии
Теплоноситель
е 1 Продукты'
f реакции
Пплоно-
ситёль
Теплоно-
ситель
Исходные
вещества
Рис. VIII.6. Типы теплообменных устройств при проведении гомогенных реакций:
с—рубашка; б—внутренние змеевики; в —внутренние теплообменные трубки; г—наружный
теплообменник; д— рубашка (на однотрубном реакторе); е — рубашка (на миоготрубном реак-
торе).
оптимальной является не какая-либо одна, фиксированная темпе-
ратура, а температурная последовательность: процесс необходимо
начинать при высокой температуре, а затем, по мере увеличения
X, температура должна снижаться.
Существует несколько способов достижения оптимального тем-
пературного режима. Для адиабатического процесса наиболее
распространенный способ заключается в том, что процесс осуще-
ствляют в несколько стадий с охлаждением реакционной смеси
после каждой стадии.
Рассмотрим способы обеспечения оптимального температурного
режима в наиболее распространенных контактных аппаратах по-
лочного типа (рис. VIII. 7).
При проведении в таком аппарате обратимой экзотермической
Реакции исходные газообразные реагенты предварительно проходят
177
все теплообменники, где нагреваются до температуры зажигания,
и поступают в первый слой контактной массы (точка 1 на
Рис. VIII.7. Полочный контактный аппарат с промежуточным охлаждением реа-
гентов холодной исходной газовой смесью и изменение температуры в нем по
слоям катализатора.
рис. VIII. 7). Здесь протекает химическая реакция и выделяется
тепло, в результате чего по толщине слоя катализатора увеличи-
вается степень превращения Х'А и повышается температура
Рис. VIII.8. Полочный контактный аппарат с'вводом холодных
реагентов между ступенями катализа и изменение температуры
в нем по слоям катализатора.
(рис. VIII. 7, линия 1—2). В слое катализатора создается режим
вытеснения, процесс — адиабатический.
Выходящий из первого слоя катализатора горячий газ охлаж-
дается в теплообменнике холодными исходными реагентами (ли-
178
ния 2—3) и поступает на второй слой катализатора (точка 3).
Затем на каждом слое повторяются те же процессы, что были опи-
саны для первого слоя.
Рис. VIII.9. Полочный
слоями катализатора и
контактный аппарат с псевдоожиженными
изменение температуры в нем по слоям
катализатора.
Слои катализатора отличаются лишь высотой, которая возра-
стает от первого слоя к последнему. Это увеличивает время пре-
бывания реагентов на каждом последующем слое и компенсирует
реакции
Рис. VIII.10. Изменение параметров в трубчатом реакторе:
«—схема движения потоков; б—T=f(H); в—Хд =йФ(Г); /—3—точки измерения температуры.
снижение скорости химической реакции. Это снижение вызвано
уменьшением концентрации исходных реагентов в газе и пониже-
нием температуры от первого слоя к последующим.
Температурный режим в подобном контактном аппарате хотя
и не полностью соответствует оптимальному, но приближается к
179
нему. Температура в реакторе колеблется вблизи линии оптималь-
ных температур (ЛОТ на рис. VIII. 7), причем, чем больше число
ступеней в реакторе, тем ближе режим к оптимальному.
В полочном аппарате с неподвижными слоями катализатора
и вводом холодных реагентов между слоями (рис. VIII. 8) темпе-
ратурный режим также близок к оптимальному, хотя характер
зависимости X = f(T) несколько иной, чем в случае охлаждения
газа в теплообменнике.
Подогретые до температуры зажигания газообразные реагенты
поступают в первый слой катализатора (точка 1); здесь увеличи-
вается степень превращения и повышается температура смеси
(линия 1—2). Между первым и вторым слоями вводятся холодные
реагенты, температура смеси снижается, но одновременно умень-
шается и степень превращения, так как к реакционной смеси до-
бавляются свежие, непрореагировавшие реагенты (линия 2—3).
Охлажденная реакционная смесь затем подается на второй слой,
где процесс повторяется аналогично первому слою.
Часто для создания определенного температурного режима
применяют комбинации аппаратов, изображенных на рис. VIII. 7
и VIII. 8.
На рис. VIII. 9 показан полочный аппарат с псевдоожиженны-
ми слоями и с отводом тепла из каждого слоя. В псевдоожижен-
ном слое создается режим смешения. Поэтому изменение степени
превращения и температуры происходит скачкообразно при входе
₽ каждый слой катализатора (точка 1 — вход газа в первый слой,
точка 2 — газ в первом слое; точка 3—-газ во втором слое и т. д.).
В трубчатом реакторе режим, близкий оптимальному, поддер-
живается при непрерывном отводе тепла (рис. VIII. 10).
Холодные исходные реагенты поступают вначале в межтрубное
кольцевое пространство трубчатого реактора и нагреваются до
температуры зажигания (точка 1) в результате соприкосновения
с горячей поверхностью труб. По выходе из межтрубного простран-
ства реагенты проходят слой катализатора. При этом их темпера-
тура вначале возрастает за счет тепла химической реакции (линия
2—3), достигает максимума, но затем снижается за счет уменьше-
ния скорости реакции (последнее обусловлено снижением концент-
рации исходного реагента). При этом выделяется меньшее количе-
ство тепла. Отвод тепла в нижней части реактора еще остается
интенсивным, так как поступающая реакционная смесь имеет низ-
кую температуру и ДГ высока.
ЧАСТЬ ТРЕТЬЯ
НЕКОТОРЫЕ ТЕХНОЛОГИЧЕСКИЕ ПРОЦЕССЬЕ
ХИМИЧЕСКИХ ПРОИЗВОДСТВ
г; Огромное разнообразие Химико-технологических процессов и
их специфические особенности создают большие трудности пр»
попытке выделить из их числа такие, которые достаточно полно
отражали бы особенности всех или хотя бы большой группы хи-
мических процессов. В связи с этим авторы выбрали для описания
достаточно сложные производства, имеющие большое значение.
Процессы, положенные в их основу, изучены наиболее полно, т. е.
для них, исходя из физико-химических закономерностей, установ-
лены зависимости между отдельными параметрами.
Одним из наиболее хорошо изученных химических производств;
является производство серной кислоты контактным методом. Эта
производство включает ряд типовых процессов, широко распро-
страненных в химической технологии как неорганических, так »
органических веществ. Так, в производстве серной кислоты осуще-
ствляются сжигание твердого, жидкого и газообразного сырья
(флотационного колчедана, серы, сероводорода); очистка газов от
взвешенных твердых и жидких частиц (аэрозолей) в электро-
фильтрах и волокнистых фильтрах, процессы физической абсорб-
ции и десорбции газов, а также абсорбция, сопровождаемая хи-
мическими реакциями.
Процесс окисления сернистого ангидрида на катализаторе, по-
ложенный в' основу производства серной кислоты контактным
методом, является типичным и хорошо изученным каталитическим
процессом. Многие общие представления о катализе получены на
основе результатов изучения процесса окисления сернистого анги-
дрида на платиновом и ванадиевом катализаторах.
При получении серной кислоты выделяется большое количе-
ство энергии, даже частичное использование которой покрывает
все энергетические затраты, связанные с этим производством.
Кроме того, части энергии может быть выдана на сторону в виде
энергетического пара иди электроэнергии. Таким образом, совре-
менное производство серной кислоты относится к энерго-техноло-
гическим процессам.
18Ь
Важным и достаточно хорошо изученным является также про-
изводство аммиака на основе конверсии метана водяным паром.
Для этого производства характерно применение высокого давле-
ния и сочетание химических взаимодействий между органическими
и неорганическими веществами, что наглядно подтверждает услов-
ность разделения химических процессов на неорганические и орга-
нические.
Среди рассматриваемых далее процессов имеется химическая
переработка топлива. Это объясняется тем, что в химической про-
мышленности топливо является важнейшим сырьем, из которого
путем глубокой термохимической переработки получают самые
разнообразные продукты и полупродукты.
Механизм взаимодействия органических веществ более сложен,
чем неорганических. Подавляющее большинство процессов сопро-
вождается протеканием параллельных и последовательных реак-
ций, при которых одновременно с основными получаются побоч-
ные продукты.
Чрезвычайное разнообразие органических процессов затруд-
няет их классификацию и изучение, поэтому в настоящем курсе
рассмотрены только отдельные, наиболее типичные и хорошо
изученные из них.
Высокомолекулярные соединения приобрели, особенно в по-
следние годы, исключительно важное значение в народном хо-
зяйстве, они относятся к наиболее сложной и менее изученной
группе органических соединений.
, Глава !Х
СЫРЬЕ, ВОДА, ЭНЕРГИЯ
1. СЫРЬЕ
Химическая промышленность характеризуется высокой мате-
риалоемкостью производства. На 1 т готовой химической продук-
ции расходуется, как правило, несколько тонн сырья и материа-
лов, отсюда следует, что себестоимость химической продукции в
значительной мере определяется качеством сырья, способами и
стоимостью его получения и подготовки, расходами на перевозку.
В химической промышленности затраты на сырье в себестоимости
продукции составляют 60—70%, а в нефтехимической промышлен-
ности — более 70%.
От вида и качества сырья существенно зависит полнота исполь-
зования производственных мощностей различных подотраслей *
химической промышленности, а также уровень производитель-
ности труда, продолжительность полезной работы оборудования,
затраты труда на изготовление готовой продукции. Свойства
сырья, содержание в нем полезных и вредных компонентов в зна-
182
чительиой мере определяют применяемую технологию его обра-
ботки; от степени совершенства технологии в свою очередь зави-
сит производительность труда.
Большая материалоемкость и крупнотоннажность химического
производства обусловливают соответственно и высокие капитало-
вложения в химическую промышленность, поэтому выбор и под-
готовка сырьевых баз, технико-экономические показатели сырья
являются важными условиями рационального размещения и эф-
фективного развития химической промышленности.
Классификация сырья
Виды сырья весьма разнообразны, их можно разделить на
следующие группы: минеральное сырье, растительное и животное
сырье. В .качестве сырья в ряде процессов используются воздух и
вода (рис. IX. 1).
Минеральное сырье — это по-
лезные ископаемые, добываемые
из земных недр; их делят на руд-
ное, нерудное и горючее мине-
ральное сырье.
Рудное сырье — это горные
породы, из которых экономически
выгодно получать металлы. При
переработке некоторых видов
рудного сырья наряду с метал-
лами получают и химические
продукты. Так, например, одно-
временно с медью, цинком, нике-
лем при переработке сульфидных
руд получают и серную кислоту.
Нерудное сырье — горные по-
Сырье
Рис. IX. 1. Виды сырья.
мате-
роды, используемые в производ-
стве химических, строительных и других неметаллических
риалов. К этому виду сырья относятся породы, содержащие серу,
фосфаты, природные калийные соли, поваренную соль, песок, гра-
вий, глины и др. Это сырье используется в производстве
удобрений, солей, кислот, щелочей, цемента, стекла, керамических
изделий и др.
Горючее минеральное сырье включает угли, нефть, торф, горю-
чие сланцы, природный и попутный газы и др., служащие источ-
ником получения разнообразнейших продуктов. Так, при перера-
ботке угля получают сырье для производства красителей, лекар-
ственных препаратов, химических волокон, пластических масс,
Удобрений и т. п. В СССР в качестве горючего минерального
сырья используются угли Донбасса, Кузбасса, Караганды, Под-
московья, Восточной Сибири и др.
Нефть — углеводородное сырье для получения различных ви-
Дов топлив (бензина, керосина и др.), а также всевозможных
183
•синтетических материалов (волокон, каучуков, пластических масс,
моющих средств и др.). Запасы нефти находятся на Кавказе,
Башкирии, Татарии и в Сибири.
Природный газ является дешевым и весьма ценным сырьем
На тысячи километров протянулись по Советскому Союзу газо.
проводы, по которым на химические предприятия со Средней
Волги, из Украины, Бухары, Северного Кавказа, Сибири идет
природный газ, необходимый для получения многочисленных хи-
мических продуктов. Он служит сырьем для получения продуктов
тяжелого органического синтеза, удобрений, пластических масс,
• синтетических каучуков, химических волокон, фармацевтических
препаратов, лаков и др.
Растительное и животное сырье подразделяется на пищевое и
техническое. К пищевому сырью относятся продукты сельского,
лесного и рыбного хозяйства, которые используются для пищевых
целей (картофель, сахарная свекла, хлебные злаки, пищевые
жиры и т. п.). Химическая и другие отрасли промышленности
потребляют техническое, растительное и животное сырье, непри-
годное для пищевых целей, которое, однако, может быть перера-
ботано в продукты или материалы бытового и промышленного
потребления. К этому виду сырья относятся хлопок, солома, лен,
конопля, китовый и тресковый жиры, кости животных и др.
Воздух и вода являются самым дешевым и доступным сырьем.
Воздух—практически неисчерпаемый источник дешевых азота и
кислорода. Вода не только служит источником непосредственного
получения из нее водорода и кислорода, но и участвует в разно-
образных химических процессах, а также применяется для раство-
рения твердых, жидких и газообразных веществ.
Сырьевые ресурсы
Экономический потенциал любой страны в современных усло-
виях в большой степени определяется природными ресурсами
полезных ископаемых, масштабами и качественной характери-
стикой их месторождений, а также уровнем развития сырьевых
отраслей промышленности.
Сырьевые ресурсы современной химической промышленности
очень разнообразны, причем с развитием техники, организацией
новых производств и внедрением более эффективных методов про-
изводства сырьевая база отрасли постоянно расширяется за счет
открытия новых месторождений, освоения новых видов сырья и
более полного использования всех его компонентов.
По степени экономической эффективности использования за-
пасы минерального сырья делятся на две группы — балансовые
и забалансовые. К балансовым относятся запасы полезных иско-
паемых, которые по своему качеству соответствуют требованиям
промышленности и по условиям залегания могут быть добыты и
484
переработаны в настоящее время экономически эффективными'
способами. Запасы сырья, характеризующиеся низким содержа-
нием полезного вещества, присутствием неблагоприятных сопут-
ствующих компонентов и сложными условиями залегания, вслед-
ствие чего они при современном состоянии техники и экономики,
не могут эффективно эксплуатироваться, относятся к забалансо-
вым. Забалансовые запасы подлежат отдельному учету и пред-
ставляют интерес как объект перспективного промышленного-
освоения.
Пи степени изученности месторождений запасы полезных ископаемых под-
разделяются на категории А, В и С.
В категорию А включаются детально разведанные, опробованные и подго-
товленные для эксплуатации запасы. Данные о запасах по категории А служат
для обоснования производственного планирования объема добычи.
По категории В учитывают установленные геологоразведочными работами,
запасы сырья, качество которых проверено лишь лабораторными исследова-
ниями. Данные о запасах категории В используются для разработки проектных
заданий.
К категории С относятся запасы, определяемые на основе геологического-
изучения по естественным обнаружениям и по данным геофизической разведки <?
частичным опробованием качества сырья. Сведения о запасах по категории С
являются предварительными и служат для обоснования перспективных планов
развития промышленности и ассигнований на последующие геологоразведочные-
работы.
Сумма запасов по категориям А + В 4- С образует промышленные запасы.
На их - основе производится расчет обеспеченности планируемого производства,
данной химической продукции запасами сырья. При определении этой обеспе-
ченности в основном принимают во внимание балансовые запасы промышленных,
категорий с учетом наличия в них необходимого количества полезных ископае-
мых категорий А и В.
Отечественная химическая промышленность имеет мощную-
сырьевую базу и располагает запасами всех необходимых ей видов-
минерального и органического сырья. В настоящее время Совет-
ский Союз занимает первое место в мире по разведанным и под-
готовленным к добыче запасам фосфора, калийных солей, пова-
ренной соли, сульфата натрия, асбеста, торфа, древесины и мно-
гим другим видам сырья. Нам принадлежит одно из первых мест
по разведанным запасам нефти и газа. В СССР находится более
половины всех мировых запасов угля, сосредоточено около 60°/о‘
разведанных запасов торфа и около 33% фосфатов и древесины..
СССР занимает также первое место в мире по разведанным запа-
сам руд железа, марганца и многих других металлов.
Благодаря интенсивно проводимым геологоразведочным рабо-
там запасы важнейших видов сырья не уменьшаются, и из года в
год увеличиваются. Например, заметно возросли запасы основного
фосфорсодержащего сырья — апатито-нефелиновых руд хибинской
группы, где определены дополнительные запасы на эксплуатируе-
мых месторождениях и открыты новые месторождения.
Балансовые запасы калийных солей увеличились за последнее-
десятилетие более чем в 2,7 раза. Резко увеличились за последние
годы ресурсы серосодержащего сырья: возросла выработка флота-
185«
ционного колчедана и увеличились разведанные запасы самород-
ной серы, увеличилось количество отходящих газов, содержащих
сернистый ангидрид, на заводах цветной металлургии и нефтепе-
рерабатывающих предприятиях. Изменилась добыча самородной
серы: около 90% ее получают теперь более дешевым открытым
способом.
Крупным источником серы является природный газ Оренбург-
ского и Бухарского месторождений: в нем содержится до 2% серо-
водорода. В стране увеличились разведанные запасы борного
сырья, особенно боросиликатов на месторождении в Приморье.
Коренные изменения произошли в базе органического сырья.
Основное место заняли природный газ, попутные газы нефтедо-
бычи и газы, получаемые в процессе нефтепереработки.
Рациональное и комплексное использование сырья
Рациональное использование сырья позволяет повысить эконо-
мическую эффективность производства, так как стоимость сырья
составляет основную долю в себестоимости химической продук-
ции. В связи с этим стремятся использовать возможно более
дешевое сырье, особенно местное сырье, для которого не требуют-
ся дальние перевозки. Например, в качестве углеводородного
сырья все шире применяют нефть и природный газ, относительная
стоимость которых значительно ниже стоимости угля. Этим объяс-
няются существенно более высокие темпы добычи нефти и газа
по сравнению с углем:
1950 г. 1960 г.
Уголь.................. 1 (206 млн. т) 1,8
Нефть и газовый конденсат 1 (37,9 млн. т) 3,9
Газ.....................1 (5,8 млрд, м3) 7,2
1980 г.
(план)
3,9
16,6
72
1970 г.
2,1
9,3
3,4
При- выборе источника сырья необходимо также учитывать
современные и перспективные условия его получения, в том числе
и местные условия. Так, например, этиловый спирт можно полу-
чать из пищевого сырья, древесины (гидролизный спирт), из от-
ходов целлюлозного производства (сульфитный спирт) и синте-
тическим способом. Себестоимость гидролизного спирта ниже
себестоимости пищевого спирта на 40%, а сульфитного и синте-
тического— на 75%. Трудовые затраты на производство 1 тспирта
из зерна и картофеля составляют 160—280 чел-ч, а из нефтехими-
ческого сырья—10 чел-ч. Удельные капитальные вложения при
замене пищевого сырья синтетическим снижаются на 400 руб/т
годовой мощности.
Экономическая обоснованность выбора сырьевой базы для хи-
мического производства особенно наглядна в азотной промышлен-
ности. С переходом на использование природного газа снизилась
себестоимость аммиака по сравнении) с ранее использовавшимися
видами сырья: на Новомосковском химкомбинате — на 42, на
186
Чирчикском электрохимкомбинате — на 52, на Руставском — на
37%. Поэтому если в 1960 г. из природного газа получали лишь
16,7% аммиака, то в 1965 г. — уже 63,7%, а в 1975 г. — свыше
80%• Р°ст производства аммиака из различных видов сырья виден
из следующих данных (млн. т.):
1960 г. 1965 г. 1970 г. 1975 г.
Природный газ . . . , . . 0,2 2,1 7,5 9,0
Коксовый газ . . . . 0,5 0,7 1,0 1,5
Твердое топливо . . . 0,5 0,5 0,7 0,6
Всего . . . 1,2 3.3 9,2 11,1
Существенное значение выбора рационального вида сырья
можно проследить также на примере производства синтетического
каучука. Раньше (до пятидесятых годов) сырьем для получения
СК служил этиловый спирт, получаемый из пищевых продуктов
(зерна, картофеля и др.). Стоимость его была высокой, а размеры
выработки не всегда обеспечивали потребность.
Сырье, стоимость которого составляла 75—80% общих затрат
на производство, определяло рентабельность промышленности
синтетического каучука и уровень цен на этот важнейший продукт.
Поэтому снижению расхода этилового спирта и его замене на
более дешевое сырье в промышленности синтетического каучука
всегда уделялось большое внимание. В настоящее время у нас и
за рубежом основным сырьем для получения СК служит бута-
диен. Например, в США удельный вес СК из бутадиена состав-
ляет 80%, в Канаде 75%.
Для выработки бутадиена в Советском Союзе в качестве исход-
ного сырья широко используют бутан и этиловый спирт. В даль-
нейшем по мере развития нефтедобывающей промышленности и
переработки нефти ресурсы бутана будут увеличиваться и доля
его в сырьевом балансе будет постоянно возрастать. Это объяс-
няется доступностью сырья, его низкой стоимостью и Относительно
простой схемой переработки. В настоящее время на всех вновь
строящихся заводах СК предусматривается получение бутадиена
из бутана.
Из сравнения затрат на получение 1 т бутадиена видны значи-
тельные преимущества бутана по сравнению с этиловым спиртом
Хв отн. единицах):
Этиловый спирт синтетический Бутан
Капитальные вложения . . 100 65
Себестоимость 100 75
Приведенные затраты . . . 100 80
Комплексная переработка сырья имеет большое народнохозяй-
ственное значение еще и потому, что при этом исключается на-
копление отходов, хранение или вывоз которых обычно сопряжены
с дополнительными затратами. Например, водный раствор хло-
ристого кальция, являющийся отходом в производстве соды, в
187
настоящее время не используется, его заливают в огромные котло-
ваны. Стоимость таких котлованов высока, так как они должны
быть герметичными и исключать утечку хлористого кальция (по-
падая в почву, он вымывается подпочвенными водами и разно-
сится на большие расстояния, отравляя все живое).
Отходом сернокислотных заводов является огарок, образую-
щийся при обжиге колчедана. Огарок еще не нашел применения,
хотя в нем содержится около
50% железа, а также некоторое
количество меди, цинка, золота,
серебра и других ценных метал-
лов.
Важное значение комплексной
переработки природного сырья
вытекает также из того, что со-
держание некоторых необходи-
мых для промышленности ве-
ществ в земной коре очень мало.
Так, на основе результатов спе-
циальных исследований были
рассчитаны запасы различных
простых веществ в земной коре
на глубине до 16 км. В зависимо-
Тис. IX.2. Схема комплексного исполь-
зования сырья на примере апатито-
вой руды.
Из приведенных данных видно, что только для нескольких эле-
ментов имеется соответствие между запасами и потреблением.
Однако для углерода, например, этого соответствия нет, углерод
по потребности находится на первом месте, а по запасам только
на одиннадцатом, его в земной коре только 0,35%. Соответствия
нет и для серы, а Си, Pb, Мп, Zn в приведенном ряде запасов
вообще отсутствуют (Си занимает 26, a Zn — 24 место). Пере-
численные 14 элементов составляют 99,5% земной коры, а осталь-
ные элементы только 0,5% земной коры.
В качестве примера комплексной переработки сырья можно
привести переработку апатитовой руды (Хибинские апатиты), до-
бываемой на Кольском полуострове (рис. IX. 2).
J88
Обогащение сырья
Применение концентрированного сырья упрощает и удешевляет
его переработку, а также позволяет получать продукты более
высокого качества. Потребляемое в химической промышленности
сырье обычно недостаточно концентрированное, поэтому перед
употреблением его в большинстве случаев обогащают, в резуль-
тате чего концентрация в нем основного вещества увеличивается.
Обогащение полезных ископаемых, как правило, сложный и
дорогостоящий процесс. Однако, несмотря на дополнительные
затраты, связанные с обогащением, оно обеспечивает значитель-
ный народнохозяйственный эффект, определяемый 1) возмож-
ностью расширения сырьевой базы и обеспечения современного
многотоннажного производства химической продукции за счет
комплексного использования сырья и вовлечения в эксплуатацию
бедных по содержанию полезных минералов и руд; 2) более пол-
ным использованием оборудования на химических предприятиях
за счет переработки высококонцентрированного обогащенного
сырья; 3) существенным улучшением качества готовой химической
продукции и 4) значительной экономией транспортных средств
вследствие уменьшения перевозок, приходящихся на долю пустой
породы.
Если обогащению подвергают твердые материалы (например,
горные породы), то полученный обогащенный продукт называют
концентратом, а отходы с меньшим содержанием полезного про-
дукта и повышенным содержанием породы называют хвостами.
В тех случаях, когда в сырье содержится несколько полезных
составляющих, его делят на отдельные части (фракции), обога-
щенные тем или иным компонентом, т. е. из сложного сырья по-
лучают несколько концентратов, что позволяет более полно (ком-
плексно) использовать сырье.
Методы обогащения твердых материалов весьма разнообразны,
они основаны на различии физических и химических свойств
веществ, входящих в состав сырья, например, прочности, плотно-
сти, твердости, растворимости, температур плавления и возгонки,
магнитной проницаемости и др.
Рассеивание (грохочение) основано на том, что мине-
ралы, входящие в состав сырья, имеют различную прочность, по-
этому при дроблении менее прочные (хрупкие) минералы
Дробятся на более мелкие зерна, чем прочные (вязкие) мате-
риалы. Если после измельчения такое сырье просеять через сита
с отверстия^! различного размера, то можно получить фракции,
обогащенные тем или иным минералом.
Гравитационное разделение основано на различии
скоростей осаждения (падения) частиц в жидкости или газе в за-
висимости от плотности или крупности этих частиц. Если осажде-
ние производят в жидкости (чаще всего в воде), его называют
мокрым гравитационным обогащением; если осаждение ведут в
189
газе (чаще всего в воздухе), его называют сухим гравитационным
обогащением.
Мокрое гравитационное обогащение осуществляется в аппа-
ратах, называемых классификаторами; схема одного из таких
классификаторов показана на рис. IX. 3. Измельченное сырье
смешивают с водой в баке с мешалкой 1, полученную пульпу
(взвесь твердого матернала в жидкости) подают в осадительные
камеры 2 (корыта — / — ///) с конусообразными днищами 3 (бун-
керами). Так как ширина камер увеличивается от входа к выходу,
Рис. IX.3. Принципиальная схема мокрого гравитационного обогащения:
1—бак с мешалкой; 2 — осадительные камеры (/—III}.
то скорость движения пульпы через камеры постепенно умень-
шается, что облегчает осаждение твердых частичек. В камере I
из воды выпадает наиболее тяжелая (крупнозернистая) фракция,
в камере 7/— средняя, в камере 111 — самая легкая (мелкозерни-
стая) фракция. В зависимости от того, на сколько фракций тре-
буется разделить сырье, аппараты-классификаторы состоят из
одной, двух и более камер. При сухом гравитационном обогаще-
нии применяют воздушные сепараторы различного устройства.
Так, например, в центробежных сепараторах частицы сырья по-
падают на диск, вращающийся в горизонтальной1* плоскости с
большой скоростью. Крупные и тяжелые частицы под действием
центробежной силы отбрасываются на более далекое расстояние,
чем легкие и мелкие частицы.
Магнитная сепарация применяется для отделения магнитно-
» восприимчивых материалов от немагнитных, а также для удале-
ния стальных предметов, случайно попавших в руду; так отделяют
190
магнитный железняк от пустой породы. Разделение руды осуще-
ствляют в электромагнитных сепараторах, схема такого сепара-
тора показана на рис. IX.4.
После измельчения материал поступает на движущийся лен-
точный транспортер 1, имеющий барабан 2, снабженный электро-
магнитом 3. При прохождении ленты транспортера над поверх-
ностью барабана частицы материала, не обладающие магнитной
восприимчивостью, ссыпаются в
бункер 4, а частицы магнитного
материала задерживаются на
ленте и продолжают двигаться до
тех пор, пока лента не пройдет
магнитную поверхность бараба-
на, после чего частицы ссылаются
в бункер 5.
Флотационный метод
обогащения основан на раз-
личной смачиваемости зерен от-
дельных минералов водой. Части-
цы несмачиваемого (гидрофобно-
го) минерала (рис. IX.5) не пре-
одолевают силы поверхностного
натяжения воды и остаются на ее
поверхности. Частички смачивае-
мого (гидрофильного) материала
обволакиваются пленкой жидко-
сти и опускаются на дно аппара-
та. Несмачиваемый минерал сни-
мают с поверхности жидкости,
отделяя от руды.
Для проведения флотации при-
меняют технологические приемы,
в частности создают условия не-
одинаковой смачиваемости водой
Рис. IX.4. Электромагнитный сепара-
тор:
1 — лента транспортера; 2—барабан транс-
портера; 3—электромагнит; 4, 5 — бункеры.
Рис. IX.5. Разделение частиц несма-
чиваемого и смачиваемого материала:
1 —несмачнвающаяся (гидрофобная) части-
ца; 2 —смачивающаяся (гидрофильная)
частица.
зерен минералов.
Флотацию проводят во флотационных машинах различного
типа, схема одной из них приведена на рис. IX. 6.
Тонкоизмельченная взвесь породы с флотореагентами посту-
пает в машину, состоящую из корытообразного резервуара — ка-
меры /, внутри которой установлены перегородки 2, а между ними
расположены трубки 3. По трубкам 3 в камеру 1 подается воздух
под давлением, который перемешивает пульпу. Его пузырьки, под-
нимаясь вверх, увлекают за собой частицы гидрофобного мине-
рала, всплывающего на поверхность воды.
Подаваемый в пульпу воздух обеспечивает циркуляцию водной
взвеси частиц (суспензии) в камере. Для успешного проведений
процесса флотации в водную взвесь вводят вещества, способ-
ствующие образованию пены — пенообразователи (сосновое масло,
191
древесный деготь и др.). Пена вместе с частицами гидрофобного
минерала сливается с поверхности жидкости через борт камеры
в желоб 4, откуда она далее поступает на сгущение (разрушение
пены) и фильтрование. Отдельные твердые частицы минерала
сушат и в виде концентрата поступают на дальнейшую перера-
ботку. Частицы, осевшие на дно камеры, выводятся в виде флота-
ционных хвостов.
Природные минералы в большинстве случаев хорошо смачи-
ваются водой, поэтому для их разделения флотацией в суспензию
вводятся специальные реагенты — собиратели, снижающие смачи-
ваемость отдельных природных минералов. К таким реагентам
Рис. IX.6. Флотационная машина с воздушным перемешиванием:
/ — резервуар; 2—перегородки; 3—воздушная трубка; 4—желоб для приема
концентрата.
относятся олеиновая кислота, нафтеновые кислоты и др. Собира-
тели покрывают поверхность частиц гидрофильных минералов
гидрофобной пленкой, частицы всплывают и выводятся из камеры.
В тех случаях, когда необходимо, наоборот, затруднить всплы-
вание частиц отдельных минералов, в суспензию вводят подави-
тели (щелочи, соли щелочных металлов и др.). Эти вещества
увеличивают гидрофильность частиц минералов, что затрудняет их
всплывание и способствует осаждению частиц на дно.
Флотационный метод обогащения получил широкое промыш-
ленное применение благодаря тому, что применение различных
флотореагентов, добавляемых в малых количествах (100 г на 1 т
породы), позволило обогащать и разделять на фракции самое
разнообразное (практически любое) сырье.
Жидкие растворы различных веществ концентрируют упари-
ванием растворителя, вымораживанием, выделе-
нием примесей в о с а д ок или в г а з о в у ю фазу. Так, на-
пример, упариванием воды из растворов получают минеральные
соли, щелочи, кислоты и др., а путем вымораживания обычно кон-
центрируют природные рассолы в зимнее время.
192
Газовые смеси разделяют на компоненты последователь-
ной конденсацией, т. е. переводят их в жидкое состояние при
постепенном понижении температуры и сжатии. Этот метод осно-
ван на различии температур конденсации компонентов газовой
смеси. В других случаях вначале газовую смесь превращают
в жидкость, а затем последовательным испарением ее разделяют
на индивидуальные компоненты. Разделение газовых смесей осу-
ществляют также поглощением отдельных газов жидкостями (аб-
сорбция) или твердыми веществами (адсорбция) с последующим
выделением их из сорбентов в концентрированном виде.
Методы концентрирования жидких растворов и разделение га-
зовых смесей широко применяются в настоящее время в химиче-
ской промышленности, поэтому они будут несколько подробнее
рассмотрены при описании отдельных химико-технологических про-
цессов.
2. ВОДА
Вода широко используется в химической промышленности, ред-
ким исключением являются процессы, в которых не участвует вода.
В одних случаях вода служит сырьем и реагентом, непосредствен-
но участвующим в основных химических реакциях, а в других —
вода употребляется как растворитель, теплоноситель или охлади-
тель, а также для других самых разнообразных физических опе-
раций.
В перспективе природная вода может использоваться как источник промыш-
ленного сырья. В воде морей и океанов содержатся почти все элементарные ве-
щества периодической системы элементов, но в небольших количествах. Так, на-
пример, в воде содержится в виде различных солей (в %):
К . . . 3,8 10“2 Ag . . . 5 -10—3
U . . . . 2-10-7 Ан . . . 4-10“10
V ... . 5-Ю-8 Ra . . . 1 • 10“14
Если учесть, что общая масса воды на земном шаре велика и составляет
1,4 • 1018 т, то содержание различных веществ в воде выражается внушитель-
ными цифрами. Например, содержание урана в воде составляет
2 • 1Г)-7
М = 1,4-Ю!8-—— = 2,8-Ю9 т
т. е. 2,8 миллиарда тонн. Достаточно извлечь из воды только 0,01% общего ко-
личества урана, его хватило бы для обеспечения энергией всего мира в течение
100 лет.
Основным источником промышленного водоснабжения в нашей
стране являются речные воды, суммарный годовой сток которых
составляет 4700 км3. Однако территориальное распределение их
крайне неравномерно: более 80% всего водного стока приходится
на-северные районы Европейской части СССР, на Дальний Восток,
Западную и Восточную Сибирь и только около 20% всех водных
ресурсов приходится на наиболее обжитые районы. В этих зонах
7 Зак, 558 - 193
дефицит в воде испытывают многие промышленные узлы и целые
районы.
В районах с дефицитным водоснабжением уже сейчас для нор-
мальной эксплуатации и расширения многих химических пред-
приятий и комплексов требуется проводить весьма сложные и до-
рогостоящие мероприятия по регулированию и переброске стока
на большие расстояния и использовать подземные воды. Все это
сопряжено со значительными дополнительными материальными и
денежными затратами и в связи с этим с ухудшением технико-эко-
номических показателей производства химической продукции.
Развитие химической промышленности вызывает огромный
рост потребности в воде (современные химические предприятия
расходуют большое количество воды, измеряемое миллионами ку-
бических метров в сутки).
Так, например, современный завод капронового волокна расхо-
дует столько же воды, сколько потребляет ее город с населением
120 тыс. человек. Специализированный завод пластмасс, вклю-
чающий производство мономеров, по потреблению воды эквива-
лентен городу с населением 400 тыс. человек. Мощный современ-
ный электрохимический комбинат по производству продуктов хлор-
органического синтеза потребляет столько же воды, сколько город
с населением 800 тыс. человек. Наряду с этим химические пред-
приятия сбрасывают большие количества сильно загрязненных вод.
Таким образом, рациональное комплексное использование вод-
ных ресурсов в условиях быстро развивающейся химической про-
мышленности и других отраслей народного хозяйства становится
крупной технологической, технической и экономической задачей.
Технологический аспект в решении водной проблемы заклю-
чается в новом подходе к разработке технологической схемы. Ра-
циональное водопотребление должно быть обязательным в каж-
дом технологическом процессе. Необходимы создание процессов,
требующих минимальных расходов свежей воды, разработка науч-
но обоснованных норм ее расхода в химических производствах и
совершенствование технологических процессов в направлении воз-
можно полного использования отходов производства, что снизит
потребность в очистных сооружениях. Насколько это важно, можно
судить по тому, что в настоящее время затраты на строительство
очистных сооружений составляют примерно 20% стоимости хими-
ческих предприятий.
Виды и качество потребляемой воды
В зависимости от назначения потребляемая вода условно под-
разделяется на промышленную и питьевую; в каждой из них со-
держание примесей регламентируется соответствующим ГОСТ.
Питьевая вода в первую очередь освобождается от бакте-
рий; к ней предъявляются особые требования в отношении вкуса,
цвета, запаха.
194
Промышленные воды не должны содержать примесей
больше допустимой нормы, которая устанавливается в зависимо-
сти от производства, на котором используется вода. Вода для пря-
моточных паровых котлов не должна содержать двуокись углерода
и кислород, вызывающих коррозионное разрушение труб, и может
содержать не более 0,2—0,3 мг/л сухого остатка. Соли в паровых
котлах, отлагаясь на внутренней поверхности труб в виде накипи,
снижают теплопроводность металла; происходит перегрев труб и
преждевременный их износ.
Повышенные требования в отношении чистоты предъявляются
к воде в производстве полупроводников, люминофоров и некото-
рых других материалов.
Качество воды характеризуется следующими показателями:
жесткостью, общим солесодержанием, прозрачностью, окисляе-
мостью и реакцией воды.
Жесткость воды — различают временную, постоянную и
общую жесткость. Временная (устранимая) жесткость обуслов-
лена наличием в воде бикарбонатов кальция и магния. Эти соли
сравнительно легко удаляются из воды при кипячении, так как
при этом они переходят в нерастворимые углекислые соли и вы-
падают в виде плотного осадка
(Са, Mg) (НСОз)2 —> (Са. Mg)CO3; + Н2О + СО2
Постоянная жесткость воды обусловлена присутствием в воде
хлоридов, сульфатов, нитратов кальция и магния. Эти соли при
кипячении не удаляются из воды.
Сумма временной и постоянной жесткости дает общую жест-
кость, измеряемую в миллиграмм-эквивалентах ионов кальция или
магния, содержащихся в виде солей в 1 л воды. Жесткость воды
равна единице, если в 1 л ее содержится 1 мг-экв ионов кальция,
что соответствует 20,04 мг кальция или 12,16 кг магния.
По жесткости (в мг-экв-л-1) природные воды подразделяют на
очень мягкую (0—1,5), мягкую (1,5—3), среднюю (3—6), жесткую
(6—10) и очень жесткую (более 10).
Общее солесодержание, или сухой остаток —
масса вещества, остающаяся после испарения воды и высушива-
ния полученного остатка при 105—110°С. Сухой остаток изме-
ряется в миллиграммах на 1 л воды (мг/л).
Прозрачность воды измеряется толщиной слоя воды, че-
рез который можно различать визуально или с помощью фотоэле-
мента изображение креста или определенного шрифта.
Окисляемость воды определяется числом миллиграммов
перманганата калия, израсходованного при кипячении 1 л воды
С избытком КМпО4 в течение 10 мин.
.Реакция воды, т. е. ее кислотность или щелочность, харак-
теризуется концентрацией водородных ионов или величиной pH.
При pH = 6,5—7,5 вода считается нейтральной, при pH < 6,5
воду называют кислой, при pH > 7,5 — щелочной.
7*
195
Очистка воды
В поступающей воде содержатся самые разнообразные при-
меси: грубодисперсные и коллоидные частицы — различные сили-
каты, гидратированная кремниевая кислота, в ней также раство-
рены газы и соли: бикарбонаты, сульфаты, хлориды, нитраты
Рис. IX.7 Схема очистки воды.
уравнению:
кальция, магния, калия, натрия
и др. Очистка воды включает
следующие операции: осветле-
ние, обеззараживание, умягчение,
дегазацию и дистилляцию
(рис. IX. 7).
Осветление воды произ-
водится с целью удаления меха-
нических примесей, оно дости-
гается отстаиванием воды в бето-
нированных резервуарах боль-
шой емкости (отстойниках) с по-
следующим фильтрованием на
песчаных фильтрах. Для осажде-
ния коллоидных примесей в от-
стойники вводят коагулянты —
сульфаты железа или алюминия.
Коагулянты гидролизуются в
воде с образованием аморфных
осадков гидроокисей железа или
алюминия, которые адсорбируют
коллоидные примеси и увлекают
их на дно резервуара. В конечном счете реакция протекает по
Al2(SO4)3 + 6Н2О =?=> 3H2SO4 + 2А1(ОН)3|
Гидроокись алюминия образуется также при взаимодействии
сульфата алюминия с присутствующими в воде бикарбонатами
кальция и магния:
A12(SO4)3 + ЗСа(НСО3)2 = 3CaSO4 + 6СО2 + 2А1(ОН)3;
Таким образом, одновременно с удалением коллоидных приме-
сей снижается и содержание солей временной жесткости.
Обеззараживание воды — удаление из нее микроорга-
низмов и бактерий путем хлорирования, озонирования или кипя-
чения.
В настоящее время для хлорирования воды используют хлор,
гипохлорит кальция и др. При обработке воды гипохлоритом каль-
ция протекает реакция
Са(СЮ)2 + СО2 4- Н2О = СаСО3; + 2НС1О
НС1О HCl-f-0
196.
Атомарный кислород обладает сильными окислительными свой-
ствами, поэтому убивает микроорганизмы и окисляет органические
примеси.
В последние годы обеззараживание питьевой воды производят
преимущественно с помощью озона, который получают путем воз-
действия тихого электрического разряда на воздух или на воздух,
обогащенный кислородом. При обработке воды озон разлагается
с выделением атомарного кислорода.
При обработке воды хлором вода приобретает запах хлора, при
озонировании запах отсутствует, что является существенным до-
стоинством метода.
Вода обеззараживается также обработкой ионами серебра и
при воздействии ультрафиолетовых лучей и ультразвуковых коле-
баний.
Умягчение воды состоит в полном или частичном удалении
из нее солей кальция и магния. Если из воды удаляются также ка-
тионы и анионы, т. е. удаляются все содержащиеся в ней соли,
этот процесс называют обессоливанием воды. Умягчение и обессо-
ливание воды являются основными процессами подготовки воды.
Способы умягчения подразделяются на физические, химические
и физико-химические.
Физические способы предусматривают термическую обработку
воды, или кипячение, дистилляцию и вымораживание. Химические
способы умягчения воды заключаются в обработке ее раство-
рами химических соединений. Наибольшее промышленное приме-
нение получили известково-содовый и фосфатный способы умяг-
чения.
Известково-содовый способ заключается в обработке воды сна-
чала известковым молоком, а затем содой, при этом кальциевые
соли превращаются в нерастворимый карбонат кальция, магние-
вые соли — в гидроокись и карбонат магния:
Са(НСО3)2 + Са(ОН)2 2Н,0 + 2СаСО3;
Mg(HCO3) + 2Са(ОН)3 = Mg(OH)2 + 2Н2О + СаСО3;
СаС12 -р Na2CO3 == 2NaCl -р СаСО3^ z
MgSO4 + 2Na2CO3 = Na2SO4 + MgCO3;
Известково-содовый способ является наиболее распространен-
ным и дешевым, но при этом достигается лишь сравнительно гру-
бое умягчение воды (примерно до 0,3 мг-экв • л-1).
Фосфатный способ состоит в обработке воды фосфатом натрия:
ЗСа(НСО3)2 -р 2Na3PO4 == 6NaHCO3 -р Са3(РО4)2р
ЗСаС12 + 2Na3PO4 = 6NaCl + Са3(РО4)4
Растворимость фосфатов кальция и магния в воде ничтожно
мала, это определяет высокую эффективность фосфатного способа
(содержание солей снижается примерно до 0,03 мг-экв-л-1).
Фосфатный способ — достаточно дорогой способ умягчения, по-
этому применяется главным образом в комбинированных схемах,
19Г
в которых основная масса солей удаляется из воды известковым
молоком или содой, а доумягчение осуществляется с помощью фос-
фатов.
Из физико-химических способов наиболее широкое практиче-
ское применение находят ионообменные способы, основанные на
свойстве некоторых труднорастворимых твердых веществ, так на-
зываемых ионитов, обменивать
свои ионы на ионы солей, рас-
творенных в воде.
Иониты подразделяются на
катиониты и аниониты. Катиони-
ты содержат подвижные катионы
натрия или водорода и соответ-
ственно называются Na-катиони-
тами и Н-катионитами. Аниониты
содержат подвижную гидро-
ксильную группу (ОН-анионита-
ми).
В качестве Na-катионитов
применяются алюмосиликаты:
глауконит, цеолит, пермутит и др.;
в качестве Н-катионитов приме-
няются сульфированный уголь и
Рис. IX.8. Схема установки для обес-
соливания воды с применением
ионообменников:
./ —Н-катиоиитовый фильтр; 2 — аиионито-
вый фильтр*, 3—дегазатор; 4—приемник
очищенной воды.
другие вещества; к ОН-анионитам относятся искусственные смолы
сложного состава, например карбамидные.
Процессы очистки воды способом ионного обмена можно пред-
ставить следующим образом.
Катионный обмен с использованием алюмосиликата состава
Na2O • А120з • 2SiO2 • пН2О протекает по уравнению
№2О[Кат] + СаС12 СаО[Кат] + 2NaCl (а)
где [Кат] — неучаствующая в обмене часть молекулы (А120з*
• 2SiO2 * пН2О).
В случае применения Н-катионита процесс протекает следую-
щим образом
Н2[Кат] + CaSO4 Са[Кат] + H2SO4 (б)
Н2[Кат] + Са(НСО3)-2 Са[Кат] + 2Н2О + 2СО2 (в)
С течением времени катиониты истощаются, их регенерируют
промывкой раствором NaCl или кислотой.
Анионный обмен можно представить в виде
2[Ан]ОН + H2SO4 [Ah]2SO4 + 2Н2О (г>
Для регенерации анионита его промывают щелочью:
[Ah]2SO4 -f- 2NaOH Na2SO4 -{- 2[Ан]ОН
При пропускании воды сначала через катионит (рис. IX. 10)
она освобождается от ионов кальция, магния и натрия в Н-катио-
нитовом фильтре 1, а затем в анионитовом фильтре 2 из нее уда-
108
ляются анионы. Далее воду выводят из фильтра, освобождают от
двуокиси углерода в дегазаторе 3, собирают в сборнике 4 и далее
направляют потребителю. Для регенерации в фильтр 1 подается
кислота, а в фильтр 2 — щелочь.
К преимуществам ионитового способа по сравнению с изве-
стково-содовым относятся компактность и простота аппаратурного
оформления, более глубокое умягчение воды (до 0,035 +
+0,07 мг-экв-л-1), простота обслуживания и контроля и самое
основное — дешевизна.
Дегазация — удаление из воды растворенных газов — про-
изводится химическим и физическим способами. При химическом
способе газы взаимодействуют с химическими соединениями и уда-
ляются из воды. Например, двуокись углерода удаляют из воды
при пропускании через фильтр, заполненный гашеной известью,
либо добавляют к воде известковое молоко. В обоих случаях об-
разующийся СаСО3 выпадает в осадок.
Для удаления кислорода воду пропускают через фильтр, за-
полненный железными опилками или стружками; кислород взаи-
модействует с железом, образующаяся нерастворимая в воде
окись железа выпадает в осадок.
Физические способы удаления газов заключаются в аэрации
или нагревании воды в вакууме.
Полная очистка воды (обессоливание), а также дегазация и
обезвреживание воды достигаются ее перегонкой, т. е. дистилля-
цией.
Предприятия химической промышленности, как правило, имеют
сложное хозяйство по водообеспечению. При этом значительно
возрастает стоимость подготовки воды. Так, при фильтровании
воды ее себестоимость увеличивается в 2,5 раза (по сравнению
с речной осветленной водой), при частичном умягчении — в 8 раз,
при обессоливании и полном умягчении — в 10—11 раз. В резуль-
тате этого доля затрат на водоснабжение и канализацию в общих
капиталовложениях на строительство заводов возрастает от 5
До 15%.
Учитывая, что на наиболее обжитые районы приходится только
около 20% всего стока речной воды в стране и что речному стоку
свойственны значительные колебания как в многолетнем, так и
в сезонном периодах, удовлетворение промышленности речной во-
дой является сложной задачей. Поэтому оценка водных ресурсов
является обязательным условием технико-экономического обосно-
вания нового промышленного строительства и расширения дей-
ствующего производства.
3. ЭНЕРГИЯ
Все химические процессы протекают с изменением энергии; вы-
делением или ее поглощением, поэтому при оформлении химико-
Технологического процесса должно быть предусмотрено использо-
®ание выделяющейся энергии для повышения экономичности
199
процесса или подвод энергии, необходимой для осуществления дан-
ного процесса. Значительная доля энергии затрачивается на про-
ведение различных вспомогательных операций; транспортирование
материалов, их дробление или фильтрование, сжатие газов и др.
Наша страна располагает большими энергетическими ресурса-
ми, которые позволяют полностью удовлетворить потребности
в них химической и других отраслей народного хозяйства. Однако
топливно-энергетические ресурсы страны распределены по ее тер-
ритории неравномерно и характеризуются весьма различными эко-
номическими показателями их использования:
Потенциаль-
Районы СССР ные запасы энергетиче- ских ресурсов всех видов, % Потребление энергоре- сурсов, %
Европейская часть . . . 8,0 64,0
Урал . 0,5 15,0
Западная Сибирь . . . 13,0 7,0
Восточная Сибирь . . . 74,0 5,0
Дальний Восток .... Средняя Азия и Казах- 2,0 3,0
стан 2,5 6,0
Итого .... 100 1С0
Аналогично обстоит дело с потреблением энергии и в химиче-
ской промышленности: в настоящее время на Европейскую часть
СССР и Урал приходится более 75% всей электроэнергии и свыше
80% всего тепла, потребляемых отраслью. В результате этого на-
блюдается резкая дифференциация отдельных районов по степени
обеспеченности энергетическими ресурсами.
Виды энергии
В химической промышленности применяются разнообразные
виды энергии: электрическая, тепловая, ядерная энергия, химиче-
ская и энергия света. •
Электрическая энергия применяется прежде всего для
приведения в движение электродвигателей, широко используемых
при проведении различных физических операций: дробления, сме-
шения, перекачивания (насосы, вентиляторы, компрессоры) и т. п.
Электрическая энергия расходуется также для проведения элек-
трохимических и электромагнитных процессов.
Расходные коэффициенты электроэнергии (в кВт • ч • т-!) на
производство различных химических продуктов изменяются в ши-
роких пределах:
Алюминий........................ 18 000
Фосфор........................ 13000
Синтетический аммиак.......... 3 000
Серная кислота .................. 100
Аммиачная селитра 10
200
Электрическую энергию производят гидроэлектростанции, теп-
ловые и атомные электростанции. В последнее время успешно ве-
дутся работы по непосредственному превращению тепловой энер-
гии в электрическую.
Тепловая энергия в химической промышленности применяется,
во-первых, для осуществления разнообразных физических процес-
сов, не сопровождающихся химическими реакциями; плавление,
сушка, выпаривание, дистилляция и др., во-вторых, для нагрева-
ния реагентов при проведении химических реакций.
Источником тепловой энергии обычно служит разнообразное
топливо, при сжигании которого образуются топочные газы. Эти
газы могут быть непосредственно использованы в качестве тепло-
носителей или же для получения водяного пара, перегретой воды
и других теплоносителей. Расход пара на производство различных
продуктов также колеблется в широких пределах от 2 МДж • т-1
(0,5 Мкал • т~!) до 520 МДж • т~! (130 Мкал • т-1).
Для получения тепла используется также электрическая энер-
гия.
Атомная энергия применяется для производства электри-
ческой энергии на атомных электростанциях.
Химическая энергия выделяется обычно в виде тепла
при проведении разнообразных экзотермических реакций. Исполь-
зование этой энергии имеет большое практическое значение, осо-
бенно в производстве многотоннажных химических продуктов.
Тепло, выделяющееся при протекании химического процесса, мо-
жет быть использовано для получения водяного пара или превра-
щено в электроэнергию. Химическая энергия преобразуется в энер-
гию электрическую также в гальванических элементах и аккуму-
ляторах. Эти источники энергии представляют большой интерес,
так как они обладают высоким коэффициентом полезного дей-
ствия.
Световая энергия находит все более широкое применение
в химической промышленности для проведения разнообразных фо-
тохимических реакций: синтеза хлористого водорода из элемен-
тарных веществ, при галоидировании органических соединений и
Других процессов.
Вторичные энергетические ресурсы представляют собой энерге-
тические отходы или побочные продукты производства: отходящие
газы, горячие жидкости, пар и др. Полноценное использование
этого вида энергии имеет большое экономическое значение, так
как влияет на снижение себестоимости готовой продукции.
Источники энергии
Химическая промышленность.является одной из самых энерго-
емких отраслей индустрии. Хотя производимая продукция состав-
ляет 5,7% общего объема производства промышленной продукции,
количество потребляемой в химической промышленности электро-
энергии равно примерно 12%.
201
• ч • КГ-1):
8,0
4,0
10,6
22,5 • 10е
Ценность различных источников энергии определяется ее коли-
чеством, которое может быть получено при использовании этих
источников. Ниже для сравнения приведена энергетическая цен-
ность некоторых источников энергии (в кВт
Каменный уголь............
Торф......................
Природный газ.............
Уран......................
Для снабжения химической промышленности энергией могут
быть использованы различные энергетические установки. Так, те-
пловую энергию в виде пара и горячей воды можно получать от
ТЭЦ или котельных установок, электрическую энергию — от кон-
денсационных тепловых электростанций (КЭС), гидростанций
(ГЭС), а также от ТЭЦ и атомных электростанций (АЭС).
В целом химическая промышленность потребляет за год тепло-
вой энергии (в эквиваленте) больше, чем электрической. Подсчи-
тано, что более 90% всех потребляемых энергоресурсов в химиче-
ском производстве получают сжиганием различных видов топлива
(углей, природного газа, мазута, торфа, горючих сланцев) и около
10% приходится на долю гидроэнергии. Все это характеризует хи-
мическую промышленность с энергетической точки зрения как
отрасль, которой необходимы в первую очередь источники деше-
вого топлива. Ниже приведены данные о потреблении химической
промышленностью различных видов энергии (% в расчете на
условное топливо):
I960 г.
1970 г. 1975 г.
38 40 'I
9 9 Ж
53 50 г i
Электрическая энергия..................... 35
в том числе поступаемая от ГЭС ... 10
Тепловая (в паре и горячей воде) .... 55
Топливо прямого, т. е. непосредственного,
использования (для целей пламенного
обогрева в процессах сушки, выпарки
и т. п.)..................................... 10 9 10
В целях наиболее экономного использования топлива для про-
мышленных и других нужд в стране все шире осуществляется
централизованное теплоснабжение как предприятий, так и горо-
дов, причем ведущая роль в этом принадлежит теплофикации, т. е.
отпуску тепла в виде пара или горячей воды от теплоэлектроцент-
ралей.
Большое значение для химической промышленности имеет при-
родный газ, поскольку он может быть использован не только для
энергетических нужд, но и в качестве сырья для получения самых
разнообразных продуктов.
Экономические показатели производства электроэнергии зави-
сят от типа электростанции (тепловые, гидравлические, атомные),
их мощности и района размещения. На тепловых электростанциях
себестоимость электрической и тепловой энергии в основном зави-
202
сит от затрат на топливо, составляющих 60—70% всех издержек
производства. Себестоимость электроэнергии на твердом топливе
значительно выше, чем на газообразном; соответствующие удель-
ные капитальные вложения в электростанции и топливную базу
примерно в 1,4 раза больше. Себестоимость и удельные капиталь-
ные затраты на гидроэлектростанциях Ангаро-Енисейского каскада
в 2—3 раза меньше, чем на Волжских и Днепровских.
В развитии энергетики в последние годы произошли коренные
изменения, направленные на дальнейшее ускорение технического
прогресса в этой отрасли. В теплоэнергетике начато строительство
крупных электростанций с блоками большой единичной мощности.
Расчеты показывают, что сооружение электростанций, оснащен-
ных блоками по 800 МВт, обеспечивает по сравнению с 300-ты-
сячными блоками экономию капитальных вложений в размере
около 10%, удельный расход топлива уменьшается на 3—4%,
а удельная численность эксплуатационного персонала — почти
в 3 раза.
Общая выработка электроэнергии в СССР характеризуется
следующими показателями (млрд. кВт-ч):
1940 г............
1960 г............
1970 г............
1975 г............
1980 г. (план) . .
Всего Тепловые электро- станции (включая АЭС), % Гидростанции, %
48,3 89 11
292 83 17
740,9 83 17
1039 86 14
1340-1380 — —
Мировые запасы урана и тория обладают энергией, превосходя-
щей во много раз потенциальную энергию разведанных запасов
угля, нефти и природного газа вместе взятых. Поэтому чрезвы-
чайно важное значение имеет использование атомной энергии
в промышленных целях.
За годы, прошедшие со времени пуска в опытную эксплуата-
цию первой в мире атомной электростанции (АЭС) в г. Обнинске
под Москвой (июнь 1954 г.), были сооружены и вошли в эксплуа-
тацию ряд крупных атомных электростанций мощностью до
1 млн. кВт.
Так как источники топлива на базе углерода ограничены, ис-
пользование атомной энергии стало ключевой проблемой для боль-
шинства стран. Считается, чтр к 2000 г. атомная энергия станет
одним из основных источников электроэнергии.
В десятой пятилетке в нашей стране намечено ввести в экс-
плуатацию новых атомных электростанций на общую мощность
13—15 млн. кВт.
Источники химической энергии (выделяющейся в виде тепла
при протекании реакции) приобретают большое практическое зна-
чение в химической промышленности. В последние годы создаются
203
энерго-технологические химические производства, в которых одно-
временно с химическими продуктами получают товарную энергию,
выдаваемую потребителям в виде энергетического пара или элек-
троэнергии. При этом вся потребность в энергии самого химиче-
ского производства полностью покрывается за счет использования
энергии химических реакций. Например, при получении 1 т серной
кислоты из серы выделяется 5 МДж тепла, а общая потребность
в электроэнергии в процессе получения 1 т серной кислоты состав-
ляет около 100 кВ • ч или 0,36 МДж, т. е. всего лишь около 7%
выделяющегося тепла химических процессов. В настоящее время
уже освоены такие энерго-технологические системы при производ-
стве серной кислоты, аммиака и др.
В дальнейшем большое значение приобретут такие источники
энергии, как солнечная, энергия ветра, приливов и отливов, тепла
земных недр.
Рациональное использование энергии
Химические предприятия потребляют большие количества энер-
гии, поэтому энергетические затраты существенно влияют на тех-
нико-экономические показатели процессов. Критерием экономич-
ного использования энергии служит коэффициент использования
энергии т]э, определяемый по уравнению:
Пэ=^100% (IX. 1>
где ТГт и ТГпр — соответственно количество энергии, затрачиваемое теоретиче-
ски и практически на получение единицы продукта.
Во многих производствах этот коэффициент очень низкий, что
свидетельствует о непроизводительном расходе энергии. Поэтому
большое практическое значение имеют мероприятия, направлен-
ные на повышение т)э.
Из всех видов потребляемой в химической промышленности
энергии первое место принадлежит тепловой энергии. Степень ис-
пользования тепла в химико-технологическом процессе опреде-
ляется тепловым коэффициентом полезного действия т]т, выражае-
мым уравнением
Пт=-^-Ю0% (IX. 2)
Чпр
где QT и Qnp — соответственно количество тепла, теоретически н практически за-
трачиваемое на осуществление химической реакции.
Использование вторичных энергетических ресурсов (отходов)
повышает коэффициент использования энергии. Энергетические
отходы могут быть использованы в химической и других отраслях
промышленности для выработки электрической и тепловой энер-
гии в утилизационных установках; отработанный пар и горячая
вода обычно расходуются непосредственно (без преобразования
204
в ДрУгие энергоносители) для отопления и горячего водоснаб-
жения.
Особенно большое значение в химической промышленности
имеет утилизация тепла продуктов реакции, выходящих из реак-
ционных аппаратов, для предваритель-
ного нагревания материалов, поступаю-
щих в эти же аппараты. Такой нагрев
осуществляют в аппаратах, называемых
регенераторами, рекуператорами, теп-
лообменниками (рис. IX.9). Реагенты
поступают в теплообменник, где нагре-
ваются за счет тепла горячих продук-
тов, выходящих из реакционного аппа-
рата, и затем подаются в реактор. По
этой схеме теплообмен между горячими
и холодными продуктами происходит
через стенки трубок теплообменника,
аппараты такого типа называют рекупе-
Продукты реакции
Рис. IX.9 Схема использо-
вания тепла продуктов реак-
ции или отходящих газов:
1—теплообменник; 2—реактор.
риторами (теплообменниками).
Регенераторы также применяются для утилизации тепла газов;
они представляют собой периодически действующие камеры, за-
полненные насадкой (обычно насадкой служат решетки из кир-
пича). Для создания непрерывного процесса необходимо иметь по
Рис. IX. 10. Схема работы регенера-
тора:
1 — 8— задвижки, А, Б — камеры регенера-
тора.
Рис. IX. 11. Котел-утилизатор:
/ — корпус; 2—трубы; 3, 4 —вентили;
5 — вл агоотделитель.
крайней мере два регенератора (рис. IX. 10). Горячий газ вначале
проходит регенератор А, нагревают его насадку, а сам охлаж-
дается. Холодный газ проходит через регенератор Б и нагревается
б результате соприкосновения с ранее нагретой насадкой. При та-
ком режиме работы должны быть закрыты нечетные задвижки 1,
5,7 и открыты четные задвижки — 2, 4,6 и 8. После нагревания
205
насадки регенератора А и охлаждения насадки регенератора Б
производят переключение и горячий газ направляют в регенера-
тор Б, а холодный газ в регенератор А. При этом четные задвижки
должны быть открыты, а нечетные — закрыты. После охлаждения
насадки регенератора А и нагревания насадки регенератора Б
вновь производят переключение. При организации такой периоди-
ческой работы регенераторов обеспечивается постоянный подогрев
холодного газа за счет тепла отходящего горячего газа.
Тепло газообразных продуктов реакции и отходящих газов ча-
сто используют для производства пара в так называемых котлах-
утилизаторах (рис. IX. 11). Горячие газы движутся по трубам, раз-
мещенным в корпусе котла. В межтрубном пространстве нахо-
дится вода, которая поступает через штуцер 3. Образующийся пар,
проходя влагоотделитель 5, выводится из котла через вентиль 4.
Тепло отходящих продуктов на химических заводах может
быть также использовано для сушки, выпаривания, дистилляции
и других процессов.
4. ОБЕЗВРЕЖИВАНИЕ СТОЧНЫХ ВОД
И ОТХОДЯЩИХ ГАЗОВ
Обезвреживание сточных вод. Производственные и бытовые
сточные воды загрязнены различными органическими и неорга-
ническими примесями, которые при сливе в водоемы отравляют их.
Действующее в СССР законодательство предусматривает строгую
санитарную охрану естественных водоемов, поэтому спуск про-
мышленных сточных вод в водоемы производится в соответствии
с санитарными правилами, в которых определено предельно до-
пустимое содержание веществ в сточных водах. Ниже приведено
допустимое содержание некоторых веществ в сточных водах
(в мг • л"1):
Азотная кислота........... 30—35
Аммиак ..................... 5
Медь.................... 0,1—0,2
Ртуть..................... 0.005
Фенолы.................. 0,001—0,002
Если концентрация примесей в сточных водах превышает пре-
дельно допустимое содержание, такие воды подвергают специаль-
ной очистке. При этом содержащиеся в воде примеси либо разру-
шаются, либо переходят в безвредную форму, либо их извлекают
для последующего использования в качестве сырья.
Многие отечественные химические комбинаты имеют очистные
установки, на которых достигается настолько высокая степень очи-
стки сточной воды, что ее можно вновь возвратить в производство,
В результате создается замкнутый цикл по воде, свежая вода по-
ступает в процесс только в таком количестве, чтобы восполнить ее
потери, неизбежные при любом производстве.
206
Очистка сточных вод химических предприятий и последующее
их использование является крупной народнохозяйственной проб-
лемой. Если в 1952 г. объем сточных промышленных вод в нашей
стране не превышал 27 млрд, м3, то уже в 1958 г. он возрос до
50 млрд, м3, а в 1965 г. — до 100 млрд, м3 в год, что составляет де-
сятую часть устойчивого речного стока и около четверти всего по-
требления воды в СССР. Поскольку количество промышленных
сточных вод все время растет, чистая вода превращается в один
из дефицитных видов природных богатств, особенно в районах, где
широко развита химическая промышленность.
Создание надежных способов очистки загрязненных промыш-
ленных стоков является весьма сложной проблемой и далека от
решения многих производств. Основное направление работ по су-
щественному улучшению очистки промышленных сточных вод яв-
ляется сооружение очистных сооружений на каждом промышленном
предприятии, что позволит резко уменьшить или даже полностью
ликвидировать сброс сточных вод после их очистки в реки и озера.
Внедрение оборотного цикла водоснабжения, замкнутой систе-
мы водопотребления с минимальным добавлением свежей воды
является одним из наиболее рациональных направлений исполь-
зования воды в промышленности. Отработанная вода после очи-
стки и извлечения из нее полезных веществ должна возвращаться
на предприятие для повторного использования. В качестве при-
мера можно привести разработанную «Гипрокаучуком» схему
внутритехнологического водооборота в производстве этилена и про-
пилена с раздельной конденсацией тяжелых (смол) и легких угле-
водородов. Такая схема в настоящее время внедрена на ряде за-
водов, в результате чего количество загрязненных сточных вод,
сбрасываемых в канализацию, сократилось с 900—1000 до 10—
30 м3/ч. В связи с этим уменьшился расход речной воды, что зна-
чительно снизило стоимость строительства и эксплуатации водоза-
борных сооружений, водоводов и сбросных канализационных кол-
лекторов.
Таким образом, внедрение системы водооборота в химических
производствах может привести не только к резкому уменьшению
вредных для водоемов сточных вод, но позволит усовершенство-
вать технологию производства, улучшить технико-экономические
показатели.
Способы обезвреживания сточных вод подразделяются на ме-
ханические, физико-химические, химические и биологические.
Механические способы включают отстаивание и фильтрацию;
эти способы используются для удаления взвешенных частиц; к фи-
зико-химическим способам относятся аэрация, адсорбция, экстрак-
ция, выделение примесей в осадок и др.; химические способы осно-
ваны на разрушении вредных примесей, их нейтрализации, осажде-
нии различными реагентами и др.; биологические способы обезвре-
живания применяются, главным образом, для очистки сточных вод
населенных пунктов с помощью некоторых микроорганизмов.
207
Вода, применяемая в качестве хладоагента, обычно не изме-
няется по составу, а только нагревается, поэтому после использо-
вания такую воду охлаждают в специальных водоемах или в гра-
дирнях, а затем возвращают обратно на производство (оборотная
вода).
В последние годы особенно остро поставлена проблема обез-
вреживания сточных вод, так как одновременно с развитием про-
мышленности возрастает и объем сточных вод. При этом в отдель-
ных случаях сильно загрязненные сточные воды, попадая в реки,
губят все живое и создают антисанитарные условия жизни.
Кроме работ по очистке производственных сточных вод, боль-
шое значение имеют проводимые в химической промышленности
мероприятия по замене водяного охлаждения воздушным. Его ши-
рокое внедрение позволит отказаться от эксплуатации многих во-
дооборотных циклов и существенно снизить расход свежей воды.
Так, например, ввод в действие девяти аппаратов воздушного
охлаждения в производстве капролактама позволил уменьшить
водопотребление почти на 15 млн. м3 в год. Внедрение таких аппа-
ратов охлаждения в производстве соли АГ даст возможность
в 3 раза сократить потребление свежей воды. Созданы и разраба-
тываются проекты многих других химических производств с ис-
пользованием воздушного охлаждения.
Обезвреживание отходящих газов. Содержание вредных приме-
сей в отходящих газах должно быть таким, чтобы после смешения
этих газов с атмосферным воздухом концентрация вредных при-
месей в приземном слое воздуха не превышала предельно допу-
стимой концентрации (ПДК). Ниже в табл. IX. 1 приведены ПДК
для некоторых газов, паров и пыли.
Таблица IX. 1. Предельно допустимые концентрации
(в мг • м-3) газов, паров и пыли
Вещества В воздухе рабочей зоны помещений В атмосферном воздухе населенных мест
максимальная, разовая среднесу- точная
Окислы азота 5 0,3 0,1
Окись углерода 20 6 1
Пыль пятиокиси ванадия 0,5 — 0,003
Ртуть металлическая 0,01 — 0,0003
Сернистый ангидрид . 10 0,5 0,15
Серная кислота, серный ангидрид 1 0,3 0,1
Хлористый водород 5 0,05 0,015
Отходящие газы обычно выводятся в атмосферу через трубы,
по выходе из которой эти газы смешиваются с атмосферным воз-
духом и концентрация вредных примесей в воздухе становится
более низкой, чем в отходящих газах. Естественно, что чем выше
208
труба, тем больше будут разбавлены отходящие газы воздухом й
соответственно, тем ниже будет концентрация вредных примесей
в приземном слое воздуха.
Расчет высоты трубы, при которой содержание примесей в при-
земном слое воздуха будет соответствовать ПДК, ведут по эмпи-
рической формуле:
где Н — минимально допустимая высота трубы, м;
М — количество вредных веществ (суммарное), выбрасываемых в атмосферу,
г • с-1;
ПДК — предельно допустимая концентрация вредных веществ в приземном
слое воздуха, максимальная разовая (табл.. IX. 2), мг • м-3;
V — объем выбрасываемых отходящих газов, м3 • с"1;
ДУ — разность между температурами выходящих газов и окружающего воз-
духа, °C;
W — число труб, через которые выводятся отходящие газы.
Если рассчитанная по формуле высота трубы получится слиш-
ком большой и, следовательно, очень дорогой, отходящие газы
подвергают специальной очистке, а затем уже выводят в атмос-
феру через трубу соответствующей высоты.
Таким образом, выбор пути достижения ПДК определяется
экономическими соображениями — ПДК должны быть обеспечены
при минимальных затратах, что достигается либо установкой до-
статочно высокой трубы, либо специальной очисткой отходящих
газов.
Однако технико-экономический анализ и опыт эксплуатации
показывают, что в большинстве случаев наиболее экономичный
путь состоит в том, чтобы основной технологический процесс про-
водился при условиях, обеспечивающих низкое содержание вред-
ных примесей в отходящих газах. При этом отпадает необходи-
мость в специальной очистке отходящих газов. Способы очистки
зависят от состава, свойств и концентрации вредных примесей в
отходящих газах. Эти способы весьма разнообразны, наиболее ча-
сто применяется абсорбция и адсорбция примесей различными
реагентами и адсорбентами.
Глава X
ОРГАНИЗАЦИЯ ХИМИКО-ТЕХНОЛОГИЧЕСКОГО
ПРОЦЕССА
Основанием для начала строительства химического производ-
ства служит проект, для выполнения которого проводится боль-
шая подготовительная работа, в частности, должны быть выпол-
нены следующие мероприятия;
209
1) разработаны химическая, принципиальная и технологиче-
ская схемы процесса;
2) выбраны оптимальные технологические параметры;
3) подобраны типы и разработаны конструкции аппаратов;
4) выбраны конструкционные материалы; ь
5) выбраны контролируемые и регулируемые параметры. 4
Рассмотрим основные особенности перечисленных мероприятий^
• <
1. ХИМИЧЕСКАЯ, ПРИНЦИПИАЛЬНАЯ И ТЕХНОЛОГИЧЕСКАЯ СХЕМЫ
F
В основу химической схемы производства положены химиче-
ские реакции, проводимые для того, чтобы из заданного сырья
получить продукт, являющийся конечной целью производства. Хи-
мическая схема разрабатывается с учетом результатов научных
исследований свойств сырья и получаемых продуктов, а также
основных и побочных реакций, которые могут протекать на от-
дельных стадиях процесса. Окончательный выбор химической схе-
мы должен быть сделан с учетом возможности осуществления хи-
мической реакции в промышленных условиях, аппаратурного
оформления процесса, подбора достаточно стойких материалов
и т. д. Определяющим критерием в выборе химической схемы
является экономичность производства.
Удачный выбор химической схемы часто является результатом
научной и инженерной интуиции отдельных специалистов и отно-
сится к области изобретательства, поэтому в настоящее время еще
не представляется возможным сделать какие-либо общие указа-
ния о методах выбора химической схемы процесса. В каждом от-
дельном случае при выборе химической схемы учитываются мно-
гие специфические особенности, которые проявляются на различи
ных этапах разработки химико-технологического процесса.
Для примера рассмотрим возможные химические схемы произ- -
водства фосфорной кислоты, содержащей 32% фосфора.
В решениях XXV съезда КПСС предусмотрено увеличить в те-
кущем пятилетии производство минеральных удобрений с 90 до
143 млн. т. в год, т. е. более чем в полтора раза, причем особое
внимание уделяется производству концентрированных фосфор-
ных и сложных удобрений, получаемых на основе фосфорной кис-
лоты.
Сырьем для получения фосфорной кислоты служат фосфаты
(минералы апатит и фосфориты), содержащие нерастворимый
в воде фторапатит Cas(PO4)3F; поэтому содержащийся в нем фос-
фор не усваивается растениями. В связи с этим процесс перера-
ботки фосфатов состоит в том, чтобы перевести фосфор в соеди-
нения, хорошо растворимые в воде. К таким соединениям в пер-'
вую очередь относится фосфорная кислота (Н3РО4).
Разложение фторапатита с образованием фосфорной кислоты
происходит при воздействии на него различными кислотами, на-
210
пример, серной кислотой
Ca5(PO4)3F + 6H2SO4 = ЗН3РО4 + 5CaSO4 + HF
соляной кислотой
Ca5(PO4)3F + юна = ЗН3РО4 + 5СаС12 + HF
пли азотной кислотой
Ca5(PO4)3F + 10HNO3 = ЗН3РО4 + 5Ca(NO3)2 + HF
(X. 1)
(Х.2)
(Х.З)
Но фосфорная кислота может быть получена из фосфата так-
же электротермическим методом, сущность которого состоит в том,
что измельченный фосфат смешивают с углем и различными до-
бавками и затем нагревают в электропечи. При температуре
1200—1300°С происходит разложение фторапатита с образова-
нием элементарного фосфора
2Ca5(PO4)3F + 15С + Н2О = ЗР2 + 15СО + ЮСаО + 2HF (X. 4)
который выделяют, а затем сжигают в токе воздуха
2Р2 + 5О2 = 2Р2О5 (X. 5)
При соединении образующегося фосфорного ангидрида с водой
получают фосфорную кислоту
P2OS + ЗН2О = 2Н3РО4 (X. 6)
Задача инженера-химика состоит в том, чтобы проанализиро-
вать все возможные химические способы получения целевого про-
дукта из заданного сырья и выбрать наиболее экономичный. В дан-
ном, частном, случае результаты научных исследований и анализ
работы промышленных установок показывают, что в настоящее
время наиболее дешевая фосфорная кислота получается при раз-
ложении фосфата серной кислотой [уравнение (X. 1)].
В районах с дешевой электроэнергией (например, вблизи мощ-
ных гидроэлектростанций) экономически выгодным может быть
также электротермический метод получения фосфорной кислоты.
В мировой промышленности около 10% фосфорной кислоты про-
изводят на основе элементарного фосфора, получаемого электро-
термическим методом.
Химико-технологических процессов очень много и, естественно,
много химических схем; они весьма разнообразны, некоторые из
них являются очень сложными, некоторые — весьма оригиналь-
ными. Примером оригинальной схемы может служить химическая
схема производства соды.
Схема получения соды аммиачным способом была разработана в 60-х годах
прошлого столетия. Этот способ состоит из нескольких весьма рационально
оформленных стадий, что обеспечивает аммиачному способу высокую экономиче-
скую эффективность.
Сода представляет собой углекислую соль натрия Na2CO3. Сырьем для по-
лучения служат природная поваренная соль NaCl и природный известняк СаСО3.
Задача процесса состоит в организации взаимного обмена нонами Na н Са.
211
Химическая схема процесса может быть представлена в таком виде:
СаСО3 —> СаО + СО2 (а)
I I * ”
-----------1 NaCl + NH3 + СО2 + Н2О NH4C1 + NaHCO3 (б)
СаО + Н2О = Са(ОН)2 (в)
* III
Са(ОН)2 + 2NH4C1 = 2NH3 + СаС12 + Н2О (г)
NaHCO3 = Na2CO3 + СО2 + Н2О (д)’
II
где I — сырье, II — целевой продукт (сода), III — отход.
Процесс состоит в том, что в результате обжига известняка СаСО3 полу-
чают негашеную известь СаО и двуокись углерода СО2 по реакции (а). Послед-
няя реагирует с NaCl и NH3 с образованием хлористого аммония NH4C1 и дву-
углекислой соды NaHCO3 по реакции (б). При нагревании NaHCO3 происходит
ее разложение по реакции (д) с образованием целевого продукта соды — Na2CO3
и СО2; двуокись углерода возвращается в процесс.
Окись кальция превращается в Са(ОН)2 по реакции (в), а затем взаимо-
действует с NH4CI по реакции (г) с образованием газообразного аммиака NH®
н хлористого кальция СаС12; NH3 возвращается в процесс, а хлористый кальций
выводится из процесса в качестве отхода.
Химическая схема производства соды является примером удач-
ного, вернее, квалифицированного оформления ХТП. Именно этим
объясняется то обстоятельство, что, несмотря на большие успехи
химической технологии, пока еще не найдены более совершенные
и, следовательно, более дешевые способы получения соды. Старый
аммиачный способ, разработанный более 100 лет тому назад,
успешно эксплуатируется и в настоящее время; большую часть
соды во всем мире получают этим способом, даже несмотря на то,
что получаемый в качестве отхода СаС12 пока не нашел примене-
ния. СаС12 выводится из процесса в виде водного раствора, его
сливают в специально вырытые котлованы, после заполнения такие
котлованы получили название «белые моря». Эти «белые моря»
отравляют прилегающую к ним почву и водоемы, куда СаС12 по-
падает вместе с подпочвенными водами.
Представление о количестве отходов СаС12 можно получить,
исходя из масштабов производства соды. В СССР в 1976 г. было
выработано около 5 млн. т соды, при этом на каждую тонну соды
выводилось 9 м3 водного раствора СаС12. Следовательно, объем
водного раствора СаС12 составил 45 млн. м3 в год; из этого коли-
212
чества создается «белое море» длиной 5 км, шириной 3 км и глу-
биной 3 м. Поскольку производство соды еще долгое время будет
проводиться по аммиачному методу, становится очевидным, на-
сколько сложной и чрезвычайно ответственной является пробле-
ма, связанная с сохранением или утилизацией огромных количеств
накапливающихся отходов хлористого кальция.
Общее графическое представление о химико-технологическом
процессе дают принципиальная и технологическая схемы; четкой
разницы между этими схемами и строгих правил для их построе-
ния не установлено.
H2SO4 Н3РО4
(побочный сроссрогипс (отход)
продукт)
Рис. X. 1. Принципиальная схема производства фосфорной кислоты
сернокислотным разложением:
/ — подготовка сырья (измельчение фосфата); 2—химическая переработка фос-
фата; 3—выделение фосфорной кислоты.
Рассмотрим основные особенности принципиальной и техноло-
гической схем в том виде, в каком они обычно используются в на-
учно-технической и учебной литературе для графического изобра-
жения отдельных узлов и всего технологического процесса в целом.
Принципиальная схема описывает главным образом связь
между основными физическими и химическими операциями, со-
ставляющими технологический процесс. Каждую из этих операций
показывают условно в виде кружков или квадратов.
Химическое производство обычно оформляют в виде несколь-
ких отдельных физических и химических операций. Это объяс-
няется тем, что при современных очень больших масштабах про-
изводства даже самый простой технологический процесс выгодно-
разбить на отдельные стадии. Это позволяет создать наиболее бла-
гоприятные условия для протекания основных и вспомогательных
процессов, составляющих химическую или физическую операцию.
Каждая из этих операций имеет свои особенности, полноценный
учет которых обычно достигается в отдельном аппарате.
Для наглядного подтверждения сравним наиболее простую
принципиальную схему получения фосфорной кислоты (рис. X. 1),.
которая применялась в период освоения этого производства, с со-
временной схемой, в которой созданы благоприятные условия для
проведения отдельных стадий процесса.
213»
Фосфат, являющийся сырьем, измельчается (что показано на
-рис. X. 1 операцией /) и направляется в реактор 2, в котором осу-
ществляется разложение фторапатита по реакции (X. 1). В реак-
тор 2 подается также серная кислота и часть продукционной фос-
форной кислоты (опытным путем установлено, что разложение
фторапатита протекает более интенсивно при совместном действии
серной и фосфорной кислот). Полученную реакционную смесь
в виде пульпы, содержащей фосфорную кислоту и взвешенные
а ней частицы гипса (CaSO4), направляют на фильтр 3, где гипс
Фоссрат
(сырье)
Н3РО4
Рис. X. 2. Принципиальная схема современного производства фос-
форной кислоты:
/—измельчение фосфора; 2—обогащение (флотация); 3, 4—химическое разло-
жение; 5—выделение фосфорной кислоты; 6—промывка фосфогипса; Z—раз-
бавление HjSO<; 8—охлаждение пульпы.
отделяется от фосфорной кислоты. Остающийся на фильтре гипс
является отходом, его называют фосфогипсом, так как в нем со-
держится небольшое количество фосфорной кислоты.
В реакторе 3 в соответствии с реакцией (X. 1) образуется
также газообразный фтористый водород (HF); этот побочный про-
дукт используют для получения различных фтористых солей.
Принципиальная схема современного производства фосфорной
кислоты более сложная, — она включает значительно большее
число операций (рис. Х.2). Фосфат измельчают (операция /), за-
тем подвергают флотации (т. е. обогащению, операция 2), в ре-
зультате чего содержание Р2О5 в фосфате возрастает от 10—15%
до 30—40%. С повышением содержания Р2О5 в фосфате соответ-
ственно увеличивается производительность всех последующих ста-
дий процесса.
Обогащенный фосфат подают в первый реактор 3, где фосфат
смешивается с фосфорной кислотой и образует пульпу, которую
далее направляют во второй реактор 4. Сюда же добавляют сер-
.214
ную кислоту. Из второго реактора 4 пульпу, состоящую из фос-
форной кислоты и сульфата кальция, направляют на первый
фильтр 5, где основное количество фосфорной кислоты отделяется
от сульфата кальция.
Часть получаемой фосфорной кислоты выводят из процесса
в виде продукционной, а остальное количество направляют в пер-
вый реактор 3.
Осадок после фильтра 5 содержит кроме сульфата кальция зна-
чительное количество фосфорной кислоты, этот осадок передают
на второй фильтр 6, где сульфат промывают водой для извлече-
ния фосфорной кислоты и выводят из процесса, как отход произ-
водства (фосфогипс).
Промывная вода, образующаяся в фильтре 6, представляет со-
бой слабый раствор фосфорной кислоты; этот раствор используют
для разбавления 93%-ной серной кислоты в смесителе 7 до кон-
центрации 70%.
Поскольку процесс отделения фосфорной кислоты на фильт-
рах 5 и 6 существенно зависит от размеров кристаллов полугид-
рата кальция, в реакторах 3 и 4 создают условия, способствующие
образованию крупных кристаллов. Для этого часть пульпы из ре-
актора 4 упаривают в вакуум-испарителе 8 и подают в первый
реактор 3; при упаривании температура пульпы снижается, а раз-
мер кристаллов полугидрата кальция увеличивается. Таким обра-
зом одновременно регулируют и температурный режим процесса,
и размер кристаллов.
В реакторах 3 и 4, а также в смесителе 7 выделяется фтори-
стый водород, используемый далее для получения фтористых
солей.
Таким образом, в современной схеме добавлена физическая
операция (2), в которой предусматривается обогащение фосфата.
Химическое разложение фосфата осуществляется в двух реакто-
рах 3 и 4, а выделение фосфорной кислоты в двух фильтрах 5 и 6.
Кроме того, предусмотрен смеситель 7 и вакуум-испаритель 8.
Несмотря на некоторое усложнение ХТП его экономичность
значительно повышается.
В настоящее время в промышленности эксплуатируется не-
сколько схем производства фосфорной кислоты, в основу которых
положен описанный выше метод. Эти схемы отличаются между
собой числом реакторов (до 4-х), числом стадий фильтрования,
а также технологическим режимом (температура пульпы, соотно-
шение серной и фосфорной кислот в реакторах, концентрация
этих кислот и др.).
Изображение технологической схемы отличается от изображе-
ния принципиальной схемы: в технологической схеме аппараты по-
казаны в виде рисунков, внешне похожих на оригинальные аппа-
раты (по возможности эти рисунки делаются в масштабе). Ино-
гда показывают разрезы отдельных аппаратов с целью пояснить
их внутреннее устройство. Параллельно работающие аппараты
215
изображаются в виде одного аппарата; потоки газа и жидкости
показывают идущими слева направо. В зависимости от назначе-
ния технологическая схема выполняется по-разному, строго уста-
новленных правил в этом отношении нет.
СаСг
Сухая
известь
а
Рис. X. 3. Принципиальная (а) и технологическая (б) схемы производства аце-
тилена из карбида кальция:
J—дробление СаС2; II—химическая реакция; III—охлаждение и промывка С2Н2; IV — очистка
С2Н2;
/ — приемный буккер; 2— автоматический затвор; 3 —буферный бункер; 4—шиек; 5—ацети-
леновый генератор; 6 — шнек для удаления извести; 7—скруббер; 8—отстойник; 9— холо-
дильник.
Сравним принципиальную и технологическую схемы производства ацетилена
из карбида кальция. Этот процесс протекает по уравнению
СаС2 + 2Н2О = С2Н2 + Са(ОН)2
Принципиальная схема включает четыре основные операции (рис. X. 3, а).
Карбид кальция измельчается (физическая операция 1) и поступает в реактор,
где осуществляется операция // — химическое взаимодействие. Полученный газо-
образный ацетилен охлаждают и освобождают от пыли (операция ///), затем
ацетилен очищают от посторонних примесей: аммиака, фтористого водорода
и др. в специальной установке (операция IV).
В технологической схеме (см. рис. IX. 3, б) показаны вспомогательные аппа-
раты 1—4 и 6, а также сделаны разрезы основных аппаратов—ацетиленового
реактора 5, где осуществляется операция II, и скруббера 7, в котором прово-
дится операция ///.
В качестве примера можно сравнить также принципиальную (см. рис. X. 2) и
современную технологическую (рис. X. 4) схемы производства фосфорной кисло-
ты сернокислотным разложением.
Технологическая схема в проектной документации должна да-
вать не только общее графическое представление о технологиче-
ском процессе, но и содержать многие важные сведения, необхо-
димые для разработки основных разделов проекта. Поэтому при
216 z
разработке проектной документации технологическая схема со-
ставляется по определенным правилам, которые записаны в спе-
циальной инструкции.
Принципиальные и технологические схемы можно разделить на
два типа: с открытой цепью и циклические. В промышленности
встречаются разнообразные комбинации этих схем.
H«SO
Фосшат
Оборотная фосфорная кислота
вода
Ю
Продукционная кислота.
13
в 13 в
К вакуум-
насосу
Рис. X. 4. Схема производства фосфорной кислоты сернокислотным разложе-
нием:
1—бункер; г—реактор; 3, 5—дозаторы фосфорной и серной кислот; 4, 10 — напорные баки;
6—вакуум-испаритель; 7—распределитель пульпы; 8— погружные насосы; 9—конденсатор;
11 — лотки карусельного вакуум-фильтра; 12—вакуум-сборник; 13— сборник фильтратов.
Схемы с открытой цепью представляют собой ряд
аппаратов, через которые все реагирующие, вещества проходят
лишь один раз (проточная схема). По открытой схеме оформляют
производства, в основе которых лежат необратимые и обратимые
процессы, идущие с большим выходом продукта. Примером схемы
с открытой цепью могут служить схемы производства ацетилена
(см. рис. Х.З), серной кислоты (см. гл. XI) и других продуктов.
Циклическая схема предусматривает многократное воз-
вращение в один и тот же аппарат всех реагирующих масс или
одной из фаз. Эту схему называют также циркуляционной. Типич-
ными примерами циклической схемы могут служить современный
синтез аммиака, синтезы спиртов, моторного топлива и др.
Схема производства аммиака состоит из трех основных операций (рис. X. 5).
Смесь газообразных азота и водорода сжимается компрессором и направляется
217
Рис. X. 5. Циклическая (циркуляционная) схема синтеза аммиака:
1 — сжатие газовой смеси; 2 —химическая реакция; 3—выделение аммиака.
Воздух*
HgS
Яоглоти-
•тельный
таствор
Воздух
Поглоти-
тельный
раствор
Воздух*
H2s
Воздух
1 г
’—” -------- , .Поглоти'
тельный
раствор
H2S тельный
раствор
Рис. X. 6. Принципиальные схемы установок для очистки воздуха от H2S:
•С,—схема с открытой цепью; б—циклическая схема: I» 2—абсорбция H2S поглотительным рао
твором; 3—десорбция H2S из поглотительного раствора.
Рис. X. 7. Принципиальные схемы химических процессов:
в—с открытой цепью; б—циклическая схема; 1 — смешение реагентов; 2— химические превра-
щения; 3 — разделение продуктов реакции; 4—теплообмен между продуктами реакции и исход-
вымя реагентами; А, В, С, D, М—исходные реагенты; К, N, S —побочные продукты; це-
левой продукт.
в реактор — колонну синтеза 2, где в присутствии катализатора протекает ос-
иовная реакция
N2 + 3H2 3NH3
Затем образующуюся газовую смесь направляют в холодильник, где аммиак
конденсируется и выводится из цикла в качестве целевого продукта, а непро-
реагировавшая часть реакционной смеси (газообразных N2 и Н2) возвращается
в цикл.
На рис. X. 6 на примере одного и того же процесса (очистки воздуха от га-
зообразных примесей СО2, SO2, H2S и др.) показаны схемы, которые нашли ши-
рокое применение в промышленности.
В схеме с открытой цепью (рис. X. 6, а) воздух, содержащий сероводород,,
проходит аппараты 1 и 2 противотоком поглотительному раствору, который аб-
сорбирует сероводород и выводит его из системы.
Во второй схеме (рис. X. 6, б) воздух (так же как и в первой схеме) после-
довательно проходит аппараты 1 и 2. Поглотительный раствор участвует в про-
цессе по циклической схеме — вытекая из аппарата 1, он возвращается в аппа-
рат 2. По мере насыщения поглотительного раствора сероводородом часть его
выводится из цикла и направляется в регенератор 3, где происходит разделение
продуктов: сероводород далее используется как побочный продукт, а поглоти-
тельный раствор либо возвращается в цикл, либо выводится из него.
На рис. X. 7 показаны более сложные схемы — с открытой цепью и цикличе-
ская.
2. ВЫБОР ПАРАМЕТРОВ ПРОЦЕССА
Показатели технологического процесса, на основе которых ве-
дется разработка проекта, выбираются таким образом, чтобы была
обеспечена наиболее высокая экономическая эффективность не
отдельной физической или химической операции, а всего произ-
водства в целом. Это объясняется тем, что иногда условия, обеспе-
чивающие минимальные затраты по одной операции, приводят
к более высоким затратам при оформлении последующих опера-
ций, в результате чего возрастают суммарные затраты на единицу
целевого продукта.
Для примера рассмотрим влияние степени измельчения фос-
фата на себестоимость фосфорной кислоты при получении ее по
наиболее простой принципиальной схеме.
Физической операцией I (см. рис. X. 1), предусматривается
дробление фосфата до частиц размером не более 0,2 мм, что со-
ответствует оптимальным условиям, при которых достигается
минимальная стоимость фосфорной кислоты. При увеличении
крупности помола (увеличение диаметра частиц фосфата) произво-
дительность размольных установок повышается, и стоимость фи-
зической операции 1 снижается. Однако при этом возрастает
стоимость последующей химической операции 2, так как одновре-
менно снижается скорость разложения фосфата: чем крупнее ча-
стица фосфата, тем дольше идет ее разложение (рис. X. 8). Из
рисунка видно, что по мере измельчения (уменьшения d) стоимость
разложения уменьшается быстрее, чем растет стоимость измельче-
ния. Суммарные затраты снижаются до тех пор, пока диаметр не
Достигнет определенного размера. Затем затраты начнут возра-
стать, так как стоимость измельчения все в большей степени будет
превышать стоимость разложения.
219
минимум (рис. X. 9);
Таким образом, зависимость суммарных затрат от размера ча-
стиц фосфата выразится кривой, имеющей минимум (рис. X. 9);
Рис. X. 8. Стоимость измельчения
фосфата А и стоимость его разло-
жения Б в зависимости от диаме-
тра частиц дробленого фосфата d.
лопт
it '
Рис. X. 9. Себестоимость фос-
форной кислоты S в зависи-
мости от степени измельчения
фосфора (от диаметра частиц
дробленого фосфата d).
<#гсюда следует, что минимальные затраты по переработке фос-
фата достигаются при некотором оптимальном диаметре частиц
фосфата.
Для химической операции 2 (см. рис. X. 1) также существуют
оптимальные условия, которые определяют скорость реакции раз-
ложения фторапатита. Эта скорость
Рис. X. 10. Зависимость степени
разложения фосфата от концен-
трации исходной сериой кислоты
CH2SO4.
зависит от многих параметров: тем-
пературы, концентрации серной
кислоты в реакционной смеси, ин-
тенсивности перемешивания этой
смеси и др. Например, разложение
фторапатита по уравнению (X. 1)
существенно зависит от концентра-
ции серной кислоты. Сложный вид
этой зависимости (рис. X. 10) объ-
ясняется тем, что в процессе реак-
ции на зерне фосфата образуется
осадок сульфата кальция. В зави-
симости от концентрации кислоты
этот осадок имеет различный состав
(по содержанию CaSO4 и Н2О) и
различную плотность, при этом со-
ответственно изменяется скорость диффузии кислоты из раствора
к поверхности частицы фосфата.
При повышении температуры скорость разложения фосфата
увеличивается. Из этого, казалось бы, должно следовать, что про-
цесс можно вести при более высокой температуре. Однако, при
220
повышении температуры увеличивается коррозионное действие сер-
ной кислоты на материал аппаратуры, поэтому в данном случае
существуют оптимальные условия, при которых суммарные затра-
ты, рассчитанные на основе производительности реактора и с уче-
том устойчивости материала аппаратуры, будут минимальными.
Для физической операции 3, т. е. для выделения фосфорной
кислоты, также существуют оптимальные условия, которые в пер-
вую очередь определяются скоростью фильтрования образующейся
пульпы.
Рис. Х.П. Зависимость себестоимости фосфорной кислоты S от различных пара-
метров:
J—подготовка сырья; 2—химические превращения: 3—разделение продуктов; d—диаметр ча-
стицы фосфата; С—концентрация серной и азотной кислот в реакционной смеси; Т)—интен-
сивность перемешивания; т —время пребывания реагентов в реакторе.
Для всех параметров, составляющих основу разрабатываемого
проекта, необходимо находить оптимум путем построения графика,
подобного приведенному выше. Тогда оптимальными значениями
будут считаться те, при которых обеспечивается минимальная се-
бестоимость конечного продукта.
Общая схема различных параметров, влияющих на себестои-
мость фосфорной кислоты при ее получении, показана на рис. X. 11.
На этом рисунке приведены лишь наиболее важные параметры;
в действительности их значительно больше, и, соответственно,
общая схема зависимости является более сложной.
Оптимальные значения параметров находят на основе матема-
тического моделирования и исследования модели на ЭВМ; при
этом могут учитывать не все параметры, влияющие на себестои-
мость. Поэтому в настоящее время достаточно полные математи-
221
ческие модели составлены только для некоторых сравнительно про-
стых и хорошо изученных химических производств.
Например, такая модель составлена для производства серной
кислоты из серы. Эта модель представляет собой решение 500 си-
стем уравнений. Решение такой громоздкой системы уравнений
хотя и возможно с помощью современных ЭВМ, но требует боль-
ших затрат, поэтому для облегчения решения предварительными
расчетами устанавливают степень влияния каждого параметра на
экономическую эффективность процесса и, по возможности, исклю-
чают из уравнений те параметры, которые оказывают незначитель-
ное влияние на экономическую эффективность. Однако следует
помнить, что такие исключения снижают точность получаемых ре-
зультатов.
Так, если в задании на проектирование указано место строи-
тельства установки по производству фосфорной кислоты и ее мощ-
ность, а также известны источники воды и электроэнергии, тогда
верхняя группа параметров, указанных на рис. Х.11, исключается.
Из общего математического описания может быть исключена
физическая операция 1 (см. рис. X. 11), так как в настоящее время
существует государственный стандарт, которым регламентируется
в дробленом и обогащенном фосфате содержание Р2О5 и влаги,
а также указывается степень измельчения фосфата и ряд других
параметров сырья. Кроме того, может быть значительно упрощено
математическое описание химической операции 2 (см. рис. X. 11),
Действительно, на основании опыта работы промышленных устано-
вок известно, что фосфорную кислоту наиболее низкой себестои-
мости получают при обработке фосфата смесью серной и фосфор-
ной кислот. В таком случае исключаются все параметры, отра-
жающие влияние химической схемы производства.
При дальнейшем анализе параметров могут быть внесены и
другие дополнительные упрощения в математическое описание про-
цесса, однако всегда надо учитывать, что упрощения снижают
точность.
3. ПОДБОР АППАРАТУРЫ
Выбор аппарата диктуется прежде всего экономическими сооб-
ражениями: аппарат должен быть максимально простым и деше-
вым, а протекающие в нем процессы должны осуществляться при
оптимальных условиях.
Химический процесс состоит из трех стадий: подготовки сырья,
химической реакции и разделения продуктов реакции (рис. 1.1).
Во многих случаях химическое производство состоит из двух
или нескольких отдельных химических процессов, в результате
каждого из них получают продукт, служащий сырьем для после-
дующего процесса. Примером этому может служить процесс полу-
чения продукта R, принципиальная схема которого показана на
рис. X. 6, а, а также процесс производства кальцинированной соды,
химическая схема которого приведена на стр. 212.
222
Из схемы видно, что в производстве соды протекает пять хими-
ческих реакций: получение СаО и СОг по реакции (а); получение
NH4CI и ЫаНСОз по реакции (б); получение Са(ОН)2 по реакции
(в), получение NH3, СаС12 и Н2О по реакции (г) и получение
Na2CO3 (целевой продукт), СО2 и Н2О по реакции (д).
В каждом из этих процессов образуются промежуточные про-
дукты, служащие сырьем для последующего процесса.
Исходя из химической схемы процесса, реактор является основ-
ным аппаратом, поскольку в нем осуществляется химическая
реакция. Без химического реактора не может быть химического
процесса. В то же время, стадии подготовки сырья и разделения
продуктов реакции, являющиеся вспомогательными, во многих
процессах отсутствуют. В таких процессах либо сырье поступает
в виде, готовом для последующей переработки непосредственно в
реакторе, либо в реакторе образуется готовый целевой продукт,
передаваемый непосредственно потребителю.
Поскольку химических производств много, а некоторые из них
состоят из нескольких химических процессов, часто весьма суще-
ственно отличающихся по своему характеру, то и реактор, и вспо-
могательная аппаратура, используемая при оформлении химиче-
ских производств, могут быть самыми разнообразными. Поэтому
не представляется возможным давать какие-либо общие указания
по выбору аппаратуры при оформлении химического процесса; в
каждом отдельном случае вопрос решается с учетом специфиче-
ских особенностей конкретного химического процесса.
В качестве примера рассмотрим выбор аппаратуры для произ-
водства фосфорной кислоты сернокислотным разложением фосфа-
тов и для производства серной кислоты из сероводорода. »
Как видно из рис. X. 2, получение фосфорной кислоты сернокис-
лотным разложением состоит в том, что измельченный фосфат,
затем фосфорная кислота и серная кислота поступают в реактор,
где в результате протекания реакции (X. 1) образуется смесь, со-
стоящая из водного раствора фосфорной кислоты и сернокислого
кальция. Последний плохо растворим в разбавленной фосфорной
кислоте, поэтому выпадает в осадок, образуя взвесь — пульпу.
Процесс может осуществляться непрерывно, поэтому целесооб-
разно применить реактор непрерывного действия, обеспечивающий
высокую интенсивность процесса (стр. 126). Для решения вопроса
о применении реактора вытеснения или реактора смешения сле-
дует учитывать специфические особенности проводимой реакции.
Так как реакция (X. 1) является простой, фактор селективности
можно не учитывать, и, следовательно, с этой точки зрения без-
различно, в каком реакторе следует вести процесс.
Наиболее важная особенность рассматриваемого процесса со-
стоит в том, что в данном случае протекает реакция между твер-
дым веществом (фосфат) и жидкими, реагентами. Причем исход-
ные реагенты должны быть тщательно перемешаны с тем, чтобы
создавалась однородная взвесь и обеспечивалась высокая скорость
223
реакции, а также чтобы было исключено осаждение твердых час-
тиц фосфата на дне реактора. Поскольку в результате реакции об-
разуется взвесь (пульпа), в завершающей стадии процесса также
необходимо обеспечить интенсивное перемешивание реакционной
смеси, чтобы исключить осаждение твердых частиц на дне реак-
тора. Указанные условия достаточно полно создаются в реакторе
смешения непрерывном.
Важная особенность рассматриваемого процесса состоит также
в том, что на первой его стадии происходит смешение измельчен-
ного фосфата только с фосфорной кислотой, а серная кислота до-
бавляется в дальнейшем, после того, как фосфат будет предвари-
тельно обработан фосфорной кислотой (см. рис. X. 2). Кроме того,
для регулирования температурного режима процесса часть реак-
ционной смеси (пульпы) направляется в вакуум-испаритель, где
в результате испарения воды пульпа охлаждается. При добавлении
охлажденной пульпы в начало процесса регулируется температур-
ный режим. Указанные условия также в наибольшей степени обес-
печиваются в реакторе смешения непрерывном. Этим объясняется
тот факт, что реактор смешения получил широкое распростране-
ние в производстве фосфорной кислоты.
Рассмотрим основные соображения, по которым производится выбор реак-
торов и вспомогательной аппаратуры в производстве серной кислоты из серово-
дорода.
Химическая схема производства серной кислоты из сероводорода изобра-
жается в виде следующих уравнений.
Газообразный сероводород окисляется кислородом воздуха до сернистого
ангидрида /
2H2S + ЗО2 = 2SO2 + 2Н2О + 1048 кДж (а)
Сернистый ангидрид окисляется иа катализаторе до серного ангидрида
SO2 + i/2O2 :?=* SO3 + 96,1 кДж (б)
Серный ангидрид реагирует с парами воды с образованием паров серной
кислоты
ЗОз(газ) + Н2О(газ) Н28О4(газ) + 125 кДж (в)
Таким образом производство серной кислоты из сероводорода состоит из трех
различных как по своему характеру, так и по аппаратурному оформлению про-
цессов.
Газообразный сероводород под давлением поступает в печь 2 на сжигание
в смеси с воздухом, подаваемым нагнетателем 1 (рис. X. 12). Из печи газ при
температуре около 1000 °C поступает в котел-утилизатор, где тепло газа исполь-
зуется для получения пара. Охлажденный газ при 450 °C поступает в контактный
аппарат 4 с промежуточным теплообменом, загруженный контактной ванадиевой
массой. Для снижения температуры газа после контактной массы его смешивают
с атмосферным воздухом (рис. VIII. 4, е).
Из контактного аппарата газ, содержащий SO3 и пары воды, поступает в
башню-конденсатор 5, заполненную кольцевой насадкой и орошаемую серной
кислотой (рис. VIII. 3, а). Температура орошающей кислоты на входе в башню
50—60 °C, на выходе из башни 80—90 °C. При охлаждении серный ангидрид и
пары воды образуют пары серной кислоты, которые затем конденсируются ча-
стично на поверхности орошающей кислоты (~65%) и частично в объеме с об-
разованием тумана, который выделяется из газа в электрофильтре 6.
Все три реактора, составляющие рассматриваемое производство, существенно
отличаются между собой. Окисление сероводорода — гомогенная высокотемпера-
224
турная некаталитическая реакция, протекающая в газовой фазе. Для этой цели
применяют полые реакторы, в которые вдувают газообразные реагенты, предва-
рительно смешанные в форсунке. По такой схеме осуществляют различные хими-
ческие реакции между газообразными реагентами, а также между жидкими и
газообразными реагентами. В последнем случае жидкий реагент распыляется
форсункой и испаряется в раскаленном реакторе, а затем уже в газообразном
состоянии вступает в реакцию. Так, например, сжигают газовое и жидкое топ-
ливо, осуществляют процесс сжигания серы с получением SOa и др.
Реакция окисления сернистого ангидрида — гетерогенно-каталитическая ре-
акция, сопровождающаяся выделением большого количества тепла. Эта реак-
ция является типичной газовой реакцией, проводимой на твердом пористом ката-
лизаторе. Наиболее эффективны для этих, процессов полочные реакторы с про-
межуточным теплообменом. Они просты, дешевы, надежны в работе и обеспе-
чивают высокую степень превращения. Такой типовой реактор и применяется в-
рассматриваемом процессе.
Рис. Х.12. Схема производства серной кислоты из сероводорода:
1— нагнетатель; 2—печь для сжигания сероводорода; ‘ 3 — паровой котел-утилизатор; 4—кон-
тактный аппарат; 5—башня-конденсатор; 6—электрофильтр; 7«-оросительный холодильник;
8 — сборник кислоты.
Наиболее важной вспомогательной аппаратурой в производстве серной кис-
лоты из сероводорода являются котел-утилизатор 3, электрофильтр 6 и холо-
дильник кислоты 7.
Поскольку выходящие из печи газы имеют высокую температуру ~1000 °C,
они могут быть использованы для получения пара высокого давления (около
4 МПа). Для этой цели и служит котел-утилизатор.
Пар является ценным продуктом и его реализация позволяет снизить себе-
стоимость целевого продукта — серной кислоты.
Электрофильтр 6 предназначен для выделения тумана серной кислоты, обра-
зующегося при конденсации паров серной кислоты в башне 5. Этот аппарат
имеет существенные достоинства перед другими аппаратами, применяемыми для
выделения аэрозолей, вообще, и тумана, в частности, (циклоны, центробежные
осадители, волокнистые фильтры и др.). Электрофильтр обеспечивает высокую
степень очистки газов от взвешенных, даже очень мелких, частиц, что особенно
важно с точки зрения охраны окружающей среды. После электрофильтра газ вы-
водят в атмосферу, поэтому содержание в нем даже небольшого количества сер-
ной кислоты приводит к загрязнению атмосферы и вызывает разрушения на тер-
ритории, прилегающей к предприятию.
Кроме того, электрофильтр создает малое гидравлическое сопротивление, он
надежен в работе и прост в обслуживании. Этими достоинствами объясняется
широкое применение электрофильтров в химической промышленности и особенно
в производстве серной кислоты.
Серная кислота, орошающая башню-конденсатор 5, нагревается за счет
тепла образования паров серной кислоты [реакция (в)], а затем за счет тепла
8 Зак, 558
225
конденсации этих паров. Существуют разнообразные конструкции холодильников
для охлаждения жидкостей. Оросительные холодильники нашли широкое приме-
нение для охлаждения коррозионно-активных жидкостей благодаря своей про-
стоте, возможности легкой смены труб при их разрушении и коррозии, деше-
визне, малому гидравлическому сопротивлению и др.
4. ВЫБОР МАТЕРИАЛОВ ДЛЯ ИЗГОТОВЛЕНИЯ
АППАРАТУРЫ
Подбор конструкционных материалов для изготовления аппара-
тов химического производства имеет важное значение, так как
удачный выбор материалов в значительной степени определяет тех-
нико-экономические показатели химического производства. При
выборе материала учитывается не только его коррозионная стой-
кость, но и прочность, термостойкость, возможность обработки, до-
ступность материала и его стоимость. Если в условиях работы
данного аппарата или технологического узла черные металлы яв-
ляются достаточно стойкими к коррозии, они используются в пер-
вую очередь как весьма прочные, доступные и достаточно дешевые
материалы.
Часто применяются легированные черные металлы (содержа-
щие особые добавки) или специальные сплавы, обладающие по-
вышенной коррозионной стойкостью. Однако, эти материалы обыч-
но дороги, поэтому они применяются только в тех случаях, когда
это экономически оправдано. В большинстве случаев используются
черные металлы, поверхность которых в зоне реакции защищают
различными достаточно стойкими покрытиями.
Химически стойкие неорганические природные материалы обыч-
но состоят из нескольких минералов и представляют собой различ-
ные соли кремниевых и поликремниевых кислот. Эти материалы
применяются в виде штучных изделий (камней) для футеровки ба-
шен, резервуаров и других аппаратов и их деталей.
В последние годы в химической промышленности все более ши-
рокое применение находит эмалированное оборудование. Эмалевые
покрытия устойчивы к действию различных кислот при повышен-
ной температуре, а коррозионно-активные газы практически не
разрушают эти покрытия.
Органические химически стойкие материалы имеют ряд преиму-
ществ по сравнению с неорганическими. Многие из этих материа-
лов можно легко обрабатывать на станках, прессовать, сваривать,
штамповать, склеивать и т. д. Кроме того, они дешевле и легче
неорганических антикоррозионных материалов и отличаются дли-
тельным сроком службы. Однако область применения этих мате-
риалов ограничена их невысокой теплостойкостью (температура,
при которой могут применяться многие органические материалы,
не превышает 100°C).
В качестве примера рассмотрим материалы для изготовления реактора в про-
изводстве фосфорной кислоты и основных аппаратов в производстве серной кис-
лоты из сероводорода.
226
В реакторе 2 (рис. X. 4) находится пульпа, содержащая фосфорную, сериую
и фтористоводородную кислоты, воду и взвешенные частицы фосфата и серно-
кислого кальция. При температуре процесса (около 80 °C) такая реакционная
смесь (пульпа) обладает очень сильной коррозионной активностью, поэтому
корпус реактора, его перегородки, мешалки, трубопроводы и другие детали за-
щищают от коррозии и истирания перемешиваемой пульпой или делают из
специальной стали. Стальной корпус реактора обкладывают резиной или поли-
изобутиленом, а затем футеруют кислотоупорным кирпичом или графитовыми
блоками. Валы и лопасти мешалок, а также детали вакуум-фильтра, трубо-
проводы и насосы для перекачивания фосфорной кислоты изготовляют из стали
ЭИ-943.
В производстве серной кислоты из сероводорода (см. рис. X. 12) создаются
условия, в которых черные металлы являются достаточно стойкими, поэтому вся
аппаратура в этом производстве изготовлена преимущественно из стали и чу-
гуна. В местах повышенной коррозионной активности среды поверхность защи-
щают коррозионно-стойкими материалами.
Печь 2 для сжигания сероводорода представляет собой стальной цилиндри-
ческий котел, внутренние стенки которого обложены листовым асбестом, а за-
тем футерованы шамотным кирпичом. Футеровка исключает проникновение реак-
ционных газов к стенкам печи и снижает потери тепла в окружающую среду.
Нагнетатель, газопроводы и арматура изготовлены из стали. Стеики контактного
аппарата 4, решетки для контактной массы, смесительные устройства и другие
детали также изготовлены из стали; внутренние стенки аппарата футерованы
кислотоупорным кирпичом, при этом исключается их разрушение серной кисло-
той, которая конденсируется при охлаждении аппарата в период остановок, неиз-
бежных в производственных условиях.
Электрофильтр 6 состоит из большого числа цилиндрических труб, в центре
которых подвешены коронирующие электроды. Очищаемые газы пропускаются че-
рез неоднородное электрическое поле, образующееся между коронирующими элек-
тродами и стенками труб (последние служат осадительными электродами). При
пропускании тока вокруг коронирующих электродов образуется область ионизи-
рованного газа. Взвешенные в газе капли серной кислоты (капли тумана) осе-
дают на ионах и, приобретая униполярный электрический заряд, движутся к оса-
дительным электродам и осаждаются на них. Стекающая с электродов серная
кислота собирается в сборнике.
При ионизации газа образуется также озон, который значительно усиливает
коррозионное действие серной кислоты; это следует учитывать при подборе ма-
териалов. Учитывая сказанное, корпус электрофильтра делают стальным, футе-
рованным кислотоупорной плиткой и кирпичом, осадительные трубы отливают из
хромистого чугуна Х28, а электроды выполняют из нихромового провода.
5. ВЫБОР КОНТРОЛИРУЕМЫХ И РЕГУЛИРУЕМЫХ ПАРАМЕТРОВ
Качественные и технико-экономические показатели химического
производства зависят прежде всего от точности соблюдения техно-
логического режима. При нарушении режима снижается произво-
дительность процесса и ухудшается качество продукции, поэтому
поддержание всех параметров технологического процесса в преде-
лах установленных норм имеет большое практическое значение.
Большинство химических процессов являются непрерывными и
состоят из ряда последовательных стадий. Поэтому при перебоях
в работе одного аппарата или одной стадии, т. е. при отклонении
от нормы какого-либо показателя процесса (объема смеси, концен-
трации исходного реагента, температуры и др.), нарушается режиУг
последующих стадий. Чтобы не допустить отклонения режима от
заданного; воздействуют на соответствующие параметры, т. е.
8*
227
регулируют процесс, в результате чего параметры принимают за-
данные значения.
Следует подчеркнуть, что устойчивая работа химического про-
цесса зависит прежде всего от постоянства исходных параметров —
количества и состава сырья и температуры. Известны случаи, когда
отдельные стадии и даже отдельные химические производства в
течение многих суток непрерывно работают' без регулирования.
Так, например, при постоянных значениях основных параметров
(количества газовой смеси, концентрации исходного реагента и
температуры) каталитический реактор с неподвижным слоем ката-
лизатора может изменить свои показатели в результате заметного
снижения активности катализатора, что иногда происходит после
длительной работы (в производстве серной кислоты I—2 года),
или же в результате резкого снижения температуры окружающего
воздуха (так как при этом изменяется теплообмен с окружающей
средой, что влияет на температурный режим процесса).
Контроль производства заключается в своевременном обнаруже-
нии отклонений от установленного режима и быстром устранении
их с целью предотвращения нарушений режима на других стадиях
процесса.
Методы контроля можно условно подразделить на ручные и
автоматические. К ручным методам контроля обычно относят
химические анализы. Вначале отбирают пробу сырья, полупро-
дукта или продукта, затем производят химическую обработку про-
бы и делают соответствующие вычисления, поэтому во многих
случаях результаты химического анализа получают через несколь-
ко часов с момента отбора пробы. В то же время при проведении
непрерывных процессов своевременное обнаружение отклонения
параметров технологического режима от установленных оптималь-
ных норм имеет решающее значение.
При автоматических методах контроля измерения
производят в основном непрерывно. Приборы автоматического кон-
троля не только указывают, но и регистрируют показания, а также
сигнализируют об отклонении измеряемого параметра от заданного
значения. При этом регистрация показаний может производиться
дистанционно — на значительном расстоянии от места замера, что
позволяет при установке измерительных приборов возле рабочих
мест одновременно сосредоточить регистрацию всех основных по-
казателей в одной точке — на контрольном пункте. Таким образом,
становится возможным одновременный контроль работы на каж-
дом участке цеха или отделения и всего цеха из контрольного пун-
кта. В связи с этим автоматический контроль производства приме-
няется все более широко и вытесняет другие методы контроля. При
этом часто используют косвенные методы измерений. Например,
концентрация серной кислоты, вытекающей из концентратора, за-
висит от ее температуры, методы измерения которой хорошо раз-
работаны, поэтому на практике предпочитают автоматически изме-
рять температуру, а не концентрацию кислоты.
- 228
Применение автоматических методов контроля технологических
параметров в химических производствах является особенно акту-
альным, потому что на основе этих методов становится возможным
внедрение автоматических методов регулирования процесса. По-
следнее имеет особенно большое значение и в тех случаях, когда
в ходе химических процессов возможно выделение вредных про-
дуктов (газы, пары, пыль и др.) в атмосферу производственных
помещений.
Однако для многих показателей отсутствуют надежные авто-
матические методы измерения, поэтому во многих химических про-
изводствах еще распространены
ручные, в частности, химические ме-
тоды контроля.
Регулирование технологического
процесса состоит в поддержании
заданного оптимального технологи-
ческого режима по каждой физиче-
ской и химической операции, состав-
ляющей этот процесс. В большин-
стве случаев регулирование техно-
логического процесса осуществляет-
ся автоматически при сочетании
трех основных приборов: измерите-
ля (или датчика), регулятора и ис-
полнительного механизма. Измери-
тель, замеряя какой-либо показа-
тель технологического режима, по-
сылает импульс регулятору, кото-
рый сравнивает значение измерен-
ного показателя с заданным, т. е.
Рис. Х.13. Схема регулирования
температуры в реакторе смешения:
1—измеритель температуры (датчик);
2—регулятор; 3—наполнительный меха-
низм (клапан).
оптимальным, и в случае отклоне-
ния посылает команду исполнительному механизму для изменения
условий процесса (рис. X. 13).
В химических производствах прибор-измеритель наиболее часто
измеряет температуру, концентрацию веществ, скорость потоков,
давление и другие параметры.
В некоторых случаях трудно или экономически нецелесообразно
применять автоматические методы регулирования процесса. Тогда
используют дистанционное или ручное регулирование.
Дистанционное регулирование — это неполное автоматическое
регулирование, при котором в качестве измерителя и исполнителя
применяются приборы, а регулятором является человек. Он наблю-
дает за показаниями приборов-измерителей и управляет процессом
на расстоянии (например, с пульта), воздействуя на исполнитель-
ные устройства по показателям измерителей.
Число - контролируемых и регулируемых параметров должно
•быть минимально необходимым, поскольку обилие точек контроля
н регулирования связано с большими затратами на приобретение
229
приборов, их монтаж и обслуживание, а также обработку резуль-
татов измерений.
Чтобы иметь представление об автоматизации химического про-
изводства в целом, на рис. X. 14 в качестве примера показана
бхема автоматического контроля и регулирования процесса произ-
водства серной кислоты из сероводорода.
С помощью регулятора а поддерживается определенное соотно-
шение между количеством поступающих в печь сероводородного
Рис. Х.14. Схема автоматизации производства серной кислоты из серово-
дородного газа высокой концентрации:
1 — нагнетатель; 2—печь для сжигания сероводорода; 3—паровой котел-утнлнзатор;
4—контактный аппарат; 5—башня-конден'сатор; 6—Электрофильтр; 7—оросительный
холодильник; 5 —сборник Кислоты; а—регуляторы соотношения НгЭ н воздуха; б—кла-
паны; в—термопары; Э— регуляторы уровня.
газа и воздуха. При изменении содержания H2S изменяется тем-
пература газа, выходящего из печи, что используется для поддер-
жания на заданном уровне концентрации сернистого ангидрида
в газе. Постоянство концентрации SO2 достигается тем, что путем
соответствующего преобразования импульса термопары, измеряю-
щей температуру газа на выходе из печи, вводится коррекция в
действие регулятора а соотношения потоков воздуха и сероводо-
родного газа. В результате этого увеличивается или уменьшается
объем воздуха, вводимого в печь на единицу объема сероводород-
ного газа.
Постоянная температура газа на входе в контактный аппарат
поддерживается при воздействии импульса термопары, измеряющей
эту температуру (через терморегулятор), на клапан, который регу-
лирует объем воздуха, разбавляющего горячий газ. Для поддер-
жания заданной температуры газа на входе во второй, третий и
четвертый слои контактной массы пц^усматривается аналогичное
воздействие термопар, измеряющих эти температуры (через соот-
ветствующие регуляторы), на клапаны, регулирующие объем воз-
духа, добавляемого в*газ на входе в'слой катализатора.
230
Поддержание постоянного температурного режима процесса
выделения серной кислоты в башне-конденсаторе 5 обеспечивается
с помощью регулятора температуры орошающей кислоты, который
воздействует на клапан, изменяющий количество воды, подавае-
мой на орошение холодильника 7. Количество кислоты, перекачи-
ваемой на склад, регулируется путем соответствующего воздей-
ствия (по импульсу от регулятора уровня) на клапан, установлен-
ный на продукционном кислотопроводе к складу.
Задачей химической промышленности в ближайшем будущем является созда-
ние автоматически действующих производств — цехов и заводов-автоматов
(стр. 19). Для иллюстрации основных приемов решения этой важной задачи
Рис. Х.15. Схема опытного цеха-автомата, работающего на концен-
трированном ангидриде:
1—фильтр; 2—нагреватель; 3—теплообменник; 4—контактный аппарат;^—элек-
трический (пусковой) подогреватель; 6, 7, 9, 10, 12, /3 —регулирующие клапаны;
в, 11 — регуляторы расхода; 14—газоанализатор; 15—18, 20, 21—термо&ары;
19—щит управления.
рассмотрим схему цеха-автомата по производству газообразного серного ангид-
рида из газообразного сернистого ангидрида, получаемого в виде смеси, содер-
жащей 8% SO3, 10% Оз, 82% и небольшое .количество паров воды. Такая га-
зовая смесь применяется для очистки нефтепродуктов. Процесс окисления
сернистого ангидрида в серный по реакции SO2 + ‘/^Оа ₽* SO3 проводится в при-
сутствии ванадиевого катализатора в реакторе с промежуточным теплообменом
(см. рис. VIII. 4, б). При этом процесс осуществляется по схеме, изображенной
на рис. X. 15.
Воздух, освобожденный от пыли в фильтре, смешивается с концентрирован-
ным сернистым газом, а затем нагнетателем 2 направляется в межтрубное про-
странство теплообменника 3, где смесь нагревается за счет тепла контактных
газов. Поступающий в систему воздух не подвергается сушке, поэтому в газах
посде контактного аппарата кроме SO3 содержатся пары воды. Для предотвра-
щения конденсации серной кислоты в трубах теплообменника 3 к газу на входе в
газодувку 2 добавляют такое количество горячего газа, чтобы температура смеси
231
была выше точки росы паров кислоты. Температуру газовой смеси регулируют
клапаном 7, на который воздействует импульс от термопары 15, измеряющей
температуру газа после нагнетателя 2. ,
Постоянная концентрация SO2 в газе достигается путем воздействия газо-
анализатора 14 на клапан 10, регулирующий объем воздуха, поступающего в си-
стему. Постоянство температуры газа на входе в первый слой контактной массы
поддерживается за счет воздействия импульса от термопары 21 на клапан 6 и
изменения объема байпасного холодного газа, поступающего в контактный аппа-
рат, минуя теплообменник 3. Температура газа на входе во второй и последую-
щие слои контактной массы регулируется также воздействием импульсов от
остальных термопар 21 на клапаны 13, изменяющие объем газа, который посту-
пает во внутренние теплообменники контактного аппарата.
Цех-автомат приводится в действие контактом «Пуск», с помощью которого
включается в сеть электродвигатель нагнетателя 2 и одновременно включается
реле времени, последнее через определенное время (когда нагнетатель приобре-
тает нормальное число оборотов) включает электрический подогреватель 5. Ре-
гулятор расхода 11 через исполнительный механизм воздействует на регулирую-
щий клапан 10, и таким образом обеспечивается определенный, строго заданный
объем воздуха на период разогрева реактора (контактного аппарата), который
несколько меньше объема пропускаемой газовой смеси при нормальной
работе.
Нагнетатель. 2 подает воздух в теплообменник 3, а затем в электрический
подогреватель 5.
Режим работы электрического подогревателя регулируется силой тока в цепи
по показаниям термопар 17 и 18. Первая из иих измеряет температуру спиралей,
Которая не должна превышать определенного предельного значения, вторая тер-
мопара измеряет и поддерживает заданную температуру газа на выходе из по-
догревателя 5 (около 500°C). Когда температура газа на входе в первый слой
контактной массы (измеряется верхней термопарой 21) достигнет 420 °C, испол-
нительный механизм постепенно открывает регулирующий клапан 9. Конечная
степень открытия этого клапана ограничивается регулятором расхода 8 в соот-
ветствии с заданной производительностью цеха.
Когда температура газа после первого слоя контактной массы станет на
30—50 °C ниже заданной для нормальной работы, исполнительный механизм в-
результате воздействия на него импульса от термопары 18 изменяет ток, питаю-
щий электроподогреватель. По мере повышения температуры в .контактном аппа-
рате термопары 21 воздействуют на клапаны 13, регулируя подачу газа в ка-
ждый слой контактной массы.
Остановка цеха-автомата осуществляется нажатием контакта или кнопки
«Остановка» либо с помощью исполнительного механизма, срабатывающего при
недостатке SO2, или аварийных устройств отдельных узлов и аппаратов. Полу-
чив импульс, исполнительный механизм закрывает клапан 6. Вследствие умень-
шения концентрации SO2 клапан 7 постепенно закрывается до тех пор, пока
объем поступающего воздуха не станет равным его объему, подаваемому при
разогреве контактного аппарата. Когда температура газа в первом слое контакт-
ной массы (измеряется термопарой 20) станет иа 20 °C ниже заданной, автома-
тически включается ток в электроподогревателе, и в контактный аппарат начи-
нает поступать горячий воздух для продувки (удаления SO3). Продолжитель-
ность продувки составляет 20—30 ч в зависимости от мощности контактного'
аппарата. По окончании продувки реле времени воздействует на соответствующие
исполнительные механизмы, которые закрывают клапан 7 и останавливают на-
гнетатель 2, нагнетающий в систему воздух.
По описанной схеме производится автоматическая остановка цеха на дли-
тельное время. В большинстве случаев остановки цеха, вызываемые неисправ-
ностью отдельных аппаратов и узлов, непродолжительны. При этом по аварий-
ному сигналу цех полностью останавливается без продувки контактного аппа-
рата с одновременным включением реле времени. Если авария ликвидирована
до истечения предельно допустимого срока, по окончании которого температура в
контактном аппарате становится ниже температуры зажигания контактной мас-
сы, приводится в действие нагнетатель н возобновляется подача сернистого газа
232
с одновременным пуском других аппаратов технологической схемы. Если же про-
должительность остановки превышает прёдельно допустимое время, реле времени
включает исполнительные механизмы, с помощью которых приводится в действие
вентилятор и остальные аппараты в таком же порядке, как при продувке кон-
тэктного аппарата.
6. ПРОЕКТИРОВАНИЕ ХИМИКО-ТЕХНОЛОГИЧЕСКОГО
ПРОЦЕССА
Проект всякого производства разрабатывается генеральной
проектной организацией, которой обычно является отраслевой про-
ектный институт. Генеральный проектировщик привлекает многие
субподрядные проектные организации, которые разрабатывают
проекты специфических узлов и аппаратов технологического про-
цесса.
Например, общий проект производства серной кислоты разра-
батывает Государственный проектный институт химической про-
мышленности (ГИПРОхим), он является генеральным проекти-
ровщиком, а разработку электрофильтров — специфического обору-
дования— ведет специализированная проектная организация
(ГИПРОгазоочистка), являющаяся субподрядной проектной ор-
ганизацией. Разработку проектов теплообменников, контактных ап-
паратов и другой сложной аппаратуры ведут другие субподрядные
организации, обычно это проектные институты химического ма-
шиностроения.
Проектирование каждой физической и химической операции, а
также всего производства в целом производится на основе много-
численных параметров, выдаваемых проектной организации соот-
ветствующими научно-исследовательскими институтами и ведом-
ствами (министерствами).
Примерными параметрами проектирования могут быть следую-
щие:
1) место строительства цеха;
2) мощность производства и ассортимент выпускаемой продук-
ции;
3) вид и качество используемого сырья;
4) химическая и технологическая схемы процессов;
5) показатели технологического режима.
В зависимости от характера химического производства и от
того, относится ли разрабатываемая проектная документация к от-
дельной операции либо ко всему производству в целом, некоторые
из перечисленных параметров могут отсутствовать; в отдельных
случаях, наоборот, приведенный перечень может быть расширен.
Организация химического производства включает в себя четыре
этапа:
1) разработку задания на проектирование;
2) составление проекта;
3) строительство цеха;
4) эксплуатацию цеха.
233
Инженер-химик-технолог принимает непосредственное участие
в первом, втором и четвертом этапах. Однако в связи с тем, что в
настоящее время развитие техники идет по пути внедрения автома-
тических методов регулирования технологических процессов и
создания цехов- и заводов-автоматов, число инженеров-химиков,
занятых вопросами эксплуатации на таких автоматизированных
предприятиях, будет невелико. Следовательно, основная деятель-
ность инженера-химика будет связана с разработкой технологиче-
ского процесса и его оформлением, т. е. с разработкой задания на
проектирование и составление проекта.
Обычно проектирование осуществляется в две стадии: разра-
ботка проектного задания и изготовление рабочих чертежей.
Пояснительная записка содержит основные данные, необходи-
мые для разработки проекта, а также обоснования этих принятых
данных. Так, например, в пояснительной записке приводятся обос-
нования выбранного места строительства цеха, данные об источ-
никах сырья, электроэнергии, а также расчеты основных процессов
и аппаратов и многие другие сведения, положенные в основу раз-
рабатываемого проекта. К пояснительной записке прилагаются
также чертежи наиболее важных аппаратов; в чертежах приво-
дится спецификация, в которой указываются размеры, масса и ма-
териал отдельных деталей.
Важной составной частью проектного задания является техно-
логическая схема; она подробно раскрывает сущность химико-тех-
нологического процесса и достаточно точно отражает оформление
отдельных аппаратов и узлов и всего процесса в целом. Так, в тех-
нологической схеме приводится краткая характеристика оборудо-
вания и его габариты, это оборудование должно быть показано
в виде чертежей с изображением его внутреннего устройства. На
чертежи наносят вводы и выводы, т. е. участки трубопроводов, со-
единяющие оборудование с внешними коммуникациями, все газо-
вые и жидкостные коллекторы, а также основные датчики и ис-
полнительные механизмы контрольно-измерительных приборов
(КИП).
При выполнении технологической схемы все аппараты показы-
вают в масштабе (обычно 1 : 100), основные технологические по-
токи выделяют жирными линиями, газовые коллекторы размещают
в верхней части схемы, а жидкостные — в нижней части схемы.
Подготовленное проектное задание, состоящее из технологиче-
ской записки и технологической схемы, направляется заказчику,
который вносит отдельные изменения и дополнения и утверждает
его. Проектное задание, утвержденное заказчиком, служит основа-
нием для разработки второй завершающей стадии проектирова-
ния— разработки рабочих чертежей.
Рабочие чертежи и другие проектные документы, которые вы-
полняются на второй стадии проектирования, поступают непосред-
ственно на строительную площадку для осуществления строитель-
ства.
234
Иногда проектирование ведут в три стадии. В этом случае
второй стадией является технический проект, который составляют
на основе утвержденного проектного задания. Разработка черте-
жей будет третьей стадией проектирования.
Глава XI
ПРОИЗВОДСТВО СЕРНОЙ кислоты
1. ОБЩИЕ СВЕДЕНИЯ
Серная кислота принадлежит к числу сильных кислот и яв-
ляется самой дешевой из них (она более чем в 2 раза дешевле
азотной и соляной). Серная кислота удобна для использования,
она не дымит, не имеет запаха, при комнатной температуре нахо-
дится в жидком состоянии и в концентрированном виде Не разру-
шает черные металлы. Этими достоинствами объясняется широкое
распространение серной кислоты. Свыше 1400 промышленных уста-
новок во всем мире вырабатывают этот ценнейший продукт хими-
ческой промышленности. Мировое производство серной кислоты
превышает 100 млн. т в год; это больше, чем вырабатывается азот-
ной, соляной, уксусной и других кислот вместе взятых.
Подобно хлебу в нашем пищевом рационе, серная кислота яв-
ляется самым необходимым и самым дешевым химическим про-
дуктом, поэтому ее часто называют «хлебом» химии.
По количеству серной кислоты, вырабатываемой на душу насе-
ления, судят о степени химизации страны.
В СССР производство серной кислоты характеризуется следую-
щими данными:
Годы.......... 1950 1960 1970 1976 1980 (план)
Млн. т- год-1 . » 2,1 5,4 12,1 20.0 35-37
По масштабам производства серной кислоты Россия занимала
в 1913 г. 13 место в мире, а ныне СССР занимает второе месте?}
наша страна опередила все страны мира, за исключением США.
Так, например, по масштабам производства (в млн. т) серной кис-
лоты в 1975 г. места распределяются следующим образом: США —
29,5; СССР—18,6; Япония —7,1, ФРГ —5,1.
Серная кислота применяется в производстве самых разнообраз-
ных веществ: минеральных солей и кислот, всевозможных органи-
ческих соединений, красителей, дымообразующих и взрывчатых
веществ и т. д. Трудно назвать какое-либо производство, в котором
не принимала бы участие серная кислота пря*мо или косвенно. Осо-
бенно большое количество серной кислоты потребляется в произ-
водстве минеральных удобрений (около 40%)',
Безводная 100%-ная серная кислота (моногидрат) представ-
ляет собой тяжелую, маслянистую и бесцветную жидкость, которая
235
смешивается с водой во всех соотношениях с выделением большого
количества тепла. Плотность H2SO4 при 20 °C равна 1,83 г/см3,
темп, кипения 286 °C, темп, замерзания 10,5 °C.
Серную кислоту, в которой растворено избыточное количество
серного ангидрида SO3, называют олеумом FUSCU-nSOs.
Температура кристаллизации серной кислоты зависит от ее кон-
центрации (рис. XI. 1). Из рисунка видно, что кривая, характери-
зующая зависимость температуры кристаллизации серной кислоты
Рнс. XI.1. Диаграмма кристаллизации системы Н2О—SO3.
от ее концентрации, имеет максимумы, соответствующие опреде-
ленным соединениям серной кислоты и воды. Аналогичная кривая
имеется и для системы T^SOv^SCh-
Чтобы уменьшить возможность кристаллизации серной кислоты
при перевозке и хранении, товарные сорта серной кислоты должны
иметь такую концентрацию, которая соответствует достаточно низ-
кой температуре ее кристаллизации (см. минимумы на кривой
рис. XL 1):
' °h H2SO4 Т. крист., °C
Башенная кислота ... 75 —41
Контактная кислота . . 92,5 —35
Аналогично выбирают товарные сорта олеума:
% SO3 (своб) Т. крист., °C
Олеум........................ 20 —11
Высокопроцентный олеум . . 65 —0,35
Для инженера-химика-технолога эти данные имеют большое
практическое значение, так как во многих процессах, а также при
производстве самой серной кислоты применяется кислота различ-
ных сортов. Поскольку основное оборудование цехов в больший»
236
стве случаев размещается на открытой площадке (вне здания),
имеется опасность кристаллизации серной кислоты.
Температура кипения серной кислоты также зависит от ее кон-
центрации. При этом состав пара жидкой и паровой фаз неодина-
ковый (рис. XI. 2). Из рисунка видно, что смесь, соответствующая
98,3%-ной H2SO4, является азеотропной, состав ее жидкой и паро-
вой фаз одинаковый. При небольших отклонениях концентрации
Рис. XI.2. Фазовое равновесие паров с жидкостью для системы
Н2О—SO3 при атмосферном давлении.
H2SO4 от азеотропной точки состав ее жидкой и газовой фаз силь-
но отличается. Так, температура кипения серной кислоты концен-
трацией менее 78% ниже 200°C, при этом в паровую фазу перехо-
дит почти исключительно вода. Лишь при увеличении концентра-
ции кислоты содержание ее в парах возрастает. Например, над
кислотой 94% H2SO4 состав пара соответствует кислоте, концен-
трация КОТОрОЙ ОКОЛО 30% H2SO4.
Пары серной кислоты при повышении температуры диссоции-
руют H2SO4 Н2О + SO3 и при температурах выше 400°C основ-
ное количество H2SO4 находится в диссоциированном состоянии.
Дальнейшее нагревание вызывает диссоциацию SO3 по уравнению
2SO3 2SO2 + О2. .
При температуре выше 1000 °C серный ангидрид диссоциирует
почти полностью. Степень диссоциации меняется при изменении
давления.
Техническая серная кислота слегка окрашена примесями в тем-
ный цвет вследствие обугливания органических веществ, которые
попадают в нее с пылью.
237
2. МЕТОДЫ ПОЛУЧЕНИЯ СЕРНОЙ КИСЛОТЫ
Исходным веществом для производства серной кислоты служит
сернистый ангидрид SO2, который получают обжигом различного
серосодержащего сырья. Переработка сернистого ангидрида в сер-
ную кислоту заключается в его окислении и присоединении воды:
SO2 4- У2О2 “ SO3
SO3 4" Н2О ?= H2SO4
Скорость взаимодействия сернистого ангидрида с кислородом
в обычных условиях очень мала. Поэтому в промышленности эту
реакцию проводят либо в присутствии катализатора—контактный
метод производства серной кислоты, — или же в качестве окисли-
теля (передатчика кислорода) применяют нитрозу — нитрозный
метод производства серной кислоты.
Основным сырьем для получения сернистого ангидрида и, сле-
довательно, серной кислоты является флотационный колчедан, со-
держащий пирит FeS2, элементарная сера и отходящие газы цвет-
ной металлургии, содержащие SO2. Из флотационного колчедана
в СССР получают 45% серной кислоты, из серы — 25%, из отходя-
щих газов — 25% и из разного сырья — 5%.
На практике для окисления сернистого ангидрида используют
контактный метод, по этому методу в СССР получают 85% всей
серной кислоты.
3. ПРОИЗВОДСТВО СЕРНОЙ кислоты КОНТАКТНЫМ МЕТОДОМ
ИЗ ФЛОТАЦИОННОГО КОЛЧЕДАНА
Химическая и принципиальная схемы производства
Химическая схема включает три основных химических процесса:
а) окисление пирита кислородом воздуха, т. е. получение сер-
нистого ангидрида
4FeS2 4-ПО2 = 2Fe2O3 4-8SO2 4-Q (XI. 1)
б) окисление сернистого ангидрида в серный ангидрид на ката-
лизаторе
SO24-‘/2O2 SO34-Q (XI. 2)
в) соединение серного ангидрида с водой и образование серной
кислоты абсорбцией серного ангидрида
SO34-H2O —> H2SO4 4- Q (XI. 3)
Принципиальная схема производства серной кислоты из флота-
ционного колчедана является схемой с открытой цепью, т. е. про-
точной, когдагаз последовательно проходит все аппараты; схема
включает 7 операций (рис. XI. 3).
Операция 7 — обжиг сырья, в процессе обжига содержащийся
в флотационном колчедане пирит вступает во взаимодействие с кис-
лородом воздуха по реакции (XI. 1). В результате образуется сер-
238
нистый ангидрид, содержащий 12—15% SO2, и огарок Ре20з- Сер-
нистый ангидрид охлаждают с использованием тепла для получения
пара (операция 2), а затем освобождают от пыли (операция 3) и
подвергают специальной очистке (операция 4 — газ охлаждают,
промывают, сушат). Очищенный сернистый ангидрид нагревают
теплом отходящих газов (операция 5) и в присутствии катализа-
тора окисляют до серного ангидрида по реакции (XI. 2) (опера-
ция 6). После катализатора газ охлаждают (операция 5) и напра-
вляют на выделение серного ангидрида абсорбцией 98,3%-ной сер-
ной кислотой (операция 7), при этом серный ангидрид соединяется
Рис. XI.3. Принципиальная схема производства серной кислоты кон-
тактным методом:
/—обжиг флотационного колчедана и получение обжигового газа; 2—охлажде-
ние газа в котле-утилизаторе; <3 —очистка газа от пыли; 4—промывка и осушка
газа; 5—подогрев газа: 6 — окисление, сернистого ангидрида в серный на ката-
лизаторе; 7 — абсорбция серного ангидрида и образование серной кислоты.
с водой (избыток ее имеется в серной кислоте) по реакции (XI. 3).
Образующуюся серную кислоту выводят из процесса в качестве
готового продукта.
Физико-химические основы и технологические
схемы отдельных стадий производства
Из принципиальной схемы видно, что производство серной кис-
лоты контактным методом из флотационного колчедана можно
разделить на четыре основные стадии:.
получение сернистого ангидрида
очистка газа от примесей
окисление сернистого ангидрида на катализаторе
абсорбция серного ангидрида.
В такой последовательности будут рассмотрены физико-хими-
ческие основы каждой из стадий.
Получение сернистого ангидрида. Флотационный колчедан со-
держит кроме пирита сульфиды других металлов, небольшое
количество мышьяка, селена и другие примеси.
239
При нагревании флотационного колчедана до 500 °C происходит
диссоциация пирита на сульфид железа (сернистое железо) и серу
2FeS2 —► 2FeS + S2 (XI. 4)
При этом сера быстро сгорает в газовой фазе
S2 + 2О2 —► 2SO2 (XI. 5)
При дальнейшем повышении температуры сульфид железа окис-
ляется по уравнению
4FeS + 7О2 —► 2Fe2O3 + 4SO2 (XI. 6)
При обжиге флотационного колчедана одновременно с окисле-
нием пирита происходит также окисление содержащихся в нем
сульфидов других металлов, а мышьяк и селен образуют газооб-
разные окислы (АэгОз и БеОг), попадающие в состав обжигового
газа; в состав этого газа входит также влага колчедана. Окислы
железа, неокисленное сернистое железо, а также инертные примеси
флотационного колчедана составляют огарок. В огарке остается от
0,5 до 2% серы.
Для обжига флотационного колчедана применяются печи раз-
личных конструкций: механические печи, печи пылевидного обжига
и печи со взвешенным (кипящим) слоем флотационного колчедана
(см. главу VIII).
В механических печах пиритный концентрат находится на не-
скольких подах и сгорает по мере перемещения его гребками с од-
ного пода на другой (рис. VIII.4,а). В печах пылевидного обжига
частицы пиритного концентрата сгорают во время падения в полой
камере. В печах обжига в кипящем слое частицы пиритного кон-
центрата поддерживаются во взвешенном (псевдоожиженном) со-
стоянии поступающим снизу воздухом и сгорают при интенсивном
перемешивании.
Механические полочные печи и печи пылевидного обжига мало-
интенсивны, поэтому в новых сернокислотных цехах устанавливают
главным образом печи кипящего слоя (КС). В связи с этим ниже
приведены данные об обжиге флотационного колчедана в печах
КС.
Обжиг пирита — гетерогенный процесс, скорость которого вы-
ражается уравнением (III. 1)
г = KF ДС
где г — скорость процесса;
•X — коэффициент скорости реакции;
F — поверхность соприкосновения фаз;
ДС — движущая сила процесса.
Процесс обжига флотационного колчедана может быть описан
с достаточной степенью точности с помощью модели частицы с не-
взаимодействующим ядром (глава III). В зависимости от условий
240
обжига каждая из стадий, составляющих эту модель (кинетика,
внешняя и внутренняя диффузии), могут лимитировать процесс.
В соответствии с установленными ранее зависимостями процесс
обжига ускоряется при повышении температуры, увеличении кон-
центрации кислорода, уменьшении размера частиц и увеличении
интенсивности перемешивания флотационного колчедана с воз-
духом.
Скорость процесса обжига пирита FeS2 определяется скоростью
окисления односернистого железа FeS по реакции (XI. 6), так как
разложение пирита по реакции (XI. 4) и окисление серы по реак-
ции (XI. 5) протекают со значительно большей скоростью, чем
Рис. XI.4. Зависимость константы
скорости реакции (1g k) от темпе-
ратуры (1/Т) для процесса горения
одиосериистого железа в кипящем
слое (степень выгорания 50%);
АВ—кинетическая область; БВ— пере-
ходная область; ВГ — диффузионная
область.
Рис. XI.5. Схема печного отделения про-
изводства серной кислоты:
1 — нагнетатель; 2—печь КС; 3—котел-утнлиза-
тор; 4 — циклон; 5 —сухой электрофильтр.
окисление FeS. Поэтому влияние различных факторов на скорость
горения пирита исследуют на реакции окисления FeS. Результаты
исследований, проведенных в условиях, близких к производствен-
ным, показывают, что при низкой температуре- процесс обжига
пирита в кипящем слое протекает в кинетической области, а при
высокой температуре в диффузионной области (рис;Х1.4).
Из рис. XI. 4 следует, что энергия активации процесса в соот-
ветствии с уравнением Аррениуса (III. 24) составляет: в кинетиче-
ской области Ек — 295, в переходной области Еп — 152, а в диффу-
зионной области Ел = 17,5 кДж-моль-1.
Процесс обжига флотационного колчедана в печах КС ведут
при температуре около 800°C (I/Т-104 = 9,5), поэтому, в соответ-
ствии с данными рис. XI.4, процесс протекает в диффузионной
области. Следовательно, для интенсификации процесса целесооб-
разно повышать скорость газового потока в кипящем слое флота-
ционного колчедана. Однако возможности для использования этого
мощного фактора интенсификации ограничены тем, что в печи КС
поддерживается такая скорость газа, при которой частицы нахо-
дятся во взвешенном состоянии. Увеличение скорости потока при-
водит к выносу частиц из печи, поэтому интенсивность печи КС
зависит также от размера частиц флотационного колчедана: чем
крупнее частицы, тем большую скорость газового потока можно
допустить в печи и тем интенсивнее будет процесс обжига. Однако
при увеличении размера частиц уменьшается их общая поверхность
(в единице объема), что снижает интенсивность процесса. Поэтому
при расчете оптимальных условий процесса обжига флотационного
колчедана следует учитывать его дисперсный состав.
Благодаря интенсивному перемешиванию частиц колчедана
воздухом процесс окисления пирита протекает в печи КС
(рис. XI. 5) с большой скоростью. Для снижения температуры
в кипящем слое размещают охлаждающие элементы, в которые
либо подается вода и при этом образуется пар, либо подается пар
и образуется перегретый пар. Коэффициент теплоотдачи от колче-
дана к поверхности охлаждающих элементов велик и составляет
800—900 кДж-м~2-ч-1-К-1, что объясняется интенсивным переме-
шиванием колчедана.
Состав газа, получаемого при обжиге флотационного колчедана
в печах КС, зависит от состава сырья и режима работы печи:
Компоненты Содержание, % (об.)
so2 13,8
so3 0,1
02 2,0
n2 79,0
н2о 5.1
Всего 100,00
Содержание As, Se и F в газе колеблется в широких пределах
в зависимости от содержания их в сырье и режима обжига, в сред-
нем оно составляет (в мг-м~3): As — 50, Se — 25 и F—15.
По выходе из печи КС в газе содержится до 300 г-м~3 пыли.
Наличие пыли в газе, поступающем на катализатор, недопустимо,
так как пыль засоряет аппаратуру (повышается гидравлическое
сопротивление), загрязняет кислоту и снижает активность катали-
затора. Очистка газа от пыли осуществляется в два этапа.
После котла-утилизатора газ поступает в циклон, где осаж-
дается основная часть пыли (содержание пыли в газе снижается
до 10—20 г-м~3). Из циклона газ направляют в сухой электро-
фильтр, состоящий из четыр'ех последовательных секций (ступе-
ней). В сухом электрофильтре достигается высокая степень очи-
стки газа — в нем остается пыли 0,05—0,1 г-м-3.
Очистка обжигового газа. После выделения пыли обжиговый
газ подвергается специальной очистке для удаления примесей, при-
сутствие которых недопустимо в газе, поступающем на катализа-
тор, так как они снижают его активность — это остатки пыли,
мышьяк, фтор и др. Одновременно из газа извлекают и такие цен-
ные примеси, как селен, теллури др.
242
образует пары серной кислоты, ко-
Газ <4?“+
печного
отделения
^>/йз 6
контентное
отделение
Такая очистка газа проводится в башнях с насадкой и в полых
башнях (рис. XI. 6), орошаемых серной кислотой различной кон-
центрации.
Обжиговый газ направляется в первую промывную полую баш-
ню 1, в верхней части которой разбрызгивается 50%-ная серная
кислота; затем газ проходит вторую промывную башню с насад-
кой, орошаемой 10—20 %-ной серной кислотой. В результате сопри-
косновения газа с кислотой он охлаждается, при этом из него вы-
деляются остатки пыли, соединения мышьяка и селена, а также
серный ангидрид и некоторые другие примеси. Однако при охлаж-'
дении часть серного ангидрида
торые конденсируются в объе-
ме в виде мельчайших капелек
кислоты (туман). Для выделе-
ния этого тумана газ направ-
ляют в мокрые электрофильт-
ры (см. рис. XI. 5), а затем в
сушильные башни для выделе-
ния влаги.
Во второй промывной баш-
не газ практически полностью
насыщается парами воды, по-
этому содержание водяных па-
ров в газе, поступающем в су-
шильные башни, велико и оно тем больше, чем выше температура
газа во второй промывной башне и ниже концентрация кислоты,
орошающей эту башню.
Осушка газа производится в насадочной (скрубберной) башне
'(см. рис. XI.6), где пары воды абсорбируются 93—95%-ной серной
кислотой, орошающей эти башни. Содержание влаги в газе, выхо-
дящем из сушильных башен, не должно превышать 0,01% (об.)’
,(0,08 г-м-3).
Процесс осушки газа рассчитывают по уравнению, которое для
практических расчетов имеет вид
Рис. XI.6. Схема очистного отделения:
7, 2—первая и вторая промывные башни;
5—мокрый электрофильтр; 4—сушильная баш-
ня; 5—холодильники кислоты.
Q = KF кр
(XI. 7)
где Q — масса паров воды, поглощаемых из газа (скорость процесса) j
К — коэффициент массопередачи;
F—поверхность соприкосновения фаз (поверхность насадки);
Др — среднелогарифмическая разность давлений паров воды в начале и 3
конце процесса (движущая сила процесса).
Коэффициент массопередачи определяют по уравнению
К = Ко®т
Где w — фиктивная скорость газа в башне (без учета в ней насадки));
Ко — константа, численно равная К при w — 1;
т — коэффициент, равный 0,5 при ламинарном потоке и 0,8 при турбулент-
V. ном потоке, --
243
При 50 °C константа Ко имеет следующие значения:
Концентрация H2SO4, % 80
Константа Ко • 104 при
давлении
в Па................. 1,9
в мм рт. ст........ 250
85 90 93 95 97 98
2,2 2,5 2,6 2,8 2,9 3,0
300 330 350 370 390 400
Окисление сернистого ангидрида на катализаторе. Окисление
сернистого ангидрида проводится в присутствии ванадиевого ката-
лизатора.
Основные характеристики реакции окисления описываются сле-
дующими уравнениями:
тепловой эффект в интервале 400—700 °C (в кДж-моль-1)
Q = 101 420 — 9,6Г (XI. 8}
константа равновесия
где значения уравнению: °о,5. (XI. 9) PsOa^SOa Кр в интервале 390—650 °C можно определить по Кр = y + В = - 4,6455 (XI. 10)
равновесная степень превращения
г=.. pso3 * (Х1 щ
Pso2 + Ро2
где Pso2> Pso3> Ро2 — равновесные парциальные давления SO2, SO3, О2.
Из уравнения (XI. 9) следует
Pso2 = 1
Pso3 Kp-yjp*Oi
Подставив это значение в уравнение (XI. 11), находим
Для окисления SO2 применяются два типа ванадиевой контакт-
ной массы (катализатора): БАВ и СВД. В состав контактной мас-
сы БАВ входит барий, алюминий, ванадий, в состав контактной
массы СВД входит сульфованадат на диатолите. Контактную мас:
су формуют, придавая ей вид таблеток, гранул и т. д-
(рис. XI. 7).
244
В соответствии с ГОСТ каталитическая активность массы БАВ
в стандартных условиях (485 °C и объемная скорость S = 4000 ч-1)
должна обеспечивать степень превращения не менее 86%.
Средний диаметр гранул массы БАВ равен 5 мм, длина 7—
15 мм. Высокими показателями обладают новые отечественные ка-
тализаторы ИК-1 (ПК означает Институт катализа), ИК-2, ИК-3-
и СВС (сульфованадат на селикагеле).
Рис. XI.7. Формы контаитной массы:
а—таблетки; б, в—гранулы: в—кольца; д—гранулы для кипящего слоя.
Скорость реакции окисления SO2 в SO3 на ванадиевом катали-
заторе (в неподвижном слое) определяется уравнением Борескова
г ± (ь _ 4) ™ (XL 13>
t/т а \ л / \ 2/1
где X — степень превращения, доли единицы;
т — время контакта, с;
k — константа скорости реакции, с-1 • атм'1;
а — начальная концентрация сернистого ангидрида, доли единицы;
Ь — начальная концентрация Оз, доли единицы.
Имеются и другие, более точные, но сложные уравнения.
Константа скорости реакции выражается уравнением Аррениу-
са. На рис. XI. 8 зависимость константы скорости от температуры
на ванадиевом катализаторе характеризуется тремя отрезками
прямых, Изломы которых соответствуют температурам 460 и 530 °C,,
что указывает на изменения энергии активации Е в уравнении Ар-
рениуса. Эта энергия равна при температуре менее 460 °C
200 кДж-моль-1, в интервале 460—530 °C — 60 кДж-моль"1 и при
температуре более 530 °C — нулю.
Ранее было показано, что для простой обратимой экзотермиче-
ской реакции, например реакции -окисления SO2, зависимость X от
Г выражается кривой, имеющей максимум. Для времени контакта
< т2 < тз < Т4 зависимость X — f(T, т) может быть выражена
серией кривых (рис. II. 10). Кривая АВ, соединяющая максимумы,
245-
является линией оптимальной температурной последовательности
(ЛОТ); она определяет оптимальные условия ведения процесса;
его следует начинать при высокой температуре, когда обеспечи-
вается большая скорость процесса, а затем, с целью достижения
высокой степени превращения, снижать температуру, ведя процесс
по ЛОТ.
Однако достижение высокой температуры в начале процесса
-связано с большими затратами энергии на подогрев газовой смеси,
поступающей на катализатор. Поэтому на практике поступают сле-
дующим образом. Температуру газа на входе в первый слой кон-
Рис. XI.8. Зависимость кон-
тактной скорости окисления
SO2 (1g k) на ванадиевой кон-
тактной массе БАВ и СВД от
температуры (1/Г).
Рис. XI.9. Диаграмма t — X при
четырехстадийном контактировании
с промежуточным теплообменом:
1—4— адиабаты; а—а—линии охлаждения
газа.
тактной массы поддерживают равной 440 °C (несколько выше тем-
пературы зажигания, которая равна 420°C). За счет тепла реакции
температура повышается по адиабате 1 [в соответствии с уравне-
нием (VII. 19)].
В дальнейшем процесс следует вести по ЛОТ, однако поддер- '
жание оптимального режима (в строгом соответствии с ЛОТ) свя-
.зано с большими трудностями, поэтому создают режим, прибли-
жающийся к оптимальному. На первом слое процесс ведут до тех
пор, пока адиабата 1 достигает кривой 0,8 rmax, спрайа от ЛОТ
(рис. XI. 9). Затем газ направляют в теплообменник (охлаждаю-
щий элемент), где происходит охлаждение газа (линия а) до тех
пор, пока линия а не достигнет кривой 0,8rmax (слева от ЛОТ).
После теплообменника газ направляют на второй слой контактной
массы и ведут процесс по адиабате 2. Затем газ снова охлаждают
и ведут процесс до тех пор, пока не будет достигнута заданная
степень превращения X.
При осуществлении процесса в неподвижном слое катализатора
применяют контактные аппараты, в которых газ охлаждается в
теплообменниках1 ^монтированных внутри контактного аппарата
246
г(см. рис. VIII. 4, в, г)', в выносных теплообменниках (рис. VIII. 4, 5)
или с помощью поддува холодного газа (рис. VIII. 4, е).
Контактные аппараты с выносными теплообменниками просты,,
удобны для регулирования процесса и позволяют использовать,
тепло реакции, поэтому в последние годы контактные отделения
сернокислотных установок оборудуются преимущественно такими
контактными аппаратами (рис. XI. 10). Число слоев катализатора
в аппаратах с промежуточным теплообменом от трех до пяти.
В четырехслойном контактном аппарате с промежуточным теплооб-
меном, который наиболее часто применяется в промышленности,,
фактическая степень превращения составляет 0,97—0,98.
Рис. ХЕЮ. Схема контактного отде-
ления производства серной кислоты:
/—нагнетатель; 2—теплообменники; 3—кон-
тактный аппарат.
Рис. XI.11. Схема абсорбцион-
ного отделения производства
серной кислоты:
7 —абсорбер; 2— сборник; 3—холо-
дильник; 4—насос.
В контактном аппарате КС охлаждйющие элементы (теплооб-
менники) размещены в самом слое массы (см. рис. VIII. 5, б).
Сюда направляют исходную, более холодную газовую смесь, воду
или другой какой-либо хладоагент. Контактные аппараты КС.
только начинают применяться в производстве серной кислоты.
Контактное отделение любой сернокислотной системы обору-
дуется подогревателем, контактными аппаратами и теплообменни-
ками. В период пуска системы, когда тепло реакции окисления SO2
До SO3 еще не начало выделяться, газ нагревают до температуры
зажигания в подогревателе. При выходе контактного узла на обыч-
ный режим работы подогреватель отключают.
Абсорбция серного ангидрида. Последней стадией процесса про-
изводства серной кислоты контактным методом является извлече-
ние серного ангидрида из газовой с^еси и превращение его в сер-
ную кислоту. Этот процесс проводят в башне с насадкой. В ниж-
нюю часть башни направляется газовая смесь, а на верхнюю часть
насадки подается серная кислота, которая, стекая вниз, смачивает-
поверхность насадки (рис. XI. 11). При соприкосновении газовой
247
смеси с серной кислотой ангидрид абсорбируется ею, а затем вза-
имодействует с содержащейся в кислоте водой по реакции
_ mSO3 + Н2О = H2SO4 • (т—1)SO3
В зависимости от количественного соотношения воды и серного
ангидрида (значения т) получают серную кислоту различной кон-
центрации. При т — 1 образуется моногидрат (100%-ная серная
кислота); при образуется водный раствор кислоты, т. е.
разбавленная серная кислота; при т > 1 образуется олеум
H2SO4 • nSO3 (где п = т — 1).
д Туман
S°3|
SOa+^o-HzSOi^HaSQ*
’н2о
SO3
H2SO4
а
so3
4s
^H2SO4
Рчс=°
SO3
777777777777777777777/
с\\г5О^98,3%
^777777777777777
C^q>98,3%
Рн&~° Psos 1
^777/777777777777/7,
Ch2SO4=98,3 %
Psos0
Рис. XI.12. Схема абсорбции SO3 серной кислотой различной кон-
центрации;
I—поверхность серной кислоты; II—диффузионный поток молекул SO3;
^НгО’ ^SC>3 н PH2S04-PaBHOBecHbie Давлення Н2О, SO3 и H2SO4 над серной
кислотой.
После абсорбции SO3 газовая смесь вместе с непоглощенным
серным ангидридом удаляется в атмосферу. Естественно, что для
уменьшения потерь SO3 с отходящими газами, а также для защиты
атмосферы степень поглощения серного ангидрида в абсорбцион-
ном отделении должна быть возможно большая. Полнота абсорб-
ции зависит от концентрации применяемой для абсорбции кислоты.
Над серной кислотой концентрацией ниже 98,3% H2SO4 равновес-
ное давление SO3 ничтожное (Pso3~^)> а равновесное давление
паров воды значительное (Рщ0 > 0), поэтому с поверхности серной
кислоты происходит испарение (десорбция) молекул воды
(рис. XI. 12).
Основное количество молекул SO3, диффундирующих к поверх-
ности серной кислоты, абсорбируется ею, но часть молекул встре-
чается с молекулами воды, испаряющимися с поверхности серной
кислоты и диффундирующими в основной поток газа (см.
рис. XI. 12). Сталкиваясь, эти молекулы соединяются с образовав
248
г .
I нием паров серной кислоты, которые затем конденсируются в объ-
еме с образованием мельчайших капель (тумана) серной кислоты
SO3 (газ) + Н2О (газ) = H2SO4 (газ) = H2SO4 (туман)
Концентрация H2SO4, %
Рис. XI.13. Степень абсорбции
серного ангидрида в моногидрат-
ном абсорбере при различной тем-
пературе:
/ — при 60 °C; 2— при 80 °C; 3— при 100 ’С;
4 —при 120 °C.
Туман плохо улавливается в обычной абсорбционной аппара-
туре и в основном уносится с отходящими газами в атмосферу.
Чем ниже концентрация серной кислоты и выше ее температура,
тем больше выделяется из нее паров воды, больше образуется ту-
мана и больше теряется SO3.
Над кислотой концентрацией выше 98,3% H2SO4 равновесное
давление SO3 p*s01 > 0, поэтому SO3 абсорбируется серной кисло-
той неполностью (см. рис. XI. 12).
В этом случае отходящие газы так-
же уносят в атмосферу часть сер-
ного ангидрида. Таким образом, при
концентрации орошающей кислоты
менее и более 98,3% абсорбция
- серного ангидрида снижается; она
тем ниже, чем выше температура,
поскольку равновесные давления
паров Н2О и SO3 над кислотой воз-
растают с повышением темпера-
туры.
При концентрации кислоты
98,3% H2SO4 равновесные давления
паров воды и SO3 малы (Pso3
Р*н2о^^)’ н0 равновесное давление
паров самой серной кислоты значи-
тельное (Ph2so4 > 0), поэтому про-
исходит испарение (десорбция) па-
ров серной кислоты с ее поверхности. Однако при температуре
ниже 100 °C, при которой на практике ведут процесс абсорбции
SO3, равновесное давление паров серной кислоты очень мало
(p^2so4= 0),поэтому в производственных условиях 98,3%-ная сер-
ная кислота' обладает наиболее высокой абсорбционной способ-
ностью по отношению к SO3 (рис. XI. 13).
, Из приведенных данных следует, что если для абсорбции SO^
используют серную кислоту концентрацией 98,3% и выше, то про-
текает обычный процесс абсорбции. В этом случае процесс описы-
вается уравнением (III. 1). Если концентрация кислоты ниже
98,3%, то уравнение (III. 1) неприменимо, поскольку часть SO3
соединяется с парами воды и образует туман серной кислоты, не
задерживаемый обычной абсорбционной аппаратурой.
В сернокислотной промышленности принято называть кислоту,
орошающую абсорбционную башню, моногидратом, несмотря на
то, что содержание H2SO4 в ней менее 100%, а башню — моногид-
ратным абсорбером.
249
В тех случаях, когда требуется обеспечить выпуск олеума, пе-
ред моногидратным абсорбером устанавливают олеумный абсор-
бер с соответствующей вспомогательной аппаратурой (сборником,
холодильником, насосом). Олеумный абсорбер устроен примерно
так же, как и моногидратный абсорбер, но орошается олеумом,
содержащим 18,5—20% SO3 (изб). В олеумном абсорбере погло-
щается часть SO3 (40—60%), остальное количество абсорбируется
в моногидратном абсорбере. По мере повышения концентрации
•олеума его разбавляют моногидратом, а накапливающийся избы-
ток олеума отводят на склад.
Поскольку в настоящее время строят контактные сернокислот-
ные установки большой единичной мощности, количество SO2, вы-
Рис. XI.14. Технологическая схема производства серной кислоты из флотацион-
ного колчедана:
/, 10 —нагнетатели; 2 —печь КС; 3—котел-утилизатор; 4—циклон; 5—сухой электрофильтр;
6» 7—1-я и 2-я промывные башни; 8—мокрый электрофильтр; 0—сушильная башня; И—тепло-
•обменники; /2 —контактный аппарат; 13, /4—моногндратные абсорберы; /5—холодильники
кислоты.
брасываемого с отходящими газами, очень велико .даже при пяти-
слойном контактном аппарате, при X = 98%. Поэтому для сниже-
ния концентрации SO2 в отходящих газах их подвергают спе-
циальной химической очистке либо применяют двойное контак-
тирование.
В основе процесса двойного контактирования лежит прием, рас-
смотренный во II главе, когда увеличение выхода целевого про-
дукта (в данном случае SO3) достигается за счет вывода его из
зоны реакции. Таким образом, двойное контактирование включает
два этапа окисления SO2 на катализаторе. На первом этапе про-
цесса степень превращения составляет около 0,90. Перед вторым
этапом контактирования из газовой смеси выделяют серный анги-
дрид. В результате этого равновесие реакции сдвигается в сторону
образования SO3, т. е. повышается равновесная степень превраще-
ния, что соответственно приводит к увеличению фактической сте-
пени превращения. При двойном контактировании фактическая
степень превращения составляет 0,995—0,997.
Общая схема производства серной кислоты контактным мето-
дом из флотационного колчедана с осуществлением процесса окис-
.250
ления SO2 на основе двойного контактирования показана на
рис. XI. 14. Из нагнетателя 10 газ проходит теплообменники 11 и
поступает на первый, а затем на второй и третий слои контактной
массы аппарата 12. После третьего слоя газ подают в промежуточ-
ный моногидритный абсорбер 13, а затем в теплообменник и в чет-
вертый слой контактной массы. Охлажденный в теплообменнике
газ проходит абсорбер 14 и далее выводится в атмосферу.
Основные показатели работы современной установки по произ-
водству серной кислоты контактным методом из флотационного
колчедана на основе двойного контактирования:
Производительность единичного агрегата, т • сут-1 серной
кислоты (100% H2SO4)....................................1 000
Температура газа, °C
на выходе из печи КС.................................. 850
на входе
в сухой электрофильтр .............................. 400
в сушильную башню . . . . - •........................ 32
в 1-й слой контактной массы......................... 420
на входе во 2-й слой контактной массы.................. 4501
на входе в 3-й слой » » .................. 430
на входе в 4-й слой » » ................. 420-
на выходе из абсорбера.............................. 60
Концентрация SO2 на входе в 1-й слой контактной массы,
% (об.).................................................... 9
Степень превращения на выходе из
1-го слоя контактной массы ...................... 0,6
2-го слоя контактной массы..........................0,85
3-го слоя контактной массы (после первой стадии кон-
тактирования) ............. ......................0,90
на выходе из 4-го слоя..............................0,995
Разрежение перед нагнетателем
Па....................................................... 9 800
мм вод. ст........................................... 1 000
Давление после нагнетателя
Па...................................................... 22 500
мм вод. ст........................................... 2 300
Технико-экономические показатели
Показатели работы различных промышленных установок по
производству серной кислоты существенно различаются, они зави-
сят от качества используемого сырья, мощности установки, совер-
шенства как технологического процесса, так и его аппаратурного
оформления, а также от многих других факторов.
Основные технико-экономические показатели:
Степень превращения SO2 на катализаторе, %............. 96 —99,5
Общее использование серы, %' . ........................85—86
Потери серы, °/о.........................:............. 5—6
Расход электроэнергии, кВт-ч •т-1........................100—110
Расход воды, м3 • т-1..................................50—60
25J
Ниже приведены элементы себестоимости 1 т серной кислоты
(100% H2SO4) при работе на флотационном колчедане (по дан-
ным одного из заводов):
Статья расхода Количество Стоимость, руб- %
Колчедан (45% S), т 0,816 10,78) кт
Контактная масса, кг 0,1 0,11/
Электроэнергия, кВт • ч 100 1,46) 7,7
Вода, м3 50 0,34/
Зарплата — 0,44 2,3
Амортизационные отчисления — 3,17 16,6
Цеховые расходы — 2,45 12,9
Общезаводские расходы Цеховая себестоимость — 1,51 8,2
побочные продукты — 20,26
(огарок, пар, селен) — 1,86 100,0
Заводская себестоимость — 18,40
Непроизводительные расходы — 0,65
Полная себестоимость — 19,05
Из приведенных данных видно, что наибольшую долю расхо-
дов (57%) составляет сырье. Стоимость контактной массы неве-
лика и составляет менее 1%. Малы также затраты на зарплату.
Это объясняется тем, что все процессы в производстве серной кис-
лоты механизированы, а большинство из них автоматизировано.
Высокие амортизационные отчисления обусловлены сложно-
стью и дороговизной аппаратуры.
Продажная цена серной кислоты составляет 32 руб-т-1. Таким
образом, прибыль для завода производительностью 360 тыс. т. в
год составляет 360 000 (32,0— 19,05) = 4 660 000 руб. в год.
4. ПРОИЗВОДСТВО СЕРНОЙ кислоты -
КОНТАКТНЫМ МЕТОДОМ ИЗ СЕРЫ
Процесс получения серной кислоты контактным методом из
элементарной серы отличается от описанного процесса получения
серной кислоты из флотационного колчедана тем, что сжигание
серы производится в более простых печах и протекает легче, чем
сжигание флотационного колчедана. Кроме того, поступающая на
сернокислотные заводы элементарная сера представляет собой вы-
сококачественное сырье с малым содержанием примесей, сгораю-
щее практически полностью. Получаемый при этом сернистый ан-
гидрид содержит мало примесей, поэтому отпадает необходимость
его очистки от пыли. Такой газ без промывки направляют непо-
средственно в контактное отделение (рис. XI. 15). Окисление сер-
ного ангидрида на катализаторе и абсорбция серного ангидрида
при работе на сере осуществляются так же, как и при работе на
флотационном колчедане.
252
Удельный вес серы в общем балансе серосодержащего сырья
как в СССР, так и за рубежом беспрерывно возрастает. Например,
в настоящее время в СССР из серы получают 25% серной кислоты,
Сера вода Вода
продукт)
Пар^
(побочный, продукт)
Рис. XI.15. Принципиальная схема производства сериой кислоты кон-
тактным методом из серы:
1 — осушка воздуха; 2— сжигание серы; 3 — охлаждение газа с использованием
тепла; 4— окисление SO3 иа катализаторе; 5 —абсорбция SO3 и образование
серной кислоты.
а к 1980 г. эта доля, по-видимому, увеличится до 30—35%. В .связи
с этим во всем мире ведутся работы по усовершенствованию про-
цесса производства серной кислоты из серы.
Рис. XI.16. Энерготехнологическая схема производства серной кис-
лоты из серы:
1 — камера сжигания серы; 2— воздушный компрессор; 3—тепловая турбина;
4—генератор электроэнергии; 5—контактный аппарат; 6—пароперегреватели;
7 —конденсатор.
Одно из наиболее интересных направлений состоит в следую-
щем. Поскольку на производство серной кислоты поступает очень
чистая сера, а теплотворная способность серы достаточно высокая
и составляет около 10 000 кДж-кг-1, представляется возможным
рассматривать ее как топливо и сжигать в тепловой турбине (гене-
ратор электроэнергии), а отходящие газы направлять для получе-
ния серной кислоты (рис. XI.16). Такое производство называется
253
энерготехнологическим, так как одновременно с серной кислотой
получают товарную электроэнергию.
5. УСОВЕРШЕНСТВОВАНИЕ ПРОИЗВОДСТВА
СЕРНОЙ КИСЛОТЫ
Одним из наиболее эффективных мероприятий, обеспечивающих
значительное улучшение технико-экономических показателей про,
изводства серной кислоты, является повышение единичной мощно-
сти агрегатов. В настоящее время типовой является контактная
система производительностью 1000 т-сут-1. Однако в ближайшие
годы можно ожидать, что оптимальной будет контактная система
мощностью 2000—3000 т в сутки. Как было показано ранее (гла-
ва 1), увеличение единичной мощности в 2—3 раза позволит сни-
зить капитальные затраты на 25—30% и себестоимость продукции
на 15—20%, при этом производительность труда возрастет при-
мерно в два раза.
Вторым важным мероприятием является повышение давления
процесса. Реакция окисления SO3 протекает с уменьшением объема
SO2 + V2O2 ** SO3, поэтому при повышении давления равновесие
процесса будет сдвигаться вправо, т. е. в сторону образования SO3.
Первая установка в мире по производству серной кислоты из
элементарной серы под давлением 0,5 МПа производительностью
575 т-сут-1 введена в эксплуатацию во Франции в 1973 г., причем^
достигнуты хорошие показатели работы: расход контактной массы
в 3 раза меньше, чем на существующих установках, а степень
превращения SO2 в SO3 составляет 0,9985. Установка полностью
снабжает себя энергией и дополнительно выдает на сторону
0,75 т-т-1 энергетического пара.
Следующее важное направление в усовершенствовании про-
цесса производства серной кислоты состоит в замене воздуха, ис-
пользуемого в настоящее время для обжига сырья, кислородом
или воздухом, обогащенным кислородом. Активным компонентом
воздуха, в котором сжигается серосодержащее сырье, является
кислород, но его в воздухе только 21 %, а азота, являющегося бал-
ластом, 79%. Этот балласт приходится перекачивать через всю
аппаратуру, в результате снижается производительность установок
и увеличивается расход электроэнергии. Применение технического
кислорода или воздуха, обогащенного кислородом, позволяет резко
увеличить интенсивность процесса (примерно в 4—5 раз при за-
мене воздуха кислородом).
В настоящее время еще отсутствуют сернокислотные установ-
ки, работающие на кислороде, поскольку получение кислорода
связано с расходом большого количества дорогой электроэнергии,
поэтому применять кислород в производстве серной кислоты пока
невыгодно. Но в дальнейшем, по мере развития техники получения
кислорода, его стоимость будет снижаться. Тогда вопрос о замене
воздуха кислородом или воздухом, обогащенным кислородом, по-
лучит надежное экономическое обоснование,
254
Примеры
Пример XI. 1. Рассчитать скрубберную башню для осушки сернистого ангид-
рида в производстве серной кислоты контактным методом (сушильную башню),
т. е. определить диаметр башни и объем насадки.
Условия.
Производительность цеха Q, т-ч-1 H2SO4............ 10
Концентрация SO2 в газе а, доли................. 0,07
Концентрация кислоты, орошающей сушильную башню,
% H2SO4......................................... 95
Температура, °C
газа (на выходе из башни) t2 ............ 32
кислоты (на входе и выходе) /к................. 49
Давление паров воды в газе На входе На выходе
в башню из башни
Па.................... 4 746 10,1
мм рт. ст............. 35,6 0,076
Разрежение в системе Рр (до газодувки)
Па...................................................... 2 933
мм рт. ст............................................... 22
Барометрическое давление Р
Па................................................... 101325
мм рт. ст.............................................. 760
Скорость газа в насадке w, м-с-1............................ 0,8
Решение.
Поверхность насадки находим по уравнению (III. 1)
К ДР
Количество паров воды В (в кг • ч-1), поглощаемых из газа:
д _ ~рГ)^н2о
(Р - - Рр) 22,4
где V — объем сухого газа, приведенного к нормальным условиям, м3 • ч-1;
22,4 л/мель — мольный объем газа, приведенного к нормальным условиям.
Если принять степень превращения X = 0,98 (стр. 247), а полноту поглоще-
ния SO3 в абсорберах z = 0,99, то при а — 0,07
А -22,4 10000-22,4 3
МИ25О4аХг ~ 98,08 • 0,07 • 0,98 • 0,99 156 U м ’ 4
Подставляя в уравнение (а) известные и найденные значения входящих в
него величин, получаем
„ 33 640 (35,6-0,076) 18 1ОСП .
В = (7бб-Ш-&Ш,-4 1360 КГ• 4
Коэффициент скорости абсорбции паров воды определяем по уравнению
(III. 1). Для 95%-ной кислоты (стр. 244) при Ко — 0,037 и давлении, выражен-
ном в мм рт, ст.
К = 0,037 • 0,8°'8 = 0,031
255
Движущую силу процесса
/ гг
Pl — Pl
// /
Р2 Р2
определяем по уравнению:
р_ (Р\-Р")(Р''-Р2) .
ЛГ--------------7 - (б)
Pl — Pi
2,3 Ig
Pl -Р2
Равновесное давление паров воды р2 над 95%-ной серной кислотой при
40 °C составляет 0,002 мм рт. ст. (принимаемр'2 = р^). Подставив значения р( =:
= 35,6; р" = 0,076; р2 = р2 = 0,002 мм рт. ст. в уравнение (б), находим:
д _ (35,6 - 0,002) - (0,076 - 0,002) _ о
ДР =---------3576 - 0,002----------------= 5,8 мм рт. ст.
43 g 0,076 - 0,002
Таким образом, поверхность насадки составляет
Из опыта эксплуатации известно, что насадка башни обычно используется
не полностью, поэтому следует ввести коэффициент запаса, равный 1,2. Тогда
поверхность насадки в рассчитываемой сушильной башне должна быть равна
F = 7580 • 1,2 = 9100 м2
Если насадка имеет вид правильно уложенных колец размером 50 X 50 (по-
верхность 1 м3 такой насадки равна НО м2), то общий объем насадки составит:
Находим внутренний диаметр сушильной башни:
4ЁТ / 4 • 33 640 (273 + 40) 760
лw 3600 Д/ з, 14 0,8 • 3600 273 (760 — 22)
где Vt — объем газа при температуре 6 и барометрическом давлении Р.
Определяем высоту насадки:
„ 4У„ 4-82,8
Н =------;— =---------5- = 6,02 м
лР2вн 3,14-4,22
Глава XII
ПРОИЗВОДСТВО АММИАКА И АЗОТНОЙ КИСЛОТЫ
1. ПРОИЗВОДСТВО АММИАКА
Аммиак (NH3) является важнейшим химическим продуктом,
так как он служит исходным сырьем для получения самых разно-
образных азотсодержащих соединений. Источников связанного
азота, имеющих промышленное значение, в природе очень мало.
256
Область
взрывоопасрост
аммиачно-Возддш-
яой смеси над Водны-
ми растворами амни-
она
Пределы взрывоопасности
сухой аммиачно-воздди-
s' ной смеси
ДОежду тем азот имеет исключительно важное значение в промы-
шленности-и природе. Наиболее крупными потребителями связан-
ного азота (а, следовательно, и аммиака) является промышлен-
ность минеральных удобрений, азотной кислоты (на ее производ-
ство расходуется около 35% всего вырабатываемого в СССР
аммиака), производство промежуточных продуктов и красителей,
взрывчатых и горючих веществ, химических волокон, пластических
масс и многих других.
Азот играет особо важную 100
роль в природе; он участвует в 90
основных биохимических процес- So
сах и образует важнейшие пита-
тельные вещества для растений, 70
животных и человека. до
Достаточно крупные место- |j
рождения связанного азота в ви- § 50
де азотнокислого натрия (нитра-
та натрия или селитры) имеются
только в Чили, они были откры- 30
ты в начале XIX в. Небольшое %
количество связанного азота (в
виде сернокислого аммония) по-
лучают при переработке коксово- О
го газа. Основную же массу азот- 1
содержащих продуктов (около
95%) производят на основе ам- рис
мйака, искусственно получаемого а»
на химических предприятиях.
Производство синтетического аммиака в 1970 г. характери-
зуется следующими данными (в млн. т): весь мир — 49,2; Запад-
ная Европа— 14,7; США—14,6; Япония — 3,7.
Аммиак представляет собой бесцветный газ с характерным рез-
ким запахом. При охлаждении до —33 °C он сжижается, а при
—78 °C затвердевает, образуя бесцветную кристаллическую массу.
Критическая температура аммиака 132,4 °C, критическое давление
111,5 атм. Аммиак хорошо растворим в воде: при 20 °C и атмо-
сферном давлении в 1 л воды растворяется 700 л газообразного
аммиака. Несколько хуже он растворяется в органических жид-
костях.
При непосредственном соединении аммиака с кислотами полу-
чают соли, например нитрат или сульфат аммония. ГТрн взаимо-
действии аммиака с СО2 образуется карбамид (мочевина), кото-
рый является одним из лучших азотных удобрений благодаря
высокой концентрации азота н хорошим физическим свойствам; на
основе карбамида получают также разнообразные химические про-
дукты.
Водные растворы аммиака (аммиачная вода), водно-аммиачные
растворы солей и жидкий аммиак широко применяются в сельском
io 18 го гг гч гв
Содержание NH3, %
.1. Области взрывоопасности
иачно.-воздушной смеви.
9 Зак, 558
257
хозяйстве для непосредственного внесения в почву в качестве
жидких азотных удобрений.
Сухие аммиак и воздух образуют взрывоопасные смеси; пре-
делы взрываемости таких смесей при 18 °C ограничены интервалом
содержания аммиака от 15,5 до 27% (об.) (рис. XII. 1). Таким
образом, смеси, содержащие менее 15,5 и более 27% (об.) NH3,
при зажигании их искрой не взрываются. При повышении темпера-
туры пределы взрываемости аммиачно-воздушных смесей расши-
ряются. Учитывая эти особенности аммиачно-воздушных смесей,
при осуществлении промышленных процессов поддерживают со-
держание аммиака до 15,5 или выше 27%.
Химическая и принципиальная схемы производства
Аммиак получают прямым синтезом из составляющих его эле*
ментарных веществ; для этого смесь азота и водорода в соотноше-
нии N2: Н2 = 1:3 (азотоводородная смесь) пропускают через ка-
тализатор. Реакция синтеза аммиака
N2 + 3H2 3NH3-|-Q (XII. 1)
На первых этапах промышленного производства аммиака азо-
товодородную смесь готовили путем смешения получаемых от-
дельно азота и водорода. В последние годы азотоводородную смесь
получают из углеродосодержащих газов и атмосферного воздуха.
Таким образом, процесс синтеза аммиака состоит из двух стадийз
приготовления азотоводородной смеси и непосредственно синтеза
аммиака.
Исходя из этого, химическая схема производства аммиака
включает реакцию (XII. 1), а также ряд важнейших химических
реакций, сопровождающих получение азотоводородной смеси.
Основным сырьем в нашей стране для получения азотоводо-
родной смеси является природный газ, содержащий 90—98% ме-
тана. Процесс состоит в такой обработке смеси природного газа,
воздуха и водяного пара, в результате которой происходит конвер-
сия (взаимодействие) метана с водяным паром (пароводяная кон-
версия)
СН4 + Н2О qpnh ЗН2 + СО - <3 (XII. 2|
и конверсия метана кислородом воздуха (кислородная конверсия)
СН4 + '/2О2 2Н2 + GO + Q (XII. 3)
При взаимодействии образовавшейся газовой смеси с парами воды
происходит конверсия окиси углерода
С0 + Н20 H2+CO2+Q (XII. 4)
При этом процесс ведут так, чтобы в конечной газовой смеси
соотношение между азотом, поступающим с воздухом, и образую-
щимся водородом составляло 1 :3. После удаления из газовой
смеси двуокиси углерода получают .азотоводородную смесь, из ко-
торой в присутствии катализатора образуется аммиак.
258
Таким образом, химическую схему производства аммиака мож-
но представить уравнениями (XII. 2) —(XII. 4) и (XII. 1)
Промышленные способы производства аммиака различаются в
зависимости от давления, при котором проводят каталитическую
реакцию синтеза (XII. 1):
1) системы низкого давления (10—15 МПа);
2) системы среднего давления (25—60 МПа);
3) системы высокого давления (60—100 МПа).
В большинстве случаев процесс осуществляют в установках,
работающих при среднем давлении; при этом обеспечивается до-
статочно высокая скорость процесса синтеза аммиака и удачно
решается аппаратурное оформление процесса выделения аммиака.
Принципиальная схема производства аммиака, осуществляе-
мого при среднем давлении, включает семь основных операций
Пар воды . Воз дух Пар .воды
Природный
'газ (СНд)
Дзотоводородная
смесь
7 Аммиак
6
5
1
3
2
Конвертированный газ
Рис. XII.2. Принципиальная схема производства аммиака под средним давлением:
I—очистка природного газа от сернистых соединений; 2, 3—конверсия метана (первая и вто-
рая ступени); 4—конверсия окиси углерода; 5—очистка конвертированного газа; 6—синтез
аммиака; 7—выделение аммиака.
[(рис. XII. 2). Природный газ очищают от сернистых соединений,
поскольку они отравляют катализаторы и замедляют реакции, со-
ставляющие химическую схему производства. К очищенному газу
добавляют водяной пар, после чего парогазовую смесь направляют
иа конверсию метана 2; здесь процесс конверсии [реакция (XII.2)]
протекает частично; достаточно полно эта реакция осуществляется
на второй стадии 3, куда поступает реакционная смесь с добавляе-
мым к ней воздухом (рис. XII. 2).
Часть метана взаимодействует с кислородом воздуха [реакция
(XII. 3)]; при этом выделяющееся тепло обеспечивает необходимый
температурный режим на второй стадии <3. Кроме того, с воздухом
вводится азот, необходимый для получения азотоводородной смеси.
После конверсии метана к реакционной смеси добавляют водя-
ной пар и направляют ее в конвертор окиси углерода 4 [реакция
(XII. 4)]. Полученный конвертированный газ содержит значитель-
ное количество примесей, снижающих активность катализатора
синтеза аммиака, поэтому газ подвергают специальной очистке,
стадия 5. Полученную азотоводородную смесь направляют в реак-
тор синтеза аммиака 6. Степень превращения азота на катализа-
торе с образованием NH3 составляет всего лишь 15—20%, поэтому
после выделения аммиака (стадия 7) непрореагировавшую азото-
водородную смесь возвращают в процесс.
9*
259
Физико-химические основы производства
Рнс. ХП.З. Равновесный состав
парогазовой смеси при конвер-
сии метана парами воды под
атмосферным давлением:
7-НгО : СН4=2 : 1% (об.); 2-
НгО : СН4 = 1 : 1% (об.).
Конверсия метана водяным паром. Конверсию метана проводят
при атмосферном или повышенном давлении с применением ката,
лизатора (каталитическая конверсия метана) или без него (высо-
котемпературная конверсия метана).
Наиболее широкое распространение
получил первый способ, рассматривае-
мый ниже.
Для наиболее полного использова-
ния сырья (остаточное содержание
СН4 в азотоводородной смеси не долж-
но превышать 0,5%) процесс конвер-
сии метана по реакции (XII. 2) жела-
тельно вести с наибольшей степенью
превращения, определяемой условия-
ми равновесия
РсоРн»
Арse * * (XII. б)
РсН.РНаО
lgKp^-Л/Т + В (ХИ. 6)
Из приведенных уравнений видно,
что сдвиг равновесия в сторону полу-
чения целевого продукта (pHJ, соот-
ветствующий увеличению равновесной
и, следовательно, фактической степе-
ни превращения может быть достиг-
нут в результате повышения концен-
трации одного или обоих исходных ре-
агентов, снижения давления и увели-
чения температуры, поскольку при
этом возрастает величина Кр (так как
перед Л/Г отрицательный знак). На
практике повышают концентрацию
только паров воды — относительно де-
шевого продукта. При этом содержа-
ние воды характеризуется коэффици-
ентом т = рНго/Рсн4-
Из рис. XII. 3 видно, что при тем-
пературе 800—900 °C и т — 2 равно-
весное содержание СН4 составляет ме-
нее 0,5%, что соответствует минимальной допустимой концентра-
ции СН4 в газовой смеси, однако скорость реакции при этой темпе-
ратуре мала. При повышении температуры возрастает скорость
побочной реакции термического разложения метана с образова-
нием молекулярного углерода.
В настоящее время процесс ведут в присутствии катализатора,
который обеспечивает достаточно высокую скорость реакции при
2в0
/температуре 800—900 °C. Для увеличения скорости реакции повы-
шают также давление процесса, несмотря на то, что в этих усло-
виях несколько возрастает равновесное содержание СН4 в реак-
ционной смеси [реакция (XII. 2) сопровождается увеличением
объема газа]. Однако с увеличением давления повышается концен-
трация реагирующих веществ, причем увеличение скорости реак-
ции значительно опережает ее снижение от увеличения объема.
Кроме того, поскольку природный газ поступает под давлением
(1—2 МПа), достигается значительная экономия энергии, так как
для осуществления синтеза ам-
миака азотоводородную смесь
подвергают сжатию.
В качестве катализатора син-
теза аммиака применяют никель,
нанесенный на окись алюминия
или на окись магния.
Конверсия метана кислородом
[реакция (XII. 3)] протекает од-
новременно со второй стадией
процесса (см. рис. XII. 2, опера-
ция 3). Эта реакция протекает с
большой скоростью и не лимити-
рует процесс, поэтому она от-
дельно не рассматривается.
Конверсия окиси углерода.
После конверсии метана [реак-
ции (XII. 2) и (XII. 3)] получен-
ная газовая смесь содержит от
20 до 40% окиси углерода. Для
Рис. ХП.4. Зависимость равновесной
степени превращения окиси углерода
от объемного соотношения Н2О и СО
и температуры:
/—при 300 °C; 2—при 400 °C; 3—при 500 °C
превращения окиси углерода в двуокись углерода и получения до-
полнительного количества водорода конвертированный газ вместе
с водяным паром пропускают через катализатор, в присутствии ко-
торого осуществляется конверсия окиси углерода [реакция (XII. 4)].
Условия равновесия этой реакции, влияющие на значение равно-
весной степени превращения, определяются уравнениями
v _ PcOj/’h,
Лр = —--------
РсоРн2о
18 Кр e А/Т + В
(XII. 7)
(XII. 8)
Из этих уравнений следует, что сдвиг равновесия в сторону
образования водорода (целевого продукта) и, следовательно, уве-
личение его выхода может быть достигнуто повышением концен-
трации исходных реагентов и снижением температуры (при умень-
шении температуры отношение А/Т увеличивается и, следовательно,
возрастает Кр).
Давление не влияет на условия равновесия, поскольку реакция
XXII. 4) протекает без изменения объема.
291
В практических условиях увеличивают концентрацию паров
воды как относительно дешевого продукта. При этом содержание
воды будет определяться коэффициентом п = рН20/рсо.
Из рис. XII. 4 видно, что равновесная степень превращения СО
возрастает при увеличении п и снижается при повышении темпе-
ратуры. Однако понижать температуру, чтобы сдвинуть равновесие
в сторону образования целевого продукта, невыгодно, так как при
низких температурах реакция (XII. 4) протекает очень медленно
даже в присутствии катализаторов. В связи с этим устанавливают
оптимальную температурную последовательность, обеспечивающую
максимальную общую скорость процесса (см. рис. II. 7 и II. 8).
Процесс конверсии СО осуществляют в присутствии железо-
хромового или цинк-хромовомедного катализатора; последний по-
зволяет проводить процесс при сравнительно низкой температуре
(250—300 °C) и достижении остаточного содержания СО в преде-
лах 0,2—0,4%. Однако низкотемпературный катализатор чрезвы-
чайно чувствителен к серосодержащим примесям, поэтому при ра-
боте на таком катализаторе особенно большие требования предъ-
являют к степени очистки газа.
Очистка природного и конвертированного газов. Углеводород-
ные газы (природный, попутные нефтяные), а также промышлен-
ные газы нефтепереработки, газификации твердых и жидких топ-
лив, конвертированный и коксовый газы содержат вредные при-
меси, способные отравлять катализаторы, вызывать коррозию и-
загрязнять аппаратуру.
К основным примесям, содержание которых в конвертирован-
ном газе недопустимо, относятся серосодержащие вещества (H2S,
меркаптаны, сероокись углерода), а также СО2 и СО.
Одной из важнейших и первых стадий в производстве аммиака
является очистка газов. Различают жидкостные (мокрые) и сухие
способы промышленной очистки. Жидкостные способы осуществ-
ляют с помощью жидких поглотителей — абсорбентов; эти способы
основаны на физической абсорбции и абсорбции, сопровождаемой
химическими реакциями. Сухие способы очистки основаны на по-
глощении веществ твердыми поглотителями. Сюда относятся спо-
собы, основанные на физической адсорбции и хемосорбции, на ка-
талитическом превращении примесей в легко удаляемые или менее
вредные соединения. В качестве адсорбентов применяют активиро-
ванный уголь, смеси активной окиси железа и соды (железо-содо-
вая масса) и др.
В зависимости от требуемой степени очистки условно разли-
чают грубую, среднюю и тонкую очистку.
Для очистки газа от H2S и СО2 наиболее широко применяются
растворы этаноламинов (аминоспиртов), которые обладают щелоч-
ными свойствами и при взаимодействии с кислотами образуют
соли.
Наиболее сильным основанием среди этаноламинов является
моноэтаноламин, МЭА (ОНСН2СН2) NH2, взаимодействие которого
262
с сероводородом и двуокисью углерода может быть представлено
следующими реакциями:
2RNH24-H2S (RNH3)2S (XII. 9)
(RNH3)2S 4- H2S 2RNH3HS (XII. 10)
2RNH2 4-Н2О 4-СО2 (RNH3)2CO3 (XII. 11)
(RNH3)2CO3 4- H2O 4- CO2 2RNH3HCO3 (XII. 12)
где R = CH2CH2OH.
Поглотительная способность абсорбента возрастает при пони-
жении температуры и повышении давления. При повышении тем-
пературы и понижении давления равновесие химических реакций
при абсорбции сдвигается влево. На этом основана регенерация
МЭА, насыщенного кислыми газами.
Обычно для очистки газов применяют 15—20%-ные водные рас-
творы МЭА.
Очистка конвертированного газа от СО2 производится, как пра-
вило, жидкими щелочными сорбентами (водными растворами кар-
бонатов натрия и калия и щелочей). Обычно абсорбцию газов
ведут при низкой температуре, что связано с уменьшением раство-
римости газов в жидкостях при повышении температуры. Так, вна-
чале газ промывают холодной водой под давлением 1,5—2,5 ДШа
в башнях с насадкой, при этом поглощается большая часть СО2.
При снижении давления до атмосферного растворимость газов сни-
жается и из воды десорбируется газ, содержащий около 80% СО2,
10% Н2, а также N2, Н2 и др. (этот газ используют далее для про-
изводства карбамида и других продуктов).
Вода после охлаждения в градирнях возвращается на орошение
башни.
Остатки двуокиси углерода удаляют из азотоводородной смеси
при промывке раствором едкого натра или других поглотителей,
имеющих большую абсорбционную емкость по СО2, чем вода.
В зависимости от способа получения и очистки от СО2 конвер-
тированный газ содержит 0,3—0,4%. (об.) СО, а также CQ2, метан,
аргон и следы кислорода. Для удаления из газа кислородсодержа-
щих примесей, являющихся ядами для катализаторов синтеза
аммиака, желательна более полная очистка газа.
Для очистки газа от СО используются методы каталитического
гидрирования (метаыирования), промывки газа жидким' азотом и
медноаммиачная очистка под давлением.
Каталитическая очистка применяется для удаления небольших
количеств СО, СО»; и О2. Основу катализаторов гидрирования со-
ставляют такие активные компоненты, как никель, железо, ме-
таллы платиновой группы, в присутствии которых протекают сле-
дующие реакции
СО4-ЗН2 Н2О4-СН4
СО2 4- 4Н2 2Н2О 4- СН4
О2 4- 2Н2 2Н2О
263
Преимуществом каталитического гидрирования является дости-
жение тонкой очистки газов. Очистка конвертированного газа от
окиси углерода часто производится абсорбцией СО водными рас-
творами комплексных солей закисной меди и аммиака, содержа-
щих анионы угольной, муравьиной или уксусной кислот, — так
называемыми медноаммиачными растворами. Поглотительная спо-
собность этих растворов в обычных условиях невелика, но она
возрастает при повышении давления и понижении температуры.
Оптимальными условиями этого
Рис. XII.5. Содержание аммиака
в равновесной смеси при различных
температурах и давлении:
/—при 10 МПа; 2—при 30 МПа; 3—при
100 МПа; 4—при 200 МПа; (I МПа =•
<« 10 кгс/см2).
процесса очистки является дав-
ление 10—32 МПа и температура
0—10°С.
Регенерацию поглотительного
раствора ведут при 75—80°C и
атмосферном давлении. Регене-
рированный раствор после охла-
ждения возвращается на абсорб-
цию, а газы после удаления ам-
миака, выделившегося при реге-
нерации, могут быть направлены
на конверсию окиси углерода.
Синтез аммиака. Как видно
из уравнения (XII. 1), синтез ам-
миака — процесс обратимый, он
протекает с уменьшением объема
и выделением тепла, следователь-
но, исходя из принципа Ле-Ша-
телье и в соответствии с уравне-
нием константы равновесия, для
смещения равновесия в сторону
образования аммиака необходимо повышать давление и понижать
температуру (рис. XII. 5).
Синтез аммиака в отсутствие катализатора протекает крайне
медленно, поэтому на практике этот процесс ведут в присутствии
катализатора при температуре выше 400 °C. Наиболее высокой
каталитической активностью в этом процессе обладают железо,
марганец, вольфрам; каталитической активностью обладает также
платина и некоторые другие металлы. Присутствие в катализаторе
небольшого количества добавок (промоторов, иди активаторов)',
повышает его активность и устойчивость. В СССР применяют же-
лезный катализатор, получаемый сплавлением оиисло-в железа
РезО4 с активаторами: А12Оа, КгО, СаО и SiO2 и последующим
восстановлением окислов желева до металлического. Этот катали-
затор обладает высокой активностью и большой стойкостью к пе-
регревам и примесям, которые в небольших количествах всегда
содержатся в азотоводороднОй смеси.
Обычно размер зерен катализатора составляет 4—6 мм; при-
мерный состав промышленного железного катализатора (до его
264
г
восстановления) следующий (в %): 31 FeO, 62 Fe2O3, 1,7 А12О3,
I 1,1 К2О, 2,5 СаО и 0,03 SiO2.
I ’ Восстановление катализатора ведут в колонне синтеза азотово-
дородной смесью, пропускаемой через аппарат в течение несколь-
| кик суток. На некоторых промышленных установках восстановлен
I ние железного катализатора производят вне колонны синтеза. Это
позволяет сократить период пуска колонны и получить более ак-
тивный катализатор.
Примеси действуют на катализатор различно. Сероводород и
другие серосодержащие соединения отравляют железный катали-
затор необратимо. Так, при содержании серы в катализаторе 0,1%
его активность снижается на 50%, в присутствии 1% серы насту-
пает практически полное отравление катализатора. Кислород и
! кислородные соединения (Н2О, СО, СО2) также отравляют ката-
лизатор, но этот процесс обратимый.
Синтез аммиака — типичный гетерогенно-каталитический про-
цесс, лимитирующей стадией которого является активированная
адсорбция реагирующих веществ на поверхности катализатора.
Выход аммиака на промышленных установках зависит от мно-!
гих параметров технологического режима: температуры, давления, |
интенсивности процесса, состава газовой смеси, активности катали-
затора, конструкции аппарата и др. При этом интенсивность ха-
рактеризуют не временем соприкосновения т, а объемной ско-
ростью S = 1/т, ч-1, которая определяется объемом газовой смеси
м3, проходящей через единицу объема катализатора м3 в час
(м3-м-3-ч~1).
На практике синтез аммиака на катализаторе по реакции
'(XII. 1) ведут в условиях, далеких от равновесных, поэтому опре-
деляющее влияние на общую скорость процесса оказывает ско-
рость прямой реакции k. Значение константы скорости этой реак-
ции зависит от ряда параметров: температуры, активности катали-
затора, размера зерен и др. Отношение значения k при повышен-
ном давлении (Р>1) к значению k при атмосферном давлении
{Р == 1) составляет следующие величины:
Р, МПа......... 0,1 20 30 40 50
ЬР>\!ЬР=} • • • • 1 0,8 0,75 0,7 0,65
Зависимость константы скорости k от температуры выра-
жается уравнением Аррениуса [уравнение (11.24)]. Энергия
активации для. промышленного катализатора равна приблизи-
тельно 168 кДж-моль-1.
Важной характеристикой химико-технологического процесса
является его интенсивность [см. уравнение (1.2)]. В рассматривае-
мом случае (как и в других химических процессах) процесс необ-
ходимо вести так, чтобы обеспечивалась оптимальная интенсив-
ность, что определяется главным образом экономическими факто-
рами. Оптимальную интенсивность определяют путем анализа
взаимного влияния отдельных параметров процесса.
26!
Для каждого значения объемной скорости содержание аммиака
увеличивается с ростом температуры до некоторого максимального
предела, называемого оптимальной температурой (рис. XII.6). При
дальнейшем повышении температуры содержание NH3 умень-
шается, так как равновесная концентрация аммиака снижается.
Из рисунка следует, что при 500 °C повышение объемной ско-
рости в 4 раза (от 1,5-104 до 6-Ю4 ч-1) приводит к уменьшению
содержания аммиака в реакционной смеси всего лишь на 20% (от
22 до 17%). Таким образом, при повышении S резко возрастает
интенсивность процесса, т. е. съем
NH3 с 1 м3 контактной массы.
При этом увеличение интенсивно-
сти может быть определено по урав-
нению
t/ = 0,77i/S (XII. 13)
Рис. XII.6. Зависимость кон-
центрации NH3 в реакционной
смеси от температуры и объем-
ной скорости при 30 МПа (со-
став исходной смеси N2 + ЗН2);
1—s = l,5.104 ч"’; 2—3=3-104 ч-1;
3—3=6-104 ч”’; 4—равновесная
кривая 3=0; 5—оптимальная кри-
вая.
где у — мольная доля аммиака в газовой сме-
си;
S — объемная скорость;
0,77 — плотность аммиака.
Положительное влияние объемной
скорости S на интенсивность процесса
обусловлено тем, что процесс идет
вдали от равновесия, т. е. в области
высоких скоростей.
Однако при увеличении S снижает-
ся содержание NH3 в реакционной
смеси и соответственно возрастает
объем непрореагировавшей азотоводо-
родной смеси, находящейся в цикле. Но это приводит к повыше-
нию расхода энергии на транспортирование газа, а также к увели-
чению размеров трубопроводов, теплообменников и конденсаторов,
при этом нарушается автотермичность процесса и снижается пол-
нота выделения аммиака из газовой смеси. Таким образом, вопрос
о выборе оптимальных объемной скорости и температуры решается
на основании результатов соответствующих экономических расче-
тов. В настоящее время промышленные установки синтеза аммиака
работают при объемной скорости от 15 000 до 30 000 ч-1.
Для реакции синтеза аммиака, как и для всякой другой экзо-
термической реакции, зависимость r — f(T, X) имеет максимум
(рис. II. 7,6), поэтому для проведения процесса с максимальной
скоростью его начинают при высокой температуре. В начале про-
цесса степень превращения низкая (процесс протекает вдали от
равновесия). В дальнейшем, по мере продвижения газа по тол-
щине слоя катализатора и повышения степени превращения, тем-
пературу понижают для того, чтобы процесс шел при максималь-
ной скорости в соответствии с ЛОТ (рис. XII. 7).
В промышленных условиях степень превращения на катали-
заторе низкая, поэтому после отделения NH3 от непрореагировав-
266
щей азотоводородной смеси ее возвращают вновь в реактор с
помощью циркуляционного компрессора. К циркулирующей смеси
добавляют свежую азотоводородную смесь в объеме, равном
объему аммиака, образовавшегося в реакторе (см. рис. XII. 2).
При такой (циркуляционной) схеме в цикле происходит нако-
пление инертных примесей, что приводит к уменьшению содержа-
ния NH3 в газовой смеси и соответственно к снижению производи-
тельности колонны синтеза (рис. XII. 8). Так, если принять S =
== 20 000 ч-1,то при увеличении содержания инертных примесей в
Рис. XII.7. Оптимальное распределе-
ние температуры по толщине слоя
катализатора при давлении 30 МПа
и объемной скорости 15 000 ч-1.
Рнс. XII.8. Зависимость выхода ам-
миака от объемной скорости при раз-
личном содержании инертных приме-
сей в исходной циркуляционной смеси
при Р = 30 МПа:
/—N2 + 3H2 без инертных примесей; 2 —
N2 + 3H2 + 7% инертных примесей; 3~N2 +
+ЗН2+10% ниертных примесей.
газовой смеси от 0 до 10%, содержание аммиака снижается от 20
до 15%. Поэтому в производственной практике для уменьшения
количества примесей в цикле часть циркулирующей газовой смеси
после выделения аммиака выводят из цикла и либо выбрасы-
вают в атмосферу в виде продувочного газа (см. рис. XII. 2), либо
направляют для извлечения ценных продуктов.
Технологическая схема производства
Технологическая схема современного производства аммиака
включает около 30 основных и большое число разнообразных вспо-
могательных аппаратов. Ниже будет дано краткое описание совре-
менной энерго-технологической схемы производства NH3, оформ-
ленной на основе приведенных выше химической и принципиальной
схем.
Природный газ под давлением, близким к атмосферному, посту-
пает в теплообменник 2 (рис. XII. 9), где нагревается до 380 °C, и
267
направляется для очистки от сернистых соединений в аппарат I,
заполненный соответствующим поглотителем; после очистки в газе
остается не более 1 мг*м~3 серы. Очищенный газ смешивают далее
с водяным паром до соотношения пар : газ = 2,5:1, нагревают в
теплообменнике 3 до 380 °C и направляют в трубчатый реактор 4,
который является первой ступенью конверсии метана. Никелевый
катализатор расположен в вертикально установленных трубах из
Рис. XII.9. Энерготехнологическая схема производства аммиака:
/—аппараты тонкой очистки газа от соединений серы; 2, 3, 5, 6, 10, 12, 18, 20-теплообмен*
иики, 4—трубчатый реактор; 7—шахтный реактор второй ступени; 8 — котел-утилизатор; 9,
11—конверторы окиси углерода; 13— адсорбционная колонна (блок тонкой очистки от двуокиси
углерода); 14—реактор метаиирования остатков окиси и двуокиси углерода; 15, /5—-турбоком"
прессоры; 17—колонна синтеза аммиака; 19, 21 — сепараторы; 22—паровая турбина.
хромоникелевой жароупорной стали. Так как процесс конверсии ме-
тана эндотермический [реакция (XII.2)] и его проводят при высо-
кой температуре, то наружные стенки труб, содержащих катали-
затор, обогревают топочными газами, образующимися при сжига-
нии природного газа.
Парогазовая смесь проходит по трубам сверху вниз, при этом
температура ее увеличивается от 380 на входе в трубы до 700 °C
на выходе из них. В трубчатом реакторе 4 метан конвертируется
приблизительно на 70%. Дальнейшая конверсия метана произво-
дится в шахтном реакторе второй ступени 7, заполненном никеле-
вым катализатором. В этот реактор вместе с реакционной смесью
подают нагретый в теплообменнике 5 воздух. За счет протекания
экзотермической реакции (XII. 3) температура в реакторе возра-
стает до 950—1000 °C. Объем воздуха, подаваемого в реактор 7,
должен быть таким, чтобы соотношение N2: На в азотоводородной
смеси составляло 1:3.
268
В реакторе 7 остаточный метан практически полностью взаимо-
действует с водяным паром и кислородом (X л; 0,99); полученный
конвертированный газ направляют вначале в котел-утилизатор 8,
где его температура снижается до 400 °C. После охлаждения газ
поступает на конверсию СО вначале в конвертор 9 с железохромо-
вым катализатором, а затем в теплообменник 10 для охлаждения
и вновь в конвертор И с цинкхромовым катализатором. После кон-
вертора 11 газ охлаждают в теплообменнике 12 и подают в
адсорбционную колонну 13 для очистки от СОг раствором метанол-
амина.
Дальнейшая очистка газа от остатков СО и Ог производится
в реакторе 14. Полученную азотоводородную смесь сжимают тур-
бокомпрессорами 15 и 16 до 30—32 МПа и направляют в колонну
синтеза 17, где в присутствии катализатора при 400—500 °C проте-
кает реакция синтеза аммиака. Из колонны синтеза 17 азотоводо-
родную смесь, содержащую 15—17% аммиака, направляют в охла-
ждаемый водой теплообменник 18, а затем в сепаратор 19, где
часть сконденсировавшегося аммиака выводят из системы как го-
товый продукт.
Из сепаратора 19 газ направляют в охлаждаемый жидким ам-
миаком теплообменник 20 и сепаратор 21, из которого также выво-
дят жидкий продукционный аммиак. Остатки азотоводородной
смеси возвращаются в цикл перед турбогенератором 16. В резуль-
тате циркуляции азотоводородной смеси в ней накапливаются при-
меси (Аг, СН4 и др.), которые снижают производительность уста-
новки, поэтому для вывода этих примесей часть азотоводородной
смеси (на выходе ее из сепаратора) выводится из цикла (проду-
вочные газы).
Как видно из рис. XII. 9, выделяющееся в процессе тепло ис-
пользуется для подогревания воды и получения пара в теплообмен-
никах и котле-утилизаторе. Этот пар перегревается в теплообмен-
нике 6 до 400 °C и поступает в паровую турбину 22, которая при-
водит в движение турбокомпрессоры 15 й 16, После паровой тур-
бины 22 пар под давлением 3,5 МПа направляется в конденсаци-
онные турбины, приводящие в движение насосы, вентиляторы и
другое вспомогательное оборудование; избыток пара выдается на
сторону в. виде товарного энергетического пара.
Основным и наиболее интересным аппаратом в производстве
аммиака является колонна синтеза. Конструкция колонны синтеза
должна быть надежной и обеспечивать безопасную и длительную
работу. Поскольку водород и аммиак, содержащиеся в газовой
смеси, при повышенной температуре вступают во взаимодействие
со сталью, снижая ее прочность, корпус колонны синтеза изготов-
ляют из литой хромованадиевой стали, которую после сверления
подвергают проковке и сложной термической и механической обра-
ботке. В последнее время получили распространение витые и свар-
ные корпуса колонн синтеза.
269
Колонна синтеза представляет собой вертикальный стальной
цилиндр со стенкой толщиной 175—200 мм и высотой 12—20 м
(рис. XII. 10). Внутренний диаметр современных колонн составляет
1,0—1,4 м. Сверху и снизу она закрыта стальными крышками 2,
укрепленными с помощью фланцев. В верхней части колонны рас-
положена катализаторная коробка, в нижней части — теплообмен-
ник 4. Снаружи колонна имеет тепловую изоляцию 5, что умень-
Рис. ХИЛО. Колонна синтеза
аммиака под средним давлением:
1—корпус колонны; 2—крышки; 3—ка-
тализаторная коробка; 4—теплообмен-,
ник; 5 —тепловая изоляция; 6 — колосни*
ковая решетка; 7—теплообменные
трубы; 8 — центральная труба.
бам нижнего теплообменника
шает термические напряжения, воз-
никающие в стенке корпуса в ре-
зультате разности температур вну-
тренней и наружной поверхностей
колонны. Изоляция также умень-
шает тепловые потери.
Азотоводородная смесь входит в
колонну синтеза сверху, спускается
вниз по кольцевому зазору (про-
странство между корпусом колон-
ны 1 и катализаторной коробкой 3)'
и поступает в межтрубное простран-
ство теплообменника 4. Благодаря
такому устройству корпус колонны
охлаждается поступающей азотово-
дородной смесью и сохраняет свою
прочность.
Предварительно подогретый газ
из межтрубного пространства теп-
лообменника 4 поднимается по его
центральной трубе в верхнюю часть
катализаторной коробки 3 и посту-
пает в двойные теплообменные тру-
бы 7, погруженные в катализатор.
Вначале газ идет по узким внутрен-
ним трубкам сверху вниз, а затем
по кольцевому пространству между
узкими и широкими трубками под-
нимается вверх и, наконец, по тру-
4 проходит сверху вниз слой ката-
лизатора, после чего покидает колонну синтеза.
Производительность колонны синтеза аммиака (в кг-ч-1) рас-
считывают по уравнению
SJMG-Cj)
у 100 4- Ci
(XII. 14)
где S — объемная скорость, г1;
V — объем катализатора, м3;
р — плотность аммиака, кг • м3 (р = 0,771 кг/м3);
Ci и С2 — содержание аммиака в азотоводородной смеси соответственно на
входе в колонну и на выходе из нее, %.
270
Технико-экономические показатели
Ниже приведены примерные расходные коэффициенты в произ-
водстве 1 т аммиака из природного газа и воздуха под средним
давлением:
Природный газ, м3................................. 860
Электроэнергия. кВт • ч......... 1 250
Вода, м3........................... 300
Водяной пар, т..................... 1,0
Катализаторы, кг
конверсии метана.................. 0,07
конверсии окиси углерода . . . 0,40
синтеза аммиака................... 0,08
Показатели работы установок по производству аммиака зави-
сят от качества исходного сырья, производительности установки,
совершенства технологического процесса и т. д. • _.
Наиболее важными направлениями в развитии производства
аммиака являются увеличение единичной мощности производ-
ственного агрегата, создание энерго-технологических установок и
снижение давления. При повышении единичной мощности произ-
водственного агрегата улучшаются технико-экономические показа-
тели производства (глава I). Это также облегчает создание энер-
го-технологических систем, когда одновременно с химическим
продуктом потребителям выдается товарная энергия.
В последние годы единичная мощность установок по производ-
ству аммиака непрерывно растет: если до 1955 г. она составляла
50—60 т-сут-1, а в 1960 г.— 100 т-сут-1, то в 1965 г. она увеличи-
лась до 600 т-сут-1, а в 1970 г. превысила 1000 т-сут-1. В настоя-
щее время освоены установки мощностью 1500 т-сут-1 и ведутся
работы по разработке агрегата мощностью 3000 т-сут-1.
С понижением давления в процессе производства аммиака
уменьшаются энергетические затраты; в настоящее время они вы-
соки и составляют на каждую тонну аммиака, получаемого из при-
родного газа и воздуха под давлением 32 МПа, 1200—1300 кВт-ч.
Кроме того, усовершенствование производства аммиака идет по
пути создания низкотемпературных катализаторов, сохраняющих
активность при температурах около 300 °C и позволяющих без
снижения выхода аммиака вести процесс синтеза при давлении
около 10 МПа.
2. ПРОИЗВОДСТВО АЗОТНОЙ кислоты
Азотная кислота принадлежит к числу важнейших кислот и по
объему производства занимает второе место после серной кислоты.
Мощность всех заводов мира, производящих азотную кислоту, в
1974 г. составляла около 40 млн. т.
Азотная кислота широко применяется в народном хозяйстве:
75—80% всей производимой в стране кислоты расходуется на по-
лучение комплексных (сложных) минеральных удобрений и самых
271
разнообразных солей (нитратов)', а 10—15%—на получение
взрывчатых веществ и синтетических красителей. Азотная кислота
-7д1_____I____।____।----1----L.J I____I—и_______I----гд
0 10 20 30 40 50 ВО 70 80 90 100
Содермание HNO3, %
Рис. ХП.11. Диаграмма кристаллизации системы HNO3—Н2О.
и жидкая четырехокись азота используются в качестве окислитель-
ных компонентов ракетного топлива, а двуокись азота находит
применение для стерилизации семян перед внесением их в почву.
Безводная азотная кислота HNO3 представляет собой тяжелую
бесцветную жидкость плотностью при 20°С р= 1,51 г-см~3, дымя-
щую на воздухе. Она замерзает при
—41,6°С и кипит при 82,6°С. Безвод-
ная азотная кислота заметно разла-
гается даже при комнатной темпера-
туре, при цагревании разложение уси-
ливается j
2HNO3 —> 2NO2 4- Н2О 4- 0,5О2
Выделяющаяся двуокись азота рас-
творяется в азотной кислоте и окра-
шивает ее в желтый или красный цвет
(в зависимости от количества окислов
азота). С водой азотная кислота сме-
шивается в любых соотношениях с об-
Концентрация HNO3, «/ofnaccj
Рис. XII.12. Диаграмма кипе-
ния системы HNO3—Н2О.
разованием гидратов (рис. XII. 11).
При упаривании разбавленной азотной кислоты ее содержание в
растворе повышается до 68,4% HNO3, что соответствует азеотроп-
ной смеси с температурой кипения 121,9°C (рис. XII. 12).
Азотная кислота является сильным окислителем. Большинство
металлов (за исключением Pt, Rh, Ir, Au) растворяется в азотной
кислоте, но на поверхности железа концентрированная азотная
кислота образует плотный слой оксида железа, который защищает
272
металл от дальнейшего разрушения. Благодаря способности же-
леза пассивироваться азотной кислотой, его часто используют для
изготовления разнообразной аппаратуры, соприкасающейся с азот-
ной кислотой, в частности, концентрированную азотную кислоту
(особенно с добавлением 10% H2SO4, такую кислоту называют
меланжем) перевозят обычно в стальных цистернах.
Азотную кислоту выпускают двух видов: разбавленную 45—
55% HNO3 и концентрированную 97—98,5% HNO3.
До XX в. азотную кислоту получали главным образом разложе-
нием натриевой селитры NaNO3 (добываемой в Чили) серной кис-
лотой.
В связи с ограниченными природными запасами селитры в ряде
стран проводились исследования по разработке способа фиксации
азота из атмосферного воздуха. Лишь в начале XX в. удалось
окислить азот воздуха в пламени электрической дуги при темпера-
туре 3500—4000 °C. Однако дуговой метод не получил распростра-
нения вследствие большого расхода электроэнергии.
В конце XIX и начале XX вв. за рубежом проводились иссле-
дования процесса получения азотной кислоты из аммиака, а также
были сделаны попытки освоить этот процесс в промышленных
условиях.
На основании исследований И. И. Андреева, Н. М. Кулепетова
и А. К- Колосова в России был запроектирован и в 1916 г. по,-
строен первый завод по производству азотной кислоты из аммиака
(г. Юзовка).
В настоящее время азотную кислоту производят из аммиака,
искусственно получаемого на химических предприятиях; в СССР
примерно 35% получаемого синтетического аммиака перераба-
тывается в азотную кислоту.
Существуют два способа производства азотной кислоты:
1) получение разбавленной азотной кислоты с последующим ее
концентрированием (в случае необходимости);
2) непосредственное получение концентрированной азотной кис-
лоты.
Наибольшее распространение получил первый способ. Ниже он
будет рассмотрен подробнее, для второго способа приводятся толь-
ко химическая и принципиальная схемы.
Химическая и принципиальная схемы производства разбавленной
азотной кислоты из аммиака
Процесс получения разбавленной азотной кислоты из аммиака
осуществляют, либо при атмосферном давлении, либо при повы-
шенном давлении; причем химическая и принципиальная схемы
этих процессов не отличаются друг от друга.
Химическая схема производства включает три основные реак-
ции: а) контактное окисление аммиака до окиси азота
4NH3 + 5О2 = 4NO 4-6H2Q (XII 'Ч
б) окисление окиси азота до двуокиси азота
2NO 4- О2 = 2NO2 4- 1240 кДж (XII. 16)
в) абсорбция двуокиси азота водой с образованием азотной
кислоты и окиси азота (которая возвращается в процесс)
3NO2 4- Н20 = 2HNO3 4- NO (XII. 17)
Суммарная реакция образования азотной кислоты из аммиака
выражается в виде уравнения
NH3 4- 2О3 — HNO3 4" Н20 (XII. 18)
Принципиальная схема производства азотной кислоты из ам-
миака включает шесть основных операций (рис. XII. 13).
Аммиак и воздух смешиваются и фильтруются (/), а затем
направляются для окисления аммиака (2). За счет тепла реакции
н2о
н2о иа2со3
nh3
Воздух
I____ь-Пар
(побочный продукт) I HNO3
{целевой продукт)
NaNO3 ц
NaNOg
(побочные
продукты)
Рис. XII.13. Принципиальная схема производства азотной кислоты:
/—очистка воздуха я аммиака; 2—окисление аммиака на катализаторе; 3, 4—охлаждение
нятрозных газов с использованием тепла н в холодильнике; 5—окисление окнслов азота и
образование азотиой кислоты; *5—очистка отходящих газов.
газовая смесь нагревается, поэтому ее направляют на охлаждение
(3 и 4), здесь происходит частичное окисление окислов азота до
двуокиси азота [реакция (XII. 16)]. Дальнейшее окисление окиси
азота осуществляется одновременно с образованием азотной кис-
лоты [реакция (XII. 17)] в процессе абсорбции двуокиси азота во-
дой (5). Отходящие газы подвергаются очистке (6), после чего
выводятся в атмосферу.
Физико-химические основы производства
Контактное окисление аммиака. Между аммиаком и кислоро-
дом могут протекать следующие реакции:
4NH3 4- 5О2 = 4N0 4- 6Н2О 4- 907 кДж (XII. 15)
4NH3 4-4О2 = 2N2O6Н2О-}-1Ю5 кДж (XII. 19)
4NH3 4- ЗО2 = 2N2 4- 6Н2О 4- 1270 кДж (XII. 20)
Эти реакции практически необратимы, поэтому направление про-
цесса определяется соотношением скоростей реакций.
В отсутствие катализатора окисление аммиака идет в основном
с образованием элементарного азота по реакции (XII. 20); эта ре-
274
акция является термодинамически наиболее устойчивой, поскольку
она сопровождается наибольшим выделением тепла. Константа
равновесия этой реакции имеет наибольшее значение, что видно
из приведенных ниже данных, (рассчитанных при 900 °C) :
/Ср _ -Р^оРн2О- = 105з
Pnh3Po2
Кр = Ао£н^ = 10«
PNH3P02
2 Л6
Кр = Р^гРн|О. = ю87
Pnh3Po2
Для получения азотной кислоты необходимо обеспечить воз-
можно более полное окисление аммиака по реакции (XII. 15), по-
этому применяют катализаторы, избирательно ускоряющие эту
НН' н н !нн нн j н н |тцс /
!/ ! \/ i \1 !/ । \/ I \1/
000000000 0/0
Рис. XII.14. Схема окисления аммиака на поверхности платины (сплошными ли-
ниями обозначены ранее возникшие связи, пунктирными — вновь образующиеся
•связи).
реакцию. Такими катализаторами могут служить платина и ее
сплавы с металлами платиновой группы (родием, палладием), а
также окислы железа и никеля с добавками окислов марганца, ко-
бальта и др. Платина и ее сплавы являются наиболее активными
катализаторами окисления аммиака, в течение длительного вре-
мени они сохраняют высокую активность, устойчивость и обладают
хорошей механической прочностью, поэтому большинство заводов,
производящих азотную кислоту, работают с применением платино-
вых катализаторов.
Процесс контактного окисления аммиака начинается со стадии
активированной адсорбции кислорода на поверхности катализатора
с образованием промежуточного соединения, затем происходит
адсорбция с образованием комплекса, который в дальнейшем раз-
рушается с освобождением катализатора и образованием NO и
Н20 (рис. XII. 14).
Чистая платина при высокой температуре процесса быстро раз-
рушается, поэтому в качестве катализатора применяют сплав пла-
тины и родия. Так как стоимость родия более высокая, чем пла-
тины, применяют сплавы, содержащие 5—10% родия. Добавление
275
к платине родия повышает не только прочность, но и активность
катализатора:
Содержание родия в сплаве с пла-
тиной, %..................... О 2 10 15
Степень превращения, % ....... 96,1 98,3 99,3 99,0
Платино-родиевые катализаторы применяют в виде сеток из
тонкой проволоки диаметром 0,06—0,09 мм. В процессе эксплуа-
тации поверхность сеток сильно разрыхляется, сетки становятся
хрупкими, а их гладкие и блестящие нити — губчатыми и мато-
выми. При этом поверхность сеток увеличивается (рис. XII. 15) и
возрастает каталитическая активность платинового катализатора.
Рис. XII.15. Зависимость выхода
окнси азота от температуры:
1— прн 105 Па; 2—при 8-106 Па.
I 100 г
<1 •-
92
88
*3
1,07 D,7Z 0,54 0,43
Время окисления г-10? с
Рис. XII.16. Зависимость степени оки-
сления аммиака от времени соприко-
сновения при 900 °C. постоянном от-
ношении Оз: NH3 « 2 и содержании
NH3= 11%.
а
Оптимальное время соприкосновения аммиачно-воздушной сме-
си с платиновым катализатором т составляет около 10~4 с. С уве-
личением т выход окиси азота по реакции (XII. 15) снижается
(рис. XII. 16) вследствие протекания побочных реакций. Для обес-
печения оптимального времени соприкосновения газа с катализа-
тором его готовят в виде сеток, наложенных друг на друга. При
этом увеличивается общая поверхность платиновых сеток и возра-
стает активность катализатора. В реакторах, работающих при ат-
мосферном давлении, достаточно 3—4 сеток, д в реакторах, рабо-
тающих под давлением 0,7—0,9 МПа,— 16—20 сеток.
Платиновый катализатор очень чувствителен к примесям, обыч-
но присутствующим в воздухе и аммиаке. Наиболее сильное влия-
ние оказывает фосфористый водород. Прй содержании его в амми-
ачно-воздушной смеси 2-10_ 5 % степень окисления аммиака
снижается на 80%, при этом отравление катализатора- является
необратимым. Вредное действие оказывают также соединения серы
и некоторые другие вещества; при содержаний в газовой,смеси 1%
сероводорода активность платины снижается на несколько про-
центов.
276
С целью уменьшения вредных примесей в воздухе, используе-
мОм для приготовления аммиачно-воздушной смеси, его подводят
через заборную трубу высотой 100—150 м, установленную вне за-
вода- Несмотря на эти меры, небольшое количество примесей все
попадает в реактор окисления аммиака, вследствие чего актив-
ность катализатора постепенно снижается. Поэтому сетки периоди-
чески подвергают регенерации (обработке) соляной кислотой кон-
центрацией 10—15% при температуре 50—70 °C. В системах, рабо-
тающих при атмосферном давлении, сетки регенерируют через
6—8 мес, а в системах, работающих при повышенном давлении, —
через каждые 15—20 сут.
В процессе работы поверхность платинового катализатора раз-
рушается и мельчайшие частицы платины увлекаются газовым
потоком. В дальнейшем некоторая доля извлекается, но значитель-
ная часть платины теряется, и эти потери тем больше, чем выше
температура и давление процесса и чем меньше содержание родия
в катализаторе. В установках, работающих при атмосферном дав-
лении и температуре 800 °C, потери платино-родиевого катализа-
тора составляют 0,04—0,06 г на 1 т HNO3 (рис. XII. 17). В систе-
мах, работающих под давлением 0,8 МПа и при 900 °C, потери
достигают 0,3—0,4 г платины на 1 т HNO3.
Когда потери сеток составят примерно 30%, их направляют на
переплавку. Платино-родиевые сетки в установках, работающих
при атмосферном давлении, служат около 1,5 лет.
Принципиальная схема современного конвертора с платино-
выми сетками показана на рис. VIII. 4, б. Платиновые сетки распо-
ложены горизонтально на колосниковых решетках или на перепле-
тенных нитях (металлических струнах); поток аммиачно-воздуш-
ной смеси направлен сверху вниз. Диаметр сеток, работающих при
атмосферном давлении, составляет от 2 до 4 м, а в аппаратах, ра-
ботающих под давлением, — от 0,5 до 2 м.
Платина является очень дорогим металлом, поэтому ведутся
настойчивые поиски неплатиновых катализаторов для окисления
аммиака. Наиболее активные из них — окисные катализаторы: же-
лезохромовый, хромоникелевый, кобальто-никелевый и др.
Поскольку степень превращения на таких катализаторах невы-
сока, применяют двухступенчатые катализаторы, состоящие из
платиновой сетки в качестве катализатора первой ступени и непла-
тинового слоя (например, железохромовый)—в качестве катали-
затора второй ступени. Толщина второго слоя 50—65 мм, он
расположен на расстоянии 10—15 мм от платиновой сетки
(рис. XII. 18).
На платиновой сетке степень окисления аммиака составляет
85—90%, общая степень окисления на комбинированном катали-
заторе достигает 97%. Если процесс ведут только на платино-ро-
Диевой сетке, выход окиси азота составляет 97,5—98%. Единовре-
менные затраты на платину при использовании двухступенчатого
Катализатора сокращаются в 3 раза.
277
720 760 BOO Bk-0 880 920
Температура конверсии,0С
Рис. XII.17. Зависимость потерь пла-
тины новых контактных сеток от тем-
пературы:
/—платиновые сетки; 2—платияо-родяевые
сетки (10% Rh).
Рис. XII.18. Контактный аппарат
с двухступенчатым катализатором;
/, 6—дырчатые диски; 2—корпус аппарата;
3 — платиновые сетки; 4—слой неплатино-
вого катализатора; 5—-слой керамических
колец; 7—-штуцер для термопары.
окнси азота от соотношения кончен- NO2. 2_5% N0 3_ш ЫОг;
трацни кислорода и аммиака. 4—20% NOz; 5—40% NO2.
Окисление аммиака на чистой платине начинается при 145 °C
с образованием элементарного азота, закиси азота и небольшого
количества окиси азота. При повышении температуры выход окиси
азота увеличивается. Оптимальный температурный режим окисле-
ния аммиака при атмосферном давлении лежит в интервале 800—
840 °C, а при проведении процесса под давлением 0,7—0,9 МПа —
в пределах 800—900°С (рис. XII. 15).
278
Источником кислорода, необходимого для осуществления реак-
ции (XII. 15), служит воздух. При этом соотношение между кисло-
родом и аммиаком (Ог: NH3) в аммиачно-воздушной смеси зави-
сит от исходных объемов воздуха и аммиака. Это соотношение
имеет важное практическое значение в производстве азотной кис-
лоты, так как от него зависит скорость и полнота реакции
(XII-15). При теоретическом содержании кислорода (О2: NH3 =
‘₽= 1,25) выход окиси азота мал (рис. XII.29), поэтому на практике
это соотношение увеличивают до 1,7—2,0, что соответствует содер-
жанию аммиака в аммиачно-воздушной смеси 10—12%. В этом
случае обеспечивается высокая скорость процесса, а избыток кис-
лорода используется в дальнейшем для окисления NO в NO2; по-
мимо того аммиачно-воздушная смесь находится за пределами
границ взрывоопасного соотношения аммиака и воздуха (см.
рис. XII. 1).
Гетерогенное окисление аммиака на платиновом катализаторе
состоит из нескольких стадий (см. рис. IV. 1). При отношении
О2: NH3 в воздушно-аммиачной смеси более 1,35 скорость процесса
контролируется скоростью диффузии аммиака к поверхности ката-
лизатора, а при отношении Ог:МН3 менее 1,35 контролирующей
становится диффузия кислорода.
При проведении процесса окисления аммиака при атмосферном
давлении и отношении О2: NH3 более 1,35 скорость реакции выра-
жается уравнением первого порядка
г — — dPmJdx — *Pnh3
По данным некоторых авторов, энергия активации в уравнении
Аррениуса реакции (XII. 15) составляет £ = 26,6 кДж-моль-1.
Окисление окиси азота. Реакция окисления окиси азота до дву-
окиси (XII. 16) —обратимая реакция, протекающая с уменьшением
объема и сопровождаемая выделением тепла, поэтому понижение
температуры нитрозных газов и повышение давления будет сдви-
гать равновесие реакции вправо, т. е. в сторону образования NO2-
Константа равновесия этой реакции
„2
К — PN02 ,vtt 0,1
Др---------
сильно зависит от температуры:
Температура, °C . 20
Кр (вычисленная) 1,2-1014 1,8-108 7,4-104 0,85 5,6-10-8
Из приведенных данных видно, что в нитрозных газах, выходя-
щих из реактора окисления аммиака при температуре около 800 °C,
Двуокись азота практически отсутствует (Кр мала). Чтобы пере-
вести NO в NO2 нитрозные газы следует охладить ниже 100 °C.
Скорость реакции (XII. 16) уменьшается при повышении тем-
пературы; таким образом, эта реакция представляет собой исклю-
, 279
h
„2 „2
PnoPo2
100 200 500 800
чение из общего правила, из которого следует, что обычно при
повышении температуры скорость реакции увеличивается.
В соответствии с одним из наиболее распространенных меха-
низмов этого процесса окисление NO в NO2 проходит через обра-
зование промежуточного продукта — димера окиси азота
2NO (NO)2 4- Q
(NO)2 4-О2 4=4= 2NO2 4- Q
Образование такого димера — процесс обратимый, он протекает
с выделением тепла, поэтому его выход и, следовательно, концен-
трация в газовой смеси уменьшаются при увеличении температуры.
Таким образом, на основе -уравнения (11.23) скорость реакции
окисления NO понижается при повышении температуры.
Скорость реакции окисления NO в NO2 — самая медленная в
процессе производства азотной кислоты, она и определяет скорость
всего процесса.
При охлаждении происходит полимеризация NO2
2NO2 N2O4 4- 57 кДж (XII. 22)
Зависимость степени полимеризации NO2 от температуры ха-
рактеризуется следующими данными:
Температура, °C ... . 200 100 70 30 0 —20
Степень полимеризации,
%.................. 0,7 2,5 38 77,8 89 92
Скорость полимеризации NO2 очень высока, поэтому в любой
момент времени соотношение NO2: N2O4 определяется условиями
равновесия, так как оно устанавливается практически мгновенно.
Абсорбция двуокиси азота. Двуокись азота взаимодействует с
водой с образованием азотной и азотистой кислот
2NO2 4- Н2о = HNO3 4- HNO2 4-116 кДж (XII. 23)
Азотистая кислота неустойчива и разлагается
3HNO2 = HNO3 4- 2NO 4- Н2О - 75,8 кДж (XII. 24)
Суммарно уравнение абсорбции двуокиси азота можно записать
в виде
3NO2 4- Н2О = 2HNO3 4- NO 4- 136,2 кДж (XII. 25)
Механизм образования азотной кислоты из двуокиси азота при
абсорбции ее водой, а затем разбавленной азотной кислотой (по
мере ее образования) можно представить следующим образом.
Двуокись азота диффундирует через пограничный слой газа
к поверхности жидкости и абсорбируется ею. При этом NO2 взаи-
модействует с водой [реакция (XII.23)]. По сравнению с диффу-
зией NO2 эта реакция протекает быстро. Азотистая кислота разла-
гается по реакции (XII. 24) сравнительно медленно. Образующаяся
цри этом окись азота плохо растворима в водных растворах азот-
ной кислоты, поэтому она выделяется в газовую фазу, где окис-
ляется кислородом.
280
Концентрация получаемой азотной кислоты определяется усло-
виями равновесия NOj над азотной кислотой (рис. XII. 20). Это
объясняется тем, что количество NO2, абсорбируемой водными рас-
творами азотной кислоты, зависит от движущей силы этого про-
цесса
АС = pN02 — pN02
где Pno,’ Pno2 ~ парциальное давление NOj в газовой фазе и равновесное дав-
ление NOz у поверхности водного раствора азотной кислоты.
С повышением температуры р*тг увеличивается, а ДС сни-
жается, соответственно уменьшается доля получаемой азотной кис-
лоты. При понижении температуры и увеличении парциального
давления NOj значение ДС растет, соответственно повышается
концентрация азотной кислоты.
Технологическая схема производства
разбавленной азотной кислоты
Технологическая схема производства азотной кислоты при ат-
мосферном давлении приведена на рис. XII. 21. Воздух поступает
через заборную трубу, проходит водяной скруббер 1 и суконный
Рис. ХП.21. Схема установки для получения разбавленной азотной кислоты при
атмбефериом давлении:
7 —водяной екигббер; 2—еуконкый фнльу^; 3— аммиачно-воздушяый вентилятод.; 4—картон-
ный фййьтр; .7—конвертор; —паровой 7—скоростной холедилыяик*; хо-
лодильнйс-кон'денёатор; $ — вентдалят^ для газов; 10— абсорбционные башни;
П — оквслйтёльная башня; /2—башня для бкйейов азота щелочами; -^холо-
дильники кислоты; Г4, 75—насбеы.
фильтр 2. Очищенный воздух и аммиак подаются в улитку венти-
лятора 3, где происходит их смешение. Полученная газовая смесь,
содержащая 10—12% NH3, вентилятором 3 направляется в картон-
ный фильтр 4, а затем в конвертор (реактор) 5, в средней части
которого размещены платино-родиевые сетки.
281
Степень окисления аммиака до окиси азота составляет при,
мерно 97—98%. Температура нитрозных газов на выходе из кон.
вертора 800 °C. Конвертор монтируют непосредственно на котле-
утилизаторе 6, в котором получают пар давлением до 4 МПа при
температуре 450 °C. Температура газбв после котла-утилизатора
составляет около 160 °C; дальнейшее охлаждение газов осуществ-
ляется в водяных холодильниках 7 и 8. Здесь же происходит ча-
стичная конденсация водяных паров и окисление окиси азота до
двуокиси азота. Степень окисления NO в холодильнике 7 незначи-
тельная, поэтому в нем получается кислота, содержащая около 5%
HNO3; в холодильнике 8 степень окисления NO более высокая, и
в нем конденсируется кислота концентрацией до 30% HNO3.
Охлажденный газ вентилятором 9 направляется в абсорбцион-
ную систему, состоящую из шести-восьми полых башен 10 (на
рисунке показаны четыре башни), орошаемых азотной кислотой.
В объеме этих башен продолжается процесс окисления NO до NO2;
одновременно NO2 абсорбируется азотной кислотой и вступает во
взаимодействие с водой, имеющейся в азотной кислоте.
Для окончательного поглощения NO2 в последнюю по ходу газа
башню подают воду; образующуюся азотную кислоту направляют
последовательно противотоком газу от последней башни к первым.
Продукционная кислота выводится из второй башни, ее концен-
трация 45—50% HNO3. Степень абсорбции нитрозных газов со-
ставляет 92—94%.
Большое число абсорбционных башен объясняется тем, что ско-
рость реакции окисления NO мала и, следовательно, мала общая
скорость процесса. Кроме того, по мере снижения концентрации
окислов азота эта скорость уменьшается еще более к концу про-
цесса, поэтому для увеличения степени окисления окиси азота и
степени абсорбции газов необходимо существеннб увеличить время
процесса, что достигается при введении большого числа абсорб-
ционных башен.
После абсорбционных башен газ направляют в окислительную
башню 11 для полного окисления NO в NO2, а затем в башни ще-
лочной абсорбции 12, орошаемые водным раствором соды. Окислы
азота (NO и NO2) абсорбируются содовым раствором с образова-
нием солей
NO + NO2 + Na2CO3 = 2NaNO2 + СО2 (XII. 26)
2NO2 + Na2CO3 = NaNO2 + NaNO3 + CO2 (XII. 27)
Получаемые растворы нитрата и нитрита натрия выводят из
первой по ходу газа башни для дальнейшей переработки. На неко-
торых заводах щелочные башни орошаются известковым молоком,
при этом получаются растворы нитрита и нитрата кальция.
Применение для окисления аммиака воздуха, обогащенного
кислородом, или чистого кислорода позволяет получать нитрозные
газы с повышенным содержанием окиси азота и соответственно
увеличить скорость последующей реакции окисления NO в NO2 и
всего процесса в целом.
282
NO + NO2 + Na2CO3 = 2NaNO2 + C02
2NO2 -f~ Na2CO3 = NaNO2 -f- NaNO3 -f- CO2
Технологическая схема производства азотной кислоты под дав-
i лением (рис. XII. 22) существенно отличается от схемы на
рис. XII. 21. Атмосферный воздух засасывается через фильтр 1 тур-
бокомпрессором первой ступени 2 и -сжимается до 0,2—0,35 МПа;
вследствие сжатия воздух нагревается до 175 °C. После охлажде-
ния до 30—45 °C в холодильнике 3 воздух поступает в турбоком-
прессор второй ступени 4, где он сжимается до конечного давления
Ватмосферу
——-- ——— --—g——..... .................-СО—*-
Рис. ХП.22. Схема производства азотной кислоты под давлением с приводом
компрессора от газовой турбины:
1—фильтр воздуха; 2—турбокомпрессор первой ступени; 3—промежуточный холодильник;
4—турбокомпрессор второй ступени; 5—газовая турбина; 6 — редуктор; 7—мотор-генератор
3—подогреватель воздуха; 9—смеситель аммиака с воздухом; 10—подогреватель воздуха;
11—поролитовый фильтр; 12—конвертор; 13, /3—котлы-утилизаторы; 14—сосуд для окисления
нитрозных газов; 15—холодильник-конденсатор; 16 — абсорбционная колонна; 17—конвертор.
0,73 МПа и нагревается до 125—135 °C. Дальнейший подогрев воз-
духа до 270 °C происходит в подогревателе 8 за счет тепла горячих
нитрозных газов, выходящих из конвертора; горячий воздух посту-
пает далее в смеситель 9.
Аммиак под давлением 1,0—1,2 МПа нагревается до 150 °C в
подогревателе 10 водяным паром и поступает в смеситель 9, где
смешивается с воздухом. Полученная аммиачно-воздушная смесь,
содержащая 10—12% ЫНз, фильтруется в поролитовом фильтре 11
и поступает в конвертор (реактор) 12, где на платино-родиевом
катализаторе при температуре 890—900 °C аммиак окисляется до
окиси азота. Тепло газов, выходящих из конвертора, используется
в котле-утилиз а торе 13 для получения пара, при этом газы охлаж-
даются до 260 °C.
Далее газы проходят фильтр для улавливания платины, распо-
ложенный в верхней части пустого сосуда 14. В сосуде 14 происхо-
дит окисление NO до NO2 (степень окисления 80%), в результате
этого газовая смесь разогревается до 300—310 °C и поступает в
подогреватель воздуха 8, где охлаждается до 175 °C. Дальнейшее
333
использование тепла нитрозных газов становится невыгодным, по.
этому они охлаждаются водой в холодильнике 16 до 50—55 °C. Од*
яовременно с охлаждением газа в холодильнике 16 происходит
конденсация паров воды и образование азотной кислоты в резуль.
тате взаимодействия воды с двуокисью азота. Концентрация обрд.
зующейся кислоты не превышает 52% HNO3, выход составляет
около 50% всей производительности установки.
Из холодильника 15 нитрозные газы поступают в абсорбцион-
ную колонну 16 с ситчатыми тарелками, где NO2 поглощается во,
дой с образованием азотной кислоты (концентрация до 55%). На
тарелках абсорбционной колонны 16 уложены змеевики (холодиль-
ные элементы), по которым циркулирует вода для отвода тепла,
выделяющегося в процессе образования азотной кислоты.
Для очистки отходящих газов от окислов азота их подогревают
до 370—420 °C, добавляют к иим небольшое количество природного |
газа и направляют в конвертор (реактор) 17. Здесь в присутствии
лалладиевого катализатора протекают следующие реакции:
2СН4 + О2 2СО + 4Н2 + Q (XII. 28)
2NO2 + 4Н2 = N2 + 4Н2О + Q (XII. 29)
2NO + 2Н2 = N2 + 2Н2О + Q (XII. 30)
Так как эти реакции идут с выделением тепла, то температура
газов повышается до 700—730 °C. Эти газы поступают под давле-
нием 0,5—0,6 МПа в турбину 5, которая приводит в движение тур-
бокомпрессоры 2 и 4, сжимающие воздух. После этого газы при
температуре около 400 °C поступают в котел-утилизатор 18, в кото-
ром получают пар низкого давления. '
Турбокомпрессоры первой и второй ступеней 2 и 4, а также га-
зовая турбина 5 представляют собой единый агрегат; турбина пер-
вой ступени 2 и газовая турбина 5 находятся на общем валу и (
соединены редуктором 6 с турбиной второй ступени 4 и электромо-
тором 7. Такой агрегат позволяет использовать основную часть
энергии, затраченную на сжатие воздуха, и таким образом значи-
тельно снизить расход электроэнергии.
Концентрирование азотной кислоты
Концентрирование разбавленной азотной кислоты осуществ-
ляется в тарельчатых барботажных колоннах или в колоннах с на- ।
садкой из колец. Так как путем упаривания воды из разбавленных
растворов HNO3 можно получить азотную кислоту концентрацией
не более 68,4% HNO3, процесс концентрирования ведут в присут-
ствии концентрированной серной кислоты — хорошего водоотни-
мающего средства.
Серная кислота связывает воду, содержащуюся в разбавленной
азотной кислоте, образуя гидраты серной кислоты, кипящие при
температуре, более высокой, чем 100%-ная HNO3. Поэтому можно
284
подобрать условия, при которых в процессе нагревания смеси, со-
держащей азотную и серную кислоты, в парах будет содержаться
дочти исключительно азотная кислота.
Схема установки для концентрирования разбавленной азотной
кислоты приведена на рис. XII. 23. Разбавленную азотную кислоту
в виде двух потоков направляют в колонну концентрирования.
Один из потоков вначале поступает в испаритель, где азотная кис-
лота испаряется. Образующиеся пары подаются на десятую та-
релку барботажной концентрационной колонны 2. Второй поток
Рис. ХП.23. Схема концентрирования разбавленной азотиой кислоты в присут-
ствии серной кислоты:
I—концентрационная колонна; 2—испаритель разбавленной азотной кислоты: 3—холодиль-
ник-конденсатор; 4—холодильник концентрированной азотной кислоты.
разбавленной азотной кислоты направляется непосредственно в
колонну (минуя испаритель) на тарелки, расположенные несколько
выше ввода в колонну паров из испарителя 1. Концентрированная
серная кислота (92—94%) подается на одну из верхних тарелок
колонны 2. Для нагревания смеси в нижнюю часть колонны
Вводят острый пар (около 250 °C).
Пары азотной кислоты с незначительным содержанием паров
воды и окислов азота, образовавшихся в результате разложе-
ния азотной кислоты, направляются из колонны в холодильник-
конденсатор 3, где азотная кислота конденсируется, а иитрозные
газы (окислы азота) идут иа дальнейшее улавливание. Часть
окислов азота растворяется в азотной кислоте, поэтому из кон-
денсатора она возвращается на верхние тарелки колонны, где
продувается, и затем отводится в качестве продукционной после
охлаждения в холодильнике 4,
285
Отработанная разбавленная серная кислота (68%) вытекает
из нижней части колонны и поступает без охлаждения непосред-
ственно на упаривание до 92—94% H2SO4. Расход серной кис-
лоты составляет 3—4 т на 1 т азотной кислоты.
Материалом для изготовления колонны служит кислотоупор-
ный чугун (ферросилид), содержащий 14—18% кремния. Он устой-
чив при повышенных температурах к действию смеси серной и азот-
ной кислот.
Прямой синтез концентрированной азотной кислоты
из окислов азота
Прямой синтез концентрированной азотной кислоты состоит
в том, что жидкая четырехокись азота взаимодействует с водой
в присутствии газообразного кислорода под давлением 5 МПаз
2N2O4 (ж) + 2Н2О (ж) + О2 (г) ч=* 4HNO3 (ж) + 59,5 кДж (XII. 31)
Наиболее сложным в этом процессе является получение жид-
кой четырехокиси азота; обычно ее получают из нитрозных газов,
образующихся при окислении амммиака. Вначале из нитрозных
газов выделяют избыточную влагу, для этого газы охлаждают
от 200 до 40 °C. Затем окисляют содержащуюся в нитрозных
газах NO до NO2 вначале кислородом воздуха в газовой фазе,
а затем концентрированной азотной кислотой
2HNO3 + NO 3NO2 + H2O (XII. 32)
После этого из нитрозных газов выделяют NO2 охлаждением га-
зов до минус 15 — минус 20 °C (под давлением не ниже 0,5 МПа)
либо до минус 10 °C и поглощением двуокиси азота концентриро-
ванной азотной кислотой (97—98% HNO3), в которой NO2 хорошо
растворяется с образованием нитролеума
nNO2 + HNO3 ч=* HNO3 • »NO2 (XII. 33)
Если в нитрозных газах присутствует 10—11% окислов азота,
получают нитролеум, содержащий до 25% NO2. При нагревании
такого нитролеума до 80 °C получают газообразную двуокись азо-
та, которая отделяется от азотноц кислоты, а затем конденсируется
и полимеризуется.
В заводских условиях конденсацию обычно осуществляют в
двух последовательно соединенных холодильниках: в первом
из них охлаждение производится водой, в во втором (до минус
8°C) —рассолом Ca(NO3)2.
Таким образом, принципиальная схема производства концентри-
рованной азотной кислоты методом прямого синтеза из нитрозных
газов, получаемых окислением аммиака воздухом, состоит в сле-
дующем (рис. XII. 24). После реактора окисления аммиака горячие
286
нитрозные газы вначале охлаждаются в котле-утилизаторе, затем
в скоростном холодильнике до 34—40 °C (операция /). Затем
слабо охлажденные нитрозные газы поступают в газовый холо-
дильник, где происходит дальнейшее охлаждение нитрозных га-
зов и выделение избыточной влаги (в виде 30 %-ной HNO3,
операция 2). Для окисления NO нитрозные газы поступают в
две последовательно включенные окислительные башни (опе-
рация 3). Дальнейшее окисление NO проводят 98%-ной азотной
кислотой. При этом количество кислоты должно быть таким, чтобы
отходящая кислота содержала не более 75% HNO3 (операция 4).
Рнс. XI 1.24. Принципиальная схема производства концентрированной азотной
кислоты прямым синтезом из окислов азота:
/—охлаждение иитрозиых газов в котле-утилизаторе; 2—охлаждение нитрозных газов в холо-
дильнике-конденсаторе; 3—окисление NO в NOj в башне с орошаемой'насадкой; 4—дооки-
сление NO 98%-иой Н> Ог; 5—охлаждение иитрозиых газов рассолом в трубчатом холодиль-
нике; 6—поглощение Noj 98%-ной HNO3; 7—обезвреживание отходящих газов; 8—выделение
NO2 из раствора в HNO3 нагреванием в отбелочной колонне; Р— охлаждение окислов азота
в холодильнике водой; 10—охлаждение N2O4 рассолом до —8 °C; И— приготовление смеси
жидкой N2O4 и HNO3; 12—окисление N2O4 до HNO3 в автоклаве; 13—десорбция избыточной
N2O4 из HNO3.
Окисленные нитрозные газы охлаждаются рассолом в труб-
чатом холодильнике до —10 °C (операция 5) и поступают в баш-
ню с насадкой на поглощение 98%-ной HNO3 (операция 6).
Раствор азотной кислоты, насыщенный NO2, отводится в отбе-
лочную колонку, где при нагревании раствора из него отгоняют
чистую двуокись азота (операция 8). Отходящие газы направля-
ются в башшр, орошаемую разбавленной азотной кислотой и
водой для выделения из них окислов азота (операция 7).
Чистая двуокись азота по выходе из отбелочной колонны
охлаждается водой до 40 °C в холодильнике (операция 9), а за-
тем рассолом до —8 °C (операция 10). Образующаяся при этом
жидкая N2O4 направляется в смеситель; сюда подается 60- и
75%-ная азотная кислота (операция 11). Полученная смесь
поступает затем в автоклав, где N2O4 взаимодей’ствует с кисло-
родом под давлением 4—5 МПа (операция 12). Полученная в
автоклаве кислота содержит около 25% избыточной N2O4; ее
выделяют нагреванием и возвращают в процесс, а 98%-ная HNO3
выводится как готовый продукт.
287
Технико-экономические показатели
Ниже приведены расходные коэффициенты в производстве
азотной кислоты, отнесенные к 1 т 100%-ной кислоты:
Система под давлением
атмосферным повышенным
Аммиак, т 0,288 0,290
Электроэнергия, кВт • ч 120 10
Платина, г 0,05 0,16
Охлаждающая иода, м3 150 150
Очищенная вода, м3 Природный газ, м3 1,9 2,7
Нет 132
Отходы пара (побочный продукт) Гкал 0,47 1,32
ГДж 1,97 5,53
На экономические показатели процесса большое влияние ока-
зывает давление, так как при увеличении давления уменьшается
объем аппаратуры, повышается интенсивность процесса и сни-
жается расход металла по системе в целом. Однако при этом
уменьшается степень окисления аммиака и возрастает расход пла-
тины. В установках, работающих под давлением, увеличивается
выход пара (побочного продукта), что существенно снижает энер-
гетические затраты и себестоимость азотной кислоты.
Элементы себестоимости 1 т разбавленной азотной кислоты
(в % от заводской себестоимости):
Система под давлением
атмосферным повышенным
Заио^екая себестоимость 100 100
Аммиак 85 70
Платина 1,15 4,1
Палладий -. . . . — 3,8
Вспомогательные материалы . 2.75 —
Электроэнергия . . 12,5 14,2
Зарплата 0,9 2,6
Амортизационные расходы 6,8 8,8
Прочие расходы 6,6 15,0
Отходы пара (побочный) продукт . 15,6 18,5
Отношение заводских себестоимо- стей (принимая себестоимость прн атмосферном давлении за 1) . . . 1,0 1,23
Себестоимость 1 т концентрированной азотной кислоты, полу-
чаемой методом прямого синтеза из окислов азота, примерно на
15% выше себестоимости кислоты, получаемой концентрированием.
Примеры
Пример XII. 1. Рассчитать производительность колонны синтеза аммиака при
работе ее на свежем катализаторе и после двухлетней работы на этом же ката-
лизаторе.
288
Условия.
Высота колонны Н, м................................. 14
Внутренний диаметр нолонны D, м..................... 0,85
Содержание и цикле инертных примесей т|, %.......... 7
Конечная температура конденсации аммиака tK, °C . . . 5
Содержание NH3 в газовой смеси на входе в колонну
С,, %..............................................._ 4
Объемная скорость S, ч—1............................ 25 000
Давление в системе Р, МПа.......................... 30
Коэффициент использования внутреннего объема (паковки)
колонны р, % ........................................ 35
Снижение активности катализатора после двухлетней ра-
боты z, %............................................ 20
Решение.
Объем катализатора, загруженного в колонну синтеза, составляет
лО2ЯР 3,14 • 0,852 • 14-35 „ . ,
° = ^710У- =---4Й00------= 2’8 М
Из рис. XII. 8 следует, что при заданных условиях (см.Р, S, т| и /к) содер-
жание NH3 в газовой смеси на выходе из колонны С2 = 17%. Подставим полу-
ченные значения в уравнение (XII. 14) н находим
„ 25 000-2,8-0,771 (17-4) . .
GH =-----------ргт—2--------= 6,75 т/ч, нлн 162 т/сут
Через два года в результате снижения активности катализатора содержание
аммиака после колонны уменьшится и составит
100-2 100-20
с Сг 100 7 юо 3,6/о
Следовательно, производительность колонны через два года будет. равна
_ 25 000-2,8(13,6- 4)0,771 _ , .
GK =--------------------------= 5 т/ч, или 120 т/сут
Таким образом, средняя производительность рассчитанной колонны синтеза
в течение 2 лет составит 141 т аммиака в сутки.
Пример XII. 2. Определить количество и состав исходной аммиачно-воздуш-
ной смеси для производства 1000 т • сут'1 азотной кислоты (100%-ной).
Условия.
П 1000 И,
Производительность установки QHNo3, т-ч-1 . —щ— = 41,7, нлн
4,17 • 104 кг ч—1
Содержание NH3 в аммиачно-воздушной смесн
Cnhs’ % ..................................... И>5
Степень окисления аммиака а.................. 0,97 '
Степень абсорбции окислов азота Р.................. 0,985
Решение.-
Расход аммиака QNHj может быть установлен на основе уравнения реакции
(XI. 4), нз которого следует, что из 1 моль аммиака (MNHj = 17) образуется
1 моль азотной кислоты (Mhnq3 = 63). Поэтому
„ _ Qhno3^nh3 _ 4,17-Ю4-17 _
Qnh3 aPMHN03 0,97 - 0,985 - 63 Ы8 -10 кг ’4
или
1,18 • 104 • 22,4 ,_к , .
°nh,=--------Гт------= 1,55-104 м3 • ч-1
10 Зак, 558
289
Расход воздуха
_ t>NH3(100-CNH3) 1,55.ю< (100.-, 11.5)
^воз ““------X - — . — —"'“'i 1 с— -- и 1,2 • 10б м3 * 4“
CNH3 lli5
В том числе
О2 = 0,21 • 1,2 • 105 = 2,54 • 10* м3 • ч_|, или 3,63 • 104 кг • ч_|
N2 = 0,79 • 1,2 • 106= 9,42 • 10* м3 • ч-1, или 11.8 • 10* кг• ч-'
Состав аммиачно-воздушной смесн:
кг м3 %
NH3 . . . . 1,18 10* 1,55- 10* 11,5
О2 .... 3,63 • Ю4 2,54-104 18,8
n2 .... 11,80 -10* 9,42-10* 69,7
1,66-105 1,35- 10s 100.0
Глава XIII
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА ТОПЛИВА
Топливо — вещество, при сжигании которого выделяется
значительное количество тепла, используемое как источник полу-
чения энергии. Топливо, содержащее органические вещества, на-
зывают углеводородным топливом, путем химической переработки
из него получают разнообразные продукты.
По происхождению топливо подразделяется на естественное
и искусственное. К естественным топливам относятся ископаемое
и растительное, а к искусственному — продукты переработки есте-
ственных топлив. По агрегатному состоянию топливо делят на
твердое, жидкое и газообразное (табл. XIII. 1).
Таблица Х111.1 Классификация топлив
Агрегатное состояние Топливо
естественное искусственное
Твердое Древесина, торф, бурый Полукокс, кокс, древесный
Жидкое уголь, каменный уголь, антрацит, сланцы Нефть уголь, смешанные брикеты Бензин, керосин, лигроин и др.
Газообразное Природный газ, попутные газы, получаемые при не- фтедобыче Генераторные газы, коксо- вый и полукоксовый газы, газы, получаемые при пе- реработке нефти и др.
Разведанные запасы нефти и газа в земной коре незначи-
тельны и составляют по расчетам геохимиков около 1% всех за-
пасов горючих ископаемых. В последние годы доля нефти и при-
290
родного газа в топливном балансе страны все время возрастает,
тогда как доля угля будет снижаться и дальше (см. стр. 186).
Все виды топлив состоят из органической массы, минеральных
веществ и воды. Твердое топливо содержит горючую органическую
массу и негорючие минеральные вещества.
Органическая часть топлива состоит из веществ, содержащих
в основном углерод и водород, а также азот и серу (табл. XIII. 2).
Таблица XIII. 2. Примгрный состав различных топлив (в %)
Топливо Органическая часть Влага Зола Сера Летучие Тепло- творная способ- ность, МДж-кг“!
с н O + N
Древесина 50 6 44 30—40 0,4 — 70 19
Торф 59 6 35 25 4,5 0,4 70 24
Бурый уголь 70 5,5 24,0 До 50 4,0 2-3 45-55 26
Каменный 82 5,0 13,0 3-8 . 6,0 2-6 8-50 34
уголь Антрацит 95 2,0 3,0 1,3 6,0 1-2 8,0 34
Горючие 55-80 6-10 7-35 10-15 30-60 1,5-11 30-65 25-33
сланцы Нефть 86-87 13 .0,2—0,3 — 0,1-0,5 0,1-5 — 39-46
Минеральная часть топлив содержит неорганические вещества:
карбонаты, силикаты, фосфаты, сульфаты и сульфиды металлов.
При сжигании или газификации топлива минеральная часть его в
основном разлагается и в виде окислов металлов переходит в золу.
Сера содержится в топливе в виде сульфидов, сульфатов и
сероорганических соединений. При горении топлива соединения
серы (кроме сульфатов) окисляются, образуя сернистый ангидрид,
а сульфаты остаются в золе.
Летучими веществами твердого топлива называются газооб-
разные и жидкие продукты (последние при высокой температуре
переходят в парообразное состояние), получающиеся при нагрева-
нии топлива без доступа воздуха. С летучими веществами из топ-
лива удаляется и вода.
Теплотворная способность топлива является его энергетиче-
ской характеристикой.
Теплотворная способность — это количество тепла, которое по-
лучают при сжигании единицы массы или объема топлива.
Методы переработки топлив зависят от его агрегатного состоя-
ния и происхождения, а также от назначения получаемых продук-
тов. Известны физические и химические методы переработки. Фи-
зические методы используют при переработке жидкого и газооб-
разного топлива. К ним относятся перегонка, ректификация, ад-
сорбция, десорбция, кристаллизация и др.
10* 291
К химическим методам относятся методы пирогенетической
переработки топлива, при которой физические и химические про-
цессы протекают при высокой температуре. Пирогенетическая
переработка топлива включает пиролиз, гидрирование, газифика-
цию и крекинг. На рис. XIII. 1 приведена схема переработки
топлива.
Сухая перегонка топлива состоит в нагревании топлива без
доступа воздуха. Этому виду переработки могут подвергаться
твердое (угли, торф, древесина, сланцы) и жидкое топливо
Рнс. ХШ. 1. Схема переработки топлив.
(нефть). В результате протекания сложных физико-химических
процессов органическая часть топлива превращается в конечном
итоге в твердый продукт (кокс, полукокс, древесный уголь) и лету-
чие продукты, состоящие из смеси газов и паров жидких про-
дуктов. -•
Пиролиз одного и того же вида сырья может проводиться при
различных температурах. Так, каменные угли в зависимости от
назначения получаемых продуктов могут перерабатываться при
средних (500—600 °C) и высоких (1050 °C) температурах. Первый
из этих процессов называется полукоксованием, а второй — кок-
сованием.
Крекинг — процесс высокотемпературной обработки топлива,
при котором происходят глубокие изменения в строении молекул
исходного сырья.
Термин «крекинг» происходит от английского слова to creak,
что означает раскалывать, расщеплять, т. е. под крекингом пони-
мают расщепление исходных молекул на более мелкие. Но при
крекинге наряду с распадом молекул происходят вторичные про-
цессы образования новых молекул и веществ,
292
Гидрирование и гидрогенизация топлива осуществляются с це-
лью получения продуктов более богатых водородом. Эти процессы
проводятся при высоких температурах и давлениях в присутствии
катализатора и водорода. В этом случае совмещают разложение
^топлива при нагревании (деструкция) со взаимодействием про-
дуктов разложения топлива с водородом под давлением.
Газификация топлива — процесс превращения органической ча-
сти топлива в горючие газы путем частичного ее окисления. В про-
цессе газификации малоценное (многозольное) топливо превра-
щается в газообразное, являющееся беззольным, транспортабель-
ным топливом и сырьем для химической промышленности. Однако
в последние годы значение генераторных газов снижается в связи
с увеличением добычи природного и попутного газов, которая за
пять последних лет возрасла почти в 1,5 раза. Однако запасы
природного газа невелики, а запасы угля огромны, поэтому есть
все основания ожидать, что в недалеком будущем роль газифика-
ции твердого топлива будет вновь возрастать.
1, переработка твердого топлива
Коксование
Коксование — процесс переработки каменных углей при их на-
гревании до 900—1050 °C без доступа воздуха. Продуктами коксо-
вания являются летучие вещества и твердый остаток — кокс.
По выходу летучих веществ каменные угли подразделяются на
следующие марки: Д — длиннопламенные, Г— газовые, Ж — жир-
ные, К — коксовые, ОС — отощенно-спекшиеся: Т — тощие, ПА —
полуантрациты, А — антрациты. Из углей марок, Д, Г, Ж, К, ОС
выделяется от 18 до 44% летучих веществ, тогда как из угля
остальных марок — от 1 до 14%. Основные залежи каменных углей
в СССР, составляющие около 2700 млрд, т, находятся в Краснояр-
ском крае и Якутской АССР (55,3%), в Сибири (31,4%), Евро-
пейской части (7,6%); 5,7% углей приходится на долю залежей,
расположенных в Закавказье, Дальнем Востоке, Казахстане, Сред-
ней Азии и других районах.
Рассмотрим процессы, происходящие при нагревании угля без
доступа воздуха.
При медленном и постепенном нагревании компоненты угля
претерпевают физические и химические превращения:
1) до 250 °C происходит выделение окиси и двуокиси углерода
и испарение воды;
2) при температуре 300—350°C в газовую фазу выделяются
пары смол и образуется пирогенетическая вода, а уголь переходит
в пластическое состояние;
3) при 500—550 °C происходит разложение пластической массы
угля с образованием первичных продуктов газа и смолы, состоя-
щей из парафиновых, нафтеновых, непредельных и ароматических
углеводородов, и затвердевание массы с образованием полукокса;
293
4) при 700 °C и выше происходит дальнейшее выделение лету-
чих продуктов и упрочнение кокса.
Летучие продукты, находясь в зоне высоких температур, под-
вергаются пиролизу. Парафиновые углеводороды разлагаются на
предельные и непредельные углеводороды
С4Н10 --► СгНв + С2Н4
Непредельные углеводороды подвергаются полимеризации и де-
гидрированию
2С2Н4 —> сн2=сн-сн=сн2 + н2
Образующиеся диеновые углеводороды взаимодействуют с олефи-
нами с образованием ароматических углеводородов
СН2=СН—СН=СН2 + СН2=СН2 —> —> -^j| + 2H2
Нафтеновые углеводороды подвергаются дегидрогенизации с обра-
зованием ароматических углеводородов
"I*
Ароматические углеводороды, являясь более стойкими в усло-
виях коксования, накапливаются в смоле. Следовательно, парога-
зовая смесь, образующаяся в процессе коксования, содержит в
основном соединения ароматического ряда и газы, содержащие
водород, метан, окись и двуокись углерода и др. Таким образом,
коксование представляет собой двухфазный процесс, включающий
процессы теплопередачи, диффузии и разнообразных химических
превращений.
Из прямого коксового газа выделяют такие ценные продукты,
как каменноугольная смола, сырой бензол, подсмольнай вода и
аммиак.
Из 1 т перерабатываемого угля в среднем выделяются про-
дукты (в кг):
Кокс................. 700-800
Смола................. 20—40
Бензол................ 8—12
Коксовый газ, м3....283—340
Аммиак ................ 2—4 4
Большая часть серы и все минеральные вещества исходных уг-
лей остаются в коксе.
Продукты коксования и их использование
Кокс — твердый, матово-черный, пористый (пористость 45—
55%), обуглероженный продукт, органическая Часть которого
содержит 94—96% углерода, 1,5% водорода, 1,5—2,0% кисло-
рода и 1,2—1,7% серы. Качество кокса определяется механической
прочностью, теплотворной способностью, горючестью (характери-
294
зует скорость горения кокса), реакционной способностью (ско-
рость восстановления им двуокиси углерода), содержанием серы,
золы, летучих веществ и влаги. При нагревании до 800 °C из
кокса не выделяются летучие вещества.
Присутствие серы резко снижает качество кокса, так как при
восстановительной плавке железных руд сера переходит в чугун,
понижая его качество. Повышенное содержание воды уменьшает
теплотворную способность кокса, поэтому количество воды должно
быть не более 5%. В состав кокса входит 10—11% минеральных
веществ, которые являются балластом и при его сгорании перехо-
дят в золу.
Кокс широко применяется в металлургии, литейном производ-
стве, как сырье для получения электродов, для газификации
с целью получения газообразных продуктов и как химический реа-
гент и топливо в ряде процессов.
Прямой коксовый газ представляет собой смесь газообразных
и парообразных веществ. При коксовании на 1 т сухой шихты об-
разуется до 340 м3 газа, состав которого в основном зависит от
температуры коксования.
Коксовые печи
Сырьем для получения кокса служат коксующиеся и спекаю-
щиеся угли марки К, образующие прочный и пористый кокс, од-
нако таких углей в недрах земли содержится мало, поэтому с
целью расширения сырьевой базы для коксования готовят тонко-
измельченную (до 3 мм) смесь — шихту, в состав которой, напри-
мер, входит по 20% углей марок Г, К, ОС и 40% углей марки Ж..
Такая смесь углей при коксовании обеспечивает получение каче-
ственного кокса.
Процесс коксования осуществляют в печах, получивших назва-
ние коксовых. Коксовые печи — камеры являются печами косвен-
ного нагрева, т. е. нагревание в них шихты происходит в результате
передачи тепла греющих газов через стенку камеры. Они футеро-
ваны огнеупорным (динасовым) кирпичом. Камеры по 60—70 штук
соединяются между собой в коксовые батареи. Между камерами
имеются пространства (простенки), в которых сжигается генера-
торный или коксовый газ. Температура в простенках печи дости-
гает 1400 °C. Так как огнеупорный кирпич и уголь являются пло-
хими проводниками тепла, а для получения кокса требуется нагре-
вать шихту до 900—1050°C, то камеры делают в виде узких
каналов: шириной — 0,4 м, длиной—13—14 м и высотой —
4—4,5 м. В такую печь загружается до 15 т шихты.
На рис. XIII. 2 показана схема коксовой печи. Она имеет две
торцевые стороны — коксовую (на рисунке показана справа), куда
выталкивается из камеры кокс (коксовый пирог), и машинную
(слева) — для ввода в камеру коксовыталкивателя 5, представ-
ляющего собой пластину, размер которой немного меньше сечения
295
камеры. При коксовании машинная и коксовая стороны камеры
плотно закрываются дверцами 3 и 4.
Через отверстия в своде камеры загружается шихта с помощью
загрузочного вагона с бункерами, который перемещается по рель.
1 2
Горячие газы, оБогреВаю-
щие камеры
Рис. ХШ. 2. Схема коксовой камеры:
/—бункеры; 2—стояк; 3, 4—дверцы камеры; 5—коксовыталкиватель.
газы
Рис. ХШ. 3. Схема нагревания ка-
мер коксовой батареи (поперечный
разрез) с перекидными каналами:
1 — камера; 2—простенки; 3—отверстия
для газа; 4—регенераторы.
совому пути, расположенному над коксовыми камерами, и обслу-
живает десятки камер. По окончании загрузки шихта в камере раз-
равнивается,- загрузочные отверстия
закрываются, и начинается процесс
коксования. Парогазовая смесь вы-
водится из камеры через стояк 2,
соединенный с газопроводом.
На рис. ХШ. 3 показана схема
нагревания шихты в камерах коксо-
вой батареи (поперечный разрез) с
перекидным над сводами камер хо-
дом топочных газов. Воздух, нагре-
тый в регенераторах 4, смешивается
с газом, который выходит из отвер-
стий 3, расположенных между ка-
мерами /, и в простенке 2 происхо-
дит сгорание газообразного топлива.
Нагретые топочные газы огибают
камеру, подогревают ее с другой
стороны и уходят через регенерато-
ры тепла, нагревая в них насадку, в
дымовую трубу. Поток газа и воз-
духа переключают через 20—30 мин
на нагретые топочными газами регенераторы, при этом поток газов
обогревает обратную сторону камеры. Такое устройство позволяет
равномерно обогревать камеру с обеих сторон. Шихта находится
в камере 13—17 ч. Выделяющиеся при коксовании газы выводятся
из камеры и подаются на переработку.
296
По окончании процесса коксования камеру отключают от газо-
хода коксового газа, открывают дверцы с обеих торцевых сторон
камеры и выталкивают кокс коксовыталкивателем в гасильный
вагон, находящийся с коксовой стороны камеры. Гасильный вагон
электровозом подается в гасильную башню, где кокс гасится во-
дой. После охлаждения кокс подают на сортировку и хранение.
Разгрузка камер проводится поочередно. Нетрудно подсчитать,
что при времени нахождения шихты в камере в течение 13—17 ч
коксовая батарея, состоящая из 60—70 камер, выдает кокс через
15—30 мин.
После разгрузки камеры ее торцевые стороны закрываются, и
цикл работы повторяется. Один коксовыталкиватель, передви-
гаясь вдоль батареи, обслуживает все камеры.
Все операции по обслуживанию коксовых печей (загрузка, съем
и закрытие дверей и люков, выдача и тушение кокса и т. д.) меха-
низированы и автоматизированы. В настоящее время ведутся ис-
следования по разработке непрерывно действующих коксовых
камер.
Переработка коксового газа
Парогазовая смесь, выходящая из коксовой камеры, назы-
вается прямым коксовым газом. В 1 м3 этого газа помимо водо-
рода, метана, окиси и двуокиси углерода и газообразных углево-
Рис. ХШ. 4. Схема переработки прямого коксового газа:
Z—газосбориик; 2—холодильник; 3—сборник; 4—электрофильтр; 5—турбогазодувка; 6—подо-
греватель; 7 —сатуратор; 8—холодильник; 9—башни.
Дородов содержится 80—130 г смолы, 30—40 г бензольных угле-
водородов, 8—13 г аммиака, 6—25 г сероводорода и других
сернистых соединений, 0,5—1,5 г цианистых соединений, 250—
450 г паров воды, 15—35 г. твердых частиц. Схема переработки
такого газа приведена на рис. XIII. 4.
Газ выходит из коксовых камер при средней температуре около
700 °C и попадает в общий для всех камер газосбориик /. в
297
который интенсивно впрыскивается охлажденная надсмольная вода4
При этом газ охлаждается до 80 °C и из него частично конденси-
руется смола. Одновременно в газосборнике из газа удаляются
частицы угля. Дальнейшая конденсация смолы происходит в труб-
чатом холодильнике 2, где газ охлаждается до 30 °C. Одновременно
в холодильнике 2 конденсируются пары воды, и в образующейся
воде частично растворяются аммиак и другие вещества; накапли-
вающуюся здесь жидкость называют надсмольной водой. Смола и
надсмольная вода стекают из холодильника 2 в сборник 3, где
разделяются за счет различной плотности.
При охлаждении коксового газа часть смолы конденсируется
в объеме с образованием тумана, для выделения которого газ на-,
правляется в электрофильтр 4-, одновременно с туманом в электро-
фильтре осаждаются взвешенные в газе твердые и жидкие час- '
тицы. Накапливающаяся в электрофильтре смола также стекает
в сборник 3. Охлажденный и очищенный газ эксгаустером 5 (на-
гнетателем) подается в подогреватель 6, а из него в сатуратор 7, ’
где аммиак поглощается раствором, содержащим серную кислоту. ’ <
В результате взаимодействия аммиака с серной кислотой обра- ; I
зуется сульфат аммония (NH^SO^ который используется как j
азотное удобрение. Вместе с аммиаком в сатураторе улавливаются *
и другие содержащиеся в коксовом газе продукты. 4 1
Из сатуратора 7 газ направляют в башню-холодильник 8, где • i
он охлаждается до 25 °C при непосредственном соприкосновении
с водой, орошающей насадку башни.
Коксовый газ, охлажденный и очищенный от аммиака, направ-
ляют на улавливание сырого бензола в скрубберы 9. В качестве
поглотителей бензола и его гомологов применяется соляровое
масло. '
Освобожденный от сырого бензола и очищенный от соединений
серы коксовый газ (так называемый обратный коксовый газ) со-
держит 54—60% водорода, 23—30% метана, 5—7% СО, 2—3%
углеводородов, примеси азота, кислорода и СОг.-
Раствор сырого бензола в соляровом масле направляют в ди- 1
стилляционную колонну, где из него отгоняют сырой бензол, а ,
масло после охлаждения вновь возвращают на орошение бензоль-
ных скрубберов.
Из приведенных данных видно, что при переработке прямого
коксового газа получают надсмольную воду, каменноугольную
смолу, сырой бензол и обратный коксовый газ. Надсмольная вода
представляет собой водный раствор, содержащий бензол, аммиак
и различные аммонийные соли: (ИН^гСОз, (NH4)2S, NH4SH,
NH4C1, NH4CNS, (NH4)2SO4 и др. Для разложения солей аммония
надсмольную воду обрабатывают гидроокисью кальция
(NH4)2SO4 +Са(ОН)2 —► 2NH3 + CaSO4 + 2Н2О (
и выделяющийся аммиак отгоняют с острым паром. Он исполь-
зуется для получения аммиачной воды и сульфата аммония.
298
Каменноугольная смола — вязкая черно-бурая жидкость, обла-
дающая специфическим запахом и содержащая около 300 различ-
ных веществ. Вначале смолу разделяют на фракции, из которых
затем получают отдельные вещества (табл. XIII. 3).
Таблица XIII. 3. Первичные фракции при разгонке каменноугольной
смолы и сырого бензина
Фракции смолы Выход, % Температура кипения, °C Фракции сырого бензола Температура кипения, °C
Легкое масло 0,5-1,0 До 180 Сероуглеродная До 80
Нафталиновое масло 6,0-8,0 210-230 Бензольная До 100
Фенольное масло 1,7—2,0 180—210 Сероуглеродная До 80
Тяжелое (поглоти- 11-13 230-270 Толуольная 100-125
тельное) масло Ксилольная 125-150
Антраценовое масло 18-25 270-360 Тяжелый бензол 150-180
(пропиточное) Пек 55-60 Выше 360 f (сольвенты) 150-180
Так, например, при охлаждении нафталиновой фракции выпа-
дают кристаллы нафталина, а антраценовой — антрацен. Фенол
извлекается путем обработки фенольного масла разбавленным
раствором NaOH
CSH6OH ч- NaOH —> C6H5ONa + Н2О
с последующим разложением образующегося фенолята двуокисью
углерода
CeH6ONa + СО2 + Н2О —> CeH6OH + NaHCO3
Из каменноугольной смолы выделяют около 100 индивидуаль-
ных веществ; бензол, толуол, ксилол, фенол, крезол, нафталин,
антрацен, фенантрен, пиридин и др., которые служат сырьем в про-
изводстве красителей, фармацевтических препаратов, инсектофун-
гицидов и др.
Сырой бензол представляет собой смесь сероуглерода, бензола,
толуола и других веществ. Каждый из этих компонентов после
выделения из сырого бензола используется как сырье в химической
промышленности.
Обратный коксовый газ является высококалорийным топливом
и нашел применение в металлургии, стекловарении, при коксова-
нии, а также используется как сырье в химической промышленно-
сти (получение Нг, сажи и др.)
Полукоксование
Для полукоксования (сухая перегонка топлив при 400—600°C)
применяют каменные угли, торф, сланцы. При полукоксовании уг-
лей получают полукокс, полукоксовый газ, смолу и надсмольную
299
воду. Полукокс используется в качестве топлива, а полукоксовый
газ (содержит СН4 и Н2) применяется в быту, для нагревания
промышленных агрегатов и как сырье для химической промышлен-
ности. Из смолы, состоящей из смеси углеводородов, извлекают
фенол и другие индивидуальные органические соединения, а оста-
ток используют как топливо.
При полукоксовании торфа получают полукокс, торфяной деготь
и надсмольную воду. Торфяной полукокс (твердый продукт, со-
держащий небольшое количество серы и фосфора) применяют в
качестве восстановителя, при приготовлении активированных уг-
лей, для газификации и т. д., а торфяной деготь служит сырьем
для получения воска, парафина, пека и других продуктов. Из над-
смольной воды извлекают уксусную кислоту и фенол.
Полукоксование горючих сланцев проводят с целью получения
смолы, которая широко используется для получения моторного топ-
лива, растворителей: Полукокс (содержит около 10% углерода и
90% минеральных веществ) применяется при получении вяжущих
веществ, а газы —для обогрева печей и в бытовых целях.
Сухую перегонку дерева проводят без доступа воздуха при
400—450 °C, при этом получают древесный уголь, древесную смолу,
водный конденсат и газы.
Древесный уголь не содержит серы и фосфора, поэтому он на-
шел применение как высококачественное топливо и восстановитель
в металлургии, для получения сероуглерода, а также как адсор-
бент и т. д.
Древесная смола используется для пропитки древесины с целью
предохранения ее от гниения, для изготовления фармацевтических
препаратов, флотационных реагентов и др. Из водного конденсата
выделяют метиловый спирт, уксусную, муравьиную и пропионовую
кислоты, ацетон, ацетальдегид, форфурол и др. Газы сухой пере-
гонки содержат 35% СО, 10% метана и его гомологов и 55% СО2.
Их используют как газообразное топливо.
Газификация твердого топлива
Газификация твердого топлива — процесс превращения органи-
ческой части топлива в горючий газ путем воздействия на него
окислителей. В зависимости от состава топлива, природы окисли-
теля, температуры и давления газы газификации различны по со-
ставу.
Для превращения твердого топлива в газообразное применяют
воздух, водяной пар, смесь воздуха и водяного пара или смесь во-
дяного пара и кислорода. В зависимости от используемого дутья,
режима и окислителей получают воздушный, водяной, паровоздуш-
ный, парокислородный и другие газы, отличающиеся друг от друга
содержанием горючих компонентов (СО, Н2, СН4 и др.).
Скорость газификации зависит от свойств твердого топлива,
размера его частиц, температуры и газифицирующего реагента и
30Q
др. Наибольшей реакционной способностью обладают древесный
уголь, торфяной кокс. Чем меньше размеры частиц топлива, тем
выше скорость газификации, так как при этом увеличивается, по-
верхность контакта топлива с окислителем.
.В зависимости от размера частиц газификацию проводят в га-
зогенераторах шахтного типа (куски от 25 до 100 мм), а также
в псевдоожиженном (кипящем) слое (тонкоизмельченное топливо)
при температуре газификации от 900 до 1200 °C, но не выше тем-
пературы плавления золы, так как
ее удаление и нарушает режим га-
зификации.
Газификацию твердых топлив
проводят в аппаратах — газогенера-
торах шахтного типа (рис. XIII. 5),
представляющих собой вертикаль-
ную шахту 3 цилиндрической фор-
мы диаметром 3,5 м и высотой 4,5 м,
футерованную огнеупорным кирпи-
чом. Нижняя часть шахты газогене-
ратора опущена во вращающуюся
чашу 5, заполненную водой для соз-
дания гидравлического затвора,
причем стенки шахты не доходят
до дна чаши и находятся в подве-
шенном состоянии. В чаше 5 кре-
пится колосниковая решетка 4, че-
рез которую подается газифици-
рующий окисляющий агент.
Газогенератор сверху периоди-
чески загружается кусковым топли-
вом через загрузочную коробку 1
при опущенном конусе затвора 2
и закрытой крышке коробки. Пр
агента через газогенератор происходит выгорание углерода из топ-
лива и оно постепенно опускается вниз. Образующаяся зола про-
ходит через отверстия колосниковой решетки и гасится водой в
чаше 5, откуда удаляется.
В качестве примера рассмотрим процесс газификации с целью
получения воздушного газа. Воздух, подаваемый в газогенератор,
проходит сквозь колосниковую решетку 4 и попадает в зону шлака
и золы, где нагревается, охлаждая при этом шлак и золу. Кисло-
род воздуха, соприкасаясь с углеродом топлива в зоне газифика-
ции I, окисляет его
С 4- О2 —► СО2 4- Q
шлакование золы затрудняет
воздух
Рис. XIII. 5. Схема газогенератора:
/—загрузочная коробка; 2 —конус за-
твора; 3—шахта; 4—колосниковая ре-
шетка; 5—чаша; 6—шлак; I — зона га-
зификации; II — зона сухой перегонки;
III — зона сушки.
и пропускании окисляющего
Двуокись углерода, поднимаясь в верхнюю часть газогенератора,
взаимодействует с углеродом с образованием окиси углерода
С4-СО2 —> 2CO-Q
301
Суммарный тепловой эффект этих реакций положительный, по-
этому газификацию можно проводить непрерывно.
Нагретые газы из зоны I поступают в зону сухой перегонки
//, где они нагревают топливо, при этом из него удаляются лету-
чие продукты. В зоне подсушки III за счет тепла проходящих га-
зов происходит подсушка топлива. Воздушный газ выводится из
верхней части газогенератора через отверстия, расположенные
в верху стенки шахты. Для поддержания оптимальной температуры
газификации (1000—1100°C) к воздуху добавляют небольшое ко-
личество водяного пара.
Теоретически воздушный газ должен содержать 34,7% 'СО и
65,3% Na, но практически в нем содержится 30—32% СО. Теплота
сгорания воздушного газа около 5000 кДж/м3.
Водяной газ получают при пропускании через нагретое твер-
дое топливо паров воды:
С + Н2о —> со + н2—Q
Так как процесс получения водяного пара эндотермический, то
вначале через газогенератор пропускают воздух для нагрева топ-
лива до 1500 °C, а затем — пар для получения водяного газа. Пар
пропускают до снижения температуры в зоне газификации до
1000 °C, после чего снова в газогенератор подают воздух, и т. д.
Теоретически водяной газ должен состоять из 50% Н2 и 50% СО,
но в результате протекания побочных реакций в газе содержатся
51% Н2, 33% СО, 9% СО2, 7% N2> Н2 и др. Топлота сгорания та-
кого газа около 10000 кДж/м3.
Паровоздушный газ получают при пропускании через твердое
топливо смеси воздуха и пара. Состав газа зависит от соотношения
пара и воздуха в смеси, подаваемой в газогенератор.
Парокислородный газ образуется при пропускании смеси кис-
лорода и пара через твердое топливо. Он не содержит азота, по-
этому обладает более высокой теплотой сгорания (12142 кДж/м3).
Получение парокислородного и паровоздушного газов прово-
дится непрерывно, так как в процессе газификации протекают од-
новременно экзотермические (преобладают) и эндотермические
реакции, поэтому температура процесса может регулироваться со-
отношением окисляющих агентов (пар — воздух, пар — кислород).
Газификацию мелкозернистого и пылевидного топлива прово-
дят в газогенераторах с кипящим слоем. Работа этих газогенера-
торов принципиально не отличается от обжига колчедана в печах
с кипящим слоем. Газогенератор — шахта высотой до 20 м и диа-
метром 5 м. Этот способ газификации очень интенсивен. Так, в га-
зогенераторе с кипящим слоем можно получить до 25 000 м3/г
парокислородного газа.
Небольшое применение в СССР получил способ подземной га-
зификации углей. Для проведения процесса газификации до пласта'
угля бурят две скважины на расстоянии друг от друга примерно
15—20 м. В одной из скважин поджигают уголь и подают в нее
302
воздух. Нагреваясь, уголь в пласте становится проницаемым для
газов. При непрерывной подаче воздуха через первую скважину
из второй скважины отсасывают образующийся воздушный газ.
Теплота сгорания его 3500—3800 кДж/м3.
Газификацию твердых топлив с целью получения высококало-
рийных газов проводят под давлением, но устройство газогенера-
торов при этом усложняется.
2. ПЕРЕРАБОТКА НЕФТИ
Нефть и продукты ее переработки широко используются в на-
родном хозяйстве. Процессы переработки нефти известны давно.
Еще в середине прошлого столетия эксплуатировались установки
для отгонки из нефти керосина: керосин использовали для осве-
щения, а полученный остаток сжигали как топливо. Развитие ав-
томобильного, а затем авиационного транспорта способствовало
увеличению спроса на бензин, что привело к разработке новых
методов переработки нефти. С усовершенствованием методов пе-
реработки нефти расширилась сырьевая база для химической про-
мышленности и, в частности, для органического синтеза.
Нефть в основном (90—98%) состоит из углерода и водорода;
в небольшом количестве в ней содержатся кислород (0,1—1,0%),
сера (0,1—3,0%), азот (0,001—0,4%), ванадий, никель, титан, ка-
лий, фосфор, германий и др.
Плотность нефтей колеблется от 820—900 кг/м3, а средняя мас-
са 1 моля — от 250 до 300. Теплотворная способность нефти состав-
ляет 40 000—44 000 кДж/кг, температура кипения обычно ниже
100 °C, а температура застывания изменяется в широких пределах
от —20 до 4-20 °C.
Нефть представляет собой смесь различных углеводородов, ко-
торые делятся на низкокипящие (до 360°C) и высококипящие
(температура кипения выше 360°C). К первой группе относятся
следующие соединения:
а) СпН2п+2 (метановые и парафиновые углеводороды);
б) СпН2п (моноциклические, полиметиленовые углеводороды,
циклопарафины, нафтены);
в) СпН2п-2 (бициклические полиметиленовые, дициклические
парафины);
г) СпН2п-4 (трициклические полиметиленовые, трициклопара-
фины);
д) СпН2п_6 (моноциклические ароматические, бензольные угле-
водороды) ;
е) СпН2п-з (нафтено-ароматические углеводороды);
ж) СпН2п_12 (бициклические ароматические углеводороды).
К высококипящим относятся высокомолекулярные парафино-
вые углеводороды, моно-, би- и трициклические циклопарафино-
вые углеводороды, би- и трициклические ароматические углеводо-
роды и др.
333,
Углеводороды парафинового ряда от С5Н12 до С15Н32 представ-
ляют собой жидкие продукты и составляют основу нефти, в кото-
рой растворены газообразные (СН4 до С4Ню) и твердые (от С16
и более атомов углерода) углеводороды. Из нафтеновых углеводо-
родов наибольшее количество приходится на циклопентан, цикло-
гексан и их производные, а из ароматических углеводородов: бен-
зол, толуол, ксилол. Кислородные соединения в нефти находятся
в виде фенолов, нафтеновых и других кислот, а сернистые — в виде
меркаптанов, сульфидов, дисульфидов, тиофенов. В нефти содер-
жатся также пиридин, хинолин, амины, т. е. соединения, содержа-
щие азот.
В зависимости от наибольшего содержания в нефтях соедине-
ний того или иного класса их принято классифицировать на мета-
новые, метан-нафтеновые, нафтеновые, нафтено-ароматические и
ароматические, а по содержанию серы на малосернистые (до
0,5%), сернистые (от 0,51 до 2,0%) и высокосернистые (более 2%)
нефти.
В зависимости от содержания парафинов нефти делят на мало-
парафинистые (до 1,5%), парафинистые (от 1,51 до 6,0%) и высо-
копарафинистые (свыше 6,0). Кроме того, в нефти содержатся ме-
ханические примеси, вода и минеральные соли. Механические при-
меси— твердые частицы песка и глины, уносимые из земли при ее
добыче, вода может содержаться в нефти в виде стойкой эмульсии
и механически примешанной, легко отделяемой от нефти. В ней на-
ходятся минеральные соли (СаС12, MgCK и др.).
После очистки в нефти должно содержаться 0,1—0,3% воды и
10—40 мг/л хлористых солей.
.Фракционный состав нефти определяется в зависимости от тем-
пературы кипения составляющих, причем фракции, кипящие выше
360 °C, разгоняются в вакууме. При разгонке нефти получают сле-
дующие фракции: бензиновую — до 170, лигроиновую — от 160 до
210, керосиновую — от 200 до 300, газойлевую — от 270 до 350 °C
и другие, которые в дальнейшем используют для получения раз-
личного вида продуктов.
В процессе переработки нефти получают топливо (жидкое и га-
зообразное), растворители, смазочные масла, консистентные смаз-
ки, твердые и полутвердые смеси углеводородов (парафины, вазе-
лины и т.д.), битумы и пеки, нафтеновые кислоты и их производные
(сульфокислоты, мылонафты и др.), индивидуальные углеводо-
роды— газообразные (этилен, пропилен и др.) и жидкие (бензол,
толуол, ксилол и др.).
В зависимости от дальнейшего использования жидкое топливо
делится на моторный бензин, тракторное, дизельное и котельное
топливо, топливо для реактивных и турбореактивных двигателей.
Моторный бензин применяют в качестве топлива для поршневых
карбюраторных двигателей с искровым зажиганием (самолеты, ав-
томобили, мотоциклы и другие машины и установки). Такой бен-
зин должен обладать определенными свойствами (фракционный со-
304
став, давление насыщенных паров, достаточная детонационная
стойкость, химическая стабильность и отсутствие коррозии аппа-
ратуры) .
Фракционный состав бензина характеризуется температурой на-
чала и конца кипения фракций, получаемых при разгонке бензина
в интервале 30—200 °C; предельное давление насыщенных паров
устанавливается ГОСТ.
Детонационная стойкость — важная характеристика бензина.
В цилиндр двигателя внутреннего сгорания поступает смесь паров
бензина с воздухом, которая сжимается поршнем и зажигается от
запальной свечи (искры). Образующиеся при горении газы дви-
гают поршень. К. п. д. двигателя тем выше, чем больше степень
сжатия смеси в цилиндре. Однако степень сжатия ограничена ха-
рактером горения смеси в цилиндре: при нормальном горении ско-
рость распространения пламени составляет 10—20 м-с-1, но при
некоторых степенях сжатия наступает детонация, при которой пла-
мя распространяется со скоростью до 2500 м-с-1. Появление дето-
нации сопровождается стуком в цилиндре, перегревом, черным
дымом на выхлопе и приводит к повышению расхода топлива;
кроме того, при этом снижается мощность двигателя, и он прежде-
временно изнашивается.
Склонность бензина к детонации характеризуется октановым
числом, которое определяется сравнением его со смесью, состоя-
щей из изооктана и н-гептана. Принято считать, что изооктан, мало
склонный к детонации, имеет октановое число 100, а н-гептан,
чрезвычайно склонный к детонации, имеет октановое число 0. Ок-
тановое число бензина будет равно процентному содержанию изо-
октана в стандартной смеси и смеси изооктана и н-гептана, кото-
рая детонирует при той же степени сжатия, что и испытуемый
бензин. Например, если детонационная стойкость бензина оказа-
лась такой же, как смеси, содержащей 80% изооктана и 20%
н-гептана, то его октановое число будет равным 80. Чем больше
октановое число, тем выше качество бензина.
Октановое число бензина изменяется в широких пределах в за-
висимости от его назначения. Например, для автомобиля «Жи-
гули» употребляется бензин с октановым числом 93, а для автомо-
биля «Волга» — с октановым числом 76. Авиационный бензин для
самолетов с поршневым двигателем выпускается с октановыми
числами 98—100.
Октановое число зависит от состава топлива: увеличению окта-
нового числа способствует присутствие в топливе изопарафинов
и ароматических соединений. Для увеличения октанового числа
к бензину иногда добавляют антидетонаторы, например тетраэтил-
свинец РЬ(С2Н5)4, который входит в состав так называемой этило-.
вой жидкости. Введение на 1 л бензина 3 мл этиловой жидкости
повышает октановое число бензина от 70 до 90. Этиловую жидкость
подкрашивают, так как она ядовита, и работа с ней, а также с эти-
лированным бензином, требует осторожности,
305
Топливо для реактивных двигателей представляет собой опре-
деленную фракцию керосина (температура кипения в пределах
150—280°C), а для самолетов с большой высотой полета применя-
ют,у тяжеленный керосин (195—315°С). В этом случае применяемое
горючее не должно содержать непредельных углеводородов, спо-
собных к образованию смол, кристаллизующихся и выпадающих
в осадок парафинов, что приводит к засорению топливной си-
стемы. В топливе должно быть минимальное содержание аромати-
ческих углеводородов, которые склонны к нагарообразованию, гиг-
роскопичности и могут образовать кристаллики льда.
Дизельное топливо — керосин, газойль и соляровый дистиллят —
характеризуется цетановым числом. Цетановое число — процентное
содержание (по объему) цетана (СчеНзч) в смеси с а-метилнафта-
лином (С10Н4—СН3), эквивалентное по самовоспламеняемости
топливу в стандартных условиях испытания. Принято, что цетано-
вое число цетана равно 100, а «-метилнафталина — 0. Цетановое
число для дизельных топлив составляет от 40 до 50.
Котельное топливо (мазут и другие остатки) сжигается в топ-
ках тепловозов, пароходов, тепловых электростанций и в промыш-
ленных печах.
Тракторным топливом является керосин, октановое число кото-
рого должно быть не менее 40.
Смазочные масла в зависимости от применения делятся на ин-
дустриальное (веретенное, машинное и др.), для двигателей внут-
реннего сгорания (автолы, авиационное масло и др.), трансмиссион-
ное, турбинное, компрессорное и масло специального назначения,
Методы переработки нефти и нефтепродуктов
Добытая на промыслах сырая нефть содержит растворенные
газы, минеральные соли, воду и механические примеси, поэтому
перед переработкой ее подвергают подготовке и очистке.
В аппаратах, называемых трапами, из нефти выделяются по-
путные газы, а- в ряде случаев и смеси легчайших углеводородов,
называемых газовым бензином. Газовый бензин отделяется от по-
путных газов абсорбцией его соляровым маслом или адсорбцией
активированным углем.
Минеральные соли удаляют в процессе обессоливания, т. е.
промывкой нефти несколькими порциями теплой воды. При дли-
тельном отстаивании от нефти отделяются вода и механические
примеси. Поскольку вода с нефтью образуют стойкие эмульсии,
для полного обезвоживания в нефть добавляют при нагревании
разрушающие эмульсии деэмульгаторы. Эффективным способом
очистки является электрообезвоживание нефти, которое заклю-
чается в том, что при прохождении пленки нагретой нефти между
электродами, питаемыми переменным током напряжением 30—
40 тыс. В, происходит разрушение эмульсии; водяные капельки
сливаются в более крупные и осаждаются.
306
Методы переработки нефти и жидких нефтепродуктов можно
разделить на физические и химические.
Физические методы переработки нефти основаны на различии
физических свойств входящих в ее состав углеводородов — темпе-
ратуры кипения, кристаллизации, растворимости и т. п.
Химические методы переработки основаны на глубоких хими-
ческих превращениях, которые претерпевают углеводороды, содер-
жащиеся в нефти или нефтепродуктах, под влиянием температуры,
давления и катализаторов. Наибольшее распространение среди
этих методов получили термические и каталитические процессы.
Аппаратура, применяемая для осуществления физических и хи-
мических процессов переработки нефти и нефтепродуктов, должна
обеспечивать, во-первых, нагревание сырья до высокой темпера-
туры, при которой процесс протекает с достаточной для практиче-
ских целей скоростью, и, во-вторых, разделение получаемых
продуктов. Нагревание нефти или нефтепродуктов производится,
главным образом, в трубчатых печах, а разделение продуктов
нефтепереработки обычно осуществляется в ректификационных ко-
лоннах. Наибольшее распространение получили колонны с барбо-
тажными колпачками.'
Каталитические Процессы на нефтеперерабатывающих установ-
ках проводят в контактных аппаратах разнообразной конструкции.
Как правило, катализатор в этих процессах быстро теряет актив-
ность, потому контактные узлы включают контактные аппараты и
регенераторы, где происходит восстановление активности катали-
затора. Наряду с основными аппаратами нефтеперерабатывающие
установки оснащены теплообменниками, конденсаторами, хранили-
щами и другой вспомогательной аппаратурой.
Физические методы
Перегонка нефти — процесс разделения нефти на части или
фракции, отличающиеся температурой кипения. Для разделения
нефти на фракции ее нагревают в трубчатой печи (рис. XIII. 6) или
в печах других типов, в которой нефть под избыточным давлением,
создаваемым насосом, движется внутри тонких труб, обогреваемых
теплом сжигаемого жидкого или газообразного топлива. Нефть
проходит по трубам так называемой конвекционной секции 3, где.
подогревается выходящими из печи топочными газами, затем по
трубам потолочного экрана 2 и фронтального экрана 1 и выходит
из печи. Нефть в трубах экранов 1 и 2 нагревается за счет конвек-
тивного тепла при сжигании топлива в форсунке 4. Нагретая нефть
Далее поступает на перегонку.
Для перегонки нефти используют одноступенчатые и двухсту-
пенчатые трубчатые установки. На рис. XIII. 7 показана схема
Двухступенчатой установки, где вначале перегонку ведут при атмо-
сферном давлении с выделением бензиновой и других высококипя-
Щих фракций; остаток — мазут — затем перегоняют в вакууме во
307
избежание расщепления углеводородов при действии высокой тем-
пературы.
Предварительно нагретая до 170—175 °C в теплообменниках з,
8 и 11 нефть поступает в трубчатую печь 1, нагревается до 350 °C
Рис. XIII.6. Схема трубчатой печи:
1, 2—экраны; 3—трубы конвекционной секции; 4—форсунки.
топочными газами и подается в ректификационную колонну 2 пер-
вой ступени, где давление снижается и происходит испарение фрак-
ций. Пары, поднимаясь в верх колонны, постепенно охлаждаются
бензин
Мазут
Лигроин
Керосин
^Саляробый.
Оистиллат
Нефть
* Веретенный
Зистиллат
^Машинный.
г Вист иллит
^Легкий цилинд-
робый диет иллит
Тяжелый цилинд*
робый Зистиллат
_ Гудрон
8
Н бакуум^насосц
Рис. ХШ.7. Схема двухступенчатой установки для перегонки нефти:
I. 6—трубчатые печи; 2, 7—ректификационные колонны; 3, 8. 11 — теплообменники; 4, 9—кон-
денсаторы; 5, 10—холодильники.
жидкостью (флегмой), стекающей сверху. При соприкосновении
паров с жидкостью происходит разделение смеси на фракции по
температурам кипения в результате многократного чередования
процессов испарения жидкости и конденсации ее паров (ректифи'
кация).
308
Пары бензина, как наиболее низкокипящая фракция, выходят
с верху колонны 2, охлаждаются в теплообменнике 3 и конденси-
руются в конденсаторе 4. Часть жидкого бензина выводится как
готовый продукт, а часть его подается на орошение колонны
(флегма). По высоте колонны выводят и другие продукты — лиг-
роин, керосин, соляровый дистиллят, которые после охлаждения
в холодильниках 5 направляют на использование.
Остаток от перегонки нефти — мазут выводят из колонны 2
снизу и направляют в трубчатую печь 6 второй ступени, работаю-
щую в условиях вакуума. Здесь мазут нагревается до 400—420°C,
образующиеся пары поступают в ректификационную колонну 7,
где по высоте колонны отбирают дистилляты — тяжелый цилиндро-
вый, легкий цилиндровый и машинный. Эти фракции охлаждаются
в холодильниках 10, орошаемые водой. Из верхней части колонны
выводятся пары веретенного дистиллята, их охлаждают в тепло-
обменнике 8 и конденсируют в конденсаторе 9. Часть веретенного
дистиллята возвращается для орошения колонны, а часть его вы-
водится как готовый продукт. Остаток от перегонки мазута — гуд-
рон выводится из колонны 7.
В зависимости от состава нефти выход дистиллятов различен
/табл. ХШ. 4).
Таблица ХШ. 4. Фракции (дистилляты) при перегонке нефти
Дистилляты Температура отбора, % Примерный выход, % (масс.)
Первая ступень
Бензиновый До 170 14,5
Лигроиновый 160-200 7,5
Керосиновый 200-300 18,0
Соляровое масло 300-350 5,0
Остаток — мазут 350 55,0
Вторая ступень
(перегонка мазута под вакуумом 60—800 мм рт. ст.)
Веретенный
Машинный
Легкий цилиндровый
Тяжелый цилиндровый
Остаток — гудрон
230-250
260-305
315-325
350-370
370
10-12
5
3
7
27-30
*
Выход бензина при прямой гонке зависит от состава нефти и
колеблется от 3 до 20%. Октановое число получаемого бензина
изменяется в пределах от 50 до 80. При добавлении этиловой жид-
кости получают бензин с октановым числом до 95.
309
Химические методы
Термические процессы переработки нефти и нефтяных фракций
связаны с расщеплением углеводородов под влиянием теплового
воздействия, которое определяется температурой, давлением и
продолжительностью пребывания сырья в зоне высокой темпера-
туры. В зависимости от исходного сырья и глубины разложения
углеводородов термические процессы проводят при 450—720 °C и
давлении до 7 МПа. К ним относятся термический крекинг, рифор-
минг, пиролиз и коксование. Эти методы отличаются друг от друга
Рис. XIII.8. Зависимость энергии об-
разования углеводородов из простых
веществ от температуры:
J—бензол; 2—гексан; 3—циклогексан; 4 —
н-гексан.
условиями и степенью расщепле-
ния углеводородов, но все они
позволяют получать дополнитель-
ные количества жидкого топлива
за счет расщепления углеводоро-
дов.
Термический крекинг разде-
ляют на крекинг, протекающий в
двухфазной системе жидкость —
пар (так называемый жидкофаз-
ный), и крекинг, протекающий
только в паровой фазе (парофаз-
ный). Жидкофазный крекинг ве-
дут при температуре 470—540 °C
и давлении до 7 МПа; парофаз-
ный— при температуре выше
550 °C и давлении, близком к ат-
мосферному.
Несмотря на сложность про-
цессов термического разложения
углеводородов можно установить
некоторые общие закономерности поведения отдельных групп угле-
водородов в этих условиях.
Сравнительную стабильность углеводородов можно определить
по зависимости от температуры энергии образования углеводоро-
дов С6 разных классов из простых веществ (рис. XIII. 8).
Энергия образования характеризует термодинамическую воз-
можность осуществления процесса разложения, так как система
всегда стремится перейти в состояние с наименьшей энергией.
Из рис, XIII. 8 видно, что термодинамическая стабильность уг-
леводородов при высокой температуре понижается в следующем*
порядке:
ароматические углеводороды —>- олефииы —>- нафтены—>-парафины
При понижении температуры порядок уменьшения .стабильно-
.сти становится иным:
парафины —> нафтены —> олефины —> ароматические углеводороды
310
Следовательно, при повышенной темепратуре в первую очередь
разрушаются парафиновые и нафтеновые углеводороды и происхо-
дит накопление ароматических углеводородов в продуктах кре-
кинга.
Зависимость скорости реакции разложения от температуры
выражается уравнением Аррениуса. Энергия активации этой реак-
ции для парафиновых и нафтеновых углеводородов составляет
£п = £„ = 200—300 кДж'-моль-1, а для ароматических Еа = 300—
400 кДж-моль-1. Из этого следует, что скорость превращения
у парафиновых и нафтеновых углеводородов более высокая, чем
у ароматических, поскольку Дп, н< Да-
Рис. XIII.9. Зависимость выхода бензина от температуры (а) и времени пребы-
вания (б) сырья в реакторе.
Скорость крекинга приближенно можно описать уравнением
первого порядка
где kcp — усредненная константа скорости реакции;
X — степень разложения.
Указанное уравнение отражает реакцию первичного расщепле-
ния парафиновых углеводородов, высших олефинов и процесса
Деалкилирования.
Усредненная константа скорости реакции kcp уменьшается по
мере углубления процесса, что объясняется замедлением расщеп-
ления устойчивых молекул сырья. Таким образом, при постоянных
температуре и давлении kcp уменьшается во времени.
Основным продуктом переработки нефти является бензин; его
выход вначале увеличивается по мере повышения температуры, так
как происходит разложение малоустойчивых тяжелых углеводоро-
дов с образованием более легких, входящих в состав бензина. Од-
нако при дальнейшем повышении температуры происходит распад
легких углеводородов с образованием газов. Таким образом, су-
ществует оптимальная температура Тоат, при которой обеспечи-
вается максимальный выход бензина (рис. XIII.9).
311
Давление, с одной стороны, подавляет реакции расщепления,
в результате которых образуются газообразные продукты, что
увеличивает выход бензина, с другой стороны, — препятствует про,
теканию вторичных реакций, при которых появляются тяжелые
продукты, уменьшающие выход бензина. Поэтому при постоянной
и достаточно высокой температуре кривая зависимости выхода бен-
зина Хб = }(Р) аналогична кривой Хв = f(T) (см. рис. XIII. 9, а),
и на ней имеется максимум, соответствующий наибольшему вы-
ходу бензина. В соответствии с этим для увеличения выхода жид.
ких продуктов крекинг проводят при повышенном давлении, а для
увеличения выхода газов его проводят при пониженном давлении.
При увеличении времени пребывания исходного сырья в зоне
высоких температур (при Р, Т — const) степень разложения тя-
желых углеводородов сначала повышается, в результате чего вы-
ход бензина возрастает. Однако, при значительном - увеличении
времени происходит расщепление легких углеводородов с выделе-
нием газов и величина Хв снижается (рис. XIII. 9, б).
В результате термического крекинга получают, помимо бензина,
газы и крекинг-остаток.
Рассмотрим на конкретных примерах распад различных углеводородов при
нагревании.
При нагревании нефтяного сырья вначале расщепляются углеводороды пара-
финового ряда с длинной цепью, например
С14Н30 —>• CyHis + С7Н14
При повышении температуры разрыв цепи сдвигается к краю цепи и возможно
выделение метана
СиНзо —> С13Н26 + СН4
Чем выше масса 1 моля парафиновых углеводородов, тем больше скорость их
расщепления. Так, если принять скорость крекинга пентана (С5Н12) при 400—
425 °C за 1, то скорость разложения октана (CsHi8) будет равна 16, а эйкозана
(С20Н42) — 120.
Газообразные парафины при термическом крекинге являются устойчивыми
соединениями.
Термическая стабильность нафтеновых углеводородов выше, чем парафино-
вых. Но при высоких температурах крекинга происходит и их расщепление. Наф-^
тены, имеющие боковые цепи, расщепляются по реакциям:
(СН2)2—СНз
+ СН2=СН2
(СН2)5-СНз
СН2—СН=СН2
+ С3Н8
СН2—СНз
+ С4Нв.
312
Нафтены расщепляются как с разрывом, так и без разрыва цикла по схе-
мам:
—► С2Н4 + СН2=СН— СН2—СН3
С2Н4 + СНг=СН—СН=СН2 + Н2
—> 2С3Нв
циклогексан
циклопентан цикло-
пеитадиен
циклогексан бензол
Конденсированные полициклические нафтены сначала деалкилируются
Ароматические углеводороды наиболее термически стабильны, однако при
высоких температурах они подвергаются расщеплению, например,
Риформинг — крекинг легких нефтяных продуктов.(низкоок-
танового бензина или лигроина) при температуре 550°C и давле-
нии до 7 МПа применяется для повышения качества бензинов (по-
вышенное содержание ароматических углеводородов), так как ок-
тановое число указанных фракций возрастает с 50 до 66—71.
313
Пиролиз — парофазный крекинг нефтяных продуктов (лиг-
роина, керосина и др.), проводимый при температуре 670—720°C
и давлении, близком к атмосферному.
В результате глубокого расщепления углеводородов и вторич-
ных процессов, образования новых соединений получают газооб-
разные непредельные и ароматические углеводороды. Газы пиро-
лиза (до 50%) содержат этилен, пропилен, бутадиен и др., а жид-
кие продукты — ароматические углеводороды, образующиеся в
результате протекания следующих реакций:
2СН2=СН— СН=СН2
^х^/СН=СН2
+ Н2
сн2=сн—сн=сн2 + с2н4
СН=СН2
Газообразные и жидкие продукты пиролиза используются для
получения продуктов органического синтеза, высокомолекулярных )
соединений и индивидуальных веществ.
Коксование — процесс термического разложения нефтяных
остатков (мазута, битума, гудрона, крекинг-остатка и др.) без до-
ступа воздуха при 450—500 °C. Коксование нефтяных остатков про-
водят с целью получения дополнительного количества жидкого
топлива (бензина и др.) и беззольного нефтяного кокса, который ;
служит топливом, сырьем при получении электродов и т. п.
Каталитические процессы проводятся в присутствии катализа- )
торов, причем переработка жидкого сырья осуществляется с боль-
шей скоростью и при более низких температурах и давлениях, по
сравнению с термическими процессами. В качестве катализаторов
применяются платина, оксиды молибдена и хрома и синтетические
алюмосиликаты.
Применение катализаторов позволяет снизить энергетические
затраты, увеличить производительность аппаратуры, качество и
количество бензинов и других продуктов.
Каталитические процессы подразделяются на каталитический
крекинг (катализатор — алюмосиликаты) и каталитический ри-
форминг (катализаторы — платина, оксиды хрома и молибдена).
В отличие от термических процессов, где расщепление углеводо-
родов протекает по радикальному механизму, при каталитических
процессах происходит ионный распад углеводородов.
Каталитический крекинг проводят в паровой фазе при
450—500°C и 0,2 МПа. В этих условиях из тяжелого нефтяного
сырья получают бензины с повышенным октановым числом (77—78).
Достоинством каталитического крекинга является возмож-
ность переработки сернистых жидких продуктов, в результате ко-
торой получают бензины с низким содержанием серы, а сернистые
соединения переходят в газовую фазу.
314
При каталитическом крекинге наряду с паро- и газообразными
углеводородами образуется твердый кокс, который оседает на
поверхность катализатора и снижает его активность. Для восста-
новления активных свойств катализатора его регенерируют, при
этом через катализатор продувают воздух при температуре 600 °C,
кокс окисляется, а восстановленный катализатор вновь направ-
ляют в процесс.
Каталитический риформинг в отличие от крекинга
проводится под давлением в среде водорода и в присутствии
Рис. XIII.10. Схема термического крекинга мазута:
/ — насос; 2 — ректификационная колонна; 3, 4—трубчатые печи; 5—редукцион-
ный вентиль; 6—испаритель', 7— конденсатор; 8— сепаратор.
катализатора (платина, оксиды молибдена, хрома и др.). В настоя-
щее время имеется много разновидностей каталитического рифор-
минга, отличающихся друг от друга условиями проведения процесса
(температурой, давлением, катализатором и др.).
Характерной особенностью процесса каталитического рифор-
минга также является возможность переработки сернистого сырья
с получением продуктов с. низким содержанием серы. При нагре-
вании в присутствии катализаторов, содержащих Со и Мо, орга-
нические соединения серы взаимодействуют с водородом по реак-
циям:
RSH + H2 —> RH + H2S
. RSR' + 2Н2 —► RH-f-R'H + H2S
</^\ + 2Н2 —> C4HI() + H2S
S
Бензины, образующиеся при каталитическом риформинге,
имеют октановое число более 80, а выход их составляет 90%.
Термический крекинг в смешанной фазе (жидкой и паровой)
проводят под давлением до 7 МПа и 350—500°C. На рис. XIII 10
316
Рис. ХШ.Н. Схема каталитиче-
ского крекинга с движущимся ка-
тализатором:
/—трубчатая печь; 2—реактор; 3—буи-
кер; 4—регенератор; 5. 7—воздуходувка;
6 — труба.
Каталитический крекинг
показана схема крекинга мазута. Мазут насосом 1 подают на одну
из нижних тарелок ректификационной колонны 2, где он смеши-
вается с тяжелой фракцией, выходящей из колонны 2. Далее смесь
поступает в трубчатую печь 3, где нагревается до 470—480 °C. Из
средней части колонны 2 выходит более легкокипящая фракция,
которая нагревается в трубчатой печи 4 до 500—510 °C. Давление
в печах составляет 5—7 МПа. Продукты крекинга из печей 3 и 4
проходят редукционный вентиль 5
и поступают в испаритель 6, где
пары отделяются от крекинг-остат-
ка, выводимого из испарителя. Па-
ры бензина и газы из испарителя
направляются в ректификационную
колонну на разделение. Здесь они
проходят конденсатор 7 и сепара-
тор 8.
Выход продуктов: крекинг-бен-
зин— 30—35%, крекинг-газы —10—
15%, крекинг-остаток, — 50—55%,
Крекинг-газы, содержащие этилен,
пропан, пропилен, бутан, бутилен
и.др., служат ценным сырьем для
синтеза органических соединений.
Крекинг-остаток является котель-
ным топливом.
Парофазный крекинг — пиролиз
проводят при 670—720 °C и атмос-
ферном давлении. В процессе пиро-
лиза жидкие продукты обогащаются
ароматическими соединениями, а
газы — непредельными углеводоро-
дами.
проводят в паровой фазе при 450—
500 °C и давлении 0,05—0,1 МПа в присутствии алюмосиликатных
катализаторов, представляющих собой твердые высокопористые ве-
щества. Углеводороды на поверхности таких катализаторов адсор-
бируются, здесь же протекают реакции расщепления и реакции
ароматизации. При каталитическом крекинге выход бензина со-
ставляет до 70%, газов 12—15% и кокса 4—6%.
На рис. XIII. 11 показана схема каталитического крекинга
с движущимся катализатором. В печи 1 сырье нагревается до
350—360 °C и затем направляется в реактор 2, в который из бун-
кера 3 поступает зернистый катализатор. Под действием собствен-
ной массы отработанный катализатор опускается в низ реактора и
перед поступлением в регенератор (самотеком) подвергается об-
работке паром.
Продукты крекинга из реактора 2 направляются на разделение
в ректификационную колонну (на схеме не показана). Для реге-
316
нерации катализатора сверху в регенератор 4 воздуходувкой 5 по-
дают воздух, который окисляет кокс, осажденный на поверхности
катализатора. Образующиеся топочные газы выводятся снизу ре-
генератора.
Из нижней части регенератора 4 под давлением сжатого воз-
духа, подаваемого воздуходувкой 7, катализатор по трубе 6 на-
правляется в бункер 3 и вновь возвращается в реактор 2.
< Бензин каталитического крекинга содержит большое количе-
: ство ароматических, нафтеновых и изопарафиновых углеводоро-
: дов, поэтому он значительно стабильнее бензина термического кре-
кинга. Газы каталитического крекинга содержат предельные и не-
предельные углеводороды.
Каталитический риформинг также получил более ши-
рокое распространение, чем термический риформинг. Различные
промышленные процессы каталитического риформинга отличаются
друг от друга температурой, при которой проводится каждый из
них, давлением, используемым катализатором и способом его реге-
нерации.
Платформинг — каталитический риформинг, используемый
для переработки бензина и лигроина прямой гонки. Процесс про-
водят в присутствии платинового катализатора (платина на окиси
алюминия). Если платформинг проводят при температуре около
500 °C и давлении до 3 МПа, то получают главным образом бензол,
толуол и ксилол. Если же процесс ведут при 5 МПа, получают бен-
зин с октановым числом (без этиловой жидкости) около 98.
Газы каталитического риформинга содержат водород, метан,
этан, пропан и другие углеводороды; их используют как сырье
для органического синтеза. Выход газов каталитического рифор-
минга такой же, как и при каталитическом крекинге и составляет
15% массы сырья.
В связи с возрастанием потребности в высококачественном бен-
зине, а также в бензине с октановым числом 98 (без этиловой
жидкости) платформинг получил более широкое распространение,
чем обычный каталитический риформинг.
Очистка нефтепродуктов
Получаемые при перегонке и крекинге нефтепродукты подвер-
гаются очистке, так как они содержат соединения, снижающие
стабильность этих продуктов. Кроме того, присутствие примесей
приводит к образованию нагара в цилиндрах двигателей при сго-
рании, они придают продуктам темный цвет и неприятный запах.
Существуют химические и физические методы очистки нефте-
продуктов. К химическим методам относятся очистка серной кис-
лотой и гидроочистка, а к физическим — адсорбционные и абсорб-
ционные методы очистки.
При сернокислотной очистке нефтепродукты смешиваются
с серной кислотой (92—94%), которая реагирует с примесями
317
содержащимися в нефтепродуктах, или растворяет их. После от-
стаивания чистый бензин сливается и поступает потребителю, а ос-
тавшийся продукт — кислый гудрон является отходом производства.
Гидроочистка заключается в воздействии водорода на очищае-
мый продукт в присутствии катализатора при температуре около
400°C и давлении 3—4 МПа. При гидроочистке водород взаимо-
действует с сернистыми, азотистыми и кислородными соедине-
ниями, образуя легкоудаляемые сероводород, аммиак и воду:
rsh + h2 = rh + h2s
RNH2 + Н2 = RH + NH3
ROH + H2 = RH + H2O
Применение гидроочистки позволяет использовать многосерни-
стую нефть для получения высококачественных нефтепродуктов.
Адсорбционный метод очистки заключается в том, что при со-
прикосновении с адсорбентами (отбеливающие глины или силика-
гель) из нефтепродуктов адсорбируются сернистые, азотистые и
кислородные соединения, а также асфальты и некоторые другие
примеси.
Абсорбционные методы очистки заключаются в растворении
вредных компонентов, содержащихся в нефтепродуктах, в нитро-
бензоле, фурфуроле, жидком сернистом ангидриде и других абсор-
бентах.
Для стабилизации нефтепродуктов к ним добавляют малые ко-
личества антиокислителей (ингибиторов), что придает нефтепро-
дуктам устойчивость при хранении. В качестве ингибиторов
применяют фенолы, ароматические амины, аминофенолы и др. Ко-
личество ингибиторов, вводимых в состав нефтепродуктов, неве-
лико и измеряется сотыми или тысячными долями процента.
3. ПЕРЕРАБОТКА ПРИРОДНЫХ ГАЗОВ
Природные газы в зависимости от их нахождения в недрах
земли делятся на попутные газы (сопровождающие нефть), выде-
ляющиеся при добыче нефти, и газы, извлекаемые непосредственно
из газовых месторождений. Следовательно, попутные газы можно
рассматривать как самую легкую фракцию нефти.
Состав природных и попутных газов очень разнообразен
(табл. XIII. 5).
Кроме того, в природных и попутных газах содержатся аргон
(0,01—0,1%) и гелий (от 0,2 до 1,25%). Из таблицы видно, что
природные газы не содержат водорода, окиси углерода, непредель-
ных углеводородов и содержат малое количество углеводородов
с числом углеродных атомов более 5, тогда как в попутных газах
содержание таких углеводородов иногда превышает 3%.
Отличие природных газов от попутных состоит также в том,
что главной составной частью в них является метан, содержание
которого достигает 98% (табл. XIII. 5).
318
Таблица XIII 5 Состав некоторых природных и попутных г’зов
[в % (об.)]
— Месторождение сн4 с2н3 с3н8 C4Hio СбНи С«Н14 со2 n2
Лужнецкое (Томской 7 85,00 Ipupod 3,60 ные га 2,96 зы 2,26 1,09 0,2 0,95 3,90
обл.) Тазовское (Тюменская 98,30 0,07 0,10 0,20 — — 0,5 0,8
обл.) Медвежье (Тюменская 98,40 0,55 0,88 0,26 — — 0,01 0,44
обл.) Окаремское(Туркмения) 95,20 2,32 1,16 0,63 0,23 0,19 0,20 —
Шатлык (Узбекистан) 95,25 1,90 0,22 0,08 0,05 0,12 1,40 1,00
Попутные газы Поволжья и Урала
Тарханское Султаиово-Заглядин- 26,0 39,4 17,7 15,0 24,6 17,3 11,8 3,8 3,8 2,1 Н2 2,9 0,6 0,2 12,8 17,0
ское
Красноярское 39,8 17,7 18,9 4,15 1,80 1,3 0,6 20,2
Журавлевско-Степанов- ское 61.8 11Л 7,5 4,9 2,0 3,1 0,5 9,25
Способы переработки природных и попутных газов зависят от
их свойств и характера получаемого целевого продукта. Они не от-
личаются от методов переработки газов, получаемых из нефти и
ее дистиллятов. Ряд этих методов были рассмотрены выше.
При пиролизе природных газов получают непредельные угле-
водороды
2СН4 —> С2Н2 + ЗН2
С2н6 —> С2Н4+Н2
с3н8 —> с3н6 + н2
широко используемые в промышленности.
Ацетилен служит сырьем для получения химических волокон,
растворителей, высокомолекулярных соединений и других продук-
тов. Этилен используется для получения полиэтилена, этилбензола
(а затем стирола), спиртов
СН2=СН2 —> I—СН2—СН2—]„
СН2=СН2 + С6Нв —> С6Н5СН2—СН3 —> С6Н5СН=СН2 + н2
этилбензол стирол
СН2=СН2 + Н2О — СН3СН2ОН
Пропилен, бутилены используются для получения бензинов, ук-
сусной кислоты и других продуктов.
Неполным окислением метана при 1200—1500 °C получают так
называемый синтез-газ, состоящий из СО + Нг. На основе синтез-
газа получают насыщенные и ненасыщенные парафины, спирты.
319
альдегиды и т. д. Коверсией с водяным паром из природных и по-
путных газов при повышенных температурах в присутствии ката-
лизаторов получают водород
СН4 + 2Н2О —> СО2+4Н2
С2Нв + 4Н2О —> 2СО2 + 7Н2
Глава XIV .
ОРГАНИЧЕСКИЙ СИНТЕЗ
В настоящее время известно более 2,5 млн. органических ве-
ществ, причем их число быстро растет, так как беспрерывно синте-
зируются все новые и новые органические соединения. Уже одна
эта цифра указывает на трудности, с которыми связана классифи-
кация химико-технологических процессов получения органических
веществ, а также их изучение и организация. Кроме того, большин-
ство химических реакций, протекающих с органическими соедине-
ниями, сопровождается побочными параллельными и последова-
тельными реакциями, поэтому не всегда удается получить надеж-
ные данные о статике и кинетике этих реакций.
Наибольшее применение в народном хозяйстве имеют такие
продукты органического синтеза, как метиловый и этиловый спир-
ты, формальдегид, окись этилена, ацетальдегид, ацетон, уксусная
кислота, фенол, анилин, фталевый ангидрид, бутадиен, стирол, хло-
рированные углеводороды и др.
Сырьем для их получения служат водород, окись углерода, ме-
тан и его гомологи, этилен, пропилен, н-бутилен, изобутилен, аце-
тилен, бензол, толуол, нафталин и др., получаемые при переработке
жидкого, твердого и газообразного топлив. В производстве синте-
тических органических продуктов используются процессы окисле-
ния и восстановления, гидрирования и дегидрирования, гидрата-
ции и дегидратации, сульфирования, нитрования, галоидирования
и др. На их основе осуществляется синтез самых различных соеди-
нений, служащих сырьем для получения полимеров, синтетических
красителей, ядохимикатов, смазочных, моющих, душистых и ле-
карственных веществ и т. д. Большинство органических процессов
протекает в присутствии катализаторов.
1. СИНТЕЗ НА ОСНОВЕ ОКИСИ УГЛЕРОДА
Органический синтез на основе окиси углерода широко приме-
няется в промышленности. Из окиси углерода получают алифати-
ческие углеводороды, спирты, альдегиды, карбоновые кислоты и
их производные и др.
Метиловыйспирт (метанол) СНзОН — токсичная жидкость
плотностью 796 кг/м3, которая смешивается с водой в любых от-
ношениях, температура кипения 64,7 °C, температура плавления —
95 °C.
320
До 1934 г. в СССР метанол получали сухой перегонкой древе-
сины (древесный спирт). В настоящее время основным методом
получения метанола является синтез его из окиси углерода и во-
дорода, протекающий в присутствии каталиазтора при температуре
230—420°C и давлении 20—32 МПа
СО + 2Н2 СН3ОН+110,8 кДж/моль
На практике применяют цинкхромовый катализатор, который по-
лучают совместным осаждением окислов цинка и хрома на носи-
теле при соотношении ZnO : Сг2О3 = 2 : 1 и выпускают в виде таб-
леток.
Образование метанола идет с выделением тепла и уменьше-
нием объема, поэтому для увеличения выхода метанола процесс
Рис. XIV.1. Зависимость равновес-
ного содержания СН3ОН в газовой
смеси от давления и температуры
(Н2 : СО = 4 : 1):
/—при 240 °C; 2—при 340 °C; 3—при 400 °C.
Рис. XIV.2. Зависимость равновес-
ного содержания метанола в газовой
смеси от мольного отношения Н2 : СО
при различном давлении:
1—при 29,4 МПа; 2—при 19,6 МПа; 3—при
9,8 МПа; 4—при 4,9 МПа.
следует вести при высоком давлении и низкой температуре. Кроме
того, на равновесный выход метанола сильно влияет состав газа,
т. е. соотношение На: СО (рис. XIV. 1 и XIV. 2).
Из рисунков видно, что при повышении температуры равновес-
ная концентрация метанола снижается и для ее увеличения необ-
ходимо повысить давление. Однако с ростом отношения Н2:СО и
с повышением давления резко снижается равновесная концентра-
ция СН3ОН, поэтому для достижения максимально возможного
выхода метанола поддерживают оптимальные условия процесса.
Чаще всего процесс получения метанола ведут при давлении
от 20 до 35 МПа, температуре 370—420°C и объемной скорости
10000—35000 ч-1. Выход метанола в этих условиях составляет
10—20% при времени контакта газа с катализатором 10—40с.
) 1 Зак, 558 321
На рис. XIV. 3 показана схема синтеза метилового спирта.
Смесь Н2 и СО в соотношении 4: 1 (применяют и другие соотно-
шения) сжимается в компрессоре до 25 МПа, смешивается в сме-
сителе 2 с непрореагировавшим циркуляционным газом, нагнетае-
мым компрессором 9, и поступает в фильтр 3, где очищается от
масла; затем в теплообменнике 4 смесь нагревается до 200 °C и
направляется в колонну синтеза метанола 5. По выходе из нее
смесь поступает в межтрубное пространство теплообменника 4,
Рис. XIV.3. Схема синтеза метилового спирта:
1—компрессор; 2—смеситель; 3—фильтр; 4—теплообменник; 5—колонна син-
теза; 6—холодильник-конденсатор; 7—сепаратор; 8 — сборник; циркуляцион-
ный компрессор.
охлаждается, нагревая газовую смесь, поступающую на синтез.
Такая организация процесса позволяет вести процесс автотер-
мично.
Из теплолообменника 4 газовая смесь, содержащая пары мета-
нола, поступает в водяной холодильник 6, а затем в сепаратор 7,
где жидкий метанол отделяется от непрореагировавшего газа; по-
следний возвращается циркуляционным компрессором 9 в смеси-
тель 2. Метиловый спирт-сырец из сепаратора сливается в сбор-
ник 8 и направляется на ректификацию для очистки от различных
органических веществ. Метанол-сырец имеет примерно следующий
состав (в %):
Метанол . . . 93—95 Диметиловый эфир.. . . 1
Вода........ 3—5 Высшие спирты .... 0,4—1
После ректификации спирта-сырца выход чистого метанола со-
ставляет 84—87%. На 1 т метанола расходуется около 700 м3
окиси углерода и 1400—2000 м3 водорода.
Чистый метанол используется для получения формальдегида
(около 50%), .метилгалогенидов, метиловых эфиров, диметилтере-
фталата, метилметакрилата и других продуктов, а также в каче-
стве жидкого топлива, растворителя и экстрагента (около 10%).
Формальдегид — альдегид муравьиной кислоты — бесцвет-
ный газ с резким раздражающим запахом, имеет температуру кон-
денсации— 19 °C при атмосферном давлении. Он хорошо растворим
822
в воде, 33—40%-ный водный раствор формальдегида называется
формалином. Водный раствор формальдегида при хранении может
полимеризоваться, во избежание этого в его состав вводят в ка-
честве стабилизатора 7—12% (масс.) метилового спирта.
Формальдегид широко используется для получения феноло-,
карбамидо-. и меламино-формальдегидных смол, полиформальде-
гида, уротропина, изопрена; он образуется в качестве промежу-
точного продукта в промышленности органического синтеза при
Рис. XIV.4. Схема производства формальдегида:
7—мерник; 2—испаритель-подогреватель; 3—контактный аппарат; 4—слой ката-
лизатора; 5—холодильник; 6 — поглотительная башня; 7. 8 —трубчатые холодиль-
ники; 9—фильтр; 10—воздуходувка.
получении бутандиола-1,4, пирролидонов, аллилового, «-пропило-
вого спиртов.
Наиболее распространенным способом получения формальде-
гида является окислительное дегидрирование метанола в течение
0,01—0,03 с в присутствии катализатора при 500—600 °C
СНзОН + 0,5О2 —> НСНО + Н2о + 156,3 кДж/моль
Наряду с основной реакцией протекают побочные процессы окис-
ления, дегидрирования и гидрирования до образования окиси и
двуокиси углерода и других продуктов.
В качестве катализатора применяются медь и серебро в виде
сеток либо осажденные на пористом носителе, например, на пемзе.
Схема получения формальдегида изображена на рис. XIV. 4.
Метанол, содержащий 10—12% воды, из сборника 1 непрерывно
поступает в испаритель-подогреватель 2. Сюда же воздуходувкой
10 подают очищенный в фильтре 9 воздух, который барботирует
через слой метанола и насыщается его парами. Для нормальной
работы -в испарительной системе поддерживается постоянные уро-
вень жидкости и температура (48—50°C). Образовавшаяся паро-
водушная смесь нагревается до НО °C в верхней части аппарата 2
11*
323
и поступает в контактный аппарат 3 с катализатором 4. Проходя
через катализатор, метиловый спирт окисляется с образованием
формальдегида, выход формальдегида 80—85% при степени кон-
версии метанола 85%.
Выходящие из контактного аппарата газы содержат 20—22%
НСНО, 36—38% N2, СНзОН, Н2, СО, СО2, СН4 и др. В холодиль-
никах 5 и 8 они охлаждаются и поступают в поглотительную
башню 6, орошаемую водой и снабженную трубчатым холодиль-
ником 7. Из поглотительной башни 6 выводится водный 33—40 % -
ный раствор формальдегида, содержащий 10—12% СН3ОН; газы,
состоящие в основном из азота и водорода, выбрасываются
в атмосферу.
Формальдегид получают также частичным окислением метана.
Этот процесс проводят в присутствии катализатора (смесь фосфата
алюминия и окиси меди) при температуре 460 °C. При окислении
метана образуются помимо формальдегида и другие продукты ре-
акции, например, НСООН, СО2, СО, Н2, поэтому нз реакционной
смеси формальдегид выделяют конденсацией или поглощением во-
дой с последующим его выделением.
Из окиси углерода и водорода под давлением 2—15 МПа и при
температуре 160—450 °C получают также углеводроды парафино-
вого ряда. В зависимости от условий процесса (температуры,
давления), состава смеси, катализатора, образуются твердые, жид-
кие и газообразные углеводороды:
NI
со + зн2 ------> сн4 + н2о
200—250 °C 4 -г 2
NI, СО
СО (2/? -р 1)Н2 ' СлН2д4-2 -р /1Н2О
Fe
2/гСО “р «Н2 С^Нгл -р лСО2
2. СИНТЕЗ НА ОСНОВЕ ПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ
Сырьем для синтеза многих органических продуктов служат
предельные углеводороды. Широкое применение из методов пере-
работки предельных углеводородов находит окисление, хлориро-
вание, сульфирование, сульфохлорирование и др.
В зависимости от условий процесса при окислении углеводоро-
дов получают спирты, альдегиды, кислоты, СО, Н2 и др. Окисле-
нием низших парафинов в газовой фазе получают формальдегид,
метиловый спирт, ацетальдегид;
CH4 + 0,SO2 —> СН3ОН + 0,5О2 —> НСНО + Н2О
С2Н6 + 0,5О2 —► C2HSOH
С2Н6 + О2 —> СН3СНО + Н2О
При окислении парафиновых углеводородов С15—Сзо кислоро-
дом воздуха получают жирные кислоты, высшие спирты, моющие и
324
поверхностно-активные вещества:
с15н32 + о,502 —> СД6Н31ОН
Сзонв2 + 2,5О2 —► 2СнН29СООН + Н2О
С,4Н29СООН + 2Н2 —► син29сн2он + н2о
Окислением нафтеновых углеводородов, например циклогек-
сана, получают циклогексанол C6Hi2O, циклогексанон СбНюО, ади-
пиновую кислоту НООС—(СН2)4—СООН. Хлорированием пре-
дельных углеводородов получают различные хлорзамещенные про-
дукты, используемые далее для производства высокомолекулярных
соединений (хлористый винил, хлоропрен, тетрафторэтилен и др.),
полупродуктов органического синтеза (хлористые метил, этил, ал-
лил и бензил, хлорбензол и др.), а также применяемые в качестве
растворителей (хлористый метилен, ССК и др.), хладоагентов
(хлороформ, хлористый этил и др.), для борьбы с вредителями
сельского хозяйства, в качестве смазочных масел и т. д.
Хлорированию подвергают жидкие (парафиновые, нафтеновые
и ароматические) и газообразные (метан, этан, пропан и др.) уг-
леводороды. Для получения хлорированных углеводородов приме-
няют термическое, фотохимическое, каталитическое хлорирование
и хлорирование, инициируемое радикалами.
Термическое хлорирование проводят при нагревании смеси угле-
водородов и хлора до температуры диссоциации хлора на атомы
(«250 °C). Чем устойчивее углеводород, тем должна быть выше
температура хлорирования, так, например, для хлорирования бу-
тана требуется нагревание до 250 °C, а метана — до 400 °C.
При фотохимическом хлорировании молекулы хлора диссоции-
руют под влияием фотонов или кванта энергии, например
Cl + nv —> Cl-
Каталитическое хлорирование ведут в присутствии хлоридов
металлов переменной валентности (Cu2Cl2, FeCl3, SbCl5).
При жидкофазном хлорировании атомы хлора образуются при
действии на молекулу С12 продуктов разложения перекиси или
азосоединений, вводимых в жидкие углеводороды, например
100 °C
С6Н6СООООСвН5 ----> 2СвН6.+2СО2
CqHs • 4- С12 ——> С3Н6С1 4- Cl-
При хлорировании метана образуются хлорсодержащие углево-
дороды; при этом протекают следующие реакции:
СН4 4- С12 —> СН3С14-НС1
СН3С1 4- С12 —> СН2С12 4- НС1
СН2С12 4- С12 —► СНС13 4-НС1
СНС13 4- С12 —> ССЦ4-НС1
Процесс осуществляют при 400—480 °C в цилиндрических реакто-
рах, футерованных диабазовыми плитками. При хлорировании об-
325
разуется смесь, содержащая хлористый метил СН3С1, хлористый
метилен CH2CI2, хлороформ СНС13 и четыреххлористый углерод
ССЦ. Выход этих продуктов [в % (мол.)] зависит от мольного со-
отношения С1г: СН4:
С12 : СН4=1 :2 С12 : СН4=3,02 :1
СНС13 62 3
СН2С12 30 15
СНС13 7 53
СС14 1 29
Выход товарных продуктов составляет 85—99% от израсходо*
ванного метана. При тщательном разделении можно получить
продукты 99—99,5 %-ной чистоты.
Хлорирование этана и пропана проводят при 300—400 °C и в
избытке углеводорода. При хлорировании этана в основном обра-
зуются хлористый этил и небольшое количество дихлорэтана
С2нв + С12 —> С2Н6С1 + НС1
С2Н6С1 + С12 —> С2Н4С12 + НС1
При хлорировании пропана образуются монохлориды (2-хлор-
попан и 1-хлорпропан) и полихлориды.
3. СИНТЕЗ НА ОСНОВЕ НЕПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ
Олефины — непредельные углеводороды СпН2п, легко взаимо-
действующие с хлором, кислородом и водородом, склонны к гид-
ратации и полимеризации.
При хлорировании этилена получают дихлорэтан
СН2=СН2 + С12 —СН2С1—СН2С1
411г-4jJ
который широко используется как растворитель жиров, каучуков,
для получения хлористого винила и др. Хлористый винил обра-
зуется по реакции:
СН2С1-СН2С1 ——> СН2=СНС1 + НС1
45'3—520
При хлорировании пропилена получают хлористый аллил и
хлориды.
Окислением непредельных углеводородов получают окиси угле-
водородов. Наибольшее применение в органическом синтезе нахо-
дит окись этилена, которую получают окислением этилена в при-
сутствии серебряных катализаторов
СН2=СН2 + 0,5О2 —> СН2— CH2 + Q
о
Окись этилена служит сырьем для получения этиленгликоля,
этаноламина, эпихлоргидрина, диоксана, ацетальдегида и др.
Олефины способны подвергаться гидратации. На этом свойстве
основаны процессы получения спиртов (например, этилового спир-
та С2Н5ОН из этилена).
326
Этиловый спирт—(этанол) кипит при 78,3°C; смесь’
С2Н5ОН [30% (об.)] с воздухом взрывоопасна; с водой спирт обра-
зует азеотропную смесь, содержащую 95,6% С2Н5ОН; кипящую
при 78,1 °C. Этиловый спирт широко применяется в пищевой и
медицинской промышленности, является компонентом жидкостного
ракетного топлива, антифризом и т. д. Особенно широко этанол
используется как полупродукт органического синтеза, и, в частности,
Рис. XIV.5, Схема производства этилового спирта сернокислотной
гидратацией этилена:
/“-тарельчатый абсорбер; 2, 3—скрубберы; 4—холодильник; 5—колонна; 6—ги-
дролизер.
для получения сложных эфиров, хлороформа, хлораля, ацетальде-
гида, уксусной кислоты, бутадиена и других продуктов. По объему
производства синтетический этиловый спирт занимает первое место
среди других органических соединений.
Ранее этиловый спирт получали из пищевого сырья — карто-
фельного крахмала и некоторых зерновых культур, однако этот
способ связан с большими затратами пищевого сырья. Кроме того,
его получают гидролизом древесины (гидролизный спирт). В на-
стоящее время этанол получают сернокислотной и прямой гидра-
тацией этилена.
При сернокислотном способе получения этанола (рис. XIV. 5)
этилен под давлением 1,5—2,5 МПа поступает в абсорбер 1 бар-
ботажного типа, орошаемый 97 %-ной серной кислотой. Темпера-
тура в абсорбере 65—75 °C. Серная кислота в этом процессе яв-
ляется катализатором и реагентом.
827
Этилен взаимодействует с серной кислотой с образованием мо-
ноэтилсульфата C2H5OSO3H и диэтил сульфата (C2HsO)2SO2
C2H4 + H2SO4 —► C2H6OS03H+Q
2С2Н4 + H2SO4 —► (C2H5O)2S02 + Q
Газы, не поглощенные в абсорбере 1, проходят водяной 2 и
щелочной <3 скрубберы и далее могут быть использованы как топ-
ливо.
Рис. XIV.6. Зависимость равновесной
конверсии этилена в спирт от темпе-
ратуры и давления:
/ — при 20 МПа; 2 — при 50 МПа; при
80 МПа; 4—при 150 МПа.
Рис. XIV.7. Зависимость равновесной
степени конверсии этилена в C2HgOH
от давления и температуры при соот-
ношении Н2О: С2Н4 = 1:1:
/—при 200 °C; 2—при 250 °C; 3—при 300 °C;
4—при 350 °C.
Этилсульфаты и серная кислота из абсорбера 1 поступают в
гидролизер 6, в который подается вода. В гидролизере при давле-
нии 1 МПа и температуре 70—90 °C происходит гидролиз этил-
сульфатов
C2H6OSO3H + H2O —> C2H6OH + H2SO4
(C2H6O)2SO2 + 2Н2О —> 2С2Н5ОН + H2SO4
Пары спирта и воды далее проходят холодильник 4, где они
конденсируются; конденсат поступает в ректификационную колонну
5 для разгонки и очистки от примесей. Разбавленная кислота
(50%) выводится из гидролизера, направляется на концентриро-
вание и снова возвращается в процесс.
При ректификации концентрация этилового спирта достигает
95—96%. По этому способу из 1 т этилена получают 1, 2 т этанола
и около 100 кг этилового эфира.
Синтез этанола прямой гидратацией этилена, как правило, про-
водят в паровой фазе в присутствии катализатора —ортофосфор-
328
ной кислоты (35—40% Н3РО4), которой пропитывают алюмосили-
каты, силикагель.
Рис. X1V.8. Влияние мольного соот-
ношения Н2О : С2Н4 иа степень кон-
версии этилена в спирт.
ЯроОолжительность к октанта, с
Рис. XIV.9. Влияние времени кон-
такта на степень конверсии С2Н4.
Реакция взаимодействия этилена с водой обратима и проте-
кает с выделением тепла
СН2=СН2 4- Н2О =F=b С2Н5ОН 4- 46,0 кДж/моль
следовательно, процесс желательно проводить при невысоких
температурах. Однако степень
превращения этилена в этанол
лимитируется скоростью реакции
и активностью катализатора.
Влияние температуры на степень
конверсии этилена показано на
рис. XIV. 6.
На практике процесс ведут
при температуре 280—290 °C.
Повышение давления сдвигает
равновесие в сторону образова-
ния этанола (рис. XIV. 7), поэто-
му в системе поддерживают дав-
ление около 7—8 МПа. Выход
Рис. XIV.10. Влияние температуры
на степень конверсии этилена в спирт
при различной объемной скорости:
/ — При 2000 ч-*; 2 —при 5100 ч—*.
проводят по циклической схеме
до 7—8 МПа, смешивается с цир-
этанола зависит также от моль-
ного соотношения Н2О: С2Н4
(рис. XI. 8), времени контакта
(рис. XIV. 9) и объемной скоро-
сти (рис. XIV. 10).
Прямую гидратацию этилена
(рис. XIV. 11).
Этилен, сжатый компрессором
куляционным газом и паром высокого давления, после чего про-
329
ходит теплообменник 1 и трубчатую печь 2, где смесь нагревается
до 280 °C и направляется в контактный аппарат 3. При прохожде-
нии смеси через катализатор образуются пары спирта. Выходящая
из контактного аппарата 3 парогазовая смесь охлаждается в теп-
лообменнике 1. Сконденсировавшиеся пары воды и спирта отде-
ляются от газа в сборнике 4, откуда спирт-сырец (15—16%'
С2Н5ОН) поступает на очистку.
Непрореагировавший этилен проходит водяной холодильник 5
и колонну с насадкой 6, орошаемую водой для полного извлечения
спирта, и поступает на смешение с новой порцией этилена.
Водный спирт на очистну
Рис. XIV.11. Схема производства этилового спирта гидратацией
этилена в паровой фазе:
/—теплообменник; 2—трубчатая печь; 3—контактный аппарат; 4—сборник;
5—холодильник; 6 — промывная колонна.
Метод прямой гидратации этилена по затратам и простоте об-
служивания более выгоден, чем сернокислотный, что видно из дан-
ных, приведенных ниже: ,
Расход , т/т С2Н5ОН При сернокислотной гндратацннн При прямой гидра- тации
этилена (100% С2Н4) . . . . 0,75 0,69
серной кислоты (100% H2SO4) 1,15 —
.едкого натра (42% NaOH) . . 0,04 0,02
ортофосфорной кислоты . . , —- 0,006
Получение синтетического спирта из этилена позволяет суще-
ственно сократить расходы пищевого сырья. Так, 1 т этилена, пе-
реработанная на этанол позволяет сэкономить 4 т зерна.
4. СИНТЕЗ НА ОСНОВЕ АЦЕТИЛЕНА
Ацетилен СН=СН — газообразное вещество, температура сжи-
жения —83,5 °C. С кислородом и воздухом образует взрывоопас-
ные смеси с очень широким пределом взрываемости, так напри-
330
мер, смесь ацетилена с кислородом взрывоопасна в пределах от
2,8 до 78% (об.) С2Н2. Для снижения взрывоопасности смеси в нее
вводят инертные газы (Н2, N2).
Ацетилен растворим в воде (в одном объеме Н2О растворяется
1 объем С2Н2), в растворах солей и особенно хорошо в органиче-
ских веществах, например, в одном объеме ацетона растворяется
23 объема ацетилена. ________________________
Химическая активность ацети-
лена обусловлена наличием в мо-
лекуле С2Н2 реакционноспособной
тройной связи. В связи с этим
ацетилен широко используется в
синтезе разнообразных химиче-
ских соединений — исходных про-
дуктов для получения химиче-
ских волокон, синтетических кау-
чуков, пластических масс и т. д.
Так, из ацетилена получают аце-
тальдегид, этиловый спирт, бута-
диен, этилацетат, хлористый ви-
нил, хлоропрен и др. Кроме того,
ацетилен нашел применение для
получения высоких температур
при сварке и резке металлов.
Ацетилен получают из карби-
Рис. XIV.12. Температурная зависи-
мость равновесной степени конверсии
этана (/) и метана (2) в ацетилен.
да кальция и термическим рас-
щеплением углеводородов. По первому способу сплавлением окиси
кальция и углерода в электродуговых печах получают карбид каль-
ция
СаО + ЗС —> СаСз + СО
который затем разлагают водой с образованием ацетилена:
СаС2 + 2Н2О —> С2Н2 + Са(ОН)2
Из 1 кг чистого карбида кальция теоретически образуется
380 л С2Н2, однако, поскольку в техническом карбиде кальция со-
держатся примеси, выход ацетилена снижается до 230—250 л.
По второму способу ацетилен получают расщеплением углево-
дородов при высоких температурах
2СН< <=> С2Н2 + ЗН2
С2Н6 < ?- С2Н2 + 2Н2
На рис. XIV. 12 показана температурная зависимость равновес-
ной степени конверсии углеводородов при 0,1 МПа. При высоко-
температурном расщеплении углеводородов выделяющиеся газы
содержат 7—14% (об.) С2Н2, последний извлекают из газов селек-
тивными растворителями (вода под давлением, жидкий аммиак,
метиловый спирт и др.).
331
В качестве примера использования ацетилена в органическом
синтезе рассмотрим синтез ацетальдегида СН3СНО.
Ацетальдегид — (альдегид уксусной кислоты) — бесцвет-
ная, легко испаряющаяся жидкость, температура кипения кото-
рого 21 °C. Ацетальдегид отличается резким запахом, хорошо сме-
шивается с водой и спиртом; с воздухом образует взрывоопасные
смеси [4—57% (об.) СН3СНО].
По масштабам производства ацетальдегид занимает одно из
первых мест среди альдегидов, он является промежуточным про-
Ртуть
Катализшпор.
на регенера-
цию
На очистку
Пар
Обратный ацетилен
Катализатор'
а регенерации
Ацетилен
сбетий.
^АцетальОргид
на ректификацию
Рис. XIV.13. Схема получения ацетальдегида гидратацией ацети-
лена:
1—гидратор; 2, 3—холодильники; 4—очистительная колонна; 5—сборник.
дуктом органического синтеза. Из него получают уксусную и мо-
лочную кислоты, эфиры акриловой кислоты, уксусный ангидрид,
кротоновый альдегид, «-бутиловый спирт и др.
По одной из схем (рис. XIV. 13) очищенный от примесей аце-
тилен смешивают с циркуляционным газом и непрерывно подают
в гидратор 1, где он нагревается до 80—100°C. Барботируя через
катализатор — жидкость, содержащую сульфаты железа и ртути
(в 1 л Н2О 200 г H2SO4, 0,4 г Hg, 40 г окислов железа), ацетилен
на 50—60% превращается в ацетальдегид
СН=СН + Н2О —► СН3—СНО+ 1416 кДж
Газы, содержащие ацетальдегид, ацетилен и примеси, охлаж-
даются сначала в холодильнике 2 (зщесъ частично конденсируются
пары воды и конденсат возвращается в гидрататор /), а затем
в холодильнике 3, где пары ацетальдегида и воды конденсируются,
332
собираются в сборнике 5 и далее направляются на ректификацию
(на схеме не показано).
Несконденсированные газы, содержащие ацетилен, поступают
в колонну 4, орошаемую водой. Здесь извлекаются остатки аце-
тальдегида, а непрореагировавший ацетилен вновь возвращается
в процесс. Для очистки оборотного газа от окислов углерода и
азота часть его (10%) непрерывно отбирается из цикла и направ-
ляется на очистку. Выход ацетальдегида составляет около 96%.
Ртуть и ее соединения, входящие в состав, катализатора, ядо-
виты, поэтому в настоящее время разрабатываются нертутные ка-
тализаторы в виде окислов Zn, Mg, Ni, СО, С.
Ацетальдегид широко применяется для получения уксусной кис-
лоты СН3СООН.
Уксусная кислота (безводная) плавится при 16,6°С и ки-
пит при 118 °C. Она растворима во многих органических веществах
и смешивается с водой в любых соотношениях. Уксусная кислота
находит широкое применение в текстильной и пищевой промыш-
ленности, а также является промежуточным продуктом при полу-
чении монохлоруксусной кислоты, сложных эфиров, винилацетата
и т. д.
Уксусную кислоту вначале получали только сухой перегонкой
древесины либо в процессе биологического окисления этилового
спирта. В настоящее время ее получают окислением ацетальде-
гида, жидкофазным окислением углеводородов С3 —С7 и синтезом
из метанола и окиси углерода.
Окисление ацетальдегида кислородом воздуха с целью получе-
ния уксусной кислоты происходит в присутствии солей марганца
(ацетат марганца) при 60—80°C. Образующаяся в качестве про-
межуточного продукта надуксусная кислота взрывоопасна, по-
этому парогазовую смесь разбавляют азотом. Образование
СНзСООН идет по следующей схеме:
О
сн3сно + о2 —> сн3—с—о—он
надуксусная
кислота
СН3СНО + СН3—С_' —> 2СН3СООН
\о—он
уксусная
кислота
Синтетическую кислоту очищают от примесей перегонкой. Тех-
ническая кислота после перегонки содержит 97—99% СН3СООН,
0,1—0,5% НСООН, 0,5—2% Н2О.
5. НИТРОБЕНЗОЛ
Нитробензол получают нитрованием бензола (С6Нв) нитрую-
щей смесью, состоящей из азотной и серной кислот:
СвНв + HNO3 —> CBH5NO2 + Н2О
Серная кислота в процессе не участвует, но по мере нитрования
связывает выделяющуюся воду, которая разбавляет реакционную
массу и резко снижает скорость процесса. Нитрование проводится
в нитраторах, изготовленных из кислотостойких чугунов или ста-
лей. Бензол и нитрующая смесь (48% HNO3, 46% H2SO4, 6% Н2О)
из дозаторов поступает в нижнюю часть нитратора, где интенсив-
но перемешивается. В нижней части нитратора при 50—60 °C начи-
нается процесс нитрования, и по мере подъема жидкой смеси
к верхней части аппарата реакция заканчивается. Время пребы-
вания смеси в нитраторе 6—12 мин.
Образовавшуюся смесь, содержащую нитробензол, 0,5% бен-
зола, 0,01 % динитробензола, серную кислоту и остатки азотной
кислоты, направляют в отстойники. В нижней части отстойника
собирается H2SO4 и HNO3, а в верхней — кислый слой нитробен-
зола, содержащий бензол. Слой нитробензола сливают, промывают
водой, раствором соды, а затем из него отгоняют непрореагировав-
ший бензол. Окончательная очистка нитробензола от примесей до-
стигается перегонкой. Выход нитробензола 98%.
Нитрованием получают самые разнообразные органические со-
единения, используемые в производстве взрывчатых веществ (три-
нитротолуол, тринитрофенол и др.), душистых веществ, раствори-
телей и т. д.
Анилин — получают восстановлением нитробензола
4C6H6NO2 + 9Fe 4-4Н2О —> 4C0H5NH2 4- 3FesO4
в присутствии соляной кислоты. Последняя, взаимодействуя с чу*
гунной стружкой, очищает ее поверхность от окислов железа и де-
лает железо более реакционноспособным. Процесс восстановления
проводят в котлах, снабженных мешалкой, куда подается нитро-
бензол, стружка и кислота. Поскольку реакция сопровождается
выделением тепла, смесь подогревают только в начале процесса.
При кипении смеси из котла выделяются пары воды и анилина.
Проходя через холодильник, они конденсируются и возвращаются
снова в котел. По окончании восстановления в котел заливают
известковое молоко для осаждения солей железа из раствора.
В котле образуются два слоя: нижний — анилин и шлам; верх-
ний— водяной слой, содержащий анилин. Анилиновый слой сли-
вается и после отстаивания отделяется от шлама и воды. Для
окончательного удаления воды анилин перегоняют в вакууме.
Анилин можно получать из нитробензола восстановлением его
водородом в присутствии катализатора (Pt, Си, Ni и др.) при тем-
пературе 200—400 °C и давлении
CeH6NO2 4" ЗН2 —> С6Н5\Н2 4" 2Н2О
Этот метод в настоящее время вытесняет метод восстановления
нитробензола железом.
Глава XV
ВЫСОКОМОЛЕКУЛЯРНЫЕ СОЕДИНЕНИЯ
1. ВВЕДЕНИЕ
Трудно найти такую отрасль промышленности или отрасль,
производящую предметы народного потребления, где бы не ис-
пользовались высокомолекулярные соединения. И это не случайно,
так как материалы, получаемые на основе высокомолекулярных
соединений, обладают самыми разнообразными свойствами: они
могут быть твердыми и мягкими, эластичными и жесткими, соче-
тать высокую прочность с низкой плотностью, могут быть звуко-
непроницаемыми и химически стойкими, а также могут легко пере-
рабатываться в изделия всеми известными методами. Кроме того,
стоимость изделий, изготовленных на основе высокомолекулярных
соединений, в большинстве случаев ниже стоимости изделий, по-
лучаемых из других матералов. Так, например, 1000 м водопро-
водных труб из пластмассы весят всего 250 кг, а из металла — 2 т,
причем затраты труда на изготовление труб в первом случае в
7—10 раз ниже, чем во втором. Применение 1 т полихлорвинило-
вого пластиката для изоляции кабеля позволяет сберечь 6 т свин-
ца, при этом повышается производительность труда и снижается
его стоимость.
Универсальность свойств высокомолекулярных соединений
обусловила применение изделий из них в самолете-, судо-, радио-
и машиностроении, в электронной промышленности, атомной и
других отраслях техники. Высокомолекулярные соединения широко
используются для защиты от коррозии изделий из металла, де-
рева и бетона, для изготовления тканей, одежды, обуви и других
предметов народного потребления. Широкое применение находят
синтетические ткани, затраты труда на производство которых зна-
чительно ниже, чем на натуральные. По своим свойствам лавсан
мало отличается от шерсти, но в 3 раза дешевле ее.
Высокомолекулярные соединения — полимеры
представляют собой особый класс органических соединений, моле-
кулы которых состоят из сотен и тысяч атомов, связанных между
собой. Такие молекулы — гиганты — принято называть макромоле-
кулами. Макромолекулы полимеров имеют неодинаковую длину, а,
следовательно, и массу 1 моля (т. е. эти соединения представляют
собой смесь макромолекул различной молекулярной массы). По-
этому молекулярную массу таких соединений характеризуют сред-
ней молекулярной массой, а неоднородность принято назы-
вать полидисперсностью по молекулярной массе.
Размеры макромолекул высокомолекулярных соединений зави-
сят от степени полимеризации и; т. е. от числа элементарных звень-
ев мономера, соединенных между собой химическими связями
в макромолекуле. Молекулярная масса полимера М связана со сте-
335
Пенью полимеризации соотношением
М = пт
где т — молекулярная масса элементарного звена (например, тетрафторэтилена
[—CFj—CF2—] в макромолекуле политетрафторэтилена [—CF2—CF2—]n).
Свойства полимеров сильно зависят от степени полимеризации.
Например, поликремнийорганические соединения при п = 100—120
йвляются вязкими жидкостями, а при п = 1000—1500 представ-
Рис. XV.1. Изменение предела
прочности при разрыве а в зави-
симости от числа поперечных свя-
зей N между макромолекулами.
ляют собой высокоэластичные мате-
риалы.
Строение макромолекул полиме-
ров различно. Большинство полиме-
ров состоит из макромолекул, пост-
роенных из одинаковых, многократ-
но повторяющихся элементарных
звеньев по принципу ...—А—А—А—
А—А . Такие полимеры назы-
ваются гомополимерами. Если ма-
кромолекула полимера состоит из
элементарных звеньев А и В разного
строения, такой полимер называют
сополимером .... —А—В—А-1—В—
А—В— •••. Простейшим сополимером является саран, получаемый
из винилхлорида СН2 — СНС1 и винилиденхлорида СН2 — СС12:
пСН2=СНС1 + пСН2=СС12=[— СН2—СНС1—СН2—СС12—]„
Макромолекулы отдельных полимеров построены чередованием
не отдельных звеньев, а целых участков молекулярной цепи (бло-
ков), отличающихся друг от друга химическим составом, напри-
мер —(— А—)п—(—Б—)т—(—А—)п—(—Б— )т ... .Сополи-
меры такого вида называют блок-со'полимерами. В настоящее
время получены блок-сополимеры крахмала и белка, ацетилцеллю-
лозы и полиакрилонитрила и др.
Атомы нли атомные группировки в макромолекуле полимера
могут располагаться различно по отношению к основной цепи:
в зависимости от этого полимеры подразделяют на линейные, раз-
ветвленные и пространственные (сетчатые). Макромолекулы ли-
нейных полимеров представляют собой вытянутые цепочки, длина
которых в 100—1000 раз н более превышает их поперечный размер.
К таким полимерам относятся полиолефины, полиэфиры, поли-
амиды, натуральный каучук и др. Полимеры этого вида, как пра-
вило, растворимы, плавятся без разложения, а их расплавы имеют
высокую вязкость. К этой группе соединений относятся также со-
полимеры, типа ... —В—А—В—А—В—А—В—А— ....
У разветвленных полимеров основная цепочка макромолекулы
имеет многочисленные ответвления. Число этих ответвлений и
длина боковых цепей различны, однако, как правило, длина боко-
938
вой цепочки значительно меньше длины основной цепи, например
А—А—А----
..—А—д—а—А—А—А—А—А—А--------или
А—А—А—А----
А—В—А—В----
---А—В—А—В—А—В—А—В—А—В------
В—А—В—А—В—А-----
Разветвленные полимеры, у которых к основной цепи линейной
макромолекулы присоединены боковые цепочки другого химиче-
ского состава, называются привитыми сополимерами, например
(В)..
•• • -(-А-)„-А-(-А-)„-А-(-А-)„з- •”
В/П2
где п1( п2> «з — степень полимеризации мономера А;
/П1, т2 — степень полимеризации мономера В. !
Разветвленные полимеры и сополимеры растворимы в органи-
ческих растворителях, размягчаются и плавятся при нагревании.
В пространственных, или сетчатых полимерах линейные це-
почки макромолекул связаны между собой поперечными химиче-
скими связями (мостиками), что приводит к образованию единой
пространственной сетки, например
---А— А—А—А—А—А—А—••
---А—А—1—А—А—А—А------
---А—А—А—А—А—А—А------
-А—А—А-----
Число поперечных связей N между молекулами может быть
различно, что влияет на свойства полимера (рис. XV. 1).
Классификация высокомолекулярных соединений >
Высокомолекулярные соединения классифицируются по не-|
скольким признакам. i
По происхождению их подразделяют на природные, синтетиче-
ские и химически модифицированные природные. К первой группе
относятся природные органические высокомолекулярные соедине-
ния (полисахариды — целлюлоза, крахмал, белки, натуральный
каучук и др.). Во вторую группу входят высокомолекулярные со-
единения, получаемые из мономеров (синтетические каучуки, смо-.
лы и др.). Третья группа включает природные полимеры, подверг-
нутые химическим превращениям (эфиры целлюлозы).
337^
По химическому составу главной цепи макромолекулы высоко-
молекулярные соединения подразделяют на карбоцепные, гетеро-
цепные и элементоорганические.
Карбоцепные полимеры — полимеры, у которых атомы в. эле-
ментарном звене, а также звенья в макромолекуле имеют один
тип соединения углерод — углерод:
К карбоцепным полимерам относятся натуральный каучук, синте-
тические высокомолекулярные непредельные, предельные и арома-
тические углеводороды и др.
Гетероцепные полимеры наряду со-связями типа углерод — уг-
лерод имеют связи типа углерод — кислород, углерод — азот и др.,
например,
К этому классу соединений относятся все (кроме натурального
каучука) важнейшие природные полимеры (белки, полисахариды,
лигнин и др.), большинство синтетически получаемых полимеров:
полиамиды, полиэфиры, полиуретаны и др.
К элементоорганическим высокомолекулярным соединениям от-
носятся соединения, содержащие химическую связь углерод — эле-
мент (титан, олово, алюминий, бор, кремний и др.). Из этой группы
полимеров широко известны кремнийорганические полимеры, ос-
новная цепь которых содержит неорганические атомы, а боковые
цепи макромолекулы состоят из органических радикалов.
По способу получения высокомолекулярные соединения подраз-
деляются на полимеризационные соединения, т. е. соединения, по-
лучаемые по реакции полиприсоединения (полиэтилен, полипропи-
лен, полистирол и др.), и поликонденсационные соединения, т. е.
соединения, получаемые по реакции полизамещения (феноло- и
карбамидоформальдегидные соединения, полиамиды, полисилок-
саны и др.).
По отношению к воздействию тепла высокомолекулярные со-
единения делят на термопластичные и термореактивные.
К термопластичным полимерам относят соединения, не утрачи-
вающие своей плавкости и растворимости при многократном плав-
лении и охлаждении. К термореактивным полимерам относят со-
единения, образующие при нагревании пространственные, сетчатые
структуры и утрачивающие при этом способность плавиться или
растворяться. s
Синтез высокомолекулярных соединений "
Для получения высокомолекулярных соединений применяют
мономеры — низкомолекулярные соединения, которые содержат не-
насыщенные связи или неустойчивые циклы, а также соединения,
338
в одной молекуле которых содержится не менее двух функциональ-
ных групп (реакционноспособные группы атомов или отдельные
атомы), например, гликоли, двухосновные кислоты, диамины и
др. При применении двух мономеров с двумя бифункциональными .
группами образующиеся полимеры имеют линейную структуру, а
при использовании смеси мономеров, из которых по крайней мере
один имеет две функциональные группы, получают сетчатые поли-
меры.
Основными методами получения полимеров из мономеров яв-
ляются полимеризация и поликонденсация.
Полимеризация — процесс образования высокомолекулярных
соединений в результате взаимодействия между собой мономеров,
имеющих двойные связи или взаимодействия гетероциклов с раз-
мыканием колец. Этот процесс протекает без выделения низкомо-
лекулярных побочных продуктов, а реакция сопровождается вы-
делением тепла и необратима. Для осуществления процесса поли-
меризации необходимо перевести мономеры, участвующие в реак-
ции, из неактивной формы в активную. В зависимости от природы
мономеров это достигается различными путями: нагреванием, при-
менением катализаторов и инициаторов, воздействием световой
энергии, ядерного излучения или ультразвука.
В общем виде процесс полимеризации можно представить сле-
дующим образом:
nA —► (А)п
где А — молекула мономера, вступающего в реакцию;
п — число присоединяющихся друг к другу молекул мономера, или степень
полимеризации;
(А)„ — молекула полимера, состоящая из п звеньев молекул мономера.
Различают цепную и ступенчатую полимеризацию, по механиз-
му цепной полимеризации получают большинство известных высо-
комолекулярных соединений.
Процесс образования полимеров цепной полимеризацией скла-
дывается из трех стадий: инициирование; рост цепи и обрыв цепи.
Скорость образования активных центров — инициирование —
протекает медленно, тогда как две другие стадии процесса идут
очень быстро, практически в течение долей секунды. Чем выше ско-
рость роста цепи полимера по сравнению со скоростью ее обрыва,
тем больше молекулярная масса образующейся макромолекулы.
Молекулярная масса полимера сильно зависит от температуры по-
лимеризации (рис. XV. 2).
По механизму образования полимера различают радикальную
и ионную полимеризацию.
При радикальной полимеризации начало роста цепи (иницииро-
вание) происходит под влиянием инициаторов, тепла, света, а-, 0-
или у-излучения. При этом происходит разрыв двойной связи или
размыкание циклов с образованием радикала, обладающего одним
неспаренным электроном. Скорость полимеризации и молекуляр-
339
ная масса образующегося полимера зависит от содержания ини-
циатора (рис. XV. 3).
Наиболее часто для инициирования процесса полимеризации
применяют инициаторы (перекиси, гидроперекиси, диазосоединения
и др.), которые при нагревании распадаются с образованием сво-
бодных радикалов.
Температура полимеризации
Концентрация'инициатора.
Рис. XV.2. Изменение относительной
молекулярной массы полимера М при
различной температуре полимери-
зации.
Рнс. XV.3. Зависимость относитель-
ной молекулярной массы М (/) и ско-
рости полимеризации Vn (2) от кон-
центрации инициатора.
Для примера рассмотрим образование радикалов при распаде перекиси
зоила:
С6НБСОООСОС6НБ —> 2С6НБСОО*
беи-
Свободные радикалы могут частично разлагаться с образованием нового ти-
па радикалов
2С6НБСОО* —> 2С6Нб + 2СО2
Радикалы по мере своего образования присоединяются к молекулам моно-
мера, давая начальные активные центры (радикалы) полимеризации.
Рассмотрим процесс полимеризации этилена в присутствии перекиси бен-
зоила. Радикал перекиси бензоила активирует молекулу этилена, присоединяя
ее с образованием новых активных центров:
с8нБсоо’ + сн2=сн2 —> с6нБсоосн2сн;
При присоединении молекул этилена к полученному активному центру растущая
молекула полимера все время остается свободным радикалом до окончания про-
цесса полимеризации
свн.соосн2сн; + сн,=сн, —> с.нБсоосн2сн„сн2сн: + сн2=сн, —>
О О & л А Л и и * 4 * Z Z 4
—> С6НБСООСН2СН2СН2СН2СН2СН2 +••• и т. д.
т. е. происходит рост цепи до образования макромолекулы полиэтилена.
В процессе полимеризации возможен и обрыв цепи полимеризации в резуль-
тате рекомбинации (взаимодействия свободных радикалов между собой), напри-
мер
С6НБСООСН2СЬЙ + *СН2СН2ООСНбСв —► С6НБСООСН2СН2СН2СН2ООСЙ^СБ
340
или диспропорционирования, когда в процессе полимеризации могут образов»
ваться две макромолекулы, одна из которых будет содержать двойную связь:
С6Н5СООСН2—сн’г + *СН2—СН2ООСН6С6 —►
4 t
—> С6Н5СООСН2=СН2 + СН3СН2ООСНбСв
Обрыв цепи растущей макромолекулы может наступать и при взаимодей-
ствии цепи с неактивной молекулой мономера, растворителя или примеси, на-
пример
----СНг—СН2 + СН2=СН2 — --------СН2—СН3 + СН2=СН
Цепная полимеризация может прекратиться и в том случае,
когда макромолекула с растущей цепью столкнется со стенкой ап-
парата, в котором происходит процесс полимеризации.
Процесс цепной полимеризации практически необратим, причем
степень полимеризации зависит от соотношения между скоростями
роста цепи и ее обрыва. Чем выше это соотношение, тем больше
степень полимеризации, а, следовательно, больше и длина цепи
макромолекулы. Так как рост цепи макромолекулы может остано-
виться в любой момент времени в результате разнообразных хими-
ческих превращений, то образующиеся макромолекулы будут иметь
различную молекулярную массу, а конечный продукт полимериза-
ции будет характеризоваться полидисперсностью.
Следует отметить, что при проведении процесса полимеризации
природа инициатора мало влияет на рост и обрыв цепи макромо-
лекулы. Однако, рост цепи, а, следовательно, и молекулярную
массу полимера можно регулировать путем поддержания оп-
тимальной температуры полимеризации.
Ионная полимеризация, в отличие от радикальной, позволяет
получить полимеры с заранее заданными свойствами. Это объяс-
няется тем, что процесс возникновения цепи и присоединения мо-
лекул мономера к растущей цепи происходит под влиянием ката-
лизаторного комплекса, состав и свойства которого можно изме-
нять, подбирая соответствующие катализаторы и растворители.
При ионной полимеризации активными центрами образования мак-
ромолекул являются ионы. В зависимости от природы применяе-
мого катализатора и заряда образующихся ионов ионная полиме-
ризация подразделяется на катионную и анионную.
Катионная полимеризация протекает в присутствии катализато-
ров— неорганических кислот, галлоидопроизводных металлов, и
неметаллов (А1С13, BF3, SnCh) и небольших количеств сокатали-
заторов (вода, органические кислоты, эфиры).
Рассмотрим механизм катионной полимеризации на примере получения по-
лиизобутилена. При смешении катализатора и сокатализатора (BF3 и Н2О) со-
ответственно образуется активный каталитический комплекс
BFs-f-HOH —> (BF3OH)'H+
841
вступающий во взаимодействие с молекулой мономера с образованием активного
центра, к которому далее присоединяется молекула мономера:
(BF3OH)~H+ +
СН2=С(СН3)2 --------СН3—С(СН3)2
СН3С(СНз)2 + СН2=С(СН3)2 —► СНз—С(СН3)2—СН2—С(СН3)2
« т. д. до образования полимера.
Обрыв цепи может быть вызван образованием двойной связи в концевом
звене макромолекулы
СНз—(СН3)2—С[—СН2—С(СН3)2—]„—СН2—С—СНз ------>
| -Н+
СНз
—> СНз—(СН3)2С[—СН2—С(СН3)2—СН2—С=СН2
СНз
Присутствие сокатализаторов в смеси уменьшает энергию ак-
тивации молекул при полимеризации, что приводит к снижению
молекулярной массы полимера.
Катионная полимеризация может проходить и в отсутствие
сокатализаторов, но в этом случае ее проводят при более высоких
температурах.
Если в качестве катализаторов применяются щелочные метал-
лы, растворы амидов металлов в жидком аммиаке, металлоорга-
нические соединения и др., полимеризация мономеров осуществ-
ляется по анионному механизму. Механизм анионной полимериза-
ции можно проиллюстрировать на примере полимеризации непре-
дельных соединений в присутствии амида калия
KNH2 k+ + nh;
nh; + ch2=chx —> nh2ch2—снх
т. e. вначале _под действием катализатора образуется активный
центр NH2CHCHX, который далее взаимодействует с другой мо-
лекулой непредельного соединения
nh2ch2chx + СН2=СНХ —> NH2CH2CHXCH2CHX И т. д.
Обрыв цепи может наступить вследствие реакции цепи с рас-
творителем, например
NH2(CH2CHX)nCHX + NH3 —> NH2(CH2—СНХ)„—СН2СН2Х + nh2
При анионной полимеризации образуются достаточно устойчи-
вые макромолекулы полимера, называемые живущими полиме-
рами, способными и дальше инициировать процесс полимеризации.
В этом состоит отличие анионной полимеризации от катионной.
Механизм ступенчатой полимеризации существенно отличается
от механизма.цепной полимеризации тем, что образование поли-
мера происходит путем последовательного присоединения молекул
342
мономера друг к другу. Например, две молекулы мономера, соеди-
няясь между собой, образуют димер, который, присоединяя еще
одну молекулу мономера, дает тример и т. д. до образования по-
лимера. Такое соединение мономеров осуществляется в результате
перехода атома водорода или какой-либо группы атомов от одной
молекулы к другой. Образующиеся промежуточные продукты легко
могут быть выделены. По механизму ступенчатой полимеризации
чаще всего реагируют мономеры с однотипными функциональными
группами, одни из которых содержат подвижный водородный атом,
способные отдать, а другие — принять его. Так получают полиуре-
таны, эпоксидные смолы и др.
В качестве примера рассмотрим образование полиформальде-
гида в присутствии воды:
СН2О + НОН —► НОСН2ОН
i 4
носн2он+сн2о —> НО—СН2—ОСН2ОН
i 4
НОСН2ОСН2ОН + СН2О —> НОСН2ОСН2ОСН2—ОН и т. д.
Из приведенной схемы видно, что вначале формальдегид при-
соединяет воду, при этом создается функциональная группа
с легкоподвижным атомом водорода, а затем происходит присо-
единение молекул формальдегида за счет перехода атома водорода
от одной молекулы к другой.
Примером ступенчатой полимеризации полифункциональных
веществ с разнотипными функциональными группами может слу-
жить образование полиуретанов из диизоцианатов и гликолей:
НО—R—ОН + O=C=N—R'—N=C=O —>
гликоль диизоцианат
HOROH, O=C=N—R'—N = C=O
—> НО—R—О—С—NH—R'—N=CO-------------------------->
II
О
Г—С—NH—R'—NH—С—О—R—О—7
II II
L о о _1„
полиуретан
Максимальная молекулярная масса полимера достигается при
эквимолекулярном соотношении компонентов. Процессы полимери-
зации ведут при нагревании смеси исходных веществ.
Сополимеризация — процесс совместной полимеризации двух
или нескольких отличающихся по химическому составу мономеров.
Сополимеризация более сложный процесс, чем полимеризация, так
Как в этом случае наблюдается влияние каждого из мономеров
Иа активность другого из них.
Процесс сополимеризации во многом аналогичен процессу по-
лимеризации, однако имеет ряд особенностей;
34а
1) сополимеризация легче протекает между молекулами моно-
меров, близких по строению;
2) ряд мономеров не способны образовывать полимеры, но
легко могут сополимеризоваться с мономерами другого состава.
Изменяя концентрацию мбномеров, можно получать полимеры,
характеризуемые определенными свойствами: растворимостью, эла-
стичностью, требуемой температурой размягчения, способностью
окрашиваться и др. Так, при увеличении содержания винилацетат-
ных групп в полиэтиленовой цепи эластичность сополимера возра-
стает по сравнению с эластичностью полиэтилена.
В макромолекуле сополимера элементарные звенья различных
мономеров могут правильно чередоваться друг с другом, например,
пЛ -|- пВ —► •••—“А—В—А—В—-А—В—
Но чаще всего элементарные звенья располагаются в макромоле-
куле хаотически, т. е. сополимеры имеют нерегулярное строение.
Такое чередование отдельных звеньев мономеров в цепи макромо-
лекулы обусловлено различной
Рис. XV.4. Схема цепей обычного по-
лимера (а), блок-сополимера (б) и
привитого сополимера (в).
скоростью полимеризации отдель-
ных мономеров, причем содержа-
ние более активного мономера в
макромолекуле выше, чем менее
активного, что ведет к изменению
состава реакционной смеси, а сле-
довательно, к изменению состава
сополимера. Поэтому возмож-
ность регулирования свойств по-
лучаемого продукта при обычных
способах сополимеризации за-
труднительна.
Для получения высокомолеку-
лярных соединений с определен-
ными свойствами применяют спе-
циальные методы сополимериза-
ции, приводящие к получению
привитых сополимеров и блок-со-
полимеров (рис. XV. 4).
Привитые сополимеры полу-
чают несколькими методами.
В основе первого метода лежит
реакция передачи цепи с превращением макромолекул полимера
(—А—)„ в макрорадикал. При наличии в реакционной смеси легко
распадающегося на радикалы инициатора и мономера В процесс
образования привитых полимеров протекает по следующей схемез
R* +--А—А—А—А—А —> RH +------А—А—А—А—А----—>
...—а—А-А—А—А--
В—В—В---
344
По второму методу в полимер вводят легко распадающиеся
группы, например, перекисные. При нагревании или облучении об'
разуются макрорадикалы, инициирующие прививку мономера
х,0
- + R-c;
•--СНз—сн-
- + НС1
со
со
I
соос;
4R
..—сн2—сн —> —сн2—сн—+ r—с:
со
;О
со
Мономер прививается по месту распада перекисных связей.
Привитые сополимеры можно получить и при наличии в цепи
макромолекул двойных связей. Таким способом прививают к нату-
ральному каучуку боковые цепи полистирола, поливинилхлорида
и др.
Блок-сополимеры, — полимеры, в макромолекулу которых вхо-
дят блоки отдельных звеньев полимеров
nA + тВ —►-----А—А—В—В—А—А—В—В------
Блок-сополимеры получают путем перевода одного полимера
в макрорадикал с последующим присоединением к нему звеньев
другого полимера.
Превращение молекул в радикалы происходит под влиянием
тепла, облучения, ультразвука, механических воздействий. Если
расщепление макромолекулы идет с образованием макрорадикалов
в присутствии мономера В, to происходит инициирование мономера
с образованием блок-сополимеров и привитых сополимеров, так
как активные центры возникают не только на концах, но и в сере-
дине цепи. Конечный продукт содержит, кроме сополимера, гомо-
полимер (—В—)то и исходный полимер (—А—)п.
Макрорадикалы получают и при введении в полимер концевых
групп, легко образующих свободные радикалы (например, в при-
сутствии галогеналкилов). Полученные макромолекулы при облу-
чении образуют радикал по схеме:
Бг(—СН2—CHR—)А—СВг3 Вг(— СН2—СН— R—СВг2 + Вг
Эти радикалы инициируют полимеризацию мономера СХ2 = СНХ
с образованием блок-сополимера
Вг (—СН2—CHR—)га— СВг2—(—СН,—СНХ—)т
Для блок-сополимеризации по методу взаимодействия концевых
функциональных групп используют низкомолекулярные полимеры
345
(степень полимеризации 10 4-50). Так, например, получают поли-
меры, содержащие на концах только карбоксильные или гидрок-
сильные группы, либо амино- или карбоксильные группы.
Блок-сополимеры получают и при использовании так называе-
мых «живущих» полимеров, представляющих собой соединения,
в макромолекуле которых концевые группы имеют свободный ак-
тивный радикал. При прибавлении к такому полимеру мономера,
способного полимеризоваться, происходит рост цепи, приводящий
к образованию блок-сополимера.
Полимеризацию мономеров проводят в блоке (или в массе),
в растворе, в эмульсии и суспензии (капельная или бисерная по-
лимеризация).
При периодическом способе блочной полимеризации мономер,
очищенный от примесей, смешивается с другими компонентами
(катализатором, инициатором, пластификатором) и загружается
в реактор, который затем нагревается до строго определенной тем-
пературы. По мере образования полимера концентрация мономера
уменьшается, а вязкость среды возрастает из-за увеличения кон-
центрации полимера в мономере. Реакция полимеризации проис-
ходит с выделением тепла, поэтому для получения полимера с не-
обходимой молекулярной массой следует поддерживать опреде-
ленную температуру, т. е. интенсивно охлаждать формы, что пред-
ставляет в этом процессе большую трудность. Как правило, из-за
местного перегрева получаемый полимер имеет высокую полидис-
персность. Этим методом получают полимеры в виде блоков, ли-
стов и т. п.
При непрерывной блочной полимеризации жидкий или газооб-
разный мономер непрерывно поступает в реактор, а образующийся
полимер непрерывно выводится из реактора в виде расплава.
Блочным методом получают полистирол, полиэтилен, полипро-
пилен, натрийбутадиеновый каучук и др.
Полимеризацию в растворителях проводят двумя путями. По
первому из них подбирают такой растворитель, в котором раство-
ряется как мономер, так и полимер, образующийся в процессе по-
лимеризации. Получаемый лак (раствор полимера в растворителе)
может использоваться сразу как готовый продукт. В тех случаях,
когда необходимо из лака выделить полимер, его осаждают.
Второй путь —это полимеризация в растворителе, который рас-
творяет только мономер, но не растворяет полимер. По мере обра-
зования полимер выпадает в осадок, и отфильтровывается. Такие
полимеры однородны по составу, так как в процессе их получения
удается строго поддерживать оптимальную температуру. Данным
способом получают бутвар, поливинилацетат и др.
Следует отметить, что при полимеризации в растворителях
большое значение для величины молекулярной массы и скорости
полимеризации имеет концентрация мономера (рис. XV. 5) и тип
растворителя.
346
Эмульсионный, или латексный способ полимеризации состоит
5 том, что мономер, смешанный с инициатором (растворимым
р воде), эмульгатором, регулятором pH и поверхностного натяже-.
ния, при перемешивании превращается в мельчайшие капельки,
навешенные в другой жидкости, чаще всего, в воде. Присутствие
эмульгатора в смеси делает эмульсию не только устойчивой, но и
существенно влияет на механизм полимеризации. Чем выше дис-
пергирующая способность эмульгатора (больше частиц мономера
в 1 см3 эмульсии), тем выше скорость полимеризации.
Рис. XV.5. Зависимость скорости по-
лимеризации Vn(-0 и относительной
молекулярной массы Л4 (2) от кон-
центрации мономера.
Рис. XV.6. Влияние времени по-
лимеризации иа количество обра-
зовавшегося полимера.
ч
Полученные эмульсии нагреваются до температуры реакции,
в результате чего происходит полимеризация мономера. Количе-
ство образовавшегося полимера зависит от времени полимериза-
ции (рис. XV. 6). При этом способе выделяемое тепло легко отво-
дится, что облегчает получение более однородного полимера с
меньшей полидисперсностью, чем полимер, получаемый блочным
способом. Недостатком этого способа является трудность выделе-
ния эмульгатора, поэтому получаемый полимер имеет низкие ди-
электрические свойства.
Суспензионная (бисерная) полимеризация состоит в том, что
мономер диспергируют в нерастворяющей или плохо растворяющей
среде, чаще всего, в воде. Для инициирования процесса полимери-
зации применяются водонерастворимые инициаторы, например пе-
рекись бензоила, но хорошо растворимые в мономере. В качестве
стабилизаторов эмульсии применяют водный раствор поливинило-
вого спирта и др.
Бисерная полимеризация протекает самостоятельно в крупной
капле мономера размером от 0,05 до 0,3 см, в отличие от размера
Капли при эмульсионной полимеризации (от 10-3 до 10~4 см). Об-
разующийся полимер выделяется в виде гранул, размер которые
347
зависит от размера исходных капель, природы и количества ста-
билизатора. Отделенный от воды фильтрованием полим.ер надрав-
ляется на использование.
Поликонденсация — процесс образования высокомолекулярного
соединения — полимера — в результате взаимодействия мономеров
между собой; при поликонденсации наряду с полимером выде-
ляются низкомолекулярные побочные продукты (вода, аммиак,
двуокись углерода и др.). В реакцию поликонденсации вступают
мономеры с би- и многофункциональными связями. Если в реак-
ции поликонденсации участвуют одинаковые молекулы, содержа-
щие две различные реакционные группы, такой процесс поликон-
денсации называется гомополиконденсацией, например, поликон-
денсация аминокислот:
H2N—(СН2)6—соон + H2N—(СН2)6СООН —>
—> Н2О + H2N(CH2)6CONH(CH2)6COOH
H2N(CH2)6CONH(CH2)sCOOH + H2N(CH2)6COOH — >
—> Н2О + H2N(CH2)6CONH(CH2)6CONH(CH2)6COOH
и т. д. до образования полимера [—HN(CH2)g— СО—]п + (га —1
— 1)Н2О.
Поликонденсация мономеров различного химического состава
получила название гетерополиконденсации. Примером гетерополи-
конденсации может служить процесс образования полимера при
взаимодействии себациновой кислоты и гексаметилендиамина
НООС(СН2)8СООН + H2N(CH2)6NH2 —>
—> Н2О + HOOC(CH2)3CONH(CH2)6NH2
HOOC(CH2)3CONH(CH2)6NH2 + НООС(СН2)3СООН —>
—> HOOC(CH2)3CONH(CH2)6NHOC(CH2)3COOH + Н2О
и т. д. до [—ОС—(CH2)8CONH(CH2)6NH—]„ + (п—1)Н2О.
-Степень поликонденсации зависит от чистоты-и соотношения
исходных веществ, температуры и времени поликонденсации
(рис. XV. 7).
При рассмотрении процесса поликонденсации можно сделать
вывод, что рост цепи макромолекулы происходит ступенчато, при-
чем образующиеся промежуточные продукты обладают стабиль-
ностью и могут быть выделены. Следует также отметить, что при
поликонденсации одновременно могут протекать процессы деструк-
ции (особенно при повышенных температурах), что приводит к
уменьшению размер-а макромолекулы, так как длинные макромо-
лекулы легче подвергаются разложению, чем короткие.
В зависимости от состава исходных мономеров образующиеся
при поликонденсации полимеры могут иметь линейное (полиамиды,
полиэфиры и др.) и пространственное строение (аминопласты, фе-
нолоформальдегидные смолы и др.).
При поликонденсации большое внимание следует уделять со-
хранению определенного соотношения исходных мономеров. При
348
нарушении этого условия поликонденсация может прекратиться на
ранних стадиях, и полимер заданной молекулярной массы не об-
разуется вследствие присоединения к обоим концам макромолекул
одинаковых реакционных групп. Следовательно, если число молей
одного мономера превышает число молей другого, или наоборот,
то избыток одного из мономеров приведет к снижению молекуляр-
ной массы, т. е. степень поликонденсации п будет определяться
избытком данного мономера
п = 16о/<у
где q — избыток одного из мономеров, % (мол.)
Особое внимание на процесс поликонденсации оказывают ката-
лизаторы и температура. При повышении температуры процесс
Рис. XV.7. Зависимость степени
полимеризации п от времени по-
ликонденсации гексаметиленди-
амида с себациновой кислотой при
различной температуре:
1 — при 185 °C; 2— при 167 °C; 3 — при
145 °C.
Время полинонвенсации
Рис. XV.8. Зависимость молеку-
лярной массы полимера М от вре-
мени поликонденсации.
поликонденсации сдвигается в сторону образования полимера, так
как в этих условиях ускоряется удаление из зоны реакции обра-
зующихся в процессе синтеза низкомолекулярных продуктов. При-
чем молекулярная масса полимера при поликонденсации зависит
от полноты удаления низкомолекулярных продуктов, что видно из
следующего уравнения
п = ''/кТа
где п — средняя степень поликонденсации;
К — константа равновесия;
а — количество воды или другого продукта в системе, % (мол.).
Молекулярная масса образующегося полимера, кроме того, за-
висит от времени поликонденсации (рис. XV. 8), т. е. от продол-
жительности пребывания полимера в зоне высоких температур.
Поликонденсацию проводят как в присутствии катализаторов
(фенолоформальдегидные, карбамидные смолы и др.), так и без
них (полиамиды и др.). В зависимости от свойств катализатора
получают полимеры с различными свойствами. Так, при взаимо-
действии формальдегида и фенола в присутствии кислого катали-
затора образуются плавкие новолачные ‘фенолоформальдегидные
смолы, тогда как в присутствии щелочного катализатора и избытка
формальдегида — термореактивные смолы резольного типа.
Поликонденсацию можно проводить в расплаве, в растворе, на
границе раздела двух фаз, в твердой фазе.
Поликонденсация в расплаве применяется тогда, когда исход-
ные мономеры и получаемый полимер могут длительное время под-
вергаться воздействию температуры без разложения в расплавлен-
ном состоянии. Этим методом удается получать полимеры высо-
кого качества (полиэфиры, полиамиды и др.). Технология получе-
ния полимеров состоит в следующем. Исходные мономеры смеши-
ваются и нагреваются в реакторе в течение нескольких часов в ат-
мосфере инертного газа во избежание их окисления. В конце про-
цесса поликонденсации с целью полного удаления низкомолеку-
лярных соединений из смолы в реакторе создается вакуум.
Получаемый продукт направляется на дальнейшее использование.
Поликонденсацию в растворе проводят в тех случаях, когда
образующийся полимер и исходные мономеры разлагаются при
температуре плавления. Тогда процесс осуществляют в раствори-
телях, которые растворяют исходные мономеры и образующийся
полимер, или в растворителях, в которых растворяются только
участвующие в реакции мономеры. При нагревании смеси взаимо-
действие мономеров между собой протекает с довольно большой
скоростью, и реакция может быть доведена до высокой степени
превращения мономеров.
Поликонденсация в растворе проводится в более мягких тем-
пературных условиях, что исключает местные перегревы смеси, не
требует применения вакуума и инертного газа, а следовательно,
и сложной аппаратуры. Однако при использовании этого метода
необходимо готовить растворы мономеров, регенерировать раство-
ритель (отмывать от полимера, фильтровать, сушить и т. д.).
Поликонденсация на поверхности раздела фаз проводится
в двух несмешивающихся жидкостях, в которых отдельно раство-
рены мономеры. Чаще всего одной из несмешивающихся жидкостей
является вода, а другой — жидкость, которая не смешивается с во-
дой и инертна к мономерам, например, углеводороды. Синтез поли-
мера происходит на границе раздела водной и углеводородной фаз.
Для ускорения процесса синтеза (для создания более полного кон-
такта между мономерами) применяют перемешивание. По оконча-
нии процесса полимер отфильтровывают, промывают и сушат.
Поликонденсация на поверхности раздела двух фаз идет очень
быстро даже при низких температурах и атмосферном давлении,
так как образующиеся продукты выделяются из сферы реакции,
350
в
м'
Рис. XV.9. Изменение предела
прочности полимера при разрыве <т
в зависимости от молекулярной
массы М.
Этим способом можно получать также полимеры с высокой тем-
пературой плавления. Однако, применение его в промышленности
весьма ограничено.
Свойства высокомолекулярных соединений
Свойства высокомолекулярных соединений зависят от многих
факторов: средней молекулярной массы, химического состава,
строения макромолекул, состояния вещества и др.
Особое влияние на свойства полимеров оказывает средняя мо-
лекулярная масса: при увеличении этого показателя до опреде-
ленного предела улучшаются прочностные свойства (рис. XV. 9),
повышается устойчивость к воздействию высоких и низких темпе-
ратур. Так, с ростом молекулярной
массы полистирола от 31200 до
62 400 температура его размягчения
увеличивается от 105 до 180 °C. •
Прочностные свойства полиме-
ров зависят и от числа поперечных
связей N между молекулярными це-
пями (см. рис. XV. 1), т. е. от сте-
пени отвердения. Светостойкость ма-
териалов можно усилить введением
в макромолекулу полимера фтора
или нитрильной группы —CN.
Для получения негорючих поли-
мерных материалов и материалов,
устойчивых к воздействию кислот и
щелочей, применяют мономеры, со-
держащие фтор и хлор.
Свойства полимеров с одинаковой геометрической формой мак-
ромолекулы зависят от их химического состава. Так, полиамиды
имеют более высокий предел прочности при разрыве, бблее высо-
кую температуру плавления и эластичность по сравнению с
уретанами.
Различное пространственное расположение атомов или групп
атомов в макромолекуле при одинаковом химическом составе резко
влияет на свойства полимеров. Применяя комплексные катализа-
торы, состоящие из металлоорганических соединений А1(С2Нз)3 и
хлоридов металлов переменной валентности (например, TiCl3,
TiCl4), получают полимеры со строго определенным расположе-
нием боковых групп в цепи макромолекулы. Такие соединения
называются стереорегулярными. Их подразделяют на изотактиче-
ские и синдиотактические. У изотактических полимеров боковые
группы расположены только по одну сторону цепи, например,
у полипропилена
... _ сн2—СН— СН2— СИ— сн2—сн-
I I I
ь СН8 СН3 СНз
поли-
351
У синдиотактических полимеров боковые группы чередуются по
обе стороны молекулярной цепи в строгом порядке:
СНз СН3
I I
----СН2—СН— СН2— СН— СН2—СН— СН,—СН-----
СНз СНз
Из приведенных данных следует, что строение макромолекулы
стереорегулярных полимеров резко отличается от строения атак-
тических полимеров, у которых метильные группы беспорядочно
расположены вдоль оси макромолекулы:
СНз СНз СНз
----СН2—СН— СН2—СН— СН2— СН— СН2—СН— СН2—(!н----
I I
СНз СНз
Правильное чередование метильных или других групп у сте-
реорегулярных полимеров обеспечивает получение материала с
лучшими физико-механическими свойствами. Такие полимеры
Рис. XV.10. Формы макромолекул:
а—вытянутая; б—изогнутая; в —свернутая;
г—разветвленная.
имеют более высокую темпера-
туру размягчения и низкую рас-
творимость по сравнению с атак-
тическими полимерами.
Большое влияние на свойства
полимеров оказывает форма мак-
ромолекул (вытянутые, изогну-
тые, сферические, разветвлен-
ные), рис. XV. 10. Чем более вы-
тянута и менее разветвлена мак-
ромолекула полимера, тем выше
вязкость его раствора, ниже его
растворимость и выше прочност-
ные свойства. На свойства поли-
меров оказывает влияние и ориентация макромолекул: чем больше
ориентированы молекулы в определенном направлении, тем выше
их механические свойства по сравнению со свойствами неориенти-
рованных макромолекул.
Химическая стойкость полимеров зависит от многих факторов.
Полимеры парафинового ряда весьма индеферентны к химическому
взаимодействию, поэтому полимеры, у которых макромолекулы
состоят из углеродных цепей, например, полиэтилен, полиизобути-
лен, устойчивы к действию кислот, щелочей, растворов солей и
слабых окислителей. Введение в полиэтиленовую цепь заместите-
лей, например, гидроксильной группы, приводит к ослаблению стой-
кости к коррозии. Так, поливиниловый спирт разрушается под дей-
ствием воды, кислот и щелочей. Исключение составляют соедине-
ния, у которых водород в полиэтиленовой цепи замещен фтором
или хлором. Такие полимеры, как политетрафторхлорэтилен и по-
352
t
ливинилхлорид и др„ устойчивы к действию щелочей, кислот, рае*
творов солей и, до некоторой степени, к окислителям.
Гетероцепные полимеры, у которых отдельные структурные
I I I
звенья соединены между собой связями типа —С—О—, —С—N.
I III
*-С—S— подвергаются воздействию химических реагентов.
Растворимость полимеров в растворителях также различна.
Неполярные полимеры (полиэтилен, полиизобутилен, полистирол)
набухают или растворяются в неполярных растворителях (бензин,
бензол, СС14), но устойчивы в полярных растворителях (вода,
спирты).
Полимеры, содержащие полярные группы, например, гидрок-
сильные (—ОН) или карбоксильные (—СООН), устойчивы в не-
полярных веществах (бензин, бензол), но набухают или раство-
ряются в полярных растворителях (фенол, вода, спирты). Нали-
чие в макромолекуле двойных связей снижает устойчивость таких
полимеров при воздействии химических реагентов и, в особенности,
окислителей.
Физико-химические свойства полимеров лежат в основе выбора
материала для изготовления изделий самого различного назначе-
ния. При изготовлении и применении изделий следует учитывать
характер и величину деформации полимера под влиянием внешних
усилий. Деформация в полимерах может быть вызвана измене-
нием межатомных расстояний (упругая деформация), способ-
ностью макромолекул изменять свою форму (высокоэластическая
деформация), а также перемещением молекул полимера относи-
тельно друг друга (пластическая деформация). Пластическая де-
формация необратима, т. е. сохраняется и после снятия нагрузки.
По видам деформаций, возникающих в полимерах под дей-
ствием внешних усилий при комнатной температуре, их подразде-
ляют на твердые полимеры, для которых характерна упругая де-
формация: эластичные полимеры, или эластомеры, которым, кроме
упругой деформации, свойственна и высокоэластическая деформа-
ция и текучие полимеры, близкие по свойствам к обычным жидко-
стям.
Свойства полимеров зависят от их строения, так как полимеры
могут находиться в аморфном или кристаллическом состоянии, или
содержать аморфные и кристаллические фазы. Соотношение между
кристаллическими и аморфными фазами в полимере характери-
зуется степенью кристалличности. Кристаллическую структуру мо-
гут иметь полимеры, макромолекулы которых строго регулярной
линейной структуры, или полимеры с редкосетчатым строением.
Полимерам, имеющим кристаллическое строение, свойственна бо-
лее высокая температура плавления, улучшенные механические и
химические свойства по сравнению с аморфными полимерами.
12 Зак. 558 3&8
При нагревании твердые полимеры сначала переходят в эла-
стическое, а затем в вязкотекучее состояние. Температура перехода
полимера из твердого состояния в эластическое, или наоборот, по-
лучила название температуры стеклования Тс, а в вязкотекучее —
температуры текучести Т?. Твердые полимеры чаще всего приме-
няются в качестве связующего при получении сложных пластмасс;
эластичные — для каучуков и некоторых резин, а для волокон ис-
пользуются полимеры, характеризующиеся умеренной деформацией
при относительно больших усилиях.
Полимеры сетчатого строения не пригодны для формования
изделий, так как они не плавятся, не растворяются и не размяг-
чаются. Изделия из них можно готовить только механической об-
работкой, что приводит к большому расходу материала и увеличи-
вает стоимость изделий, поэтому при синтезе стремятся получать
полимеры линейной структуры, которая либо сохраняется в гото-
вом изделии, либо переходит под действием тепла или химических
реагентов в пространственную структуру в процессе переработки.
Как указывалось выше, полимеры под действием кислорода
воздуха, температуры и других факторов утрачивают свои перво-
начальные свойства. Так, каучуки могут полностью разрушаться
при длительном хранении. Особенно сильно подвержены воздей-
ствию кислорода воздуха полимеры аморфного строения. Для тор-
можения или полного подавления процессов окисления в состав
полимеров вводят малые количества веществ, называемых стаби-
лизаторами, или антиоксидантами.
Антиоксиданты, присутствующие в полимере, сами не реаги-
руют с кислородом, но соединяясь с радикалами, образующимися
в процессе окисления полимера, замедляют реакцию окисления.
Следовательно, антиоксиданты необходимо вводить в полимер до
начала окисления. Наибольшее применение при стабилизации по-
лимеров находят ароматические амины (фенил-р-нафтиламин, сме-
си гомологов и производных дифениламина), бутилфенол, фосфиты,
фенолы и др. Наиболее дешевым и универсальным стабилизатором
резин является сажа.
Высокомолекулярные соединения служат основой для получе-
ния полимерных материалов. Наиболее широкое применение в тех-
нике из этих материалов находят пластические массы, химические
волокна, целлюлоза, лаки, пленки и др.
2. ПРОИЗВОДСТВО ПЛАСТИЧЕСКИХ МАСС
Материалы, получаемые из природных или синтетических высо-
комолекулярных соединений, способные при нагревании и давле-
нии приобретать и устойчиво сохранять форму после охлаждения
или отверждения, получили название пластических масс.
Пластические массы сочетают в себе ряд ценных свойств. Они
имеют малую плотность, устойчивы к атмосферной коррозии, а
многие из них к действию агрессивных сред, являются хорошими
354
теплоизоляционными материалами и диэлектриками, некоторые из
них могут быть оптически- и радиопрозрачными, упругими или
эластичными. Оии легко формуются в изделия, а некоторые из них
по удельной прочности превосходят углеродистые стали, цветные
металлы и их сплавы. Стоимость 1 м3 отдельных пластических
масс приближается к стоимости черных металлов.
Пластмассы, наряду с указанными достоинствами, обладают и
некоторыми недостатками: низкой теплостойкостью, теплопровод-
ностью, малой твердостью и жесткостью, а также подвержены ста-
рению.
Пластические массы по составу подразделяют на простые (не-
наполненные) и сложные, или композиционные.
Простые пластмассы состоят только из одного высокомолеку-
лярного соединения (полиэтилен, полипропилен, полистирол, фто-
ропласты и др.). В состав сложных пластмасс входят связующие
наполнители, пластификаторы, красители, отвердители, стабилиза-
торы и другие добавки, равномерно распределенные в связующем.
Свойства пластмассы определяются свойствами основного ком-
понента— полимера (связующего), который обеспечивает пропитку
и соединение частиц, входящих в состав смеси, в однородную
массу. За счет связующего достигается монолитность и сохраняется
заданная форма готового изделия.
Наполнители — твердые вещества, придающие или усиливаю-
щие определенные механические или диэлектрические свойства
пластмасс, снижающие усадку при формовании, горючесть и стои-
мость изделий, улучшающие внешний вид и т. д. Для получения
пластических масс в качестве наполнителей используются мате-
риалы органического и неорганического происхождения: древесная
мука (тонко измельченная древесина хвойных пород), бакелито-
вая мука, каолин, графит, плавиковый шпат, асбест и др. Некото-
рые наполнители увеличивают дугостойкость (слюда, плавиковый
шпат), улучшают полупроводниковые свойства (графит), тепло-
стойкость (асбест) изделий из пластмасс.
Наполнители подразделяются на порошкообразные (древесная
и кварцевая мука, асбест, графит, тальк и др.), волокнистые (ас-
бестовое и стеклянное волокна, хлопок и др.) и листовые (бумага,
древесный шпон и др.). В зависимости от применяемого наполни-
теля изменяются и свойства пластмасс.
Пластификаторы — органические соединения, повышающие пла-
стичность, эластичность композиции. Их применяют только для
ограниченного числа полимеров, например, пластифицируют поли-
меры и сополимеры хлористого винила, эфиров целлюлозы, каучук,
полиамиды и т. п.
На рис. XV. 11 схематично показана структура непластифици-
рованного и пластифицированного полимеров.
Введение большого количества пластификаторов в композицию
уменьшает предел прочности изделия при растяжении и сжатии.
В качестве пластификаторов применяют малолетучие вещества,
12*
355
а
главным образом, касторовое масло, эфиры фталевой илиортофос-
форной кислот, реже адипиновой и себациновой.
Красители — вещества, придающие пластмассам требуемую
окраску. В качестве красителей применяют как органические, так и
минеральные вещества, обладающие высокой термической стой-
костью и светопрочностью. Красители вводят либо непосредственно
в связующее, либо добавляют в смесь при смешении. Они должны
совмещаться или смешиваться со связующим либо растворяться
в нем. Вводимые в смесь красители должны сохранять свой цвет,
как в процессе формования изделий,
так и при их эксплуатации.
Отвердители — вещества, способ-
ствующие переводу линейной струк-
туры макромолекул полимера в про-
странственную в результате сшива-
ния (образования химических свя-
зей) макромолекул между собой.
В качестве отвердителей исполь-
зуют уротропин, дикарбоновые кис-
лоты, диамины, ненасыщенные мо-
номеры и др. Часто наряду с отвер-
дителями в состав композиции вво-
дят ускорители отверждения (окись
кальция или магния). '
Кроме того в зависимости от На-
значения изделий в их состав вво-’
(стеараты кальция, цинка и др.),
облегчающие удаление изделий из формы; парообразователи — ве-
щества, обеспечивающие получение легких пластмасс (пено- и по-
ропластов); стабилизаторы — вещества, способствующие сохране-
нию в пластмассе первоначальных свойств; фунгициды — веще-
ства, вводимые для защиты пластмасс от разрушающего действия
плесени и др.
В настоящее время выпускается большое количество синтети-
ческих смол и пластмасс на их основе. В зависимости от типа свя-
зующего пластические массы подразделяют на следующие классы:
1) пластмассы на основе полимеров, получаемых цепной поли-
меризацией;
2) пластмассы на основе полимеров, получаемых поликонден-
сацией и ступенчатой полимеризацией;
3) пластмассы на основе химически модифицированных при-
родных полимеров;
4) пластические массы, получаемые на основе природных и
нефтяных асфальтов и смол — продукта деструкции различных ор-
ганических веществ.
По масштабу производства пластические массы можно разбить
на три группы. В первую группу входят полимеры, выпуск кото-
рых составляет более 75% производимых полимеров и исчисляется
б
Рис. XV.11. Цепи обычного (а) и
пластифицированного (б) поли-
мера.
дят смазывающие вещества
356
миллионами тонн (полиолефины, полистирол, поливинилхлорид,
фено- и аминопласты). Ко второй группе относятся акриловые,
эпоксидные, кремнийорганические полимеры, полиамиды, поли-
эфиры и др., выпуск которых составляет тысячи и десятки тысяч
тонн, К третьей группе относятся полифенилены, полисульфаты,
фторопласт-4, физиологически активные биополимеры и др., выпуск
которых достигает десятки и сотни тонн.
Для примера рассмотрим получение наиболее широко приме-
няемых полимеров.
Пластические массы на основе полимеров,
получаемых цепной полимеризацией
К этой группе пластмасс относятся полиэтилен, полипропилен,
полиизобутилен, полистирол, полимеры и сополимеры хлористого
винила, фторпроизводных этилена, полиакрилаты и др. Такие
пластмассы выпускаются без наполнителя; они термопластичны,
обладают хорошими диэлектрическими свойствами, высокой удар-
ной вязкостью (за исключением полистирола), устойчивы к дей-
ствию многих агрессивных сред, но большинство из них имеет
низкую теплостойкость.
Полиэтилен [—СН2 — СН2—]п — насыщенный углеводород па-
рафинового ряда с молекулярной массой (в зависимости от метода
получения) от 18000 до 800000. Это роговидный продукт, выпускае-
мый в виде гранул.
Сырьем для производства полиэтилена служит этилен, полу-
чаемый высокотемпературным пиролизом нефтяных фракций или
высокотемпературным крекингом пропана и бутана при 800 °C в
трубчатых печах. Для полимеризации применяют этилен высокой
степени чистоты (99,99% СгНД, так как присутствие примесей мо-
жет,привести к обрыву полимерной цепи и снижению массы моля
полимера. Особенно опасны примеси в сырье, поступающем на
полимеризацию по радикальному механизму.
Для получения чистого этилена газовую смесь пропускают че-
рез систему охлаждения, работающую при температуре от —ПО
до —130 °C и давлении от 0,5 до 5 МПа. При этом все примеси (за
исключением ацетилена и олефинов) извлекаются из этилена. Аце-
тилен и олефины удаляются гидрированием в присутствии ко-
бальтмолибденового катализатора при температуре 250 °C и дав-
лении 1,5 МПа.
В настоящее время полиэтилен получают тремя способами:
полимеризацией этилена при низком давлении (0,5—0,8 МПа) и
температуре 70—80 °C в присутствии комплексных катализаторов,
состоящих из четыреххлористого титана TiCU и триэтилалюминия
А1(С2н5)3;
полимеризацией этилена в растворителе при 130—170 °C и сред-
нем давлении 3,5—4,0 МПа в присутствии окислов металлой пере-
менной валентности (окислов хрома, ванадия и др.);
полимеризацией этилена при высоком давлении (130—250 МПа)'
и температуре 200—270 °C в присутствии кислорода (0,005—
0,008% в смеси).
В зависимости от методов получения полиэтилена свойства его
различны (табл. XV. 1).
Таблица XV. 1. Свойства полиэтилена
Показатели Полиэтилен
низкого давления среднего давления высокого давления
Масса 1 моля 80 000-400000 70 000—400 000 70 000—350 000
Температура плавления, С° 120-125 125-137 105-108
Плотность, 103-кг/м3 Предел прочности, МПа 0,945—0,955 0,95—0,97 0,916—0,935
при растяжении 22—32 27—33 12-16
при изгибе 20-35 25-40 12-17
Относительное удлине- ние при разрыве, % 400-800 200-800 150-600
Модуль упругости при изгибе, МПа 550-900 — 150-250
Удельное объемное электрическое сопро- тивление, Ом • м 10>4—10!5 1015 J015
Тангенс угла диэлектри- ческих потерь при 106 Гц (2+5) • 10-4 (2+3) • 10-4 (2+3) • 10“*
Диэлектрическая прони- цаемость при 106 Гц 2,1-2,4 2,2-2,4 2,2-2,3
Электрическая проч- ность, мВ/м 45-60 45-60 45-60
Морозостойкость, °C До -70 До -70 До -70
Полиэтилен высокого давления (рис. XV. 12) получают в при-
сутствии кислорода. Процесс полимеризации протекает по ради-
кальному механизму. Свежий этилен высокой степени чистоты из
газгольдера под давлением 0,8—1,1 МПа поступает в смеситель
(на схеме не показан) для смешения с кислородом (от 0,005 до
0,008%) и возвратным этиленом, после чего подается в систему
компрессоров 1, где сжимается сначала до 25 МПа, а затем (после
очистки от масла) до 15 МПа. Причем, чем выше давление, тем.
выше скорость полимеризации (рис. XV. 13).
Пройдя систему очистки и смазкоотделитель 2, этилен посту-
пает в реактор 3 трубчатого типа на полимеризацию. Реактор яв-
ляется аппаратом идеального вытеснения. Он состоит из наклонно
расположенных труб диаметром до 25 мм и общей длиной до 300 м
и имеет три зоны: зону подогревания этилена до 200 °C; зону по-
лимеризации, где температура поддерживается в пределах 200—
225°C, и зону охлаждения реакционной массы (НО—125°C). На-
ЗЕ8
гревание этилена и охлаждение реакционной массы осуществляется
водой. Из реактора 3 образующийся полиэтилен вместе с этиленом,
не вступившим в реакцию, через систему редукторов (на схеме не
Рис. XV.12. Схема установки Для получения полиэтилена высокого
давления:
/—компрессор; 2—маслоотделитель; 3—реактор; 4—сепаратор; 5 —приемник;
6 — ловушка.
Рис. XV.13. Изменение скорости поли-
меризации полиэтилена Vn в зави-
симости от давления.
показано) проходит сепаратор 4 и поступает в приемник 5, где
после снижения давления происходит разделение этилена и поли-
этилена. Этилен, пройдя ловушку
6 и после промывки, снова воз-
вращается на полимеризацию. Из
приемника 5 расплавленный по-
лиэтилен направляется на стаби-
лизацию, окрашивание (если не-
обходимо) и грануляцию. В каче-
стве стабилизатора применяется
смесь, состоящая из фенил-наф-
тиламина и дифенил-фениленди-
амина и др.
Гранулирование осуществ-
• ляют несколькими методами и, в
частности, продавливанием смеси
полиэтилена и стабилизатора че-
рез фильеру гранулятора. Выхо-
дящие жгуты разрезаются вращающимся ножом на гранулы раз-
мером 2—3,5 мм. Готовый полиэтилен упаковывают в мешки и по-
ставляют потребителю.
Степень конверсии этилена за одну стадию составляет 8—12%,
а при неоднократной циркуляции газа достигает 95—97%.
359
Являясь термопластичным полимером, полиэтилен не раство-
ряется в органических растворителях, но набухает в них, раство-
ряется при температуре выше 70 °C в хлорированных углеводоро-
дах, устойчив к действию концентрированных кислот, щелочей и
растворов солей, но разрушается под действием окислителей, осо-
бенно при нагревании. Полиэтилен устойчив при нагревании без
доступа воздуха до 290 °C, но при температуре 350—400 °C раз-
лагается с образованием жидких и газообразных продуктов.
Полиэтилен перерабатывается в изделия литьем под давле-
нием, экструзией и прессованием; изделия из полиэтилена под-
даются сварке. Он' используется для изготовления пленки и ли-
стов, литьевых изделий, труб, для изоляции кабеля и других
изделий, широко применяющихся во многих отраслях народного
хозяйства и в быту. Особенно широко применяется полиэтилен для
защиты металлических изделий от коррозии.
Поливинилхлорид [—СН2 — СНС1—]п относится к высокомо-
лекулярным галогенопроизводным углеводородам. Получают его
лаковым, эмульсионным и блочным способами. Полимеризацию
лаковым, эмульсионным и блочным способами. Полимеризацию
хлористого винила ведут по радикальному механизму, но она мо-
жет протекать и по ионному типу. Наиболее распространенным
методом получения поливинилхлорида является суспензионная
полимеризация хлористого винила в водной среде.
В эмалированный реактор емкостью 10—20 м3, снабженный
мешалкой и рубашкой для обогревания или охлаждения смеси ре-
агентов, подается вода, раствор стабилизатора эмульсии (поливи-
ниловый спирт, желатина и др.), раствор инициатора например
Йерекись бензоила и другие компоненты, нерастворимые в воде.
После их загрузки реактор продувают азотом и при перемешива-
нии вводят жидкий винилхлорид. Реакционную смесь нагревают
до 40—50 °C, подавая в рубашку реактора горячую воду. Процесс
полимеризации длится 20—30 ч при температуре 40—80 °C и дав-
лении 0,5—1,4 МПа. В процессе полимеризации давление в реак-
торе снижается, что указывает на окончание реакции.
Образующуюся суспензию перекачивают в емкость, сюда же
вводят раствор щелочи для разрушения эмульгатора, инициатора
й низкомолекулярных продуктов, а затем подают острый пар для
Нагревания массы до 94—96 °C. Полимер охлаждают, промывают
й подают на центрифугу для отделения от раствора. Полученный
Продукт, содержащий около 25% влаги, поступает в аппарат для
сушки в кипящем слое, а затем на рассев и затаривание в мешки.
Поливинилхлорид, полученный этим способом, отличается от
любого другого поливинилхлорида более высокой степенью чи-
стоты, лучшими диэлектрическими показателями, высокой водо-
й термостойкостью. Это объясняется однородностью его состава
£имеет определенную молекулярную массу, что обусловлено под-
даржанием строго определенной температуры в самих каплях мо-
номера, т, е, как бы в микроблоке).
960
Технический поливинилхлорид — белый полидисперсный поро-
шок, молекулярная масса его 30000—150000 и степень полимери-
зации от 100 до 2500. Он не воспламеняется и не горит, нераство-
рим в воде, спирте, бензине и многих других растворителях, но при
нагревании растворяется в хлорированных углеводородах, аце-
тоне, циклогексаноне и др., устойчив к воздействию сильных и сла-
бых кислот и щелочей, смазочных масел.
На основе поливинилхлорида получают пластические массы
двух типов: жесткие, не содержащие пластификаторов (вини-
пласт), и мягкие, содержащие пластификаторы (пластикат).
Винипласт получают из поливинилхлоридной смолы, смешан-
ной при повышенной температуре (160 °C) со стабилизаторами
(стеаратами кальция, бария, углекислым свинцом и др.) и сма-
зывающими веществами. Из полученной смеси вальцеванием, ка-
ландрованием, экструзией, формованием при 150—170 °C получают
пленки, листы, трубы, вентили и другие изделия.
Винипласт поддается механической обработке, хорошо свари-
вается и склеивается. Механические свойства винипласта доста-
точно высоки:
Плотность, 103 кг/м3...................................... 1,38—1,43
Температура размягчения, °C............................... 80
Предел прочности, МПа
при изгибе............................................ 90
при сжатии.......................................... 80—100
при растяжении.................................. 40—60
Относительное удлинение, %............................... 10—25
Удельная ударная вязкость, кДж/м2......................... 120
Удельное объемное электросопротивление, Ом • м . . . 1012—1014
Электрическая прочность, МВ/м............................ 15—45
Винипласт используется для изготовления трубопроводов, вен-
тилей, кранов, барабанов центрифуг, колонн для поглощения окис-
лов азота и других агрессивных газов. Из листового материала го-
товят крупные емкостные аппараты, сложные фильтры, вентиля-
ционные системы в помещениях с агрессивной средой и т. д.
Пластикат готовят из смеси смолы, наполнителя (каолин), ста-
билизатора (стеараты и карбонаты кальция, свинца и др.) и пла-
стификатора (фталаты, себацинаты, трикрезилфосфаты и другие
малолетучие жидкости и их смеси), вводимого в количестве от 30 до
60% от массы смолы. Пластификаторы улучшают эластические и
пластические свойства поливинилхлорида. Из полученной однород-
ной массы вальцеванием, калайдрованием или экструзией полу-
чают пленки, листы, трубы и различные другие изделия.
Пластифицированный материал обладает эластическими свой-
ствами, но не является эластомером. Он используется для получе-
ния пленки, линолеума, приводных ремней, армированных тканью,
транспортных лент, в качестве заменителя кожи.
Для защиты от коррозионного разрушения аппаратуры пла-
стикат используется реже, так как обладает меньшей химической
стойкостью, чем винипласт,
361
Полистирол г—СН2—СН—"I представляет собой твердый
L С6Н5 Jrt
аморфный продукт, молекулярная масса которого 200000—400 000.
Обладая ценными техническими свойствами, полистирол и сопо-
лимеры стирола по объему производства занимают третье место
среди пластмасс, уступая только полиэтилену и поливинилхло-
риду.
Исходным сырьем для получения полистирола служит стирол
СбН5СН = СНг, образующийся в процессе дегидрирования этил-
бензола в присутствии катализатора:
СН2—СНз сн=сн2
6 -О +н<
Реакционная смесь, содержащая стирол и примеси побочных
продуктов, подвергаются разделению. От стирола отделяют непро-
реагировавшие углеводороды, а затем вымораживают воду.
Полимеризацию стирола проводят блочным, эмульсионным и
суспензионным способами. В зависимости от способа получения
свойства полистирола различны (табл. XV.2).
Таблица XV. 2. Свойства полистирола
Показатели Полистирол
блочный эмульсион- ный суспензионный
Температура размягчения, °C 80-90 80—90 80—90
Плотность, 103 кг/м3 Предел прочности, МПа 1,05-1.08 1,05—1,08 1,05-1.06
при растяжении 35 36—40 30
при изгибе 37,5-55 50-60 —•
Относительное удлинение при раз- 1,5 1,5 1,5
рыве, % Тангенс угла диэлектрических по-
2- 10 4 4-10 4 3-10“4
терь при 106 Гц 2,5-2,7
Диэлектрическая проницаемость при 2,5-2,6 2,5—2,6
106 Гц
Электрическая прочность, МВ/м 20-25 —• —
Твердость по Бриннелю, МПа 1,4-1,5 1,4-1,5 1,5
Полимеризацию стирола в блоке (массе) осуществляют по ра-
дикальному механизму как в присутствии инициатора (перекись
бензоила), так и без него. Чаще полимеризацию проводят без ини-
циатора, только под действием тепла, так как получаемый при
этом стирол обладает хорошими диэлектрическими свойствами.
В настоящее время применяют непрерывную термическую полиме-
362
ризацию стирола в реакторах колонного типа без перемешивания
и в каскаде реакторов с перемешиванием.
Технологический процесс получения полистирола (рис. XV.14)
состоит в следующем. Химически чистый жидкий стирол из храни-
лища центробежным насосом непрерывно подается в реакторы
предварительной полимеризации 1, представляющие собой цилинд-
рические аппараты, изготовленные
из алюминия и снабженные мешал-
ками и рубашками для нагревания
или охлаждения массы в зависимо-
сти от условий процесса. В реакто-
рах при 80—82 °C процесс образо-
вания полимера проходит частично.
После того как содержание поли-
стирола в реакторах достигнет 30%,
жидкую смесь сливают в верхнюю
часть полимеризационной колон-
ны 3 состоящей из шести секций.
Каждая секция снабжена рубаш-
кой 4 и змеевиком, изготовленным
из нержавеющей стали, для поддер-
жания в секциях определенной тем-
пературы (от 100 до 200°C). В ниж-
ней части колонны имеется обогре-
ватель 5. Диаметр колонны 600—
1500 мм, высота — 5—11 м. В ка-
честве теплоносителя применяется
смесь 26,5% дифенила и 73,5% ди-
фенилоксида, называемая дини-
лом.
По мере движения стирола по
колонне сверху вниз заканчивается
его полимеризация. Степень превра-
щения стирола (рис. XV. 15) и молекулярная масса образующегося
полистирола зависят от температуры (рис. XV. 16).
Из реактора и колонны пары стирола поступают в холодильник
2, где они конденсируются, и стирол возвращается в верхнюю
часть полимеризационной колонны 3. Полимеризацию стирола
в реакторах /ив колонне 3 ведут в среде азота во избежание
окисления полистирола кислородом воздуха. Расплавленный поли-
стирол из нижней конической части колонны поступает в шнек-
пресс и в виде прутков подается на охлаждение, а затем на даль-
нейшую переработку.
Блочный способ получения полистирола имеет ряд преимуществ
перед другими методами: процесс прост в аппаратурном оформ-
лении, непрерывен, а получаемый продукт является самым деше-
вым. В полистироле отсутствуют примеси, снижающие его свой-
ства, особенно электрические.
Стирол
ВоОа^^
Азот
Вова
1ВО°С
190°С
200 °C
Полистирол
Азот Азот:
вова.
Мономер
к - 'шректи-
\100-110С ршацинг-
]l10-/20°C
1150 °C
Рис. XV. 14. Схема непрерывной
полимеризации стирола блочным
• методом:
1—реакторы предварительной полиме-
ризации; 2—холодильник; 3—колонна;
4—нагревательная рубашка; 5—элекро-
обогрев.
363
Эмульсионную полимеризацию стирола проводят в эмалиро-
ванных аппаратах или аппаратах, изготовленных из нержавею-
щей стали (с мешалкой и рубашкой) емкостью 6—15 м3. В реак-
тор заливают стирол, воду, эмульгаторы (натриевые соли сульфо-
кислот), раствор едкого натра и инициаторы (надсернокислый ка-
лий, перекись водорода и др,), В течение 30 мин смесь перемеши-
вают и нагревают до 70—90 °C. Полимеризация стирола длится
5—6 ч, в результате чего образуется мелкодисперсная смесь —
латекс, который под действием кислой среды, создаваемой кисло-
тами или кислыми солями [например, Al^SO^s], разрушается.
Рис. XV. 15. Зависимость степени пре-
вращения стирола от температуры
полимеризации блочным методом:
1—при 131,5 °C; 2—при 79,5 °C.
Рис. XV.16. Зависимость средней моле-
кулярной массы М от температуры
полимеризации.
После промывки горячей водой, полистирол отделяют от воды, су-
шат при 60—70 °C, а затем направляют для разделения на фракции.
Получаемый по этому методу полистирол содержит небольшие
количества эмульгатора и имеет высокую дисперсность, поэтому
он используется главным образом для производства пенополисти-
рола.
Полимеризацию полистирола можно проводить и по непрерыв-
ной схеме.
Изотактический полистирол получают в присутствии триэтил-
или триизобутилалюминия и треххлористого титана и др. Образую-
щийся полимер имеет кристаллическую структуру со строго регу-
лярным расположением боковых цепей, например, все группы
С6Н5 — расположены с одной стороны, главной цепи макромоле-
кулы:
Н НН НН н
C6HS Н CeHs Н CjHa н
364
Изотактический полистирол имеет более высокие механические
свойства.
Лаковый и суспензионные методы получения полистирола при-
меняются реже.
Полистирол относится к термопластичным полимерам. Он устой-
чив к воздействию минеральных кислот и щелочей, спиртов, масел;
но разрушается азотной кислотой, растворяется в ароматических
и хлорированных углеводородах, алифатических эфирах и во мно-
гих кетонах. Полистирол поддается всем видам механической об-
работки и применяется для получения изделий прессованием,
литьем под давлением, экструзией. Из полистирола получают нити,
пленочные материалы и различного вида фасонные изделия. Осо-
бенно широко полистирол используется для изготовления товаров
народного потребления.
Пластические массы на основе полимеров, получаемых
поликонденсацией и ступенчатой полимеризацией
Фенолоальдегидные смолы и пластмассы на их основе находят
широкое применение в технике. В качестве сырья для их получения
используют фенолы (крезол, ксилолы, фенол и др.) и альдегиды
(формальдегид, фурфурол). Наибольшее применение для получе-
ния смол находят фенол и формальдегид.
Фенол представляет собой кристаллическое вещество, плавя-
щееся при 40,9 °C. В основном его получают синтетическим мето-
дом и лишь в небольшом количестве (~3,5°/о) из каменноуголь-
ной смолы. Он смешивается в любых соотношениях со спиртом,
водой при нагревании свыше 66 °C, растворим в эфире, глицерине,
сероуглероде и др.
Смолы, полученные на основе фенола и формальдегида, полу-
чили название фенолоформальдегидных. Они находят наибольшее
применение из других смол этой группы. В зависимости от соотно-
шения между фенолом и формальдегидом и применяемого катали-
затора получаемые смолы могут быть термопластичными (ново-
лачные) и термореактивными (резольные).
Новолачные смолы получают в водной среде при конденсации
избытка фенола и формальдегида в присутствии кислоты (соля-
ная, щавелевая, серная кислоты) по схеме:
(n + 1)С6Н6ОН + пСН2О —> Н—[—С6Н3(ОН)СН2—]П-С6Н4ОН + пН2О
Обычно взаимодействие протекает при мольном соотношении
фенола и формальдегида, равном 7:6. Новолаки являются низко-
молекулярными соединениями (п = от 4 до 8). При нагревании
они плавятся, легко растворяются в спирте. Новолачные смолы
применяют для получения пресспорошков, абразивных изделий,
365
газонаполненных материалов. Для перевода этих смол из термо-
пластичного состояния в термореактивное в качестве отвердителя
используют уротропин,
Резольные смолы получают из фенола и небольшого избытка
формальдегида в присутствии щелочных катализаторов NH4OH,
NaOH, Ва(ОН)2.
Процесс образования смолы состоит из двух стадий: реакции
фенола с формальдегидом с образованием фенолоспиртов и кон-
денсации фенолоспиртов с образованием резола (смесь полифе-
нолов) :
Фенолоспирты реагируют между собой с образованием резолов
Резолы представляют собой смесь линейных и разветвленных
полимеров общей формулы
Н—[—CSH2(OH) (СН2ОН)СН2—Jw-[— СбН3(ОН)СН2—]„—он
где п = 2ч-5, а т + « = 4-5-10.
Резольная смола является низкомолекулярным соединением
(от 400 до 1000). При нагревании смолы происходит дальнейшая
конденсация макромолекул, в результате чего образуется резит,
366
представляющий собой неплавкий и нерастворимый полимер сле-
дующего строения:
Процессы получения' новолачной и резольной смолы практиче-
ски не отличаются друг от друга и состоят в следующем
(рис. XV. 17). В реактор 1 вводят
фенол, формальдегид, различные
добавки и катализатор и смесь
нагревают в реакторе до 70—
75 °C с помощью пара, подавае-
мого в рубашку реактора. Поли-
конденсация фенола с формаль-
дегидом протекает с выделением
тепла. Оптимальная температура
в реакторе должна быть не выше
80 °C, поэтому по достижении
этой температуры отключают по-
дачу пара и ведут охлаждение
смеси, подавая в рубашку реак-
Рис. XV. 17. Схема установки для
получения фенолоформальдегидной
смолы;
/—реактор; 2—холодильник; 3—противень.
тора воду. В процессе поликон-
денсации пары воды поступают в
трубчатый холодильник 2 и кон-
денсируются. Образовавшаяся
смола плавится и в жидком со-
стоянии выдавливается из реактора по трубе сжатым воздухом
в противень 3, здесь она затвердевает и измельчается в порошок.
Фенолоформальдегидные смолы находят широкое применение
в качестве связующего при получении пресспорошков, волокни-
стых материалов, слоистых пластиков, поропластов. Кроме того,
они используются для получения лаков и клеев.
Пластические массы, получаемые на основе фенолоформальде-
тидных смол, получили название фенопластов. Литой резит отно-
сится к ненаполненным фенопластам, так как он состоит только
367
из одной смолы. Он имеет низкие физико-механические свойства,
трудно перерабатывается в изделия, поэтому практического зна-
чения не имеет.
Композиционные фенопласты в зависимости от наполнителя
делятся на прессовочные, волокнистые и слоистые материалы.
Изделия из пресспорошков. Выпускаемые промышлен-
ностью пресспорошки классифицируются в зависимости от назна-
чения, по виду сырья, применяемого для получения смол; по пове-
дению смолы при цагревании. Ассортимент выпускаемых порош-
ков очень велик (примерно 100 марок).
Пресспорошки в зависимости от связующего подразделяются
на новолачные и резольные.
Новолачные пресспорошки готовят из смеси твердых феноло-
формальдегидных смол, наполнителей (древесная мука, каолин,
тальк, слюда, графит, кварц, асбест и др.), отвердителей (уротро-
пина, окиси магния, кальция и др.), смазки (олеиновая, стеари-
новая кислоты и их соли), красителей и пигментов (мулия, охра,
литопон и др.). Получают их вальцеванием и шнековым ме-
тодом.
Резольные пресспорошки отличаются по составу от новолач-
ных только тем, что при их получении применяются фенолоформ-
альдегидные смолы резольного типа. Пресспорошки перед прес-
сованием таблетируют.
Свойства изделий, получаемых из пресспорошков, зависят от
свойств и количества применяемых связующего и наполнителя.
Например, тепло- и жаростойкость изделий можно повысить, вводя
в пресспорошки кварц, слюду, асбест.
Технологические свойства пресспорошков зависят от типа
смолы, рода наполнителя и его содержания, количества отверди-
теля. В табл. XV. 3 приведены некоторые свойства изделий из
пластмасс, получаемых из пресспорошков.
Изделия из пресспорошков готовят методом горячего прямого
и литьевого прессования при 160—200 °C под давлением и другими
специальными методами.
Рассмотрим примеры использования пресспорошков для изго-
товления изделий.
Изделия общетехнического назначения получают из пресспо-
рошков, содержащих фенолоформальдегидные смолы и древесную
муку. К ним относятся ненагруженные армированные и неармиро-
ванные детали и изделия широкого потребления (розетки, ру-
коятки, корпуса проигрывателей и др.).
Радиодетали и другие изделия, работающие при высоких тем-
пературах, готовят из пресспорошков, содержащих смолы, асбест
или слюду. Изделия, устойчивые к действию воды и кислот, гото-
вят из пресспорошков, имеющих в своем составе новолачные смо-
лы, совмещенные с поливинилхлоридом, каучуком и другими поли-
мерными соединениями. В качестве наполнителей применяют кокс,
графит, каолин и др.
368
13 Зак. 658
<
Вол ок ниты — прессовочные материалы, получаемые на ос-
нове волокон различного типа и растворов фенолоформальдегид-
ных или других смол.
Из волокнита (наполнитель — хлопковая целлюлоза) прессуют
детали общетехнического назначения, работающие на изгиб, исти-
рание, кручение, а также изделия, обладающие антифрикцион-
ными свойствами (переключатели, фланцы, рукоятки, шестерни и
т. д.). Изделия из них устойчивы в воде, слабых кислотах и ос-
нованиях, но разрушаются при действии сильных кислот и ще-
лочей.
Асбоволокнит готовят прессованием асбеста и других со-
ставляющих, пропитанных фенолоформальдегидной или другой
смолой. Материал применяют для изготовления деталей электро-
технического назначения и изделий, обладающих фрикционными
свойствами, высокой теплостойкостью и механической прочностью,
например, тормозные колодки экскаваторов, подъемных кранов.
Фаолит получают прессованием асбеста или смеси графита
и асбеста, пропитанных фенолоформальдегидной смолой. Он
устойчив в растворах соляной кислоты любой концентрации,
в 40%-ной серной, в 50%-ной ортофосфорной и уксусной кислотах,
в хлорированных углеводородах, но, разрушается иодом, бромом,
щелочами, ацетоном, спиртом и кислотами, обладающими окисли-
тельными свойствами.
Из фаолита при обычной температуре, без давления готовят
крупногабаритные изделия: трубы, ванны, адсорберы, различного
вида емкости и т. д., которые затем отверждаются при температуре
60—130°C. Теплостойкость фаолита около 145°C.
Фенопласты, содержащие стеклянное волокно в качестве на-
полнителя, называются стекловолокнитами. Эти материалы обла-
дают высокой удельной прочностью, жесткостью, устойчивы к ви-
брационным нагрузкам, к многим химическим реагентам и микро-
организмам, имеют хорошие диэлектрические и теплоизоляцион-
ные свойства (см. табл. XV. 3).
Слоистые пластики — прессматериалы, содержащие ли-
стовой наполнитель: хлопчатобумажную ткань (текстолит), бумагу
(гетинакс), асбестовую (асботекстолит) или стеклянную (стекло-
текстолит) ткань, стеклянный и древесный шпон (древеснослоис-
тые пластики ДСП). Получают слоистые материалы горячим прес-
сованием уложенных правильными рядами слоев листового напол-
нителя, пропитанного резольной смолой. Полученный материал об-
ладает хорошими физико-механическими и химическими свой-
ствами (см. табл. XV. 3).
Текстолит используется для изготовления изделий, обладаю-
щих стойкостью к вибрационным нагрузкам; износостойкостью и
электроизоляционными свойствами. Из него готовят шестерни,
вкладыши, детали, работающие в условиях трения, декоративные
и отделочные материалы.
370
Гетинакс широко применяется в электро- и радиотехниче-
ской промышленности и, в частности, в производстве печатных
схем для радио и телевизоров, переключателей и т. п.
Стеклотекстолиты выпускаются в виде листов различных
размеров длиной до 2,5 м, шириной до 1 м и толщиной 0,5—15 мм.
Они широко применяются для изготовления фюзеляжей самоле-
тов, кузовов автомобилей, деталей машин, лодок, судов, самоле-
тов и т. д. Теплостойкость стеклотекстолита 200 °C при про-
должительной эксплуатации и 300 °C при кратковременной ра-
боте.
Стекловолокнистый анизотропный материал
(СВАМ) готовят горячим прессованием стеклянного шпона с на-
несенным на его поверхность связующим (совмещенные феноло-
формальдегидные смолы с поливинилбутиралем или с эпоксид-
ными олигомерами). Прессование проводят в гидравлических прес-
сах при температуре 150—160°C и давлении 4 МПа с последующей
вытяжкой изделий, продолжительность вытяжки 6 мин на 1 мм
толщины получаемого изделия.
СВАМ, подобно фанере, обладает упругими анизотропными
свойствами и в зависимости от укладки волокон может иметь вы-
сокую прочность вдоль расположения стекловолокон и низкую
в поперечном направлении. Из него готовят трубы и другие изде-
лия, устойчивые к действию химических реагентов, детали кате-
ров, лодок, конструкций кораблей, кузовов автомашин, прицепов,
цистерн, хранилищ и т. д., а также применяют как электроизоля-
ционный материал (электрощиты, электроаппаратура). СВАМ
нашел широкое применение в строительстве: из него готовят плиты
перекрытий и несущие панели стен, тепло- и звукоизолирующие
строительные элементы, шахтную крепь и кузова шахтных вагоне-
ток и др.
Древесно-слоистые пластики (ДСП) используются для изготов-
ления мебели, спортивных лодок, деталей катеров, вкладышей под-
шипников, шестерен, опорных рам, фрикционных шкивов, втулок
небольшого диаметра и других деталей.
Эпоксидные смолы получают взаимодействием эпоксисоединений
с веществами, содержащими подвижные атомы водорода, или вве-
дением эпоксидных групп —СН—СН2 в ненасыщенные соединения
О
под действием надкислот, например, надуксусной кислоты
СНзСОООН.
Наиболее широко применяется первый метод получения эпок-
сидных смол, где в качестве сырья используют мономеры, содер-
жащие эпокси-группы, и многоатомные фенолы (дифенилолпропан,
гидрохинон, пирогаллол и др.).
Образование эпоксидной смолы при взаимодействии эпихлор-
гидрина и дифенилолпропана в присутствии щелочи происходит по
13*
37f
схеме^
эпихлоргидрин дифенилолпропан
диглицидный эфир
+ 2NaCl + H2O
Кроме диглицидного эфира в смеси содержатся полимеры боль-
шей молекулярной массы (т. е. с числом звеньев от 2 до 16), ко-
торые имеют следующую структуру:
Эпоксидные смолы такого строения имеют линейную структуру,
поэтому они плавятся и растворяются в органических раствори-
телях.
Для примера рассмотрим синтез смолы Э40. В реактор с рубашкой, мешал-
кой и обратным холодильником загружаются дифенилолпропан и эпихлоргидрин
в строго определенном соотношении; смесь перемешивают в течение 1 ч до об-
разования однородной суспензии. Затем в реактор очень медленно при переме-
372
шивании в теченце 3—5 ч вводят 15%-ный раствор едкого натра (строго опреде-
ленное количество NaOH).
Реакция образования смолы идет с выделением тепла, поэтому смесь охла-
ждают водой, при этом температура в реакторе должна поддерживаться не вы-
ше 50 °C. Затем увеличивают скорость подачи раствора щелочи, доводят темпе-
ратуру в реакторе до 75 °C, и смесь выдерживают при данной температуре в те-
чение 3 ч.
По окончании синтеза образовавшуюся эпоксидную смолу растворяют в то-
луоле при перемешивании и нагревании до 30—55 °C, и при этой температуре
смесь выдерживают в течение 3—6 ч. Затем от раствора смолы отделяют над-
смольную воду, а оставшуюся в реакторе массу обрабатывают двуокисью угле-
рода до нейтральной реакции, после чего из раствора отгоняют воду. По окон-
чании отгонки воды раствор смолы сушат в вакууме до прозрачной пробы ди-
стиллята. После сушки из раствора смолы выделяют кристаллы поваренной соли,
а затем в вакууме (600—650 мм рт. ст.) и при температуре 45—95 °C отгоняют
толуол. Окончание процесса определяют по вязкости раствора смолы, которая не
должна превышать 19—25 с. После отгонки толуола смолу охлаждают и направ-
ляют на использование.
Низкомолекулярная смола Э40 широко используется для получения высоко-
молекулярных эпоксидных и модифицированных смол, шпатлевок, клеев и др.
Низкомолекулярные эпоксидные смолы представляют собой жидкие продукты в
отличие от высокомолекулярных смол, являющихся твердыми веществами с тем-
пературой плавления до 150 °C.
Эпоксидные смолы получают и при взаимодействии эпихлор-
гидрина с аминами (например, с анилином), гликолями (напри-
мер, этиленгликолем) и др.
По второму методу эпоксидные смолы можно получать при
взаимодействии непредельных углеводородов (например, диви-
нила) с надуксусной кислотой:
2СН3СОООН
СН2=СН—СН=СН2 —----------->- СН2—СН—СН—СН2 + 2СН3СООН
\) ХО/
Образующиеся соединения могут далее реагировать с мономе-
рами по реакциям, приведенным для первого способа.
Эпоксидные смолы представляют собой олигомеры, содержа-
щие свободные концевые эпокси-группы, которые при взаимодей-
ствии с некоторыми веществами (отвердителями) и между собой
образуют полимеры сетчатого строения. В качестве отвердителей
применяются гексаметилендиамин, этиленполиамин, меламин, смо-
лы, фталевый ангидрид, кислоты и др. Эпоксидные смолы допу-
скают высокую степень наполнения (50%) их различными напол-
нителями (кварцем, стеклом, тальком, асбестом, графитом и др.).
Присутствие наполнителей способствует увеличению твердости и
теплостойкости, снижению усадки и стоимости пластмассы, при ис-
пользовании же армирующих наполнителей в виде волокна и тка-
ней можно получать высокопрочные материалы.
Эпоксидные смолы и компаунды широко применяются для изо-
ляции и герметизации изделий; в машиностроении (изготовление
штампов холодной вытяжки, прессформ, литейных моделей и т. п.);
для изготовления клеев, исправления дефектов литых изделий, ре-
монта металлических деталей и трубопроводов и для изготовления
373
различного вида фасонных изделий. Так, из стеклопластиков и
СВАМ, полученных из эпоксидных смол, готовят корпуса моторных
лодок, разнообразные детали, используемые в авиа-, суде- и раке-
тостроении и т. д. Обладая высокой адгезией ко многим материа-
лам, прочностью и химической стойкостью к растворителям, кисло-
там, щелочам и растворам солей, эпоксидные смолы широко ис-
пользуются для защиты металлов, сплавов и других материалов от
коррозии.
3. ПРОИЗВОДСТВО КАУЧУКА И РЕЗИНЫ
Среди большого числа высокомолекулярных соединений в со-
временной технике и технологии каучук находит особенно широкое
применение.
Каучук является основным компонентом при изготовлении рези-
новых, резино-тканных и резино-металлических изделий, используе-
мых в промышленности, сельском хозяйстве, на транспорте и в до-
машнем обиходе. На основе каучуков получают более 40000 наиме-
нований резиновых изделий, к их числу относятся автомобильные
и авиационные шины, приводные ремни, потребность в которых ис-
числяется в десятках миллионов квадратных метров в год, гибкие
шланги и рукава, детали машин ^механизмов, предметы санитарии
и гигиены и т. д. Такое широкое применение резин объясняется
тем, что они обладают уникальной способностью к обратным дефор-
мациям в сочетании с высокой прочностью, эластичностью, сопро-
тивляемостью к истиранию.
Резины устойчивы к действию многих химических реагентов, что
позволяет использовать их для футеровки химической аппаратуры
и при изготовлении деталей уплотнений. Они газо- и водонепрони-
цаемы, имеют высокие диэлектрические свойства и являются неза-
менимым материалом при производстве кабеля, аэростатов, надув-
ных лодок, скафандров и т. п. Новые быстро развивающиеся от-
расли техники требуют разработки современных типов каучуков и
резин, выдерживающих температуры от —100 до +350 °C, устойчи-
вых к действию окислителей, бензина, масел, растворителей, облу-
чения и т. п.
Сырье и материалы резинового производства
Сырьем для получения резины служат сырой каучук, синтетиче-
ские латексы и смолы, регенерат, вулканизующие вещества, ускори-
тели вулканизации, активаторы или замедлители вулканизации,
красители, пластификаторы, противостарители, активные наполни-
тели, армирующие и вспомогательные материалы. Основным
сырьем, определяющим свойства резин, является каучук.
Каучуки являются высокомолекулярными соединениями. Они
имеют линейное строение и обладают способностью к обратной де-
формации в сочетании с высокой прочностью.
874
Для изготовления резин применяется натуральный и синтети-
ческие каучуки.
Натуральный каучук относится к каучукам общего назначения,
т. е. к каучукам, применяемым для изготовления резиновых изде-
лий широкого потребления (шии, ремней, обуви и т. д.).
Синтетические каучуки в зависимости от строения макромоле-
кулы подразделяются на органические, элемеитоорганические и не-
органические. Они могут быть гомополяриыми, т. е. макромоле-
кулы их состоят из звеньев одного мономера, и сополимерными,
когда макромолекулы содержат два или несколько мономерных
звеньев.
Натуральный каучук до 1932 г. был единственным сырьевым
материалом, применяемым для производства резины. Получают
натуральный каучук (НК) из млечного сока некоторых каучуко-
носных растений («Као-чу», т. е. «слезы дерева»), преимущественно
из тропического дерева гевеи, из которого выделяют 99% всего
мирового каучука. Млечный сок (латекс) дерева гевеи содержит
частички каучука (до 30%), взвешенные в воде (до 60%), белко-
вые вещества (2—2,7%), смолы (1,65—3,4%), сахаристые (1,5—
4,2%) и минеральные (0,2—0,7%) вещества. На состав латекса
влияют возраст дерева, время года, климатические условия.
При обработке млечного сока на месте его добычи кислотами,
например уксусной, происходит коагуляция сока с образованием
гелеобразного продукта. Этот продукт пропускают через вальцы,
сушат, коптят (для предохранения от загнивания при хранении)
и в виде листов, упакованных в кипы, направляют потребителю.
Г СН3 -I
Макромолекулы натурального каучука L—СН2—С=СН—CH2J„,
состоят из элементарных звеньев изопрена; он имеет малую плот-
ность (917—937 кг/м3) и большую молекулярную массу (от 150000
до 500000), что соответствует длине макромолекулы 10000—
40000 А при поперечном сечении 3 А. Такие длинные нитевидные
гибкие макромолекулы и определяют физические и механические
свойства каучука. Он обладает высокой эластичностью, прочностью,
малой гигроскопичностью и теплопроводностью и является превос-
ходным изолятором. Прочность при разрыве его составляет от 1
до 4 МПа, а в вулканизированном состоянии — до 30 МПа.
НК является термопластичным полимером. При нагревании вы-
ше 200 °C он разлагается с образованием изопрена и других низ-
комолекулярных соединений, а при радиационном облучении вы-
деляется водород. Натуральный каучук растворяется в бензине,
бензоле, сероуглероде и др. с образованием вязких растворов, ко-
торые используются как клеи.
Поскольку в макромолекуле каучука имеются двойные связи, он
вступает во взаимодействие с галогенами, тиоспиртами, тиокисло-
тами, кислородом, озоном и др. При взаимодействии с серой и орга-
ническими перекисями линейная структура макромолекул иату-
375
рального каучука превращается в сетчатую (процесс вулканиза-
ции). Это свойство каучука лежит в основе получения резины.
Натуральный каучук в основном применяется для изготовления
резины, причем большая его часть используется для получения ав-
томобильных шин и только около 1 % применяется в обувной про-
мышленности и для получения резинового клея.
Синтетические каучуки (СК) в настоящее время занимают ве-
дущее место (около 2/3 всего производимого в мире каучука) при
производстве резины.
В зависимости от применения СК подразделяют на каучуки об-
щего назначения: бутадиеновый (СКВ), бутадиен-стирольный
(СКС), бутадиен-метилстирольный (СКМС), изопреновый (СКИ);
каучуки специального назначения: бутадиен-нитрильный (СКН),
хлоропреновый (наирит), бутилкаучук, тиоколовый, силиконовый
(СКТ) и др.
Первый в мире синтетический каучук был получен в ССС-Р в
1932 г., несколько позже каучук стали получать в Германии
(1938 г.) и США (1942 г). Процесс получения синтетического кау-
чука включает две стадии: синтез мономеров и полимеризацию или
поликонденсацию мономеров.
Для получения СК и латексов применяют так называемые кау-
чукогенные мономеры: бутадиен, стирол, изопрен, хлоропрен, изо-
бутилен и др.
Натрий-бутадиеновый каучук (СКВ) является первым каучу-
ком, полученным синтетическим путем по методу акад. Лебе-
дева С. В. полимеризацией бутадиена в присутствии металличе-
ского натрия. Бутадиен для этой цели получили из этилового
спирта в присутствии катализаторов. В настоящее время его полу-
чают в основном, дегидрированием бутана.
Натрий-бутадиеновый каучук обладает низкими адгезионными
свойствами и эластичностью, характеризуется невысокими прочно-
стными свойствами и морозостойкостью по сравнению с натураль-
ным и другими синтетическими каучуками, поэтому в настоящее
время он практически не применяется.
Бутадиен-стирольные каучуки (СКС, СКМС) получают из бу-
тадиена СН2 = СН — СН = СН2 и стирола CgHjCH = СН2(СКС),
бутадиена и метилстирола СбН5ССН3 — СН2 (СКМС). Процесс
сополимеризации приведенных выше мономеров проводят эмуль-
сионным способом по непрерывной схеме.
В зависимости от содержания стирола бутадиен-стирольные
каучуки маркируют различными числовыми индексами, например,
каучук типа СКС-10, содержит 10% стирола и 90% бутадиена. При
увеличении в каучуках стирола или метилстирола ухудшаются
клеющая способность, эластические свойства, морозостойкость, но
улучшаются показатели прочности.
Вулканизированные каучуки СКС и СКМС имеют низкую проч-
ность, которая резко возрастает (в 10—12 раз) при введении в ре-
зиновую смесь газовой сажи. Диэлектрические свойства этих кау-
376
чуков приближаются к свойствам натурального, а молекулярная
масса увеличивается от 10000 до 100000.
На рис. XV. 18 показана схема установки для получения бута-
диен-стирольного каучука. Бутадиен из сборника 1 и стирол из
сборника 2 поступают в смеситель 3. В аппарате 4 готовится вод-
ный раствор эмульгатора (олеат натрия, канифольное мыло и др.),
Рис. XV. 18. Схема установки для получения бутадиен-стирольного каучука-:
сборники; 3—смеситель; 4—аппарат для приготовления эмульгатора; 5 —полимеризатор;
б—сборник латекса; 7—вакуум-насосы; 8—компрессор; 9—отпарная колонка; 10,11— кон-
денсаторы; 12—дроссельный вентиль.
его подают в батарею полимеризаторов 5. Сюда поступает смесь
мономеров из смесителя 3, а также вводятся инициаторы (персуль-
фаты) и регуляторы. В полимеризаторах смесь перемешивается и
проходит все аппараты батареи со скоростью, достаточной для
того, чтобы за время нахождения мономеров в реакторах проре-
агировало примерно 60% исходного сырья с образованием латекса.
Смесь латекса и непрореагировавших мономеров направляют да-
лее через дроссель 12 в сборник 6, где большая часть мономеров
отделяется от латекса и вакуум-насосом 7 и компрессором 8 по-
дается на конденсацию. Латекс поступает в колонну 9, работаю-
щую при разряжении, для окончательной отдувки мономеров во-
дяным паром.
377
Бутадиен и стирол из конденсаторов 10 и 11 снова возвра-
щаются в процесс.
Латекс, отделенный от мономеров, далее поступает на коагуля-
цию, которую проводят смешением его с электролитами, т. е. с рас-
творами солей (NaCl) или кислот (СН3СООН). Образовавшийся
гелеобразный продукт промывают водой, обезвоживают и подаюг
на лентоотливочную машину. Латекс в вйде ленты выходит из нее,
просушивается горячим воздухом, припудривается тальком и сво-
рачивается в рулоны. Рулоны упаковывают в мешки из ткани, про-
питанной нитролаком, или в мешки из поливинилхлорида и отправ-
ляют на склад готовой продукции.
Из этой группы каучуков наибольшее применение находят кау-
чуки СКС-10/СКС-30, СКМС-10, СКМС-30. Каучуки СКС-30 и
СКМС-30 относятся к универсальным каучукам. Они используются
для изготовления шин, транспортерных лент, обуви и других рези-
новых изделий. Каучуки СКС-10 и СКМС-10 обладают высокой
прочностью при истирании, твердостью, растворяются в бензине,
бензоле и других углеводородах, хорошо смешиваются с различ-
ными ингредиентами резиновых смесей. Прочность на разрыв не-
наполненных резин на основе этих каучуков невысока. Для повы-
шения механических свойств в состав резин вводят наполнители,
например 50 масс, частей сажи. Такие резины имеют прочность на
разрыв 20—25 МПа и температуру стеклования от —72 до —77°C.
Синтетический изопреновый каучук (СКИ), полученный впер-
вые в Советском Союзе, находит широкое применение для получе-
ния резины. Его готовят полимеризацией изопрена (~15%) в рас-
творе изопентана или другого растворителя непрерывным методом
при температуре 18—25 °C в присутствии комплексных катализа-
торов (металлического лития, тетрахлорида титана, триизобутил-
алюминия, триэтилалюминия и др.):
пСН2=С—СН=СН2 —> г—сн,—с=сн—сн2—
I ' I
СНз L СНз
СКИ обладает высокой прочностью и клеющей способностью,
сохраняет свои свойства при нагревании до +100 °C. По своему
строению, физическим, технологическим, эластическим и эксплуа-
тационным свойствам СКИ близок к НК, поэтому, в отличие от
других каучуков общего назначения, он вполне может заменить на-
туральный каучук при изготовлении резины.
Резины, получаемые из СКИ, набухают в маслах, бензине и
других органических веществах, окисляются кислородом воздуха,
морозостойки и газонепроницаемы.
Хлоропреновый каучук (наирит) получают эмульсионной поли-
меризацией хлоропрена:
пСН2=С—СН=СН2 —> Г—СН2—С=СН— сн2 —
I I
ci L ci
878
Процесс проводят в водной среде в присутствии эмульгатора
Г(олеинат натрия). Получаемый латекс коагулирует при взаимо-
действии с кислотами и солями, после чего гелеобразный продукт
промывают, сушат и превращают в листы, блоки, бруски. Средняя
молекулярная масса такого каучука ~100000. Наирит не требует
специальной пластификации, так как обладает высокой клейкостью.
Это упругое, но более жесткое по сравнению с натуральным каучу-
ком вещество.
Прочностные свойства его близки к свойствам натурального
каучука. Он нерастворим в углеводородах жирного ряда, но рас-
творяется в хлорированных и ароматических углеводородах, устой-
чив к воздействию света, озона, кислорода, огнестоек.
При получении резины каучук вулканизируют в присутствии'
окислов цинка, ртути и др. Резины, получаемые на его основе, об-
ладают высокой прочностью от 22 до 35 МПа при относительном
удлинении 800—1000%.
Наирит неустойчив при изменении температур, твердеет при
хранении, а при нагревании от него отщепляется хлористый водо-
род. Применяется он главным образом в производстве ремней,
транспортерных лент, клеев, для изготовления кабеля.
Кремнийорганические (силиконовые) каучуки получают при по-
ликонденсации циклических силоксанов или линейных силоксан-
диолов. Полимеры имеют линейное строение, например
—S i—О—S i—О— S i—О— * • •
III
R' R' R
где R, R'— метильные, этильные, фенильные, винильные и другие
группы.
Кремнийорганические каучуки могут быть высокомолекуляр-
ными (молекулярная масса от 500000 до 1000000) и низкомолеку-
лярными— от 20000 до 100000. Каучуки озоно-, морозе- и термо-
стойки, но механические их свойства, масло- и нефтестойкость
хуже, чем любого другого каучука. В отличие от других видов
вулканизацию кремнийорганических каучуков ведут в присутствии
органических перекисей (например перекиси бензоила) при нагре-
вании до 200 °C. Силиконовые каучуки используются при получении
резин с повышенной морозе- и теплостойкостью, а также для из-
готовления деталей, работающих на сжатие. Из резины готовят
жароупорные прокладки, уплотнения, клапаны, электроизоляцию
и т. д.
В настоящее время выпускается несколько видов каучуков: ди-
метилсилоксановый СКТ, метилвинилсилоксановый СКТВ, этил-
силоксановый СКТЭ, фенилсилоксановый СКТ ФВ, бор- и фосфор-
силоксановые, низкомолекулярные каучуки СКТН, СКТН-1 и др.
379
Резины, изготовленные на основе силиконовых каучуков, начи-
нают’разлагаться при температуре 600—700 °C, но в течение не-
скольких секунд они могут выдерживать температуру 3000 °C.
Вспомогательные материалы. Синтетические латексы — слож-
ные коллоидные системы, содержащие до 35% каучуков с части-
цами размером от 5-Ю-6 — 5-10~4см. Они устойчивы в воде бла-
годаря присутствию в системе эмульгатора.
Латексы получают эмульсионной полимеризацией и сополиме-
ризацией. Наибольшее применение находят бутадиен-стирольные,
хлоропреновые, бутадиен-нитрильные, бутадиен-винилиденхлорид-
ные латексы. Они широко используются в производстве губчатых
изделий, нетканных материалов, тонкостенных изделий,* для про-
питки волокон, в производстве бумаги, кожи, красок и т. д. Осо-
бенно незаменимы латексы прц изготовлении изоляционных изде-
лий и как пропиточный материал для тканей, используемых при
изготовлении шин и резино-технических изделий, что позволяет
усилить прочность корда и тем самым увеличить срок эксплуата-
ции шин.
Синтетические смолы вводят в резиновую смесь и в пропиточ-
ные составы как добавки к каучуку для улучшения обработки ре-
зиновой смеси, повышения ее износостойкости и прочности резино-
вых изделий. Наибольшее применение из полимеров находят поли-
изобутилен, полипропилен, поливинилхлорид, фенолоформальде-
гидные смолы.
Регенерат — материал, получаемый в результате переработки
отходов резинового производства, резин, изношенных шин и других
изделий. Получают регенерат тепловой обработкой тонко измель-
ченной резины в течение 10—16 ч при 150—200 °C в присутствии
пластификаторов и других веществ. В результате нагревания про-
исходит девулканизация резины и материал превращается в пла-
стичную массу.
Регенерат широко применяется в резиновой промышленности,
так как он в 3—5 раз дешевле каучука. Кроме того, введение реге-
нерата в резиновую смесь облегчает ее обработку и изготовление
изделий, увеличивает химическую стойкость резин и их сопротив-
ление старению.
Армирующие материалы, применяемые при изготовлении рези-
новых изделий, позволяют им сохранять свои размеры под нагруз-
кой или при нагревании. При работе вводимый в резиновые изде-
лия каркас из ткани или металла воспринимает всю механическую
нагрузку, регулирует деформируемость изделия, а резина придает
ему необходимую эластичность. В зависимости от назначения из-
делий армирующими материалами могут быть стальные тросы и
проволока, природные и химические волокна, различные ткани и
Т. д.
Ингредиенты резиновых смесей. Резина — материал, способный
к большим высокоэластичным деформациям. Получают ее из кау-
чука (или регенерата) или из смеси каучука и других веществ, по-
380
лучивших название ингредиентов. Число веществ, входящих в со-
став резиновой смеси, зависит от условий работы резины и изделий
на ее основе и колеблется от 5 до 20.
Как указывалось ранее, макромолекулы каучука имеют ли-
нейное или разветвленное строение, поэтому для превращения ли-
нейной структуры в пространственную (сетчатую) в резиновую
смесь вводят, вулканизующие вещества (серу, окислы и перекиси
металлов и др.), которые образуют поперечные мостики между
длинными цепями макромолекул (процесс вулканизации). Вулка-
низация может быть вызвана ядерным облучением, в этом случае
вулканизатор не требуется.
Процесс вулканизации можно ускорить введением в резиновую
смесь ускорителей вулканизации (полисульфиды, каптакс и др.),
которые способствуют также улучшению физико-механических
свойств резины. В ряде случаев при получении резины для ускоре-
ния или замедления действия ускорителей в смесь вводят актива-
торы, а при необходимости и замедлители процесса вулканизации.
Механические и другие свойства резины улучшают введением
в смесь активных наполнителей (сажи, двуокиси кремния, титана,
окиси цинка и др.) в виде тонкоизмельченных порошков, количе-
ство которых колеблется от 15 до 100 масс, частей и более на
100 масс, частей каучука. Неактивные наполнители (мел, тальк,
каолин и др.) незначительно повышают прочностные свойства ре-
зины, но они улучшают обрабатываемость сырой резины и сни-
жают ее стоимость.
Для улучшения обрабатываемости сырой резины, равномерного
распределения ингредиентов в смеси, повышения ее эластических
свойств в состав смеси вводятся мягчители или пластификаторы,
которые несколько снижают прочностные, но повышают пластиче-
ские свойства резины. При использовании каучуков общего назна-
чения в качестве пластификаторов применяются углеводороды (ог
5 до 30 масс, частей), органические жирные кислоты (1—2 масс,
части), смолы (3—10 масс, частей), а в каучуки специального на-
значения— эфиры (дибутилфталат) и синтетические смолы (фено-
лоформальдегидные и др.).
Изделия из каучука и резины под действием кислорода воздуха
даже в состоянии покоя стареют, становятся хрупкими и ломкими.
Для предотвращения или замедления этого процесса в состав ре-
зиновой смеси вводят противостарители (фенолы, ароматические
амины и др.). Для придания цвета изделиям из резины приме-
няют наполнитель — коллоидальную кремневую кислоту (белую
сажу) или окись цинка и красители. Пористую резину (резину, со-
держащую ячейки, заполненные газом) получают введением в смесь
порообразователей (углекислого аммония, диазосоединений и др.),
разлагающихся при нагревании с выделением газов. Кроме пере-
численных выше, в изделия добавляют резиновые клеи, смазки,
растворители, пудры, (для снижения слипания полуфабрикатов)
И др.
381
Изготовление резиновых изделий
Процесс изготовления резиновых изделий состоит из несколь-
ких стадий:
1) приготовление сырой резиновой смеси из ингредиентов;
2) изготовление или формование заготовок или изделий из сы-
рой резиновой смеси;
3) вулканизация изделий;
4) отделка изделий.
Приготовление резиновой смеси включает операции, связанные
с подготовкой сырьевых материалов (развеска, дозировка сырья,
прорезинивание тканей, пластикацию каучука под действием тепла
и др.) и их смешение.
Смешение составляющих резиновой смеси осуществляют в ре-
зиносмесителрх, червячных прессах или на вальцах (рис. XV. 19).
Рис. XV.19. Схема приготовления
смеси на вальцах:
1 — передний валок; 2—задний валок;
3—резиновая смесь.
Рис. XV.20. Схема работы каландров:
1,2 — каландры; 3—транспортеры.
Резиновая смесь подается в вальцы между двумя валками,
укрепленными в станине. Поскольку передний валок вращается
медленнее заднего валка, окружные скорости вращения валков
различны. При прохождении резиновой смеси через зазор между
валками в ней возникают деформации сдвига, что приводит к рав-
номерному смещению ингредиентов.
Для изготовления заготовок, деталей, трубок, прорезиненных
тканей и т.д. применяются червячные прессы (шприц-машины), ка-
ландры.
Схема работы каландра показана на рис. XV. 20. Изделия
сложного профиля собирают из предварительно заготовленных ча-
стей и деталей (прорезиненные ткани, металлические каркасы и
т. п.), соединяя их между собой склеиванием или подпрессовкой.
Отдельные изделия получают из латексов, например пористые ре-
зины (вспенивание латекса), а тонкостенные изделия (перчатки и
др.) получают методом окунания формочек в латекс, с последую-
щей его коагуляцией, сушкой и вулканизацией получаемых изде-
лий.
Вулканизация является заключительной стадией изготовления
резиновых изделий, основное назначение которой состоит в закреп-
382
лении полученной формы изделием и придании изделию свойств
резины.
Количество вулканизатора, вводимого в резиновую смесь, ока-
зывает сильное влияние на свойства резины (табл. XV. 4).
Таблица XV.4. Примерный состав резиновых смесей
Состав смеси, масс, части на 100 масс,
частей каучука
Резины Тип каучука вулкани- затор ускори- тель наполни тель противо- ст зри- тель пласти фикатор
Мягкая НК 2,75 1,5 56 1,0 4,0
Мягкая скс-зо 2,0 0,6 55 0/45 7,0
Мягкая С КН-40 1,5 0,8 50 —— 1,5
Твердая (эбонит) НК, СКС СКИ и др. 25-35 0,2 30 2
1 Большое число резиновых, резино-тканевых и других изделий
подвергают вулканизации при температуре 125—180 °C в аппара-
тах, работающих под давлением в ат-
мосфере насыщенного пара, горячего
воздуха и др.
Продолжительность вулканизации
изделий зависит от температуры, дав-
ления, габаритов изделия, состава ре-
зиновой смеси, греющей среды, аппа-
ратуры, применяемой при вулканиза-
ции. Должен поддерживаться опти-
мальный режим вулканизации, так как
от этого в значительной степени зави-
сят прочностные свойства резины.
Вулканизацию резиновых изделий
проводят в аппаратах различного ти-
па, устройство которых зависит от
сложности изготовляемых изделий и
типа их конструкции. Наибольшее при-
менение находят вулканизационные
прессы, автоклавы, камеры, аппараты
барабанного типа и другие.
Рассмотрим процесс вулканизации
резиновой покрышки с применением
автоклава (рис. XV. 21). В вулканиза-
ционные камеры 5 (14—22 шт.), распо-
Рис. XV.21 Схема автоклава-
пресса:
/ — варочная камера; 2 — барабан;
3— плунжер; 4 — стол; 5 — вулканиза-
ционные камеры; >— покрышки.
ложенные на столе 4 и перемещающиеся внутри барабана с по-
мощью плунжера 3, укладываются покрышки 6 или шины с вло-
женными в них варочными камерами /, внутрь которых подается
перегретая вода под давлением. Варочная камера, представляющая
383
Таблица XV. 5. Основные виды каучуков и некоторые свойства резины иа их основе Характеристика резин Основные свойства Набухают в маслах, бензине, морозо' стойки до —50 °C, окисляются ки- слородом Высокая эластичность, морозостой- кость и прочность при истирании, повышенная окисляемость, набу- хает в бензине, маслах и др. Малая эластичность, легкость окисле- ния и низкая химическая стойкость Высокая прочность при истирании, ограниченная эластичность и моро- зостойкость, легко окисляется Высокая прочность при истирании, низкая морозостойкость и эластич- ность, резко окисляются Не набухают в жидком топливе и маслах, низкая морозостойкость и эластичность, легко окисляются Малая набухаемость в бензинах и маслах, высокая атмосферо- и хи- мическая стойкость Высокая атмосферостойкость и газо- непроницаемость, набухают в жи- дком топливе и маслах Высокая газонепроницаемость, моро- зостойкость, эластичность, кисло- родостойкость. набухают в бен- зине и маслах Химическая инертность, морозо- и кислородостойкость, масло- и беи- зиностойкость
Теплостой- кость, °C О О 0-000 о о •—О о о —» сч о ю ) 1 S ) 1 1 1 § ) ) ОО О О ООО о 00 tO ООООО’-ч to о —< —< сч сч
Относнтель- | иое удлине- ние, °/о оо ооооооо о ю to to to о to о о о to 00 00 *3* о о о ь. О оо to 1 I I 1 1 1 1 1 1 1 оо ооооооо о ОО tO to О Ю О О СЧ tO СО СО CQ to b- to CD to сч сч
Предел проч- ности нри растяжении, МПа о о ю to сч о to сч to со со 1 СЧ сч 00 00 СЧ 00 сч II 1 1 1 1 1 1 1 1 о ю о О о Г* О to *3* о СЧСЧ »-’СЧ’—’СЧСЧ’—’ сч
Плотность каучука 103кг/м3 to Q0 00 00 о о CD о о СЧ СЧ О о»-« — о о $£ сч сч ’-7 ] CD о 1 t 1 О 1 Д , О . о Д Д О о ’ ’ _ ' О О СО Q0 о о “ о
Исходные ‘ . s я я * , ® о, сх. оз н s ч х ш « 2 о .о «ч я S a S «К и щ я Е? * S 2 ° У о, р, я я щ <D<y о я s р о. я сь Ч 3 сХ о а 5 S’ S «о 5s 5о & О О н X н г* оз Я О 9 X 2 ef ? Я tj с ss и и и и s ч е
Каучук , >s 5 . й н О Ч д я js' »s« s о. g „ с, 2 з »к S3 u’S Ь м гая g S§ g, V ? i? £ g gs s§ S S' C3 H B4 ’S s я я о S H w Я я «j >> gg £-§53 SS .S3 & . g § ди диииддс е
Примечания: Кроме указанных видов находят применение полнизобутиленовый, полисульфндные, уретановые, каучуки типа СКД, бута,
ен-метилвинил-пиридиновые, акриловые, пипериленовые, хлорсульфополиэтиленовые, этилен-пропиленовые каучуки. Наполнителем резин j
ляется сажа.
собой тонкостенную резиновую оболочку, прижимает покрышку 6
к стенкам вулканизационной камеры, обогреваемую снаружи па-
ром под давлением. Такое устройство автоклава позволяет обеспе-
чить равномерный процесс вулканизации резиновых изделий и по-
лучить изделия высокого качества.
Изделия, полученные после вулканизации, полностью теряют
свои пластические свойства, но приобретают эластичность, повы-
шенные прочностные свойства и износоустойчивость и др.
После вулканизации все резиновые изделия подвергаются от-
делке. При этом устраняются заусенцы, производится зачистка, об-
точка, шлифовка изделий, а в ряде случаев и их окраска с целью
предохранения от старения (автомобильные шины) или для улуч-
шения внешнего вида (галоши, мячи и др.). Все изделия для опре-
деления сортности подвергаются осмотру с целью выявления де-
фектов и выяснения возможности их устранения без изменения
качества изделий.
Определение свойств и качества изделий осуществляется лабо-
раторными, стендовыми, эксплуатационными испытаниями. При
этом определяются физико-механические свойства, тепло- и моро-
зостойкость, долговечность, сопротивление старению и истиранию.
В настоящее время выпускается большой ассортимент резино-
вых изделий, свойства и назначение которых определяются в ос-
новном свойствами применяемого каучука (табл. XV. 5).
В зависимости от свойств и применения резины их делят на
следующие группы:
1) резины общего назначения (шины, обувь, ремни и др.), экс-
плуатируемые при интервале минус 50 — плюс 110°С; такие резины
изготавливаются иа основе каучуков НК, СКВ, СКИ, СКС, СКМС
и др.;
2) теплостойкие резины (детали самолетов, машин и др.), по-
лучаемые из силиконовых каучуков и фторкаучуков;
3) резины, устойчивые к действию низких температур, получае-
мые из силиконовых и фторкаучуков;
4) резины, устойчивые в окислительных средах, в растворах
кислот, щелочей и солей и т. д.; их готовят на основе наирита,
фторкаучуков, бутилкаучуков и др.;
5) маслостойкие резины, устойчивые к действию нефти и про-
дуктам ее. переработки, получаемые из фторкаучуков, бутадиеи-
нитрильных и других каучуков.
Наряду с этими резинами находят применение газонаполненные
резины, стойкие к воздействию радиации, обладающие высокими
диэлектрическими свойствами.
Клава XVI
ЗАКЛЮЧЕНИЕ
Цель всякого производства состоит в получении продукции при
минимальных материальных затратах, т. е. с максимальной эко-
номической эффективностью.
385
Это может быть достигнуто с одной стороны за счет усовершен-
ствования и интенсификации существующих процессов, а с другой,
за счет использования новых приемов, в оформлении ХТП.
Задача состоит в том, чтобы с возможно большей точностью
установить связи между многочисленными параметрами ХТП, и
составлении математического описания, что позволит с помощью
математического моделирования в сочетании с вычислительной
техникой установить оптимальные значения параметров проектиро-
вания.
Насколько трудна задача по составлению математического опи-
сания, можно судить по выводу уравнений для расчета химических
реакторов (гл. VI и VII). В основу этих выводов положены диф-
ференциальные уравнения конвективного массообмена (уравнение
[VI. 6]) и теплообмена (уравнение [VII. 7]), причем эти уравнения
рассматриваются только применительно к установившемуся ре-
жиму, поскольку он имеет наибольшее практическое значение.
Между тем, неустановившийся режим также имеет практическое
значение, поскольку такой режим наблюдается при пуске и оста-
новке технологических процессов, а также при колебаниях отдель-
ных его параметров, что всегда имеет место в реальных условиях
и должню учитываться при решении вопросов, связанных с авто-
матическим регулированием процесса. Что касается идеального
режима, то производственные процессы приближаются к нему, но
всегда несколько отличаются, так как в реальных реакторах вы-
теснения обычно наблюдается движение реакционной смеси во
всех направлениях, а в реакторах смешения параметры процесса
могут изменяться по объему реактора.
В настоящее время более точные методы решения указанных
дифференциальных уравнений интенсивно разрабатываются при-
менительно к различным типам реакторов с учетом режимов их
работы; полученные уже результаты отражены в разнообразной
специальной литературе. Однако теория химических реакторов,
являющаяся важнейшим разделом химической технологиии, на-
ходится в стадии становления, имеющиеся рекомендации еще не
всегда могут быть использованы для расчета промышленных ап-
паратов.
Приведем некоторые примеры, отражающие имеющиеся воз-
можности для усовершенствования ХТП за счет использования но-
вых приемов.
1. ПЛАЗМОХИМИЧЕСКИЕ ПРОЦЕССЫ
При сильном нагревании любого вещества наступает термиче-
ская ионизация, когда молекулы газа начинают разлагаться на со-
ставляющие их атомы, которые затем превращаются в ионы
(рис. XVI. 1). Таким образом, плазма — это частично или пол-
ностью ионизированный газ, содержащий заряженные частицы
(свободные электроны и газовые ионы).
386
Ранее отмечалось, что температура является мощным фактором
интенсификации химических процессов, поэтому можно ожидать,
что в будущем, по мере совершенствования техники получения вы-
соких температур, плазменные процессы будут находить широкое
практическое применение.
Плазму условно разделяют на низкотемпературную (Т « 103—
10s К) и высокотемпературную (Т « 10е—108 К).
Свойства высокотемпературной плазмы в настоящее время ин-
тенсивно исследуются, а промышленные методы ее получения
только разрабатываются. Методы получения низкотемпературной
плазмы достаточно разработаны, такая
плазма уже используется в промыш-
ленности для получения отдельных хи-
мических продуктов.
Плазменные реакторы, в которых
осуществляются химические процессы,
состоят из трех основных элемен-
тов: плазмотрона, реактора и закалоч-
ного устройства. На рис. XVI. 2 пока-
заны схемы двух типов плазменных
реакторов с прямоточными (рис.
XVI. 2, а) и встречными струями
(рис. XVI. 2, б).
В плазмотроне 1 образуется плаз-
ма, т. е. поток ионизированного газа
(Ar, Не, N2 и др.) при высокой темпе-
Температура, Н
Рис. XVI.1. Зависимость сте-
пени ионизации водорода от
температуры.
ратуре, с помощью дугового, высокоча-
стотного или другого способа. Плазма
направляется в реактор 2, куда по-
дают также исходные реагенты. Из
реактора реакционная смесь направляется в закалочное устрой-
ство, в котором обеспечивается настолько быстрое охлаждение
смеси (ее закалка), что целевая реакция не успевает пройти в
обратном направлении, т. е. в сторону образования исходных реа-
гентов.
Высокие скорости плазмохимических процессов (их продолжи-
тельность составляет 10-2—10-5 с) позволяют уменьшить размеры
промышленной аппаратуры. Например, для осуществления плазмо-
химического пиролиза метана плазменный реактор производитель-
ностью 25000 т/год должен иметь диаметр 15 см и длину 65 см.
Плазмохимические процессы легко управляемы; они хорошо мо-
делируются и оптимизируются. В промышленном и полупромыш-
ленном масштабах реализованы многие плазмохимические про-
цессы: получение ацетилена и технического водорода из природ-
ного газа; получение ацетилена, этилена и водорода из углеводоро-
дов нефти; производство синтез-газа для получения винилхлорида;
получение пигментной двуокиси титана и других продуктов химиче-
ской промышленности.
387
Рассмотрим в качестве примера плазменный метод получения ;
азотной кислоты на основе атмосферного азота.
Существующие методы производства азотной кислоты окисле-
нием аммиака являются громоздкими и связаны с затратой при-
водного газа ценного углеводородного сырья, мировые запасы ко-
Рис. XVI.2. Схема плазмохимических
агрегатов с реактором прямоточного
типа (д) и со встречными струями (б);
- / — плазмотрон; 2—плазменный реактор;
3—закалочное устройство; 4 — узел улав-
ливания и обработки продуктов.
торого ограничены. Между тем
окружающий нас воздух содер-
жит азот и кислород, из которых
состоит молекула окиси азота. По-
этому процесс получения окиси
азота из воздуха с последующей
переработкой ее в азотную кис-
лоту (стр. 273) давно привлекает
внимание исследователей.
Степень окисления азота по
реакции
N2 + O2 2NO (а)
может достигать достаточной для
практических целей величины
при температуре выше 3000 К, что
видно из следующих эксперимен-
тальных данных о равновесной концентрации окиси азота, получае-
мой из атмосферного воздуха:
Температура, К . . . . 1810 2033 2680 3000
Концентрация NO, %
(об.)................... 0,37 0,64 2,05 3,57
Для промышленных целей важное значение имеет скорость до-
стижения равновесия, так как этим определяется интенсивность
процесса. Для рассматриваемой реакции эта скорость резко воз-
растает при увеличении температуры:
Температура К ..............
Время, в течение которого
устанавливается равнове-
сие, с......................
1700 2000 3000 4000
140 1,0 1,4-Ю-4 1,5-10"8
Одна из возможных схем получения окиси азота следующая.
Атмосферный воздух, сжатый в компрессоре, подогревается в
теплообменнике до 1500—1700 К и поступает в плазменную печь.
Полученный нитрозный газ при 3000—3300 К проходит далее
в смеситель, где смешивается с циркулирующим нитрозным га-
зом, охлаждается до 1700—1900 К и далее поступает в теплооб-
менник. Здесь газ отдает тепло поступающему воздуху, охлаж-
дается до 1100 К и затем подается в газовую турбину. Часть-газа
после турбины идет на абсорбцию, а остальное количество комп-
рессором подается в систему в качестве циркуляционного газа.
388
При получении окиси азота из аммиака общие затраты энергии
на производство аммиака и окиси азота составляют около
8 МВт-ч-т-1. Ожидается, что при прямом синтезе окиси азота под
давлением 3 МПа по схеме, изображенной на рис. XVI. 3, затраты
энергии будут также около 8 МВт-ч-т-1, что позволяет сравнивать
этот метод с аммиачным. Однако, есть все основания ожидать, что
при дальнейшем усовершенствовании метод прямого синтеза бу-
дет экономичнее аммиачного за счет более полного использования
тепла отходящих газов и интенсификации процесса [при темпера-
туре выше 3000 К реакция (а) протекает практически мгновенно].
2. РАДИАЦИОННО-ХИМИЧЕСКИЕ ПРОЦЕССЫ
При воздействии на вещества ионизирующих излучений высо-
ких энергий происходят различные химические превращения. Ис-
пользование этого явления для совершенствования ХТП имеет
большое практическое значение, так как в технике получения мощ-
ных излучений за последние годы имеются существенные достиже-
ния и эта область усиленно развивается. Таким образом, есть все
основания ожидать, что радиационно-химические процессы имеют
большое будущее.
Ионизирующей способностью обладают как электромагнитные
излучения (рентгеновские лучи, у-лучи и др.), так и заряженные
частицы (электроны, протоны и др.).
Процессы, протекающие при облучении вещества, разделяются
на три основные стадии. На первой, физической, стадии происхо-
дит столкновение заряженной частицы с молекулами вещества,
в результате чего химическая энергия частицы передается молеку-
лам, что приводит к изменению их энергетического состояния. При
этом возникает большое число «активированных» молекул, неста-
бильных в состоянии возбуждения.
Вторая стадия состоит в распадении возбужденных (нестабиль-
ных) молекул либо во взаимодействии их с другими молекулами.
В результате образуются ионы, атомы и радикалы, т. е. промежу-
точные активные частицы.
На третьей стадии, образовавшиеся активные частицы вступают
в химическое взаимодействие с другими окружающими молеку-
лами или друг с другом с образованием конечных химических про-
дуктов.
Реакции активных частиц с молекулами отличаются от реакции
невозбужденных молекул друг с другом. Для того чтобы невоз-
бужденные молекулы вступили во взаимодействие, им необходимо
сообщить некоторую избыточную энергию е (энергию активации),
что достигается, например, повышением температуры. Реакции ра-
дикалов с молекулами требуют преодоления относительно неболь-
шого энергетического барьера (20—40 кДж-моль-1). В результате
этого радиационно-химические реакции протекают с большой ско-
ростью даже при низкой температуре; в отличие от обычных хими-
389
ческих реакций скорость радиационных процессов мало зависит от
температуры.
Радиационно-химический процесс характеризуется радиацион-
ным выходом G, равным числу превратившихся (или образовав-
шихся) молекул вещества на 100 эВ поглощенной энергии. Для
обычных реакций выход G составляет от 1 до 20 молекул. При
этом энергия расходуется непосредственно на осуществление про-
цесса взаимодействия. Такие процессы имеют ограниченное приме-
нение, поскольку требуют больших затрат энергии. В цепных ра-
диационно-химических процессах электромагнитное излучение иг-
рает роль инициатора, поэтому радиационный коэффициент дости-
гает большой величины (G — 103 * *—106). Среди процессов, в кото-
рых излучение инициирует протекание нецепных реакций, практи-
ческое осуществление нашли радиационно-химические процессы
сшивания отдельных макромолекул при облучении высокомолеку-
лярных соединений. Так, например, в результате сшивания поли-
этилена повышается его термостойкость и прочность, а для каучука
обеспечивается его вулканизация. На этой основе разработано ра-
диационно-химическое производство упрочненных и термостойких
полимерных пленок, труб, кабельной изоляции, процесс вулкани-
зации резино-технических изделий и др.
К числу интенсивно излучаемых и практически осуществляе-
мых цепных радиационно-химических процессов относятся различ-
ные процессы полимеризации, а также синтеза ряда низкомолеку-
лярных соединений. Важное практическое значение приобрели ра-
диационные методы отверждения связующих (полиэфиры и др.)'
в производстве стеклопластиков и при нанесении лакокрасочных
покрытий на металличеекие, деревянные и пластмассовые изделия.
Большой интерес представляют также радиационно-химические
процессы модифицирования простых материалов (древесины, бе-
тона, торфа и т. д.) путем пропитки их мономерами (метилмет-
акрилатом, стиролом и др.) и последующей поляризацией этих мо-
номеров с помощью у-излучения.
Для разработки и внедрения в промышленность экономичных
радиационно-химических процессов возникла радиационно-химиче-
ская технология, а также радиационно-химическое аппаратуро-
строение, в разработку теоретических основ которых вложен ог-
ромный труд советских ученых.
3. БИОХИМИЧЕСКИЕ ПРОЦЕССЫ
В природе под воздействием микроорганизмов, а также в при-
сутствии различных природных катализаторов протекают всевоз-
можные биохимические процессы, многие из которых еще до сих
пор не изучены и которые не удается воспроизвести в искусствен-
ных условиях. Характерным для биохимических процессов яв-
ляется то, что многие из них протекают при атмосферных усло-
виях с очень высоким коэффициентом полезного действия.
390
В микробиологии — науке о деятельности микроорганизмов —
недавно возникло новое направление — техническая микробиоло-
гия, в которой рассматривается химическое поведение микробов и
возможность использования их деятельности в химической про-
мышленности и в различных отраслях техники.
Сейчас техническая микробиология только начинает разви-
ваться, но уже с самого начала перед ней открываются огромные
возможности, которые трудно переоценить. С помощью микроорга-
низмов можно с большой экономией труда и средств прозводить
самые разнообразные продукты. Продуктивность микробиологиче-
ского синтеза не зависит от географического размещения пред-
приятия, от почвы и климатических условий. Все процессы проте-
кают с минимальным расходом энергии при атмосферном давлении
и комнатной температуре. Применение микроорганизмов удешев-
ляет и упрощает производство.
Расчеты показывают, что при воспроизведении лабораторных
экспериментов в промышленных условиях бактерии способны в те-
чение нескольких десятков дней выдать «на-гора» несколько сот
тысяч тонн продуктов.
За последние годы из стадии лабораторных исследований пере-
даны производству микробиологические процессы, в том числе син-
тез антибиотиков, витамина Bi2, аминокислот, гормонов и других
биологически активных веществ.
Сейчас в мире не хватает белковых продуктов, и одним из наи-
более реальных путей восполнения этого является организация
производства белков с помощью микроорганизмов. На основе ис-
следований, проведенных учеными различных стран, были разра-
ботаны промышленные биохимические методы получения белка.
В настоящее время работают заводы, выпускающие ценный кормо-
вой продукт (используется на птицефабриках) из парафиновой
фракции нефти, получаемый с помощью бактерий. На животновод-
ческих фермах применяются биологические корма, содержащие бо-
лее 15% микробного белка. Весьма перспективным сырьем для по-
лучения кормового белка является метан. В нашей стране, богатой
природным газом, этот процесс представляет особенно большой
интерес.
Области применения биохимических процессов в промышленно-
сти весьма разнообразны: от сбраживания углеводородов, получе-
ния спиртов и кислот — первого этапа технической микробиоло-
гии— до развитой микробиологической промышленности витами-
нов и ферментов, ферментативных катализаторов и др.
В последние годы стали известны бактериологические способы
окисления серы до сернистого ангидрида и серной кислоты. Такие
процессы уже находят практическое применение, например для
очистки нефти и нефтепродуктов от серы. Бактерии перерабаты-
вают присутствующую в нефтепродуктах серу в сернистый ангид-
рид, легко удаляемый затем из очищаемых продуктов.
391
Существуют также .бактерии, способствующие восстановлению
сернистого ангидрида, сульфатов и других серосодержащих про-
дуктов до элементарной серы. Особый практический интерес пред-
ставляют результаты промышленных опытов по бактериологиче-
скому превращению сернистых соединений, содержащихся в про-
изводственных сточных водах, в элементарную серу (в этом случае
одновременно с получением серы достигается очистка, сточных
вод), а также по разработке микробиологического выщелачивания
металлов: алюминия, золота, кадмия, кобальта, меди, мышьяка,
никеля, олова, рения, селена, титана, урана, цинка из минералов.
Еще несколько лет тому назад казалась фантастической даже
сама идея применения микроорганизмов в качестве активных «про-
изводителей» металлов. А сегодня химическая деятельность этих
«невидимых металлургов» начинает находить широкое применение.
Полнее всего разработан метод микробиологического выщела-
чивания меди из различных минералов, в которых медь соединена
с серой. Медь вымывается из руды раствором, содержащим суль-
фат окиси железа, серную кислоту и тионовые бактерии. В резуль-
тате деятельности бактерий и действия сульфата окиси железа, на-
ходящихся в растворе, руда выщелачивается, при этом образуются
серная кислота и медцый купорос, переходящий в раствор. Этот
раствор поступает в цементационную установку, где находятся
куски железа. Происходит обменная реакция и образуется суль-
фат закиси железа, а чистая медь выпадает в осадок.
На одном .из рудников построена промышленная установка по
бактериальной добыче меди. Бактериальный раствор, подаваемый
по трубопроводу в скважину, окисляет металл и он переходит
в раствор, который откачивается на поверхность. Получаемый
этим способом металл почти втрое дешевле меди, извлекаемой
другими способами. Есть все основания ожидать, что будущее за
биохимическим способом получения меди, который со временем
позволит исключить доменные и медеплавильные печи.
Очень интересными являются результаты исследований по раз-
работке метода повышения производительности истощаемых нефтя-
ных скважин. В скважину вводится суспензионный раствор, в со-
став которого входят микроорганизмы и питательная смесь для них.
Попав в подземное нефтяное озеро, бактерии начинают быстро
размножаться. В результате их жизнедеятельности вырабатывается
большое количество газов, повышается давление и скважина ожи-
вает.
Мир микробов пока еще изучен гораздо хуже мира животных
и растений. Научный поиск полезных бактерий в сущности только
начинаете?. Одной из важнейших проблем ближайшего будущего
является поиск и направленное выделение микробов, обладающих
высокой активностью.
392
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Общая
1. Вольфкович С. И., Егоров А. П., Эпштейн Д. А. Общая химическая техноло-
гия, Т. 1. М.—Л., Госхимиздат, 1953. 632 с.
2. Вольфкович С. И. и др. Общая химическая технология. Т. 2. М. — Л., Госхим-
нздат, 1959. 848 с.
3. Жаворонков Н. М.— Горизонты химической технологии. — «Наука и жизнь»,
1970, № 4, с. 71.
4. Кафаров В. В. Методы кибернетики в химии и химической промышленности.
М., «Химия», 1976. 464 с.
5. Касаткин А. Г. Основные процессы и аппараты химической технологии. Изд.
9-е, испр. М., «Химия», 1973. 750 с.
6. Лебедев Н. Н., Манаков М. Н., Швец В. Ф. Теория технологических процес-
сов основного органического синтеза. М., «Химия», 1975. 478 с.
7. Левеншпиль О. Инженерное оформление химических процессов. Пер. с англ.
Под ред. М. Г. Слинько. М., «Химия», 1969. 622 с.
8. Лукьянов П. М. Краткая история химической промышленности СССР. М. —
Л., изд. АН СССР, 1959. 463 с.
9. Мухленов И. П. и др. Общая химическая технология. Части I, II. М., «Выс-
шая школа», 1977.
10. Практикум по общей химической технологии. Под ред. И. П. Мухленова. М.,
«Высшая школа», 1973. 424 с.
11. Расчеты' химико-технологических процессов. Под ред. И. П. Мухленова. Л.,
«Химия», 1976. 301 с.
12. Хайлов В., Брандт Б. Введение в технологию основного органического син-
теза. Л., «Химия», 1969. 560 с.
К первой части
1. Боресков Г. К. Катализ. Части I, II. Новосибирск, «Наука», 1971.
2. Бахирко Б. А., Колосов А. Ф., Москвин В. Ф. Экономика химической промыш-
ленности. М., «Высшая школа», 1975. 480 с.
3. Катализ в кипящем слое. Под ред. И. П. Мухленова. Л., «Химия», 1971.
312 с.
4. Кафаров В. В. Основы массопередачи. М., «Высшая школа», 1972. 494 с.
5. Киреев В. А. Курс физической химии. М., «Химия», 1975. 776 с.
6. Мухленов И. П. и др. Технология катализаторов. Под ред. И. П. Мухленова.
М.—Л., «Химия», 1974. 326 с.
7. Экономика химической промышленности. Под ред. В. Д. Якобсона. М., «Выс-
шая школа», 1975. 352 с.
Ко второй части
1. Безденежных А. А. Математические модели химических реакторов. Киев,
«Техника», 1970. 176 с.
2. Брайнес Я. М. Введение в теорию и расчеты химических и нефтехимических
реакторов. М., «Химия», 1976. 232 с.
393
3. Вэйлас С. Химическая кинетика и расчеты промышленных реакторов. Изд. 2-е,
испр. Пер. с англ. Под ред. П. А. Семенова. М. — Л., «Химия», 1967. 416 с. '
4. Денбиг К. Г. Теория химических реакторов. Пер. с аигл. Под ред. Н М Жа-
воронкова. М., «Наука», 1968. 192 с.
5. Крамере X., Вестертерн К. Химические реакторы, расчет и управление ими.
Пер. с англ. Под ред. Г. М. Панченкова. М., «Химия», 1967. 263 с.
6. Кунин Д:, Левеншпиль О. Промышленное псевдоожижение. Пер. с англ. Под
ред. М. Г. Слинько и Г. С. Яблонского. М., «Химия», 1976. 448 с.
К третьей части
1. Амелин А. Г. Технология серной кислоты. М., «Химия», 1971. 496 с.
2. Андреев Ф. А. и др. Технология связанного азота. М., «Химия», 1974. 464 с.
3. Атрощенко В. И., Каргин С. И. Технология азотной кислоты. М., «Химия»,
1970. 494 с. '
4. Боресков Г. К. Катализ в производстве серной кислоты. М. — Л., Госхимиз-
дат, 1954. 348 с.
5. Ганз С. И., Пархоменко В. Д. Получение связанного азота в плазме. Киев,
«Высшая школа», 1976. 191 с.
6. Гринберг Я. И. Проектирование химических производств. М., «Химия», 1970.
269 с.
7. Гуревич И. Л. Общие свойства и первичные методы переработки нефти и
газа. М. — Л., «Химия», 1972. 360 с.
8. Кульский Л. А. Химия и технология обработки воды. Киев, изд. АН УССР
1960. 359 с.
9. Кушелев В. П. Основы техники безопасности на предприятиях химической
промышленности. М., «Химия», 1977. 280 с.
10. Лебедев И. И. и др. Теория технологических процессов основного органиче-
ского и нефтехимического синтеза. М., «Химия», 1975. 478 с.
11. Лейбуш А. А. и др. Производство технологического газа для синтеза аммиа-
ка и метанола из углеродных газов. М., «Химия», 1972.
12. Меньковский М. А. Комплексное использование горючих и нерудных иско-
паемых. М., Гостоптехиздат, 1962. 282 с.
13. Рогозин 3. А. Основы химии и технологии химических волокон. М. «Химия»,
1974 с.
14. Справочник азотчика. Т. 1, 2. М. — Л., «Химия», 1967—1969.
15. Справочник по пластмассам. Т. 1, 2. М., «Химия», 1975.
16. Справочник резинщика. М., «Химия», 1971. 607 с.
17. Справочник сериокислотчика. М., «Химия», 1971. 744 с.
18. Швец И. Т. и др. Энергетика. М., Машгиз, 1961. 501 с.
19. Шур А. М. Высокомолекулярные соединения. М., «Высшая школа», 1971.
519 с.
20. Юрлевич Я., Росиньский С. Пер. с польск. Под ред. М. Г. Фельдбрина. Угле-
химия. М., «Химия», 1973. 360 с. J
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбция
коэффициент ускорения 93
моделирование 89 сл.
нефтепродуктов 318
серного апгидрнда 247 сл.
сероводорода и двуокиси углерода 262,
263
Автоклав-пресс 383
Автоматизация производства H2SO4 230 сл«
Автотермический процесс 160
Адиабатический реактор вытеснения 160
Адиабатический режим 147, 148
Адипиновая кислота 68
Адсорбенты 262
Адсорбция
кислорода на катализаторе 275
нефтепродуктов 318
Азотная кислота
безводная 272
концентрирование 284 сл.
применение н свойства 271 сл.
производство см. Производство азотной
кислоты
Активаторы (Промоторы) 105
Активность катализатора 96. 97
Алюминий 200
Аммиак
выход 265
каталитическое окисление 98
пределы взрывоопасности 257, 258
применение 257, 258
производство 14, 187 см. также Произ-
водство аммиака
расход электроэнергия 200
свойства 257
синтез 264
содержание в смеси 264
степень окисления 276, 277
Аммиачная селитра 200
Анилии 334
Антидетонаторы 305
Антрацит, состав 29i
Аппаратура, критерии выбора 222 сл.
Армирующие материалы 380
Асбоволокниты 370
Ацетилен
получение 331
применение 332
производство из СаС2 216
свойства 330 , 331
Бензин 304, 305
выход прн крекинге нефти 3ll
каталитического крекинга 317
— риформинга 315
.Биохимические процессы 390 сл.
Бутадиен 187
Ванадиевая контактная масса 244, 245
Взрывоопасные смесн
аммиака с воздухом 257, 258
ацетилена с кислородом 330, 331
Винипласт 361
Вода(ы) 184
виды 194, 195
качество 195
методы очистки 196 сл.
состав 193
сточные см. Сточные воды
Водород, равновесное давление 49
' Водяной гйз, получение и состав 302
Воздух 184
Волокниты 369, 370
Время пребывания в реакторах 133, 134
Вспомогательные материалы 380
Вторичные энергетические ресурсы 201, 204,
205
Вулканизаторы 381
Вулканизация
каучука 381
резиновых изделий 382 сл.
Выбор параметров процесса 219 сл.
Высокомолекулярные соединения
гетероцепные 353
классификация 337, 338
применение 335, 354 сл.
прочностные свойства 35L 353
свойства 336
синтез см. Синтез высокомолекулярных
соединений
средняя молекулярная масса 351
стереорегулярные 351
строение 336, 337, 351 сл.
форма 351, 352
химическая стойкость 352, 353
Газификация
подземная угля 302, 303
твердого топлива 300 сл.
Газы коксования, состав 294
Газогенераторы 301, 302
Гетерогенные процессы
время взаимодействия 88
движущая сила 77, 78
коэффициент скорости 70 сл.
механизм 69
область протекания 89
поверхность контакта фаз 76, 77
скорость 70
Гетерогенный катализ 99
кинетика 101 сл.
окисление NH3 279 ,
—*NO2 279 сл.
элементарные стадии 100
Гиббса энергия 42
Гидратация олефинов 326
Гидроочистка нефтепродуктов 318
Гомогенные процессы 66 сл.
Горение угля 69
математическое моделирование 82 сл_
Горючее минеральное сырье 183
Горючие сланиы '
полукоксование 300
состав 291
Гравитационное разделение 190, 191
Графические расчеты
выхода продукта 130, 131
каскада РИС 122
последовательных реакторов 128
реактора идеального смешения ИЗ
степени превращения 128
Двуокись азота
абсорбция кислотой 278, 280, 281
получение 280
степень полимеризации 280
Двуокись углерода 63, 64
Движущая сила гетерогенного процесса
77. 78
Детонационная стойкость 305
Диаграммы кристаллизации и кипения
HNO3 272
Динамическая характеристика реактора
134 сл.
395
Дистилляты, состав 308, 309
Древесина, состав 291
Древесная смола 300
Древесно-слоистые пластики 371
Единичная мощность установки 34, 36
.Жесткость воды 195
Жидкое топливо 304, 306
Завод-автомат, схема 231 сл.
Изготовление резиновых изделий 382 сл.'
Изобарно-изотермический потенциал 42, 43
Изотермический режим 148
Интенсивность -
катализатора 104
производства 21
процесса
в пенных аппаратах 171
синтеза аммиака 265, 266
в системе г — ж 91
характеристика реактора 106, 107
Каландры 382
Каменноугольная смола 294
состав 299
Каменный уголь
марки 293
состав 291
темпы добычи 186
энергоемкость 202
Капитальные затраты 33, 34
Карбонат натрия (Сода), аммиачный спо-
соб производства 211, 212
Каскад реакторов
.аналитический расчет 123 сл.
вытеснения с промежуточным подогре-
вом 160, 161
графический расчет 122, 123
. идеального вытеснения 121
— смешения 121
относительный общий объем 124
с промежуточным вводом реагентов 161,
162
смешения с промежуточным подогре-
вом 160, 161
Катализаторы *
активность 96, 97
ванадиевая масса 244, 245
влияние на кинетику 63
— температуры 103
восстановление 265
выделение 99
изготовление 105
интенсивность 104
конверсии СО 262
метанирования 263
механизм действия 102
окисления аммиака 275 сл.
отравление 97
платиновые 276 сл.
распределение температуры в слое 267
свойства 105
селективность 97, 98
синтеза аммиака 264, 265
температура зажигания 97
— в—неподвижном слое 175
Каталитические процессы
влияние температуры 103
гетерогенные см. Гетерогенный катализ
контактное окисление NH3 247 сл*
крекинг 314, 315
метаиирование 263 сл.
окисление SO2 244 сл.
очистки газов от окислов азота 284
платформинг 317
риформинг 315 сл.
«ручуки
виды и свойства 384
Каучуки
натуральный 375
синтетические см. Синтетические кау*
чуки
Качество
воды 195
продукции 31, 32
Квазигомогенная модель процесса 81 сл*
Кинетика процесса
гетерогенных 71 сл., 101 сл.
гомогенных 68, 69
' лимитируемая внешней диффузией
83 сл.
— внутренней диффузией 83 сл.
— химической реакцией 87, 88
макро- и микроуровень 50 сл*
необратимого, первого порядка 73 сл.
обжига колчедана 240, 241
окисления
аммиака 275, 276
двуокиси серы 244 сл.
окиси азота 279, 280
Классификация .
высокомолекулярных соединений 337.
338
нефтей 304
пластических масс 356, 357
реакторов 109, 14J
резни 385
сырья 183 сл.
топлив 290
химических производств 13
— процессов 17, f8, 66
— реакций 40 с л,
Кокс
образование 315 •
состав и свойства 294, 295
Коксование
нефтяных остатков 314
применение 295
продукты 294
Коксовые печи 295 сл.
Коксовый газ 279 сл.
Колонна синтеза 269, 270
Комплексная переработка сырья 187, 188
Конверсия
метана 47, 258 сл.
низших углеводородов 331
окисн углерода 47 сл., 258, 261, 262
этилена 327, 328
Константа
равновесия реакции 45 сл.
скорости реакции 58
Конструкционные материалы, подбор 226
227
Контактный аппарат
с выносным теплообменником 249
с двухступенчатым катализатором 278
Контроль и регулирование процесса 227 сл«
Котел-утилизатор 205, 206
Коэффициент
использования эйергии 204
массопередачи при осушке 243, 244
скорости гетерогенного процесса 70 сл,-.-
76
тепловой полезного действия 204
ускорения реакции 93
Кривые отклика 134 сл.
Латексы 380
Лимитирующая стадия 70
Линия оптимальных температур 162, 245,
246
Магнитйая сепарация 190, 191
Мазут 309
Математическое моделирование 79 сл.,
111 сл., (15 сл., 119 ел.
Материалоемкость химического производ-
ства 182, 183
396
Материальный баланс
обжига пирита 27 сл.
печи КС 29
реактора 107 сл«
Метан 258 сл.
Метаннроваяив 263
Метиловый спирт 320 сл.
Механическая технология 12
Минеральное сырье
балансовое 184 сл.
забалансовое 185
Моделирование систем
г — ж 89 сл.
г — т 81 сл.
Модель частиц с невзаимодействующим
ядром 81 с л.
Мощность 21
Надсмольная вода 298
Наполнители 355
Натуральный каучук 375, 376
Необратимая параллельная реакция 60,
61
Нерудное сырье 183
Нефтепродукты
бензин 309, 31 f
дистилляты 309
мазут 309
очистка 317, 318
Нефть
классификация 304
продукты переработки 304 z
свойства 303, 304
состав 291, 303
темпы добычи 186
Нитробензол 333, 334
Новолачные смолы 365, 366
Носители 105
Обезвреживание
отходящих газов 208, 209
сточных вод 206 сл.
Обжиг колчедана 240 сл.
Обжиговый газ 242, 243
Обогащение сырья
газового 193
гравитационное разделение 189 сл.
жидкого 192
рассеивание (грохочение) 189
флотация 191, 192
Окись азота
выход 276, 278
окисление 279, 280
плазменный способ получения 388, 389
Окись углерода
конверсия 258, 261, 262
синтезы на основе 320 сл.
степень превращения 63, 64, 261, 262
Октановое число 305
Олеум 236 , 237
Оптимальный температурный режим 56
в контактных аппаратах 177 сл.
в реакторах 175, 176
в трубчатом аппарате 180
Органический синтез
на основе ацетилена 330 сл.
— — непредельных углеводородов
326 сл.
“ — окиси углерода 320 сл.
— — предельных углеводородов 324 сл.
Отвердители 356
Отходы производства 187, 188
Отходящие газы, обезвреживание 208, 209
Очистка
воды 196 сл.
газов 242, 243, 262 сл.
— отходящих 284
нефтепродуктов 317, 318
Параметрическая чувствительность
реактора с неподвижным слоем ката-
лизатора 156. 157
РИВ 155 сл.
слоя катализатора 157
Переработка нефти
двухступенчатая 308
дистилляты 309
каталитическая 314 сл.
обессоливание и обезвоживание 306
физические процессы 307 сл.
химические процессы 310 сл.
Переработка твердого топлива
газификация 293, 300 сл.
коксование 293 сл.
полукоксование 299, 300
Печи КС 241, 242
Пиролиз 314
Питьевая вода 194
Плазма 386, 387
Плазмотрон 387, 388
Плазмохимические процессы 14, 386 сл.
Плотность
метилового спирта 320
нефтей 303
полиэтилена-358
серной кислоты 236
Пластикат 361
Пластификаторы 355, 356
Пластические массы
классификация 356. 357
композиционные 355
на основе фенолоформальдегидных
смол 365 сл.
— — эпоксидных смол 371 сл.
поливинилхлорид 360 сл.
полистирол 362 сл.
полиэтилен 357 сл.
простые 355
свойства 354, 355
Поверхность контакта фаз 76, 77
Подземная газификация угля 302, 303
Поливинилхлорид ЗбО, 361
Поликонденсация
гомо'-" и гетеро-процесс 348
на поверхности раздела фаз 350. 351
в расплаве 350
Полимеризация
блочная 346
ионная 341, 342
радикальная 339 сл.
в растворителях 346
совместная 343 сл.
ступенчатая 342, 343
суспензионная 347, 348
Полистирол 362 сл.
Политропический режйм 144 сл.
Полиэтилен 357 сл.
Полукоксование 299, 300
Попутные газы, состав 318, 319
Поршневой режим движения реагентов
114
Порядок реакции 41, 42. 98, 99
Последовательные реакции 61, 62
Предельно допустимые концентрации ве-
ществ 208
Предельные углеводороды 324 сл.
Пресспорошки 367 сл.
Приготовление резиновых смесей 381
Примеры расчетов
выхода продуктов 65
графических см. Графические расчеты
зависимости X*==f(Kp) 63, 64
исходной аммначно-воздушиой смеси
289, 290
максимума зависимости r=f(T) 64, 65
объема РИВ 137, 138
скорости подачи растворов в РИС-Н
138, 139
скрубберной башни 255, 256
397
Примеры расчетов
степени превращения
аммиака 38, 39
двуокиси серы 37, 38
в каскаде 139, 140
температуры нагревания 164. 165
тепла, отводимого в РИС-Н 162, 163
— и поверхности в РИС-Н 163, 164
удельных затрат 39, 40
ускорения окисления SOz 93, 94
ускоряющего действия катализаторов 96
Принципиальная схема производства
аммиака 259
ацетилена из СаСг 216
очистки воздуха 218
разбавленной азотной кислоты 274
серной кислоты из колчедана 238, 239
----------серы 253
типы 218
циклическая, аммиака 218
Природный газ 184
конверсия 320
пиролиз 319
состав 318, 319
темпы добычи 186
энергоемкость 202
Проектирование химико-технологического
процесса 233 сл.
Производительность 20. 21
труда 36, 37
Производство азотной кислоты
разбавленной из аммиака 273 сл,
схемы 273, 274, 281
физико-химические основы 274 сл.
Производство аммиака
аппаратура 269, 270
интенсивность 265, 266
промышленные схемы 259
прямой синтез 258
сырье 258
технико-экономические показатели 271
физико-химические основы 260 сл.
Производство серной кислоты 181
абсорбция SO3 247 сл.
выбор аппаратуры 224 сл.
катализаторы 244, 245
из колчедана 250
конструкционные материалы 226, 227
контактное нз серы 252 сл.
в мире 235
обжиг сырья 239 сл.
окисление SO2 244 сл.
очистка газа 242 сл,
принципиальные схемы 238, 239
разной концентрации 248
расход электроэнергии 200
из сероводорода 224 сл.
сырье 238
технологические показатели 251, 252
усовершенствование 254
химическая схема 238
Промышленные воды 195
Простая необратимая реакция 59, 63
Простая обратимая реакция 60
Противостарители 381
Прочностные свойства
винипласта 361
макромолекул 336
пластмасс на основе феиолоформальде-
гидных смол 369
полимеров 351, 352 '
полистирола 362
полиэтилена 358 *
Прямой коксовый газ 294
Равновесие реакций 44 сл., 48 сл.
Радиационно-химические процессы 389, 390
Рассеивание (Грохочение) 189
Растительное и животное сырье 184
398
Расходные коэффициенты 21 . ’ L
на 1 т азотной кислоты 288
на 1 т аммиака 271
s на 1 т этилового спирта 330
пути снижения 52
Расчет высоты выхлопной трубы 209
Реактор(ы)
вращающийся барабан 168
для гетерогенно- каталитических про-
цессов 171 сл.
— гетерогенно-некаталитических про- ‘
цессов 167 сл.
—- гомогенных процессов 168, 169
с движущимся катализатором 174. 175
динамическая характеристика 134 сл^
истинное время пребывания 133, 134
классификация 109
колонны 170
контактные с неподвижным катализа-
тором 172 сл.
’ кривые отклика 136
кубовые 166
материальный баланс 107 сл.
непрерывного действия
идеального вытеснения 114 сл.
— смешения 119 сл. ?
периодические 109, ПО
полочная печь 168
полочный контактный 178
полунепрерывного действия 125
противоточные 168
с псевдоожиженным слоем 77
с различным тепловым режимом 141 сл„
144 сл., 149 сл., 157 сл.
расчеты 106 сл., 111 сл., 115 сл., 119 слв
селективность процессов 128 сл.
для систем г — т и ж — т 167 сл.
----г — ж и ж — ж 169 сл.
сравнение 125 сл.
теплообменные устройства 177
типы и характеристики 132
труба Вентури 171
трубчатого типа 166, 167, 179, 180
устройство 165
Регенерат 380
Регенераторы 205, 206
Резиновые смеси 382, 383 _ .
Резины
ингредиенты 380, 381
классификация 385
на основе силиконовых каучуков 380
свойства 374
сырье 374 сл.
Резольиые смолы 366, 367
Рекуператоры тепла 205
Риформинг 313
Рудное сырье 183
СВАМ 371
Себестоимость
азотной кислоты 288
влияние единичной мощности 36
серной кислоты 252.
фосфорной кислоты 35, 219 сл.
элементы 34
энергии 203 4
Селективность
катализатора 97, 98
процесса 24
процессов в различных реакторах 128 сл.
Сера 252, 253
Серная кислота 18
абсорбция SO3 247 сл.
диаграмма кристаллизации 236
обезвоживание азотной кислоты 284, 285
очистка нефтепродуктов 317. 318
применение 235
производство *см. Производство сериой
кислоты
свойства 235, 236
Сернистый ангидрид
каталитическое окисление 244 сл.
получение 27 сл.
Серный ангидрид
абсорбция 247 сл.
получение 239
Синтез аммиака см. Производсто аммиака
Синтез высокомолекулярных соединений
339 сл., 348 сл. см. также Полимерн-
зация, Поликонденсация
Синтез-газ 319, 320
Синтетические каучуки
бутадиен-стирольный 378 сл.
изопреновый 378
наирит 378, 379
силиконовый 379
СКВ 376
Синтетические смолы 380
Скорость
гетерогенного процесса 70
диффузии газов 71
каталитической реакции 101, сл.
объемная 104
потока 72
химической реакции 24 сл.
Слоистые пластики 369, 370
Смазочные масла 306 '
Смола Э40 372, 373
Сополимеры 344, 345
Состав природных вод 193
Стабильность углеводородов 310 сл.
Степень превращения 37 сл.
влияние температуры 46, 47, 66, 57
в каскаде 124
равновесная 46, 47
реагентов 21 сл.
в реакторах различного типа 127
Сточные воды 206 сл.
Сульфат аммония, производство 67
Сухая перегонка
древесины 321
угля 300
Сырье
классификация 183 сл.
обогащение 189 сл.
в органическом синтезе 320
для производства Н?РО4 210
ресурсы 184 сл.
экономичность выбора 186, 187
Твердое топливо, газификация 300 сл.
Текстолит 370
Температура
зажигания 87
кипения
метилового спирта 320
серной кислоты 236, 237
кристаллизации
олеума 236, 237
серной кислоты 236
плавления
метилового спирта 320
полиэтилена 358
Тепловой баланс
обжига пирита в печн КС 30. 31
реакторов 142 сл.. 148
Теплота сгорания воздушного газа 303
Теплотворная способность
нефтей 303
топлива 291
Термический крекинг
выход продуктов 316
с движущимся катализатором 316, 317
кинетика 311
Технологическая схема производства
абсорбции 50з 247
аммиака 267 сл.
ацетилена из СаС2 218
бутадиен-стирольного каучука 377, 378
контактного окисления SO2 247
Технологическая схема производства
концентрированной HNO3 285
крекинга с движущимся катализатором
— мазута 315
метилового спирта 322
печного отделения 241
полиэтилена высокого давления 359
разбавленнной HNO3 281 сл.
среной кислоты из колчедана 250
формальдегида 323
фосфорной кислоты 217
этилового спирта 327, 330
Технологические газы, получение 300 сл.
Топливо
классификация 290
состав 291
схема переработки 292
твердое 291
химическая переработка см. Химиче-
ская переработка топлива
Торф
полукоксование 300
состав 291
энергоемкость 202
Трубчатые печи 307, 308
Удельные капитальны- затраты 39. 40
Уксусная кислота 333 •
Умягчение воды 197 сл.
Уран, энергоемкость 202
Утилизация тепла 205
Фаолит 370
Фенол 365
Фенолоформальдегидиые смолы 365 сл.
Флотационная машина с воздушным пере-
мешиванием 192
Флотационное обогащение 191, 192
Флотационный колчедан 27
обжиг 240 сл.
Формальдегид 322 сл.
Фосфат 219, 220
Фосфин 137, 138
Фосфор 200
Фосфорная кислота
получение 210, 211
себестоимость 219 сл.
принципиальная схема производства
213, 214 '
производство, выбор аппаратуры 223,
224
фосфорные удобрения 31, 32
Химико-технологический процесс
с выводом продукта реакции 48, 49
интенсификация 58
контроль и регулирование 227 сл.
проектирование 233 сл.
простейшая схема 20
селективность 57, 58
циркуляционная схема 48, 49
Химическая переработка топлива см. так-
же Переработка иефти, Перера-
ботка твердого топлива
газификация 293
гидрирование и гидрогенизация 293
крекинг и пиролиз 292
сухая перегонка 292
Химическая промышленность
объем производства 16, 17
экономическая эффективность 33 сл.
Химическая реакция
вероятность 43
кинетика 52 сл.
классификация 40 сл.
условия равновесия 43, 44
экзотермическая 161
эндотермическая 159, 160
399
Химическая технология 13
классификация процессов 17, 18, 46
Хлорирование углеводородов 67, 325, 326
Экономическая эффективность процесса
капитальные затраты 33
производительность труда 36, 37
себестоимость 34 сл.
Электромагнитный сепаратор 191
Энергетический баланс 29
Энергия
активации 58
виды 200, 201
запасы и потребление 200
источники 201 сл.
коэффициент использования 204
Энергия
общая выработка 203
потребление 202
рациональное использование 204 сл.
себестоимость 203
Энерготехиологическая схема
производства H2SO4 253
синтеза аммиака 268
Эпоксидные смолы 371 сл.
Этаноламииы 262, 263
Этилен
конверсия 327, 328, 359
прямая гидратация 328 сл.
хлорирование 326
Этиловый спирт 327, 328
Этилсульфат, разложение 68
ОБЩАЯ
ХИМИЧЕСКАЯ
ТЕХНОЛОГИЯ
АНАТОЛИИ ГАВРИЛОВИЧ АМЕЛИН
АФАНАСИЙ ИВАНОВИЧ МАЛАХОВ
ИЯ ЕВГЕНЬЕВНА ЗУБОВА
ВЛАДИМИР НИКОЛАЕВИЧ ЗАЙЦЕВ
Редактор И. В. Лебедева
Технический редактор Г. И. Косачева
Художественный редактор Н. В. Носов
" Художник Е. А. Сумнительный
Корректоры Л. А. Волкова, М. С. Хрипунова
ИБ № 578
Т-12881. Сдано в наб. 8/IV 1977 г. Подп. к печ. 22/VIII 1977 г. Формат бум.
60X9J‘/is. Бумага тип. № 3. Усл. печ. л. 25. Уч.-нзд. л. 27,34. Тираж
27 400 экз. Заказ 558. Изд. № 733- Цена 1 р. 20 к.
Издательство «Химиях 107076, Москва, ул. Стромынка, д. 13.
Ордена Трудового Красного Знамени Ленинградская типография № 2
имени Евгении Соколовой Союзполиграфпрома прн Государственном
комитете Совета Министров СССР по делам издательств, полиграфии
и книжкой торговли. 198052, Ленинград, Л-52, Измайловский
проспект, 29,