/
































































































































































































































































































Author: Ключников Н.Г.
Tags: химия аналитическая химия издательство просвещение практические занятия
Year: 1972




Text
Н. Г. Ключников
ПРАКТИЧЕСКИЕ
ЗАНЯТИЯ
ПО
ХИМИЧЕСНОЙ
ТЕХНОЛОГИИ
Утвержден
Министерством просвещения СССР
в качестве учебного пособия
для педагогических институтов
ИЗДАНИЕ ТРЕТЬЕ,
ПЕРЕРАБОТАННОЕ
ИЗДАТЕЛЬСТВО «ПРОСВЕЩЕНИЕ»
Москва 1972
6П7
К52
Ключников Н. Г.
К 52 Практические занятия по химической технологии.
Учеб, пособие для пед. ин-тов. Изд. 3-е, перераб.
М., «Просвещение», 1972.
296 с. с ил.
Пособие составлено в соответствии с программой практикума по
химической технологии для студентов химических и биолого-химических
факультетов педагогических институтов, В третье издание книги вклю¬
чены работы по исследованию материалов, применяемых в химическом
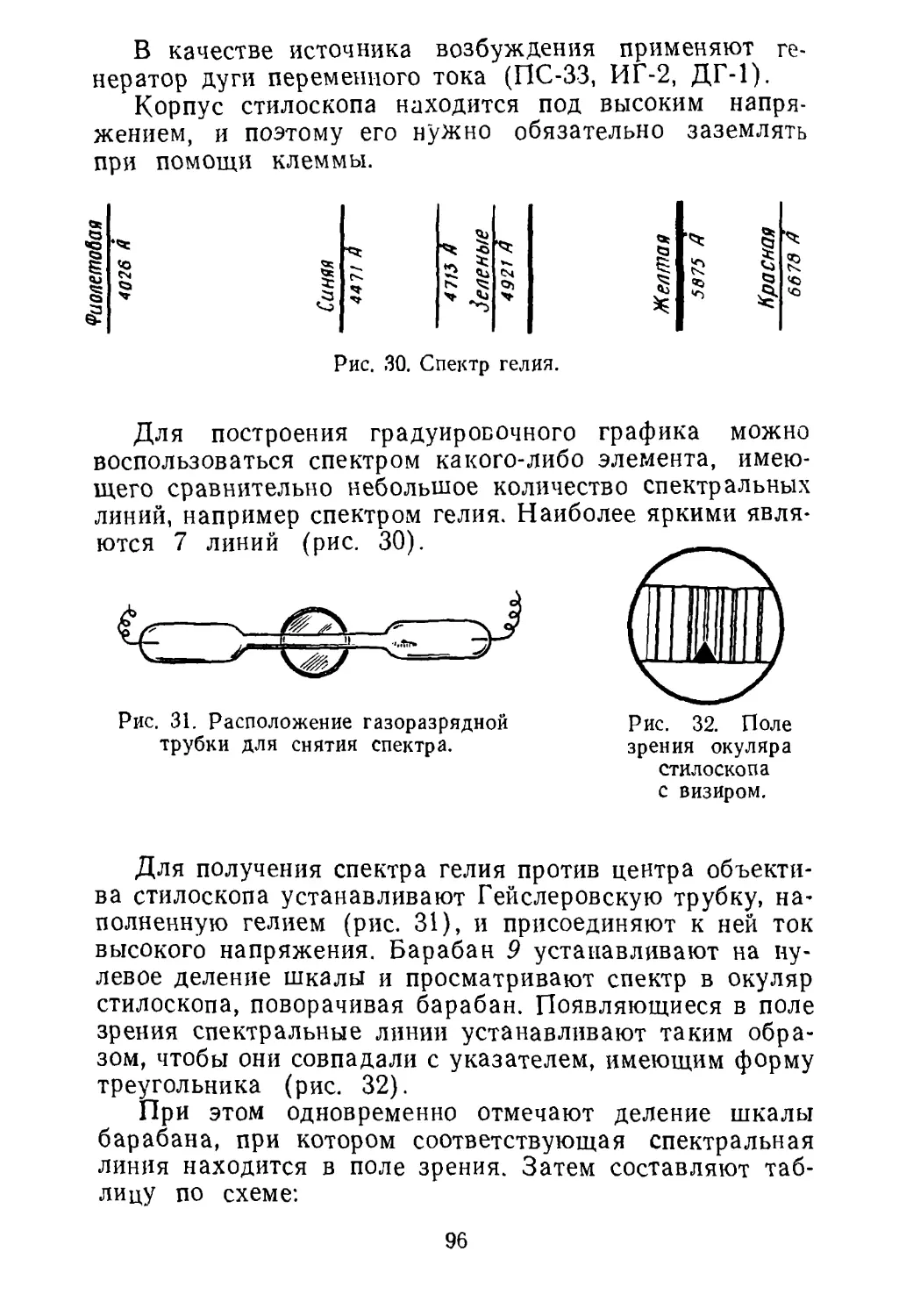
машиностроении. Практические работы помогут студентам приобрести
прочные практические навыки в изготовлении приборов и установок,
проведении расчетных задач по составлению материального баланса.
2-5-2
34-72
6П7
ГЛАВА I
МАТЕРИАЛЬНЫЙ И ЭНЕРГЕТИЧЕСКИЙ
БАЛАНСЫ ТЕХНОЛОГИЧЕСКИХ ПРОЦЕССОВ
§ 1. МЕТОДИКА СОСТАВЛЕНИЯ МАТЕРИАЛЬНОГО
И ЭНЕРГЕТИЧЕСКОГО БАЛАНСОВ
Под материальным технологическим балансом под¬
разумевается расчет количества использованных (вве¬
денных или заданных) веществ, количества полученного
продукта, а также побочных веществ. Результаты расче¬
тов выражают в виде графиков, уравнений, таблиц или
диаграмм. Количество веществ, введенных в производ¬
ство, должно равняться количеству полученных веществ.
Это вычисляется на основании стехиометрических урав¬
нений, описывающих отдельные стадии производства и
побочные процессы.
Материальный и энергетический балансы позволяют
составить наиболее рациональную схему производства,
установить удельное значение выходов продукции, рас¬
хода сырья, энергии, определить необходимые размеры
аппаратуры, ее экономические показатели, степень со¬
вершенства соответствующих процессов. При составле¬
нии материальных балансов необходимо знать состав
сырья, продуктов и полупродуктов, а иногда их некото¬
рые физико-химические свойства и их изменения в зави¬
симости от внешних условий.
В том случае, когда после расчета количество вве¬
денных в процесс веществ не равно количеству получен¬
ного продукта и побочных веществ, вычисляют эту раз¬
ницу, которая называется невязкой. Ее значение также
помещают в таблицу материального баланса. Необходи¬
мо стремиться, чтобы невязка имела минимальное зна-
3
чение. Результаты материального баланса записываются
по определенной форме (табл. 1).
Таблица 1
Материальный баланс
Задано
Масса
Получено
Масса
(В г) | (в %)
(в г) | (в %)
1. Количество ве¬
ществ, введенных
в процесс, в ап¬
парат, или в од¬
ну из стадий
(mi+m2+^3 +■ •
= in
1. Количество по¬
лученных про¬
дуктов произ¬
водства
2. Количество не¬
прореагировав¬
ших веществ
3. Количество по¬
бочных продук¬
тов реакции
4. Количество по¬
терь
5. Невязка
Итого
Итого
Расчеты по материальному балансу чаще составляют¬
ся на 100 или 1000 кг сырья или готового продукта.
Энергетический баланс обычно составляется по дан¬
ным материального баланса. Он также рассчитывается
на определенное количество сырья или продукта и выра¬
жается в тепловых единицах (джоулях), иногда в элек¬
трических (киловатт-часах).
Тепловой, или энергетический, баланс выражается
уравнением:
Qi + Q2 + Q3 + Q* — Qb i- Q6>
где Q1 —теплота, введенная в процессе с исходными ве¬
ществами,— физическая теплота; Q2 — теплота экзотер¬
мических реакций; Qz — теплота физических превраще¬
ний веществ, имеющих положительное значение; если же
некоторые превращения эндотермические, то значение
Qz берется с отрицательным знаком; Q4 — теплота, вве¬
4
денная в процесс извне, например поступающая с горя¬
чей водой, нагретыми газами и т. д.; Q5 — теплота, вы¬
веденная из процесса с продуктами реакции; Qq — по¬
тери теплоты в окружающее пространство.
Qi рассчитывается по формуле:
где с — средняя теплоемкость исходных веществ при
температуре их поступления в технологический процесс;
т — количество исходных веществ (кг, мъ, моль и т. д.);
t — температура исходных веществ; Q2 рассчитывается
на основании справочных (экспериментальных) данных
или на основании термохимического расчета с использо¬
ванием закона Гесса; Q3 рассчитывается на основании
справочных данных по теплоте превращения, испарения,
возгонки и т. д.; Qi рассчитывается аналогично по теп¬
лосодержанию газообразного, жидкого или твердого но¬
сителя; Qs рассчитывается аналогично Qi и Q4; Qe — теп¬
ловые потери — определяются экспериментально или
рассчитываются исходя из существа происходящих про¬
цессов.
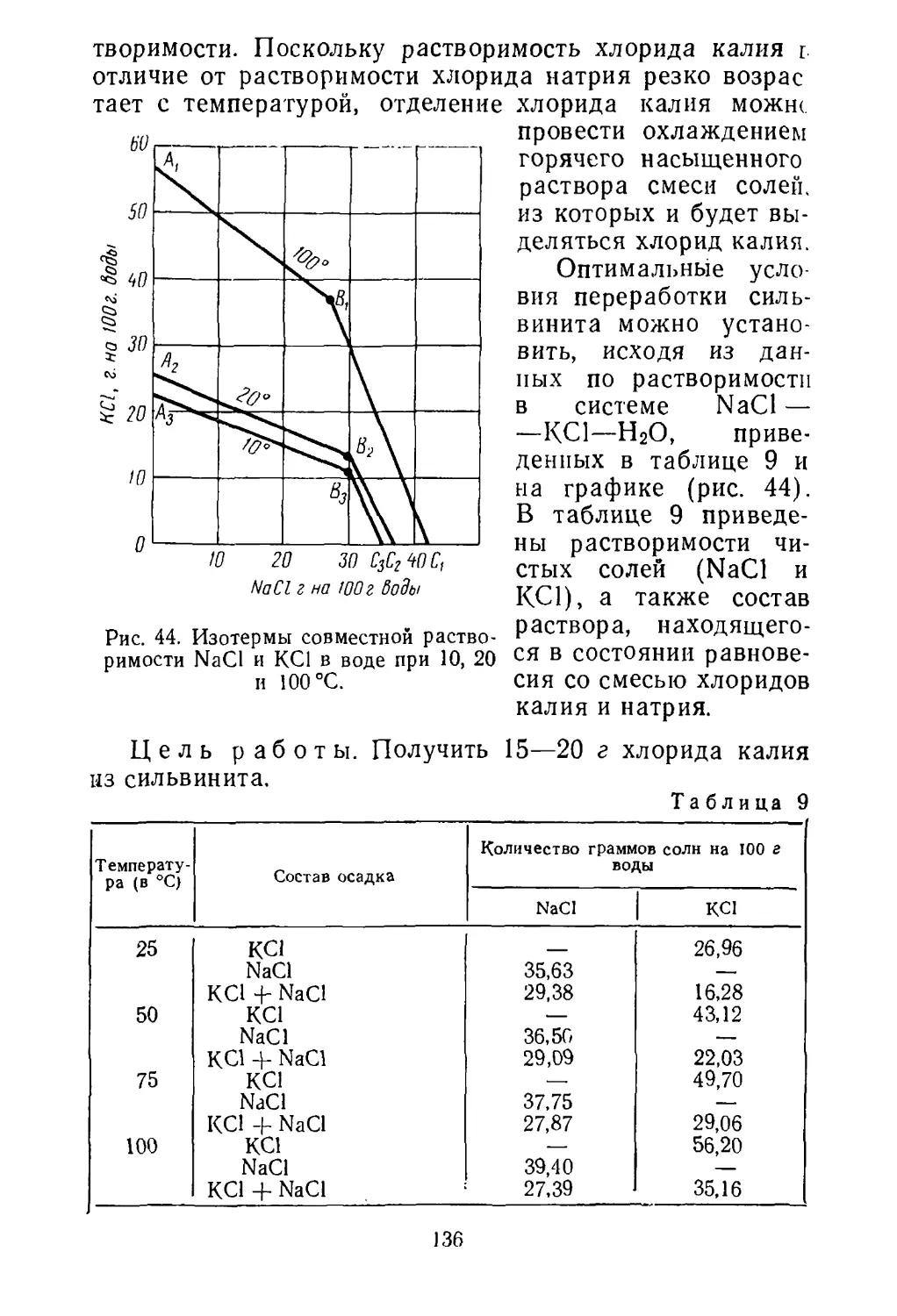
При определении теплоты, теряемой через стенки ап¬
паратуры (Q), пользуются законом Фурье:
Согласно этому закону Q пропорционально поверхности
стенок 5, разности температур между поверхностями
стенки t\ — t2, времени х и обратно пропорционально тол¬
щине стенок Ь,Х — коэффициент теплопередачи, завися¬
щий от материала и температуры стенки (дж/м ■ г • град).
При расчете потерь за счет теплоизлучения приме¬
няют закон Стефана — Больцмана:
где К — постоянная излучения; в — коэффициент чер¬
ноты; S — поверхность; Т— абсолютная темпера¬
тура.
Результаты теплового баланса составляются по опре¬
деленной форме (табл. 2).
Невязка как при материальном, так и при тепловом
балансе может иметь положительное и отрицательное
Qx = cmt,
Q — ^ S т (/t /2).
5
Таблица 2
Тепловой баланс
Задано
Получено
Статьи баланса
дж
%
Статьи баланса
дж
%
1. Физическая теп¬
лота
2. Теплота экзотер¬
мических реакции
3. Теплота физичес¬
ких превращений
веществ
4. Теплота, введен¬
1. Теплота, выве¬
денная из про¬
цесса с продук¬
тами реакций
2. Потери теплоты
в окружающее
пространство
3. Невязка
ная извне
Итого
Итого
значение. Анализ знака невязки позволяет иногда
вскрыть какие-то неучтенные процессы. Например, при
получении молибдена алюминотермическим способом не¬
вязка имеет положительное значение — масса молибдена
и шлака оказывается несколько меньше массы исходных
веществ (молибденового ангидрида и алюминия). Вы¬
яснение причин этого явления позволило установить, что
часть молибдена испаряется в виде молибденового ан¬
гидрида. При получении кальция электролизом хлорида
кальция в материально-энергетическом балансе невязка
имеет положительное значение, а в тепловом — отрица¬
тельное. Выяснение этого явления позволило установить,
что частично получаемый кальций вступает в реакцию
с электролитом и образуется субхлорид:
Са + СаС12 = 2СаС1,
который на аноде окисляется до хлорида. Это снижает
выход кальция по току, но увеличивает количество вы¬
деляющейся теплоты — она выделяется не только за счет
прохождения тока через электролит, но и за счет реак¬
ции окисления субхлорида.
Из результатов теплового баланса можно определить
коэффициент использования энергии — это процентное
б
отношение количества энергии, которое необходимо тео¬
ретически затратить на получение массовой или объем¬
ной единицы продукта Дот, к количеству практически за¬
траченной энергии шпр:
= 100%.
^пр
Если речь идет о тепловой энергии, то указанное от¬
ношение называют тепловым к. п. д.
В примерах по составлению материальных и энерге¬
тических балансов для облегчения решения задач дают¬
ся готовые формы таблиц с перечислением статей ба¬
ланса.
ГЛАВА II
МЕТОДЫ ОБОГАЩЕНИЯ РУД
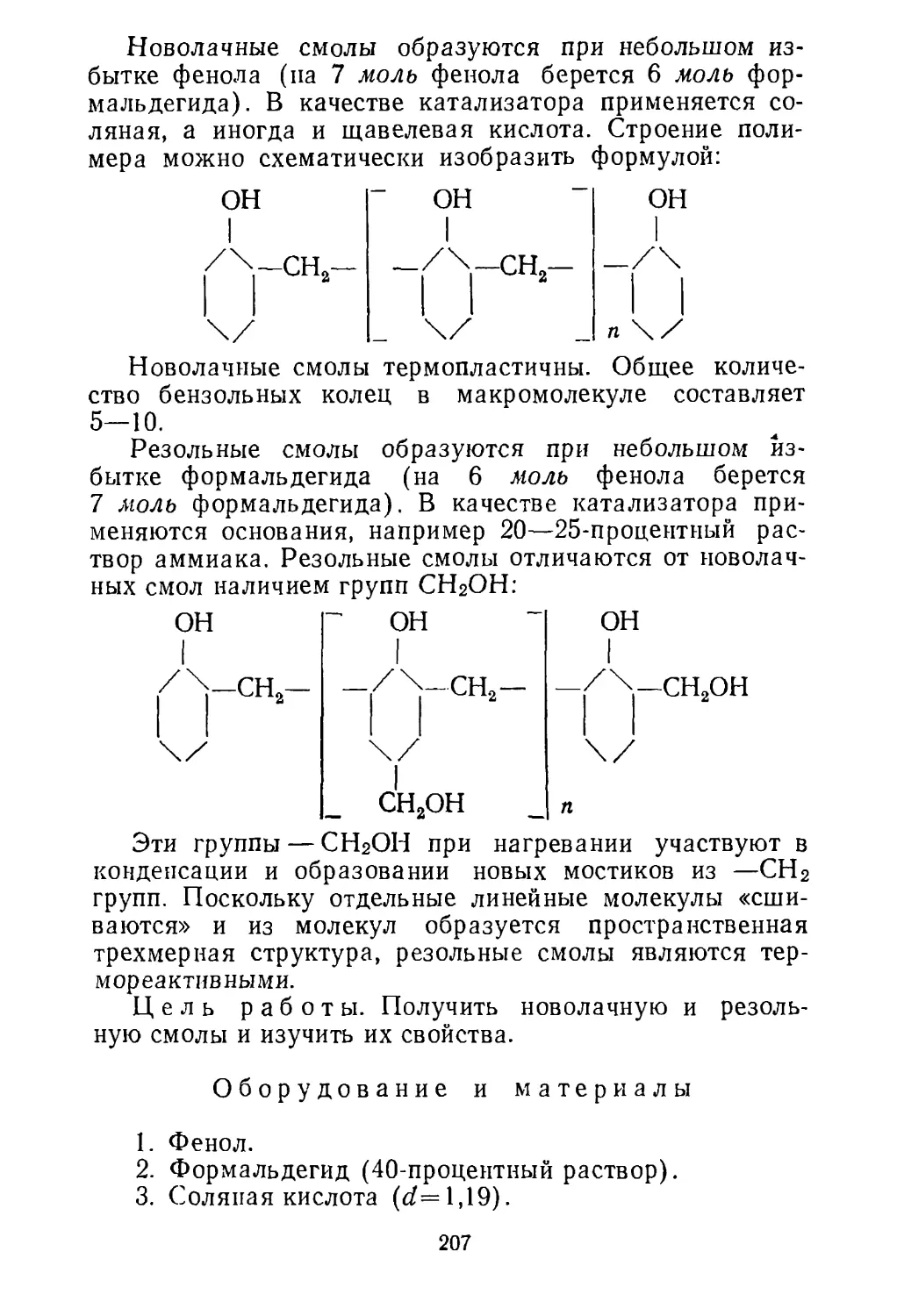
В настоящее время непосредственной переработке
подвергают очень ограниченное число руд и большин¬
ство из них предварительно обогащают, т. е. полезные
минералы по возможности отделяют от пустой породы.
Это позволяет использовать бедные руды, содержащие
небольшие количества полезного элемента.
При обогащении руд используется их способность
смачиваться определенными веществами, магнитные
свойства, плотность и т. д. Метод обогащения эффек¬
тивен в том случае, когда те или иные свойства отдель¬
ных минералов и пустой породы различны.
В настоящее время для обогащения наиболее часто
применяют флотацию; меньшее место занимают магнит¬
ная и электрическая сепарация и гравитационный метод.
В результате обогащения руд получают концентра¬
ты. Отходы производства, содержащие пустую породу и
промежуточные продукты, подвергают вторичной пере¬
работке. Концентраты содержат 20—30%, а иногда и
больше необходимого минерала.
При обогащении руд большое экономическое значе¬
ние имеют выход концентрата, степень извлечения и сте¬
пень концентрации ценного элемента.
Выходом концентрата называется процентное отно¬
шение массы полученного концентрата к массе взятой ру¬
ды. Степенью извлечения называется процентное отно¬
шение количества извлекаемого элемента в концентрате
к его количеству в руде. Степенью концентрации назы¬
вается отношение содержания элемента в концентрате
к его содержанию в руде.
8
Пример. При обогащении 6 т руды, содержащей
2% цинка, получено 350 кг концентрата, содержащего
25% цинка. Определить выход концентрата, степень из¬
влечения цинка и степень концентрации. Выход концен¬
трата составляет:
350 юо
6000
5,8
В 6 т руды имеется 120 кг цинка, а в концентрате —
87,5 кг. Отсюда степень извлечения цинка в концентрате
составляет:
87,5 • 100
120
72,9 (%).
Степень концентрации цинка равна:
= 12,5 раза.
§ 2. ФЛОТАЦИОННОЕ ОБОГАЩЕНИЕ
Флотационное обогащение основано на избиратель¬
ной смачиваемости поверхности руды поверхностно¬
активными веществами, называемыми флотореагентами.
Частица руды, адсорбировавшая флотореагент, пере¬
стает смачиваться водой и при продувании взвеси «при¬
липает» к пузырькам воздуха. Поскольку пузырьки
воздуха вместе с прилипшими частицами минералов
обладают в среднем меньшей плотностью по сравне¬
нию с окружающим раствором, они всплывают наверх,
образуя пену.
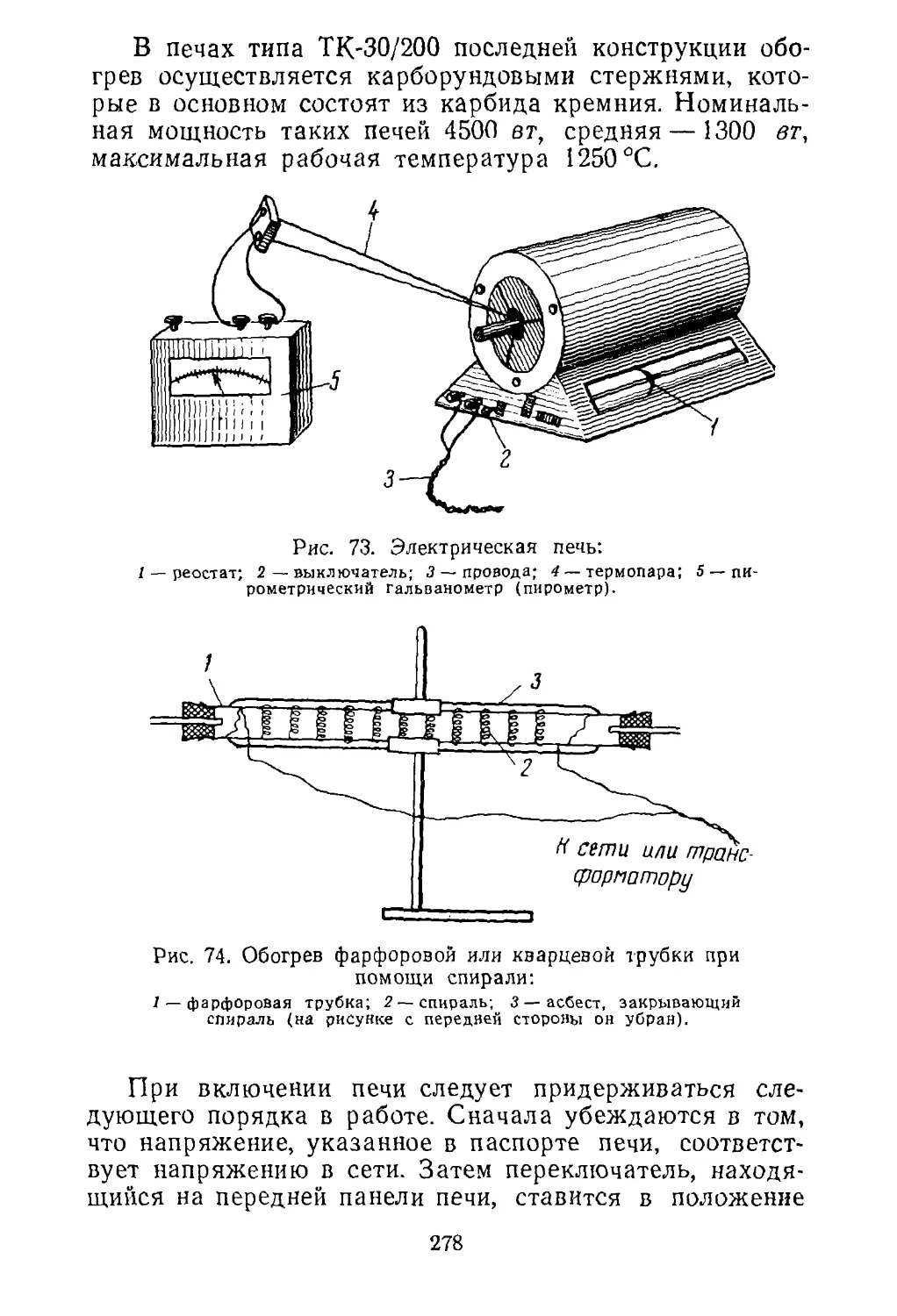
В состав флотореагента входят вещества, называе¬
мые коллекторами и пенообразователями. Коллекторы
адсорбируются поверхностью флотируемого минерала.
Молекулы пенообразователя адсорбируются на поверх¬
ности пленок, образующих воздушные пузырьки, резко
увеличивают устойчивость последних и предупреждают
их слияние и разрушение на поверхности воды. При
соприкосновении минеральной частицы с таким воздуш¬
ным пузырьком и происходит их взаимное прилипание
(см. рис. 1).
Эффективность и скорость флотации будет повы¬
шаться при достаточно сильном продувании пульпы
воздухом.
9
В качестве флотореагентов используются различные
органические вещества и их смеси. Хорошо действуют
пихтовое, сосновое, эвкалиптовое масла, которые можно
примпять без добавок дру¬
гих веществ.
В качестве флотореаген¬
тов можно использовать
также некоторые органиче¬
ские кислоты, например оле¬
иновую кислоту, продукты
окисления парафина, содер¬
жащие карбоновые кислоты
и оксикислоты, ксантогена-
ты ROCSSNa, аэрофлоты
(RO)2 PSSNa др.
Флотореагенты расходу¬
ются в очень небольших количествах (10—200 г на 1 т
руды), так как на поверхности флотируемых частичек
минерала часто создается только мономолекулярная
пленка флотореагента.
Как указывалось, частички минерала удерживаются
в образующейся пене. Пенообразователи: сапонины, не¬
очищенный крезол и др.— снижают поверхностное натя¬
жение воды. Однако сильное снижение поверхностного
натяжения воды, которое происходит при наличии в во¬
де таких пенообразовате¬
лей, как мыла, часто при¬
водит к тому, что пена
оказывает «пустой», т. е.
не содержит частичек
флотируемого минерала.
Происходит это потому,
что снижение поверхност¬
ного натяжения на гра¬
нице вода — воздух вызывает лучшее смачивание части¬
чек минерала водой, что и препятствует флотации. Пра¬
вильный подбор пенообразователя также имеет большое
практическое значение.
Частички флотируемой руды, адсорбировавшей фло-
тореагент, могут удерживаться на поверхности воды и
без пенообразователя за счет плохой смачиваемости их
водой и за счет своеобразного «продавливания» поверх¬
ности воды (рис. 2).
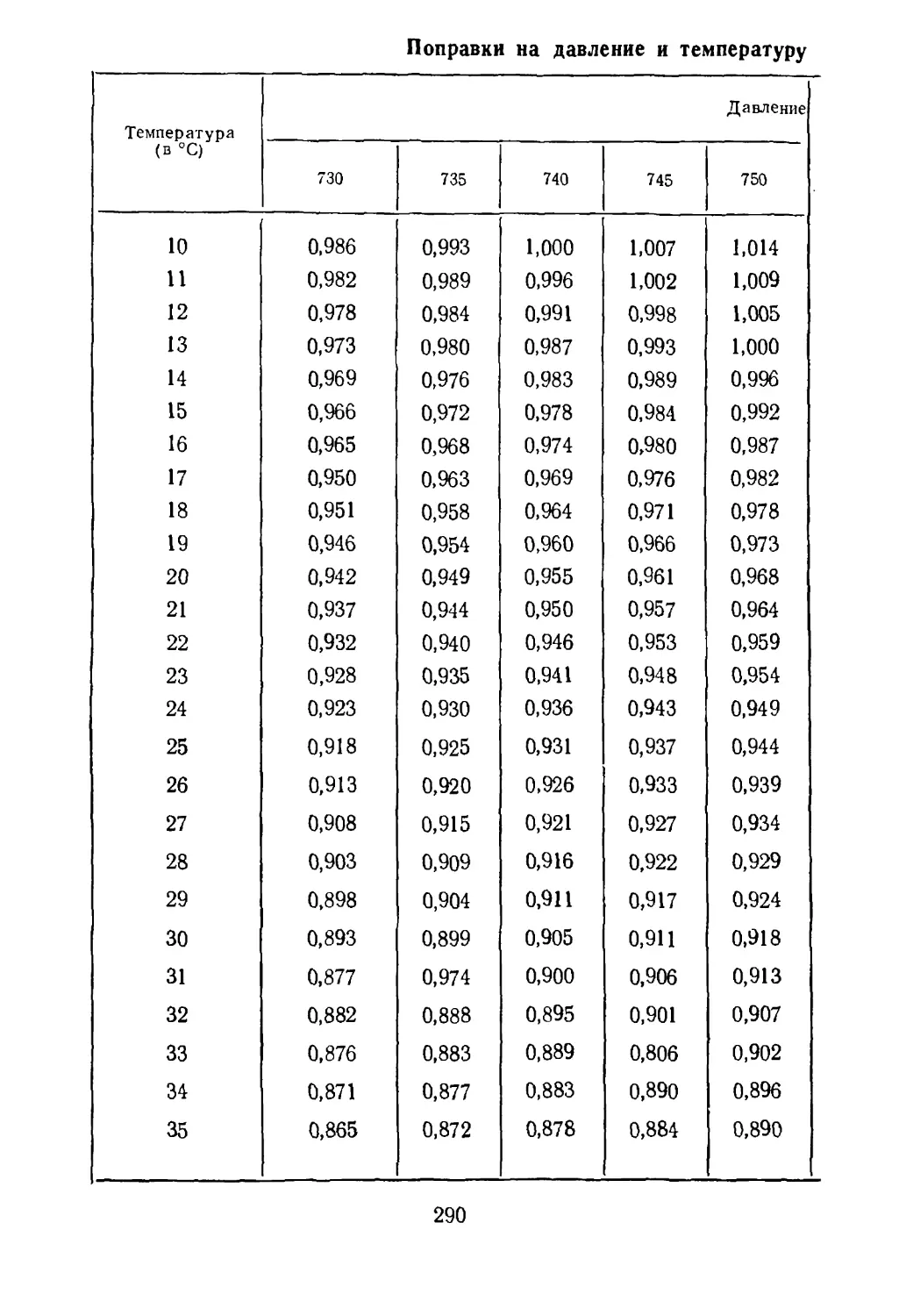
Рис. 2. Частицы флотационной ру¬
ды, плавающей на поверхности
воды.
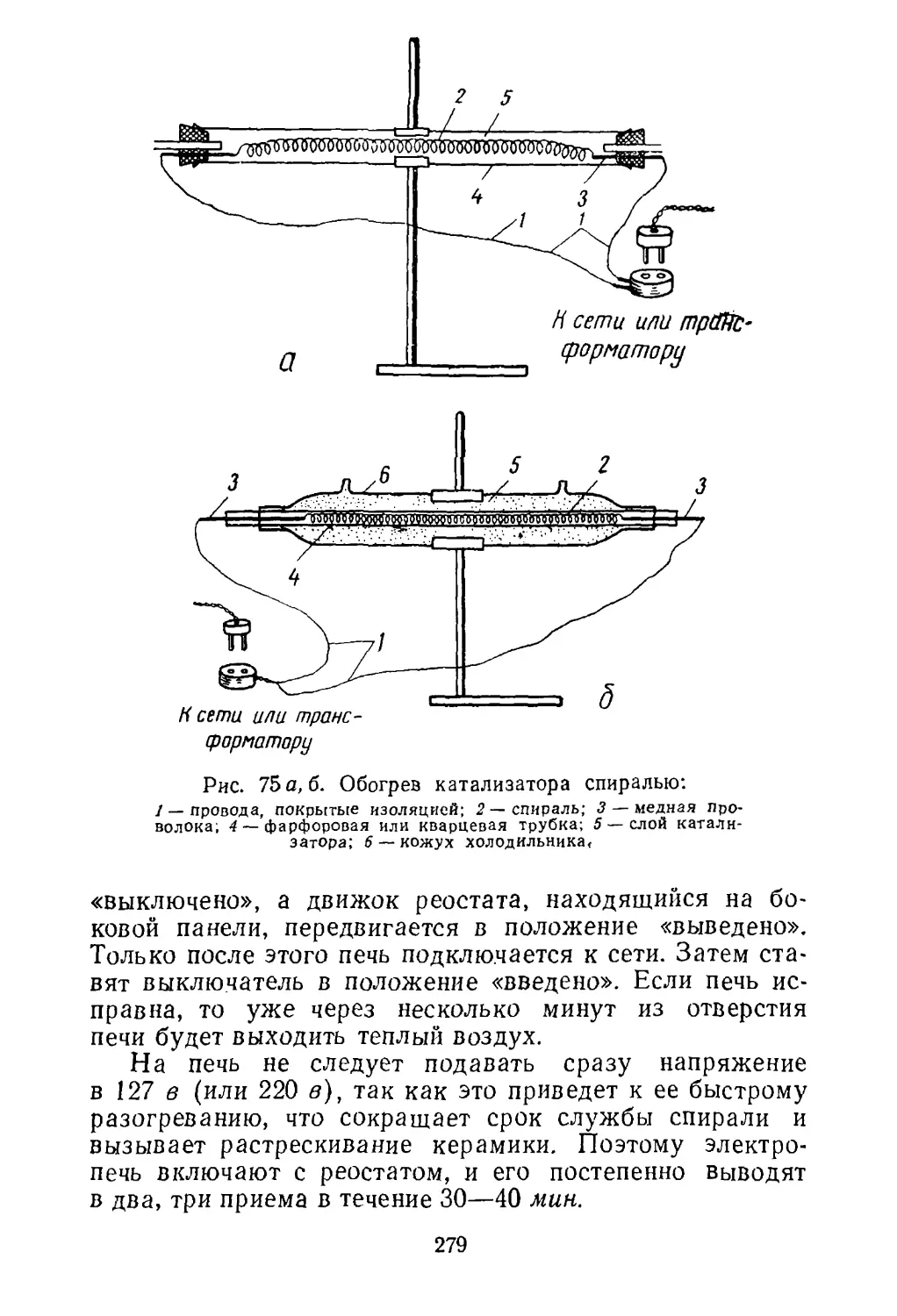
Рис. 1. Схема флотации:
1 — пузырек воздуха, окруженный
молекулами поверхностно-активно¬
го вещества; 2 — частицы флоти¬
руемой руды; 3 —молекулы по¬
верхностно-активного вещества.
10
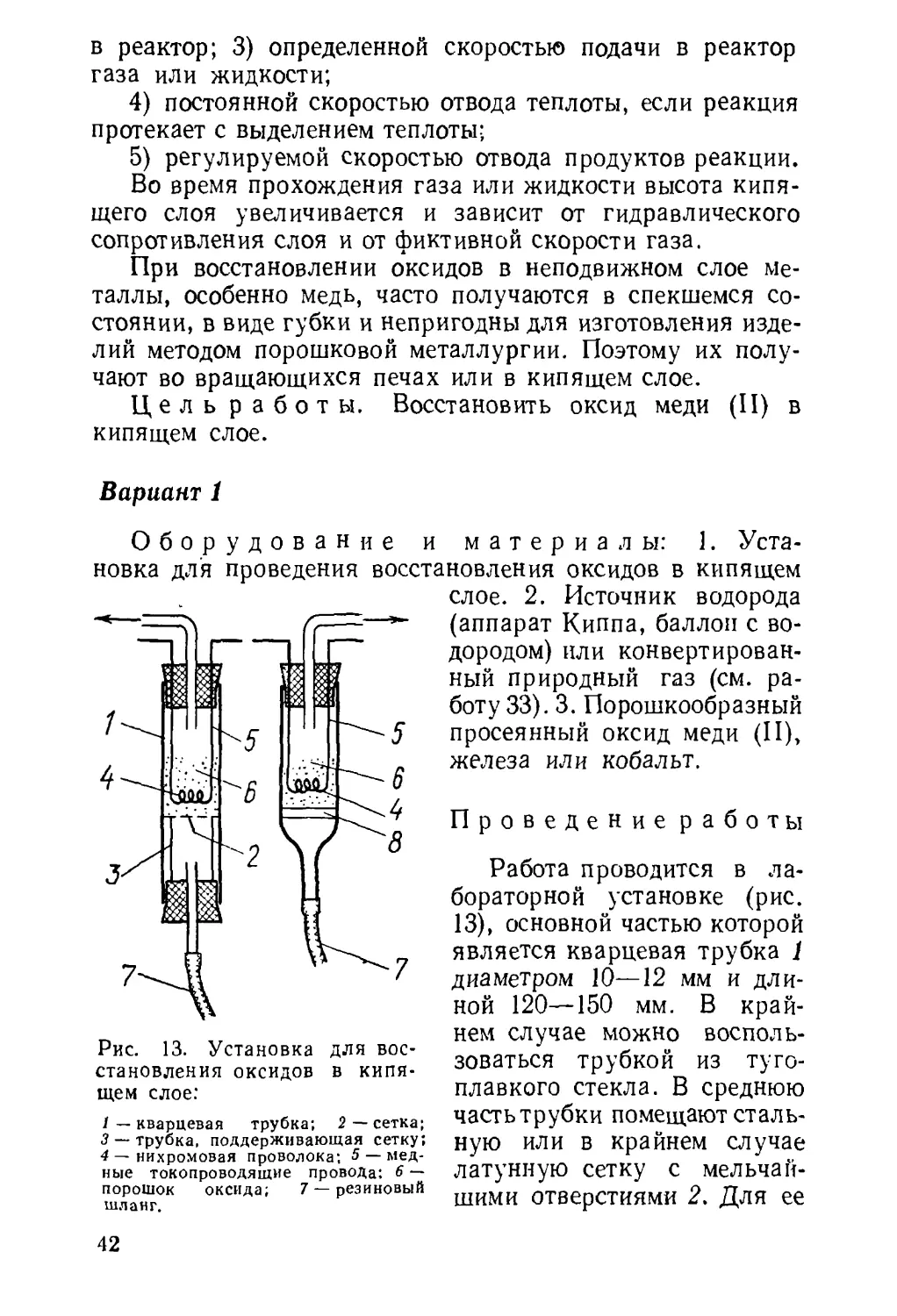
Работа 1
Флотационное обогащение медной сульфидной руды
Цель работы. Провести флотацию железного или
медного колчедана. Определить выход концентрата, сте¬
пень извлечения металла и степень концентрации.
Оборудование и материалы
1. Сосуд для флотации — колба или цилиндр на
250 мл с пробкой.
2. Флотореагент (репейное или сосновое масло, ксан-
тогенат или азрофлот).
3. Сульфидная руда (медная или железная) или
искусственно приготовленная смесь сульфида с песком.
4. Металлическая и фарфоровая ступки.
5. Сито с отверстиями 0,1—0,05 мм.
6. Аптекарские или технохимические весы с разно¬
весом.
7. Воронка Бухнера с отсосом.
8. Сушильный шкаф.
Проведение работы
Крупные куски руды измельчают в металлической
ступке, а затем растирают в фарфоровой до тончайшего
порошка. Полученный порошок просеивают через мел¬
кое сито. Частицы руды размером 0,1 мм подвергают
повторному дроблению.
Для флотации берут 2,5—3 г руды, содержащей
4—6% сульфида меди или железа 1.
Руду высыпают в цилиндр на 250—300 мл, наливают
100—150 мл воды и добавляют 2—3 капли флотореа-
гента — репейного или соснового масла, или ксантогена-
та, или азрофлота 1 2.
1 При отсутствии руды применяют искусственно приготовлен¬
ную смесь (см. ниже).
2 Рекомендуемый ииогда в методической литературе скипидар
обладает по отношению к сульфидам плохими флотационны¬
ми свойствами. Но его можно применять для флотации гра¬
фита.
11
Цилиндр закрывают пробкой и смесь энергично
взбалтывают в течение 0,5—1 мин до тех пор, пока не¬
которая часть сульфидов не всплывет на поверхность
водного слоя. Через 1—2 мин в сосуд по стенке нали¬
вают воду, которая смывает сульфиды в воронку с пори¬
стой пластинкой или в воронку Бухнера с фильтроваль¬
ной бумагой. Нужно стремиться, чтобы песчинки, кото¬
рые иногда пристают к стенкам сосуда, не попадали на
фильтр. Поскольку после первого взбалтывания всплы-
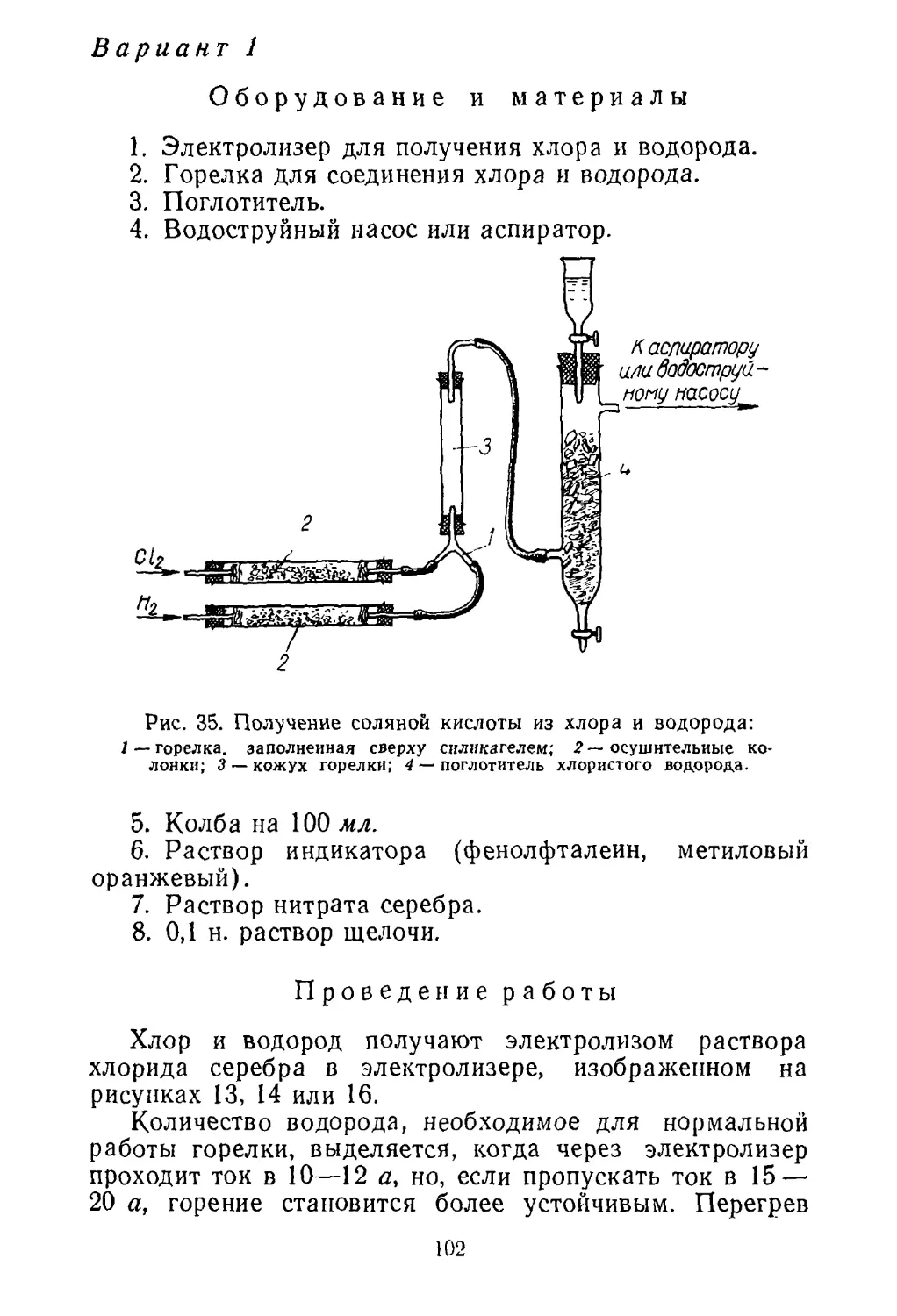
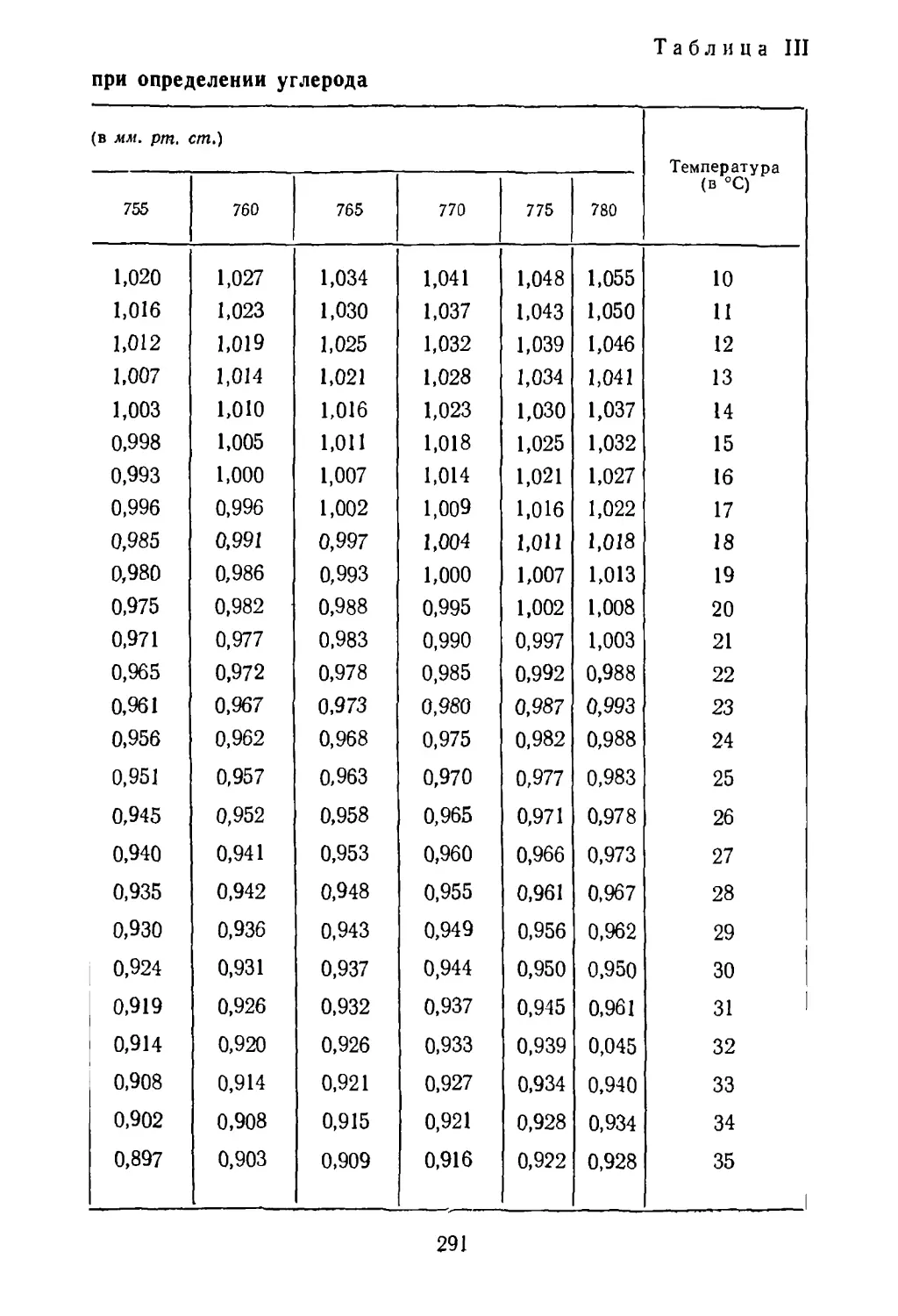
Рис. 3. Флотация руды:
1 — сосуд для флотации; 2— колба с воронкой для фильтрования;
3 — осушительные колонки с хлоридом кальция; 4 — иасос для со¬
здания вакуума.
вает только часть сульфидов, операцию флотации повто¬
ряют еще два-три раза. Необходимый вакуум на ворон¬
ке Бухнера создают водоструйным насосом или насосом
Комовского. Общий вид установки для фильтрования
приведен на рисунке 3.
Отфильтрованные сульфиды вместе с фильтром вы¬
сушивают при 80—100° С, взвешивают и проводят не¬
обходимые расчеты.
Результаты опыта оформляют в виде таблицы.
Оформление результатов работы
Взято руды (в г)
Получено
Выход
Степень извле¬
чения металла
(в %)
Степень кон¬
центрации (во
сколько раз)
концентрата
(в г)
концентрата
(в %)
12
Данные о процентном содержании сульфида в руде,
необходимые для расчета, берут у преподавателя. Со¬
держание металла в концентрате определяют расчетным
путем, считая, что отфильтрованное вещество является
чистым сульфидом.
При отсутствии природной руды можно приготовить искусст¬
венную смесь, состоящую из сульфида меди или железа и пустой
породы, например кварцевого песка. Для получения сульфида меди
составляют смесь из 63 массовых частей порошкообразной меди или
меди, нарезанной кусочками размерами в 1—2 мм (медная проволо¬
ка или листовая медь), и 32 массовых частей растертой серы. Эту
смесь помещают в пробирку и нагревают до расплавления серы. Ре¬
акция протекает с выделением теплоты. Часть серы, не вступившая
в реакцию, выделяется из реакционной смеси и оседает на холод¬
ных стенках пробирки. После того как в реакцию вступит основ¬
ная часть меди и серы, пробирку еще раз нагревают до 200—300 °С
в течение 3—4 мин, смесь охлаждают и пробирку разбивают. К по-
лученнолгу продукту после отделения стекла добавляют 12—15 мас¬
совых частей серы, смесь растирают в ступке и нагревают в про¬
бирке при указанной температуре в течение 5—10 мин. После этого
пробирку разбивают, сульфид меди еще раз растирают в ступке до
тончайшего порошка, просеивают и смешивают с чистым песком,
беря его в таком количестве, чтобы в полученной смеси песка было
90—95%.
Вопросы и задачи
1. Что такое флотореагенты, как они классифицируются и ка¬
кое назначение имеет каждая группа?
2. Для чего при флотации через пульпу продувают воздух?
3. С какими флотореагептами производится флотация сульфид¬
ных руд, окислов металлов и нерудных ископаемых?
4. Капля воды на поверхности сульфидной руды не растекает¬
ся. Что произойдет, если в эту каплю добавить некоторое количест¬
во депрессора? Объясните наблюдаемое явление.
5. Как проводится флотация полиметаллических руд, например
свинцово-цинковых руд и медно-свинцово-цинково-пиритных руд?
6. При обогащении 10 тп медной сульфидной руды, содержащей
1,5% меди, получено 400 кг концентрата, содержащего 30% меди.
Определить степень извлечения меди и степень концентрации.
Ответ. Степень извлечения меди 80%, степень концентрации
20 раз.
7 При флотации 5 пг цинковой руды, содержащей 3% цинка, по¬
лучено 340 кг концентрата, содержащего 22% цинка. Определить
выход концентрата, степень извлечения цинка и степень концен¬
трации.
Ответ. Выход концентрата 6,8%, степень извлечения цинка 48%,
степень концентрации 7,3 раза.
ГЛАВА III
МЕТАЛЛЫ И НЕМЕТАЛЛЫ
§ 3. ПОЛУЧЕНИЕ ПРОСТЫХ ВЕЩЕСТВ ВОССТАНОВЛЕНИЕМ
ОКИСЛОВ
Окисные и сульфидные руды — сырье для получения
металлов и некоторых неметаллов. Сульфидные руды
перед выделением металла переводят в окисные. Для
этого измельченную руду обжигают в присутствии кис¬
лорода воздуха, например:
2ZnS + 302 = 2ZnO + 2S02.
При выделении простых веществ из окислов учиты¬
вают физические и химические свойства исходных и
получаемых веществ. Выбор восстановителя зависит от
прочности окислов. Данные по теплотам образования
для сравнения рассчитывают на 1 г-экв окисла (см.
табл. 3).
Наиболее прочными окислами являются окислы под¬
группы скандия, магния и щелочноземельных металлов.
Элементы с переменной валентностью образуют не¬
сколько окислов; наиболее прочные из них — окислы
низшей валентности, и поэтому возможность получения
металла или неметалла из окисла определяется проч¬
ностью низшего окисла. Например, хромовый ангидрид
находится в начале приведенного ряда прочностей и
должен легко восстанавливаться, что и наблюдается на
практике. Однако в результате восстановления полу¬
чается очень прочная окись хрома, которая может быть
восстановлена только такими активными восстановите¬
лями, как алюминий, магний и т. д.
14
Таблица 3
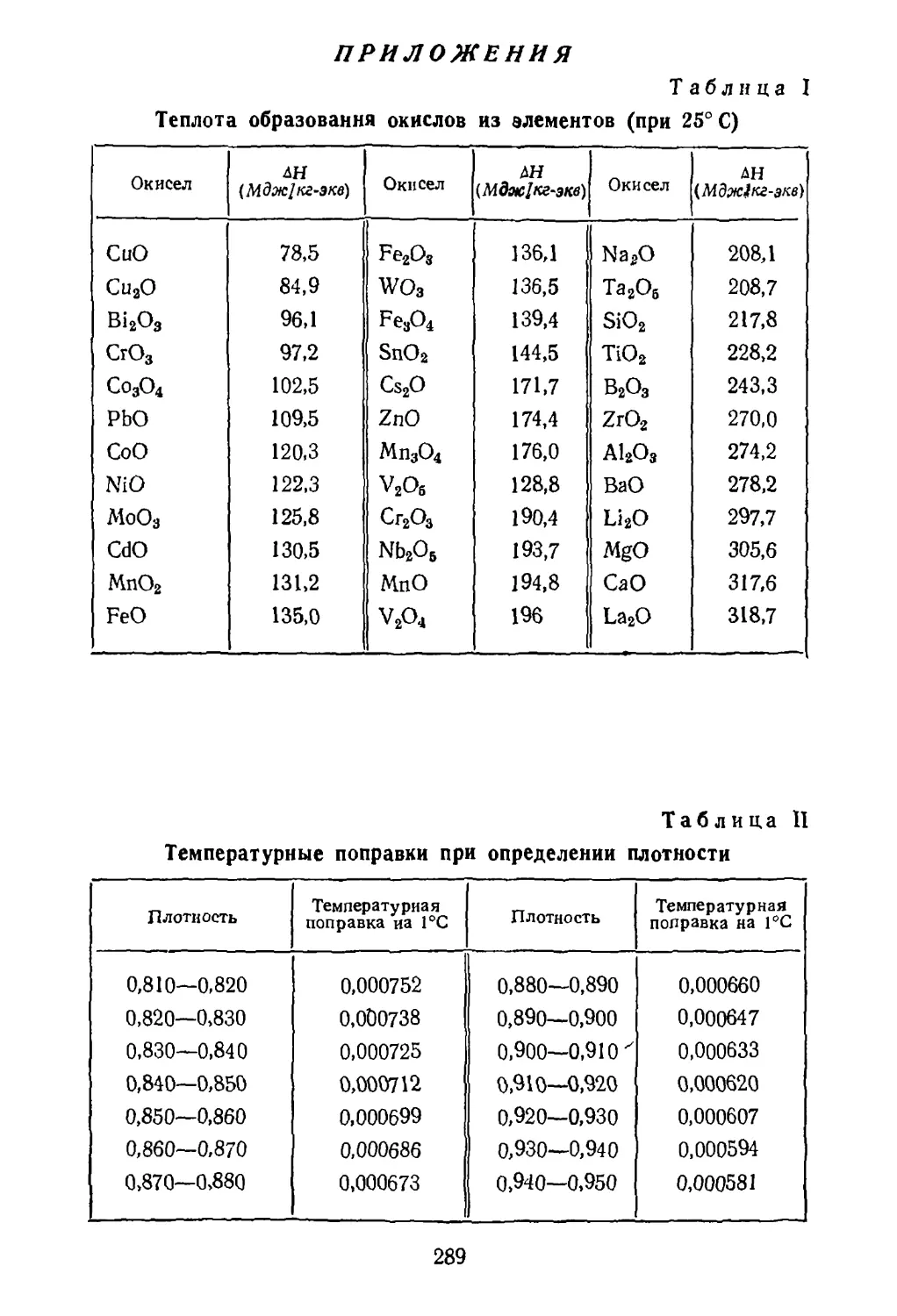
Теплота образования окислов из элементов (при 25иС)
Окисел
АН (в
кал/г-экв)
Окисел
AH
(в кал/г-экв)
Окисел
ж
(в кал/г-экв)
СиО
18 750
Fea03
32 530
Na20
49 730
Си20
20 300
WO3
32 620
Ta205
49 900
Bi203
22 970
Fe304
33 310
Si02
52 060
Сг03
23 220
Sn02
34 530
Te02
54 530
С03О4
24 500
Cs20
41 050
B203
58 170
РЬО
26 180
ZnO
41 680
Zr02
64 530
СоО
28 750
Mn304
42 060
A1203
65 550
NiO
29 200
v2o5
43 700
BaO
66 500
Мо03
30 070
Cr203
45 500
Li20
71 150
CdO
31 180
Nb206
46 300
MgO
73 050
Мп02
31 350
MnO
46 550
CaO
75 900
FeO
32 250
V204
46 880
La203
76 170
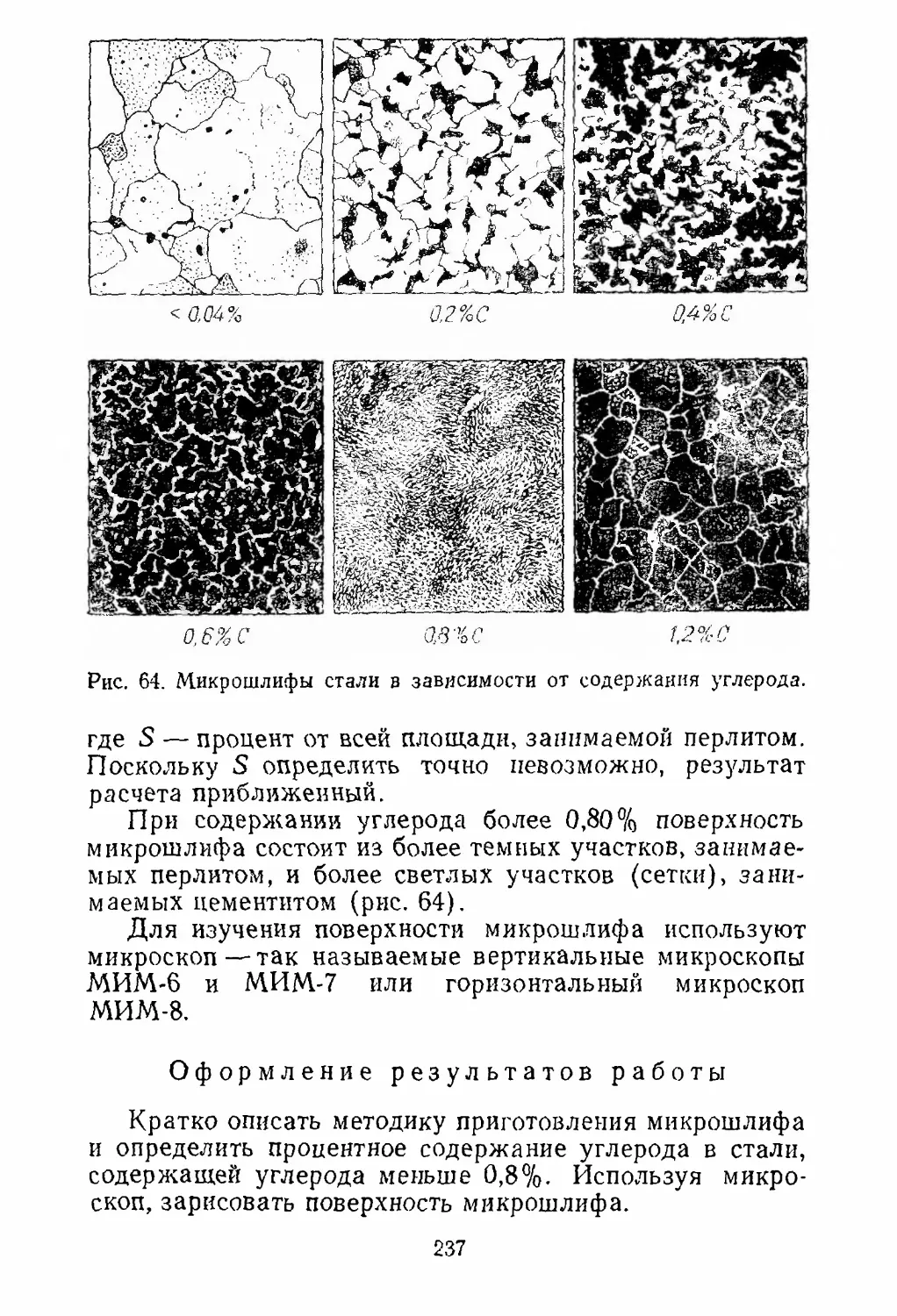
Двуокись марганца — непрочный окисел, однако при
восстановлении ее до металла образуются последова¬
тельно Мп20з, Мп304, МпО. Поскольку закись марган¬
ца— прочный окисел, марганец даже из такого непроч¬
ного окисла, как двуокись марганца, получить довольно
трудно, несмотря на то что двуокись марганца отщеп¬
ляет кислород при простом нагревании. Исключением
из этого правила является закись железа (см. ряд проч¬
ностей окислов), которая при обычных температурах
менее прочна по сравнению с другими окислами железа.
Особенно резко изменяется прочность окислов при
переходе из одного физического состояния в другое вос¬
станавливаемого окисла или получаемого вещества
(плавление, испарение, переход в другую кристалличе¬
скую модификацию), так как при этих процессах проис¬
ходит изменение свойств окислов или получаемых ве¬
ществ.
Прочность окислов меняется в зависимости от на¬
личия в окисле примесей других окислов. Например,
прочность окиси хрома в условиях ее восстановления
в смеси с окисью железа снижается. Объясняется это
15
тем, что железо, легко образующееся из его окиси, рас¬
творяет хром и уводит его из сферы реакции. Тем самым
равновесие смещается в сторону восстановления хрома.
Ниже рассматривается получение металлов из их
окислов с использованием в качестве восстановителей
алюминия, углерода и водорода.
Работа 2
Получение металлов и сплавов
металлотермическим методом
Реакции металлов с окислами, протекающие с выде¬
лением теплоты, называются металлотермическими.
В таблице 1 окислы расположены по увеличению их
прочности. Восстановительные свойства металлов меня¬
ются также в указанной последовательности.
Практическое применение в качестве восстановите¬
лей нашли наиболее активные элементы: кальций, маг¬
ний, алюминий и кремний. Реакции с участием этих
восстановителей соответственно называются кальцио-,
магнио-, алюминие- и силикотермическими.
Характер протекающих реакций определяется коли¬
чеством выделяющейся теплоты и температурами плав¬
ления получающихся продуктов. Если теплоты, выде¬
ляющейся во время реакции, не хватает для нагревания
получающихся продуктов выше температуры их плав¬
ления, то в результате реакции получается смесь окисла
и металла. Это наблюдается при использовании в каче¬
стве восстановителя магния или кальция, окислы кото¬
рых плавятся при 2800 и 2570 °С. Получающиеся ча¬
стички металла как бы изолированы тугоплавкими
окислами.
При восстановлении окислов алюминием теплоты
выделяется меньше, но окись алюминия по сравне¬
нию с окислами кальция и магния плавится значи¬
тельно ниже, при 2050 °С, поэтому она при восстанов¬
лении многих окислов получается в расплавленном виде.
Большинство металлов имеет более низкие температуры
плавления и большую плотность (по сравнению с
окисью алюминия), поэтому они плавятся, оседают на
дно тигля, при охлаждении получается кусок металла,
называемый корольком. То же наблюдается и при ис¬
6
пользовании кремния, хотя он как восстановитель зна¬
чительно слабее алюминия.
Магний и кальций довольно дорогие восстановители,
и их применяют для выделения титана и циркония из
двуокисей или хлоридов этих металлов.
Алюминий можно применять для восстановления
Fe203, F304, СоО, С03О4, NiO, Мп304, Мо03 и V205.
Сравнительно нестойкие окислы Мп02, Сг03 при вос¬
становлении алюминием разлагаются, и реакционная
смесь сильно разбрасывается.
Для получения двухкомпонентных сплавов берут
смесь двух окислов, например, для получения сплава
марганца с железом нужно взять смесь Fe203 и Мп304
или смесь Fe304 и Мп304. Количество алюминия рассчи¬
тывается в зависимости от массы и состава смеси окислов.
При восстановлении алюминием прочных окислов,
как например Ti02, Zr02, Nb2Os, Ta205, Cr203, Si02, B203,
добавляют легко восстанавливаемый окисел, который по¬
вышает тепловой эффект реакции. В таблице 4 приво¬
дятся данные о составе смеси окислов, которые при
алюминотермическом восстановлении дадут проплавлен¬
ный королек сплава. Легко восстанавливаемые окислы
взяты в минимальном количестве. Поэтому реакции,
как правило, проходят весьма медленно и значительная
Таблица 4
N° п/п
Состав
N° п/п
Состав
1
11 % СЮз+89% Сг203
12
55% Со304 + 45% ТЮ2
2
37% МоОз+63% Сгг03
13
50% Fе202 + 50% Ti02
3
37% Fe203+63% Сг203
14
60% Fe304 + 40% Ti02
4
50% СозО4+50% Сг203
15
60% V206 + 40% Ti02
5
50% СоО+50% Сг203
16
55% NiO + 45% Ti02
6
55% NiO+45% Сг203
17
70% Mn304 + 30% Ti02
7
75% Fe203+25% В203
18
70% M0O3 + 30% Si02
8
80% Fe3O4+20% В203
19
75% CuO + 25 Si02
9
75% V205+25% В203
20
80% V205 + 20 Si02
10
80% СоО+20% В203
21
80% NiO + 20% Si02
11
85% Мп304+15%В203
22
90% Mn304 + 10% Si02
17
часть сплава всегда остается в шлаке. Увеличение ко¬
личества легко восстанавливаемого окисла на 3—5%
значительно улучшает процесс.
Количество выделяющейся теплоты рассчитывается
на 1 г шихты и называется удельным тепловым эффек¬
том реакции и обозначается буквой q.
Пример. Определить q реакции получения ферро¬
хрома из шихты, состоящей из 45% Fe203 и 55% Сг203.
Теплота образования окиси алюминия, окиси хрома и
окиси железа соответственно равна 1670, 1128 и
822,2 кдж/моль. При восстановлении 1 моль окиси хрома
Сг203 + 2А1 = 2Сг + А1203
теплоты выделяется 1670—1128 = 542 (кдж).
На 1 г шихты выделяется:
542 : (2 26,97 + 152,02) = 2,63 (кдж).
При восстановлении 1 моль окиси железа теплоты
выделяется 1670 — 822,2 = 847,8 (кдж) или на 1 г шихты
847,8: (2-26,07+ 159,7) =4 (кдж.).
Поскольку при получении феррохрома использова¬
лась шихта, в которой было 45% окиси железа, удельный
тепловой эффект реакции равен:
g = + 2,63 = 3,24 (кдж).
Точный состав полученного сплава можно устано¬
вить химическим анализом. Состав с точностью до 1 —
2% можно рассчитать теоретически, исходя из состава
взятой смеси окислов. Например, из смеси окислов, со¬
ставленной из 25 г Fe203 и 25 г Сг203, теоретически по¬
лучится 17,49 г железа и 17,11 г хрома и сплав, следо¬
вательно, будет состоять на 50,55% из железа и на
49,45% из хрома. Практически хрома в сплаве будет
примерно на 2% меньше, так как хром восстанавлива¬
ется несколько труднее железа и окись хрома в неболь¬
ших количествах остается в шлаке.
Состав двухкомпонентного сплава можно рассчитать
по формуле:
А2 п2 %Ока
%М*1 = '~А1 щ %0Kl А2 п2 %Ок2 100,
М1 + м2
18
где МI и М2— молекулярные массы первого и второго
окислов;
Aj и А2— атомные массы первого и второго метал¬
лов;
п 1 и п2 — количество атомов в молекуле первого
и второго окислов;
%Oki и %Ок2 — процентное содержание первого и вто¬
рого окислов в шихте.
В промышленности с помощью алюминотермии по¬
лучают безуглеродистые металлы: хром, молибден и ва¬
надий и ферросплавы хрома, молибдена, вольфрама, ва¬
надия, ниобия, титана и циркония.
Цель работы. Получить алюминотермическим
методом один из металлов или двухкомпонентный сплав.
Определить выход в процентах металла или сплава.
Рассчитать теоретический состав сплава и удельный
тепловой эффект реакции. Приготовить глиняный тигель
для проведения реакции.
Оборудование и материалы
1. Прокаленный окисел выбранного вами металла 1.
2. Порошкообразный алюминий.
3. Шамотный или глиняный тигель объемом
50—70 см3.
4. Перекись бария.
5. Лента или проволока магния.
6. Подставка с песком.
7. Аптекарские весы с разновесом.
Проведение работы
Работа по алюминотермическому получению метал¬
лов и сплавов включает следующие операции:
1. Подготовка материалов.
2. Отвешивание шихты.
3. Проведение реакции.
4. Определение выхода продукта.
Для опыта нужно брать 20—50 г сухих и измельчен¬
ных окислов, отдельные частички которых должны быть
1 Конкретное задание для получения металла (Fe, Со, Ni, Мп,
Cr, Mo, V) или сплава необходимо взять у преподавателя.
19
не более 0,5 мм (высушивание окислов проводится при
200—300 °С).
Если получают двухкомпонентный сплав, то количе¬
ство окислов берется с учетом данных, приведенных
в таблице 2. Например, для получения феррохрома (на
20 г смеси) необходимо взять 7,4 г Fe203 и 12,6 2 Сг203,
что и составляет 37% Fe203 в смеси. Увеличение количе¬
ства легко восстанавливаемого
окисла в смеси облегчает про¬
ведение реакции. Число грам¬
мов алюминия рассчитывается
в соответствии с равенством
реакции. Например, на 20 г
смеси 50% Fe203 и 50% Сг203
нужно взять 6,92 г алюминия.
Вещества отвешиваются на
технохимических или аптекар¬
ских весах с точностью до
0,01 г, тщательно перемешива¬
ются шпателем на листе бума¬
ги (или в ступке) и пере¬
сыпаются в тигель. Верх ших¬
ты засыпается тонким сло¬
ем (1—2 мм) зажигательной
смеси (рис. 4), которая состав¬
ляется из 9 массовых частей
перекиси бария и 1 массовой части порошкообразного
алюминия. За неимением перекиси бария зажигательную
смесь составляют из 2,25 массовых частей растертой в
ступке калиевой селитры и 1 массовой части алюминие¬
вой пудры. Зажигательную смесь перемешивают и пере¬
тирают в ступке.
Загорается смесь от ленты магния, которую поджи¬
гают горелкой или лучинкой. Тигель помещают на слой
песка.
Реакция проходит с сильным разбрасыванием раска¬
ленных частичек, поэтому ее нужно проводить в вы¬
тяжном шкафу, убрав из него все воспламеняющиеся
жидкости и материалы. Вызвать реакцию можно и дру¬
гим путем: зажечь ленту магния, зажав ее щипцами, и
тут же положить горящий магний на смесь.
После охлаждения полученных продуктов тигель
разбивают и королек металла или сплава, находящийся
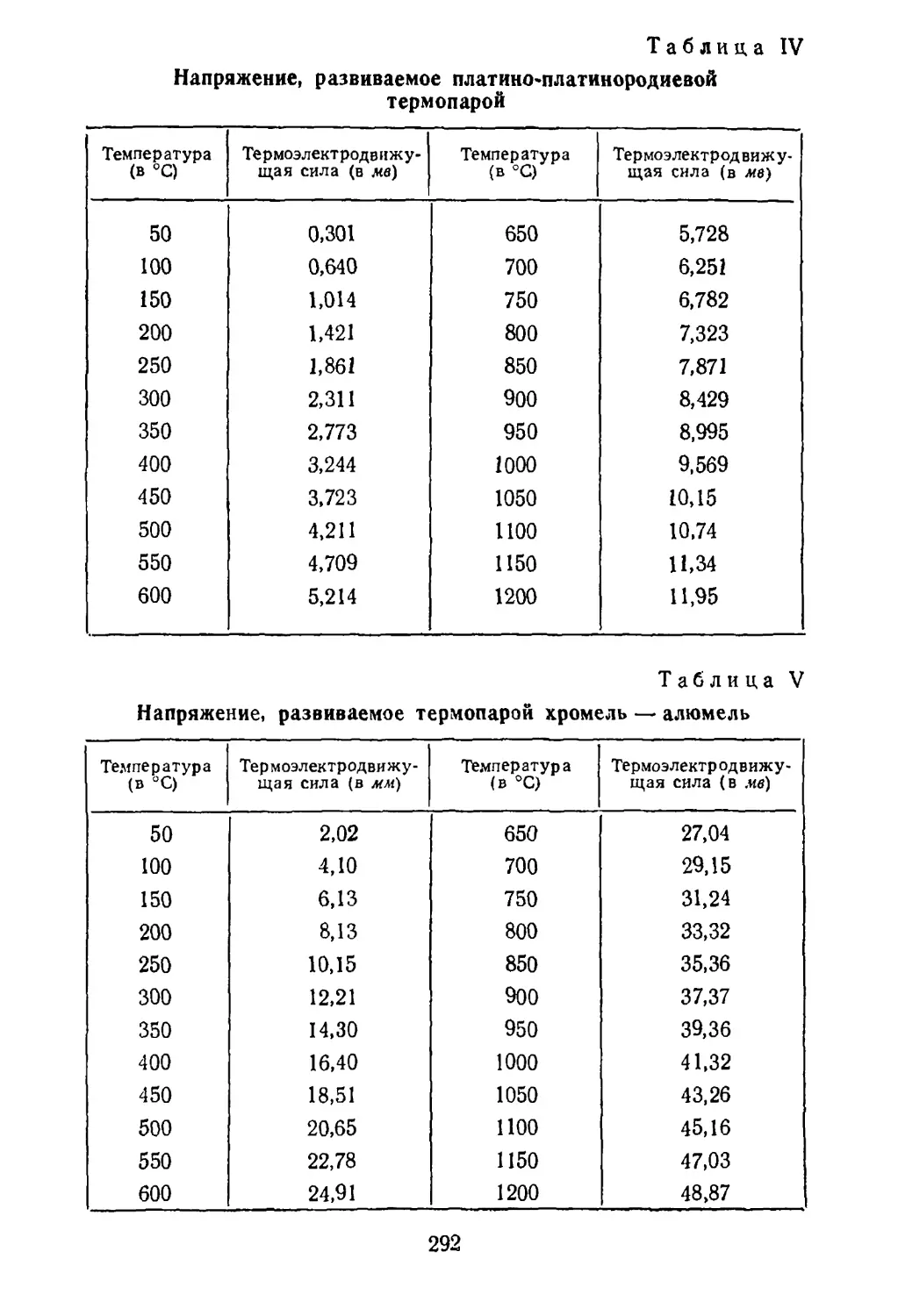
Рис. 4. Получение метал¬
лов и сплавов алюмино
термическим способом:
/ — тигель; 2 — шихта;
3 — зажигательная смесь;
4 — лента магния.
20
на дне тигля, отбивают молотком па бруске железа от
шлака и взвешивают. Если сплавы при ударе расколо¬
лись, то собирают и взвешивают отдельные их кусочки.
Выход продукта составляет обычно 60—80% от тео¬
ретически возможного. Для определения выхода про¬
дукта полученный металл или сплав взвешивают и на
основании количества взятых окислов проводят вычисле¬
ния. Например, из 50 г смеси 37% Fe203 и 63% Сг203
получается 23,9 г сплава, а должно теоретически полу¬
читься 33,32 г. Отсюда выход составляет 71,71% от
теоретически возможного.
Результаты опытов оформляют в виде таблицы.
Состав смеси
Взято алю¬
миния (в г)
Получено
сплава (в г)
Должно
получиться
сплава (в г)
Выход
сплава
(в %)
q
реак¬
ции (в
кал (г)
(в %)
1 (В г)
Окислы, необходимые для проведения работы, можно пригото¬
вить из солей н других веществ, имеющихся в лаборатории.
Закись-окись марганца приготовляется прокаливанием двуокиси
марганца около 950 6С. Молибденовый ангидрид готовится прокали¬
ванием молибдата аммония при 600—700 °С. Если на дне тигля при
накаливании получается коричневый порошок (двуокись молибде¬
на) или порошок темно-синего цвета (пятиокись молибдена), то
образующийся продукт нужно смочить небольшим количеством азот¬
ной кислоты и еще раз прокалить, чтобы окислить эти окислы в мо¬
либденовый ангидрид, имеющий светло-желтый цвет. Пятиокись ва¬
надия получается прокаливанием ванадата аммония при 500—
550 °С. Если же на дне тигля порошок будет иметь черный цвет
(примесь четырех- и трехокиси ванадия), то его нужно перемешать
и еще раз прокалить до образования порошка светло-коричневого
цвета.
Закись кобальта и закись-окись кобальта готовятся прокалива¬
нием окиси кобальта при 500 и 950 °С. Закись никеля получается
прокаливанием окиси никеля при 950—1000 °С. Двуокись марганца,
окислы никеля и кобальта для получения сплавов металлотермиче¬
ским путем не применяют, так как во время реакции сильно раз¬
брасывается смесь.
Для работы наиболее пригодны шамотные тигли.
Заменить их можно глиняными самодельными тиглями.
Для их изготовления глину нужно предварительно замо¬
чить, добавив в нее !/з (примерно) песка, и все тща¬
тельно перемешать. Глина должна быть пластична.
Работа производится ручным способом.
21
Сделанные тигли подсушиваются на воздухе в тече¬
ние 2—3 дней, а затем прокаливаются в сушильном
шкафу при температуре не ниже 150—200 °С
Вопросы и задачи
1. Чем определяется восстановительная способность металлов
по отношению к окислам и по отношению к ионам металлов в
растворе?
2. При нагревании смеси натрия и окиси лития устанавливается
равновесие:
2Na + L i20^> Na20 + 2Li.
В какую сторону смещено равновесие? При ответе учтите те¬
плоты образования окислов.
3. Что произойдет при нагревании смеси порошкообразного желе¬
за с хромовым ангидридом?
4. Протекает реакция:
ЗСаО + 2AI ^ А1203 + ЗСа.
В какую сторону сместится равновесие при высоких темпера¬
турах (~1200°С) в вакууме и при нормальном давлении?
5. Какие металлы можно получить алюминотермическим путем?
6. В результате реакций между окисью хрома и алюминием по¬
лучается смесь, состоящая из окиси алюминия и мельчайших кусоч¬
ков хрома. Металлический хром получается в виде куска только
в том случае, если окись хрома предварительно нагреть до 500—
600 °С или добавить к ней определенное количество хромового ан¬
гидрида. Какую роль играет добавление хромового ангидрида в
шихту или ее предварительный нагрев?
7. В результате алюминотермического восстановления окиси
железа получается (теоретически) по массе 54,42% железа и 45,58%
окиси алюминия. Определите: 1) среднюю удельную теплоемкость
полученной смеси, если средняя удельная теплоемкость железа в
пределах от 0 до 2500 °С равна 0,000878 дж/г, а окиси алюминия
1,67 дж/г\ 2) количество теплоты, выделяющееся на 1 г смеси;
3) температуру горящего термита, если тепловые потери составля¬
ют 15%.
Ответ. 0,00124 дж/г\ 3,88 дж/г\ 2650 °С.
8. При восстановлении окислов железа, хрома и других метал¬
лов магнием или кальцием теплоты выделяется больше, чем при вос¬
становлении их алюминием. Однако при восстановлении указанных
окислов магнием или кальцием металлы получаются в- виде неболь¬
ших крупинок, а при восстановлении алюминием — в виде сплавлен¬
ного куска. Чем объясняется данное явление? При ответе нужно
учесть, что окись алюминия плавится при 2050 °С, окись магния —
при 2800 °С, а окись кальция — при 2570 °С.
9. Сколько нужно взять алюминия для восстановления 50 г сме¬
си окислов, состоящей из 40% окиси железа и 60% окиси хрома?
Сколько должно теоретически получится феррохрома?
Ответ. А1 — 17,39 г; Fe—Сг — 34,50 г. 11 Изготовленный тигель для сушки передается лаборанту, а
взамен его студент получает готовый просушенный тигель.
22
10, Сколько выделится теплоты при восстановлении алюминием
100 г смеси окислов, составленной из 50% окиси железа и 50% дву¬
окиси титана?
Ответ. 2,74 дж/г.
Примеры на составление материального баланса.
Составьте материальный баланс реакции алюминотермического
восстановления 10 кг смеси, состоящей из 40% окиси железа и 60%
окиси хрома.
В результате реакции получено 13,390 кг продуктов реакции. Из
шлака выделено 5,412 кг феррохрома. При расчете необходимо
учесть, что некоторая часть сплава остается в шлаке в виде мель¬
чайших включений, а часть шлака во время реакции разбрызгивает¬
ся и испаряется.
1) Определяем количество алюминия, необходимое для восста¬
новления 10 кг смеси окислов.
Смесь состоит из 4 кг окиси железа и 6 кг окиси хрома. Исходя
из уравнения реакций восстановления окислов, находим, что на вос¬
становление окиси железа необходимо 1,351 кг алюминия и на вос¬
становление окиси хрома — 2,129 кг алюминия.
2) Из 4 кг окиси железа и 6 кг окиси хрома должно получиться
по уравнению реакции восстановления 6,901 кг сплава (феррохрома).
3) Потери сплава в шлаке составляют: 6,901 — 5,412= 1,489 (кг).
4) Из 3,480 кг алюминия должно получиться 6,576 кг окиси алю¬
миния. Всего получено шлака — окиси алюминия 13,390—
— (5,412+1,489) =6,489 (кг).
5) Для определения количества шлака, которое не учли, из об¬
щего количества взятых веществ вычитаем количество полученных
веществ. Неучтенные потери при составлении материального балан¬
са называется невязкой. Невязка составляет (см. таблицу стр. 23):
13,480 - 13,390=0,090 (кг).
11. Составьте материальный баланс реакции алюминотермнче-
ского восстановления 100 кг смеси, состоящей из 50% окиси железа
и 50% двуокиси титана. К указанной смеси для снижения вязкости
шлака добавлено 5 кг окиси кальция.
Масса продуктов реакции составляет 143 кг. После разделки
продуктов реакции выделено 40 кг ферротитана.
Таблица 5
Материальный баланс
Задано (в кг)
Получено (в кг)
Окись железа
4
Сплав
5,412
Окись хрома
6
Окись алюминия
6,489
Алюминий
1,351
Потери металла в шлаке
1,489
Алюминий
2,129
Невязка
0,090
Итого
13,480
Итого
13,480
23
Продолжение
Задано (в /СсД
Получено (в кг)
Окись железа
Двуокись титана
Алюминий
Окись кальция
Ферротитан, в том числе:
Железо
Титан
Алюминий
Шлак, в том числе:
Трехокись титана
Окись алюминия
Невязка
Итого
Итого
При ответе учтите, что часть титана остается в шлаке в виде
закиси титана ТЮ и трехокиси титана Т^Оз, которые с окисью алю¬
миния дают соответствующие алюминаты.
Для расчета нужно принять:
I) вся окись железа восстанавливается до железа; 2) весь не¬
восстановленный титан остается в шлаке в трехвалентном состоянии;
3) алюминий, который берется в теоретически необходимом количе¬
стве (вследствие частичного восстановления титана), остается в
свободном виде и переходит в виде металла в титан, в котором его
находится 5%. Неточность отнести за счет окиси алюминия.
12. Определите состав ферромарганца (в кг), если восстанов¬
лению углеродом подвергалось 2070 кг руды состава (в %):
Принимается, что 8% Мп перейдет в шлак, 12% испарится и
80% перейдет в металл. Расчет проведите на I т 79,2%-иого фер¬
ромарганца, содержащего 6,3% углерода. Восстановлению, помимо
марганца, подвергается железо, кремний и фосфор.
Ответ. Мп — 792 кг\ Fe — 136 кг; С—63 кг; Si — 6 кг; Р — 3%.
13. Сколько выделится теплоты при восстановлении 10 кг ших¬
ты, в состав которой входит алюминий и окислы, взятые в соотно¬
шении 10% СгОз и 90% Сг20з. Ответ выразите в кдж и ккал \
Ответ. 30 450 кдж; 4349 ккал. 11 1 калория равна 4,184 джоуля.
Мп — 47,92
Fe — 6,72
Р — 0,14
02 —25,55
Si02 — 6,50
остальное — 6,93
А1203 - 1,54
СаО —3,79
MgO —0,35
ВаО —0,56
24
14. Сколько выделится теплоты при восстановлении двуокиси
титана:
ТЮ2 + 2Са = 2СаО + Ti
Количество шихты 1 кг.
Ответ. 2026 кдж.
15. Расположите в убывающий ряд щелочные металлы на осно¬
вании их восстановительной активности по отношению к окислам
металлов.
16. Расположите в убывающий ряд литий, алюминий, кальций,
магний па основании их восстановительной активности по отноше¬
нию к окислам металлов. Для этого рассчитайте теплоты образо¬
вания окислов указанных элементов на 1 г-жа.
17. Нагрели смесь окиси кальция с барием и смесь кальция с
окисью бария. В каком случае прошла реакция и почему?
Ответ. Кальций восстанавливает окись бария.
18. При промышленном получении ферротитана восстановле¬
нием смеси закиси-окиси железа и двуокиси титана алюминием в
шихту добавляют около 10% окиси кальция, в противном случае
шлак содержит титан в виде Т120з и TiO. Частично он остается в
шлаке, а сплав содержит значительное количество алюминия. Какую
роль играет добавление окиси кальция?
19. Какие достоинства и недостатки имеются у таких восстано¬
вителей; как алюминий, кальцин, магшгй, кремний? При ответе учти¬
те теплоты образования указанных окислов и их температуры плав¬
ления.
20. Хромовая руда одного из месторождений имеет состав
60,91% Сг203, 14,67% FеО, 8,28% А/203, 11,51% NlgO и 2,15% SiOz.
Если к такой руде добавить 10% СгОз, то ее можно непосредственно
восстанавливать алюминием без обогащения. Определите коли¬
чество алюминия, необходимого для восстановления 1 m смеси руды
с хромовым ангидридом. Какой состав имеет феррохром, если в
шлаке теряется 6% окиси хрома и 1% закиси железа? Какое коли¬
чество феррохрома должно получиться из указанного количества ис¬
ходных веществ?
При ответе учтите, что Si02, MgO и А120з переходят в шлак.
Ответ. 281,7 кг\ 506,1 кг феррохрома; 79,92% хрома.
Работа 3
Получение металлов
действием газообразных восстановителей.
Приготовление изделия
методом порошковой металлургии
Газообразные восстановители Н2, С02, СН2 и т. д.,
в отличие от жидких и твердых, имеют большое преиму¬
щество: продукты их окисления Н20 и С02 можно
легко вывести из зоны реакции, а концентрацию восста¬
25
новителя повысить. Это позволяет восстанавливать мно¬
гие окислы, которые более прочны по сравнению с па¬
рами воды и двуокисью углерода.
В промышленности восстанавливают водородом
только окислы таких тугоплавких металлов, как воль¬
фрам и молибден, а также германий.
В последние десятилетия газообразные восстанови¬
тели, особенно окись углерода и природный газ, стали
применять для получения металлов в виде порошков,
используемых в металлокерамической промышленности.
Из металлических порошков, особенно железа, путем
прессования в формах получают различные детали. Из¬
готовление деталей этим методом экономически более
выгодно по сравнению с изготовлением их механической
обработкой компактных металлов; к тому же при из¬
готовлении деталей не получается отходов.
Практически восстановлением окислов водородом
можно получить следующие металлы: железо, кобальт,
никель, молибден, вольфрам, сурьму, висмут, германий,
олово, свинец, галлий, индий, таллий, кадмий, медь.
Из неметаллов этим методом иногда получают теллур.
Если восстановление проводится выше температуры
плавления металла, to продукт получается в сплавлен¬
ном виде, при более низких температурах — в порошко¬
образном виде.
Цель р а б о ты. Получить восстановлением соот¬
ветствующих окислов один из следующих металлов:
железо, кобальт, никель, молибден, вольфрам, сурьму 1,
висмут, олово, свинец, кадмий, медь. Металлокерамиче¬
ским методом получить образец металла.
Оборудование и материалы
1. Фарфоровая или кварцевая трубка длиной 50—
60 см.
2. Электрическая трубчатая печь на 1100 — 1200 °С.
3. Источник водорода (электролизер, аппарат Киппа,
баллон с водородом).
4. Промывная склянка с серной кислотой.
1 Сурьма, обладающая амфотерными свойствами, часто отно¬
сится к неметаллам. Однако по ряду свойств (например, ха¬
рактер изменения электрического сопротивления с температу¬
рой) ее следует относить к металлам.
26
5. Фарфоровая лодочка.
6. Соединительные резиновые трубки, резиновые или
корковые пробки, газоотводные трубки, сверла.
7. Окислы соответствующих металлов.
Проведение работы
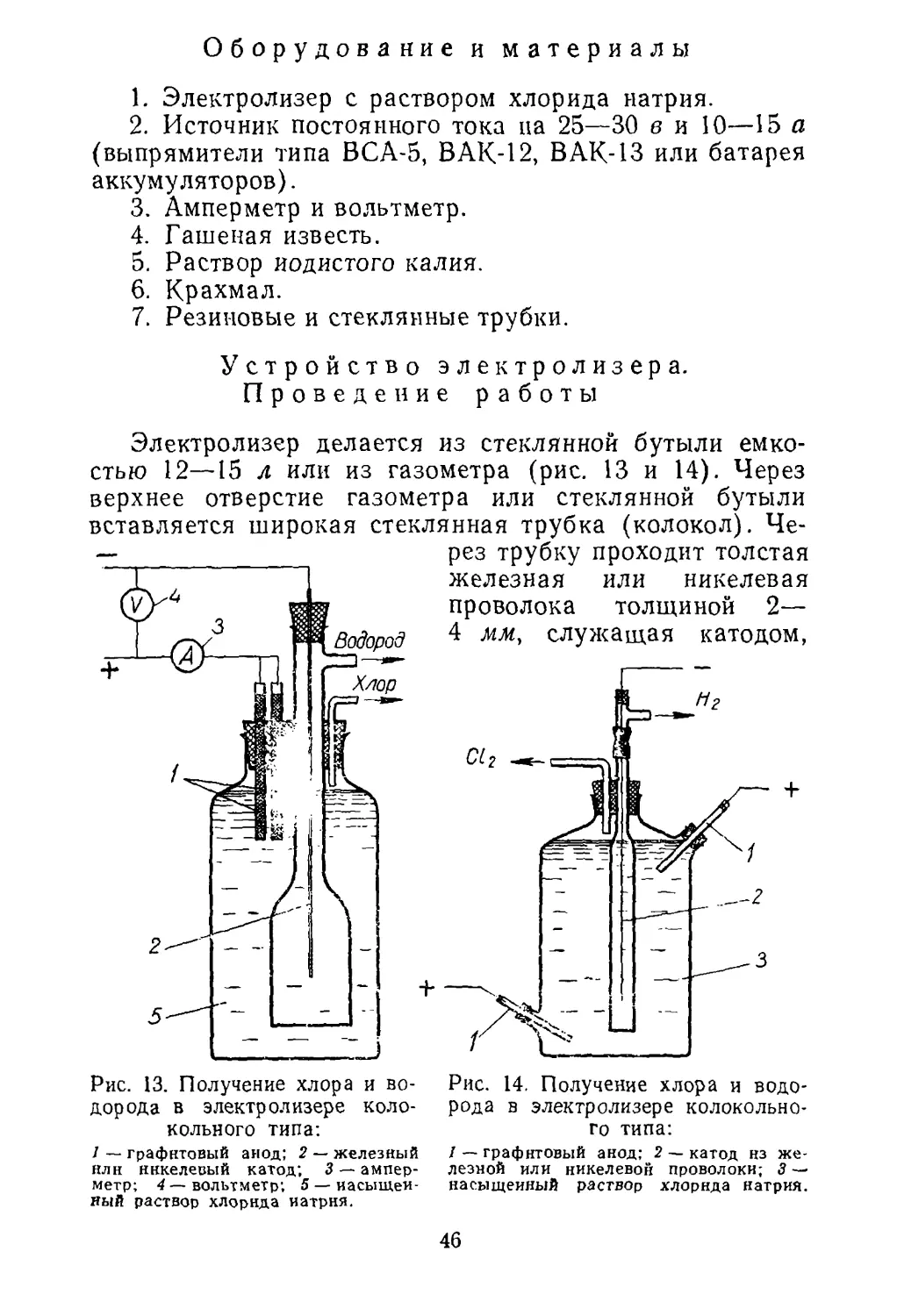
Восстановление окислов водородом проводится в при¬
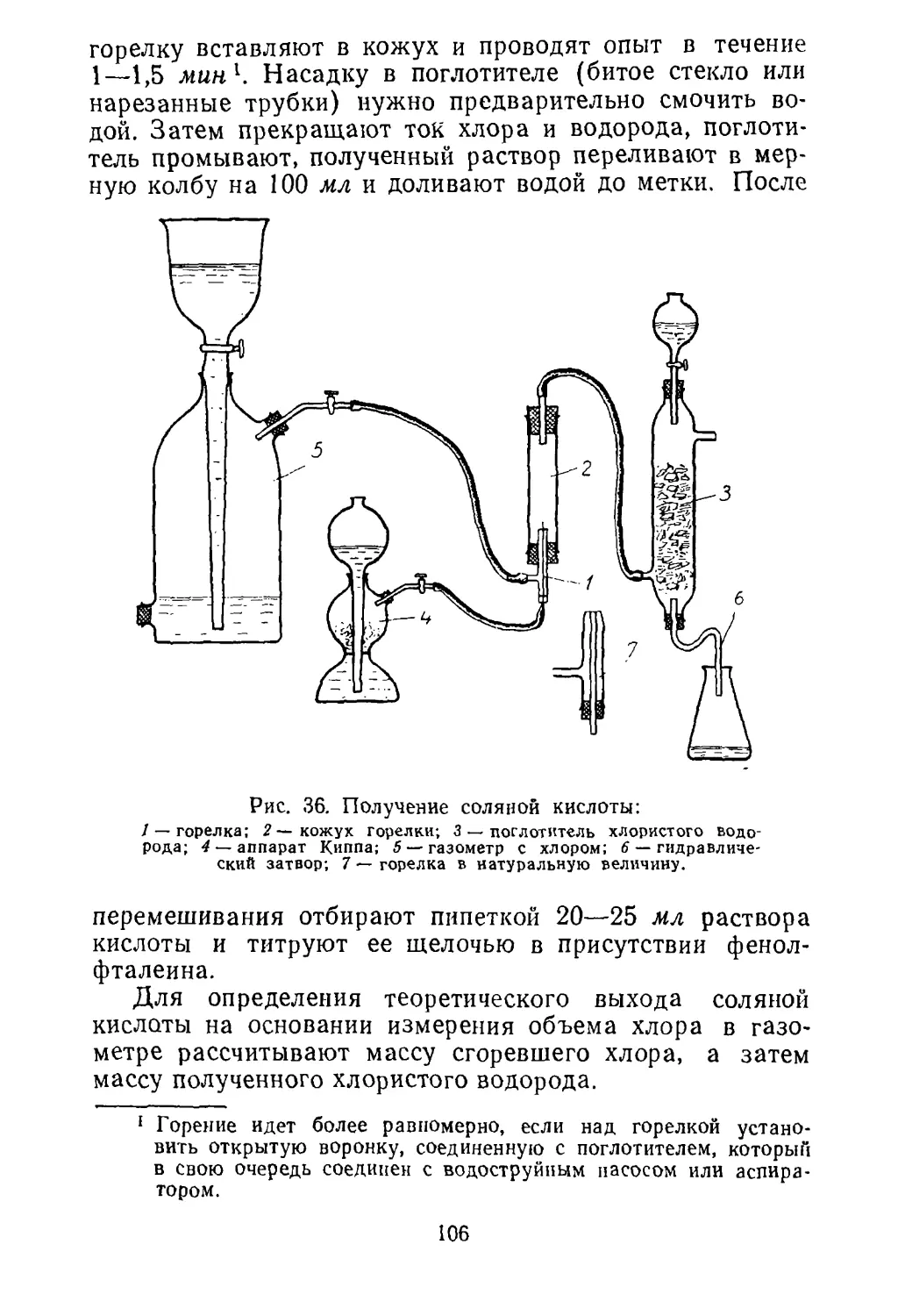
боре, приведенном на рисунке 5. Прибор состоит из квар¬
цевой или фарфоровой трубки 2, закрытой пробками, че¬
рез которые пропущены стеклянные трубки.
6
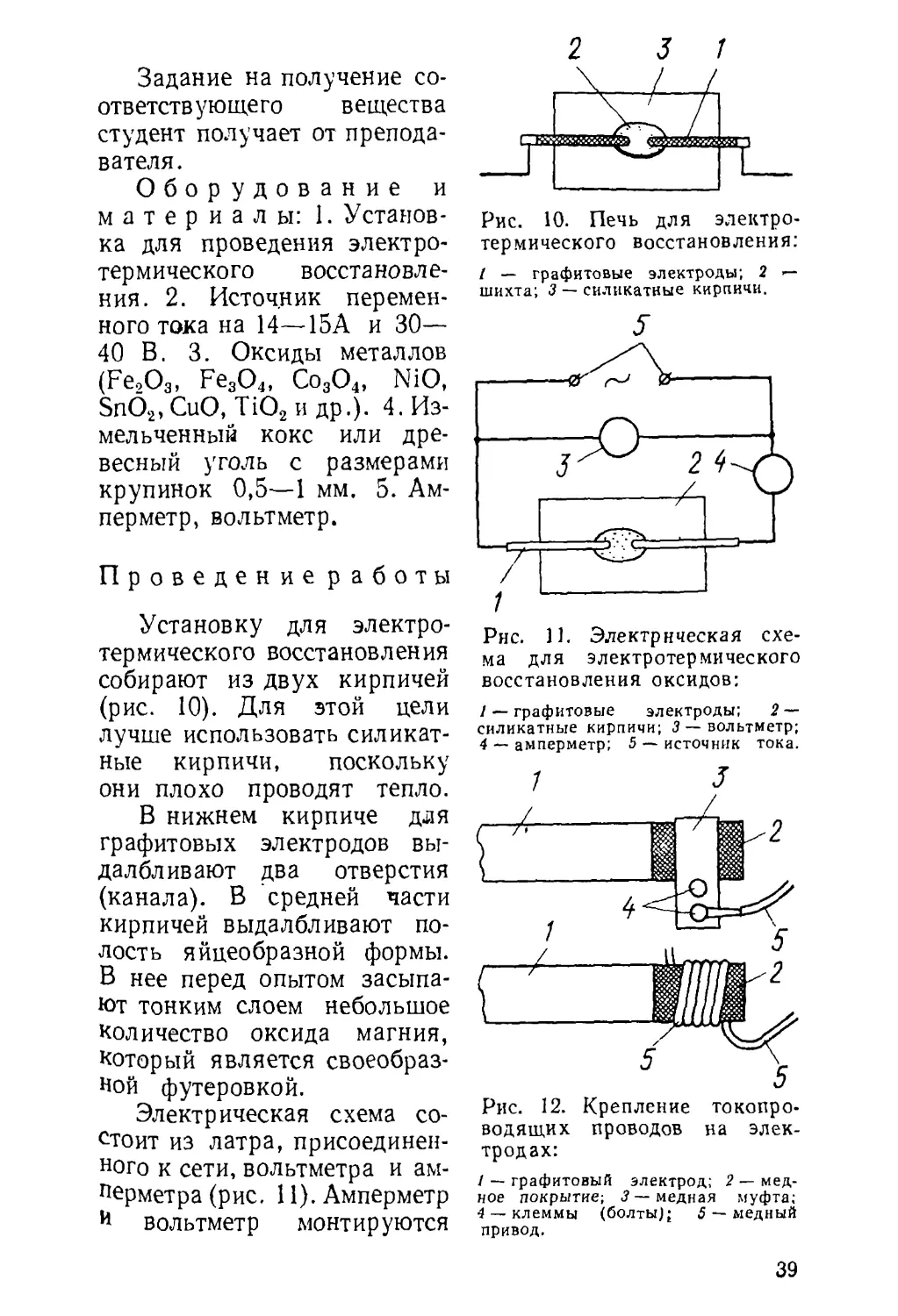
Рис. 5. Получение металлов из их окислов восстановлением
водородом:
/ — промывалка (счетчик пузырьков) с серной кислотой нлн ще¬
лочью; 2 —фарфоровая или кварцевая трубка; 3 — лодочка с окис¬
лом металла; 4 — электропечь (или газовый обогрев); 5 —термопара;
6 — пирометрический гальванометр.
Водород, получаемый электролизом раствора щелочи
или действием кислот на цинк, предварительно сушится
серной кислотой в промывной склянке 1 и направляется
в трубку для восстановления. Окисел помещается в фар¬
форовой или кварцевой лодочке 3 в середину трубки.
Температура измеряется термопарой 5, конец кото¬
рой помещается в непосредственной близости от лодоч¬
ки. Перед опытом необходимо проверить прибор на
герметичность. Для этого конец газоотводной трубки
опустите на 3—4 см в кристаллизатор с водой и про¬
пустите водород. При герметичности прибора пузырьки
водорода проходят через воду. Водород пропускайте до
полного вытеснения воздуха из фарфоровой трубки.
Проверьте водород на чистоту, собрав его в пробирку
(над водой). После этого, не прерывая тока водорода,
27
включите электропечь и следите за показаниями пиро¬
метра.
Время восстановления определяется количеством взя¬
того окисла и скоростью прохождения водорода. Если
берется 3—4 г окисла и водород пропускают в промы-
валке со скоростью 2—
3 пузырька в секунду,
то восстановление за¬
канчивается примерно
через 15—20 мин. По¬
сле восстановления
печь выключают и да¬
ют ей остыть, не преры¬
вая тока водорода.
Несоблюдение этого
правила может привести к взрыву.
Температуру восстановления выбирают в зависимо¬
сти от природы восстанавливаемого окисла. При темпе¬
ратуре 600—700 °С хорошо восстанавливаются окислы
железа, кобальта, никеля, сурьмы, висмута, олова, свин¬
ца и меди. Восстановление молибдена, вольфрама и гер¬
мания проводится при температуре не ниже 800—850 °С.
Если при восстановлении вольфрама и молибдена полу¬
чен порошок коричневого или синего цвета, то это ука¬
зывает на неполное восстановление окислов.
Необходимо отметить, что приведенные температуры
восстановления указаны ориентировочно. Многие из ука¬
занных окислов восстанавливаются и при более низких
температурах: например, окислы меди — при 150—200 °С,
железа — при 270 °С. Однако реакции при низких темпе¬
ратурах идут очень медленно.
Полученный металл можно запаять в пробирке
(рис. 6).
При отсутствии в лаборатории электропечи обогрев
трубки можно проводить газовой горелкой или даже
спиртовкой. При применении спиртовки, температура бу¬
дет невысокой, поэтому вольфрам, молибден и олово
получать таким способом не рекомендуется. При отсут¬
ствии в лаборатории фарфоровой или кварцевой трубки
восстановление можно проводить и в стеклянной.
Во избежание прилипания лодочки к стеклу окисел
необходимо положить непосредственно в трубку; темпе¬
ратуру ориентировочно определяют по внешнему виду
I
Рис. 6. Пробирка для запаивания:
1 — место запаивания; 2 — вещество.
28
трубки: при 600 °С трубка имеет отчетливо видимое крас¬
ное каление, и большинство окислов при этой темпера¬
туре хорошо восстанавливается.
Описанный метод применим и для получения спла¬
вов. Например, если взять смесь окислов олова и свинца,
Рис. 7. Прессформа для получения изделий из
порошкообразных металлов:
а — отдельные части прессформы; б — прессформа во
время прессования; в — прессформа во время вытал¬
кивания спрессованного изделия.
1 — матрица; 2—верхний пуансон; 3 — нижний пуан¬
сон; 4 — порошкообразный металл; 5 — кольцо.
то после восстановления получится сплав олова со свин¬
цом. При восстановлении смеси окислов железа и ни¬
келя получится порошок, состоящий из сплава железа
с никелем.
Описанная методика работы применима и для полу¬
чения металлов с использованием природного газа. Ме¬
тан более сильный восстановитель по сравнению с водо¬
родом, но для получения металлов необходимо приме¬
нять температуру на 50—100° С выше указанных. Желе¬
зо, молибден и вольфрам получаются несколько загряз¬
ненными углеродом. Из полученных порошкообразных
металлов изготовляют изделия спеканием прессованного
порошка. Такие изделия называются металлокерамиче¬
скими.
Для прессования можно использовать школьный ги¬
дравлический пресс, к которому нужно сделать пресс-
форму. Одна из возможных конструкций прессформы
приведена на рисунке 7. Она позволяет получать образ¬
цы в виде круглых шайб. Порошкообразный металл, на¬
пример железо, насыпается в матрицу на нижний пуан¬
сон. Сверху порошок прижимается верхним пуансоном,
29
вся форма вставляется в гидравлический пресс, и поро¬
шок сжимается. Затем форма несколько освобождается
из пресса и сверху на нее накладывается круглое коль¬
цо. При повторном сжатии пресса нижний пуансон вы¬
талкивает спрессованный металл в виде таблетки. Для
того чтобы спрессованный порошок не рас сыпался, нуж¬
но по возможности применить наибольшее давление. Это
достигается при небольшой площади отверстия матрицы,
например при диаметре 5—7 мм.
После прессования с полученной заготовки снимают
верхний пуансон, осторожно помещают ее в фарфоровую
лодочку, которую вставляют в фарфоровую или кварце¬
вую трубку. Из трубки вытесняют воздух водородом,
прокаливают заготовку при соответствующей темпера¬
туре.
Таблица 6
Режим прокаливания1
Металлы
Пористые
изделия
Компактное изделие
время про¬
каливания
(в ч)
температу¬
ра (в °С)
время про-1
наливания
(в ч) 1
температура
(в °С)
Медь
0,5
600
1
900
Железо
700
1000—1100
Медь + железо
700
1000—1100
Медь +10% углерода
700
1000—1100
Кобальт или никель
700
1000—1100
При длительном прокаливании металл полностью
спекается, почти не имеет пор и обладает такими же ме¬
ханическими свойствами, как и компактный металл.
Школьный гидравлический пресс. Его
внешний вид приведен на рисунке 8. Пресс смонтирован
на подставке 1. В большой цилиндр 2 вставлен пор¬
шень 3, который под давлением масла может поднимать¬
ся кверху. Максимальная сила давления, сознаваемая
поршнем, равна 3960 кг. На стенке цилиндра находится
вентиль (на рисунке не виден) для выпуска воздуха из
цилиндра. 11 Студент получает конкретное задание от преподавателя. Тем¬
пература спекания определяется на основании данных, приве¬
денных в таблице.
30
Малый цилиндр с поршнем служит для нагнетания
масла. Масло поступает из бака, находящегося в основа¬
нии пресса и при давлении рукоятки поступает через
всасывающий клапан в малый цилиндр, а затем через
нагнетательный клапан — в большой цилиндр. В кресто¬
образной колонке спереди укреплен манометр 6 для из¬
мерения давления. Вертикальная часть колонки — же¬
лезная трубка — имеет вверху винт 7, а сзади вентиль 8
Рис. 8. Гидравлический пресс:
/ — основание пресса; 2 — большой цилиндр; 3 — большой пор¬
шень; 4 — поршень малого цилиндра; 5 — рукоятка; 6 — манометр;
7 — винт предохранительного клапана; 8 — вентиль для слива
масла из большого цилиндра в бак; 9 — плита.
для сливания масла из цилиндра в бак. Винт связан
с предохранительным клапаном. Винт нужно так отре¬
гулировать, чтобы при создании давления в 150 кг/см2
наружу через клапан выступало бы не больше несколь¬
ких капель масла. Если же масло выступает при мень¬
шем давлении, то нужно несколько ввинтить винт и сно¬
ва проверить клапан на давление.
Для прессования прессформу помещают на плиту 9,
вентиль 8 завинчивают и при помощи рукоятки 5 нака¬
чивают масло в цилиндр 2, что вызывает поднятие пли¬
ты. После прессования открывают вентиль 8 для того,
чтобы часть масла из цилиндра 2 перешла в бак для
масла. При этом давление уменьшается, плита поршня
опускается книзу и прессформа освобождается. Затем
31
спрессованную заготовку выталкивают из прессформы
способом, описанным выше. Вентиль 8 нужно при этом
снова завинтить.
Оформление результатов работы
Описать получение металлов методом порошковой
металлургии.
Вопросы и задачи
1. Водород, содержащий некоторе количество паров воды, про¬
пустили через нагретые фарфоровые трубки, наполненные стружка¬
ми различных металлов: кальция, натрия, магния, железа, меди.
В каком из случаев удаления паров воды будет наиболее полным,
а в каком — наименее полным?
2. Окись хрома при высоких температурах постепенно восста¬
навливается при пропускании над ней сухого водорода. Почему при
наличии в водороде следов воды реакция не идет? Что можно ска¬
зать о значении константы равновесия реакции восстановления оки¬
си хрома?
3. Железо при высоких температурах легко окисляется парами
воды до закиси-окиси железа Fe304. Для того чтобы окислить этот
окисел до окиси железа, над ним нужно пропустить большое коли¬
чество паров воды. Что можно сказать на основании этих опытов о
значениях константы равновесия реакций:
ЗРвдОз ^2 ^ 2Fe304 -}- Н20;
Fe304 + 4Н* ^ 3Fe + 4Н20.
4. В фарфоровую трубку поместили две лодочки, одна из кото¬
рых была наполнена порошкообразным железом, другая — окисью
меди. Затем трубку плотно закрыли, поместили в трубчатую печь п
постепенно нагрели до 500 — 600 °С. Через 2—3 ч трубку охладили.
Оказалось, что железо окислилось, а окись меди восстановилась.
Как можно объяснить наблюдаемое явление? Какие два газа практи¬
чески полностью исчезли из воздуха, находящегося в трубке, и ка¬
кой газ вошел в его состав?
5. Над 100 г окиси меди, находившейся в нагретой трубке, про¬
пустили водород, который при нормальных условиях занимал объем
22,4 л. В результате реакции образовалось 17,6 г воды. Составьте
материальный баланс данного процесса.
Задано (в г)
Получено (в г)
Окись меди
Медь
Водород
Окись меди
Вода
Водород
Итого
Итого
32
6. Для получения железа над 130 г нагретой окиси железа бы¬
ла пропущена окись углерода, которая при нормальных условиях
занимала объем 31 л. Составьте материальный баланс восстановле¬
ния окиси железа.
Задано (в г)
Получено (в г)
Окись железа
Окись углерода
Железо
Двуокись углерода
Окись углерода
Итого
Итого
7. Над 50 г окиси свинца было пропущено 2 л окиси углерода.
На восстановление использовалось 98% газа. Составьте материаль¬
ный баланс восстановления окиси свинца до свободного свинца по
схеме:
Задано (в г)
Получено (в г)
Окись свинца
Окись углерода
Свинец
Окись свинца
Двуокись углерода
Окись углерода
Итого
Итого
8. Изобарно-изотермический потенциал закиси меди при 800
и 1000°К равен 118 кдж/моль и 203 кдж/моль и воды при указан¬
ных температурах 203 кдяд/моль и 192 кдж/моль. Рассчитайте кон¬
станту равновесия реакции:
Сц20 + Н2«>2Си + Н20.
Ответ. Кр8оо=4,7-103 и KPIOoo=4,57- IО4.
9. Определите константу равновесия реакции
СоО -f Н2 ^ Со -J- Н20
при 1000 % если изобарно-изотермический потенциал образования
СоО равен 162 кдж!/моль и Н20 192 кдж/моль. Сколько потребует¬
ся водорода для получения 100 г кобальта восстановлением закиси
кобальта?
Ответ. /(Рюоо = 3,4* 10; 4,3 Н2.
2 Н. Г. Ключников
33
10. Почему многие металлы, например кальций, магнии, алюми¬
ний, титан, цирконий, ванадий и т. д., нельзя получить восстанов¬
лением их окислов водородом?
11. Сколько требуется молен водорода для получения I г воль¬
фрама
W Оэ + ЗНа = W + ЗН20,
если степень использования водорода составляет 40% ?
Ответ. 7,5 моль.
12. Почему при восстановлении окиси серебра водородом ино¬
гда происходит взрыв?
13. Почему реакции восстановления окислов меди, железа, ко¬
бальта, никеля при комнатной температуре не проходят, несмотря
на то, что они термодинамически вероятны?
14. Почему при восстановлении железа водородом при 270 °С
оно получается пирофорным?
15. Константа равновесия реакции
Н20 + СО С02 + Н2 + 41,8 кдж
при 830 °С равна единице и восстановительная активность водорода
и окиси углерода при этой температуре одинакова. Какое вещество
более активно как восстановитель выше 830 °С?
16. Изобарно-изотермические потенциалы закиси железа при
1000 °К и 1500 °К равны 190 кдж/моль и 167 кдж/моль; воды при
указанных температурах—192 кдж/мъль и 164 кдж/моль. Рассчи¬
тайте константу равновесия реакции при 1000 и 1500°К:
FeO + H2<£Fe + Н20.
Определите процентный состав газовой фазы в состоянии равнове¬
сия.
Ответ. /Срюоо“0,34; /Cpisoo”0,135; 74,7% Но при 1000 °К и 88,3%
Н2 при 1500 °К.
17. Расход угля при электрохимическом получении алюминия
составляет на I m металла от 450 до 550 кг. В каких пределах ко¬
леблется содержание окиси и двуокиси углерода (в %) в отходя¬
щих газах?
Теоретический расход углерода на 1 m полученного алюминия
составляет 333 кг, когда он окисляется до двуокиси углерода, и
667 кг при его окислении до окиси углерода. Кроме алгебраического
решения возможно простейшее графическое решение (из зависи¬
мости % СО от количества окисленного углерода).
Ответ. 35% и 65% СО.
Работа 4
Получение металлов и сплавов
из окислов действием углерода
Углерод при высоких температурах обладает силь¬
ными восстановительными свойствами. Он может вос¬
станавливать самые прочные окислы, например окислы
алюминия, магния, титана, циркония и т. д. Однако
34
многие из получаемых металлов активно соединяются
с углеродом и дают прочные карбиды, например СаС2,
TiC, ZrC и т. д., поэтому восстанавливать углеродом
можно только такие металлы, которые не дают карби¬
дов. Практически из прочных окислов восстанавливают
углеродом только один магний, который испаряется из
реакционного пространства, и пары его улавливаются.
Цель работы. Восстановить углем один из сле¬
дующих металлов: свинец, олово, висмут — или сплавы
этих металлов с медью или друг с другом.
Оборудование и материалы
1. Высокий фарфоровый, или графитовый, или ша¬
мотный тигель емкостью в 50—60 см3.
2. Тигельная электропечь на 1100° С.
3. Термопара.
4. Тигельные щипцы.
5. Подставка для тигля (кирпич и т. п.).
6. Древесный уголь.
7. Окислы металлов: SnOo, РЬО или РЬзО^ В^гОз,
СиО или Си20.
8. Аптекарские весы с разновесом.
Проведение работы
Для проведения реакции древесный уголь размель¬
чают до мелкой крошки и отсеивают от угольной пыли.
Его количество берется с избытком, так как часть угля
окисляется за счет проникающе¬
го в тигель воздуха. Расчет ве¬
дут на 5—10 г окислов, считая
(для упрощения расчета), что
уголь окисляется до окиси угле¬
рода и не имеет примесей. Для
получения металлов берут соот¬
ветствующие окислы, а для полу¬
чения сплавов — смесь окислов,
например 4 г окиси меди и 1 а
окиси олова и т. д. На дно тигля
насыпают слой угля, а затем
смесь окислов и снова слой угля
Рис, 9. Получение метал¬
лов восстановлением
окислов углем.
2*
35
(рис. 9). Сверху смесь засыпают слоем угольной крошки
толщиной в 3—4 см, плотно закрывают тигель крышкой
и помещают его в электропечь. В случае отсутствия
крышки тигель нужно плотно закрыть куском асбеста и
сверху засыпать кварцевым песком.
Реакцию проводят при П00°С в течение 10—12 мин.
Затем тигель вынимают и после его охлаждения (поста¬
вить на песок или кирпич) отделяют полученный металл
от угля.
При избытке угля восстановление проходит практи¬
чески полностью, однако вследствие некоторых потерь,
за счет неполноты отделения металла, выход далек от
100%. Для определения выхода металл взвешивают и
на основании количества взятого окисла вычисляют про¬
центное отношение массы полученного металла к теоре¬
тическому. Часто металл или сплав прочно приплавля-
ется к фарфору и тигель для отделения металла прихо¬
дится разбивать.
Оформление результатов работы
Описать получение металла или сплава и определить
его выход в процентах.
Вопросы и задачи
1. Для чего сульфидные руды перед восстановлением обжигают
и переводят в окислы?
2. Какая химическая связь более прочная': связь металлов с ки¬
слородом или связь металлов с серой? На каких простейших опы¬
тах это можно показать?
3. В две фарфоровые трубки поместили смесь окиси железа с
углем. Воздух из первой трубки- вытеснили аргоном. Затем трубки
нагрели до 800 °С. Восстановление окиси железа прошло во второй
трубке. В первой трубке смесь осталась практически без изменения.
Объясните наблюдаемое явление.
4. В прошлом веке обнаружили, что в газах, отходящих из до¬
менной печи, всегда содержится значительное количество окиси угле¬
рода. С целью улучшения использования окиси углерода в Англии
была построена доменная печь на несколько метров выше обычных.
Однако и в том случае количество окиси углерода в доменном га¬
зе не уменьшилось. Чем это можно объяснить?
5. Почему при восстановлении окислов железа коксом окись
углерода иногда называют переносчиком электронов от кокса к
Руде?
6. Чем объяснить, что реакции окислов с водородом идут при
более низких температурах, чем реакции с углеродом?
7. В фарфоровую трубку поместили две лодочки, одна из ко¬
торых была наполнена порошкообразным железом, а другая —
36
окисью свинца. В трубку поместили маленький кусочек угля, в ко¬
личестве, недостаточном для восстановления окислов, трубку плотно
закрыли и нагрели до 800—900 °С. Через 2—3 ч- железо окислилось,
а окись свинца восстановилась до свинца. Как можно объяснить
наблюдаемое явление? Какое участие принял в реакциях углерод?
8. За счет каких процессов в мартеновской печи и конверторе
происходит уменьшение содержания углерода в чугуне?
9. Почему при увеличении давления газа, подаваемого в до¬
менную печь, ускоряется плавка?
10. Объясните, как и почему влияет подача воздуха, обога¬
щенного кислородом, в доменную печь: а) на скорость плавки;
б) на температуру в доменной печи; в) на качество чугуна; г) на
расход топлива; д) на экономичность процесса.
11. Почему при конверторном методе получения стали количе¬
ство фосфора в ней уменьшается при наличии основной футеровки,
в состав которой входят окислы кальция и магния? Почему при на¬
личии кислой футеровки (ЭЮг) количество фосфора практически не
уменьшается?
12. Передельный чугун, перерабатываемый на сталь в конвер¬
торе с использованием воздуха, содержит повышенное количество
кремния, марганца, углерода. Почему использование в бессемеров¬
ском процессе воздуха, обогащенного кислородом, дало возмож¬
ность перерабатывать на сталь чугун любого состава?
13. Почему при разливке стали в вакууме качество ее повы¬
шается?
§ 4. ПОЛУЧЕНИЕ МЕТАЛЛОВ И НЕМЕТАЛЛОВ
ЭЛЕКТРОЛИТИЧЕСКИМ МЕТОДОМ
Электролиз растворов и расплавов применяется для
получения веществ и различных защитных и декоратив¬
ных покрытий. Во многих случаях это почти единствен¬
ный метод выделения некоторых металлов, например
алюминия, натрия, калия, ниобия, тантала из их соеди¬
нений.
Электролиз позволяет:
1. Получить металл или неметалл в чистом виде.
2. Выделить металл или неметалл из смеси веществ
сложного химического состава.
3. Получить металлы в виде порошка, чешуек и т. д.
По первому закону Фарадея количество веществ, вы¬
деляющихся на электродах, прямо пропорционально ко¬
личеству электричества, прошедшего через электролит,
т. е. пропорционально силе и времени прохождения
электрического тока.
Согласно второму закону Фарадея одинаковые коли¬
чества электричества выделяют на электродах эквива¬
лентные количества веществ. При этом 96 500 к, т. е.
37
26,8 а • ч, выделяют 1 г-экв вещества. Количество веще¬
ства, выделяющегося при прохождении через электролит
I а>ч электричества, называется электрохимическим
эквивалентом вещества. Следовательно, для определения
электрохимического эквивалента нужно значение 1 г-экв
вещества разделить на 26,8 а- н. Значения электрохими¬
ческих эквивалентов некоторых веществ приведены в
таблице 7.
Таблица 7
Вещество
Электрохимический
эквивалент (в г)
Вещество
Электрический
эквивалент (в г)
Водород
0,0376
Железо
1,0419
Кислород
0,2985
Никель
1,9477
Хлор
1,3230
Медь
1,1354
Едкий натр
1,4925
Серебро
2,0127
Натрий
0,8581
Алюминий
0,3354
Зная силу тока, время прохождения электричества че¬
рез электролит и электрохимические эквиваленты, легко
подсчитать количества веществ, выделяющихся на элек¬
тродах. Однако практически при электролизе вещества
часто выделяются в несколько меньших количествах, так
как часть электрического тока расходуется на побочные
электрохимические процессы.
Выделение веществ на электродах происходит только
при определенной разности потенциалов, называемой на¬
пряжением разложения. Напряжение разложения скла¬
дывается из анодного п катодного потенциала и сопро¬
тивления электролита. Анодным потенциалом называет¬
ся разность потенциалов между анодом и раствором,
а катодным — разность потенциалов между катодом и
раствором. Практическое напряжение разложения всегда
больше теоретического на величину, называемую перена¬
пряжением. Величина перенапряжения зависит от кон¬
кретных условий ведения электролиза, особенно от
материала электрода, а также от плотности тока, приро¬
ды электролита, продолжительности электролиза. Пере¬
напряжение уменьшается при применении электродов
с матовой поверхностью, при снижении плотности тока
и повышении температуры электролита.
38
При промышленном применении электролиза боль-
Шое значение для экономики производства имеет выход
вещества по току и расход электрической энергии. Вы¬
ходом по току называется процентное отношение количе¬
ства выделенного вещества к тому количеству его, кото¬
рое должно было бы выделиться согласно законам
Фарадея. Выходом по энергии называется процентное
отношение количества электроэнергии, теоретически не¬
обходимой для проведения данного процесса к действи¬
тельно затраченной при электролизе.
При проведении электролиза стремятся создать наи¬
более оптимальные условия, при которых выход по току
и энергии максимален. Для этого уменьшают сопротив¬
ление ванны и проводов, так как это позволяет умень¬
шить напряжение потребляемого тока. В конкретных
случаях эти оптимальные условия различны. Для сниже¬
ния напряжения на ванне по возможности уменьшают
расстояние между электродами. Однако сильное сближе¬
ние электродов часто затрудняет разделение анодных и
катодных продуктов.
Пример. При электролизе раствора хлорида натрия при на¬
пряжении 3,4 в и силе тока 1500 а в течение 36 ч было получено
67,94 кг хлора. Определить выход по току и степень использования
электрической энергии, если теоретическое напряжение разложения
равно 2,19 в.
Теоретически при указанных условиях должно получиться
хлора:
1,323 1500 36 = 71 440 (г), или 71,44 (кг).
Выход по току равеи:
ВТ—6™
100
71,44
= 95,1(96).
Теоретический расход энергии (обозначенный через ТР) при по¬
лучении 67,94 кг хлора равен произведению теоретического напря¬
жения разложения (2,19 в) на количество затраченных ампер-ча¬
сов. Это количество также можно определить как частное от деле¬
ния количества вещества, полученного при электролизе, на химиче¬
ский эквивалент. Отсюда
ТР = 2,19
1
1,323
67 940
"ТООО
— 112,5 (кет
ч).
Практический расход энергии (ПР) при напряжении на ванне
в 3,4 в н выходе по току в 95,1% равен:
ПР = 3,4 1323 Гооо" 95,1 ' = *83,690 (*<?"* <*)■
Выход по энергии:
ТР 100 112,5-100
ПР " 183,6
61,27(96).
39
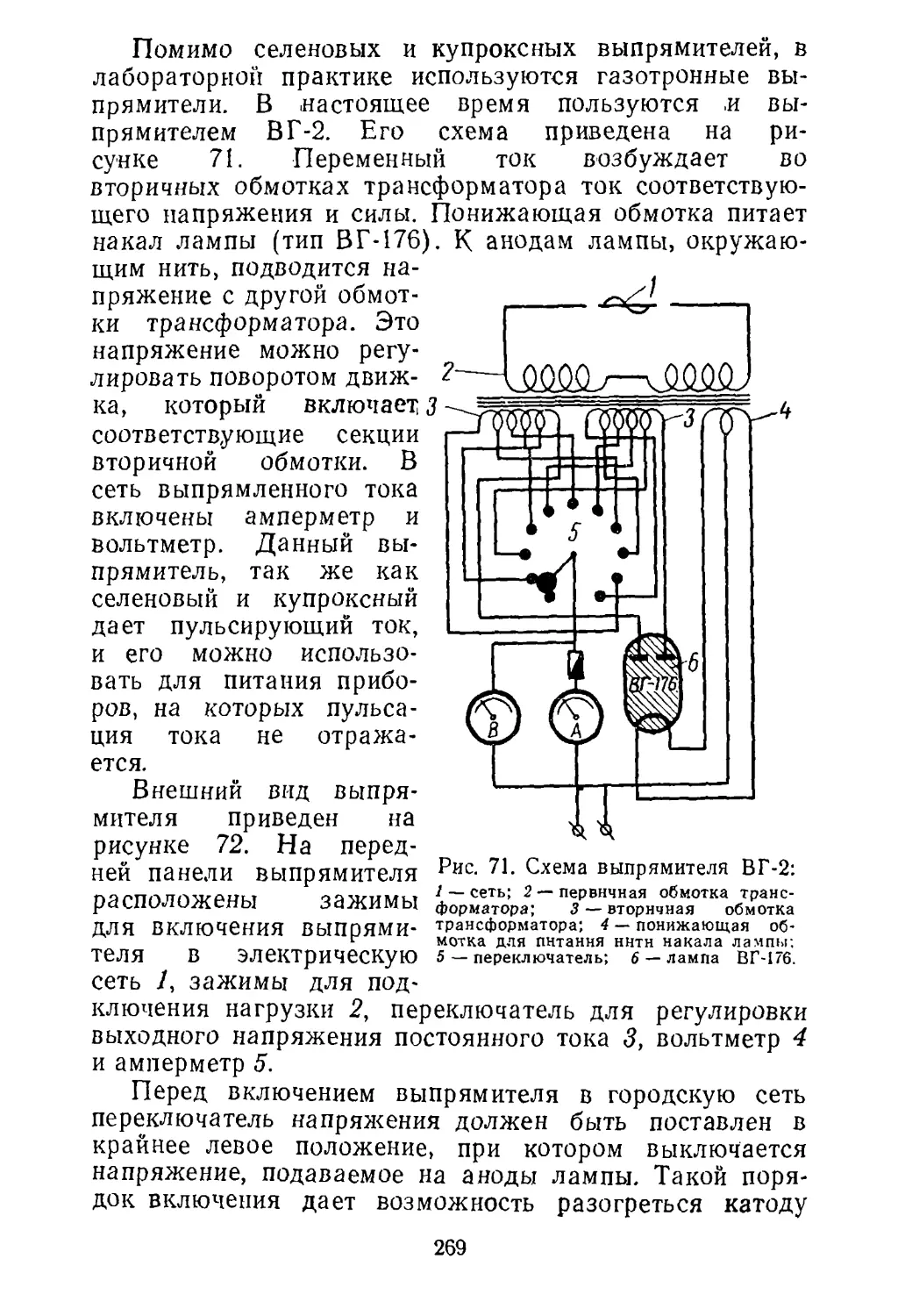
Работа 5
Получение никелевого покрытия
электролитическим методом
Никелевые покрытия применяются для защиты ме¬
таллов и сплавов от коррозии и для изготовления худо¬
жественных изделий.
Нормальный электродный потенциал никеля равен
0,25 в. Однако в атмосферных условиях благодаря об¬
разованию пассивной пленки никель длительное время
сохраняет свой блеск.
В контакте с железом никель служит катодом, и
поэтому он защищает железо чисто механически. При
наличии пор и других нарушений покрытия довольно
быстро развивается коррозия железа, приводящая к от¬
слаиванию никеля. Для уменьшения пористости никеле¬
вого покрытия и увеличения прочности сцепления его со
сталью на последнюю предварительно наносят тонкий
слой меди.
Существует много рецептов приготовления электроли¬
тов для получения никелевых покрытий. Наиболее рас¬
пространены ванны на основе сульфата никеля. Для
увеличения электропроводности в электролит добавляют
сернокислые соли натрия и магния. Для повышения ста¬
бильности pH раствора в него вводят борную кислоту,
а иногда и ацетат натрия.
Для лучшего растворения никелевых анодов в элек¬
тролит вводят в небольших количествах ионы хлора в
виде хлорида натрия, хлорида калия или хлорида ни¬
келя.
На работу электролита при никелировании большое
влияние оказывает значение pH раствора. В сильнокис¬
лых растворах выделение никеля вообще не происходит,
и на катоде выделяется только водород. Если же pH
электролита будет выше 6,3, то из электролита начинает
выпадать гидроокись никеля и покрытия становятся тем¬
ными. Для определения рецептуры никелевых ванн и для
определенной плотности тока выбирается определенное
значение pH раствора. Обычно pH раствора поддержива¬
ется в пределах от 4 до 6,3. При pH, равном 4, покрытия
металла получаются блестящими, но они сравнительно
плохо сцеплены с металлом и содержат значительное ко¬
личество пор и трещин.
40
При нормальной работе ванн убыль никеля из элект¬
ролита должна восполняться за счет растворения нике¬
левого анода. Однако нормальное растворение никеле¬
вого анода часто нарушается вследствие поляризации.
Внешним признаком ухудшения растворимости анода яв¬
ляется потемнение пластин и выделение на них пузырь¬
ков кислорода.
Электролиты для никелирования весьма чувствитель¬
ны к загрязнениям, особенно ионами меди, цинка и же¬
леза. Поэтому содержание примесей в солях, применяе¬
мых для приготовления электролита, должно быть мини¬
мальным.
Цель работы. Покрыть стальную пластинку
слоем никеля.
Оборудование и материалы
1. Стеклянный электролизер объемом 2—3 л (прямо¬
угольная стеклянная банка).
2. Реостат на 8—10 ом.
3. Амперметр и вольтметр постоянного тока на 5—
8аи 5—10 в.
4. Выпрямитель или батарея аккумуляторов на 10 в.
5. Стальные и никелевые электроды.
6. Сульфат никеля и другие реактивы, необходимые
для приготовления ванн никелирования.
Проведение работы
Никелирование складывается из следующих опера¬
ций: подготовка поверхности металла или сплава, нике¬
лирование и полировка никелевого покрытия.
Для опыта наиболее пригодны
прямоугольные железные пластинки
с общей поверхностью в 10—15 см2
с отверстиями (рис. 10). Площадь
пластинок замеряется штангенцир¬
кулем или в крайнем случае линей¬
кой. Пластинки предварительно за¬
чищаются крупной наждачной
шкуркой, а затем мелкой. При пере¬
ходе к более мелким номерам необ- Рис 10 Железный
ходимо полностью сошлифовать катод.
41
^ Рис. 11. Приспособление для шлифовки и полировки ме¬
таллических пластинок:
/ — место для помещения пластинки; 2 — винт для зажима пла
стннки н металлическая рамка из тонкого железа.
царапины, оставшиеся от более крупных номеров шку¬
рок. Для очистки пластинок и их шлифовки удобно при¬
менять зажимное приспособление, изображенное на ри¬
сунке 11. Оно состоит из двух металлических деталей,
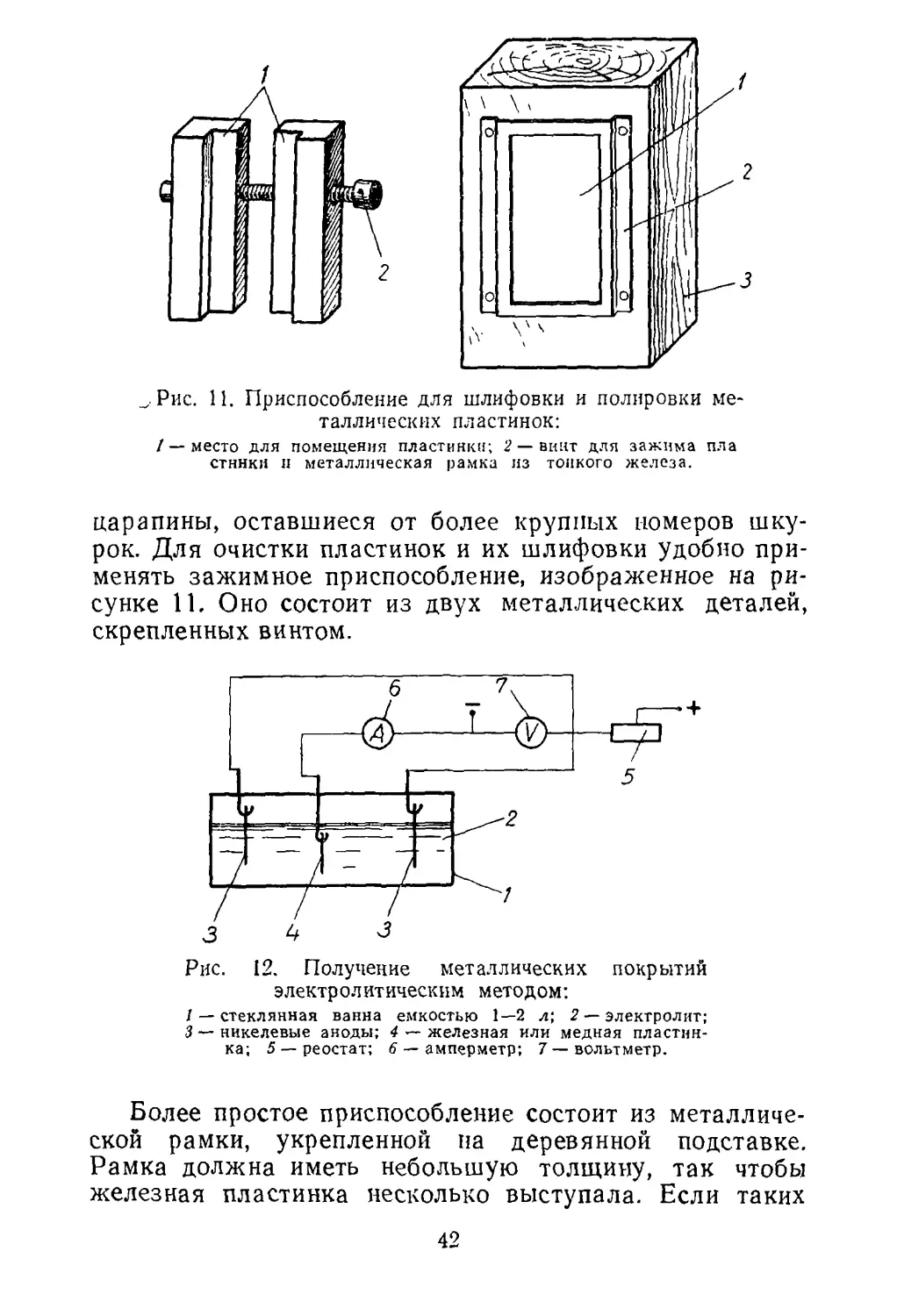
скрепленных винтом.
Рис. 12. Получение металлических покрытий
электролитическим методом:
/ — стеклянная ванна емкостью 1—2 л; 2 — электролит;
3 — никелевые аноды; 4 — железная или медная пластин¬
ка; 5 — реостат; 6 — амперметр; 7 — вольтметр.
Более простое приспособление состоит из металличе¬
ской рамки, укрепленной на деревянной подставке.
Рамка должна иметь небольшую толщину, так чтобы
железная пластинка несколько выступала. Если таких
42
приспособлений нет, то можно набить на небольшую
деревянную доску железные полоски, о которые и будет
упираться пластинка при шлифовке.
Для получения ровного блестящего покрытия пластин¬
ку после шлифовки полируют порошком окиси хрома или
алюминия, который наносится на сукно, набитое на де¬
ревянный брусок. Для обезжиривания пластинку натира¬
ют гидроокисью кальция, нанесенной на зубную щетку.
Пасту удаляют дистиллированной водой и щеткой. Пла¬
стинку в это время удерживают пинцетом. После обез¬
жиривания пластинку ополаскивают дистиллированной
водой, помещают примерно на 1 мин в 3—5-процентный
раствор серной кислоты, снова промывают водой и поме¬
щают в ванну для никелирования (рис. 12).
При кратковременном травлении железа серной кис¬
лотой па поверхности выявляется кристаллическая
структура железа, вследствие чего улучшается контакт
покрытия с железом.
Состав электролита для никелирования (в г/л):
сульфат никеля — 150—200
сульфат натрия — 70
сульфат магния — 10
борная кислота — 30
хлорид натрия — 5
Никелирование покрываемой площади проводится при
плотности тока 0,5—1 а/дм2 в течение примерно 10 мин.
Напряжение на электродах зависит от сопротивления
электролита и от расстояния между ними. При никелиро¬
вании нужно следить за показаниями амперметра, так
как превышение силы тока выше указанной ухудшает
качество покрытия. Сила тока регулируется реостатом.
После никелирования постоянный ток выключается,
пластины промывают водой и полируют.
Оформление результатов работы
Зарисовать ванну для никелирования. Описать поря¬
док проведения работы, состав электролита, режим нике¬
лирования.
43
Вопросы и задачи
1. Для чего перед нанесением электролитического покрытия по¬
лированную поверхность изделия обычно подвергают кратковре¬
менному травлению в кислоте?
2. С целью предохранения от коррозии железо покрывают цин¬
ком, оловом, хромом или никелем. Что будет подвергаться коррозии
в первую очередь, если на покрытие нанести глубокую царапину,
затрагивающую железо: основной металл или металл покрытия?
Дайте объяснение.
3. В растворе имеются сернокислые соли серебра, меди, никеля
и цинка. В какой последовательности (теоретически) должны выде¬
ляться металлы на катоде при электролизе?
4. Почему при электролизе солей цинка на катоде выделяется
цинк, а не водород, несмотря на то что электродный потенциал цин¬
ка более отрицателен по сравнению с водородом?
5. Теоретически напряжение разложения воды во время электро¬
лиза (при 17 °С и давления в 1 am) равно 1,23 в. Почему практи¬
чески напряжение разложения составляет 1,9—2,5 в?
6. Рассчитайте рабочее напряжение, имеющееся между анодом
и катодом ванны для производства алюминия, если падение напря¬
жения складывается из следующих величин: 1) анодная шина —
0,2 в; 2) переходное сопротивление анодная шина (штырь) —0,06 в;
3) штыри—0,04 в; 4) контактный штырь (анод) —0,1 в; 5) анод —
0,17 в; 6) электролит (металл) — 1,5 в; 7) подовый блок — 0,095 в,
8) контактный блок (катодный стержень) —0,005 в; 9) катодные
стержни — 0,16 в; 10) контактный катодный стержень (катодная ши¬
на)— 0,01 в; 11) катодная шина — 0,11 в; 12) напряжение разложе¬
ния глинозема — 1,7 в.
Ответ. 4,15 в.
7. Сколько теоретически должно выделиться алюминия в эле¬
ктролитической ванне при расходе энергии в количестве 1 кал!ч>
Ответ. 0,336 кг.
8. Сколько за 1 ч выделится алюминия, если выход по току
составляет 86%, а сила расходуемого тока — 60 000 а?
Ответ. 17,34 кг.
9. Сколько выделится алюминия за сутки, если выход по току
составляет 87%, а сила расходуемого тока — 55 000 а?
Ответ 385,6 кг. 1010. Составьте энергетический баланс работы электролизной ван¬
ны для получения алюминия (в ккал и дж) на основании следую¬
щих данных: 1) сила тока 35 000 а; 2) напряжение 4,34 в\ 3) при
окислении анодов выделяется 24 200 ккал/ч\ 4) затрачивается на
разложение глинозема 73 700 ккал/ч\ 5) уносится с удаляемым алю¬
минием 3000 ккал/ч\ 6) уносится с отходящими газами 1100 ккал/ч;
7) потери через анод и анодные штыри 22 400 ккал/ч\ 8) потерн че¬
рез боковые стенки, корку электролита, цоколь ванны 55 800 ккал\ч.
При подсчете прихода теплоты нужно принять, что 1 вт/ч экви¬
валентен 0,861 ккал.
Ответ. Приход и расход теплоты равен 15 600 ккал1ч или
65 270 кдЖ/ч.
44
Ра бота 6
Получение хлора и водорода в электролизе
колокольного типа
В водном растворе хлорида натрия присутствуют
ионы: Na+, Н+, С1“ и ОН". При пропускании постоянного
тока через раствор хлорида натрия к аноду движутся
отрицательно заряженные ионы С1~ и ОН~, а к катоду —
ионы Na+ и Н+ Нормальный разрядный потенциал иона
натрия равен —2,71 в, а обратимый потенциал водорода
равен —0,415 0, поэтому на катоде будет выделяться
только водород, а ионы натрия будут скапливаться около
катода.
Обратимый потенциал разряда гидроксильных ионов
равен +0,83 0, а потенциал разряда иона хлора равен
+ 1,33 в, поэтому на аноде должны были бы в первую
очередь разряжаться гидроксильные ионы. Практически
же разряду подвергаются ионы хлора ввиду большого
перенапряжения кислорода, которое смещает потенциал
выделения кислорода в более положительную сторону.
В то же время хлор выделяется на аноде (особенно на
платиновом) почти без перенапряжения. На угольном
аноде перенапряжение хлора довольно высоко, и поэто¬
му становится возможным выделение не только хлора,
но и кислорода, особенно при обеднении электролита
ионами хлора.
Продукты электролиза необходимо отделять друг от
друга, так как они между собой вступают в реакции.
Хлор дает в водном растворе хлорноватистую кислоту,
которая может взаимодействовать с едким натром с об¬
разованием гипохлорита:
НС10 + NaOH = NaCIO + Н20.
В промышленности отделяют катодное и анодное про¬
странства пористыми асбестовыми или цементными ди¬
афрагмами.
В лабораториях наиболее просто построить электро¬
лизер колокольного типа, в котором анодное и катодное
пространства почти не отделяются и вследствие этого
значительная часть продуктов реакции вступает во взаи¬
модействие.
Цель работы. Получить хлор и водород в элект¬
ролизере колокольного типа и использовать их для полу¬
чения синтетической соляной кислоты и хлорной извести.
45
Оборудование и материалы
1. Электролизер с раствором хлорида натрия.
2. Источник постоянного тока на 25—30 в и 10—15 а
(выпрямители типа ВСА-5, ВАК-12, ВАК-13 или батарея
аккумуляторов).
3. Амперметр и вольтметр.
4. Гашеная известь.
5. Раствор йодистого калия.
6. Крахмал.
7. Резиновые и стеклянные трубки.
Устройство электролизера.
Проведение работы
Электролизер делается из стеклянной бутыли емко¬
стью 12—15 л или из газометра (рис. 13 и 14). Через
верхнее отверстие газометра или стеклянной бутыли
вставляется широкая стеклянная трубка (колокол). Че-
Рис. 13. Получение хлора и во¬
дорода в электролизере коло¬
кольного типа:
1 — графитовый анод; 2 — железный
нлн никелевый катод; 3 — ампер¬
метр; 4 — вольтметр; 5 — насыщен¬
ный раствор хлорида натрия.
рез трубку проходит толстая
железная или никелевая
проволока толщиной 2—
4 мм, служащая катодом,
Рис. 14. Получение хлора и водо¬
рода в электролизере колокольно¬
го типа:
/ — графитовый анод; 2 — катод нз же¬
лезной или никелевой проволоки; 3 ■—
насыщенный раствор хлорида натрия.
46
Рис. 15. Крепление токо
проводящих проводов на
графитовом электроде.
нижний конец которой не должен доходить до конца труб¬
ки на 4—6 см. При более сильном погружении водород мо¬
жет попадать в анодное пространство и загрязнять хлор.
Угольный анод вставляется через пробку или через
боковое отверстие газометра. Для подвода тока к
электроду применяют медную муфту; в крайнем случае
угольный стержень плотно обкручивают медной проволо¬
кой (рис. 15).
Необходимо отметить, что
уголь ввиду своего значительного
сопротивления допускает прохож¬
дение лишь сравнительно сла¬
бых токов. Между поперечным
сечением угольного электрода и
максимально допустимой силой
электрического тока существует
определенное соотношение. При
употреблении выпрямителя ВГ-2,
дающего силу тока 6 а, угольный электрод должен иметь
диаметр не менее 7—8 мм. При большей силе тока, пода¬
ваемого на электролизер, электрод будет сильно разогре¬
ваться. Если же питание электролизера осуществляется
через выпрямитель ВСА-5, то нужно не менее трех уголь¬
ных электродов указанного диаметра или один электрод
диаметром около 2 см. В этом случае диаметр пробки,
закрывающей бутыль, должен быть достаточно широким,
чтобы разместить аноды. Лучше употреблять несколько
тонких стержней, чем один большого диаметра.
Если же употребляется один стержень в качестве
анода, то при значительной силе тока довольно быстро
наступает обеднение приэлектродного пространства
ионами хлора и вследствие этого получаемый хлор силь¬
но загрязняется кислородом.
Объем электролизера зависит от силы подаваемого
на него электрического тока. Если объем электролизера
мал, а сила подаваемого тока велика, то он быстро на¬
гревается. Практически, если для питания электролизера
применяется выпрямитель ВГ-2, дающий ток до 6 а, ми¬
нимальный объем электролизера должен быть равен
4—5 л. При использовании выпрямителя ВСА-5, который
дает ток до 12 а, объем электролизера должен быть
не менее 10—15 л.
Работа с готовым электролизером сравнительно про¬
47
ста. На электролизер подается ток нужной силы и уже
через несколько минут из газоотводных трубок начина¬
ют выходить хлор и водород. 26,8 а • ч дают 1 г-экв хло¬
ра, т. е. 11,2 л при нормальных условиях; ток силой в 1 а
в течение минуты выделяет 6,9 мл хлора. Хлор и водород
используют или для получения соляной кислоты (см.
раб. 16), или (хлор) для получения хлорной извести.
Для получения хлорной извести хлор направляют в
хлоркальциевую трубку, наполненную гашеной из¬
вестью:
2Са(ОН)2 + 2С12 - СаС12 + Са(С10)2 + 2Н20.
Отвешивают 2—3 г гашеной извести и рассчитывают
время хлорирования. Для этого по уравнению реакции
сначала определяют весовое количество хлора, необхо¬
димое для реакции с гашеной известью. Затем узнают,
сколько граммов хлора выделяется за единицу времени
при пропускании через электролизер постоянного тока
(силу тока узнают по показателям амперметра).
Полученную хлорную известь испытывают на присут¬
ствие активного хлора. Для этого часть извести присы¬
пают к раствору иодида калия и приливают раствор
крахмала.
Оформление результатов работы
Зарисовать электролизер и описать его работу. По
силе электрического тока рассчитать время, необходимое
для хлорирования 2—3 г хлорной извести.
Электролизеры колокольного типа могут дать сравнительно не¬
большое количество хлора, который к тому же довольно сильно за¬
грязнен кислородом, особенно при значительных плотностях тока
на аноде.
Для получения больших количеств хлора и водорода электро¬
литическим методом более пригодны электролизеры на 50—100 л с
вертикальной диафрагмой. Ниже приводится описание устройства
подобного электролизера (рис. 16).
Электролизер представляет собой деревянный ящик длиной 50—
80 см шириной 35—40 см и высотой 25—35 см. Внутри электролизе¬
ра делают цементную пористую диафрагму. Для ее устройства вну¬
три ящика устанавливают два листа фанеры и пространство между
ними заполняют цементной массой. Для ее приготовления берут 10
массовых частей цемента, 3—4 массовые части кварцевого песка и
1—2 массовые части хлорида натрия. Указанную смесь перемеши¬
вают и заливают водой до вязкой консистенции. После схватыва¬
ния цемента, примерно через сутки, фанеру убирают. Через 15—20
48
9
Рис. 16. Лабораторный электролизер большой емкости:
/ — деревянный ящик; 2 — цементная диафрагма; 3 — гудроновая
обмазка; 4 — пластмассовая крышка; 5—графитовые стержни; 6 —
железные стержни; 7 — переливная стеклянная трубка; 8 — сифон
для слива отработанного электролита; 9 — воронка; 10 — электролит.
суток заканчивается твердение цемента. Диафрагма делается тол¬
щиной 7—8 см. Поскольку цементные диафрагмы обладают сравни¬
тельно низкой фильтрующей способностью, в стенку диафрагмы
вставляют переливную стеклянную трубку диаметром 0,5—1 см.
Внутреннюю часть ящика обмазывают толстым слоем разогре¬
того гудрона. Места соединения цементной диафрагмы с деревом
также промазывают гудроном. Электролизер заливают водой для
растворения поваренной соли, находящейся в цементе. Через 5—10
суток выливают воду и электролизер заливают концентрированным
раствором хлорида натрия. Катодное и анодное пространство за¬
крывают крышками с вставленными в них электродами. В крышке
просверливают отверстия соответствующего диаметра, в которые
и вставляются электроды. Место соединения электрода с крышкой
промазывают клеем БФ-2 или густым раствором органического стек¬
ла в дихлорэтане. Общая поверхность анодов должна быть равна
600—1000 см2. Если для анодов применяются графитовые стержни
диаметром 1—2 см, то их устанавливают по 8—10 штук.
Крышки не должны пропускать хлора и водорода. Наиболее
удобно их делать из листового органического стекла. Края стенок
электролизера и верхнюю поверхность цементной диафрагмы про¬
мазывают горячим гудроном, па который накладывают крышку.
Затем места соединения крышки со стенками электролизера еще
раз промазывают горячим гудроном.
Так же устанавливают катоды. Если они графитовые или уголь¬
ные, то их количество должно быть равно числу анодов. Катоды
можно делать и из железа. Ввиду того что электропроводность же¬
леза во много раз больше электропроводности графита, общая по¬
верхность катодов может быть меньше поверхности анодов. Для
установки катодов в крышке просверливают 5—6 отверстий, в кото¬
рые и вставляют железные стержни диаметром 5—10 мм.
49
В крышку около анодного пространства вставляют воронку,
через которую наливают раствор хлорида натрия. Конец ее опу¬
скают в раствор электролита. В верхнюю часть стенки катодного
пространства (см. рис. 16) вставляют сифон 8 для слива отработан¬
ного электролита. Во время работы сифон должен быть заполнен
электролитом.
Расстояние от поверхности электролита до крышки равно
2—3 см. На таком же расстоянии от крышки устанавливают и
сифон.
В крышки вставляют стеклянные трубки для отвода хлора и
водорода.
Перед работой необходимо проверить герметичность прибора.
Для этого перекрывают резиновые трубки зажимами и в электроли¬
зер наливают в избытке электролит. При герметичности прибора
уровень электролита в воронке будет несколько выше уровня в
электролизере. Устранить негерметичность можно повторной про-
мазкой мест соединений гудроном и соответствующим клеем. Если
возникнет необходимость снять крышку, то для этого места соеди¬
нения осторожно прогревают горелкой до размягчения гудрона и
крышку снимают.
После проверки электролизера на герметичность подают посто¬
янный электрический ток силой 20—30 а и напряжением 20—40 в.
Напряжение па электролизерах зависит от сопротивления электро¬
лита, диафрагмы и находится опытным путем.
Использовать водород и хлор можно уже через 2—3 мин по¬
сле включения тока. Водород перед испытанием обязательно нуж¬
но проверить на чистоту, собрав его в пробирку над водой.
Необходимо следить, чтобы электролизер во время работы не
перегревался: крышка электролизера должна быть холодной.
Электролизер описанной конструкции дает хлор, всегда загряз¬
ненный кислородом. Если кислорода в хлоре имеется значительное
количество, то в электролит (в анодное пространство) нужно доба¬
вить 10—15 л концентрированного раствора хлорида натрия. Для
определения содержания кислорода в хлоре надо набрать в про¬
бирку газа и опустить ее в концентрированный раствор иодида ка¬
лия. Хлор поглотится раствором иодида калия. Непоглощенный
объем газа приходится на кислород.
Хлор и водород идут на приготовление соляной кислоты.
Вопроси и задачи
1. Обратимый потенциал разряда ионов ОН“ равен +0,88 в, а
обратимый потенциал разряда ионов С1~ в насыщенном растворе
хлорида натрия равен +1,33 в. Следовательно, при электролизе
хлорида натрия на аноде должен выделяться кислород. Объясните,
почему на большинстве электродов, в том числе и на угольном, пре¬
имущественно выделяется хлор?
2. Почему при электролизе горячего раствора хлорида натрия
без разделения анодного и катодного пространства в основном по¬
лучается бертолетова соль, а при электролизе холодного раствора
получается раствор гипохлорита натрия?
3. Для чего электроды в промышленных электролизерах распо¬
лагают по возможности наиболее близко друг от друга?
50
4. При электролизе раствора хлорида натрия иа ваине, рабо¬
тающей при силе тока 1000 а, в течение суток выделилось 30 кг
хлора. Сколько хлора должно выделиться теоретически и чему равен
выход по току?
Ответ. 31,8 кг и 94,3%.
Работа 7
Получение хлора и водорода
в электролизере с ртутным катодом
При электролизе водных растворов хлорида натрия
или хлорида калия на твердом катоде (угольном или ме¬
таллическом) всегда выделяется водород. Однако, если
электролиз проводится с ртутным катодом, на нем выде¬
ляется не водород, а металлический натрий, дающий
с ртутью амальгаму натрия. Объясняется это тем, что
выделение водорода на ртутном катоде происходит с
большим перенапряжением благодаря поляризации рту¬
ти. Перенапряжение водорода на ртутном катоде около
1 в и зависит от плотности тока, концентрации ионов
в растворе и от температуры.
Ванны с ртутным катодом, применяемые в промыш¬
ленности, состоят из собственно электролитической ван¬
ны, где проводится электролиз, и разлагателя. В элек¬
тролитической ванне происходит выделение хлора (на
угольном аноде) и натрия (на ртутном катоде). Ртуть
наливается на дно ванны.
Получающаяся амальгама ртути направляется в раз-
лагатель, где образуются едкий натр и водород за счет
взаимодействия с водой амальгамы натрия.
Цель работы. Получить хлор, водород и щелочь
в ванне с ртутным катодом. Получить хлорную известь.
Оборудование и материалы
1. Электролизер с ртутным катодом.
2. Выпрямитель типа ВГ-2 или батарея аккумулято¬
ров на 5—6 а и 15—20 в.
3. Хлоркальциевая трубка.
4. Гашеная известь.
5. Иодид калия и крахмал.
51
Устройство элекролизера и его работа
Электролизер с ртутным катодом (рис. 17) делается
из стеклянной банки с тубусом. Электрический ток под¬
водится к ртути при помощи медной проволоки, встав¬
ленной в стеклянную трубку. Положительным полюсом
является угольный стержень, пропущенный через резино¬
вую пробку. Электролизер соединен с разлагателем при
помощи двух стеклянных трубок. После сборки электро¬
лизера и разлагателя стеклянные банки помещают в
железный поддон и наливают ртуть, промытую разбавлен¬
ной азотной кислотой и водой. Ртути нужно налить такое
количество, чтобы после небольшого наклона электроли-
Рис. 17. Получение хлора в варше с ртутным ка¬
тодом:
1 — электролизер; 2 — разлагатель; 3 — раствор хлорида
натрия; 4 — вода; 5 — ртуть; 6 — резиновый клапан; 7 —
трубки для переливания ртути и раствора.
зера в сторону разлагателя, верхняя переливная трубка
в электролизере оказалась бы приподнятой над поверх¬
ностью ртути. Затем в электролизер наливают насыщен¬
ный раствор хлорида натрия, а в разлагатель — чистую
воду. Раствор соли и воду нужно наливать одновремен¬
но, чтобы их уровень был все время примерно одинаков.
На одну из переливных трубок полезно надеть резино¬
вый клапан — тонкую полоску резины, закрывающую
отверстие трубки. Концы резинки плотно прикручивают¬
ся к трубке ниткой. Полоска резины не должна быть
52
слишком широкой. Работу клапана необходимо прове¬
рить перед заполнением электролизера электролитом.
При наклоне электролизера в сторону разлагателя ртуть
несколько оттесняет резиновую пленку от окончания
трубки и переливается в разлагатель. Когда же электро¬
лизер займет свое прежнее положение, клапан давлени¬
ем ртути закроется, и часть ртути перейдет из разлагате¬
ля в электролизер по верхней трубке. Необходимо сле¬
дить, чтобы пробки были вставлены в тубус достаточно
плотно.
Перед работой трубку, отводящую хлор, соединяют
с хлоркальциевой трубкой, в которую помещают 2—3 г
гидроокиси кальция. Затем включают электрический ток
силой 5—6 а. Выделение хлора происходит на угольном
электроде.
Время, в течение которого нужно пропускать электри¬
ческий ток через электролизер, определяется количе¬
ством гашеной извести, взятой для хлорирования 1.
Когда закончится опыт, выключают ток, из электро¬
лизера переливают ртуть в разлагатель. Операцию пере¬
мешивания ртути повторяют 2—3 раза. Водород будет
выделяться не только в разлагателе, но и в электролизе¬
ре, так как частично амальгама натрия остается и в
электролизере.
Полученную хлорную известь пересыпают в колбу
с раствором иодида калия, к которому добавляют крах¬
мального клейстера.
Наличие раствора щелочи в разлагателе доказывают
в отдельной пробе при помощи фенолфталеина.
Ртуть в электролизере постепенно покрывается слоем
хлоридов ртути, чем нарушается контакт слоя ртути
с раствором хлорида натрия. В этом случае нужно вы¬
лить растворы и ртуть из разлагателя и электролизера,
промыть ртуть разбавленной азотной кислотой и водой и
снова налить в электролизер. Электролизер и разлага¬
тель необходимо поставить в поддон.
Оформление результатов работы
Зарисовать электролизер с ртутным катодом и опи¬
сать его устройство. Доказать наличие хлорной извести.
1 О методе расчета смотри предыдущую работу по получению
хлора в электролизере колокольного типа.
53
Вопросы и задачи
1. Почему при электролизе раствора хлорида натрия на желез¬
ном или графитовом электроде выделяется водород, а при электро¬
лизе с ртутным катодом выделяется натрий?
2. В электролизере с ртутным катодом образующаяся амаль¬
гама практически не взаимодействует с раствором. Почему ее раз¬
ложение легко протекает в разлагателе?
3. Почему для ускорения разложения амальгамы натрия в раз¬
лагателе ее приводят в контакт с графитом и для этого на дно раз-
лагателя укладывают графитовые плиты?
4. Для чего в промышленности графитовый анод располагают
по возможности на более близком расстоянии от ртутного катода?
Работа 8
Получение водорода
электролизом раствора едкого натра
В лабораторных условиях равномерный ток чистого
водорода удобнее всего получать электролитическим ме¬
тодом. Хорошо сделанный электролизер может работать
в течение многих лет.
Водород, получаемый действием кислот на цинк, в
отличие от электролитического водорода загрязнен и при
проведении ряда работ нуждается в тщательной очистке.
Устройство и работа электролизеров
Количество водорода, получаемого при электролизе,
определяется силой потребляемого тока. Как указыва¬
лось, 26,8 а • ч дают 1 г-экв водорода, т. е. 1 г или 11,2 л
при нормальных условиях. Таким образом, указанное
количество водорода можно получить в течение 1 ч, про¬
пуская ток силой в 26,8 а, или в течение 2 пропустив
ток силой 13,4 а, и т. д.
Лабораторные электролизеры имеют сравнительно
небольшие размеры, через них вследствие большого со¬
противления и разогревания нельзя пропускать ток боль¬
шой силы. Опыт показывает, что через электролизер,
устроенный из бутыли на 15—18 л, можно длительное
время пропускать ток в 12—15 а. Устройство такого
электролизера приведено на рисунке 18. Электролитом
служит концентрированный раствор щелочи. Чем мень¬
ше будет концентрация щелочи, тем больше будет сопро-
54
тивление электролита и тем больше будет выделяться
теплоты во время электролиза.
Анодное и катодное пространство отделены в элек¬
тролизере стеклянным колоколом. Электроды делают из
- железной проволоки диаметром
3—4 мм. Токоподводящие мед¬
ные провода припаивают к элек¬
тродам.
А/р
Рис. 19. Осушка водорода:
1 — промывалка с раствором щелочи; 2 —
колонка с хлоридом кальция; 3 — колонка
с твердой щелочью.
Глубина погружения электро¬
дов определяет сопротивление
электролизера и до известной
степени чистоту получаемого во¬
дорода. При небольшой глубине
погружения сопротивление электролизера возрастает, что
увеличивает выделение теплоты во время работы. При
большом заглублении электродов облегчается попадание
водорода в анодное пространство, а кислорода — в ка¬
тодное. Смешивание продуктов реакции происходит
вследствие того, что они несколько растворяются в элек¬
тролите.
Электроды должны находиться на расстоянии"
4—5 см от нижнего края колокола.
Питание электролизера наиболее удобно проводить
через выпрямитель (селеновый или газотронный) с регу¬
лируемым напряжением. После подключения электроли¬
Рис. 18. Получение водо¬
рода электролизом щело¬
чи:
I — раствор щелочи; 2.—стек¬
лянный колокол (илн широ¬
кая трубка) для сбора водо¬
рода; 3 — резиновая трубка,
соединяющая стеклянный ко¬
локол с тройником; 4 — ка¬
тод; 5 — анод; 6 — резиновая
пробка.
55
зера к выпрямителю регулятор силы и напряжения тока
нужно повернуть так, чтобы сила тока в случае примене¬
ния селенового выпрямителя типа ВСА-5 дошла до 12 а
или до 6 а в случае применения газотронного выпрями¬
теля. Напряжение на электродах обычно доходит до
20—30 в. Для питания электролизера можно воспользо¬
ваться также и батареей аккумуляторов, дающей напря¬
жение 20—30 е, включив в цепь низкоомный реостат.
Вновь собранный электролизер проверяют на герме¬
тичность. Для этого газоотводную трубку, отводящую
водород, погружают на 5—6 см в воду и включают
электролизер, дав на электроды ток 5—10 а. Через не¬
сколько минут из газоотводной трубки должны выходить
пузырьки водорода. Отсутствие выделения водорода
указывает на негерметичность в катодном пространстве
или в газоотводящей системе. После этого проверяют
герметичность анодного пространства.
Получаемый водород содержит некоторое количество
паров воды и следы кислорода. Водород пропускают че¬
рез промывалку с раствором щелочи (промывалка слу¬
жит в основном для наблюдения за током водорода),
затем для осушки через трубку с безводным хлоридом
кальция и, наконец, через трубки с кусочками едкого
кали (рис. 19). Лучшим осушителем является фосфор¬
ный ангидрид. Однако для подавляющего большинства
работ осушка твердой щелочью вполне достаточна. Для
специальных работ, требующих водород высокой чисто¬
ты, его пропускают через фарфоровую или кварцевую
трубку, наполненную стружками магния. Трубку нагре¬
вают до 550—600 °С, и благодаря большому сродству
кислорода к магнию пары воды и кислород удаляются
практически полностью.
Для того чтобы колонки с осушителями не заменять
в течение нескольких месяцев, их делают размером око¬
ло 0,5 м в длину и диаметром 3—4 см.
Вновь построенный электролизер, а также тот, кото¬
рым не пользовались в течение длительного времени,
перед употреблением нужно включить на 3—4 ч, чтобы
вытеснить воздух из катодного пространства и из осуши¬
тельной системы. Затем водород нужно проверить на
чистоту путем сжигания его в пробирке.
Описанный электролизер может работать бесперебой¬
но в течение 1—2 лет. Через 4—6 месяцев работы в него
56
добавляют раствор щело¬
чи. Через 1—2 года рабо¬
ты стеклянный колокол
заменяют новым.
В зависимости от име¬
ющейся посуды и количе¬
ства необходимого водо¬
рода электролизер можно
сделать из большой U-об-
разной трубки (рис. 20) и
т. д.
При работе с электро¬
лизерами необходимо со¬
блюдать следующие меры
предосторожности:
1. Не изменять на¬
правление тока.
2. Не допускать про¬
никновения воздуха в
систему для очистки водо¬
рода.
Рис. 20. Получение водорода элек¬
тролизом раствора щелочи:
/ — U-образная трубка с раствором ще¬
лочи; 2 — катод; 3 — анод; 4 — про-
мывалка с раствором щелочи; 5 —
вольтметр; 6 — амперметр; 7 — деревян¬
ная подставка.
3. Перед началом ра¬
боты проверять водород на чистоту.
4. Следить за исправностью осушительной системы.
Вопросы и задана
1. При прохождении тока одинаковой силы через электролизеры,
наполненные разбавленной и концентрированной щелочью, теплоты в
первом случае выделяется больше. Чем это можно объяснить?
2. Почему водород, полученный электролитическим методом,
обычно содержит следы кислорода?
3. Для удаления следов кислорода из водорода, полученного
электролитическим методом, газ пропускают через нагретый платини¬
рованный асбест. Какую роль играет платина в данном случае?
4. Какое количество водорода (по массе и объему) получится,
если через электролизер в течение 3 ч, пропускали ток силой 6 а?
Ответ. 0,677 г и 7,52 л.
5. В электролизере, через который проходил ток силой 10 а, вы¬
делилось 12 л водорода. Сколько времени работал электролизер?
Ответ. 2,87 ч.
6. Через электролизер в течение 6 ч проходил электрический
ток силой 10 а. Сколько водорода выделилось по массе и объему?
Ответ. 25,4 л и 2,28 г.
7. За сколько времени выделится 3 л водорода, если через
электролизер пропускать ток силой 12 а?
Ответ. 0,598 ч.
57
§ 5. ТЕХНИЧЕСКИЙ АНАЛИЗ МЕТАЛЛОВ.
ЦЕЛИ И МЕТОДЫ ТЕХНИЧЕСКОГО АНАЛИЗА
В настоящее время в промышленности используют
самые разнообразные металлы и сплавы. Химический
состав сплавов регламентируется ГОСТами, и главной
целью технического анализа является установление мар¬
ки или сорта металла или сплава, которые и определя¬
ют качество и назначение материала.
Наиболее распространенным методом технического
анализа является химический анализ, при помощи кото¬
рого устанавливают содержание отдельных элементов
в материале. Реже в задачу химического анализа входит
установление состава смесей веществ, например опреде¬
ление содержания окислов в шлаках и т. д. (фазовый
анализ). Помимо химических методов, используют чисто
физические методы, например установление плотности,
спектральный анализ и т. д. Довольно часто применяется
физико-химический анализ.
Наряду с указанными методами в техническом ана¬
лизе иногда используют чисто эмпирические и упрощен¬
ные методы, относящиеся к так называемым технологи¬
ческим методам, например искровая проба, проба на
изгиб, проба напильником твердости поверхности. На¬
пример, цвет и характер искры, получаемой от наждач¬
ного круга, зависит от количества легирующих элемен¬
тов в металле; хрупкость титана, зависящая от примеси
кислорода, азота, определяется многократным перегиба¬
нием листового титана и т. д.
При практическом использовании методов техниче¬
ского анализа первостепенное значение имеет точность
и скорость выполнения анализа, а также назначение
анализа, т. е. требования производства.
При контроле технологического процесса на его про¬
межуточных стадиях (например, при выплавке стали и
др.) обычно применяются экспрессные (быстрые) мето¬
ды. Быстрота анализа достигается как за счет ускоре¬
ния обычных операций (фильтрование под вакуумом,
автоматическое взвешивание и др.), так и за счет приме¬
нения новейших достижений в области аналитической
химии (применение спектрального анализа, фотоколори-
метриц, реактивов, специфичных только для данного
элемента, и т. д.). Экспрессные методы анализа метал¬
58
лов выполняются за 5—15 мину что позволяет следить за
ходом технологического процесса.
Для установления марки металла или сплава приме¬
няются так называемые маркировочные методы, которые
отличаются от экспрессных повышенной точностью.
Обычно эти методы более длительны.
В случае возникновения споров между заводом-по-
ставщиком и заводом-потребителем, а также для провер¬
ки результатов маркировочных и экспрессных анализов
применяют так называемые арбитражные анализы, отли¬
чающиеся повышенной точностью.
Разнообразные требования к точности и быстроте
анализов, большое число методов побудили стандарти¬
зировать сами методы и ввести ГОСТы.
В практике работы аналитических лабораторий ши¬
рокое применение находят так называемые стандартные
образцы (сокращенно СО). Стандартные образцы име¬
ют строго постоянный химический состав. По ним про¬
веряют точность методов химического анализа, а также
используют для установления эмпирических титров рас¬
творов.
Вычисление результатов анализа следует проводить
с такой точностью, чтобы последняя вычисленная цифра
была бы первой, точность которой недостаточна. Как
известно, величина ошибки выражается двумя путями.
Абсолютная ошибка показывает, насколько найденная
величина больше или меньше истинной. Относительной
ошибкой называется отношение абсолютной ошибки
к истинной величине. Это отношение обычно умножают
на 100 и получают значение относительной ошибки в про¬
центах.
Вычисление результатов анализа должно произво¬
диться с такой же относительной точностью, с какой про¬
изведен сам анализ. В то же время точность анализа
зависит от точности отдельных измерений. Например,
точность взвешивания обычных аналитических весов со¬
ставляет ±0,0001 г, что при навеске в 0,1 г составляет
±0,1%. Следовательно, все вычисления нужно делать с
точностью до 0,1%, т. е. до четвертой значащей цифры.
Если же после взвешивания был проведен ряд операций,
например приготовлен раствор, проводилось титрование,
то точность вычисления может зависеть от характера
этих операций. Например, если из навески было приго¬
59
товлено 100 мл раствора и объем жидкости измерялся с
точностью до 1 мл, т. е. с точностью до 1%, то и резуль¬
таты работы следует определять с точностью до трех
значащих цифр.
При обычных рядовых анализах металлов, при
навеске в 1 г, точность взвешивания в 1 мг вполне
достаточна.
Работа 9
Определение углерода
Углерод в стали находится как в виде карбида желе¬
за, так и в растворенном состоянии.
Для быстрого определения общего количества угле¬
рода применяют так называемый газометрический (во-
люмометрический) метод. Помимо этого, применяют
объемный баритовый'и весовые методы.
Для определения общего количества углерода по
волюмометрическому методу навеску стали или чугуна
сжигают в токе кислорода в электрической печи при
1100—1300 °С. Железо при этом сгорает до окиси желе¬
за, а углерод — до двуокиси углерода. Образующийся
углекислый газ вместе с избытком кислорода после
охлаждения направляют в газоизмерительную бюретку
аппарата Виртца — Штролейна, которая предварительно
наполняется жидкостью. Затем газ проходит через по¬
глотительную склянку со щелочью, где поглощается угле¬
кислый газ. Оставшийся кислород снова направляют
в газоизмерительную бюретку, где замеряется его объ¬
ем. По разности объемов (кислорода и двуокиси углеро¬
да) определяют количество углерода в стали. Это
определение облегчается благодаря особой градуировке
делений шкалы измерительной бюретки, по которой
непосредственно находят ответ о процентном содержании
углерода в стали. Поскольку эта градуировка приводит¬
ся для определенных условий окружающей температуры
и давления, в ответ вносят поправку на рабочее давление
и температуру.
Точность этого метода при содержании углерода
0,1% и более равна ±0,05%.
60
Вариант 1
Цель работы. Определить содержание углерода
в стали волюмометрическим методом.
Оборудование и материалы
1. Баллон с кислородом и редукционным вентилем.
2. Газометр с водой.
3. Склянка Тищенко, наполненная раствором 4-про¬
центного раствора перманганата калия в 40-процентном
растворе едкого кали.
4. Колонка с сухим поглотителем, наполненная снизу
натронной известью, сверху — стеклянной ватой.
5. Электрическая трубчатая печь на 1200 °С.
6. Реостат.
7. Термопара с пирометрическим гальванометром.
8. Амперметр переменного тока на 15 а.
9. Дугообразная трубка — фильтр, наполненный для
задержания пылевидных окислов стеклянной или обык¬
новенной ватой.
10. Аппарат Виртца — Штролейна.
11. Фарфоровая трубка и фарфоровые лодочки (не-
глазурованные, прокаленные при 1200 °С).
12. Крючок для вдвигания и извлечения лодочек из
трубки; изготовляется из мягкой, или малоуглеродистой,
стали.
13. Стальная стружка с известным содержанием уг¬
лерода (нормаль).
14. Стружка с неизвестным содержанием углерода
(набор).
Проведение работы
Перед проведением анализа на углерод необходимо
познакомиться с устройством и работой аппарата Вирт¬
ца— Штролейна (рис. 21).
Газ из печи для сжигания поступает в холодильник 3,
а затем в газовую бюретку 4 (эвдиометр) емкостью
150 мм. Иногда бюретку помещают в стеклянный кожух,
пространство между бюреткой и кожухом заполнено
водой, что устраняет резкие колебания температуры и,
следовательно, предупреждает изменение объема газа.
61
Рис. 21. Аппарат для определения углерода
в стали:
; — кран; 2 — трехходовый кран; 3 — холодильник;
4 — газовая бюретка (эвдиометр); 5, 6 — газопо*
глотательные сосуды; 7 — шкала; 8 — край уравни¬
тельной склянки; 9 — уравнительная склянка; /0—
груша; 11 — термометр.
При помощи каучуковой трубки газовая бюретка
соединена с уравнительной склянкой 9. Ее наполняют
25-процентным раствором хлорида натрия, к которому
добавлено несколько капель серной кислоты и метилово¬
го оранжевого. Появление желтой окраски указывает,
62
что в уравнительную склянку попала щелочь. В этом
случае эвдиометр надо промыть и раствор в уравнитель¬
ной склянке подкислить. Для наполнения газоизмери¬
тельной бюретки раствором служит груша 10. Во время
Рис. 22. Общий вид установки для определения углерода волюмо-
метрическим методом:
/ — кран; 2 — трубка для сжигания; 3 — амперметр; 4 — электропечь; 5 — пи¬
рометрический гальванометр; 6 — реостат; 7 — фильтр; 8 — газоизмерительная
бюретка; 9 — кран; 10 — трехходовой кран; 11, /«? — поглотительные склянки;
12 — уравнительная склянка.
накачивания воздуха кран 8 закрывают. Иногда для
наполнения бюретки раствором используют уравнитель¬
ную склянку без груши.
В верхней расширенной части бюретки находится тер¬
мометр 11. На нижней суженной части бюретки
закреплена приставная передвижная шкала 7. Для упро¬
щения расчета по ней дано процентное содержание угле¬
рода. Наверху имеется стеклянная трубка с трехходовым
краном 2У соединенная с газопоглотительными сосудами
5 и 6, наполненными концентрированным раствором ед¬
кого кали. Кран 1 служит для отделения бюретки от
атмосферного воздуха и трубки для сожжения, а также
для соединения бюретки с атмосферным воздухом. Об¬
щий вид установки приведен на рисунке 22.
Кислород в систему поступает из баллона при помо¬
щи специального редуктора, показывающего давление
63
кислорода в системе. Избыток кислорода поступает в га¬
зометр. Во время работы кислород должен время от
времени выходить через воронку газометра. Поэтому
в газометр наливают сравнительно небольшое количе¬
ство воды. Предварительно кислород проходит через
промывалки, которые одновременно служат счетчиками
пузырьков. В фарфоровую трубку для сжигания 2 кисло¬
род поступает по каучуковой трубке через кран 1. Труб¬
ка помещена в электропечь 4 типа «Марс», нагретую до
1200 °С. В печь вставлена термопара, соединенная с пи¬
рометрическим гальванометром 5, по показаниям кото¬
рого судят о температуре в печи. Во избежание разруше¬
ния керамики печь нагревают до 1200 °С в течение часа,
постепенно выводя реостат 6. За изменением силы тока,
потребляемого электропечью, следят по показаниям ам¬
перметра 3, укрепленного на щитке. Ввиду значительной
силы тока, потребляемого электропечью, электропечь
включают через рубильник.
При сгорании стали получается некоторое количество
дыма, состоящего из частичек окислов, и поэтому для его
улавливания ставят фильтр 7 из ваты. Газ из печи посту¬
пает в прибор для определения процентного содержания
углерода в стали.
Перед выполнением анализа аппаратуру предвари¬
тельно проверяют на герметичность. Для этого нижний
конец змеевика соединяют с выходным концом трубки
для сжигания. Кран 9 устанавливают на соединение
прибора с атмосферным воздухом и поднятием уравни¬
тельной склянки 12 (или при помощи груши) заполняют
газоизмерительную бюретку 8 раствором, выпуская
находившийся в ней воздух через кран 9. Затем бюретку
соединяют при помощи этого крана с системой и опуска¬
ют уравнительную склянку. Уровень жидкости в бюрет¬
ке в первые секунды опускается, а затем, если система
герметична, остается на одном уровне. После этого сис¬
тему отсоединяют и сосуды 11 и 13 заполняют щелочью.
При помощи трехходового крана 10 сосуды 11 к 13 со¬
единяют с бюреткой. За счет этого жидкость в бюретке
несколько опустится. Бюретку снова заполняют жид¬
костью до метки. Для сжигания фильтр 7 отсоединяют от
трубки 2, открывают вентиль и регулируют ток кислоро¬
да, с тем чтобы он проходил со скоростью 3—4 пузырька
в секунду.
64
Затем перекрывают кран 1, при помощи проволочно¬
го крючка вдвигают в раскаленную трубку лодочку с
навеской 1 г и трубку закрывают пробкой. Кислород в
это время выходит через газометр. Через 1 мин поворо¬
том крана 9 бюретку 8 соединяют с трубкой для сжига¬
ния. Как только жидкость опустится до нулевого деле¬
ния, кран 9 перекрывают, вынимают на асбестированную
сетку лодочку из трубки при помощи проволочного
крючка и перекрывают вентиль баллона с кислородом.
Передвигая шкалу, ставят ее нулевое деление на уровень
с жидкостью и проводят поглощение двуокиси углерода
следующим образом: бюретку соединяют при помощи
трехходового крана с газопоглотительным сосудом 11 и
поднимают уравнительную склянку до вытеснения из нее
газа. Затем ее снова опускают и бюретку соединяют
с газопоглотительным сосудом 13. Подобным же обра¬
зом проводят повторное поглощение двуокиси углерода
и замеряют при помощи уравнительной склянки новый
уровень жидкости.
Содержание углерода вычисляют по формуле:
где а — показания шкалы бюретки (1 деление соответ¬
ствует 0,05% С при навеске в 1 г);
k — поправка на температуру и давление (см. при¬
ложение) ;
т — навеска стали или чугуна в граммах.
Оформление результатов работы
Кратко записать методику работы и принципиальную
схему расположения приборов. Полученные данные за¬
нести в таблицу.
Навеска (в г)
Температура
(в °С)
Давление
(в мм)
% углеро¬
да по шка¬
ле
Поправка
% углеро¬
да в стали
3 Н. Г. Ключников
65
Вариант 2
Определение углерода весовым методом
Весовой метод заключается в сжигании навески угле¬
рода в токе кислорода. Образующуюся двуокись углеро¬
да поглощают патронной известью в предварительно
взвешенных трубках. Количество углерода в стали рас¬
считывают по привесу поглотительных трубок.
Цель работы. Определить содержание углерода
в стали весовым методом.
Оборудование и материалы
1. Трубчатая печь на 1200 °С со сжигательной труб¬
кой.
2. Баллон или газометр с кислородом.
3. Три трубки с натронной известью.
4. Две промывалки с серной кислотой и промывалка
с хромовой смесью.
5. Трубка со стеклянной ватой.
6. Термопара с пирометрическим гальванометром.
7. Медный или стальной крючок для вдвигания лодо¬
чек в трубку.
Проведение работы
Собирают установку по рисунку 23. Для проверки
герметичности пускают ток кислорода, закрыв выходное
отверстие у последней промывной склянки. Если прибор
герметичен, то прохождение газа через непродолжитель¬
ное время прекращается.
После проверки системы на герметичность присоеди¬
няют поглотительные трубки, пропускают в течение
10 мин ток кислорода (2—3 пузырька в 1 сек), отсоеди¬
няют поглотительные трубки и взвешивают их на анали¬
тических весах. Затем трубки снова присоединяют и в
трубку, нагретую до 1150—1200 °С, выдвигают при помо¬
щи медного или стального крючка лодочку с навеской
стальной стружки ( \ 1 г). Через 1 мин пускают ток кис¬
лорода (3—4 пузырька в 1 сек). Во время сгорания ста¬
ли ток кислорода постепенно замедляется, что видно по
уменьшению количества пузырьков в промывных склян¬
66
ках. Сжигание проводят в течение 1 мин. Для полного
вытеснения образовавшейся двуокиси углерода газа че¬
рез систему еще пропускают кислород примерно в тече¬
ние Ю мин. После этого лодочку вынимают на асбести-
рованную сетку, отсоединяют поглотительные трубки и,
выдержав их в эксикаторе в течение 10 мин, взвешива¬
ют на аналитических весах. Перед взвешиванием краны
у поглотительных склянок на 1—2 сек открывают и сно¬
ва закрывают.
Рис. 23. Схема установки для определения углерода весовым ме¬
тодом:
/ — склянка с раствором едкого кали; 2 — трубка с натронной известью; 3 —
фарфоровая трубка; 4 — лодочка с анализируемой сталью; 5 — термопара с пи¬
рометрическим гальванометром; 6 — фильтр; 7 — склянка с хромовой смесью;
8 — склянка с серной кислотой; 9, 10 — поглотители с натронной известью.
Содержание углерода вычисляют по формуле:
о/г — а' 0*2727 100
/0 VV — ,
т
где а — привес поглотителей;
т — навеска стали.
Оформление результатов работы
Дать описание устройства и действия установки по
определению углерода. Рассчитать процентное содержа¬
ние углерода в стали.
Работа 10
Определение серы
Сера находится в сталях главным образом в виде
сульфидов железа и сульфида марганца. Содержание
серы в стали определяют объемным иодометрическим ме-
3*
67
то дом (метод сжигания), методом отгонки (в виде серо¬
водорода) и весовым методом.
Объемный иодометрический метод определения серы
заключается в сжигании навески стали в токе кислоро¬
да с последующим определением количества образую¬
щейся двуокиси серы.
Двуокись серы образуется при окислении сульфидов
железа, марганца и некоторых других металлов, присут¬
ствующих в стали:
4FeS + 702 =*- 2Fe203 + 4S02.
Двуокись серы вместе с избытком кислорода пропу¬
скают через сосуд с водой, где образуется сернистая ки¬
слота. Ее количество определяют титрованием раствором
иода:
H2S03 + I2 + Н20 = H2S04 + 2HI.
Содержание серы рассчитывается на основании коли¬
чества раствора иода, пошедшего на титрование. Как
видно из уравнения реакции, 1 г-экв иода, т. е. 126,9 г,
отвечает 16 г серы.
Точность метода ±0,001%.
Цель работы. Определить в стали количество се¬
ры объемным иодометрическим методом.
Оборудование и материалы
1. Баллон с кислородом.
2. Газометр.
3. Промыва л ка с серной кислотой (счетчик пузырь¬
ков).
4. Колонка с хлоридом кальция.
5. Печь «Марс» па 1200 °С.
6. Фарфоровая трубка длиной 600 мм и диаметром
18—20 мм.
7. Прокаленные фарфоровые лодочки.
8. Бюретки с титрованным раствором иода (примерно
0,01 н.) в иодиде калия.
9. Водный раствор крахмала.
10. Крючки из медной (стальной) проволоки, служа¬
щей для вдвигания лодочек и для вытаскивания их из
трубки.
11. Термометр с пирометрическим милливольтметром.
68
12. Два стеклянных цилиндра высотой 250 мм и диа¬
метром 40 мм.
13. Аналитические весы.
Проведение работы
Схема установки для определения серы приведена на
рисунке 24. Кислород из баллона поступает в промывал-
ку /, наполненную серной кислотой, и в колонку 2, на¬
полненную хлоридом кальция. Промывалка одновремен¬
но является счетчиком пузырьков. В случае создания в
системе повышенного давления кислород попадает в га¬
зометр и выходит через его воронку. С этой целью в газо¬
метр нужно наливать небольшое количество воды. Затем
кислород поступает в сжигательную трубку 3, где нахо¬
дится навеска стали. Образующаяся двуокись серы вме¬
сте с избытком кислорода проходит через раствор иода
с крахмалом, находящийся в цилиндре 5. Рядом находит¬
ся цилиндр 4 со стандартным раствором иода. При сжи¬
гании стали образуется небольшое количество дыма,
который мешает определению серы. Поэтому газовую
смесь предварительно пропускают через U-образную
трубку 6, наполненную ватой или асбестом. Нагревание
трубки проводят в трубчатой печи «Марс». За темпера¬
турой печи следят по показаниям пирометрического мил¬
ливольтметра 3, соединенного с термопарой.
Электропечь включают от щитка за 1—1,5 ч до рабо¬
ты; в течение этого времени температура постепенно до¬
водится до 1200 °С.
На аналитических весах отвешивают около 1 г анали¬
зируемой стали или 0,2—0,4 г чугуна (в виде стружки)
и рассыпают навеску по дну лодочки. Одновременно в
другую лодочку помещают около 1 г стальных стружек с
известным содержанием серы. Аппаратуру проверяют на
отсутствие сернистых соединений и других восстанови¬
телей (рис. 24). Для этого сначала в цилиндр наливают
6 мл воды, прибавляют несколько капель иода, крахмала
и смесь перемешивают. Раствор должен быть окрашен в
синий цвет. Одновременно готовят такой же раствор иода
в другом цилиндре и оба цилиндра ставят рядом. Затем
из баллона через систему пропускают ток кислорода.
Электропечь в это время должна быть нагрета до 1200 °С.
При отсутствии баллона кислород можно пропустить из
69
кислородной подушки, надавливая на нее рукой. При
пропускании кислорода через раствор иода не должно
наблюдаться ослабления окраски по сравнению со стан¬
дартным раствором. Прекращают ток кислорода, выни¬
мают пробку, быстро в середину трубки вдвигают при
помощи проволочного крючка лодочку с навеской стан¬
дартной стали, и трубку снова закрывают. Затем через
трубку пропускают ток кислорода. Пузырьки его должны
/ — промывалка с серной кислотой; 2 — колонка с хлоридом кальция; 3 — пи¬
рометрический милливольтметр; 4, 5 — цилиндры с растворами иода; 6 — U-об-
разная трубка; 7—термопара; 8 — фарфоровая трубка.
следовать непрерывной струей. Чтобы не исчезала окрас¬
ка иода, нужно все время из бюретки приливать титро¬
ванный раствор иода, следя за тем, чтобы окраска его в
цилиндре равнялась окраске стандартного раствора.
Процесс заканчивается, когда прекращается изменение
окраски раствора иода. После этого кислород пропуска¬
ют еще в течение минуты. Лодочку вынимают при помо¬
щи крючка и помещают на асбестированную сетку.
Затем рассчитывают титр раствора иода, выраженный
в граммах серы. Допустим, что на титрование пошло
15 мл раствора иода, а содержание серы в нормали равно
0,03%. Следовательно, 15 мл иода соответствуют 0,0003 г
серы, а 1 мл откроет 0,0003: 15 = 0,00002 г серы иТ,^$ =
= 0,00002 г. Таким образом, титр иода можно определить
по формуле:
70
%Sm
1QQ . мл I2 ’
где мл Ь — количество миллилитров раствора иода, по¬
шедшее на титрование двуокиси серы;
т — навеска стали в граммах;
% S — процент серы в стали.
Находят количество серы в анализируемой навеске,
т. е. определяют количество миллилитров иода, пошед¬
шего на титрование двуокиси серы, полученной при сжи¬
гании стали или чугуна.
При анализе чугуна в раствор для поглощения может
попадать значительное количество окислов, загрязняю¬
щих его. Поэтому при анализе чугунной стружки кисло¬
род впускают в прибор спустя 2 мин после помещения
лодочки в трубку. Плохо сгорают мягкие стали, поэтому
при их сжигании поверх пробы в лодочку еще помещают
0,5 г стружек олова.
Перед пуском кислорода лодочку с навеской выдер¬
живают в трубке в течение 0,5—1 мин. После окончания
анализа трубку открывают, вынимают из нее крючком
раскаленную лодочку и кладут ее на толстый кусок ас¬
беста. Отмечают количество иода, пошедшее на титро¬
вание, и рассчитывают количество серы по формуле:
%S =
_ 1 i/s
V 100
где Tys— титр раствора иода, выраженный в граммах
серы;
V — количество миллилитров раствора, пошедшее
на титрование;
т— навеска стали или чугуна в граммах.
Оформление результатов работы
Записать методику анализа. Результаты анализа
оформить в виде таблицы.
Навеска стали
(в г)
Т
I/S
Количество нода
(в мл)
Количество серы в
стали (в %)
71
Вопросы и задачи
1. Во время определения серы при сжигании стали в кислороде
образуются окислы железа, являющиеся катализатором для окис¬
ления двуокиси серы в серный ангидрид, который не восста¬
навливает раствор иода. Почему при описанных условиях не сле¬
дует опасаться заметного перехода двуокиси серы в серный
ангидрид?
2. Каково назначение газометра в установках по определению
серы и углерода?
3. Определите титр иода по сере, если на титрование пошло
7,8 мл иода; иавеска нормали, содержащей 0,02% серы, равнялась
0,853 г.
Ответ. 1 мл иода открывает 0,000219 г серы.
4. Навеска стали 0,9500 г. На титрование двуокиси серы, полу¬
ченной при сжигании нормали, пошло 17 мл иода. Навеска нормали
равна 1 г, а количество серы в ней 0,03%. Рассчитайте количество
серы в анализируемой стали, если на титрование пошло 12 мл иода.
Ответ. 0,000211 г, или 0,0222%,
5. Количество иода, пошедшего на титрование двуокиси серы,
пропорционально процентному содержанию серы и массе взятой на¬
вески. Выведите формулу, по которой можно непосредственно под¬
считать содержание серы, без расчета титра иода по сере.
При этом %Si — процентное содержание серы в нормали;
%S2 — процентное содержание серы в анализируемом
образце;
V\ и V2—объем раствора иода, пошедшего на титрова¬
ние двуокиси серы, полученной из нормали и
анализируемого образца;
rtii и тп2— навеска нормали и анализируемого образца.
Ответ.
п/ С _ °/oSi V2 • mi
70 02 = w —
Vi • щ 6 7 8 96. Можно ли в установке по определению серы в стали опре¬
делять содержание серы в сульфидных рудах?
7. Можно ли для определения серы отходящие из печи для
сжигания газы пропустить через воду и серную кислоту, затем
оттитровать раствором иода?
8. Почему абсолютное количество (объемное) раствора иода с
крахмалом не имеет особого значения при определении серы? Влия¬
ет ли на результаты анализа разница в объемах титруемого раство¬
ра иода с крахмалом и раствора, взятого для колориметрирования
(стандартного)?
9. Взвешивание стали при определении серы достаточно про¬
водить с точностью до тысячных долей грамма. Почему прл расчете
содержания серы достаточно получить три значащие' цифры, при¬
чем последняя из них будет сомнительной?
72
Работа 11
Определение кремния
Для определения кремния в сталях и чугунах при¬
меняют солянокислый, хлорнокислый, колориметриче¬
ский, спектральный и другие методы. Ниже описывается
солянокислый метод — один из наиболее простых по ис¬
полнению и достаточно точный.
Кремний присутствует в стали главным образом в ви¬
де твердого раствора. При его содержании выше 14%
в стали образуются различные силициды: FeSi, Fe3Si2
и т. д.
Навеску стали вначале растворяют в соляной кисло¬
те. При растворении стали кремний переходит в кремние¬
вую кислоту. Образующиеся промежуточные продук¬
ты— силан SiH4, тетрахлорид кремния SiCl4— являются
нестойкими веществами и также дают кремниевую кисло¬
ту. Потери кремния при растворении стали в кислоте не
происходит.
Кремниевая кислота получается в коллоидном состоя¬
нии и при фильтровании может проходить через фильтр.
Поэтому раствор упаривают досуха. При этом кремние¬
вая кислота обезвоживается и переходит в силикагель.
Полученные окислы железа и оксихлориды растворяют
в соляной кислоте, а силикагель отфильтровывают и про¬
каливают.
По массе полученной двуокиси кремния рассчитывают
содержание кремния в стали.
Цель работы. Определить количество кремния в
стали.
Оборудование и материалы
1. Фарфоровая чашка диаметром 10—12 см.
2. Соляная кислота (d=l,19).
3. Азотная кислота (d= 1,4).
4. Беззольные фильтры с красной лентой.
5. Воронка для фильтрования, промывалка, тигель,
стеклянная палочка с резиновым наконечником.
6. Электрическая муфельная печь.
7. 5-процентный раствор роданида калия или рода¬
нида аммония.
8. Часовое стекло.
73
Проведение работы
2 г стали или 0,2 г чугунной стружки помещают в фар¬
форовую чашку и растворяют при нагревании в 30—40 мл
соляной кислоты. Чашку закрывают часовым стеклом.
После растворения стали на дне чашки остается взвесь
карбидов и графита (если был взят чугун). Для их окис¬
ления к горячему раствору прибавляют 2—3 мл азотной
кислоты, раствор выпаривают досуха и слегка подсуши¬
вают (около 130 °С) на асбестированной сетке до разло¬
жения хлоридов и появления светло-бурой окраски, за¬
висящей от наличия гидратированных окислов железа.
После охлаждения часовое стекло обмывают из промы-
валки водой и к осадку приливают 10—15 мл соляной
кислоты и 100 мл горячей воды. Раствор нагревают до
полного растворения окислов и солей. Теплый раствор
фильтруют через беззольный фильтр, сливая его на
фильтр по палочке.
Чашку из промывалки обмывают отдельными порци¬
ями горячей воды, подкисленной соляной кислотой (1 : 20),
протирают оплавленной стеклянной палочкой с резино¬
вым наконечником, сливая воду на фильтр. Осадок на
фильтре (он обычно не видим) нужно хорошо промыть
горячей водой, подкисленной соляной кислотой. Промы¬
вать осадок нужно до тех пор, пока промывные воды с
раствором роданида калия или аммония не перестанут
давать розового окрашивания. Фильтр с осадком поме¬
щают в фарфоровый тигель и после подсушивания про¬
каливают в муфельной печи при температуре 900—
1000 °С до полного сгорания фильтра. Тигель охлаждают
в эксикаторе, после чего снова тигель взвешивают с
осадком на аналитических весах. Затем взвешивают пус¬
той тигель. При правильно проведенном анализе осадок
двуокиси кремния должен быть белого цвета. Наличие
красноватой окраски указывает на наличие в ней окис¬
лов железа. Процентное содержание кремния в стали
рассчитывается по формуле:
о/ с; _ т . 0,4672 . 100
/оЬ1 - а ’
где т — масса двуокиси кремния в граммах;
а — навеска стали (или чугуна);
0,4672 — фактор пересчета двуокиси кремния на крем¬
ний.
74
При проведении арбитражных анализов к осадку добавляют
2 з капли серной кислоты и прокаливают в платиновом тигле.
Затем охлаждают в эксикаторе и взвешивают тигель. К осадку на¬
ливают 1 мл плавиковой кислоты и 2—3 капли серной кислоты и
проводят повторное прокаливание и взвешивание. Уменьшение мас¬
сы после прокаливания с плавиковой кислотой дает массу двуоки¬
си кремния. Расчет проводят, как указано выше.
Оформление результатов работы
Записать методику анализа. Цифровые данные офор¬
мить в виде таблицы.
Навеска стали (в г)
Масса двуокиси кремния
(в г)
Содержание кремния в
стали (в %)
Вопросы и задачи
1. Почему раствор, полученный при растворении навески, не
фильтруют для отделения кремниевой кислоты, а предварительно
выпаривают до образования сухих солей? Что при этом происхо¬
дит с кремниевой кислотой?
2. Какая ошибка будет внесена в результаты анализа, если
карбиды не будут окислены азотной кислотой? Напишите уравне¬
ние реакции окисления карбида железа (цементита).
3. Каким образом можно рассчитывать значение фактора пере¬
счета 0,4672 двуокиси кремния на кремний?
4. Завышен или занижен результат анализа, если двуокись
кремния после прокаливания имеет кирпично-красный цвет?
5. Какие химические реакции протекают при добавлении плави¬
ковой кислоты к осадку двуокиси кремния, загрязненному окислами
железа?
6. Платиновый тигель с прокаленной двуокисью кремния имел
массу 13,4282 г. После добавления плавиковой кислоты и повторно¬
го прокаливания его масса стала 13,4128 г.
Рассчитайте процентное содержание кремния в стали, если на¬
веска стали равнялась 1 г.
Ответ. 0,719%.
Работа 12
Определение фосфора
Наиболее распространенным методом определения
фосфора в стали является так называемый объемный или
алкалиметрический метод.
Навеску стали растворяют в кислотах, обладающих
сильными окислительными свойствами, а иногда и в цар¬
75
ской водке. При этом фосфор, находящийся в стали глав¬
ным образом в виде фосфидов, окисляется до фосфорной
кислоты:
3FesP + 4IHNO3 =
= 9Fe(N03)3 + ЗН3Р04 + 14NO + 16Н20.
Для предупреждения возможности образования кис¬
лот с меньшей валентностью фосфора, например фосфо¬
ристой кислоты, в раствор прибавляют перманганат
калия:
5Н3Р03 + 2КМп04 + 6HN03 =
= 5Н3Р04 + 2KN03 + 2Mn(N03)2 + 3H20.
Раствор перманганата калия прибавляется в избытке,
поэтому иногда за счет разложения перманганата калия
образуется темно-коричневый осадок двуокиси марганца:
2КМп04 + 2HN03 = 2KN03 + Н20 + 2Мп02 + 30.
Осадок двуокиси марганца растворяют сульфатом
двухвалентного железа:
Mn02 -f 2FeS04 + 8HN03 =
= Mn(N03)2 + 2Fe(N03)3 + 2H2S04 + 2Ha0.
Для определения фосфора фосфорную кислоту осаж¬
дают раствором молибдата аммония:
Н3Р04 + 12(NH4)2 Мо04 + 21HN03 =
= (NH4)3P04 12Мо03 + 2INH4N03 -f 12Н20.
Осаждение фосфорномолибденовокислого аммония
проводят из слабокислого раствора, в который добавля¬
ют 5—10% нитрата аммония для понижения раствори¬
мости осадка. Слишком высокая концентрация азотной
кислоты может привести к разрушению комплексного со¬
единения, а пониженное содержание может привести к
загрязнению осадка молибденовой кислотой, которая
плохо растворима в воде и в разбавленных кислотах.
Оптимальной температурой для осаждения является 60—
70 °С. Выпадение осадка задерживается при наличии в
растворе больших количеств сульфатов, соляной и фто¬
ристоводородной кислот. Мешают определению мышья¬
ковая и кремниевая кислоты, так как они также дают
комплексные соединения с молибдатом аммония.
76
Полученный осадок фильтруют, отмывают от кисло¬
ты и титруют щелочью:
1 (NH4)3P04 I2Mo03 + 23NaOH =
= (NH4)2HP04 + NH4NaMo04 + 1 lNa2Mo04 + 11H20.
По количеству щелочи, пошедшей на титрование, рас¬
считывается количество фосфора.
Осадок можно также высушить при 105 °С и взвесить
в виде (NH4)3P04- 12Мо03. При прокаливании (темпе¬
ратура 400—500 °С) это вещество разлагается и дает
вещество состава РгОб- 12МоОз-
Цель работы. Определить количество фосфора
в стали.
Оборудование и материалы
1. Азотная кислота (1:2).
2. 2-процентный раствор перманганата калия.
3. Насыщенный раствор сульфата двухвалентного
железа.
4. 20—25-процентный раствор аммиака.
5. Раствор молибдата аммония (молибденовая жид¬
кость).
6. 1-процентный раствор (нейтральный) нитрата ам¬
мония или калия.
7. 0,02-процентный водный раствор метилоранжа.
8. Титрованный раствор азотной кислоты.
9. Титрованный раствор едкого кали или едкого
натра.
10. Раствор фенолфталеина.
11. Бюретка, колбы, мерные цилиндры, фильтры.
Проведение работы
2 г стали помещают в коническую колбу на 250 мл
и приливают к ней 45 мл азотной кислоты, разбавлен¬
ной 1 2 (тяга!). Кислоту приливают вначале неболь¬
шими порциями, так как реакция протекает бурно. За¬
тем раствор нагревают до полного растворения стали.
К горячему раствору прибавляют раствор перманга¬
ната калия до появления устойчивого малинового окра¬
шивания, на что требуется около 5 мл раствора. Раствор
кипятят 5 мин, и к горячему раствору для растворения
выпадающей двуокиси марганца прибавляют по каплям
77
раствор сульфата двухвалентного железа. После этого
раствор еще кипятят 1—2 мин, охлаждают до 60—70 °С,
нейтрализуют раствором аммиака до образования не¬
большого осадка гидроокиси железа. Осадок растворя¬
ют в концентрированной азотной кислоте, которую при¬
бавляют в раствор по каплям. Затем прибавляют еще
5 мл азотной кислоты. К прозрачному раствору, имеюще¬
му температуру около 60—70 °С, приливают 30 мл рас¬
твора молибдата аммония, смесь энергично взбалтывают
в течение 1—3 мин до появления желтого осадка и
оставляют стоять в течение 0,5—1 ч для более полного
осаждения фосфорномолибденовокислого аммония. Жел¬
тый осадок отфильтровывают через двойной фильтр с си¬
ней лентой, промывают колбу и осадок на фильтре 5-про-
центным раствором нитрата аммония или нитрата калия
до нейтральной реакции. Для этого время от времени
фильтрат собирают в пробирку и прибавляют несколько
капель метилоранжа. Промытый осадок вместе с филь¬
тром переносят в колбу, в которой проводилось осажде¬
ние, приливают 20 мл титрованного раствора щелочи и
взбалтывают до тех пор, пока фильтр "не распадется на
волокна и желтый осадок полностью не растворится.
Щелочь должна быть в некотором избытке, на что ука¬
зывает окраска фенолфталеина. При отсутствии окраски
в раствор прибавляют еще 10—15 мл титрованного рас¬
твора щелочи. Затем в колбу прибавляют 20 мл свеже-
прокипячениой воды (не содержащей двуокиси углеро¬
да) и, обмыв стенки колбы водой, раствор титруют
раствором азотной кислоты до исчезновения малиновой
окраски.
Содержание фосфора в стали рассчитывается по
формуле:
п/т, _ Гр(У~-УУС).Ю0
^ т *
где Тр — титр раствора щелочи по фосфору;
V —количество миллилитров раствора щелочи, взя¬
той для растворения осадка;
Vj — количество миллилитров азотной кислоты, по¬
шедшей на титрование избытка щелочи;
С — соотношение между концентрациями щелочи и
азотной кислоты.
Рекомендуется, чтобы значение С было равно или
приближалось бы к единице. Это соотношение находим
78
путем титрования кислоты щелочью. Оно представляет
частное от деления количества миллилитров щелочи на
количество миллилитров кислоты.
Указанная формула для вычисления количества фос¬
фора выводится на основании следующих рассуждений:
по уравнению реакции разложения фосфорномолибде¬
новокислого аммония 1 г-атом фосфора, т. е. 30,98 г
фосфора, открываются 23 моль щелочи (т. е. 1290,3 г
едкого кали или 1610 г едкого натра). Допустим, что
титр применяемого раствора едкого кали равен 0,008.
Следовательно, титр едкого кали по фосфору равен:
1290,3 — 30,98
0,008 —х
0,008 ■ 30,98
1290,3
0,0001921.
Таким образом, если титр едкого кали равен Гкон,
то титр едкого кали по фосфору равен:
Т’кон ■ 30-98
1290,3
Допустим, на титрование осадка фосфорномолибде¬
новокислого аммония пошло 10 мл едкого кали. Тогда
тр • Ю 100
%Р = —^
где m — навеска стали.
В действительности к осадку приливают не 10 мл
щелочи, а некоторый ее избыток, например 25 мл. Для
определения истинного количества щелочи, пошедшей
на взаимодействие с осадком, из 25 мл щелочи нужно
вычесть количество миллилитров щелочи, не вошедшей
в реакцию. Для этого вычитают количество миллилит¬
ров кислоты, пошедшей на титрование избытка щелочи,
умноженное на соотношение между щелочью и кислотой.
Оформление результатов работы
Записать методику работы. Полученные цифровые
данные занести в таблицу.
Навеска стали
(в г)
Количество ще¬
лочи (в мл)
Количество
кислоты (в мл)
Титр щелочи
Содержание
фосфора (в %)
79
Реактивы, необходимые для анализа
Титрованный раствор щелочи. 300 г едкого натра нлн 420 а ед¬
кого кали отвешивают на технических весах, растворяют .в фарфо¬
ровом стакане, добавляют 1 г окиси бария, разбавляют до 2 л водой
и оставляют стоять на сутки. Прозрачный раствор щелочи осторож¬
но декантируют сифоном с осадка карбоната бария в стеклянную
бутыль и плотно закрывают резиновой пробкой. Этот раствор (ис¬
ходный) разбавляют так, чтобы 1 мл соответствовал 0,0002 г фос¬
фора (или 0,02% фосфора при навеске стали в 1 г). В этом случае
разложение фосфорномолибденовокислого аммония идет в наиболее
благоприятных условиях и расчеты значительно упрощаются.
Согласно уравнению реакции при разложении фосфорномолиб¬
деновокислого аммония щелочью, 23 моль щелочи отвечают 1 г-атом
фосфора, а 1 моль щелочи отвечает 1,35 г фосфора. Поэтому
разбавленный раствор должен содержать в 1 л 8,33 г едкого кали
или 5,939 г едкого натра и иметь нормальность, равную 0,1481.
В 1 мл приготовленного раствора (для титрования) содержится
0,15 г едкого натра или 0,21 г едкого кали. Чтобы получить 1 л рас¬
твора с нормальностью 0,1481, требуется 39,6 мл крепкого исход¬
ного раствора щелочи, к которому необходимо добавить 960,4 мл
воды, т. е. разбавить ее до 1 л.
Полученный раствор щелочи сливают в бутыль. Бутыль закры¬
вают пробкой с двумя отверстиями, соединенными с хлоркальцневой
трубкой, наполненной натронной известью, и с бюреткой для ти¬
трования. Титр раствора щелочи устанавливается по образцу стали
с известным содержанием фосфора, или по янтарной кислоте, или
иным способом.
Для определения титра раствора щелочи по янтарной кислоте
отвешивают на аналитических весах 0,2 г кислоты, растворяют ее в
конической колбе в 250 мл кипяченой воды, прибавив 2—3 капли
фенолфталеина. Раствор оттитровывают до появления неисчезаю¬
щей розовой окраски (колбу помещают на лист белой бумаги).
Если раствор щелочи содержит указанное количество едкого
натра или едкого кали, то на 0,2 г янтарной кислоты должно пойти
22,9 мл раствора. В том случае, когда на титрование идет больше
или меньше щелочи, раствор подгоняют к заданной величине титра,
добавляя в него воду или крепкий раствор щелочи.
Титр щелочи можно установить по стали с известным содержа¬
нием фосфора. Для этого проводят определение фосфора в стали,
как это описано выше. Титр рассчитывается по формуле:
r %Р ' m
1р ~ V— Vx С ’
где % Р — содержание фосфора в стали в процентах;
m — масса стали в граммах;
V—количество миллилитров щелочи, прибавленное для
разложения осадка фосфорномолибденовокислого ам¬
мония;
V\—количество миллилитров азотной кислоты, пошедшее
на титрование избытка щелочи;
С — отношение количества миллилитров раствора едкого
натра к количеству миллилитров азотной кислоты.
80
Значение С находится титрованием азотной кислоты щелочью
в присутствии фенолфталеина.
Титрованный раствор азотной кислоты. Для определения фос¬
фора используют 0,1481 н. раствор кислоты нли раствор, концен¬
трация которого близка к указанному.
раствор молибдата аммония. Для осаждения фосфора 50 г мо¬
либдата аммония растворяют в смеси, составленной из 180 мл во¬
ды и 30 мл аммиака (d=0,91).
Аммиачный раствор отдельными порциями вливают в разбав¬
ленную азотную кислоту, приготовленную из 260 г азотной кислоты
(*/=1,4) и 300 мл воды. При добавлении аммиачного раствора вна¬
чале выпадает осадок, который исчезает при взбалтывании. Затем
раствору дают отстояться, после чего его отфильтровывают через
слой асбеста.
Вопросы и задачи
1. Почему для анализа стали на фосфор нельзя применять со¬
ляную кислоту?
2. Почему осадок фосфорномолибденовокислого аммония не ти¬
труют непосредственно щелочью, а щелочь прибавляют в избытке
и избыток оттитровывают кислотой?
3. Какое количество едкого кали нужно взять для открытия
0,003 г фосфора в виде фосфорномолибденовокислого аммония?
Ответ. 0,155 г.
4. Каков титр едкого кали по фосфору, если его титр равен
0.0072?
Ответ. 0,000173.
Работа 13
Определение хрома
Одним из наиболее распространенных методов
определения хрома в стали является серебряно-персуль-
фатный метод. Сталь растворяют в кислоте, и трехва¬
лентный хром окисляют персульфатом аммония в при¬
сутствии ионов катализатора серебра:
Cr2(S04)3 + 3 (NH4)2S208 + 8Н20 =
= 2Н2СЮ4 + 3 (NH4)2S04 + 6H2S04.
После окисления хрома происходит окисление двух¬
валентного марганца до марганцовой кислоты, которая
окрашивает раствор в малиновый цвет. Появление этой
окраски указывает на окончание процесса окисления
хрома. Для разрушения марганцовой кислоты к раство¬
ру добавляют небольшое количество раствора хлорида
натрия:
81
2HMn04 + lONaCl + 7H2S04 =
= 2MnS04 + 5Na2S04 + 5Ci2 + 8H20.
Затем к раствору прибавляют избыточное количе¬
ство титрованного раствора сульфата железа (II), При
этом происходит восстановление хрома до трехвалент¬
ного состояния:
2Н2Сг04 + 6FeS04 + 6H2S04 =
= Cr2(S04)3 + 3Fe2(S04)3 + 8H20.
Избыток двухвалентного железа оттитровывают рас¬
твором перманганата калия:
10FeSO4 + 2КМп04 + 8H2S04 =
= 5Fe2(S04)3 + 2MnS04 + K2S04 + 8H20.
Количество хрома в стали рассчитывают на основа¬
нии количества двухвалентного железа, пошедшего на
титрование шестивалентного хрома.
Для растворения стали используют смесь серной и
фосфорной кислот. Фосфорная кислота с ионом трех¬
валентного железа дает бесцветное комплексное соеди¬
нение.
Цель работы. Определить серебряно-персуль-
фатным методом количество хрома в стали.
Оборудование и материалы
1. Смесь для растворения стали: 100 мл серной кис¬
лоты (^=1,84) вливают в 850 мл воды и добавляют
50 мл фосфорной кислоты (d—1,7).
2. 0,6-процентный раствор нитрата серебра.
3. 15-процентный раствор персульфата аммония.
4. Азотная кислота (d = 1,4).
5. Титрованные растворы соли Мора и перманганата
калия.
6. 5-процеитный раствор хлорида натрия.
7. Бюретка и пипетка на 25 мл.
Проведение работы
Отвешивают на аналитических весах около 1 г сталь¬
ной стружки, растворяют ее при нагревании в 50 мл
смеси серной и фосфорной кислот в конической колбе
82
емкостью 500 мл, Карбиды, находящиеся на дне колбы,
и двухвалентное железо окисляют концентрированной
азотной кислотой, которую по каплям приливают к го¬
рячему раствору (до прекращения выделения окислов
азота). Раствор кипятят до удаления окислов азота и
прибавляют к нему 100 мл воды, 5 мл раствора нитрата
серебра и 15 мл персульфата аммония. Раствор выдери
живают некоторое время при 60—80 °С, а затем нагре¬
вают до кипения и выдерживают в горячем состоянии
до полного разрушения персульфата аммония, что узна¬
ется по прекращению выделения пузырьков кислорода.
Затем к раствору при кипячении прибавляют 5—10 мл
раствора хлорида натрия до исчезновения малиновой
окраски марганцовой кислоты. На дно колбы выпадает
белый осадок хлорида серебра. При большом содер¬
жании марганца осадок иногда бывает окрашен в ко¬
ричневый цвет за счет двуокиси марганца. В этом слу¬
чае раствор необходимо еще прокипятить некоторое
время.
Подготовленный раствор снимают с электроплитки,
прибавляют к нему около 250 мл холодной воды и,
охладив до комнатной температуры, приливают к нему
пипеткой 20 или 25 мл раствора соли Мора. После
взбалтывания раствор должен иметь светло-зеленую
окраску, без малейшего желтого оттенка, что указывает
на полное восстановление хромат-иона. Затем раствор
титруют перманганатом калия до появления слабо-ро¬
зового окрашивания, не исчезающего при взбалтывании
в течение 1 мин.
После титрования устанавливают соотношение меж¬
ду раствором перманганата калия и раствором соли Мо¬
ра. Для этого в колбу с оттитрованным раствором, где
проводилось определение хрома, прибавляют пипеткой
10—20 мл раствора соли Мора и титруют пермангана¬
том до появления устойчивой розовой окраски.
Количество хрома в стали рассчитывается по фор¬
муле:
О/ Гг - ^KMnOj/Cr (V ' С У\) 1QQ
где ТкмпО^/сг — титр раствора перманганата калия, выра~
женный в граммах хрома;
V — число миллилитров раствора соли Мора,
83
добавленное для восстановления шести¬
валентного хрома;
V\ — число милилитров раствора пермангана¬
та калия, пошедшее на титрование избыт¬
ка соли Мора, не вступившей в реакцию
с шестивалентным хромом;
С — соотношение между растворами соли
Мора и перманганата калия. Оно равно
числу миллилитров раствора перманга¬
ната калия, пошедшего на титрование со¬
ли Мора, деленному на число миллилит¬
ров раствора соли Мора;
т — масса образца в граммах.
Титр перманганата калия по хрому обнаруживается
по навеске стали, содержащей известное количество хро¬
ма, или по оксалату натрия. Когда нормальность перман¬
ганата калия известна, его титр по хрому рассчитывает¬
ся исходя из следующих рассуждений.
Во время титрования протекают обычные окисли¬
тельно-восстановительные процессы и грамм-эквивалент
перманганата калия эквивалентен грамм-эквиваленту
хрома. Поскольку марганец меняет свою валентность на
пять единиц, а хром—на три, то грамм-эквивалент пер¬
манганата калия равен его молекулярной массе, делен¬
ной на пять, а хрома — его атомной массе, деленной на
три. Следовательно, 1 л 1 н. раствора перманганата ка¬
лия, в котором находится 1 г-экв перманганата калия,
отвечает 52,01 3 = 17,336 г хрома. Тогда 1 мл перманга¬
ната калия, если раствор его 1 н., отвечает:
гр 17,336 н. /Ч
iKMn04/Cr = jQQQ (2) Хрома.
Оформление результатов работы
Записать методику выполнения работы. Полученные
цифровые данные при анализе занести в таблицу.
Навеска (в г)
Соль Мора
(в мл)
Перманганат
(в мл)
Соотношение
Fe'/МпОд
Содержание
хрома (в %)
81
Реактивы, необходимые для анализа
1. Раствор соли Мора. 200 г FeS04 • (NH^SCU • 6Н2О растворя¬
ют в 900 мл воды, прибавляют в раствор 100 мл концентрирован¬
ной серной килоты, разбавляют его до 5 л и перемешивают.
2. Титрованный раствор перманганата калия. Для приготовле¬
ния титрованного раствора перманганата калия 10 г соли помещают
в стакан емкостью 500 мл и приливают туда около 200 мл воды.
После размешивания раствор сливают в бутыль емкостью 5 л, окра¬
шенную снаружи черной краской. К оставшейся в стакане соли при¬
ливают еще 200—300 мл воды, снова все размешивают и сливают
раствор в бутыль. Эту операцию повторяют до растворения соли.
Раствор в бутыли доливают водой до 5 л и после перемешивания
оставляют на несколько дней. Затем его профильтровывают через
слой чистого асбеста. Титр перманганата калия устанавливают по
навеске оксалата натрия или по стали с известным содержанием
хрома.
Для установления титра по оксалату натрия 0,15—0,2 г соли
растворяют в конической колбе на 300 мл, прибавляют 10 мл кон¬
центрированной серной кислоты и, нагрев раствор до 80 °С, титруют
приготовленным раствором перманганата калия до исчезающей в
течение 1 мин слабо-красной окраски.
Расчет титра проводится на основании уравнений протекающих
реакций. Из уравнения реакции
5Na2C204 + 2КМп04 + 8H2S04 =
= 5Na2S04 + K2S04 + 2MnS04 + 8H20 + ЮС02
следует, что 5Ыа2Сг04 соответствует 2КМп04, а 1 г-экв Na2C204, т. е.
67,0 г, соответствует 1 г-экв КМп04.
Из вышеприведенного уравнения реакции восстановления шести-
валеитиого хрома следует, что 1 г-экв железа соответствует 1/з
г-атом хрома, или 17,33 г хрома, и хром к оксалату натрия отио-
J7,38 л осп
сится как ——— = 0,259.
Ь7
Следовательно,
^KMnOJCr = ~ * °>2£>9,
где Гкмпо^/сг — титр перманганата калия по хрому;
пг — масса оксалата натрия в граммах;
V—объем перманганата калия, пошедшего на титрова¬
ние навески оксалата натрия.
При установлении титра перманганата калия по стали с извест¬
ным содержанием хрома навеску стали растворяют в кислоте и про¬
водят все операции, которые были выше описаны при анализе стали
на хром. Титр перманганата калия рассчитывают по формуле:
'г а • m
KMnO./Cr — у с _ [ад -
где а — содержание хрома в стали в процентах;
иг — масса стали;
V — количество миллилитров раствора соли Мора, прибав¬
ленное для восстановления хромовой кислоты;
85
С — соотношение между раствором соли Мора и перман¬
ганатом калия;
V\— количество миллилитров перманганата калия, пошед¬
шее на обратное титрование избытка двухвалентного
железа.
Вопросы и задачи
1. Почему появление малиновой окраски указывает на полноту
окисления хрома? При ответе учтите значения окнслительио-восста-
новительцых потенциалов соответствующих реакций.
2. Насколько изменятся результаты анализа при определении
хрома, если не удалить марганцовую кислоту хлоридом натрия?
3. Для чего после растворения стали в смеси кислот к раствору
прибавляют небольшое количество азотной кислоты? При ответе
учтите большое сродство хрома, находящегося в стали, к углероду.
Как может измениться результат анализа, если азотная кислота не
будет добавлена?
4. Соотношение между солью Мора и перманганатом калия ре¬
комендуется устанавливать в той же колбе, где проводилось опреде¬
ление хрома, не выливая имеющийся там оттитрованный раствор.
Почему не рекомендуется устанавливать это соотношение в отдель¬
ной колбе?
5. Для определения титра перманганата калия по хрому было
взято I г нормали, содержащей 2,1% хрома. После растворения
стружки и окисления хрома к раствору было прибавлено 25 мл соли
Мора, на обратное титрование которой пошло 10 мл перманганата
калия. Соотношение между перманганатом калия и солью Мора
равно 24 25, т. е. равно 0,96. Рассчитайте титр перманганата калия
по хрому.
Ответ. 1 мл КМп04 открывает 0,0015 г хрома.
6. Выведите формулу для определения титра перманганата калия
по хрому на основании проведения работы с нормалью. Дайте ей
обоснование.
7. Определите содержание хрома в стали, если в раствор было
прибавлено 25 мл соли Мора и на титрование ее избытка пошло
2 мл перманганата калия, 1 мл которого открывает 0,001762 г хрома.
На титрование 20 мл соли Мора пошло 19 мл перманганата калия.
Навеска стали равнялась 0,8500 г.
Ответ. 4,49%.
Работа 14
Определение марганца
Одним из наиболее распространенных и быстрых ме¬
тодов определения марганца в сталях, при его содержа¬
нии не более 1,5%, является персульфатный метод.
Сталь растворяют в смеси серной и фосфорной кислот и
двухвалентный марганец окисляют персульфатом аммо¬
86
ния в присутствии ионов серебра в качестве катализато¬
ра до марганцовой кислоты:
2MnS04 + 5 (NH^SA + 8Н20 =
- 5 (NH4)2S04 + 2HMn04 + 7Н£0^
Ионы серебра осаждают также ионы хлора, которые
могут присутствовать в кислотах в виде следов. Их при¬
сутствие мешает окислению марганца, так как ионы хло¬
ра в кислой среде являются восстановителем по отноше¬
нию к семивалентному марганцу.
Полученный раствор титруют раствором тиосульфата
натрия. В результате сложной реакции происходит вос¬
становление марганцовой кислоты до соли двухвалентно¬
го марганца, и вследствие этого раствор обесцвечивается.
Сталь растворяют в смеси серной и фосфорной кис¬
лот. Фосфорная кислота дает с ионом трехвалентного
железа бесцветное комплексное соединение и поэтому
предупреждает появление бурой окраски, вызываемой
ионом трехвалентного железа.
Взаимодействие марганцовой кислоты с тиосульфа¬
том носит сложный характер. Поэтому титр тиосульфата
устанавливают по навеске стали с известным содержа¬
нием марганца.
Цель работы. Определить количество марганца
в стали.
Оборудование и материалы
1. Смесь для растворения стали составляется из
940 мл воды, 50 мл концентрированной серной кислоты,
5 мл концентрированной фосфорной кислоты и 5 мл
0,6-процентного раствора нитрата серебра.
2. Азотная кислота (d=l,4).
3. 15-процентный раствор персульфата аммония.
4. Раствор тиосульфата натрия готовят так: 0,65 г
Na2S203 * 5Н20 растворяют в 100 мл воды, разбавляют
раствор до 1 л и выдерживают его в течение нескольких
дней.
Проведение работы
При содержании марганца менее 1 % на аналитиче¬
ских весах взвешивают 0,2—0,3 г стали. При большем
содержании марганца навеску уменьшают до 0,1—0,05 г.
87
Навеску пересыпают в колбу на 150 мл, растворяют при
нагревании в 25 мл смеси кислот. Колбу нагревают на
асбестированной сетке или водяной бане. Раствор при
этом кипятить не следует. После растворения на дне
колбы остается небольшой осадок карбидов. Для окисле¬
ния карбидов к раствору прибавляют по каплям азот¬
ную кислоту. Для удаления окислов азота раствор еще
некоторое время нагревают. Затем к раствору прибавля¬
ют 40 мл теплой воды и около 10 мл раствора персуль¬
фата аммония. Раствор некоторое время выдерживают
горячим, а затем нагревают до начала закипания. Если
розовая окраска не появится, то к раствору приливают
еще около 3—5 мл персульфата аммония. После окисле¬
ния марганца и появления розовой окраски марганцовой
кислоты раствор снимают с плиты, несколько раз взбал¬
тывают и оставляют стоять до прекращения выделения
пузырьков кислорода из персульфата аммония. Затем
его охлаждают и титруют раствором тиосульфата натрия
до исчезновения розовой окраски.
Одновременно в тех же условиях проводят анализ
стали с известным содержанием марганца. По количест¬
ву миллилитров раствора тиосульфата, пошедшего на
это второе титрование, определяют титр раствора тио¬
сульфата натрия по марганцу. Количество марганца
пропорционально количеству миллилитров раствора
тиосульфата натрия, пошедшего на титрование, и его
количество рассчитывается по формуле:
%Ш = У г %Мп Эт >
V 2
где %МпЭт — процентное содержание марганца в эта¬
лоне;
V\—количество миллилитров раствора тио¬
сульфата натрия, пошедшего на титрова¬
ние марганца в анализируемой стали;
V2 — количество миллилитров раствора тио¬
сульфата, пошедшего на титрование мар¬
ганца в стали известного состава.
Формула применима в том случае, если были взяты
одинаковые массы анализируемой стали и эталона.
В противном случае вычисляют титр тиосульфата по
марганцу.
Допустим, в стали было 0,6% марганца. На титро¬
вание пошло 3 мл тиосульфата натрия. Следовательно,
88
3 мл при массе стали в 0,2 г открывает 0,0012 г мар¬
ганца, а 1 мл соответствует 0,0004 г марганца. Отсюда
титр тиосульфата натрия по марганцу равен 0,0004.
Титр тиосульфата натрия по марганцу определяется
по формуле:
т %Мп т
1 Na2S203/Mn — юо . мл Na2s203 ’
где %Мп — процентное содержание марганца в стали;
мл ЫагЭгОз — количество миллилитров тиосульфата
натрия, пошедшее на титрование;
т — масса стали в граммах.
Количество марганца в анализируемой стали в этом
случае определяется по формуле:
%Мп =
^Na8SaQ3/Mn • МЛ Na2S2Q3
т
100.
Оформление результатов работы
Записать методику работы. Полученные данные
оформить в виде таблицы.
Навеска стали
(в г)
Количество тиосульфата натрия,
израсходованного на титрование (в мл)
Количество Мп в ста¬
ли (в %)
нормаль | образцы
Вопросы и задачи 11. Почему в присутствии соляной кислоты (или хлоридов) двух¬
валентный марганец не окисляется персульфатом аммония? При от¬
вете учтите свойства марганцовой кислоты и ионов хлора.
2. Почему, зная количественное содержание тиосульфата нат¬
рия в растворе, нельзя рассчитать его титр по марганцу?
3. Для чего к серной кислоте, применяемой для растворения
навески, прибавляют фосфорную кислоту? Какую роль она играет
при титровании марганцовой кислоты тиосульфатом натрия?
4. На титрование 0,25 г нормали, содержащей 0,58% марганца,
пошло 4,2 мл раствора тиосульфата натрия. Рассчитайте титр тио¬
сульфата натрия по марганцу.
Ответ. 0,0002452.
5. На титрование 0,2742 г нормали, содержащей 0,5% марганца,
пошло 3,8 мл тиосульфата натрия, а на титрование 0,2472 г анали-
89
зируемой стали пошло 4,5 мл тиосульфата натрия. Рассчитайте со-
держание марганца в анализируемой стали.
Ответ. 0,657.
6. На титрование одинаковых навесок нормали и анализируемой
стали пошло соответственно 3,5 и 4,2 мл тиосульфата натрия. Рас¬
считайте количество марганца в анализируемой стали, если нормаль
содержала 0,72% марганца.
Ответ. 0,86%.
7. Можно ли проводить описанным методом анализ на марганец
при значительном содержании в стали хрома? В виде какого соеди
нения будет находиться хром после окисления персульфатом ам¬
мония?
8. Почему при анализе на марганец для растворения стали при¬
меняют серную, а не соляную кислоту?
9. Напишите уравнение реакции тиосульфата натрия с марган¬
цовой кислотой, считая, что в результате основной реакции сера
окисляется до шестивалентного состояния.
10. Почему иногда после титрования марганцовой кислоты по¬
лученный бесцветный раствор постепенно снова приобретает мали¬
новую окраску? На какую неточность в работе это указывает? Какое
из прибавляемых во время анализа веществ осталось неразложнв-
шимся?
11. Какое влияние оказывает на окраску раствора присутствие
фосфорной кислоты?
Работа 15
Знакомство со стилосколом СА-10. Построение
градуировочного графика
Для уяснения основных принципов спектрального
анализа необходимо вспомнить некоторые законы гео-
Рис. 25. Схема преломления
н отражения светового луча.
метрической оптики: 1. За¬
кон прямолинейного распро¬
странения света. 2. Закон от¬
ражения света. 3. Закон пре¬
ломления света. Согласно
первому закону луч света в
однородной среде распрост¬
раняется прямолинейно. Од¬
нако, если луч света падает
на границу раздела двух
сред, он претерпевает изме¬
нение. Часть светового луча
на границе раздела отража¬
ется, причем угол падения
луча а равен углу отраже¬
ния (рис. 25).
90
Кроме того, часть светового потока входит во вторую
среду, преломляясь в ней. Закон преломления говорит,
что:
sin а ,
—■-Q- = const = Л,
smfi
где п — показатель преломления для данных двух сред.
Кроме того, при прохождении световой волны через
границу раздела двух сред наблюдается явление дис¬
персии света, сущность которого заключается в том, что
скорость света не является постоянной величиной и в
различных средах зависит от частоты электромагнит¬
ных колебаний.
Рис. 26. Преломление света в трехграиной
призме.
Изменения в скорости распространения различных
волн приводят к отклонению направления луча от
первоначального. Представим себе, что на границу раз¬
дела двух сред под некоторым углом падает монохрома¬
тический, т. е. состоящий из волн одной длины, свето¬
вой поток. В этом случае произойдет преломление
света, т. е. отклонение его от первоначального направле¬
ния. Если мы теперь направим на границу раздела
двух сред не монохроматический световой поток, а сме¬
шанный, то лучи, имеющие различные частоты колеба¬
ний, будут распространяться во второй среде с различ¬
ными скоростями, а следовательно, будут отклоняться
от первоначального направления на различные углы.
Произойдет разложение смешанного светового потока в
спектр. Дисперсией обладают все прозрачные среды,
кроме вакуума. В вакууме скорость распространения
электромагнитных волн не зависит от частоты.
Очень удобно производить такое разложение света в
спектр при помощи трехгранной призмы, которая яв¬
91
ляется до настоящего времени основной деталью боль¬
шинства спектральных приборов (рис. 26).
Причиной возникновения излучений и спектров яв¬
ляется неравномерно ускоренное движение электриче¬
ских зарядов (движение с переменным ускорением).
Так, при резком торможении пучка электронов, па¬
дающих на твердую поверхность, возникает рентгенов¬
ское излучение. При перескоке электронов с одной ор¬
биты на другую в атомах возникают атомные спектры.
При колебаниях более крупных зарядов (молекул,
групп атомов) возникают спектры, расположенные в
инфракрасной области. Энергия, переносимая излуче¬
нием, зависит от частоты колебаний:
Е = h * v.
Отсюда следует, что наибольшими энергиями обла¬
дают лучи, имеющие наибольшие частоты, или, иначе
говоря, наименьшие длины волн. В порядке убывания
энергий излучения лучи можно расположить в следующий
ряд: рентгеновские, ультрафиолетовые, видимые, ин¬
фракрасные, радиоволны.
Кроме, указанного деления спектров по длинам волн,
все спектры можно разделить на спектры испускания
(эмиссионные) и спектры поглощения (абсорбцион¬
ные). Кроме того, для удобства спектры делят на атом¬
ные и молекулярные. Эмиссионные спектры возникают
при переходе электронов в атомах с более высоких
энергетических уровней на более низкие уровни. При
этом излучается квант энергии:
Еi Е% = h • vf
причем Е{>Е2.
При переходе частицы с более низкого энергетиче¬
ского уровня на более высокий энергия поглощается.
В случае движения электрона по орбите не наблю¬
дается ни излучения, ни поглощения энергии. Испуска¬
ние света веществом наблюдается при сообщении ему
энергии, например, если через газообразный водород
пропустить электрический разряд, возникает свечение.
Это свечение можно разложить в спектр. Спектр водо¬
рода является наиболее простым. Он состоит из ряда
линий, расположенных сериями (рис. 27). Таких серий
несколько. Впервые закономерность в расположении
92
линий одной из серий спектра водорода обнаружил в
1885 г. Бальмер. Он предложил формулу, описывающую
положение линий в этой серии. В настоящее время фор¬
муле Бальмера придается такой вид:
1
X
/I2
)■
где п — целое число, R — постоянная Ридберга.
I
6500 6000 5500 5000 4500
Рис. 27. Спектр водорода.
4000 5500
Затем были описаны при помощи аналогичных фор¬
мул и другие линии водородного спектра. Общей форму¬
лой, описывающей линии всех серий, является следую¬
щая:
Теоретическое объяснение спектра водорода было да¬
но Бором в 1913 г. на основании предложенных им по¬
стулатов.
Спектры более тяжелых элементов имеют более
сложное строение и насчитывают сотни различных ли¬
ний. Каждый элемент имеет свой характерный спектр.
Это позволяет проводить качественный анализ веществ.
Количественное содержание вещества влияет на интен¬
сивность (яркость) спектральных линий. На основании
яркости соответствующих линий можно провести коли¬
чественный спектральный анализ.
Цель работы. Ознакомиться со стилоскопом
СА-10. Построить градуировочный график.
Оборудование и материалы
1. Стилоскоп СА-10.
2. Индуктор высокого напряжения (катушка Рум-
корфа).
3. Гейслеровская трубка с гелием.
93
4. Источник постоянного тока на 10—12 в (аккуму¬
лятор, выпрямитель).
5. 10—15-процентный раствор хлорида натрия и та¬
кой же концентрации раствор хлорида лития.
6. Асбест и кусок иихромовой спирали.
Проведение работы
Оптическая схема стилоскопа СА-10 приведена на
рисунке 28. Свет от источника (электрическая дуга, иск¬
ровой разряд и т. д.), пройдя осветительную линзу 1У
концентрируется на щели
2, пропускающей узкий
пучок света. Призма 3 по¬
ворачивает световой пу¬
чок, идущий от щели, и
направляет его на объек¬
тив 4. По выходе из объ¬
ектива свет попадает на
трехгранные диспергиру¬
ющие призмы 5 и 6, кото¬
рые образуют спектр. Вто¬
рично пройдя объектив 4У
свет поворотной приз¬
мой 7 направляется в
окуляр 5.
Все части стилоскопа
(рис. 29) смонтированы
на плите, служащей осно¬
ванием прибора. Основны¬
ми частями стилоскопа являются: столик для анализиру¬
емых материалов 1, конденсорная трубка 2, головная
часть 3, объективная трубка 4У призменная коробка 5,
окуляр 6. Для защиты глаз наблюдателя от яркого света
на трубке 2 имеется щиток 7. Диспергирующие призмы
находятся в призменной коробке. Одна из них неподвиж¬
на, другая может поворачиваться под некоторым углом,
вследствие чего спектр перемещается в поле зрения оку¬
ляра. Поворот этой призмы осуществляется с помощью
маховика 8У соединенного с барабаном 9У имеющим шка¬
лу. Для наведения линий на резкость в различных облас¬
тях спектра служит кольцо 10у поворотом которого осу¬
ществляется фокусировка.
Рис. 28. Оптическая схема стило¬
скопа СА-10:
1 — осветительная линза; 2 — щель; 3 —
призма; 4 — объектив; 5, 6 — дисперги¬
рующие призмы; 7 — поворотная
призма; 8 — окуляр.
94
Стилоскоп имеет два окуляра. При рассматривании
спектров с большим количеством линии (сталь, тяже¬
лые металлы) применяется окуляр с двадцатикратным
Рис. 29. Внешний вид стилоскопа:
/ — столик; 2 — конденсорная трубка; 3 — головная часть; 4 — объективная
трубка; 5 — призменная коробка; 6 — окуляр; 7 — щиток; 8 — маховик;
9 — барабан; 10 — кольцо; 11 — держатель электродов; 12 — дисковый элек¬
трод; 13, 14 — шиуры питания.
увеличением; для рассматривания спектров цветных ме¬
таллов применяется окуляр с 12Уз увеличением.
Анализируемый образец помещается на столик /,
против вспомогательного электрода (медный диск или
стальной цилиндрический образец), зажимаемый держа¬
телем 11. При использовании дискового электрода его
перед каждым анализом поворачивают при помощи ма¬
ховика. Электрический ток подводится через контакты
13 и 14.
Дисковый электрод изготовляется из чистой меди.
Цилиндрический электрод имеет длину 100 мм и диа¬
метр не более 7,5 мм. Содержание хрома в нем должно
быть не более 0,1%, марганца—не более 0,3—0,4% и
никеля — не более 0,2—0,3% при отсутствии молибдена,
вольфрама, ванадия, кобальта.
95
В качестве источника возбуждения применяют ге¬
нератор дуги переменного тока (ПС-33, ИГ-2, ДГ-1).
Корпус стилоскопа находится под высоким напря¬
жением, и поэтому его нужно обязательно заземлять
при помощи клеммы.
«at
I
<о
см
са
5 1
См
> ^
>
1
I
Г*
2Р
Рис. 30. Спектр гелия.
Для построения градуировочного графика можно
воспользоваться спектром какого-либо элемента, имею¬
щего сравнительно небольшое количество спектральных
линий, например спектром гелия. Наиболее яркими явля¬
ются 7 линий (рис. 30).
Рис. 31. Расположение газоразрядной
трубки для снятия спектра.
зрения окуляра
стилоскопа
с визиром.
Для получения спектра гелия против центра объекти¬
ва стилоскопа устанавливают Гейслеровскую трубку, на¬
полненную гелием (рис. 31), и присоединяют к ней ток
высокого напряжения. Барабан 9 устанавливают на ну¬
левое деление шкалы и просматривают спектр в окуляр
стилоскопа, поворачивая барабан. Появляющиеся в поле
зрения спектральные линии устанавливают таким обра¬
зом, чтобы они совпадали с указателем, имеющим форму
треугольника (рис. 32).
При этом одновременно отмечают деление шкалы
барабана, при котором соответствующая спектральная
линия находится в поле зрения. Затем составляют таб¬
лицу по схеме:
96
„ 0
Длина волн (в А)
Деление шкалы барабана
6678
5875
5015
4921
4713
4471
4026
После этого на основании данных таблицы строят
градуировочный график (рис. 33).
Точность построения графика может быть проверена
по спектральным линиям какого-либо элемента, имею¬
щего характерные спектральные линии, например по
спектру натрия и лития. Для этого перед объективом сти-
лоскопа создают пламя, окрашенное солями натрия или
лития: зажигают горелку и вносят в нее кусок асбеста,
смоченный раствором хлорида натрия или хлорида ли¬
тия. Пламя должно находиться на некотором расстоянии
от объектива стилоскопа, чтобы теплота не разогревала
стекла.
Поворотом градуированного барабана находят яр¬
кую желтую линию натрия. В действительности эта
яркая линия состоит из двух близко расположенных ли¬
ний (дублет), имеющих соответственно длины волн,
о о
равные 5896 А и 5889 А. Замечаем, при каком делении
шкалы барабана видна данная желтая линия натрия.
Затем проверяем правильность заранее построенного
графика. Для этого от
оси координат из точки,
соответствующей длине
о
волны, равной 5896 А,
восстанавливаем пер¬
пендикуляр до пересе¬
чения с кривой и из
точки пересечения опу¬
скаем перпендикуляр
на ось абцисс. Место
пересечения перпенди-
/Г
5000
4000
5000
2000
50
то
150
4 Н, Г, Ключников
Рис. 33. Градуировочный график.
97
куляра должно соответствовать делению шкалы бараба
на, которое наблюдалось при появлении желтого дубле
та натрия.
Подобным же образом для проверки правильности
построения графика можно воспользоваться яркой ли¬
нией спектра лития, расположенной в длинноволновой
части и имеющей красную окраску. Ее длина волны рав¬
на 6708 А.
Работа 16
Определение хрома, никеля и кремния
в стали
Качественное обнаружение элементов в стали про¬
водится на основании характерных спектральных ли¬
ний. При анализе сплавов интенсивность линий (яркость)
соответствующего элемента определяется количеством
этого элемента в сплаве. Чем больше определяемого
элемента в сплаве, тем более интенсивны его спектраль¬
ные линии. Такая зависимость между количеством эле¬
мента и яркостью линий позволяет количественно, с из¬
вестной точностью, определить его содержание в сплаве.
Поскольку яркость спектральных линий основного метал¬
ла, например железа, постоянна, то эти линии являются
стандартом для определения яркости линий другого ме¬
талла.
Цель работы. Провести полуколичественный ана¬
лиз стали на хром, никель, кремний, марганец.
Оборудование и материалы
1. Стилоскоп СА-10.
2. Генератор.
3. Набор легированных сталей.
Проведение работы
Исследуемый образец помещают на столик стило-
скопа, и между образцом и вспомогательным электро¬
дом зажигается дуга, спектр которой рассматривается
в окуляр стилоскопа.
Для определения хрома в качестве вспомогательного
электрода используют армко-железо или сталь-10, не
9й
содержащую хрома. При содержании хрома в пределах
4—15% пользуются одной линией хрома и двумя линия*
ми железа. Длина воли этих линий и их условные обо¬
значения приведены в таблице 8.
Таблица 8
Элемент
Длина линий спектра
(а А)
Условное обозначение
линий
Хром
4922,0
1
Железо
4924,7
2
Железо
4918,9
3
Фотография участка спектра железа с указанными
линиями приведена на рисунке 34.
Вначале, включив стилоскоп, находят участок спект¬
ра, приведенный на фотографии. Для этого, пользуясь
градуировочным графиком, находят, при каком делении
шкалы барабана должны появиться в поле зрения оку¬
ляра линии железа и хрома, приведенные в таблице.
Затем устанавливают барабан на нужном делении шка¬
лы, включают дугу и, пользуясь фотографией, отыски-
35
См
&
§
<о ^ СО Со
Ч 'О
П ^ ^ ч
'■П СС$ >-_•
со аь со со
ап со та ао
V ^ ^ ^
Ml М \1
QQ
if*
с- СО
С\1
со irj
8
со со со
^ >
СО
«О ^
S5 ^
СО
м-
I \/ \ I
5
40 NO
> CSj
^ со
Ц-О V
to
Со со
со
см*
\/ I
/\
1
1
i\
/\
$
Cs
QQ
81?
Ьо С5
59 £
§1
t4T
§
"«ч.-
5;
*rS >'
СМ СМ
со со
to to
О) со
'***
м- V
'W V
$ I
Со csj
to to
СО Со
Рис. 34. Дуговой спектр железа.
99
вают нужный участок спектра. Затем находят линию
хрома 1 и сравнивают ее интенсивность (яркость) с ли¬
ниями железа 2 и 3. Если линия хрома 1 по интенсив¬
ности равна линии 3, то количество хрома в стали ле¬
жит в пределах 10—15%. Если первая линия по интен¬
сивности равна или несколько ярче второй линии, то
количество хрома равно 4% или несколько больше. На¬
конец, если линия 1 слабее линии 2, то количество хро¬
ма равно примерно 1%.
Оформление результатов работы
Составить градуировочный график стилоскопа. За¬
писать результаты анализа стали.
ГЛАВА IV
кислоты
§ 6. ПОЛУЧЕНИЕ КИСЛОТ
Работа 17
Получение соляной кислоты из хлора и водорода
Получение синтетической соляной кислоты расчле¬
няется на три стадии:
1. Получение хлора и водорода.
2. Получение хлористого водорода.
3. Получение соляной кислоты растворением хлори¬
стого водорода в воде.
Хлор и водород можно получать электролизом или
при помощи химических реакций.
Соединение хлора и водорода проводится в промыш¬
ленности в специальных кварцевых или металлических
горелках. В лабораторных условиях, ввиду того что ис¬
пользуются небольшие количества хлора и водорода,
горелка может быть стеклянной.
На заводах растворяют хлористый водород в погло¬
тительных башнях.
Цель работы. Получить раствор соляной кисло¬
ты. Ознакомиться с работой установки по получению
хлора, водорода и соляной кислоты. Рассчитать ко¬
личество соляной кислоты, получаемое теоретически в
зависимости от силы и времени прохождения электри¬
ческого тока. Определить выход кислоты.
101
Вариант 1
Оборудование и материалы
1. Электролизер для получения хлора и водорода.
2. Горелка для соединения хлора и водорода.
3. Поглотитель.
4. Водоструйный насос или аспиратор.
Рис. 35. Получение соляной кислоты из хлора и водорода:
1 — горелка, заполненная сверху силикагелем; 2—осушительные ко¬
лонки; 3 — кожух горелки; 4 — поглотитель хлористого водорода.
5. Колба на 100 мл.
6. Раствор индикатора (фенолфталеин, метиловый
оранжевый).
7. Раствор нитрата серебра.
8. 0,1 н. раствор щелочи.
Проведение работы
Хлор и водород получают электролизом раствора
хлорида серебра в электролизере, изображенном на
рисунках 13, 14 или 16.
Количество водорода, необходимое для нормальной
работы горелки, выделяется, когда через электролизер
проходит ток в 10—12 а, но, если пропускать ток в 15 —
20 а, горение становится более устойчивым. Перегрев
102
электролита можно устранить определенным соотноше¬
нием между силой тока, подаваемого на электролизер,
и объемом электролизера. Минимальный объем элект¬
ролизера в литрах примерно равен силе подаваемого
тока (в амперах).
Горелка 1 (рис. 35) делается из тройника. Выходное
отверстие для смеси газов должно быть около 0,1 мм.
В противном случае горение хлора и водорода будет не¬
устойчивым. Водород и хлор смешиваются в самой горел¬
ке, которая заполнена порошкообразной окисью хрома,
алюминия или силикагеля. Чтобы окислы не провали¬
лись вниз горелки, нижнюю ее часть заполняют рыхлым
слоем асбеста. Соединение хлора и водорода происходит
в слое окислов и называется беспламенным горением.
При введении горелки в кожух часто горение прекра¬
щается, но этого не наблюдается, когда в кожух
подается воздух. На резиновой пробке, надетой на
горелку, делают напильником небольшие продольные
прорези для подачи воздуха. Соединение хлора идет бес¬
прерывно, если вместо закрытого кожуха над горелкой
установить открытую воронку (колокол), соединенную
с поглотителем.
Хлор и водород, получаемые в описанном электроли¬
зере, насыщены парами воды, поэтому их предвари¬
тельно сушат хлоридом кальция. Применение жидких
осушителей, например серной кислоты, мало эффектив¬
но, так как газы проходят через промывные склянки в
виде пузырьков, что вызывает кратковременные пере¬
рывы в подаче газов в горелку и приводит к прекраще¬
нию горения.
При промышленном получении хлористого водорода
в горелку подают некоторый избыток водорода по срав¬
нению с хлором.
При работе на описанной установке избыток водоро¬
да получается за счет того, что хлор частично раство¬
ряется в воде и взаимодействует со щелочью, получае¬
мой на катоде.
Поглотитель для хлористого водорода делается стек¬
лянным. Внутрь его помещается насадка в виде битого
стекла или коротких трубок для увеличения поверхно¬
сти соприкосновения хлористого водорода и воды.
При получении соляной кислоты нужно соблюдать
следующий порядок в работе.
Сначала промыть поглотитель дистиллированной во¬
дой от соляной кислоты, капли которой могут остаться
от предыдущего опыта. Кожух горелки протирают.
Затем включают электролизер, следя за тем, чтобы
сила подаваемого тока равнялась 10—15 а (в зависи¬
мости от его объема). После этого следует проверить
водород на чистоту. Убедившись в чистоте водорода,
трубки обтирают марлей, присоединяют к горелке и
пропускают хлор и водород в течение 0,5—1 мин.
Поджигают водород и хлор горящей лучинкой или
горелкой. При этом необходимо надеть защитные очки.
Убедившись, что горение идет нормально, горелку встав¬
ляют в кожух, включают водоструйный насос, замечают
время начала синтеза и силу подаваемого тока. В по¬
глотитель необходимо предварительно налить из капель¬
ной воронки 10—12 мл воды, чтобы смочить насадку.
Пропускают хлористый водород в течение 3—4 мин.
После этого выключают электролизер, замечают время
окончания синтеза и, открыв кран поглотителя, промы¬
вают его 3 раза по 20—30 мл воды, сливая полученную
кислоту в мерную колбу. Затем кислоту оттитровывают.
Теоретический выход соляной кислоты1 легко под¬
считать на основании времени прохождения тока и его
силы: при прохождении тока в 26,8 а-ч на электродах
выделяется по 1 г-экв хлора и водорода, т. е. можно по¬
лучить 1 моль хлористого водорода. Если же через
электролизер шел ток силой а ампер и в течение t ча¬
сов, то хлористого водорода теоретически должно по¬
лучится:
НС1 (г) =
М • а . t
26^8 ’
где М — молекулярная масса хлористого водорода.
Необходимо отметить, что описанная установка весь¬
ма капризна в работе и горение хлора часто прекраща¬
ется. Электролит при небольшом объеме быстро обед¬
няется ионами хлора около анода, вследствие чего по¬
ступление хлора в горелку падает. Поэтому его
приходится время от времени перемешивать и насыщать
хлоридом натрия. При многократном получении соля¬
1 Практически выход соляной кислоты обычно незначителен,
так как наряду с хлором выделяется большое количество ки¬
слорода.
104
ной кислоты этим способом нужно использовать элект¬
ролизеры большой емкости с несколькими анодами
(см. главу II).
Оформление результатов работы
Зарисовать установку для получения соляной кис¬
лоты и описать ее работу. Заполнить таблицу по форме.
Время
электроли¬
за (в мин)
Сила тока
(в а)
Количество
электроэнер¬
гии (в а-ч)
Теоретически
получится
НС1 (в г)
Получено
НС1 (в г)
Выход
(в %)
Вариант 2
Цель работы. Получить раствор соляной кислоты
из хлора и водорода.
Оборудование и материалы
1. Установка для получения соляной кислоты, со¬
бранная по схеме, приведенной на рисунке 36.
2. 0,1 н. раствор щелочи.
3. Раствор фенолфталеина.
Проведение работы
Устройство и работа установки по получению соля¬
ной кислоты даны на рисунке 36. Предварительно в гра¬
дуированный газометр, заполненный насыщенным рас¬
твором хлорида натрия, собирают хлор, полученный дей¬
ствием соляной кислоты на перманганат калия или
двуокись марганца. Затем, вынув горелку, пускают из
аппарата Киппа ток водорода, который предварительно
проверяют на чистоту. Водород поджигают и затем пус¬
кают струю хлора, наблюдая за изменением цвета пла¬
мени. Воду в газометр впускают медленной струей, так
чтобы ток хлора был слабым и водород был в неболь¬
шом избытке. После того как цвет пламени изменится и
станет желто-зеленым, замечают исходный объем хлора,
105
горелку вставляют в кожух и проводят опыт в течение
1—1,5 мин1. Насадку в поглотителе (битое стекло или
нарезанные трубки) нужно предварительно смочить во¬
дой. Затем прекращают ток хлора и водорода, поглоти¬
тель промывают, полученный раствор переливают в мер¬
ную колбу на 100 мл и доливают водой до метки. После
Рис. 36. Получение соляной кислоты:
/ — горелка; 2 — кожух горелки; 3 — поглотитель хлористого водо¬
рода; -/ — аппарат Киппа; 5 — газометр с хлором; 6 — гидравличе¬
ский затвор; 7 — горелка в натуральную величину.
перемешивания отбирают пипеткой 20—25 мл раствора
кислоты и титруют ее щелочью в присутствии фенол¬
фталеина.
Для определения теоретического выхода соляной
кислоты на основании измерения объема хлора в газо¬
метре рассчитывают массу сгоревшего хлора, а затем
массу полученного хлористого водорода.
1 Горение идет более равномерно, если над горелкой устано¬
вить открытую воронку, соединенную с поглотителем, который
в свою очередь соединен с водоструйным насосом или аспира¬
тором.
106
Выход соляной кислоты определяется как процент¬
ное отношение практически полученного хлористого во¬
дорода (растворенного в воде) к теоретическому коли¬
честву.
Оформление результатов работы
Описать работу установки по получению соляной
кислоты. Привести цифровые данные о выходе полу¬
ченной соляной кислоты.
Вопросы и задачи
1. Почему при получении синтетической соляной кислоты водо¬
род берут в некотором избытке?
2. Какое значение при синтезе хлористого водорода имеет на¬
личие влаги в исходных газах?
3. Для чего хлористый водород, идущий для наполнения балло¬
нов, подвергают тщательной осушке?
4. Сколько соляной кислоты теоретически можно получить за
сутки из электролитического хлора и водорода, если сила тока,
подаваемого на электролизер, равна 1500 а. Соляная кислота содер¬
жит 37,23% хлористого водорода и имеет плотность 1,19.
Ответ. 131,5 /сг и 115,0 л.
5. Определить количество хлора и водорода для получения
1000 л соляной кислоты, содержащей 37,2% хлористого водорода
(г/ = 1,19). Исходная смесь газов содержит избыток примерно +3%
водорода по сравнению с теоретически необходимым количеством.
Ответ. 430,417 кг, или 135,955 нм3 хлора; 12,236 кг, или
140,033 нм3 водорода.
Работа 18
Получение серной кислоты контактным
способом
Получение серной кислоты контактным способом
включает следующие стадии:
1. Получение сернистого газа.
2. Очистка сернистого газа.
3. Окисление сернистого газа до серного ангидрида.
4. Улавливание серного ангидрида серной кислотой.
В лаборатории обжиг серного колчедана и сжигание
серы проводят в токе кислорода, так как при использо¬
вании воздуха обжиговый газ содержит пониженное ко¬
личество сернистого ангидрида.
107
Очистка сернистого газа, имеющая большое значение
в промышленности, в лабораторных условиях может
быть ограничена осушкой концентрированной серной
кислотой.
Окисление, протекающее по уравнению реакции:
2S02 + 02 2S03 + Qf
идет с уменьшением объема газов н выделением тепло¬
ты. Например, при 450 °С на 1 моль выделяется
22 660 кал. Для повышения скорости окисления двуоки¬
си серы применяют катализатор — пятиокись ванадия.
При большой скорости окисления катализатор нагре¬
вается, что смещает равновесие реакции в сторону рас¬
пада серного ангидрида, поэтому в промышленности
применяют отвод теплоты от катализатора. В лаборато¬
рии можно применять значительные количества катали-
затора, заполнив им трубку, и пропускать газ с неболь¬
шой скоростью. В этих условиях перегрева катализатора
не происходит и можно создавать оптимальный темпера¬
турный режим окисления с большим выходом серного
ангидрида. Так, при 450 °С степень контактирования
сернистого газа доходит до 97,6%.
Для поглощения серного ангидрида в промышлен¬
ности используется 98,3-процентная серная кислота, так
как давление паров воды над ней минимальное. Если же
для поглощения применять воду, то серный ангидрид,
находящийся в газовой смеси, дает с парами воды ту¬
ман, который водой не поглощается. Но в лабораторных
условиях, когда требуется определить выход серной
кислоты или доказать ее наличие, используется вода, в
которой растворяется часть серного ангидрида. Выходя¬
щий из поглотителя туман серной кислоты можно уло¬
вить мокрым фильтром из стеклянной ваты или электро¬
фильтром.
Вариант 1
Цель работы. Получить раствор серной кислоты
контактным способом с использованием ванадиевого ка¬
тализатора. Определить степень контактирования и вы¬
ход серной кислоты в расчете на серу. Ознакомиться
с методикой лабораторного приготовления катализатора.
108
Оборудование и материалы
1. Газометр с кислородом или воздухом.
2. Трубчатая электропечь на 800—900 °С или 500—
600 °С.
3. Термопара с пирометрическим гальванометром.
4. Кварцевая или фарфоровая трубка.
5. Трубка из тугоплавкого стекла.
6. Четыре промывалки.
7. Выпрямитель или аккумулятор на 2—3 а и 10—
12 в.
8. Индукционная катушка.
9. Лабораторный электрофильтр.
10. Бюретки.
11. 0,1 н. раствор щелочи и кислоты.
12. Сера.
13. Асбест и пятиокись ванадия.
14. Аптекарские весы с разновесом.
15. Резиновые пробки, соединительные трубки, газо¬
подводящие и газоотводящие стеклянные трубки.
Проведение работы
Схема установки иля получения серной кислоты кон¬
тактным способом приведена на рисунке 37. Сжигание
серы проводится в трубке 3 в токе сухого кислорода из
газометра. Для осушки кислород пропускают через ко¬
лонку с хлоридом кальция и через промывалку с концен¬
трированной серной кислотой. При сжигании небольшая
часть серы возгоняется и ее пары улавливают в стеклян¬
ном шаре 4. Вместо стеклянного шара можно применить
колбу Вюрца. Получающийся сернистый газ после до¬
полнительной осушки серной кислотой в промывалке 1
поступает в трубку 5 с катализатором, обогреваемым
электропечью 6. При недостаточной осушке сернистого
газа в трубке с катализатором может конденсироваться
серная кислота. Серный и сернистый ангидриды погло¬
щаются в склянках Дрекселя водой. Образующийся ту¬
ман серной кислоты улавливают электрофильтром 8.
Трубка для сжигания серы должна иметь диаметр
не более 0,9 см, так как пары серы иногда взрываются
при большем диаметре ее. Концы трубки с катализато¬
ром должны быть нагреты электропечью, чтобы преду¬
предить конденсацию серного ангидрида в трубке.
109
Устройство электрофильтра видно на рисунке 37. Егс
можно сделать также нз стеклянной трубки длиной 30—
35 см с отверстиями для входа и выхода газов. Через
пробку, расположенную сверху, вводится электрод (ос¬
винцованная железная или медная проволока), на конце
которого подвешен груз (свинцовая гиря). Лучше этот
ю
Рис. 37. Получение серной кислоты контактным способом из серы:
1 — промывалка с серной кислотой; 2 — осушительные колонки с хлоридом
кальция; 3 — трубка для сжигания серы; 4 —шар для улавливания несго¬
ревшей серы; 5 — трубка с катализатором; 6 — электропечь; 7 — поглотитель
с водой; 8 — электрофильтр; 9 — термопара; 10 — пирометр; И — индукци¬
онная катушка; /2— аккумулятор,
электрод поместить в стеклянную трубку, чтобы преду¬
предить отклонения. Другим электродом является мед¬
ная проволока или алюминиевая фольга, которая в виде
спирали наклеивается на наружную поверхность элект¬
рофильтра.
В трубку 5 помещают катализатор. Контактную
массу готовят из пятиокиси ванадия, которая переме¬
шивается с асбестом так, чтобы порошок вещества по
возможности проник между отдельными волокнами ас¬
беста. Для повышения активности катализатор смачи¬
вают 0,5-процентными растворами нитрата серебра,
нитрата калия и нитрата бария, подсушивают и прока¬
ливают при 650—670 °С.
При отсутствии пятиокиси ванадия ее можно приго¬
товить из вацадата аммония, который прокаливают при
500—650 °С в фарфоровом стакане или тигле. При этом
часто наблюдается образование низших окислов вана¬
110
дия темного цвета. Для перевода этих окислов в пя-
тиокись полученную смесь смачивают азотной кислотой
(1:2), подсушивают в фарфоровой чашке и снова про¬
каливают при 500—550 °С.
Другим хорошим катализатором является платинированный ас¬
бест или кварц.
Для приготовления платинового катализатора нужно не менее
0,3—1,0 г платины. Ее помещают в коническую колбочку и заливают
20—30 мл царской водки и растворяют при небольшом нагревании.
Компактная платина растворяется медленно, и если жидкости оста¬
ется 5—7 мл, а платина еще не растворилась, необходимо добавить
новую порцию царской водки. Высокодисперсная платиновая чернь
часто вспенивается при растворении. В этом случае к платине, поме*
щенной в коническую колбу, следует предварительно добавить 5—
6 мл воды, подогреть, а затем при нагревании добавлять отдельны¬
ми порциями царскую водку. После растворения платины раствор
следует выпарить до 3—4 мл, добавить в него 3—4 мл концентри¬
рованной соляной кислоты и еще раз выпарить до указанного объе¬
ма. Полученную платинохлориетоводородиую кислоту нужно разба¬
вить до 10—15 мл и смочить ею асбест или крупный кварцевый пе¬
сок. Затем асбест или кварц подсушивают в фарфоровой чашке и
прокаливают при 600—700 °С.
Пятиокись ванадия можно заменить окисью железа,
двуокисью марганца или молибденовым ангидридом,
перемешанным с асбестом или даже битым кирпичом,
содержащим окись железа.
Катализатором заполняют всю трубку, без просве¬
тов, но не слишком плотно, чтобы не создать препят¬
ствия прохождению газа.
Серу в количестве 0,2—0,3 г помещают в трубку.
Для проверки установки на герметичность включают
электропечь и, когда температура в ней достигает
500 °С, из газометра пускают слабый ток кислорода:
если установка герметична, то в приемнике и в промы-
валках должны проскакивать пузырьки газа. При по¬
явлении в поглотителе тумана включают электрофильтр
и оставшийся серный ангидрид вытесняют кислородом
или воздухом. Электрофильтр перед опытом промывают
водой и высушивают.
Трубку с серой нагревают горелкой, продолжая про¬
пускать кислород — около 1—2 пузырьков в 1 сек.
После того как сера загорится, горелку отставляют и
усиливают ток кислорода. Если по каким-либо причи¬
нам сера погаснет, то снова нагревают трубку до вос¬
пламенения серы. После воспламенения серы необхо¬
111
димо включить электрофильтр. При нормальной работе
электрофильтра (длина 40 см) из газоотводной трубки
сернокислотный туман не выходит.
Когда вся сера сгорит, через установку при той же
температуре необходимо пропустить еще несколько лит¬
ров кислорода до прекращения появления тумана в
промывалке. Серный ангидрид также можно вытеснить,
присоединив установку к газометру с воздухом. Затем
включают электропечь и воду из приемников перели¬
вают в стакан, приемники ополаскивают водой и про¬
мывную воду присоединяют к основной части раствора
серной кислоты. Электрофильтр промывают 2—3 раза
водой, выливают воду в стакан с полученной серной кис¬
лотой. Затем растворы кислот, полученные из приемни¬
ка и электрофильтра, переливают в мерную колбу на
250 мл, доливают водой до метки, отмеряют пипеткой
25 мл и титруют его 0,1 н. раствором щелочи в при¬
сутствии фенолфталеина. Для определения сернистой
кислоты отбирают 50 мл раствора, подкисляют 5—8 мл
серной кислоты (1 4) и титруют 0,1 н. раствором пер¬
манганата калия.
Первое титрование позволяет определить общую кис¬
лотность, т. е. количество сернистой и серной кислот
в расчете на серную. Число граммов серной кислоты
можно рассчитывать только после определения количе¬
ства сернистой кислоты.
Пример. Сожгли 0,3 г серы. Двуокиси серы должно
получиться:
-0,= °>5993 <*>•
При титровании перманганатом калия нашли, что
получено 0,08 г сернистой кислоты. Это количество со¬
ответствует:
82Ф9 = 0’062 (2>SCV
Следовательно, в серный ангидрид превратилось:
0,5993 (г) — 0,062 г = 0,5373 aSOa.
Отсюда степень контактирования равна:
0,5373 • 100 00 кс/п/\
—05993“ = 89,65(%).
Практически серный ангидрид может частично
оставаться в катализаторе, сера иногда сгорает не пол¬
112
ностью, и, следовательно, данный расчет часто не соот¬
ветствует действительности.
Допустим, что при титровании щелочью общая кис¬
лотность в расчете на серную равнялась 0,932 г. Сле¬
довательно, чистой серной кислоты получено:
0,932 —0,08 = 0,852 (г).
Это количество кислоты соответствует:
0,852 • 64,07
98,09
= 0,5617 (г) S02.
Отсюда степень контактирования равна:
0,5617 100
0,5993
91,08(%).
Расчет нужно вести по второму методу.
Выход серной кислоты определяется как процентное
отношение полученной серной кислоты к тому ее коли¬
честву, которое должно теоретически получиться из
взятой навески серы. Выход серной кислоты сравнитель¬
но небольшой ввиду неполного сгорания серы. Но сте¬
пень контактирования достаточно высока и доходит до
80—90%.
Оформление результатов работы
Зарисовать установку для получения серной кислоты
и описать методику проведения опыта. Результаты
оформить в виде таблицы.
Взято серы
(в г)
Получено
Выход серной
кислоты (в %)
Степень контакти¬
рования (в %)
серной кислоты
(в г)
сернистой кис¬
лоты (в г)
Вариант 2
Цель работы. Получить серный ангидрид и сер
ную кислоту контактным способом.
113
Оборудование и материалы
1. Газометр или баллон с кислородом.
2. Источник двуокиси серы (колба, капельная ворон¬
ка, серная кислота, медные стружки или бисульфит нат¬
рия).
3. Склянки Дрекселя и склянка Вульфа.
4. Фарфоровая или кварцевая трубка.
5. Катализатор (пятиокись ванадия и асбест).
6 U-образная трубка.
7. Две пробирки.
8. Стакан с охлаждающей смесью или снегом.
9. Электропечь.
10. Пирометрический гальванометр с термопарой.
11. Электрофильтр.
12. Индукционная катушка.
13. Аккумулятор.
Ниже описывается более простой вариант получения
серного ангидрида и серной кислоты (рис. 38). Выход
серной кислоты не определяется.
Сернистый газ получается взаимодействием медных
стружек с концентрированной серной кислотой в колбе /.
Для осушки сернистый газ проходит через осушитель¬
ную склянку 2 с серной кислотой, а затем поступает
в смеситель 3. Туда же из газометра поступает кисло¬
род, который предварительно проходит через счетчик
пузырьков — промывную склянку, наполненную серной
кислотой, и колонки с хлоридом кальция. В смесителе
газы подвергаются дополнительной осушке.
Газовая смесь поступает в фарфоровую или кварце¬
вую трубку 4, наполненную ванадиевым катализатором.
Для его приготовления берут порошкообразную пяти¬
окись ванадия, которую перемешивают с асбестом.
После контактирования серный ангидрид поступает
в U-образный приемник 5, опущенный в охладительную
смесь. Частично серный ангидрид поступает в пробирки
с подкисленным раствором хлорида бария 7 и с раство¬
ром индикатора 8, позволяющего обнаружить наличие
ионов водорода.
Перед началом работы включают электропечь и про¬
пускают небольшое количество кислорода (~0,5 л).
Когда температура печи достигает 500 °С, в колбу
с медными стружками наливают серной кислоты и по¬
114
догревают до получения равномерного тока сернистого
газа. Одновременно пропускают кислород и включают
электрофильтр. Газы должны проходить со скоростью
1—2 пузырька в 1 сек. Необходимо следить за показа¬
ниями пирометра, так как перегрев печи приводит к сни¬
жению выхода серного ангидрида и к порче катализа¬
тора, а охлаждение уменьшает выход серного ангидрида.
Рис. 38. Получение серного ангидрида и серной кислоты контактным
способом:
1 — колба для получения сернистого газа; 2 — осушительная скляика с серной
кислотой; 3 — смеситель — промывалка с серной кислотой; 4 — трубка с вана¬
диевым катализатором; 5—приемник для серного ангидрида; 6 — охладитель¬
ная смесь; 7 — пробирка с хлоридом бария; 8 — пробирка с индикатором;
9 — термопара; 10 — пирометр; 11 — электропечь; 12— электрофильтр; 13 — ин¬
дукционная катушка; 14 — аккумулятор.
В U-образной трубке постепенно накапливается сер¬
ный ангидрид, в первой пробирке образуется осадок
сульфата бария, а во второй — изменяется окраска ин¬
дикатора. После прекращения подачи газа выключают
печь. К серному ангидриду добавляют 1 —1,5 мл серной
кислоты и получают олеум.
Примечание. Трубку с катализатором можно обогревать
пламенем газовой горелки или 2—3 спиртовками. Можно также на
трубку навить спираль от электроплитки и подключить к электро¬
осветительной сети.
В простейшем случае можно взять стеклянную труб¬
ку диаметром 1 см и нагревать спиртовкой.
В этом опыте можно воспользоваться кислородом
115
воздуха. Скорость прохождения пузырьков воздуха
должна быть в 3—4 раза больше скорости прохождения
двуокиси серы. При использовании воздуха серный ан¬
гидрид сильно разбавлен азотом и потому его конден¬
сация происходит с трудом: большая часть серного ан¬
гидрида проходит в пробирки, а затем в виде тумана
поступает в электрофильтр. Для получения значительных
количеств серного ангидрида нужно использовать кис¬
лород.
Оформление результатов работы
Зарисовать установку для получения серного ангид¬
рида и серной кислоты и описать ее работу.
Вопорсы и задачи
1. Двуокись марганца, молибденовый и ванадиевый ангидриды
сравнительно легко отщепляют при нагревании кислород. Напри¬
мер, двуокись марганца при 600 °С переходит в окись марганца
Мп203. Как можно объяснить каталитические свойства этих окислов
при окислении двуокиси серы в серный ангидрид?
2. Почему при окислении двуокиси серы в серный ангидид стре¬
мятся поддерживать по возможности умеренную температуру, на¬
пример 500 °С? Почему не применяют более высокую температуру,
несмотря на то что скорость реакции в этом случае повышается?
3. Почему сера сгорает до двуокиси серы, а получающаяся дву¬
окись серы содержит только следы серного ангидрида?
4. Какими явлениями можно объяснить задержку в электро¬
фильтре тумана серной кислоты (см. описание работы по получе¬
нию серной кислоты)?
5. В каких аппаратах при получении на производстве серной
кислоты осуществляется принцип противотока?
6. Химический анализ показал наличие в серном колчедане 44%
серы. Определить: а) сколько процентов в колчедане чистого дву¬
сернистого железа; б) сколько из 1 т колчедана указанного соста¬
ва получится двуокиси серы, если при его обжиге в двуокись серы
переходит 98% серы.
Ответ. 82% FeS2; 862 кг S02.
7. Определить процентное содержание двуокиси серы в обжи¬
говом газе, если в результате обжига получается окись железа и
двуокись серы. При расчете необходимо учесть, что 3/п всего ки¬
слорода расходуется на образование окиси железа, а 8/п — на
образование двуокиси серы.
Ответ. 16,2%.
8. Подсчитать количество теплоты, выделяющейся при обжиге
1 кмоль чцстого серного колчедана. Колчедан и вещества, образую¬
щиеся при обжиге, имеют следующие теплоты образования:
116
Fe203 + ^22,0 дж;
Fe304 + 1116,9 дж.
FeS2+ 177,8 дж;
S02 -f 296,8 дж;
Ответ. 826,6 дж.
9. Какое количество двуокиси серы необходимо затратить по
массе и объему для получения 1 m серной кислоты?
Ответ. 653,2 кг, 228,4 нмъ.
10. Какое количество обжигового газа, содержащего 7% дву¬
окиси серы (по объему), необходимо затратить для получения 1 m
серной кислоты, если степень использования двуокиси серы состав¬
ляет 95 %?
Ответ. 3433 мнъ.
Работа 19
Получение серной кислоты нитрозным способом
Окисление двуокиси серы при получении серной кис¬
лоты нитрозным способом осуществляется двуокисью
азота.
Ранее этот процесс проводился в больших свинцовых
камерах в присутствии паров воды. Производительность
аппаратуры была невелика, так как реакция шла в га¬
зовой фазе и камеры приходилось делать объемом в не¬
сколько сот кубических метров.
В современном башенном способе окислителем яв¬
ляется нитроза и реакцию проводят в жидкой фазе,
что резко повысило производительность аппаратуры.
Нитроза образуется при растворении окислов азота в
серной кислоте:
no + no2^n2o3;
N203 + 2H2S04 ^ 2HSN05 + Н20.
Нитроза представляет собой раствор нитрозилсерной
кислоты
° = N-°W°
н—0/
в серной кислоте.
Нитрозилсериая кислота в растворе находится в рав¬
новесии с азотистой кислотой:
HSNOb + Н20 ^ H2S04 + HN02.
117
При растворении двуокиси серы в нитрозе азотис
тая кислота окисляет сернистую кислоту до серной кис
лоты:
S02 + Н20 = H2S03;
H2S03 + 2HN02 = H2S04 + 2NO + H20.
По-видимому, нитрозилсерная кислота также при
нимает участие в окислении двуокиси серы.
Окись азота, образующаяся в продукционных баш
иях, направляется в окислительную башню, где он;,
частично окисляется до двуокиси азота. Окись и дву
окись азота находятся в равновесии с ангидридом азо¬
тистой кислоты:
NO + N02 ^ N203.
Смесь газов направляют в поглотительные башни,
в которых трехокись азота поглощается серной кисло¬
той с образованием нитрозилсерной кислоты. Ее раст¬
вор (нитрозу) снова направляют в продукционную
башню на окисление двуокиси серы.
Цель работы. Получить серную кислоту нитроз-
ным методом.
Оборудование и материалы
1. Установка для получения серной кислоты.
2. Медные или латунные стружки или медная про¬
волока.
3. Концентрированная азотная кислота.
4. Сульфит или бисульфит натрия.
5. Концентрированная серная кислота.
6. 0,1 н. раствор щелочи.
7. Мерная колба на 250 мл, пипетка на 20—25 мл.
Проведение работы
Получение серной кислоты нитрозным методом про¬
водят в установке, схема которой приведена на рисун¬
ке 39. Двуокись азота получают в колбе 1 взаимодейст¬
вием 7—10 мл концентрированной азотной кислоты с
медными стружками. Двуокись серы получают в колбе 2
при взаимодействии 3—5 г сульфита или бисульфита
натрия (взвешивают с точностью до 0,01 г) с избыт¬
118
ком серной кислоты (1 1), взятой в количестве 15—
20 мл.
Газы проходят через промывалки с разбавленной
серной кислотой и поступают в реактор, который выпол¬
няет роль продукционной башни. Приливать обе кисло¬
ты нужно небольшими порциями, лучше по каплям, что¬
бы получались примерно одинаковые объемы обоих га¬
зов, о чем отчасти можно судить по количеству
Рис. 39. Получение серной кислоты нитрозным методом:
I — колба для получения двуокиси азота; 2 — колба для получения серни¬
стого газа; 3 — реактор; 4— поглотитель.
пузырьков, проходящих через промывалки. Реактор за¬
полняют короткими стеклянными трубками, которые
смачивают водой. В результате взаимодействия двуоки¬
си серы, двуокиси азота и воды в реакторе образуется
серная кислота.
Образующаяся окись азота выходит из реактора и
поступает в поглотитель, заполненный насадкой из на¬
резанных стеклянных трубок, которые смочены кон¬
центрированной серной кислотой.
119
В поглотитель должна поступать смесь равных
объемов окиси и двуокиси азота. Поскольку в реакторе
образуется окись азота, для ее окисления через трой¬
ник во время опыта вводят из газометра отдельными
порциями по 3—5 мл воздуха. Для того чтобы окисление
двуокиси серы и поглощение окислов азота проходило
наиболее полно, подачу газов нужно проводить мед¬
ленно, примерно в течение 20—25 мин. Необходимо сле¬
дить, чтобы из поглотителя не выходили окислы азо¬
та, в противном случае нужно прекратить подачу газов
в реактор, смочить насадку небольшим количеством
серной кислоты, пропустить небольшое количество воз¬
духа и только после этого продолжать синтез серной
кислоты.
Несмотря на предпринимаемые предосторожности,
часть окислов азота, особенно при быстром токе га¬
зов, попадает в атмосферу лаборатории. Поэтому уста¬
новку следует собирать под тягой.
Во время работы нужно следить, чтобы окислы азо¬
та и двуокись серы поступали в реактор небольшими
порциями, по 1 пузырьку в 2—3 сек. Наличие бурой
окраски допустимо только в нижней части реактора.
Если же весь реактор будет заполнен бурыми парами
двуокиси азота, то подачу окислов азота нужно прекра¬
тить и несколько усилить подачу двуокиси серы.
После окончания работы небольшое количество об¬
разовавшейся серной кислоты нужно слить из реактора
в стаканчик, промыть реактор 3—4 раза отдельными
порциями воды, по 10—12 мл, сливая промывные воды
в тот же стаканчик. Затем раствор серной кислоты пе¬
релить в мерную колбу на 200 или 250 мл, долить до
метки водой и после перемешивания оттитровать 0,1 н.
раствором щелочи.
Рассчитать количество полученной серной кислоты.
На основании массы взятого сульфита или бисульфита
натрия рассчитать выход серной кислоты. Для этого
сначала нужно определить количество двуокиси серы,
которое образуется из взятой навески соли. В поглоти¬
теле остается нитроза. Ее сливают в стаканчик и погло¬
титель промывают небольшим количеством серной кис¬
лоты, подготовив тем самым установку к следующему
опыту.
120
Оформление результатов работы
Зарисовать установку, записать порядок работы и
уравнения реакций. Заполнить таблицу.
Взято сульфита
или бисульфита
(В 2)
Количество серной кислоты
Выход H2SO,
(В %)
получено (в г)
должно получить¬
ся (в г)
Вопросы и задачи
1. У какого вещества: у окиси азота или двуокиси серы —
больше химическое сродство к кислороду при одинаковых условиях?
2. За счет какого процесса образуется нитрозилсерная кислота?
3. В одном случае для приготовления нитрозилсерной кислоты
была взята концентрированная серная кислота, а в другом — раз¬
бавленная. В каком случае концентрация раствора нитрозилсерной
кислоты будет выше?
4. Почему для нормальной работы продукционной башни нитро¬
за должна содержать воду?
5. ^ Почему окись азота, поступающую на поглощение серной ки¬
слотой, окисляют до двуокиси азота не полностью? Почему смесь
газов окиси азота и двуокиси азота, находящаяся в отношении I 1,
поглощается почти полностью?
6. Для синтеза серной кислоты в описанной выше установке
было взято 3 г безводного сульфита натрия. Какое количество сер¬
ной кислоты должно получиться теоретически?
Ответ. 2,33 г.
7. Для чего при вышеописанном методе получения серной ки¬
слоты в реактор подается воздух?
Работа 20
Получение аммиака
Синтетический аммиак в промышленности получают
из азота и водорода:
N2 + ЗН2 ^ 2NH3 + Q.
Эта реакция не доходит до конца, так как между
исходными и полученными веществами устанавливает¬
ся равновесие. Согласно принципу Ле Шателье, выход
аммиака будет увеличиваться при увеличении давле-
121
ния, так как реакция идет с уменьшением объема п
при уменьшении температуры.
Для ускорения реакции применяются катализаторы:
платина, железо и другие металлы. Широкое применение
в промышленности нашел катализатор — мелкозернистое
железо, содержащее небольшое количество активаторов
(окислы алюминия, калия и некоторых других метал¬
лов). Перегрев катализатора (до 650—700 °С) быстро
выводит его из строя. Сернистые и фосфористые соеди¬
нения отравляют катализатор, поэтому азотоводородная
смесь должна быть тщательно очищена. Быстро выходит
катализатор из строя и в присутствии паров воды. Одна¬
ко пропускание через него сухой азотоводородной смеси
регенерирует катализатор.
В лабораторных условиях реакцию проводят при ат¬
мосферном давлении и выход аммиака составляет всего
около 0,4%, для обнаружения которого нужно приме¬
нять фенолфталеин или реактив Неслера.
При поступлении небольшого количества газовой
смеси перегрева катализатора обычно ие происходит,
поэтому катализатор можно не охлаждать.
Вариант 1
Цель работы. Получить водный раствор аммиа¬
ка. Ознакомиться с лабораторным способом получения
азота, водорода (железо-паровым способом). Опреде¬
лить выход аммиака.
Оборудование и материалы
1. Трубка из фарфора, кварца или стекла.
2. Железная трубка с железными стружками.
3. Электропечь.
4. Термопара с пирометрическим гальванометром.
5. Градуированный газометр.
6. Установка для получения азота.
7. Две промывалки со щелочью.
8. Приемник для улавливания аммиака.
9. Соединительные трубки.
10. Железный катализатор.
11. Бюретка.
12. 0,01 н. раствор соляной кислоты.
13. Раствор метилоранжа.
122
Описание установки. Проведение работы
Азотоводородную смесь получают, пропуская воз¬
дух, насыщенный парами воды, через раскаленную труб¬
ку, наполненную железными стружками (рис. 40).
Пары воды и кислород дают с железом закись-окись
железа и водород. Для того чтобы в отходящих газах
соотношение азота и водорода равнялось 1 :3, воздух
насыщают парами воды при 80—85 °С. Это достигается
пропусканием из газометра 1 воздуха через промывал-
ку 2, помещенную в водяную баню 3. Для того чтобы
Рис. 40. Получение аммиака:
/“Газометр с воздухом; 2—промывалка с водой; 3 — водяная баня; 4 — же¬
лезная трубка; 5, 6 — электропечи; 7 — промывалка с концентрированной ще¬
лочью; 8 — стеклянная нли фарфоровая трубка, наполненная железным ката¬
лизатором; 9— приемник; 10 — асбест; 11 — термопара; 12 — пирометрический
гальванометр.
пары воды не конденсировались в соединительных труб¬
ках, выводную трубку промывалки соединяют непосред¬
ственно с железной трубкой 4. Эта трубка нагревается
электропечью или газовыми горелками до красного ка¬
ления (750—800 °С).
Градуированный газометр можно сделать из обыч¬
ного газометра. Для этого в него последовательно вли¬
вают по 0,5 л воды и ставят на его поверхности каран¬
дашом метки или наклеивают бумажные полоски с де¬
лениями.
Аммиак получают, пропуская смесь азота и водорода
через трубку 8, наполненную катализатором. Катализа¬
тор помещают в трубку вместе с асбестом так, чтобы на¬
греваемая часть трубки была им заполнена. Трубку на-
123
гревают в электропечи или на газовой горелке. Оптималь¬
ная температура синтеза лежит в пределах 450—480 °С.
За температурой нужно следить по показаниям пиромет¬
ра или термометра.
Железный катализатор готовят из чистой окиси или
чистой гидроокиси железа. Окись железа пропитывают
раствором нитратов калия, алюминия и бария. На сто
частей окиси железа берут примерно по одной части каж¬
дой из указанных солей. После этого окись железа высу¬
шивают и прокаливают при 700—800 °С. При этом про¬
исходит разложение нитратов и образование ферритов,
например Ba(Fe02b. Полученный порошок помещают в
количестве 20—30 г в трубку и восстанавливают водоро¬
дом при 500—600 °С. В качестве катализатора можно
брать и обыкновенное порошкообразное железо, но его
активность несколько ниже активности катализатора,
приготовленного описанным способом.
Для поглощения аммиака служит приемник 9, напол¬
ненный водой с 2—3 каплями метилового оранжевого,
раствор которого нужно предварительно подкислить 1—2
каплями разбавленной уксусной кислоты до появления
красного окрашивания К
Перед началом работы проверяют прибор на герме¬
тичность. Для этого открывают кран газометра и про¬
пускают воздух через всю систему. Наличие пузырьков
газа в промывалках и приемнике указывает на то, что
прибор герметичен. Если пузырьки газа не проходят,
необходимо проверить места соединений и добиться гер¬
метичности. После проверки кран у газометра закры¬
вают, включают электропечь 5 и нагревают ее до
750—800 °С, после чего пускают слабый ток воздуха со
скоростью 1—2 пузырька в 1 сек (в промывалках),
К этому времени вода в водяной бане должна быть на¬
грета почти до кипения. Азотоводородную смесь про¬
пускают в течение 10 мин (для вытеснения воздуха из
всей системы) и только после этого включают печь 6
с катализатором. Когда она нагревается до 450—480 °С,
замечают уровень воды в газометре и присоединяют при¬
емник. При указанной температуре пропускают около 11 Если количественный выход аммиака не определяется, то в
приемник лучше добавить не метиловый оранжевый, а фенол¬
фталеин.
124
двух литров воздуха, объем которого определяют по под¬
нятию уровня воды в газометре. После появления жел¬
того окрашивания индикатора, что указывает на наличие
аммиака, разъединяют приемник с раствором аммиака,
включают печь 6 и продолжают пропускать азотоводо¬
родную смесь до охлаждения печи (150—200 °С). Разби¬
рать прибор во избежание окисления катализатора
ложно только после полного охлаждения печи. После
охлаждения катализатора до указанной температуры
выключают вторую печь, где получается азотоводород¬
ная смесь, и прекращают ток воздуха.
Разбавленный раствор аммиака из приемника пере¬
ливают в колбу на 100 мл, добавляют 2—3 капли рас¬
твора метилового оранжевого и титруют 0,01 н. раство¬
ром соляной кислоты до изменения окраски индикатора.
Объем азота и водорода, поступивший на синтез, под¬
считывается по количеству воздуха, пропущенного через
систему.
Пример. Из газометра было пропущено 3,5 л воз¬
духа. Считаем, что азота в воздухе 78%, т. е. было взято
2,73 л азота. При указанной температуре в воздухе было
такое количество паров воды, которое дало теоретически
необходимое количество водорода, т. е. считаем, что во¬
дорода получилось: 2,73-3 = 8,19 (л). В действительности
водорода может получиться или несколько меньше, или
несколько больше. Но эти отклонения будут сравнитель¬
но небольшие. Затем подсчитывается масса азота и во¬
дорода, поступивших на синтез. Для этого используется
уравнение Менделеева — Клайперона:
где т— масса газа в граммах;
р — давление в атмосферах;
V — объем в литрах;
R — газовая постоянная, равная 0,082-г'-^;
Т — абсолютная температура;
М — молекулярная масса газа.
Выход аммиака подсчитывается по уравнению:
m NH3. 100
tri N2 MW2
125
где mNH3 — масса аммиака в граммах (находится thi
рованием);
mN2— масса азота до синтеза в граммах;
тН2— масса водорода до синтеза в граммах.
Необходимо отметить, что данный опыт иногда не уда¬
ется вследствие проскоков кислорода через железные
стружки, особенно когда они окислились или когда они
лежат рыхлым слоем.
Оформление результатов работы
Зарисовать и описать установку для синтеза аммиа
ка. Числовые данные оформить в виде таблицы.
Взято возду¬
ха (в л)
Давление
(в am)
Температу¬
ра (в °С)
Масса (в г)
Выход ам¬
миака (в %)
водорода
аммиака
Вариант 2
Цель работы. Получить водный раствор аммиа¬
ка. Ознакомиться с методом получения азота из воздуха
и электролитическим методом получения водорода. Оп¬
ределить выход аммиака по водороду.
Оборудование и материалы
1. Две трубчатые электропечи.
2. Газометр.
3. Железная или фарфоровая трубка с железными
стружками.
4. Фарфоровая или стеклянная трубка с железным
катализатором.
5. Две промывалки с раствором щелочи.
6. Поглотитель для аммиака.
7. Две термопары с пирометрическими гальваномет¬
рами.
8. Раствор метилового оранжевого.
9. 0,01 н. раствор соляной кислоты.
10. Колба, бюретка.
126
Описание установки. Проведение работы
Схема прибора приведена на рисунке 41. Воздух из
газометра пропускают через фарфоровую или железную
трубку У, наполненную железными стружками. Трубку
нагревают до темно-красного каления (до 750—800 °С).
Водород впускают в систему из электролизера (см. гл. II,
раб. 8) Синтез аммиака проводится в трубке 2 (см. вы-
uie, вариант 1). Обогрев трубок осуществляется электро¬
печами. За током газов следят по количеству пузырьков
газов, проходящих через щелочь в промывалках 8—9.
Для предупреждения попадания брызг щелочи в трубку
2 помещают асбест 7.
Установку проверяют на герметичность. Для этого из
газометра пускают слабый ток воздуха и наблюдают за
прохождением пузырьков в промывалке и поглотителе.
После этого поглотитель отсоединяют, продолжая пропу¬
скать слабый ток водорода. Включают печи 3 и 4 и откры¬
вают зажим 11, зажим 12 закрывают. Для вытеснения
воздуха из трубки 2 включают электролизер. После того
как печь 3 нагреется до 750—800 °С, из газометра пускают
слабый ток воздуха. Получающийся азот вытеснит из си¬
стемы воздух. Зажим 11 закрывают, а зажим 12 откры¬
вают. При этом азот пойдет в трубку 2, которая к этому
времени будет нагрета и заполнена водородом, поступа- 1Рис, 41. Получение аммиака:
/ — трубка с железными стружками; 2 — трубка с железным катализатором;
4 — электропечи; 5, 6 — термопары с пирометрами; 7 — асбест; 8, 9 — про-
мывалка с раствором щелочи; 10 — приемник для поглощения аммиака;
11, 12 — зажимы.
1 При отсутствии электролизера водород можно получить из
аппарата Киппа. В этом случае расчет по определению выхо¬
да аммиака не проводится.
127
ющим из электролизера или аппарата Киппа. Водород в
промывалке 9 должен проходить со скоростью 1—2 пу¬
зырька в 1 сек, а азот в промывалке 8 — в три раза мед¬
леннее. После того как печь 4 нагреется до 480 °С, присо¬
единяют поглотитель для аммиака и отмечают силу тока
и время. Азотоводородную смесь пропускают через ката¬
лизатор в течение 10—15 мин.
Для обнаружения аммиака в поглотитель добавляют
2—3 капли метилового оранжевого, подкисленного 1—2
каплями разбавленной уксусной кислоты. Замечают вре¬
мя окончания синтеза, отделяют приемник от системы,
выключают печь 4, пропуская водород до ее-охлаждения
(/=150—200 °С).
Раствор аммиака переливают в колбу и титруют
0,01 п. раствором соляной кислоты в присутствии мети¬
лового оранжевого до появления розового окрашивания.
Выход продукта подсчитывается по количеству полу¬
ченного аммиака и пропущенного водорода. Например,
синтез длится 0,5 ч. Через электролизер шел ток силой
5 а. Следовательно, водорода получилось:
Чёт1 = °>09 («)•
Подсчитаем, сколько азота вступит в реакцию с 0,09 г
водорода. Определим выход аммиака.
В данной работе азот можно получить и другим, бо¬
лее простым способом. Воздух из газометра пропускают
через две промывалки, наполненные щелочным раство¬
ром пирогаллола, который поглощает кислород, или раст¬
вором гидросульфита натрия Na2S2C>4. Для осушки азот
нужно пропустить через промывалку с концентрирован¬
ным раствором щелочи или через колонку с хлоридом
кальция.
Оформление результатов работы
Зарисовать схему установки для синтеза аммиака и
описать ее работу. Определить выход аммиака. Расчет¬
ные данные оформить в виде таблицы.
Время опыта (в ч)
Сила тока (в а)
Количество водо¬
рода (в г)
Выход аммиака
(в %)
128
Опыт проходит при соблюдении определенных условий.
1. Азот, выходящий из трубки с железными стружками, не дол¬
жен содержать паров воды и кислорода. Проскоки паров воды и
кислорода наблюдаются в том случае, когда температура печи не¬
достаточно высока и когда железные стружки помещены в трубку
слишком рыхлым слоем.
2. Железные стружки не должны содержать загрязнений и по
возможности в них должно содержаться минимальное количество
углерода. В противном случае к азоту будет примешана двуокись
углерода, которая частично может «проскакивать» через промы-
валку со щелочью и будет нейтрализовать получающийся аммиак.
Стружки нужно предварительно промыть бензином для удаления
масла.
3. Необходимо учитывать, что сернистые соединения отравляют
катализатор. Сернистые соединения могут попадать в трубку с ка¬
тализатором из загрязнений, имеющихся в железных стружках, а
также при нагревании резиновых пробок, если употребляются труб¬
ки недостаточной длины.
Сернистые соединения часто содержатся в порошкообразном
железе и даже в окиси железа. Для проверки на отсутствие серни¬
стых соединений в приемник для аммиака наливают раствор иода
с крахмалом (слабо окрашенный). При наличии сернистых соедине¬
ний раствор иода через некоторое время обесцвечивается.
Лучше опыт по синтезу аммиака удается, когда между труб¬
кой, содержащей железные стружки, и трубкой, содержащей ката¬
лизатор, помещается промывалка со слабым раствором иода.
Вопросы и задачи 1 2 3 4 51. Как влияет изменение давления и температуры на выход ам¬
миака при его синтезе из азота и водорода?
2. При синтезе аммиака в промышленных условиях смесь азота
и водорода предварительно проходит по трубам через катализатор-
ную коробку и только после этого пропускается через катализатор.
Для чего это делается? При ответе учтите принцип Ле Шателье.
3. При медленном пропускании трех объемов аммиака и одного
объема азота через слой порошкообразного железа, нагретого до
500 °С, в отходящей газовой смеси будет находиться около 0,4%
аммиака. Сколько будет находиться аммиака в газовой смеси, если
через слой порошкообразного железа пропустить при указанных
условиях чистый аммиак?
4. Сколько литров азота и водорода нужно взять для получения
1 г аммиака при нормальном давлении? Выход аммиака составля¬
ет 0,4%.
Ответ. 165 л N2 и 494 л Н2.
5. При 400 °С и давлении 300 am в равновесии с азотоводород¬
ной смесью находится 47% аммиака (по объему). Исходя из урав¬
нения реакции: N2-f 3H2^2NH3, определить количество аммиака,
азота и водорода, находящееся при указанных условиях в 1 м3 га¬
зовой смеси.
Ответ. 470 л NH3; 397,5 л \\2 и 132,5 л N2.
5 Н. Г. Ключников
129
6. Колонна синтеза аммиака имеет производительность 200 т
аммиака в сутки. Синтез проводится при 450 °С и давлении в 300 сип.
При этих условиях в газовой смеси находится 35,8 объемного про¬
цента аммиака. Определить, сколько получится аммиака за сутки ц
сколько водорода и азота не вступит в реакцию.
Ответ. 263 520 нм3 NH3; 354 426 нм3 Н2 и 118 142 нм3 N*
Работа 21
Получение азотной кислоты
При окислении аммиака кислородом в присутствии
определенных катализаторов можно получить окись
азота:
4N=H3 + 502 4NO + 6Н20 + Q.
В промышленности катализатором служит сетка из
платины (5—10% родия); в лабораторных условиях мож¬
но применять окислы железа, хрома, кобальта, марганца,
ванадия.
Окисление аммиака на платиновом катализаторе про¬
текает с большой скоростью, примерно в 100 раз быстрее
по сравнению со скоростью реакций при использовании
других катализаторов, но водородные соединения серы
и фосфора необратимо отравляют платиновый катализа¬
тор.
Получаемые в результате окисления аммиака нитроз-
ные газы содержат до 10—11% окиси азота. Переработ¬
ка нитрозных газов в азотную кислоту включает две ос¬
новные стадии: окисление окиси азота кислородом воз¬
духа до двуокиси и поглощение последней водой:
2N03 + Н20 - HN02 + HN03.
Азотистая кислота в кислых растворах и при достаточной
концентрации разлагается на азотную кислоту и окись
азота. При избытке кислорода воздуха окись азота снова
окисляется до двуокиси и азотной кислоты.
Цель работы. Получить водный раствор азотной
кислоты окислением аммиака. Определить выход азот¬
ной кислоты.
Оборудование и материалы
1. Фарфоровая или кварцевая трубка с катализатором.
2. Колба с 12-процентным раствором аммиака.
130
3. Приемник для улавливания окислов азота водой.
4. Градуированный газометр.
5. Соляная кислота—0,05 н. раствор.
6. Щелочь — 0,05 н. раствор.
7. Индикатор (фенолфталеин или метиловый оранже¬
вый).
8. Раствор дифениламина.
Проведение работы
Вариант 1
Собирают прибор по рисунку 42. Воздушноаммиач¬
ную смесь получают просасыванием воздуха через колбу
с 12-процентным водным раствором аммиака. Через
4—5 опытов раствор аммиака следует заменить свежим
раствором. Для создания тока газа применяют газометр,
соединенный с колбой 3.
Воздушноаммиачная смесь освобождается от влаги
в колонке с твердым едким кали. В кварцевую или фар¬
форовую трубку 5 помещают ванадиевый ангидрид, пе¬
ремешанный с волокнистым асбестом, которым нужно за¬
полнить трубку без просветов. При его отсутствии в ка¬
честве катализатора можно использовать окись хрома,
тигельная колонка с твердым едким кали; 5—трубка с ванадиевым катали¬
затором; 6 — электропечь; 7 — термопара; 8 —> пирометр; 9—поглотитель азот¬
ной кислоты.
5*
131
полученную разложением бихромата аммония. Окислы
азота поглощают в приемнике 9 с 10—20 мл воды.
Для определения концентрации аммиака в газовой
смеси наливают в поглотитель 20 мл 0,05 н. раствора со¬
ляной кислоты и через систему просасывают 0,5 л возду¬
ха. Затем избыток кислоты оттитровывают в присутствии
метилового оранжевого щелочью и рассчитывают коли¬
чество аммиака (в г), содержащееся в 1 л газовой
смеси.
После этого включают электропечь, доводят ее тем¬
пературу до 750—800 °С, пропускают через нее около
0,5 л аммиачной смеси, затем, прекратив подачу воздуха
из газометра, к системе присоединяют приемник для азот¬
ной кислоты, в который нужно налить 20 мл дистиллиро¬
ванной воды. Из газометра пускают медленной струей
воздух (2—3 пузырька в 1 сек). Пропустив 1 л смеси
воздуха с аммиаком, приемник отделяют от трубки и вы¬
ключают электропечь ].
Количество азотной кислоты определяют титрованием
ее 0,1 н. раствором щелочи.
Для расчета выхода азотной кислоты количество ам¬
миака, находящееся в 1 л газовой смеси, пересчитывается
на азотную кислоту.
Оформление результатов работы
Зарисовать установку для получения азотной кисло¬
ты, описать ее действие.
Определить выход азотной кислоты.
Вариант 2
Работа по второму варианту проводится без количе¬
ственных расчетов по определению выхода азотной кис¬
лоты.
Для получения азотной кислоты собирают прибор
(рис. 43). Воздушноаммиачная смесь получается прбса-
сыванием воздуха через колбу с 12-процентным раство¬
ром аммиака. Создание необходимого тока воздуха до- 11 Вместо электропечи можно использовать две горелки с на¬
садкой. В этом случае лучше применять трубки из тугоплав^
кого стекла.
132
стигается или при помощи газометра, или водоструйным
насосом.
Сначала нагревают электропечь до 750—800 °С, за¬
тем пропускают через трубку с катализатором воздушно¬
аммиачную смесь, через некоторое время приемник на¬
полняется бурыми парами двуокиси азота. В поглотитель
А
Рис. 43. Получение азотной кислоты окислением аммиака:
I — градуированный газометр: 2 — промывалка (счетчик пузырьков): 3 —- колба
с 12-процентным раствором аммиака; 4 — осушительные колонки с твердым
едким калн; 5 —трубка с ванадиевым катализатором; 6 — электропечь; 7 —
термопара; Я — пирометр; 9 — приемник; 10 — поглотитель азотной кислоты.
пускают воду: 1—2 капли в 1 сек. Через 5—10 мин ток
воздуха прекращают и азотную кислоту выливают из
приемника в колбу или стаканчик. Колбу 9 и приемник
10 промывают 2—3 раза водой (по 15—20 мл), сливают
раствор кислоты и промывные воды в мерную колбу на
250 мл и титрованием определяют количество получен¬
ной кислоты.
Оформление результатов работы
Зарисовать установку для получения азотной кислоты
и описать ее действие. Определить выход кислоты.
Вопросы и задачи
1. Какие катализаторы применяются в промышленности и в ла¬
бораторных условиях для окисления аммиака в окись азота?
2. Какие продукты получаются при сгорании аммиака?
133
3. Почему при окислении аммиака вначале получается окись
азота?
4. Почему при получении двуокиси азота газовую смесь, выхо
дящую из контактного аппарата, охлаждают?
5. Для чего при поглощении двуокиси азота водой к ией дает¬
ся избыток кислорода?
6. Чем объяснить, что при многократном поглощении двуокиси
азота водой в присутствии кислорода в конечном итоге получается
азотная кислота, а при поглощении двуокиси азота раствором ще¬
лочи получается смесь нитратов и нитритов?
7. Сколько теоретически должно получаться азотной кислоты
(d— 1,4) из 1 т аммиака?
Ответ. 5666 кг, или 4047 л.
8. На синтез поступило 568 кг аммиака, из которого получено
2280 л азотной кислоты, содержащей 62,23% HN03. Определить
выход азотной кислоты (d= 1,390).
Ответ. 96,4%.
9. Через контактный аппарат для окисления аммиака пропусти¬
ли газовую смесь, содержащую 11% аммиака. Сколько получится
азотной кислоты из 20 нмг газовой смеси, если общие потери в рас¬
чете на азотную кислоту составляют 2% ?
Ответ. 6,052 кг.
10. В продукционные башни на 1 пг получаемой серной кислоты
подается 20 мг нитрозы (d=l,7), содержащей от 10—15% окислов
азота в расчете на азотную кислоту. Определите, сколько подается
окислов азота (в расчете на HNO3) на 1 пг серной кислоты.
Ответ. От 3 до 5 пг.
И. Определите, сколько теоретически нужно нитрозы (в расче¬
те на N2O3) для получения 1 пг серной кислоты. Фактический расход
нитрозы составляет около 40% от теоретического.
Ответ. 11Ь кг.
ГЛАВА V
СОЛИ И ТВЕРДЫЕ РАСТВОРЫ
§ 7. РАЗДЕЛЕНИЕ СОЛЕИ ЗА СЧЕТ ИХ РАЗЛИЧНОЙ
РАСТВОРИМОСТИ
В природных условиях соли обычно смешаны с дру¬
гими солями или минералами. Отделение солей друг от
друга осуществляется растворением их в воде с последу¬
ющим фильтрованием и кристаллизацией. Дальнейшая
очистка солей проводится путем дробной кристаллиза¬
ции.
Для выделения веществ из раствора находит приме¬
нение метод политермической кристаллизации, который
основан на различной растворимости солей в зависимо¬
сти от температуры. Если растворимость одной из солей
увеличивается с повышением температуры более резко
по сравнению с растворимостью второй соли, то выде¬
ление первой соли можно осуществить охлаждением рас¬
твора солей, приготовленного при повышенной темпе¬
ратуре. Этим методом проводят выделение хлорида ка¬
лия из сильвинита.
При кристаллизации соли учитывают самые разнооб¬
разные факторы: скорость кристаллизации, температуру,
степень очистки солей от примесей, использование теп¬
лоты, получаемой при охлаждении растворов, и т. д.
Работа 22
Выделение хлорида калия из сильвинита и его
анализ
Хлорид калия получают из сильвинита. Разделение
хлоридов калия и натрия основано на их различной рас-
135
творимости. Поскольку растворимость хлорида калия г
отличие от растворимости хлорида натрия резко возрас
тает с температурой, отделение хлорида калия можт.
провести охлаждением
горячего насыщенного
раствора смеси солей,
из которых и будет вы¬
деляться хлорид калия.
Оптимальные усло¬
вия переработки силь¬
винита можно устано¬
вить, исходя из дан¬
ных по растворимости
в системе NaCl —
—КС1—Н20, приве¬
денных в таблице 9 и
на графике (рис. 44).
В таблице 9 приведе¬
ны растворимости чи¬
стых солей (NaCl и
КС1), а также состав
Рис. 44. Изотермы совместной раство- РаСТВОра, находящего-
римости NaCl и КС1 в воде при 10, 20 ся в СОСТОЯНИИ равнове-
и 100 °С. сия со смесью хлоридов
калия и натрия.
Цель работы. Получить 15—20 г хлорида калия
из сильвинита.
Таблица 9
Температу¬
ра (в °С)
Состав осадка
Количество граммов солн на 100 г
воды
NaCl
| КС1
25
К С1
26,96
NaCl
35,63
—
КС1 +- NaCl
29,38
16,28
50
КС1
—
43,12
NaCl
36,50
—
KC1 -Ъ NaCl
29,09
22,03
75
KCI
—
49,70
NaCl
37,75
—
KCI + NaCl
27,87
29,06
100
KCI
—
56,20
NaCl
39,40
—
KCI + NaCl
27,39 1
35,16
136
Оборудование и материалы
1. Два фарфоровых или стеклянных стакана на 200—250 г.
2. Мешалка.
3. Термометр.
4. Воронка для горячего фильтрования.
5. Раздробленный сильвинит или искусственная смесь
эквивалентных количеств хлорида калия и хлорида натрия.
6. Технические весы с разновесом.
7. Мензурка на 100—150 мл.
8. Раствор кобальтнитрита натрия.
Проведение работы
Растворение сильвинита проводится в стакане, снаб¬
женном стеклянной мешалкой и термометром (рис. 45) \
Выщелачивание сильвинита проводится щелоком.
Для его получения отвешивают в стакане 50 г сильви¬
нита, наливают 100 мл воды, нагревают до кипения и
после перемешивания стеклянной палочкой охлаждают.
Холодный раствор декантируют с выпавшего осадка
в стакан, в который добавляют 50 г сильвинита, и смесь
при перемешивании нагревают до 100 °С. Затем раствор
отделяют от осадка через воронку для горячего филь¬
трования. Можно также горячий раствор быстро про¬
фильтровать через воронку Бухнера с отсосом. Для филь¬
трования можно применить обычный бумажный фильтр.
Оставшуюся смесь солей сдают лаборанту. Охлаждают
раствор до 25 °С, выпавший осадок хлорида калия от¬
фильтровывают через воронку с пористой стеклянной
пластинкой или через бумажный фильтр, осадок высу¬
шивают и взвешивают.
Для анализа продукта отвешивают на аналитических
весах 0,1—0,2 г соли, растворяют ее в 10—12 мл воды,
подкисляют 1—2 мл уксусной кислоты и приливают рас'
твор кобальтнитрита натрия до прекращения выделения
желтого осадка K2Na[Co(N02b] • Н2О. Фильтруют оса¬
док через фильтровальный тигель или через воронку с
пористой пластинкой и асбестовым волокном. Осадок про¬
мывают сначала 10-процентным раствором уксусной кис¬
лоты до появления бесцветного фильтрата, а затем спир¬
том. Осадок высушивают при 110 °С, взвешивают и рас- 11 При отсутствии мешалки растворение сильвинита можно ве¬
сти при перемешивании раствора стеклянной палочкой.
137
считывают процентное содержа¬
ние хлорида калия во взятой
навеске. Аналитический фак¬
тор для определения количе¬
ства хлорида калия по массе
K2Na[Co (N02) б] • Н20 = 0,1642.
На основании количества по¬
лученного хлорида калия и коли¬
чества взятого сильвинита рассчи¬
тывают выход хлорида калия.
Фильтрат, оставшийся после
отделения хлорида калия, можно
использовать в дальнейших рабо¬
тах по выщелачиванию хлорида
калия из сильвинита.
Количество солей, перешедших
в раствор, определяют теорети¬
ческим путем на основании коли¬
чества взятой воды и раствори¬
мости солей. Количество хлорида
™ — калия рассчитывается на основа-
НИТа; нии результатов анализа и общей
1 - мотор; 2 - мешалка; 3 —МЗССЫ ПОЛучеННОГО Осадка СОЛеЙ.
стакан; 4 — термометр.
Оформление результатов работы
Результаты опыта оформляют в виде таблицы.
Взято воды
(в г)
Взято солей (в г)
Выпало солен из
раствора (в г)
Выход соли
(в %)
КС1 | NaCI
КС1 | NaCI
§ 8. ПОЛУЧЕНИЕ СОЛЕЙ ДЕЙСТВИЕМ КИСЛОТ
НА МИНЕРАЛЬНОЕ СЫРЬЕ
Кислоты в настоящее время довольно широко при¬
меняют для разложения минералов. Наиболее часто для
этой цели применяют серную кислоту, так как концент-
133
рироваиыая серная кислота инертна к чугуну и, пользу¬
ясь серной кислотой, можно проводить реакции кислот¬
ного расщепления при значительном нагревании.
Наибольшее промышленное значение кислотное рас¬
щепление имеет при производстве фосфорных удобрений
и при переработке руд редких элементов, например ти¬
тана, циркония, лития. Под действием серной кислоты
разрушается естественная структура минералов и обра¬
зуются соли серной кислоты, которые выщелачиваются
водой и подвергаются соответствующей химической пере¬
работке.
Работа 23
Получение суперфосфата и его анализ
Суперфосфат получается при взаимодействии апа¬
тита или фосфорита с серной кислотой:
2Ca5F(P04)3 + 7H2S04 - 3Ca(H2P04)2 + 7CaS04 + 2HF;
Ca3(P04)2 + 2H2S04 = Ca(H2P04)2 + 2CaS04.
Поскольку это взаимодействие осуществляется между
жидким веществом и твердым, реакция протекает до¬
вольно медленно. В первую очередь реакция идет на по¬
верхности крупинок с образованием фосфорной кисло¬
ты и сульфата кальция, например:
Ca5F(P04)3 + 5H2S04 + 2,5Н20 =
= 5 (CaS04 0,5Н2О) + ЗН3Р04 + HF.
В дальнейшем масса постепенно загустевает, так как ос¬
тавшиеся серная и фосфорная кислоты постепенно вза¬
имодействуют с непрореагировавшей частью апатита или
фосфорита:
Ca5F(P04)3 + 7Н3Р04 + 5Н20 =
= 5Са(Н2Р04)2 H20 + HR
Указанные реакции, а также кристаллизация веществ
протекают довольно медленно. Полное разложение фос¬
фата проходит в течение 20—30 суток и заканчивается
на складе. Помимо однозамещенного фосфата кальция
Са(Н2Р04)2-Н20, в суперфосфате в небольших количе¬
ствах присутствует также и двухзамещенный фосфат
139
кальция СаНР04*2Н20, а также и свободная фосфор¬
ная кислота.
Оптимальные условия переработки —температура
концентрация кислоты и время переработки—определя¬
ются составом сырья и его физико-химической структу¬
рой.
При анализе суперфосфата определяют общее содер¬
жание фосфора (в расчете на Р2О5) и количество фос¬
фора, который усваивается растениями. Для определения
последнего суперфосфат обрабатывают водой, а затем
аммиачным раствором цитрата аммония. В водный рас¬
твор переходит свободная фосфорная кислота и одно-
замещенный фосфат кальция. В цитрате аммония раст¬
воряется главным образом двухзамещеппый фосфат
кальция, который в воде плохо растворим. Содержание
влаги определяют высушиванием 10 а суперфосфата при
100—102 °С в течение трех часов.
Из раствора ион НР042~ осаждают аммиачным рас¬
твором хлорида магния:
HPOJT + Mg2+ + NH4OH = MgNH4P04 + Н20.
Выпавший осадок магний-аммоний фосфата отфильтро¬
вывают и прокаливают. При этом он переходит в пиро¬
фосфат магния. По массе пирофосфата магния рассчиты¬
вают количество фосфора (в расчете на пятиокись), со¬
держащегося в суперфосфате.
Цель р а б о т ы. Получить 15—20 г суперфосфата
и провести его анализ.
Оборудование и материалы
1. Фосфорит.
2. Концентрированная серная кислота.
3. Железная ступка.
4. Сито с отверстиями в 0,2—0,3 мм.
5. Аптекарские или технохимические весы.
6. Стеклянный цилиндр.
7. Ареометр.
8. Фарфоровая чашка со стеклянной палочкой.
9. Магнезиальная смесь (см. ниже).
10. Соляная кислота (d=l,19).
11. Раствор Патермана (см. ниже).
12. Фенолфталеин.
140
13. 25-процеитный раствор аммиака.
14. 2—3-процентный раствор аммиака.
15. Воронки, фильтры, колбы.
Проведение работы
Фосфорит размалывают в металлической ступке, а за*
тем растирают в фарфоровой ступке и просеивают. От*
вешивают около 20—25 г фосфоритной муки на техно*
химических весах. Для реакции используют 62—64-про*
центную серную кислоту.
Количество кислоты (в мл), необходимое для разло¬
жения фосфорита или апатита, рассчитывают на ос¬
новании реакции образования суперфосфата, считая, что
состав фосфорита или апатита соответствует теоретиче¬
скому. Поскольку в фосфорите всегда имеются примеси,
кислоты берут на 15% меньше теоретически рассчитанно¬
го количества. Лучше для работы использовать чистый
фосфат кальция Са3(Р04)2.
Кислоту наливают в фарфоровый стакан и подогре¬
вают до 50—60 °С. Затем в стакан отдельными порциями
насыпают фосфоритную муку или апатитовый концент¬
рат. Смесь перемешивают стеклянной палочкой в тече¬
ние 3—4 мин и ставят в сушильный шкаф при 110—150 °С
на 1 ч. Затем ее охлаждают и проводят анализ суперфос*
фата. При использовании аппатита все работы по его ки¬
слотному расщеплению нужно проводить под сильной тя¬
гой ввиду выделения фтористого водорода.
Для анализа отвешивают на аналитических весах
2—2,5 г суперфосфата и растирают в фарфоровой ступ¬
ке до уничтожения комочков. В ступку приливают 25 мл
воды, смесь растирают и после отстаивания раствор
фильтруют в мерную колбу (на 250 мл). В колбу предва¬
рительно наливают 5—6 капель соляной кислоты, которая
предупреждает гидролиз солей кальция. Остаток в ступке
еще три раза растирают с водой, прибавляя каждый раз
воды по 20—25 млу и каждый раз декантируют раствор
на фильтр. Затем нерастворившийся остаток переносят
на фильтр и промывают его водой, заполняя колбу до
метки. Раствор тщательно перемешивают. Он содержит
водорастворимую пятиокись фосфора Р2О5.
Фильтр с нерастворившнмися остатками переносят
в другую мерную колбу емкостью в 250 мл} приливают
141
100 мл раствора Патермана и, закрыв колбу пробкой,
сильно взбалтывают (фильтр распадается на волокна).
Колбу нагревают до 60 °С в течение 15 мин,. Нагревание
лучше проводить в термостате. Затем содержимое колбы
охлаждают, доливают дистиллированной водой до метки,
тщательно перемешивают и фильтруют через сухой
фильтр в колбу. Первые порции фильтра отбрасывают.
Раствор содержит так называемую цитраторастворимую
пятиокись фосфора Р2О5.
Для определения общего количества цитрато- и водо-
растворимой фосфорной кислоты из водной вытяжки и
цитратного раствора берут по 50 мл и выливают эти
растворы в стакан. В стакан наливают 25 мл раствора
Патермана, перемешивают и нейтрализуют 2—3-процент¬
ным раствором аммиака по фенолфталеину. Для осаж¬
дения фосфат-иона к раствору приливают 25—35 мл
магнезиальной смеси, а затем 10—25 мл 25-процентного
раствора аммиака. Содержимое стакана перемешивают
в течение 30 мин или оставляют стоять в течение 5—15 ч.
Выпавший осадок магний-аммоний фосфата фильтруют
через плотный беззольный фильтр и промывают 2—3-
процентным раствором аммиака. Затем фильтр с осад¬
ком переносят в тигель, высушивают, озоляют, прокали¬
вают в муфеле при красном калении и взвешивают.
Содержание фосфорной кислоты в расчете па фосфор¬
ный ангидрид вычисляют по формуле:
%РА -
0,6377 500
Ь 100
100. a Q1Q a
у~ь~’
где a — масса пирофосфата магния в граммах;
Ь — навеска суперфосфата в граммах;
0,6377—множитель для пересчета Mg2P20? на Р2О5.
Оформление результатов работы
Описать метод получения суперфосфата.
Провести расчет и оформить результаты анализа.
Реактивы, используемые при анализе супер¬
фосфата. 1. Магнезиальная смесь приготовляется растворением
55 г хлорида магния и 70 г хлорида аммония в 1 л воды.
2. Реактив Патермана (цитрат аммония) должен содержать в
1 л раствора 173 г невыветрившейся лимонной кислоты, которую
растворяют в 300 мл горячей воды. На это количество отмеряют та¬
142
кое количество 12—13-процентного раствора аммиака, чтобы в нем
содержался 51 г аммиака. Для этого к концентрированному раство¬
ру аммиака прибавляют равный объем дистиллированной воды и
по плотности определяют его концентрацию. По концентрации рас¬
считывают количество миллилитров раствора, необходимое для ней¬
трализации лимонной кислоты и содержащее 51 г аммиака.
Раствор аммиака наливают в мерную колбу емкостью 1 л и
туда же при непрерывном помешивании и охлаждении приливают
небольшими порциями раствор лимонной кислоты.
Затем раствор цитрата аммония доливают водой до метки и
перемешивают.
Вопросы и задачи
1. Трехзамещенный фосфат кальция в воде практически не рас¬
творим. Однако его можно с успехом применять на кислых почвах.
На чем основано это применение?
2. Во сколько раз в двойном суперфосфате больше фосфора по
сравнению с простым? Расчет ведется на Р2О5. Влажность удобре¬
ния не учитывается; сульфат кальция в суперфосфате находится
главным образом в виде гипса.
Ответ. В 3,02 раза.
3. Одним из наиболее ценных удобрений считается удобрение,
получаемое при взаимодействии азотной кислоты с фосфоритом. На¬
пишите уравнение реакции и подсчитайте процентное содержание
азота и фосфора в этом удобрении.
При вычислении нужно учесть, что кислотное расщепление идет
до однозамещенного фосфата кальция, а влажность удобрения не
учитывается.
Ответ. 11,2% N и 12,7% Р.
4. Почему при анализе фосфорных удобрений обычно учитывают
водорастворимый и цнтратнын фосфор?
5. Сколько расходуется по массе и объему 66-процентной серной
кислоты (d= 1,57) на приготовление 1 т суперфосфата.
Ответ. 577,7 кг или 368 л.
6. Почему перед отправкой потребителю суперфосфат около
20 дней храпят на складе?
7. Почему при анализе фосфоритов на фосфор применяют сер¬
ную кислоту, а при анализе стали используют азотную кислоту?
8. Почему при получении суперфосфата применяют серную кис¬
лоту определенной концентрации? Почему нельзя использовать раз¬
бавленную или сильно концентрированную кислоту?
9. Сколько теоретически нужно 65-процентной серной кислоты
для получения 10 г суперфосфата из фосфорита, содержащего 5%
карбоната кальция?
Ответ. 6,212 г.
10. Напишите уравнение реакции осаждения фосфат-иона магне¬
зиальной смесью.
11. Сколько процентов SO2 должен теоретически содержать об¬
жиговый газ, если кислород воздуха полностью вступит в реакцию,
а исходные вещества FeS3 и ZnS не содержат примесей?
Ответ. 16,2% и 15%.
143
12. Степень использования двуокиси серы при получении серной
кислоты составляет в среднем 95%. Определите расход двуокиси
серы на 1 т кислоты, если обжиговый газ содержит 7—9% SO2.
Ответ От 2674 нм3 до 3437 нм3.
13. Сколько теоретически потребуется (кг и нм3) двуокиси серы
для получения 1 m серной- кислоты?
Ответ. 653,1 кг, 228,6 нм3.
§ 9. СТЕКЛО И ВЯЖУЩИЕ ВЕЩЕСТВА
Промышленное получение стекла, вяжущих, огнеупор¬
ных, керамических материалов основано на переработке
природных силикатов и алюмосиликатов, конечной ста¬
дией которой является обжиг и сплавление, вызывающие
те или иные химические процессы и приводящие к обра¬
зованию новых веществ.
При получении силикатных изделий и материалов ха¬
рактерные реакции протекают в твердой фазе при тем¬
пературе (0,8 или 0,9^), гДе t — температура плавления
наиболее легкоплавкого из получаемых компонентов. На¬
пример, при совместном нагревании карбоната кальция
и окиси кремния силикаты кальция получаются уже при
700—800 °С, хотя самые легкоплавкие соединения в си¬
стеме СаО — Si02 плавятся около 1000 °С.
Наибольшее влияние на скорость протекающих хи¬
мических реакций в твердой фазе оказывают темпера¬
тура и степень измельчения веществ.
При высокотемпературном обжиге смесей силикатов
реакции часто протекают не только в твердой, но и в
жидкой фазе, образующейся за счет плавления получен¬
ных веществ и за счет плавления специально добавляе¬
мых веществ. Это наблюдается, например, при обжиге
фарфора и цемента. Жидкая фаза образуется на границе
раздела твердых веществ; при охлаждении обожженно¬
го материала она застывает и обеспечивает сцепление
между частицами.
Силикаты и алюмосиликаты плавятся при самых раз¬
нообразных температурах; наряду с силикатами магния,
кальция и других металлов, многие из которых плавятся
при 1500—1600 °С, имеются легкоплавкие силикаты, на¬
пример свинца, плавящиеся при 700—800 °С.
144
Работа 24
Приготовление легкоплавких стекол
Вещества, образующие стекла, называются стеклооб-
разователями. К ним относятся следующие окислы: В2О3,
Ge02, Si02, Р205, As205, А1203, V205 — и некоторые дру¬
гие. Окислы типа Э20 и ЭО могут входить в состав стек¬
ла, но в индивидуальном состоянии они стекло не дают.
При образовании стекол сложного состава катионы
одно- и двухвалентных металлов располагаются в сили¬
катной (или другой) решетке в соответствующих пус¬
тотах, не нарушая расположения атомов кремния и
кислорода. Таким образом, стекла имеют своеобразный
каркас, построенный из молекул стеклообразователя
(Si02, В203 и др.), в который входят ионы щелочных или
щелочноземельных металлов.
Свойства стекла определяются его составом. Напри¬
мер, BaO, Ge02, РЬО повышают показатель преломле¬
ния стекла (оптическое стекло), А1203 увеличивает меха¬
ническую прочрюсть (тарная посуда) и. т. д. Многие
окислы, растворяясь в стекле, сообщают ему соответствую¬
щую окраску. В наибольших количествах готовится про¬
стое силикатное стекло, которое имеет примерный сос¬
тав (оконное стекло) Na20 * СаО • 6Si02.
Приготовление стекла включает следующие стадии:
1) дробление или помол окислов и их просев; 2) дозиров¬
ка окислов; 3) перемешивание окислов; 4) варка стекла.
В лабораторных условиях варку стекла удобно прово¬
дить в электрических печах. Особенно легко готовятся
легкоплавкие стекла — свинцовые и борсвинцовые, так
как для их изготовления требуются сравнительно невы¬
сокие температуры. На базе этих стекол можно легко
приготовить и окрашенные стекла.
Цель работы. Получить 15—20 г легкоплавкого
цветного стекла.
Оборудование и материалы
1. Шамотный, корундовый или фарфоровый тигель
объемом 50—70 см3.
2. Кварцевый песок.
3. Борный ангидрид или борная кислота.
145
4. Безводные поташ или сода.
5. Тигельные щипцы.
6. Тигельная электропечь.
7. Жаровня с песком.
8. Прокаленные окислы металлов РЮ, СоО иль
С03О4, NiO, V2O5, СГ2О3, МпОг, CuO, С112О, S11O2, БегОз.
Проведение работы
Для приготовления прозрачных и легкоплавких сте¬
кол — борсвинцовосиликатных — можно рекомендовать
следующие смеси (табл. 10).
Таблица 10
№
н/л
Состав (в %)
Температура
плавления
(в СС)
РЬ 1
| В2О3
Si02
1
84,5
11,0
4,5
484
2
86,0
10,6
3,4
486
3
87,5
11,4
1,1
488
4
75,0
15,0
10
540
Наиболее легкоплавкие борсвинцовые и свинцовоси¬
ликатные стекла имеют состав (табл. 11).
Таблица 11
№
п/п
Состав (в %)
Температура
плавления
(в "С)
РЬО
BjOa
$Ю2
1
92,7
7,3
—
565
2
86,6
13,4
—
497
3
93,7
6,3
—
560
4
61,4
38,6
—
768
5
70,4
—
29,6
732
6
88,1
—
11,9
723
7
91,8
—
8,2
714
Для того чтобы варка стекла проходила успешно, вы¬
бирают те смеси (по указанию преподавателя), темпе¬
ратура плавления которых примерно на 50—110 °С ниже
146
температуры печи. Расчет ведут на 30—35 г шихты. Пред¬
варительно окислы растирают в ступке и просеивают
через сито с мелкими отверстиями так, чтобы размер
частичек был не более ОД мм. Окислы взвешивают на
аптекарских весах с точностью до 0,01 г и тщательно пе¬
ремешивают на листе бумаги или в ступке. Затем 8—10 г
смеси всыпают в тигель и помещают его в нагретую
печь. Вследствие вспенивания расплавленная масса мо¬
жет переливаться через край тигля и испортить футеров¬
ку печи. Особенно вспенивание наблюдается при исполь¬
зовании борной кислоты. Полученная стекломасса должна
быть совершенно однородной. Для этого ее выдерживают
в расплавленном состоянии в течение 10—15 мин. Варка
стекла ускоряется при перемешивании стекломассы про¬
волокой. Однако если для этой цели применяется не пла¬
тиновая проволока, а, например, железная, то стекло'не¬
много загрязняется окислами железа. После окончания
варки тигель вынимают из печи и стекло выливают на
чистую железную или никелевую пластинку или керами¬
ческую плитку.
Варку 5—8 г наиболее легкоплавких стекол можно
проводить и в стеклянных пробирках на пламени газо¬
вой горелки 3—4 2 стекла можно приготовить и на пла¬
мени спиртовки. Приготовление стекла на пламени га¬
зовой горелки занимает не более 5 мин. Нагрев нужно
проводить до полного расплавления стекломассы. Затем
ее быстро выливают на керамическую плитку или на чис¬
тый железный лист. Если для варки использовать обыч¬
ные пробирки, то при выливании стекло часто застывает
на холодных стенках пробирки. Поэтому нужно брать
более короткие пробирки (длиной 4—5 см) и перед вы¬
ливанием стенки пробирки необходимо прогреть. Если
варка стекла проводится на пламени газовой горелки, то
для приготовления стекла можно также взять неболь¬
шой фарфоровый тигель.
Для варки стекла окись свинца РЬО можно приготовить прока¬
ливанием сурика, карбоната или нитрата свинца при 500 °С в те¬
чение 15—20 мин. Поташ и сода должны быть подсушены и прока¬
лены при 500—600 °С. Хранить эти вещества нужно в плотно за¬
крытых склянках. За неимением борного ангидрида для варки
стекла можно воспользоваться борной кислотой. Поскольку она при 11 Этот метод получения стекла можно с успехом рекомендо¬
вать в качестве демонстрационного опыта в школе.
147
нагревании сильно вспенивается, ее лучше перевести предваритель¬
но в борный ангидрид. Для этого ее нужно прокалить небольшим!
порциями (по 2—10 г) в платиновой или фарфоровой чашке npi
^~650 °С. Вязкую массу выливают на железную, никелевую или
керамическую плитку. Остывшую хрупкую массу разбивают на ку
сочки. Дальнейшее обезвоживание нужно проводить небольшим!
порциями, по 2—3 г, в фарфоровой чашке или тигле при ^ — 650 °С
За обезвоживанием необходимо все время наблюдать, так ка>.
вследствие вспенивания масса может вылиться из чашки и испор
тить муфель. Хранить борный ангидрид нужно в плотно закрыто!
склянке, так как он гигроскопичен. Порошок борного ангидрида
вводимого в шихту, приготовляется растиранием его в фарфоровой
ступке. Ввиду того что кусочки борного ангидрида при этом раз¬
брасываются, на пестик нужно надеть лист бумаги, закрывающий
ступку.
Приготовление окрашенного стекла.
На основе вышеперечисленных прозрачных стекол мож¬
но приготовить окрашенные стекла. Для этого к приго¬
товленной шихте, дающей прозрачные стекла, нужно до¬
бавить окисел, сообщающий стеклу соответствующую
окраску (табл. 12).
Таблица 12
№
п/п
Краситель
Количество окис¬
лов (в %)
Окраска
1
Закись железа
0,2—0,3
Сше-зеленая
2
Окись железа
0,3—0,5
Желто-зеленая
3
Закись кобальта
0,003—0,1
Синяя
4
Закись никеля
ОД—0,2
Сине-зеленая
5
Окись меди
0,1—0,2
Голубая до синей
6
Окись хрома
0,05—0,1
Зеленая
7
Окись марганца
0,01—0,05
Дымчато-красная
8
Серебро (в виде ни¬
трата)
0,1—0,3
Желтая
9
Сера
1-2
Черная
10
Двуокись олова
5-6
Молочное стекло
11
Тальк
5—6
» »
Интенсивность окраски зависит от количества добав¬
ленного окисла. Для равномерного распределения кра¬
сители перетирают с шихтой, но их можно добавить и в
148
сплавляемую массу при осторожном покачивании тигля
или пробирки.
Серу для получения черного непрозрачного стекла
нужно вводить после сплавления первых порций стекла
прямо в жидкое стекло. Если же серу ввести в начале
плавки, то она иногда выгорает.
В качестве красителей можно брать не только те окис¬
лы, которые указаны в таблице, но также и другие. Вме¬
сто закисей кобальта и никеля можно брать любые окис¬
лы кобальта и никеля, а также их соли, которые в усло¬
виях варки стекла разлагаются и переходят в закиси;
вместо окиси марганца можно брать двуокись марганца
и перманганат калия. Необходимо учесть, что окись мар¬
ганца в условиях варки легко переходит в закись, кото¬
рая окрашивает стекло в желтоватый цвет. Поэтому с
целью предупреждения восстановления трехвалентного
марганца в шихту полезно добавить 1—2 крупинки се¬
литры.
Для варки стекла наиболее пригодны корундовые тиг¬
ли, так как они не загрязняют стекло и термостойки. Вар¬
ку стекла можно проводить и в фарфоровых тиглях, но
они иногда трескаются. Если тигель развалится в печке
или опрокинется, нужно немедленно выключить печь,
снять для более быстрого охлаждения печи крышку и вы¬
нуть тигель. Расплавленное стекло сильно разъедает фу¬
теровку печи, а при попадании на спираль быстро вы¬
водит ее из строя, поэтому стекло после охлаждения пе¬
чи нужно полностью удалить. Тигель лучше ставить на
подставку, сделанную из кирпича. Ее можно сделать и из
обычной глины, которая после подсушивания и обжига
приобретает необходимую механическую прочность.
Оформление результатов работы
Записать состав шихты для получения наиболее лег¬
коплавких стекол и методы их окраски. Описать методи¬
ку приготовления стекла.
Вопросы а задачи
1. Перечислите вещества, способные давать стекла.
2. Почему стекло иногда называют переохлажденной жидко¬
стью?
149
3. Почему при медленном охлаждении расплавленного стекла
оно иногда становится мутным?
4. Какие лучи поглощает стекло, окрашенное закисью кобальта -
5. Для чего перед изготовлением изделий из стекла расплавлен
ную стекломассу несколько охлаждают?
6. Какую роль при изготовлении стекла выполняют осветители
и глушители?
7. Сколько нужно взять кальцинированной соды, мела и квар¬
цевого песка для приготовления 100 кг оконного стекла состава
Na20-Ca0-6Si02?
Ответ. 22,16 кг Na2C03; 20,92 кг СаС03 и 75,32 кг Si02.
8. Наиболее легкоплавким борсвинцовосшшкатным стеклом
(тем. пл. 484 °С) является стекло следующего состава: РЬО 84%,
В203 11,5% и Si02 4,5%. Сколько нужно взять сурика (РЬзСМ,
борной кислоты и кварцевого песка для приготовления 10 г стекла
указанного состава?'
Ответ. 8,65 г РЬ304; 1,78 г В(ОН)3 и 4,5 г Si02.
Работа 25
Получение вяжущих веществ и изучение их свойств
К вяжущим веществам относятся материалы? которые
после смешения с водой образуют пластичную массу,
превращающуюся через некоторое время в искусствен*
ный камень. В первый момент, когда масса начинает
твердеть, но еще не обладает значительной механиче¬
ской прочностью, происходит так называемый процесс
схватывания. В дальнейшем прочность медленно возрас¬
тает, причем этот процесс длится иногда в течение мно¬
гих дней, недель или даже лет. Этот процесс называют
твердением.
Эти свойства вяжущих веществ используются в стро¬
ительном деле для связывания друг с другом различных
деталей, кирпичей, камня и т. д. Обычно вяжущие веще¬
ства применяются в смеси с песком, гравием и т. д. На
основании отношения к воде их делят па воздушные, спо¬
собные твердеть только на воздухе, и на гидравлические,
которые могут затвердевать не только на воздухе, но и
под водой. К воздушным относится воздушная известь,
гипс, к гидравлическим — цемент. 11. Получение и применение цементного
строительного раствора
В фарфоровой чашке отвешивают 10 г цемента, добав¬
ляют 30 г сухого песка и перемешивают до получения
150
вполне однорододной смеси. Эту смесь переносят на фа¬
янсовую плитку или фанеру, добавляют к ней около 5 мл
воды и тщательно перемешивают палочкой. Из части по¬
лученного цементного раствора при помощи железной
или стеклянной формы, покрытой внутри слоем парафи¬
на, формуют лепное изделие. Другую часть раствора ис¬
пользуют для связывания друг с другом двух небольших
кусков кирпича. Куски кирпича смачивают в воде и скре¬
пляют на плитке тонким слоем раствора.
На следующем занятии испытывают прочность скре¬
пления кусков на разрыв.
2. Получение и свойства строительного
гипса
Гипс, используемый в строительном деле, а также для
художественных отливок, является полуводным сульфа¬
том кальция.
При добавлении к гипсу воды (явление называется
затворением) гипс превращается в двуводный кристал¬
логидрат.
Ввиду худшей растворимости двуводного гипса в во¬
де он перекристаллизовывается из пересыщенного раст¬
вора и постепенно превращается в твердый камень.
Для приготовления строительного гипса гипсовый ка¬
мень сначала измельчают молотком, а затем растирают
небольшими порциями в ступке в тонкий порошок и от¬
вешивают в фарфоровой чашке 10 г. Чашку помещают
в сушильный шкаф и нагревают ее при 130—140 °С до
постоянной массы (первое взвешивание — после 20 мин,
последующие — после 10 мин нагревания). Следует из¬
бегать перегревания гипса, так как уже выше 150°С он
постепенно превращается в безводный гипс. После обез¬
воживания гипса чашку вынимают из шкафа и опреде¬
ляют убыль в массе. Содержание воды выражается как
процентное отношение количества удаленной воды к те¬
оретическому.
Отвешивают 20 а строительного (полуводного) гипса,
помещают его на фаянсовую плитку или на фанеру, до¬
бавляют 9 мл воды, перемешивают и, когда тесто немного
загустеет, нажатием железной формы, покрытой внутри
тонким слоем парафина, готовят изделия. Определяют
время схватывания гипса. Для этого к полузатвердевше¬
15)
му изделию прикасаются тупым концом ланцета и наблю¬
дают заполнение образовавшейся- ямки водой; гипс счи¬
тается затвердевшим, когда ямка не будет заполняться
водой. Замечают время с момента приливания воды дс
момента затвердевания.
Оформление результатов работы
Описать приготовление вяжущих веществ и резуль¬
таты опытов.
Вопросы и задачи
1. Что происходит с дву водным гипсом CaS04-2H20 при его на¬
гревании до 140 °С?
2. Какие явления происходят при твердении гипса?
3. Какое количество воды необходимо для гашения 100 кг оки¬
си кальция, содержащей 40% посторонних примесей?
Ответ. 30,8 кг.
4. Какое количество воды теоретически необходимо для гашения
100 кг окиси кальция, содержащей 5% окиси магния и 3%. посторон¬
них примесей, не взаимодействующих с водой.
Ответ. 31,48 кг.
5. Как будет влиять изменение давления на реакцию термиче¬
ского разложения карбоната кальция?
6. Рассчитайте количество продукта, полученного при обжиге
1 m известняка, содержащего 90% СаСОз, 6% MgC03 и 4% SiC>2.
Ответ. 573,2 кг.
ГЛАВА VI
ТОПЛИВО И ВОДА
§ 10. ПЕРЕРАБОТКА ТВЕРДОГО ТОПЛИВА
Работа 26
Полукоксование твердого топлива и анализ газообразных
продуктов 1
При нагревании каменного угля без доступа воздуха
происходят сложные химические процессы, в результате
которых образуются газообразные, жидкие и твердые
продукты.
Коксование угля протекает при 1000—1100 °С, при
этом удаляются почти все летучие продукты и получае¬
мый кокс содержит их не более 1%- Полукоксование про¬
водится при 500—600 °С, и ему подвергают главным об¬
разом бурые угли, торф, сланцы. Количество и состав
продуктов коксования в значительной степени определя¬
ется температурой процесса. При полукоксовании выход
смолы, называемой первичной, значительно выше и до¬
стигает в отдельных случаях 50—60% от массы топ¬
лива, а выход газообразных продуктов около 80 м3 на 1 от.
При коксовании эта первичная смола подвергается при
высоких температурах пирогенетическому разложению и
выход газообразных продуктов увеличивается до 300—
350 ж3 на 1 от топлива, а выход жидких продуктов снижа¬
ется. Низкотемпературная смола содержит значительное
количество различных фенолов и углеводородов жирного
1 Анализ газообразных продуктов проводится по указанию
преподавателя.
153
ряда. Твердый продукт, получаемый при полукоксовании,
легко рассыпается в порошок и содержит 5—8% лету¬
чих продуктов. Кокс обладает значительной механиче¬
ской прочностью, что связано с реакциями пиролиза пер¬
вичной смолы в толще угля.
Цель работ ы. Изучить условия полукоксования
твердого топлива и определить выход полукокса, первич¬
ной смолы и газообразных продуктов. Провести анализ
газообразных продуктов.
Оборудование и материалы
1. Железная реторта для полукоксования.
2. Тигельная электропечь на 700—800 °С.
3. Термопара с пирометрическим гальванометром.
4. Градуированный приемник смолы.
5. Бутыль на 3 л.
6. Мерный цилиндр на 1 л.
7. Каменный или бурый уголь, торф.
8. Технохимические весы с разновесом.
Проведение работы
Полукоксование проводят в железной сварной ре¬
торте (рис. 46). Сверху реторта 1 закрыта крышкой 2,
в которую вварена труба 3 для термопары и труба 4,
отводящая газообразные продукты. Крышка укрепляется
на реторте с помощью болтов. Для создания герметич¬
ности между крышкой и флянцем реторты прокладыва¬
ется асбестовая прокладка. Реторта помещается в элек¬
тропечь 6. Газоотводная трубка реторты соединена с
приемником <5, погруженным в охладительную смесь.
Боковая трубка приемника жидких продуктов соединена
с бутылью 9.
Реторта загружается мелкими кусочками топлива
примерно на половину объема. Для переработки можно
брать каменный или бурый уголь, а также торф и слан¬
цы. Затем реторту плотно закрывают крышкой, вставля¬
ют ее в электропечь и присоединяют к ней приемники для
жидких и газообразных продуктов. Последний должен
быть полностью заполнен водой.
Для проверки герметичности прибора включают на
5—8 мин электропечь и наблюдают за поднятием уров¬
154
ня воды в трубке, отводящей воду и газы из приемни¬
ка 9 Если вода в трубке не поднимается, несмотря на
то что реторта немного нагреется, нужно более плотно
закрыть крышку или сменить асбестовую прокладку.
Если же прибор герметичен, включают электропечь и
доводят температуру в реторте до 100 °С. При этом часть
Рис. 46. Полукоксование твердого топлива:
/ — реторта; 2 — крышка; 3 — трубка для термопары; 4, 5 — газоотводные
трубки; 6 — электропечь; 7 — охлаждающая смесь; 8 — приемник жидких про¬
дуктов; 9 — приемник газообразных продуктов.
воды выливается из приемника газообразных продуктов.
Затем печь выключают и приемник газообразных про¬
дуктов снова наполняют доверху водой. Подготовив та¬
ким образом установку, включают печь, нагревают ее до
500—550 °С в течение 10—25 мин. Жидкие продукты кок¬
сования собираются в приемнике 8, а газообразные — в
приемнике 9, При этом вода из приемника переливается
по сифону в мерный цилиндр.
После окончания опыта печь выключают и реторта
остывает до 100 °С или несколько ниже. При этом часть
воды из мерного цилиндра по сифону снова переливается
в приемник газообразных продуктов. После этого раз¬
бирают прибор и определяют выход продуктов. Количе¬
ство полукокса определяется его взвешиванием на тех-
Для собирания газообразных продуктов можно использовать
также и газометр (см. раб. 27).
155
ническнх весах, количество жидких продуктов (вода и
смола) —по массе и объему, газообразных — по объему
воды, поступившей в цилиндр из приемника газообраз¬
ных продуктов.
Реторта для полукоксования может иметь и другую
конструкцию. В частности, нагревательные элементы
можно расположить непосредственно на ее поверхности.
Для этого реторту обертывают листовым асбестом, по¬
верх которого наматывается нихромовая проволока.
Сверху ее снова закрывают асбестом и помещают в же¬
лезный или керамический кожух. Можно применить и
газовый обогрев реторты. Сбор газа можно проводить в
газометре, выпуская воду (по мере поступления газа) че*
рез нижнее отверстие газометра.
Оформление результатов работы
Зарисовать установку для полукоксования и описать
ее работу. Результаты опытов занести в таблицу.
Взято топлива (в г)
Получено
полукокса (в г)
жидких продук¬
тов (в г)
!
газообразных про¬
дуктов (В см3)
Анализ газообразных продуктов
Анализ газообразных продуктов проводят при помо¬
щи газоанализатора. В настоящее время нашей промыш¬
ленностью выпускаются самые разнообразные приборы
позволяющие проводить анализы сложных газовых сме¬
сей. Один из простейших газоанализаторов — прибор
марки ГХ-1, общий вид которого приведен на рисунке 47
схема устройства — на рисунке 48. Этот прибор позволя¬
ет производить непосредственный отбор газа из систе¬
мы. Основные части прибора: газоизмерительная бюрет¬
ка U уравнительная склянка 2 и поглотительные сосуды
3, 4, 5, которые соединены с гребенкой 6. Газоизмеритель¬
ная бюретка окружена водяной рубашкой, что предохра¬
няет газ от колебаний температуры. Сосуды служат для
поглощения соответствующих газов. В сосуд 3 наливают
156
щелочь, в сосуд 4 — раст¬
вор пирогаллола или гид¬
росульфита натрия
Na2S204, в сосуд 5 —мед¬
ноаммиачный раствор. В
уравнительную склянку
при анализе дымовых га¬
зов или газов, образую¬
щихся при сухой перегон¬
ке, наливают 1-процент¬
ный раствор серной кисло¬
ты, насыщенный дву¬
окисью углерода и под¬
крашенный метиловым
оранжевым. Для этого к
раствору серной кислоты
прибавляют немного би¬
карбоната натрия. Затем
в полученную смесь при¬
бавляют несколько капель
фенолфталеина, чтобы
следить за реакцией раст¬
вора— он не должен быть
щелочным. На гребенке
имеется трехходовой кран
7, который служит для
соединения прибора с ем¬
костью, содержащей анализируемый газ, или с атмосфе¬
рой. Чтобы легче разобраться в положении крана, на его
рукоятке имеется цветной прилив, который указывает на
положение просвета в кране. Для работы прибор при по¬
мощи трубки 8 соединяют с емкостью, содержащей ана¬
лизируемый газ. Затем поворачивают кран 7 в положение
в и поднимают уравнительную склянку 2 и заполняют
бюретку 1 жидкостью. Воздух из нее выходит в атмосфе¬
ру. Открывают кран 9 и ставят кран 7 в положение а.
Опуская уравнительную склянку, добиваются того, чтобы
Жидкость из баллона 12 перелилась в сосуд 3 и поднялась
в нем до метки. Эту операцию повторяют и с жидкостями,
находящимися в сосудах 4 и 5.
После этого проводят анализ газа. Кран 7 ставят в
положение б (рис. 48) и, опуская бюретку, набирают газ
в прибор. Поскольку он смешивается с воздухом, имею¬
Рис. 47. Общий вид газоанализа¬
тора ГХ-1:
1 — газонзмерительная бюретка; 2 —
уравнительная склянка; 3, 4, 5 — погло¬
тительные сосуды; 6 — гребенка; 7 —
трехходовый кран; «9 — трубка.
157
щимся в бюретке, его выпускают в атмосферу. Эту
операцию повторяют трижды. Затем заполняют бюретку
жидкостью и набирают в нее газ до нижней метки па
бюретке. Для обнаружения двуокиси углерода поднима¬
ют уравнительную склянку и тогда вытесняется газ в со¬
суд 3. Кран 9 необходимо открыть. Поднимают и опуска¬
ют уравнительную склянку до прекращения уменьшения
объема газа. (Необходимо следить, чтобы кислота не по¬
пала в гребенку.) Опуская уравнительную склянку, под¬
нимают щелочь в сосуде 3 до метки и закрывают кран 9
(следите, чтобы щелочь не попала в гребенку). Коли¬
чество двуокиси углерода в объемных процентах опреде¬
ляется по уменьшению объема жидкости в бюретке.
Также обнаруживают кислород, поглощая его раствором
пирогаллола. Окись углерода поглощают аммиачным
раствором C112CI2. Оставшийся объем приходится на
метан и азот 1.
Рис. 48. Схема газоанализатора ГХ-1:
1 — газоизмерительиая бюретка; 2 — уравнительная склянка; 3, 4,
5 — поглотительные сосуды; 6 — гребенка; 7 — трехходовой кран;
трубка; 9, 10, 11 — краны; 12, 13, 14 — баллоны с поглотитель*
иыми жидкостями; 15 — водяная рубашка.
1 Существуют газоанализаторы аналогичного типа, позволяю¬
щие определять количество метана и непредельных углеводо¬
родов. Эти газоанализаторы имеют четыре поглотительных
баллона и приспособление для сжигания метана и других го¬
рючих газов.
153
По окончании газового анализа остатки газа удаля¬
ют в атмосферу. Краны на поглотительных баллонах от¬
крывают. Уравнительную склянку ставят на подставку.
Затем все краны закрывают.
Оформление результатов работы
Описать принцип действия газоанализатора и запи¬
сать результаты анализа газа.
Вопросы и задачи
1. При пропускании воздуха в смеси с водяным паром через
раскаленный кокс образуются водород, окись и двуокись углерода;
С02 + С = 2СО — Q;
СО + Н20 = С02 + Н2 + Q.
Как будет меняться состав продуктов реакции, если реакцию про¬
водить в одном случае при 900 °С, а в другом — при 1100 °С?
2. При газификации твердого топлива при нормальном давле¬
нии образуется около 2,7% метана, а при 20 am его образуется
22,2%. Чем это объясняется?
3. При коксовании из 1 т угля образуется около 300 нмъ газа.
В 1 нм3 газа содержится около 40 г паров бензола и около 10 г ам¬
миака. Определите, сколько аммиака и бензола получается из 1 т
каменного угля.
Ответ. 12 кг бензола и 3 кг аммиака.
4. Почему для выплавки чугуна применяют кокс, а не каменный
уголь?
5. Почему для коксования пригодны не все сорта каменного угля,
а только так называемые коксующиеся, содержащие большое ко¬
личество смолистых, высокомолекулярных органических веществ?
Работа 27
Сухая перегонка (полукоксование) дерева
При нагревании древесины без доступа воздуха полу¬
чаются древесный уголь, уксусная кислота, метиловый
(древесный) спирт, ацетон, деготь, вода, метан, двуокись
углерода.
Жидкие продукты перегонки образуют два слоя —
смолу и водный конденсат. Продукты перегонки листвен¬
ных пород дают тяжелую смолу и надсмольную воду.
Хвойные породы дают более легкую смолу, в состав ко¬
торой входят терпены, фенолы, различные алифатические
углеводороды. Водный конденсат скапливается под смо¬
лой и называется подсмольной водой.
159
Характер выделяющихся продуктов определяете*
температурой нагрева древесины.
Практически сухую перегонку дерева проводят npi
450—500° С, так как дальнейшее повышение температу
ры почти не увеличивает выход жидких продуктов.
Продукты перегонки дерева — ценное химическое
сырье.
Цель работы. Провести сухую перегонку дерева
и определить выход твердых, жидких и газообразных
продуктов. Определить количество уксусной кислоты.
Провести анализ полученных газообразных продуктов.
Оборудование и материалы
1. Железная герметически закрывающаяся реторта
(можно заменить колбой).
2. Тигельная электропечь на 500 °С.
3. Термопара с пирометром или термометр на 500 °С.
4. Приемники для жидких и газообразных продуктов.
5. Древесные опилки или кусочки дерева.
6. 0,1 н. раствор щелочи.
7. Фенолфталеин.
8. Известковая вода.
9. Бромная вода.
Проведение работы
Для сухой перегонки дерева можно воспользоваться
железной ретортой (рис. 46) или в крайнем случае лит¬
ровой стеклянной колбой (рис. 49). Реторту загружают
до половины ее объема предварительно взвешенными
опилками или кусочками дерева (около 30—40 г). Со¬
бранную установку испытывают на герметичность, т. е.
открывают зажим 8. Если прибор герметичен, то вода вы¬
текает непродолжительное время, а затем ее ток прекра¬
щается. Когда ток воды прекратится, закрывают зажим 8,
резиновый шланг отсоединяют от приемника 4 и нагрева¬
ют колбу до 100—110 °С, выпуская воздух в атмосферу.
Шланг присоединяют к приемнику 4, открывают зажим
8 и нагревают колбу до 300 °С, постепенно повышая тем¬
пературу до 450—500 °С. Жидкие продукты перегонки со¬
бираются в стеклянном приемнике, а газообразные — в
газометре. Количество газообразных продуктов опреде-
160
ляется по объему воды, вытекшей из газометра. После
окончания опыта выключают электропечь и отделяют
приемник, чтобы жидкие продукты не засосались в реак¬
тор.
Замеряют объем жидких продуктов мерным цилинд¬
ром, переливают их в колбу, соединенную с холодильни¬
ком, и содержимое колбы постепенно нагревают до 120 °С.
Вначале происходит отгонка метилового спирта, затем
Рис. 49. Сухая перегонка дерева:
/ — колба с кусками дерева; 2 — термометр; 3 — газоотводная трубка; 4 — при¬
емник; 5 — охлаждающая смесь; 6 — газометр — приемник газообразных про¬
дуктов; 7 — сосуд для приема вытекающей воды; 5 —зажим; 9 — асбест,
воды и уксусной кислоты. Прибор для перегонки должен
иметь небольшие размеры: колба на 20—30 мл, холодиль¬
ник длиной 20 см.
Для определения уксусной кислоты дистиллят пере¬
ливают в стакан и оттитровывают 0,1 н. раствором щело¬
чи в присутствии фенолфталеина. Для качественного
анализа газообразные продукты вытесняют из газометра
водой. Наличие двуокиси углерода доказывается пропу¬
сканием его через раствор гидроокиси кальция. Наличие
непредельных соединений доказывают пропусканием
собранного газа через бромную воду.
Газообразные продукты горючи. Это можно показать,
поджигая газ, выходящий из газометра. Для этого в га¬
6 Н. Г. Ключников
161
зометр наливают воду и газ, выходящий из газоотводной
трубки, поджигают. Анализ газообразных продуктов в
газоанализаторе проводится по указанию преподавателя.
Оформление результатов работы
Зарисовать установку для сухой перегонки дерева и
кратко описать ее работу. Цифровые данные занести в
таблицу.
Взято дерева
(в г)
Получено при перегонке
уксусной кис¬
лоты (в г)
жидких про¬
дуктов (в мл)
газообразных
продуктов (в л)
угля (в г)
Вопросы и задачи
1. При сухой перегонке березовой древесины при 450—500 °С
в среднем получается древесного угля 32%, газа 14%, смолы 16%,
воды 28%, уксусной кислоты 7%, метилового спирта 1,6%, ацето¬
на 0,9%; полученный газ содержит 46% двуокиси углерода, 33%
окиси углерода, 17% метана, 2% этилена и 2% водорода. Сколько
получается неорганических и органических веществ из 100 массовых
частей березовой древесины?
Ответ. Органических — 28 массовых частей, неорганических —
72 массовые части.
2. Почему количество органических веществ и двуокиси угле¬
рода в продуктах сухой перегонки уменьшается при повышении тем¬
пературы (например, до 800—900 °С), а количество окиси углерода
и водорода увеличивается?
3. Для получения активированного угля древесный уголь на¬
гревают под давлением с водяным паром. Какие физические про¬
цессы и химические реакции при этом протекают?
4. За счет каких реакций при сухой перегонке древесины обра
зуется окись углерода и двуокись углерода?
5. Теоретический расход воздуха, подсчитанный по уравнениям
реакций горения твердого и жидкого топлива, определяется так:
V = 0.089С + 0,267Н -f- 0,033 (S — О) яж3//сг топлива,
где С, Н, S, О — содержание углерода, водорода, серы и кислорода
в массовых процентах. На основании каких соображений выведе¬
но данное уравнение?
6. Для определения теплотворной способности топлива
Д. М. Менделеевым выведена формула:
Q = 81С + 246Н — 26 (О — S) — 6Н20 ккал/кг,
162
где С, Н, О, S и Н20 — содержание углерода, водорода, кислорода,
серы и паров воды в топливе в массовых процентах. Используя теп¬
лоты образования соответствующих окислов, определите, каким пу¬
тем Д. И. Менделеев вывел приведенную формулу?
§ 11. АНАЛИЗ ТВЕРДОГО ТОПЛИВА
Твердое топливо имеет сложный состав и включает
воду, органические и минеральные вещества. В состав ор¬
ганических веществ топлива входят углерод, водород,
азот, кислород, сера. В состав минеральных веществ, яв¬
ляющихся балластом, входят главным образом элементы:
кальций, магний, железо, алюминий — в виде карбонатов,
силикатов, фосфатов и т. д.
Технический анализ сводится к определению влаж¬
ности, зольности и количества летучих продуктов. Ре¬
зультаты анализа относят к топливу, не подвергавшему¬
ся подсушке (рабочее топливо), или к воздушно-сухому,
или к абсолютно сухому. Воздушно-сухим топливом на¬
зывается топливо, получаемое высушиванием при 70—
75 °С в сушильном шкафу, а затем при комнатной темпе¬
ратуре на воздухе. Количество влаги, удаляемой при та¬
кой подсушке, будет определяться не только абсолют¬
ным содержанием в топливе, но также и гигроскопично¬
стью последнего и влажностью окружающего воздуха.
Абсолютно сухим называется топливо, получаемое высу¬
шиванием его при 105 °С до постоянной массы.
Во всех случаях при анализе топлива необходимо
брать среднюю пробу. Для этого измельчают несколько
кусков угля (сланца, антрацита и др.) и просеивают че¬
рез сито с отверстиями 0,2 мм. Фракцию, не прошедшую
через сито, снова измельчают и просеивают.
Работа 28
Определение влажности твердого топлива
Цель работы. Определить влажность топлива.
Оборудование и материалы
1. Твердое топливо (каменный уголь, антрацит, сла¬
нец).
в*
163
2. Аналитические весы с разновесами (точность взве¬
шивания 0,001 г).
3. Бюкс высотой 25—30 мм и диаметром 35—40 мм.
4. Сушильный шкаф с термометром.
5. Ступка с пестиком.
6. Сито с диаметром отверстий 0,2 мм.
7. Эксикатор.
Проведение работы
Каменный уголь, антрацит или сланец предварительно
измельчают в ступке и просеивают. Берут 1—2 г топлива
и помещают в предварительно взвешенный бюкс, распре¬
деляют топливо ровным слоем и высушивают при 102—
105 °С около 2 ч до постоянной массы. Крышка бюкса
при высушивании должна быть полуоткрыта. Высушен¬
ное топливо вынимают из сушильного шкафа, закрывают
бюкс крышкой, охлаждают сначала на воздухе, а затем
в эксикаторе и взвешивают. Содержание влаги рассчи¬
тывается по формуле (в %):
М_100-
т ’
где М — убыль в массе после высушивания в граммах;
т — навеска топлива в граммах.
После первого определения влаги проводят повтор¬
ное просушивание при 104—105 °С в течение 30 мин.
Потеря в массе не должна превышать 0,01 г. Если же
потеря в массе окажется более 0,01 г, то высушивание
повторяют до тех пор, пока убыль массы не окажется
меньше 0,01 г.
Оформление результатов работы
Кратко записать методику работы и полученные дан¬
ные занести в таблицу.
Навеска топлива (в г)
Убыль в массе
(в г)
Количество влаги (в %)
до высушивания |после высушивания
164
Работа 29
Определение количества летучих веществ в топливе
Количество летучих веществ, выделяющихся при на*
греваиии топлива без доступа воздуха, в значительной
степени определяется условиями анализа и зависит от
скорости и температуры нагревания, измельченности топ¬
лива, сосуда, в котором проводится анализ, и т. д. Поэ¬
тому для получения сопоставимых результатов анализ
нужно проводить в одинаковых условиях.
Цель работы. Определить количество летучих ве¬
ществ в твердом топливе.
Оборудование и материалы
1. Твердое топливо (каменный уголь, антрацит, сла¬
нец и т. д.).
2. Фарфоровый тигель с крышкой (высота 40 мм, ди¬
аметр 30 мм).
3. Муфельная печь на 900 °С с термопарой.
4. Аналитические весы с разновесами.
5. Эксикатор.
6. Тигельные щипцы.
Проведение работы
Навеску измельченного воздушно-сухого твердого
топлива в количестве около 1 г отвешивают в предва¬
рительно просушенный и взвешенный тигель. Тигель
закрывают крышкой и помещают в электрическую печь,
нагретую до 850 °С. Тигель ставят на жароупорную
подкладку (например, сделанную из глины), чтобы его
дно находилось от дна муфеля на расстоянии 10—20 мм.
Тигель выдерживают в печи в течение 7 мин. При этом
происходит удаление влаги и летучих органических ве¬
ществ. Затем тигель вынимают и охлаждают, не снимая
крышки, сначала на воздухе 5 мин, а затем в эксикаторе
до комнатной температуры. В тигельную печь тигель
можно также опускать при помощи проволоки. На конце
ее сделано кольцо, в которое вставляется тигель.
165
Охлажденный тигель взвешивают и количество летучих
веществ рассчитывают по формуле:
% ЛВ = ^—
т
где р—потеря топлива в массе после прокаливания;
т — навеска топлива;
НгО — влага, имевшаяся в навеске угля.
Количество влаги вычисляется, исходя из процентно¬
го содержания влаги, имевшейся в угле. Следовательно,
прежде чем определить количество летучих веществ,
нужно определить количество влаги (см. предыдущую
работу). Например, для определения количества летучих
веществ было взято 1,3468 г угля, после прокаливания
масса стала 1,1122 а. Количество влаги было равно 1,2%.
Следовательно, в 1,3468 г топлива влаги будет:
1,3468 • 1,2
100
0,0161 (г).
Количество летучих веществ равно:
(0,2346 — 0,0161) • 100
% лв =
1,3468
16,3.
Тот же результат можно получить непосредственно по
формуле:
% ДБ = -Р-. 100—% W,
т
где р — потеря топлива в массе после прокаливания;
%W— процентное содержание влаги в топливе (см.
предыдущую работу).
Оформление результатов работы
Кратко описать методику работы и полученные циф¬
ровые данные внести в таблицу.
Навеска топли¬
ва (в г)
Убыль в массе
(в г)
Влажность,
перечисленная
к навеске (в г)
Количество летучих веществ
(в г)
(в %)
166
Работа 30
Определение зольности твердого топлива
Минеральные вещества, присутствующие в твердом
топливе, имеют двоякое происхождение. С одной сторо¬
ны, это песок, глина и другие виды пустой породы, попав¬
шей в топливо при его добыче. Другая часть минераль¬
ных веществ связана с углем. Независимо от происхож¬
дения наличие минеральных веществ ухудшает качество
топлива, а при высоком их содержании часто затрудняет
его применение.
При определении зольности топлива большое значе¬
ние имеет отбор средней пробы. Уголь нужно измельчить
до небольших кусков, тщательно перемешать, отобрать
пробу в несколько десятков граммов, а затем эту пробу
растереть в ступке и снова перемешать.
Оборудование и материалы
1. Твердое топливо (каменный уголь, антрацит, сла¬
нец и т. д.).
2. Фарфоровый тигель.
3. Муфельная электрическая печь с термопарой.
4. Тигельные щипцы.
5. Аналитические весы (точность взвешивания
0,001 г).
6. Эксикатор.
Проведение работы
В предварительно прокаленный и взвешенный фарфо¬
ровый тигель или лодочку отвешивают 1—2 г топлива и
помещают его в муфельную печь, которая предваритель¬
но может быть нагрета до 250—300 °С. Затем температу¬
ру печи постепенно повышают до 800—825 °С в течение
1—2 ч и выдерживают при указанной температуре 1 ч.
После этого тигель (или лодочку) вынимают из муфель¬
ной печи, охлаждают 5 мин на воздухе и переносят в эк¬
сикатор, где он охлаждается до комнатной температуры.
После взвешивания тигля зольность рассчитывается по
формуле:
167
о /n/v Af 100
Зольность (%) — -—,
где М — масса зольного остатка в граммах;
т — масса топлива.
Для проверки правильности определения зольности
тигель с зольным остатком можно еще раз прокалить.
Между двумя определениями разница не должна превы¬
шать 0,001 г.
Оформление результатов работы
Записать методику определения зольности топлива.
Полученные результаты записать в таблицу.
Масса
топлива
(в г)
золы
(в г)
(в %)
§ 12. ПЕРЕРАБОТКА ЖИДКОГО ТОПЛИВА
Работа 31
Крекинг нефтепродуктов
Цель работы. Провести каталитический крекинг
керосина. Определить выход бензина, тяжелых фракций
и газообразных продуктов.
Оборудование и материалы
1. Электропечь.
2. Железная трубка с катализатором.
3. Бюретка на 50 или 100 мл.
4. Аллонж.
5. Приемник-колба на 200—250 мл.
6. Градуированный газометр.
7. Прибор для разгонки продуктов крекинга.
8. Стеклянные и резиновые трубки, пробки.
168
Проведение работы
Крекинг проводится в приборе, изображенном на ри¬
сунке 50. Керосин подается из бюретки 1 в железную
трубку 1 с катализатором, обогреваемую электропечью 3.
Жидкие продукты крекинга собираются в приемнике 4У
помещенном в охлаждающую смесь, газообразные про¬
дукты поступают в газометр 6.
Для работы пользуются керосином, не содержащим
непредельных углеводородов. Непредельные соединения
в керосине обнаруживают бромной водой.
Для очистки от непредельных соединений наливают 100—200 мл
керосина в делительную воронку и сильно встряхивают в течение
5 мин с 15—20 мл концентрированной серной кислоты; при этом во¬
ронку нужно время от времени переворачивать и открывать кран
для удаления газообразных продуктов реакции. Отстоявшийся ниж¬
ний слой выпускают из воронки и очистку повторяют с повой пор¬
цией серной кислоты. Керосин очищают щелочным раствором пер¬
манганата калия и промывают водой. Для удаления воды в керо¬
син добавляют безводный хлорид кальция, встряхивают в течение
10—15 мин и фильтруют через складчатый фильтр.
Катализатор, употребляемый для проведения крекин¬
га, готовится из окислов алюминия и кремния. Из при¬
родных соединений пригодны хорошие сорта глин, не со¬
держащие по возможности примесей. Для очистки глину
перемешивают с соляной кислотой, разбавленной в отно¬
шении 1 1, и смесь выдерживают в течение нескольких
дней. Соляную кислоту и соли отмывают водой деканта¬
цией, а затем на воронке Бухнера. Промытую глину под¬
сушивают на воздухе, а потом в сушильном шкафу при
150—200 °С, смешивают с асбестом и загружают в реак¬
тор. В качестве катализатора можно также применить
обожженную глину в виде кусочков размером 3—4 мм
или битый кирпич.
Установку проверяют на герметичность. Для этого
открывают кран 7 для выпуска воды, конец резинового
шланга необходимо держать ниже уровня газометра на
10—15 см. При герметичности прибора вода не будет
выливаться из шланга. Если же он будет опущен значи¬
тельно ниже, то в системе создается значительное
Трубка должна иметь наклон в сторону приемника.
169
разрежение и в таких условиях герметичность прибора
трудно поддержать.
Убедившись в герметичности прибора, нагревают
электропечь до 600 °С. Воздух выпускают в атмосферу.
Присоединяют газометр, открывают кран 7 и впускают
керосин по 2—3 капли в 1 сек. Когда через трубку прой¬
дет 50—80 мл керосина, печь выключают и проводят
Рис. 50. Каталитический крекинг нефтепродуктов:
/ — бюретка с нефтепродуктом; 2 — трубка с катализатором; 3 — электропечь;
4 — приемник для жидких нефтепродуктов; 5 — охлаждающая смесь; 6 — газо¬
метр для приема газообразных нефтепродуктов; 7 — кран для выпуска воды;
8 — термопара; 9 — пирометр.
разгонку продуктов крекинга. Замеряют объем жидких
продуктов и переливают их в колбу, соединенную с холо¬
дильником. Дистиллят собирают в мерный цилиндр. За¬
меряют объем полученного бензина (выкипающего до
170 °С) и тяжелых фракций (выкипающих от 170 до
230°С). Для сравнения проводят разгонку керосина.
В полученном продукте всегда имеются непредельные
соединения, наличие которых доказывается реакцией
с бромной водой.
Объем газообразных продуктов крекинга определяет¬
ся по уровню воды в газометре или по объему воды, вы¬
текающей из газометра.
170
В газообразных продуктах крекинга также содер¬
жатся непредельные углеводороды. Для их обнаружения
газ собирают из газометра в цилиндр с водой, добавляют
немного бромной воды и, закрыв цилиндр стеклом,
встряхивают.
Во время работы на катализаторе накапливается
кокс, поэтому после 3—4 опытов катализатор нужно за¬
менить или выжечь кокс. Для выжигания кокса печь
нагревают до 800 °С, трубку присоединяют к газометру
или к водоструйному насосу и в течение 15—20 мин че¬
рез трубку пропускают слабый ток воздуха.
Крекинг можно вести и при более высокой темпера¬
туре, например при 650—700 °С. В этом случае количе¬
ство газообразных продуктов увеличивается.
Оформление результатов работы
Зарисовать установку для крекинга и описать ее ра¬
боту. Результаты опытов оформить в виде таблицы.
Вопросы и задачи
1. Почему при высокотемпературном крекинге (600—700 °С)
газообразных продуктов образуется больше, чем при низкотемпера¬
турном?
2. Напишите уравнение реакции термического распада гептана
при крекинге на два углеводорода с меньшей молекулярной массой.
3. Напишите уравнение реакции дегидрирования бутана при кре¬
кинге.
4. Почему увеличение давления при крекинге снижает выход
газообразных продуктов?
§ 13. АНАЛИЗ НЕФТЕПРОДУКТОВ
Работа 32
Определение плотности нефтепродуктов
Применение различных нефтепродуктов определяется
их плотностью, вязкостью, температурой вспышки, окта¬
новым числом, содержанием влаги и различных приме¬
сей, в том числе сернистых соединений, и т. д.
Плотность фракций нефти и различных нефтепродук¬
тов является характерной величиной. В большинстве
171
случаев она возрастает с увеличением молекулярной
массы веществ, входящих в состав нефтепродуктов.
Плотность повышается при увеличении в нефтепродук¬
тах ароматических углеводородов.
Плотность измеряется массой тела, заключенной в
единице его объема. Плотность нефтепродуктов обычно
измеряется при температуре 20 °С. Если отнести ее
к плотности воды при 4 °С, то получают относительную
плотность. Для определения плотности нефтепродуктов
и нефти используют следующие три метода: 1) ареомет-
рический; 2) пикнометрический; 3) взвешивание на ве¬
сах Вестфаля.
Цель работы. Определить плотность нефтепро¬
дуктов с помощью ареометра.
Оборудование и материалы
1. Ареометр для определения плотности нефтепро¬
дуктов (нефтеденсиметры).
2. Стеклянный цилиндр с диаметром не менее 5 см.
3. Нефтепродукты с вязкостью не более 2 * 10~4 м2/сек
при 50 °С (бензин, керосин, трансформаторное масло
и т. д.).
Проведение работы
Для определения плотности нефтепродуктов исполь¬
зуют специальный ареометр, снабженный в нижней ча¬
сти термометром. Он отградуирован с точностью до
0,0005, и его показания дают непосредственное значение
плотности, т. е. он показывает отношение массы нефте¬
продуктов при 20 °С, отнесенной к массе такого же объ¬
ема воды при 4 °С.
Нефтепродукт наливают в мерный цилиндр. Темпера¬
тура нефтепродукта не должна отклоняться от темпера¬
туры окружающей среды более чем на 5°С. Затем в него
погружают сухой ареометр, не касаясь стенок сосуда.
В маловязких продуктах в положение равновесия
ареометр приходит через 2—5 мин. Если же вязкость
нефтепродуктов значительна, то ареометр приходит в
равновесие через 10—15 мин. Отсчет плотности прово¬
дят при температуре 20 °С по верхнему краю мениска.
Глаз необходимо держать на уровне мениска. Расхожде¬
172
ние в значениях плотности между двумя определениями
не должно превышать 0,001. Если температура нефтепро¬
дукта ниже или выше 20 сС, то в значение плотности
вносится поправка (о ее нахождении см. ниже).
Оформление результатов работы
Записать методику определения плотности. При на¬
личии температурной поправки провести соответствую¬
щие вычисления.
Рис. 51. Виды пикнометров.
Цель работы. Определить плотность нефтепро¬
дуктов при помощи пикнометра.
Пикнометрический метод определения плотности
применяется: 1) когда имеется небольшое количество
нефтепродуктов, 2) когда требуется высокая точность
определения, 3) когда нефтепродукт имеет высокую
вязкость (например, битумы).
Наиболее распространенными типами пикнометров
(рис. 51) являются пикнометр с меткой и пикнометр
с капилляром в крышке. Точность измерения увеличива¬
ется с увеличением объема пикнометра, так как в этом
случае повышается точность взвешивания и точность из¬
мерения объема нефтепродукта.
173
Пикнометрический метод измерения плотности неф¬
тепродуктов основан на определении массы нефтепро¬
дукта и воды в объеме пикнометра при одинаковых тем¬
пературах.
Оборудование и материалы
1. Пикнометр.
2. Аналитические весы с разновесами.
3. Жидкие или твердые нефтепродукты.
4. Дистиллированная вода.
Проведение опыта
Для определения водного .числа пикнометра, т. е.
массы воды в объеме пикнометра, взвешивают на анали¬
тических весах сначала тщательно высушенный в су¬
шильном шкафу и охлажденный до 20 °С пустой пикно¬
метр. Затем пикнометр заполняют несколько выше метки
дистиллированной водой и ставят его в термостат на
20—30 мин с температурой до 20 °С. Если температура
окружающего воздуха равна 20 °С, то пикнометр можно
оставить на воздухе (20—30 мин), чтобы вода приобрела
температуру воздуха. Затем избыток воды удаляют по¬
лоской фильтровальной бумаги, отсчитывают по нижне¬
му мениску. При помощи фильтровальной бумаги нужно
также удалить воду, которая иногда остается на внут¬
ренних стенках горлышка пикнометра. В пикнометрах
с капилляром в крышке избыток воды удаляется через
капилляр при помощи фильтровальной "бумаги. Водное
число пикнометра равно разности между массой пикно¬
метра с водой и массой пустого пикнометра. Затем опре¬
деляют массу нефтепродукта в объеме пикнометра.
Испытуемый нефтепродукт предварительно выдержи¬
вают в сухом закрытом сосуде при 20 °С, наливают его
в пикнометр до метки и взвешивают. Плотность нефте¬
продукта рассчитывают по формуле:
1 0.9^70 + 0,0012,
где М[ — масса пустого пикнометра;
М2 — масса пикнометра с нефтепродуктом;
М3 — водное число пикнометра;
0,9970 — плотность воды при 20 °С;
174
0,0012 — плотность воздуха при 20 °С и 760 мм рт. ст.
При определении плотности вязких продуктов их
предварительно нагревают до 50—70 °С. После заполне¬
ния пикнометра его перед взвешиванием охлаждают до
20 °С.
Пикнометром можно определить плотность твердых
нефтепродуктов, например битумов. Для этого мелкими
кусочками нефтепродукта заполняют примерно полови¬
ну пикнометра и последний помещают в термостат при
90—100 °С до полеюго расплавления нефтепродукта.
После охлаждения пикнометра его заполняют несколько
выше метки водой, выдерживают в течение 20—30 мин
при 20 °С и, удалив избыток воды (см. выше), взвешива¬
ют. Плотность нефтепродукта рассчитывагот по фор¬
муле:
d
20
4
— Mt
~М3-(М4-М2)
0,997 + 0,0012,
где М± — масса пикнометра с нефтепродуктом и водой;
остальные обозначения прежние.
Оформление результатов работы
Записать методику работы и полученные цифровые
данные занести в таблицу.
Объем пикномет¬
ра (в см3)
Масса
Плотность
пустого
пикнометра (в г)
заполненного
пикнометра (в г)
Цель работы. Определить плотность жидкости
с помощью весов Вестфаля.
Оборудование и материалы
1. Весы Вестфаля.
2. Стеклянный цилиндр с диаметром не менее 5,5—
4 см.
3. Нефтепродукты (бензин, керосин, масла).
175
Проведение работы
Для определения плотности используют специальные
весы (рис. 52). На штативе 1 укреплено коромысло 2,
опирающееся на призму, находящуюся в вилке 3. Одно
плечо коромысла имеет десять делений. Десятое деление
соответствует крючку, на котором укреплен ареометр 5
с постоянным объемом. Другое плечо заканчивается ос¬
трием 6. В случае равновесия оно устанавливается про¬
тив острия 7.
Рис. 52. Весы Вестфаля:
/ — штатив; 2 — коромысло; 3 — вилка; 4 — крючок; 5 —
ареометр; 6, 7 — острия; 8—12 — разновесы; 13 — цилиндр.
Весы имеют пять разновесов (8—15), два из которых
одинаковы и масса каждого равна массе вытесненной
ареометром (поплавком) воды. Другие три разновеса
имеют массу в 10, 100 и 1000 раз меньшую. При навеши¬
вании первого или второго разновеса на крючок масса
их равна единице, третьего — 0,1, четвертого — 0,01 и пя¬
того — 0,001.
При навешивании разновесов на вырезки плеча коро¬
мысла их масса уменьшается. Например, если масса пер¬
вого (или второго) разновеса на крючке равна 1, то на
девятом делении она равна 0,1.
Перед работой прибор устанавливают на нулевую
точку, т. е. подвижное острие 6 должно совпадать с не¬
подвижным 7. Затем по стеклянной палочке осторожно
176
наливают в цилиндр 13 жидкость до погружения в нее
поплавка на глубину 5 мм от поверхности жидкости. На¬
рушенное равновесие масс восстанавливают с помощью
разновесов. Например, равновесие было достигнуто,
когда первая разновеска находилась на девятом делении,
вторая — на шестом, третья — на пятом, четвертая — на
четвертом. В этом случае так называемая видимая плот¬
ность di равна 0,9654. Затем измеряют температуру жид¬
кости. Плотность жидкости при температуре испытания
рассчитывается по формуле:
d{ = (0,99823 — 0,0012) -f 0,0012 = 0,99703 + 0,0012.
Для пересчета плотности жидкости при соответст¬
вующей температуре на d‘l° применяют метод, описан¬
ный в следующем разделе.
Оформление результатов работы
Записать результаты расчетов по определению и
Нахождение поправки на плотность
нефтепродукта при его определении
ниже или выше 20 °С
Плотность нефтепродуктов определяется при темпе¬
ратуре 20° С. Если температура нефтепродукта ниже или
выше указанного значения, то в значение плотности
необходимо внести поправку. В этом случае плотность
рассчитывается по формуле:
d\° = d[ - А (20-/),
где dl° — значение плотности при 20° С;
d\ — значение плотности при температуре опыта;
А — поправка (см. таблицу II).
Пример. Плотность, найденная на опыте приЗО°С,
т. е. d\°, равна 0,9114. Значение dl° равно:
р4° = 0,9114 — 0,00062 (20 — 30) = 0,9114 + 0,0062 = 0,9176.
Вопросы и задачи
1. На каком известном законе физики основано определение
плотности нефтепродуктов?
177
2. Почему температура нефтепродукта при определении плотно¬
сти не должна сильно отличаться от тем пера туры окружающего воз¬
духа?
3. Почему определение плотности при помощи пикнометра дает
более точные результаты, чем при измерении ареометром?
4. Как изменится плотность нефтепродукта, содержащего неко¬
торое количество воздуха (воды), при пикнометрическом способе и
определении с помощью ареометра по сравнению с плотностью чи¬
стого нефтепродукта?
5. Почему в формулу для определения плотности нефтепродук¬
тов при помощи пикнометра входит значение плотности воздуха?
Почему значение плотности взято со знаком «плюс»?
Работа 33
Определение температуры вспышки и воспламенения
нефтепродуктов
Нефтепродукты — сложная смесь углеводородов.
При определенной температуре легкокипящие фракции
создают над поверхностью нефтепродукта такую кон¬
центрацию паров углеводородов, при которой возможно
загорание при поднесении пламени. Эго горение (вспыш¬
ка) продолжается некоторое время и прекращается пос¬
ле окисления паров горючих углеводородов. Температу¬
ра, при которой нефтепродукт, нагреваемый в строго
определенных условиях, выделяет такое количество
паров, которое образует с окружающим воздухом смесь,
вспыхивающую при поднесении к ней пламени, называ¬
ется температурой вспышки. Температура вспышки
меняется в зависимости от поверхности нефтепродукта,
смены газовой фазы над его поверхностью, температуры
и размера пламени, которым вызывают вспышку. Поэто¬
му определение температуры вспышки должно прово¬
диться в строго определенных условиях.
При дальнейшем повышении температуры из нефте¬
продукта начинают испаряться менее летучие фракции.
При какой-то определенной температуре наступит мо¬
мент, когда при поднесении пламени к парам углеводо¬
родов вслед за вспышкой наступит горение. Температу¬
ра, при которой продукт при поднесении к нему пламени
загорается и продолжает гореть, называется температу¬
рой воспламенения. Отвод теплоты, недостаток кислоро¬
да могут привести к тому, что пламя может погаснуть.
Поэтому при определении температуры вспышки не учи¬
тывают длительность горения.
178
Повышение атмосферного давления приводит к
уменьшению концентрации паров над поверхностью неф¬
тепродукта. Поэтому температура вспышки и температу¬
ра воспламенения повышаются. Повышение давления
на 1 мм рт. ст. повышает температуру вспышки на
0,033—0,030 °С. Таким образом, температура вспышки и
температура воспламенения зависят от условий проведе¬
ния опыта и от конструкции прибора, в котором прово¬
дятся эти определения.
Цель работы. Определить температуру вспыш¬
ки и температуру воспламенения нефтепродукта.
Оборудование и материалы
1. Прибор для определения температуры вспышки
и прибор для определения температуры воспламенения.
2. Нефть и различные нефтепродукты: керосин, неф¬
тяные масла.
Проведение работы
Температура воспламенения определяется в приборах
открытого типа, так как во время определения к поверх¬
ности нефтепродукта необходим непрерывный приток
воздуха.
Для определения температуры вспышки существуют
два типа приборов: с закрытым и открытым тиглем.
В тиглях закрытого типа температура вспышки на 20—
30° С ниже, чем в приборах открытого типа.
Для определения температуры вспышки применяют
закрытый прибор типа ПВН (рис. 53). Снаружи прибо¬
ра имеется металлическая рубашка /, окружающая чу¬
гунную баню 2, в которую вставлен резервуар 3 для неф¬
тепродукта. Сверху резервуар закрыт крышкой, через
которую проходят термометр 4, мешалка 5 на гибкой
ручке и зажигательная лампочка 6. Рукоятка 7 переме¬
щает лампочку через соответствующее отверстие в
крышке в пространство внутри резервуара 3.
Нефтепродукт наливают в чистый и сухой резервуар 3
до кольцевой метки на его стенках. Последний поме¬
щают в чугунную баню, закрывают крышкой, вставляют
термометр и нагревают при периодическом перемешива¬
нии нефтепродукта. Его температура должна повышать-
179
4
Рис. 53. Прибор для определения темпе¬
ратуры вспышки:
/ — металлическая рубашка; 2 — чугунная ба¬
ня; 3—резервуар для нефтепродукта; 4—тер¬
мометр; 5 —мешалка; 6 — зажигательная лам¬
почка; 7 — рукоятка.
ся со скоростью 10—20 °С в 1 мин, если температура
вспышки выше 150 °С. Если температура вспышки ниже
150 °С, то его температура должна повышаться со ско¬
ростью 5—8°С в 1 мин.
За 25—30 °С до ожидаемой температуры вспышки
180
нагревание ослабляют. Температура должна повышаться
со скоростью 2 °С в 1 мин.
Одновременно проводят испытание на зажигание.
С этой целью, прекратив перемешивание, поворачивают
рукоятку 7, которая перемещает зажигательную лампоч-
ку в пространство над неф¬
тепродуктом. Лампочка в
парах нефтепродукта дол¬
жна находиться не более
2 сек. Это испытание прово¬
дят каждый раз, когда тем¬
пература поднимается па
2 °С. После появления пер¬
вой вспышки температуру
повышают еще па 2°С и
проводят повторное зажига¬
ние. Если пары нефтепро¬
дукта не вспыхнут, то обо¬
грев удаляют и, охладив не¬
сколько нефтепродукт, по¬
вторяют испытание. Расхож¬
дение между двумя опреде¬
лениями не должно состав¬
лять более 2°С.
Для определения темпе¬
ратуры вспышки в открытом
приборе (рис. 54) использу¬
ют железный тигель / (диа¬
метр 64±1 мму высота 47±
± 1 мм, толщина стенки
I мм), помещенный в песоч¬
ную баню 2 (высота 50 ±
±5 мм, диаметр 100±5 мм).
Между дном тигля и чашкой
необходимо насыпать слой песка толщиной 5—8 мм. Уро¬
вень песка в бане должен быть одинаков с уровнем жид¬
кости в тигле. Песочную баню устанавливают в кольцо
штатива; термометр 4 погружают в жидкость на глубину
45 мм. Уровень жидкости должен находиться на расстоя¬
нии 12 мм от края тигля, если температура вспышки неф¬
тепродукта ниже 210 °С, и на расстоянии 18 мм, если тем¬
пература вспышки 210 °С. Для определения уровня
жидкости относительно края тигля можно воспользо¬
Рис. 54. Прибор для опре¬
деления температуры вспыш¬
ки и температуры воспла¬
менения:
1 — железный тигель; 2 — пе¬
сочная баня; 3 — песок.
181
ваться узкой линейкой или специальным шаблоном,
имеющим выступы на 12 и 18 мм.
Для определения температуры вспышки нагревают
песочную баню, наблюдая за показаниями термометра.
Температуру нефтепродукта можно вначале повышать
на 10 °С в минуту, а за 40 °С до ожидаемой температу¬
ры вспышки — на 4 °С в минуту. Примерно за 10 °С до
ожидаемой температуры вспышки скорость нагревания
еще замедляют и через каждые 2°С проводят зажигание
пламенем микрогорелки по краю тигля. При этом дела¬
ют два оборота: один — по часовой стрелке, а другой —
против. Поскольку пламя горелки несколько нагревает
поверхность нефтепродуктов, длина его должна быть
3—4 лш, длительность испытания — не более 2—3 сек.
За температуру вспышки принимается температура,
которая была отмечена перед появлением над продуктом
пламени, обычно сопровождаемого небольшим хлопком.
Расхождение между двумя определениями для продук¬
тов с температурой вспышки выше 150 °С не должно
составлять более 6°С, а для продуктов с температурой
вспышки ниже 150 °С находится в пределах 4°С.
Для определения температуры воспламенения 1 неф¬
тепродукт нагревают со скоростью 4 °С в 1 мин и через
каждые 2°С повышения температуры проводят пламя
горелки над поверхностью жидкости. За температуру
воспламенения принимают температуру, при которой
продукт загорается и продолжает гореть не менее
5 сек. Расхождение между двумя определениями не дол¬
жно превышать 6°С. В противном случае испыта¬
ния повторяются.
Оформление результатов работы
Записать методику работы и полученные цифровые
данные.
§ 14. АНАЛИЗ ВОДЫ И ЕЕ УМЯГЧЕНИЕ МЕТОДОМ ИОННОГО
ОБМЕНА ИЛИ ИЗВЕСТКОВО-СОДОВЫМ МЕТОДОМ
Природная вода в своем составе всегда содержит
различные примеси — соли и газы, механические приме¬
си, находящиеся во взвешенном состоянии, иногда орга¬
1 Работа проводится только в присутствии преподавателя.
182
нические вещества и т. д. Соли, присутствующие в воде,
вызывают жесткость воды.
Бикарбонаты кальция и магния обусловливают кар¬
бонатную жесткость. При нагревании воды растворенные
бикарбонаты разлагаются и карбонатная жесткость при
этом сильно снижается. Полного устранения карбонат¬
ной жесткости не происходит вследствие того, что карбо¬
нат кальция и особенно магния, несколько растворим
в воде.
Наряду с понятием «карбонатная жесткость» сущест¬
вует еще понятие об устранимой, или временной, жестко¬
сти; это величина жесткости, на которую она понижается
при десятиминутном кипячении воды. Жесткость, остав¬
шаяся после кипячения воды, называется постоянной
жесткостью.
Хлориды и сульфаты кальция и магния обусловлива¬
ют некарбонатную жесткость воды. Сумма карбонатной
и некарбонатной жесткости составляет общую жест¬
кость. Общую жесткость выражают в виде суммы милли¬
грамм-эквивалентов ионов кальция и магния, содержа¬
щихся в 1 л воды. 1 мг-экв жесткости отвечает содержа¬
нию 20,04 мг/л Са 2+ или 12,16 мг/л Mg2+.
По величине жесткости различают очень мягкие, мяг¬
кие, среднежесткие, довольно жесткие, жесткие и очень
жесткие воды. Мягкая вода содержит 1,5—3,0 мг-экв Са2+,
жесткая — 6,5—11,0 мг-экв.
Жесткость воды определяется аналитически.
Устраняется жесткость воды при помощи ионитов и
известково-содовым методом (см. ниже, работу 35). Дру¬
гие методы находят более ограниченное применение.
Работа 34
Определение жесткости воды
Современным методом определения жесткости явля¬
ется титрование воды раствором трилоиа Б в присутст¬
вии специальных индикаторов — хромогенов, а именно
кислотного хрома темно-синего или кислотного хрома
черного. Титрование проводится в аммиачной среде при
значении pH раствора в пределах 9—10.
Хромогены образуют с ионами магния и другими
ионами комплексные соединения, окрашенные в красно-
183
фиолетовый цвет. При титровании трилоном содержа¬
щиеся в воде ионы кальция и магния, а также ионы ме¬
ди, цинка, марганца, кадмия, никеля, алюминия, двух-
и трехвалентного железа и некоторые другие реагируют
с ним и образуют малодиссоциированные соединения.
В конце титрования ионы магния, кальция и другие пере¬
ходят от комплексного соединения с хромогеном к трило-
ну и раствор окрашивается в сине-фиолетовый цвет —
цвет самого красителя, что указывает на окончание тит¬
рования.
■—оос—сн2
\
N — (СН2)2 — N<
ноос—сн.
СН,—СОО—'
сн2—соон
Na,
Трилон Б
0 = С—On
/О—С=0
I
сн2
СН2 Са
X ><
СН2 СН2-СН2 сн2
—о—с
I!
О
С-О—
II
о
Na,
Комплексное соединение трилона с кальцием
Для определения железа в качестве индикатора при¬
меняют роданид аммония, дающий вишнево-красное
окрашивание с ионом трехвалентпого железа. Для опре¬
деления меди и кальция в качестве индикатора применя¬
ют пурпуреат аммония, называемый также мурексидом.
Определение каждого иона нужно проводить при таком
значении pH среды, при котором трилон Б дает с опре¬
деляемым ионом более прочное соединение по сравнению
с индикатором. Например, жесткость воды, т. е. опреде¬
ление содержания кальция и магния, проводится при pH
184
выше 9, железа—при pH 1—2, кальция в присутствии
индикатора мурексида — при pH 12.
Содержание ионов кальция определяют также окса¬
латным методом. Ион кальция осаждают в виде нерас¬
творимого оксалата кальция:
Са2+ + ОД- = СаОД.
Затем из оксалата кальция выделяют щавелевую кис*
лоту:
СаОД + НОД = CaS04 + Н2ОД,
которую оттитровывают перманганатом калия:
5Н2ОД + 2КМп04 + ЗНОД -
= КОД + 2MnS04 + ЮС02 + 8Н20.
Количество кальция определяют на основании затрачен¬
ного объема перманганата калия.
Содержание ионов магния определяют объемно-ана¬
литическим или колориметрическим методом. Объемно-
аналитический метод заключается в том, что бикарбона¬
ты и карбонаты при титровании соляной кислотой пере¬
водятся в хлориды. Образующуюся двуокись углерода
удаляют из раствора кипячением. Затем ион магния
осаждают избытком раствора гидроокиси кальция:
Mg2+ + Са(ОН)2 = Mg(OH)2 + Са2+.
Гидроокись магния отфильтровывают, а избыток гид¬
роокиси кальция оттитровывают 0,1 н. раствором соля¬
ной кислоты. Таким образом, общую жесткость можно
определить как титрованием воды раствором трилона Б,
так и путем определения содержания кальция и магния
в растворе. Для вычисления общей жесткости концент¬
рации ионов кальция и магния, выраженные в милли¬
граммах на литр, перечисляют в концентрации, выражен¬
ные в миллиграмм-эквивалент па литр. Затем определя¬
ют общую жесткость.
Карбонатную жесткость определяют титрованием во¬
ды соляной кислотой:
Са(НС03)2 + 2НС1 - СаС12 + 2Н20 + 2С02;
Mg(HC03)2 + 2НС1 = MgCl2 + 2Н20 + 2С02.
185
Некарбонатную жесткость находят по разности меж¬
ду общей и карбонатной жесткостью.
Содержание двуокиси углерода в воде определяют
путем титрования щелочью в присутствии фенолфталеи¬
на до достижения окраски эталонного раствора.
Проба воды должна характеризовать действитель¬
ный ее состав, поэтому при отборе пробы из водопровода
воду спускают в течение 10—15 мин. Когда склянка
наполнится, воду некоторое время переливают через
край.
Из рек и ручьев воду отбирают на глубине 0,75 м
в нескольких местах, около берегов и в середине реки.
Отдельные пробы смешивают вместе. Анализ воды про¬
водится немедленно после взятия пробы или в крайнем
случае при соответствующем хранении спустя несколько
часов.
Цель работы. Определить общую карбонатную
и некарбонатную жесткость воды.
Определить количество ионов кальция, магния и ко¬
личество двуокиси углерода.
Оборудование и материалы
1. Бюретки, пипетки на 100 мл или мерные цилинд¬
ры, колбы на 250 мл и 500 мл, стаканы на 200—250 млу
мерные колбы на 200 мл, фильтры.
2. Водяная баня.
3. Соляная кислота (1 3).
4. Хлорид аммония, 10-процентный раствор.
5. Оксалат аммония, содержащий 50 г соли в 1 л.
6. Перманганат калия, 0,05 н. раствор.
7. Серная кислота (10 мл концентрированной H2S04
в 1 л воды).
8. Соляная кислота, 0,1 н. раствор.
9. Едкий натр, 0,1 н. раствор.
10. Метиловый оранжевый, 0,1-процентный раствор.
11. Фенолфталеин, 1-процентный раствор.
12. Специальные реактивы: раствор трилона Б, рас¬
твор кислотного хрома темно-синего,, аммиачная смесь,
раствор оксалата кальция и эталонный раствор. Описа¬
ние приготовления этих реактивов приводится в конце
работы.
186
Проведение работы
Определение ионов двухвалентных металлов. Внача¬
ле проверяют кислотность воды, так как комплексное
соединение кальция с трилоном Б в кислой среде не¬
устойчиво. Для этого к пробе воды, налитой в пробирку,
приливают 3—4 капли раствора метилового оранжевого.
Если раствор будет окрашен в желтый цвет, то можно
проводить трилонометрическое определение жесткости.
В противном случае воду нужно нейтрализовать разбав¬
ленным раствором щелочи (примерно 0,1 н.) в присут¬
ствии метилового оранжевого. Для этого к 200—300 мл
воды приливают 2—3 капли щелочи и после перемешива¬
ния отбирают пробу воды в пробирку и приливают 3—
4 капли раствора метилового оранжевого. Щелочь при¬
ливают до тех пор, пока метиловый оранжевый будет
окрашиваться в желтый цвет. После этого определяют
жесткость воды.
В коническую колбу емкостью на 250 мл отмеряют
100 мл воды, добавляют 5 мл аммиачного буферного
раствора, 10 капель индикатора хромогена и титруют по
1 капле 0,1 н. раствором трилона Б до перехода красно¬
фиолетовой окраски раствора в сине-фиолетовую.
Жесткость воды рассчитывается по формуле
(в мг-экв/л):
Ж = Ю00 - ну^,
где с — количество миллилитров раствора трилона Б,
израсходованного на титрование;
н.— нормальность трилона Б;
V — объем воды, взятый для титрования;
К — поправочный коэффициент трилона Б к данной
его нормальности.
Если, например, раствор был 0,1 и. и при хранении
его концентрация изменилась, то для определения по¬
правочного коэффициента истинную нормальность рас¬
твора необходимо разделить на 0,1. 0,1 н. раствор трило¬
на Б рекомендуется применять при значительной жест¬
кости воды, например 20 мг-экв/л. При жесткости от 0,5
до 20 мг-экв/л следует пользоваться 0,05 н. раствором и,
наконец, при жесткости ниже 0,5 мг-экв/л применять
0,01 н. раствор.
187
Определение иона кальция. К 100 мл анализируемой
воды прибавляют соляной кислоты (1 9) до кислой ре¬
акции на лакмус. Раствор нагревают до кипения, при¬
бавляют пипеткой 10 мл 10-процеитпого раствора хлори¬
да аммония и при перемешивании 15 мл раствора окса¬
лата аммония. Через 15 мин прибавляют 3—4 капли
раствора аммиака до слабого запаха. Ставят раствор
с осадком на два часа в кипящую баню К После этого
осадок отфильтровывают, смывая осадок со стенок ста¬
кана раствором оксалата кальция. Осадок на фильтре
промывают раствором оксалата кальция, определяя
полноту промывания 5-процентным раствором нитрата
серебра. Отмытый осадок растворяют на фильтре серной
кислотой, на что требуется около 10—12 мл кислоты,
собирают фильтрат в тот же стакан, в котором проводи¬
лось осаждение. Фильтрат нагревают до кипения и тит¬
руют раствором перманганата калия до появления розо«
вой окраски. После этого фильтр переносят в тот же ста¬
кан, взбалтывают и в случае исчезновения розовой
окраски титруют перманганатом калия.
Концентрацию ионов кальция в миллиграммах на
литр подсчитывают по формуле:
Концентрация Са2+ (мг/л) — 7"кмпо4/саН-
V 1000 . 100
100
где Ткмпо*/са2+ — титр пермаганата калия, выражен¬
ный через ион кальция. При его под¬
счете необходимо учесть, что согласно
уравнению реакции 2 моль перманга¬
ната калия соответствуют 5 г-атом
кальция;
V — объем раствора перманганата калия,
взятого на титрование.
Определение иона магния. В коническую колбу на
250 мл отбирают пипеткой 100 мл анализируемой воды
и, прибавив к ней 3 капли раствора метилового оранже¬
вого, воду оттитровывают 0,1 н. раствором соляной кис¬
лоты до оранжевой окраски. Одновременно так же отти¬
тровывают 100 мл дистиллированной воды до такой же
окраски. 11 При проведении рядовых анализов это время можно сократить
до 10—15 мин.
188
Колбы с растворами нагревают до кипения, а затем
растворы переливают в мерные колбы на 200 мл. В кол¬
бы добавляют по 50 мл насыщенного раствора гидро¬
окиси кальция и доливают до нижней части горлышка
свежепрокипяченной дистиллированной водой. Содер¬
жимое колбьг перемешивают и нагревают на водяной
бане 30 мин. При этом гидроокись кальция вступает в
реакцию с солями магния и как более сильное основание
осаждает малорастворимую гидроокись магния. Затем
растворы в колбах охлаждают водой, доливают дистил¬
лированной водой до метки, перемешивают и тут же от¬
фильтровывают выпавшую в осадок гидроокись магния
через складчатые фильтры. Фильтраты собирают в сухие
конические колбы. Из каждой колбы отбирают по 100 мл
раствора и титруют оставшийся избыток гидроокиси
кальция 0,1 н. раствором соляной кислоты в присутствии
фенолфталеина.
Количество магния в 1 л воды подсчитывается по фор¬
муле:
ГГ ЛЛ 9-U / / ч 12,16 2 (н. Kl — h. V2) ЮОО
Количество Mg2+ (мг/л) = щ ,
где 12,16 — грамм-эквивалент магния;
н. — нормальность соляной кислоты;
V\ — количество миллилитров соляной кислоты,
пошедшей на титрование раствора, получен¬
ного из дистиллированной воды;
V2 — количество соляной кислоты, израсходован¬
ной на титрование раствора, полученного из
анализируемой воды.
Для определения общей жесткости (количество
мг/экв Са24* и Mg2+ в 1 л) нужно концентрации магния
и кальция (см. выше), выраженные в мг-экв/л, разделить
на соответствующие эквивалентные массы, затем полу¬
ченные концентрации сложить:
Жесткость воды ----
Са мг-экв/л
2004
Mg мг-экв/л
Шб
Определение карбонатной и некарбонатной жестко¬
сти, В коническую колбу на 250 мл отбирают 100 мл
анализируемой воды и титруют ее 0,1 н. раствором соля¬
ной кислоты с индикатором метиловым оранжевым до
появления розового окрашивания.
189
Карбонатная жесткость рассчитывается по формуле:
iL . Ю00 (мг-экв/л),
Гг
где н. — нормальность соляной кислоты;
Vi — объем кислоты, затраченной на титрование
(в мл);
V2 — объем воды, взятой для титрования (в мл).
Определение содержания двуокиси углерода. В круг¬
лодонную колбу отмеряют 200 мл анализируемой воды,
прибавляют 0,2 мл 1-процентного раствора фенолфта¬
леина и перемешивают. Если вода окрасилась от фе¬
нолфталеина и эта окраска более интенсивна по сравне¬
нию с окраской эталонного раствора, то вода не содер¬
жит свободной двуокиси углерода. Если же вода не
окрасилась от фенолфталеина или окрасилась слабее,
чем раствор эталона, то ее титруют 0,01 н. раствором
едкого натра до окраски эталонного раствора.
Количество двуокиси углерода определяется по фор¬
муле:
СОа {мг/л) 2оо *
где н.— нормальность и V — объем (в мл) едкого нат¬
ра, израсходованного на титрование.
Оформление результатов работы
Описать ход анализа и провести соответствующие
расчеты.
Реактивы, необходимые для проведения
работы
1. о,1 н. раствор трилона Б1. Навеску трилона Б в количестве
18,613 г растворяют в 1 л дистиллированной воды и фильтруют, ес¬
ли он окажется мутным. Установка точного титра триллона Б про¬
водится по фиксоналу MgS04*7H20, который продается вместе с
набором. Установку титра можно проводить и но точной навеске
свежеперекристаллизованного сульфата магния (см. определение
жесткости воды). Раствор может храниться до года.
1 Реактивы, необходимые для колориметрического определения
жесткости воды, имеются в продаже.
190
При отсутствии фиксонала — сульфата магния, используемого
для проверки титра трилона Б, можно воспользоваться навеской
сухой соли. Для приготовления 0,01 н. раствора отвешивают
1,1315 г сульфата магния MgS04‘7H20 и растворяют соль в мер¬
ной колбе на 1 л. Титрование раствора трилоном Б проводится в
присутствии индикатора, как это описано при определении жестко¬
сти воды.
2. Раствор индикатора. 0,5 г кислотного хрома темно-синего
или 0,5 г кислотного хрома черного растворяют в 20 мл аммиачной
смеси (необходимо следить, чтобы не оставалось нерастворенных
крупинок вещества) и добавляют до 100 мл спирта-ректификата.
3. Аммиачная смесь. 100 мл 20-процентного раствора хлорида
аммония смешивают с 100 мл 20-процентнсго раствора аммиака и
добавляют до 1 л дистиллированной воды.
Вода, используемая для приготовления вышеупомянутых реак¬
тивов, не должна содержать солей, вызывающих жесткость. Для
проверки воды на жесткость к 100 мл ее прибавляют 1 мл аммиач¬
ной смеси и 6—7 капель индикатора; при этом должна появиться
синяя окраска.
4. Оксалат кальция. Для приготовления раствора оксалата
кальция к 10 мл 2-процентпого раствора хлорида кальция прили¬
вают 100 мл 5-процентного раствора оксалата аммония, кипятят
30 мин и дают отстояться. Осадок фильтруют, промывают водой до
удаления нона хлора, помещают в 500 мл воды, взбалтывают в те¬
чение 5—10 мин и отфильтровывают. Фильтрат является насыщен¬
ным раствором оксалата кальция,
5. Гидроокись кальция. 20 г гидроокиси кальция помещают в
мерную колбу на 1 л, наливают дистиллированной воды до метки,
взбалтывают и дают отстояться. Концентрацию раствора определя¬
ют титрованием 0,1 и. раствором соляной кислоты в присутствии
фенолфталеина. Для анализа отбирают пипеткой раствор так, что¬
бы не взмутить осадка. Хранить гидроокись кальция надо в тщатель¬
но закрытой склянке.
6. Эталонный раствор. В колбу наливают 200 мл дистиллиро¬
ванной воды, прибавляют 0,5 мл 10-процентного раствора едкого
натра, взбалтывают и прибавляют 0,2 мл раствора фенолфталеина.
Для приготовления последнего берут 2 мл 1-процентного раствора
фенолфталеина и разбавляют дистиллированной водой до 100 мл.
Эталонный раствор окрашен в розовый цвет. Хранить раствор нуж¬
но в плотно закрытой склянке.
Вопросы и задачи 11. При определнии карбонатной жесткости воды используют
индикатор. Почему для этого определения нельзя использовать фе¬
нолфталеин?
2. Почему при определении свободной двуокиси углерода при¬
меняют в качестве индикатора фенолфталеин, а не метиловый оран¬
жевый?
3. При титровании 100 мл воды 0,1 н. раствором трилона Б его
пошло 2 мл. Рассчитайте жесткость воды.
Ответ. 2 мг-экв!л.
191
4. При титровании осадка оксалата кальция, полученного при
осаждении ионов кальция оксалатом аммония, пошло 10 мл 0,01 н.
раствора перманганата калия. Определите количество ионов каль¬
ция, содержащихся в 1 л. воды.
Ответ. 0,04 г Са2+ в 1 л.
Работа 35
Умягчение воды методом ионного обмена или изветково-
содовым методом 1
Умягчение воды производится с целью предотвра¬
щения образования в паровых котлах и теплообменпых
аппаратах накипи.
Умягчение воды может быть достигнуто:
1. Осаждением иона кальция (обычно совместно
с ионом магния) и последующим удалением осадка.
2. Удалением иона кальция (и магния) методом ион¬
ного обмена.
Метод ионного обмена основан на способности иони¬
тов поглощать из растворов одни ионы и отдавать вза¬
мен другие. Иониты, способные обменивать находящиеся
в растворе катионы, называются катионитами. К катио¬
нитам относятся: алюмосиликаты, сульфированные угли,
синтетические смолы. Характерной особенностью катио¬
нитов является наличие в них большого числа кислотных
групп: силикатных, карбоксильных и сульфогрупп. Эти
кислотные группы содержат ионы водорода, которые
подвижны и могут быть заменены па различные ка¬
тионы.
Иониты, обменивающие анионы с находившимися в
растворе ионами солей и кислот, называются анионита¬
ми. Аниониты представляют собой аминосмолы, содер¬
жащие амино(—NH2) и имино(—NH) группы. Способ¬
ность этих групп образовывать соли с различными анио¬
нами и используется для анионного обмена.
Для умягчения воды применяются обычно катиониты
в водородной (Н-катиониты) и натриевой (Na-катионн-
ты) форме. Если обозначить весь катионит, кроме под-
движного иона (например, натрия), буквой R, то реакции
ионного обмена в жесткой воде (содержащей ноны каль- 11 Умягчение воды делается одним из методов по указанию пре¬
подавателя.
192
ция и магния) могут быть выражены следующими урав¬
нениями:
Na2R + (Са, Mg) (HC03)2 ^ (Са, Mg) R + 2NaHC03;
Na2R + (Са, Mg)S04 ^ (Ca, Mg)R + Na2S04;
Na2R + (Ca, Mg)Cl2 ^ (Ca, Mg)R + 2NaCl.
Количество катионов кальция и магния, которое мо¬
жет быть поглощено определенным количеством катио¬
нита, называется емкостью поглощения Е и выражает¬
ся в грамм-эквивалентах на 1 мг ионита. Емкость погло¬
щения некоторых катионитов приводится ниже:
сульфоуголь 1000 г-экв1мъ\
эспатит 1700 г-экв/м*\
алюмосиликаты 850 г-экв/м*.
После длительной работы, т. е. после замены иона
натрия на кальций, ионит регенерируют, т. е. пропускают
через него насыщенный раствор хлорида натрия. При
этом ионы кальция вытесняются ионами натрия.
Другой распространенный способ умягчения воды —
известково-содовый. Для очистки воды по известково-со¬
довому методу используют смесь соды и гашеной из¬
вести. При действии извести соли, дающие карбонат¬
ную жесткость, и растворенная в воде двуокись угле¬
рода переводятся в осадок:
Са(НС03)2 + Са(ОН)2 = 2СаС03 + 2НаО;
Mg(HC03)2 + Са(ОН)2 = MgC03 + СаС03 + 2Н20;
С02 + Са(ОН)2 - СаС03 + Н20.
Одновременно магниевая жесткость заменяется на каль¬
циевую:
MgCl2 + Са(ОН)2 = Mg(OH)a + СаС12.
Сода удаляет соли, обусловливающие наличие не-
карбопатной жесткости:
CaS04 + Na2C03 = CaC03 + Na2S04;
СаС12 + Na2C03 - CaC03 + 2NaCl.
xk7 Н. Г. Ключников
193
Карбонаты выпадают в виде взвеси. После фильтрова¬
ния вода содержит соли, не дающие накипи, поэтому
вполне пригодна для многих промышленных целей.
Цель работы. Умягчить водопроводную воду ка-
тионитовым или известково-содовым методом; опреде¬
лить жесткость воды до и после умягчения. Ознакомить¬
ся с методом приготовления катионита, приготовить ка¬
тионит.
Оборудование и материалы
1. Катионитовая колонка.
2. 0,1 н. раствор трилона Б (18,61 г в 1 л раствора).
3. 0,5-процентный раствор индикаторов кислотного
¥ хрома темно-синего или кислотного хрома
черного.
4. Мерный цилиндр на 250 мл.
5. 6—8-процентный раствор поваренной
6. Аммиачный буферный раствор.
Рис. 55. Ио-
нитовая ко¬
лонка:
1 — стеклян¬
ная трубка;
2 — стеклянная
пластинка с от¬
верстиями или
стеклянная ва¬
та; 3 — ионит.
Проведение работы
В качестве катионитовой колонки при¬
меняют стеклянную трубку диаметром 2—
3 см и длиной 80—90 см.
Для того чтобы катионит не высыпался,
в нижнюю часть трубки впаивают стеклян¬
ную пластинку с отверстиями и конец труб¬
ки оттягивают. Можно также использовать
и обычную трубку, закрыв ее нижнюю часть
пробкой с узкой стеклянной трубкой.
Ионитовую колонку наполняют катио¬
нитом так, чтобы высота слоя составляла
50—60 см (рис. 55).
Приготовляют 6—8-процентный раствор
хлорида натрия (относительная плотность
1,06—1,085) и пропускают 600—700 мл его
через колонку. Затем избыток соли отмыва¬
ют дистиллированной или водопроводной
водой до практически полного исчезновения
реакции на хлор-ион (проба с нитратом се¬
ребра). Обычно на отмывку расходуется
191
около 1000 мл воды. После отмывки катионита
через него пропускают 200 мл воды, подлежащей умяг¬
чению, например водопроводной. Затем в профильтро¬
ванной (умягченной) воде и сырой водопроводной воде
определяют жесткость. Для этого в коническую колбу
емкостью 250 мл отмеряют 100 мл анализируемой воды
и 5 мл буферного раствора и титруют 0,1 н. раствором
трилона Б в присутствии индикатора до перехода виш¬
нево-красной окраски раствора в сине-фиолетовую (см.
выше).
При умягчении воды известково-содовым методом
предварительно определяют карбонатную жесткость во¬
ды, некарбонатную, магниевую и количество двуокиси
углерода. На основании этих данных подсчитывают ко¬
личество извести и соды, необходимых для устранения
жесткости. Например, найдено, что карбонатная жест¬
кость равна 5 мг-экв/л, некарбонатная — 3 мг-экв/л,
магниевая — 2 мг-экв/л и двуокиси углерода содержит¬
ся 11 мг/л. Для умягчения воды необходимо:
1) извести
на устранение карбонатной жесткости 5 мг-экв/л
» » магниевой » 2 мг-экв/л
на нейтрализацию двуокиси углерода 11 .^=0,5мг-экв,л
Всего 7,5 мг-экв/л,
где -~j*2 Фактор пересчета С02 в эквивалентах на литр.
Всего извести нужно:
7,5*37 = 277,5 (мг), или 0,277 а на 1 л воды.
2) Соды
на устранение некарбонатной жесткости 3 мг-экв/л, или
3 • 53= 159 (мг), или 0,159 а на 1 л воды.
После расчета отвешивают с точностью до 0,01 а
известь и соду и растворяют их в 1 л воды, налитой
в двухлитровую колбу. Содержимое колбы взбалтывают
в течение 2—3 мин, дают отстояться и фильтруют через
складчатый фильтр. Первые порции фильтрата отбрасы¬
вают. Отфильтровав 200 мл воды, определяют ее карбо¬
натную жесткость, некарбонатную, магниевую и количе¬
ство двуокиси углерода.
7г7*
195
Оформление результатов работы
Описать методику устранения жесткости (при по¬
мощи катионита или известково-содовым методом) и
провести соответствующие расчеты.
Катиониты продаются в магазинах, но их можно приготовить
в лаборатории. Для приготовления сульфоугля применяются мало¬
зольные, спекающиеся и коксующиеся угли марок ПС и ПСЖ.
Уголь дробят в кусочки размером 1—5 мм и смешивают в фар¬
форовом стакане с 18—20-процентным олеумом. На 1 массовую
часть угля берется 5 массовых частей олеума. При смешении
угля с олеумом температура смеси повышается до 180—200 °С.
Смесь выдерживается в течение 6—12 ч. Затем полученный суль-
фоуголь отфильтровывают через пористую стеклянную пластинку,
отмывают водой от избытка кислоты и просушивают на воздухе
между листами фильтровальной бумаги. Вместо олеума в качестве
исключения можно брать концентрированную серную кислоту. Про¬
цесс образования сульфоугля в этом случае проходит более дли¬
тельное время, в течение 2—3 суток. Для облегчения сульфирования
смесь нужно нагреть до 180—200 °С.
В лабораторных условиях можно использовать и другой ка¬
тионит, состав которого отвечает формуле Na2rH4Al2Si2Oiol, для при¬
готовления его глину высушивают при 110—120 °С, тщательно из¬
мельчают и смешивают с безводной содой. На 26 массовых частей
глины берут 10 массовых частей безводной соды. Смесь тщательно
перетирают в ступке, помещают в тигель и прокаливают при 900 °С
в электрической печи. Полученную пористую массу после охлажде¬
ния измельчают до кусочков размером 1—5 мм и промывают не¬
сколько раз водой, подкисленной уксусной кислотой. После этого го¬
товый катионит высушивают и отсеивают кусочки размером 1—Змм
от пыли.
Вопросы и задачи
1. Для удаления карбонатной жесткости применяют одно из сле¬
дующих веществ: едкий натр, гашеную известь, тринатрийфосфат.
Какое из названных веществ окажется наиболее эффективным, если
взять их одинаковые количества? Ответ иллюстрируйте уравнения¬
ми реакций и соответствующими расчетами.
2. Почему для регенерации катионита его промывают раствором
хлорида натрия, а затем водой? Можно ли применить для регене¬
рации катионита раствор хлорида кальция?
3. Вода содержит 0,12 г/л растворенного бикарбоната кальция.
Сколько нужно прибавить извести к 10 л такой воды, чтобы осадить
бикарбонат в виде карбоната?
Ответ. 5,48 г.
ГЛАВА VII
АНАЛИЗ ПИЩЕВЫХ ПРОДУКТОВ
§ 15. АНАЛИЗ МОЛОКА
Качество молока и его свежесть определяются на
основании его плотности, кислотности, количества жира
и сахара. Иногда определяется также его загрязнен’
ность.
Работа 36
Определение плотности молока
Плотность молока определяется ареометром, назы¬
ваемым лактоденсиметром. На средней и узкой части
лактоденсиметра нанесены деления с цифрами, показы¬
вающими второй и третий знаки плотности молока (гра¬
дусы Кевена). При измерении плотности молока в гра¬
дусах Кевена единица отбрасывается. Например, плот¬
ность молока равна 1,031, следовательно, значение плот¬
ности молока в градусах Кевена равно 31 град.
Определение плотности молока проводят при тем¬
пературе 20 °С. Для ее измерения лактоденсиметр снаб¬
жен термометром.
Поскольку вокруг лактоденсиметра образуется ме¬
ниск, для нахождения истинного значения плотности
молока к найденному его значению прибавляют 0,2.
Если температура молока ниже или выше 20 °С, то в
показания лактоденсиметра вносят поправку. Для этого
на каждый градус выше 20 °С к найденной величине
прибавляют 0,2. Если же температура ниже 20 °С, от
найденной величины на каждый градус вычитают 0,2.
7 H. Г. Ключников
197
Например, показание лактоденсиметра равно 31,0; тем¬
пература молока +15°С. Истинное значение плотности
молока (в градусах Кевена) будет равно:
31,0 + 0,2—(0,2 5) = 30,2.
Плотность молока равна 1,030.
При отсутствии лактоденсиметра для определения
плотности молока можно воспользоваться соответствую¬
щим ареометром и термометром.
Цель работы. Определить плотность молока.
Оборудование и материалы
1. Лакто денсиметр.
2. Молоко (200 мл).
3. Стеклянный цилиндр на 250—300 мл.
Проведение работы
Молоко наливают в чистый и сухой цилиндр (темпе¬
ратура молока должна лежать в пределах от +10 до
+ 20 °С) и погружают в молоко чистый и сухой лакто¬
денсиметр до 30—20-го деления, не допуская прикоснове¬
ния прибора к стенкам цилиндра. Через минуту, после
того как лактоденсиметр остановится, отмечают деле¬
ния шкалы и температуру. После этого проводят рас¬
четы по определению плотности молока в градусах Ке¬
вена с учетом поправок на температуру и мениск.
Оформление результатов работы
Записать методику работы и данные по определению
плотности молока.
Работа 37
Определение кислотности молока
Кислотность молока обусловливается в основном
наличием белков, однозамещенных фосфорнокислых со¬
лей и молочной кислоты, образующейся в результате
расщепления лактозы, возникающей под влиянием дея¬
тельности бактерий молочнокислого брожения. Количе¬
198
ство молочной кислоты резко увеличивается при хране¬
нии молока. Кислотность молока выражается в граду¬
сах Тернера. Градусы Тернера показывают количество
миллиметров децинормалыюй щелочи, идущей на ней¬
трализацию 100 мл молока в присутствии фенолфта¬
леина. Кислотность свежего молока колеблется в пре¬
делах 16—18 °Т. Кислотность несвежего молока — 23 °Т
и выше, разбавленного или имеющего соду — ниже
16° Т.
Цель работы. Определить кислотность молока.
Оборудование и материалы
1. Молоко.
2. Пипетка на 10 мл.
3. Мерный цилиндр на 20 мл.
4. 2-процентный раствор фенолфталеина.
5. 0,1 н. раствор едкого натра.
6. Коническая колба для титрования.
Проведение работы
Для титрования при помощи пипетки отмеряют в
коническую колбу 10 мл молока и добавляют к нему
при помощи мерного цилиндра 20 мл прокипяченной
и охлажденной воды. Затем к раствору добавляют
5—6 капель раствора фенолфталеина и после перемеши¬
вания взбалтыванием проводят титрование.
После титрования слабо-розовая окраска должна со¬
храняться в течение 2 мин. Если же окраска за это вре¬
мя исчезает, то раствор дотитровывают.
Для нахождения кислотности по Тернеру число мил¬
лилитров 0,1 н. щелочи, израсходованного на титрова¬
ние, умножают на 10, поскольку расчет ведется на 100 мл
молока. Если щелочь не строго децинормальная, то по¬
лученный ответ умножают на поправку, учитывающую
концентрацию щелочи.
Оформление результатов работ ы
Записать методику определения кислотности молока
и произвести соответствующие расчеты по результатам
титрования.
7*
199
Работа 38
Определение жирности молока ацидобутирометрическим
методом
Определение жирности молока ацидобутирометриче¬
ским методом проводится путем выделения из молока
жира и измерения его объема. Жир в молоке находит¬
ся в виде маленьких жировых капель, окруженных тон¬
ким слоем адсорбированного белка, в частности казеи¬
ната кальция. Для разрушения эмульсии и коагуляции
жира к молоку прибавляют концентрированную серную
кислоту, которая разрушает казеинат кальция, вытес¬
няя из него казеин. Изоамиловый спирт, добавляемый
при анализе в смесь молока и серной кислоты, также
способствует выделению жира. Частично изоамиловый
спирт растворяет жир и одновременно дает с серной
кислотой эфир, растворимый в серной кислоте, при этом
жир, растворенный в спирте, снова выделяется. Жир
отделяют центрифугированием.
Цель работы. Определить жирность молока аци¬
добутирометрическим методом.
Оборудование и материалы
1. Серная кислота (d—1,81—1,82).
2. Амиловый спирт.
3. Молоко.
4. Жирометр (бутирометр).
5. Центрифуга.
6. Пипетка на 11 мл, 10 мл, 1 мл.
Проведение работы
В жирометр, находящийся в штативе, наливают
10 мл серной кислоты, затем 11 мл молока и I мл изо-
амилового спирта. Если же сначала налить молоко, а
затем серную кислоту, то в шейке жирометра может
образоваться пробка свернувшихся белков, которая в
дальнейшем будет мешать работе.
Молоко в жирометр нужно наливать из пипетки мед¬
ленно, чтобы оно по возможности не смешивалось с сер¬
ной кислотой и находилось вверху. Затем в жирометр
200
наливают изоамиловый спирт. Точность определения
жирности молока в значительной степени определяется
точностью, с которой был отмерен объем молока. При
наполнении жирометра следует следить, чтобы жидкость
не попадала на его шейку, так как при дальнейшем на¬
гревании пробка может выскочить из шейки жирометра.
Наполненный жирометр закрывают пробкой и энер¬
гично взбалтывают до образования однородной массы.
Затем его нагревают в течение 5 мин на водяной бане
при 60—66 °С, вытирают наружную поверхность поло¬
тенцем и помещают в центрифугу. Для равновесия в
центрифугу вставляют другой жирометр, наполненный
водой. Затем смесь подвергают в течение 5 мин центри¬
фугированию со скоростью 800—1000 об/мин.
После центрифугирования жирометр снова помеща¬
ют в баню вниз пробкой. Столбик жира в жирометре
должен быть ниже уровня воды в водяной бане. Че¬
рез 5 мин жирометр вынимают из бани и быстро отсчи¬
тывают размер столбика жира от верхней его границы
до нижней. Жирометр при отсчете нужно держать проб¬
кой вниз на уровне глаз а. Если нижняя граница столби¬
ка жира не совпадает с каким-либо делением шкалы,
то нужно на пробку, закрывающую жирометр, надавить
пальцем, чтобы столбик жира передвинулся до ближай¬
шего деления. Каждое деление шкалы соответствует
0,1% жира.
При неполном выделении жира его отсчет вести труд¬
но. В этом случае жирометр энергично встряхивают,
помещают в баню, центрифугируют, снова помещают в
баню и определяют процент жира по высоте столбика.
Оформление результатов работы
Записать методику определения жира и отметить
жирность молока.
Работа 39
Выделение из молока казеина и молочного сахара
Белок молока — казеин — выделяют кислотой, кото¬
рая вызывает его свертывание. После отделения белка
в фильтрате остаются различные растворимые вещест¬
201
ва, в том числе соли и углеводы, главнейшим из кото¬
рых является лактоза (молочный сахар).
Цель работы. Выделить из молока казеин и
лактозу.
Оборудование и материалы
1. Молоко (20 мл).
2. Пробирки, фарфоровая чашка или тигель на 15—
20 мл.
3. 20—30-процентный раствор уксусной кислоты.
4. Воронка с фильтром.
5. 10—15-процентный раствор аммиака.
6. 0,5—1-процентный раствор нитрата серебра.
7. Концентрированная кислота.
Проведение работы
Половину пробирки заполняют молоком, к которому
добавляют 3—4 капли раствора уксусной кислоты. Мо¬
локо нагревают примерно до 90 °С. При этом наблю¬
дается створаживание молока. Когда закончится ство¬
раживание, раствор профильтровывают через воронку с
фильтром с красной лентой или через марлю, сложен¬
ную вдвое. На фильтре остается творожистая масса, со¬
стоящая из казеина и жира.
Для того чтобы убедиться в наличии белков, ОД-
ОД г казеина помещают в пробирку и прибавляют к
нему 0,5 мл концентрированной азотной кислоты. Появ¬
ление желтого окрашивания указывает на наличие бел¬
ков.
Лактозу выделяют из фильтрата. Если раствор мут¬
ный, то его нужно хорошо прокипятить и отфильтро¬
вать.
Для обнаружения восстановительных свойств лакто¬
зы (С12Н22О11) 1—2 мл фильтрата испытывают в про¬
бирке с аммиачным раствором нитрата серебра — по¬
явление серебряного зеркала указывает на наличие аль¬
дегидной группы.
Для выделения лактозы оставшуюся часть фильтра¬
та переливают в фарфоровую чашку или небольшой
тигель и осторожно выпаривают до появления кристал¬
лов молочного сахара (вместе с минеральными солями).
Затем чашку ставят в холодное место.
202
Оформление результатов работы
Записать методику выделения казеина и молочного
сахара из молока.
§ 16. АНАЛИЗ ХЛЕБА И ДРУГИХ ПИЩЕВЫХ ПРОДУКТОВ
Работа 40
Определение кислотности хлеба
Каждый сорт хлеба имеет определенную кислотность,
и всякие отклонения от нормы указывают на неправиль¬
ный способ его изготовления или хранения. Кислотность
хлеба выражают в условных градусах, показывающих
количество миллилитров 1 н. раствора щелочи, израсхо¬
дованного на титрование водной вытяжки, полученной
из 100 г хлеба.
Цель работы. Определить кислотность хлеба.
Оборудование и материалы
1. Хлеб (10—15 г).
2. 0,1 н. раствор едкого натра.
3. Стакан или коническая колба па 100 мл.
4. Раствор фенолфталеина.
Проведение работы
Отвешивают 10 г хлебного мякиша и помещают его
в стакан или в коническую колбу, куда добавляют
25 мл дистиллированной воды. При помощи стеклянной
палочки с резиновым наконечником хлеб тщательно
растирают, а затем взбалтывают в течение 3—4 мин
для экстрагирования органических кислот. В колбу до¬
бавляют 9—10 капель раствора фенолфталеина и тит¬
руют 0,1 н. раствором щелочи до появления розовой
окраски, не исчезающей в течение 2—4 мин.
Поскольку титрование велось 0,1 н. раствором ще¬
лочи, а расчет необходимо вести на 1 н. раствор, для
определения кислотности хлеба в условных градусах
полученное число нужно разделить на 10 и, поскольку
бралось 10 г, а не 100, то необходимо умножить на 10.
203
Оформление результатов работы
Записать методику проведения работы и результа¬
ты анализа.
Работа 41
Определение кислотности муки
Кислотность муки является одним из показателей ее
качества, и она зависит от сорта муки и условий ее хра¬
нения.
Кислотность муки выражают в условных градусах,
которые показывают количество миллилитров 1 н. ра¬
створа щелочи, взятой на нейтрализацию органических
кислот водной вытяжки, полученной из 100 г муки.
Цель работы. Определить кислотность муки.
Оборудование и материалы
1. Мука (7—10 г).
2. 0,1 н. раствор щелочи.
3. Раствор фенолфталеина.
4. Коническая колба на 100 мл.
Проведение работы
Отвешивают 5 г муки, пересыпают ее в коническую
колбу, в которую наливают 40 мл воды и добавляют
5—6 капель раствора фенолфталеина. Взвесь титруют
0,1 н. раствором щелочи до появления розового окраши¬
вания, не исчезающего в течение 2 мин.
При вычислении кислотности муки в условных гра¬
дусах вычисляют, какое количество 1 н. щелочи пойдет
на титрование кислот, содержащихся в 100 г муки.
Оформление результатов работы
Записать методику проведения работы и полученные
результаты.
ГЛАВА VIII
СИНТЕЗ НЕКОТОРЫХ ОРГАНИЧЕСКИХ ВЕЩЕСТВ
§ 17. СИНТЕЗ И КАЧЕСТВЕННЫЙ АНАЛИЗ
ВЫСОКОМОЛЕКУЛЯРНЫХ ВЕЩЕСТВ
Как известно, высокомолекулярными соединениями
или полимерами называются соединения, имеющие боль¬
шие молекулярные массы, доходящие до сотен тысяч
углеродных единиц, и имеющие в составе их макромоле¬
кул большое количество элементарных звеньев, состоя¬
щих большей частью из одних и тех же групп атомов.
Получение высокомолекулярных веществ, т. е. сое¬
динение друг с другом большого количества звеньев в
макромолекулы, осуществляется реакциями двух типов:
1. Реакции поликонденсации, заключающиеся b кон¬
денсации большого количества молекул с выделением
в качестве побочного продукта какого-либо веществу с
небольшой молекулярной массой, например воды.
2. Реакции полимеризации, заключающиеся в сое¬
динении молекул низкомолекулярного вещества (моно¬
мера) с образованием макромолекул полимера, причем
каких-либо других веществ при этих реакциях не обра¬
зуется.
Чаще всего используется цепная полимеризация сое¬
динений, содержащих в молекулах мономера одну
двойную или две сопряженные двойные связи. Разно¬
видностью полимеризации является сополимеризация,
заключающаяся в совместной полимеризации двух или
большего числа различных мономеров с образованием
сополимера:
А + Б — А — А — Б — А — Б — Б — А —
205
Большое практическое значение за последние годы
получили синтетические смолы и пластмассы, получае¬
мые на их основе.
Составными частями смесей, используемых для фор¬
мирования пластмасс, являются:
1. Связующее вещество (основная часть пластмассы),
связывающее составные части в единое целое.
2. Пластификаторы — вещества, повышающие плас¬
тичность смол и облегчающие формование из них из¬
делий.
3. Наполнители — инертные вещества, придающие
пластмассам те или иные механические свойства и
уменьшающие содержание связующего вещества.
4. Растворители — вещества, снижающие вязкость
связующего вещества и испаряющиеся при формовке из¬
делий.
5. Стабилизаторы — вещества, обеспечивающие хи¬
мическую стабильность пластмассы, т. е. препятствую¬
щие протеканию реакций окисления, разложения и т. д.
В тех случаях, когда для изготовления изделий или
полупродуктов наполнитель в смолу не вводится, поня¬
тия «смола» и «пластмасса» совпадают.
Синтетические смолы на основании их свойств и ус¬
ловий переработки делятся на две группы:
1. Термопластичные смолы, которые при нагревании
переходят в эластичное, а затем в вязко-текучее состоя¬
ние; после охлаждения они снова затвердевают. Нагре¬
вание и охлаждение этих смол не вызывают изменений
в их структуре, так как нагревание и охлаждение не со¬
провождаются какими-либо реакциями.
2. Термореактивные смолы при нагревании вследст¬
вие протекающих химических реакций подвергаются
сильным изменениям. В частности, они перестают раз¬
мягчаться при повторном нагревании.
Работа 42
Получение фенолформальдегидных смол
Фенолформальдегидные смолы получаются при кон¬
денсации фенола и формальдегида. В зависимости от
соотношения фенола и формальдегида получаются так
называемые новолачные и резольные смолы.
206
Новолачные смолы образуются при небольшом из¬
бытке фенола (на 7 моль фенола берется 6 моль фор¬
мальдегида). В качестве катализатора применяется со¬
ляная, а иногда и щавелевая кислота. Строение поли¬
мера можно схематически изобразить формулой:
ОН
I
\/
он
-/\-СН2-
\/
он
п \/
Новолачные смолы термопластичны. Общее количе¬
ство бензольных колец в макромолекуле составляет
5—10.
Резольные смолы образуются при небольшом из¬
бытке формальдегида (на 6 моль фенола берется
7 моль формальдегида). В качестве катализатора при¬
меняются основания, например 20—25-процентный рас¬
твор аммиака, Резольные смолы отличаются от новолач-
ных смол наличием групп СН2ОН:
ОН
I
|/чрсн2
\/
он
/\-СН2—
\/
I
СН2ОН
он
I
—'СН2ОН
\/
Эти группы — СН2ОН при нагревании участвуют в
конденсации и образовании новых мостиков из —СН2
групп. Поскольку отдельные линейные молекулы «сши¬
ваются» и из молекул образуется пространственная
трехмерная структура, резольные смолы являются тер¬
мореактивными.
Цель работы. Получить новолачную и резоль-
ную смолы и изучить их свойства.
Оборудование и материалы
1. Фенол.
2. Формальдегид (40-процентный раствор).
3. Соляная кислота (d— 1,19).
207
4. Уротропин (гексаметилентетрамин).
5. Этиловый спирт или ацетон.
6. Круглодонная колба.
7. Водяная баня.
Проведение работы
В круглодонную колбу емкостью 50 мл помещают
7,5 г фенола и 5 мл 40-процентного раствора формаль¬
дегида. Смесь взбалтывают до растворения фенола.
После этого добавляют 2—3 капли концентрированной
соляной кислоты, колбу закрывают пробкой с вставлен¬
ным в нее обратным холодильником и нагревают на
водяной бане при 90—100 °С до разделения водного и
смоляного слоев. Смесь выливают в предварительно
взвешенную фарфоровую чашку. После отстаивания
верхний водный слой сливают и смолу промывают теп¬
лой водой до нейтральной реакции по метилоранжу.
Для высушивания смолы чашку помещают в сушиль¬
ный шкаф и нагревают до 180—200 °С. Для определе¬
ния выхода продукта чашку со смолой взвешивают.
Для установления линейного строения полимера 1 г
полученного вещества измельчают, помещают в про¬
бирку, приливают 10 мл этилового спирта или ацетона
и нагревают. Смесь взбалтывают через каждые 5—
10 мин в течение 1—1,5 ч. Смолы, имеющие линейную
структуру, растворяются.
Для перевода новолачной смолы в полимер, имею¬
щий трехмерное строение, к измельченной смоле добав¬
ляют 10—15% (от веса смолы) уротропина и смесь на¬
гревают в пробирке до 160°С в масляной бане до ее
отвердения. После охлаждения пробирку разбивают,
полимер измельчают и проверяют его растворимость в
спирте или ацетоне.
Для получения резольной смолы в круглодонную
колбу на 50 мл помещают 5 г фенола, 10 мл 40-процент -
ного раствора формальдегида и 0,5 мл 40-процентного
раствора едкого натра. Колбу закрывают пробиркой с
вставленным в нее воздушным холодильником. После
встряхивания смесь нагревают на асбестированной сет¬
ке, медленно повышая температуру. Если смесь сильно
закипает, то нагревание на некоторое время прекраща¬
ют, а затем нагревают до кипения.
208
Нагревание продолжают примерно 1 ч до образова¬
ния вязкой массы, окрашенной в красный цвет. Горя¬
чую вязкую массу выливают во взвешенную фарфоро¬
вую чашку или тигель. Для окончания реакции чашку
помещают в сушильный шкаф и выдерживают при
100—120 °С до отвердения полимера. После охлаждения
взвешиванием определяют выход полимера. Для уста¬
новления наличия пространственной структуры полиме¬
ра несколько небольших его кусочков помещают в про¬
бирку и изучают растворимость в спирте или ацетоне.
Работа 43
Определение азота, серы, хлора, фтора в полимерах
При качественном анализе полимеров на указанные
элементы полимер сплавляют с калием или натрием.
При этом происходит глубокое разложение полимера:
водород частично выделяется в свободном виде, а
частично дает гидроокись калия; кислород дает окись
калия; азот, сера, хлор и фтор дают соответственно
цианистый калий, сульфид, хлорид и фторид калия. Эти
вещества открываются обычными химическими мето¬
дами.
Цель работы. Определить в полимере наличие
азота, серы, хлора, фтора.
Оборудование и материалы
1. Набор полимеров в виде небольших кусочков.
2. Пробирки с пробками, имеющими отверстия для
выхода газов.
3. Азотная кислота (1 10).
4. Соляная кислота (1 : 10).
5. Металлический калий или натрий.
6. 5-процентные растворы сульфатов двух- и трех¬
валентного железа.
7. 5-процентный раствор нитрата серебра.
8. 5-процентный раствор хлорида кальция.
9. 5-процентный раствор ацетата свинца.
Открытие азота. В сухую пробирку помещают кусок
калия размером с полгорошины и такое же количество
209
полимера. Пробирку закрывают пробкой с небольшим
отверстием и сильно нагревают смесь до ее полного
сплавления. При этом обычно получается черная спек¬
шаяся масса. Затем еще горячую пробирку опускают
(тяга!) в небольшую фарфоровую чашку с 10—15 мл
воды. При проведении этой операции глаза нужно защи¬
щать очками, так как иногда протекает бурная реакция
между водой и остатками непрореагировавшего калия.
При наличии в полимере азота образуется цианид
калия. Для его открытия раствор отфильтровывают,
1/4 часть его переливают в пробирку и приливают неболь¬
шое количество растворов солей двух- и трехвалентного
железа, например FeS04 и FeCl3. Затем раствор нагре¬
вают до кипения, охлаждают и подкисляют 10—15-про¬
центным раствором соляной кислоты. При наличии азота
раствор окрашивается в синий цвет за счет образования
берлинской лазури, т. е. гексацианоферрата трехвалент¬
ного железа:
FeS04 + KCN = Fe(CN)2 + K2S04;
Fe(CN)2 + 4KCN = K4[Fe(CN)6];
3K4[Fe(CN)6] + 4FeCl3 = Fe4[Fe(CN)6]3 + 12KCI.
При стоянии раствора на дне пробирки образуется
синий осадок берлинской лазури. Эту реакцию дают
азотсодержащие полимеры: карбамидные, полиамидные,
аминоформальдегидные, полиуретаны и т. д.
Открытие серы. 1/4 часть раствора, оставшуюся от
анализа на азот, переливают в пробирку и приливают к
нему раствор уксуснокислого свинца. При наличии в по¬
лимере серы образуется черный осадок сульфида свинца:
K2s + Pb(CH3COO)2 = PbS I + сн3соок.
От действия перекиси водорода осадок становится
белым:
PbS + 4Н202 = PbS04 + 4НаО.
Сульфид калия образуется за счет отнятия серы от
полимера при его сплавлении с калием.
210
Реакцию на серу дают серусодержащие полимеры,
например тиокарбамидные, сульфофенольные и т. д.
Открытие хлора. часть раствора, оставшуюся по¬
сле открытия азота, подкисляют 5—10-процентным раст¬
вором азотной кислоты и прибавляют несколько капель
5—10-процентного раствора азотнокислого серебра. Об¬
разование белого осадка хлорида серебра указывает на
наличие хлора. Осадок хлорида, в отличие от осадков
бромида и иодида серебра, растворим в растворе ам¬
миака.
Открытие фтора. lU часть раствора, оставшуюся от
открытия азота, переливают в пробирку, подкисляют
разбавленной соляной кислотой и прибавляют несколь¬
ко капель хлорида кальция. При наличии фтора посте¬
пенно выпадает белый хлопьевидный осадок фторида
кальция. Иногда осадок становится заметным только
через 1—1,5 ч.
Оформление результатов работы
Записать методику и результаты анализа.
Работа 44
Определение кремния и фосфора в полимерах
Для определения наличия в полимерах кремния и
фосфора указанные элементы переводят путем глубо¬
кого химического разложения полимера в силикаты и
фосфаты. С этой целью полимер сплавляют с содой и
перекисью натрия. Для открытия силикатов и фосфа¬
тов используют обычные химические методы.
Цель работы. Провести качественное определе¬
ние наличия в полимере кремния и фосфора. Ознако¬
миться с методикой работы.
Оборудование и материалы
1. Набор полимеров.
2. Безводная сода.
3. Перекись натрия.
4. Никелевый или платиновый тигель.
211
5. Соляная кислота (1 1).
6. Фарфоровая чашка.
7. Воронки для фильтрования, фильтры.
8. 0,1-процентный раствор метиленового голубого,
растворенного в 10-процентном растворе уксусной кис¬
лоты.
9. Раствор молибдата аммония.
Открытие кремния. 0,05—0,12 г полимера смешива¬
ют в платиновом тигле с 6—10-кратным количеством
безводной соды и нагревают в муфельной печи при
850—900° С до получения однородного расплава. Сплав¬
ление смеси соды с перекисью натрия можно вести в
никелевом тигле. Затем тигель переносят в фарфоровую
чашку с водой, кипятят до отделения сплава от тигля.
Вынув тигель из фарфоровой чашки и обмыв его водой,
добавляют к раствору соляную кислоту (1 2) до пре¬
кращения выделения двуокиси углерода. Затем раствор
выпаривают досуха, чтобы подвергнуть частичному обез¬
воживанию кремниевую кислоту, прибавляют к осадку
10—15 мл разбавленной (1 15) соляной кислоты, раст¬
вор профильтровывают и фильтр промывают дистилли¬
рованной водой. Кремниевая кислота остается на филь¬
тре. Для ее обнаружения на фильтр наливают 0,1-про¬
центный раствор метиленового голубого, а затем фильтр
промывают водой. В присутствии кремниевой кислоты
появляется интенсивно-синяя окраска за счет адсорбции
красителя силикогелем. При отсутствии метиленового
голубого для обнаружения кремния можно применить
малахитовый зеленый или софранин.
Открытие фосфора. 0,05 г полимера смешивают в
никелевом тигле с 2 а безводной соды и 2 г перекиси
натрия. Тигель нагревают на пламени горелки или в
муфельной печи до расплавления массы. Смесь выдер¬
живают в расплавленном состоянии еще 10 мин, затем
охлаждают и содержимое растворяют в воде. Раствор
фильтруют, фильтрат подкисляют азотной кислотой и
добавляют раствор молибдата аммония. Осадок иногда
выделяется спустя 10—15 мин. Выделение из раствора
желтого осадка указывает на наличие фосфора.
Оформление результатов работы
Записать методику и результаты анализа.
212
Работа 45
Открытие в полимерах функциональных групп
Открытие в полимерах функциональных групп при
наличии результатов качественного анализа во многих
случаях позволяет установить примерный состав и строе¬
ние полимера. Открытие функциональных групп обыч¬
но проводится после декструктивного разложения поли¬
мера путем его нагревания с последующим исследова¬
нием продуктов разложения. Иногда это разложение
осуществляется путем воздействия химических реаген¬
тов.
Цель работы. Открыть функциональные группы,
входящие в состав полимера; учитывая результаты эле¬
ментарного анализа (см. выше), дать характеристику
химического строения полимера.
Оборудование и материалы
.1. Набор полимеров в виде небольших кусочков.
2. Пробирки с газоотводными трубками. Пробирки,
часовые стекла.
3. 10-процентный раствор нитрата серебра, едкий
натр (1 10), раствор аммиака.
4. 30-процентный раствор анилина в уксусной кис¬
лоте.
5. Концентрированная серная кислота.
6. Реактив Миллона.
7. Фелингова жидкость.
8. 1-процентный раствор пирогаллола.
9. 0,5-процентный раствор резорцина.
10. 30-процентный раствор едкого натра.
11. Насыщенный раствор бихромата калия в 10-про¬
центном растворе серной кислоты.
12. Насыщенный раствор хлорной извести.
13. Бромная вода.
14. Насыщенный раствор хлорида железа (III).
15. Раствор хлористоводородного бензолдиазония.
Проведение работы
Для термического разложения полимера 10—12 г
его помещают в пробирку из тугоплавкого стекла, за¬
крывают пробкой с изогнутой газоотводной трубкой, ко¬
213
нец которой помещают в другую пробирку (приемник),
содержащую 5 мл воды. Во избежание засасывания
воды газоотводную трубку держат у поверхности или
погружают ее в воду не более чем на 2—3 мм. Пробир¬
ку с полимером сильно нагревают до полного разло¬
жения вещества, а уходящие пары улавливают в прием¬
нике. Водный раствор продуктов разложения может
содержать анилин, фенол, альдегиды, фурфурол, уксус¬
ную кислоту, стирол и метилметакрилат.
Для открытия анилина к xh .части полученного раст¬
вора прибавляют 1 мл бихромата калия в 10-процентном
растворе серной кислоты. Появление синей, а затем
черной окраски указывает на наличие анилина. В при¬
сутствии водного раствора хлорной извести раствор окра¬
шивается при наличии анилина в фиолетовый цвет.
Открытие фенола. Приготовляют водный раствор
продуктов термического распада (см. выше) и в нем
открывают обычными реакциями фенол: 1) при наличии
фенола раствор при добавлении к нему раствора хлори¬
да железа (III) окрашивается в фиолетовый цвет; 2) при
добавлении избытка бромной воды выпадает объеми¬
стый осадок трибромфенола; 3) при добавлении не¬
скольких капель щелочи и хлористоводородного бензол-
диазония выпадает желтый осадок оксиазобензола.
Открытие альдегидов. Приготовляют водный раствор
продуктов термического распада, к нему прибавляют
аммиачный раствор нитрата серебра и раствор подогре¬
вают до 70—80° С. При наличии альдегидов на стенке
пробирки образуется серебряное зеркало:
2 [Ag(NH3)2]OH + ЗН20 + RCHO -
= RCOOH + 2Ag + 4NH4OH.
Наличие альдегидов можно также открыть с по¬
мощью фелинговой жидкости. После приливания к вод¬
ному раствору продуктов термического разложения по¬
лимера и нагревания выпадает красный осадок закиси
меди:
RCHO + 2Cu(OH)2 = RCOOH + Си20 + 2Н20.
Приготовление фелинговой жидкости. 14 г едкого
натра и 35 г невыветрившейся сегнетовой соли раство¬
ряют в 50—60 мл воды и раствор доводят до объема
100 мл. Растворяют 6,93 г сульфата меди в 50—60 мл
214
воды и раствор доводят также до объема 100 мл. Затем
эти растворы смешивают.
В тех случаях, когда необходимо идентифицировать
альдегиды, использованные для получения полимера,
проводят следующие реакции.
Для открытия формальдегида к 1 мл 1-процентного
раствора пирогаллола прибавляют 2—3 капли испытуе¬
мого раствора и 2 мл концентрированной серной кисло¬
ты. Появление белого осадка бис-(триоксифенил)-мета¬
на указывает на наличие формальдегида:
СвНа(ОН)3
СНаО + 2С6Н3(ОН)3 = СН / + нао.
\СвНа(ОН)з
Бис-(триоксифенил)-метан окисляется и дает окра¬
шенные продукты красного цвета.
Весьма чувствительной является реакция формаль¬
дегида с резорцином. В 1 мл водного раствора продук¬
тов разложения полимера прибавляют 1 каплю 0,5-про¬
центного раствора резорцина и в эту смесь, наклонив
немного пробирку, по стенке приливают 2—3 мл кон¬
центрированной серной кислоты, чтобы образовалось два
слоя. При наличии формальдегида на границе между
нижним, сернокислотным, слоем и верхним, водным, по¬
является красно-фиолетовое кольцо, а затем — белый
осадок, постепенно изменяющий свой цвет в красно-фио¬
летовый.
Если раствор содержит ацетальдегид, подобная же
реакция с резорцином дает ярко-зеленое кольцо. Для
открытия ацетальдегида можно также провести его
конденсацию в присутствии едкого натра. Для этого
к прозрачному 20-процентному раствору едкого натра
прибавляют водный раствор продуктов разложения по¬
лимера и смесь нагревают. При наличии ацетальдегида
раствор сначала желтеет, а затем становится темным за
счет образования продуктов конденсации ацетальдегида.
Бензальдегид можно легко открыть по своеобразно¬
му запаху горького миндаля. Можно также несколько
капель раствора поместить на часовое стекло и оста¬
вить на 10—15 мин. За счет окисления бензальдегида
на поверхности раствора постепенно образуются белые
кристаллы бензойной кислоты, которые плохо раствори¬
мы в воде. При добавлении нескольких капель метило¬
215
вого спирта кристаллы должны перейти в раствор. Хло¬
рид железа (III) дает желтый осадок гексабензоата
железа [Ре(СбН5СОО)б](ОН)3. В ацетоне осадок раство¬
ряется.
Для открытия фурфурола приготовляют водный ра¬
створ термического разложения полимера и прибавляют
к нему несколько капель 30-процентного раствора ани¬
лина в уксусной кислоте. При наличии фурфурола на¬
блюдается красное окрашивание.
Открытие уксусной кислоты. Приготовляют водный
раствор продуктов термического разложения полимера.
Присутствие уксусной кислоты открывается по запаху.
Можно также к отгону прибавить двойное количество
этилового спирта и несколько капель концентрирован¬
ной серной кислоты. При нагревании в присутствии ук¬
сусной кислоты появляется запах уксусноэтилового
ткрытие стирола и метилметакрилата. 3—4 г по¬
лимера нагревают на пламени в пробирке с газоотвод¬
ной трубкой и продукты разложения собирают в прием¬
нике-пробирке, погруженном в холодную воду или охла¬
ждающую смесь.
Стирол и метилметакрилат открывают по запаху, а
также по их температурам кипения. С этой целью про¬
бирку с жидким продуктом разложения закрывают проб¬
кой с вставленным термометром. Ртутный шарик тер¬
мометра должен находиться на расстоянии 5 мм от по¬
верхности жидкости. В пробке нужно сделать отверстие
для выхода паров.
Для определения температуры кипения пробирку
нагревают до кипения жидкости и наблюдают за пока¬
заниями термометра. Температура кипения полистирола
146 °С, метилметакрилата— 100,8 °С.
Открытие нитросоединений. На кусочек полимера на¬
носят раствор дифениламина в серной кислоте. Появле¬
ние синего окрашивания указывает на наличие нитро¬
соединений.
Вопросы и задачи
1. При открытии в полимере азота обычно используют лабора¬
торный раствор соли двухвалентного железа. Почему в данном слу¬
чае допускается отступление от методики?
а.
216
2. При определении в полимере серы к приготовленному раство¬
ру прилили перекись водорода, а затем нитрат свинца. При этом
выпал белый осадок. Напишите уравнение реакции.
3. К раствору при определении хлора прилили нитрат серебра,
затем аммиак и раствор профильтровали. К фильтрату прилили
азотную кислоту, и при этом выпэл белый осадок. Напишите урав¬
нение реакции.
4. Почему при определении в полимерах фосфора полимер
сплавляют не с калием, а с содой? Какое соединение образует фос¬
фор, входящий в состав полимера, при сплавлении с калием?
§ 18. ФИЗИЧЕСКИЕ МЕТОДЫ ИЗУЧЕНИЯ ПОЛИМЕРОВ
Практическое применение полимеров определяется не
только их химическими свойствами, но также и физи¬
ческими. К числу физических методов изучения поли¬
меров относят рентгенографический, определение раст¬
воримости, определение температурных характеристик
(температуры текучести, стеклования, размягчения, кап-
лепадения, плавления) и других различных физических
свойств.
Работа 46
Определение температуры размягчения
Существует несколько методов определения темпе¬
ратуры размягчения. Для этой цели служат специаль¬
ные приборы, позволяющие проводить испытания об¬
разцов под постоянной нагрузкой. Температура размяг¬
чения зависит от методики работы.
Наиболее простым методом является определение
температуры размягчения на металлической плите.
Цель работы. Определить температуру размяг¬
чения полимера.
Оборудование и материалы
1. Металлическая плита с термометром.
2. Электрическая плитка.
3. Набор измельченных полимеров.
4. Шпатель.
217
Проведение работы
1—2 г измельченного полимера помещают на метал¬
лическую плиту, обогреваемую снизу горелкой или элек¬
трической плиткой, включенной через реостат. Металли¬
ческая плита имеет сбоку отверстие, в которое вставляет¬
ся термометр. Определение проводится при постепенном
нагревании плиты, а следовательно, и полимера, который
необходимо все время перемешивать стеклянной палоч¬
кой или шпателем. За температуру размягчения прини¬
мают температуру, при которой отдельные крупинки
полимера начинают слипаться в комки.
Оформление результатов работы
Записать методику работы и полученные цифровые
данные.
Работа 47
Определение температуры каплепадения
Температурой каплепадения называется температура,
при которой капля полимера отделяется под действием
силы тяжести от полимера.
Цель работы. Определить в приборе Убеллоде
температуру каплепадения полимера.
Оборудование и материалы
1. Прибор Убеллоде.
2. Колба с глицерином.
3. Набор полимеров.
Проведение работы
Прибор Убеллоде представляет собой термометр, на
ртутном шарике которого при помощи металлической
гильзы укреплена стеклянная чашечка с отверстием
в дне.
Термометр находится вместе с чашечкой в стеклян¬
ной пробирке, которая в свою очередь помещена в кол¬
бу с глицерином. Перед работой стеклянную чашечку
вынимают из гильзы, ставят на стекло, наливают в нее
218
расплавленный полимер и опускают шарик термометра
в расплав. Чашечку укрепляют в обойме гильзы и со¬
бирают прибор. Затем колбу с глицерином нагревают,
повышая температуру со скоростью 1 °С в 1 мин.
За температуру каплепадения принимается темпера¬
тура, при которой капля расплавленного полимера про¬
ходит через дно чашечки и падает на дно пробирки.
Оформление результатов работы
Описать методику работы и полученные цифровые
данные.
Р а б о т а 48
Определение температуры плавления полимера
Цель работы. Определить температуру плавле¬
ния полимера в капилляре.
Оборудование и материалы
1. Капилляры диаметром около 1 мм.
2. Набор тонкоизмельченных полимеров.
3. Термометр, вмонтированный в пробирку.
4. Колба с глицерином.
Проведение работы
Капилляр запаивают с одного конца и, обломив ку¬
сок длиной 5—6 мм, набивают в него порошкообразный
полимер в виде столбика. Высота столбика 3—4 мм.
Капилляр при помощи резинового кольца укрепляют в
непосредственной близости к ртутному шарику термо¬
метра. Затем термометр помещают в пробирку, кото¬
рую в свою очередь укрепляют в колбе, наполненной
глицерином. При нагревании колбы с глицерином, ко¬
торое производится со скоростью 1 °С в 1 мин, происхо¬
дит размягчение полимера, а затем и расплавление.
За температуру плавления принимается такая тем¬
пература, при которой содержимое капилляра перейдет
в жидкость.
Некоторые полимеры при плавлении на воздухе окис¬
ляются. В этом случае открытый конец капилляра перед
определением запаивают.
219
Оформление результатов работы
Описать методику работы и полученные цифровые
данные.
§ 19. СИНТЕЗ КАРБОНОВЫХ КИСЛОТ
Работа 49
Получение уксусной кислоты синтетическим способом
Синтез уксусной кислоты из ацетилена является ин¬
тересным примером получения органического вещества
из неорганических.
Получение уксусной кислоты включает следующие
стадии:
1. Получение ацетилена.
2. Гидратация ацетилена по реакции Кучерова.
3. Окисление уксусного альдегида кислородом воз¬
духа до уксусной кислоты в присутствии ацетата мар¬
ганца.
Окисление уксусного альдегида проходит довольно
медленно, и в промышленности неокисленный альдегид
направляют на повторное окисление. Осуществить окис¬
ление уксусного альдегида в лабораторных условиях по
циркуляционной схеме довольно трудно.
Окисление быстро идет в присутствии перманганата
калия, который в основном действует как окислитель:
2СН3СНО + КМп04 = СН3СООК + СН3СООН + Мп02.
Наряду с уксусной кислотой и двуокисью марганца
образуется также и некоторое количество ацетата мар¬
ганца, ‘который действует как катализатор для реакции
окисления альдегида.
Цель работы. Получить раствор уксусной кис¬
лоты, исходя из карбида кальция.
Оборудование и материалы
1. Колба Вюрца с капельной воронкой.
2. Промывная склянка.
3. Колба с газоотводными трубками и термометром.
4. Пробирка с газоотводной трубкой.
5. Реактивы: хлорид натрия, окись ртути, серная
кислота, перманганат калия, карбид кальция.
220
Проведение работы
Работу проводят в приборе, собранном по схеме, при¬
веденной на рисунке 56. Ацетилен получают, приливая
по каплям к карбиду кальция концентрированный раст¬
вор хлорида натрия. О количестве получаемого ацети-
Рис. 56. Получение уксусной кислоты:
1—колба с карбидом кальция; 2 — промывалка с водой (счетчик пу¬
зырьков); 3 — колба с раствором солей ртути; 4 — колба или пробирка
с раствором перманганата.
лена судят по количеству пузырьков в промывной склян¬
ке. Ацетилен поступает в колбу с насыщенным раствором
сульфата ртути, где и происходит его гидратация и обра¬
зование уксусного альдегида. Для предупреждения гид¬
ролиза сульфата ртути в колбу прибавляют 1—2 мл
концентрированной серной кислоты. В случае отсутствия
готового сульфата ртути его легко можно приготовить
растворением окиси ртути в серной кислоте при нагре¬
вании. Для этого нужно взять около 1 г окиси ртути
и 30 мл серной кислоты (разбавленной 1 4).
221
Водный раствор сульфата ртути нагревают до 70 °С и
пропускают ацетилен. За температурой следят по пока¬
заниям термометра.
Уксусный альдегид, получающийся за счет гидрата¬
ции ацетилена под влиянием катализирующего действия
соли ртути, испаряется и поступает в пробирку с раз¬
бавленным раствором перманганата калия, где происхо¬
дит его окисление до уксусной кислоты.
Ацетилен следует пускать в прибор в небольших ко¬
личествах, около одного пузырька в секунду (в промы-
валке).
Пары альдегида перед поступлением в пробирку с
перманганатом следует смешать с воздухом, который по¬
дается из газометра через тройник. Для наблюдения за
током воздуха между тройником и газометром включа¬
ют промывалку с водой. Воздух пускают со скоростью
один пузырек в секунду.
Опыт продолжают в течение 10 мин до исчезновения
окраски перманганата калия и образования осадка дву¬
окиси марганца. После этого прекращают ток ацетилена
и воздуха и отсоединяют пробирку с раствором уксусной
кислоты.
Для отделения раствора уксусной кислоты от двуоки¬
си марганца раствор профильтровывают в пробирку. На¬
личие уксусной кислоты определяют по реакции с хлори¬
дом железа (III) или по образованию уксусноэтилового
эфира. Для этого к полученному раствору приливают
раствор хлорида железа и нагревают. Появляется окра¬
ска, напоминающая цвет крепкого чая.
Оформление результатов работы
Описать методику получения уксусной кислоты в ла¬
бораторных условиях.
Вопросы и задачи
1. Почему при окислении парафина получается смесь карбоно¬
вых кислот разной молекулярной массы? Какие продукты еще мо¬
гут получаться при окислении парафина?
2. Каким методом в продуктах окисления парафина можно об¬
наружить наличие альдегидов?
3. Напишите уравнение термического разложения пермангана¬
та калия и уравнение окисления какого-либо углеводорода перман¬
ганатом.
ГЛАВА IX
ПРИГОТОВЛЕНИЕ ПОЛУПРОВОДНИКОВЫХ
МАТЕРИАЛОВ И ИЗУЧЕНИЕ ИХ СВОЙСТВ
§ 20. ВЫПРЯМИТЕЛЬНЫЕ СВОЙСТВА ПОЛУПРОВОДНИКОВ
Работа 50
Получение селенида (или сульфида) меди и изучение
его вентильных свойств
Селенид меди представляет собой черное вещество
с фиолетовым оттенком, плавящееся при 680 °С. В кон¬
такте с магнием и некоторыми другими сильными вос¬
становителями при пропускании постоянного тока (маг¬
ний является анодом) возникает слой высокого сопротив¬
ления. При этом протекает реакция:
2CuSe + Mg = MgSe + Cu2Se,
в результате которой слой, примыкающий к магнию, при¬
обретает электронную проводимость \ Селениды одно- и
двухвалентной меди имеют дырочную проводимость.
Вследствие образования электронно-дырочного перехода
после формовки селенид меди, находящийся в контакте
с магнием, становится по отношению к переменному току
выпрямителем.
Подобными же свойствами обладает и сульфид меди.
Цель работы. Получить селенид (или сульфид)
меди и снять его вольт-амперную характеристику. 11 По-видимому, селенид магния содержит незначительный избы¬
ток магния.
223
Оборудование и материалы
1. Медь в виде крупки, порошка.
2. Селен или комовая сера.
3. Источники тока на 15—20 в и 4—5 в и 2—3 а.
4. Амперметры на 5—10 а и миллиамперметр на 150—
200 ма.
5. Вольтметры на 2—3 в и 15—20 в.
6. Низкоомный реостат.
Проведение работы
Для получения селенида меди берут 3—4 г меди, на¬
резанной в виде небольших кусочков (или порошок), до¬
бавляют к ней порошкообразный селен в соответствии
с уравнением реакции:
Си + Se = CuSe.
Затем смесь помещают в пробирку из тугоплавкого
стекла и нагревают на пламени газовой горелки до рас¬
плавления полученного селенида меди. Во время реакции
на бл юд ается в ы де¬
ление теплоты и про¬
бирка может лоп¬
нуть.- Поэтому, если
для реакции исполь¬
зуют порошкообраз¬
ную медь, селен нуж¬
но брать в виде ку¬
сочков. После ох¬
лаждения пробирки
с селенидом меди ее
разбивают, сплав¬
ленный кусок зажи¬
мают в тиски и опи¬
ливают в виде ци¬
линдра. Одну из сторон цилиндра зачищают на листе
наждачной бумаги.
Для изучения выпрямительных свойств и снятия
вольт-амперной характеристики селенид меди зажимают
между медной и магниевой пластинками в приспособле¬
ние, устройство которого видно на рисунке 57. Для фор¬
мовки магниевую пластинку присоединяют к положи-
Рис. 57. Приспособление для включения
селенида меди в цепь:
/ — селенид меди; 2 — магниевая пластинка;
3 — медная пластинка; 4 — винт.
224
Рис. 58. Электрическая схема для снятия обрат¬
ной вольт-амперной характеристики.
Рис. 59. Электрическая схема для снятия
прямой вольт-амперной характеристики.
а/смг
5
4
3
2
15 10 5 1
I l l I I I I I I I |
0.1 03 05 03 0.9 0.11
5
\
10
15
-
20
та/см2
Рис. 60. График для нанесения еольт-амперной
характеристики,
тельному полюсу, а медную — к отрицательному полюсу
источника постоянного тока и через селенид начинают
пропускать ток небольшого напряжения, около 1 в, по¬
степенно увеличивая в течение 30—60 мин силу тока.
При формовании вентиля происходит искрение в месте
контакта с магниевой пластинкой, во время которого
сила тока возрастает. Формование вентиля заканчивают,
когда напряжение достигнет 12—13 в.
Для снятия обратной вольт-амперной характеристики
собирают установку по схеме, приведенной на рисунке
58. Измерения проводят сразу же после окончания фор¬
мования, начиная с небольшого напряжения, около 1 в.
Силу тока замеряют через каждые 2 в. Таким же обра¬
зом проводят снятие прямой характеристики (рис. 59).
Полученные данные о зависимости силы тока от напря¬
жения наносят на график (рис. 60).
Оформление результатов работы
Приготовить сульфид или селенид меди и начертить
график его вольт-амперной характеристики.
§ 21. ЭЛЕКТРИЧЕСКОЕ СОПРОТИВЛЕНИЕ
ПОЛУПРОВОДНИКОВ
Работа 51
Приготовление окислов в спеченном состоянии для
физико-химических исследований
Беспористые образцы окислов получают путем прес¬
сования порошкообразных окислов с последующим от¬
жигом при соответствующей температуре. При проведе¬
нии работы необходимо учитывать следующее:
1. Исходная смесь должна быть однородна по со¬
ставу.
2. Зерна смеси должны иметь минимальные размеры.
3. Порошок окислов нужно насыпать в прессформу
равномерным слоем по толщине. Особенно это относится
к прессованию заготовок, имеющих значительные раз¬
меры.
4. Нагрев и охлаждение при обжиге необходимо про¬
водить при постепенном изменении температуры.
226
Температура обжига и его продолжительность в каж¬
дом конкретном случае находится опытным путем. Для
окислов, имеющих высокие температуры плавления, она
лежит на 200—300 °С ниже температуры плавления. При
соблюдении ряда условий и удлинении времени спекания
температуру спекания можно снизить. Окислы, темпера¬
тура плавления которых 1000 °С и ниже (например,
Си20, РЬО), обычно спекаются в сравнительно узком ин¬
тервале температур, который близок к температуре их
плавления.
Чистые окислы, как правило, спекаются с трудом, и
часто они имеют невысокую механическую прочность.
Для облегчения спекания трудноспекаемых окислов к
ним иногда добавляют (около 0,5%) так называемые
плавни — легкоплавкие вещества (например, борный ан¬
гидрид) или вещества, вступающие в химическую реак¬
цию с основным окислом и дающие сравнительно легко¬
плавкие соединения. Часто в состав плавня вводят окись
алюминия, которая вследствие амфотериости взаимодей¬
ствует как с основными так и с кислотными окислами.
Плавни дают на границе кристаллитов окислов стекла.
Это как бы цементирует образец и придает ему большую
механическую прочность. Для получения беспористых
образцов наиболее тугоплавких окислов часто применя¬
ют двукратный обжиг, т. е. спеченный образец разбива¬
ют, тщательно растирают до тончайшего порошка, прес¬
суют и снова обжигают. Для облегчения прессования
порошок предварительно смачивают небольшим количе¬
ством какой-либо жидкости, например спирта, масла,
бензина. Во многих случаях наиболее пригодным для
этой цели является чистый бензин, бензол или толуол.
Цель работы. Получить в спеченном состоянии
окись железа, закись железа, закись никеля и т. д.
Оборудование и материалы
1. Окись железа, свежевосстановленное железо, окис¬
лы никеля и др.
2. Прессформа.
3. Школьный гидравлический пресс.
4. Трубчатая электропечь на 1200 °С.
5. Фарфоровая или агатовая ступка с пестиком.
227
Проведение работы
Приготовление окиси железа. 15—20 г окиси железа
перетирают с небольшим количеством бензина в фар¬
форовой ступке в течение 10—15 мин. Затем из получен¬
ной массы на школьном гидравлическом прессе делают
круглую заготовку по методу, описанному в работе 8;
затем ее подсушивают в сушильном шкафу при ПО—
120 °С для удаления основной части бензина и прокали¬
вают в течение 3—4 ч при 1200 °С. С этой целью заго¬
товку помещают на фарфоровую лодочку, которую вдви¬
гают в фарфоровую трубку, помещенную в трубчатую
электропечь. Затем включают электропечь и постепенно
повышают температуру. После выдержки образца при
указанной температуре печь выключают. Образец можно
вынимать из печи (лучше вместе с фарфоровой трубкой)
только после того, как температура снизится до 500—
600 °С. Лучше же дать ему остыть вместе с печью.
Образцы, приготовленные при 1200 °С, имеют неболь¬
шое количество пор. Для приготовления беспористых
образцов прокаливание нужно вести при 1350 °С.
Приготовление закиси-окиси железа. На технохими-
ческих весах отвешивают прокаленную окись железа и
свежевосстановленное порошкообразное железо. Реак¬
тивные препараты железа часто в той или иной степени
бывают несколько окислены. Поэтому такое железо сле¬
дует в течение 10—15 мин прокалить при 500° С в токе
водорода (см. работу 3). Железо и окись железа отве¬
шивают в соответствии с уравнением реакции:
4Fe203 + Fe = 3Fe304.
Затем смесь тщательно перемешивают в ступке с не¬
большой добавкой бензина, прессуют и прокаливают,
как описано выше, при 1200°С в течение 3—4 ч. Для
того чтобы железо преждевременно не окислилось, через
фарфоровую трубку пропускают из баллона слабый ток
азота. Азот сначала пропускают через промывалку, на¬
полненную концентрированной серной кислотой (счетчик
пузырьков), а затем для удаления следов кислорода—
через трубку, наполненную магниевыми стружками.
Трубку нагревают электропечью или газовой горелкой
228
до 500—550 °С. Охлаждение образцов необходимо вести
также в токе чистого азота.
Приготовление закиси железа. Берут смесь порошко¬
образного железа и окиси железа в соответствии с урав¬
нением реакции:
Fe203 + Fe = 3FeO.
В остальном поступают так же, как и при получении
закиси-окиси железа.
Приготовление закиси никеля. Для получения закиси
пригоден любой из окислов никеля, так как при высоких
температурах происходит отщепление кислорода с обра¬
зованием низшего окисла никеля.
После прессования окисла, как описано выше, заго¬
товку обжигают при 1200 °С в течение 3—4 ч и охлаж¬
дают.
Окисел содержит кислорода несколько больше, чем
это соответствует формуле NiO.
Работа 52
Определение удельного электрического сопротивления
полупроводников и установление зависимости
сопротивления от температуры
Для неметаллов удельная электропроводность с по¬
вышением температуры увеличивается вследствие увели¬
чения количества носителей электрического тока.
Окислы и некоторые соли, например ферриты, по фи¬
зическим свойствам примыкают к неметаллам.
Цель работы. Определить удельное сопротивле¬
ние окисла и установить зависимость сопротивления от
температуры.
Оборудование и материалы
1. Спеченные окислы (FeO, Fe304, Fe203, СоО и др.).
2. Паста для серебрения.
3. Термостат.
4. Источник постоянного тока, вольтметр, миллиам¬
перметр, соединительные провода.
229
Проведение работы
Определение удельного сопротивления проводится на
установке, состоящей из термостата и электроизмеритель¬
ной схемы (рис. 61).
Рис. 61. Установка для определения сопротивления
полупроводников:
1—образец; 2—сопротивление для обогрева; 3 — водяная
рубашка; 4 — металлический сосуд; 5 — электрическая схема
для определения сопротивления.
Рис. 62. Приспособление для укрепле¬
ния образцов при определении сопро¬
тивления :
/ — образец; 2 — вннт; 3 — шайба нз веще¬
ства с большим сопротивлением (текстолит
фарфор и т. д.); 4 — электроды.
230
Для лучшего контакта окисла с электродами его по¬
верхность покрывают тонким слоем серебра. С этой
целью на боковые поверхности наносят ровным тонким
слоем пасту для серебрения и после подсушивания при
100 °С образец прокаливают при 250 °С, затем зажимают
в специальное приспособление (рис. 62).
Поместив образец в масляную баню, замеряют тем¬
пературу и, замкнув цепь, с помощью вольтметра и мик¬
роамперметра или миллиамперметра замеряют падение
напряжения на образце и силу тока, проходящего через
образец. Затем повышают температуру масляной бани
на 10—15 °С и проводят повторное измерение.
Промер сопротивления проводят до 140—150°С.
Оформление результатов работы
Полученные данные занести в таблицу.
Диаметр
образца
(в см)
Толщина
образца
(в см)
Температу¬
ра (в °С)
Напряже¬
ние (в в)
Сила тока
(в а)
Удельное
сопротив¬
ление
(в ом)
ГЛАВА X
МЕТОДЫ ИСПЫТАНИЯ И ЗАЩИТЫ МАТЕРИАЛОВ
§ 22. ДИАГРАММА СОСТОЯНИЯ ЖЕЛЕЗО-УГЛЕРОД
Работа 53
Упражнения по диаграмме состояния железо-углерод
Упражнения по диаграмме состояния железо-углерод
заключается в рассмотрении самой диаграммы, состав¬
лении ответов на вопросы и в сопоставлении данных, по¬
лученных при микроскопическом исследовании конкрет¬
ных сплавов железа (работа 54).
В современной диаграмме железо-углерод приводят¬
ся данные как для равновесной системы (стабильной),
так и для неравновесной. При наличии в сплаве цементи¬
та Fe3C возможен его распад на железо и углерод, кото¬
рый выделяется в виде графита. После полного распада
цементита система приобретает стабильность и на диа¬
грамме линии раздела фаз изображаются пунктирными
линиями. Большое практическое значение имеет метаста-
бильная (неравновесная) диаграмма состояния Fe—РезС
с нераспавшимся цементитом (рис. 63). По оси абсцисс
отложено содержание углерода от 0 до 6,67%, по оси
ординат — температура от 0 до 1539°С. Линия АВС
называется линией ликвидуса, и сплавы выше этой ли¬
нии всегда находятся в жидком состоянии. Линия AFBO
называется линией солидуса, и сплавы ниже этой линии
находятся в твердом состоянии. Цементит Fe3C, обра¬
зующийся из жидкого расплава, называется первичным.
Его кристаллы выделяются ниже линии ВС. Ниже линии
АВ происходит выделение аустенита — твердого раство-
232
Рис. 63. Диаграмма состояния железо-углерод.
ра углерода в у-железе. При содержании 4,3% углерода
сплав затвердевает ниже 1130°С полностью, без выделе¬
ния твердой фазы из жидкой.
Точка В называется эвтектической. Ей соответствует
при метастабилыюм состоянии эвтектический сплав
аустенита с цементитом — ледебурит, а при стабильном
состоянии — аустенит и графит. Аустенит, входящий в
состав сплава, содержит максимальное количество угле¬
рода— 2,14%. Сплавы, содержащие 2,14% углерода и
меньше, называются сталями. При большем количестве
углерода сплав называется чугуном. При полном распа¬
де цементита (в стабильном состоянии) y-Fe растворяет
углерод в меньшем количестве, около 1,7% С. Поэтому
иногда эту границу в содержании углерода также назы¬
вают границей между сталями и чугунами. При сниже¬
нии температуры растворимость углерода ву-Fe снижа¬
ется и при 723 °С доходит до 0,8—0,83% С. Поле IV огра¬
ничивает существование аустенита.
Точка S является эвтектойдной. Стали, содержащие
менее 0,8% С, называются доэвтектойдными, более —
заэвтектойдными. Аустенит ниже линии GS неустойчив и
8 Н* Г£ Ключников
233
постепенно распадается. В зависимости от скорости
охлаждения стали из аустенита образуются следующие
продукты распада: перлит, сорбит, троостит и мартен¬
сит. При небольших скоростях охлаждения стали (около
40 /сек) образуется перлит — механическая смесь фер¬
рита и цементита. Феррит — это твердый раствор угле¬
рода в a-железе. При 723 °С в железе растворяется до
0,03% углерода. При 0,83% получается чисто перлитная
структура. Цементит Fe^C, образующийся при распаде
аустенита, называется вторичным. При больших скорос¬
тях охлаждения (около 50 град!сек) образуется сорбит.
Это тоже распавшийся аустенит, но зерна феррита и це¬
ментита мелкие и едва обособились. Тростит — смесь
феррита и цементита очень высокой степени дисперснос¬
ти. Он получается при скорости охлаждения около
80 град!сек.
Мартенсит — фаза, образующаяся (начальные ста¬
дии распада аустенита) при большой скорости охлажде¬
ния стали (180—200 град /сек), и представляет собой пе¬
ресыщенный твердый раствор углерода в a-железе. Мар¬
тенсит характеризуется большой твердостью, В то же
время он хрупок.
Чугун, так же как и сталь, может кристаллизовать¬
ся как по метастабильной, так и по стабильной системе.
При медленном охлаждении образуется серый чугун,
содержащий графит. При этом в зависимости от харак¬
тера превращений чугун может иметь структуру перлит¬
ного типа (перлит +графит), ферритно-перлитного (фер¬
рит + перлит + граф ит), ферритного (феррит + графит).
При быстром охлаждении, когда некоторые фазы не
успеют разложиться, образуется белый чугун (перлит+!
+ цементит+ледебурит) или чугун другого типа
(перлит+ледебурит+цементит+гр афит).
Оформление результатов работы
Зарисовать по памяти диаграмму состояния железо-
углерод и ответить на вопросы.
Вопросы и задачи
1. Чем отличается стабильная диаграмма состояния железо-
углерод от метастабильной (неравновесной)?
2. Каково процентное содержание углерода в цементите и в ка¬
кой точке (на диаграмме) выделяется чистый цементит?
234
3. Объясните, почему иногда сталями называют сплавы железа,
содержащие 1,7% С, а иногда сплавы, содержащие 2,14% С?
4. При каких температурных условиях и при каком содержании
углерода образуется аустенит?
5. Каков состав продуктов распада аустенита и при каких усло¬
виях они образуются?
6. Какие виды чугуна получаются в зависимости от скорости его
охлаждения?
Работа 54
Микроскопическое изучение сплавов железа с углеродом
и сопоставление полученных данных с диаграммой
состояния
Микроскопический метод исследования металлов и
сплавов, называемый микроанализом, заключается в изу¬
чении структуры (строения) металлов с помощью мик¬
роскопа на специально приготовленных образцах — мик¬
рошлифах. Микроанализ позволяет определить форму и
размеры зерен и включений, структуру металла после
термообработки, деформации и других видов технологи¬
ческой обработки.
Цель работы. Приготовить микрошлиф из угле¬
родистой стали, зарисовать под микроскопом структур¬
ные составляющие и определить приблизительно содер¬
жание углерода.
Оборудование и материалы
1. Образцы сталей, содержащих 0,2—0,8% С.
2. Наждачный круг, различные номера наждачной бу¬
маги и полировальные пасты.
3. Растворы для травления микрошлифов (5-про¬
центный раствор HN03 в этаноле).
4. Металлографический микроскоп.
Проведение работы
Для приготовления микрошлифа от образца стали
отрезают кусок, стороны которого равны 10—15 мм. Вы¬
сота микрошлифа — 10—15 мм. При небольших разме¬
рах образцов (лист, проволока) их заливают легкоплав¬
ким сплавом (Вуда, Розе) или полимером (бакелит,
8*
235
полистирол и т. д.) так, чтобы небольшая часть образца
была свободной. Вырезанный кусок затачивают на абра¬
зивном круге с мелким зерном с периодическим охлаж¬
дением. Затем его шлифуют наждачной бумагой с
убывающей зернистостью. Бумагу кладут на стекло или
другую ровную, твердую поверхность и образцом водят в
одном направлении по поверхности бумаги. При переходе
к другому номеру бумаги абразив с шлифа нужно счи¬
щать ватой и менять направление шлифовки на 90° до
полного удаления рисок от предыдущей шлифовки.
Заканчивать шлифовку следует на бумаге, с микронной
зернистостью (20 мк). Затем микрошлиф полируют на
сукне или фетре, который смачивается водной суспен¬
зией из окиси хрома. Полировка должна проводиться до
зеркального блеска. Полированный шлиф для удаления
загрязнений тщательно протирают ватой, смоченной
спиртом, а затем опускают на 1—2 мин в 5-процентный
спиртовой раствор азотной кислоты, промывают водой
и высушивают путем прикладывания травленой по¬
верхности к фильтровальной бумаге.
Поверхность микрошлифа по составу неоднородна, и
ее отдельные части имеют различные потенциалы. Участ¬
ки с более низким потенциалом будут травиться легче.
Картина, получаемая после травления, хорошо видна
при рассматривании микрошлифа с помощью металло¬
графического микроскопа. Хорошо протравливаются гра¬
ницы зерен, поскольку они обогащены различными при¬
месями. Поэтому на границах зерен образуются углуб¬
ления, видимые в виде темных извилистых полос.
При содержании углерода более 0,03%, но менее
0,80% сталь состоит из смеси феррита и перлита (рис.
64). Перлит на микрошлифе выявляется в виде более
темных участков (при 0,80% сталь состоит целиком из
перлита). Из соотношения площадей, занимаемых пер¬
литом и ферритом, можно ориентировочно определить
полуколичественно процентное содержание углерода.
Для этого составляем простейшую пропорцию:
100% перлита содержат 0,8% С
S% перлита » Х% С,
- 0,8-5
отсюда х%С~ -fQQ-,
236
Рис. 64. Микрошлифы стали в зависимости от содержания углерода.
где S — процент от всей площади, занимаемой перлитом.
Поскольку S определить точно невозможно, результат
расчета приближенный.
При содержании углерода более 0,80% поверхность
микрошлифа состоит из более темных участков, занимае¬
мых перлитом, и более светлых участков (сетки), зани¬
маемых цементитом (рис. 64).
Для изучения поверхности микрошлифа используют
микроскоп—так называемые вертикальные микроскопы
МИМ-6 и МИМ-7 или горизонтальный микроскоп
МИМ-8.
Оформление результатов работы
Кратко описать методику приготовления микрошлифа
и определить процентное содержание углерода в стали,
содержащей углерода меньше 0,8%. Используя микро¬
скоп, зарисовать поверхность микрошлифа.
!37
§ 23. ОПРЕДЕЛЕНИЕ КОРРОЗИОННОЙ СТОЙКОСТИ
МЕТАЛЛОВ В АТМОСФЕРНЫХ УСЛОВИЯХ И МЕТОДЫ
ИХ ЗАЩИТЫ
Лабораторные испытания коррозионной стойкости
металлов в той или иной степени имитируют условия
эксплуатации изделий из этих металлов, что позволяет
оценивать их коррозионное поведение. В тех случаях,
когда лабораторные условия полностью имитируют про¬
мышленные, сроки хранения изделий можно оценивать
по лабораторным. Обычно применяют ускоренные испы¬
тания, в которых используют более агрессивные атмо¬
сферные среды. В этом случае прогнозирование возмож¬
ной коррозионной стойкости в естественных условиях
должно проводиться с большой осторожностью.
При выборе метода таких испытаний необходимо учи¬
тывать следующее:
1. Выявить главный фактор (контролирующий), ко¬
торый оказывает первоочередное влияние на данный
процесс, например солевой туман (морская атмосфера),
сернистый газ (промышленная атмосфера), повышенная
влажность и температура, солнечная радиация (влаж¬
ные тропики) и т. д. При ускоренных испытаниях в пер¬
вую очередь необходимо усиливать воздействие именно
главного фактора (повысить концентрацию солевого ту¬
мана или сернистого газа и т. д.).
2. Повышение скорости коррозионного процесса, про¬
водимого в лабораторных условиях, не должно менять
его механизма в условиях эксплуатации изделий. На¬
пример, если коррозионный процесс протекает при учас¬
тии влаги и кислорода воздуха (т. е. с кислородной депо¬
ляризацией), то применение кислых сред для ускоренных
испытаний неприемлемо: металлы более стойкие в кис¬
лых средах могут оказаться менее стойкими (по сравне¬
нию с другими) в атмосферных условиях, так как в кис¬
лых средах коррозионный процесс протекает с водород¬
ной деполяризацией.
3. Имитация климатических перепадов температур
и влажности также важнейший элемент ускоренных ис¬
пытаний.
Для некоторых металлов на кривой «коррозия —
влажность» были обнаружены две критические точки,
названные первичной и вторичной критическими точками
238
влажности. При первичной критической влажности про¬
исходит адсорбция молекул воды и незначительное уско¬
рение коррозионного процесс а. При вторичной критиче¬
ской точке влажности на продуктах коррозии конденси¬
руется вода, которая образует тончайшую невидимую
для глаза пленку. Тогда сильно ускоряется коррозионный
процесс. Конденсация
влаги происходит не
при 100-процентной
влажности, а значи¬
тельно ниже (около
80%). У железа эти
критические точки на¬
ходятся соответственно
при 60% и 80% отно¬
сительной влажности.
При атмосферной кор¬
розии наиболее часто
используют следующие Рис. 65. Определение коррозионной
показатели* стойкости металлов в атмосферных
,\ ' * Л условиях.
1) визуальный, по- J
луколичественный ме¬
тод, заключающийся в осмотре поверхности образцов и
установлении характера коррозии;
2) весовой (гравитационный);
3) установление глубины коррозионных поражений.
Этот метод следует применять в тех случаях, когда кор¬
розия носит неравномерный питинговый характер, т. е.
на поверхности металла имеются глубокие язвы;
4) изменение физико-химических свойств поверхности
образцов, а также свойств металла: уменьшение отража¬
тельной способности, уменьшение электрического сопро¬
тивления, изменение механических свойств, определение
электрического сопротивления окисных пленок и т. д.
Коррозионную стойкость металлов определяют или
в специальных коррозионных камерах, где можно имити¬
ровать соответствующую атмосферу, или в эксикаторах
(рис. 65), куда на стеклянных подставках с помощью
стеклянных крючков подвешиваются образцы. Для под¬
вески можно также использовать капроновую нить (ле-
ску).
Для создания в эксикаторе определенной влажности
на дно эксикатора наливают воду или помещают кри¬
239
сталлогидраты солей с известной упругостью пара. При
использовании воды давление паров приближается
к 100% (оно равно 98—99-процентной влажности). Ниже
в таблице 13 приводятся данные об относительной влаж¬
ности над водными растворами глицерина.
Таблица 13
Относительная влажность воздуха над водными растворами
глицерина
Содержание
глицерина
(в %)
Относительная влаж¬
ность (в %) при
Содержание
глицерина
(в %>
Относительная влаж¬
ность (в %) прн
25° С
| 50°С
25° С
| 50° С
10,4
98,0
98,2
64,1
70,0
71,3
13,6
96,0
96,4
69,0
65,0
66,4
22,0
95,0
95,5
73,2
60,0
61,4
34,9
90,0
90,7
77,0
55,0
56,5
44,1
85,0
85,9
80,3
50,0
51,5
52,0
80,0
81,1
83,4
45,0
46,6
59,5
75,0
76,2
88,0
40,0
41,6
При визуальной оценке степени коррозии описывает¬
ся характер коррозионных поражений и определяется
(ориентировочно) их площадь коррозионных поражений.
Работа 55
Определение коррозионной стойкости металлов
Цель работы. Определить коррозионную стой¬
кость железа, цинка, алюминия, меди в атмосферных
условиях.
Оборудование и материалы
1. Металлические пластинки.
2. Эксикатор с подвесками для металлических пла¬
стинок.
3. Металлические пластинки с отверстиями.
4. Наждачная бумага.
240
Проведение работы
Металлические пластинки зачищают с помощью наж¬
дачной бумаги разных номеров, опускают на несколько
минут в спирт для удаления жировых загрязнений и пе¬
реносят в эксикатор с хлоридом кальция, затем пластин¬
ки помещают на фильтровальную бумагу. Коррозионные
испытания желательно проводить через сутки, так как
за этот срок происходит полное удаление спирта и фор¬
мирование естественной окисной пленки. Спирт можно
удалить, поместив пластинку между листами фильтро¬
вальной бумаги, а затем на 15—20 мин в сушильный
шкаф, нагретый до 40—50 °С.
Пластинки подвешивают в эксикаторе, на дно кото¬
рого наливают воду или помещают реактивы, создающие
определенную влажность (рис. 65). За состоянием по¬
верхности металла наблюдают через сутки, двое суток,
затем реже, через пять и далее через десять суток. Ре¬
зультаты наблюдений заносят в таблицу 14. Для этого
на пересечении горизонтали «характер коррозии» и вер¬
тикали «время» ставят отметку (например, крестик и
подчеркивают соответствующий балл коррозии).
На основании полученных данных строят график
Таблица 14
Оценка коррозионной стойкости металлов при неравномерной
коррозии
Описание характера коррозии
Сутки
Балл
1
2
3
5
10
корро¬
зии
Отсутствие коррозии
0
Незначительное потускнение
1
Появление первых коррозионных пора¬
жений
2
Коррозионные поражения занимают
10—20% поверхности
3
Коррозионные поражения занимают
40—60% поверхности
4
Сплошная коррозия
5
241
«коррозия — время». Характер равномерной коррозии
выразить в каких-то условных единицах довольно труд¬
но, и поэтому следует дать простое описание состояния
поверхности.
Более строго коррозионную стойкость можно опреде¬
лить весовым способом. С этой целью металлические
пластинки взвешивают до опыта и после опыта, удалив
предварительно продукты коррозии. Снимают их при по¬
мощи мягкой резинки. Прочно сцепленные с поверх¬
ностью продукты коррозии травят кислотами в присут¬
ствии ингибиторов. С меди и ее сплавов отложения сни¬
мают в 5—7-процентном растворе серной кислоты, а с
железных и стальных деталей — в слабокислом (рН =
= 4—3) 10-процентном растворе цитрата аммония при
40—50 °С. Необходимое значение pH достигается добав¬
лением лимонной кислоты или аммиака. Для того чтобы
полностью исключить возможное небольшое растворение
металла, в раствор желательно добавить ингибитор
(0,1% катапина + 1% каптакса, катапин можно заменить
ОП-7 или ОП-Ю). Можно также применить 10-процент¬
ный раствор серной кислоты с добавлением 1 % ингиби¬
тора (4 М или равной смеси К1+уротропин).
После удаления продуктов коррозии образцы промы¬
вают в воде с щеткой, в спирте, высушивают и взвеши¬
вают. Скорость коррозии рассчитывается по формуле:
где Wj и /«2 — масса образца до опыта и после опыта;
S — площадь образца (в см2 или м2); t — время в часах
или годах.
У нас принята десятибалльная шкала коррозионной
стойкости металлов (ГОСТ 5142-50).
Коррозия измеряется проницаемостью — толщиной
металла (в мм), прокорродировавшего в течение года.
Рассчитывают проницаемость на основании потерь
массы металла по формуле:
я = -^ ю-3,
т
где П — проницаемость {мм • год); р — скорость корро¬
зии (а/л«2-год); у — плотность металла (г/см3).
242
Таблица 15
Группа стойкости
Скорость коррозии (г{мг-год)
Балл
Совершенно стойкие
Менее 0,001
1
Весьма стойкие
От 0,001 до 0,006
2
0,005 до 0,01
3
Стойкие
0,01 до 0,05
4
0,05 до 0,1
5
Пониженно стойкие
0,1 до 0,5
6
0,5 до 1,0
7
Малостойкие
1,0 до 5,0
8
5,0 до 10,0
9
Нестойкие
10,0 и более
10
Оформление результатов работы
Кратко описать методику работы, заполнить табли¬
цы, приведенные в работе, или провести расчеты, харак¬
теризующие коррозионную стойкость металла.
Работа 56
Изучение защитных свойств ингибиторов атмосферной
коррозии
К ингибиторам коррозии металлов относятся веще¬
ства, замедляющие коррозию. Существуют следующие
методы применения ингибиторов атмосферной коррозии:
1. Завертывание изделий в бумагу, пропитанную ин¬
гибиторами.
2. Нанесение ингибиторов в виде водных, спиртовых
или загущенных растворов на поверхность металла.
3. Для защиты емкостей или изделий, имеющих по¬
лости, ингибиторы помещают внутрь.
Для консервации внутренних поверхностей труб и
емкостей иногда применяют воздух, насыщенный парами
ингибитора, который продувают через емкость.
4. Для определения радиуса действия ингибитора
над ним на разных расстояниях помещают металличе-
243
ские пластинки и определяют, на каком расстоянии про¬
является защитное действие ингибитора. Опыты прово¬
дят в цилиндрических сосудах.
Для получения данных по защитным свойствам инги¬
биторов атмосферной коррозии ставят опыты по корро¬
зии металла с ингибитором и в аналогичных условиях
в его отсутствие.
Оборудование и материалы
1. Пластинки металлов (сталь, медь, алюминий,
цинк).
2. Ингибиторы: нитрит дициклогексиламина (НДА),
моноэтаноламин, хромат уротропина, нитрит натрия,
уротропин, адипинат или бензоат натрия, бензтриазол.
3. Оберточная бумага.
4. Эксикаторы с подвесками для пластинок.
5. Наждачная бумага.
Проведение работы
Вариант 1. Пластинки металлов заворачивают в
ингибированную бумагу и помещают в эксикатор над
водой. В другом эксикаторе подвешивают аналогичную
пластинку, завернутую в такую же бумагу, но не содер¬
жащую ингибитора. Через несколько дней образцы
осматривают и отмечают состояние поверхности по табли¬
це. Осмотр продолжают проводить еще в течение некото¬
рого времени и заканчивают, когда образцы полностью
прокорродируют. Для определения защитных свойств ин¬
гибитора сравнивают время до начала появления пер¬
вых очагов коррозии в контроле и на образцах, заверну¬
тых в ингибированную бумагу.
Вариант 2, Чашку или тигель с ингибитором по¬
мещают в эксикатор, на дно которого наливают воду.
Металлические пластинки подвешивают над ингибито¬
ром.
В другой эксикатор помещают контрольные образцы
без ингибитора. За состоянием поверхности проводят
систематическое наблюдение, фиксируя время, через ко¬
торое появляются первые очаги коррозии, когда прокор-
родирует примерно 10% поверхности (табл. 14).
244
Для защиты стали применяют один из следующих
ингибиторов: нитрит дициклогексиламина (НДА), моно-
этаноламин, хромат уротропина, равную смесь уротропи¬
на с нитритом натрия. В качестве ингибитора при корро¬
зии меди применяют бензотриазол, бензоат или адипи¬
нат натрия. Защита цинка и алюминия осуществляется
хроматами уротропина или циклогексиламина. Адипинат
и бензоат натрия, бензотриазол и хроматы аминов следу¬
ет применять только для приготовления ингибированной
бумаги, ввиду их небольшого радиуса действия.
Оформление результатов работы
Кратко записать методику работы и заполнить таб¬
лицу.
Масса образцов (в г)
Ингибитор
Время
сушки
Скорость
коррозии
до опыта
после опыта
уменьше¬
ние
массы
§ 24. ПАССИВАЦИЯ И ОКСИДИРОВАНИЕ МЕТАЛЛОВ
Под пассивностью металлов понимают состояние вы¬
сокой коррозионной устойчивости, вызванное преимуще¬
ственным торможением анодного процесса растворения
металла. Высокая коррозионная стойкость металлов
объясняется термодинамической стабильностью, т. е.
химической инертностью металлов (Pt, Pd, Ir, Au), тор¬
можением катодного процесса электрохимической корро¬
зии, диффузионными затруднениями при подводе
агрессивных агентов и т. д., однако пассивное состояние
обусловлено торможением только анодной стадии корро¬
зионного процесса. Пассивное состояние характеризуется
сильным смещением потенциала корродирующего метал¬
ла в положительную сторону (у железа от —0,4 до
+ 0,5 в)у что указывает на торможение анодного процес¬
са, приводящего к резкому снижению коррозии. Перевод
металлов в пассивное состояние можно осуществить дей¬
ствием соответствующих окислителей или анодной поля¬
ризацией.
245
При анодной поляризации на электрод (изделие) по¬
дается ток, чтобы сместить потенциал металла в область
положительных значений, при которых наступает пас¬
сивное состояние. Например, нержавеющая сталь, совер¬
шенно неустойчивая в 50-процентной серной кислоте при
анодной поляризации током плотностью 2,5 а/см21 может
быть переведена в устойчивое пассивное состояние; при
этом скорость коррозии снижается в тысячи раз.
Окислители, вызывающие пассивное состояние метал¬
ла, называются пассиваторами. К ним относятся различ¬
ные хроматы, молибдаты, вольфраматы, пертехнаты, нит¬
риты, а также некоторые кислоты, например азотная по
отношению к железу, алюминию, титану.
Пассивное состояние металла может в большей или
меньшей степени сохраняться и после снятия причин,
вызывающих пассивацию. Например, железо, запассиви-
рованное концентрированной азотной кислотой или рас¬
твором бихромата калия, сохраняет на некоторое время
свою стойкость в воде, атмосферных условиях и более
разбавленной азотной кислоте.
Пассивное состояние металлов в настоящее время
объясняется пленочной и адсорбционной теориями. Со¬
гласно пленочной теории это состояние обусловлено воз¬
никновением на металле тончайшей (десятки или сотни
ангстрем) защитной пленки, состоящей из продуктов
взаимодействия металла с внешней средой. Чаще всего
эта пленка представляет собой какое-то кислородное со¬
единение металла (Рез04 или FegOu на железе в азот¬
ной кислоте или Сг203 и Сг02 на хроме в кислой среде).
Такая пленка служит фазовым барьерным слоем, кото¬
рый отделяет металл от окружающей его коррозионной
среды. Таким образом, коррозионные свойства металла
по отношению к окружающей его среде в значительной
степени определяются физико-химическими свойствами
этой защитной пленки.
Адсорбционная теория пассивности предполагает воз¬
никновение на поверхности металла мономолекулярных
адсорбционных слоев кислорода, окислителя или других
веществ. Эти слои или насыщают валентности поверх¬
ностных атомов металла и тем самым снижают его ак¬
тивность, или вызывают резкое изменение кинетики анод¬
ного процесса растворения металла. Эти теории не про¬
тиворечат, но лишь взаимно дополняют друг друга.
246
По мере того как адсорбционная пленка, постепенно
утолщаясь, переходит в фазовую, на торможение анод¬
ного процесса будет накладываться также торможение
коррозии, вызванное диффузионными затруднениями за
счет образовавшейся защитной пленки.
Пассивное состояние металла может быть нарушено:
1) катодной поляризацией (т. е. наложением внешне¬
го катодного тока, который будет восстанавливать
пассивирующий слой); 2) введением в раствор ряда
ионов-депассиваторов (СГ, Г, Br', SO/'); 3) механиче¬
ским нарушением пассивирующей пленки; 4) повыше¬
нием температуры.
Пассивность металлов имеет большое практическое
значение при защите металлов от коррозии. Она опреде¬
ляет коррозионную стойкость в естественных условиях
большинства конструкционных металлов, например алю¬
миния, магния, хрома, нержавеющих сталей. Торможение
коррозионного процесса может быть успешно осуществ¬
лено за счет формирования более толстых (0,5—3 мкм)
окисных пленок, видимых невооруженным- глазом. Такие
пленки, создавая своеобразный барьер, выполняют экра¬
нирующую роль и изолируют металл от коррозионной
среды.
Процесс формирования окисной пленки на металле
с целью защиты от коррозии или придания изделию де¬
коративного вида называется оксидированием. Оно на¬
шло применение при обработке стальных, магниевых и
алюминиевых изделий. Существуют различные способы
оксидирования, из них наиболее распространенными яв¬
ляются химический и электрохимический. Для железа
в настоящее время применяют химический способ с ис¬
пользованием щелочных растворов и добавлением окис¬
лителей. Толщина образующейся в этих растворах оксид¬
ной пленки в зависимости от условий формирования
(температуры раствора, концентрации щелочи и окисли¬
теля) составляет 2—3 мкм.
При химическом оксидировании металл, погруженный
в оксидирующий состав, взаимодействует с едким натром
и окислителем:
Fe + О + 2NaOH = Na2Fe02 + Н20;
2Fe + 30 + 2NaOH = Na2Fe204 + H20.
247
Соединения Na2Fe02 и Na2Fe204 реагируют между со¬
бой с образованием магнитной закиси-окиси железа:
Na2Fe02 + Na2Fe204 + 2Н20 = Fe304 + 4NaOH.
При этом закись-окись железа отлагается на поверх¬
ности металла. Может происходить и гидролиз Na2Fe204,
в результате чего выпадает осадок Fe203 • mH20, причем
в зависимости от температуры раствора степень гидрата¬
ции окиси железа оказывается различной.
Поскольку концентрация Na2Fe02 и Na2Fe204 наи¬
большая у поверхности железа, то здесь же достигается
и пересыщение раствора продуктами их взаимодействия
и гидролиза. В зависимости от условий могут преимуще¬
ственно кристаллизоваться Fe304, безводный Fe203 в ви¬
де гематита или безводной у-Ре20з. Характер кристал¬
лизации оказывает влияние на плотность, цвет и корро¬
зионную устойчивость пленки.
Работа 57
Пассивация цинка и стали в хроматных растворах
Хроматы уже много лет применяются для замедления
коррозии различных металлов. Их используют в вод¬
ных системах различного назначения, маслах, красите¬
лях, пленках, рассолах. Важным преимуществом хрома-
тов является способность их защищать все практически
важные металлы. Особенно широкое распространение
получил процесс обработки стали, меди, цинковых и кад¬
миевых гальванических покрытий водными растворами
хроматов с последующей сушкой на воздухе (хроматиро-
вание). При этом на поверхности металла образуется
тонкая защитная пленка, повышающая его коррозион¬
ную устойчивость. Толщина такой пленки (0,5 мкм) за¬
висит от времени обработки и сушки. Пленка состоит
в основном из окислов металла и окиси хрома.
Цель работы. Получить пассивную хроматную
пленку на стали и цинке, изучить ее защитные свойства.
Оборудование и материалы
1. Химические стаканы на 500 мл.
2. Наждачная бумага.
248
3. Сушильный шкаф.
4. Образцы углеродистой стали и цинка.
5. Камера для коррозионных испытаний (или эксика¬
торы).
6. Бихромат калия или натрия и другие реактивы,
необходимые для пассивирования.
Проведение работы
Пассивирование цинка и цинковых покрытий. Обез¬
жиренные образцы цинка или цинковых покрытий про¬
мывают в холодной воде, опускают на 3—5 сек в 3-про¬
центный раствор азотной кислоты, промывают в холод¬
ной воде и погружают в пассивирующий раствор состава:
Na2Cr207 — 200 г/л;
H2S04 (плотность 1,84) — 8—10 мл!л.
Температура раствора 15—20 °С, продолжительность
обработки — 5—10 сек. Цинковые образцы предваритель¬
но зачищают наждачной бумагой. Бихромат натрия мо¬
жет быть заменен бихроматом калия или аммония.,После
пассивирования образцы промывают в холодной проточ¬
ной воде и сушат на воздухе при комнатной температуре
или в сушильном шкафу при 100—120°С. Обработан¬
ное таким способом цинковое покрытие или цинк приоб¬
ретает зеленовато-желтую окраску с радужным оттен¬
ком. При длительном воздействии пассивирующего рас¬
твора (30—60 сек) получаются более рыхлые пленки
коричневого цвета, обладающие худшими защитными
свойствами. Сушка при 100—120 °С способствует получе¬
нию более плотных пленок с глубокими и яркими цвета¬
ми побежалости. Хроматная пленка состоит из ZnCr04>
Сг(0Н)Сг04, Сг203 • пН20.
Пассивирование стальных образцов проводят анало¬
гично пассивированию цинка с той лишь разницей, что
раствор не содержит серной кислоты, которая ухудшает
защитные свойства защитной пленки. Для пассивации
стали применяют обычно 10-процентный раствор би¬
хромата калия. Длительность обработки в этом растворе
при 20 °С составляет 60 мин, а при температуре раствора
60 °С — 20 мин. После такой обработки образцы промы¬
вают в проточной холодной воде и сушат на воздухе при
249
комнатной температуре. Образующаяся при этом пленка
имеет небольшую толщину и бесцветна.
Пассивированные цинковые и стальные образцы под¬
вергают затем коррозионным испытаниям. Для этого в
эксикатор, на дно которого налита дистиллированная
вода, подвешивают на стеклянных подвесках хроматиро-
ванные образцы. Для контроля в эксикатор помещают
также непассивированные образцы стали и цинка. Испы¬
тания проводят в течение двух или трех суток. По исте¬
чении времени испытаний образцы вынимают из эксика¬
тора и определяют визуально степень коррозионного по¬
ражения.
При отсутствии образцов с цинковым покрытием его
получают гальваническим путем из электролита сле¬
дующего состава:
ZnS04 7Н20 —200—300 г/л;
A12(S04)3 18Н20 —30 г/л;
Na2S04 ЮН20 —500—100 г/л;
Декстрин — 8—10 г/л.
Температура электролита 18—25 °С, рН = 3,5—4,5.
Катодная плотность тока — 2—5 а/дм2.
Оформление результатов работы
Кратко описать методику проведения работы и ре¬
зультаты коррозионных испытаний запасснвироваииых
металлов.
Работа 58
Пассивация стали в нитритных растворах
Пассивация в растворах нитрита натрия использует¬
ся главным образом для защиты от ржавления при меж¬
операционном хранении, а также при длительном хране¬
нии и транспортировке изделий. Обработка в растворах
нитрита натрия позволяет улучшить качество продукции,
уменьшить объем работ, связанный с очисткой деталей
от продуктов коррозии перед сборкой, консервацией и
окраской, заменив в ряде случаев трудоемкую и слож¬
ную операцию защиты металлов смазками. Растворы
250
с малыми концентрациями нитрата вызывают коррозию
стали. При достаточной концентрации нитрата на по¬
верхности образуется пленка, хорошо защищающая ме¬
талл от коррозии.
Обработка цинка, цинковых сплавов, оцинкованного
железа нитритом вызывает интенсивную коррозию.
Цель работы. Провести пассивацию стали рас¬
твором нитрита натрия.
Оборудование и материалы
1. Химические стаканы на 500 мл.
2. Наждачная бумага.
3. Образцы стали.
4. Эксикаторы для коррозионных испытаний.
5. Нитрит натрия.
Проведение работы
Предварительно зачищенные, обезжиренные и про¬
травленные образцы тщательно промывают в течение
1 мин в воде и тут же погружают в пассивирующий 10—
12-процентный раствор нитрита натрия. Время обработ¬
ки— 1—2 мин, температура раствора 20 °С. После обра¬
ботки образцы сушат на воздухе до полного удаления
влаги и проводят коррозионные испытания.
Оформление результатов работы
Описать порядок проведения работы и режим пасси¬
вирования. Сравнить коррозионную стойкость пассивиро¬
ванных и непассивировацных образцов.
Работа 59
Оксидирование черных металлов
Цель работы. Ознакомиться с процессом окси¬
дирования и получить окисное покрытие на образцах
стали.
251
Оборудование и материалы
1. Ванна для оксидирования.
2. Образцы стали.
3. Эксикатор для коррозионных испытаний.
4. Стеклянные крючки для подвески образцов.
5. Термостат.
6. Мыльный раствор.
7. Едкий натр и другие реактивы, необходимые для
оксидирования.
Проведение работы
После очистки и обезжиривания образцы погружают
в ванну оксидирования:
едкий натр — 700—800 г/л
нитрат натрия — 200—350 г/л
нитрит натрия — 50—70 г/л
сульфат железа— 3—5 г/л
Температура раствора в начале оксидирования 138—
140 °С. Продолжительность оксидирования 20—30 мин.
К концу процесса, вследствие испарения воды, концент¬
рация раствора возрастает и температура повышается
до 142—146 °С. В процессе оксидирования рекомендуется
через каждые 10—15 мин вынимать образцы из ванны,
промывать в холодной воде и снова помещать в оксиди¬
рующий раствор.
После окончания процесса оксидирования образцы,
имеющие черную поверхность, тщательно промыть в хо¬
лодной проточной воде для более полного удаления ос¬
татков оксидирующего раствора (2—3 мин). Для конт¬
роля наносят на образец 1—2 капли спиртового раствора
фенолфталеина. Если индикатор не окраншвается в ро¬
зовый цвет, отмывка хорошая.
Полученная оксидная пленка не обладает достаточно
высокой коррозионной стойкостью. Поэтому оксидиро¬
ванные изделия обычно подвергают дополнительной об¬
работке с целью повышения их коррозионной устойчиво¬
сти. Эта операция заключается в обработке деталей рас¬
твором мыла, в сушке, промасливании и протирке.
Дополнительную обработку оксидированных образ¬
цов проводят в мыльном растворе, содержащем 20—30 г
252
мыла в 1 л воды, в течение 3—5 мин. После обработки
в мыльном растворе образцы просушивают в сушильном
шкафу при 80—100 °С, протирают ветошью и помещают
в эксикатор для проведения коррозионных испытаний.
Оформление результатов работы
Дать описание процесса оксидирования и сравнить
коррозионную стойкость оксидированных и неоксидиро-
вапных образцов.
§ 25. ОПРЕДЕЛЕНИЕ КОРРОЗИОННОЙ СТОЙКОСТИ
МЕТАЛЛОВ В НЕЙТРАЛЬНЫХ СРЕДАХ И МЕТОДЫ
ИХ ЗАЩИТЫ
Коррозия металлов в нейтральных средах протекает
по электрохимическому механизму, и ее скорость опреде¬
ляется скоростью двух сопряженных электрохимических
реакций — анодной и катодной. Анодная реакция заклю¬
чается в переходе ион-атомов металла из кристалличе¬
ской решетки металла в раствор в виде растворимого
или нерастворимого соединения. В кристаллической ре¬
шетке атомы металла находятся в ионизированном со¬
стоянии и между ионами находятся хаотически движу¬
щиеся электроны. При кратковременных столкновениях
электронов с ионами последние могут на короткое время
превращаться в атомы. Вследствие наличия взаимодей¬
ствия между ионами металла и свободными электронами
ион-атомы обозначаются в виде Ме*+ • tie,‘ где Мел+— ион
металла с зарядом пF—количество полусвободных
электронов. Анодный процесс в конечном итоге заключа¬
ется в разрыве связи между ионами металла и полусво¬
бодными электронами и образовании свободных ионов
металла:Мея+ • п&—>-Me"+ + nF. Чем прочнее связь между
ионами металла и «электронным газом», тем более кор-
розионно стойкий металл. На переход ионов металла в
раствор оказывает влияние также энергия гидратации
свободного иона металла или энергия кристаллической
решетки образующихся нерастворимых продуктов кор¬
розии.
Освобождающиеся электроны расходуются на восста¬
новлен не ионов водорода (гидроксония) электролита или
кислорода, растворенного в электролите. Химическая ре¬
253
акция, связанная с этой так называемой ассимиляцией
электронов, выражается одним из следующих уравне¬
ний:
нз0+ + в = Н + Н20;
02 + 4Н+ + 4ё = 2НаО;
02 + 2Н+ + 42 = 20Н“;
02 + 2Н20 + 4ё = 40Н“
Первая реакция, приводящая в конечном итоге к вы¬
делению водорода на поверхности металла, носит назва¬
ние реакции с водородной деполяризацией. Остальные —
реакции с кислородной деполяризацией. При участии
кислорода могут также протекать реакции с образова¬
нием перекиси водорода.
С водородной деполяризацией в нейтральных средах
корродируют только наиболее активные металлы. Боль¬
шинство металлов корродирует со смешанной деполяри¬
зацией, с преобладанием кислородной деполяризации.
В качестве стандартного раствора для изучения
коррозионной стойкости металлов и защитных свойств
замедлителей коррозии, называемых ингибиторами, ча¬
сто применяется 3-процентный раствор хлорида натрия.
Для оценки коррозионной стойкости металлов приме¬
няется весовой метод. О скорости коррозии можно также
судить по количеству металла, перешедшего в раствор,
а в некоторых случаях, например при коррозии магния,
по количеству выделившегося водорода.
Работа 60
Определение коррозионной стойкости стали
в нейтральных средах
(3-процентный раствор хлорида натрия)
Оборудование и материалы
1. Стальные пластинки с отверстиями.
2. Стеклянные стаканчики на 100—150 мл (с 3-про¬
центным раствором хлорида натрия).
3. Стеклянные крючки для подвешивания пластинок.
4. Наждачная бумага, спирт и эфир.
254
Проведение работы
Стальные пластинки зачищают мелкозернистой наж¬
дачной бумагой, выдерживают для обезжиривания 2—
3 мин в спирте, а затем в эфире. Для высушивания пла¬
стинки обтирают фильтровальной
бумагой, удерживая их пинце¬
том, и выдерживают в сушиль¬
ном шкафу в течение 10—15 мин
при 30—40 °С. Затем пластинки
взвешивают на аналитических
весах (пластинки брать только
пинцетом) и опускают в раствор
хлорида натрия па специальных
стеклянных подвесках (рис. 66).
Для этой же цели можно восполь¬
зоваться капроновой нитью (лес¬
кой). Через 4—5 дней пластинки
вынимают, очищают от продуктов
коррозии мягкой резинкой, про¬
мывают в спирте, эфире и после
высушивания пластинки взвеши¬
вают. Скорость коррозии рассчитывают по формуле:
Рис. 66. Определение кор¬
розионной стойкости ме¬
таллов в нейтральных
средах.
т2 — ггц
р= 'S-1~
где гп\ и тг — масса пластинок (до и после опыта); 5 —
площадь пластинок (м2); t — время; р —скорость кор¬
розии (г/м2-ч).
Для определения площади пластинки замеряют
штангенциркулем ее ширину (а), длину (&) и толщину
(с). Площадь рассчитывают по формуле:
S = 2 (ab + be -f- са).
Оформление результатов работы
Кратко записать методику работы и полученные ре¬
зультаты занести в таблицу.
255
Опы¬
ты
Масса пластинок
(в г)
Уменьше¬
ние
массы
(в г)
Площадь
пласти¬
нок
(в м2)
Время
опыта
(в ч)
Скорость
коррозии
(в г[м2-ч)
До
опыта
после
опыта
1
2
3
Работа 61
Определение защитных свойств ингибиторов в
нейтральных средах (3-процентный раствор хлорида
натрия) по отношению к стали
В настоящей работе рекомендуется исследовать за¬
щитные свойства следующих ингибиторов:
1. Силикат натрия в виде его 1-процентного раствора
в хлориде натрия. Защитные свойства силиката натрия
обусловлены тем, что он в водном растворе подвергается
частичному гидролизу и в приэлектродном слое (в ко¬
нечном итоге на поверхности пластинки) образуется не¬
которое, практически невидимое количество силикагеля.
Он также содействует образованию более плотной плен¬
ки Рез04. По-видимому, кратковременно образующиеся
на активных участках силикаты FeSi03 и Fe2(Si03)3 под¬
вергаются гидролизу. Гидроокиси железа в момент обра¬
зования имеют повышенный запас энергии и вступают
между собой в реакцию:
Fe(OH)a + 2Fe(OH)3 - Fe304 + 4Н30.
2. Смесь силиката натрия и нитрита дициклогексила-
мина.
3. Адипинат натрия (NaOO(CH2)5COONa) или себа-
цинат натрия (NaOO(CH2)eCOONa). Нейтральный рас¬
твор этих солей содействует образованию на поверхно¬
сти железа плотной, невидимой для глаза пленки
Ч-FeOOH. Если же берется слабокислый буферный рас¬
твор адипината, то в состав защитной пленки входит ади¬
пинат железа.
256
4. Нитрит натрия в достаточных концентрациях яв¬
ляется хорошим ингибитором. При недостатке он вызы¬
вает опасную питинговую коррозию.
Нитрит"натрия содействует образованию Fe304. Это
связано с тем, что нитрит натрия подвергается в раство¬
ре слабому гидролизу:
NaN02 + Н20 £ NaOH + Ш02.
Образующаяся азотистая кислота является окислителем
и окисляет поверхность металла:
Fe + HN02 Fe304 + Н20 + NO.
Окись азота в присутствии кислорода и воды снова дает
азотистую кислоту. Следовательно, следы азотистой
кислоты являются активатором и своеобразным перенос¬
чиком кислорода к поверхности металла.
Подобным же образом действует и нитрит дицикло-
гексиламина, который является ингибитором атмосфер¬
ной коррозии, хорошо защищает сталь и в нейтральной
среде.
5. Этаноламин H2NC2H4OH защищает сталь, но вы¬
зывает усиленную коррозию цветных металлов. Защита
стали обусловлена тем, что этот ингибитор содействует
образованию плотной защитной пленки Fe304. При не¬
значительной концентрации ингибитора и при длитель¬
ном воздействии на поверхности металла получается
тончайший слой многоядерного комплексного соединения
железа, включающего этаноламин. ч Смеси ингибиторов
в большинстве случаев обладают лучшими защитными
свойствами.
Цель работы. Определить защитные свойства од¬
ного из ингибиторов коррозии в 3-процентном растворе
хлорида натрия по отношению к стали.
Оборудование и материалы
1. Стальные пластинки.
2. Ингибиторы: НДА, адипинат натрия, силикат нат¬
рия, нитрит натрия.
3. Стаканчики с 3-процентным раствором хлорида
натрия. Подвески для пластинок.
4. Наждачная бумага, спирт, эфир.
257
Проведение работы
Зачищенные и обезжиренные пластинки взвешивают
и опускают в 3-процентный раствор хлорида натрия, в
котором предварительно растворен один из следующих
ингибиторов: 1) силикат натрия (1%); 2) силикат нат¬
рия (1%) и НДА (0,1—0,3%); 3) адипинат или себацинат
натрия (1%); 4) адипинат натрия и нитрит натрия (1 %
и 0,5%); 5) раствор нитрита натрия (3%).
Для определения эффективности действия ингибито¬
ра в аналогичных условиях ставят контрольный опыт —
стальную пластинку помещают в раствор хлорида нат¬
рия без ингибитора. Опыт заканчивают через семь дней.
Образцы очищают от продуктов коррозии мягкой резин¬
кой, промывают в спирте и эфире, высушивают и взве¬
шивают. Расчет коррозионной стойкости проводят по
формуле, приведенной в предыдущей работе.
Количественную оценку защитных свойств ингибито¬
ра выражают через ингибиторный эффект:
где Кк — скорость коррозии металла в контроле (без
ингибитора);
Ки — скорость коррозии с ингибитором.
Оформление результатов опыта
Кратко описать методику работы. Определить инги¬
биторный эффект.
§ 26. ОПРЕДЕЛЕНИЕ КОРРОЗИОННОЙ СТОЙКОСТИ МЕТАЛ¬
ЛОВ В КИСЛЫХ СРЕДАХ И МЕТОДЫ ИХ ЗАЩИТЫ
Коррозия металлов протекает по электрохимическому
механизму главным образом с водородной деполяриза¬
цией. При катодной реакции происходит проникновение
иона гидроксония через двойной электрический слой, его
разряд с выделением атомарного водорода, молизация
водорода и десорбция с поверхности металла. Анодная
реакция связана с адсорбцией активных анионов на по¬
верхности металла и образованием промежуточных ком¬
плексов. Заключительной стадией анодного процесса яв-
258
ляется переход иона металла в раствор в гидратирован¬
ном состоянии.
Вещества, замедляющие коррозию, адсорбируясь на
катодных или анодных участках, замедляют анодный или
катодный процесс или оба одновременно. Некоторые ин¬
гибиторы дают на поверхности металла фазовые защит¬
ные пленки. Например, замедление растворения титана
в соляной кислоте в присутствии купферрона (реактив,
применяемый в аналитической химии для осаждения ти¬
тана) объясняется образованием на поверхности тонко¬
го, но малопроницаемого и нерастворимого слоя купфер-
роната титана и т. д.
Наиболее простым и в то же время надежным мето¬
дом определения коррозионной стойкости металла явля¬
ется весовой (особенно, когда наблюдается равномерная
коррозия).
Коррозия может идти с замедлением (накопление на
поверхности шлака и т. д.) или с ускорением процесса
(накопление в растворе активных продуктов реакции,
усиление работы микрогальванических пар за счет при¬
месей, имевшихся в чистых металлах, и т. д.). В этих слу¬
чаях весовой метод позволяет определить среднюю ско¬
рость коррозии за измеряемый промежуток времени.
Опыты проводят в лабораторных стаканах, пластинки ме¬
таллов подвешиваются на стеклянных крючках (рис. 66).
Объемный метод заключается в измерении объема
водорода, выделяющегося во время реакции металла
с кислотой. Этот метод значительно точнее весового
(примерно в 10—100 раз). Однако при несоблюдении
некоторых условий результаты получаются несколько
заниженными. Это связано с тем, что часть кислорода,
растворенного в кислоте, вступает в реакцию с атомар¬
ным водородом, образующимся на поверхности металла,
вследствие чего количество замеренного водорода
оказывается несколько меньше по сравнению с тем его
количеством, которое должно было бы выделиться на
основании весовых измерений. Если реакция растворения
металла протекает со значительной скоростью и поверх¬
ность кислоты соприкасается с воздухом, то с водород¬
ной деполяризацией растворяется около 93—95% стали.
В этом случае результаты опыта будут занижены на 5—•
7%. Если реакция протекает медленно (в разбавленной
кислоте или в присутствии ингибиторов), то количество
259
металла, растворившегося с водородной деполяризацией,
сильно снижается и может составлять всего 20—50%
(будет допущена большая ошибка). В этом случае рас¬
творенный кислород нужно
удалить из кислоты и преду¬
предить его диффузию в
кислоту. Для этой цели че¬
рез кислоту пропускают
медленный ток водорода в
течение 10—15 мин и приме¬
няют коррозиметр, в кото¬
ром предупреждается про¬
никновение кислорода в ки¬
слоту (рис. 67). Воронка
коррозиметра должна дохо¬
дить до дна стаканчика, что
будет предупреждать про¬
никновение кислорода к по¬
верхности образца.
Работа 62
Определение коррозионной
стойкости металлов
в кислых средах
Рис. 67. Определение коррозий¬
ной стойкости металлов в кис- При определении корро-
лых средах. зионной стойкости металлов
в стационарных условиях
нужно соблюдать следующее:
1. Пластинки металла должны иметь определенные
легко измеряемые размеры, лежащие в пределах 15—
30 см* 1 2 3. При измерении небольших пластинок ошибки
увеличиваются и точность результата снижается.
2. На пластинку указанных размеров необходимо
брать около 150 мл кислоты. При меньшем количестве
кислоты, особенно если она имеет небольшую концентра¬
цию, будет наблюдаться снижение ее концентрации
вследствие взаимодействия с металлом.
3. В кислоте пластинка должна находиться такое
время, чтобы уменьшение ее массы равнялось 0,1000—
0,3000 г. При меньших потерях массы точность опытов
снижается.
к
260
Цель работы. Определить коррозионную стой¬
кость стали в соляной или серной кислоте.
Оборудование и материалы
1. Стальные пластинки 10—20X30—40x1—2 мм.
2. Лабораторные стаканы на 150—200 мл, стеклян¬
ные подвески.
3. Кислоты (серная, соляная).
Проведение работы
Три стальные пластинки зачищают наждачной бума¬
гой, обезжиривают и взвешивают на аналитических ве¬
сах и помещают на подвесках в соляную или серную
кислоту (рис. 66) определенной концентрации (1, 3, 5,
7 н.). Через несколько часов (2—4) пластинки вынима¬
ют из кислоты, промывают водой, разбавленным раство¬
ром соды, снова водой, спиртом и эфиром. После высу¬
шивания пластинки взвешивают и рассчитывают скорость
коррозии по формуле, приведенной в работе.
Оформление результатов работы
Кратко описать методику работы. Полученные дан¬
ные занести в таблицу.
Масса образцов (в г)
Уменьше¬
ние
массы (в г)
Площадь
(в см2)
Время
(в ч)
Скорость
коррозии
(в г/м2* ч)
до опыта | после опыта
I
Работа 63
Определение защитных свойств ингибиторов кислотной
коррозии
Ингибиторы кислотной коррозии замедляют раство¬
рение металлов в кислотах и мало изменяют скорость
261
растворения окислов. Поэтому ингибиторы применяют
при кислотном травлении металлов с целью удаления
с них окалины. Добавляют 0,5—1% ингибитора в кис¬
лоту.
Цель работы. Определить защитные свойства од¬
ного из ингибиторов кислотной коррозии.
Оборудование и материалы
1. Лабораторные стаканы объемом на 150—200 мл.
2. Ингибиторы: 4 М, ПБ-5, катании, различные про¬
дукты конденсации аминов с альдегидами, равная смесь
иодида калия с уротропином.
3. Стальные пластинки.
Проведение работы
Работа проводится аналогично предыдущей работе,
но для испытания применяют кислоту, содержащую ин¬
гибитор.
Ввиду небольшой скорости взаимодействия стали с
ингибированной кислотой опыт продолжается два-три
дня. Одновременно ставят опыт с неингибированной кис¬
лотой. После определения скорости коррозии рассчиты¬
вают ингибиторный эффект (см. работу 64).
Работа оформляется аналогично предыдущей ра¬
боте.
Сведения об ингибиторах
1. Ингибитор марки ЧМ (черная металлургия). Данная добав¬
ка является ингибитором коррозии главным образом углеродистых
сталей в серной кислоте. Ингибитор ЧМ состоит из регулятора трав-
вления и пенообразователя. Регулятор — это сульфированные тяже¬
лые фракции пиридиновых оснований, вязкая темная жидкость с
характерным запахом, трудно растворимая в воде. Пенообразо-
тель — сульфитцеллюлозная щелочь, липкая, темная, почти без за¬
паха жидкость, легко растворимая в воде. В промышленности
регулятор вводят в 10—15-процентный раствор серной кислоты в ко¬
личестве от 0,5 до 1,5 кг на 1 ж3 раствора для уменьшения страв¬
ливания металла. Перед внесением в травильную ванну регулятор
предварительно растворяется в небольшом количестве горячей сер¬
ной кислоты. Пенообразователь вводится в травильную ванну для
предотвращения образования кислотного тумана, концентрация его
составляет 0,5 кг на 1 ж2 поверхности зеркала (площади) травиль¬
ного раствора.
262
Ингибитор ЧМ добавляют в раствор в количестве 0,5%. Он за¬
медляет скорость растворения углеродистых сталей при 60—80°С
в 10—15 раз и почти полностью предохраняет металл от наводоро-
живания.
2. Ингибитор ПБ-5 — продукт взаимодействия анилина с уро¬
тропином. Применяется как присадка в количестве 0,8% к 20-про¬
центному раствору кислоты при травлении металлов, снятии котель¬
ной накипи и обработке нефтяных скважин. При температурах трав¬
ления 40—50 °С вносится в кислоту в количестве от 0,5 до 1,0%.
С повышением температуры и концентрации кислот эффективность
ингибитора падает.
Ингибиторы (ПБ-5, ЧМ) при температуре выше 60 °С снижают
свои ингибиторные свойства, выпадают в осадок в присутствии
солей железа.
3. Катапин. Это катионоакгивное вещество, получаемое хлорме-
тилированием алкилбензола или углеводородов, содержащихся в ке¬
росиновой фракции нефти, с последующей конденсацией их с пи-
ридином. В зависимости от применяемого исходного сырья разли¬
чают катапин двух марок: катапин А (при использовании алкилбен¬
зола) и катапин К (при использовании естественных ароматических
углеводородов).
Каталины марки А и К применяются в качестве ингибитора кис¬
лотной коррозии стали для соляной и плавиковой кислот (до 20-
процентной концентрации) при 60—80 °С, для серной кислоты (до
40—50-процентной концентрации) при 80—100 °С, для фосфорной
кислоты (до 80-процентной) при температуре до 100 °С. Концентра¬
ция ингибитора в соляной кислоте применяется от 1 до 3 г/л, в сер¬
ной, в фосфорной ■— несколько больше. Одним из недостатков ката-
пина является неустойчивость в сильно разбавленных и очень кон¬
центрированных растворах соляной и серной кислот.
4. БА-6, БА-12. Эти продукты конденсации бензиламина соответ¬
ственно с уротропином и параформальдегидом обладают высоким
защитным действием на стали в соляной кислоте в интервале тем¬
ператур 20—60° С. Оптимальная концентрация этих ингибиторов
0,5-процентпая. При этом они почти полностью защищают углероди¬
стую сталь в 30-процентной соляной кислоте.
5. Синтез некоторых продуктов конденсации формальдегида
аминами;
а) с п-анизидином. К 0,1 моль п-анизидина, предварительно рас¬
творенного в небольшом количестве спирта, добавить 0,1 моль вод¬
ного раствора формальдегида (40%) при комнатной температуре. При
перемешивании смесь разогревается и через 20—25 мин после на¬
чала опыта выпадает кристаллический осадок белого цвета, который
отфильтровывают и перекристаллизовывают из спирта. Температура
плавления вещества 130 °С. Выход — 56%. Это тример метилен-я-
анизидина; 1, 3, 5-три (п-метоксифенил) гексагидро-я-триазин:
б) с циклогексиламином. К 0,1 моль 30-процентного водного
раствора формальдегида медленно по каплям добавить 0,1 моль ци-
клогекснламина, при этом образуется масло, которое вскоре затвер¬
\
CeH4—N = СН2/я, п=3;
263
девает. Полученное вещество перекристаллизовывается из спирта в
виде белых игл с температурой плавления 73 °С. Выход — 48%.
Продукт представляет собой тример метиленциклогексиламина или
1,3,5-три (циклогекснл) тексагидро-з-трназнна:
(C6HUN - СН2)3;
в) с р-нафтиламином. 0,1 моль р-нафтиламина растворить в
200 г ледяной уксусной кислоты, раствор охладить до 3 °С и мед¬
ленно добавлять 0,1 моль 30-процентного водного раствора форм-
альдегида. Через час после начала опыта выпадает осадок, который
отделяют фильтрованием. Для полного извлечения продукта кон¬
денсации к фильтрату можно прилить избыток спирта, нагреть до
кипения и охладить; при этом получается дополнительное количе¬
ство продукта. Синтезированный продукт — мелкокристаллическое ве¬
щество (в виде небольших призм), светло-желтого цвета, темпера¬
тура плавления 208 °С. Выход — 47%. Это тример метилен р-наф-
тиламина; 1,3,5-три (р-нафтил) гексагндро-Б-триазин.
Все синтезированные вещества являются ингибиторами корро¬
зии стали, никеля и кобальта в соляной кислоте. Оптимальная кон¬
центрация их 0,5—0,7%. С повышением концентрации соляной ки¬
слоты и повышением температуры до 60 °С защитное действие доба¬
вок возрастает. В серной кислоте все добавки защищают сталь ху¬
же, чем в соляной. Максимальное защитное действие синтезирован¬
ных ингибиторов проявляется на стали в 7 н. растворе соляной и
8 н. растворе серной кислотах.
ГЛАВА XI
ЭЛЕКТРИЧЕСКИЕ ПРИБОРЫ
§ 27. ВЫПРЯМИТЕЛИ
Селеновый выпрямитель типа ВСА-5 (рис. 68). Вы¬
прямитель предназначается для получения постоянного
тока из переменного с напряжением ПО, 127 и 220 в.
Рис. 68. Селеновый выпрямитель типа ВСА-5:
J — сигнальная лампа; 2—амперметр; 3 — вольтметр;
4 —* выключатель; 5 — переключатель на I и II ступень;
6 — отрицательный полюс; 7 — положительный полюс;
3 — заземление; 0 — ручка регулировки силы тока и на¬
пряжения.
9 Н, Г, Ключников
265
Преобразование переменного тока осуществляется
селеновыми выпрямительными элементами, которые
представляют собой железные диски, покрытые с одной
стороны тонким слоем селена, поверх которого нанесен
катодный слой из легкоплавкого сплава. Выпрямление
переменного тока осуществляется на границе между се¬
леном и катодным слоем (рис. 68).
Направление движения
Рис. 69. Выпрямление переменного тока в селеновом выпрямителе:
а — образование запорного слоя; б — прохождение электрического тока;
+дырки; —электроны.
В состав катодного слоя входят образующиеся за
счет взаимодействия селениды висмута и кадмия, кото¬
рые, как и металлы, имеют электронную проводимость,
а сам селен ввиду недостатка электронов имеет так
называемую «дырочную» проводимость. Вследствие это¬
го, когда на легкоплавкий слой подается положитель¬
ный потенциал, а на селен — отрицательный, электроны
и дырки расходятся в разные стороны и создается слой,
в котором отсутствуют носители электрического тока.
В этом случае электрический ток через выпрямитель не
проходит. При смене направления электрического тока
запирающий слой исчезает и ток проходит через вы¬
прямитель.
Каждый выпрямительный элемент (шайба) в выпря¬
мителе имеет диаметр 100 мм и допускает нагрузку
до 4 а. Допустимое напряжение на шайбу равно 18 в.
Для того чтобы получить постоянный ток большего на¬
пряжения, отдельные выпрямительные элементы соеди¬
няют последовательно.
266
При работе выпрямительные шайбы нагреваются до
60—70 °С. При перегрузке выпрямителя, т. е. при снятии
с него тока более 12 а, слой селена может нагреться
выше допустимой температуры и выпрямитель испор¬
тится.
Выпрямитель, выпускаемый с завода, можно вклю¬
чить в сеть с напряжением 220 в. Если в сети напря¬
жение ПО или 127 в, то следует снять верхнюю крыш¬
ку выпрямителя и установить перемычки на панели
Рис. 70. Включение выпрямителя с нерегулируемым на¬
пряжением:
I выпрямитель; 2 — реостат; 3 — амперметр; 4 — вольтметр;
5 — прибор, потребляющий электрический ток,
трансформатора в положение, соответствующее напря¬
жению сети.
Для включения выпрямителя (рис. 68) - переклю¬
чатель I и II ступени 5 ставят на I ступень, выключа¬
тель 4 ставят на положение «выключено», ручку регу¬
лировки тока и напряжения 9 доводят до отказа в ле¬
вую сторону и вставляют вилку шнура, расположенного
на задней панели выпрямителя, в розетку. Затем пере¬
водят выключатель 4 в положение «включено». При
этом должна загореться сигнальная лампа 1.
Зажимы 6 и 7 перед включением нужно соединить
с прибором, потребляющим постоянный ток, а зажим 5
соединить проводом с заземлением. Затем ручку 9 мед¬
ленно поворачивают вправо. При этом амперметр будет
показывать потребляемый постоянный ток, а вольт¬
метр — напряжение. Если ток недостаточен, то регуля¬
тор 9 нужно вывести, переключатель 5 поставить на
II ступень и снова повернуть ручку 9 вправо до получе¬
ния нужного тока.
Выпрямитель ВСА-5 наиболее удобен для лаборатор¬
ных целей, так как он позволяет плавно менять ток от
0 до 12 а и напряжение от 0 до 64 в.
267
Необходимо отметить, что при включении выпрями¬
теля, который длительное время не работал, обычно на¬
блюдается потрескивание и даже некоторое искрение
в выпрямительных шайбах. Это явление указывает на
то, что выпрямительный слой расформировался. Через
несколько минут работы этот слой отформируется и вы¬
прямитель станет работать нормально.
Нашей промышленностью выпускаются и другие ти¬
пы выпрямителей, которые не допускают плавного регу¬
лирования напряжения и тока. Однако их также можно
использовать для лабораторных целей. Включение по¬
добных выпрямителей проводится по схеме (рис. 70).
Сначала к выпрямителю присоединяют реостат, ампер¬
метр, вольтметр и прибор, потребляющий электрический
ток. Движок реостата ставят в положение максимально¬
го сопротивления. Затем включают выпрямитель в сеть
переменного тока и наблюдают за показаниями ампер¬
метра. Если амперметр показывает ток больше допусти¬
мого по паспорту выпрямителя, то его следует тут же
отключить от сети, а реостат заменить на другой с боль¬
шим сопротивлением.
Описанные в практикуме электролизеры и другие при¬
боры имеют сравнительно большое сопротивление, поэто¬
му реостат можно вообще не включать. Заранее рас¬
считать сопротивление реостата можно в том случае,
когда известно сопротивление потребителя тока, сопро¬
тивление выпрямителя и его характеристика. Лучше и
быстрее сопротивление реостата подбирать опытным
путем.
Помимо селеновых выпрямителей наша промышлен¬
ность выпускает купроксные выпрямители. Выпрями¬
тельные элементы в купроксном выпрямителе выполнены
из медных шайб, имеющих на поверхности слой закиси
меди.
Электрический ток подводят к медной шайбе и
свинцовой, которая накладывается на слой закиси меди
во избежание нарушения его целостности. Элек¬
троны в купроксном выпрямителе могут проходить толь¬
ко в одном направлении — от свинцовой к медной
шайбе.
Включение и работа с купроксными выпрямителями
проводится таким же путем, как и с селеновым выпря¬
мителем.
263
Помимо селеновых и купроксных выпрямителей, в
лабораторной практике используются газотронные вы¬
прямители. В настоящее время пользуются ,и вы¬
прямителем ВГ-2. Его схема приведена на ри¬
сунке 71. Переменный ток возбуждает во
вторичных обмотках трансформатора ток соответствую¬
щего напряжения и силы. Понижающая обмотка питает
накал лампы (тип ВГ-176). К анодам лампы, окружаю¬
щим нить, подводится на-
пряжение с другой обмот¬
ки трансформатора. Это
напряжение можно регу¬
лировать поворотом движ¬
ка, который включает з
соответствующие секции
вторичной обмотай. В
сеть выпрямленного тока
включены амперметр и
вольтметр. Данный вы¬
прямитель, так же как
селеновый и купроксный
дает пульсирующий ток,
и его можно использо¬
вать для питания прибо¬
ров, на которых пульса¬
ция тока не отража¬
ется.
Внешний вид выпря¬
мителя приведен на
рисунке 72. На перед¬
ней панели выпрямителя
JKKKb—'sJHJjKL
Рис. 71. Схема выпрямителя ВГ-2:
ПЯРПОЯПЖРНМ ояотмм { — сеть; 2 — первичная обмотка транс-
рдС1Ш«лиЖсяЫ ЗаЖИМЫ форматора; 3 — вторичная обмотка
ДЛЯ включения выпрями- трансформатора; 4 - понижающая об-
г мотка для питания ннтн накала лампы;
ТеЛЯ В ЭЛеКТрИЧеСКуЮ 5 — переключатель; 6 — лампа ВГ-176.
сеть Л зажимы для под¬
ключения нагрузки 2, переключатель для регулировки
выходного напряжения постоянного тока 3, вольтметр 4
и амперметр 5.
Перед включением выпрямителя в городскую сеть
переключатель напряжения должен быть поставлен в
крайнее левое положение, при котором выключается
напряжение, подаваемое на аноды лампы. Такой поря¬
док включения дает возможность разогреться катоду
269
лампы (нити накала) до подачи напряжения на аноды
лампы. При этом предотвращается разрушение нити
накала лампы. Время подогрева нити равно 40—50 сек.
После этого поворотом ручки переключателя* вправо
увеличивают выпрямленное напряжение на нагрузке до
требуемого. Напряжение и ток контролируют по вольт¬
метру и амперметру. Не допускается включать выпря¬
митель в сеть с напряжением, не соответствующим ука-
Рис. 72. Внешний вид выпрямителя ВГ-2:
1 — зажимы для включения выпрямителя в
сеть; 2 — зажимы для снятия постоянного то¬
ка; 3 — переключатель напряжения; 4 — вольт¬
метр; 5 — амперметр.
занному в паспорте прибора. Перегрузка прибора током
свыше 6 а также приводит к быстрой порче выпрями¬
теля. Описанные выпрямители пригодны для питания
электролизеров, получения металлопокрытий, а также
для зарядки аккумуляторов. Аккумуляторы следует
включать через реостат, устанавливая ток в пределах,
допустимых для выпрямителя и аккумуляторов.
270
§ 28. АККУМУЛЯТОРЫ
В химических лабораториях часто используют кис¬
лотные и щелочные аккумуляторы. В электрическую ха¬
рактеристику аккумулятора входят: электродвижущая
сила (э.д.с.), напряжение, внутреннее сопротивление, ем¬
кость и скорость саморазряда.
Электродвижущей силой аккумулятора называется
разность потенциалов положительного и отрицательного
электродов. Э.д.с. зависит от электродных потенциалов
пластин, т. е. от их состава. Э.д.с. несколько меняется
в зависимости от температуры и от концентрации элек¬
тролита и измеряется компенсационным способом, когда
от аккумулятора энергия не берется, т. е. без тока на¬
грузки.
Напряжением аккумулятора называется разность по¬
тенциалов между положительной и отрицательной пла¬
стинами при прохождении через аккумулятор электри¬
ческого тока. Благодаря наличию внутреннего сопро¬
тивления при разрядке аккумулятора его напряжение
меньше э.д.с. на величину падения напряжения в акку¬
муляторе; при зарядке аккумулятора его напряжение
больше э.д.с.
Внутреннее сопротивление аккумуляторов определя¬
ется конструкцией электродов, плотностью электролита
и зависит от величины зарядного и разрядного токов.
При зарядке и разрядке щелочных аккумуляторов
плотность электролита, а следовательно, и концентра¬
ция щелочи не меняются. Поэтому не изменяется и их
сопротивление. При разрядке кислотных аккумуляторов
плотность электролита уменьшается за счет уменьше¬
ния концентрации кислоты. Вследствие этого сопротив¬
ление разряженного кислотного аккумулятора больше,
чем заряженного.
Внутреннее сопротивление кислотных аккумуляторов
значительно меньше, чем щелочных. Поэтому кислотные
аккумуляторы допускают более значительные" токи раз¬
ряда.
Емкость аккумуляторов, являющаяся их важнейшей
характеристикой, определяется по формуле:
Q=I-tf
271
где /— сила разрядного тока в амперах;
t — время разряда в часах.
Таким образом, емкость аккумулятора определяется
количеством электричества, которое может отдать пол¬
ностью заряженный аккумулятор при разряде его то¬
ком с определенной силой до определенного допустимого
напряжения. В течение срока службы емкость аккуму¬
ляторов несколько меняется. В первое время, в течение
гарантийного срока, она несколько возрастает. В процес¬
се эксплуатации емкость некоторое время не меняется,
а затем начинает падать.
Кислотные и щелочные аккумуляторы подвержены
саморазряду. Саморазряд кислотных аккумуляторов за
сутки достигает 1 % от их емкости, а у щелочных —
за 30 суток 7% их емкости.
Кислотные аккумуляторы
В настоящее время выпускается большое количество
различных типов аккумуляторов. Для учебных целей
практически пригодны аккумуляторы любой системы, так
как они допускают как последовательное, так и парал¬
лельное соединение, что позволяет подбирать необходи¬
мые емкость и напряжение батареи аккумуляторов.
Аккумуляторные батареи имеют условные обозначе¬
ния из букв и цифр, например З-СТ-60, З-МТ-14.
Первая цифра в этих обозначениях указывает на ко¬
личество аккумуляторов, из которых составлена бата¬
рея. Буквы указывают назначение батареи (СТ — стар¬
терная, МТ — мотоциклетная) и материал сосуда (П —
пластмассовый, Э — эбонитовый). Числа, стоящие после
букв, указывают на номинальную емкость в ампер-
часах. Батареи, собранные в деревянном ящике, имеют
маркировку СТЭН, а в эбонитовом сосуде — СТЭА. На¬
личие буквы М, стоящей после цифры, обозначающей
емкость, указывает на то, что в качестве сепаратора
(материал, отделяющий пластины) использован мипор.
Ниже, в таблице 16, приведены некоторые общие дан¬
ные о выпускаемых стартерных батареях.
Срок службы аккумуляторов в значительной степени
определяется правильным приведением аккумуляторов
в рабочее состояние и правильным режимом эксплуа¬
тации.
272
Таблица 16
Данные о некоторых стартерных и мотоциклетных
аккумуляторных батареях
Обозначение
Номи¬
наль¬
ное
напря¬
жение
(в в)
Ем¬
кость
(в а-ч)
Габариты (в мм)
Масса
с элек¬
троли¬
том
(в кг)
длина
ширина
высота
З-МТ-14
6
10
124
100
181
4,2
З-МТМ-14
6
12
125
81
189
5,2
З-СТ-60
6
60
179
178
237
15,0
З-СТ-70
6
70
257
194
230
16,0
З-СТ-84
6
84
272
188
230
19,0
З-СТ-98
6
98
308
188
245
21,5
З-СТ-112
6
112
340
188
245
24,5
Приведение аккумуляторов в рабочее состояние.
Электролитом для свинцовых аккумуляторов служит
чистая, так называемая аккумуляторная кислота, раз¬
бавленная дистиллированной водой. Особенно вредной
примесью в кислоте являются соли железа, которые
сильно ускоряют саморазряд аккумулятора.
Для заливки сухозаряженных батарей используется
раствор кислоты, плотность которого в зависимости от
климатических условий лежит в пределах от 1,25 до
1,31, т. е. применяют электролит эксплуатационной плот-
Таблица 17
Плотность электролита заряженных
аккумуляторных батарей в зависимости от минимальной
рабочей температуры
Районы эксплуатации
Минимальная
окружающая
температура
(в °С)
Плотность
электролита
при -f-15°C
Южные
До —20
1,25
Центральные
&
0
1
со
О
1,27
Северные
&
0
1
wb.
о
1,29
Районы с резко континентальным
климатом
Ниже —40
1,31 (зимой)
1,27 (летом)
273
ности (табл. 17). Для приведения таких батарей в рабо¬
чее состояние требуется их заряжать в течение 3—25 ч
в зависимости от емкости аккумулятора и срока предва¬
рительного хранения. После заливки батареи должны
постоять 2—3 ч для пропитки пластин. При этом плот¬
ность электролита обычно снижается и часто доходит до
1,1 —1,05. Сила тока и время зарядки указаны в таб¬
лице 18.
Таблица 18
Зарядные токи (первая зарядка) и время зарядки батарей
с сухозаряженными пластинами
Тнп батарей
Зарядный ток
(в а)
Продолжительность заряда
после хранения батарей (в ч)
до 1 года
от 1 года до
2 лет
6-СТЭН-140М
12
3-5
3-5
6-МСТ-140
12
3-5
3—5
З-СТ-70
6
5
До 25
З-СТ-98
9
5
До 25
6-СТМ-128
10
5
До 25
6-СТ-110
12—20
3
3
б-МСТ-110
12—20
3
3
Во время зарядки аккумулятора необходимо сле¬
дить, чтобы температура электролита не поднималась
выше 45 °С. В противном случае аккумулятор отсоеди¬
няют от источника тока, чтобы электролит охладился
до 30 °С.
Окончание заряда узнается по повышению плотности
электролита, которая должна отличаться от эксплуата¬
ционной плотности (при 15 °С) не более чем на 0,005.
После окончания зарядки наблюдается электролиз, со¬
провождающийся бурным выделением кислорода и во¬
дорода. Если плотность электролита в конце зарядки
окажется выше нормы, то в аккумулятор следует доба¬
вить некоторое количество воды.
274
Батареи с сухозаряженными пластинами заливают
электролитом плотностью 1,12 (при +15°С). Зарядку
проводят в два приема. Батареи З-МТ-14 и З-МТМ-14
заряжают в течение 35—45 ч (до обильного выделения
газа) соответственно током 2 и 3 а. Затем батареи нуж¬
но в течение 10 ч разряжать через сопротивление током
1,0—1,2 а до конечного напряжения на аккумуляторе
1,7 в. Разряженные батареи нужно еще раз зарядить
током 1 —1,5 а и снова разрядить до конечного напряже¬
ния на аккумуляторе в 1,7 в. Батареи, прошедшие два
цикла заряд — разряд, снова заряжаются током двух
степеней и поступают в эксплуатацию.
Радионакальные батареи (РНП) заливаются элект¬
ролитом плотностью 1,21. Через 2—3 ч после пропитки
пластин их ставят на заряд. Первый заряд батарей
PH-40, РН-60 и РН-80 проводится соответственно током
силой 4 а, 6 а и 8 а. Заряд продолжается непрерывно
в течение 36 R. После трехчасового перерыва зарядку
батарей продолжают тем же током в течение 12 #.
Эксплуатация кислотных аккумуляторов
Летом батареи следует разряжать не более чем на
50%, зимой — не более чем на 25%. Независимо от
степени разрядки батареи ее нужно заряжать один раз
в месяц. В аккумуляторы при испарении электролита
разрешается наливать только дистиллированную воду.
Зарядный ток должен находиться в соответствии
с емкостью аккумуляторной батареи. Для определения
силы зарядного тока в амперах нужно емкость батареи
(в ампер-часах) разделить на 10. Например, зарядный
ток батареи З-МТ-14, емкость которой составляет 10 а-ч,
примерно равен 1 а. Зарядку следует проводить до
обильного выделения газа. Время зарядки ориентировоч¬
но можно определить на основании емкости аккумуля¬
тора, с учетом того, что некоторая часть тока теряется
непроизводительно. Например, батарею вышеуказанно¬
го типа следует заряжать теоретически 10 ч, практически
11 —12 ч током в 1 а. Во время эксплуатации следует
проверять напряжение. Когда оно упадет до 1,7 н, акку¬
мулятор нужно ставить на зарядку.
275
Щелочные аккумуляторы
Щелочные аккумуляторы имеют ряд преимуществ по
сравнению с кислотными: в частности, они не боятся
перегрузок и коротких замыканий, прочны, работают в
более широком диапазоне температур. В зависимости от
состава активной массы они делятся на железо-никеле¬
вые, серебряно-цинковые, кадмиево-никелевые, цинково¬
никелевые и т. д. Активная масса щелочных аккумуля¬
торов заключена в стальные перфорированные пакеты,
или ламели, а ламели впрессованы в стальные стойки.
В качестве электролита в настоящее время приме¬
няется раствор гидроокиси калия и гидроокиси лития
с плотностью 1,19—1,21 при эксплуатации аккумулято¬
ров в пределах от —20 до +50 °С. При эксплуатации
ниже —20 °С применяется раствор едкого кали (плот¬
ность 1,25—1,27). Наличие в растворе гидроокиси лития
повышает емкость аккумулятора. Его содержание равно
около 20 г на 1 л раствора.
Электролит следует менять один раз в год. Для этого
аккумулятор нужно разрядить через сопротивление до
1 в (на аккумуляторе), вылить электролит, промыть
аккумулятор подщелоченной дистиллированной водой,
залить снова электролитом и зарядить.
Для определения силы тока в амперах, необходимой
для зарядки аккумулятора, нужно его емкость (в а • ч)
разделить на 4, а зарядку вести в течение 7 ч. При этом
аккумулятору сообщается около 175% его емкости.
Аккумуляторы и батареи, находившиеся в длитель¬
ной эксплуатации и потерявшие частично емкость, под¬
вергаются усиленному заряду, что способствует восста¬
новлению активной массы. Для этого их подвергают
заряду в течение 6 ч током вдвое меньшей силы. Уси¬
ленному заряду подвергаются аккумуляторы после про¬
мывки, при вводе их в эксплуатацию и после 10—12 цик¬
лов нормальной работы.
Предельная температура составного электролита рав¬
на +45 °С, а для электролита, содержащего только гид¬
роокись калия, +30 °С. Напряжение аккумуляторов
нужно измерять вначале через 3 ч, а под конец заряд¬
ки — через 2 ч.
Напряжение кадмиево-никелевых аккумуляторов бы->
стро повышается до 1,43 в, затем постепенно увели¬
276
чивается до 1,5 в и, наконец, до 1,7—1,8 в. Напря¬
жение у железо-никелевых аккумуляторов быстро под¬
нимается до 1,6 в, а затем постепенно увеличивается
до 1,75 в.
Перезаряд для щелочных аккумуляторов не вреден.
Показателем окончания зарядки является наличие оп¬
ределенного напряжения на клеммах аккумулятора.
Для железо-никелевого аккумулятора оно равно 1,8—■
1,9 н, а для кадмиево-никелевого— 1,75—1,85 б. При
низких температурах это напряжение может быть выше
указанного.
Нормальный разрядный ток щелочных аккумулято¬
ров равен номинальной емкости, деленной на 8. Увели¬
чение силы разрядного тока сокращает срок службы
аккумуляторов. Разрядка аккумуляторов ниже номиналь¬
ного конечного напряжения также уменьшает их
емкость. Однако определить номинальное конечное на¬
пряжение довольно трудно, так как оно зависит от режи¬
ма разряда. При разрядке нормальным разрядным током
это напряжение равно около 1,0 в. После падения напря¬
жения до указанной величины аккумуляторы нужно
подвергать зарядке.
§ 29. ЭЛЕКТРОНАГРЕВАТЕЛЬНЫЕ ПРИБОРЫ
Наиболее удобны в обращении электрические нагре¬
вательные приборы, так как они позволяют получать в
течение длительного времени строго контролируемую
температуру.
В лабораторной практике чаще всего приходится
пользоваться трубчатыми печами и тигельными печами.
Трубчатые печи позволяют получать температуру до
1200 °С. Нагревательный элемент таких печей делается
из специального жаростойкого сплава, выполненного
в виде спирали из проволоки или лепты. Печи старой
конструкции имеют понижающий трансформатор, ко¬
торый включают в сеть. Пониженное напряжение (обыч¬
но 36 в) подается на ленту из жаростойкого сплава
и разогревает ее. Поворотом движка на боковой стенке
печи можно добиться нужной температуры печи. В дру¬
гих печах (рис. 73) на боковой поверхности имеется рео¬
стат, который позволяет регулировать температуру.
277
В печах типа ТК-30/200 последней конструкции обо¬
грев осуществляется карборундовыми стержнями, кото¬
рые в основном состоят из карбида кремния. Номиналь¬
ная мощность таких печей 4500 вт, средняя — 1300 вт,
максимальная рабочая температура 1250°С.
Рис. 73. Электрическая печь:
/ — реостат; 2 — выключатель; 3 — провода; 4 — термопара; 5 — пи¬
рометрический гальванометр (пирометр).
Рис. 74. Обогрев фарфоровой или кварцевой трубки при
помощи спирали:
I — фарфоровая трубка; 2 — спираль; 3—асбест, закрывающий
спираль (на рисунке с передней стороны он убран).
При включении печи следует придерживаться сле¬
дующего порядка в работе. Сначала убеждаются в том,
что напряжение, указанное в паспорте печи, соответст¬
вует напряжению в сети. Затем переключатель, находя¬
щийся на передней панели печи, ставится в положение
278
Рис. 75 а, б. Обогрев катализатора спиралью:
/ — провода, покрытые изоляцией; 2— спираль; 3 — медная про¬
волока; 4 — фарфоровая или кварцевая трубка; 5 — слой катали¬
затора; 6 — кожух холодильника,
«выключено», а движок реостата, находящийся на бо¬
ковой панели, передвигается в положение «выведено».
Только после этого печь подключается к сети. Затем ста¬
вят выключатель в положение «введено». Если печь ис¬
правна, то уже через несколько минут из отверстия
печи будет выходить теплый воздух.
На печь не следует подавать сразу напряжение
в 127 в (или 220 в), так как это приведет к ее быстрому
разогреванию, что сокращает срок службы спирали и
вызывает растрескивание керамики. Поэтому электро¬
печь включают с реостатом, и его постепенно выводят
в два, три приема в течение 30—40 мин.
279
При включении печи, рассчитанной на напряжение
220 в, в сеть с напряжением 127 в развивается темпе¬
ратура примерно в два раза меньше расчетной. При
включении печи, рассчитанной на 127 в, в сеть с напря¬
жением 220 в она разогревается выше допустимой тем¬
пературы. Такую печь можно включить в сеть с напря¬
жением 220 в только через реостат или понижающий
трансформатор.
Посторонние вещества — асбест, многие окислы, со¬
ли— вызывают при высоких температурах быстрое пе¬
регорание спирали.
Правила работы с тигельными печами по существу-
не отличаются от правил работы с трубчатыми. Темпе¬
ратуру печей необходимо контролировать термопарой
с пирометром.
Обогрев многих приборов можно проводить при по¬
мощи спиралей для электрических плиток. Использовать
их можно различными способами: помещением спирали
внутрь прибора или снаружи. Некоторые возможные
конструкции обогрева приведены на рисунках 69, 70 а, б.
Спираль' от электроплитки при использовании соот¬
ветствующей тепловой изоляции обогреваемого прибора
может повысить в приборе температуру до 700—800 °С,
что не всегда бывает желательно. В этом случае под¬
вод тока нужно осуществлять через реостат или соответ¬
ствующий понижающий трансформатор. При напряже¬
нии в сети 127 в можно также применить спираль на
220 в, с помощью которой можно осуществить нагрев
прибора до 200—300° С. Для повышения температуры
обогрева спираль нужно несколько укоротить.
§ 30. РЕГУЛЯТОРЫ НАПРЯЖЕНИЯ
Приборы, выпускаемые нашей промышленностью,
обычно рассчитаны на 127 в и 220 в. В лабораториях,
как правило, имеется какое-то одно напряжение. Если
соответствующий прибор не допускает переключения на
различное напряжение, его нужно включить через по¬
вышающий или понижающий трансформатор. Таким
трансформатором с плавной регулировкой напряжения
и являются регуляторы напряжения, позволяющие регу¬
лировать напряжение от 0 до 250 в.
280
Регуляторы напряжения во многих электрических
схемах заменяют реостаты. Например, если электриче¬
ская печь рассчитана на 1200 °С, то для получения более
низких температур нужно в цепь последовательно вклю¬
чить реостат. Если же на печь дать напряжение меньше
расчетного, то печь также будет нагрета ниже 1200 °С.
Изменяя напряжение с помощью регулятора напряже¬
ния, можно регулировать температуру электропечи.
Рис. 76. Регулятор напряжения:
1 ручка для регулирования напряжения; 2 — вольт¬
метр; 3 — клеммы для подключения нагрузки; 4 —
переключатель напряжения.
Нашей промышленностью выпускаются два типа ре¬
гуляторов напряжения — РНШ-55 и РНШ-56 (рис. 76).
Они весьма просты в обращении и удобны в работе.
К сети регулятор напряжения присоединяется вывод¬
ным шнуром. Он допускает включение в сеть с напря¬
жением и 127 ей 220 б, причем необходимо переставить
вставку на соответствующее напряжение — на 127 в
(если в сети 127 в) и на 220 в (если в сети 220 б). При¬
бор присоединяется к зажимам с надписью «нагрузка».
Вольтметр показывает напряжение в сети (если пере¬
ключатель поставлен на «сеть»). При положении пере¬
ключателя «нагрузка» вольтметр показывает напряже¬
ние, подаваемое на прибор. На верхней крышке регу¬
лятора напряжения имеется ручка, поворотом которой
можно плавно регулировать напряжение (от 0 до 250 в),
подаваемое на прибор. Включив прибор в сеть, нужно
поставить переключатель на положение «сеть». Стрелка
вольтметра должна показывать напряжение в сети. За¬
281
тем переключатель нужно поставить1 в положение «на¬
грузка». Поворотом ручки, расположенной на верхней
крышке регулятора напряжений, убедиться, что стрелка
вольтметра передвигается от 0 до 250 в. Затем напря¬
жение нужно уменьшить до нуля, отключить регулятор
напряжения от сети и присоединить к зажимам «нагруз¬
ка» соответствующий прибор. После этого подключить
регулятор напряжения снова к сети и поворотом ручки
дать необходимое напряжение, следя за показаниями
вольтметра.
Перегрузка регулятора напряжения выше нормы при¬
водит к его порче. Ниже (табл. 19) приводятся данные
о регуляторах напряжения при включении в сеть с на¬
пряжением 220 в.
Таблица 19
Тип регулятора
Пределы регулирования (в а) при
0—220 в
220—250 в
РНЩ'55
9
8
РНШ-56
2
2
При включении на длительные сроки допустимый то к
снижается на 20%. При включении в сеть с напряжением
127 в снимаемый ток должен быть снижен (табл. 20).
Таблица 20
Тип регулятора
Пределы регулирования (в а) при
до 140 в
140—220 в
РНШ-55
8
6
РНШ-56
2
1,2
Ввиду отсутствия в регуляторе напряжения ампер¬
метра, для измерения снимаемого тока в цепь надо
включить амперметр переменного тока. Нужно следить,
чтобы регулятор напряжения не перегревался. Значи¬
тельный нагрев регулятора напряжения указывает на
то, что с него снимается ток больше допустимого.
282
ГЛАВА XII
КОНТРОЛЬНО-ИЗМЕРИТЕЛЬНЫЕ ПРИБОРЫ
Контрольно-измерительные приборы в настоящее
время весьма широко применяются в промышленности.
Контролируются температура, давление, состав газов и
жидкостей, расход веществ. Показания приборов обычно
используются для автоматического управления произ¬
водством.
Ниже описываются простейшие приборы для изме¬
рения температур и расхода газов, которые могут найти
применение при постановке вышеописанных работ.
§ 31. ПРИБОРЫ ДЛЯ ИЗМЕРЕНИЯ ТЕМПЕРАТУР
При проведении ряда работ по химической техноло¬
гии возникает необходимость в измерении высоких тем¬
ператур. Для этой цели особенно пригодны термопары.
Как известно, при нагревании места спая (контакта)
проводников, различных по составу, в месте спая воз¬
никает термоток (рис. 77). Поместив место спая в го¬
рячую часть прибора и изменив развиваемое напряже¬
те, можно определить и температуру места спая.
Рис. 77. Термопара с пирометрическим гальванометром:
/ — термопара; 2— место спая термопары; 3 — провода; 4 — пирометри¬
ческий гальванометр.
283
Появление в металлических термопарах термотока
объясняется особой внутренней структурой металлов.
В металлах атомы в значительной степени ионизированы
и электроны находятся в хаотическом движении. При
нагревании металла концентрация электронов в его хо¬
лодном конце несколько увеличивается, а в горячем —
падает. Поскольку концентрация электронов в горячих
и холодных концах термопары различная, электроны бу¬
дут перемещаться из одной ветви в другую, где их кон¬
центрация меньше и тепловая энергия будет переходить
в электрическую. Металлические термопары развивают
термоток 0,01—0,1 мв на 1 °С.
Для сравнения термотоков, развиваемых различными
металлами, берут чистую платину. В таблице 21 приво¬
дятся значения термо-э.д.с., развиваемые различными
материалами в паре с платиной.
Таблица 21
С
с
2
Темпе¬
рату¬
ра
(в СС)
Термо-э. д. с. (в милливольтах)
платина-
хромель
платина-
алюмель
платина-
медь
платина-
железо
платнна-
константан
плати¬
на-ман¬
ганин
1
0
0
0
0
0
0
0
2
100
+2,81
—1,29
0,76
+ 1,89
-3,51
+0,61
3
200
+5,96
—2,17
+1,83
+3,54
—7,46
+ 1,55
4
300
+9,32
—2,89
+3,15
+4,85
—11,71
+2,77
5
400
+ 12,75
—3,64
+4,68
+5,88
—16,19
+4,25
6
500
+ 16,21
—4,43
+6,41
+6,79
—20,79
+5,95
7
600
+ 19,62
—5,28
+8,34
+7,80
—25,47
+7,84
8
700
+22,96
—6,18
+ 10,49
+9,12
—30,10
—
9
800
+26,23
—7,08
+12,84
+ 10,86
—34,86
—
10
900
+29,41
—7,95
+ 15,41
+ 12,84
—39,45
—
11
1000
+32,52
—8,79
+ 18,20
+ 14,30
—43,92
—
По таблице можно определить величину термотока,
которая развивается термопарой при температуре хо¬
лодного конца, равной 0. Например, при 400 °С термо¬
пара, составленная из манганина и копстантана при
400°С, разовьет э.д.с. 4,25—(—16,19) =20,44 (мв).
284
Таблица 22
Вещество
Точка
Градусы
шкалы
Кадмий
^Свинец
Калий двухромовокислый
Цинк
Хлорид калия
Хлорид натрия
Сульфат натрия
Сульфат калия
Плавления
Плавления
Плавления
Плавления
Кристаллизации
Кристаллизации
Плавления
Кристаллизации
320,9
327.4
397.5
469,45
770
800,4
884,7
1069,1
Наибольшее практическое значение имеют термо¬
пары, у которых между температурой горячего спая и
термо-э.д.с. имеется прямолинейная зависимость. Это
позволяет градуировать шкалу милливольтметра, соеди¬
ненного с термопарой, в градусах.
В настоящее время пользуются платино-платиноро¬
диевой термопарой, одна из ветвей которой изготовлена
из чистой платины, а другая — из сплава платины с 10%
родия. Этой термопарой можно измерять температуру
до 1600 °С. Платино-платинородиевую термопару нельзя
применять в восстановительной среде, особенно при вы¬
соких температурах, так как платина в этих условиях
может насыщаться кремнием, углеродом и другими ве¬
ществами и приобретает хрупкость.
Распространенным видом термопар являются хро-
мель-алюминиевая и хромель-копелевая. При длитель¬
ном пользовании хромель-алюминиевую термопару мож¬
но применить до 1000 °С; при кратковременном включе¬
нии— до 1250°С. Хромель-копелевой термопарой можно
пользоваться до 600 °С, а при кратковременном включе¬
нии — до 800 °С.
Для определения термо-э.д.с., развиваемой термопа¬
рой, можно применить обычный высокоомный милли¬
вольтметр и по значению напряжения, пользуясь таб¬
личными данными, определить температуру горячего
спая. Более удобны для измерения температуры пиро¬
метрические гальванометры (пирометры) типа МПП-054,
285
которые имеют две шкалы — верхнюю, показывающую
напряжение, и нижнюю — температуру. Их выпускают
трех типов: МПП-054, МПП-154, МПП-254. После под¬
ключения термопары к пирометру необходимо поворо¬
том корректора, имеющегося на лицевой части корпуса,
поставить стрелку на 20 °С.
Правильность показаний термопар проверяется до
350—400 °С при помощи проверенных ртутных термомет¬
ров. Для этого термопару и
ртутный термометр поме¬
щают в фарфоровую труб¬
ку, находящуюся в элек¬
тропечи. Если показания
ртутного термометра и тер¬
мопары не совпадают, то в
показания пирометра нужно
ввести соответствующую по¬
правку. Для проверки до¬
статочно проверить хотя бы
одно из показаний темпера¬
туры, лучше высокую. Для
калибровки термопар мож¬
но также рекомендовать ве¬
щества, данные о которых
приведены в% таблице 21.
В вещество, например
хлорид натрия, помещают
конец термопары и ставят
в электрическую печь. В мо¬
мент плавления или кри¬
сталлизации температура
вещества остается на некоторое время без изменения, что
заметно по показанию пирометра. При использовании со¬
лей температуру определяют, обычно в момент их плав¬
ления. Когда же берутся металлы, то температуру часто
замечают в момент кристаллизации. Необходимо также
следить, чтобы холодные концы термопары не нагрева¬
лись и чтобы место спая термопары находилось в том
месте прибора, температура которого измеряется.
Для измерения и автоматического регулирования
температуры печи весьма удобны электронные регуля¬
торы, например типа ЭРМ-47. Электронный регулятор
включается в цепь нагревательного прибора.
Рис. 78. Реометр:
1 — капиллярная трубка; 2—мано¬
метр; 3 — шкала; 4 — подставка.
286
§ 32. ПРИБОРЫ ДЛЯ ОПРЕДЕЛЕНИЯ РАСХОДА ГАЗОВ
В химической промышленности, где имеют дело с
газами, всегда контролируют количество расходуемого
газа. Для измерения расхода газов применяют реометры
различных типов, газовые часы, ротаметры. Газовые ча¬
сы имеют циферблат, на котором непосредственно отсчи¬
тывается объем газа, прошедший за определенное время.
Рис. 79. Калибрование реометра:
1 — трехходовой кран; 2 — уравнительная склянка; 3 — сосуд — с во¬
дой; 4 — кран; 5 —- газометр; 6 — реометр.
Наиболее просто устроен реометр. Реометры могут
найти применение и при постановке химического практи¬
кума, например при синтезе аммиака.
Реометр (рис. 78) состоит из манометра и капилляр¬
ной трубки, соединяющей колена манометра. При про¬
хождении газа через капилляр вследствие сопротивле¬
ния создается перепад давлений, который измеряется
манометром. Чем больше скорость прохождения газа,
тем больше будет разность давлений. Точность показа¬
ний манометра определяется не только диаметром ка¬
пилляра, но также и плотностью жидкости. Реометр
предварительно необходимо проградуировать (рис. 79).
287
Для этого кран 1 ставят в положение Л, чтобы система
соединялась с атмосферой. Уравнительную склянку 2
ставят в такое положение, чтобы в сосуде 3 было не¬
большое количество воды, и через реометр пропускают
воздух. Избыток воздуха проходит через жидкость в со¬
суде 3. Убедившись в том, что в манометре создалась
определенная разность давлений, кран 1 поворачивают
в положение Б и открывают кран 4. Одновременно заме¬
чают время по секундомеру. Воздух, прошедший через
реометр, поступает в приемник газа 5. О количестве газа
за прошедшее время судят по опусканию уровня жидко¬
сти. Одновременно замечают разность давления в мано¬
метре. Затем при помощи уравнительной склянки ме¬
няют уровень воды в сосуде 3 и снова проводят градуи¬
ровку'реометра.
На основании наблюдений находят объем воздуха,
поступающего в газометр (проходящего через реометр)
в единицу времени. Многократно изменяя скорость воз¬
духа, на шкале реометра получают ряд отметок, кото¬
рые соответствуют определенным значениям скорости
прохождения воздуха.
Из широко распространенных приборов для измере¬
ния скоростей прохождения газов следует отметить ро¬
таметр.
Ротаметр представляет собой расширяющуюся квер¬
ху трубку, в которой имеется поплавок. Газ пропускает¬
ся снизу прибора и поднимает поплавок кверху на опре¬
деленную высоту, отсчитываемую на шкале прибора.
Шкала показывает объемную скорость газа. Правила
пользования ротаметром описаны в инструкции, прила¬
гаемой к прибору.
ПРИЛОЖЕНИЯ
Таблица I
Теплота образования окислов из элементов (при 25° С)
Окисел
АН
(Мдж/кг-экв)
Окисел
AH
(Мдж/кг-экв)
Окисел
AH
{МджЗкг-зкв)
СиО
78,5
Fe203
336,1
Na*0
208,1
СиаО
84,9
W03
136,5
Та2Об
208,7
В12О3
96,1
Fe304
139,4
Si02
217,8
Сг03
97,2
S11O2
144,5
ТЮ*
228,2
С03О4
102,5
Cs20
171,7
B203
243,3
РЬО
109,5
ZnO
174,4
Zr02
270,0
СоО
120,3
Mn304
176,0
AlfcOg
274,2
NiO
122,3
v2o6
128,8
BaO
278,2
М0О3
125,8
Сг2Оэ
190,4
Li20
297,7
CdO
130,5
Nb205
193,7
MgO
305,6
Mn02
131,2
MnO
194,8
CaO
317,6
FeO
135,0
v2o4
196
La20
318,7
Таблица II
Температурные поправки при определении плотности
Плотность
Температурная
поправка иа 1°С
Плотность
Температурная
поправка на 1°С
0,810—0,820
0,000752
0,880—0,890
0,000660
0,820—0,830
0,000738
0,890—0,900
0,000647
0,830—0,840
0,000725
0,900—0,910 х
0,000633
0,840—0,850
0,000712
0,910—0,920
0,000620
0,850—0,860
0,000699
0,920—0,930
0,000607
0,860—0,870
0,000686
0,930—0,940
0,000594
0,870—0,880
0,000673
0,940—0,950
0,000581
289
Поправки на давление и температуру
Температура
(в °С)
Давление
730
735
740
745
750
10
0,986
0,993
1,000
1,007
1,014
11
0,982
0,989
0,996
1,002
1,009
12
0,978
0,984
0,991
0,998
1,005
13
0,973
0,980
0,987
0,993
1,000
14
0,969
0,976
0,983
0,989
0,996
15
0,966
0,972
0,978
0,984
0,992
16
0,965
0,968
0,974
0,980
0,987
17
0,950
0,963
0,969
0,976
0,982
18
0,951
0,958
0,964
0,971
0,978
19
0,946
0,954
0,960
0,966
0,973
20
0,942
0,949
0,955
0,961
0,968
21
0,937
0,944
0,950
0,957
0,964
22
0,932
0,940
0,946
0,953
0,959
23
0,928
0,935
0,941
0,948
0,954
24
0,923
0,930
0,936
0,943
0,949
25
0,918
0,925
0,931
0,937
0,944
26
0,913
0,920
0,926
0,933
0,939
27
0,908
0,915
0,921
0,927
0,934
28
0,903
0,909
0,916
0,922
0,929
29
0,898
0,904
0,911
0,917
0,924
30
0,893
0,899
0,905
0,911
0,918
31
0,877
0,974
0,900
0,906
0,913
32
0,882
0,888
0,895
0,901
0,907
33
0,876
0,883
0,889
0,806
0,902
34
0,871
0,877
0,883
0,890
0,896
35
0,865
0,872
0,878
0,884
0,890
290
Таблица III
при определении углерода
(в мм. pm. cm.)
Температура
(в °С)
755
760
765
770
775
780
1,020
1,027
1,034
1,041
1,048
1,055
10
1,016
1,023
1,030
1,037
1,043
1,050
11
1,012
1,019
1,025
1,032
1,039
1,046
12
1,007
1,014
1,021
1,028
1,034
1,041
13
1,003
1,010
1,016
1,023
1,030
1,037
14
0,998
1,005
1,011
1,018
1,025
1,032
15
0,993
1,000
1,007
1,014
1,021
1,027
16
0,996
0,996
1,002
1,009
1,016
1,022
17
0,985
0,991
0,997
1,004
1,011
1,018
18
0,980
0,986
0,993
1,000
1,007
1,013
19
0,975
0,982
0,988
0,995
1,002
1,008
20
0,971
0,977
0,983
0,990
0,997
1,003
21
0,965
0,972
0,978
0,985
0,992
0,988
22
0,961
0,967
0,973
0,980
0,987
0,993
23
0,956
0,962
0,968
0,975
0,982
0,988
24
0,951
0,957
0,963
0,970
0,977
0,983
25
0,945
0,952
0,958
0,965
0,971
0,978
26
0,940
0,941
0,953
0,960
0,966
0,973
27
0,935
0,942
0,948
0,955
0,961
0,967
28
0,930
0,936
0,943
0,949
0,956
0,962
29
0,924
0,931
0,937
0,944
0,950
0,950
30
0,919
0,926
0,932
0,937
0,945
0,961
31
0,914
0,920
0,926
0,933
0,939
0,045
32
0,908
0,914
0,921
0,927
0,934
0,940
33
0,902
0,908
0,915
0,921
0,928
0,934
34
0,897
0,903
0,909
0,916
0,922
0,928
35
I
291
Таблица IV
Напряжение, развиваемое платино-платинородиевой
термопарой
Температура
(в °С)
Термоэлектродвижу¬
щая сила (в мв)
Температура
(в °С)
Термоэлектродвижу¬
щая сила (в мв)
50
0,301
650
5,728
100
0,640
700
6,251
150
1,014
750
6,782
200
1,421
800
7,323
250
1,861
850
7,871
300
2,311
900
8,429
350
2,773
950
8,995
400
3,244
1000
9,569
450
3,723
1050
10,15
500
4,211
1100
10,74
550
4,709
1150
11,34
600
5,214
1200
11,95
Таблица V
Напряжение, развиваемое термопарой хромель — алюмель
Температура
(в °С)
Термоэлектродвижу¬
щая сила (в мм)
Температура
(в °С)
Термоэлектр одвижу-
щая сила (в мв)
50
2,02
650
27,04
100
4,10
700
29,15
150
6,13
750
31,24
200
8,13
800
33,32
250
10,15
850
35,36
300
12,21
900
37,37
350
14,30
950
39,36
400
16,40
1000
41,32
450
18,51
1050
43,26
500
20,65
1100
45,16
550
22,78
1150
47,03
600
24,91
1200
48,87
292
ОГЛАВЛЕНИЕ
Глава I
Материальный и энергетический балансы технологических процессов
§ 1. Методика составления материального и энергетического
балансов 3
Глава II
Методы обогащения руд
§ 2. Флотационное обогащение . ... 9
Работа 1. Флотационное обогащение медной сульфидной
руды 11
Глава III
Металлы и неметаллы
§ 3. Получение простых веществ восстановлением окислов . 14
Работа 2. Получение металлов и сплавов металлотермиче¬
ским методом 16
Работа 3. Получение металлов действием газообразных
восстановителей. Приготовление изделия методом порош¬
ковой металлургии ........ . . 25
Работа 4. Получение металлов и сплавов из окислов дей¬
ствием углерода . . 34
§ 4. Получение металлов и неметаллов электролитическим ме¬
тодом . ... . 37
Работа 5. Получение никелевого покрытия электролитиче¬
ским методом . 40
Работа 6. Получение хлора и водорода в электролизе ко¬
локольного типа . . 45
Работа 7. Получение хлора и водорода в электролизере
с ртутным катодом ... .51
Работа 8. Получение водорода электролизом раствора
едкого натра . ... 54
§ 5. Технический анализ металлов. Цели и методы техническо¬
го анализа ..... 58
Работа 9. Определение углерода 60
Работа 10. Определение серы 67
Работа 11. Определение кремния 73
Работа 12. Определение фосфора 75
Работа 13. Определение хрома 81
Работа 14. Определение марганца ... 86
Работа 15. Знакомство со стилоскопом СА-10. Построение
градуировочного графика . 90
Работа 16. Определение хрома, никеля и кремния в стали 98
293
Глава IV
Кислоты
§ 6. Получение кислот . 101
Работа 17. Получение соляной кислоты из хлора и во¬
дорода ....... —
Работа 18. Получение серной кислоты контактным спо¬
собом .... 107
Работа 19. Получение серной кислоты нитрозным спо¬
собом ...... 117
Работа 20. Получение аммиака . . 121
Работа 21. Получение азотной кислоты 130
Глава V
Соли и твердые растворы
§ 7. Разделение солей за счет их различной растворимости
Работа 22. Выделение хлорида калия из сильвинита и его
анализ ... .135
§ 8. Получение солей действием кислот на минеральное сырье 138
Работа 23. Получение суперфосфата и его анализ 139
§ 9. Стекло и вяжущие вещества 144
Работа 24. Приготовление легкоплавких стекол 145
Работа 25. Получение вяжущих веществ и изучение их
свойств 150
Глава VI
Топливо и вода
§ 10. Переработка твердого топлива . . 153
Работа 26. Полукоксование твердого топлива и анализ га¬
зообразных продуктов . . —
Работа 27. Сухая перегонка (полукоксование) дерева 159
§ И. Анализ твердого топлива . . 163
Работа 28. Определение влажности твердого топлива —
Работа 29. Определение количества летучих веществ
в топливе . 165
Работа 30. Определение зольности твердого топлива 167
§ 12, Переработка жидкого топлива . 168
Работа 31. Крекинг нефтепродуктов —
§ 13. Анализ нефтепродуктов . 171
Работа 32, Определение плотности нефтепродуктов —
Работа 33. Определение температуры вспышки и воспла¬
менения нефтепродуктов . 178
§ 14. Анализ воды и ее умягчение методом ионного обмена или
известково-содовым методом . 182
Работа 34, Определение жесткости воды . . 183
Работа 35. Умягчение воды- методом ионного обмена или
известково-содовым методом 192
294
Глава VII
Анализ пищевых продуктов
§ 15. Анализ молока ... 197
Работа 36. Определение плотности молока 197
Работа 37. Определение кислотности молока . 198
Работа 38. Определение жирности молока ацидобутиро-
метрическим методом 200
Работа 39. Выделение из молока казеина и молочного
сахара . .... . 201
§ 16. Анализ хлеба и других пищевых продуктов 203
Работа 40. Определение кислотности хлеба —
Работа 41. Определение кислотности муки 204
Глава VIII
Синтез некоторых органических веществ
§ 17. Синтез и качественный анализ высокомолекулярных ве¬
ществ .......... . 205
Работа 42. Получение фенолформальдегидных смол 206
Работа 43. Определение азота, серы, хлора, фтора в по¬
лимерах ...... ... . 209
Работа 44. Определение кремния и фосфора в полимерах 211
Работа 45. Открытие в полимерах функциональных групп 213
§ 18. Физические методы изучения полимеров . . . 217
Работа 46. Определение температуры размягчения —
Работа 47. Определение температуры каплепадения . 218
работа 48. Определение температуры плавления полимера 219
§ 19. Синтез карбоновых кислот .... .... 220
Работа 49. Получение уксусной кислоты синтетическим
способом , , —
Глава IX
Приготовление полупроводниковых материалов и изучение их свойств
§ 20. Выпрямительные свойства полупроводников .... 223
Работа 50. Получение селенида (или сульфида) меди
и изучение его вентильных свойств ... —
§ 21. Электрическое сопротивление полупроводников 226
Работа 51. Приготовление окислов в спеченном состоянии
для физико-химических исследований —
Работа 52. Определение удельного электрического сопро¬
тивления полупроводников и установление зависимости
сопротивления от температуры , 229
Глава X
Методы испытания и защиты материалов
§ 22. Диаграмма состояния железо-углерод ...» 232
Работа 53. Упражнения по диаграмме состояния железо-
углерод ....... . . . —
Работа 54. Микроскопическое изучение сплавов железа
с углеродом и сопоставление полученных данных с диа¬
граммой состояния в . 235
295
§ 23. Определение коррозийной стойкости металлов в атмос¬
ферных условиях н методы их защиты . . 238
Работа 55. Определение коррозионной стойкости металлов 240
Работа 56. Изучение защитных свойств ингибиторов ат¬
мосферной коррозии 243
§ 24. Пассивация н оксидирование металлов 245
Работа 57. Пассивация цинка н стали в хроматных рас¬
творах * . . . . 248
Работа 58. Пассивация стали в ннтрнтиых растворах . 250
Работа 59. Оксидирование черных металлов , 251
§ 25. Определение коррозионной стойкости металлов в ней¬
тральных средах и методы их защиты . . 253
Работа 60. Определение коррозионной стойкости стали
в нейтральных средах (3-процентный раствор хлорида
натрия) . . . 254
Работа 61. Определение защитных свойств ингибиторов в
нейтральных средах (3-процентный раствор хлорида нат¬
рия) по отношению к стали . . . 256
§ 26. Определение коррозионной стойкости металлов в кислых
средах и методы нх защиты . . 258
Работа 62. Определение коррозионной стойкости метал¬
лов в кислых средах 260
Работа 63. Определение защитных свойств ингибиторов
кислотной коррозии 261
Глава XI
Электрические приборы
§ 27 Выпрямители , 265
§ 28. Аккумуляторы , . , 271
§ 29. Электронагревательные приборы 277
§ 30. Регуляторы напряжения 280
Глава XII
Контрольно-измерительные приборы
§ 31. Приборы для измерения температур . 283
§ 32. Приборы для определения расхода газов 287
Приложения s • • * , . 289
Николай Григорьевич Ключников
ПРАКТИЧЕСКИЕ ЗАНЯТИЯ ПО ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
Редактор О. П. Федорович. Художественный редактор Г. А. Жегин. Техниче¬
ский редактор М. И, Смирнова. Корректор Т. А, Кузнецова.
Сдано в набор 12/1V 1971 г. Подписано к печати 6/Ш 1972 г. 84XI08Vs2. Ти¬
пографская № 2. Печ. л. 9,25. Условн. печ. л. 15,54. Уч.-изд. Л. 15,02. Тираж
20 тыс. экз. (Пл. 1972 г.—№ 34). А. 07148.
* * *
Издательство «Просвещение» Комитета по печати при Совете Министров
РСФСР. Москва, 3-й проезд Марьиной рощи, 41.
Полиграфкомбинат им. Я. Коласа Государственного комитета Совета
Министров БССР по печати. Минск, Красная, 23. Заказ № 732*
Цена без переплета 42 к., переплет 10 к»
J-)oSLO^<-fzjC/<oC< J?
Ц1
О.
первого взбалтывания всплывает только часть сульфидов,
операцию флотации повторяют еще раз. Необходимый
вакуум на воронке Бухнера создают водоструйным насо¬
сом или насосом Комовского. Общий вид установки для
фильтрования приведен на рисунке 3’.
Отфильтрованные сульфиды вместе с фильтром высу¬
шивают при 80—100 °С, взвешивают и проводят необхо¬
димые расчеты.
Результаты опыта оформляют в виде таблицы.
Оформление результатов работы
Взято руды (в г)
Получено,
концентрата
(в г)
Выход
концентрата
(в %)
Степень
извлечения
металла (в %)
Степень
концентрации
(ВО сколько
Раз)
Данные о процентном содержании сульфида в руде,
необходимые для расчета, берут у преподавателя. Содержа¬
ние металла в концентрате определяют расчетным путем,
принимая, что отфильтрованное вещество является чистым
сульфидом.
При отсутствии природной руды можно приготовить искусствен¬
ную смесь, состоящую из сульфида меди (II) или железа и пустой
породы, например кварцевого песка. Для получения сульфида
меди (II) составляют смесь из 63 мае. ч. порошкообразной меди или
меди, нарезанной кусочками размером 1—2 мм (медная проволока
или листовая медь), и 32 мае. ч. растертой серы. Эту смесь помещают
в пробирку и нагревают до расплавления серы. Реакция протекает
с выделением теплоты. Часть серы, ие вступившая в реакцию, выде¬
ляется из реакционной смеси и оседает иа холодных стенках про¬
бирки. После бурной реакции пробирку еще раз нагревают до
200—300 °С в течение 3—4 мин, смесь охлаждают и пробирку раз¬
бивают. К полученному продукту после отделения стекла добавля¬
ют 12—15 мае. ч. серы. Смесь растирают в ступке и нагревают в
пробирке при указанной температуре в течение 5—10 мин. Затем
пробирку разбивают. Сульфид меди (II) еще раз растирают в ступ¬
ке до тончайшего порошка, просеивают и смешивают с чистым пес¬
ком (90—95% от смеси).
Для флотации удобно использовать воздуходувку,
к которой через резиновый шланг присоединяется стеклян¬
ная трубка. Ее конец запаивают и раздувают в виде не¬
большого шара, в котором делают несколько мелких от¬
14
верстий. При выходе во флотируемую смесь воздух разби¬
вается на мелкие пузырьки и увлекает частички руды.
Вопросы и задачи
1. Что такое флотореагенты, как они классифицируются и
какое назначение имеет каждая группа?
2. Для чего при флотации через пульпу продувают воздух?
3. Какие флотореагенты используют для флотации сульфид¬
ных руд, оксидов металлов и нерудных ископаемых?
4. Капля воды на поверхности сульфидной руды ие растека¬
ется. Что произойдет, если в эту каплю добавить некоторое количе¬
ство депрессора? Объясните наблюдаемое явление.
5. Как проводится флотация полиметаллических руд, на¬
пример свинцово-цинковых и медно-свинцово-цинково-пиритных?
6. При обогащении 10 т медной сульфидной руды, содержащей
1,5% меди, получено 400 кг концентрата, содержащего 30% меди.
Определите степень извлечения меди и степень ее концентрации.
Ответ. Степень извлечения меди —80%, степень концентра¬
ции в 20 раз.
7. При флотации 5 т цинковой руды, содержащей 3% цинка,
получено 340 кг концентрата, содержащего 22% цинка. Опреде¬
лите выход концентрата, степень извлечения цинка и степень кон¬
центрации.
Ответ. Выход концентрата — 6,8%, степень извлечения цин¬
ка — 48%, степень концентрации — 7,3 раза.
Работа 2
Флотация сильвинита
Сильвинит представляет собой смесь хлоридов натрия
и калия. Помимо разделения этих веществ на основании
их различной растворимости используют также флота¬
ционный метод. В качестве флотореагента применяют смесь
аминов жирного ряда, в углеродной цепочке которых име¬
ется 7—12 атомов углерода. Получают их нитрованием
парафина азотной кислотой под давлением с последую¬
щим восстановлением смеси нитросоединений до аминов.
Амины лучше адсорбируются на ионе калия и поэто¬
му флотируется хлорид калия, а хлорид натрия идет в
хвосты.
Цель работы. Провести флотацию сильвинита
и определить выход концентрата, степень извлечения хло¬
рида калия и степень его концентрации.
Оборудование и материалы: 1. Сосуды
для флотации — колба и цилиндр на 150—200 мл с пробкой.
2. Флотореагент (амины жирного ряда). 3. Сильвинит.
4. Металлическая или фарфоровая ступка. 5. Сито с отвер¬
15
стиями 0,1—0,05 мм. 6. Технохимические весы. 7. Воронка
Бухнера с отсосом. 8. Сушильный шкаф. 9. Термометр.
Проведение работы
Приготовляют так называемый щелок. Для этого в колбу
наливают 100 мл воды, присыпают 15—20 г сильвинита
(или искусственно приготовленную смесь) и готовят при
сильном взбалтывании насыщенный раствор солей. Затем
раствор фильтруют. При 25 °С в 100 г воды растворяется
3,56 г хлорида натрия и 2,69 г хлорида калия.
Раствор переливают в цилиндр, всыпают в него 1,5=-2 г
тщательно измельченного сильвинита и 1—2 капли флото-
реагенга и проводят флотацию, т. е. пропускают во взвесь
сильную струю воздуха. Во время флотации в раствор
нужно опустить термометр и следить за постоянством тем¬
пературы.
Всплывший хлорид калия вместе с частью раствора сли¬
вают на бумажный фильтр, который предварительно взве¬
шивают, и затем фильтр с хлоридом калия высушивают и
взвешивают.
Рассчитывают выход концентрата.
Для определения степени извлечения и степени кон¬
центрации необходимо провести анализ полученного порош¬
ка хлорида калия, как это описано в работе 23
Оформление результатов работы
Описать методику проведения работы и полученные дан¬
ные оформить в виде таблицы.
Взято сильвинита
(в Г)
Получено
хлорида калия
(в г)
Выход
концентрата
<» %>
Степень
извлечении
(в %)
Степень
концентрацни
(в %)
Примечание. После приготовления щелока до
фильтрования осадка хлорида калия температура раствора
должна оставаться неизменной. Лучше раствор сильвинита
готовить заранее, чтобы он принял температуру окружаю¬
щего воздуха. При некотором повышении температуры
раствора во время флотации навеска сильвинита будет
растворяться, а при понижении температуры будут выпа¬
дать хлориды и результаты будут завышенными.
16
Г Л А В A III
МЕТАЛЛЫ И НЕМЕТАЛЛЫ
§ 4. ПОЛУЧЕНИЕ ПРОСТЫХ ВЕЩЕСТВ
ВОССТАНОВЛЕНИЕМ ОКСИДОВ
Оксидные и сульфидные руды — сырье для получения
металлов и некоторых неметаллов. Сульфидные руды перед
выделением металла переводят в оксидные. Для этого из¬
мельченную руду обжигают в присутствии кислорода юз-
духа, например:
2ZnS + 302 = 2ZnO + 2SOa
При выделении простых веществ из оксидов учитывают
физические и химические свойства исходных и получаемых
веществ. Выбор восстановителя зависит от прочности окси¬
дов, которая определяется значением их изобарно-изотер¬
мических потенциалов. В. первом приближении прочность
оксидов определяется их теплотами образования, рассчи¬
танными на 1 г-экв (табл. 5).
Наиболее прочными оксидами являются оксиды под¬
группы скандия, магния и щелочноземельных металлов.
Элементы, проявляющие разную степень окисления, обра¬
зуют несколько оксидов. Наиболее прочные из них — окси¬
ды, в состав, которых входят элементы в низшей степени
окисления. Поэтому возможность получения металла или
неметалла из оксида определяется прочностью низшего
оксида. Например, оксид хрома (VI) находится в начале
приведенного ряда прочностей и должен легко восстанав¬
ливаться, что и наблюдается на практике (табл. 5). Однако
в результате восстановления получается очень прочный
17
7. В фарфоровую трубку поместили две лодочки, одна из
которых была наполнена порошкообразным железом, а другая —
оксидом свинца (II). В трубку поместили кусочек угля в количе¬
стве, недостаточном для восстановления оксидов; трубку плотно
закрыли и нагрели до 800—900 °С. Через 2—3 ч железо окислилось,
а оксид свинца (II) восстановился до свинца. Как можно объяс¬
нить наблюдаемое явление? Какое участие принял в реакциях уг¬
лерод?
8. За счет каких процессов в мартеновской печи и конверторе
происходит уменьшение содержания углерода в чугуне?
9. Почему при увеличении давления газа, подаваемого в до¬
менную печь, ускоряется плавка?
10. Объясните, как и почему влияет подача воздуха, обога¬
щенного кислородом, в доменную печь: а) на скорость плавки;
б) на температуру в доменной печи; в) на качество чугуна; г) на
расход топлива; д) на экономичность процесса.
11. Почему при конверторном методе получения стали коли¬
чество фосфора в ней уменьшается при наличии основной футеров¬
ки, в состав которой входят оксиды кальция и магния? Почему при
наличии кислой футеровки (Si02) количество фосфора практически
не уменьшается?
12. Передельный чугун, перерабатываемый на сталь в конвер¬
торе с использованием воздуха, содержит повышенное количество
кремния, марганца, углерода. Почему использование в бессемеров¬
ском процессе воздуха, обогащенного кислородом, дало возможность
перерабатывать на сталь чугун любого состава?
13. Почему при разливке стали в вакууме качество ее повыша¬
ется?
Работа 6
Получение металлов, сплавов и карбида кальция
электротермическим методом
Электротермический метод в настоящее время применяют
для получения многих сплавов, а для получения карбида
кальция и кремния он является единственным.
Преимуществом электротермического метода является
то, что в реакционной зоне можно быстро создать высокую
температуру, которая особенно нужна для восстановления
таких прочных оксидов, как оксиды кальция, бора, крем¬
ния, хрома и Т: д.
Восстановителем оксидов является измельченный кокс,
который одновременно служит и проводником электриче¬
ского тока.
Цель работы. Получить электротермическим ме¬
тодом один из металлов (железо, кобальт, никель, олово,
медь), или сплав (феррохром, феррокремний, ферротитан
и т. д.), или карбид кальция.
38
Задание на получение со¬
ответствующего вещества
студент получает от препода¬
вателя.
Оборудование и
материалы: 1. Установ¬
ка для проведения электро¬
термического восстановле¬
ния. 2. Источник перемен¬
ного тока на 14—15А и 30—
40 В. 3. Оксиды металлов
(Fe203, Fe304, Со304, N40»
Sn02, CuO, Ti02 и др.). 4. Из¬
мельченный кокс или дре¬
весный уголь с размерами
крупинок 0,5—1 мм. 5. Ам¬
перметр, вольтметр.
Проведение работы
Установку для электро¬
термического восстановления
собирают из двух кирпичей
(рис. 10). Для этой цели
лучше использовать силикат¬
ные кирпичи, поскольку
они плохо проводят тепло.
В нижнем кирпиче для
графитовых электродов вы¬
далбливают два отверстия
(канала). В средней части
кирпичей выдалбливают по¬
лость яйцеобразной формы.
В нее перед опытом засыпа¬
ют тонким слоем небольшое
Количество оксида магния,
Который является своеобраз¬
ной футеровкой.
Электрическая схема со¬
стоит из латра, присоединен¬
ного к сети, вольтметра и ам¬
перметра (рис. 11). Амперметр
и вольтметр монтируются
2 J 7
Рис. 10. Печь для электро¬
термического восстановления:
/ — графитовые электроды; 2 —
шихта; 3 — силикатные кирпичи.
5
Рнс. 11. Электрическая схе¬
ма для электротермического
восстановления оксидов:
1 — графитовые электроды; 2 —
силикатные кирпичи; 3 — вольтметр;
4 — амперметр; 5 — источник тока.
Рис. 12. Крепление токопро¬
водящих проводов на элек¬
тродах:
/ — графитовый электрод; 2 — мед¬
ное покрытие; 3— медная муфта;
4 — клеммы (болты); 5 — медный
привод.
39
на плите из текстолита или другого подобного материа¬
ла. Можно латр присоединить и непосредственно к элект¬
родам, без электроизмерительных приборов. В этом слу¬
чае предварительными опытами устанавливают, при каком
положении ручки латра создается ток, необходимый для
проведения процесса.
Необходимо обратить внимание на крепление проводов
с электродами. Их концы предварительно омедняют элект¬
ролитически в растворе медного купороса. Затем на элект¬
род надевают медную или латунную муфту с креплением
для токопроводящего провода (рис. 12). Место прикрепле¬
ния провода обвязывают шнуровым или в крайнем случае
листовым асбестом с жидким стеклом.
Можно также конец электрода обмотать мягким медным
проводом и для закрепления провести его пайку.
На технохимических весах отвешивают 8—10 г соответ¬
ствующего оксида, а для получения сплава металлов —
смесь оксидов и рассчитывают необходимое количество
кокса или угля. А восстановитель отвешивают, предполагая,
что он окисляется до оксида углерода (II). Его следует
брать в небольшом избытке (около 5%).
Для проведения реакции электроды сближают почти
до соприкосновения и шпателем засыпают реакционную
смесь. Левый электрод нужно несколько сдвинуть влево.
Затем включают ток, поворачивая переключатель латра до
создания напряжения около 40 В, и приближают правый
электрод до соприкосновения с другим электродом и возник¬
новения электрической дуги. Через 1—1,5 с этот электрод
постепенно отодвигают примерно на 2—2,5 см. В это время
и происходит восстановление и плавление продуктов реак¬
ции. Весь процесс длится 1—1,5 мин. Затем электрический
ток выключают и после охлаждения отделяют полученный
металл, сплав или карбид кальция от шлака осторожным
постукиванием молотком на металлической плите.
Оформление результатов работы
Записать проведение работы и заполнить таблицу.
Взято (8 г)
Получено
металла
или
сплава
(в г)
оксидов
угля или
кекса
Теоретически
Должно полу¬
читься
(в г)
Практический
выход
(в %)
40
Вопросы и задачи
1. На что затрачивается электроэнергия при проведении
электротермических процессов, в частности при получении карбида
кальция? Перечислите шесть позиций.
2. Какой вклад в тепловой баланс вносит реакция восстановле¬
ния оксидов при электротермическом получении металлов и карби¬
дов?
3. Согласно ГОСТу 460—46 1 кг технического карбида каль¬
ция первого сорта дает 250—280 л ацетилена (при 20ЛС и 101,33 кПа),
а второго сорта 230—260 л. Определите процентное содержание
(минимальное и максимальное) чистого карбида кальция в техни¬
ческом продукте.
4. При электротермическом получении цинка углерод окисля¬
ется главным образом до оксида углерода (II). Если же восстановле¬
нию подвергнут оксид меди (II), то в основном получается оксид
углерода (IV). Чем это можно объяснить?
5. При электрохимическом восстановлении оксида кремния
(IV) получается в основном карбид кремния SiC, а не кремний.
Чем это можно объяснить?
6. Чистый ацетилен не имеет запаха. Почему ацетилен, полу¬
чаемый из технического карбида кальция, имеет запах? Какие при¬
меси в нем присутствуют?
Работа 7
Восстановление оксидов некоторых металлов
в «кипящем слое»
Проведение реакции в «кипящем слое» было предложено
еще Д. И. Менделеевым.
Сущность метода заключается в том, что реагирующий
газ или жидкость проходит через мелко раздробленное
твердое вещество, помещенное в специальный реактор на
решетку, диаметр отверстий которой несколько меньше
крупинок вещества. Газ, проходя снизу через слой веще¬
ства, разрыхляет и перемешивает его. Насыпной объем
вещества увеличивается и создается впечатление кипящей
жидкости. Благодаря интенсивному перемешиванию возни¬
кает соприкосновение газа с твердым веществом, увеличи¬
вается производительность аппаратуры.
Реакции в кипящем слое обеспечивают также непрерыв¬
ность процесса: в реактор непрерывно подается реагирующая
смесь и отводятся продукты реакции. При заданном режиме
работы необходимо соблюдение определенного, постоянного
теплового и материального балансов, это обеспечивается:
1) размерами частиц, которые должны быть определенной
толщины помола; 2) определенной скоростью подачи частиц
41
в реактор; 3) определенной скоростью подачи в реактор
газа или жидкости;
4) постоянной скоростью отвода теплоты, если реакция
протекает с выделением теплоты;
5) регулируемой скоростью отвода продуктов реакции.
Во время прохождения газа или жидкости высота кипя¬
щего слоя увеличивается и зависит от гидравлического
сопротивления слоя и от фиктивной скорости газа.
При восстановлении оксидов в неподвижном слое ме¬
таллы, особенно медь, часто получаются в спекшемся со¬
стоянии, в виде губки и непригодны для изготовления изде¬
лий методом порошковой металлургии. Поэтому их полу¬
чают во вращающихся печах или в кипящем слое.
Цель работы. Восстановить оксид меди (II) в
кипящем слое.
Вариант 1
Оборудование и материалы: 1. Уста¬
новка для проведения восстановления оксидов в кипящем
слое. 2. Источник водорода
(аппарат Киппа, баллон с во¬
дородом) или конвертирован¬
ный природный газ (см. ра¬
боту 33). 3. Порошкообразный
просеянный оксид меди (II),
железа или кобальт.
Проведение работы
Работа проводится в ла¬
бораторной установке (рис.
13), основной частью которой
является кварцевая трубка 1
диаметром 10—12 мм и дли¬
ной 120—150 мм. В край¬
нем случае можно восполь-
г,,";:: зеваться тру»™# из туго-
щем слое: плавкого стекла. В среднюю
/—кварцевая трубка; 2—сетка; ЧВСТЬ ТрубКИ ПОМещаЮТ СТаЛЬ-
3 -трубка, поддерживающая сетку; НуЮ ИЛи В Крайнем СЛуЧае
4 — нихромовая проволока; 5 — мед- J 1 J
ные токопроводящие провода; 6— ЛЗТуНИуЮ СвТКу С МеЛЬЧЗИ-
пш°Ра°ншгок оксида; 7-резиновый шими отверстиями 2. Для ее
П .. . 10 Л Т. ЛП П ЛЛ
42
укрепления можно использовать различные методы. В пер¬
вом случае на трубке делают незначительную перетяжку,
на которую опираются края сетки. Во втором случае в
трубку плотно вставляют другую трубку 3 меньшего диа¬
метра, которая служит опорой для сетки. Можно в ниж¬
нюю часть трубки вставить стальную спираль (пружинку),
а поверх ее — сетку.
Для обогрева порошка оксида в трубку вставляют кусок
нихромовой проволоки 4 от плитки, соединенной с медными
проволоками 5, которые подключены к понижающему тран¬
сформатору или латру. В реактор насыпают просеянный
оксид меди (II) слоем в 15—20 мм, который не должен про¬
сыпаться через отверстия сетки. Затем через прибор для
вытеснения воздуха пропускают 1—1,5 л водорода или
конвертированного газа и включают электрический ток
от понижающего трансформатора или от латра (5—б В).
Силу тока нужно заранее отрегулировать так, чтобы нижняя
часть нихромового обогревателя нагрелась до 650—700 °С
(ярко-красное каление). Газ-восстановитель пропускают
до тех пор, пока не изменится черный цвет оксида меди (II)
в розовый, характерный для меди. Необходимо отметить,
что оксид меди (I) также розового цвета. Поэтому после
появления розовой окраски восстановитель нужно пропу¬
скать еще в течение 2—3 мин.
При слабом токе восстановителя наблюдается только
некоторое перемещение частиц оксида относительно друг
друга. Для создания сильного тока водорода нужно иметь
свежезаряженный аппарат Киппа, по возможности больших
размеров. Только в этом случае создается впечатление
кипящего слоя. Необходимо иметь оксид меди (II) доста¬
точно тонкого помола, который необходимо заготовить
в достаточном количестве и хранить в отдельной банке.
Оксид меди (II) с достаточной скоростью восстанавлива¬
ется водородом уже при 350—400 °С. Оксиды железа, ко¬
бальта и никеля — при более высоких температурах —
при 500—550 °С.
Они менее удобны для работы, так как труднее устано¬
вить окончание реакции восстановления, поскольку цвет
оксида и порошкообразного металла отличается незначи¬
тельно.
Прибор можно видоизменить, намотав нихромовую
проволоку на верхнюю часть реактора и изолировав ее
асбестом. Такой прибор будет иметь наружный обогрев.
43
При использовании для восстановления конвертирован¬
ного газа (работа 33) работу проводят под тягой, так как
в нем может содержаться некоторое количество оксида угле¬
рода (II).
Перед работой необходима предварительная отладка
установки: использование порошка оксида меди (II) с
постоянной тониной помола, подбор постоянной темпера¬
туры обогрева (постоянное, установленное напряжение),
определение скорости тока восстановителя и известное
время прохождения реакции.
В описанной установке можно получать также некоторые
оксиды с низшей степенью окисления металла, например
оксид марганца (II). Для этого в качестве исходного веще¬
ства используют порошок оксида марганца (IV). Работу
проводят, как описано выше. Об окончании реакции судят
по изменению цвета — оксид марганца (II) серо-зеленова¬
того цвета. Восстановление проходит легко уже при 500—
550 °С.
Оформление результатов работы
Зарисовать установку по получению металлов в «кипя¬
щем слое». Указать преимущества и недостатки проведения
реакций в «кипящем слое».
Вариант 2
При работе по первому варианту, при недостаточной
скорости прохождения газа-восстановителя, кипящий слой
не получается вследствие значительной удельной плотности
оксида меди (II). В этом случае оксид меди (II) можно на¬
нести на измельченный силикагель или волокнистый асбест
в виде мелко дисперсного порошка. При восстановлении
получающаяся медь покрывает тонким розовым слоем
носитель.
Цель работы. Восстановить оксид меди (II), на¬
несенный на порошкообразный носитель — асбест или сили¬
кагель.
Оборудование и материалы: 1. Установ¬
ка для проведения восстановления оксидов в «кипящем
слое». 2. Источник водорода (аппарат Киппа, баллон с во¬
дородом, конвертированный природный газ). 3. Порошко¬
образный силикагель или асбест с нанесенным оксидом ме¬
ди (II).
44
Проведение работы
Для измельчения силикагеля или асбеста их растирают
в ступке, просеивают и отбирают фракцию с размером ча¬
стиц 0,5—0,2 мм. В установке для проведения восстанов¬
ления проверяют, насколько отобранные частицы удержи¬
ваются в «кипящем слое». При этом высота слоя носителя
при пропускании газа должна увеличиваться в объеме
в 1,5—2 раза, а частицы не должны уноситься.
Для нанесения оксида меди (II) порошок носителя (си¬
ликагель, асбест) смачивают 5—7%-ным раствором нитрата
меди (II), прокаливают при температуре около 500 °С до
появления черно-серого цвета поверхности асбеста или
силикагеля. После этого порошок снова просеивают и по¬
мещают в реактор высотой 5—6 см. Пропустив некоторое
количество восстановителя для вытеснения воздуха, включа¬
ют обогрев, пропуская восстановитель. Обогрев выключа¬
ют после появления розового налета меди на носителе. За¬
тем, после охлаждения продукта реакции, прекращают
ток восстановителя.
Оформление результатов работы
Зарисовать установку и описать работу по восстановле¬
нию оксидов в «кипящем слое».
§ 5. ПОЛУЧЕНИЕ МЕТАЛЛОВ И НЕМЕТАЛЛОВ
ЭЛЕКТРОЛИТИЧЕСКИМ МЕТОДОМ
Электролиз растворов и расплавов применяется для по¬
лучения веществ и различных защитных и декоративных
покрытий. Во многих случаях это почти единственный ме¬
тод выделения некоторых металлов, например алюминия,
натрия, калия, ниобия, тантала из их соединений.
Электролиз позволяет: 1) получить металл или неметалл
в чистом виде;
2) выделить металл или неметалл из смеси веществ слож¬
ного химического состава;
3) получить металлы в виде порошка, чешуек и т. д.
По первому закону Фарадея количество веществ, вы¬
деляющихся на электродах, прямо пропорционально ко¬
личеству электричества, прошедшего через электролит,
т. е. пропорционально силе и времени прохождения элект¬
рического тока.
45
2. Посмотрите положение хрома в электрохимическом ряду
напряжений металлов. Почему хром весьма устойчив во влажной,
агрессивной атмосфере и в нейтральных растворах электролитов?
3. Почему соляная кислота активно взаимодействует с хромом,
а азотная практически с ним не взаимодействует?
4. Для определения наличия пор в хромовом покрытии на
него накладывают бумагу, смоченную растворами хлорида натрия
и гексациано-(Ш) феррата калия. На чем основано применение
этого раствора?
Работа 10
Получение никелевых и других покрытий
химическим способом
Помимо электролитических имеются также химические
способы нанесения металлических покрытий, осуществляе¬
мые без наложения электрического тока. Восстановление
ионов металлов осуществляется обычно таким активным
восстановителем, как гипофосфит натрия NaH2P02. Иногда
для восстановления применяют и более слабые восстано¬
вители, например формальдегид, гидразин и т. д. Реакция
восстановления очень сложная: наряду с выделением метал¬
ла идет образование водорода, фосфора и других продуктов.
В упрощенном виде восстановление никеля или другого
металла можно представить в виде следующего ионного
уравнения:
Ni+2 + 2Н2РОГ + 2Н20 = Ni + 2Н2РОГ + 4Н+
Восстановлению данным способом могут подвергаться
ионы никеля, кобальта, меди, серебра, олова, свинца, пла¬
тиновых металлов и т. д. Возможно также одновременное
осаждение двух или трех металлов.
Реакции осуществляются лучше, если поверхность по¬
крываемого металла активирована следами солей палла¬
дия. Для этого изделия предварительно опускают в сильно
разбавленный раствор солей палладия.
Восстановление хорошо проходит при определенных
значениях pH и в присутствии комплексообразователей.
Таким образом, растворы для получения металлических
покрытий должны содержать следующие компоненты:
1. Соли металлов, из которых образуется покрытие.
2. Восстановитель.
3. Комплексообразователи (комплектны, органические
кислоты, например лимонная, различные оксикислоты
и т. д.).
54
4. Буферные добавки, поддерживающие значение pH в
определенных пределах.
Получаемые никелеаые покрытия содержат 6—10%
фосфора, и поэтому они тверже электролитических покры¬
тий. Эти покрытия не имеют пор и, следовательно, более
коррозионно стойкие.
Химический метод никелирования в промышленности
особенно часто применяют для покрытия мелких деталей
сложной формы.
Цель работы. Получение никелевого покрытия
на стали, меди и ее сплавах и изучение некоторых свойств
покрытий.
Оборудование и материалы: 1. Медные
и стальные пластинки с отверстиями и стальными подвес¬
ками. 2. Стакан на 150—200 мл, термометр на 150 °С. 3. Ре¬
активы: гипофосфит натрия, сода кальцинированная, ди-
.хромат калия или натрия, серная, соляная и азотная кис¬
лоты, силикат натрия, гидроксид натрия (калия), эфир,
ацетат и цитрат натрия, натрий фосфат. Приготовление
растворов описано в конце работы.
Проведение работы
Работа состоит в подготовке образцов, приготовлении
растворов для никелирования, получении никелевого по¬
крытия и изучении некоторых свойств покрытия.
Стальные или медные образцы в виде пластинок пло¬
щадью 15—20 см‘2 шлифуют и полируют, как это описано
в работе 7. Проводят обезжиривание, опуская образцы на
стальных или стеклянных крючках на 5 мин в соответст¬
вующие растворы. Предварительно надо измерить длину,
ширину и толщину образцов. После,этого их промывают
в проточной воде и опускают на 1—2 мин в ванну декапи¬
рования. При этом происходит растворение оксидных пле¬
нок и некоторое выявление микрорельефа поверхности.
Затем образцы промывают проточной водой, опускают в
эфир или спирт, с помощью пинцета снимают с крючков
и для высушивания помещают на фильтровальную бумагу
на 10—15 мин. Сушку лучше вести в сушильном шкафу
при 30—35 °С. С помощью пинцета образцы помещают на
чашку аналитических весов и взвешивают. Для никели¬
рования образцы с помощью пинцета надевают на крючки
и помещают на 60 мин в раствор для никелирования. Мед¬
55
ные образцы перед никелированием опускают в раствор
хлорида палладия PdCl2 для активации.
После нанесения покрытия образцы промывают водой,
опускают в стаканчик с эфиром или спиртом и сушат в
сушильном шкафу на фильтровальной бумаге в течение
5—10 мин при 50—60 °С.
Образцы повторно взвешивают, определяя по увеличе¬
нию массы массу осажденного никеля.
Толщину покрытия в микронах определяют по формуле;
; (т2 — тх) 1000
где т1 и тг — масса образца до и после никелирования;
S — поверхность образца (в см2); р — плотность металла
покрытия (р никеля 8 г/см3).
Для определения адгезии (сцепление) упрощенным мето¬
дом покрытие прорезается бритвой (до основного металла)
в виде сетки. Расстояние между линиями должно составлять
около 1,0 мм. Нарезанные квадратики не должны отслаи¬
ваться, если адгезия покрытия к основному металлу хо¬
рошая.
I. Растворы для обезжиривания (в г/л)
и их рабочая температура
Обезжиривание стальных деталей
А. Сода кальцинированная 20
Дихромат калия 1
Температура 80 °С
Б. Тринатрийфосфат 20
Сода кальцинированная 15
Гидроксид натрия (калия) 10
Температура 80 °С
Обезжиривание медных деталей
Тр и н ат р и йфосфат 30
Сода кальцинированная 20
Силикат натрия 5
И. Растворы для декапирования
Декапирование стальных и медных деталей
А. Серная кислота
Температура
Б. Соляная кислота
Температура
5-10%
20—25 °С
5—10%
20—25 °С
56
Декапирование медных деталей
Азотная кислота (конц.)
Серная кислота (конц.)
Соляная кислота (конц.)
Сажа газовая
100 мл
100 мл
1 мл
1 г
Вода
до 1 л
(Сажу предварительно смешивают с азотной кислотой
и добавляют в травильный раствор.)
Вопросы и задачи
1. Для чего перед нанесением электролитического покрытия
полированную поверхность изделия обычно подвергают кратко¬
временному травлению в кислоте?
2. С целью предохранения от коррозии железо покрывают
одним из следующих металлов: цинком, оловом, .хромом или нике¬
лем. Что будет подвергаться коррозии в первую очередь в каждом
случае, если на покрытие нанести глубокую царапину, затрагиваю¬
щую железо: основной металл или металл покрытия? Дайте объяс¬
нение.
3. В растворе имеются сульфаты серебра, меди, никеля и цинка.
В какой последовательности (теоретически) должны выделяться
металлы на катоде при электролизе этих солей?
4. Почему при электролизе солей цинка на катоде выделяется
цинк, а не водород, несмотря на то что электродный потенциал цинка
более отрицателен по сравнению с водородом?
5. Теоретически напряжение разложения воды во время электро¬
лиза (при 17 °С и давлении 101,3 кПа) равно 1,23 В. Почему практи¬
чески напряжение разложения составляет 1,9—2,5 В?
6. Рассчитайте рабочее напряжение, имеющееся между анодом
и катодом ванны, для производства алюминия, если падение на¬
пряжения складывается из следующих величин: 1) анодная шина —
0,2 В^ 2) переходное сопротивление анодная шина (штырь) —
0,06 В; 3) штыри —0,04 В; 4) контактный штырь (анод) ^-0,1 В;
5) анод — 0,17 В; 6) электролит (металл) — 1,5 В; 7) подовый
блок — 0,095 В; 8) контактный блок (катодный стержень) —
0,005 В; 9) катодные стержни —0,16 В; 10) контактный катодный
стержень (катодная шина) —0,01 В; 11) катодная шина —0,11 В;
12) напряжение разложения глинозема — 1,7 В.
Ответ, 4,15 В.
7. Сколько теоретически должно выделиться алюминия в
электролитической ванне при расходе энергии в количестве
1,84 Дж/ч?
Ответ. 0,336 кг.
8. Сколько за 1 ч выделится алюминия, если выход по току
составляет 86?/о, а сила расходуемого тока — 60 000 А?
Ответ. 17,34 кг.
9. Сколько выделится алюминия за сутки, если выход по току
составляет 87%, а сила расходуемого тока -= 55 000 А?
Ответ. 385,6 кг.
57
готового сульфата ртути (II) его легко можно приготовить
растворением оксида ртути (II) в серной кислоте при на¬
гревании. Для этого нужно взять около 1 г оксида ртути (II)
и 30 мл серной кислоты (разбавленной 1 4).
На технохимических весах отвешивают 1 — 1,5 г карби¬
да кальция и помещают его в сухую колбу 1. Затем нагре¬
вают раствор сульфата ртути (II) до 70 СС, следя за тем¬
пературой по показаниям термометра. Раствор хлорида
натрия приливают по каплям.
Ацетальдегид, получающийся за счет гидратации ацети¬
лена под влиянием катализирующего действия соли ртути,
испаряется и поступает в пробирку с разбавленным раство¬
ром перманганата калия, где происходит его окисление
до уксусной кислоты.
Пары альдегида перед поступлением в пробирку с пер¬
манганатом следует смешать с воздухом, который подается
из газометра через тройник. Для наблюдения за током
воздуха между тройником и газометром включают промы-
валку с водой. Воздух пускают со скоростью один пузырек
в секунду.
Опыт продолжают до прекращения появления пузырьков
газа в промывалке 1. После окончания реакции отсоединяют
пробирку с раствором уксусной кислоты.
Для отделения раствора уксусной кислоты от оксида
марганца (IV) раствор профильтровывают, переливают в
мерную колбу на 100 мл, доливают до метки, перемешивают
и титруют 0,05 н. раствором щелочи.
Выход уксусной кислоты считают, предполагая, что
карбид кальция не имеет примесей. В действительности он
всегда имеет загрязнения, и если из чистого карбида долж¬
но согласно ГОСТу получиться 370 л ацетилена (20 °С,
101,3 Па), то из технического получается 230—280 л.
Потери продукта происходят также и за счет неполноты
гидратации ацетилена и окисления ацетальдегида.
Выход уксусной кислоты рассчитывают как процентное
отношение полученного количества уксусной кислоты к
тому ее количеству, которое должно получиться теорети¬
чески.
Оформление результатов опыта
Зарисовать и описать действие установки по получению
уксусной кислоты. Цифровые данные занести в таблицу.
204
Взято карбида
кальция (в г)
Количество
щелочи на титро¬
вание (в МЛ)
Уксусная кислота (в г)
Выход (в %)
ДОЛЖНО
получиться
получи¬
лось
Вопросы и задачи
1. В техническом карбиде кальция содержалось 80% чистого
карбида. Из 100 г карбида кальция получено 75 кг уксусной кисло¬
ты. Определите выход уксусной кислоты.
Ответ. 87,90%.
2. Как теоретически объяснить, почему соли металлов, прояв¬
ляющих несколько степеней окисления, катализируют процесс
окисления ацетальдегида в уксусную кислоту?
3. Как изменяют энергию активации соли ртути (II) при реак¬
ции гидратации ацетилена?
4. Одним из способов очистки ацетилена от примесей серово¬
дорода и фосфида является его промывка хромовой кислотой. На¬
пишите уравнения'реакций, происходящих при очистке ацетилена.
Р а б q т а 47
Получение карбоновых кислот
окислением парафина
Окисление парафина вследствие инертности углеводо¬
родов протекает с небольшой скоростью, и в промышлен¬
ности его проводят в течение 15—20 ч путем продувки воз¬
духа через расплавленный при ПО—120 °С парафин в при¬
сутствии катализатора — перманганата калия. Реакция
является цепной радикальной реакцией. В присутствии
катализатора происходит отрыв атома водорода от углево¬
дорода и образующийся радикал присоединяет молекулу
кислорода, образуя радикал гидроперекиси, который, всту¬
пая в реакцию с другой молекулой, дает гидроперекись, на¬
пример RC(OOH)CH2R' Ее распад проходит с образовани¬
ем кетонов, вторичных спиртов. Кетоны, окисляясь, дают
кислоты с меньшим числом углеродных атомов в цепи. В
результате этих сложных реакций происходит образование
сложной смеси различных жирных кислот, эфиров, окси-
кислот, альдегидов.
В течение 2—3 ч окисляется только незначительная часть
парафина, но процесс можно ускорить, проводя окисление
205
Рис. 49. Каталитическое окисление парафина:
/ — газометр с воздухом; 2 — трубка; 3 — катализатор; 4 — кусочки парафина;
5 — электропечь; 6 — приемник продуктов окисления; 7 — термопара (или высо¬
котемпературный термометр); 8 — пирометрический гальванометр.
парафина в газовой фазе, при более высоких температурах.
При этом, по-видимому, некоторая часть веществ окис¬
ляется до оксида углерода (IV) и состав продуктов окисле¬
ния еще более усложняется.
Цель работы. Провести окисление парафина в
газовой фазе и дать качественную идентификацию получен¬
ных продуктов.
Проведение работы
Окисление проводится в установке (рис. 49). В кварце¬
вую или стеклянную трубку (реактор) помещают катали¬
затор. Для его приготовления мелкую кирпичную крошку
или силикагель (диаметр крупинок 1,5—2 мм) пропитывают
концентрированным раствором перманганата калия и вы¬
сушивают при 80—100 °С. Трубку заполняют катализато¬
ром без просвета. На расстоянии 10—15 см от входа в
электропечь помещают 8—10 г парафина в виде кусочков.
Нагревают печь до 270—280 °С, в реактор впускают, из газо¬
метра воздух и одновременно нагревают парафин. Парафин
должен поступать постепенно в течение 20—30 мин. Поэто¬
му нагревание нужно вести постепенно, вначале чу часть
парафина, которая расположена ближе к входному отвер¬
206
стию печи. Расплавленный парафин постепенно стекает в
горячую зону, где и происходит его окисление. За это время
в реактор из газометра нужно пропустить 5—б л воздуха.
Для облегчения определения количества воздуха, идущего
на окисление, газометр градуируют, например, деления на¬
носят через каждые 0,5 л. В этом случае каждые 2,5 мин
в газометр нужно вливать примерно 0,5 л воды.
Продукты окисления будут частично собираться в виде
полужидкой массы в отводящем конце трубки. Его немного
подогревают горелкой, чтобы продукты окисления собрались
в приемнике — небольшом стакане. Его предварительно
взвешивают на технохимических весах. Затем по привесу
определяют количество полученного продукта.
Для определения качественного состава продуктов в
колбу приливают примерно равное количество воды и смесь
нагревают до кипения. После охлаждения осторожно сли¬
вают водный слой, в котором содержатся низшие спирты,
альдегиды и кислоты. Наличие кислот определяют с помощью
универсального индикатора, альдегидов — по реакции обра¬
зования серебряного зеркала (с частью раствора). К нераст-
ворившемуся в воде остатку, содержащему неокисленный
парафин, высшие кислоты, спирты, альдегиды, приливают
1 н. раствор щелочи (в количестве 10—15 мл). Раствор
нагревают до кипения, охлаждают и сливают с нерастворив-
шегося остатка. Затем к нему приливают избыток разбав¬
ленной серной кислоты. Выделение из раствора нераство¬
римых в воде жирных кислот указывает на окисление
парафина.
Оформление результатов опыта
Описать проведение опыта по окислению парафина и
качественному определению составных частей полученных
продуктов.
Вопросы и задачи
1. Почему при окислении парафина получается смесь карбоно¬
вых кислот с разной молекулярной массой? Какие продукты еще
могут получаться при окислении парафина?
2. Каким методом в продуктах окисления парафина можно
обнаружить наличие альдегидов?
3. Напишите уравнение термического разложения перманга¬
ната калия и уравнение окисления какого-либо углеводорода пер¬
манганатом.
207