/



















































































































































Author: Мюнстер А.
Tags: термодинамика энергетика физика химия физическая химия учебник по химии издательство мир
Year: 1971




Text
А.МЮНСТЕР
ТЕРМОДИНАМИКА
CHEMSSCHE
THERMODYNAMIK
VON
ARNOLD MUNSTER
Dr. rer. nat., o. Professor und Direktor des Instituts fiir theoretische physikalische Chemie an der Universitat
Frankfurt/Main
Akademie — Verlag
Berlin 1969
А. МЮНСТЕР
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
Перевод с немецкого
канд. хим. наук Е. П. Агеева
Под редакцией
чл.-корр. АН СССР Я. И. Герасимова
Москва, 1971
УДК 536.7
Автор книги — директор института теоретической физической химии при университете во Франкфурте-на-Майне, обладая многолетним опытом преподавания, сумел сравнительно кратко, четко и доступно изложить основы химической термодинамики. Теоретические рассуждения сопровождаются многочисленными примерами, часто в виде схематических диаграмм, что облегчает понимание предмета и способствует приобретению навыков применения термодинамических знаний.
Книга предназначена в качестве учебника термодинамики для студентов и преподавателей химических вузов. Она представляет несомненный интерес и для специалистов данной области.
Редакция литературы по химии
Инд.
2-5-4
88-71
ПРЕДИСЛОВИЕ
Книга А. Мюнстера «Химическая термодинамика» отличается от большей части оригинальных и переводных учебных пособий по химической термодинамике, имеющихся на русском языке. В основу данного курса химической термодинамики положен дедуктивный и математический метод изложения, причем большое внимание уделяется математической структуре термодинамических уравнений и совокупности этих уравнений. Вся формальная структура книги основана на выводе термодинамических уравнений из трех основных положений: фундаментального уравнения, условия равновесия и условия стабильности.
Показано, как формализованная система уравнений при наличии освоенной системы преобразований этих уравнений позволяет легко и надежно применять термодинамические закономерности к любому виду равновесий. Такое применение рассмотрено для ряда проблем: электрохимические системы (растворы электролитов, мембранные равновесия, гальванические ячейки), системы в поле тяготения и в центробежном поле.
Интересно сравнительно подробное изложение аксиоматики второго закона термодинамики, вводящее читателя в логические тонкости и трудности этой проблемы. Анализ теплового закона Нер-нста—Планка дает представление о значении этого закона и его месте в системе термодинамики, несколько изменяющее и дополняющее представления, даваемые в обычных учебниках.
Книгу Мюнстера целесообразно изучать, уже владея химической термодинамикой в объеме небольшого курса физической химии. Изучение этой книги поможет пытливому читателю, имеющему склонность к обобщенной математической трактовке явлений, глубже понять логику, математическую структуру, возможности классической термодинамики и границы ее применимости и с большей легкостью и точностью использовать этот важный и хорошо разработанный метод исследования.
Я. Гер асимов
ПРЕДИСЛОВИЕ АВТОРА
Данная книга создана на основании материала лекций, которые я читал в течение 15 лет в университете во Франкфурте-на-Майне. По замыслу эта книга в концентрированной форме, соответствующей уровню современных требований, должна разъяснить читателю формальную структуру термодинамики и технику ее применения таким образом, чтобы в результате он мог самостоятельно применять теорию. Основная концепция, которая возникла при многолетнем изучении предмета и дидактического опыта, состоит в том, что чисто математически все здание термодинамики можно вывести из трех соотношений: фундаментального уравнения, условия равновесия и условия стабильности. Таким образом эти соотношения играют здесь такую же роль, как и уравнения Максвелла в электродинамике. Несомненно, при таком способе изложения происходит некоторое отступление от наглядности в обычном смысле. Но отказ от наглядности будет щедро возмещен более глубоким пониманием, а также легкостью и надежностью применения теории к конкретным проблемам.
Из основной концепции следует непосредственное построение книги. Прежде всего будет показано, как простые опытные факты приводят к упомянутым трем основным соотношениям (гл. I). Затем они будут объяснены и исследованы с учетом их формальных свойств (гл. II и III). Наконец, они будут применены к ряду общих проблем (гетерогенные равновесия, химические равновесия, критические фазы, электрохимические системы, поле тяготения и центробежное поле). Связанный предусмотренным объемом книги, я вынужден был ввести некоторые ограничения, при которых в первую очередь учитывал интересы химиков и физико-химиков.
Предисловие автора
7
План книги исключает систематическое обсуждение классов веществ (газы, жидкости и т. д.), а также рассмотрение формул для термодинамических функций, обоснованных эмпирически или статистически, и числовые расчеты. Однако я старался многочисленными примерами (часто в виде схематических диаграмм) облегчить понимание и одновременно иметь в виду применение к специальным проблемам. В этой связи с особой тщательностью будет проведено обсуждение некоторых вопросов, которые, по-моему, представляют трудность для начинающих при применении их к теории (например, понятие внутренних параметров, особенно числа пробегов реакций, нормировка термодинамических функций, потенциалы отдельных электродов). С этой точки зрения также подробно рассмотрен тепловой закон Нернста.
fee Книга предполагает знания химии, физической химии и физики примерно в объеме, который имеется в большинстве курсов лекций для начинающих. Из математических знаний необходимы основы дифференциального и интегрального исчислений, особенно уравнения в частных производных для функций многих переменных. Некоторые дальнейшие вспомогательные математические средства кратко рассматриваются в этой книге.
Я благодарен за неоценимую помощь при подготовке рукописи к изданию доктору Е. Люкс и А. Тюпкер. Кроме этого я благодарю издательство «Химия» за хорошую и полную взаимопонимания работу.
А. Мюнстер
Франкфурт-на-Майне, Декабрь, 1968 год
СПИСОК ОСНОВНЫХ ОБОЗНАЧЕНИЙ
А —сродство;
Су — мольная теплоемкость при постоянном объеме;
Ср — мольная теплоемкость при постоянном давлении;
-—►
Е — напряженность электрического поля;
§ —константа Фарадея;
F —свободная энергия Гельмгольца;
G — свободная энергия Гиббса, свободная энтальпия;
Н —энтальпия;
Кр—константа равновесия, выраженная через парциальные давления;
Mi — молекулярный вес компонента Z;
Р — давление;
Pl —интенсивный параметр;
Q —теплота, подведенная к системе;
R — газовая постоянная;
S — энтропия;
Т —абсолютная температура;
U —внутренняя энергия;
V — объем;
W —работа, совершенная над системой;
Xi — экстенсивный параметр;
Yj — рабочий коэффициент, обобщенная сила;
Z —функция состояния;
а1 —активность компонента i;
Ci~ n/V — концентрация
[моль-см-3];
fl — коэффициент активности;
q — осмотический коэффициент (§ 50);
g— ускорение свободного падения (§ 53);
tn — число компонентов;
П[ — число молей;
Pi — парциальное давление;
t — эмпирическая температура;
X/— мольная доля;
У] — рабочая координата;
П — осмотическое давление;
Ф — электродвижущая сила;
Ф/г — функция Массье—Планка, зависящая от k интенсивных параметров;
— термодинамический потенциал, зависящий от k интенсивных параметров; а — термический коэффициент расширения;
х — изотермическая сжимаемость;
х5 — изэнтропийная сжимаемость;
— химический потенциал;
Pl — электрохимический потенциал ионов вида i;
<з — число фаз;
ср — электрический потенциал.
Мольные величины для индивидуальных веществ обозначены^ строчными буквами (например, и, s) или прописными со звездочкой (например, U*, S*),
§ 1. Введение
Термодинамика относится к группе феноменологических теорий физики, т. е. к той же группе, в которую входят гидродинамика и электродинамика. Эти теории имеют следующие общие черты:
а) они не рассматривают атомную структуру материи; б) вследствие этого они используют величины, которые могут быть определены только для макроскопической системы;
в) аксиомы этих теорий основаны на известных макроскопических эмпирических фактах, которые представлены в соответствующей математической форме {уравнения Навъе—Стокса, Максвелла);
г) специфические свойства веществ выражены в форме характеристических параметров (вязкость, диэлектрическая проницаемость).
Применяя обычную терминологию, предмет термодинамики можно предварительно определить как область физических явлений, в которой существенную роль играет теплота и температура. Фактически термодинамика имеет дело только с частью этой области. Она ограничивается рассмотрением состояний равновесия и таких изменений состояний, которые представляют собой непрерывную последовательность равновесных состояний (квазистатиче-ские изменения состояний). Такие изменения состояний, строго говоря, могут происходить только бесконечно медленно. Поэтому их нельзя представить как функцию времени. В некоторых случаях нужно учитывать также существование нестатических изменений состояний, но они не являются собственно предметом данной теории. По этой причине для названия этой теории неоднократно предлагали термин «термостатика», однако он не привился. Раз-
10
Введение
виваемая в последнее время область — термодинамика необратимых процессов — связана именно с термодинамикой, но имеет совсем другую структуру. В этой книге она не рассматривается.
Из сказанного уже ясно, что структура термодинамики существенно отличается от остальных феноменологических теорий, и прежде всего тем, что в термодинамике нет производных по времени и по координатам физического пространства, так как чаще всего термодинамические величины в состоянии равновесия не являются функциями пространственных координат. Системы, рассматриваемые в термодинамике, не обязательно должны быть гомогенными (пример, система жидкость — пар). Пространственное расположение гомогенной области не имеет значения. Ситуация несколько меняется, если учитывать влияние внешних полей (гравитационного, электрического и магнитного) или границ раздела. В конце книги (§53 и § 54) будут коротко рассмотрены эти специальные случаи, но основная структура термодинамики при этом не изменится.
Эту структуру негативно можно охарактеризовать тем, что термодинамика в противоположность гидродинамике и электродинамике не является теорией поля. Ее соотношения можно геометрически представить только в некотором абстрактном пространстве. Отсюда следуют свойства, которые делают термодинамику, с одной стороны, простой, но с другой — все же трудно доступной наукой.
Таким образом, в термодинамике не встречаются типичные дифференциальные уравнения математической физики с частными производными по времени и пространственным координатам. Фактически математический аппарат, кроме некоторых специальных случаев, очень прост. Он ограничивается методами частного дифференцирования и обычными дифференциальными уравнениями простого типа. В противоположность этому основные понятия термодинамики чрезвычайно абстрактны и в этой абстрактности, собственно, и заключена трудность. Долгое время пытались избежать эту трудность за счет обманчивой наглядности рассуждений. Однако оказалось, что этим только затрудняется глубокое понимание предмета. Поэтому надо заранее признать указанную выше характеристику термодинамики и затем проанализировать развитие основных поня
Введение
И
тий из опытных фактов, а также структуру математического аппарата.
Отсюда следует непосредственное построение книги. В первой главе обсуждаются основные понятия, в последующих разрабатывается формализм. Автор рассматривает примеры, показывающие применение выводов термодинамики, но не приводит систематического обсуждения различных классов веществ. Это вполне оправдано, поскольку термодинамические соотношения не зависят от специфических свойств веществ. Для термодинамических расчетов они должны быть всегда взяты из эксперимента. Теоретическим изучением свойств веществ занимается статистическая термодинамика.
Статистическая термодинамика в явной форме исходит из атомной структуры материи, и это облегчает дедуктивное обоснование аксиом, введенных в термодинамику на основании макроскопических опытных фактов.
Исторически начало развития термодинамики связано с изучением коэффициента полезного действия тепловых машин, откуда и происходит само название.
1824. Появилась статья Карно ,,Размышления о движущей силе огня**; в ней был сформулирован принцип, в соответствии с которым производительность тепловой машины не зависит от рабочего вещества, а только от разницы температур.
1834. В последующих заметках Карно четко сформулировал принцип энергии, эквивалентность теплоты и механической энергии.
1834. Клапейрон применил результаты Карно к равновесию жидкость — пар и вывел соотношение, названное позднее уравнением Клаузиуса — Клапейрона*. Соотношение содержало неизвестную функцию температуры, которую вскоре Клаузиус идентифицировал как абсолютную температуру.
1840—1845. Джоуль экспериментально доказал эквивалентность теплоты и механической работы. Результаты были опубликованы в 1845 г.
1842. Роберт Майер сформулировал принцип энергии.
1848. Томсон (лорд Кельвин) на основе работы Карно ввел понятие абсолютной (т. е. независящей от термометрического вещества) температурной шкалы.
1850. Клаузиус опубликовал работу ,,О движущей силе теплоты и законах учения о теплоте, которые отсюда можно вывести**.
* В советской литературе принято называть это уравнение уравне-
нием Клапейрона — Клаузиуса. — Прим. ред.
12
Введение
Работа в основном представляла собой объединение принципа энергии и принципа Карно и содержала предпосылки для формулировки второго закона термодинамики. По мнению Гиббса — крупнейшего термодинамика последующего времени,— эта статья знаменует собой эпоху в истории физики и является началом термодинамики как науки.
1851. Томсон на основе работ Карно, Джоуля и Клаузиуса сформулировал оба основных закона термодинамики.
1854. Клаузиус ввел понятие энтропии и дал новую формулировку второго закона термодинамики.
1865. Клаузиус ввел термин энтропия. Эта работа содержит знаменитую фразу: ,,Энергия мира постоянна. Энтропия мира стремится к максимуму”.
1869. Массье ввел первые характеристические функции, из которых дифференцированием можно получить все термодинамические свойства.
1873. Хорстман впервые рассчитал химические равновесия (закономерности диссоциации СаСО3 и РС15).
1875. Гиббс опубликовал работу ,,О равновесии гетерогенных веществ”, в которой применил общие термодинамические представления к гетерогенным системам и химическим реакциям, вывел из общей формулировки условия равновесия для различных специальных случаев и ввел характеристические функции. Эпиграфом статьи было приведенное выше высказывание Клаузиуса.
1882. Гельмгольц независимо от Гиббса ввел представление о свободной энергии и вывел уравнение, известное под названием уравнение Гиббса—Гельмгольца.
1886. Дюгем вывел уравнение, названное позднее уравнением Гиббса — Дюгема.
1887. Планк разбил изменение состояния на два класса: обратимые и необратимые процессы.
1906. Нернст опубликовал свою новую тепловую теорему.
1909. Каратеодори дал новое аксиоматическое обоснование термодинамики.
В более поздних работах были обсуждены вопросы аксиоматики, усовершенствован формальный аппарат и рассмотрено применение термодинамики к специальным проблемам.
Глава I
Основные законы термодинамики
§ 2. Определения
Так называемые основные законы термодинамики представляют собой аксиомы. Они развивают взятые из известных опытных фактов понятия, которые служат для создания формального аппарата. Однако формулировка основных законов является результатом исторического процесса. С логической точки зрения они не представляют собой полноценной системы аксиом. Следует учесть, что в термодинамике используются также и опытные факты, не содержащиеся в основных законах. При случае это положение будет рассмотрено еще раз.
Начнем с некоторых определений.
Системой называют ограниченную каким-либо образом часть физического мира, которая составляет предмет исследования. В большинстве случаев системой является образец вещества, изучаемый в лаборатории. Но система может иметь и более сложную структуру (тепловая машина, электросеть).
Состояние системы определяется набором опытных данных, обладающих тем свойством, что каждый последующий результат измерения можно из них рассчитать. Разумеется, из этого формального определения на первых порах мало что можно извлечь. Однако опыт показывает, что полный набор параметров можно представить как совокупность вспомогательных наборов, которые, по крайней мере, приблизительно являются самостоятельными. Это значит, что в каждом таком вспомогательном наборе из определенных экспериментальных величин можно рассчитать относящиеся к нему остальные значения. Таким образом, опытные данные, необходимые для расчета остальных
14
Глава I
Основные законы термодинамики
15
величин, определяют состояние во вспомогательном наборе. Это (связанное с некоторой идеализацией) разложение соответствует различным областям теоретической физики. Так, механическое состояние классической системы определяется из точечных масс при помощи обобщенных координат и импульсов (или скоростей), состояние квантовомеханической системы — через волновую функцию, электромагнитное состояние — через электрические и магнитные силы поля в зависимости от координат пространства. Полное разложение можно провести только для идеальной системы (несжимаемая жидкость, вакуум). В известной мере можно учесть связь через подходящие параметры. При дальнейшем развитии различные области сливаются в более общие теории (необратимая термодинамика), которые в большинстве случаев занимаются особыми проблемами.
Термодинамика как феноменологическая теория имеет дело только с макроскопическими величинами. Эти величины либо определимы только для макроскопической системы (точечная масса не имеет температуры), либо, по крайней мере, структура материи не входит в их определения (в этом смысле постоянная решетки кристалла не является макроскопической величиной). Величины, которыми оперирует термодинамика, уже частично определены в механике*, частично в самой термодинамике, в ее основных законах.
Существенное упрощение происходит благодаря тому, что термодинамика ограничивается рассмотрением состояний равновесия (и квазистатических процессов). Так как существование и свойства равновесия тесно связаны со вторым законом термодинамики, то предварительно определим равновесие как состояние, к которому самопроизвольно стремится полностью изолированная от внешнего мира система, или как состояние, в котором термодинамические параметры системы не зависят от времени. Опыт показывает, что число параметров, полностью описывающих равновесное состояние, меньше, чем в любом неравновесном
* Некоторые термодинамические величины определяются в электродинамике. Так как такого рода проблемы в этой книге не рассматриваются, автор ограничивается лишь этим замечанием.
состоянии. Так, равновесное состояние данного количества идеального газа полностью описывается любыми двумя переменными (давление, объем и температура), в то время как для описания неравновесного состояния необходимы еще градиенты температуры и плотности.
В дальнейшем будем называть (термодинамическими) параметрами состояния набор переменных, характеризующий термодинамическое состояние при равновесии. Термодинамическое состояние при равновесии назовем просто (термодинамическим) состоянием и систему, полностью описываемую с этой точки зрения (термодинамической), — системой. Величина, дифференциал которой является полным дифференциалом переменных состояний, называется функцией состояния. Абстрактное пространство, образуемое параметрами состояния, называется пространством состояния. Каждое термодинамическое равновесное состояние системы обратимо и однозначно является точкой в пространстве состояния.
Система называется закрытой, если она может обмениваться с окружающей средой энергией и не может обмениваться веществом (жидкость и ее пар представляют собой открытую систему). Изолированная система не может обмениваться с окружающей средой ни веществом, ни энергией. Тело называется гомогенным, если внутри него в каждой точке соблюдается постоянство температуры, давления, концентрации и остальных макроскопических физических свойств (кристаллической структуры, показателя преломления и т. д.). Следует отметить, что такое определение имеет смысл только с макроскопической точки зрения. Если абстрагироваться от величины и формы тела, то говорят о фазе (пар, жидкость, кристалл). Гомогенная система содержит только одну фазу. Две фазы называются сосуществующими, если они, имея плоскую границу раздела, могут находиться в равновесии между собой, которое не обусловлено лишь «торможениями» (это имеет место, например, если два разных кристалла спрессованы при комнатной температуре). Гетерогенная система состоит из двух или более сосуществующих фаз. Число (независимых) компонентов системы т в смысле термодинамики является одновременно числом видов веществ (или сортов частиц) в смысле химии с минус число уравнений реакций г, их
16
Глава I
связывающих, и возможных уравнений связи Ь. Следовательно, имеем
т = с — г — Ь. (2.1)
Пример. Водный раствор поваренной соли содержит виды веществ Н2О, NaCl, Na+, С1“. Но имеется уравнение реакции
Na+ + Cl“ NaCl (2.2)
и, если обозначить число молей вещества сорта А через па , уравнение связи
%а+=ПС1-* (2’^
В соответствии с уравнением (2.1) система с точки зрения термодинамики имеет два (независимых) компонента (бинарная система). При этом предполагается, что реакции, соответствующие уравнениям типа (2.2), (2.4), при данных условиях действительно протекают.
Пример. Система Н2, О2, Н2О в общем случае является двухкомпонентной, потому что существует реакция
2Н2 + О2 2Н2О. (2.4)
Однако при комнатной температуре и нормальном давлении эта реакция не может протекать даже в присутствии катализатора. Поэтому при этих условиях система будет трехкомпонентной (тройной системой). Приведенные определения важны. Пренебрежение ими ведет к многочисленным недоразумениям и ошибкам.
В дальнейшем ограничимся прежде всего определенным классом систем, которые назовем простыми. Простые системы характеризуются тем, что каждая фаза имеет постоянную массу и состав и что их состояние определяется двумя независимыми переменными. Как будет видно в дальнейн , для определения термодинамического состояния необходимы по крайней мере две переменные состояния, поэтому из рассмотрения исключаются твердые тела, внешние поля, поверхностные явления и т. д. Далее из определения следует, что не должен происходить переход вещества между фазами и в окружающую среду. Следовательно, система в целом и каждая фаза сама по себе являются за
Основные законы термодинамики
17
крытыми. Наконец, этим определением исключается также протекание химических реакций. Этот вопрос будет рассмотрен в разд. В данной главы.
А. КЛАССИЧЕСКАЯ ФОРМУЛИРОВКА ОСНОВНЫХ ЗАКОНОВ ТЕРМОДИНАМИКИ
§ 3. Первый закон. Внутренняя энергия
При классической формулировке основных законов термодинамики понятия температуры и теплоты берутся из непосредственного жизненного опыта и подробно не анализируются. Возможность их измерения предполагается априори. Более подробное обсуждение понятия температуры и теплоты будет дано в разд. Б данной главы.
Эмпирическое обоснование первого закона термодинамики дается опытами Джоуля (1840—1845), который показал, что всегда требуется одна и та же механическая работа, чтобы нагреть определенное количество воды на 1°. Этот результат представляет собой так называемый принцип эквивалентности, который Томсон сформулировал следующим образом: если из термических источников получается или в результате термических эффектов уничтожается одно и то же количество механической работы, то исчезает или возникает одно и то же количество теплоты.
Существует также другая формулировка принципа эквивалентности: невозможно построить машину, которая производила бы механическую работу, не затрачивая при этом эквивалентного количества теплоты (принцип невозможности вечного двигателя первого рода).
Пусть Q будет теплотой, подведенной к системе, a W — произведенной работой. Рассмотрим процесс, в течение которого система за счет (положительного или отрицательного) подвода теплоты AQ и соответственно (положительной или отрицательной) совершенной работы A IF проходит через ряд последовательных стадий и вновь возвращается в исходное состояние (круговой процесс или цикл). Согласно принципу эквивалентности,
2AQ=-£AF. (3.1)
18
Глава I
Если ввести теперь величину
Д[/ = AQ4-AIF, (3.2)
то из (3.1) следует
(j) dU = 0, (3.3)
где интеграл по замкнутому контуру охватывает пространство состояния. Но это значит, что
dU = d'Q + d'W (3.4)
есть полный дифференциал и U является функцией состояния. Выражение (3.4) представляет собой в рамках классической теории математическую формулировку первого закона термодинамики. По предложению Клаузиуса функция U была названа внутренней энергией системы. Как следует из определения, внутреннюю энергию можно вычислить с точностью до константы интегрирования.
Следует помнить, что в общем случае d' Q и d' W не являются полными дифференциалами. Значение выражений 2 2
Jd'Qn ^d'W зависит от пути процесса в пространстве 1 1
состояния, в чем легко можно убедиться на простых примерах.
Для гомогенной простой системы в случае квазистати-ческого процесса работа равна
dW — —PdV, (3.5)
где V — объем, а Р — давление. Тогда выражение для внутренней энергии примет вид
dU^d'Q — PdV. (3.6)
Из сказанного следует, что внутренняя энергия изолированной системы постоянна.
§ 4. Второй закон термодинамики. Энтропия и абсолютная температура
Эмпирическое обоснование второго закона термодинамики сформулировано Клаузиусом следующим образом: невозможен самопроизвольный (т. е. происходящий в изолиро
Основные законы термодинамики
19
ванной системе) переход теплоты от менее нагретого тела к более нагретому.
Формулировка, использованная Томсоном и позднее Планком, гласит: невозможно построить периодически действующую машину, которая бы только охлаждала тепловой резервуар и производила механическую работу (принцип невозможности вечного двигателя второго рода).
Обе формулировки на первый взгляд имеют мало общего, но фактически они эквивалентны.
Доказательство
а. Предположим, что верна формулировка Томсона и неверна Клаузиуса. Тогда из теплового резервуара R за счет какого-нибудь процесса часть теплоты могла бы быть переведена к более высокой температуре и, таким образом, был бы создан второй тепловой резервуар R' с более высокой температурой. Из его запаса часть теплоты могла бы быть превращена в работу, а оставшееся количество приведено к температуре резервуара R. Таким образом R' исчез бы и в результате из запаса теплоты резервуара R была бы получена работа. Этот процесс можно было бы повторять сколь угодно часто, он представлял бы собой вечный двигатель второго рода и противоречил бы формулировке Томсона. Следовательно, если формулировка Томсона верна, то должна быть верна и формулировка Клаузиуса.
б. Предположим, что верна формулировка Клаузиуса и неверна Томсона. Тогда можно было бы построить вечный двигатель второго рода и извлекать из единственного теплового резервуара определенное количество теплоты, которое затем переводить в работу. Полученную работу можно было бы без ограничений вновь превращать в теплоту при более высокой температуре. Таким образом, рассматриваемое количество теплоты переводилось бы без компенсации от более низкой температуры к более высокой, что противоречило бы постулату Клаузиуса. Если он справедлив, то должен быть также верен и постулат Томсона.
Из постулата Томсона следует, что периодически действующая машина, превращающая теплоту в работу (тепловая машина), будет работать только в том случае, если
20
Глава I
рабочее вещество (например, водяной пар) совершает круговой процесс между двумя тепловыми резервуарами, находящимися при различных температурах. Оно забирает от более горячего резервуара количество теплоты (здесь и в дальнейшем имеется в виду абсолютное количество) и отдает более холодному резервуару количество <?2, Раз‘ ность Qt—Q2 превращается в работу. Поэтому коэффициент полезного действия (к. п. д.) кругового процесса можно определить следующим уравнением:
Qi
(4.1)
Из постулата Клаузиуса следует теорема:
коэффициент полезного действия полностью обратимого кругового процесса К не может быть больше коэффициента полезного действия любого другого цикла, который протекает с тем же рабочим телом между теми же температурами.
Доказательство
Предположим, что теорема не верна. Тогда между теми же температурами существует цикл К', для которого выполняются соотношения
W' = W, Q2<Q2- (4.2)
Пусть обе машины работают одновременно и в противоположных направлениях. В соответствии с условиями (4.2) результирующая работа должна бы быть равной нулю, но вследствие того, что Q' i<Qi, дополнительное количество теплоты должно перейти к более нагретому резервуару, что противоречит постулату Клаузиуса.
Аналогичным образом доказывается теорема Карно: коэффициент полезного действия обратимого цикла зависит только от температуры тепловых резервуаров и не зависит от природы рабочего тела.
Теорема Карно, как показал Томсон, может быть использована для определения термодинамической или абсолютной температуры. Согласно (4.1),
-Tj. (4.3)
Основные законы термодинамики
21
Из теоремы Карно следует
= tj,
41
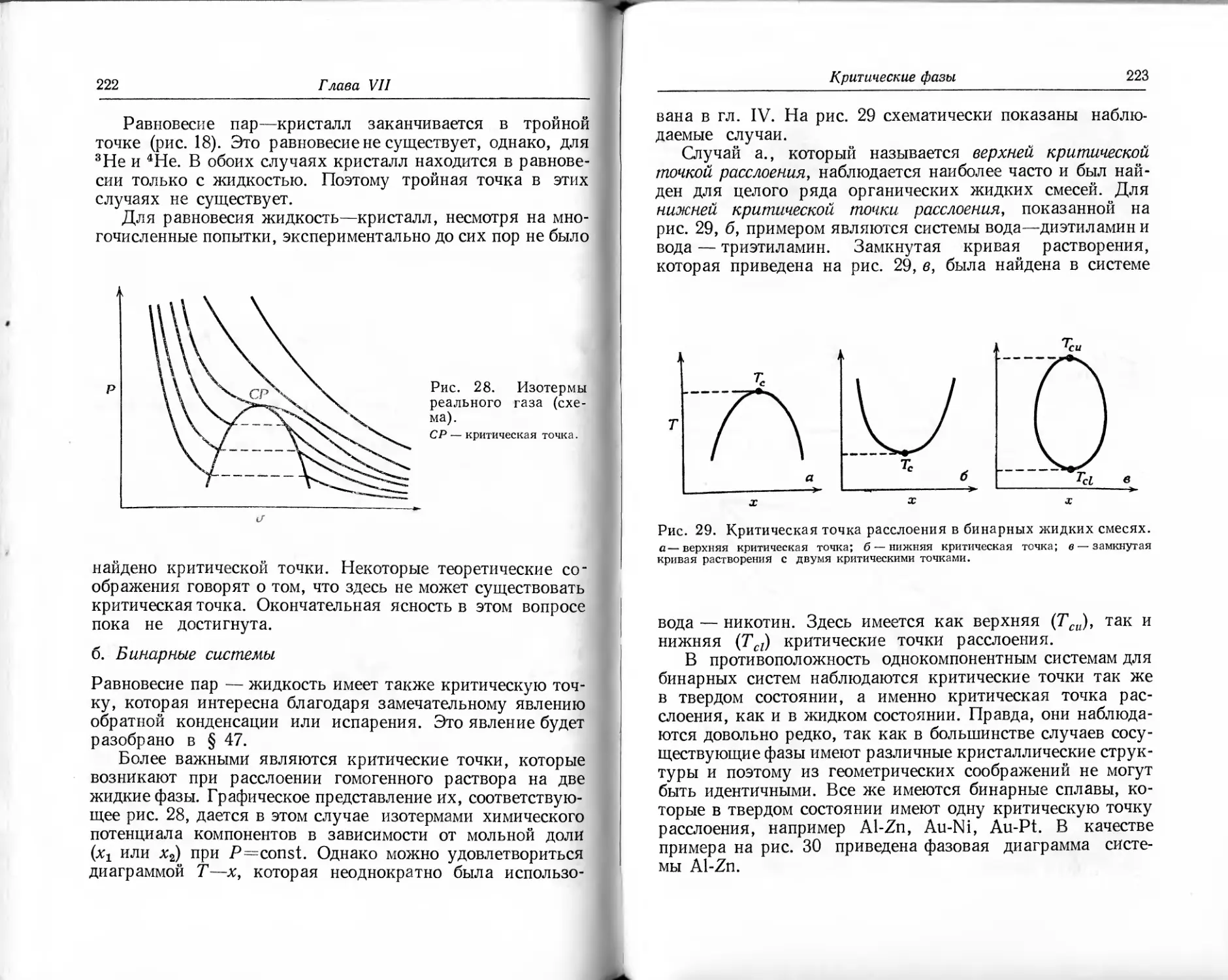
(4.4)
где /j и /2 — температуры обоих резервуаров в какой-нибудь эмпирической шкале. Пусть имеется третий тепловой резервуар с температурой /3, причем > /3 > ^2- Далее представим себе два обратимых цикла К и К', из которых один (К) работает между температурами и /3, другой (К') — между температурами t3 и t2. О™ связаны таким образом, что К отдает резервуару с температурой /3 такое же количество теплоты, которое К' из него забирает. Тогда результирующий к. п. д. спаренных процессов, согласно приведенным теоремам, должен быть таким же, как и в цикле между температурами и /2. В обоих случаях отдается более холодному резервуару количество теплоты Q2. Тогда
(4.5) 41
= (4.6)
4з
Из (4.4)—(4.6) следует
О2 __ Q2/Q1 __ f(^i> ^2)
Оз Q3/Q1 f (G, ^3)
(4.7)
(4-8)
и, таким образом
/(<1. <2) /(A. t3)'
ж
Коэффициент полезного действия цикла между температурами t3 и /2> согласно теоремам, должен не зависеть от выбора температуры tt. Поэтому из равенств (4.7) и (4.8) следует
/2) = -^ = ^-, (4.9)
Qs <t (t3)
где <p — новая функция. Абсолютную температуру Т определяют уравнением
(4.Ю)
22
Глава 1
где С — универсальный коэффициент пропорциональности. Тогда, согласно выражению (4.9),
= (4.Ц)
Qi Л
Уравнения (4.10) и (4.11) представляют собой определение абсолютной температуры по Томсону. Чтобы получить температурную шкалу, рассматривают обратимый цикл между температурами плавления и кипения воды при нормальном давлении (1 атм) и делят температурный интервал на 100 единиц*. Тогда
Qi __ х 4-100 /4 12)
Q2 х v '
Измерения дают
х = 273,15°К. (4.13)
Таким образом установлена абсолютная шкала температур, или шкала Кельвина.
Введем два новых определения. Процесс, проходящий при условии Т = const, называется изотермическим процессом', процесс, проходящий при условии d' Q =0, называется адиабатическим. Следовательно, для протекания адиабатического процесса система должна быть термически изолирована от окружающей среды. Рассмотрим гомогенную систему и обратимый цикл, состоящий из двух адиабат и двух изотерм (цикл Карно).
Если записать теплоты со знаками, то, согласно условию (4.11), для цикла Карно
* В настоящее время по международному соглашению основой температурной шкалы является не деление на 100 частей температурного интервала между нормальными точками плавления и кипения воды, а приравнивание нормальной температуры плавления воды величине 273,15° (точно) по абсолютной шкале температур. В соответствии с этой новой основой температурной шкалы нормальная температура кипения воды не равна 373,15° по абсолютной шкале, а может изменяться при совершенствовании измерительной техники, и в 1960 году была, например, равна 373,148°. Впрочем, в современной практической температурной шкале это небольшое отклонение нормальной температуры кипения (н. т. к.) воды от 100°С игнорируется и н. т. к. воды, как и раньше, приравнивается 373,15°К.— Прим. ред.
Основные законы термодинамики
23
р- + ^ = °- (4-14)
Если рассмотреть любой обратимый цикл в координатах р и V, то можно показать, что его всегда можно представить как сумму бесконечно малых циклов Карно.
Поэтому должно соблюдаться соотношение
ф_^. = 0. (4.15)
Если ввести величину
dS=-^-, (4.16)
то из уравнения (4.15) следует, что dS является полным дифференциалом, aS — функцией состояния. Эта функция
Рис. 1. Цикл Карно.
была названа Клаузиусом (1865) энтропией системы. Таким образом, абсолютная температура является интегрирующим делителем неполного дифференциала d'Q*. Это утверждение является математической формулировкой второго закона термодинамики для обратимых процессов. Из определения следует, что энтропия может быть вычислена с точностью до постоянной. Из выражения (4.16) следует, что в адиабатическом обратимом процессе энтропия остается постоянной. Такие процессы называются изэнтропий-ными.
* Для упрощения формул в дальнейшем не будем особо отмечать неполные дифференциалы и будем писать dQ и dW.
24
Глава I
Из (4.16) и (3.6) получим
TdS = dU + PdV. (4.17)
Рассмотрим теперь гетерогенную систему и обозначим фазы индексом а. По определению для простой системы исключен обмен веществ с окружающей средой и между фазами. Если между фазами невозможен теплообмен, то очевидно
dS = 5dS<a>. а
(4.18)
Следовательно, изменение энтропии всей системы в целом равно сумме изменения энтропий частей системы и dS вновь определяется формулой (4.16). Если сделать стенки между
Рис. 2. Произвольный цикл.
фазами теплопроницаемыми и предположить, что все части системы имеют одинаковую температуру, то, как будет подробно доказано в разд. Б, опять будут справедливы те же утверждения.
Особого обсуждения требует случай, когда части системы имеют разные температуры. Ради простоты рассмотрим адиабатически изолированную систему, состоящую из двух частей а и (3, разделенных адиабатической перегородкой и имеющих температуру и , причем
(4 J9)
Энтропия в этом состоянии (которое обозначим Л) в соответствии с равенством (4.18) будет иметь вид
(4.20)
Основные законы термодинамики 25
Если сделать теперь стенки теплопроницаемыми, то произойдет выравнивание температур, а именно, согласно по-стулату Клаузиуса, теплота перейдет от а к |3. В конечном состоянии В энтропия будет равна
SB = S£>+Sf>. (4.21)
Процесс является адиабатически необратимым; для него, согласно второму закону, =#5в. Для системы, изолированной в целом, d'Q = 0. Поэтому уравнение (4.16) в данном случае не применимо. Этого результата следовало ожидать, так как был рассмотрен необратимый процесс. Этот пробел в теории можно восполнить при помощи принципа Клаузиуса. Предположим, что переход теплоты между фазами протекает существенно медленней, чем выравнивание температур внутри фазы. Тогда во время необратимого процесса каждая фаза сама по себе находится во внутреннем равновесии, т. е. в термодинамическом состоянии, в то время как обе фазы не находятся друг с другом в равновесии. Поэтому для каждой фазы во время процесса существуют функции состояния U и S и изменения энтропии всей системы в целом можно определить по уравнению (4.18) после введения адиабатической перегородки между частями системы. Рассмотрим бесконечно малые изменения процесса, при котором количество теплоты
— dQ™ = dQ^ = dQt (4.22)
переходит от а к ₽. Температуры обеих частей системы можно считать постоянными. Соответствующее изменение энтропии, если нет работы расширения (dV — 0), согласно сказанному выше и уравнению (4.17), дается выражением
T(o)dS(a) = dt/(a); Т(₽) dS(₽) = dUm . (4.23)
Используя общее уравнение (3.4) (для d'W = 0), получим dSM = —И; dSm == . (4.24)
г(«) т(^ '
Отсюда видно, что применять уравнение (4.16) можно только в том случае, если рассматриваемая фаза находится во
26 Глава I
внутреннем равновесии, т. е. в термодинамическом состоянии, и не требуется, чтобы сам процесс перехода тепла был обратимым. Общее изменение энтропии, вызванное необратимым процессом, будет
dSt = dS™ + dSm (4.25)
или, используя (4.22) и (4.24),
dS, = dQ, - Др) . (4.26)
Поэтому из принципа Клаузиуса следует фундаментальный результат [вследствие условия (4.19)]
dSz>0. (4.27)
Изменение энтропии, вызванное необратимым процессом теплопроводности, может быть только положительным или (в крайнем случае) равным нулю.
Приведенные рассуждения можно распространить на другие процессы выравнивания (обмен веществ, химические реакции). Уравнения тогда получаются, естественно, более сложными. Если система как целое не является изолированной от внешней среды, то могут идти диссипативные процессы (необратимое превращение работы в теплоту, например, за счет трения или электрического тока). Наконец, можно рассмотреть также непрерывные процессы, сводя фазы к элементам объема и считая различия соседних элементов объема бесконечно малыми. Оказывается, что уравнение (4.27) всегда выполняется для всех необратимых процессов. В дальнейшем ради простоты будем учитывать только рассмотренный выше случай. Однако существенные результаты имеют общий характер. Чтобы получить общее изменение энтропии системы, необходимо рассматривать теплообмен с окружающей средой. При этом должно быть учтено, что обе фазы, согласно предположению, имеют различные температуры. Поэтому общая теплота, подводимая извне, должна быть разложена на
Q = Q<a> + Q(P). (4.28)
Если обозначить прирост энтропии, вызываемый за счет подвода теплоты извне, через dSa, то
Основные законы термодинамики 27
dS = dSa + dSt, (4.29)
или, используя выражения (4.16) и (4.26), можно записать
dS = + -^2. _f_ dQi (—--------—'j. (4.30)
т(«) г(₽) г(₽) r(«) J ' '
Чтобы получить наглядные соотношения, предположим, что dQ(a) = и введем эффективную темпе-
ратуру
2_ = 7Ч«) + Г(Р)
Т Г(*) Г(3) ’ к • /
Далее введем
. , тЧа)_уЧР)
dQ = 2dQt -L-—1—. (4.32)
rp(a) i j.(p)
Тогда из условия (4.30) следует
= (4.33)
Эта формулировка Клаузиуса имеет самый общий характер. Если система находится при однородной температуре (т. е. если ограничиться необратимыми процессами переноса вещества, химическими реакциями и диссипативными процессами), то эффективная и истинная температуры совпадают. Величина dQ' названа Клаузиусом некомпенсированной теплотой. Ее значение для теории необратимых процессов впервые было понято Дюгемом (1911). Сравнение (4.33) и (4.29) с учетом (4.27) дает соотношение
dQ' > 0, (4.34)
имеющее общий характер. Следует обратить внимание, что Уравнение (4.33) представляет собой не что иное, как символическую запись уравнения (4.30) для теплопроводности. Сохраним этот способ написания до конца параграфа, чтобы получить простые формулировки. В основу расчетов, конечно, должно быть положено уравнение (4.30).
28
Глава I
Из соотношений (4.33) и (4.34) следует совершенно общий результат
dQ^TdS. (4.35)
Это важное соотношение называется неравенством Клаузиуса. При отсутствии необратимых процессов оно сводится к уравнению (4.16).
Рассмотрим теперь адиабатически изолированную систему. Тогда в соответствии с уравнением (4.29) dS = dSt и с учетом (4.27)
dS>>0 (адиабатически изолированная система). (4.36) Таким образом, энтропия изолированной системы никогда не может уменьшаться. В крайнем случае, если протекают только обратимые процессы, она остается постоянной. Отсюда следует, что энтропия изолированной системы, находящейся в термодинамическом равновесии (т. е. в состоянии, в котором все возможные необратимые процессы в системе уже прошли), имеет наибольшее значение, возможное при данных условиях. Это значение в математическом смысле в большинстве случаев является стационарным. Приведенные рассуждения дополняют второй закон и являются основой для формулировки условий равновесия (гл. II). Если рассматривать вселенную как изолированную систему, то приходим к утверждению Клаузиуса, приведенному в § 1.
§ 5. Холодильные машины и тепловые насосы
Классическое развитие второго закона термодинамики неразрывно связано с решением специальных технических проблем. Поэтому оно представляет удобную возможность для постановки аналогичных задач. Рассмотрим коротко две такие задачи. Как и в § 4, ограничимся рассмотрением идеализированного случая полностью обратимых круговых процессов.
Цикл, рассмотренный в начале § 4, представляет собой принцип работы любой тепловой машины и дает максимальный, т. е., согласно второму закону, наибольший из вообще возможных, коэффициент полезного действия tj, который
Основные законы термодинамики 29
достигается только в предельном случае полностью обратимого процесса. В соответствии с уравнениями (4.3) и (4.11) уравнение для коэффициента полезного действия можно записать в виде:
Так как Т2 практически всегда зафиксировано (температура окружающей среды), то оказывается, что к.п.д. тепловой машины тем больше, чем выше рабочая температура (что достигается применением перегретого пара, двигателей внутреннего сгорания).
Можно также заставить протекать рассматриваемый процесс в обратном направлении таким образом, что из более холодного резервуара будет взято количество теплоты Q2, затем над рабочим телом произведена работа W и наконец рабочее тело передаст более горячему резервуару количество теплоты Такое устройство называется холодильной машиной, если поставлена техническая задача охладить более холодный резервуар. Если происходит дальнейшее нагревание горячего резервуара, то такую машину называют тепловым насосом. Таким образом, в основу работы обеих машин заложен один и тот же принцип, они различаются только с технической точки зрения.
Простым примером холодильной машины является обычный бытовой холодильник. Его внутреннее пространство представляет собой более холодный резервуар (температура Т2). окружающая среда — более горячий резервуар (температура 7\), работа производится электрическим током. Форма записи для выражения коэффициента полезного действия зависит, естественно, от самого процесса. Поэтому для холодильной машины к. п. д. определяют как отношение полученного холода (т. е. теплоты Q2, отнятой от более холодного резервуара) к затраченной работе. Следовательно,
Так как предыдущие рассуждения относились к обратимому круговому процессу, то, согласно равенству (4.11),
Основные законы термодинамики
31
30
Глава I
если написать теплоты с включением знаков всех величин, получим
~ + ~ = (5.3)
1 1 1 2
По первому закону термодинамики
^ + Q2 + Qx=O, (5.4)
и тогда
= (5.5)
1 2 i 1
Из выражений (5.2) и (5.5) следует выражение для к.п.д. холодильной машины
Отсюда видно, что к.п.д. холодильной машины тем-больше, чем меньше разница Т\—Т2 (в летнюю пору холодильник потребляет больше электроэнергии). С другой стороны, к.п.д. тем меньше (при данном Тг), чем ниже Т2. В этом заключается одна из трудностей получения очень низких температур.
Тепловые насосы основаны на том же принципе. Практически внутреннее помещение дома должно было бы представлять собой более горячий резервуар, свежий воздух более холодный, а работа должна была бы производиться за счет электроэнергии. Коэффициент полезного действия в этом случае равен
ew= » (5-7)
что вместе с уравнением (5.4) дает
£w = ^. (5.8)
Следовательно, коэффициент полезного действия тем меньше, чем больше разница температур 7,—Т2. Если Тг—Т2= =const, то к.п.д. увеличивается с повышением температуры более горячего резервуара.
В принципе такое устройство можно реализовать, если выставить открытый холодильник в открытое окно (эффект, правда, будет минимальный). Тепловые насосы вследствие их чрезвычайно большого к. п. д. представляют принципиальный интерес для целей отопления. Из уравнения (5.8) следует, что, например, при Тг = = 289° К и Т2 = 273° К 18-кратное количество потребляемой электрической энергии (в идеальном случае) переходит в теплоту*. Реализация тепловых машин вследствие высокой себестоимости и эксплуатационных расходов до сих пор не осуществлена, однако применение маленьких агрегатов уже в настоящее время может оказаться экономически целесообразным**. В связи с этим представляет особый интерес использование эффекта Пельтье в полупроводниках.
Б. АКСИОМАТИКА КАРАТЕОДОРИ
§ 6. Определения
Изложение законов термодинамики в соответствии с их историческим развитием особенно целесообразно для введения в круг основных понятий термодинамики, однако с логической точки зрения такой способ изложения является не вполне удовлетворительным (некритическое введение понятий температуры и теплоты, неясное разграничение чисто физических опытных фактов и некоторых чисто математических элементов). Рассмотрим теперь изложение законов термодинамики в более строгой форме, следуя формулировкам Каратеодори (1909).
Не обращая внимания на повторения, сопоставим прежде всего необходимые важнейшие определения. Без уточнения будем пользоваться понятиями механики, такими, как масса, объем, давление, работа и т. д. Рассмотрим гомоген-
* Как видно из уравнения (5.8), к. п. д. тепловых насосов принципиально не может быть меньше единицы. Величина е^> 1 означает лишь то, что энергию можно извлекать из окружающей среды сверх той энергии, которая необходима для осуществления самого процесса превращения работы в теплоту. — Прим, перев.
** На самом деле это не так. В Англии уже существует тепловая машина для отопления Вестминстерского дворца водой Темзы. Аналогичная установка есть и в Цюрихе. В Америке (Индиана) в качестве низкотемпературного источника теплоты для теплового насоса, отапливающего здание, используется земля. Полезные сведения о тепловых насосах можно найти в книге проф. П. К. Ощепкова «Жизнь и мечта», изд. «Моск, рабочий», 1967.— Прим, перев.
32
Глава I
ное изотропное тело, находящееся в равновесии при отсутствии внешних полей. Согласно гидростатике, объем V однозначно задается давлением Р. Обратимся теперь к первому опытному факту, который одновременно служит определением.
Эмпирический закон и определения
Существуют процессы, в которых при постоянном Р изменяется V. Такие процессы называются термическими процессами, к ним относятся, например, нагревание и охлаждение. Из сказанного следует, что в термодинамике для определения состояния рассматриваемой системы необходимы две независимые переменные. Выберем в качестве переменных состояния величины Р и V, определяемые в механике. Тогда каждому состоянию равновесия будет соответствовать точка на плоскости Р—И.
Оболочка, окружающая тело, называется адиабатической, если состояние тела может изменяться только за счет нетермических, т. е. механических, электрических и других воздействий (движение частей оболочки, включение электрического тока и т. д.). Каждая неадиабатическая оболочка называется диатермической или теплопроводящей. Изменение состояния называется квазистатическим, i если оно проходит через непрерывную последовательность, состояний равновесия. Поэтому оно протекает бесконечно медленно и не является функцией времени (см. § 1).
Пример. Расширение газа в цилиндре за счет медленного движения поршня.
Любое другое изменение состояния называется нестатическим. Промежуточные состояния в нестатических процессах требуют для своего описания дополнительных переменных. Рассмотрение нестатических состояний, за исключением некоторых общих следствий, выходит за рамки классической термодинамики, поэтому ограничимся квазистати-1 ческими процессами.
Следует принять во внимание, что понятие «квазистати-ческие изменения состояния» перекрывается по своему содержанию понятием «обратимых изменений состояния», но логически они различны. Понятия обратимый и необратимый будут введены позднее при обсуждении второго закона термодинамики.
Основные законы термодинамики
33
§ 7. Эмпирическая температура
Рассмотрим гомогенное, адиабатически изолированное тело, находящееся в состоянии Ро, Vo. Пусть существует теперь квази статическое изменение состояния, при помощи которого можно достигнуть состояния Р, V. Так как согласно условию, термические процессы исключены из рассмотрения, то все состояния, достигаемые квазистатически-адиабатическим путем (начиная от начального Р0Р0), должны быть расположены на одной кривой в плоскости Р—V, Эта кривая называется адиабатой тела, проходящей через PgVq. Совокупность адиабат тела образует однопараметрическое семейство кривых в плоскости Р—V, плотно покрывающее определенную область этой плоскости. Таким образом, через каждую точку плоскости Р—V проходит одна и только одна адиабата.
Уравнение адиабаты символически можно записать в виде
s(P, V) = s, (7.1)
где s означает параметр.
Пример. Уравнение адиабаты идеального газа
PVX = s, (7.2)
где х— постоянная, характерная для данного вещества, и s=P0VS. (7.3)
Рассмотрим теперь адиабатически изолированную систему, состоящую из двух фаз ' и " с множеством адиабат
s'(P', V') = s'- sf'(P", V")=s". (7.4)
Если обе фазы разделены адиабатической перегородкой, то не существует никакой связи между Р', V', с одной стороны, и Р", Е" — с другой. В состоянии равновесия могут существовать любые пары значений. Если же перегородка диатермическая, то, как показывает опыт, это уже не имеет места.
2-235
34
Глава 1
Эмпирический закон и определение
Между двумя телами, адиабатически изолированными от окружающей среды и отделенными друг от друга диатермической перегородкой, существует равновесие только в том случае, если выполняется уравнение вида
F(P', Г; Р", У") = 0. (7.5)
Это равновесие называется термическим равновесием, поскольку оно, согласно определению, может возникнуть только в результате протекания термического процесса.
Приведенный закон еще ничего не говорит о том, связано ли выполнение равновесия (7.5) со специфическими свойствами разделенного на части тела или в основе лежит общее свойство, которым в принципе обладают все тела. Разъясним это на примере. Включим в систему еще одну, третью (без штрихов) фазу таким образом, чтобы она (за счет превращения адиабатической перегородки в диатермическую) могла прийти в термический контакт как с первой, так и со второй фазами. Предположим, что между фазой без штриха и фазами ' и " существует термическое равновесие, следовательно, выполняются равенства
F(P, V; Р', К') = 0; Р(Р, V; Р", Г') = 0. (7.6)
Приведенный закон не дает никаких критериев для определения, существует термическое равновесие между фазами ' и " или нет. Необходимо привлечь новый опытный факт.
Эмпирический закон и определение
Если в трехфазной системе фазы, обозначенные ' и ", находятся в термическом равновесии с третьей фазой, то они также находятся в состоянии равновесия между собой. Каждая фаза обладает поэтому измеримым свойством t (Р, V) такого рода, что из t = /' и t = t" следует t' =
Это свойство называется эмпирической температурой.
1 Приведенный закон позволяет лишь установить, является температура двух тел одинаковой или разной. Для того чтобы получить температурную шкалу, которая давала бы возможность количественно сравнивать различные температуры, должен быть изучен вид функции .
Основные законы термодинамики
35
Z(P, V)=^t.
(7.7)
Для этого фиксируют две хорошо воспроизводимые температуры (термометрические опорные точки, например точка плавления льда и точка кипения воды при нормальном давлении) и измеряют для подходящего термометрического вещества изменения соответственно выбранного свойства в этом интервале (например, изменение объема для ртути или спирта). Придавая температурам опорных точек опреде
Рис. 3. К определению эмпирической темпе-
ратуры.
ленное числовое значение (0 и 100° для шкалы Цельсия}, устанавливают таким образом температурную шкалу. С теоретической точки зрения наиболее целесообразно в качестве термометрического вещества применять идеальный газ, т. е. измеренные значения произведения РУ, экстраполированные к Р->0 для наиболее низко конденсируемых газов (Н2 или Не). В этом случае функция t (Р, V) известна до трансформации шкалы и необходима только одна опорная точка. Реальная газотермометрическая шкала очень точно совпадает с термодинамической шкалой (§ 4).
Уравнение (7.7) называют термическим уравнением состояния, для идеального газа оно имеет вид
PV=f(t), (7.8)
где функция f (/) зависит только от эмпирической температуры. В газотермометрической шкале f (/) совпадает с температурой с точностью до сомножителя*, и значение для точки плавления льда равно
273,15°. (7.9)
Из уравнения (7.8) (для одного моля) следует
_ PV = RT* , (7.10)
* См. примечание редактора на стр. 22. — Прим, перев.
2*
Основные законы термодинамики
37
36
Глава I
где —так называемая газовая постоянная, а Т* —температура, численно совпадающая с температурой по шкале Кельвина (§ 4).
Уравнение (7.7) представляет собой уравнение однопараметрического семейства кривых в плоскости Р—V и называется изотермой вещества.
§ 8. Первый закон термодинамики
В теории Каратеодори первый закон термодинамики формулируется иначе, чем в классической теории. Внутренняя энергия здесь вводится при помощи понятий механики, а для определения понятия теплоты используют опытные данные.
Эмпирический закон и определение
Для того чтобы перевести гомогенную или гетерогенную систему адиабатическим путем из начального состояния 1 в конечное состояние 2, всегда необходимо затратить одинаковую работу (механическую, электрическую и т. д.) независимо от того, какие равновесные или неравновесные состояния проходит при этом система. Работу, совершенную в таком процессе, называют приростом внутренней энергии системы.
Следовательно,
2
U2— U1=^dW (адиабатически). (8.1)
По этому определению внутренняя энергия является функцией состояния, которую можно вычислить с точностью до константы интегрирования.
Применимость этого определения (которое одновременно может служить руководством для измерения) зависит, естественно, от того, будет ли рассматриваемый процесс реализоваться для всех пар состояний 1, 2. Это обстоятельство является слабой стороной теории Каратеодори, поскольку уже здесь надо учитывать некоторые следствия второго закона термодинамики. Они утверждают, что фактически не всегда возможно из произвольного состояния 1 адиабатическим путем достигнуть произвольного состоя
ния 2. Например, невозможно перевести гомогенную систему при постоянном объеме адиабатическим путем в состояние с меньшей внутренней энергией, так как такой процесс был бы обратным диссипативным процессам (например, производство теплоты за счет трения). В таких случаях всегда можно адиабатически провести обратный процесс 2->1 и, таким образом, сохранить общий характер определения (8.1).
Если процесс 1->2 проводить любым путем, кроме адиабатического, то затраченная работа зависит от пути процесса, т. е. от последовательности промежуточных состояний. Однако разность значений внутренней энергии должна быть такой же, как и в случае адиабатического изменения, так как эта разность зависит только от начального и конечного состояний.
Определение
Для произвольного процесса 1->2 разность между приростом внутренней энергии и затраченной работой называется подведенной теплотой.
Таким образом имеем
2 2
J d'Q = (U2 — иг) — J d'W (любое изменение состояния) 1 1
(8.2) или, в дифференциальной форме,
dU = d'Q, + d'W. (8.3)
Приведенные определения вместе с уравнениями (8.1)— (8.3) представляют собой первый закон термодинамики в формулировке Каратеодори. Рассмотрим кратко некоторые специфические случаи и начнем с гомогенных систем. Для квазистатического процесса можно написать
dW = — PdV. (8.4)
Вместе с тем
dU = dQ — PdV. (8.5)
Для адиабатического процесса имеем
dQ = 0; dU = dW\ (8.6)
38
Глава I
для адиабатического квазистатического процесса
dQ = 0; dU = — PdV. (8.7)
Формулы легко можно обобщить для гетерогенных систем, так как величины, входящие в выражения (8.3), аддитивны по отношению к вкладу каждой фазы. Ради простоты ограничимся рассмотрением двух фаз ' и ". Тогда для квази-статического процесса можно записать
dU' + dU" = dQ' + dQ" — P'dV' — P"dV", (8.8) а для адиабатического квазистатического процесса будет справедливо
dQ' 4-dQ" = 0; dU' + dU" = — P'dV' — P"dV". (8.9)
Выберем в качестве переменных состояния t и V, тогда для гомогенной системы, согласно первому закону термодинамики,
dU
(8.10)
Из уравнений (8.7) и (8.10) для адиабатического квазистатического процесса следует
+ P]dV + (—dt—O. Lw )t J \ dt ]v
(8П)
Для двухфазной системы, фазы которой находятся в термическом равновесии, аналогичным образом можно получить
V"
г/ dU'\
[\ dt )v’,
dt — O. (8.12)
Появляющаяся в приведенных уравнениях производная внутренней энергии по температуре называется теплоемкостью при постоянном объеме. Если эта величина отнесена к одному молю индивидуального вещества, то ее называют мольной теплоемкостью при постоянном объеме Cv.
Основные законы термодинамики
39
§ 9. Дифференциальные выражения Пфаффа
Приведем некоторые вспомогательные сведения из математики, которые будут нужны при рассмотрении теории Каратеодори. Как следует из § 8, первый закон термодинамики можно представить уравнениями следующего вида:
п
dQ^^Xidxi, (9.1)
Z=1
где — функции одного или нескольких независимых переменных. Уравнения такого типа называются дифференциальными выражениями Пфаффа. Уравнение (9.1) можно проинтегрировать вдоль некоторого пути в и-мер-ном пространстве, причем значение интеграла в общем случае зависит от выбора этого пути. Если обозначить границы интегрирования через 1 и 2, то, следовательно, сам интеграл нельзя представить в виде Q(2)—Q(l). Поэтому говорят, что dQ является неполным или неточным дифференциалом. Рассмотрим два вопроса:
а) при каких условиях dQ является полным дифференциалом;
б) при каких условиях существует интегрирующий делитель т такого вида, что dQ/т является полным дифференциалом.
а) Условие, при котором дифференциал dQ является полным.
Условие того,что dQ является полным дифференциалом, можно записать следующим образом:
dQ = df(x19...9 хп), (9.2)
где df (хг, .*. , хп) является дифференциалом функции f(xlt , хп).
Тогда
2
JdQ=f(2)-/(l). (9.3)
1
Но по определению
df=yj~dxi (9.4)
40 Глава I
Основные законы термодинамики
41
и вследствие условия (9.1)
х, = . (9.5)
ОХ/
Отсюда следует
= = (9.б)
dxj dxidxj dxi dxftxi
Необходимым и достаточным условием того, что dQ является полным дифференциалом, будет
^- = ^-(1, /=1..........И). (9.7)
dxj dxi
В векторном виде уравнение (9.1) можно записать
dQ = R • dr. [(9.8)
Таким образом условию (9.7) для п = 3 можно придать наглядную форму*
rot R = grad х R = 0. (9.9)
Если R можно представить как градиент скалярной функции, то это будет необходимым и достаточным условием равенства нулю интеграла от R вдоль замкнутой кривой, б) Условие существования интегрирующего делителя**.
Предположим, что dQ не является полным дифференциалом, и будем искать условие существования интегрирующего делителя т такого рода, что dQ/т будет^полным дифференциалом.
Если назвать
dQ = O (соответственно VXjdXj =0) (9.10)
* Для п>3 условие (9.7) можно записать аналогичным образом в тензорной форме.
’* Читатель, не интересующийся математическими подробностями, может пропустить доказательства приведенных теорем
дифференциальным уравнением Пфаффа, принадлежащим к dQ, то можно сформулировать следующую теорему.
Теорема 1
Дифференциальное выражение Пфаффа dQ обладает интегрирующим делителем т тогда и только тогда, когда дифференциальное уравнение Пфаффа, принадлежащее к dQ, имеет решение в виде
х„) = о. (9.11)
Доказательство
Проще всего доказательство провести геометрически. Решение условия (9.11) определяет в п-мерном пространстве однопараметрическое семейство гиперповерхностей. Рас-
смотрим две бесконечно близкие соседние поверхности о и 04-do, и пройдем путь от Л до С один раз через Въ другой через В2. На каждой из обеих поверхностей по определению dQ = 0 и do = 0. Согласно предположению, изменение dQ зависит от пути. Так как оно на каждой из обеих поверхностей равно нулю, то, следовательно, оно может зависеть только от точки пересечения В, поэтому должно быть
dQ = x(B)da. (9.12)
Но так как по определению da является полным дифференциалом, то имеем
“ т(хх, .... хп)
(9.13)
что и доказывает существование интегрирующего делителя.
I
Основные законы термодинамики 43
42 Глава I
Теорема 2
Если дифференциальное выражение Пфаффа имеет один интегрирующий делитель, то оно имеет также бесконечное множество интегрирующих делителей.
Доказательство
Если обозначить одно-однозначную функцию от о через S (о), то из (9.11) следует
SHxp..., x„)] = S. (9.14)
Используя (9.13), получим
clS = — (9.15)
da da т
Если принять
T(xv .... x„) = т(х1э.... х„)£, (9.16)
то из (9.15) следует
dS(xv..., х,)~ — . (9.17)
I (Х1, ...» хп)
Таким образом, Т (хь ... , хп) также является интегрирующим делителем, что и доказывает теорему.
ТеоремаЗ(1 теорема Каратеодори)
Если дифференциальное выражение Пфаффа имеет инте-грирующий делитель, то в окрестности каждой точки Р0(х10, ••• > хло) имеются сколь угодно близко расположенные от нее точки (хХ1 , ... , хп1), которые из Ро нельзя достигнуть путем, состоящим только из отрезков dQ = 0.
Доказательство
Согласно теореме 1, существование интегрирующего делителя связано с существованием семейства поверхностей (9.11). На каждой поверхности в соответствии с выражениями (9.10) и (9.11) da = 0 и dQ = 0. Пусть Ро будет точкой на такой поверхности. Тогда путем dQ = 0 можно достигнуть только точек, лежащих на этой же поверхности. Теорема доказана.
Если не существует интегрирующего делителя, то можно из точки Ро попасть в любую точку Pt путем dQ = 0, что можно легко-проверить . на примерах. ... . .. ...... ..
Теорема 4 (2 теорема Каратеодори)
Если дифференциальное выражение Пфаффа dQ обладает тем свойством, что сколь угодно близко от каждой точки пространства Ро находятся другие точки Рг, к которым нельзя прийти из Ро путем dQ = 0, то для dQ существует интегрирующий делитель.
Эта теорема является обратной теореме 3. Однако доказательство ее более сложное, и поэтому здесь оно приведено не будет.
Теорема 5
Для существования решения (9.11) дифференциального уравнения Пфаффа необходимо и достаточно, чтобы уравнение
X ________дА) \ । х.(д-^____I _
k dxt dxj ) }\dxk dxt ) 1 dxj dxk) ~
= 0 (9.18)
выполнялось для всех значений троек i, k, j.
Доказательство
Ограничимся доказательством, что это условие является необходимым. Поэтому предположим, что решение (9.11)
существует. Если для упрощения подставить и, то
из условия (9.13) с учетом (9.1) следует
u(xlt..., x)Xt = (9.19)
dxi ' '
и так как da является полным дифференциалом, получим
После дифференцирования приходим к выражению
и № _ = х i - X.
\ dxi dxj j dxj J dxt и соответственно
]
44
Глава I
/ dXj __ dXk \____du _______du_
\ дхь dxi ) k dxt 1 дх^ ’
/dXk dXA __ qu du .
I dXj dx^ j 1 dxk k dXj
(9.21)
Умножая первое из уравнений (9.21) на Xk, второе на Xj и третье на Xt и складывая, получим уравнение (9.18). Таким образом, показано, что условие (9.18) необходимо. Можно также показать, что оно является достаточным.
В случае трех независимых переменных (9.18) можно записать в векторном виде
?* —
7? • grad х 7? = О (три переменные). (9.22)
Рассмотрим теперь подробно особенно важные случаи двух и трех независимых переменных.
а. Две независимые переменные.
Теорема 6
Дифференциальное выражение Пфаффа для двух независимых переменных всегда обладает интегрирующим делителем.
Доказательство
Если подставить в уравнение (9.18) k = j, то следует, что условие для двух независимых переменных выполняется всегда. Поэтому с учетом теоремы 1 приходим к теореме 6.
Рассмотрим дифференциальное уравнение Пфаффа
dQ = Xdx + Ydy^O. (9.23)
Согласно теореме 6, существует решение
а(х,у) = а, (9.24)
представляющее собой однопараметрическое семейство кривых в плоскости х—у. Вдоль каждой кривой
dQ = 0 (9.25)
и
cb = — dx + — dy = 0. (9.26)
dx dy
Основные законы термодинамики
45
Из уравнения (9.26) получаем dy dajdx dx да/ду *
и из (9.23) следует dy_____________________________Х_
dx Y '
Из условий (9.27) и (9.28) находим
X Y / ч
=-----— v (х, у), да/дх---------да/ду-' v’
(9.27)
(9.28)
(9.29)
где т (х, у) — функция, зависящая от х и у.
В соответствии с выражениями (9.1) и (9.13) можно записать
do = = 2Ldx + — dy. (9.30)
т (х, у) X Т
Поэтому интегрирующий делитель т (х, у) всегда дается выражением (9.29).
|3. Три независимые переменные
Рассмотрим дифференциальное уравнение Пфаффа
dQ = Xdx + Ydy + Zdz = 0. (9.31)
В этом случае условие (9.18) не всегда выполняется, что легко можно показать на примерах. Поэтому уравнение (9.31) не обязательно имеет решение в виде
о (х, у, Z) = а (9.32)
и, таким образом, dQ не всегда имеет интегрирующий делитель.
Предположим, что решение (9.32) существует. Оно определяет однопараметрическое семейство поверхностей в трехмерном пространстве. На этих поверхностях
do= — dx + — dy + — dz. (9.33)
dx dy dz
Отсюда следует
dy ___ da/dx t dz ___ da/dx . dz I __ da/dy
dx z da/dy ’ dx v dajdz ’ dy L da/dz
(9.34)
46
Глава I
dz_
Z 9 dy
Но, согласно (9.31), dy ________ X ' dz
dx z Y ’ dx
Из условий (9.34) и (9.35) получаем
—— = —— = = х(х, у, z), (9.36)
д<з)дх да/ду дъ/дг
и видно, что т (%, у, z), как и прежде, является интегрирующим делителем неполного дифференциала dQ. Этот вывод легко можно обобщить для числа независимых переменных, большего чем три.
у- (9.35)
§10. Второй закон термодинамики для квазистатических процессов
Опытные данные, лежащие в основе второго закона термодинамики, были введены Каратеодори в форме, которая была уже использована при обсуждении первого закона термодинамики (§ 8).
Эмпирический закон (принцип Каратеодори)
Сколь угодно близко от любого состояния гомогенной или гетерогенной системы существуют соседние состояния, не достижимые адиабатическим путем.
Один пример уже был приведен в § 8. В данном параграфе ограничимся рассмотрением квазистатических процессов. Очевидно, вышеупомянутый закон должен быть справедлив для этого специального класса изменений состояний. Примером может служить адиабатическое сжатие или расширение идеального газа, которое осуществляется изменением величин Р и V, связанных уравнением (7.2).
Рассмотрим сначала соотношение между принципами Каратеодори и Клаузиуса (соответственно Томсона) (§ 4). Сразу видно, что принцип Каратеодори вытекает из принципа Клаузиуса. Обратное несправедливо, так как принцип Каратеодори ограничивается утверждением, что существуют вообще нереализуемые адиабатические процессы, в то время как принцип Клаузиуса указывает, какие процессы адиабатически нереализуемы.
Основные законы термодинамики
47
Поэтому с точки зрения аксиоматики принцип Каратеодори следовало бы предпочесть, если бы из него можно было вывести все следствия второго закона термодинамики. Как будет видно, на самом деле это не так и фактически необходим еще один эмпирический закон (§ 13).
Рассмотрим еще раз квазистатические процессы. Для гомогенной системы дифференциальное выражение Пфаффа dQ зависит только от двух независимых переменных. Существование интегрирующего делителя, а также энтропии является, согласно теореме 6 §9, чисто математическим следствием, для которого не нужны дополнительные опытные данные. С этой точки зрения интересен случай с тремя независимыми переменными. Кроме того, идентификация интегрирующего делителя с температурой требует наличия термического равновесия, которое при ограничении двумя независимыми переменными невозможно. По обеим причинам начнем с анализа системы, состоящей из двух фаз ' и ", разделенных друг от друга диатермической перегородкой и находящихся в термическом равновесии. В качестве независимых переменных выберем V' , V" и t.
г % Из принципа Каратеодори в сочетании с теоремой 4 §9 непосредственно следует существование интегрирующего делителя для dQ. Поэтому
dQ = dQ' + dQ" = т(Г, V", t)da(V', Г, t). (10.1)
Для обеих фаз можно написать
dQ' = г' (Г, t) da' (Г, 0; dQ" = т" (Г, t) da" (V", t).
(Ю.2)
Из условий (10.1) и (10.2) следует
'zda = v'da' 4- т!' da". (10.3)
Выберем теперь независимыми переменными о', о" и t. Так как do является полным дифференциалом, то из (10.3) получим
А = о. (10.4) дс' т дъ" т dt
Функция о не зависит, таким образом, от t
а = 0« а"). (10.5)
48
Глава 1
Поэтому, согласно (10.4), должно быть
Диффер енци р ов ани е дает
1 дт' 1 дт" 1 дт
•t' dt т" dt т di
(10.6)
(Ю.7)
Но т' зависит только от t и о', т"— ТОЛЬКО ОТ t И О'". Поэтому первый знак равенства выполняется только тогда, когда логарифмические производные от т' и т" по t не зависят ни от о', ни от о", а только от t. Поэтому должно выполняться соотношение
dlnx' д1пт" д 1п т ,,х
где g (/)—общая для всех находящихся в термическом равновесии фаз функция эмпирической температуры, не зависящая от всех специфических свойств фаз. Интегрирование (10.8) для всей системы и соответственно для обеих фаз дает
(10.8)
In т = j g (t) dt + In Ф (</, о"), In = J g (t) dt + In Ф' (</), 1пт" = Jg(Z) d/ + ln Ф" (</'),
или
T = exp[j g(O//q • Ф« о"), х' = exp J g (t) dt • Ф' (o'), T"=exp[Jg(/)d/| • Ф"(о").
(10.9)
(10.10)
Для системы, фазы которой находятся между собой в термическом равновесии, интегрирующий делитель для всей системы и каждой фазы в отдельности распадается на два фактора, один из которых зависит только от общей эмпирической температуры t, в то время как второй является функцией индивидуальных переменных состояния (о' и а" для системы в целом, а' для ' и а" ддя ").
Основные законы термодинамики
49
Абсолютную температуру определяют по уравнению
T = C.exp[jg(O<ft], (10.11)
где С—произвольная константа, которая фиксируется установлением шкалы. При этом считают, что между двумя опорными точками (точки плавления и кипения воды) температурный интервал равен 100°. Теперь в уравнение (10.11) не входит произвольная константа. Нулевая точка по Т определена таким образом физически. Используя уравнение (10.11), из (10.1), (10.2) и (10.10) получим
dQ = Tda = Т ~ da, (10.12)
dQ' = r'do' = Т — do', dQ"=K"do" = T —do". (10.13) c c
Рассмотрим теперь отдельную фазу (например, ') и определим энтропию фазы равенством
S' = -L С Ф' (o') do' + const. (10.14)
По определению энтропию (так же как и внутреннюю энергию) можно рассчитать с точностью до произвольной аддитивной постоянной. Используя (10.14), из выражения (10.13) получим
dQ' = TdS'; dQ" = TdS". (10.15)
Таким образом, с учетом только квазистатических изменений состояний было доказано существование для каждой фазы функции состояния — энтропии и выведено основное уравнение (4.16). Рассмотрим теперь систему в целом. Из уравнения (10.12) с учетом (10.3) и (10.10) следует
<£>do = ФШ + Ф'^о". (10.16)
Покажем, что Ф можно представить в виде уравнения
Ф = Ф(а), (10.17)
т. е. зависящей от а' и о" только через а. Продифференцировав (10.17), получим
50
Глава I
дФ дФ да дФ дФ да
~ГГ==~Г--Т7^ ~ТТ = “7“ Т7‘ (10.18)
да' да да да" да да 4 7
Исключая дФ/ch, получим
А-=о, (10.19)
да' да" да" да'
или в записи, обычной для определителей Якоби,
^лЛ. = /(фг д) = 0. (10.20)
Функция Ф зависит в явном виде только от о, если определитель Якоби J (Ф,о) равен нулю. Это условие выполняется и в данном случае. Из (10.16) следует
Ф — = Ф'; Ф — = Ф". (10.21)
да' да"
По определению Ф' зависит только от о', Ф"—только от о". Дифференцирование (10.21) по о"(соответственно по о') дает
дФ да ф д2а
да" да' да'да"
дФ да ф д2а
да' да" да'да"
Отсюда следует фактически уравнение (10.20), и, таким образом, (10.17) доказано. Поэтому энтропию всей системы в целом можно определить по уравнению
S = -— J Ф (о) do + const. (10.23)
А для всей системы в соответствии с выражением (10.12)
dQ = TdS.
(10.24)
Если подставить в уравнение (10.15) выражение для первого закона термодинамики в виде условия (8.8), то получим для каждой фазы
T'dS' = dU' + P'dV' (10.25)
и для всей системы в целом
dS^dS' + dS". (10.26)
Основные законы термодинамики
51
Таким образом, при ограничении круга вопросов рассмотрением квазистатических процессов удалось доказать существование абсолютной температуры и энтропии как функции состояния для каждой фазы, определенной уравнениями (10.15) или (10.25), и показать, что энтропия всей системы в целом складывается аддитивно из энтропий фаз.
Из существенных следствий второго закона термодинамики отсутствует пока свойство возрастания энтропии, сформулированное в условии (4.36), которое является фундаментальным для формулировки условий равновесия. Этот вопрос будет рассмотрен в § 13.
Заметим еще, что в последнее время а называют эмпирической, aS — метрической энтропией. При этом эмпирическая переменная установлена при любых согласованных и непрерывных трансформациях шкал, в то время как метрическая переменная допускает только линейную трансформацию шкал (расширение масштаба и смещение нулевой точки). Существование эмпирической энтропии следует из теоремы 6 §9, а также из принципа Каратеодори. Введение термической связи служит для того, чтобы сконструировать метрическую энтропию и таким образом выделить среди всех возможных (см. теорему 2 §9) пар переменных a, t одну определенную пару.
§ 11. Эмпирическое определение U, S и Т
Рассуждения, приведенные в § 10, доказывают существование температуры и энтропии. Так как функции g (t) и Ф (о) не известны, то нельзя построить Т и S из непосредственных эмпирических данных.
Рассмотрим закрытую гомогенную систему с адиабатами
и изотермами
s(P, V) = s
Z(P, 17) = /.
(И.1)
(Н.2)
В качестве независимых переменных выберем s и /, которые должны быть измерены в какой-либо эмпирической шкале.
Следовательно, имеем
лт/ 3V л . dV ..
dV =------ds 4------dt
ds dt
(11.3)
52
Глава I
и dU =ds +dt. (11.4)
ds dt
Далее справедливо
dU = T(f)dS(s) — PdV. (11.5)
Таким образом,
dU = Г P— 1 ds — P — dt. (11.6)
L ds ds J dt
Так как U является функцией состояния, то из выражений (11.5) и (11.6) следует
—Гт(0 dS(s)- — Р—1 = —( —Р . (11.7) dt L ds ds J ds \ dt )
После дифференцирования получим
dS(s) e dT (t) = dv dP _ dV dp = d(V, P) J g. ds dt ds dt dt ds d (s, t)
Таким образом, определитель $коби J (V, Р), как и всегда, является произведением двух функций, одна из которых зависит только от s, а другая только от t. Следовательно, можно написать
J(V, p) = a(s).p(^ (П-9)
где величины а ($) и 0 (/) необходимо рассматривать как эмпирически известные. Уравнение (11.8) распадается, таким образом, на два отдельных уравнения, и после интегрирования получим
S (s) = -bj a (s) ds + So, T(f) = C^(t)dt + T0.
И наконец, из (11.6) следует
U(s, t) = f(s,t) + U0,
(11.10)
(11-11)
(11.12)
где С, So, То и Uo — константы. Из них, как показано, So и Uo выбирают совершенно произвольно (т. е. им не придают какой-либо физический смысл), в то время как С определена выбором температурной шкалы (см. уравнение 10.11)). Однако нулевая точка Т однозначно устанавлива
Основные законы термодинамики ' 53
ется из физических соображений. Константу То нельзя определить произвольно. Она должна быть определена экспериментально. Уравнение (И. И) показывает, что это нельзя сделать при помощи квазистатических процессов, лежащих в основе уравнений (11.1) и (11.2). Поэтому необходимо применять нестатические процессы. Но так как абсолютная температурная шкала является универсальной, то достаточно провести такого рода опыт с любым веществом (например, опыт Гей-Люссака по расширению идеального газа в вакуум), чтобы определить То.
Можно показать, что значения S, Т и U не зависят от выбора эмпирических шкал для s и /.
Описанный метод является не чем иным, как конкретным способом построения метрической энтропии S из эмпирической энтропии s, причем предполагается существование S и термическая связь вводится в неявном виде за счет использования изотерм (ср. § 7). Интересно отметить, что хотя доказательство существования можно провести, ограничиваясь рассмотрением квазистатических процессов, однако рассмотренный выше метод требует нестатического опыта.
Применимость этого рассуждения рассмотрим на примере идеального газа. В этом случае (ср. § 7) уравнение изотермы имеет вид
PV = t, (11.13)
уравнение адиабаты записывается следующим образом:
РГ = РУ1+т = s, (Н-14)
где х и у — константы. Проведя логарифмическое диффе-
ренцирование, получаем
1 ds _ ж . 1 ds _ 1 . (11.15)
s dV “ V ’ s dP ~ P ’
1 dt = 1 , 1 dt = (11.16)
t dV “ V ’ t dP ~~ P
Отсюда следует
d(s, t) = ds dt ds dt v (11-17)
d (V, P) ~ KT ~dP _= dP dV *
54
Глава I
Согласно общему соотношению,
д (х, у) d(z, w) __ д(х,у) д (z, w) д (и, v) д (и, v) (11.18)
и, согласно (11.17),
<НУ_, Р) _ 1 д (s, t) (11.19)
Функции, определенные выражением (11.9), имеют, таким образом, вид
(11.20)
— о > г
Из (11.10), (11.11) и (11.20) следует
$(«)- r bs+S0, (П-21)
T(t) = Ct-\-T0. (11.22)
Если подставить это выражение в уравнение (11.5), то по-
лучим
dU = (Ct + То) -±-d In s — PdV, (11.23)
или, используя (11.13) и (11.14),
dU = -^-d Ins + — d In (sV~T) 1
Q т — -^-d In s 4- — dt. ci t J (11.24)
Интегрируя (11.24), получим
U = -^ Ins + — + Uo, Q 7 (11.25)
или, используя еще раз (11.13) и (11.14),
t/ = -A-ln(^) + —+ J70. С7 т (11.26)
I Для того чтобы определить То, очень разреженный газ расширяют в вакуум (опыт Гей-Люссака). При этом было найдено
Основные законы термодинамики
55
=0. (11.27)
\ dv It
Согласно условию (11.26), это возможно только при То=О. Поэтому
S = —L_ in S ч-So, (11.28)
Cl
T=Ct, (11.29)
{/ = — + U0. (11.30)
7
В соответствии с равенством (11.29) абсолютная температура и температура идеального газового термометра отличаются произвольным множителем. Поэтому их легко можно идентифицировать. Для этого пишут уравнение (11.13) в форме (7.10). Тогда имеем
t = RT*. (11.31)
Учитывая, что С = 1/7?, получим
Т = (11.32)
Для одноатомного газа у = 2/3. Отсюда из выражения (11.28), используя (11.13), (11.14), (11.31) и (11.32), получим известные формулы
S = -|-#lnT + 7?lnV + So (П-33)
(где So — новая константа) и
U = ~RT + Uo. (11.34)
§12. Измерение предельно низких температур
Принципиально важным способом установления абсолютной температурной шкалы является, как было показано, обратимый цикл между опорными точками (§ 4) и идеальный газовый термометр (§ 11). При очень низких температурах (примерно Т < ГК) оба способа практически нереализуемы. Это связано с тем, что количество теплоты, переходящее при таком циклическом процессе, становится
56
Глава I
настолько малым, что его нельзя измерить с достаточной точностью. С другой стороны, в этой области конденсируются все газы, так что от газометрического метода тоже приходится отказаться. Таким образом в распоряжении есть только эмпирическая шкала, и для низкотемпературной физики возникает важная проблема пересчитать эмпирическую шкалу на абсолютную температуру. Решение этой проблемы содержится в общем формализме § 11. Приве-
P P2
Рис. 5. К измерению крайне низких температур.
(Callen Н., Thermodynamics, New York, 1960, Wiley, p. 83, Fig. 4.9.)
дем это решение для специального случая в явном виде, так как здесь обычные способы несколько отличаются от общей схемы § 11.
Предположим, что абсолютная шкала известна вплоть до температуры То. Пусть будет установлено, что температура ниже То пропорциональна объему жидкого термометрического вещества. Коэффициент пропорциональности нужно выбирать таким образом, чтобы эмпирическая шкала Т* при То совпадала с абсолютной, т. е. чтобы То = То. Выберем в качестве независимых переменных состояния Т (соответственно Т*) и Р и определим связь между Т и Т* на прямой Р = 0.
а. Первый шаг заключается в доказательстве того, что для всех точек (Т*, 0) значения энтропии могут быть определены независимо от эмпирической шкалы.
Для этого предположим, что энтропия известна в обычных единицах для точки (То, 0). Обозначим ее через So. Если провести теперь изотермическое сжатие до давления Р2. то энтропия в точке (Те, Р2) будет*равна
Основные законы термодинамики
57
о
Как будет показано в § 24,
/ dS \ _ / дУ \
\ дР )т \ дТ )р
Если ввести еще термический коэффициент
1 / dV \ а =---------- ,
V \ дТ )р
то из (12.1) получаем
Рг
S1 = S0 — §VadP.
О
(12.1)
(12.2)
расширения
(12.3)
(12.4)
Таким путем можно рассчитать Sb если известны объемы и термический коэффициент расширения для То в области давления 0->Р2.
Исходя из точки (То, Pz), проведем квазистатическое адиабатическое расширение, в результате которого эмпирическая температура станет равной Тр Для этого процесса dS = 0, т. е. энтропия имеет то же значение, что и в точке (То, Р2). Этим показано, что на прямой Р=0 можно определить энтропийную шкалу, не зависящую от эмпирической шкалы Т*.
б. Вторым шагом будет показано, что при помощи энтропийной шкалы, установленной на прямой Р = О, Т* можно пересчитать на абсолютную температуру Т для той же прямой.
Исходим из уравнения
dU = TdS — PdV, (12.5)
которое при Р = 0 сводится к
dU = dQ = TdS. (12.6)
Отсюда следует
/ dU. \ / dQ \ ___ гр / dS. \ (12 7)
\ dr* ,'р^о ~ \ dr* /р==о ~ \ dT* '
58
Глава I
Величина (dQJdT* )р==0 экспериментально вполне доступна и является теплоемкостью при постоянном давлении, измеренной в эмпирической шкале (которую ради простоты будем рассчитывать на один моль и обозначим через Ср). Таким образом,
dS \-1 dT* )р=о
(12.8)
Но так как зависимость S от Т" известна, то для каждого Т* можно рассчитать соответствующее Т.
Практически в качестве термометрического вещества применяют определенные парамагнитные соли. Используе-
Рис. 6. Нестатические процессы для идеального газа.
(Falk G., Jung Н., Axiomatik der Thermodynamik, Handbuch der Physik, Bd. III2, p. 169, Fig. 5s Berlin 1959. Springer-Verlag.)
мый адиабатический процесс является адиабатическим размагничиванием и давлению Р = 0 соответствует сила магнитного поля Н = 0. Эмпирическую шкалу определяют при помощи магнитной восприимчивости.
§ 13. Второй закон для нестатических процессов*
Уже было упомянуто (§ 10), что из принципа Каратеодори нельзя вывести увеличение энтропии при адиабатической изоляции (4.36). На этот факт обращал внимание еще Планк (1926). Если хотят получить все следствия вто
* Этот параграф не нужен для понимания последующих, и поэтому его можно пропустить
Основные законы термодинамики
59
рого закона, сформулированные в разд. А, то для этого необходимо привлечь дальнейшие опытные факты. Сформулируем
Теорему 1
Каждая поверхность адиабаты s разлагает пространство состояния на два полупространства такого рода, что все состояния z, достижимые адиабатическим путем из расположенного на поверхности состояния z0, находятся в том же полупространстве.
Доказательство
Теорема непосредственно вытекает из принципа Каратеодори. Предположим, что недостижимые состояния г' расположены по обе стороны от s. Так как они, согласно принципу Каратеодори, должны быть расположены сколь угодно близко от z0, то отсюда должно было бы следовать, что s вообще нельзя покинуть адиабатическим путем; достижимость г' подразумевает именно достижимость всех состояний, которые расположены на поверхности адиабаты s', проходящей через гг. Но тогда это следствие должно было бы противоречить названному принципу, что и доказывает теорему.
Для идеального газа соотношения понятны из рис. 6. Ради простоты ограничимся рассмотрением гомогенной системы с двумя неизвестными переменными состояния. Каратеодори вводит в качестве определения
Теорему 2
Адиабатические переходы, исходящие из г0 и оканчивающиеся в V=V', покрывают полупрямую V'-линии, расположенной с одной стороны s.
Так как V' можно варьировать как угодно, то отсюда следует
Теорема 3
Множество всех состояний, достижимых из s адиабатическим путем, дается одним полупространством, образованным s.
60
Глава 1
Наконец имеем еще
Теорему 4
Адиабата s", расположенная между s и s', не может быть достигнута адиабатическим путем из s и из s'.
Доказательство
Теорема вытекает непосредственно из принципа Каратеодори. Это видно из рис. 7, на котором с двух сторон от адиабаты s" проведены адиабаты sx и s2.
Если из s можно было бы достигнуть адиабатическим путем s", то тогда, согласно теореме 3, это должно быть
Рис. 7. К доказательству теоремы 4.
справедливым также и Лдля s2. Поэтому s2 было бы также адиабатически достижимо из s". Если бы из s' можно было адиабатическим путем достигнуть s", то в соответствии с теоремой 3 это должно было быть также справедливым и для sx. Поэтому sx было бы также адиабатически достижимо из s".
Таким образом, в любой бесконечно малой области состояния г", расположенного на s", не существовало бы никаких состояний, которые были бы недостижимы из г" адиабатическим путем, что противоречит принципу Каратеодори. s" можно достигнуть адиабатически либо только из s, либо только из s'.
Теорема 4 утверждает, что адиабатически доступные полупространства всегда расположены на той же стороне возникшей адиабаты.
Из приведенных теорем следует общее утверждение, что существуют адиабатически необратимые процессы. Если,
Основные законы термодинамики
61
следовательно, адиабатический процесс 1->2 невозможен, то всегда возможен адиабатический процесс 2->1, что уже было использовано в § 8. Далее оказывается, что эмпирическая энтропия, определенная через семейство адиабат [уравнения (10.1) и (10.2)], при условии адиабатического изолирования может только или увеличиваться, или уменьшаться. Причем опыт не дает ответа на вопрос, как изменяется эмпирическая энтропия, поскольку направление, в котором она увеличивается, выбирается произвольно. Другими словами, можно сказать, что оба полупространства, образованные одной адиабатой, нельзя различить при помощи эмпирической энтропии. Поэтому выбор между двумя упомянутыми возможностями является чисто условным. Введем его посредством следующего определения.
Определение
При условии адиабатической изоляции эмпирическая энтропия никогда не может уменьшаться.
Отметим еще раз, что это высказывание экспериментально не поддается проверке и поэтому в принципе могло бы быть заменено на противоположное. Свойство возрастания энтропии при адиабатической изоляции, рассмотренное в § 4, нельзя, следовательно, сформулировать с помощью эмпирической энтропии. Поэтому нужно привлечь метрическую энтропию, которая, согласно (10.23), дается выражением
dS=— Ф(а)с1с. (13.1)
С
В правой части этого уравнения С, согласно определению абсолютной температуры [ур. (10.11) и (11.11)], является условно положительной константой. Согласно приведенному выше определению, da также является положительной величиной, в то время как в соответствии с условиями (10.10) знак Ф (су) такой же, как у интегрирующего делителя т. Вывод выражения (4.36) требует в рамках нашего рассмотрения ответа на следующие вопросы:
а) можно ли физически различать полупространства, образованные адиабатой;
Основные законы термодинамики
63
62
Глава I
б) приведет ли физическое различие полупространств к перемене знака интегрирующего делителя;
в) какие опытные факты определяют знак т.
На вопрос а) отвечает
Теорема 5
Полупространства, образованные адиабатой, физически различаются тем, что на каждой линии V = V' монотонно изменяется внутренняя энергия U.
Доказательство
Так как для гомогенной простой системы U, V являются полным набором переменных состояния, то на линии V = V' U не может дважды принимать одинаковых значений. Если бы это имело место, то, как видно из рис. 6, должны были бы существовать два состояния, которые обладали бы двумя одинаковыми значениями U и V, но разными значениями Р. Это должно было бы противоречить предположению, что U и V являются полным набором переменных состояния. Поэтому U на каждой линии V — V' должна монотонно изменяться. Каждый адиабатический процесс, который протекает вдоль линии V = V', связан поэтому для одного полупространства с увеличением и для другого полупространства с уменьшением внутренней энергии*. Оба полупространства, возникшие через s, являются поэтому физически различными. Таким образом, теорема доказана.
На вопрос б) отвечает
Теорема 6
Если для возможных адиабатических процессов знак da по определению положительный (см. выше), то интегрирующий делитель т имеет также положительный знак, если вдоль линии V = V возможные адиабатические процессы сопровождаются увеличением внутренней энергии. Если они сопровождаются уменьшением внутренней энергии, то т имеет знак минус.
* Для случая, изображенного на рис. 6, легко можно показать, что процессы, приводящие в верхнее полупространство, связаны с увеличецием внутренней энергии.
Доказательство
Так как, согласно § 10, эмпирическая энтропия а является функцией состояния, то а, V должны быть полным набором переменных состояния. Преобразование U, VV должно быть однозначно обратимо. Отсюда следует, что для всех состояний либо (д1Лда)у >0, либо (dU/da^y <0. Согласно уравнению (10.1),
= т. (13.2)
\ За /у
Интегрирующий делитель т поэтому для всех состояний может быть либо только положительным, либо только отрицательным. Так как левая часть выражения представляет собой изменение внутренней энергии при адиабатическом процессе, протекающем вдоль линии V = V, и изменение эмпирической энтропии для возможного процесса по определению является положительным, то из этого непосредственно следуют утверждения теоремы 6.
Таким образом, удалось свести проблему к вопросу, связаны ли адиабатические процессы, протекающие вдоль линии V = V с увеличением или с уменьшением внутренней энергии. Рассмотрение этих специальных процессов приводит к общему результату, потому что, согласно теоремам 1 и 3, все состояния, которые можно получить/исходя из данной адиабаты, расположены в том же полупространстве. Как уже было упомянуто в § 10, на поставленный вопрос нельзя ответить с помощью принципа Каратеодори, так как в нем ничего не сказано о том, какое из обоих полупространств адиабатически недостижимо. Сформулируем следующий
Эмпирический закон
Адиабатический процесс, протекающий вдоль линии V = = V, всегда связан с увеличением внутренней энергии.
Примерами таких процессов являются возникновение теплоты трения за счет механической работы или возникновение джоулева тепла за счет электрического тока. Очевидно, что в обоих случаях обратные процессы невозможны. Они противоречили бы принципам Томсона и Клаузиуса (§ 4). Фактически приведенный эмпирический закон пред-
64
Глава I
ставляет собой новую формулировку старых принципов, более подходящую для наших целей. Как и они, он подразумевает принцип Каратеодори. Поэтому чисто логически эмпирический закон (или эквивалентное высказывание) должен образовывать эмпирическую основу теории Каратеодори. Несмотря на это, автор избрал другой путь, соответствующий историческому развитию, потому что при этом многие точки зрения выступают более отчетливо.
Следствия, вытекающие из эмпирического закона, можно развить. Прежде всего находят, используя определение и теорему 6, что интегрирующий делитель т всегда положителен. Из уравнения (10.10) далее следует
Ф'(о')>0; Ф"(о")>0; Ф(°)>0. (13.3)
С учетом равенства (13.1) для адиабатически изолированной системы получаем общее соотношение
dSj> 0 (адиабатически изолированная система). (13.4)
Таким образом, выведены все следствия из разд. А в рамках теории Каратеодори.
В. РАСПРОСТРАНЕНИЕ ВТОРОГО ЗАКОНА ТЕРМОДИНАМИКИ НА ОТКРЫТЫЕ СИСТЕМЫ И ХИМИЧЕСКИЕ РЕАКЦИИ
§ 14. Постановка проблемы
В предыдущих случаях рассмотрение было ограничено простыми системами, определение которых дано в § 2. Обсудим теперь вопрос, как устранить эти ограничения и как можно сформулировать законы термодинамики в общем виде, удобном для применения.
До сих пор в качестве рабочей координаты были рассмотрены лишь объемы фаз. Свойства твердых тел, действие внешних полей, поверхностные эффекты и т. д. можно легко учитывать с помощью дополнительных рабочих координат. Последние в общем виде определяются таким образом, что дифференциал рабочей координаты dt/j после умножения на рабочий коэффициент или на обобщенную силу Yj дает работу, которая придается закрытой фа
Основные законы термодинамики
65
зе при квазистатическом изменении состояния. Эти рабочие координаты, как и объем, адиабатически свободно подвижны. Их введение поэтому ничего не изменяет в структуре теории, изложенной в разделе Б, и приводит лишь к повышению числа мерности пространства состояния. Вместо уравнения (8.4) для квазистатического изменения состояния теперь имеем
dW = — Pdv+ S Yfiy — £ Yjdyj. (14.1) /=2 /=1
Ситуация, однако, становится совсем иной, если попытаться рассмотреть открытую систему и химические реакции. Прежде всего следует заметить, что определение фундаментальных понятий работы и теплоты здесь сталкивается с трудностями. Более подробное рассмотрение существа дела выходит за рамки этой книги. Ограничимся поэтому рассмотрением двух простых примеров.
а. Пусть одна фаза будет окружена полупроницаемой мембраной, а все рабочие координаты у^ будут зафиксированы. Далее пусть вещество будет введено снаружи и пройдет через полупроницаемую мембрану. При этом, очевидно, производится работа сжатия, но объем остается постоянным. Поэтому в общем случае невозможно однозначно определить объемную работу в открытой фазе. Кроме этого нельзя также определить подведенную теплоту согласно § 8. Несколько более точно можно суть дела сформулировать таким образом, что по определению в открытой фазе нельзя совершить адиабатическую работу, как это требовалось в § 8.
б. Рассмотрим химическую реакцию, протекающую внутри закрытой фазы, которую символически запишем в виде
М = 0, (14.2)
где V. — стехиометрический коэффициент вещества i. Вследствие (14.2) изменения числа молей всех веществ, участвующих в реакции, полностью определяются изменением числа молей определенного вещества nt. Поэтому химическую реакцию можно записать в виде:
3—235
66
Глава 1
dn^^di, (14.3)
где $ — число пробегов реакции.
Величина $ не является переменной состояния. Ее равновесное значение полностью определено двумя переменными состояния системы (например, Т и V). Речь идет скорее о «внутреннем параметре» (ср. § 16), который лишь при отклонении от состояния равновесия получает самостоятельное значение. Поэтому в формализме раздела Б можно допустить внутри каждой (закрытой) фазы наличие химических реакций, так как они в явном виде вообще не входят в рассмотрение. Естественно, что таким образом нельзя получить никаких сведений о химическом равновесии.
Можно было бы подумать о том, чтобы сделать из В квазистатически-адиабатическую свободно подвижную координату за счет произвольного торможения или ускорения реакции положительным или отрицательным катализатором. Рассмотрим для этого следующий процесс*. Пусть в начальном состоянии (7\, Vlf Bi) реакция заторможена, а. За счет квазистатического изменения Т и V при постоянном система переводится в равновесное состояние, относящееся к
б. Реакция растормаживается и протекает квазистатически до значения £2. Это возможно только в том случае, если Т и V изменяются также квазистатически.
в. Реакция тормозится при значении $2, и система за счет квазистатического изменения Т и V при постоянном $2 переводится в конечное состояние (Т2, Р2, В2).
Трудность, однако, заключается в том, что сама по себе работа, совершенная в этом квазистатическом процессе, если состояния 1 и 2 расположены друг от друга сколь угодно близко, в общем случае не сводится к бесконечно малому выражению. Фактически приходят вновь к предыдущему результату, если написать форму Пфаффа
dW = — PdV — Adt,
(14-4)
* Haase R.t Thermodynamik der Mischphasen, Springer, Berlin 1956.
Основные законы термодинамики
67
где А является так называемым сродством. Это обсуждается в § 34, здесь же будет использован только тот факт, что в состоянии химического равновесия А = 0. Состояния, связанные с квазистатически-адиабатическими процессами, расположены тогда на поверхности решений дифференциального уравнения Пфаффа
dU + PdV + Adi = 0. (14.5)
Рис. 8. Квазистатическое проведение химической реакции.
Но пример показывает, что во всех частичных процессах либо А = 0, либо di = 0, так что последний член исчезает для всех квазистатически-адиабатических процессов и, таким образом, i не является рабочей координатой.
§15. Общая формулировка второго закона термодинамики
Причина трудностей, о которой говорилось в § 14, заключается в том, что были введены массы (или числа молей) компонентов не как величины состояния. В рамках теории Каратеодори это приводит к трудностям, потому что масса является адиабатически заторможенной величиной. Поэтому методом, использованным в разделе Б, нельзя показать, что энтропия зависит от масс.
Отсюда видно, что речь идет о настоящем расширении теории. Это расширение и вытекающие из него следствия являются главным содержанием термодинамики Гиббса. В феноменологических теориях такое расширение всегда требует новых эмпирических обоснований. Для этого можно пойти двумя путями. Первая возможность состоит в том, чтобы сначала построить систему аксиом таким образом, чтобы стало возможным введение массы как переменной состояния. Такая попытка была предпринята в последнее
68
Глава I
время Фальком и Юнгом. Обычный метод состоит в том, что обобщают предыдущие результаты путем аналогий и рассматривают потом совокупность экспериментально поддающихся проверке выводов как эмпирическую основу.
Выберем средний путь, отказавшись от строгой дедукции из эмпирических аксиом, но попытаемся разъяснить, какие эмпирические факты существенны для расширения теории и оправдывают этот путь.
Предыдущие исследования показывают, что для каждой закрытой фазы существует функция состояния — энтропия, для которой при ограничении одной рабочей координатой справедливо соотношение
TdS = dU + PdV. (15.1)
Каждая из величин состояния U и V в правой части равенства связана теперь с установлением «контактного равновесия». Для <7 контактным равновесием является термическое равновесие, установление которого возможно через диатермическую стенку, для V — равенство давлений (механическое равновесие), которое может установиться через подвижную перегородку, двигающуюся без трения. Обозначим обе части системы, между которыми возникло контактное равновесие, вновь через ' и ", переменные U и V вместе назовем экстенсивными параметрами Xt. Тогда можно сформулировать следующие существенные свойства контактного равновесия:
а. Каждое контактное равновесие можно получить и поддерживать за счет адиабатической изоляции всей системы в целом.
б. Для экстенсивных параметров справедлив при этом закон сохранения:
Х; + Х; = const. (15.2)
в. Процессы, ведущие к равновесию, являются адиабатически необратимыми.
Поэтому для данных значений X/, Х;- энтропия всей системы в целом как функция X/ при равновесии имеет максимум.
г. Каждое контактное равновесие определяет один интенсивный параметр Р* такого рода? что в состоянии равновесия
Основные законы термодинамики
69
p;' = pf (15.з)
и
= (15.4)
В уравнении (15.1)
р: = —, р; = —. (15.5)
1 гр ’ Z у X '
Приведенные формулировки являются описательными и не отвечают на вопрос, являются ли отдельные высказывания независимыми друг от друга.
В соответствии с полученными результатами энтропия является функцией состояния, причем до сих лор применяли только определенные типы переменных состояния. Если рассматривать приведенные выше высказывания как достаточное и необходимое условие существования переменной состояния Хь то можно сформулировать в качестве основы для расширения теории следующий
Эмпирический закон
Для массы mk (соответственно числа молей nk) каждого компонента существует контактное равновесие, которое обладает перечисленными выше свойствами (а.—г.). Примером этому служат все равновесия между открытыми фазами.
Таким образом непосредственно получаем общую формулировку второго закона термодинамики.
Для каждой фазы а, содержащей т компонентов, существует функция состояния
= S(«) pip yMt t yO)t г nO)y (15 6)
называемая энтропией фазы, которая обладает следующими свойствами.
а. Дифференциал энтропии дается выражением
TMdS{a)=dUw +PwdV<a) — £ yja)dy}a)—
1=2 т
-S V^dn™. (15.7)
/-1
70
Глава I
б. Для энтропии всей системы в целом справедливо соотношение
S = £S(a). (15.8)
а
в. При адиабатической изоляции всей системы в целом
dS >^0 (адиабатически изолированная система). (15.9)
г. Величина является универсальной функцией эмпирической температуры фазы.
Уравнение (15.7) называется фундаментальным уравнением Гиббса. Величина, введенная Гиббсом.
Т \дп<р (15Л0)
называется химическим потенциалом вещества i в фазе а. В феноменологической термодинамике химический потенциал рассчитывают на один моль. В принципе его можно рассчитывать на единицу массы (Гиббс) или на молекулу (статистическая механика). Применимость уравнения (15.7) не ограничивается обратимыми процессами, хотя в основном будут рассмотрены именно эти процессы. Здесь справедливы аналогичные рассуждения, которые уже были приведены при обсуждении уравнения (4.24). Критерием применимости является то, что внутреннее состояние фазы в каждой стадии изменения состояния полностью задается приписанными переменными. Даже если это не имеет места, то справедлива также интегральная форма уравнения (15.7), если начальное или конечное состояния процесса описываются при помощи этих переменных.
Остается еще вопрос, как сформулировать первый закон термодинамики в рамках приведенного расширения теории. По причинам, объясненным в § 14, для открытой системы являются беспредметными как классическая точка зрения (эквивалентность теплоты и работы), так и точка зрения Каратеодори (определение теплоты). Можно, правда, сохранить обычную формулировку, если вновь независимо и произвольно определить подведенную теплоту. В нашем изложении такие рассуждения не представляют интереса. Тогда остается только высказывание, что внутренняя энер
Основные законы термодинамики
71
гия фазы является функцией состояния. Простые рекомендации § 8, естественно, теперь больше не применимы, и вопрос экспериментального определения требует более подробного изучения.
Рассмотрим для этого адиабатически изолированную систему, состоящую из двух частей ' и ". Если зафиксировать определенные стандартные состояния сравнения, то можно по способу, описанному в § 8, определить внутренние энергии U, U' и U”. Удалим теперь адиабатическую перегородку между ' и ", но будем поддерживать адиабатическую изоляцию всей системы в целом и исключим также производство внешней работы. Тогда части системы ' и смешиваются при изменении температуры и давления, причем общая энергия останется постоянной. Этот процесс можно представить как изменение состояния открытой фазы', для которой изменение внутренней энергии равно
kU' = U — U'. (15.11)
Так как в правой части этого уравнения находятся только измеримые величины, то и изменение внутренней энергии открытой фазы в принципе является измеримой величиной.
Глава II
Общие условия равновесия и стабильности
§ 16. Обсуждение понятия равновесия. Внутренние параметры
В § 2 предварительно было обсуждено понятие термодинамического равновесия. Теперь уточним его в
Определении
Изолированная система находится в термодинамическом равновесии, если в ней с измеримой скоростью не происходит никаких изменений состояния.
Существование равновесия непосредственно следует из принципа Клаузиуса и его обобщений на другие процессы выравнивания. Поэтому изменения состояний, происходящие в изолированной системе, необратимы, т. е. они могут протекать (при отсутствии внешних воздействий) только в одном направлении. Отсюда уже следует, что такие процессы должны асимптотически стремиться к конечному состоянию. Этот вывод станет более наглядным, если учесть (факт, содержащийся в принципе Клаузиуса), что рассматриваемые процессы являются процессами выравнивания, «движущая сила» которых равна разности интенсивных параметров, стремящихся с течением процесса к нулю (ср. § 15).
Добавление «с измеримой скоростью» означает, что о равновесии можно говорить, только учитывая определенный процесс и конкретные экспериментальные условия. Пример 1
Пусть в объеме V находятся Н2, О2 и Н2О (пар) с неравномерным распределением вещества и температуры. При комнатной температуре и в отсутствие катализатора протекают процессы теплопроводности и диффузии с измеримой, а
Общие условия равновесия и стабильности
73
превращение гремучего газа с неизмеримо малой скоростью. После выравнивания разности температур и концентраций система для любого соотношения масс трех веществ находится в термодинамическом равновесии. При наличии катализатора или достаточно высокой температуры протекает реакция между гремучим газом с измеримой скоростью. Поэтому система находится лишь тогда в термодинамическом равновесии, когда установится также и химическое равновесие.
Пример 2
Известно, что распределение нуклонов в ядрах не соответствует термодинамическому равновесию. Но так как при условиях, с которыми обычно имеет дело термодинамика, термические ядерные процессы протекают с бесконечно малой скоростью, то этим фактом можно полностью пренебречь.
Понятие «абсолютного равновесия» или «равновесия с учетом всех мыслимых» процессов не имеет физического смысла. Особую трудность представляют некоторые твердые вещества (например, стекла, сплавы). В условиях опыта происходят изменения состояния неизмеримо медленно, но свойства системы зависят от ее предыстории. Вопрос применимости термодинамики требует в этом случае более детального изучения. Эта проблема здесь рассматриваться не будет.
Формализм, развитый в § 15, дает возможность описать равновесные и неравновесные состояния между различными фазами. Остается еще вопрос, как следует описывать установление термодинамического равновесия в каждой однокомпонентной системе. Если такая система не находится в термодинамическом равновесии (здесь не учитывается наличие внешних полей), то она имеет в общем случае местные неоднородности и поэтому, согласно общему определению § 2, не является фазой. Тогда нужно расширить следствия § 15 и применить их к бесконечно малым элементам объема, считая их гомогенными. В этом случае величины состояния § 15 станут величинами поля, что приводит к формализму термодинамики необратимых процессов. В обычной термодинамике в явном виде этот формализм не применяют. Следовательно, фазы всегда рассматривают как
74
Глава II
макроскопические. Тогда остаются еще некоторые возможности отклонения от термодинамического равновесия, которые описываются при помощи внутренних параметров, независимых от координаты.
Важнейшим примером внутреннего параметра является число пробегов реакции В, введенное в § 14. Существенные свойства внутренних параметров заключаются в следующем:
а. Внутренние параметры не являются переменными состояния в смысле определения § 2.
б. Каждый внутренний параметр можно выразить через переменные, появляющиеся в фундаментальном уравнении (15.7), с учетом известных дополнительных условий.
Свойство а. следует непосредственно из факта (см. § 14), что в равновесии интенсивный параметр (сродство А), сопряженный с $ по уравнению (15.4), становится равным нулю.
Свойство б. для числа пробегов реакции непосредственно вытекает из уравнений (14.2) и (14.3). Большинство внутренних параметров можно свести к «обобщенным химическим реакциям», т. е. их можно выразить с помощью уравнений, которые формально идентичны (14.2) и (14.3). В этих случаях внутренние параметры можно выразить через числа молей и соответствующие дополнительные условия. Может также случиться, что внутренний параметр выразится через другие переменные уравнения (15.7) и соответствующие дополнительные условия.
В соответствии с положением а. внутренние параметры являются, таким образом, независимыми переменными только вне равновесия. Это просто соответствует факту, уже упомянутому в § 2, что число величин, полностью описывающих состояние в равновесии, меньше, чем в каждом неравновесном состоянии. Согласно б., набор переменных фундаментального уравнения (15.7) может сохраняться также и для этого случая, если введены дополнительные условия. Но так как энтропию определяют как функцию состояния, т. е. в предположении внутреннего равновесия, то остается ответить еще на вопрос, можно ли определить энтропию для упомянутого отклонения от равновесия.
Общие условия равновесия и стабильности
75
Разъясним проблему на простом примере гомогенной реакции:
А-^В + С. (16.1)
Пусть система, которая должна содержать только компоненты А, В и С, будет адиабатически изолирована, и пусть объем будет единственной рабочей координатой. Предположим, что реакция (16.1) заторможена отрицательным катализатором, так что с уверенностью можно утверждать, что энтропия существует в виде
S = S(t7, V, иА, ив, ис). (16.2)
Пусть катализатор теперь будет удален на короткое время, при этом произойдет бесконечно малое превращение, и затем снова будет введен. Согласно второму закону термодинамики, энтропия при этом превращении увеличится. Так как система после этого превращения вновь приходит во внутреннее равновесие, то, согласно выражениям (15.7) и (16.1), для приращения энтропии можно написать
TdS = pAdnA— PBdnB — Pcdnc. (16.3)
((/ = const, V = const).
Если в соответствии с § 14 ввести число пробегов реакции $ и сродство А, то уравнение (16.3) переходит в
TdS = Adi (U = const, V = const). (16.4)
Таким образом определено изменение энтропии для гомогенной системы также в отсутствие химического равновесия.
Приведенные рассуждения легко можно распространить на другие внутренние параметры. Они основаны на том, что, с одной стороны, о равновесии можно говорить, только учитывая конкретный процесс, с другой стороны, введение торможения повышает число переменных состояния.
Было бы, конечно, абсурдным предположить, что в приведенных рассуждениях энтропия каким-либо образом «создается» за счет использования торможения. Введение понятия торможения является только мысленным средством, которое делает возможным определение энтропии вне равновесия в приложении к современным результатам. Это аналогично тому, как определяют переменную ско
76
Глава II
рость проведением касательной к кривой путь — время. Вопрос, возможно ли торможение в каждом конкретном случае, для дальнейшего не имеет значения.
Введение внутренних параметров не ограничивается гомогенными фазами. Легко увидеть, что понятие числа пробегов реакции можно обобщить также на гетерогенные реакции. В других случаях можно также ввести для гетерогенной системы один или несколько внутренних параметров, если вариации переменных состояния ограничены дополнительными условиями. В § 17 будет пояснено это на простом примере.
Теперь определим энтропию также для всех неравновесных состояний, которые требуется рассмотреть. Таким образом, свойство возрастания энтропии можно использовать для формулировки условий термодинамического равновесия. Эту проблему обсудим в следующем параграфе.
§ 17. Условия равновесия Гиббса
Одной из главных проблем термодинамики является конкретная формулировка условий термодинамического равновесия для различных специальных случаев (например, равновесие жидкость — пар, осмотическое равновесие, химическое равновесие). Старые методы решения этой проблемы (которые были распространены в Европе вплоть до 1930 г.) состояли в том, что для каждой конкретной задачи конструировали обратимый цикл.
Этот метод дал важные результаты. Однако, несмотря на то что он разработан детально, применение его часто приводит к ошибкам. Теперь он повсюду заменен методом Гиббса, который исходит из общей формулировки условий равновесия, и отсюда чисто математическим путем можно вывести все специальные случаи.
Формулировка условий равновесия Гиббса связана с экстремальными принципами механики чисто формально. Поэтому эти условия, как и принципы механики, можно рассматривать с аксиоматической точки зрения. Не будем далее прослеживать эту точку зрения, она прояснится, когда из формулировки Гиббса будут получены следствия, которые были введены в гл. I, как эмпирические данные. В дальнейшем будет дана сначала математическая формули
Общие условия равновесия и стабильности
77
ровка условий равновесия, затем некоторые объяснения и доказательства, поскольку они не содержались в гл. 1
Условия равновесия по Гиббсу заключаются в следующем.
Для равновесия изолированной системы, рабочие координаты которой зафиксированы, необходимо и достаточно, чтобы либо
(85)у<0, (17.1)
либо
(8J7)S > 0. (17.2)
Пояснения
а. Символ 8 означает виртуальное смещение в смысле аналитической механики. Следовательно, здесь представляют мысленное изменение состояния, которое является бесконечно малым первого порядка и удовлетворяет следующим условиям:
а) изменение состояния возможно, т. е. согласовано с общими условиями существования системы (ср. б.); Р) оно не является функцией времени;
у) энтропия определена также и для измененного состояния.
б. Условия существования системы прежде всего следуют из определения закрытой системы с фиксированными рабочими координатами. Сверх этих условий индекс U указывает, что при изменении внутренняя энергия должна оставаться постоянной. Отсюда следует также, что условие (17.1) для энтропии фактически относится к изолированной системе. Условие а. а) исключает из рассмотрения:
а) изменения состояний, которые предотвращаются путем торможений (например, реакция гремучего газа при комнатной температуре в отсутствие катализатора);
Р) изменения состояний, при которых уменьшается энтропия адиабатически изолированной части системы.
В случае выполнения условий равновесия (17.2) система не является закрытой, так как здесь дополнительные условия требуют постоянства энтропии, что не подразумевает адиабатическую изоляцию. Впрочем, равновесие (17.2) для закрытой системы бессмысленно, так как здесь невозможна вариация внутренней энергии.
78
Глава II
в. Знак равенства в уравнении (17.1) означает, что энтропия как функция рассматриваемых в состоянии равновесия переменных имеет стационарное значение. Так как уравнение (17.1) содержит только вариации первого порядка, то нельзя сделать никаких заключений о том, является ли стационарное значение максимумом или минимумом. К этому вопросу вернемся еще в § 18.
Знак неравенства (часто вызывающий недоразумения) относится к случаю, при котором возможны только односторонние виртуальные смещения и поэтому нельзя решить, является ли равновесное значение энтропии стационарной точкой в математическом смысле. Разъясним соотношения на примере* и рассмотрим для этого распределение вещества между двумя фазами ' и ". В частности, можно представить распределение вещества между двумя несмешивающи-мися растворителями (например, бензол и вода). Распределение может быть описано с помощью параметра
п,.
$ = -Ч- . (17.3)
ni
Следует различать два случая:
а. Вещество I находится в равновесии в обеих фазах. Равновесное значение £равн тогда положительно и конечно. (Пример: распределение бензойной кислоты в воде и бензоле.) Поэтому функция S(£) имеет физический СМЫСЛ Как ДЛЯ $ < ^равн, так и для $ > £равн. Возможны также виртуальные смещения по обе стороны от $равн. В этом случае уравнение (17.1) свидетельствует о том, что £Равн является стационарной точкой функции S($) (рис. 9). Для стационарной точки должно быть
Поэтому при постоянном значении всех остальных переменных вариация первого порядка равна
85 = /'—') 8$ = 0. (17.5)
\ /равн
* См. сноску на стр. 66.
Общие условия равновесия и стабильности
79
Р. Вещество i находится в равновесии только в одной фазе, например в ". Равновесное значение $равн равно тогда нулю. (Пример: приблизительно это имеет место при распределении нафталина между водой и бензолом.) Тогда кривая S($) для $ < £равн не имеет физического
смысла. Поэтому возможны только смещения > О (рис. 10). В этом случае из уравнения (17.1) следует, что при возможном смещении oS или равно нулю, или отрицательно. Из рисунков отчетливо видно, что производная в точке $равн может быть либо равной нулю, либо отрицательной. Вместе с тем также возможно, но не необходимо, что $равн является стационарной точкой.
Следовательно,
<о (17.6)
\ /равн
и, таким образом, 8^0. \ дб /равн
(17.7)
80
Глава II
Величина В для всей системы в целом является внутренним параметром в смысле определения § 16. На этом простом примере можно легко увидеть, как ее можно свести к переменным состояния § 15. Выберем в качестве переменных числа молей и п\. Так как система закрытая, то справедливо условие, что при всех возможных изменениях сумма щ + п\ должна оставаться постоянной. Поэтому случай а. представляется теперь следующим образом:
a'. 83 = 83' + 83" = (8П;+ (Ъп. \ ^ni / равн \ ^ni /равн
с дополнительным условием
(17.8)
Ъп. + 8п". = 0.
В случае (3. имеем
(17.9)
Р'. 83=83'+83"=/-^ Ъп. (17.10)
\ ^ni /равн 1 с дополнительными условиями
Ъп. + W. = 0, Ъп. > 0, Ъп. < 0.
(17.11)
Эти формулировки ясно показывают, что в общем виде условия равновесия имеют характер экстремальной задачи с дополнительными условиями.
В качестве простого примера выберем распределение вещества. Фактически случай [3. на практике не играет никакой роли. В дальнейшем при конкретных расчетах, как правило, его не будем учитывать (хотя он подробно был рассмотрен Гиббсом). Он представляет интерес для химических реакций (§ 36) и появляется также при некоторых внутренних параметрах, которые встречаются в статистической термодинамике (например, степень дальнего порядка бинарных смешанных кристаллов). Поэтому его обсуждение оправдано не только историческими причинами.
Доказательства*.
Условие равновесия (17.1) непосредственно следует из второго закона термодинамики, следствия (15.9) и опреде
Общие условия равновесия и стабильности
81
ления равновесия в § 16. Поэтому опустим доказательство, что условие необходимо и достаточно. Следует принять во внимание, что в случае знака равенства условие (17.1) содержит только часть следствий условия (15.9), так как утверждается лишь, что в равновесии энтропия должна принимать стационарное значение. Возможность такого разложения, проведенного впервые Гиббсом, основана на формулировке в виде экстремального принципа. Она оказалась чрезвычайно плодотворной для термодинамики.
Условие равновесия (17.2) не следует непосредственно из (15.9). Поэтому докажем, что оно представляет собой формулировку, эквивалентную (17.1). Доказательство использует следующие факты:
а. По определению система не является закрытой. Если не наложены дополнительные условия [как в случае формулировки (17.1)], то, следовательно, всегда возможен подвод и отнятие теплоты.
р. Согласно условию а., всегда можно за счет подвода или отвода теплоты одновременно увеличить либо уменьшить энергию и энтропию системы.
Доказательство эквивалентности уравнений (17.1) и (17.2)
а. Допустим, что (17.1) не справедливо. Тогда должно существовать виртуальное смещение, для которого
8S>0; W = 0. (17.12)
Из этого измененного состояния II можно за счет одновременного уменьшения энергии и энтропии достигнуть состояния III, для которого (если его рассматривать как вариацию начального состояния I) справедливо
8S = 0; W <0. (17.13)
Но таким образом нарушается условие (17.2) (рис. 11, а).
б. Допустим, что условие (17.2) не выполняется.
Тогда должна существовать вариация, для которой
W <0; 8S = 0. (17.14)
Исходя из этого измененного состояния II за счет одновременного увеличения энергии и энтропии можно достигнуть состояния III, для которого (если его рассматривать как вариацию начального состояния I)
82
Глава II
справедливо
W = 0; 8S>0.] (17.15)
Таким образом, условие (17.1) также нарушено, следовательно эквивалентность условий (17.1) и (17.2) доказана.
Тот факт, что следствия второго закона термодинамики можно также представить с помощью внутренней энергии, если выбрать энтропию в качестве переменной состояния, был установлен Гиббсом. Он показывает, что расчленение
Z
Отвод
теплоты $S>0
III 6S~0. 1
6U<0
Подвод теплоты
SU=0, 6S>0
Il <5U<0, I $S=0>
III
Рис. 11. К доказательству эквивалентности уравнений (17.1) и (17.2).
на первый и второй законы термодинамики в принципе произвольно и его можно понять исключительно с исторической точки зрения (ср. обсуждение первого закона термодинамики в § 8). Использование внутренней энергии как функции состояния и энтропии как независимой переменной состояния приводит, как будет видно в гл. III, к упрощению формального аппарата.
§ 18. Условия стабильности
Уже неоднократно было отмечено, что условия равновесия (17.1) и соответственно (17.2) (в случае знака равенства) содержат лишь то, что в равновесии при данных условиях энтропия и соответственно внутренняя энергия имеют стационарное значение. Эта ситуация точно соответствует соотношениям механики, где определение равновесия оставляет открытым вопрос, является ли оно стабильным, нестабильным, нейтральным или метастабильным.
Общие условия равновесия и стабильности
83
Формально эти ограничения основаны на том, что уравнения (17.1) и (17.2) содержат только вариации первого порядка, т. е. разложения Тейлора (17.5) и (17.8) прерываются на линейном члене. Чтобы использовать все следствия второго закона термодинамики для формулировки условий равновесия, необходимо рассмотреть также вариации более высокого порядка. Фактически практическое значение для термодинамики, как будет видно из дальнейшего (гл. VI), имеют только вариации второго порядка. Пока это не будем учитывать и напишем для возможного смещения, определяемого с учетом вариаций более высокого порядка, символ А. По аналогии с механикой различают следующие случаи.
а. Стабильное равновесие
Для всех возможных смещений справедливо
(AS)y<0 (18.1)
ИЛИ
(At/)s > 0. (18.2)
Таким образом при стабильном равновесии для данных условий энтропия имеет максимум (соответствующий наибольшему значению), а внутренняя энергия минимум (соответствующий наименьшему значению). Это является следствием второго закона термодинамики.
б. Нестабильное равновесие
Существуют виртуальные смещения, для которых
(AS)y>0 (18.3)
или
(Д[/)5<0. (18.4)
В этом случае для рассматриваемого смещения стационарное значение энтропии имеет минимум, а внутренней энергии — максимум.
Нестабильное равновесие физически не реализуемо. Это утверждение нередко подвергается сомнению на основе феноменологических рассуждений, однако его
84
Глава II
можно доказать методами статистической термодинамики.
в. Нейтральное равновесие
Существуют возможные изменения, для которых
(AS)„ = 0 (18.5)
или
(Д[/)5 = 0. (18.6)
Этот случай принципиально возможен, но будет рассмотрен позднее (гл. IV).
г. Метастабилъное равновесие
Осуществляется тогда, когда равновесие стабильно по отношению к бесконечно малому соседнему состоянию, но нестабильно по отношению к конечно удаленному состоянию. Известным примером этого служит переохлажденная жидкость. В рамках теории термодинамической стабильности этот случай не имеет самостоятельного значения. Суть дела лучше можно уяснить через понятие торможения.
Г л аеа 111
Термодинамические потенциалы и функции Массье—Планка
§19. Преобразование Лежандра. Гомогенные функции и теорема Эйлера
Для построения формального аппарата термодинамики необходимы еще некоторые математические методы, которые будут рассмотрены в данном параграфе.
а. Преобразования Лежандра
Рассмотрим следующую задачу: пусть задана функция
У = ....Хп). (19.1)
Найдем преобразование, которое
а) вводит в качестве независимых переменных одну или несколько производных типа
(3) является однозначно обратимым.
Условие |3. требует, чтобы из трансформированной функции можно было однозначно восстановить первоначальную функцию (19.1). Другими словами, требуется, чтобы при преобразовании сохранялось математическое содержание (19.1) (или соответствующая физическая информация).
Рассмотрим эту задачу и ее решение на примере одной переменной. Тогда имеем исходную функцию
86 Глаза HI
Термодинамические потенциалы и функции Массье — Планка 87
У = У(х). (19.3)
Это уравнение представляет собой кривую в плоскости х,у, наклон которой для каждого значения х равен
р = _^/(х). (19.4)
ах
Можно было бы решить задачу, просто исключив х из уравнений (19.3) и (19.4). Тогда действительно получим у как функцию р:
У = У.(р)- (19.5)
Но если захотим вновь получить из (19.5) начальную функцию (19.3), то видно, что эта задача решается неоднозначно и, таким образом, нарушается условие |3).
Формально это основано на том, что выражение (19.5) является дифференциальным уравнением первого порядка,
Рис. 12. Кривая как огибающая семейства ее касательных.
решение которого представляет собой семейство кривых в плоскости х,у. При переходе от (19.3) к (19.5) частично теряется математическое содержание (19.3). Решение задачи, удовлетворяющее условиям а) и |3), основано на том, что кривую в плоскости х, у, с одной стороны, можно представить как геометрическое место точек (х,у), удовлетворяющих уравнению (19.3), с другой — как огибающую семейства ее касательных.
Уравнение, соответствующее семейству касательных, математически эквивалентно уравнению (19.3). Если обозначить наклон касательной через р, соответствующий ей отрезок ординаты через ф, то уравнение
Ф = Ф(р) (19.6)
формально будет представлять собой решение задачи и каждому значению р будет соответствовать одно значение ф. Так как именно (19.6) является семейством касательных к кривой (19.3), то р определяется по уравнению (19.4) и, таким образом, выполняется условие а); далее, выражения (19.3) и (19.6) по отношению друг к другу однозначно обратимы и, таким образом, выполняется также условие |3). Остается еще выразить в явном виде функцию (19.6) из (19.3). Рассмотрим для этого касательную к кривой (19.3) в точке (х,р), которая имеет наклон р и отсекает отрезок ординаты ф. Тогда по определению
X
(19.7)
ИЛИ
— у — рх. (19.8)
Если теперь определить из уравнений (19.3), (19.4) и (19.8) величины х и р, то получим искомое соотношение между ф и р. Легко понять, что из (19.6) при обращении процесса снова получается (19.3). Из условия (19.8) после дифференцирования и учета (19.4) получим
йф = — pdx — xdp + dy = — xdp
(19.9)
или
(19.10)
х _ 44 dp
Если из уравнений (19.6), (19.8) и (19.10) определить переменные ф и р, то снова получим уравнение (19.3). Определение переменных возможно только тогда, когда р является функцией х. Разлагая (19.4) в ряд Тейлора
Ьр ~ JdL &х + ..., dx
(19.11)
получим в качестве достаточного условия
0
dx
(19.12)
88
Глава III
ИЛИ
(19.13) dx2 dp2
Эти условия достаточны для большинства применений. Некоторые специальные случаи, в которых они не выполняются, требуют особого обсуждения.
Представим еще раз схематически описанный метод:
У = У(х), dy dx ty = —px + y, 1 -е- II п II * , -е- Ей + |-G- 3^ (19.14)
Исключение х и у дает Исключение р и ф дает
Ф = Ф (Р) у = у(х)
Преобразования у(х) ф (р), определенные с помощью приведенных уравнений, называются преобразованиями Лежандра. ф(р) — результат преобразования Лежандра функции р(х). Преобразования Лежандра являются частным случаем преобразований прикосновения. Они встречаются в классической механике при переходе от формулировок Лагранжа к формулировкам Гамильтона. Важными для нас являются следующие свойства.
Преобразования Лежандра однозначно и обратимо превращают не каждую точку плоскости х,у в точку плоскости ф, р, а каждую точку кривой у(х) в точку кривой ф (р).
Обобщение этого рассуждения на функцию (19.1) п независимых переменных требует перенесения рассмотрения с плоскости в (и + 1)-мерное пространство, что, впрочем, не представляет трудностей. Не будем это приводить подробно, а дадим лишь формулы. Рассмотрим особенно важный для применения случай преобразования только поднабора (хь ..., х^ полного набора (хь ..., хл). Геометрически это значит, что преобразование проводится в (k + 1)-мерном подпространстве (и + 1)-мерного пространства, причем, естественно, подпространство должно содержать координату у. При таком А-кратном преобразовании Лежандра переменные xk+i, ..., хп следует рассматривать как параметры. В результате получаем схему, аналогичную (19.14):
Термодинамические потенциалы и функции Массье — Планка 89
у=у(х1,...,хк,..., х„),ф=ф(р1, ...,pk,xk+vx,
йУ = l^Pidxi’
Z=1
k
^=У~ ^Pixi>
Z=1
—xt = (j < k),
1 dp, V =
Pj =
J dxj k (Ц = — 2 XidPi +
Z=1
+ 2 pJdxi’ z=H-i
k
у=ф+ 2 x^-
Z=1
(19.15)
Исключение у и xlt ... , xk Исключение ф и pv pk
дает
дает
Ф = Ф (Р1> Pk’ Xk+v-’ Хп) У = У (Х1’ -> xk’ •••> Хп)-
В этом общем случае достаточным условием для существования преобразования будет
d(Pi, Pfe)
d(xlt .... xk)
д^у I dXjdXj J
^0 (f, j<k). (19.16)
Определители $коби pt и хг должны, следовательно, отличаться от нуля.
б. Гомогенные функции и теорема Эйлера Определение
Если функция
? = ф(х1, , хй) (19.17)
удовлетворяет уравнению
<p(axlt ах„) = az<p(xp х„), (19.18)
где I — положительное целое число, то она называется гомогенной функцией /-й степени.
Термодинамические потенциалы, и функции Массье — Планка 91
90 Глава III
Теорема Эйлера
Для гомогенной функции /-й степени выполняется соотношение
п дер
lcp(xlt .... хп) = V xt. (19.19)
Z=1
Доказательство
Если продифференцировать уравнение (19.18) по а, то получим
п
У ^^=«-4(4. <19-20)
d(axi) да
Z=1
Это соотношение должно выполняться для каждого значения а. Если подставить а = 1, то получим уравнение (19.19). В термодинамике имеют значения только гомогенные функции первой степени, где / = 1.
§ 20. Фундаментальное уравнение. Экстенсивные и интенсивные параметры. Уравнения состояния. Уравнение Гиббса—Дюгема
Рассмотрим теперь более подробно фундаментальное уравнение Гиббса (19.7). Ради простоты объем будем считать единственной рабочей координатой. Индексы фаз в данном случае не важны, и они будут опущены. Напишем фундаментальное уравнение в виде
т
dS=-^ dU+ JLdV- dnt (20.1)
Z=1
или в интегральном виде
S = S(U, V, nv ... , nJ. (20.2)
Ранее уже было показано для условий равновесия и стабильности (§ 17 и 18), что эквивалентное изображение возможно с помощью внутренней энергии. Придадим соответствующую форму фундаментальному уравнению. Так как по определению (§ 10)
/ dS \
UU.........»,>0- <20-3>
то энтропия при постоянстве остальных величин состояния представляет собой однозначную, непрерывную и дифференцируемую функцию внутренней энергии. Поэтому уравнение (20.2) можно однозначно решить относительно U, в результате чего получим
U = U(S,V,nv ... ,пт) (20.4)
или в дифференциальной форме
dU = TdS— PdV + j? (20.5)
Z=1
Это уравнение также называется фундаментальным уравнением Гиббса. Уравнение (20.1) называется энтропийным выражением, уравнение (20.5) — энергетическим выражением. В современной термодинамике в основном используют энергетическое выражение, в то время как энтропийное выражение имеет значение прежде всего для термодинамики необратимых процессов и статистической механики. В дальнейшем будем учитывать энтропийное выражение только при некоторых общих рассуждениях.
Переменные состояния в фундаментальном уравнении по своей конструкции распадаются на два класса. Для переменных первого класса характерно, что при составлении из двух частей ' и " системы в целом (без штриха) выполняется соотношение
х;. + х; = х.. (20.6)
Эти переменные состояния, к которым (что совершенно ясно) прежде всего относятся объем и число молей, называются экстенсивными параметрами.
Переменные второго класса, как подробно показано в § 15, можно определить через контактное равновесие таким образом, что при равновесии
Р'< = Рг (20.7)
Эти переменные, примерами которых являются температура, давление и химический потенциал, называются интенсивными параметрами. Из выражений законов термодина-
92
Г лава III
мики следует [см. уравнения (8.8) и (10.26)], что внутренняя энергия и энтропия также являются экстенсивными параметрами. Теперь можно сформулировать важнейшие свойства фундаментального уравнения. Но прежде всего их надо сопоставить и затем дать некоторые разъяснения.
Свойства фундаментального уравнения
а. Фундаментальное уравнение в энтропийном и энергетическом выражениях является функцией, зависящей только от экстенсивных параметров.
б. Фундаментальное уравнение в энтропийном и энергетическом выражениях является однородной гомогенной функцией первой степени всех независимых переменных.
в. Фундаментальное уравнение в энтропийном и энергетическом выражениях является характеристической функцией, т. е. оно содержит все термодинамические сведения о данной системе.
г. Определение интенсивных параметров в энтропийном выражении имеет вид
р;= (20.8)
и Л/
и в энергетическом выражении
Л = (20.9)
ОЛ £
д. Каждый интенсивный параметр можно представить как функцию тех же независимых переменных, которые имеются в соответствующей форме фундаментального уравнения. Это уравнение, которое в общем виде записывается как
Рг = Рг(Х1,...,Х„)> (20.10)
называется уравнением состояния. Функция (20.10) является гомогенной функцией нулевой степени.
е. Уравнения состояния не являются независимыми друг от друга, так как между интенсивными параметрами имеется дополнительное соотношение, дифференциальная форма которого называется уравнением Гиббса — Дюгема. В энтропийном выражении оно имеет вид
Термодинамические потенциалы и функции Массъе — Планка 93
m
(20.11)
и в энергетическом выражении
SdT — VdP + 2 ntdv-i = 0. (20.12)
Z=1
Объяснения и доказательства
К пункту а. Это следствие вытекает из уравнений (20.1) и (20.5) в связи с законами термодинамики и определением экстенсивных параметров.
К пункту б. Если предположить, что в уравнении (20.6) части системы ' и " равны, то следствие вытекает из а. и уравнения (19.18).
К пункту в. В этом высказывании содержатся следующие свойства:
а. Если фундаментальное уравнение известно для каждой гомогенной части системы, то можно полностью и в явном виде рассчитать термодинамическое равновесие всей системы подстановкой в (17.1) соответственно (17.2). р. Из (18.1) — (18.6) получаются все данные о стабильности равновесия.
у. Первые частные производные фундаментального уравнения дают интенсивные параметры. В частности, в энергетическом вы р ажен ии
I dU \ \ ds л» «1.....пт
7 dU \
\ dV «1» ••• .
7 dU \
\ dnt V, п.
(20.13)
(20.14)
(20.15)
3. Вторые частные производные дают дальнейшие измеримые величины и на основе общего соотношения для полного дифференциала df
d*f d*f
дх ду ду дх
(20,16)
94
Глава III
уравнения, которые связывают эти величины между собой. Приведем пример только для энергетического выражения, так как вторые производные подробно обсудим в § 24 и 25. В соответствии с (20.5) и (20.13)
. (20.17)
am dv \ ds ) \ dv )st n
Эта величина представляет собой прирост температуры на единицу объема при квазистатически-адиабатическом расширении. Согласно (20.5) и (20.14),
д2и _ д ( dU \ __ _ / дР \ dSdV ~ dS [ dV ) \ д8 Jv, п ’
(20.18)
Величину, стоящую справа, можно также написать в виде T(dP/dQ)v. Она означает повышение давления при подводе единицы количества теплоты при постоянном объеме.
Согласно (20.16),
' дТ X _ _ / дР \
, dV )s, п \ 08 /у. п *
(20.19)
Соотношения такого типа называют соотношениями Максвелла. Они будут подробно рассмотрены в § 24.
Существенно, что свойства характеристической функции присущи не энтропии и внутренней энергии как таковым, но только выбранному набору переменных в фундаментальном уравнении. Это хорошо видно на противоположном примере. Внутреннюю энергию можно представить как функцию переменных Т, V, и (для однокомпонентной системы). Тогда получим
dU = (—} dT + dv + (—) dn. (20.20)
\ дТ )v, n \ dV /т.п * \ dn )v,t
Это уравнение вполне корректно и, согласно первому закону термодинамики, dU также в этом представлении является полным дифференциалом. Функция
U = U(T, V, п) (20.21)
не является характеристической функцией. Из нее нельзя ни рассчитать термодинамическое равновесие, ни
Термодинамические потенциалы и функции Массье — Планка 95
получить дифференцированием интенсивные параметры Р и |л. В основе этого лежит тот факт, что в уравнении (20.21) энтропия определена не однозначно. Фактически сравнение с уравнением (20.13) показывает, что уравнение (20.21), если рассматривать его исходя из фундаментального уравнения, представляет собой дифференциальное уравнение первого порядка в частных производных, решение которого содержит произвольную функцию. Следовательно, из фундаментального уравнения (20.4) можно получить уравнение (20.21), но обратный процесс невозможен. Другими словами, это значит, что выражения (20.4) и (20.21) не эквивалентны и (20.21) содержит меньше информации, чем (20.4). Уравнение (20.20) в принципе уже знакомо из § 8. Как будет видно, оно имеет существенное значение и непосредственно дает определение мольной теплоемкости пр и постоянном объеме Cv. С учетом того, что и = U/n, получим
с . (20.21а)
v \дТ )v.n v
К пункту г. Экстенсивные параметры в энтропийном и энергетическом выражениях одинаковы, но как только энтропия и энергия поменяются ролями, то определение интенсивных параметров в явном виде, как видно из (20.1) и (20.5), для обоих выражений является различным. Большое преимущество энергетического выражения, которое объясняет его предпочтение в термодинамике, состоит в том, что в этом случае интенсивные параметры (Т, —Р, |if) являются непосредственно измеримыми величинами, в то время как это не имеет места для интенсивных параметров в энтропийном выражении (1/Т, Р/Т — ^/Т).
К пункту д. Для однокомпонентной системы уравнения состояния в явном виде в энтропийном выражении записываются следующим образом:
V’ (20-22)
V, п),
(20.23)
96
Глава III
V, n); (20.24)
в энергетическом выражении
Т = T(S,V,ri), (20.25)
Р = Р (S, I/, п), (20.26)
[1 = |л (S, V, и). (20.27)
В старой, но все еще часто применяемой терминологии уравнение
Р = Р(Т, V, п) (20.28)
называется термическим уравнением состояния, уравнение
и = и (Г, V, п) (20.29)
называется калорическим уравнением состояния. Сравнение с выражениями (20.22) и (20.23) показывает, что (20.28) и (20.29) относятся к энтропийному выражению и являются преобразованиями систематически более ясных уравнений (20.22) и (20.23). Термическое уравнение состояния идентично уравнению изотермы [уравнение (7.7)1, введенному в § 7. В уравнении (20.28) нужно лишь заменить эмпирическую температуру на абсолютную.
То, что уравнение состояния является гомогенной функцией нулевой степени, следует из б. с учетом определений (19.18) и (20.8) соответственно (20.9). Тогда
д (aS) _ д (аХ<) SS . р* dXt 1 ' (20.30)
соответственно
д (a U) = (20.31)
dXt ”
поэтому = Рг (Х1(, Xr). (20.32)
PftaX,, ...,аХг)
Термодинамические потенциалы, и функции Массье — Планка 97
Отдельное уравнение состояния не полностью определяет термодинамические свойства системы. С другой стороны, знание всех уравнений состояния эквивалентно знанию фундаментального уравнения. Из свойства б. следует, что для однокомпонентной системы знание двух уравнений состояния полностью определяет термодинамические свойства системы. Если в уравнение (20.32) подставить (с г = 3) а = Х31, то получим три уравнения состояния в виде
Pt = Pt(^-, 0. (20.33)
\ ^3 Л3 /
Из этих трех уравнений можно исключить переменные Xi/X3 и Х2/Х3, что дает уравнение между тремя интенсивными параметрами Pt. Если даны два уравнения состояния, то из них можно рассчитать третий интенсивный параметр.
Переменные в уравнении (20.33) являются в энтропийном выражении мольными величинами
п
или плотностями и
V 9
(20.34)
(20.35)
в энергетическом выражении мольными величинами
п или плотностями V ’ ; v = —, (20.36) п Р = у- (20.37)
Мольные величины и плотности не зависят от размера системы, т. е. они не обладают свойствами экстенсивных параметров, определяемых по уравнению (20.6). Поэтому в литературе их часто называют интенсивными величинами состояния. Эта терминология ошибочна и ее следует избегать. Мольные величины и плотности именно не обладают фундаментальными свойствами интенсивных параметров, определяемых уравнением (20.7), 4—235
98
Глава III
что видно на простейшем примере мольной плотности для равновесия жидкость — пар.
Приведенные рассуждения можно провести также в несколько другой форме. Согласно б. и (19.18), для фундаментального уравнения (20.4) для однокомпонентной системы можно записать
at/= [7 (aS, aV, an). (20.38)
Если подставить a = 1/п и использовать определение (20.36), то получим
u = u(s, v, 1), (20.39)
или в дифференциальной форме
du = — ds + — dv. (20.40)
ds dv
Но, согласно (20.31),
— = —= T, — P. (20.41)
ds dS dv dV v ’
Вместе с тем из (20.40) следует
du — Tds — Pdv. (20.42)
Уравнения (20.39) и (20.42) называются фундаментальными уравнениями, рассчитанными на один моль. Если даны уравнения состояния для Т и Р, то подстановкой в (20.42) и интегрированием получают в явном виде фундаментальное уравнение, рассчитанное на один моль. Соответствующие рассуждения справедливы также для энтропийного выражения.
К пункту е. Дополнительное соотношение между интенсивными параметрами, которое представляет собой следствие из б., можно дать в явном виде в дифференциальной форме. Из б. с использованием теоремы Эйлера и уравнений (20.13)—(20.15) следует, что фундаментальное уравнение (20.4) можно записать следующим образом:
U = TS — PV + ^itii. (20.43)
/=]
Отсюда после дифференцирования получим
Термодинамические потенциалы и функции Массье — Планка 99
dU = TdS + SdT — PdV — VdP + ^idnl + i—1
+ (20.44)
z=i
Сравнение (20.44) с (20.5) дает
SdT — VdP + J^ nt di^ = 0. (20.45)
/=!
Это чрезвычайно важное соотношение называется уравнением Гиббса — Дюгема.
В энтропийном выражении уравнение Гиббса —Дюгема имеет вид
m
(20.46)
Вместо метода, описанного под рубрикой «К пункту д.», уравнение Гиббса — Дюгема может также служить для получения фундаментального уравнения из двух уравнений состояния. На примере идеального газа можно легко проверить применимость различных методов.
§21. Термодинамические потенциалы
Ранее было показано, что фундаментальное уравнение в энтропийном или энергетическом выражении содержит полную термодинамическую информацию о системе. Развитие этой информации в явном виде в рамках формализма, описанного в § 20, часто сталкивается с очень большими трудностями, потому что в фундаментальном уравнении в качестве независимых переменных используются только экстенсивные параметры. Но экстенсивные параметры очень трудно непосредственно измерять и контролировать, а чаще всего это вообще невозможно сделать. Так, не существует прибора, при помощи которого можно непосредственно измерить энтропию, и нет приспособления, при помощи которого можно было бы поддерживать ее постоянной; для конденсированной фазы практически невозможно поддер-4*
100
Глава III
живать постоянным объем и т. д. С другой стороны, как уже было упомянуто, в энергетическом выражении интенсивные параметры являются непосредственно измеряемыми величинами, которые можно легко контролировать экспериментально. Это основано на том, что интенсивные параметры определяют при помощи контактного равновесия. Поэтому возникает проблема расширения формального аппарата термодинамики, которую можно сформулировать следующим образом.
Фундаментальное уравнение должно быть преобразовано таким образом, чтобы один или несколько интенсивных параметров были введены в качестве независимых переменных и при этом сохранялась полная информация фундаментального уравнения.
Последнее требование говорит о том, что трансформируемая функция должна так же обладать свойствами характеристической функции. Сразу видно, что это является постановкой задачи, приведенной в § 19, и что проблема может быть решена при помощи преобразований Лежандра фундаментального уравнения.
Чтобы это формальное развитие по возможности было более наглядным, будем в дальнейшем применять использованные ранее обобщенные величины состояния и Pf. Преимущество такого способа написания заключено в том, что для многих термодинамических соотношений существенным является только различие между экстенсивными и интенсивными параметрами. Поэтому этим путем можно свести многочисленные однотипные соотношения в одно уравнение. Но нельзя не заметить, что энергия и энтропия наряду с общими свойствами экстенсивных параметров обладают еще индивидуальными свойствами, вытекающими из законов термодинамики. Если это понадобится, будем записывать энергию в явном виде в энтропийном выражении и энтропию в энергетическом выражении. Аналогичным образом химические потенциалы среди интенсивных параметров занимают особое положение, которое становится понятным из способа их введения (§ 15). В то время как именно определения Т и Р не содержат произвольных констант, химические потенциалы, как видно из уравнения (21.40), можно определить с точностью до члена а+ +Ь-Т, где а и b—произвольные константы.
Термодинамические потенциалы и функции Массье — Планка 101
Преобразование Лежандра можно применять как к энтропийному выражению, так и к энергетическому выражению фундаментального уравнения, что приводит к двум рядам характеристических функций. В этом параграфе ограничимся рассмотрением энергетического выражения, которое в рамках термодинамики имеет несравненно большее значение.
Фундаментальное уравнение в обобщенных величинах состояния имеет вид
и - и (Х4, ..., Хг) (г > m + 2). (21.1)
Интенсивными параметрами являются
(21.2)
Величины Xt и Pt, связанные между собой уравнением (21.2), называются сопряженными параметрами. В дифференциальной форме фундаментальное уравнение имеет вид
dU = У Pt dXt.
(21-3)
Результаты преобразования Лежандра фундаментального уравнения в энергетическом выражении называют термодинамическими потенциалами. Поэтому общее определение термодинамических потенциалов записывается k
^k = U-^PiXi. (21.4)
Z=1
Условием, достаточным для существования такого преобразования, будет
d(Ar, ... ,Xk)
Дифференцированием (21.4) с использованием (21.3) получим k г
<wk = -'%xidpi+ 2 pjdXi- (21-6) г=1 /=/г+1
102
Глава III
При k < г все термодинамические потенциалы по их определению являются характеристическими функциями. Они являются гомогенными функциями первой степени экстенсивных параметров. Уравнение (20.43) в обобщенных параметрах имеет вид
[/=2РгХг. (21.7)
«=1
Подстановка в (21.4) дает
^=2 pjxr (2L8)
/=^+1
откуда, согласно теореме Эйлера, следует утверждение о гомогенности термодинамических потенциалов. Частные производные термодинамического потенциала по интенсивным параметрам Pt дают (с отрицательным знаком) сопряженные экстенсивные параметры Xf. Следовательно, имеем
= — Xt. (21.9)
dPt 1 v ’
Частные производные по экстенсивным параметрам Ху • дают, как и для фундаментального уравнения, сопряженные интенсивные параметры Ру. Следовательно, имеем
ОЛу
Особое место представляет случай k = г. Из уравнений (21.4) и (21.7) получаем
(21.11)
z=i «=1
и, согласно (21.6),
«г=УХ((й>( = 0. (21.12)
i=\
Эго не что иное, как уравнение Гиббса—Дюгема в обобщенных величинах состояния. Вначале поражает тот факт,
Термодинамические потенциалы и функции Массье — Планка 103
что полный (r-кратный) результат преобразования Лежандра фундаментального уравнения, т. е. термодинамический потенциал ЧР*Г, равен нулю. Однако, как показывает вывод, этот факт математически основан на том, что фундаментальное уравнение является гомогенной функцией первой степени. Физически это объясняется тем, что для определения системы необходим по крайней мере один экстенсивный параметр.
Рассмотрим теперь важный для термодинамики специальный случай уравнения (21.4), причем будем вновь использовать запись величин состояния в явном виде*.
Термодинамический потенциал
U^U(S, V, Hp...,nJ (21.13)
является фундаментальным уравнением в энергетическом выражении, которое подробно было рассмотрено в § 20. В дальнейшем мы будем использовать эту функцию для некоторых общих рассуждений. Однако для обсуждения специальных проблем вместе с экспериментальными данными эта функция по причинам, упомянутым в начале параграфа, нецелесообразна.
Термодинамический потенциал
Я = Я(5, Р, Пр... ,nj (21.14)
называется энтальпией или теплосодержанием.
Уравнение (21.4) в этом случае имеет вид
H=U + PV, (21.15)
а уравнение (21.6)
dH = TdS + VdP + 2 Hi dni- (21-16) i=\
Частные производные записываются следующим образом:
' дН \
< dS /р, п
дН \ =
дР JS, п
f дН \
, dnt /3, Р, п.
(21.17)
Обсудим кратко физическое значение энтальпии. Естественно, что каждая наглядная физическая интерпретация
* Символы для энтропии и важнейших термодинамических потенциалов установлены международным соглашением.
104
Глава III
выхватывает только один определенный аспект и не представляет собой полного содержания термодинамического потенциала. Поэтому такое толкование нельзя отождествлять с самой функцией.
Напомним о двух результатах, которые были подробно обсуждены в гл. 1. Если обозначить обратимую работу, совершаемую системой, через IF* = —IF, то получим
— (dU)Sn = dW*. (21.18)
Работа, произведенная закрытой системой при постоянной энтропии, т. е. квазистатически-адиабатическим путем, равна уменьшению внутренней энергии. С другой стороны,
(сШ)у' п = TdS = dQ. (21.19)
Теплота, подводимая к закрытой системе при постоянном объеме, равна увеличению внутренней энергии.
Чтобы получить аналогичные следствия об энтальпии, представим систему, соединенную с резервуаром, поддерживающим постоянное давление. При этих условиях может быть совершена работа расширения, но она служит лишь для поддержания постоянного давления и не может быть отдана наружу. Если поэтому хотят выполнить условие, аналогичное (21.18), то необходимо принять по крайней мере еще одну рабочую координату. Сделаем такое предположение, которое ради простоты введем не в общем виде, и предположим далее адиабатическую изоляцию системы и резервуара (индекс R). Обратимая работа, совершенная системой и резервуаром, согласно (21.18), равна
dIF* = -d(t7 + C^). (21.20)
Но так как в резервуаре и в системе давление постоянно и одинаково, то
— dU R = PdVR = —PdV = — d(PV). (21.21)
Из (21.15), (21.20) и (21.21) следует
~(dH)s>Ptn = dW\ (21.22)
Термодинамические потенциалы и функции Массье — Планка 105
Работа, совершенная закрытой системой в обратимом процессе при постоянной энтропии и постоянном давлении, равна уменьшению энтальпии.
По аналогии с (21.19) имеем
(d#)Pt/x = dQ. (21.23)
Теплота, подведенная к закрытой системе при постоянном давлении, равна увеличению энтальпии. Так как тепловые эффекты различных процессов (процессы смешения, химические реакции) измерены в большинстве случаев при постоянном давлении, то из равенства (21.23) непосредственно следует значение энтальпии для теоретической интерпретации таких измерений.
Значительную роль играет энтальпия в теории эффекта Джоуля — Томсона, поскольку этот процесс протекает при постоянной энтальпии. Теория этого эффекта будет приведена лишь в § 24, так как для этого необходимы еще некоторые формальные вспомогательные средства.
Применение энтальпии как термодинамического потенциала затрудняется тем, что она содержит в качестве независимой переменной энтропию как функцию состояния. Энтальпию можно представить как функцию каждого полного набора переменных состояния, но тогда она больше не является термодинамическим потенциалом. Особое значение имеет представление, аналогичное (20.20) и (20.21):
Н = Н(Т, P,ni9..., пт) (21.24)
вместе с
dH =
dT + (™-\ дТ ]р, п \дР )т.
dP + п
(21.25)
Причина предпочтения этого представления будет разъяснена в § 24. Заметим здесь, что выражение (21.25) непосредственно дает определение мольной теплоемкости при постоянном давлении Ср. Учитывая, что h = Н/п для однокомпонентной системы и используя (21.23), получаем
106
Глава III
Ср = (» • (21-26)
* \дТ /р, п
Производная (dH/dn^r^p^j , однако, не является химическим потенциалом.
Термодинамический потенциал
F = F(T, ...,nm) (21.27)
называется свободной энергией Гельмгольца.
Уравнение (21.24) тогда запишется следующим образом:
F = U — TS, (21.28)
а уравнение (21.26)
dF = — SdT — PdV + 2 dnf (21 29)
z=i
Частные производные равны:
/ dF \ Q. ( dF \ п ( dF \ /о i
---- =—о:- =—Г> - = (21.30) \dT]v,n { dV )т,п \dni]T,Vtn. гг v 7
При определенных условиях можно дать наглядное толкование свободной энергии Гельмгольца. Рассмотрим систему в термическом равновесии с резервуаром (термостат), который поддерживает температуру постоянной. Пусть система и термостат вместе будут адиабатически изолированы. Диатермическая перегородка между системой и резервуаром предполагается неподвижной, так что в этом случае резервуар сам по себе не совершает работы. Работа, произведенная системой в обратимом процессе, согласно условию (21.18), равна
dW* = — d(U + UR}. (21.31)
Так как резервуар не производит никакой работы, то уменьшение его внутренней энергии обусловлено только за счет перехода теплоты, необходимой для поддержания постоянной температуры. Поэтому вследствие Т = const
— dUR = —dQR=dQ = TdS = d (TS). (21.32)
Из выражений (21.28), (21.32) следует
-(dF)Tfl = dW*. (21.33)
Термодинамические потенциалы и функции Массье — Планка 107
Работа, совершенная закрытой системой в изотермическом обратимом процессе, равна уменьшению свободной энергии Гельмгольца. По этой причине применяют ведущие к недоразумениям термины «функция работы» и «максимальная работа» (для —F), так же как используемый часто в американской литературе символ А.
Свободная энергия Гельмгольца как термодинамический потенциал весьма полезна в тех случаях, когда объем легко можно контролировать (в основном для газовой фазы). В этой связи представляет интерес то, что второе из уравнений (21.30) представляет собой ранее упоминавшееся термическое уравнение состояния (20.28). В старой литературе свободную энергию Гельмгольца многократно привлекали для рассмотрения конденсированных фаз. Объясняется это тем, что вначале интересовались зависимостью от температуры и, принимая во внимание очень слабую зависимость от давления (в области давлений, доступных для обычных методов измерения), пренебрегали различием между постоянным объемом и постоянным давлением. Современное значение свободной энергии Гельмгольца основано прежде всего на том, что этот термодинамический потенциал особенно подходит для расчетов методом статистической термодинамики.
Термодинамический потенциал
G = G(T,P,ni9...9nm) (21.34)
называется свободной энергией Гиббса.
Уравнение (21.4) запишется следующим образом:
G = U — TS + PV^H — TS = F + PV, (21.35)
а уравнение (21.6)
dG = — SdT + VdP +2 dnt.
Z—1
(21.36)
Частные производные равны
( dG \ q I dG \ T/. [ dG \ q x
---- =—о - =V, ---------- = (21.37) \дТ ]р,п \ дР )т,п \ дщ )T. P. 1 v 7
Наглядную интерпретацию можно провести аналогичным путем, как и в предыдущих случаях. Рассмотрим систему,
108
Глава HI
соединенную с двумя резервуарами R и R', один из которых поддерживается при постоянной температуре (термостат), другой при постоянном давлении (маностат). Как и в случае энтальпии, здесь необходимо рассмотреть дополнительные рабочие координаты. Работа, совершенная системой обратимым путем, равна
dW* = —d (U + UR + URf). (21.38)
Однако dU# теперь дается уравнением (21.32), dU^ — уравнением (21.21). Используя (21.35), получим
= (21-39)
Работа закрытой системы в обратимом изобарно-изотермическом процессе равна убыли свободной энергии Гиббса.
Среди термодинамических потенциалов свободная энергия Гиббса находит самое широкое применение. Особое положение ее объясняется тем, что здесь экстенсивными параметрами являются только числа молей, которые сравнительно легко измерить и контролировать. Отсюда следует, что свободную энергию Гиббса можно полностью сконструировать из числа молей и также хорошо поддающихся измерению химических потенциалов. Уравнение (21.8) в этом случае имеет вид
т
G = ^V.tnt. (21.40)
i=l
Это уравнение показывает, что для однокомпонентной системы химические потенциалы равны мольной свободной энергии Гиббса. Это совпадение не должно вводить в заблуждение, так как фактически химические потенциалы имеют более общее значение.
Термодинамический потенциал
2 = 2(Т,У,Н,...,М (21.41)
обычно называют большим потенциалом, однако ни название, ни символ его однозначно еще не установлены. Уравнение (21.4) запишется т
Q = U — TS —
¥1*4,
(21.42)
Термодинамические потенциалы и функции Массье— Планка 109
а уравнение (21.6)
dQ = — SdT — PdV — (21.43)
Частные производные равны
( dQ \ =_s. ( \ = _ р. ( dQ \
\ дТ /V, |1 \ dv )т, р- \ dpi /Л v, ixz
= -п1. (21.44)
Из уравнений (21.28), (21.35), (21.40) и (21.42) находим
2 = — PV. (21.45)
Поэтому вместо большого потенциала часто применяют величину PV.
В обычной термодинамике большой потенциал почти не исполь зуют, но он часто применяется в статистической термодинамике, так как его можно рассчитать при помощи большого канонического ансамбля. Отсюда и происходит его название.
§ 22. Функции Массье—Планка
Рассмотрим теперь коротко характеристические функции, выведенные из энтропийного выражения фундаментального уравнения. В обобщенных величинах состояния фундаментальное уравнение имеет вид
S = S (Хо ..., Хт) (r>jn + 2). (22.1)
Интенсивными параметрами здесь являются
<'+>> <22-2>
Тогда фундаментальное уравнение в дифференциальной форме запишется
ds = 2 dXi- <22-3)
/=1
Результат преобразования Лежандра фундаментального уравнения в энтропийном выражении называют функциями Массье — Планка. Общее определение функций Массье — Планка записывают следующим образом:
по
Глава III
^k = S-itP]Xt. (22.4)
Z=1
Условием, достаточным для существования преобразования, является
= ан.......(22'5)
Из (22.4), используя (22.3), получим
d Ф* = - S XtdP’. + £ Р*. dXr (22.6)
Z=1 /=^+1
Общие формальные свойства функций Массье — Планка полностью соответствуют свойствам термодинамических потенциалов. Поэтому можно сослаться на объяснения § 21 и дать здесь лишь формулы для двух специальных случаев, которые представляют известный интерес в термодинамике.
Функция Массье — Планка
= Oi
Т
V, nlt..., nm
(22.7)
как легко заметить, соответствует свободной энергии Гельмгольца. По предложению Гуггенгейма ее следует называть функцией Массье. Уравнение (22.4) теперь имеет вид
Ф4 = S-----^и, (22.8)
а уравнение (22.6)
dO1 = — Ud(—Н \ Т )
Частные производные равны
/ дФх \ ____
\ д(ЦТ) )V,n
\ дгц Jur.v.n
( д®! \ __ Р .
\ dV Ji/т. п~ Т ’
=------у-. (22.10)
Термодинамические потенциалы и функции Массье — Планка 111
Подобным же образом функция Массье — Планка
у » j. ’ ••• >
(22.11)
соответствует свободной энергии Гиббса. Гуггенгейм назвал ее функцией Планка.
Уравнение (22.4) запишется теперь следующим образом:
Ф2 = 5-----X-U — -yV, (22.12)
а уравнение (22.6)
d<D, = — Ud А
Частные производные равны
(22.14)
В термодинамике эти функции представляют прежде всего исторический интерес, потому что они были первыми характеристическими функциями, которые были введены (Массье, 1865), и потому что Планк широко использовал в своих исследованиях функцию Ф2. Их современное значение для практики основано на двух особенностях: во-первых, в таком представлении появляются в явном виде калорические величины U и Н в качестве переменных (что в полной мере будет показано в § 24), во-вторых, Фх и Ф2 находятся в простой связи с соответствующими термодинамическими потенциалами (что не является общим для функций Массье — Планка). Сравнение выражений (22.8) с (21.28) и (22.12) с (21.35) показывает, что
ф1=_2_; (22.15)
112
Глава Ш
Поэтому в этих случаях можно легко заменить термодинамические потенциалы на функции Массье — Планка, не вводя последних в явном виде, и, таким образом, делать полезными преимущества обоих представлений. Вернемся к этому в § 24.
Как уже было упомянуто, общее значение функции Массье— Планка имеют для термодинамики необратимых процессов и для статистической термодинамики. Но это здесь рассматриваться не будет.
§ 23. Преобразование условий равновесия и стабильности
Так как при преобразовании Лежандра полностью сохраняется физическая информация, то можно сформулировать общие условия равновесия и стабильности также при помощи результата преобразования Лежандра фундаментального уравнения, т. е. термодинамических потенциалов или функций Массье — Планка. Проведем эти преобразования для термодинамических потенциалов, а для функций Массье — Планка, поскольку доказательство производится аналогично, дадим лишь конечный результат.
а. Условия равновесия
В формулировке (17.2) дополнительные условия экстремальной задачи выражены через экстенсивные параметры всей системы в целом, относящиеся к представлению энергии. Поэтому можно ожидать, что при формулировке условий равновесия при помощи результата преобразования Лежандра внутренней энергии одно или несколько дополнительных условий можно выразить через интенсивный параметр всей системы в целом. Это предположение (правильность которого будет доказана) ясно показывает природу задачи, которая здесь возникает. В то время как именно для гетерогенной системы каждый экстенсивный параметр равен сумме соответствующих экстенсивных параметров фазы, интенсивные параметры, согласно § 15, определены только для каждой фазы, но не для всей системы в целом. Определение экстенсивных параметров для всей системы в целом основано на фундаментальном свойстве (20.6). Аналогичным образом определение интенсивных параметров основано на фундаментальном свойстве (20.7), т. е. оно предполагает, что соответствующие интен
Термодинамические потенциалы и функции Массье — Планка 113
сивные параметры имеют одинаковое значение для всех сосуществующих фаз. Поэтому формулировка условия равновесия с помощью термодинамического потенциала должна предполагать, что каждый из ^-интенсивных параметров, которые для являются независимыми, имеет одинаковое значение для всех фаз системы. Так, при использовании свободной энергии Гельмгольца предполагается, что все фазы имеют одинаковую температуру, т. е. находятся в термодинамическом равновесии. Каждая формулировка условия равновесия при помощи результата преобразования Лежандра фундаментального уравнения предполагает, следовательно, некоторые необходимые данные о равновесии. В дальнейшем этот вопрос будет рассмотрен еще раз.
Вывод преобразованных условий равновесия состоит из двух частей. Сначала условие (17.2) приводят в эквивалентную форму, которая больше не содержит дополнительных условий, а содержит их в формулировке экстремальной задачи. Далее, из этой формулировки при помощи преобразования Лежандра (21.4) выводят условия равновесия для термодинамических потенциалов. Прежде всего предположим, что между параметрами Xj(j > k) не существует никаких дополнительных соотношений.
Теорема 1
Для равновесия системы, в которой интенсивные параметры ... , Pk зафиксированы и во всей системе поддерживаются постоянными, необходимо и достаточно, чтобы
k
W — £ PZ8XZ:> О, (Ху = const для j>k). (23.1) Z=1
Доказательство
В случае знака равенства (23.1) непосредственно следует из фундаментального уравнения. Общее доказательство проводится таким образом, что доказывается эквивалентность (17.2) и (23.1) с учетом сделанных предположений. Для этого используют вытекающее из фундаментального уравнения следствие, что всегда можно провести изменения состояния, для которых
114
Глава III
W — £РгЗХг = О, (Xy-= const для />Л). (23.2)
Ради простоты ограничимся случаем k = 1 и Xr = 3. Тогда из уравнения (23.2) следует, что (за счет подвода или отвода теплоты) всегда можно осуществить изменения состояния, при которых
817 — T8S = О, (V, п = const).
(23.3)
Используя этот факт, докажем, что для системы с одинаковой температурой условие
W _ nsО, (V, п = const)
(23.4)
эквивалентно условию (17.2). При этом нужно различать следующие случаи:
8S = 0.
В этом случае (23.4) непосредственно переходит в (17.2).
W = 0.
Уравнение (23.4) переходит в (17.1), эквивалентность которого (17.2) доказана в § 17.
8(7>O; 8S>0.
Согласно неравенству (23.4), тогда 8(7 > TdS. За счет отвода теплоты можно всегда достигнуть состояния, для которого, если его рассматривать как вариацию начального состояния, W > 0; 8S = 0. Таким образом, условие (17.2) выполнено.
8(7 <0; 8S<0.
По уравнению (23.4) в этом случае (8(7^ |T8S|. Поэтому за счет подвода теплоты всегда можно достигнуть состояния, для которого, если его рассматривать как вариацию начального состояния, 8(7 = 0; 8S 0. Таким образом, условия (17.1) и, следовательно, (17.2) выполнены.
8(7>O; 8S<0.
Термодинамические потенциалы и функции Массье — Планка 115
За счет отвода теплоты можно достигнуть состояния, для которого, если его рассматривать как вариацию начального состояния, W — 0; 8S < 0.
Поэтому условие (17.1) и, следовательно, (17.2) выполнены.
С. W <0; BS>0.
В этом случае уравнение (23.4) не выполнимо. За счет отвода теплоты можно всегда достигнуть состояния, для которого, если его рассматривать как вариацию начального состояния, W < 0; = 0. Поэтому в этом случае и (17.2)
не выполнимо.
Аналогичным путем всегда можно доказать, что возможность вариации, которая противоречит (23.4), необходимо подразумевает возможность вариации, которая противоречит условию (17.2). Таким образом, полностью доказана эквивалентность (17.2) и (23.4). Для общего случая условия (23.1) доказательство проводят аналогичным образом.
Хотя условие (17.2) формально сохраняется за счет частных случаев (случаи а. и |3.), но фактически оно не представляет частного случая, а, как показывает доказательство, является эквивалентной формулировкой.
Теорем а 2
Для равновесия системы, в которой интенсивные параметры Pi, ... , Pk зафиксированы и поддерживаются на протяжении всей системы постоянными, необходимо и достаточно, чтобы
РЧ'Ч.................<23.5)
Доказательство
Так как Xk+ii ... , Хг постоянны, для рассматриваемого случая условие (23.1) может быть записано
k
^)Pit х >о, (i<k,j>k). (23.6)
* J . , 1 ]
1=1
Из уравнения (21.4) следует
W — f Pt 8Хг = 8¥Л + Л Xi 8Рi (23.7)
1=1 i=l
116
Глава III
и, далее, для Ръ ... , Pk = const k
=mfi. .. (23.8) 7 Z=1 7 7
Подстановкой равенства (23.8) в (23.6) приходим непосредственно к утверждению теоремы 2.
Важнейшими специальными случаями (23.5) являются: Условие равновесия при постоянной энтропии, постоянном давлении и постоянных числах молей
№. р. „ > 0. (23.9)
Условие равновесия при постоянной температуре, постоянном объеме и постоянных числах молей
(SF)r.v.„>0. (23.10)
Условие равновесия при постоянной температуре, постоянном давлении и постоянных числах молей
(8G)r,P,„>0. (23.11)
Условие (23.5) является менее общим, чем условия (17.1), (17.2) и (23.1), поскольку из них нельзя получить следствий о внутренних равновесиях, которые описываются параметрами ... , Pk. Так из (23.10) нельзя определить условия термического равновесия внутри системы. Физически это объясняется тем, что при формулировании условия (23.5) заранее предполагается внутреннее равновесие по отношению к параметрам ... , Pk, Формально сравнение выражений (23.7) и (23.8) показывает, что при выводе (23.5) исключена часть возможных для (23.7) вариаций. Для функций Массье — Планка аналогичным образом получают
Теорему 3
Для равновесия системы, в которой интенсивные параметры Pi , ... ,Pk зафиксированы и поддерживаются постоянными по всей системе, необходимо и достаточно, чтобы
(W,-......Р- х.......х<0. (23.12)
1 k &41 Г
Термодинамические потенциалы и функции Массье — Планка 117
До сих пор предполагалось, что между параметрами (j> k) не существует никаких дополнительных условий. Важнейшим случаем, в котором это предположение не выполняется, является химическая реакция, для которой изменение числа молей определяется стехиометрическими соотношениями, следующими из уравнения реакции. Формально аналогичные соотношения могут появляться также между другими переменными состояния. Во всех случаях такого рода можно, как показано в § 16 и 17, предложить два пути. Первый путь заключается в том, что вводят соответствующий внутренний параметр. Экстенсивный параметр, связанный через дополнительные условия, не появляется больше в дополнительных условиях, и возникает экстремальная задача, при которой изменение внутреннего параметра ограничено лишь оставшимися дополнительными условиями (например, в случае химической реакции постоянство температуры, давления и числа молей компонентов, которые не принимают участия в реакции). Другой метод состоит в том, что не уменьшают числа переменных (следовательно, в случае химической реакции числа молей всех участников реакции в фундаментальном уравнении сохраняются), однако для экстремальной задачи вводят новые побочные условия, следующие из дополнительных соотношений. Приведенный выше вывод таким обобщением не нарушается. Но так как общая формулировка для таких случаев нецелесообразна, оставим обсуждение химических реакций до § 33 и 36.
б. Условия стабильности
Условия (23.5) и (23.12) для практически важного случая знака равенства только показывают, что в состоянии равновесия термодинамический потенциал (соответственно функция Массье — Планка Ф^) для любых данных дополнительных условий принимает стационарное значение. Суждения о виде этого стационарного значения требуют учета вариации более высокого порядка. Как уже было упомянуто, в термодинамике практически имеют значение только вариации второго порядка. Отложим более подробный анализ этой проблемы до гл. VI, а здесь удовлетворимся, как и в § 18, схематическим утверждением, что в состоянии стабильного равновесия стационарное значение термо
118
Глава III
динамического потенциала есть минимум, а функции Массье — Планка — максимум,
В виде формул это выглядит следующим образом:
(ДФДрх>0, (23.13)
(ДФ*)р.х<0. (23.14)
§ 24. Уравнения Гиббса—Гельмгольца и Максвелла
соотношения
До сих пор термодинамические потенциалы рассматривались как результат преобразования Лежандра внутренней энергии. Конечно, можно также при помощи преобразований Лежандра перейти от термодинамического потен-потенциалу
циала к другому термодинамическому Ф* (k > /). Тогда
k
i=l+\
Согласно (21.9),
djk dPt
-Xt.
Таким образом, получим
(24.1)
(24.2)
(24.3)
Для практического применения важным является только случай k — I— 1. Тогда
(24.4)
oPk
В этой форме пока еще не зависит в явном виде от Pk, а зависит от Xk. Из ЧТ, получают, однако, уравнение состояния
р.-4?-=р* лл............(24-S)
при помощи которого можно исключить Xk.
Термодинамические потенциалы и функции Массье — Планка 119
Все величины в уравнении (24.4) являются функциями Pk\ получаем линейное дифференциальное уравнение первого порядка, которое позволяет рассчитать если
дано Для свободной энергии уравнение (24.4)
впервые было выведено Гиббсом и затем позднее и независимо от него Гельмгольцем. Поэтому оно называется уравнением Гиббса — Гельмгольца.
Для термодинамики прежде всего имеют значения следующие частные случаи.
a. H = U + P(—'j (24.6)
\ дР Js, п *
Это уравнение позволяет рассчитать энтальпию, если известна внутренняя энергия как функция давления.
б. F = U + Т . (24.7)
\ дТ )v. п
Это уравнение позволяет рассчитать свободную энергию Гельмгольца, если известна внутренняя энергия как функция температуры.
в. G = H + t(—. (24.8)
\дТ )р,п '
Это уравнение позволяет рассчитать свободную энергию Гиббса, если известна энтальпия как функция температуры.
г. 6 = 77 + р(—. (24.8а)
\ дР )т, п
При помощи этого уравнения можно рассчитать свободную энергию Гиббса из свободной энергии Гельмгольца, если последняя известна как функция давления.
Эти уравнения прежде всего иллюстрируют сделанные ранее замечания о том, что может иметь значение также представление U и Н в наборе переменных, для которых они не являются термодинамическими потенциалами (ср.§ 20 и 21). Так как из приведенных примеров случай в. является наиболее важным, то дальнейшие рассуждения в основном будут посвящены ему.
Особое значение уравнения (24.8) объясняется следующим:
120
Глава 1П
а. Уравнение применимо для наиболее часто встречающихся условий эксперимента Р = const.
|3. Функцию Н (Т) можно получить из уравнения (21.26) интегрированием, если известна экспериментально легко доступная величина — мольная теплоемкость при постоянном давлении.
7. Функцию G рассчитывают из уравнения (24.8), согласно (23.11), при Т = const, Р = const; она полностью определяет все равновесия между компонентами, включая химические равновесия.
Формальное решение уравнения (24.8) получается сразу, если его записать в виде
' д (G/Т) Я
. д(1/Т) Jr, п
(24.9)
Тогда получим
G = Т ^Hd (-Ь),
(24.10)
причем константу интегрирования следует еще определить. Легко заметить, что эта формулировка, собственно, означает переход к функциям Массье — Планка. Дополнительные условия частного дифференцирования и функция Н следуют из формализма термодинамических потенциалов. Аналогичный метод для уравнения (24.7) ведет полностью к формализму функций Массье — Планка и просто дает первое из уравнений (22.10).
Упомянем, наконец, еще одну проблему, которая встречается в статистической термодинамике. Статистический расчет свободной энергии Гиббса в явном виде в принципе возможен, но практически сталкивается с непреодолимыми трудностями. Поэтому часто поступают таким образом, что рассчитывают статистически свободную энергию Гельмгольца и термодинамически переходят к свободной энергии Гиббса. Для этого используют уравнение (24.8а). Расчет интеграла, аналогичного (24.10),
G = Р
I га(т)
(24.10а)
Термодинамические потенциалы и функции Массье — Планка 121
требует, чтобы в выражении для свободной энергии Гельмгольца V было исключено при использовании термического уравнения состояния.
Интегрирования можно избежать, если использовать систему уравнений, параллельную уравнению Гиббса — Гельмгольца. Ограничимся случаем k— I = 1. Тогда из (24.1) и (24.5) следует
Wk = ^-Xk^-, (24.106)
dXk
с важнейшим частным случаем
G = F — V
dF \
аг ]т. п ‘
(24.10в)
Если F известна из статистических расчетов, то производная в правой части может быть легко определена и остается лишь исключить объем.
Ранее уже было отмечено, что из того факта, что характеристические функции необходимо являются функциями состояния, т. е. полными дифференциалами переменных состояния, можно получить соотношения между их вторыми производными (ср. § 20). Проделаем это в общем виде для термодинамических потенциалов. Из уравнения (21.6) сразу
получаем д2 Wk _ dXt ~ dPi " (24.11)
dPtdXj ” dXj
d2 _ dXt OX,, (24.12)
SPidPm dPm dPt ’
(I, tn ^k; j, n~Z >k). (24.13)
dXjdXn dXn dXj '
Эти уравнения называются соотношениями Максвелла, Важнейшими частными случаями (для однокомпонентной системы) являются
а. Внутренняя энергия [здесь встречается только тип (24.13)1
дТ\ [дР\
дУ )s, п \ )у, п ’
(24.14)
122
Глава III
(^L\ = , (24.15)
\ dn ]s, V \ OS )vt n
— (—'l = (—• (24.16)
\ On )s, v \dV )s, n
б. Энтальпия [встречаются только типы (24.11) и (24.13)]
г дУ \ = / к 0S /р, П ' ' дТ\ k дР )s, п ’ (24.17)
/ дУ \ \ On )s, р / 0(1 \ \ дР )s, п ’ (24.18)
( дТ\ = \ On )s. р / Ори \ \ 0S /р, п (24.19)
энергия Гельмгольца [также только (24.11) и
в. Свободная (24.13)]
(24.20)
(24.21)
(24.22)
г. Свободная энергия Гиббса [только (24.11) и (24.12)]
дУ \ _ ( 0|л \
On /г, р \ дР )т, п’
/ 0S \ ( дУ \
\ дР )т, п \ дТ )р, п ’
/ 0S \ / 0ц \
\ On /г, р VoF/р.л ’
(24.23)
(24.24)
(24.25)
§ 25. Пересчет частных производных. Метод определителей Якоби. Эффект Джоуля — Томсона
При термодинамическом рассмотрении некоторых специальных вопросов часто приходится иметь дело с частными производными, которые непосредственно нельзя изме
Термодинамические потенциалы и функции Массье — Планка 123
рить. Поэтому для термодинамики пересчет таких производных на экспериментально легко доступные величины играет чрезвычайно важную роль. В дальнейшем обсудим некоторые применяемые при этом методы, ограничившись однокомпонентными системами.
Распространение на многокомпонентные системы формально не представляет трудностей, но использует понятия, которые будут введены в § 28. Так как в § 20 показано, что для однокомпонентных систем число молей в качестве переменных является тривиальным, то будем их с самого начала считать постоянными. Таким образом, все функции состояния зависят только от двух независимых переменных.
Разъясним эту задачу более подробно. Если дана функция состояния
z = г (х, у), (25.1)
то можно написать
dz = (—} dx+(—) dy, (25.2)
\ дх /у \ ду )х
и вместе с тем, если w — еще одна переменная состояния, то
Комбинацией трех уравнений типа (25.3) можно выразить любые частные производные через (максимально) три произвольно выбранные другие частные производные. Но только для 10 переменных состояния число частных производных составляет 720 и число соотношений типа (25.3) порядка 1010. Отсюда непосредственно следует значение рациональных методов таких расчетов.
Первое основное упрощение заключается в том, что все частные производные выражают через одинаковый стандартный набор трех независимых частных производных. По предложению Бриджмена выбирают для этого экспериментально легко доступные величины:
Мольную теплоемкость при постоянном давлении
СР = Т
ds \
~дТ]р'
(25.4)
124
Глава Ш
Термический коэффициент расширения
1 I dV \ а = — ----- •
V \дТ )р
Изотермическую сжимаемость
1 ( dV\ X =----------
V \дР )т
(25.5)
(25.6)
В большинстве случаев такой выбор достаточен для практических целей. Если необходимы другие соотношения, то их можно легко вывести из уравнений для стандартных наборов (25.4)—(25.6).
а. Метод Бриджмена (метод последовательных исключений)
Метод Бриджмена использует следующие, известные из теории уравнений в частных производных соотношения:
дх \ / ду V1 ду Jz \ дх )z’
дх \ _ (dx/dw)z ду L (dy/dw)z дх \ = _ (дг/ду)х ду )z (дг/дх)у
(25.7)
(25.8)
(25.9)
Бриджмен делит 720 частных производных по постоянному параметру z на десять классов.
Согласно уравнению (25.8) для каждого класса достаточно знать девять величин (dx!dw)z, чтобы сконструировать все частные производные класса. Таким образом, задача сводится к нахождению 90 уравнений. Использование соотношений Максвелла (§ 24) уменьшает это число еще вдвое, так что остается 45 основных уравнений, которые Бриджмен свел в таблицы.
Покажем теперь кратко, как по этим методам, не используя таблицы, можно провести расчеты. При этом дадим в форме рецепта* отдельные этапы и разъясним каждый этап, не приводя полных расчетов.
* Callen Н. В., Thermodynamics, Wiley, New York, 1960.
Термодинамические потенциалы и функции Массье — Планка 125
а. Если данная производная содержит термодинамический потенциал, то его при помощи выражений (25.7) или (25.9) переводят в числитель и определяют по уравнению (21.6).
Пример.
(25.10)
Результирующее выражение больше не содержит термодинамических потенциалов. Встречающиеся производные порознь отыскивают ниже описанными методами.
|3. Если производная содержит химический потенциал, то его переводят в числитель и определяют при помощи уравнения Гиббса — Дюгема (20.45).
Пример.
дрь \
дТ \ дУ )s
дР \
дУ /s*
(25.11)
+ v
у. Если производная содержит энтропию, то ее переводят в числитель и или исключают при помощи соотношений Максвелла, или посредством выражения (25.8) (при w — Т) приводят к стандартным производным.
Пример 1.
дТ \ дР Is
(dS/dP)T (дУ/дТ)р (dS/dT)p ~ (п/Т)с [с (25.9)] [с (24.2^4)]
(25.12)
Пример 2.
as\ (ds/dpp дУ )р (дУ/дТ)р
(П/Г)-- [с (25.9)]. (25.13)
(дУ/дТ)р 1 v v
В этом случае энтропию нельзя исключить посредством соотношений Максвелла [ср. (24.17)].
8. Объем переводят в числитель.
126
Глава III
Пример.
дТ\ дР ]у
(dVjdP)^^^ (25.9)]. (25.14)
(dV/dT)p a v 7J v 7
Мольная теплоемкость при постоянном объеме определяется при помощи уравнения (которое будет выведено в дальнейшем).
CP-CV = T^. (25.15)
/ k X
Таким образом, данная производная сводится к стандартным производным.
б. Метод определителей Якоби
Более общим и более изящным, чем метод Бриджмена, является метод определителей Якоби, разработанный Шоу. Приведем без доказательства свойства определителей Якоби, необходимых в дальнейшем, и ограничимся двумя независимыми переменными. Определитель Якоби от х и у для независимых переменных и и v по определению равен
J (х, у) =
У) __ д (и, о)
(25.16)
Из определения следует
J (и, V) = — J (у, и) = 1,
J (х, х) = О J (k, х) = 0 (k = const), J(x, у) = Цу, — х) = J(— у, х) = — J(у, х).
(25.17)
Можно записать
д (х, у) д (z^ w) д[х[ у) д(г, w) д(и, v) д(и, v)
(25.18)
Термодинамические потенциалы и функции Массье — Планка 127
Для у = v из (25.16) следует
Каждую частную производную, таким образом, можно записать как определитель Якоби.
Метод основан на следующем рассуждении. Согласно (25.19), уравнение (25.2) можно записать
dz = dx + d(z’ x) dy. (25.20) d (x, y) ' d(y, x) *
Если ввести новые независимые переменные г и s и сокращенную запись J (г, у) = д (г, у)/д (г, s), то из уравнения (25.20) с использованием (25.18) следует
dz = -J dx -ь /(г’ x) dy.
J(x, у) J(y, х)
(25.21)
Умножая на J (х, у) и учитывая (25.17), получим
J (z, y)dx + J(x, z)dy + J(y, x)dz = 0. (25.22)
Это соотношение является основным уравнением метода. Не будем останавливаться на общей формулировке (которую можно привести в табличной форме) и рассмотрим только наиболее простые частные случаи, которые проиллюстрируем примерами. Если подставить в уравнение (25.22) dy = 0, то получим
Цг, y) + J(y,x) =0, \дх )у
(25.2 3)
или, используя (25.17), в явном виде
дг \
дх)у
д(у, z)/d(r, s) д(у, х)/д(г, s) ’
(25.24)
Это уравнение представляет собой преобразование от независимых переменных х, у к переменным г, s. Представление частных производных через стандартные производные (25.4) —(25.6) означает только преобразование к независимым переменным свободной энергии Гиббса, т. е. к Т и Р. В уравнении (25.24) поэтому всегда г ==Т, s = Р.
128
Глава III
Пример 1
Решим пример (25.10) методом детерминантов Имеем
Якоби.
/ дР_\ _ d(G, P)/d(T, Р) =
\dU )G “ d(G,U)/d(T, Р)
_ _______________(dG/dT)P______________
~ (dGldT)p (dU/dP)T — (dG/dP)T (dU/dT)p
Таким образом, получаем
/ dP'
(25.25)
S_________
\dU )g S (dU/dP)T 4- V (dU/dT)p Выражение в правой части можно легко свести
(25.26)
Выражение в правой части можно легко свести к стандартным производным, используя (25.3), (25.7)—(25.9), а также соотношение Максвелла (24.20).
Пример 2
Мольная теплоемкость при постоянном объеме должна быть выражена через стандартные производные. Согласно (25.24), имеем
с =т (= т d(Vt р)
V \dT)v d(v, T)/d(T, Р) ’ или, используя (25.19) и (25.26),
Cr=—L их d(T, Р)
Раскрытие детерминантов дает
~ Т Г/ ds \ / dv \ / ds \ / dv
V и х Н dT /р \ дР /г \ dP )т \ dT
(25.27)
(25.28)
т
VK
Ср -----V*
Т
21
(25.29)
причем вновь было использовано соотношение Максвелла (24.24). Таким образом, получаем
С„ — CV = T—. (25.30)
Р V X
Термодинамические потенциалы и функции Массье — Планка 129
Эта формула очень важна и ее часто используют в приложениях термодинамики.
в. Эффект Джоуля — Томсона
Теперь можно развить теорию эффекта Джоуля — Томсона, упомянутого при обсуждении энтальпии в § 21. Экспериментально эффект выглядит следующим образом (рис. 13).
Цилиндрическая трубка закрыта с обоих концов подвижными поршнями и в середине разделена пористой перегородкой на две части / и II. Перегородка должна быть
Рис. 13. Схема эксперимента по изучению эффекта Джоул я —Т омсон а.
сделана таким образом, чтобы она препятствовала спонтанному выравниванию давлений между I и II за счет конвекции или диффузии, но допускала проход газа, если он вытесняется за счет движения поршней из части с более высоким давлением Pi в часть с более низким давлением Рц. Подходящим материалом для такой перегородки является, например, вата или стеклянное волокно. Вся аппаратура должна быть адиабатически изолирована. Предположим, что в части I находится моль газа, в то время как в части // поршень прилегает к перегородке. Рассмотрим перенос этого количества газа из части I в часть //, причем давления Pj и Рц должны оставаться постоянными. Так как процесс протекает адиабатически, то, согласно первому закону термодинамики [уравнение (8.1)],
ип =ui + PJvI — Рц (25.31)
или, используя (25.15), получим
hj = hu. (25.32)
^Следовательно, в процессе энтальпия остается постоянной. Поэтому его называют изоэнтальпийным расширением. 5—235
130
Глава III
Следует обратить внимание на то, что, хотя в частях I и II осуществляется равновесие, внутри перегородки равновесия нет. Как подчеркнуто в конце § 15, это обстоятельство не имеет значения, если ограничиться рассмотрением начального и конечного состояния и описывать его через термодинамические переменные состояния.
Эффектом Джоуля — Томсона называют тот факт, что в описанном опыте в общем случае Tj =£Тц, а именно может быть T/j. Поэтому задача теории состоит в расчете изменения температуры из заданного изменения давления.
Если давление изменяется на бесконечно~малую величину, то вследствие (25.32) исходное уравнение имеет вид
dT = (—\ dp. \дР )ц
(25.33)
Преобразуем частную производную при помощи (25.24) от переменных Р, Н к стандартным переменным Т, Р. Тогда получим
(дН/дР^ dp (дН!дТ)р
(25.34)
Так как число молей постоянно, то использование (21.16) дает
Т (dS/dT)p к
При помощи соотношения Максвелла (24.24) и определений (25.4) и (25.5), наконец, получаем
dT = — (Та — 1) dP. (25.36)*
Ср
Это уравнение показывает, что для идеального газа (так как в этом случае а 1/Т) dT = 0. Эффект Джоуля — Томсона, следовательно, связан с отклонением от идеаль-
* Уравнение (25.36) описывает так называемый дифференциальный эффект Джоуля—Томсона. Величину Ъ=(дТ/дР)н называют яо-эффициентом Джоуля—Томсона.
Термодинамические потенциалы и функции Массье — Планка 131
кого газового состояния. Поэтому можно использовать измерения эффекта Джоуля — Томсона, чтобы пересчитать эмпирическую температуру реального газового термометра (§ 7) на абсолютную температуру. Более точные исследования показывают, что dT при высоких температурах положительно, при низких температурах отрицательно. Температуру инверсии определяют по уравнению
Та = 1, (25.37)
решение которого относительно Т требует знания термического уравнения состояния газа.
Эффект Джоуля — Томсона является важным вспомогательным средством в низкотемпературной технике. Его практическое использование требует, с одной стороны, чтобы описанный процесс был непрерывным, с другой стороны, газ должен быть предварительно охлажден до температуры ниже инверсионной*. Тогда эффект Джоуля — Томсона можно использовать вплоть до температуры сжижения газа.
§ 26. Средние мольные и парциальные мольные величины
Как уже было упомянуто в § 21, самой важной из всех характеристических функций для применения в термодинамике является свободная энергия Гиббса. В дальнейшем будут приведены еще некоторые понятия и соотношения, которые полезны при применении этих функций к многокомпонентным системам.
Вернемся к рассуждениям § 20 (пункту д). Определение плотности можно легко расширить на многокомпонентные системы, но в равновесной термодинамике это мало используется. Напротив, понятие мольных величин нелегко обобщить, так как, очевидно, нет смысла рассчитывать функции состояния на моль одного компонента.
* Для большинства газов предварительное охлаждение не требуется, так как инверсионные температуры часто расположены выше комнатной температуры (например, Tt — 621rfK для N2). Предварительное охлаждение требуется в особо важных для низкотемпературной физики случаях Н2 (7/ = 195° К) и Не (Tz = = 23,6° К).
5*
132
Глава III
Поэтому вводят новое понятие следующим образом. Из уравнения (21.40) и теоремы Эйлера следует
aG = G (Т, Р, anv ..., мт). (26.1) т
Далее, подставляют а = 1/J]nz и пишут
Z=1
G* = G* (Т, Р, хт^). (26.2)
Величины
хг = (26.3)
2 ni z=»l
называют мольными долями. Очевидно,
2 Xt = 1 (26.4)
Z=1
так что только т — 1 мольных долей являются независимыми переменными. Функция G*, определенная по уравнению (26.2), называется средней мольной свободной энергией Гиббса,
Из уравнений (21.37) и (24.8) видно, что при использовании свободной энергии Гиббса также появляются экстенсивные величины состояния S, V и Н как функции 7, Р и ni9 ... , пт. Поэтому ясно, что приведенный способ образования понятия можно обобщить. Итак, пусть Z будет экстенсивной функцией состояния независимых переменных 7, Р, п4, ... , nm. Тогда
(26.5)
называется средней мольной величиной состояния. Величины, впервые введенные Льюисом,
z. = (26.6)
\dni}T,Ptn.
называются парциальными мольными величинами компонента i. Они играют важную роль в термодинамике много
Термодинамические потенциалы и функции Массье — Планка 133
компонентных систем*. Сравнение (21.37) с определением (26.6) показывает, что химический потенциал формально можно рассматривать как парциальную мольную свободную энергию Гиббса. Это само по себе корректное высказывание часто рассматривается как определение химического потенциала и затемняет поэтому более общее значение, которое следует из фундаментального уравнения и результата преобразования Лежандра для этого уравнения.
Из (21.37) и определений (26.2), (26.3) и (26.5) следует
/ dG* \
\дТ )р, х
dG*\
дР )т, х
(26.7)
Поэтому из (26.2) получаем
dG* = — S*dT + V*dP + J
dxt. (26.8)
Производная от G* по мольной доле не является химическим потенциалом. Таким образом, возникают вопросы: а) какое значение имеет производная (dZ*ldx^r, р, х.\ б) какая связь между средними мольными и парциальными мольными величинами.
Оба вопроса тесно связаны между собой и в дальнейшем будут кратко обсуждены.
Согласно оп ределению,
т
dZ =
I drit.
Т,Р, п.
(26.9)
По определению экстенсивных параметров Z является гомогенной функцией первой степени от чисел молей. Поэтому теорема Эйлера вместе с определением (26.6) дает
* Для однокомпонентных систем как средние мольные величины, так и парциальные мольные величины сводятся к мольным величинам, которые до сих пор [уравнения (20.34) и (20.36)] обозначали строчными буквами. В дальнейшем для мольных величин будем также применять символ Z*
134
Глава III
(26.10)
После дифференцирования (26.10) и сравнения с (26.9) получаем
dT — (—\ dP = 0. \дР )т, п
(26.11)
Это соотношение, формально идентичное уравнению (20.43), называют обобщенным уравнением Гиббса — Дюгема. Обобщение основано на том, что само уравнение Гиббса — Дюгема следует лишь из факта, что фундаментальное уравнение является гомогенной функцией первой степени. Используя (26.3) и (26.5), уравнение (26.11) можно записать
dT — (—} dP = 0. X \дР )т, х
(26.12)
Особенно важным частным случаем этого уравнения является изотермически-изобарный процесс (dT = 0, dP — 0). Для него имеем
2 Xtdzi = 0 (dT = 0, dP = 0). z=i
(26.13)
Но для dT = dP = 0
(26.14)
и, таким образом,
(26.15)
Термодинамические потенциалы и функции Массье — Планка 135
—Г—-----------------------------------------------------
В соответствии с (26.4) т—\ величин dXj не зависят друг от Друга. Уравнение (26.15) может быть выполнено только тогда, когда
т
i—1
= 0.
р. xk
(26.16)
Эту форму уравнения Гиббса—Дюгема часто применяют при изучении изотермически-изобарных процессов.
Если написать уравнение (26.10) также для средних мольных величин, то получим т
Z*=^ziXi. (26.17)
Согласно уравнению (26.4), имеем
^- = — 1. (26.18)
dXj
Дифференцирование (26.17) по Xj дает
+ (26Л9) дх; dXj J
J Z==l 7
или вследствие (26.16)
-^- = Zj-zm (/=l,...,m-l). (26.20)
Это уравнение отвечает на вопрос а) о значении производной (dZ*ldX^T,P,Xj- Частично оно отвечает также на вопрос б). Выведем еще соотношения, которые выражают парциальные мольные величины через производные (dZ*ldXfri\P,Xj.
Решим уравнение (26.20) относительно zy и подставим это значение в уравнение (26.17). Тогда получим
z*=2Xt +IXiZ-+х^- (26-21) /=1 1 i=\
136
Глава III
Учет равенства (26.4) дает
т— 1
zm = Z*-V хг^-. (2€>.22)
Z—1 z
Таким образом, получен полный ответ на вопрос б). Важнейшим частным случаем уравнения (26.22) является m = 2 (бинарная система). Поэтому получаем
21 = Z*-xa^, (26.23)
z2 = Z* — Xj — . (26.24)
дх-±
При помощи этих уравнений можно рассчитать парциальные мольные величины из средних мольных величин. Это
Рис. 14. К объяснению метода касательной.
имеет большое значение для практического применения, так как в некоторых случаях (например, объем, энтальпия) средние мольные величины экспериментально доступнее, чем парциальные мольные величины. Для этого обычно используют так называемый метод касательной (рис. 14).
Уравнение касательной к кривой в точке х, у с наклоном dyldx имеет вид
= + (26.25)
ах
Отсюда при В = 0 получают отрезок ординаты. Сравнение с (26.23) и (26.24) показывает, что при данном способе отрезки на ординатах дают парциальные мольные величины в точке Р, Сопоставим еще некоторые частные соотно
Термодинамические потенциалы и функции Массье — Планка 137
шения для свободной энергии Гиббса, которые часто бывают необходимы при практическом применении. Согласно (26.6), по определению для парциальных мольных величин имеем
f dG \ < < dnt )т, р, п. ' ( дН\ 7 k dnt )т, Р, п. ( dS\ 1 — 4» dnt )т, р, п. 1 ЭУ \ = Vi- \ dn-L )т, Р, п. (26.26)
Отсюда, используя (21.36), получим
= = v (26.27)
\ дТ )р, п \дР ]т,п '
Далее, согласно (21.35) и (26.26), имеем
14 — Ts}.
(26.28)
Из (26.27) и (26.28) вытекает уравнение, аналогичное уравнению Гиббса — Гельмгольца:
14 = ht-\- Т
днг\
дТ )р, п
(26.29)
Если в жидкой многокомпонентной системе один компонент, который и в чистом состоянии также является жидким, находится в избытке, то его обычно называют растворителем. Принято величины, отнесенные к растворителю, обозначать индексом 1. Величина
Д[ЛХ = [11 — p-ю,
(26.30)
где р-10, химический потенциал чистого растворителя при той же температуре и давлении, называют свободной энергией разведения. Аналогичным образом определяют теплоту разведения Д/ц и энтропию разведения Ast. Из уравнений (26.27)—(26.29) получают часто применяемые соотношения
ДР1 = ДЛХ— ТДзр
д(ДР1/Т) д (МТ)
(26.31)
(26.32)
— Miv
138
Глава III
Д^Д^-7^^ п. (26.33)
Большое значение свободной энергии разведения для термодинамики растворов основано на том, что эту величину (и вместе с ней также Д$! и Д/ч ) можно легко определить разными экспериментальными методами. В частности, для бинарных растворов термодинамические свойства полностью определены, если известен Др^ как функция мольной доли х2 во всей области концентраций. Из изотермиче-ски-изобарной формы уравнения Гиббса—Дюгема следует
(1 — х2) + *2 — = о, (Т = const, Р = const) (26.34)
дх2 дх2
и вместе с этим
A^=f— (26.35)
V Хо
Xi
или, после частичного интегрирования,
Д|г2 = ? dxt — . (26.36)
J 1 — Xi 1 —
Средняя мольная свободная энергия Гиббса, отнесенная к чистым компонентам (которая называется также свободной энергией смешения), согласно (26.17), равна
ДО* = Др4 + х2Д^2- (26.37)
Глава IV
Гетерогенные равновесия без химических реакций
§ 27. Общие условия равновесия гетерогенных систем
Рассмотрим закрытую систему с фиксированными рабочими координатами, состоящую из т компонентов и о фаз. Фазы будем обозначать строчными греческими буквами, причем одновременно будем применять а как текущий индекс. Не будем рассматривать влияние внешних полей, поверхностные эффекты, растворы электролитов и химические реакции. Поскольку фазы являются твердыми телами, то предположим, что на них действует со всех сторон гидростатическое давление и нет никаких напряжений по осям или тангенциальных напряжений. Поэтому объем является единственной рабочей координатой. Фазы должны находиться во внутреннем равновесии и быть друг по отношению к другу полностью открытыми, т. е. для каждой фазы энтропия, объем и все числа молей являются переменными величинами. Для условий равновесия и дополнительных условий примем с учетом рассуждений § 17, что они даны в виде уравнений.
Учитывая эти допущения, фундаментальное уравнение (20.5) для каждой фазы запишется следующим образом:
§£у(а) __ J^(a) §g(a)
_Р(«) 80«> + 2^а) Ма) *
Z=1
(27.1)
= _ р(’) §у(°) 2 ;Л0> 8п*.о) .
1=1
Согласно общему условию равновесия (17.2), должно быть
140
Глава IV
2 = о, (27.2)
й=«1
с дополнительными условиями
о
а=1 = 0, (27.3)
а=1 = 0, (27.4)
а=1 = 0 (i=l,. .., т). (27.5)
Такую экстремальную задачу с дополнительными условиями можно решить методом неопределенных множителей Лагранжа. Умножим дополнительное условие (27.3) на неопределенный, но постоянный коэффициент (27.4) на Х2 и (27.5) на Х3/. Затем вычтем эти уравнения из (27.2). Используя (27.1), получим
V _ Хх) oS(a) + 2 (^(а) — Хг) 8V(a> + а а
т
+ 22(^>“Ч^“> = 0’ (27.5a)
Z=M а
Далее предположим, что множители определены таким образом, что в суммах по а каждый раз одно выражение, стоящее в скобках, становится равным нулю. Тогда становятся свободными остающиеся вариации и уравнение (27.5а) может выполняться только в том случае, когда все остальные скобки также равны нулю. Поэтому для каждого а и для каждого i должно выполняться
Т(а) = Хх; Р(а) = Х2; ..(а) __ > (27.56)
Отсюда следует
р(а) _ р(р) __ _ р(°) (27.6)
р(а) __ р(£3) =... __ (27.7)
Гетерогенные равновесия без химических реакций
141
„( ) _ „(₽) _ _ „(?) Г/ — rZ — ... — р/ ,
(i = 1,..., m).
(27.8)
С учетом сделанных предположений в состоянии термодинамического равновесия все фазы гетерогенной системы должны обладать одинаковой температурой и одинаковым давлением и химические потенциалы каждого компонента во всех фазах должны быть равны. Уравнение (27.6) содержит условие термического равновесия, (27.7) — механического равновесия и (27.8) — равновесия между веществами.
Уравнениями (27.6)—(27.8) полностью определено термодинамическое состояние системы. Из фундаментального уравнения с учетом выражения (15.8) следует, что состояние задано (т+2)о независимыми переменными S(a>, У(а), n(d). В качестве уравнений состояния (см. § 20 к пункту д.) служат уравнения (27.6)—(27.8) в количестве (т+2)(о—1). Дополнительные условия (27.3)—(27.5) дают далее (zn+2) уравнений, так что имеется столько же уравнений, сколько и неизвестных. Фактически условия (27.6)—(27.8) как опытные факты были введены уже в гл. 1 при обсуждении первого закона термодинамики. В выводе из общего условия равновесия Гиббса выражен аксиоматический аспект последнего, о чем уже упоминалось в § 17.
Условия равновесия (27.8) можно сформулировать также при помощи средней мольной свободной энергии Гиббса. Рассмотрим ради простоты две фазы а и (3. Подстановка выражений (26.20) и (26.22) в (27.8) дает
/ = / (.= b ? m_l)t (27 9)
m—1
(«)
У?' c)G* 2Xt dXi
(₽)
. (27.10)
§ 28. Мембранные равновесия. Осмотическое
давление
Плодотворность экстремального принципа, сформулированного в § 17, проявляется лишь при применении к сложным случаям. Приведем пример, имеющий большое значение
142
Глава IV
для теории жидких смесей. Рассмотрим вновь закрытую систему с фиксированными рабочими координатами, которая состоит из т компонентов и а фаз. Сделаем те же предположения, что и в § 27, но теперь, наоборот, будем считать, что все фазы отделены друг от друга твердой полупроницаемой мембраной, которая проницаема только для s компонентов (s<m). Таким образом, установлен объем каждой фазы. Далее, для всех фаз вариации т—s чисел молей равны нулю. Поэтому дополнительные условия для экстремальной задачи (27.2) имеют вид
=0, (28.1)
а—1
8У(а) = 0; 8VW) = 0,...; 8V(o) = 0, (28.2)
2Ца)=0 (i = 1,..., s), (28.3)
а—1
8п)а) = 0; 8п<₽) = 0 ...; 8п}а) = 0
(/ = « + 1,..., т). (28.4)
Отсюда в сочетании с (27.1) и (27.2) получаем условия равновесия
|4₽>= ...= p,<3). (i=l,..., S). (28.6)
Наличие твердой полупроницаемой перегородки приводит к тому, что сосуществующие фазы могут находиться при различных давлениях и что равенство химических потенциалов требуется только для s компонентов, которые могут проходить через мембрану.
Простейшим и важнейшим примером применения этого рассуждения является осмотическое равновесие (рис. 15).
В этом случае а=2. Одна фаза состоит из чистого растворителя (компонент 1), вторая фаза из раствора, который ради простоты будем считать бинарным. Таким образом, мембрана является проницаемой только для растворителя.
Гетерогенные равновесия без химических реакций
143
Условия равновесия (28.5) и (28.6) в этом случае имеют вид
pf _______ fjprr _____ р
(28.7)
(28.8)
h(T, Р') = ^(т, Р", xj.
Так как обе фазы в отсутствие мембраны не находятся в равновесии, т. е. р(7\ Р, 1) =/= (У, Р, xj, то (28.8) может
быть выполнено только в том случае, если
Р"фР'.
(28.9)
Условия равновесия не дают возможности судить о том, являются ли Р'<Р" или Р’>Р". На этот вопрос можно ответить при помощи условий стабильности, значение которых для термодинамики становится наглядным на этом простом примере. Как позднее будет показано в гл. VI, из условий стабильности следует
dfa\ дхг )т.
>0. р
(28.10)
Далее, учитывая (26.27), для чистого растворителя можно написать
дР )т
= v'1>0.
(28.11)
Так как при неравенстве (28.10) для Р'—Р" то, согласно выражению (28.11), условие (28.8) может быть выполнено только в том случае, если Р"^>Р'. Избыточное давление
П = Р" — Р',
(28.12)
144
Глава IV
при котором раствор находится в осмотическом равновесии, называется осмотическим давлением.
Осмотическое давление находится в простом соотношении со свободной энергией разведения, определяемой по уравнению (26.30). Из уравнения (28.11) (если Р' идентифицировать с нормальным давлением Ро) следует
Л>+п
Hi (Ро + п, х2) = Н! (Ро> + J VjdP. (28.13)
Вследствие (26.30) и (28.8) получаем
ЛН-и
Д^= — j* V1dP, (28.14)
р0
или
Si
(28.15)
где — средний парциальный мольный объем между давлениями PQ иР0+П. Таким образом, осмотическое давление в основном является мерой свободной энергии разведения.
Осмотические измерения имеют самостоятельное значение только для очень разбавленных растворов. Порядок величины П в этой области для низкомолекулярных растворов 1—10 атм (можно также отметить, что для них такие измерения выполнимы только в исключительных случаях, так как практически отсутствуют пригодные полупроницаемые мембраны); для макромолекулярных растворов порядок величины П 10“4 —10"3 атм. Вследствие незначительной сжимаемости жидкостей почти всегда можно пренебречь зависимостью парциального мольного объема от давления.
§ 29. Правило фаз
Рассмотрим систему, находящуюся в термодинамическом равновесии и состоящую из т компонентов и о фаз. Условия равновесия содержатся в уравнениях (27.6)—(27.8). Исследуем теперь вопрос, какие возможны изменения ве
Гетерогенные равновесия без химических реакций
145
личин состояния, если требуется, чтобы сохранялось первоначально данное гетерогенное равновесие. Следовательно, число сосуществующих фаз должно оставаться постоянным. Вопрос можно упростить, если выяснить сколько переменных состояния можно варьировать в вышеупомянутом смысле для заданных значений т и а.
В качестве независимых переменных выберем температуру, давление и химические потенциалы. Из условий равновесия следует, что при рассматриваемых изменениях для каждой фазы должно быть
dT(a) = dT; dP(a) = dP; d^a) = d^ (все а, все i). (29.1)
Как было показано в § 20, справедливость фундаментального уравнения подразумевает выполнимость уравнения Гиббса—Дюгема для каждой фазы. Напишем фундаментальное уравнение в форме (26.12) и, используя (29.1), получим систему уравнений
S*(а) dT — V*(а) dP + 2 d^i = °> Z==l
S*<а) dT — V*(а) dp 4- V dpi = 0.
/=1
(29.2)
Таким образом, имеем т+2 независимых переменных, вариации которых ограничены а уравнениями (29.2). Поэтому число свободных переменных равно
f = т + 2 — о.
(29.3)
Это уравнение является правилом фаз Гиббса, f называют числом термодинамических степеней свободы.
По значению величины f равновесия делятся на нонва-риантные, моновариантные, бивариантные и поливариант-ные.
Из уравнения (29.3) следует, что число термодинамических степеней свободы уменьшается на 1, если число сосуществующих фаз увеличивается на 1.
Как видно из вывода, правило фаз в форме уравнения (29.3) связано с предположениями, сделанными в § 27. Сопоставим здесь еще раз эти предположения, так как недо
146
Глава IV
пустимое применение уравнения (29.3) приводит к кажущимся противоречиям между теорией и экспериментом. В основе уравнения (29.3) заложены следующие допущения: а. Поверхностными эффектами можно пренебречь.
б. Химические реакции не происходят.
в. Единственной рабочей координатой является объем.
г. Поверхность раздела между фазами является теплопроводной, деформируемой и проницаемой для всех компонентов. Это значит, что для каждой фазы энтропия, объем и число молей свободно варьируются в рамках, данных дополнительными условиями (27.3)—(27.5).
§ 30. Фазовые реакции
В § 27 было показано, что состояние гетерогенной системы полностью определено условиями равновесия (27.6)—(27.8) вместе с дополнительными условиями (27.3)—(27.5). Остается только выяснить, возможны ли и если возможны, то при каких условиях, изменения состояния, при которых зафиксированы только уравнения (27.5)—(27.8), в то время как энтропия и объемы фаз и всей системы в целом изменяются. Так как химические потенциалы, согласно (26.2), (26.20) и (26.22) можно представить как функции Г, Р и xt, то вопрос означает, возможны ли и при каких условиях изменения состояния, при которых температура, давление и мольные доли фаз остаются постоянными и лишь масса определенных фаз увеличивается за счет массы других фаз при сохранении общей массы каждого компонента. Изменение состояния такого рода называется фазовой реакцией. Она всегда обратима, так как при этом в каждый момент сохраняется гетерогенное равновесие.
То, что фазовые реакции в принципе возможны, видно на примере равновесия жидкость — пар в однокомпонентной системе. При постоянных Т и Р можно за счет подвода или отвода теплоты при одновременном изменении объема перевести любое количество жидкости в пар и пара в жидкость. То, что фазовые реакции возможны не при любых условиях, показано на рис. 16, представляющем равновесное испарение бинарной системы при const, т. е. диаграмму кипения. В этом случае компонент 2 в жидкости всегда обладает более высокой концентрацией, чем в сосу-
Гетерогенные равновесия без химических реакций
147
шествующем паре. Если пар конденсируется, то уменьшается концентрация компонента 2 в паровой фазе и одновременно изменяется равновесная температура, как это видно из диаграммы. Фазовая реакция при этом не может происходить.
Для того чтобы вывести условия для фазовой реакции в общем виде, рассмотрим закрытую систему, находящуюся
Рис. 16. Диаграмма кипения t бинарной жидкой смеси.
в равновесии и состоящую из т компонентов и а фаз. Температура, давление и мольные доли фаз должны быть зафиксированы. Пусть в исходном состоянии общее число молей отдельной фазы будет равно
= (30.1)
i
За счет фазовой реакции достигается состояние, в котором при тех же значениях Г, Р, х^ числа молей фаз равны и(к) + = X «Г1 + 2 V -а). (30.2)
где является, таким образом, изменением числа молей фазы а при фазовой реакции. Так как мольные доли в фазах остаются постоянными, то должно быть
п<а) п<а) +
• (30-3)
Решением этого уравнения является
vja) = х(Р v(a)- (30.3а)
148
Глава IV
Так как при фазовой реакции по определению общее количество каждого компонента в системе остается постоянным, то, используя (30.3а), получим систему уравнений
S v<“> = 0, а=1
(30.4)
S >(а) = 0.
а=1
Фазовая реакция поэтому возможна тогда и только тогда, когда гомогенная линейная система уравнений (30.4) обладает для у(а> неизвестных решением, отличным от нуля.
Формально различают следующие случаи:
а.
Число уравнений меньше, чем число неизвестных. Поэтому всегда существует решение, отличное от нуля.
б. т — а.
Имеется столько же уравнений, сколько неизвестных. Тогда решение, отличное от нуля, существует тогда и только тогда, когда для определителя коэффициентов выполняется соотношение
|4“>| = о.
(30.5)
т > а.
Число уравнений больше, чем число неизвестных. Система поэтому является избыточной и имеет решение, отличное от нуля только тогда, когда все уравнения совместимы друг с другом. Это имеет место, если равен нулю каждый из детерминантов степени о, образованный матричными коэффициентами. Так как число этих детерминантов равно 1+т—а, то существует 1-фт—а уравнений для разрешимости системы (30.4). Из этого чисто математического факта вытекают следующие физические следствия:
а'. т < а.
Гетерогенные равновесия без химических реакций
149
Согласно (29.3), имеет место нонвариантное или мо-новариантное равновесие. В обоих случаях фазовые реакции неограниченно возможны.
б'. m = а.
Согласно (29.3), имеет место бивариантное равновесие. Фазовая реакция может происходить только для определенного состава фаз, при котором выполняется уравнение (30.5), т. е. исчезают детерминанты, образованные из мольных долей.
в'.
m > а.
Согласно (29.3), имеет место поливариантное равновесие. Фазовые реакции возможны только для таких составов, при которых 1+т—а детерминантов степени а, образованных мольными долями, равны нулю.
Условия равновесия, упомянутые в пунктах б. и в., в термодинамике называют условиями индифферентности. Рассмотрим два простых примера:
а". Равновесие системы, состоящей из двух фаз и т компонентов.
т—1 условий индифферентности имеют вид
у(«) у(3)
у(«)
-А. у Л-у
= 0 (i, / = 1, 2, ..., m; (30.6)
Эти условия выполняются только хГ> = хР, .... х<?>
в том случае, если
= х^. (30.7)
Поэтому фазовая реакция протекает тогда и только тогда, когда обе фазы имеют одинаковый состав. Тогда говорят об азеотропных смесях.
б". Трехфазное равновесие тройной системы. Имеет место бивариантное равновесие и в соответствии с пунктом б/ имеем единственное условие индифферентности
х?
4“’
„(₽)
Xi
х^
4Т> хр’ х£>
= 0.
(30.8)
150
Глава IV
Это уравнение геометрически означает, что при изображении состава фаз в треугольных координатах Мёбиуса должна получаться прямая линия (рис. 17). Частный случай, вытекающий отсюда, состоит в том, что уравнение (30.7) выполняется, если две сосуществующие фазы имеют одинаковый состав (совпадение двух точек в геометрическом представлении).
Рис. 17. Изображение условия индифферентности для трехфазного равновесия в тройной системе.
в". Из примеров легко получается следующее общее
Правило:
При би- и поливариантных равновесиях для протекания фазовой реакции всегда является достаточным одинаковый состав двух сосуществующих фаз, в случае сосуществования только двух фаз это является также необходимым условием.
Из определения нонвариантного равновесия следует, что здесь фазовая реакция может протекать только в соответствующей точке фазовой диаграммы. Так как при моно-вариантных равновесиях на фазовую реакцию не накладывается никаких ограничений, то она может протекать везде на кривой Р(Т). Такая кривая называется индифферентной кривой.
При би- и поливариантных равновесиях т+2—о степеней свободы фазовой реакции ограничены l-j-m—о условиями индифферентности. Поэтому для фазовой реакции остается еще. одна степень свободы и соответствующая кривая Р(Т), как и при моновариантных равновесиях, является индифферентной кривой. В частности, при двухфазовых равновесиях кривая, на которой расположены азеотропные смеси, так называемая азеотропная кривая, является индифферентной кривой.
Гетерогенные равновесия без химических реакций
151
§ 31. Нонвариантные и моновариантные равновесия
Если в равновесии сосуществуют т+2 фазы, то, согласно уравнению (29.3), /=0. Такое равновесие возможно только при специальном наборе значений Т, Р и в (т+1)-мерной фазовой диаграмме оно изображается точкой. Тогда говорят о нонвариантном равновесии.
Для однокомпонентной системы нонвариантное равновесие называется тройной точкой. Известными примерами являются тройная точка лед — вода — водяной пар, тройная точка лед I—лед II —вода или три тройные точки моноклинная — жидкая — парообразная сера, ромбическая — моноклинная — жидкая сера, ромбическая — моноклинная — парообразная сера.
В бинарных системах при нонвариантных равновесиях сосуществуют четыре фазы. Поэтому это равновесие называется четверной точкой. Примерами таких равновесий являются системы соль — кристаллогидрат — водный раствор — водяной пар, при которых газовая фаза, раствор и две твердые фазы находятся в равновесии.
Неограниченная возможность фазовых реакций при нонвариантных равновесиях, о которых говорилось в § 30, становится наглядной на примерах.
Гетерогенное равновесие, при котором сосуществуют т-j-l фаз, имеет, согласно (29.3), одну степень свободы и называется поэтому моновариантным равновесием. Наиболее простым примером является двухфазовое равновесие однокомпонентной системы, которое схематически представлено на рис. 18.
Так как на кривых сосуществуют по две фазы, то они называются кривыми сосуществования.
Выведем дифференциальное уравнение кривых сосуществования. При продвижении вдоль кривых сосуществования, согласно (27.8) и соответственно (29.1), можно записать
d^ = (31.1)
Так как для однокомпонентной системы химический потенциал и мольная свободная энергия Гиббса совпадают, то перепишем (31.1)
dG*<a> = dG*^ .
(31.2)
152
Глава IV
Рис. 18. Двухфазное равновесие однокомпонентной системы (схема).
Используя (26.8), (27.6) и (27.7), получим
— S*W dT + dP = — dT + V*<₽) dP (31.3)
или
dP __ S*(a) — S*(P) y*(a) _____________ y*(₽)
(31-4)
Из (27.8) с учетом (21.35) следует
#*(“) _ fs*(«) = #*(₽) _ 7S*(₽) . (31.5)
Подстановка в (31.4) дает
dP _ //*<“> — Я*(₽)
^7 т (у*(я) ____у*(₽))
(31.6)
Это соотношение называется уравнением Клаузиуса— Клапейрона. Оно представляет собой дифференциальное уравнение кривой сосуществования для двухфазного равновесия в однокомпонентных системах. Разность энтальпий в числителе правой части является в соответствии с уравнением (21.23) теплотой фазовой реакции в расчете на один моль. Целесообразно обозначать через а фазу с большей мольной энтальпией и писать
L = #*(«) — #*(3) .
(31.7)
Величина L называется мольной теплотой превращения, в частности мольной теплотой испарения, мольной теплотой плавления и т. д. Если написать
ДУ* = У*(а)_______у*(₽),
(31.8)
Гетерогенные равновесия без химических реакций
153
то уравнение Клаузиуса—Клапейрона примет более компактный вид
dP __ L
dT “ TAV* ’
(31.9)
Так как по определению £>0, то наклон кривой Р(Т) имеет тот же знак, что и разность объемов ДК*.
Рассмотрим теперь общий случай моновариантного равновесия. Тогда, согласно (29.3), о=/п4-1 и, согласно § 30, кривая Р(Т) является индифферентной кривой. Чтобы вывести дифференциальное уравнение этой кривой, будем исходить из системы уравнений (29.2), которую теперь запишем
у*(а) ___ V н __ С* (а)
dT 1 dT
Z1
(31.10)
m
|/*(m+l) ALL____V - — = S*(OT+D
dT dT
Z=1
причем все производные должны быть взяты вдоль кривой равновесия. Система (31.10) является негомогенной линейной системой уравнений с m-J-1 неизвестными dP/dT, d^JdT, ..., d^/dT. Решение для dP/dT имеет вид
dP Ds
~dT = Т\'
(31.11)
где
S*(a) X(!a)
(31.12)
) v(m+l) y(m+l)
л 1 ... Лт
V*(a) x{A
(31.13)
V Л\ ... Лщ
154
Глава IV
Разложение Лапласа обоих детерминантов по элементам первого столбца дает, если обозначить сомножители (в обоих случаях одинаковые) через D<a)*,
Ds = 2 D^S*W , (31.14)
Dv = 2 D(a) V*(a). (31.15)
a
Напишем теперь общую систему уравнений (30.4) для специальной задачи в виде
tn
2 x(ia) v(a> = - xim+!)
m
2Г(") _ r(m+D
/ — лт v
(31.16)
Если заранее известно то (31.16) представляет собой негомогенную линейную систему уравнений с т неизвестными у(«). Используем введенные выше детерминанты £)(а) (т. е соответствующие сомножители элементов и у*(а)от Ds и Dy). Решение (31.16) в этом случае имеет вид
__2^2— (31 17)* ** n(m4-D ’ \ /
Образуем теперь выражение
= — , (31.18) 2 £)<“> V*<“> a
* Применяемые в дальнейшем правила знаков для D(<0 состоят в следующем: £)(«) положительно для нечетного а, отрицательно для четного a. положительно для четного т, отрицательно для
D(a) .
нечетного т_______отрицательно, если величины а и т обе четные
£)(Лп+1)
или обе нечетные,в противном случае обе величины положительны.
** Число перестановок, которые переводят первоначально стоящие в числителе детерминанты в D(a), является нечетным, если величины а и т обе четные или обе нечетные, в противном случае это число четное.
Гетерогенные равновесия без химических реакций
155
и поделим справа числитель и знаменательна—D(m+X\ тогда, учитывая (31.17), получим
У v(a) £*(«) £s_ = ______________
Dv 2 ^(а) г*(а)
a
(31.19)
По определению v<a) числитель справа представляет собой изменение энтропии AS, знаменатель — изменение объема А V для рассматриваемой фазовой реакции. Таким образом, из уравнения (31.11) получим
dP __ k S dT ~ kV '
(31.20)
dP _ L
dT ~ T^V '
Изменение энтальпии при фазовой реакции равно
L = АН = 2>(a)H*(a) . (31.21)
В связи с тем, что dT=Q, dP=0, dnL=0 для фазовой реакции
AG = АН — TAS = L — TAS = 0. (31.22)
Окончательно получаем
(31.23)
Это соотношение, выведенное впервые Гиббсом, представляет собой обобщенное уравнение Клаузиуса—Клапейрона для любого поливариантного равновесия. При пг=1, а= =2 уравнения (31.20) и (31.23) переходят в обычные уравнения Клаузиуса—Клапейрона (31.4) и (31.9).
Уравнение (31.23) имеет особо важное значение при равновесии жидкость — пар в многокомпонентных системах. Если система состоит из m компонентов и m жидких фаз находятся в равновесии с паром, то уравнение (31.23) представляет температурную зависимость общего давления пара, соответственно зависимость температуры кипения от давления.
Важные особые случаи моновариантных равновесий представляют собой так называемые эвтектические смеси, при которых две твердые фазы находятся в равновесии с жидкой фазой.
156
Глава IV
На рис. 19 показано такое равновесие в изобарных условиях* **. Зависимость эвтектической температуры от давления дается уравнением (31.23).
Рис. 19. Фазовая диаграмма системы Sn—Bi (схема) Е — эвтектическая точка.
§ 32. Би- и поливариантные равновесия
В соответствии с § 30 при би- и поливариантных равновесиях как существенно новый элемент добавляются условия индифферентности. Интересен вывод дифференциального уравнения для индифферентной кривой Р(Т), на которой могут протекать фазовые реакции.
Будем исходить опять из системы уравнений (29.2), которую теперь целесообразно записать (для т>р)\
а—1 tn
V*(«) dP — 2 4e) dpt = S*(a) dT -f- 5 x^d рг,
а—1 т
у*(а) dP — 2 d'^t = S*(a) dT + x^d^i.
(32.1)
* Фаза II на самом деле не состоит из чистого висмута, а содержит в области эвтектических температур примерно 0,1—0,2 ат. % Sn. Область существования этой фазы в масштабе рисунка не обозначена.
** При рассмотрении изобарных диаграмм состояния сплавов можно вообще не рассматривать газовую фазу, так как постоянное давление поддерживается при помощи инертного газа (например, аргона), в то время как давлением пара металла в большинстве случаев можно пренебречь. Учет газовой фазы не изменял бы поэтому числа термодинамических степеней свободы.
Гетерогенные равновесия без химических реакций
157
Рассмотрим (32.1) как негомогенную линейную систему уравнений для а неизвестных dP, dplt..., ь Используем вновь определения (31.12) и (31.13), причем индексы т+1 и m заменим на о и а—1. Далее определим
(32.2)
Эти определители, если не обращать внимание на знаки, которые здесь не существенны, идентичны определителям степени о, приведенным в § 30, в., которые можно построить из коэффициентов матрицы (30.4). Используя формулу
Лц + Ви Л12 _ Ап Л12 7^21 “Ь ^21 ^23 ^21 ^22
Ви Л12 ^21 ^22
(32.3)
получим решение (32.1) для dP
m
Dy dP = Ds dT -p Did p--,
Z=a
(32.4)
Согласно § 30, в., фазовая реакция возможна только тогда, когда Dl равны нулю. Как следует из уравнения (31.19), Dv п Ds по физическим соображениям конечны и вообще не могут быть равны нулю. Отсюда следует необходимое и достаточное условие для протекания фазовой реакции в случае m>ai
или
дР \
д и Д’
дТ \
= 0 (i, / = о, о + 1, ..., m; i =/= j) (32.5)
= 0 (f, j = а, а + I, ..., m; i j). (32.6)
Равновесное давление в изотермических условиях (соответственно равновесная температура в изобарных условиях) является по условию (29.3) функцией m-j-l—а независимых
158
Глава IV
переменных, для которых можно выбрать т+1—а химических потенциалов. Таким образом, из (32.5) и (32.6) получим следующий
Закон:
При би- и поливариантных равновесиях равновесная температура при постоянном давлении и равновесное давление при постоянной температуре имеют стационарное значение, если может протекать фазовая реакция.
Этот закон был сформулирован для частных случаев Гиббсом и Коноваловым и поэтому называется обобщенным законом Гиббса—Коновалова. В общем виде он впервые был доказан Сорелем.
Рис. 20. Диаграмма кипения системы бензол—этиловый спирт (схема).
Дифференциальное уравнение индифферентной кривой по условию (32.4) имеет вид
dP D.
~dT = D^ (32-7)
Расчет правой части можно проводить так же, как и в § 31. Нужно только подставить вместо (31.16) (а—1) первых уравнений из системы (30.4). Тогда получим
dT bV (oz.o)
или
dT ~ Т Д V ’ (32.9)
Гетерогенные равновесия без химических реакций
159
Эти выражения являются обобщенным уравнением Клаузиуса—Клапейрона для индифферентной кривой би- и полива-риантных равновесий.
Простейшим примером рассмотренных здесь общих закономерностей является равновесие жидкость — пар в бинарных жидких смесях. На рис. 20 приведена схема диаграммы кипения системы бензол—этиловый спирт в изобарных условиях.
В качестве независимой переменной выбрана мольная доля этилового спирта. Стационарная точка, в которой в соответствии с обобщенным законом Гиббса— Коновалова может протекать фазовая реакция и в которой, согласно
Рис. 21. Трехфазное равновесие тройной системы этанол — бензол— вода (схема).
X — паровая фаза.
§ 30 (правило в"), обе сосуществующие фазы имеют одинаковый состав, называется (как уже было отмечено в § 30) азеотропной точкой. Уравнение (32.9) является дифференциальным уравнением азеотропной кривой. Диаграмма подразумевает следующие закономерности равновесного кипения бинарных жидких смесей, которые называются законами Коновалова'.
I. При постоянном давлений температура кипения как функция состава имеет стационарную точку тогда и только тогда, когда жидкость и пар имеют одинаковый состав.
II. При постоянном давлении температура кипения повышается при добавлении того компонента, концентрация которого в паре меньше, чем в жидкости.
III. При изменении температуры кипения при постоянном давлении состава жидкости и сосуществующего пара изменяются в одинаковом направлении.
160
Глава IV
Глава V
Эти законы можно доказать также аналитически. Закон I является просто частным случаем обобщенного закона Гиббса — Коновалова. Для изотермических условий справедливы аналогичные законы.
Технически важным для получения чистого этилового спирта является равновесие в тройной системе этанол— бензол—вода, схематически приведенное на рис. 21. Состав трех сосуществующих фаз расположен, как это следует из § 30, на прямой, и давление пара при постоянной температуре имеет максимум, который одновременно является наибольшим давлением пара на общей поверхности давления пара системы.
Химические равновесия
§ 33. Общие условия равновесия
В главе IV химические реакции не рассматривались. Однако химическое равновесие является особым случаем внутреннего равновесия, так как оно устанавливается также в гомогенной системе. Так как в §27 для каждой фазы предполагалось существование внутреннего равновесия, то внутри фаз можно допустить протекание химических реакций, если предположить полное химическое равновесие и ввести в условия равновесия только числа молей независимых компонентов в смысле определения § 2. Ранее полученные результаты останутся тогда неизменными, но, естественно, не будут содержать сведений об условиях химического равновесия. Реакций между фазами (гетерогенные реакции) можно допустить, предполагая равновесие и ограничиваясь независимыми компонентами:
а) если уравнение реакции не входит в обсуждение проблемы;
б) если фазовые реакции идентичны химическим реакциям. Примером для пункта а) служат так называемые диалектические точки, т. е. максимум изобарной кривой плавления, при котором бинарный расплав имеет тот же состав, что и твердое химическое соединение, не смешивающееся с чистыми компонентами. Как показывает рис. 22, на диаграмме должны появляться тогда две эвтектические точки Ег и Е2. Вопрос, происходит ли химическая реакция при переходе соединения АВ из твердой фазы в жидкую, в данном случае не играет роли так же, как и реакция между тремя твердыми фазами (которая вследствие торможения вообще не может протекать). Дистектическая точка имеется, например, в системе Sn—Mg. Твердым интерметаллическим соединением является здесь SnMg2.
Примером для пункта б) служит разложение карбоната кальция
6—235
162
Глава V
Химические равновесия
163
СаСО3 С02 + СаО. (33.1)
В этом случае имеем два независимых компонента и три фазы, т. е., согласно условию (29.3), моновариантное равновесие. При чисто физическом способе рассмотрения СО2 и СаО будут независимыми компонентами, в то время как СаСО3 появляется как смешанная фаза, которая сосуществует с двумя другими фазами. Согласно § 30 а', фазовые реакции
Рис. 22. Фазовая диаграмма бинарной системы с дистек-тической точкой D и двумя эвтектическими точками и
возможны тогда без ограничения и уравнение (33.1) представляет собой просто фазовую реакцию в химическом способе написания, как это следует из сохранения общего количества каждого компонента. Поэтому равновесное давление СО2 («давление разложения») задается обобщенным уравнением Клаузиуса—Клапейрона (31.22).
Аналогичным образом можно рассмотреть также реакцию
2NiO О2 + 2Ni. (33.2)
Но если никель находится в виде сплава Ni-Pt, то имеем три независимых компонента и три фазы и, следовательно, бивариантное равновесие. Поэтому химическая реакция не может быть отождествлена с фазовой реакцией, которая, согласно § 30 и 32, возможна только при определенных составах. Поэтому методы главы IV к этой проблеме не применимы.
Чтобы вывести условия химического равновесия в общем виде, нужно в фундаментальное уравнение ввести в качестве независимых переменных числа молей всех веществ, которые встречаются в рассматриваемых нами уравнениях реакций. Числа молей фазы т не являются более незави
симыми друг от друга в смысле определения § 2. Ради простоты предположим, что имеется единственная реакция
аА + ЬВ уУ, (33.3)
причем безразлично, протекает она внутри фазы или между различными фазами, или имеют место оба случая. В этом случае можно применять фундаментальное уравнение (27.1) и условие равновесия (27.2). Соединим оба в уравнение
2 8 S<a> — jjpw +
a a
tn
+ 2 2^)Ma)
a L i = 1
= 0.
(33.4)
Здесь также справедливы дополнительные условия
2 8S<“) = 0, (33.5)
a
2 = о. (зз.б)
a
Далее справедливо без ограничения
2 &4а) = о (i А, В, Y). (33.7)
a
Однако, так как А, В и Y связаны уравнением реакции (33.3), то нет больше необходимости в том, чтобы
2 S«Aa) = 0; 2 Ma) = 0; 2 S4a) = 0. (33.8) a a a
Вместо этих дополнительных условий появляются теперь
уравнения + 7 S s«Va) = o,
a a + 7 S Ma) = o. (33.9)
6* ОС
164
Глава V
Эти условия свидетельствуют о том, что вариации чисел молей А, В, Y, суммированные по всем фазам в том случае, если, согласно (33.8), не каждая вариация сама по себе равна нулю (что соответствует просто сдвигу веществ между фазами), должны учитывать стехиометрические соотношения задаваемые уравнением реакции (33.3). Теперь условия (33.8) представляют собой специальный случай, который содержится в более общих условиях (33.9). Вариации, возможные согласно (33.8), удовлетворяют поэтому также (33.9). Если принять их во внимание, то следует, что ранее выведенные условия равновесия (27.6)—(27.9) необходимы также и здесь. Но за счет возможности протекания химической реакции (33.3) появляются еще новые условия. Если подставить (27.6)—(27.8) в (33.4), то с учетом (33.5)—(33.7) получим
раХ &4а) +р-в 2 &«ва) + py 2 Ма) = о. (зз.ю) а а а
Если при помощи уравнения (33.9) исключить ВпА и Впв, то получим
— а Ра X — Ьрв 2 Ч-уру 2 8пу)=0. (33.11) а а а
Так как вариации 8п(у являются произвольными, т. е. может быть Впу <0 (ср. § 17), то (33.11) будет выполнено только тогда, когда
а на + Ь цв = У Ну . (33.12)
Аналогичным путем можно показать, что для каждой произвольной химической реакции
аА + ЬВ + сС + ... ^yY + zZ + ... (33.13) в состоянии термодинамического равновесия должно быть выполнено условие
а На 4“ b нв 4" с Нс 4- ••• У Ну Ч~ Hz 4~ ••• (33.14)
Уравнение (33.14) представляет собой общее условие химического равновесия, справедливое как для гомогенных, так и для гетерогенных реакций. Реакцию (33.13) можно записать в общем виде
Химические равновесия
165
SvfXf = 0, (33.15)
причем получающиеся продукты имеют положительный знак. Тогда соответствующее выражение для условий равновесия имеет вид
2 0. (33.16)
§ 34. Гомогенные реакции. Закон действия масс
В этом параграфе рассмотрим химические реакции, протекающие внутри фазы (соответственно гомогенной системы).
а. Гомогенные реакции е газовой фазе
Парциальное давление компонента i в газовой фазе дается уравнением
Pi = xtP- (34.1)
По закону Дальтона для идеальной газовой смеси
Pi = RT. (34.2)
Химический потенциал компонента i идеальной газовой смеси равен
Нг = RT\nPi + (Т), (34.3)
где р/(Г) — стандартный химический потенциал, рассчитанный при рг=1. Уравнения (34.1) и (34.3) можно записать
рг = RT\nXi + RT\nP + $ (Т). (34.4)
Предположим, что в идеальной газовой смеси протекает реакция (33.13). Подстановка выражения (34.3) в условие равновесия (33.14) дает
рУ р* .
........с-- = Кр (П (34.5) Ра Рв Рс •••
в то время как подстановка (34.4) в (33.14) дает
хК Ху...
„а~~Ть~7.с- = Кх (Т, Р). (34.6)
166
Глава V
Уравнения (34.5) и (34.6) являются аналитическим выражением закона действия масс (ЗДМ), который был впервые выведен Гульдбергом и Вааге из кинетических соображений. Константы равновесия КР и Кх связаны соотношением
Кх (Т, Р) = ра+b+c...-y-z... Кр (34.7)
Наконец ЗДМ можно сформулировать таким образом, чтобы ввести при помощи (34.2) в качестве концентраций величины сг=иг/У. Тогда
4 с*
Д е » (Л (34.8)
СА СВ сС
И
Кс = (Я7у+ь+с.. .-y-z... Кр9 (34>9)
Константа равновесия Кс в этом случае также зависит только от Т и не зависит от Р. Здесь была рассмотрена идеальная газовая смесь не только по историческим причинам, но и прежде всего потому, что приведенные формулы в большинстве случаев дают достаточно хорошее приближение для практических целей, если общее давление не превышает нескольких атмосфер.
Если необходимо учитывать отклонения от идеального газового состояния, то можно сохранить аналогичную формулировку введением летучестей вместо парциальных давлений. Летучести р] определяются уравнением
= RT In р- + р* (Т), (34.10)
и кроме этого
lim -Д = lim Д- = 1 (34.11)
р->0 Г Р->0 Pl
(закон Гиббса — Дальтона). Тогда получают
*у *z
Д ---------- = %р (Л- (34.12)
Ра Рв Рс-
Следует обратить внимание, что хотя справа вновь стоит функция только температуры, однако летучести не являются чистыми концентрационными переменными, а являются функциями всех переменных состояния.
167
Химические равновесия .-----------------------------------
Для большей наглядности напишем еще раз различные формы ЗДМ в компактной записи уравнений (33.15) и (33.16), которая была до сих пор использована. Имеем
Пр/ = Кр (Т),
Пхр = КЛТ,Р), (34.13)
Пер = кс(Т)
и для реальных газов
= к' (Т).
(34.14)
Теперь необходимо подвергнуть исследованию константы равновесия. Чтобы получить по возможности более общие результаты, покажем прежде всего, что стандартные значения химических потенциалов в уравнениях (34.4) и (34.10) идентичны. Согласно уравнениям (26.27) и (34.10), имеем
(д fl; \ ( д 1п р,- \
дР /Т.Х \ дР ) т. х
Так как
/ dln(Pxz) \ _ RT
К \ дР )т,х ~ Р '
то уравнение (34.15) можно переписать
(34.15)
(34.16)
din (p*/Pxz) дР
Если проинтегрировать это уравнение при постоянных температуре и составе, то получим
р
* О / n/т» \ *
In I ( vt-------) dP + lim In . (34.18)
P*i RT J V P / p-о Pxi о
Используем теперь тот опытный факт, что при исчезающе малых давлениях все реальные газовые смеси ведут себя, как идеальные. Тогда, согласно (34.16),
168
Глава V
lim р->о
Pvi ~RT
/ (34.(9)
чем и обеспечивается существование интеграла. Да^ее из опытных фактов в сочетании с (34.2) следует уравнение (34.11), согласно которому последний член в (34.18) равен нулю. Учитывая (34.18), уравнение (34.10) можно переписать
р
Иг = ЯТ1п (Рхг) + J (vf----------p-j dP + р* (Т). (34.20)
О
Отсюда получаем для стандартного значения
Н* (Г) = lim [|Х{ — RT In (PXi)].
Р->0
(34.21)
Граничное значение первого члена по эмпирическому закону дается уравнением (34.4). Таким образом, имеем
(Т) = (Т). (34.22)
Из уравнений (34.13), (34.14) и (34.22) следует
Кр(Г) = ^(Т) (34.23)
и
RT In Кр = — 3 р? = — A G° (34.24) или
Кр = e-(^/RT)t (34.25)
причем было использовано уравнение (21.40). По определению AG0 является изменением свободной энергии Гиббса, соответствующим уравнению реакции, если все участники реакции находятся в стандартном состоянии.
Выведем теперь зависимость констант равновесия от температуры и давления.
Дифференцирование уравнения (34.24) по температуре дает
din К. V d ( Т )
--р— = — 2j — —(34.26)
dT . R dT v 7
Используя уравнение (26.29), получим
Химические равновесия
169
dlnKp _ у
dT RT2 '
(34.27)
Если определить энтальпию реакции для стандартного состояния АЯ° по уравнению
ДЯ°=5*гЛ°. (34.28)
i
то из (34.27) получим
Это соотношение называется уравнением Вант-Гоффа. Для идеального газа оно непосредственно дает температурную зависимость константы равновесия. Так как в этом случае парциальные мольные энтальпии не зависят от состава и равны стандартным значениям, то А//0 является истинной (измеренной) энтальпией реакции. Для реальных газов соотношения получаются более сложными, и они здесь не будут рассматриваться.
Для остальных констант равновесия можно вывести аналогичные соотношения. Ограничимся рассмотрением идеальных газов*. Если подставить для сокращения Ev.= v, то для изобарно-изотермического изменения объема будем иметь
Р А V = vRT. (34.30)
Для энтальпии реакции справедливо
ДЯ = (А17)г,р+ Р(ДЕ)лр
= (АЯ)Г,У + Р(АЕ)г,р= ДЯ + РАЕ, (34.31) причем использовано условие, что внутренняя энергия идеальной газовой смеси не зависит от объема и давления. Согласно уравнению (21.19), АЯ представляет собой теплоту реакции при постоянном объеме. Наконец из уравнения (34.9) имеем
In Rc = In Кр — v In RT. (34.32)
* Индекс0, который может приводить к недоразумениям, в дальнейшем опустим.
170
Глава V
Из уравнений (34.29)—(34.32) следует
Д£/ dT “ RT2 ‘
(34.33)
Это уравнение также называется уравнением Вант-Гоффа или изохорой реакции.
Из выражений (34.6) и (34.29) получаем
( д In Кх Д Н
I дТ Ур = "№“•
Из уравнений (34.6) и (34.30) следует
( д In Кх ДУ
\ дР )т = RT~'
(34.34)
(34.35)
Это соотношение впервые было выведено Планком и Ван Лааром. Оно показывает [так же, как и уравнение (34.6)], что константа равновесия Кх только тогда зависит от давления, когда v=^=0.
б.Гомогенные реакции в жидкой фазе
Гомогенные реакции в жидкой фазе описываются в основном теми же методами, что и гомогенные реакции в газовой фазе. Здесь также приходят к ЗДМ и к уравнениям зависимости констант равновесия от температуры и давления. Поэтому можно ограничиться более кратким рассмотрением. Существенное отличие от реакций в газовой фазе состоит в выборе переменных концентрации и стандартного состояния. Для теоретического рассмотрения здесь целесообразно выбрать мольные доли*. Поэтому для химического потенциала компонента i пишем
= 7?Т1п(А^) + и? (Т, Р), (34.36)
где величина xlf ..., называется коэффици-
ентом активности. Обычно применяют два способа нормировки стандартного состояния
* В экспериментальйой термодинамике в качестве переменных концентрации часто используют число молей на 1000 г растворителя (моляльность) и число молей на литр раствора (молярность). Последняя величина имеет тот недостаток, что она зависит от температуры.
Химические равновесия
171
«• Н? = Hio(T, Л (* = 1» •••> tn), (34.37)
где рго— химический потенциал чистого компонента i.
Pi (Г, Р) = И10 (Т, Р),
(4 (Т, Р) = lim (;тг — RT In xt) (i — 2, tri).
Xi->1
(34.38)
Компонент 1 называется растворителем. (Ср. § 26). Способ а. имеет преимущество полной симметрии по отношению к компонентам.
Нормировка р. тесно примыкает к теории газов. Ее следует использовать, если хотят сохранить аналогичную теории газов формулировку ЗДМ. Выбор стандартного состояния подразумевает соответствующую нормировку коэффициентов активности.
Получаем:
Для а:
lim ft = 1. (34.39)
Для ₽:
lim fi — 1; lim ft = 1 (i = 2, ..., m). (34.40) Xi = l
Подстановка (34.36) в (33.16), если ввести активности
fiXit (34.41)
дает
П a\i = К (Т, Р), (34.42)
где
[1? = — Д G0. (34.43)
С нормировкой р. для идеально разбавленного раствора получим*
* Под этим понимают раствор, для которого справедливы уравнения, вытекающие из (34.36) совместно с (34.38) и (34.40) для Хр-И. Формально речь идет при этом о граничном законе, применимость которого к реальным системам зависит от природы системы и точности измерения.
172
Глава V
(34.46)
(34.46)
(34.47)
Пхр = К(Т, Р). (34.44)
Теперь получаем зависимость констант равновесия от температуры и давления
а In = А 77° дТ )р, ~ RT*
и, используя (26.27), / a In М _ _ А У° \ дР )т ~ RT ’
где
Дуо =
Рассмотренный в этой главе выбор стандартных состояний для химических потенциалов имеет общее значение для термодинамики жидких смесей. Как легко установить, он лежит также в основе определения свободной энергии разбавления (§ 26).
Для твердых смешанных фаз используют в основном такой же формализм. Однако здесь применяют всегда нормировку а., так что для чистого твердого тела всегда Л=1.
§ 35. Гетерогенные реакции
Как уже было упомянуто, общее условие равновесия (33.16) справедливо как для гомогенных, так и для гетерогенных реакций. Однако дальнейшие рассуждения §34 не позволяют рассмотреть в общем виде гетерогенные реакции. Поэтому ограничимся некоторыми замечаниями, которые проясняют природу появляющейся здесь проблемы.
Рассмотрим вновь реакцию (33.15) и предположим теперь, что все участники реакции находятся не только в газовой фазе, но также в одной (или нескольких) конденсированных фазах. Химическую реакцию в газовой фазе можно рассматривать таким образом, как в § 34а. Предполагая идеальное газовое состояние, формально вновь приходим к уравнению (34.13). При данных условиях для каждого компонента устанавливается не только химическое рав
Химические равновесия 173
новесие, но и равновесие жидкость — пар. В то время как для ЗДМ стоящие слева выражения являются функциями Т9 соответственно Т и Р, каждое парциальное давление pt является теперь функцией Т и Р, которая определяется равновесием жидкость — пар. Преимущество ЗДМ для гомогенных реакций, с одной стороны, состоит в том, что значение констант равновесия является мерой продвижения реакций при данных начальных условиях, с другой стороны, в том, что, изменяя начальную концентрацию отдельных компонентов, можно влиять на ход реакции. Примером последнего является торможение диссоциации HI
2HI — Н2 + 12 (35.1)
за счет добавки 12. Многочисленные другие примеры можно найти в методах химического анализа. Оба упомянутых преимущества отпадают, если парциальные давления в выражении ЗДМ будут равновесными давлениями. Формализм ЗДМ становится тогда бессмысленным и нужно вновь применять общие условия равновесия (33.16).
Несколько иными получаются соотношения, если в рассматриваемой области состояния некоторые участники реакции находятся только в газовой фазе. В качестве примера рассмотрим равновесие Будуарда
Ств. 4- СО2газ 2СОгаз. (35.2)
Так как зависит практически только от температуры, то в этом случае, предполагая идеальное газовое состояние, ЗДМ можно записать в виде
2
= Кр (Т). (35.3)
Рсо2 р
Для константы равновесия уже больше не выполняется уравнение (34.24), а (в рассматриваемом случае)
RT In Кр = рсП+ Рсо2 - 2 ;хсо • (35.4)
Аналогичное рассмотрение можно провести, если углерод растворен в (твердом или жидком) железе. Используя (34.36) и нормировку а, получаем
174
Глава V
Р X (35.5)
Рсо2хс 'С
где К вновь определяется уравнением (35.4). Из уравнений (35.3) и (35.5) следует
f _ (Рсс/ Рсо2) СПЛЯВ /qr п\
ХС /С = / 2 / „-----\----• (00.0}
\ ’ СО ^СО2) графит
Подобным же образом можно рассмотреть окисление никеля, упомянутое в § 33. При окислении чистого никеля имеем
Ро2 = К/Т), (35.7)
где
-RTln/Cp = 2[tNiO — Ро2 — 2[xNi. (35.8)
Но если никель находится в виде сплава Ni-Pt, то уравнение (34.36) и нормировка а приводят к
Ро,
-^2- = Хр (Т), (35.9)
XNi/Ni Р
где Кр вновь дается уравнением (35.7). Из (35.6) и (35.8) получаем
4 й, = . (35.10)
”О2 никель
Уравнения (35.6) и (35.10) показывают, что коэффициенты активности конденсированных смешанных фаз можно определить из измерений гетерогенных равновесий.
Если изучать гетерогенные равновесия в более широкой области изменения состояния, то можно встретить два случая:
а. Парциальное давление достигается только для одного вначале газообразного компонента и при данных условиях происходит его конденсация.
б. Для компонента, находящегося вначале в конденсированной фазе, давление становится меньше равновесного И конденсированная фаза исчезает.
Химические равновесия
175
В обоих случаях представление всей области состояния при помощи ЗДМ невозможно и необходимо вернуться к общим условиям равновесия (33.16).
В заключение сделаем замечание о правиле фаз для случая химической реакции. Способ подсчета в принципе такой же, как в § 29. Следовательно, здесь вновь имеется о уравнений Гиббса—Дюгема. Но суммы теперь распространяются на все вещества (виды частиц). Для этого имеем дополнительно г уравнений реакций типа (33.15) и возможно еще b уравнений связи. Поэтому число термодинамических степеней свободы равно
f == т + 2 — о — г — Ь. (35.11)
Сравнение с определением независимых компонентов уравнения (2.1) показывает, что уравнение (35.11) переходит в обычную форму правила фаз (29.3), если под т понимать число независимых компонентов.
Если реакция протекает до полного исчезновения одного из компонентов, то т уменьшается на единицу и г также на единицу, так как соответствующее уравнение реакции не может быть больше учтено. Поэтому число степеней свободы остается неизменным. Лишь при сингулярных составах, которые соответствуют стехиометрическим соотношениям, т может уменьшаться сильнее, чем г, и, таким образом, можно уменьшить /.
§ 36. Число пробегов реакции. Сродство
Рассмотрим кратко одну, формально несколько отличную от метода § 33 интерпретацию химических равновесий, которая представляет собой второй из способов, разобранных в § 16, 17, 23. В виде исключения используем еще раз общую форму условия равновесия
^U)s.v.n> о. (36.1)
Рассмотрим закрытую гомогенную систему, в которой может протекать реакция
(36.2)
176
Глава V
Согласно условию знаков § 33, возникающие вещества считаем положительными. Поэтому условие равновесия имеет вид
T&S — P8V+ 5 (36.3)
Z = 1 “
с дополнительными условиями
BS = О, &V = 0, = 0 (36.4)
для всех i, которые не встречаются в уравнении (36.2).
Поэтому могут отличаться от нуля только вариации чисел молей участников реакции. Но среди них, как показано в § 33, только один является независимым. Поэтому можно, как уже было упомянуто в § 14, ввести новую независимую переменную по уравнению
dnt = (36.5)
Величина $ называется числом пробегов реакции. Тогда условие равновесия имеет вид
(36.6)
Если все участники реакции присутствуют в системе, то может быть 8В^0. Согласно § 17, тогда должен осуществляться знак равенства и в соответствии с уравнением (33.16) получаем
2 ЬЧ — 0 (присутствуют все участники реакции). (36.7) i
Если продукты реакции отсутствуют, то должно быть Тогда из (36.6) следует
2 vz Hz = 0 (отсутствуют продукты реакции). (36.8) i
Если, наоборот, присутствуют только продукты реакции, то должно быть <0 и имеем
2 Н 0 (присутствуют только продукты реакции). (36.9)*
Наглядное объяснение условий (36.7) — (36.9) следует из (37.3).
Химические равновесия
177
Величину
л = = (36.Ю)
I
де Донде назвал сродством реакции. Если присутствуют все участники реакции, тогда условие равновесия выглядит просто
4 = 0. (36.11)
Приведенные рассуждения можно легко распространить на гетерогенные системы. Следует заметить, что $ является внутренним параметром в смысле § 16 и не является термодинамической переменной состояния.
§ 37. Термодинамический расчет химических реакций
Конкретные термодинамические расчеты химических реакций требуют, естественно, знания в явном виде соответствующих термодинамических функций. Для большинства индивидуальных веществ термодинамические функции имеются в виде таблиц. Здесь не будут поясняться методы, используемые для их определения. Ограничимся поэтому только некоторыми общими замечаниями.
Принципиально термодинамический расчет химических реакций можно проводить двумя путями:
а. Для данной реакции можно, согласно уравнению (33.16), задать, для каких значений величин состояний наступает химическое равновесие.
б. Так как, согласно §23, G при T=const, Р=const в состоянии равновесия минимально, то для данных значений величин состояния можно предсказать, будет ли в принципе протекать реакция (т. е. если отвлечься от возможных торможений).
Пункт б. имеет особое значение во многих случаях, в которых химическая реакция вообще не приходит к равновесию между участниками реакции, а протекает полностью до исчезновения одного из компонентов. В первую очередь к таким реакциям относятся реакции между чистыми конденсированными фазами, например
2Ag + Hg2Cl2 -> 2AgCl + 2Hg, (37.1)
178
Глава V
и большая часть реакций органических соединений, но имеются такие случаи и для гетерогенных реакций, в которых присутствует газовая фаза, как, например,
TiC + О2 -> TiO2 + С. (37.2)
Вообще справедливо*
AG = О, AG<0, AG> О,
А = О
Л >0
Л <0
Химическое равновесие, Реакция возможна, Реакция не возможна.
(37.3)
Поэтому термодинамические‘расчеты химических реакций требуют в каждом случае расчета AG и, таким образом, знания химических потенциалов всех участников реакции при данных значениях величин состояния. Рассмотрим кратко важнейшие специальные случаи:
а. Чистые конденсированные фазы
Химические потенциалы равны мольным свободным энергиям Гиббса. Поэтому имеем
AG - 2 я;-7 (37.4)
i i
H*i иХ^- можно взять из таблиц. Однако часто в таблицах даны только стандартные значения лг- и S*0 при 298,15°К и Р=1 атм. В соответствии с (21.16)
/ дкН -) = /р р
\ дТ
и, согласно (25.4), [ dkS \ __ дср
\ дТ )р Т '
где
лс, = 2 »,С„.
i
Поэтому
(37.5)
(37.6)
(37.7)
* Соотношения (37.3) делают условия равновесия (36.7) — (36.9) более понятными.
Химические равновесия
179
AG = АЯ° — TAS0 + j \CpdT — T j dT. (37.8) 70 70
Интегралы можно рассчитать либо по таблицам, либо при помощи интерполяционных формул. Уравнение (37.8) является, конечно, не чем иным, как решением уравнения Гиббса—Гельмгольца (24.8). При частичном интегрировании уравнения (37.8) или непосредственно из уравнения (24.10) получим выражение
т т
KG = &Н° — ТД8° — Т j &CpdT". (37.9) 70 T'O
Непосредственное экспериментальное определение AG возможно, если реакция протекает в обратимом гальваническом элементе (ср. § 52).
р. Гомогенные реакции в газовой фазе
Для расчета пригодны те же принципы. Однако следует обратить внимание на то, что табличные значения Н? и £>z относятся к идеальному газовому состоянию при Р~ 1 ашм. Поправка на отклонение от поведения идеальных газов при расчетах химических равновесий в большинстве случаев пренебрежимо мала. Из таблиц, соответственно при помощи выражения (37.8), рассчитывают непосредственно
AG0 = — RT\nRp. (37.10)
Принципиально всегда можно направить реакцию за счет соответствующего выбора начальных парциальных давлений. Но практически, конечно, лимитируют экспериментальные возможности. Если хотят рассчитать AG для данных начальных условий, то нужно к значениям р-, рассчитанным из таблиц, добавить еще RTlnpL.
у. Конденсированные смешанные фазы
Расчет химических реакций, в которых участвуют конденсированные смешанные фазы, сталкивается даже в
180
Глава V
простейшем случае гомогенной реакции в идеально разбавленном растворе (§ 34, б.) с существенно большими трудностями, чем в предыдущих случаях, потому что стандартное состояние при нормировке [3. (которое приводит к ЗДМ) зависит от растворителя и поэтому нельзя составить систематических таблиц. Если обойти эту трудность выбором нормировки а., то придем к обобщенному ЗДМ уравнения (34.42). Константы равновесия можно теперь рассчитать при помощи таблиц по уравнениям (34.43) и (37.8), но при этом для расчета AG должны быть известны еще коэффициенты активности, для которых систематическое табулирование практически невыполнимо.
§ 38. Тепловой закон Нернста. Недостижимость абсолютного нуля температуры. Энтропия при абсолютном нуле
Термодинамическое исследование гетерогенных реакций привело в 1906 году Нернста к формулировке общей закономерности, которая называется тепловым законом Нернста. Обнаружилось, однако, что первоначальную формулировку этого закона нельзя сохранить, она приводит к трудным задачам, исследование которых возможно только на основе квантовой статистики и до сих пор не проведено полностью. С другой стороны, тепловой закон Нернста имеет исключительно большое значение для многочисленных применений термодинамики. Поэтому в дальнейшем в основном будем рассматривать эту сторону в первую очередь и лишь кратко осветим вытекающие из теплового закона проблемы. Подробное теоретическое рассмотрение его можно найти в учебниках статистической термодинамики.
а. Тепловой закон Нернста
Рассмотрим изобарно-изотермический процесс между чистыми конденсированными фазами*, который символически обозначим
(38.1)
* Этим сказано, что из рассмотрения исключаются смешанные фазы.
Химические равновесия
181
При этом речь может идти о гетерогенной реакции между чистыми кристаллами, например:
РЬ + 21->РЫ2, (38.2)
Pb + Hg2Cl2 -> 2Hg + РЬС12, (38.3)
или о превращении аллотропных модификаций кристаллов
Sn (белое) -> Sn (серое), (38.4)
S (ромбическая) -> S (моноклинная), (38.5) или о процессе плавления, например:
Не (твердый) -> Не (жидкий). (38.6)
Ради краткости обозначим в дальнейшем (38.1) в общей форме как реакцию. Сделаем следующее
Предположение 1
При реакции (38.1) не происходит никакого процесса, в результате которого изменяется значение внутреннего параметра внутри фазы.
Опыт показывает, что для многих веществ внутреннее равновесие для определенных процессов при низких температурах не устанавливается. Внутреннее состояние, т. е. определенное значение данного внутреннего параметра, которое соответствует внутреннему равновесию при более высокой температуре, сохраняется при низких температурах, или, как обычно говорят, внутреннее состояние заморожено. Важнейшим примером замороженных фаз являются определенные молекулярные кристаллы при очень низких температурах (например, СО, NO, N2O, Н2О), а также неорганические и органические стекла*. Предпо
* Замороженная фаза не обязательно является метастабильной в смысле § 18. Обычный полистирол, например, не может кристаллизоваться. Поэтому он никогда не может быть переохлажденной жидкостью. Напротив, при температуре, которая хорошо воспроизводится, наступает замораживание, превращающее полистирол в «стекло». В противоположность этому силикатные стекла или глицерин появляются сначала во внутреннем равновесии как переохлажденные жидкости, которые замораживаются лишь при более низкой температуре. В этих случаях замороженная фаза является одновременно по отношению к кристаллам метастабильной.
182
Глава V
ложение 1 исключает, таким образом, процессы, связанные с «расплавлением» замороженной фазы.
Разность значений функции состояния Z в состояниях а и (3 напишем в виде
AZ = Z(₽) — Z (а). (38.7)
Для химических реакций эта форма записи более подробно объясняется уравнениями (36.10), (37.4) и (37.7). Тогда, согласно (24.8), имеем
ДО = ДЯ + Т • (38.8)
Учитывая § 37, решение этого уравнения формально можно записать
ДО = ДН0— ТД50 + j \CpdT — Т y-^dT, (38.9) о о
или Т Т'
ДО = ДН0 — ТД80 — Т ^-^-^ДС/Т", (38.10)
О о
где АЯ0 — энтальпия реакции и AS0 — энтропия реакции при абсолютном нуле. Эти величины определяются уравнениями
т
ДН = ДН0 + J дс/т, о
(38.11)
(38.12)
AS = AS0
Так как при абсолютном нуле и в непосредственной близости от него нельзя проводить измерения*, то остается еще объяснить значение интегралов, стоящих в правой части уравнений (38.9)—(38.12). Для этого сделаем
* В большинстве случаев нижняя граница, доступная для измерений, лежит приблизительно в интервале 1—20° К-
Химические равновесия
183
Предположение 2
Для всех фаз, участвующих в процессе (38.1), существуют интегралы
т т
lim f CndT= \CrdT, ] p J p 1
т°->о у© о
(38.13)
(38.14)
lim r°->o
т т
dT= f— dT.
J т J т
T° 0
Требуемое при этом обращение удельной теплоемкости в нуль при Т->0* можно строго доказать на основе кван-
Рис. 23. Атомные теплоты ТЬ при крайне низких температурах.
(Rosenberg Н. М., Low Temperature Solid State Physics, Oxford, 1963, Clarendon Press, p. 31, fig. 1.13.)
товой статистики для простых моделей (например, модель кристалла Дебая). Согласно общим знаниям конденсированных фаз, эти модели можно рассматривать в общем слу-
Для сходимости интеграла достаточно, чтобы при Т—>0 Ср при п>0. Таким образом, не требуется, чтобы п>У.
грп
184 Глава V
чае как вполне справедливые*. Однако обращение удельной теплоемкости в нуль в общем случае экспериментально не подтверждается, как показывает типичный пример на рис. 23.
Предположение 2 является прежде всего только формальным определением и должно быть дополнено методикой расчета. Существенная трудность заключена здесь в факте, установленном экспериментально и теоретически, что в многочисленных случаях удельная теплоемкость в области Т<1 °К стремится к нулю немонотонно, так что эти данные, как видно, например, из рис. 23, не дают никаких указаний для экстраполяции. Так как такие аномалии** могут встречаться уже при температурах, лежащих ниже современных возможностей низкотемпературной техники***, то экспериментально проверяемую закономерность можно сформулировать, только если вообще пренебречь упомянутыми аномалиями****. Поэтому сделаем еще
Предположение 3
Уменьшение удельной теплоемкости до нуля, которое не обходимо согласно предположению 2, можно с достаточной точностью отобразить простым степенным законом, описывающим последние экспериментальные данные.
Практически для неметаллических кристаллов используют закон Дебая Т3, в то время как для металлов с учетом электронной теплоты пишут
Ср = аТ9 + ^Т. (38.15)
Из экспериментальных данных и теоретических оценок можно заключить, что для обычных температур ошибка, введенная в результате предположения 3 с учетом точности определения интеграла (38.13), пренебрежимо мала. Для
* Это тем более оправдано, поскольку сходимость интегралов обусловлена также экспериментальными фактами, что АЯ и AS конечны.
** Такие аномалии следует ожидать прежде всего в парамагнитных и ферромагнитных кристаллах. Однако они могут проявляться также и в диамагнитных кристаллах.
*** Для некоторых аномалий, которых следует ожидать теоретически, температурная область оценивается в 10~6 °К.
**** фактически этим только «признан» метод, который применяли раньше.
Химические равновесия 185
интеграла (38.14) ошибка может быть существенно больше и в неблагоприятных случаях достигает 5% и более.
Вернемся вновь к интегрир°ванию уравнения Гиббса— Гельмгольца. Так как величина АЯ непосредственно измерима, то по уравнению (31.11) можно рассчитать АЯ0 из калорических данных. В правой части уравнений (38.9) или (38.10) остается тогда в качестве неизвестной константы интегрирования величина Д50. Тепловой закон Нерн-ста является общим способом предсказания этой константы интегрирования.
Рассуждения Нернста связаны с принципом, установленным в середине XIX в. Томсеном и Бертло, согласно которому протекание реакции между конденсированными фазами определяется тепловым эффектом, следовательно, должно иметь место равенство Из уравнения (38.8)
видно, что этот закон не выпоЛняется как общий термодинамический принцип. Однако в ряде случаев при определенных приближениях он справЗДлив и именно, как экспериментально показал в 1903 г- Ричардс, тем больше, чем ниже температура. Поэтому Нсрнст предположил, что при этом речь идет о граничном заКоне лля низких температур таким обоазом, что при T-+-Q не только AG=A/7 (что уже следует из уравнения (38.8)*), но что обе кривые по крайней мере имеют первый порядок касания**. Таким образом, имеем
Тепловой закон Нернста
С учетом предположений 1—3 для изобарно-изотермических процессов между чистыми конденсированными фазами выполняется соотношение
lim = Jim . (38.16)
т->о дТ дт
График уравнения (38.16) схематически приведен на рис. 24.
Величина, стоящая в правой части уравнения (38.16), согласно (37.5), является граничным значением ДСр при * Формально при этом должны предположить, что при 7->0 AS не является бесконечной.
** Две кривые f(x) и <р(х) в точке Хо имеют порядок касания п, если в точке х0 производные f(x) и Равны между собой до порядка и, а производные порядка (n-pl) различны.
186
Глава V
Рис. 24. К тепловому закону Нернста.
T-+-Q, которое, согласно предположению 2, должно быть равно нулю. Поэтому, используя (21.37), получаем
Тепловой закон Нернста (альтернативная формулировка) С учетом предположений 1—3 для всех изобарно-изотермических процессов между чистыми конденсированными фазами
ДХ0 = 0. (38.17)
Эта формулировка ясно показывает связь с интегрированием уравнения Гиббса—Гельмгольца, так как при выполнении (38.17) ДО можно рассчитать из чисто калорических данных.
Сделаем еще следующие замечания:
а. Величины Д/70 и ДХ0 определяются как граничные значения, которые сохраняются при экстраполяции опытных данных в экспериментально недоступную область. Поэтому они имеют физическое значение не сами по себе, но только в связи с тепловым законом Нернста*.
|3. Физическое утверждение, содержащееся в тепловом за_ коне Нернста, можно переписать следующим образом, в температурной области 20° и 1 °К величина ДХ настоль^
* Смысл, возможно, станет более понятным за счет аналогии. Для осмотического давления бинарного раствора (§ 28) при очень больших разбавлениях можно написать
РТ
П = сё (закон В ант-Гоффа),
где Л42 — молекулярный вес растворенного вещества, cg — концентрация в а/см3. Для П->0. Граничное значение ИгпП/с^.
с —>0 g
является конечным и определяет молекулярный вес М%. Однако его нельзя измерить, но можно получить экстраполяцией в экспериментально недоступную область.
Химические равновесия
187
ко мала, что экстраполяция на Т=0, проводимая в соответствии с предположением 3, находится внутри границ ошибок эксперимента (38.17).
у. В приведенных выше формулировках были исключены из рассмотрения смешанные фазы. Но фактически всегда имеют дело со смесями изотопов. На этой проблеме здесь не будем останавливаться. Ограничимся только замечанием, что все формулировки остаются справедливыми, если в результате процесса (38.1) не изменяется изотопный состав участвующих элементов*.
Дляэкспериментальнойпроверки теплового закона Нернста проводят измерения в обратимом гальваническом элементе (§ 52) и изучают фазовые превращения в конденсированных системах. Установлено, что внутри границ экспериментальных ошибок уравнение (38.17) всегда выполняется. В качестве примера приведем данные превращения S (ромбическая)->5 (моноклинная), так как в дальнейшем придется еще раз к этому вернуться. Было найдено**,*** (/?—газовая постоянная)
kH/R = 47,5 ± 5°К, (Т = 368,6 °К)
AS/# =0,12 ±0,01, (Т = 368,6°К) (38.18)
kS0/R — 0,01 ±0,05.
б. Недостижимость абсолютного нуля
С тепловым законом Нернста тесно связана другая закономерность, которую в 1912 г. сформулировал Нернст. Речь идет о
Принципе недостижимости абсолютного нуля
Невозможно охладить систему до абсолютного нуля за счет какого-нибудь процесса с конечным числом операций.
* Также для водорода и других веществ, смеси которых содержат орто- и пара-модификации, возникают осложнения, объяснения которых можно найти в учебниках статистической термодинамики.
** Eastman Е. D., McGavock W. С., J. Am. Chem. Soc., 59, 145 (1937).
*** Более поздние измерения West E. D. [J. Am. Chem. Soc., 81, 29 (1959)] дают несколько другие значения температуры превращения, ДЯ и Д5. Но так как он не приводит результаты низкотемпературных измерений, то эти значения авторами не использованы из соображений согласованности.
188
Глава V
Этот закон нужно рассматривать как опытный факт, независимый от основных законов термодинамики*. Чтобы сделать наглядным эмпирическую основу и связь с тепловым законом Нернста, рассмотрим вновь процесс в конденсированных фазах
а->ф, (38.19)
который теперь определим несколько иначе, чем в разделе а. Прежде всего ясно, что достижение абсолютного нуля было
Рис. 25. Недостижимость абсолютного нуля температуры.
------— квазистатически-адиабатический процесс; .... нестатический адиабатический процесс .
бы возможно только при адиабатическом процессе. Согласно второму закону термодинамики, в нестатическом адиабатическом процессе энтропия увеличивается. Из рис. 25 хорошо видно, что для достижения абсолютного нуля квазистатически-адиабатический процесс всегда был бы предпочтительней, чем нестатический адиабатический процесс. Поэтому сразу предположим, что процесс (38.19) является квазистатическим и адиабатическим, т. е. обратимым. В частности, при этом речь может идти об изменении рабочей
* Принцип недостижимости абсолютного нуля был выведен Нернс-том из второго закона термодинамики при использовании кругового процесса. Этот вывод был раскритикован Эйнштейном. Разногласия по этим вопросам (которые здесь нельзя проследить) продолжаются до сих пор. Наиболее правильно, хотя и очень абстрактно, суть проблемы сформулирована в аксиоматике Ландсберга [Rev. Mod. Phys, 28, 363 (1956)]. По этой аксиоматике при обсуждении принципа недостижимости абсолютного нуля речь идет о положении краевой точки открытого множества точек, что в общем виде должно быть математически особо сформулировано. Справедливость вывода Нернста зависит от этой формулировки, которая, во всяком случае, представляет собой дополнительный постулат.
Химические равновесия
189
координаты гомогенной конденсированной системы, о химических реакциях в обратимом гальваническом элементе или о фазовых превращениях в конденсированном состоянии. Последний процесс рассматривать не будем. Если обозначить через Т' начальную температуру, через Т" конечную температуру, то энтропия системы в обоих состояниях равна
(38.20)
(38.21)
где Са и Ср — теплоемкости. Для квазистатически-адиаба-тического процесса должно быть
7*т Тп
о о
(38.22)
Поэтому при Т"~0 получаем
Т' S03-Ste= f-^-dT. Up т
0
(38.23)
Если Sop—Soa> 0 и так как должно быть Са >0*, то всегда имеется температура Т', которая удовлетворяет (38.23), т. е., исходя из температуры, Т' можно достигнуть за счет процесса (38.19) абсолютного нуля. Поэтому предположение должно быть неправильным и можно заключить, что 50р<^
Soa. Если выбрать состояние р при температуре Т' в качестве исходной точки, чтобы достигнуть при квазистати-ческом и адиабатическом процессе абсолютного нуля, то должно быть
s.— s03 = f-^-dr.
Ua Up | гр
О
(38.24)
Проведя аналогичные рассуждения, найдем, что Soa <^5ор.
Доказательство приведем в § 41.
190
Глава V
Поэтому недостижимость абсолютного нуля подразумевает soa = S^. (38.25)
На рис. 26 схематически представлены эти соотношения.
Рис. 26. Энтропия как функция абсолютной температуры при крайне низких температурах (схема).
а — абсолютный нуль температуры достигается в результате квазистатически-адиабатического процесса; б — абсолютный нуль температуры недостижим.
Рассмотрим кратко три важнейших частных случая, а. Процесс (38.19) состоит в изменении рабочей координаты у, В этом случае уравнение (38.25) можно записать lim (—'l =0 (38.26)
т->о \ ду )т
или вследствие соотношения Максвелла, аналогичного (24.20),
lim =0. (38.28)
т->0 \ дт )у
Если подставить y~V, Y=—Р, то из (25.5), (25.6)
и (25.9) получим
Предполагая, что сжимаемость при Т->0 остается конечной, из (38.28) получаем
lima = 0. (38.30)
Химические равновесия
191
Согласно экспериментальным данным, это условие выполняется для твердых тел и жидкого гелия.
Если подставить у=М, Y=H, где М — общая намаг-
——>
ниченность и Н — напряженность магнитного поля, то получим
дН дТ
(38.31)
__ ( дМ/дТ А? ( дМ/дн)т
Предполагая, что знаменатель правой части при Т->0 остается конечным, из (38.28) получаем
lim L = 0. (38.32)
г-м> \ дТ )ТГ ' '
Согласно экспериментальным данным, это условие также выполняется.
Проверка уравнений (38.30) и (38.32) составляет собственную эмпирическую основу принципа недостижимости абсолютного нуля. В этой связи уравнение (38.32) имеет гораздо большее значение, так как оно относится к адиабатическому размагничиванию, которое было упомянуто в § 12 и которое представляет собой единственный известный способ получения очень низких температур. Теоретический анализ молекулярного механизма, на котором здесь нет возможности остановиться, приводит также к результату, показывающему, что этим путем нельзя достигнуть абсолютного нуля.
р. Процесс (38.19) представляет собой химическую реакцию между чистыми конденсированными фазами.
В этом случае уравнение (38.25) идентично тепловому закону Нернста (38.17). Требование, чтобы процесс (38.19) протекал квазистатически и адиабатически, подразумевает предположение 1, так как «плавление» замороженной фазы по необходимости является адиабатически необратимым процессом.
I- Процесс (38.19) представляет собой фазовый переход в конденсированной системе.
В этом случае обратимое превращение возможно только вдоль кривой сосуществования, поэтому приведенные выше уравнения не применимы. Более подробное
192
Глава V
рассмотрение, которое в последнее время провел Хаазе, ведет к аналогичному результату. При этом должно быть предположено, что кривая сосуществования простирается при конечных давлениях до Т=0. Это условие выполняется, например, при равновесном плавлении гелия, однако с уверенностью можно сказать, что не выполняется при упомянутом превращении S (ромбическая)-^ (моноклинная). В этом случае тепловой закон Нернста нельзя вывести из принципа недостижимости абсолютного нуля, хотя он, как показано, соответствует экспериментальным данным.
Принцип недостижимости абсолютного нуля, с одной стороны, исходит из теплового закона Нернста, так как для каждого простого вещества в конденсированном состоянии он требует существования нулевой энтропии, независимой от рабочих координат. С другой стороны, он не перекрывает область применимости теплового закона Нернста, так как последний можно вывести для фазовых переходов только при ограничивающих условиях. Следствие, вытекающее из теплового закона Нернста, что нулевая энтропия не зависит от кристаллической модификации, не может быть поэтому получено в общем виде из принципа недостижимости абсолютного нуля. Таким образом этот принцип нельзя отождествлять с тепловым законом Нернста, а нужно рассматривать как самостоятельный эмпирический закон.
в. Нулевая энтропия
Одним из важнейших применений закономерностей, рассмотренных в пунктах а. и б., является построение таблиц термодинамических функций, упомянутых в § 37а. Рассмотрим здесь кратко это применение и дополним в некоторых пунктах предыдущие рассуждения.
Обратимся прежде всего к энтальпии. Согласно первому закону термодинамики, функция Н содержит произвольную константу интегрирования. Измерять можно только разность
\Н = Щ~ На, (38.33)
где аир — два равновесных состояния системы, которые связаны между собой любым процессом*. Если при этом а * Здесь и в дальнейшем исключим любой вид ядерных превращений.
Химические равновесия
193
будет означать стандартное состояние, которое нужно определить и для которого известна константа интегрирования, то таким образом, согласно (38.33), она определена также для всех состояний |3, которые можно достигнуть из а за счет какого-нибудь процесса. Поэтому достаточно определить аддитивную константу для каждого элемента в стандартном состоянии. Следующие соглашения являются в настоящее время общепринятыми и положены в основу таблиц:
Стандартное состояние энтальпии
Т = 298,15°К, Р = 1 атм. (38.34)
Нормировка энтальпии
Для каждого элемента (38. 34) в его форме, стабильной в стандартном состоянии,
Н*° = 0. (38.35)
Таким образом, стандартная энтальпия всех химических соединений численно равна энтальпии образования (38.33), которую можно измерить экспериментально. Для стандартных энтальпий имеются наиболее полные таблицы. Поскольку другие значения энтальпии не табулированы, они должны быть рассчитаны по методике, изложенной в § 37, сс. Аналогичным образом рассчитывают из стандартных значений энтальпию нулевой точки Яо. Для мольных величин справедливо
298,15
Н*° = Н* + J &CpdT (Р = 1 атм). (38.36) О
Для энтропии существует та же исходная ситуация. Согласно второму закону термодинамики, S содержит произвольную константу и экспериментально можно определить только разность
AS = Sp — Sa. (38.37)
Нормировка (38.35) здесь нецелесообразна, так как, с одной стороны, энтропию образования нельзя непосредственно измерить*, с другой—этим самым затруднялся бы учет теп
* Только в равновесии ДО = 0 и таким образом &ШТ — Д5. 7—235
194
Глава V
лового закона Нернста. Поэтому приняли следующее соглашение:
Стандартное состояние для энтропии
Р = 1 атм.
Нормировка энтропии
Для каждого элемента в форме при Т->0
s; = o.
стабильной конденсированной
(38.39)
Эта нормировка не зависит от теплового закона Нернста. Из нее следует, что для всех химических соединений нулевые энтропии численно равны энтропии образования при абсолютном нуле. Для каждого индивидуального вещества
298,15 с
= J —£-dT. (38.40)
о
Единственной экспериментально доступной величиной в этом уравнении является интеграл правой части. Нормировка (38.39) поэтому не достаточна, чтобы численно зафиксировать стандартную энтропию S*0.
Эту трудность можно преодолеть, привлекая тепловой закон Нернста. Этим объясняется его чрезвычайно большое значение для термодинамики. Из (38.17) и принципа недостижимости абсолютного нуля следует в сочетании с нормировкой (38.39) формулировка, данная впервые Планком.
Тепловой закон Нернста (формулировка Планка)
Для всех простых веществ в конденсированном состоянии, для всех стабильных фаз независимо от значений рабочих координат, кристаллических модификаций и агрегатных состояний
s; = o. (38.41)
Используя (38.40) и (38.41), можно рассчитать стандартную энтропию S*0 из чисто калорических данных. Расчет энтропии для других условий был разъяснен в § 37,а. Величину So называют условной нулевой энтропией.
Химические равновесия
195
Сделаем еще некоторые замечания к формулировке (38.41). Прежде всего она позволяет, как показал Джок, при определенных предположениях провести существенно более точную экспериментальную проверку, чем предыдущая формулировка. Предположим, что некоторое вещество можно перевести без разложения из кристаллического состояния в состояние разбавленного газа*. Тогда для энтропии газа можно написать
т
s; (Т, Р) - s; = lim . (38.42)
Здесь dQ означает подведенное количество теплоты, включая теплоты превращения. Путь интегрирования является обратимым путем от кристалла при Т° и Р->0 до газа при температуре Т и давлении Р. Предельный переход Т°->0 должен быть проведен также способом, разъясненным в пункте а. (предположение 3). Тогда можно определить правую часть уравнения (38.42) при помощи калориметрических измерений. Величину S*g(T, Р) — S*o называют поэтому калориметрический энтропией**. С другой стороны, величину Sg (Т, Р), как показано в статистической термодинамике, можно рассчитать для разбавленного газа при некоторых предположениях*** из спектроскопических данных. Поэтому ее называют спектроскопической энтропией. Согласно (38.42), сравнение калориметрической и спектроскопической энтропии дает непосредственно проверку уравнения (38.41).
* Это предположение не выполняется, с одной стороны, для тугоплавких кристаллов (например, С, А1гО3,Т1С), которые не имеют измеримого давления пара, с другой —для органических соединений, которые разлагаются при нагревании (например, тростниковый сахар).
** Это понятие, естественно, не ограничивается газовым состоянием.
*** Для выяснения подробностей нужно использовать учебники статистической термодинамики и молекулярной спектроскопии. Здесь следует только заметить, что предположения не выполняется особенно для многоатомных молекул с внутренним вращением (например, для этана).
7*
196
Глава V
Проверка уравнения (38.42) была проведена примерно на 30 веществах (двухатомные и простые многоатомные молекулы). В большинстве случаев оказалось, что уравнение (38.41) выполняется в пределах точности эксперимента*. Однако для разных веществ экспериментально установлено отклонение от уравнения (38.41). Таким образом, формулировка Планка теплового закона Нернста не выполняется как точное утверждение. Речь идет об отклонениях в основном замороженных молекулярных кристаллов, которые были упомянуты в пункте а. в связи с предположением 1. Фактически при формулировке (38.41) предположение 1 вообще не учитывается. Поэтому предложено два способа для превращения (38.41) в точный закон. Первый состоит в том, что для рассматриваемого вещества дополнительно требуют внутреннее равновесие при Т->0, в то время как во втором способе в правой части уравнения (38.41) нуль заменяется на положительную конечную величину In W. Против первой формулировки свидетельствует то, что понятие внутреннего равновесия имеет смысл только по отношению к определяемым процессам**. При второй формулировке из сравнения калориметрической и спектроскопической энтропии известно, что либо W~ 1, либо по порядку величины 1^=2. Это сравнение выполнимо только для относительно малого числа веществ. В других случаях приходится ограничиваться только предположениями. Практически всегда исходят из уравнения (38.41) и учитывают, что нормальная энтропия, рассчитанная таким образом, имеет неточность порядка R In 2. Этот способ тем более обоснован, так как неточность, обусловленная экстраполяцией при 7->0 (разд, а., предположение 3), того же порядка. Для большинства применений величина этого порядка не играет
* Различие калориметрической и спектроскопической энтропии составляет <0,5%.
** Даже при ограничении определяемыми процессами требование внутреннего равновесия делает иллюзорным практическое использование уравнения (38.41). Это легко понять на следующем ,примере: кристаллы TiO имеют при комнатной температуре концентрацию пустых мест примерно 15%, которая довольно хорошо воспроизводится. Не известен, однако, путь, для того чтобы решить, соответствуют ли при Т~>0 концентрация и пространственное распределение пустых мест внутреннему равновесию.
Химические равновесия 197
существенной роли. Формулировка в виде «правила»*, которая на практике выполняется с достаточной точностью, соответствует более всего не только характеру теплового закона Нернста, как это следует из рассуждений пункта а., но и современному состоянию статистической термодинамики. Как показали исследования Клейна и Казимира, более старые попытки на^ основе квантовой статистики сформулировать «третий закон термодинамики» оказались несостоятельными. До сих пор еще не приходится говорить о статистическом понимании теплового закона Нернста, которое только и могло бы лечь в основу точной формулировки (соответствующего закона).
* Важнейшим примером такого «правила», которое выполняется неточно, но практически очень важно, является теорема соответственных состояний, согласно которой для реального газа существует универсальное термическое уравнение состояния, если оно выражено в безразмерных («приведенных») величинах состояния (например, Т!ТС, Р!РС, V/Vc, где индекс с относится к критической точке). В статистической термодинамике показано, что эта теорема не может быть точной, однако она выполняется для многих веществ часто с достаточной точностью.
Глава VI
Условия стабильности
§ 39. Постановка проблемы. Критерий Гиббса
В предыдущих рассуждениях был использован второй закон термодинамики (за некоторыми исключениями, например, в §37) только в виде высказывания, что термодинамические потенциалы в состоянии равновесия принимают стационарное значение. Дальнейшее высказывание, что это стационарное значение является минимумом, составляет, как уже было кратко отмечено в § 18 и 23, содержание условий стабильности. Задача данной главы полностью аналогична той, которая обсуждалась в гл. IV и V для условий равновесия. Теперь речь идет о том, чтобы из общей формулировки условий стабильности в § 18 и 23 при помощи фундаментального уравнения вывести в явном виде следствия. Этим ограничивается задача. Формально нужно теперь исследовать вариации термодинамических потенциалов более высокого порядка. В рамках термодинамики для четкой трактовки рассматривают, как и в случае условий равновесия, только такие возможные возмущения, которые можно выразить через величины состояния. Это ограничение допускает для гомогенной системы при условиях равновесия лишь обсуждение равновесий, которые можно представить через внутренние параметры. Для условий стабильности гомогенной системы даже при исключении внутреннего равновесия постановка вопроса оказывается не тривиальной. Фактически, как будет видно, остальные проблемы стабильности, если отвлечься от химического равновесия, можно свести к проблеме стабильности гомогенной фазы. Вопрос стабильности химического равновесия является сравнительно простым, и позднее можно будет удовлетвориться некото
Условия стабильности
199
рыми замечаниями. Исключим пока рассмотрение химической реакции*.
Итак, рассмотрим гомогенную систему, состоящую из щ компонентов, в которой не протекают химические реакции и которая находится в состоянии термодинамического равновесия. Перечисленные выше ограничения приводят к следующей постановке задачи.
Каким условиям должна удовлетворять система, чтобы она не стала гетерогенной, т. е. не распалась бы на две отличные от первоначальной (макроскопические) фазы, в результате сколь угодно малых локальных возмущений.
Эта постановка задачи (если отвлечься от рассмотрения химических реакций) составляет основную проблему термодинамической теории стабильности. Возникает вопрос, не препятствует ли приведенное выше ограничение общности выводов. На самом деле это не так, потому что понятие стабильности, так же как и понятие равновесия, имеет физический смысл только по отношению к рассматриваемому процессу. Общность была бы лишь тогда сомнительна, если бы были возможны смещения, которые можно представить через величины состояния и которые бы не содержались в приведенной выше формулировке или по крайней мере к ней не сводились бы.
Приведенная формулировка проблемы оставляет открытыми две возможности:
а. Новые фазы отличаются от первоначальных только бесконечно малым изменением величин состояния (соседние фазы).
б. Новые фазы отличаются от первоначальных конечным (дискретным) изменением величин состояния.
Возможность б. приводит к упомянутому в § 18 понятию метастабильного равновесия. Знание диаграммы состояния позволяет сказать, какие фазы по отношению к другим являются метастабильными. Однако общие закономерности здесь получить нельзя. Ограничимся поэтому случаем а., который связан непосредственно с принципом
* Ради простоты используем это положение здесь и в дальнейшем для всех процессов, которые можно представить через внутренние параметры.
200
Глава VI
возможных смещений и который составляет собственно содержание термодинамической теории стабильности.
Как и в гл. II, предположим, что рассматриваемая система является закрытой и что значения рабочих координат зафиксированы. Первоначальную фазу обозначим через ', обе новые фазы, возникающие в результате возможных сме^ щений, — через * и **. Для первоначальной фазы в соответствии с предположением и уравнением (20.43) можно написать
т
U' — TS' + PV' — 2 ^п\ = 0? (39.1)
Предположим, что для обеих новых фаз выполняются соотношения
(/* _ TS* + PV* — £ vtn* > 0, Z=1
и** _ TS** + PV** — Y ^п** > 0.
Z=1
При этом имеем еще дополнительные условия £*-]_£** = 5', у* у** = у', п* + nz* ~ nt G = •» m)-
(39.2)*)
(39.3)
Если теперь в уравнении (39.2) будет знак равенства, то, учитывая (39.1) и (39.3), получим
[7* + [/** = U'. (39.4)
При образовании новых фаз и при выполнении дополнительных условий (39.3) внутренняя энергия остается постоянной. Согласно уравнению (18.6), это значит, что по отношению к образованию новых фаз имеется нейтральное равновесие,
*) Величины со звездочкой не являются здесь средними мольными
величинами.
Условия стабильности
201
т. е. они могут сосуществовать с первоначальными фазами. Если в (39.2) будет знак неравенства, то
+ U**>U'. (39.5)
При образовании новых фаз и при соблюдении дополнительных условий (39.3) в этом случае внутренняя энергия возрастает. По второму закону термодинамики такой процесс не может протекать самопроизвольно. Первоначальная фаза, согласно (18.2), является стабильной по отношению к новым фазам. Если, наконец, подставить в (39.2) знак < вместо >, то получим
U* +
(39.6)
Образование новых фаз при соблюдении дополнительных условий (39.3) уменьшает внутреннюю энергию. Поэтому первоначальная фаза в соответствии с (18.4) является нестабильной по отношению к новым фазам. Первоначально гомогенная система не способна к существованию и сразу должна стать гетерогенной.
Итак, соблюдается правило:
Если в выражении m
U-TS + PV-'Zw (39.7)
Z=1
интенсивным параметрам Т, Р, можно приписать такие значения, что это выражение для рассматриваемой фазы станет равным нулю, а для каждой другой (образованной из тех же компонентов) фазы будет положительным, то рассматриваемая фаза по отношению к распаду на другие фазы является абсолютно стабильной. Если выражение (39.7) для некоторых других фаз равно нулю, то рассматриваемая фаза по отношению к ним находится в нейтральном равновесии. Они могут сосуществовать с первоначальной фазой и Т, Р, являются общими значениями температуры, давления и химических потенциалов, которые требуют условия равновесия.
Это правило является критерием стабильности Гиббса.
Сделаем два замечания. Прежде всего в уравнении (39.2), возможно, покажется произвольным, что интенсивные параметры новых фаз имеют такие же значения, как и парамет
202
Глава VI
ры первоначальной фазы. Фактически это непосредственно следует из предположения, что первоначальная фаза находится в термодинамическом равновесии. Тогда именно, согласно § 17, для всех возможных смещений вариации первого порядка равны нулю. Так как в принципе безразлично, имеется ли уже фаза или она в результате смещения возникла вновь, то можно непосредственно применять метод § 27, из которого следует, что для равенства нулю вариаций первого порядка необходимо и достаточно равенство интенсивных параметров для всех фаз (реальных или виртуальных) (ср. § 40).
Далее следует принять во внимание, что критерий стабильности Гиббса представляет собой очень общую и поэтому очень абстрактную формулировку (она охватывает также метастабильные равновесия, так как при этом в явном виде не используется ограничение пункта а.). Его значение заключается в том, что он, с одной стороны, за счет простых рассуждений получается из второго закона термодинамики, с другой — делает возможным вывод всех остальных общих и частных условий стабильности чисто математическим путем.
§ 40. Условия стабильности в энергетическом выражении
Ограничимся теперь возможностью а., упомянутой в § 39, и рассмотрим простой пример, который позволит лучше понять связь между общими формулировками. Так как обсуждаются только соседние фазы, то можно не рассматривать нейтральное равновесие (сосуществование соседних фаз).
Рассмотрим гомогенную однокомпонентную систему, находящуюся в термодинамическом равновесии, и изучим возможный распад на две фазы с объемами У2У, энтропией ^(S+BS), V2(5—85) и числом молей x/2(n+Sn), 1/2(п— —Зп). Уравнения (39.2) тогда примут вид
L/*--tT(S + 85) + -L PV — -Lfx(n + 8n)>0, Л Л £
1 1 1 (40.1)
^** --уГ(5-85)4--1-РУ—|-1х(п-8п)>0. J
Условия стабильности
203
Дополнительные условия (39.3) уже учтены в (40.1). Поэтому имеем
и* = — Пу + — 88 + — 8n + — (8S)2 4-
2 L 9S дп 2 dS2 V ’
+ 888/г + -у 4т (И2 + - 1 . (40.2)
dSdn 2 дп2 J
соответственно
U** = J_ Г и _ 8S JL_^L (8S)2
2 L 9S дп 2 dS2
+ bSbn + — (8n)2 — ... 1 . (40.3)
dSdn 2 dn2 J
Если сложить уравнения (40.2) и (40.3), то сокращаются члены первого порядка. Если после подстановки в (40.1) составить разность по отношению к (39.1), то получим
[/* + [/**>[/' (40.4)
тогда и только тогда, когда
d2U
dS2
(88)2 + 2 888/г + (8и)2 > 0. (40.5)
dSdn дп2
Таким образом, условия стабильности утверждают, что квадратичные формы, стоящие в неравенстве (40.5), должны быть положительными. В теории квадратичных форм показывается, что это возможно тогда и только тогда, когда
d2U д2и dS2 dndS d2U d2U dSdn дп2
со всеми главными минорами>0. (40.6)
Это является формой условия стабильности, наиболее удобной для дальнейшего обсуждения.
Прежде чем разъяснять дальнейшие детали, выведем условия (40.6) в общем виде и для систем, состоящих из
204
Глава VI
т компонентов.
Будем исходить из того, что, согласно критерию Гиббса, для каждой фазы, соседней к рассматриваемой, должно быть
U* _ TS* + PV* — V ^п* > 0. (40.7)
Z—1
Если из (40.7) вычесть уравнение (39.1), то для возможных возмущений получим
&U > TkS — PAV + 2 vAni> (40.8)
где символ Д означает вариацию любого порядка и для независимых переменных естественно Дх->8х. Выберем в качестве независимых переменных S, V, nt. Тогда в соответствии с фундаментальным уравнением
W = ns — PW + 2 (40.9)
i=l
В неравенстве (40.8), таким образом, исчезают вариации первого порядка. Предварительно предположим, что члены второго порядка отличаются от нуля. Тогда по отношению к ним остальные члены являются бесконечно малыми более высокого порядка; поэтому ими можно пренебречь. Таки образом, получаем
W(S, V, (40.10)
Обсуждение фундаментального уравнения (§ 20) показало, что термодинамика гомогенной системы требует только т+1 независимых переменных. Приведенный выше пример ясно показывает, что одна из независимых переменных (именно V) служит лишь для того, чтобы описывать количественное соотношение фаз, образованных при возможных смещениях, а в условия стабильности вообще не входит, так как это количественное соотношение вообще не имеет ничего общего с проблемой стабильности. Все это можно просто учесть таким образом, что в (40.8) и (40.9) V, пт
Условия стабильности
205
или ^nt поддерживаются постоянными.
Выберем здесь эту последнюю возможность, которая (как это следует из § 20) появляется потому, что используется фундаментальное уравнение для средних мольных величин и вместо числа молей m в качестве независимых переменных имеем m—1 мольных долей. Таким образом, из (40.10) (в записи § 26)* имеем
W*(S*, V*, xf)>0. (40.11)
Итак, условие стабильности говорит о том, что квадратичная форма, образованная из вариаций второго порядка, должна быть положительной. Это возможно тогда и только тогда, когда
d2U* d2U*
dS*2 dxm_idS*
d2U*
dS*dV*
• со всеми главными (40.12)
минорами > 0.
д2и* д2и*
dS^dxm^..........
Это является общей формулировкой условий стабильности в энергетическом выражении. Отсюда видно, что условия стабильности состоят в предсказаниях знака вторых производных характеристических функций, которые должны выполняться для каждой стабильной фазы. Далее, (40.12) показывает, что для гомогенной системы, состоящей из пг компонентов, имеется 1 независимых условий стабильности.
Рассмотрим два частных условия, которые вытекают из условия (40.12). Прежде всего непосредственно следует
д2и* \
dS*2 /у*, х
0.
(40.13)
* Отсюда и до конца параграфа величины со звездочкой обозначают средние мольные величины.
206
Глава VI
Согласно фундаментальному уравнению
d2U \ _ / дТ \
OS*2 Jy*, х \ dS* Jv*, х
(40.14)
Используя (25.7), соотношение (40.13) можно написать в виде
>0. (40.15)
\ дТ )v*.x V ’
Из фундаментального уравнения и определения (20.21а) следует
=с (40.16) \ дТ )V*,x \ дТ )v\x V
Так как личиной, Т по определению является положительной вето окончательно имеем
Су>0. (40.17)
Таким образом, для стабильной фазы мольная теплоемкость при постоянном объеме всегда положительна. Выражение (40.17) называют условием термической стабильности.
Так как при построении детерминантов (40.12) можно выбрать любую последовательность переменных, то должно быть
>0. (40.18) \ dV*2 s\ х
Используя фундаментальное уравнение и формулу (25.7),
получим >о. (40.19) V дР )s*,x
Величина 'j (40.20) s V \дР )s.x ' '
называется адиабатической или (что более правильно) изэнтропийной сжимаемостью. Поэтому
(40.21)
Условия стабильности
207
Для стабильной фазы изэнтропийная сжимаемость всегда положительна. Неравенство (40.21) называется условием механической стабильности.
Вывод остальных следствий (которые имеют не только формальный интерес) возможен, но довольно громоздок. Причина этого заключается в структуре фундаментального уравнения, рассмотренной в § 21. Поэтому возникает аналогичная § 23 задача выразить условия стабильности через результат преобразования Лежандра для фундаментального уравнения.
§41. Преобразование условий стабильности
Выразим теперь условия стабильности при помощи любой характеристической функции. Ради простоты ограничимся расчетом для энергетического выражения и дадим для (аналогично рассматриваемого) энтропийного выражения только конечный результат.
Согласно общим предположениям, высказанным в § 39, виртуальная фаза также находится в термодинамическом состоянии, т. е. во внутреннем равновесии. Поэтому должно выполняться фундаментальное уравнение, причем интенсивные параметры теперь определяются не [как в (40. 7)] из первоначальной фазы, а при помощи фундаментального уравнения возможной фазы. Поэтому в интегральной форме можно написать*)
U* = T*S* — P*V*+ р*и*.
г=1
(41-1)
Условие (40.7) можно переписать
—TS* + PV*— 2 V-ini + T*S* — p*v* +
/=1
(41.2)
*) Здесь и в дальнейшем «со звездочкой» обозначим вновь виртуаль ную фазу.
208
Глава VI
Если фаза «со звездочкой», согласно равенству (41.1), находится во внутреннем равновесии, то можно ее также рассматривать как первоначальную фазу и ставить вопрос о ее стабильности по отношению к спонтанному образованию фазы «без звездочки». Условия для этого получаются непосредственно из (41.2) обменом местами «со звездочкой» и «без звездочки». Таким образом, имеем
_ + P*V—2 Щ + TS — PV +
tn
+ 2 Vini > °’
(41-3)
Суммирование (41.2) и (41.3) дает
ATAS — APAV + V Др.гАпг > 0.
(41-4)
1=1
Выражение, стоящее слева, представляет собой вариацию второго порядка S2t/. Поэтому (41.4) является лишь иной формой записи, удобной для преобразования условия (40.10). Как и в §21, введем обобщенные величины состояния XL и Pt и, таким образом, условия стабильности (41.4) можно записать
2 ДРгАХг > 0.
(41.5)
Эту абстрактную формулировку выразим теперь через вторые производные термодинамического потенциала
фй = ^(Рг, ху)(// = k4-1....,г). (41.6)
Для этого нужно ДХ5($=1.....k) и ДРД^=А4-1, •••» г)
представить как функции Рг и Xj. Так как в (41.5) ограничивались только вторым порядком, то получим линейную систему уравнений
Условия стабильности
209
k г
= 2 + S "ИГ
/=1 1 j=k+l
др,_У-^-др|+ у -2й_дх..
‘ дР, 1 дХ, J
z=i /=fe+i 7
(41.7)
Согласно уравнению (21.9),
dXs =
dPi dPsdPi ’
и, согласно (21.10), имеем
dPt =
dXj dXtdXj
Далее, из соотношения Максвелла (24.11) следует
дХ, = dPt
dXj dPt
Если теперь подставить (41.7) в (41.5), то возникает квадратичная форма ДР,- и ДХу, коэффициенты которой являются вторыми производными Из уравнений (41.8)—(41.10) вытекают следующие совершенно общие закономерности: а. Сокращаются все члены формы ДТ^ДХ,-, т. е. члены, составленные из интенсивных и экстенсивных параметров. Поэтому первоначальное выражение распадается на две квадратичные формы, одна из которых зависит только от интенсивных, другая — только от экстенсивных параметров.
б. Квадратичная форма интенсивных параметров имеет отрицательный знак, а экстенсивных параметров — положительный знак.
Таким образом, получаем
[82^(Ху)]Р/-^ФИР»)]ху > 0. (41.11)
Это неравенство в общем случае выполняется тогда и только тогда, когда первая квадратичная форма определена положительной, а вторая — отрицательной. Если исключить
210
Глава VI
также вариации, которые изменяют лишь массу виртуальной фазы, поставив Xr=const, то получим условия стабильности в форме, пригодной для всех термодинамических потенциалов
(41-12)
рт*(**+1.....Vi)]pz,xr>°- (41-13)
Эквивалентная и практически более важная формулировка гласит
r С 0 для главных миноров '
I I нечетного порядка, /4114.4
| dPtdPs I >0 для главных миноров (41.14) четного порядка,
I d2Wk
"лу для всех главных миноров >0. (41.15)
Использованный метод вывода принадлежит Гиббсу, общая формулировка (41.12), (41.13), а также закономерностей а. и б. впервые дана Шоттки, Улихом и Вагнером.
Рассмотрим более подробно свободную энергию Гиббса, важнейший частный случай условий (41.14) и (41.15). В явном виде условия (41.14) (для средних мольных величин)* будут иметь вид
d2G* d2G*
дТ2 дРдТ d2G* d2G* дТдР дР2
d2G* дТ2
d2G* дР2
(41.16)
Среди этих условий, как легко заметить, только два являются независимыми. Так, например, третье неравенство вытекает из первого и второго.
Второе неравенство вместе с (26.7) и (25.4) дает
Ср>0. (41.17)
Для стабильной фазы мольная теплоемкость при постоянном давлении всегда положительна (термическая стабильность).
Из третьего неравенства вместе с (25.5) следует
* Отсюда и далее величины со звездочкой вновь означают средние
мольные величины.
Условия стабильности
211
х>0.
(41.18)
Для стабильной фазы изотермическая сжимаемость всегда положительна (механическая стабильность).
Первое неравенство, (26.7) и (25.4)—-(25.6) и (25.30) дают
d2G* d2G* _ / d2G* У _ ™Су о
дТ2 дР2 \ дТдР} ~ Т * ,
Отсюда, учитывая (41.18), следует
Cv>0,
(41.20)
что уже было выведено в § 40 при помощи внутренней энергии.
Из приведенных неравенств вытекают еще некоторые замечательные следствия. Из (41.17), (41.18) и (41.20) вместе с уравнением (25.30) получим
Ср > Су •
(41.21)
Для стабильной фазы мольная теплоемкость при постоянном давлении всегда больше, чем мольная теплоемкость при постоянном объеме, если а=^=0. Это условие, например, не выполняется для воды при 4° С, потому что в этом случае а = 0.
При помощи тождеств [ср. уравнение (25.9)]
' дУ* \
. дР )s*
I дУ* \
\ дР /г
(<3S*/dP)v.
(dS*/dV\p ’ (дТ/дР^. (dT/dV*)p
(41.22)
(41.23)
из определений (25.6) и (40.20), используя (25.8), получаем
ъ = (dS^dTfy* т. ~ (dS*/dT)p
Из (24.4) и (40.16) следует
х Ср
(41.24)
(41.25)
212
Глава VI
Вследствие (41.21) окончательно получим
z>zs. (41.26)
Для стабильной фазы изотермическая сжимаемость всегда больше, чем изэнтропийная сжимаемость, если а=^0. Рассмотрим теперь условия стабильности (41.15). При этом несколько изменим общую формулировку тем, что не Хг, а Е nt будем сохранять постоянной. Для ясности приведем эти выкладки. Прежде всего запишем (41.13) без ограничения Xr — const в виде т
[AG]r p> S HAnf. (41-27)
Z—1
Отсюда для Е n—const следует
т
[ДС*]ГР> (41.28)
z=l
или вследствие (26.4)
т—1
[ДС*]г,Р> S (н-^)Лхг. (41.29)
1=1
Так как, согласно (26.20),
dG* \
dxt )T,P,Xj~V't ^т’
(41.30)
то правая часть (41.29) представляет собой вариацию первого порядка от G* при постоянных Т и Р. Таким образом, следует соотношение, эквивалентное (41.13):
[ВЧгЧхр... ,хЯ!_1)]ЛР>0 (41.31)
или
I дЮ* для всех главных миноров >0 32)
I dxtdxj (I, / = 1, ... , Ш — 1).
Для бинарной системы (41.32) сводится к условию
/J2G*_\ 0 41 33)
I ах2
\ ил1 /Т,Р
Условия стабильности
213
Из уравнений (26.16) и (41.30) следует / d2G* \ _ 1 / др4 \
к д^1 JT.P l~X1 \ дХ1 )т.р
(41.34)
Поэтому условие стабильности (41.33) можно написать в виде
>о, (41.35)
\ Эх! ]т,р
что уже было использовано в § 28.
Для тройной системы условия стабильности (41.32) имеют вид
82G* дЮ* dx, d2G* d2G* дхгдх!
22-ХИ >0.(41.36)
дх% дх2
Здесь также только два условия независимы, так как, например, третье неравенство следует из первых двух. Из этих неравенств следует, что при T=const, Р= const поверхность G* выпукло-выпуклая по отношению к плоскости хг—х2. Другими словами, каждое перпендикулярное сечение через поверхность G* дает выпуклую книзу кривую.
Полностью аналогичные формы получаются для энтропийного выражения фундаментального уравнения и функций Массье—Планка. Они отличаются от выражений, пригодных для термодинамических потенциалов, в основном за счет перемены знака. Следовательно, для энтропии имеем
PS(XP ... (41.37)
и для функций Массье — Планка
pOJPp ...,Pft)]Xy>0, [82ФЙ (Xft+1,..., Хг_х)]р^ х? < 0.
(41.38)
(41.39)
214
Глава VI
§ 42. Условия стабильности для гетерогенных систем
В предыдущих параграфах условия стабильности рассматривались для гомогенных систем. Покажем теперь, что гетерогенные равновесия с необходимостью являются стабильными, если условия стабильности выполнены для отдельных фаз. Ради простоты ограничимся рассмотрением однокомпонентной системы с двумя фазами а и |3. Возможные возмущения, изменяющие лишь количество фаз, определим таким образом, что числа молей фаз и(а) и n(f3) остаются постоянными. Следовательно, предположим, что фазы отделены диатермической перегородкой, двигающейся без трения. Выберем энергетическое выражение и рассмотрим возможные возмущения At/ с дополнительными условиями
8п<а) = 0; 8п(₽) = 0, (42.1)
n(“)Ss(“) + n(₽>8s<₽) = о, (42.2)
nMZv{a) + п(₽)8и(₽) = 0, (42.3)
где было использовано определение (20.36). Так как при этом предполагалось равновесие, то вариации первого порядка равны нулю и в результате получаем условие стабильности
84/ = — П(а)2
2
' 1 d2M(a)
ч ds^dv^
1 д2ц(а)
.<«>
1 d2u(a) n<“> ds™'
1
____1 d2u(P) п<₽>
д2ц(Р)
1 а2и<р>
п<₽> №<₽>*
(8о(а))2
0. (42.4)
Поэтому квадратичная форма должна быть положительной. Так как это должно быть справедливым для любых значений n(a) и п<₽>, то, подставивn(a)=n(|5) = l, получим условия стабильности в явном виде
д2ц<к) ds^‘
д2«(р)
д2ц(а) dv^‘
(42.5)
(42.6)
д2»^> 0
Условия стабильности
215
d2u<a) . 52u(₽) \( д2им д2и(р) \
5s<a)2 ds^2 /\ 5t/a>2 5t>^>2 /
_ / даы<а> 52ц(Р) \ 0
\ds(“) ds^5t>^ /
(42.7)
Для стабильности всей системы в целом необходимым условием является стабильность отдельных фаз. Следовательно, согласно (40.12), должно быть
d2»(tt) •0; a2u<^ _ >0, (42.8)
5s<“>2 as<₽>2 "
52«(а) •0; a2M<p) o, (42.9)
5t><«2 >
сРи^ д2и^ ( a2»(a) \ 1 >0,
дз(а)г до(а)2 \ 5s /
52ы<₽) д2ит ( 52»<P> \ 2 I >0. (42.10)
5s<₽>2 dv<№
Видно, что из неравенств (42.8) и (42.9) следуют неравенства (42.5) и (42.6).
Справедливо следующее равенство:
(Oj + а2)(сх + с2) — (bt 4- 62)2 = (1 + (агс2 — 62) +
+ + -у-) (aici —&1) + (аД — аА)2/аха2. (42.11)
Таким образом, оказывается, что из (42.8)—(42.10) вытекает также неравенство (42.7). Итак, получаем
Правило:
Для стабильности гетерогенного равновесия необходимо и достаточно, чтобы условия стабильности выполнялись для отдельных фаз.
§ 43. Принцип Ле Шателье—Брауна
С условиями стабильности тесно связано положение, которое часто называют принципом наименьшего принуждения. Истоки этого принципа берут начало в XVIII столетии
216
Глава VI
(Даламбер, Гаусс). Современная формулировка дана Ле Шателье (1884) и Брауном (1887). Поэтому ее называют принципом Ле Шателье — Брауна.
Простейшая формулировка (которую часто называют принципом Ле Шателье) гласит:
Система, находящаяся в равновесии, реагирует на внешнее воздействие таким образом, чтобы уменьшить это воздействие.
Легко понять, что упрощенные условия стабильности охватываются этим принципом. Если к системе подводится
Рис. 27. Принцип Ле Шателье—Брауна.
теплота, то возникает движущая сила разности температуры между системой и теплоподводящим резервуаром. Поэтому движущая сила уменьшается, если подвод теплоты увеличивает температуру, что требует, чтобы в согласии с (40.17) и (41.17) было Су >0, соответственно Ср>0. Аналогичным образом условие (41.18) можно свести к этому принципу. Другие примеры будут упомянуты в связи со стабильностью химического равновесия.
В простейших случаях принцип Ле Шателье просто эквивалентен условиям стабильности. Как чисто качественную формулировку его часто применяют в химии. Однако он может привести к неправильным заключениям, причина которых кроется в неточной формулировке. Так, высказывание: «Экзотермическое растворение вызывает уменьшение растворимости с повышением температуры», может быть неправильным. Температурная зависимость растворимости определяется именно условием стабильности (41.35), но не интегральной теплотой растворения, а дифференциальной
Условия стабильности
217
теплотой растворения в точке насыщения («последняя теплота растворения»), которые могут иметь противоположные знаки. Более сложные условия стабильности, как, например, неравенства (41.36), вообще нельзя получить из принципа Ле Шателье. Поэтому для термодинамики он не имеет значения и должен быть упомянут только вследствие его частого применения. Более важным является собственно принцип Ле Шателье—Брауна.
Прежде чем его сформулировать, объясним его содержание на простом примере. Пусть цилиндр, наполненный газом, находится над подвижным поршнем и имеет одинаковое давление с резервуаром I. В первом опыте пусть температура цилиндра контролируется диатермической стенкой и тепловым резервуаром II. Изменение давления dP вызывает тогда лишь изменение объема dYV. Во втором опыте пусть цилиндр будет адиабатически изолирован. Одинаковое изменение давления вызывает тогда изменение объема d2V=7^iV, так как кроме этого возникает еще изменение температуры d2T. Принцип Ле Шателье—Брауна утверждает, что
d2V<d1V. (43.1)
В правильности этого положения для частных случаев можно легко убедиться на изотермах и адиабатах газа.
Обычная качественная формулировка принципа Ле Шателье—Брауна говорит о том, что приведенные рассуждения можно обобщить для любых пар переменных состояния. Количественный анализ, проведенный Эренфестом, показал, что принцип в такой общей формулировке не выполняется. Он должен быть дополнен положением, что один из параметров должен быть экстенсивным, другой — интенсивным. Приведем доказательство Эренфеста, которое дает корректную формулировку принципа.
Пусть Pj— интенсивный параметр, регулируемый при помощи резервуара I; Xj— экстенсивный параметр с ним сопряженный, на который Pj влияет непосредственно; Pi— интенсивный параметр, который в опыте I поддерживается постоянным за счет резервуара II, в то время как в опыте 2 dXt =0, и поэтому за счет изменения Pj происходит изменение Pz.
Согласно принципу Ле Щателье—Брауна,
218
Глава VI
|d2Xy|<KXy|. (43.2)
Доказательство'.
Имеем
/ дР,- \ dP.^l—L] d.X,, (43.3)
1 \dXi )PL 1
I dP, \ / dP, \
dP, = — d2X< + — I d2Pt. (43.4)
' \dXj )P. 2 \dpjxj
Для второго опыта, кроме того, должно быть
d X. = Щ d2X, + d2pt = 0. (43.5)
2 \ dXj )p. 2 J 1 \ dPt )x.
Исключение dPj и d2Pz дает
или, используя соотношения Максвелла (24.11),
dXj dPt
dXj У ' dXJ kt.
dxJ hi
d2Xt~
\ / dXj
dXj )P. dPt
Xi
d.Xj.
(43.7)
Согласно условиям стабильности (41.14) и (41.15), с учетом
(21.9) получим
dXj
dXi
dPi Xi
(43.8)
Отсюда непосредственно следует неравенство (43.2).
Принцип Ле Шателье—Брауна наглядно показывает, что индуцированное изменение параметра d2Pt «разгружает» непосредственно влияемый параметр Х7-. Хотя принцип в приведенной выше формулировке строго справедлив, для термодинамики он не получил большого значения. Однако он оказался плодотворным для теории необратимых процессов.
Условия стабильности
219
§ 44. Стабильность химического равновесия
Рассмотрим теперь кратко стабильность₽химического равновесия и для упрощения предположим, что 7=const, Р = =const. Тогда условия стабильности можно сформулировать, используя свободную энергию Гиббса, и для реакции (33.15), согласно § 36, единственной независимой переменной будет число пробегов реакции L
Тогда неравенство (41.27) имеет вид
(44.1)
Правая часть, согласно (36.6), представляет собой вариацию первого порядка. Поэтому условие стабильности будет
> 2 ’ >*
0.
(44.2)
Выведем отсюда некоторые следствия, иллюстрирующие принцип Ле Шателье. Рассмотрим сдвиг химического равновесия (положение которого характеризуется значением ?е) с изменением температуры при постоянном давлении и с изменением давления при постоянной температуре. Будем исходить из уравнения
dG = - SdT + VdP + (2 Wi) (44.3)
причем в состоянии равновесия
(44.4)
Из (44.3) для полного дифференциала от (dG/d'i)r,p следует
=^(2^) = -® dT +
\ dt )т, р \dt )т, р
। ( ^ \ лп । (d2G\ ...
+ —- dP + [--- dt (44.5)
\ ае /т, р \ d& ]т,р V
При равновесии левая часть равна нулю и для dP=0 получаем
220
Глава VI
die 'j _ (dS/d£)r,/> dT )p ~ (d2G/di2)Tt p '
Согласно (24.8),
(dG\ = ( d2L \ _ т (—'I
\ di )t, p \ di )t, p \ di )r, p
(44.6)
(44.7)
В равновесии левая часть равна нулю и величина (дН/дЧ)т,р, согласно (21.23), представляет собой теплоту реакции при постоянном давлении или энтальпию реакции. Таким образом, из (44.6) следует
\ dT )р Т (d2G/di2)Tt р V
Так как, согласно условию стабильности, знаменатель всегда положителен, то из уравнения (44.8) следует:
Повышение температуры при постоянном давлении сдвигает химическое равновесие в направлении, в котором теплота реакции положительна, т. е. реакция эндотермическая. Это положение согласуется с принципом Ле Шателье.
Аналогичным образом получают
/зм _ тт, Р _ (449)
\дР )т (д26/д?)т,р ' ’
Повышение давления при постоянной температуре сдвигает химическое равновесие в направлении, в котором изменение объема в результате реакции (при 7=const, Р=const) отрицательно. Это положение также согласуется с принципом Ле Шателье.
Следует обратить внимание на то, что приведенные положения являются совершенно общими и могут быть получены из уравнений Вант-Гоффа, соответственно Планка— Ван Лаара, только для частного случая идеального газа. Это основано на том, что при химическом равновесии в идеальной газовой смеси условие стабильности (44.2) выполняется автоматически.
Глава VII
Критические фазы
§ 45. Определение и свойства критических фаз
Экспериментальная практика показывает, что часто две сосуществующие фазы при изменении величин состояния изменяются таким образом, что они в конце концов становятся идентичными. Фаза, в которой это происходит, называется критической фазой. Критические фазы в однокомпонентных системах, как будет видно, не имеют термодинамических степеней свободы. Поэтому в пространстве состояния они являются сингулярными точками, которые называют критическими точками. Если, как это часто бывает, не рассматривается зависимость критической фазы от величин состояния, то так же говорят о критических точках в многокомпонентных системах. Приведем обзор наиболее важных случаев.
а. Однокомпонентные системы
Равновесие жидкость — пар имеет всегда одну критическую точку. Выше определенной температуры Тс, которая называется критической, невозможно сконденсировать газ. Происходит, как это впервые было открыто в 1869 г. Эндрюсом на примере СО2, непрерывный переход от газообразного в жидкое состояние. Соотношение проще всего проследить на изотермах диаграммы Р—V, которая для реального газа схематически приведена на рис. 28. Кривые, на которых расположены сосуществующие фазы жидкости и пара, называются кривыми сосуществования или бинодалями. Пунктирные линии, соединяющие две сосуществующие фазы, называются коннодами. Эти обозначения используют также при других гетерогенных равновесиях.
222
Глава VII
Равновесие пар—кристалл заканчивается в тройной точке (рис. 18). Это равновесие не существует, однако, для 3Не и 4Не. В обоих случаях кристалл находится в равновесии только с жидкостью. Поэтому тройная точка в этих случаях не существует.
Для равновесия жидкость—кристалл, несмотря на многочисленные попытки, экспериментально до сих пор не было
Рис. 28. Изотермы реального газа (схема).
СР — критическая точка.
найдено критической точки. Некоторые теоретические соображения говорят о том, что здесь не может существовать критическая точка. Окончательная ясность в этом вопросе пока не достигнута.
б. Бинарные системы
Равновесие пар — жидкость имеет также критическую точку, которая интересна благодаря замечательному явлению обратной конденсации или испарения. Это явление будет разобрано в § 47.
Более важными являются критические точки, которые возникают при расслоении гомогенного раствора на две жидкие фазы. Графическое представление их, соответствующее рис. 28, дается в этом случае изотермами химического потенциала компонентов в зависимости от мольной доли (Xi или х2) при Р=const. Однако можно удовлетвориться диаграммой Т—х, которая неоднократно была использо
Критические фазы
223
вана в гл. IV. На рис. 29 схематически показаны наблюдаемые случаи.
Случай а., который называется верхней критической точкой расслоения, наблюдается наиболее часто и был найден для целого ряда органических жидких смесей. Для нижней критической точки расслоения, показанной на рис. 29, б, примером являются системы вода—диэтиламин и вода — триэтиламин. Замкнутая кривая растворения, которая приведена на рис. 29, в, была найдена в системе
Рис. 29. Критическая точка расслоения в бинарных жидких смесях. а—верхняя критическая точка; б — нижняя критическая точка; в — замкнутая кривая растворения с двумя критическими точками.
вода — никотин. Здесь имеется как верхняя (Тси), так и нижняя (Тс1) критические точки расслоения.
В противоположность однокомпонентным системам для бинарных систем наблюдаются критические точки так же в твердом состоянии, а именно критическая точка расслоения, как и в жидком состоянии. Правда, они наблюдаются довольно редко, так как в большинстве случаев сосуществующие фазы имеют различные кристаллические структуры и поэтому из геометрических соображений не могут быть идентичными. Все же имеются бинарные сплавы, которые в твердом состоянии имеют одну критическую точку расслоения, например Al-Zn, Au-Ni, Au-Pt. В качестве примера на рис. 30 приведена фазовая диаграмма системы Al-Zn.
224
Глава VII
в. Тройные системы
В первую очередь представляют интерес критические точки, которые появляются при расслоении жидкой фазы. Здесь нет возможности останавливаться на деталях, а поэтому сделаем в этой связи два замечания общего характера.
Рис. 31. Изотермически-изобарные G* поверхности тройных систем.
СР — критические фазы.
Ранее было отмечено, что в однокомпонентных системах критическая температура является наивысшей температурой, при которой система может стать гетерогенной. Для
Критические фазы
225
бинарных систем критическая фаза имеет одну степень свободы. Но если, как это обычно бывает в эксперименте, давление будет постоянным, то критическая температура вновь является наибольшей (в случае нижней критической точки расслоения — наименьшей) температурой, при которой гомогенный раствор (или гомогенные смешанные кристаллы) может стать гетерогенным. В системах, состоящих из трех и более компонентов, существует совершенно другая ситуация. Критическая фаза тройной системы имеет, как будет
Рис. 32. Расслоение тройных систем при х1/х2= const.
СР — критическая точка* Р' — фаза с наибольшей температурой расслоения; Р" — фаза, сосуществующая с Р'•
показано, две степени свободы. Если давление задано, то соотношение можно представить в пространственной диаграмме, нанося над плоскостью хг—х2 изотермические поверхности G*.
Такая диаграмма приведена на рис. 31. Видно, что теперь к каждой изотермической поверхности G* принадлежит бинодаль с критической точкой и, таким образом, понятие критической температуры вообще больше не имеет смысла. Однако если после задания давления использовать оставшуюся степень свободы, так чтобы соотношение x^fx2 было фиксировано, то снова можно определить Тс. Получающаяся «критическая температура» не является теперь наивысшей температурой, при которой система может стать гетерогенной. Это видно при простейшем использо-
1/29—235
226
Глава VII
вании треугольных координат Мёбиуса (рис. 32), введенных в гл. IV.
На диаграмме показаны бинодали, относящиеся к различным температурам, критические точки которых отмечены черными точками. Все растворы, для которых компоненты Л и В смешиваются в заданном соотношении х^/х^ расположены на прямой FC, Она может пройти только через одну критическую точку, которая, таким образом, определена однозначно. Наибольшая температура, при которой система может стать гетерогенной, соответствует не этой бинодали, а скорей бинодали, к которой прямая FC является касательной. Соответствующая фаза показана при помощи точки касания Р'. Видно, что она в общем случае не является критической точкой, а находится в равновесии со второй фазой Р”. Приведенное рассуждение, которое провел Том-па, имеет очень большое значение для теории растворимости макромолекул.
Второе замечание относится к часто встречаемому в экспериментальной практике случаю применения смеси двух органических жидкостей в качестве растворителя. В более старой литературе такие системы рассматривали как бинарные системы, у которых свойства растворителя каким-нибудь образом рассчитывали из свойств компонентов. Этот способ недопустим прежде всего тогда, когда (как это в большинстве случаев бывает) растворитель в сосуществующих фазах имеет различный состав. Особенно недопустимо определять критическую точку расслоения простым расчетом для бинарных систем. Скорее следует применять гораздо более сложные уравнения для тройных систем, которые в дальнейшем будут выведены. Это замечание также имеет значение прежде всего для растворов высокомолекулярных соединений.
Обратимся теперь к законам термодинамики и докажем установленный Гиббсом
Закон
В системе, состоящей из т компонентов, критическая фаза, которая не сосуществует с остальными фазами, имеет т—1 термодинамических степеней свободы.
Критические фазы
227
Доказательство
Рассмотрим тройную систему с двумя сосуществующими фазами. Согласно правилу фаз [уравнение (29.3)], она имеет три степени свободы. Две из них имеются за счет постоянства давления и температуры. Поведение системы изображается тогда соответствующей поверхностью G* на рис. 31. Так как имеется еще одна степень свободы, то можно, например, изменять химический потенциал рч и при этом ни одна из фаз не будет исчезать. Как показывает рис. 31, эти изменения принадлежат к последовательному ряду сосуществующих фаз, который заканчивается в критической точке. Если величинам Т и Р придать другие значения, которые отличаются от первоначальных на бесконечно малые величины, то аналогичным образом получим последовательность пар фаз, которая снова заканчивается в критической точке. Ее положение отличается от первоначального также на бесконечно малые величины*. Таким образом, можно идти дальше и сделать вывод, что в рассматриваемом случае критическая фаза имеет две степени свободы.
В общем случае для системы из т компонентов рассмотрим вновь две сосуществующие фазы. Гетерогенная система тогда имеет, согласно правилу фаз, т степеней свободы. Выберем в качестве независимых переменных т величин из набора интенсивных параметров Т, Р, и зафик-
сируем т—1 из них тем, что припишем им значения, которые они имеют в критической точке из совокупности рассматриваемых критических фаз. Оставшиеся интенсивные параметры (давление, температуру или один из химических потенциалов) можно варьировать, не уничтожая при этом одну из фаз. Таким путем получим снова последовательность сосуществующих фаз, которая заканчивается в критической точке.
Изменим теперь на бесконечно малые величины значения зафиксированных прежде т—1 интенсивных параметров и за счет вариации свободного параметра вновь получим последовательность сосуществующих фаз. Теперь
* Так как рис. 31 является изобарной диаграммой, то наглядными являются только вариации по Т,
1/а9*
228
Глава VII
каждая пара сосуществующих фаз в первой последовательности должна соответствовать паре, отличающейся от нее на бесконечно малую величину во второй последовательности, и наоборот. Поэтому вторая последовательность должна заканчиваться критической точкой, положение которой отличается от критической точки первой последовательности только на бесконечно малую величину. Таким образом, можно вновь идти дальше. Если значения т—1 интенсивных параметров, задаваемых положением критической точки, свободно варьируются, то каждому набору измененных значений снова принадлежит одна и только одна критическая фаза. Следовательно, в системе из т компонентов критическая фаза обладает т—1 термодинамическими степенями свободы. Таким образом, теорема доказана.
Из теоремы следует ранее использованное следствие о том, что в однокомпонентной системе критическая фаза является сингулярной точкой в пространстве состояния, в то время как критическая фаза бинарных систем обладает одной степенью свободы, которая в общем случае для конденсированных систем определяется давлением*. Для тройных систем имеется уже две степени свободы, чем соотношения, как будет показано выше, значительно усложняются. Как уже отмечалось, приведенное высказывание справедливо только в том случае, если ни одна из остальных фаз не сосуществует с критической фазой. Так как, согласно правилу фаз, каждая новая фаза уменьшает число степеней свободы на единицу, то следует, что с критической фазой однокомпонентной системы вообще не может сосуществовать никакая другая фаза, а с критической фазой бинарной системы может сосуществовать только одна фаза. Вообще критическая фаза может сосуществовать самое большое с т—1 других фаз. Как показывает сравнение с уравнением (29.3), при подсчете числа степеней свободы критическую фазу нужно учитывать трижды.
* Для конденсированных бинарных смешанных фаз зависимость критической температуры расслоения от давления очень мала. Поэтому при обычных экспериментальных условиях в большинстве случаев ею можно полностью пренебречь.
Критические фазы
229
§ 46. Уравнения Гиббса для критических фаз
В дальнейшем не будем рассматривать возможность сосуществования критической фазы с остальными фазами. Так как, согласно правилу фаз, обычная фаза имеет т-|-1 степеней свободы и, согласно приведенной теореме, критическая фаза имеет ш—1 степеней свободы, то отсюда следует, что критическая фаза определяется двумя уравнениями.
Для однокомпонентной системы получают эти уравнения из рис. 28 с помощью простого рассуждения. Согласно условиям равновесия (27.6) и (27.7), сосуществующие фазы должны быть расположены на одной и той же изотерме и конноды должны проходить параллельно оси абсцисс. Так как критическая точка определяется по совпадению сосуществующих фаз, то она должна быть точкой перегиба изотермы с горизонтальной касательной. Отсюда следует
—'i =0; } =0. (46.1)
dv )т \ dv2 )т
Для критической точки расслоения бинарной системы, как уже было отмечено, получаются изотермы диаграммы р-—х, подобные рис. 28. Поэтому при аналогичных рассуждениях для критической точки расслоения бинарной системы получают
дН1\ дХ! )т, Р
d2Hi \
у у* р
(46.2)
Для системы, состоящей более, чем из двух компонентов, соотношения получаются менее наглядными и, как уже отмечал Гиббс, необходимо формулировать теорию в более строгом и общем виде. Исходной точкой этой формулировки является теория стабильности, рассмотренная в гл. VI. Рассуждения можно провести тогда различным образом и получить соответствующие уравнения для критической фазы в различных формах. Разберем в этом параграфе метод, предложенный Гиббсом.
Рассмотрим прежде всего однокомпонентную систему (рис. 33) и заметим, что на каждой конноде, соединяющей две сосуществующие фазы, обязательно находятся состояния, для которых гомогенная система является абсолютно нестабильной. Это положение станет наглядным, если предполо-8-235
230
Глава VII
жить, что изотермы во всей области состояния являются аналитическими функциями. Тогда они обязательно должны иметь вид, известный из уравнения состояния Ван-дер-Ваальса, который изображен на рис. 33.
Рис. 33. Изотермы реального газа с «.петлей Ван-дер--Ваальса» (схема).
На каждой изотерме между минимумом и максимумом обязательно расположены только нестабильные состояния. Кривая, на которой расположены минимумы и максимумы и которая поэтому ограничивает нестабильную область, называется спинодалью. Область между спинодалью и бинодалью соответствует тогда метастабильным состояниям (перегретая жидкость, пересыщенный пар). Такое представление в термодинамике неоднократно использовали как полезное вспомогательное средство. Однако здесь такое представление не будет использовано для логики рассуждений, а будет введено лишь для наглядности, так как исходная гипотеза, как можно показать, несостоятельна*.
Представим обе сосуществующие фазы расположенными сколь угодно близко к критической точке. Так как тогда между ними должны быть расположены абсолютно нестабильные состояния, то отсюда следует, что критическая
* Математически строго можно доказать, что точный статистический расчет термодинамических функций ни при каких обстоятельствах нельзя провести для нестабильных состояний. Рассчитанные таким путем изотермы никогда не могут давать «петлю Ван-дер-Ваальса», они дают скорее всегда картину рис. 28.
Критические фазы
231
точка расположена на границе между стабильной и абсолютно нестабильной областью. В терминах, введенных выше, это значит, что в критической точке касаются бинодаль и спинодаль.
Граница между стабильной и нестабильной областью, во всяком случае, определяется тем, что по крайней мере одна из вторых производных внутренней энергии равна нулю. Это значит, что в этом месте квадратичная форма S2&(s, v) является положительно полуопределенной. Как следует из линейной алгебры, для этого необходимо и достаточно, чтобы выполнялось условие
д2и д2и
ds2 dsdv
dvds dv2
Поэтому уже уравнение (46.3) является первым уравнением для критической точки.
Чтобы вывести второе уравнение, зададим температуру и рассмотрим вариацию критической фазы на изотермах. Из рис. 28 или рис. 33 следует, что такая вариация ни при каких обстоятельствах не может привести в нестабильную область. Это значит, что изменение детерминанта D, определенного уравнением (46.3), не может быть отрицательным. Но оно не может быть также и положительным, так как в противном случае за счет простой перемены знака независимых переменных оно стало бы отрицательным. Таким образом, для рассматриваемой вариации имеем оба уравнения
dT = (—ds + И dv = 0, (46.4)
\ds jv \ dv )s
dD = (—ds + (—dv = 0. (46.5)
\ ds )v \ dv )s
Это гомогенная линейная система уравнений с неизвестными ds и dv. Условием ее решения будет:
8*
232
Глава VII
Это второе уравнение критической точки.
Уравнения (46.3) и (46.6) основаны в конечном счете на фундаментальном уравнении и имеют поэтому много раз упомянутые недостатки. Особенно громоздко непосредственное доказательство идентичности с уравнениями (46.1). Поэтому возникает мысль выбрать сначала в качестве исходного пункта формулировку условий стабильности при помощи подходящего термодинамического потенциала. Рассматриваемой проблеме лучше всего соответствует свободная энергия Гельмгольца. Для нее условия стабильности, согласно (41.14) и (41.15), имеют вид
—'j >0; (—'j <0. (46.7)
дТ )v \dv )т
Видно, что первое из этих условий не имеет значения для определения критической точки. Если провести со вторым условием полностью аналогичные рассуждения, то получим уравнение (46.1).
Обобщение приведенных выводов для системы, состоящей из пг компонентов, не представляет трудностей. Для сокращения напишем
d2U* d2U* a2:/*
as*2 as* av* as^a*^
d2U* .
D* = dV*dS*
o2U* a2^*
dxm_! dS*
(46.8)
Далее, обозначим через D*7 детерминант, отличающийся от D* тем, что элементы произвольно выбранных строчек заменены на частные производные, стоящие в выражении
dD* = js* + dV* + ^~dx1+ ...
dS* dV* dxx 1
... + ^-dxm.r (46.9)
°xrn-i
Критические фазы
233
Граница области стабильности задана тем, что квадратичная форма (40.11) является положительно пол у определенной. Для этого необходимо и достаточно, чтобы детерминант D* обращался в нуль, чем уже задается первое уравнение для критической фазы.
Для того чтобы получить второе уравнение, нужно снова проварьировать критическую фазу. При этом, согласно дополнительным условиям, из m-pl производных типа
dU* dU* dU* dU*
dS* ’ dV* dxL
m величин поддерживаются постоянными. Выбор последних является произвольным. Они определяют, как видно из уравнений (27.6)—(27.8), в каждом случае путь на m-мерной гиперповерхности, который связывает две сосуществующие фазы. Вариация критической фазы при этих условиях ни при каких обстоятельствах не может вести в нестабильную область. Если, например, предположить, что первые m производных из (46.10) постоянны, то для вариации критической фазы справедливы дополнительные условия:
d2U*
d
= ds* + ----- dv* +
dS*2 dS*dV*
-^L.dx,+ ...+ № o, dS* dxr 1 dS* dx^ m 1
, ... dS. + ' dv. +
dV* dS* dV*2
А2П* №11*
- — — dxr+.... + dxm^ = 0,
dV* dxr 1 dV*dxm_1 m 1
.^ds»+ - i
dx^S* dxt dV*
+ ^~dX1+- + l^L-dX^ = °-
C/Xj UXlOXffi—i
(46.11)
d
пг-2(
dx± + ... +
• - w .dS. + .„.w .
_2 / dxm_2dS* dxm_2 dV*
- SUL- dX1 + ... + dXm_t _ 0.
dXm-i
234
Глава VII
Так как вариация не может вести в нестабильную область, то при помощи рассуждений, полностью аналогичных проведенным ранее, можно показать, что также должно выполняться равенство
dD* = — dS* + — dV* + — dX1 + ...
dS* dV* dx,. 1
... + ^L^_1==0. (46.12)
Однородная линейная система уравнений (46.11), (46.12) имеет нетривиальное решение только тогда, когда детерминанты коэффициентов обращаются в нуль. Таким образом, получим общие уравнения критической фазы для системы, состоящей из т компонентов:
D* = 0; £)*'= 0. (46.13)
Другой выбор дополнительных условий (46.11) ведет, естественно, к другой форме детерминанта D*'. Эту возможность нужно учитывать уже при определении, так что уравнение (46.13) этим не затрагивается. Легко понять, что для однокомпонентной системы уравнение (46.13) сводится к уравнениям (46.3) и (46.6). Другие эквивалентные формулировки можно получить, если для начальных условий (40.11) и (40.12) выбрать другой способ написания, например (как это сделал Гиббс) поддерживать объем постоянным.
§ 47. Преобразование уравнений для критических фаз. Дальнейшие свойства критических фаз
В общем случае для конкретных задач предпочтительно получать уравнения для критических фаз не из фундаментального уравнения, а из соответствующих термодинамических потенциалов. Эта возможность основана на том факте, что определенные условия стабильности остаются справед-
Критические фазы
235
ливыми также на границе стабильной области. Разделение, появляющееся при преобразовании условий стабильности (§ 41) на условия для интенсивных параметров и на условия для экстенсивных параметров, позволяет отбросить сразу сомнительные условия и, таким образом, упростить уравнения для критической фазы. Приведем два примера. Как уже выше было отмечено, для равновесия жидкость — пар в однокомпонентных системах условие термической стабильности Су >0 остается справедливым также на границе области стабильности. То же справедливо для равновесия жидкость — пар в многокомпонентных системах. Поэтому в данном случае наиболее целесообразно применение свободной энергии Гельмгольца, которая позволяет провести разделение этих условий. С условием стабильности для остающихся интенсивных параметров V*,Xi, xm__\ проводят рассуждение, полностью аналогичное приведенному выше. Поэтому достаточно привести лишь конечный результат. Введем
d2F* dV^dx^
d2F* d2F*
dV*2 dV* дХ1 d2F* ’ .
dx± dV*
d2F* d2F*
dxm^dV* dxm-l
(47.1)
и обозначим через Df детерминант, который получается из (47.1), если заменить элементы любой строки на частные производные
dD"F dV* ’
дРр дхг
дРр дхт-1
(47.2)
Тогда уравнения для
критической фазы будут иметь вид
Df — 0; Df = 0.
(47.3)
236
Глава VII
При этом следует принять во внимание, что появляющиеся здесь частные производные нужно брать при постоянной температуре. Для однокомпонентных систем уравнения (47.3) сводятся к уравнениям (46.1).
При расслоении конденсированных фаз происходит дальнейшее упрощение, так как здесь (в противоположность равновесию жидкость — пар) на границе области стабильности выполняется также условие механической стабильности (d2F/dV2)T >0. Поэтому в этих случаях используют формулировку при помощи свободной энергии Гиббса, которая дает начальные условия (41.31) и (41.32). Сам вывод проводят таким же образом. Введем
d2G* d2G* d2G*
dx% dxtdx2
d2G* .
dx2dx±
d2G* d2G*
dxm^dxr dxm-\
и обозначим через Dg детерминант, который получается из (47.4), если элементы любой строки заменить на частные производные
5D* dD* dD*Ct dD*
—(47.5) дхг дх2 дх3 дхт-л
В этом случае уравнения для критической фазы запишутся
Dg = 0; Dg = 0. (47.6)
При этом нужно брать все производные по мольным долям при постоянных Т и Р. Уравнения (47.6) отличаются от предыдущих тем, что их нельзя свести к уравнениям для однокомпонентных систем. В случае т~3 уравнения нашли
Критические фазы
237
интересные применения в теории растворов макромолекулярных соединений.
Уравнения (46.13) и (47.6) (в несколько другой форме) были выведены уже Гиббсом. Формулировка при помощи свободной энергии Гельмгольца [уравнение (47.3)] была использована, в частности, Ван-дер-Ваальсом для изучения равновесия жидкость — пар.
Критические фазы особенно интересны потому, что они показывают ряд замечательных явлений, которые обычно называют «критическими явлениями». Приведем здесь только несколько примеров. Прежде всего уже для однокомпонентных систем поразительно то, что при приближении к критической точке мольная теплоемкость при постоянном давлении, термический коэффициент расширения и сжимаемость стремятся к бесконечности. Эти факты следуют из положения критической точки на границе области стабильности. Поведение сжимаемости приводит к тому, что точные измерения вблизи критической точки вследствие влияния гравитации наталкиваются на громадные трудности.
В бинарных системах вблизи критической точки испарения можно встретить явления «обратного испарения» и «обратной конденсации». Более подробное разъяснение этих явлений выходит за рамки данной книги. Поэтому объясним понятие только кратко и покажем на простых
Рис. 34. Обратная кон- Р денсация.
примерах, как могут происходить такие явления. Рассмотрим сначала однокомпонентную систему, температура которой Т<ТС пусть будет зафиксирована (рис. 28). Если газ
238
Глава VII
сжимают, то движутся по изотерме. При определенной плотности наступает конденсация, с дальнейшим сжатием увеличивается количество жидкой фазы до тех пор, пока наконец не исчезнет газовая фаза. Вблизи критической точки бинарной смеси может произойти следующее. Если смесь изотермически сжимают, то достигают давления, при котором появляется жидкая фаза. При определенных температуре и составе дальнейшее сжатие вызывает уменьшение и затем исчезновение жидкости. Это явление, которое на примере смеси СО2— воздух впервые наблюдал Кальете, называется обратной конденсацией первого рода. Рис. 34, на котором схематически приведена изотермическая диаграмма Р—х, показывает, как может происходить это явление. СР — критическая точка. Сжатие происходит вдоль пунктирной линии А—В. В точке А происходит конденсация, при дальнейшем сжатии сначала увеличивается количество жидкой фазы, затем снова уменьшается и в точке В полностью исчезает. Аналогичное поведение наблюдают, если при постоянном давлении изменяют температуру. Тогда говорят об обратной конденсации второго рода.
Но, пожалуй, самым замечательным критическим явлением будет так называемая критическая опалесценция, которая для однокомпонентных систем была открыта Авенариусом (1874) уже через несколько лет после открытия критической точки. Если газ охлаждать при критической плотности, то он при температуре примерно на один градус выше критической начинает излучать голубоватый свет опалесценции, интенсивность которого сильно увеличивается с приближением к критической точке, хотя система все еще остается гомогенной. Это явление основано на том, что при приближении к критической точке сильно возрастает прежде всего в прямом направлении интенсивность рассеяния света. Такие же явления наблюдаются в критической точке расслоения жидких и твердых систем. В последнем случае для доказательства нужно, конечно, использовать рентгеновские лучи. Критическая опалесценция является, как показывает теоретический анализ, непосредственным следствием того факта, что критическая точка расположена на границе области стабильности.
Глава VIII
Электрохимические системы
§ 48. Определение и общие свойства электрохимических систем
Опыт показывает, что водные растворы определенных веществ, которые были названы электролитами, являются сравнительно хорошими проводниками электрического тока. Речь идет о веществах, которые в химии называются кислотами, солями и основаниями. Электролитическая электропроводность отличается от металлической проводимости следующими характерными свойствами:
а. Прохождение тока связано с макроскопическим переносом материи.
б. Прохождение тока сопровождается химической реакцией, в. Электрическая цепь, содержащая электролитический проводник, обязательно является гетерогенной системой, состоящей из нескольких открытых фаз.
Такие системы называются электрохимическими системами, причем ради простоты в это понятие будем включать также гомогенные растворы электролитов*.
Отмеченные выше свойства электрохимических систем впервые были объяснены в 1887 г. Аррениусом при использовании (в дальнейшем экспериментально вполне подтвержденной) гипотезы о том, что электролиты в водных растворах диссоциируют на электрически заряженные ионы. Поэтому электрохимические системы можно определить также при помощи утверждения, что в них при прохожде. нии тока происходит перенос заряда ионами, в то время как в металлах и полупроводниках — электронами.
Наряду с приведенными примерами водных растворов электролитов к электрохимическшм системам относятся
Многие авторы различают эти два понятия.
240
Глава VIII
растворы электролитов в некоторых неводных растворителях, расплавы солей и твердые соли. Изучение электрохимических систем представляет собой обширную область физической химии, которая выходит далеко за рамки термодинамики. В соответствии с планом данной книги ограничимся разъяснением принципиальных вопросов, которые встречаются при применении термодинамики к этому классу систем, а в остальном отошлем читателя к многочисленным монографиям в этой области. Так как для такой постановки вопроса различия всевозможных типов электрохимических систем не имеют значения, то в основном рассмотрим водные растворы электролитов, которые к тому же лучше всего теоретически исследованы. Введем при этом некоторое упрощение. До сих пор еще ничего не было сказано о степени электролитической диссоциации. Условно различают сильные и слабые электролиты, однако такое деление, естественно, довольно нечетко. Количественные показатели в каждом отдельном случае требуют основательного изучения термодинамических, электрических и оптических свойств системы. Для данного рассмотрения эта проблема имеет вспомогательное значение.
Поэтому не будем на этом останавливаться и предположим, что электролит полностью диссоциирован и, таким образом, раствор содержит только ионы и нейтральные молекулы воды*. Это предположение выполняется с достаточной точностью, в частности, для разбавленных растворов галогенидов и перхлоратов щелочных металлов, щелочноземельных металлов и некоторых переходных металлов. Впрочем, учет присутствия нейтральных молекул электролита не представляет трудности.
Разъясним некоторые общие свойства электрохимических систем, которые будут важны для дальнейшего рассмотрения. Прежде всего заметим, что в отсутствие разностей электрического потенциала для раствора электролита сохраняется условие электронейтральности. Если обозначить числа молей ионов через пь их (положительные или отрицательные) валентности через zz, то получим
2*^ = 0, (48.1)
__________ Z
* Гидратацию ионов по причинам, упомянутым в § 2 и 33, можно здесь не рассматривать.
Электрохимические системы
241
где сумма берется по всем видам ионов. Если раствор содержит только один бинарный электролит*, который диссоциируют на v+ катионов и v_ анионов, то имеем
z+n+ + = 0 (48.2)
и
z+v+ + = 0. (48.3)
Из приведенных соотношений (которые без труда можно обобщить на другие типы электролитов) в сочетании с определением § 2 следует, что раствор отдельного электролита имеет только два независимых компонента. Поэтому его состав устанавливается одной концентрационной переменной**. Однако целесообразно и при определенных задачах необходимо*** ввести в качестве компонентов ионы (при необходимости также недиссоциированную часть электролитов) и нейтральный растворитель и учитывать дополнительно условия нейтральности.
Если имеется разность электрических потенциалов (что возможно только в гетерогенных системах), то условие электрической нейтральности теперь уже выполняется не строго. Но отклонения для рассматриваемых разностей потенциалов настолько незначительны, что они расположены далеко ниже границы аналитических методов доказательства****. Эти факты оправдывают приближение, которое составляет формальную основу термодинамического обсуждения электрохимических систем. Если пренебречь изменением состава, связанным с разностью потенциалов, то изменение внутренней энергии при виртуальных смещениях можно разложить на две части, первая из которых (что доказывается ранее рассмотренным образом) зависит от изме
* Бинарными электролитами называют электролиты, которые диссоциируют на два вида ионов.
** Это справедливо также в случае неполной диссоциации.
*** Здесь имеем аналогичную ситуацию, как и для химической реакции: ср. § 36.
**** Помещенный в вакуум, однородно заряженный шарик радиусом 1 см, который имеет положительный избыток зарядов в 1 моль одновалентных ионов, имеет на своей поверхности потенциал ~1017 в. Потенциалы, рассматриваемые в электрохимических системах, во всяком случае, меньше, чем 107 в. Отклонение от строгой нейтральности составляет поэтому меньше, чем 10-10щоль/см
242
Глава VIII
нений переменных состояния, в то время как вторая представляет собой электростатическую работу, совершаемую в результате переноса ионов через скачок потенциала. В противоположность диэлектрическим системам для электрохимических систем электростатическую работу нельзя ввести в виде рабочей координаты в смысле определения (14.1). Причина этого заключена в том, что здесь электростатическая работа обязательно является переносом ионов между открытыми фазами, что несовместимо с определением рабочих координат.
Наконец, для термодинамики электрохимических систем имеет значение факт, на который впервые указал Гиббс и в последнее время вновь отметил Гуггенгейм. Разность потенциалов электрических проводников измерима только между средами одинакового химического состава (например, между двумя частями медной проволоки). Напротив, разность потенциалов между двумя проводящими средами различного химического состава (например, между раствором электролита и погруженным в него электродом) не является измеримой величиной. С этим, как будет видно, непосредственно связан тот факт, что химический потенциал отдельных видов ионов нельзя измерить. Причины, которые, несмотря на это, оправдывают использование этой величины в рамках феноменологической теории, будут ясны из следующих параграфов.
§ 49. Общие условия электрохимического равновесия
Чтобы вывести общие условия равновесия для гетерогенной электрохимической системы, необходимо провести соответствующее обобщение фундаментального уравнения. Согласно § 48, для вариации внутренней энергии можно написать
W = W + W{e\ (49.1)
где U — полная внутренняя энергия и — электрическая часть ее. Как было показано, величина U зависит от переменных состояния и задается уравнениями (20.4) и (20.5)*. Чтобы вывести в явном виде выражение для W!<е>,
* Читатель, незнакомый с векторным исчислением, может пропустить последующие расчеты и продолжать чтение с уравнения
Электрохимические системы
243
используем известное из электростатики соотношение U{e) = A- J E2dV. (49.2)
Необходимым условием термодинамического равновесия является то, что внутри проводника электрическое поле равно нулю, так как в противном случае протекал бы необратимый процесс электропроводности. Интеграл (49.2) распространяется поэтому на все пространство вне проводника, которое ради простоты будем считать вакуумом. Из (49.2) следует
8(/с) = _L С Е. BE dV =------ f grad <р • bEdV, (49.3)
4u J 4к J
где ср — электрический потенциал. Тогда имеем
J grad ? bEdV = J div (<pSE) dV — JcpdivBEdV. (49.4)
Второй интеграл правой части равен нулю, так как для вакуума
divE-0. (49.5)
Первый интеграл можно преобразовать при помощи теоремы Гаусса в интеграл по поверхности проводника и бесконечно удаленной поверхности. Последний интеграл равен нулю, так как сила поля на бесконечности достаточно сильно стремится к нулю. Таким образом, из уравнения (49.3), вследствие того что ср на поверхности проводника постоянно, получаем
ъи{е) = — ф f BE . df. (49.6) 4к J
Интеграл правой части равен изменению заряда проводника, умноженному на 4к. Таким образом, имеем
Ш(е> = (49.7)
где е — общий заряд проводника. Тогда е = (49.8)
Z где g = 96 487 кулон-молт1 (49.9)
244
Глава VIII
— константа Фарадея, т. е. заряд одного моля одновалентных ионов (заряд эквивалента), и суммирование распространяется на все электрически заряженные виды частиц*. Поэтому
g[7(c> = § 1>г8пг. (49.10)
L
Из (49.1) и (49.10) получаем искомое обобщение фундаментального уравнения в виде**
dU = TdS — PdV + 2 (^ + <р) (49.11)
4=1
Величина
Н = Н + (49.12)
названа Гуггенгеймом электрохимическим потенциалом компонента i. Для электрически нейтральных компонентов, для которых zz =0, он сводится к химическому потенциалу.
Рассмотрим теперь закрытую систему с фиксированными рабочими координатами, состоящую из т компонентов и а фаз. Предположим, что по крайней мере одна фаза является раствором электролита и что эта фаза находится в непосредственном контакте по крайней мере с одной электрически заряженной фазой. В остальном делаем такие же предположения, как и в § 27. В частности, фазы должны быть полностью открытыми, так чтобы энтропия, объем и числа молей каждой фазы можно было варьировать, однако с тем ограничением, что не обязательно каждый компонент должен присутствовать в каждой фазе. Далее, исключаем диссоциацию электролитов (которая, согласно предположениям, рассматривается только как гетерогенная реакция), а также химические реакции в собственном смысле. Диссоциацию рассмотрим в § 50, случай твердых поверхностей раздела фаз—в § 51, химические реакции — в § 52.
* Если рассматриваемая фаза является электронным проводником, то в (49.8) необходимо также учитывать электроны.
** Для твердых тел предполагаем всестороннее гидростатическое давление, а также отсутствие одноосевых нормальных напряжений и тангенциальных напряжений.
Электрохимические системы
245
Обозначим фазы, как и в § 27, строчными греческими буквами и одновременно будем употреблять а как текущий индекс. Тогда для рассматриваемого случая общее условие равновесия (17.2) будет иметь вид
2 &£/(а) = о
(49.13)
с дополнительными условиями
2BS(a) = o, а (49.14)
28V(a) = 0, а (49.15)
2М°) = 0 (все о а (49.16)
По методу, описанному в § 27, из (49.13)— (49.16) получаем
условия равновесия в явном виде
(49.17)
р(а) __ р(₽) __ __ р(а) (49.18)
-(а) -(₽) ~(а) (Л. = = ... = . (49.19)
Уравнения (49.17) и (49.18) идентичны (27.6) и (27.7), в то время как (49.19) для нейтральных молекул сводится к (27.8). Условия (49.17)—(49.19) отличаются от (27.6)—(27.8) тем, что для ионов вместо химических потенциалов появляются электрохимические потенциалы. Существенная трудность при этом заключена в том факте, что электрохимические потенциалы не находятся в простом соответствии с измеряемыми величинами. Так как эта проблема довольно сложна, то в следующих параграфах она будет постепенно рассмотрена на примерах. При этом сформулируем также условия равновесия для некоторых случаев, которые при предыдущем выводе не учитывались, но которые существенны для обсуждения проблемы.
246
Глава VIII
§ 50. Растворы сильных электролитов
Введем сначала некоторые определения, повсеместно используемые в современной термодинамике электрохимических систем. При этом речь идет в основном о приложении определений § 34 к специальной проблеме растворов электролитов.
Химический потенциал растворителя (который обозначим индексом 1) равен
+ Р), (50.1)
причем
lim Д = 1, (50.2)
где — коэффициент активности растворителя в растворе и Pi0(T,P) — химический потенциал чистого растворителя в стандартном состоянии.
Для растворенных ионов уравнение (49.12) записывают в виде*
Кi = ЯПп Xifi + g ф + (7, Р), (50.3)
где
U° (Т, Р) = lim (|if - RT In хг) (50.4)
И
lim/f= 1. (50.5)
Выбор стандартных состояний и нормировка коэффициентов активности соответствуют случаю |3. § 346.
Рассмотрим теперь водные растворы электролитов и ", находящиеся в контакте и содержащие вид ионов i. Тогда в состоянии равновесия, согласно (49.19) и (50.3),
2.д((р"_ф') = РПп^ф. (50.6)
Xi ‘ i
* В экспериментальной электрохимии в качестве концентрации используют молярность (моль/л раствора) и моляльность (жмь/1000 г растворителя). Коэффициенты активности тогда определяют формально аналогично.
Электрохимические системы
247
Разность потенциалов ф"—ф',как было упомянуто в § 48, нельзя измерить. Коэффициенты активности также нельзя измерить ни порознь, ни в комбинации 1пД- —1пД. Поэтому уравнение (50.6) не представляет собой экспериментально проверяемого соотношения между измеряемыми величинами. Однако в принципе Д. можно рассчитать методами статистической термодинамики, и можно показать, что разность потенциалов ф"—ф' определяется уравнением (50.6) как величина, имеющая физический смысл. Правда, практически ситуация несколько иная, поскольку до сих пор точный расчет Д. удается провести только для предельного случая бесконечного разбавления. Для разбавленных растворов электролитов существуют приближенные формулы, при помощи которых можно примерно определить ф"—ф'. Для концентрированных растворов электролитов в настоящее время нужно ограничиваться утверждением, что ф"—ф', по крайней мере в принципе, является физически определяемой величиной. Аналогичные рассуждения справедливы в особенно важном случае, когда одна фаза является раствором электролита, а другая металлическим проводником. Тогда разность потенциалов называется потенциалом отдельного электрода. Этот вопрос будет рассмотрен в § 52.
Чтобы получить величины, подлежащие измерению, рассмотрим комбинацию различных видов ионов, удовлетворяющих условию нейтральности
2^ = о, (50-7)
где V. — целые числа. Соответствующая линейная комбинация электрохимических потенциалов, согласно (50.3), будет иметь вид
S = RT 2 In Xi 4- RT £ In fl + 2 W? (T, P).
(50.8)
Члены, содержащие потенциал ф, здесь выпадают вследствие (50.7). Комбинации электрохимических потенциалов типа стоящих в левой части уравнения (50.8), как будет показано, можно отнести к измеряемым величинам. Отсюда следует, что также комбинации ионных коэффициентов активности типа
248
Глава VIII
ПЛ
(50.9)
[где v. удовлетворяют уравнению (50.7)] экспериментально определимы и, таким образом, термодинамически определимы однозначно. На этой основе Льюис и Рендалл вводят средние коэффициенты активности электролита^ которые обозначим через f±. Ради простоты ограничимся бинарными электролитами, диссоциирующими на v_f. катионов валентности z+ и v_ анионов валентности г__. Для этого случая условие нейтральности (50.7) запишем:
М+ + v_z_ = 0. (50.10)
Средний коэффициент активности определяется тогда по уравнению
(бол)
где f+ и Д — коэффициенты активности катионов и соответственно анионов, а
v = v+-f-v_. (50.12)
Определение (50.11) является частным случаем (50.9). Поэтому средние коэффициенты активности определены термодинамически однозначно. Уравнение (50.11) приводит к примечательному следствию, что в растворе нескольких электролитов, которые имеют общими определенные виды ионов, средние коэффициенты активности электролитов зависят друг от друга. Если, например, имеются в растворе два одновалентных катиона А+ и В+, а также два одновалентных аниона С" и D" , то для средних коэффициентов активности из уравнения (50.11) получаем
/а с __ fb с /ad /в D
(50.13)
В этом смысле коэффициентам активности ионов можно приписать физическое значение также и в рамках термодинамики.
По аналогии с уравнением (50.11) определяют среднюю мольную долю электролита по уравнению
V v+4-v_ V+ v_
= х± = .
(50.14)
Электрохимические системы
249
При этом используют мольную долю электролита, которая определяется уравнением
х =-------, (50.15)
«1 + п+ + п_
где п2— число молей, рассчитанное из химической формулы электролита. При этом следует обратить внимание на то, что Но (при полной диссоциации) справедливы
простые соотношения
х+ = v+x; х_ = х. (50.16)
Из (50.14) и (50.16) следует
x±==v±x’ (50.17)
где
= (50.18)
Наконец, используют еще стехиометрическую мольную долю электролита
х2=—. (50.19)
Л1 + П2
Хотя здесь x1+x2=zl, но не справедливы соотношения (50.16).
Из уравнения (33.16) непосредственно следует, что в случае гомогенного равновесия диссоциации химический потенциал недиссоциированного электролита [i2 связан с электрохимическими потенциалами ионов уравнением
Р2 = V+H+ + М*- (50.20)
Правая часть этого уравнения является линейной комбинацией типа, появляющегося в уравнении (50.8). Поэтому можно определить эту линейную комбинацию также при полной диссоциации формально как химический потенциал электролита. То, что это определение имеет физический смысл, будет показано ниже. Соответственно запишем
Р° (Т, Р) = v+ IX» (Т, Р) + v_ (Т, Р). (50.20а)
250
Глава VIII
При помощи уравнений (50.11), (50.12), (50.14), (50.20) и (50.20а) для химического потенциала электролита из (50.8) получим
Н2 = ^71пх±/± + ^(7, Р). (50.21)
Используя (50.17) и (50.18), получим альтернативную форму
|Л2 = vRT In х f ± + нГ (Л Р), (50.22)
причем
(7, Р) + vRT In v± = pf (7, P). (50.23)
Для химического потенциала растворителя вместо коэффициента активности часто используют осмотический коэффициент g, введенный Бьеррумом*. Он определяется по уравнению
Pi = gRT In + Рю (7, Р) (50.24)
с нормировкой
limg= 1. (50.25)
-Ч-Я
Связь между осмотическим коэффициентом и коэффициентами активности ионов при постоянной температуре и постоянном давлении дается уравнением Бьеррума
(50.26)
где суммирование производится повеем видам ионов. Уравнение (50.26) вытекает из уравнения Гиббса—Дюгема (20.43) с учетом (50.3) и (50.24) и условия нейтральности (50.7).
Для объяснения приведенных определений рассмотрим два простых примера гетерогенных равновесий. Прежде всего рассмотрим распределение бинарного электролита между двумя несмешивающимися растворителями ' и ". Предположим, что для обеих фаз существуют одинаковая температура, одинаковое давление, полная диссоциация и электрическая нейтральность. Тогда нужно учитывать
* Термин «осмотический коэффициент» объясняется упомянутым в конце § 28 соотношением между —р.1О (Т,Р) и осмотическим давлением.
Электрохимические системы
251
только вариации чисел молей ионов и условие равновесия
(49.13) сводится к следующему выражению* **:
2 (и/ + Z; g ф') Вп/ + 2 (+ Zi S ф") = о (50.27)
/
с дополнительными условиями
Вп/ + 8п/ = 0 (все i) (50.28)
2 2i Ч = 0; S 2i = 0. (50.29)
Из (50.29) следует, что в уравнении (50.27) сокращаются члены, содержащие электрический потенциал <р. Далее, второе из условий (50.29) не является независимым. Поэтому в дальнейшем оно не будет учитываться .{Таким образом, для бинарного электролита получаем
Р-+ Ъп+ + Н- + и* %п+ + р-2 bnL = 0 (50.30)
с дополнительными условиями
^п'+ + оп+ — 0; + 8п2 — 0,
z. = 0-
+ + • ~
(50.31)
Используем вновь метод неопределенных множителей Лагранжа***. Умножим первое дополнительное условие (50.31) на второе на Х2, третье на Х3 и сложим эти уравнения с (50.30), тогда получим
(Р+ + Х3) Ъп+ + ( 4" 2- ^з) §п_ +
+ ( Рч + ^i) %п+ + (Р- + ^2) ~ (50.32)
Неизвестные сомножители Х2, Х3 выберем таким образом, чтобы выражения, стоящие в первых трех скобках, были равны нулю. Вариация является тогда независимой,
* Таким образом, фактически используем условие равновесия (23.11), сформулированное при помощи свободной энергии Гиббса.
** В дальнейшем индекс I относится только к ионам.
*** Объясним применение этого метода, неизбежного для сложных случаев, еще раз на двух простых примерах, хотя обычное исключение независимых переменных здесь скорее приводит к цели.
252
Глава VIII
и (50.32) может выполняться только тогда, когда четвертая скобка также равна нулю. Таким образом, имеем
IS 4- 4- z+ (50.33)
Р- + ^2 4~ “ 9, (50.34)
Рч 4~ = 0, (50.35)
Р- 4~ — 9. (50.36)
Умножим теперь уравнения (50.33) и (50.35) на v+, уравнения (50.34) и (50.36) на v_. Тогда сложение (50.33) и (50.34) вследствие условия (50.10) дает
v+ 4“ v- 4“ v+ 4~ v- \ = 0- (50.37)
Используя (50.35) и (50.36), наконец получим
v+p4 + У- V- = v+ р" + v- (50.38)
или, если ввести химический потенциал электролита, определяемый по уравнению (50.20),
Иг = Р-2 • (50.39)
Таким образом, равновесие распределения определено равенством химических потенциалов электролита. Если даже предположить, что фаза ' является очень разбавленным раствором, для которого коэффициенты активности ионов теоретически известны достаточно точно, то можно было бы определить экспериментально из равновесия распределения только средний коэффициент активности фазы" и то только с точностью до сомножителя, зависящего от Т и Р. Правда последнее ограничение для нашей постановки вопроса не имеет значения, так как оно основано лишь на различии стандартного значения для обеих фаз, появляющегося в уравнении (50.21). Это видно еще более отчетливо, если, используя (50.11), (50.14) и (50.21), написать уравнение (50.39) в виде
(<)’*( »5)-( 4)’ ’ I» '
Электрохимические системы
253
где
RT\nK(T, Р) = ^(Т9 Р)-р%(Т, Р). (50.41)
Величину К(Т, Р) можно определить экстраполяцией измерений на бесконечное разбавление. Случай, когда К(Т, Р)= = 1 и в результате этого отпадает упомянутое ограничение, будет описан в § 51.
В качестве второго примера рассмотрим равновесие жидкость — пар водного раствора летучего бинарного электролита. При этом предположим, что электролит в растворе, который обозначим через ', полностью диссоциирует и что в паровой фазе, которую обозначим через ", электролит существует только в виде нейтральных молекул. Эти предположения выполняются достаточно точно для разбавленных водных растворов НС1, НВг и HI. Как и в предыдущем примере, предположим, что в обеих фазах одинаковая температура и одинаковое общее давление. Электролитическую диссоциацию нужно теперь рассматривать как гетерогенную реакцию. Тогда условие равновесия (49.13) имеет вид
( Н+ + z+ S ф') Ъп'+ + ( р/ + § <р') 8nL + pj Ц +
-F Р-2 Вп2 + гЧ = ° (50.42)
с дополнительными условиями
= 0, (50.43)
8< + v+8«; = 0, (50.44)
8nl + v_8n; = O. (50.45)
Условия (50.44) и (50.45) полностью аналогичны условиям (33.9) для химических реакций. При помощи неопределенных множителей Лагранжа получаем
р; + к, = 0, (50.46)
р; + = о, (50.47)
+ 2+ S ф' -ма = °> (50.48)
pL + z_ g Ф' 4- Хз = 0, (50.49)
Р2 4~ k2 4~ v_k3 = 0. (50.50)
Из уравнений (50.46) и (50.47) следует
254
Глава VIII
= (50.51)
Если умножить (50.48) на v+, (50.49) на v_ и сложить оба уравнения, то вследствие (50.10) сократятся члены, содержащие электрический потенциал. Используя (50.50), получим
\.|4 + v-Ij'- = P'2- (50.52)
Так как раствор содержит только два независимых компонента, то равновесие полностью определено уравнениями (50.51) и (50.52). Линейная комбинация, стоящая в левой части уравнения (50.52), даже в случае полной диссоциации должна быть интерпретирована как химический потенциал электролита, что уже ранее было использовано.
Если паровую фазу с достаточной точностью можно рассматривать как идеальный газ, то из (50.51), (34.3) и (50.1) для парциального давления растворителя следует
Pi = PioXifi, (50.53)
где р10—давление пара чистого растворителя при той же температуре. Поэтому коэффициент активности растворителя в растворе электролита можно экспериментально определить, измеряя парциальное давление рг*. Вывод формулы для парциального давления электролита (вследствие появляющейся при этом проблемы нормировки) является несколько громоздким, и он здесь приведен не будет. Заметим лишь, что соотношение между парциальным давлением р2 и средним коэффициентом активности электролита в растворе содержит множитель, зависящий от температуры, который (как и в предыдущем примере) можно определить экстраполяцией измерений на бесконечное разбавление.
§51. Мембранные равновесия растворов
электролитов
Рассмотрим теперь равновесия между двумя водными растворами электролитов, разделенными между собой твердой полупроницаемой мембраной. Обе фазы обозначим вновь через ' и ". Ради простоты предположим, что фаза ' со дер-
Этот метод почти не имеет практического значения.
Электрохимические системы
255
жит только бинарный электролит, катионы и анионы которого могут проходить через мембрану. Кроме этого, пусть фаза 'содержит еще полиэлектролиты*, катионы которых идентичны катионам первого электролита. Для макромолекулярных анионов мембрана пусть будет непроницаемой. Тогда из общей теории мембранных равновесий (§ 28) и теории электрохимического равновесия (§ 49) получим условия равновесия
Г = 7", (51.1)
(все i),
(51.2)
причем индекс i относится теперь ко всем компонентам, которые могут проходить через мембрану. Условие (49.18) здесь выпадает, т. е. для существования равновесия равенство давлений для обеих фаз не является необходимым. Возможно ли различие давлений или это даже необходимо для равновесия, зависит лишь от того, выполняются ли при данных условиях уравнения (51.2).
а. Неосмотическое равновесие
Предположим, что мембрана является непроницаемой также для растворителя и что давления равны. В этом случае условия (51.2) примут вид
RT In %; f'+ + z+ g ф' = RT In x* Д + z+ g ф", (51.3)
ЯТ1ПХ1Д + ф' = WnZj:+ ?_йф". (51.4)
Члены для стандартного состояния в обеих фазах равны и сокращаются. Если исключить члены, содержащие электрические потенциалы, при помощи условия нейтральности (50.10), то, используя (50.11), получим
(xi)’+(Z)-(4)-+- -Е
(51.5)
* Полиэлектролитами называют макромолекулы с большим числом групп, которые электролитически диссоциируют в водном растворе. Многочисленные физиологически важные вещества (например, белки, нуклеиновые кислоты), а также некоторые синтетические вещества (например, полиакриловая кислота) являются полиэлектролитами.
256
Глава VIII
Уравнение (51.5) показывает, что при данных предположениях мембранное равновесие возможно при равенстве давлений в обеих фазах. Тогда говорят о неосмотическом мембранном равновесии. Согласно уравнению (51.5), оно полностью определено химическими потенциалами проникающих электролитов. Если известен средний коэффициент активности для фазы ', то для фазы " его можно определить из мембранного равновесия по условию (50.5).
Величина ср" — <рЛ, равная
ф — Ф •=—— 1п-4-£- = —— In-TT^-, (51.6)
2+& \f+ X_f_
называется мембранным потенциалом. Уравнение (51.6) является естественно частным случаем (50.6), и предыдущие выводы справедливы в основном также для данного случая. Поэтому сделаем еще лишь два дополняющих замечания. Прежде всего мембранный потенциал, хотя его непосредственно нельзя измерить, при подходящих условиях можно, по крайней мере приблизительно, определить экспериментально. С другой стороны, не существует пути, допускающего (хотя бы приблизительно) расчет коэффициентов активности фазы ", содержащей макромолекулярные анионы.
б. Осмотическое равновесие (равновесие Доннана)
Рассмотрим систему, такую же как и в пункте а., но теперь предположим, что мембрана проницаема не только для ионов бинарного электролита, но также и для растворителя. В этом случае для равновесия вообще является необходимым, чтобы обе фазы находились при различных давлениях*. Простые примеры таких равновесий были рассмотрены впервые Доннаном. Поэтому они обычно называются равновесием Доннана. Общая теория их была разработана Доннаном и Гуггенгеймом.
* Это сразу видно, если рассмотреть экстремальный случай, когда полиэлектролит имеет очень высокую, а проникающий электролит — очень низкую концентрацию. Количество последнего тогда недостаточно, чтобы понизить химический потенциал растворителя в фазе '[ср. уравнение (28.10)] настолько, чтобы он имел то же значение, что и в фазе ".
Электрохимические системы 257
Из § 28 и 49 получим условия равновесия
К (т, у, х\) = {л; (л р", xj), (51.7)
(Т, Р', х+, х) =^" (Т, Р", х’, xj , (51.8)
~^_(Т,Р', х'+,х_) = ^(Т, Р", х’+, х_у (51.9)
Предположим, что фаза ' находится при нормальном давлении Ро и напишем Р"=Р0+П. Тогда, согласно уравнению (26.27),
!Л1о (^> + Щ = |х10 (Т, Ро) + j vю dP =
р0
= Ро)+^1ОП, (51.10)
где г10 — среднее значение парциального мольного объема в интервале разности давлений П. Поэтому, учитывая (50.1), уравнение (51.7) можно записать
П = — 1п-Д4-. (51.11)
flo Xl f 1
Для проникающих ионов имеем
(Т, р0 + П) = (Т, Ро) + Р J+ п щ dP = £ (Т, р0) +
р.
+ »?П, (51.12)
где — парциальный мольный объем ионов вида i в стандартном состоянии, определяемом уравнением (50.4). Используя (50.3), уравнения (51.8) и (51.9) можно записать
ф" — Ч>' = (in (51.13)
z+& \ х+/+ RT )' V ’
Ф^_Ф'=_^_(1П±^_ + ЗЛ). (51.14) г_8 \ x-f- ' RT ) V
Если определить средний парциальный мольный объем электролита в стандартном состоянии по уравнению
258
Глава VIII
t»°_ = *+ »+ + > (51.15)
то из (51.13) и (51.14), используя (50.10) и (50.11), получим
п = __in
4 (^Г+(<)’-( 4)’++’-
(51.16)
Исключение П из уравнений (51.11) и (51.16) окончательно дает
((z)- (,у.+- _ ((£)•-(,у.+-
(W (W '(Ll7)
где _ _
r=4/v10. (51.18)
В данных уравнениях все коэффициенты активности фазы " определены для давления Р04-П. Проведем пересчет коэффициентов активности на нормальное давление на примере растворителя. Согласно (26.27), (50.1) и (51.10), имеем
^ = v1 = v10 + RT^^.. (51.19)
Интегрирование в пределах Ро и Ро4-П дает
RT In f ” (Ро + П) = RT In f ” (Ро) + fa - v10) П, (51.20)
где f { (Ро)— коэффициент активности, определенный для температуры и состава фазы ", но для давления Ро. Так как практически рассматривается очень небольшая разность давлений, то вторым членом правой части, а также зависимостью коэффициента активности от давления часто можно пренебречь.
Разность давления П, определяемая уравнением (51.11) или (51.16), как и в случае, обсужденном в § 28, называется осмотическим давлением. Соответственно равновесие Доннана в противоположность случаю, рассмотренному в пункте а., называют осмотическим мембранным равновесием. Разность потенциалов, определенная уравнением (51.13) или (51.14), как и для случая, рассмотренного в пункте а., называется мембранным потенциалом. Замечания,
Электрохимические системы
259
которые были сделаны в пункте а. о физическом значении этого понятия, естественно, справедливы также для осмотического равновесия.
§ 52. Гальванические элементы
Теперь нужно еще также обсудить вопрос, как связаны с измеряемыми величинами разности потенциалов соседних фаз ф"—ф', которые нельзя непосредственно измерить. Для этого рассмотрим закрытую систему с фиксированными рабочими координатами, состоящую из о фаз и отвечающую следующим условиям:
а. Все фазы являются электрическими проводниками.
б. Фазы, которые снабдим твердой нумерацией, расположены таким образом, что каждая из фаз с нумерацией от 2 до о—1 находится в контакте точно с двумя другими фазами*. Фаза 1 находится в контакте только с фазой 2, фаза о только с фазой о—1.
в. Конечные фазы 1 и о являются химически одинаковыми металлическими проводниками.
г. Между фазами 2 и о—1 находится по крайней мере один ионный проводник.
д. Каждая пара из граничащих между собой фаз имеет общим по крайней мере один электрически заряженный вид частиц (ионы или электроны), для которых граница раздела фаз является проницаемой.
Такая система называется гальваническим элементом. Она представляет собой в целом электрический проводник, через который, если цепь замкнута, может течь электрический ток от фазы 1 через фазу 2 до о—1 к фазе о. Так как ток в одной части фаз вызывается ионами, в других фазах электронами, то он обязательно связан с протеканием химических реакций.
Так как обе конечные фазы 1 и о химически идентичны, то разность потенциалов ф<а> — ф^> является измеримой величиной. В термодинамике рассматривается только равновесное значение этой величины. Поэтому ее надо определять в
* Не будем учитывать здесь контакты с непроводящими фазами (например, с паровой фазой), которые возможны, но несущественны для последующего рассмотрения.
260
Глава УШ
каждом случае в состоянии без тока, так как электрический ток всегда является необратимым процессом. Исключим также все прочие необратимые процессы из элемента. Это прежде всего значит, что химические реакции, протекающие в элементе, определенным образом должны быть связаны с электрическим током, так что при изменении направления тока на обратное все реакции протекают в обратном направлении. Далее исключены стационарные необратимые процессы (например, диффузия, теплопроводность, термодиффузия)*. Последнее требование выполняется не для всех гальванических элементов. Поэтому различают
а'. Обратимые гальванические элементы, у которых все процессы, протекающие в элементе, при перемене направления тока на противоположное также меняют свое направление.
б'. Необратимые гальванические элементы-, для них существенно наличие стационарных необратимых процессов, направление которых не зависит от направления тока.
Строгая теория необратимых гальванических элементов может быть развита только на основе термодинамики необратимых процессов, поэтому в дальнейшем ограничимся рассмотрением обратимых гальванических элементов.
Прежде всего следует более точно определить, что понимают под термодинамическим равновесием гальванического элемента. При этом существенным является то, что, согласно предположению, общая химическая реакция, протекающая в элементе, определенным образом связана с электрическим током. Поэтому различают следующие три случая:
а". Элемент является открытым, т. е. между конечными фазами 1 и о не существует непосредственно электропроводящего контакта. Поэтому электрический ток не может протекать и, следовательно, химическая реакция также не идет. Система может находиться в термодинамическом равновесии, но химическая реакция является лишь заторможенной. Поэтому она не происходит (так как невозможны соответствующие вариации) в условиях
* Нестационарные необратимые процессы не приводят к экспериментальным результатам, не зависящим от времени, и поэтому заранее должны быть исключены из рассмотрения.
Электрохимические системы
261
равновесия (ср. § 17). Соответственно при этих условиях разность потенциалов ф(а> — <р(1> является неизмеримой величиной.
б". Конечные фазы 1 и о накоротко замкнуты через проводник одинакового химического состава. В этом случае химическая реакция идет под действием электрического тока как необратимый процесс до тех пор, пока либо не установится химическое равновесие, либо не исчезнет один из участников реакции. Поэтому в начальном состоянии термодинамическое равновесие невозможно.
в". К конечным фазам 1 и о, за счет подключения потенциометра, приложена внешняя разность потенциалов Ф'. Тогда при |Ф'|>|ф(а)—(р(1)| электрический ток потечет в определенном направлении, причем будет идти химическая реакция. При |ФЛ| < |ф(а)—Ф(1)| изменяется направление тока и химическая реакция пойдет также в обратном направлении. Поэтому случай |Фх|= |ф<°>—ф(О| соответствует состоянию без тока. Оно представляет собой термодинамическое равновесие, так как в системе не могут протекать необратимые процессы. Химическая реакция, однако, в этом случае не заторможена, так как она может быть проведена в обоих направлениях за счет бесконечно малого изменения Ф' как квазистатический соответственно обратимый процесс.
Строгое определение термодинамического равновесия гальванического элемента, с одной стороны, очевидно должно теперь содержать в условиях равновесия химическую реакцию и разность потенциалов ф(а>—ф(1\с другой стороны, должно быть согласовано с общими определениями § 16 и 17. Случаи а". и б", поэтому исключаются и равновесие, описанное в в"., следует определить как термодинамическое равновесие гальванического элемента. Величина
|Ф'| = |ф(о) — <р(1)| = Ф (52.1)
называется электродвижущей силой (э. д. с.) гальванического элемента.
Сделаем еще два замечания к приведенному определению. Прежде всего легко заметить, что приведенные выше рассуждения полностью аналогичны термодинамическому рассмотрению газа, находящемуся в цилиндре с подвижным поршнем, двигающемся без трения. Если поршень
262
Глава VIII
арретирован, то происходит распространение заторможенного процесса и давление газа неизмеряемо (случай а".). Если поршень свободно подвижен и снаружи на него не действует давление, то происходит необратимое расширение газа (случай б".). Если на поршень снаружи действует давление, которое равно общему давлению газа в цилиндре, то имеем термодинамическое равновесие и равновесное давление является измеримой величиной. За счет бесконечно малых отклонений внешнего давления от давления газа можно провести изменение объема газа в обоих направлениях квазистатическим путем (случай в7'.). Существенное различие между обеими задачами заключается в том, что в случае газа объем является рабочей координатой в смысле § 14, в то время как в рассматриваемом здесь случае электрическая работа не может быть описана при помощи рабочих координат, потому что гальванический элемент определен как гетерогенная система, состоящая из открытых фаз.
Второе замечание относится к природе термодинамического равновесия гальванического элемента. При этом речь идет об электрохимическом равновесии, которое следует отличать от химического равновесия рассматриваемой реакции (если такое существует). Соответственно отличается также обратное протекание реакции, описанное в случае в"., от протекания реакции при сохранении химического равновесия, рассмотренного в § 14.
Вывод условия равновесия проведем двумя разными методами. Первый метод вводит только наблюдаемые величины и не имеет никакого отношения к деталям механизма. Так как по § 50 и 51 все равновесия веществ между соседними фазами определяются химическими потенциалами не-диссоциированного электролита и нейтральных молекул, то рассмотрим их как компоненты системы и напишем соответствующую общую реакцию (ср. § 33)
(52.2)
где Х}— электрически нейтральные химические соединения и vy—стехиометрические коэффициенты. Общая реакция, описываемая уравнением (52.2), не обязательно является
Электрохимические системы
263
химической реакцией в собственном смысле*. Так как появляется одна и только одна измеряемая разность потенциалов ф<а) — ср*1*, то дополнительно должен быть введен один электрически заряженный компонент. Его выбор устанавливается тем, что конечные фазы 1 и о считаем электронными проводниками. Число молей электронов в фазе а обозначим через Поэтому наряду с химическими потенциалами нейтральных компонентов в фундаментальном уравнении появляются электрохимические потенциалы электронов Для того чтобы сформулировать связь химической реакции с электрическим током, напишем для г компонентов, которые принимают участие в реакции, изменение чисел молей, согласно § 36, в виде
Sny = v;. 8£, (52.3)
где $ — число пробегов реакции. Для бесконечно малого тока в фазе о исчезают Ъпе молей электронов, которые появляются в фазе 1 (или наоборот). Тогда связь выражается уравнением
Sn<? = v*8£, (52.4)
где v*— коэффициент пропорциональности, зависящий от реакции, применяемой для осуществления элемента.
Предположим, что энтропию и объем каждой фазы можно варьировать**. О проницаемости границы раздела фаз не делаем никаких особых предположений, выходящих за пределы определения гальванического элемента (пункт д.). Однако далее будет показано, что, во всяком случае, не все пределы раздела фаз могут быть проницаемыми для всех компонентов. Поэтому суммирование по вариациям чисел молей следует понимать с данными ограничениями, что можно выразить через символ Е'. Наконец предположим также, что все компоненты, принимающие участие в реакции
* Реакция (52.2) может, например, заключаться в переходе компонента i из фазы' в фазу " с другим составом. Пример такого элемента приведем в конце параграфа.
** Предположение об объеме не является необходимым и сделано здесь только для упрощения. Существенным для последующих рассмотрений является лишь то, что обе конечные фазы 1 и о находятся при одинаковом давлении.
264
Глава VIII
(52.2), присутствуют в системе. Таким образом, имеем ус-
ловие равновесия (ср. § 33)
2 Тм 8S(a) — 2 P(a) 8V(a) -{- 2 a a a +^°’ 4<a>] = ° _z—1 (52.5)
с дополнительными условиями
У 8S<a) = 0. 28V(a) =0> a (52.6) (52.7)
S. ^ a) =0 для компонентов, которые не при-а нимают участие в реакции (52.2), (52.8)
== для г компонентов, которые при- а ’ нимают участие в реакции (52.2), g/7<a) =0 (а =2,3, ...,о — 1), (52.9) (52.10) (52.11)
Из (5215) — (52.8) получаем
уЧ₽) у^(ст) 9 (52.12)
=z Р^ Hz =Нг. 9 (52.13) (52.14)
причем ' и " означают каждую пару соседних фаз, общая граница раздела которых является проницаемой для компонента L Используя (52.9)—(52.11), уравнение (52.5) можно свести к условию
(52.15)
Так как, согласно предположению, I можно произвольно варьировать, то из (52.15) следует
Электрохимические системы
265
XX = (52.16)
а /=1
Так как обе конечные фазы 1 и о, согласно предположению, химически идентичны и находятся при одинаковых температуре и давлении, то их химические потенциалы также равны. Поэтому в соответствии с уравнением (49.12) имеем
^и-иГ’ = -8 б"’- •
Из (52.16) и (52.17) следует
v* $ (ф(1) — ф<0)) = X X V; p.ja). а /=1
(52.17)
(52.18)
Уравнение (52.18) представляет собой общее условие равновесия гальванического элемента, в котором при обратимом прохождении тока протекает гетерогенная обобщенная реакция (52.2). Если, как это часто бывает, каждый участник реакции (52.2) находится только в одной фазе, то (52.18) можно записать в виде
**$(ф(1)-ф(а))= X W (52.19)
/=1
Если ввести изменение свободной энергии AG*\ определяемое по уравнению (36.10), и э. д. с. элемента, определяемую уравнением (52.1), то из (52.18) получим
— v*g<D = AG. (52.20)
Электродвижущая сила, которая получается, если все участники реакции находятся в стандартных состояниях, называется стандартным значением электродвижущей силы для данного элемента. Если обозначить эту величину через Ф°, то из (34.36), (34.41), (34.43) и (52.18) получим
Ф = Ф°-----?^-1п ЦП«Р
'3' « /=1' 1 >
(52.21)
*) Отнесем здесь в соответствии с предыдущим определением AG к химической реакции. В литературе AG часто относят к эквивалентному превращению, что в нашей записи соответствует величине AG/v*.
10—235
266
Глава VIII
где
фо = _ AG° = — £ £ («). (52.22)
а /-1
Если все участники реакции находятся в чистых фазах, то тогда просто Ф=Ф°.
Приведенный метод является строгим и общим. Но он имеет тот недостаток, что с его помощью нельзя установить связь с материалом предыдущих параграфов и нет никаких соотношений между гальваническими элементами, которые имеют общими отдельные частные реакции. Его также не следует применять в теории необратимых элементов. Кратко приведем теперь второй метод вывода условий равновесия, который лишен этих недостатков. Принцип его состоит в том, что обобщенная реакция (52.2) разлагается в локализованные равновесия веществ между фазами, а также локализованные гомогенные химические равновесия внутри фаз. Такое разложение, однако, не вводит измеряемые величины и поэтому не всегда является свободным от произвола. Так как общая формулировка метода затруднительна, то объясним ее на простом примере. Итак, рассмотрим элемент
(1) (2) (3) (4) (5) (6) (7)
Pt|M(TB.) |МХ2(тв.) |KX(aq)|AgX(TB.)|Ag(TB.)| Pt, (52.23)
где X — галоген, который, как с серебром, так и с двухвалентным металлом М образует в воде труднорастворимую соль*. KX(aq)—водный раствор соответствующего галогенида калия. Предположим, что границы раздела фаз 1—2 и 6—7 проницаемы только для электронов, 3—4 и 4—5 только для ионов X", 2—3 только для ионов М2+ и 5—6 только для ионов Ag+. Тогда, согласно (49.12) и (49.19), для равновесий между веществами имеют место равенства
* Предположение, что соль гтруднорастворима, является необходимым, чтобы пренебречь диффузией и необратимыми химическими реакциями.
Электрохимические системы
267
25 = р-2. W -»*•>)-fg-rf». 8(ч>,п-т|в) = 1>™,
Для гомогенных равновесий внутри фаз, имеем ,.(2) 4- 9о.<2) — оЯ 1 ,.(3) л_ 9ti(3) — J3> „,(5) 4_ и.(5) _ „(5) !Ag+ + Р'х- — P'AgX ’ 0,(6) 1 J6) = о.(6) ^Ag+ Ag ' согласно
(52.24)
(52.25)
(33.16),
Если сложить уравнения (52.24) и ввести, используя (52.25), химические потенциалы нейтральных компонентов, то получим
(<р( * J) = Р'мхг + Ag 21хAgX . (52.26)
Обобщенная реакция, протекающая при прохождении тока в элементе, имеет вид
М + 2AgX -> МХ2 + 2Ag. (52.27)
Используя (36.10) и (52.1), получим снова уравнение (52.20) с v*=2.
Приведенный вывод прежде всего разъясняет, что разности потенциалов между соседними фазами, неоднократно упомянутые в предыдущих параграфах, измеримы не порознь, а только в комбинации в соответствующей гальванической цепи. В частности, это справедливо для упомянутых в § 50 потенциалов отдельных электродов, которые появляются в примере (52.23) в виде <р(3> — <р(4) и ф(4> —
10*
268
Глава VIII
—Ф<5). Для того чтобы можно было сравнить различные электродные потенциалы, комбинируют полуэлементы, соответствующие электродному потенциалу, с полуэлементами стандартного электродного потенциала в гальванические элементы, электродвижущую силу которых можно измерить. В качестве стандартного электрода используют платину, насыщенную водородом при 1 атм* ** *** и погруженную в раствор, содержащий ионы Н+ с активностью растворенного электролита, равной единице. Значения, измеренные таким образом, называют потенциалами отдельных электродов, или, точнее, стандартными значениями электродвижущей силы данного полуэлемента. Для всех важных полуэлементов они протабулированы. Разъясним использование их значений на частном случае (52.23), который представлен в виде элемента
Pt | Pb (тв.) | РЬС12 (тв.) | КС1 (aq) | AgCl(TB.) | Ag(TB.) | Pt. (I) Стандартные значения электродвижущей силы обоих полуэлементов определяются через элементы
Pt 1 Pt-H2 раствор, содержащий H+ раствор, содержащий С1" AgCl (тв.) | Ag (тв.) | Pt, (П)
Pt 1 Pt-H. 1 & раствор, содержащий Н+ раствор, содержащий ci- PbCl2(TB.)|Pb(TB.)|Pt. (III)
При этом следует предположить, что в элементах (II) и (III) оба соседних раствора имеют практически одинаковый состав**. Согласно последнему описанному методу, получаем***
* С теоретической точки зрения для определения стандартного электрода лучше было бы использовать водород с летучестью, равной единице.
** Поэтому элементы (II) и (III) пишут обычно только с раствором электролита. Приведенная выше форма записи Гуггенгейма теоретически более наглядна.
*** Так как здесь введены ионы, то индексы фаз не нужны.
Электрохимические системы
269
2 %Ф°п = - 2ц°н+ + 2nAgC1 - 2р.Ag - 2р°с1_ , (52.28)
“ ^н2 2р-н+ + P-jpbcia ^рь 2нС1- • (52.29)
Из (52.26), (52.28) и (52.29) следует
2Oj = 2Ф° — Ф°п , (52.30)
так как, согласно (52.26), электродвижущая сила цепи (I) определена только через чистые фазы. Таким образом, можно рассчитать электродвижущую силу всех элементов, составленных из полуэлементов, стандартные значения которых известны.
В некоторых случаях можно, по крайней мере приблизительно, экспериментально определить разность потенциалов между соседними фазами, если интересующая разность потенциалов велика по отношению к сумме всех остальных разностей потенциалов, встречающихся в гальваническом элементе. По этому принципу можно приблизительно определить мембранные потенциалы. На подробностях здесь нет возможности останавливаться.
Вывод уравнения (52.26) показывает, что хотя между двумя соседними фазами всегда имеется электрохимическое равновесие, но между двумя конечными фазами нет равновесия, так как Для открытых элементов
это объясняется тем, что одна или несколько границ раздела фаз не пропускают электронов. Но если разность потенциалов компенсируется внешней разностью по-
тенциалов Ф', введенной здесь в явном виде, то между фазами 1 и о также существует электрохимическое равновесие.
Определенные здесь локальные равновесия играют значительную роль в теории необратимых гальванических элементов. Подробности можно найти в учебниках по термодинамике необратимых процессов.
Рассмотрим теперь кратко некоторые термодинамические аспекты обратимых гальванических элементов. Если скомбинировать основное уравнение (52.20) с уравнением Гиббса—Гельмгольца (24.8), то для температурной зависимости электродвижущей силы получим соотношение, выведенное впервые Гельмгольцем:
270
Глава VIII
Электрохимические системы
271
Ф — Т | = —Д/7. (52.31)
L \ дТ ]р]
Далее, согласно (52.20) и (21.37), имеем
(52.32) I
Здесь А// и AS — изменения энтальпии и энтропии, которые, согласно (52.2), соответствуют уравнению химической реакции. Таким образом измерением электродвижущей силы и ее температурной зависимости можно определить величины AG, АЯ и AS для реакции (52.2). Так как все три величины являются функциями состояния, то их значения не зависят оттого, протекает ли реакция (при постоянной температуре и постоянном давлении) необратимо (случай б".) или обратимо (случай в".). Напротив, теплота, принятая системой (которая зависит от пути в пространстве состояния), при необратимом протекании равна АЯ, при обратимом процессе равна 7AS, в то время как в последнем случае, согласно (52.31), АЯ равна сумме подведенной теплоты и электрической работы, подведенной потенциометром к системе. Термодинамическое исследование гетерогенной реакции с помощью обратимых гальванических элементов играет также важную роль при экспериментальной проверке теплового закона Нернста (§ 38).
Измерение электродвижущей силы гальванических элементов часто используется для определения средних коэффициентов активности электролитов. Разъясним это на простом примере и рассмотрим элемент
Pt | М (тв.) | MCI (aq) | AgCl [ Ag | Pt, (52.33) где М — одновалентный металл, который образует растворимый в воде хлорид*. Суммарной реакцией этого элемента является
М (тв.) + AgCl (тв.) -> MCI (aq) + Ag (тв.). (52.34)
При этом v* = l. Так как каждый участник реакции находится только в одной фазе, то, согласно условию (52.21), для электродвижущей силы элемента можно написать
* Нужно предположить, что вследствие малой растворимости AgC. необратимая реакция Ag+-|-M->Ag-|-M+ практически не протекает
ф = ф» + _«L. In 1" > . (52.35;
5 аМС1 (aq) aAg (тв.)
Активности чистых твердых тел по определению равны единице (§34,6.). Поэтому, используя (50.22) (cv=2, v±=l), можно написать (52.35)
ф = ф’ - 4- ш омс, = ф» - In . (52.36)
U о
где х— мольная доля, рассчитанная по уравнению (50.15), и /± — средний коэффициент активности электролита MCI, определенный по уравнению (50.11). Если ввести теперь величину
ф* = ф+^211пх> (52.37)
8'
то, учитывая (52.36), получим
ф* = ф0 — in f . (52.38)
S
Так как правая часть уравнения (52.37) содержит только измеримые величины, то можно по экспериментальным данным представить Ф* как функцию х*/». Так как по определению lim/± = l, то по (52.38) ордината экстраполирован-х->0
ной кривой равна величине Ф°. Тогда из кривой, согласно (52.38), получают непосредственно /± как функцию Надежность экстраполяции можно существенно повысить, если использовать теоретическую формулу для /±, приведенную в § 50.
Наконец, измерение электродвижущей силы гальванических элементов является одним из немногих методов, при помощи которых можно определить активности и коэффициенты активностей в твердых растворах, в частности в бинарных сплавах*. В качестве примера рассмотрим элемент
* Другими методами являются исследование гетерогенных химических равновесий, рассмотренное в § 35, и измерение парциальных давлений. Однако последний метод применим только в^том случае, если в интересующей температурной области по крайней мере один компонент является достаточно летучим. При более высоких температурах это имеет место, например, для систем Ag—Zn и Си—Zn.
272
Глава VIII
Pt | Al (тв.) IA1C13- NaCl (распл.) | Al - Zn (tb.) | Pt, (52.39) где AlCl3-NaCl (распл.) —расплав солей, который служит электролитом, и Al-Zn (тв.) — твердый бинарный сплав. Общая реакция электролита в этом случае состоит просто в переносе одного моля А1 из чистой фазы в бинарный сплав, что символически можно записать в виде
(52.40)
Так как здесь v*~3, то из (52.19) для электродвижущей силы элемента получаем
3ft Ф = - (!ХА1 - l\i-zn ) = - Д^А1 = - 1п аА1 =
= -RTlnxMfAl. (52.41)
Таким образом, измерение электродвижущей силы дает непосредственно разность химических потенциалов А1 в сплаве и чистом металле или, что следует отсюда же, активность и коэффициенты активности А1 в сплаве.
Наряду с научным интересом гальванические элементы имеют чрезвычайно большое техническое значение. Они служат, с одной стороны, как источники тока (например, аккумуляторы), с другой стороны, для проведения химических реакций, которые осуществляются трудно или в других условиях вообще не осуществляются. Известными примерами таких процессов, которые технически проводят в большом масштабе, является электролиз хлоридов щелочных металлов, электролитическое производство алюминия и электролитическое осаждение металлов в виде поверхностных слоев (гальванические покрытия).
Глава IX
Поле тяготения.
Центробежное поле.
Определение молекулярного веса
§ 53. Системы в поле тяготения
В формулировке теории до сих пор не учитывалось, что почти все рассматриваемые здесь измерения проводятся в поле тяготения Земли. Остается еще исследовать вопрос, как изменяются условия равновесия при наличии поля тяготения. Для этого существенны следующие свойства поля тяготения*:
а. Сила тяжести по величине и направлению не зависит от времени и координаты.
б. Присутствие системы не изменяет поле тяготения.
в. Поле тяготения действует только на массу системы независимо от химического состава и термодинамического состояния.
Поэтому поле тяготения задается потенциалом
<|>(z) = gz, (53.1)
где g — ускорение свободного падения иг — высота над произвольно выбранным уровнем отсчета (например, лабораторным столом). Таким образом, эквипотенциальные поверхности поля тяготения параллельны поверхности отсчета.
Из (53.1) следует, что для системы, которая в вертикальном направлении распространена конечно, вклад, относящийся к полю тяготения, по отношению к общей энергии непрерывно изменяется с высотой над уровнем отсчета. Поэтому, если обозначить часть массы системы между двумя бесконечно малыми соседними эквипотенциальными
Отклонения от условий а. и б. лежат гораздо ниже величины ошибок термодинамических измерений.
274
Глава IX
поверхностями через dm, то общий энергетический вклад поля тяготения будет равен
= ^dm. (53.2)
Так как для системы из т компонентов общей массы т можно написать
т = f nh (53.3)
/=-1
где Mi— молекулярный вес* компонента f, то выполняется следующее соотношение:
dm = J Mi dnt, (53.4)
где — число молей компонента г, которое находится между двумя бесконечно малыми соседними эквипотенциальными поверхностями.
£ Так как вклад в общую энергию системы, обусловленный полем тяготения, является функцией пространственных координат, то поэтому все величины состояния должны быть рассмотрены как функции пространственных координат. Это приводит к следующим выводам:
а'. Определения гомогенного тела и фазы, данные в § 2, в строгом смысле не реализуемы.
в'. Для вариации энергии нужно учитывать вариацию ^пространственного распределения масс компонентов.
К пункту а'.
С этого момента гомогенным телом будем называть тело, негомогенность которого обусловлена исключительно полем тяготения. Тогда фаза определяется таким же образом, как в § 2. Эта терминология оправдывается тем, что обусловленные полем тяготения негомогенности, поскольку ими не полностью следует пренебрегать (как для твердых тел), являются непрерывными функциями пространственных координат, учет отличия которых от прерывных негомо-генностей, встречающихся на границах раздела фаз, в общем не представляет трудностей**. Она является целесо
* Более подробно о молекулярных весах и их определении см. § 55,
** Вблизи критической точки конденсации возникают трудности, которые здесь не могут быть рассмотрены.
Поле тяготения. Центробежное поле
275
образной, потому что терминология, использованная в предыдущих главах, может быть сохранена без изменения.
К пункту в'.
Так как при новом определении гомогенного тела больше не требуется постоянства величин состояния в пространстве, то для гомогенных тел без химических реакций условия равновесия должны быть выведены в явном виде. Обобщение для гетерогенных систем не представляет трудностей, но с точки зрения термодинамики мало интересно.
Поэтому на этом вопросе в дальнейшем останавливаться не будем.
Рассмотрим закрытую флюидную систему с фиксированными рабочими координатами, состоящую из m компонентов и являющуюся гомогенной в приведенном выше смысле. Другие внешние поля и химические реакции исключаем из рассмотрения. Если написать
и* = и + , (53.5)
где U — внутренняя энергия в отсутствие поля тяготения и определена по уравнению (53.2), то условие равновесия (17.2) (в случае знака равенства) будет иметь вид
(8£7*)5 = 0 (53.6)
или
+ 8 J 'jdm = 0 (53.7)
с дополнительными условиями
a J dS =0, (53.8)
8 j* == 0 (i=l,2.....tn). (53.9)
Единственные смещения, которые возможны при принятых предположениях, состоят в вариациях энтропии и чисел молей в каждом слое, ограниченном двумя бесконечно малыми соседними эквипотенциальными поверхностями. Поэтому, используя (20.5) и (53.4), условие равновесия можно записать
276
Глава IX
т
f T(2)8 (44 + V [Hi (z)8 (44dz +
J \ dz J J \ dz ]
+2 J ф <*) Mt8 (44dz=°- (53-10)
Z —1
Соответственно дополнительные условия имеют вид
[ 8 f J!L\dz = 0, (53.11)
J \ dz )
C 8 (dz = 0 (i = 1, 2........m). (53.12)
J \ dz )
Умножим (53.11) на неопределенный сомножитель Хо, (53.12) на Xz и вычтем затем эти уравнения из (53.10). Тогда получим
f [Т (г)-Х0] 8(44^2 +
J \ dz )
m
+ 2 f H) + W)
z=l
-М8(-
^Mdz = 0. (53.13) dz ) v
В этом уравнении вариации не подчиняются больше дополнительным условиям. Поэтому в общем случае они будут выполнены только тогда, когда выражения в квадратных скобках равны нулю. Таким образом, для всех z получаем
T(z) = k0 = 7\ (53.14)
(1г (2) + Mt ф (2) = Хг = |х* (i = 1, 2,..., т) (53.15)
ИЛИ
-^Т_ = 0, (53.16)
dz
d^
-Л- = 0 (i= 1,2,... ,m). (53.17)
dz
Уравнения (53.14) и (53.15), соответственно (53.16) и (53.17), представляют собой выраженные в явном виде условия равновесия для гомогенной флюидной системы без
Поле тяготения. Центробежное поле
277
химических реакций, находящейся в поле тяготения. Видно, что формальное влияние поля тяготения проявляется в том, что вместо химических потенциалов появляются величины p<z*, определяемые уравнением (53.15). Поэтому сразу можно ответить на вопрос, будет ли поле тяготения влиять на химическое равновесие. Согласно методу, описанному в § 33, в качестве условия химического равновесия получим 1>г!\ = 0, (53.18)
i
где суммирование, как и в § 33, проводят по всем участникам реакции. Но так как при химической реакции массы остаются постоянными, то имеем
Х>£Мг = 0. (53.19)
i
Вследствие (53.15) уравнение (53.18) сводится к условию 1>гН(г) = 0. (53.20)
i
Для гомогенных реакций в газовой фазе уравнение (53.20), согласно (34.14) и (34.23), можно записать следующим образом:
n[p*z(z)p=Kp(T). (53.21)
Так как правая часть этого уравнения зависит только от температуры, то, согласно (53.16), поле тяготения не влияет на константу равновесия То же самое справедливо для константы равновесия /Сс, определяемой уравнением (34.13). Напротив, константа равновесия а также константа равновесия для гомогенных реакций в жидкой фазе К(Т,Р) зависят от давления. Так как давление в направлении z не остается постоянным (что будет видно из дальнейшего), то это справедливо также для названных выше констант равновесия. Однако при обычных условиях в обоих случаях влиянием поля тяготения можно пренебречь.
Чтобы получить выражение для изменения давления в направлении z, исходим из уравнения Гиббса—Дюгема (20.43), которое вследствие (53.16) можно записать в виде
VdP = f п^. (53.22)
Z=1
278
Глава IX
Используя (53.15), получим т
= (53.23)
dz dz
г=1
или, используя (53.1) и (53.3), будем иметь
dP т dtp dtp /со п..
где р — средняя плотность системы. Уравнение (53.24) является общим условием гидростатического равновесия.
Чтобы исследовать изменение мольной доли в направлении г, исходим из соотношения*
d^t = — stdT +vtdP+ Xf—dx.. (53.25) \ dx/ t. p.xtJ, J /=2 J
Если для сокращения написать
Gi}=(-5М . (5з-26)
к дх} )t. p. xk+l
то из (53.16), (53.17) и (53.25) следует
m
dP . . dtp vi dxt
^-dT + M‘-dr+ = ° (1’=1>2.......m)- (53-27)
/==2
Из этих уравнений можно исключить давление при помощи уравнения (53.24). Но при этом нужно обратить внимание, что условия (53.24) и (53.27) не являются независимыми одно от другого. Если умножить уравнение (53.27) на nz, просуммировать по всем i и учесть соотношения, вытекающие из (26.10) и (26.16),
Е «i Vt = V, (53.28)
Z=1
£ fii Gtj = 0, (53.29)
___________ z==l
* Учитывая последующие применения, суммирование в уравнении (53.2 5) проведем от / =2 до т.
Поле тяготения. Центробежное поле 279
то получим уравнение (53.24). Поэтому, если использовать уравнение (53.24), то еще пг—1 уравнений (53.27) будут независимы одно от другого. Тогда исключение давления дает
пг
+ = (53.30)
/=2
Поэтому вместе с уравнением (53.24) имеем вновь систему из пг уравнений, которые называются уравнениями седиментационного равновесия.
Рассмотрим еще кратко применение общей теории к однокомпонентным системам. В этом случае равновесие полностью определено уравнением (53.24). Если обозначить через М молекулярный вес и через v мольный объем, то
р = (53.31)
Для идеального газа
Pv = RT. (53.32)
Подстановка (53.31) и (53.32) в (53.24) дает
— = _ (/ф =----gdz. (53.33)
Р RT ' RT ь v
Отсюда интегрированием в пределах от 0 до А получим барометрическую формулу
RT In (р(Л) / Р(о) ) = — (ф(Л) — ф<0)) = — Mgh. (53.34)
Для несжимаемой жидкости из уравнения (53.24) непосредственно следует
р(Л) _ р(0) = _ р (ф(Л) _ ф(0)) = _ ряА> (53.35)
Из уравнения (53.34) можно заключить, что для газов разность давления, обусловленная полем тяготения, а также, согласно (53.32), разность плотностей только тогда имеют значение, когда либо рассматриваются большие разности высот, либо сжимаемость существенно больше, чем у идеального газа. Последнее прежде всего имеет место вблизи критической точки конденсации (ср. § 47). Для жидких систем при рассматриваемых разностях давлений можно полностью пренебречь сжимаемостью и изменением плот
280
Глава IX
ности. Однако разность гидростатического давления, задаваемая уравнением (53.35), играет важную роль, например, при обычных методах измерения осмотического давления (ср. § 28).
В случае идеальной газовой смеси, как легко показать, для парциального давления каждого компонента справедливо соотношение, полностью аналогичное уравнению (53.34). Вследствие различия молекулярных весов более легкие газы накапливаются в верхних слоях атмосферы. В лаборатории этот эффект не играет вообще никакой роли, потому что поле тяготения слишком слабое.
§ 54. Системы в центробежном поле
Рассмотрим жидкую систему, состоящую из т компонентов, которая вращается в цилиндрическом сосуде вокруг оси цилиндра с постоянной угловой скоростью о). В противоположность силе тяжести интенсивность центростремительной силы*, действующей на единицу массы, не является постоянной по пространственной координате, а пропорциональна расстоянию от оси вращения г. Впрочем центробежное поле обладает свойствами поля тяготения а., б. и в., перечисленными в § 53. Поэтому если представить центробежное поле в виде потенциала
ф(г) =----i-<oar2, (54.1)
то общие выводы § 53 можно просто перенести на центробежное поле. В частности, поэтому уравнения (53.24) и (53.30) справедливы также для седиментационного равновесия в центробежном поле, если для ф подставить выражение (54.1). Эти уравнения определяют пространственное распределение компонентов как функцию расстояния от оси вращения г.
Объясним эти соотношения более подробно на примере бинарной системы. Так как теория практически применима только к разбавленным растворам, то растворитель можно рассматривать однозначно определенным. Обозначим его,
* В состоянии термодинамического равновесия сила Кориолиса равна нулю.
Поле тяготения. Центробежное поле
281
как обычно, индексом 1. Тогда уравнения седиментационного равновесия будут иметь вид
dP = $w?rdr, (54.2)
(ТИ2 — pv2) Ш2г
\ dx2 dx2 JT.P dr
(54.3)
Так как в соответствии с (41.35) частная производная в правой части всегда положительна, то знак градиента концентрации зависит только от выражения, стоящего в скобках в левой части. Знак можно легко определить, если ввести парциальные удельные объемы
(54.4)
Mi ’ М2
и плотности компонентов pi и р2- Тогда, используя теорему Эйлера (§ 19 б.), получим
(М2 - = М2 [ 1 - (Р1 + Р2) = Л42Р1 . (54.5)
Поэтому выражения в скобках положительны, если парциальный удельный объем растворителя больше, чем растворенного вещества, что осуществляется практически всегда. При таких условиях в состоянии равновесия концентрация растворенного вещества увеличивается с расстоянием от оси вращения. Градиент также растет с расстоянием от оси вращения и пропорционален квадрату угловой скорости.
Используя (34.36), уравнение (54.3) можно записать в виде
ЛПХ2 /пл \
—2 = (Л4а — pt»2)
д In М-i
д In х2)
(О2 г
~RT~ ’
причем коэффициент активности нормирован § 34,6. ₽. Так как
lim /2 = 1,
(54.6)
согласно
(54.7)
то уравнение (54.6) для граничного случая бесконечного разбавления можно сразу проинтегрировать. Тогда получают формулу, впервые выведенную Сведбергом:
282
Глава IX
ЯТЧп ±(54.8) \Х2 )
Таким образом, седиментационное равновесие растворенного вещества в центробежном поле при упомянутых условиях аналогично седиментационному равновесию идеального газа в поле тяготения, определяемому по уравнению (53.34).
Чрезвычайное значение центробежного поля для физики и физической химии основано на том факте, что в ультрацентрифугах, сконструированных впервые Сведбергом (1924), можно достигнуть ускорений примерно до 106 g. При этих условиях седиментационное равновесие, не имеющее значения в поле тяготения, используется для того, чтобы либо разделить компоненты смеси (препаративная ультрацентрифуга), либо по уравнению (54.8) определить молекулярный вес (аналитическая ультрацентрифуга). По экспериментальным причинам для последней цели используют почти исключительно измерение скорости седиментации. Теория этого последнего метода основана на термодинамике необратимых процессов. Поэтому не будем здесь останавливаться на подробностях и отошлем читателя к специальным учебникам.
§ 55. Определение молекулярного веса
В данной книге, как общепринято в современной термодинамике, в качестве единицы массы всегда использовали моль. При составлении или анализе системы массы компонентов обычно определяют взвешиванием, т. е. в граммах. Так как масса компонента mh измеренная в граммах, связана с числом молей по уравнению
mt — Mi щ
(55.1)
то приведенные формулировки подразумевают, что известны молекулярные веса всех компонентов.
Использование чисел молей дает большие преимущества, особенно при применении термодинамики к химическим и электрохимическим проблемам, а также для связи феноменологической и статистической термодинамики. Но оно имеет следующие недостатки:
а. В рамках термодинамики молекулярный вес невозможно определить. Он должен быть введен через дополнительные опытные факты или через статистическую термодинамику.
Поле тяготения. Центробежное поле
283
б. Молекулярный вес можно определить только для двух граничных состояний, а именно для бесконечно разбавленного однокомпонентного газа и (в виде молекулярного веса растворенного вещества) для бесконечно разбавленного бинарного раствора. Однако в общем случае можно предположить, что для соседних состояний (например, для умеренно сжатого реального газа) понятие молекулярного веса вполне сохраняет физический смысл. Для сравнительно удаленных состояний, особенно если они отделены от определяемого состояния одним или несколькими фазовыми переходами, молекулярный вес является условной расчетной величиной. Например, молекулярный вес NaCl в достаточно разбавленной паровой фазе можно определить, в то время как для кристалла NaCl молекулярный вес в принципе определить нельзя.
в. Многочисленные вещества (например, графит, алмаз, А12О3, силикаты, пространственно разветвленные полимеры) вообще нельзя перевести в состояние, в котором можно измерить молекулярный вес.
Из предыдущего следует, что для феноменологической термодинамики как с логической, так и с практической точки зрения массы компонентов являются естественными или первичными переменными состояния. Поэтому вопрос, как распределена размерность массы на оба фактора правой части уравнения (55.1), для термодинамики имеет подчиненное значение. Новые международные соглашения, которые сформулированы в рекомендациях Международного союза чистой и прикладной физики (ЮПАП)* и Международного союза чистой и прикладной химии (ЮПАК)**, вводят в единую систему в качестве новой основной величины количество вещества. Соответствующей основной единицей является моль, который определяется как количество вещества системы, которое состоит из стольких же молекул (или ионов, или атомов, или электронов, или других интересующих нас в конкретном случае частиц), сколько атомов содержится в точно 12 г чистого изотопа
* ШРАР Symbols, Units and Nomenklatura in Physics, 1965.
** IUPAC, Information Bulletin Number 24.
284
Глава IX
углерода 12С. Тогда из этого определения, согласно уравнению (55.1), следует, что молекулярный вес нужно определять как массу единицы количества вещества.
Вернемся теперь к вопросу, как вводится молекулярный вес в термодинамику, и обсудим затем важнейшие методы экспериментального определения молекулярного веса, поскольку они основаны на измерении термодинамических равновесий. Так как требуется объяснить только основные точки зрения, то в последующем изложении исключим из рассмотрения электролиты и химические реакции (особенно ассоциации и диссоциации).
В основе определения молекулярного веса лежат следующие уравнения статистической термодинамики, точно выполняемые при самых общих предположениях*:
Термическое уравнение состояния однокомпонентного газа
р = Л1_рГ14-В_Р_4_с(±-У + £)(-^-У + ...1, (55.2)
м L м \м) \.м] 1
где р означает плотность (г/см?).
Уравнение для осмотического давления бинарного рас-
твора
— ГТ Г I Сгг \2
(55.3)
где cg— концентрация в г!см? и индекс 2 относится к растворенному веществу**.
Поэтому термодинамическое определение молекулярного веса в газовой фазе будет
М = RT lim — р->о Р
(55.4)
* Непосредственное введение молекулярного веса из эксперимента является менее удовлетворительным, потому что оно не имеет широкой эмпирической основы, как основные уравнения статистической механики.
** Разложения (55.2) и (55.3) называются вириальными разложениями, коэффициенты — вириальными коэффициентами.
Поле тяготения. Центробежное поле
285
и для растворенного вещества бинарного раствора Л4а = ЯТИт-^- . (55.5)
11
Существование пределов доказывается в статистической термодинамике. Уравнения (55.4) и (55.5) показывают, что экспериментальное определение молекулярного веса требует в основном экстраполяции экспериментальных данных на нулевые плотности (соответственно бесконечное разбавление), и, таким образом, измерения нужно проводить при нескольких плотностях, соответственно концентрациях*. Для этого в основном используют следующие методы:
а. Измерение давления и плотности однокомпонентного газа Этот метод, для которого существуют различные формы выполнения, представляет собой непосредственное применение определения (55.4). Его применяют только в том случае, если данное вещество испаряется без разложения или давление насыщенного пара в экспериментально доступной температурной области не слишком низкое.
(3. Измерение осмотического давления бинарного раствора Этот метод представляет собой непосредственное применение уравнения (55.5). Для низкомолекулярных веществ он практически не имеет значения, так как здесь только в исключительных случаях удается подобрать подходящую полупроницаемую мембрану. Для макромолекулярных веществ различие в размерах между растворенными молекулами и молекулами (низкомолекулярного) растворителя настолько значительно, что упомянутые трудности отпадают. Поэтому в этой области измерение осмотического давления длительное время было одним из важнейших методов определения молекулярного веса**. Однако в последнее время он отступает на задний план по сравнению с дру
* Если возможны измерения при одной плотности, соответственно концентрации, при которых отклонения от граничных законов (55.4) и (55.5) еще не очень значительны, и не требуется высокая точность, то можно удовлетвориться измерением при одной плотности, соответственно концентрации.
** При определении молекулярного веса полиэлектролитов необходимо учитывать равновесие Доннана (§ 51).
286
Глава IX
гими методами, которые являются более точными и более простыми в исполнении. Практически важнейшие методы определения молекулярного веса в растворе основаны на том, что, согласно уравнению (28.15), осмотическое давление в основном является мерой свободной энергии разведения. Поэтому в принципе для определения молекулярного веса на основе уравнения (55.5) может быть использован каждый эффект, который определяется свободной энергией разведения или ее производной по концентрации.
у. Понижение давления пара растворителя
Предположим ради простоты выполнимость законов идеальных газов и обозначим через парциальное давление растворителя над раствором, через р10 парциальное давление чистого растворителя. Тогда при равновесии жидкость — пар из уравнения (27.8) в сочетании с (26.30) и (34.3) следует
= RT 1п-^. (55.6)
Рю
Из уравнений (28.15) и (55.6) получим
Р.+П _
f vtdP = vt П = RT In . (55.7)
p! Pi
Поэтому, используя (55.3), приходим к
-g-_exp[ —-+ В* + ...)] (55 8) Разложение экспоненциальной функции и переход к пределу cg->0 окончательно дают
.. Р10— Р1 1 /гг-
lim —-— = ---------. (55.9)
Cg~*Q Рю Cg
Так как на границе бесконечного разбавления сц-^V/ni и х2->п2/П1, то уравнение (55.9) может быть записано в более компактной форме
lim_Pio_-Pi. = 1. (55.10)
х2-*0 Рю ^2
Это уравнение называется законом Рауля. Таким образом,
Поле тяготения. Центробежное поле
287
молекулярный вес растворенного вещества можно определить из данных измерения понижения давления пара растворителя. Однако проще и точнее измерение повышения температуры кипения, которое будет обсуждено в следующем разделе.
Уравнение (55.9) в противоположность уравнению (55.5) содержит парциальный мольный объем и, таким образом, в неявном виде молекулярный вес растворителя, который, согласно предыдущим высказываниям, не является измеримой величиной. Однако вывод уравнения (55.9) показывает, что при использовании условия равновесия растворителю в жидкой фазе приписывают такой же молекулярный вес, как и в паровой фазе. Но последняя величина, согласно уравнению (55.4), является экспериментально доступной, чем обеспечивается однозначность молекулярного веса, определяемого по уравнению (55.9).
8. Повышение температуры кипения растворителя Предположим, что только растворитель имеет измеримое давление пара, и примем, что внешнее давление поддерживается постоянным (примерно около 1 агпм). Исследуем зависимость температуры кипения растворителя от концентрации раствора. Если обозначить через химический потенциал растворителя в паровой фазе*, то условие равновесия будет
^’ = Н- (55.11)
Если при постоянном давлении и при сохранении равновесия изменять температуру и состав, то должно быть
(55.12)
или, используя (26.27),
— dT = — SldT + dx2. (55.13)
дх2
Так как, согласно правилу фаз (29.3), система имеет только одну термодинамическую степень свободы, то изменением температуры однозначно определено изменение кон-нентоапии. Используя (26.28) и (55.11), уравнение (55.13)
* Ради простоты в дальнейшем будем писать величины, относящиеся к жидкой фазе, без верхнего индекса.
288
Глава IX
можно записать в виде
Л1 — h\v^
2-----l—d.T=-—-dx„,
Т дх2 2
(55.14)
или, если ввести мольную теплоту испарения
Lv = ft<0) — (55.15)
то уравнение (55.14) примет вид
----Ь-dT = ^-dx2. (55.16)
Это соотношение, которое является не чем иным, как дифференциальным уравнением кривой сосуществования в плоскости Т, х2, является точным, но для практических целей непригодным, так как величина Lv зависит от концентрации. Для того чтобы ее преобразовать, напишем теплоту испарения в виде
Lv=L°v+hi0-hi, (55.17)
где L°— мольная теплота испарения чистого растворителя. Таким образом, из (55.16) получим
_ dT _ dT = d (55 J 8)
Т Т дх2 2 v '
При постоянном давлении всегда выполняются соотношения
(d^p = - SidT + dx2, (55.19)
(^И'ю)р “ — sio dT. (55.20)
Из (55.18)—(55.20) в сочетании с (26.28) следует
Г о / А \
-----v-dT=d . (55.21) т2 \ т )р 4
Искомое повышение температуры кипения получается интегрированием этого уравнения от точки кипения чистого растворителя То до точки кипения раствора Т.
Для температурной зависимости теплоты испарения чистого растворителя имеем уравнение Кирхгоффа
Поле тяготения. Центробежное поле
289
__ __Q
dT р р'
(55.22)
Оно непосредственно следует из определений (21.26) и (31.7). Так как для обсуждаемой здесь проблемы рассматриваются только очень небольшие температурные интервалы, то температурной зависимостью правой части уравнения (55.22) можно пренебречь. Поэтому если обозначить через Lqv теплоту испарения чистого растворителя в точке кипения То, то после интегрирования уравнения (55.22) получим
L° = L°o+ (с£> - Ср) (Т - То). (55.23)
Если подставить это выражение в уравнение (55.21) и проинтегрировать от То до температуры кипения раствора Ть то получим
=L° (—--------+ — Ср) X
Л Оо\ Л То ) \ р р)
X (1п^+ 1 — (55.24)
Если ввести повышение температуры кипения
— TQ (55.25)
и разложить правую часть уравнения (55.24) по степеням кТ/Т^ то, ограничиваясь квадратичным членом, будем иметь
П
Лр,1
RTt
— lnxJi =
L° 4)v rt20
Д7 +
, f___ ^Qp ._____1 — Cp \ / ДУ \2
X 2 R ) ’
v55.26)
Уравнение (55.26) показывает связь повышения температуры кипения со свободной энергией разведения, осмотическим давлением и коэффициентами активности растворителя. Таким образом, последний может быть определен измерением повышения температуры кипения. Так как для расчета молекулярного веса растворенного вещества нужно экстраполировать на бесконечное разведение, то для этого
290
Глава IX
можно сразу пренебречь вторым членом правой части уравнения (55.26). Поэтому можно записать
Д7 =
Rtf
4,
RTt. ‘
(55.27)
Если теперь ввести определение (55.5), то получим
ДТ RT20 1 rt2 Р10
lim-------= —~--------------- lim-----= —тг-------------•
Cg-*° Cg ^0v Cg ^0v ^2
(55.28)
В правой части появился молекулярный вес растворителя. Поэтому принято вводить теплоту испарения, рассчитанную на единицу массы (удельную теплоту испарения)
(55.29)
г * _ Qv
Ov~ 9
и использовать в качестве переменных состояния величину
т* Юз-^-. (55.30)
Так как для то получаем lim * ш2~>0 Величина очень больших разведений MjV « у10 = "Ч _L. т2 1°3LOV ^-0 Cg 103^ М2 е v WL'to (55.31) (55.32) (55.33)
называется эбуллиоскопической константой^ для важней-
ших растворителей она протабулирована.
г. Понижение температуры замерзания растворителя
Если жидкий бинарный раствор находится в равновесии с твердой фазой чистого растворителя, то можно провести рассуждения, полностью аналогичные рассуждению предыдущего раздела. Поэтому приведем лишь конечный ре
Поле тяготения. Центробежное поле
291
зультат. Если обозначить через То температуру плавления чистого растворителя, через 7\ температуру плавления раствора и через
ДТ = Т0 —(55.34)
понижение температуры плавления, то получим
.. дт RT20 \
lim —г- = — — — т*-.о «2 103L0f М2
(55.35)
где Lof является удельной теплотой плавления чистого растворителя.
Величина
= М
f 103^
(55.36)
называется криоскопической константой. Она также прота-булирована для важнейших растворителей.
В заключение упомянем еще два метода определения молекулярного веса, которые также основаны на уравнении (55.5), но практически (так же как непосредственное измерение осмотического давления) применяются только для растворов макромолекулярных соединений. Первым из них является рассмотренное в §54 седиментационное равновесие в ультрацентрифуге. Этот метод, как было упомянуто, не имеет пока большого значения. Второй метод использует измерения рассеяния света растворами. Общие основы теории изложены в более подробных работах по статистической термодинамике, в то время как применение к растворам макромолекулярных соединений следует искать в специальной литературе.
ЛИТЕРАТУРА
Монографии и учебники по общим вопросам термодинамики
Гиббс Дж. В., Термодинамические работы, Гостехиздат, 1960.
Ван-дер-Ваалъс И., Констамм Ф., Курс термостатики, Изд. хим. литературы, 1936.
Schottky W., Ulich Н., Wagner С., Thermodynamik, Springer, Berlin, 1929.
Планк M.t Термодинамика, ГИЗ, 1925. (Planck М., Vorlesungen iiber Thermodinamik, 11 Aufl., Gruyter, Berlin, 1964.)
Льюис Дж., Рендалл M., Химическая термодинамика, ОНТИ, Химтеорет., 1936. (Lewis G. N., Randall M.t Thermodynamics, 2nd ed., McGraw-Hill, New York, 1961.)
Zemanski M. W., Heat and Thermodynamics, 4th ed., McGraw-Hill, New York, 1957.
Haase R., Thermodynamik der Mischphasen, Springer, Berlin, 1956.
Callen H., Thermodynamics, Wiley, New York, 1960.
Kirkwood J. G., Oppenheim I., Chemical Thermodynamics, McGraw-Hill, New York, 1961.
Гуггенгейм Э. А., Современная термодинамика, Госхимиздат, 1941.
Аксиоматика термодинамики
Land# A., Die Caratheodory’sche Axiomatik (Geiger-Scheel Hand-buch der Physik, Bd. IX), Springer, Berlin, 1926.
Falk G., Jung H., Axiomatik der Thermodynamik (Handbuch der Physik, Herausgeber S. Fliigge, Bd. Ill, 2), Springer, Berlin, 1959.
Landsberg P. T., Thermodynamics, Interscience-Wiley, New York, 1961.
Giles R., Mathematical Foundations of Thermodynamics, Pergamon, Oxford, 1964.
Buchdahl H. A., The Concepts of Classical Thermodynamics, Cambridge University Press, Cambridge, 1966.
Tisza L., Generalized Thermodynamics, M. I. T. Press, Cambridge (Mass.), 1966.
Гетерогенные равновесия
Bakhuis Roozeboom H. W., Die heterogenen Gleichgewichte, Vieweg, Braunschweig, 1901—1911.
Hansen M., The Constitution of Binary Alloys, 2nd ed., McGraw-Hill, New York, 1958.
Литература
293
Тепловой закон Нернста
Nernst W., Theoretische und experimentelle Grundlagen des neuen Warmesatzes, 2. Aufl., W. Knapp, Halle, 1924.
Bennewitz K-, Der Nernst’sche Warmesatz (Geiger-Scheel Handbuch der Physik, Bd. IX), Springer, Berlin, 1926.
Simon F.t Ergebnisse der exakten Naturwissenschaften, 9, 222 (1930).
Simon F., Science Museum Handbook, 3, 61 (1937).
Wilks J., Der dritte Hauptsatz der Thermodynamik, Vieweg, Braunschweig, 1963.
Электрохимические системы
Фалъкенгаген Г., Электролиты, Химтеорет., Л., 1935. (Falkenha-gen Н., Elektrolyte, Hirzel, Leipzig, 1953.)
Харнед Г., Оуэн Б., Физическая химия растворов электролитов, ИЛ, 1952. (Hamed Н. S., Owen В., The Physical Chemistry of Electrolytic Solutions, 3rd ed., Reinhold, New York, 1958).
Robinson R. A., Stokes R. H., Electrolyte Solutions, 2nd ed., Butterworth, London, 1959.
Kortum G., Lehrbuch der Elektrochemie, 2. Aufl., Verlag Chemie, Weinheim, 1957.
Статистическая термодинамика
Фаулер P., Гуггенгейм Э., Статистическая термодинамика, ИЛ, 1949.
Munster A., Statistische Thermodynamik, Springer, Berlin, 1956.
Справочники
Rossini F., Selected Values of Chemical Thermodynamic Properties, Government Printing Office, Washington, 1952.
Kelley К. K., Entropies of Inorganic Substances, Bur. of Mines Bull., 477 (1950).
Kelley К. K-, High Temperature Heat Content, Heat Capacity and Entropy Data, Bur. of Mines Bull., 476 (1949).
Landolt-Bornstein, Zahlenwerte und Funktionen, 6. Aufl., Bd. II, 2—4 Teil, Springer, Berlin, 1956—1964.
Глушко В. П. и др., Термические константы веществ, 10 вып., ВИНИТИ АН СССР, вып. I—V, 1965—1971.
Гурвич Л. В., Хачкурузов Г. А. и др., Термодинамические свойства индивидуальных веществ, т. I, II., изд. АН СССР, 1962.
Математическое пособие
Margenau Н., Murphy G. М., The Mathematics of Physics and Chemistry, 2nd ed., Van Nostrand, New York, 1956—1964.
СОДЕРЖАНИЕ
Предисловие 5
Предисловие автора ..................................... 6
Список основных обозначений ................... 8
§ 1. Введение .......................................... 9
Глава 1. Основные законы термодинамики ... 13
§ 2. Определения ...................................... 13
А. Классическая формулировка основных законов термодинамики............................................... 17
§ 3. Первый закон. Внутренняя энергия ............... 17
§ 4. Второй закон термодинамики. Энтропия и абсолютная температура ............................... 18
§ 5. Холодильные машины и тепловые насосы ... 28
Б. Аксиоматика Каратеодори . . 31
§ 6. Определения .............................. 31
§ 7. Эмпирическая температура 33
§ 8. Первый закон термодинамики ............. 36
§ 9. Дифференциальные выражения Пфаффа ... 39
§ 10. Второй закон термодинамики для квазистатических процессов ........................... 46
§11. Эмпирическое определение £7, S и Т ..... 51
§ 12. Измерение предельно низких температур ... 55
§ 13. Второй закон для нестатических процессов . . 58
В. Распространение второго закона термодинамики на открытые системы и химические реакции................ 64
§ 14. Постановка проблемы ............................. 64
§ 15. Общая формулировка второго закона термодинамики 67
Глава II. Общие условия равновесия и стабильности 72
§ 16. Обсуждение понятия равновесия. Внутренние параметры 72
§ 17. Условия равновесия Гиббса ...................... 76
§18. Условия стабильности 82
Глава III. Термодинамические потенциалы и функции Массье—Планка ...................... .... 85
§ 19. Преобразование Лежандра. Гомогенные функции и
теорема Эйлера .................................. 85
§ 20. Фундаментальное уравнение. Экстенсивные и интенсивные параметры. Уравнения состояния. Уравнение Гиббса—Дюгема 90
§ 21. Термодинамические потенциалы 99
§ 22. Функции Массье—Планка 109
§ 23. Преобразование условий равновесия и стабильности 112
§ 24. Уравнения Гиббса—Гельмгольца и соотношения Максвелла ₽ . ,............................................118
Содержание 295
§ 25. Пересчет частных производных. Метод определителей
Якоби Эффект Джоуля—Томсона .... 122
§ 26. Средние мольные и парциальные мольные величины 131
Глава IV. Гетерогенные равновесия без химических реакций 139
§ 27. Общие условия равновесия гетерогенных систем 139 § 28. Мембранные равновесия. Осмотическое давление 142
§ 29. Правило фаз 144
§ 30. Фазовые реакции ..............146
§ 31. Нонвариантные и моновариантные равновесия ... 151
§ 32. Би- и поливариантные равновесия .... 156
Глава V. Химические равновесия ...................161
§ 33. Общие условия равновесия .......................161
§ 34. Гомогенные реакции. Закон действия масс . . . 165
§ 35. Гетерогенные реакции ...................172
§ 36. Число пробегов реакции. Сродство ...............175
§ 37. Термодинамический расчет химических реакций . 177
§ 38. Тепловой закон Нернста. Недостижимость абсолютного
нуля температуры. Энтропия при абсолютном нуле 180
Глава VI. Условия стабильности....................198
§ 39. Постановка проблемы. Критерий Гиббса . . . 198
§ 40. Условия стабильности в энергетическом выражении 202 § 41. Преобразование условий стабильности . . . 207
§ 42. Условия стабильности для гетерогенных систем 214 § 43. Принцип Ле Шателье—Брауна ...........215
§ 44. Стабильность химического равновесия .... 219
Глава VII. Критические фазы ...........221
§ 45. Определение и свойства критических фаз . . . 221 § 46. Уравнения Гиббса для критических фаз . . . 229
§ 47. Преобразование уравнений для критических фаз. Дальнейшие свойства критических фаз ......................234
Глава VIII. Электрохимические системы . . 239
§ 48. Определение и общие свойства электрохимических систем ...............................239
§ 49. Общие условия электрохимического равновесия . . 242
§ 50. Растворы сильных электролитов ..................246
§ 51. Мембранные равновесия растворов электролитов. . 254
§ 52. Гальванические элементы ................259
Глава IX. Поле тяготения. Центробежное поле. Определение молекулярного веса .......................273
§ 53. Системы в поле тяготения .......................273
§ 54. Системы в центробежном поле ....................280
§ 55. Определение молекулярного веса . . . . 282
Литература . . ... .... . 292