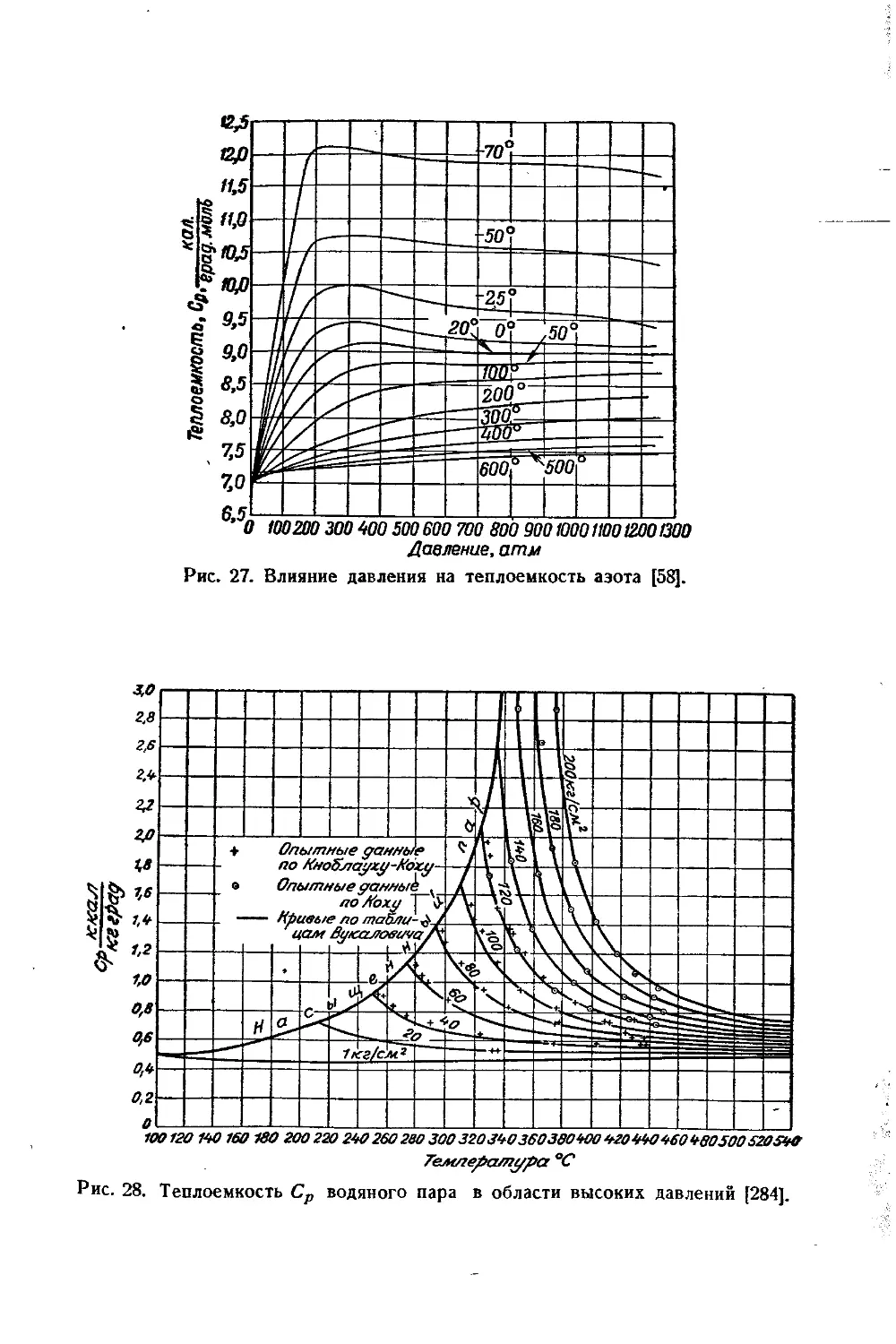
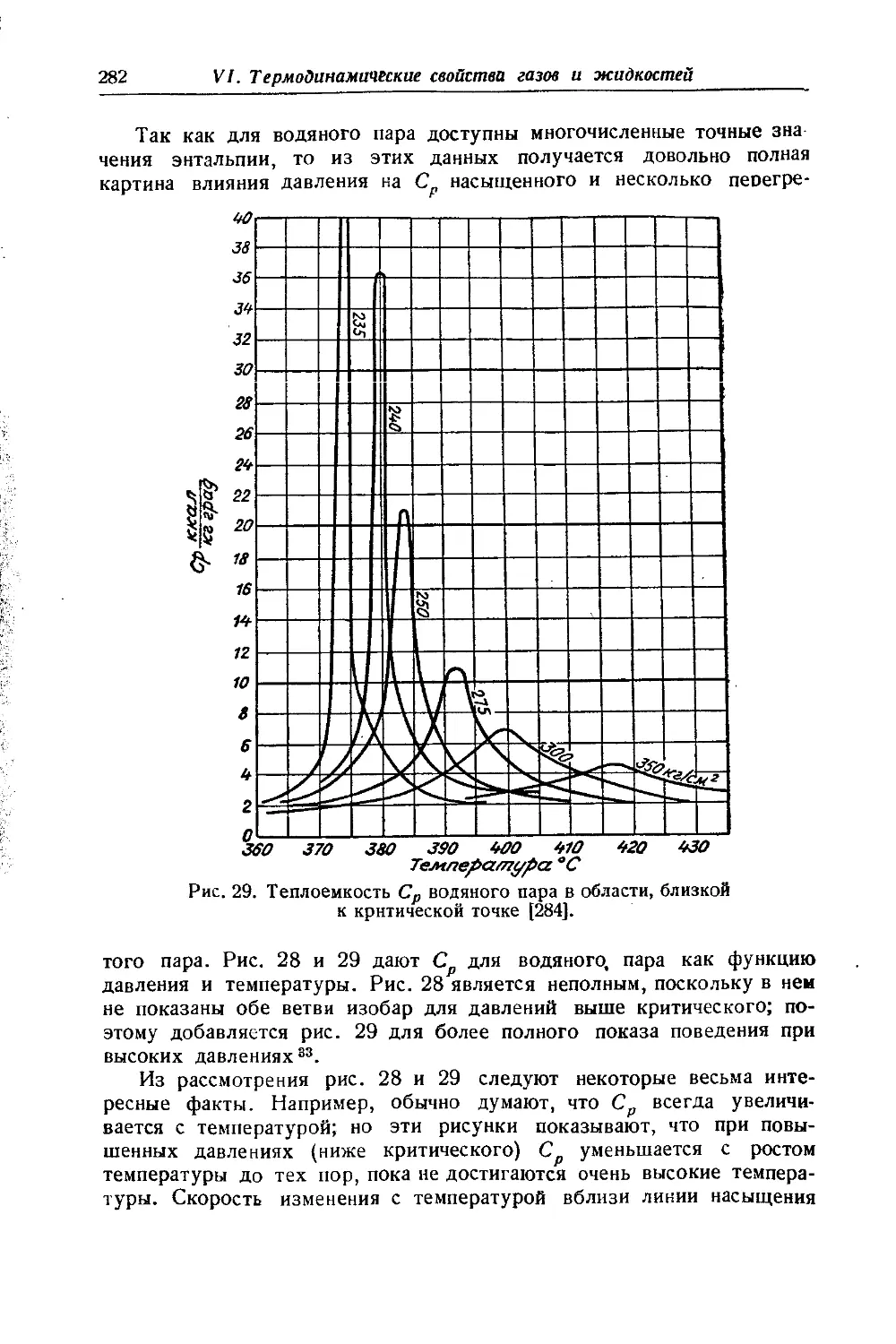
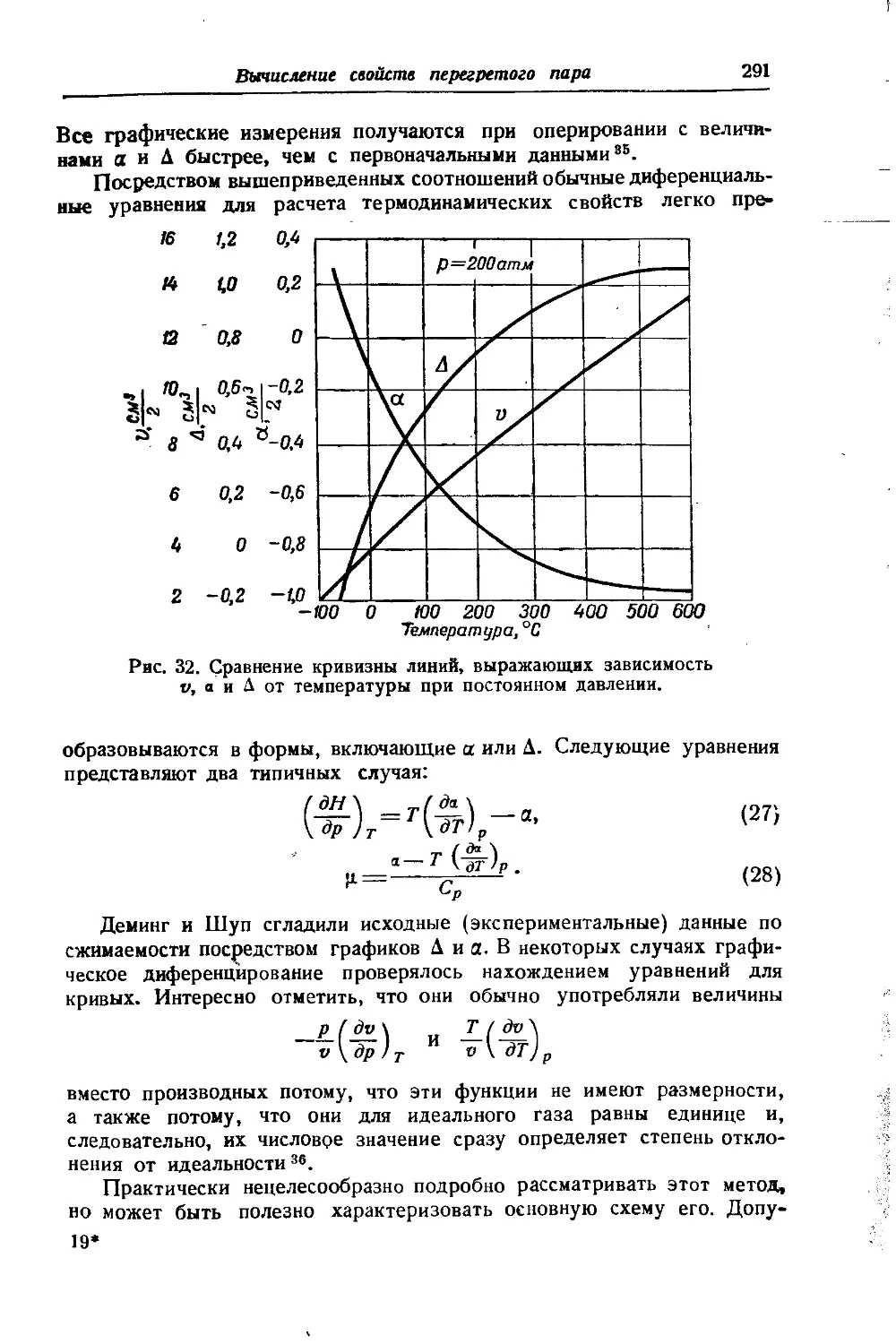
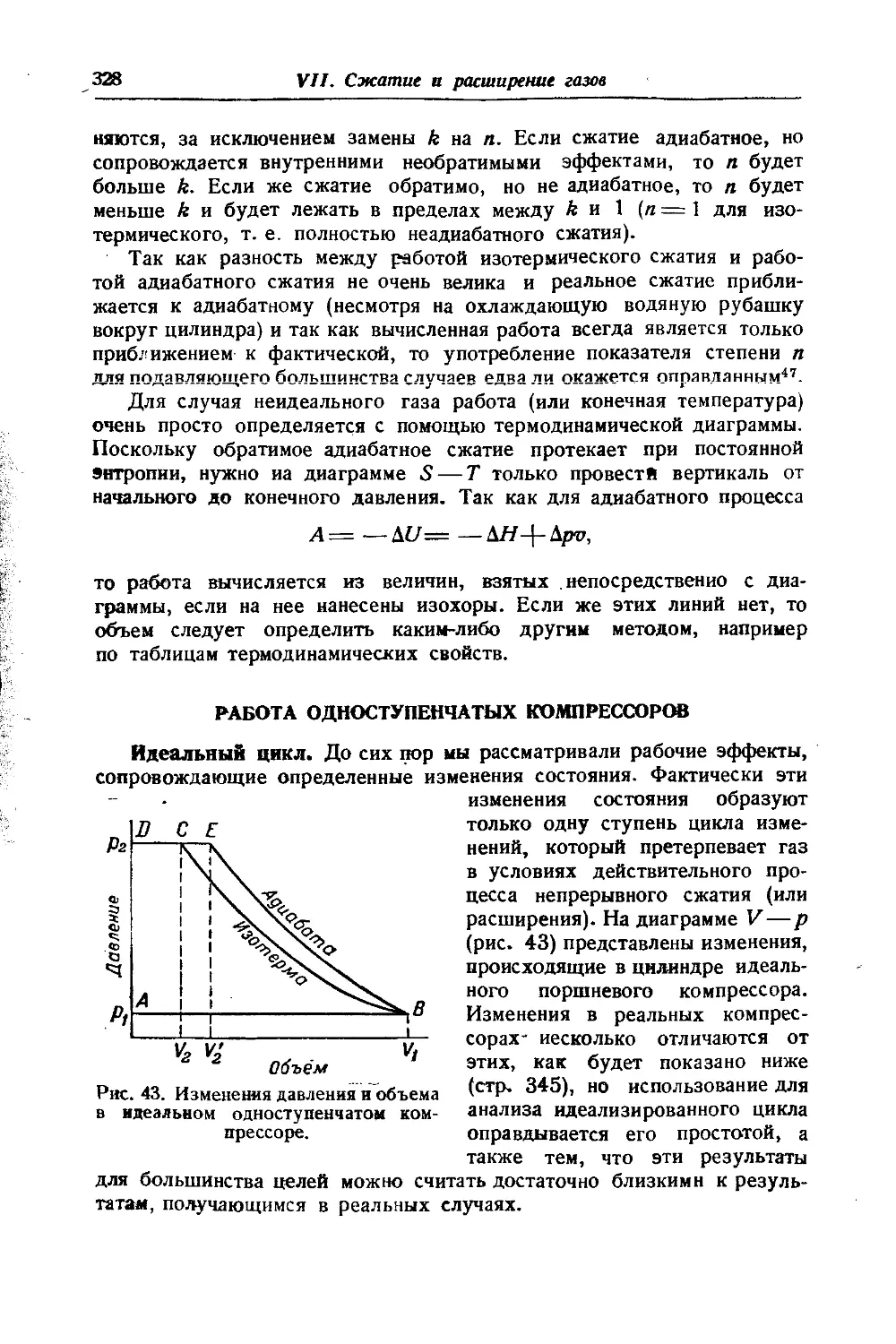
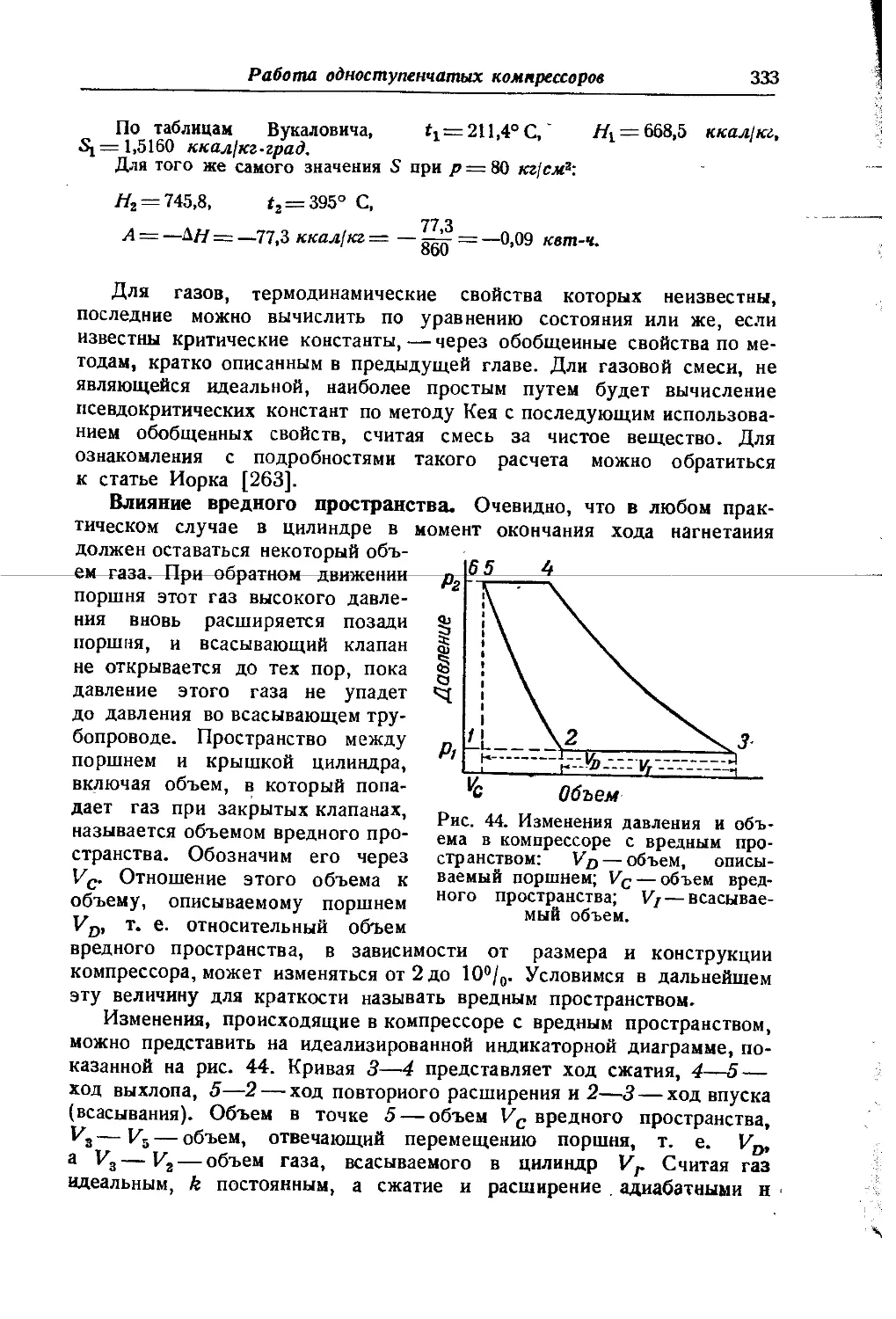
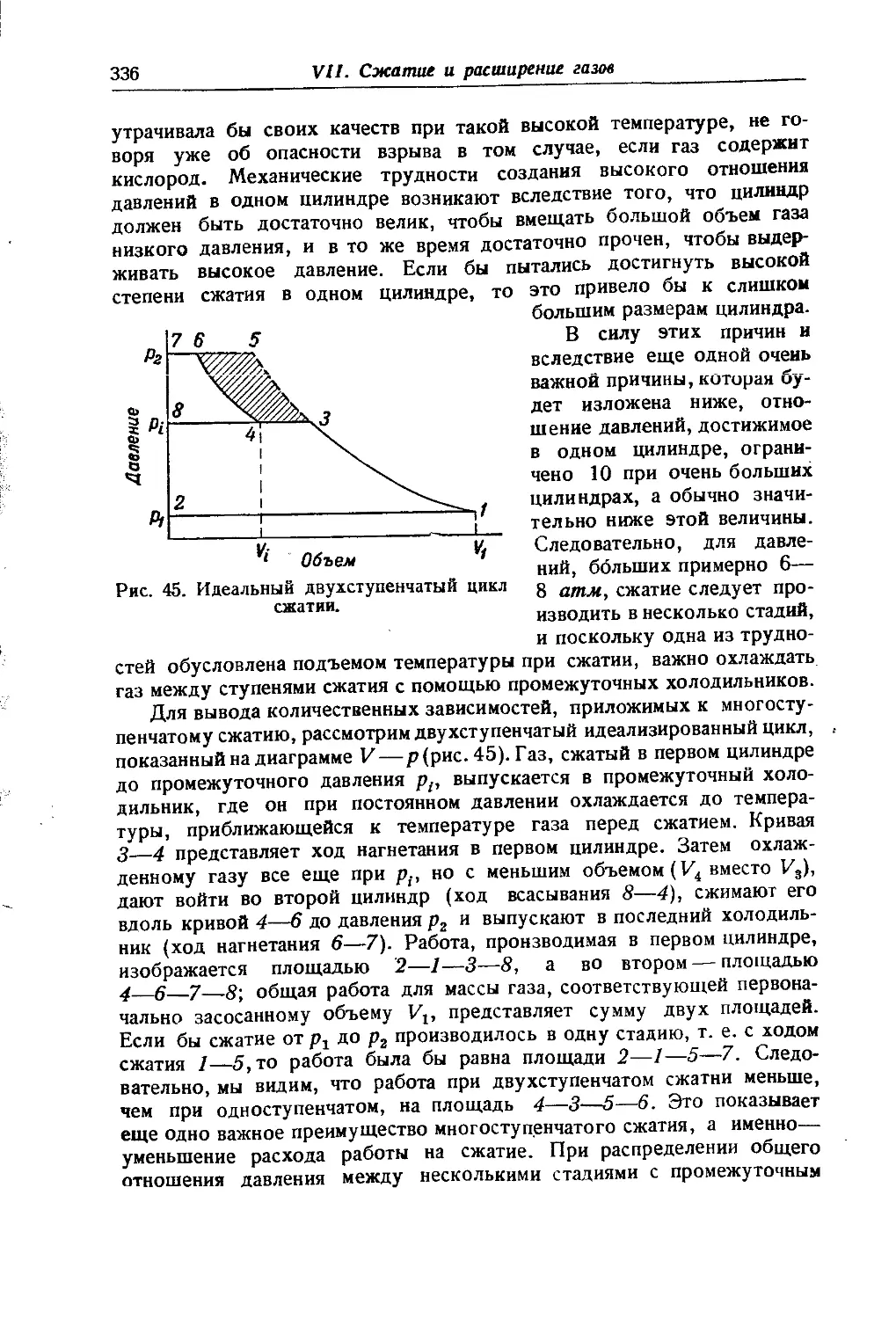
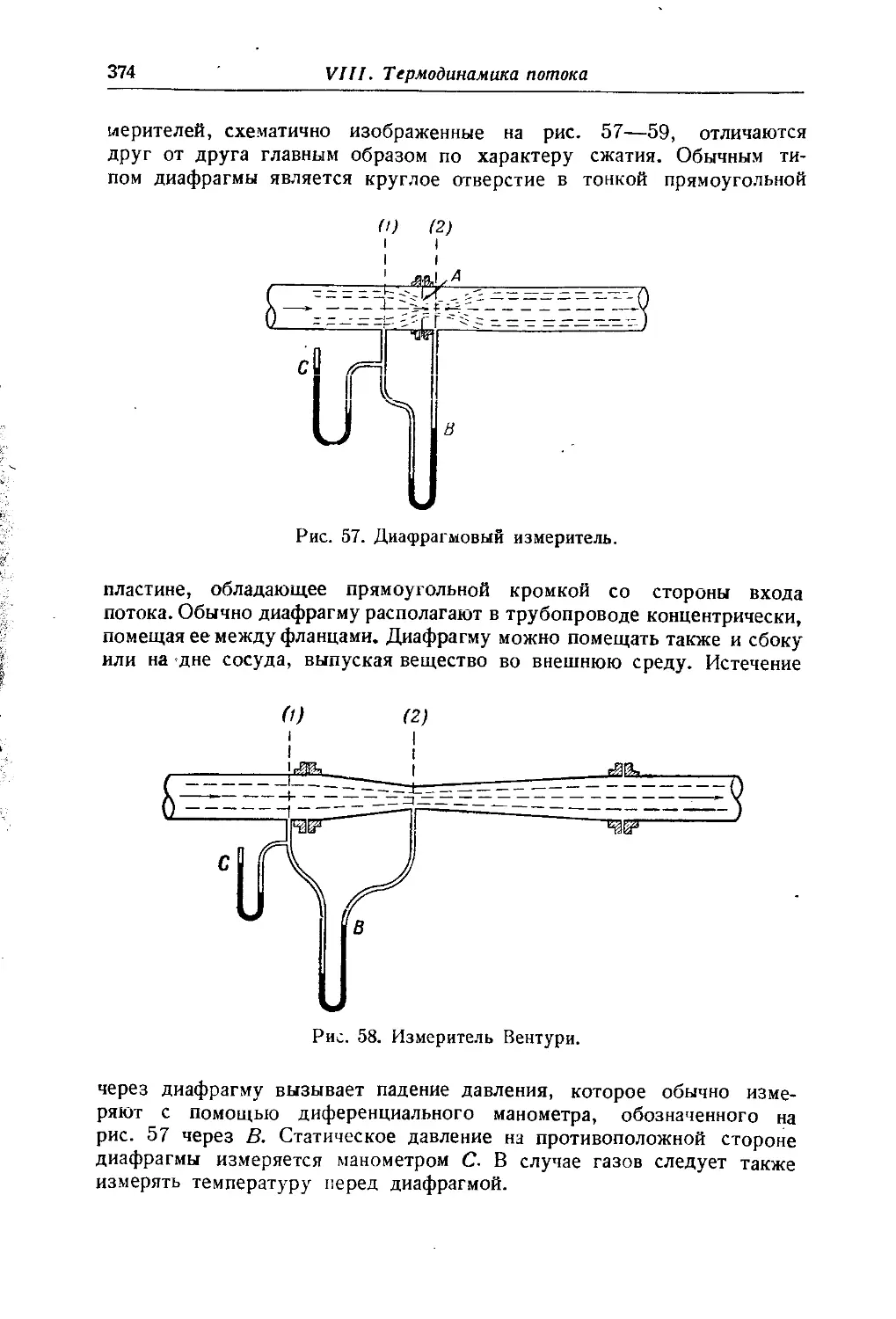
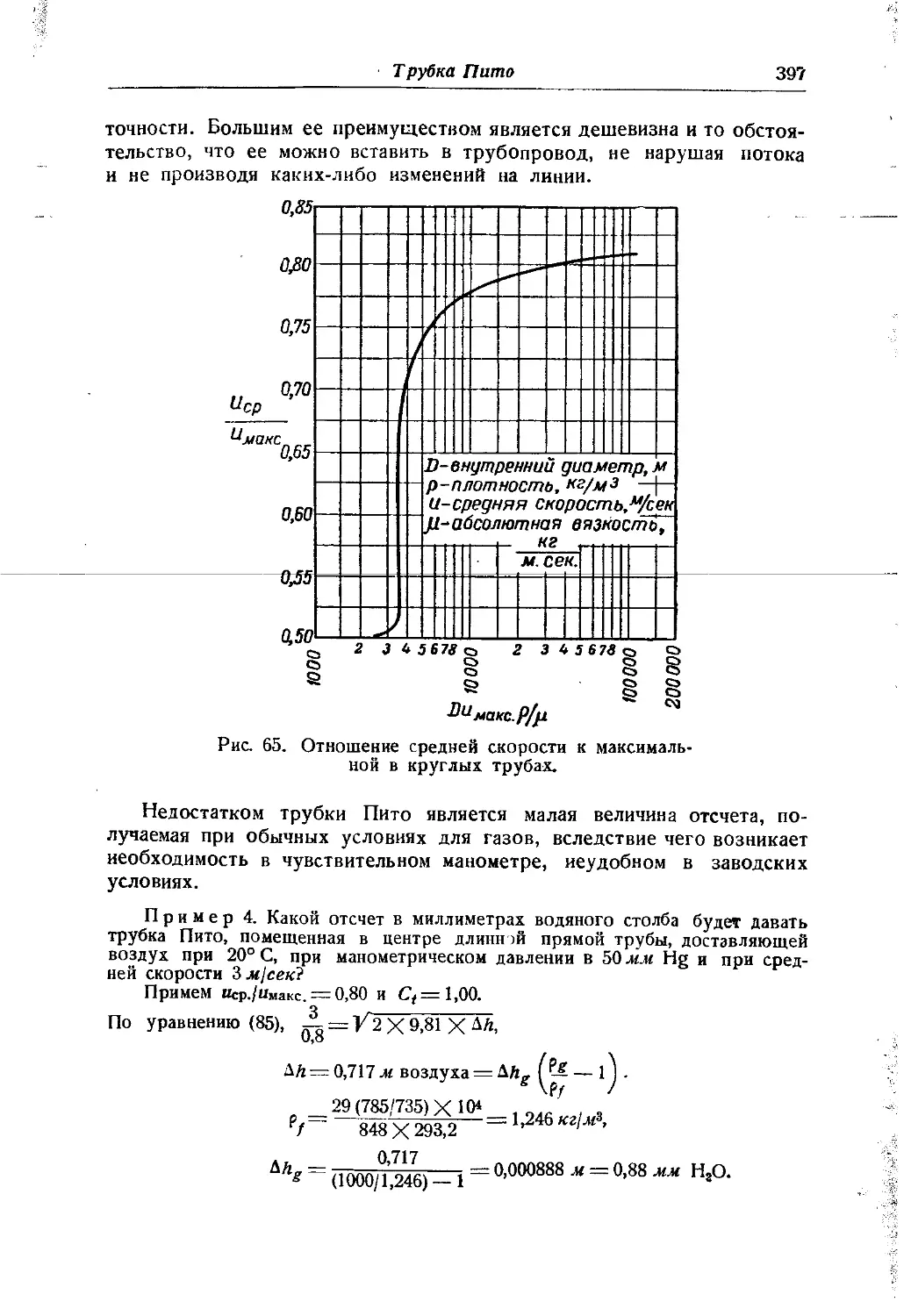
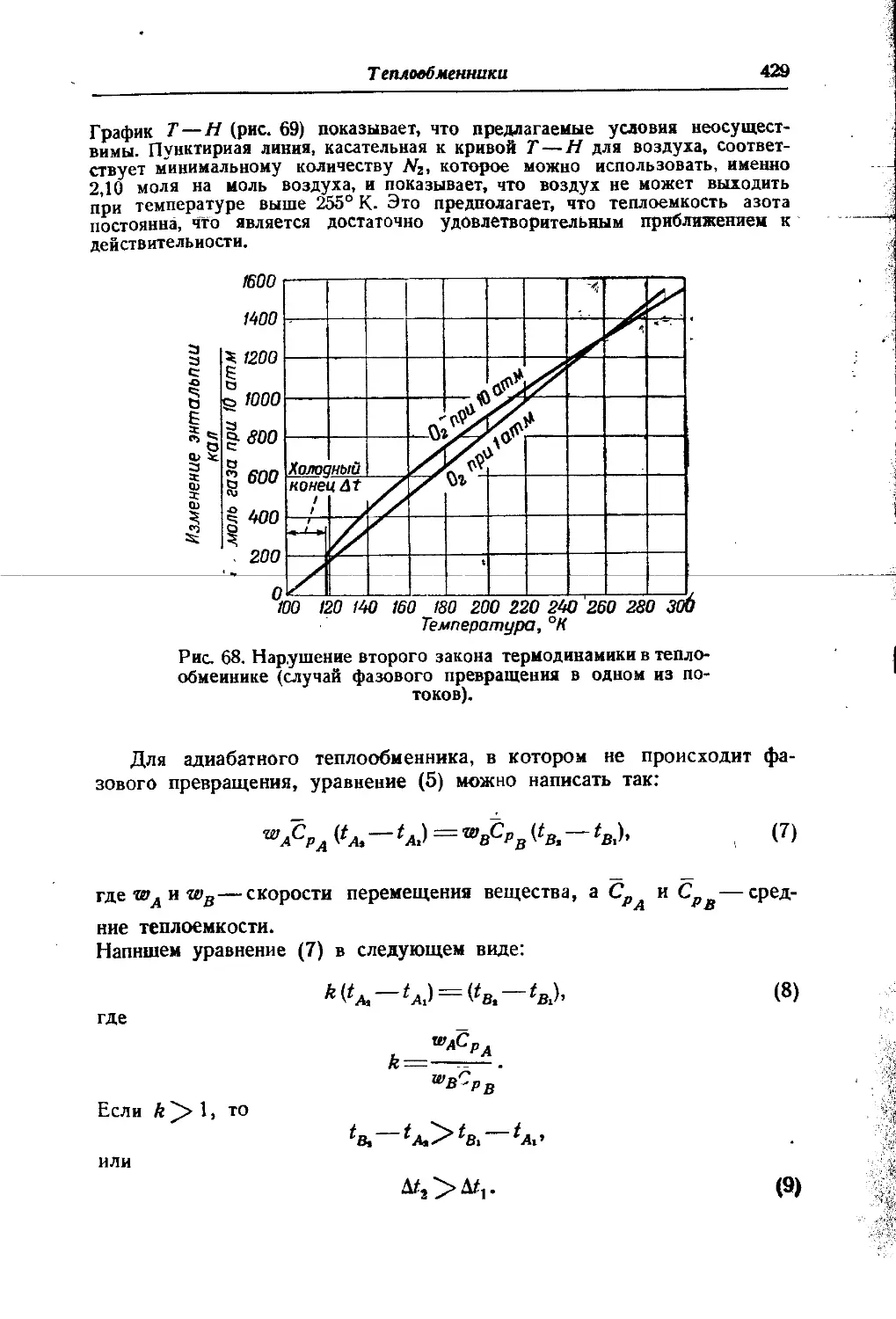
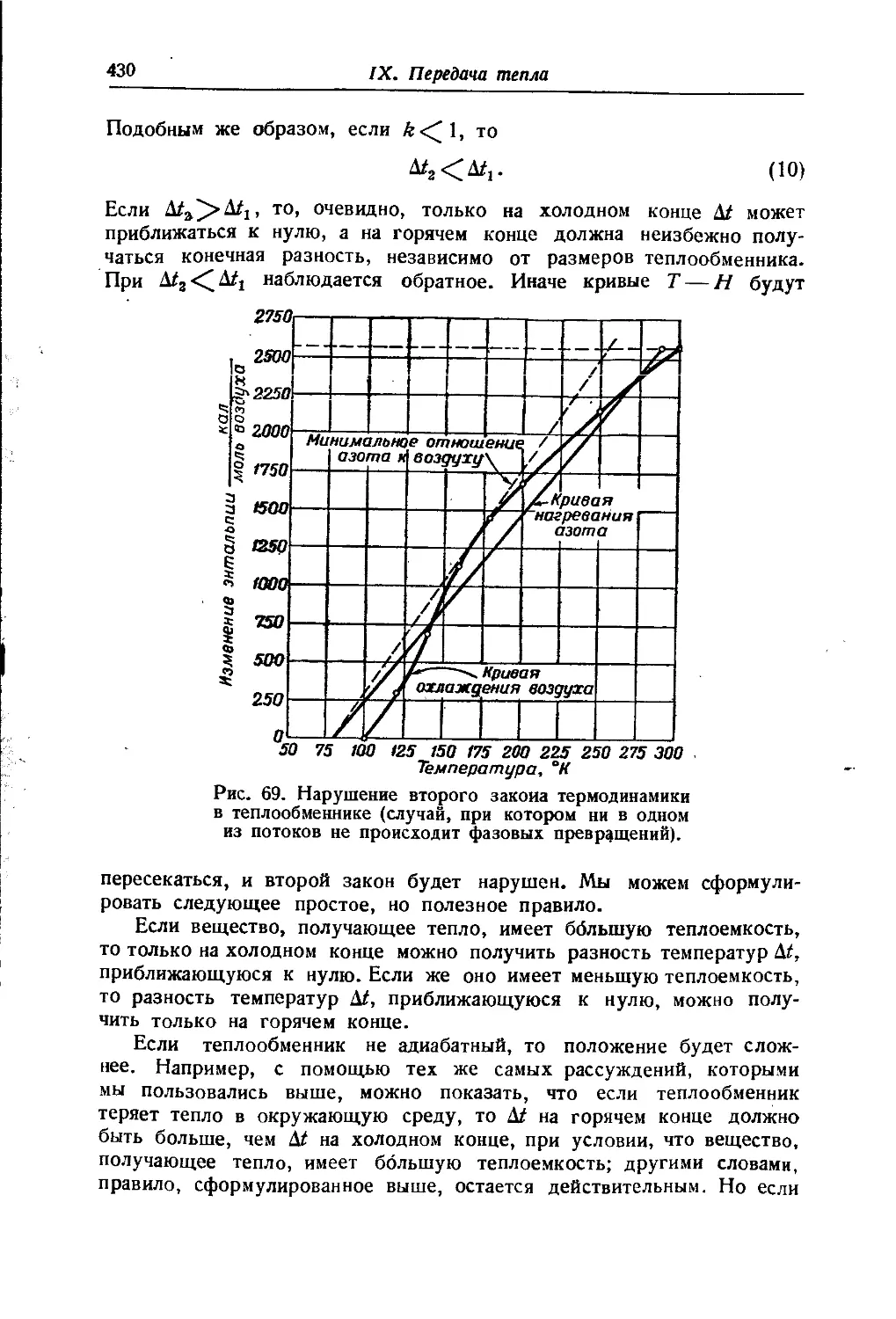
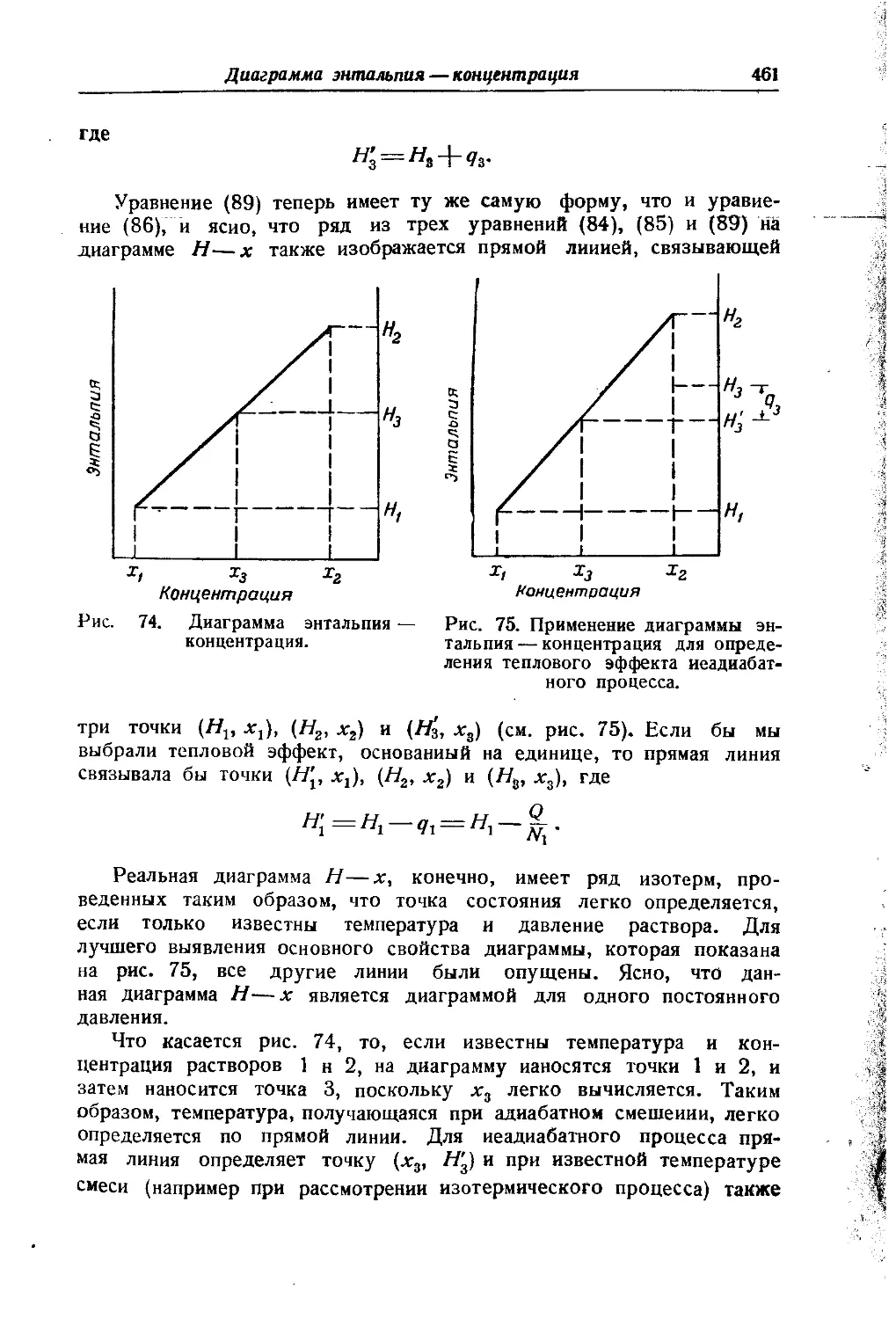
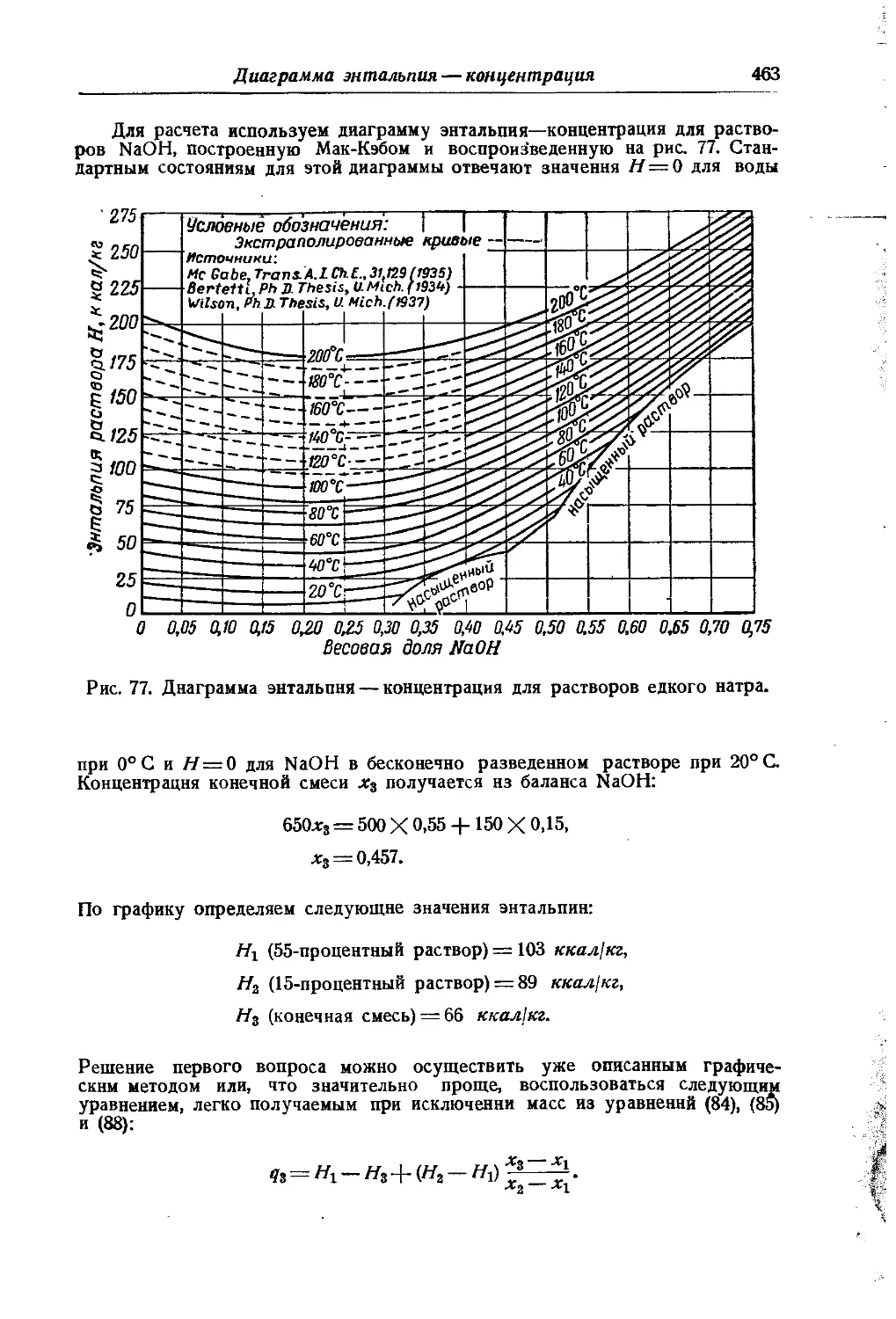
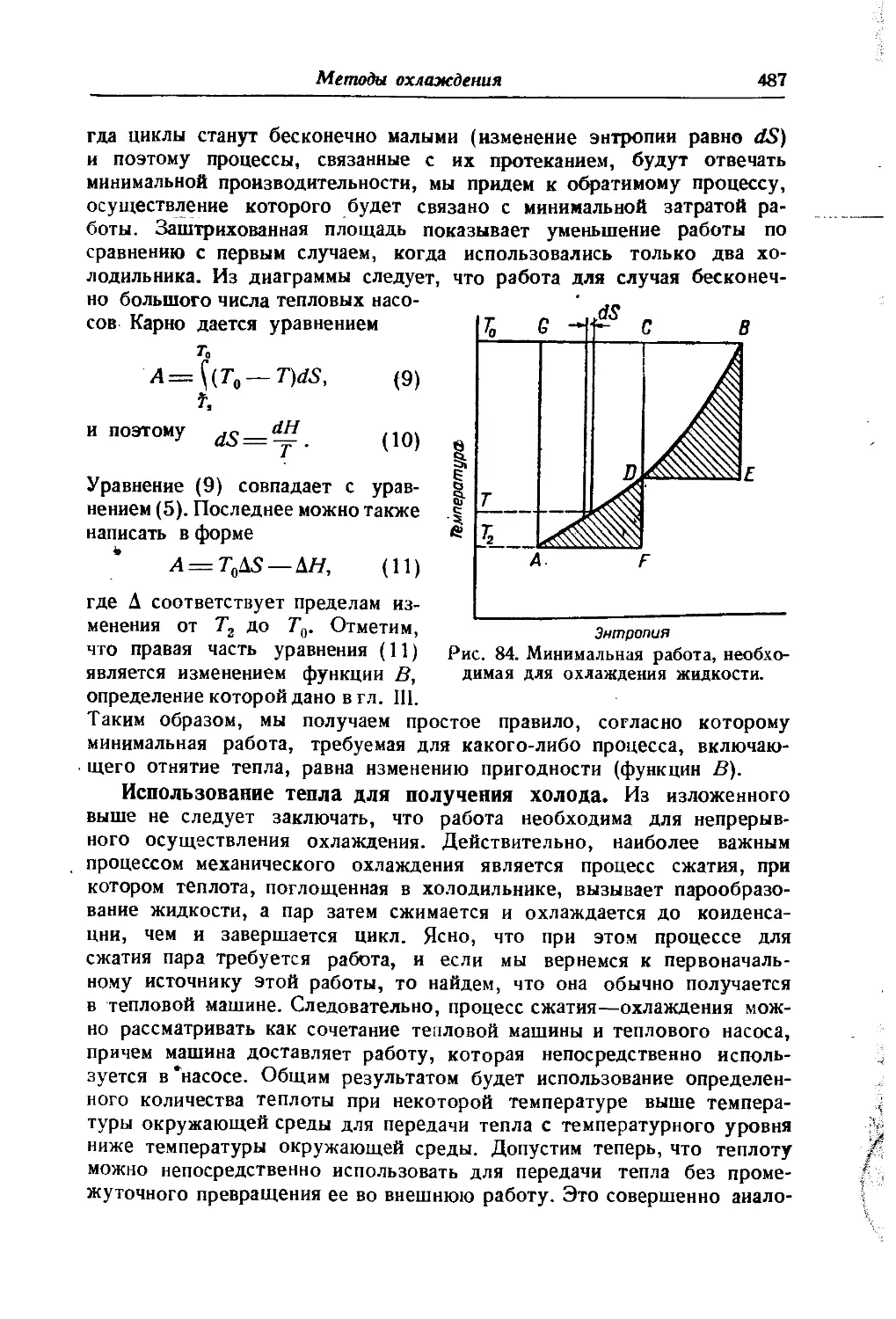
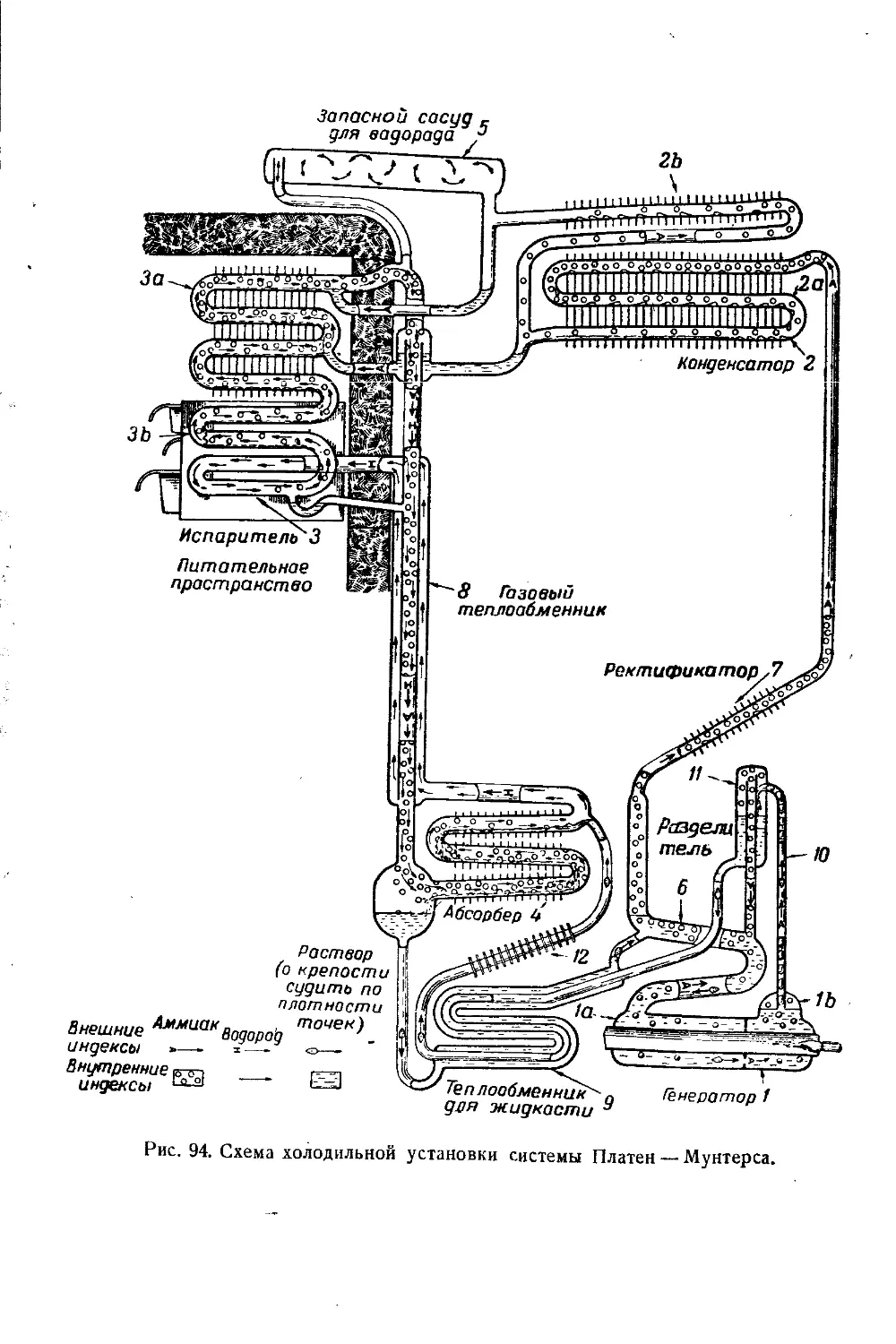
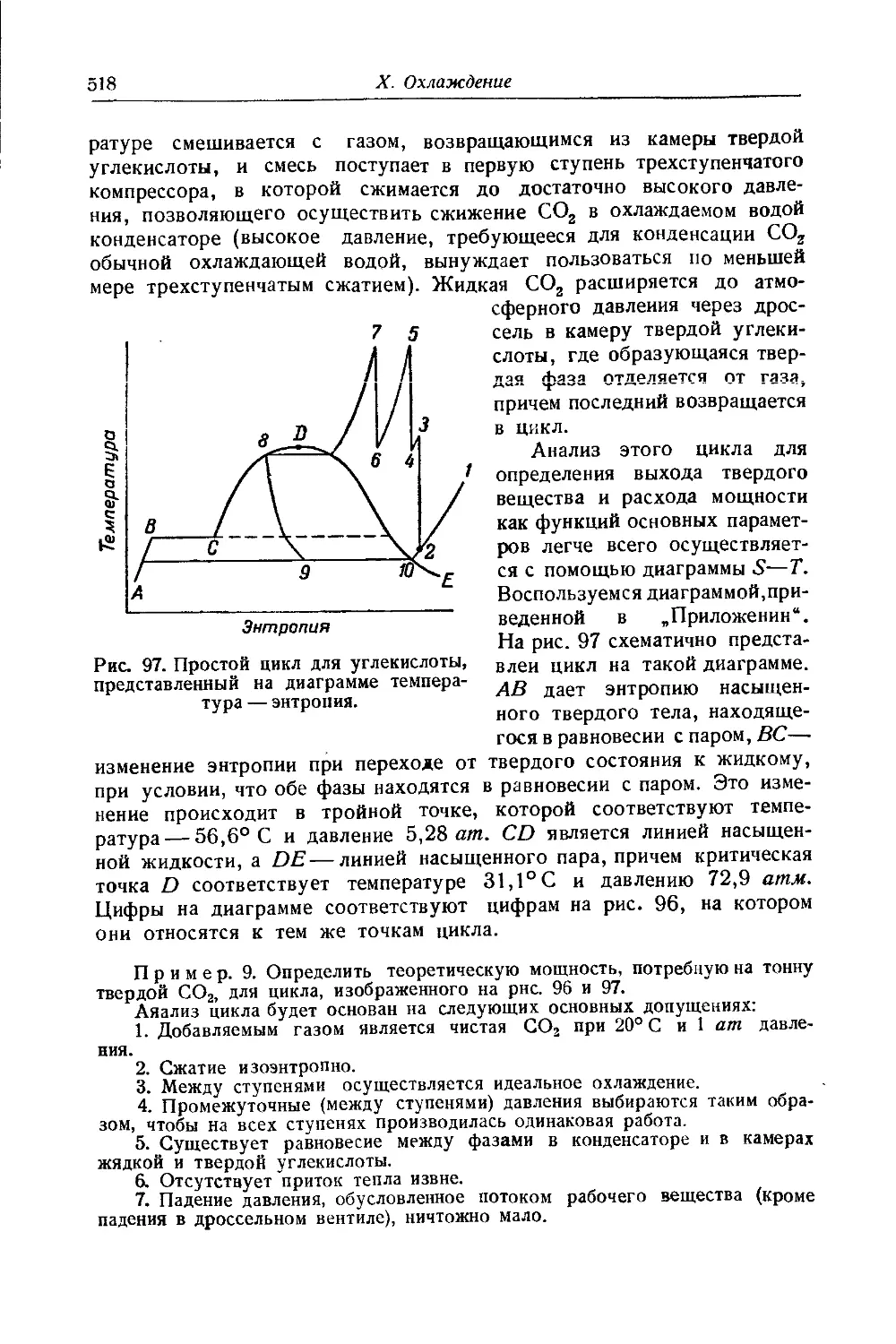
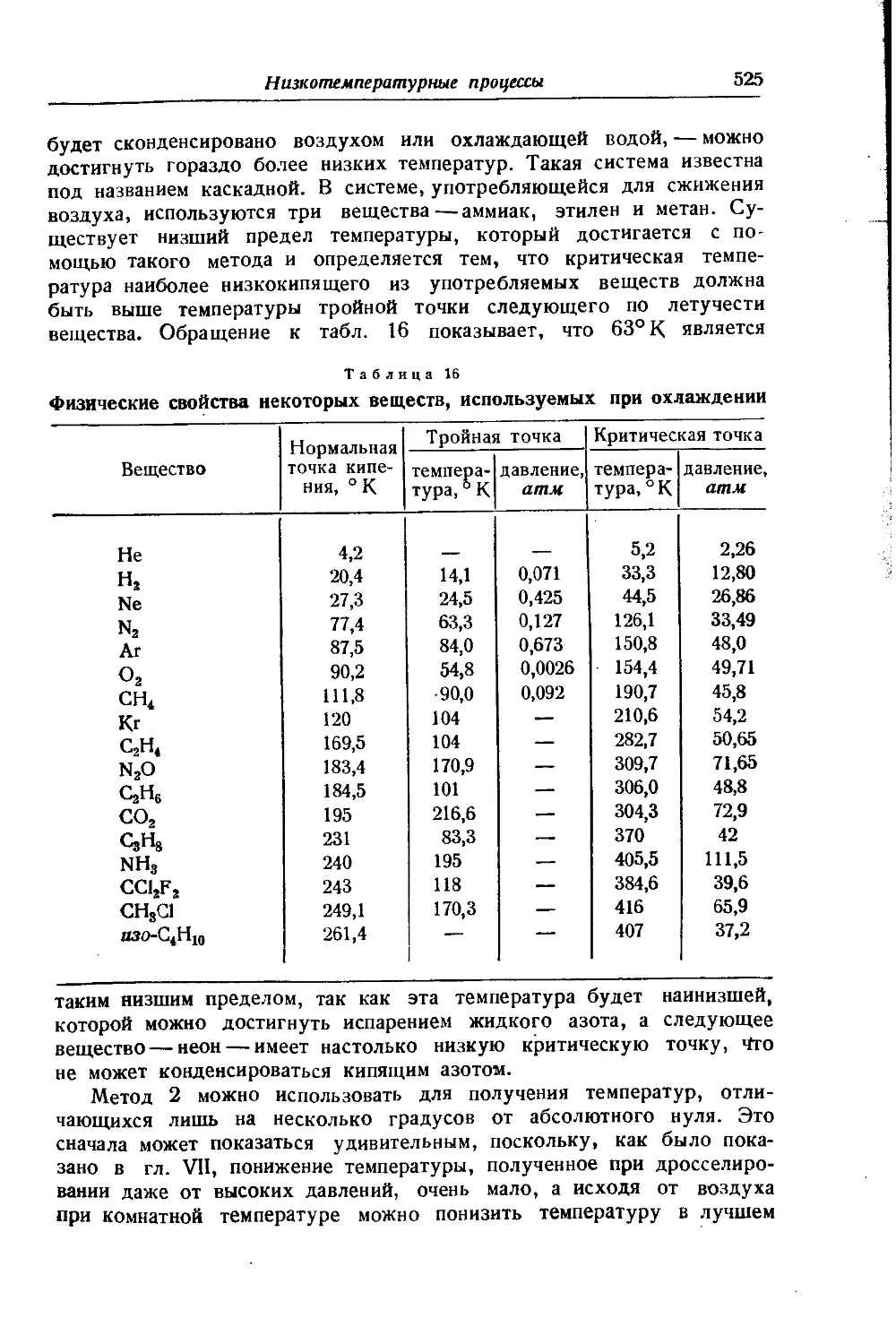
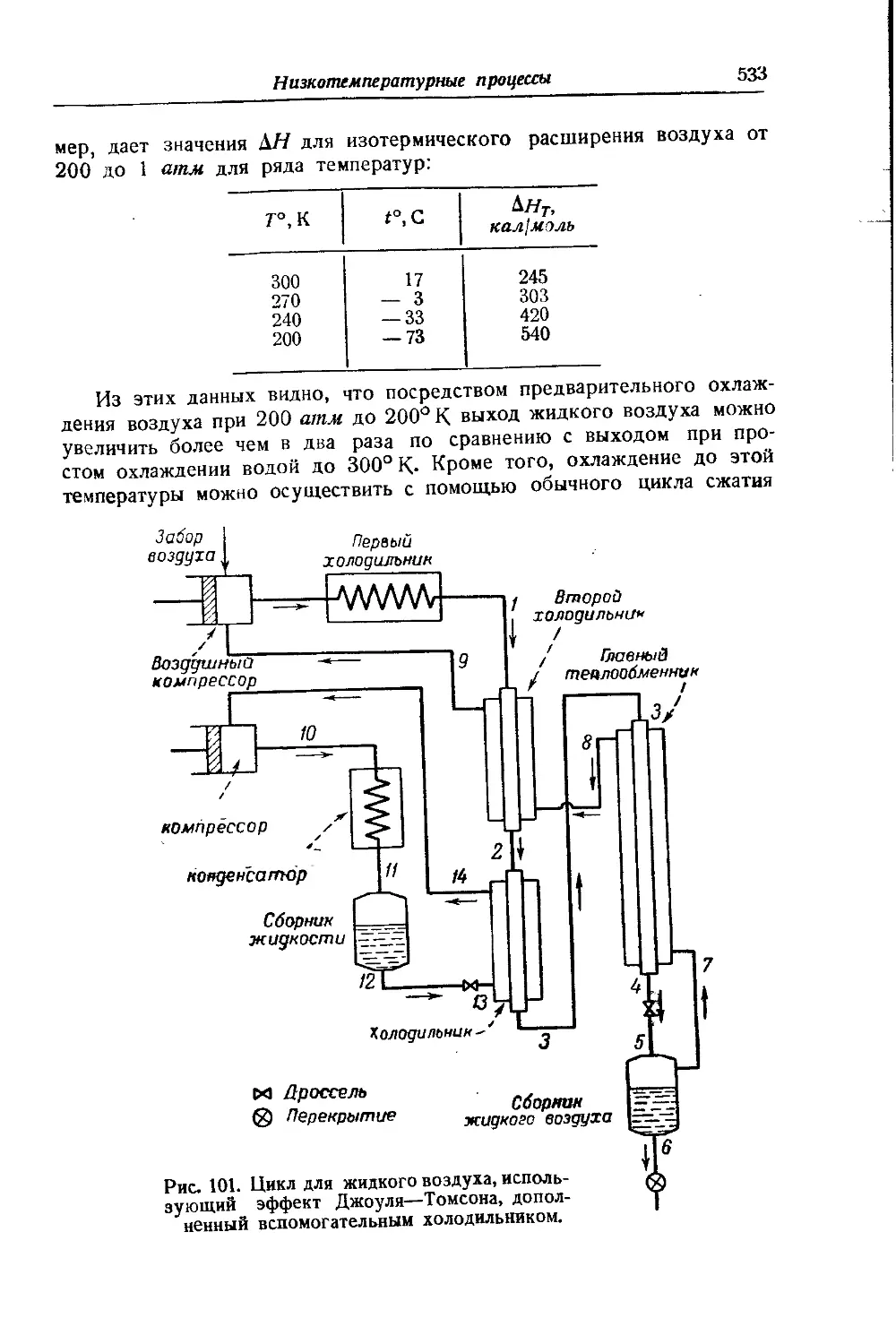
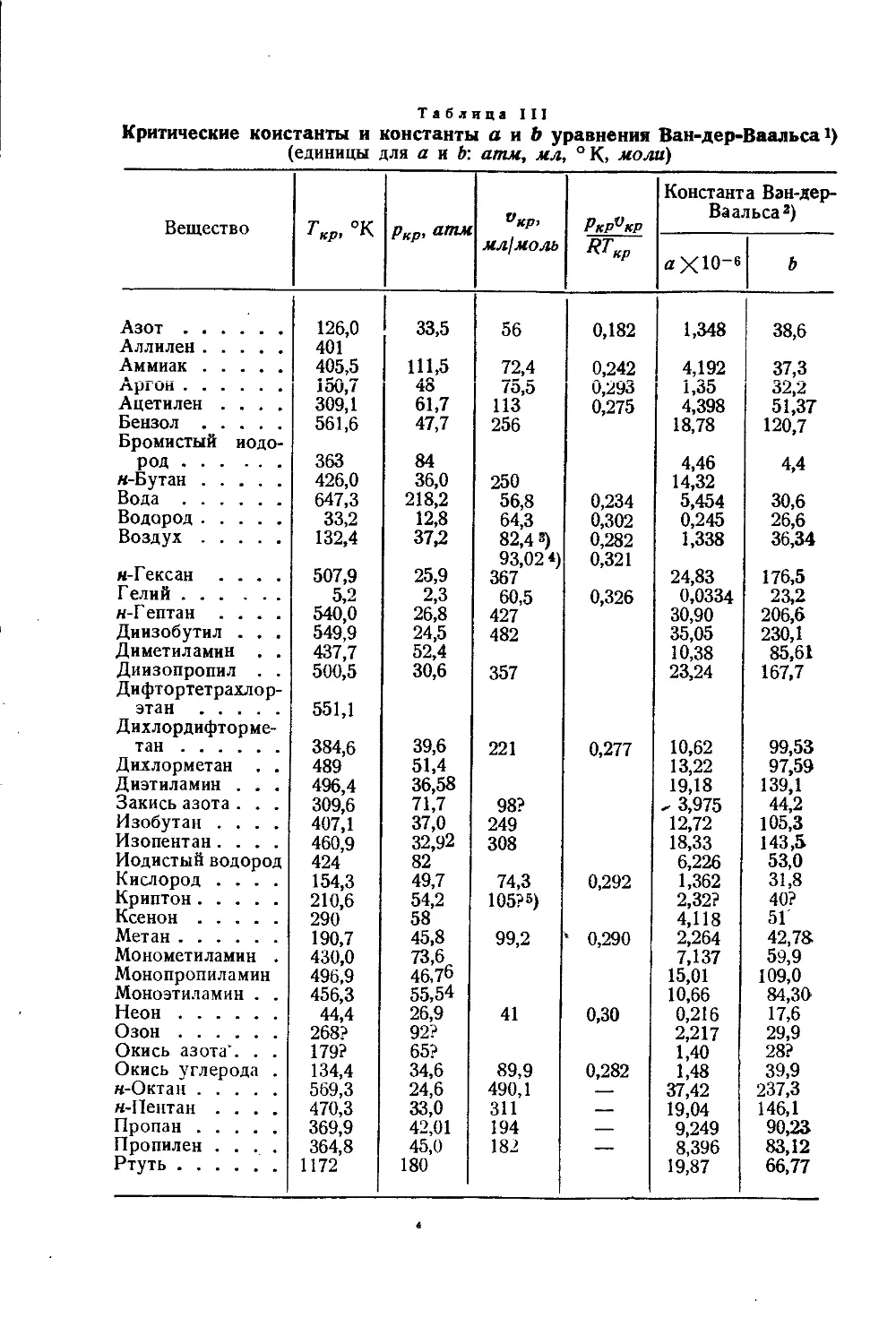
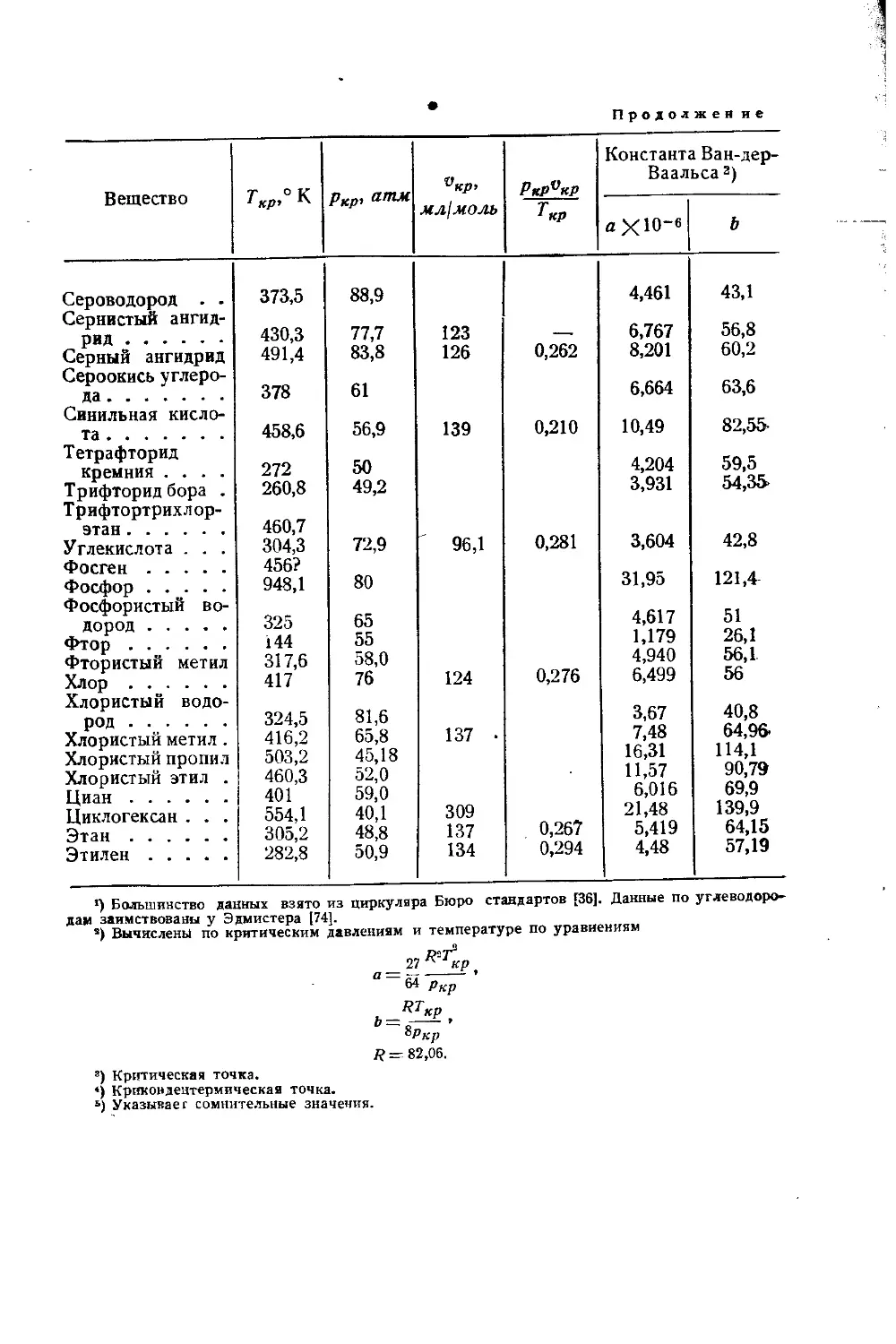
/








































































































































































































































































































































































































































































































































































































































































































































Author: Додж Б.Ф.
Tags: физика химия термодинамика физическая химия переводная литература учебник по химии издательство иностранной литературы
Year: 1944
Text
и* л
Из дате лъст во
1остранно литературы
CHEMICAL ENGINEERING THERMODYNAMICS
by BARNETT F. DODGE Professor of Chemical Engineering Yale University
FIRST EDITION FOURTH IMPRESSION
New York, London
1944
¥
Б. Ф. ДОДЖ
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
В ПРИМЕНЕНИИ
К ХИМИЧЕСКИМ ПРОЦЕССАМ
И ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
Перевод с английского
М. Л. КАРАПЕТЬЯНЦ
Под редакцией п р о ф. В. А. КИРЕЕВА
Вступительная статья и примечания
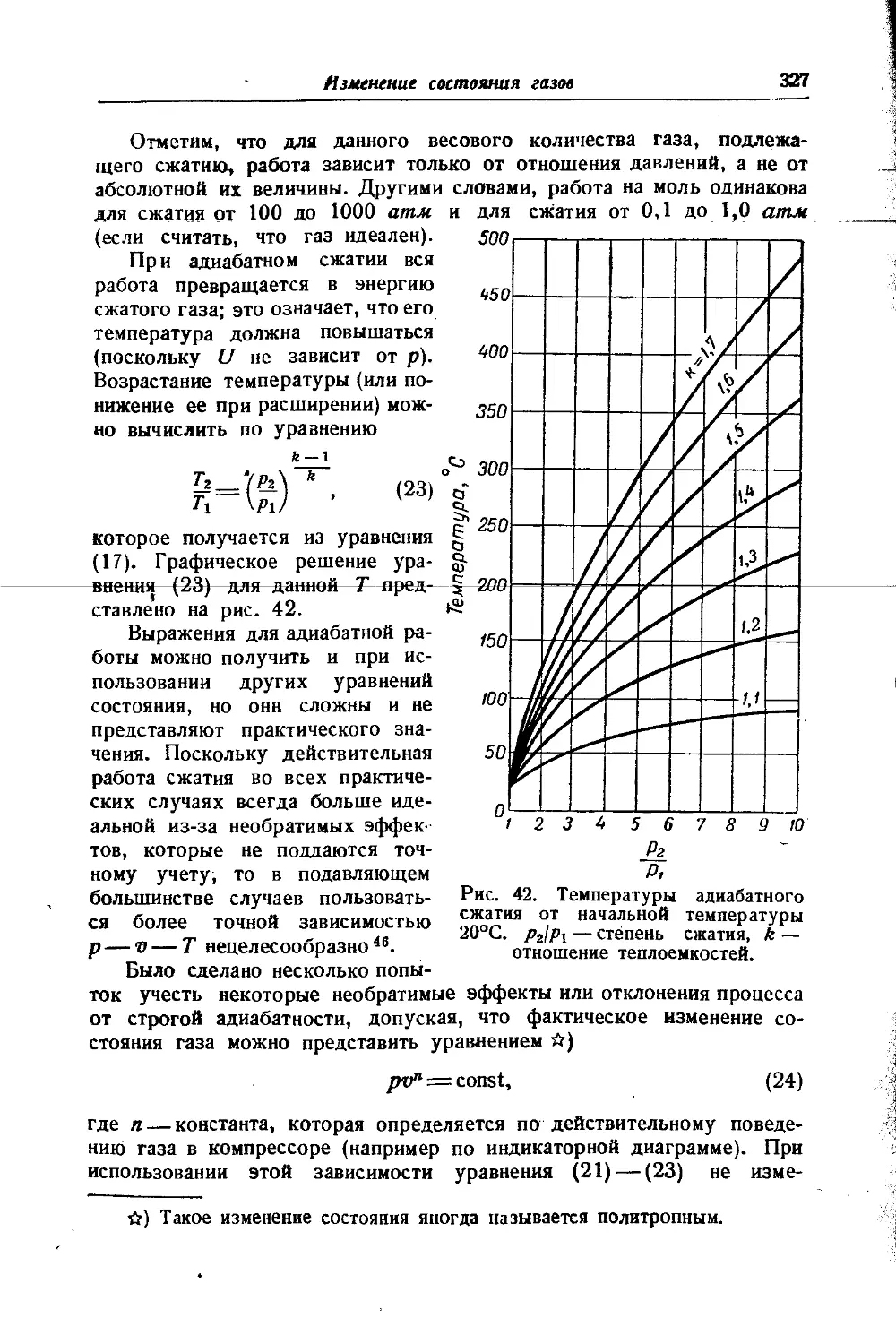
В. А. КИРЕЕВА и М. X. КАРАПЕТЬЯНЦА
1950
Издательство ИНОСТРАННОЙ ЛИТЕРАТУРЫ Москва *
ПРЕДИСЛОВИЕ РЕДАКТОРА
В настоящее время элементы термодинамики широко применяются в различных областях науки и техники. Термодинамика как наука при сохранении единства своих основ весьма сильно диференцирова-лась в прикладной части. Книга Доджа „Химическая термодинамика* посвящена применению термодинамики к решению различных проблем» встречающихся при химических экспериментах и в инженерно-техни" ческой практике.
Советские и русские ученые сделали много почетных открытий в термодинамике, в частности в химической термодинамике. Достаточно вспомнить работы Д. П. Коновалова, Н. А. Умова, С. А. Богуславского, Н. Н. Шиллера, Б. Б., Голицына, Г. Г. Гесса, В. Ф. Лу-гинина. Термодинамические школы Московского университета, институтов Академии наук, Теплотехнического института, Института азота и другие группы советских термодинамиков обеспечили дальнейшее быстрое развитие этой науки в настоящее время.
У нас в СССР создан ряд серьезных монографий и курсов по химической термодинамике, дающих глубокое и четкое освещение современного ее состояния. Хорошо известны книги Н. А. Колосов-ского, А. Ф. Капустинского, К. А. Путилова, А. И. Бродского, А. В. Раковского, М. М. Попова, И. Р. Кричевского.
Издание книги Доджа „Химическая термодинамика* представляется целесообразным вследствие весьма интересного, своеобразного подбора и расположения материала, широко освещающего современное состояние этой науки в США. В книге содержится критически представленный материал большого числа новых экспериментальных работ американских авторов по химической и инженерно-химической термодинамике. Кроме того, в книге имеется много примеров практических расчетов. Додж является автором ряда исследований по термодинамике; в течение 15 лет он читал курс химической термодинамики в Йельском университете.
При переводе книги потребовалось внести ряд изменений. Имеющиеся неточности и ошибочные утверждения автора исправлены в примечаниях (например, автор, выходя в описании из рамок термодинамики при интерпретации закона эквивалентности массы и энергии, подменяет понятие „масса* словом „материя", что приводит его к явно ошибочным физическим и философским выводам). Все расчеты в основном тексте и в примерах переведены на метрическую систему, а относящиеся к водяному пару, кроме того, перестроены применительно к нашим новым таблицам Вукаловича вместо используемых автором
6
Предисловие редактора
более старых таблиц Кинена и Кейеса. Опущены некоторые ссылки автора на источники заимствования.
Ввиду того что в книге, за единичными исключениями, не отражены работы русских и советских авторов, при переводе помимо примечаний, помещенных в конце книги, была написана вступительная статья. Этот материал не следует считать исчерпывающим обзором работ в данной области, — здесь указываются лишь некоторые из наиболее крупных и интересных исследований, а также работы обзорного характера и работы, относящиеся к различным частным вопросам. Кроме того, редактор, пользуясь имеющимся у него материалом, показал в примечаниях приоритет советских и русских ученых, не отраженный у автора.
Термодинамическая терминология при переводе приведена в соответствие с действующим ГОСТом 3270 — 46, с учетом проекта „Терминология технической термодинамики”, разработанного Комитетски технической термодинамики Академии наук СССР и изданного в 1948 г. Это же относится и к буквенным обозначениям.
Указатель литературы дан раздельно для иностранных авторов, упоминаемых в оригинале, и для русских авторов, упоминаемых во „Вступительной статье” и в „Примечаниях”. Авторы иностранных работ, введенных при переводе, отмечены в указателе звездочкой *.
Звездочкой ☆ отмечены ссылки на примечания автора. Примечания, сделанные при переводе, отмечены надстрочной цифрой.
В квадратных скобках указаны ссылки иа литературу.
Книга предназначена для научных и практических работников в области химии и химической технологии—для исследователей, инженеров-производственников, проектировщиков, конструкторов и профессорско-преподавательского состава.
Редактор считает своим долгом выразить признательность профессору, доктору технических наук Н. И. Гельперину за ряд ценных замечаний по главам X и ХШ, доценту технических наук Л. Г. Скриц-кому за просмотр перевода гл. VIII и кандидату технических наук А. М. Розену за просмотр перевода гл. VII и за ряд ценных замечаний.
Проф. В. А. Киреев.
В. А. Киреев, М. X. Карапегпьянц.
ВСТУПИТЕЛЬНАЯ СТАТЬЯ
Редактор перевода счел необходимым предпослать книге вступительную статью, освещающую некоторые вопросы, недостаточно подробно или совершенно не затронутые автором, в первую очередь отражающие успехи развития термодинамики в СССР за последние годы. Естественно, что такая статья по своему характеру не может представлять собой нечто однородное, так как в ней затрагиваются различные и непосредственно не связанные между собой разделы термодинамики. Тем не менее редактор перевода полагает, что даже краткое изложение этих вопросов будет полезно для советского читателя.
В настоящей статье рассмотрены следующие вопросы: расчеты свойств реальных газов, расчеты давления насыщенного пара и расчеты химического равновесия, растворимость веществ в сжатых газах, ограниченная взаимная растворимость газов, свойства азеотропных смесей; кроме того, дано описание диаграммы энтропия — энтальпия. В статье сделаны указания на соответствующие места текста книги, так же как и в тексте книги имеются ссылки на относящиеся к нему разделы вступительной статьи.
1. РАСТВОРИМОСТЬ ЖИДКОСТЕЙ В СЖАТЫХ ГАЗАХ
Явление растворимости жидкого (твердого) вещества в сжатых газах и газовых смесях приобрело в последнее время значительный интерес в связи со все возрастающим внедрением в промышленность высоких давлений. С этим явлением приходится иметь дело:
1) в промышленности неорганического и органического синтеза (в синтезе аммиака, например, высокое давление приводит к уменьшению концентрации аммиака в сжатой азотоводородной смеси);
2) при эксплоатации компрессоров высокого давления, где к механическому увлечению смазочных масел прибавляется растворимость их в газе;
3) в паросиловых установках высокого.давления, где растворимость солей в водяном паре высокого давления приводит к ухудшению работы турбин вследствие осаждения твердых солей на их лопатках (за счет снижения давления).
Растворимость жидкости в сжатом газе зависит от температуры, давления, природы жидкости и газа и определяется совместным действием эффекта Пойнтинга (см. стр. 200) и межмолекулярных сил.
Для расчета концентрации вещества в сжатом газе надо знать его летучесть в газовой фазе. Вычисление по точному уравнению (85, гл. IV),
8
Вступительная статья
из-за отсутствия данных по сжимаемости смеси газ — пар в настоящее время не осуществимо. Расчет по уравнению 115 (гл. IV) ошибочен, так как это уравнение относится к идеальным растворам, тогда как рассматриваемые смеси в общем случае содержат компоненты, обладающие не одинаковыми силовыми полями и не равными молярными объемами. Закон Дальтона здесь, разумеется, также неприменим, так как растворимость паров вещества в газе зависит от природы последнего.
Интересные работы, посвященные теории растворимости веществ в сжатых газах, принадлежат Кричевскому и его сотрудникам.
Попытка решения задачи путем вывода уравнения для смеси пар — газ, исходя из закона взаимодействия между молекулами, не приводит к надежным результатам, так как хотя с помощью известных допущений и удается получить приближенное уравнение, однако наличие в нем нескольких констант не позволяет при имеющемся фонде опытного материала получать количественное решение [333].
Для случая растворимости полярного вещества в неполярном газе 1) Кричевскому и Хазановой [340] удалось установить приближенную полуэмпирическую закономерность.
Представляет интерес проследить характер зависимости растворимости паров в сжатых газах от давления (при /= const).
Если для упрощения анализа считать, что конденсированная фаза является чистым веществом, а газообразная обладает свойствами бесконечно разбавленного раствора, то уравнение (138, гл. IV) примет следующий вид:
д log х_v — о ...
др ~~RT‘
Из этого уравнения следует, что при малых давлениях растворимость х с ростом давления будет уменьшаться * 2), так как парциальный мольный объем пара в сжатом газе v будет близок к мольному объему идеального газа и будет значительно превышать мольный объем вещества в конденсированной фазе V, затем кривая х = <р(р) достигнет минимума (так как с ростом давления v будет уменьшаться при практически неизменном v); после этого растворимость начнет возрастать (г'|<^1'), причем как перед минимумом, так и в непосредственной близости за ним кривая будет выпукла к оси абсцисс: вторая производная уравнения (1) по давлению при 7’ = const и при условии равновесия дает
D™ d2lnx __/ dv\ (ди \ .
~др^~ ~\д7>)т \др)г^’ ()
') Этот случай является практически наиболее важным, так как от охватывает растворы паров воды в сжатых воздухе, а зотоводородной смеси, N2, О2, Н2, СО2 и т. д., растворы паров метанола в сжатых Н2, СО, СО2, N2, СН4 и другие.
2) Кривая х = у(р); она при данной температуре начинается при давлении, равном давлению насыщенного пара данного вещества.
Вступительная статья
9
и хотя в этом уравнении и уменьшаемое и вычитаемое отрицательны, но по абсолютному значению первая величина меньше второй.
При еще ббльших давлениях v будет мало отличаться от v, таю как растворение вещества в сильно сжатом газе должно вызывать увеличение объема (вследствие значительного влияния при малых объемах отталкивательных сил), почти равное объему вещества в конденсированной фазе; следовательно, кривая растворимости, приобретая выпуклость к оси ординат, будет стремиться к горизонтали, когда растворимость практически не будет зависеть от давления.. С этим связан рост v на возрастающей ветви изотермы растворимости (начиная от точки перегиба)—-обстоятельство, получившее экспериментальное подтверждение в работе Кричевского и Гамбурга [335].
Подробному изложению рассмотренного вопроса посвящена третья глава монографии Кричевского [333].
2. О РАСЧЕТЕ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ РЕАЛЬНЫХ ГАЗОВ
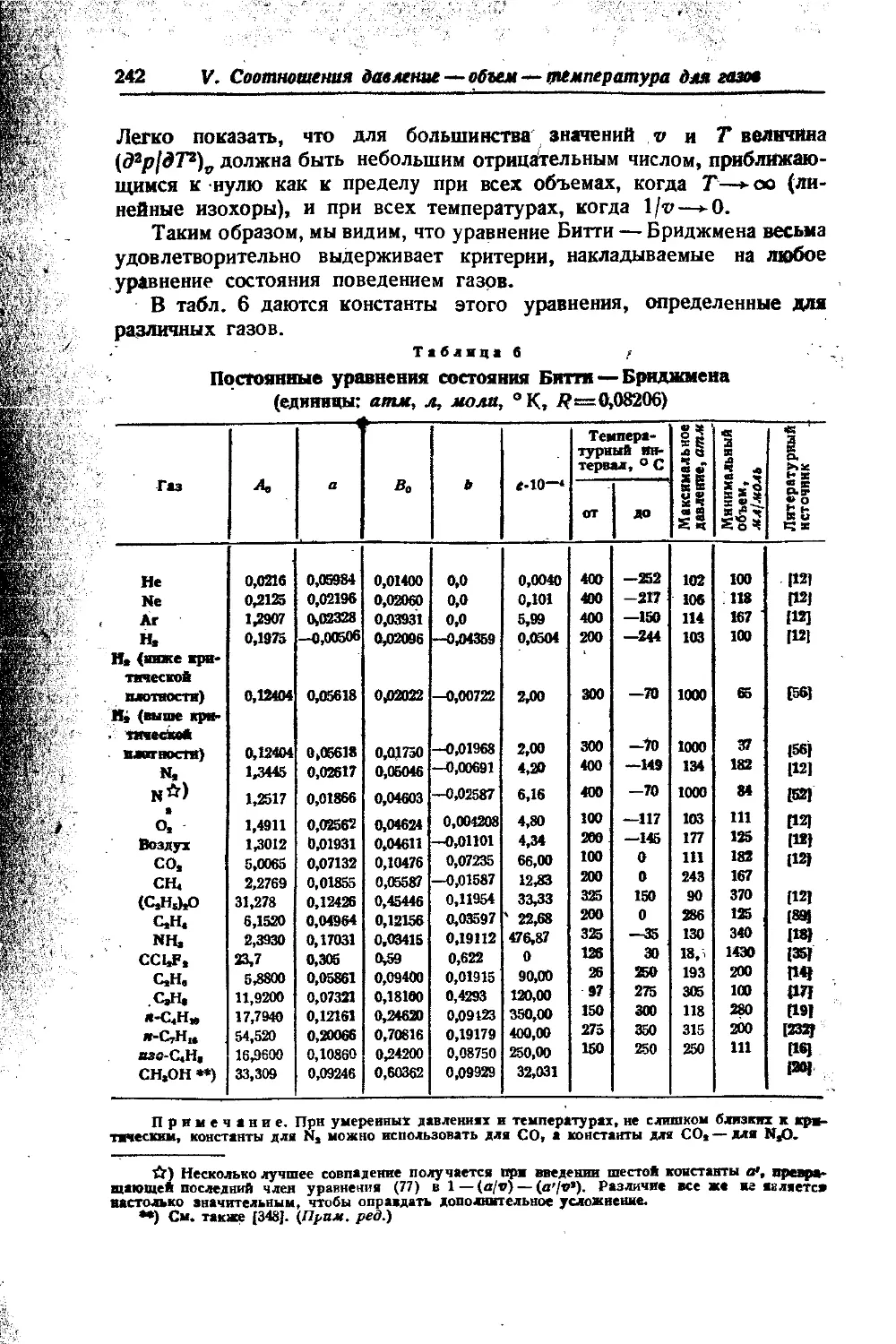
Вряд ли существует возможность построить аналитическую техническую термодинамику реальных газов с помощью уравнения состояния. Это связано не только с большой трудоемкостью подобных расчетов или ненадежностью результатов, ио подчас и с непреодолимыми трудностями. Так, сравнительно точные уравнения Битти — Бриджмена. (77, гл. V) и Кейеса (70, гл. V) для адиабатного процесса не интегрируются. В этом смысле можно согласиться с замечанием Доджа на стр. 290. Метод же летучестей ничего не дает для неизотермических процессов.
Однако, как показал Розен [370], существует другая возможность-получения результатов в аналитической форме, связанная с непосредственным применением экспериментальных данных по сжимаемости газов. Одной лишь объемной характеристики реального газа С — vlvwll=pvlRT для этого недостаточно, так как в основные термодинамические уравнения (88, 95, 103, гл. III) входят производные параметров р и Т. Поэтому в основу метода, разработанного Розеном, кладутся коэфициенты отклонения представля-
ющие собой соответственно отношения производных (dp/dT)v, {ди]дТ)р и (dv/dp)r реальных газов к тем же производным для идеального газа, т. е. (pjT, R/p) и (—RT/p2). Значения рр, рг при т^>2 сравнительно близки к единице: например, для азота при р = 2000 ат и/ = 20° С, рр= 1,172, цг=0,937 и pv=pp/'xr=l ,252г. тогда как z^>3.
С помощью этого метода было показано, что для реального газа можно сохранить — во всяком случае по форме — ряд простых зависимостей термодинамики идеальных газов, например, уравнение Пуассона (15, гл. VII) для адиабатного процесса. В этом отношении метод коэфициентов отклонения аналогичен методу летучестей для. изотермических процессов.
ао
Вступительная статья
Ниже изложены некоторые результаты работы Розена.
Адиабатный процесс. Изменение темпеоатуры определяется уравнением
Нр р
^2 _ (ср г । ,
Л \pJ \pj ’ ' '
где k = Cp]Cv. [Здесь, как и во всех последующих уравнениях, черта -над буквой означает среднее интегральное значение соответствующей величины.] Однако, несмотря на значительное изменение k н с р, величина - оказалась сравнительно постоянной. Поэтому целесообразно сохранить форму уравнения идеального газа (23, гл. VII):
тде в отличие от k
у — ^Р ________ Ср
Ср R?p
(3)
Величина (Cp)„s имеет физический смысл теплоемкости, отвечающей уравнению (15, гл. VII); она отличается от С^), так как для реального газа уравнение (15, гл. VII) несправедливо. Вплоть до очень высоких давлений х много меньше k и даже меньше k„a. Так, для NH3 при р —1000 ат и £=150 х = 1,094 (см. табл. 1). Ввиду того, что изменения k и / с р имеют противоположные знаки, то использование экспериментальных значений Cp/Cv при высоких давлениях в формуле (23, гл. VII), как это неоднократно практйковалось, приводит не к уточнению, а к ухудшению результатов, о размерах которого, согласно уравнению (1), можно судить по величине Так, например, для N2 при р = 500 ат и / = 0°С 1,7. Поэтому
мы приходим к парадоксальному выводу: в тех случаях, когда неизвестны значения х, лучше пользоваться уравнением (23, гл. VII) с Ацд, Чем ^реал-
Изменение объема определяется уравнением
pvk* = const, (4)
-сходным с уравнением (15, гл. VII), но в уравнении (4)
__С*
Р \dv)T ‘
(S)
Величина kv уменьшается с повышением температуры (особенно значительно при низких температурах) и сильно увеличивается с давлением. Так, для N2 при р — 3000 ат и / = 25° С АО = 5,25
1) Для реального газа Ср— Cv~ Rv-оУр, откуда = Ср — Ry-pV-v
Вступительная статья
11
вместо &„ = 1,40 (лишь при к > 100 kv почти перестанет зависеть от р). Поэтому кривые сжатия и расширения в последней ступени компрессора высокого давления становятся очень крутыми; вместе с этим уменьшается влияние вредного пространства.
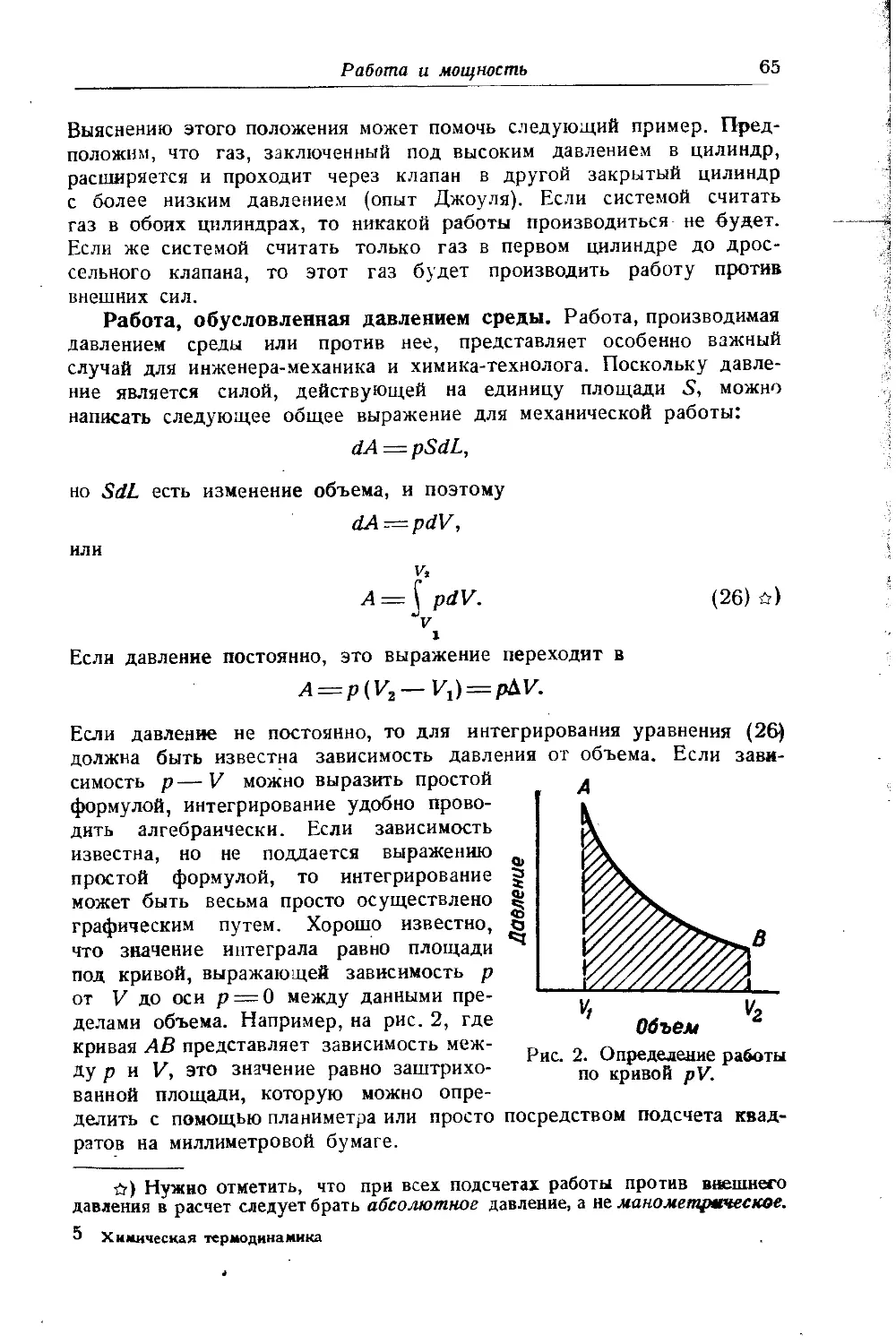
Расчет работы сжатия. Изотермический процесс. Работа определяется уравнением
(Ареал )у== Ну- (Аид)т'» (6)
в котором (Дил)т' вычисляется по уравнению (6, гл. VII). Если вести расчет не иа моль (или кг), а на более удобную безразмерную величину /реал = Лреал 1р0Щ, т. е. на работу, выраженную в единицах Амага, так, что A=pQvol = RTQl = 273,27?/, то уравнение (6) примет вид
(^реал)т- — (^ид)у—Мт 273,2^ ‘
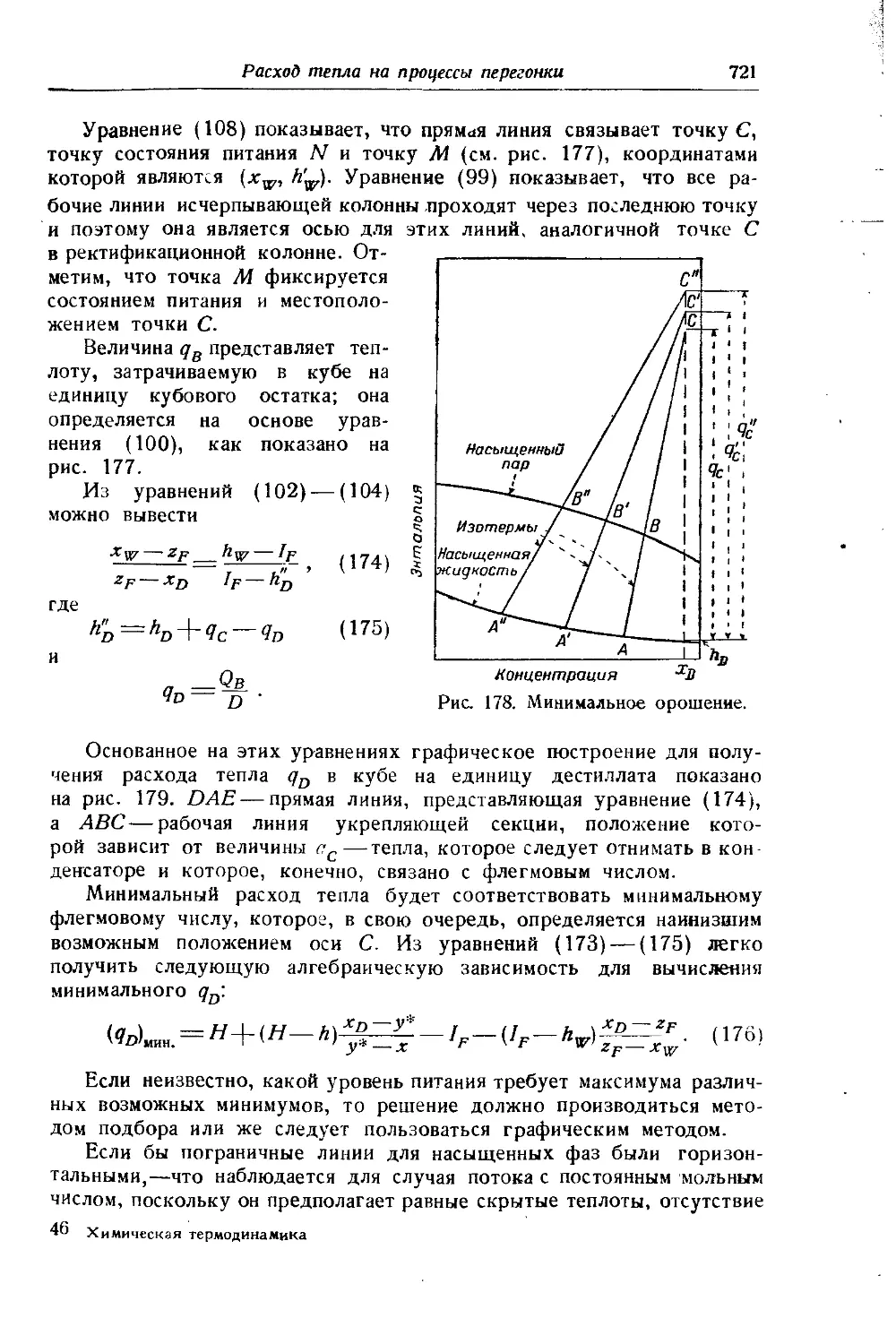
Работа цикла компрессора с изотермическим сжатием в соответствии с уравнением (29, гл. VII) равна
(Ареал)?, цикла = RTIn = RTIn -ф- ЪТ (р2 — р1)
или
(^реал)г, цикла -273 2 (Р2 ---Нт (^ид)г, цикла “^2 >
где
(8)
(9)
bT = v — у , $Т=ЬТ\р^^ЬТ\Я.7\
и z2h2j — коэфициенты сжимаемости Амага [см. уравнение (15, гл. V)], соответственно, при давлениях нагнетания и всасывания. Значения р7. для некоторых газов приведены в табл. 2. В случае отсутствия следует пользоваться вторым вариантом формулы, подсчитывая как среднее арифметическое значение цг, приведенных в табл. 3.
Дополнительной работой сверх ,идеальной“ Ьт(р2 — Pi) можно пренебрегать лишь до давлений, меньших 100 ат, так как при более высоких давлениях относительная величина ее сильно возрастает. Так, например, для азотоводородной смеси 1:3 при р[ =300 ат и степени сжатия г = — = 3 она составляет 40°/0 от (Аид)т-1ЦИКЛа, а Pi
прн pj=800 ат она составляет уже 100°/0 от этой величины.
Адиабатный процесс. Работа цикла компрессора с адиабатным сжатием равна:
х—1
(Ареал)\циыа==^^'1 । (,Р1) "Ь == ^нл) ^.чикла-!-
+ Мр2—Pi) (10)
F
12
Вступительная статья
или
х
X
(^реал)з, цикла1—273 2 1
---(^ид) 5,цикла ~Ps (?2 Pl)----------ИуХ^ид) Т, цикла Z±, ( 11)
где Zj и д2 берутся при одной и Значения jJs приведены в табл. 4. также получить соотношение
той же температуре всасывания. С помощью уравнения (4) можно
(12)
1 |
4
(^реал) 3,цикла — Pl^l — ,
— 1
При высоких давлениях это уравнение менее удобно, чем предыдущие, так как ko сравнительно сильно меняется с давлением и температурой и надежное усреднение затруднительно; поэтому при расчете работы можно сделать ошибку до 3—-5%. Однако уравнение (12) наглядно показывает, что расчет по уравнению (32, гл. VII) даже при замене RT величиной pv, взятой из опытных данных, дает заниженный расход работы. При низком же давлении (примерно до 30—50 ат, т. е. до к = 2/3), когда ko сравнительно близко к &ид (например, для расчета аммиачных и углекислотных компрессоров), уравнение (12) достаточно точно. Его наиболее удобно применять в следующей форме: f^pea.i уз,цикла == ^-'Дид JS,цикла. (13)
Политропный процесс. Сохраняются все формулы адиабатного процесса с заменой у_ на
(сХд-с (14)
где С — теплоемкость политропного процесса. При расчете работы по уравнениям (13) с помощью табличных значений можно положить:
(15)
Отметим также, что в случае, когда для данного газа известны коэ-фициенты уравнения (836, гл. V), работа цикла компрессора может быть рассчитана по уравнению
/реал = 273,2 С/ия + (а — 273,2 c)lng-[-^p2 —р^ . (16)
Ограничиваясь изложением лишь некоторых основных результатов, мы отсылаем читателя к оригинальным статьям [370, 371], в которых можно найти подробные выводы перечисленных уравнений, а также многие теоретические и прикладные вопросы, относящиеся к рассмотренным процессам. В работе Розена даются также расчет различных политропных процессов и числовые значения коэфициентов отклонения и других термодинамических величин (в широком интервале
Вступительная статья
13
(температур и давлений) для ряда важнейших газов и газовых смесей Й2, Н2, СО, CHt, NH3, N24-3H2, СО4-2Нг, воздух). Кроме того, рассматриваются методика вычисления и ряд примеров расчета компрессоров высокого давления. В отдельной работе разбираются вопросы расчета калорических величин по данным р — v — Т.
Метод Розеиа позволяет обойтись без термодинамических диаграмм (в частности без диаграммы S — Г), что весьма существенно не только в тех случаях, когда таких диаграмм нет (или они построены, но не охватывают нужного интервала р и Т), но и при наличии их, так как подобные диаграммы часто оказываются менее удобными для вычислений, чем аналитические методы (например, при различных сравнительных расчетах и в частности при отыскании оптимальных условий компрессии).
Преимущество метода — не только в простоте расчета и точности результатов, но и в наглядности качественной стороны дела, поскольку отклонения от законов идеальных газов фигурируют в явной форме
Таблица 1
Показатель адиабаты х~ х— различных газов при высоком давлении
Газ °C р, ат
1 100 200 300 600 800 1000
20 1,410 1,416 1,400 1,379 1,345 1,340 1,346
n2 100 1,406 1,418 1,426 1,419 1,377 1,372 1,373
200 1,400 1,409 1,409 1,408 1,387 1,380 1,374
25 1,404 1,407 1,408 1,407 1,402 1,394 1,390
н, 100 1,398 1,399 1,400 1,401 1,396 1,393 1,388
200 1,396 1,397 1,398 1,399 1,396 1,394 1,392
25 1,400 1,433 1,414 1,394 1,349 1,344 1,341
со 100 1,400 1,422 1,424 1,422 1,395 1,390 1,390
200 1,399 1,407 1,415 1,422 1,408 1,403 1,398
25 1,32 1,36 1,28 1,24 1,22 1,21 1,21
сн4 100 1,272 1,30 1,30 1,28 1,25 1,23 1,22
200 1,233 1,263 1,252 1,247 1,237 1,235 1,234
150 1,3 1,335 1,086 1,073 1,079 1,083 1,094
14 Пз 300 1,2 1,252 1,286 1,286 1,216 1,187 1,179
N2+3H2 25 1,405 1,407 1,406 1,404 1,397 1,393 1,395
Таблица 2
Коэфициенты fr-lO3 уравнения (9) для расчета работы изотермического
сжатия при степени сжатия — = 3
Pi
Газ Температура всасывания, °C Давление всасывания, ат Точ-ность В %
100 200 300 400
N2+3H2 25 0,660 0,719 0,759 0,782 0,5
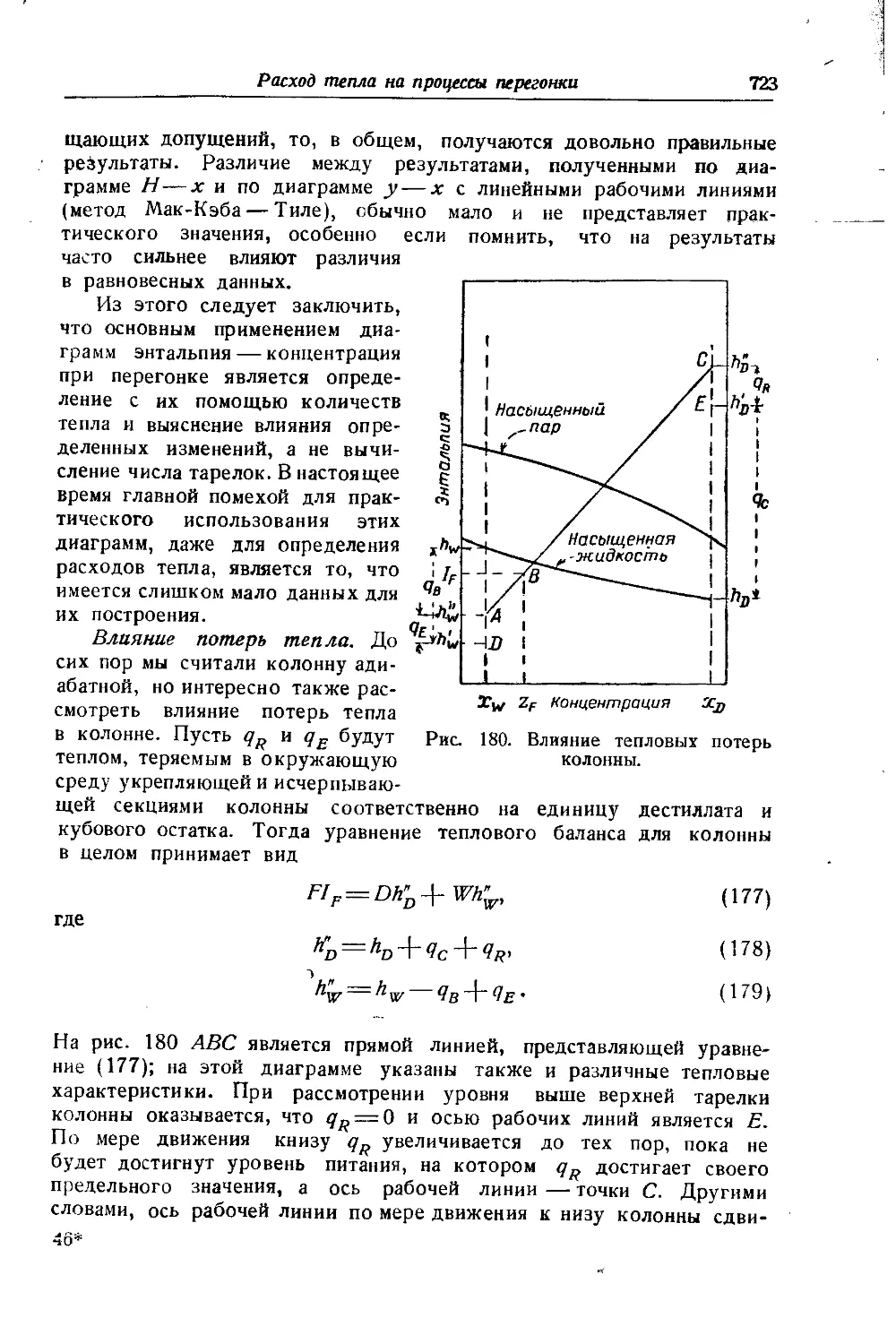
СО+2Л2 25 0,607 0,693 0,752 0,789 1,0
Воздух 25 0,179 0,565 0,776 0,899 1,0
n2 20 0,282 0,654 0,858 0,974 0,5
Таблица 3
Коэфициент отклонения рт=
р2 (dv\ RT \др) т
различных газов
Газ С°С р, ат
100 200 300 400 600 800 1000
20 0,972 0,906 0,823 0,788 0,750 0,730 0,774
n2 100 0,984 0,960 0,921 0,897 0,862 0,839 0,859
200 0,993 0,986 0,974 0,960 0,933 0,921 0,926
25 0,997 0,993 0,992 0,992 0,993 1,000 1,010
Н, 100 0,998 0,996 0,991 1,000 1,003 1,010 1,024
200 0,999 0,998 1,001 1,003 1,010 1,014 1,027
25 0,971 0,888 0,820 0,763 0,729 0,730 0,775
СО 100 0,997 0,954 0,915 0,875 0,842 0,838 0,858
200 0,995 0,983 0,966 0,947 0,925 — 0,928
25 0,963 0,734 0,542 0,526 0,496 0,520 0,573
сн4 100 0,970 0,898 0,810 0,736 0,676 0,656 0,694
200 1,000 0,946 — 0,901 0,908 — —
150 1,143 0,160 1,108 0,106 0,109 0,110 0,111
NH, 200 1,017 1,023 0,459 0,288 0,220 0,201 0,211
300 0,910 0,827 0,764 0,731 0,752 0,825 0,922
25 0,996 0,987 0,977 0,966 0,956 0,949 0,945
100 0,998 0,996 0,990 0,983 0,973 0,971 0,969
200 0,999 0,999 0,998 0,997 0,994 0,992 0,989
Вступительная статья
15
Таблица 4
Коэфициент Ps-103 уравнения (11) для расчета работы
адиабатного сжатия при степени сжатия — = 3 Pi
Газ Температура всасывания, °C Давление всасывания, ат Точность В % ч
100 ' 200 300 400
N2+3H2 25 0,713 0,753 0,781 0,799 0,5
СО+2Н2 25 0,680 0,737 0,777 0,802 1,5
Воздух 25 0,446 0,682 0,838 0,943 1,0
N,* 20 0,539 0,781 0,931 1,025 0,5
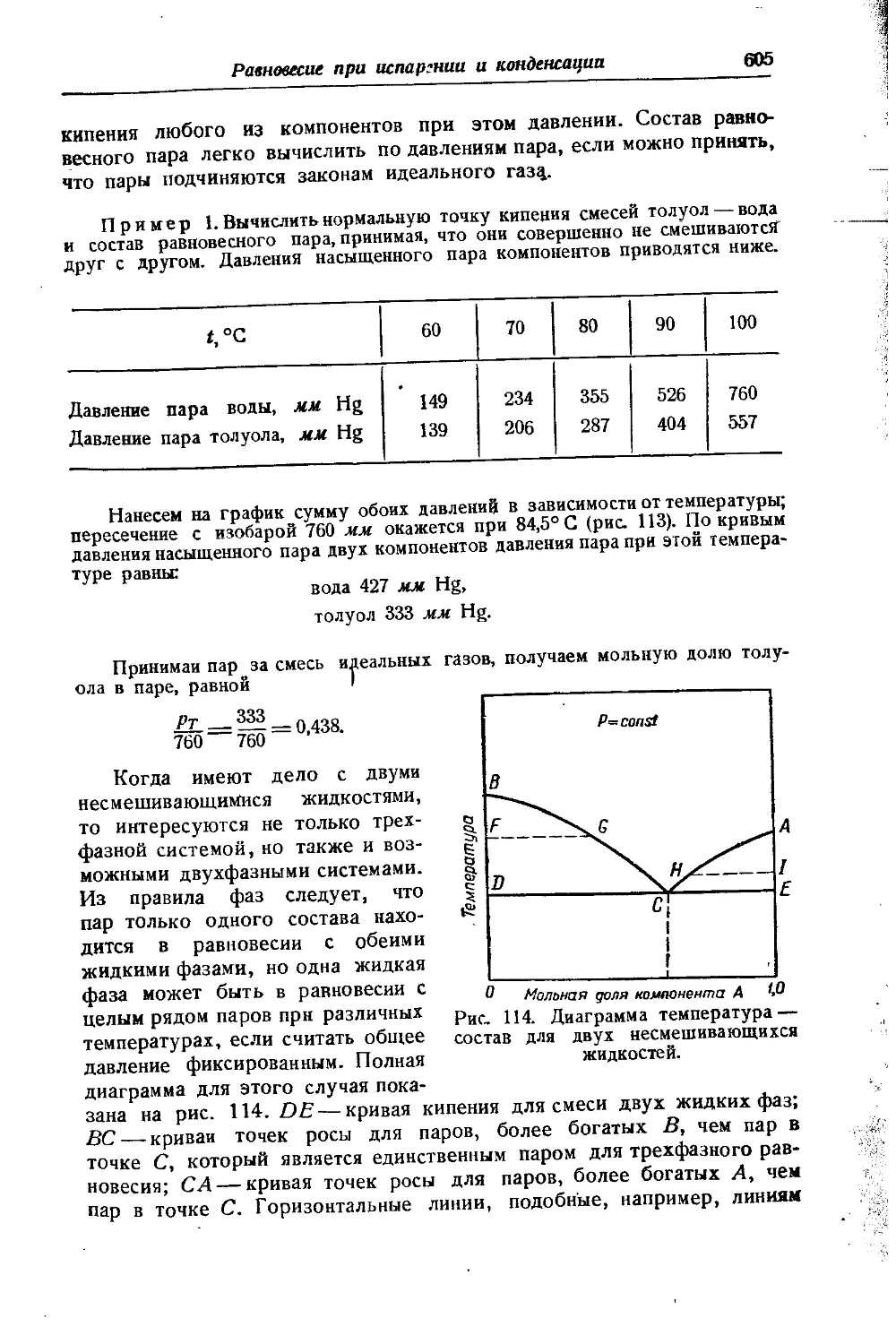
& ОБ ОГРАНИЧЕННОЙ ВЗАИМНОЙ РАСТВОРИМОСТИ ГАЗОВ ПРИ ВЫСОКИХ ДАВЛЕНИЯХ
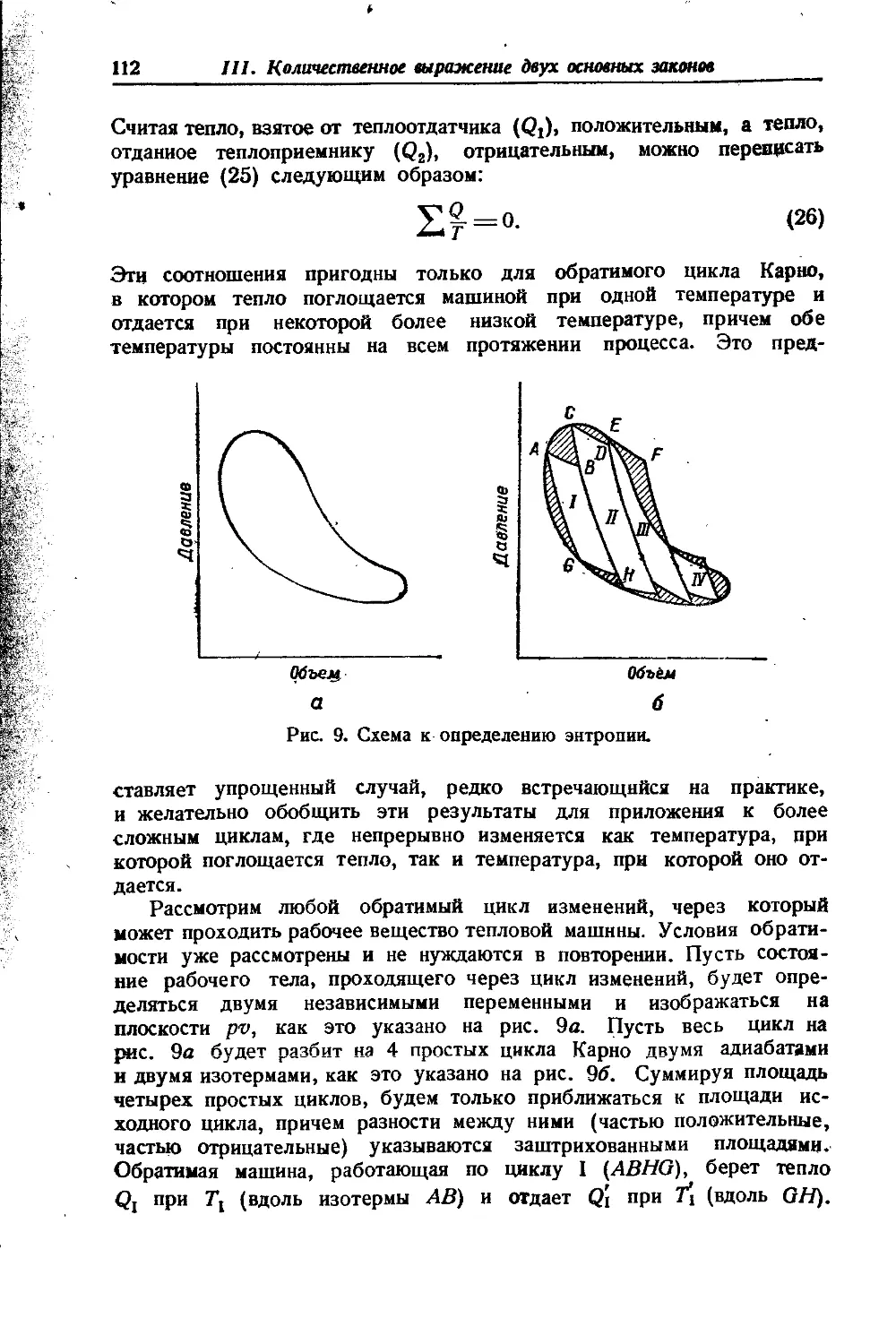
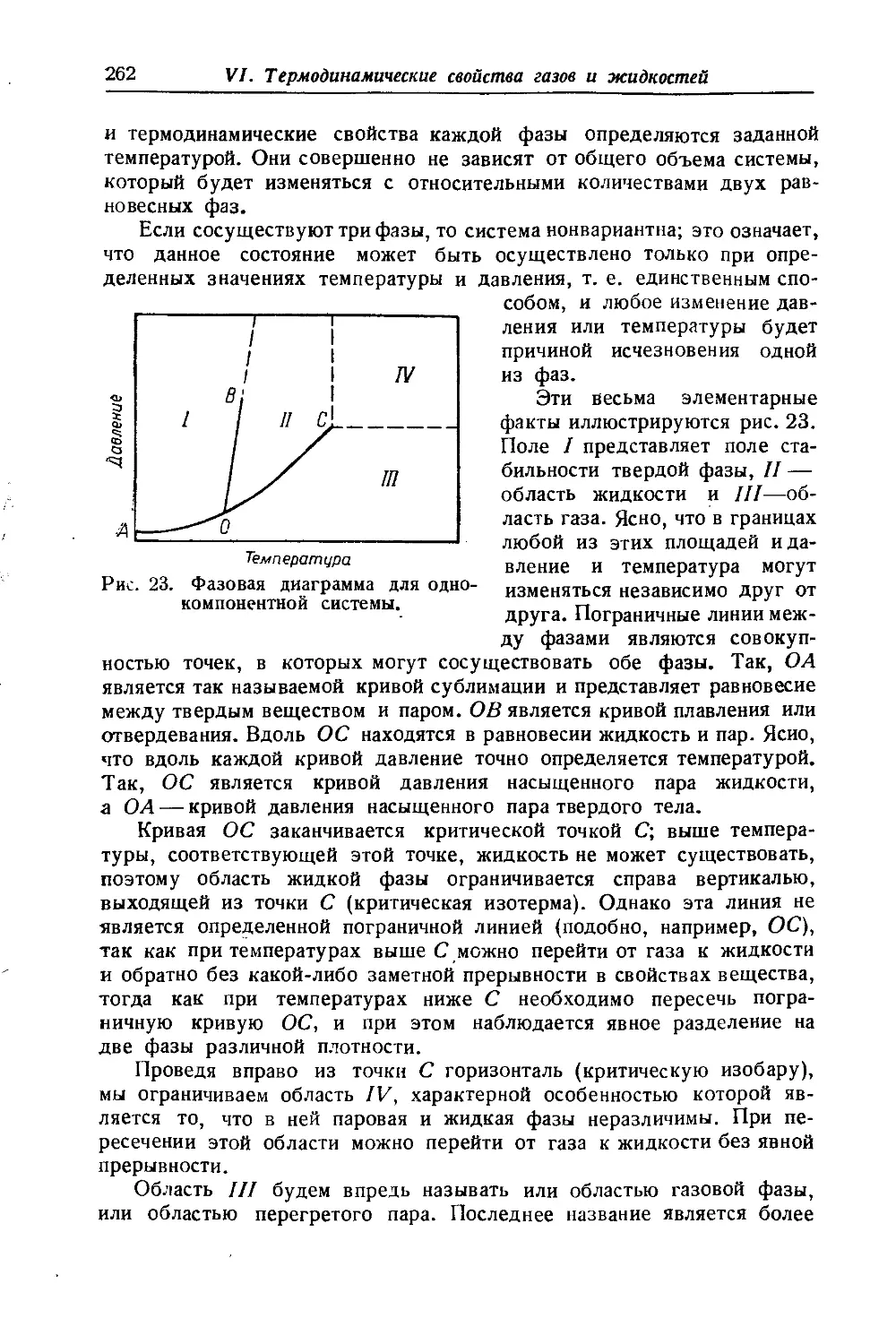
Положение о неограниченной смешиваемости газов, основанное на допущении энергетической независимости газов в смеси, оказы-
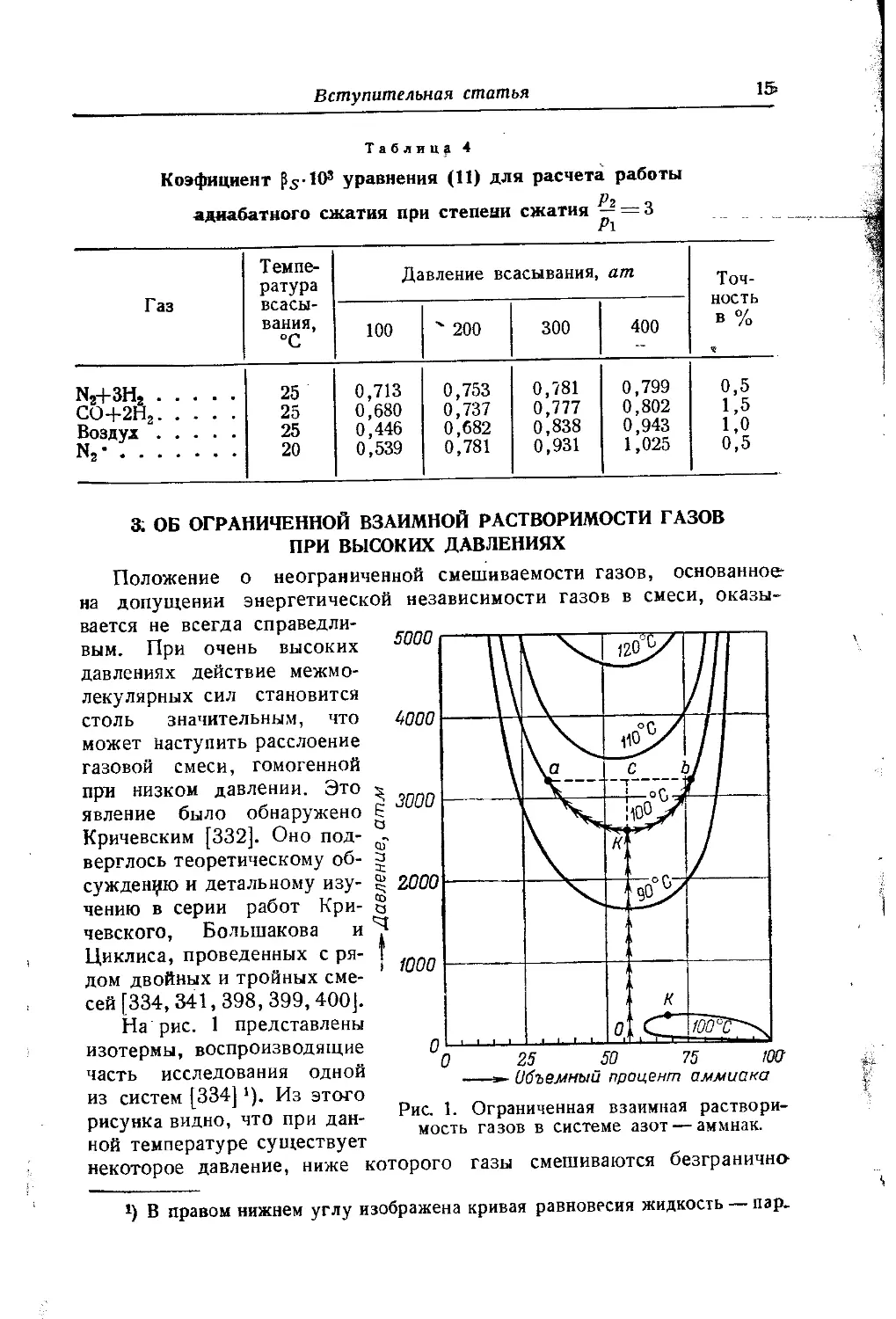
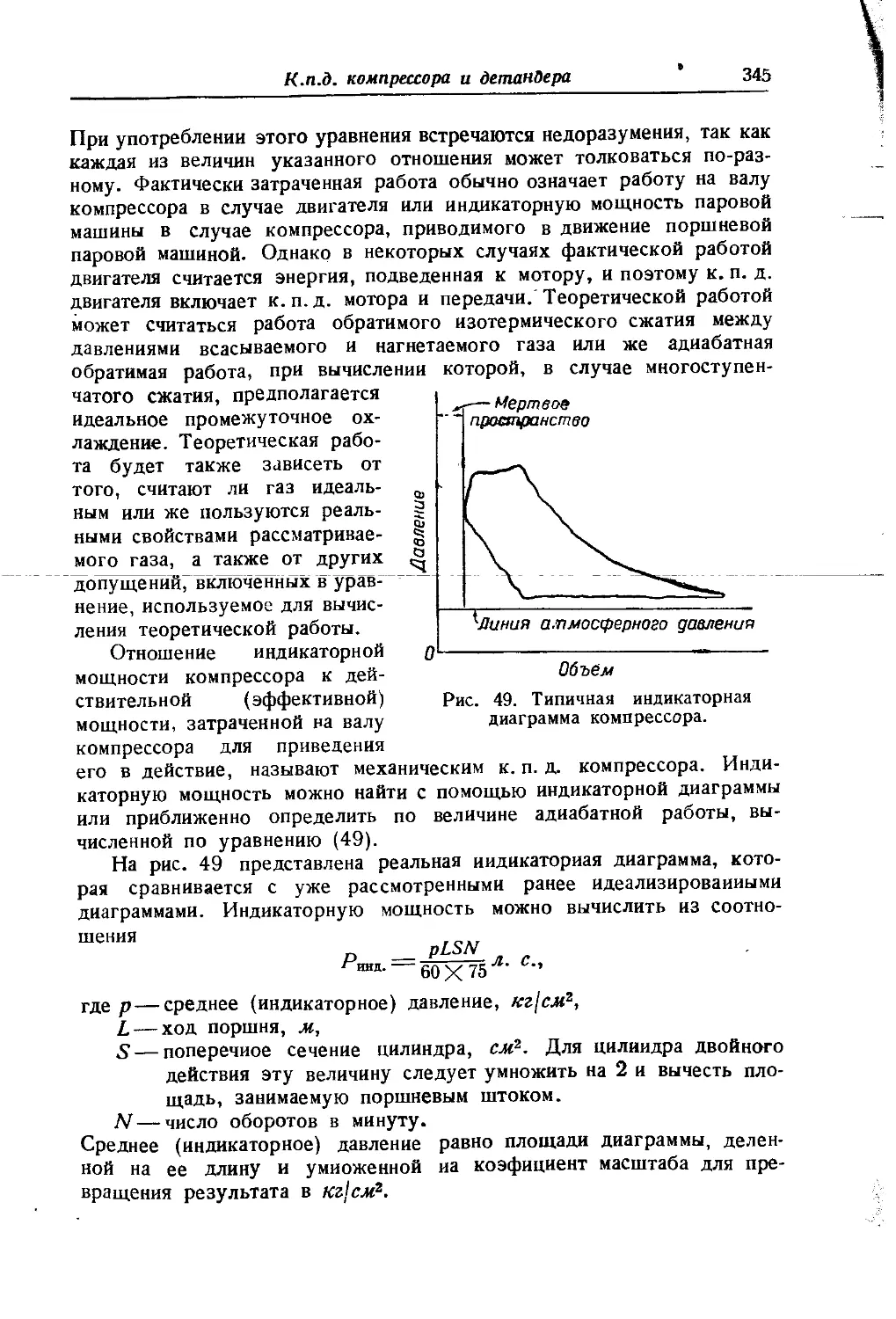
вается не всегда справедливым. При очень высоких давлениях действие межмолекулярных сил становится столь значительным, что может наступить расслоение газовой смеси, гомогенной при низком давлении. Это явление было обнаружено Е Кричевским [332]. Оно под-верглось теоретическому об- g суждению и детальному изу- § чению в серии работ Кри- § невского, Большакова и Циклиса, проведенных с ря- I дом двойных и тройных смесей [334, 341,398, 399,400J.
На рис. 1 представлены изотермы, воспроизводящие часть исследования одной из систем [334]'). Из этого рисунка видно, что при данной температуре существует
некоторое давление, ниже которого газы смешиваются безгранично-
!) В правом нижнем углу изображена кривая равновесия жидкость — пар.
16
Вступительная статья
(критические точки А')- При более высоких давлениях наступает г расслоение смеси на две фазы, причем с ростом давления расслоение охватывает все больший интервал концентраций, в соответствии с чем области взаимной растворимости суживаются и состав равновесных фаз становится все более различным. Так, если при /=100°С подвергнуть сжатию смесь, состоящую из 57°/0 NH3 и 43% N2 (точка О), то до р = 2600 атм (точка К) смесь будет гомогенной. При дальнейшем повышении давления наступит расслоение, и, например, при р = 3200 атм смесь будет состоять из двух фаз, в одной из которых "будет 33°/0 NH3 (точка в), а в другой — 17°/0 NH3 (точка Ь). Соотношение между фазами при /= 100°С и р — 3200 атм в соответствии с правилом рычага (см. стр. 671) определяется отношением
Количество фазы, более богатой NH3_отрезок ас
Количество фазы, более богатой N3 отрезок Ъс '
[Если смесь, подвергаемая изотермическому сжатию, сильно отличается к от смеси, отвечающей критической точке, то с начала расслоения ‘ обе фазы будут значительно отличаться по составу.
С ростом температуры область расслоения суживается и переме-?! щается в район высоких давлений; при этом состав критических фаз
f изменяется незначительно.
у Термодинамический анализ [334] приводит к заключению, что
критическая точка в бинарных системах может быть охарактеризована уравнениями
(^L=° <’>'?
и
- = °> (2)
; I дх2 т
т. е. критической точке на изотерме/= ср (х) должна соответство-' с вать точка перегиба, имеющая горизонтальную касательную.
Диференцируя уравнение (88, гл. IV) по х (при р, Т= const) и приравнивая нулю, согласно уравнению (1), производную (д In fldx)ptT, получим
о
Следовательно, распад на две фазы возможен только для тех смелей, для которых
дх/Р,т
0.
(4)
1
Вступительная статья
17
К сожалению, в настоящее время не представляется возможным произвести расчет по уравнению (3) с целью определения давления, при котором наступит расслоение, так как данные о численном значении производных (ду1дх)р>Т при давлениях порядка нескольких тысяч атмосфер отсутствуют. Лишь для газовой смеси NH8 — N2 одного состава (с привлечением р — v—Т данных для чистых NH8 и N2) удалось оценить значение (dvldx)ptT. Опытные данные подтвердили результаты расчета [333, 341]. Косвенным подтверждением служит аналогия с жидкими растворами (если принять, что вплоть до очень высоких давлений производная dvjdx остается неизменной).
Существование расслоения в газовых смесях позволило дать общую картину фазовых равновесий в бинарных смесях, объединяющую равновесие газ — газ, жидкость — газ и твердое тело — газ [333, 341, 398, 399, 400].
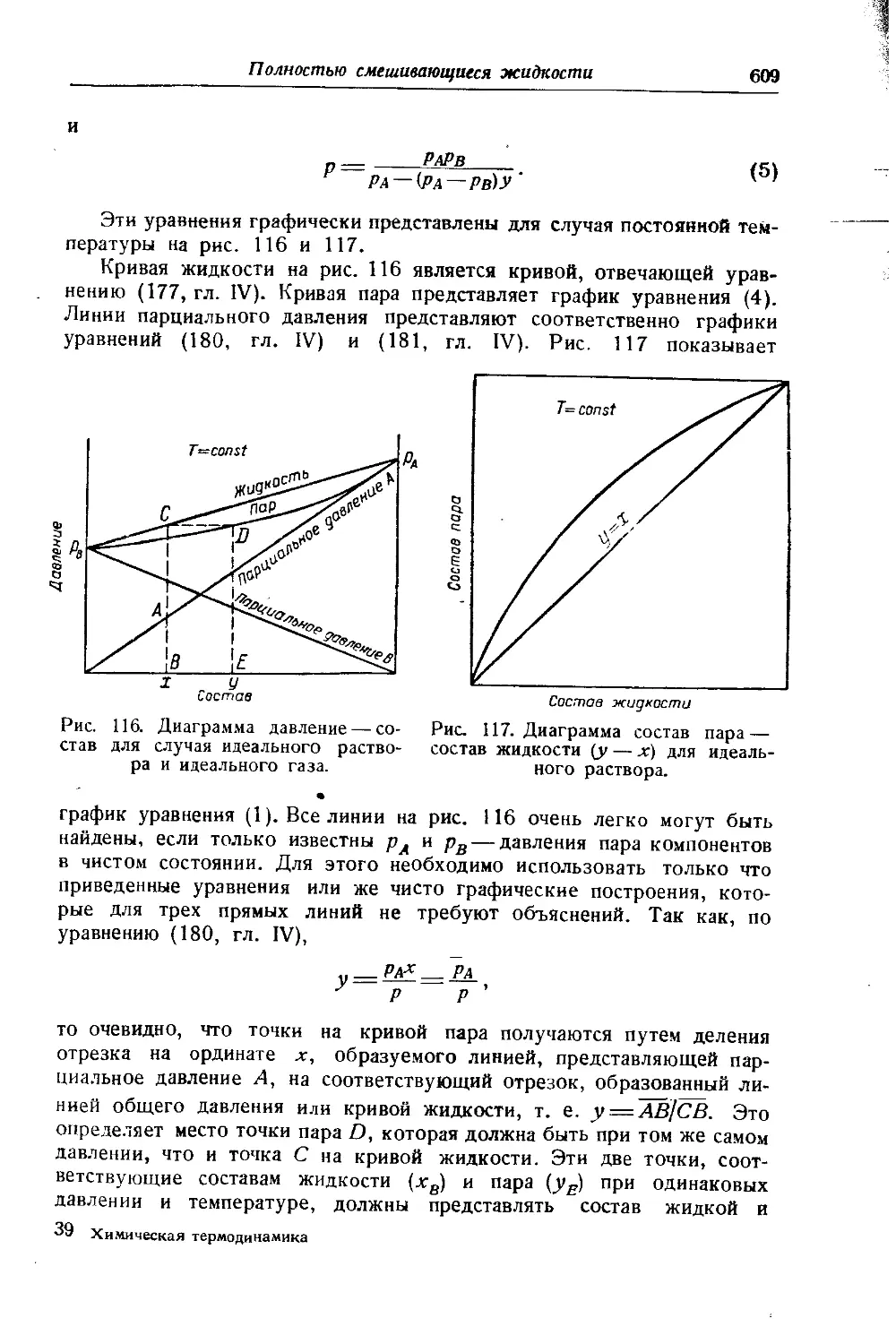
4. О ДИАГРАММЕ ЭНТРОПИЯ — ЭНТАЛЬПИЯ
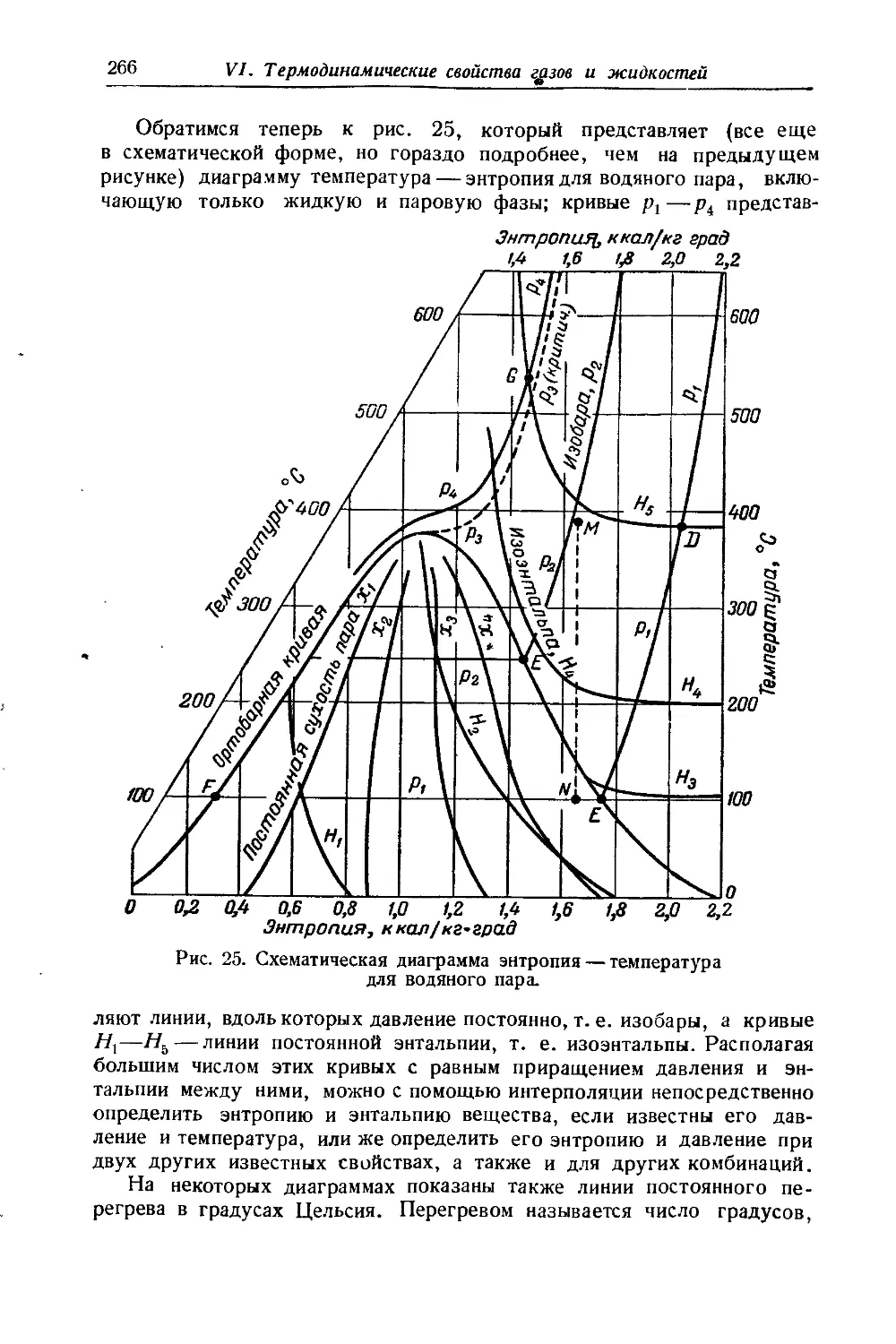
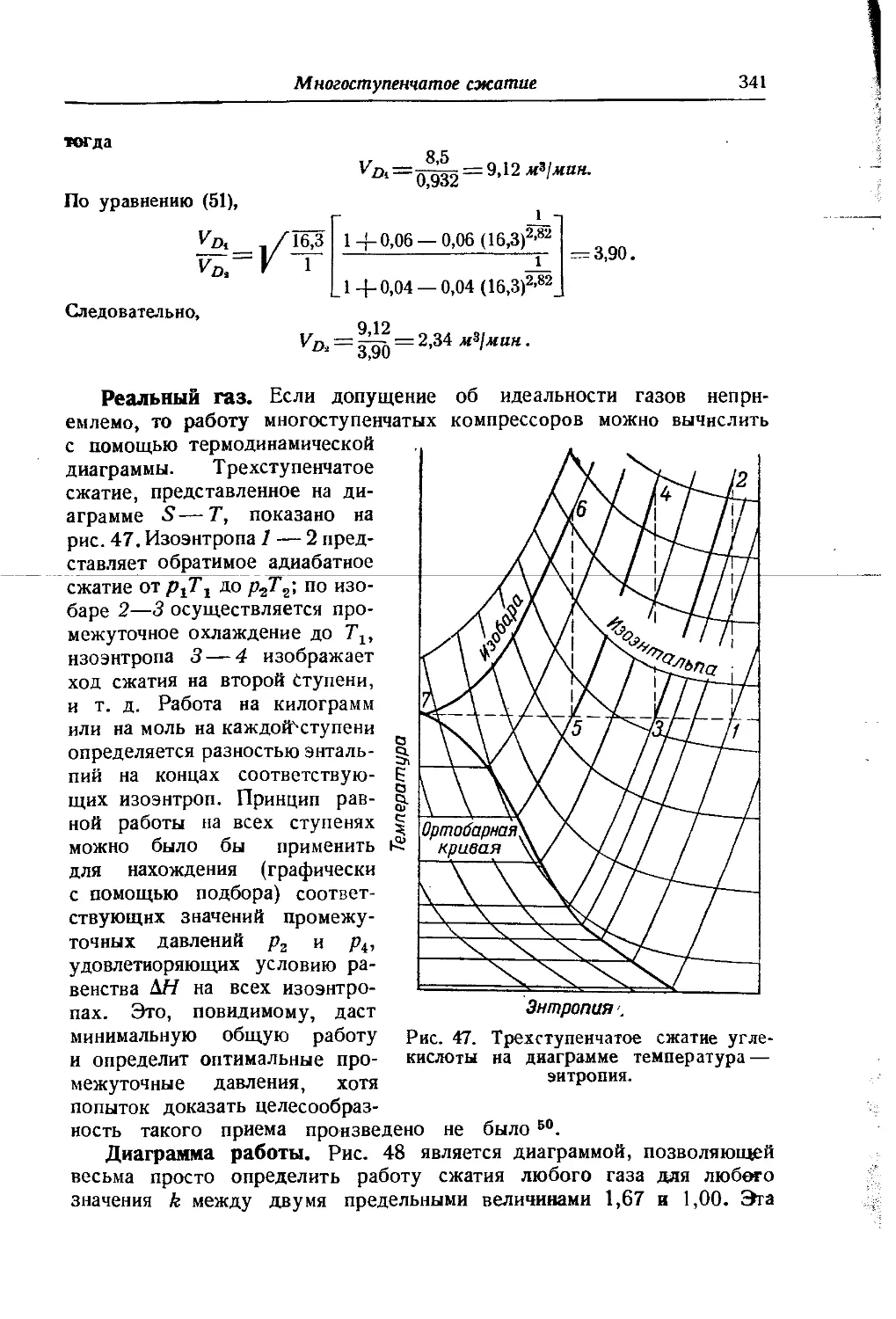
Схематическое изображение 5 — Н диаграммы дано на рис. 2. Кривая жидкости АС начинается в начале координат; этим принимается, что при Н'= 0, S’=0, и наоборот. Кривая жидкости АС и кривая пара CF смыкаются в критической точке С. Обе кривые строятся по табличным данным. Так, координатам точки В (S', И’) соответствуют графы (7) и (10) таблицы 7 (гл. VI), а координатам точки Ч ,D(S”, Н")—графы (11) и (8) той же таблицы. Ортобарная кривая заключает область влажного пара. Часть диаграммы, находя-щаяся правее и выше кривой CF, отвечает перегретому пару, часть же ее, лежащая левее кривой АС, — жидкости, которая не нахо-дится в равновесии с паром.
Если изотермы-изобары влажного пара в диаграмме S — Т представлены прямыми, параллельными оси S (рис. 25), то в диаграмме S — Н они изображаются наклонными линиями, так как сухой насыщенный пар обладает большей по сравнению с жидкостью энтропией (на величину L/T) и энтальпией (на величину L). Ввиду того, что переход жидкости в сухой насыщенный пар вдоль изотермы-изобары связан только с изменением сухости пара, то в соответствии с уравнениями (42 и 44, гл. VI) Н и S возрастают пропорционально х; поэтому изотермы-изобары являются прямыми линиями (например, BD и др.). Наклон изотерм-изобар отвечает температуре кипения при данном давлении, так как в соответствии с уравнением (32 и 9 гл. Ill) тангенс угла наклона этих прямых равен f " 7,
\dS)р, т~ -г.?/ 'f 4 Ск u 1 4а
- L'~ * " > л н оте к ai
По мере перехода к возрастающему давлению (перемещение по направлению к точке С) крутизна линий возрастает, поскольку повы-
Химическая термодинамика
18
Вступительная статья
шается температура кипения. Так как предельная изотерма-изобара имеет наклон, равный Ткр, то в отличие от 5 — Т диаграммы здесь критическая точка расположена в восходящей части кривой жидкости.
Линии постоянной сухости пара сходятся в критической точке и делят изотермы-изобары на части, пропорциональные значениям сухости х (это следует из уравнений 42 и 44, гл. VI), т. е. так же, как и в диаграмме S — Т.
В области перегретого пара изотермы и изобары не совпадают. Так как вдоль изотерм энтальпия изменяется незначительно, то они располагаются полото и по мере отдаления от кривой пара приближаются к горизонталям, в соответствии с тем, что свойства пара приближаются к свойствам идеального газа, энтальпия которого от давления не зависит. Изобары, наоборот, поднимаются круче и по мере отдаления от ортобарной кривой приближаются к вертикалям, так как пересекают изотермы, помеченные возрастающими значениями Т.
На диаграмме 5—Н для облегчения вычислений иногда наносятся изохоры (пунктирные линии на рис. 2). Они расположены несколько круче изобар и в области насыщенного пара представляют почти прямые линии.
Так как в практических расчетах нижней частью диаграммы обычно не пользуются, то она опускается (см. рис. 26). Это позволяет увеличить масштаб диаграммы и повысить точность вычислений.
В качестве примеров применения диаграммы 5 — Н рассмотрим дросселирование и истечение газа.
Процесс дросселирования согласно уравнению (65, гл. VII) условно изображается в виде горизонталей1).
В соответствии с уменьшением давления и необратимостью процесса (система изолирована) линии процесса идут слева направо.
Рассмотрение рис. 2 приводит к следующим выводам:
1) Дросселирование влажного пара приводит к увеличению его сухости (примером служит процесс ab).
При значительном перепаде давлений влажный пар перегревается (т. е. переходит через состояние насыщения) (процесс cde}.
2) Дросселирование перегретого пара вызывает увеличение степени перегрева, так как температура насыщения при низком давлении будет меньше, чем при высоком (процесс fg).
Условность изображения вызвана тем, что процессне является изоэнталь-пийным, так как в суженном сечении возникают завихрения, приводящие к увеличению кинетической энергии потока, что в соответствии с адиабатностью процесса вызывает уменьшение энтальпии. Лишь после прохождения сужения энтальпия приобретает первоначальное значение (если пренебречь разностью кинетической энергии по обе стороны сопротивления). Таким образом, изображение процесса горизонталью является лишь графическим приемом, позволяющим по значениям pi,t^ и t2 найти остальные свойства вещества после его дросселирования. Однако изменение Н, происходящее между исходным и конечным состояниями, не может отразиться на результате расчета, так как энтальпия является свойством системы.
Вступительная статья
19
3) При высоком давлении дросселирование перегретого пара может вызвать его конденсацию (процесс Л/), а дросселирование
Рис. 2. Схема диаграммы S—Н.
влажного пара может привести к уменьшению х (процесс kl); тем савшм могут быть случаи, когда х2=хг (процесс km).
4) Дросселирование жидкости вызывает частичное испарение ее. (процесс по).
2*
20
Вступительная статья
5) Понижение температуры при одинаковом перепаде давления для перегретого пара значительно меньше, чем для влажного (сравнить процессы ab и fg).
В значительном отдалении от кривой пара температура перегретого пара при дросселировании практически не будет изменяться. При очень больших р и Т возможен даже нагрев, так как изотермы приобретут наклон вниз. Это не наблюдается для водяного пара, так как в пределах практического его применения (р<^300 атм и /<^550° С) инверсионные точки не достигаются х).
Расчет дросселирования по S—Н диаграмме иллюстрируется примером 2, гл. VI, и примерами 14 и 15, гл. VII.
Процесс истечения на диаграмме 5—Н изображается линией, направленной сверху вниз, так как работа, затрачиваемая на кинетическую энергию пот ока, совершается за счет убыли энтальпии газа. Если считать истечение процессом обратимым, то линия процесса будет вертикальна* 2).
Рассмотрение рис. 2 приводит к следующим выводам:
1) При истечении влажного пара сухость его уменьшается (процесс рг).
2) При истечении перегретого пара наблюдается уменьшение его перегрева (процесс st).
3) В известных условиях перегретый пар может стать насыщенным (процесс uw) и даже влажным (процесс uz).
Зная два параметра вещества, находящегося в резервуаре (например, Р] и и давление в окружающей среде (р2), легко найти по диаграмме остальные свойства вещества в точке выпуска из резервуара. Если г<^гкр и если вытекание происходит не через расширяющуюся насадку, то давление газа в месте вытекания из резервуара будет равно гкр-р}-, тогда перпендикуляр из точки (1) надо опустить на критическую изобару (г^-р,).
Иногда вместе cS—Н диаграммой дается номограмма (см. рис. 2), позволяющая по изменению энтальпии сразу определить скорость истечения, не прибегая к уравнению (43, гл. VIII).
Расчет истечения по 5 — //-диаграмме иллюстрируется примером 3, гл. VI (см. также примеры 10 и 11, гл. VII).
5. О ПРАКТИЧЕСКИХ МЕТОДАХ РАСЧЕТА ДАВЛЕНИЯ НАСЫЩЕННОГО ПАРА
В основном тексте книги хорошо рассмотрены некоторые аналитические методы расчета давления насыщенного пара жидкостей, однако среди них почти не представлены методы сравнительного рас
*) Примером перегретого пара, нагревающегося при дросселировании, служит воздух при высоких температурах (см. [293], стр. 77).
2) Наличие потерь на трение в действительном процессе вызывает отклонение от адиабаты вправо, так как процесс сопровождается возрастанием энтропии. Следовательно, в этом случае при одинаковых начальных условиях конечное состояние будет отвечать изобаре р2> расположенной выше, чем при обратимом процессе.
Вступительная статья
21
чета, кроме метода Дюринга, который хотя и применяется широко в США, но отнюдь не является лучшим из них. Здесь мы рассмотрим и сопоставим различные методы сравнительного расчета в их аналитической форме и опишем некоторые графические формы их применения.
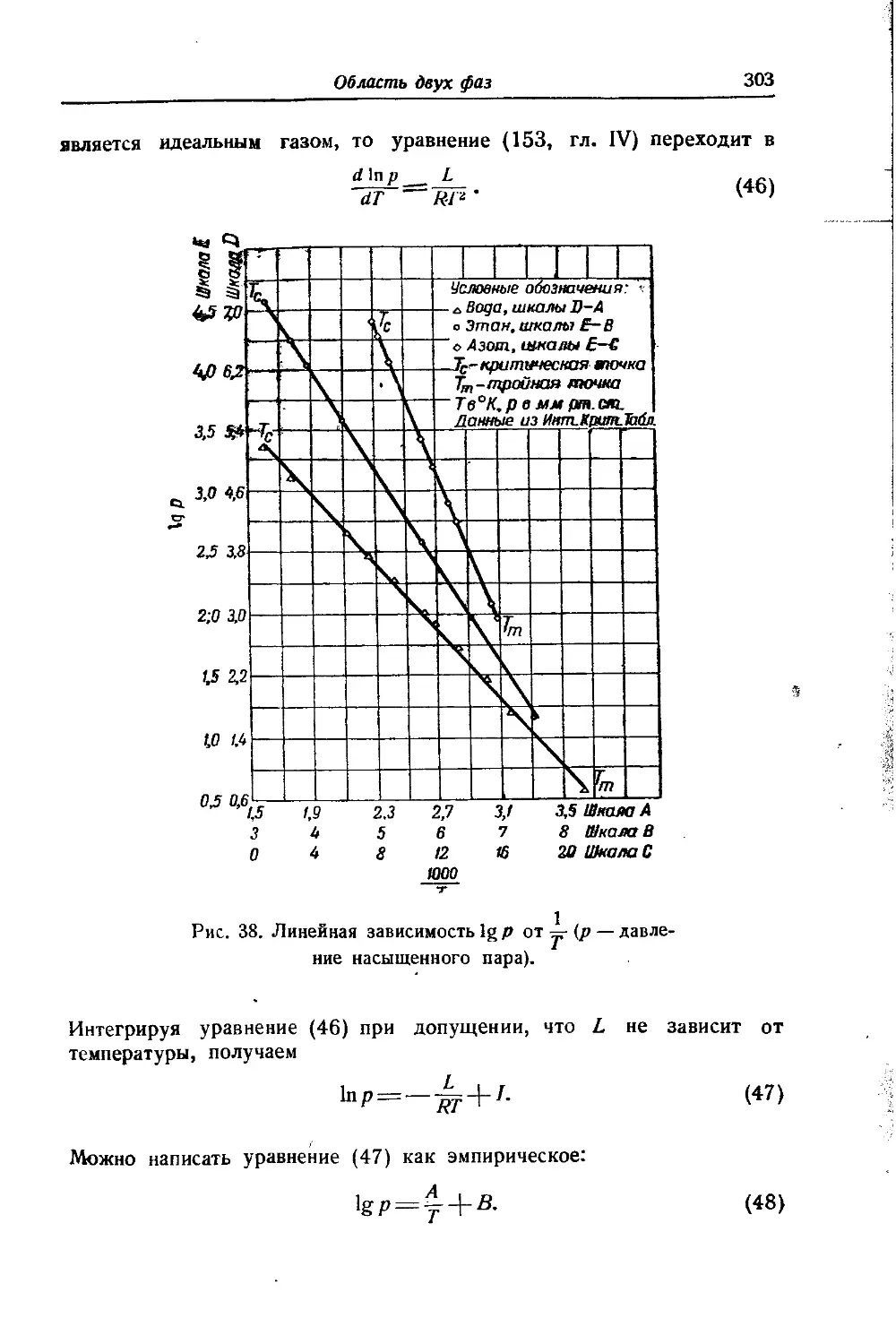
Все методы сравнительного расчета основываются на том, что кривые, выражающие зависимость давления насыщенного пара жидкости от температуры, подобны друг другу, в особенности для веществ, не слишком различающихся между собой по своему химическому характеру и по температурам кипения. Вывод уравнений основывается на применении уравнения Клаузиуса — Клапейрона для двух веществ А и В, для одного из которых (положим, для В) зависимость давления насыщенного пара от температуры известна. Для упрощения рассуждений и большей наглядности мы проведем здесь вывод этих уравнений лишь для области низких давлений пара, пользуясь уравнением Клаузиуса — Клапейрона (46,гл. IV) в форме
(1)
Однако все они могут быть выведены и для области более высоких давлений пара, как это показано для одного из них в примечании39 на стр. 739.
Самое сравнение уравнений (1) для рассматриваемых веществ А и В можно произвести двумя путями: или сравнивать температуры кипения их при равных давлениях (рд = рв), или, наоборот, сравнивать давления пара при одинаковых температурах (ТА=ТВ).
Рассматривая сначала первый путь и производя в уравнении (1) соответствующую замену переменных, получаем уравнение
L Т2
dTA = ~^dTB, (2)
1 bla
дающее искомую зависимость в диференциальной форме.
Интегрирование этого уравнения может быть произведено в трех различных вариантах. Во-первых, допуская, что отношение не зависит от температуры давлений пара отношения порциональны между собой.
L-^-k
и что, следовательно, в условиях равных обоих сравниваемых веществ А и В про-Во-вторых, допуская, что отношение
J bla
(3)
не зависит от температуры и что, следовательно, в условиях равных давлений пара отношения ~ обоих сравниваемых веществ А к В пропорциональны между собой.
22
Вступительная статья
В-третьих, допуская, что отношение
Й=*. <5>
не зависит от температуры и что, следовательно, в условиях равных давлений пара сами мольные теплоты испарения сравниваемых веществ пропорциональны между собой. Эти три варианта допущений, очевидно, не одинаково строги. Наиболее строгим из них является допущение (4). В самом деле, отношение у представляет собой так называемый коэфициент Трутона, и относительно его обычно принимается допущение, что его значение в условиях равных давлений пара для различных веществ в первом приближении4 постоянно или, точнее, является некоторой слабой функцией температуры. Естественно, что допущение (4), требующее не равенства, а только пропорциональности значений его для двух веществ, будет значительно более строгим. Большая строгость этого допущения была показана по другому поводу в работах Герца [107] и Киреева [316]. Допущения (3) и (5) по сравнению с ним значительно менее строги.
Интегрирование уравнения (2) при этих трех допущениях приводит к следующим трем уравнениям. Применяя допущение (3), мы получаем уравнение
+ (6)
дающее линейную зависимость температуры кипения одного вещества от температур кипения другого вещества в условиях равных давлений. Постоянная и постоянная интегрирования Ct, очевидно, могут быть определены по двум известным точкам для А (или kx может быть определено по теплотам парообразования обоих веществ, если они известны). В первом случае, обозначая одним и двумя штрихами температуры, относящиеся к двум различным давлениям, величину kr можно выразить соотношением
и, определяя Сг по уравнению (6), получить готовую для решения формулу
Та = ~^(7в--Г’в) + Та- (8)
‘в ‘в
Это уравнение было установлено Дюрингом чисто эмпирическим путем и описано на стр. 305 основного текста книги тоже без вывода его. А между тем именно вывод его, проводимый параллельно с выводом уравнений других методов сравнительного расчета, позволяет наиболее просто установить значительно меньшую строгость его.
Вступительная статья
23
Пользуясь допущением (4), после интегрирования и перехода от натуральных логарифмов к десятичным, аналогично предыдущему, можно получить уравнение
igTA=k2\gTB + C2, где Л2 и С2 определяются подобно предыдущему, причем
А, =----?-----» •
lgT’B-lgTB
Это приводит к следующей формуле для решений:
Та=(lg Т'в}+lg т'л • lg TB — \gTB
(9)
(Ю)
(П)
Это уравнение было предложено Генглейном [105], который вывел его несколько иным путем, пользуясь допущением о независи-„ L
мости от температуры отношений ----, т. е. для рассматриваемого
L
участка давлений — отношений данного вещества. Такое допуще-i\ 1
ние, очевидно, является довольно грубым, и с ним не согласовывалась хорошая точность самого уравнения. Приведенный здесь вывод, найденный значительно позднее [106], показывает, что это допущение не является необходимым н уравнение (9) основывается по существу лишь на допущении (4), достаточно строгом *).
В третьем варианте, пользуясь допущением (5), можно получить аналогичным путем уравнение
J___k X-J-е
(12)
(13)
откуда
, _Т^в Та-Л
Т’АТ"А'Тв-Гв
Это уравнение несколько иным путем было выведено также Генглейном [106] и позднее вновь предложено Уайтом. Вследствие малой точности его мы не будем на нем останавливаться.
Все три уравнения (6), (9) и (12) допускают упрощения путем применения правила Трутона как в одной из его развернутых форм, например уравнения академика В. А. Кистяковского (35, гл. IX), так и в более упрощенных. Так, например, полагая ~^ = 1, получаем 1 al в
Ь Т$А JL 1 л ТВВ .
, l~TSB' *’ 3'“Г5д’ <14>
!) Аналогичное изменение вывода может быть произведено и для области более высоких давлений пара. Этот вывод, кроме того, показывает значение выбора сравниваемого вещества, чего не мог дать старый вывод.
24
Вступительная статья
откуда все три уравнения приводят к одной формуле:
ТА^~, (15)
7 в где Т' = Ts.
Уравнения (6), (9) и (12) выведены нами путем сравнения двух веществ в условиях равных давлений пара. Если же проводить сравнение в условиях равных температур, то из (1) можно получить аналогичное (2) уравнение
d\npA^L^-d\npB (16)
1aL-b
или, так как ТА Тв,
dhpA^fyilnpg.
(17)
Так как в условиях равных температур все три допущения, аналогичные допущениям (3), (4) и (5), идентичны между собой, т. е.
LA_ LATB
Lb TaLb
LaT2b
- v S const, T\LB ‘
(18)
то этим путем можно получить лишь одно уравнение. Интегрируя уравнение (17) при допущении (18) и переходя от натуральных логарифмов к десятичным, мы получаем уравнение
Ъра=Ь№рв+с»
(19)
где kk и могут быть подобно предыдущему определены по двум известным точкам илн может быть определено по теплотам парообразования, если они известны для обоих веществ. Применяя первый из этих вариантов, получаем для соотношение
, 1&Ра~ >gРл
#4 =-----------
Ig/'B— ^Рв
(20)
откуда формула для расчета получается в следующем виде:
= ~^(IgPfi-lgpB)4-l&Pn. (21)
1&Рв~ 1&Рв
Это уравнение выведено Киреевым [319], показавшим одновременно хорошую практическую применимость его для ряда систем. Позднее оио было вновь выведено и предложено Отмером (см. стр. 442). Легко видеть, что, подставляя в него вместо 1gрд принимаемого стандартного вещества В выражение температурной зависимости 1gрв, если
Вступительная статья
25
таковая известна, нетрудно выразить в явной форме и зависимость 1g рА от температуры.
Все четыре рассмотренных уравнения (6), (9), (12) и (19) дают линейное соотношение между некоторыми простейшими функциями давления насыщенного пара или температур кипения двух сравниваемых веществ. Каждое из них для своего решения удовлетворяется знанием двух точек для рассчитываемого вещества или одной точки для него и теплот парообразования обоих веществ. Каждое из них допускает использование и большего числа точек и определения наиболее вероятного значения постоянных k и С методом наименьших квадратов. Все они могут быть применены не только для расчетов давления пара или температур кипения, но также и для расчетов соответствующих тепловых эффектов. Все они в равной степени допускают возможность наглядной графической обработки результатов. Однако эти общие свойства, очевидно, отнюдь не делают их равноценными в отношении практического применения. Приведенный выше вывод и сопоставление практической применимости их показывает, что наиболее удобными и простыми в обращении в зависимости от исходных данных и постановки задачи являются уравнения (6) или (19), а наиболее точными по результатам расчета в зависимости от выбора стандартного вещества и условий расчета являются уравнения (9) или (19).
Таблица 5
Давления насыщенного пара (бензола в мм ртутного столба) по экспериментальным данным н рассчитанные по уравнениям (6), (9) и (19)
t, °C Эксперимент. Рассчитанные по гексану Рассчитанные по воде
ПО уравн. (6) ПО уравн. (9) ПО уравн.(19) ПО уравн. (6) ПО уравн. (9) ПО уравн.?(19)
1 2 3 4 5 6 7 8
10,0 45,43 45,43 45,43 45,43 45,43 45,43 45,43
20,0 74,66 75,9 75,8 74,6b 73,0 73,7 74,48
30,0 118,24 120,4 120,2 118,51 113,7 115,3 116,16
40,0 181,08 184,6 183,9 181,10 172,7 175,6 179,93
50,0 268,97 274,1 273,6 268,22 255,7 260,2 266,99
60,0 388,6 395,2 394,5 386,7 370,3 376,2 385,7
70,0 547,4 552,9 552,0 548,1 525,4 526,4 543,8
80Д 753,6 764,6 762,2 752,8 731,2 138,0 749,7
90,0 1016,1 1032,0 1031,0 1014,1 1000 1004 1013,2
100,0 1344,3 1344 1344 1344 1344 1344 1344
110,0 1748,2 1778 1777 1752 1781 1768 1754
120,0 2238Д 2272 2272 2247 2335 2287 2253
130,0 2824,9 2858 2861 2839 2997 2933 2854
1 1,01845 0,98618 1,05930 1,11869 1,19457 0,76760
С 4-4,50 4-0,04862 — 0,32891 — 63,09 — 0,52320 4-0,91725
26
Вступительная статья
Для практических расчетов по всем им правильный выбор стандартного вещества, по которому производится расчет, всегда бывает существенным. Допущения, использованные при выводе всех рассматриваемых уравнений, являются наиболее строгими для сопоставления веществ, близких между собой. Полярные вещества (вода, спирты, кислоты, аммиак, ацетон и другие) дают лучшие результаты при
расчете по одному из полярных веществ, и для них в качестве стандартного вещества пользуются большей частью водой. Неполярные
и слабо полярные вещества (углеводороды, галоидопроизводные,
эфиры и другие) рассчитывают по одному
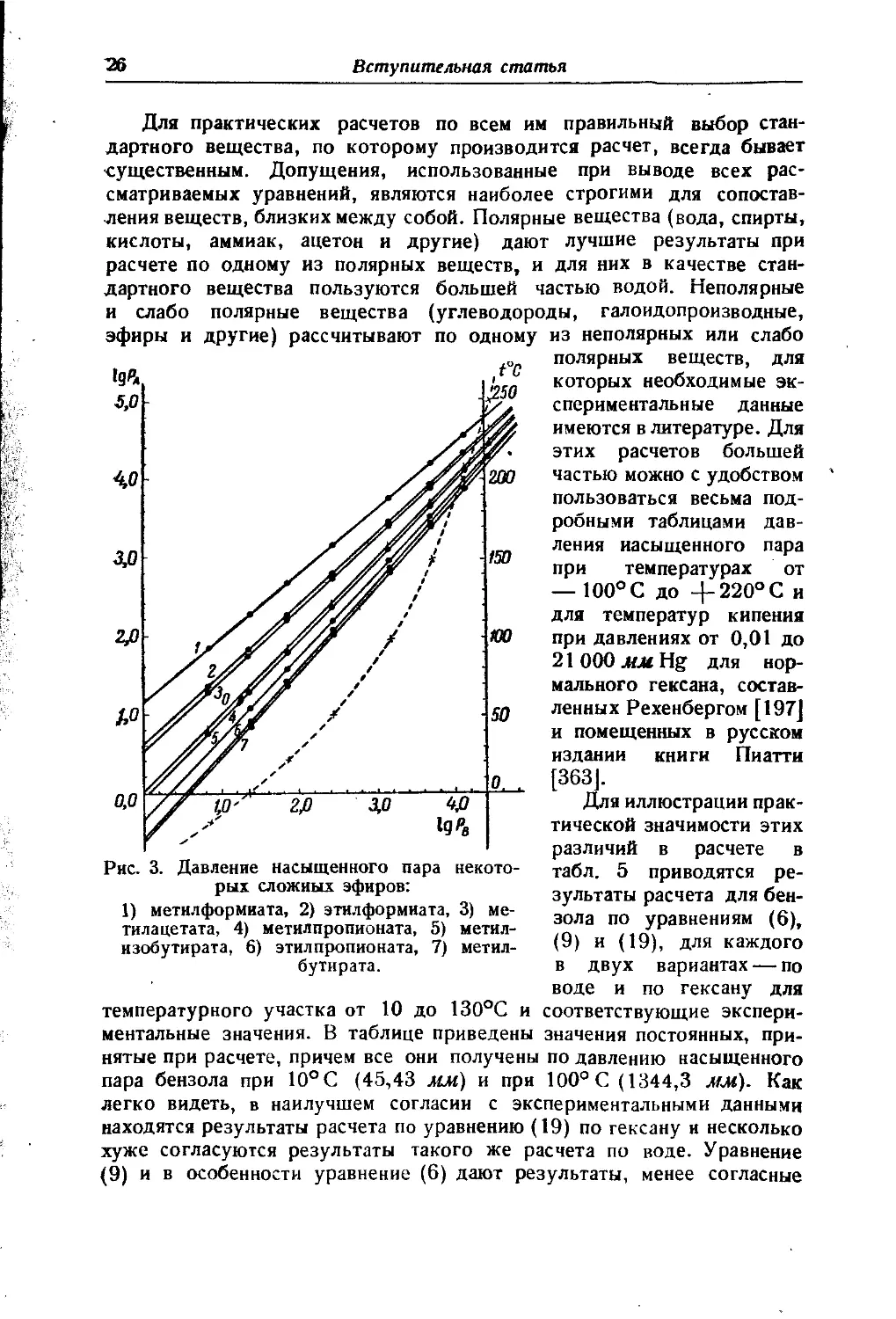
Рис. 3. Давление насыщенного пара некоторых сложных эфиров:
1) метилформиата, 2) этилформиата, 3) метилацетата, 4) метилпропионата, 5) метилизобутирата, 6) этилпропионата, 7) метилбутирата.
температурного участка от 10 до 130°С и ментальные значения. В таблице приведены
из неполярных или слабо полярных веществ, для которых необходимые экспериментальные данные имеются в литературе. Для этих расчетов большей частью можно с удобством пользоваться весьма подробными таблицами давления насыщенного пара при температурах от — 100° С до 4-220° С и для температур кипения при давлениях от 0,01 до 21 000 мм Hg для нормального гексана, составленных Рехенбергом [197] и помещенных в русском издании книги Пиатти [363].
Для иллюстрации практической значимости этих различий в расчете в табл. 5 приводятся результаты расчета для бензола по уравнениям (6), (9) и (19), для каждого в двух вариантах — по воде и по гексану для соответствующие эксперизначения постоянных, при
нятые при расчете, причем все они получены по давлению насыщенного пара бензола при 10°С (45,43 мм) и при 100° С (1344,3 мм). Как
легко видеть, в наилучшем согласии с экспериментальными данными находятся результаты расчета по уравнению (19) по гексану и несколько хуже согласуются результаты такого же расчета по воде. Уравнение (9) и в особенности уравнение (6) дают результаты, менее согласные
Вступительная статья
27
с опытом, причем для обоих их расчет по гексану, как и следовало ожидать, дает лучшие результаты, чем расчет по воде. Впрочем, не всегда уравнение (19) дает лучшие результаты, чем (9), однако в общем расхождения незначительны. Для веществ, менее близких между собой, наблюдается и обратная картина. Преимущество применения того или другого из них нередко зависит еще от соотношения в температурах и давлениях, для которых имеются данные для стандартного вещества и для которых требуется получить сведения для рассчитываемого вещества.
Графические расчеты. Наряду с применением в аналитической форме все эти методы могут быть применены также и для графических расчетов. На рис. 3 приведены для примера две формы применения графического метода для расчетов по уравнению (19). На первом из них по оси абсцисс отложены 1g рв стандартного вещества (в данном случае этилацетата), а по оси ординат Ig рА некоторых других сложных эфиров. Для каждого вещества опытные данные дают одну прямую до области, непосредственно прилегающей к критической точке. Вертикальные прямые отвечают постоянному давлению и отсекают на прямых различных веществ точки кипения их под этим давлением. Горизонтальные прямые соответствуют, наоборот, постоянным температурам и отсекают на прямых различных веществ значения, характеризующие давления насыщенного пара их при этих температурах. Пунктирная кривая дает зависимость для стандартного вещества Т = у (1gрв) в масштабе, указанном с правой стороны. Она дает возможность связать температуру и давление пара рассчитываемого вещества путем несложных графических построений. Как следует из приведенного выше вывода, углы наклона каждой прямой определяются отношением теплот парообразования сравниваемых веществ. Вследствие этого прямые для веществ, близких между собой по химическому характеру, обладают некоторыми общими признаками. Одним из таких свойств является взаимная пересекаемость их почти в одной точке. Точка эта располагается обычно далеко за пределами реального существования жидкости и физического смысла не имеет, но она дает возможность при ориентировочных расчетах удовлетворяться для веществ одного класса знанием одной 'экспериментальной точки для построения прямой, что нередко бывает весьма ценно.
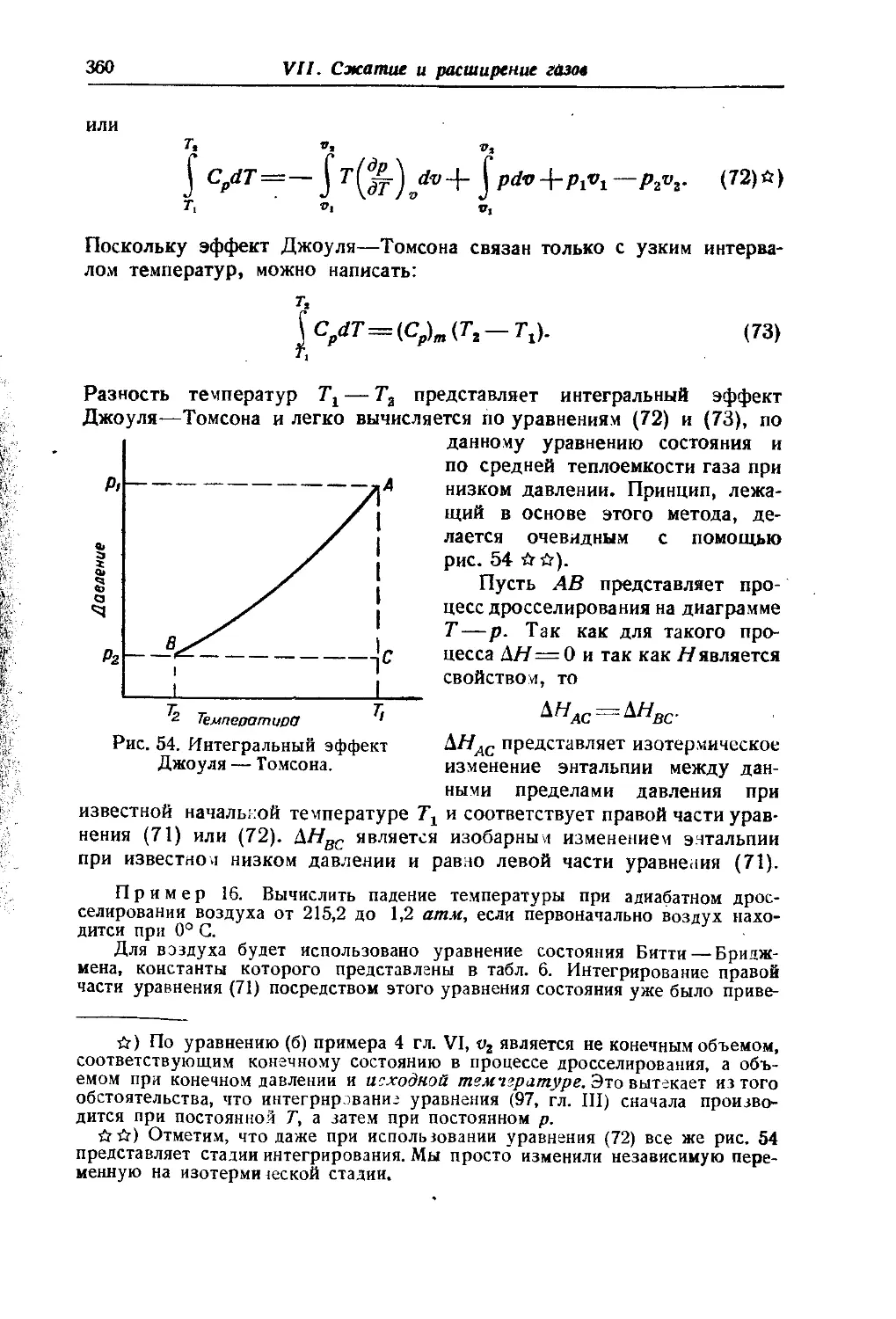
Для практических расчетов различных веществ в особенности удобным оказывается применение графика в параллельных шкалах. Соответствующая номограмма представлена на рис. 4 в комбинированной форме для расчетов по воде и по гексану. В середине ее помещена общая для обеих частей ее шкала давлений и по бокам ее — шкалы температур по гексану и по воде1).
!) Обширный и весьма ценный материал по применению номографических методов расчета можно найти в .Атласе номограмм по физической химии*, составленном Виноградовым и Красильщиковым [277].
28
Вступительная статья
В этой номограмме каждому веществу соответствует одна точка, и любая прямая, проходящая через нее, пересекает оси температуры и давления при значениях их, соответствующих друг другу, т. е. непосредственно связывает температуру с отвечающим ей давлением насыщенного пара данного вещества. Так, например, прямая MN показывает, что хлорбензол (точка № 33) обладает при температуре 60°С давлением насыщенного пара, равным 61 мм рт. ст. Само положение точки данного вещества определяется 'по двум известным значениям давления насыщенного пара его при каких-нибудь температурах как место пересечения двух прямых, что видно на показанном на номограмме определении положения точки бензола (№ 24) по известным данным — 45,4 мм при 10°С (прямая CD) и 269 мм при 50°С (прямая ЕР).
Точность получаемых результатов может быт^ иллюстрирована тем, что данные двух авторов (Юнга и Ворингера) дают для нормального октана точки №№ 32 и 31, заметно отличающиеся друг от друга, так что ошибка в расчете по этой номограмме в данном случае не превышает расхождения между данными двух авторов.
На номограмме приведены точки для 61 различного вещества и могут быть построены для других. При этом углеводороды,
Значение точек на рисунке 4
1. СН3—СН3 22. и-С6Ни 43. С10Н8
2. CH s СН 23. ССЦ 44. л-Крезол
3. SiH3 — СН3 24. С6Нв 45. С6Н8 —С6Н8
4. СН3СН = СН2 25. СН3СООС2Н5 46. а-Бромнафталин
5. СН3СН2СН3 26. СН2С1СН2С1 47. а-Нафтол
6. СН2=С = СН2 27. C6H6F 48. р-Нафтол
7. СН3С1 28-. Я-С7Н16 49. NH3
8. СН2 = СНС1 29. Диоксан 50. CH3NH2
9. а-Бутилен 30. С6Н8СН3 51. СН3СОСН3
10. 1,3-Бутадиен 31. н-С8Н18 52. СН3ОН
11. я-C^Hjq 32. н-С8Н18 53. С2Н5ОН
12. fi-Бутилен 33. С6Н5С1 54. Н2О
13. С,Н5С1 34. л-С,;Н4 (СНз)2 55. СНдСООН
14. Изопрен 35. С6Н5Вг 56. С2Н8СООН
15. (С2Н8)2О 36. я-С10Н22 57. изо-С3Н?СООН
16. НСООСНз 37. C6H5NO2 58. Бутиленгликоль
17. н-С8Н12 38. Декалин 59. СН2ОН— СН2ОН
18. С,Н8Вг 39. С6Н81 60. СН2ОН —СНОН—
19. СН2С1, 40. C6H5NH2 — СН2ОН
20. НСООС2Н8 41. о-Крезол 61. Hg
21. СНС13 42. Тетралин
Рис. 4. Номограмма для расчета давления насыщенного пара жидкостей, построенная по уравнению lgpA = A4lgpa4-C4(19).
А — В—общие давления пара растворов NH« в воде, содержащих 0, 10, 15, 20, . . . 100 вес. ®/0 NH3.
30
Вступительная статья
галоидопроизводные, эфиры и прочие располагаются в левой части номограммы для расчета их по гексану, а спирты, кислоты, аммиак, ацетон, ртуть и другие — в правой для расчета их по воде.
Весьма удобным для приближенных расчетов оказывается использование того свойства этой номограммы, что точки нормальных жидкостей одного класса располагаются практически на одной прямой. Так, например, точки углеводородов и их галоидопроизводных почти точно лежат на прямой RS. Это позволяет при приближенных расчетах для этих соединений удовлетворяться знанием одной температуры кипения при каком-нибудь давлении, определяя полржеиие точки данного вещества по пересечению прямой данной температуры кипения с прямой RS. Когда же известной является, температура кипения при атмосферном давлении, как это бывает в большинстве случаев, то последнего построения можно и не производить, пользуясь непосредственно шкалой температур кипения при 760 мм, специально для этой цели нанесенной на линии RS с правой стороны ее.
В группе полярных жидкостей наблюдается подобная же закономерность. Близко к прямой R'S1 располагается большинство точек соответствующих веществ. Это позволяет для ориентировочных расчетов использовать эту закономерность и для этих веществ *).
Хотя мы рассматриваем здесь только расчеты давления пара чистых веществ, одиако попутно следует отметить, что все три описанные уравнения (6), (9) и (19) могут применяться и к расчетам давлений пара растворов как общего, так и парциальных, так как исходное для них уравнение Клаузиуса—Клапейрона относится, как известно, и к этим случаям. Так, в частности, номограмма рис. 4 тоже может применяться и для этих систем. Кривая АВ, показанная иа ней, соответствует точкам водно-аммиачных растворов различного состава для расчета общего давления пара. Она дает возможность, так же легко, как и для чистых жидкостей, определить давление пара для раствора любого состава. Например, прямая KL показывает, что 38-проц. раствор аммиака должен иметь при 20°С давление пара, равное 880 мм. Подобные же кривые для парциальных давлений не показаны на рисунке во избежание его чрезмерного загромождения.
х) Точность результатов, получаемых при номографических расчетах, зависит, очевидно, как от масштаба номограммы, так н от точности того уравнения, по которому она построена. Последнее всегда следует иметь в виду, и применение номограмм, построенных в большом масштабе, но по недостаточно точному уравнению, не может быть ничем оправдано, так как время, затрачиваемое на расчет по ним, одинаково. К сожалению, и в наше время для расчетов давления насыщенного пара нередко применяют номограммы, построенные по менее точным уравнениям. Так, в частности, недавно была опубликована [377а] сводка результатов номографического расчета давлении насыщенного пара различных химических соединений, выполненная по номограмме, построенной в большом масштабе, но по уравнению вида уравнения (47, гл. VI), которое является значительно менее точным, чем описанные здесь уравнения (9) и (19).
Вступительная статья
3t
В заключение полезно остановиться на одном чисто эмпирическом уравнении, предложенном Антуаном [4], которое благодаря его удачной математической форме находит и в настоящее время широкое применение в качестве интерполяционной формулы. Оно содержит три индивидуальные постоянные А, В и С н имеет Следующий вид:
т
Обладая большой гибкостью, оно дает вместе с тем возможность не только рассчитывать давление пара для различных температур, но и производить легко обратные расчеты путем преобразования его в форму
Преимущества этого уравнения недавно подробно рассматривались Томсоном [246], и оно было положено в основу расчета интерполированных значений в сводке давлений насыщенного пара углеводородов, опубликованных Тиличеевым [39Q] и др.
Для частного случая, когда С = 230, оно переходит в уравнение (59, гл. VI).
6. К РАСЧЕТУ РАВНОВЕСИЯ НА ОСНОВАНИИ ТРЕТЬЕГО ЗАКОНА ТЕРМОДИНАМИКИ
I
Темкин и Шварцман [388] предложили упрощенный расчет равновесия, позволяющий полностью обойти вычисление константы интегрирования I уравнения (14, гл. XI) (а при отсутствии значения Д//298 — и константы Д/70). Этот способ заключается в следующем: если уравнение AZ£=<p(7’) написать в виде т т
^ = A^98-^098-T pC,rfT (1)
298 298
и ввести обозначение т т
мп=\Ц^Тпат, (2)
298 298
то получим: для п — 0
34g In 2 ~I- f 1 и для
М Т“ I 298’2°+1 298-2° /41
» «(л+Ь^^+О Т п ’ ' >
32
Вступительная статья
Тогда уравнению (1) можно придать следующий вид:
AZ® = ДН298 — TAS0g8 — Т (М^а++ ^1) (5>
[если зависимость Ср = ср (Т) выражена уравнениями типа (15, гл. IX)] или
= ДЯ293 — ТАЗ^ — Т (М0Ьа + М^Ь 4- М'^с) (6)
[если зависимость Ср—щ(Т’) выражена уравнениями типа (16, гл. IX)].
Удобство применения уравнений (5) и (6) обусловлено возможностью свести в таблицы значения Л10, М1г Л12 и М'2 для различных температур [388]. Приведенные уравнения позволяют не только легко найти зависимость А2° = 'д (Т), но при наличии значений AZ0 (илн/Со) при различных температурах определить величины Д//298 и ASjgg. Для этого следует построить график, откладывая по оси абсцисс ЦТ, а по оси ординат (AZ0/?”) 4-7H0Aa-|-• • • • Тогда наклон прямой определит величину Д//298, а отрезок, отсекаемый на оси ординат, — величину AS°gg.
Поэтому, если ставится задача нахождения величин Д/7298 и Д^2эз’ т0 б°лее целесообразным следует считать не одновременное определение А//о и I (см. стр. 558), а только что рассмотренный способ.
II
Необходимые для расчета равновесия значения «йв1) неизученных веществ можно найти с помощью различных закономерностей. Это имеет большое значение^ так как такой путь позволяет значительно расширить фонд данных по энтропиям без непосредственного измерения равновесия и без трудоемкой низкотемпературной калориметрии.
Если для органических соединений на основании сопоставления значений AS, вызванных различными изменениями молекулярной структуры, удалось обнаружить лишь известные закономерности для некоторых классов соединений, то для неорганических веществ, как показал Киреев [322], можно установить некоторые общие закономерности.
В основу расчета положены: 1) изменение энтропии ASa при образовании данного соединения в его нормальном состоянии из элементов, находящихся в гипотетическом состоянии идеального одноатомного газа, и 2) изменение энтропии &S{ при образовании данного соединения в состоянии идеального газа из элементов, находящихся в состоянии идеального одноатомного газа. Тогда изменение энтропии при
!) Для простоты обозначения символ $J)gg в дальнейшем заменен символом S.
Вступительная статья
33
образовании данного соединения из элементов будет
Д5=Л5Д [У,Пэл $ал — ^Пал =
= AS, + (5 — S,.) — — (5аД]> (7)
где S и 5,-— энтропии вещества соответственно в нормальном и идеализированном состояниях и (Ss^)z — энтропия элемента в идеализированном состоянии.
Величина Д5а определяется в основном количеством атомов в молекуле соединения. Объединение существующих данных в группы с одинаковым числом атомов в молекуле показывает, что только в единичных случаях указанное влияние оказывается перекрытым. Если в каждой группе ASa изменяется в сравнительно широких пределах, то, разделив вещества на подгруппы по признаку однотипности молекул (по составу и структуре), удается сузить интервал колебаний Д5а. Внутри подгруппы однотипных соединений в* свою очередь обнаруживаются закономерности,, обусловленные видом металла, кислотного остатка и положением соответствующего элемента в периодической системе. >
В результате закономерность изменения ASa оказывается столь отчетливой, что создается возможность предсказать отсутствующие значения ASO. По этим данным легко вычислить стандартные энтропии еще неизученных веществ, так как необходимые для пересчета изменения энтропии, связанные с переходом грамм-атома элемента из нормального (в стандартных условиях) состояния в состояние идеального одноатомного газа, т. е. (5эл)г— Sajt, известны почти для всех элементов [322].
В отличие от 5 и AS величины Д*$а вследствие исключения влияния агрегатного состояния элементов обнаруживают определенные правильности; еще большие правильности обнаруживают A5Z, так как в этих величинах полностью исключается влияние агрегации самого соединения. Поэтому внутри групп показывают меньшие колебания, и между группами имеется более резкая граница, чем в случае ASa. К сожалению, ограниченность экспериментального материала для соединений (в отношении теплот парообразования, теплот возгонки и зависимости давления пара от температуры), а также невозможность в ряде случаев их нахождения (из-за неустойчивости некоторых соединений) не позволяет широко применять расчет AS,.
Капустинский и Яцимирский [313] недавно предложили метод расчета энтропии соединений ионного типа, основанный на аддитивности энтропии иоиов.
Киреев [322а] в развитие разработанного им метода расчета энтропии показал, что можно добиться хороших результатов при вычислении, рассматривая реакцию образования соединения не из простых веществ, а из более сложных составных частей (например, карбонатов металлов из окислов металлов и углекислоты, кристаллогидратов из безводных солей и Н2О и т. д.). При этом не только исключается 3 Химическая термодинамика
34
Вступительная статья
влияние различия в валентном состоянии, но и в значительной степени исключаются различия в структуре, так как сравниваются однотипные соединения.
Если считать, что для однотипных реакций можно принять среднее значение AS, то, зная Д/f, можно рассчитать величину AZ0, а затем /Са. При этом ошибка расчета не превышает погрешности экспериментального определения тепловых эффектов, так как последние большей частью известны с недостаточной точностью, и поэтому неточность в энтропии не сказывается иа величине AZ0.
Разность в AZ° двух однотипных реакций не изменяется с температурой.
7. ОВ АЗЕОТРОПНЫХ СМЕСЯХ
Азеотропные смеси по своей природе и внутреннему строению в общем случае ничем ие выделяются из числа других жидкостных смесей. Однако они имеют большое практическое значение, так как создают значительные затруднения в процессах разделения смесей путем перегонки, и поэтому будет целесообразно рассмотреть некоторые свойства их более детально, чем это сделано в основном тексте книги.
Образование азеотропы, т. е. возникновение максимума или минимума на изотерме общего давления пара (или на изобаре температур кипения) смесей в данной системе, определяется соотношением двух факторов: 1) различия в давлениях пара (или соответственно температур кипения) компонентов в чистом состоянии и 2) степени отклонения системы от законов идеальных смесей, т. е. от линейной зависимости давления пара от состава. Как показывает рис. 5, если компоненты смеси в чистом состоянии мало различаются по
А в давлению пара, то даже весьма незначительное
Рис. В. Образо- отклонение от идеальности приводит к образова-ваиие азеотроп- нию азеотропы. Система бензол — циклогексан мо-иых смесей. жет служить примером такого поведения1). При большем различии компонентов для этого требуется уже большее отклонение от идеальности.
Положение азеотропной точки по составу, как легко видеть по тому же рисунку, будет в общем случае тем ближе к эквимолекулярной
*) Интересно отметить, что в системе втиленхлоргидрин— пропилея-хлоргидрин, судя по результатам ориентировочных определений, приведенных в работе Киреева, Каплана и Злобина [323], компоненты обладают также практически одинаковыми температурами кипения и смесь подчиняется законам идеальных смесей. В такой системе для любой смеси состав равновесного пара будет практически одинаков с составом дайной смеси.
Вступительная статья
35
смеси, чем ближе по давлению пара компоненты в чистом состоя-нии. Оно будет отличаться от нее в сторону большего содержания более летучего компонента в системах, обладающих максимумом давления пара, и в обратную сторону в системах с минимумом давления пара.
Сама по себе природа отклонений от идеальности не определяет в общем случае возникновения и положения азеотропы. Все формы взаимодействия, вызывающие эти отклонения,—различия в интенсивности и характере ван-дер-ваальсовского притяжения между молекулами, образование молекулярных соединений (в частности путем образования водородной связи) или уменьшение степени ассоциации одного из компонентов — часто одновременно в различных сочетаниях имеют место в данной системе, и обычно мы не можем разделить эти влияния. Однако при значительном преобладании какой-нибудь из форм это иногда оказывается возможным, и в этих случаях можно установить, что образование соединений между молекулами компонентов усиливает тенденцию к образованию минимума на кривой давления пара, а уменьшение степени ассоциации действует в обратном направлении.
В последнем случае, при образовании смесей сильно ассоциированных жидкостей с неполярными компонентами, когда тенденция к расслоению достигает в системе значительной степени, максимум становится все более плоским, как это показано на верхней кривой рис. 5 (в соответствии с тем, что при достижении расслоения здесь будет располагаться горизонтальная часть кривой общего давления пара). В таких системах со значительной тенденцией к расслоению положение азеотропной точки по составу может легко варьировать под действием разных факторов, и под их влиянием могут возникать отклонения от указанной выше общей закономерности.
Изменение температуры в общем случае изменяет положение точек максимума или минимума на кривых давления пара (так же, как изменение давления влияет на положение азеотропных точек на кривых температур кипения). Чувствительность к этим воздействиям может быть весьма различной. Она определяется, с одной стороны, формой максимума или минимума (чем более острыми они будут, тем, очевидно, меньшими будут изменения в их положении по составу при изменении температуры или давления), с другой стороны — различием в мольных теплотах парообразования компонентов, так как при малом их различии йзменение температуры будет примерно в одинаковой степени изменять давления пара компонентов и, следовательно, мало влиять на положение азеотропы по составу. При более значительной разнице в теплотах парообразования у компонента с более высокой теплотой парообразования давление насыщенного пара будет сильнее возрастать с повышением температуры, чем у другого компонента, и его содержание в парах и, в частности, в азеотропной смеси будет относительно увеличиваться. Хотя, строго говоря, в этом 3*
36
Вступительная статья
случае должны сопоставляться мольные теплоты парообразования компонентов из смеси, однако практически большей частью можно пользоваться значениями их теплот парообразования в чистом состоянии.
В количественной форме эти соотношения рассматривались в работах Вревского [279,280,282] и Киреева [320,321]. Позднее эти вопросы также подвергались исследованию в работах Редлиха и Шутца [195] и Кульсона и Герингтона [49].
Практический интерес представляет также вопрос о влиянии введения третьего компонента на положение азеотропной точки. Для небольших количеств третьего компонента эти влияния в качественной форме определяются (как и в отношении влияния изменения температуры или давления) тем, что чувствительность положения азеотропной точки по составу будет тем большей, чем менее острым будет максимум или минимум в дайной системе, так как это увеличивает чувствительность его к изменению различия в давлениях пара компонентов в чистом состоянии. С другой стороны, эти влияния зависят также от того, насколько различны будут понижения давления пара основных компонентов над смесью, вызываемые добавками третьего компонента. Это зависит уже от химической природы компонентов.
Мы говорили до сих пор об азеотропных (постоянно кипящих) смесях, т. е. о смесях, в которых при равновесии состав пара одинаков с составом жидкости. При больших скоростях испарения, когда равновесие по составу не успевает достигаться, подобную же роль играют так называемые постоянно испаряющиеся смеси. Исследованию основных соотношений между составами азеотропных (постоянно кипящих) смесей и постоянно испаряющихся смесей были посвящены интересные работы Скляренко и Баранаева [268, 373, 374, 375, 376, 377]. Однако рассмотрение этих работ, как равно и других их работ, посвященных изучению вопросов, связанных со скоростью испарения жидкостей, выходит за рамки термодинамики.
ИЗ ПРЕДИСЛОВИЯ АВТОРА
Нас могут вполне законно спросить: „Зачем еще одна книга по термодинамике? Разве сейчас по этому вопросу не имеется книг уже в достаточном количестве?* Термодинамика является методом разрешения ряда проблем, методом разносторонним и широко применимым. Она находит многочисленные приложения в физике, химии, машиностроении и химической технологии и в других областях науки. Часто в каком-нибудь высшем учебном заведении в США ведутся три или даже четыре курса термодинамики, относящихся к различным кафедрам. По общему признанию, основы термодинамики одинаковы для всех областей, но различны способы приложения и точки зрения. Что же оправдывает выпуск автором новой книги по термодинамике?
Когда начиналась работа над этой книгой, не существовало ни одного произведения, посвященного специально приложениям термодинамики к проблемам химии и химической технологии. С тех пор, как известно автору, появилась книга, трактующая исключительно об этом предмете, и несколько других, кратко разбирающих его наряду с другими темами. Автор уверен, что найдется место по меньшей мере и еще для одной книги.
Данная книга выросла из лекций, чтение которых было начато в Пеле более 15 лет тому назад. Хотя она предназначается прежде всего для химиков и инженеров-химиков, ио может оказаться полезной также для ученых и техников иного профиля, поскольку при пользовании ею не требуется никаких предварительных знаний по термодинамике, а каждый вопрос излагается на основе общеобразовательных курсов, обязательных в любом хорошем учебном заведении, готовящем инженеров или естествоиспытателей.
Бесспорно, план настоящей книги является своеобразным, по крайней мере в некоторых отношениях. Первые две главы посвящены развитию основных концепций и особенно идей, лежащих в основе двух великих законов термодинамики. Это сделано намеренно с минимальным использованием математики. После того, как эти концепции освещены, к их развитию привлекается математика — такой метод принят в гл. III. Первая математическая обработка ограничивается очень простыми типами систем. Следующей логической ступенью является распространение выводов на более сложные системы, чему и посвящена гл. IV. Идея, лежащая в основе этой главы, представляет развитие с помощью вычислений концепций двух законов в общие уравнения состояния, из которых при особых условиях почти автоматически получаются многие специальные уравнения. Эти уравнений
38
Из предисловия автора
обычно выводятся в различных главах несколькими независимыми способами. Выбранный путь имеет то преимущество, что показывает их взаимосвязь. Однако он имеет и недостаток, заключающийся в том, что главы, усеянные математическими формулами и не оживленные каким-либо практическим приложением, становятся слишком сухими. Однако в них заключается полезный запас зависимостей, к которым можно обращаться впоследствии, когда возникнет надобность.
После вывода диференциальных уравнений, связывающих термодинамические свойства с переменными состояния, уравнения следует интегрировать. Для этого необходимы рг»/-даиные. Поэтому следующей ступенью является обзор этих данных и уравнений состояний, выражающих их. Это составляет предмет гл. V. Гл. VI вводит диференциальные уравнения и вместе с ними средства для их интегрирования и излагает числовые расчеты термодинамических свойств.
Первые шесть глав закладывают фундамент для приложения термодинамики к специальным операциям и процессам, составляющим главный предмет остальных семи глав. Выбор приложений всегда можно оспаривать. Автор никоим образом не считает, что его выбор является наилучшим, он просто выбрал тот материал, который казался для него наиболее интересным и который он разработал иа основе собственного опыта. Опущено много важвого, что автор охотно включил бы в книгу, если бы время и размер книги позволили это.
Автор старался постоянно развивать доминирующую мысль, что термодинамика должна быть полезным орудием для химика и инженера-химика, и это нужно непрестанно демонстрировать решением небольших специальных задач практического типа. Хотя предлагаемая книга в этом отношении далека от идеала, все же в нее включено 135 примеров с решениями. Надо надеяться, что эти примеры откроют читателю пути к разрешению проблем, с которыми он встретится в своей практической деятельности.
Многие лица косвенно участвовали в составлении этой книги, помогая автору сформулировать его мысли, но нет никакого другого способа оплатить им долг, кроме как выразить общую благодарность. Автор благодарит своего коллегу д-ра Гардинга Блисса, который критически рассмотрел рукопись и дал много ценных предложений. Наконец, автор хочет выразить свою давнюю признательность Гервею Н. Девису, ранее профессору машиностроения в Гарвардском университете, затем президенту Стивенсовского технологического института, а в настоящее время директору Бюро по производству исследований и разработок при Управлении военного производства США. Г. Девис многие годы назад ввел автора в предмет термодинамики и возбудил в нем интерес к ней, которому автор обязан созданием этой книги.
Нью-Гавен, Коннектикут.
Апрель, 1944 г.
СПИСОК ВАЖНЕЙШИХ ОБОЗНАЧЕНИЙ
1. ЛАТИНСКИЕ ПРОПИСНЫЕ БУКВЫ
А — Работа.
Эмпирическая константа. Компонент в реакции.
Ао — Передаваемая работа (работа на валу).
В — Эмпирическая константа. Компонент в реакции. «Пригодность*.
С — Мольная теплоемкость (С относится к средней теплоемкости или к парциальной мольной теплоемкости).
Эмпирическая константа.
рр
Коэфициент сжимаемости^;.
Коэфициент расхода при истечении.
Со —Гидравлический коэфициент.
Са—Адиабатный коэфициент.
Ст — Коэфициент, основанный на средней плотности.
Концентрация, моль) л. Компонент в реакции. Константа интегрирования.
D — Диаметр.
Эмпирическая константа.
Степени свободы.
Компонент в реакции.
Число молей дестиллата.
Е— Электродвижущая сила (э.д.с.). Эмпирическая константа.
Полный коэфициент фракционирования или отношение обогащения (дестилляция).
F—Свободная энергия (С—TS) на единицу массы или на моль ♦). Сила, общая нагрузка.
Энергия, затрачиваемая на трение.
Эмпирическая константа.
Число молей питания в процессе перегонки.
G — Общее термодинамическое свойство иа единицу массы или иа моль &).
Скорость массы в единицу времени на единицу площади сечения.
G — Парциальное значение свойства dQ на единицу массы ч— или
dG
на один моль vrr, где oNi
N/—число молей какого-либо компонента и тг- — масса какого-либо компонента в г или кг.
Следовательно,
— dF _ dV
fi — dNt' v»—dNt’ дСр CPi — dNt
я т. д.
Н— Энтальпия на единицу массы или на моль *) (применяется для энтальпии пара в тех случаях, когда желательно различать энтальпию пара и энтальпию жидкости).
Нр — Вес насадки в насадочной колонне.
I—Константа интегрирования. Сила тока.
Интенсивность намагничивания. Энтальпия смеси жидкости и пара.
J— Механический эквивалент тепла. Интенсивность видимого излучения.
А"— Константа равновесия химической реакции (с различными индексами).
А"о— Константа равновесия. ' выраженная через активности.
Общее значение каждого нз экстенсивных свойств, за исключением объема, будет обозначаться символом, напечатанным жирным шрифтом.
40
Список важнейших обозначений
Хс — Константа равновесия, выраженная через концентрации. Ду—Константа равновесия, выраженная через летучести.
Хр — Константа равновесия, выраженная через парциальные * давления.
Кр° — Предельное значение Кр при Р ^0.
Хх — Константа равновесия, выраженная через мольные доли.
JC-j—Константа равновесия, выраженная через коэфициенты активности = —.
Кинетическая энергия.
Константа равновесия при фазовом равновесии жидкость— У пар=-.
Характеристический фактор для нефтяных продуктов.
Константа интегрирования.
Л—Длина или расстояние.
Число молей жидкости прн процессе перегонки.
Скрытая теплота парообразования на моль (или на единицу массы).
Компонент в реакции.
М — Молекулярный вес. Компонент в реакции.
N—Число молей.
Число компонентов.
О — Число молей жидкости, перетекающей в колонне.
Р—Мощность (работа за единицу времени).
Q — Количество тепла.
Количество электричества или электрический заряд.
/? — Газовая постоянная.
Термическое сопротивление.
Отсчет по измерителю потока (с индексом).
Флегмовое число.
ре—Число Рейнольдса.
S — Энтропия на единицу массы или на моль ☆).
Площадь.
Т—Термодинамическая температура вообще или в °К.
Тд — Точка кипении по термодинамической шкале.
Т/г—Точка плавления по термодинамической шкале.
U — Энергия на единицу массы или на моль *).
Общий коэфициент теплопередачи.
V—Общий объем.
Число молей пара в процессе перегонки.
IF—Число молей кубового остатка в процессе перегонки.
X — Соотношение молей в жидкой фазе.
Y—Соотношение молей в паровой фазе.
Коэфнциент расширения в потоке.
Z — Изобарный (изобарно-изотермический) потенциал (И—TS) на единицу массы или на моль *).
Число фаз.
2. ЛАТИНСКИЕ СТРОЧНЫЕ БУКВЫ
а — Эмпирическая константа.
Активность (а для компонента раствора).
Поверхность насадки на единицу объема.
Стехиометрический коэфициент.
Ъ — Эмпирический коэфициент.
Стехиометрический коэфициент.
Постоянная закона Стефаиа — Больцмана.
☆) См. сноску на стр. 39.
Список важнейших обозначений
41
С — Эмпирический коэфициент.
Объемная концентрация.
Скорость света.
Условная химическая постоянная.
Вредное пространство в ком-прессоре.
Теплоемкость.
d — Оператор диференцирования.
е — Основание натуральных логарифмов.
Коэфициент полезного действия (к. п. д.).
Эмпирическая константа.
Стехиометрический коэфициент.
f — Летучесть (/ для летучести компонента раствора).
Коэфициент трения в уравнении Фаннинга.
Поправочный фактор.
Доля потока, следующего по одному направлению после разветвления.
g—Ускорение силы тижести.
h — Пленочный коэфициент теплопередачи.
Напор.
Энтальпия жидкости (в том случае, когда желательно разграничить энтальпию пара и энтальпию жидкости).
i—Химическая постоянная. Константа интегрирования.
k — Коэфициент теплопередачи.
Отношение теплоемкостей. Эмпирическая константа. Константа закона Генри.
I — Стехиометрический коэфициент. Эмпирическая константа.
Доля жидкости в питании ректификационной колонны.
т—Средний гидравлический радиус. Мольность.
Масса.
Стехиометрический коэфициент. Наклон рабочей линии.
п—Показатель степени.
Число оборотов в единицу времени.
Мольная скорость истечении. Эмпирическая константа.
Число ступеней при сжатии.
Показатель степени в политропном расширении или сжатии.
Число ступеней или единиц переноса массы в колонне.
р — Давление; общее давление.
Давление насыщенного пара в том случае, если употребляется с индексом.
р—Парциальное давление в том случае, если употребляется с индексом.
q—Количество тепла, передаваемого в единицу времени.
Объемная скорость истечения. Количество тепла при перегонке (с различными индексами) на единицу потока.
Отношение количеств тепла, употреблиемых в теории ректификации.
Теплоты растворения.
г — Радиус.
Весовая доля.
Степень расширения или сжатия.
Отношение стехиометрических коэфициентов.
t — Температура (отличная от термодинамической).
и — Линейная скорость.
v — Удельный объем или мольный объем.
w — Скорость массы или потока.
х — Мольная доля.
Мольная доля в жидкой фазе. Общая независимая переменная. Сухость пара.
у — Мольная доля в газовой фазе. Общая независимая переменная.
z — Высота выше нулевого уровня. Коэфициент сжимаемости Ама-
Мольная доля компонента при смешанном (жидкость -|- пар> питании ректификационной колонны.
42
Список важнейших обозначений
3. ГРЕЧЕСКИЕ БУКВЫ
я (альфа)— Эмпирическая константа. Относительная летучесть при равновесии жидкость — пар.
Остаточный объем-----v
Р
$ (бета) — Эмпирическая константа. Отношение диаметра канала к диаметру трубки измерителя напора.
Холодильный коэфициент.
7 (гамма) — Эмпирическая константа. Коэфициент активности
Д (дельта) — Конечная разность, Остаточное количество.
i (дельта) — Эмпирическая константа.
е (эпсилон) — Показатель степени при адиабатном сжатии.
Интенсивность общего излучении.
С (дзета) — Обобщенная сила.
Испарившаяся или сконденсировавшаяся доля материала.
Доля материала, претерпевшая превращеняе при химической реакции.
Н (эта) — Магнитное силовое поле.
Я (тета) —Время.
Температура но шкале идеального газа.
Характеристическая температура.
, . _ 1 ди
х (каппа) —Сжимаемость = .
х$ — для адиабатной сжимаемости.
xj- — для изотермической сжимаемости.
X (лямбда) — Длина волны.
Скрытая теплота фазового превращения.
Скрытая теплота парообразования на единицу массы (только в 'гл. XIII).
Ji (ми) —Диференцяальный дроссель-эффект.
Абсолютная вязкость. Химический потенциал.
» (ни) — Отношение объема к
критическому объему.
« (пи) — Отношение давления к
критическому давлению.
Отношение длины окружности к диаметру.
Особая функция р, и н Т, равная
р°
? (ро) — Плотность.
S (сигма) — Оператор суммирования. 2л — Сумма стехиометрических коэфициен-тов химической реакции — а— — ь —...
с (сигма) — Поверхностное натяжение.
т (тау) ? (ф”) — Отношение температуры к критической температуре. — Знак функции.
X (хи) — Общая перегруппировка.
1 (хи) — Знак функции. Магнитная проница- емость.
f (пси) — Знак функции.
Список важнейших обозначений
43
4. НИЖНИЕ ИНДЕКСЫ (только общеупотребительные)
А, В, С,..., N— Относятся к компонентам.
В — Относится к температуре кипения или к кубу в ректификационной колонне.
С — Относится к конденсатору в ректификационной колонне или к процессу конденсации.
D — Относится к дестил-лату при процессе перегонки.
Е — Относится к детан-’ деру или к исчерпывающей секции ректифика ционной колонны.
Е—Относится к температуре затвердевания или к питанию при процессе перегонки.
L — Относился к жидкой фазе или к притоку тепла к низкотемпературной системе.
R — Относится к вспомогательной холодильной системе.
V— Относится к паровой фазе или к процессу испарения.
I, j — Относятся к какому-либо компоненту или к точке пересечения рабочих линий.
т — Относится к среднему значению, к смеси или к исчерпывающей секции
ректификационной колонны.
п — Относится к укрепляющей секции ректификационной колонны.
р, v, Т — Относятся к постоянному объему, давлению или температуре. Так же и для других свойств, таких, как S, Н или U.
s — Относится к стандартным условиям для объема газа, к насыщенной фазе или к раствору.
t — Относится к касательной.
W—Относится к кубовому остатку в ректифика ционной колонне.
кр — Относится к критическому состоянию.
0,1,2,... — Относятся к различным состояниям или к различным точкам процесса или употребляются для того, чтобы различить константы, имеющие одинаковые символы. О обычно относится к стандартному состоянию или к точке плавления льда, или к абсолютному нулю, или к наиниз-шей температуре окружающей среды, при которой могут отдаваться большие количества тепла.
44
Список важнейших обозначений
5. ВЕРХНИЕ ИНДЕКСЫ
о
00
in
Jg (г),(ж),(т)
д
•С-
Относится к стандартному состоянию при низком давлении, когда газы являются идеальными.
Относится к состоянию бесконечного разведения.
* — Относится к состоянию равновесия.
z" — Относятся к различным фазам, в частности (') относится к жидкой фазе, а (")—к паровой фазе.
6. СОКРАЩЕНИЯ И ОСОБЫЕ ОБОЗНАЧЕНИЯ
Натуральный логарифм.
ДесЬтичный логарифм.
Относятся к состоянию вещества (соответственно—газ, жидкость, твердое тело) при записи химической реакции.
• Оператор частного ди-ференцирования.
Градус Цельсия.
°К — Градус Кельвина.
(водн.) — Относится к разбавленному раствору, не имеющему определенной концентрации. Часто подразумеваетси одио-мольный раствор.
— Приближение к значению.
-----Черточка над экстенсивным свойством обозначает парциальную реличину. (См. пункт О в разделе 1.)
ГЛАВА I
ОПРЕДЕЛЕНИЯ И ОСНОВНЫЕ ПОНЯТИЯ
ЗАДАЧИ ТЕРМОДИНАМИКИ В ПРИМЕНЕНИИ К ХИМИЧЕСКОЙ ТЕХНОЛОГИИ
Термодинамика в широком смысле слова имеет дело: 1) с энергией и ее превращениями и 2) с процессами и состояниями равновесия в системах, в которых имеют место тепловые эффекты. „Чистая” термодинамика базируется на двух основных законах, которые выведены и всесторонне испытаны во второй половине XIX в. Третий закон установлен уже в текущем столетии; несмотря на присущий ему характер основного закона, его применение значительно более ограничено, чем применение двух других законов. Третий закон находит главное приложение в области химического равновесия и будет особо рассматриваться в главе, посвященной этому вопросу. Здесь же достаточно отметить, что тогда как первый и второй законы приводят к определению функций, третий закон в сущности просто ограничивает значение одной из них (энтропии).
Практически вся термодинамика, в обычном смысле этого термина, представляет собой „прикладную термодинамику”, поскольку в основном она является приложением этих трех законов, соединенных с некоторыми фактами и принципами математики, физики и химии, к проблемам из различных областей науки и технологии. Вполне понятно широкое применение термодинамики в физике, химии, машиностроении и химической технологии.
При изучении химическая технология обычно подразделяется на некоторое число отдельных операций, главным образом физических по характеру, как, например, теплопередача, течение жидкости, испарение и фильтрование, а также различные процессы, являющиеся сочетанием этих физических операций с химическими изменениями, как катализ, нитрование и этерификация. К изучению всех этих операций и процессов можно подойти с трех совершенно различных точек зрения, определяя:
1) предельные условия для операции и вовлеченных в нее количеств энергии;
2) скорость перехода энергии или материала;
3) оборудование, материалы, механизмы и т. д.
Термодинамика в приложении к химической технологии включает только вопросы, относящиеся к первой из этих трех групп.
Возьмем конкретный пример из области перегонки. Термодинамика интересуется пределом или максимальной степенью разделения, которой
46
1. Определения и основные понятия
можно достигнуть при перегонке в данных условиях давления, температуры и состава системы независимо от примененного механизма. Другими словами, термодинамика связана прежде всего с равновесными соотношениями, управляющими данной системой, подвергающейся перегонке, и с требованиями минимальной затраты энергии. Главной целью ее приложения является установление идеальных предельных стандартов, при сравнении с которыми можно судить о работе реального и несовершенного механизма. Детали практического приложения термодинамики для получения предельных условий не отличаются от условий, используемых в химической термодинамике. Следовательно, она должна быть связана с соотношениями фазового равновесия, с давлениями пара, скрытой теплотой парообразования и т. д. — проблемами, обычно рассматриваемыми в этой области. Основное и наиболее существенное различие заключается в разных точках зрения. В случае термодинамики в химической технологии все сосредоточивается на приложении ее к некоторым технологическим задачам, и это обстоятельство может значительно изменять детали. Известны, например, случаи, когда положения, наиболее важные с чисто теоретической точки зрения, превращались во второстепенные и даже совсем незначительные с точки зрения практического применения. Кроме того, термодинамика в химической технологии вообще идет на ступень дальше и после установления предельных условий рассматривает степень приближения к пределу в применяемой аппаратуре, т. е. ее эффективность и возможные улучшения для большего приближения к идеалу.
Вопрос о соотношении между термодинамикой н скоростью процесса иногда вызывает путаницу ~н поэтому заслуживает хотя бы краткого обсуждения. Скорость любого процесса определяется двумя факторами: движущей силой процесса и сопротивлением. Термодинамика может быть применена для определения только движущей силы, но ничего не может сказать о сопротивлении. Следовательно, на основе одной термодинамики нельзя заранее судить о скорости изменения. Например, при химической реакции с помощью термодинамики можно определить максимально возможный выход в данных условиях при неограниченном времени для протекания реакции, но вопрос о действительной скорости, с которой будет происходить реакция для получения этого выхода, термодинамикой не рассматривается. Подобным же образом в таких процессах, как теплопередача или абсорбция газа, термодинамика определяет движущую силу, необходимую для изменения, требующуюся энергию, минимально необходимое количество поглощающей жидкости и связанные с этим величины; скорость же перехода включает также сопротивление жидкости и твердого тела потоку тепла или материала, что требует изучения механизма процесса, т. е. явления, выходящего за пределы термодинамики.
Температура
47
ТЕМПЕРАТУРА
Качественное представление. Температура является, вероятно, Ч наиболее важной переменной, используемой во всех термодинамических обсуждениях, и поэтому представляется существенным с самого начала -----
получить по возможности ясное представление о ее значении. В от- j
личие от давления — другой важной переменной — температуру нельзя ?
выразить простыми основными величинами, а ее приходится измерять только косвенным путем, и выражение результатов этих измерений в величинах, имеющих абсолютное значение, вызывает многочисленные осложнения. Для многих практических целей температуру лучше всего рассматривать как движущую силу тепловой энергии. Одно тело находится при более высокой температуре, чем другое, если тепло переходит от первого ко второму. Если устанавливается равновесие и не происходит никакого перехода тепла между двумя сопри- к касающимися телами, то считают, что эти два тела имеют одинаковую температуру. Измерение температуры основано на допущении, что все вещества, находящиеся в соприкосновении и остающиеся в покое, ? в конце концов придут к термическому равновесию и примут одинаковую температуру.
С точки зрения кинетической теории температура отражает ? кинетическую энергию молекул. Кинетическая теория газов устанавливает, что молекулы данного вещества находятся в быстром поступательном движении и обладают весьма различными скоростями. Поэтому температура является статистической величиной, связанной со средней скоростью.
Количественное определение. Количественное определение тем- i пературы может быть основано на любом свойстве веществ, изменяющемся с температурой, например на термическом расширении, электрическом сопротивлении, контактной электродвижущей силе » (э. д. с. спая) или давлении пара. Для установления числовой шкалы температуры прежде всего необходимо определить величину градуса. Это обычно производится путем'выбора установленного и легко воспроизводимого интервала температуры, например интервала между температурами замерзания и кипения чистой воды при атмосферном давлении, и делением этого интервала на произвольное число градусов (например 100° в случае шкалы Цельсия). Следовательно, выбор нулевой точки совершенно произволен. Любая температура определяется простым уравнением, которое для стоградусной шкалы имеет следующий вид:
100(G —Go) Gioo — Go ’ где t — любая температура по стоградусной шкале;
G — числовое значение данного свойства при температуре /; О0 и Gloo — числовое значение того же свойства, соответственно, при 0 и 100°.
48
I. Определения и основные понятия
Это уравнение предполагает между свойством вещества и его температурой простейшую зависимость, именно—линейную. Нет никаких оснований для допущения более сложного соотношения. Свойства, применяющиеся для технических измерений и регулирования температуры, следующие:
1. Объемное расширение газов, жидкостей и твердых тел при постоянном давлении.
2. Увеличение давления жидкостей и газов при постоянном объеме.
3. Давление насыщенного пара жидкостей.
4. Электрическое сопротивление металлов (в частности платины).
5. Э. д. с. спая двух различных металлов.
Из определения температуры по уравнению (1) следует, что не существует двух шкал, определенных вышеописанным способом, которые совпадали бы при какой-либо другой температуре, кроме двухосновных — 0 и 100°. Табл. 1 иллюстрирует этот очень важный факт. Подсчеты производились по уравнений (1) при использовании данных, полученных из Интернациональных критических таблиц.
Таблица 1
Расхождение между температурными шкалами, основанными на свойствах веществ
Свойство Отсчет при следующих температурах в °C по термодинамической шкале
50° 200°
Расширение ртути ♦) 50,0 202,2
Давление насыщенного пара этилового спирта . . 23,3 1320
Э д. с. термопары платина — платина-родий . . . 46,4 222,5
Сопротивление платины 50,25 195,7
*) Расширением резервуара пренебрегают.
Ясно, что ч«м дальше отступать от основного интервала, тем больше будут отклонения. Если в качестве термометрического вещества используется газ, подобный водороду или азоту, то поправки к абсолютной шкале значительно уменьшаются. Например, при — 200 и 200° С поправки к водородной шкале постоянного объема соответственно равны лишь —0,06 н -|- 0,02° С. При 1000° С водородная шкала постоянного объема имеет поправку -|-0,7оС.
Абсолютная шкала. Само собой очевидно большое преимущество применения шкалы, не зависящей от свойств отдельного вещества, т. е. абсолютной шкалы. Второй закон термодинамики, как будет показано дальше, дает основу для абсолютной шкалы и, следовательно, дает возможность определения нулевой точки, или .абсолют
Температура
49
ного нуля", как его обычно называют. Нельзя сконструировать какого-нибудь „абсолютного термометра", но с помощью законов термодинамики можно вычислить поправки, которые следует внести в отсчеты практически применяемого термометра для выражения результатов в градусах абсолютной шкалы. Эта работа установления абсолютной температурной шкалы проведена такими учреждениями, как Национальное бюро стандартов в США, и соответствующими институтами и лабораториями в других странах. Для установления этой шкалы обычно используются газовые термометры с гелием, азотом или водородом в качестве термометрического вещества.
Температурная шкала, являющаяся абсолютной (т. е. независящей от свойств какого-либо индивидуального вещества), может быть основана на использовании идеального газа как термометрического вещества, и такую шкалу часто называют „температурной шкалой идеального газа". Поскольку в действительности не существует идеального газа, эта шкала не является практической, но значение ее заключается в том, что она идентична с термодинамической шкалой и достаточно точно воспроизводится в некоторых областях реальными газами.
Верхний предел использования газового термометра при установлении термодинамической или абсолютной шкалы температур определяется свойствами резервуара для газа. Не найдено ни одного резервуара, который давал бы возможность превысить 1500° С. Практически газовые термометры применяются для установления абсолютной шкалы не выше точки плавления золота (1063° С). Конечно, часто бывает желательно измерять более высокие температуры. При этих повышенных температурах можно использовать различные другие свойства, например расширение некоторых твердых тел или термоэлектрическую силу спая неодинаковых металлов, но опять возникает вопрос об установлении абсолютной шкалы в этой области температур. Это осуществляется на основе законов радиации, которые точно известны при температурах ниже 1500° С и могут быть с достаточной точностью экстраполированы выше этой температуры.
Нижним пределом температуры является температура абсолютного нуля, которая, как точно известно, на 273,2° С ниже точки плавления льда. В недавно проведенных исследованиях оказалось возможно получить и измерить температуры, которые расположены лишь на несколько сотых градуса выше абсолютного нуля. Повидимому, не существует верхнего предела температуры. Астрономы говорят о звездах, имеющих температуры в миллионы градусов, но это связано с очень большой экстраполяцией известных фактов. Наивысшими земными температурами являются температуры вольтовой дуги, которые были с помощью различных методов определены как лежащие в пределах от 3800 до 10 000° К.
Международная шкала [37]. Эта шкала различными стандартизирующими учреждениями мира единодушно считается для практических целей наилучшим приближением к термодинамической шкале. 4 Химическая термодинамика
50
I. Определения и основные понятия
Она основана на следующих ориентировочных точках (точки фазового изменения), взятых при давлении в 1 атм;
°C
Точка кипения кислорода . . . .—182,97
> плавления льда.............. О^ОООТ
> кипения воды.............. 100,000/
> плавления серы............ 444,60
> > серебра .... 960,5
> > золота..................1063
по определению
Интерполяции посредством термометра с платиновым сопротивлением от —190 до 660° С и платиново — платино-родиевой термопары от 660 до 1063° С согласуется с соответствующей интерполяционной формулой. Выше точки плавления золота международная шкала определяется с помощью оптического пирометра, отсчеты по которому связаны с температурой посредством уравнения
1п^- = —2
Ji X
1
1336
1 *2-f-273.
(2)
где J2 — интенсивность монохроматического видимого излучения черного тела при неизвестной температуре;
Jj — интенсивность излучения той же самой длины волны от черного тела при температуре плавления золота;
>.— длина волны в см;
С2 — эмпирическая константа, равная 1,432 см/° С.
Для использования при калибровке инструментов, измеряющих температуру, пригодны вторичные фиксированные точки, указанные в табл. 2.
Таб лица 2
Вторичные фиксированные точки для.калибровки приборов, измеряющих температуру [37, 203]
Вещество Температура в °C по международной температурной шкале Вещество Температура в °C по международной температурной шкале
Точка сублимации угле- Точка плавления свинца . . 327,3
КИСЛОТЫ — 78,5 > > цинка . . 419,4
Точка плавления ртути . . — 38,87 > > сурьмы 630,5
> превращения Na-SCL- > > алюминия 660,1
•ЮН2О 32,38 > > эвтектики
Точка кипения нафталина . 217,96 Си—Ag . 778,8
> плавления олова . . 234,9 > > меди . . 1083,0
» кипения бензофенона 305,9 > > палладия . 1555
> плавления кадмия . . 320,9 > > платины . 1773
» > вольфрама 3400
Давление
51
Не существует ни одной общепринятой в международном масштабе температурной шкалы ниже 90° К. Температуру между 90 и 14° К можно измерять термометром с платиновым сопротивлением, для которого Бюро стандартов США недавно [109] установило шкалу путем сравнения с гелиевым газовым термометром. В этой области имеются две фиксированные точки, а именно'—точка кипения водорода (20,39° К) и тройная точка водорода (13,96° К). Температуры между 14° К и абсолютным нулем требуют специальных методов, рассмотрение которых выходит за пределы этой книги ’).
ДАВЛЕНИЕ
Давление является второй наиболее важной переменной при любом термодинамическом анализе. Эта величина представляет значительно более простое понятие, чем температура, так как давление можно непосредственно связать с основными единицами массы, длины и времени.
Давление определяется как сила, действующая на единицу площади. В абсолютной системе единиц давление выражают в динах на квадратный сантиметр (1 грамм на квадратный сантиметр = g динам на квадратный сантиметр, где g—ускорение силы тяжести в см1сек2). В технических работах наиболее употребительной единицей давления является 1 кг]см2, называемой технической атмосферой {ат).
Давление часто измеряется весом столба жидкости, который уравновешивает давление. Соотношение между высотой Л столба жидкости над данной поверхностью и давлением р у этой поверхности дается простым уравнением p = h.p, где р — плотность жидкости. В очень точной работе h следует относить к некоторой определенной температуре, так как р изменяется с температурой. Кроме того, для превращения в абсолютные единицы, необходимые для сравнения точных измерений, произведенных в различных лабораториях, следует принимать во внимание географическую широту и высоту над уровнем моря.
Другой общеупотребительной единицей давления является атмосфера, которая, как показывает самое название, представляет собой давление, производимое весом земной атмосферы. Поскольку оно не является постоянным, эту единицу давления следует определить более точно. Нормальная атмосфера определяется как давление, создаваемое вертикальным столбом чистой ртути высотой в 760 мм и плотностью в 13,5951 г!мл (при 0°С) в местности, лежащей на широте 45° и на такой высоте, где £=980,665 см1сек2. Нормальная атмосфера (атм) несколько более технической: 1 атм = 1,03323 кг/см2.
В технике давление измеряется не непосредственно в абсолютных единицах, а путем измерения разности между давлением в системе и внешним давлением атмосферы по манометру. Такое давление называется сверхбарометрическим или манометрическим давлением. Абсолютное давление равно сумме отсчитанного по манометру
!) См., например, [242]. {Прим, ред.)
4*
52
I. Определения и основные понятия
давления (по отношению к атмосферному) и барометрического давления в этом месте. Пространство, из которого удалены все газы, является идеальным вакуумом и находится под нулевым давлением. Лучшие вакуум-насосы в состоянии дать весьма глубокий вакуум (до 10-9лл ртутного столба). Хотя такое давление является очень малым, все же оно очень далеко от того, которое требуется для достижения пустоты, так как при этом давлении и 0°С газ все же будет содержать 2 X Ю8 молекул на каждый миллилитр. Наибольшее приближение к абсолютному вакууму существует в межзвездном пространстве, где плотность определяется величиной порядка 10~30 г{мл, что отвечает одной молекуле Н2 на каждые 30 м3.
Давления ниже атмосферного большей частью измеряют тоже по отношению к внешнему (атмосферному) давлению. Разность между внешним давлением и давлением внутри системы называется вакуум-метрическим давлением (разрежением). Оно выражается обычно в миллиметрах или сантиметрах ртутного столба или в долях атмосферы. Абсолютная величина в этом случае равна разности между атмосферным и вакуумметрическим давлениями. Давление в системе удобнее характеризовать в этом случае абсолютным давлением, так как применение для этой цели вакуумметрического давления нередко приводит к недоразумениям, например 711,2-миллиметровое разрежение означает одно, если барометр показывает 711,4 мм, и совершенно другое, если барометр показывает 774,7 мм. Первое соответствует очень большому разрежению (711,4 — 711,2 = 0,2 мм), недостижимому с помощью обычных механических насосов, тогда как второе (774,7—711,2 = 63,5 мм) достигается очень легко. Сверхбарометрн-ческое и вакуумметрическое давления являются величинами вполне определенными, если только они отнесены к определенному барометрическому давлению, но так как часто барометрическое давление не указывается, то лучше пользоваться абсолютным давлением.
Давления до 150 атм относительно часто применяются в технике, давления около 1000 атм используются в промышленности; давления порядка 3500 атм обычны в артиллерии, а давления порядка 250 000 атм получаются при некоторых экспериментальных работах в лабораториях.
ОПРЕДЕЛЕНИЕ СОСТОЯНИЯ
В этой главе мы попытаемся определить некоторые общие термины и понятия, которые находят широкое применение, но с трудом поддаются строгому определению. Условимся с самого начала, что формулируемые определения являются только общими вехами и что всегда имеются специальные случаи, к которым они не могут строго применяться и для которых потребуется некоторое изменение.
Макросостояние и микросостояние. Указывая свойства вещества или смесн веществ, крайне важно отметить состояние, в котором находится данная система. Нам часто придется иметь дело с веще
Определение состояния
53
ствами в различных состояниях, и желательно сначала кратко рассмотреть, что понимается под состоянием вещества в термодинамическом смысле слова.
Если взять некоторое количество газа при определенных температуре и давлении, то другие его свойства, как плотность, вязкость, показатель преломления и теплопроводность, при этом вполне определяются. Таким образом мы определяем его состояние. Любой образец того же газа, взятого в таком же количестве и при той же температуре и том же давлении, будет обладать теми же свойствами. Но газ состоит из очень большого числа отдельных частиц (молекул), находящихся в быстром движении. Если рассматривать эти отдельные частицы, то необходимо определить положение, скорость и направление движения каждой из них. Это является методом, используемым при кинетической трактовке, и состояние, определяемое таким образом, условно называется „ микроскопическим “. Очевидно, что для трактовки системы этим путем требуется очень большое число переменных. Математические методы, используемые дедуктивно для получения измеримых величин, обычно объединяются под общим названием статистической механики.
С другой стороны, состояние, с которым приходится иметь дело при термодинамических исследованиях, является „макроскопическим* состоянием, т. е. таким, которое определяется статистической средней величиной из всех отдельных микроскопических состояний. Мы используем некоторые переменные, являющиеся результатом действия микроскопических изменений. Таким образом, давление и температура рассматриваются как такие статистические величины. Эта точка зрения очень важна, так как является сущностью всего термодинамического метода решения различных проблем. Нельзя заранее сказать, каким образом будут себя вести отдельные частицы в данных условиях, но в отношении большой группы частиц это обычно можно сделать с достаточной точностью. Это определяется статистической вероятностью; таким образом, очевидна тесная связь термодинамики со статистикой и вероятностью.
Важно отметить также, что этот метод трактовки не требует каких-либо допущений относительно структуры объекта, с которым имеют дело. Последний рассматривается скорее как непрерывная среда, чем как система, состоящая из дискретных частиц, какой мы ее знаем в действительности.
Системы, компоненты и переменные. Теперь перейдем к некоторым общим терминам и понятиям, которыми будем пользоваться в дальнейшем очень часто. Основным приемом термодинамики является выделение интересующей иас совокупности тел, называемой системой, и противопоставление ее окружающим телам, образующим внешнюю среду. Гомогенной системой называется такая система, свойства которой в макроскопическом смысле одинаковы во всех точках или меняются непрерывно при переходе от одной точки к другой,
54
I. Определения и^осювные понятия
т. е. не существует области, где наблюдалась бы прерывность свойств или резкое изменение их. Такая гомогенная система называется „фазой*. Гетерогенной системой называется система, содержащая больше одной фазы, причем на границах фаз происходит резкое изменение свойств. Системы, состоящие только из газов или паров, всегда гомогенны £?). В случае жидкостей обычно имеются две фазы, в случае твердых тел возможно любое число различных фаз в границах, установленных другими условиями.
Раствор является гомогенной (т. е. однофазной) системой, содержащей больше одного компонента. Растворы, в которых отдельные частицы обладают молекулярными размерами, часто называются „истинными* растворами, в отличие от коллоидных растворов, в которых отдельные частицы по крайней мере одного из компонентов представляют большие агрегаты и приближаются по свойствам к отдельным фазам. Здесь имеется много промежуточных случаев, и невозможно провести резкого разграничения, но мы будем употреблять термин „раствор*, относя его к истинным растворам.
Термин „компонент* имеет большое значение во всех термодинамических выводах; необходимо поэтому рассмотреть несколько примеров для более четкого понимания его смысла. Возьмем такую простую систему, как чистая вода, Н2О. Нам известно существование нескольких различных химических разновидностей, которые могут образоваться изН2О. Могут происходить, например, следующие реакции:
Н2О = Н+4-ОН", 2Н2О = (Н2О)2.
Следовательно, мы можем наблюдать присутствие четырех различных химических составляющих, именно: ионов Н+, ионов ОН~, молекул Н2О и молекул (Н2О)2; однако все же система состоит только из одного компонента, так как остальные составляющие непосредственно получаются из единственной разновидности — Н2О — и не являются независимыми переменными.
Обычно вода содержит малые количества окиси дейтерия D2O— химического индивидуума, по свойствам очень близкого к Н2О, но содержащего тяжелый изотоп водорода. Окись дейтерия образуется не из Н2О, и поэтому обыкновенная вода не является, строго говоря, однокомпонентной системой; но поскольку D2O присутствует в этой воде в очень малой концентрации, являющейся к тому же постоянной, то для практических целей обыкновенную воду можно считать системой однокомпонентной. Однако, если нужно исследовать свойства воды, получаемой при добавлении различных количеств D2O
☆ ) Надстрочными цифрами обозначены примечания сделанные при переводе и помещенные в конце книги. (Ред.)
Определение состояния
55
к обыкновенной воде, то такая система должна рассматриваться, конечно, как двухкомпонентная.
При определении числа компонентов часто возникают ошибки вследствие того, что это число может изменяться в зависимости от обстоятельств даже тогда, когда система содержит те же самые химические вещества. Например, система, содержащая Н2О, Н2 и О2, может иметь два или три компонента в зависимости от условий. При' изучении растворимости в воде смеси водорода и кислорода, взятых в различных пропорциях, система является трехкомпонентной, поскольку каждый химический индивидуум является независимой переменной, и состояние системы можно определить только посредством определения содержания каждого из них.
С другой стороны, при изучении химического равновесия между водяным паром, водородом и кислородом при высоких температурах система с теми же самыми тремя веществами является только двух-компонентной, так как любой состав равновесной смеси может быть получен с помощью каких-нибудь двух из этих трех веществ. Здесь на систему наложено добавочное ограничение, и поэтому стало невозможным произвольно изменять содержание всех трех веществ.
Теперь рассмотрим систему, состоящую из водного раствора, в котором присутствуют следующие ионы: К+» Na+, Cl- и Вг_. Такая система может получиться при растворении в воде КС1, NaCl, КВг и NaBr, и с первого взгляда может показаться, что это — пятикомпонентная система. Однако это не так, поскольку некоторые составляющие можно получить из других составляющих по реакции NaCl + КВг = NaBr + КС1.
Рассматриваемая система может быть получена, например, из Н2О, NaCl и КВг, поэтому она будет трехкомпонентной. Но при этом получится определенное соотношение между NaBr и КС1, и для получения любого желаемого состава нужно исходить из воды и из трех этих солей. Из этого можно заключить, что система в целом может получиться посредством независимого изменения количества любого из четырех составляющих и поэтому является четырехкомпонентной.
К этому же выводу можно притти и другим путем. Обычные методы химического анализа обнаруживают присутствие в растворе молекул Н2О и ионов Н+, ОН-, К+, Na+, С1~ и Вг~. Ионы Н+ и ОН-могут получаться только из молекул Н2О и, таким образом, не являются независимыми переменными. Ионы К+, Na+, Cl- и Вг— не все являются независимыми переменными, поскольку необходимо, чтобы раствор был электронейтральиым, и это ограничивающее условие оставляет только три независимых переменных из четырех составляющих ионов.
Это можно резюмировать формальным определением числа компонентов как наименьшего числа независимо изменяемых химических веществ, из которых можно получить данную систему в любых вариантах ее состава.
56
I. Определения и основные понятия
Свойства определенным образом связаны с состоянием вещества. Если состояние определено, то будут определены (но ие обязательно известны) и все свойства. Переменными, применяемыми для определения состояния, обычно служат давление, объем, температура и массы компонентов в каждой фазе. Не все эти переменные являются независимыми. Например, для случая чистого газа при определенном объеме, массе и температуре определяется и давление. Оно является зависимой переменной и зависит от состояния, определяемого объемом и температурой так же, как и любое другое свойство. Число независимых переменных или число переменных, значениями которых точно определяется состояние рассматриваемой системы, называется „степенями свободы* ее. Только что рассмотренная система (чистый газ) имеет две степени свободы, иначе говоря, она является „бивариантной*. С другой стороны, вода в равновесии со своим паром имеет только одну степень свободы, или является „уинварнант-иой* системой. Если определена любая из двух переменных—-температура или давление, то этим определяется и состояние, а следовательно, и все свойства. Если бы мы попытались произвольно установить и температуру и давление, то одна из фаз могла бы исчезнуть, и система стала бы бивариантной. Система, & которой сосуществуют в равновесии три фазы одного компонента, будет системой нонвариант-ной, т. е. она ие будет иметь степеней свободы. В этом случае и давление и температура строго определены, и ни одно из них не может изменяться без того, чтобы не исчезла какая-либо фаза или две какие-либо фазы. В следующей главе будет выведено исключительно важное соотношение между числом компонентов, фаз и степеней свободы для любой системы, известное под названием „правила фаз*.
Состав раствора. Состав раствора можно определить просто указанием массы каждого составляющего, например NA молей А и молей В. Однако удобнее выражать состав посредством отношения масс. Например, в растворе, содержащем тА кг А, тд кг В и т. д., состав будет изображаться в весовых долях следующим образом:
Г ---------тА______ I
А «л + 'лв + «с+---’
где гА—весовая доля вещества А.
Подобное же уравнение выражает мольную долю хА через общее число молей и число молей А. В некоторых случаях, например в процессах абсорбции, удобно выражать состав бинарного раствора посредством, отношения массы одного компонента к массе другого.
В системе из W компонентов будет только (Л/— 1) независимых переменных состава, вследствие того, что весовые доли компонентов связаны между собой следующим соотношением:
хл + хвЧ-хс + • • 4"xjv= (4)
Основные уравнения, связывающие свойства и параметры состояния 5?
Состав иногда выражают в объемных долях. Если при смешении f
наблюдается изменение объема, то это может вызвать значительные j
недоразумения, так как объемный процент может толковаться различно и изменяющееся процентное содержание всех компонентов может не составить в сумме 100%. " ""J
В случае газов за объемную долю обычно принимают частное от деления величины, на которую уменьшается объем вследствие $
удаления из смеси данного компонента, на первоначальный объем смеси. Поскольку газы образуют практически идеальные растворы, $
т. е. при смешении их не происходит сколько-нибудь значительных I
объемных изменений, это определение не поведет к недоразумениям.
ОСНОВНЫЕ УРАВНЕНИЯ* СВЯЗЫВАЮЩИЕ СВОЙСТВА i
И ПАРАМЕТРЫ СОСТОЯНИЯ
Если G представляет какое-либо свойство системы, состояние которой определяется тремя независимыми переменными р, t и х, то мы можем написать, что
G = tp(p, t, х).
Это означает, что величина G точно определяется во всех случаях когда определены р, t и х. Путь, по которому меняется G при изменении значений этих переменных, дается хорошо известным уравнением в диференциальной форме
“= (f ),.,л+ (ж),.,**- <5>
Общее изменение можно считать результатом изменения трех неза-висимых переменных, причем первое изменение происходит от изменения одного только р при постоянных t и х, второе — от измене- j
ння t при постоянных р и х, а третье — только от изменения х при постоянных р и t. В последующем изложении х для простоты будет считаться постоянным.
Замена независимой переменной. Часто дается изменение свойства, выраженного как функция одного ряда независимых переменных, а желательно выразить его как функцию другого ряда. Например, предположим, что
G = ^ (/?,/) (6)
И
P=:^2{V, t), (7)
где давление теперь является зависимой переменной. Предположим, что нам нужно выразить G, принимая в качестве независимых переменных v и t, причем р становится зависимой переменной. Из уравнения (6) имеем
‘г0=(^)/'’ + (ат)/'- <8>
58
I. Определения и основные понятия
Поскольку p = 'oz(v, t), то можно написать
dp=ff\ dt. (9)
\OVJt 1 \dt!v
Подставляя значение dp из уравнения (9) в уравнение (8), получим
“°-(f), [(*)/’+ (?) л]+(£)л=
=(1Ш>+ [(f), $).+№ “»>
Рассматривая G = tpa (v, t), можно, аналогично (8), написать
dG=(^'} dv-\-(^} dt. (11)
Xdv/t 1 \dtjp ' '
Теперь, поскольку изменение G имеет только одно значение для данного изменения v и t, коэфициенты при dv и dt в уравнениях (10) и (11) должны быть равны. Следовательно,
(f),= (f),S). 02)
(£).=(£),+($М 03)
Другие родственные уравнения могут быть получены из уравнений (12) и (13) просто путем перемены индексов. Так, подставив р вместо t в уравнении (12), получаем
fdG\ _fdG\ (/14Л
\ dvJ р \dtjp\dvjp’ ' '
Изменение при постоянном G. Предположим, что мы интересуемся процессом, во вр мя которого свойство G остается неизменным. В таком случае dG = 0, и уравнение (8) можно написать в виде
(fL^+CsV^0- "5>
Индекс G при dp и dt показывает, что эти два изменения происходят при постоянном О. (В дальнейшем при записи уравнений, содержащих частные производные, будем ставить индекс только в тех случаях, когда не сразу видно, что именно принимается постоянным при диференцировании.) Уравнение (15) можно написать также в следующей форме:
(др\ ' (dG/dt)p
\dtja (dGldp)t'
Основные уравнения,свяэывоющие свойства и параметры состояния 59
Таким же путем можно вывести и следующее, полезное в некоторых случаях, уравнение
(fy\ (ШУ (?£\ (17^
\dtJa\dvjG \dvja' ' '
♦
Для особых случаев, когда G~v, уравнение (16) получает следующую форму:
m i^\ — __ (i8)
\dt) о \др] t \dvjp ' '
Равенство вторых производных. Поскольку свойство зависит от данного состояния, изменение свойства при переходе системы от состояния 1 к состоянию 2 является постоянной величиной, не зависящей от пути изменения. Эта чрезвычайно простая и почти очевидная формулировка представляет собой очень важный принцип,
приложение которого можно легко не распознать во многих реальных случаях. Для случая двух независимых переменных его можно выразить условно в виде следующего уравнения:
*G___&G ]qj
Opdt dtdp' к '
Это уравнение означает только, что значение второй производной не зависит от порядка диференцирования. Его смысл можно выяснить полнее, если дать для него еще один простой вывод. Рассмотрим изменение G при переходе от 4 к В по пути АВ (см. рис. 1). Для
Рис. 1. К выводу уравнения (19).
простоты рассмотрим только
бесконечно малое изменение, например GB — GA^=dG. Поскольку изменение не зависит от выбранного пути, будем считать его проис-
ходящим по пути ADB. Так как изменение от А до D является изме-
нением при постоянном давлении, то
(20)
Подобным же
образом для пути DB (постоянная температура) имеем
Gb = Gd -4- (^] dp. в D ' \ др J t
(21)
Диференцируя GD по давлению и подставляя в уравнение (21), получаем:
OB = OA+(^\<lt+^dP + ^plU. (22)
60
I. Определения и основные понятия
Совершенно также для пути АСВ получим
«„ = <;,+ (*2»)г (23)
Поскольку Ол — Ол должно быть одинаковым для обоих путей, необходимо, чтобы уравнение (19) было правильно, что видно из сопоставления уравнений (22) и (23).
Конечные изменения. В практических случаях наибольший интерес представляют конечные изменения, например изменение О при переходе от состояния А к состоянию В. Рассматривая попрежнему только две независимые переменные, получим следующую интегральную форму уравнения (8):
°.-°л==](f ),#+]' (")/'• W
Pl il.
Для большей конкретности рассмотрим какое-нибудь термодинамическое свойство, например энтальпию Н. Для чистого вещества, как будет показано позже, правильны следующие уравнения:
Подставляя значения этих производных в уравнение (24), получаем
л Л
Интегрирование можно производить как графически, так и аналитически, в зависимости от обстоятельств. Важно отметить, что интегрирование может совершаться двумя различными путями, причем каждый из них требует несколько различных данных. Например, можно интегрировать сначала при постоянной температуре Т1 и определить первый интеграл. Для этого нужно знать, как объем v и наклон изобары v — Т изменяются только при данной температуре 7\ в пределах давлений от рг до р2. НА представляет значение Н в начальной точке (pj, 7\) и должно быть известно заранее. Результатом первого интегрирования явится значение Н при р2 и 7\. Второе интегрирование проводят затем при постоянном давлении р2, для чего необходимы данные по изменению Срс температурой при постоянном давлении р2. Интегрирование также можно начинать и со второго интеграла (при постоянном давлении), причем в этом случае необходимо знать зависимость Ср от Т, но уже при постоянном начальном
Воспроизводимость состояний
61
давлении р}. Тогда определение первого интеграла должно быть произведено при постоянной температуре Т2 вместо Т,. Эти два пути показаны на рис. 1. Первый путь соответствует перемещению по АСВ, а второй — по ADB. Из природы свойства должно быть ясно, что оба пути приводят к одинаковому результату и что выбор их зависит от того, какими данными располагают.
ВОСПРОИЗВОДИМОСТЬ состояний
В предыдущих разделах предполагалось, что если определить все независимые переменные, то состояние системы (макроскопическое) будет точно определено. Другими словами, если какое-либо вещество данного состава подвергается целому ряду изменений, а затем независимые переменные определяются при их начальном значении, то вещество в конечном состоянии будет иметь те же свойства, что и в начальном. Иначе говоря, свойства зависят только от состояния системы, но не от ее предистории. Это является строго правильным для газов и в большинстве случаев для жидкостей, но далеко от истины в случае твердых тел. Поскольку молекулы в газе и жидкости могут свободно двигаться, они моментально достигают равновесного состояния (не статического равновесия, а динамического, представляющего собой статистическое равновесие), и свойства ие будут изменяться со временем. В случае твердых тел, когда не существует такой свободы движения, могут происходить структурные изменения, не являющиеся обратимыми, т. е. твердое тело при условиях, совпадающих с первоначальными, может не иметь полностью тех же свойств, которые оно имело. Эти изменения происходят очень медленно, и при достаточном времени твердое тело, повидимому, достигает расновесного состояния. Стабильным состоянием для твердого тела будет кристаллическое состояние, т. е. состояние, при котором устанавливается полностью упорядоченное расположение частиц и идеалом является твердое тело в виде монокристалла.
Для наглядности приведем несколько конкретных примеров. Свойства металлов могут сильно изменяться от горячей или холодной обработки; следовательно, материал данного состава при данном давлении и температуре может обладать совершенно различными свойствами в зависимости от своей предистории. Углерод можно получать в виде сажи или графита. Такой металл, как медь, может быть получен в виде крупных кристаллов при медленном охлаждении расплавленной меди или в совершенно другой форме путем восстановления окиси меди при сравнительно низкой температуре. Эти две формы меди выглядят совершенно различно и обладают резко отличающимися свойствами. Одна форма, например, пирофорна и будет активно действовать на прохождение некоторых химических реакций, тогда как другая в этом отношении совершенно инертна. Это различие не определяется механическим измельчением, так как
62
/. Определения и основные понятия
неосязаемая медная пудра, полученная посредством тончайшего измельчения плавленой меди, все же совершенно отлична от меди, полученной при низкотемпературном восстановлении окиси.
Конечно, величина поверхности является важным фактором для этих эффектов. Хорошо известно, что соль в форме очень мелких кристаллов несколько лучше растворима в воде, чем в форме крупных кристаллов. В этом проявляется влияние поверхностной энергии; хорошо известно, что любое тело с сильно развитой поверхностью стремится измениться так, чтобы уменьшить поверхностную энергию. С другой стороны, одна величина поверхности не решает полностью дела. Работы по гетерогенному катализу достаточно убедительно показали, что в поверхностях наблюдаются важные качественные различия, вызывающие совершенно различные свойства даже при приблизительно одинаковых поверхностях.
Вопрос о воспроизводимости состояния твердого тела усложняется также тем обстоятельством, что некоторые твердые тела крайне трудно получить в чистом состоянии и что даже следы примесей могут сильно изменять их свойства. На примере металлов можно также видеть, что очень малые количества примесей, в особенности неметаллических, как кислород, сера й фосфор, глубоко изменяют свойства металлов. Многие свойства, обычно ассоциируемые с данным металлом, часто вызываются главным образом примесью. При получении углерода посредством термического разложения углеводородов или при получении окиси металла посредством осаждения раствора наравне с вопросом о структуре возникает и вопрос о составе.
Все вышеизложенное означает, что, имея дело с термодинамикой процессов, включающих твердые тела, необходимо осознать эти особенности и стараться привести все твердые тела в равновесное кристаллическое состояние или по меньшей мере учесть наличие таких различий в свойствах: если говорят о теплоте сгорания углерода, то обязательно нужно указать, какую именно форму углерода имеют в виду. При изготовлении гальванического элемента посредством введения в реакцию некоторых твердых веществ и имея твердые вещества в качестве электродов, нужно принимать во внимание, что э. д. с. элемента будет изменяться в зависимости от предистории этих твердых веществ, если они не были приведены в состояние равновесия. В данной книге всюду, кроме специально оговоренных случаев, предполагается, что твердое вещество находится в своем наиболее стабильном состоянии или по меньшей мере настолько к нему приближается, что различиями в термодинамических свойствах можно пренебречь.
СТАНДАРТНЫЕ СОСТОЯНИЯ
Так как мы будем часто сталкиваться со свойствами, которые ие имеют известных абсолютных значений, и будем пользоваться разностями свойств между двумя состояниями, то, давая числовые
Стандартные состояния
63
значения свойств веществ и систем, будет удобно принимать некоторые состояния вещества как стандартные. Выбор стандартного состояния является совершенно произвольным и может изменяться в зависимости от применения. Во многих случаях стандартное состояние относится только к исходному давлению или к концентрации по изотерме и не имеет отношения к какой-либо определенной температуре. В других случаях выбирается определенная температура. Следовательно, указывая значение некоторых термодинамических функций, как энтальпии или энтропии, принимают равным нулю их значение в условном стандартном состоянии при 0° С и 1 атм давления, если вещество находится в агрегатном состоянии, отвечающем комнатной температуре; в некоторых случаях можно предпочесть другие стандартные состояния. Так, для СО2 стандартным состоянием будет газ при 0° С и 1 атм, а для Н2О — жидкая вода.
При представлении данных по теплоте химических реакций, или по изобарно-изотермическим потенциалам, или по свободной энергии реакции и по другим подобным изменениям свойств обычной стандартной температурой выбирается 25° С (298,2° К), и каждое вещество принимается как находящееся в своей наиболее стабильной форме и в чистом состоянии при 1 атм.
Таким образом, для реакции
СО Н2О (ж) = СО2 Н2
ДО будет представлять изменение рассматриваемого свойства при 25° С. Тогда получим
Д0 = ОНз-1-Осо2 — Осо —Он2о>
где каждое значение О является значением этого свойства для соответствующего чистого вещества при 25° С и 1 атм давления. Поскольку мы имеем дело со свойствами, действительный путь, по которому проходит реакция, абсолютно несущественен. Необходимо лишь точно определить начальное и конечное состояния.
Для иллюстрации возьмем воду в жидком состоянии, которое для данных условий является стабильным. В практике обычно даются значения для той же самой реакции с водой в газообразном состоянии. Так как вода при такой температуре и давлении не существует в виде газа в стабильном равновесии, то выбранное состояние является гипотетическим. Для определения свойства нужно произвести экстраполяцию от известного значения в стабильном состоянии (например, от 25° С и от давления насыщенного пара при этой температуре) до гипотетического состояния. Принципиально это неправильно, но часто оправдывается соображениями удобства, и при небольшой экстраполяции ошибка не может быть значительной.
Для жидких и твердых тел вообще не всегда бывает необходимо устанавливать стандартное давление, так как их свойства очень мало изменяются с давлением. В случае же газов это очень важно. Обычным.
64
/. Определения и основные понятия
стандартным состоянием, которое выбирается для газа при любой данной температуре, является давление р°, достаточно низкое для того, чтобы газ можно было считать идеальным. Для практических целей это давление обычно принимается равным 1 атм.
Для растворов в качестве стандартного состояния любого компонента можно выбирать чистое вещество при той же температуре и давлении, равном 1 атм, но иногда, как мы увидим ниже (стр. 174), удобнее в качестве стандартного выбрать некоторое совершенно фиктивное состояние. Причины этого лучше выявятся при изучении конкретных случаев.
РАБОТА И МОЩНОСТЬ
Определение работы. Работа производится всякий раз, когда система действует против внешней силы. Она выражается как произведение фактора интенсивности на фактор емкости. Для обычной механической работы фактором интенсивности является сила, а фактором емкости — расстояние, на котором действует сила. Поэтому А (работа) — F (сила) X (расстояние); или, если сила является пере-
менной, то А — FdL, откуда А можно определить, если F можно
L,
выразить как функцию от L.
Кроме механической работы, связанной с действием механических сил, можно иметь и другие виды работы в зависимости от природы действующей силы. Работа может производиться действием электрической силы или, точнее, электродвижущей силы (э.д. с.), обозначаемой буквой Е. В этом случае фактором емкости является Q — количество (или заряд) электричества, обычно выражаемое в кулонах.
Л*
Поэтому электрическая работа — \ EdQ. Аналогичными выражениями Q.
для работы будут следующие:
s.
Работа поверхностного натяжения — j sdS,
s, где а — поверхностное натяжение, а 5 — площадь поверхности.
h,
Работа преодоления силы тяжести = mgdh, л, где т—масса, g—-ускорение силы тяжести, h — высота в гравитационном поле по отношению к некоторому условно принятому нулевому уровню.
В термодинамическом понимании работа связана только с противодействием силам, являющимися внешними по отношению к данной системе. Не существует никакой .внутренней* работы, и употребление этого термина только увеличивает количество недоразумений.
Работа и мощность
65
Выяснению этого положения может помочь следующий пример. Предположим, что газ, заключенный под высоким давлением в цилиндр, расширяется и проходит через клапан в другой закрытый цилиндр с более низким давлением (опыт Джоуля). Если системой считать газ в обоих цилиндрах, то никакой работы производиться не будет. Если же системой считать только газ в первом цилиндре до дроссельного клапана, то этот газ будет производить работу против внешних сил.
Работа, обусловленная давлением среды. Работа, производимая давлением среды или против нее, представляет особенно важный случай для инженера-механика и химика-технолога. Поскольку давление является силой, действующей на единицу площади S, можно написать следующее общее выражение для механической работы:
dA = pSdL,
но SdL есть изменение объема, и поэтому
dA = pdV, или
к,
А = ( pdV. (26) д)
1
Если давление постоянно, это выражение переходит в
A = p(V2—
Если давление не постоянно, то для интегрирования уравнения (26) должна быть известна зависимость давления от объема. Если зави-
симость р — V можно выразить простой формулой, интегрирование удобно проводить алгебраически. Если зависимость известна, но не поддается выражению простой формулой, то интегрирование может быть весьма просто осуществлено графическим путем. Хорошо известно, что значение интеграла равно площади под кривой, выражающей зависимость р от V до оси р — 0 между данными пределами объема. Например, на рис. 2, где кривая АВ представляет зависимость между р и V, это значение равно заштрихованной площади, которую можно определить с помощью планиметра или просто ратов на миллиметровой бумаге.
Рис. 2. Определение работы по кривой pV.
посредством подсчета квад-
☆ ) Нужно отметить, что при всех подсчетах работы против внешнего давления в расчет следует брать абсолютное давление, а не манометрическое.
б Химическая термодинамика
бб
/. Определения и основные понятия
Индикаторная диаграмма. В случае непрерывно действующей машины, например газового компрессора или паровой машины, система претерпевает ряд изменений, возвращаясь в исходное состояние. На диаграмме рУ (рис. 3) представлен несколько идеализированный цикл, состоящий из четырех различных изменений, происходящих в течение двух ходов поршня ☆ ). Так как соответствующая работа для каждого изменения равна площади под каждой р—V линией до оси р = 0, то общая работа равна алгебраической сумме четырех членов. Два изменения происходят при движении поршня справа налево и представляют (в случае компрессора) работу, производимую поршнем над газом, тогда как при двух других изменениях поршень движется в противоположном направлении и работа производится газом над поршнем. Следовательно, два члена работы при алгебраическом суммировании следует писать со знаком, противоположным знаку двух других членов. Из геометрических соотношений легко показать, что сумма отдельных площадей равна заштрихованной площади, ограниченной контуром цикла
(Площадь DCCD' Ц- площадь ADD'А' — площадь АВВ'А'— — площадь ВССВ' = площади ABCD.)
Это приводит к выводу, что для любого цикла, представленного на диаграмме, работа равна площади, ограниченной контуром цикла.
Рис. 3. Определение работы цикла по pZ-диаграмме.
Посредством приспособления, известного как „индикатор*, двигатели могут автоматически вычертить цикл рУ-изменений, происходящих в цилиндре, и по этой индикаторной диаграмме можно сразу определять работу. Работа, определяемая таким путем, обычно называется „индикаторной*. Она будет равна работе за цикл и, при умножении на число циклов за данное время, будет давать работу, произведенную за данный период времени. Цикл, показанный на рис. 3, может быть двух вйдов, именно: это может быть зам-
кнутый цикл или цикл, представляющий протекающий процесс. В первом случае масса одинакова во всех частях цикла, и изменение объема представляет изменение состояния системы. В протекающем процессе загрузка в цилиндр производится на отрезке АВ, и изменение
☆) Следует, однако, отметить, что количества вещества не одинаковы во всех частях цикла и что новая загрузка производится на протяжении каждого цикла только один раз. Другими словами, мы рассматриваем незамкнутый цикл.
Работа и мощность
67
объема вызывается увеличением массы и ие представляет изменения состояния. Работу на таком отрезке будем называть „работой всасывания". Аналогично этому отрезок CD изображает выход материала из цилиндра, и работа на его протяжении будет называться „работой выхлопа". Отрезки ВС и DA представляют совершенно те же изменения состояния при постоянной массе, как и при замкнутом цикле. Выражение для общей работы цикла будет одинаково для обоих случаев при условии, что пользуются общим объемом.
Мощность. Мощность есть работа, произведенная за единицу времени, т. е.
(27),
Для электрической работы это уравнение переходит в следующее;
поскольку сила тока I по определению равна dQ/d^. Для механической работы преодоления давления среды
Р=РЖ- <28>
Легко заметить, что относительно небольшая работа может быть связана с большой мощностью при очень малом промежутке времени. Общеизвестными примерами являются взрыв или удар молнии.
В некоторых случаях мощность связывают с общей работой, произведенной за тот период времени, когда мощность расходовалась с определенной скоростью. Из уравнения (27) можно получить
о»
(29)
и, пользуясь этим, можно найти А, зная зависимость мощности от времени.
Некоторые инструменты, например ваттметр, измеряют мощность непосредственно, и определение общей энергии, затраченной на производство работы, может быть выполнено посредством уравнения (29 , пользуясь кривой мощность—время. Счетчик ватт-часов производит это интегрирование автоматически.
Если мощность затрачивается с равномерной скоростью, то
А = РД9, или Р=~
До
Таким образом, равномерный расход мощности для сжатия 100 м3 воздуха за час при данных условиях был бы равен работе сжатия этого объема, деленной на интервал времени.
5*
68
/. Определения и основные понятия
Единицы работы и мощности. Механическая работа выражается в килограмметрах (расстояние, умноженное на силу), кубометр-ат-мосферах (произведение pV), лнтр-атмосферах и других подобных единицах, которые еще не упоминались выше. Механическая мощность будет выражаться в единицах работы, деленной на время, или в килограмметрах в минуту, литр-атмосферах в час и т. д. Лошадиная сила произвольно определяется равной 75 кгм[час. Поскольку сила, умноженная на время, равна работе, работа часто выражается в единицах мощность—время, например лошадиная сила-час. Электрическая работа будет выражаться в вольт-кулонах (называемых также „джоулями*) или вольт-эквивалентах (эквивалент основан на электрохимических законах Фарадея и равен числу кулонов, отвечающих 1 грамм-эквиваленту иона), а мощность — в вольт-кулонах в секунду или вольт-амперах, обычно называемых „ваттами*. Аналогично механической работе электрическая работа может также выражаться в ватт-часах и других подобных единицах. В табл. II „Приложения* даются переводные коэфициеиты для различных единиц энергии &). Эквиваленты мощности будут такими же, за исключением различных единиц измерения, которые могут быть использованы в различных случаях.
ТЕПЛОТА
До XIX в. теплота считалась невесомой материальной субстанцией, известной под названием теплорода. Опыты Румфорда- в конце XVIII в. по изучению теплоты от трения тупого сверла и эксперименты Дэви, в которых лед полностью плавился от трения, нанесли решительный удар этому представлению и привели к установлению понятия о теплоте как о форме энергии, обусловленной движением отдельных молекул, из которых состоит вещество. Но только после Джоуля, проведшего примерно в середине прошлого века свои знаменитые опыты по количественному определению зависимости между теплотой и другими формами энергии, старые представления были полностью отвергнуты, и теплота была твердо признана одной из форм энергии (см. прим.2).
Термодинамическое определение. В термодинамике термин „теплота* употребляется в более узком смысле, и для полного понимания предмета важно с самого начала ясно представить себе это более ограниченное значение термина. Энергию, сохраняющуюся в системе, даже ту ее часть, которая возникает от молекулярного движения, не следует ассоциировать с термином „теплота*. Теплотой в термодинамическом смысле является энергия, которая передается от одной системы к другой посредством хаотического движения молекул.
☆) „Энергия* обычно употребляется как общий термин, охватывающий как теплоту, так и работу, а также характеризует способность системы совершать работу или передавать тепло. Более точное определение термодинамического свойства, называемого „энергией* или „внутренней энергией*, будет дано в следующей главе.
Т еплота
69
Энергия, передаваемая излучением, обычно также считается теплотой, поскольку подобно теплоте она не связана с действием против внешней силы, и результаты получаются в сущности одинаковые. Другими словами, теплота есть форма энергии, находящаяся в состоянии перехода от одной системы к другой. Мы будем стараться употреблять термин в этом ограниченном смысле и благодаря этому избегнем многих недоразумений, возникающих при обычном небрежном определении теплоты в термодинамических дискуссиях.
Тепловой источник. Тепловой источник является обычным термином, когда имеют дело с тепловыми двигателями, но термин несколько несовместим с только что высказанными представлениями. Строго говоря, теплота всегда перемещается и никогда не сохраняется в источнике. Термин относится к некоторой массе, которая может передавать или воспринимать энергию в форме теплоты от другой системы (или энергию излучения, эквивалентную теплоте). При некоторых термодинамических выводах удобно представлять себе очень большие источники тепла, например океан или атмосферу, к которым или из которых могут перемещаться конечные количества теплоты без заметного влияния на температуру.
Адиабатный процесс. Нам часто придется рассматривать изменения, при которых не происходит теплообмена между системой и окружающей ее средой. Такой процесс называется „адиабатным*. Он может происходить только тогда, когда 1) не существует никакой разности температур или 2) когда система термически изолирована от окружающей среды. Поскольку неизвестно вещество, дающее идеальную тепловую изоляцию, практически можно только в большей или меньшей степени приближаться к истинному адиабатному процессу. Регулируя температуру среды, непосредственно окружающей систему, таким образом, чтобы она была всегда равна (конечно, в некоторых пределах) температуре системы, можно осуществить почти полное приближение к адиабатному процессу.
Количественное выражение теплоты. Количество теплоты можно определить посредством вызываемого ею изменения температуры того вещества, которому она передается. Обычно в качестве такого вещества выбирается вода; так как количество тепла, необходимое для повышения температуры единицы массы на данную величину, не одинаково при различных температурах, то следует указывать температуру веществ, когда требуется высокая точность. Обычно единицей тепла служит калория. Грамм-калория, или малая калория, равна количеству тепла, необходимого для повышения температуры 1г воды на 1°С; она является „15-градусной калорией*, если температура меняется от 14,5 до 15,5° С. Средняя калория равна одной сотой количества тепла, необходимого для повышения температуры 1 г воды от 0 до 100° С.
В последние годы наблюдалось некоторое стремление к более или менее произвольному определению тепловых единиц посредством
70
I. Определения и основные понятия
других единиц энергии, особенно посредством электрических единиц. Например, интернациональная калория определяется как интер-OUV
национального киловатт-часа или грамм-калория — как 4,1833 интернациональных джоулей. Основы таких определений будут яснее после изучения следующей главы. Причиной такого выражения является стремление избежать того, чтобы единица основывалась на каком-либо свойстве вещества, которое может подвергнуться пересмотру. Это важнее в теоретической работе, чем в технологической. Кроме того, в современной калориметрии тепловые величины непосредственно измеряются в электрических величинах, и поэтому удобнее определять их на этой основе. Выбор числовых значений может быть •совершенно произвольным, но естественно, что желательно их выбирать таким образом, чтобы по возможности меньше изменять величины единиц, уже находящихся в употреблении.
Теплоемкость. Передача теплоты веществу часто приводит к повышению температуры, которое зависит от массы и специфического свойства вещества. Для бесконечно "малого изменения температуры это можно выразить с помощью следующего уравнения:
dQ = mcdt, (30)
где Q — количество теплоты, т— масса вещества, с — константа, характеризующая вещество и способ осуществления нагревания, с называется „удельной теплоемкостью0 и определяется этим уравнением. Произведение тс будет называться теплоемкостью, а если т представляет молекулярный вес М, то произведение Мс будет называться мольной теплоемкостью и будет обозначаться буквой С.
Из предшествующего изложения должно быть ясно, что теплота не является свойством веществ, и тем более С не является их свойством. Значение С зависит от того специального способа, которым нагревается система. Если давление поддерживается постоянным, то С изображается как Ср и называется изобарной теплоемкостью (или теплоемкостью при постоянном давлении). Подобным же образом можно определить и изохорную теплоемкость Cv (или теплоемкость при постоянном объеме). Обе эти величины представляют собой свойства, что будет очевидно из гл. III. Возможны и многие другие виды теплоемкости, но они не находят широкого применения, хотя одна из них будет определена в дальнейшем. Необходимо еще раз подчеркнуть, что теплоемкость вообще не является свойством в обычном смысле этого термина; это только величина, определяемая по приведенному выше уравнению. Однако, как только указано определенное условие, например постоянное давление, теплоемкость становится свойством системы.
Величину е, определенную по уравнению (30), можно назвать „истинной теплоемкостью0, причем это — величина, недоступная непосредственному термическому измерению. (Ее, однако, можно опреде
Самопроизвольные или необратимые процессы
71
лить непосредственно из измерения скорости звука.) На практике измеряют не истинную, а среднюю теплоемкость, определяемую по уравнению
где t2 — ty — конечная разность температур. Соотношение между двумя этими теплоемкостями дается следующим уравнением:
cdt
Теплоемкость имеет размерность теплоты, деленной на массу и на температуру, т. е. калория на грамм на градус. Поскольку теплота выражается также и в других единицах энергии, можно и теплоемкость выразить, например, в джоулях на грамм на градус (1 кал)г-град = 4,183 джоуля/г-град).
Истинная теплоемкость при постоянном р или постоянном V представляет функцию температуры, и для большинства практических целей эта функция обычно изображается простым рядом
С=а4-^ + т^ + а/3+... (33)
Так как уравнение (33) является чисто эмпирическим уравнением, его не следует экстраполировать за пределы температур, для которых были определены константы. Несоблюдение этой предосторожности приводит ко многим ошибочным выводам.
Скрытая теплота. В некоторых случаях сообщение или отнятие теплоты от системы не приводит к какому-либо изменению ее температуры. Например, это наблюдается при фазовом изменении чистого вещества при постоянном давлении. Такая теплота называется „скрытой теплотой". Обычными примерами такой теплоты служат теплота испарения, теплота плавления и теплота сублимации. Теплота химической реакции также является скрытой теплотой, поскольку она обычно ассоциируется с изотермическими условиями. Она выражается в калориях на грамм или грамм-моль.
САМОПРОИЗВОЛЬНЫЕ ИЛИ НЕОБРАТИМЫЕ ПРОЦЕССЫ
Все естественно происходящие процессы являются необратимыми в том смысле, что они стремятся происходить в определенном направлении и, предоставленные самим себе, никогда не обращаются, т. е. не идут в противоположном направлении. Подобно часам они стремятся израсходовать завод и не могут сами „завестись". Известными примерами необратимых процессов являются:
1. Передача теплоты посредством теплопроводности и конвекции при конечном температурном перепаде.
72
I. Определения и основные понятия
2 Расширение газа через дроссель от высокого давления к низкому.
3. Смешение двух газов.
4. Падение воды?
5. Химические реакции, как, например, горение водорода.
Что смешанные газы не разделяются самопроизвольно, или что вода самопроизвольно не разлагается при комнатной температуре на кислород и водород, или что теплота всегда переходит от тела с более высокой температурой к телу с более низкой температурой,— все это настолько хорошо известно из обычного опыта, что представляется почти ненужным упоминать о таких очевидных фактах. Тем не менее, второй закон термодинамики не представляет ничего более как обобщенную формулировку этого обычного опыта, и чтобы понять его значение, необходимо более детально рассмотреть зависимость между реальным или необратимым процессом и некоторым идеализированным процессом, который будет называться обратимым.
Все вышеупомянутые изменения могут быть обратимы. Можно передавать теплоту из места с низкой температурой в место с более высокой температурой, можно разделять газовую смесь на ее составляющие, можно заставить воду течь вверх и можно разложить воду при комнатной температуре на водород и кислород. Но важно то, что все это можно сделать только при затрате работы, производимой некоторой другой системой, которая сама при этом уменьшает свою работоспособность. Для обращения этих изменений следует произвести работу, а работу можно получить, только давая возможность произойти самопроизвольному изменению где-нибудь в другом месте. Иными словами, уничтожить результат данного самопроизвольного изменения можно только за счет какого-либо другого изменения.
Можно получить полезную работу только в том случае, если имеется стремление к самопроизвольному изменению. Есть нечто парадоксальное в том, что изменение не может быть совершенно необратимым, если оно служит для получения полезной работы, но должна существовать тенденция к самопроизвольному изменению. Так, если рассматривать большие количества воды при одинаковой температуре, то не существует стремления к переходу теплош и нельзя получить работы, хотя система обладает большим запасом энергии. Однако, если отдельные части воды находятся при различных температурах, то имеется стремление к самопроизвольному изменению-— к выравниванию температуры. Располагая надлежащим механизмом, можно использовать стремление к самопроизвольному изменению для получения работы; поступая таким образом, мы, по существу, избе.аем полной необратимости и можем1 представить механизм в пределе обратимым.
Стремление к изменению обусловлено определенными „движущими силами", и эта концепция встречается при рассмотрении почти
Равновесие и обратимый процесс
73
всех операций и процессов в химической технологии. Движущей силой при переходе тепла является разность температур, при переходе вещества — разность концентраций и для потока жидкости или газа — разность давлений. Везде, где есть такая движущая сила, имеется стремление к изменению. Может казаться, что изменения не происходят, так как сопротивление делает изменение столь медленным, что его даже незаметно. Если знать о существовании движущей силы, то можно предсказать направление изменения и знать, что можно получить полезную работу. При переходе тепла, при движении потока или при каком-либо другом чисто механическом процессе движущие силы легко распознаются, но это далеко не так просто для химических систем. Для таких случаев предложена особая функция — изобарный иотенциал, которая будет подробнее рассмотрена в следующей главе.
РАВНОВЕСИЕ И ОБРАТИМЫЙ ПРОЦЕСС
Истинное равновесие. Конечным результатом любого самопроизвольного процесса является состояние равновесия, при котором силы уравновешены и не имеется никакого дальнейшего стремления к изменению, никакой движущей силы. Важно различать истинное (или устойчивое) равновесие с уравновешенными силами и кажущееся, ложное равновесие, при котором стремление к изменению еще существует, но вследствие большого сопротивления скорость изменения настолько мала, что практически не происходит никаких изменений. Чтобы различить истинное и кажущееся равновесия, существует простое испытание. При истинном равновесии, поскольку силы уравновешены, малое (точнее, бесконечно малое) увеличение или уменьшение одной из сил будет вызывать сдвиг в положении равновесия.
Например, представим поршень в вертикальном цилиндре с давлением газа, действующим на нижнюю поверхность поршня, и с уравновешенным грузом, расположенным на поршне. При полном отсутствии трения даже очень маленький добавочный груз, накладываемый на поршень, будет заставлять его опускаться. Движение может быть очень медленным, и, чтобы проследить его, могут понадобиться чувствительные приспособления, но оно все же происходит. При столь же незначительном увеличении давления газа движение вниз может быть приостановлено, а дальнейшее (незначительное) увеличение давления даже изменит направление движения поршня. Наличие значительного трения между поршнем и стенкой цилиндра вызывает необходимость наложения на поршень гораздо большего груза для опускания поршня или, соответственно, гораздо большего увеличения давления газа, для того чтобы вызвать обратное движение. В этом случае прекращение движения не указывает на истинное равновесие. В действительности трение никогда нельзя полностью
74
/. Определения и основные понятия
устранить, и поэтому никогда не достигается истинное состояние равновесия, но посредством тщательного подбора условий можно подойти к равновесию очень близко, и, чтобы представить себе состояние истинного равновесия, потребуется лишь очень небольшая экстраполяция. Состояние истинного равновесия, подобно идеальному газу, является концепцией, а не реальным состоянием, встречающимся в практике; тем не менее эта концепция имеет громадное практическое значение при изучении процессов *).
Рассмотрим другой пример. Смешение при комнатной температуре газообразного кислорода, водорода и паров воды не приводит к заметному изменению в составе смеси, независимо от того, как долго вещества остаются вместе. Тем не менее эта система не находится в состоянии химического равновесия. Существует стремление к изменению, но оно очень сильно сдерживается некоторыми силами, аналогичными трению в механических системах. Ложность равновесия можно показать несколькими путями. Если система находится в истинном равновесии, то при прибавлении небольшого количества водорода или кислорода должна произойти реакция, приводящая к образованию большего количества водяного пара. В действительности при комнатной температуре ни добавка водяного пара, ни добавка водорода не вызывает каких-либо реакций; с другой стороны, при введении относительно ничтожного количества энергии в виде электрической искры может произойти значительное изменение, свидетельствующее о том, что в системе существовало сильное стремление к изменению. Если рассматривать ту же самую систему при температуре в 2000° С, то положение совершенно меняется. Незначительно малые добавки водорода или водяного пара будут вызывать немедленную реакцию, а пропускание искры не будет оказывать заметного действия. Таким образом, мы имеем определенный критерий истинного равновесия, и при экспериментальных исследованиях равновесий всегда лучше достигать равновесия с двух сторон, чтобы не быть введенными в заблуждение ложным равновесием.
Про систему в равновесии говорят, что она находится в стабильном состоянии; напротив, неравновесная система будет являться нестабильной. Так, вода может охлаждаться ниже 0°£ при давлении в 1 атм, но это не будет стабильной формой воды при этих условиях. Твердые тела вследствие сопротивления внутренним перегруппировкам, присущего их структуре, могут в течение долгого времени существовать в нестабильных формах. Известным примером является
*) Эта точка зрения неправильна. Во многих реакциях, протекающих с большой скоростью, как, например, во многих ионных реакциях в растворах, равновесие достигается очень быстро, и если подходить к нему с обоих противоположных направлений, то в обоих случаях конечное состояние системы оказывается одинаковым с любой заданной степенью точности. Такое состояние является реальным равновесным состоянием. Впрочем, несколько дальше автор и сам высказывает такую же точку зрения. (Прим, ред.)
Равновесие и обратимый процесс
75
стекло, которое с термодинамической точки зрения является нестабильной системой.
Частичное равновесие. Очень часто, даже почти всегда, имеют дело с частичным (неполным) равновесием, особенно в случае химических реакций. Под частичным равновесием понимается такое, при котором истинное равновесие достигается по отношению к какому-нибудь отдельному изменению, а не по отношению к другим возможным изменениям. Можно иметь дело с равновесием растворимости водорода и кислорода в воде при таких условиях, когда система далека от химического равновесия. Скорость достижения химического равновесия в данном случае настолько мала по сравнению со скоростью достижения равновесия растворимости, что ею можно полностью пренебречь. Это является крайним случаем. Имеется много случаев, особенно при органических реакциях, когда этот вопрос имеет большое значение. Предположим, например, что желательно определить состояние равновесия реакции
СО 4- 2Н2 = СН3ОН
при данных давлении и температуре. Главная трудность заключается в том, что одновременно могут происходить и другие реакции. Укажем только на некоторые из них:
СО-(-ЗН2 = СН4-4-Н2О, со + н2о=со2 + н2, 2СН3ОН = (СН3)2О + Н2О.
В общем мы не интересуемся полным равновесием по отношению ко всем возможным реакциям, которое может быть совершенно отличным от частичного равновесия по отношению к реакции синтеза метанола, если другие полностью подавляются. Если скорость других или побочных реакций не равна в действительности нулю, то нельзя достигнуть истинного равновесия в основной реакции. Практически все, на что можно рассчитывать, заключается в таком выборе условий, чтобы свести к возможному минимуму скорости побочных реакций. Этим путем в одной реакции можно достигнуть вполне хорошего для всех практических целей приближения к истинному равновесию.
Равновесие и движущая сила. Хотя уже указывалось, что термодинамика связана с равновесными состояниями, а совсем не со скоростями изменения, тем не менее имеется важное связующее звено между равновесием и скоростью через движущую силу. Скорость, при которой происходит любое изменение, в общем определяется расстоянием от равновесного состояния, и поэтому для определения движущих сил необходимо знать равновесные состояния. Так, для вычисления размеров абсорбционной башни нужно вычислить среднюю
76
I. Определения и основные понятия
движущую силу, которая будет равна разности действительной и равновесной концентраций растворенного вещества. Подобные соображения можно приложить к передаче теплоты, к сушке, к химическим реакциям и ко многим другим процессам, с которыми имеет дело химик-технолог.
Обратимый процесс. До сих пор мы рассматривали изолированные, т. .е. несвязанные, равновесные состояния и установили, что критерием равновесия является обратимость. Рассмотрим теперь связанный ряд равновесных состояний, каждое из которых представляет только бесконечно малое смещение по отношению к соседнему, но все вместе приводят к конечному изменению; в этом случае мы имеем обратимый процесс. Все реальные или самопроизвольно протекающие процессы могут путем соответствующего выбора условий в большей или меньшей степени приближаться к обратимому процессу, но, подобно абсолютному нулю температуры и идеальному газу, строго обратимый процесс является только концепцией, помогающей при анализе некоторых задач. Однако приближение реальных процессов к этому идеальному пределу может быть настолько близко, насколько мы этого пожелаем. Степень приближения вообще ограничена скорее техно-экономическими факторами, чем чисто физическими. Истинно обратимый процесс потребовал бы для своего совершения бесконечного времени, а мы обычно требуем, чтобы наши процессы полностью заканчивались в возможно короткий срок.
Обратимый переход тепла. Выше рассматривались некоторые необратимые процессы. Теперь рассмотрим те же самые процессы в обратимой форме. Вместо того, чтобы допустить самопроизвольный переход теплоты посредством теплопроводности из теплового источника при tx (теплоотдатчик) в другой источник при более низкой температуре t2 (теплоприемник), можно заставить тепло передаваться газу, сжатому посредством поршня, сила которого уравновешена другой силой, передаваемой от механизма, совершающего работу. Затем можно дать возможность газу расшириться, поддерживая при этом температуру постоянной посредством передачи тепла от теплоотдатчика при tx. Для обеспечения такого перехода температура газа должна быть несколько ниже температуры теплоотдатчика, но разность может быть как угодно малой, если мы не связаны скоростью прохождения процесса. Если разность равна величине dt, то бесконечно малое увеличение температуры газа будет прекращать передачу тепла, и дальнейшее бесконечно малое увеличение будет менять ее направление. Другими словами, это необходимое и достаточное условие для обратимого проведения процесса.
При расширении газа его давление падает, и уравновешивающая сила, действующая в противоположном направлении через поршневой шток, должна также уменьшиться. Мы не будем детально рассматривать механизм, нужный для совершения этого процесса. Достаточно
Равновесие и обратимый процесс
/
Объем
4. Цикл, иллюстрирующий обратимый переход тепла.
сказать, что он может быть осуществлен с помощью маховика. Из ранее сказанного ясно, что для обратимости процесса необходимо, чтобы сила, передаваемая от совершающего работу механизма, была все время только на бесконечно малую величину меньше, чем сила, обусловленная давлением газа.
В определенный момент хода поршня удалим цилиндр из соприкосновения с теплоотдатчиком и изолируем его термически от окру-' жающей среды. Поскольку продолжается расширение, температура газа будет уменьшаться (адиабатное расширение). Когда температура понизится до t2, цилиндр приводится в соприкосновение с теплоприемником при t2, и направление движения поршня делается противоположным посредством поддерживания силы, действующей через поршневой шток от махового механизма на уровне, § немного превышающем (иа вели- ® чину dp, если выражать ее через g давление) силу, обусловленную Ч давлением газа. Затем газ изотермически сжимается, причем постоянство температуры поддерживается передачей тепла тепло- ] приемнику при t2. Можно завершить цикл, дав системе возмож
ность воспринять больше тепла из теплоотдатчика при затем вновь термически изолировать цилиндр от окружающей среды и заставить газ сжаться до начального давления, причем его температура возрастет до (адиабатное сжатие).
Цикл изменений, которым был подвергнут газ, можно изобразить на pV-диаграмме (рис. 4). Процесс 12 является изотермическим расширением, во время которого газ получает тепло из теплоотдатчика при .температуре tv Процесс 23 является адиабатным расширением, 34 — изотермическим сжатием и 41 — адиабатным сжатием. Общим результатом процесса в целом является переход определенного количества теплоты от теплоотдатчика при в теплоприемник при t2; в то же самое время производится некоторая работа против внешних сил, которой не было бы, если переход тепла происходил бы путем непосредственной передачи тепла от одного резервуара к другому. Следует отметить еще одно различие между двумя этими процессами.
В случае обратимого процесса количество тепла, переданного тепло-приемнику при t2, будет меньше количества тепла, взятого из теплоотдатчика при на величину, эквивалентную произведенной работе.
Рассмотренный круговой процесс можно считать типичным для изменений, происходящих в тепловых машинах. Вообще говоря,
78
I. Определения и основные понятия
тепловая машина определяется как механизм, превращающий теплоту в работу. В идеализированной тепловой машине, цикл которой рассматривался выше, мы намеренно избегали рассмотрения деталей механизма, поскольку они привели бы только к запутанности в доказательствах и помешали бы ясному пониманию основных принципов. Это является обычным приемом для большинства термодинамических рассуждений.
Если сила, противодействующая поршню, отличается от уравновешивающей ее силы только на бесконечно малую величину и если температура тепловых источников не отличается более чем на бесконечно малую величину от температуры газа, заключенного в цилиндр, то бесконечно малое изменение температуры или давления будет вызывать обращение процесса, и можно пользоваться передачей теплоты от t2 к tv Так как условия одинаковы как для прямого, так и для обратного процесса (в пределах бесконечно малых величин), то работа, требуемая от механизма для передачи тепла от t2 к в точности равна (для одного и того же количества тепла в каждом тепловом источнике) той работе, которая производилась бы, если тепло передавалось бы в обратном направлении. Это означает, что если бы процесс осуществлялся посредством отдельного цикла или некоторого количества циклов, передаю цих тепло от tr к t2, и произведенная работа использовалась бы для накопления энергии в такой форме, которую можно было бы легко и полностью вновь употребить (можно представить это в виде подъема груза), то, позволив грузу упасть, можно было бы с помощью накопленной энергии возвратить тепловые источники к их исходному состоянию. Это очень важный принцип. В общем это означает, что полностью обратимый процесс будет давать достаточное количество энергии в легко доступной форме (т. е. в виде работы) для того, чтобы все свойства системы могли быть возвращены к своим исходным значениям без обращения к помощи какой-либо внешней системы.
Степень необратимости. Мы рассмотрели процесс, проведенный двумя предельными способами — полностью необратимым и полностью обратимым. Между этими пределами возможно бесконечное число промежуточных ступеней. Это означает, что имеются различные степени обратимости и необратимости, и важно учитывать это обстоятельство, говоря о необратимых процессах.
Рассмотрим простой цикл паровой машины, изображенный на рис. 4, при условии, что газ в цилиндре во время хода’ 12 будет при t\, что ниже на конечную величину М, или же давление газа во время того же самого хода будет на величину Др меньше силы, действующей через поршневой шток (и деленной на площадь поршня). Каждое из этих условий привело бы к уменьшению площади, заключенной в пределах цикла для одинакового интервала изменения. Поскольку замкнутая площадь, как мы уже показали, является
Равновесие и обратимый процесс
79
мерой работы, любое необратимое действие, например конечные Др или St, будет приводить к уменьшению выигрыша в работе (или к увеличению работы, требуемой для процесса, протекающего в противоположном направлении). Следовательно, полностью обратимым процессом будет процесс, дающий в данных условиях максимальное количество работы (или требующий минимального количества работы).
Обратимый процесс как стандарт для сравнения. Рассмотренные соотношения привели к выяснению важности понятия обратимости и обратимого процесса в термодинамике. По общему признанию, обратимый процесс является идеализированным процессом, недостижимым на практике. Это следует понять совершенно ясно. В процессе, который мы только что рассмотрели, обратимость требует полного отсутствия трения, материалов, не проводящих тепла, и бесконечно малую скорость изменения (поскольку допустимы только бесконечно малые движущие силы). Практически экономичная операция может потребовать значительного отклонения от этих идеализированных условий, но нет никаких внутренних физических трудностей, которые препятствовали бы выбору соответствующих условий, чтобы настолько приблизиться к этому идеалу, что незначительная экстраполяция привела бы к нему полностью. Единственной причиной введения понятия об обратимом процессе является установление стандарта для сравнения реальных процессов. Обратимым процессом является процесс, дающий максимальную работу или требующий наименьшей работы для своего осуществления. Он говорит о максимальной производительности, к которой можно стремиться, но которой никогда нельзя достигнуть полностью. Без такого абсолютного стандарта или масштаба все попытки инженеров улучшить процессы будут уподобляться стрельбе в темноте без определенной цели. С обратимым процессом как со стандартом мы встречаемся каждый раз, когда реальный процесс уже достаточно эффективен или же он слишком непроизводителен и поэтому может быть значительно усовершенствован.
Понятие обратимого процесса представляет громадное значение при анализе сложных процессов, состоящих из большого числа "стадий. Умеющий распознавать обычные необратимые процессы может сразу определить причины незначительного коэфициента полезного действия, так как несомненно, что любое необратимое изменение означает уменьшение эффективности. Многочисленные примеры, иллюстрирующие это положение, будут даны в следующих главах.
Обратимое расширение газа. Мы уклонились от цели, поставленной несколькими страницами выше и состоящей в рассмотрении обратимого проведения самопроизвольных процессов, упомянутых на стр. 71—72. После детального рассмотрения первого процесса для других потребуется только краткий обзор. Обратимый процесс расширения газа входит в простой процесс тепловой машины и не нуждается в дальнейшем рассмотрении. Попутно можно указать, что адиабатное
so
I. Определения и основные понятия
расширение реального (в отличие от идеального) газа через пористую втулку или дроссель при совершении полезной внешней работы не является, как это иногда думают, полностью необратимым процессом;
в результате его прохождения создается разность температур, которую можно использовать для получения работы в тепловой машине, как только что было показано, и эту работу можно использовать
для частичного повторного сжатия газа.
Обратимое смешение газов. Если два газа привести в соприкосновение и смешать с помощью диффузии, то процесс будет
•необратимым, так как не существует возможности разделения смеси без помощи какого-либо внешнего
:Рис. 5. Обратимое смешение двух газов.
источника. Обратимое смешение (или разделение) газов легко совершается (мысленно) с помощью полупроницаемой мембраны, т. е. мембраны, полностью проницаемой для одного газа и абсолютно непроницаемой для всех других. Это является идеализированным видом аппаратуры, подобно поршню без трения и твердому телу, лишенно-
му теплопроводности; но в настоящее время уже изготовляются мембраны, приближающиеся к идеалу .(например мембраны, употребляющиеся при изучении^ осмоса, и тонкая палладиевая фольга, в высшей степени проницаемая для водорода и лишь
незначительно проницаемая для всех других газов), и нужна только
незначительная экстраполяция до предела, т. е. до идеальной мембраны *).
Рассмотрим резервуар А (рис. 5), содержащий одинаковое число молей кислорода и азота при давлении 1 атм. Пусть D — цилиндр, содержащий поршень, который подвергается атмосферному давлению и един--ственной задачей которого является увеличивать объем А (при добавлении газа) для поддержания в ием постоянного давления. Цилиндр В содержит кислород, а С — азот, и оба они снабжены поршнями, связанными
с соответствующими механизмами, уравновешивающими давление газа при всех положениях поршня. Пространство справа от поршней представляет абсолютный вакуум. Е и F — выдвигаемые заслонки, которые по желанию можно заменить полупроницаемыми мембранами. Для простоты все газы будут считаться идеальными, но это ограничение не влияет на интересующий нас принцип. В целом аппарат мыслится окруженным термостатом, действующим как тепловой источник.
*) Гидратированная соль в соприкосновении с влажной средой представляет хороший пример фильтрующих свойств полупроницаемой мембраны. Водяной пар проходит через поверхность газ — твердое тело, а другие газообразные компоненты через иее не проходят.
Равновесие и обратимый процесс
81
В начале процесса резервуары В и С заполнены газом при давлении 1 атм и в точках Е и Р вставлены непроницаемые заслонки. Предоставим каждому газу расширяться изотермически и обратимо до давления, равного его парциальному давлению в резервуаре. Затем заменим заслонки полупроницаемыми мембранами, одна из которых проницаема только для кислорода, а другая — только для азота, и протолкнем оба газа при постоянном давлении в А. Благодаря полупроницаемости мембран давление, противостоящее проталкиванию каждого газа, равно только 1/2 атм, т. е. парциальному давлению газа, для которого мембрана является проницаемой. Процесс может быть обращен в любой момент цикла посредством бесконечно малого изменения сил, иначе говоря, работа, которая могла бы быть получена при смешении данного количества газов, может быть полностью испбльзована для разделения того же количества газа.
Обратимое падение воды. Падение юды можно сделать обратимым посредством отвода потока с верхнего уровня в трубу, подающую воду к турбине, расположенной на нижнем уровне. В турбине воду под давлением заставляют течь через соответствующие сопла, превращающие ее потенциальную энергию в кинетическую, которая в свою очередь превращается в кинетическую энергию вращающегося колеса. Эту энергию можно сохранить для последующего использования или же употребить непосредственно для подачи воды иа ее первоначальный уровень при условии, что каждая стадия проводится обратимо, т. е. без трения.
Обратимая «мимическая реакция. Даже химическую реакцию можно провести обратимо при наличии соответствующего механизма, которым может быть гальванический элемент. Не входя в подробное рассмотрение этих элементов, нужно лишь указать, что из большинства реакций, происходящих между ионами, можно получить э. д. с. между двумя электродами, и если эта э. д. с. противоположна э. д. с., приложенной извне, то реакция может быть проведена обратимо. Практический пример обратимой реакции можно видеть в применении гальванического элемента в потенциометрической цепи. Необходимо, Чтобы этот элемент работал практически обратимо.
Даже некоторые газовые реакции, как, например, реакцию, уже рассмотренную в качестве примера необратимого процесса, можно сделать ионными реакциями при подборе подходящего материала для электродов. Так, водород и кислород можно соединять необратимо при обычном горении или же соединять обратимо в электрохимическом элементе, если обратная э. д. с. на бесконечно малую величину меньше э. д. с. элемента. При медленном возрастании обратной э. д. с. реакция будет итти в противоположном направлении, причем вода будет разлагаться на водород и кислород. При практической работе такого элемента, как и при всех других рассмотренных процессах, неизбежны некоторые необратимые эффекты, которые снижают э. д. с., пригодную для внешней работы, когда реакций б Химическая термодинамика
82 I. Определения и основные понятия
проходит в направлении образования воды или увеличивает э. д. с., необходимую для разложения воды сверх значения для идеального (обратимого) случая.
Химическую реакцию между газами, например реакцию ЗН2 + N2 = 2NH3, можно мысленно провести обратимо посредством механизма, уже использованного для иллюстрации обратимого смешения газов. Пусть на рис. 5 резервуар А, названный «ящиком равновесия* Вант-Гоффом, который предложил эту схему, содержит смесь N2, Н2 и NH3 в равновесии при некотором данном давлении р и температуре t, причем и р и t остаются постоянными. Цилиндры В, С и D при некотором данном давлении р0 содержат, соответственно, Н2, N2 и NH3, а непроницаемые затворы предохраняют их содержимое от соприкосновения с газами в ящике. Заметим, что D теперь считается цилиндром с поршнем, связанным с рабочим механизмом, так же как и цилиндры В п С. При смешении газов с помощью полупроницаемых мембран Н2 и N2 расширяются изотермически и обратимо от своих исходных давлений до таких давлений, которые они имели бы, находясь с помощью полупроницаемых мембран в равновесии со смесью в ящике. (Для идеальных газов давление было бы равно парциальному давлению соответствующего газа в ящике.) Затем, так же как и раньше, непроницаемые затворы заменяют полупроницаемыми мембранами, и газы нагнетаются в ящик. Так как это нарушает равновесие в ящике, то будет образовываться NH3, и с той же скоростью, с которой он образуется, он проникает в D через полупроницаемую мембрану. Благодаря этому состав в ящике всегда останется постоянным. Наконец, аммиак можно изотермически сжать от парциального давления, под которым он находится в ящике, до р0. Общим результатом будет превращение данного количества Ы2 и Н2 при давлении р0 в NH3 при р0 посредством изотермического процесса. Обратимость этого процесса становится очевидной при приложении обычного критерия. Вопрос о суммарной величине работы такого процесса будет рассмотрен в следующей главе. Конечно, существуют определенные тепловые эффекты, сопровождающие операции в ящике. Во время сжатия теплота должна передаваться от газа в окружающую среду, и это будет являться тепловым эффектом, сопровождающим реакцию в ящике, причем он может быть положительным или отрицательным. Поскольку эти переходы тепла происходят между системой и окружающим ее термостатом с температурой, отличающейся от температуры среды только на dT, переходы будут обратимыми.
Замечания по поводу обратимых процессов. Обратимый процесс представляет очень трудно определимое понятие, и идея обратимости редко сразу усваивается. Тем не менее эта концепция очень полезна; для ясного ее понимания могут служить примеры обратимых процессов, приводимые в последующих главах.
Равновесие и обратимый процесс
83
Полезно дать краткий обзор затруднений, возникающих при попытках слишком строго придерживаться любого формального определения. Рассмотрим расширение газа, находящегося под поршнем, на который он оказывает давление, постоянно уравновешенное противоположной силой в поршневом штоке. Такой процесс будет обратим по одному из критериев, данных для обратимого процесса, но предположим, что сила поршневого штока полностью израсходована в виде теплоты за счет приведения в действие гидравлического тормоза. Согласно другому, данному ранее критерию, процесс в целом является бесспорно необратимым, так как нет способа для повторного сжатия газа без привлечения на помощь другой внешней системы.
Пойдем дальше и предположим, что газ, заключенный в цилиндр-сборник, расширяется через дроссель до более низкого давления. В целом этот процесс является определенно необратимым, но, поскольку газ находится в цилиндре, считается, что нет никакой разницы, проталкивается ли газ против поршня или только против других молекул газа. Следовательно, можно рассчитать изменение состояния газа в цилиндре с помощью уравнений, выведенных для равновесного или обратимого процесса.
Возьмем другой простой пример из области теплопередачи. Предположим, что вещество нагревается от 30 до 90° С путем протекания через противоточный теплообменник, содержащий вещество, охлаждающееся от 120 до 40° С. И нагрев и охлаждение сами по себе можно считать обратимыми изменениями, и обычно так и делается при термодинамических рассуждениях. Чтобы не возникло представление, что тепловой поток при бесконечно малой разности температур может быть только обратимым, достаточно вообразить тепловой источник, температура которого всегда остается на dt градусов выше (или ниже) температуры нагреваемого (или охлаждаемого) вещества. С другой стороны, это не создает путаницы относительно процесса в целом. Он бесспорно является необратимым процессом, поскольку тепло передается с конечной разностью Д/. При некоторых выводах полезным оказывается другой подход к анализу обратимости процесса. Вкратце его можно характеризовать следующим образом. Если представить достижение всех равновесных состояний только как бесконечно малый переход от соседних состояний, то обратимый процесс можно изобразить графически непрерывной линией на диаграммах состояния (pv или pt ит. д.). Необратимый же процесс так изобразить нельзя. В случае необратимого процесса можно отметить начальное и конечное состояния и указать общее направление изменения; но природе необратимого процесса присуще, чтобы полный путь изменения был неопределенным, и поэтому он не может быть изображен в виде линии на термодинамической диаграмме.
6*
ГЛАВА И
ПЕРВЫЙ И ВТОРОЙ ЗАКОНЫ ТЕРМОДИНАМИКИ
Исторический очерк. Опыты Румфорда [217] были первой, хотя и очень незрелой, попыткой количественного определения зависимости между тепловой и другими формами энергии8. Он измерял возрастание температуры и количество металлических стружек при сверлении пушек тупым сверлом с помощью конной тяги. Этим путем Румфорд показал, что количество получаемого тепла не ограничено и зависит только от длительности опыта. Количество выделявшегося тепла было так велико, а количество высверленного металла так мало, что не могло быть и речи о существовании зависимости между ними. Из своего опыта Румфорд заключил, что тепло не может быть материальным потоком, а должно быть движением. Этот вывод положил начало ниспровержению теории теплорода3. Относящиеся примерно к тому же времени опыты Дэви по таянию льда от трения дали результаты, котор ле также были совершенно несовместимы с теорией теплорода и привели к заключению, что теплота обусловлена движением маленьких частиц и что это движение может быть возбуждено процессом трения. Несмотря на эти опыты, многие ученые все же цеплялись за старую теорию теплорода и оставили ее только после прекрасно проведенных количественных опытов Джоуля в 184Э—1850 гг., непоколебимо установивших новую теорию. Нельзя было закрыть глаза на точные цифры, собранные Джоулем. Он показал, что, где бы ни производилась механическая или электрическая работа при условии, что в системе не было никакого накопления энергии, количество выделившейся теплоты всегда находится в определенном соотношении с произведенной работой, причем соотношение между ними зависит только от выбранных единиц. Это явилось решающим доказательством того, что теплота является формой энергии4. Другие ученые рашше Джоуля высказывали правильные идеи об эквивалентности между теплотой и работой и даже давали приближенные значения эквивалента, но их работы не были так убедительны, как исслечования Джоуля, вследствие малого количества опытных данных, на которых они обосновывали свои доказательства. Следует, впрочем, упомянуть Майера, который очень ясно выразил сущность идеи в 1842 г.
Идея сохранения энергии, т. е. что энергия не может создаваться и не может уничтожаться, была уже установлена физиками в при
Первый закон а понятое энергии
85
менении к чисто механическим системам. Распространение этого принципа на другие формы энергии составляет первый закон термодинамики.
Тогда как первый закон был в сущности выводом из опыта, второй закон возник почти из чисто индуктивных положений. Карно поставил себе задачей определить максимальное количество работы, которое можно получить в тепловой машине из данного количества теплоты. Его «Размышления о движущей силе огня и о машинах, пригодных для получения такой снлы“, опубликованные в 1824 г., содержат принцип Карно (основную базу любого анализа тепловых машин), согласно которому производительность идеальной тепловой машины зависит только от температур теплоприемника и теплоотдатчика и совершенно не зависит от механизма и примененного рабочего тела. Доказательство своей теории Карно отчасти основывал на теории теплорода, позднее совершенно опровергнутой работой Джоуля.
Кельвин и Клаузиус независимо друг от друга показали, что принцип Карно действителен и на основе новой теории теплоты. Аксиомы, провозглашенные ими, превратили доказательство Карно в ту форму, которая теперь является общепринятой краткой формулировкой второго закона. В это же время Клаузиус ввел функцию «энтропия*, которая оказала чрезвычайно сильное влияние на количественное развитие второго закона и является необходимой при его применении, хотя и доставляет до настоящего времени огорчения всем начинающим изучение термодинамики.
Второй закон был неохотно принят учеными всего мира, и сомнения в его спр; ведливости выражались даже в очень недавние годы. Однако он остается весьма широкой и надежной основой опытных данных и может , считаться аСсолютно точным законом, не имеющим ни одного исключения. Со времени Карно, Кельвина и Клаузиуса сильно расширилось его применение в области тепловых машин; действительно, можно сказать, что второй закон лежит в основе почти всех физических явлений. Следует упомянуть еще Вилларда Гиббса, заложившего основы применения термодинамики к химическим системам.
ПЕРВЫЙ ЗАКОН И ПОНЯТИЕ ЭНЕРГИИ
Работа, эквивалентная теплоте. Если взять статическую, т. е. покоящуюся, систему, причем безразлично, насколько она сложна по составу, и провести ее через цикл изменений, во время которых работа, совершаемая над системой, производится силами, действующими со стороны окружающей среды (или наоборот), а теплота переходит к системе от среды (или от системы к среде), то опытным путем устанавливается, что между алгебраической суммой всех,работ и суммой всех тепловых эффектов имеется очень простое соотношение, а именно
2A = J%Q.
О)
86
II. Первый и второй законы термодинамики
Чтобы все рабочие эффекты измерялись в одинаковых единицах, примем, что все они одной природы, т. е. представляют механическую работу. Тепловые эффекты, конечно, должны измеряться в термических единицах, например в калориях, поскольку их измерение в других единицах тесно связано с иллюстрируемым здесь принципом.
Это простое обобщение является прямым результатом уже упомянутых опытов Джоуля, но константа пропорциональности J была гораздо точнее определена позднейшими исследователями. Она зависит только от выбора единиц для измерения А и Q, а никоим образом не зависит от выбранной системы, от примененного механизма или от других причин. Для механической работы константа пропорциональности известна под названием „механического эквивалента теплоты”. Можно также получить и „электрический эквивалент теплоты”. Поскольку опыт показал, что имеется такая же эквивалентность между различными видами работы (например электрический эквивалент механической работы), можро откинуть ограничение относительно одинаковости рабочих эффектов, так как различные виды их легко взаимно превращаются (это обстоятельство учитывается при всех дальнейших рассуждениях).
Понятие о внутренней энергии. Зависимость, выраженная уравнением (1), строго действительна только тогда, когда рабочее вещество или система, с которой мы экспериментируем, проходит через полный цикл изменений, т. е. возвращается в свое исходное состояние. При рассмотрении таких процессов, как испарение жидкости, химическая реакция или сжатие газа, не существует простой, общей для всех процессов, зависимости между теплотой и работой. Поскольку теплота и работа являются внешними эффектами, получающимися вследствие изменений внутри системы, и поскольку эти эффекты должны считаться возникающими из уже существующих действий или, другими словами, должны иметь причину, то следует ввести понятие энергии. Теплоту и работу следует считать формами энергии и в более узком термодинамическом смысле — внешним выражением накопленной энергии4.
Всякий раз, когда теплота и работа не эквивалентны, как это вообще бывает в некруговых процессах, это обстоятельство принимается во внимание, и говорят, что в системе накопилось определенное количество энергии, или же, наоборот, что некоторая накопленная энергия утрачена системой и проявилась как теплота, или работа, или как то и другое одновременно. Энергия может сохраняться в системе или может легко передаваться от одной системы к другой в виде теплоты или работы. При всех этих превращениях она остается количественно неизменной. Это является принципом сохранения энергии, образующим основу первого закона термодинамики. Он покоится главным образом на опытных фактах, обобщенных уравнением (1).
Энергия, накапливаемая в системе, является определенным свойством системы и изображается символом U. Ее называют внутренней
Первый закон и понятие энергии
87
энергией, в отличие от энергии, которая может ассоциироваться с системой, но не является ее свойством. Внутренняя энергия обусловлена конфигурацией частиц вещества, составляющего систему, и их движением. Энергия, происходящая от движения системы в целом или от ее положения в поле силы тяжести, или во внешнем электрическом или магнитном поле, не включается в величину U, так как эти виды энергии не являются свойствами системы. Данная система при данных условиях обладает определенной внутренней энергией. Это предполагает, что каждый раз, когда система после ряда изменений возвращается в исходное состояние, внутренняя энергия становится точно такой же, как и прежде. Это предполагает также, что при других условиях мы будем иметь другое значение внутренней энергии. Последнее в общем является правильным, но не для всех случаев. Например, идеальный газ, как мы увидим ниже (стр. 150), имеет одинаковую внутреннюю энергию при данной температуре независимо от давления.
Математическая формулировка первого закона для статических систем. Для включения этих выводов уравнение (1) может быть представлено в виде
(2)*)
Изменение внутренней энергии системы AZ7 легко определяется уравнением (2). Для кругового процесса Д17= 0, и уравнение (2) переходит в уравнение (1). Для некругового процесса оно просто определяет разность между тепловыми эффектами и работами, обусловленными накоплением или истощением энергии системы в количествах, определяемых исходным и конечным ее состояниями.
Следует отметить, что мы имеем дело только с изменениями количества энергии, а не с абсолютным количеством ее, о котором мы в настоящее время знаем очень мало.
Уравнение (2) предполагает, что AZ7 измеряется в тех же единицах, что4 и A. AU можно также измерить в тепловых единицах, для чего эту величину следует умножить на J. Поскольку только что рассмотренные представления предполагают эквивалентность и полную взаимопревращаемость всех видов энергии, все члены уравнения (2) удобно выражать в одинаковых единицах и исключить J из всех термодинамических уравнений. Формулировка первого закона, данная уравнением (2), приложима только к статическим системам. Для динамической, т. е. для движущейся, системы вводятся еще два члена энергии, что будет показано подробнее ниже, в гл. VIII,
Справедливость первого закона. Для всех обычных явлений и, в частности, для явлений, встречающихся в инженерной практике,
й) в конечном состоянии и — U в исходном состоянии. Тепло,
подводимое к системе, и работа, произведенная системой, считаются положительными.
88
II. Первый и второй законы термодинамика
первый закон является строго справедливым в пределах точности наиболее совершенных измерений, которые в состоянии произвести современные исследователи.
Однако попутно интересно отметить, что для объяснения некоторых внутриатомных, а также звездных явлений необходимо отказаться от идеи сохранения энергии и постулировать возможность возникновения энергии из материи, и наоборот, — материи из энергии. Соотношение между ними дается теорией относительности в виде U=c2m, где с — скорость света. Так как с2 очень велико, то при разрушении ничтожных количеств материи возникают громадные количества энергии. Таким образом, 1 г любого вещества эквивалентен 25000 000 квтч., для получения которых существующими методами потребовалось бы сжечь 12 500 т угляБ.
В настоящее время никто не имеет определенного представления о том, как добывать и контролировать громадные источники энергии, которые заключены в материи согласно этой теории ☆ ).
Классификация энергии. Энергию иногда разделяют на различные виды, выражаемые терминами „потенциальная*, „кинетическая*, „механическая*, „химическая*, „электрическая*, „атомная* и т. д. Такая классификация приводит скорее к недоразумениям, чем к выяснению вопроса, но эти термины имеют всеобщее применение и удобны для некоторых целей, а поэтому заслуживают упоминания. Потенциальной энергией является энергия, которой обладает система вследствие своей особой конфигурации. Простейшим примером этого служит любой предмет, рассматриваемый с точки зрения его положения выше уровня поверхности земли. Для поднятия любого тела выше этого уровня нужно произвести работу, и про энергию, доставляемую такой работой, говорят, что она сохраняется в системе (тело -(- земля) как потенциальная энергия. Потенциальной она называется потому, что способна дать работу в том случае, если телу дать возможность • падать на землю. Подобно всякой энергии, она не имеет абсолютного значения и зависит от выбора нулевого уровня.
Потенциальная энергия данной системы также может быть „внутренней*, вследствие особой конфигурации молекул, атомов, электронов и rip., из которых она состоит. Между молекулами вещества имеются силы притяжения, и так как расстояние между ними изменяется при расширении газа или испарении жидкости, то в потенциальной энергии происходят изменения, аналогичные тем, которые получаются, если тело изменяет свое положение в поле земного тяготения.
☆ ) Недавнее исследование по делению изотопа урана с атомным весом 235, во время которого были установлены относительно очень большие количества освобождающейся энергии (в действительности очень малые из-за ничтожных количеств участвующих в процессе веществ), подало надежду, что, может быть, найдутся способы для вскрытия запасов атомной энергии.
Первый закон и понятие энергии
89
Недоразумения, к которым приводит опрометчивая, слишком буквальная интерпретация некоторых из этих терминов, можно показать на следующем примере. При изотермическом сжатии газа от рг до pt представление о потенциальной энергии может привести сначала к заключению, что газ при давлении р2 обладает большей энергией, чем при рр благодаря его способности совершать работу при расширении. Такой взгляд совершенно ошибочен, как будет показано ниже; в действительности правильно обратное: внутренняя энергия газа уменьшается с ростом давления при постоянной температуре.
Кинетической энергией называется энергия, связанная с движением тела или частицы относительно некоторого произвольно выбранного стандартного тела. Для простого случая движения с постоянной скоростью она определяется количественно хорошо известным уравнением К. = ^тиг, где т—масса и v — относительная скорость. Кинетическая энергия может быть связана как с движением больших масс, так и с движением частиц, например молекул или атомов. Вся энергия, не являющаяся кинетической, считается потенциальной.
Энергия, как потенциальная, так и кинетическая, связанная с относительно большими массами (по сравнению с первичными частицами, из которых состоит вещество), обычно относится к механической энергии. Энергию, связанную с движением или с относительным положением молекул, часто называют .термической" или .тепловой" энергией. Следует упомянуть, что этот удобный образный термин относится к любому роду накопленной энергии, которую можно передать системе или отнять от нее благодаря разности температур.
Энергия, связанная с э. д. с., называется электрической и получается благодаря движению сравнительно подвижных электронов, присутствующих в разной степени во всех веществах.
При химических реакциях потенциальная энергия накапливается или освобождается. Точная форма накопления этой энергии в деталях неизвестна; она бесспорно должна быть связана с конфигурацией атомов и электронов в молекуле. Поскольку эта энергия связана с химическим изменением, ее удобно называть химической энергией.
Атомная (точнее ядерная) энергия является формой потенциальной энергии, происхождение которой еще недостаточно изучено. Она может происходить от сил, действующих между электронами и протонами внутри ядра атома®, и отличается от сил, действующих между внешними электронами соседних атомов и приводящих к химическим реакциям. Как уже указывалось, она может происходить от действительных превращений материи в энергию. Каков бы ни был источник атомной энергии, количество ее исключительно велико, и все наши источники энергии являются по сравнению с ней карликовыми.
Мы не имеем средств для различения видов энергии, содержащихся в системе. Мы не можем сказать, сколько в ней содержится
90
II. Первый а второй законы термодинамики
химической, потенциальной Или кинетической энергии. Поэтому все виды внутренней энергии объединяются вместе в одну общую величину U.
Превращение энергии. Редко располагают энергией именно в таком виде, который является желательным для данной цели; обычно ее приходится превращать из одного вида в другой. Конечно, очень важно перевести как можно большее количество исходной энергии в желательную форму, и большая часть' деятельности инженера связана с факторами, влияющими на к. п. д. процессов превращения энергии. ’ Специальное поле деятельности инженера-химика включает превращения, в которых участвует так называемая .химическая энергия*. Во всех химических реакциях происходит или переход накопленной химической энергии в окружающую среду в виде теплоты (экзотермическая реакция) или обратный процесс (эндотермическая реакция). Во многих случаях переход не является простым превращением химической энергии в какой-нибудь один вид энергии, но происходит более сложный процесс, в котором могут участвовать также механическая и электрическая энергии. Например, при прохождении химической реакции в гальваническом элементе часть химической энергии может проявляться как тепло, отдаваемое окружающей среде, а другая часть, если вследствие реакции элемента происходит увеличение объема, проявляется как механическая работа, совершаемая против постоянного давления атмосферы; наконец, главная часть химической энергии будет проявляться как электрическая работа.
Прежде чем окончательно получить желательный вид энергии, может оказаться необходимым провести ее через ряд превращений. Для обозначения такого ряда изменений можно использовать термин .цепь превращений*. Рассмотрим, например, следующую цепь превращений. Химическая энергия топлива освобождается посредством химической реакции, известной как .горение*, и превращается в тепловую энергию продуктов горения. Эта энергия переходит в виде теплоты посредством теплопроводности, конвекции и лучеиспускания в энергию пара, которая, в свою очередь, превращается в механическую энергию (работу) в паровой машине или турбине. В этой стадии процесса можно иметь примерно 2О°/о исходной химической энергии больших масс, находящихся в движении. Остальные 8О°/о представляют .потери*. В действительности энергия не теряется, но просто находится не в той форме, которую хотят получить, и по причинам, которые будут ясны при рассмотрении второго закона термодинамики, она может быть .обесценена*, т. е. неспособна превращаться в какую-либо полезную форму. Большую часть этих 8О°/о представляет энергия, еще существующая в паре после использования его в двигателе и передаваемая в конденсационную систему. Для продолжения цепи механическую энергию можно превратить в электрическую с помощью генератора, а электрическую энергию — в химическую в аккумуляторной батарее. Таким образом, общим результатом будет переход
Второй закон термодинамики
91
от химической энергии в одной системе к химической энергии в другой системе, причем об:ций к. п. д. перехода не превышает 15°/0. Это показывает возможность тех или иных улучшений на некоторых стадиях такой цепи превращений; мы рассмотрим это подробнее в следующих главах.
Может показаться, что выбранный путь является очень неудачным и непрямым для перехода от химической энергии одной системы к той же самой форме энергии в другой системе. В практике возможно выбрать более короткий путь и перейти прямо от химической энергии топлива к электрической энергии; это можно осуществить и в действительности, хотя еще нет практически экономичного пути для осуществления этого в большом масштабе7.
Рассмотрим теперь эту цепь до конца в обоих направлениях. Каким образом попала в топливо химическая энергия, заключенная в нем? Это произошло вследствие превращения лучистой энергии солнца в химическую энергию роста растений посредством процессов, о которых в настоящее время известно еще мало. Эти процессы также являются крайне неэффективными, поскольку они касаются превращения общей лучистой энергии, падающей на данную площадь земной поверхности, в химическую энергию растения; при благоприятных условиях к. п. д. составляет около 3°/0 й). Теплота и давление, возможно, с помощью бактерий, вызывают химические реакции в веществе растения, которое в конце концов превращается в уголь.
Для продолжения цепи в другом направлении химическую энергию системы в аккумуляторной батарее можно вновь превратить в электрическую энергию, а последнюю, в свою очередь, — в лучистую, в лампе накаливания. Доля исходной химической энергии, окончательно переходящей в желаемый вид (свет), очень мала, не выше 2°/0. Наконец, лучистая энергия превращается в тепловую при температуре среды и в этой форме в пределах данной системы больше непригодна для дальнейших превращений.
Описанная схема типична для всех цепей превращения энергии. Первичным источником всей земной энергии является солнце, а конечной формой накопления ее является энергия веществ, находящихся на поверхности земли. Достигнув последней стадии, энергия полостью теряется для дальнейших превращений, поскольку больше не имеется движущей силы.
ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ
Ограничения первого закона. Термодинамические процессы можно рассматривать с двух точек зрения: 1) Какое изменение энергии связано с процессом? 2) Произойдет ли изменение самопроизвольно,
й) Конечно, главной причиной низкой производительности является то обстоятельство, что для фотосинтеза эффективна только относительно малая часть спектра солнечного излучения.
92
11. Первый и второй законы термоданамики
а если произойдет, то в какой мере? Следовательно, имея дело с такими величинами, как тепловой баланс, теплота реакции или растворения и работа изотермического сжатия, мы связаны с вопросами, относящимися к группе (1), При рассмотрении же химического равновесия, работы, получаемой за счет теплоты, или работы, требуемой для охлаждения, вводятся вопросы, относящиеся к группе (2).
Рассмотрим несколько подробнее случай химической реакции. Сколько выделится тепла и как сильно это количество будет изменяться при изменении давления и температуры системы, если реакция от данного исходного состояния реагирующих веществ до требуемого конечного состояния продуктов полностью проходит при данной температуре? Эти вопросы принадлежат к 1-й группе. Будет ли реакция проходить самопроизвольно от данного исходного состояния до требуемого конечного состояния? Какое состояние является рав-новесным состоянием, при котором не будет происходить никаких дальнейших изменений, и как действуют на него изменения температуры и давления? Если реакция не будет проходить самопроизвольно в желательном направлении и в желательной степени, то сколько энергии в форме работы следует приложить для ее осуществления? Это—вопросы второй группы. Ясно, что первый закон термодинамики применим для ответов на вопросы первой группы, но первый закон сам по себе не применим для решения вопросов второй группы, и тогда следует применять другой общий принцип — второй закон термодинамики.
Некоторые самопроизвольные или необратимые процессы уже были рассмотрены в гл. I. Первый закон имеет дело с количествами различных видов энергии, участвующих в этих процессах, но не связан с направлением изменения. В первом законе нет ничего, позволяющего отрицать возможность движения воды снизу вверх или переход тепла от тела с низкой температурой к более нагретому телу, или отрицать, что смешанные газы могут самопроизвольно разделяться. Второй закон имеет дело именно с этими вопросами, и он просто является формулировкой выводов из опытных наблюдений в отношении самопроизвольных процессов или тенденции к изменению.
Причины необратимости. Интересно хотя бы кратко рассмотреть основные причины необратимости изменений. Конечно, потери энергии при этом не происходит, но энергия переходит в обесцененную форму, неспособную к совершению работы. Пока энергия связара с направленными силами или потенциалами, например с давлением потока или э. д. с., или с разностью температур, она может действовать в определенном направлении и пригодна для превращения в работу, ио как только энергия превращается в форму хаотического движения молекул, она сразу становится непригодной для этого, так как шансы на то, чтобы беспорядочное движение могло снова стать достаточно ориентированным для получения направленной силы, исключительно малы.
Второй закон термодинамики
93
Это не означает, что все изменения энергии, связанные с переходом тепла, всегда непригодны для совершения работы. Если переход тепла соединяется с направленной силой, например с давлением газа или с э. д. с., то тепло может частично превращаться в работу. Но в отличие от натянутой пружины, поднятого груза или сжатого газа в самой теплоте не имеется никакой направленной силы, и когда под действием силы получается теплота, действие не может стать обратимым без помощи некоторого внешнего агента.
Короче говоря, основной причиной необратимости является наша неспособность оперировать с отдельными атомами и молекулами.
Для иллюстрации некоторых из этих выводов рассмотрим какой-нибудь конкретный случай, например падение воды. Если 1 кг воды свободно падает с высоты 427 м, то кинетическая энергия в месте падения превращается (если не учитывать трения воздуха) в одну килокалорию теплоты (первый закон), и эта энергия полностью находится в форме беспорядочного, т. е. ненаправленного, движения молекул воды. Этого количества тепловой энергии достаточно для поднятия 1 кг воды вновь до исходного уровня, и нет ничего несогласного с первым законом в предположении, что эту теплоту можно использовать для возвращения воды в исходное состояние, т. е. для обратного процесса. Единственным ограничением, налагаемым первым законом, является требование, чтобы вода во время этого процесса охладилась на 1°С. Трудность заключается в том, что обратный процесс требует направленной силы, а также в том, что шансы для ориентирования всего множества молекул с их беспорядочным движением в каком-либо данном направлении (что необходимо для получения направленной силы, способной обеспечить необходимую работу) так исключительно малы, что такой результат можно считать практически невозможным. Однако, строго говоря, это является только невероятным. Этот вопрос несколько ниже будет рассмотрен подробнее, так как он вскрывает основы второго закона.
Твердые тела на первый взгляд представляются* несколько противоречащими представлениям, что самопроизвольные процессы протекают в направлении от упорядоченного состояния к хаотическому. В случае твердых тел всегда имеется тенденция изменяться в противоположном направлении от беспорядочного состояния к состоянию идеального кристалла, представляющего идеальное упорядочение. Но в этом случае на первый план выступает вопрос о поверхностной энергии. Твердое тело стремится перейти в монокристалл вследствие того, что последний обладает наименьшей поверхностной энергией.
Вероятность и второй закон. Второй закон является утверждением необратимости самопроизвольных процессов. Основной причиной необратимости является переход системы от „упорядоченного* (направленного) движения к хаотическому (беспорядочному). Следовательно, происходит переход от менее вероятного состояния
94
II. Первый и второй законы термодинамики
к значительно более вероятному. Для пояснения этого рассмотрим простую аналогию с колодой карт, розданной четырем игрокам по 13 карт каждому. Всего имеется 635 000 000 000 различных возможных группировок или сдач. Наиболее упорядоченная группировка, при которой все 13 карт у каждого игрока будут одной масти, не является более невероятной, чем любое другое размещение, которое может случиться при тщательной перетасовке; но имеются только 4 такие возможные одномастные группировки и около 1,37 X Ю11 возможных группировок, когда у каждого игрока будет по 4 карты одной какой-нибудь масти, по 4 — другой, по 3 — третьей и по 2 — четвертой. Таким образом, шансы получения 13 карт одной масти из тщательно перетасованной колоды по сравнению с 4 — 4 — — 3 — 2 сдачей равны только 1 :(3,4-1О10). При переходе от перетасованных карт к беспорядочной смеси молекул можно для оценки вероятности того или другого состояния применить такой же подход, и он легко показывает, что шансы появления упорядоченных группировок, возникающих из беспорядочной смеси молекул, бесконечно малы.
Предположим, что мы имеем два сосуда, соединенных трубкой. В один поместим газообразный азот и в другой — кислород, а затем дадим обоим газам смешаться. В относительно короткий срок мы получаем, судя по анализу, совершенно равномерную смесь. Благодаря непрерывному движению молекул, перемешивающему газы, шансы возобновления первоначального распределения молекул по сосудам настолько ничтожны, что мы можем полностью исключить их из рассмотрения и сказать, что процесс этот является совершенно необратимым.
Точка зрения, согласно которой процесс является необратимым больше из-за его невероятности, чем из-за невозможности упорядочения хаотической системы, представляет скорее философский и теоретический интерес. Но прежде чем оставить рассмотрение этого предмета, упомянем об остроумной математической демонстрации, данной Лотка [159]. Система из 26 маятников с различными периодами колебаний у каждого приводится в движение в определенный момент времени. Система кажется беспорядочной, поскольку дело касается распределения маятников данных размеров по отношению к нулевой линии, т. е. к линии равновесия. Однако в действительности система в целом имеет определенный период, который можно вычислить, и наблюдатель, который мог бы ждать 7385 лет, нашел бы, что спустя это время будет воспроизведена исходная конфигурация.
Максвелл был одним из первых, заметивших, что причиной необратимости является только наша неспособность оперировать с отдельными молекулами. Он мысленно представил себе такое фантастическое существо (его стали называть позднее «демоном Максвелла*), которое при своих достаточно малых размерах могло
Второй закон термодинамики
95
бы иметь дело с отдельными молекулами и, следовательно, легко могло бы нарушать подчинение второму закону. Если такой демон контролировал бы открытие и закрытие очень маленького отверстия между двумя сосудами, содержащими газ при одинаковой температуре, то он мог бы вызвать разность температур путем рассортировки .горячих” и .холодных" молекул надлежащими манипуляциями $ задвижкой или мог бы разделить смесь двух газов. Некоторые бактерии при разделении рацемических смесей способны исполнять роль, приближающуюся к роли демона Максвелла.
Формулировки второго закона. Второй закон отрицает возможность обращения самопроизвольного процесса без внешнего воздействия. Клаузиус в 1851 г. дал следующую формулировку: .Невозможно для машины, действующей без помощи какого-либо внешнего агента, переносить тепло от данного тела к другому телу, обладающему более высокой температурой".
Эта формулировка на первый взгляд представляется более ограниченным положением, поскольку она касается только частного необратимого процесса, именно — перехода тепла. В действительности эта формулировка является совершенно общей.
В самом деле, как вывод из ранее рассмотренного ясно, что если бы один из необратимых процессов был обратим, то могли бы быть обратимы и другие. Мы не будем доказывать этого здесь, но для иллюстрации можно привести следующий пример.
Предположим, что газ изотермически расширяется без совершения какой-либо внешней работы из данного резервуара при данном давлении в другой резервуар с меньшим давлением. Ясно, что этот процесс является необратимым. Его можно провести вновь, но только при помощи внешнего воздействия, а тогда мы уже имеем дело с другой системой. Однако предположим, что имеется тепловой резервуар при температуре, равной температуре газа. Если мы не отрицаем возможности того, что тепло может передаваться от какого-нибудь данного тела к другому телу, имеющему более высокую температуру, без помощи внешнего воздействия, тогда можно представить, что имеется некоторый род механизма, допускающий получение разности температур в нашем тепловом резервуаре. Как результат этой разности температур некоторое количество тепла в источнике можно использовать для получения работы посредством тепловой машины, и эту работу можно употребить на возвращение расширенного газа, посредством изотермического сжатия, к его исходному давлению. Работа сжатия проявляется как теплота, возвращающаяся в резервуар. Общим результатом будет обращение необратимого расширения газа без каких-нибудь изменений во внешней среде.
Если, однако, вместе с Клаузиусом отрицать переход тепла от тела с более низкой температурой к телу с более высокой температурой, то вся эта схема рушится, и расширение газа должно быть в этих
96
Il. Первый и второй законы термодинамики
условиях необратимым. Вследствие того, что подобным образом можно связать любые два необратимых процесса, отрицание возможности возобновления любого из них будет автоматически распространяться и на все прочие процессы, и поэтому формулировка Клаузиуса является совершенно общей.
Кельвин независимо от Клаузиуса высказал следующее положение: „Невозможно посредством неодушевленного материального деятеля получить от какой-либо массы вещества механическую работу путем охлаждения ее ниже температуры самого холодного из окружающих предметовИ эта формулировка встретила возражения. Например, если газ при температуре воздуха подвергается давлению в термически изолированном цилиндре, а затем ему дают расшириться, преодолевая внешнюю силу, то будет производиться работа, и газ охладится значительно ниже температуры среды. Однако такой процесс не может беспрерывно давать работу; любая попытка сделать «го непрерывным означает, что газ должен быть возвращен к своему исходному состоянию (т. е. процесс должен быть круговым), а это -будет требовать затраты определенного количества работы, по меньшей мере равного работе, получаемой при расширении.
Планк дал формулировку, являющуюся видоизменением формулировки Кельвина: „Невозможно построить периодически действующую машину, все действие которой сводилось бы к поднятию некоторого груза и охлаждению теплового источника". Это просто означает, что энергия, содержащаяся в данном тепловом источнике, не может быть использована для получения полезного действия, если только в нашем распоряжении нет другого источника с более низкой температурой. Если бы было возможно обратное, то любой пароход, поезд или автомобиль могли бы двигаться без топлива, так как окружающая вода или атмосфера доставляла бы почти неистощимые количества энергии. Машину, реализующую такую возможность, иногда называют „вечным двигателем второго рода". Формулировки Кельвина и Планка свидетельствуют о невозможности •осуществить вечный двигатель этого рода. Любую из этих формулировок можно связать с формулировкой Клаузиуса; это видно из того, что если работу можно было бы непрерывно получать из теплового источника, то теплоту можно было бы передавать от более низкой к более высокой температуре без охлаждения какого-либо внешнего агента. Также ясно, что любой необратимый процесс в этом случае можно было бы повторять без необходимости остаточных изменений в какой-либо другой системе.
Для отдельных явлений можно дать много специальных формулировок второго закона. Одна из наиболее важных вводит в рассмотрение количества работы, которое можно получить от данного количества тепла, переходящего к рабочему телу машины. Это приводит к рассмотрению принципа очень большой практической важности и очень интересного с исторической точки зрения, именно — принципа Карно;
Второй закон термодинамика
91
Принцип Карно. Этот принцип, провозглашенный Карно в 1824 г., служит основанием для последующего развития второго закона if является фундаментом, на котором построена теория тепловых машин. Его можно формулировать следующим образом: «Максимально возможный к. п. д. тепловой машины, работающей между двумя температурными уровнями, является функцией только этих двух температур и никак не зависит от применяемого механизма и от рабочего тела”. Рабочим телом является просто газ, подобный воздуху, водяному пару или аммиаку, который проходит через цикл изменений в машине. К. п. д. тепловой машины равен произведенной работе, деленной на подведенное тепло. Наипростейшей из мыслимых тепловых машин является такая, в которой вся получаемая газом теплота берется из одного источника с постоянной температурой tA и вся отдаваемая теплота затем передается в один теплоприемник, поддерживаемый при постоянной более низкой температуре /2. Обратимое проведение такого цикла было подробно описано в гл. I. Этот цикл известен под названием «цикла Карно”, так как Карно вывел свой принцип из рассмотрения подобного цикла. В настоящее время цикл Карно представляет главным образом исторический интерес. Для анализа работы тепловых и холодильных машин, компрессоров и т. д. использованы другие идеальные циклы, более близкие к реальным, чем цикл Карно. Все же он сохраняет свое значение для предварительного анализа и как введение к более сложным циклам. Так как он очень прост, то основанные на нем следствия оказываются более понятными и не затемненными усложнениями практического характера.
Доказательство принципа Карно. Доказательство, этого принципа, данное самим Карно, основано на теории теплорода, полностью ниспровергнутой работами Джоуля. Клаузиус и Кельвин, работая независимо друг от друга, поняли-, что доказательство Карно перестало быть действительным, но что его принцип согласуется с опытом и без сомнения истинен. Они дали новые доказательства, согласующиеся с новым представлением о теплоте как о форме энергии (см. прим.4). Аксиомами, положенными ими в основу доказательства, были уже цитированные формулировки второго закона. Ниже будет дано краткое описание доказательства принципа Карно, предложенного Кельвином.
Пусть имеются два источника тепла при постоянных температурах и t2 (рис. 6). Эти температуры можно измерить с помощью любой шкалы; для данной цели необходимо только, чтобы они были постоянны и отличались друг от друга. Пусть t2. Рассмотрим тепловую машину I, которая действует между этими двумя температурами по простейшему возможному циклу, а именно — по циклу Карно. Допустим, что она берет из источника количество тепла Q] при и отдает Q2 в источник при t2. Рассмотрение операций этого цикла, данное в предыдущей главе, показывает, что необходима отдача тепла при 7 Химическая термодинамика
98
II. Первый и второй законы термодинамики
более низкой температуре. Здесь не нужно никаких ограничений, относящихся к природе механизма или рабочего тела, использованных в машине. Теперь, согласно первому закону, произведенная работа А{ будет равна Q, — Q2. Если машина работает в идеальных условиях, т. е. рассмотренным ранее обратимым способом, то эта работа будет максимально возможной при выбранном цикле и выбранном рабочем теле. (Следует напомнить при этом, что способ предполагает неко-
торые практически неосуществимые условия, допуская, например, применение поршня без трения, не проводящих тепла материалов
и других термодинамических аксессуаров, необходимых для избежания необратимых эффектов.) Рассмотрим теперь другую машину II, работающую по какому угодно 4д циклу и использующую любое рабочее тело, но берущую тепло QJ при той же температуре и отдающую тепло при температуре 1^. Выберем такое количество
Рис. 6. Схема к доказательству принципа рабочего тела, чтобы Ql Карно. было равно Q'. Теперь
предположим, что машина II более эффективна, чем машина I, т. е. что при работе оиа дает бдльшую работу, чем машина I, с тем же количеством тепла. Выразим это уравнением
Ац —Ai —Д; (3)
так как по первому закону Ai=q;—<z2 и Ai q2,
Qr1 — Q’2 = Q1~Qlt+^ (4)
Теперь, так как машина I обратима, то ее можно заставить работать в обратном направлении при приложении работы Aj, причем машина будет отнимать тепло Q2 из источника при и передавать Q, тепловому источнику при tv Пусть машина II, действующая как тепловая машина, приводит в движение машину I, действующую как тепловой насос. Так как Q1 = Qi, то
(5)
Результатом этих процессов является только получение работы за счет поглощения эквивалентного количества тепла из источника
Второй закон термодинамики
99
ври t2. В этом нет никакого противоречия с первым законом. Однако это противоречит формулировке второго закона, данной Кельвином или, вернее, Планком и специально отрицающей возможность осуществления такого процесса. Следовательно, если мы принимаем эту аксиому, то машина II не может быть более эффективна, чем машина I, так же как и никакая другая машина, работающая между теми же самыми температурными пределами.
Подобным же образом ни одна машина, если она обратима, не может быть менее эффективна, чем I, что может быть доказано следующим рассуждением. Предположим, что машина II будет обратимой, но менее эффективной, чем I. Пусть I действует как тепловой насос и приводит в движение машину II и пусть
А =Ап, (6)
т. е.
Qt-Qt=Q[- Q'T (7).
Но если к. п. д. II меньше, чем к. п. д. I, то
Qi — Qt
При сочетании с уравнением (7) видно, что
(9) Следовательно,
Q'2>Q2. (10)
Таким образом, единственным результатом соединения машин является переход тепла от тела при t2 к телу при flt а это именно и невозможно согласно постулату Клаузиуса.
Следовательно, все обратимые машины, работающие между данными температурными уровнями, имеют одинаковую производительность и эта производительность максимальна и зависит только от температур.
Это доказательство по своему характеру настолько обще и абстрактно, что не может удовлетворить никого, кто привык оперировать более конкретными представлениями. Другим же это может показаться настолько очевидным, что и не требует доказательств. В сущности доказательство принципа каким-либо подобным фор маль-ным образом не является уже необходимостью, поскольку из всей массы экспериментальных фактов и заключений, построенных на нем, не найдется ни одного, оказывающегося в противоречии с этим принципом. Тем не менее доказательство представляет большой интерес, так как этот принцип, доказанный таким простым способом, имеет универсальное значение и лежит в основе многих областей науки 7*
100 И. Первый и второй законы термодинамики
и техники. Хотя этот принцип чрезвычайно прост по своей формулировке, однако не так легко охватить все его применения, и нужен большой опыт в применении его к различным проблемам, прежде чем оценить его поразительную разносторонность.
Следует всегда помнить, что все заключения о переходе тепла в работу относятся к круговому процессу. Заключения эти не действительны для части цикла и верны только для цикла в целом. Так, при изотермическом расширении газа практически все получаемое тепло превращается в работу; если же процесс этот сопровождается адиабатным расширением, то мы заключаем, что производится значительно большее количество работы, чем было взято тепла. Компенсация происходит при возвращении рабочего тела в исходное состояние. Это требует выполнения работы, и когда цикл будет завершен, то общее количество произведенной работы окажется значительно меньше, чем количество, эквивалентное затраченному теплу.
ГЛАВА Ш
КОЛИЧЕСТВЕННОЕ ВЫРАЖЕНИЕ ДВУХ ОСНОВНЫХ ЗАКОНОВ. ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ
В предыдущей главе идеи и факты, лежащие в основе обоих законов термодинамики, были изложены и развиты с качественной стороны. Для применения этих принципов к решению конкретных числовых задач им следует дать точное количественное выражение. Это удобнее всего сделать с помощью некоторых функций или свойств, уже определенных ранее и являющихся общеупотребительными. К сожалению, при этом возникают некоторые недоразумения вследствие того, что символы, применяемые для обозначения этих функций, не унифицированы8.
Материал этой главы будет относиться главным образом к простым статическим, т. е. не меняющимся во времени, системам, состоящим только из одной фазы и одного компонента или из смеси постоянного состава. Общие принципы, конечно, можно приложить и к более сложным системам. Однако желательно сначала иллюстрировать эти принципы на простейших случаях, а затем уже перейти к более сложным.
ОБЩИЕ ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ
Функции удобно разделить на две группы: 1) функции, связанные только с первым законом, и 2) функции, связанные с обоими законами.
Первой из функций, относящихся к группе 1, является внутренняя энергия U, которая была уже до некоторой степени рассмотрена. Другой функцией группы I является энтальпия Н, или .теплосодержание”, как ее иногда называют. Эту величину не следует смешивать с количеством превращенного тепла, которое изображается символом Q. Н является определенным свойством системы, тогда как Q им не является. Для уменьшения возможности недоразумений многие авторы называют Н „энтальпией* вместо старого термина „теплосодержание*, и мы будем придерживаться этого названия.
Функциями, связанными со вторым законом, являются термодинамическая, или абсолютная, температура Т, энтропия S, свободная энергия Р (называемая также „максимальной работой*, хотя она в действительности может быть минимальной из всех рассматриваемых работ) и изобарный потенциал Z. Абсолютная температура впервые
102
Ill. Количественное выражение двух основных законов
определена Кельвином в 1848 г., а энтропия введена Клаузиусом в 1851 г. Функция F названа Гельмгольцем .свободной энергией” и до сих пор называется так большинством европейских авторов. Функция Z впервые определена и. использована Виллардом Гиббсом в 1875 г., хотя несколькими годами ранее Массье определил родственную функцию. Она была названа Гиббсом .термодинамическим потенциалом” и обозначена буквой С. Существуют и некоторые другие функции, несколько менее основного характера, оказавшиеся полезными при решении некоторых проблем; ссылки на иих будут сделаны ниже.
Все упомянутые функции, за исключением абсолютной температуры, являются экстенсивными свойствами, т. е. зависят от массы системы. Следует вообще усвоить, что символ для каждой функции относится или к единице массы, или к одному молю ее, причем чаще к последнему. При необходимости представить общее значение одного из этих свойств для системы в целом будет использован тот же самый символ, но напечатанный жирным шрифтом.
Остальная часть этой главы посвящена выводу соотношений между этими функциями и обычными параметрами состояния, которые необходимы для числовых расчетов.
ВНУТРЕННЯЯ ЭНЕРГИЯ
В предыдущей главе было показано, что определение внутренней энергии, которая является свойством любой системы, представляет сущность первого закона термодинамики. Уравнение (2) можно написать в диференциальной форме следующим образом:
dU = dQ — dA, (1)
где dU — полный диференциал, так как он является свойством системы, a dQ и dA обозначают просто бесконечно малые количества теплоты и работы, но не являются производными какой-либо функции. Это означает, что при рассмотрении конечного изменения от состояния 1 к состоянию 2 можно написать
2 2
J du = и2 — и. = J (dQ — dA). (2)
Другими словами, dU можно интегрировать сразу, без учета природы процесса, поскольку U является свойством, и его изменения зависят только от исходного и конечного состояний, ио не от пути процесса. Второго же интегрирования нельзя произвести, пока мы не будем точно знать пути, по которому происходит изменение, и, таким образом, пока не сможем выразить dQ и dA через определенные свойства. Другими словами, как Q, так и А зависят от пути перехода, а не определяются только исходным и конечным состояниями.
Энтальпия
103
Мы будем иметь дело только с разностями U, так как об абсолютном значении этой функции ничего неизвестно. Для удобства U принимают равной нулю при некотором произвольном стандартном состоянии, но чаще принимают Н=0, а затем из этого условия определяют U. В соответствии с уравнением (19, гл. I) можно написать
(3) ахду дудх ’ '
где х и у — две независимые переменные, определяющие состояние системы. Как это, так н подобные ему уравнения для других функций полезны при некоторых выводах, что будет показано ниже.
В последующих разделах книги окажутся полезными некоторые специальные формы уравнения (1). Например, если единственной силой, действующей на систему, является давление среды, то уравнение (1) можно написать в виде ☆)
dU =dQ — pdv. (4)
Если, кроме того, изменение происходит при постоявном объеме, то dU=dQ = Cvdt,
где Cv — изохорная теплоемкость.
Внутренняя энергия является экстенсивным свойством, т. е. она строго пропорциональна массе системы. Кроме того, внутренняя энергия системы аддитивна, т. е. она равна сумме внутренних энергий отдельных частей ее; однако следует заметить, что при смешении двух систем может произойти взаимодействие с окружающей средой, приводящее к изменению энергии, так что внутренняя энергия смеси не является суммой внутренних энергий компонентов, за исключением того случая, когда система изолирована от внешней среды.
ЭНТАЛЬПИЯ
Определение. Энтальпия представляет сложную функцию, образованную из других, более простых, и определяемую уравнением ☆&)
H = U-±pv. (€)
Эта функция введена потому, что при многочисленных практических приложениях часто производится суммирование внутренней энергии U
☆ ) Важно отметить, что использование уравнения dA = pdv,
где р относится к давлению самой системы, предполагает обратимый процесс. Если же существует какая-либо степень необратимости, то dA < pd».
it £?) Заметим, что U и pv следует привести к одинаковым единицам перед тем, как производить суммирование.
104
III. Количественное выражение двух основных законов
и произведения давления и объема pv, и поэтому удобно рассматривать эту сумму как целое. Хотя в общем изменение Н не идентично Q — количеству передаваемого тепла, но в некоторых специальных случаях обе величины делаются равными друг другу, и в этом заключается одна из главных причин использования этой функции. По определению эта функция является свойством, и поэтому изменение ее значения определяется только исходным и конечным состояниями системы.
При некоторых процессах, например при изотермическом расширении идеального газа, наблюдается передача значительного количества тепла, но нет изменения в Н. Наоборот, при адиабатном дросселировании неидеального газа и Q и Д/7 равны нулю. При обратимом адиабатном расширении газа Q равно нулю, а Н уменьшается. Это отмечается здесь для того, чтобы еще раз подчеркнуть, что теплота Q и энтальпия Н являются двумя совершенно различными величинами.
Подобно U, Н является экстенсивным свойством и, очевидно, не имеет определенного абсолютного значения. Мы будем иметь дело только с изменениями энтальпии Д2/, и для удобства для некоторого стандартного состояния будет принято произвольное значение Н.
Процесс при постоянном давлении. Диференцируя уравнение (6), можно получить
ан=аи-]-ра^-]-т1р. (7)
Для процесса при постоянном давлении (изобарный процесс) уравнение (7) переходит в следующее:
dH—dU-\-pdv. (8)
Из сравнения с уравнением (4) и из определения Ср ясно, что
dH=dQ — Cpdt (9)
для любого изобарного процесса, в котором единственной переменной является температура. В интегральной форме уравнение (9) приобретает следующий вид:
с
&H—Q—\Cpdi. (10)
t.
Это уравнение чрезвычайно важно с точки зрения практического применения. В словесной формулировке оно выражает, что изменение функции Н является мерой теплового эффекта при постоянном давлении. Многие важные технологические процессы происходят при постоянном давлении, и для нахождения количества теплоты, выделяемой или поглощаемой при таком процессе, необходимо только
Энтальпия
105
вычесть значение энтальпии в начале процесса (Ht) из его значения в конце процесса (У2). Это является основой всех тепловых диаграмм и таблиц и значительно упрощает вычисление тепловых эффектов для всех систем, для которых значения Н сведены в таблицу или нанесены на график.
Пример 1. Сколько тепла следует сообщить 1 кг воды при температуре 164,2°С и давлении 7,0 кг/см2 для превращения ее в перегретый пар при 260°С и при том же самом давлении?
По таблицам Вукаловича [284] (табл. III, стр. 44) находим, что Н для перегретого пара при 260°С и давлении 7,0 кг/см2 равно 710,0 ккал/кг.
Н для воды при 164,2°С и 7,0 кг/сж2= 165,6 ккал/кг (там же, табл. II, стр. 23). Следовательно, Q = ДУ =710,0—165,6 = 544,4 ккал/кг.
Следует указать, что соотношение (10) действительно только для изобарных процессов. Однако из этого не следует заключать, что функция Н полезна только для изменений при постоянном давлении. Позднее мы будем иметь случай применить ее к очень важным процессам, отличающимся от изобарных.
Соотношение между ДУ и ДУ. Из уравнения (5) следует, что тепловые эффекты при постоянном объеме равны изменению внутрен-ней энергии, т. е. что теплота, выделенная или поглощенная в процессе, протекающем при постоянном объеме, равна разности U, так « же как тепловой эффект при постоянном давлении равен разности Н. )
Если известны давление и объем в начальной и конечной точках превращения, то можно легко получить ДУ из ДУ или ДУ из ДУ по уравнению
ДУ=ДУ4-Д(рт). (11)
Рассмотрим, например, испарение жидкости. Если жидкость испарялась при постоянном давлении в калориметре, то необходимое для этого тепло определяется ДУ, т. е. разностью между энтальпией пара и энтальпией жидкости. ДУ можно вычислить по уравнению (11), если известны удельные объемы жидкости и пара. Очевидно, *
,что во всех случаях, когда вещество находится в конденсированном, *
т. е. жидком или твердом состоянии, Д(/то) будет небольшим, и разность между ДУ и ДУ будет невелика, а во многих случаях даже ничтожно мала.
Пример 2. Жидкий пентан при температуре, соответствующей температуре его кипения 77,8°С под давлением 3,40 атм, имеет удельный объем 0,00177 м2/кг и энтальпию 42,6 ккал!кг. Насыщенный пар при том же давлении имеет объем 0,10437 м'Чкг и Н— 118,4. Чему равно изменение энергии, сопровождающее процесс парообразования 1 кг пентана?
По уравнению (11) —Д(ро)= 118,4— 42,6 = 75,8 ккал;
д (/то) = р (v2 — Vj) = 3,40-1,033.104 (0,10437—0,00177)=
3604 „ .
= 3604 кгм = Qgj- = 8,4 ккал,
поэтому ДУ = 75,8 — 8,4 = 67,4 ккал
106
III. Количественное выражение двух основных законов
АБСОЛЮТНАЯ, ИЛИ ТЕРМОДИНАМИЧЕСКАЯ, ТЕМПЕРАТУРА
Основные положения. Желательность установления абсолютной температурной шкалы, не зависящей от свойств какого-нибудь отдельного вещества, уже была указана в гл. I. Температурная шкала, основанная на идеальном газе, является по самому своему смыслу абсолютной шкалой, но чисто гипотетической, поскольку идеальный газ не обладает практической реальностью. Идеального газа не существует, но, конечно, многие газы в некоторых областях температуры и давления очень мало от него отличаются по свойствам. При очень низких температурах нет веществ, которые хотя бы даже приближенно отражали поведение, свойственное идеальному газу.
Кельвин в 1848 г. нашел, что принцип Карно дает надежную основу для определения абсолютной шкалы температур, так как он утверждает, что к. п. д. машины является функцией только температур теплоотдатчика и теплоприем-ника н совершенно не зависит от природы используемого рабочего тела. Другими словами, машину Карно можно причем при данных температурах теп-
РиС. 7. Схема абсолютной
к определению температуры.
как термометр,
(12)
использовать
лоотдатчика и теплоприемника она будет всегда показывать одинаковые отсчеты независимо от того, будет ли она наполнена ртутью, спиртом, каким-нибудь газом или другим веществом.
Определение функции 7*. Принцип Карно математически формулируется следующим образом:
к.п.д. =Л = 21=-2?= ср
где Qj — количество теплоты, поглощенной обратимой тепловой машиной из теплоотдатчика, находящегося при постоянной температуре 1г;
Q2 — тепло, отданное в теплоприемник, находящийся при температуре U2;
— некоторая неизвестная функция двух температур, зависящая от способа определения температурной шкалы.
Так как (Q1 — QS)IQ1 = 1можно также написать
= (13)
Пользуясь схемой рис. 7, допустим, что имеются тепловые источники при трех постоянных температурах t2 и t3 и три тепловые
Абсолютная, или термодинамическая, температура , 107
машины, работающие по циклу Карно с данным рабочим тело^, причем:
машина I берет тепло Qj при tx и отдает тепло С?2 при /2;
машина II берет тепло Q2 (тепло, отданное I) и отдает Q3 при /3;
машина III берет Q, при и отдает тепло при ts.
Теперь, так как все обратимые машины, работающие между равными температурными пределами, имеют одинаковый к. п. д., то машины I и II, работающие совместно, имеют тот же самый к. п. д., что и машина III, и поэтому должны отдавать то же самое количество теплоты при ta, а именно Qs. Если бы это не соответствовало истине, то был бы нарушен второй закон.
Для трех машин можно написать выражения, аналогичные уравнению (13):
21 = (р1(,1Л), (14)
^ = <рг(/2Л3), (15)
^ = ?3(^Л3). (16)
Деля уравнение (16) на уравнение (15) и сопоставляя с уравнением
(14), получаем
Это может быть верно только при условии, что 'М'-.У-К- о»)
Подобные выражения правильны для других двух функций. Точный вид этих функций неизвестен и будет зависеть от способа определения шкалы температур. Определим теперь новую шкалу темпера-
тур уравнением
21—11
— Тг'
(19)
где Т1 = ф1(/1) и Т2 = ф2(<2). Это определяет шкалу температур таким образом, что отношение любых двух температур равно отношению теплот, полученных и отданных обратимой тепловой машиной, действующей между двумя этими температурами. Выбор этой специальной формы функции, как отметил Кельвин, произволен. Он мог выбрать ет £г), или 1g Т, или любую другую форму. Причина выбора,
й) В сущности абсолютная шкала, предложенная Кельвином, первоначально была основана на функции этого вида, но никогда не была общеупотребительной. Она имеет некоторые преимущества перед обычной шкалой Кельвина при работе в областях, близких к абсолютному нулю обычной шкалы.
108
III. Количественное выражение двух основных законов
сделанного им, вполне очевидна, так как такой выбор дает шкалу, совпадающую со шкалой идеального газа, и, следовательно, реальные газы в большинстве случаев будут давать с ней хорошее совпадение.
Для доказательства идентичности термодинамической шкалы Кельвина со шкалой идеального газа надо просто оперировать циклом Карно, используя в качестве рабочего тела идеальный газ. Температура 0 по шкале идеального газа определяется соотношением
pv = Z?0, и легко показать, что 21 — Q2 ~ в2 ’
где Qj и Q2 — теплота, взятая и отданная в машине Карно, работающей на идеальном газе между источниками, температуры которых равны 0t и 02 по шкале идеального газа.
Температурные интервалы. Из уравнения (19) для машины I
01-0»Т\-Г2
01 Л
(20)
Если другая машина (машина II на рис. 7) воспринимает Q2 (тепло, отданное первой машиной) и отдает Qs другой машине при Т8, то
Qi — Qs —7*3
02 Т’з
(21)
Деля уравнение (21) на уравнение (20) и помня, что QilQ1 = T2/T1, получаем
01—02 __А — 7"2_Aj
02-0з ~Т2-Т3 — Аа-
В словесной формулировке это означает, что любые два интервала на этой абсолютной шкале относятся друг к другу, как относятся между собой количества работы, произведенной обратимыми машинами, действующими в соответствующем температурном интервале.
Длина градуса совершенно произвольна. Если мы имеем 100 машин Карно и первая из них берет тепло из источника, поддерживаемого при нормальной температуре кипения воды, а последняя отдает тепло резервуару, поддерживаемому при температуре замерзания воды, причем каждая берет тепло, отданное предыдущей машиной, работающей при более высокой температуре, и если все эти машины производят одинаковое количество работы, тогда уравнением (22) можно определить ТОО равных температурных интервалов между двумя конечными точками. Каждый интервал представляет градус абсолютной шкалы или шкалы Цельсия. Подобным образом можно определить 80 разных интервалов между теми же двумя точками, и каждый из них будет представлять абсолютный градус Реомюра.
Абсолютная, ила термодинамическая, температура
№9
. Абсолютный нуль. Так как то ясно, что Тг = 0,
если Q2 = 0. Это означает, что нуль абсолютной шкалы является температурой теплового источника, которому обратимая тепловая машина не отдает никакого тепла, а все имеющееся в наличии тепло полностью превращается в работу ☆). Это дает реальный физический смысл понятию абсолютного нуля, которым оно не обладает, будучи определено только на основании поведения идеального газа. Исходя из второго закона термодинамики и изменения определенных свойств с температурой, возможно определить местонахождение абсолютного нуля где-то между 273,1 и 273,2°С ниже точки замерзания воды. Его значение фиксируется измерениями, произведенными при температурах, далеко отстоящих от абсолютного нуля. Если, например, измерить при изотермическом расширении газа значения отношения pf/Pi'»! и нанести эти значения на диаграмму, выражающую зависимость их от р, а затем экстраполировать их до р = 0, то мы получим значения Povo/Pivi- Пусть это значение для данного газа при /1 будет кР Пусть соответствующее значение для t2 и для того же самого первоначального значения pxvx будет равно kj. Поскольку произведение povQ представляет предельную величину для р = О, когда газ является идеальным, то
1г 02 Тг h ®i Л.
Измерения, произведенные для большого числа различных газов при ^==0°С и f2=100°C, дают для этого отношения среднее значение 1,36607. Из соотношений
7’2/7’i = 1,36607 и Тг — Т1=100 получаем
Тг(1 = 0 на стоградусной шкале) = 273,16°К-
Точное значение все еще не установлено. Мы произвольно выберем значение —273,2°С. Почти для всех технических расчетов эти значения можно округлить до —273°С.
Интересно отметить, что за последнее время в научных кругах наблюдается значительная активность, направленная к получению и использованию температур ниже 1°К. Эти низкие температуры особенно интересны в связи с третьим законом термодинамики. Такие низкие температуры, как 1,5°К, можно получить методами, по принципу подобными применяемым в обычной холодильной практике, с гелием в качестве рабочего вещества. Ниже 1,5°К используется совершенно другой метод — адиабатное размагничивание, впервые предложенное Джиоком [84]. Этим путем была получена даже такая низкая температура, как 0,005°К. Для дальнейших деталей по теории
й) Переход более 1ОО°/о теплоты в работу свидетельствовал бы, конечно, об отклонениях от первого закона.
110
III. Количественное выражение двух основных законов
и технике этого интересного метода рекомендуется обзорная статья Джиока [85].
Связь абсолютной шкалы со шкалами, основанными на свойствах веществ. Относительная простота формы термодинамических уравнений происходит от способа, которым Кельвин определил абсолютную температуру. При использовании температурной шкалы, основанной иа свойствах каких-либо конкретных веществ, все эти уравнения были бы гораздо сложнее. Во всех обычных термодинамических формулах всегда применяется абсолютная (термодинамическая) температура и никакая иная. В этбм смысле любая термодинамическая формула или уравнение могут считаться определением абсолютной температуры. Это, однако, ие означает, что термодинамические формулы являются исключительно предметом определения без реальной фактической освовы, так как вполне достоверно, что абсолютную температуру можно определить, а это и есть сущность второго закона термодинамики, из которого выведены многие точные соотношения и ни одно из них не оказалось противоречащим практике.
Температуры следует измерять на основе изменения некоторых свойств с температурой, а затем эти температуры следует определенным образом связать со шкалой Кельвина. Например, требуется определить функцию уравнения
Т=<Р(О- (23)
Это можно сделать путем использования любого общего термодинамического уравнения, связывающего Т с другими свойствами. Для дальнейших деталей см. [33, 2б6].
ЭНТРОПИЯ
Общая формулировка. Эта функция введена физиком Клаузиусом в 1851 г. Вследствие абстрактного характера ее она всегда служила пугалом для начинающих изучение термодинамики; написано много томов в попытках объяснить ее физическое значение и придать ей более конкретный смысл. В данной книге мы будем рассматривать энтропию прежде всего как математическую функцию, дающую простейший путь для количественных приложений второго закона. Сначала рассмотрим классический вывод Клаузиуса, данный почти с той же точки зрения. Более поздние работы Больцмана, Планка, Льюиса и других обнаружили связь энтропии с вероятностью и придали ей ббльшую физическую определенность. Эта трактовка будет рассмотрена очень кратко; но автор уверен, что для целей прикладной термодинамики достаточно считать энтропию удобной математической функцией, а не делать упор на ее физическую интерпретацию. Инженер, который интересуется главным образом переходом тепла в полезную работу, может найти удобным рассматривать энтропию как меру той части превращенной энергии, которая является „беспо
Энтропия
111
—^-Кислород
при taiTM и 20 С
Воздух яри 45 отм и 20°С
—«- Азот при !атм и 20°С
Рис. 8. Термодинамически невозможный процесс.
лезной* для работы, т. е. не может быть превращена в работу. Эта точка зрения также будет рассмотрена.
Из предыдущей главы известно, что второй закон представляет собой просто обобщенную формулировку опыта относительно самопроизвольных процессов или стремления к изменению. Для превращения этой формулировки в количественную требуется некоторая функция, которая во время самопроизвольного (необратимого) процесса будет всегда изменяться определенным образом и поэтому будет характеризовать такое изменение. Функция U определена для того, чтобы придать количественный характер первому закону, и представляет основную функцию первого закона, между тем как Н выведена из нее просто как удобная функция. При самопроизвольном процессе U не изменяется каким-либо характерным обра
зом и поэтому не имеет основного значения при установлении второго закона. Основной функцией при развитии второго закона является энтропия (в соединении, конечно, с понятием абсолютной температуры, от которой завнснт ее определение), и наиболее целесообразно рассматривать ее как математическую величину, значительно упрощающую количественное развитие и приложение второго закона.
Проиллюстрируем кратко эти представления более конкретным примером. Рис. 8 представляет результат воображаемого процесса разделения газа. Поскольку дело касается термодинамической возможности процесса, мы не нуждаемся в рассмотрении того, что происходит внутри камеры, но мы должны рассмотреть все переходы энергии и вещества между камерой и окружающей ее средой. Если допустить, что все газы идеальны, то предполагаемый процесс не включает никаких изменений внутренней энергии U, и с точки зрения первого закона нет причин для невозможности этого процесса. Однако с помощью энтропии можно легко показать, что рассматриваемый процесс невозможен, так как его протекание связано с уменьшением энтропии системы без соответствующего увеличения энтропии окружающей среды.
Количественное определение. Мы уже показали, что принцип Карно в соединении с определением абсолютной температуры (Кельвина) приводит к простому уравнению
или
Оз___Оз
Гз — Тг'
21—2?
Л А
= 0.
(24)
(25)
112
III. Количественное выражение двух основных законов
Считая тепло, взятое от теплоотдатчика (Qi), положительным, а тепло, отданное теплоприемнику (Q2), отрицательным, можно переписать уравнение (25) следующим образом:
Е^ = 0. (26)
Эти соотношения пригодны только для обратимого цикла Карно, в котором тепло поглощается машиной при одной температуре и отдается при некоторой более низкой температуре, причем обе температуры постоянны на всем протяжении процесса. Это пред-
Рис. 9. Схема к определению энтропии.
ставляет упрощенный случай, редко встречающийся на практике, и желательно обобщить эти результаты для приложения к более сложным циклам, где непрерывно изменяется как температура, при которой поглощается тепло, так и температура, при которой оно отдается.
Рассмотрим любой обратимый цикл изменений, через который может проходить рабочее вещество тепловой машины. Условия обратимости уже рассмотрены и не нуждаются в повторении. Пусть состояние рабочего тела, проходящего через цикл изменений, будет определяться двумя независимыми переменными и изображаться на плоскости pv, как это указано на рис. 9а. Пусть весь цикл на рис. 9а будет разбит на 4 простых цикла Карно двумя адиабатами и двумя изотермами, как это указано на рис. 96. Суммируя площадь четырех простых циклов, будем только приближаться к площади исходного цикла, причем разности между ними (частью положительные, частью отрицательные) указываются заштрихованными площадями. Обратимая машина, работающая по циклу I (ABHG), берет тепло Qj при (вдоль изотермы АВ) и отдает Qj при 71 (вдоль GH).
Энтропия
ИЗ
Подобным образом машина, работающая по циклу II, берет Qn при Тп и т. д. для остальных циклов. Для каждого из этих циклов можно
написать уравнения, подобные уравнению (25): Qi Qi q
Л Qn га
(27)
§- = о ^1
(28)
и т. д.
Таким образом, машина, работающая по ступенчатому циклу ABCDEF..., совершает работу, представленную суммой четырех площадей, образованных изотермами и адиабатами; поэтому работа цикла равна замкнутой площади на /го-диаграмме. Кроме того, поскольку машина работает иа тех же самых изотермах, что и отдельные машины, то и теплота, взятая и отданная этой машиной, должна быть равна алгебраической сумме соответствующих теплот для отдельных машин. Таким образом, можно считать, что машина, работающая по чатому циклу, эквивалентна четырем отдельным машинам; для нее можно написать
Qi Qn
V
Qi Qu
= 0
ii
или сокращенно
о,
ступен-поэтому
(29)
(30)
охватывая суммированием весь ступенчатый цикл. При проведении более близких друг к другу адиабат и при использовании большого числа ступеней бесспорно будут оставаться действительными те же самые соотношения, а заштрихованная площадь будет становиться меньше. Если перейти теперь к пределу и взять адиабаты настолько близкими друг к другу, чтобы изотермы отличались только на величину dT, то заштрихованная площадь сделается исчезающе малой, а ступенчатый процесс совпадет с циклом, показанным на рис. 9а. Согласно хорошо известному методу вычисления, имеем в пределе
(31)
С точки зрения прикладной термодинамики это является исключительно важным уравнением. В словесной формулировке оно может быть выражено так: для любого цикла, связанного с переходом теплоты и совершением работы, как бы он ин был сложен, если он осуществляется вполне обратимым способом, алгебраическая сумма всех тепловых эффектов, деленная на соответствующие абсолютные температуры, при которых происходят переходы тепла, равна нулю. 8 Химическая термодинамика
114
III. Количественное выражение двух основных законов
Из рассмотрения функций и свойств, изложенного в гл. I, должно быть ясно, что dQfT представляет изменение свойства, т. е. полный диференциал. В круговом процессе изменение величины, элементарное приращение которой равно dQ/Т, согласно уравнению (31), равно нулю; это просто означает, что эта величина имеет определенное значение, характеризующее данное состояние системы, и поэтому является ее свойством. Это свойство, диференциал которого равен dQ/T, названо Клаузиусом энтропией; оно будет изображаться буквой S. Таким образом,
dS=^r (32)
м 2
45=^ — ^ = ]^. (33)
1
Уравнения (32) и (33) можно считать количественными определениями энтропии £).
Энтропия является экстенсивным свойством, и поэтому она пропорциональна массе, но следует отметить, что это не означает, что энтропия раствора представляет сумму энтропий компонентов, поскольку, как мы увидим ниже (стр. 167), должно возникать изменение энтропии при смешении.
Изменение энтропии в необратимом процессе. Следует подчеркнуть, что указанные определения приложимы только к полностью обратимым изменениям. Для необратимого процесса изменение энтропии Д5 не равно dQ/Т. С первого взгляда это представляется серьезным ограничением полезности функции, потому что в действительности мы интересуемся необратимыми процессами, так как все реаль-шае процессы необратимы, но по существу здесь нет никаких ограничений. Так как энтропия представляет свойство, то ее изменения совершенно не зависят от действительного пути, по которому происходит процесс, а зависят только от исходных и конечных состояний системы. Следовательно, обратимые и необратимые процессы между одинаковыми исходными и конечными состояниями отвечают одному и тому же изменению энтропии.
Вычисление изменений энтропии. Иллюстрируем числовыми примерами некоторые из изложенных выводов, относящихся к энтропии.
Пример 3. Чему будет равно изменение энтропии, если 1 кг-моль идеального газа при 20°С и давлении в 10 атм расширяется через дроссель до давления в 1 атм, причем во время процесса эти давления поддерживаются постоянными посредством соответствующих поршней. В процессе не
☆) Может быть, следует подчеркнуть то обстоятельство, что тогда как dQ/Г является полным диференциалом, dQ не является им. Нет свойства Q, но есть свойство, производная которого будет dQ)T.
Энтропия
115
совершается никакой внешней работы, кроме той, которая производится против поршня постоянным давлением.
Рассматриваемый процесс, очевидно, необратим, так как газ нельзя возвратить к его исходному давлению, не вызывая изменения во внешней среде. При расширении, подобном рассматриваемому, газ не будет испытывать никаких общих температурных изменений. Местные температурные изменения будут происходить в результате превращения внутренней энергии в кинетическую энергию расширяющегося газа, но при рассеивании последней общим результатом будет изотермичиость процесса.
Вычислим теперь изменение энтропии, сопровождающее этот процесс. Для этого нужно сначала придумать обратимый процесс между данными состояниями. Это можно осуществить, заключая газ в цилиндр и медленно сжимая его поршнем, свободным от трения, поддерживая температуру газа постоянной и отводя теплоту сжатия (путем соответствующего охлаждения) сразу же при ее образовании. Условия обратимости такого процесса уже были детально рассмотрены в гл. I. Для вычисления AS используется уравнение (33), и поэтому нужно сначала определить тепловой эффект сжатия. Как будет показано ниже, для изотермического сжатия идеального газа изменение U равно нулю и поэтому, согласно первому закону, A = Q, т. е. работа сжатия равна теплоте сжатия, если обе величины выражены в одинаковых единицах. Таким образом,
dA=pdv=dQ,
и так как для 1 моля идеального газа pv = RT, то
dQ = RT — = — RT~ » Р
(так как Т = const),
2 ps
откуда С НО. — — R С = — R In Р? = AS.
J Т J р Pi
1 Pi
Подставляя числовые значения, получаем
AS = — 1,987 X 2,303 X 1g Ю = — 4,57.
AS будет выражено, очевидно, в тех же единицах, что и R; и так как R выражаем в ккал на кг-моль на градус или, что одно и то же, в кал/моль-град, то энтропия будет выражена в тех же единицах ☆). Уменьшение энтропии газа на 4,57 единицы явилось результатом обратимого сжатия, и поэтому необратимое расширение приведет к увеличению энтропии газа на ту же самую величину.
Пример 4. Чему будет равно изменение энтропии, если 250 ккал тепла передаются посредством теплопроводности от теплоотдатчикй, обладающего постоянной температурой 100°С, к теплоприемнику с постоянной температурой 0°С.
Этот необратимый тепловой поток можно было бы передать обратимым способом, если бы тепло переходило при элементарных температурных разностях от источника с температурой 100°С к рабочему веществу в машине Карно. Машина будет отдавать меньшне количества теплоты при более низкой температуре источника. Уменьшение энтропии теплоотдатчика равно QiJT\ — 250/(100 -)- 273,2) = 0,670 ккал/град. Из уравнения (24) сразу
£?) Единицам энтропии не присвоено никакого названия; единица энтропии обычно обозначается символом э. е. (энтропийная единица). Предлагалось назвать ее „клаузиус*. 8*
116
Ill. Количественное выражение двух основных законов
видно, что увеличение энтропии теплоприемника численно равно уменьшению энтропии теплоотдатчика, или, другими словами, общее изменение S для всего процесса равно нулю. Для обратимого процесса характерно, что все, что претерпевает тепловые изменении, входит в систему. С другой стороны, если бы переход теплоты был необратим, то теплоприемник получил бы 250 ккал при 0°С, и так как действительный переход к источнику может считаться происходящим при бесконечно малой температурной разности и поэтому может рассматриваться как обратимый, то увеличение энтропии равно 250/273,2 = 0,915 ккал!град. Другими словами, общий процесс теплопередачи приведет к увеличению энтропии на 0,915 — 0,670 = = 0,245 ккал/град.
Можно и другим путем рассмотреть этот необратимый процесс. Пусть 250 ккал теплоты, переходящей к теплоприемнику с температурой 0°С, возвращается обратно с помощью обратимого теплового насоса Карно. Это дает в обоих резервуарах изменение энтропии, равное 0,915, но теплота, отдаваемая источнику с более высокой температурой, будет равна 250 ккал плюс произведенная извне работа. Следовательно,
<?1 = <?2 у- = 250 = 341,5 ккал
Таким образом, в этом источнике накапливается излишек тепла в 91,5 ккал как общий результат всего процесса, состоящего из необратимого самопроизвольного перехода теплоты и обратимого возвращения ее тепловым насосом. Это соответствует возрастанию энтропии на 91,5/373,2 = 0,245 ккал/град.
Пример 5. Чему будет равно изменение энтропии, если 1 кг сухого воздуха при постоянном давлении, равном 1 атм, нагревается от 20 до 60°С?
Так как мы интересуемся только изменением энтропии самого воздуха, а не внешней среды, которая доставляет тепло, то мы можем представить фактический переход тепла к воздуху как происходящий таким образом, что температура внешней среды отличается от температуры воздуха все время на величину dT. Следовательно, поскольку дело касается только воздуха, процесс нагревания будет обратимым. Поэтому
2 AS = f ^2.
J Г 1
Другими словами, изменение энтропии какой-нибудь системы при любом процессе нагревания или охлаждения ее определяется выражением, отвечающим изменению энтропии в обратимом процессе, если температуру относить к рассматриваемой системе, а не к окружающей среде.
Для изобарного процесса
dQ = ipdT
и Л
Г,
Интегрирование легко осуществить, если ср можно выразить как функцию абсолютной температуры. Для данной цели принимаем, что ср — постоянная величина, равная 0,24 ккал/кг-град. Тогда
AS = 0,24 In £ = 0,24X2,303 ]g ^9 + = 0,0307 ккал/кг-град.
Пример 6. Чему будет равно изменение энтропии, если 4 кг жидкой воды при 260°С адиабатно смешиваются с 12 кг воды при 20°С?
Энтропия
117
Процесс необратим, так как он не дает никакой работы, которую можно было бы использовать для приведения системы в исходное состояние. Тем не менее, охлаждение горячей воды само по себе может считаться обратимым процессом; то же самое справедливо и для нагревания холодной воды (см. гл. I). Следовательно, общее изменение энтропии определяется уравнением £?)
/4-273,2 /4-273,2
Д5 = 4 | ^-^4-12 J ср^.
2604-273,2 204 273,2
Считаем ср постоянной и равной единице. Из теплового баланса получаем 4 (260 — t) = 12 (t — 20), откуда t ~ 80°С.
Используя данные по энтальпии из таблиц Вукаловича, находим t 82,5°С. Решая уравнение, находим
Д8 = 4 X 2,303 1g |||| + 12 X 2,303 lg = 0,60
(по таблицам AS = 0,63).
Пример 7. Чему будет равно изменение энтропии для химической реакции, протекающей при постоянной температуре?
Рассмотрим простую реакцию
Нй(ж)+у С1г(г) = Н§С1(т).
Если эта реакция проводится при постоянном давлении в калориметре с по стоянной температурой 25°С, то выделяется тепло в количестве 31580 кал1моль образующегося HgCl, т. е. Д// = —31580. Так как рассматриваемый процесс необратим, то изменение энтропии нельзя получить делением величины выделяющегося тепла на температуру, при которой происходит процесс. Для вычисления Д8 необходимо рассмотреть обратимый путь проведения реакции. Как уже было указано в гл. 1, химическая реакция может быть обратимой, если ее осуществить в гальваническом элементе так, чтобы была произведена максимально возможная электрическая работа. После конструирования такого элемента, в котором может итти только одна вышеуказанная реакция, будет обнаружено, что выделяющееся при 25°С тепло равно только 6450 кал/моло образующегося HgCl. Эта величина является теплотой обратимой реакции, и поэтому уменьшение энтропии равно 6450/298,2 = 21,63 э. е. В действительности измеряется не выделяемое тепло, а э. д. с. элемента, и из последней величины, пользуясь первым законом, можно вычислить теплоту обратимой реакции по уравнению
bU = Q — А.
При обратимом процессе работа состоит из двух частей: 1) из электрической работы Ае и 2) из работы, произведенной постоянным давлением окружающей среды />До. Поэтому
bU=Q — (AE+pbv)
или
Q = bH+AE.
☆) Вода при 260°С должна находиться под повышенным давлением, но влиянием давления на ее энтропию можно пренебречь.
i 118 III. Количественное выражение двух основных законов
Э. д. с. элемента, в котором происходит эта реакция при 25°С, равна 1,0894 вольта. Поэтому АЕ — 1,0894 X 23066 й) = 25130 кал/моль и Q = —31580-|-25130 = —6450 (выделяющаяся теплота). Можно отметить, что в некоторых случаях теплота обратимой реакции по знаку противоположна , обычной калориметрической теплоте реакции.
Энтропия и обесцененная энергия. Мы видели, что когда теплота с помощью тепловой машины превращается в работу, то превращение никогда не бывает полным. Определенная доля тепла отдается теплоприемнику, и эту долю следует считать бесполезной для получения работы. Чем ниже температура теплоприемника, тем меньше будет эта обесцененная доля; но практически температурой теплоприемника будет температура окружающей атмосферы или большой массы воды; и та и другая в сущности тоже представляют теплоприемники бесконечных размеров. Нецелесообразно отдавать теплоту какому-нибудь телу конечных размеров при более низкой температуре, потому что эту более низкую температуру нужно поддерживать, и для этого необходимо затрачивать работу, по меньшей мере равную той, которая была бы выиграна при использовании более низкой температуры.
В соответствии с уравнением (24) обесцененная теплота Qo = , =Qj (To/Tj), где Qj—общее количество теплоты, полученное маши-
ной, а Г, и То соответственно абсолютные температуры теплоотдат-7 чика н теплоприемника. Если теплота, отобранная от вещества во
I , время изменения его состояния, такова, что температура Тг может /* * непрерывно изменяться (это относится к теплоте, переходящей от конечного теплового источника к бесконечно большому), то тогда это уравнение можно обобщить, именно:
2
<м>
1 или 2
Qo — П J* у == (35)
i
= 7'0AS, (36)
2
так как dQjT — изменение энтропии вещества, отдающего теплоту
1
во время процесса. Мы можем формулировать результат, выраженный уравнением (36), следующим образом. Прн любом переходе определенного количества теплоты к веществу или от вещества (при обратимом изменении его состояния) доля этого количества теплоты, бесполезная в отношении превращения в работу в круговом процессе, равна изменению энтропии, сопровождающему данное изменение
й) Число калорий на вольт-эквивалент.
Энтропия
119
состояния, умноженному на наииизшую доступную абсолютную температуру, при которой могут быть отданы большие количества теплоты. Поэтому имеется прямая пропорциональность между изменением энтропии в таком процессе и количеством обесцененной энергии. По уравнению (36) легко вычислить обесцененную теплоту для любого данного изменения состояния, если только известны свойства участвующего в процессе вещества.
Понятие об обесцененной энергии полезно также и при необратимых процессах. При таких процессах говорят, что некоторое количество энергии стало „бесполезным* для работы. Примем, что мерой бесполезного изменения энергии в процессе служит минимальное количество работы, необходимое для возвращения всей системы в состояние, существовавшее до необратимого изменения. Пусть эта работа равна До. Так как система возвращается к своему исходному состоянию, то Д£/=0, и поэтому работа Ао должна равняться теплоте Qo в тепловом источнике. Единственный источник, которому мы можем продолжать отдавать любое количество теплоты без заметного изменения его температуры, это — атмосфера или большие массы воды при температуре То. Между прочим, если бы эта теплота отдавалась источнику при какой-либо температуре более высокой, чем наинизшая температура Т9 окружающей среды, то можно было бы получать большее количество работы, а для минимальной работы получается отдача тепла при То. Возрастание энтропии этого источника равно AS=Q0/T0, что также равно AS системы, поскольку обратный процесс считается обратимым. Следовательно, возвращенная работа равна Qo=TohS. Таким образом, мы видим, что понятие обесцененной энергии приводит к тем же самым математическим соотношениям, как и полученные выше.
Обесцененную энергию можно определить как произведение изменения энтропии на абсолютную температуру Тв теплового источника при иаинизшей естественной температуре окружающей среды. В приложении к изменению состояния, включающему переход теплоты, это означает, что доля теплоты, которую следует отдать тепловому источнику при То, равна теплоте, которую следует превратить в работу в цикле тепловой машины. При приложении к тепловому процессу это будет означать наименьшее количество работы, необходимое для возвращения системы в состояние, в котором она существовала до необратимого процесса.
Несколько числовых примеров помогут уяснению некоторых из этих представлений.
Пример 8. 1кг воды нагревается при постоянном давлении 15 кг/см-от 20°С до температуры кипения и затем полностью испаряется при тем же давлении. Какая доля передаваемого тепла бесполезна для превращения в работу в машине, если наинизшая температура, при которой может быть отдано тепло, равна 10°С?
Значения для свойств пара заимствуем из таблиц Вукаловича.
Температура кипения при данном давлении = 197,4°С.
120 *111. Количественное выражение двух основных законов
- - ч “ - . -
Скрытая теплота парообразования = 466 ккал!кг.
т,
Общее количество передаваемого тепла =^ cpdT-\-L,
где L — скрытая теплота парообразования.
Эту сумму можно найти непосредственно из таблиц для пара по разностям энтальпий, принимая, что энтальпия жидкости не зависит от давления.
Тогда Н2 (энтальпия в конечном состоянии) — 666,6.
//1 = 20,4.
Д//(передаваемая теплота) = 646,1.
Изменение энтропии =
Эту сумму можно найти непосредственно из таблиц водяного пара как разность энтропий
S2=l,5406 и $! = 0,0707;
AS=S2 —$!= 1,4699.
Обесцененная энергия= 7'0AS = (10 + 273,2) • 1,4699 = 416,3 ккал, т. е. (416,3/646,1) X 100 = 64,4% теплоты, переходящей к воде, бесполезно для превращения в работу.
Пример 9. Предположим, что теплота для испарения воды в примере 8 передается от топки при температуре 1200° С. Это отвечает уменьшению энтропии на 646,1/(1200-J-273,2) = 0,439 ккал/кг.
Увеличение энтропии воды было равно 1,470, и поэтому общее возрастание энтропии, обусловленное необратимым переходом тепла, равно 1,470 — 0,439=1,031.
Увеличение бесполезной энергии, сопровождающее этот переход по отношению к тепловому источнику при 10° С, равно
7’0Д$=283,2 X 1,031 = 292,8 ккал.
Это количество энергии можно рассматривать 1) как разность между работой, получаемой от обратимой машины, Действующей между теплоот-датчиком при 1200° С и теплоприемником при 10° С, и работой, передаваемой от тепловой машины, получающей теплоту от воды, когда она изменяет свое состояние от состояния насыщенного пара при 15 кг/см2 до состояния воды при 20° С, и отдающей тепло при 10° С, или 2) как наименьшее количество работы, необходимое для возвращения системы в исходное состояние, причем изменения допускаются только вне системы, а отдача теплоты только при 10° С.
Увеличение количества бесполезной энергии как результат любого необратимого процесса, рассмотренного на стр. 71 и 72, получается сразу умножением вычисленного изменения энтропии на принятую температуру То теплового источника. В случае примера 3 (стр. 114) AS = 4,57, и для температуры теплового источника в 20° С энергия, становящаяся бесполезной, равна 293,2 X 4,51 = 1340 ккал/хг-моль. Если расширение совершалось при 90° С вместо 20° С, то бесполез
Энтропия
121
ная энергия, согласно принятому нами определению, была бы точно такой же. Тем не менее количество работы, получаемое при обратимом расширении, было для этих двух случаев различно. Таким образом, из уравнений в примере 3 очевидно, что
Л = ЯЛп
Pi у
и поэтому потерянная работа пропорциональна абсолютной температуре, при которой происходит изотермическое расширение.
Были попытки определить изменение энтропии как отношение бесполезной энергии, обусловленной процессом, к абсолютной температуре теплового источника при иаинизшей температуре окружающей-среды. Это представляется малооправданным, так как не существует вполне определенного и однозначного определения обесцененной энергии, приложимого ко всем процессам различного вида, с которыми мы будем встречаться. С другой стороны, энтропия является более основным понятием и может быть определена очень простым соотношением. Поэтому целесообразнее определять увеличение обесцененной энергии на основе энтропии, чем пытаться проводить рассуждение в обратном направлении.
Пригодность. Рассмотрим 1 кг любого вещества в данном состоянии и предположим, что вещество действует как теплоотдатчик, дающий теплоту обратимой тепловой машине. Машина принимает теплоту при переменной температуре вещества на данной стадии, но отдает теплоту всегда при наинизшей достижимой температуре То. Максимальная работа, получаемая за счет тепла, доставляемого таким источником, дается выражением, основанным на принципе Карно [см. уравнение (20)] ☆):
4 = (37>
или, переходя к диференциалам, поскольку при данной температуре воспринимается только бесконечно малое количество теплоты,
. A = —^T-~^-dQ= (38>
X
Д к
= —^ dQ-\-T0^~ = (39)
т, т,
= _[Q-7’0AS]£. (40)
☆ ) Q относятся к теплоте, отдаваемой веществом, и поэтому оно отрицательно.
122
III. Количественное выражение двух основных законов
Член T^S представляет собой то, что мы назвали бесполезной энергией. Интерпретация этого уравнения очень проста. Для рассматриваемого изменения общая теплота перехода Q состоит из двух частей, одна из которых — Т0Д5— бесполезна для работы, а другая в идеально работающей машине может полностью превращаться в полезную ра-боту.
Если изменение состояния вещества является изобарным, то уравнение (40) переходит в
л = (41)
В этом частном случае работа является функцией исходного состояния вещества (для данного источника с температурой То) и поэтому аналогична свойству, которому было дано наименование «пригодность*1 л которое изображается символом В. В последнее время оно получило распространение в некоторых термодинамических анализах. Для любого перехода вещества из состояния 1 в состояние 2 изменение пригодности дается уравнением
—АВ=[ЛЯ—(42)
Кинен [127] дал для статических процессов несколько более общее «определение функции В. Он считает пригодность максимально полезной работой, которую могло бы произвести вещество в данном состоянии, действуя не только как источник тепла для обратимой тепловой машины Карно, но также и производя ту работу, которая является результатом его собственных рт-изменений. Если и давление и температура вещества снижаются до температуры и давления окружающей среды, то для работы не существует более никакой движущей силы. Максимально возможная работа, получающаяся от этих двух эффектов между исходным состоянием ргТг и состоянием, отвечающим p0TQ, равна
о о
Д = —+ (43)
1 1
Но из этой суммы следует отнять величину р^о, которая является просто работой против атмосферного давления и не может считаться полез-•ной работой. Поэтому нетто-работа, или пригодность, дается уравнениями
о о
В= — J Т—тГ-° dQ-]-§p dv — p^v, 1 i
о о
о
§ pdv — PqAv, i
(44)
(45)
I
Энтропия
123
о
В = — Q4-T0(Sa — 5j) + §pdv — p0(ve — (46)
1
По первому закону
о
=UQ — U1 = Q — J pdv.
i
Поэтому
B = -(U0 -U,) + Ta (Sg - Sy) —Pg (Vg ~ Vy ) , (47)
В = (Ц +P0^ - T^) - (Ug+PgVg - TgSg). (48)
Последняя величина при постоянных р0 и Та, очевидно, является функцией состояния 1 и поэтому — свойством системы.
Изменение пригодности при переходе из состояния 1 в состояние 2 дается уравнением
ДВ = (U2 +рЛ - T0S2) - (Uy +PgVl - TgSJ. (49)
Эти два определения функции пригодности иногда порождают недоразумения; в действительности пригодность применяется главным образом к системам, изменяющимся непрерывно, и для таких систем пригодность, определяемая Киненом по уравнению (48), приводится к более простой, определяемой уравнением (41), если можно пренебречь действием кинетической энергии и потенциальной энергии силы тяжести.
Пример 10. Пусть предполагаемый солнечный двигатель поглощает солнечное излучение оргавической жидкостью, температура которой будет увеличиваться от 100 до 150° С при постоянном давлении. Чему равна максимальная доля поглощенной лучистой энергии, соответствующая пригодности по отношению к окружающей среде с температурой 15° С?
Общая энергия, поглощенная иа 1 кг жидкости, равна ЬН = ср(Т2— Ту).
Пригодность = Mi — T0AS = ср |\тг — Ту) — Г0 In pj .
„ , То , Тг , 288,2 423,2
Степень пригодности = 1— =—~ 1п =1----In = 0>276.
12 “““ * 1 *1 3v
Кроме количественного приложения, понятие пригодной энергии очень удобно для описания некоторых эффектов. Некоторые виды энергии, например кинетическая энергия движущейся массы, полностью пригодны для совершения работы в идеальном механизме, но в реальном процессе имеются потери, приводящие к тому, что получается только часть (скажем, 7О°/о) возможной работы. ЗО°/о не представляют потерю энергии, так как, согласно первому закону, энергия не может быть потеряна. Тем не менее потеря, в смысле второго закона, имеется, и она является именно потерей' пригодной энергии.
Изменение энтропии в изолированных системах. Практически не могут существовать полностью изолированные системы; но представ
124
III. Количественное выражение двух основных законов
ление о такой системе, подобно представлению об обратимом процессе, является полезным орудием анализа. Мы определим изолированную систему как систему, включающую все тела, в которых происходят какие-либо изменения, связанные с данным рассматриваемым изменением. Строго говоря, это понятие включает всю вселенную, однако для практических целей можно произвольно выбрать гораздо более ограниченную систему. Конкретная иллюстрация сделает это значительно более ясным. Предположим, что химическая реакция происходит в замкнутом сосуде постоянного объема, помещенном в термостат. Если последний хорошо изолирован от среды, так что происходит лишь незначительный теплообмен, то система из реакционного сосуда и термостата очень близко подходит к представлению об изолированной системе.
Материал, образующий химически реагирующую систему, сам по себе будет рассматриваться как „отдельная система" в отличие от изолированной системы, которая включает также и термостат. Если реакция происходит в гальваническом элементе и дает э. д. с., то всю электрическую систему, внешнюю по отношению к реакционному сосуду, следует включать в Изолированную систему. Фабричный корпус с, полностью закрытыми дверями и окнами, в котором машины приводятся в действие электрической энергией, получаемой от аккумуляторной батареи, был бы изолированной системой, если ие считать небольшого обмена теплотой и светом с окружающей средой.
Истинно изолированной была бы система, вокруг которой можно было бы создать оболочку, непроницаемую ни для материала, ни для энергии. Конечно, такая система не могла бы подвергаться наблюдению вследствие очень сильного воздействия наблюдателя, нарушающего изоляцию. Тем не менее представление об изолированной системе оказалось полезным в термодинамических рассуждениях и, как мы увидим ниже (стр. 130), привело к широко применяемым результатам. Наши так называемые „изолированные системы" будут содержать в общем три составляющие: 1) „отдельные системы", в которых происходят изменения состояния; 2) тепловые источники, достаточно большие для того, чтобы в них не происходило никаких изменений состояния, и 3) механизм, производящий работу.
Если рассматриваемая отдельная система является рабочим телом тепловой машины, работающей по циклу Карно, то изолированная система должна включать как тепловую машину, действующую по циклу Карио, так и механизм, производящий работу. Не требуется детально рассматривать природу этого механизма, который для простоты будем рассматривать как поднимающий груз и как свободный от трения. Это не будет связано ни с какими изменениями энтропии. Изменения энтропии будут происходить в рабочем теле и в двух тепловых источниках. Если рабочее тело и источник находятся всегда при одинаковой температуре и обмениваются теплотой, то увеличение энтропии рабо
Энтропия
125
чего тела будет в точности компенсироваться изменением энтропии источника, и наоборот. Если машина сама по себе обратима, то не будет происходить никаких изменений энтропии вследствие какой-либо направленной силы, и энтропия совершенно не будет изменяться при условии отсутствия трения (или другого сопротивления). Таким образом, в любой изолированной системе, в которой функционирует обратимая машина Карно, суммарная энтропия системы в целом остается постоянной.
Этот результат легко можно обобщить для более сложного цикла. Ясно, чтоД5=0 для рабочего тела „отдельной системы", так как оно
проходит через цикл изменений. При каждое увеличение 5 рабочего тела будет точно компенсироваться уменьшением 5 теплового источника, и наоборот. Это означает, что рабочее тело по температуре никогда не отличается больше, чем на бесконечно малую величину от лю- § бого источника, с которым оно .5 обменивается теплотой, и, таким g образом, для них обоих ин-теграл ^dQjT численно ограничен одинаковыми пределами, но с противоположным знаком. Это
условии обратимости процесса
Объём
Рис. 10. Необратимость в цикле тепловой машины.
заключение ие должно ограничиваться случаем полного замкнутого цикла изменений. Оно действительно также и для лю-
бого изменения, не возвращаю-
щего систему к исходному состоянию. Следовательно, в любой
изолированной системе, в которой происходят только обратимые процессы, энтропия остается постоянной. Теперь рассмотрим случай, когда происходит некоторый необратимый процесс. Для облегчения представления рассмотрим простой цикл Карно, подразумевая, что те же самые принципы приложимы и к более сложному случаю. Пусть на рис. 10 ABCD представляет цикл изменений, через которые проходит рабочее тело. Оно получает теплоту Qj вдоль изотермы 7\ от теплоот-
датчнка при постоянной температуре Г,. Подобным же образом рабочее тело отдает тепло Q2 вдоль изотермы DC при Т’2, ио тепло передается
к теплоприемнику при Т2. Два тепловых потока при конечной разности температур дают необратимые эффекты. Необратимость такого процесса в целом графически доказывается тем, что максимально возможная работа, представленная площадью ABCD, меньше, чем максимально возможная работа, которую можно было бы произвести, т. е. меньше
.126
III. Количественное выражение двух основных законов
площади А’В'С'1У. -Тепловой источник при Т{ претерпевает уменьшение энтропии, равное QilTlt а тепловой источник при Т2— увеличение, равное Q2/T2. Так как рабочее тело само проходит полный цикл, то его AS должно быть равным нулю, т. е.
Qi_ 0г = 0
Т'2
Так как и Т'2~^>Т2, то —(Q1/7’1)-f-(Q2/7’2)^>0, и общим ре-
зультатом всего процесса в изолированной системе (поднятие груза не связано с каким-либо изменением энтропии) является увеличение энтропии Л).
Вместо необратимого теплового потока мы могли бы рассмотреть трение в машине. Подобный ход рассуждений привел бы к тому же результату, т. е. к общему увеличению энтропии.
Можно было бы привести и другие примеры, включающие различные типы необратимых изменений, например, расширение газа без совершения полезной работы, смешение двух газов, химическая реакция без совершения электрической работы, испарение перегретой жидкости, растворение твердого тела в жидкости, но нет необходимости далее разбирать специальные случаи, так как наши рассуждения можно обобщить для приложения к любому необратимому процессу. Из рассмотрения необратимых процессов в гл. 1 очевидно, что где бы ни происходил такой процесс, «отдельная система*, участвующая в нем, может быть полностью возвращена к своему исходному состоянию только при использовании работы, получаемой от некоторой внешней системы.
Теперь представим некоторую изолированную систему, в «отдельных системах* которой происходит один или несколько процессов с некоторыми, однако, слабыми признаками необратимости. При этом может производиться различная работа, но примем, что в конечном результате производится чисто механическая работа поднятия груза. Так же будут происходить и переходы теплоты между „отдельными системами* и тепловыми источниками, причем все совершается внутри изолированной системы. Если грузу дать упасть, то системы можно частично возвратить к их исходным состояниям, но не целиком, так как имела место некоторая необратимость.
Предположим далее, что в общей среде, составляющей часть изолированной системы, имеется поднятый груз, который теперь можно опустить для получения работы, необходимой для возвращения всех «отдельных систем* в их исходные состояния. Для всех систем, кроме
☆) Перегородки, разделяющие тепловые источники и рабочие тела, должны претерпевать некоторые изменения энтропии, особенно поскольку они или нагреваются, или охлаждаются, но в идеальном случае мы считаем, что этими изменениями можно пренебречь. Последнее равноценно допущению, что эти перегородки имеют ничтожно малую теплоемкость.
Энтропия
127
тепловых источников, возвращающихся к своим исходным состояниям, Д£/=0 и Л5 = 0; таким образом, произведенная работа будет полностью переходить в теплоту, что связано с увеличением энтропии. Следовательно, общим результатом необратимого процесса в изолированной системе будет увеличение энтропии системы в целом. ______
Иногда приводят формулировку, согласно которой энтропия всегда возрастает при любом необратимом процессе. Это является слишком произвольным утверждением, вернее, просто неправильным. Многие химические реакции проходят необратимо с уменьшением энтропии, поскольку дело касается самой реагирующей системы, но всегда имеется более чем компенсирующее возрастание энтропии среды, так что для изолированной системы (химически реагирующая система плюс среда) общим результатом всегда является увеличение энтропии.
Общая математическая формулировка второго закона. Клаузиус кратко изложил все это посредством своей известной формулировки: „Энтропия мира стремится к максимуму* &).
Мы можем суммировать все наши рассуждения об энтропии следующей краткой математической формулировкой:
dS 0 для изолированных систем. (50)?
В изолированной системе общая энтропия остается неизменной для любого бесконечно малого обратимого изменения; но для любого процесса, включающего необратимые изменения, общая энтропия изолированной системы возрастает.
Уравнение (50) составляет сущность всех наших рассуж дений о втором законе термодинамики и является наиболее общей из всех возможных крлйчественных формулировок его. Оно также дает общий критерий равновесия и направления самопроизвольного' изменения.
й) Распространение принципа возрастания энтропии на вселенную привело бы к выводу о необходимости тепловой смерти ее, так как при достижении максимума энтропии во всех частях вселенной установилась бы одинаковая температура, и движущая сила процессов исчезла бы. Такой вывод является совершенно неверным.
Ключом к опровержению концепции тепловой смерти является статистическое толкование второго начала, доказывающее неосновательность распространения принципа возрастания энтропии на вселенную. Подобно тому, как нельзя прилагать этот принцип к микросистемам, где процессы, протекающие с уменьшением энтропии (флюктуации), представляют существенное значение и где при наличии немногих частиц само понятие энтропии лишается своего физического содержания, так я применение его к процессам космического масштаба лишено всякого основания.
Распространения свойств систем, доступных нашему наблюдению, на вселенную является столь значительной экстраполяцией, что при этом могут быть пропущены качественно новые закономерности, которые делают возможным протекание процессов, приводящих к .облагораживанию* энергии. Это утверждение может быть подтверждено также тем, что в беспредель-
128
III. Количественное выражение двух основных законов
Числовые значения энтропии. Энтропия имеет размерность энергии, деленной на температуру, и для ее выражения пригодны различные единицы в зависимости от способа выражения энергии и использованной температурной шкалы. В данной книге ее выражают в килокалориях на килограмм (или, соответственно, на киломоль) на градус Цельсия.
Во многих задачах имеют дело только с разностью энтропии без учета ее абсолютного значения, и в таких случаях принимают обычно, что в каком-либо состоянии, например при 0° С и 1 атм, S = 0. В результате установления третьего закона термодинамики, кратко рассматриваемого в гл. XI, можно определить абсолютное значение энтропии; эти значения существенны для исчерпывающей характеристики химического равновесия.
Физический смысл энтропии. Исчерпывающее рассмотрение этого вопроса завело бы нас слишком далеко, но кратко упомянуть об этом сле-' дует. Из изложенного в гл. II мы видели, что причиной, лежащей в основе необратимости, является тот факт, что при всех необратимых процессах система переходит от состояния меньшей вероятности к состоянию большей вероятности или от более упорядоченного расположения к менее упорядоченному, и шансы на перегруппировку для возвращения к исходному состоянию бесконечно малы, вследствие очень большого числа участвующих индивидуумов. Рассмотрим простой случай — два газа, имеющие разные температуры. При термическом соприкосновении один газ охлаждается, а другой нагревается до тех пор, пока ие будет достигнуто равновесное состояние, отвечающее равенству их температур. Из опыта известно, что эта система сама по себе никогда не дает возникнуть разности температур. Согласно кинетической теории, мы знаем, что газ при температуре, одинаковой во всех его -частях, в действительности состоит из молекул, очень сильно отличающихся друг от друга по своей скорости, а следовательно, и по ных просторах вселенной состояние материи резко отличается от состояния ее в обычных для нас системах. Успехи современной науки подтверждают существование процессов, приводящих к .облагораживанию* энергии.
Неосновательным также является применения к вселенной в целом тех основных термодинамических понятий, которые фигурируют в принципе возрастания энтропии. Нельзя, повидимому, говорить об энтропии вселенной как о сумме энтропий отдельных ее частей.
Следовательно, проблема тепловой смерти вселенной является мнимой и лженаучной проблемой, возникшей в результате неправильного применения второго начала. Однако самым существенным является следующее.
Независимо от способов доказательства несостоятельности рассматриваемой концепции всегда следует помнить, что допустить возможность тепловой смерти означало бы отказаться от основ материалистической диалектики, отказаться от того, что движение нетворимо и нерушимо, как сама материя, т. е. занять идеалистическую и реакционную позицию. Допустить возможность тепловой смерти означало бы также принятие возникновения вселенной в результате некоторого творческого акта.
Вот почему термодинамика, с одной стороны, является чисто технической наукой, а с другой стороны, смыкается с философией, т. е. ответственна за верное естественно-научное мировоззрение. (Прим, ред.)
»' - ‘t-f S , ' ,f * > Л'£‘
> ’ V v" “ 'г ч 1 1 . - ” * ’> > -
, ?' „ • » * ' г < i •
Энтропия 129
кинетической энергии. Имеются „горячие* молекулы (большая скорость) и „холодные* молекулы (малая скорость) и все градации между ними. Как мы видели по аналогии с колодой карт, наиболее вероятным расположением является полное перемешивание всех молекул, различных по скорости, по той простой ттричине, что число конфигураций, отве^ чающих беспорядочному расположению, несравненно больше числа конфигураций, в которых имеется приближение к упорядоченности. Любое расположение, в котором горячие молекулы преобладают в каком-нибудь месте в достаточной степени, чтобы допустить видимое различие в температуре между какими-либо макроскопическими частями газа, является настолько невероятным, что не может существовать.
Какова связь всего изложенного с энтропией? Энтропия является мерой вероятности данного состояния. Чем беспорядочнее система, тем больше ее энтропия, и наоборот, упорядоченная конфигурация является конфигурацией с низкой энтропией. Это дает нам определенный ключ к тому, что энтропия должна иметь нулевое значение и является абсолютной величиной. Когда вещество достигает состояния, при котором исчезает всякая беспорядочность, оно должно иметь нулевую энтропию. Чистое кристаллическое вещество при температуре абсолютного нуля должно представляться полностью удовлетворяющим этому условию и, таким образом, должно бы иметь энтропию, равную нулю (если исключить некоторую возможную хаотичность, которая может существовать во внутренней структуре самих атомов).
При приложении теории вероятности к молекулярным системам, — к которым ее можно применять совершенно строго вследствие громадного числа участвующих частиц, — Больцман показал, что имеется простая зависимость между энтропией данной совокупности молекул и вероятностью ее осуществления. Вывод этого соотношения и его дальнейшая разработка, позволяющая вычислить абсолютные энтропии, выходят из рамок этой книги ☆).
Комбинированная формулировка первого и второго законов. В уравнении (50) 5 для простоты относится к энтропии изолированной системы, т. е. „отдельной системы* плюс среда (непосредственно ее окружающая). При любом термодинамическом анализе желательно сосредоточить внимание на „отдельной системе* и изъять из рассмотрения среду. Для этого общую энтропию можно разделить на две части: 5 — энтропию изучаемой „отдельной системы*, и S'— энтропию окружающей среды, которую следует включить для получения изолированной системы. Уравнение (50) тогда переходит в
dS—dS'^O. (51)
Любой обмен энергией между „отдельной системой* и средой должен состоять из перехода теплоты или совершения работы благодаря
[185]^ ^ЛЯ боЛее П0ДР°®Н0Г0 ознакомления с этим вопросом см. гл. XVII ® Химическая термодинамика
130 Ш. Количественное выражение двух основных законов
действию некоторой силы (между ними никакого обмена веществом не допускается). Так как нас не интересуют детали того, что происходит в окружающей среде, то для анализа мы вправе допустить простейший случай, а именно, что все изменения в окружающей среде являются обратимыми. Следовательно, энтропию окружающей среды можно изменить только посредством перехода тепла, т. е. можно написать, что
dS’ = =^-. (52)
Знак минус перед dQ означает, что теплота относится к ,отдельной системе' и поток тепла от системы считается отрицательным. По первому закону
dQ=dU-{- dA.
Подставляя это выражение в уравнение (52), получаем
dS' = ~dU~dA . * (53)
Если принять, что все тепловые потоки между системой н окружающей ее средой являются обратимыми, что можно сделать, поскольку мы не связаны с деталями процесса, происходящего в среде, то
Г=Г, (54)
где Т—температура отдельной системы на любой стадии процесса. Ив сочетания уравнений (51), (53) н (54) имеем
dS — dU+dA g0, (55)
TdS—dU—<MgO. (56)
Все величины, входящие в это уравнение, относятся к „отдельной системе', причем dS и dU представляют соответственно приращения ее энтропии и ее энергии, a dA — работу, произведенную системой. Равенство справедливо для обратимого процесса, происходящего в системе, а неравенство относится к изменению, включающему некоторое необратимое действие без учета его величины. Это, вероятно, наиболее важное уравнение во всей книге; оно является комбинированной формулировкой обоих основных законов, и все последующие зависимости будут основываться на нем. В этом смысле оно в самом себе содержит все термодинамические основы для полной последующей обработки, за исключением тех случаев, которые относятся к третьему закону. Вероятно, среди выведенных до сих пор уравнений не имеется ни одного, содержащего так много возможностей для широкого применения.
Свободная энергия
131
СВОБОДНАЯ ЭНЕРГИЯ
Уравнение (56) в общем не поддается интегрированию, так как TdS и dA не являются полными диференциалами. Для интегрирования его при применении к конечному изменению было бы необходимо выразить dA в значениях свойств вдоль некоторого частного пути и, следовательно, знать, как связаны между собой S и Т. Перенося dA вправо и изменяя знаки, получаем
dU— TdS — dA. (57)
Для частного случая изотермического изменения левая сторона этого уравнения является диференциалом функции U — TS, как можно легко убедиться посредством диференцирования этой функции при постоянной температуре. Данная функция, представленная символом F, была названа Гельмгольцем свободной энергией. Изменение функции F обычно называют максимальной работой, но это несколько неудачно, так как в зависимости от направления изменения она может характеризовать и минимальную работу. Кроме того, так называемая „чистая" работа, измеряемая изменением изобарного потенциала AZ, может быть больше максимальной работы, как это и будет сейчас показано. При рассмотрении функции F не возникает никаких недоразумений, если помнить, что ее изменение равно алгебраической сумме всех работ, связанных с изотермическим изменением.
По определению,
F=U—TS. (58)
При постоянной Т
dF=dU—TdS^ — dA. (59)
Интегрируя, получаем
F2 - F, = (U2 — TSa)—(ОЛ -TS^- 2Д. (60)
Символ S введен для того, чтобы подчеркнуть, что уравнение содержит больше одного члена работы в зависимости от действующих сил, и 2Й4 представляет их алгебраическую сумму. Например, может существовать работа, вызываемая действием давления, и работа, вызываемая действием э. д. с., причем они могут иметь и противоположные знаки.
Согласно уравнению (60), если какая-нибудь система претерпевает обратимое изменение при постоянной температуре, то общая работа, произведенная системой, равна уменьшению свободной энергии, тогда как при изотермическом, но необратимом изменении произведенная работа будет меньше, чем уменьшение свободной энергии F. Возможно, что это будет яснее, если уравнение (60) написать в виде л— F,^A. (61) ☆)
Сг) Заметим, что изменение знаков по обеим сторонам неравенства изменяет знак самого неравенства.
9*
132 ill. Количественное выражение двух основных законов
Если Fj]>F2, то происходит уменьшение F, и работа производится над системой. Из этого видно, что изменение F является мерой максимальной работы, которую может совершить система, претерпевая изотермическое изменение в том направлении, в котором она работает на среду. Очевидно, зная значения U и 5 для различных возможных состояний, мы могли бы предсказать максимальную работу, получаемую для какого-либо предложенного изменения, при условии, что оно всегда будет изотермическим.
Очевидно, что F является свойством, и при любом изменении состояния происходит определенное изменение F, независимо от того, будет ли оно изотермическим или нет; но AF выражает работу только для изотермического Изменения.
Наоборот, если для совершения работы над системой используются внешние силы, приводящие к изменению ее состояния, то возрастанием F для системы измеряется минимальная работа, требуемая для совершения данного изменения [см. уравнение (60)]. Минимальная работа будет в обратимом процессе, а для необратимого процесса, как легко показать из уравнения (60), работа будет больше увеличения F для системы. - •
Преобразуя уравнение (60), получаем
(£4 —t/j) —T(S2—L4, (62)
(63)
— £/j представляет общее изменение энергии, сопровождающее данный процесс, а T(S2— Sj)— тепловой эффект в'том случае, если процесс проводится обратимо. Из уравнения (63) легко показать, что максимальная работа, которую способен дать процесс, может быть больше или меньше общего изменения энергии в зависимости от знака AS. Если в результате изменения энтропия системы уменьшается, тогда величина TAS будет положительна, теплота будет переходить от системы к окружающей среде, и работа будет меньше уменьшения энергии. С другой стороны, при возрастании энтропии теплота будет переходить к системе от среды, и произведенная работа будет больше общего изменения энергии именно на это количество теплоты.
В случае химической реакции числовое значение At7 большей частью бывает значительно больше TAS, и поэтому AF обычно имеет тот же знак, что и At7, и тот же порядок величин. Это обстоятельство можно использовать для грубо приближенного определения работы или равновесия при отсутствии данных по энтропии. Очевидно, что для частного случая, когда нет изменения энтропии,
AF=Af7,
и равновесие можно количественно рассчитать по изменению U.
Если процесс необратим, то тепловой эффект может быть совершенно иным и даже иметь противоположный знак. Например, в случае
Изобарный потенциал
133
возрастания энтропии, при обратимом процессе тепло поступает в систему, причем все At/ превращается в работу. Если же процесс необратим, то часть At/ не будет превращаться в работу и поэтому будет передаваться среде в виде теплоты, причем это количество теплоты может быть больше обратимого Теплового эффекта TAS’, в результате чего теплоты выделяется больше, чем поглощается. Например, реакция
РЬ (т) -ф- 2Hg Cl (т) — РЬС12 (т) -ф- 2Hg (ж)
будет проходить с поглощением тепла, равным 1990 кал, если она проводится обратимо и изотермически в гальваническом элементе; но при изотермическом проведении в калориметре (без обратной э. д. с.) будет выделяться 22730 кал (все теплоты даются при 25° С).
С первого взгляда можно подумать, что имеется нечто парадоксальное и противоречащее второму закону в том, что теплота, передаваемая системе от термостата, полностью превращается в работу, как это происходит при химической реакции, при которой 5 возрастает, или же при обратимом изотермическом расширении газа. В последнем случае At/=O, если газ является идеальным и вся работа обусловлена тепловым эффектом TAS. Однако следует указать, что нет никакого противоречия второму закону в полном превращении теплоты в работу в любом процессе, не являющемся круговым. Для физической интерпретации этого можно представить работу получающейся не из энергии беспорядочного движения молекул, что совершенно недостижимо при отсутствии разности температур, а из потенциальной энергии, обусловленной направленными силами самого вещества; поскольку эта энергия превращается в работу, температура будет стремиться к уменьшению, но это падение компенсируется тепловым потоком из термостата. При круговом проведении процесса, обусловленного отсутствием общего изменения энергии в системе, работа не может производиться, если нет разности температур. Другими словами, при любом изотермическом обратимом цикле общая работа равна нулю. Этот результат легко выводится из уравнения (60), так как для цикла AF должно равняться 0 и поэтому 2А равно 0. Этот вывод представляет значительную историческую ценность, так как он лежал в основе некоторых важных следствий.
₽ ИЗОБАРНЫЙ ПОТЕНЦИАЛ
В предшествующем разделе мы рассмотрели особый тип изменения, именно — изотермическое, и нашли, что для таких случаев оказалась особенно полезной функция F. Теперь рассмотрим другой частный случай, представляющий еще бблыпую практическую важность, а именно — случай постоянной температуры и постоянного давления. Многие практически важные изменения происходят приблизительно в этих условиях, причем постоянное давление часто считается
134 IfI. Количественное выражение двух основных законов
равным атмосферному. Для этого случая удобно алгебраически разделить общую работу 24 на две части: 1) работу, производимую постоянным давлением среды для бесконечно малого изменения, н 2) работу, производимую против других сил, иногда называемую „максимально полезной работой" и обозначаемую через А'. Тогда
dA = dA' -J- pdv (64)
или, для конечного изменения,
24 = 4' 4-рДгг. (65)
В случае химической реакции, проводимой в элементе, А' есть электрическая работа, a pbtu — механическая работа, произведенная против атмосферного или другого постоянного давления среды. Может произойти недоразумение из-за терминов, обычно употребляемых для 24 и А', так как в зависимости от знака До максимально полезная работа А’ может быть больше или меньше общей работы 24. Пока юна связана с увеличением объема, сопровождающим совершение системой полезной работы, толкование ее совершенно ясно, но при уменьшении объема, как в обратимом элементе, производящем жидкую воду из водорода и кислорода, так называемая „максимальная" работа— ДГ равна 55670 кал)моль при 25° С, тогда как максимально полезная работа или — AZ равна 56560 кал/моль.
Подставляя уравнение (64) в уравнение (57) и перенося pdv в левую сторону уравнения, получаем для изотермического изобарного процесса следующее выражение:
dU-TdS-^pdv^ — dA'. (66)
Так как и р и Т постоянны, левая часть этого уравнения является полным диференциалом функции U—TS-\-pv, обычно обозначаемой через Z.
Эта функция впервые была широко использована Гиббсом и названа им „термодинамическим потенциалом". Мы будем его называть изобарным потенциалом, пользуясь этим сокращенным терминой вместо полного и более точного — „изобарно-изотермический потенциал".
Интегрируя уравнение (66), получаем
Д7^ — А'. (67)
fi словесной формулировке: Д2, илн изменение изобарного потенциала, является мерой максимально полезной работы в обратимом изотермическом изобарном процессе. В одном направлении она измеряет максимальную полезную работу, которая может быть получена как результат изменения, а н другом направления измеряет минимальную работу, требуемую для совершения изменения. При наличии в изменении известной необратимости работа будет меньше в одном направлении
Основные диференциальные уравнения
135
и больше в другом. Уравнение (67) можно также написать следующим образом:
дг=ц — иг — T(S2 —SJ+piVz—А', (68)
AZ=F2 —л + ^)^—Л', ___(69Х
bZ = H2 — Н, — T(S2— 51)^ — А'. (70)
Таким образец, максимальная (или минимальная) работа для любого процесса, проводимого при постоянном давлении и температуре, может быть определена заранее, если известны значения U, S и v для исходного и конечного состояний, причем она не будет зависеть от реального механизма, применяемого для совершения изменения. При отсутствии объемного изменения AZ будет равно AF, и так как член ptico обычно бывает относительно .мал, то AZ почти во всех случаях будет приблизительно равно АЛ.
ОСНОВНЫЕ ДИФЕРЕНЦИАЛЬНЫЕ УРАВНЕНИЯ
Для удобства применения функций, определенных в этой главе, предлагаются некоторые уравнения, устанавливающие связь этих функций с обычными параметрами состояния. Целью этого раздела является вывод таких уравнений; вывод всех основных положений, уже введенных при определении функций, будет осуществлен посредством элементарных математических манипуляций. Пока мы ограничимся выводом для следующего частного случая: 1) гомогенная система содержит только один компонент (или многокомпонентные системы постоянного состава, подобные воздуху); 2) единственной действующей силой является равномерное гидростатическое давление; 3) система находится в равновесии. (В следующей главе уравнения будут обобщены для приложения к более сложным системам.) В рассматриваемом случае состояние системы можно представить уравнением (обычно называемым „уравнением состояния“) следующей общей формы:
®(р, V, Т, т) = 0. (71)
Для дайной постоянной массы m состояние полностью определяется двумя независимыми переменными, следовательно,
Р=<Р(У,Л;
подобные уравнения могут быть применены при использовании другой пары независимых переменных (р и V или р и Т).
Уравнения для U и S, выраженные через v и Т. Так как состояние системы определяется двумя переменными, то U, Н, S или какие-либо другие термодинамические свойства могут быть выражены посредством двух независимых переменных ☆), например,
Ц=<р(Т, ©).
£•) В остальной части этой главы все экстенсивные свойства, подобные объему и энергии, будут относиться к одному молю или к единице массы.
136
III. Количественное выражение двух основных законов
По хорошо известным методам вычислений, представленным в гл. I,
но по первому закону
dQ = dU-\-pdv ☆), поэтому
‘'«=(»г)г,я’+[(^)г+ <’]*•
По второму закону
dS=^.
Подставляя в уравнение (73), получаем
,с 1 (dU\ . 1 Г fdU\ . ] .
dS — Т\дТ )dT+ Т [ {dUJг+ Р]dv' откуда
/68\
\dT)v— Т\дТ1ъ
Теперь используем то обстоятельство, что № _ № dTdv dviT ’
поэтому
Так как d^U!dTdv= cPUi&vdT,
то последнее уравнение переходит в
(73)
(74)
(75)
(76)
(19, гл. I)
Из первого закона следует, что
/сНМ _ (^О\ — с
\dT}v—\dTjv—^^
£г) Во всех уравнениях этой главы опускается механический эквивалент тепла J. Уравнение первого закона следует точно писать в виде
dQ — dU 4- у pdv,
если U измеряется в тепловых единицах. При использовании уравнений для числового решения следует в надлежащем месте включать J.
Основные дифференциальные уравнения
137
Произведя соответственную подстановку в (72), получаем dU=CvdT-\-^T(^ —pjdv. (79)
Выражения для двух основных законов можно объединить уравнением
TdS=dl/-}-pdv. (80)
При постоянной температуре
Т\^)г— {toJT+P- <81>
Диференцируя по Т при постоянном v, получим
1 \dvdTj ' \dv)т dvdT'\dTjv'
При постоянном объеме dQ = dU=CvdT, н поэтому вообще dQ=TdS, С*~ <83>
нли
Сг>~Т ((/г)v’ (84)
Диференцируя уравнение (84) по v при постоянном Т, найдем, что
Из уравнений (82) и (85) ^L—т^-дТди дТди ’ (£)г=@/ <86>
Теперь, так как 5 является свойством, то
ds= !~SyT+(*)/’• (87>
Из уравнений (80) и (87)
Подстановка значений диференциальных коэфициентов из уравнений (84) и (86) дает
dU= CvdT-\- (79)
138 П1. Количественное выражение двух основных законов
Соответствующее уравнение для энтропии получается непосредственно из уравнений (79) и (80):
"=сЛ+(эт)/г-
(88)
Уравнения, содержащие в качестве независимых переменных р и Т. Для вывода аналогичных уравнений с независимыми переменными р н Т нужно применить функцию Н следующим образом: из определения
dH=dU-\- pdv-\-vdp. (7)
Сочетая уравнение (7) с уравнением (80), получим
TdS=dH—vdp. (89)
Деление на dp при постоянной Т дает
TfdS\ __(дН\ \др)т \др) т
(90)
Диференцируя уравнение (90) по Т при постоянном р, получаем
т #s 1 дрдТ /dSA _ _ \др / т дрдТ fdv\ \dfjp (91)
При постоянном p dQ = CpdT=TdS
или (dS\ _Ср [дт)р— Т ’ (92)
Но г — (дн\ Ьр-\дт)р- (9)
Следовательно, r(dS\ _ (дН\ \dr)p— \дт)р' - . (93)
и поэтому r &S _#Н дТдр дТдр'
Подставляя это уравнение в уравнение (91), найдем, что
Так как S является свойством, то dS=№\ dp.
\dTJp 1 \dpJT r
Основные дифференциальные уравнения
139 -
Подставив значения соответствующих частных производных из уравнений (92) и (94), получаем
dS=Cpd/-(^dp. (95)
Из уравнений (89) и (95) можно непосредственно получить аналогичное уравнение для Н:
dH=CjdT-\-\v—T(^\}dp. (96)
L \°‘ / pJ
Постоянные И и S. При постоянном Н из уравнения (96) получается важное уравнение
I дТ\ Т (dvldT)D — v
• (97) \др)н Ср-------------------------------' '
Коэфициент (дТ1др)н известен как коэфициент Джоуля — Томсона и обычно обозначается через р. х
При постоянной 5 уравнение (95) можно превратить в
или
dp)s~ Ср
с ___T(dv/dT)p
Р 1(дТ1др)5-
(98)
(99)
Уравнение (99) приложимо для измерения теплоемкости при постоянном давлении.
Аналогично из уравнения (88) получают
С _______-г (др /аТ)о
(dTldv)s •
(100)
Замена независимых переменных. Для замены двух переменных р и v обычно применяют соотношения между диференциальными коэ-фициентами:
as \ fdS\ ) ran
дя) р <dv)p' (101)
dS\ _ ras\ РдТ \
др)„ \др)в (Ю2)
Эти уравнения основаны на уравнении (12, гл. 1). Соответвтвующие подстановки непосредственно дают
Со(дТ\ , Св/дТ\
"“тЫ/’+тЫ*- »>03)
Здесь следует отметить, что функция U полезна главным образом \ в тех случаях, когда независимыми переменными являются Т и vt и
140 III. Количественное выражение двух основных законов
что функция Н особенно полезна тогда, когда независимыми переменными являются Тир. Располагая диференцнальным коэфициентом, например (йи[дТ)р, иногда превращают его в коэфициент с другим фиксированным параметром, например в (dU[dT)p. Этого можно достигнуть посредством уравнения (13, гл. I).
Связь между теплоемкостями Ср и Ср. Полезные уравнения, включающие Ср и Ср, легко выводятся следующим образом: применяя
сРи _ d'lU дйдГ~дТдо
к уравнению (79), получаем
/<ЧГ(<^Г)о-р]| \ди]т 1 дТ = т(^\ . (104)
Подобным же образом при применении соотношения > а2// _ д2// дрдТ дТдр
к уравнению (96) получаем (дСД [дги — =—т ( — \ др / т W р [105)
Теперь из уравнений (88) и (95) следует, что
И dQ=TdS=CvdT-\- Т ( TdS=CpdT—T^~^ •0р\ дТ) ь dp-, р dv, (Ю6) (107)
и так как то и v=’f(p, Т), , (<to\ । (<to\ dv=\^)dP+\lyr}pdT fdp\ _^(dvldT)p \dTJv~ (dvjdp)T' (Ю8) (Ю9)
Подставляя уравнение (108) в уравнение (106), находим, что
и, используя уравнение (109), имеем
т^)^- (111)
Основные диференциальные уравнения 141
сравнения уравнений (111) и (107) очевидно, что с».=с.+т(ет),(г),- <и2> Н|
При применении уравнения (109) можно легко представить ура вне- ‘"""3
ние (112) в следующем виде: Я
С"-С-=-Т(я)‘,(^1т (,13)/ Я
Уравнения (112) и (ИЗ) можно использовать для вычисления Ср по ' ’Л
Cv (или наоборот) для любого вещества при условии, что для него 3
известны соотношения между р, v и Т. 1
Из уравнений (99) и (100) и из уравнений 1
МП МП __(др\ ’J
\д°) s -4
и • '
(др\ да — да \dT)p\dv)p \dvjf *•
которые следуют непосредственно из уравнений (12, гл. I) и (16, гл. I), можно получить
Cp_(dpldu)s_.s
Ср — (др1дО)т— *т’ '***'
где
— 1 (^У
*s v\dp)s и
х =—-да
г я\др)т'
xs и хт представляют соответственно адиабатную и изотермическую ‘у 1
сжимаемость. Уравнения для F и Z. Уравнения, связывающие свойствами, легко выводятся следующим образом. F=U—TS. 3 F и Z с другими По определению
Отсюда, диференцируя, получим ' -Я
dF=dU— TdS — SdT, но dU — TdS=—pdv, или dF=—pdv—SdT. Поэтому (£),=-* (115) (80) (116) (117) ’
142 ill. Количественное выражение двух основных законов
и
'1,8>
Уравнения для F, выраженные через другие пары переменных, н» так просты, как эти, и не находят особенного применения. Другими словами, функция F полезна только в том случае, когда независимыми переменными являются Т и ф.
Подобным образом, по определению,
Z=U— TS^-pv
И '
dZ = dU—TdS—SdT-\-pd,b-\-‘odp.
Но
' dU—TdS-\-pdv—O,
поэтому
dZ=— SdT-{-vdp, (119)
откуда
(»),“-* <<20>
Й
($,“’• <’2’>
Функцию Z следует применять во всех случаях, когда независимыми переменными являются р и Т.
Выводы при использовании F и Z. Интересно проследить, насколько упрощаются ранее произведенные выводы уравнений для U, Н н S при употреблении функций F и Z. Для этого мы вернемся несколько назад.
Начнем с уравнений (116) и (119). Из этих уравнений и уравнения (19, гл. I) непосредственно следует, что
(86)
(94)
Но
TdS=dQ—CvdT (при постоянном объеме),
= CpdT (при постоянном давлении),
или
dS\ IdS\ _СР
дГ)~Т И \dt)p~~T
Основные диференциальные уравнения
143
Из этого следует, что dS=~dT-}-(^vdv (88)
н тд&\ ----~—
dS=/dT — (ЭЙ)
И из уравнений TdS=dU-\-pdv (80)
и TdS=dH—vdp (89)
легко можно получить соответствующие уравнения для U н Н.
Изобарный потенциал как функция температуры. Из уравнения (120) и из того, что
Z=H—TS, (122)
получаем
(Я=^-Тогда TdZ—ZdT=— HdT (при постоянном давлении) и TdZ—ZdT HdT pt pt *
нлн
diZ/T) дТ Н т*‘ (124)
Интегрируя, получаем t=-Ih^+z’ (125)
где [—константа интегрирования.
Н легко выразить как функцию Т посредством теплоемкостей, а последующее интегрирование дает очень важное уравнение, связывающее изобарный потенциал с температурой. Таким образом,
или
/дЯА _г
\дт)р~г'
H=H„+^CpdT,
(9)
где Но—константа интегрирования. Очевидно, что она должна быть значением Н при абсолютном нуле, если уравнение интегрируется с помощью данных Ср, Справедливых вплоть до Т==0.
Примем
Ср = а+?Т+у^. (126)
144 1Н. Количественное выражение двух основных законов
Тогда » •
Я=Я0 + а7'+1₽Г-|-4у74. (127)
Подставляя уравнение (127) в уравнение (125) и интегрируя, получим или
Z=HQ—aTln'T—4 gfT’ + IT. (128)
Также
Z=/f— aTlnT— ₽Т— 1уТ8+(/—а)Т. (129)
При такой формулировке это уравнение приложимо только к гомогенным однокомпонентным системам; во позже оно будет распространено н на более сложные системы. Заметим, что оно дано для изменения Zc 7 при постоянном давлении.
Z как функцию Т можно получить нз соотношения ,
Z—H—-TS, (122)
выражая 5 н Н как фувкции Т при постоянном р. Так как dS=C^r, то
S=jc/)dln7’4-50; (130)
50 будет энтропией при Т=0, если уравнение для Ср справедливо вплоть до Т=0.
Используя уравнение (126), получим
S=f(a+₽r+7T8)7+50=alnT4-pT4-lYr + S0. (131)
Подставляя уравнения (127) и (131) в уравнение (122), найдем, что
Z=/f0-—aTln 7—-i- ir-±-tI* + (a — S0)T. (132)
Из этого метода вывода яснее видно, что константа интегрировании непосредственно связана с энтропией и ее можно было бы определить, если бы теплоемкость была известна до 7=0 и если бы была известна энтропия при абсолютном нуле.
Прежде чем закончить рассмотрение этих двух функций, интересно отметить, что энтальпию Н можно вычислить при условии, если Z известна как функция давления и температуры. Это следует
F и Z как критерии равновесия
145
(133) v и Т
(134)
изме-
из уравнения (124), которое можно также написать в форме WIT)P '
Подобным же путем и U можно вычислить из F как функцию с помощью соотношения
F И Z КАК КРИТЕРИИ РАВНОВЕСИЯ
Всякий раз, когда имеется тенденция к самопроизвольному
нению, с помощью процесса можно получить работу при условии наличия соответствующего механизма. Максимальная работа получается в том случае, когда процесс происходит с уравновешенными силами, т. е. обратимо. Для изотермического процесса максимальная работа равна уменьшению функции F. Подобным же образом для процессов, протекающих при постоянных р н Т, максимальная полезная работа равна уменьшению функции Z. Другими словами, F и Z при самопроизвольных процессах всегда уменьшаются, и поэтому их можно применить в качестве критериев, указывающих, будет или не будет происходить любой предполагаемый процесс.
Рассмотрим, например, некоторую систему в исходном состоянии 1 *
и предположим, что ее изобарный потенциал Z известен. Допустим, что мы хотим знать, будет ли (или, точнее, может ли) происходить переход к другому состоянию 2 сам по себе, при условии постоянных р и Т, или же для этого следует затратить энергию. В принципе это является одним из наиболее обычных типов вопросов, возникающих при рассмотрении химических систем. Ответ весьма прост, если мы знаем значение изобарного потенциала в состоянии 2. Рассматриваемое изменение будет (или, точнее, может) происходить в том случае, если Z2<^Zj, другими словами, если происходит уменьшение изобарного потенциала ☆). Если Z2'^>Zi, то изменение не может происходить самопроизвольно. Следовательно, F и Z являются критериями направления самопроизвольного изменения. По этой причине они иногда называются „потенциальными функциями*, так как потенциал представляет применяющуюся в теоретической механике величину, значение которой всегда уменьшается в направлении самопроизвольного изменения.
й) С практической точки зрения не совсем правильно сказать, что изменение будет происходить. Правильнее было бы говорить о тенденции к изменению, так как некоторые сопротивления, аналогичные механическому трению, *
могут создавать такое уменьшение скорости изменения, что для практических целей изменение может не иметь места. Кроме того, надо отметить, что мы намеренно отклонились от строгой математической трактовки для большей простоты аргументов. Точнее, при подобной трактовке имеют дело только с диференциалом, а не с конечным изменением.
16 Химическая термодинамика
146
111. Количественное выражение двух основных законов
Уравнение (56) в виде равенства п’а.'ложимо ко всем системам, находящимся в равновесии, и является в этом случае совершенно общим; то же самое справедливо для выведенных уравнений (116) и (119). Из уравнения (116) при постоянных v и Т имеем
dF=0, (135)
и ив уравнения (119) при постоянных р и Т имеем
dZ = Q. (136)
Эти два уравнения обычно считаются важными критериями равновесия. Приложенные к однокомпонентной однофазной системе, они не имеют смысла, так как в подобной системе невозможны какие-либо изменения, если две переменные состояния фиксированы. Однако при рассмотрении более сложных систем, где возможны и другие изменения, например переход массы из одной фазы в другую или химические реакции, эти уравнения можно применить для вывода некоторых важных уравнений равновесия. Это будет показано в следующей главе.
ЛЕТУЧЕСТЬ И АКТИВНОСТЬ
Эти специальные функции были введены Льюисом в 1901г. [149] для облегчения трактовки случаев, к которым не приложимы законы идеального газа и идеального раствора. Эти функции бывают особенно полезны, когда имеют дело с растворами, а не с однокомпонентными системами, но мы здесь определим их сначала для более простого случая, а затем в гл. IV обобщим определение, включив случай системы, состоящей из нескольких компонентов.
Определение летучести. Из уравнения (119) следует, что при постоянной температуре
dZ — vdp. (137)
В особом случае, когда вещество является идеальным газом, v = RTip и
dZ = RTd]np. (138)
Для сохранения формы этого уравнения в тех случаях, когда система ие является идеальным газом, Льюис ввел специальную функцию, определяемую с помощью уравнения
dZ=RTd\nf = vdp (139)
и названную летучест!ю вещества.
Интегрируя уравнение (139) при постоянной температуре между двумя состояниями с различными давлениями р и р°, получим
р
Z — Z° = RT In f =jvdp, (140)
Р°
где р° — давление в состоянии, выбранном за стандартное.
Летучесть и активность
147
Уравнение (140) просто определяет отношение летучестей в двух изотермических состояниях и недостаточно для определения абсолютного значения летучести. Сравнение уравнений Ц138) и (139) показывает, что для идеального газа летучесть пропорциональна давлению. С чисто теоретической точки зрения любое вещество — твердое, жидкое или газообразное — может быть доведено до состояния идеального газа, если при постоянной температуре снижать давление над ним. Все жидкости и даже твердые тела в конце концов испарятся, и все газы будут стремиться к идеальному состоянию ио мере снижения давления. Это подтверждается экспериментальными наблюдениями над. поведением газовых систем при понижении давления. Большинство уравнений состояния, устанавливающих соотношение между р, v и Т для газов, превращается в уравнение состояния идеального газа (pv = RT) в пределе, когда р = 0 или v = oo.
Определение летучести удобно дополнить, сделав ее численно равной давлению, когда вещество находится в состоянии идеального газа. Пусть состояние, представленное и /°, является таким состоянием, и поэтому р°==/°.
Для всех практических целей р° можно принять равным некоторому определенному низкому давлению, например 1 атм или, лучше, 0,1 атм, когда для большинства систем отклонения от законов идеального газа малы и, вероятно, лежат в пределах ошибок опыта. В практике является обычным принимать р?=\атм, что иногда приводит к некоторой путанице, так как во многих случаях это является чисто гипотетическим состоянием. Для примера обратимся к газовой реакции при повышенной температуре, включающей конденсирующийся пар, например водяной. Некоторые основные данные доступны для 25° С, и поэтому является необходимым принять стандартное состояние при этой температуре, несмотря на то, что водяные пары не существуют как газ при р—\. Проще экстраполировать свойства в неустойчивую область, чем изменять стандартное состояние до более низкого давления. Несмотря на возможность незначительных ошибок, возникающих при подобной экстраполяции, она полностью оправдывается большим удобством при практическом применении. Для математических целей прибегают к пределу, когда р—>-0. Как указал Тенелл [248] в исчерпывающем и строгом исследовании по определению летучести, уравнение (140) не определяет летучести во всех состояниях вдоль данной изотермы, так как в пределе, когда р°—»-0, Z° приближается к —со. Таким образом, нельзя строго математическим путем доказать правильность уравнения
Пт <= 1 р Р
(U1)
10*
148
III. Количественное выражение двух основных законов
Тенелл предполагает, что нижеприводимое уравнение является более удовлетворительным определением летучести:
р
RTlnf=RTlnp— a dp, (142)
где а представляет собой функцию (RT/p)— v.
Это выражение в пределе не приводит к неопределенным значениям при принятии гипотезы о том, что а стремится к постоянному конечному значению прн р = О-Сс). Эта гипотеза базируется на фактическом наблюдении изменения а с р (см. гл. V).
Определение Тенелла не меняет числовой оценки летучести, н для ее вычисления Льюисом и Рендаллом н всеми последующими исследователями применялось уравнение (142). Кроме того, как определение Льюиса, так и определение Тенелла приводят к совершенно одинаковому выражению для вычисления летучести нз уравнений состояния.
Уравнение (140) можно написать, как
RTlaf—7?7’ln/° = j vdp— RTinpP, и так как р=р?, то
\nf=±^vdp. (143)
Влияние давления и температуры на летучесть. Изменение
летучести с давлением дается следующим уравнением, получаемым нз
уравнения (139):
__ v
\ dp )t~RT'
(144)
Изменение летучести с температурой можно вывести следующим путем.
Диференцированне первой части уравнения (140) прн постоянном давлении приводит к уравнению
® — (%£) =/?ln t+RT 1- (145)
\дТ)р \дТ)р* /° 1 \Л дТ JР \ J Р°\ ' '
Производя следующие подстановки в уравнение (145):
<>23>
I Z Z0 г
R In = у— у [из уравнения (140)]
£г) Может показаться противоречивым, что тогда как pv = RT при р—> 0, (RT/p) — v не приближается к нулю. Однако отсутствие противоречия в этом случае лучше всего показывается с помощью уравнения состояния, что и будет сделано в гл. V.
Применение к идеальным газам
149
/д In/® \
\ дТ )Р°
— О
(так как по определению летучести /° = р°), получаем
<Нп/\
<?Г )Р
RT* '
(146)
Нй—Н представляет разность между энтальпией вещества в исходном состоянии при низком давлении р° и энтальпией в данном состоянии, причем обе берутся при одной и той же температуре.
Определение активности. В большинстве случаев удобнее иметь дело с отношением летучести в данном состоянии к летучести в произвольном стандартном состоянии при той же температуре. Это отношение, по Льюису, называется „активностью" и изображается символом а. Следовательно, по определению,
2
/в’
а поэтому из уравнения (140)
Z — Z° = RT\n а.
(147)
На основании этого уравнения активность можно определять непосредственно, вместо того чтобы связывать ее с летучестью.
Для нахождения числового значения активности следует определить стандартное состояние. Практически функция а применяется только при рассмотрении растворов, и так как в этой главе мы имеем дело с чистыми веществами, дальнейшее рассмотрение этой функции будет несколько отложено.
ПРИМЕНЕНИЕ К ИДЕАЛЬНЫМ ГАЗАМ
Основным содержанием этой главы являлось развитие различных общих зависимостей, которые можно было бы в дальнейшем приложить к частным задачам, но все же представляется желательным закончить наше обсуждение рассмотрением приложения некоторых из этих уравнений к наиболее простому случаю, а именно — к идеальному газу.
Для данной цели определим идеальный газ как газ, подчиняющийся следующему уравнению состояния:
pv = RT.
(148)
150
III. Количественное выражение двух основных законов
При диференцировании фициенты: уравнения (148) получаются следующие коэ- = *., (149) \дТ/v v ’ ' , (150) \dT]P Р 1 (₽). = “• <'51) (₽)„ = » <152>
При применении этих уравнений, которые, как следует указать, приложимы только к идеальному газу, уравнения (79), (88), (95), (96), (104), (105), (112), (116) и (119), соответственно, превращаются в следующие уравнения:
dU=CvdT, (153)
dS = C^+R^, (154)
dS=Cp^— R^-, dH—CpdT,
=0,
\ dv
(dCp} =0,
\ dp ]t
Cp — CO = R,
dF= — RT— — SdT, v ’
dZ — RT— — SdT.
(155)
(156)
(157)
(158)
(159) *)
(160)
(161)
Из этих уравнений мы можем сделать следующие важные качественные выводы: 1) энергия, 2) энтальпия, 3) теплоемкость при постоянном объеме, 4) теплоемкость при постоянном давлении идеального газа являются функциями одной только температуры. Изменения давления или объема при постоянной температуре не изменяют этих величин.
Пример 11. Вычислить энергию, энтальпию, энтропию, мольную теплоемкость при постоянном объеме, мольную теплоемкость при постоянном давлении 1 моля азота при t = 500°C ир = 100 атм, считая, что он является идеальным газом и что имеются следующие данные:
£?) Заметим, что это применимо только для мольной теплоемкости. Эквивалентным уравнением для удельной теплоемкости является Ср — CO = R/M, где М — молекулярный вес.
Применение к идеальным газам
151
1. Мольная теплоемкость при 1 атм как функция температуры дается уравнением
Ср = 6,5 4-0,001 Т,
где Г — температура в градусах Кельвина, а Ср — теплоемкость в каламом-град.
2. Энтальпия произвольно принимается равной 0 при t = 0 и р = 1.
3. Энтропия азота при 25° С и 1 атм равна 45,8 э. е.
При t — 500 и р = 1
Ср = 6,50 4- 0,001 • 773,2 = 7,273.
По уравнению (159), прн тех же условиях
Cv = Ср — R = 7,273 — 1,987 = 5,286.
По уравнениям (157) и (158), Ср и С„ идеального газа не зависят от давления, и поэтому найденные значения приложимы также и при 100 атм.
По уравнению (156),
Г 773,2
Н — Но+ \Cpdt — 04- j (6,504-0,001 Г)dT = 3512.
273,2
Найденная величина является энтальпией N2 при ( = 500 и р=1, но так как мы уже видели, что энтальпия не зависит от давления, то она является также энтальпией при 500° С и 100 атм. Из уравнения (153) имеем
т т
U^Ua+\CvdT = U^\(Cp — R)dT.
*0 *0
Мы могли бы применить и это уравнение, ио проще вычислить (/ из соотношения
U = Н—pv = И — RT = 3512 — 1,987 • 773,2 = 1973.
Для идеального газа U также не зависит от давления (или объема) при постоянной температуре.
По уравнению (154),
т Р
S^S^Cp^-R^- (1)
То 773,2 = 45,84- J ^6,50^4-0,OOlrfF^—1,9871п 100 = 298,2
= 43,3.
По уравнению (161),
р т
Z-Za = RT^- — \sdT.
152
111. Количественное выражение двух основных законов
Интегрируя сначала при постоянном давлении, при Ро~ 1, и затем при постоянной температуре Т и используя соотношение
S = S0+|‘cp^,
т„
получаемое из (1) прн р —const, имеем
In Т
ИТ.
(2)
Вместо уравнения (2) для вычисления Z в этом случае проще применить непосредственно уравнение, определяющее Z, именно
Z — H — 7’5=3512 — 773,2-43,3 = —29990.
Эта величина не имеет абсолютного значения, так как она основана на произвольном значении Н§.
Важно отметить, что тогда как величины Д/7 или Д77, или AS, т. е. разности значений этих функций между любыми двумя состояниями, являются совершенно независящими от выбранных начальных состояний, это несправедливо для 6Z. Из уравнения (2) очевидно, что значение AZ будет зависеть от значения 50; и так как для данного изменения состояния должно быть определенное значение AZ, то ясно, что AZ можно вычислить из термических данных только тогда, когда энтропии можно придать абсолютное значение.
ГЛАВА IV
ОБЩИЕ УРАВНЕНИЯ РАВНОВЕСИЯ
В предыдущей главе выведено много важных уравнений для случая относительно простой системы — системы, состоящей из одного компонента и содержащей только одну фазу, подверженную лишь равномерному давлению окружающей среды. В этой главе мы расширим рассмотрение, включив системы, содержащие более одного компонента в данной фазе (растворы), и системы, состоящие из нескольких фаз. Кроме того, желательно показать, как можно распространить эти уравнения на системы, на которые действуют другие силы, например электрическая и магнитная. При этом мы ограничимся рассмотрением так называемых „замкнутых систем“, понимая под этим системы, общая масса которых остается неизменной. Кроме того, мы исключим жидкие растворы, в которых происходит электролитическая диссоциация, так как они требуют специального рассмотрения.
Нашей целью является показать, что многие типы равновесий, часто трактуемые как независимые друг от друга, в действительности тесно связаны и что все они могут быть выведены из некоторых общих уравнений, которые в свою очередь базируются непосредственно на основных понятиях термодинамики, выраженных различными функциями. Хотя вывод общих уравнений может показаться более трудным и сложным, чем менее строгие выводы частных уравнений, тем не менее мы уверены, что этот предлагаемый нами метод является в действительности самым простым. Общие уравнения являются связующей нитью для массы явлений, а частные уравнения почти автоматически вытекают из общих при условии, что на последние накладываются определенные ограничения.
Сначала допустим, что общие уравнения сами по себе обычно не могут быть применены к каким-либо частным задачам. Это обусловлено тем обстоятельством, что интегральные формы значительно сложнее, а также тем, что данные, необходимые для их интегрирования, редко * бывают доступны. Несмотря на это, общие уравнения ценны в качестве исходных для получения частных уравнений, оказавшихся полезными во многих случаях. Например, можно указать следующие частные зависимости, легко получающиеся из общих уравнений:
1. Уравнение Клаузиуса — Клапейрона.
2. Закон Рауля.
154
IV. Общие уравнения равновесия
3. Закон Генри.
4. Уравнение Дюгема.
5. Уравнение для расчета понижения точки замерзания в разбавленных растворах.
6. Правило фаз.
7. Закон действующих масс для химического равновесия.
Эта глава почти полностью математическая и не содержит числовых примеров. Мы считаем ее главным образом введением к большому числу зависимостей, которые могут оказаться полезными при решении конкретных задач. Многие уравнения этой главы, но отнюдь не все, будут использованы в последующих главах. При применении их мы будем ссылаться на эту главу.
ОБЩИЕ УРАВНЕНИЯ ДЛЯ ЛЮБОГО ЭКСТЕНСИВНОГО СВОЙСТВА
Пусть О представляет общее значение какого-либо экстенсивного свойства, например объема, энтальпии, изобарного потенциала, гомогенной системы или раствора, состоящего из N компонентов. Для того чтобы сосредоточить внимание целиком на изменении G как иа результате изменений количеств различных компонентов раствора, примем, что другие независимые переменные, например давление и температура, остаются постоянными. Таким образом, мы можем написать
G = <f(NA, NB, (1)
Дифереицирование уравнения (1) дает
<° = <‘гЛ,л + ^"в+...+^^ (2)
Понятно, что каждая из частных производных берется при постоянных р и Т (или каких-либо иных внешних переменных) и при постоянном значении мольной массы, т. е. числа молей каждого из остальных компонентов. Мы могли бы, конечно, написать это уравнение в единицах обычной массы, но в общем проще иметь дело с мольной массой. Однако следует учитывать, что последнее требует принятия определенного и постоянного (хотя возможно и произвольного) молекулярного веса для каждого компонента в растворе. Это может в некоторых случаях вызвать известные недоразумения, но в общем этот способ ☆) оказывается наиболее простым.
Величины dGjdNt обычно называются парциальными мольными величинами и условно обозначаются символом G;. Таким образом, мы
й) Большинство уравнений этого раздела будет идентичным независимо от того, употреблены ли мольные свойства и мольные доли или удельные свойства, основанные на единице массы или весовой доле, но практически вычисления будут производиться в мольных единицах.
Общие уравнения для любого экстенсивного свойства
155
можем написать уравнение (2) в следующем виде.’
dG = GAdNi-\-GBdNB+...-\-GNdN!r (3)
Природа экстенсивного свойства такова, что при увеличении всей массы раствора в одинаковой степени, т. е. без изменения состава или относительных масс, значение свойства возрастает в той же мере. Другими словами, 2 кг-моля любого раствора должны иметь объем или энтальпию, в точности равные удвоенным значениям этих свойств для 1 кг-моля при условии, что давление, температура и состав остаются неизменными. Если мы удваиваем общую массу при постоянных р и Т, но не сохраняем массы отдельных компонентов в том же соотношении, то значение О может измениться иначе.
Эти простые, практически очевидные факты можно сформулировать более кратко, если сказать, что О является однородной функцией массы первой степени. Из математической теоремы, известной как теорема Эйлера, следует, что коэфициенты dG/dNj являются функциями массы нулевой степени, или, другими словами, не зависят от массы, а зависят только от соотношения масс, т. е. от состава.
Интегрируя уравнение (3) при постоянном составе, получим
0 = Од^4-О^в+...+ОЛ *4)
Константы интегрирования равны нулю, как можно показать посредством рассмотрения физической картины интегрирования. Интегрирование просто соответствует прибавлению друг к другу небольших количеств компонентов данного раствора с целью получения конечного количества того же раствора.
Уравнение (4) теперь можно диференцировать, не прибегая к специальному ограничению постоянства состава; это дает
dG = GAdNA-yNAdGA^-GBdNB^
+ NBdGB+...+Gy/^+^GJV. (5)
Сравнивая уравнения (3) и (5), получаем
+n^gb +... 4- =о- (6)
Вместо того, чтобы оперировать со значениями экстенсивных свойств, основанных на общей массе, для большинства наших рас-суждений будет удобнее иметь дело с различными свойствами, отнесенными к одному молю или к единице массы, и тогда уравнения (2), (4) и (6) переходят соответственно в
dG = GAdxА-\-G^fixв-\- ..,G ^1х (7)
G == xAGA 4~ xBGB 4- ... 4~ xjPk> (8)
ХАаОА-*ГХ1^в+"- 4-ХА=0’ (9)
150
IV. Общие уравнения равновесия
где х—мольная доля различных компонентов Д). Из того обстоятельства, что
• • • +*#= 1, (Ю)
получаем
dxN= — dxA — dxB— dxN_}. (11)
Исключая dXx из уравнения (7) посредством уравнения (11), получим dG = (Ол- ОЛ) dxA + (Ъд—ОЛ) dxB+
+ •.-+(0^1— ON)dxN_v (12)
Так как все х теперь являются независимыми переменными, мы можем написать
(13)
(14)
(15)
(16)
(17) послед-
Из уравнений (8) и (10) находим
G= (Ga G^)xa-\- (Gb Gn) xb-\- ... + • • • + (Gn-i Gjj) xN_i G№ или
G (GA Gn) xA (GB GN) xB ...
(^JV-1 ®лг) X№—1'
Подставляя уравнение (13) в уравнение (15), получаем
-р. „ dG dG дО
G„=G — х,-^--------х„-т-----...—х.. ,-т------
N Адхл в дхв
и сравнивая с (13), имеем
п . дО дО дО дО
1 ' дхг А дхА вдхв n-i dxN_x
Во всем этом разделе мы будем руководствоваться двумя
ними уравнениями. Уравнение (17) связывает с мольными свойствами и составом раствора парциальные мольные свойства всех компонентов, кроме одного, мольная доля которого была исключена. Уравнение (16) дает то же самое для компонента N, мольная доля которого и была исключена при применении уравнения (10).
Бинарные растворы. Чтобы получить более конкретное представление о парциальных мольных величинах, рассмотрим очень кратко частный случай бинарного раствора с мольными долями хи 1 — х и в качестве свойства возьмем объем при постоянных р и Т. Тогда
й) Необходимо отметить, что так как не все х являются независимыми, то О} не равно dG[dxt.
Общие уравнения для любого экстенсивного свойства
157
уравнения (16) и (17) соответственно примут вид:
= V —xg (18)
и = U— х)^- (19)
Значение парциальных мольных объемов можно легко найти алгебраическим или графическим методом по зависимости мольного объема раствора от мольной доли. Графический метод будет показан в гл. IX для частного случая диференциальной теплоты растворения, которая является парциальной мольной энтальпией.
Типичное уравнение для парциального мольного свойства. Многие уравнения, выведенные в предыдущей главе и устанавливающие зависимость экстенсивного свойства одного компонента (чистого вещества), легко можно распространить на соответствующие парциальные мольные величины. Рассмотрим, например, следующее
уравнение:
(£) = V_T(%\ , \др)т \дТ/р
которое непосредственно вытекает из уравнения (96, гл. Ш). Это уравнение одинаково хорошо прнложнмо к мольным свойствам н к общим свойствам гомогенной системы. Применяя это уравнение для общих свойств и диференцируя его по Nt (числу молей какого-либо данного компонента) при постоянных р и Т, получим
&Н дУ _
dpdNi dNj 1 dTdNi'
Это можно представить в виде
д t<Na Nb ... __ f д У\
дР JT,»,... р, T,NA,Ng,...
d (dVIdNpp' г, Na> Nb ]
L dT или (dfft\ - (доД
\ "Р / т, х, • • • \ дТ / р, х, —
(20)
Как мы уже видели, парциальные мольные величины являются функциями не отдельных масс, а состава. Следовательно, частная производная от парциальной мольной величины по р или Т должна быть взята прн постоянном составе. Индекс х,_____представляет (W— 1)
переменных состава.
158
IV. Общие уравнения равновесия
УРАВНЕНИЯ РАВНОВЕСИЯ ДЛЯ ЛЮБОГО ЧИСЛА КОМПОНЕНТОВ И ФАЗ
Химические потенциалы. Основное уравнение равновесия для однокомпонентной гомогенной системы, в которой действуют лишь механические силы, дается следующим соотношением
dU=TdS—pdv (80, гл. Ill)
или в эквивалентной форме при использовании изобарного потенциала
dZ= — SdT~\-vdp. (119, гл. Ill)
Эти уравнения одинаково хорошо приложимы к любому гомогенному раствору постоянного состава. Рассмотрим раствор, содержащий NA, молей компонентов A, соответственно.
Число молей любого компонента можно изменять независимо от других (по определению компонента), и такие изменения будут вызывать изменения энергии или изобарного потенциала смеси или раствора. Так как эти изменения независимы друг от друга, можно написать, что
Л, = 7-‘»-^+^^ + ^даг+...+Ддаж, <21>
где частные производные берутся при постоянных S и V и всех других N, кроме данного. Подобным же образом можно написать
(22)
Так как, по определению,
Z = U— TS4-pV, то dU — TdS + pd V = rfZ + SdT — Vdp,
и коэфициенты при членах dN в уравнениях (21) и (22) одинаковы, т. е.
(А /231
\dN,Js, v,KA, ... \dMi)p,t,na,---- 1
Так как U и Z представляют экстенсивные свойства, то эти коэфициенты являются величинами, уже ранее названными нами „парциальными мольными величинами". Эти специальные величины особенно важны для равновесных состояний; Гиббс [87] первый указал их значение и обозначил символом ;л; они были названы „химическими потенциалами". Химический потенциал можно определить четырьмя
Уравнения равновесия дм любого числа компонентов и фаз
159
различными способами в зависимости от выбора независимых переменных:
ydwj s,v,na,... \s, р, na, ...
_/<?z \
\dNj v, t,na,... \dNiJр, т, na, ..."
Из рассмотрения парциальных мольных величин очевидно, что химические потенциалы являются интенсивными свойствами, аналогично давлению и температуре, причем зависят от состава раствора, но не зависят от общей массы его. Химический потенциал любого компонента фазы определяется изменением общего изобарного потенциала этой фазы (который отличается от его мольного значения) с изменением количества этого компонента при постоянных давлении, температуре и количестве всех остальных компонентов фазы. Он также определяется изменением общей энтальпии фазы при изменении количества данного компонента, когда поддерживаются постоянными энтропия, давление и количество остальных компонентов и т. д.
Так как давление и температура являются двумя независимыми переменными, обычно рассматриваемыми во всех практических задачах, то парциальный мольный изобарный потенциал является практически единственно применимым при исключении других химических потенциалов.
Критерий равновесия. В своей работе „О равновесии гетерогенных веществ“ Гиббс предложил в качестве общего критерия равновесия зависимость
dU = 0 при S,V = const. (24)
Для строгого доказательства мы отсылаем читателя к этой работе [87]. U является общей энергией, содержащейся в системе, которая может состоять из нескольких компонентов и из нескольких фаз. Гиббс также показал, что уравнение
dZ = 0 при постоянных р и Т (25)
является эквивалентным критерием равновесия. Оба эти условия вытекают непосредственно из уравнений (80, гл. III) и (119, гл. III), если примем (без строгого доказательства), что эти уравнения применимы к любой системе, независимо от того, является ли она сложной или состоит из одного компонента и одной фазы. (Заметим, однако, что мы все еще пренебрегаем всеми силами,«кроме давления р.)
Так как обычно имеют дело с системами, которые по условиям своего существования ближе подходят к системам, находящимся при постоянном давлении и температуре, чем к системам, изолированным при постоянных S и V, то критерий, выраженный уравнением (25), является наилучшим для использования. Z равен общему изобарному
160
IV. Общие уравнения равновесия
Рис. 11. Функция Z как критерий равновесия.
непрерывно изменяться, как показано
потенциалу системы; он может изменяться с изменением любых возможных переменных, за исключением р и Т.
Постараемся теперь несколько яснее показать значение этого уравнения. Система может находиться в термическом равновесии (без изменения Т) или в механическом равновесии (без изменения р), ио все же может не достигнуть химического или фазового равновесия. Другими словами, все еще возможны различные изменения, например химические реакции или переход вещества из одной фазы в другую. Все подобные изменения будут связаны с уменьшением изобарного потенциала системы, и при достижении равновесия Z приобретает минимальное значение. Это математически выражается уравнением (25).
Для большей наглядности значения уравнения (25) рассмотрим простую гомогенную химическую реакцию
2А = А2, где состав системы можно выразить единственной переменной х —мольной долей мономера. При данных давлении и температуре следует ожидать, что изобарный потенциал будет ia рис. 11, причем минимум на
кривой отвечает равновесному составу. Интересно отметить, что и два различных неравновесных состояния, например хг и xs, могут иметь равный изобарный потенциал; поэтому вообще неверно писать в качестве критерия равновесия 4Zp т = 0. Кроме того, даже при Zj >Z2 система не будет самопроизвольно изменяться от состояния 1 к состоянию 2, так как они расположены по различные стороны от состояния равновесия.
Фазовое равновесие. Уравнения, подобные уравнениям (21) и (22), можно написать для любого числа фаз, из которых может состоять рассматриваемая нами система. Таким образом, применяя один, два, три и т. д. штриха для обозначения различных фаз, можно написать:
dZ' = — S’dT + V’dp + fl’ dN' + g’dN’ 4-... 4 g' dN\„ A dZ" = - S’dT + Vdp + p’ dN”A + 4-... 4- } (26)
и так далее столько раз, сколько имеется в системе фаз. Очевидно, что в случае равновесия давление и температура всюду должны быть одинаковы (это предполагает отсутствие каких-либо специальных
Уравнения равновесия для любого числа компонентов и фаз
161
мембран, которые могут допустить существование различных давлений), н поэтому для этих параметров штрихи были опущены. Если Z представляет общий изобарный потенциал всей многофазной системы, то
z=Z'4-z'+z"' + ... + zz.
(27)
Диференцирование дает
dL — <ZZ' + <7Z' + rfZ"'+ ...-4- rfZz. (28)
Сочетая уравнения (26) и (28) и применяя критерий равновесия, выраженный уравнением (25), мы приходим к зависимости
Р ’а^Л + V-'b^B + . . . + Р'Л +
рж+и^мв+... +
pzrfA^4-pzrfA^4-... +ц^/Л^=0. (29)
Так как общая масса каждого компонента остается неизменной (мы рассматриваем замкнутую систему), то
dN’A-\-dN”A-\-dN,"-\-dNzA = Q. (30)
Подобное уравнение справедливо для каждого из остальных компонентов. Для удовлетворения уравнений (29) и (30) необходимо и до-статочнЬ, чтобы соблюдались следующие условия:
Рл=Р4=Рл=---=РЬ
Рд = Рд ==Рв== • • • ==Рд>
Рдг — Pv •••—Р^- (31)
Другими словами, необходимым условием равновесия является равенство химических потенциалов каждого данного компонента во всех фазах. Это указывает на то, что химический потенциал является видом движущей силы для передачи массы между фазами или между частями данной фазы (гомогенное равновесие), и равновесие достигается тогда, когда разность потенциалов для каждого данного компонента уменьшается до нуля.
При рассмотрении равновесия в одной фазе (гомогенное равновесие) применение критерия, выраженного уравнением (25), к одному из уравнений (26) дает
рЛ+рв^вЧ-...+рл^=о, (32)
что является основным уравнением для этого случая.
11 Химическая термодинамика
162
ZF. Общие уравнения равновесия
Для получения серии уравнений (31) специально рассмотрим случай, когда все компоненты находятся во всех фазах. Однако часто этого не бывает, и уравнения (31) потребуют в этих случаях небольшого видоизменения, характер которого совершенно очевиден. Например, если компонент А присутствует в фазе ['] и отсутствует в фазе [*], то уравнение р/4 = р." следует выпустить из серии уравнений и подобным же образом поступить во всех случаях, когда какой-нибудь компонент не присутствует в какой-либо фазе.
Уравнения (31) и (32), выведенные в этой общей форме Гиббсом, образуют основу для всех выводов, имеющих отношение к равновесию в химических системах. Их дальнейшее развитие, необходимое для применения к специальным случаям, заключается в выражении различных р. через параметры состояния, например давление, температуру и состав.
Связь летучести и активности с химическим потенциалом. Многие авторы предпочитают рассматривать химическое равновесие с помощью специальных функций, введенных Льюисом [149], а не применять непосредственно химических потенциалов, и поэтому представляется желательным несколько отвлечься для установления соотношений между ними.
В гл. Ш были определены функции — летучесть и активность — для одного компонента. Летучесть любого компонента раствора fi можно определить уравнением
dp.~RTd\afi- s (33)
Интегрирование между двумя изотермическими состояниями, как и в случае одного компонента, дает
р,. — р’ = /?Пп4> (34)
h
где pj и f* представляют соответственно химический потенциал и летучесть компонента I в некотором произвольно выбранном начальном стандартном состоянии в отношении давления и концентрации. Следовательно, они являются функциями одной только температуры. Например, пусть рассматриваемая система будет смесью газов при некотором высоком давлении р и температуре Т и пусть х(. будет мольной долей какого-то компонента I раствора. Теперь представим, что система изотермически расширяется до некоторого более низкого давления р°, при котором газовая смесь может считаться идеальным газом. По аналогии с определением летучести чистого компонента можно дать определение летучести компонента смеси, приравняв ее
☆) В тех случаях, когда необходимо отличать летучесть или активность компонента раствора от тех же функций чистого компонента, мы будем для случая компонента раствора ставить над символом черту.
Уравнения равновесия для любого числа компонентов и фаз
163
к парциальному давлению данного компонента в условиях, когда смесь расширена до состояния идеального газа, т. е. й)
= (35)
Учитывая уравнения, которые будут выведены в этой же главе несколько дальше, напишем
р.° = КТ\пх{-\-7°. (79)
Вводя уравнения (35) и (79) в уравнение (34), получим следующую зависимость между летучестью и химическим потенциалом:
RT\np°, (36)
где Zf— мольный изобарный потенциал чистого компонента i при давлении
Если рассматриваемая система является жидким раствором, все же можно представить раствор изотермически испарившимся и расширившимся до состояния идеального газа и приложить к нему те же уравнения. Даже имея твердые тела, можно применить те же самые стандартные состояния, так как любое твердое тело можно рассматривать как обладающее некоторым давлением насыщенного пара, и необходимо лишь принять давление р° достаточно низким, для того чтобы превратить твердое тело в идеальный газ. Обычно в случае растворов, содержащих жидкости и твердые тела, удобнее бывает выбрать другие стандартные состояния, что будет рассмотрено ниже. 11
Активность компонента в растворе можно определить уравнением
~at = h (37)
Ji или уравнением
— p.'i = RT\nat, (38)
которое является сочетанием уравнений (34) и (37). Следует отметить, что активность в стандартном состоянии всегда равна единице. Главной причиной введения функции активности является то, что в некоторых случаях можно определить отношение летучестей, когда нельзя найти числового значения ни для сдной из них; поэтому представляется удобным присвоить этому отношению определенное название.
£?) Штрих над f{ опускается, так как летучесть компонента в смеси идеальных газов прй давлении та же, что и летучесть чистого компонента при давлении Xjjfi. Это следует из понятия идеального газа. 11*
164
IV. Общие уравнения равновесия
При подстановке уравнений (36) и (38) в уравнение (32) получим для гомогенного равновесия следующие уравнения:
(RT\nfA-RT\nр«Za) dNA-\-(RTlnfB—RT]ap°-\-ZB) dNB-\-...
‘ ...+(/?ЛпЛу-RTln p° + Z°N)dNN=0, (39)
(RT In ал -j- Цд) dNA -(- (RT In aB p.B) d^!B~]~ • • •
Ч- • • • -J- (RT In Hjy) dNN 0. (40)
Если стандартным состоянием является какое-либо состояние чистого вещества, то ясно, что
где Z(— мольный изобарный потенциал чистого компонента I, и в этом случае можно подставить Z°. вместо р°. в уравнения (38) и (40).
Подстановка уравнения (36) в уравнение (31) дает для фазового равновесия
fA=fA=--=fZN HD
с подобными же уравнениями для других компонентов. Иначе говоря, согласно уравнению (41) условием равновесия является равенство летучести каждого данного компонента во всех фазах. Так как актив-. ности пропорциональны летучестям или химическим потенциалам, то очевидно, что активности данного компонента при равновесии не обязательно равны во всех фазах, и только в тех случаях, когда для данного компонента выбрано одно стандартное состояние для всех , фаз, активности будут равны, и можно написать уравнение (41), заменив летучести активностями.
ИЗОБАРНЫЙ ПОТЕНЦИАЛ ГОМОГЕННОГО РАСТВОРА
Следующим этапом вывода общих уравнений равновесия будет выражение изобарного потенциала гомогенного раствора с помощью параметров состояния. Теперь мы покажем метод, посредством которого это можно осуществить.
Для мольного изобарного потенциала любой фазы при постоянных температуре и составе из уравнения (26) имеем
(IZ. "Ud.p
ИЛИ
р
Z=§vdp-^-Z°. (42)
р*
Как и раньше, р0 является давлением, достаточно низким для того, чтобы вся фаза, независимо от того, являлась ли она вначале газом,
Изобарный потенциал гомогенного раствора
165
жидкостью или твердым телом, при этом давлении превратилась в смесь идеальных газов.
Определенный интеграл в уравнении (42) представляет площадь под кривой v — р, так как данная фаза расширяется при постоянной температуре от давления р до р°. В том случае, когда исходная система представляет собой газовую смесь, интегрирование можно произвести алгебраически, если зависимость между v и р можно выразить с помощью уравнения состояния. В случае исходного твердого или жидкого раствора объем не является непрерывной функцией р. Разрыв происходит в той точке, где впервые появляется газовая фаза, и в точке, где исчезают жидкая или твердая фаза; тем не менее и в этом случае интегрирование может быть осуществлено графически.
Теперь определим Z0 — изобарный потенциал смеси идеальных газов. Для внутренней энергии любой гомогенной фазы можно написать
U— иАХА + • • • + (43)
где U—мольная энергия всей фазы; UA, UB и т. д. — мольные энергии компонентов А, В и т. д. в чистом состоянии; Д(7 — изменение энергии, получающееся от смешивания при постоянной температуре и общем давлении. Это уравнение не основывается ни на чем другом, кроме первого закона и того обстоятельства, что U является экстенсивным свойством. При смешении идеальных газов при постоянной температуре из самого определения идеального газа очевидно, что Д77=О (см. также гл. V). Для идеального газа справедливо также уравнение
v = vaxa-\tvbxb+ (92, гл. V)
где v—мольный объем смеси, vA, vB и т. д. — мольные объемы чистых компонентов при давлении и температуре смеси. Умножая обе части этого уравнения на общее давление, получаем
pv = pvAxA + pvBxB+ •+pvNxN- (44)
Складывая уравнения (43) и (44) и принимая Д77=О, находим
U + pv=Wа + PVA) ха + (ив + Pvb) хв Ч~ - • • + (UN + pvN) xN (45) или
//= НАх^ 4- Нвхв-\- . .. Н^х№ (46)
т. е.
Н= 2 H.Xi. (47)
Все Н отдельных компонентов берутся при температуре и давлении смеси. По аналогии с уравнением (43),
5 = 5лхл-)-5вхв4- ... -j-SjyXjy+AS. (48)
166
IV. Общие уравнения равновесия
Из природы энтропии ясно, что AS вообще ие будет равно нулю при смешении идеальных газов при постоянных давлении и температуре.
Как уже было выяснено в гл. 1, непосредственное смешивание газов является необратимым процессом. Для того, чтобы найти изменение энтропии при смешении, необходимо применить обратимый процесс, и тогда будет приложимо уравнение
2
= (33, гл. Ill)
1
где 1 относится к исходному состоянию (до смешения), а 2—к конечному состоянию (после смешения). Такой процесс, связанный с применением полупроницаемой мембраны, уже был разобран в гл. I, и анализ его здесь повторяться не будет. Для этого специального случая камера смешения будет иметь столько поршней и цилиндров, сколько газов следует смешать. Максимальная работа, производимая каждым газом при изотермическом расширении от исходного давления р до его парциального давления в камере смешения, равна
о*
Af = J pdvit
V
где »* — объем газа, когда он находится при этом парциальном давлении. Интегрируя, получаем
A =N.RT In-
= — N.RT\n^ р
= —NiRT\n/-*<
= — N{RT In xt. (49)
Общая работа для всех газов равна
2 А= —/?Т(^1пхд + ^1пхв4- ... -|-^1пхЛ), или на моль смеси
yiA=~RTyixi\axl. (50)
Механическая работа проталкивания всех газов в камеру смешения, после которой происходит расширение газов до их парциального давления, точно уравновешивается работой противоположного знака, производимой при выталкивании поршня общим давлением; следовательно, эти работы взаимно компенсируются и при алгебраическом суммировании всех работ могут быть опущены.
Изобарный потенциал гомогенного раствора
167
По первому закону
£X = 2Q — дц
и так как для всех изотермических изменений идеальных газов kU=O, то
2JQ=-/?7-S^-l°xr (51)
При использовании уравнения (33, гл. III) получаем
Д3 = — /?3х,1п*г (52)
Это уравнение представляет возрастание энтропии в том случае, когда 1 моль смесн получается из чистых идеальных газов при постоянной температуре и постоянном общем давлении. При сочетании уравнений (52) и (48) энтропия идеальной газовой смеси дается уравнением
5 = SAxA + SgXg 4-... 4- S„xR (xA In xA 4- xB In xB 4"
4-... 4-x^nx^),
илн в сокращенном виде
S — ^x{S{ — R^XjlnXf. (53)
Теперь, так как
Z = H—TS, то Z°=//) —KS°
и
= S xrf — Г£х^ +RT^ Xt In (54)
или
Z* = 5Xzi 4- RT S In xt. (55)
Наконец, подставляя уравнение (55) в (42), приходим к искомому выражению для мольного изобарного потенциала любой гомогенной фазы:
е
Z~ vdp-\-'^ixiZ'i RT^Xf In х, (56)
р"
Отметим, что при постоянной температуре j vdp = <o(p) — <р(рв), р“
и так как мы допускаем идеальность газов при р°, то
<р(р°)=/?7’ 1пр°;
поэтому
^vdp=^vdp—RTlnp?, (57)
р“
168
tV. Общие уравнения равновесия
и уравнение (56) можно представить в форме
Z = j vdp + 2 -j- RT J] xt In x{ — RT In pP. (58)
Следует отметить, что неопределенный интеграл берется вдоль изотермы.
ЭНЕРГИЯ, ЭНТАЛЬПИЯ И ЭНТРОПИЯ ГАЗОВОЙ СМЕСИ
Прежде чем продолжать развитие уравнений равновесия, мы несколько отвлечемся для обобщения некоторых уравнений гл. III, с тем чтобы их можно было применить к газовым смесям. Любое из этих уравнений, как уже было установлено, так же хорошо применимо к смеси, как и к чистому газу, при условии, что рассматривается смесь постоянного состава. Сейчас нашей целью является введение состава системы в качестве переменной.
Интегрирование уравнения энергии
(»)г=7'(эт),-/’ (78, гл. Ш)
по изотерме между объемом v и очень большим объемом ©со, при котором газы являются идеальными, дает
V
и-и°= J [г (g)„ dv. (59)
Так как СТ® представляет энергию смеси идеальных газов, то можно написать [см. уравнение (43)]
+^в +... = (во)
и для каждого отдельного газа
т
и°1 = ^с'о ат + и\, (61)
где То—произвольная стандартная температура и —энергия любого отдельного газа при этой температуре и низком давлении, a Ua, Uв — мольные энергии при температуре и мольном объеме смеси.
Сочетание уравнений (59), (60) и (61) дает общее выражение для энергии любой газовой смеси, которое можно интегрировать с помощью уравнения состояния (или р — v—Т данных) для смеси, зная изобарные теплоемкости при низком давлении.
Энергия,- энтальпия и энтропия газовой смеси
169
Совершенно аналогичным способом можно найти соответствующие уравнения для энтальпии и энтропии путем сочетания следующих уравнений:
«=«"+И’-т® »]*• ,б2>
р"
H0 = ^Xitri, (47)
= + (63)
р
•s=s’-f &),,“? <«>
р"
S° = ^xl^i — R^xl\nxl, (53)
Т
s'{=\ C'pdXnT + ^r (65)
Сочетая уравнения (64), (53) и (65), получаем уравнение
р т
S=— J (^\ dp — R^xibxl-}-'^lxi^c!‘pd]nT-{-^lxl^t, р°Р Т,
где Soj — энтропия компонента смеси при условии, что чистый компонент взят при pQ и То.
Путем сложения и вычитания R]n(plp?) из уравнения (64) и сочетания его с уравнением (53) получается следующее уравнение (для изотермических состояний):
р
S=f + (66)
Эта форма имеет то преимущество, что весь интегральный член исчезает для системы, представляющей смесь идеальных газов и, будучи по природе поправочным членом, не требует очень точного определения. Это обстоятельство является особенностью, которую часто и с выгодой используют.
Для более строгих и полных выводов уравнений для расчета U, Н, S и Z газовых смесей и для доказательства, что нижним пределом может быть принят предел, когда р° приближается к 0, следует обратиться к статье Битти [10].
170
IV. Общие уравнения равновесия
ХИМИЧЕСКИЕ ПОТЕНЦИАЛЫ И ЛЕТУЧЕСТИ
Химический потенциал. Так как изобарный потенциал является экстенсивным свойством, можно применить к нему общие уравнения (16) и (17) и получить
<?Z _ dZ dZ dZ
dJVff A dxA 4 дхв dxN_x ’ ' '
7 . dZ dZ dZ dZ
~ Z + dxt ~~ xAdxA ~XN-1 (?Хлг_1 • (68)
С помощью уравнения (56) можно теперь получить выражения для различных коэфициентов этих уравнений.
При диференцировании уравнения (56) после записи его в развернутой форме получим
a (J »<//>)
= ~4--------\-RT\r^-\-^i — ^n,
dxt дХ[ 1 xN ' ‘
(69)
где XN= 1 — ХА — Хв— ... — xN_v р
Обозначая для краткости (1/7? Т) vdp через п0, имеем
р°
(70)
Уравнение (70) можно также ваписать в форме
% 4 s,*,+Rri"^+z;- 17,1
р*
Для бинарного раствора уравнение (70) переходит в
g. = RT(g + bi4-l)+Zi-zi. (72)
Если вместо уравнения (56) воспользоваться уравнением (58), то можно получить уравнение, по форме одинаковое с уравнением (70), но с п вместо п0, где п = (1/7?7’) J vdp. С помощью уравнения (58) и уравнения, аналогичного уравнению (70) с п, уравнения (67) и (68) можно развить далее; это дает соответственно
• — хлг-1 +
4- RT in xt 4- Zj — RT lnp°
(73)
Химические потенциалы и летучести
171
и гл *Т' | Лс
Ц. ., = /</ lit Х.-л-------ХдЗ
\ АдхЛ вдхв
-xv-1^)+^7'lnxJV+
-{-Z‘N—RT\n р9.
(74)
Уравнения, выраженные через п, будут одинаковы с этими двумя уравнениями, если опустить член ЯТЧпр0. Для специального случая бинарных растворов уравнения (73) и (74) переходят в
Ид = /?т[п+(1— x)g] +/?Tlnx+Z;— RT\np9, (75) p.B — RT (n — +/?Tln(l — x) +Zb — RTlnpP. (76)
Соответствующие уравнения для цг и в несколько более простой форме можно получить из уравнения, аналогичного уравнению (56), для общего изобарного потенциала молей любой фаз'ы. Умножая обе части этого уравнения на и учитывая, что xl = Nil'^N{, получаем р
Z = \vdP + ^Ntz; -t-RT^h^. (77)
р°
Так как
dZ dN, ’
то при диференцировании уравнения (77) имеем
л
Hz = J ^р + RT In x^zi, p*
(78)
и уравнение для Рд, будет иметь совершенно уменьшается и приближается к предельному газы считаются идеальными, уравнение (78) щее уравнение:
pz = /?Tlnxz
такую же форму. Если р значению р9, когда все превращается в следую-
(79)
которое является специальной формой уравнения (78), применимой к идеальным газовым смесям.
Эквивалентность уравнения (78) и уравнения, аналогичного (73), выраженного через п0, легко показать следующим образом. Из уравнения (17) очевидно, что
—__ dV__ । до до до
i~dty-~v'~dxl Хлдх^~Хвдхв
до
(80)
172
/V. Общие уравнения равновесия
Уравнение (78) можно написать так:
+ 7?7'lnx.+Zz.
Так как
р
7?7тг0 = ^ vdp ро
и
~XN~xd^~^ dpJf
(81)
(82)
(83)
р
то уравнение (81) легко превращается в уравнение (73), выраженное через тг0.
Летучесть и активность. Из уравнений (36) и (73) имеем
, -г 1 • । I дч дч дк дк
ln/т— lnXf + ’t + ^~ ХА^~ ХВд^~~ XN-tdX^~[- (84)
Из уравнений (36) и (74) получается соответствующее уравнение для ln/лг
Из уравнений (36) и (78) получаем
р
1п^ = £?• J dP +1п xt + 1пЛ (8§)
. р*
Уравнения (84) и (85) являются просто двумя формами одного и того же уравнения. Можно получить и другие формы, которые могут оказаться полезными в отдельных случаях. Например, мы можем несколько сдвинуть нижний предел интеграла уравнения (85), что и было сделано в случае уравнения (56).
Так как газ при давлении рй является идеальным, то дУ _ _RT dNi —Ч—р» ’
и функция нижнего предела сводится к —1п/>°, и уравнение (83) превращается в
Для чистого компонента i при давлении р и температуре Т р
In А = f ^dp. (140, гл. IIJ)
Химические потенциалы и летучести
173
Сочетая уравнение (140, гл. III) с уравнением (85) и учитывая, что ft
— р, получаем
р
In 7Z = -g? [ (Oi — v{) dp 4- in ft 4- In x.. (87)
. p°
Уравнение (85) также легко превращается в
р
1п Л = J ) dP + ln Pxi- (88)
рО
Уравнение (88) для летучести компонента смеси аналогично уравнению (142, гл. III) для летучести чистого вещества; в сущности оно превращается в уравнение (142, гл. 111) в пределе, когда х — 1. И уравнение (87) и уравнение (88) представляют весьма удовлетворительный путь для определения летучести компонента смеси, так как величины — v.) t— pj- \
и -------—j, как уже было показано [88], приближаются к ко-
нечным значениям, когда р стремится к нулю, подобно функции а, применяемой для определения летучести чистого вещества. Они не стремятся к нулю, как к пределу, что иногда ошибочно допускают. Это можно показать или путем экстраполяции существующих данных по сжимаемости газовых смесей, или с помощью уравнения состояния для смесей.
Из уравнения (84) или (85) или других форм этих уравнений легко заметить, что для случая, когда xt =1,
ln/z = irz или
= (89 е)
Уравнения для активности, соответствующие различным уравнениям для летучести, можно получить или из уравнения (89) и уравнения (37), или непосредственно из уравнения (38). Например, уравнением, отвечающим уравнению (85), является
р
1п а/ = -Бг f VidP- (90)
Ki J
PQ
Замечание о стандартных состояниях. Кажется очевидным, что можно было бы избежать недоразумений в том случае, если для одного вещества всегда придерживаться одного стандартного состояния и никогда не пользоваться каким-либо другим стандартным состоянием. Наиболее естественным стандартным состоянием было бы состояние чистого вещества при данной температуре и р — 1.
174
IV. Общие уравнения равновесия
К сожалению, оттого что для некоторых систем доступны только весьма ограниченные данные, из-за чего невозможно приблизиться к пределу, отвечающему чистому состоянию компонента, необходимо избирать другие стандартные состояния, и это иногда является источником большого числа недоразумений. Например, при работе с относительно разбавленными спиртовыми растворами при повышенных температурах недостаточно данных для перехода от данной концентрации к чистому спирту при той же самой температуре. В таком случае обычно основывают активность на экстраполяции до другого предела, именно — до бесконечно разбавленного раствора. Следовательно, можно закончить определение активности, частично данное уравнением (38), написав
lim 4=1. (91)
х -»0 •*
Вместо х — мольной доли — можно также пользоваться мольностью т или мольной концентрацией.
Из уравнения (37) очевидно, что активность в стандартном состоянии равна единице, а вследствие этого стандартное состояние, принятое при определении, основанном на уравнении (91), очевидно, не является состоянием бесконечного разбавления. Для уяснения этого обстоятельства возьмем простой частный случай, именно — летучее растворенное вещество в разбавленном растворе, подчиняющееся закону Генри, и примем также, что пар является идеальным газом. Тогда, так как для идеального газа летучесть равна давлению, то
(92)
Для бесконечно разбавленного раствора
— р00
ас°=уГ’ (93)
но по уравнению (91) с00 = х00; поэтому
; (94)
рсо
но по закону Генри
р СО и поэтому
a=kp. (95)
Поскольку в стандартном состоянии а=1, это состояние является единственным, для которого парциальное давление . = 1/&.
Химические потенциалы и летучести
175
Раствор этилового спирта в воде, в котором мольная доля спирта равна 0,0177, имеет парциальное давление пара С2Н5ОН, равное 3,65 мм при 25° С, и точно следует закону Генри. Поэтому
, 0,0177 1
k = „ gg- или -г = 20b мм.
о,65 к
Давление пара чистого C2HgOH при 25° С = 59 мм, и поэтому оказывается, что состояние пара при 206 мм является фиктивным ☆ ).
Так как для любого раствора до тех пор, пока он подчиняется закону Генри, xjp = k, то из этого следует, что до того же самого предела а — х (нли а = т, если концентрация выражена через мольность). Тот факт, что все сводится к простому соотношению, является одной из главных причин для определения стандартного состояния на основе уравнения (91). Можно было бы избежать фиктивных стандартных состояний, приблизив а‘[х к 100, а не к 1,0, как в уравнении (91), но все же лучше было бы удовлетвориться выбором стандартного состояния при некоторой низкой концентрации, например х=0,01 или х = 0,001, при котором закон Генри можно считать достаточно точным для практических целей.
Адамс [1] предложил соединить уравнения (38) и (91), дав следующее единственное уравнение, полностью определяющее активность недиссоциирующих растворенных веществ:
а, = lira —--------7^=.,
Е1)/ЛГ
(96)
где —активность любого растворенного вещества при данной концентрации Xj, когда химический потенциал равен jir Взяв ряд концентраций х, которым соответствуют потенциалы р, определив (ц— и найдя предел (аналитическим или графическим способом), получаем значение av Для дальнейших подробностей и для рассмотрения вопроса о системах, содержащих диссоциирующие растворенные вещества, см. статью Адамса.
Зависимость химического потенциала, летучести и активности от температуры, давления и состава. Изменение химического потенциала с составом легко получается при диференцировании уравнения (78).
Это приводит к уравнению
р°
й) Если раствор идеальный, то стандартное состояние совпадает с состоянием чистого компонента.
176
IV. Общие уравнения равновесия
Другая форма получается при диференцировании уравнения (81):
Так как
или
то
д(П\
\dNt)P, т
др )т,х
др
л(д7Л
/ Т, х dNt
(98)
д^Л _dV__~ др )т,х dNi Vi‘
(99)
Это уравнение также следует непосредственно из уравнения (78) при диференцировании его по р.
Подобным же образом, так как dZjdT=—S, то ______________________<^Z _ dS \дТ )р, х dN(dT dNi
Далее, так как Z — Н — 7S, то при диференцировании получим ®=*=й,-гё,.
Сочетая с уравнением (100), находим
^ = 1(HZ-Hz).
Sz.
(100)
(101)
Из соотношения между летучестью и химическим потенциалом, выраженным уравнением (36), легко заметить, что
(£) =RTd-^, (102)
= RTd-^l, (103)
\др )т,х др и
{df)p,x=RT^ + Ria7+^~Riaffi- (104)
Из уравнений (102) и (97) вытекает, что
Химические потенциалы и летучести
177
Из уравнений (103) и (99) очевидно, что
Из (104) и (100) и из того обстоятельства, что-^- =— 5°, следует, что
= —Sf —Rin Л + +К1П/Л (107)
Но так как Z H — TS, то
Z = p. = H— TS. (108)
Уравнение (107) переходит в уравнение
RTd-~jll = ^ — %- — Rin/ — y+^ + Rlnp8.
Но по уравнению (36)
f — R 1п/ — + R In р* = 0,
поэтому
д 1п // Hi — H“/
(109)
Диференцируя уравнение (38) и сочетая с уравнением (100), получаем аналогичное уравнение для изменения активности с температурой, а именно
<?1па/ Н/ — H*i
(ИО)
Уравнения для изменения активности с изменением р и х те же самые, что и соответствующие уравнения для летучести, так как летучесть в стандартном состоянии является функцией только температуры. Однако температурный коэфициент несколько отличается, как это только что было показано.
Уравнение (ПО) понятно, когда стандартным состоянием является состояниечистого компонента и Н^ = Н°(, но если стандартное состояние определяется по уравнению (91), то диференцирование становится неопределенным, и смысл Н° не ясен без дальнейшего рассмотрения. Таким образом, не только стандартное состояние является фиктивным, но фиктивные концентрации различны для каждой температуры, и поэтому смысл д]з.]дТ при постоянной концентрации, конечно, не ясен. Если стандартное состояние определяется некоторой произвольно 12 Химическая термодинамика
178
IV. Общие уравнения равновесия
выбранной концентрацией (см. стр. 175) для всех температур, тогда /// является парциальной мольной энтальпией при этой концентрации. С другой стороны, если а определяется по уравнению (96), то по-прежнему получается уравнение (ПО), и Л? относится к предельному значению, приближающемуся к Н., когда х стремится к нулю.
Интересно отметить, что уравнение (ПО) можно получить непосредственно также из уравнения (96) путем диференцирования его.
ИДЕАЛЬНЫЕ РАСТВОРЫ
Если уравнение (8), относящееся к любому экстенсивному свойству, применить к объему, то можно написать, что
v = vaxa + vbxb+ +‘йлхг О’*)
Вообще парциальные мольные объемы являются функциями состава раствора; но в некоторых частных случаях изменение их с составом незначительно, и для простоты можно установить особый тип раствора, в котором эти величины остаются постоянными независимо от состава. Такие растворы известны как идеальные. Понятие идеального раствора, подобно понятию идеального газа, представляет предельный случай, к которому все реальные растворы могут только в большей или меньшей степени приближаться в зависимости от условий.
Если различные компоненты идеального раствора устойчивы в чистом состоянии при температуре и давлении раствора, то
где v. — мольный объем чистого компонента i при р и Т. В этом случае уравнение (111) переходит в
V = ^AXA-YVBXB+’--+VNXrA (*12)
т. е. объем раствора аддитивно складывается из объемов компонентов. Следовательно, уравнение (П2) можно применять к смеси газов, например азота и водорода, при комнатной температуре и любом давлении, но нельзя применить к смеси водорода и аммиака при любом давлении, превышающем давление насыщенного пара аммиака при рассматриваемой температуре. Обобщая, можно сказать, что уравнение (112) применимо к любой газовой смеси при любом давлении при условии, что данная температура смеси выше критической температуры всех компонентов. Если температура ниже критической температуры какого-либо компонента, то уравнение (112) приложимо- только до давления пара этого компонента. Оно прило-жимб и к жидкой смеси только тогда, когда температура ниже тем-
Идеальные растворы
179
пературы кипения (при рассматриваемом давлении) наиболее летучего компонента, так как при более высокой температуре этот компонент не может существовать как жидкость в свободном состоянии при давлении смеси. Очевидно, уравнение (112) никогда нельзя применить точно к случаю фазового равновесия жидкость — пар, поскольку всегда для какого-нибудь компонента будет нарушаться какое-нибудь из этих условий. Следовательно, для бинарного равновесия жидкость — пар каждый компонент по необходимости устойчив только в одной из фаз, находясь в чистом состоянии при давлении и температуре смеси. В частном случае равновесия жидкость — пар при температуре ниже критической температуры всех компонентов можно определить идеальный жидкий раствор как такой, для которого уравнение (112) приложимо тогда, когда vA, vB и т. д. принимаются равными объемам чистых компонентов при температуре раствора, но каждый при своем соответствующем давлении пара. Это допустимо вследствие очень малого влияния давления на объем жидкости. Ясно, что подобное определение для газовых растворов было бы абсурдным.
Для специального случая идеальных растворов, когда рассматриваемый компонент устойчив в данной фазе при давлении и температуре раствора, уравнение (78) переходит в
р
р,- — J vtdp -|- RT In xt
р* но так как
р
<\jvidp = Zi — Z''t, р*
р. = 7.-}-7?Ппх,., (113)
и уравнение (87) переходит в
In ft = 1п/г -|- In хр (114)
или
Л=/Л- (115)
Эта простая зависимость широко применяется, но всегда следует помнить, что она приложима лишь тогда, когда объемы аддитивны и когда компоненты устойчивы в чистом состоянии при давлении и температуре смеси. Она аналогична соотношению, обычно называемому „законом Рауля*, а именно pi = xlpi, которое применимо для идеальных жидких растворов, когда пар является идеальным газом.
Из уравнений (115), (109) и (146, гл. III) находим, что — и отсюда
"=ЯЛхЛ4-ЯвХв+.... ’ (Н6)
12*
180
IV. Общие уравнения равновесия
Другими словами, идеальный раствор является раствором, для которого теплоты смешения равны нулю, поскольку энтальпии аддитивны.
Из уравнений (38) и (113) легко видеть, что для идеального раствора
аг=кх{, (117)
и если стандартным состоянием, на котором основывается активность, является состояние чистого компонента при той же температуре и общем давлении, то k= 1. Это употребляется обычно лишь для жидких растворов, когда изменение активности с давлением невелико и им можно пренебречь. Отклонение ajx от единицы при уменьшении х от 1,0 является удобной мерой отклонения раствора от идеальности. Поэтому некоторые авторы применяют уравнение (117) в качестве определения идеального раствора.
Из уравнения (111), в случае, когда xN исключается посредством выражения через другие мольные доли, имеем
("»>
и из уравнения (112) следует, что
Д = (119»
Из уравнений (83) и (119) получаем р
Й = <,2°’
р*
с подобным же уравнением для дтЦдх. При диференцировании уравнения (120) и соответствующего уравнения, выраженного через тг, имеем
= (121)
дх\ дх]
СПЕЦИАЛЬНЫЕ СЛУЧАИ ФАЗОВОГО РАВНОВЕСИЯ
Правило фаз. Уравнения (31) определяют общие условия равновесия в системе, состоящей из N компонентов и Z фаз, при условии, что каждый компонент присутствует во всех фазах. Для каждой фазы в соответствии с уравнением (26) на первый взгляд кажется, что имеется (?/-|~2) независимых переменных; ио если все мольные массы компонентов увеличатся в равной степени, то не произойдет ни изменения состава, ни изменения состояния, а только изменение общего количества. Ясно, что для изменения состояния фазы должны изменяться отношения масс, т. е. составы, и будет (N—1) независимых отношений масс или переменных состава. Поэтому общее число
Специальные случаи фазового равновесия
181
параметров, определяющих состояние всей системы из N компонентов, находящихся в Z фазах, равно Z(N—1)-}-2. Уравнения равновесия (31) налагают (Z—1) условий, которым должен удовлетворять каждый данный компонент, а для системы в целом (для W компонентов) это число условий будет равно N (Z—1). Общее число параметров, за вычетом общего числа условий, налагаемых уравнениями, равно D—степеням свободы системы, т. е.
Z{N— 1)4-2 — N{Z— !) = £>, откуда
D = N — Z-4-2. (122)
Это и является правилом фаз, впервые установленным Гиббсом в 1875 г. Данный нами вывод совпадает в принципе с выводом Гиббса, но мы ради простоты до некоторой степени пожертвовали точностью.
Если в какой-нибудь фазе отсутствует какой-либо компонент, то в этой фазе будет меньше переменных состава, а следовательно, и одним условием меньше, так что правило остается действительным для всех частных случаев этого типа.
Диференциальные уравнения для двухкомпонентных двухфазных систем. Наше дальнейшее обсуждение фазового равновесия ограничится системами, наиболее сложные из которых будут состоять из двух компонентов и двух фаз. Поэтому и дальше мы будем выводить диференциальные уравнения, имеющие отношение к этому случаю, для того чтобы они могли служить исходным пунктом для дальнейшего перехода к специальным задачам. Для этого случая общее условие равновесия, выраженное уравнением (31), переходит
В = (123)
= (124)
Пусть А будет компонентом, мольная доля которого равна х. Из
того обстоятельства, что д является функцией р, Т и х, имеем
+ + + (125)
Подставляя значения коэфициентов, даваемые уравнениями (97), (99) и (100), можно получить общее диференциальное уравнение, связывающее давление, температуру и состав обеих фаз в двухкомпонентной двухфазной системе, а именно:
(J dp + dx' — v'Adp — $ ’А<1Т~
p9
=(Jdp+5)dx"+”Adp—s"AdT- (i26>
182
IV. Общие уравнения равновесия
Этому уравнению иногда даются и другие формы, отличающиеся от данной. Например, иная форма получается при применении уравнений (98) и (17). Также и при использовании уравнения (101) можно сочетать члены с dT для получения выражения
Полезная форма уравнения (126), включающая диференциал состава
одной из фаз, получается следующим образом:
Из уравнений (67) и (68) получаем соответственно
_ dZ V-B~Z х dx (127)
И
^=Z+(l-x)g. (128)
Затем, так как
Ив = Ив» (129)
Ил=и;, (130)
то
7» -х’ I z х z х ~дх” (131)
и
z-+(i-ng=z-+(i-x-)g. (132)
Сравнивая уравнения (131) и (132), получаем
dZ" dZ*
bx? дх*' (133)
Диференцируя уравнение (131), находим
,.dZ' dZ’ , ..dZ” dZ" . . dZ xd dx> dxfdx —d^ ^d~3x” dx”dx?’ (134)
Пользуясь уравнением (133) и тем, что
dZ=vdp—SdT 4- g dx, (135)
уравнение (134) можно написать в форме
(Vя — v')dp — (S’ —S’) dr—(x" — x')d^ = 0. (136)
Подстановка уравнения
.dZ'__&Z' &Z' . d*Z’ , d dx? dx'dp dp + dx'dT dT^ dx'i dx (137)
Специальные случаи фазового равновесия
183
в уравнение (136) дает искомое уравнение, именно ।
[v’ — v> _ (Х" _ x')^j dp — —
— [s' —S’ — (x” — x')g] dT — {x" — x')^f2dx'^=0. (138)--------
Если исключить из уравнения (134) dZ'[dx' вместо dZ'Idx", то по-лучим аналогичное уравнение для другой фазы:
(х'-г') др]
_[^__^_(Лл_х')^;]йГГ-(х'—x')gdr' = 0. (139) 1
Выражение v"— v' — (х"— х') (dv'/dx') имеет простой физический смысл; оно представляет изменение объема, сопровождающее конденсацию 1 моля второй фазы ["] в таком большом количестве первой фазы ['], что состав последней не изменяется заметным образом. > Это можно доказать следующим путем. При добавлении 1 моля второй фазы к N молям первой, мольный объем последней будет стремиться измениться вследствие изменения состава, и после окончания процесса объем первой фазы можно представить выражением
w'-f-^Ax'-j- ....
1 дх' 1
Если мы примем N очень большим по сравнению с 1, то высшими членами этого многочлена можно пренебречь.
Ах' = конечному составу первой фазы минус начальный состав ее =
Wx'-j-x’ Xя — х* — JV-J-1 Х ~ W4-1 ' , Поэтому
конечный мольный объем первой фазы=г>'
х” — х’ до'
n-4-Г а?
и общий объем ее —(А/-|-1) v'-4-(x" — Х')^р.
Так как вторая фаза полностью конденсируется, то эта величина является также и конечным общим объемом.
Первоначальный общий объем
Следовательно, Ат», т. е. общее изменение объема в процессе, выражается соотношением
Av = (N -4- 1) v' -4- (х* — х') —
[v’~ Vr — (X* — х')^].
184
IV. Общие уравнения равновесия
Подобным же образом [5" — S' — {х” — х’) (dS’jdx')] равно —Д-S, т. е. изменению энтропии в том случае, когда 1 моль второй фазы конденсируется в большом (в пределе бесконечно большом) количестве первой фазы. Наоборот, Д5 является изменением энтропии в том случае, когда 1 моль фазы ["] испаряется из очень большого количества первой фазы, и 7'Д5 называется „диференциальной скрытой теплотой парообразования0, когда рассматриваемые фазы представляют жидкость и пар.
Из уравнения, аналогичного уравнению (72), выраженного с помощью тг, и уравнения (133) имеем
ЯТ + =RT
\дх' 1 1 — х'/ \<?х" 1 1 — Xе)
или
„ I дк’ Ля»
х — Х Удх' ~~ дх»
1— Xя 1—
Это является общим уравнением, непосредственно связывающим составы фаз, сосуществующих в равновесии.
Из уравнений (76) и (129) следует, что
7Г’ + 1п(1 -X') — Л'^ = 1Т’ + 1п(1— х’) — (141)
Сравнивая уравнения (140) и (141), получаем
х" = + О-*') gi -[*’ + О-»*) ^1 (142)
X*
Легко показать, что Л = (NA Ng) тг, т. е. что тг является экстенсивным свойством, и поэтому
— Лс I ,, . д*
djy: —Тс + О Х^дх'
Следовательно, уравнение (142) можно представить в форме
Следует заметить, что
г”
л___0*А~ М
X’
^A = ^f^AdP^‘
(143)
Специальные случаи фазового равновесия
185
Термодинамические методы определения фазового равновесия. Обычным методом экспериментального исследования фазового равновесия является приведение в соприкосновение двух фаз и проведение над ними определенных наблюдений в период их сосущест-
вования. Между различными методами имеется много отличнй в деталях,
но все зависит от установления [2J разработал косвенный метод, в котором примени- 3,
ются некоторые уравнения, §
представленные в предыду- §
щем разделе. Он является J
косвенным потому, что рав-новесие между фазами в нем с
не достигается. Этот метод з
имеет определенные преиму- «
щества, особенно в случае з
равновесия при высоких да-влениях. к
Рассмотрим случай рав-
равновесия между фазами. Адамс
t= const, х= const
Давление
новесия между твердой фа-
зой и жидким раствором и
Рис. 12. Косвенный метод определения
влияние давления на это
фазового равновесия.
равновесие при постоянных
температуре и составе раствора. Из уравнений (123) и (99) имеем
и
=
Отсюда
р —
= Ч- j vAdp. р°
Измеряя объем каждой фазы для ряда давлений и составов, можно определить кривую pi—р для этой фазы. Если нанести затем две кривые, то их пересечение в соответствии с уравнением (123) определит точку равновесия (см. рис. 12). Необходимо отметить, что неизменность твердой фазы на всем протяжении изменения давления должна быть установлена каким-нибудь другим способом. Например, в исследованном Адамсом случае взята система NaCI — Н2О, и стабильной твердой фазой при этих условиях был лед VI.
Уравнение Дюгема. Для бинарной системы уравнение (9), выражаемое через изобарный потенциал, переходит в
xdV-A = — О— xjdV-e- О44)
186
IV. Общие уравнения равновесия
Теперь, так как ,хл и цв можно выразить как функции Т и х (р является зависимой переменной), то при постоянной температуре
-‘>(3?)г <145>
или х/^) = (1—X) • (146)
\дх]т ' ’ [_<?(!—x)Jr
Из определения летучести и активности компонента легко найти, что
Э1п/д_ п dinfB (147)
х дх х)д(1 — х) '
И
<Ип«д п . ^1ПЯд (148)
х дх —I1 х> д{1 — X)' '
Уравнения (146), (147) и (148) являются формами уравнения, обычно называемого уравнением Дюгема или иногда уравнением Дюгема — Маргулеса. Уравнение (148) можно также написать в форме
dlnaA = — L=^dlnaB. (149)
Легко показать, что
dlnx ——l=^dln(l— х). (150)
Вычитая уравнение (150) из уравнения (149), приходим к
dln^= — (151)
т. е. к той форме уравнения Дюгема, которая оказалась очень удобной для графических расчетов. Уравнения (144—151) применимы к любой гомогенной системе или фазе.
Однокомпонентная система. Для этого случая х" = х', и уравнение (139) может быть представлено в следующем виде А):
(tf—v')dp = [Sr — S'}dT, (152)
или
й) Предполагая, что различные производные стремятся к ковечвому значению, когда х" и х' стремятся к единице.
Специальные случаи фазового равновесия
187
где X — скрытая теплота фазового превращения и Дп— изменение объема, сопровождающее фазовое превращение. Это очень важное уравнение, показывающее, как изменяется равновесное давление с температурой для любого фазового равновесия в однокомпонентной системе. Это уравнение известно как уравнение Клаузиуса — Клапейрона.
Здесь желательно несколько отклониться в сторону, чтобы показать, как можно очень простым способом непосредственно вывести уравнение (153). Общий изобарный потенциал системы из двух фаз представляет сумму изобарных потенциалов отдельных фаз, или
Z = N'Z' + MZ".
Необходимым условием для двух фаз, находящихся в равновесии при постоянных р и Т, является
dZ = 0. (25)
Единственным возможным изменением при постоянных р и Т является переход массы из одной фазы в другую. Пусть dN молей переходит из фазы ['] в фазу [’]; тогда изменение изобарного потенциала dZ равно
dZ = (Z' — Z')dN.
Применяя критерий равновесия, получаем
Z* = Z*.
Так как при изменении давления и температуры изобарный потенциал обеих фаз должен измениться таким образом, чтобы это равновесие сохранилось, то
dZ" = dZ’, но
dZ = д4- dp-4- ^.dT = vdp — SdT, op r 1 oT r
поэтому
v"dp — S’dT=v'dp—S'dT,
что совпадает с уравнением (152).
Раствор постоянного состава. При постоянном х’ уравнение (138) переходит в
dp___S"~S’ — (xr — x') (dS’ldx’}
dT v’ — v' — (x* — x7) (dv'/dx7) ’
или
rfp__
(154)
(155)
188
IV. Общие уравнения равновесия
где АН выражает диференциальную скрытую теплоту парообразования, если ["] обозначает парообразную фазу; Av—изменение объема.
При низких давлениях v' и dv’jdx’ ничтожно малы по сравнению с v", и тогда знаменатель превращается в if.
Если один из компонентов нелетуч, то х" — 1, и знаменатель превращается в
— p + U— = if — v‘,
а числитель становится равным
S’ — р4-(1—= S° — S'.
Разность энтропии, содержащуюся в правой части этого равенства при данных р и Т, можно определить соотношением
H’ — tf
и в результате получить
dp_ H’—Jf dT~ Т(if — t?)
(156)
Эго — специальная форма уравнения Клаузиуса — Клапейрона, связывающая температуру с давлением насыщенного пара растворителя в растворе, содержащем нелетучее растворенное вещество.
Фаза, состоящая из идеальных газов. Для этого случая
1 р
if dp — In 4 р° (157)
И л” Р дх"~~ Rt\ dx’dp~0. (158)
Г
и Эх1* и (159)
Подобно этому if = }пр (160)
и ^-=0 дх" (161)
Специальные случаи фазового равновесия
189
При диференцировании уравнения (72) получаем
&Z _ Г*«о I 1 1
дхг * [<**2’Г х(1— х)]’
которое для рассматриваемого случая переходит в
<PZ”__ RT
dx"i х’(1—х")'
Отметим, что
(162)
(163)
что v' по сравнению с v* можно пренебречь и что v" = ^. Р
Уравнение (139) превращается в
RTy~ [5* — S' — (x? — х')^] dT—RTJ£^r}dx”==Q. (164)
Если мы снова примем, что х"=х' (один компонент), то уравнение (164) превращается в
RTd In р = ASdT = у dT,
ИЛИ
(165)
— специальную, очень полезную форму уравнения Клаузиуса — Клапейрона.
Аналогичное уравнение можно получить для раствора постоянного состава. Так, уравнение (138) для случая идеальных газов и постоянного х' переходит в
RTd^= Is* — S' — (х’ — x')gl dT. (166)
р L ' ,дх’\
Выражение в скобках = где L — диференциальная скрытая теплота парообразования жидкого раствора. Поэтому
dlnp = ^rfT.
В случае постоянной температуры dT = 0 и уравнение (164) превращается в dlnp х" — х’
^F = x-(l^)- <167)
190
IV. Общие уравнения равновесия
Это — частная форма уравнения Дюгема.
По определению парциального давления, имеем
рЛ=х>,
Рв = ^—
Рл~\~Рв—Р'
При введении этих выражений в уравнение (167) его можно превратить в следующую форму:
_______ dlnpg Э1пх' д!п(1—х')'
(168)
Это уравнение также непосредственно вытекает из уравнения (147), поскольку f'i=fi=p{y когда парообразная фаза является идеальным газом.
При использовании уравнения (158) уравнение (140) превращается в
. , д*'
(169)
Введение уравнений (160) и (161) в (141) превращает его в уравнение
тс'4-1п(1— х') — х'^ = 1пр4-1п(1— х"), (170)
ИЛИ
J, д*’
р(1—х") = (1—x')Z Х дх’. (171)
Подстановкой (1—х')/(1—х*) из уравнения (169) получим
р^ = Х’еХ'+(1~Х'^'. (172)
Складывая уравнения (171) и (172), получаем
, .к д*' Эя*
рх"+р(1— х")—р = х'е е*' +(1— х')е <*'. (173)
Это уравнение связывает давление пара бинарного раствора с составом жидкой фазы при постоянной температуре, когда пар можно считать идеальным газом.
Если х'=1, то уравнение (173) переходит в
Специальные случаи фазового равновесия
191
где if, — значение тг' для чистого компонента А в жидкой фазе. Подобным же образом, если х' = 0, то
Р=Рв = е*В-
Используя эти соотношения, можно уравнение (173) представить в форме
р=рАх'е*'+ +рв(1— (174)
где рА и рв—давления насыщенного пара двух компонентов при данной температуре в чистом состоянии.
Идеальный газ во второй фазе и идеальный раствор в первой фазе. Для бинарного идеального раствора уравнение (118) переходит в
до — —
з- = v. — о дх А в
, дя 1 Г — —. .
поэтому ] J (®А — Wb) dP'
Av
Тх^а-^в (175)
и
тг = TtAx + (1—X). (176)
Подстановка уравнений (175) и (176) в уравнение (174) дает
Р=Рлх'+рв^—*). U77)
Подобным же образом уравнение (169) превращается в
Уравнения (177) и (178) представляют простые полезные соотношения, справедливые при постоянной температуре. Они, очевидно, строго применимы лишь в тех случаях, когда вторая фаза является смесью идеальных газов, а первая — идеальным раствором. Так как
P = Pa + pb=^"+p(1-^). (179)
то из уравнения (177) следует, что
Р_а=Рах^ (I80)
Рв = Рв^— Х'У (181)
Соотношение, выраженное уравнением (180) или (181), обычно из-
вестно как закон Рауля, который открыл его опытным путем.
192
IV. Общие уравнения равновесия
Диференцирование уравнения (177) no Т прн постоянном х' и последующее деление на р дает
, 1— x’dpB ZIQQ,
pdT~~pdT' р dT ’ '1й '
ио _______________________________хГ
Р~РА и 1—х'_1—х* р~рв Подставляя эти уравнения в уравнение (182), получим 1 dp х” dpA 1 — Xя dpB
pdT—pAdfT рв ~dT’ но по уравнению(165) I dpA _ La pAdT~RT2'
где La — скрытая теплота парообразования чистого компонента А при температуре смеси. Поэтому
Idp__хяТА+(1—хя)1д
р dT RT2
(184)
(185)
(186)
Для случая идеального жидкого раствора и идеальных газов при допущении, что объем жидкости ничтожно мал по сравнению с объемом газа, уравнение (138) превращается в
RT^- — Г$’ — S’ — (Xя — х')~| dT— р [ WJ
~кт Ьчй?)] Л-°
или, при постоянном х\
р dT~ RT
Сравнивая уравнения (184) и (186), получаем
х"La + (1 - х") LB = Т [s* - S’ — (Xя - x')g] или
Л?1пр\ _х"£д + (1— x")LB_ Lm
V дТ )х> RT2 RT2’
(187)
(188)
где Lm — средняя скрытая теплота парообразования смеси. Это уравнение аналогично уравнению (165), относящемуся к случаю одного компонента.
Специальные случаи фазового равновесия
193
Правая часть уравнения (187), как уже было показано, должна быть диференциальной скрытой теплотой парообразования жидкой фазы. Вообще испарение раствора можно представить себе как бы состоящим из двух последовательных стадий: 1) разделения жидкости и 2) испарения чистых компонентов. В общем случае обе эти стадий включают изменение энтальпии, и поэтому. тепловой эффект, связанный со стадией 1, обычно называют „теплотой растворения“. Так как левая часть уравнения (187) включает в себя только скрытые теплоты парообразования чистых компонентов, то допущение идеальности газов и идеальности растворов требует отсутствия теплоты растворения.
Следует отметить, что зависимость Т—х' при постоянном давлении, в отличие от зависимостей р — х' и р — Т, не переходит для этого случая в какую-нибудь простую форму.
Идеальные растворы в обеих фазах. Уравнение (143) можно записать так:
Если компонент А устойчив в паровой фазе при давлении и температуре раствора, то для идеального раствора
Va = VA’ и
vA" dp = е-А" =/^.
По аналогии можно написать va — Va и
— Г г> г dp VA> ' =
Но это вызывает ряд серьезных возражений: так как А в стабильном равновесии не существует как чистая жидкость при давлении и температуре раствора, то и v'A и f’A являются гипотетическими величинами. Эти допущения превращают уравнение (143) в
(189)
где fA и f’A — соответственно летучести чистого А в паровой фазе и в жидкой фазе при давлении и температуре раствора. f'A определяется путем экстраполяции в нестабильную область.
13 Химическая термодинамика
194
JV. Общие уравнения равновесия
Для идеального раствора va постоянно, и если принять его при умеренных давлениях не зависящим от давления, то уравнение (143) можно написать так:
kAP
~ = (190)
* fA
где kA— постоянное значение vA-
Аналогичными уравнениями для компонента В являются:
1— хГ 1—X' _ в f"B (191)
И 1—х"_ 1—X* - kgp ’ (192)
и в этом случае представляет гипотетическую величину, которую можно определить посредством экстраполяции.
Уравнение (138) для частного случая изотермических условий и идеальных растворов превращается с помощью уравнения (111) в уравнение
[х" й — гл)4-(1 — х')(^—*B)J dp = RTdx’. (193)
Л. Л )
Аналогичным уравнением, получающимся из уравнения (139), будет уравнение
[х’ (v'a — Va) + (1 — х') (Vg — v'b)] dp = RT dx”. (194)
•* X1 /
Эти уравнения можно считать частными формами уравнения Дюгема, когда и жидкий и парообразный растворы идеальны. Рассмотрим специальный случай, для которого уравнение (193) становится интегрируемым, именно—слабо растворимый газ в растворителе, который можно считать нелетучим (например, азот в воде при обычных температурах). Для этого случая х"=1 и va=^va; производя эти подстановки в уравнение (193), получаем
(195)
Интегрированн е при допущении, что не зависит от р, дает
\vAdp — v’Ap = RT\nx' + c (196)
Специальные случаи фазового равновесия
195
или
RTlnfA — vAp = RTlnx’ + c. (197)
При р = 1 примем f—р; тогда fA = 1 и хг = х\ — растворимость при общем давлении в 1 атм. Производя соответствующую подстановку в уравнение (197), получаем
1пх' = 1пх'1-|-1п/д+-^(1—р), (198)
или
= ' (199)
Если показатель степени равен нулю, то уравнение (199) переходит в x' = x\fA или
x' = kfA, (200)
т. е. в уравнение, аналогичное обычной форме закона Генри, но с давлением, замененным летучестью. Такое уравнение не согласуется с данными для действительных систем, тогда как уравнение (199) прилагается вполне успешно12.
Другим интересным частным случаем является летучая конденсированная фаза, находящаяся под давлением газа, который можно считать нерастворимым, например жидкость, подобная воде, или твердое тело, например BaCl2-8NH3, подвергающееся давлению водорода. Если х является мольной долей инертного газа, то х'— Q и = и уравнение (194) превращается в
(v"B — v'B)dp^ — RT^^p. (201)
Это уравнение представляет относительно простую зависимость между мольной долей менее летучего компонента (например, воды или аммиака) в газовой фазе и давлением.
Для случая идеальных растворов уравнение (140) переходит в
= 1 х' , е± [j* (*в> -С^а “’в>(202)
Величины v не зависят от х, и v' можно также считать независимым от р, по крайней мере при умеренных давлениях.
Применение допущения об идеальности растворов к уравнению (126) дает
RT + v'a dp — Sa dT = RT-|- vA dp — SA dT. (203)
13*
196
IV. Общие уравнения равновесия
При постоянном давлении можно написать
или dx" dx* < — S'. <204) d^-d4-=^^dT <205>
Обозначая х")х' через К (константа равновесия) и 7/д— /Л через Д/7д, получим
d In А"_ /907\
дТ RT* ’ 2U/
Если допустить, что Д/7д— постоянная, не зависящая от температуры (допущение идеальности раствора уже предполагает независимость от х), то уравнение (207) можно интегрировать; тогда получим
1П Ki___Ы1А Tj — Тх
(208)
Это уравнение полезно при вычислении теплоты растворения по данным растворимости. В частном случае, когда одна фаза содержит почти чистый компонент А, например случай растворимости газа, подобного СО2, в воде, уравнение (208) переходит в
]п х2____ Tj — 7\
Xi R ТХТ2 ’
(209)
где х2 и X; — концентрация газа в таком растворе при двух температурах 12а.
Уравнение (207) аналогично уравнению Вант-Гоффа для изменения константы химического равновесия с температурой или уравнению Клаузиуса — Клапейрона для давления пара как функции температуры.
Разбавленные растворы. Для большинства целей можно считать, что термин „разбавленный раствор* соответствует раствору, в котором один из компонентов бинарной системы присутствует в количестве, равном или меньшем 5 мольных процентов. Так называемые „законы разбавленных растворов* являются предельными законами, когда мольная доля стремится к нулю или к единице. При увеличении мольной доли растворенного вещества от нулевого значения простые предельные законы становятся все менее и менее точны. Эти законы обычно очень просты, и во многих случаях ими удобно пользоваться как приближениями и до сравнительно больших
Специальные случаи фазового равновесия
197
концентраций. Очевидно, степень отклонения от этих законов будет зависеть от условий, и нельзя дать общих заключений, определяющих ее.
Случай I. Пар является идеальным газом. Если х приближается к 1 или 0, то член 1/х(1—х) уравнения (162) становится очень большим, тогда как д2и'дх2 остается незначительной величиной. (Заметим, что д2п)дх2 связано с отклонением от идеального раствора и поэтому никогда ие бывает большой величиной. Для идеального раствора оно равно 0.) Также н ду"1дхя = 0, поскольку пар является идеальным газом. Эти специальные условия плюс допущение, что объемом жидкости по сравнению с объемом пара можно пренебречь, переводят уравнения (138) и (139) соответственно в
RT^-L^-RTJ^-^r-dx' = 0 (210)
RT^ — Ld/ — RTjc\-x'dx’^=Q, (211)
1 Л I X Л I
где L — скрытая теплота парообразования чистого растворителя (т. е. компонента, мольная доля которого приближается к 1).
При постоянной температуре
dp х" — х' . , — — Z7V йХ , р X* (1—X*) ’ Лр _ X" — Xf р х"(1 — X") (212) (213)
или dx* _ dx" (214)
х'(1—X') х"(1—х*) ’
Интегрирование дает соотношение
= (215)
1 — х" 1 — х” ' '
где k—эмпирическая константа (для дайной температуры).
Подставляя значение х", даваемое уравнением (215), в (213), получаем уравнение
dp__ (k — 1) dx'
у— 1+(Л-1)х'-
Интегрирование уравнения (216) дает
1пр = In [1 +(А— 1)х']—|—с.
Если х’ = 0, то р=рв. Подстановка этих предельных условий и исключение k с помощью уравнения (215) приводят к
р(1— Х”)=р£(1— х'). (218)
уравнение
(216)
(217)
198
IV. Общие уравнения равновесия
Это тот же закон Рауля, и мы видим, что он приложим только к растворителю в разбавленном растворе, если пар является идеальным газом. Для компонента А можно получить аналогичное уравнение для предельных условий х'=1 и р—рА.
Из уравнений (215) и (218) находим, что
рх" = kpgX' — ex', (219)
т. е. парциальное давление растворенного вещества пропорционально его мольной доле, но константа пропорциональности в этом случае чисто эмпирическая. Это соотношение обычно называется законом Генри, который открыл его опытным путем на основании результатов изучения растворимости газов в жидкостях.
То же самое уравнение для случая разбавленного раствора некон-денсирующегося газа можно очень просто получить из уравнения (212), принимая х"=1 (т. е. пренебрегая содержанием пара растворителя в газообразной фазе). Таким образом, мы имеем
dp___dx’
~р ’
или
х’ = ср, (220)
где с — константа закона Генри.
Для случая разбавленного раствора нелетучего растворенного вещества в летучем растворителе х" (мольная доля растворенного вещества в паровой фазе) = 0, и уравнение (212) переходит в
dp dx' d(l —х') р 1—х' (1—х')
Интегрируя и принимая во внимание предельные условия х' = 0, р=рв, получим
х'=\— (р/рв), или
Это уравнение является законом Рауля для понижения давления пара, и оно совпадает с уравнением (218) для случая, когда х" = 0.
При возвращении к общим уравнениям для разбавленных растворов (210) и (212) и при разборе частного случая постоянного давления очевидно, что уравнение (215) также приложимо к этому случаю. '
При постоянном давлении уравнение (210) превращается в
<222>
Специальные случаи фазового равновесия
199
Подставляя значение х" из уравнения (215), имеем
-^2 -г Г*,~\ , dx'. (223)
RT 14-(Л—l)x'
Интегрируя при допущении постоянства L и включая предельное условие Т= Тв (температура кипения чистого растворителя) при х' = 0, имеем
(224)
Если х' мало, то 1п[14-(&—1)х']— как можно показать посредством разложения в ряд — стремится к значению (k—1)х'. Поэтому уравнение (224) можно написать так:
у — —(&— 1)х
или
тв— T=~&-(k — 1)х', (225)
откуда, полагая, что ТТВ=^ Т2В (это является законным приближением), получаем
RT2B
Т-Тв = с-~ х', (226)
где с — эмпирическая константа. Это уравнение выражает закон изменения точки кипения— понижение или повышение (в зависимости оттого, какой компонент присутствует в меньшем количестве) — для раствора летучего растворенного вещества в летучем растворителе.
Если растворенное вещество нелетуче, то х" = 0, и уравнение (222) переходит в
---= V* (227) /?Г2 (1—-*') '
Интегрируя в пределах от Тв до Т, получаем
[228>
или, когда х’ мало по сравнению с 1,
Т— Тв = —^х'. (229)
Это является обычным уравнением, выражающим повышение точки кипения разбавленного раствора нелетучего растворенного вещества.
Общие уравнения приложимы так же хорошо к равновесиям твердев тело — жидкость и твердое тело — газ, как и к равновесию
200
IV. Общие уравнения равновесия
жидкость — газ. Например, принимая, что фаза ['] является твердой и что чистое твердое тело В выделяется из жидкой фазы (случай смеси, образующей эвтектику), х" в уравнении (222) = 0, и L будет скрытой теплотой плавления чистого В. Этим путем получаем уравнение для понижения температуры замерзания
RT^
Т—Тр=-~^х', (230)
где Тр—точка замерзания чистого В при данном давлении и Т— точка замерзания раствора ☆ ).
Случай II. Пар не является идеальным газом. Для случая постоянной температуры уравнения (138) и (139) переходят соответственно в
<231)
И
и*—т/ — (х* — х>) g j dp = dxT. (232)
Для частного случая летучего растворенного вещества в нелетучем растворителе х’—»-0, х"—►!, гГ—«•од и »'—► пд. Кроме того, dv’/dx' можно считать постоянным в широком интервале значений х'. Тогда уравнение (231) превращается в
(®д — vB — c)dp=RT^, (233)
где c = dv’ldx'. Пусть vB-\-c—k-, поступая как в случае уравнения (195), получим
x' — XifAe^T (1 Р\ (234)
Это по существу то же, что и уравнение (199).
Соотношение Пойнтинга. Представим двухфазную систему при постоянном составе и постоянной температуре, но с различным давлением над каждой фазой. Для такого случая уравнение (126) переходит в
VAdp' = VAdpf (235)
или, если обе фазы являются однокомпонентными, то в
v'dp' — v"dff. (236)
☆) Это, конечно, относится не только к рассматриваемому случаю, когда нар является идеальным газом. На самом деле уравнение (222), из которого выведено уравнение (230), правильно, независимо от того, будет ли фаза ['] идеальным газом или нет (см. прим.11).
Химическое равновесие
201
Это уравнение используется для вычисления влияния давления на давление насыщенного пара твердого тела или жидкости. Оно включает и допущение о полупроницаемой мембране, необходимой для поддержания различных давлений над двумя фазами. На самом деле этот эффект, известный как эффект Пойнтинга, по имени ученого, его открывшего, никогда не наблюдается один, но всегда совместно с другими эффектами, вследствие того, что единственным практическим способом увеличения давления над конденсированной фазой, находящейся в равновесии со своим паром, является применение газа, а газ никогда не бывает совершенно инертным. Он распространяется в конденсированной фазе и также оказывает притягательное действие на молекулы параи.
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
Константа равновесия. Рассмотрим следующую общую реакцию, происходящую в данной фазе, жидкой или газообразной:
аА-{-ЬВ-{- ... .... (237)
где а,Ь,... представляют число молей реагентов А, В,..., a Z, т, ... — число молей продуктов реакции L, Л1 ..., и допустим, что дается достаточное время для достижения состояния равновесия. Уравнение (32) выражает общее условие для любого гомогенного равновесия при постоянных давлении и температуре, и для данного частного случая его можно написать как
- p.AdNA — - ... + Л + ИлЛ 4- ... = 0. (238)
Знак минус указывает на то, что при прохождении реакции слева направо количества А, В и т. д. уменьшаются, a L, М и т. д. увеличиваются. Конечно, совершенно условно, считать ли, что реагирующие вещества и, следовательно, уменьшающиеся по количеству находятся в левой части уравнения (237) или в правой части его, так как, поскольку мы имеем дело с истинным равновесием, реакция обратима и в состоянии равновесия ее можно заставить проходить в любом направлении с помощью ничтожно малого изменения внешних условий.
Из стехиометрических соотношений имеем
dNA а йМд а
<ШВ b dNi I • д"
Тогда уравнение (238) можно представить в форме
— Ц1А — Ьцв— ... + + • • • =°- (239)
202
IV. Общие уравнения равновесия
Вводя уравнение (38) в (239), имеем
— а(^7’1пал4-рд) — />(7?Т1па34-}1в) —
-f-1(7? Tin aL -}- |1£) -j-m (RTln ам Щи) . =0 ( 240)
или
(арЦам)"1... =~^Tt (241)
(аА)а (а 1})ь • • • где
= . — ар.‘А — Ьр.в—.... (242)
Поскольку р° отвечает определенным значениям давления и концентрации, величина правой части уравнения (241) является функцией только температуры и обычно называется „константой равновесия". Следовательно,
_ ар-”
е ^т = Ка, (243)
или
Др° = — RT\nKa. (244)
Если стандартным состоянием каждого компонента является чистое вещество, то уравнение (244) переходит в
Д2° = — RT\nKa. (245)
Как принято, мы будем во всех случаях употреблять уравнение (245), хотя в некоторых случаях было бы более точно применять уравнение (244).
Вывод уравнения (241) мы относили к гомогенной реакции, но это ограничение легко снимается, и уравнение становится приложимым к любому химическому равновесию (независимо от присутствующих фаз). Если реагирующая система наряду с газообразной фазой содержит твердую и жидкую фазы, то ясно, что когда система в целом будет находиться в равновесии, то будут иметь место и химическое равновесие в газообразной фазе и фазовое равновесие между различными фазами. Другими словами, любую реакцию можем условно рассматривать как гомогенное равновесие в газовой фазе, поскольку можно считать, что все вещества обладают некоторым давлением насыщенного пара. Кроме того, для равновесия между фазами мы имеем уравнения Г If flf
V-i = V-l = Р-/ = ... и
7?Т1пя/ = 7?Т1па/ -ф-с"-, (246)
где Ct — константа, зависящая от выбора стандартного состояния. Если стандартное состояние компонента сохраняется постоянным,
Химическое равновесие
203
независимо от фазы, то с,=0. Другими словами, активности компонента в различных фазах равны или пропорциональны между собой.
Поэтому активность в уравнении (241) может быть активностью в любой фазе, и уравнение является общим уравнением закона действующих масс для любого химического равновесия. Так как числовое значение константы равновесия зависит от выбора стандартного состояния, то следует соблюдать осторожность при использовании уравнения для вычисления активностей или связанных с ними величин, иначе нельзя быть уверенным в том, что не произошло никаких изменений в стандартном состоянии.
Так как
a~f°
и для газа
/° = р°,
то можно написать уравнение (241) для газового равновесия в форме
= ipo\sn К (247)
Для практических целей р° берется равным 1 атм, и (р°)Еп=1. Константа равновесия в уравнении (247) иногда изображается символом КрО.
Идеальные газы. Уравнение (86) для случая идеальных газов превращается в
ft = pxt. (248)
Подставляя в уравнение (247), имеем
(хдр)а (хврУ> ...
(249)
Именно в форме этого уравнения наиболее часто применяется закон действующих масс. Из вывода очевидно, что это уравнение является лишь предельным случаем, к которому приближается более общая закономерность, по мере того как давление стремится к нулю.
Идеальный раствор. Для таких растворов уже раньше было показано, что а{ = х., если стандартным состоянием является состояние чистого компонента при давлении и температуре раствора. При приложении этого условия к жидким растворам уравнение (249) переходит в
М“(хв)ь...
(250)
204
IV. Общие уравнения равновесия
Для случая газовых растворов сочетание уравнений (247) и (115) дает
(xrf№(xBfB}b ... r
(251)
Это можно также написать, как
{х^(хв)ь .
(252)
где Кх — константа интегрирования, являющаяся функцией как давления, так и температуры, но не зависящая от состава.
Определение Кр и Кг Определим функцию Кр уравнением
(Х^ЦХддР)'»...
<хАр)а(хвр}ь...
(253)
Кр в действительности не является „константой” равновесия, потому что она зависит от температуры, давления и состава; в этом легко убедиться, если сравнить уравнение (253) с уравнением (247), помня, что Kf зависит только от температуры. Когда давление уменьшается и стремится к нулю, К приближается к Кр« или Kf как к предельному значению. Сочетая уравнения (251) и (253),
получаем
К/___(пР(Гм)т--- ___
KP~(rA)a(rB)b... ~
(254)
иногда называется коэфициентом активности (или летучести). Кч—функция давления и температуры, но не зависит от состава.
Влияние температуры на константу равновесия. Из уравнения (243) следует, что
Диференцируя по температуре, получаем
<*1пЛо__Др" 1 d(V )
dT RT2 RT dT
(255)
Необходимо отметить, что величина dinKJdT ие является частной производной при постоянных р и х, как иногда пишут, так как она является функцией только температуры. По уравнению (100),
________™ dT
Г(/л)°(/в)* ••• Ч(/д'(/ж)я’-..
Обобщенные силы
205
(256)
(257)
(258)
(259)
и
<*(Д|* ) Аъ> dT
Кроме того, из определения Z и р. имеем
тг{ и
Д|1° = Д77° —ГДЛ
Подставляя уравнения (256) и (257) в (255), получаем
dln/Ca_Д/7°
dT RT2 ' где
Д77° = ///“ + тН°м + ... — аТГА — ЬН°В — ...
H°t представляет парциальную мольную энтальпию любого компонента, принимающего участие в реакции, при выбранном частном стандартном состоянии, отвечающем определенным значениям давления и состава. 7?, зависит только от температуры.
Если стандартным состоянием каждого компонента является состояние чистого вещества, то уравнения (258) и (259) переходят в
—(260) dT RT2 ' 1
И
Д№=/Я’ аН°А — ЬН°В —... . (261)
ОБОБЩЕННЫЕ СИЛЫ
При всех наших выводах в этой и предыдущей главах принималось, что единственной действующей силой было равномерное давление окружающей среды. Другими словами, мы исключали все силы, подобные поверхностному натяжению, электростатическим силам и э. д. с. Имеются некоторые важные случаи, особенно в области электрохимии, когда желательно включать действие других сил, и поэтому мы закончим эту главу очень кратким рассмотрением общего метода, применяемого в тех случаях, когда следует учитывать и другие силы.
Первый и второй законы термодинамики при применении их к любой замкнутой системе, находящейся в равновесии, приводят к уравнению
TdS — dU-\-dA,
(262)
206
IV. Общие уравнения равновесия
и если давление является единственной силой, то
Теперь примем, что С является обобщенной силой или фактором интенсивности и X — соответствующим обобщенным перемещением или фактором емкости; тогда уравнение (262) переходит в
+ (263)
С этим уравнением в качестве исходного можно обобщить многие уравнения, выведенные в гл. III для включения влияния силы £. Например, по определению изобарного потенциала,
Z=U— TS-^pv.
Диференцирование и сопоставление с уравнением (263) дает
dZ = — SdT-\-vdp^dX, (264)
£>~ \дт)Р,х
и
- (dZ\ \дх)Р,т
Подобным же образом
Z = "+T(W,.X- <265>
Это уравнение имеет важные приложения в электрохимии.
При постоянном давлении
dZ= — SdT-\-£dX, и поэтому
— (266)
\.<?Х ) Р,т \аТ / р,х ' ’
Для некоторых приложений желательно обобщить определение известных функций посредством включения обобщенной силы и перемещения. Например, определим вновь Z с помощью уравнения
Z=U— TS + pv-J-CX. (267)
Тогда
dZ=vdp — 5dT4-XdC. (268)
Из этого уравнения получим
Обобщенные силы
207
При применении этого уравнения к веществу, находящемуся в магнитном поле, получаем уравнение
= (—} (270)
так как С равно напряжению Н магнитного поля и X равно интенсивности намагничивания /. Магнитная проницаемость % (аналогична электропроводности) определяется уравнением
I
X-* Н •
При постоянном Н
dI=Hdy.
Сочетая это уравнение с уравнением (270), получаем
(Й) '
\<?Н/ р, т \дТ) р, н
(271)
Это уравнение имеет важное применение при получении очень низких температур в области, близкой к абсолютному нулю. Для дальнейших подробностей см. статью Джиока [85]126.
ГЛАВА V
СООТНОШЕНИЯ ДАВЛЕНИЕ—ОБЪЕМ—ТЕМПЕРАТУРА ДЛЯ ГАЗОВ
Вычисление термодинамических свойств любого газа с помощью уравнений, выведенных в гл. Ш и IV, требует знания зависимости р — v — Т. Это необходимо для интегрирования диференциальных соотношений, связывающих свойства с параметрами состояния. Кроме того, зависимость р — v—Т необходимо зиать для решения многих важных технических задач, например для определения размеров газохранилищ, вычисления падения давления, обусловленного потоком газа, для измерения потока и определения времени, необходимого для соприкосновения в реакционных аппаратах. Целью этой главы является обзор и суммирование наших сведений о том, каким образом объем (или плотность) газов изменяется с температурой и давлением, а в случае смесей—и с составом. Изложение будет ограничено газами, поскольку данные для жидкостей не так существенны и гораздо менее обширны. Первая часть нашего анализа будет посвящена только чистым газам, но позднее рассмотрение будет распространено и на газовые смеси. Соотношения могут быть представлены в графической форме или в виде уравнения. Уравнение, представляющее зависимость р— v—Т для газов, обычно называется уравнением состояния. Такое уравнение является удобным способом для отражения большого числа данных и выражения их в форме, полезной для различных приложений.
ИДЕАЛЬНЫЙ ГАЗ
Давно уже исследования поведения газов привели к двум простым законам:
pV=kt (закон Бойля) (1)
и
V=k2T (закон Шарля), (2)
где kx—константа для данной температуры, k2— константа для данного давления ☆).
й) Следует отметить, что закон Шарля в этой форме связан с вопросом определения температурной шкалы. В действительности обычно определяют температуру по шкале газового термометра постоянного давления. Дли идеального газа определенная таким образом шкала оказалась идентичной с термодинамической шкалой, определенной в гл. III; если не будет особо оговорено, все наши рассуждения будут основываться на этой шкале.
Идеальный газ
209
Сочетание этих двух уравнений приводит к зависимости
pV=kT, (3>
где k зависит от единиц измерения р, V и Т, от массы и от природы рассматриваемого газа; но если уравнение относить к 1 молю газа, то k превращается в универсальную константу, не зависящую от природы газа, а зависящую только от единиц, выбранных для выражения р, V и Т. Тогда мы получаем уравнение Клапейрона
pv = RT. (4)
Для любого количества газа
pV = NRT, (5)
или
pV=^RT. (6)
Сразу же было обнаружено, что простое поведение, требуемое уравнением (5), является только приближением к действительному поведению газов 13.
При больших плотностях отклонения от уравнения (5) могут быть весьма значительны; в общем, чем меньше плотность, т_м в большей степени реальные газы приближаются к поведению, отвечающему уравнению (5). Другими словами, это уравнение отвечает предельному случаю, к которому приближаются все газы по мере уменьшения давления или повышения температуры.,Это приводит к понятию идеального (или совершенного) газа, поведение которого точно воспроизводится уравнением (5). Это понятие приносит громадную практическую пользу вследствие простоты связанных с ним уравнений и вследствие того обстоятельства, что оно с достаточной для практических целей точностью отвечает поведению реальных газов при весьма разнообразных условиях. Однако следует помнить, что оно является только предельны.м условием, к которому все реальные газы могут приближаться в большей или меньшей степени, но никогда его не достигают.
С кинетической точки зрения идеальный газ представляет собой газ, между молекулами которого отсутствуют силы притяжения или отталкивания и в котором молекулы настолько малы по сравнению с расстояниями между ними, что их собственным объемом вполне можно пренебречь. Из этого простого представления с помощью кинетической теории можно вывести уравнение состояния идеального газа. Поскольку мы заинтересованы скорее в применениях закона идеального газа, чем в его теоретическом обосновании, этот вопрос в дальнейшем обсуждаться не будет.
С термодинамической точки зрения идеальный газ может быть определен посредством одного из следующих способов:
1. По уравнению (5).
14 Химическая термодинамика
210
V. Соотношения давление — объем — температура для газов
2. По закону Бойля и закону Джоуля. Закон Джоуля утверждает, что энергия газа не зависит от его давления или его объема, т. е. является функцией только температуры. Формулируя это математически, получаем
Так как давление во время изменения объема при постоянной температуре является зависимой переменной, то это уравнение предполагает также, что энергия не зависит от давления. В общем, как мы уже видели в гл. III, энергия газа зависит от двух независимых переменных, например от объема и температуры. Из простого кинетического представления об идеальном газе следует, что поскольку силы, действующие между молекулами, отсутствуют, одно изменение объема не может вызвать изменения внутренней энергии.
3. По закону Бойля и нулевому эффекту Джоуля — Томсона. Эффект Джоуля — Томсона будет более подробно рассматриваться в гл. VII; для настоящей цели достаточно сказать, что он заключается в изменении температуры, происходящем от расширения газа от одного постоянного давления до другого, тоже постоянного давления, при условии отсутствия иной внешней работы, кроме работы поддержания постоянного давления, и при условии рассеивания всех временных энергетических эффектов, обусловленных скоростными факторами. Расширение этого типа называется процессом Джоуля—Томсона, по имени двух физиков, впервые применивших его при изучении газов. Как будет показано ниже, во время этого процесса функция Н остается постоянной14. Следовательно, изменение температуры с давлением определяется в этом случае коэфициентом (дТ1др)н. Нулевой эффект Джоуля — Томсона означает, что
при всех давлениях и температурах. Для всех реальных газов этот коэфициент не равен нулю, за исключением ограниченного числа точек (см. гл. VII), и в зависимости от условий расширения может быть как положительным, так и отрицательным.
Что закона Бойля и закона Джоуля достаточно для установления идеальности газа, можно показать следующим образом:
(78.ГЛ.Ш)
и, следовательно, при применении закона Джоуля получим
’Ш~г,=0’ <®>
или
Tdp— pdT = Q (при постоянном объеме), (10)
Идеальный газ
211
или
dp___dT
~Р~~Т'
Интегрируя (11), получим
р = ЛТ,
(П)
(12)
где константа k является функцией объема. Сочетание уравнений (12) и (1) непосредственно приводит к уравнению состояния идеального газа в форме уравнения (3).
Подобным же образом в случае эффекта Джоуля — Томсона имеем
— v
др)н
dv \
дт)Р
(97, гл. III)
С
р
и посредством операций, аналогичных только что использованным, приходим к уравнению
v = kT, (13)
где k — функция только давления. Это уравнение также сочетают с уравнением (1) для того, чтобы получить уравнение состояния идеального газа. Другими словами, мы показали, что все три определения идеального газа эквивалентны.
Если объем выражается в литрах, давление в атмосферах, температура в градусах Кельвина и масса в грамм-молях, то по уравнению (5)
*=^=4^=0’08206-
Объем, занимаемый 1 молем идеального газа при нормальных условиях (1 атм и 0°С или 273,16°К), равен 22,414 л. Произведение pv можно выразить также и в других единицах, поскольку оно имеет размерность энергии. Так, мы можем выразить pv в кал)моль и тогда
= 1’987> 2/0,10
где
542 5 — 10333 Х °-02241
РЕАЛЬНЫЕ ГАЗЫ
Молекулы реальных газов занимают некоторый определенный объем, которым, по сравнению с общим объемом, занимаемым газом, в некоторых случаях можно (а в некоторых случаях и нельзя) пренебречь; кроме того, они обладают силовыми полями, в результате чего 14*
212
V. Соотношения давление — объем — температура для газов
происходит взаимное притяжение между ними. Оба эти условия приводят к тому, что ио мере приближения молекул газа друг к другу (т. е. по мере повышения плотности газа) газ все больше и больше отличается от идеального. Если температура газа ниже некоторой температуры, известной как критическая температура, то возрастание давления в конце концов приведет к конденсации газа в жидкость. Выше критической температуры газ можно сжимать безгранично, не вызывая появления жидкости. Этим способом некоторые газы были доведены до плотностей выше нормальных плотностей жидкости или даже твердого тела. Например, Бриджмен [272] сжимал водород при 65° С и примерно при 15 000 атм до плотности 0,1301 г ]мл. Нормальная плотность жидкого водорода составляет около 0,071 и твердого водорода — 0,081. Подобные соотношения получены и для азота. Интересно также отметить, что при указанных давлении и температуре объем азота в 16 раз больше, чем он был бы в случае идеального газа в этих условиях.
Критическая точка является единственной точкой, в которой жидкая и газовая фазы тождественны. Выше критической температуры возможна только одна фаза. Все газы обладают определенной критической точкой, которая может быть определена экспериментально. Эта точка имеет большое практическое и теоретическое значение1S.
Иногда, даже при относительно низких давлениях, наблюдаются необычайно большое отклонение от законов идеального газа. Считают, что эти отклонения должны обусловливаться химическими изменениями, приводящими к ассоциации молекул. Двумя классическими примерами являются пары уксусной кислоты и двуокись азота, в которых происходят соответственно следующие реакции:
2СН3СООН = (СН3СООН)„
2NO2 = N2O4.
В случае уксусной кислоты при 118°С и 0,94 атм давления объем, вычисленный по закону идеального газа на основе молекулярного веса, равного 60,03, в 1,65 раза больше объема, определяемого путем опыта.
Коэфициенты сжимаемости. Поведение любого реального газа часто изображают на диаграмме посредством нанесения объема как функции давления с температурой в качестве постоянного параметра. Несравненно более удобным методом изображения поведения газов является использование диаграммы коэфициент сжимаемости — давление с температурой в качестве постоянного параметра. Коэфициент сжимаемости, которым мы будем пользоваться, определяется уравнением
~~ RT'
(14)
Реальные газы
213
С для любого идеального газа при всех условиях имеет значение, равное единице, и, следовательно, такой график сразу показывает степень отклонения газа от идеального состояния при данных давлении и температуре. Уравнение (14), соединенное с выражением, дающим С как функцию давления и температуры для данного газа, дает уравнение состояния для этого газа. Зависимость между С, р и Т обычно дается в графической форме. Такой график для газообразного азота представлен на рис. 13. Подобные графики можно построить
Рис. 13. Коэфициенты сжимаемости для газообразного азота [7, 8].
и при использовании других пар независимых переменных, а именно — T,v и р, v. Выбор вида графика зависит от того, какие даны условия; например, если следует определить объем и даются давление, температура и масса, то рис. 13 может быть непосредственно использован для получения С, и из уравнения (14) вычисляется мольный объем, а затем общий объем из уравнения
V=Nv.
Если нужно вычислить давление при заданных массе, общем объеме и температуре, то для непосредственного отсчета С нужен был бы график в координатах С — мольный объем с температурой в качестве постоянного параметра. С другой стороны, всегда возможно, как для этого случая, так и для другого, использовать график р—Т, применяя метод подбора, как показывает следующий пример.
214
V. Соотношения давление — объем — температура для газов
Пример 1. Чему будет равно давление, если 127 кг азота при 200° С хранится в объеме 424,5 л?
CNRT р~ у •
Для первого подбора примем 6=1. Тогда
1 •12 vjj— 0,08206 • 473,2 «О Jiff
р =----------^24Д----------= 415 атм-
По рис. 13,
6=1,245.
Второй подбор при использовании 6=1,40 дает /» = 580; при этом давлении 6= 1,37. Из этого очевидно, что правильное давление, лежит между 415 и 580 атм, причем много ближе к последнему из этих значений.
Примем 6=1,35, р = 560 и по чертежу найдем 6=1,345. Это достаточно точное совпадение, и поэтому искомое давление равно 560 атм.
Эту задачу можно также решить графически, без процесса подбора, следующим способом:
-С- РХ424.5 _
NRT 4536 X 0,08206 X 473,2 ’ Р'
Это уравнение представляет прямую линию, проходящую на рис. 13 через начало координат (р = 0, 6 = 0) и через точку с координатами р = 800, 6=1,935. В рассматриваемом случае вместо начала координат, не приведенного на рис. 13, может быть включена другая точка, например точка с координатами 6 = 0,800, р = 330; подобная замена, очевидно, не может исказить результата. Там, где эта линия пересекает изотерму £ = 200° С, можно непосредственно отсчитать давление, равное 560 атм.
Часто употребляется другой коэфициент сжимаемости, определяемый уравнением ☆)
где psv3—произведение pv в нормальных условиях, т. е. при 0°С и при давлении в 1 атм. Если давление выражается в атмосферах; а объем — в единицах объема при нормальных условиях, то можно написать
Z~pV.
Соотношение между двумя коэфициентами сжимаемости дается уравнением
С=гСяЦ, (16)
где То —точка плавлении льда, а С3—коэфициент сжимаемости при нормальных условиях, определенный по уравнению (14). Cs
☆) Это отношение иногда изучением сжимаемости газов.
называют числом Амага, который занимался
Реальные газы
215
очень мало отличается от единицы, и для большинства технических расчетов мы можем написать уравнение (16) в форме
(17)
Для получения наибольшей точности нельзя принимать, что законы идеального газа сохраняются при 1 атм, но следует считать их справедливыми лишь в пределе, когда р приближается к 0. Плотность различных газов измерялась при давлениях меньше 1 атм, и результаты экстраполировались до р = 0 посредством уравнения pv = а 4~ Ьр ср2. (18)
Этим путем получены значения р0о0 — произведения pv в пределе, когда р = 0. Отношение ро©о/РЛ часто обозначаемое через 1 Л, для большого числа газов при 0° С дается в табл. 3. Для большинства газов различие между рото и произведением pv при 1 атм ничтожно мало, но в некоторых случаях оно достигает 4°/0.
Таблица 3
Сжимаемость газа при давлениях ниже 1 атм (данные из Интернациональных критических таблиц) &
Газ 9 Предельное значение z при 0° С, когда р-»0 Газ Предельное значение z при 0°С, когда /м-0
Аг 1,0009 NO 1,0011
н2 0,9993 NH3 1,015
Не 0,9995 СО 1,0005
n2 1,0004 со2 1,0070
о2 1,0009 СН4 1,0024
на 1,0074 С2н4 1,0078
HI 1,015 (СН3)2О 1,0254
so2 1,024 н-Бутан 1,042 [117а)
£? См. также Справочник физических, химических и технологических величии в Технической энциклопедии. (Прам. ред.)
Приведенные координаты. Графики коэфициентов сжимаемости (рис. 13) различны для каждого газа, но все они имеют некоторый общий характер. Существует физико-химический закон, который утверждает, что все газы ведут себя одинаково, если они находятся в соответственных состояниях, т. е. если любые две переменные из трех — давление, объем и температура — находятся в одинаковом
216
V. Соотношения давление — объем — температура для газов
отношении к их критическим значениям. Этот закон, называемый законом соответственных состояний, не точен, но служит очень полезным приближением. Следовательно, если определить приведенное давление, приведенную температуру и приведенный объем следующим образом:
п = /-, (19)
Л кр
z = ~, (20)
'«л
где индекс кр относится к критическому состоянию, то закон этот утверждает, что любые два газа, находящиеся при одинаковых п и т, будут иметь одинаковое значение V, а также одинаковый коэфициент сжимаемости и одинаковые значения других термодинамических свойств.
Ньютон [175] показал, что лучшее совпадение для водорода, гелия и неона получается при изображении величины ftp как функции п и т при использовании специальных приведенных условий для этих газов, определяемых следующим образом: _______________________________ Т
Х~Ткр + *’
(22)
(23)
При изображении коэфициентов сжимаемости этих газов мы также будем пользоваться этими видоизмененными определениями.
Для более наглядного объяснения смысла закона соответственных состояний рассмотрим следующий пример: водород, который имеет критическую температуру 33,2° К и критическое давление 12,8 атм, будет иметь при—190,7°С (т= 2,0) и 2,08 атм давления (п = 0,10) приблизительно те же самые значения термодинамических свойств, что углекислый газ при 335,3° С (т==2,0) и при 7,30 атм давления (тг= 0,10).
Использование приведенных координат позволяет значительно упростить изображение свойств реальных газов, так как если нанести С как функцию приведенных давления и температуры, то можно использовать один график для всех газов. На рис. 14 дается такой график для приведенных давлений 10, а на рис. 15 расширены пределы давлений до больших значений тг. Эти графики основаны на литературных данных по сжимаемости газов. В них включены данные для 20 различных газов.
Отклонения коэфициентов сжимаемости, основанных на экспериментальных данных, от значений их, даваемых этими графиками, были
Коэфициент сжимаемости
Рис. 14. Зависимость коэфициента сжимаемости от приведенного давления для ряда приведенных температур (в области низких давлений).
218 V. Соотношения давление — объем — температура для газов
подвергнуты широкому исследованию. Для 263 отдельных случаев, относящихся к 18 различным газам и охватывающих все пределы графиков, среднее отклонение оказалось меньше 2°/0. Наихудшее совпадение давал аммиак, для которого найдено отклонение выше 15°/0;
Рис. 15. Зависимость коэфициента сжимаемости от приведенного давления для ряда приведенных температур (в области высоких давлений).
среднее отклонение для этого газа составляет 7°/0. По этим чертежам можно с достоверностью заключить, что такие обобщенные графики будут давать достаточную точность для большинства технических расчетов. Графики, подобные рис. 14 и 15, были впервые предложены Копом, Льюисом и Вебером [46] и Брауном, Саудерсом и Смитом [31] для углеводородов, а позднее были распространены Доджем [61] и на другие вещества.
Реальные газы
219
Другой иногда применяемой корреляцией является график тг — т при постоянном значении V. Он обладает тем преимуществом, что приведенные изохоры, т. е. линии постоянного приведенного объема, почти линейны, и поэтому интерполяция оказывается несколько более легкой, но этот график имеет большой недостаток, так как данные по критическим объемам очень скудны и не очень достоверны.
Эти графики сразу показывают, в какой степени тот или иной газ отклоняется от идеального состояния. При низких давлениях все газы приближаются к состоянию идеального газа (С=1,00). При температурах ниже критической (т<^ 1) или при температурах, не слишком превышающих критическую, и при умеренных давлениях все газы занимают объем, меньший, чем объем идеального газа (или, что одно и то же, имеют большую плотность). По мере возрастания давления отклонение, сохраняя свой знак, увеличивается по абсолютной величине, пока не будет достигнут минимум коэфициента сжимаемости, после которого отклонения становятся меньшими, т. е. коэфициент сжимаемости увеличивается. Наконец, при определенном приведенном давлении объем становится равным объему идеального газа, и тогда, так как давление продолжает возрастать, все отклонения приобретают противоположный знак, т. е. объем реального газа превосходит объем идеального газа. Если приведенная температура больше 2,5 (приблизительно), то при всех давлениях объем реального газа будет больше объема идеального газа. В двух областях, именно в той, в которой отклонения отрицательны (реальный объем меньше идеального), и в той, в которой они положительны, отклонения приближенно характеризуются тем, что в первой области преобладающим фактором являются силы притяжения между молекулами, а во второй — собственный объем молекул. Температура, при которой происходит переход от значения С 1 к значению С > 1 при низком давлении (как предел при р = 0), известна под названием „точки Бойля“. Она различна для всех газов, но при выражении через приведенные единицы она равна примерно 2,5 для всех газов.
Определение точки Бойля дается уравнением:
f д (pv)l „ п
У = =0, когда р—>0.
L др Jr
(24)
Так как pv=CRT, то
дС 1 д (pv) др КТ др ’
и поэтому в этой точке дС)др также равно нулю. Точка Бойля не имеет другого практического значения, кроме удобства для сравнения при рассмотрении отклонения свойств газов от законов идеального газа.
При температурах выше точки Бойля изотермы по мере роста температуры сначала увеличивают наклон, а затем уменьшают его.
220
V. Соотношения давление — объем — температура для газов
При приведенном давлении 10 все газы отклоняются примерно на одну и ту же величину, независимо от температуры. Наиболее отчетливо это видно на диаграммах для данного газа. Например, для СО все изотермы от—70 до 200° С пересекаются очень близко к одной точке — точке с координатами С= 1,217 нр = 375 атм[8].
Высказываемое иногда утверждение, что все газы достигают идеального состояния, если температура повышается при постоянном давлении, очевидно, неправильно. Оно верно при температурах ниже точки Бойля и также выше этой точки при высоких давлениях, но с приведенными давлениями ниже 10 и при температуре выше точки Бойля имеется область, где С больше 1,0 и увеличивается с повышением температуры.
Максимальное отклонение от идеальности, поскольку дело касается этих частных графиков, наблюдается в критической точке, окр примерно в 3,7 раза меньше г'идеал- Степень отклонения, однако, не вполне независима от природы газа, и для гелия, например, она равна 3,05, а для цианистого водорода составляет 4,78. Ниже критической изотермы линии резко обрываются при приведенном давлении, соответствующем давлению пара, выше которого газ не может более существовать в стабильном состоянии. Интересно отметить, что во всех этих областях отклонения от идеального состояния относительно невелики.
Графики рис. 14 и 15 в сочетании с таблицами критических параметров можно использовать для приближенного определения объема любого газа. Критические параметры даны в „Приложении" вместе с константами уравнения Ван-дер-Ваальса.
Пример 2. Определить объем 1 моля этилена при 60°С и 200 атм. и сравнить с объемом идеального газа.
Из уравнений (19) и (20) и данных табл. II „Приложения" находим:
273,2 + 60
Т- 282,8 -1’18’
200 ч оч тс=5ад=3’93-
Интерполяцией по рис. 14 находим:
С =0,605,
CRT 0,605-0,08206-333,2 „ „о„,,
» = -у =-------------non--------= 0,08271 л.
200 0 08271
Объем идеального газа = ’ ... = 0,1367л.
О,ООО
Экспериментальные данные по сжимаемости дают v = 0,08298.
Пример X Какое давление получится, если азот, взятый при нормальных условиях в количестве 30 л3, сжать до 0,075 ж3 при температуре 50°С?
Реальные газы
221
Из уравнения (14) имеем
Рг—Pi
= 1
СгУгТ2___
С\р21 j
(273,24-50) 30 -Сг 0,075-273,2
(при постоянном количестве)
= 473 С2(С'1 принимается равным 1,00).
Так как />к/, = 33,5, то
Рг == 33,5 tc«j = 4731?2, или сг = 0,0710 П2-
Это уравнение на графике С — л отвечает прямой, проходящей через начало координат. Пересечение этой прямой с приведенной изотермой
126,0 ’
дает значение я.
Используя рис. 15, получаем
к =24,5; р = 24,5 X 33,5 = 821 атм.
Эти методы вычисления, коэфициентов сжимаемости, Для более точного воспроизведения зависимости р — v — Т следует или построить такие же графики для каждого из изучаемых газов, или пользоваться каким-либо другим методом.
Определение критической температуры. Вследствие важности критического состояния для корреляции данных по сжимаемости и трудности измерения критической температуры важно разработать метод для вычисления ее из других, более легко определяемых свойств. Одним из таких методов служит метод, предложенный Ватсоном [252], согласно которому критическая температура вычисляется по эмпирическому уравнению
основанные на обобщенных графиках очень удобны, но не очень точны.
Рис. 16. График для определения критической температуры.
у _____ 7в4~ А
*Р 0,283(Л1/рв)0.18
(25)
где М—молекулярный вес, рв—плотность жидкости при нормальной температуре кипения, Д— поправочный фактор, равный Те — Тв,
222
V. Соотношения давление — объем — температура для газов
и Те — температура, при которой вещество находится в равновесии с насыщенным паром, концентрация которого равна 1 молю на 22,4 л. Поправочный фактор определяется по нормальной температуре кипения Тв с помощью рис. 16, взятого из статьи Ватсона. Этот метод проверен Ватсоном на очень многих веществах и имеет точность около 2°/0 для неполярных или малополярных веществ. Для полярных веществ, подобных воде, уксусной кислоте и аммиаку, этот метод не рекомендуется.
Критическое давление, т. е. давление насыщенного пара при критической температуре, можно определить измерением давления пара по одному из методов, рассмотренных в гл. VI.
Мейсснер и Реддинг [168] дают следующие эмпирические уравнения для предсказания критических температур, объемов и давлений для полярных и неполярных жидкостей.
Для соединений, кипящих ниже 235° К, и для всех элементов
Ткр=1,70Тв-2,
где Т— критическая температура, °К,
Тв —нормальная температура кипения, °К.
Для соединений, кипящих выше 235° К:
1. Соединения, содержащие галогены или серу:
^=’.41^ + 66- 11F,
где F—число атомов фтора.
2. Ароматические соединения и нафтены (не содержащие галогенов и серы): *
Ткр= 1,417^4-66 —г (0,3837^ —93),
где г—отношение числа нециклических атомов углерода к их общему числу.
3. Все" соединения (не содержащие галогенов и серы), кроме ароматических и нафтенов:
1,027 Тв 4-159.
Для всех веществ:
^ = (0,377Р4-11,0)1,25,
где Р —парахор,
v— критический объем, мл [моль к.
Парахор вычисляется по уравнению Сегдена
п Afo7,
где р£ и pv—плотности, соответственно жидкости и пара в граммах на миллилитр при комнатной температуре. Парахор можно также
Реальные газы
223
вычислить, зная структурную формулу органического соединения и пользуясь аддитивными константами (см. табл. 4). Критическое давление вычисляется по уравнению
_ 20,87^
Р °кр —8’
где Ткр дано в градусах Кельвина.
Вычисленные значения для 42 веществ сравнивались с экспериментальными значениями. В случае Т максимальное отклонение оказалось равным 5°/0. Для v получилось хорошее совпадение, за исключением двух случаев, одним из которых была вода, которая вообще относится к аномальным веществам, поскольку дело касается общих зависимостей. Вычисленные критические давления менее точны; но за исключением 5 веществ максимальная ошибка не превышала 11°/017-
Таблица 4
Атомные и структурные парахоры
Элемент Парахор , Структурный элемент Парахор
С 4,8
Н 17,1 Двойная связь .... 23,2
N 12,5 Тройная связь .... 46,6
Р 37,7 Трехчленное кольцо . . 16,7
О 20,0 Четырехчленное кольцо 11,6
S 48,2 Пятичленное кольцо . . 8,5
F 25,7 Шестичленное кольцо . 6,1
С1 54,3 О2 в сложных эфирах . 60,0
Вг 68,0
I 91,0
Диаграмма извлекаемого объема. Одним из наиболее общих типов задач, встречающихся при работе с газами, находящимися под давлением, является вычисление объема газа, выпускаемого из хранилища данных размеров или нагнетаемого в него. Если для данного газа нужно произвести много таких расчетов, то целесообразно построить диаграмму с непосредственным отсчетом. Из уравнения (14) следует, что
СгГгрг (ЪРЛ
Если берется равным 1 м3 и выбираются некоторые стандартные условия для р2 и Т2, то объем v2 при низком давлении определяется двумя переменными рг и 7\. Удобная диаграмма получается при нанесении v2 как функции р2 в виде ряда изотерм. Такой график для
224
V. Соотношения давление — объем — температура для газов
азота в ограниченных пределах дается на рис. 17. Так, непосредственно из диаграммы можно определить, что если азот находится в хранилище при 200 атм манометрического давления и при 20° С, то из каждого литра емкости хранилища будут извлекаться количества азота, которые при 1 атм и 15° С займут объем в 174 л. Для хранилища любых других размеров извлекаемый объем будет прямо пропорционален его емкости ☆).
Рис. 17. График для расчета объема азота, извлекаемого из резервуара (данные из Интернациональных критических таблиц).
УРАВНЕНИЯ СОСТОЯНИЯ 18
Общая форма. Вообще уравнениями состояния называются уравнения вида
<р(р, V, Т, т) = 0. (27)
Обычно имеют дело с 1 молем, и поэтому обыкновенной формой является уравнение
ср (р, v, ТУ = 0. (28)
Для практических целей наиболее полезным уравнением состояния является уравнение, определяющее объем, например
v = и(р, Т), (29)
☆) Подобные диаграммы для других газов и различные полезные сведения о свойствах газов см. [36].
Уравнения состояния
225
поскольку р и Т — наиболее общеупотребительные независимые переменные. Однако большинство уравнений состояния было выведено на основе кинетической теории, а это обычно приводит к уравнению, определяющему давление. Уравнения могут быть также чисто эмпирическими, как, например, уравнения вида
и
pv = а Ьр ср2 -j- dp3 -|- ... , (31)
где А, В, а, Ь и т. д. являются функциями температуры, представленными, в свою очередь, в чисто эмпирической форме такими уравнениями, как
С точки зрения практического применения все уравнения состояния в сущности следует считать эмпирическими. Даже когда их можно вывести с помощью приложения кинетической теории, константы следует определять из экспериментальных данных. Кроме того, нельзя полагаться ни на одно уравнение состояния, когда дело касается точного представления данных по сжимаемости далеко за пределами давления и температуры, для которых определены константы. Другими словами, они являются эмпирическими уравнениями, полезными для интерполяции, но по ним нельзя надежно экстраполировать. Вывод общей формы уравнения из кинетической теории, возможно, имеет то преимущество, что дает уравнение, представляющее большое количество данных с минимальным числом констант, меньшим, чем может понадобиться для уравнения, полученного полностью на эмпирической основе.
Некоторые уравнения состояния выведены на термодинамической, а не. на кинетической основе. Например, можно начать с основного уравнения для эффекта Джоуля — Томсона, а именно:
T(^\-V = }lCP’ <97’ гл‘ Ш>
где
Принимая, что ji и Ср находятся в определенной зависимости от р и Т, можно проинтегрировать диференциальное уравнение. Будем считать С„ постоянным, а р не зависимым от давления, но связанным с температурой посредством уравнения
а
И 1
15 Химическая термодинамика
226
V. Соотношения давление — объем — температура для газов
где а и п — константы. Подставляя это выражение в уравнение (97, гл. Ш) и несколько преобразуя его, получаем
Tdv — vdT___ аСр
рг j-n+2al
или
Интегрируя при постоянном давлении, имеем
v _
У
___Q-Ср_ (П+\}ТП^
<р(р)-
(32)
Если мы примем, что рассматриваемый газ приближается к состоянию идеального газа по мере приближения Т к бесконечности по изобаре, то
v R
тогда г
и уравнение (32) переходит в
Это уравнение иногда называют уравнением состояния Каллендара, поскольку оно было им использовано в работе по свойствам водяного пара.
Предельные условия. Имеются определенные предельные условия, которые должны удовлетворяться любым уравнением состояния. Наиболее важные из них следующие:
1. pv = RT, когда р—>0 при любой температуре. Другими словами, уравнение состояния при низких давлениях должно переходить в уравнение идеального газа. С другой стороны, следует отметить, что величина а, которая равна (RTjp)— v, не обязательно достигает нуля, когда р—>0 (при постоянной температуре). Рис. 18 показывает ход изотерм а — р для окиси углерода и метана.
Тот факт, что pv—>-RT, но (RT/p) — v не достигает нуля, кажется с первого взгляда парадоксальным, но, вероятно, станет более ясным из следующего простого рассуждения.
о у*
По определению, ——v — a,
где а— конечная величина.
Умножая обе части уравнения на р, получаем
RT — pv = ар,
Уравнения состояния
227
если р = 0, ар = О и pv = RT, даже если а^О. То же легко можно выявить и с помощью следующего уравнения, которое, как было показано [110], точно воспроизводит изотермы трудно сжимаемых газов до 100 атм:
(34)’
pv — RTарЬр2ср^
Рис. 18. Изотерма зависимости а от р.
'v = ~- + а + Ьр-\-ср2. (35)
Если р = 0, то, по уравнению (34), pv = RT и, по уравнению (35),
2. Предельное значение vfT вдоль изобары представляет величину, которая, как мы убедимся ниже, встречается в некоторых задачах. По уравнению идеального газа можно ожидать, что
,. v R
Г/ ~ Р ‘ „(36)
15*
.228
V. Соотношения давление — объем — температура для газов
Большинство уравнений состояния, как можно видеть по уравнениям, приводимым далее в этой главе, выражается в форме, определяющей -объем; в пределе, когда 1/Т—>-0, они превращаются в уравнение (36); I однако анализ некоторых экспериментальных данных не подтверждает этого правила. Следовательно, этот вывод нельзя еще считать определенно установленным; поэтому мы пока можем только принять, что уравнение (36) выражает предельное поведение по изобаре.
3. Наклон кривых pv—р (или кривых коэфициент сжимаемости — давление) при постоянной температуре для идеального газа равен нулю при всех температурах. В случае реального газа этот наклон при р, стремящемся к нулю, может быть равен нулю и может быть больше или меньше нуля, в зависимости от температуры (см., например, рис. 15). Выразим это математически:
РЧйгП (когда р-0). (37)
L °F Jr
Если эта производная становится равной нулю, мы имеем критерий для точки Бойля. При всех температурах выше этой точки производная будет больше нуля.
4. Критическая изотерма, т. е. кривая, связывающая давление и объем при критической температуре, должна иметь точку перегиба, что отвечает условиям
(ж), =° (38>
кр
И
Шг =»• ™
«р
5. Кривые р—Т (изохоры) линейны, за исключением участков, соответствующих очень большим плотностям, т. е.
(М=° (40>
приближенно для большинства условий и точно, когда Т—>-оо. При большой плотности
(41)
т. е. изохоры выгнуты вверх по отношению к оси Т.
Уравнение Ван-дер-Ваальса. Одной из наиболее ранних попыток представить поведение реальных газов посредством уравнения
Уравнения состояния
229
является работа Ван-дер-Ваальса, относящаяся примерно к 1873 г. Он предложил следующее уравнение, известное под его именем:
— b) = RT, (42)
или
ЯГ а ....
0 —-----к----7- (43)
г V — О V2 ’ '
Оно отличается от уравнения идеального газа добавлением члена а/©2 к давлению и вычитанием величины b из объема, ajv2 является поправкой к давлению, обусловленной силами притяжения между молекулами; ее называют внутренним или, иногда, кохезионным давлением. Другими словами, реальное давление р, оказываемое газом на стенки сосуда, меньше идеального давления рг на величину a/v2, или р=рг — (а/®2)- Константа b является поправкой к общему объему, обусловленной действительным объемом, занимаемым самими молекулами. а и b—константы, характерные для данного газа, тогда как R представляет и в этом случае универсальную газовую постоянную.
Посмотрим теперь, как это уравнение согласуется с пятью упомянутыми выше критериями. Относительно критерия 1 сразу видно, что, когда v—>оо, уравнение дает pv — RT.
Уравнение можно записать в форме
RT । к а Ь 1лл\
v =-----\-b-----------5, (44)
р 1 pv pv2
или
ЯГ а . b .
-----v — a = 5 — b. р----pv 1 pv2
При >0 = (45)
т. е. а приближается к конечному значению, когда р—>-0 по изотерме. Делением уравнения (44) на Т получаем
v R । Ь а b pvT~pH2T'
v R
Когда при постоянном р 1)Т—»-0, то -^ = —, т. е. удовлетворяется критерий 2.
Диференцирование произведения pv по р дает
Определяя правую сторону из уравнения состояния и принимая предел при р—»-0 или v—»-оо, находим:
Р(/М 1 __ъ_
L дР Jr RT'
230
V. Соотношения давление — объем — температура для газов
Приравнивая это соотношение нулю, получаем
Т= — bR'
(46)
Рис. 19. Изотермы Ван-дер-Ваальса для углекислоты вблизи критической точки.
где Т-—точка Бойля. При температурах выше этого значения наклон d(pv))dp при р = 0, как легко заметить, больше 0, а при всех значениях Т ниже Т бойля он’ меньше 0. Следовательно, уравнение Ван-дер-Ваальса отвечает условию 3.
Как показывает простое преобразование, уравнение Ван-дер-Ваальса содержит объем в третьей степени. Другими словами, имеются три значения объема для данных р и Т. С другой стороны, не все эти корни могут быть действительны; это легко доказать тем, что выше определенного значения Т имеется только один действительный корень и два мнимых. Ниже этого значения имеются три действительных корня, и при этом определенном значении Т
все три корня равны между собой (точка перегиба на кривой р — v). Все это отражается уравнением Ван-дер-Ваальса, графически представленным на рис. 19. Вдоль изотерм и Т2 имеется только один действительный корень, т. е. только одно значение и для данного р. По мере понижения температуры достигается температура Ткр, для которой имеется точка перегиба на кривой р — v т. е. критическая точка. При более низких температурах, например при Т4, существуют некоторые давления, для которых имеются три действительных и неравных объема, например v1, v2 и -v3. Это поведение, предсказанное для температур ниже критической, конечно, не согласуется с наблюдаемыми фактами, по крайней мере для равновесных состояний. В действительности вместо непрерывного изменения, указанного
уравнением, в некоторой точке, например в точке v}, имеется резкое изменение вследствие появления второй фазы. Начиная с этой точки, давление остается постоянным, тогда как объем уменьшается
Уравнения состояния
231
до тех пор, пока не достигнет значения, отвечающего точке vs, и не исчезнет первая фаза; после этого при увеличении объема давление падает. Следовательно, уравнение Ван-дер-Ваальса предсказывает критические явления и согласуется с общим характером р — w-кривых выше критической температуры.
Из уравнений (38) и (39) имеем
др\ ____Q=_ — RT . 2я
ди) Т (и — Ь)2 и3 ’
сРр\ _п_______ 2RT 6а
dv2) г (и — b}:l v4'
(47)
(48)
Из этих двух уравнений и уравнения Ван-дер-Ваальса получим = (49)
а
(51)
Ркр 21Ь2
Эти уравнения дают возможность вычислить координаты критической точки из констант уравнения а и Ь. Практически производят обратную операцию и вычисляют а и b нз экспериментально определен-
ных значений из уравнений
р и Т используя следующие уравнения, полученные (50) и (51):
__27 «X
—64 Ркр ’ А__RTkP
*Ркр'
(52)
(53)
Значения а и Ь, вычисленные из этих уравнений, даются в табл. Ш „Приложения*. Конечно, можно представить а и b как эмпирические константы и определить их наилучшие значения для данного ряда величин. Это в общем будет давать уравнение, представляющее эти частные случаи более точно, чем уравнение с константами, определенными по уравнениям (52) и (53). Однако следует помнить, что уравнение Ван-дер-Ваальса является в лучшем случае приближением; его основное значение заключается в возможности производить предварительные подсчеты тогда, когда доступны лишь немногие данные и когда точность приносится в жертву удобству.
Применяя критерий 5, находим, что
дт)и
R v — Ь
следовательно, уравнение требует линейности изохор.
232
V. Соотношения давление — объем — температура для газов
Мы видели, что уравнение Ван-дер-Ваальса правильно представляет некоторые общие соотношения; остается еще показать, насколько близко оно согласуется количественно с поведением реальных газов. Из уравнений (49), (50) и (51) находим, что
Ркр vxp 3
(54)
т. е. коэфициент сжимаемости в критической точке имеет одно и то же значение для всех газов. Обращение к табл. 111 „Приложения* показывает, что этот коэфициент значительно отклоняется от указанного значения и никоим образом не является одинаковым для всех газов. Так как для идеального газа коэфициент сжимаемости должен быть равным 1,00, то ясно, что это уравнение является значительно лучшим приближением к действительности, чем уравнение идеального газа.
Подставляя для а и b в уравнение (46) значения, полученные из уравнений (52) и (53), имеем для точки Бойля
7бойля = 3,375 7’к/,.
(55)
В действительности Тбойля не сохраняет постоянного отношения к критической температуре для всех газов, а значительно изменяется при среднем значении около 2,5.
Вычисление давления посредством уравнения Ван-дер-Ваальса при данных объеме и температуре или вычисление температуры прн данных объеме и давлении относительно просто и не требует подробных объяснений. Следует заметить, что v в уравнении представляет удельный или мольный объем. Следовательно, если нужно вычислить давление, создаваемое в сосуде емкостью в V литров, когда в нем заключено т граммов газа, то необходимо только вычислить удельный объем Щт или мольный объем MV/m, а затем подставить это значение в уравнение. Если дан не вес, а исходный объем при низком давлении, то вес можно вычислить с достаточной точностью из закона идеального газа при условии, что давление не очень отличается от атмосферного. Если желательна ббльшая точность, то уравнение Ван-дер-Ваальса можно приложить к обоим состояниям газа; но это уточнение недостаточно оправдывается вследствие приближенности самого уравнения.
Вычисление объема по уравнению Ван-дер-Ваальса при данных давлении и температуре наиболее просто осуществляется методом подбора.
Пример 4. Чему будет равен объем, занимаемый 4 кг СО2 при 50 атм и 100° С?
По табл. III „Приложения* константы а и b соответственно равны 8,604 и 0,0428. При отсутствии каких-либо ориентировочных данных относи-
Уравнения состояния
233
телыю объема первый подбор удобно произвести, используя объем идеального газа, который легко определяется следующим образом:
v
RT Р
0,08206 X 373,2
50
= 0,612 л/моль.
Подстановка этого значения v в уравнение (43) дает р = 44,2,
что является слишком низким значением. Из этого первого подбора очевидно, что следует взять меньший объем. Ниже показаны результаты нескольких подборов:
v = 0,50 л р~ 52,62 атм ,=0,524 . ,=50,48 »
, = 0,527 , ,=50,23 .
, = 0,5297 . . = 50,03 .
. = 0,530 . .=50,00 .
Конечный результат можно получить с достаточной точностью путем линейной интерполяции между близкими заданными величинами:
Объем 4 кг СО2 = —тт- X 0,530 = 48,2 л.
44
Степень точности, достигаемая при использовании уравнении Ван-дер-Ваальса с константами, найденными нз критических давления и температуры, показана в табл. 5 для нескольких случаев, умышленно выбранных таким образом, чтобы получить более жесткие условия испытания.
Таблица 5
Приложимость уравнения Ван-дер-Ваальса
Газ Т,°К р, атм Г Объем, л) моль
наблюдаемый по уравнению Ваи-дер-Ваальса идеальный (по уравнению Клапейрона)
СО2 313,1 72,16 100,0 1,03 0,99 1,387 0,200 0,0693 0,209 0,0893 0,356 0,257
273,1 200 1000 2,165 5,96 29,8 0,116 0,0463 0,111 0,0539 0,1120 0,0224
н2 273,1 200 1000 8,22 15,6 78,0 0,1272 0,0384 0,130 0,0467 0,1121 0,0224
NH3 323,1 20,06 0,795 0,180 1,077 1,193 1,321
Из табл, 5 видно, что во всех случаях уравнение Ван-дер-Ваальса представляет значительный шаг вперед по сравнению с уравнением идеального газа; исключение составляют данные дли азота при 200 атм\ эти данные приходятся около точки (см. рис. 13), где азот подчиняется уравнению идеального газа, хотя он в действительности и далек от идеального состояния. Конечно, для любого газа имеется громадное число отдельных состояний, в которых газ
234
V. Соотношения давление — объем — температура для газов
ни в коем случае не является идеальным, но в точности подчиняется уравнению идеального газа (положение всех точек опреде ляется условием С=1). Во всех рассмотренных случаях, за одним исключением, плотность была порядка критической плотности; только в случае аммиака мы имели дело с насыщенным паром.
Перед тем как закончить обсуждение точности уравнения Ван-дер-Ваальса, следует указать, что получающаяся степень совпадения зависит от того, какие именно величины вычислялись. Из табл. 5 видно, что уравнение Ван-дер-Ваальса для вычисления объема N2 при 0° С и 1000 атм значительно точнее, чем закон идеального газа; но если мы возьмем измеренный объем при 0° С и по нему вычислим давление, то оно по уравнению идеального газа окажется равным 484, а по уравнению Ван-дер-Ваальса — 2293 атм. Оба значения содержат весьма значительную ошибку, сильно отклоняясь от 1000 атм, и трудно сказать, какое из этих уравнений дает лучшие результаты.
Приведенные уравнения состояния. При подстановке в уравнение Ван-дер-Ваальса значений а, b и R, полученных из уравнений (52), (53) и (54), получается следующее уравнение:
(tt+v4)(3v— 1) = 8т, (56)
где тг, > и т— определенные ранее приведенные переменные состояния. Это уравнение не содержит никаких произвольных констант, характеризующих индивидуальные газы, т. е. оно представляет универсальное уравнение состояния, приложимое ко всем „ван-дер-вааль-совским газам". Можно сказать, что все газы подобны друг другу, если они находятся в состояниях, при которых их параметры составляют одинаковые доли критических параметров; в этом случае говорят, что газы находятся в соответственных состояниях. Следовательно, теория соответственных состояний, которой мы уже успешно пользовались в этой главе для характеристики реальных газов, основывается главным образом на применимости уравнения Ван-дер-Ваальса или других уравнений состояния, содержащих не более 3 констант (включая газовую постоянную /?)19. Так как ни одно из этих уравнений не точно для всей области изменений р — v— Т, то ясно, что закон соответственных состояний и основанные на нем выводы являются только приближенными. Тем не менее это приближение вносит громадное упрощение, а точность его достаточна для многих технических целей.
Применим теперь приведенное уравнение состояния (56) для получения некоторых выводов о поведении газов. Рассмотрим, например, изменение произведения pv с давлением при постоянной температуре (т. е. отклонение от закона Бойля). Подставляем х = тп» в уравнение (56) и получаем
(1 +§}(3х-тг) = 8т. (57)
Уравнения состояния
235
Продиференцируем уравнение (57) при постоянной температуре и положим, что (Ох/дтг) = О. Тогда получается, что
х(9 —х) = 6тг.
Это соотношение между irv и тг, нанесенное на рис. 20, определяет геометрическое место всех точек минимума на кривой pv — р. Каждая точка этой кривой соответствует различной температуре, причем последняя возрастает по мере увеличения pv. Если тг —0, то irv = O и 9. Если тп» —9/2, то тг = 3,375, т. е. максимальному значению тг. Из уравнения (57) имеем
т= 1,90, если ttv = 9/2 и тг = 3,375, т=3,375, если ir> = 9,0 и тг = О.
Рис. 20. Положения минимумов на кривых, выражающих зависимость л» от л.
В применении к поведению реальных газов это означает, что при повышении температуры минимумы на кривых pv — р сдвигаются вправо до тех пор, пока они не достигнут, наконец, изотермы т=1,90 как предела; для всех более высоких температур минимумы сдвигаются влево; при г = 3,375 (точка Бойля) минимум приходится на р = 0; для всех более высоких температур нет минимума. При применении этих выводов к реальным газам при температурах порядка комнатной этилен (т = 0,95 при 25°С) должен показывать на изотерме pv—р сдвиг минимума вправо, азот (т = 2,37 при 25° С) — сдвиг
влево, а водород (т = 8,98 при 25° С) не должен показывать никакого минимума. Эти выводы согласуются с наблюдаемыми фактами. Нельзя, конечно, ручаться за точность количественных предсказаний, как это уже указывалось выше. Таким же путем можно вывести определенные качественные заключения об эффекте Джоуля — Томсона в газах, но метод, аналогичный этому, был уже дан для точек Бойля и поэтому здесь излагаться не будет.
Уравнения Вертело и Дитеричи. Было предложено очень большое количество (свыше 100) уравнений состояния, но только немногие из них стали общеупотребительными. Рассмотрим здесь очень кратко несколько уравнений, а затем подробнее два из них.
236
V. Соотношения давление — объем — температура для газов
Две ранние попытки усовершенствовать уравнение Ван-дер-Ва-
альса привели к следующим уравнениям:
(уравнение Дитеричи), (59)
Р v — b
RT а_ Р v — b Tv2 (уравнение Вертело). (60)
Оба эти уравнения, как и уравнение Ван-дер-Ваальса, содержат
по две константы; поэтому константы а и b можно выразить через критические параметры тем же самым методом, как это было сделано для уравнения Ван-дер-Ваальса, и получить приведенные уравнения. Соотношения для констант уравнений Дитеричи и Вертело соответственно следующие:
а = РкРе2 ’ (61)
Ь- _ RTkP . Ркр^ (62)
а- _27 R2T3 64 рКр (63)
Ь = — ^ТкР *РКр' (64)
Для уравнения Вертело следующие эмпирические уравнения дают на практике наилучшие результаты:
_^32
Ркр^кр 9 ’
ь_ 9 RTкр 128 Ркр •
(65)
(66)
Уравнение для а остается таким же, как н уравнение (63).
Не может быть дано никаких применимых во всех случаях положений о точности, с которой эти два уравнения представляют значения р — v—Т различных газов по сравнению с уравнением Ван-дер-Ваальса. Каждое из этих уравнений может оказаться лучшим в том или другом случае в зависимости от природы газа или от условий его применения. На рис. 21 и 22 сравниваются резуль таты расчета по трем уравнениям с опытными данными для двух различных газов (азота н этилена) при 25° С. Эти газы выбраны потому, что один из них находится вблизи своей критической температуры, а другой—много выше ее. Константы уравнения Вертело вычислены по уравнениям (63) и (64). Для более детального сравнения
Уравнения состояния
237
Рис. 21. Сравнение уравнений Ван-дер-Ваальса, Вертело и Дитеричи С опытными данными для азота при 50° С [36].
Давление, атм
Рис. 22. Сравнение уравнений Ван-дер-Ваальса, Вертело и Дитеричи с опытными данными для этилена при 40° С [36].
этих трех уравнений можно обратиться к циркуляру № 279 Бюро стандартов США [36].
Уравнение Воля. Это уравнение
RT_______а с
v — b v(v — b)' v3
238
V. Соотношения давление—объем — температура для газов
интересно тем, что имеет три константы, которые можно определить из критических констант. Для этого служат следующие уравнения:
a = 6v2KppKp,
с — 4vKp ркр.
Критический объем редко бывает точно известен, и его можно определить с помощью отношения
По исследованию, произведенному Волем [260], уравнение дает хорошие результаты в широких пределах, и если это действительно так, то оно заслуживает большего внимания, чем то, которое до сих пор ему оказывалось.
Некоторые уравнения, определяющие объем. Все до сих пор рассмотренные уравнения определяли давление и принадлежали к линейно-изохорному типу. Для некоторых приложений гораздо удобнее располагать уравнением, определяющим объем. Уравнением этого типа, полезным в технических работах, является уравнение Гудинафа
® = ^ + у-^-(1+За^), (67)
где с, т, п и а — эмпирические константы, а также уравнение Линде
р (С-4-Ер) , pv = AT — P- 4-р(Д+Др), (68)
где А, С, D, Е и F—также эмпирические константы. (Заметим, что А должно совпадать с газовой постоянной R.) Уравнение Гудинафа в несколько измененной форме использовано при корреляции термодинамических свойств водяного пара [53]. Уравнение Линде, первоначально предложенное им для перегретого водяного пара, использовано для вычисления термодинамических свойств некоторых дестил-лятов нефти [6].
Бюро стандартов США [36] для вывода термодинамических свойств перегретых паров аммиака использовало следующее уравнение:
АТ В C-\-Dp Еръ
v р Ts Тп Г19 (69)
где А, В, С, D, Е и Е—эмпирические константы, а (р) определяется уравнением
<р (р) = а Ьр + ср2 -]- dp3.
Уравнения состояния
239
Уравнение (69) содержит 10 эмпирических констант, и мы привели его главным образом для иллюстрации того, что точное изображение зависимости р— v—Т для газов, даже в довольно ограниченных пределах, может потребовать слишком сложного уравнения со многими взаимно согласованными константами. Несмотря на эту сложность, уравнение состояния оправдывается большими удобствами, даваемыми им в процессе вычисления термодинамических свойств. При вычислении новых значений термодинамических свойств водяного пара Кейес с сотрудниками [132] нашел необходимым вывести уравнение с 10 эмпирическими константами, и даже тогда оно не было правильно для насыщенного пара при объемах, меньших 10 мл^г.
Уравнение Кейеса. Уравнение, воспроизводящее в широких пределах экспериментальные данные с очень большой точностью и связанное с небольшим числом входящих в него констант, предложено Кейесом [131]; оно имеет следующий вид:
RT А (/0)
где а, р, А и I — эмпирические константы. При рассмотрении уравнения (70) становится очевидным, что оно представляет видоизмененную форму уравнения Ван-дер-Ваальса. Подобно последнему, оно требует линейности изохор, но имеет более двух произвольных констант, и поэтому можно было бы ожидать, что оно будет более точно воспроизводить опытные данные. В силу того, что это уравнение содержит большое число констант, его нельзя дать в приведенной форме. Другим существенным его недостатком является большая трудоемкость вычислений, так как оно неудобно для определения некоторых термодинамических свойств вследствие того, что интеграл ^dvj(v—$е ®) не может быть определен как простая функция.
Нахождение констант уравнения Кейеса сравнительно просто, и в процессе расчетов видно, как хорошо уравнение соответствует опытным данным, что является его большим практическим преимуществом. Кроме того, имеется еще одно практическое преимущество — требование лишь ограниченного количества исходных данных. Ниже будет даио краткое описание метода вычисления, для того чтобы наглядно показать некоторые важные его особенности. Значения р— v — Т сначала следует представить в форме изохор, т. е. ряда давлений и температур при постоянном объеме. Экспериментальный метод, предложенный Кейесом, предоставляет эти данные прямо в нужной форме, но и данные в виде значений р — v вдоль изотермы можно графически обработать для приведения их к изохорной форме. Уравнение изохор имеет следующий вид:
р = ^Т~ (72)
240
V. Соотношения давление — объем — температура для газов
где
Ф=Л <73>
И
<74>
Таким образом, ф и <р являются функциями только объема, и их значение для каждой изохоры легко определяется обычными методами нахождения констант уравнения прямой линии.
Из уравнения (74) получаем
. I У /1 >
+ (75)
Таким образом, график v— 1/V<p будет также линейным, причем наклон равен ]/ д, а отрезок на оси ординат равен I. Из уравнения (73) вычисляется значение § для соответствующих значений v и ф; и так как S и v связаны уравнением
lnS=lnjl —-(76)
то график 5 (или 1g 5)—1/v представляет прямую линию, по которой легко найти а. и jl. Следовательно, все константы уравнения определяются из прямолинейных графиков.
Уравнение Битти—Бриджмена. Несколько более сложное, но исключительно полезное уравнение предложено Битти и Бриджменом [12j:
LL^2r+в Л _ Ml _а_\ (77)
v2 L * \ v / J \ v) '
где а, Ао, Ь, Во и с — эмпирические константы, значения которых следует определить из экспериментальных данных. Все константы, кроме с, определяются из прямых линий способом, подобным примененному для уравнения Кейеса. Для деталей следует обратиться к оригинальной статье. Уравнение очень точно до тех пор, пока его применение ограничено условиями, для которых определялись константы. Битти и Бриджмен первоначально применили его к данным по 10 газам до максимальных давлений от 100 до 200 атм и до плотностей, приближающихся к критической, — среднее отклонение вычисленных давлений от наблюденных оказалось равным только 0,18°/0. Другими словами, можно считать результаты расчета лежащими в пределах точности эксперимента.
Исследуем теперь это уравнение с точки зрения приведенных выше критериев. Сначала преобразуем уравнение следующим образом:
/п' = Я7' + -|’+^ + ^, (78)
•Уравнения состояния
241
где (i, Y и 8—функции температуры:
₽=/?ТВ0-Д0-^, - (79)
7 = -/?ТВ0& + аД0-^, W
. _ RBfic °- Т2 ’ (81)
У равнение (78) известно как „внрнальная* форма уравнения состояния.
Если р—*0 и v—и все члены, содержащие v в знаменателе, будут ничтожно малы, то уравнение примет форму уравнения идеального газа
pv=RT.
Из уравнения (78) следует, что
RT -----v = a —
Р
J_____I____L
pv pv2 pv2
Так как Ijv—>0, то pv—>-RT н
а =
Следовательно, мы видим, что а не равна нулю, а в согласии с опыт-’ ними фактами является конечной величиной и функцией температуры.
Днференцируя уравнение состояния по р при постоянной Т и полагая v — oo или р = 0, получаем
lira
РСР»)] __ ? __о 4)_______£.
L Ф Jr-' RT~~ ° RT Л *
(82)
При принятии этого предела равным нулю получающееся кубическое уравнение имеет только один действительный корень, и он является температурой точки Бойля. Когда 7’^>7’бойля, производная больше нуля, и наоборот, при ТТбойля она меньше нуля.
Приложение критериев для критической точки [уравнения (38) и (39)] приводит к двум уравнениям, которые, повидимому, дают единственное решение для температуры н объема (следовательно, и для давления), и благодаря этому критерий удовлетворяется.
При диференцировании уравнения состояния иаходнм
(83)
16 Химическая термодинамика
242 V. Соотношения давление—объем—температура для газм
Легко показать, что для большинства значений v и 7* величина (dipldTz)v должна быть небольшим отрицательным числом, приближающимся к нулю как к пределу при всех объемах, когда Т—► сю (линейные изохоры), и при всех температурах, когда 1/v—<-0.
Таким образом, мы видим, что уравнение Битти — Бриджмена весьма удовлетворительно выдерживает критерии, накладываемые на любое уравнение состояния поведением газов.
В табл. 6 даются константы этого уравнения, определенные для
различных газов.
Таблица б
* Ki jfe? Постоянные уравнения состояния Битти — Бриджмена (единицы: атм, л, моли, ° К, 0,08206)
Газ а Во й Д.10-‘ Температурный интервал, ° С Максимальное давление, атм ! Минимальный объем, мл/моль Литературный 1 источник
от до
Не 0,0216 0,05984 0,01400 0,0 0,0040 400 —252 102 100 (12)
Ne 0,2125 0,02196 0,02060 0,0 0,101 400 -217 106 118 (12J
Аг 1,2907 0,02328 0,03931 0,0 5,99 400 —150 114 167 (12]
* К??' Н, И. (ниже критической 0,1975 -0,00506 0,02096 —0,04359 0,0504 200 —244 103 100 (121
плотности) УН (выше крн-. пгаеошй 0,12404 0,05618 0,02022 -0,00722 2,00 300 —70 1000 65 £56]
33* V. влотностн) 0,12404 0,05618 0,01750 —0,01968 2,00 300 —70 1000 37 156]
N, 1,3445 0,02617 0,05046 —0,00691 4,20 400 —149 134 182 (12]
И*) 1,2517 0,01866 0,04603 —0,02587 6,16 400 —70 1000 84 152)
о, 1,4911 0,02562 0,04624 0,004208 4,80 100 —117 103 111 [12]
Воздух 1,3012 0,01931 0,04611 —0,01101 4,34 200 —145 177 125 (12)
г СО, 5,0065 0,07132 0,10476 0,07235 66,00 100 0 111 182 (14
СН. 2,2769 0,01855 0,05587 -0,01587 1233 200 0 243 167
(СУЩ) 31,278 0,12426 0,45446 0,11954 ззлз 325 150 90 370 [12]
С.Н. 6,1520 0,04964 0,12156 0,03597 ' 22,68 200 0 286 125 1891
NH, 2,3930 0,17031 0,03415 0,19112 476,87 325 —35 130 340 £18)
CCl,F, 23,7 0,305 0£9 0,622 0 126 30 18,, 1430 £35]
с,н. 5,8800 0,05861 0,09400 0,01915 90,00 26 250 193 200 ПЧ
С,н, 11,9200 0,07321 0,18100 0,4293 120,00 97 275 305 100 ЦП
«-с,н„ 17,7940 0,12161 0,24620 0,09123 350,00 150 300 118 280 £191
и-СтН,, 54,520 0,20066 0,70816 0,19179 400,00 275 350 315 200 £232}
изо-С,Н, СН.ОН **) 163600 33,309 0,10860 0,09246 0’4200 0,60362 0,08750 0,09929 250,00 32,031 150 250 250 111 (16) )20(
Примечание. Прн умеренны! давлении! и температура!, не слишком близких к критическим, константы для Na можно использовать для СО, а константы для СО, — для N/X
&) Несколько лучшее совпадение получается при введении шестой константы в*, превращающей последний член уравнения (77) в 1—(п/п) — (л'/*’). Различие все же не являете» настолько значительным, чтобы оправдать дополнительное усложнение.
*♦) См. также (348]. (Прим. ред.)
Уравнения состояния 243
Необходимо указать, что не следует ожидать, чтобы уравнение было точным при использовании его вне тех пределов, для которых определены константы. Например, если первое уравнение для азота, которое основано на максимальном давлении в 134 атм, экстраполи-ровать до 1000 атм при 0°С, то окажется, что оно представляет только грубое приближение, незначительно превосходящее в этом отношении уравнение Ван-дер-Ваальса. В любом данном случае лучше всего руководствоваться мольным объемом (или плотностью). В общем, уравнения не будут точны в области, где мольные объемы меньше минимума, указанного в табл. 6.
В отличие от большинства упомянутых уравнений, уравнение Битти — Бриджмена не требует линейности изохор. Если, однако, принять константу с равной нулю, то это приведет к простейшей линейной изохорной форме, которая может оказаться полезной в специальных случаях. Одни из таких случаев представляет холодильный агент дихлордифторметан, константы которого даны в табл. 6. Уравнение в этой форме также применялось к данным по трем другим фторхлорпроизводным метана и к одному фторхлорпроиз-водному этана [22].
Деминг и Шуп [55, 56] применили уравнение Битти — Бриджмена для азота и водорода в более широком интервале условий, чем выбранные Битти и Бриджменом при их проверке уравнения, и нх константы также включены в табл. 6. Введением шестой произвольной константы они добились воспроизводимости наблюденных давлений азота с максимальным отклонением в 1,4°/0 до приведенного объема около 0,94. При больших плотностях совпадение оказалось много худшим; отклонение в одном случае достигает 43°/0. В случае водорода они расширили предел использования уравнения применением различных значений констант выше и ниже критической плотности (0,0154 мл[моль).
Марон и Торнбулл [165] разработали метод определения констант уравнения Битти — Бриджмена для любого газа на основании его критических параметров и констант уравнения состояния стандартного газа. Этот метод основан на допущении, что все газы имеют одинаковые коэфициенты летучести при одинаковом приведенном давлении и одинаковой приведенной температуре. В сущности этот метод является применением закона соответственных состояний, приводящим к алгебраическому, а не к графическому результату.
При применении как уравнения Кейеса, так и уравнения Битти — Бриджмена объем должен быть найден процессом подбора, в точности подобным тому, которой был описан для случая уравнения Ван-дер-Ваальса.
Уравнение Битти. Битти [11] привел уравнение Битти — Бриджмена к следующему виду, определяющему объем:
® = (п+В)(1-е)-^, (84)
16*
244 V. Соотношения давление — объем — температура для газов
где
Константы — те же самые, что и в уравнении Битти — Бриджмена, и можно пользоваться теми же числовыми значениями. Уравнение (84) не так точно, как уравнение Битти — Бриджмена, но совпадение с наблюденными данными достаточно хорошо, если его использование ограничить «состояниями, лежащими много выше критической области, и плотностями, много меньшими, чем критическая. Причиной вывода менее точного варианта уравнения Битти — Бриджмена является то, что форма уравнения, определяющая объем, для некоторых расчетов более удобна20.
ГАЗОВЫЕ СМЕСИ
В промышленной практике чаще приходится иметь дело со смесями газон, чем с чистыми газами. Поэтому важно проследить, каким образом представления и зависимости, справедливые для чистых газов, можно обобщить для охвата более сложного случая смесей. Можно сказать, что положение в этом вопросе в настоящее время далеко не удовлетворительно, так как по смесям произведено сравнительно мало работ. Наша цель в этой главе — дать обзор сведений, имеющихся н настоящее время, причем следует помнить, что картина еще очень неполна и может быть обрисована только в общих чертах.
Под термином „ смесь “ будем понимать молекулярно-гомогенную смесь, т. fe. истинный раствор. Методы выражения состава растворов •уже рассматривались в гл. I. Некоторые смеси, представляющие громадную практическую важность, например встречающиеся при очистке нефти, настолько сложны, что их состав нельзя дать в простых выражениях. Такие смеси скорее будут характеризоваться некоторыми свойствами, которыми они обладают, чем установлением присутствующих в них химических индивидуумов.
Если известен состав смеси, то идеальным путем для определения * ее р — v — Т соотношений является выражение их через эти же соотношения для чистых компонентов. Следовательно, если имеют уравнение состояния для каждого компонента смеси, то простейшим путем для описания свойств смеси было бы некоторое сочетание констант уравнений для компонентов. К сожалению, такое положение еще не достигнуто, ио все же в этом направлении наблюдается значительный прогресс.
Газовые смеси
245
Смеси идеальных газов. Из кинетических представлений об идеальном газе ясно, что поскольку речь идет об изменениях р — v— Т, смесь идеальных газов будет вести себя так же, как и чистый газ. Так как в основе учения об идеальном газе лежит представление об отсутствии сил притяжения между отдельными молекулами и представление о том, что на свойствах газа не сказывается, находятся ли в нем молекулы вещества А или вещества В, то для свойств газа является совершенно несущественным вопрос, окружены ли данные молекулы А молекулами того же вещества А или же вместо молекул А вводятся молекулы В, С и т. д. Другими словами, каждый газ в смеси идеальных газов ведет себя так, как если бы он в данном объеме был один. Конечно, мы предполагаем при этом, что между компонентами газовой смеси не происходит химического взаимодействия.
Рассмотрим смесь трех идеальных газов А, В и С, заключенных в объем V при общем давлении р и температуре Т по абсолютной шкале. Количество каждого из этих газов, выраженное числом молей, обозначим через NA, NB и Nc. Каждый газ, если бы он был один, занимал бы весь объем при той же температуре и оказывал бы определенное давление, меньшее р и вычисляемое из законов идеального газа. Следовательно,
PaV=KaRT, p^NJT, pcV=NcRT. (85)
Если смесь идеальна, то для нее
pV=NRT, (86)
где М=:.Л/л-]-Л7в-]-?/с. Отсюда следует, что
Р = Ра + Рв + Рс- (87)
Деля любое из уравнений (85) на уравнение (87), получаем
p-N-x‘
ИЛИ
р,. = рхг. (88)
Давление р{, определяемое этим уравнением, известно как парциальное давление и будет в дальнейшем изображаться символом pt. На этом основании можно переписать уравнение (87) в форме
Р = Ра + РвА-Рс- (89>
Это уравнение справедливо для любого числа компонентов при обязательном условии, что вся смесь будет считаться идеальным газом. Факт, выражаемый уравнением (87), известен как закон аддитивности
246 V. Соотношения давление— объем— температура для газов
давлений, или закон Дальтона, а уравнение (89) представляет частный случай его применения только к смеси идеальных газов.
Термин .парциальное давление" часто употребляется без точного определения, что приводит к многочисленным недоразумениям в случаях смесей реальных газов. Парциальное давление нами везде будет определяться уравнением (88).
Теперь рассмотрим трн идеальных газа, которые находятся под одинаковым давлением р, но взяты в различных мольных количествах, так что объемы нх различны; для них можно написать
pVA—NART, pV^NgRT, (90)
pVc — NcRT.
Теперь пусть эти три газа смешаются, причем температура и общее давление останутся постоянными. Смесь также должна быть идеальной, и мы можем написать для нее уравнение (86). Сравнивая уравнение (86) с (90), видим, что
1^=КД4-УВ+КС. . (91)
Другими словами, для идеальных газов трн постоянных температуре и давлении объемы аддитивны. Следует отметить, что объемы в уравнении (91) являются общими объемами всего взятого количества газа. При записи через мольные объемы уравнение переходит в
v=vaxa+v^xb+vcxc- (92)
Этот закон аддитивности объемов также известен как закон Амага или закон Ледюка. Аналогично парциальному давлению можно определить парциальный объем по уравнению
vt = xp>. (93)
По аналогии со случаем отдельного идеального газа можно написать для любой смеси идеальных газов
где Мт — средний молекулярный вес газа и т — общий вес всех компонентов. Сравнивая уравнения (94) и (86), получаем
м ___т___MANA । MBNB MCNC ।
Mm — N— N + N "T N "T • • •
ИЛИ
Мт = ХАМАЛ-ХВМв + ХСМс+ • • • • <96>
По уравнениям (94) и (96) сложная смесь постоянного состава может рассматриваться так, как если бы она была чистым газом с молекулярным весом, равным среднему молекулярному весу смеси.
‘ . Г * / . Н 1
И Газовые сжса 247
‘ я
Пример 5. Вычислить плотность газовой смеси, состоящей из 5Оо/оН», ЗО®/оNj, 18®/о СО, и2% Н2О (объемные проценты), если t = 25° С ир = 1,5атм.
Мт = 0,500 X 2,016 4- 0,300 X 28,00 + 0,18 X 44,0 + 0,020 X 18,00 = 17,69.
--------------- п т-----рМт 1,5X17,69 , лол—7— По уравнению (94) плотность = у= = 0/)82^х = 1,084 г/л.
В этом случае принято допущение, что объемный процент газа равен его мольному проценту. Это строго только для идеальных газов, как можно показать делением одного из уравнений (90) на (86); при этом получается соотношение
'"I
'i
которое в общем несправедливо для любого идеального газа.
Закон аддитивности давлений. Мы все же можем -принять закон аддитивности давлений (закон Дальтона), даже когда газ не является идеальным. Тогда для смеси газов при постоянных объеме и температуре можно написать
Р=Ра+Рв+Рс> (98)
рА, Рв и т- Д- в данном случае означают давления, которые газы А, В и т. д. оказывали бы, занимая каждый весь общий объем при данной температуре. Эти давления не следует рассматривать как парциальные давления, потому что в общем онн не находятся в такой зависимости от общего давления, которая требуется уравнением (88), принятым нами для определения парциального давления.
Для частного случая, когда зависимость р — v — Т для каждого отдельного газа может быть представлена уравнением состояния в форме, определяющей давление, уравнение (98) можно представить в виде
p=^(v, T,xA) + y2(v,T,xB)+ , (99)
где w (и, Т, xt) — уравнение состояния в форме, определяющей давление для данного чистого компонента, занимающего мольный объем смеси при той же самой температуре, что и смесь. Даже если бы уравнения состояния отдельных газов могли применяться в форме, определяющей объем, можно было бы применить закон аддитивности давлений, выраженный уравнением (98), но его применение менее удобно, так как для отдельных давлений оно требует решения методом подбора. Для идеального газа у (у, Т, х() имеет вид х{ЦТ]ф. Если в чистом состоянии газы подчиняются уравнению Ван-дер-Ваальса, то мы имеем для любого газа
NtRT
р V — Nfii Vs ’
248 V. Соотношения давление—объем —температура для газоо
где V— общий объем. Это уравнение получается непосредственно из уравнения (43) подстановкой
V
Если общее число молей смеси равно единице, то переходит в
__ X[RT *iai
Р t>— Xjb/ v2 ’
уравнение (100)
(101)
где v—мольный объем смеси. Правая сторона уравнения (101) представляет собой <р (v, Т, хг), как требуется для уравнения (99); подстановка его значения в уравнение (99) дает
г=* • )-?^+vb+- •). <Ю2)
где аА и ЬА — константы Ван-дер-Ваальса для компонента А. Подобные же уравнения могут быть получены прн сочетании с уравнением (98) любого другого уравнения состояния, определяющего р.
Другая форма закона аддитивности давлений ☆) может быть написана следующим образом:
Р=хаРа + хвРвЛ~ , (ЮЗ)
где рА, рв...—давления, которые должны были бы оказывать отдельные компоненты при температуре смеси н при мольном объеме, совпадающем с объемом смеси. Формулировки закона аддитивности давлений, данные уравнениями (98) н (103), эквивалентны для идеальных, но не для реальных газов. Например, если компоненты подчиняются уравнению Ван-дер-Ваальса, то при использовании уравнения (103) получаем для смеси
/>=ет(^+^+...)-^(«лхл+«Л+...), («и)
что не совпадает с уравнением (102)21.
Закон аддитивности объемов. Аналогичным образом можно обобщить закон аддитивности Амага и написать уравнения (91) и (92) для случаев, когда смесь не является идеальным газом. Это просто частный случай понятия идеального раствора, развитого в предыдущей главе. Объемы отдельных компонентов, конечно, относятся к дав-
£?) Эта форма может быть названа законом аддитивности давлений Бартлетта, так как этот закон впервые был использован, повидимому, Бартлеттом с сотрудниками.
Газовые смеси
249
ленню и температуре смеси. Можно написать уравнение (92) в форме
v=xA'bl(p,T)-YxB^2(p,T)-\r ... , (105)
где ф (р, Т) — уравнение состояния в форме, определяющей объем данного чистого компонента при общем давлении ры температуре смеси, a v — мольный объем смеси. Для смеси идеальных газов х;ф (р, Т) имеет вид х^Г'р. Для реальных же газов можно использовать любое уравнение в форме, определяющей объем, например уравнение (84).
Ниже; дается несколько числовых примеров использования этого уравнения для газовых смесей как для того, чтобы показать метод их применения, так и для того, чтобы установить согласие с опытными данными.
Пример 6. 7,075 м3 азотоводородной газовой смеси, содержащей 25 мольных процентов азота при давлении 1 атм и температуре Ю°С, следует сжать в пустом цилиндре емкостью в 0,0283 лГ. Какое надо приложить давление, если после сжатия температура в цилиндре должна равняться 50°С?
Сравним следующие способы расчета этого давлении с опытными данными Бартлетта, Капплса и Тримерна [7]:
1. Закон идеального газа.
2. Сочетание закона аддитивности давлений [уравнение (98)] с уравнением Ван-дер-Ваальса.
3. Сочетание закона аддитивности объемов с уравнением Вав-дер-Ваальса.
4. Сочетание закона аддитивности давлений [уравнение (98)] и уравнении состояния Битти — Бриджмена.
5. Сочетание закона аддитивности объемов и уравнения Битти—Бриджмена.
Экспериментальные данные представлены в виде коэфициента сжимаемости z как функции давления при постоянной температуре. Ниже даны некоторые значения при 50° С.
р, атм 1 50 100 200 300 400
Z 1,1831 1,2144 1,2495 1,3282 1,4034 1,4862
Интерполяция может быть осуществлена графическим путем; но так как в пределах, которыми мы интересуемся, зависимость является приблизительно линейной, то используем метод линейной интерполяции. Считая законы идеальных газов применимыми к состоянию при 1 атм, получаем
„ pv IX 7075 . .
N ~ ЯГ ~ 0,08206 X 283,2 ~ 304,4 М0ЛЯ"
При высоком давлении мольный объем
0,0283-103 ^-^04Л-^0,093 Л-
Из определения z [см. уравнение (15)]
22,42 р = CJ—z — —-— z,
'250 у. Соотношения давление—объем—температура для газов
тде 22,42 — произведение p3v3
/7075-273,2 \ \304,4-283,2/’
этого частного случая
22,42г
0,093
240,8 г.
; Теперь решение может быть осуществлено посредством простого подбора:
Принимаем/> = 300; тогда г = 1,4034; />вычисл. = 338.
*' Принимаем /> = 350; тогда г =1,4448; Рвичисл. = 348.
! Правильное значение лежит между этими двумя значениями и много ближе
к большей величине. Один или два подбора показывают, что />=348 алые (что достаточно близко). Это значение можно назвать наблюденным значением.
£.' , NRT 304,4 X 0,08206 X 323,2
|1-Р=-^ =......................... 28J-"----= 285®km.
2. Константы Ван-дер-Ваальса для водорода и азота даются в табл. III , Приложения*.
Подставляя соответствующие величины в уравнение (102), имеем:
п ляэло \s Ч9Ч 9 (____0,750_________I________0,250_______\
р— и,игмио X « V _ 0)750 х 0 02658 "Г о,093 — 0,250 X 0,03856)
S? “(OoU5- 0>2447 (0,750)2+l,348(0,250f = 326,0 атм.
3. По уравнению (92)
7 * 0,093 = 0,75t>ff, 4- 0^5од>,
«ли
Од, = 0,124—О.ЗЗЗод,-
Решение заключается в подборе пробных значений а#? получении из этогб
уравнения vffj и вычислении затем давления для каждого газа по уравнению
Ван-дер-Ваальса. Критерием правильности принятых значений Од, является > равенство давлений для двух газов.
1 Примем, например,
vNi = 0,0936 л/моль,
*' тогда
щО. ' vffi = 0,0928 л/моль.
Давления, вычисленные по Ван-дер-Ваальсу, равны
pNt = 328 атм,
Рн, = 372 атм.
После нескольких подборов были выбраны как достаточно близкие к правильным следующие значения:
vNl = 0,08797, = 0,09421.
Из этих значений объемов находим
/>д, = 363, />д, = 364.
Искомое общее давление равно 363,5 атм.
Газоли смеси
251
4. Мольные объемы отдельных газов в расчете
1,00 X 28,3 .
0,75 X 304,4 0,124
3 X 0,124 = 0,372 л
на литр смеси равны
(для Н2)
(для N2)___________
Подставляя эти значения в уравнение Битти — Бриджмена и используя константы Деминга н Шуп, данные в табл. 6, находим:
Рн, = 246,5, /^,= 70,9.
Давление смеси равно 317,4 атм.
5. Ход решения тот же, что и в случае 3. Конечные результаты решения путем родбора таковы:
— 0,0932 л/мом, vN1 = 0,0924 л1мом, р = 343,5 атм.
Из рассмотрения различных методов вычисления давления следует заключить, что любой из них превосходит по точности метод, основанный на применении закона идеального газа, и что закон аддитивности объемов дает для этой частной системы несколько лучшие результаты, чем закон аддитивности давлений.
Коэфициент сжимаемости газовых смесей. Можно определить коэфициент сжимаемости для смеси так же, как для чистого газа, т. е. по уравнению
pv=CmRT. (106)
Ст—функция не только р и Т, но также и состава смеси. Для данного состава можно использовать графики сжимаемости так же, как это было сделано для одного компонента. Для представлении этим способом свойств всех днухкомпонентных смесей пришлось бы прибегнуть к значительному усложнению, и в большинстве случаев результаты вонсе не были бы достигнуты. Как первое приближение для смеси можно принять, что
Ст = хАСА —хвСя . (107)
т А А • о а • ' '
Если Ст, СА, Св и т. д. относятся к одной и той же температуре и общему давлению, то из уравнения (107) и определения коэфи-циента сжимаемости получаем
AT А “г" лв “Г •
или
(108)
(109)
Другими словами, этот метод сочетания коэфициентов сжимаемости основан на использовании закона аддитивности объема. С другой стороны, если коэфициенты сжимаемости для смеси и для отдельных компонентов берутся при одной и той же температуре и при том же
252 V. Соотношения давление—объел — температура для газов
самом общем объеме, то
с —РАН 1 RT ’
и поэтому
откуда
Подставляя значения Ct в уравнение (107), получаем
Рт—Рл~\~Рв~\~- •• • U10)
Другими словами, этот метод сочетания коэфициентов сжимаемости основан на законе аддитивности давлений. Подобным же образом можно показать, что если коэфициенты сжимаемости компонентов берутся при той же температуре и том же мольном объеме, как для смеси, то также получается уравнение (107). Следовательно, это уравнение можно использовать для изображения трех различных законов аддитивности в зависимости от того, как были получены коэфициенты сжимаемости отдельных компонентов.
Те же самые методы сочетания применимы к другому коэфициенту * сжимаемости z, и можно показать, что они также основываются на законе аддитивности объемов н законе аддитивности давлений при условии, что для стандартного давления можно принять закон идеального газа.
Использование обобщенной диаграммы коэфициента сжимаемости в сочетании с законом аддитивности объемов или с законом аддитивности давлений дает относительно простой путь для характеристики газовой смеси. Для иллюстрации этого мы продолжим пример 6 (стр. 249).
Пример 6 (продолжение).
& Использование закона аддитивности объемов и обобщенной диаграммы коэфициента сжимаемости.
Приведенные давления и температуры по критическим данным табл. Ill .Приложения* соответственно равны:
__273,2-|-50
33,24-8 ~7’84, <для Н2>
_ _ Р __ Р .
12,8 4-8 — 20,8’
273,24- 50 nr,- . ...
=—Г26,0~'=2,56, Na)
_____Р
ЗЗЛ’ ч
ч
Газовые смеси
253
Поскольку давление неизвестно, следует использовать метод подбора. Если принять /> = 350 атм, то
it#, = 16,8 и «//,= 10,44.
По диаграмме коэфициента сжимаемости (рис. 15)
С//, = 1,21; CN,= 1,18.
Подставляя в уравнение (107), получаем
Ст — 0,75 X 1.21 + 0.25 X 1.18 = 1.204.
Тогда из уравнения (106)
1,204 X 0,08206 X 323,2 р=----------ода---------= 343’
Этот результат настолько близок к принятому значению, что дальнейшее уточнение путем подбора едва ли необходимо. Несколько лучшее совпадение получается при р = 340 атм, что и будет принято в качестве решения.
Т. Использование закона аддитивности давлений и обобщенной диаграммы коэфициента сжимаемости.
Примем сначала, что следует использовать закон аддитивности давлений Дальтона. Тогда мольные объемы будут следующие:
t,m = 0,093; =^ = 0,124 и ^. = ^ = 0,372.
• U,/О - VjZU
Решение было бы очень просто, если были бы доступны обобщенные диаграммы, выраженные через » и т. Использование рис. 14 и 15 потребует для получения коэфициента сжимаемости процесса подбора:
Рн ''н 0,124/?//
omx3k2=0*/
Примем рн — 250, тогда it = 250/20,8 = 12,0. Из рис. 15, при т = 7,84 и я = 12,0, С =1,105. ’Но
С = 0,00468 X 250 = 1,170.
После нескольких подборов рн оказывается равным 234:
Рк vn 0,372/>м
Cn= ~ RT *=0,08206 X 323,2 = °’01403^,-
Путем подбора, подобного использованному дли Нг, получаем
p^ = 72.
Общее давление = 234 Ц- 72 = 306 атм.
Если мы примем для закона аддитивности давлений форму Бартлетта, то
Ст = С АхА 4- Свхв, где СА и Сд являются коэфициентами сжимаемости компонентов при мольном объеме смеси в 0,093 л и при 50°С. Следовательно,
Са = 0,08206 X 323,2 ~ °’00351^
Подобным образом
Cq = 0,00351^
254 V. Соотношения давление—объем—температура для газов
вли так как р—р я, то мы имеем
СА = 0,0730ял (водород),
Св = 0,1175г.в (азот).
При нанесении этих уравнений в виде прямых линий иа график С — к и при нахождении их пересечения с приведенными изотермами получим
СА = 1,205, Св= 1,19, откуда
Ст=0,75Сл 4- 0,25Св = 1,202, „ Ст/?Г_ 1,202X0,08206X323,2 .... P — ОДОЗ-------— МЗатк.
Комбинирование констант уравнений состояния. Как опытным путем, так и на основании теоретических соображений было показано, что уравнения состояния, использованныё для одного газа, могут также использоваться и для смесей. При наличии экспериментальных данных константы для данной смеси можно определить тем же способом, что и для чистого газа. Поскольку экспериментальные данные по смесям очень ограничены, желательно располагать несколькими способами комбинирования констант отдельных газов для получения констант смеси при использовании того же самого уравнения состояния.
В случае бинарной смеси принято на основе кинетической теории, что константа смеси связана с константами отдельных газов уравнением
=^аХа "4“ ~Ь (Ш)
где kAB—так называемая константа взаимодействия. Было предложено несколько методов для определения константы взаимодействия, причем два наиболее простых следующие:
(112) и
(113)
Сочетание уравнений (112) и (113) с уравнением (111) приводит, соответственно к
(114>
И
4*=^Хд+А>Хв. (115)
Метод комбинирования, отвечающий уравнению (114), обычно называется линейным, а метод уравнения (115) — линейным квадратного корня. Другой метод, предложенный Лоренцом, широко используется
Газовые смеси
255-
и поэтому также должен быть упомянут. Согласно этому методу,.
-—что в сочетании с уравнением (111) дает ----------
А«=*а^+т(^+^)’жажв+М-
Битти и Икехара [15] предприняли тщательное исследование методов комбинирования констант и нашли, что наилучшим способом является использование линейного сочетания для всех констант, содержащих объем в первой степени, и линейного метода квадратных корней для имеющих объем во второй степени. Следовательно, константу а уравнения Ван-дер-Ваальса следует сочетать по линейному методу квадратных корней [уравнение (115)], а константу & — по уравнению (114). В случае уравнения Бчтти—Бриджмена следует принять линейный метод квадратных корней для Ао и линейное сочетание для всех других констант. Этот метод испытан при использовании уравнения Битти — Бриджмена на следующих смесях: аргон — этилен, кислород— этилен, азот—водород, азот—метан и водород — окись углерода.. Во всех случаях данные представлены удовлетворительно и большей частью лежат в пределах возможной ‘ошибки опыта. Совпадение оказалось много лучше, чем по закону аддитивности объемов или по закону аддитивности давлений.
Деминг и Шуп [56] применили уравнение Битти — Бриджмена к данным Бартлетта по смеси Н2—N2 в соотношении 3 к 1 и могли их удовлетворительно воспроизвести при использовании двух рядов констант, одного ряда для плотностей ббльших критической (около 0,0135 моль 1мл), а другого — для плотностей меньших критической. Константы для смесей, выведенные из констант для отдельных компонентов, дали хорошее соответствие до плотности, равной 0,0070 моль]мл. В противоположность найденному Битти и Икехара они нашли, что для Во линейное комбинирование по методу квадратных корней дало лучшие результаты, чем линейное комбинирование.
Общее уравнение состояния в форме, данной Битти — Бриджменом для смеси, можно написать следующим образом:
P = RT(^^ + B)-^, (П8>
л=(2х№р(1-^), 5=2^и(1-^), у
Е = —-
»Г3 V ___
/ЛсЭ* = А>« — (хА VАйА + хв УАж + . . .)*, S ХА = = ХАаА + ХВ° В + • • •» ‘
256 V. Соотношениядавление — объем—температура для газов
с подобными же выражениями для и Это
уравнение основано на мольном объеме; но если желают использовать общий объем, необходимо только заменить х{ на Nt и добавить множитель W перед RT.
Пример 6 (продолжение).
8. Сочетание констант уравнения состоинйя Битти — Бриджмена.
При использовании констаит Деминга и Шуп, линейного сочетания квадратных корней для Ао и простого линейного сочетания для других кон- стант найдем, что:
(S xi = (0,75 У 0,12404 = 0,25 V1,2517)2 = 0,29576,
v 2 xtat = 0,75 (0,05618) + 0,25 (0,01866) = 0,04680,
S xiB& = 0J5 X 0,02022 + 0,25 X 0,04603 = 0,02668,
2 xfii — 0,75 (— 0,00722) + 0,25 (— 0,02587) =— 0,01189, . •
g х/с, = 0,75 (2,00 X 10*) + 0,25 (6,16 X 10*) = 3.04 X 10*;
А = 0,14686,
3 = 0,030092, '
в = 0,0000969, 7? =0,08206.
Подставляя в уравнение (118) значения этих констант и р= 0,093, получаем
. /> = 360,5 атм.
Джиллиленд [91] предложил для обработки сжимаемости газовых смесей метод, основанный на использовании уравнения состояния ,в линейной изохорной форме и на различных схемах сочетания кон-
стант. Например, Рт==<9т^'т Фт’ (ПЭ)
где и ф являются функциями только объема и
' —= —X, + — Хв (120)
. Гт ГА А Гз В 1 .
Tm — +'Рв^В + • •• ’ (121)
ФЯ. = ФлХл + ФвХв+—’ (I22)
Фт = (*л1/Ъ+-*вИ>в + ---)2- (123)
Константы для отдельных газов берутся при температуре н мольном объеме смеси. Из различных сочетаний уравнений (120) — (123) с уравнением (119) получаются четыре различных уравнении состоянии для смеси. Джиллиленд, проверивший все четыре метода по данным сжимаемости, нашел, что сочетание констант посредством уравнений (121) и (123), которое он называет аддитивностью внутренних давлений, дало наилучшне результаты. Этот метод лучше, чем применение закона аддитивности объемов или закона аддитивности давлений.
Газовые смеси
257
Хотя он, невидимому, дает точные результаты, но он довольно затруднителен в применении, и некоторые другие методы, данные в этой главе, много удобнее в том случае, если хотят произвести лишь немного вычислений22.
Битти и Штокмайер [20] недавно дали обстоятельный обзор уравнений состояния как для чистых газов, так и для газовых смесей23.
Согласие газовых законов с опытом. По смесям произведено сравнительно мало исследований, поэтому трудно сделать какие-либо общие заключения о приложимости к ннм тех или иных законов. Мы можем выйти из положения, сказав, что почти любой из рассмотренных методов, относящихся к газовым смесям, более точен, чем закон идеального газа.
Для смесей газов, находящихся при температуре значительно более высокой, чем критическая, например для азота и водорода при температурах, близких к комнатным, закон аддитивности объемов оказывается вполне точным. Например, все данные Бартлетта и сотрудников по системе Н2 — N2 можно воспроизвести по закону аддитивности объемов с точностью до 2°/0 и выше. Закон аддитивности давлений, выраженный уравнением (98), значительно менее точен. С другой стороны, частная форма этого закона, выраженная уравнением (103), как показал Бартлетт, давала для смеси N2 — Н2 такую же точность, как и закон аддитивности объемов. Однако этот закон так мало исследован, что относительно него нельзя сделать никаких обобщений.
Если температура выше критической температуры одного из компонентов, но близка к ней, то закон аддитивности давлений представляется более точным, как можно судить, если обобщить результаты, полученные Мэссоном и Доллеем [167] на системах аргон—этилен и кислород — этилен. Максимальное отклонение от закона аддитивности давлений составляло около 9°/0, а от закона аддитивности объемов — 32°/0.
Использование уравнения состояния для смеси с константами, полученными соответствующим сочетанием констант чистых компонентов, является, вероятно, наиболее точным *из всех методов для двух рассмотренных общих случаев.
Если температура ниже критической температуры одного или нескольких компонентов, тогда все -правила для различных смесей нарушаются, поскольку один или несколько компонентов не могут существовать в виде пара в чистом состоянии выше давления насыщенного пара. Для этого случая практически не было произведено никаких исследований, и поэтому нельзя высказать никаких определенных утверждений, но возможное приближение состоит в сочетании констант уравнения состояния, даже если один (или несколько) компонентов не существуют в виде пара при общем давлении смеси. Для проверки этого и других возможных методов расчета желательно располагать большим количеством экспериментальных данных.
17 Химическая термодинамика
258 V. Соотношения давление — объем — температура для газов
Применение приведенных координат. Этот метод оказался настолько полезным в случае чистых компонентов, что был тотчас же проверен и на смесях. Если для вычисления приведенных параметров используется истинная критическая точка смеси, то кривая значительно отклоняется от кривой для чистых компонентов, особенно в критической области [243]. Экспериментальные данные для смеси, особенно фиктивные значения критического давления и критической температуры, можно выбрать так, чтобы кривые коэфициента сжимаемости для смеси, выраженные через приведенные коэфициенты, совпадали с кривыми для чистых компонентов. Эта точка была названа Кеем псевдокритической точкой; он вычислил ее для большого числа сложных смесей углеводородов на основании экспериментальных данных по их сжимаемости, используя данные по этилену и изопентану для нахождения кривых чистых компонентов. Кей также показал, что для смесей низших углеводородов, состав которых известен, псевдокритические давление и температуру можно вычислить с большой точностью по критическим давлениям и температурам чистых компонентов посредством простого (т. е. линейного) правила смешения при использовании мольных долей. В случае сложных смесей углеводородов неизвестного состава эти методы тоже могут быть применены при использовании „характеристического коэфициента11 К, предложенного Ватсоном и Нельсоном [254] и определяемого уравнением
T'g
(124)
где Тв—средняя мольная точка кипения жидкой фракции, определенная по стандартной перегонке, и d—удельный вес ее при 15,5°/15,5С в кг\л. Кей [124] дает кривые, связывающие псевдокритические давления и температуры с молекулярным весом при постоянных значениях К. Следовательно, зная среднюю температуру кипения, удельный вес и молекулярный вес любой смеси нефтяных углеводородов, ‘можно, по крайней мере приближенно, определить объем при давлении и температуре, близких к встречающимся на практике. Недавно Смит и Ватсон [235] произвели более широкое изучение критических, свойств смесей; они дали ряд графиков, по которым можно определить как истинные, так и псевдо-ьритические давления и температуры углеводородных смесей по удельным весам, определенным Американским нефтяным институтом, и по некоторым средним температурам кипения.
Для природных газов неизвестного состава псевдокритические давление и температуру можно связать графически с теплоемкостью газа, как показано Брауном (см. пример 7), и это дает простой метод для вычисления объема с удовлетворительной точностью.
Газовые смеси
259
Пример 7. Природный газ имеет следующий состав:
Компонент Мольный %
сн4 С2Н6 С3Н8 изо-С4Н10 я-С4Н]0 изо-С5Н12 w-CgHjj С6Н14 83,19 8,48 4,37 0,76 1,68 0,57 0,32 0,63
Вычислить удельный объем при 38° С и 140 атм.
Вычисления среднего молекулярного веса, псевдокритической температуры и псевдокритического давления показаны в нижеследующей таблице:
Н) (2) (3) (4) (5) (6) (7) (8)
Кожпо- Мольная Молеку- Крити- ческая Крити- ческое
нент ДОЛЯ лярный (2) Х(3) темпера- (2)Х(5) давление, (2)Х(7)
тура, °К атм.
СН; 0,8319 16,04 13,31 191,1 159,0 45,8 38,1
С2Н6 0,0848 30,07 2,54 305,2 25,9 48,8 4,14
Q+s 0,0437 44,09 1,92 369,9 16,2 42.01 1,84
изо-С4Н1п 0,0076 58,12 0,44 407,1 3,09 37,0 0,281
я-С4Н10 0,0168 58,12 0,97 426,0 7,16 36,0 0,605
изо-С6Н12 0,0057 72,15 0,41 460,9 2,63 32,92 0,188
Я-С5Н12 0,0032 72,15 0,23 470,3 1,51 33,0 0,106
0,0063 86,17 0,54 507,9 3,20 29,5 0,186
2 = 20,36 2 = 218,7 2=45,45
По рис. 14,
37.8 + 273,2 _
218,7 “ ’ ’
^ = 2,991.
45,45
С = 0,765, v _ CR7^_ 0,765X0.08206X311 _ v - Рмт - —20,36X 136,1— ~ °’00705 л г-
Экспериментальная величина, найденная СэджемиЛэси, равна 0,00663 л/г. Браун [29] решил эту задачу, используя специальную диаграмму коэфициента сжимаемости для природных газов, и получил v = 0,00664. Объем идеального газа был бы равен 0,00921.
17*
Г Л А В A VI
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ГАЗОВ И ЖИДКОСТЕЙ
Общее рассмотрение свойств. При изучении различных процессов инженеру необходимо иметь данные по термодинамическим свойствам участвующих в процессе газов и жидкостей, объединенные в определенной форме, удобной для непосредственного употребления. В гл. III мы в диференциальной форме вывели некоторые основные зависимости между обычными термодинамическими свойствами — объемом v (или плотностью р), энергией U, энтальпией Н, энтропией 5, свободной энергией F, изобарным потенциалом Z, летучестью / и активностью а и параметрами состояния — давлением и температурой. В гл. IV мы распространили эти соотношения с однокомпонентных однофазных систем на более сложные системы, введя в качестве переменной также и состав. В гл. V мы более подробно рассматривали одно из свойств, именно—-объем, и исследовали его изменение с давлением, температурой, составом и природой газа. В настоящей главе мы покажем, как посредством интегрирования уже выведенных уравнений определяются числовые значения других свойств, и дадим некоторые наиболее часто применяемые методы определения комплекса важнейших термодинамических свойств данного газа или жидкости в форме, удобной для использования. Инженер более связан с применением этих свойств, чем с их определением. Но для сознательного пользования ими он должен иметь основные сведения о методах их вычисления; нередко случается так, что он сам должен и определить их.
Рассматриваемое вещество может быть или жидкостью, если давление выше давления насыщенного пара при данной температуре, или паром (газом), если температура выше температуры насыщения, соответствующей данному давлению (перегретый пар), или насыщенной жидкостью, или насыщенным паром, если температура равна температуре насыщения, т. е. температуре, при которой обе фазы находятся в равновесии при данном давлении; наконец, это может быть смесь жидкости и насыщенного пара (так называемый „влажный" пар). И газовая и жидкая фазы могут состоять из нескольких компонентов, но принимая во внимание размер этой книги, мы ограничимся рассмотрением главным образом случая одного компонента или растворов и смесей постоянного состава, подобных воздуху.
Изобарный потенциал Z используется главным образом при рассмотрении химического равновесия. Поскольку в этой главе мы
Введение
261
касаемся только физических изменений, Z далее рассматриваться не будет. То же самое относится и к функции F.
Следует сразу привлечь внимание к тому обстоятельству, что объем, энергия, энтальпия и энтропия являются экстенсивными свойствами; поэтому их числовые значения зависят от массы системы. Мы будем иметь дело почти исключительно с удельными (относящимися к единице массы) или с мольными (относящимися к одному молю) величинами.
Некоторые свойства не приводятся в обычных таблицах термодинамических свойств, между тем они имеют большое значение. Например, скрытая теплота парообразования жидкости является весьма полезным свойством, хотя ока представляет собой лишь разность между двумя значениями других свойств, как энергии или энтальпии, в зависимости от способа определения скрытой теплоты. Изохорная теплоемкость Cv, изобарная теплоемкость Ср и коэфициент Джоуля — Томсона (дТ/др^ являются важными для определения других свойств, а также и для некоторых сопоставлений и расчетов, как будет показано ниже.
Конечно, желательно определить совокупность свойств по минимальному числу опытных измерений, и здесь особенно полезны соотношения, подобные выведенным в гл. III. Осборн с сотрудниками [78,178, 179,180] разработал метод вычисления основных термодинамических свойств вещества, а также давления его пара на основании непосредственного определения разности энтальпии, осуществляемого в простой калориметрической установке совместно с определением давления насыщенного пара. Кейес с сотрудниками [132, 233, 234] (в Массачузетском технологическом институте) специализировался на измерении объема и мог вывести большинство свойств из данных по объему в сочетании с давлением насыщенного пара и теплоемкостью при низком давлении.
Применение правила фаз. При рассмотрении однокомпонентных систем правило фаз указывает, что в присутствии только одной фазы имеется две степени свободы; одна степень свободы имеется, когда сосуществуют две фазы, и нет ни одной степени свободы при сосуществовании трех фаз. Это означает, что если вещество целиком находится в жидком состоянии подобно воде или полностью в газообразном состоянии подобно пару, то необходимо определить две независимые переменные, прежде чем окончательно определить состояние системы. Например, одной температуры недостаточно для определения объема, или энтальпии, или какого-нибудь другого свойства пара; для этого следует также указать и давление. Когда присутствуют две фазы — это может быть газ и жидкость, или жидкость и твердое тело, или газ и твердое тело,—то существует только одна независимая переменная, т. е. только температура или только давление, которая полностью определяет состояние системы. Следовательно, если имеется смесь воды и пара, находящихся в равновесии, то давление
262
VI. Термодинамические свойства газов и жидкостей
и термодинамические свойства каждой фазы определяются заданной температурой. Они совершенно не зависят от общего объема системы, который будет изменяться с относительными количествами двух равновесных фаз.
Если сосуществуют три фазы, то система нонвариантна; это означает, что данное состояние может быть осуществлено только при опре-
деленных значениях температуры и давления, т. е. единственным спо-
Рис. 23. Фазовая диаграмма для однокомпонентной системы.
собой, и любое изменение давления или температуры будет причиной исчезновения одной из фаз.
Эти весьма элементарные факты иллюстрируются рис. 23. Поле / представляет поле стабильности твердой фазы, II — область жидкости и III—область газа. Ясно, что в границах любой из этих площадей и давление и температура могут изменяться независимо друг от друга. Пограничные линии меж-
ду фазами являются совокупностью точек, в которых могут сосуществовать обе фазы. Так, ОА
является так называемой кривой сублимации и представляет равновесие между твердым веществом и паром. ОВ является кривой плавления или отвердевания. Вдоль ОС находятся в равновесии жидкость и пар. Ясно, что вдоль каждой кривой давление точно определяется температурой.
Так, ОС является кривой давления насыщенного пара жидкости, а О А — кривой давления насыщенного пара твердого тела.
Кривая ОС заканчивается критической точкой С; выше темпера-
туры, соответствующей этой точке, жидкость не может существовать, поэтому область жидкой фазы ограничивается справа вертикалью, выходящей из точки С (критическая изотерма). Однако эта линия не является определенной пограничной линией (подобно, например, ОС), так как при температурах выше С можно перейти от газа к жидкости и обратно без какой-либо заметной прерывности в свойствах вещества, тогда как при температурах ниже С необходимо пересечь пограничную кривую ОС, и при этом наблюдается явное разделение на две фазы различной плотности.
Проведя вправо из точки С горизонталь (критическую изобару), мы ограничиваем область IV, характерной особенностью которой является то, что в ней паровая и жидкая фазы неразличимы. При пересечении этой области можно перейти от газа к жидкости без явной
прерывности.
Область III будем впредь называть или областью газовой фазы, или областью перегретого пара. Последнее название является более
Введение
263
подходящим вследствие того, что при постоянном давлении любой газ, охлаждаясь в этой области, будет достигать состояния насыщенного пара, в котором при дальнейшем охлаждении он начнет переходить в другую фазу, жидкую или твердую. Отметим, что этого не происходит при постоянном давлении, если охлаждать вещество в том состоянии, в котором оно находится в области, ограниченной пунктирным углом.
Для кривой ОВ предел еще не найден. Некоторые вещества подвергались достаточно высокому давлению при температуре выше критической, и жидкость целиком превращалась в газ. Таким образом, мы имеем область, в которой, подвергая газ высокому давлению, можно заставить его конденсироваться непосредственно в твердое состояние, но никогда не в жидкое. В некоторых случаях происходит переход в другую форму твердого вещества, и может существовать несколько таких точек перехода; в этих случаях утверждение об отсутствии предела кривой ОВ остается действительным, если оно относится к твердому состоянию, которое стабильно при наиболее высоком давлении.
Точка О, в которой существуют совместно все три фазы, называется тройной точкой. Обычно она лежит при давлении в 1 атм или меньше, но имеются интересные исключения. Из продуктов промышленного значения такое исключение обнаруживает, например, углекислота СО2, тройная точка которой находится при — 56,6°С и 5,1 атм, т. е. немного выше нормальной точки кипения — 78,5°С, которая, таким образом, становится точкой сублимации.
Упомянем еще одно исключение: кривая ОВ на рис. 23 для некоторых веществ (и, в частности, для воды) имеет отрицательный наклон, т. е. увеличение давления в этих случаях понижает температуру плавления, вместо того чтобы повышать ее. Из уравнения Клаузиуса — Клапейрона
dp _ УН dT ~~ ТУи
(153, гл. IV)
видно, что этот наклон связан с изменением объема, происходящим при плавлении вещества. Если при плавлении происходит увеличение объема (жидкость менее плотна, чем твердое тело), то наклон будет положительным; если же наблюдается обратное, как в случае воды, то наклон будет отрицательным.
На рис. 23 представлена только одна твердая фаза. В действительности многие вещества могут иметь несколько твердых фаз, но это большей частью не имеет технического значения. Например, в случае воды, наряду с обычной тройной точкой при 0,01°C и 0,00602 атм, известны еще 5 других тройных точек, соответствующих образованию других модификаций льда; но даже наиболее низколежащая (по давлению) тройная точка отвечает давлению примерно в 2000 атм.
264
VI. Термодинамические свойства газов и жидкостей
Гелий представляет исключение в двух отношениях: 1) он имеет две различные жидкие фазы и 2) при давлении в 1 атм и ниже не может быть превращен в твердое состояние ни при одной из до сих пор полученных низких температур. Его можно превратить в твердое состояние при более высоких давлениях, и, повидимому, его кривая плавления (ОВ иа рис. 23) должна пересечь ось давлений при более высоком давлении, чем соответствующее точке А, и поэтому для него не существует тройной точки.
ТЕРМОДИНАМИЧЕСКИЕ ДИАГРАММЫ И ТАБЛИЦЫ СВОЙСТВ
Общие типы диаграмм. В случае чистого вещества, состояние которого большей частью полностью определяется двумя независимыми переменными, большинство термодинамических свойств относительно просто можно представить посредством таблицы или графика при использовании только двух координат. График особенно удобен и прост для применения. Любой график, представ ляющий данные по свойствам, перечисленным в начале этой главы, может быть назван термодинамической диаграммой. Выбор координат совершенно произволен и диктуется главным образом удобством в зависимости от характера рассматриваемой задачи. Наиболее употребительны следующие типы диаграмм, обозначаемые по выбранным координатам: энтропия—температура S— Т, энтропия — энтальпия 5 — Н; давление— энтальпия р — Ни энтальпия — температураН —Т. Диаграммы, в которых одной из координат является энтальпия, особенно диаграмма S — Н, обычно называются диаграммами Молье.
Диаграммы, в которых одной из координат является объем или функция объема, например коэфициент сжимаемости, рассматривались в предыдущей главе (стр. 211).
Полезными при решении отдельных задач могут оказаться многие специальные диаграммы. Например, может оказаться необходимым нанести Ср против Т при различных давлениях, или р против Т при постоянной Н (так называемые „кривые дросселирования”), или какие-либо другие кривые.
Положение становится значительно сложнее, если присутствует несколько компонентов. Даже если рассматривать только бинарную систему, то максимальное число степеней свободы достигает трех, и для полного представления такой системы необходимы пространственные диаграммы. Во всех случаях для систем, имеющих более одного компонента, обычно рассматривают одновременно только ограниченную часть поля, т. е. ограниченный ряд свойств по сравнению с тем рядом свойств, которые можно было бы представить на одной диаграмме для однокомпонентной системы. Следовательно, на одной диаграмме можно рассмотреть определенные свойства при данном давлении или данной температуре и получить отдельные диаграммы для
Термодинамические диаграммы и таблицы свойств
265
других давлений или температур; или же можно выбрать один какой-нибудь состав, т. е. рассматривать систему как чистый компонент (примером такой системы может служить воздух).
Схема диаграммы температура — энтропия. Прежде чем перейти
к детальному рассмотрению различных свойств и диаграмм, их пред-
ставляющих, полезно рассмотреть сначала одну из них в общих чер-
тах. Одной из наиболее общеупотребительных диаграмм является диаграмма S— Т, показанная схематично на рис. 24 и 25. Они представляют частную форму диаграммы для вещества, подобного воде нли СО2. Диаграммы для других веществ сходны, но обладают некоторыми важными отличиями, одно из которых будет отмечено ниже при рассмотрении теплоемкости насыщенных паров.
На рис. 24 ACF представляет пограничную кривую между однофазными областями II и III и гетеро-
Рис. 24. Схематическая диаграмма S—Т.
генной областью VI, где со-
существуют жидкая и паровая фазы (так называемая ортобарная кривая); область II является областью жидкости, соответствующей II на рис. 23; подобным же образом область III является областью перегретого пара, соответствующей III на рис. 23. Поле / также соответствует / на рис. 23. Области IV, V и VI, в которых существуют две фазы, на рис. 23 оказываются просто кривыми, причем IV соответствует OB, V — ОА, а VI—ОС. Точка С, в которой горизонтальная линия представляет касательную к пограничной кривой, является
критической точкой. Ветвь кривой АС известна как кривая жидкости, a CF— как кривая сухого насыщенного пара. ACG является критической изобарой ☆), ветвь которой CG служит для произвольного разграничения поля III. Температура, соответствующая BAF, представляет температуру тройной точки, а область ниже нее, ограни-
ченная также BD и FH, является гетерогенной областью, в которой присутствуют твердое тело н пар. Отметим, что нет ни одной точки, соответствующей тройной точке на рис. 23.
☆ ) Вследствие слабой сжимаемости жидкости изобары для умеренных давлений в этой области отличаются лишь незначительно, и поэтому для целей данной иллюстрации их можно считать совпадающимя с кривой жидкости.
266
VI. Термодинамические свойства г^зов и жидкостей
Обратимся теперь к рис. 25, который представляет (все еще в схематической форме, но гораздо подробнее, чем на предыдущем рисунке) диаграмму температура — энтропия для водяного пара, включающую только жидкую и паровую фазы; кривые pt — р4 представ-
Знтропи^ъ ккал/кг град /,4 1,5 1JB 2,0 2,2
Рис. 25. Схематическая диаграмма энтропия — температура для водяного пара.
ляют линии, вдоль которых давление постоянно, т. е. изобары, а кривые Нх—IT— линии постоянной энтальпии, т. е. изоэнтальпы. Располагая большим числом этих кривых с равным приращением давления и энтальпии между ними, можно с помощью интерполяции непосредственно определить энтропию и энтальпию вещества, если известны его давление и температура, или же определить его энтропию и давление при двух других известных свойствах, а также и для других комбинаций.
На некоторых диаграммах показаны также линии постоянного перегрева в градусах Цельсия. Перегревом называется число градусов,
Термодинамические диаграммы и таблицы свойств 267
на которое температура перегретого пара превосходит температуру пара, насыщенного при одном и том же давлении. Это является важной величиной, так как ее числовое значение непосредственно указывает, насколько близко пар приближается к своей точке конденсации (т. е. к точке росы). Изобара, проходящая через критическую точку В, называется критической изобарой, и область, лежащая от кривой насыщенного пара выше и справа, но ниже этой изобары, является областью перегретого пара, отмеченной цифрой III на рис. 23. Таким образом, если взять какую-нибудь точку в этой области, например точку D, то давление будет равно рг и энтальпия —Нъ. При изобарном охлаждении этого пара, что равносильно перемещению вдоль изобары jt?j, изменение будет непрерывным, пока мы не достигнем точки Е. В этой точке пар становится насыщенным, и начинает выделяться жидкость как фаза. Эта точка также известна как „точка росы“ хотя этот термин чаще употребляется для соответствующей точки в системах, содержащих больше одного компонента.
До тех пор пока тепло отнимается непрерывно, состояние системы представляется горизонтальной линией EF, так как, поскольку имеется только одна степень свободы, изобара является также и изотермой. Если в этой области теплота отнимается при постоянном давлении, то состояние каждой фазы остается постоянным, а изменяется только их относительное количество. Следовательно, перемещаясь от Е к F, мы переходим от системы, содержащей 1ОО°/о пара, к системе, содержащей 1ОО°/о жидкости. Точка на линии насыщенной жидкости называется иногда „точкой пузырьков" вследствие того, что она представляет равновесие между относительно большим количеством жидкости и последним пузырьком пара. (Этот термин чаще употребляется для систем, содержащих больше одного компонента.) Для быстрого определения относительных количеств двух фаз в любой точке этой области проводятся линии постоянной сухости пара (линии хг— х4 на рис. 25). Любая из этих линий связывает все точки, для которых одинаковы относительные количества жидкости и пара. Так, х = 0,80 означает, что 1 кг смеси содержит 0,80 кг пара и 0,20 кг жидкости, и все точки вдоль линии х = 0,80 будут представлять смеси с тем же самым соотношением двух фаз. Все линии постоянной сухости сходятся в критической точке, где обе фазы становятся тождественными.
Охлаждение кипящей жидкости (точка F) вдоль изобары означает переход вдоль кривой, которая лежит настолько близко к кривой жидкости, что практически при масштабе этой диаграммы она будет неотличима от нее. Действительно, при постоянной температуре изменение энтропии с давлением определяется уравнением
<?S х ____ fdv х
дР)Т~~~ ySfjp'
(94, гл. Ill)
268
VI. Термодинамические свойства газов и жидкостей
и так как для жидкостей изменение объема с температурой (за исключением области, близкой к критической точке) очень мало, то ее энтропия лишь незначительно изменяется с давлением. Следовательно, для большинства случаев изобара в области жидкости, находящейся при более низкой температуре, чем температура кипения, по существу совпадает с кривой жидкости24.
Вещество в точке, подобной точке G, лежащей при давлении и температуре, превышающих критические значения, находится в области, обозначенной IV на рис. 23. Если вещество в состоянии, отвечающем точке G, охлаждается при постоянном давлении, то оно не достигает ортобарной кривой (точки насыщения), где выделяется вторая фаза; вместо этого оно проходит через ряд непрерывных изменений от газообразного до жидкого состояния, и хотя начальное и конечное состояния совершенно различны, но невозможно выбрать ни одной промежуточной точки, для которой можно было бы сказать, что изменение резко прерывается. Следовательно, вся область выше и левее критической изобары является особой областью, в которой жидкости постепенно становятся неотличимыми от газов, и наоборот.
Выгодная особенность диаграммы, в которой одной из координат является энтропия, следующая: так как процесс обратимого адиабатного расширения или сжатия является процессом изоэнтропным, т. е. протекающим при постоянной энтропии, то такой процесс очень легко нанести на эту диаграмму; он изобразится просто вертикальной линией. Если вещество первоначально находится при температуре и давлении, отвечающих точке М (рис. 25), и расширяется адиабат-но (т. е. без теплообмена с окружающей средой) и обратимо (т. е. идеально), то путь изменений, который оно проходит, будет изображаться вертикальной пунктирной линией, идущей от точки М. Если расширение продолжается до тех пор, пока давление не упадет до р1( то точка W показывает состояние вещества в конце расширения. Его температура, сухость и энтальпия легко определяются по диаграмме.
Диаграммы Молье. Другой широко употребляемой диаграммой является диаграмма энтропия—энтальпия, или диаграмма Молье, которая схематично показана на рис. 26. Эта диаграмма имеет не только преимущество диаграммы 5 — Т, заключающееся в том, что по ней легко проследить изоэнтропный процесс, но она также очень удобна для точного определения разностей энтальпии или для расчета изо-энтальпийного процесса, т. е. процесса при постоянном значении энтальпии25. Конечно, те же результаты можно получить и с помощью других диаграмм, выбор которых зависит целиком от соображений удобства.
Диаграмма энтропия—энтальпия является только одним из нескольких типов диаграмм, предложенных Молье [172] и включающих энтальпию в качестве одной из координат. 5—//-диаграмма с обычными прямоугольными координатами несколько неудобна по
' Термодинамшеские диаграммы и таблицы свойств
269
форме и особенно трудна для чтения в нижней части, поэтому многие предпочитают пользоваться наклонными координатами, предложенными Молье. Другие широко употребляемые диаграммы Молье основываются на следующих парах координат: v— Н, lg v — Н,
Рис. 26. Схематическая диаграмма энтропия — энтальпия для водяного пара.
р— Н, 1gр — Н. Последний тип особенно ценится при расчетах по охлаждению. Диаграммы этого типа для аммиака [36], кислорода [171] и азота [171] можно найти в соответствующих справочных изданиях 26.
Таблицы термодинамических свойств. Для некоторых задач удобнее пользоваться таблицами термодинамических свойств, а не графиками. Из таблиц значения свойств обычно можно получить гораздо точнее; кроме того, в таблицах дается объем, которого обычно нет в диаграммах, потому что он сильно усложняет их и увеличивает трудность чтения. Стандартные таблицы термодинамических свойств имеются для нескольких наиболее важных веществ, употребляемых в промышленной практике, например для воды, углекислоты и аммиака. В этом разделе мы намереваемся дать краткое описание таких таблиц для тех, кто не имел случая встречаться с ними раньше. В качестве примера выберем таблицы водяного пара27.
270
VI. Термодинамические свойства газов и жидкостей
Таблицы для системы жидкость — пар обычно подразделяются на три части следующим образом: табл. 1—свойства сухого насыщенного тара с температурой в качестве независимой переменной; табл. 2 — свойства сухого насыщенного пара с давлением в качестве независимой переменной; табл. 3 — свойства воды и перегретого пара 28.
ПРИМЕРЫ, ИЛЛЮСТРИРУЮЩИЕ ПОЛЬЗОВАНИЕ .
ТЕРМОДИНАМИЧЕСКИМИ ДИАГРАММАМИ И ТАБЛИЦАМИ
Здесь мы будем оперировать с простыми изменениями состояния 29.
Пример 1. Вода, находившаяся при 20° С и атмосферном давлении (это исходное состояние будем обозначать индексом I), накачивается в котел, испаряется при давлении в 22 кг/см2 и перегревается до 320° С при постоянном давлении (это конечное состояние обозначим индексом 2). Сколько тепла следует затратить на 1 кг воды?
За исключением стадии накачивания воды в котел, весь процесс происходит при постоянном давлении; для такого процесса мы имеем по уравнению (10, гл. III)
Q = \H=H2 — Нг.
Влияние давления на энтальпию жидкости очень незначительно, и можно принять, что Н для жидкой воды при 20° С и давлении насыщения равно Н при 20° С и давлении 22 кг/см2. По таблицам
#2 = 731,0 ккал/кг,
Н{ — 20,5 ккал/кг, 0 = 710,5 ккал/кг.
Пример 2. Насыщенный водяной пар при 15 кг/см2 непрерывно расширяетси через дроссель до постоянного давления 1 кг! см2. Каким будет состояние расширившегося пара и чему будет равно изменение энтропии при процессе, если нет потерь тепла в окружающую среду (адиабатный процесс) и если вся кинетическая энергия, обусловленная потоками большой скорости, рассеивается?
Как будет показано в следующей главе, дросселирование является расширением при постоянной энтальпии. Это дает ключ к нахождению конечного состоянии пара, что легче всего осуществляется при использовании диаграммы Молье (S — Н). Определим на диаграмме местонахождение точки, в которой изобара в 15 кг/см2 пересекает кривую пара. Это будет исходное состояние. По координатам диаграммы мы получаем для него значения
H-i — 666,6 и 5г= 1,541.
Затем проведем на диаграмме горизонталь (//= const) до пересечения с изобарой 1 кг!см2. Это будет конечное состояние. По диаграмме находим:
S, = 1,828, / __1 кто с
AS = 1,828 — 1,541 = 0,287.
Температуры определяются интерполяцией между линией постоянной температуры и линиями постоянного перегрева зо.
Ту же самую задачу можно почти так же легко решить с помощью таблиц. Определим место исходной точки по табл. 2; затем перейдем к
Вычисление свойств перегретого пара
271
табл. 3 и проведем вертикаль от р = 1 кг/см2 до энтальпий, наиболее близких к найденному значению 666,6. С помощью линейной интерполяции между двумя значениями Н по обе стороны данного значения получим конечные t и S. По таблицам можно также получить объемы, которые иа диаграммах обычно не указываются.
Пример 3. Пар при 15 кг]см2, перегретый на 30° С, расширяется адиабатно и обратимо до давления 1 кг/см2. Охарактеризовать конечное состояние и найти изменение И.
Обратимое адиабатное изменение является изменением при постоянной энтропии. Определяем положение исходной точки, как и раньше, с помощью интерполяции между линиями постоянного перегрева
S,= 1,579, /А = 685.
Проводим вниз вертикаль вдоль линии постоянной S до пересечения с изобарой. Конечное состояние отвечает двухфазной области. Оно является смесью пара и воды, причем количество последней составляет 12,5 весовых % от общего количества, т. е. сухость пара равна 0,875. Н2 — 573 и \Н=—112. Эту задачу также легко можно решить с помощью таблиц при наложении услови я
Ss =Sj= 1,579,
где каждое S относится к 1 кг общего материала в данном состоянии. Тогда задача сводится к нахождению конечного состояния, для которого давление равно 1 кг/см2 и энтропия равна 1,579. Обращение к табл. III (у Вукаловича) показывает, что все энтропии при этом давлении больше 1,579; отсюда мы заключаем, что конечное состояние пара отвечает не области перегретого пара, а области равновесия двух фаз. Следовательно, можно написать
S2 = S2 4“ ^t2AS, (43)
где S, — энтропия насыщенной жидкости,
AS — изменение энтропии, обусловленное испарением, х2 — сухость пара.
Так как данное состояние отвечает двухфазной области, то мы имеем дело с системой, содержащей жидкость и насыщенный пар при давлении в 1 кг/см2, и из таблиц получаем следующие значения:
S2 = 0,3096, AS =1,4491, откуда
1,579 = 0,3096 + 1,4491 х, Х = 0,876, а также
Н2 = /4 + х2АЯ= 99,12 + 0,876 X 539,4 = 571,6,
»2 = t>2+x2Av = 0,0010 + 0,876 X 1,725= 1,512 л^/кг смеси.
ВЫЧИСЛЕНИЕ СВОЙСТВ ПЕРЕГРЕТОГО ПАРА
Рассмотрев схематически совокупность термодинамических свойств, остановимся более подробно на некоторых методах вычисления отдельных свойств. В этом разделе будет сделан упор на область перегретого пара (поле III на рис. 23). Однако следует уяснить себе, что приводимые ниже соотношения одинаково хорошо приложимы
272
VI. Термодинамические свойства газов и жидкостей
к любой однофазной области, например к жидкости (поле II на рнс. 23).
Аналитические методы. Уравнения гл. 111 совместно с соответствующими уравнениями состояния позволяют вычислить термодинамические свойства чистых веществ в области перегрева. Это, вероятно, является наиболее простым методом, если известны константы уравнения состояния, но следует помнить, что такой метод является только приближенным, даже при лучшем уравнении состояния.
Для вычисления U удобнее всего непосредственно пользоваться уравнением состояния, определяющим р; для вычисления Н простейшим является уравнение, определяющее v. При определении 5 нет большой разницы от того, какой тип уравнения используется. Уравнения, определяющие р, являются более употребительными из этих двух типов, и с помощью простого преобразования, которое будет показано ниже, их можно использовать для вычисления энтальпий.
Робинсон и Блисс [198] дали законченное аналитическое выражение для вычисления изотермического изменения U, Н и 5 по трем уравнениям состояния, определяющим р, а именно—по уравнениям Ван-дер-Ваальса, Воля и Битти — Бриджмена. Сравнение вычисленных данных для трех различных газов со значениями, найденными из данных p—v—Т графическим методом, показывает, что уравнение Битти — Бриджмена является наилучшим по сравнению с двумя остальными, в особенности для вычисления энтальпии. Но даже с этим ' уравнением отклонение все же составляло 7°/0 для энтропии и 12°/0 (а в некоторых случаях и более) — для энтальпии.
Энтальпия. Энтальпия растет с температурой и уменьшается с увеличением'давления. Это — удобное правило для общего руководства, хотя в некоторых случаях и имеются исключения.
В рассматриваемой области энтальпия обычно вычисляется по измерениям других свойств, хотя положение кривой пара часто определяется калориметрическим измерением скрытой теплоты парообразования. Недавно произведены обширные непосредственные измерения энтальпий водяного пара во всей области перегрева ] 103]. Принцип такого измерения весьма несложен. Пар при выбранных исходных давлении и температуре непрерывно расширяется через дроссель до атмосферного давления и затем полностью конденсируется в калориметрическом конденсаторе. Выделившееся тепло измеряется повышением температуры охлаждающей воды. Тепло, полученное охлаждающей водой с поправками на тепловые потери, равно разности между энтальпией пара высокого давления и энтальпией конденсата. Количество пара определяется взвешиванием конденсата.
Основным уравнением для вычисления энтальпии любого гомогенного вещества, если независимыми переменными являются р и Т, служит уравнение
dH^CodT-\- L— т№\ Idp. (96, гл. Ш) L \у* j р J
Вычисление свойств перегретого пара
273
Интегрирование дает
т р
н=нй+ fc/T+Jk-Т(g) ~\dp, (1)
h Р„ р
где Но — начальное, произвольно выбранное значение Н при некотором значении р и Т. В обычной практике принимают Нй~0 для насыщенной жидкости при 0°С. Нй в этом уравнении будет просто разностью энтальпий пара при р0 и То и насыщенной жидкости при 0° С. Если рв—давление насыщенного пара при То, а То равно 0°С, то Нв равно скрытой теплоте парообразования при 0° С.
Как было выяснено в гл. I, это уравнение можно интегрировать двумя путями. Так как при высоких давлениях данные по теплоемкости редко имеются в распоряжении, то в рассматриваемом случае интегрирование сначала следует производить по температуре. Применение этого уравнения иллюстрируется следующим примером.
Пример 4. Вычислить энтальпию азота при—100° С и 50 атм.
Так как значение Но в уравнении (1) совершенно произвольно, то его можно принять равным нулю или какой-нибудь другой величине; но для сравнения вычисленных нами результатов с данными, полученными из существующих компиляций по термодинамическим свойствам азота, мы примем Л/0 = 269б£-) кал/моль, если 70 = 273,2° и р=1 атм. Для того чтобы найти первый интеграл, требуется знать Ср как функцию температуры при низком давлении. Бринкворт [27] дает следующее уравнение для мольной теплоемкости азота при 1 атм и температуре ниже 0° С:
Ср = 7,40 — 0,00405 T-J- 8,272 X Ю"6 Т\
Примем это уравнение для определения первого интеграла в уравнении (1) т т
CpdT = j (7,40 — 0,004057 8,272 • 10“6 Г2) dT=
= 7(40 (Г _ Го) ф (7* _ Л) + 8-’-27-^-10-6 (Л - Т*).
Подставляя 7=173,2 и 70 = 273,2, получаем значение интеграла, равное —692.
Для определения второго интеграла необходимо располагать соотношением р — v—Т для азота в рассматриваемой области. Если ряд числовых значений объема и коэфициента (ди]дТ)р приведен в таблицах и имеется в распоряжении, то интегрирование можно выполнить графически. Это потребует знания v как функции р при температуре 7 (173,2° К в этом случае), а также v как функции 7 в рассматриваемом интервале давления для вычисления коэфициента (до/дТ)р. Графическое определение термодинамических свойств будет иллюстрироваться в одном из последующих разделов этой главы (стр. 290).
Простейшим путем нахождения второго интеграла является определение его с помощью уравнения состояния. Это будет точным способом при усло-
й) Величина взята из табл. 6, см. [171].
18 Химическая термодинамика
274
VI. Термодинамические свойства газов и жидкостей
вии, что применяемое уравнение состояния оказывается точным в исследуемом интервале температур и давлений. Простое уравнение состояния pv—RT дает
( RT^-— С=
J Р J Р
ра р°
Этого результата можно было ожидать из определения идеального газа, именно из того, что его энергия, а поэтому и энтальпия не зависят от давления.
Уравнение Битти — Бриджмена оказалось достаточно точным; константы для азота известны. Для определения §vdp удобно располагать уравнением состояния, определяющим v как функцию р. Битти преобразовал уравнение состояния, чтобы дать его в этой форме (84, гл. V), но оно менее надежно, чем исходная форма, и поэтому для нашей задачи примем, что можно использовать исходную форму, т. е. уравнение (77, гл. V). Для преобразования §vdp примениется следующая операции *):
d (pv) — pdv vdp,
р v
vdp ~pv~ pav0 — J pdv.
(a)
(6)
Кроме того, коэфициент (dvjdT)p неудобен для использования в случае уравнения, определяющего не объем, а давление, и для этого случая его можно преобразовать следующим образом:
dv\ /др\ (д&\ дТ)р \дт)ъ\др)т’
При постоянной температуре
(в)
Используя уравнения (б) и (в), находим, что
При диференцироваиии уравнения Битти — Бриджмена (77, гл. V) получаем
др\ — i Io iJo
дТJv> v^vs^ v* ’
(Д>
#) Методом, заменяющим этот метод (и равноценным ему), является применение коэфициента (dHldv)T, который связан с параметрами состояния следующим образом:
дЛ} =v(&\ 4-Г^
dv J т \до)т' \дТ) v
Это выражение легко найти с помощью уравнения состояния, определяющего р.
Вычисление свойств перегретого пара
275
где
о __2RcB§
Ро у,3
RBf>b,
®о= 7?/?оЧ
2/?с \ Т3 ’
r2RcbB<} То
При использоиании уравнения состояния в форме уравнения (78, гл. V) получаем
₽, f и 3 являются температурными функциями, значения которых даны на стр. 241:
jpdv = [/?T in V - I - - ±,] ’. (ж)
Из уравнения (д) имеем
dv — о
ЯГ In»— »2
2»2 Зо»
(з)
Подстановка уравнений (ж) и (з) в уравнение (г) дает
Г/1 1 \ । 0 ( 1 1 \ /I 1 \1
—pv — povo — «1 ( —— - l+₽i 75 — )—71 —-----з .
L \v v^j \v* v^j ^»з Vq/]
(и)
где
3Rc T2 ’
_ 3RcRq a40
™ 2Г2 2 ’
RBffic
71 *
v следует определять по уравнению состояния методом подбора. В рассматриваемом случае и является объемом при Т — 173,2 и р —50 атм, и окончательно подобранное значение его, полученное с помощью констант,, указанных в табл. 6, равно 0,233 л)моль. »0 можно получить с достаточной точностью 18*
276
VI. Термодинамические свойства газов и жидкостей
по уравнению Клапейрона:
0,08206 173,1 ,
va= - =-------------— = 14,21 л[моль;
Ро *
^=1,690; h = — 0,00892; Т1 = —0,0000400.
Подстановка этих значений в уравнение (и) дает для числового значения второго интеграла —9,53. Это число дано в механических единицах (литр-атмосфера на моль), н его необходимо перевести в калории, чтобы привести к единицам, в которых выражены и другие члены уравнения (1). Тогда получим
—9,53X24,20 =—231 кал.
Окончательно Н при —100° С и 50 стил< = 2696—692—231 = 1773 кал)моль. (Для сравнения укажем, что значение, полученное интерполяцией по табл.6 [171], равно 1780.)
Другим методом решения этой задачи является нахождение ДТ/ с помощью уравнения (79, гл. III) с последующим вычислением Д// из уравнения
ДЯ=ДТ/4-Д (/>«). (Н, гл. Ill)
Этот метод по существу совпадает с только что рассмотренным, так как включает те же самые интегралы.
Внутреиияя энергия. С практической точки зрения внутренняя энергия является гораздо менее важной величиной, чем энтальпия, и значения ее редко приводятся в сводках термодинамических свойств. С другой стороны, величина U требуется для отдельных задач. Простейшим путем определения ее служит вычисление из энтальпии по уравнению
U=H—pv. (6, гл. Ill)
Пример 5. Чему равна внутренняя энергия перегретого водяного пара при 15 кг/см2 и 320° С?
По таблицам Вукаловича, имеем
/7=735,1 ккал! кг,
« = 0,1800 м9/кг.
Поэтому
„ 15,0-0,1800 10* _
U = 735,1 ------J27------— 671,9 ккал 1кг.
Пример 6. Закрытый сосуд емкостью в 0,283 м2 наполнен насыщенным водяным паром при давлении в 19,5 кг/см2. Чему будет равно давление в сосуде, если сконденсировать 25°/0 заключенного в нем пара? Какое количество тепла следует отнять для того, чтобы произвести эту конденсацию?
«1 = 0,1041 м2/кг (по таблицам водяного пара).
0 283
Количество пара = = 2,72 кг.
После конденсации остается 0,75X2,72 = 2,04 кг пара. Пренебрегая объемом жидкости «2, получаем конечный удельный объем =0,283/2,04 = 0,1387. Из таблиц рг~ 14,5 кг^см2. Поскольку рассматриваемый процесс протекает при постоянном объеме, тепловой эффект Q определяется величиной Д(7, а не Д/7.
Вычисление свойств перегретого пара
ТП
По уравнению U=H—pv из таблиц получаем
Ux = 668,3
14,5-10*-0,1388
427
621,2;
U2 = U жидкости -f-0,75177 парообразования 7/жидкости -[-0,75(7/ пара — — U жидкости)= 198,9-[-0,75-(б21,2—193,9) = 515,6,
177= 621,2—515,6 = 105,6 ккал/кг, откуда
Q = 2,72X105,6 = 287,2 ккал.
Влияние давления на теплоемкость. Имея дело с переходом тепла от одного вещества к другому под давлением, инженер обычно пользуется значениями энтальпии и не рассматривает теплоемкости. Однако встречаются обстоятельства, при которых удобно пользоваться данными по теплоемкости веществ под давлением для вычисления тепловых эффектов; кроме того, это свойство часто бывает полезно при различных сопоставлениях и расчетах, а также как способ проверки других термодинамических свойств. Сделано очень немного непосредственных измерений теплоемкости веществ прн повышенных давлениях, и поэтому желательно кратко рассмотреть пути, по которым можно вычислить изменение теплоемкости с давлением. Следующие точные уравнения образуют основу для вычисления влияния давления на теплоемкость:
,2>
р°
dv, (3)
J J \ui J V
С--С. = 7(|).(Й)/ (112, гл. Ill)
р° (или v°°) относится к состоянию при низком давлении (обычно при 1 атм), ниже которого теплоемкость практически не зависит от давления. Уравнения (2) и (3) получаются непосредственно при интегрировании уравнений (105, гл. Ill) и (104, гл. III), причем механический эквивалент J введен для того, чтобы не пропустить его при числовом подсчете.
Для идеального газа мы уже видели, что
= ° (157, гл. Ill)
и
^2^=0. (158, гл. III)
Другими словами, для идеального газа теплоемкость при постоянном объеме и постоянном давлении не изменяется с давлением (или
278
VI. Термодинамические свойства газов и жидкостей
с объемом); поэтому вопрос о влиянии давления на теплоемкость реальных газов непосредственно связан с вопросом об их отклонениях от закона идеальных газов. Это является полезным качественным выводом, так как показывает, что для многих вычислений можно игнорировать влияние давления на теплоемкость, так как оно становится значительным лишь при больших отклонениях от идеального состояния.
Кроме того, для идеального газа (или, приближенно, для любого газа при низком давлении)
Ср — Cv— R. (159, гл. Ill)
Полезны следующие приближенные наблюдения о влиянии давления на теплоемкость: увеличение Ср сверх значения при 1 атм может легко достигнуть даже 25°/0 для газов выше их критической температуры и при давлении в 1000 атм. Чем выше температура, тем меньше действие давления; при температурах, близких к критической, увеличение Ср становится весьма большим, так как в критической точке Ср = оо. Влияние давления на Cv значительно слабее; это легко видеть из уравнения (3) и из того факта, что изохоры газов очень близки к прямым, за исключением изохор при очень больших плотностях.
Из уравнения (2) очевидно, что Ср при высоком давлении можно легко определить, если известны данные р — v—Т для рассматриваемого газа и его изобарная теплоемкость С„ при низком давлении. Вычисление можно произвести графически или алгебраически, причем последний путь проще, если известно уравнение состояния. В любом случае для нахождения надежных значений Ср должны быть доступны очень точные данные по сжимаемости или точное уравнение состояния, так как приходится пользоваться второй производной.
Пример 7. Вычислить изобарную теплоемкость паров нефти при t ~ 300 и р = 20 кг/см2, если:
1. Константы уравнения состояния Линде [уравнение (68), гл. V] равны:
А = 0,000717, С = 447370, 0 = 0,000723, Е = 89700, 0=0,00267,
если р выражено в килограммах на квадратный сантиметр, v — в кубических метрат на килограмм и Т—в градусах Кельвина.
2. Теплоемкость пара прн 1 атм дается уравнением
Ср = 15,57 + 0,03992t,
где Ср выражено в килокалориях на килограмм на градус Цельсия. При диференцироваиии уравнения состояния получим
[&v\ 12 (С —Ер)
— Т\ — =----------------.
\дТ2/р Т<
Вычисление свойств перегретого пара
279
Из уравнения (2) имеем
f. __f. I 12 J f „ . p , . — , 12CJ । 6EJ (
Cp — Cpt~h -yi I + EP)dP — {P ~Pd + \p —Pi)’
p,
Cpi = 0,03992 X 300 +15,57 = 27,546.
Подставляя значения 7=4^7’ полУчаем
Ср = 27,546 + 0,711 + 0,003 = 28,26.
Произиодная (Л’/дТ'2) может быть исключена, так как она затрудняет обращение с уравнением состояния, определяющим р; поэтому для этого случая проще вычислить Ср путем сочетания уравнений (3) и (112, гл. III).
пример 8. Вычислить теплоемкость Ср воздуха при 0° С и 200 атм, если известны константы уравнения состояния Кейеса и Ср при 1 атм — = 0,240 кал/г-гра').
Уравнение состояния Кейеса
2,833 Г А
р = -----------------------.
v — 3 (о — Z)2
где
а — $е -в .
Для воздуха
а — 0,682, ₽= 1,589,
Л = 1605,3, Z = —0,088, атмосферах, v — в миллилитрах на грамм и Т — в гра-
если р выражено в дусах Кельвина.
При диференцировании уравнения состояния получаем
/<?р\ _ 7?
\дГ) 3 v — 8’
Поэтому, по уравнению (3), Cv не зависит от давления.
При диференцировании уравнения состояния получаем dv \ v — 8___________________________1____ , .
дТ)р — ~Т~ . аЗ 2Л (о —3)2 W
v2 RT(v — Z)3 и
Т (э?) Р ( dt) р = - is 2Л (о — 8)2 = 7 ~ <б)
1 t>2 RT(v — Гр
Так как при низком давлении члены, содержащие v, представляют лишь небольшие поправочные члены, то будет достаточно точно воспользоваться объемом идеального газа.
82,06 X 273,2 ,
V = 29,0-- = 773 МЛ! г’
280
VI. Термодинамические свойства газов и жидкостей
где 29,0 — средний молекулярный вес воздуха.
Для рассматриваемого случая уравнение (б) можно упростить до
р
J (Ср - Cv) = x_(2AiRrv} = 2.845 мл атм]г, ИЛИ
Ср ~ = 2,845 X 0,02422 = 0,0683 кал/г,
откуда = 0,240 _ 0 068 = 0д72 (при j атм^
Поскольку Ср не зависит от давления, это является также значением Cv при 200 атм.
При решении уравнения Кейеса методом подбора находим, что объем при 200 атм и 0° С равен 3,900 мл/г.
Снова используя уравнение (б), получаем
2,833
~ (1-0,0599- 0,431) ’ ’
откуда
Ср = 0,172 4- 0,135 = 0,307.
Сравнительно простое выражение для теплоемкости как функции давления можно получить, если опустить некоторые члены из уравнений, которые получаются при интегрировании диференциальных уравнений, подобных уравнению (2), с помощью уравнений состояния. Так, Битти [10] получил следующее уравнение при использовании этого приема и применении к уравнению состояния Битти — Бриджмена: с,=с,-+(^+?)'’’ <4’ где Ай и с — константы уравнения состояния. Это уравнение, конечно, только приближенно, но оно дает хорошие результаты до тех пор, пока давления не слишком высоки и температура значительно выше критической.
Деминг и Шуп вычислили влияние давления на теплоемкость водорода [57], азота [58] и окиси углерода [59] с помощью уравнения (2), применяя графические методы для определения второй производной и интеграла 31. Их значения для азота приведены на рис. 27. Эта диаграмма может дать общее представление о том, каким образом влияет давление на Ср в случае пара, который очень сильно перегрет, т. е. находится далеко от состояния насыщения. Мак-Кей и Кразе [163] измерили Ср азота при давлениях до 800 атм для нескольких температур, и совпадение между вычисленными и экспериментальными значениями, повидимому, находится в пределах точности экспериментальных измерений32.
Недавно опубликованы результаты вычисления влияния давления на Ср для восьми газов с помощью уравнения состояния Битти — Бриджмена [76].
Значения изобарной теплоемкости можно легко определить из энтальпии по уравнению
г — (дн\ р~~ \дт)р-
Рис. 27. Влияние давления на теплоемкость азота [58].
Рис. 28. Теплоемкость Ср водяного пара в области высоких давлений [284].
282
VI. Термодинамические свойства газов и жидкостей
Так как для водяного пара доступны многочисленные точные зна чения энтальпии, то из этих данных получается довольно полная картина влияния давления на Ср насыщенного и несколько пеоегре-
Рис. 29. Теплоемкость Ср водяного пара в области, близкой к критической точке (284].
того пара. Рис. 28 и 29 дают Ср для водяного, пара как функцию давления и температуры. Рис. 28 является неполным, поскольку в нем не показаны обе ветви изобар для давлений выше критического; поэтому добавляется рис. 29 для более полного показа поведения при высоких давлениях33.
Из рассмотрения рис. 28 и 29 следуют некоторые весьма интересные факты. Например, обычно думают, что Ср всегда увеличивается с температурой; но эти рисунки показывают, что при повышенных давлениях (ниже критического) Ср уменьшается с ростом температуры до тех пор, пока не достигаются очень высокие температуры, Скорость изменения с температурой вблизи линии насыщения
Вычисление свойств перегретого пара
283
также заслуживает внимания. Ниже критической точки теплоемкость увеличивается очень быстро по мере приближения К этой точке и становится бесконечно большой в критической точке, как и можно было ожидать из следующих рассуждений.
Из уравнения (109, гл. III) вытекает, что
(<Цр\ __ (<?/>/<«")„ \dvJT (dv/dT)p‘
Из свойств газов, рассмотренных в гл. V, очевидно, что (dp[dT)v является в критической точке конечной величиной. По уравнению (38, гл. V),
(д/\ =о.
V W/ Т
Поэтому
Наконец, по уравнению (112, гл. III), Ср=оо.
Выше критического давления (225,5 кг)см2} кривая Ср—Т быстро поднимается к максимуму и затем падает, причем при возрастании давления максимум становится менее острым.
Другим важным приложением уравнений (2) и (3) является проверка совместимости данных по термодинамическим свойствам. Если имеется ряд значений объема некоторого вещества, а из другого источника известен ряд значений Ср, то оба ряда можно сравнить с помощью указанных уравнений. Это дает очень чувствительный критерий точности экспериментальных данных по объему.
Определение объема по Ср. Возможность использования данных по теплоемкости для нахождения объемов не получила еще общего признания. Это является не только одним из интересных применений термодинамики, но также одним из практически важных ее применений. Таблицы водяного пара Кноблауха [325] основаны именно на таком расчете. Если дважды проинтегрировать уравнение (105, гл. III), то мы получим
^ = <P(p)+ri(p)-jjl^J7’rf7’_ (5)
где <р(р) и <Ь(р) являются только функциями р. Если газ можно считать идеальным при некоторой начальной высокой температуре, то дСр1др = 0 и
?(р) + 7’ф(р) = у^-
Существующие экспериментальные данные указывают, что подобное предельное допущение неточно. Однако мы знаем, что при любой
284
VI. Термодинамические свойства газов и жидкостей
температуре v = RT[p при р—*0, и это условие удовлетворяется, если Ф(Д)=у+Ф1(р)
и если <р! (р) и <р(р) остаются конечными при р—*0. Эта функции следует определить по экспериментальным данным ☆ ). Комбинированные члены (р) 4- Т’ф (р) уравнения (5) просто равны объему при исходной температуре интегрирования и легко определяются, если известно одно значение объема для каждой изобары (например объем насыщения).
Двойное интегрирование можно выполнить графически, зияя изме-нение Ср с р и Т. Этот метод был применен Якобом [117], тогда как Планк [189] пытался отыскать формулы для Ср как функции р и Т, чтобы интегрирование можно было выполнять аналитически. Хаузен [100] дал уравнение с девятью константами для данных по теплоемкостям Кноблауха и Коха [137] и из него вывел уравнение для v, Н и S как функций р и Т. Упомянутые выше таблицы для водяного пара основываются на этих формулах Хаузена для Ср. Этим путем, по крайней мере теоретически, можно получить очень точные значения объема из данных Ср, даже если последние обладают лишь умеренной точностью, так как начинают расчет со вторых производных и приходят к значениям интеграла.
Определение объема по энтальпии. При непосредственном измерении энтальпии вещества объем можно вычислить с помощью уравнения (96, гл. Ш.) Так как
то
vdT — Tdv
Интегрируя вдоль изобары, получаем
dT т
(6)
(7)
(8)
(9)
(дН]др)т определяется непосредственно по экспериментальным данным и может быть выражено как функция Т графически или аналитически. Значения v0 должны быть известны по нескольким независимым измерениям во всем интервале давлений, но только при одной температуре То. 34 _
•&) Описание метода см. [325].
Вычисление свойств перегретого пара
285
Рис. 30. Кривые постоянной энтальпии.
Свойства, определяемые по измерению эффекта Джоуля — Томсона. Эффект Джоуля—Томсона подробно рассматривается в следующей главе, но для нашей цели достаточно установить, что он заключается в изменении температуры, которым сопровождается адиабатное расширение газа от одного постоянного давления до другого постоянного давления таким образом, что не производится никакой внешней работы (кроме работы инжекции и эжекции) и не происходит превращения внутренней энергии в кинетическую энергию движения массы. Из результатов таких измерений можно определить удельные объемы, теплоемкости и энтальпии. Этот метод исключает необходимость непосредственных измерений объема и массы, точное измерение которых очень затруднительно; с другой стороны, о применение его связано с труд-ностью измерения средней Тем- q
пературы потока и поддержания §•
адиабатности процесса. Нельзя сказать, будет ли этот метод определения термодинамических свойств лучше или хуже непосредственного измерения объема, но он, по крайней мере, является важной заменой его.
Из измерения эффекта Джоуля — Томсона непосред
ственно получается или ряд кривых Т — р при постоянном Н, как показано на рис. 30, или ряд значений коэфициента Джоуля — Томсона g как функции давления и температуры, из которых при интегрировании можно найти зависимость Т — р. Значение//для каждой Т—/?-кривой получается экстраполяцией до оси р = 0, и вдоль этой оси можно применить зависимость т
H=H, + \cpdT. (10)
Зная теплоемкость при низком давлении как функцию температуры и произвольно выбирая значение Но, можно определить значение Н для каждой точки, где кривая Т—р пересекает ось р=0. С другой стороны, для небольшого температурного интервала можно написать
ср— [ьт)р и считать это за Ср при средней температуре. Тогда Ср __ ^Н^Г)р Ср0 —<ДН1ЬТ)р0 ’
(И)
(12)
286
VI. Термодинамические свойства' газов и жидкостей
и так как значения Д/7 одинаковы для всех точек между любыми двумя линиями постоянного Н, например Нг и Н2 (рис. 30), то
ср _*Тр°
СР<> мр
(13)
Можно получить &Тро и Д7^ непосредственно из графика, как показано на рис. 30- Зная Ср», можно легко найти Ср при различных температурах и давлениях. Для дальнейших деталей применения этих метод'ов см. статьи Кинена [127] и Ребука [199].
Поскольку
Г^(НСО)1 \др)т дТдр L дТ Jp’
Сэдж, Кенеди и Лэси [220] применили это уравнение для вычисления теплоемкости пропана под давлением по эффекту Джоуля—Томсона. Так как переменные нельзя разделить для проведения непосредственного интегрирования, то это должно быть осуществлено посредством соответствующих приближений.
Если известны значения Ср и р. как функции р и Т, то удельные объемы можно определить по измерениям эффекта Джоуля—Томсона следующим путем. Из уравнения (97, гл. Ш) получаем
<15>
Начиная с этого места ход расчета совпадает с ранее примененным для вычисления объема по измерениям энтальпии, причем конечным уравнением является
7 = (16)
1 1 0 J 1
Та
Этот метод также требует определения объема как функции давления при одной температуре То.
Энтропия. Это свойство нельзя измерить непосредственно, но оно вычисляется из других свойств. В общем энтропия увеличивается с возрастанием температуры и уменьшается с увеличением давления при постоянной температуре. Количественное соотношение дается уравнением
dS = Ср dp, (95, гл. III)
которое при интегрировании переходит в уравнение
+ dp. (17)
т, р„ р
Вычисление свойств перегретого пара
287
So—произвольное значение энтропии при состоянии р0, То, если только энтропия вещества не была уже зафиксирована при некотором другом стандартном состоянии. (Например, на практике довольно часто принимают So=0 в точке плавления льда.)
Для температур ниже критической максимальным значением верх
него предела второго интеграла является, конечно, давление пара при рассматриваемой температуре. Числовое определение энтропии с помощью уравнения (17) совершенно аналогично определению энтальпии по уравнению (1).
Иногда удобно вычислить энтропию из известных значений энтальпии. Примем, что имеется ряд значений Н как функции р и Т в графической форме или в виде таблиц. При постоянном давлении- уравнение (17) можно представить в виде т
S = dT. (18)
Т ?
Если известны наклоны изобар И — Т, это уравнение можно проин
тегрировать, чтобы найти разность энтропий вдоль ряда изобар. Для
получения абсолютных значений, связанных с некоторыми выбранными данными, необходимо иметь ряд значений S при различных давлениях вдоль одной изотермы или значения на кривой насыщенного пара.
Рис. 31, на котором представлена схема диаграммы энтропия — температура, может помочь разъяснению этих двух методов вычисления энтропии при приложении ко всей области перегретого пара. Кривая АСВ
Рис. 31. Расчет энтропии перегретого пара.
представляет ортобарную
кривую, а кривые рй— рА — изобары. Первый метод устанавливает значения S для различных температурвдоль изобары р0, выраженные через исходное значение So для насыщенного пара, которое, в свою очередь, основывается на значении S' для жидкости при р0, Tv При
няв значения энтропии вдоль рь за исходную величину, идут затем вдоль изотермы; две из них показаны на чертеже (линии DE и ДО). Ниже критической точки С все изотермы заканчиваются на кривой насыщенного пара. t
По второму методу даются значения энтропии вдоль каждой изобары, отсчитанные от некоторого исходного значения, например о г
288
VI. Термодинамические свойства газов и жидкостей
значения на кривой сухого насыщенного пара, которое связано с величиной S' изменением энтропии от испарения и изменением энтропии вдоль кривой жидкости. Для изобар выше критической изобары связь должна получаться или со значениями 5 (причем последние должны быть известны вдоль какой-либо изотермы выше критической температуры), или от принятия в уравнении (18) за нижний предел То температуру ниже критической и от приведения, на основании известных удельных объемов жидкости, энтропии к энтропии насыщенной жидкости.
Летучесть. Как показано в гл. III, летучесть определяется жо уравнениям
р
= (140, гл. Ill)
р°
и
f°=p° при р°—>0,
причем интегрирование производится вдоль изотермы. Из этих уравнений и уравнения состояния легко определяется числовое значение летучести.
Пример 9. Вычислить летучесть СО2 при 50° С и 100 атм, принимая, что СО2 подчиняется уравнению состояния Ван-дер-Ваальса
RT а
Р=-^~-------(42, гл. V)
v — b v2
Для нахождения интеграла можно переменить независимые переменные (р на v), как было сделано и примере 4, но мы используем эту возможность для иллюстрации другого метода. Диференцируя уравнение состояния при постоянной температуре, получим
RTdv ,2adv
dP- ~ •
тогда
vdp
RTvdv । 2a (v—b^’v2 v
RTv (v — b
dv.
p
v и и00 являются пределами объема, соответствующими давлениям р и /А Подставляя их в уравнение (140, гл. III) и интегрируя, получаем
1п / — 1п /° = Г — -4---— In (о — Z>)1 =
’ ’ [ RTv 1 v — b ' J ъ=°
b Ь 2а . 2а , , ,, > . , оо
------------------------------— — 1п(д — />)4-1п(и —Ь}.
v — b Vх— b RTv RTv™
Вычисление свойств перегретого пара
289
Если р 0, то р°° оо, поэтому
~’=0 и Ь = «°° = —Г.
Производя эти подстановки, находим
ln/_in/o=_±__^__in(y_z,) + in^ =
= In
RT b v — b'v — b
2a
RTv
lnp°.
Но так как из определения летучести /° = р° при р° -> 0, то окончательно получаем
, , . RT . b 2а .
1п/ = In---я-----Г—== . (а)
1 v — b'v — b RTv
По табл. III .Приложения" д = 3,604, Ь — 0,0428 (в атмосферах, литрах, градусах Кельвина и молях), R— 0,08206, Т = 273,2 -р 50 = 323,2° К, «(полученное из уравнения состояния методом подбора) = 0,1054. Подставляя эти величины в уравнение (д), получаем
1п/ = 4,153, /=63,1 атм.
Это, конечно, лишь приближенный результат, поскольку уравнение Ван-дер-Ваальса не вполне точно описывает соотношения для СО2. Однако может случиться и так, что приближение в этом случае будет очень хорошим, как это видно при применении более точного уравнения состояния.
Уравнение состояния Битти — Бриджмена приводит к следующему уравнению для летучести:
. z —, RT , 2? . Зу . 43
1п/_in + RTv + 2RTo2 + 3RTv3- ()
(Значения у и 8 указаны на стр. 241.) Для рассматриваемого примера это уравнение дает / = 62,1.
Льюис и Рендалл [351] разработали следующий полезный приближенный метод вычисления летучести. Примем, что в уравнений для определения летучести р
In t=--L^adp (142, гл. Ill)
о
величина а постоянна; тогда интегрирование дает
J--=e (19)
Для значений aplRT, которые являются малыми по сравнению с единицей, правая часть этого уравнения превращается в следующее выражение:
, аР pv
RT~ RT’
поэтому
/=^- (20)
19 Химическая термодинамика
290
VI. Термодинамические свойства газов и жидкостей
Следует помнить, что уравнение является точные только при относительно низких давлениях.
Термодинамические свойства, полученные из данных по сжимаемости графическими методами. Если должны определяться точные значения термодинамических свойств в широком интервале р и Т, ю применение обычного уравнения состояния в общем является неудовлетворительным, так как ни одно из уравнений состояния в широком интервале температур и давлений не бывает достаточно точно для того, чтобы дать возможность точно определить хотя бы первые (не говоря уже о вторых) диференциальные коэфициенты. Тогда за помощью следует обратиться к графическим методам обработки экспериментальных данных. Ниже приводимое краткое рассмотрение подобных методов основывается главным образом на ряде статей Деминга и Шуп [57, 58, 59], которые вычислили термодинамические свойства по данным сжимаемости водорода, азота и окиси углерода.
При вычислении 5, И, Ср, ц и т. д., если независимыми переменными будут р и Т, следует определить коэфициенты (dvjdT)p и (д^ъ/дТ2^. При нанесении на график зависимости v от Т кривые получаются почти линейными, и непосредственного определения наклона при различных температурах с достаточной точностью произвести нельзя. Если, однако, наносится на график не сам объем, а некоторая связанная с ним величина, то кривизна значительно увеличивается и поддается точному определению. Деминг и Шуп пользовались двумя величинами, именно:
а = (21)
Д = г'(^-1)- (22)
Рис. 32 показывает большие различия в кривизне между v—Т, Ь — Т и а — Т.
Из определения идеального газа ясно, что для такого газа обе величины а и Д будут равны нулю, и поэтому они являются важными поправочными коэфициентами, характеризующими отклонения от идеального состояния. Посредством диференцирования из уравнений (21) и (22) получаются, соответственно, следующие уравнения:
f dv\ _ R f да \
\dTjp~ Т \дт)р’
(дл) р= ~{дТ2\' <24>
(dv\ _ (pvfRT) (vID - (д±1дТ)р
\dTjp-- 1+(2д/«) ’
1 _?р
\дТг)р~ (2д/ц)+1 \\дТ*]р RT [(2д/и) +1 f
Вычисление свойств перегретого пара
291
Все графические измерения получаются при оперировании с величинами а и А быстрее, чем с первоначальными данными35.
Посредством вышеприведенных соотношений обычные диференциаль-
Температура, °C
Рис. 32. Сравнение кривизны линий, выражающих зависимость v, а и Д от температуры при постоянном давлении.
образовываются в формы, включающие а или А. Следующие уравнения представляют два типичных случая:
(27)
(28)
Деминг и Шуп сгладили исходные (экспериментальные) данные по сжимаемости посредством графиков А и а. В некоторых случаях графическое диференцирование проверялось нахождением уравнений для кривых. Интересно отметить, что они обычно употребляли величины
-£(£) и
v \др / Т v \ дТ J р вместо производных потому, что эти функции не имеют размерности, а также потому, что они для идеального газа равны единице и, следовательно, их числовое значение сразу определяет степень отклонения от идеальности36.
Практически нецелесообразно подробно рассматривать этот метод, но может быть полезно характеризовать основную схему его. Допу-19*
292
VI. Термодинамические свойства газов и жидкостей
стим, что следует вычислить энтальпию метана как функцию давления и температуры. Квалнес и Гэдди [14-0] составили таблицу для коэфициента сжимаемости Амага для давлений от 1 до 1000 атм и для температур от-—70 до 200°С. Из этой таблицы легко вычисляются удельные объемы v и с помощью уравнения (21) — значения а. Затем наносятся на график изобары а — Т, и путем графического диферён-цирования определяются наклоны кривых, т. е. (da/дТ)^. Наконец, в таблицу сводятся числовые значения (да/дТ)? как функции р и Т. Из уравнения (27) получаем
р
Н= Н.Л- f ГТ (//О
-- -V 1 J \ dTJp J г-
р°
Рассмотрим специальный случай, допустив, что желательно найти Н "при 100° С и 200 атм, причем Н равно нулю при 0°С и 1 атм. Зна-
чения а и (да/дТ)? для различных давлений вдоль изотермы 100° С будут считываться из заранее приготовленной таблицы, после чего вычисляется величина
Эта величина наносится затем на график как функция давления; получается кривая, площадь под которой между двумя данными давлениями равна указанному интегралу. Его значение пересчитывается в термические единицы и прибавляется к константе интегрирования Но, которая дается уравнением
373,2
Но= j Cp°dT,
Z13,2
где Cpi — теплоемкость при ,1 атм.
Этот путь слишком длинен и утомителен, если желательно получить только несколько значений энтальпии, но он очень полезен, если нужно составить диаграмму или таблицу свойств, охватывающую значительную область. Эдмистер [73] этим путем вычислил Ср, Ср—Cv, pi, S и Н для метана и свел в таблицы значения этих функций для ряда температур и давлений.
Метод вычисления летучести посредством использования а впервые введен Льюисом и Рендаллом и впоследствии применялся многими другими учеными. Так как из определения / и a
р
\nf=\np—~ [adp, (29)
'о
то для вычисления летучести на график наносятся изотермы a—р, и кривые экстраполируются до р = 0; площадь под кривой дает значение интеграла.
Вычисление свойств перегретого пара
293
Другой метод вычисления изотермических изменений термодинамических свойств, основанный на применении остаточной величины, подобной а и Д, дан Йорком [262]. Используемой остаточной величиной является отклонение от линейной зависимости при изохорном изменении. Для деталей следует обратиться к оригинальной статье.
Обобщенные термодинамические свойства. В гл. V было показано, что объем любого газа или пара можно удобно представить на обобщенной диаграмме коэфициентов сжимаемости путем нанесения коэфициента сжимаемости C = pvjRT как функции приведенного давления тс{=р[ркр) с приведенной температурой х(=Т[Ткр).
Этим путем можно представить приближенное поведение всех газов на одной диаграмме. Уравнение, выражающее С как функцию пит, будет обобщенным уравнением состояния. Из такой диаграммы или из соответствующих уравнений можно вычислить различные термодинамические свойства, например / Н и S.
Таким образом, уравнение (140, гл. Ill) можно видоизменить в
р
\g-^ = ^Cd\gp. (30)
р°
Значения С можно считать с диаграммы для различных значений п при данной т и нанести в зависимости от 1g р. Площадь ниже кривой до некоторого определенного давления, например р° = 1 атм или р° = 0,1 атм, дает значение lg///°; принимая f°=pP, получают значение f.
Для уточнения вычислений берут в качестве предела р° = 0, но при р°—Д) значение интеграла становится неопределенным. Эту труд-
р
ность можно избежать посредством вычитания^ dpfpиз обеих сторон - р°
уравнения (30); тогда, полагая, что p°=f°, получим
lg/=lg/4
р
1 С С~1 л
2,303 J р dp‘ р"
^1)
Легко показать, что (С—1)/р приближается к конечному значению при р —> 0.
Пример 10. Вычислить летучесть СО2 при 50° С и 100 атм, используя диаграмму коэфициента сжимаемости
323,2 _
304,2 — 1’062,
100_____j о™
73,0 1,37‘
294
VI. Термодинамические свойства газов и жидкостей
Считывая значения С с рис. 14, можно составить следующую таблицу:
р, атм 1С С С—1 Р
1 0,0137
5 0,0685 0,990 —0,002
10 0,137 0,970 —0,003
20 0,274 0,930 —0,0035
30 0,411 0,885 —0,0042
40 0,548 0,830 —0,00425
50 0,685 0,770 —0,0646
60 0,823 0,710 —0,00484
70 0,960 0,650 —0,0050
80 1,095 0,600 —0,0050
90 1,233 0,525 —0,00527
100 1,370 0,430 —0,0057
На графике, выражающем зависимость (С—1)/р от р, находим площадь под кривой между р = 100 и р = 0, равной —0,44.
Из уравнения (31) получаем: 2,303 lg/ = 2,303 1g 100—0,44=1,807,
/ = 64,2 атм.
Отношение f\p [175] называется коэфициентом активности (летучести) по аналогии с тем же термином, примененным Льюисом и Рендаллом для жидких растворов и является функцией пит. Ньютон вычислил летучесть большого числа газов графическим методом из p — v—7'-данных и представил средние результаты на трех графиках (рис. 33—35). По этим графикам практически при любых условиях можно легко определить коэфициент активности у, а следовательно, и летучесть для вещества, критические данные которого известны.
Из определения f и С имеем при постоянной Т d\nf=Cd\np.
Вычитая из обеих частей d In р н заменяя р на ркрп, получаем
ГС
In — = lny=J(C—1)йГ1птг, (32)
ж“
где я® — приведенное давление в стандартном состоянии, когда газ можно считать идеальным. С помощью этого уравнения можно получить коэфициент активности как функцию и и т на обобщенной
Рис. 33. Коэфициенты активности (летучести) чистых газов (область низких давлений) [175].
Приведенное давление, Л
Рис. 34. Коэфициенты активности (летучести) чистых газов (область средних давлений) [175].
296
VI. Термодинамические свойства газов и жидкостей
диаграммы коэфициента сжимаемости. Этот метод использован Ватсоном и Смитом [255], проводившими интегрирование графически.
Таким же способом, с помощью обобщенных данных по коэфи-циенту сжимаемости, можно приближенно определить влияние давления
Рис. 35 Коэфициенты активности (летучести) чистых газов (область высоких давлений) [175].
на другие свойства. Например, уравнение (96, гл. III) для случая
постоянной температуры можно представить в форме
дН\ ___RT2 (дС\
др/Т Р
(33)
и это выражение можно проинтегрировать с помощью значений С, полученных по обобщенной диаграмме коэфициента сжимаемости.
Коп, Льюис и Вебер [46] и Льюис и Люк [155] отчасти с помощью алгебраических зависимостей и отчасти с помощью графических представили С для углеводородов как функцию тс и т и из найденной зависимости получили уравнение для Н и v как функций давления. Эти уравнения сложны, но их решение упрощается, если дать некоторые функции в графической форме.
Вычисление свойств перегретого пара
297
Ватсон и Нельсон [254] вывели относительно простые уравнения для С как функции тг и т для углеводородов в ограниченных пределах и применили эти уравнения состояния для интегрирования уравнения (33), окончательно представив результаты на графике (№ — /7)/Т— я при различных значениях т.
Такой график представляет очень простое средство для приближенного вычисления энтальпии любого вещества, если известны его
Рис. 36. Обобщенный график разности энтальпий. ДЯ— разность между энтальпией при атмосферном давлении и при повышенном давлении по изотерме ккал!моль [255].
такого типа, путем:
который получен Ватсоном и Смитом [255] следующим
Н» — Н
RT2
Д//°
RT2 '
(146, гл. Щ)
Н—мольная энтальпия при данных р и Т и /7° — мольная энтальпия при той же самой Т, но при стандартном давлении р°, когда газ является идеальным и Н не меняется с давлением. Так как f='iP, то
/д In А __ /’сМп т \
298
VI. Термодинамические свойства газов и жидкостей
и так как Т=тТкр, то
_ птТ Л? In т \ Т —кр \Т дх) *
1 \* кр°^Р
р (д 1п -А Uinv®’
(34)
(35)
Наклоны определены графически диференцированием по графику 7—т с и в качестве параметра.
Пример 11. Чему равна разность между энтальпией 1 кг водяного пара при 100 кг; см2 и 400° С и при той же самой температуре, но низком
давлении? т = 400 4-273,2 = Ь47,3 100-1,033 218,2 -°’473-
По рис. 36 «= .,2, ад=И2^?=44,9. 1о
Соответствующее значение по таблице Вукаловича равно 43,0. Это является хорошим совпадением, учитывая приближения, связанные с рис. 36, и аномальные свойства водяного пара.
Другие термодинамические свойства можно обобщить аналогичным способом. Например, следующие уравнения легко выводятся посредством строгих термодинамических методов:
_ Н» — Н , 1 д [(Н0 — Н)/Т] 1
Т ”*1 д 1п т /я’
Ркр t
(36)
(37)
Ясно, что если известны критические давление и температура, то с помощью рис. 36 можно вычислить теплоемкость и коэфициент Джоуля— Томсона при повышенных давлениях. Ватсон и Смит [255] применили эти уравнения и для удобства дали результаты в виде графиков.
Додж [61] обобщил влияние давления на теплоемкость путем построения графика, на котором нанесено Ср/Ср>— отношение теплоемкости при любом давлении к теплоемкости при низком давлении, приближающемся к нулю (обе при одинаковой температуре), как функция приведенного давления с приведенной температурой в качестве параметра. Ватсон и Смит [255] обработали тем же путем разность С—Ср»-, их график воспроизводится на рис. 37, так как
Вычисление свойств перегретого пара
299
он несколько удобщ$ для применения, чем график Доджа. Надежных данных для проверки такой корреляции не очень много, и этот график менее точен, чем график для коэфициента сжимаемости. Отклонения в 25°/0 и более от наблюденных ☆ ) значений нередки. С другой стороны, следует отметить, что влияние давления невелико, и можно сделать значительную ошибку в ДС без большого влияния на С . Р
р
Пример 12. Определить мольную изобарную теплоемкость метана при 136 атм и 37,8° С, азота при 272 атм и 37,8° С, СО2 при 136 атм и 121,1° С и водорода при 340 атм и —17,8° С.
Решение представим в виде следующей таблицы.
Газ Приведенное давление, атм Приведенная температура, °C Ср £р» по рис. 37 СрО СР Ср .наблюденное*
СН4 2,96 1,63 3,6 8,79 12,4 12,17
n2 8,1 2,46 1,8 6,96 8,8 9,05
со2 1,87 1,30 6,5 10,14 16,6 16,20
Н2 26,6 13,8 0,1 6,79 6,9 7,16
Значение Сро и наблюденное берутся из [76J.
Другой метод обобщения термодинамических свойств (в применении к углеводородам) дан Эдмистером [74]. Он основан на приведенном уравнении состояния в графической форме при использовании величины
а,
а акр
где а определяется по уравнению (21). Последнее уравнение можно легко преобразовать В
V КГк„1
• -----------------(38)
акр Ркракр^
Отношение RTKfJpKpaKp не имеет размерности и почти постоянно для всех углеводородов; оно равно 1,37. Уравнение (38) в сочетании с графиком дж—кс т в качестве параметра дает основу для
☆) Непосредственные экспериментальные измерения очень немногочисленны. »Наблюденными* в данном случае названы значения, вычисленные каким-либо другим путем, например по уравнению состояния. Такие значения сами по себе могут быть не очень точными, так как часто приходится экстраполировать уравнение состояняя.
300
VI. Термодинамические свойства газов и жидкостей
вычисления термодинамических свойств. Такой график, основанный на опубликованных р—v— Т-д энных для девяти углеводородов, дан в статье Эдмистера. Метод вычисления влияния давления на свойства
Рис. 37. Обобщенный график влияния давления на мольную теплоемкость прн постоянном давлении [255].
можно кратко иллюстрировать на примере расчета энтальпии. Из уравнений (1) и (38) имеем при постоянной температуре
К
^Н= — Ркр^кр f — X J f39)
6
Интегрирование можно произвести графически с помощью значений ая, определенных по графику. Эдмистер дал обширную таблицу «приведенных термодинамических функций*, по которым можно определить летучесть, теплоемкость, энтальпию и энтропию любого углеводорода, для которого известны критические давление и температура.
Марон и Торнбулл [166] недавно дали обзор различных методов расчета и представили обобщенные аналитические корреляции, осно
Область двух фаз
301
ванные на уравнении состояния Битти — Бриджмена и на эмпирическом уравнении состояния в форме уравнения (31). Коротко говоря, их метод состоит в исключении р и Т из уравнения, определяющего v, выраженное через ркр, тг, Т и т, и в последующем определении констант, характеризующих рассматриваемый газ с помощью данных для азота. Этим способом они получили обобщенное уравнение, являющееся точным для любого газа в пределах точности теоремы соответственных состояний. В их статье детально рассмотрено несколько примеров.
Свойства газовых смесей. Невозможно входить в детальное рассмотрение этой очень обширной темы, и мы ограничим наше рассмотрение простым изложением некоторых доступных методов. Если уравнение состояния для смеси можно вывести по способам, указанным в гл. V, то можно вычислить различные свойства, например Н, S и /, с помощью методов, подобных указанным в этой главе для случая одного компонента. Этот метод является строгим, и точность результатов будет зависеть только от того, насколько хорошо уравнение состояния воспроизводит объемное изменение смеси37.
Эмпирическим методом, достаточно хорошим для многих случаев, является нахождение псевдокритических давления и температуры смеси и затем рассмотрение смеси как чистого вещества, обладающего этим критическим состоянием. Такие методы нахождения свойств посредством применения теоремы соответственных состояний уже были рассмотрены в этой главе.
Сэдж, Олдс и Лэси [223] разработали метод вычисления энтальпии смеси углеводородов, заключающийся в рассмотрении смеси как четырехкомпонентной системы, состоящей из метана, этана, н-бутана и н-пентана. Энтальпия смеси вычисляется по обычному уравнению, связывающему ее с парциальными энтальпиями различных компонентов [уравнение (8, гл. IV)], причем последние величины определяются из приведенных в таблицах экспериментальных данных по парциальным энтальпиям бинарных растворов. Этот метод вычисления сравнивался с экспериментальными измерениями для двух природных газов и с вычисленными значениями, основанными на псевдокритических константах и на законе соответственных состояний. Их метод дал несколько более близкое совпадение с экспериментальными данными, чем последний из упомянутых способов, но сомнительно, оправдывает ли выигрыш в точности большую сложность метода.
ОБЛАСТЬ ДВУХ ФАЗ
Рассмотрение главным образом будет ограничиваться равновесием жидкость — пар, но следует отметить, что ббльшую часть общих зависимостей можно также хорошо применить и к равновесиям жидкость —’ твердое тело, твердое тело — пар и твердое тело — твердое тело.
302
VI. Термодинамические свойства газов и жидкостей
Рассматриваемая нами область в отношении давления и температуры ограничена тройной точкой снизу и критической точкой сверху (см. рис. 23). Она показана как область VI на рис. 24.
Объем, энтальпия и энтропия. Любая точка в этой области представляет смесь насыщенных пара и жидкости, и так как все эти три свойства экстенсивны, то их значения для смеси просто равны сумме отдельных значений для двух фаз. Следовательно:
v = v’ Ц-х (v"— v'), (40)
H = Н’}, (41)
И— Н’-\- xL, (42)
5 = S' + x(S" —5'), (43)
5 = (44)
где х — сухость пара, т. е. доля смеси, присутствующая в виде пара. Свойства насыщенного пара (') можно считать просто предельными значениями свойств в области перегрева, если давление равно давлению насыщенного пара при данной температуре или температура равна температуре кипения при рассматриваемом давлении. Подобным же образом свойства насыщенной жидкости (') являются предельными значениями для жидкой области, если жидкость находится при температуре кипения. Из уравнения (43) очевидно, что сухость пара в области двух фаз на диаграмме равна расстоянию по горизонтали отш данной точки до кривой жидкости, деленному на общее расстояние по горизонтали между двумя пограничными кривыми. Это служит основой для проведения линии постоянной сухости пара на такой диаграмме.
Внутренняя энергия определяется уравнением
U=H—pv — lf—/н>'-|-х[(/Г— И’)—р(чГ — »')]. (45)
Соотношения для давления насыщенного пара. Давление насыщенного пара как функция температуры является Одним из наиболее важных термодинамических свойств и должно определяться экспериментально. Для интерполяции и экстраполяции экспериментальных данных, а также для сведения к минимуму числа требуемых данных, весьма полезны уравнения давления насыщенного пара, и мы дадим обзор некоторых из них, являющихся наиболее важными. Исходным для большинства уравнений по давлению насыщенного пара служит уравнение Клаузиуса — Клапейрона; в своей диференциальной форме оно совершенно строго [уравнение (153, гл. IV)], но для интегрирования его вводятся различные допущения.
Линейная зависимость 1g р от 1/Т. Если пренебречь объемом жидкости по сравнению с объемом пара и допустить, что пар
Область двух фаз
303
является идеальным газом, то уравнение (153, гл. IV) переходит в
юоо
Рис. 38. Линейная
1 , от jr (р — давле-
зависимость1g р ние насыщенного пара).
(46)
Интегрируя уравнение (46) при допущении, что L не зависит от температуры, получаем
(47>
Можно написать уравнение (47) как эмпирическое:
lgP = y + 5-
(48)
304
VI. Термодинамические свойства газов и жидкостей
Это очень полезное уравнение, поскольку, пользуясь им, можно получить данные по давлению насыщенного пара практически для всех веществ в довольно широком интервале температур с приемлемой точностью, и для определения констант требуются только две экспериментальные точки. Рис. 38 показывает, насколько хорошо согласуются с этим простым соотношением измеренные давления паров воды, этана и азота. Для азота совпадение почти полное во всем интервале сосуществования жидкости и пара. Это особенно замечательно потому, что ни одно из трех допущений, на которых основывается уравнение, даже приближенно не является действительным для столь большого интервала. Копсон и Фролих [471 представили диаграмму давления пара для восьми низших углеводородов в интервале примерно от 0,001 атм до критического давления при использовании координат 1g р и 1/Т, причем отклонение от прямой линии оказалось небольшим.
Другие уравнения. При допущении, что скрытая теплота парообразования связана с температурой уравнением
L = Z.Q а.Т, (49)
интегрирование уравнения (46) дает следующее полезное эмпирическое уравнение для давления насыщенного пара:
л lgp = ^+B\gT+C. (50)
Выбор уравнения давления пара в каждом конкретном случае целиком зависит от данного вещества и от желательной точности расчета, и поэтому нельзя дать никакого общего правила. Ниже приводятся некоторые уравнения, применяющиеся для представления данных по давлению насыщенного пара:
lgp = ^ + B+CT+DT2+..., (51)
= 4-B-Ll,751g7’ + C7’, (52)
А
lgP = y + 5 + CIgT-|-r>T+..., (53)
< Ркр ( Ткр
^~р~—1/’ (54)
, РКР Ткр-Т[а+Ь(Ткр-Т)+с{Ткр-Т}3 + е{Ткр^Т)*1
= — [ Г+^ТКР-Т)-----------J’ <55>
Уравнение (52) известно как формула Нернста; с помощью уравнения (55) Смит, Кейес и Джерри [234] весьма точно представили свои
Область двух фаз 305
данные для воды от нормальной точки кипения до критической точки; уравнение (54) предложено Ван-дер-Ваальсом.
Правило Дюринга. Это правило даёт простой и очень точный метод для Интерполяции и экстраполяции давления насыщенного пара. Правило выражается уравнением ----------
<57>
где tA и tB —соответственно температуры кипения веществ А я В при одинаковом давлении ри tA и tB —те же величины для некоторого другого давления р2, а с — константа. А — вещество, давление пара которого определяется, а В—стандартное вещество, давление пара которого известно во всем рассматриваемом интервале температур и которое, по возможности, должно быть химически подобно веществу А. Для определения с требуются два значения давления пара вещества А. Уравнение (57) можно также представить в форме
(58)
где tA и tB — температуры кипения веществ А к В при одинаковом давлении их насыщенных паров. По этой форме уравнения можно видеть, что если нанести на график tA в зависимости от tB, то получится прямая линия, и правило обычно применяется в этой графической форме. В какой бы форме правило не применялось, несколько неудобно то, что оно должно применяться в соединении с кривой давления пара или с таблицей для стандартного вещества. Если эти данные отсутствуют, то следует решить, имеет ли это правило преимущества по сравнению с применением уравнения, отвечающего линейной зависимости 1g р от 1/Г [уравнение (48)]38.
Рэл [202] установил, что это правило применимо также и к водным растворам, и показал, как можно связать константы и k2 с концентрацией растворов, а также показал связь этих констант с числом углеродных атомов в гомологических рядах органических соединений39.
Диаграмма Кокса. Кокс [50] предложил метод нанесения данных по давлению насыщенного пара, который дает прямые линии и, кроме того, позволяет в некоторых случаях определить полную кривую давления пара по одной экспериментальной точке. Его метод заключается в следующем. На ось ординат наносится логарифмическая шкала давления, и проводятся горизонтальные координатные линии. Затем около центра листа проводится прямая линия снизу вверх направо под углом около 45° к горизонтали. Эта линия выбирается в качестве линии давления насыщенного пара стандартного вещества, обычно воды (или ртути для температур выше критической , температуры воды). С помощью данных по давлению насыщенного
20 Химическая термодинамика
306
VI. Термодинамические свойства газов и жидкостей
пара воды на абсциссе строится шкала температур в соответствии с выбранной линией давления пара и проводятся вертикальные координатные линии. Если нанести давления пара любого вещества на полученную таким путем систему координат, то получается примерно прямая линия. Кроме того, оказалось, что группы родственных веществ, как парафиновые углеводороды, спирты и металлы, дают линии, сходящиеся в общей точке. Последнее означает, что для члена подобной группы, для которой известна точка пересечения, можно определить всю линию давления пара по одному значению давления пара, например в нормальной точке кипения.
Калингерт и Девис [39] показали, что применение диаграммы Кокса эквивалентно использованию следующего уравнения:
^SP = A г-|-230 ’
где Л и В — эмпирические константы, a t выражено в градусах Цельсия. Ясно, что прямая линия здесь будет результатом нанесения 1g р в зависимости от 1/(/-}-230), и для нахождения линии достаточно двух экспериментальных значений. Это по существу то же, что и применение линейной зависимости 1g р от 1/Т [уравнение (48)], за исключением эмпирической константы 230, выбранной вместо точки затвердевания воды на абсолютной стоградусной шкале.
Скрытая теплота парообразования. .Если известно давление ' насыщенного пара рассматриваемого вещества, удельные объемы насыщенной жидкости и пара, то скрытую теплоту можно точно определить по уравнению (153, гл. IV).
Пример 13. Измерения давления насыщенного пара метана, произведенные Кейесом, Тейлором и Смитом [133J, представлены следующим уравнением:
Igp (атм) = — 595^546 4-8.09938 — 4,04175X Ю-2Т+ 1,68655-10-Т2 — -2,51715-10-77®.
Удельные объемы пара и жидкости при 165° К найдены равными соответственно 34,49 и 3,0496 мл/г. Вычислить скрытую теплоту парообразования при этой температуре.
Напишем уравнение давления насыщенного пара в общей форме:
1g Р = у 4- В + СГ4- DT2 4- ЕТЗ.
Диференцируя, получаем
й®7=(-7-+с+2ОГ+зетг)''7', или
^£ = 2,3026/7 —^4-С4-2£>Г4-3£72У
Т = 165° К, р = 19,22, До = 34,49—3,05 — 31,44 мл[г,
^ = 0,73018 атм1°К-аТ
Область двух фаз
307
Поэтому, на основании уравнения (153, гл. IV), имеем:
L = 165 X 31,44 X 0,73018 = 3788 мл-атм.г X 0,02420 — 91,68 кал/г.
При низком давлении можно принять, что пар является идеаль-ным газом и что удельным объемом жидкости по сравнению с удельным объемом пара можно пренебречь; в этом случае скрытую теплоту парообразования можно вычислить по уравнению (46).
Пример 14. Давление пара азота дается уравнением
lg/> (атм) = — Р04^ + 3,93352.
Вычислить скрытую теплоту парообразования в нормальной точке кипения.
По этому уравнению, если р — 1,000, то Т— 77,41° К. Напишем уравнение в общей форме:
lgp = -y + -B-
Тогда
1 dp 2,30264 L . ,.й„
~pdf= Л ~^7~г1до Уравнению (46)],
или £ = 2,302647? = 2,3026 X 304,49 X 1.987=1393 к ал]моль. (а)
Разность между этим числом и наблюденной величиной Дана [52J (1335 кал1молб) больше, чем следовало бы ожидать на основании отклонения от законов идеального газа. В других случаях отмечаются еще большие расхождения между наблюденными и вычисленными величинами. Например, уравнение
1gр (мм) = — _|_ 8,7764,
которое очень хорошо соответствует данным Рамзая и Юнга для давления пара метанола, дает при использовании уравнения (46) для скрытой теплоты парообразования при нормальной точке кипения 9115 кал1моль, тогда как экспериментальная величина равна 8420.
Этн расхождения указывают на важное обстоятельство. Хотя уравнения, выражающие зависимость 1gр от 1/Т в линейной форме, могут очень хорошо представлять данные по давлению насыщенного пара, но они могут оказаться неудовлетворительными для вычисления наклона dp/dT. На это указывает тот факт, что такое уравнение для давления насыщенного пара в сочетании с уравнением состояния идеального газа приводит к уравнению (а) примера 14, основанному на допущении о независимости скрытой теплоты от температуры, что явно противоречит фактам.
Теплоемкости насыщенных жидкости и пара. Теплоемкость вообще определяется уравнением
r—dQ
G dT
20*
308 VI. Термодинамические свойства газов и жидкостей
и зависит от пути изменения температуры. Теплоемкость насыщенной фазы (жидкости или пара) представляет частное от деления теплового эффекта, отвечающего переходу от одной точки на кривой насыщения к другой точке, лежащей на той же кривой на бесконечно малом расстоянии от первой, — на бесконечно малое приращение температуры. Пусть на рис. 39 А и В будут двумя точками на кривой давления насыщенного пара, представляющими два состояния насыщенного пара, бесконечно мало отличающихся по р и Т. Переход из А в В можно представить как результат двух независимых изменений, одного при постоянном давлении (отрезок АС), а другого при постоянной температуре (отрезок СВ). По уравнению (95, гл. Ш) имеем
TdS = dQ=CpdT-T^dp (60)
И
dQ „ 'г dP
dT~cs—cp т \дт)рат’ <61)
наклона кривой давления пара. Это урав-
где dpjdT—тангенс угла
Рис. 39. Теплоемкость насыщенного пара.
нулю. В случае водяного пара тельна. Это
нение можно применить к любой фазе. Повидимому, теплоемкость кипящей жидкости С$ практически совпадает с теплоемкостью Ср жидкости, взятой в том же самом температурном интервале, так как коэфициент (дъ[дТ)р для нее очень мал (за исключением области, близкой к критической точке).
В зависимости от относительной величины двух членов правой части уравнения (61) теплоемкость насы
щенного пара С$ может быть положительна, отрицательна или равна она в большинстве случаев отрица-почтн всегда кажется удивительным всем, кто привык
думать только об изобарной или изохорной теплоемкости, которые всегда положительны. То, что С$ для водяного пара отрицательна, легко можно показать по значениям, взятым из таблиц для двух
состояний насыщенного пара, которые достаточно близки друг к другу, чтобы мы могли принять Др/АТ = dpjdT. Подобным же образом это
можно показать и для других производных.
Пример 15. Вычислить теплоемкость насыщенного водяного пара при 175°С.
Из таблиц Вукаловича заимствуем следующие величины: tx = 174° С, р± — 8,888 кг/см2, = 0,2215 м3/кг, t2=:176oC, р2 = 9,317 кг]смг, о2 = 0,2118 ж3/«г,
Область двух фаз
309
Для нахождения Ср и (dv/dTfp возьмем следующие два состояния вдоль изобары:
t3=180°, ps = 9,0, v3 = 0,2226, r=665,8 ккал!кг,
<4=190°, p4=9,0, p4 = 0,2291, ккал/кг,
„ _671,5 - 665,8,.
p~\iT ) p~~ 190 — 180 ~ ’ ’
/Av A = 02291 -02226 =
J p 1 v
Ар ч 9,317—8,888
ду, вдоль кривой насыщения = __щ- =0,2145.
Из уравнения (61) имеем: С^ = 0,57— (175-)-273,2)-0,00065-0,215-23,42 = = —0,90 (23,42 представляет тепловой эквивалент работы при примененных в данном случае единицах).
Физическая интерпретация отрицательной теплоемкости очень" проста. Действительно, для перехода вдоль отрезка АС (см. рис. 39) следует подвести тепло, вследствие чего пар становится несколько перегретым. Чтобы возвратить пар в состояние насыщэния, его следует изотермически сжать от С до В, «то связано с отнятием тепла. Если это тепло больше тепла, сообщенного вдоль АС, что имеет место в выбранных условиях в случае насыщенного водяного пара, то теплоемкость будет отрицательна ☆).
Интересным следствием этого является заключение о том, что происходит, когда насыщенный водяной пар при этих условиях расширяется адиабатно и обратимо. Для такого случая dQ =0 и dS = 0, н поэтому
Теперь, так как для изменения от В до А, как мы только что видели, Т(dv]dT)p dp больше, чем CpdT, или, другими словами, так как вдоль ВС сообщается больше тепла, чем отнимается вдоль СА, то ясно, что после точки А для сохранения адиабатности процесса в целом тепло должно отниматься от водяного пара, т. е. он должен частично конденсироваться. Другими словами, при адиабатном и изоэнтропном расширении насыщенный водяной пар становится влажным; наоборот, при сжатии он становится перегретым. По вышеприведенному доказательству эта формулировка строго верна только для расширения или
*) По существу нет ничего удивительного в отрицательной теплоемкости насыщенного пара. Рассмотрим идеальный двухатомный газ при 0° С и 1 атм, который изменяет свое состояние до 25° Си 1,50 атм. Легко видеть, что средние теплоемкости на двух путях, по которым может происходить изменение, являются отрицательными.
310
VI. Термодинамические свойства газов и жидкостей
другие вещества, причем
Энтропия
Рис. 40. Схематическая диаграмма температура—энтропия для углеводородов.
сжатия при бесконечно малом интервале давлений, но практически ее можно распространить на конечное изменение.
Отрицательной теплоемкостью насыщенного пара обладают многие чными примерами являются СО2, NHS и О2, что приводит к диаграмме 5—Т, показанной в общем виде на рис. 25.
Другие вещества, например многие углеводороды, имеют положительную теплоемкость насыщенного пара, по крайней мере в некоторых областях, и это приводит к противоположному, по сравнению с водяным паром, поведению при нх сжатии и расширении. Типичная диаграмма 5—Т для такого вещества схематично показана на рис. 40. АВ указывает путь адиабатного расширения или сжатия и показывает, что перегретый пар при сжатии становится
влажным, что как' раз противоположно поведению водяного пара *°.
Изменение скрытой теплоты парообразования с температурой 4I. По уравнению Клаузиуса2—Клапейрона
L = T&v~ (153, гл. IV)
скрытую теплоту парообразования можно вычислить как функцию температуры, если известны давление пара и удельные объемы обеих фаз во всем температурном интервале. Хотя dpjdT и увеличивается с температурой, но До уменьшается с большей скоростью, и общим результатом является уменьшение скрытой теплоты с увеличением температуры, причем значение скрытой теплоты достигает нуля в критической точке, где До = 0.
Ниже приводимые уравнения для изменения L с температурой интересны, но не представляют большой практической ценности, хотя некоторые из них и использовались для проверки надежности термодинамических данных. Из определения теплоемкости насыщенной фазы и из первого закона термодинамики следует, что
Г ___dO _dU , dv _
dT dT dT' ^62)
Прилагая это уравнение к обеим фазам и вычитая одно уравнение из другого, приходим к уравнению
_ d(U”-U’) ,dW-v'}
Cs — Cs—-------------F р----—— . (63)
Диференцируя уравнение
L = H" — Н' — W—U' -\-p(v'r — vr) (64)
Область двух фаз 311 ।
по Т, получаем ;
dL d(Ur—(/'} , d(v"~v') . . , ,.dp _ J
dT=- dt~ +P—UT +(« (65) ~ —
Из уравнений(бЗ) и (65) находим, что ---------—-г
' <3-Л=§<**)
По уравнению Клаузиуса—Клапейрона а
{V" — V')d± = ^. (153, гл. IV) ;
Поэтому ]
с;-6 = ^-4- (67) ' 4
При принятии C's = Ср уравнение (67) переходит в
й = ^+я—т- <68>
____Пример 16. Используя уравнение (68), вычислить C”s для того же са-мого случая, что и в примере 15, а именно — для насыщенного водяного иара ири 175° С.
По таблицам Вукаловича,
t — 174° С, L = 486,0 ккал/кг,
t = 176° С, L = 484,3 ккал) кг, А£ 1,7 л ос I А —~ = 0,9а ккал/кг-град.
При 175° С
L = 485,2, = 1,05,
откуда
CS = 1,05- 0,85- gg = -0,88.
Если учесть приближенность вычисления, то совпадение с результатами, полученными в примере 15, является достаточно хорошим.
Пользуясь уравнениями (67), (61) и (154, гл. IV), можно легко показать, что
dL__с' L Г 1
dT— т ‘~ ₽ V" — p'Lk^/p Ur/pJ
(69)
(70)
Уравнение (70) можно также написать в форме dL_^ । Г(дН"\ /дН'\ 1 dp
ат~ р ₽_* Lk^/r \^p)т\aT,
(71)
312
VI. Термодинамические свойства газов и жидкостей
в которой оно было использовано Сэджем, Эвансом и Лэси [218] для вычисления скрытых теплот парообразования пропана и я-пентана. Если имеются данные по объемам насыщенных фаз, то уравнение Клаузиуса — Клапейрона является гораздо более простым средством для вычисления L, чем это уравнение.
Общая зависимость
(^),=дс₽ <72>
иногда применяется и к случаю испарения, но следует помнить что она является точным соотношением лишь для случая постоянного давления. В случае фазового равновесия давление представляет функ-цию температуры и не может поддерживаться постоянным при изме-- ; нении температуры. С другой стороны, при уменьшенных давлениях
J- даже для пара теплоемкость при постоянном давлении не изменяется
В значительно с давлением, и поэтому &Ср в вышеприведенном урав-
Jf. нении можно принять с хорошим приближением равным разности
> между Ср пара и Ср жидкости при атмосферном давлении42,
. Если написать
ДСр=Да4-Д37’4-ДуГ, (73)
г где Да = апара— ®жвдкости (с подобными же выражениями и для дру-
гих коэфициентов , и связать его с уравнением (72) в форме
*74>
то после интегрирования в пределах от Т до Тв (нормальная точка кипения) получим
£г=£в + Да(Т— ТВ)+±Щ(-П— 7^) + 1дТ(7^-7^). (75)
z По этому уравнению можно вычислить скрытую теплоту парообразо-
* вания при любой температуре (при условии, что давление ие будет
’ слишком высоким) из скрытой теплоты парообразования прн нормаль-
ной точке кипения Тв, если имеются данные по изобарным теплоемкостям двух фаз.
При сочетании уравнений (75) и (46) и интегрировании в пределах от Т до Тв, приходим к следующему полезному уравнению для интерполяции и экстраполяции данных по давлению насыщенного пара:
1пр= ~ + 51пТ4-СТ + £)7^4-Е-, (76)
Совокупность термодинамических свойств
313
где
LB Ыв ^1%
А— r л ад з/? * о______Аа
-------
с-^-
£=М—#1пГв’ ф
Это уравнение совпадает с уравнением (53), но можно вычислить все константы уравнения (76), если известны теплоемкости двух фаз как функции температуры, нормальной температуры кипения и скрытой теплоты парообразования в этой точке*8.
СОВОКУПНОСТЬ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
---Совокупность всех наиболее важных термодинамических свойств данного вещества, в частности v, Н, U и S, как функций давления и температуры будем называть комплексом термодинамических свойств. Эта совокупность обычно представляется в виде серии таблиц или в виде одной или нескольких термодинамических диаграмм, уже рассмотренных выше. Получение полного комплекса свойств из минимального количества экспериментальных данных и проверка их надежности составляют проблему значительной важности, требующую большого труда и изобретательности. Хотя мы и не можем входить в детали, тем не менее целесообразно в самых общих чертах рассмотреть некоторые методы, использованные в отдельных случаях, без всяких попыток дать исчерпывающие результаты для какого-нибудь одного случая.
Паровые таблицы Кинена— Кейеса [129] (исключая на время область, близкую к критической точке) основываются по существу на следующих данных:
1. Давление насыщенного пара воды.
2. Удельный объем водяного пара во всем интервале давлений и температур.
3. Теплоемкость водяного пара как функция температуры при нулевом давлении, рассчитанная по полосатым спектрам.
4. Удельный объем жидкой воды.
5. Скрытая теплота парообразования при одной температуре.
6. Энтальпия насыщенной жидкости во всем интервале температур.
Энтальпия пара была найдена с помощью уравнения (1), причем интегрирование производилось алгебраически, с помощью степенных рядов функции для Ср0 и уравнения состояния, которое очень точно воспроизводит измеренные объемы перегретого пара. Объемы насы
314
VI. Термодинамические свойства газов и жидкостей
щения получались экстраполяцией данных по объему до известного давления насыщения при данной температуре, причем использовалось специальное уравнение состояния. Произвольная константа интегрирования определялась из энтальпии при 100° С и 1 атм, полученной из калориметрических измерений, произведенных в Бюро стандартов США [179].
Энтропия пара получена тем же самым путем, как и энтальпия, т. е. с помощью уравнения (17). Константа интегрирования определялась из энтропии при 1 атм и 100° С, также полученной по калориметрическим измерениям Бюро стандартов.
Данные по объему жидкости служили для определения энтальпии и энтропии жидкости вдали от линии насыщения, причем интегрирование производилось при сочетании алгебраических и графических методов. Энтальпия насыщенной жидкости взята из работы Осборна с сотрудниками. Энтропии жидкости найдены вычитанием AS парообразования из энтропии насыщенного пара, причем AS парообразования были определены из Д/7.
Для получения свойств в области, близкой к критической точке, были использованы несколько отличные методы, так как выведенные формулы неудовлетворительны в этой области, где производные изменяются так быстро. Для определения свойств в этой области служили измерения энтальпии, произведенные Хавличеком и Мисковским и связанные с давлением и температурой полуграфическими формулами. Объемы и энтропии вычислялись из значений Н с помощью ранее представленных соотношений.
Надежность свойств, выведенных по вышеприведенным методам, и их совпадение с родственными данными, полученными другими исследователями, могут быть проверены несколькими путями, типичными примерами которых являются нижеследующие:
1. Вычисление энтальпий и сравнение их с энтальпией, измеренной непосредственно в Бюро стандартов США.
2. Теплоемкости можно вычислить из энтальпий с помощью зависимости
г —(дН\ Ср~\дТ)р
и сравнить с непосредственными измерениими.
3. Коэфициенты Джоуля — Томсоиа можно вычислить из уравнения энтальпии посредством диференцирЬвания = и сравнить с непосред-
ственно измеренными значениями.
4. Уравнение Клаузиуса — Клапейрона дает средство для проверки надежности следующих данных:
а) давление насыщенного пара;
б) удельные объемы насыщенных жидкости и пара;
в) разность энтальпий насыщенных пара и жидкости.
Пример 17. Измерения термодинамических свойств н-бутана дают следующие значения при 32° С:
плотность насыщенной жидкости = 0,5635 г/мл,
плотность сухого насыщенного пара = 0,00756 г!мл,
Совокупность термодинамических свойств
315
скрытая теплота парообразования = 85,2 кал/г.
Давление пара как функцвя температуры представлено уравнением
lgp(amM) — 1,751g Т-[-сТ,
где
а = 1,7568, Ь = — 1337,8, с = — 0,004070.
Выясним термодинамическим путем, являются ли вышеприведенные значения надежными илн нет.
Диференцирование уравнения давления пара и подстановка Т = 305,2 дают:
= 0,029462р,
при
Г =305,2, р^= 3,006, и поэтому
* ^ = 0,08856.
dT
При использовании уравнения Клаузиуса — Клапейрона
dp L
dT ТЛи
85,2 X 41,33 —
ЧЛК 1 ( 1_________1
’ \о,00756 0,5635
= 0,08831.;
Совпадение находится в пределах 0,3°/о, что вполне удовлетворительно Ч
Термодинамические свойства пропана [224] найдены по следующим данным, причем все они измерялись в одной лаборатории.
1. Удельный объем жидкости и пара как функция давления и температуры.
2. Давление насыщенного пара. _
3. Теплоемкость жидкости как функция давления и температуры. Все диференцирования и интегрирования производились графически. AS парообразования вычислялась по уравнению Клаузиуса—-Клапейрона.
Составление паровых таблиц Кинена— Кейеса и Вукаловича иллюстрирует случай, когда термодинамические свойства в основном выводятся из очень полных и весьма точных измерений объема, получе иных в одной лаборатории, где были доступны и многочисленные другие измерения для проверки точности результатов. Чаще для получения комплекса термодинамических свойств приходится соединять данные, взятые из большого числа различных источников, данные, которые часто являются неполными и не всегда согласованными между собой. Пример такого рода дает работа Миллара и Сюлли-вана[171] по термодинамическим свойствам кислорода и азота. Их метод можно кратко описать следующим образом.
Имевшиеся в наличии данные (в частности, относящиеся к кислороду) могут быть сведены в следующие группы:
i
316 VI. Термодинамические свойства газов и жидкостей
1. Теплоемкость Ср жидкости при 1 атм от нормальной точки кипения N2 до нормальной точки кипения О2.
2. Теплоемкость Ср газа при 1 атм.
3. Давление насыщенного пара.
4. Теплота парообразования при 1 атм.
5. Плотность насыщенного пара.
6. Плотность насыщенной жидкости.
7. Отдельные р— v—Т-данные в области перегрева.
Энтальпия и энтропия для обеих жидкостей принимаются равными нулю в нормальной точке кипения азота (77,4° К)-
Данные группы 1 служили для определения Н и S жидкого кислорода при его нормальной точке кипения. Затем, по данным группы 4, определялись Н и S насыщенного пара, которые с помощью данных группы 2 распространялись на область перегрева вдоль изобары 1 атм. Значения Н и S вдоль других изобар в области перегрева можно получить по данным группы 7. Значения их на линии насыщеннцро пара определялись по объемам пара, которые получались экстраполяцией изохор перегретого пара до кривой давления пара. Затем можно получить Н и 5 насыщенной жидкости вычитанием скрытых теплот парообразования из соответствующих значений для насыщенного пара, причем скрытые теплоты получались по уравнению Клаузиуса— Клапейрона при использовании данных групп 3, 5 и 6.
Сглаживание данных, диференцирование и интегрирование производились большей частью графически. Для составления термодинамических диаграмм изготовлялись графики Т — Н при постоянном р, р—Н при постоянной Т и Т—р при постоянной Н, и принимались меры для получения плавных кривых на всех трех диаграммах и для получения соответствующих изменений Ср (наклон изобар на диаграмме Т—И) с температурой. Затем таким же способом строилась диаграмма S— Т, причем линии постоянного р определялись по изобарам на диаграмме Т—Н посредством графического интегрирования уравнения . ?
dS=~.
При постоянном давлении конечные результаты всех трех вычислений объединялись в таблицах свойств и на диаграмме, связывающей \gpuH.
ГЛАВА VII
СЖАТИЕ И РАСШИРЕНИЕ ГАЗОВ
При многих химических процессах вещества подвергаются изменениям давления; поэтому для инженера-химика важно уметь связать такие изменения с изменениями других переменных состояния и с сопровождающими их энергетическими эффектами. В особенности в течение последних 10—20 лет давление приобретает все большее значение в качестве переменной, с помощью которой воздействуют на течение химической реакции или физического изменения. Каждый год мы являемся свидетелями развития одного или нескольких новых процессов, в которых влияние давления представляется наиболее важ-
газов. Целью этой главы является вывод и иллюстрация, посредством применения к частным задачам, количественных соотношений, управляющих различными типами сжатия и расширения, которые могут претерпевать газы. Это сделано для того, чтобы инженер мог ответить на следующие типичные вопросы:
Каким образом влияют иа величину работы такие факторы, как тип цикла сжатия, исходная температура, вид газа?
Какое количество тепла следует отнять при сжатии или какая будет
1. Какое количество работы требуется для сжатия газа от одного давления до другого?
2. ”
а)
б)
в)
3.
достигнута температура, если теплота при сжатии не отводится?
4. В каких случаях предпочтительнее многоступенчатое сжатие газа?
5. Каким образом меняется состояние газа или пара при его дроссели-
ровании? * 1
СНИМАЮЩИЕ УСТРОЙСТВА.
ОБЩЕЕ ОПИСАНИЕ И КЛАССИФИКАЦИЯ
Во всей этой книге мы старались по возможности избегнуть описания оборудования. Однако представляется целесообразным сделать несколько общих замечаний по поводу оборудования, применяемого . для сжатия газов.
Всякая машина, увеличивающая давление газа, называется компрессором; однако применение этого термина обычно ограничивается определенными классами машин, а именно, машинами, работающими
318
VII. Сжатие и расширение газов
при давлениях выше нескольких атмосфер. Давления, которые обычно применяются в промышленности, могут изменяться от высокого вакуума до 1000 ат и более. Машина, в которой давление всасывания много ниже атмосферного и которая выталкивает газ примерно при атмосферном давлении, обычно называется вакуум-насосом или эксгаустером. Такие машины по принципу не отличаются от машин, всасывающих газ при атмосферном давлении и выталкивающих его при повышенном давлении, и при рассмотрении общих принципов мы не будем делать между ними различий.
Отчасти на основании интервалов давления, отчасти по принципу действия можно дать следующую грубую классификацию сжимающих устройств: 1) центробежные вентиляторы, 2) ротационные воздуходувки, 3) центробежные компрессоры, 4) поршневые (возвратно-поступательные) компрессоры и 5) пароструйные компрессоры. Центробежные вентиляторы действуют на основании принципа придания газу высокой скорости с последующим совершением работы сжатия газа кинетической энергией, обусловленной этой скоростью. Они обычно работают на перепаде давлений между всасыванием и выхлопом около 0,04 атм.
Пределы давления от 0,3 атм примерно до 4 атм обслуживаются ротационными машинами, которые создают давление с помощью вытесняемого объема, а не за счет придания высокой скорости.
Центробежные компрессоры по принципу действия тождественны центробежным вентиляторам, но более мощны и работают на более высоких скоростях, благодаря чему развивают более высокие давления— до 1 атм на одну ступень. При соединении на одном валу двух или нескольких отдельных ступеней с выходными лопатками между ними, для превращения кинетической энергии можно получить еще более высокие давления; многоступенчатые машины этого типа изготовляются для давлений до 10 ат. Такой многоступенчатый центробежный компрессор подобен турбине водяного пара как по принципу действия, так и по общей конструкции. Давление, приходящееся на ступень, зависит от размеров и скорости вращения; обычно максимальное отношение давлений на ступень составляет около 1,2. Известны машины даже с 30 ступенями. Преимуществами этого типа компрессора по сравнению с поршневыми компрессорами являются: 1) компактность, 2) отсутствие клапанов, 3) отсутствие больших изнашивающихся частей, 4) отсутствие пульсации у выпускаемого газа, 5) более простое регулирование объема, 6) небольшие эксплоатационные расходы, 7) возможность непосредственного соединения с турбиной.
Пароструйный компрессор отличается от остальных отсутствием движущихся частей. Для совершения работы используется кинетическая энергия струи высокой скорости одного вещества, причем эта работа затем используется для сжатия другого вещества. В качестве первого чаще всего применяется водяной пар.
Наиболее важной машиной для давлений выше 3,5 ат, с которой мы будем главным образом иметь дело в этой главе, является
Сжимающие устройства
319
поршневой компрессор, схематическое изображение которого дается на рис. 41. Главными его частями являются: цилиндр А, поршень и поршневой шток В, всасывающий клапан С, нагнетательный клапан D и водяная рубашка Е. (Отметим, однако, что некоторые компрессоры охлаждаются воздухом.) Если сжатие происходит только с одной стороны поршня, то компрессор называется компрессором простого (ординарного) действия; если с обеих сторон, то — компрессором двойного действия. Ниже приводится следующая, более подробная классификация • поршневых компрессоров:
1. Одноступенчатые, двухступенчатые и многоступенчатые. Одноступенчатым компрессором является такой, в котором все сжатие, начиная от забора газа и кончая достижением конечного давления, совершается в одном цилиндре, т. е. за одну ступень. В двух- Рис, 41. Схема простого поршневого ком-или многоступенчатых ма- прессора,
шинах имеется два илн несколько цилиндров, в каждом из которых газ сжимается только на часть общего давления. Между цилиндрами (ступенями) газ охлаждается в промежуточных холодильниках с помощью воды или воздуха.
2. Вертикальные, горизонтальные и угловые. Они различаются по тому, является ли ось цилиндра вертикальной, горизонтальной или же используется комбинация того и другого расположения.
3. С механическим приводом, приводимые в движение водяным паром и приводимые в движение газом. Компрессор с механическим приводом работает от электромотора и присоединяется к нему с помощью ременной или цепной передачи или же соединяется непосредственно с валом мотора. Компрессор, приводимый в движение паром, действует от паровой машины поршневого типа с непосредственной связью между поршнем компрессора и поршнем паровой машины. Паровая машина может быть или простой (пар расширяется на одной ступени) или компаунд-машиной (пар расширяется на нескольких ступенях). Компрессор, приводимый в движение газом, является компрессором, действующая сила для которого доставляется двигателем внутреннего сгорания, использующим в качестве горючего газ.
4. Прямолинейный компрессор и сдвоенный компрессор. В прямолинейном компрессоре все цилиндры, в которых сжимается газ или получается движущая сила, расположены горизонтально и на одной оси. Сдвоенным компрессором является машина, имеющая два ряда
320 VII. Сжатие и расширение газов
сжимающих ступеней, приводимых в движение общим коленчатым валом.
Детандер представляет собой машину, по действию обратную компрессору; в нем газ забирается при высоком давлении и расширяется до более низкого давления, давая определенное количество работы. Паровая машина и турбинного и поршневого типа представляет собой детандер, используемый преимущественно для получения работы. Детандер в том значении, в котором это слово будет применяться в данной книге, означает машину турбинного или поршневого типа, используемую главным образом для целей охлаждения.
Бдльшая часть материала этой главы будет относиться к поршневому компрессору. Теория центробежного компрессора, подобная во многих отношениях теории поршневого компрессора, отличается во многих важных деталях и выпадает из рамок этой книги. Для дальнейших сведений по этому типу компрессоров см. специальную литературу [126].
ИЗМЕНЕНИЕ СОСТОЯНИЯ ГАЗОВ
Основные уравнения. Диференциальные уравнения, характеризующие изменение состояния любого вещества, выраженные с помощью обычно применяемых независимых переменных, выведены в гл. III из первого и второго законов термодинамики; три из иих вновь привадятся здесь для удобства ссылок.
dQ = TdS = C„dT + Т dv, (88, гл. Ill)
\u* J V
dQ = TdS=CpdT— тШ dp, (95, гл. Ill) \ / p
dQ = TdS=Cp^dv-\-Ca^dp. (ЮЗ.гл.Ш)
Эти уравнения являются совершенно общими; они применимы к любому веществу и ко всем видам изменения состояния в гомогенной системе, на которую действуют только механические силы. Если считать, что эти уравнения относятся только к свойствам, присущим самой системе, то вопрос об обратимости отпадает. Однако он возникает , при рассмотрении взаимодействия системы с окружающей средой. Если мы условимся, что изменение является обратимым по отношению к окружающей среде, то можем написать
TdS=dQ.
По первому закону,
dQ — dU-\~dA,
и если процесс обратим, то
dA = pdv.
Изменение состояния газов
321
Снова подчеркиваем, что теплота и работа не являются свойствами, а являются внешними эффектами взаимодействия системы и окружающей среды. Если силы (употребляя этот термин в общем смысле для любой движущей силы) уравновешены, то мы имеем обратимый процесс, и только для этого случая можно вычислить теплоту и работу, располагая лишь свойствами системы. Это, конечно, представляет идеальное положение, которое на практике не достигается, но к которому можно приблизиться как к пределу. При всех действительных изменениях имеются необратимые эффекты, обусловленные механическим трением газов или жидкостей, разностью температур и другими подобными причинами; следовательно, фактические теплота и работа будут отличаться от идеальной теплоты и работы на некоторую неопределенную величину. Идеальные величины, которые мы вычисляем, полезны в качестве стандарта для сравнения, а также в качестве приближений к действительным величинам, и их можно превратить в последние посредством некоторых коэфициентов, которые обычно называют к. п. д. (эффективности) и которые должны определяться путем измерений. Во избежание недоразумений, мы в некоторых случаях будем пользоваться термином .теоретическая работа* для обозначения идеальной обратимой работы, вычисленной по свойствам системы, -и~ тер-мином „действительная работа" для обозначения работы, определенной в действительной машине с учетом необратимых эффектов.
Для частного случая, когда рассматриваемое вещество можно считать идеальны:.! газом, диференциальные коэфициенты общих уравнений легко определяются из уравнения состояния
pv = RT (на моль).
Тогда вышеприведенные уравнения примут следующий вид (см. гл. III, раздел „Применение к идеальным газам"):
TdS = dQ = C ,dT-\-pdv, (1)
TdS = dQ = CpdT—RT^, (2)
TdS=dQ = CpT^-[-CvT^. (3)
Изобарный процесс. Для этого частного случая
т,
Q=\CpdT
г(
и
А = \ pdv — p(v2— ©i), (4)
21 Химическая термодинамика
322
VII. Сжатие и расширение газов
где V] и v2 представляют объемы (удельные или мольные) в начале и в конце процесса. Уравнением, совпадающим с уравнением (4), именно уравнением
А=Р (К2 — VJ, (5)
можно пользоваться для другого изобарного процесса, при котором не происходит никакого изменения состояния и для которого работа является просто механической работой инжекции (или эжекции) газа или жидкости в пространство, в котором поддерживается постоянное давление; с этим приходится сталкиваться при рассмотрении многих круговых или непрерывно протекающих процессов. Объемы V1 и V2 не представляют характеристики различных состояний, а представляют просто два предельных объема, отвечающих двум емкостям. Отметим, что давление в обоих уравнениях является абсолютным.
Изотермический процесс. Для идеального газа о, р,
А= ^pdv==RT^ ^==—RT^ Vi pi
или
A = — RTln = —2,303/?Лр^ (на моль). (6)
Pi Pi
Это выражение справедливо как для сжатия, так и для расширения. Для сжатия р2>Р1, и работа отрицательна (работа производится над системой). Для расширения рг р2, и работа положительна. По уравнению (1) или (2)
dQ = dA.
(Это равенство следует также из определения идеального газа, согласно которому dU=Q при постояннойТ.) Таким образом, мы видим, что при изотермическом сжатии идеального газа от него следует отнять количество тепла, эквивалентное затраченной работе. Другими словами, вся произведенная работа рассеивается в виде теплоты в окружающую среду. С первого взгляда может показаться, что это очень неэффективный процесс. В действительности это не так, потому что всю затраченную работу (теоретически) можно вернуть, дав газу расшириться и поддерживая его при постоянной температуре с помощью подвода того же самого количества тепла, которое перед этим было отдано в окружающую среду.
Для неидеального газа изотермическую работу можно найти различными способами. Если располагают данными по сжимаемости газа, можно построить кривую v—р\ тогда работа определится графически, как площадь под этой кривой.
Если располагают уравнением состояния, то теплоту сжатия можно найти посредством интегрирования уравнения (88, гл. III) или (95, гл. Ill),
Изменение состояния газов
323
а работу — посредством интегрирования величины pdv. Например, если газ подчиняется уравнению Ван-дер-Ваальса, то
771
= RT]nv-^-, (7)
щ — ъ
А = pdv =
о, V*
^RT\^-a\d4 =
J v — b J v2
vt Vi
= -------------(8)
--------------------Vj^b \Vj V2) ' ’
Если для рассматриваемого вещества располагают диаграммой энтропия— температура, то теплота и работа вычисляются из определения энтропии для обратимого процесса
Q = 7AS,
где AS—разность энтропий между двумя точками пересечения данной изотермы с двумя изобарами, представляющими пределы сжатия или расширения. По первому закону,
A = Q — bU=TkS — №= — ДЛ (9)
Значения функции F обычно не приводятся в таблицах и не наносятся на термодинамические диаграммы, но работу можно получить вычитанием Д£7 из TAS, причем At7, в свою очередь, определяется из Д/Т и Др».
В случае газа, значительно отличающегося от идеального, разность между теплотой и работой может быть очень большой, хотя для многих практических целей эти величины обычно можно считать равными.
Пример 1. Вычислить: а) теоретическую работу, необходимую для изотермического сжатия 1 кг1моль СО2, находящегося при 20° С, и 1 кг1см2 до давления 35 KzjcM.2, и б) теплоту, подлежащую отнятию для трех случаев: 1) если СО, считать идеальным газом, 2) если СО» подчиняется уравнению Ван-дер-Ваальса и 3) если пользоваться свойствами, приведенными на диаграмме^— Т:
•4 = 2,303/?Т 1g — = — 4,575 • 1000-293,2 lg^ = —2071 ккал = ^^ =
Pl 1 641,7
= — 3,23 л. с.-ч., где 641,7 — коэфициент для перевода ккал в л. с.-ч.
Q = A — — 2071 кал.
324
VII. Сжатие и расширение газов
2. По табл. III «Приложения*,
а = 3,604, 6 = 0,0428,
(из уравнения идеального газа) = 24860 л,
v2 (путем подбора из уравнения Ван-дер-Ваальса) = 590 л.
Подставляя в уравнение (8), получаем
4 = 4,575-1000.293,2 lg2g^g_3,604 (-[—) X 0,0242 *).
Из уравнения (7) находим:
Q = —2220.
3. Из диаграммы S — Т [190] (воспроизведена в .Приложении”)
Sj= 1,418, S. = 1,231 ккал/кгХград,
Нх = 173, Н2 = 163,3 ккал/кг,
v1==560 л/кг = 24640 л/кг-моль, v2= 13 л/кг = 572 л/кг-моль,
... (35-13 —1-560) ПОООТ „,о .
= 44 (163,3— 173)—'------- ? '------ =—319 ккал/моль.
По уравнению (9),
4 = 293,2 (1,231 — 1,418) 44 + 319= —2093 ккал/моль и
Q=T^S = —2412 ккал/моль.
Разность между теплотой и работой составляет 15% величины самой работы; интересно отметить, что при повышенном давлении объем СО2 примерно иа 2О°/о отличается от объема идеального газа.
Изотермическая работа янляется абсолютным минимальным количеством работы, которую в данном интервале давлений следует затратить для сжатия газа (нлн, наоборот, максимальной работой, которую можно получить при расширении газа в том же интервале давлений). На практике изотермическое сжатие не реализуется, потому что практически невозможно достаточно быстро удалять теплоту сжатия. В действительности, в компрессорах удаляется только относительно небольшой процент тепла, и сжатие скорее приближается к другому идеальному случаю, именно — к адиабатному сжатию.
Адиабатный процесс. Для обратимого адиабатного изменения состояния
TdS = dQ = Q. (10)
Для идеального газа, по уравнениям (3) и (10),
Са— =0, (11)
р v 1 ° р ' ’
или
£r) 1 л~Хатм = 0,0242 ккал.
Изменение состояния газов
325
Интегрирование при допущении, что Ср]С„ является константой, дает уравнение Пуассона
А1п®Ц-1пр = Л' (константа интегрирования) (13)
или
Р^ = Кг, (14)
И
..., (15)
где
k=cpicv.
Уравнения, связывающие величины р, Т и v, Т, можно получить интегрированием соответственно уравнений (2) и (1) или, что проще, сочетанием уравнения (14) с уравнением состояния идеального газа. Эти уравнения имеют следующий вид:
Tvk~^ = K2, (16)
i — k
Тр * = КВ. (17)
Уравнения (14), (16) и (17) связывают изменения—соответствующих переменных состояний, которые сопровождают обратимое адиабатное расширение или сжатие идеального газа. Они не являются точными даже для идеального газа, так как отношение теплоемкостей k не является постоянным, хотя изменения k в обычно встречающемся сравнительно узком интервале температур невелики.
Подстановка в общее выражение для работы значения р, данного уравнением (15), приводит к уравнению
•р,
(18)
V1 отсюда шт
. (19)
Поскольку неизвестно, оно может быть исключено с помощью уравнения
£
(20) \>*2/
тогда
‘<=й?г[1-(Й)—] <2’>
326
V//. Сжатие и расширение газов
или
А—
А— Л-1
к — 1
(22)
Уравнения (21) и (22) дают теоретическую адиабатную работу на моль при сжатии или расширении при условии, что для любого процесса pi всегда относится к исходному давлению; k принимается постоянным, не зависящим от конкретных условий, но изменяющимся в зависимости от сложности молекул газов. Ниже дается несколько ouauauutt Ь гтпы nfSuruuuTv трмпрпятУПР и ттяпгтрниы*
L?11U 1V**AA*A *«г ллрглл л — Г'*4 “ J Г*" *^*»**<* •
Одноатомные газы (Не, Аг и т. д.)................1,67;
Двухатомные газы (Н2, СО, N2 и т. д.)............1,40 (приблизи-
тельно);
Трех-, четырех- и пятиатомные газы (СО2, СН4 и т. д.) 1,30 (при-
близительно),
а для газов с ббльшим числом атомов в молекуле значения k еще меньше. В общем k уменьшается с температурой и увеличивается с давлением; поэтому можно принять, что при адиабатном процессе эта величина остается постоянной. Нижеприводимая таблица вычисленных значений k для воздуха дает некоторое представление о той степени изменения k, которую можно ожидать45.
t, °C Давление, ат
0 100 200 300
0 1,40
100 1,39 1,49 1,51 1,63
200 1,38 1,44 1,48 1,52
300 1,37 1,44 1,44 1,46
Эдмистер [75] вычислил значения k для 17 углеводородов из данных по Ср при 1 атм и из уравнения состояния и представил результаты на графике как функцию приведенных давления и температуры. В случае чистых газов, для которых отсутствуют значения А, или для газовых смесей можно вычислять k из соотношения
Для газовой смеси Ср можно вычислить с достаточной точностью ио правилу аддитивности.
Изменение состояния газов
327
Отметим, что для данного весового количества газа, подлежа-
щего сжатию, работа зависит только от отношения давлений, а не от
абсолютной их величины. Другими словами, работа на моль одинакова
для сжатия от 100 до 1000 атм и для
(если считать, что газ идеален).
При адиабатном сжатии вся работа превращается в энергию сжатого газа; это означает, что его температура должна повышаться (поскольку U не зависит от р). Возрастание температуры (или понижение ее при расширении) можно вычислить по уравнению
к — 1
которое получается из уравнения (17). Графическое решение уравнения (23) для данной Т пред-ставлено на рис. 42.
Выражения для адиабатной работы можно получить и при использовании других уравнений состояния, но онн сложны и не представляют практического значения. Поскольку действительная работа сжатия во всех практических случаях всегда больше идеальной из-за необратимых эффек-
сжатия от 0,1 до 1,0 атм
Рг
Р,
Рис. 42. Температуры адиабатного сжатия от начальной температуры 20°С. PalPi — степень сжатия, k — отношение теплоемкостей.
тов, которые не поддаются точному учету, то в подавляющем большинстве случаев пользоваться более точной зависимостью р—v — Т нецелесообразно 46.
Было сделано несколько попы-
ток учесть некоторые необратимые эффекты или отклонения процесса от строгой адиабатности, допуская, что фактическое изменение состояния газа можно представить уравнением й)
/w" = const,
(24)
где п — константа, которая определяется по действительному поведению газа в компрессоре (например по индикаторной диаграмме). При использовании этой зависимости уравнения (21) — (23) не изме-
й) Такое изменение состояния яногда называется политропным.
328
VII. Сжатие и расширение газов
няются, за исключением замены k на п. Если сжатие адиабатное, но сопровождается внутренними необратимыми эффектами, то п будет больше k. Если же сжатие обратимо, но не адиабатное, то п будет меньше k и будет лежать в пределах между k и 1 (/z = 1 для изотермического, т. е. полностью неадиабатного сжатия).
Так как разность между работой изотермического сжатия и работой адиабатного сжатия не очень велика и реальное сжатие приближается к адиабатному (несмотря на охлаждающую водяную рубашку вокруг цилиндра) и так как вычисленная работа всегда является только приближением к фактической, то употребление показателя степени п для подавляющего большинства случаев едва ли окажется пппавланным47.
Для случая неидеального газа работа (или конечная температура) очень просто определяется с помощью термодинамической диаграммы. Поскольку обратимое адиабатное сжатие протекает при постоянной энтропии, нужно иа диаграмме 5 — Т только провести вертикаль от начального до конечного давления. Так как для адиабатного процесса
— Д/74-Дрт,
то работа вычисляется из величин, взятых непосредственно с диаграммы, если на нее нанесены изохоры. Если же этих линий нет, то объем следует определить каким-либо другим методом, например по таблицам термодинамических свойств.
РАБОТА ОДНОСТУПЕНЧАТЫХ КОМПРЕССОРОВ
Идеальный цикл. До сих тор мы рассматривали рабочие эффекты, сопровождающие определенные изменения состояния. Фактически эти изменения состояния образуют
Рис. 43. Изменения давления и объема в идеальном одноступенчатом компрессоре.
только одну ступень цикла изменений, который претерпевает газ в условиях действительного процесса непрерывного сжатия (или расширения). На диаграмме V—р (рис. 43) представлены изменения, происходящие в цилиндре идеального поршневого компрессора. Изменения в реальных компрессорах- несколько отличаются от этих, как будет показано ниже (стр. 345), но использование для анализа идеализированного цикла оправдывается его простотой, а
также тем, что эти результаты для большинства целей можно считать достаточно близкими к резуль
татам, получающимся в реальных случаях.
Работа одноступенчатых компрессоров
329
В соответствии с этой диаграммой предполагается, что когда поршень идет к концу своего хода после сжатия газа, то в цилиндре между поршнем и выпускным клапаном газа не остается. Это отвечает допущению об отсутствии вредного (мертвого) пространства. Допустим, что в рассматриваемый момент поршень готов двинуться обратно и всосать новую порцию газа. Другими словами, начнем с точки А на диаграмме (рис. 43); рх является давлением всасывания. Будем считать, что когда поршень движется вправо, всасывающий клапан сейчас же открывается, и газ всасывается при постоянном давлении рг (линия АВ] до тех пор, пока в цилиндр не будет забран общий объем V, (что происходит в конце хода поршня). Когда поршень начинает двигаться влево, всасывающий клапан закрывается, и газ сжимается, причем изменение его состояния изображается линией ВС или BE в зависимости от рассматриваемого случая. Когда давление достигает величины р2 (которая представляет давление на линии выпуска из компрессора), то нагнетательный клапан автоматически открывается, и газ выталкивается из цилиндра при постоянном давлении р2 до тех пор, пока не будет достигнута точка нулевого объема D. Поскольку и А и О являются точками нулевого объема, то различие в давлениях не имеет значения, и мы можем представить изменение от D до А происходящим мгновенно, после чего система готова к повторению рассмотренного цикла.
Работа цикла. Общая работа цикла компрессора представляет сумму работы для четырех ступеней, т. е.:
Ашкла = А АВ + Л ВС ^~АС£>~ГАПА’ (25)
ААВ = р1У1 (работа всасывания),
V.
Авс = { pdV (работа сжатия), V,
Acd = —РтУг (работа выталкивания), АдЛ = 0 (изохорный процесс).
Следовательно,
V ,
Аикла =Р1 — рг V2 + \ pdV. (26)
Vi
Так как
Г’ рс
\Pav'\~PiV1—p2V2 =— } Vdp, (27)
% р,
ж»
Лцикла = — \VdP- (28)
330
VII. Сжатие и расширение газов
Это уравнение можно решить алгебраически или графически. В гл. I показано, что замкнутая площадь на диаграмме V—р (площадь ABCD или ABED) непосредственно дает работу цикла. Этим путем, по диаграмме (индикаторной диаграмме), автоматически вычерчиваемой механизмом, прикрепленным к компрессору или детандеру, определяют действительную работу, получающуюся в цилиндре.
Изотермическое сжатие. Третий член правой части уравнения (26) является работой сжатия (или расширения); он уже был определен для большого числа частных случаев. Так, если сжатие изотермическое, а газ идеальный, то работа сжатия дается уравнением (6); так как для идеального газа при постоянных температуре и массе р1И1=ргИг, то уравнение (6) вместе с тем дает и работу цикла на моль газа.
Для неидеального газа можно поступить следующим образом. По уравнению (140, гл. III),
= f vdp = ^ f Vdp, р, р,
где N—число молей.
Следовательно,
* ' -
Таким образом, для неидеального газа изотермическая работа цикла легко вычисляется в том случае, если известны летучести. При постоянной температуре
dZ = vdp,
и работа цикла на моль будет
A = ^Z = ZJ — Zl,
причем &Z берется по изотерме. Значения Z обычно не наносятся на термодинамические диаграммы; но так как Z = H—TS, то можно написать
А = Н2 — Нг — T(S2 — SJ, (30)
т. е. работа на моль определяется непосредственно из значений, взятых с диаграммы S—Т или подобных ей диаграмм.
Адиабатное сжатие. Поскольку при адиабатном сжатии температура повышается, то объем при заданном давлении будет больше, чем при изотермическом сжатии, и ход адиабатного сжатия будет изображаться кривой, подобной BE на рис. 43. Легко заметить, что адиабатная работа для данного количества газа и данных пределов давления несколько больше, чем изотермическая работа, причем графически эта разность выражается площадью ВЕС.
Работа одноступенчатых компрессоров
331
Для идеального газа при добавочном допущении постоянства k работа адиабатного цикла дается подстановкой уравнения (21) в уравнение (26) и использованием соотношения
А —1
Pi®!— P2V2=PiVi [1 — к ].
L \Р1/ J
которое следует нз уравнений (19) и (21) ☆).
Тогда получаем
А —1 [>-(£) *] on
А? —1
Л_=^[1-(Й) * ]. (32)
Из уравнений (19) и (26) можно также получить уравнения вникла ~ £_________________________1 (Pi Рг^г)» (33)
(34)
являющиеся полезными в специальных случаях видоизменениями уравнения (31). В уравнении (31) V\ представляет емкость цилиндра или объем, забираемый за цикл в цилиндре ординарного действия. Если пй обозначает число циклов в минуту, то общий объем, засасываемый в минуту, равен «o^i- Используя это для замены V1 в уравнении (31), получаем работу цикла в единицу времени. В случае цилиндра двойного действия работа за цикл будет в два раза больше той, которую дает уравнение (31), если является всасываемым объемом, так как за цикл происходят два всасывания.
Работа адиабатного цикла очень легко определяется по термодинамической диаграмме следующим образом.
На основании уравнения (26) и первого закона,
^цикла (на кг) Plvl P2V2 (^2 ^1)»
24цикла (на кг)= (^4 4“Pl^l) (^4 “Ь Рг^гК
^цнкла (на кг) —
(35)
(36)
(37)
Для расчета нужно только прочесть с диаграммы значения Н на концах изоэнтропы (т. е. линии постоянной энтропии), которая связывает две точки, соответствующие давлениям рг и р2. Применение диаграммы S—Т или подобных ей особенно желательно в тех случаях, когда сжатие или расширение связано с переходом через
☆) Уравнения (19) и (21) применяются для мольного объема, но уравнения в той же самой форме приложимы и для общего объема.
332
VII. Сжатие и расширение газов
область двух фаз, потому что в этом случае (например, сжатие влажного пара) применение закона идеального газа и любых других уравнений состояния может привести к большим ошибкам.
Пример 2. Вычислить теоретическую мощность в лошадиных силах, требуемую для сжатия до 10 ат 3 м3 СО2 в минуту при — 50° С и 1 ат. Сжатие осуществляется адиабатно в идеальном (т. е. работающем обратимо и без вредного пространства) компрессоре ординарного действия.
Если допустить, что газ идеален и что отношение теплоемкостей равно постоянной величине (1,3), то мощность вычисляется с помощью уравнения (31):
. 1,30-1-3-10000 Г. /1О\о.24'1 019fin
оабота пикла = —1т----------- 1 — — I — — 91260 кгм
0,60 L \ * / J
91260
и мощность — ——20,28 л. с.
60-75
Интересно отметить, что в этом случае изотермическое сжатие не имело бы практического значения, так как давление нагнетания выше давления пара при данной температуре, и поэтому СО2 в цилиндре полностью конденсировалась бы в жидкость и затем сжималась бы как жидкость до конечного давления.
Вычислим теперь требуемую мощность с помощью диаграммы S — Т для COS (см. .Приложение') и сравним ее со значением, найденным при допущении идеальности газа.
По диаграмме S—Т, для СО2 #1=159,2 ккал/кг, Н2 = 189,3 ккал/кг, и
к1 = 0,43 ж3/кг.
Поэтому мощность, потребная для адиабатного сжатия, равна
3 О 427
- (189,3 - 159,2) X X 60/75= ~ 19’93 с-
Поскольку при одноступенчатом сжатии отношение давлений (степень сжатия) обычно меньше 10, из этого расчета можно заключить, что допущение идеальности газов дает достаточно точные результаты для всех практи-ческгах целей в случае одноступенчатого компрессора с давлением всасцрания ниже атмосферного.
Пример 3. Чему будет равна температура газа в цилиндре в конце адиабатного сжатия для случая, рассмотренного в примере 2?
По уравнению (23), Г2 = 223,2 • 10-°>231 = 379,7° К — 106,5° С. По диаграмме S — T,t2= 102° С.
Пример 4. Чему будет равна минимальная работа (выраженная в киловатт-часах на 1 кг водяного пара), необходимая для адиабатного сжатия насыщенного водяного пара при 20 кг/см2 до давления 80 кг/см2 посредством непрерывного процесса?
В настоящем примере практически существенно использование термодинамической диаграммы. В данном случае применение закона идеального газа ие только приводит к серьезной ошибке, но и неизвестно, какое значение следует принять для А48.
Работа одноступенчатых компрессоров
333
По таблицам Вукаловича, =211,4° С,' Sj= 1,5160 ккал/кг-град.
Для того же самого значения S при р = 80 кг/см1:
Нг = 668,5
ккал [кг.
Я2 = 745,8, Т2 —395° С,
А = —&Н — —77,3 ккал/кг =
77,3
860
•0,09 квт-ч.
Для газов, термодинамические свойства которых неизвестны,
последние можно вычислить по уравнению состояния или же, если известны критические константы, — через обобщенные свойства по методам, кратко описанным в предыдущей главе. Дли газовой смеси, не являющейся идеальной, наиболее простым путем будет вычисление псевдокритических констант по методу Кея с последующим использованием обобщенных свойств, считая смесь за чистое вещество. Для
ознакомления с подробностями такого расчета можно обратиться к статье Иорка [263].
Влияние вредного пространства. Очевидно, что в любом прак-
тическом случае в цилиндре в момент окончания хода нагнетания
должен оставаться некоторый объ-ем газа. При обратном движении поршня этот газ высокого давления вновь расширяется позади поршня, и всасывающий клапан не открывается до тех пор, пока давление этого газа не упадет до давления во всасывающем трубопроводе. Пространство между поршнем и крышкой цилиндра, включая объем, в который попадает газ при закрытых клапанах, называется объемом вредного пространства. Обозначим его через Vc. Отношение этого объема к объему, описываемому поршнем VD, т. е. относительный объем
Рис. 44. Изменения давления и объема в компрессоре с вредным пространством: Vo — объем, описываемый поршнем; Vc — объем вредного пространства; V/— всасываемый объем.
вредного пространства, в зависимости от размера и конструкции компрессора, может изменяться от 2 до 1О°/о. Условимся в дальнейшем
эту величину для краткости называть вредным пространством.
Изменения, происходящие в компрессоре с вредным пространством, можно представить на идеализированной индикаторной диаграмме, показанной на рис. 44. Кривая 3—4 представляет ход сжатия, 4—5 — ход выхлопа, 5—2 — ход повторного расширения и 2—3 — ход впуска (всасывания). Объем в точке 5 — объем Vc вредного пространства, Vs — — объем, отвечающий перемещению поршня, т. е. Гр,
a Va—V2 — объем газа, всасываемого в цилиндр Vr Считая газ идеальным, k постоянным, а сжатие и расширение адиабатными и
334
VII. Сжатие и расширение газов
обратимыми, можно легко притти к выражению для работы цикла посредством суммирования работ отдельных его ходов. Однако наиболее просто это выражение получается на основании того, что площадь 2—3—4—5 представляет разность площадей 1—3—4—6 и 1—2—5—6, каждая из которых представляет цикл без вредного пространства; работа для каждого цикла дается уравнением (31).
Это приводит к уравнению
1
А для цикла 2—3—4—* j . (381
Другими словами, вредное пространство не влияет на работу при условии, что в уравнении (31) вместо объема всасывания пользуются' действительным объемом забираемого газа.
£
Так как, по уравнению (20), V2=V5 (ft)*’ Т°
2 у __у — V ____КI р-г V
кз 2 2 к5\рх) ’
и так как
Уз-Уз У/
Уз- Уз Уд ’ то
1
У/_ Уз Уз /м*
Уо Уз-Уз Уз-Узкл/ ’
Обозначая относительное вредное пространство через с, получаем
Уз—Уз’
и имеем
£
с (?>)*]’ 1391
соотношение, с помощью которого можно вычислить теоретически засасываемый объем Vt, зная степень сжатия, размеры цилиндра и величину вредного пространства. Выражение в скобках, которое представляет отношение фактически всасываемого объема V, к объему всасывания, называется объемным коэфициентом полезного действия компрессора. Чтобы отличать его от других коэфициентов, которые мы будем рассматривать ниже, будем называть его теоретическим объемным к. п. д.
Многоступенчатое сжатие
335
Влияние вредного пространства на объемный к. п. д. при изотермическом сжатии больше, чем при адиабатном; но поскольку цикл с изотермическим сжатием не представляет большого промышленного значения, его количественная обработка рассматриваться не будет.
Пример 5. Чему должна быть равна степень сжатия при адиабатном сжатии воздуха для того, чтобы теоретический объемный к. п. д. был равен 5ОО/о, если вредное пространство равно 5°/0?
По уравнению (39),
1
Vr МО
ev = = 0,50 = 1 4- 0,05—0,05г •
VD
Решая это уравнение, находим степень сжатия
г = 29.
Если бы г было равно 71, то е„ = 0, и в результате не происходило бы никакого всасывания свежего газа компрессором. Другими словами, было бы невозможно непрерывно сжимать любой двухатомный газ от 1 кг/см2 до давления, несколько превышающего 70 кг1см2 в одном цилиндре при вредном пространстве, большем 5°/о-
Пример 6. Одноступенчатый компрессор двойного -действия с цилинд-______ром в 250—300 мм А) и вредным пространством в бА^работаюший при 175 обо-ротах в минуту, следует использовать для сжатия метана от сверхбарометрического давления в 0,07 ат до сверхбарометрического давления в 7, 5 ат, если Т=15°С. Вычислить максимальную производительность компрессора, выраженную в кубометрах в минуту при начальных условиях, и теоретическую адиабатную мощность.
Если пренебречь объемом, занимаемым штоком поршня, то объем всасы-,, 3,14X0,252X0,30X2X 175 ...
вания VD—-— ----------------———------= 5,15 м^минс,
1
Г / 8 5 \ Wi 1
Vz= 5,15 1 4-0,06—0,06 ( ) =3,97 м21мин
и, по уравнению (31),
„ _1,31Х 1,07X 10000 X3,97 Г. /8,5\о,2371 _
аЭ 0,31 X60X75 [ М-07/ J -».» л. с.
МНОГОСТУПЕНЧАТОЕ СЖАТИЕ
Обшие принципы. Пример 5 показывает, что вредное, пространство ставит предел степени сжатия, которой можно достигнуть в одном цилиндре. В других случаях были отмечены гораздо более низкие пределы. Из уравнения (23) ясно, что адиабатное сжатие сопровождается значительным подъемом температуры. Для двухатомного газа, забираемого в цилиндр при 20° С, температура в конце хода адиабатного сжатия при степени сжатия, равной 10, была бы более 290° С. Трудно найти смазку, которая не
й-) При указании размеров цилиндра первая цифра всегда означает внутренний диаметр, а вторая — длину поршня.
336
VII. Сжатие и расширение газов
утрачивала бы своих качеств при такой высокой температуре, не говоря уже об опасности взрыва в том случае, если газ содержит кислород. Механические трудности создания высокого отношения
давлений в одном цилиндре возникают вследствие того, что цилиндр должен быть достаточно велик, чтобы вмещать большой объем газа
низкого давления, и в то же время достаточно прочен, чтобы выдерживать высокое давление. Если бы пытались достигнуть высокой
степени сжатия в одном цилиндре, то
Рис. 45. Идеальный двухступенчатый цикл сжатии.
это привело бы к слишком большим размерам цилиндра.
В силу этих причин и вследствие еще одной очень важной причины, которая будет изложена ниже, отношение давлений, достижимое в одном цилиндре, ограничено 10 при очень больших цилиндрах, а обычно значительно ниже этой величины. Следовательно, для давлений, ббльших примерно 6— 8 атм, сжатие следует производить в несколько стадий,
и поскольку одна из трудностей обусловлена подъемом температуры при сжатии, важно охлаждать
газ между ступенями сжатия с помощью промежуточных холодильников.
Для вывода количественных зависимостей, приложимых к многоступенчатому сжатию, рассмотрим двухступенчатый идеализированный цикл, показанный на диаграмме V—р(рис. 45). Газ, сжатый в первом цилиндре до промежуточного давления р,., выпускается в промежуточный холодильник, где он при постоянном давлении охлаждается до температуры, приближающейся к температуре газа перед сжатием. Кривая 3—4 представляет ход нагнетания в первом цилиндре. Затем охлажденному газу все еще при р{, но с меньшим объемом (вместо Р3), дают войти во второй цилиндр (ход всасывания 8—4), сжимают его вдоль кривой 4—6 до давления р2 и выпускают в последний холодильник (ход нагнетания 6—7). Работа, производимая в первом цилиндре, изображается площадью 2—1—3—8, а во втором — площадью 4—6—7—8\ общая работа для массы газа, соответствующей первоначально засосанному объему представляет сумму двух площадей. Если бы сжатие от рх до р2 производилось в одну стадию, т. е. с ходом сжатия 1—5, то работа была бы равна площади 2—1—5—7. Следовательно, мы видим, что работа при двухступенчатом сжатии меньше, чем при одноступенчатом, на площадь 4—3—5—6. Это показывает еще одно важное преимущество многоступенчатого сжатия, а именно— уменьшение расхода работы на сжатие. При распределении общего отношения давления между несколькими стадиями с промежуточным
Многоступенчатое сжатие
337
охлаждением между ними можно достигнуть еще большей экономии в работе. Пусть на рис. 46 кривая 2—9 представляет адиабатное сжатие от рг до р2, а кривая 2—11—изотермическое сжатие. Ступенчатая линия 2—3—4—5—6—7—8—10 представляет адиабатное
сжатие с промежуточным охлаждением до начальной температуры вса-
сываемого газа вдоль изотермы 2—11. Заштрихованная площадь
представляет экономию работы, получаемую при употреблении не-
скольких ступеней. Предел экономии работы, которого можно достигнуть применением нескольких ступеней, определяется площадью 2—11—9—2, представляющей разность между адиабатным и изотермическим сжатием; этот предел, очевидно, требует бесконечно большого числа ступеней. Таким образом/ видим, что экономия работы, обусловленная при-менением нескольких ступеней, полностью зависит от
степени приближения к изо- Объём
термическому сжатию. С этой рИС- 4g ррафик, иллюстрирующий макситочки зрения желательно мальную экономию в работе при многобольшое число ступеней, но ступенчатом сжатии.
некоторые практические со-
ображения ограничивают их число в большинстве случаев шестью; этот вопрос будет кратко рассмотрен ниже (см. раздел „Число ступеней*).
Работа многоступенчатых компрессоров. Из рассмотрения рис. 45 видно, что работа двухступенчатого цикла равна сумме работ двух одноступенчатых циклов и может быть представлена уравнением:
а—1 а—1
= * ]+*b[,_(g) *]. (40)
Это уравнение включает обычные допущения, из которых наиболее важными являются: 1) идеальность газа; 2) постоянное отношение теплоемкостей; 3) адиабатность и обратимость сжатия. Если мы теперь будем считать, что промежуточное охлаждение является идеальным, т. е. что сжатый газ охлаждается до температуры забираемого газа, то можем написать
22 Химическая термодинамика
рУг—рУс
338
VII. Сжатие и расширение газов
тогда уравнение (40) превращается в следующее: А-1 Л-1
А (2 ступени) = ^^^2—* —(-) * 1. (41)
« *|_ \Р1/ \PiJ J
Обобщая уравнение (41) на случай п ступеней и полагая при этом справедливыми допущения, сделанные при его выводе, получим
~ wL—— г_^ .Г!, (42)
А—1( \ptJ \pij \pi,J LPz(n-i)J J
где
Pi,'Pi, Pip ••Pi(n-V>—язвления нагнетания co ступеней l,2,3...n—1.
Эти промежуточные давления могут иметь любые значения между 0! и р2, но, очевидно, существует единственный ряд этих величин, соответствующий минимальной работе (и следует пользоваться именно этим рядом). Для нахождения условий, отвечающих затрате минимальной работы, будем считать каждое промежуточное давление изменяющимся независимо от других и приложим обычный критерий минимума функции, именно,
дА п дА п
3—=0, ч—=0 и т. д. (43)
др^ ' ’
Диференцируя уравнение (42) и применяя уравнение (43), получаем
дА _ к 1 \ .Pl '
поэтому или S _ S — 1 £ 1 S Pi, Pi, =pi, Pi , P2i,=PiPi,- (44)
Подобным получаем образом из условий dA!dpii==O, дА]др._ = О и т. д.
^=Pi, А3, (45)
p\=Pi'Pie (46)
А?(П-1) =Л(я-2)Рг- (47)
На основании этих четырех уравнений можно заключить, что
Ph _ г
A Pit Pi, Pi(n-l)
Многоступенчатое сжатие 339 |
-----------------—-------—------------------------------------- i
т. е. отношение давлений г одинаково на всех ступенях. Следовательно, условие минимальной работы заключается в том, чтобы отношение да- J
влений, а следовательно, и работа были одинаковы на всех ступенях48.
Так как ___. ]
ПХ^Х^з X й
ТО J
РГ
и
(48)
Подстановка уравнения (48) в уравнение (42) дает
fe—1
А (п ступеней) _"* J . (49)
Компрессоры обычно конструируют таким образом, чтобы они давали приблизительно одинаковую работу на всех ступенях не только потому, что это приводит к минимальной работе, но также потому, что это желательно и с чисто механической точки зрения.
Влияние вредного пространства. Так же, как и для одноступенчатых компрессоров, можно показать, что величина вредного пространства не влияет на работу, если вместо объема всасывания используется фактически засасываемый объем.
Объемный (теоретический) к. п. д. многоступенчатой машины определяется главным образом величиной вредного пространства на первой ступени, потому что весь газ, забранный в цилиндр первой ступени, должен пройти через другие ступени. Следовательно, из уравнения (39) можно получить
1
eVi (объемный к. п. д. первой ступени) == 1 с1— ,
где р; — давление нагнетания первой ступени.
Если все емкости и вредные пространства соответствующим образом пропорциональны в согласии с принципом наименьшей работы, то общий объемный к. п. д., равный к. п. д. на первой ступени, дается уравнением
1
^=1-гс1 —ci (50)
где Cj — величина вредного пространства на первой ступени, а р2/р1— общая степень сжатия для многоступенчатого компрессора. Чтобы это
22*’
340
VII. Сжатие и расширение газов
соответствовало истине, должна существовать определенная зависимость между производительностью, величиной вредного пространства и давлениями на любых двух ступенях. Так, для первых двух ступеней нужно иметь
Yd,
Vd,~
г /„Л2*"
1+с2 —сЦ-J
(51)
rwn 1 »» О /-ХТЧТГЛлЛЛ'Т>пп'Я/<'ГПП11Г|/-> те rtnnnnH I» Т»*»»ЛГ»лй РТ1’ПО11ПЧ X ZAV Л *1 X/ Uinvtnitn tUUlDCI tiotnnv Л lltpoun Г1 DlUjjun v i j Ht.n/1 iH, a p2 — давление нагнетания второй ступени. Это уравнение непосредственно следует из того, что отношение всасываемых двумя ступенями объемов равно обратному, отношению давлений.
Пример 7. Сравнить теоретическую мощность, необходимую для адиабатного сжатия 30 м3 гелия в минуту от нормальною атмосферного давления и 25° С до 15 атм сверхбарометрического давления при 1) одноступенчатом и 2) двухступенчатом цикле.
Для одноатомного газа k = 1,67:
Р (одна ступень)
_ 1,67 X 1 X ЮЗЗОХЗО
— 0,67X60X75
Р (две ступени) =
2X 1.67X1X10 330X30
0,67 X 60 X 75
Знак минус, согласно принятому ранее условию, показывает, что работа произведена над системой.
Пример 8. Водород следует сжать в четыре стадии от 1 атм до 200 атм сверхбарометрического давления. Чему будет равно давление между ступенями?
Примем обычные упрощающие допущения, именно — идеальность газа, адиабатность и обратимость сжатия, полное промежуточное охлаждение и т. д.
4 /” 201
По уравнению (48), г на ступень — у — = 3,77.
Промежуточные давления равны:
Первый промежуточный холодильник.. 3,77 X 1 = 3,77 атм,
Второй промежуточный холодильник. 3,77 X 3,77 = 14,2 атм. Третий промежуточный холодильник 3,77X 14,2 = 53,4 атм.
Пример 9. Двухступенчатый компрессор должен забирать водород со скоростью 8,Ьм3[мин при 25°С и нормальном атмосферном давлении и выдавать его при 15,3 атм сверхбарометрического давления. Чему будет равна производительность каждой ступени цилиндра (в кубометрах в минуту), если вредное пространство в первом цилиндре равно 0,04, а во втором — 0,06?
При обычных для задач с компрессорами допущениях объемный к. ж. л. первой ступени определяется уравненяем (50)
1 /16.3\ 2X1,41 = 1 + 0,04 — 0,04 () = 0,932,
Многоступенчатое сжатие
341
тогда
vDi = — 9112 м*!ман-
По уравнению (51),
VDl ,/ТбЗ 1 4-0,06 — 0,06 (16,3)2^
vD,-V 1 -L -
.14-0,04 —0,04 (16,3)2’82.
Следовательно,
О,«7 V
Реальный газ. Если допущение об идеальности газов непри
емлемо, то работу многоступенчатых компрессоров можно вычислить
с помощью термодинамической диаграммы. Трехступенчатое сжатие, представленное на диаграмме 5— Т, показано на рис. 47. Изоэнтропа 1 — 2 пред-ставляет^обратимое адиабатное сжатие от до р^Г2’, по изобаре 2—3 осуществляется промежуточное охлаждение до 7\, изоэнтропа 3—4 изображает ход сжатия на второй Ступени, и т. д. Работа на килограмм или на моль на каждойчступени определяется разностью энтальпий на концах соответствующих изоэнтроп. Принцип равной работы на всех ступенях можно было бы применить для нахождения (графически с помощью подбора) соответствующих значений промежуточных давлений р2 и р4, удовлетворяющих условию равенства ДА/ на всех изоэнтропах. Это, повидимому, даст минимальную общую работу
Рис. 47. Трехступенчатое сжатие угле-
и определит оптимальные про- кислоты на диаграмме температура — межуточные давления, хотя энтропия.
попыток доказать целесообраз-
ность такого приема произведено не было50.
Диаграмма работы. Рис. 48 является диаграммой, позволяющей
весьма просто определить работу сжатия любого газа для любого значения k между двумя предельными величинами 1,67 и 1,00. Эта
342
VII. Сжатие и расширение газов
диаграмма построена на основании ранее выведенных уравнений для случая обратимого адиабатного расширения идеального газа. Диаграмма дает непосредственный отсчет только для одноступенчатых компрессоров и для одной температуры засасываемого газа; но для
Рис. 48. Вычисленная работа сжатия для идеального газа в идеальном одноступенчатом компрессоре.
нескольких ступеней и других температур существуют простые правила превращения величин, считанных с диаграммы, в искомые величины. Нижеследующий пример иллюстрирует применение этой диаграммы.
При мер 10. Решить часть 2 примера 7 с помощью диаграммы (рис.48).
г = 1/Гб = 4,0.
на ступень у 1
По диаграмме Л = 1,70 л. с.-ч.'кг-мом. Корректируя результат на двухступенчатую работу и 25° С, получаем
А = 1,70 X ^94 X 2 = 3,45 л. с.-ч./кг-моль;
1X10X30
848 X 298
= 1,188 кг-моль.
Общая работа = 1,188 X 3,45 = 4,1 л. с.-ч.1мин, откуда 4,1X60 = 246 л. с.
Многоступенчатое сжатие
343
Так как в рассмотренном случае следует производить различные вычисления, то от использования этой диаграммы здесь выигрыш невелик, но в некоторых случаях она экономит работу по вычислению. Можно построить диаграмму с непосредственным отсчетом, охватывающую все рассматриваемые здесь переменные, но ее сложность препятствует ее применению.
Число ступеней. Число ступеней для любого случая определяется соотношениями между такими показателями, как снижение расходуемой мощности, повышение к. п. д. и соответственным увеличением стоимости оборудования.
Нельзя дать никакого определенного правила, но обычно максимальное отношение давлений на любой ступени большого многоступенчатого компрессора лежит между 3 и 5; несколько большее отношение у одноступенчатых машин. Ниже указаны ориентировочные пределы применяемых давлений (конечные давления в кг{см2).
Одна ступень 2 ступени 3 ступени 4 ступени
1—8 5—100 20—200 60—20<Г
300-атмосферные компрессоры, используемые при некоторых современных процессах высокого давления, являются обычно шестиступенчатыми сдвоенными машинами, хотя также употребляются четырех- и пятиступенчатые машины. Наиболее высокое давление, для которого построены промышленные компрессоры, составляет 1000 кг]см2. (Это давление используется в некоторых современных процессах синтеза под высоким давлением.) Они обычно являются шести- или семиступенчатыми машинами; иногда употребляется даже девять ступеней.
Небольшие компрессоры для экспериментальных целей обычно употребляются при значительно более высоких отношениях давлений, поскольку для них не представляет значения экономия, обусловленная более низкой мощностью и увеличенным объемным к. п. д. Так, небольшие компрессоры от 70 до 85 атм давления имеют только две ступени, а трехступенчатые машины сжимают до таких высоких давлений, как 300 атм. Компрессор Гофера в 5 ступеней дает до 1000 атм. Построена четырехступенчатая машина, работающая при максимальном давлении в 4000 атм.
Из сказанного о влиянии вредного пространства очевидно, что нельзя ожидать, чтобы одноступенчатый поршневой компрессор давал очень высокий вакуум. Строятся двухступенчатые поршневые вакуум-насосы, дающие вакуум порядка от 5 до 10 мм Hg. Вследствие очень большой емкости, требуемой для них, нецелесообразно пользоваться более чем двумя ступенями; для более высоких вакуумов применяются ротационные или пароструйные насосы.
344
VII.‘Сжатие и расширение газов
К. П. Д. КОМПРЕССОРА И ДЕТАНДЕРА
До сих пор для упрощения анализа мы рассматривали идеализированные компрессоры. Теперь мы остановимся вкратце на некоторых наиболее важных причинах, которые приводят к тому, что реальные компрессоры отклоняются от принятого нами идеального поведения, и укажем влияние этих отклонений на затрачиваемую работу и на объемный к. п. д.
Наиболее важные необратимые эффекты, сопровождающие сжатие газа в поршневом компрессоре, сводятся к следующим видам:
1. Механическое трение между движущимися и неподвижными частями компрессора, например между поршнем и цилиндром, поршневым штоком и коробкой сальника, крейцкопфом и его основанием и т. д.
2. Дросселирование газа через клапаны и отверстия.
3. Утечка газа из поршневых колец и закрытых клапанов.
4. Теплообмен между газом и стенками цилиндра.
Эффекты 1 и 2 бесспорно приводят к увеличению работы по сравнению с работой, вычисленной для идеального цикла. Эффект 3 также влияет на работу газа, но сильнее сказывается в снижении объемного к. п. д. Здесь мы имеем дело с фактическим объемным к. п. д., который носит название коэфициента подачи компрессора н отличается от ранее применявшихся идеального и объемного к. п. д.; но последний можно определить как отношение объема газа, засасываемого в цилиндр низкого давления при давлении и температуре на подающей магистрали, к емкости цилиндра. Эту величину можно также определить по объему нагнетаемого газа, отнесенному к определенным условиям всасывания. Коэфициент подачи определяется действительными измерениями и несколько отличается от ранее рассмотренного идеального объемного к. п. д. вследствие утечки, наличия разности температур и давлений между, газом в подающей магистрали и газом в цилиндре и других отклонений от идеального цикла. Так, тепло, сохраненное клапаном и стенками цилиндра после хода сжатия, передается забираемому газу во время хода всасывания, что приводит к уменьшению объемного к. п. д.51
Эффект 4 снижает работу сжатия на килограмм газа вследствие уменьшения показателя степени, отвечающего кривой сжатия (линии v — р прн ходе сжатия), до значения, меньшего, чем отношение теплоемкостей. Выигрыш от этого эффекта небольшой, так как охлаждение газа в цилиндре с помощью водяной рубашки не очень эффективно, вследствие чего фактическое сжатие скорее приближается к адиабатному, чем к изотермическому.
К. п. д. компрессора может быть определен уравнением
____ теоретически необходимая работа w фактически затраченная работа
К.п.д. компрессора и детандера
345
Рис. 49. Типичная индикаторная диаграмма компрессора.
При употреблении этого уравнения встречаются недоразумения, так как каждая из величин указанного отношения может толковаться по-разному. Фактически затраченная работа обычно означает работу на валу компрессора в случае двигателя или индикаторную мощность паровой машины в случае компрессора, приводимого в движение поршневой паровой машиной. Однако в некоторых случаях фактической работой двигателя считается энергия, подведенная к мотору, и поэтому к. п. д. двигателя включает к.п.д. мотора и передачи. Теоретической работой может считаться работа обратимого изотермического сжатия между давлениями всасываемого и нагнетаемого газа или же адиабатная обратимая работа, при вычислении которой, в случае многоступенчатого сжатия, предполагается идеальное промежуточное охлаждение. Теоретическая работа будет также зависеть от того, считают ли газ идеальным или же пользуются реальными свойствами рассматриваемого газа, а также от других допущений, включенных в уравнение, используемое для вычисления теоретической работы.
Отношение индикаторной мощности компрессора к действительной (эффективной) мощности, затраченной на валу компрессора для приведения
его в действие, называют механическим к. п. д. компрессора. Индикаторную мощность можно найти с помощью индикаторной диаграммы или приближенно определить по величине адиабатной работы, вычисленной по уравнению (49).
На рис. 49 представлена реальная индикаторная диаграмма, которая сравнивается с уже рассмотренными ранее идеализированными диаграммами. Индикаторную мощность можно вычислить из соотно-
шения
, _ pLSN
60X75С'
где р—среднее (индикаторное) давление, кг/см2,
L — ход поршня, м,
S — поперечное сечение цилиндра, см2. Для цилиндра двойного действия эту величину следует умножить на 2 и вычесть площадь, занимаемую поршневым штоком.
N— число оборотов в минуту.
Среднее (индикаторное) давление равно площади диаграммы, деленной на ее длину и умноженной иа коэфициент масштаба для превращения результата в кг1см2.
346
VII. Сжатие и расширение газов
Из этого краткого рассмотрения производительности компрессора очевидно, что любые числовые значения этой величины, если мы хотим их точно интерпретировать, должны сопровождаться указанием, определяющим, какой вид к. п. д. подразумевается в данном случае.
При определении работы или мощности, требуемой для сжатия, как элемента химического производства, с чем обычно сталкивается инженер-химик, удобно рассматривать суммарный к. п. д. Для компрессора, приводимого в движение мотором, мы будем считать его равным отношению работы, вычисленной для изоэнтропного сжатия [по уравнению (49), если допущение идеальности газа не дает серьезной ошибки], к расходу электроэнергии в первичном двигателе. Эта величина будет изменяться в довольно широких пределах, но ее средняя величина будет, вероятно, 75°/0 для компрессоров рациональных размеров. То же самое значение будет действительно для машины, приводимой в движение водяным паром, если электрическую мощность заменить индикаторной мощностью паровой машины. Зная для применяемого типа паровой машины среднюю скорость водяного пара, т. е. число килограммов пара на индикаторную лошадиную силу-час, можно определить количество пара, требуемого для процесса.
Для большого центробежного компрессора отношение теоретической адиабатной работы к работе на валу в среднем будет также составлять около 75°[0.
В случае детандера, независимо от того, будет ли он поршневого или турбинного типа, использует ли водяной пар или какой-нибудь газ, к. п. д. определяется уравнением
(52)
где А — работа, производимая машиной на 1 кг вещества,
As — работа, которую производила бы адиабатная и обратимая машина, действующая между данным начальным состоянием и данным конечным давлением.
Для адиабатной машины с ничтожно малыми скоростями на линиях всасывания и нагнетания газа можно написать
Если машина не является адиабатной, то производимая работа может быть больше или меньше Л/7 в зависимости от того, поглощает ли машина теплоту из окружающей среды или же теряет ее. В случае машины, которую следует использовать главным образом для охлаждения, повидимому, лучше всего определять к. п. д. по уравнению (53), независимо от того, является ли он в действительности адиабатным или нет.
Пароструйные компрессоры
347
ПАРОСТРУЙНЫЕ КОМПРЕССОРЫ
За последние годы паровые эжекторы получили широкое распространение для нагнетания больших объемов пара или газа при низких давлениях. Такое применение имеет место, например, при вакуумной перегонке, вакуумной кристаллизации, рефрижерации и кондиционировании воздуха. Принцип пароструйного насоса или эжектора иллюстрируется рис. 50. Водяной пар заставляют расширяться в сопле А, из которого он с большой скоростью поступает в камеру смешения С, где он передает некоторое количество движения газу или пару низкого давления, засасываемому через В. В диффузоре D смесь сжимается, причем кинетическая энергия превращается в работу сжатия, и смесь выпускается через Е в конденсатор или же во вторую сту-
Рис. 50. Схема пароструйного компрессора.
пень такого же насоса. Общим результатом является сжатие газа от низкого давления в точке В до более высокого давления в точке Е за счет затраты энергии водяного пара.
Действие эжектора можно уяснить путем рассмотрения его с количественной точки зрения. Даваемая здесь трактовка в основном отвечает выводам, найденным Калустианом [120]. Пусть Нг будет энтальпия применяемого рабочего водяного пара при давлении рг в начальном состоянии, Нг — энтальпия рабочего водяного пара после обратимого адиабатного расширения до р2 при условии, что газ входит в эжектор из эвакуированного пространства,
Н'<2, — энтальпия рабочего водяного пара после фактического расширения в сопле,
тогда /7] — Н2 будет теоретическая работа, получаемая при расширении рабочего водяного пара,
Н}—Н% —фактическая работа и к. п. д. сопла
^-//2
(54)
*1 — Нг-Н2
348
VII. Сжатие и расширение газов
Пусть Н3— энтальпия смеси (смесь рабочего вещества с газом, подлежащим сжатию) в начале сжатия в диффузоре,
Н±— энтальпия сжатой смеси на выходе из диффузора, если принять, что сжатие является идеальным изоэнтропным процессом, т. е. идеальная работа сжатия = /74— Н3, пусть е2— к. п. д. сжатия, тогда
фактическая работа = —5.
Все вышеуказанные величины даны на килограмм газа.
Пусть тг — вес (в кг) рабочего водяного пара и
т2 — вес (в кг) забираемого газа или пара (в нижеприводимом анализе водяного пара).
Фактическая работа, получаемая при расширении рабочего водяного пара, меньше теоретической не только вследствие трения о сопло, которое учитывается величиной но и вследствие других потерь при передаче количества движения от струи с высокой скоростью к относительно медленно движущемуся засасываемому газу. Если обозначить к. п. д. этой передачи через е„ то общая (доступная) работа струи будет = /л1 ег е3(Н1—Н2).
Приравнивая эту величину к действительной работе, требуемой для сжатия, т. е. к величине (mj-|-m2)[(H4 — Н3)/е2], получим
— (действительное)=777 й\———-гй ег? • (55)
от2' ’ {H1 — H2)ele2e3 — (Hi — H3) ' '
Это уравнение следует решать методом подбора, так как Н3 и /74 неизвестны до тех пор, пока не будет известна сухость пара в состоянии 3, а это, в свою очередь, зависит от величин тх и т2. Полагая, что перегрева не происходит и что смешение протекает без изменения энтальпии, получаем баланс процесса смешения:
х3 [т1 -j- т2) = Х2/Я1 + х4/я2, или
___X2W1хУПг (56) 3 mi + «2
ft
где х2— сухость рабочего пара после расширения до давления на линии всасывания р2 и после потери кинетической энергии в процессе забора, а х4 — сухость забираемого водяного пара; х2 связано с х% (сухостью пара после расширения, но до стадии забора) уравнением
(1— e2)(Hj — Н2) = (х2~ X'2)L, (57)
Пароструйные компрессоры
349
где L — скрытая теплота парообразования при низком давлении расширившегося газа. Величина х2, в свою очередь, таким же образом связана с сухостью пара после изоэнтропного расширения х2, т. е. уравнением
(1 — в1) (Нг — Н2) = (х2 — х2) L. (58)
Уравнения (57) и (58) выводятся из энергетических балансов соответствующих процессов. Следовательно, величина (1—е3){Н1—Н2 ) представляет кинетическую энергию, превращенную во внутреннюю энергию при передаче количества движения. Эта энергия используется при высушивании пара и поэтому равна (х2— x2)L.
Благодаря широко поставленным испытаниям сопел турбин к. п. д. сопла хорошо известны. В зависимости от условий они могут изменяться от 0,85 до 0,95; е2 можно принять около 0,80; е3 можно вычислить из принципа сохранения количества движения. Полученные таким образом значения е3 хорошо совпадают с результатами испытаний по расходу водяного пара в эжекторах.
Минимально необходимое количество рабочего пара дается уравнением(55) при условии? что е2 и е3—1. Однако для этого частного случая уравнение можно упростить, чтобы избежать решения методом подбора, если учесть, что работа, полученная при расширении рабочего пара от давления в конце диффузора до давления забора, равна работе, которую следует произвести для сжатия пара в диффузоре, поскольку в этом идеальном случае все изменения считаются обратимыми. Следовательно, общий результат будет одинаков как в случае, если рабочий водяной пар расширяется только до давления в конце диффузора, так и в случае, если только сжимается забираемый пар. Другими словами, работа, необходимая для сжатия забираемого пара от давления р2 до давления ходе из насоса, получается за счет расширения рабочего пара от pij до давления при выходе из насоса. Тогда имеем
Ш2
при вы-водяного
(59)
где Нг — та же величина, что и раньше,
Н3 — энтальпия рабочего водяного пара после изоэнтропного расширения до давления сжатой смеси, покидающей диффузор, Н6 — энтальпия забираемого газа (пара),
Н1—энтальпия забираемого газа (пара) после изоэнтропного сжатия от давления, при котором он входит в пароструйный насос, до давления после диффузора.
Конечно, в пароструйном насосе в качестве рабочего вещества, кроме водяного пара, можно использовать и другие вещества. Для исследования этой возможности, особенно интересной инженерам-химикам,
350
VII. Сжатие и расширение газов
сделано очень мало. Следует предположить, что расчеты и экспериментальные данные из этой области могут оказаться весьма полезными ☆).
Пример 11. Следует испарить воду при 7°С, сжать ее струей пара, используя насыщенный водяной пар при 7 кг/см2, н сконденсировать при 0,07 кг/см2. 1) Чему будет равен минимальный расход водяного пара на 1 кг испаренной воды? 2) Чему будут равны а) теоретическая скорость истечения водяного пара нз сопла и б) фактическая скорость? 3) Определить фактический расход пара.
1. Абсолютное давление в 0,07 кг/см2 соответствует температуре в 38,66°С, которая легко достигается при обычном охлаждении водой.
Все свойства водяного пара взяты из таблиц Вукаловича и из диаграммы S — Н по Вукаловичу:
H-t — (энтальпия исходного рабочего водяного пара при сухости пара, равной 1,00) = 659,4 ккал/кг,
Hs — (энтальпия рабочего водяного пара после изоэнтропного расширения до 0,07 кг/см2) = 497,3,
Н$ — (энтальпия забираемого пара, в качестве которого принимается насыщенный водяной пар при 7°С) = 600,2,
Нч— (энтальпия забираемого пара после изоэнтропного сжатия до 0,07 кг/см2) = 678, к
По уравнению (59),
^1 = 0,48 кг рабочего водяного пара на кг забираемого пара.
2. а) Теоретическая скорость струи водяного пара, выходящей из сопла, будет определяться уравнением (43), которое можно написать так:
н2 = 91,53 V//x — Т/2,
если и2 дается в м/сек, а Н в ккал/кг.
б) Для фактической скорости можно написать
и2 (фактическая) = 91,53 Vei^//;
//j — 659,4 и Нг = 448,8 (изоэнтропное расширение до 0,01 кг/см2 давления насыщенного пара при 7°С):
и2 (теоретическая) = 91,53 V659,4— 448,8 м/сек =1330 м/сек. Если к.п.д. Сопла равен 85%, то действительная скорость будет равна 1220 м/сек.
3. Для определения фактического расхода водяного пара используем следующие к. п. д.:
«?! = 0,85, е2 = 0,80, е3 = 0,65.
х, (сухость рабочего водяного пара при 0,001 кг/см2 после изоэнтропного расширения) = 0,739.
//1 = 659,4; //2 = 448,8; Н’2 (по уравнению (54)) = 480,4
х' (по уравнению (58)) = 0,731,
Ху (по уравнению (57)) = 0,837.
☆ ) После окончания работы над книгой появилась статья У орка и Гедриха [261], в которой приводятся некоторые данные по эжекторам, использующим в качестве движущего вещества некоторые органические пары. Эта статья также отсылает читателя к некоторым более ранним статьям, посвященным рассматриваемому вопросу.
Эффект Джоуля—Томсона (дросселирование)
351
Примем х3 — 0,920, тогда
//, = 552,5.
ZZ4 (после изоэнтропного сжатия от состояния 3) = 614. По уравнению (55),
«2
Принимаем х4=1,00 (х3, по уравнению (56), равно 0,91) и выбираем эту величину как окончательную, полученную методом подбора.
Заметим, что действительный расход водяного пара бывает примерно в пять раз больше теоретического.
ЭФФЕКТ ДЖОУЛЯ — ТОМСОНА (ДРОССЕЛИРОВАНИЕ)
Основные принципы. До сих пор обсуждение расширенна ограничивалось обратимым процессом, противоположным сжатию. Мы очень мало говорили о расширении, но все формулы, выведенные в этой главе, так же приложимы к обратимому расширению, как и к сжатию. Мы скажем больше о расширении этого типа в последующих главах, в которых рассматриваются вопросы, связанные с охлаждением и получением очень низких температур. В данном разделе мы будем иметь дело с необратимым типом расширения, который, естественно, не является процессом, полностью противоположным сжатию.
Если газ, хранящийся при повышенном давлении в резервуаре постоянного объема, перешел в пространство более низкого давления, например в атмосферу, то он не совершил полезной работы. Тот же самый газ мог бы расширяться при таких условиях, что получалась бы полезная внешняя работа, и эту работу можно было бы использовать для нового сжатия газа. Расширение называется „свободным", если отсутствует механизм, в котором расширяющийся газ мог бы совершать работу; такое расширение явно необратимо.
Один такой вид расширения (периодический) называется джоу-левским (эффект Джоуля). Родственное ему расширение, обычно называемое эффектом Джоуля — Томсона и происходящее в непрерывно движущейся системе, имеет гораздо больше значения в промышленности. Мы пока отложим обсуждение эффекта Джоуля (стр. 363) и перейдем к рассмотрению эффекта Джоуля—Томсона (известного также как процесс дросселирования).
Этот тип расширения несколько раз уже упоминался, но ни разу не рассматривался подробно; расширение в данном случае происходит таким образом: вещество, которое непрерывно поддерживается при постоянном давлении рг, претерпевает падение давления, перемещаясь через клапан, пористую перегородку или другое препятствие на своем пути, и переходит в пространство, в котором непрерывно поддерживается постоянное давление р2, причем процесс протекает адиабатно. Сначала рассмотрим процесс, несколько более простой с теоретической точки зрения, а именно — процесс,
352
VII. Сжатие и расширение газов
в котором на пути газа за дросселем следует теплообменник, в котором веществу сообщается (или от него отнимается) такое количество теплоты, чтобы оно приняло исходную температуру. Этот процесс называется дросселированием при постоянной температуре. Очевидно, что он не является истинным изотермическим процессом, поскольку вещество только в исходном и конечном состояниях находится при одинаковой температуре. Для целей термодинамического анализа такой процесс может быть представлен схемой, подобной изображенной на рис. 51. А представляет хранилище для газа при давлении ри из которого газ непрерывно вытекает через дроссель В. Для поддержания в А постоянного давления служит поршень, С
Рис. 51. Изотермическое дросселирование.
который приводится в движение вправо как раз с такой скоростью, чтобы компенсировать вытекание газа. (В любом действительном случае поршень или эквивалентное ему перемещающее объем приспособление может быть удален от действительной точки расширения, но обязательно должен быть где-либо связан с системой, иначе давление не будет оставаться постоянным. Для любой системы с перемещающимся газом — или жидкостью — обычно представляют себе поршень, проталкивающий газ в некоторую изолированную для целей изучения секцию, даже если поршня в данной точке нет. Этот прием способствует наглядному представлению о всех видах энергии, подлежащих рассмотрению.) Газ перемещается от дроссельного вентиля В через теплообменник D в хранилище С, в котором давление с помощью соответствующего движения поршня поддерживается постоянным и равным рг. В действительности эта часть системы может быть просто атмосферой, но воображаемый поршень служит для упрощения анализа.
Применим теперь к этому процессу первый закон:
Р = Д£7+Л. (2, гл. II)
Работа, произведенная поршнем в Л на моль газа, протекающего от Л к С, равна — РУи где рх— абсолютное давление газа в точке Л, а Vj — его мольный объем. Подобным же образом работа, произведенная поршнем в точке С, равна р2®2' Если Ux и U2 представляют мольные энергии газа в состояниях 1 и 2, то подстановка этих величин в уравнение (2, гл. II) дает
Q = Ц + РгЪ ~ Pivi > (60)
Эффект Джоуля — Томсона (дросселирование)
353
или
Q^H2 — HV (61)
Тепловой эффект, сопровождающий расширение от Pj до р2, легко вычисляется по уравнению (61), данному либо в виде уравнения состояния, либо в виде термодинамической диаграммы. Так, по уравнению (96, гл. Ill), имеем при постоянной температуре:
Р* Р*
№=-§T(ffldp + §vdp, (62)
Pl Pl
или
Д/7=J? ^dv — ^pdv — p2v2 — p(or. (63)
Для идеального газа из уравнений (61) и (62) непосредственно следует, что Q = 0. Лэгко заметить, что для газа, подчиняющегося уравнению Ван-дер-Ваальса,
Q = а + Pzvz —РЛ- (64)
Качественные выводы. Обращение к основному уравнению (60) позволяет сделать некоторые интересные качественные выводы о тепловых эффектах и температурных изменениях, сопровождающих рассматриваемый процесс. Так как для идеального газа Uявляется функцией только температуры и так как pv при постоянной температуре постоянно, то сразу видно, что при изотермическом расширении идеального газа не будет наблюдаться теплового эффекта. Ясно, что для изотермического расширения реального газа U2 всегда больше Ц. Во всех газах в любом состоянии между молекулами имеются силы притяжения и, поскольку молекулы при расширении перемещаются против этих сил, в газе должна накапливаться энергия, которая поступает к нему от окружающей среды. Следовательно, поскольку дело касается только изменения U, всегда должен существовать приток тепла в систему или, если система термически изолирована, должно происходить падение температуры.
При рассмотрении изотермического изменения мы имеем три возможных случая, а именно:
1. PiWi=p2v2,
11. ?)•»!< P2V2,
111. Р^УРЯ-
Случай I, который имеет место для всех реальных газов, находящихся в определенном состоянии, очевидно, должен приводить к 23 Химическая термодинамика
354
VII. Сжатие и расширение газов
охлаждению. Случай II, который для всех газов наблюдается при температурах ниже точки Бойля данного газа и при невысоком давлении, приводит к положительному значению Д(/гп) и поэтому, согласно уравнению (60), — к еще большему Q, т. е. к охлаждению. При переходе к случаю III найдем, что величина Д(/щ) будет иметь знак, противоположный знаку Д£/, н будут существовать три подгруппы, приводящие к различным результатам в зависимости от относительного числового значения этих двух величин.
Случай (а):
— Рги2 = и2 -т. е.
Q = 0.
Здесь не происходит ни нагревания, нн охлаждения газа. Поэтому в этом частном случае газ ведет себя как идеальный.
Случай (б):
Pivi— Рг°г<.и2 — и1‘
Тогда Q является положительным, и для адиабатной системы все же имеет место охлаждение.
Случай (в):
Рг°2>и2~~ ui-
Для этого случая по уравнению (60) Q отрицательно; следовательно, будет наблюдаться нагревание, т. е. для поддержания изотермичности процесса тепло должно отводиться. Последний случай отвечает дросселированию таких газов, как водород и гелий, при комнатной температуре и при невысоком давлении.
Поправка на кинетическую энергию. При всех этих рассуждениях о необратимом расширении мы принимали, что газ обладает только той энергией, которая обусловлена его состоянием и определяется его давлением, объемом и температурой. В действительности текущий газ обладает кинетической энергией, обусловленной движением его массы. До тех пор пока скорость движения газа остается малой, кинетическая энергия настолько мала, что различие в кинетической энергии между газом в состоянии / ив состоянии 2 (рис. 51) ничтожно мало по сравнению с Д/7.
Пример 12. Воздух при 25°С и 100 атм давления перемещается со средней линейной скоростью в 0,3 м/сек, расширяясь в сопле до давления в 1 атм, и выходит из сопла со средней скоростью в 6 м/сек. К системе подводится тепло для поддержания температуры в 25°С. Чему будет равна ошибка при определении эффекта охлаждения, если пренебречь кинетической энергией?
По диаграмме 5 — Г для воздуха [292] при изотермическом расширении ДЛ/ = 4,8 ккал/кг.
Разность кинетической энергии — т ( и£ — и?) (36—0,09) —
2 v ' 2 X У»” *
— 1,83 кгм/кг ~ 0,0043 ккал/кг.
Эффект Джоуля — Томсона (дросселирование)
355
Мы видим, что пренебрежение кинетической энергией в этом случае дает ошибку только в 0,1°/0. Однако летке может случиться, что газ на выходе из сопла обладает значительно большей скоростью. Так, если бы она была равна 30 м[сек или больше, то влияние кинетической энергии стало бы заметным. Во всех наших рассуждениях о необратимом расширении мы будем принимать, что все мгновенные высокие скорости струи и соответствующие им значительные кинетические энергии рассеиваются, благодаря чему кинетические энергии в этих состояниях ничтожно малы. При экспериментальном изучении рассматриваемого процесса для осуществления падения давления обычно пользуются пористой перегородкой и, таким образом, сводят к минимуму образование струй высокой скорости.
Эффект Джоуля — Томсона. Если расширение, показанное на рис. 51, проводится адиабатно, то уравнение (61) переходит в
ДН=0 (65)
или, для бесконечно малого изменения, dH=0. (66)
Это уравнение обычно называется основным уравнением эффекта Джоуля — Т омсона.
Из уравнений (66 и 96, гл. III) получаем
\др)ц Ср '
Коэфициент Джоуля — Томсона р является функцией состояния газа. Интегрирование уравнения (97, гл. III) приводит к выражению для конечного перепада температуры ДТ, получающегося в результате расширения при конечном перепаде давления. ДТ будет называться интегральным эффектом Джоуля—Томсона или просто эффектом Джоуля — Томсона. Для идеальных газов определение правой части уравнения (97, гл. Ш) по уравнению pv = RT приводит в результате к g = 0^). Так как ц должно быть равно
£г) Здесь возникает кажущееся противоречие, которое заслуживает краткого обсуждения. Правая часть уравнения (97, гл. III) при условии использования уравнения идеального газа превращается в величину
ср
Но числитель этого уравнения является величиной а, которая, как уже было показано, при р—>-0 не делается равной нулю; поэтому ц приближается не к нулю, а к конечной положительной или отрицательной величине. Это обстоятельство подтверждается как экспериментальными данными, так и использованием уравнения состояния (см. пример 13). Для объяснения этого можно только сказать, что нет строго идеального газа. Идеальный газ является абстракцией, введенной для удобства, и не следует ожидать, что реальный газ будет обладать свойствами, постулированными для идеального газа, даже когда р приближается к нулю.
23*
356
VII. Сжатие и расширение газов
к
нулю для всех состояний идеального газа, последний не будет обнаруживать эффекта Джоуля — Томсона. Для реального же газа мы можем
написать
То, что р=0, не обязательно предполагает, что газ является идеальным или близким к идеальному. В главе V мы показали, что условие
Рис. 52. Кривая инверсии эффекта Джоуля — Томсона для азота [201].
[1 = 0 просто приводит к равенству
~ = const — у (р).
Другими словами, любой газ, для которого объем вдоль изобары линейно изменяется с температурой, будет обладать нулевым коэ-фициентом Джоуля — Т ом-соиа. Подобным же образом любой реальный газ может иметь нулевой эффект Джоуля— Томсона для некоторого частного конечного изменения давления, как показано на стр. 353.
Точки инверсии. Точка, в которой [1 = 0, известна
под названием точки инверсии эффекта Джоуля — Томсона. Это не единственная
точка, так как существует целый ряд соответствующих друг другу давлений и температур, при которых р = 0. На рис. 52 показана кривая инверсии, т. е. геометрическое место точек инверсии для азота, основанная на измерениях эффекта Джоуля — Томсона Ребуком и Остербергом [201]. Лишь эти данные и некоторые данные по аргону тех же исследователей дают достаточно полные кривые инверсии. Область внутри кривой между р = 0 и р=375 атм отвечает состояниям, где р положительно (охлаждение). Вся область вне кривой представляет состояния, где [1 отрицательно. При любом давлении ниже 375 атм имеются две температуры инверсии — верхняя, где р прн понижении Т переходит от отрицательного значения к положительному, и нижняя, где происходит обратное изменение. Выше 375 атм превращения не происходит ни при каких температурах. Кривую инверсии можно найтн с помощью уравнения состояния,
Эффект Джоуля — Томсона (дросселирование}
357
но если уравнение не воспроизводит поведения газа с исключительной точностью, то результат может быть только приближенным. Действительно, такое вычисление, если его результаты можно сравнить с экспериментальными данными, является очень строгой проверкой уравнения состояния.
Пример 13. Вычислить температуру инверсии для азота при 100 атм, принимая, что азот подчиняется уравнению состояния Битти (84, гл. V).
По уравнению (97, гл. III) и уравнению состояния
г ( о । 2А0 4с\ . __ЗЛ0я , 5В0С\ (ЬВ$с\
|iCp вОт Т3) "* у Rf К2Т2' КТ* / \R2TiJP~
В качестве первого приближения можно откинуть член, содержащий р-, т. е. принять, что
-В0 + (2Л0//?Г) + (4с/Л) Р (2B0b!RT} - (АА^КЧ2) + (5Вос1КП)'
Это соотношение представляет зависимость между давлением и температурой инверсии. При подстановке различных значений Т легко получаются следующие решении (константы даны в табл. 6):
/>=Ю0 атм, Г—589 и 44°К, -----------------р — 1 атм,-------Т = 656°К.----------- -------------
Совпадение с экспериментальной кривой на рис. 52 оказалось прекрасным для высокой температуры, но очень плохим для более низкой. Однако расхождение в последнем случае не является неожиданным, если учесть, что нижняя точка инверсии в действительности находится в области жидкости и уравнение состояния, очевидно, не приложимо к таким условиям. При 1 атм уравнение дает только одну температуру инверсии, между тем как наблюдаются две температуры. В этом отношении уравнение Ван-дер-Ваальса лучше представляет факты, чем 'уравнение Битти.
Полуколичественные представления о кривой инверсии для любого вещества получаются из закона соответственных состояний. Принимая в уравнении (97, гл. III) р = 0 и выражая затем это уравнение в приведенной форме, получаем
t(SV~v=0- (67)
Диференцирование приведенного уравнения Ван-дер-Ваальса (56, гл. V) и подстановка в уравнение (67) дают
_3(3v—I)2 Т 4>-
где т — приведенная температура инверсии. Исключая т из уравнения (68) и приведенного уравнения состояния, получаем
^^-1). (69)
358
VII. Сжатие и расширение газов
Сочетая уравнения (68) и (69), прн исключении >, получим урав-
нение кривой инверсии для любого газа, выраженное в приведенных 27 3
параметрах. Когда тт —> 0, то это уравнение дает т = и , следо-
вательно, имеется максимальное значение тт = 9,0. Поэтому приведенная
кривая инверсии представляет параболу, которая схематически изображена на рис. 53. Сравнение с рис. 52 показывает,что этот метод
вполне удовлетворительно представляет общую картину.
С количественной стороны совпадение
Рис. 53. Приведенная инверсионная кривая.
только очень приблизительно. Так, для азота максимальное давление, при котором происходит инверсия, равно 376 атм, т. е. 71=11,2 вместо 9,0, предсказанного теорией соответственных состояний. Кроме того, температуры инверсии для азота при низком давлении равны 103,1 и 621° К, что соответствует приведенным температурам 0,82 и 4,93 вместо предсказанных теорией соот-
ветственно 0,75 и 6,75. Подобные грубые предсказания качественного
порядка можно легко дать и для других веществ.
Интегральный эффект Джоуля — Томсона. При любом реальном процессе всегда больше интересуются конечными изменениями, чем диференциальными, а поэтому непосредственный интерес представляет интегральный эффект. При этом возникает задача следующего характера: дается определенное вещество в некотором исходном состоянии при повышенном давлении; требуется найти его состояние в результате адиабатного расширения до более низкого давления. Это очень г.росто решается с помощью термодинамической диаграммы (52), что может быть иллюстрировано следующими двумя примерами.
Пример 14. Насыщенный жидкий аммиак при 25° С расширяется через дроссель до постоянного давления 2 кг1см2.
Каким будет состояние аммиака при понижевном давлении?
По диаграмме S—Т для NH3 [293] определяем местоположение начальной точки при пересечении изотермы 25° С и линии нагыщенной жидкости. Затем перемещаем по изоэнтальпе (52) до пересечения с изобарой 2 кг/см2 и считываем значение сухости пара. В рассматриваемом случае удобнее определить сухость пара по уравнению (41, гл. VI), сочетая его с уравнением (65) и используя диаграмму S — Т:
Н, =71,5,
И., = 339,6 х + 22,3 (1 — х).
Эффект Джоуля — Томсона (дросселирование) 359
Так как T/j = //2, то х = 0,136.
Другими словами, в результате процесса дросселирования испарилось 13,6°/0 жидкого аммиака.
Пример 15. Влажный водяной пар при 18 кг/см2 расширяется через дроссель до давления 1,4 кг/см2. Температура расширившегося водяного пара равна 140° С. Чему была равна сухость пара, подвергшегося дросселированию?
Эта задача иллюстрирует принцип дроссельного калориметра для определения сухости влажных паров. Этот принцип основан на том, что адиабатное дросселирование влажных паров приводит, по крайней мере при многих условиях, к перегреву пара; поэтому его состояние однозначно определяется давлением и температурой.
По значениям конечных давления и температуры определим на диаграмме $ — Н (или S — Т) положение точки, соответствующей этому состоянию, и затем идем по изоэнтальпе до пересечения с изобарой 18 кг/см2. Исходная сухость пара, как легко прочесть на диаграмме, равна 0,977.
Эту задачу также легко решить без диаграммы, полозуясь таблицей термодинамических свойств, как было сделано в предыдущем примере.
Ясно, что существует определенный предел сухости пара, который можно определить с помощью этого метода. Наинизшей возможной является сухость пара, соответствующая состоянию расширив-шегося пара до кривойнасыщения; пока расширившееся вещество остается смесью, его состояние не будет точно определяться его давлением и температурой. При условиях, соответствующих примеру, легко заметить, что 0,972 представляет наиболее низкую сухость пара, которую можно измерить с помощью данного расширения. Интересно отметить, что при высоком давлении, например при 140 кг]см2, насыщенный водяной пар при дросселировании становится «влажным*, а не перегретым82.
Для вычисления изменения температуры при дросселировании при отсутствии диаграмм или таблиц можно использовать различные методы. Из уравнения (97, гл. III) имеем:
ДТ= y.dp. (70)
Pi
Приведенные в таблицах значения р как функции р и Т можно найти постепенным интегрированием, используя средние значения g в узких пределах давлений и получая в конце концов ДТ для расширения в целом.
При отсутствии данных для ц, ДТ можно вычислить с помощью уравнения состояния по следующему методу, непосредственно связанному с вычислением энтальпии (см. гл. VI).
Из уравнения (97, гл. Ш) имеем:
Т, pt Pt
f CpdT = J dp- j vdp, (71)
Ti й P Pi
360
VII. Сжатие и расширение газое
или
Г» v-i
J CpdT=— J^pdv-b-p^-p^. (72)*)
Г, vt v,
Поскольку эффект Джоуля—Томсона связан только с узким интервалом температур, можно написать:
г,
^CpdT = (Cp)m(Tt — Tl). (73)
Разность температур 7\ — Т2 представляет интегральный эффект Джоуля—Томсона и легко вычисляется по уравнениям (72) и (73), по
Рис. 54. Интегральный эффект Джоуля — Томсона.
данному уравнению состояния и по средней теплоемкости газа при низком давлении. Принцип, лежащий в основе этого метода, делается очевидным с помощью рис. 54 ☆ *).
Пусть АВ представляет процесс дросселирования на диаграмме Т—р. Так как для такого процесса Д/7=0 и так как //является свойством, то
А НАС == кН ВС ’
&НАС представляет изотермическое изменение энтальпии между данными пределами давления при
известной начальной температуре Т1 и соответствует правой части уравнения (71) или (72). &НВС является изобарным изменением энтальпии при известном низком давлении и равно левой части уравнения (71).
Пример 16. Вычислить падение температуры при адиабатном дросселировании воздуха от 215,2 до 1,2 атм, если первоначально воздух нахо-дитси при 0° С.
Для воздуха будет использовано уравнение состояния Битти — Бриджмена, константы которого представлены в табл. 6. Интегрирование правой части уравнения (71) посредством этого уравнения состояния уже было приве-
☆ ) По уравнению (б) примера 4 гл. VI, v2 является не конечным объемом, соответствующим конечному состоянию в процессе дросселирования, а объемом при конечном давлении и исходной температуре. Это вытекает из того обстоятельства, что интегрирование уравнения (97, гл. III) сначала производится при постоянной Т, а затем при постоянном р.
☆ ☆) Отметим, что даже при использовании уравнения (72) все же рис. 54 представляет стадии интегрирования. Мы просто изменили независимую переменную на изотерми зеской стадии.
Эффект Джоуля — Томсона (дросселирование)
361
деао в примере 4 предыдущей главы. При использовании полученных там результатов уравнение (71) переходит в
Т1—Тг= 1 --fрл>г — + «г (------— \ +
W1 р2/
, . / 1 1 \ _ / 1 1 \1
^2 / J
(а)
v1 определяется из уравнения состояния методом подбора. Это делается очень просто, потому что по диаграмме коэфициента сжимаемости можно выбрать почти правильное значение для первого подбора; р2 с достаточной точностью можно подсчитать по уравнению состояния идеального газа:
t>j = 0,1047 л)моль,
к2= 18,70 л)моль,
(^)ш = 6,95 кал/моль
J= 0,04133 л/атм-кал.
Подставляя эти значения в уравнение (а), получаем
ДГ=44,6 или
t2 = — 44,6° С.
Если более низкое давление значительно ниже атмосферного, среднюю Ср можно определить по одному из методов, описанных в предыдущей главе.
Рассмотренный метод оказывается полезным для вычисления эффекта Джоуля—Томсона и в том случае, если в нем участвуют газовые смеси. Если известны константы уравнения состояния для отдельных газов, то константы для смеси можно определить по одному из методов, описанных в гл. V. После этого эффект Джоуля—Томсона вычисляется совершенно так же, как для чистого газа.
Вычисление эффекта Джоуля—Томсона, как мы сейчас выяснили, прежде всего представляет вычисление изменения Н при изотермическом расширении. Этот вопрос уже обсуждался в предыдущей главе, и теперь мы проиллюстрируем применение нескольких методов, рассмотренных выше. Так, мы можем использовать рнс. 36 для решения задачи, поставленной в примере 16. По критическим данным табл. Ш „Приложения" мы находим, что при 0°С т = 2,06. Значения тг равны соответственно 5,78 и 0,032. По рис. 36 находим
1,15,
1,15 X 273 = 314.
Приравнивая эту величину к —(С^т{Тг—Тг}, получаем Тг— 1\=— 45,2° С, что является хорошим совпадением со значением, полученным и» уравнения Битти—Бриджмена.
362
VII. Сжатие и расширение газов
Для определения эффекта Джоуля—Томсона непосредственно из диаграммы коэфициента летучести можно также использовать уравнение (35, гл. VI):
= (Ср)т^Т= RT\ (35, гл. VI)
ИЛИ
г) •т'З
ДТ (эффект Джоуля—Томсона)=^Д^/= =-. -t -(74)
r^pimx и? Jk \>'р)т*кр\ Vх / к
Наклон с достаточной точностью определяется из диаграммы при допущении, что
din f__Д1пу
—<h~ Дт ’
и при выборе двух, значений т (т* и тг), близких к данной начальной температуре. Тогда уравнение (74) принимает следующий вид:
/?Т[ 1п5
&Т = .7^т-.----. (75)
(Ср)пАх1 z^)TKp
Пример 17. Определить понижение температуры при адиабатном расширении воздуха через дроссель от 185,3 атм. до 1,2 стиле,'если вначале t = 3,2° С.
Из вывода уравнения (75) очевидно, что оба значения у относятся к исходному давлению, которое отвечает it = 4,98. Данная температура соответствует т = 2,09; можно произвольно принять = 2,20 и г, = 2,00. По диаграмме коэфициентов активности у, = 0,972 и у2 = 0,922. Подставляя найденные значения в уравнение (75), получаем
О 979
(1,987) (276.3И (2,303) 1g ду__ ________________________49 so р
(6,95) (2,20 — 2,00) (132,4)
Экспериментальное значение Ребука [200] равно 39,6° С.
Ньютон и Додж [176] с большим успехом применили уравнение (75) в семи случаях расчета эффекта Джоуля—Томсона. Они также предложили видоизмененное ураннение, которое можно использовать в тех случаях, если более низкое давление значительно выше 1 атм, принимая во внимание, что уравнение (75) ограничено такими случаями, когда газ при низком давлении можно считать идеальным.
Сравнение эффекта охлаждения при обратимом и необратимом расширении. Падение температуры в результате обратимого расширения, при котором совершается полезная внешняя работа, например в цилиндре машины, легко вычисляется по уравнению (23) при допущении идеальности газа. Так, если азот при 20° С и 4 атм адиа-батно и обратимо расширяется до 1 атм, то
1,40—1
Т2 = 293,2^ 1,40 = 293,2 X 0,673= 197,2° К, или—76°С.
При этих условиях газ вследствие расширения охлаждается на 96° С. С другой стороны, если бы тот же самый газ (конечно, не идеальный) расширялся бы необратимо по способу Джоуля — Томсона, то
Эффект Джоуля
363
применение уравнения (а) примера 16 показало бы охлаждение меньше чем на 0,5° С. Другими словами, имеется очень большое различие эффектов охлаждения, получающихся в результате этих двух предельных случаев расширения. Любая необратимость при расширении, связанном с работой, будет снижать падение температуры, и можно представить настолько неэффективное расширение в цилиндре машины, что не будет совершаться никакой внешней работы, и результат будет тот же, что и при эффекте Джоуля — Томсона.
ЭФФЕКТ ДЖОУЛЯ
Этот тип расширения представлен схематически на рис. 55. Процесс может заключаться в расширении из цилиндра под постоянным давлением в хранилище постоянного объема или же в расширении из хранилища постоянного объема в цилиндр под постоянным
Дроссель
Цилиндр
Резервуар_
для хранения
Рис. 55. Джоулевское расширение.
давлением. Количественная теория процесса выводится крайне просто при приложении первого закона, если допустить, что процесс является адиабатным, или, другими словами, если пренебречь теплообменом между веществом и стенками хранилища. Для вывода будут использованы следующие обозначения:
N—число молей (или килограммов) газа, VT— общий объем сосуда.
Индексы Тис относятся соответственно к сосуду и цилиндру. Индексы (') и (") относятся к исходному и конечному состояниям. На основании первого закона, при допущении адиабатности процесса
С2 = М74-Д = 0.
Для рассматриваемого процесса
N’rU'r 4- N"cU"c — N’tUt— N’eU'e ,
U”c = U'c = Ue, A = pcvc (Nt — N't), rf' — N’^N’t — N'r,
Тогда
364
VII. Сжатие и расширение газов
Если вещество представляет идеальный газ и если начальные и конечные температуры в хранилище и в цилиндре принимаются равными, то справедливы следующие соотношения:
U= ^CvdT-\-C,
"v А —1 ’
и=^+с,
H=U-L-RT==^-tRT-[-C,
г __P\vt
"___P'VT
т RT2 ’
где — начальное давление в хранилище,
Г] и Т2 — начальная и конечная температуры в хранилище.
Подстановка их в уравнение (76) дает
-т* _-т* _kPc
J2-4{k_i)pi+pc-
(77)
Если р! мало по сравнению с рс, то уравнение (77) принимает вид
Т2 = ЛТ1. (78)
Применение этих уравнений иллюстрируется нижеследующими примерами.
Пример 18. Цилиндр для хранения воздуха полностью эвакуируется, и ему даот принять комнатную температуру, равную 20° С. Затем внезапно открывается клапан, и воздух перемещается до тех пор, пока давление в цилинтре не достигнет атмосферного. Считая воздух идеальным газом и пренебрегая переходом тепла от воздуха к стенкам цилиндра, определить температуру воздуха в цилиндре после выравнивания давлений.
В этом случае рг настолько мало по сравнению с ре, что можно воспользоваться уравнением (78); тогда
Г2= 1,41 X 293,2 = 413,4° К или 140,2° С.
Теплоемкость цилиндра значительно больше теплоемкости воздуха, и фактическая температура будет много меньше найденной, так как процесс далек от адиабатного; но чем больше судет сосуд, тем более правильные результаты будут получаться по уравнению. Об экспериментальной проверке правильности предсказанного повышения температуры см. статью Эштриха [177а].
Пример 19. 1. Хорошо изолированный сосуд емкостью в 150 м3 предназначен для хранения водяного пара. Вычислить, сколько можно хранить в нем водяного пара, если он, будучи первоначально наполнен насыщенным водяным паром при давлении в 1 кг/см2, соединяется затем с источником насыщенного водяного пара при постоянном давлении в 10 кг/см2 и наполняется до выравнивания давления. Каковы будут свойства водяного пара в сосуде? Тепловыми потерями при расчете следует пренебречь.
Эффект Джоуля
365
В рассматриваемом случае приложимо уравнение (76); пользуясь им в расчете на 1 кг пара, получим (см. таблицы Вукаловича):
/7С = 663 ккал1кг,
=598,2.
427
Лу и U"T определяются методом подбора. Принимаем конечную температуру водяного пара t2 = 250° С.
v2 = 0,2374 м31кг, ^=7O216 io><^xoaz.4
^=0»4=етЛ
Подставляя в уравнение (76), получаем
7,26 # 4,05.
Еще несколько подборов дает решение:
t» = 267°C; о, = 0,2452; л£ = 609,3.
Водяного пара хранится 522,3 кг.
2. Примем, что тот же самый сосуд заполнен на 0,75 своего объема водой при 100° С, причем оставшийся объем занят насыщенным водяным паром при 1 атм.. Пусть сосуд, как и раньше, соединен с источником насыщенного водяного пара при 10 кг/см* 2 и пусть водяной пар при входе в сосуд тщательно смешивается с водой так, чтобы он в сосуде всегда находился с ней в равновесии. Сколько водяного пара может перейти в сосуд при этих условиях, если давления будут равны? Тепловыми потерями снова пренебрегают.
Если пренебречь массой водяного пара в сосуде по сравнению с массой воды, то к этому случаю приложимо уравнение (76), причем UT будет внутренней энергией жидкой воды
/4 = 663,
77^=100,
U” = 179 4=s—
Нт= 125400 кг.
Количество хранищегося водяного пара =17600 кг. Общий объем воды в сосуде в конце процесса — 125400 X 0,0011262= 141,2 м3.
Этот расчет иллюстрирует принцип аккумулятора водяного пара, являющегося важным устройством во многих отраслях промышленности, в которых расход пара сильно изменяется во времени. Аккумулятор восполняет несоответствие между медленной подачей из котлов и колеблющимися требованиями в процессе.
ГЛАВА VIH
ТЕРМОДИНАМИКА ПОТОКА
Количественное рассмотрение потока, охватывающее перемещение в трубопроводе, перемещение через различные измерительные устройства (например диафрагмы, сопла, трубы Вентури) или перемещение, служащее для получения работы, может непосредственно основываться на первом и втором законах термодинамики. Целью этой главы и является показать, как можно приложить эти законы, и особенно первый закон, к различным задачам, относящимся к потоку. С самого начала следует уяснить себе, что приложение термодинамики может связать только различные виды энергии, связанные с процессом, и этим установить идеальные условия истечения. Термодинамика не может дать никаких сведений, относящихся к механизму потока, например сведений о потере напора, обусловленной трением. Подробное обсуждение влияния трения, так же как и детальное описание оборудования, относится скорее к книге, в которой изучаются отдельные процессы химической технологии, и в этой книге большей частью рассматриваться не будет.
Строгий термодинамический анализ потока не требует введения каких-либо рассуждений, относящихся к его механизму. Принимают, что все вещества перемещаются совершенно без трения, поэтому можно игнорировать вязкость, которой обладают все реальные вещества, как жидкие, так и газообразные. Теоретические уравнения, выведенные на основании этого допущения для описания свойств реальных веществ, требуют некоторого видоизменения. Так как нашей целью является расширение термодинамической трактовки за пределы чисто теоретических уравнений, что дает возможность решать практические задачи, то необходимо включить некоторые кинетические рассуждения. Будем полагать, что читатель уже знаком с такими понятиями, как вязкость, ламинарный поток и турбулентный поток, а также с характеристикой каждого из них и с применением безразмерного отношения (критерий или число Рейнольдса — Re) для характеристики условий истечения.
ОБЩИЕ УРАВНЕНИЯ ЭНЕРГИИ
Рис. 56 представляет в схематической форме секцию трубопровода, через которую протекает поток вещества. При всех наших рассуждениях будем принимать, что поток находится в установив
Общие уравнения энергии
367
шемся состоянии, т. е. будем считать, что весовое количество вещества (жидкости или газа), проходящего в единицу времени через любое сечение трубопровода, остается повсеместно одинаковым и со временем не изменяется. М представляет некоторое устройство, например насос или турбину, с помощью которого перемещающееся вещество производит работу над окружающей средой или же внешняя система производит работу над ним. Так
Рис. 56. Простая схема потока.
как такая работа обычно передается к системе или от системы посредством вала, то она для отличия от других видов работ часто именуется работой на валу; z представляет среднюю высоту вещества на каком-нибудь участке линии выше произвольно выбранного нулевого уровня.
Потенциальная и кинетическая энергия. Выше мы выразили первый закон посредством уравнения
ц—и.=а—а, (1)
где U—общая внутренняя энергия, характеризующая состояние системы,
Q—теплота, передаваемая системе от окружающей среды, А — работа, производимая системой над окружающей средой.
{7 включает только такие формы энергии, которые связаны с силами, действующими между атомами, молекулами, электронами и т. д., т. е. между элементарными частицами, а также кинетическую энергию, обусловленную их движением. Общий запас такой внутренней энергии однозначно определяется, если известны значения соответствующего числа обычных переменных состояния. С другой стороны, в случае движущейся системы имеются две формы энергии, которыми обычно пренебрегают во многих термодинамических выводах и которые следует принимать во внимание при полной формулировке закона
368
VIII. Термодинамика потока
сохранения энергии. Эти две формы по характеру совершенно отличны от форм энергии, входящих в величину U, так как они связаны с большими массами вещества, а не с первичными частицами. Этими макроскопическими формами энергии являются: 1) потенциальная энергия, обусловленная положением системы в поле действия силы тяжести, и 2) кинетическая энергия, обусловленная движением системы в целом. Само собой очевидно, что при переводе любой массы от какого-нибудь одного уровня к другому, более высокому, должна затрачиваться работа; наоборот, насколько можно судить,
изменение положения не изменяет состояния вещества, поэтому не изменяется и величина U. Тем не менее процесс связан с совершением работы, и так как трактовка силы и энергии требует, чтобы работа проявлялась как некоторая форма энергии, и наоборот, чтобы работа, получаемая при перемещении массы сверху вниз, поступала от некоторого источника энергии, то мы ввели новую форму энергии, назвав ее „потенциальной” энергией. В дальнейшем мы не будем подробно исследовать, каким образом накапливается или расходуется эта форма энергии. Количество энергии, накопленной в этой форме,
измеряется произведением веса системы на высоту по вертикали над любым произвольно выбранным уровнем.
Очевидно также, что движущаяся масса обладает кинетической энергией; эту энергию необходимо четко отличать от кинетической энергии, обусловленной движением отдельных частиц. Последней формой энергии обладают и системы, находящиеся в состоянии покоя;
она и определяется параметрами состояния и поэтому входит в величину U. Кинетическая же энергия движущейся массы возникает без какой-либо связи с переменными состояния и рассматривается как отдельная составляющая общей энергии; значение ее дается хорошо известным уравнением:
кинетическая энергия = у (масса) (скорость)2 = ,
(2)
где т — общая масса перемещающейся системы, и — скорость по отношению к неподвижному телу, например трубопроводу, по которому перемещается поток, g—ускорение силы тяжести ☆).
Средняя скорость. Вышеприведенная формула кинетической энергии строго правильна только для определенных идеальных условий. Для ясного понимания этого необходимо несколько отвлечься в сторону и более подробно рассмотреть, что означает член z/zzz2/2. Хорошо известно, что осевая составляющая линейной скорости движущегося вещества изменяется по поперечному сечению трубопровода. Напри-
☆ ) Обозначение g вводится вследствие того, что в уравнениях, подлежащих выводу, будет использоваться система единиц CGS. В этой системе единиц масса равна mjg, причем т является абсолютной, или инерциональной, массой.
Общие уравнения энергии
369
мер, при круглом поперечном сечении скорость потока у стенок равна нулю и увеличивается до максимума в центре, причем кривая зависимости скорости от расстояния, до центра изменяется в зависимости от режима потока и степени турбулентности. Среднюю линейную скорость обычно определяют уравнением:
UOV U=~S' где w — весовая скорость истечения, кг/сек, v — средний удельный объем вещества в рассматриваемой точке, м31кг,
S—площадь поперечного сечения канала, м2.
Средняя скорость, определяемая по уравнению (3), не дает средней кинетической энергии в уравнении Д 2). Для частного случая ламинарного истечения истинную среднюю кинетическую энергию можно получить суммированием мгновенных значений в каждой точке поперечного сечения. Так, в случае цилиндрической трубы кривая распределения скоростей может быть точно определена с помощью теории, и можно показать, что кинетическая энергия при определе-________нии и по уравнению (3), точнее выражается величиной таг)^, чем mu2i2g. При турбулентном потоке, который наиболее типичен для большинства случаев, имеется дополнительное усложнение, заключающееся в том, что вещество обладает не только кинетической энергией в направлении перемещения, но также и кинетической энергией, обусловленной завихрением и наличием потоков в направлении, перпендикулярном к основному движению. Поэтому уравнение (2) справедливо только для идеального (направленного) потока, не сопровождающегося изменением скорости по сечению канала. Рассмотрение действительного потока будет включать некоторые поправочные члены; однако последние почти полностью сокращаются в том случае, когда баланс энергии составляется между двумя сечениями, и общий результат получается почти такой же, как если бы уравнение (2) правильно воспроизводило кинетическую энергию в фактических условиях истечения. В дальнейшем изложении допускается, что среднюю кинетическую энергию любого перемещающегося вещества можно с достаточной точностью представить уравнением (2), если использовать среднюю линейную скорость, определяемую уравнением (3) :?).
В случае потока, связанного с передачей тепла, также вносится известная неопределенность, так как величина v ^уравнение (3)] изменяется по поперечному сечению; поэтому следует пользоваться некоторым средним удельным объемом.
й) Некоторые авторы предпочитают пользоваться более общим выражении2
нием: кинетическая энергия =---, где а зависит от режима потока,
(а=1,0 для ламинарного потока и приближается к 2,0 для турбулентного потока).
24 Химическая термодииа мика
370
VIII. Термодинамика потока
Общие уравнения энергии. Вновь возвращаясь к рассмотрению уравнения (1), мы встречаемся не только с формами энергии, входящими в U, но и с только что рассмотренными потенциальной и кинетической энергиями. Q представляет суммарный переход тепла между рассматриваемым трубопроводом и окружающей средой. Эта величина включает не только теплоту, сообщаемую или отводимую с помощью теплообменника, как показано на рис. 56, но и все потери тепла в самом трубопроводе. Работу А можно условно разделить на две части: 1) на работу, производимую потоком или над потоком при его входе или выходе из рассматриваемой секции трубопровода, выражаемую произведением pv, и 2) на работу, получаемую от машины М или подводимую к ней (работа на валу); последняя величина будет обозначаться через Ао и относиться к килограмму перемещающегося вещества. Так как в системе может быть больше одной машины, то Ао должна считаться алгебраической суммой всех работ на валу или, иначе говоря, общей работой на валу.
Применим теперь первый закон к секции трубопровода между сечениями 1 и 2 на рис. 56. Тогда получим уравнение
(л«з + + ^) — Gw + + ф = Q + и! — иг — А, (4) в котором каждый член отнесен к 1 кг вещества и все они выражены в одинаковых единицах энергии. Если U и Q выражены в термических единицах, то их следует умножить на J. Ло положительна, если М является турбиной или другим устройством, производящим работу, и отрицательна, если М представляет насос, р представляет абсолютное давление. Уравнению (4) также можно придать следующий вид:
и? — и?
^4 4- Zz zi~] 2^ =Q ^0' (5)
Его можно дать также в следующей диференциальной форме, целесообразной для некоторых приложений:
dt/~]-dz-[-pdv-[-vdp ~[-^^ = dQ— dA0. (6)
Теорема Бернулли. Во многих случаях машина, производящая внешнюю работу, отсутствует, и поэтому члены общего уравнения, включающие величины Q и U, будут ничтожно малы. Для этих случаев уравнение (4) можно написать в более простом виде;
/ . t “2 \ / । , и1 \
|*2+p2v2 + 2^; — J=0, (7)
включающем только члены, связанные с механической энергией. Это уравнение, обычно называемое уравнением Бернулли, было хорошо
Общие уравнения энергии
371
известно в гидродинамике задолго до установления первого закона. Оно основано на предположении, что поток лишен трения, поскольку всякое трение привело бы к некоторому переходу в теплоту других форм энергии, а это может быть учтено только членами &U и Q в общем уравнении. Кроме того, оно строго приложимо только к несжи-маемым веществам.
Баланс механической энергии. Для случая сжимаемого вещества, когда давления в двух сечениях различны, баланс энергии, представленный уравнением (7), приведет к неправильным результатам. Для некоторых приложений многие авторы предпочитают давать общее уравнение энергии (6) в следующем виде:
dz-\-vdpdFdA0 —О, (8)
или в интегральной форме:
г Гц2 —и2
д2 ----рА — 0- (9)
1 “s i
Член, характеризующий трение, выражается в виде суммы, поскольку часто желательно рассматривать трение отдельно. Уравнение (8) можно получить из уравнения (6) следующим путем:
По первому закону dQ = dU dA. Для обратимого процесса dA—pdv, и поэтому dQ = dU -\-pdv.
Подстановка этого уравнения в уравнение (6) дает уравнение (8) без члена dF. Для приложения уравнения к потоку в трубопроводе и к другим случаям, когда трением нельзя пренебрегать и поток не является обратимым, включается член dF. Целесообразнее всего рассматривать его как член, необходимый для составления уравнения баланса. Необходимость в нем легко показать при приложении уравнения (8) без члена dF к процессу дросселирования. Это приведет к следующему, явно неправильному результату
vdp — 0.
Поскольку член dF определяется лишь ориентировочно, уравнением (8) следует пользоваться с осторожностью, так как оно легко приводит к неправильному результату. Одной из трудностей, связанных с применением уравнения (8), является то обстоятельство, что для его интегрирования необходимо располагать точной зависимостью р — v. Но определенная зависимость между р и v в каждой точке предполагает обратимое изменение, а присутствие члена F указывает на необратимость процесса.
24*
372
VIII. Термодинамика потока
Уравнение, выраженное через напор потока. Так как каждый член в уравнении (4) должен быть выражен в одинаковых единицах, то ясно, что все они могут быть выражены в единицах потенциальной энергии, т. е. через вес, умноженный на высоту, или, если вес принимается равным единице, через высоту в метрах. Таким образом, U можно назвать напором, обусловленным внутренней энергией; z — потенциальным или гравитационным напором; u2j2g— скоростным напором, pv — статическим напором или напором давления, а Ао — напором, создаваемым насосом, который, в случае приложения к нагнетающей или всасывающей системе, называется „общим напором*. Так как напор, эквивалентный величине £7, не представляет ясного физического значения, то, повидимому, лучше ограничить применение термина „напор* механико-энергетическими членами. Напор давления представляет просто напор вертикального столба данного вещества. В случае потока жидкости в трубопроводе напор будет равен расстоянию по вертикали от оси трубопровода до высоты подъема жидкости в трубке с открытым концом, вставленной в трубопровод таким образом, чтобы открытая плоскость трубки была параллельна направлению потока. В случае газового потока напор обычно измеряется давлением, уравновешивающим столб жидкости с последующим пересчетом в эквивалентный напор газа. Эквивалентный напор любого вещества равен высоте его вертикального столба, который будет уравновешиваться данным столбом стандартного вещества. Имеется следующая простая зависимость
Ар = Лоро, (Ю)
где й0 и р0 соответственно равны напору и плотности стандартного вещества, ай— эквивалентному напору, выраженному через плотность другого вещества^ Так, эквивалентный напор водорода при 20°С и
м.
помощью трубки „Трубка Пито*),
причем он выражается через эквивалентный напор вещества, использованного в манометре. Интересно отметить, что для жидкости скоростной напор равен высоте, с которой она должна свободно падать под влиянием силы тяжести для приобретения данной скорости.
Теорему Бернулли, выраженную уравнением (7), теперь можно сформулировать следующим образом: общий напор перемещающегося вещества остается постоянным. Общий напор в этом случае представляет сумму гравитационного, статического и скоростного напоров. Конечно, при этом полностью пренебрегают трением вещества, поскольку, как уже было указано, в уравнении (7) пренебрегают членами Q и Д£Л
3 кгсм2 для напора воды в 150 мм будет равен t 0,150 X Ю00 X 848 X 293,2 „. „ „
h =----ЗХ1ОХШ6--------= 616’7
Скоростной (динамический) напор измеряется с Пито, которая будет рассмотрена ниже (см. раздел
Приложения к измерению скорости потока жидкости
373
Из определения статического напора hp и скоростного напора ha очевидно, что можно написать следующие простые уравнения: _
Р 0 ---45*
ИЛИ 1
р = (12) j
= ОЗ)
ИЛИ
« = Р2^. (14)
Пример 1. Чему будет равен напор масла с удельным весом 0,85, эквивалентный статическому давлению в 50 мм рт. ст.?
По уравнению (11),
50 X 1000 X 9,81
Й ~ 735 X 0,85 X Ю00
= 0,78 м.
Пример 2. Чему будет равен скоростной напор СО2 при 25°С и 0,7 кг/см2 манометрического давления, перемещающегося со средней линейной скоростью в 3 м!сек? Чему будет равен эквивалентный скоростной напор воды?
З2
По уравнению (13), hu = = 0,459 м СО2, рсо_ (принимая идеаль-
44Xl,7-10i
НЫЙ Га3> = 1X^48X29X2 =2’96 Кг<МЗ-
По уравнению (10), эквивалентный напор = = 0,00136 м Н2О.
ПРИЛОЖЕНИЯ К ИЗМЕРЕНИЮ СКОРОСТИ ПОТОКА ЖИДКОСТИ (ИЗМЕРИТЕЛИ НАПОРА)
Типы измерителей напора. Этот класс измерителей включает наиболее употребляемые и наиболее важные типы устройств, измеряющих поток вещества, а именно, диафрагму (насадку), трубу Вентури, трубку Пито и сопло. Общий принцип для всех случаев одинаков: поток с помощью соответствующего препятствия на линии заставляют давать поддающуюся наблюдению разность давлений, т. е. напор, который можно связать со скоростью потока. Отношение между скоростью потока и разностью напоров получается при применении одного из общих уравнений первого закона.
Действие как диафрагмы, так и трубы Вентури и сопла основано на местном сжатии перемещающегося вещества, вследствие чего мгновенно увеличивается его скорость. Получаемое таким образом увеличение кинетической энергии или скоростного напора компенсируется уменьшением статического напора, который можно измерить и затем связать со скоростью потока. Три упомянутых выше типа из-
374
VIII. Термодинамика потока
иерителей, схематично изображенные на рис. 57—59, отличаются друг от друга главным образом по характеру сжатия. Обычным типом диафрагмы является круглое отверстие в тонкой прямоугольной
О (2)
। I
। ।
Рис. 57. Диафрагмовый измеритель.
пластине, обладающее прямоугольной кромкой со стороны входа потока. Обычно диафрагму располагают в трубопроводе концентрически, помещая ее между фланцами. Диафрагму можно помещать также и сбоку или на дне сосуда, выпуская вещество во внешнюю среду. Истечение
через диафрагму вызывает падение давления, которое обычно измеряют с помощью диференциального манометра, обозначенного на рис. 57 через В. Статическое давление на противоположной стороне диафрагмы измеряется манометром С. В случае газов следует также измерять температуру веред диафрагмой.
*1
Приложения к измерению скорости потока жидкости
375
Рис 59. Сопловый измеритель.
Труба Вентури отличается от диафрагмы тем, что имеет постепенное сужение к участку меньшего диаметра, известному под названием горловины, за которым следует еще более постепенное увеличение поперечного сечения до размеров первоначального диаметра.
Сопло по существу является диафрагмой с постепенным сужением, как показано на рис. 59. Существует много типов насадок, но общий принцип их во всех случаях одинаков.
Трубка Пито, схематично изображенная на рис. 60, имеет отверстие статического давления В, плоскость которого параллельна направлению потока, и отверстие кинетического давления С, плоскость которого перпендикулярна направлению по-
тока. Отводы от обоих отверстий будут передавать статическое давление на плоскость А (различиями в подъеме для газов можно прене-бречь, а для жидкостей трубка Пито редко употребляется), но отвер-стие С будет также давать давление, соответствующее скоростному напору и2/2^, вследствие чего разность между этими двумя величинами— диференциальное давление — является давлением, обусловленным только скоростным (динамическим) напором. Другими словами, с помощью трубки Пито измеряется скоростной напор при определенном расположении трубки в потоке.
Истечение жидкостей через трубы Вентури, диафрагмы и сопла. Поскольку жидкость можно считать несжимаемым веществом, формулы истечения для жидкостей проще, чем для газов; поэтому мы сначала рассмотрим этот случай, а затем перейдем к более сложному случаю. Отправной точкой будет служить общее
уравнение (4). Пусть сечение 1 со- ^ис' Схема трубки Пито, ответствует давлению до диафрагмы, а сечение 2 — давлению после нее. Примем, что поток является адиабатным и лишен трения и что диафрагма или другое приспособление дифе-ренциального (разностного) давления не обязательно должно располагаться в горизонтальной трубе. Очевидно, что Ul = U2, поскольку состояние несжимаемой жидкости может изменяться только в результате изменения температуры, а это может быть результатом только перехода тепла или же превращения (посредством трения) некоторой части механической энергии в тепло. Обе эти возможности исключаются
376
VIII. Термодинамика потока
сделанными допущениями. Так как Q = 0 и Ло = О, то уравнение (4) превращается в
ц2 2
^'2 + ^1— Z2- О5)
Так как жидкость несжимаема, то
г?— и
- ^-' = —+^1—^2 = ^ (16)
ар = рг—р2 и известно как „разностное давление"; Д/z является разностным напором, выраженным в килограммах перемещающейся жидкости, или же разностью уровней жидкости в двух вертикальных трубках, вставленных в сечениях 1 и 2, как показано на рис. 61.
В подавляющем большинстве случаев г1=£,2 и ДА = Др/р.
Уравнение (16) можно превратить в
|/" «2 — «1 = V 2g\h.
(И)
Так как поток считается стационарным, то объем жидкости, проходящей в единицу времени через любые два сечения, должен быть оди
наковым; поэтому можно написать
= u2S2, (18)
где Sj и S2 — площади поперечного сечения 1 и 2. Это уравнение иногда называется уравнением непрерывности потока, так как мы при-
нимаем, что поток яв-
ляется стационарным, т. е. между двумя секциями нет увеличения или уменьшения потока, и что обе секции полностью заполнены жидкостью.
В случае остроугольной диафрагмы, отвод давления по течению струи располагается на некотором расстоянии ниже диафрагмы, и, строго говоря, площадь S2 должна считаться действительной площадью поперечного сечения потока в этой точке. Хорошо известно, что струя жидкости или газа, вытекающая из такой диафрагмы, сжимается после выхода из нее, достигает минимального поперечного сечения и затем расширяется в поперечнике. Точка, соответствующая минимальной площади, известна под названием „vena. contracta“ (точка сжатого сечения); отвод низкого давления обычно помещают как раз в этой точке. Площадь в этой точке измерить нелегко, и поэтому для практических целей площадь S2 принимается равной площади диафрагмы,
Приложения к измерению скорости потока жидкости
377
учитывая, что Svena contraeta~aSz (величину а можно назвать коэфи-циентом сжатия).
Поскольку для круглого трубопровода
S2
51
(19)
где £)j и D2 соответственно — диаметры трубопровода и диафрагмы или горловины,
Ш’=(§)*=?*> »2о * * 23>
тогда уравнению (17) можно придать следующий вид:
"2 1/1 — (“f]2 = V2gM = V2gb(pv), (21) г \**2/
ИЛИ
(22)
Величина V 1 — j4 iih or да называется- фа ктором пр и б л иже н ия.
Конечно, в действительности при измерении интересуются не ли-
нейной скоростью потока, а объемной или весовой скоростью его. Мы можем записать
W
fS,
___q
~ S,
(23)
р дается без индекса, поскольку для несжимаемого вещества нет различия между этой величиной в сечениях 1 и 2. Тогда уравнение (22) можно записать так:
4 = s2 - (24)
ИЛИ
w = ^2? pZTJT ’ (25)
/W- <26>
Коэфициент расхода. Сравнение величин, найденных посредством этих уравнений, с результатами опыта показывает, что они отвечают имеющемуся изменению объемной скорости истечения (пропорционально квадратному корню разностного напора), но числовые значения в общем не совпадают с уравнением. Главные причины этого следующие: 1) поток не полностью лишен трения; 2) происходит сжатие струи, которое особенно резко сказывается в случае остроугольной диафрагмы. Отклонения от теории вследствие этих или каких-либо дру-
378
V///. Термодинамика потока
гих причин обычно заставляют вводить коэфициент й-), который будет называться коэфициентом расхода. Коэфициент расхода определяется уравнением
С _ действительная скорость истечения _
теоретическая скорость истечения '
и представляет, конечно, безразмерную величину. Скорость истечения можно выразить через вес или объем; в последнем случае оба объема должны относиться к одинаковым условиям, особенно если перемещающимся веществом является газ. (Следует отметить, что значение С, естественно, зависит от теоретического уравнения, использованного для вычисления потока.) При введении коэфициента расхода уравнение (24) переходит в
= (28)
то же самое имеем для уравнения (25).
Иногда пользуются другим коэфициентом, определяемым уравнением
q^CV^h, (29)
в котором
Этот коэфициент иногда называется коэфициентом расхода с учетом фактора приближения. Если представляет незначительную ве-
личину, то р4 в уравнении (28) будет ничтожно малым, и ему можно придать форму уравнения (29). Это равносильно пренебрежению скоростью «j в уравнении (17).
Пример 3. Чему должно быть равно максимально допустимое значение Ds/Dl для того, чтобы при определении объемной скорости истечения пренебрежение величиной 1 — р* не приводило к ошибке более 1°/0? Принимаем турбулентный режим потока, благодаря чему коэфипиент расхода можно считать не зависящим от отношения диаметров.
Пусть qt — истинная скорость потока, даваемая уравнением (28), a qa—приближенная скорость, даваемая уравнением (29) при замене С' на С:
^==0,01, 4t
или
1 — —0,01.
й) Некоторые пользуются двумя коэфициентами, одни из которых учитывает сжатие, а другой — прочие отклонения от теории. Мы будем следовать более простой и обычной практике, объединяя их в одну величину.
Приложения к измерению скорости потока жидкости
379
По уравнениям (28) и Следовательно,
или
(29) — V1 — ?*.
—2 =0,0199,
Wl/
% = 0,376.
Таким образом, мы имеем следующее правило: если диаметр сопла составляет 3/8 (или меньше) диаметра трубы, то пренебрежение величиной 1 — [И будет давать ошибку не более 1°/о-
{(оэфициент расхода для жидкостей. Числовое значение коэфициента С не зависит от единиц измерения при условии использования согласованных единиц. Анализ зависимости С от различных величин приводит к выводу, что этот коэфициент зависит от следующих факторон:
2. Размеры измерителя.
3. Скорость потока.
4. Свойства вещества, в особенности его плотность и вязкость.
5. Форма измерителя.
Размеры могут определяться величиной Dx или D2; скорость потока определяется и, q или w. Форма измерителя характеризуется отношениями различных линейных размеров к какому-нибудь одному размеру, например к D1. Наиболее важными факторами, характеризующими измеритель, являются:
1. Длина подающей и отводящей трубок.
2. Диаметр диафрагмы или горловины.
3. Толщина пластинки диафрагмы.
4. Расстояние до места измерения давления.
5. Диаметр отверстия для отвода давления.
6. Шероховатость стенок трубопровода.
Факторы 1, 3, 4 и 5 обычно для данного типа измерителя делаются постоянными с помощью некоторых конструктивных деталей.
Дальнейший анализ вопроса показывает, что коэфициент для измерителя, сконструированного в соответствии с некоторыми стандартными условиями, является функцией и, р, р, D2, Dr и [J. Приложение анализа размерностей приводит к зависимости
C = v(Re, р, DJ. (31)
Число Рейнольдса обычно определяется условиями, существующими в устройстве сужения, т. е диафрагмы или горловины, причем плотность и вязкость вещества в этой точке принимаются одинаковыми
380
VIII. Термодинамика потока
с этими же величинами в сечении перед диафрагмой. Для установления точной природы этой функциональной зависимости было проведено много экспериментов, причем результаты были представлены как в графической, так и в аналитической форме. Здесь будут упомянуты лишь некоторые из существенных фактов; для дальнейших деталей рекомендуется обратиться к сообщению Комитета по исследованию измерителей потоков Американского общества инженеров-механиков [80].
В случае измерителей Вентури в ’ пределах, встречающихся на практике, С обычно считают не зависящим от f и £),, а зависящим только от числа Рейнольдса. В области вязкого истечения С быстро изменяется с Re, однако значения нельзя считать точно установленными. В турбулентной области С асимптотически увеличивается с Re приблизительно от 0,90 примерно до 0,99 при очень высоких значениях Re. Для большинства случаев, невидимому, хорошей средней величиной для использования будет 0,98.
Для остроугольных диафрагм и турбулентного потока С совершенно не зависит от величин Re, и О,; в большинстве случаев среднее значение 0,605 будет давать точность в пределах 1—2°/0 при условии тщательного изготовления диафрагмы и установки ее в соответствии со стандартными требованиями. Предельными значениями С для изменений Re от 30 000 до 600 000, от 0,1 до 0,8 и D\ от 50 до 350 мм являются 0,594 и 0,620. Те немногие данные, которые имеются для ламинарной области, показывают, что С значительно изменяется с Re и и поэтому его значение нельзя точно предсказать.
Из этого можно заключить, что коэфициент расхода для остроугольной диафрагмы можно точно предсказать, благодаря чему калибровка ее становится не обязательной. Отказ от калибровки требует тщательной установки диафрагмы в соответствии с определенными условиями. Главными требованиями являются правильная круглая форма диафрагмы, тщательное центрирование ее в трубопроводе при достаточно длинном прямом отрезке его до и после диафрагмы и установление в определенных точках кранов для измерения давления. Для вышеуказанных значений коэфициентов отводы давлений должны находиться или 1) во фланцевом соединении, или 2) в соединении горловины.
В первом случае отверстия для отводов статического давления высверливаются во фланцах, поддерживающих пластинку диафрагмы, причем центры этих отверстий должны находиться примерно на расстоянии 25 мм от наружной поверхности пластинки. Во втором случае кран для давления выше горловины должен быть расположен от диафрагмы на расстоянии диаметра трубы, а кран ниже горловины в „vena contract а“, которая находится от диафрагмы примерно на расстоянии 0,5 диаметра трубы. Оба эти варианта установки дают практически одинаковый результат.
Определение разностного давления. Применение только что выведенных уравнений требует краткого анализа того, каким образом
Приложения к измерению скорости потока жидкости
381
вычисляется разностное давление (напор). Например, величину Ай в уравнении (28) обычно не отсчитывают по манометру разностного давления на измерителе, а получают расчетом. На рис. 62 разностное давление между сечениями / и 2 измеряется манометром, наполненным жидкостью большего удельного веса, чем перемещающееся вещество. Предполагают, что отводы давления над манометрической жидкостью заполнены веществом, перемещающимся в трубопроводе. Составляя баланс статического давления при равновесии, достигаемом на низшем уровне манометрической жидкости, имеем
Pi + А1?/ + ^g?f = ^g + А1Р/ + Рг>
ИЛИ
Pl— p2=bp=Mg(?g—
и
Др [ \
Л=АЙ = Дй Н— 1 , (32)
?f е \?f /
Дй— разностный напор (разность уровней перемещающегося вещества), — плотность манометрической жидкости, а р?—плотность переме-
Остаточное давление. Остроугольная диафрагма представляет гораздо более простое и дешевое устройство, чем труба Вентури,
но последняя обладает важным преимуществом, заключающимся в том, что вызываемое ею постоянное падение давления много меньше, чем в случае диафрагмы. В последней резкое увеличение сечения от сечения диафрагмы к сечению трубопровода приводит к рассеиванию большого количества кинетической энергии вследствие ее превращения в тепло, тогда как в трубе Вентури постепенное возрастание поперечного сечения (от горловины) вызывает превращение кинетической
с. 62. Диферепциальный манометр.
энергии в механическую (изобра-
жаемую членом pv). Величина потери первоначального давления потока зависит от многих обстоятельств, но в хорошо изготовленной трубе Вентури при нормальных условиях остается примерно от 80 до 9О°/о считанного разностного давления, тогда как в диафрагме, в зависимости от отношения диаметра диафрагмы к диаметру трубы, остается только от 5 до 5О°;о. Постоянные потери напора для сопел являются промежуточными между потерями в трубе Вентури и в остроугольной диафрагме.
382
VIII. Термодинамика потока
Расход из хранилища при переменном напоре. В случае хранилища, из которого жидкость через диафрагму, расположенную на его боковой поверхности нли на дне, поступает в воздух, можно написать общее уравнение (24) в виде
dV t.dI ,-----
cI=^=^CV2£h > / (33)
где h — напор выше центра диафрагмы в данный момент, a dV/db— скорость истечения в тот же момент. Теперь, поскольку
dV=Sdh, где 5—среднее поперечное сечение сосуда, уравнение (33) переходит в
dh kdI , z--------
= • (34)
Принимая 5 постоянным, получаем
где
К=------45-----•
Интегрирование уравнения (35) в пределах от ht при 6 = 0 до А2 при 0 = 0 дает
Л? — Й2/з = 4 9- (36)
Нижний предел h следует выбирать при некотором значении больше нуля, так как, во-первых, уравнение диафрагмы перестает быть справедливым при очень малых напорах; во-вторых, имеются трудности, обусловленные образованием завихрений и влиянием поверхностного натяжения.
Поскольку У = 5 (7ц—А2), уравнение (36) переходит в
V=-------4----9-----325-
Уравнение (36) также можно написать в виде
(VTh+Vhz\
* 4 \ 2 ]
или
r^CV^ghYp
(37)
(38)
Измерение газового потока
383
где ___
^ = № + 2/м2 + А2)/4-
Помимо приложения к истечению вещества, это уравнение интересно тем, что оно дает некоторую среднюю величину, отличающуюся от арифметической и логарифмической средней.
Значение С для диафрагмы, выпускающей жидкость в пространство, наполненное газом, не так хорошо установлено, как для случая погруженной диафрагмы, но для остроугольной диафрагмы в тонкой пластинке хорошим приближением будет С = 0,60. Для короткой трубки, действующей подобно диафрагме (отношение длины к диаметру около 3), С равно приблизительно 0,80 [25].
ИЗМЕРЕНИЕ ГАЗОВОГО ПОТОКА
Общие теоретические уравнения. Здесь приложимы те же самые общие принципы, что и в случае измерения потока жидкостей, но имеется несколько важных отличий, требующих учета. Это обстоятельство вытекает из того, что вещество в данном случае является сжи-маемым и при изменении условий быстро изменяет свой объем. Сле-довательно, для фиксирования плотности следует определить давление и температуру перемещающегося вещества (в случае влажных паров должна быть известна и сухость пара); кроме того, если диференци-альный напор составляет значительную долю абсолютного давления, то будет наблюдаться изменение плотности в самом измерительном приборе, что следует учесть для получения точных результатов.
Возвращаясь к общему уравнению (4), основанному на первом законе термодинамики, мы попрежнему принимаем адиабатный режим истечения, и тогда, конечно, Ло = О. Можно также принять г1=г2, поскольку для газа разность весового напора почти всегда очень мала. С этими допущениями уравнение (4) переходит в следующее *):
«2—
> = + —— (39)
Как и раньше, индексы 1 и 2 относятся к сечениям, в которых расположены краны для отвода давления с уже отмеченным исключением для случая остроугольной диафрагмы, когда S2 относится к площади самого измерителя.
Теперь, так как скорость вещества постоянна, то
w = WjSjPj = w252p2, (40)
или
ui Рг fQz\2 Е?
SiPi \Dj Р1-
й) / в этом случае вводится во избежание недоразумений при последующем рассмотрении.
*
384
VIII. Термодинамика потока
Подставляя
уравнение (41) в (39) и перегруппировывая, получаем
2gJ±H — В4 (
\Р1/
(42)<г)
Если D2ID1 мало (0,4 или меньше), то уравнение (42) переходит в
zz2 = (43)53
Теоретические объем или весовой расход" получаются сочетанием уравнения (42) с уравнениями
Я 2 = ^2^2’ или
® = и2<$, р2.
Следовательно, уравнением массы потока будет
® = S2p2|/—(44)
\?1 /
При употреблении этого уравнения можно прямо прочесть по таблице термодинамических свойств, а Н2 получить таким же способом после допущения, что вещество изоэнтропно расширяется от рг ДО рг.
Частный случай идеального газа. Если перемещающимся веществом является идеальный газ, то {дН1др)г = 0, и
<45>
но так как для изоэнтропного расширения идеального газа
k—i
/„ \ k
'2 = Т1 км
2 1 \Р1/
то
1
__ kR7\ Г. (р-Л
Л4(Л-1)1 \pj
Подстановка уравнения (46) в уравнение (43) дает
(23, гл. VII)
(46)
(47)
а
Л) Отметим, что в этом выводе ^Н=НХ — Н2 и отнесено к 1 кг.
Измерение газового потока
385
или поскольку
RT\ =
МрхУх J
то, если R выражено в термических единицах, уравнение (47) можно
превратить в
(48)
или если нельзя пренебречь фактором приближения, то
А —1
— (Т'г/Л) k ]
(49)
Уравнение весового расхода, соответствующее уравнению (49), при использовании того, что
= P2v2 (15, гл. VII)
и, следовательно,
£ ра, __ I Pz \ & t __________________________Fl /--------------------------- переходит в уравнение
w = 1)К'2~Г, (50)
1 — (гР
(51)
1 _ Р«(Г)Г
где r = p2jp1. Уравнения (50) и (51) дают теоретическую скорость истечения; чтобы найти действительную скорость, надо, как и в случае потока жидкости, ввести эмпирические коэфициенты. Специальный коэфициент, предназначенный для использования с этими уравнениями, будет обозначаться через Са и будет называться адиабатным коэфи-циентом расхода. Опыт показывает, что для измерителей Вентури, сопел, круглых диафрагм и других устройств, в которых минимальное поперечное сечение струи вещества ограничено определенным каналом, Са является фактически постоянным, и его значение для большинства случаев близко к единице. Однако это не верно для остроугольного сопла, для которого Са значительно изменяется с отношением давления г.
Приведение к простым формулам гидравлики. Пусть р2 =рх — &р, и поэтому p2/pl=i—(Ap/pi). Разлагая в ряд, получим
а—1
Л Д/A * _______. k~ 1Д», k —
к Рх ) * й* 2А V kj{PJ
25 Химическая термодинамика
386
VIII. Термодинамика потока
В пределе, когда Др—>0, это уравнение приводится к виду
!k—\bp Л Pi ’
k—i
и поэтому член 1—(Pa'Pi) * в уравнении (48) при очень малом падении давления Др превращается в
k — 1 Др рх '
Подстановка последней величины в уравнение (48) дает
и2 = V^gv^p (52)
илн при введении поправки на скорость приближения и при опускании индекса v, поскольку плотность в этом предельном случае не изменяется, имеем
наконец, так как w = usS2^, то
(54)
Уравнения (52) — (54) совпадают с уравнениями, уже выведенными для жидкостей. Поскольку они основаны на допущении, что Др представляет очень малую величину и поэтому изменение плотности газа практически будет ничтожно мало, их можно вывести непосредственно из теоремы Бернулли, как это и было сделано для уравнения (22) &). Это означает принятие допущения, что плотность газа не изменяется, т. е. что газ ведет себя практически как жидкость. Кроме того, так как уравнение (52) или (54) строго правильно только, когда Др—>0 (и поэтому плотность постоянна), то с теоретической точки зрения будет несущественно, берут ли для v удельный объем до диафрагмы, или после нее, или же пользуются средней величиной. Использование последней, как будет показано ниже, имеет практические преимуще-
<г) Возможен также и другой вывод:
dH = 4- TdS. (89, гл. III)
Для изоэнтропного расширения
(если можно пренебречь изменением объема). Подстановка в уравнение (43) дает уравнение (52). Отметим, что уравнение (52) не включает допущения об идеальности газа.
Измерение газового потока
387
ства. Так как в большинстве случаев будет наблюдаться небольшое, но существенное различие между этими тремя значениями плотности, то эмпирический коэфициент расхода С, который следует применять с уравнением (54), будет зависеть от выбранной плотности. Указывая величину С, всегда следует отмечать, какое именно значение плотности было использовано.
Если сохранить член, содержащий ( — I , так же как и член в первой степени для расширения, то получится уравнение, являющееся несколько более простым, чем уравнение (48), но все же достаточноточным до тех пор, пока р21рг больше 0,75.
Коэфициент расширения. Общему уравнению (51) иногда придают форму простого гидравлического уравнения:
™ = j/' <55>
где У]— коэфициент расширения, основанный на плотности вещества до диафрагмы, величина которого в соответствии с уравнениями (51) и (55) должна быть равной _______
2 k — 1
(W-1)10-)* (!-(/-) * ](1-р<) . (56)
[1-Р(г)*](1-г)
Если меньше 0,35, то член, включающий это отношение, можно опустить, причем это ие вызывает изменения значения Y больше чем на 1°/0. Коэфициент расширения также может основываться иа использовании в уравнении (55) плотности после диафрагмы или средней плотности. Легко показать, что между этими различными коэфи-
циентами имеются следующие соотношения:
У2 (коэфициент, основанный на р2) =
(57)
и
Ym (коэфициент, основанный на средней плотности) = У]
/ТТ7- (58>
Коэфициент расширения зависит главным образом 1) от отношения теплоемкостей k, 2) от отношения диаметров р и 3) от отношения давлений г. Подробный анализ показывает, что коэфициент расширения можно выразить как функцию двух безразмерных отношений Aplkpt (иногда называемую акустическим отношением) и f), что подтверждается опытом [231].
Уравнения для практического применения. Для учета влияния трения и других отклонений от идеальных условий, принятых при выводе уравнений для газового потока, необходимо также включать в них коэфициент расхода, который должен определяться опыт-25*
388
VIII. Т ермодинамика потока
ним путем. Мы уже определили адиабатный коэфициент расхода Са для использования его в уравнении (51). Уравнение (55) можно написать, 'как
(59)
где Со — гидравлический коэфициент расхода, т. е. коэфициент, который измеряется опытным путем, когда Уг приближается к 1,0. Это уравнение можно считать универсальным для любого случая протекания через измеритель напора. Оно включает два эмпирических коэфициента— Со и которые обычно связываются с условиями исте-
чения посредством опытных данных. *Их лучше рассматривать раздельно, поскольку они зависят от разных величин. Со, как мы уже видели, зависит от Du Re и тогда как Kj зависит от Ap/pjfe и
Уравнение (59) можно дать в более удобной форме:
w= 0,000348 У (60)
где w — скорость истечения, кг) сек;
D2 — диаметр сопла или горловины, мм;
Ар— разностное давление, кг) см2;
р — плотность потока выше сопла, кг)м .
Так как Со представляет функцию не отдельных свойств вещества, а числа Рейнольдса, то оно будет иметь значения, уже приведенные для случая жидкостей, независимо от того, какое вещество является объектом измерения. Во многих случаях хорошей средней величиной Со для прямоугольной диафрагмы будет 0,605.
Значения для труб Вентури, сопел и круглых диафрагм можно с достаточной точностью вычислить по уравнению (56), но значения для остроугольных диафрагм должны определяться опытным путем. Это различие вызвано тем, что расширение газа в остроугольной диафрагме представляет собой более сложное явление, чем расширение в трубе Вентури или в сопле, вследствие искривления потока, вызываемого диафрагмой. Для газа имеется <tvena contracta-ь, так же как и для жидкостей, но тогда как для жидкостей отношение площади струи в точке наибольшего сжатия к площади диафрагмы практически постоянно, для газов оно, без сомнения, переменно и зависит от отношения давлений и от других факторов.
Рис. 63 представляет в графической форме значения Ylt вычисленные из уравнения (56) для труб Вентури, сопел и круглых диафрагм, как функции k, г и [L Для случая прямоугольных диафрагм, прикрепленных к горловине, Букингем [34] нашел, что в пределах ^ = 0,20— 0,75 применимо следующее уравнение:
#(0,41 +0,35^). (61)
Измерение газового потока
389
В результате широко поставленных испытаний по истечению воздуха через остроугольные диафрагмы сотрудники Бюро стандартов США [9] нашли, что использование в простом гидравлическом уравнении средней плотности (плотность при температуре до диафрагмы и при среднеарифметическом давления до и после нее) приводит к коэфициенту расхода, отличающемуся только на несколько процентов от коэфициента
Рис. 63. Коэфициент расширения для измерителей Вентури, патрубков и круглых сопел [80J.
расхода для воды во всем интервале от г= 1 до г-0,50. Этот коэфициент определяется уравнением
и» = 0,000348 Ст£>| )/~
(62)
Рис. 64, взятый из вышеупомянутой статьи, показывает измене-нения Ст, Са и Со с изменением г (основанные как на плотности до диафрагмы, так и на плотности после диафрагмы) для случая, когда —>-0 *). Естественно, что все три коэфициента приближаются к одинаковому значению — в данном случае 0,597,'—когда г—>1,0. Инте-
i?) Для этого случая коэфициент расхода не зависит от местоположения кранов для измерения давления. Для больших значений f>, Ст также почти не зависит от г, ио уже зависит от места расположения кранов.
390
VIII. Термодинамика потока
ресно отметить, что Са изменяется даже больше, чем Со. Повидимому, уравнение (62) с Ст, равным 0,600, можно использовать в качестве универсального уравнения для всех веществ, перемещающихся через остроугольные диафрагмы с кранами в горловине при условии, что допустимая погрешность не превышает 2°/0. Для газов следовало бы также ограничиться случаями, при которых отношение давлений больше критического значения. Молстед и Варга [173] дали номограмму для упрощения решения этого уравнения.
Так как для случая идеального газа
, ___MiPi+Pz)
?т~ 2RTy
то уравнение (62) можно также дать в форме
w = 0,037336CmZ)2 l/..fL,
(63)
если Ту выражено в градусах Кельвина, а остальные единицы те же, •что и в уравнении (60).
Максимальный расход и критическое отношение. Выведенные
• в предыдущих разделах урав-
Рис. 64. Изменения коэфициентов расхода для остроугольного сопла в зависимости от г при ₽—>-0 [9].
или w. Исследование этого уравнения
нения газового потока справедливы только тогда, когда отношение давлений больше определенной критической величины, к определению которой мы сейчас перейдем.
Для данного газа н данных условий истечения и для случая, когда скоростью приближения можно пренебречь (знаменатель под корнем превращается в единицу), уравнение (51) можно написать в следующей упрощенной форме:
1 *+1
w2 = ./iC[r*—г * ],(64)
где —константа, считающаяся независимой от г показывает, что w = 0 как
при ру=р2(г— 1), так и прн р2 = 0(г=0), т. е. при выпуске газа в вакуум. Исследуем теперь, как изменяется w при уменьшении г от предельного значения 1,0. Естественно, что w будет сначала
Измерение газового потока
391
возрастать, но не следует ожидать, что это будет продолжаться безгранично. Действительно, оказалось, что w возрастает до максимума в тот момент, когда достигается определенное значение г, называемое критическим отношением и обозначаемое через гкр. Это обстоятель- ___________
ство можно предсказать по уравнению, пользуясь обычными методами диференциального исчисления.
Диференцирование уравнения (64) дает
2-wd.w = K 14 г * — • (65)
dr Lk k J
Принимая dm] dr = О и решая относительно г, получаем
, 2 \ —
• <66>
Подстановка этой величины в уравнение (48) и исключение p±v± с помощью уравнения адиабатного расширения в форме
i-fe
------------------------Pi^i=P2‘V2 (г) -------------------------(67)-------- дает
и,, . = 1^%kp,v-. (68)
2(макс.) г г j 2 \ /
Это является хорошо известным соотношением для скорости распространения звука в идеальном газе. Получение такого результата совершенно закономерно; весьма существенно то обстоятельство, что это можно так просто предсказать с помощью термодинамики. Это следовало ожидать, поскольку скорость звука представляет ту максимальную скорость, с которой действие давления может распространяться через газ. При понижении давления после диафрагмы или сопла скорость в горловине возрастает до тех пор, пока не будет достигнуто критическое давление, при котором скорость в горловине будет равна скорости звука. Дальнейшее снижение давления в камере после диафрагмы не будет оказывать влияния на давление в горловине, где будет сохраняться критическое значение. (Одтако отметим, что в расширяющихся соплах могут быть созданы скорости, превышающие скорость звука.)
Критические отношения давлений, вычисленные по уравнению (66) при 15° С, равны 0,49 для одноатомных газов, 0,53 — для двухатомных газов и несколько выше—для газов с большим числом атомов , в молекуле. Для умеренно перегретого водяного пара гкр можно при-нять равным 0,55, а для насыщенного водяного пара — равным 0,58. ?
Если г всегда принимать равным отношению давления в горловине
к давлению перед диафрагмой, то уравнение (51) и связанные с ним « уравнения будут давать правильные результаты. Однако принято счи- ’ тать, что при расширении, происходящем от данного давления рг перед j
392
VIII. Термодинамика потока
диафрагмой до конечного давления р2 после диафрагмы, использование этого уравнения приведет к ошибкам, если p2/Pi будет меньше гкр. Следовательно, лучше применять уравнение (51) только тогда, когда р21Р1 равно или больше критического значения. Если р2'рг меньше критического значения, то в уравнение (51) вместо г подставляют критическое значение [уравнение (66)]; пренебрегая поправочным членом для скорости приближения и вводя коэфициент расхода, получают следующее уравнение
w=сА gkpi p1 (гп) ’ (69)
EJttlJ TXT'V ПТЛТТОСТ п Л IlVUttiU 1UO
Г> с ,/ ( 2 V+l (7m
у (70)
Уравнениями (69) и (70) следует пользоваться во всех случаях, когда р2 (измеренное или предположенное) меньше rKppv Следует отметить, что критические явления наблюдаются в круглых диафрагмах и соплах, но не в остроугольных диафрагмах. Для последнего типа диафрагм поток продолжает возрастать, когда отношение давлений уменьшается ниже значения, даваемого уравнением (66). Однако было проведено очень мало работ в этих условиях с прямоугольными диафрагмами; поэтому можно сделать только некоторые общие замечания.
Неидеальные газы. Все уравнения, начиная с уравнения (44), основываются на допущении идеальности газов. При значительных отклонениях от идеальности считают, что все же можно получить по этим уравнениям достаточно точные результаты, если для pj (= I/t^) пользоваться действительной плотностью. Это, конечно, является тем случаем, когда Др представляет малую долю рх, и можно применять простую гидравлическую формулу (54). Точность такого метода вызывает некоторые сомнения, если коэфициент расширения Y значительно отклоняется от единицы, но и тогда он может служить первым приближением. Методы вычисления плотностей газов, которые нельзя считать идеальными, были изложены в гл. V.
Влияние изменений состояния на измерение газового потока. Для анализа этого вопэоса примем, что простое гидравлическое уравнение можно применять и с плотностью до диафрагмы, как в уравнении (59) с У, = 1,00, и с средней плотностью, как в уравнении (62). Для данного измерительного устройства (диафрагма, труба Вентури или сопло) можно написать
™-=СмУ^р, (71)
где См — константа измерителя, которую можно сразу определить для всех случаев и которая в пределах, обычно встречающихся во всех практических случаях, не зависит от величин w, &р или р.
Измерение газового потока
393
См определяется или калибровкой, или, что более распространено, расчетом по известным размерам и известному значению Со при средних условиях для измерительного устройства. Если пользоваться уравнением (59), то
(72)
Поток рассчитывается с помощью уравнения (71) по считанным или записанным значениям Др и по значению р, полученному из наблюденных давления и температуры при условии, что свойства измеряемого газа известны. Для измерения непосредственным отсчетом
а> = С'мУГ^р,
(73)
и поскольку См включает плотность, отсчеты измерителя правильны только для одного состояния газа, а для других состояний следует применять поправочный множитель. Рассмотрим теперь, какие поправки легче всего произвести.
Плотность газа может изменяться в результате изменения: 1) температуры, 2) давления, 3) состава сухого газа и 4) влагосодержания. При принятии закона идеального газа
Если р и t — абсолютное давление и температура в градусах Цельсия перед диафрагмой и если MD — молекулярный вес сухого газа, а х — мольная доля водяного пара, то
__р\Мц (1—х) -I- 18х] /? (t +273,2)
(74)
а См для данных условий потока перед диафрагмой можно вычислить из уравнений (72) и (74).
Пусть индекс 0 представляет условия потока перед диафрагмой, для которых известна константа См, и пусть символы без индекса представляют ряд других условий. Применяя для упрощения записи уравнения абсолютные давление и температуру и молекулярный вес влажного газа, получим
Если w0 — вес газа, протекающего через данный измеритель при плотности р0, тогда но — вес газа для того же самого измерителя, но для других условий, будет дан уравнением
(76)
394
VIII. Термодинамика потока
или
«' = «'о/РХ/гХЛи-
(77)
Чтобы вносить поправки с наименьшими затруднениями, эти поправочные множители можно нанести на график или свести в таблицу для всего диапазона изменений, который может встретиться в любом данном случае.
Может оказаться необходимым выразить поток через объем, а не через вес. При использовании индекса s для стандартных условий р и Т, к которым относится объем, получаем
Подстановка этих величин в уравнение (71) дает
(78)
Для данного измерителя потока заданного режима перед измерителем и данного стандартного состояния это уравнение превращается в
qs= СмУ^р.
(79)
Для данного ряда условий См, так же как и С'м, можно определять или непосредственной калибровкой, или расчетом. Если состояние газа после диафрагмы или трубы Вентури изменяется от р0, Тй и Л40, для которого применяется коэфициент С"м, то по уравнению (78) имеем:
(80)
Отметим, что поправочный множитель для состава представляет обратную величину множителя, употребляемого в случае поправки для весового потока.
Следует отметить, что если измеряемый газ является влажным или сухим насыщенным паром, то он при выбранных стандартных условиях может не целиком быть газом. Другими словами, если газ действительно привести к стандартному состоянию, то может произойти частичная или полная его конденсация. Обычно этим пренебрегают и пользуются фиктивным стандартным объемом, основанным на допущении, что конденсации не происходит.
В лабораториях диафрагмы часто калибрируются посредством измерения расходуемого объема для данного ряда условий и нанесения объема на график в зависимости от квадратного корня из показания манометра или непосредственно в зависимости от отсчета по манометру на логарифмической бумаге. Затем объем, считанный с графика, еле-
Трубка Пито
395
дует корректировать для изменений давления, температуры или удельного веса по их значениям, которые поддерживались постоянными во время калибровки. Лучшим методом является следующий: уравнение (71) можно написать в виде
а — С 1
Ч — |/ рМ ,
(81)
где q— объем при условиях данной диафрагмы, См — константа измерителя, a Rm — отсчет по манометру разностного давления. По указанию Уайтвелла [257], удобно наносить qVpMjT в зависимости от так как при этом естественно учитываются поправки на изменяющиеся условия.
ТРУБКА ПИТО
С теоретической точки зрения трубка Пито представляет наиболее простой измеритель напора, но так как ее принцип действия значительно отличается от других типов измерителей потока, мы рассмотрим ее отдельно.--------------------------------------------
Общее уравнение для трубки Пито получается из уравнения первого закона термодинамики, если принять Q и Л0 = О. В этом случае индекс 1 относится к условиям ненарушенного потока вблизи ударной трубки, т. е. трубки, изогнутой под прямым углом и имеющей отверстие, расположенное навстречу течению, а индекс 2 относится к условиям выхода из этой трубки; и] имеет особое значение в том смысле, что не является средней скоростью всего потока, а только средней скоростью вещества в малом цилиндре, прегражденном ударной трубкой. В пределе, когда диаметр ударного отверстия приближается к нулю, скорость будет равна скорости в данной точке. Так как в этом случае в ударной трубке нет потока, то а2 = 0, и расчетное уравнение переходит в
uj = / 2g(A/74-Az). (82)
За исключением редких случаев очень большой скорости, сжатие, получающееся от удара, будет ничтожно малб, даже в случае газового потока, и поэтому уравнение (82) принимает вид
«1= + (83)
Если же пренебречь разностью весового напора, то получим
“1 = }/ 2gy - yf 4gMi. (84)
Это уравнение обычно дается для трубки Пито. Как видно из уравнения, трубка Пито, по существу является приспособлением для изме
396
VIII. Термодинамика потока
рения скоростного напора при заданной небольшой площади поперечного сечения потока. Если ввести коэфициент для учета отклонений от теории вследствие нарушения потока, вызываемого самой трубкой, арматурой, поворотами на линии или какими-либо другими причинами, то получим
а = Ct V~2gbh, (85)
или если скоростной напор измеряется напором вещества в манометре, имеющего плотность р^, то, по уравнению (32),
“ = Ct У2^Дй/^-1)’ (86>
или для вещества, перемещающегося в круглой трубке,
= ct У, (87)
где D — диаметр трубы.
Коэфициент Ct будет в некоторой степени зависеть от конструкции трубки, но для надлежащим образом изготовленной трубки он обычно принимается близким к единице.
Было отмечено, что трубка Пито измеряет скорость для небольшого элемента площади поперечного сечения. Для определения массы или объема потока для канала в целом обычно пользуются двумя способами: 1) помещают трубку ударного давления в центре трубы, где скорость является максимальной, и из этой максимальной скорости («макс.) получают среднюю скорость (аср.) по ранее определенной зависимости между ними; 2) пользуются подвижной ударной трубкой и измеряют скорость во многих точках поперек диаметра, соответствующих одинаковым площадям, и берут среднюю арифметическую отдельных скоростей.
Анализ размерностей показал, что отношение «ср./«макс. должно быть функцией числа Рейнольдса Du. р/ц; на рис. 65 даиа эта функциональная зависимость для нормального распределения скоростей на прямолинейном отрезке трубопровода, не имеющем каких-либо приспособлений или пре штствий на протяжении по меныпей мере 50 диаметров до диафрагмы против направления потока. Это представляется чисто эмпирическим исключением для области ламинарного истечения (Re 2100)Б4, где из теории можно получить значение «ср./«маке. = 0,50. Если поток полностью находится в турбулентной области, то для большинства целей среднее значение Оср./амакс. = 0,80 достаточно точно.
Трубка Пито редко употребляется для промышленных измерений вследствие недостаточной надежности, но она очень удобна для экспериментальных или испытательных работ, когда не требуется высокой
Трубка Пито
397
точности. Большим ее преимуществом является дешевизна и то обстоятельство, что ее можно вставить в трубопровод, не нарушая потока и не производя каких-либо изменений на линии.
Рис. 65. Отношение средней скорости к максимальной в круглых трубах.
Недостатком трубки Пито является малая величина отсчета, получаемая при обычных условиях для газов, вследствие чего возникает необходимость в чувствительном манометре, неудобном в заводских условиях.
Пример 4. Какой отсчет в миллиметрах водяного столба будет давать трубка Пито, помещенная в центре длиннэй прямой трубы, доставляющей воздух при 20° С, при манометрическом давлении в 50л.и Hg и при средней скорости Зм]сек?
Примем нср./имакс. = 0,80 и ^=1,00.
По уравнению (85), Д, = /2X9,81 X v,o
Дй = 0,717л воздуха = Дй» ( — 1 ).
\Р/ J с _ 29 (785/735) X 10< , ,
р/ 848 X 293,2 “'1,246 Кг1м ’
= (1000/1,246) — 1 = 0,000888 М = 0,88 ММ Н’°-
398
VIII. Термодинамика потока
Отсчет пропорционален квадрату скорости и первой степени плотности газа. Например, водород будет давать очень маленький отсчет, если только скорость его не будет ненормально высокой. Величина отсчета по трубке Пито возрастает при замене трубки статического давления трубкой с той же самой осевой плоскостью, как и ударная трубка, но повернутой по направлению потока. Ее иногда называют трубкой обратного типа. О коэфициенте для такого измерителя известно очень мало, и его следует калибрировать при действительных условиях. Отсчет никогда не бывает равен удвоенному отсчету, полученному по обычной трубке Пито, но бывает больше него примерно на 4О°/о.
ЧИСЛОВЫЕ ПРИМЕРЫ ПРИМЕНЕНИЯ УРАВНЕНИЙ ПОТОКА
Применение некоторых выведенных уравнений будет показано на ряде числовых примеров. Необходимо отметить, что при пользовании уравнениями следует применять совместимые единицы; в противном случае коэфициенты, входящие в уравнение, не будут истинными, и их значения будут изменяться с единицами. Так, если при использовании уравнения (59) 52 выражено в квадратных метрах, g—в метрах в секунду за секунду, Др —в килограммах на квадратный метр и р — в килограммах на кубический метр, то w будет выражено в килограммах в секунду. В уравнении, подобном уравнению (78) [соединенном с уравнением (72)], те же самые единицы для S2, g и Др в сочетании с.о значением /?, включающим величину v в кубических метрах, дадут qs в кубических метрах в секунду. Заметим, однако, что р и Т могут быть даны в любых единицах при условии использования соответствующего значения R.
Пример 5. Прямоугольная диафрагма в тонкой пластинке была про-калибрирована по сухому воздуху при 20° С и примерно при стандартном атмосферном давлении, после чего был построен график, по которому можно непосредственно считывать объем воздуха в минуту, отнесенный к 0° С и стандартному барометрическому давлению. Требуется выяснить, получаются ли заниженные или завышенные результаты при использовании этого измерителя для замера потока сухого газообразного СО2 при 20° G и атмосферном давлении при условии, что поток отнесен к тем же самым стандартным условиям. Следует использовать поправочный множитель уравнения (80).
Так как углекислота имеет большую плотность, чем воздух, то меньший (по количеству газа) поток СО2 дает тот же отсчет по манометру, который был получен для данного потока воздуха. Следовательно, при употреблении СО, отсчет по измерителю будет выше.
По уравнению (80),
^воздух /Исо.
4s (СО2) = qs (воздух)
— qs (воздух) ]/ 0,812 <7,, (воздух).
Для получения объема СО2 нужно умножить объем, полученный по диаграмме, на 0,8'2.
Числовые примеры применения уравнений потока
399-
N2
750/0 Н2, 25°/0 25° С
50 мм рт. ст.
30° С
нормальное
Пример 6. Прямоугольная диафрагма в тонкой пластинке вставлена в стандартную 6-дюймовую трубу для измерения потока газа при следующих принятых средних условиях:
Объемный состав газа (на основе сухого газа) Точка росы .............................. Манометрическое давление................. Температура ............................. Барометрическое давление ................
Диафрагма имеет 3 дюйма в диаметре и вставлена в горловину трически между фланцами с кранами. Принять, что перепад давления
концен-в сопле никогда не будет составлять больше 1°/0 абсолютного давления потока.
1. Определить константу измерителя, которая при умножении на квадратный корень из отсчета по манометру в миллиметрах ртутного столба будет давать объем в кубических метрах в минуту при стандартных условиях (15° С и нормальном барометрическом давлении). Принять турбулентный режим потока при числе Рейнольдса, по меньшей мере равном 30 000.
2. Вычислить поправочный множитель, на который следует умножить объем, полученный из коэфициента (1), если средними условиями потока будут следующие:
Объемный состав’ газа, пересчитанный на сухой газ
Точка росы............................. . . . .
Манометрическое давление......................
Температура............... .__._
Барометрическое давление .....................
78°/0 Н2, 22°/0 N2
10° С
100 мм рт. ст.
20° С______________
750мм рт. ст.
1. Так как перепад мал, то коэфициент расширения можно принять равным единице. Условия таковы, что можно считать газы идеальными и поэтому можно принять коэфициент диафрагмы равным 0,610. Константа измерителя, по уравнению (72), равна
/ 2-981
Сл} = 0,610 X 0,785 X 7,622 / f-(3^f6:(W = 0,0121.
Искомым коэфициентом является С"м. По уравнениям (78) и (79),
„ Ts у ум-
Давление насыщенного водяного пара при 25° С — 0,03229 кг1см2.
0 03229
Мольная доля пара Н2О в газе — —р j — 0,0293.
MD — 0,75 X 2,016 -|- 0,25 X 28,00 = 8,510.
М = 0,9707 X 8,510 + 0,0293 X 18 = 8,788.
С" - о 0121 288’2 - 1 /848 X МО XW _
—0,0121 (760y735j 10< у 303,2X8,788 °’020'
Это значение будет давать объем в кубических метрах в секунду, если Др выражено в килограммах на квадратный метр. Для получения qs в кубических метрах в минуту при Др, выраженном в мм ртутного столба, С"м следует умножить на 221.
Окончательно, qs — 4,421^Др, где qs выражено в кубических метрах в минуту, а Др — в мм рт. ст.
400
VIII. Термодинамика потока
2. Новыми являются следующие условия:
М = 7,545, р = 1,157, Т= 293,2.
Поправочный множитель по уравнению (80) будет
1,107 303,2 1/8-788 —
1,10 У 293,2 У 7^4а —
1,114.
Отметим, что даже незначительные изменения условий перед диафрагмой могут вызвать значительную ошибку в объеме, если не будут внесены соответствующие поправки.
Пример 7. Газ с молекулярным весом 58 при 32,5° С и 12,5 ат протекает через остроугольную концентрическую 2-дюймовую диафрагму и претерпевает падение давления в 3,5 атм. Отношение диаметра диафрагмы к диаметру трубы равно 0,300. Чему будет равна скорость истечения газа в килограммах в секунду?
Степень расширения значительно меньше 1,00, но больше критического отношения. Если принять справедливым закон идеального газа, то можно воспользоваться уравнением (62). Поправкой на величину (1 — [И) можно пренебречь. Принимаем Ст = 0,600, тогда
ртМ _ (12,5 + 9) X104 Х58 _ 24 g ?т НТ 2 X 848 X 305,7
w — 0,000349 X 0,600 X 50,82 /24X 3,5 = 4,95 кг/сек.
Молстед и Варга [173] решили подобную задачу с помощью предложенной ими номограммы.
Пример 8. Поток метана в стандартной 4-дюймовой трубе следует измерить с помощью измерителя Вентури с диаметром горловины в 2 дюйма. Чему будет равна максимальная, поддающаяся измерению, скорость истечения в кг/сек, если давление потока равно 1,5 кг/см2, а температура 30° С? Максимально возможная разность равна 250 мм рт. ст. Примем коэфициент Вентури равным 0,980.
При таком г нельзя пренебречь коэфициентом расширения. Если принять k =1,30, то коэфициент расширения Y [уравнение (56) или рис. 63] = 0,86.
Pi
_ 1,5X НИХ 16,03 ~ 848 X 303,2
= 0,935 кг)м\
^р—^= 0,34 kz'icm’, t Ou
Подставляя эти величины в уравнение (59), получаем
w = 0,980 X 0,785X5,082X0,861/ 2 Х--81 Х °’34- = 0,440 кг[сек.
Т и>У4
Числовые. примеры применения уравнений потока
г
Si
‘л’
г Пренебрежение коэфициентом расширения, невидимому, вызывает большую 4. ошибку. Использование средней плотности в обычном гидравлическом урав-и даст ш—0,49,. х.е...все же значительную, ошибку.. . ............
Пример 9. Газообразный этилен, почти совершенно чистый и сухой, :__измеряется с помощью остроугольной диафрагмы в танкой пластинке с кра-
нами в горловине. Диафрагма имеет диаметрвГ,03дюйма и вставлена в 2-дюймовую особо прочную трубу. Среднее, разностное давление потока и температура за сутки, полученные по регистрирующему указателю, были соответственно равны 267 мм рт. ст., 57 кг/см3 и 65° С. Определить расход этилена за сутки.
Частичное падение давления настолько незначительно, что расширением можно пренебречь, но следует учесть отклонение от законов идеального газа. Плотность этилена при этих условиях можно вычислить любым из методов,. описанных в гл. V. Мы будем пользоваться обобщенной диаграммой коэфициента сжимаемости (рис. 14).
Ткр = 282,8° К, ркр = 50,9 атм, 338,2 5,7X 1,033
Т = 282Д = 1*20’ * =----50Д----- =1’16-
Коэфициент сжимаемости =0,79.
_________________ _ рМ 57 X 10*X28,0 р ~ CRT~ 0,79 X 848 X 338,2 ’ 1
Примем коэфициент равным 0,61.
5г (площадь диафрагмы) = 0,785 X 2,62 = 5,4 гл2,
Др = =Z = 0,363 кг/см2, 7 л)
По уравнению (59),
= 0,0811.
w = 24 X 3600 X 5,4 X 0,61 v
= 66 450 кг/сутки.
981 Х70.4Х 10-6Х 0.363 _
При использовании закона идеального газа результат следовало бы умножить на ХЬ,79 = 0,89, т. е. ошибка составляла бы около 11°/о- Для большей точности следует проверить принятый коэфициент расхода. Re — 2,7-106, и из таблиц б'о определяется равным 0,602 [80].
Пример 10. Воздух при 20°С и напоре в 0,6 м водяного столба сверх нормального атмосферного давления выпускается через круглую диафрагму в пространство, где с помощью вакуум-насоса поддерживается давление в 400 мм рт. ст. Определить, на сколько процентов возрастает объемная скорость истечения, отнесенная к условиям потока до диафрагмы, если давление в эвакуированном пространстве упадет до 200 мм рт. ст.
р1 = 760+= 804 мм.
Следовательно,
26 Химическая термодинамика
- = ^ = 0,497.
Pi 805
402 VIII. Термодинамика потока
Эта величина меньше критического отношения давления для воздуха, и потому скорость потока достигает максимума. Любое дальнейшее снижение давления после диафрагмы не будет влиять на'поток. Этот принцип можно использовать для поддержания постоянного потока газов при постоянном иысоком давлении и изменяющемся более низком давлении [186].
Пример 11. Для измерения выхода из азотного компрессора газ высокого давления (100 кг/см2) дросселируется через ресивер, из которого азот выпускается в атмосферу через диафрагму. В ресивере следует поддерживать манометрическое давление в 3,5кг/сл2. Чему будет равен диаметр горловины сопла, если газ входит в сопло при этом давлении и температуре 25°С и если производительность компрессора равна 35 ms/muh (при отнесении к неизменным условиям забора при 40° С и давлении в 150 мм водяного столба по манометру и насыщении газа водяным паром)? Барометр показы-одл'р 737 л»л» рт ст
Этот пример представляет случай, когда отношение давлений меньше критического, и поэтому применяется уравнение (70). Давление насыщенного водяного пара при 40° С равно 0,0752 кг/см2. Общее давление при входе 737-(-(150/13,6) ,
в компрессор равно ------ -------=1,02 кг/см2.
Мольная доля Н2О = =0,0737.
Г аз высокого давления можно считать сухим, поскольку бблыпая часть воды' конденсируется в промежуточном холодильнике.
Объем сухого азота при стандартных условиях забора равен
(35 X 0,9263) /оЛуУУб=28 м^ман’
А ,1/00 01О,«ы по
- W = 224X60Х 28 = °’583кг1сек-
Примем k — 1,41, Св = 0,98.
Подстановка в уравнение (70) дает
2Л1
9,81 X MIX 28/ .2 \о,« 848 X 298,2” \2&) ’
$2 = 0,000755 л2, D2 = 31 мм.
Пример 12. Воздух, хранящийся в сосуде при манометрическом давлении 70 атм, выходит с такой скоростью, что по истечении ! часа давление падает до 63 атм. Чему было бы равно давление в сосуде после часового истечения газа, если бы сосуд был наполнен водородом при той же самой температуре?
Представляется целесообразным предположить, что уравнение (70) можно применить и к этому случаю, и если мы примем, что температура практически остается постоянной, то можем написать, что
т = КР\ У М 9,
Где К — константа,
т — общий вес, вытекающий за время 9,
Pi — постоянное давление в сосуде. .
Поскольку давление меняется, уравнение следует дать в диференциальной форме _
dm = KpVМ dO.
9
Термические измерители
403
Допуская идеальность газов, получим Мр\ т~ RT ’
где V—общий объем сосуда. Так как V и Г остаются постоянными, то dm = K'Mdp.
JCMdp = KpVMdb dp м p V м
64
Следовательно,
или
= — 0,565.
71
Для случая водорода:
2,303 =
S 71 ^2,016
р = 48 атм.
ТЕРМИЧЕСКИЕ ИЗМЕРИТЕЛИ
По принципу термический измеритель очень прост, но разработке его в полезный инструмент препятствуют различные практические трудности. Подвод теплоты будет вызывать подъем температуры перемещающегося вещества, который связывается со скоростью истечения уравнением
q = ,w\H='w J
(88)
(89)
(90)
или, для узкого температурного интервала, w =-----------------------------------.
Cp(t2-^i)
В единственной, имеющей промышленное значение, форме такого измерителя (измеритель Томаса) теплота подводится в виде электрической энергии, и для этого случая уравнение (89) переходит в 0,000948
где w— скорость истечения, кг/сек, Uw—расход энергии, ватты, Ср—теплоемкость, ккал1кг1град, t — температура, °C.
Точное определение w требует знания теплоемкости рассматриваемого газа и точного измерения малого подъема температуры — порядка 0,01°. Измерение средней температуры потока с необходимой для расчета степенью точности является исключительно трудной за
404
VIII. Термодинамика потока
дачей и осуществляется в измерителе Томаса при использовании термометров сопротивления в виде проволочной сетки, протянутой поперек канала истечения. В этом измерителе расход энергии для поддержания постоянного повышения температуры изменяется, благодаря чему скорость истечения становится прямо пропорциональной -потребляемой электрической энергии, и задача автоматического интегрирования для получения общего потока значительно упрощается. Следует помнить, что жидкость, попавшая в газовый поток, благодаря поглощению тепла при парообразовании, может вызвать серьезные ошибки.
ИСТЕЧЕНИЕ ГАЗОВ И ПАРОВ ЧЕРЕЗ СОПЛА
Тогда как диафрагма и труба Вентури используются для измерения потока, сопла употребляются главным образом для создания струй высокой скорости, которые применяются для получения мощности в турбине или для засасывания газов и паров в инжектор или эжектор. Детали конструкции сопел выходят из рамок этой книги, и мы лишь кратко обсудим некоторые основные термодинамические принципы, которым подчиняются потоки в соплах.
Теоретическую скорость, развиваемую газом, испытывающим изо-энтропное расширение, можно подсчитать по ранее выведенным уравнениям (47), (48) или (49), если перемещающимся веществом является идеальный газ, и по уравнению (42) или (43), если закон идеального газа не является достаточно хорошим приближением. В табл. 10 приведен ряд скоростей и2, вычисленных по уравнению (48) для расширения воздуха от данного исходного давления в 10,5 кг1см2 и температуры в 37,5° С до более низких давлений. Табл. 10 включает также мольный объем газа при низком давлении, вычисленный по уравнению (15, гл. VII), и площадь поперечного сечения сопла, необходимую для получения скорости потока в 0,4536 кг-моль!сек.
Таблица 10
Результаты расчета потока через сопло
р2, кг/см2 а2, м1сек v%m2[k2-mom S2, см2
10,55 ничтожно мала 2,50
8,8 178 2,85 72,8
7,03 261 3,34 58,1
5,98 306 3,75 55,7
5,59 (Крит.) 322 3,93 55,4
5,27 335 4,10 55,6
3,51 410 5,47 60,6
1,76 500 8,95 81,4
1,05 586 12,9 99,6
Истечение газов и паров через сопла
405
z/zZZZZZZZZ<^
Рис. 66. Схема суживающегося и расширяющегося сопла.
Анализ этих результатов показывает, что скорость увеличивается и площадь поперечного сечения уменьшается до тех пор, пока не будет достигнуто критическое давление (в данном случае 0,53 X Pt); затем, при более низких давлениях, скорость продолжает возрастать, но одновременно увеличивается и сечение. Такой же результат получается с любым газом или паром, независимо от того, является ли он идеальным газом или нет, и это приводит к заключению, что нужной формой сопла для получения струи высокой скорости является суживающийся переход к горловине с минимальным сечением с последующим расширением, как это схематично показано на рис. 66. Точную форму, желательную для данного ряда условий, нельзя предсказать на основе термодинамики, — она определяется большей частью эмпирически. После того как дана определенная форма, по термодинамическим уравнениям можно вычислить теоретическое давление и распре Заметим, что площадь горловины полностью определяется исходными условиями и желательной скоростью истечения и не зависит от низкого давления.
Конечно, фактическое расширение газа в сопле не строго изоэн-тропно, так как сопровождается трением, приводящим к возрастанию энтропии. Для сравнения характеристики различных сопел их производительность определяют по уравнению
е =
( **2 ц1) действительная
( а2 И1 ^идеальная
(91)
Таким образом, к. п. д. определяется отношением действительной кинетической энергии, развиваемой струей, к теоретически вычисленной по уравнениям; для экспериментального измерения изобретены специальные приспособления. Хорошо сконструированные сопла дают к. п. д. 0,95 или даже еще выше.
К. п. д. можно также выразить уравнением
„ (/Д — Я2) действительное
что можно подтвердить сравнением уравнений (91) и (43). (//,— H2)s является изменением энтальпии для идеального расширения при постоянной энтропии. Пренебрегая скоростью приближения, действительную скорость можно вычислить по уравнению
д2 (действительная) — 2gJ (93)
406
VIII. Термодинамика потока
ПОТОК В ТРУБОПРОВОДАХ
Жидкости. Для жидкостей, благодаря их несжимаемости, зависимости являются наиболее простыми. К жидкостям мы можем также присоединить и газы, если относительное падение давления невелико, и поэтому плотность практически постоянна. Если принять изотермическое истечение (следовательно, = отсутствие рабочего механизма в данной секции трубопровода и v1=v2 = v, то уравнение (4) переходит в
Zi — A>W + 12g“2 = —Q- (94)
Q представляет возникающее при трении в трубе тепло, которое необходимо полностью передать в окружающую среду, поскольку были приняты изотермические условия.
То же самое уравнение получается из механико-энергетического баланса, выраженного уравнением (9), с тем исключением, что вместо — Q мы имеем ^F; это представляет известное преимущество, привлекая внимание непосредственно к силе трения, а не к конечному । результату в виде теплоты.
Выше показано, как далеко можно итти при рассмотрении потока в трубопроводах с помощью одной термодинамики. Уравнение (94) выражает только связь общей потери на трение с изменениями статического давления, весового напора и скоростного напора. Если трубопровод горизонтален и имеет одинаковое поперечное сечение на обоих концах, то работа, произведенная для преодоления ч । трения, равна Д(/гп), т- е. уменьшению напора статического давления.
Если трубопровод не горизонтален, то напор давления между секциями । может увеличиваться, уменьшаться или оставаться постоянным в | зависимости от относительной величины потери на трение и изменения потенциального напора. 1
Вычисление потери иа трение как функции геометрических размеров трубопровода, скорости истечения и свойств вещества свя- < зано с рассмотрением механизма истечения, что является вопросом кинетики и не входит в область чистой термодинамики. Здесь это ' ! будет рассмотрено лишь очень кратко. (
В случае вязкого потока в прямой цилиндрической трубе по- ! стоянного поперечного сечения, работа преодоления сил трення _ ;
легко находится по коэфициенту вязкости р. из уравнения
Р 128|х£<?
(95)
Если труба горизонтальна и имеет постоянное поперечное сечение, т. е. если z2 = zr и а2 = ап то сочетание уравнений (94) и (95) дает уравнение
. 128ц/. в
(96)
Поток в трубопроводах 407
которое представляет одну из форм хорошо известного уравнения Пуазейля
(Др.= Р1 —р2).
В случае турбулентного потока в канале без резких изменений поперечного сечения, обычно считают потери на трение пропорциональными квадрату скорости, длине трубы, смачиваемому периметру и обратно пропорциональными площади поперечного сечения. Это приводит к одной из форм уравнения Фанниига
• |97'
где Pw — смачиваемый периметр, или для круглой трубы:
(98) gD ' ’
Если изменяется диаметр, а поэтому и скорость, то следует рассматривать бесконечно малый отрезок трубопровода, а также исключить с помощью уравнения
ap=G (99)
величину и, что дает ------
dP=?f&dL (ЮО)
gD? ' '
Если пренебречь изменениями скоростного и весового напоров, то сочетание уравнений (94) и (98) дает
= <101)
И
<102>
Константа пропорциональности, или так называемый „коэфициент трения* /, как можно показать анализом размерностей, должна быть функцией числа Рейнольдса. Для установления вида этой функции проведено много экспериментов. Ее можно дать или в графической форме, или в виде уравнения. Одним из наиболее простых уравнений является уравнение Женеро [83]
/= 0,04/?е~016, (103)
которое и рекомендуется, так как оно оказалось надежным для проектирования.
Только что приведенные уравнения приложимы только к трубопроводам без фиттингов или резких изменений поперечного сечения. Потеря напора, обусловленная протеканием через фиттинги, обычно учитывается с помощью эквивалентного по длине отрезка прямой
408
VIII. Термодинамика потока
трубы того же самого диаметра, который будет вызывать такое же действие трения. Механическая энергия, рассеивающаяся на трение при внезапных расширениях или сужениях трубопровода, вычисляется по уравнениям
J2_j
Fе =—л—(расширение), (104)
Fe==~2g (сУжение)>
(Ю5)
где К — эмпирическая константа, выражаемая обычно как функция отношения площадей S2\SX. Если S2/Sx—>0, то Л’=0,5. Оба эти уравнения основаны на очень скудном экспериментальном материале и могут рекомендоваться только в качестве грубого приближения, полезного для целей определения.
Приложение теоремы Бернулли к потоку жидкости в трубе можно теперь суммировать в уравнение
4 —
*2 ~+ P1W4 2g
+ 006)
Где 1 и 2 относятся к конечным точкам всего трубопровода, a F , Fj, Fe и Fe представляют механическую энергию, превращенную в теплоту трением соответственно в прямой трубе, в приспособлениях, при внезапном сжатии и внезапном расширении.
Пример 13. Жидкость с удельным весом 0,85 и вязкостью в 10 сантипуазов55 вытекает из вертикального цилиндрического сосуда диаметром в 1,8 м через трубопровод, состоящий из стандартных 2-дюймовых труб, со скоростью 0,2 м3/мин. Требуется определить статическое давление в точке трубопровода, отстоящей на 60 м от сосуда и на 15 м ниже уровня жидкости в сосуде. Фиттинги на линии привять эквивалентными длине трубы в 45 м. Манометрическое давление в сосуде непосредственно над уроввем жидкости равно 1,5 кг]см2.
их принимается ничтожно малой,
0,2
“2 — 60 X 0,785 X (0,0508)2 ~ 1,64 м1сек’ гх— г2~ 15 м,
w=7=oi>=1’176
Pi = 1,5 кг] см2 по манометру,
/ |Ур..„ме ,l.3)1 = o.04r^^x;«x.q-°«„0,OM7i
Поток в трубопроводах 40&
^Х»Хус№, -
Fc=ПГОТГ = 0,0685 кгм-
По уравнению (106), 15 + (1,5—/>2)10*Х 1,176 — 0,137 = 0,548 + 0,0685, р2 = 1,9 кг/см2 (по манометру).
Газы. Предположим, что поток является изотермическим (это будет обосновано в дальнейшем) и что различиями в уровнях можно пренебречь. При этих допущениях и при добавочном допущении, что в рассматриваемой части трубопровода не производится работы на валу, уравнение (5) принимает следующий вид:
al—а?
^~=Q. (107)
Это уравнение показывает, что общий энергетический эффект на линии является источником получения тепла, вызывающего увеличение скоростного напора. Для нахождения зависимости между энергией, соответствующей внутреннему трению, и другими членами, отражающими механическую энергию, мы будем пользоваться балансом, данным уравнением (8), которое для этого частного случая переходит в
= (108)
Принимая, что все трение обусловлено турбулентным потоком в прямой трубе или эквивалентно ему, можно соединить уравнения (108) и (100) н получить
+ + vd q (Ю9)
g£)₽2 ~Г g I Г VI
При делении на ®2 (или умножении на р2) уравнение (109) переходит в
2/G2 , . G2dv । п
~^dL+-g- +pdp = O. (110)
Для идеального газа уравнение (НО) можно написать так:
2/G2 . G2dv . М , n L
-~dL-\--------\--Tr=pdp = 0. (Ill)
gD 1 gv 1 RTK r ' '
Коэфициент трения является функцией величины Z?zzp/jx, которая практически постоянна для данной линии и данной скорости истечения массы.
410
VIII. Термодинамика потока
При интегрировании уравнения (111) для секции длиной L получим
+ + = t"2)
или, поскольку мы приняли газы идеальными,
(Ш)
Это уравнение позволяет вычислить падение давления рг—р2 при т*лг,Ж»ттг1глг1*пл *пл»лчг1г«лН »гл/»Л1т
допппл лиакршдмспхс ipcnnn, tnupuvin icKjii^cn mavvnt, luviaot д add (или его удельном весе) и длине трубопровода. Вторым членом уравнения (ИЗ) почти во всех случаях можно пренебречь *), как и следует ожидать из того обстоятельства, что он представляет изменение скоростного напора; тогда уравнение (113) переходит в
. <"<)
Поскольку число молей в секунду n=n£>2O/44f, уравнение (114) можно дать в другой форме, именно:
» _2 MfRTn*LM ...г.
= <115>
Интересно отметить, что уравнение (114) совпадает с уравнением
Фаннинга при применении его к среднеарифметической плотности.
Следовательно,
Л1 #1 /11 (П
Р"— Rf—(И6)
и подстановка этого выражения в уравнение (102) дает уравнение (114).
Если поток не является изотермическим, все же можно пользоваться уравнением Фаннинга, но тогда применяется средняя плотность, определенная по уравнению
. м (117.
Рт 2R\T1^T2) ' ’
Уравнение Уеймута, широко употребляемое для задач, связанных с потоком натурального газа в длинных трубопроводах, является просто уравнением (114), соединенным со следующим уравнением
£?) Ошибка при пренебрежении скоростным напором невелика, если скорости не превышают 30 м/сек. Для этих же специальных случаев Лобо, Фрайнд и Скаперда [158] предложили удобный для Применения графический метод решения уравнения [113] для перепада давления. В примере решения, давиом ими, пренебрежение скоростным напором вызывает ошибку в 25%, но следует отметить, что скорость впуска газа была равна 135 м/сек.
Поток в трубопроводах
411
для коэфициента трения:
<=0,008-0 3.
Максимальный поток. Так же, как и в случае истечения через патрубки, существует максимум для скорости потока в трубе, который можно легко найти с помощью критерия
<Х} , п
— при постоянном Pi и <=0.
Производя эту операцию над уравнением (113), получаем
/gMpl
(И8)
_ рЛ &Ркр (119)
Г vKp
(Индекс с употребляется для обозначения случая максимального по-тока.)____________________________________________________________
Так как
> и — Gv,
то ________
“Kp = V gPKpvKp- (120)
Уравнение (120) является уравнением акустической скорости в изотермическом случае; отсюда можно заключить, что максимальная скорость, достигаемая при истечении через трубу, равна скорости звука.
Сочетая уравнения (ИЗ) и (118), получим:
1 -J-In (121)
Для случая высокой скорости f в сущности является постоянной величиной, равной 0,0040. Пользуясь этим значением, уравнение (121) можно легко решить посредством подбора рс — давления на выходе из трубы, которое будет больше, чем давление в пространстве, в которое ведет трубопровод. При этом значении рс максимальный поток можно легко вычислить по уравнению (113), которое принимает следующий вид: о п
О2 _ (РХ-РК1№ макс,— 2RT[\nPllpKp-}-(2fL!D)] •
В сущности, различие между уравнением (ИЗ) ср2, равным р и рг, равным давлению в месте выпуска, почти во всех случаях очень, малб и, вероятно, лежит в пределах ошибки, связанной с различными допущениями и с выбором значения для f. Другими словами, вопрос
412 VI11. Термодинамика' потока
о максимальном потоке в трубопроводе представляет интерес скорее академический, чем практический.
Сравнение изотермического и адиабатного потоков. При выводе зависимостей для потока в условиях большого перепада давления принимались изотермические условия. Примем теперь другое предельное допущение, а именно — адиабатные условия, и посмотрим, какое получится различие. Тогда для идеальных газов уравнение (4) превращается в
(123)
и2 — И1
JC^T. — T^
или, для узкого температурного интервала, “2 —“1 25-
Так как a1/u2=v1/v2 и р^/Т^ =p2v2/T2, то
РтТ2
U2 = U.t-^, 2 1 дЛ
U|
JCp(T\-T2)= +
.или
Р1^~гА 2
(124)
Примем 7’] = 278°К, и1=3 м/сек, р, = 35 кг/см2, р2 = 1 кг/см2. Решая уравнение относительно Т2 методом подбора, легко показать, что Tj — Т2 меньше 0,6° С. Другими словами, допущение изотермических и адиабатных условий приводит, в сущности, к одинаковому результату. Это не является неожиданным, поскольку адиабатный поток в длинном трубопроводе представляет, в сущности, не что иное, как дросселирование, т. е. расширение при постоянной Н. С помощью уравнения (123) легко показать, что это может происходить до тех пор, пока скорость не будет слишком велика. Для идеального газа и приближенно для любого другого газа необратимое адиабатное расширение является также изотерми--ческим. Если их и рх\р2 относительно велики [уравнение (124)], то Это перестает быть правильным.
Лапл [142] недавно детально рассмотрел зависимость между изотермическим и адиабатным истечением веществ через трубопроводы. Уравнение для адиабатного потока можно вывести следующим способом, исходя из уравнения (108):
— + vdp + £?-dL = 0. (125)
5 2gm ' '
Зависимость const нельзя использовать для интегрирования члена vdp, так как она справедлива только для обратимого адиабат.
Мощность, требуемая для накачивания
«3
ного расширения, но можно записать
vdp = d (pv) — Pv~~
(126)
(127)
(128)
при по-
(129)
й затем соединить с общим уравнением (6) в частной форме, ценной к этому случаю (dz, dQ и dAo = O), а именно с уравнением
dU= ь~\.а№>,
которое представляет просто специальную форму уравнения dU=CvdT;
это дает
d(pv)
k — \ k
Сочетанием этого уравнения с уравнениями (125) и (126) и следующей перестановке получают
_________(l+fe)^-[2fegp1v1 + (fe-l)^J$ + ^^ = °-
Это уравнение можно непосредственно интегрировать, и Лапл дает несколько интегральных форм. С помощью обычного метода нахождения максимума легко показать, что максимальный поток получается опять-таки тогда, когда скорость на выходном конце трубопровода
равна скорости звука для случая адиабатного сжатия (или расширения) звуковой волны.
Очевидно, что уравнения изотермического и адиабатного потока в подавляющем большинстве случаев дают практически одинаковые результаты. Для очень коротких труб и относительно большого перепада давлений скорость адиабатного выпуска будет больше,
но максимально возможное различие между двумя случаями составляет только 2О°/о.
. МОЩНОСТЬ, ТРЕБУЕМАЯ ДЛЯ НАКАЧИВАНИЯ
Несжимаемые вещества. Если принять изотермичность процесса, то
Ц = Ц и
Др
m —/w> = — у •
При этих допущениях уравнение (4) переходит в
щ — «; Др
->i0 = -Q+^ + -^rJ+7- (130)
414
VIII. Термодинамика потока
Мы уже показали, что —Q = F, механической энергии, которая превращается в теплоту посредством трения. Величину, стоящую в правой части уравнения (130), можно назвать общим разностным напором ДА; она является суммой напора трения, скоростного напора и напора статического давления.’ Будем считать, что F обозначает работу преодоления трения всюду, за исключением самого насоса. — До представляет теоретическую работу, затрачиваемую на килограмм вещества. Теоретическая мощность в лошадиных силах будет выражаться уравнением
(131) /о х
Теоретическая работа является работой, действительна доставляемой веществу. Потребная работа (или мощность) на накачивание будет больше вследствие потерь на трение в насосе. Обычно это учитывается с помощью к. п. д., который приблизительно определен для насосов различных типов и размеров.
Пример 14. Раствор удельного веса 1,25 должен накачиваться со скоростью 0,2 ms/muh из открытого хранилища в верхнюю часть абсорбционной башни. Ои выпускается в башню через отверстия, суммарная площадь которых равна сечению трубы в 1 дюйм. Давление в башне равно 4,5 кг по манометру. Точка выпуска расположена на 24 м выше уровня раствора в хранилище. Насос забирает раствор в 2-дюймовую т'рубу, расположенную на 1,8 м ниже уровня раствора в хранилище, и выпускает его через 2-дюймовую трубу примерно на том же самом уровне. Напор давления во всасывающей линии равен 0,6 м водяного столба, а на линии выпуска — 3,6 м водяного столба. Какую мощность следует дать на вал насоса, если его к.п. Д. равен 70%? Какое давление будут показывать манометры на входе и выходе из иасоса?
Напор давления должен определяться по уже рассмотренным методам из данной скорости истечения и известных геометрических размеров системы. Будем считать, что это уже было произведено, причем получились вышеприведенные цифры.
4 2
Общий напор давления = 3,36 м раствора.
Гравитационный напор = 24 м.
' Скорость в стандартной 1-дюймовой трубе равна
60 X 0,785 X (0,0254)2 = 6,58 м1сек-
Скорость в сосуде для хранения = 0 (принимается).
6 582
Скоростной напор = g 81 == 2’2^ м'
Напор давления = 6 м.
Общий напор = 35,57 м.
0,2X 1,25X 1000 „ ------------=
4,16 кг/сек.
По уравнению (131), теоретическая мощность равна —X 35,57 _ ^gg / о
] 98 Действительная мощность равна
2,83 л. с.
в. ... ' , J ' ..££.-4-'
Мощность, требуемая для накачивания 415
Вычисление давлений легко производится путем определения энергетического баланса между двумя сечениями, одно из которых расположено в точке, где следует определить давление, а другое — на уровне раствора в хранилище (для давления забора) или в точке выпуска в башню (давление выхода). Таким образом,
ДР=/>2— Р1.
Д-г = г2 — zlt &u* = i$—ul,
причем сечение 2 принимается находящимся в насосе. Для линии всасывания А=^ = 0,48л, Дл =—1,8 м, й2 /1,135\* 6158 (Д07 2g 2X9,81
Р
---------------------а 1,21 X 1.25 X 1000--------------, ,
Др — -—-------------------------------------= 1,51 кг/см2.
= 0,108,
10*
Давление при входе в насос будет равно 1,51 кг/см2 по манометру. Применив то же самое уравнение к линии выпуска, получим
— ^ = 2,82 4- 25,8-4-0,55 = 29,17,
. 29,17X 1.25X 1000 , ,
— Др = р1 — р2 =---------—---------= 3,64 кг/см2.
Следовательно, рг = 3,64 -[-4,5 = 8,14 кг [см2 (по манометру). Отметим, что теоретическая мощность насоса в лошадиных силах равна ®др/75р, где Др — общаи разность давлений в насосе:
(8,14 —2,21) 10*
И~ 75 X 1.25 X Ю00
0,63 л. с.
Это дает возможность проверить некоторые вычисления. 3
Сжимаемые вещества. Если сжатие считать адиабатным, что d оказывается практически достаточно хорошим допущением, то теоре- | тический расход мощности для сжатия на одной ступени дается ,3 уравнением (31, гл. VII), видоизмененным следующим образом £г): Да
k—t.
Р== 4500^ — 1) [ (pj) ~11Л' с-’ (132> ijj
й) Строго говоря, если придерживаться прежнего условия относительно • -ЯД знака работы, то эта величина должна быть отрицательной. И здесь, и везде, ЗЯи где нет необходимости различать положительные и отрицательные величины, .^-Ж| вопрос о знаке обычно игнорируется. *g§sM
416
V/П. Термодинамика потока
где Vj — общий объем, в кубических метрах в минуту, забираемый в компрессор при давлении рг. Если степень сжатия г мала (Строго говоря, когда г—*1,0), то это уравнение переходит в уравнение
Р
__(Рг—Р1> V_p(r—1) v 4500 4500 ' '
(133)
которое совпадает с уравнением для жидкостей.. Пределы приложимости этого уравнения можно значительно расширить, если использовать средний объем, отвечающий изотермическим условиям; это дает
Л. 1/ , « гй ' I I />—НЧ"----1Д л с
4500 (2г)
(134)
В табл. 11 сравниваются эти три уравнения в применении к двухатомному газу при различных отношениях давления, причем резуль-таты выражены как отношение мощности, найденной по уравнениям (133) и (134), к мощности, вычисленной по уравнению (132).
' Таблица 11 L'<, Сравнительная точность уравнений (133) и (134) в применении к двухатомному газу при раз- ' V личных степенях сжатия
В' . ' т Уравнение (133) Уравнение (134)
< . 1,01 1,05 1.10 ’ 1,25 1,50 2,00 3,00 Т 5,00 < 1,005 1,02 1,035 1,088 1,16 1,31 1,56 1,95 <0,995 0,995 0,990 0,977 0,969 0,977 1,038 1,170
Из данных, приведенных в этой таблице, следует, что уравнение (133) обладает точности» до 5°/0 только до степени сжатия около 1,15, а уравнение (134), даже при таких больших значениях, как 3,00, дает расхождения, не превышающие 5°/0.
Пример 15. Газовая смесь, со средним молекулярным весом, равным 18.0, находящаяся в газгольдере при манометрическом давлении в 150 мм водяного столба и имеющая температуру 20° С, забирается из него ротационным насосом и направляется через скруббер в реакционную камеру, в которой манометрическое давление равно 25 мм рт. ст. Измерения в условиях газгольдера дали 150 м31мин. Перепад давления, обусловленный трением на всей линии и в башне, равен 750 мм вод. ст. Чему равна теоретическая
Мощность, требуемая для накачивания
417
мощность газового двигателя (в лошадиных силах)? Принять, что диаметр трубы, ведущей из газгольдера в реактор, на всем ее протяжении одинаков. Принимаем нормальное барометрическое давление.
Будем считать весовой напор ничтожно малым.
Разность скоростных напоров равна 0.
„ (771/735) 1(Р X18 ,
Плотность газа в газгольдере = —ооч о— = 0,758 кг)м2. о4о ZjOiZ
. 750X104 , (25X13,6—150)Х 104 ,„.Л
Общий разностный 735 X 0,758 + 13,6 Х 735 ХО^ = 1240
Теоретическая мощность = 150 =31>3 с-
/О ьи
Пример 16. Природный газ, который практически можно считать метаном, накачивается через 150-километровую секцию трубопровода с внутренним диаметром в 40 дюймов со скоростью 4250000 ж3 в сутки, причем скорость измерена при 15° С и при стандартном атмосферном давлении. Газ доставляется к насосу практически при атмосферном давлении и при 15° С и должен иметь на выпускном конце линии давление по манометру в 4,5 KtjcM2. Какая мощность потребуется для накачивания?
„ 4250000 X 273,2 „Ло ,
Количеством - х х ^2,08 Кг-моль1сек.
Скорость массы G = = 41,07 к^сек-м2.
(число Рейнольдса) — 4090000.
/ (коэфициент тренвя) — 0,0027 [69].
По уравнению (115),
2 ’ 2 _ 64 X 0.0027 X 848 X 288,2 X (2,08)2 X 150 X 1000 X 16
Р1 р2~ (3,14)2X9,81 X (1.016)2 —
= 1,667.10» (кг/см2)*,
R760 + 4,5 X 735) ИМ! 2 1П9
р2 = ---------------------- = 3,062 • 10»,
Pi = 6,876 X 104 кг/м2 = 6,876 кг/см2.
По уравнению (132)
1,30X1 X104 X 4250000 Г/6,876X0,231 1
теоретическая мощность = Q Ц— J =1] =
— 15950 л. с.
%? Химическая термо динамика
ГЛАВА IX
ПЕРЕДАЧА ТЕПЛА И ТЕПЛОВЫЕ ЭФФЕКТЫ
Подавляющее большинство операций и процессов, которые интересуют инженера-химика, независимо ог того, являются ли они по своей природе физическими или химическими, сопровождается тепловыми эффектами. Поэтому между рассматриваемой системой и окружающей средой должен существовать теплообмен, другими словами, должен происходить процесс нагревания или процесс охлаждения. Единственным исключением является адиабатный процесс, который, по определению, протекает без тепловых эффектов.
Передача тепла может осуществляться посредством одного из трех указанных ниже способов или их сочетания. Эти способы сле-^ дующие: 1) теплопроводность, 2) конвекция и 3) излучение56.
Теплопроводность связана с передачей тепла посредством движения и столкновения атомов и молекул, из которых состоит вещество. Она аналогична процессу диффузии, при котором с помощью подобного же механизма происходит передача материала. Конвекция является переносом тепла посредством движения больших агрегатов молекул, т. е., в сущности, подобна процессу смешения. Очевидно, что теплопередача путем конвекции может происходить только в жидкостях и газах, тогда как теплопроводность является основным видом теплопередачи в твердых телах. В жидкостях и газах, наряду с конвекцией, наблюдается также и теплопроводность, однако первая является значительно более быстрым процессом и обычно полностью маскирует второй процесс. И теплопроводность и конвекция требуют материальной среды и не могут происходить в полном вакууме. Этим подчеркивается основное различие между этими двумя процессами и процессом излучения, который лучше всего происходит в пустоте. Точный процесс, которым осуществляется передача энергии излучением через пустое пространство, еще не установлен, но для нашей цели будет удобно считать его происходящим посредством волнового движения в чисто гипотетической среде (эфире). Считается, что внутренняя энергия вещества передается волновому движению эфира; это движение распространяется во всех направлениях, и когда волна сталкивается с веществом, энергия может передаваться, отражаться или поглощаться. При поглощении она может увеличить внутреннюю энергию тела тремя способами: 1) вызвав химическую реакцию, 2) увеличив кинетическую энергию молекул (подъем температуры),
Передача тепла
419
3) увеличив потенциальную энергию, обусловленную конфигурацией системы (вызвав, например, парообразование). Способ (1) является редким случаем, и его можно в дальнейшем не рассматривать. При поглощении энергии другими двумя способами мы будем называть процесс в целом теплопередачей.
Интересно отметить, что интенсивность излучения полностью замкнутого пространства совершенно не зависит от природы материала, из которого сделаны его стенки, а зависит только от температуры. Уравнение, выражающее для этого случая зависимость энергии излучения от температуры, является одним из основных законов природы и может быть найдено с помощью термодинамики, как показывает следующий упрощенный вывод.
Примем, что лучистая энергия в данном пространстве может термодинамически трактоваться как внутренняя энергия U материального вещества и что общая лучистая энергия, деленная на объем, дает плотность излучения или его интенсивность, которую мы будем1 обозначать через е. Согласно электромагнитной теории (это также можно показать и опытным путем), излучение будет оказывать давление на твердую непрозрачную стенку,_______находящуюся—на его
пути5Т, и это давление связывается с энергией совершенно так же, как давление идеального газа связывается с кинетической энергией его молекул, а именно: илн
Р = 4- 12)
О
Сочетание уравнения
с уравнениями (1) и (2) дает
___Т dz е
~3dT~ Т ’ или
de . dT
Поэтому при интегрировании
получаем In е = In Т'* 4 —const, или
г — ЬТ*.
(3)
Это уравнение представляет закон Стефана — Больцмана, который строго приложим только к так называемому „абсолютно черному телу*. Для данной цели абсолютно черное тело представляет просто тело, которое целиком поглощает все падающие на него лучи. Только 27*
420
IX. Передача тепла
(4)
полностью замкнутое пространство, имеющее во всех своих частях одинаковую температуру, действительно является абсолютно черным телом. ।
Для простого случая теплопроводности в стационарном состоянии в одном направлении скорость теплопередачи дается уравнением
АГ
т. е. количество теплоты, передаваемой за единицу времени, равно «отношению перепада температуры к сопротивлению. Не делая большой ошибки, можно сказать, что это уравнение обычно применяется ко всем практическим случаям передачи тепла, даже если они гораздо сложнее, чем простая линейная теплопроводность. При линейной теплопроводности Д£ представляет разность температур двух параллельных ловерхностей, перпендикулярных к направлению потока тепла, а сопротивление дается уравнением
где L — расстояние между поверхностями, S—площадь одной поверхности, a k—коэфипиент теплопередачи. С другой стороны, в более сложных случаях, когда теплопередача происходит н нескольких направлениях или через несколько материалов, из которых не все могут быть газами и жидкостями, и R становятся сложными ' функциями. Дальнейший анализ величин R и М (за исключением случаев, связанных с нарушением второго закона) выходит за пределы данной книги, и мы сконцентрируем наше внимание главным образом на количестве теплоты, связанной с различными физическими и химическими изменениями, что является важным для практических задач, относящихся к теплопередаче. Кроме рассмотрения количества теплоты, некоторое внимание будет уделено нагревающим и охлаждающим средам, а также к. п. д. и действию теплообменников. Способы достижения температур ниже температуры окружающей среды являются особой областью теплопередачи, которая будет рассмотрена в следующей главе.
МЕТОДЫ НАГРЕВАНИЯ И ОХЛАЖДЕНИЯ. ТЕПЛОНОСИТЕЛИ68
Допустим, что мы имеем сосуд, содержащий систему, к которой следует подвести теплоту или от которой следует отнять ее. Так как вопрос о том, является ли рассматриваемый процесс нагреванием или охлаждением, в принципе связан только со знаком разности температур А/, то мы будем все процессы условно считать процессами нагревания. Рассматриваемой системой может быть система, в которой происходит химическая реакция или чисто физический процесс, например парообразование. Мы будем рассматривать только методы, пригодные для передачи требуемого количества теплоты с соответствую
Методы нагревания и охлаждения. Теплоносители
421
щей скоростью. Рассмотрим вкратце с точки зрения общих принципов следующие теплоносители и методы нагревания:
1. Горячая вода.
2. Водяной пар.
3. Горячее масло.
4. Органические пары.
5. Ртуть.
6. Расплавленные соли.
7. Топочные газы.
8. Электроиагрев.
Полезно вспомнить, что в конце концов источниками всякой энергии для нагревания являются 1) топливо и 2) напор воды (за исключением второстепенных источников, как непосредственное использование солнечной энергии, энергии ветра, силы приливов и отливов и некоторых других источников энергии). Первые шесть теплоносителей только переносят теплоту, получаемую ими от топочных газов или от источника излучения, причем энергия получается непосредственно от только что упомянутых основных источников. Главной причиной использования косвенных методов нагревания является то, что при их применении удается осуществить более точную регули-ровку температуры в системе, подлежащей нагреванию, а это часто бывает очень важно для успеха процесса.
Одним из наиболее важных вопросов при любом методе нагревания и в действительности самым главным из интересующих нас здесь является температурный уровень, при котором энергия может сделаться пригодной для передачи. Очевидно, что температурный уровень должен быть выше температуры, до которой следует нагреть вещество; разность температур будем называть „термическим потенциалом*. Величина этого потенциала непосредственно связана не только со скоростью теплопередачи, но во многих случаях также и с термическим к. п. д. Эта величина определяет ту долю энергии теплоносителя, которая может быть передана к нагреваемой системе. Если теплоносителем является газ, то термический потенциал уменьшается по мере передачи теплоты, и это ограничивает долю энергии, которую можно использовать. Например, если процесс следует провести при 1100° С и теплоносителем являются топочные газы при 1200° С, то непосредственно можно использовать только около 10°/о энергии газов. При непрерывном процессе некоторое количество энергии, которое теряется с отходящими газами, можно передать материалам, поступающим в систему, благодаря чему к. п. д, повышается. С другой стороны, если теплоносителем является насыщенный пар, то ббльшая доля энергии передается при постоянном потенциале, что является выгодным как с точки зрения термического к. п. д., так и с точки зрения регулирования температуры.
Горячая вода в качестве теплоносителя имеет ограниченное применение, будучи особенно полезной в пределах от 40 до 100° С для
422
IX. Передача тепла
нагрева чувствительных к перегреву материалов, которые не следует нагревать выше указанного температурного предела.
Насыщенный водяной пар является наиболее удобным теплоносителем в пределах от 100 до 300° С. Он прост для использования и регулирования температуры, свободен от загрязнений, доступен и дает равномерную температуру по всей поверхности и большую скорость теплопередачи. Температура в данной точке легко регулируется посредством варьирования давления. Последнее обстоятельство в то же время является и главным недостатком насыщенного водяного пара, так как высокие температуры могут быть достигнуты только при высоких давлениях. Следующие цифры, заимствованные из таблиц Вукаловича [284], дают представление о температурах, достижимых при различных давлениях, при применении насыщенного водяного пара:
Давление, ат Температура, °C
15 50 100 150 224 (критич.) 197,4 262,7 309,5 340,6 373,6
Критическая температура является абсолютным верхним пределом, ио в настоящее время, вследствие ряда причин, она не является практически применяемой температурой. Для давления в 100 атм употребляются паровые котлы, но на большинстве химических заводов обычно применяются более низкие давления, так что практический предел нагревания водяным паром близок к 200—230° С. Применение более высоких давлений значительно увеличивает трудности конструирования нагревающих поверхностей, особенно для установок рубашечного типа, так как при высоких давлениях водяной пар должен использоваться в змеевике или в другой трубчатой системе. „Термокойл“ (торговое название) представляет интересное сочетание рубашки и змеевика, в котором змеевик подводит водяной пар высокого давления в цельную отливку, составляющую одно целое со стенками сосуда, подлежащего нагреву.
Перегретый пар можно получать при повышенных температурах, не прибегая к высоким давлениям, но он редко употребляется в качестве теплоносителя, поскольку он не обладает двумя большими преимуществами насыщенного пара, а именно — равномерной температурой и высокой скоростью теплопередачи. Следует также помнить, что при температуре 480° С и выше происходит химическая реакция между водя-гым паром и железными поверхностями.
Методы нагревания и охлаждения. Теплоносители
423
В небольших установках иногда оказывается целесообразно повышать давление водяного пара с помощью поршневого компрессора. Как было показано в гл. VI, сжатие насыщенного водяного пара приводит к образованию перегретого пара, который, как было только что отмечено, нежелателен в качестве теплоносителя. Поэтому за сжатием насыщенного пара должно следовать охлаждение. Это легко осуществляется посредством соприкосновения водяного пара с водой.
Для температур более высоких, чем достижимые с помощью водяного пара и лежащих примерно выше 500° С, существует три метода (кроме электронагрева), имеющих промышленное применение в тех случаях, когда требуются равномерные температуры и их точное регулирование. Этими методами являются: циркуляция горячего масла в рубашках или змеевиках, конденсация насыщенного пара органического соединения и конденсация паров ртути. В системах с горячим маслом используется фракция нефти с высокой температурой воспламенения; она применяется главным образом в пределах 175—300° С, причем верхний предел определяется началом разложения масла. Масло циркулирует с помощью насоса между труб-чатым нагревателем, где оно нагревается топочными газами,”И рубаш-кой сосуда, к которому подводится тепло. Так как используется не скрытая теплота парообразования, то температура на поверхности не равномерна, причем ее изменение является функцией тепловой нагрузки, связанной со скоростью циркуляции масла.
Аналогично водяному пару употребляются органические жидкости с более высокой точкой кипения, чем вода. Этим путем можно получить температуры, превышающие те, которых можно достигнуть с помощью водяного пара и при значительно более низком давлении. Однако существует очень мало органических соединений, которые были бы жидкими при комнатной температуре и все же достаточно устойчивыми для практического употребления при повышенных температурах. Единственными соединениями, используемыми в некоторой степени в промышленности, являются ароматические соединения— дифенил и дифенилоксид. Особенно полезной является эвтектическая смесь этих двух соединений, известная под названием даутерма. Она содержит 73,5°/0 дифенилоксида и имеет точку плавления в 12° С, между тем как точка застывания для дифенилоксида Составляет 27°С и для дифенила 70,5°С59.
Даутерм применяется в пределах от 200 до 400° С, хотя выше 370° С происходит заметное разложение, и непрерывная работа выше этой температуры требует периодической очистки и добавления нового теплоносителя. Его нормальная точка кипения 258° С, и получаемые при указанных предельных температурах давления пара соответственно равны 0,287 и 10,5 кг/см2. Пар получается в котле при сжигании угля, нефти или газа и используется подобно водяному пару. Главными недостатками паров даутерма по сравнению с водяным
424
IX. Передача тепла
паром является более низкая температура разложения, меньшая скорость теплопередачи и то обстоятельство, что он является горючим материалом, и поэтому его применение создает опасность пожара и необходимость более плотных соединений в циркуляционной системе, поскольку для предотвращения потерь материала утечка должна быть минимальной.
Ртутные пары могут применяться в пределах от 300° С (давление пара около 250 мм рт. ст.) до 550° С (8 атм), причем высший предел определяется давлением и температурой, которым может противостоять материал аппарата. Поскольку ртуть является элементом, она абсолютно устойчива по отношению к температуре, и поэтому не возникает вопроса о ее разложении. Дополнительные преимущества ее применения заключаются в очень высокой скорости теплопередачи и отсутствии опасности пожара, но высокая стоимость, вредность для здоровья, если только не принимается соответствующих предосторожностей, и необходимость крайней тщательности для обеспечения отсутствия утечки из системы составляют ее серьезные недостатки.
Расплавленные соли в качестве теплоносителей применяются давно, поскольку дело касается лабораторной практики; в относительно небольших ваннах для термической обработки металлов они используются уже в течение долгого времени, но промышленное применение их в большом масштабе еще совершенно ново. В настоящее время существуют действующие установки с таким большим количеством соли в циркуляционной системе, как 450000 кг. Во многих установках используется смесь, состоящая из 4О°/о NaNO2, 7°/0 NaNO3 и 53°/0 KNO3, обозначаемая в дальнейшем HTS. Ее можно употреблять в пределах от 150 до 550° С, а в некоторых случаях — даже до 600° С. Эта смесь особенно полезна при температурах, превышающих верхний предел для горячего масла и для даутерма. Ее точка застывания лежит около 143° С, т. е. достаточно низка для того, чтобы соль можно было легко расплавить водяным паром, имеющимся в наличии на большинстве заводов. Соль совершенно устойчива до 425° С; выше этой температуры происходит очень медленное разложение, не сильное даже при 600°С. Коэфициент теплопередачи очень велик, причем указывались даже значения выше 9760 ккал'м2 X ч X грпй-Другим преимуществом этого теплоносителя является то, что он употребляется практически при атмосферном давлении, и так как он имеет ничтожное давление насыщенного пара, то давление при изменении температуры не изменяется. Для более полных сведений по свойствам HTS см. статью Кирста, Нэгла и Кастнера [135}. Попутно интересно отметить, что в настоящее время главное применение этой смеси состоит в использовании ее в качестве холодильного агента в процессе каталитического крекинга Гудри.
Так как практически вся энергия, передаваемая в виде теплоты, первоначально получается из химической энергии топлива, то наиболее
Методы нагревания и охлаждения. Теплоносители
425
прямым методом передачи тепла является использование продуктов сгорания топлива. Максимальная температура, достижимая с помощью продуктов сгорания, изменяется в зависимости от большого числа факторов, таких, например, как природа топлива и применяемый избыток воздуха; верхний предел лежит около 2200°С, а практический предел для обычно применяемых топлив — ниже 1700° С. Более высокие температуры можно получить при сжигании топлива в кислороде, но в настоящее время этот способ применяется только в небольших масштабах для сварки и резки металлов. Методы вычисления максимальной температуры горения топлива изложены ниже (см. пример 15).
Главными недостатками передачи тепла от продуктов сгорания являются: плохое регулирование температуры, загрязнение нагреваемых поверхностей и большой объем газа, с которым приходится работать (последнее обстоятельство объясняется его низкой теплоемкостью). Кирст, Нэгл и Кастнер [135] произвели сравнительный расчет для использования в качестве теплоносителей воздуха при 3 атм и HTS. Опыт был поставлен так, что оба теплоносителя циркулировали в однодюймовых трубках при перепаде температур в 28° С и при одинаковомперепаде давления, обусловленном потоком.Результаты показали, что HTS, превосходя по теплопередаче воздух в 48^2 раз, требует для циркуляции только 1/1700 часть мощности, затрачиваемой на циркуляцию воздуха. Кроме того, коэфициент теплопередачи для соли оказался в 30 раз больше, чем для воздуха.
Несмотря на недостатки нагрева с помощью непосредственного сжигания топлива, этот способ остается единственным средством для передачи тепла при температурах выше 600° С, если исключить электронагрев, имеющий ограниченное применение.
Электронагрев связан с непосредственным превращением электрической энергии в другие виды энергии, которые можно передавать в виде теплоты и которые мы объединили под общим названием термической энергии. По первому закону, 1 квт-ч электрической энергии эквивалентен 860 ккал теплоты. Имеются три метода превращения: 1) посредством сопротивления; 2) посредством электрической дуги и 3) посредством индукции. Материал, который следует нагреть, может сам служить сопротивлением; благодаря этому тепло выделяется непосредственно в точке его приложения, и проблемы передачи его отпадают. В других случаях можно использовать специальные сопротивления, например проволоку из хромовых сплавов, графитовые или карборундовые формы; тогда тепло, получаемое в сопротивлении, передается в точку приложения с помощью излучения или конвекции, или посредством их сочетания. Скорость нагрева сопротивлением дается простой зависимостью:
9 = 0,8614 PR,
426
IX. Передача тепла
где q — скорость перемещения тепла, ккал!час,
1 — средняя сила тока, амп,
R — сопротивление, ом.
Нагрев с помощью электросопротивления, иногда называемый „нагревом инфракрасными лучами”, за последние годы получил значительное промышленное распространение для сушки и прокаливания покрытий и отделочных слоев. Теплота выделяется при прохождении электрического тока через нити накала, работающие при относительно низких температурах (меньше 2500°К), и посредством соответствующего отражения инфракрасного излучения от нитей накала направляется к обогреваемой поверхности. Лучистая энергия, падая на поверхность, поглощается (степень поглощения зависит от природы поверхности).
При индукционном методе материал, подлежащий нагреванию, или сосуд, в котором он находится, действует как вторичная обмотка понижающего трансформатора, первичной обмоткой которого служит виток проволоки, к которому подается переменный ток; получающийся в материале или в сосуде вихревой ток низкого напряжения {токи Фуко) полностью», превращается в теплоту. Частота используемого переменного тока должна быть низкой для магнитных материалов и значительно более высокой (порядка 10 000 герц) для немагнитных проводников. Недавно был предложен метод электронагрева для непроводящих материалов, который может разрешить многие трудные проблемы обогрева. В отличие от прежних индукционных методов, он не использует принципа трансформатора, но материал, подлежащий нагреванию, служит диэлектриком конденсатора в цепи ультравысокой частоты. Молекулярные нарушения, получающиеся при быстром изменении электрического поля в диэлектрике, вызывают нагревание. Используется частота порядка 106—107 герц.
При дуговом методе электрическая дуга образуется между двумя электродами, обычно угольными, после чего электроды можно раздвинуть, а дуга поддерживается благодаря прохождению тока через пары, получаемые из материала электродов. Материал, подлежащий нагреванию, располагается на пути дуги или в непосредственной близости от нее.
Единственным пределом для температур, достижимых при электронагреве, является стойкость материалов, используемых в качестве сопротивлений или электродов. Наиболее высокой температурой, используемой в промышленности, является температура вольтовой дуги (около 3600°С); этот верхний предел, повидимому, определяется точкой кипения углерода. Следовательно, электрообогрев особенно целесообразен в области очень высоких температур, таких, например, как применяющиеся при производстве синтетических абразивов и карбида кальция. Он также используется и во всем интервале применяющихся температур, конкурируя с другими методами нагрева, благодаря своим определенным преимуществам, именно:
Т еплообменники
427
1. Тепло можно непосредственно применять там, где требуется, часто без необходимости передачи его.
2. Легкость регулирования температуры.
3. Чистота.
4. Удобство расположения установки и компактность ее.
5. Регулирование атмосферы в печи.
6. Высокий к. д. меньше
тепловые потери среду излучением
к. п.
п. д. (в общем единственной причиной 1ОО°/о являются в окружающую
и конвекцией).
1орячий коней
'в
получеПйя
ТЕПЛООБМЕННИКИ
Мы. не будем рассматривать вопросов, связанных с коэфициентами теплопередачи и производительностью теплообменников, которые обычно рассматриваются в учебниках по теплопередаче, а разберем вопросы, которыепредставляют существенное значение, но обычно в таких учебниках не затрагиваются. Мы будем рассматривать главным образом термический к, п. д. теплообменников и предельные условия их работы.
Способ действия. Рассмотрим приложение двух основных законов
термодинамики к простому противоточному теплообменнику, схематично изображенному на рис. 67 и работающему при постоянном давлении с любыми двумя газами или жидкостями. Первый закон требует, чтобы удовлетворялось следующее уравнение:
fA,
Рис,
Колодный конец
67. Схема противоточного теплообменника.
Na (На, — НаУ4- NB (НВ1 — НВг) + Q = 0, (5)
а второй закон требует, чтобы во всех секциях теплообменника удовлетворялось бы неравенство
(6)
(если А является нагревающимся веществом).
Хотя каждому человеку, обладающему только элементарным знанием физики, ясно, что это должно быть правильно, все же удивительно, как легко допускаются нарушения второго закона при некоторых расчетах теплообменников. Это будет показано на нескольких числовых примерах.
428
/X. Передача тепла
Пример 1. Газообразный кислород поступает в конденсатор при 10 атм и 300° К и покидает его с 15% жидкости (сухость пара равна 0,85). Охлаждение и частичная конденсация кислорода осуществляются с помощью противоположно направленного потока кислорода, входящего в теплообменник при 100° К и 1 атм. Нужно, чтобы кислород выходил из теплообменника при 290° К. Чему будет равно отношение массы потока низкого давления к массе потока высокого давления, если теплообменом с окружающей средой можно пренебречь?
По уравнению (5),
Na нв,~нвг
Nb
Энтальпии определяем по диаграмме S — Т для кислорода [293].
Нв = 111,7,
Нв = 0,85 X 68 0,15 X 26,5 = 61,8,
НА = 67,5, /7^ = 109,5.
По уравнению,
Na___моли газа при 1 атм____। jg
NB моли газа при 10 атм ’ ’ tB= 119,7° К,
At на холодном конце = 19,7° С,
At на горячем конце = 10,0° С.
Так как конечные разности температур положительны, то кажется, что теплообменник должен работать, но если нанести кривые для охлаждаемого и нагреваемого кислорода, как это сделано на рис. 68, то будет очевидно, что найденные условия не осуществимы, поскольку по высоте теплообменника встречаются отрицательные разности температур. Ввиду того, что дли адиабатного теплообменника значения Д/7 для обоих потоков должны быть равны от каждой точки до любого уровня, то разность температур для любой части теплообменника определяется расстоянием по горизонтали между двумя кривыми60.
Особенно легко впасть в нарушение второго закона при расчете теплообменника, в котором происходит фазовое превращение. Но это может случиться и при отсутствии фазового превращения, как показывает пример 2.
Пример 2. Воздух при 75 атм и 300°К требуется охладить в противоточном теплообменнике до 100° К азотом, поступающим при 1 атм и 80° К. На 1 моль воздуха приходится 1,75 моля азота. При какой температуре будет выходить азот, если пренебречь потерей тепла в теплообменнике?
Энтальпии воздуха и азота [171] равны
И в, = 2854, HBi =280, НА> = 1352.
Из теплового баланса
Ял = 2822,
% = 291 ° К,
А1^ —9° С, Д^ = 20°С.
Т еплообмеюшки
429
График Т — Н (рис. 69) показывает, что предлагаемые условия неосуществимы. Пунктирная линия, касательная к кривой Т—Н для воздуха, соответствует минимальному количеству N2, которое можно использовать, именно 2,10 моля на моль воздуха, и показывает, что воздух не может выходить при температуре выше 255° К- Это предполагает, что теплоемкость азота постоянна, что является достаточно удовлетворительным приближением к действительности.
Рис. 68. Нарушение второго закона термодинамики в теплообменнике (случай фазового превращения в одном из потоков).
Для адиабатного теплообменника, в котором не происходит фазового превращения, уравнение (5) можно написать так:
WA^PA ^а» ^А^ ~ в ^В, *в)> (7)
где wA и юв—скорости перемещения вещества, а Ср^ и Ср&—средние теплоемкости.
Напишем уравнение (7) в следующем виде:
= (8)
где _
wacpa
R =---—--- .
WB^PB
Если k 1, то
*В, *А,>*В> *А,’
или
(9)
430
IX. Передача тепла
Подобным же образом, если k 1, то
&2<&1«
(Ю)
Если Д/3^>Д/], то, очевидно, только на холодном конце № может приближаться к нулю, а на горячем конце должна неизбежно получаться конечная разность, независимо от размеров теплообменника. При Д/3 <С наблюдается обратное. Иначе кривые Т — Н будут
Рис. 69. Нарушение второго закона термодинамики в теплообменнике (случай, при котором ни в одном из потоков не происходит фазовых превращений).
пересекаться, и второй закон будет нарушен. Мы можем сформулировать следующее простое, но полезное правило.
Если вещество, получающее тепло, имеет большую теплоемкость, то только на холодном конце можно получить разность температур bt, приближающуюся к нулю. Если же оно имеет меньшую теплоемкость, то разность температур А/, приближающуюся к нулю, можно получить только на горячем конце.
Если теплообменник не адиабатный, то положение будет сложнее. Например, с помощью тех же самых рассуждений, которыми мы пользовались выше, можно показать, что если теплообменник теряет тепло в окружающую среду, то М на горячем конце должно быть больше, чем № на холодном конце, при условии, что вещество, получающее тепло, имеет большую теплоемкость; другими словами, правило, сформулированное выше, остается действительным. Но если
Т еплообменники
431
в теплообменнике имеется приток тепла, то At на горячем конце может быть больше, равно или меньше At на холодном конце, в зависимости от количества тепла, получаемого от окружающей среды.
К. п. д. теплообменника. К. п. д. определяется разностью температур на одном из концов теплообменника. Чем меньше эта разность, тем эффективнее будет теплообменник; теплообменник, у которого температуры разнились бы только на dt, обладал бы 100-процентной эффективностью. Но мы только что видели, что второй закон накладывает определенные ограничения на возможное сближение температур. Например, из предыдущего рассуждения видно, что при определенных условиях нельзя получить тесного сближения температур ни на одном конце; при других условиях можно получить тесное сближение или на горячем, или на холодном конце, но не одновременно на обоих. НЬ если мы ограничим наше рассуждение условием линейной зависимости Н от t как для нагреваемого, так и для охлаждаемого вещества, то мы будем иметь два простых случая:
Случай (а). Теплоемкость вещества, получающего теплоту (вещество Д), меньше теплоемкости вещества, сообщающего теплоту. В этом случае At па горячем конце—может—приближаться к нулю, и
К _______ ___фактическое увеличение температуры вещества А_
максимально возможный подъем температуры
*В, — tA,
(И)
Иногда предпочитают выражать результаты посредством величины
1 —е
^в,— ^’
(12)
Случай (б). Теплоемкость вещества, получающего теплоту, больше теплоемкости вещества, сообщающего теплоту.
At на холодном конце теперь может приблизиться к нулю, и мы имеем:
*в, — tBl tB, — lA,
Пример 3. Примем, что 50 кг I час вещества В выходит из перегонного куба при 200° С. Желательно сконструировать теплообменник, который будет передавать теплоту этого вещества другому веществу А, находящемуся при 25° С и перемещающемуся со скоростью 40 кг)час. Вещество В имеет теплоемкость 0,55 ккал/кг-град, а А — 0,48 ккал/кг-град. К. п. д. теплообменника должен быть равен 98%. Чему будут равны конечные At, если тепловыми потерями можно пренебречь?
432
IX. Передача тепла
По уравнению (12),
Д*2 = 0,02 (200—25) = 3,5° С, = 196,5.
Тогда
40X0,48
*“50 X 0,55-°’698-
По уравнению (8),
tBt = 200—0,698 (196,5—25) = 80° С, At1 = 55°C.
ТЕПЛОВЫЕ ЭФФЕКТЫ ФИЗИЧЕСКИХ ПРОЦЕССОВ
Разделим все тепловые эффекты на две категории: 1) эффекты, сопровождающие физические процессы, и 2) эффекты, сопровождающие химические процессы. В этом разделе мы рассмотрим только физические процессы, классифицируя тепловые эффекты следующим образом:
1. Тепловые эффекты, сопровождающиеся изменением температуры, а) Чистые газы.
б) Чистые жидкости.
в) Чистые твердые тела.
г) Растворы.
2. Теплота фазового превращения (при постоянной температуре в случае одного компонента).
а) Твердое тело — твердое тело.
б) Твердое тело — жидкость.
в) Твердое тело — пар.
г) Жидкость — пар.
Тепловые эффекты, сопровождающиеся изменением температуры. В этом разделе мы будем касаться только влияния температуры на теплоемкость и энтальпию чистых веществ (т. е. одного компонента). Влияние давления уже было рассмотрено в гл. VI. Строго говоря, мы имеем дело со значениями Ср> и FP при нулевом давлении, но практически нам не нужно делать никаких различий между этими величинами при р = 0 и при р — 1 атм. Этн величины при нулевом давлении можно вычислить из спектроскопических измерений, причем, если спектры хорошо известны и молекула относительно проста, они, вероятно, будут более точны, чем значения, взятые из термических данных. Методы подобных вычислений см. в книге Винера [276]61.
Для одноатомного газа, согласно кинетической теории, Ср не зависит от температуры и равняется 5/27? или 4,96 кал.’моль-град, и это вполне подтверждается экспериментом. Теплоемкости многоатомных газов возрастают с температурой. Для эмпирического воспроизведения истинной теплоемкости как функции температуры
Тепловые эффекты физических процессов
433
пользуются, в частности, следующими уравнениями:
Ср = а+^ + Т^+ .... (15)
С„ = а4-«4-сг!-2, (16)
Р
Cp = A-\-Bt-\-Ci К (17)
Температуру можно выражать или по стоградусной шкале или по абсолютной. Соответствующие уравнения для Н получаются интегрированием, причем уравнение, отвечающее уравнению (15), будет следующее:
7/=770 + а/+1^ + -|Т^+..., (18)
где Но—константа интегрирования, которая определяется произвольной фиксацией Н при некоторой температуре. Обычно принимают Н=0 при /=0°С. Нижеследующие уравнения для средней Ср в пределах температур и t2 также легко получаются из уравнения истинной Ср, если использовать уравнение (32, гл. 1):
---------= Д + + 4) + у Y (4 + 44 +4М—т----------(19)
^ = а + 4м4 + 4) + Г^, (20)
(21)
На вопрос о том, какое из этих эмпирических уравнений является наилучшим, нельзя дать ответа для . всех случаев. Например, Томсон [245], сравнивая эти уравнения для газообразных СО2, СО, Н2О, Н2 и О2, нашел, что уравнение (15) оказалось наиболее точным для СО и Н2О, уравнение (16) —наиболее точным для Н2 и уравнение (17) — для СО2 и О2.
Удобно принять в качестве исходной температуры для средней теплоемкости / = 0° С; тогда уравнение (19) примет вид
= 2" • • •> .(22)
а ДН между любыми двумя температурами будет равно
ЬН=Ср12—Ср^. (23)
Значения Д/У очень легко определяются из графика, отвечающего уравнению (18). Если нужно найти более точные значения, чем те, которые можно определить с помощью обычных диаграмм небольшого масштаба, то удобнее пользоваться таблицами для Н или С , чем применять диаграммы больших размеров.
28 Химическая термодинамика
434
IX. Передача тепла
Константы уравнения (15) для большого числа газов даны Истменом [71], Спенсером и Юстисом [239] и Брианом [32]. Константы уравнения (18) для Ср, заимствованные из сводки Бриана, приведены в табл. 12.
Таблица 12
Константы уравнения энтальпия — температура, основанного на уравнениях теплоемкостей Бриана
Единицы: Ср, кал/моль-град, Т в °К; пределы 300 — 2000°К,
Т Г_ ГУ __ Т* Л'ТО ГЧ Г> ТЛ"
/7— v при 1 —
Газ a pXio3 ТХЮ7 «0
о2 6,26 2,746 — 7,70 —1808
n2 6,30 1,819 — 3,45 —1787
Н2 6,88 0,066 2,79 —1880
со- 6,85 8,533 —24,75 —2173
со 6,25 2,091 — 4,59 —1782
н,о 6,89 3,283 3,43 —2007
NO 6,21 2,436 — 6,12 —1784
HC1 6,64 0,959 — 0,57 —1850
НВг 6,30 1,819 — 3,45 —1787
СН4 3,38 17,905 —41,88 —1563
HI 6,25 2,091 — 4,59 —1782
HjS 6,48 5,558 —12,04 —1970
so2 8,12 6,825 —21,03 —2459
HCN 7,01 6,6 —16,42 —2150
COS 8,32 7,224 —21,46 —2528
CS, 9,76 6,102 —18,94 —2881
NH3 5,92 8,963 —17,64 —1940
СгН2 8^8 10,50 —26,44 —2636
Хек [104] составил таблицы в пределе от 60 до 2700°К для истинных Ср и Н для N2, О2, СО, Н2, СО2 и Н2О, причем его значения основаны на спектроскопических данных. Таблицы для средней С р особенно целесообразны для вычисления тепловых эффектов; такая таблица для большого числа газов дана Юсти и Людером [119] и гюспронзводится частично в табл. 13 ☆). С помощью данных, приведенных в этой таблице, легко найти точные значения теплового эффекта при нагревании или охлаждении газа.
☆) Значения Юсти и Людера для D2, HD, ОН и D2O опущены.
Тепловые эффекты физических процессов
435
Таблица 13
Средние теплоемкости газов между О и t° С при нулевом давлении Единицы: кал/моль
ГС н2 n2 о2 СО NO Н2О со2 n2o so2 Воздух
100 6,92 6,97 7,05 6,97 7,14 8,03 9,17 9,79 9,74 6,96
200 6,95 7,00 7,15 7,00 7,17 8,12 9,65 10,12 10,15 7,01
300 6,97 7,04 7,26 7,06 7,22 8,22 10,06 10,45 10,52 7,06
400 6,98 7,09 7,38 7,12 7,30 8,34 10,40 10,74 10,84 7,13 .
500 6,99 7,15 7,49 7,19 7,38 8,47 10,75 11,02 11,11 7,20
600 7,01 7,21 7,59 7,27 7,46 8,60 11,03 11,24 11,35 7,27
700 7,03 7,27 7,68 7,34 7,54 8,74 11,28 11,50 11,55 7,34
800 7,06 7,35 7,77 7,43 7,62 8,89 11,50 11,71 11,72 7,42
900 7,09 7,42 7,85 7,50 7,70 9,04 11,70 11,90 11,88 7,49
1000 7,12 7,49 7,92 7,57 7,76 9,18 11,00 12,07 /,эо
1100 7,15 7,56 7,98 7,64 7,83 9,32 12,05 12,21 12,13 7,62
1200 7,20 7,62 8,04 7,70 7,89 9,45 12,19 12,35 12,23 7,68
1300 7,24 7,67 8,11 7,76 7,94 9,58 12,32 12,48 12,33 7,73
1400 7,28 7,73 8,16 7,81 7,99 9,72 12,45 12,60 12,41 7,78
1500 7,32 7,78 8,20 7,85 8,03 9,84 12,56 12,69 12,48 7,84
1600 7,36 7,82 8,24 7,90 8,08 9,96 12,66 12,78 12,55 7,88
1700 7,40 7,86 8,28 7,94 8,12 10,09 12,75 12,88 12,61 7,92
1800 7,45 7,91 8,33 7,98 8,15 10,20 12,84 12,95 12,67 7,96
1900 7,49 7,94 8,38 8,02 8,19 10,30 12,92 13,01 12,71 7,99
2000 7,53 7,98 8,42 8,05 8,22 10,41 12,99 13,09 12,77 8,03
2100 7,57 8,01 8,45 8,09 8,26 10,52 13,06 13,17 12,81 8,06
2200 7,62 8,05 8,48 8,12 8,29 10,61 13,13 13,21 12,85 8,08
2300 7,66 8,08 8,52 8,15 8,31 10,71 13,19 13,28 12,89 8,12
2400 7,70 8,10 8,56 8,18 8,34 10,79 13,24 13,33 12,93 8,14
2500 7,74 8,14 8,59 8,21 8,36 10,87 13,30 13,38 12,96 8,18
2600 7,78 8,17 8,63 8,24 8,38 10,96 13,34 13,42 12,99 8,20
2700 7,81 8,19 8,65 8,26 8,40 11,03 13,39 13,46 13,02 8,23
2800 7,85 8,22 8,68 8,28 8,42 11,11 13,43 13,51 13,04 8,25
2900 7,89 8,24 8,72 8,30 8,44 11,18 13,48 13,55 13,07 8,27
3000 7,92 8,26 8,76 8,32 8,45 11,23 13,52 13,59 13,10 8,29
28*
436
IX. Передача тепла
Пример 4. Сколько тепла следует сообщить 1 кг воздуха для изменения его температуры от 500 до 2250° С?
По табл. 13, tx — 500, Ср =7,20; t2 = 2250, Срз = 8,10
По уравнению (23), 1
Q = ~ (8,10X2250—7,20X500) = 504 ккал.
Значения первых трех констант в уравнении (18) для различных углеводородов основаны на данных Эдмистера [74] и приводятся в табл. 14.
Константы уравнения энтальпия — температура для различных углеводородов й)
Газ а
СН4 4,60 0,0133 —1753
С2Н, 7,51 0,01115 —2466
с.2Н4 3,84 0,022 —1870
С,Н6 3,84 0,0298 —2161
С3Н6 4,09 0,0372 —2506
С3Н8 4,09 0,0432 —2730
С4Н8 4,60 0,0515 —3179
с4н10 4,60 0,0562 —3354
^5^12 5,11 0,0692 —3979
С6Н14 5,62 0,0822 —4603
С7Н16 6,13 0,0952 —5228
С6н6 6,61 0,0526 —3646
£?) Рекомендуемый температурный интервал 250—600° К-
Эдмистер нашел, что Ср для парафиновых и олефиновых углеводородов, содержащих три и более атомов углерода, можно связать с числом атомов углерода н водорода с помощью уравнения
С = 2,56 0,51л + (0,0013л2 + 0,0044л — 0,00065/шг-[-
Р 0,00495/л — 0,0057) Т, (24)
где п — число атомов углерода, т — число атомов водорода, Т — градусы Кельвина.
Спенсер и Фланаган [238] представили константы уравнений (15) и (1€) для 61 газа с трехатомными и многоатомными молекулами, основанные на вычислениях по спектроскопическим данным.
Тепловые эффекты физических процессов
437
Важно подчеркнуть эмпирическую природу уравнений, представленных для Ср и Н, и обратить внимание на то обстоятельство, что ими не следует пользоваться для экстраполяции. Они, например, являются совершенно ошибочными для температур ниже 0° С, а в некоторых случаях уравнения типа уравнения (15) обнаруживают максимумы, отсутствующие в действительности. Следует также твердо помнить, что многие из имеющихся данных по теплоемкости не очень точны. Уравнения, представляющие зависимость Ср от Т для одного и того же газа, могут по результатам отличаться друг от друга на 10°/0, а в некоторых случаях и больше.
Зависимость Ср от структуры органических молекул. При рассмотрении проблем нагревания и охлаждения мы можем иметь дело с любым из многочисленных органических соединений, и для упрощения задачи нахождения термодинамических свойств желательно связать свойства веществ с их структурой. Беневитц и Роснер [211 предложили метод вычисления Ср для органических молекул, структура которых включает семь различных связей между С, Н и О. Фугаси и Руди [82] упростили этот метод, а Добратц [60] для увеличения точности внес в него видоизменения и добавил данные для связей, включающих N, S и галогены. Уравнение Добратца имеет следую-щий вид
Ср (при р = 0) = 3R + + £ 9гСу, + , (25)
где а — число связей, допускающих свободное вращение (С—С или С — О групп в эфирах и сложных эфирах), — число валентных связей всех типов,
С»: и Со; —функции Эйнштейна ☆), п — число атомов в молекуле.
2^г-СЧ; (или S^zCst) представляет сумму произведений числа связей данного типа на соответствующее значение функции Эйнштейна для этой связи. Cv— изохорная теплоемкость. Функции Эйнштейна определяются из частот колебаний и для удобства применения изображаются как функции температуры уравнениями следующего вида:
С, (или Сг) = А-\-ВТ 4-СТ2. (26)
Константы этого уравнения для каждой связи сведены в таблицу в статье Добратца. Из значения С^, полученного этим методом, можно вычислить Ср способами, рассмотренными ранее (см. гл. VI). Учитывая различные неточности, связанные с этим методом, можно предположить, что употребление каких-либо более сложных уравнений, чем уравнение Ср—CP = R, не оправдывается.
й) Производная энергии по температуре, другими словами, теплоемкость при постоянном объеме.
438
IX. Передача тепла
Изложенный метод весьма прост для применения. Рассмотрим, например, пары ацетона. В этой молекуле имеется шесть связей С — Н, две связи С — С и одна связь С = О, т. е. 1'</. = 9, а = 2 и п =10. Константы уравнения (26) для двух частот (v и S) берутся из таблицы (для каждой связи) и умножаются на число связей данного типа. Подстановка в уравнение (25) дает уравнение для Cv той же формы, что и уравнение (26). Для дальнейших деталей см. цитированную статью.
Жидкости. Изменения теплоемкости жидкостей с температурой менее существенны, чем изменения теплоемкости газов, гак как жидкости и нагреваются и охлаждаются в значительно более узких пределах. В общем можно сказать, что Сп возрастает с температурой, и обычно употребляется уравнение (15),' хотя редко оправдывается применение более двух констант. Громадное большинство жидкостей имеет удельную теплоемкость, лежащую в пределах 0,40—0,50. Исключения составляют более высокие значения для воды, аммиака и метана и более низкие значения для ртути и хлористых соединений в2.
Теплоемкость твердых тел. Классическая теория теплоемкости предсказывает максимальное значение атомной Ср простых веществ, равное 3/?, или 5,96, что соответствует Ср около 6,2, т. е. значение для элементов при комнатной температуре согласуется с правилом Дюлонга и Пти. Это правило очень хорошо оправдывается во всех случаях, за исключением элементов с атомным весом, меньшим 40. Для легких элементов значение Ср меньше значения, вытекающего из этого правила, как и указывается ниже. Для соединений в качестве первого приближения можно использовать правило Коппа, а именно, что Ср соединения =^Ср атомов или, в большинстве случаев, числу атомов, умноженному на 6,2. Но для некоторых легких элементов вместо 6,2 следует пользоваться нижеприводимыми величинами: С—1,8; Н — 2,3; В — 2,7; Si — 3,8; О — 4,0; F — 5,0; Р — 5,4; S — 5,4. Теплоемкость твердых тел меньше теплоемкости жидкостей и также возрастает с температурой. Зависимость обычно выражается уравнением (15); Келли [130] предпочитает уравнение (16).
Теплоемкость твердых тел при низких температурах не имеет большого практического значения, но представляет существенный теоретический интерес и важна для понимания третьего закона термодинамики и определения абсолютной энтропии. Поэтому желательно кратко рассмотреть этот вопрос.
Многие исследователи приступали к решению проблемы зависимости теплоемкости от температуры с помощью квантовой теории. В частности, Эйнштейн и Дебай вывели соотношения между Cv и Т, включающие частоты колебаний атомов и молекул. Эти соотношения неудобны для практического применения, но некоторые вытекающие из них упрощенные зависимости могут оказаться полезными для
Тепловые аффекты физических процессов
439
Инженера. Например, зависимость С от Т для одноатомного твердого тела можно выразить в форме
<27>
где <р — одна и та же функция для всех твердых тел данного класса, а 6 — характеристическая температура, различная для разных веществ. Другими словами, достаточно знать одно значение Cv для определения значения 0, а поэтому и всей кривой C,z — Т, если только эта кривая была установлена для какого-нибудь одного вещества данного класса. Это одно значение Cv должно определяться при температуре, значительно более низкой, чем та, при которой C^-SR. На практике оказалось более удобным наносить на график в зависимости от lg(T/6).
Льюис и Гибсон [150] распространили уравнение (27) на элементы более сложной структуры и даже на соединения, придав ему форму
( т \ я
(т) ’
(28)
где функция ср — та же самая, что и в уравнении (27), а п доба вочная характеристическая константа, которая всегда меньше единицы. Записывая уравнение (28) в форме
легко показать, что
Ce = <p 1g 4),
а1— аг
(29)
(30)
и
IgO^lgT^-S,
где (ij и а2 — два значения n\g(Tfb\, соответствующие двум измеренным значениям Cv, которые можно прочесть с графика, применимого к предыдущему классу (поскольку функции одинаковы).
При очень низких температурах, приближающихся к абсолютному нулю, уравнение Дебая для Cv как функции температуры принимает следующий вид
Cv=al*. (31)
Из этого уравнения видно, что С., = 0 при Т = 0 (поскольку а является константой) ®8.
Из уравнения (31) легко можно вывести, что
S=~aT\ (32)
О
440
IX. Передача тепла
формулировку третьего закона; от-тесно связан с теплоемкостями твер-
и поэтому S=0 при Т=0. При соответствующем ограничении последнее уравнение представляет сюда мы видим, что этот закон дых тел вблизи 7'=0.
Для исчерпывающего обзора данных по теплоемкости неорганических соединений как выше, так и ниже комнатной температуры см. две сводки Келли [130].
Теплота фазового превращения (один компонент). Уравнение Клаузиуса— Клапейрона является основным уравнением, связывающим скрытую теплоту любого типа фазового превращения с другими величинами, но для вычисления скрытой теплоты требуются точные данные, связывающие равновесное давление с температурой, и значения объемов сосуществующих фаз. Если бы все эти величины всегда имелись в распоряжении, то не было бы необходимости продолжать обсуждение вопроса; в действительности же эти данные редко бывают доступны. Поэтому возникает необходимость в некоторых эмпирических соотношениях для вычисления скрытых теплот с точностью, достаточной для технических целей- В этом разделе мы будем рассматривать главным образом такие зависимости для теплоты парообразования.
Скрытая теплота парообразования в нормальной точке кипения. Одним из наиболее старых и наиболее простых соотношений является правило Трутона:
— const.
Это правило ограничено нормальной точкой кипения. „Константа* в действительности значительно изменяется, но для большого числа веществ в среднем составляет около 21. Ниже приводятся некоторые типичные значения LB/T, показывающие степень вила:
отклонения от пра-
Водород ........
Азот ... • . . .
Аммиак..........
Авилин .........
Этилацетат . . . Этиловый спирт . Вода..........•
Октан...........
10,6
17,3
23,2
21,1
22,0
26,8
26,0
20,4 •«
Более сложная зависимость, также приложимая кипения, дается следующим уравнением: 7?1пркрГ1 1 1
LB=^----------
к нормальной точке
Ркр 1-2в Ткр
(3S)
которое оказалось очень точным для различных веществ, но требует знания критических констант.
Тепловые эффекты физических процессов
441
Кистяковский [324] нашел, что очень простое уравнение
LB — v\nv (34)
является исключительно хорошим в нормальной точке кипения, причем L дается в миллилитр-атмосферах, если v— мольный объем в миллилитрах. Применение к пару законов идеального газа превращает это уравнение в более обычную форму
— =8,75 + 4,571 lg Т, (35)
где LB выражена в кал {моль, а Т—в градусах Кельвина. Кистяковский для 60 неполярных веществ нашел совпадение в пределах 3°/0.
Скрытая теплота парообразования как функция температуры. Гильдебранд [108] заметил, что изменение энтропии при парообразовании является функцией мольной концентрации пара. Льюис и Вебер [157] и Мак-Адамс и Морелл [160] развили эту идею посредством нанесения значений LBiT в зависимости OTlgp/T, поскольку р[Т пропорционально концентрации пара для идеальных газов. Можно провести две кривые, которые оудут примерно линенными, - одну для неполярных жидкостей и другую для полярных жидкостей, подобных воде и спиртам. Зависимость, представленная этими линиями, известна как „функция Гильдебранда“ и может применяться в широком интервале температур, но нарушается при приближении к критической области 6В.
Ватсон [252] нанес/ {L/T) в зависимости от т, где /—константа, характеризующая каждое вещество, и нашел, что данные для всех жидкостей, как полярных, так и неполярных, попадают на одну кривую. График можно применять для отыскания относительных значений LfT. Ту же зависимость можно написать в следующей алгебраической форме [253]:
L ____ /1 — т \ 0,38
М U — т1/
(36)
Значение L1 можно получить одним из методов, рассмотренных выше для нормальной точки кипения. Этот метод для нахождения скрытых теплот при всех температурах требует только знания критической температуры.
Из уравнения Клапейрона — Клаузиуса и уравнения давления пара (59, гл. VI) Ватсон вывел следующее уравнение для скрытой теплоты в нормальной точке кипения
L,= 0,95RB ++)’,
(37,,
которое затем сочетается с уравнением (36) в целях получения уравнения для вычисления L при любой температуре, из ркр, Т и Тв
442
IX. Передача тепла
[В — константа уравнения (59, гл. VI), которую можно определить по этим трем величинам].
Отмер [181] исходил из уравнения Клаузиуса—Клапейрона в форме
= (38)
Записав это уравнение для двух веществ Л и В и разделив друг на друга получающиеся уравнения для случая заданной температуры, он получил уравнение
интегрирование которого дает
1^Рд = ^1?/’в + с- (40)
Если логарифм давления пара нескольких веществ нанести в зависимости от логарифма давления пара стандартного вещества, то получаются прямые линии, наклон которых, согласно вышеприведенному уравнению, равен отношению скрытых теплот парообразования исследуемого и стандартного веществ при данной температуре. Таким образом, если известна скрытая теплота парообразования стандартного вещества как функция температуры, то возможно вычислить скрытую теплоту парообразования других веществ, если для них дано давление пара при двух температурах (одна из них может быть нормальной точкой кипения)66.
Позднее Отмер [182] дал видоизменение своего метода; он также исходил из уравнения (38), но внес во все соотношения критические параметры, т. е. пользовался равенствами р = ркргс и Т— Ткрт. Тогда делением при одинаковых т получим
d In к Z.7t„
Ж = (41>
Штрихом отмечены величины, относящиеся к стандартному веществу. Интегрируя, получим
(42)
По имеющимся опытным данным, константа интегрирования С оказалась равной нулю. Следовательно, можно окончательно написать, что
Ткр^
(43)
Тепловые эффекты физических процессов
443
где л и я' — приведенные давления пара соответственно исследуемого и стандартного вещества при условии, что оба эти вещества взяты при одинаковой приведенной температуре. В качестве последней можно выбрать нормальную точку кипения вещества. L и L' также берутся для одинаковой приведенной температуры. С помощью этого уравнения можно вычислить скрытую теплоту парообразования при любой температуре, если даны только нормальная точка кипения, критическая температура и критическое давление67.
Мейснер f 1’69j, исходя из уравнения Клаузиуса — Клапейрона, введя коэфициенты сжимаемости и приняв линейную зависимость igp—1]Т для давления пара до критической точки, пришел к уравнению
L
In тг---------------
R(CT— Сх)Ткр
(44)
в котором тг и т соответственно — приведенное давление пара и температура кипения, а Сг и Сж соответственно — коэфициенты сжимаемости пара и жидкости. Так как Сг и Сж можно представить как функции пит, то LfTKp является однозначной функцией этих двух переменных. Мейснер дал график этой функции, по которому можно определить А, если—известны давление пара______и критическая
температура вещества. Давление пара в широком температурном интервале можно определить по двум известным значениям с помощью линейной зависимости 1g р— 1/У. Этот график был использован Мейснером при вычислении L для различных жидкостей при различных условиях, и сравнение с экспериментальными данными показало среднее отклонение меньше 5% с максимальным отклонением только в + 9%68.
Применение различных методов расчета иллюстрируется следующим примером.
Пример 5. Вычислить скрытую теплоту парообразования: 1) этилового спирта при 0°С; 2) аммиака при 100° С; 3) н-бутана при 115,7° С. Результаты расчета сопоставить с опытными данными (соответственно 10110, 3050 и 2980 кал1моль).
Необходимые для расчета свойства указанных веществ прив едены ниже
Вещество rB, ° к гкр, ° к р , атм Давление пара при рассматриваемой температуре, атм
с,н5он 351,5 516,3 63,1 0,016
NH3 240,0 405,5 111,5 61,8
h-C^Hjq 272,7 426,2 36,0 20,4
Метод Кистяковского. Этот метод не является строго приложимым ни к одному из этих случаев, но будет использован для этилового спирта, чтобы показать, какова может быть ошибка, есля не соблюдать ограничения.
L = 273,2 (8,75 + 4,5711g 273,2) = 5440,
г. е. ошибка около 50% ю.
444
IX. Передача тепла
Использование функции Гильдебранда. При использовании графика, данного в статье Мак-Адамса и Моррела для бензола при расчете для бутана, и графика для воды в случае расчета для аммиака получаем следующие значения:
Вещество 1000-^4^ l L
CjHgOH 0,059 37 10100
NHj 165 12 4470
w-C4Hj0 52 10,5 4090
Этот метод достаточно точен для низких давлений, но дает слишком высокие результаты при высоких давлениях.
Метод Ватсона [сочетание уравнений (36) и (37)]. Константу В в уравнении
1пр = А— В-дь (59, гл. VI)
1 --
вычислим по нормальной точке кипения и критической температуре.
Уравнение для этого случая будет
2,3031g (ркс/рв* [1/(7в — 43)] — [1/Ткд — 43)] ’
Значения В, Lg и L следующие:
Вещество В LB L
C2H6OH 3690 9050 10500
NH3 2040 5700 3030
и-С^Ню 2130 5650 3300
Совпадение для всех трех случаев очень хорошее.
Методы Отмера. При использовании первого из методов Отмера мы вычислим только скрытые теплоты парообразования этилового спирта и бутана, используя воду в качестве стандартного вещества для первого и аммиак для второго. Вода непригодна в качестве стандартного вещества ни для аммиака, ни для бутана, если нормальные точки кипения выбираются для одного из известных давлений пара, потому что температуры слишком низки. В добавление к нормальной точке кипения мы выбираем давление пара при температуре, при которой следует определить скрытую теплоту парообразования. Величины, которые следует нанести на логарифмические координаты, следующие:
1. С2Н5ОН:
f = 78,3°C, Рс2н5он = 760 мм, />н,о~332 мм, t — 0° С. T^c^HgOH — 12,2 мм. ^н2о — 4,58 мм.
2. Бутан:
t = —0,5° С, Рс.Ню — 760 мм, Pnh3 = 4,23 атм, t = 115,7° С. Рсл.» = 2®’4 Pnh3 = атм.
Тепловые эффекты физических процессов
445
Практически лучше получить наклоны из соотношения
Д IgPa наклон = . , д , AlgPB чем при использовании графика.
Для этилового спирта:
2,881 — 1,086 наклон = ----7,= = 0,965.
2,522 — 0,661 ч
L для воды при 0° С = 10760.
L для СгН5ОН = 0,965X 10760 =10390.
Дли бутана:
наклон = 1,013,
L для NH3 при 115,7° С = 2160,
L для бутана = 1,013 X 2160 = 2190.
Совпадение хорошее в случае спирта, но много хуже в случае бутана. Без сомнения, совпадение было бы лучше при использовании другого стандартного вещества. Это обстоятельство иллюстрирует одну из главных трудностей метода, именно — надлежащий выбор стандартной жидкости.
Второй метод Отмерз будет детально рассмотрен для этилового спирта;
-двух других веществ- будет приведен только конечный результат. 351,5 ' этилового спирта при нормальной точке кипения = _ -Dlu,O
0,680.
0,01586.
г. этилового спирта при нормальной точке кипения = =
По условию,
откуда
тс»н5он — тн5о>
rHso = -(^)н2о = °’680 X 647,3 = 440° К.
Давление пара Н2О при 440° К = 7,3 атм.
_ Р _ 7,3 _ 'н=° 217,7 ~
0,0336;
— 1,7997
— 1,4737
1,221.
FIq упяр.црнИЮ (43),
При 0°С
^ = ^Х 1,221 =0,997. L 64/
*с.,н5он — 51 gi3 — °-529 — тн,о> rHs0 = 0,529 X 647 = 342° К, /.н„0 при 342° К =10 060, £Cjhsoh = °>977 X Ю 060 = 9850.
446
/X. Передача тепла
По тому же самому методу получим
Z.NHi при 100° С = 3080,
£с4н,„ при 115,7° С = 3160.
Совпадение между вычисленными и экспериментальными значениями вполне удовлетворительное во всех трех случаях.
Метод Мейснера. Этот метод требует знания давления пара при температуре, для которой желательно определить скрытую теплоту парообразования. При отсутствии кривой давления пара можно использовать линейную зависимость 1gр—l/Т, определяя две константы по нормальной точке кипения и по одной точке, в качестве которой может быть взята и критическая точка. Для этого примера мы будем пользоваться данными, взятыми из указанной выше литературы. Значения приведенных давления и температуры для трех веществ указаны в первых двух графах следующей таблицы. В третьей графе указаны значения LjTKp, считанные с графика Мейснера, а в последней — вычисленное значение L.
Вещество 1С т L 1 кр L
С2Н5ОН 2,54 X 10"4 0,529
NH3 0,555 0,920 1А 3000
h-C4H10 0,566 0,912 6,2 2640
В случае C2HSOH давление настолько низко, что Сг — Сж в уравнении (44)= 1, и L можно вычислить непосредственно, не обращаясь к графику. Результат равен 9550. Во всех трех случаях совпадение с экспериментом является достаточно удовлетворительным.
Теплота плавления. Эту величину, являющуюся разностью энтальпий 1 моля жидкости и 1 моля твердого тела, можно вычислить по данным для температуры затвердевания растворов при применении уравнения (206, гл. IV) в форме
(<?1п——\
= (45)
где х"—мольная доля А в жидкой фазе и х’—мольная доля А в твердой фазе. Уравнение предполагает идеальность растворов в обеих фазах. Принимая за постоянную величину, получаем
Согласно этому уравнению, график lnx“/x'—1/Т (где 1/Т—обратная температура затвердевания) будет давать прямую линию, наклон которой равен —&HAR.
Тепловые эффекты физических процессов
447
Для случая, когда твердая фаза является чистым компонентом или когда концентрация одного компонента в твердом растворе невелика, уравнение (46) переходит в
_д и
1вх’ = —^Ц-С. (47)
KJ
Келли [130с] широко использовал эти уравнения для вычисления скрытой теплоты плавления неорганических веществ. Уравнение (47) также полезно для вычисления растворимости чистого твердого компонента в идеальном жидком растворе, если известна теплота плавления компонента.
Эятальпия растворов. Постоянный состав. Случай раствора, нагреваемого или охлаждаемого при постоянном составе, в сущности совпадает со случаем одного компонента. Конечно, требуются данные по теплоемкости раствора, причем желательно было бы вычислить их по теплоемкостям чистых компонентов, но в этом отношении достигнут лишь незначительный успех; поэтому следует обратиться к экспериментальным данным. Так как теплоемкость является экстенсивным свойством, то применяют общие зависимости, справед-ливые для такого свойства; тогда для рассматриваемого случая получим соотношение
cp=s«A+5.,N>+--
(в данном случае мы будем рассматривать изобарную теплоемкость? или для теплоемкости на моль (или на единицу массы)
^=Ср/д + %хв+... . (48)
Парциальные мольные величины являются функциями -температуры, природы компонента, состава раствора и в меньшей степени — давления. Из уравнения (116, гл. IV) очевидно, что для компонента идеального раствора
^ = СА’
что означает независимость Ср от состава. Следовательно, для этого специального случая теплоемкость раствора крайне просто вычисляется по теплоемкостям чистых компонентов. Для неидеальных растворов в настоящее время мы не имеем путей вычисления теплоемкости. Допущение идеальности растворов является удовлетворительным приближением в одних случаях, но может привести к совершенно ошибочным результатам в других. Если, например, по имеющимся данным для теплоемкости водных растворов азотной кислоты вычислить парциальные мольные теплоемкости, то результаты показали бы, что парциальная мольная теплоемкость воды при комнатной температуре изменяется в пределах от 0,76 до 1,42 нал)г-град. До тех пор, пока не будет найден метод вычисления парциальных мольных теплоемкостей из каких-либо других величин, кроме теплоемкости для данного
448
IX. Передача тепла
раствора, они совершенно бесполезны, потому что, располагая такими данными, мы уже не будем нуждаться в парциальных мольных величинах. То же самое в большей или меньшей степени справедливо в настоящее время для всех парциально-мольных величин, между тем как у многих авторов имеется тенденция преувеличивать их важность.
Теплоты растворения и разбавления. Существует много важных промышленных операций, в которых растворы либо получаются из компонентов, либо смешиваются, разбавляются и концентрируются, причем такие изменения связаны с тепловыми эффектами, вызываемыми изменением концентраций. В этом разделе мы будем разбирать именно такие тепловые эффекты и, более кратко, их зависимость от состава и температуры.
Если смешиваются две чистые жидкости, например спирт и вода, или растворимое твердое тело и жидкость, например соль и вода, то наблюдается тепловой эффект, который может быть положительным (тепло выделяется) или отрицательным (тепло поглощается). Если два компонента имеют одинаковую температуру и образующийся раствор приводится к той же температуре, так что весь процесс является изотермическим, то тепловой эффект называют интегральной теплотой растворения. Для получения определенной величины она должна быть отнесена к данной массе какого-либо из компонентов или к массе раствора. Обычно если одним из компонентов является твердое тело, то выбирается единица массы или один моль этого компонента; если оба компонента являются жидкостями, то обычно теплоту растворения относят к единице массы или к одному молю раствора. Эта величина в общем представляет функцию давления, температуры и концентрации образующегося раствора. Большинство теплот растворения определено при стандартном давлении в 1 атм и стандартной температуре в 18 или 25° С. Влиянием давления обычно можно пренебречь, так как оно очень малб в случае жидкостей, а в случае газов теплота растворения является второстепенным эффектом, исключая случай очень высоких давлений.
Изменения интегральной теплоты растворения с температурой иллюстрируются некоторыми данными для растворов этиловый спирт— вода, приведенными на рис. 70.
На моль растворенного вещества теплоты растворения могут достигать таких больших величин, как 20 000 кал, или, другими словами, они могут быть величинами такого же порядка, как большинство теплот химических реакций. Во многих случаях при процессе растворения происходит или химическое взаимодействие, или процесс, очень близкий к нему. Имея дело с такими процессами, как образование раствора серной кислоты : ли олеума из SO, и воды, удобно трактовать их так, как если бы они были чисто физическими процессами, т. е. игнорировать взаимодействие.
Тепловые эффекты физических процессов
449
Если один из компонентов добавляется к уже образовавшемуся
раствору, то тепловой
вается теплотой разведения. Очевидно, что разность между любыми двумя теплотами растворения для двух различных концентраций представляет просто теплоту разведения между этими двумя пределами концентраций (отнесенными, конечно, к равной массе). Практически теплота растворения обычно измеряется путем определения теплового эффекта при образовании концентрированного раствора из двух чистых компонентов и последующего измерения те
эффект изотермического процесса назы-
Рис. 70. Интегральная теплота растворения в системе вода — этиловый спирт как функция температуры и состава.
Na
"в
Рис. 71. Интегральная теплота растворения.
плоты разведения.
Если к раствору двух компонентов добавляется бесконечно малое количество одного из компонентов, то тепловой эффект для этого случая называется диференциальной теплотой растворения.
Так как теплоты растворения или разведения представляют собой изобарные тепловые эффекты, то они могут быть выражены как разности энтальпий. Зависимость между энтальпией раствора, энтальпиями чистых компонентов и интегральной теплотой растворения можно очень легко
определить при рассмотрении процесса смешения, показанного на рис. 71. Применяя первый закон, имеем
^ + ^b-Q = (^ + Nb)/7s, (49)
где N — число молей компонента А,
NB — число молей компонента В,
НА—мольная энтальпия чистого А при данных давлении и температуре,
Нв— та же величина для В,
29 Химическая термодинамика
450
IX. Передача тепла
Q—тепловой эффект изотермического процесса (положительный, если теплота выделяется),
Н§—энтальпия получающегося раствора.
Посредством деления на NA и перегруппировки получаем уравнение
Hs = xHA-\-(\—x)HB—xqA, (50)
в котором qA — интегральная теплота растворения на моль компонента А, а х — его мольная доля.
Подобно этому, мы можем получить
Hs=xHA-\-(l—x)HB — qs, (51)
где qs—интегральная теплота растворения на моль раствора. Эти уравнения позволяют вычислить энтальпии растворов вдоль изотермы по измеренным значениям интегральной теплоты растворения. Имея дело с любым физическим процессом, можно выбрать энтальпии отдельных компонентов совершенно независимо друг от друга; представляется удобным считать как НА, так и Нв равными нулю на основной (стандартной) изотерме. В некоторых случаях имеются в распоряжении данные по энтальпии растворов и двух чистых компонентов, но они основаны на различных величинах, и задача состоит в нахождении соотношения между ними. Это легко осуществляется прн применении уравнения (51) к некоторому состоянию, для которого известна теплота растворения или для которого ее можно принять равной нулю. Это будет давать вычисленную Hs, а разность между вычисленными и табличными значениями Hs является константой и может быть использована в качестве поправочного члена для целого ряда данных.
Пример 6. Недавно опубликованы таблицы термодинамических свойств воздуха [259]. Желательно найти поправочный член для приложения к значениям энтальпии в этих таблицах для приведения их к той же самой основе, что и данные для кислорода и азота Миллара и Сюлливана [171]; это дает возможность использовать все три ряда данных в одной задаче.
При давлении в 1 атм и температуре, близкой к комнатной, воздух можно считать идеальной смесью. Поэтому при данных давлении и температуре
//воздуха = 0,217/q^0,79/Zn,. (а)
При t=15° С и р — 1 атм. //воздуха по таблице Вильямса = 122,4 ккал/кг или 122,4 X 29 = 3550 кал/моль. Значения для О> и N2 при тех же самых условиях и в тех же самых единицах равны
//о, = 3094,
//я, = 2727.
Подставляя в (а), получаем
//воздуха = 2808.
Тепловые эффекты физических процессов
451
Поправка = 3550— 2808 = 742. Другими словами, любое значение энтальпии в таблице Вильямса может быть приведено к данным таблиц Миллара — Сюл-ливана вычитанием величины 742 из значения, данного в кал! моль или 742/29 = 25,6 из действительных табличных значений в ккал/кг.
Если процесс смешения (рис. 71) проводится только с бесконечно малым количеством А, т. е. с dNA, то тепловой эффект является ди-ференциальной теплотой растворения. Количественная зависимость получается путем диференцирования уравнения (49) по NA.
=? -и _м _(W д_м)Ж . (52)
\0Na)t,nb а а 5 ' А 1 B'\dNAJr,NB х '
Исключение dNA посредством соотношения
dNA = (NA-}-NB)1^ дает
— НА Щ ) Т. (53)
Диференцируя уравнение (51), получаем
— Нв- (54)
Сочетая уравнения (51), (53) и (54), получаем
<7.4 = <7.<?~М1 —х)
Уравнение (55) можно найти непосредственно из общих уравнений для экстенсивных свойств, приведенных в гл. IV. Так, если рассматриваемым свойством будет энтальпия, то
и
(56)
(57)
Уравнение (55) можно вывести из уравнения (56), поскольку теплота растворения является разностью двух энтальпий. Так как
Я5 = Яах + Яв^~*)> (58)
то уравнение (51) можно видоизменить, придав ему следующий вид:
Л5 = х//д + (1 — х)Нв — qAx— qB{\~ х). (59)
29*
452
IX. Передача тепла
Рис. 72 иллюстрирует на диаграмме энтальпия — концентрация связь между некоторыми рассмотренными величинами. АВС—изотерма,
Рис. 72. Соотношение между энтальпией и теплотой растворения (при х = 0,77=0).
связывающая энтальпию с концентрацией компонента А в бинарном растворе. Из уравнения (51) очевидно, что ВО является qs, т. е. интегральной теплотой растворения на моль раствора для раствора состава xt; в этом случае она положительна (теплота выделяется). EF—касательная к изотерме при Хр следовательно, АЕ— ди-ференциальная теплота растворения компонента В в растворе данного состава, a CF—та же величина для компонента А.
Это ясно видно из следующих рассуждений, основанных на данных рис. 72.
Наклон EF равен BGjEG и BQ/EG ==BGjxl.
Следовательно, при хп
н, по уравнению (57), — ВО ==ЕО = Н„. О в
Но по уравнениям (53) и (56),
Ча =Иа иа, и, подобно этому, _ _
Чд — Ид Ид.
Следовательно, так как __ _____
АО — ЕО — АЕ, то __
Ид Ид — АЕ = qB.
(60)
(61)
Такая диаграмма, как показанная на рис. 72, предполагает полную смешиваемость во всем интервале составов от чистого В до чистого А. В тех случаях, когда А является твердым телом, будет наблюдаться ограниченная растворимость, и изотерма, так же как и кривая АВС,
Тепловые эффекты физических процессов
453
должна обрываться при составе, отвечающем концентрации насыщенного раствора.
Использование уравнения (51) для вычисления энтальпии раствора предполагает, что стандартным состоянием для обоих компонентов будет состояние чистого вещества. Однако не всегда удобно выбирать это стандартное состояние; например, если имеются данные по теплоте разведения, но нет данных по теплоте растворения, то нет способа для перехода при расчете от состояния чистого А к состоянию какого-нибудь раствора. В таких случаях обычно в качестве стандартного состояния для растворенного вещества выбирают бесконечно разбавленный раствор и полагают, что для этого раствора при некоторой одной основной температуре //д = 0.
В этом случае для вывода уравнения, аналогичного уравнению (51), поступают следующим образом.
Рассмотрим исходный раствор, содержащий NА молей А и Л7В молей В, мольная энтальпия которого равна Hs. Добавим растворителя для разбавления раствора в такой степени, чтобы раствор содержал Л/в молей В. По первому закону
QD={NA+NB)Hs^^B — NB)HB—(NA — N'B)Tfs, (62)
где Qd—общая теплота разведения.
Так как
(^ + Ув)/4 = Л/л7/д + ^Нв. (63)
то уравнение (62) переходит в
QD= (^д + »В) Hs + ^вНв — NBHB — NAH'A— N'bH'b. (64)
Если конечное состояние является состоянием бесконечно разбавленного раствора, то
Qo== Qoo
И X
Н'В = НВ.
Поэтому уравнение (64) примет вид
Q^ = (Na^Nb)Hs — NJI'a — NbHb. (65)
Теперь, если выбрать в качестве стандартного состояния для А бесконечно разбавленный раствор и допустить, что
то при стандартной температуре получим
qO0=(na+nb)hs—nbhb (66)
454
/X. Передача тепла
или, относя теплоту бесконечного разведения к 1 молю растворенного А, получим
= = (67>
или
/7s = x9oo + (l— х)Нв. (68)
Теплоту бесконечно большого разведения можно получить при нанесении на график теплоты растворения на моль растворенного вещества в зависимости от числа молей добавленного растворителя. ТУ* тч т v тч п п Лжг т1 л’т» пгч t »Л тт т л»г» тг кАПИПАитп ттт тт/чЛ о л»»ж»«тТ‘Т,/Ч1’О Лчгтжгч'т’
напоил 1UV71 л i u^nouuia.iunun atninuх v/х v,
величина q^. Более удовлетворительный метод экстраполяции данных по теплоте растворения указан Россини [205], который показал, что теория Дебая — Гюккеля ведет к следующему предельному закону (когда концентрация достигает нуля):
?D=a/^
где qD — теплота разведения от данной концентрации с до бесконечно разбавленного раствора на моль растворенного вещества;
a — константа, равная 435 для одноодновалентных электролитов при 18° С, если qD дается в калориях на моль, а с является мольностью.
При нанесении экспериментальных данных на график qD— у/с прямая линия, представляющая предельный закон, позволяет экстраполировать до с = 0.
Пример 7. 100 кг раствора олеума, содержащего 15,4°/0 свободного SO3, следует развести чистой водой так, чтобы приготовить 30,8-процентный водный раствор H2SO47 Какое количество тепла будет выделяться при этом, если исходная и конечная температуры равны приблизительно 18° С?
Эта задача была разрешена Моргеном [174] при исследовании парциальных мольных величин. Здесь она будет решена непосредственно с использованием данных по интегральной теплоте растворения, заимствованной из его статьи.
Основание: 100 кг олеума.
15,4 . 84,6 . _7
Моли SOs=-^+-gg-= 1,057.
Моли Н2О = 0,860. » 9о
Мольная доля SO3 — 0,55.
Мольная доля SO3 в конечном растворе равна _______________________________W8)________________= (30,8/98) 4- (69,2/18) ф- (30,8/98) и’
Общее количество молей конечного раствора =1^5Z= 15,09.
Тепловые эффекты физических процессов
455
По таблице в. статье Моргена данные по интегральной теплоте растворения равны следую щим величинам:
Мольная доля SO3
0,55
0,07
4s 10100 2620
Выделяющаяся теплота представляет разность между интегральными теплотами растворения исходного и конечного растворов. Следовательно, выделяется 15,09X2620 — 1,917X 10Ю0 = 20230 ккал теплоты. Этот метод много проще метода, использующего парциальные мольные величины, и, возможно, более точен, поскольку результаты не зависят от определения наклонов.
Пример 8. 23,2-процентный раствор H2SO4 (раствор 1) следует укрепить до содержания 80,6% H2SO4 (раствор 3) добавлением олеума (раствор 2), содержащего 41,2% свободной SO3. Сколько тепла следует отнять на 100 кг исходной кислоты, чтобы температура приняла первоначальное значение в 18° С (пример совпадает с примером в статье Моргена) [174]. Пусть Qt — тепловой эффект при изотермическом образовании раствора 1 из SO3 и Н2О;
Q2—тепловой эффект при изотермическом образовании раствора 2;
<?3— тепловой эффект при смешении растворов 1 и 2 для образования раствора 3;
<?4 — интегральная теплота растворения для раствора 3.
Так как все эти тепловые эффекты являются значениями Д/7, то образование раствора 3 из компонентов через промежуточные стадии образования растворов 1 и 2 и последующее смешение их должны давать тот же самый тепловой эффект, что и непосредственное образование раствора 3 из компонентов.
Следовательно, Qi + + Qs = Gi- (а)
Возьмем за основу 100 кг раствора 1.
13 - 1 23,2 I 76’8 Л
Число молей раствора 1 = X 2 -|—= 4,738.
Уо 1о
Мольная доля SO3 в каждом растворе легко получается по способу, указанному при решении примера 7. Результаты таковы:
Раствор 1 Раствор 2
Мольная доля SO3 = 0,050 0,650
Раствор 3 0,302
Пусть W—число молей добавляемого олеума.
Тогда 4,738 X 0,050 + 0,650 0,302 (W+4,738)
N —3,43.
Из таблицы, помещенной в статье Моргена, теплоты растворения на моль раствора соответственно равны:
ХН1 = —1890, Д Н2 - — 8520, Д % = — 8780.
В соответствии с уравнением (а) получаем
Qs=8780 X 8,167 — 1890 X 4,738 — 8520 X 3,93 = 33570 ккал.
456
IX. Передача тепла
Теплота парообразования. Различают диференциальную и интегральную теплоту парообразования бинарного раствора. Первая является тепловым эффектом при испарении бесконечно малого количества жидкости с получением равновесного пара и непосредственно связана с давлением пара раствора, как показано в гл. IV. Следовательно, с помощью уравнения (155, гл. IV) можно вычислить дифереициаль-ную скрытую теплоту парообразования, если известны данные по давлению пара раствора при постоянном составе и по парциальным объемам двух фаз. Для идеального раствора применяется уравнение (187, гл. IV), и для этого специального случая можно написать, что.
7. (диференциальная) —- LАу -J— LB (1 —_)’). (69)
Интегральная теплота парообразования представляет теплоту полного парообразования жидкости при постоянном составе, причем существует два вида теплоты — один для процесса при постоянном давлении, а другой — для процесса при постоянной температуре. Мы будем иметь дело только с первой величиной. Она может быть связана с другими величинами следующим образом.
Для пара и жидкости соответственно:
Н3=уНА-[-(1-у)Нв, (70)
= + (71)
Пусть Hs — hs = Lp, х = интегральной теплоте парообразования при постоянных давлении и составе.
Отметив, что y=zx, можно записать
LptX=^x(HA A^)-|-(l х)(Нв hg). (72)
По уравнению (60)
На = на—9а=нЛ>+ ( cPAdT^-7A< (73)
ТА — температура кипения чистого А при давлении раствора, —температура точки росы раствора, С„ —теплоемкость чистого А в паре, и q"A — диференциальная теплота смешения А в паре.
Аналогичным уравнением для жидкости будет
= ( C'pdT— q (74)
< А А
где Т2 — температура точки пузырьков70.
Тепловые эффекты физических процессов
457
Вычитая уравнение (74) из уравнения (73) и отмечая, что
LA'—Ha,, — Ад, имеем
Яд-йд = Лд + ? (C"p-C'p)dT + \c"pdT^q’-q" (75) .1 A A J А л л
Та
и подобное же уравнение для компонента В.
Подстановка уравнения (75) и аналогичного уравнения для В в уравнение (72) и использование уравнения (58) и аналогичного ему уравнения для пара приводят к уравнению
т,
Ap,x=W+zBo(i-x)4-xj (С; -с; )</г+ ч, А А
Т2 Ту
+(!-*)? «’’+* J cP/r+
Ту +(1-х>и/7'+<“'£ (76)
# в
Мы можем без заметной ошибки считать пар идеальной смесью,, а это приведет к уравнению
с;="с;л+(1-х)С^’
<=°-
Кроме того, температурные пределы настолько малы, что все С можно считать постоянными. При этих допущениях уравнение (76) переходит в уравнение
L„, х = LAox + LBo (1 - х) + q ; + х (С"^ — С'^) (Т2 - 7д) +
+ <1-х)(С; -с; )(7'2-7'в,) + С;(7’1-7'2). (77>
в в
По этому уравнению можно вычислить скрытую теплоту растворения из скрытых теплот парообразования чистых компонентов, из теплоемкостей чистых компонентов и из интегральной теплоты растворения. Если раствор является идеальным, то ^—0; если пренебречь членами, содержащими теплоемкости, поскольку они малы па сравнению со скрытыми теплотами парообразования, то в качестве первого приближения будем иметь
Lp,x = Laox-]-Lbo(1— х). (78>
458
/X. Передача тепла
Имеется очень немного данных по проверке и применению этих соотношений. Хотя они во многих случаях и не могут прилагаться с достаточной точностью, все же они оказываются полезными в качестве приближений и для качественных заключений. Например, уравнение (77) показывает, что скрытая «теплота парообразования раствора приблизительно равна сумме теплот парообразования компонентов плюс интегральная теплота растворения71.
Для случая раствора нелетучего вещества подобным путем можно вывести уравнение
LB=hBo-YLBo j + x2)hB,~ x2q2\ —
*В
[XlflA, + (1 — Xj) hB1 — X^J,
(79)
где Lb—изобарная скрытая теплота парообразования растворителя, в котором растворена мольная доля хг для получения раствора с мольной долей х2;
hB„ — энтальпия чистого жидкого В (растворитель) в точке его - кипения Тв;
Lb„—скрытая теплота парообразования чистого В при давлении, равном давлению над раствором;
С" —мольная теплоемкость пара В;
рв
На, — энтальпия растворенного вещества в точке кипения раствора 2;
Ив,— энтальпия растворителя в точке кипения раствора 2;
йдиАд— аналогичные величины для раствора 1;
q2 — интегральная теплота растворения для раствора 2 на моль растворенного вещества;
qx— интегральная теплота растворения для раствора 1 на моль растворенного вещества.
Пар растворителя принимается выделяющимся при температуре Т2.
Если мы пренебрежем разностью в теплотах, вызывающих изменение температуры, то уравнение (79) примет вид уравнения
i8 = U + /^(?i-?2), (80)
которое для большей части целей оказывается достаточно точным.
Энтальпия растворов по данным давления насыщенного пара или данным э. д. с. Если имеется только ограниченное число данных по теплотам растворения или разведения, то можно восполнить
Тепловые эффекты фазических процессов
459
этот пробел, используя данные по давлению насыщенного пара. В гл. IV было показано, что
д In ai
дТ~
Hi —
(ПО, гл. IV)
Заменим на /j-//? и будем считать, что —/в (если имеем дело с растворителем) и что стандартным состоянием является состояние чистого жидкого растворителя; тогда уравнение (ПО, гл. IV) переходит в
dln-^- = —HB~”B dT, (81)
или, для случаев, когда пары можно считать идеальными газами,
d In = — Нв ~ъ-Ц dT. (82)
Рв RT2 '
Из графика, представляющего зависимость In (Рв1рв) от Т при постоянном х, можно определить наклоны, а следовательно, Нв — Нв вдоль изотермы. Затем, на основании известных значений Нв, вычисляем Нв. Из Нв можно найти НА, если использовать обычную зависимость для парциальных мольных величин [см. уравнение (9, гл. IV)], которая для данного случая примет вид
^dHA=~^^-dHB. (83)
Затем с помощью уравнения (70) или (71) легко получить энтальпию раствора. Для иллюстрации использования данных по давлению насыщенного пара при вычислении энтальпии раствора можно сделать ссылку на статью Хальтенбергера [99], который подсчитал несколько изотерм энтальпия — концентрация для растворов соды с помощью зависимости Дюринга для давления насыщенного пара. Энтальпию раствора для случая ионизующего растворенного вещества можно получить из измерений э. д. с. соответствующего гальванического элемента. Из этих данных можно вычислить активность растворенного вещества, а затем, применив уравнение (ПО, гл. IV), найти разность энтальпий Н{ — Н9, или, как ее обычно называют, относительную мольную энтальпию. Такие данные по растворам НС1 при температуре от 0 до 50° С и для концентраций до мольности, равной 16, были получены Акерлофом и Тиром [3] и использованы ими для вычисления различных термодинамических свойств, включая вышеупомянутую разность энтальпий.
460
IX. Передача тепла
N,
Рис. 73. Простой процесс смешения.
ДИАГРАММА ЭНТАЛЬПИЯ —КОНЦЕНТРАЦИЯ
Приложение к задачам смешения. Если требуется решить большое число задач, включающих тепловые эффекты, сопровождающие изменения концентраций данного бинарного раствора, то оказывается очень удобным пользоваться диаграммой энтальпия — концентрация. Некоторые авторы называют ее „диаграммой Меркеля", по имени инженера [170], который применил ее для задач с тепловыми эффектами. Эту диаграмму также соединяют с именами Поншона [191] и Савяри Г225], которые применили ее к расчетам по перегонке. Наиболее значительное развитие ее применения дал Бошнякович [24], к книге которого следует обратиться, поскольку она содержит значительно больше материала, чем можно дать здесь.
Прежде чем остановиться на подробном анализе построения такой диаграммы, кратко рассмотрим некоторые ее свойства. Рассмотрим простой процесс смешения, представленный на рис. 73. Составим три нижеприводимых баланса для случая адиабатного процесса:
N,
N-
Q
M+^ = 7V3, (84)
AZjX, Ц- N2x2 — N3x3, (85)
ад + ад = ад. (86)
Можно пользоваться либо весами и весовыми долями, либо молями и мольными долями. Исключение масс трех компонентов дает уравнение
хз — ___7/3 — Нх .g_.
х2-хх~Н2-Нх' ' 9
Это уравнение на диаграмме, координатами которой являются Н и х, изображается как прямая линия, проходящая через три точки (Нх, х,), (Н2, х2) и (Н3, х8), как показано на рис. 74. Это положение непосредственно следует из подобия соответствующих треугольников на рис. 74.
Теперь рассмотрим случай неадиабатного процесса смешения. Первые два уравнения баланса не изменяются, но третье принимает следующий вид:
+ ад=ад + q. (88)
Пусть q3 = Q/N3 == тепловому эффекту на единицу веса (или на 1 моль) потока 3. Тогда уравнение (88) можно написать в виде
ад+ад=ад, (89)
Диаграмма энтальпия — концентрация
461
где
= +
Уравнение (89) теперь имеет ту же самую форму, что и уравнение (86), и ясно, что ряд из трех уравнений (84), (85) и (89) на диаграмме Н—х также изображается прямой линией, связывающей
Концентрация
Рис. 75. Применение диаграммы энтальпия — концентрация для определения теплового эффекта иеадиабат-ного процесса.
Концентрация
Рис. 74. Диаграмма энтальпия —
концентрация.
три точки (Hj, хД (Т/2, х2) и (Из, х3) (см. рис. 75). Если бы мы выбрали тепловой эффект, основанный на единице, то прямая линия связывала бы точки (//', xj, (Н2, х2) и (Н3, х3), где
Реальная диаграмма Н—х, конечно, имеет ряд изотерм, проведенных таким образом, что точка состояния легко определяется, если только известны температура и давление раствора. Для лучшего выявления основного свойства диаграммы, которая показана на рис. 75, все другие линии были опущены. Ясно, что данная диаграмма Н—х является диаграммой для одного постоянного давления.
Что касается рис. 74, то, если известны температура и концентрация растворов 1 н 2, на диаграмму наносятся точки 1 и 2, и затем наносится точка 3, поскольку х3 легко вычисляется. Таким образом, температура, получающаяся при адиабатном смешении, легко определяется по прямой линии. Для иеадиабатного процесса прямая линия определяет точку (х3, Н'3) и при известной температуре смеси (например при рассмотрении изотермического процесса) также
462
IX. Передача тепла
определяется точка (х3, Н3), а из нее сразу получается q3, как показано иа чертеже.
Пример 9. Требуется решить пример 7 с помощью графического построения на диаграмме энтальпия — концентрация.
Так как рассматривается только одна изотерма и так как энтальпию обоих компонентов можно произвольно принять равной нулю, то диаграмма для этого случая (рис. 76) представляет просто график интегральных теплот
растворения, нанесенных как функция концентрации. Изотерма построена по данным, приведенным в статье Моргена [174]. Прямая линия связывает точки Xi = 0, Нг = () (чистая вода) и х3 = 0,55, Нг —10100. Н, и Н3 расположены, как показано на чертеже, и их разность q3 = 1,35 ккал/моль конечного раствора. Поскольку УУ3—15,09, Q— 15,09 X 1,35 X 1100 = = 20300 ккал.
Пример 10. 500 кг 55-процентного (по весу) раствора NaOH при 40° С смешивается с 150 кг 15-процентного раствора при 100° С и получающийся раствор охлаждается до 35° С. 1). Сколько тепла следует отвести во время процесса и чему должна быть равна конечная температура смеси, если процесс будет адиабатным?
Диаграмма энтальпия — концентрация
463
Для расчета используем диаграмму энтальпия—концентрация для растворов NaOH, построенную Мак-Кэбом и воспроизведенную на рис. 77. Стандартным состояниям для этой диаграммы отвечают значения /7=0 для воды
Рис. 77. Диаграмма энтальпия — концентрация для растворов едкого натра.
при 0°С и Н—0 для NaOH в бесконечно разведенном растворе при 20° С. Концентрация конечной смеси х3 получается из баланса NaOH:
650*3 = 500 X 0,55 + 150 X 0Д5,
*з = 0,457.
По графику определяем следующие значения энтальпии:
Л/i (55-процентный раствор) = 103 ккал/кг,
Н2 (15-процентный раствор) = 89 ккал/кг,
Н2 (конечная смесь) = 66 ккал1кг.
Решение первого вопроса можно осуществить уже описанным графическим методом или, что значительно проще, воспользоваться следующим уравнением, легко получаемым при исключении масс из уравнений (84), (85) и (88):
Чг= Hi — T7S+ (//2 — ^1) г _г,‘
•*2 Л'1
464
/X. Передача тепла
Подставляя значения /7/ и xt, получаем
^ = 33,7 ккал на 1 кг конечного раствора.
Следовательно, Q3 = 650 X 33,7 — 21900 ккал.
2. Этот вопрос почти также легко решить с помощью уравнения, как и при графическом методе решения.
Из уравнения (87) получаем
/73 = 99,7.
На рис. 77 точка с координатами х3 = 0,457 и /73 = 99,7 лежит на изотерме t = 78, и следовательно, это и есть искомая температура конечного раствора.
в гл. XIII.
nijarnauuLi
гтгчтлтза ттлiti ж
Построение диаграммы энтальпия — концентрация. Как мы уже видели, некоторые задачи по смешению крайне просто решить непосредственно по диаграмме энтальпия — концентрация (или на основе эквивалентных данных в форме таблиц), но, к сожалению, таких данных в настоящее время существует очень немного. Поэтому желательно дать метод построения диаграмм и показать, какие именно данные необходимы.
Методы построения такой диаграммы в известной степени зависят от данных, доступных для изучаемой системы. Рассмотрим сначала систему из двух полностью смешивающихся жидкостей А и В (например этилового спирта и воды) и примем, что диаграмма применима только к области жидкой фазы. Примем, что имеются следующие данные:
1. Интегральная теплота растворения и разведения для одной изотермы, например для t = 20° С.
2. Теплоемкости чистых компонентов и нескольких растворов различных концентраций как функции температуры.
Будем считать, что как НА, так и Нв равны нулю при некоторой температуре, например при 0° С. Тогда две конечные точки расположатся на основной изотерме (той, для которой имеются теплоты растворения), что видно из соотношения
(90)
/7о = О для обоих компонентов по определению,
/0— температура основной изотермы в ° С.
Это определяет место точек Л и В на рис. 78. На основании данных по теплоте растворения вычисляют Н по уравнению (51) и наносят определенные таким образом значения на основную изотерму АСВ. Затем из соотношения
t
= (91)
*0
Диаграмма энтальпия — концентрация
465
где (Ср)х — удельная или мольная теплоемкость любого раствора, получают значение Ht на любой другой изотерме. Таким путем,
например, по исходной точке С определяется местоположение точки D на рис. 78. Для проверки полезны данные по теплотам растворения на других изотермах, кроме основной.
Если бы имелись в распоряжении: (1) теплоемкости двух чистых компонентов и (2) теплоты растворения при нескольких температурах, то данные (1) определяли бы конечные точки нескольких изотерм вдоль осей х = 0 и х = 1, а промежуточные точки тогда могли бы быть определены, по уравнению (51), из данных по теплоте растворения. Другие изотермы можно было бы интерполировать при нанесении на график Н—t сглаженных кривых для постоянных х.
Если бы система состояла из соли
данными по (1) теплоемкостям воды функций температуры и (2) по теплотам разведения при одной температуре t9, то тогда ход расчета несколько отличался бы от рассмотренного, так как мы не могли бы найти положения точек на оси х = 1. В этом случае можно использовать уравнение (68), определяющее значения вдоль оси х = 0, и, как уже было показано, можно получить (fa из данных по теплотам разведения.
Рассмотрим теперь случай двух полностью смешивающихся жидкостей, если в диаграмму следует включить как область жидкой фазы, так и область пара. Кроме уже указанных данных для этой диаграммы требуется: 1) скрытые теплоты паро-
Концентрация
Рис. 78. Построение диаграммы энтальпия — концентрация для случая двух полностью смешивающихся жидкостей.
и воды и мы располагали бы
и некоторых растворов как
Рис. 79. Диаграмма энтальпия — концентрация для областей жидкой и паровой фаз.
образования для обоих чистых компонентов; 2) теплоемкости чистых паров; 3) точки кипения как функ-
ции состава; 4) точки росы для раз-
личных смесей. Если доступны данные по теплоте парообразования при постоянном х, то могут оказаться излишними или (3), или (4). Как и раньше, НА и Нв могут быть приравнены к нулю при некоторой
30 Химическая термодинамика
3300
3200
3100
3000
2900
2800
2700
2600
2500
2400
2300
2200
2100
2000
1900
1800
^1700 1*1600 ** 1500
1400 1300
1200
1100
1000
900 800
700
600
500
400
300
200
юо
о
<R
о;
ззоо 3203 ЗЮО 3000 2900 2800 2700 2600 2500 2400 2300 2200 2100 2000 1900 1800 1700^ 1600 1500 1400 1300 1200 1100 то 900 800 700 600 500 400 300 200 яОЦ'ЮО , , щь|
0,10 0,200^0 0,400.50 OJBO 0,70 ЦК Q&IJX)0 Мольная доля кислорода
Рис. 80. Диаграмма энтальпия — концентрация для смесей кислорода и азота при давлении в 1 атм.
Базис: Н = 0 как для жидкого кислорода, так и для жидкого азота при нормальной температуре кипения азота. Тот же самый базис использовали Миллар и С юл Ливан [171J.
о Е
g
Тепловые эффекты химических процессов
467
температуре, которая может соответствовать или переохлажденной жидкости, или перегретому пару; для данного примера мы выберем первый случай. Изотермы в области жидкости до tA (точки кипения более летучего компонента, см. рис. 79) определяются по уже описанному методу. Выше этой температуры компонент А при данном постоянном давлении не будет устойчив как жидкость; но так как влияние давления на энтальпию жидкости может считаться ничтожным, то значение НА можно определить по давлениям выше давления насыщенного пара. Конечно, если изотермы определяются по значениям теплоемкости, а не по теплотам парообразования, то величина НА не будет необходима. В любом случае линия насыщенной жидкости легко определяется по точкам пересечения изотерм и вертикальных линий до составов, связанных с этими температурами линией точки кипения на диаграмме х—t.
Например, точка А на рис. 79 найдена пересечением изотермы и ординаты х, причем t2 и х являются координатами точек на линии точек кипения. Положение точек В и С на линии насыщенного пара найдено прибавлением скрытых теплот парообразования чистых компонентов к значениям Н на двух концах линии жидкости. Другие точки на линии насыщенного пара находятся подобным же путем, если доступны данные по скрытым теплотам парообразования при постоянном составе. Обычно такие данные недоступны, и для определения этой линии должны применяться другие способы. Теплоты расширения газов и паров при низких давлениях малы и без серьезной ошибки могут быть приняты равными нулю. Из уравнения (51) очевидно, что изотермы Н—х в области пара линейны, если qs=0, и поэтому они определяются только на основании данных для двух чистых компонентов. Ниже tB (точка кипения менее летучего компонента) паровая фаза неустойчива для смеси, богатой В, но изотерму все же можно провести, если принять, что давление более низко и что Н не зависит от давления. Точки на линии насыщенного пара легко определяются по данным для точки росы. Так, если t6 и у являются координатами точки на линии точек росы, то пересечение изотермы t6 и ординаты у определит точку на кривой насыщенного пара. Диаграмма Н—х для смесей кислорода и азота, охватывающая двухфазную область перегретого пара, дается на рис. 80. Линия, идущая между пограничными кривыми для двух насыщенных фаз, является соединительной линией (коннодой) для фаз, сосуществующих при равновесии.
ТЕПЛОВЫЕ ЭФФЕКТЫ ХИМИЧЕСКИХ ПРОЦЕССОВ
Определение теплоты реакции. Все химические реакции сопровождаются изменением энергии в результате действия сил химической связи. Если реакция протекает адиабатно, то изменение энергии, обусловленное реакцией, может привести к повышению или Зв*
468
IX. Передача тепла
понижению температуры системы в зависимости от того, является ли общим результатом уменьшение или увеличение химической энергии системы. Если последняя уменьшается, то должен быть соответствующий выигрыш какой-нибудь другой формы энергии; это означает, что должна увеличиваться кинетическая энергия молекул или потенциальная энергия, обусловленная их положением, или могут изменяться оба эти вида энергии одновременно. Если реагирующая система не изолирована от окружающей среды, то некоторая часть энергии проявляется в виде теплоты, переходящей к окружающей среде или от нее. Для частного случая, когда конечная-реакционная систвмз доводится до той же температуры, что и исходная система, тепловой эффект, сопровождающий это изменение, носит название теплоты реакции. Для того чтобы придать этой величине количественное значение, необходимо определить точное исходное и конечное состояния всех участвующих в реакции веществ и установить, происходит ли изменение при постоянном объеме или при постоянном давлении. Соотношения между этими величинами, уже рассмотренные в гл. III, являются важными, поскольку многие теплоты реакций измеряются при постоянном объеме. В нашем обсуждении мы ограничимся рассмотрением реакций, протекающих при постоянном давлении, так как они важнее в промышленности. Очевидно, что любая теплота реакции при постоянном давлении представляет разность энтальпий системы в двух ее состояниях. Мы можем, следовательно, определить теплоту реакции как изотермическую разность энтальпий продуктов реакции и веществ, в нее вступающих, причем все они должны быть взяты в некотором определенном стандартном состоянии, которое характеризуется указанием агрегатного состояния, давления и (в случае растворов) концентрации. Теплотам реакции обычно даются специальные названия в зависимости от типа реакции, например теплота образования, сгорания, диссоциации, гидратации или изомеризации. Теплоты образования, которые являются тепловыми эффектами любой реакции между простыми веществами с образованием данного соединения, особенно важны, так как из них сложением и вычитанием можно найти теплоты для реакций многих типов.
Теперь мы разберем эти положения на специальных примерах. Рассмотрим реакцию
SO2 + yO2=SO3, проведенную в несколько стадий, схематично изображенных на рис. 81. Допустим, что молей SO2 непрерывно изотермически при постоянном давлении в 1 атм смешивается с N2 молей кислорода. Любой процесс изотермического смешения дает тепловой эффект, даже если он, кдк в рассматриваемом случае (QJ, и очень мал. Затем смесь переводят в реакционный сосуд, где с помощью некоторых методов, описание которых для нас не является обязатель-
Тепловые эффекты химических процессов
469
ним, осуществляется реакция и выделяется или поглощается теплота Q2, что приводит к образованию смеси продуктов при температуре Т2. Затем эта смесь приводится к начальной температуре Ти что сопровождается тепловым эффектом Q3. Допустим, что реагирует Ns молей SO2 и что полученная смесь изотермически разделяется
fx Qz fe fl*
Рис. 81. Стадия простой химической реакции.
на Ns молей SO3 и —Ns молей SO2, и тогда (n2— кислорода остаются непрореагировавшими, причем этот процесс будет сопровождаться тепловым эффектом. Примем, что в конце концов весь SOs получается в твердом состоянии, которое будет равновесным для этих условий. Энергетический баланс для всего процесса отвечает равенству
N1H1 ^2^2 — Qi + Q2 + Сз + Qt + (wi — ^з) Нг -|-
+ (ед!^)едед,
в котором Нг, Н2 и Hs соответственно — мольные энтальпии SO2, О2 и SO3 (твердого) при Тг и 1 атм.
Тогда
дед=(М - Ч) Н. + (лг2 - 4 ^з) н2+ед - N.H. - n2h2 = = Ns (яз - Нг -1Я2) = - (Q1 + Q2 + Qg + Q4).
&Н на моль одного из реагирующих веществ или продуктов, например на моль SO2 или SO3, равно
^Нрбщ,____01 Ч~Ог Ч~Оз Ч~ Qt
А3 — Аз
Эта величина называется „теплотой реакции". Так как эта величина представляет разность энтальпий, то она не должна зависеть от отдельных стадий и от соответствующих им тепловых эффектов72. Поэтому фактическая температура в реакционном сосуде, т. е. температура Т2 продуктов реакции, будет несущественна. Однако эта
470
IX. Передача тепла
температура будет зависеть от исходного и конечного состояний системы, т. е. от состояния реагирующих веществ. Например, если SO3 будет выделяться в газообразном состоянии вместо твердого, то Н3 будет другой, и в результате получится иная теплота реакции. В рассматриваемом частном случае теплоты Q, и Q4 будут совершенно ничтожны, и ими можно пренебречь при экспериментальном определении теплоты реакции. Состояния реагирующих веществ и продуктов, к которым относятся данные теплоты реакции, обычно определяются следующим способом:
Н2(г, 1 атм) ±О2 (г, 1 атм) = Н2О (ж, 1 атм),
Д/У298 ~~-68318 КИЛОМОЛЬ,
где г означает газообразное состояние, а ж — жидкое.
Величина ДН298 означает, что в исходном и конечном состояниях температура равна 25° С или 298° К- В рассматриваемом примере очевидно, что реагирующие вещества следует взять в виде газов при 1 атм (или достаточно близко к 1 атм, чтобы изменение энтальпии, обусловленное изменением давления, было незначительно) ☆), и можно не указывать состояний, но все это не относится к продукту. При этой температуре и этом давлении жидкое состояние является единственным устойчивым состоянием воды, но при более высоких температурах устойчивым состоянием должно быть газообразное.
Задачи, связанные с определением теплоты реакции, обычно охватывают целый ряд температур, и при решении их удобно рассматривать любое данное вещество в стандартном состоянии. Например, может представить интерес реакция
со+н2о=cq2-i-н2
при 500° С и 1 атм, когда вода находится в газообразном состоянии. Теплота этой реакции определяется из теплот реакций, относящихся к 25° С и 1 атм, когда вода существует в виде жидкости. Однако можно рассматривать АН для этой реакции при 25° С с Н2О в газообразном состоянии и, следовательно, при давлении, равном или меньшем давления насыщенного нара, благодаря чему можно пренебречь Д//, обусловленной любой разностью в давлении водяного пара.
В некоторых случаях желательно выбрать одно или несколько стандартных состояний, поскольку это состояние скорее является состоянием раствора, чем состоянием чистого вещества. Например, при реакции
SO3(r) HjCK-k) = H2SO4(bow.)
£?) В некоторых случаях, особенно если значения энтальпии яли энтропии получаются из спектроскопических данных, может оказаться желательным учесть различие между этими величинамя при нулевом давлении и давлении в 1 атм. Это можно легко осуществить с помощью методов, рассмотренных в гл. VI.
Тепловые эффекты химических процессов
471
предполагается, что состояние продукта является состоянием настолько разбавленного раствора H2SO4 в воде, что дальнейшее разбавление не произведет уже заметного теплового эффекта.
Как уже было указано в гл. I, для определения состояния твердого тела не всегда достаточно знать переменные состояния. Это имеет значение при расчетах теплот реакций, включающих твердые тела. Классический пример представляет углерод, который может существовать в нескольких различных твердых формах; эти формы, за исключением кристаллических форм алмаза и графита, не поддаются точному определению. Теплота образования СО2 входит в определение теплоты образования почти всех органических соединений, и поэтому теплота сгорания углерода является одной из основных термохимических констант. Во всех современных термохимических экспериментальных работах употребляется графит, поэтому мы будем считать, что эта форма углерода и будет стандартной для всех термохимических уравнений.
Суммирование теплот реакции. Теплота любой реакции представляет алгебраическую сумму теплот образования всех реагентов. Рассмотрим, например, реакцию
аА 4- ЬВ = IL 4- тМ. (92)
Теплота реакции ДНу на моль А дается уравнением
где Д77л, ДНВ ит. д. — теплоты образования 1 моля соответствующего вещества. Это следует из природы свойства Н, и предполагает ся, что все Д77 относятся к одинаковой температуре и что все элементы, участвующие в реакции образования, всегда берутся в одном и том же стандартном состоянии. Если одно ир реагирующйх веществ в уравнении (92) является простым веществом, то его теплота образования, по определению, конечно, равна нулю. Поскольку все теплоты реакции могут быть весьма просто получены из теплот образования, то наиболее полезной формой сводки этих данных является таблица теплот образования при стандартной температуре, например при 25° С. Энтальпии всех простых веществ в стандартном состоянии при этой температуре принимаются равными нулю. Теплота образования, конечно, является энтальпией соединения в выбранном стандартном состоянии.
Как источник термодинамических данных особенно рекомендуется обширная компиляция Быховского и Россини [23], охватывающая все неорганические вещества, для которых имеются данные, и органические соединения с одним и двумя атомами углерода. Более современная компиляция для 27 газообразных углеводородов опубликована Россини [206].
IX. Передача тепла
472
Пример 11. Вычислить теплоту реакции
2Н2О (г) + СН4 = СОг+4Н2
при 25°С, если даны следующие теплоты образования (также при 25°С):
АН образования СО2 =—94030 кал/моль, &Н образования Н2О(г) =—57798 кал1моль, образования СН4 =—17865 кал!моль, ЬН для реакции на моль СН4 равна —940304-4X0—(—2X57798) —(—17865) — 4-39430 кал.
Таким образом, реакция сопровождается поглощением теплоты, т. е. является эндотермической.
Очень немногие теплоты реакции определяются непосредственно измерением, так как этот путь очень неудобен и в подавляющем большинстве случаев практически неосуществим, но благодаря свойству Н теплоты большинства реакций можно вычислить косвенно, путем комбинирования теплот реакций, являющихся стадиями рассматриваемой реакции. Например, теплоты образования органических соединений, содержащих только углерод, водород и кислород, могут быть вычислены из теплоты сгорания соединения (принимая полное сгорание до Н20 и СО2) и теплоты сгорания углерода и водорода.
Так, теплота реакции
С + ^О24-2Н2 = СН3ОН(г) (ДНх)
получается алгебраическим суммированием теплот реакций
СН3ОН (ж)4-1 у О2 = со2 4- 2Н2О (ж), (ДЯ2)
СН3ОН (ж) = СН3ОН (г), (Д/73)
с4-о2=со2, (дя4)
•н24-1о2=н2О(ж). (дя5)
При сочетании последних четырех уравнений таким образом, чтобы получилось первое уравнение, приходим к равенству
Д/7Х = ДЯ3 — ДН2 + ДН4 + 2ДН6.
Однако надо иметь в виду, что многие теплоты реакции очень малы по сравнению с теплотами сгорания; поэтому трудно получить результат даже приблизительной точности без очень сложной и трудоемкой работы. Так, теплота сгорания к-гексана примерно в 25 раз больше теплоты его образования, и поэтому любая ошибка в первой в 25 раз увеличивается в последнейтз. Еще более разительным примером является следующая реакция изомеризации:
к-гексан
3-метилпентан.
Тепловые эффекты химических процессов
473
Теплота изомеризации составляет только около О,1О°/о теплоты сгорания любого гексана. Несмотря на это, Россини с сотрудниками *) достиг большой точности, позволившей определить теплоты изомеризации с возможной ошибкой примерно в -4-20°Рассмотренный случай, конечно, является крайним;" он выбран лишь для того, чтобы подчеркнуть значение этого вопроса.
Теплоты реакции из данных о равновесии. Теплоты реакции могут быть вычислены также из результатов измерений равновесия при применении соотношения
^H=RT^(d-^s\ . (260, гл.IV)
Если известны значения Ка как функции температуры, то значение (д\пКа1дТ)р можно получить алгебраическим или графическим способом. Этот метод вполне точен в случае ионного равновесия, когда Ка получается из измерений э. д. с., но является только приблизительным для реакций при повышенных температурах, где точное определение Ка бывает скорее исключением, чем правилом.
Если принять ДУУ в узких пределах температуры, не зависящей от ее изменений, то уравнение (260, гл. IV) переходит в уравнение
Следовательно, для определения ДУУ достаточно располагать двумя значениями Ка при двух различных температурах (не слишком близких друг к другу). В этом случае ДУУ принимается равным теплоте реакции при среднеарифметической температуре.
Если равновесные данные можно представить соотношением
№ = 4 + В’
то тогда
д In Ка 2,303 А
дТ ~ Т2
и
ДУУ= — 2,303 AR. (95)
Этот метод использовался для многих случаев, но точность результатов всегда была небольшой.
Пример 12. Используя данные, приведенные на рис. ПО, вычислить тепловой эффект реакции
СЛ + Н2О (г) = С2Н5ОН (г).
См. список статен [138, 192].
474
/X. Передача тепла
Экспериментальные данные представлены уравнением
= ------6,241,
где Т дается в градусах Кельвина.
Тогда, в соответствии с уравнением (95), получаем тепловой эффект реакции при средней температуре в 500° К:
ДЯ = —2,303X2132Х1,987 = —9750 кал]моль.
Найденное значение Д/7 не очень хорошо совпадает со значением, полученным из термических данных [188], именно с величиной —11235.
Теплота реакции и структура. Количественная зависимость АН реакции от структуры была бы крайне полезна прн определении те-плот образования органических соединений, для которых не имеется экспериментальных данных. Хотя этому вопросу было уделено значительное внимание, все же получено еде мало результатов, имеющих практическое значение. Если мы рассматриваем простую реакцию образования, как, например, реакцию
4С + 5Н2 = С4Н10,
то мы можем считать общим результатом образование 3 связей углерод— углерод и 10 связей углерод — водород. Если бы можно было придать определенную энергию каждой из этих связей, то мы были бы в состоянии вычислить энергию образования этого или любого другого подобного соединения путем простого сложения. Эксперимент показывает, что в действительности дело обстоит не так просто. Энергии связи не постоянны, но зависят от конфигурации молекулы. Например, если бы для энергий связи имелась простая аддитивная зависимость, то все изомеры данного соединения, например такого, как пентан, должны были бы иметь одинаковую теплоту образования, однако эксперимент не подтверждает этого74.
Начало этому было положено нахождением соотношений для случая гомологических рядов, позволяющих сделать определенные предсказания. Так, Россини [207] показал, что для трех гомологических рядов углеводородов — и-алканов, и-алкенов и, главным образом, и-алкилспнртов — для теплоты образования действительна следующая зависимость:
ЬН = А-{-Вп-[-Ь, (96)
где А — константа, характеризующая конечную группу ряда [Н для нормальных парафинов (алканов) и ОН для спиртов],
В—универсальная константа для всех трех рядов, п — число атомов углерода,
Д— отклонение от линейной зависимости (небольшое после п — 2 и равное нулю для л^>5).
Эту зависимость можно использовать для предсказания значений Д// при образовании тех членов рядов, для которых отсутствуют данные.
Тепловые эффекты химических процессов
475
Влияние температуры на теплоту реакции. Все теплоты химической реакции, даваемые в обычных сводках, являются стандартными теплотами, относящимися к температурам, близким к комнатной температуре,— для современных определений обычно к 25°С. Во многих случаях возникает необходимость пользоваться теплотой при какой-нибудь другой температуре; это легко осуществить путем вычисления по данным о теплоемкости. Метод вычисления возвращает нас к основной зависимости
Если мы приложим это уравнение к каждому веществу, участвующему в реакции, представленной в общем виде уравнением (92), то, произведя алгебраическое суммирование, получим
<97>
где ДН—теплота реакции на моль А;
и
4СР = 4 <СЛ- + 4 <СА —Ьа <СЛ - <СЛ-
Каждый Н представляет теплоту образования рассматриваемого вещества.
На основании уравнения (97) можно сделать следующие качественные заключения. Если теплоемкость продуктов реакции больше, чем теплоемкость реагирующих веществ, то с повышением температуры эндотермическая теплота реакции будет уменьшаться. Другими словами, если реакция экзотермическая (Д/7—отрицательно), то числовое значение Д/7 будет уменьшаться с повышением температуры. Если продукты и реагирующие вещества имеют одинаковую общую теплоемкость, то теплота реакции не будет зависеть от температуры. Интегрируя уравнение (97), получаем
т
ЬНт = ЬНТо+ 5 ^CPdT‘ (98)
С помощью этого уравнения теплоту реакции при любой температуре ДНу. легко получить из теплоты реакции при основной температуре ДНг0 при условии, что в рассматриваемом температурном интервале для каждого вещества Ср известна как функция температуры. Если мольная теплоемкость каждого вещества представлена уравнением, подобным уравнению (15), то уравнение (98) можно
476
IX. Передача тепла
преобразовать в
ДЯГ=ДЯЛ + Д<* (Т- Т’о) + ~ д? (^ - 7$) +1 Ду (7’3 - То), (99) где
~ ам + "7 а1 — "^“в— аА' (10°)
и подобные соотношения определяют Д^ и Ду.
Взяв неопределенный интеграл от уравнения (97), получим
ДЯ=ДЯ0 + Да7’+-1д^ + уДуТ8; (101)
Д/7е — константа интегрирования и не равна теплоте реакции при какой-либо температуре.
Влияние давления на теплоту реакции. Этот вопрос значительно менее важен, чем вопрос о влиянии температуры, и может быть удовлетворительно разобран в одном разделе. Он представляет известное значение в связи с газовыми реакциями при высоком давлении, где влияние давления велико. Из уравнения (96, гл. 1”'
Ж =V-7’® .
\др } т \дТ) р
Поступая так же, как и в случае анализа влияния температуры, получим
\дР Jr или р р
Mi= + J Ыр — Т J dp, р° р- Р
где
bv = vL + vM — vA — vB,
если стандартным состоянием для каждого вещества будет состояние чистого компонента при р°.
Для вычисления Д/7 при повышенном давлении требуется знать уравнение состояния или эквивалентные ему данные для каждого вещества, участвующего в реакции75.
Общий тепловой эффект реакции. До сих пор мы были связаны главным образом с частным тепловым эффектом, называемым теплотой реакции. Теперь представляется желательным несколько обобщить уравнения для введения любого теплового эффекта. Даже уравнения для теплоты реакции [например уравнения (97)—(101)] ограничены случаями, исключающими скрытые теплоты, поскольку в уравнении (97) предполагаются только такие тепловые изменения, которые вызывают изменение температуры. Следует дать более общее уравнение для теплового эффекта, охватывающее скрытые теплоты, отношения реагирующих веществ, отличающиеся от стехиометрических,
Тепловые эффекты химических процессов
477
инертные вещества, случай неполной реакции и все тепловые эффекты между двумя предельными случаями изотермической и адиабатной реакций. Для получения такого уравнения рассмотрим цикл изменений, представленных на рис. 82. Термин „реагирующие вещества* будет означать все вещества, входящие в реакционную камеру или присутствующие в ней в некоторый начальный период независимо от того, участвуют ли они в реакции или нет. Термин „продукты* будет объединять все вещества, покидающие камеру или присутствующие в ней, когда реакция считается законченной. Для цикла: — Q1 + Q + Q2—Q3=a. ‘ (102)
Теперь каждая Q для ступеней, включающих температурное изменение, состоит в общем из суммы теплоты, вызывающей изменение температуры и скрытых теплот. Например, для компонента А будем иметь
т
Q = NAMiA = NA J CPAdT + NaLq. (103)
Рис. 82. Круговой процесс химической реакции.
Интеграл представляет изменение теплосодержания компонента А обусловленное изменением температуры (от основной температуры до температуры Т). Lo представляет скрытую теплоту фазового превращения компонента, если любое такое превращение происходит между рассматриваемыми температурами при основной температуре То. В действительности, конечно, любое па
рообразование или конденсация, происходящие в реакционной смеси, будут происходить выше данных пределов температуры, и практически ни одна из них не происходит при основной температуре. Однако мы считаем законным рассматривать все процессы парообразования и конденсации как происходящие при основной температуре, так как Д/7 не зависит от действительного пути, и поэтому мы свободно можем выбрать наиболее простой и наиболее удобный путь, поскольку все они эквивалентны. При приведении всех скрытых теплот к стандартной температуре (обычно 25°С) мы имеем дело с теплоемкостями только одной фазы данного компонента. Кроме того, неважно, что путь, выбранный нами для определения Д/7, не будет строго изобарным, если все давления будут достаточно низки, то можно сделать обычные допущения об идеальности газа, предполагающее независимость 1Н от давления.
478
IX. Передача тепла
Следовательно, мы можем выразить Qj и Q2 в уравнении (102) как сумму ряда уравнений, подобных (103), для реагирующих веществ и продуктов. — Q3 представляет общий тепловой эффект для обратной реакции при основной температуре, т. е. равняется &Нт0— теплоте реакции при TQ, умноженной на число молей реагирующего вещества (принимается компонент А), на котором основана теплота реакции, и на £— реагирующую долю его. Мы можем поэтому придать уравнению (102) следующий вид:
Л т,
(5 + SpM ( j CpdT+L0) +
1 о То
+^АД//г0 + <?==0, (104)
где 2/? обозначает суммирование по всем реагирующим веществам, а —то же по всем продуктам, причем каждый член \CpdT-\-L0 умножается на число молей данного компонента. Это включает все инертные вещества, которые могут присутствовать как в реагирующих веществах, так и в продуктах, а продукты также включают все излишки реагирующих веществ.
Для удобства при последующих применениях напишем это уравнение в следующей сокращенной форме:
S^+Wa^70 + Q=0. (105)
Наиболее важным случаем, при котором следует учитывать скрытые теплоты, является случай сгорания топлива, содержащего водород. Посредством исключения члена ДГдД/7г0 можно избежать необходимости учета скрытой теплоты конденсации водяного пара при суммировании произведений. Другими словами, из теплоты сгорания можно исключить скрытую теплоту парообразования воды. Тогда получим величину, которая называется низшей теплотворностью топлива. Если для представления данных по теплоемкости выбирается уравнение (15), то уравнение (104) можно интегрировать, что дает
[мл - Т-о) + 4 ?(- т? ) +1Y (7f - 7^)] - XrNiLo+ + ЕрЧ[а(Л-Л)+4м^-^) + |у(Л3-7’о)]+ (106)
+ ЕрМА + + Q = 0.
Sp/ЛЗ и SpM-Y представляют суммы констант а, Р и у уравнений для Ср всех реагирующих веществ, умноженных на соответствующее число молей. y^oN'-a и т. д. являются такими же величинами
Тепловые эффекты химических процессов
479
для всех продуктов., Для большей ясности напишем два из этих выражений:
== Нд&а Ч~ ^вав Ч~ ^iai> =n^l++л^, где индекс 1 относится к инертным компонентам.
Уравнение (106) полезно для вычисления Т2 — температуры, достигаемой при реакции, если даются исходная температура реагирующих веществ Тр теплота реакции при основной температуре Та, теплота, отдаваемая окружающей среде, и теплоемкости реагирующих веществ. Когда Tj = Т2 = Т и $ = 1, Q — — NA&HT, и уравнение (106) совпадает с уравнением (99).
Пример 13. Вычислить теплоту реакции
СН4 +2Н2О (г) = СО2 +4Н2 ' при 500° С.
Из примера 11 Д//Го = 39430 кал)моль. Из табл. 12
Да = 6,85+4 (6,88) —2 (6,89) —3,38=17,21,
Др = [8,533 + 4 (0,066) —2 (3,283) —17,905] X Ю'3 = —15, 674 X Ю"3, Д7 = [ — 24,75 + 4 (2,79) — 2 (3,43) + 41,88] X Ю“7 = 21,43 ХЮ’7.
Подставляя в уравнение (99), получаем
Д//7 = 39430 +17,21 (773—298) — Х1()-з [(773)2 _ (298)2]
21 43
+ £Д2±-Х10-1 [(773)3 — (298)3] = 43950. о
Пример 14. Газовая смесь, состоящая из 94°/0 водорода, 3«/0 азота и 3®/в окиси углерода, должна быть освобождена от СО посредством прохождения через соответствующий катализатор для проведения реакции
СО + ЗН2 = СН4 + Н2О (г).
Предположим, что газ подходит к катализатору при 500° С и 1 атм, что эта реакция является полной и не сопровождается побочными реакциями и что от окружающей среды не получается и к ней не передается теплота. Определить температуру газа, покидающего катализатор.
Д77 образования СО при 25° С = —26394. Используя другие теплоты образования из примера 11, получаем
Д//Го = —57798—17865+26394 = —49270 кал'моль СО.
Количество реагирующих веществ и продуктов на моль СО составляет:
Газ Реагирующие вещества Продукты
СО 1 0
н2 31,3 28,3
Ыг 1 1
СН4 0 1
Н2О 0 1
480
IX. Передача тепла
Решение можно найти при использовании уравнения (104) с Т2 в качестве неизвестного. Г0 = 298°К и 7\ —773°К- Вместо определения интегралов посредством уравнений Ср = <р (Г) на этот раз мы воспользуемся средней Ср из данных Юсти и Людера (табл. 13) и следующими средними данными по Ср для СН4, приводимыми ими же:
t, ° с Ср, кал/моль t,°C Ср, кал/моль
0 8,24 600 12,03
100 8,66 700 12,62
200 9,40 800 13,17
аии 10,10 900 13,68
400 10,77 1000 14,17
500 11,41
При использовании уравнения (23) первый член уравнения (104) определяется следующим образом:
Г,
2^VZ J CpdT= 7,19 (500) — 6,97 (25)4-31,3 [6,99 (500) — 6,92(25)] 4-
4- 7,15(500) — 6,97 (25) = 110 800.
Второй член уравнения (104) определяется методом подбора.
Примем t2 = 700°С. Считывая среднее Ср из таблицы, получаем г»
CpdT= 28,3(7,03(700)—6,92(25)] 4-7,27 (700)—6,97 (25) 4-12,62(700)—
— 8,34 (25)] 4- 8,74 (700) — 8,03 (25) = 153570.
Подстановка в уравнение (104) дает
— 110 800 4-153 570 — 49 270 4- 0 0, или
— 160 0704~ 153 570 ;£0.
Очевидно, принятая температура слишком низка. Еще несколько подборов покажет, что правильное значение лежит около t = 727° С.
Пример 15. Вычислить максимальную температуру горения при сжигании метана с 20°/о избытка воздуха, если и воздух и метан подогреты до 300° С.
Максимальная температура горения реакции горения принимается равной температуре, которая достигалась бы в том случае, если бы реакция была адиабатной. Действительная температура горения несколько меньше, но разность достаточно мала, и вычисленные значения являются удовлетворительными для большинства технических целей £?).
☆) Джонс, Льюис, Фриуф и Перротт [118] определили температуру горения различных газообразных углеводородов, сжигаемых в воздухе, пользуясь спектроскопическим методом исследования, и сравнили их с вычисленными значениями. Наблюденные температуры оказались на 50—100° ниже вычисленных. Различие, вероятно, вызывается излучением от пламени.
Тепловые эффекты химических процессов
481
Реакцию можно написать следующим образом:
СН4 4- 2,4002+ ^X2,40N2 = СО2 + 2Н2О (г) + 0,4002 + ^ X 2,40N2, Д//?-о= —212790 кал1моль.
Эта величина указывает на полное сгорание и отсутствие каких-либо других реакций. При решении уравнения (106) будут использованы данные по теплоемкости (табл. 12):
= 9,03 (6,30) + 2,40 (6,26) + 3,38 = 75,30,
= [9,03 (1,819) + 2,40 (2,746) -j-17,91 I X 1Х3 = 0,04094,
Г^Л = [9,03( —3,45) + 2,40( —7,70) —41.88JX 10~7= — 0.915Х 1»-*, ZpNfl = 9,03 (6,30) + 0,40 (6,26) + 2 (6,89) + 6,85 = 80,13, ЕрЛЭД = [9,03 (1,819) + 0,40 (2,746) + 2 (3,283) + 8,533] X Ю~3 = 0,0326, SpW/T = [9,03 (— 3,45) + 0,40 (—7,70) + 2 (3,43) — 24,75] Х11Н= — 0.522ХЮ-6, {г определяется опять путем подбора.
Пусть f2=2000°C.
Подставляя в уравнение (106), получаем
— 25130 + 241900 — 212790 — 0 4 0.
Очевидно, принятая температура несколько высока.
Пусть £а=1950°; тогда — 237 900 + 235 300 4 0. Очевидно, правильная величина лежит между этими двумя, и посредством грубой интерполяции можно получить для нее 1970° С. Это достаточно близко к правильному решению. При таком вычислении не следует стремиться к большой точности, поскольку этот метод сам по себе не очень точен. При высоких температурах теплоемкость неточно известна, и при таких температурах будет происходить в некоторой степени диссоциация СО2 и водяного пара. Например, при 2200° К и общем давлении в 1 атм водяной пар примерно на 1,5% диссоциирован на Н2 и О2, а СО2 примерно на 5°/0 диссоциирован на СО и О2. В топочных газах, получающихся при горении, степень диссоциации будет значительно больше благодаря низкому парциальному давлению этих газов. Диссоциацию этих газов можно учесть при вычислении по методу, рассмотренному в гл. XI.
Интересно отметить, что теоретические температуры горения для сжигания газов в воздухе без избытка последнего не изменяются значительно для различных газов в противоположность очень большим различиям в теплоте сгорания. Например, Н2, обладая теплотворностью в 2840 ккал'м3 (при 15° С и 1 атм) имеет теоретическую температуру горения около 2200° С, тогда как бензол с теплотворностью в 11,5 раза большей, т. е. 32 700 ккал\м\ дает температуру в 2240°С. Это, конечно, обусловлено тем обстоятельством, что газы с большей теплотворностью соответственно требуют также большего количества воздуха для сжигания. Большинство вычисленных температур горения для полного сгорания в воздухе без избытка его и без подогрева воздуха или газа лежит между 2000 и 2400° С. Температура может увеличиваться при подогреве или при использовании кислорода или обогащенного воздуха вместо обычного воздуха. Последняя возможность крайне интересна, так как на ней могут базироваться дальнейшие достижения в получении высоких температур.
31 Химическая термодинамика
ГЛАВА X
ОХЛАЖДЕНИЕ
Хотя охлаждение обычно относят к кругу вопросов, которыми занимается инженер-механик, но существует много способов, с помощью которых низкие температуры можно с пользой применять в химической промышленности, и поэтому для инженера-химика важно ознакомление с общими принципами охлаждения и знание пределов применения и преимуществ процессов охлаждения и их аппаратурного оформления. Это поможет инженеру-химику принять разумное решение в тех случаях, когда он столкнется с задачами, включающими применение температур ниже температуры окружающей среды.
Краткий обзор случаев применения охлаждения к промышленным процессам должен включать такие производства, как сжижение хлора, получение твердой углекислоты, рекуперацию растворителей, получение бензина из натурального газа, конденсацию паров летучих жидкостей, подобных сероуглероду, этиловому эфиру и четыреххлористому углероду, кристаллизацию солей из раствора, дегидратацию газов и удаление загрязнений из них, кондиционирование воздуха при производстве вискозы, фотографической пленки, желатина и кон-фект, выделение парафинового вара из нефти; регулирование скорости реакции для таких органических реакций, как нитрация и диазотирование; получение кислорода и азота из воздуха и водорода из газа коксовых печей и из других газов, сжижение и хранение природного газа.
Методы охлаждения. В широком значении слова охлаждение является наукой и искусством получения и поддержания температур ниже температуры окружающей среды. Низких температур можно достигнуть различным путем, а именно: 1) с помощью фазовых превращений, сопровождающихся поглощением тепла, например посредством парообразования воды или аммиака, плавления льда или растворения соли; 2) расширением сжатого газа или пара, при котором совершается внешняя работа; 3) дросселированием; 4) десорбцией газа; 5) размагничиванием твердого тела; 6) пропусканием электрического тока через спай двух металлов (эффект Пельтье). Действительно, любое обратимое изменение, включающее затрату работы, можно использовать для отвода теплоты и получения низких температур. Метод 1 чаще всего применяется для промышленного
Методы охлаждения
483
охлаждения. Он лежит также в основе использования большого числа охлаждающих смесей, наиболее распространенной из которых является смесь соли и льда, полезная для осуществления периодического охлаждения в малых масштабах. Методы 2 и 3 полезны для достижения очень низких температур в промышленном масштабе. Методы 4 н 5 используются для получения очень низких температур в лаборатории, в частности, метод 5 употребляется для достижения температур, приближающихся к абсолютному нулю. Метод 6 упоминается как возможный, так как он никогда не был использован на практике. Любой из перечисленных процессов может происходить: 1) адиабатно, причем в этом случае будет падать температура самой системы; 2) изотермически, если процесс сопровождается притоком тепла из окружающей среды; 3) при сочетании 1 и 2.
Непрерывное получение холода чисто механическим способом, например путем использования сжатия в цикле, включающем попеременную конденсацию пара в жидкость и парообразование, известна как „механическое охлаждение". Этот способ представляется наиболее важным для поддержания низких температур, и рассмотрение его будет главной темой настоящей главы. Методы достижения температур несколько ниже температуры внешней среды с помощью испарения воды важны для кондиционирования воздуха и охлаждения воды; однако обзор этих методов не входит в задачи настоящей главы. Методы достижения очень низких температур, например ниже —70° С, несколько специфичны, но будут кратко рассмотрены в этой главе.
Идеальный цикл охлаждения. Поддержание любой температуры । иже температуры окружающей среды требует непрерывного отвода теплоты при данной температуре и подвода ее при несколько более высокой температуре. Таким образом, охлаждение в сущности является процессом, включающим перевод теплоты с одного температурного уровня на другой, более высокий. Это толкование имеет очень большое значение, позволяя ясно представить существенные элюенты любого процесса охлаждения. В гл. II, при рассмотрении второго закона термодинамики, было показано, что любую обратимую тепловую машину можно рассматривать как тепловой насос. Как было показано, машина, работающая по циклу Карно, забирающая теплоту при абсолютной температуре 7\ и отдающая ее при Т2, имеет максимальный к. п. д. из всех, какие могут быть достигнуты любой тепловой машиной, работающей между данными температурными пределами. К.п. д., который является отношением произведенной работы к теплоте, поглощенной при 7\, дается уравнением ☆)
А _7\-Т,
Qi Л
(1)
☆) В этой главе для упрощения во всех уравнениях опускается механический эквивалент теплоты J.
31*
484
X. Охлаждение
Для обоснования этого уравнения можно обратиться к гл. II и III. Поскольку мы рассматриваем обратимый цикл, можно представить машину Карно действующей в обратном направлении, причем в ней к рабочему телу подводится теплота Q2 от холодного источника Т2 и отводится теплота Q, в горячий источник при Тг. В этом случае приложимо то же самое уравнение; но так как при сообщении тепла мы заинтересованы в том, чтобы знать, сколько тепла можно отнять при более низкой температуре с затратой данного количества работы, то уравнение (1) обычно пишется в форме ☆)
Здесь Т2 относится к холодильнику при температуре ниже температуры окружающей среды, а 7\— к теплоприемнику при температуре, равной или превышающей наинизшую температуру любой большой массы в окружающей среде (например атмосферы или большой массы воды). В дальнейшем эта температура будет обозначаться символом Т{1. Уравнение (2) непосредственно следует из уравнения (1), если принять во внимание равенство
Д = — Q2, (3)
вытекающее из первого закона.
Величина Q2M, которая является мерой эффективности охлаждения на единицу затраченной работы, называется холодильным коэ-фициентом jj. Уравнение (2) устанавливает максимальную производительность любой холодильной машины, работающей в данных температурных пределах, и не может существовать машина, с помощью которой можно было бы осуществить отнятие тепла с меньшей затратой работы. Уравнение (2) предполагает также, что все обратимые холодильные машины, работающие в данном температурном интервале, будут иметь одинаковые холодильные коэфициенты, независимо от природы рабочего тела и механизма. Другими словами, холодильный коэфициент зависит только от температур теплоотдатчика и теплоприемника. Эти положения, в сущности, являются не чем иным, как формулировками второго закона термодинамики.
Подчеркнем здесь, что уравнение (2) представляет выражение, наглядно показывающее, что может дать любой холодильный процесс. Вследствие неизбежных необратимых эффектов в любом реальном процессе для отнятия данного количества теплоты потребуется бблыпая затрата работы, чем это следует из указанного уравнения. Уравнение (2) полезно при установлении абсолютного стандарта для сравнения и для упрощения анализа реального процесса. Сопоставле-
й-) Условия, что работа, произведенная над системой, является отрицательной, мы будем придерживаться только в тех случаях, когда алгебраическое сложение нескольких рабочих членов требует условного знака.
Методы охлаждения
485
ние фактического процесса с идеальным указывает путь для возможных усовершенствований. Уравнение (2) также полезно для ориентировочного определения работы, необходимой для осуществления заданного охлаждения. Рассматриваемое уравнение указывает также на следующие важные факты:
1. Работу, затрачиваемую на единицу охлаждения, нельзя значительно изменить заменой одного рабочего вещества другим. В идеальном цикле она не зависит от природы рабочего вещества, но в. реальном цикле будет существовать различие между рабочими веществами, хотя оно в большинстве случаев и незначительно.
2. Работа будет возрастать по мере понижения температуры холодного источника.
3. Работа будет увеличиваться по мере повышения температурь^ теплоприемника.
Пример 1. Чему будет равна минимальная работа, необходимая для осуществления непрерывного отнятия 100 ккал тепла в минуту от вещества, температура которого должна поддерживаться в —10° G, если температура окружающей среды равна 15° С?
Теплоту, отбираемую при температуре Г2, следует привести, по меньшей мере, к температуре 15° С, раньше чем ее можно будет передать от системы к окружающей среде. Следовательно, Г1 = 273-|-15 = 2980К> и, в соответствии с уравнением (2),
А = 100 X !,^Т~ГГ~= 9’5 ккал/мин,
Л! о -J— (—1м)
что отвечает мощности в
Уравнение (2) предполагает постоянную температуру Т2 в холодильнике и также постоянную температуру То в месте отдачи тепла. Для случая, когда То является постоянной, но температура холодильника Т является переменной, уравнение (2) принимает более общую форму, именно:
т,
A=^=ldQ, (4>
т,
где Тх и Т2 — пределы изменения Т. Поскольку для вещества, охлаждающего при постоянном давлении, dQ = dH, уравнение (4) можно также написать, как
A = ^=^dH. (5)
т.
Пример 2. Чему будет равна минимальная затрата работы (в киловатт , часах) иа охлаждение 1 кг-моля СО2 при 1 атм от 20 до — 20° С, если / в распоряжении имеется большой запас охлаждающей воды при 20° С? «
486
X. Охлаждение
Примем
Ср = а~9.
Поскольку dH= CpdT, то, по уравнению (5),
А = Ja T<>~-dT = I aTQ In Т £*• т, *
Подставляя пределы и учитывая, что Г1 = Г0> получаем А = аТй\п^.
Т. — 9Q4O
Г2 — 253° К,
386 7
А = 386,7 ккал/кг-моль — -ggg- = 0,45 £вт-ч/кг-моль.
Так как
то
Рис. 83. Простой холодильный цикл Карно.
На основании первого и
Простой холодильный цикл Карно, к которому приложимо уравнение (2), можно представить на диаграмме температура— энтропия, как показано на рис. 83. АВ представляет изотермическое поглощение тепла, ВС — адиабатное сжатие рабочего вещества, CD — изотермическую отдачу тепла и DA — адиабатное расширение, возвращающее рабочее вещество в исходное состояние.
второго законов термодинамики
Q2 = T2(SB — SA), (6)
Q1 = То (Sc — SD) = т0 (SB — SA), (7)
_X==qi_q2 = (7’0 — T2) (Sb—Sa). (8)
Из этих уравнений видно, что площадь под прямой АВ (до оси Г— 0) представляет Q2. Подобно этому, площадь под DC соответствует а площадь ABCD — работе.
Для охлаждения вещества от Тй до Т2 по пути ВА (рис. 84) тепло следует отводить при различных температурах. Примем сначала, что это производится с помощью двух холодильных машин, действующих по циклам Карно BCDE и CGAF. Очевидно, что переход тепла будет происходить необратимо, поскольку рабочее вещество везде (кроме двух точек) будет иметь более низкую температуру, чем теплоотдатчик. Это приведет к затрате некоторого количества работы, определяемого площадями указанных прямоугольников. Если использовать большее число машин, причем каждая из них будет иметь меньшую производительность, чем первоначально взятая, то заштрихованная площадь будет уменьшаться. При переходе к пределу, ко
Методы охлаждения
487
гда циклы станут бесконечно малыми (изменение энтропии равно dS) и поэтому процессы, связанные с их протеканием, будут отвечать минимальной производительности, мы придем к обратимому процессу, осуществление которого будет связано с минимальной затратой работы. Заштрихованная площадь показывает уменьшение работы по
сравнению с первым случаем, когда использовались только два холодильника. Из диаграммы следует, что работа для случая бесконеч-
но большого числа тепловых насосов Карно дается уравнением т«
A=\(T0 — T)dS, (9) т,
и поэтому (10)
Уравнение (9) совпадает с уравнением (5). Последнее можно также написать в форме
A = TQkS — AH, (11) где Д соответствует пределам изменения от Т2 до Тй. Отметим, что правая часть уравнения (11) является изменением функции В, определение которой дано в гл. III.
Рис. 84. Минимальная работа, необходимая для охлаждения жидкости.
Таким образом, мы получаем простое правило, согласно которому минимальная работа, требуемая для какого-либо процесса, включающего отнятие тепла, равна изменению пригодности (функции В).
Использование тепла для получения холода. Из изложенного выше не следует заключать, что работа необходима для непрерывного осуществления охлаждения. Действительно, наиболее важным
процессом механического охлаждения является процесс сжатия, при котором теплота, поглощенная в холодильнике, вызывает парообразование жидкости, а пар затем сжимается и охлаждается до конденсации, чем и завершается цикл. Ясно, что при этом процессе для сжатия пара требуется работа, и если мы вернемся к первоначальному источнику этой работы, то найдем, что она обычно получается в тепловой машине. Следовательно, процесс сжатия—охлаждения можно рассматривать как сочетание тепловой машины и теплового насоса, причем машина доставляет работу, которая непосредственно используется в ‘насосе. Общим результатом будет использование определенного количества теплоты при некоторой температуре выше температуры окружающей среды для передачи тепла с температурного уровня ниже температуры окружающей среды. Допустим теперь, что теплоту можно непосредственно использовать для передачи тепла без промежуточного превращения ее во внешнюю работу. Это совершенно аиало-
488
X. Охлаждение
гично абсорбционному процессу охлаждения, который будет описан ниже.
Из высказанных здесь представлений и из уравнений (1) и (2) легко заключить, что
2? Л (Л — 7*о) /191
<ЗГ~т\(Тв-Т2)- ' '
Индексы 1, 2 и 0 относятся соответственно к источнику тепла, холодильнику и теплоприемнику. Q2/Qj можно считать коэфициентом производительности холодильного цикла, в котором для получения охлаждения непосредственно используется теплота, а не работа.
Холодопроизводительность. Представляется удобным пользоваться единицей холодопроизводительности, с помощью которой можно дать оценку холодильной машине, подобно тому, как паровую машину оценивают по ее мощности в лошадиных силах. Единицей холодопроизводительности является „тонна охлаждения® за 24 часа, обычно сокращенно называемая „тонной®. Она произвольно выбрана так, что отвечает удалению 55,5 ккал/мин ☆) без учета условий отвода теплоты. Производительность любой действующей холодильной установки будет сильно изменяться в зависимости от условий работы, и по этой причине необходимо установить основные переменные для того, чтобы стандартизировать меру производительности. Мощность, необходимая для осуществления охлаждения, часто дается в лошадиных силах или киловаттах на тонну. Так, количество лошадиных сил на тонну охлаждаемого материала в случае примера 1 равно 0,90^1,8 = 0,53; Очевидно, мощность на тонну будет сильно изменяться в зависимости от условий.
ОХЛАЖДЕНИЕ СЖАТИЕМ
Реальные холодильные циклы. При рассмотрении холодильного цикла Карно (идеального цикла) ие обязательно обращаться к деталям, связанным с механизмом процесса. Действительно, громадным преимуществом этого метода анализа является его простота, обусловленная тем, что он не зависит от механизма. В действительности не существует процесса охлаждения, равноценного идеальному процессу Карно. Следующей нашей задачей будет рассмотрение реальных циклов и определение степени их отклонения от идеального. Реальные холодильные циклы отличаются от идеального цикла Карно двумя основными признаками. Во-первых, сам цикл, даже если механизм для его совершения является идеальным, имеет определенные, присущие ему необратимые эффекты, которые делают его менее производи-
☆) Исторической основой для этой единицы служит тот факт, что для плавления 1 т льда при 0° С требуется 80 000 ккал т. Это количество теплоты за 24 часа эквивалентно 55,5 ккал/мин76.
Охлаждение сжатием
«О
тельным, чем цикл Карно. Во-вторых, имеются неизбежные необратимые эффекты, обусловленные трением, несовершенной тепловой изоляцией и необходимостью разности температур для обеспечения передачи тепла. Все они устранимы лишь в предельном (идеальном) процессе; в любом же реальном процессе они оказывают влияние в большей или меньшей степени.
Процесс расширения газа. В этом процессе, схематично показанном на рис. 85, в качестве рабочего вещества используется газ.
например воздух, который не конденсируется на протяжении всего процесса. Газ входит в компрессор, в котором он сжимается до более высокого давления и h то же время (в результате адиа-батности сжатия) до более высокой температуры. Сжатый газ охлаждается при постоянном давлении до температуры охлаждающей воды и затем в цилиндре детандера расширяется до давления, которое в идеальном случае должно быть равно давлению при
Рис. 85. Процесс охлаждения посредством расширения газа.
всасывании в компрес-
сор. Детандер обычно связан с валом компрессора, благодаря чему работа
расширяющегося газа непосредственно используется для сжатия, причем двигатель сообщает только недостающее количество работы. Холодный газ, получающийся при расширении, пропускается через холодильник, где он поглощает тепло при постоянном давлении, а нагретый газ для завершения цикла возвращается к месту ввода в компрессор. При некоторых видоизменениях этого процесса газ, выходящий из холодильника, может и не проходить вновь через цикл, однако принцип метода остается тот же.
Важно отметить, что сжатый газ должен совершать внешнюю работу при расширении в машине. Из рассуждений, приведенных в гл. VII, должно быть ясно, что дросселирование будет давать значительно меньшее охлаждение и будет приводить к крайне неэффективному циклу по сравнению с циклами при использовании расширения в машине. Правда, дроссель-эффектом как единственным источником охлаждения можно пользоваться для процессов, проте
490
X. Охлаждение
кающих при очень низких температурах, но это требует значительно более высоких давлений.
Идеализированный цикл представлен на диаграмме X—Т рис. 86. Го—температура охлаждающей воды, а Т2—температура, которую следует поддерживать в холодильнике (обе температуры для упрощения анализа принимаются постоянными), АВ — адиабатное (и изо-энтропное — в идеальном случае) сжатие газа, ВС—изменение при
Рис. 86. Цикл охлаждения посредством расширения газа на диаграмме температура — энтропия.
постоянном давлении в водяном холодильнике, CD — адиабатное расширение, a DA — поглощение тепла н холодильнике при постоянном давлении. Ясно, что проведенный таким образом цикл менее эффективен, чем цикл Карно, осуществленный в данных температурных пределах (Го и Г2). Работа, требуемая для отвода теплоты, соответствующей площади ниже DA до оси Г—0, дается площадью ABCD. В цикле Карно удаляется большее количество тепла (площадь под £4) при меньшей работе (площадь AECF), Для работы по циклу Карно необходимо было бы проводить часть
сжатия и расширения изотермически, что очень трудно осуществить в любой реальной машине.
Если принять теплоемкости постоянными, то поглощенная и отданная теплоты даются соответственно уравнениями
Qz — ?a ^d) >
Qj = wCp (Тв Тс),
(13)
(14)
где w— количество газа, циркулирующего в единицу времени (в кг). Теоретическая затрата работы, требуемой в единицу времени, т. е. мощность, для адиабатного сжатия дается уравнением (31, гл. VII), которое для данного случая можно записать так:
'> k—i
А (сжатие) = ^41 (^RTa [!~(fj) * ]• (15)
Поскольку, по уравнению (59, гл. III),
Охлаждение сжатием
491
то уравнению (15) можно также придать следующий вид:
1
-4=«сл4,-(й * ]• <1б>
Подобно этому, теоретическая работа, получаемая за счет расширения, равна
k—i
A = ~wCpTD[\-^ k ], (17)
причем является во всех случаях минимальным давлением. Общая работа цикла определяется алгебраической суммой работ, даваемых уравнениями (16) и (17), т. е.
a—i
А (общая) = ™Ср(Тл-То)[1-^) * ]. (18)
Общая работа может также выражаться уравнением
Q2, (3)
и поэтому
A = wCp[(TB-Tc)-{TA-TD)]. (19)
Из уравнений (13), (18) и (19) получаются следующие два выражения для холодильного коэфициента:
Р _ (ГА-Тр)-(ТВ-Тс) ’ (20)
₽ =-----(21)
LPilPi) к —1
Цикл расширения газа в настоящее время не представляет большого практического значения, но заслуживает краткого рассмотрения, поскольку он иллюстрирует некоторые важные принципы и поскольку представляется существенным показать, почему его вытеснил процесс сжатия пара, несмотря на значительные преимущества использования в качестве рабочего вещества воздуха, который доступен и не обладает отрицательными свойствами многих холодильных агентов, например корродирующей способностью, воспламеняемостью, раздражающим и отравляющим действием. Кроме того, тот же общий принцип находит применение в процессах низкотемпературного разделения газов и в некоторых процессах рекуперации растворителей. Нижеследующий конкретный пример послужит для выявления основных трудностей этого цикла.
492
X. Охлаждение
Пример 3. Для поддержания в помещении температуры в —10° С следует использовать воздушную холодильную машину, причем она должна поглощать тепло со скоростью 500 ккал1мин. Охлаждающая вода подается при 20° С. Чему будут равны требуемая мощность, холодильный коэфициент и производительность компрессора, если компрессор забирает воздух при нормальном барометрическом давлении и сжимает его до 5 атм?
В идеальных условиях не должно быть разности температур между воздухом и охлаждаемым пространством в той точке, где воздух покидает холодильник, н между воздухом и охлаждающей водой в той точке, где воздух покидает водяной холодильник; это обстоятельство фиксирует 7д и Тс. Следовательно,
ТА = 273,2 4- (— 10) = 263,2° К
Гс = 273,2 + 20 = 293,2° К-
По уравнению (23, гл. VII),
1,40 — 1 / 5 \ 1,40
Тв = 263,2 (у} = 416,3° К,
1,40—1 / 1 \ 1,40
TD = 293,2 ( у \ = 185,4° К = — 87,8° С.
Принимая Ср для воздуха постоянным и равным 0,24, из уравнения (13) получаем вес воздуха, циркулирующий в минуту, равный
500
W = 0,24(263,2—185,4)= 26,8 кг1мин-
Общаи теоретическая работа по уравнению (19) равна
А = 26,8 X 0,24 [(416,3 — 293,2) — (263,2 — 185,4)] =
27 7
= 291 ккал1мин = 21,7 л. с.= -д- =3,1 л. с./т.
Холодильный коэфициент = 500/291 = 1,72. Холодильный коэфициент для цикла Карно в тех же самых пределах равен
Таким образом, рассматрииаемый цикл даже при идеальной работе значительно отличается от цикла Карно.
Объем воздуха, забираемого компрессором, равен
Х 22’4 Х = 19’94 м31мин-
Если пренебречь влиянием вредного пространства, то получим, что производительность на «тонну охлаждения' равна
19,94 „ „„ „
—д— = 2,22 м^мин.
В целях получения более показательных дли практических условий результатов примем 5 как минимальную разность температур между холодильником и водяным холодильником и примем к. п. д. компрессора равным 8О°/о,
Охлаждение сжатием
493
а детандера 70% (детандер менее эффективен вследствие низкой температуры).
Тогда
ТА = 258,2 Тс = 298,2.
Примем, что 85% общего (теоретического) падения температуры осуществляется в детандере. Если вычислить теоретический перепад температуры по уравнению (23, гл. VII) и взять от него 85%, то получим
Гд = 205.
По уравнению (13),
w = 39,2 кг/мин.
Работа сжатия, вычисленная по уравнению (16) и умноженная на 1 /0,80, равна
А = 1780 ккал/мин.
Работа расширения, вычисленная по уравнению (17) и умноженная на 0,70, равна
А = 790 ккал/мин.
Общая работа = 990 ккал/мин = 94 л. с.
₽=9^ = 0,505.
По порядку величин это, без сомнения, совпадает с результатами, получаемыми на практике, хотя имеющиеся данные по системам воздушного охлаждения очень скудны. Интересно отметить очень сильное влияние на холодильный коэфициент нескольких учтенных при расчете необратимых эффектов. Как будет показано ниже, аммиачная холодильная машина, совершающая ту же самую работу, обладает значительно бблыпим холодильным коэфициентом.
Из уравнения (21) очевидно, что холодильный коэфициент увеличивается при уменьшении степени сжатия. Предел достигается при
k k
Рг _ (Тс\ 1 (То\
Pl \Та) Vz/
(22)
что соответствует холодильному коэфицненту цикла Карно. При этом пределе Тв—Тс и TD=TA, и мы видим из уравнений (13) и (14), что должно циркулировать очень большое (теоретически бесконечно большое) количество газа. Другими слонами, увеличение холодильного коэфициента этого цикла должно осуществляться за счет увеличения производительности компрессора и детандера. Как будет показано ниже, производительность в примере 3 уже будет высока по сравнению с производительностью для цикла сжатия пара. Эту трудность можно частично преодолеть с помощью работы цикла при более высоком давлении. Так, при выборе компрессора с давлением всасывания в 5 атм и напорным давлением в 26,85 атм, холодильный коэфициент будет тем же самым, что и для цикла в примере 3, ио его производительность должна быть только на одну пятую больше. Холодильная система,
/
494
X. Охлаждение
использующая воздух при низком и высоком давлении примерно при 4,5 и 17 атм, известная под названием „система Аллена с плотным воздухом*, одно время широко использовалась для морских рефрижераторов, но теперь почти полностью вытеснена системами с сжатием пара77.
Сжатие пара или двухфазный процесс. Хладагент (рис. 87) сжимается до такого давления, что конденсируется при изобарном
Рис. 87. Схема, показывающая элементы простого цикла охлаждения посредством сжатия пара.
охлаждении его водой. Жидкость, покидающая конденсатор, временно накапливается в сборнике, из которого течет к дроссельному вентилю, где дросселируется до более низкого давления, которое поддерживается в змеевике испарителя. При этом давлении жидкость, поглощая тепло, испаряется, а пар возвращается в всасывающую линию компрессора и вновь проходит цикл. Теплота, которую следует отнимать в испарителе, может непосредственно поглощаться испаряющейся жидкостью нли промежуточным хладагентом, например рассолом, непрерывно циркулирующим между поверхностью, подлежащей охлаждению, и испарителем или холодильником для рассола. Непосредственно действующая система всегда предпочтительнее, если ее можно применить, но в тех случаях, когда следует избегать всякой возможности проникновения охлаждающего агента в охлаждаемое пространство или когда одна холодильная машина служит для охлаждения нескольких удаленных друг от друга точек, следует пользоваться системой непрямого действия.
При помощи рис. 88 можно проследить идеальный холодильный цикл на диаграмме S—Т. Точка А представляет насыщенную жидкость при температуре охлаждающей воды То и при давлении р2, т. е. в условиях, при которых жидкость проходит к дроссельному вентилю. Дросселирование до давления в испарителе р2 изображено
Охлаждение сжатием
495
изоэнтальпой АВ ☆). Из диаграммы сразу видно, что расширение приводит к частичному парообразованию жидкости, причем сухость определяется отношением FB к FC. Остающаяся жидкость
затем испаряется в испарителе при постоянном давлении всасывания
в компрессор и, следовательно, при постоянной температуре, как показано линией ВС. В этом случае пар, покидающий испаритель,
является насыщенным. Однако если бы он был слегка влажным или перегретым, то это не вызывало бы изменения общей картины. CD представляет адиабатное сжатие пара до первоначального давления рг. Затем перегретый пар охлаждается при постоянном давлении вдоль DE и конденсируется вдоль ЕА, завершая цикл. Два давления в цикле фиксируются соответствующими темпе-
Рис. 88. Цикл сжатия пара на диаграмме температура — энтропия.
ратурами, которые, в свою очередь, фиксируются температурой охлаждающей воды и температурой, ко-
торую следует поддерживать в испарителе.
Сравнение этого цикла с циклом Карно показывает две причины его меньшей эффективности, именно: 1) необратимое расширение АВ и 2) перегрев до температуры, превышающей Тй. Первую причину можно устранить, по крайней мере теоретически, посредством расширения в машине (линия XG) вместо дросселирования. Вторая причина устраняется осуществлением парообразования в испарителе только до С и, следовательно, впуском в компрессор влажного пара; тогда, в результате адиабатного сжатия, будет происходить изменение состояния, отмеченное линией СЕ.
Такое „влажное сжатие* применяется, но практический выигрыш слишком мал и не возмещает затруднений при осуществлении этого процесса.
Можно частично преодолеть снижение эффективности, обусловленное расширением вдоль АВ, если расширение производить в несколько ступеней, отделяя каждый раз пар от жидкости и вновь сжимая раздельно каждую порцию пара.
Действительно, необратимые эффекты в идеальном цикле сжатия аммиака, работающем при умеренных давлениях, вызывают увеличение мощности по сравнению с циклом Карно лишь на 15—2О°/0.
Поэтому возможности для снижения мощности бывают не так велики, и экономия обычно более чем уравновешивается усложнением аппаратуры.
☆ ) Это показано пунктирной линией, потому что действительный процесс, представляющий необратимое изменение, не известен точно. Известны только исходное и конечное состояния системы.
496
X. Охлаждение
Анализ цикла сжатия пара. Интересно проанализировать этот цикл количественно и сравнить его как в идеальных, так и в практических условиях с циклом сжатия газа.
Для этой цели диаграмма Молье несколько удобнее, чем диаграмма 5— Т; обычно применяется диаграмма Молье с координатами Igр и Н. На такой диаграмме (рис. 89) схематично показан цикл сжатия пара, причем различные точки обозначены теми же буквами, что и аналогичные точки на диаграмме 5—Т (рис. 88).
Область жидкости
А
В'-
Область двух фаз
Конденсация
^Дроссельное расширение Испарение
Область перегретого пара
Е Л
Сжатие
Знтальпия
Рис. 89. Процесс сжатия пара иа диаграмме Молье.
Используя индексы соответствующих точек на рис. 88 и 89, мы будем иметь дело со следующими величинами:
Базис расчета: 1 кг вещества.
Тепло, поглощенное в испарителе = Q2 = Нс — Нв. (23)
Тепло, отданное в конденсатор =Q1 = HD—НА. (24)
(Отметим, что при сжатии влажного пара точки D и Е могут совпадать.)
Необходимая работа = Q,— Qz = Нс— Нв— HD Нд= Нс — HD (25)
(так как НА — НВ).
Холодильный коэфициент — цС . (26)
“о—-“с
Значения энтальпии легко найти из термодинамических таблиц или диаграмм. Если отсутствуют обычные таблицы или графики термодинамических величин, то количество тепла можно вычислить из теплоемкости и скрытой теплоты, а работу — из уравнений, выведенных в гл. VII. Количество хладагента (рабочего вещества), необходимое для циркуляции иа .тонну охлаждения" в минуту, определяется уравнением
Охлаждение сжатием
497
(если энтальпия выражена в ккал]кг). Производительность компрессора (если пренебречь вредным пространством) на тонну дается уравнением
V (м3)мин) = wtic,
где vc—удельный объем хладагента в том состоянии, в котором он поступает в компрессор.
„ 3300 /ооч
Мощность 7.рл’ с- на тонну охлаждения. (28)
С помощью уравнений (23) — (28) и таблиц термодинамических величин можно количественно сравнить друг с другом различные холодильные циклы, основанные на процессе сжатия пара, а также сравнить их с процессом расширения газа. Можно изучить влияние таких факторов, как 1) природа рабочего вещества; 2) температура охлаждающей воды; 3) температура; которую желательно поддерживать в испарителе, и 4) влажное и сухое сжатие. Хотя расчеты производятся для идеальных циклов, тем не менее общие выводы будут справедливы для практических циклов. Отклонения от идеального процесса будут кратко разобраны ниже.
Пример 4. Сравнить процесс сжатия пара при применении аммиака в качестве холодильного агента с процессом расширения газа (условия те же, что и в примере 3).
При —12° С давление насыщенного пара NH3 —2,96В атм, и это будет давлением за дроссельным вентилем. Высокое давление будет давлением насыщенного пара при 20° С; оно равно 8,74 атм.
Для аммиака [293] //д = 65,3 ккал)кг = Нв, Нс = 340,5 (при предположении, что из испарителя поступает сухой насыщенный пар);
Sc = 1,308;
HD — энтальпии перегретого пара при давлении 8,74 атм и с энтропией в 1,308 = 378,2;
340,5 - 65,3
378,2 — 340,5 ~ ’
В минуту циркулирует
340,5—65,3~ 1,82 KZ NHs’
Требуется
1,82(340,5 — 378,2)427 _
------------------------= 6,51 л. с.,
75X60
= 0,41 м3]кг.
Производительность компрессора = 1,82 X 0,41 = 0,746 msImuh.
Большое преимущество процесса сжатия пара по сравнению с процессом расширения газа сразу видно из следующей таблицы, суммирующей некоторые результаты решения примеров 3 и 4;
32 Химическая термодинамика
498
X. Охлаждение
Показатель Сжатие пара при применении NH3 Расширение воздуха с забором при атмосферном давлении
Холодильный коэфициент 7,30 1,72
Мощность, л. с............ 27,7 114
Производительность компрессора, мЦмин 0,746 19,94
Эти числа относится к идеальному процессу, но результат сравнения не будет значительно изменяться в действительных условиях работы.
При обычной системе воздушной машины производительность воздушного компрессора будет, конечно, много меньше.
Сравнение холодильных агентов. Пользуясь только что рассмотренными методами, можно произвести сравнение различных холодильных агентов, которое приводит к некоторым интересным выводам.
В табл. 15 дается анализ следующего частного случаи ☆):
1. Температура парообразования—15°С.
2. Температура конденсации 30°С.
3. Жидкость подходит к дроссельному вентилю без переохлаждения.
4. Пар, входящий в компрессор, является насыщенным.
Предполагается изоэнтропное сжатие.
Большинство термодинамических констант заимствовано из таблиц „Справочника инженера-химика" Перри. Данные по метиленхлориду взяты из статьи Уотерфилда [251]. Отдельные данные взяты из „Справочника по холодильным данным" [196[. О подобных таблицах см. статью Макинтайра [ 162].
При работе по циклу Карно, конечно, не существовало бы разницы в к. п. д. для различных рабочих веществ. Поскольку цикл сжатия пара незначительно отличается от цикла Карно, не следует ожидать больших различий между расходами мощности на получение „тонны охлаждения" для различных хладагентов. Табл. 15 показывает справедливость этого заключения для всех случаев, кроме углекислоты, где отклонение от цикла Карно велико вследствие того, что температура конденсации близка к критической температуре СО2. Существуют различные пути, с помощью которых можно увеличить
й) Эти условия мало отличаются от стандартных, выбранных Американским обществом инженеров холодильной промышленности для оценки холодильных машин. Этот стандарт имеет такие же температуры парообразования и конденсации, что и указанные выше, но жидкость, идущая к дроссельному вентилю, охлаждается до—12,8° С, а пар, входящий в компрессор, подогревается до— 12,8° С.
32*
Таблица 15
Сравнение холодильных агентов
(данные относятся к 1 «тонне охлаждения* для идеального цикла Карно с температурой парообразования —15° С и температурой конденсации 30° С)
Холодильный агент Давление парообразования, ат Давление конденсации, ат Удельный объем всасываемого газа, м3/кг Циркулирует в минуту, кг Производительность компрессора, м31мин Холодильный коэфициент Мощность, л. с. К. п. д. по отношению к циклу Карно, °/о Примечание
Любое вещество в цикле Карно . 5,74 0,821 100
Углекислота . 23,51 73,04 0,0167 1,60 0,0266 2,56 1,84 44,7
Аммиак 2,41 11,90 0,509 0,187 0,0951 4,85 0,973 84,5
Пропан . 2,95 10,92 0,155 0,748 0,116 4,88 0,967 85,0
Дихлордифторметан 1,86 7,59 0,0927 1,78 0,165 4,72 0,997 82,3 Известен также как „фреон-12* (F-12)
Метилхлорид 1,47 6,71 0,283 0,644 0,183 4,67 1,007 81,7
Сернистый газ 0,830 4,68 0,401 0,640 0,256 4,74 0,995 82,5
Этилхлорид . 0,327 1,91 1,07 0,635 0,679 5,32 0,888 92,7
Диэтиловый эфир 0,105 0,865 2,18 0,717 1,57 4,86 0,971 84,6
Метиленхлорид 0,0823 0,710 3,16 0,671 2,09 4,90 0,965 85,0 Известен также как
Дихлорэтилен 0,0576 0,492 3,98 0,794 3,14 5,14 0,918 89,4 Известен также как
Трихлорэтилен 0,0110 0,128 14,4 0,989 14,3 5,09 0,928 88,5 ДИЭЛИН Известен также как триэлин
500
X. Охлаждение
эффективность цикла с СО2, но все они осуществляются за счет усложнения процесса. Этот вопрос будет разобран более подробно ниже.
Изучение таблицы показывает, почему для промышленного охлаждения почти исключительно применяется аммиак. Он имеет хороший практический к. п. д. и требует самой незначительной производительности компрессора (за исключением углекислоты, которая обычно выпадает из промышленного применения вследствие высоких давлений и низкого практического к. п. д.). Следует отметить, что фреон-12 (ди-хлордифторметан), который получает все большее распространение, по своим термодинамическим свойствам подобен аммиаку и имеет преимущество, заключающееся в большей безопасности применения, поскольку он является нераздражающим, неядовитым и невзрывоопасным. Пропан также очень напоминает аммиак, но образует с воздухом взрывчатые смеси.
Водяной пар с точки зрения безопасности и дешевизны является идеальным холодильным агентом, ио его нельзя использовать для температур ниже 5°С, и поэтому он Me включен в таблицу. При использовании для охлаждения в пределах от комнатной температуры до 5°С он может конкурировать по холодильному коэфициенту с другими веществами, но очень большие объемы, подлежащие обработке,—13,5 лс3 в минуту на >,тонну охлаждения" при температуре парообразования в 5°С и температуре конденсации в 30°С, — требуют применения центробежных или пароструйных компрессоров.
Для небольших установок, например применяемых в домашнем хозяйстве, аммиаку предпочитают SO2, этилхлорид и метилхлорид, вследствие меньшего давления их насыщенного пара, меньшего раздражающего действия и того обстоятельства, что при конструировании машин можно пользоваться сплавами, содержащими медь.
Следует отметить, что на стороне низкого давления системы желательно иметь давление несколько выше атмосферного, чтобы предотвращать проникновение воздуха, который будет вызывать затруднения как сам по себе, так и из-за сопутствующего ему водяного пара. Это является одним из недостатков SO2 и других холодильных агентов с низким давлением насыщенного пара. Это требование делает также невыгодным использование аммиачного охлаждения ниже — 32°С. Для парообразования аммиака значительно ниже этой температуры необходимо давление всасывания ниже атмосферного, а это в прошлом обычно считалось невыгодным для практики ☆ ).
В ряде случаев, например при кондиционировании воздуха в театрах, общественных помещениях и т. д. или при охлаждении в морском
£г) Однако в последние годы разработаны эффективные непроницаемые уплотнения для валов, и теперь вполне обычны небольшие установки с давлением всасывания значительно ниже атмосферного. Холодильные машины с фреоном работают даже при таких низких давлениях, как 0,07 кг1см2, и дают такие низкие температуры, как — 74°С, а в некоторых случаях и еще ниже.
Охлаждение сжатием
501
флоте, такие холодильные агенты, как NH3 и SO2, не могут применяться вследствие их токсичности, взрывоопасности, раздражающего действия и других свойств, представляющих серьезные неудобства. В таких случаях прежде широко пользовались углекислотой, мети-ленхлоридом и этилхлоридом, но, вероятно, они будут в значительной мере вытеснены хлорфторпроизводными метана (дихлордифторметан является только одним из этих галоидозамещенных метана, пригодных в качестве холодильных агентов).
Сравнение действительного и теоретического расхода мощности. При действительном холодильном процессе следует учитывать необходимые разности температур для выгодной передачи тепла, приток тепла из окружающей среды, трение и другие потери в компрессоре и прочие необратимые эффекты. В большинстве случаев можно предположительно взять разность температур и определить температуры, при которых хладагент должен испаряться и конденсироваться. Если теплота поглощается не непосредственно испаряющимся холодильным агентом, а косвенно — посредством использования циркулирующего рассола, что иногда бывает необходимо, то общая разность температур, а следовательно, и производительность будут возрастать.
Расход мощности на сжатие, вычисленный по обычным формулам или полученный из термодинамических диаграмм при допущении изоэн-тропного сжатия, приближается к действительной индикаторной мощности компрессора. Принимая 8О°/о к. п. д. при переходе от мощности на валу к индикаторной мощности и 93°/0 — для мотора и двигателя, получим для компрессора общий к. п. д., равный 75°/0,— величина, которая и предлагается для использования при приближенном определении требуемой мощности.
При мер 5. Используя прямое расширение аммиака, следует охладить за час от —14 до — 18°С 6 л3 раствора с удельным весом 1,15 и теплоемкостью в 1,05 ккал/кг-град. Охлаждающая вода при наиболее тяжелых условиях подается при 18°С и в конденсаторе вагреваетси на 8°С. Определить требуемые размеры холодильной установки и мощность, выраженную в киловаттах.
Тепло, которое следует поглотить=6Х 1,15 X 1000 X 1,05 X 4 = 28 980 ккал/ч
Кроме этого Апла имеется еще некоторое количество его, поступающее из окружающей среды. Его можно определить в том случае, если известны детали данной установки. Однако если система хорошо изолирована, то этот приток тепла будет незначителен, и им можно пренебречь. Округлим полученную выше величину до 30 000 ккал)ч, что в известной степени компенсирует это пренебрежение. Примем минимальную разность температур в 5°С как в аммиачном конденсаторе, так и в холодильнике. Это будет совершенно произвольным допущением, но для данной цели оно достаточно приближается к практическим условиям. Это допущение фиксирует температуру конденсации аммиака при 23аС и температуру парообразования при — 23°С. Соответствующими давлениями будут 9,6 и 1,67 ат те.
502
X. Охлаждение
Энтальпия жидкого NH3 при 23°С = 69 ккал кг.
Энтальпия паров NHS при — 23°С = 338.
В час должно циркулировать
30 000 мн
338 — 69 — 111,5 КНз‘
Энтальпия перегретых паров NH3 при давлении 9,6 атм, имеющих одинаковую энтропию с насыщенным паром при — 23°С, равна 398.
Теоретическая мощность, требующаяся для сжатия = (338 — 398)111,5 =
= 6690 ккал!ч
_ 6690
~ 862
= 7,76
кет.
Действительная
7,76 мощность — ——
0,75
= 10,35 кет.
Теоретическая мощность для цикла Карно, работающего между заданными пределами температур, была бы равна
30 000 Х36_
862 X 255 ~
4,9 кет.
Следовательно, для быстрого ориентировочного определения расхода мощности на данный холодильный процесс рекомендуется вычислить расход мощности для цикла Карно, приняв в качестве температурных пределов наиниэшие из нужных температур холодильника и охлаждающей воды. Затем для получения действительной мощности (реальный цикл) следует увеличить мощность цикла Карно на 4О°/о.
Оценка холодильного оборудования. Так как номинальная производительность любой холодильной установки обычно будет зависеть от условий процесса, то необходимо принять некоторые произвольные условия в качестве стандарта, на котором будет основываться оценка. Стандартные условия, предложенные Американским институтом инженеров холодильной промышленности, приведены в разделе „Сравнение холодильных агентов1*, стр. 498. Для потребителя такого оборудования важно ясно себе представлять, насколько значительно может изменяться производительность в зависимости от условий работы, и что если эти условия будут значительно отличаться от стандартных, то нельзя будет получить номинальной производительности. Это лучше, всего проиллюстрировать на чнслоном примере.
Пример 6. Аммиачная холодильная установка имеет номинальную производительность в 50 „тонн охлаждения”. Компрессор имеет цилиндр двойного действия размерами в 250 X 300 мм, число оборотов 180 в минуту, вредное пространство равно 5°/0. Чему будет равна холодопроизводительность установки в „тоннах охлаждения”, если температура парообразования аммиака равна — 25°С, а температура конденсации 40°С? Примем, что жидкость в дроссельном вентиле переохлаждается и в компрессор входит насыщенный пар.
Варианты простого процесса сжатия пара
503
При —25°С:
При 40°С:
Давление = 1,55 ат, v насыщенного пара = 0,77 м^кг, Н насыщенного пара = 337,5, ккал 1кг.
Давление —15,85 ат, И жидкости = 89.
Охлаждающий эффект — 337,5 — 89 = 248,5 ккал/кг.
Производительность цилиндра = 0,785 X 0.2502 X 2 X 0,300 X 180 = = 5,30 м^мин.
По уравнению (39, гл. VII)
1_ 1
объемный к. п. д. = 1-|-<г — с =14-0,05—0,05 =0,76;
забираемый объем = 0,76 X 5,30 = 4,03 м3/мин, 4,03 ,
^уу = 5,23 кг/мин,
5,23 X 248,5 .
------------= 23,4 «тонны охлаждения'.
55,5
Принятое изменение в рабочих условиях по сравнению со стандартным определением вызывает падение номинальной производительности с 50 до 23,4 т.
ВАРИАНТЫ ПРОСТОГО ПРОЦЕССА СЖАТИЯ ПАРА
Многоступенчатое сжатие и расширение. Химическая промышленность все более и более испытывает потребность в таких низких температурах, как —45°С и ниже. Если приходится пользоваться аммиачной системой охлаждения при температурах парообразования ниже примерно — 25°С, то степень сжатия будет настолько высока, что становится желательным сжатие более чем в одну ступень. Так, при температуре парообразования в —26°С и температуре конденсации в 30°С степень сжатия в компрессоре должна равняться 8,. что является пределом для одноступенчатого сжатия. Причины применения многоступенчатого сжатия, в сущности, те же самые, что и в случае сжатия газа (см. гл. VII). В данном случае добавочным преимуществом является то обстоятельство, что охлаждение достигается при нескольких температурах. Система отличается от системы, применяемой при сжатии газа, тем, что сжатый газ невозможно охлаждать в промежуточном холодильнике вновь до начальной температуры, и тем, что редко применяется более двух ступеней. Двухступенчатые системы употребляются примерно до —56°С, а трехступенчатые — для охлаждения до таких низких температур, как — 84°C.
Как и при сжатии газа, холодильная система двухступенчатого сжатия может состоять просто из двух цилиндров с промежуточным водяным охлаждением. Степень промежуточного охлаждения, осуществляемого с помощью воды, конечно, ограничена тем- / пературой воды, но иногда осуществляют и дальнейшее охлаждение г
504
X. Охлаждение
инжекцией жидкого аммиака в сжатый пар. Дальнейший выигрыш возможен при расширении жидкого аммиака в две стадии вместо одной, что снижает количество пара, которое следует сжать на ступени низкого давления. Схема двухступенчатой системы при использовании всех этих возможностей показана на рис. 90. Пар из конденсатора J
А
Рис. 90. Двухступенчатое сжатие и расширение.
входит в первую ступень компрессорах — цилиндр низкого давления; сжатый пар охлаждается водой в промежуточно*м холодильнике В и затем охлаждается в отделителе С посредством парообразования инжектируемого в него жидкого аммиака. Пар, выходящий из С, сжимается в цилиндре высокого давления D (вторая ступень компрессора), охлаждается и конденсируется с помощью водяного охлаждения в конденсаторе Е и после дросселирования попадает в сборник G, в котором поддерживается давление, среднее между двумя ступенями. Пар, образующийся при дросселировании, возвращается в цилиндр D, а жидкость разделяется, причем одна часть ее расширяется через Н в холодильнике J для получения требуемого охлаждения, а другая часть отводится для использования в качестве охлаждающей среды в С. Первая ступень сжатия часто осуществляемся в отдельной машине, называемой „вспомогательным
Варианты простого процесса сжатия пара
505
конденсатором”, поскольку она допускает ббльшую гибкость при выборе условий.
Анализ идеального цикла (рис. 90) для определения возможной производительности при различных холодильных агентах и при различных условиях работы представляет интересную задачу, которую можно легко решить с помощью таблицы или диаграммы термодинамических свойств рассматриваемого холодильного агента. В статье Макинтайра [162] количественно анализируется такой процесс для частного случая использования аммиака в качестве холодильного агента.
Бинарный цикл. Если следует достигнуть температур ниже примерно — 25°С, то можно использовать ранее описанную систему многоступенчатого сжатия. Теоретически нижний предел температуры определяется только тройной точкой холодильного агента (при этой температуре образуется твердое тело), которая для аммиака лежит около—78°С. Однако практически могут встретиться трудности в том случае, если давление на стороне всасывания компрессора ниже атмосферного давления; это кладет предел температуре парообразования аммиака в — 33°С. Этот предел можно понизить использованием таких холодильных агентов, как СО2 (тройная точка около—57°С), но тогда при применении охлаждающей воды в конденсаторе будет очень высокое давление. Последнего можно избежать при использовании цикла с двумя холодильными агентами, иногда называемого „раздельно-ступенчатым сжатием” ☆). Он состоит из двух простых циклов сжатия, действующих совместно таким образом, что холодильный агент с более высоким давлением насыщенного пара конденсируется, причем теплота конденсации используется для испарения другого холодильного агента, который, в свою очередь, конденсируется с помощью охлаждающей воды. Так, если следует достигнуть температуры в — 50°С, а температура охлаждающей воды должна быть такой, чтобы была возможна конденсация при 30°С, то можно использовать комбинацию циклов с СО2 и NH3. Считая, что парообразование СО2 происходит при — 50°С, a NH3 — при — 20°С, и допуская, что разность между температурами конденсирующейся СО2 и испаряющегося аммиака равна 5°С, получим, что для бинарного цикла по сравнению с цик-
Цикл Давление всасывания, ат Давление в конденсаторе, ат
Бинарный цикл:
давление СО, 6,75 23,3
давление NH3 1,94 11,9
Цикл СО2 6,75 65
Цикл NHg 0,42 И ,9
й-) При использовании для получения очень низких температур трех лии более охлаждающих сред этот вид цикла обычно называется .каскадом'.
506
X. Охлаждение
лами, использующими каждый холодильный агент в отдельности, давления будут представлены следующими величинами. (См. таблицу на стр. 505.)79
Пример 7. Сравнить с помощью термодинамического анализа двухступенчатую систему, изображенную на рис. 90, в которой используется в качестве холодильного агента аммиак, с бинарным циклом, осуществляемым с аммиаком и углекислотой. Условии и допущения, которые следует принять дли данного случая, таковы:
Холодильный агент должен испаряться при — 50°С.
Вещество покидает конденсатор водяного охлаждения или холодильник при 30°С.
Сжатие является изоэнтронным.
Пары, входящие в компрессор, насыщены (в условиях всасывания).
Разность температур между конденсирующейся СО2 и испаряющимся NH3 в бинарном цикле равна 5°С.
Давление между ступенями при двухступенчатом процессе должно быть таким, чтобы на обеих ступених производилась приблизительно равная работа.
Пренебречь притоком тепла и уменьшением давления, обусловленным треиием, за исключением падения давления при дросселировании.
В бинарном цикле аммиак должен испаряться при — 20°С-
Свойства углекислоты взяты из диаграммы S — Т (см. .Приложение').
1. Двухступенчатое сжатие
Базис расчета: 1 кг NH3, проходящий через змеевики холодильника.
Даиление всасывания = 0,42 ат.
Давление выпуска = 11,9 ат.
Давление между ступенями дли получения равной работы можно вычислить путем подбора. Несколько подборов показывают, что приблизительно правильной величиной будет 2,2 ат. Эта величина и будет принята.
Воспользуемся следующими свойствами аммиака:
Состояние Р t S Н Примечание
1. Насыщенный пар . . 0,42 — 50 1,47 328
2. Перегретый пар. . . 2,2 56 1,47 380 Изоэнтропное сжатие от состояния 1
3. Перегретый пар . . . 2,2 30 ... 366
4. Насыщенный пар . . 2,2 — 17,5 1,335 340
5. Насыщенная жидкость 2,2 — 17,5 ... 24
6. Перегретый пар . . . 11,9 103 1,335 398 Изоэнтропное сжатие от состояния 4
7. Насыщенная жидкость 11,9 .. • 77
8. Насыщенный пар . . 1,94 — 20 1,344 339
9. Перегретый пар . . . 11,9 ... 1,344 402 Изоэнтропное сжатие от состояния 8
Примечание. Номера состояний соответствуют номерам на рис. 90. Состояния 8 и 9 являются состояниями в бинарном цикле.
Варианты простого процесса сжатия пара
507
Охлаждение в холодильнике = НХ — НЬ = У28 — 24 = 304 ккал/кг NH3. Тепло, удаляемое в С (рис. 90),
— Н$— — 340 = 26 ккал.
Для найденного охлаждения требуется
%-/Л %-%
346^24 = 0’0822 кг NH3.
Пусть х—сухость пара NH3 после расширения в F.
Тепловой баланс для^:Я7 = —х) Нъ,
77 = 340x4-24(1 —х), х = 0,167.
Пусть т — количество NH3, входящего во вторую ступень, тогда т = 1,00 4- 0,0822 4- 0,167 т, т — 1,3 кг.
Работа в первой ступени — Нг — Н1 = '?№ — 328 = 52 ккал.
Работа во второй ступени = т (%—%) = 1,30 (398— 340) = 75,4.
Общая работа =127,4 ккал/кг NH3.
304 Холодильный коэфициент = j^y-^=2,39.
Производительность первой ступени компрессора при 100% объемном к. п. д. на „тонну охлаждения* =2,6х|^ = 0,475 м^/мин.
2. Бинарный цикл
Базис расчета: 1 кг СО2.
Свойства углекислоты таковы:
Состояние Р t 5 н Примечания
10. Насыщенный пар 6,75 —50 1,264 155
11. Перегретый пар 23,3 —15 1,264 168 Изоэнтропное сжатие от состояния 10
12. Насыщенная жидкость .... 23,3 —15 * • • 92
Состояние 10 является состоянием СО2, выходящей из холодильника и входящей в компрессор.
Состояние 11 является состоянием СО2, выходящей нз компрессора.
Состояние 12 является состоянием жидкой СОа, выходящей из конденсатора для охлаждения аммиака и входящей в дроссельный вентиль.
Охлаждающий эффект = Hw — Н12 =155 — 92 = 63 ккал.
Работа компрессора СО2 = /7ц— А7М=168—155=13.
508
X. Охлаждение
Тепло, отнимаемое в конденсаторе для охлаждения аммиака, = = Нп — //12 = 168 — 92 = 76.
Тепло, поглощенное 1 кг аммиака = Н%— /7, = 339— 77 = 262, ,
OT = 2^T —0>29 кг МН3/кг СО,
Работа компрессора NH3 = т (Н9 — Н&) = 0,29 (402—339) = 18.
Общая работа = 31 ккал.
Холодильный коэфициент = ^| = 2,03,-о!
Теоретическая производительность компрессора СО2 на „тонну 55 5
охлаждения“= 0,058 = 0,051 л^/мин.
ио
Из этих вычислений можно заключить, что двухступенчатый цикл, в котором используется аммиак, имеет определенное термодинамическое преимущество перед бинарным циклом, в котором используются аммиак и углекислота. Следует отметить, что в последнем цикле мы произвольно выбрали температуру, при которой будет конденсироваться углекислота. Эта температура может изменяться, что приводит к изменению расхода мощности, но вычисления показывают, что минимальная мощность получается в пределе, когда аммиак несет полную нагрузку. С другой стороны, бинарный цикл имеет ряд практических преимуществ, так как его применение не требует давлений ниже атмосферного, допускает меньшие размеры компрессора и позволяет легко осуществить охлаждение при двух различных температурах.
АБСОРБЦИОННОЕ ОХЛАЖДЕНИЕ
Абсорбционное охлаждение раньше широко использовалось для получения относительно низких температур, но современное развитие компрессоростроения привело почти к полному отказу от абсорбционного процесса в промышленности, хотя он все еще используется даже на некоторых современных заводах. Это объясняется тем, что оборудование значительно более громоздко, чем при схеме, использующей сжатие, менее гибко и требует больше осторожности и внимания. Оно имеет известное преимущество, допуская использование отработанного пара, чем и можно объяснить его продолжающееся применение. Оно и до сих пор имеет значение для небольших установок, например для холодильников в домашнем хозяйстве. Несмотря на его относительно малое значение для промышленности в настоящее время, этот процесс будет кратко рассмотрен, потому что никогда нельзя уверенно отрицать возможности новых усовершенствований, которые снова могут дать стимул к его применению.
Абсорбционное охлаждение
509
Периодический цикл. В своей наиболее простой форме абсорбционная система не требует никакой механической энергии и не имеет движущихся частей. Ее основной принцип показан на рис. 91. Охлаждение осуществляется посредством испарения таких жидкостей, как NH3 или SO2, совершенно так же, как в процессе сжатия, но давление, необходимое для сжижения холодильного агента, достигается не с помощью механической энергии, а за счет затраты тепла в процессе перегонки. В простом периодическом процессе, показанном на рис. 91, сосуд А, действующий попеременно как перегонный
Рис. 91. Схема периодически действующей абсорбционной установки.
куб и как абсорбер, заменяет компрессор. Во время нагрева холодильный агент отгоняется от абсорбента в А посредством водяного пара или непосредственного обогрева при таком давлении, что он будет конденсироваться при охлаждении водой в холодильнике В. Из резервуара для жидкости С холодильный агент расширяется через дроссель D в змеевики холодильника Е, где он испаряется, поглощая тепло. Эта стадия охлаждения в цикле происходит после завершения перегонки, причем в этой стадии абсорбент в А охлаждается водой, благодаря чему он поглощает испарившийся холодильный агент. В качестве абсорбента может применяться жидкость или твердое тело. Обычно используют аммиак и воду; употреблялись также такие твердые абсорбенты, как силикагель или хлориды щелочноземельных металлов.
Наиболее простым практическим приложением этого процесса была домашняя холодильная установка, показанная на рис. 92. Она состоит из двух сосудов А и В, связанных трубкой. Сосуд А попеременно действует как генератор и как абсорбер, а. В — как конденсатор и
510
X. Охлаждение
раствор аммиака.
А
Рис. 92. Метод охлаждения с помощью .ледяного шара".
холодильник. Холодильным агентом служит аммиак, а абсорбентом — рообразующей стадии цикла А нагревается горелкой, а В помещается в сосуд с холодной водой. В охлаждающей стадии цикла В помещают в пространство, которое следует охладить, а А оставляют при комнатной температуре.
Непрерывные процессы. Непрерывно действующая абсорбционная система, использующая аммиак в качестве рабочего вещества и воду в качестве абсорбента, показана на рис. 93. Две функции сосуда А в периодическом процессе теперь разделены между генератором, в котором аммиак непрерывно отгоняется из
крепкого аммиачного раствора, и абсорбером, в котором аммиак непрерывно абсорбируется слабым аммиачным раствором, охлаждаемым водой. Так как эти дра сосуда находятся при различных давлениях и аммиачный раствор нужно передавать от абсорбера к генератору, то необходим насос. Система содержит также теплообменник для передачи тепла от горячего раствора, покидающего генератор, к охлажденному раствору, поступающему из абсорбера, и так называемый разделитель, который просто представляет собой короткую секцию ректификационной колонны, служащую для удаления большей части водяиогр пара, отгоняемого от аммиака.
Термодинамический анализ этой системы можно произвести с помощью данных по давлению насыщенного пара аммиачно-водного раствора, по составу равновесного пара как функции температуры, по составу жидкого раствора и по теплотам растворения.
Прежде чем закончить рассмотрение абсорбционного охлаждения, следует упомянуть о системе Платен—Мунтерса, используемой для домашних установок. Этот процесс еще не применяется в промышленности, ио крайне удачное применение простого принципа делает -его очень интересным, причем возможна разработка и иных важных приложений этого принципа.
Установка представляет собой непрерывно действующую систему, использующую аммиак и воду. Необходимость в насосе для раствора устраняется следующим остроумным способом. В обычной аммиачной системе абсорбер и испаритель действуют при одном давлении, например около 2 ат, а генератор и конденсатор действуют при значительно более высоком давлении — около 12 ат. Однако в системе Платен — Мунтерса вся установка находится практически при давлении, имеющемся в конденсаторе, а необходимая разность давлений между конденсирующимся и испаряющимся аммиаком поддер
Абсорбционное охлаждение
511
живается с помощью газообразного водорода. Аммиак все же может испаряться при низкой температуре даже при общем давлении в 12 ат, потому что он испаряется в поток водорода, а парциальное давление NH3 можно поддерживать ниже необходимых 2 ат.
Рис. 93. Схема непрерывно действующей аммиачной абсорбционной установки [188 а]
/—редукционный клапан; 2—предохранительный клапан; 3—генератор; 4—крепкий раствор аммиака; 5—теплообменник; б—крепкий раствор аммиака; 7—перепускной клапан; 8—насос; 9—холодная вода; /О—абсорбер; 11—слабый раствор аммиака; 12—разделитель; 13— предохранительный клапан; 14— газ; 15— холодная вода; 2d—конденсатор; 17—дроссельный вентиль; 13—жидкий аммиак; 19— холодный рассол для охлаждения хранилища или для получения льда; 20—газ; 21—холодильник с рассолом или испаритель; 22—возврат теплого рассола; 23— слив охлаждающей воды; 24— предохранительный клапан.
Водород циркулирует только между абсорбером и испарителем, где поддерживается низкое парциальное давление NH3; попадание воздуха в генератор илн конденсатор, где пар NH3 должен находиться при общем давлении, предотвращается жидким уплотнением. Источником движущей силы системы является теплота газового пламени (по уже описанному здесь способу).
Схема установки показана на рис. 94. Циркуляция раствора от генератора 1 к абсорберу 4 и обратно происходит благодаря действию силы тяжести; необходимая разность уровней возникает в генераторе благодаря подъемной силе пара в среде кипящей жидкости, которая проталкивает жидкость через трубку 10 к вертикальной
Запасной сосуд для вадорада /
^ri^TiTl'Trhri
Йи1Н111ШГ
, □ <
j’ 0°f
Испаритель 3
Питательное пространст во
Раствор (о крепости судить по плотности Внешние водород точен) индексы *— i~~у
Внутренние ртг> индексы Li-al ' ’
2Ь
-uxiiniHiHiinxM^^
*w.r°TirinifliTliini.iii‘MT
-о U о Ч-»=>-1 О О 0-
""J“1ММ111" "'-'"-"I"*
ДИ»
iiiiiiiiiiiimiiijjijjiiHiig
тТттттн'ГтгпitTi11111иjmг <
Конденсатор
2
8 Газовый теплообменник
Ъ Вооо->->.
Абсорбер 4
Ректификатор. 7
If
1b
Раздели тель
12
la
ппт^"сппик 'а Генератор 1 для жидкости у
Рис. 94. Схема холодильной установки системы Платен — Мунтерса.
Вакуумное охлаждение
513
трубке 11, из которой она течет под действием силы тяжести к абсорберу и затем вновь к генератору. Циркуляция водорода и паров аммиака между испарителями За и ЗЬ и абсорбером 4 осуществляется благодаря разности гидростатического напора между смесью Н2 с NH3 во внутренней трубке теплообменника 8 и значительно более легким водородом в кольцевом пространстве. Пары из генератора проходят через разделитель 6 и ректификатор 7, функцией которых, как и в обычной абсорбционной системе, является удаление водяного пара. Аммиак конденсируется в конденсаторе с воздушным охлаждением 2 и течет, благодаря силе тяжести, в змеевики испарителя, где встречается с противоположным потоком водорода из 8, который испаряет аммиак при температуре, значительно более низкой, чем та, которая соответствует общему давлению в системе. Разделение конденсатора и испарителя на две секции объясняется прежде всего конструктивными соображениями и, кроме того, стремлением обеспечить более точную регулировку условий в кабине.
Водород, содержащий NHg, входит в абсорбер 4, где NHg абсорбируется встречным потоком слабого раствора. Теплота абсорбции удаляется воздухом, обтекающим ребра абсорбера. Запасной сосуд для водорода 5 действует как автоматический регулятор для компенсации изменений комнатной температуры, которая в противном случае оказывала бы значительное влияние на работу системы.
ВАКУУМНОЕ ОХЛАЖДЕНИЕ
Холодильные системы, работающие при давлениях, значительно более низких, чем атмосферное, за последние несколько лет получили большое распространение в промышленности. Они особенно удобны для кондиционирования воздуха — процесса, получающего все возрастающее значение. Широко употребляются две системы, причем в одной в качестве холодильного агента применяется водяной пар и она снабжена паровым инжектором; в другой используются центробежные компрессоры с водяным паром или парами органических соединений.
Охлаждение водяным паром. Системы, в которых в качестве рабочего вещества используется водяной пар, являются одними из наиболее старых в технике получения холода, но и до сих пор они не особенно популярны среди производственников. В первых установках применен и принцип охлаждения сжатием и принцип абсорбционного охлаждения, причем в них в качестве абсорбента обычно пользуются серной кислотой. Трудность процесса сжатия при применении водяного пара заключается в том, что поршневой компрессор для работы большими объемами совершенно непрактичен. Современное развитие охлаждения водяным паром посредством сжатия обусловлено совершенством высокопроизводительных пароструйных компрессоров. 33 Химическая термодинамика
514
X. Охлаждение
Принцип действия пароструйной вакуумной системы прост, как можно видеть на рис. 95. Вода, подлежащая охлаждению, впрыскивается в холодную камеру, где частично переходит в пар, а оставшаяся вода охлаждается до температуры насыщения, соответствующей давлению, поддерживаемому в испарителе. Пар низкого давления
Охлаждаемая вода при 13°С
Пусковые -'-*1
Холодный резервуар
Коллектор водяного пара Рабочие сопла
щие трубы
воздуха
Конденсатор
Эжектор 2-ои ступени
Насос
-тдля охлаж-'даемай воды
Насос для конденсата'. Выход кон-Л geHcqma,-f™&3
37 С ______
Охлаждаемая вода Спускной кран Конденсатор
{^водяной пар высокого давления мнаеюзторц ,~оастУпвни Е53 Водяной пар низкого давления *
ЁЕЗ Охлаждающая вода
Конденсат
Вспомогательны
Контроль уровн жидкости
Охлаждаемая вода вы
зжект Разбрыз-
^Вакуум
ATi.Yll 7Ю ел ч пт гч
Вода, выходящая из конденсатора
Эжектор ,1-ой 'ступени
конденсатор охлаждающей воды, 27°С
Рис. 95. Вакуумная холодильная установка.
сжимается в вспомогательном эжекторе посредством пара, выходящего из сопел в эжектор при очень высоких скоростях (порядка 1200 м[сек). Необходимо сжать пар, но не до атмосферного давления, а только до такого, при котором его можно сконденсировать охлаждающей водой, как показано на рис. 95. Из конденсатора, который может быть конденсатором как непрямого соприкосновения (поверхностным), так и прямого соприкосновения, конденсат удаляется насосом. Неконденсирующиесн газы, которые всегда присутствуют в результате незначительных подсосов, или как газы, растворенные в охлаждающей воде, удаляются вакуум-насосом, работающим при давлении, промежуточном между давлением в конденсаторе и атмосферным давлением. Вакуум-насос может быть обычным механическим иасосом или же каким-нибудь паровым эжектором специального типа.
Условия работы такой системы будут яснее на следующем примере.
Вакуумное охлаждение
515
Пример8. С помощью системы с водяным паром следует получить 100 „тонн охлаждения'. Охлажденная вода должна иметь температуру 5° С, и в процессе выделения тепла она нагревается до 15° С. Охлаждающая вода берется ирн 25° С и нагревается в конденсаторе до 40° С. Вычислить: 1) давление в испарителе; 2) давление в конденсаторе; 3) количество циркулирующей холодной воды; 4) количество доливаемой воды и 5) объем пара, подлежащего сжатию. Принять идеальные условия работы.
1. Давление в испарителе равно давлению насыщенного пара воды при 5° С, которое по паровым таблицам равно 0,00889 ат.
2. Давление в конденсаторе равно давлению насыщенного пара при 40° С, или 0,0752 ат.
В обоих случаях пренебрегаем второстепенными эффектами, например присутствием неконденснруемого газа и падением давления, обусловленным потоком.
3. В минуту должно циркулировать 55,5 X 100/(15 — 5) = 555 кг НаО.
4. Мгновенное парообразование воды является процессом при постоянной энтальпии.
Для воды при 15° С Н— 15,04 ккал)кг.
При 5° С 77=5,03.
Н насыщенного пара при 5° С = 599,4.
Тогда для постоянной Н 15,04 = 599,4х 4-5,03(1—х), где х — доля испарившейся воды;
х = 0,0168.
Доливается воды 0,0168X555 = 9,35 кг/мин.
5. Считаем пар идеальным газом; тогда
, 9,35X848X278,2 ,ооп
объем — 0 00889 104 х 18 — 1380 м !мин-
Из этой последней величины ясно, что поршневой компрессор непригоден.
Можно сделать и дальнейшие вычисления, чтобы показать, что производительность такой системы сильно изменяется в зависимости от температуры холодной воды. Большое преимущество рассмотренного процесса заключается в его простоте. Кроме насосов для жидкости нет никаких движущихся частей, не требуется никаких специальных хладагентов. Он главным образом применяется для получения охлаждения примерно до 1,7° С. Более низкие температуры могут быть достигнуты при употреблении соляных растворов, но это не иашло практического применения.
Расход пара сильно изменяется в зависимости от условий, причем главными переменными являются давление водяного пара, объем и температура охлаждающей воды и температура охлаждаемой воды. Джэксон [116] дает опытные данные по расходу водяного пара как функции четырех упомянутых выше переменных для насыщенного пара при сверхбарометрическом давлении в 7 ат, т. е. при давлении, которое обычно применяется на практике. Потребление водяного пара быстро увеличивается по мере понижения давления. Его очень точно можно определить по методу, иллюстрированному в гл. VII. 33*
516
X. Охлаждение
Было показано, что для водяного пара при давлении в 7 ат, при температуре охлаждения в 7° С и при температуре конденсации в 38,7° С (0,07 ат) требуется 2 кг рабочего пара на 1 кг испарившейся воды. Примем, что в качестве добавленной воды используется конденсат при 35° С; тогда общий охлаждающий эффект на 1 кг испарившейся воды = энтальпии насыщенного пара при 7° С; энтальпия воды при 35° С равна 565,2 ккал. (Это можно легко показать посредством энергетического баланса для системы, включая холодный сосуд и циркулирующую холодную воду.)
В час на «тонну охлаждения" расходуется
2ссГс = И,8 кг водяного пара.
5оо,о
Это количество пара, использованное для приведения в движение поршневого или центробежного компрессора, произвело бы большее охлаждение, потому что пароструйный компрессор относительно неэффективен, но имеет другие преимущества, которые во многих случаях значительно компенсируют меньшую его экономичность.
В применении водяного пара высокого давления нет необходимости. Можно пользоваться отработанным водяным паром при атмосферном давлении, но тогда, как легко показать расчетом, потребуется большее количество его.
Система центробежного сжатия. К этому типу может принадлежать система, в которой в качестве холодильного агента используется водяной пар, но пароструйный компрессор заменен центробежным компрессором. Характеристики центробежных компрессоров уже рассматривались в гл. VII. Было показано, что центробежные компрессоры применимы для сжатия больших объемов при низком или умеренном давлении. Действительно, можно утверждать, что для успешного использования таких компрессоров особенно благоприятны большие объемы. Давление, получаемое в любой ступени при данной скорости вращения, приблизительно пропорционально квадрату диаметра ротора; поэтому небольшая машина потребовала бы излишнего числа ступеней. Так как увеличение давления в одной ступени центробежного компрессора зависит непосредственно от плотности сжимаемого газа, то предпочтительнее пользоваться газами, имеющими более высокую плотность, чем водяной пар. К веществам, употребляемым в вакуумных системах вместо водяного пара, относятся дихлорметан (СН2С12), дихлорэтилен (С2Н2С12) и фреон-И (CClaF).
Дихлорэтилен при тех же самых температуре и давлении обладает примерно в пять раз большей плотностью; чем водяной пар, и поэтому данный центробежный компрессор при использовании паров дихлорэтилена потребует в одном и том же пределе давлений в пять раз меньше ступеней для сжатия, чем при работе на водяном паре. В действительности основной интерес представляют заданные пределы температур; давления же для различных хладагентов будут совер
Процессы с твердой углекислотой
Ъ\1
шенно различны, поэтому лучше иметь дело со степенью сжатия. Из теории центробежного сжатия легко показать, что степень сжатия увеличивается с увеличением молекулярного веса пара независимо от абсолютного давления. Кроме того, степень сжатия для данного предела температур для большинства органических газов меньше, чем для воды. Например, для предела температур от 4,5 до 37,5°С степень сжатия для воды равна 7,8 и только 3,9 для СН2С12. Результатом влияния обоих этих факторов будет то, что для центробежного охлаждения с органическим паром можно пользоваться более простым компрессором, чем тот, который применяется при использовании водяного пара.
Центробежный компрессор дороже парового эжектора, но последний, вероятно, потребует несколько большего расхода водяного пара и будет давать большую нагрузку на конденсатор, делающую необходимым пользование конденсатором ббльших размеров и требующую большего количества охлаждающей воды. Оба эти вакуумных процесса гораздо легче осуществимы, чем обычный процесс сжатия, и они дают возможность экономично работать с меняющейся нагрузкой, которая типична, например, при кондицгонировании воздуха.
ПРОЦЕССЫ С ТВЕРДОЙ УГЛЕКИСЛОТОЙ
Важным приложением холодильных циклов янляется производство твердой СО2. Краткое рассмотрение этого процесса желательно, так
Рис 96. Простая установка для производства твердой углекислоты.
как изучение различных процессов дает многие интересные приложения термодинамических принципов.
Очень простой процесс с углекислотой схематически показан иа рнс. 96. Чистая СО2 при атмосферном давлении и комнатной темпе-
518
X. Охлаждение
Рис. 97. Простой цикл для углекислоты, представленный на диаграмме температура — энтропия.
ратуре смешивается с газом, возвращающимся из камеры твердой углекислоты, и смесь поступает в первую ступень трехступенчатого компрессора, в которой сжимается до достаточно высокого давления, позволяющего осуществить сжижение СО2 в охлаждаемом водой конденсаторе (высокое давление, требующееся для конденсации СО2 обычной охлаждающей водой, вынуждает пользоваться по меньшей мере трехступенчатым сжатием). Жидкая СО2 расширяется до атмосферного давления через дроссель в камеру твердой углекислоты, где образующаяся твер-дая фаза отделяется от газа* причем последний возвращается в цикл.
Анализ этого цикла для определения выхода твердого вещества и расхода мощности как функций основных параметров легче всего осуществляется с помощью диаграммы 5—Т. Воспользуемся диаграммой,приведенной в „Приложении". На рис. 97 схематично представлен цикл на такой диаграмме. АВ дает энтропию насыщенного твердого тела, находящегося в равновесии с паром, ВС— твердого состояния к жидкому,
изменение энтропии при переходе от
при условии, что обе фазы находятся в равновесии с паром. Это изменение происходит в тройной точке, которой соответствуют температура— 56,6° С и давление 5,28 am. CD является линией насыщенной жидкости, a DE—линией насыщенного пара, причем критическая точка D соответствует температуре 31,1° С и давлению 72,9 атм. Цифры на диаграмме соответствуют цифрам на рис. 96, на котором они относятся к тем же точкам цикла.
Пример. 9. Определить теоретическую мощность, потребную на тонну твердой СО2, для цикла, изображенного на рис. 96 и 97.
Анализ цикла будет основан на следующих основных допущениях:
1. Добавляемым газом является чистая СО2 при 20° С и 1 ат давления.
2. Сжатие изоэнтропно.
3. Между ступенями осуществляется идеальное охлаждение.
4. Промежуточные (между ступенями) давления выбираются таким образом, чтобы на всех ступенях производилась одинаковая работа.
5. Существует равновесие между фазами в конденсаторе и в камерах жидкой и твердой углекислоты.
6. Отсутствует приток тепла извне.
7. Падение давления, обусловленное потоком рабочего вещества (кроме падения в дроссельном вентиле), ничтожно мало.
Процессы с твердой углекислотой
519
8. Охлаждающая вода имеет температуру 20° С.
9. Минимальная конечная разность температур в теплообменниках равна 5° С.
Мы будем исходить из жидкой СО2 в точке 8 и вычислим выход твердой углекислоты на килограмм расширяющейся жидкой СО2. Принимаем, что сконденсированная жидкость не переохлаждается; ts = 25° С на линии насыщенной жидкости. Следуя по линии постоянной энтальпии до 1 ат (8—9), найдем сухость пара равной 0,765, т. е. 23,5%СО2 получается в виде твердого вещества. Остающийся пар (точка 10} смешивается при постоянном давлении с добавляемым газом (7), и получающаяся смесь (2) поступает в компрессор. Условия для смеси находятся нз уравнения теплового баланса
0,745 (Н2 — Hw) = 0,255 (Яг — Я2).
Следовательно,
Н2 = 158,5 и t2 = — 53,5° С.
Определение условий сжатия осуществляется путем постепенного подбора до уравнивания количества работы на всех трех ступенях. Следующая таблица дает условия, которые с достаточной точностью отвечают этому критерию.
Наименование 1-я ступень 2-я ступень 3-я ступень
Температура всасывания, °C. • . . Температура выпуска, °C — 53,5 25 25
45,5 117 126
Давление всасывания, ат 1 5,0 17,9
Давление выпуска, ат 5,0 17,6 65,7
Степень сжатия 5,0 3,51 3,64
Энтальпия при всасывании, ккалкг 158,5 173,2 170,0
Энтальпия при выпуске, ккал<кг . . 177,7 192,2 189,0
ЬН или работа, ккал!кг 19,2 19,0 19,0
Общаи работа сжатия = 57,2 ккал/кг =
57,2X 1000
— „ X о_, = 260 квт-ч/т твердой углекислоты. v,2oo оо2
Линии 2—3—4—5—6—7 на рис. 97 представляют процесс сжатия, а 7—8— охлаждение с сжижением СО2.
Так как твердая СО2 должна получаться достаточно дешевой, то очевидно, что расход энергии (или водяного пара при использовании компрессоров, приводимых в движение водяным паром) является важным фактором, и поэтому желательно рассмотреть все изменения цикла, которые могут привести к более высокому выходу СО2 на единицу затраченной энергии. Возможность использования холодного газа из камеры твердой углекислоты для переохлаждения жидкой СО2 показана на рис. 98.
Применив первый закон к теплообменнику, получим
Я8-Я9 = х(Я12-Яи),
где х—сухость пара расширившейся смеси в точке 10. Затем фиксируем 7712, учитывая температурную разность на горячем конце
520
X. Охлаждение
теплообменника, равную 5° С. Это оставляет неизвестными х и Н9, и требуется еще одно уравнение. Оно получается из баланса энтальпии для камеры твердой углекислоты:
/У9 = X#!j + (1 — *) #13'
Совместное решение этих уравнений дает
//8-//13_ 118,5-16,7
//12— //13 — 173,2 — 16,7 — и’
Так как компрессор теперь забирает газ при 20° С вместо — 53,5° С, то расход работы увеличится до 65 ккал/кг, но выход
Конденсатор
для твердой углекислоты
Твердая J I Г/3 углекислота ’ ’ '
Рис. 98. Цикл для твердой углекислоты, в котором холодный расширяющийся газ используется для переохлаждения жидкости.
будет настолько значительнее, что работа на тонну твердой углекислоты уменьшится до 206 квт-ч. Недостатком является то, что размер компрессора увеличивается примерно в 1,3 раза.
Другие возможности снижения расхода энергии заключаются в применении многоступенчатого расширения жидкости, а также в применении бинарного цикла (NHS -|~ СО2). Можно также использовать нх различные сочетания. Анализ любого предложенного видоизменения простого цикла можно производить с помощью ранее рассмотренных методов. (Для дальнейших деталей по термодинамике цикла твердой СО2 см. статью Стикни [241].)
Минимальный расход энергии для получения 1 кг твердой СО2 из газообразной СО2 при 1 ат и 20° С при допущении, что тепло может передаваться при 20° С, дается уравнением ( И). Из диаграммы
Холодильный цикл как метод нагревания
521
5 — Т следует, что
ЬН= 156,5, AS = 0,787.
Следовательно,
А = 1000 (298,2 X 0,787 — 156,5) =
— 78 200 ккал!т = = 91 квт-ч]т.
В наиболее эффективном из циклов, анализированных Стикни (бинарный цикл, в котором для конденсации СО2 используется аммиак), имел место наименьший расход пара, превышающий примерно в 1,4 раза этот минимум. Поэтому, принимая за основу цикл Карно, можно сказать, что к. п. д. рассмотренного процесса равен 72°/0.
Интересно отметить, что если в камере твердой углекислоты поддерживать давление, равное 5,28 ат (тройная точка), т. е. немного больше, чем в рассмотренном случае, то расширение приведет к образованию смеси жидкости и твердого вещества, которая легче уплотняется в твердый лед—СО2, чем твердая углекислота в виде снега. После того как образовалось необходимое количество смеси, давление для превращения жидкости в твердое вещество поднимается до 1 ат. Это является основой важного процесса производства СО2.
ХОЛОДИЛЬНЫЙ ЦИКЛ КАК МЕТОД НАГРЕВАНИЯ
Кельвин в 1852 г. предложил применить холодильный цикл как средство отопления зданий. Несмотря на простоту принципа и на то, что он представляется очень заманчивым с точки зрения экономии энергии, практического применения он до самого последнего времени не нашел. Неизвестно, может ли он оправдать себя экономически как чисто нагревательный метод, но он может открыть значительные возможности в качестве комбинированной схемы для нагревания зданий и кондиционирования воздуха летом.
Для идеальной тепловой машины Карно или холодильного цикла имеем уравнение
A = Qi — Q2~Q1 . (29)
Вели
1\ = 293,2° К(20° С) и Т2 = 273,2° К (0° С), то
<?! _ 293,2
А ~~ 293,2 — 273,2 ,0
Таким образом, затрата 1 ккал работы в идеальной системе сделает возможным получение почти 15 ккал тепла при 20°С, если температура внешней среды будет равна 0° С. Это представляется экономически целесообразным средством использования электрической
522
X. Охлаждение
энергии для отопления жилых домов и больших помещений. Электрическая энергия, используемая необратимо в виде сопротивления для получения тепла, может давать только эквивалентное количество, но при применении ее для работы теплового насоса теплота, выделяющаяся на верхнем температурном уровне, может в несколько раз превосходить количество, эквивалентное затраченной электрической энергии, как и показывает уравнение (29). Однако следует также помнить, что при получении электрической энергии из угля большая часть химической энергии становится бесполезной и при сжигании угля в лучшем случае выделяется в виде теплоты только 25°/0 энергии. Таким образом, в любой системе нагрева с помощью электрической энергии возникает прежде всего эта потеря, что и следует учитывать при сравнении с непосредственным использованием топлива. С другой стороны, не следует упускать из вида того обстоятельства, что топливо можно сжечь гораздо эффективнее на большой центральной станции, чем в обычных маленьких тепловых установках.
Существует много практических трудностей, препятствующих промышленному развитию этой интересной идеи. Прежде всего, вследствие неизбежности трения в любой практической холодильной системе и вследствие необходимости создания разности температур для передачи тепла холодильный коэфициент QJA будет гораздо меньше идеального. Хорошая средняя величина эффективности холодильной системы, работающей посредством сжатия, при использовании ее в качестве нагревающей установйй составляет около 6О°/о по отношению к циклу Карно, работающему между теми же температурными уровнями *). Для температуры наружного воздуха в 0°С и температуры помещения в 20° С можно принять, что температуры охлаждения и конденсации соответственно равны — 6,5 и 37,5°С. На этой основе, т. е. при указанных выше производительности и величине ДЛ холодильный коэфициент будет равен только 4,2 вместо 14,66. При холодном климате, отвечающем температуре окружающей среды в — 18°С, и принятой температуре охлаждения в — 26° С, холодильный коэфициент практически равен 2,9.
Другая трудность возникает от того, что при изменении температуры окружающей среды система реагирует как раз противоположно тому, чего от нее хотят. Так, при понижении температуры охлаждения (той температуры, при которой поглощается тепло) производительность обычной системы охлаждения посредством сжатия быстро падает, и поэтому подается значительно меньше тепла в тот момент, когда его требуется больше. Если система спроектирована с учетом более тяжелых условий, то она будет значительно более крупной и менее экономичной, чем это необходимо для средних условий.
й) Это означает, что практический холодильный коэфициент равен 60% от идеального.
Холодильный цикл как метод нагревания
523
Один из методов преодоления этой трудности заключается в извлечении низкотемпературного тепла от источника с равномерной температурой, например от воды на большой глубине. Другой схемой является накопление тепла в воде в теплое время года с последующим использованием его в холодное время.
Первое практическое осуществление нагревательной машины Кельвина, повидимому, было произведено инженером-электриком Холда-ном [98], который описал удовлетворительно работавшую в его собственном доме установку80. Это, в сущности, была обычная аммиачная холодильная система с конденсатором, работающим как нагреватель горячей воды для радиаторов. С температурой парообразования в — 6,5° С и температурой конденсации в 37,5° С был достигнут холодильный коэфициент в 2,5. Спорн и Мак Ленеган [240] подробно описали установку, которая успешно работала как нагревающее устройство зимой и как система для кондиционирования воздуха летом. Источником тепла в этом случае была глубинная вода, имевшая практически постоянную температуру 13,5° С. Средний холодильный коэфициент получился равным 3,9 (с учетом работы водяного насоса эта величина понизится до 3,5).
Установка значительных размеров была создана лишь позднее. В ней используется метилхлорид, и она дает 480 „тонн охлаждения“ для кондиционирования воздуха летом и 15000 ккал^мин для нагревания зимой. Тепло поглощается из воды, которая охлаждается от 4,5 до 1,7° С, причем вода нагревается снова непосредственным соприкосновением с воздухом в башне с принудительной вентиляцией. Нагрев здагия осуществляется воздухом, который, в свою очередь, нагревается водой из конденсатора метилхлорида. Испытания установки дали холодильный коэфициент, основанный на общем расходе мощности компрессоров и всех вспомогательных устройств от 1,45 до 1,98. При испытаниях, в результате которых был получен коэфициент 1,98, наружная температура была равна 9° С, и воздух нагревался до 23,5° С, так что теоретический холодильный коэфициент был равен 20. Это указывает на значительные возможности усовершенствования метода.
Наиболее современным применением, привлекшим внимание автора, является установка, описанная в статье Лоулесса [145], но в ней не представлены данные по производительности. Эта установка является комбинированной системой для нагревания и для кондиционирования воздуха с большим количеством воды при постоянной температуре 12,8° С; она служит как источник тепла для зимнего нагревательного цикла. Нагревательная система обратного охлаждения была спроектирована для минимальной наружной температуры в —6,5° С; при падении температуры ниже этого уровня получается добавочное тепло от системы для накопления горячей воды. Вода в этой системе нагревается непосредственно за счет электрической энергии.
524
X. Охлаждение
НИЗКОТЕМПЕРАТУРНЫЕ ПРОЦЕССЫ
Процессы сжижения и последующего разделения газов приобретают все большее значение в промышленности. Производство кислорода, азота и аргона из воздуха с помощью низкотемпературных методов осуществляется давно и хорошо освоено, но будущие возможности для увеличения применения кислорода и обогащенного кислородом воздуха настолько велики, что желание получить более дешевые и более надежные методы разделения стимулируют непрерывную активность в этой области. Получение водорода низкотемпературными методами из водяного газа и газа коксовых печей хорошо известно за границей, но лишь в ограниченной степени практикуется в Америке. Значительным достижением в этой области является получение гелия из природных газов. Очень недавним усовершенствованием является сжижение и хранение природного газа для удовлетворения увеличивающейся в нем потребности зимой. Из этих немногих примеров очевидно, что область низких температур имеет такое техническое значение, что заслуживает большего места, чем мы мржем посвятить ей в этой книге. Мы ограничимся только кратким ознакомлением с этой интересной областью.
Методы получения низких температур. Для нашей цели низкие температуры могут быть произвольно определены как температуры ниже 200° К. К охлаждению в этой области применимы те же самые общие принципы, которые были уже разобраны, но существуют некоторые важные различия в деталях. Сейчас известны три общих метода, которые применяются в практике для получения охлаждения до этой или до еще более низкой температуры. Эти методы следующие:
1. Испарение жидкости.
2. Использование эффекта Джоуля — Томсона.
3. Расширение в машине с отдачей внешней работы.
Современные процессы могут использовать или только один из этих методов, или комбинацию их. Рассмотрим теперь несколько подробнее каждый из этих методов в отдельности.
Из свойств жидкости очевидно, что если используется только одна жидкость, то применение метода 1 не может привести к очень большому перепаду температур. Максимально возможный перепад будет ограничен критической и тройной точками, причем во многих случаях давление в тройной точке настолько низко, что нецелесообразно использовать весь "этот интервал. Наинизшая температура, достижимая по этому методу с помощью одного вещества (т. е. по уже рассмотренному процессу сжатия пара), равна примерно 200° К- Однако при использовании двух или нескольких веществ в такой последовательности, что вещество с наиболее низкой точкой кипения конденсируется благодаря охлаждающему действию, вызываемому испарением следующего по точке кипения вещества,—и так до тех пор, пока вещество с наиболее высокой точкой кипения не
Низкотемпературные процессы
525
будет сконденсировано воздухом или охлаждающей водой, — можно достигнуть гораздо более низких температур. Такая система известна под названием каскадной. В системе, употребляющейся для сжижения воздуха, используются три вещества — аммиак, этилен и метан. Существует низший предел температуры, который достигается с помощью такого метода и определяется тем, что критическая температура наиболее низкокипящего из употребляемых веществ должна быть выше температуры тройной точки следующего по летучести вещества. Обращение к табл. 16 показывает, что 63° К является
Таблица 16
Физические свойства некоторых веществ, используемых при охлаждении
Вещество Нормальная точка кипения, ° к Тройная точка Критическая точка
температура, ° к давление, атм температура, ° к давление, атм
Не 4,2 5,2 2,26
н2 20,4 14,1 0,071 33,3 12,80
Ne 27,3 24,5 0,425 44,5 26,86
n2 77,4 63,3 0,127 126,1 33,49
Аг 87,5 84,0 0,673 150,8 48,0
о2 90,2 54,8 0,0026 154,4 49,71
сн4 111,8 •90,0 0,092 190,7 45,8
Кг 120 104 — 210,6 54,2
С2Н4 169,5 104 — 282,7 50,65
n2o 183,4 170,9 — 309,7 71,65
СЛ 184,5 101 — 306,0 48,8
со2 195 216,6 — 304,3 72,9
СЛ 231 83,3 — - 370 42
NH3 240 195 — 405,5 111,5
CC12F2 243 118 — 384,6 39,6
СН3С1 249,1 170,3 — 416 65,9
озо-С4Н10 261,4 — — 407 37,2
таким низшим пределом, так как эта температура будет наинизшей, которой можно достигнуть испарением жидкого азота, а следующее вещество—неон—имеет настолько низкую критическую точку, Что не может конденсироваться кипящим азотом.
Метод 2 можно использовать для получения температур, отличающихся лишь на несколько градусов от абсолютного нуля. Это сначала может показаться удивительным, поскольку, как было показано в гл. VII, понижение температуры, полученное при дросселировании даже от высоких давлений, очень мало, а исходя от воздуха при комнатной температуре можно понизить температуру в лучшем
526
X. Охлаждение
случае примерно на 50° С. Однако при сочетании расширения с теплообменом, как показано на рис. 99, можно осуществить аккумулирование незначительных охлаждающих эффектов. Газ высокого давления при комнатной температуре входит в низкотемпературную систему в точке 1; окружающей среды,
если вся система находится при температуре то
Рис. 99. Использование эффекта Джоуля — Томсона для низкотемпературного охлаждения.
газ достигает точки 4 все еще при комнатной температуре; затем он дросселируется, испытывая падение температуры, после чего вновь поступает в теплообменник, перемещаясь навстречу входящему газу высокого давления. Это вызывает охлаждение газа высокого давления, благодаря чему он подходит к точке 4 более холодным и становится еще холоднее после охлаждения в точке 5. Очевидно, температура в точке 5 будет продолжать падать, если только не имеют места компенсирующие эффекты; если пренебречь притоком тепла в систему из окружающей среды, то в результате рассматриваемого процесса должно наступить частичное сжижение в точке 5, сопровождающееся разделением на две фазы. Так как только часть вещества возвращается в теплообменник, то охлаждение, достигаемое за счет охлаждения газа высокого давления, уменьшается, и в конце концов получается стационарное состояние, которое характеризуется энергетическим равновесием.
Для этого метода охлаждения также имеется предел, так как начальное состояние вещества должно быть таким, чтобы при изотермическом расширении его энтальпия увеличивалась. Это условие при комнатной температуре удовлетворяется всеми веществами, за исключением водорода и гелия. Как следует ожидать на основании теоремы соответственных состояний, даже эти два газа можно сжижить с помощью дросселирования, если начальная температура поддерживается иа некотором более низком уровне посредством внешнего охлаждения (около 100° К для водорода и 20° К для гелия).
Метод 3 уже упоминался в связи с охлаждением при более высоких температурах. Его применение при низких температурах задерживалось конструктивными трудностями, связанными с необходимостью поддерживания нормального режима работы поршневых машин при температурах, при которых жидкие смазочные материалы непригодны. Эта задача впервые была разрешена Клодом, который разра
Низкотемпературные процессы
527
ботал специальное уплотнение из кожи, остающееся гибким при температурах жидкого воздуха. Последующие исследователи употребляли другие остроумные способы для преодоления этой трудности, например поршневые кольца Микарта, детандер плунжерного типа с малым вредным пространством и использованием плотно входящего поршня с желобками, наполненными газом и действующими как поршневые кольца для предотвращения контакта между поршнем и цилиндром. С помощью детандера, принадлежащего к последнему из упомянутых типов, Капица [121] достиг сжижения гелия без использования цикла сжижения водорода для предварительного охлаждения. Позднее Капица [311] преодолел трудности, связанные со смазкой, и добился других преимуществ посредством применения турбодетандера.
Следует отметить, что даже изоэнтропное сжатие (отличное от изоэнтальпийного) не является само по себе пригодным средством для непрерывного сжижения такого газа, как воздух. В практическом процессе расширение соединяют с теплообменом, чтобы аккумулировать охлаждение, как в методе 2.
Минимальная работа для сжижения или разделения газов. Из первого и второго начал термодинамики ясно, что изменение состояния, связанное со сжижением газа или разделением газовой смеси, должно сопровождаться уменьшением энтропии. Так как энтропия изолированной системы самопроизвольно никогда не уменьшается, то для осуществления рассматриваемых процессов должна затрачиваться работа; одновременно будет происходить переход тепла в окружающую среду. Таким образом, установившийся процесс сжижения или разделения (поскольку дело касается общих энергетических эффектов) можно представить простой схемой, приведенной на рис. 100. Вся аппаратура для проведения процесса (за единственным исключением, упоминаемым ниже) помещена внутри камеры, изображенной в виде прямоугольника (в данном случае детали не существенны). В камеру входят вал для передачи работы и две трубы (или больше) для подачи веществ в систему и вывода их из нее. Камера расположена в среде с постоянной температурой То, и единственной связью между аппаратурой и окружающей средой является теплообменник. По первому закону
— А = Ш— Q. (30)
Если процесс обратим, то по второму закону
Д£ = Д£г
= (31)
и
Д5<Д5е = ^, (32>
528
X. Охлаждение
если процесс необратим; Д5 выражает изменение (уменьшение) энтропии вещества, а Д5в — изменение (увеличение) энтропии среды. Подставляя значение Q из уравнения (31) в уравнение (30), получаем
— Л = T<AS. (33)*)
Поступление
вещества
выход рабочего вещества
Н,-Общая энтальпия входящего потока
Нг-Общая энтальпия выходящего потока
Рис. 100. Энергетический баланс непрерывного процесса сжижения или разделения воздуха.
Минимальная работа процесса, выраженная уравнением (33), совершенно не зависит от применяемого механизма, а зависит только от свойств веществ.
Пример 10. Чему равна минимальная работа, необходимая: 1) для сжижения азота, находившегося первоначально при 300° К и 1 атм, чтобы получить жидкий азот при 1 атм, и 2) для разделения воздуха на жидкий кислород и газообразный азот, если оба вещества должны находиться при давлении 1 атм, а азот, кроме того, при 300° К- Результат выразить в лошадиных силах на 1 кг.
Нижеследующие значения Н и S взяты из статьи Миллара и Сюлли-вана [171], исключение составляют особо отмеченные случаи:
при 300° К и 1 атм = 2884.
Н жидкого N2 при нормальной точке кипения = 0.
' SNj при 300° К и 1 атм = 26,70.
S жидкого N2 при 300° К и 1 атм — 0.
Н воздуха при 300° К и 1 атм = 2963.
S воздуха при 300° К и 1 атм — 28,08.
Но при 300° К и 1 атм = 3251.
£•) Это уравнение, конечно, совпадает с уравнением (11), и вывод их в сущности одинаков. Оправданием для этого кажущегося повторения является то, что при изучении термодинамики опыт показывает желательность представлять одни и те же выводы с разных точек зрения.
Низкотемпературные процессы
529
So при 300° К и 1 атм = 28,41.
Н жидкого О2 при нормальной точке кипения = 158.
S жидкого О» при нормальной точке кипения == 1,90.
Энтальпия воздуха вычисляется из энтальпий Oj и N2; энтальпия смешения принимается равной нулю {идеальный раствор). Следовательно,
Нвозлу1а = 0,790 Нк 4-0,210 Но
= 0,790 X 2884 4-0,210 X 3251=2963.
Энтропия воздуха вычислена по уравнению (53, гл. IV) (допуская идеальность газов).
воздуха = 0,790 X 26,70 4- 0,210 X 28,41 — 1,987 (0,7901п 0,7904-
4-0,210 In 0,210) = 28,08.
(1) Л=(2884— 0) — 300(26,70 — 0) = — 5136 код/моль =— 0,29 л. с.-ч[кг.
(2) А = 2963 — 0,79X2884—0,21X158 — 300 (28,08 — 0,79 X 26,70—0,21X1,90) =
= 1324 кал/моль
= 0,072 л. с.-ч)кг воздуха
= 0,343 л. с.-ч!кг кислорода.
Вследствие различных необратимых эффектов действительные процессы потребуют большей затраты работы, чем минимальная работа, найденная в этом примере; отношение минимальной (обратимой) работы к практической работе реального процесса будет называться термодинамическим к. п. д. Оказались полезными и два других к. п. д., определяемых следующим образом:
к. п. д. цикла =
________________обратимая работа________________
теоретическая работа для цикла идеального действия’
практический
к. п. д. =
теоретическая работа для цикла идеального действия практическая работа для цикла
Ясно, что произведение двух последних к. п. д. равно термодинамическому к. п. д. Идеальное действие цикла означает отсутствие необратимых эффектов, за исключением внутренних, присущих данному циклу. Следующие необратимые эффекты обычно ветре чаются при низкотемпературных циклах: 1) приток тепла из окружающей среды; 2) теплопередача в теплообменниках при конечной Д/; 3) трение вещества в трубопроводах и в приспособлениях; 4) механическое трение в машинах; 5) дросселлирование и 6) обмен веществом между фазами, осуществляемый не при равновесии. Приток тепла всегда можно считать в пределе сводящимся к нулю; то же справедливо и в отношении трения; но дросселирование может быть необходимо для данного частного цикла. В отдельном случае может оказаться необходимой определенная разность температур, между тем как в пределе эта разность иногда может уменьшаться до нуля (см. гл. IX).
34 Химическая термодинамика
530
X. Охлаждение
Можно принять за правило, не имеющее исключений, что всякий раз, когда в какой-нибудь части низкотемпературного процесса происходит необратимый процесс, он неизбежно должен компенсироваться определенным количеством затраченной работы. Это является очень полезным руководящим правилом, которое следует помнить при сравнении различных низкотемпературных процессов.
Сжижение газов с помощью дросселирования (процесс Линде). Наиболее важные элементы этого процесса уже были представлены на рис. 99. Достижение в процессе стационарного состояния отвечает равенству
/7. 4-<7. = ГЛ/_-1-(1—Г) Н8, (34)
вытекающему из первого начала термодинамики. Из уравнения (34) следует, что
где Ни Нг н так далее — энталЪпии вещества в точках, обозначенных на схеме цифрами;
С—доля входящего газа, которая удаляется в виде жидкости;
^ — количество тепла, поступающего в аппарат из окружающей среды на единицу входящего газа.
Интересно отметить, что степень сжижения определяется разностью энтальпий на горячем конце теплообменника, а не условиями в точке, где фактически происходит расширение.
С помощью этого простого уравнения и таблицы свойств можно легко определить долю любого газа, которая будет конденсироваться, а также зависимость этой доли от давления, температуры, притока тепла, к. п. д. теплообменника и других факторов. Нижеследующий пример может иллюстрировать этот метод.
Пример 11. Следует сжижить воздух с помощью процесса Лииде. Примем, что воздух входит в низкотемпературную систему при температуре 300° К и давлении 100 ат и расширяется до 1 ат. Скорость потока воздуха равна 1,5 м^мин при 15° С и 1 атм. Требуется:
1. Вычислить долю сжижаемого воздуха, количество килограммов, получаемых в час, и к. п. д. цикла. Предполагаются идеальные условия работы, т. е. отсутствие притока тепла, нулевая разность температур на горячем конце теплообменника ☆ ) и обратимое адиабатное расширение.
2. Определить уменьшение производительности (в °/0) в том случае, если имеется приток тепла в 2 ккалкг-моль входящего воздуха и если на горячем конце теплообменника температура достигает 5° С.
3. Определить увеличение производительности (в °/о) по сравнению со случаем 1, если давление равно 200 ат.
☆ ) Из обсуждения, приведенного в гл. IX, должно быть очевидно, что можно фиксировать разность температур только на горячем конце. Разность температур на холодном конце при этом процессе неизбежна.
Низкотемпературные процессы
531
(1) Из диаграммы S — Т для воздуха 81 получаем значения Нх = 117,3, Нг = 22, /7j = 122,3.
Подстановка в уравнение (35) дает С = 0,05;
, с „ 1,5X60X1X10000X29 ,п, , .
1,5 м 1мин 848 2^2 — 107 кг/ч,
жидкости за час = 0,050 X 107 = 5,35 кг.
Работа сжатия (по рис. 48 принимаются три ступени)
= 5,4 л. с.-Ч] кг-моль воздуха
= 108 л. с.-чкг-моль жидкости.
Работа обратимого процесса [по уравнению (33)] =(117,3 — 22) —
— 300 (0,915 — 0) = — 179,2 ккал!кг
= 8,25 л. с.-ч1кг-моль.
к. п. д. цикла = X 100 = 7,65°/в.
lUo
(2) Т еперь Н9 = 127,5. По уравнению (35),
С = 0,03.
Уменьшение выхода жидкости = 4О°/о. Этот расчет указывает на значение, которое имеют тщательная изоляция и рациональная конструкция теплообменника, так как принятые необратимые эффекты на практике могут стать значительно больше, если не заботиться об их снижении.
(3) 7/1=114, С = 0,08.
Увеличение выхода = 60%. Этот расчет ясно показывает, что давление является очень важным параметром.
Увеличение давления оказывает значительное влияние на выход жидкости, н, естественно, возникает вопрос: будет ли выход продолжать увеличиваться с давлением или же будет существовать максимум? На этот вопрос легко ответить следующим образом.
Из уравнения (35) видно, что выход бывает максимальным при максимальной разности Н3 — Нх. Hs, естественно, фиксируется при возможном наинизшем давленнн и возможной наивысшей температуре, и поэтому должно быть минимальным. Критерием этого будет условие
или
34*
532
X. Охлаждение
Это является основным уравнением для кривой инверсии эффекта дросселирования (см. гл. VII, раздел „Точки инверсии"), и из этого мы заключаем, что максимальная степень сжижения будет наблюдаться тогда, когда исходным давлением для данной температуры газа, входящего в теплообменник, будет давление, отвечающее инверсии. Это, конечно, дает ряд давлений и температур (кривая инверсии), при которых наблюдаются максимумы; но, принимая температуру фиксированной при 300° К, получим для сжижения азота оптимальное давление в 375 атм (см. рнс. 52).
В случае водорода и гелия легко показать с помощью расчетов, описанных в гл. VI, что изотермическое Д/7 расширения при атмосферном давлении является отрицательной величиной, и поэтому невозможно сжижать эти газы только посредством данного метода. Из обсуждения, приведенного в гл. VII, очевидно, что точка инверсии для этих двух газов лежит ниже комнатной температуры. Например, для водорода легко вычислить, что при давлении в 1 атм температура инверсии равна примерно 200°Jf. Следовательно, для сжижения этого газа посредством дросселирования необходимо предварительно охладить его по меньшей мере до этой температуры, потому что для более высокой температуры значения р. будут отрицательны, и из соотношения
ЬНТ=\р.Ср<1р (37)
Pi
ясно, что в результате должно получиться отрицательное значение изотермического ДЛЛ чДля любого данного давления предельная температура, выше которой сжижение будет невозможно, находится вычислением из зависимости р —v— Т или из уравнения состояния й).
Сжижение водорода обычно осуществляется посредством предварительного охлаждения сжатого газа жидким воздухом с последующим его пропусканием через систему, состоящую из теплообменника и дроссельного вентиля, как показано на рис. 99. Гелий нельзя сжижать таким образом, потому что его температура инверсии лежит, невидимому, ниже 50° К, т. е. для сжижения гелия требуется предварительное охлаждение его жидким водородом.
Из исследования общей формы кривой инверсии очевидно также, что для данного давления газа ниже максимального давления инверсии оптимальная температура не будет температурой инверсии, но будет лежать несколько ниже ее. Это также легко показать, определив значения Д/7 по диаграмме S — Т. Следующая таблица, напри-
£г) Однако следует отметить, что этот способ скорее является серьезной проверкой уравнения состояния, так как результат будет очень приближенным, если только уравнение не будет очень точно для всего рассматриваемого интервала.
Низкотемпературные процессы
533
мер, дает значения АН для изотермического расширения воздуха от 200 до 1 атм для ряда температур:
то,к С°,С Дяг, кал! моль
300 17 245
270 — 3 303
240 — 33 420
200 — 73 540
Из этих данных видно, что посредством предварительного охлаждения воздуха при 200 атм до 200° К выход жидкого воздуха можно увеличить более чем в два раза по сравнению с выходом при простом охлаждении водой до 300° К- Кроме того, охлаждение до этой температуры можно осуществить с помощью обычного цикла сжатия
Забор воздуха
Первый холодильник
СО Дроссель
® Перекрытие
Сборник жидкого воздуха
Рис. 101. Цикл для жидкого воздуха, использующий эффект Джоуля—Томсона, дополненный вспомогательным холодильником.
534
X. Охлаждение
пара с использованием таких холодильных агентов, как аммиак или фреон-12. Поскольку термодинамический к. п. д. такого процесса значительно больше, чем для цикла, основанного на эффекте дросселирования, имеется также значительная экономия в расходе энергии на килограмм получающейся жидкости.
Компресем
Холодильник
Вспомогательный компрессор
Г/7ЛМ
Переохладитесь жидкости
Вменив таплообманюк
Сборник промежуточного , давления
Рис. 102. Цикл сжижения газа, использующий двухступенчатое дросселирование.
Забор газа
Сборник низкого давления
Схема цикла, в котором используются эти принципы, показана на рис. 101. Отметим, что, кроме холодильника, в котором воздух высокого давления охлаждается вспомогательным холодильным агентом, имеется также дополнительный холодильник, в котором в качестве охлаждающего агента используется отработанный газ низкого давления. Без этого добавочного холодильника вспомогательная система охлаждения была бы значительно больше, и отработанный
Низкотемпературные процессы
535
воздух выпускался бы при гораздо более низкой температуре, чем это необходимо; это означало бы снижение к. п. д.
Температура, при которой испаряется вспомогательный холодильный агент, определяется давлением всасывания в компрессоре. Если холодильным агентом является фреон-12, держивается равным 0,1 атм (р14), то 200° К, a ts, учитывая приемлемую разность температур для передачи тепла, может быть равной —70,5° С. Если атмосферное давление будет наинизшим желательным давлением всасывания, то предварительное охлаждение примерно до — 27° С будет наилучшим из всех, которого можно достигнуть с помощью этого холодильного агента. Затем можно определить долю сжиженного газа с помощью баланса для этой части системы, включающей только главный теплообменник, дроссельный вентиль и сборник для жидкости; это даст уравнение, совпадающее с уравнением (35). Затем с помощью описанных методов можно определить расход мощности для обоих компрессоров и найти термодинамический к. п. д. этого цикла.
При расширении сжатого газа, подлежащего сжиже-
нию в две ступени (рис. 102), расход энергии можно значительно снизить. Это видно по тому факту, что охлаждающий эффект &Нт при расширении от 200 до 20 атм при 26,7° С составляет около 9О°/о охлаждения, получаемого при полном расширении до 1 атм, между тем как работа для сжатия от 20 до 200 атм много меньше, чем для сжатия от 1 до 200 атм. Таким образом, бесспорно выгоднее рас-
а давление Лз бУДет
всасывания подпримерно’ равна
Охлаж-
Рис.
103. Процесс сжижения газа, использующий детандер.
ширять сначала до некоторого промежуточного давления и отделять получающуюся жидкость от газа, а затем расширять эту жидкость до 1 атм для удаления ее из системы. При этом способе большая часть газа должна сжиматься только от 20 до 200 атм, а меньшая часть —
536
X. Охлаждение
во всем интервале давлений. Количественный анализ такого цикла для определения оптимального режима работы и расхода мощности легко произвести, пользуясь ранее изложенными методами.
Сжижение с помощью машины для расширения (детандера) (процесс Клода). Простая форма цикла Клода показана на рис. 103. Как и в случае цикла Карно, для аккумулирования холода необходимо сочетание машины и теплообменника. Отметим также, что в машине расширяется только часть газа, причем остаток его дросселируется; поэтому такой процесс охлаждения осуществляется комбинацией дроссель-эффекта и расширения. Если бы весь газ расширялся в машине, то было бы необходимо получить в машине и жидкость, а это, вероятно, было бы нежелательно, потому что привело бы к уменьшению к. п. д. Следовательно, часть газа высокого давления проходит через теплообменник (часто' называемый сжижителем) в направлении, противоположном газу, возвращающемуся из сборника жидкости, и расширившемуся газу, и сжижается ими.
Анализ цикла, изображенного на рис. 103, значительно сложнее, чем анализ цикла рис. 99, но он не' включает никаких новых принципов. Метод анализа будет изложен в следующем примере.
Пример 12. Определить практические условия стационарного процесса сжижения кислорода посредством цикла, показанного на рис. 103, и определить расход мощности иа 1 кг получающейся жидкости. Базис расчета: 1 моль кислорода, входящего в первый теплообменник.
Пусть С — сжиженная доля кислорода,
/—часть газа, идущая в детандер, е — термодинамический к. п. д. детандера, — уменьшение энтальпии в детандере при изоэитропном расширении.
Индексы будут относиться к местам, обозначенным на рисунке соответствующими цифрами. Для предварительного анализа будем считать приток тепла ничтожно малым.
Применение первого закона к различным частям установки приводит
к следующим уравнениям. Тепловой баланс для первого теплообменника:
Л4-Я4 = (1-0(Я8-Я6). • (а)
Балансы для второго теплообменника:
1. Общий тепловой баланс: (1-/)(Я4-Я,) = (1-С)(Я5-Я10). (б)
2. От уровня сжижения до дна: = (в)
Баланс для сборника жидкости: (1-/)я7=ся,+ (1-/-г:)я9. (г)
По определению термодинамического к. п. д. детандера, Hi = Ht + e^Ht. (д)
Низкотемпературные процессы
537
Баланс в точке смешения перед вторым теплообменником:
/Яв + О—/~0Я9 = (1—С)//10. (е)
Если принять, что давление в сборнике и давление отработанного газа всегда будут равны 1 ат, то независимыми переменными для этих шести уравнений являются Нх, Я3, Нь Нъ, Щ, Нъ Hw, С, / и е, т. е. всего 11. Поскольку имеется шесть независимых уравнений, можно фиксировать 5 из вышеупомянутых переменных и затем решить уравнения для оставшейсн переменной. Существует известная произвольность при выборе того, какие именно переменные фиксировать, но предположим, что мы выбрали для фиксации следующие условия: р1г tlt ts, е, t4 и tK, которыми определяются 5 переменных: Я3, Н%, Н^ HVi и е. Основанием для фиксации t12 является то, что на уровне точки росы поток высокого давления будет находиться прн минимальной разности температур в этом теплообменнике, и так как tu становится известной сразу после фиксации р1г то можно фиксировать при несколько более низком значении для получения приемлемой разности температур: допустим, что At равняется 5° С.
Решая шесть уравнений, получаем:
_ (Я3 - Н\) (Н< - Нп - e\Hs) 4- (Ну - Яп) (Н3-Н2}(Н4-Нп-е^Нх) + (Н12-Н^е^Н3 ’ ' ’
, ^Н3-Н2)-(Н,-Н3}
Т ~ еЬН3
Примем следующие частные условия:
Р! = 20 атм
Л =300° К 7*5 = 295° К е = 0,70
7*4=175°К й)
/>6 = 1 атм
Т12= 7*ц —5=133 —5= 128° К
Из таблиц Миллара и Сюлливана [171] найдем следующие значения:
/4=3212; 24=158; /4 = 3215; /24 = 2285; /2П = 1859;
Нп — 2041; SHs — 660; /76=1823 [по уравнению (д) это соответ-
ствует пару, перегретому примерно на 5° С]; /7Э=1788.
По уравнению (ж)
По » (з)
По . (а)
По » (в)
С = 0,122
/= 0,8(КГ
/4 = 2160 и Г6=142°К
Нч= 800 7*7=133 (смесь жидкости и пара).
Принимая двухступенчатое адиабатное сжатие и идеальность газа, по рис. 48, получим:
теоретическая работа =3,46 л. с.-ч/кг-моль сжатого газа.
Принимая общий к. п. д. компрессора равным 75°/о, получаем:
практическая работа = 4,62 л. с.-ч/кг-моль.
й) Этот выбор производится при допущении, что газ, выпускаемый из машины, должен быть близок к насыщению или, что предпочтительнее, будет слегка перегрет.
538
X. Охлаждение
Работа на валу детандера
= fe\Hs = 0,800X0.70X660
= 369 ккал
= 0,584 л. с.-ч на кг-моль сжимаемого О2.
Принимая, что с помощью газового компрессора можно утилизировать 90% этой работы, найдем, что
общая работа, требуемая на 1 кг жидкого кислорода, равна
4,62—0,90X0,584 ,
- 0Л22Х32- -1>05 х ,-ч.
Термодинамический к. и. д. — X 100 = 22,4%.
0,235 л. с.-ч является минимальной (обратимой) работой для сжижения 1 кг кислорода, вычисленной по ранее указанным методам.
Это определяет одно из условий, при которых, как можно ожидать, будет работать данный цикл. Влияние таких переменных, как приток тепла, начальное давление, давление выпуска из детандера, температура входа н детандер и температура газа низкого давления, покидающего систему, легко можно исследонать с помощью показанного здесь метода. Интересно отметить, что чем ниже давление, тем ниже должна быть температура при входе в детандер для достижения оптимального к. п. д. Можно получать жидкость с приемлемым к. п. д. при таких низких давлениях, как 5 атм, хотя по мере повышения давлений к. п. д. будет увеличиваться. Капица [311] описал сжижитель воздуха, работающий при низких давлениях и включающий несколько новых особенностей, например турбину расширения и регенеративный тип теплообменника как для передачи тепла, так и для очистки.
Если итти в другом направлении и увеличить давление, то температура входа в детандер может увеличиваться до тех пор, пока примерно при 200 атм она не достигнет комнатной температуры. Это служит основой процесса Гейланда, являющегося по существу сочетанием процессов Линде и Клода. Никаких сведений о деталях детандера Гейланда не .опубликовано, *о если расширение проводится до 1 атм, то ясно, что следует пользоваться по меиьшей мере двухступенчатым расширением, но лучше применять три ступени.
Использование машины для расширения делает также возможным сжижение гелия без цикла предварительного охлаждения водорода, как это необходимо при использовании дроссель-эффекта. Необходимость во вспомогательном охлаждении полностью не исключается, поскольку в сжижителе, разработанном Капицей [121, 141], сжатый гелий перед входом в самый цикл гелия охлаждается жидким воздухом или азотом. Без этого было бы необходимо применять несколько детандеров. Капица разработал поршневой детандер очень остроумной
Низкотемпературные процессы
539
конструкции, который, по некоторым сведениям, работает при следующих условиях:
Давление входа......................... 30 ат
Давление выпуска...................... 2,2 ат
Температура входа...................... 19° К
Температура выпуска.................... 10° К
Эти данные соответствуют термодинамическому к. п. д. в 60°/в на основании 5— Т’-диаграммы Кизома для гелия.
Каскадный метод. Хотя этот метод является одним из наиболее ранних методов, употреблявшихся для сжижения так называемых „постоянных газов*, он только недавно начал применяться в промышленных масштабах ☆).
По крайней мере, один большой завод в Америке использует этот процесс для сжижения и разделения воздуха, и наиболее современным применением его является сжижение природного газа для целей хранения [43].
В случае двух разобранных сейчас методов сжижения газа, газ, подвергаемый процессу, действует сам как собственная охлаждающая среда, и обычно внешнее охлаждение не употребляется, хотя, конечно, имеются исключения из этого правила. В случае каскадного процесса охлаждение обычно получается почти полностью от внешней системы (циркуляции), и только небольшое количество поступает от самого газа. На рис. 104 показан каскад из трех веществ: из аммиака, этилена и метана для сжижения воздуха, азота или кислорода. Если сжимаемым газом является кислород, то необходимо сжать его примерно до давления в 7 ат, чтобы сконденсировать метаном, кипящим при 1 ат-, дальнейшее охлаждение до нормальной точки кипения будет достигаться за счет испарения самого жидкого кислорода. Метан сжижается испаряющимся этиленом, который, в свою очередь, сжижается испаряющимся аммиаком; последний сжижается охлаждающей' водой.
В процессе, использованном в Кливленде [43] для сжижения натурального газа, имеется каскад из двух веществ — этилена и аммиака, которые охлаждают газ примерно до—88° С. Так как жидкость хранится лишь при незначительном положительном давлении при —161°С, то ясно, что в этом случае значительная доля охлаждения должна получаться за счет самого газа. Для сжижения посредством испарения этилена последний сжимается до 42,2 ат, затем жидкость испаряется в две ступени (для достижения конечной температуры), причем газ, испарившийся с обеих ступеней, используется для охлаждения жидкости перед первым дроссельным вентилем. Двухступенчатое расширение экономит мощность, потому что часть испарившегося газа должна
☆) В течение многих лет Лейденская холодильвая лаборатория производила жидкий воздух посредством каскадного процесса, используя в качестве холодильных агентов метилхлорид, этилен и кислород.
540
X. Охлаждение
сжиматься только от этого промежуточного давления (3,88 ат в этом случае) до давления в 42,2 ат, вместо того чтобы сжиматься от атмосферного давления.
Полный анализ каскадных циклов для определения приемлемых условий работы и расходов мощности можно произвести, пользуясь
Компрессоры
Рис. 104. Каскадный цикл для сжижения газа.
соответствующими видоизменениями с помощью указанных ранее методов. Недостаток места не позволяет дать детальную разработку какого-либо частного конкретного случая. Основное преимущество каскадного метода заключается в том, что создается возможность обходиться без какой-либо низкотемпературной машины, и так как расширению подвергают только жидкости, то процесс будет менее необратим, чем процесс Линде, и поэтому для своего осуществления будет требовать меньшего расхода мощности. В будущем для очень больших заводов по разделению воздуха каскадный цикл может оказаться одним из очень желательных, поскольку стоимость энергии будет одной из наиболее важных составляющих баланса.
Процессы разделения газов. Одним из лучших методов разделения смеси газов является использование различия в составе сосуществующих жидкой и паровой фаз, как это применяется в процессах дефлегмации и ректификации. Такие методы, очевидно, должны осуществляться ниже критических температур и включать сочетание методов сжижения, рассмотренных в предыдущем разделе этой главы, с методами, связанными с взаимодействием двух фаз и обычно объединяемыми под названием перегонки. Этот метод является в настоящее
Низкотемпературные процессы
541
время единственным практически возможным для применения в большом масштабе при разделении воздуха на кислород, азот, аргон, иногда с выделением некоторых из редких газов. Он в известной
степени применялся для разделения различных промышленных
газов, например водяного или коксового газа, на водород как главный продукт и на некоторые полезные побочные продукты, например этилен, окись углерода и метан. Компетентные инженеры, опытные в этой области, предсказывают в ближайшем будущем значительное расширение применения низкотемпературного разделения газов [214]. Обширное применение кислорода или, вернее, воздуха, обогащенного кислородом, в металлургии и газовых процессах может явиться результатом улучшенных методов разделения воздуха.
Общие принципы. Разделение газа требует совершенно такого же метода охлаждения, как и сжижение газа, поскольку, несмотря на приток тепла, должны поддерживаться низкие температуры и, кроме того, должна компенсироваться йеидеальная работа теплообменников и другого оборудования, работающего при температурах ниже комнатной. Следовательно, все рассмотренные выше методы сжижения газа годятся для разделения газа при дополнительном применении ректификационной колонны или другого приспособления для осуществления постепенного изменения состава.
Получающийся азот
Получающийся газообразный мислород
Рис 105. Адиабатная ректификация воздуха.
Даже если бы не существовало
притока тепла в аппаратуру и теплообмен был бы идеальным, все же из уравнения (33) очевидно, что для разделения газов должна затрачиваться работа. Хотя Д/У практически равно нулю, но ДА всегда больше для раствора, чем для чистых компонентов.
Минимальная работа, необходимая для разделения воздуха на газообразные продукты при 26,7°С и 1 атм, легко вычисляется по уравнению (33); она равна 0,0526 квт-ч на 1 кг кислорода или 0,0685 квт-ч на 1 ж3 кислорода (0,354 квт-ч1кг-моль воздуха). Это
542
X. Охлаждение
значительно меньше, чем работа, требующаяся для сжижения 1 кг чистого кислорода (0,176 квт-ч} кг), потому что при идеальном процессе разделения не имеется никакой нагрузки охлаждения, если только процесс осуществляется при стационарном режиме. С другой стороны, разделение воздуха на жидкий кислород и газообразный азот требует большей затраты энергии (0,2279 квт-ч[кг О2), чем получение жидкого кислорода из газообразного, потому что, кроме условий, необходимых для сжижения, при разделении газа предполагается еще необходимость сообщения теплоты. Лучшие современные заводы для разделения воздуха требуют около 0,42 квт-ч на 1 м3 кислорода, и поэтому их термодинамический к. п. д. составляет около 16°/0, между тем как более обычным расходом для большого завода является 0,67 квт-ч или к. п. д., равный 1О°/о. Из этого не следует заключать, как это делают некоторые, что относительно легко достигнуть значительного снижения расхода энергии путем правильного применения основных принципов. Существует много необратимых эффектов, которые нельзя полностью устранить ни в одном практическом процессе; и незначительный к. п. д. таких процессов обусловлен, как ясно показано Доджем и Хосумом ☆) [67], увеличением числа членов с большим или умеренным к. п. д.
Сравнение обратимой а адиабатной ректификации. Замечательным примером положений, изложенных в предыдущем разделе, является сама ректификационная колонна. Ниже (гл. XIII) будет показано, что адиабатная колонна (это относится собственно к колонне, кипятильник и конденсатор исключаются) обратимо работать не может, как бы идеально она ни действовала, потому что обратимость предполагает существование фазового равновесия на всех уровнях колонны, а это требует изменяющегося потока жидкости и пара в колонне вместо почти постоянного мольного потока, характерного для адиабатной колонны. Другими словами, обратимая ректификация требует определенного распределения потока, который, в свою очередь, требует, чтобы тепло подводилось извне на всех уровнях идеальной колонны ниже впуска питания и отнималось бы на всех уровнях выше него. Для дальнейших деталей этого вопроса можно обратиться к статье Доджа и Хосума и к серии статей Ван Нуиса [249]. Следующий пример поможет иллюстрировать это утверждение.
Пример 13. Вычислить термодинамический к. п. д. идеально действующей адиабатной ректификационной колонны для разделения воздуха на газообразные кислород и азот. Определить термодинамический к. п. д. при практической работе.
При решении этой задачи необходимо применить некоторые зависимости для ректификационной колонны, которые выведены в гл. ХШ. Примем, что пар воздуха, насыщенвый при 1 атм и содержащий 21% О2 и 79% N2, входит в колонну (рис. 105) и разделяется на чистые азот и кислород, при-
☆ ) К этой статье можно обратиться также для рассмотрения других фаз процесса разделения воздуха.
Низкотемпературные процессы
543
чем оба газа являются насыщенными парами, имеющими давление 1 ат. Так как колонна принимается адиабатной, то необходим приток тепла QB в кубе и удаление тепла Qc в конденсаторе. Это положение справедливо для любой адиабатной колонны независимо от температурного уровня; механизм, которым осуществляются эти переходы тепла, нас не интересует.
По уравнению (86, гл. XIII), применяющемуся к уровню питания, получаем
Ор _ Ур>— Ур
D Ур—xF
(а)
где Op/D — отношение числа молей перетекающей жидкости на уровне, на котором подается питание, к числу молей получающегося азота. В гл. XIII будет показано, что это отношение является максимальным для уровня питания; поэтому этот уровень и определяет отношение для колонны. Минимально возможное отношение наблюдается тогда, когда разность между уР и хр будет наибольшей яли когда обе фазы находятся в равновесии. При использовании равновесных данных Доджа и Дэнбара [65], суммированных Доджем [62], получим при подстановке в (а)
Ор 0—0,210 _
D ~~ 0,210—0,523 — 0,6 L
Ос — поток жидкости, получающийся в конденсаторе, определяется по уравнению (114, гл. ХШ) из Ор. Из доступных данных по энтальпии кислородно-азотных смесей £?) находим, что
£-с = 1,175. Up
На 1 кг-моль получающегося кислорода
79
Qc= 1,175X0,671X1335X^ = 3960 ккал.
Из общего теплового баланса для всей ректификационной системы (пряток тепла принимается равным нулю) на моль воздуха имеем
QB-Qc=Hf-\-Q,79Hd+Q,2\Hv. (б)
Имеющиеся данные свидетельствуют о том, что правая часть этого уравнения практически равна нулю, и поэтому
Qb — Qc- (8)
Общим термодинамическим требованием для адиабатной колонны является удаление из конденсатора 3960 ккал или подведение того же количества тепла к кубу при расчете на 1 кг-моль получающегося газообразного кислорода. Так как эта операция заключается в сообщении системе тепла, то для осуществления ее потребуется работа, минимум которой дается уравнением
A = QT0( fr)
\'я, jo,/
оно легко выводится из приведенного ранее цикла Карно. То представляет наинизшую температуру, при которой производится отдача тепла (примем ее равной 300° К); Ти 7O, — точки кипения азота и кислорода. Подставляя
it) См. рис. 80.
544
X. Охлаждение
числовые значения соответствующих величин, получим
4 = 3960X300^-^) =2160 ккал—
= 102 квт-ч/1 000 жз О2 при 300° К и 1 ат.
Ван Нуисом [249J показано, что теплообмен между входящим воздухом и Выходящими продуктами может совершаться обратимо без затраты какой-либо работы; следовательно, вышеприведенная величина представляет наименьшее количество работы, необходимое для разделения воздуха с помощью адиабатной колонны, при условии, что все остальные части системы работают обратимо. Другими словами, никакой процесс, в котором используется адиабатная колонна, не может иметь термодинамического к. п. д., превышающего
63,5 пп___
102/4‘uu~ и' '°-
Теперь, если допустить, что орошение на 20% превышает минимальное (минимальное орошение требует бесконечно большой высоты колонны) и что средняя разность температур между кубом и конденсатором, необходимая для обеспечения нужной скорости теплопередачи, равна 3° С, то термодинамический к. п. д. будет равен
1452
3960X1.2X300 [(1/74,3) — (1/93,2)Г Х 10° = 37,5°/о'
(Величина 1452 является минимальной работой разделения в калориях на моль кислорода.)
Отсюда можно заключить, что система для разделения иоздуха, включающая адиабатную колонну с соответствующими условиями работы, имеет к. п. д. меньше 40%, даже если вся система охлаждения является обратимой. Поэтому ре удивительно, что даже в лучших процессах к. п. д. бывает меньше 20°/а.
Рис. 106. Полный энергетический и материальный баланс процесса разделения воздуха.
Уравнения общего баланса. Общий результат любого процесса разделения воздуха схематически изображен на рис. 106. В этом общем случае рассматриваются все три метода охлаждения при низких
Низкотемпературные процессы
545
температурах. Можно написать, что + (38)
МУ1 = ^2 + ^з?з> (39) *)
= N2H2-\-1\/3Н3-[-АЕ-[-QR— Qj. (40)
Если принять, что смеси, представленные двумя продуктами, являются идеальными растворами, причем оба продукта газообразны, то
^2 = ^OiJ2 + ^№(l—Л), (41)
^ = "OjV3 + HV3(l-v3)- (42)
где Н01 и //№ и подобные им члены с индексом 3 соответственно выражают энтальпию чистого кислорода и азота.
Теперь, пусть р2=р3 и Т2=Т3; тогда
ЯОз=ЯОз = Я0’ = ^з = ^N'
Алгебраическое сочетание этих уравнений дает
^yAHa-HN) + HN+qA, (43)
где
Из уравнения (43) видно, что если АЕ и QR равны нулю, то Ну определяется только притоком тепла и состоянием газа, покидающего низкотемпературную систему; если фиксировать эти два фактора, то фиксируется и рабочее давление. Если одним из продуктов является жидкий кислород, то положение будет совершенно иное, поскольку давление уже не определяется этими двумя переменными; наоборот, оно является функцией состава уходящего азота, который, в свою очередь, связан с выходом жидкости. Это доказывается следующим образом.
Одновременное решение трех уравнений баланса, соединенное с уравнение.! (42), дает
__ У1 WN. — Н.,)~ х, (HN,— Ну ^д)
Ь — U2 -Л) («о, - Я№) + Hy-H2-qA ’
и мы видим, что Ну (а следовательно, и рх) при фиксировании qA и состояния в точке 3 является функцией у3. Выход жидкого или газообразного кислорода определяется как отношение количества кислорода в продукте к общему содержанию кислорода в воздухе и дается уравнением
выход — о / — 100-*2 ( 0.210 —Уз \ о / /4g\
Д 0,21^ ' 0,210 < х2 — у3 ) ‘О’ (
й-) Для получения жидкого кислорода у2 следует заменить на х2.
35 Химическая термодинамика
546
X. Охлаждение
Простой процесс Линде. Такой процесс для разделения воздуха на газообразный кислород и азот представлен на рис. 107. Воздух сжимается и охлаждается водой и затем дополнительно охлаждается посредством теплообмена с азотом и кислородом, покидающим колонну. Холодный сжатый воздух (вместе с незначительным количеством жидкости, необходимой для компенсации притока тепла в колонну) поступает в теплообменник кипятильника, где он сжижается, сообщая этим теплоту кипящему жидкому кислороду. Затем жидкий воздух расширяется в верхней части колонны, и жидкость ректифицируется обычным способом. Следует отметить, что азот, покидающий верхнюю часть колонны, не будет чистым, так как наименьшее количество кислорода, которое он может содержать, представляет содержание его при фазовом равновесии с жидким воздухом и равно 6,4°/0. В действительности благодаря мгновенному парообразованию в дроссельном вентиле, а также другим причинам, азот по чистоте не будет превышать 92°/0, а максимальное извлечение кислорода составит около 65°/0. Цикл для получения жидкого кислорода будет таким же, причем единственным от-
личием на диаграмме будет то, что жидкий кислород вытекает из кипятильника, и нет обратного потока кислорода в теплообменнике. Нижеприводимый пример выявит некоторые различия между процессами получения газообразного и жидкого кислорода, а также покажет применение выведенных выше уравнений.
Пример 14. Определить приемлемые рабочие условия простого процесса Линде для разделения воздуха, производящего газообразные продукты. Повторить то же для процесса, дающего жидкий кислород. Указать также потребную мощность в киловаттах на 1 кг кислорода и термодинамический к. п. д. процесса. Принять приток тепла в низкотемпературную си-
Впуск 1 воздуха
) Охлаждаю- | щая вода |
Азот
^Кислород
Теплообменник
6
Ректификационная колонна
4
Рис. 107. Простой цирл Линде для разделения воздуха.
Низкотемпературные процессы
547
стему равным 50 кал/моль воздуха и чистоту получающегося кислорода равной 99,50/0-
Основные допущения:
1. Воздух входит в низкотемпературную систему при 300° К.
2. Минимально допустимая Д/ в теплообменнике равна 5° С.
3. Максимальное практически возможное извлечение кислорода в ка-честве продукта в колонне типа, показанного на рис. 107, равно 65%
4. Оба продукта покидают систему при 1 ат.
5. Можно пренебречь падением давления, обусловленным перемещением вещества.
6. Общий к. п. д. компрессора по мощности, основанный на изоэнтроп-ном сжатии, равен 75P/q.
7. В качестве базиса расчета принимается 1 кг воздуха.
Газообразный кислород. //0 = 3215; /7^ = 2850 и —— 50 (заметим, что Ае и Qr для этого цикла равны 0).
По уравнению (43), /7г = 0,210(3215 — 2850) + 2850 — 50 = 2877.
По диаграмме S— Т для воздуха, р2 = 59 ат.
Следует отметить, что это значение очень сильно зависит от величины притока тепла и будет совершенно иным для другого количества тепла.
Принимая трехступенчатое сжатие, находим по рис. 48, что теоретическая работа сжатия —4,62 л. с.-ч/кг-моль или практическая работа = 4,60 квт-ч)кг-моль.
По уравнению (46), у3 = 0,0852.
Минимальная работа разделения на продукты указанного выше состава равна 0,18 квт-ч/кг-моль воздуха, и поэтому термодинамический к. п. д. равен только 3,9%.
Для определения условий, при которых работает теплообменник, можно напвсать для него следующий баланс:
Nr (Нг - Н4) + Ql, = N2 (Н2 - Н6) + N3 (Н3 — Н2). (а)
Принимаем, что ЗО°/о от общего количества тепла попадает в теплообменник и 70% — в колонну, или
(?£1/М=15,
^=0,137 и ^ = 0,863;
tf2= 3215, Н3 = 0,0852 X 3215 4- 0,9148 X 2849 = 2884, /76=1788, /79= 1379.
Подставляя эти значения в (а), получаем
/4 = 1397;
и из диаграммы видим, что температура Т4 равна 149° К. Т4 значительно выше, чем Т6 или 7», и поэтому можно с достаточным основанием сказать, что нигде в теплообменнике нет отклонений от второго закона.
Для проверки расчетов целесообразно составить тепловой баланс колонны;
1397 4- 35 = 0,863X1379 4-0,137X1788; 1432 = 1434.
Жидкий кислород. Для этого случая существует не одно рабочее давление, а ряд давлений, связанных с выходами жидкости. Для иллюстрации этого выберем только одно условие, а именно, рг = 200 ат. 35*
548
X. Охлаждение
Подставляя соответствующие величины в уравнение (45), получаем _ 0,210 (2849-158)—0,995 (2849—2721—50)
Уз ~ (0,995—0,210) (3215—2849) + 2721 —158 + 50 ’ ’
По уравнению (46), выход жидкости = 24%
Принимая четырехступенчатое сжатие, получаем теоретическую работу сжатия =8,45 л. с.-ч/кг-моль воздуха или практическую работу = = 8,42 квт-ч[кг-моль.
По уравнению (33), минимальная работа разделения =613 кал)моль воздуха =0,324 квт-ч. Термодинамический к. п. д. =8,5%.
Из баланса для теплообменника находим:
= 1326, откуда = 170° К.
Цикл жидкого кислорода не будет работать при всех давлениях, пригодных для цикла газообразного кислорода. Даже при высоком давлении в 200 ат выход очень низок, и расход воздуха на единицу полученной жидкости будет чрезмерным. Как было установлено выше, имеется несколько путей для увеличения выхода при жидком цикле и при снижении давления. Двумя обычными методами являются: 1) использование вспомогательного охлаждения; 2) использование машины расширения. Анализ таких циклов выходит за рамки этой книги, но его легко осуществить описанным выше методом.
Ректификация при низких температурах. Хотя в общем процесс ректификации при низких температурах подобен ректификации при температурах выше комнатной, однако он отличается несколькими важными особенностями, интересными с термодинамической точки зрения. Прежде всего, расход тепла в кипятильнике и отдача тепла в конденсаторе при обычной ректификации совершенно не зависят друг от друга, а при низкой температуре они должны быть вполне определенно связаны вместе в обычном цикле, в котором охлаждающей средой является газ, подлежащий разделению. В последнем случае, если желают получить эффективную работу, то подачу тепла в кипятильник нельзя производить от любого источника, так как теплота должна удаляться из системы. Это сводится к следующему: тепло, отводимое в конденсаторе для получения флегмы, является единственным теплом, которое можно сообщать кипятильнику. Во-вторых, получение флегмы из жидкого азота, существенное для полного разделения, связано с известными трудностями и вызвало появление некоторых специальных конструкций колонн, которые представляются заслуживающими рассмотрения с точки зрения их принципов.
Так называемая одиночная колонна является одной из тех, в которых воздух при повышенном давлении входит в теплообменник, погруженный в резервуар с жидким кислородом, расположенный ниже колонны, и сжижается там, отдавая свое тепло для кипения кислорода. Получающаяся таким образом жидкость подается затем на верхнюю тарелку колонны, которая является укрепляющей колонной,
Низкотемпературные процессы
549
обычной для практики перегонки; очевидно, что она не может дать чистого азота или уловить высокий процент кислорода из воздуха.
Существует примерно пять различных способов, по которым можно получить почти чистую флегму из жидкого азота для секции обогащения колонны. Способы эти следующие: 1) испарение жидкого
Получающийся азот
Колонна атмосферного давления
Колонна 4-5 атмосфер
Получающийся кислород
Питание воздухом
Дроссель
Конденсатор-испаритель с орошением
Жидкий кислород
Жидкий азот
Рис. 108. Разделительная колонна Линде двойной ректификации.
кислорода при пониженном давлении; 2) использование внешнего цикла для сжижения азота; 3) использование вспомогательного хладагента, как гелия или неона; 4) частичная конденсация воздуха, питающего конденсатор-кипятильник, посредством разделения его на две фракции, одна из которых обогащена азотом, и 5) употребление двухколонного разделительного аппарата. Методы 1, 2 и 3 находили применение раньше, но в настоящее время представляют второстепенное значение и поэтому не будут рассматриваться подробнее. Метод 4 является основой оригинального („возвратного0) конденсатора, предложенного Клодом, который до сих пор находит применение. Метод 5 наиболее важен с практической точки зрения и будет кратко рассмотрен с помощью рис. 108, иллюстрирующего принцип действия двухколонного аппарата, впервые разработанного Линде.
550
X. Охлаждение
Аппарат состоит из колонны, работающей при повышенном давлении, над которой находится колонна атмосферного давления. Испаритель верхней колонны является в то же время конденсатором орошения для обеих колонн. Газообразный воздух с достаточным количеством жидкости для предотвращения притока тепла в колонну (конечно, требуется больше жидкости, если вытекает получающийся жидкий кислород) входит в теплообменник у основания колонны и конденсируется, отдавая теплоту кипящей жидкости и создавая для этой колонны поток пара. Жидкий воздух, как показано, входит в среднюю по высоте часть колонны. Пары, поднимаясь по этой колонне, частично конденсируются с образованием флегмы, а некон-денсированный пар идет к внешнему ряду труб, где полностью конденсируется, причем жидкий азот собирается, как показано, в кольце. При работе такой колонны при давлении от 4 до 5 от жидкий кислород, кипящий при 1 ат, получается достаточно холодным для конденсации чистого азота. Жидкость, собирающаяся на дне нижней колонны, содержит около 45°/0 О2 и служит питанием верхней колонны. Такая сдвоенная колонна может давать очень чистый кислород с высоким процентом улавливания его или очень чистый азот с кислородом умеренной чистоты. Она не может производить одновременно оба продукта высокой чистоты, главным образом вследствие того, что воздух в действительности представляет собой тройную смесь и аргон должен удаляться с одним из продуктов.
Для дальнейших подробностей по разделению воздуха, сжижению газа и связанным с этим вопросам см. Руеманн М. [215], Вин и Хармс [258] и Руеманн М. и Руеманн Б. [216] 82.
ГЛАВА XI
РАВНОВЕСИЕ ХИМИЧЕСКИХ РЕАКЦИЙ
Инженер-химнк, связанный с проектированием или с эксплоата-цией аппаратуры, в которой осуществляются химические реакции, или разрабатывающий новые процессы, включающие химические изменения, должен не только понимать влияние основных факторов, управляющих реакциями, но и знать также, как применять эти сведения при решении задач, которые могут возникать на практике. Целью этой главы является рассмотрение наиболее важных зависимостей, относящихся к равновесию в химических реакциях, и применение принципов и уравнений, выражающих эти зависимости, к типичным реакциям, важным для промышленности в настоящее время или представляющим интерес в будущем.
РАВНОВЕСИЕ И СКОРОСТЬ РЕАКЦИИ
Практически все вопросы, связанные с любой химической реакцией, можно разделить по двум основным признакам: 1) выход при достижении равновесия, 2) скорость реакции. Важно с самого начала ясно разделить эти группы. Пусть дана, например, реакция
СО + Н2О = СО2 + Н2, (1)
и требуется узнать, в какой степени она будет проходить за определенное время при данных условиях. Наиболее важными факторами, которые может регулировать технолог, являются: 1) температура, 2) давление, 3) количественное соотношение между реагирующими веществами и 4) катализатор. Действие этих факторов можно понять лишь при разделении их по двум различным признакам: 1) влияние на скорость реакции и 2) влияние на равновесие. Если не сделать этого разделения, то наблюдаемые факты, относящиеся к данной реакции, могут представляться взаимопротиворечащими.
Очевидно, что скорость, с которой будет проходить реакция, является с промышленной точки зрения важнейшим фактором. Реакция, проходящая очень медленно, будет требовать такого громоздкого и дорогого оборудования для получения достаточной производительности, что может оказаться совершенно неэкономичной. Но одинаково важно знать, чему равна максимально возможная степень
552
XI. Равновесие химических реакций
превращения, которая может получаться при данных условиях. В прошлом было бесполезно затрачено много времени в попытках ускорить реакции, которые не могут произойти в желательной степени при какой угодно большой скорости.
Примем, что любая реакция обратима, т. е. в зависимости от условий она может проходить в определенной степени в прямом или обратном направлении. Возможно, что точнее было бы сказать, что каждая реакция имеет определенное стремление проходить в том или другом направлении. Другими словами, рассматривая в качестве примера реакцию (1), следует сказать, что при любых условиях совместное введение чистой СО и чистой Н,0 приведет к образованию некоторого количества СО2 и Н2. Количества, образующиеся в измеримый отрезок времени, могут быть слишком малыми для обнаружения их наиболее тонкими аналитическими методами, но, тем не менее, можно считать их действительно существующими. Подобно этому, при совместном введении чистых СО2 и Н2 можно принять, что образуются некоторые количества СО и Н2О.
При химических реакциях в ряде случаев бывает исключительно трудно опытным путем отличить, находится ли система в истинном равновесии, или она просто инертна вследствие тормозящего действия внутреннего сопротивления, аналогичного трению в механических процессах, вследствие чего скорость изменения слишком мала, чтобы реакцию можно было проследить. Иногда скорость изменения может быть настолько незначительна, что его нельзя обнаружить за измеримый отрезок времени. Если попытаться увеличить скорость посредством повышения температуры, то мы не сможем сказать, какая доля наблюдаемого эффекта обусловлена изменением скорости и какая доля обязана сдвигу равновесия. Термодинамика дает нам методы предсказания движущих сил, а следовательно, и равновесных состояний, благодаря чему, для того чтобы различить влияние этих двух факторов, мы совершенно не зависим от эксперимента. Развитие и использование этих методов и является основной темой данной главы.
Умение провести это различие представляет значительную практическую важность, так как если реакция за данный промежуток времени при определенных условиях не проходит в сколько-нибудь значительной степени, то всегда существует возможность найти способ заставить ее итти, если лимитирующим фактором является скорость; ио если реакция не идет вследствие того, что система близка к состоянию равновесия, то тогда необходимо изменить условия таким образом, чтобы сдвинуть равновесие в нужном направлении.
Как уже упоминалось в гл. I с общей точки зрения и как будет снова подчеркнуто здесь для частного случая химической pg-акции, скорость, с которой проходит какая-нибудь химическая реакция, не находится ни в какой определенной зависимости от движущих сил, т. е. от тенденции к изменению. Понимание роли обоих этих факторов существенно для интерпретации фактов, действительно
Равновесие и скорость реакции
553
наблюдающихся в тех случаях, когда условия для реакции благоприятны. Между тем термодинамика, будучи полезной для определения движущих сил и влияния на них различных факторов, не может дать никаких сведений о скоростях реакции. Это обстоятельство следует твердо усвоить. Недостаточная оценка этого привела к многочисленным недоразумениям и неверным заключениям. Тот факт, что термодинамика не может дать никаких сведений о скорости процесса,
очевиден из того, что она, в сущности, имеет дело с различиями
между функциями состояния, но т. е. с путем, по которому происходит изменение между двумя состояниями системы.
Из четырех факторов, упомянутых выше в качестве управляющих течением реакции, первые три оказывают влияние как на скорость, так и на движущие силы, но четвертый — катализатор — будет влиять только на скорость реакции. Это положение нуждается в некотором уточнении; оно строго правильно только для случая гетерогенного ка
не связана с механизмом реакции,
Рис. 109. Влияние равновесия и скорости реакции на течение реакции.
тализа, т. е. когда катализатор
представляет собой фазу, отличную от реагирующих веществ. В случае же гомогенного катализа катализатор может действовать на дви
жущие силы и, следовательно, на состояние равновесия, так как он будет влиять на активность реагирующих веществ.
Соотношение между скоростями и движущими силами можно выяснить путем анализа наблюдений, относящихся к течению реакции и показанных на рис. 109. Это — данные наблюдений над реакцией СОЦ-2Н2 = СН3ОН, но то же самое практически приложимо ко всем реакциям. Почти во всех случаях, за исключением очень редких и еще недостаточно хорошо изученных, скорость реакции возрастает с температурой. С другой стороны, движущая сила может возрастать или уменьшаться. Практически все реакции синтеза (образование соединений из более простых веществ или из элементов) показывают уменьшение движущей силы с ростом температуры; для реакций же разложения наблюдается обратное. Ниже определенной температуры скорость реакции может стать настолько ничтожной, что за данный промежуток времени, несмотря на значительную движущую силу, не произойдет заметного превращения реагирующих веществ в продукты реакции. По мере роста температуры, при условии, что другие факторы остаются неизменными, скорость возрастает очень быстро и, как показано на рис. 109, превращение увеличивается, хотя движущая сила уменьшается. Наконец, достигается
554
XI. Равновесие химических реакций
такой температурный уровень, при котором действие движущих сил, направленных на снижение степени превращения, становится преобладающим над противоположным действием скорости, вследствие чего степень превращения проходит через максимум. Повидимому, эта общая тенденция характерна для всех реакций, однако форма кривой и температура, отвечающая максимуму, могут от случая к случаю значительно отличаться.
Как дальнейшую иллюстрацию важности ясного понимания влияния этих двух факторов рассмотрим кратко толкование некоторых экспериментальных наблюдений над реакцией синтеза аммиака. Пусть объяснению подлежат следующие факты:
1. При совместном введении N2 и Н2 при атмосферном давлении и комнатной температуре не образуется заметных количеств NH3 как в присутствии катализатора, так и в отсутствии его.
2. При совместном введении N2 и Н2 при 450°С и 1 атм в присутствии катализатора образуются следы NH3.
3. При совместном введении N2 и Н2 при 450° С и 100 атм при отсутствии катализатора не образуется NH3.
4. Если же условия пункта 3 реализуются в присутствии катализатора, то образуется около 16°/0 NH3 при условии стехиометрического соотношения между газами и при достаточной длительности контакта.
Объяснение этих фактов с точки зрения равновесия и скорости реакции заключается в следующем.
В первом случае регулирующим фактором является скорость процесса, и если реагирующие вещества будут каким-нибудь способом активированы, произойдет почти полное превращение.
Во втором случае регулирующим фактором является равновесие. Скорость значительна, но максимальное превращение соответствует содержанию NH3 в газе, равному примерно О,2°/о.
В третьем случае регулирующим фактором является скорость, поскольку, как показано в случае 4, может происходить заметная реакция.
В четвертом случае важны как скорость, так и равновесие. Последнее ограничивает выход NH3 до 16°/0, но при недостаточвом времени контакта реагентов с катализатором выход может значительно снизиться.
Не располагая данными о равновесии реакции, мы не могли бы правильно оценить подобные результаты, и легко можно было бы потратить много времени и энергии, пытаясь достигнуть невозможного.
Зная равновесные состояния для какой-нибудь химической системы как функции трех независимых переменных — давления, температуры и исходного соотношения между реагирующими веществами, можно предсказать равновесную, т. е. максимально возможную степень превращения. Максимальная степень превращения будет достигаться только тогда, когда система придет в состояние равновесия, на что потре
Основные зависимости
555
буется бесконечно много времени. Поэтому максимальное превращение является асимптотическим пределом, к которому фактическая степень превращения может приближаться, но которого она никогда не может полностью достигнуть. Тем не менее, знание максимального превращения практически очень важно, поскольку оно устанавливает абсолютный предел, перейти который не представляется возможным, и поскольку оно дает масштаб для сравнения различных методов проведения данной реакции.
Близость к достижению равновесия в каком-нибудь реальном случае будет зависеть от многих причин и будет различной для каждого отдельного случая. Если реакции рассматриваются с целью осуществления их в промышленном масштабе, то играют роль и экономические и чисто технические факторы. Часто жертвуют степенью превращения для получения за данное время большего количества продукции. Несмотря на это, знание максимально возможного превращения при какой-нибудь реакции в различных условиях является чрезвычайно важным.
В этой главе мы будем рассматривать только самопроизвольно идущие превращения. Реакции, вызванные специальными факторами, например э. д. с., исключаются из рассмотрения.
ОСНОВНЫЕ ЗАВИСИМОСТИ
Для удобства ссылок здесь собраны некоторые из уравнений химического равновесия, выведенные в гл. IV. Эти уравнения относятся к обобщенной реакции
a A -f- ЬВ Ц-... = IL -f- тМ
(2)
Основными уравнениями закона действующих масс, приложимыми как к гомогенным, так и к гетерогенным равновесиям, являются:
уравнение
(3)
которое получается из уравнений (241, гл. IV) и (243, гл. IV), и уравнение
Дц° = — RT\nKa. (244, гл. IV)
Если стандартное состояние для каждого вещества, участвующего в реакции, то же, что для чистого компонента (при данных р и ^), то уравнение (244, гл. IV) принимает вид
Д/о = — RT\nKa. (245, гл. IV)
Для частного случая гомогенных газовых реакций, когда стандартное состояние каждого газа отвечает такому его состоянию, при котором
556
XI. Равновесие химических реакций
значение летучести равно единице, имеем
(Л)Чли)'"---
(fAW--- г
(247, гл. IV)
Это уравнение совпадает с уравнением (3), поскольку для газа, по определению стандартного состояния, a=-f.
Уравнением, аналогичным уравнению (245, гл. IV), является
AZ° = — RT \nKf.
(4)
Для частного случая реакции в идеальной газовой смеси
/<•_ _Лт
(ХА/а)Л (ХВ ?
(251, ГЛ. IV)
Для реакции в идеальном жидком растворе (xi)1 (хм)т - ••_________________________у
(хА>а(хВ>Ь--- *'
(250, гл. IV)
Для реакции между идеальными газами, в соответствии с уравнением (249, гл. IV),
(хд1 (хм)т- __ (хл)а (хв)ь-- •
р-'&К?.
(5)
Сочетая уравнения (251, гл. IV) и (253, гл. IV), получим
где
Rf к (yi)z (тм)т • • •
ЯР (тд)а(Тв)*--- ’ l=flp (или а/р).
(254, гл. IV)
Отметим, что К не зависит от состава равновесной смеси, но представляет функцию р и Т. является полезной величиной, дающей представление о степени отклонения от простого случая идеального газа, для которого К при всех температурах и давлениях должна быть равна единице.
При низких давлениях, когда все реальные газы можно считать идеальными, уравнения (3), (247, гл. IV), (251, гл. IV) и (5) при применении к гомогенной газовой реакции становятся идентичными, поэтому
Кр=К^=Кг=Ка,
причем р° отвечает предельному давлению, при котором все газы можно считать идеальными.
Для разбавленного раствора активности можно принять пропорциональными концентрации (см. гл. IV), и общее уравнение (3) для этого случая превращается в уравнение (251, гл. IV).
Другая константа равновесия определяется уравнением
____(Сд1 (Cju)m с ~ (С А)а (С B)b
(6)
Влияние температуры
557
Для идеальных газов
_(У/_Pi___Pxi I7\ V RT^RT' - [ '
Из уравнений (5)—(7) имеем для случая идеальных газов
Kp=Kc(RT^. (8)
Поэтому для любого раствора
С,. = х,4. (9)
(W)
Уравнение (250, гл. IV) применительно к идеальному жидкому раствору можно написать как
(С1)г(Сл)'”-.._^ \N) (СА)а(Св)Ь...
Если раствор является разбавленным по отношению ко всем компонентам, то величина V/N не будет изменяться так значительно, как изменяются отдельные концентрации, и эту величину можно скомбинировать с константой, что дает уравнение, совпадающее с уравнением (6).
Зависимость константы уравнения от температуры обычно дается уравнением
dT
(258, гл.IV)
в котором
(259, гл. IV)
Если стандартным состоянием каждого вещества будет состояние чистого вещества при данных значениях р и Т, то уравнение (258, гл. IV) превращается в
Ма_ № dT ~~ RT2 ’
(260, гл. IV)
ВЛИЯНИЕ ТЕМПЕРАТУРЫ
Из общих принципов, относящихся к равновесию, можно вывести, что если какая-либо из переменных, определяющих состояние равновесия, изменяется, то равновесие будет сдвигаться таким образом, чтобы содействовать уничтожению изменения. Это положение иногда называют принципом Ле-Шателье—Брауна. В том случае, когда переменной является температура, смещение равновесия при повышении температуры будет происходить в том направлении процесса, при котором поглощается теплота. Следовательно, эндотермической реакции будет благоприятствовать возрастание температуры, и наоборот, экзотермической— понижение температуры.
558
XI. Равновесие химических реакций
Количественно влияние температуры дается уравнениями (258, гл. IV) и (260, гл. IV); поскольку в общем виде оба уравнения тождественны, мы в дальнейшем остановимся на последнем.
Из гл. IX имеем &)
Д/7=Д/70 + Да7’ + 1д^ + уДу7’3, (101, гл.1Х)
Да=/а£ + таЛ1 + ... —аад ——... (11)
и подобные же уравнения для других коэфициентов уравнения (101, гл. IX). Д/70 представляет константу интегрирования, которая определяется по одному известному значению Д/У.
Подставляя уравнение (101, гл. IX) в уравнение (260, гл. IV), получим
>12>
Интегрирование дает
taX.=-^+f 1п7+1^г+|^л+а (13>
Из уравнений (245, гл. IV) и (13) находим, что
AZ° = AZ/0 —ДаПпТ—^Д^Г2 —^-Ду7’3-1-/7’, (14)
где 1=—RC. С и / — константы интегрирования, значения которых определяются по одному известному значению Ка или Д2°.
Из этих уравнений очевидно, что если точно известны теплоемкости всех реагирующих веществ как функции температуры и теплота реакции при одной какой-либо температуре, то для вычисления константы равновесия при любой температуре достаточно иметь значение ее при какой-либо определенной температуре, поскольку одного значения Ка достаточно для нахождения С (или /).
Если теплота реакции не известна или точность ее сомнительна, но значения Ка определены в широком интервале температур, то полезен следующий метод:
Перегруппируем уравнение (13) в
In/Со+Да 1пТ + 1д^Т’+-А-ДуТ2. / л о
Нанесем правую часть этого уравнения на график как функцию и проведем по точкам наилучшую из возможных прямых линий. Наклон этой линии даст величину Д/70, а пересечение ее с осью орди-
*) Верхний индекс [°], указывающий стандартное состояние, во всех последующих уравнениях ради простоты опускается.
Влияние температуры
559
нат—константу I. Этим путем мы получим константу равновесия как функцию Т и одновременно теплоту реакции.
Пример 1. Ньютон и Додж [177] определили константу равновесия Кр для реакции СО4~2Н2 = СН3ОН при достаточно низком давлении для того, чтобы можно было без большой ошибки считать все реагирующие вещества идеальными газами. Большое число определений при 250° С дало среднее значение ^\р — 2,32X10“3. Чему будут равны изменение стандартного изобарного потенциала и константа равновесия дли этой реакции при 500° С?
Для определения Л//о и коэфициентов Ля, Д₽, Д-у имею тся следующие данные:
Теплоты сгорании при 25° С:
Н2, Д/У=— 68 313 кал1, моль, СО, Д//=— 67 623 кал/мом, СНдОН(г), Д//=—182 560 кал/мом.
(Н2О во всех случаях считается находящейся в жидком состоянии.)
Из этих данных следует, что Д// для реакции синтеза метанола при 25° С = — 21 690.
Мольные теплоемкости при постоянном давлении соответственно равны:
Н2, Ср = 6,65 4- 0,00070 Т, СО, Ср = 6,89 4-0,00038 Т, СН3ОН, Ср = 2,0 4- 0,03 Т.
Ср даны в калориях на градус Цельсия и Т — в градусах Кельвина.
Да = 2,0 — 2 (6,65) — 6,89 = 18,19,
Др = 0,03 — 2 (0,00070) — 0,00038 = 0,0282, Д-{ = 0.
Используя эти данные и уравнение (101, гл. IX), получаем
Д/Уо = —17 530.
Поскольку мы приняли идеальность газов, то K„ — Ra. Подстановка ЛГД=2,32Х Ю“3 при 7 = 523,2 в уравнение (13) дает
С = 30,67,
/ = — 1,987 X 30,67 = —60,94.
Из уравнений (13) и (14) при 500° С получаем
Ка =1,60 ХЮ"6, AZ® = 20 510.
Если принять, что Д/У не зависит от температуры, то интегрирование уравнения (260, гл. IV) дает
1п*а = -^+С, (15)
или если взять определенный интеграл, то
1 S =
П^, R Ui Тг
(16)
560
XI. Равновесие химических реакций
Допущение постоянства Д/7 эквивалентно допущению, что суммарная теплоемкость продуктов реакции равна суммарной теплоемкости исходных реагирующих веществ. Это допущение является достаточно хорошим в очень многих случаях, и уравнения (15) и (16) весьма полезны для корреляции и экстраполяции экспериментальных данных по константам равновесия.
Уравнение (15) можно также записать в виде »
(17)
или
AZ“-—Д/7„„„ —ТД5. /2Ув
(17а)
В некоторых случаях может оказаться удобнее применение
Рис. НО.Сопоставление констант равновесия при различных температурах.
урав-чисто
нений (15) и (17) в эмпирической форме, а именно, в виде
, ,, Л
(18)
(19)
Ка илн
Д/7 является линейной функцией Т, то ческие коэфициенты)
Любые два значения Д£° будут служить для определения констант. Эти два уравнения часто применяют вместо уравнений (13) и (14).
Другая полезная формула для интерполяции может быть получена при применении иных упрощенных форм уравнения (101, гл. IX). Так, например, если принять, что получим (применяя эмпирн-
Ig^-y+^lgT’ + C.
(20)
н
Рис. 110 иллюстрирует применение линейной зависимости 1gКр — 1/Т для корреляции данных по константе равновесия реакции
С2Н4 + Н2О(г) = С2НБОН(г).
Уравнением наиболее близко проведенной прямой линии (метод средних значений), представляющей эти данные, является уравнение
1g = ^-6,241.
Влияние давления
561
Соответствующим уравнением для изобарного потенциала будет
Д2° = 28,607’ — 9740.
Уравнение (16) полезно также для приближенного определения во;можности реакции, для которой имеются данные при 25 °C, но не при более высоких температурах.
Пример 2. По новейшим данным по теплотам образования и абсолютным энтропиям изменение стандартного изобарного потенциала для реакции С2Н4 4- H2O(r) = C2HsOH(r) при 25° С равно —2030 кал/моль.
Теплота реакции при 25° С = —11 000. Определить константу равновесия для этой реакции при 600° К.
Из уравнения (245, гл. IV) при 25° С = 30,9.
Из уравнения (16)
КР2 — 11 000 /1 1 \
lg 30?9 = 4,575 600,2/ ’
7^ = 2,76 X Ю-з.
Экспериментальное изучение этого равновесия дает значения 1(р около 2Х Ю~3 (см. рис. ПО). При использовании уравнений для теплоемкости, данных Парксом [187], и уравнения (13) найдем, что
^=2,36 ХЮ"3-
Этот метод экстраполяции констант равновесия, начиная от комнатной температуры до практически недостижимой, нельзя считать точным, однако следует иметь в виду, что порядок величины часто достаточен для того, чтобы решить, возможна ли данная реакция или нет.
ВЛИЯНИЕ ДАВЛЕНИЯ
Из общих принципов равновесия можно вывести, что возрастание давления будет смещать равновесие в том направлении, в котором объем системы уменьшается. Следовательно, дли реакции
N2 4- ЗН2 = 2NH3
можно сразу сказать, что повышение давления благоприятствует образованию NHS. Многие важные органические реакции проходят с уменьшением объема, и для них, следовательно, давление также будет благоприятным фактором. Это обстоятельство лежит в основе современного развития синтеза при высоких давлениях.
При переходе к вопросу о количественном влиянии давления нужно различать влияние давления на равновесие и влияние его на константу равновесия. Константы Ка и Kf, определяемые соответственно по уравнениям (3) и (247, гл. IV), зависят только от температуры, и поэтому их значения не должны изменяться с давлением. С другой стороны, активность и летучесть изменяются с давлением; поэтому изменение давления должно вызывать сдвиг равновесия (для того чтобы константа равновесия осталась неизмен-36 Химическая термодинамика
562
XI. Равновесие химических реакций
ной). Даже для частного случая, когда ^п = 0, как, например, для
реакции
СО + Н2О(г) = СО2 + Н2,
давление все же будет оказывать некоторое влияние на равновесие. Это не противоречит общим принципам, изложенным выше, так как в реальной системе должно быть изменение объема, обусловленное отклонениями от законов идеального газа.
Константу равновесия Кр можно определить для любой газовой системы уравнением
<хй1(хм)т---
но эта константа, за исключением случая идеальных газов, будет функцией давления. При низких давлениях, когда газы приближаются к идеальному состоянию, Кр, в сущности, будет постоянной и равной К? или Ка. Ее изменение с давлением можно точно подсчитать по уравнениям (21) и (3), если активности компонентов можно выразить как функции давления и состава. Данных для этой цели обычно бывает недостаточно, но в качестве приближения вместо уравнения ,(3) можно, как будет показано ниже (стр. 563), применять уравнение (251, гл. IV). Наилучший пример влияния давления на Кр дается равновесными измерениями реакции синтеза аммиака, произведенными Ларсоном и Доджем [144] и Ларсоном [143].
Результаты работ вышеупомянутых исследователей приведены в табл. 17. Легко заметить, что возрастание давления от 1 до 1000 атм приводит примерно к четырехкратному увеличению Кр-
Таблица 17
Влияние давления на константу равновесия реакции 1/2NZ —|~ 3/2 H, = NH3 при 450° С
Давление, атм
(XNH,)P~J
(ЧЛЫ7
1 10 зо 50 100 300 600 1000
0,00659 0,00676
0,00690 0,00725 0,00884 0,01294 0,02328
Приложение точного уравнения (3) к газовой реакции требует данных по зависимости р — v—Т, по которым можно определить активность. Таких данных недостаточно даже для простейших систем;
Влияние давления
563
поэтому это уравнение, будучи абсолютно строгим, является практически неприменимым, по крайней мере для газовых реакций. В качестве первого приближения можно считать газы идеальными и пользоваться уравнением (5). Лучшее совпадение получается при применении уравнения (251, гл. IV), которое соответствует допущению, что смешение газов происходит в соответствии с законом образования идеальных смесей, хотя сами компоненты и не являются идеальными газами.
Пример 3. Вычислить константу равновесия Ду для реакции синтеза аммиака при 450° С и 300 атм, если дано значение Кр при 1 атм и той же самой температуре.
Примем, что для этого случая справедливо уравнение (251, гл. IV), и будем пользоваться им в форме уравнения (254, гл. IV). Летучести газов, как было показано в гл. VI, можно найти различными методами, но мы воспользуемся обобщенной диаграммой коэфициента активности (см. рис. 35 и 36). Необходимые данные представлены в табл. 18.
Таблица 18
Критические константы и приведенные температура и давление водорода, азота и аммиака при 450°С и 300 атм
Газ Критическая температура, °К Критическое давление, атм Приведенная температура й) Приведенное давление ту)
н2 33,2 12,8 17,53 14,4
N2 126,0 33,6 5,73 8,94
NH3 406 111,6 1,78 2,69
4) Для водорода т = ТЦТкр -J- 8) и it = р!(ркр + 8).
По этим данным,
7Na=l,14 и tNHj = 0,91,
1 (м^-а.оэ)’'*
Константа равновесия при 1 атм не была измерена, но можно произвести небольшую экстраполяцию на основании данных Ларсона и Доджа [144] для 10 атм., пользуясь эмпирическим уравнением
2679,354-1,1184л
1g Кр =--------------- — (5,8833 + 0,001232/»),
выведенным Джиллеспаем (90] для применения данных Ларсона и Доджа к давлениям ниже 100 атм. Значение Ду полученное из этого уравнения для 1 атм и 450° С, равно 0,00664.
Наблюденное значение Кр, как было вычислено Джиллеспаем [90] из экспериментальных данных Ларсона [143], равно 0,008770. Вычисленное 36*
564
XI. Равновесие химических реакций
значение Кр получается следующим образом:
К& 0,00664 ^=<=таг="даз5’ 0,00664
наблюденная = q QQgjj — 0,757.
В табл. 19 значения К^ для реакции синтеза аммиака, вычисленные с помощью только что рассмотренного метода, сравниваются с наблюденными значениями. Последние найдены по уравнениям Джил-леспая для К = Р)> которые сглаживают экспериментальные дан-
ные Ларсона и Доджа и Ларсоиа.
Совпадение ниже 600 атм является очень хорошим и остается хорошим даже при 600 атм. Заметное отклонение при 1000 атм, вероятно, обусловлено нарушением правила аддитивности объемов, на котором основаны уравнения (251, гл. IV) и (254, гл. IV). Хотя это правило оказалось вполне удовлетворительным для азотно-водородной смеси даже при 1000 атм, присутствие NH3 в количестве, близком к 70°/о, при этом давлении, повидимому, было причиной значительного отклонения от правила.
Таблица 19
Совпадение между наблюденными и вычисленными значениями Кт для реакции синтеза аммиака
Давление, атм к, Т е мператур а, °C
325 350 375 400 425 450 475 500
10 Вычисленная 0,981 0,983 0,984 0,986 0,987 0,988 0,990 0,992
Наблюденная 0,986 0,987 0,988 0,990 0,991 0,992 0,993 0,994
30 Вычисленная — 0,960 0,963 0,965 0,967 0,969 0,971 0,973
Наблюденная — 0,963 0,968 0,972 0,974 0,978 0,982 0,985
50 Вычисленная — 0,935 0,942 0,946 0,950 0,953 0,956 0,959
Наблюденная — 0,937 0,946 0,954 0,958 0,965 0,970 0,978
100 Вычисленная — — 0,889 0,895 0,900 0,905 0,910 0,914
Наблюденная — — 0,894 0,907 0,918 0,929 0,941 0,953
300 Вычисленная — — .—. —_ 0,750 0,768 0,788
Наблюденная — — — — — 0,757 0,765 0,773
600 Вычисленная — — — 0,573 0,594 0,612
Наблюденная — — — — — 0,512 0,538 0,578
1000 Вычисленная — — — — 0,443 0,473 0,487
Наблюденная 1 — — — — 0,285 0,334 0,387
Сопаденне между наблюденными и вычисленными мольными долями аммиака в равновесной смеси лучше, чем совпадение значений К. Например, при 450°С и 600 атм вычисленное и наблюденное значения для мольной доли NHS составляют соответственно 0,516 и
Вычисление констант равновесия на основании термических данных 565
0,536, т. е. различие <>авно только З.Э0^ по сравнению с разницей в 11,9°/0 для значений К?
Уравнение (251, гл. IV) дает гораздо лучшее приближение к опытным данным, чем уравнение (5), сноваонное на допуще
нии идеальности газов; это сразу видно из того, что на основании последнего уравнения все значения Должны быть равны 1,0.
В некоторых случаях влияние давления на константу равновесия невелико даже тогда, когда газы значительно отклоняются от иде-
альности. Например, это имеет место для реакции
С2Н4 + Н2О(г) = С2Н5ОН(г), если мы примем, что для нее приложимы уравнения (251, гл. IV) и (254, гл. IV). В этом случае отклонения от идеальности компенсируют друг друга, вследствие чего при высоких давлениях практически равна единице.
Этот метод определения влияния давления на константу равновесия особенно полезен в случае органических реакций, где данных по зависимости р — v—Т обычно недо
Рис. 111. Значения Д' для реакции синтеза метанола.
статочно даже для отдельных
компонентов. С другой стороны, если известны критические константы, то летучести можно найти по обобщенным диаграммам, после чего можно вычислить Ку. На рис. 111 приведены значения Д' для реакции
CO-f-2H2 = CHsOH, полученные таким способом [176]. В этом случае весьма заметно отклонение от равновесия в системе идеальных газов.
ВЫЧИСЛЕНИЕ КОНСТАНТ РАВНОВЕСИЯ НА ОСНОВАНИИ ТЕРМИЧЕСКИХ ДАННЫХ
Определение констант равновесия представляет часто встречающуюся задачу; экспериментальное же нахождение их связано нередко с большими трудностями. Поэтому желательно иметь возможность определять их вычислением. Существует много реакций, особенно в области органической химии, которые -практически трудно провести Д° конца. Прежде чем начать осуществлять длительную и дорогостоящую программу исследования катализаторов и влияния условий
566
XI. Равновесие химических реакций
на осуществление реакции, желательно иметь некоторую уверенность в том, что равновесие достижимо, т. е. что реакция в нужной степени термодинамически возможна. В прошлом было затрачено немало усилий на поиски катализаторов и других средств ускорения реакций, которые, как можно было бы показать на основе термодинамического анализа, не могли оказывать достаточного действия в желаемом направлении.
Осуществимость реакции. Иногда говорят, что данная реакция термодинамически невозможна. Это является неверным утверждением, потому что оно не имеет смысла при отсутствии уточняющих данных. Например, любая реакция, начинающаяся с чистым веществом, т. е. при отсутствии загрязняющих его примесей, будет иметь стремление продолжаться, хотя степень превращения может оказаться лишь бесконечно малой. Так, например, прн 25° С реакция
J
Н2О(г) = Н24-4 О2
в некоторой степени проходит; мы можем даже с значительной уверенностью вычислить процентное содержание водяного пара, который должен разложиться. По точно известному значению AZ0 для этой реакции при 25° С константа равновесия примерно равна 1 X Ю-4в, и степень разложения будет ничтожно мала, но вполне определенна.
По изменению стандартного изобарного потенциала для любой реакции можно составить мнение об осуществимости реакции, не прибегая к дальнейшим вычислениям. Так, если AZ° = O при данной температуре, то К =\, и ясно, что реакция до достижения равновесия должна проходить в значительной степени. Положение становится менее благоприятным, когда AZ0 растет в положительном направлении, но иет определенного значения, которое можно было бы выбрать как ясно указывающее на то, что реакция с точки зрения промышленного использования будет неосуществима. При 600° К для реакции синтеза метанола AZ0 равна -|- 11 000, и все же при этой температуре реакция бесспорно возможна. В этом случае неблагоприятное изменение изобарного потенциала в стандартном состоянии частично нейтрализуется высоким давлением, применяющимся для смещения равновесия. Для этой цели можно также употребить и другие средства, например изменение соотношения между реагирующими веществами или изъятие одного из продуктов реакции.
Для быстрого, но только приближенного решения вопроса — возможно ли протекание интересующей нас реакции при данной температуре, могут оказаться полезными следующие правила:
1) если AZ°<^0, то реакция возможна;
2) если AZ°^>0, но <^-|- 10 000, то возможность реакции сомнительна, однако вопрос об осуществимости реакции заслуживает дальнейшего изучения;
Вычисление констант равновесия на основании термических данных 567
3) если AZ0 + 10 000, то условия весьма неблагоприятны, и i
реакция может оказаться пригодной только при исключительных условиях. ~~1
Необходимо иметь в виду, что этот способ дает лишь прибли- i
женный критерий, полезный для предварительного выяснения -..............—
вопроса.
Аддитивность значений AZ. Изменение изобарных потенциалов можно Определить путем сложения их значений для реакций, алгебраическая сумма которых образует рассматриваемую реакцию. i
Основой для этого является то, что AZ зависит только от начального и конечного состояний, а не от промежуточных стадий, через которые достигается конечное состояние. Например, реакцию
с+|о2 = со (22)
можно считать состоящей из следующих стадий:
С-|-СО2 = 2СО, (23)
Н24-| О2 = Н2О(ж), (24)
СО + Н20(ж) = СО2 + Н,. (25)
Сложение уравнений дает
(23) + (24) + (25) = (22).
Следовательно,
AZ°(22) = AZ°(23) + AZ°(24) + AZ°(25).
Чтобы результаты сложения были правильны, необходимо следить, чтобы все вещества, которые участвуют более чем в одной реакции, находились в одинаковом состоянии. Другими словами, они должны быть при одинаковых температуре, давлении и агрегатном состоянии; в противном случае изобарные потенциалы нельзя сокращать.
Простейшим путем нахождения изобарного потенциала для любой реакции является алгебраическое суммирование изобарных потенциалов образования различных веществ, участвующих в реакции. Так, для реакции
СО + ЗН2 = СН4 + Н2О(г).
AZ(peaKiiHii) = AZ (образования СН4) + AZ (образования газообразной воды) — AZ (образования СО).
Поскольку Н2 является простым веществом, а не соединением, то его AZ образования равен нулю. Изобарный потенциал образования любого соединения представляет собой просто изменение изобарного потенциала для реакции, по которой соединение образуется из соответствующих простых веществ. Например, изобарный потенциал образова-ния жидкого метанола будет изменением изобарного потенциала реакции
С + |02 + 2Н2 = СН30Н(ж). С
668
XI. Равновесие химических реакций
Изобарные потенциалы образования обычно даются для случая, когда все участвующие вещества находятся в стандартных состояниях при 25° С. Если в имеющихся таблицах есть изобарные потенциалы образования при 25° С для всех соединений, участвующих в интересующем нас ряде реакций, то можно простым сложением найти AZ0 для любой данной реакции. Затем с помощью данных по теплоемкостям можно найти по уравнению (14) значения AZ0 при любой температуре и, наконец, из уравнения (245, гл. IV) — константу равновесия. Главной причиной, которая мешает универсальному применению этих простых методов, является недостаток необходимых данных. Нужно продолжительное время, прежде чем работники этой области будут в состоянии собрать достаточные данные, позволяющие предсказать равновесия для большого числа случаев.
AZ0 образования органических соединений, установленное по их структуре. Задачу определения стандартного изобарного потенциала образования органических веществ можно значительно упростить с помощью структурных зависимостей, которые позволяют предсказать большое число значений AZ0 по немногим измеренным значениям. В этом направлении удалось достигнуть пока незначительных, но все же существенных результатов. Например, Ивелл [77], сделав критический обзор существующих данных по Д//° и Д5° для парафиновых и олефиновых углеводородов, разработал некоторые простые правила для определения и Згэв разветвленных парафинов, 1-олефинов и разветвленных и непредельных олефинов по известным значениям AZ0 для нормальных газообразных парафинов с одинаковым числом атомов углерода. Правила эти заключаются в аддитивности некоторых фиксированных величин, зависящих от типа структурного изменения. Такие обобщения обычно считаются грубыми, но они в известной степени полезны при отсутствии экспериментальных данных. Они были использованы для предсказания выходов в реакциях углеводородов, представляющих большое промышленное значение, например при реакциях изомеризации, полимеризации и алкилирования.
Третий закон термодинамики. Этот закон кратко упоминался в предшествующих главах, но его обсуждение было отложено. Ограничим его рассмотрение лишь несколькими важнейшими пунктами.
Тепловая теорема Нернста, из" которой вырос третий закон, явилась завершением длинного ряда попыток химиков вычислить равновесие химических реакций из термических данных. В гл. III было показано, что этот вопрос связан с изменением энтропии при абсолютном нуле. На основании данных о характере изменения AZ и Д/7 с температурой при температурах, далеких от абсолютного нуля, Нернст сделал допущение о тенденциях в изменении этих величин, когда температура приближается к абсолютному нулю. Это допущение условно эквивалентно
AS и ДСр = 0 при Т = О,
Вычисление констант равновесия на основании термических данных 569
где Д относится к какому-нибудь физическому изменению или к химической реакции. Если все вещества имеют одинаковую энтропию и одинаковую теплоемкость при Т= 0, то будет естественно предположение, что эти величины также равны нулю. Это и явилось постулатом Планка, который часто рассматривается как третий закон термодинамики. Поскольку Ср необходимым образом равно нулю, если Т— 0, то этого достаточно для утверждения, что энтропия отдельных фаз при абсолютном нуле равна нулю.
Эта формулировка закона является слишком общей и требует некоторых дальнейших ограничений, поскольку известны случаи, когда энтропия при Т= 0 больше нуля. Со статистической точки зрения энтропия становится равной нулю только тогда, когда вещество находится в стабильном равновесии на всех стадиях процесса охлаждения, и достигает 7'=0, когда энергетическое состояние всех его частиц приходит к наинизшему уровню. Планк выразил эту идею с помощью уравнения
S = R In g,
где g представляет число конфигураций, в которых может существовать система. Для единственного состояния наинизшей энергии g = 1 и S=0. Если система не приходит к этому равновесному состоянию при Т =0, то она будет иметь положительную энтропию, обусловленную сложностью конфигураций. Следовательно, можно ожидать, что все жидкости (за исключением гелия), поскольку они находятся в нестабильном переохлажденном состоянии (подобно стеклу при обычных температурах), будут иметь положительную энтропию, что можно подтвердить и экспериментом. Подобным образом растворы должны иметь энтропию смешения, что также подтверждается опытом. В случае некоторых газов, как Н2, СО, N2O, Н2О и нескольких других, различие между абсолютными энтропиями, вычисленными из спектроскопических данных, и энтропиями, вычисленными на основании калориметрических измерений, должны были бы привести к признанию существования известной „сложности* конфигураций для некоторых молекул, которые при столь низких температурах являются „замороженными*; поэтому единая равновесная конфигурация наинизшей энергии не может быть получена, и калориметрическая энтропия будет слишком низкой. Это, в частности, показано для водорода, который, благодаря различию в ядерных спинах, имеет две различные формы молекул—пара- и орто-формы. Когда водород охлаждается до низких температур, то получающиеся кристаллы представляют твердые растворы двух форм, и перехода в устойчивую пара-форму ге происходит. Показано, что для получения истинной абсолютной энтропии к калориметрической энтропии нужно прибавить разницу в 4,39 энтропийных единиц. Однако также показано, что энтропии ядериого спина уничтожаются в реакциях при обычных и повышенных температурах, и правильная энтропия, которую следует применять
570
XI. Равновесие химических реакций
при вычислениях равннвесия, будет так называемой „ практической “ энтропией, исправленной на энтропию, отвечающую ядерному спину при повышенных температурах. Так как многие элементы представляют собой смесь изотопов, то они будут обладать энтропией смешения при Т— 0, но этой величиной при практическом применении можно пренебречь, потому что пропорция изотопов не изменяется при обычных физических изменениях или при химической реакции.
Следует заметить, что тогда как первые два закона термодинамики приводят к определению новых величин фундаментальной важности,— первый закон — к понятию энергии, а второй закон — к термодинамической температуре и энтропии, — третий закон не вносит никакой новой величины, но лишь устанавливает предел одной из указанных ранее величин. Поэтому некоторые авторы предпочитают не возводить положение о существовании абсолютной энтропии в ранг столь же общего закона, как первый и второй законы термодинамики.
Экспериментальное подтверждение третьего закона заключается в предсказании константы равновесия при использовании абсолютных энтропий, вычисленных из калориметрических или спектроскопических данных, и в сравнении предсказанного значения с экспериментально установленным. Сравнение абсолютных энтропий, найденных двумя путями, т. е. и спектроскопическим и термохимическим, является также средством проверки правильности третьего закона. Коротко можно сказать, что для всех реакций, исследованных в конденсированных системах, третий закон в пределах ошибки опыта является справедливым. В случае газовых реакций или при сравнении энтропий из двух различных источников имеются определенные расхождения, но ббльшая часть их удовлетворительно объясняется. Поскольку дело касается практического применения, мы можем с уверенностью заключить, что все химические индивидуумы обладают некоторым значением абсолютной энтропии, которым можно пользоваться для предсказания равновесия. Для относительно простых молекул эти энтропии можно вычислить из спектроскопических данных с помощью статистической механики, и значения, определенные таким образом, будут, вероятно, более точны, чем полученные по термодинамическим измерениям. Для большинства веществ должен применяться калориметрический метод; он состоит в определении теплоемкостей и скрытых теплот для всего диапазона температур. Охлаждать до абсолютного нуля практически невозможно, но если охлаждение осуществляется с помощью жидкого водорода примерно до 12° К, то остаточная энтропия мала, и экстраполяцию до 7=0 можно провести с достаточной уверенностью. Метод экстраполяции был намечен в гл. IX. Конечным результатом подобных измерений является составление таблиц абсолютных энтропий элементов, ионов и соединений. Такие таблицы ещ? далеко не полны, но все же имеются величины для большого числа веществ, представляющих промышленное значение.
Вычисление констант равновесия на основании термических данных 571
В этом кратком обзоре третьего закона упомянуты лишь немногие основные положения. Для ознакомления с деталями читатель отсылается к обзору Истмена [72а].
Применение третьего закона81. В конце концов все значения изобарного потенциала определяются либо на основании измерения равновесия, либо с помощью уравнения
= — TAS. (26)
Это уравнение при сочетании с третьим законом термодинамики, который позволяет определять абсолютное значение энтропии, дает возможность рассчитать равновесие на основании чисто термических измерений. Оно является крайне важным уравнением, и его применение будет показано довольно подробно с помощью числовых примеров.
Для предсказания константы равновесия Ка для реакции в газовой фазе при любой температуре необходимо располагать по крайней мере следующими данными:
1. Абсолютная энтропия каждого вещества в его стандартном состоянии прн данной стандартной температуре.
2. Теплота реакции прн стандартной температуре.
3. Теплоемкость каждого вещества в пределах от стандартной до рассматриваемой температуры.
4. Скрытая теплота парообразования и давление пара веществ, которые при стандартной температуре находятся в твердом или жидком состоянии. Могут быть использованы и другие эквивалентные этим данные. Например, вместо абсолютной энтропии каждого вещества можно было бы также использовать энтропии образования всех веществ, участвующих в реакции. Энтропии образования определяются сочетанием абсолютных энтропий соединения и энтропий соответствующих простых веществ. Теплоту реакции можно получить из теплот образования, которые, в свою очередь, обычно определяются (по крайней мере для органических соединений) путем сочетания теплот сгорания рассматриваемых соединений и составляющих их простых веществ.
Для реакций, включающих жидкую фазу, обычно необходимы дополнительные данные, связывающие равновесные концентрации в газообразной н жидкой фазах. В случае реакций при повышенных давлениях необходимы данные по сжимаемости газов, если желают ^определить К а не Ка (Ка не зависит от давления), или если концентрации вычислены из значения Ка.
Пример 4. Найти с помощью термических данных для реакции синтеза метанола
СО-4-2Н2 = СН3ОН(г) (1)
зависимость константы равновесия Кр от температуры при давлении в 1 атм.
Можно непосредственно применить уравнение (26) при 25° С, используя соответствующие теплоты образования газообразного СН3ОН и СО и зная
572
XI. Равновесие химических реакций
абсолютные энтропии СО, Н2 и СН3ОН. Используем следующие данные:
S жидкого СН3ОН при 25°С = 30,3 э. е. [130а], Sc0 при 25°С и 1 я/и_и = 47,3 э. е. [44], 3Нп при 25°С и 1 атм= 31,25 э. е. [1306].
Энтропия СНдОН найдена на основании измерений теплоемкости и скрытой теплоты, между тем как энтропии СО и Н2 получены из спектроскопических измерений. Д/7 реакции (1) получается сочетанием Д/Удля следующих
превращений:
СН3ОН(ж)+11/2О2=СО2 + 2Н2О(ж), (2)
Н2 -j-1 /2 О2 = Н2О(ж), (3)
CO -j-1 /2 О2 = СО2, (4)
СН3ОН(ж) = СН3ОН (г, 0,163 атм), (5)
СН3ОН (г, 1 атм) = СН3ОН (г, 0,163 атм), (6)
(0,163 атм—давление насыщенного пара СН3ОН при 25°С), Д7Уг = 2ДЯ34-Д/У4 — Д//2-]_Д//5_ Д/у6. (7)
Значения ДУ/ в калориях на моль при 25°С й):
Д/У:> = —68 313 [208], Д/У4 = —67 623 [209], Д/У.. — — 173 630 [209«], Д/У3 = — 8 930 [79].
Примем, что Д//6 будет ничтожно мало. Эта величина равняется нулю для идеального газа, и ошибка от принятия паров метанола за идеальный газ при этих низких давлениях будет, вероятно, очень небольшой.
Д/У по (7) = —21 690,
8930
ДЗ парообразования СН3ОН при 0,163 атм — ^^g-^ = 30,0, ДЗ для процесса СН3ОН (г, 0,163 атм)—► СН3ОН (г, 1 атм)
S паров СН3ОН при 1 атм = 30,3 30,0 — 3,60 = 56,7,
ДЗ! = 56,7 — 47,3 — 62,5 = — 53,1, наконец,
Дг? = —21 690—298,2 (—53,1) = —5850.
Возражение, которое может возникнуть при анализе этого расчета, заключается в том, что пары метанола не могут существовать в стабильном равновесии при 25°С и 1 атм, и поэтому стандартное состояние, выбранное для метанола, является чисто гипотетическим. Это является несколько искусственным. Мы можем избежать использования этого гипотетического состояния или приняв в качестве стандартной температуру, равную либо превышающую нормальную точку кипения, или выбрав в качестве давления,
й) С тех пор, как впервые была решена эта задача, появились более точные термические данные, но применение этих данных изменяет решение лишь в незначительной степени; поэтому автор считает не обязательным прибегать к пересмотру расчета.
Вычисление констант равновесия на основании термических данных 573
определяющего стандартное состояние, давление более низкое, чем 1 атм, например 0,1 атм. Однако очень удобно относить все термодинамические данные к стандартному состоянию при 25° С и 1 атм вместо принятия отдельных, каждый раз отличающихся друг от друга стандартных состояний, Тем более, что небольшая экстраполяция в нестабильную область, являющаяся вынужденной в случаях, подобных рассмотренному, приводит к ничтожно малой ошибке. Другими словами, хотя в принципе и неверно применять гипотетическое состояние, однако это все же оправдывается большим удобством.
Для мольных изобарных теплоемкостей мы будем пользоваться уравнениями, приведенными на стр. 559, по которым А//о =— 17 530.
Подставляя это значение в уравнение (14), получаем
= —17 530+18,197 In Т~ 0,0141 Г3 + /Т.
Так как
Д2^98 = — 5850, то /=—60,4.
Окончательно имеем
lg9,150lg Т+ 0,00308Т+ 13,20. (8)
При 250°С (Т = 523,2) уравнение дает Кра~ 2,ОХЮ-3- Эту величину можно сравнить с экспериментальным значением в 2,ЗХЮ-3, полученным Ньютоном и Доджем [177]. Совпадение для данных в этой области необычайно хорошее.
Интересно сравнить этот результат с результатом, полученным по приближенному уравнению (16). Используя для Д// значение при 25°С, имеем
Кп 21 690/ 1 1 \
й 20 000 4,575 \298,2 523,2 J’
или
Кр= 2,76X10'3.
Совпадение между приближенным и более точным методом получается в этом случае относительно хорошее и может быть даже лучшее, чем можно было бы ожидать.
Рассмотренный метод предсказания констант равновесия из термических данных является ценным орудием в руках исследователя; однако в настоящее время в большинстве случаев точные данные отсутствуют. Поэтому невозможно ожидать результатов, дающих больше, чем только порядок величин. Как показывает вышеприведенный пример, изменение изобарного потенциала реакции может оказаться небольшой величиной по сравнению с некоторыми термическими величинами, используемыми в расчете, поэтому ошибка в одной из последних величин, незначительная в процентном отношении, может дать значительно бблыпую процентную ошибку в AZ реакции и еще более значительную в величине Кр 84. Обращаясь снова к примеру 4, мы видим, что 1-процентное изменение в теплоте сгорания метанола при постоянстве прочих величин изменит К при 250° С до 1,07X10“*» т. е. почти в пять раз.
574
XI. Равновесие химических реакций
Было время, когда предсказанные константы равновесия для реакции синтеза метанола около 600° К отличались в 10 и даже в 200 раз от лучших экспериментальных данных. Это различие в течение некоторого времени оставалось непонятным, пока не было предположено [63], что многие из этих расхождений легко могут быть объяснены неточностью в термических данных, особенно в теплоте сгорания метанола. Когда теплота сгорания метанола вновь была определена Россини [210], то это предположение подтвердилось. В настоящее время для этой реакции имеется хорошее совпадение экспериментальных и вычисленных констант равновесия.
Как было указано в гл. IX в связи с рассмотрением теплот реакций, ошибки очень сильно возрастают в случае таких реакций, как изомеризация углеводородов, когда Д/7 реакции составляет очень небольшую долю от Д// сгорания. В этих случаях даже для грубого предсказания равновесия требуются термические данные очень высокой точности.
Предсказание констант равновесия из термических данных по описанному методу было проверено с помощью экспериментальных определений для большого числа случаев, благодаря чему мы можем быть достаточно уверены, что при условии доступности точных термических данных этот метод является надежным.
Для ознакомления с наиболее современным применением третьего закона к вычислению изобарного потенциала химической реакции и для обсуждения вычисления энтальпий и изобарных потенциалов из спектроскопических дацных можно обратиться к статье Астона [5].
Мы дадим теперь другой пример реакции, в которой участвуют как жидкая, так и газообразная фазы и в расчете применяются различные стандартные состояния.
Пример 5. Вычислить по термическим данным константу равновесия для реакции между газообразными этиленом и водой с образованием этилового спирта при 254° С, если давление достаточно высоко для того, чтобы присутствовала жидкая фаза.
Конечно, все три составляющие присутствуют как в газовой, так и в жидких фазах, и в системе будут существовать три одновременных равновесия: 1) химическое равновесие в жидкой фазе; 2) химическое равновесие в газовой фазе и 3) фазовое равновесие. Мы можем или непосредственно рассматривать химическое равновесие в жидкой фазе, или считать его сочетанием двух других равновесий и трактовать его так, как если бы реакция целиком происходила в газовой фазе. Так как изменение изобарного потенциала для любой реакции совершенно не зависит от механизма или промежуточных стадий процесса, то между этими методами практического различия нет.
Напишем реакцию в форме
С2Н4(г) 4* Н20(ж) = С2Н5ОН(ж).
Для вычисления константы равновесия, определяемой уоавнением
/Г = ас ° аАаВ’
(1)
Вычисление констант равновесия на основании термических данных 575
где ад, ад и ас соответственно активности этилена, воды и этанола, следует иайти изменение изобарного потенциала для реакции в условиях, когда каждый из реагентов находится в стандартном состоянии. Реакция, написанная выше, предполагает, что избранными стандартными состояниями будут следующие:
Этилен в виде чистого газа при давлении в 1 атм (более строго с летучестью = 1 атм).
Вода в виде чистой жидкости прн 1 атм.
Этанол в виде чистой жидкости при 1 атм.
На протяжении всего примера температура, если не будет оговорено особо, принимается равной 25° С.
А/7 для реакции (1) можно вычислить сочетанием A/у для следующих реакций:
С2Н4 + ЗО2 = 2СО2 + 2Н2О(ж),
ДЛУ2 = —337 280 [212], (2>
С2Н5ОН(ж) 4- ЗО2 = 2СО2 4- ЗН2О(ж),
Д/у3 —— 326 660 [211], (3>
Atfj = Д/у2 — Д/у3 = — 10 620.
AS для реакции (1) получается из следующих величин:
Энтропия жидкого С2Н5ОН = 38,40 [130а].
Энтропия газообразной Н2О при 1 атм=45,10 [93].
Теплота парообразования Н2О при 25° С =10 500 [86].
Давление насыщенного пара Н2О при 25° С = 0,0312 атм.
Следовательно, энтропия жидкой воды = , 1 10500 то
— 45,10 — 4,5751g о^оз|2 — 298,2 — 16,8’
Энтропия С2Н4 = 52,3 [188]. *
AS для реакции (1)=38,40—16,8 — 52,3= —30,7,
AZ® для реакции (1) = — 10 620 + 30,7X298,2 = — 1470.
Так как
— AZ»
"4,5757’
то при Т — 298,2
/Со=12,0.
Для приближенного вычисления Ка ПРИ 527,2° К примем, что УН является постоянной величиной. Тогда, используя соотношение
— УН
]g Ко = 4575 7+ С’
по значению Ка при 298,2° К определяем С = —6,72 и находим, что при 527,2° К
Ка = 5,0ХЮ-з.
Вероятно, более точное значение Ка при 527,2° К можно было бы получить в том случае, если бы имелись данные по теплоемкости, позволяющие связать УН с температурой. Однако существуют определенные затруднения, связанные с нахождением Ка для этой реакции при выбранной нами температуре, которые следует рассмотреть более подробно, поскольку они вообще имеют существенное значение для выбора стандартного состояния. Во-первых, критическая температура этанола ниже 527,2° К, и поэтому стандартное состояние, выбранное для него при этой температуре, является гипотетическим, — этанол ие может существовать в виде жидкости при 527,2° К.
576
XI. Равновесие химических реакций
В данном случае имеется еще одна добавочная трудность: при попытках связать Д/7 с температурой требуется знать, каким значением Ср нужно воспользоваться. Теплоемкость же Ср всех газов в критической точке становится бесконечно большой.
Даже если температура, при которой желают определить Ка, ниже критической, то все же стандартное состояние будет неустойчивым, потому что этанол и вода не существуют в устойчивом состоянии как жидкости при 1 атм и повышенных температурах. Мы могли бы обойти это затруднение, выбрав стандартное состояние при таком давлении, чтобы оба эти вещества были при данной температуре жидкими. С практической точки зрении это может ивиться и ненужным уточнением, потому что влияние давления на изобарный потенциал жидкостей относительно невелико. Тогда для процесса
С2Н5ОН (ж, 1 атм) — C2HjOH (ж, 100 атм).
&Z = j* vdp.
Принимая объем не зависящим от давления, получаем = при 25° С v = 57,5 мл)моль,
&Z = 55,7X99 = 5693 млХатм = 138 кал.
Хоти эта величина составляет заметную долю от AZ реакции, однако она лежит, вероятно, в пределах ошибок опыта при определении некоторых термических величин, использованных при вычислении AZ.
Кроме того, чтобы использовать константу равновесия для вычисления равновесных условий, требуются данные по давлению насыщенного пара этанола над растворами в пределах от почти чистого этанола до равновесной концентрации, которая может соответствовать бесконечному разведению.
Этих трудностей можно избежать или, по крайней мере, сделать их менее значительными, если выбрать дли этанола другое стандартное состояние, именно— состояние разбавленного раствора. Тогда уравнение (1) превращается в уравнение
С2Н4 (г) Н2О (ж) = С2Н5ОН (водн.). (4)
AZ для этой реакции является суммой AZ для реакции (1) и AZ для процесса
С2Н5ОН (ж) = С2Н5ОН (водн.). (5)
AZ для процесса (5) легко получить, представив, что он протекает изотермически и обратимо через следующие промежуточные процессы:
1. Испарение этанола п^и давлении его насыщенного пара рс.
2. Изотермическое расширение паров этанола от рс до его парциального давления рс над водным раствором.
3. Конденсация пара через полупроницаемую мембрану, находящуюся между паром и раствором, благодаря которой конденсация происходит равновесно при давлении рс.
Стадии 1 и 2 не связаны с изменением Z. Для стадии 3, принимая пар идеальным газом, получаем
4Z = /?nn^-.
Рс
Эта величина в то же время является AZ для реакции (5). Следуя обычной практике, примем в качестве стандартного состояния 1М раствор. Для такого раствора (мольная доля С2Н5ОН = 0,0177) парциальное давление рс равно 3,65 мм.
рс — 59,0 мм.
Вычисление констант равновесия на основании термических данных 5П
Следовательно,
AZ = /?7'ln|^ = —1650.
59,0
Тогда AZ для реакции (4) —— 1470 — 1650 = —3120.
Ка ПРН 298,2° К =-195. ПН для реакции (5) будет приближенно равна теплоте растворении 1 моля С2Н6ОН в воде с образованием бесконечно разбавленного раствора, т. е. —2500 кал. Тогда Д//4 =—13100. Считая эту величину константой, находим, что дли реакции (4) Ка при 527,2° К = 0,014.
Экспериментальные результаты по жидкофазному равновесию для этой реакции скудны и не очень согласованы друг с другом, но результаты Джил-лиленда, Ганнеса и Боулса [92] могут служить в качестве грубой проверки вычисленной константы равновесия. Из данных при 527,2° К можно вычислить Ка на основе следующих допущений:
1. Концентрация этанола мала, и для жидкой фазы можно воспользоваться законом Генри. Так как стандартным состоянием является 1-мольный раствор, <?с.и5он = тС,н5он •
2. Так как мольная доля Н2О равна примерно 0,9, то можно считать справедливым закон Рауля и “н,о = хнаО’
3. Пар можно считать идеальным раствором, т. е. принять, что f—yf, где / — летучесть чистого С2Н4 при данной температуре и общем давлении; / определяется по обобщенной диаграмме коэфициентов активности.
Результаты вычислений приведены ниже:
№№ граф Общее давление, атм Мольная доля в жидкости Мольная доля сл в паре Активности Ка
С2Н5ОН Н2О CjH6OH с2н4 н2о
7 82,6 0,014 0,956 0,475 0,813 37,3 0,956
8 196,9 0,060 0,918 0,380 3,63 67,3 0,918 0,0589
9 264,2 0,084 0,881 0,250 5,29 59,1 0,881 0,101
10 129,7 0,0445 0,937 0Д26 2,63 27,3 0,937 0,103
Средняя Ка~ 0,071.
Совпадение между вычисленной Ка и экспериментальными значениями настолько хорошо, насколько этого можно было бы пожелать, учитывая допущение о постоянстве Д/7 и то обстоятельство, что сами экспериментальные значения не очень хорошо совпадают друг с другом.
Пример 6. Вычислить по термическим данным константу равновесия для синтеза муравьиной кислоты из СО и воды при наличии жидкой фазы, если температура равна 156° С и если принять следующие стандартные состояния:
Для СО — газ при 1 атм.
Для Н3О — чистая жидкость.
Для муравьиной кислоты — 1-мольный водный раствор.
Мы решили дать добавочный пример, потому что он включает фактор, не вошедший в предыдущий, а именно — полимеризацию пара. Рассматриваемая реакция представляется уравнением
СО Н2О(ж) = НСООН(водн.) (а)
Д-^298 дла реакции СОН2О(ж] = НСООН(ж) (б)
37 Химическая термодинамика
578
XI. Равновесие химических реакций
вычисляется из изобарных потенциалов образования каждого чистого компонента на основании третьего закона. Известно, что:
для образования СО — — 32 700 кал[моль,
для образования Н2О (ж) = — 56 560,
^^298 для образования НСООН (ж) = — 85 300,
Д^98 для реакции (б) =—3960.
Для того чтобы рассчитать реакцию (а), необходимо располагать Изменением изобарного потенциала для реакции
НСООН (ж) — НСООН (водн.); (в)
его можно вычислить из данных по давлениям насыщенного пара чистой муравьиной кислоты и ее водных растворов, учитывая следующее равновесие, существующее в паровой фазе:
(НСООН),, — 2НСООН. (г)
Рамспергер и Портер [193] исследовали "то равновесие при 25° С и нашли парциальное давление мономолекулярного пара над чистой жидкостью и над одномолярным водным раствором, соответственно равными 1,17Х10-2л/ил< и 7.89Х10-6 атм. Согласно этому, Ди для реакции (3) будет равно
Следовательно, Д|1 для (а) равно 3960—2960 = 1000;
ДЯ для (а) = —4920;
последняя величина определяется из следующих данных:
ДЯ образования жидкой муравьиной кислоты =—99750
ДЯ образования жидкой воды =—68310
ДЯ образования СО = —26617
ДЯ растворения НСООН = — 100.
Если принять ДЯ постоянной, то Ка при 156° С = 1,45 X Ю-2. Бранч [26] исследовал эту реакцию экспериментально, и его результаты привели к значениюКа— 1,18X Ю-2 при 156°. Это является прекрасным совпадением, особенно если учесть, что ошибка в AZ образования НСООН в О,17°/о уже достаточна, чтобы вызвать это различие.
Если нужно определить константу равновесия для реакции в парообразной фазе между веществами, часть которых при стандартной температуре (25° С) является жидкими, то для точного вычисления необходимы данные по скрытым теплотам парообразования при 25° С. Подобные данные обычно не имеются в распоряжении, но можно вычислить скрытую теплоту по давлению насыщенного пара методом, рассмотренным в гл. VI, или приближенно определить ее одним из методов, рассмотренных в гл. IX. Во многих случаях достаточно будет принять, что скрытая теплота парообразования прн 25°С равна таковой при нормальной точке кипения, так как эта теплота обычно чаще имеется в распоряжении. Часто случается и так, что затрата времени на отыскание точного значения
Вычисление констант равновесия на основании термических данных 579
не будет оправдываться, потому что приближенные величины будут служить цели так же хорошо, как и точные. Действительно, при вычислении константы равновесия всегда следует правильно оценивать пользу, которую могут принести точные расчеты. Часто это может предотвратить затраты очень большого количества времени на излишние улучшения. Следующий пример иллюстрирует некоторые из этих соображений.
Пример 7. Требуется определить константу равновесия реакции сгН<+С6Н6(г) = С2Н5С6Н5 (г) для выяснения, осуществима ли данная реакция в газовой фазе при доступных условиях.
Следующие давные взяты из различных источников:
Вещество А-^298 ^^298
Этилбензол (ж) .... Бензол (ж) Этилен (г) 26 300 29 400 15 820 — 5 070 11630 11 975
Скрытые теплоты парообразования можно определить несколькими методами, рассмотренными в гл. IX; используя уравнение Кистяковского
— = 8,75-j-4,5711g Тв, (35, гл. IX)
получим следующие значения:
Вещество Нормальная точка кипения, Тв, ° К Мольная теплота парообразования
Бензол 353,2 7200
Этилбензол . . . 408 8450
Для вычисления скрытой теплоты при 25° воспользуемся методом Ватсона [252]. Применение этого метода требует знания критических температур, так же как и значения L при одной температуре. Для бензола Ткр — 561,6° К. Для этилбензола Ткр, определенная по методу Ватсона [252], оказывается равной 624° К-
Вычисленные значения L при 25° С равны: для этилбензола 9800 и для бензола 8000 ☆).
Давление насыщенного пара бензола при 25° С = 0,1235 атм..
Данные по давлению пара бензола очень скудны и не согласованы друг с другом, но по данным, приведенным в Интернациональных критических таблицах, при 25° оно определяется примерно равным 20 мм.
☆ ) Колосовский и Меженин [329] дают для скрытой теплоты парообразования этилбензола при 26° С значение 9900. Фиок, Джиннинг и Холтон [79J дают для бензола 8050.
37*
580
XI. Равновесие химических реакций
Для процесса
С6Н5С2НВ (ж) —► CgHjCaHg (г, 1 атм),
Д7298 = RTIn = 2160 кал/мом.
ли
AZ298 образования С4НВС2НВ (г) = 28 460.
Подобно этому,
Д2298 для С6Н6 — 30 640.
Наконец, Д2298 для рассматриваемой реакции
= 28 460 — 15 820 — 30 640 = — 18 000,
и так как
то
1,6-1013.
На осноиании значений Д/У, приведенных в предыдущей таблице, и значений для скрытых теплот парообразования находим, что для реакции равна—26900.
Принимая Д/У не зависящим от температуры, имеем
8 KPl 4,575 ^71 Тгг
Тх = 298,2, Xpi = 1,6 X 101»,
Т2 = 523,2 (250° С); ХРз = 5 X Ю*. .
На основании результатов расчета можно заключить, что, по крайней мере, при любой температуре до 250° С реакция имеет тенденцию итти практически полностью. Более строгий расчет на основании точных данных, вероятно, не приведет к изменению константы в такой степени, чтобы повлиять на это заключение. Даже стократное изменение константы равновесия как в сторону ее увеличения, так и в сторону ее уменьшения не может привести к существенной разнице.
Приближенный метод Нернста. Метод, разработанный Нерн-стом для предсказания равновесия, основывается на так называемой тепловой теореме Нернста (которая позже развилась в третий закон термодинамики) и на использовании так называемых химических постоянных. Этот метод был впоследствии почти полностью вытеснен более простыми и более логическими методами, использующими третий закон. Приближенное уравнение, данное Нернстом, таково:
ig 1 ’75 lg7+^пС’
С названа Нернстом условной химической постоянной в отличие от истинной химической постоянной, применяемой при более строгой трактовке, широко употреблявшейся нами ранее для предсказания химических равновесий. Но от последнего уравнения нельзя ждать большего, чем определение порядка величины Кр, и его применение следует всюду, где возможно заменять более надежными методами,
Равновесное превращение
581
изложенными и иллюстрированными выше. Поскольку рассматриваемый метод Нернста дает достаточно простой путь для нахождения приближенной константы равновесия, его применение в некоторых случаях все же можно оправдать. В силу этой причины рассмотрим один пример, иллюстрирующий его применение.
Пример 8. Вычислить, пользуясь приближенным методом Нернста, константу равновесия реакции синтеза метанола при 250° С и 1 атм. &Н (принимается постоянным и равным значению при 25° С) = — 22 000 (приближенно). С (химическая постоянная) для Н2=1,6, для СН3ОН = 3,5 и для СО = 3,5 й). Ел = — 2, SnC= 3,5 — 3,5 — 2 X 1,6 = — 3,2.
. „ 22 000 _ „„
’g Кр = 4^757 — 3,50 1g Г — 3,2.
Т= 523,2, /Ср = 3,2Х Ю"4-
Из решения примера 4 величина, основанная на третьем законе и на точных экспериментальных данных, равна примерно 2-10~й. Совпадение будет достаточно хорошим, если речь идет только об определении того, осуществима ли данная реакция при данных условиях или неосуществима. На самом деле величина, полученная по приближенному методу Нернста, гораздо ближе к действительной величине, чем многие другие, полученные на основании третьего закона лишь несколько лет тому назад.
Необходимо признать, что приближение Нернста приводит во многих случаях к неожиданно хорошим результатам. Основным затруднением является то, что нельзя сказать, насколько можно доверять найденным значениям в каком-нибудь отдельном случае. В настоящее время мы рекомендовали бы применение этого приближения только в тех случаях, когда данные, позволяющие пользоваться более точными методами, отсутствуют85.
РАВНОВЕСНОЕ ПРЕВРАЩЕНИЕ
С точки зрения инженера-химика измерение или предсказание константы равновесия представляет лишь ступень к решению более важной задачи — определению того, как далеко может итти реакция
&) Значения некоторых условных химических постоянных даются в различных физико-химических таблицах. Для их вычисления могут оказаться полезными также следующие эмпирические уравнения:
С = 0,14^,
1в
С=\,1 \%Ркр>
где L — скрытая теплота парообразования, кал/моль',
Тв — нормальная точка кипения, °К;
ркр — критическое давление, атм.
582
XI. Равновесие химических реакций
в данных условиях. Еще раз подчеркнем, что мы будем касаться вопроса максимально возможной степени проведения реакции, а не того, как далеко она будет итти в действительности. Максимально возможное превращение точно определяется из термодинамических соображений; но действительное изменение включает рассмотрение вопросов о катализаторе, энергии активации и о других факторах, относящихся к области химической кинетики и совершенно выпадающих из области термодинамики.
Гомогенная газовая реакция. Сначала рассмотрим гомогенную газовую реакцию, протекающую или в токе газа, или в замкнутой системе и представленную уравнением
аА-\-ЬВ-\- ...
Пусть £—доля любого данного реагента, например А, которая подвергается превращению, когда под влиянием данных условий в системе устанавливается равновесие. Пусть NA, NB, NL, NM и т. д. будут числом молей различных реагентов, присутствующих в исходной смеси. Допустим, что в дополнение к этим веществам исходный газ содержит Nt молей газа, который инертен по отношению к рассматриваемым реагентам.
г, Ь I т
Пусть rB, rL, гм и т. д. соответственно равны —, —, — ит. д.
При достижении равновесия различные составляющие будут присутствовать в следующих количествах:
NA(l—С) молей A = N*a,
NB — rBNмолей B~N*B, NL + rLN£ молей £ = л1-NM+rMNA' М0ЛеЙ М=^М-Nj молей инертного газа.
Пусть SV—общее число молей в равновесной смеси. Мольная доля каждого компонента в равновесной смеси равна числу молей данного компонента, деленному иа T.N. Допустим, что реагенты образуют идеальную газовую смесь; тогда, применяя уравнения (5) и (254, гл. IV), найдем, что
(27) VOWj)» ...
Каждый член в скобках представляет число молей данного газа, присутствующего в равновесной смеси. В частном случае идеальных газов К^== I. Полученное уравнение для каждого отдельного ряда условий можно решить относительно С, т. е. относительно превращенной доли А.
Равновесное превращение
583
Пример 9. Газовую смесь, содержащую 6О°/о Н2, 20% Ы2 и 20°/о инертного газа, следует пропустить над соответствующим катализатором для получения аммиака. Чему будет равно максимальное содержание водорода, превращенного в NHS, и содержание последнего в выходящем газе при однократном прохождении смеси над катализатором, если давление равно 50 атм и конечная температура 400° С? При решении принять идеальность газов.
Подлежит рассмотрению реакция
3/2H2 + l/2N2 = NHs, в = 3/2, *=1/2, 1=1, гв=113, rL=2(K 2я=1 —1/2 —3/2 = —1.
Кр, при 400° С (из уравнения на стр. 563) =0,0125. Возьмем за основу 100 молей исходной газовой смеси; пусть t — доля превращенного водорода. Тогда в равновесной смеси
Н2 = 60 (1 — С) молей
N2 = 20(l- 0 .
ЫН, = 4О4 инертного газа = 20.
Общее число молей XV= 100 — 40С.
Подстановка этих значений в уравнение (27) дает
40t
5 i60(i-orM20(i-№-<100 - 40 Э=50 X °’0125-
Это уравнение лучше всего решается методом подбора. Пусть 8—разность между обеими частями уравнения. Тогда 8 = 0 для правильного значения С.
Подбираемые значения:
t = 0,100 8 = 0,399 С = 0,200 8= 0,075 С = 0,227 8 = — 0,035 С = 0,218 8 = 4-0,005 X. = 0,220 8 = — 0,006 С = 0,219 8= 0,000
Следовательно, в аммиак превращается 21,9®/о водорода, ж процентное содержание NH3 в выходящем газе равно
(40С) 100 100 — 40С
= 9,6%.
Пример 10. Г азовую смесь, содержащую 25% СО, 55% Н2 и 20% инертного газа (мольные проценты), следует использовать для синтеза метанола. Какова степень превращения окиси углерода при давлении в 300 атм и температуре 350° С, если газы, покидая контактный аппарат, находятся в химическом равновесии по отношению к реакции
CO + 2H2 = CHsOH
Примем, что равновесная смесь является идеальней.
584
XI. Равновесие химических реакций
Из уравнения (8) в примере 4 Kf — 4,9 ХЮ-5- По рис. 69, ^ = 0,35. Примем за основу 100 молей начальной газовой смеси.
л£о = 25(1 — С),
^ = 55 — 50:, ^сн,он = 2^> &V=100 —50С
Подставляя эти значения в уравнение (27), получаем
С(100 — 50<Г)2 _ 4,9X10-4300)2 Ц—С)(55—50С)2- 0,35
.откуда £<=0,610, т. е. будет превращаться около 60% СО. Если бы газы считались идеальными, то С было бы равно 0,441.
Реакция в газовой и жидкой фазах. Проблема нахождения равновесного состава жидкого раствора, получающегося при реакции, когда присутствуют как газовая, так и жидкая фазы, является более трудной. Обычно ее можно решить только после того, как будет сделано большое количество упрощающих допущений. Сначала мы укажем метод решения в общей форме, а затем иллюстрируем его на частных случаях.
Рассмотрим реакцию
аА-\-ЬВ = сС
»
и допустим, что мы хотим вычислить равновесную концентрацию С в жидкой фазе при данных давлении и температуре. Примем, что в качестве стандартного состояния каждого вещества выбрано состояние чистого газа. Для любой данной температуры можно записать следующие уравнения:
__Г /Ооч
(«А)в(ав)*~ °* ( '
оа=&(Р>Уа> У в* У<± (29)
ав = ?2 ^Р’ У А' У&> У(У> (30)
“с^ЦР'Уа’Ув’УсУ (31)
Все они являются уравнениями для активности в газовой фазе. Подобные же уравнения можно записать для жидкой фазы, например,
<ft=4>’t(P, ХА, Хд, Хс).
Поскольку мы должны иметь фазовое равновесие, то ц;=ц,-.
и для данного стандартного состояния для любого вещества
а. = а,г
Равновесное превращение
585
Следовательно, мы имеем следующие уравнения:
<fi (Р> У А’ У В’ Ус) = (Р> ХА> ХВ’ ХД <32>
?2 (Р> У А’ У В’ Ус) = <?2 (Р’ ХА’ ХВ’ Хс)’ (33)
®3 (Р’ У А’ у В, Ус) = ?з ха- ХВ’ Хс)- (34)
Очевидно также, что справедливы следующие уравнения:
Ха + Хв + ХС=1’ (35)
-Уд+.Ув+Л^1- (36)
Таким образом, мы имеем минимум девять уравнений и девять неизвестных, и если известны Ка и различные функции, обозначенные через <р, то решение этих уравнений является возможным. Оно будет очень сложно для тройной системы, даже если мы имеем необходимые данные для представления их в графической форме.
Если считать каждую фазу идеальным раствором, то уравнения (29) — (34) значительно упрощаются, и числовое решение становится практически возможным.
Так, например, уравнение (29) переходит в следующее:
ал=/л = ?(р)^л; (37)
то же самое справедливо для активностей других компонентов.
tp(p) является летучестью чистого А при данных давлении и температуре и определяется с помощью ранее рассмотренных методов. Уравнение (32) переходит в
Уа?а = хаЪ' (38)
где /а и соответственно летучести чистого А при данной температуре и общем давлении в виде жидкости и в виде пара. При применении этого уравнения, как было выяснено в гл.IV, приходится сталкиваться с необходимостью определения летучести в нестабильном состоянии.
Уравнения еще более. упрощаются, если принять, что газы идеальны. При этом допущении уравнение (29) переходит в
аА=/А=УАР’ (39)
и уравнение (32) в
УаР=хаРа- (40)
Применение уравнений (28), (35), (36), (39) и (40) дает _ (г Рс\с
KjT1- = ___________,
|1 ~xcPf — т <хс) J в[? (хс}\ь где
<о(х \Ра — Р — хс^Ра—РсГ\
586
XI. Равновесие химических реакций
Это уравнение легко решить для хс, если известны давления пара компонентов при рассматриваемой температуре.
Пример 11. Определить максимальный мольный процент этилового спирта в водном растворе, получающемся при взаимодействии этилена и жидкой воды при 254° С в присутствии соответствующего катализатора при условии, что общее давление поддерживается постоянным посредством соединения реакционного сосуда с источником этилена при 100 атм. Примем, что концентрация катализатора достаточно мала для того, чтобы он не действовал на активность реагирующих веществ й).
Сначала следует подсчитать Ка для рассматриваемой реакции, а это включает выбор стандартных состояний. Примем в качестве такового для каждого вещества состояние чистого газа при 1 атм и допустим, что газы гтпи атгч!# поптоиш» полпглт/’л пт.ит.т*<тл I/f a ттаиичт rrnuooirauut.1T
на стр. 575, и из скрытой теплоты парообразования этилового спирта, равной 10 120 кал1моль [211], легко найти, что для реакция
С2Н4 -|- Н2О (г) = С2Н5ОН (г) при 25° С ДЯ = — 11 000,
AS = — 30,1, AZ = — 11 000 + 298,2 X 30,1 = — 2030.
Из этого значения и из уравнений теплоемкости получаем, следуя Парксу [187]:
AZ® = — 9674 + 6,43 Т In Т — 0,00665 Г2 — 9,01 Г, Г = 527,2, Д£0 = 4980, Ка = 0,0085.
Для рассматриваемого случая нельзя непосредственно применить уравнение (41), потому что температура 254° С выше критической температуры как этилового спирта, так и этилена. Поскольку она значительно выше критической температуры этилена, мы будем считать, что можно пренебречь концентрацией CjH4 в жидкой фазе или что х = 0. Это приводит уравнение (41) к
К — ______________хсРс______________ ,п
а [р— ХСРС — (1— *с)Рв](1— хс}рв'
рс — давление пара этилового спирта при 527,2° К — можно найти посредством весьма незначительной экстраполяции кривой давления насыщенного пара, относящейся к области температур выше критической точки (Г =516,3; р = 61,3 атм). При использовании линейной зависимости р от 1/Г (см. гл. VI) экстраполяция дает
Рс = 76 атм.
Решая уравнение (1) методом подбора, получаем
хс= 0,195.
Экспериментальные результаты Джиллилэнда и сотрудников, данные на стр. 577, указывают на значительно меньшую концентрацию спирта. Возможно, что допущение идеального раствора для спирта вносит значительную ошибку. Этого можно частично избежать, выбрав для него другое стандартное состояние, а именно — состояние разбавленного раствора, для которого можно считать справедливым закон Генри. Приняв в качестве стандартного состояния для воды состояние чистой жидкой воды, имеем, согласно данным на стр. 156,
ЛГв = 0,014
ас
(«д) («в) ’
й) Эта задача взята из статьи Доджа [64].
Равновесное превращение
587
Опять принимая идеальность газов, отсутствие CjH4 в жидкой фазе и считая жидкий раствор идеальным по отношению к воде, получим
(Зависимость между т и х вытекает из их определений.)
aA=J'AP>
о-в = хв = 1 — ХС' Уа—^~Ув~Ус z=l~?BPB—fcx р
= 1_n~^d£g_fe Р с
k является константой закона Генри для раствора этилового спирта. Следовательно,
55,5хс/(1 —хс)_______= о 014
О — (1 — хс) рв — pkxc\ (1 — хс)
При отсутствии данных, необходимых для определения k, будем считать, что давление пара спирта над раствором дается законом идеальных растворов, т. е.
k=P-£.
Р
Отметим, что хотя это и является тем же самым допущением, которое было сделано при предыдущем решении примера, оно теперь входит только в поправочный член и не оказывает большого влияния на значение хс.
Решая методом подбора, находим, что хс — 0,015.
Этот результат прекрасно совпадает с ранее указанными экспериментальными данными. Конечно, оба метода вычисления включают большое количество приближений и могут дать результаты, только определяющие порядок величины.
Сравнение изотермической и адиабатной реакций. Измерение констант равновесия реакции всегда производится в условиях, приближающихся к изотермическим. Когда для оценки возможности превращения пользуются равновесными данными, то иногда забывают то обстоятельство, что все реакции сопровождаются тепловыми эффектами. Если поддерживается изотермический режим, то теплота должна либо выделяться, либо поглощаться. При экспериментировании в небольшом масштабе в лаборатории, относительно просто осуществить рассеивание или поглощение тепла; поэтому обеспечить изо-термичность процесса нетрудно. Если же реакция проводится в значительно большем масштабе, то подвод или отвод тепла становится серьезной проблемой, и реакции во многих случаях проводятся в условиях, более близких к адиабатным, чем к изотермическим. Поскольку дело касается вычисления равновесного превращения, не имеет
588
XI. Равновесие химических реакций
значения, какие термические условия существовали при проведении реакции, если конечное равновесное состояние устанавливается при некоторой определенной и известной температуре.
При применении констант равновесия следует твердо помнить, что начальное состояние реагирующих веществ может по температуре совершенно отличаться от конечного состояния продуктов. Имеется два предельных случая: 1) изотермическая реакция и 2) адиабатная реакция; все действительные случаи должны лежать между этими пределами, но для целей вычисления обычно принимается достаточное приближение к тому или другому пределу. В случае газовых реакций, ЛЛА/УРцип Г7ГЧТ1 тгпптппА nauuu irrxu'T'jtz'T’uг»г«г» t/ота туизяТаПЯ ттажжа
»-»%. V'-' vimv j 11U1 IIC/HIUIIIUUIV nu 1 U»4*k/U 1 v w , '-*•* V>*»1**
подвода и отвода тепла является особенно трудной. В больших аппаратах подобные реакции обычно проводят в условиях, сравнительно близк х к адиабатному процессу.
Важность изложенных соображений иллюстрируется следующим примером.
Пример 12. Газ, состоящий из 2 молей водорода на 1 моль окиси углерода и не содержащий инертных газов, входит в контактный аппарат для получения метанола при 275° и 200 атм. Считать, что происходит только одна реакция:
СО2Н2 = СН3ОН.
Чему равен максимально возможный процент превращения СО в СН3ОН, 1) если реакция адиабатная и 2) если реакция изотермическая?
1. Адиабатный процесс. Примем, что газы, покидающие катализатор, находятся в химическом равновесии; это допущение позволяет сразу установить зависимость между степенью превращения и температурой. Мы будем выражать эту зависимость общим уравнением
С = *(Л,
(а)
где С — доля превращенного СО.
Т — абсолютная температура газов, покидающих катализатор. Форма функции известна из уже рассмотренных соображений, но имеется два неизвестных, и для решения требуется другое уравнение, связывающее С и Т.
Оно получается при применении первого закона, выраженного уравнением (105, гл. IX), которое для этого частного случая переходит в
Sp Н — Sp Н+ г д НТо = 0.
Этому уравнению можно придать вид
С=Т2(Г).
Число молей продуктов реакции = (1—С) молей СО
(б)
(в)
-1-2(1—С) молей Н2 Z молей СН3ОН.
Для решения используем теплоемкости, приведенные в примере 1. Они даны при условии, что влиянием давления на Ср можно пренебречь.
Приняв в качестве основной температуру, при которой реагирующие вещества входят в реакционную зону, именно 548,2° К, можем написать, что
Spf/=0.
Равновесное превращение
589
Тогда г т
2р7/ = (1—С) Г (6,89 + 0,00038 Г) dT = 2(1 — С) f (6,65 -)- 0,00070 7) dT 4-
548,2 548,2
Т
4-t (2,0 4- 0,03 7) dT. (г)
548,2
Интегрирование и преобразование дают
Sp /7= С (0,0141 Т2 — 18,19 Т 4- 5726) 4- 0,00089 Т2 4- 20,19 Т — 11322. (д)
Д/7 для реакции при 275° С получается из уравнения (99, гл. IX) при использовании данных, приведенных в примерах 1 и 4:
Д//= — 17530 — 18,19 7'4-0,0141 Т2,
Д//548,2 = — 23250 (на моль),
или, для реакции с С молями СО, Д/7 = — 23 250 С. Подставляя это значение в уравнение (б) и решая последнее относительно получаем
_ —0,00089 72 — 20,19 7Д- 11 322
0,0141 72—18,19 7— 17 524 ‘ (е'
Это уравнение является уравнением (в) в явной форме. Теперь найдем, какой вид имеет уравнение (а).
Если принять, что равновесная смесь представляет идеальный раствор, то, пользуясь уравнением (27), получим
£(3—2Г)2 _ pWf 4(1-О3
(ж)
Ку и /<4 являются функциями температуры, и поэтому уравнение (ж) будет явной формой уравнения (а).
Ау дается уравнением (8) примера 4, а дается как функция Т на рис. 111. Найденные уравнения для зависимости j от Т, т. е. уравнения (е) и (ж), легко решаются совместно методом подбора; прн решении получаются следующие значения:
7= 702,2° К = 429° С, t = 0,140.
2. Изотермический процесс.
7=548,2; /Су =7,25X10-* и = 0,32.
Решая уравнение (ж), получаем £ = 0,84.
Различие между двумя этими случаями разительно. Даже незначительное охлаждение реактора вызывает существенное увеличение степени превращения. Другим средством, которое применяется для увеличения степени превращения, является проведение реакции в несколько стадий с промежуточным охлаждением. Это особенно широко применяется при контактном способе производства серной кислоты.
Автотермические реакции. Теплоту, выделяемую в результате химической реакции, можно использовать для того, чтобы сделать
590
XI. Равновесие химических реакций
реакцию автотермической, т. е. саморегулируемой при определенных термических условиях. Хотя, например, в случае синтеза метанола в начале процесса следует подвести теплоту для того, чтобы повысить температуру реагирующих веществ до необходимого уровня, однако, как только реакция начнется, продукты будут иметь доста
точно высокую температуру, чтобы при использовании соответствующего теплообменника обеспечить нагревание реагирующих веществ.
В связи с автотермическими реакциями заслуживает краткого рассмотрения другой интересный вопрос: он заключается в термической ।----------------------------- стабильности этих реакций и в воз-
можности их регулировки. Рассмотрим схематически представленную на рис. 112 экзотермическую реакцию, происходящую в замкнутом пространстве, которая посредством теплообменника становится саморегулирующейся; допустим, что реакционная система далека от состояния равновесия. Пусть Нх представляет общую энтальпию реагирующих ве-
Рис. 112. Схема автотермического ществ, вступающих в систему, а процесса. /72— суммарную энтальпию продук-
тов, покидающих систему. Есди теплообменник системы был бы идеальным, то продукты реакции снова охлаждались бы до начальной температуры, и Нх — Н2 была бы равна теплоте реакции при начальной* температуре реагирующих веществ перед предварительным нагревом их; поэтому Q = HX — Нъ, т. е. система в целом должна была бы выделять в окружающую среду теплоту, равную теплоте реакции. В действительности, конечно, продукты будут покидать систему, имея большую энтальпию, чем Нг, и Q будет меньше, чем теплота реакции; но вышеприведенное соотношение должно быть действительно для всех систем, находя-
щихся в устойчивом состоянии.
Теперь предположим, что по причине некоторого изменения условий скорость реакции возрастает и возникает стремление к большей степени превращения. Принимая, что температура конечных продуктов, покидающих систему, остается неизменной, получим возрастание (//] — Н2), а поэтому н Q для сохранения баланса должно увеличиться. Это означает, что температура в системе должна повышаться, что явится причиной дальнейшего повышения скорости реакции, а это, в свою очередь, вызовет большую степень превращения, и так до тех пор, пока не будет достигнуто равновесие. Другими словами, подобная система будет термически неустойчивой н выйдет из-под контроля. Это может случиться только тогда, когда система вначале далека от равновесного состояния. Если же она находится в состоянии равновесия, то стремление к возрастанию температуры нейтрализуется
Равновесное превращение
591
в результате меньшего превращения. Другими словами, в равновесии автотермнчески реагирующая система будет и саморегулирующейся.
Примером практического использования этих соображений может служить конвертор в синтезе метанола, где, кроме реакции
СО4-2Н2 = СН3ОН,
могут проходить и другие реакции, в особенности реакция
СО + ЗН2 == СН4 + Н2О.
Первая реакция приближается к состоянию равновесия, и так как состояние равновесия очень быстро изменяется с температурой, конвертор по температуре относительно стабилен. Вторая реакция обычно проходит лишь в слабой степени; но если бы были созданы благоприятные обстоятельства путем некоторого изменения условий, то она могла бы выйти из-под контроля и стать причиной чрезмерно высокой температуры в конверторе вследствие того, что до тех пор, пока не будет достигнута значительно более высокая температура, уравновешивающее действие химического равновесия не остановит реакции.
Максимальный выход продуктов реакции. Иногда возникает вопрос, в каких соотношениях следует смешивать реагирующие вещества, чтобы добиться максимального выхода данного продукта. Например, может возникнуть вопрос об отношении О2 к N2, обеспечивающем максимальный выход NO при реакции
N2 -j- О2 = 2NO,
другими словами, о максимальной концентрации NO в газах, получающихся при равновесии. (Отметим, что это не то же самое, что максимальный процент превращения.) Дадим ответ для более общего случая, рассматривая гомогенную реакцию
aA-\-bB = lL-\-mM.
Примем, что исходная смесь реагирующих веществ содержит только А и В и что имеется гв молей В на моль А.
Если взят 1 моль А, то в равновесной смеси будет
NA молей А,
гв — (1 — IVА) молей В,
^- (1 — IVА) молей L,
~ —NA) молей М.
592
XI. Равновесие химических реакций
Пусть N—общее число молей,
' х— максимальная мольная доля М в равновесной смеем.
Nx^(\-Na},
ИЛИ UA = 1— — Nx. m
и N= 14-^x ’
где k = _!L + A_±_i. m 1 m m
С помощью этих уравнений можно выразить мольную долю каждого компонента в равновесной смеси через х. Допущение идеальности газов и подстановка в уравнение (5) дают
/14- fex а Т° Ггй (1 4-М & 1 *= К> (42)
\14-Гв т J L 14-/В т J
Для того чтобы найти гв, когда х становится максимальным, необходимо лишь воспользоваться критерием
-^- = 0. (43)
Диференцируя уравнение (42) и объединяя члены, получаем
дх__ K^+kx^+r^bX^X^-aX^X^ (44)
дгВ~ Р
F представляет сложное алгебраическое выражение, детализировать которое нет необходимости, а
v 14- kx а
Л, = -------х
1 14-гв т
rB(l+M Ъ
Л*~ 1+гв ~~ тХ '
Применяя уравнение (43), получаем
=аХ%~х или
а Хх ‘
Подставляя значения Xt и Х2 и решая по частям, получаем
Лв=|- (45)
Одновременно протекающие реакции
593
Следовательно, для получения максимальной концентрации продуктов в равновесной смеси реагирующие вещества должны быть взяты в стехиометрическом отношении. Хотя мы доказали это только для простого случая двух реагирующих веществ, однако можно утверждать, что результат остается тем же самым для любого числа реагентов.
Следует отметить,* что этот вывод применим не только к случаю идеальных газов, но и к любому случаю, когда можно принять, что смесь является идеальным раствором.
ОДНОВРЕМЕННО ПРОТЕКАЮЩИЕ РЕАКЦИИ
Во всех предыдущих анализах химического равновесия внимание сосредоточивалось на одной химической реакции. На практике чаще имеют дело с двумя или несколькими одновременно проходящими реакциями. Это особенно важно для органической химии, в которой при данных исходных веществах возможны многие различные реакции. Например, рассматривая лишь два реагирующих вещества — окись углерода и водород, — можно написать множество реакций, из которых типичными являются следующие:
СО-j- Н2 = НСОН
СО-j- 2Н2 = СН3ОН со4-зн2=сн44-н2о 2СО 4- 5Н2 = С2Н6 4- 2Н2О ЗСО 4- 6Н2 = С3Н7ОН 4- 2Н2О
(46)
(47)
(48)
(49)
(50)
Очевидно, реакции, подобные (50), едва ли протекают непосредственно, так как требуют одновременного столкновения девяти молекул, что является крайне невероятным случаем. Несомненно, каждая из этих реакций, кроме первой, происходит в несколько стадий, но это уже вопрос механизма, на который законы термодинамики не проливают света.
Когда говорят о равновесии в реакции, подобной (50), которая является результатом нескольких превращений, то полагают, что концентрации четырех реагентов удовлетворяют закону действующих масс, т. е. равновесному соотношению даже в том случае, если присутствуют и другие реагенты. В общем нельзя рассчитать максимального выхода любого соединения, получающегося из СО и Н2 при допущении, что реакция, которая может происходить между этими соединениями и данными реагирующими веществами, является единственной. Строго говоря, следовало бы рассматривать одновременно равновесия для всех возможных реакций, происходящих между всеми реагирующими веществами. Это, конечно, практически невозможно, но во многих случаях мы можем уменьшить число реакций, подлежащих рассмотрению.
38 Химическая термодинамика
594
XI. Равновесие химических реакций
Например, при рассмотрении равновесия реакции синтеза метанола мы поступаем так, как будто происходит только реакция (47). Эта реакция проходит в две стадии:
со+н2=нсон . нсон4-н2=сн3он
(51)
(52)
и возможны многие побочные реакции. Наши вычисления равновесия метанола включают два подразумеваемых допущения:
1. Все побочные реакции проходят с ничтожной скоростью по сравнению со скоростью рассматриваемой реакции или ео скоростью промежуточных реакций.
2. Все промежуточные продукты так неустойчивы, что их концентрации при равновесии ничтожны по сравнению с концентрациями основных продуктов.
Эти допущения подразумеваются при всех вычислениях химических равновесий; важно осознать это обстоятельство, так как во многих случаях они недостаточно хорошо отвечают действительности. В случае метанольного равновесия они представляются хорошо обоснованными, так как формальдегид при принятых условиях весьма неустойчив и так как был найден катализатор, способствующий практически только метанольной реакции. В случае реакции, подобной (50) и многим другим, включающим ряд промежуточных стадий, вероятно, ни одно из подобных допущений не будет справедливо, и мы не можем ожидать обеспечения высокого выхода продукта, даже если изменение изобарного потенциала для реакции будет указывать на благоприятное значение константы равновесия.
Так как изменение изобарного потенциала суммарной реакции равно сумме изменений изобарных потенциалов отдельных ее стадий, то из этого следует, что константа равновесия реакции равна произведению констант составляющих реакций. Так, считая реакцию (47) проходящей в две стадии — (51) и (52), можно написать, что
^(47)=^(51)Х^,(52).
Предположим, исключительно ради иллюстрации, что Кр (47) при доступной температуре составляет величину порядка 10~3. При этом /Гр(51) и /^(52) могут иметь самые разнообразные значения и все же удовлетворять условию, что их произведение должно быть равно 10~3.
Например,
а) 10-3= 10-10Х10’.
б) 10-3= 10-1Х10-2, в) 10-з= 107Х Ю-1°.
Одновременно протекающие реакции
595
а) соответствует случаю, когда равновесные концентрации промежуточных продуктов (в данном случае формальдегида) очень малы, и правильный результат получается при рассмотрении только общей реакции (синтез метанола). В случае б) имелось бы значительное количество промежуточных веществ, и нельзя было бы их игнорировать. В случае в) в смеси присутствовали бы главным образом промежуточные продукты и ничтожное количество основного продукта.
Реакцию, подобную (50), можно представить состоящей нз шести промежуточных реакций. Из только что рассмотренного видно, что весьма благоприятное значение константы равновесия для реакции (50) не обязательно предполагает большой выход пропилового спирта при равновесии; в действительности выход может быть небольшим.
Количественные зависимости. Чтобы показать, как следует рассчитывать равновесие для ряда одновременно проходящих реакций, рассмотрим простой случай двух реакций с общим реагирующим веществом и с общим продуктом, причем продукт первой реакции является реагирующим веществом для второй реакции, т. е. в общем виде процесс будет следующим:
aA-\~bB = cC-\-dD, (53)
cC-\-b'B = eE-\-d'D. (54)
Мы исходим из начальной смеси, состоящей из 1 моля А и гв молей В.
Пусть Na — число молей А, реагирующих по уравнению (53), и Nc— число молей С, реагирующих по уравнению (54).
При равновесии по отношению к обеим реакциям будут присутствовать:
(1 — Na) молей А,
(г„ — — N. — — молей В, \ в а А с J (~ N. — молей С, \а А с)
(£ Nл _L - мУ молей D, \а а 1 с с)
— Nr молей Е-, е с
общее число молей
N = 1 + г + N ' + + N .
’ о 1 л л 1 с с
Допуская идеальность газов, имеем при равновесии для реакции (53)
/ е \с / d d' \d
{-Na-Nc} {-Na + -Nc}
= ~------(55)
°-NA-±Nc]
2Я*
596
XI. Равновесие химических реакций
и, при равновесии для реакции (54),
К =_______Xе ! \ а с >______к-Ъ.
лр" — (с \cf ь ь' х*v ; {^nA-Nc) IrB—^XA-^Nc)
(56)
оба эти уравнения должны удовлетворяться одновременно.
Пример 13. Какой максимальный процент метанола можно ожидать в газах, покидающих контактный аппарат, если смесь из 2 молей Н, и 1 моля СО проходит над катализатором, который позволяет достигнуть равновесия по отношению к двум реакциям:
СО -f- 2Hj = CI I3OH (г)
СН3ОН (г) + Н3 = СН4+Н2О (г)
(а)
(б)
и если 7’=600°К и р = 100 атм.
Для стандартного изменения изобарного потенциала реакции (а) можно воспользоваться уравнением, найденным при решении примера 4. Это дает для 600°К
Кр = 8,3X10-».
Для нахождения константы равновесия реакции (б) будем пользоваться уравнением Истмена [72] для температурной зависимости изобарного потенциала реакций образования СО и Н2О и уравнением Томаса, Эглоффа и Моррела [244] для реакции образования СН4 совместно с уравнением для изменения изобарного потенциала реакции (а); тогда получим
— 29 120, Кр при 600° С — 7,86 X ЮЧ
Даже не записывая уравнений закона действующих масс, из констант равновесия очевидно, что процентное содержание метанола в продуктах реакции будет ничтожным. Этим подчеркивается интересный факт, что успех синтеза метанола зависит от подавления реакции (б). Употребляемые катализаторы должны быть как раз такими, чтобы эта реакция происходила в ничтожной степени.
П р Н м е р 14. Предположим, что в процессе производства водорода метан и водяной пар пропускаются над катализатором при атмосферном давлении и газы, покидающие реактор, находятся в равновесии при 600° С. Требуется определить процент разложившегося метана и состав сухого выходящего газа, если в реактор поступает смесь водяного пара и метана в отношении 5:1.
В рассматриваемой системе возможны многие реакции, например реакции
и так далее.
СН4+Н2О = СО4-ЗН2 (а)
СН4+2Н2О = СО24-4Н2 (б)
СН4 = С4-2Н2 (в)
СО + Н2О = СО24-Н2 (г)
СО2 + С = 2С0 (д)
СО2 = С + О2 (е)
2СО = 2С4-О2 (ж)
н3о=н24-|о2 (з)
Н2О+С=Н, + СО (и)
2СН4 = С2Н64-Н2 (к)
С2Н6 — С2Н4 Н2 (л)
Одновременно протекающие реакции
597
(Все вещества, кроме углерода, принимаются находящимися в газообразном состоянии.)
Это создает впечатление о сложности рассматриваемого случая, однако J
простой анализ делает возможным произвести значительное упрощение. j
Во-первых, не все эти реакции являются независимыми, и на этом основании (
можно исключить из рассмотрения некоторые из них. Так, (б)—(а)--(-(г), ;
(ж) = (е)— (д), (и) = (з)— 1/2 (ж) и (а) = (в)+(и). В результате этих зависимостей можно исключить четыре уравнения, например (б), (в), (ж) и (и). Конечно, нет никаких специальных указаний, предписывающих сократить именно эти уравнения. Исключить можно было бы любые четыре из девяти. Реакциями (е) и (з) можно пренебречь, поскольку рассмотрение их изобарных потенциалов показывает, что при этой температуре они могут протекать только в очень слабой степени. Также можно показать, что реакция (к) не будет итти в заметной степени, а это автоматически исключает реакцию (л). Мы произвольно исключаем из рассмотрения реакцию (д) и объясним это решение ниже. Поэтому остается рассмотреть только реакции (а) и (г). Уравнения стандартного изобарного потенциала для этих двух реакций будут основываться на следующих данных. Теплоты сгорания при 25° С (при расчете на образование жидкой воды) соответственно равны
Н2, Д// = —68 313,
СО, Д// = —67 623,
С, Д/7= —94 240,
СН4, Д//=—212 790.
Энтропии при 25° С в состоянии идеального газа равны:
Н2, 5= 31,23,
СО, 5 = 47,32,
СО,, 5 = 51,10, Н2б, S = 45,17, СН4, 5 = 44,46.
Теплоемкоств как функции температуры при 1 атм. равны:
Н2, Ср = 6,947 — 0,200 X Ю-з У + 4,808 X Ю“7 Т\ СО, Ср = 6, 342+ 1,836 X IO-3 Т — 2,801 X Ю-7 Т\ Н2О, Ср = 7,187 + 2,373 X IO-3 ?+2,084 X Ю~7 Т2, СО* Ср — 6,396+ 10,193 X IO-3 Т— 35,193 X 10~’ Т2, СН4, Ср = 3,38 + 17,903 X Ю-з 7—4,188X Ю"6 Т2.
Единицами во всех случаях являются калории, моли и градусы Кельвина.
Из этих данных выводятся следующие уравнения:
Для реакции (а):
Д2» = 45 118 — 16,616 7 In 7 + 9,52 X Ю”» Г2 — 8,57X Ю"773+ 54,40 Т.
Для реакции (г):
ДЛ° = —9985 + 0,186Tin 7 — 2,892X Ю'3 Г+ 4,945X ТЗ + 10,51 Т.
При T=873,2(t=600°C),Д2°(а)=1060,Afp(a)=0,54,AZ°(r) = —1585, JCp(r)=2,49.
598
XI. Равновесие химических реакций
В уравнениях (55) и (56)
a = b = c = lf — e = d' = 1,
d = 3, N — 5, для уравнения (55) —2, для уравнения (56) = 0, N=6-|-22Ул. , Подставляя в уравнения (55) и (56), получаем
Wa-Nc№a + NcP . 05„ U-NA)(5-Na-Nc)(64-2N4)2 ’ ’
____Nc(3Na4-Nc) 249 (Na-Nc)(5-N4-Nc) “ ’ *
Решая уравнения (м) и (н) методом подбора, получаем
Na = 0,911, Nc — 0,653.
Следовательно, будет разлагаться 91 % метана.
Общее число молей сухого выходящего газа = = 64-2Na- (^rB-^NA- = 4,386;
молей СН4 =1 — Na = 0,089;
молей СО ="~ Na — Nc — 0,258;
молей Н2 = -~ Na 4- у Nc = 3,386;
молей СО2— Nс = 0,653.
Состав сухого газа:
(м)«
(И)
77,2% Н2
14,90/в СО2
5,9% СО
2,ОО/о СН<
Вернемся к рассмотрению реакции (д) и посмотрим, возможно ли выделение углерода.
Для этой реакции
Кр хсо
Р ~ *со, ‘
Для выделения углерода значение -*со/хСОа должно быть равно или больше Кр/p. Из уравнения изобарного потенциала для этой реакции, данного Льюисом и Рендаллом [351], Кр при 600° = 0,083.
лсо ------ в равновесной смеси = 0,0234.
хсо2
Следовательно, выделение углерода при установлении равновесия в этой । системе невозможно, и исключение уравнения (д) оправдано.
Для дальнейших деталей применения термодинамики к химическим реакциям можно обратиться к статье автора [64], в которой также дается большое количество ссылок на равновесные данные для ряда реакций й).
й) Только что решенный пример также взят из этой статьи.
ГЛАВА ХИ
РАВНОВЕСИЕ ПРИ ИСПАРЕНИИ И КОНДЕНСАЦИИ
Группу отдельных операций, которую иногда называют процессами испарения и которая включает такие важные и широко применяемые операции, как перегонка, ректификация, конденсация, испарение, увлажнение и абсорбция, можно успешно трактовать с термодинамической точки зрения. Классификация различных процессов по этим рубрикам полезна, но далека от точности и связана с некоторой неопределенностью в терминологии. Однако все эти процессы испарения имеют общее в том отношении, что все они включают обмен веществом между соприкасающимися фазами (обычно между газообразной и жидкой, хотя в адсорбции и сублимации может участвовать и твердая фаза) и что скорость этого обмена в большей или меньшей степени (при испарении, например, меньше, чем при абсорбции) можно регулировать с помощью диффузии. Если происходит переход из жидкой фазы в пар, то процесс часто называют испарением, а обратный процесс — конденсацией. Из кинетической теории известно, что оба процесса происходят одновременно всякий раз, когда жидкость и пар находятся в соприкосновении, и что наблюдаемый эффект является результатом соотношения скоростей двух противоположных процессов. Если скорости этих процессов равны и в результате не происходит перехода вещества, тогда считают, что система находится в равновесии. В этом случае незначительное изменение одного из параметров состояния — давления, температуры или концентрации — будет вызывать продолжение процесса в том или другом направлении, а значительное изменение одного из этих параметров будет вызывать преобладание одного из этих процессов, благодаря чему общим результатом явится испарение или конденсация.
Мы будем считать испарение и конденсацию основными процессами, происходящими при всех упомянутых выше отдельных операциях, и поэтому этн процессы и включены в название главы. При изложении будет сделан упор на процессы перегонки и ректификации, при которых жидкий раствор разделяется на две или несколько фракций, которые могут быть или почти чистыми компонентами, или же смесью их. Мы будем также рассматривать и случай сжижения одного или нескольких способных к конденсации веществ из смеси, содержащей неконденсирующиеся газы.
ООО
XII. Равновесие при испарении и конденсации
Все процессы, включающие переход вещества между фазами — безразлично, газообразными, жидкими или твердыми, — в сущности, подобны друг другу, и можно вывести соотношения, достаточно общие для обработки любого из них при условии наложения специальных условий. В этом направлении уже предпринимались отдельные иопытки (см., например, статьи Рендалла и Лоиг-тена [194]), но при настоящем состоянии развития этого общего метода мы считаем более надежным придерживаться обычной практики разделения темы на более или менее произвольные группы и рассматривать каждую из них отдельно.
Приложение термодинамики к этим процессам связано главным оюразсми со следующим кругом вопросов:
1. Изучение различных типов фазового равновесия для нахождения предельных условий разделения.
2. Расход энергии для осуществления процесса.
3. Эффективность и распределение потерь.
4. Число единиц (тарелок и т. д.), необходимых для заданного разделения.
Переход массы данного компонента между фазами происходит только при различии в значениях химического потенциала этого компонента. Если все его потенциалы равны между собой, то между фазами существует равновесие. Удаленность от состояния равновесия является движущей силой происходящего изменения; поэтому скорость перехода будет зависеть от степени удаленности от состояния равновесия86. Давление, температура и состав фаз при равновесии являются основными параметрами для анализа процессов испарения. Рассмотрение различных типов равновесия, в частности, встречающихся при перегонке, составляет содержание этой главы. Самые процессы перегонки будут рассмотрены в следующей главе.
Правило фаз. Правило фаз представляет полезное и важное средство изучения сложного многообразия данных, собранных в настоящее время по многим системам. Оно настолько просто и в настоящее время считается настолько само собой разумеющимся, что трудно представить, до какой степени все наши уравнения по фазовым равновесиям основываются на нем. Хотя правило фаз уже было рассмотрено в гл. IV в связи с изучением термодинамических свойств, желательно снова рассмотреть его здесь с этой точки зрения даже с риском некоторого повторения.
Правило фаз (вывод см. в гл. IV) формулируется так:
D = N~Z+2, (122, гл. PV)
где D — число степеней свободы или число независимых переменных, N — число компонентов, Z — число фаз.
Если мы имеем двухкомпонентную систему, например этиловый спирт и воду, и две фазы—жидкость и пар, — то такая система обладает двумя степенями свободы. Это значит, что мы можем фиксировать
Равновесие при испарении и конденсации
601
любые два параметра состояния более или менее произвольно (конечно, В известных пределах, так как если бы температура превышала температуру кипения менее летучего компонента при выбранном давлении, то жидкая фаза не могла бы существовать), и если мы это осуществили, то состояние системы будет точно определено. Например, в случае спирта и воды, если мы устанавливаем давление в 1 атм, то температуру можно произвольно выбрать в любой точке между 100 и 78,5°С. Если фиксированы обе величины, то состав фаз также фиксирован. Если бы мы попробовали изменить состав любой фазы, поддерживая неизменными давление и температуру, то одна из фаз должна была бы исчезнуть. Если бинарная система состоит из двух жидких фаз и паровой фазы, то система будет унивариантной (одна степень свободы), и ее состояние определяется только температурой, или давлением, или составом одной из фаз. Если же присутствует также Четвертая фаза, например твердая, то. система будет нонвариантной, т. е. это состояние может существовать только в одной точке — только при одном сочетании параметров (температуры, давления и состава) — и невозможно изменить ни одного из параметров, не вызвав исчезновения какой-нибудь из фаз.
Наиболее важными параметрами состояния при перегонке являются давление, температура, состав 'жидкой фазы и состав паровой фазы. Для бинарных двухфазных систем, которые мы главным образом и будем рассматривать, согласно правилу фаз возможны следующие виды кривых фазового равновесия:
1) р—t при постоянном х;
2) p~t при постоянном у;
3) р — х при постоянном /;
4) р — х при постоянном у;
5) р—у при постоянном t;
6) р—у при постоянном х;
7) t — x при постоянном р;
8) t-У при постоянном р;
9) t — х при постоянном у;
Ю) t—у при постоянном х;
И) у — х при постоянном р-
12) у—х при постоянном t.
Зависимости (4), (6), (9) и (10) маловажны и не будут рассматриваться. Другие восемь соотношений будут иллюстрированы ниже. Зависимости при постоянном давлении наиболее важны с практической
602
XII. Равновесие при испарении и конденсации
точки зрения, так как большинство процессов перегонки осуществляется при р = const. С другой стороны, обычно в лаборатории несколько удобнее поддерживать постоянную температуру, и поэтому большинство опубликованных данных по фазовому равновесию получено при изотермических условиях. В некоторых трудах по фазовому равновесию (например, в книге Куэнена [139]) в качестве одной из координат при графической интерпретации равновесия используется объем. Применение этого параметра имеет некоторое преимущество для рассмотрения систем при повышенных давлениях, особенно в критической области, но мы не будем пользоваться им в этой книге.
Пограничная кривая р — t называется кривой давления пара, хотя этот термин обычно ограничивают жидкой ветвью или кривой р — t при постоянном х. Кривые р — х и t — х являются «линиями жидкости* или «кривыми точек пузырьков*, поскольку эти кривые связывают давление (или температуру) с составом всей системы, если жидкость находится в равновесии с ничтожно малым количеством (единственный пузырек) пара. Кривая t — х также называется «кривой точек кипения*. Кривые р—у или t—у называются «кривыми пара* или «кривыми точек росы*, так как эти кривые связывают давление (или температуру) с составом системы в целом, если пар находится в равновесии с ничтожно малым количеством жидкости. Эти пограничные кривые также ограничивают области, в которых устойчивы определенные фазы или смеси фаз (гетерогенная система).
Кроме пограничных кривых, определяющих области устойчивости фаз, которые появляются иа диаграммах фазового равновесия, необходимы также и линии, связывающие составы двух сосуществующих фаз; подобные линии называют «соединительными линиями* или конно-дами. В случае диаграмм давление — состав или температура—состав соединительные линии всегда горизонтальны (поскольку сосуществующие фазы должны быть при одинаковом давлении и температуре), и поэтому их нецелесообразно показывать; но на некоторых других диаграммах, например на таких, как энтальпия —концентрация, объем — концентрация или на тройных диаграммах, соединительные линии целесообразно наносить.
Заметим, что в вышеприведенном анализе р всегда относилось к общему давлению, которое представляет единственный вид давления, поддающийся экспериментальному наблюдению и контролю. В некоторых случаях удобно иметь дело с парциальным давлением, но следует помнить, (что это чисто математически определяемая величина, которая не является параметром состояния.
Растворимость. В практике перегонки встречаются три основных случая растворимости. Первым и наиболее простым будет случай, при котором оба рассматриваемых компонента не способны взаимно растворяться в жидком состоянии. Вероятно, не существует жидкостей, совершенно не растворимых друг в друге, но имеется много пар (в част
Равновесие при испарении и конденсации
603
ности, многие органические жидкости с водой), которые смешиваются столь незначительно, что для всех практических целей их можно считать взаимно нерастворимыми. Поэтому нерастворимость является предельным случаем, к которому действительные системы приближаются более или менее значительно, но которого никогда не достигают. Такие системы, как ртуть—вода, вплотную приближаются к этому предельному случаю, и, конечно, для всех практических целей эти две жидкости можно считать нерастворимыми. Хотя мы могли бы с той же простотой, как мы это только что сделали для бинарной системы, рассмотреть и многокомпонентную систему, в которой каждый компонент образует чистую жидкую фазу, однако имеется лишь незначительное число случаев, в которых сосуществуют несколько жидких фаз, и ни один из них не представляет практического значения.
Наиболее обычным и наиболее важным будет такой случай, при котором жидкие компоненты полностью смешиваются в широком интервале состава, и поэтому может существовать только одна жидкая фаза. Подавляющее большинство органических жидких смесей принадлежит именно к этой категории.
Промежуточным между этими двумя предельными случаями будет случай ограниченной растворимости, когда имеется только одна жидкая фаза для некоторой области концентраций и две — для остальных.
Граница между указанными случаями не является резкой. Вещества, которые совершенно не смешиваются при данной температуре, становятся частично смешивающимися при повышении температуры, а многие, частично смешивающиеся, становятся полностью смешивающимися выше (а в некоторых случаях ниже) определенной критической температуры.
Степень смешиваемости, очевидно, связана с полями сил, окружающими молекулы. Если два вещества имеют одинаковые силовые поля, то следует ожидать, что они будут смешиваться во всех отношениях. Там, где они очень различны, как в случае полярной жидкости, подобной воде, и неполярной жидкости, подобной бензолу, мы получим почти полную несмешиваемость. Если органические жидкости стремятся к полярному типу, то мы обнаруживаем более высокую степень смешиваемости с водой; наконец, такое соединение, как этиловый спирт, полностью смешивается с водой. Жидкости, являющиеся очень сходными, например бензол и толуол или метиловый и этиловый спирты, должны смешиваться с минимальным изменением свойств раствора по сравнению с ожидаемым на основании строгой аддитивности (идеальные растворы).
Большинство случаев частичной смешиваемости наблюдается тогда, когда одной из жидкостей является вода или какая-нибудь другая неорганическая жидкость, но существует много случаев, когда и органические жидкости в определенных пределах только частично смешиваются. Например, частичную смешиваемость показы
604
XII. Равновесие при испарении и конденсации
вают следующие пары жидкостей: сероуглерод — метиловый спирт; гексан — этиловый спирт; амилен — анилин; уксусная кислота — бензол; метиловый спирт—ацетон. При изменении температуры почти для любой пары следует ожидать достижения точки, при которой жидкости будут только частично смешиваться; однако обычно раньше достижения этой температуры или появляется твердая фаза, или достигается критическая точка.
Рис. 113. Давление пара в системе толуол — вода.
Неемешивающиеся жидкости. В случае взаимно нерастворимых жидкостей равновесные зависимости даже в многокомпонентных системах относительно просты, так как существует только одна степень свободы и все жидкие фазы содержат только чистые компоненты. Так, зависимость р — t является единственной, не зависящей от относительных количеств различных компонентов. Давление пара любой смеси будет равно сумме давлений пара отдельных компонентов. Температура кипения не зависит от соотношения компонентов и представляет температуру, при которой сумма давлений пара чистых компонентов равна внешнему давлению. Она, очевидно, будет ниже температуры
Равновесие при испарении и конденсации
605
кипения любого из компонентов при этом давлении. Состав равновесного пара легко вычислить по давлениям пара, если можно принять, что пары подчиняются законам идеального газа.
Пример 1. Вычислить нормальную точку кипения смесей толуол — вода и состав равновесного пара, принимая, что они совершенно не смешиваются друг с другом. Давления насыщенного пара компонентов приводятся ниже.
t,°G 60 70 80 90 100
Давление пара воды, мм Hg 149 234 355 526 760
Давление пара толуола, мм Hg 139 206 287 404 557
Нанесем на график сумму обоих давлений в зависимости от температуры; пересечение с изобарой 760 мм окажется при 84,5° С (рис. 113). По кривым давления насыщенного пара двух компонентов давления пара при этой температуре равны:
вода 427 мм Hg,
толуол 333 мм Hg.
Принимав пар за смесь идеальных ола в паре, равной I
Рт __
ТОО —760-°1438-
Когда имеют дело с двуми несмешивающиМися жидкостями, то интересуются не только трехфазной системой, но также и возможными двухфазными системами. Из правила фаз следует, что пар только одного состава находится в равновесии с обеими жидкими фазами, но одна жидкая фаза может быть в равновесии с целым рядом паров прн различных температурах, если считать общее давление фиксированным. Полная
Рис, 114. Диаграмма температура — состав для двух несмешивающихся жидкостей.
диаграмма для этого случая пока-
зана на рис. 114. DE — кривая кипения для смеси двух жидких фаз; ВС—-кривая точек росы для паров, более богатых В, чем пар в точке С, который является единственным паром для трехфазного равновесия; СА — кривая точек росы для паров, более богатых А, чем пар в точке С. Горизонтальные линии, подобные, например, линиям
606
XII. Равновесие при испарении и конденсации
FG и HI, являются соединительными линиями, связывающими сосуще-‘ствующие фазы. Ясно, что любой пар, состав которого лежит слева от С, будет находиться в равновесии с чистым жидким В, а любой пар, состав которого лежит справа от С, будет в равновесии с чистым жидким А ☆ ).
ПОЛНОСТЬЮ СМЕШИВАЮЩИЕСЯ ЖИДКОСТИ
Наиболее обычным случаем при перегонке и связанных с ней процессах является разделение смесей веществ (точнее, растворов), которые смешиваются в любых соотношениях. Подавляющее большинство всех органических жидкостей образует растворы этого типа. Приводимый ниже анализ, за исключением особо указанных случаев, будет ограничен бинарными растворами.
Идеальные растворы. Идеальный раствор представляет предельный случай, для которого равновесные соотношения между жидкостью и парами особенно просты, и поэтому представляется удобным наше обсуждение начать именно с них. (Определение идеального раствора дано в гл. IV.) Следует ожидать, что пары жидкостей, подобных по химической структуре, как, например, некоторые типы изомеров и соседние члены гомологических рядов, образуют почти идеальные растворы. Типичными будут следующие пары жидкостей: бензол — толуол; гексан — гептан; я-октан— 2, 2, 4-триметилпентан; этиловый спирт — метиловый спирт; этилацетат — этнлпропионат. Можно ожидать, что растворы, компоненты которых отличаются только по составу изотопов, например обычная вода Н2О и тяжелая вода D2O, будут очень точно следовать законам идеальных растворов.
Существует три критерия, полезных для экспериментальной проверки идеальности раствора, именно:
1) Ничтожно малое изменение объема при смешении компонентов.
2) Ничтожно малое изменение температуры при смешении компонентов (отсутствует теплота смешения).
3) Общее давление пара представляет линейную функцию состава. Критерий 1) вытекает из определения идеального раствора; критерии 2) и 3) логически следуют из 1), как было показано в гл. IV, при условии, что пар можно считать идеальным газом. Ниже приводятся некоторые типичные данные, характеризующие изменения объема и температуры при смешении (при комнатной температуре) эквивалентных количеств двух жидкостей [264]:
☆) Лица, знакомые с диаграммами равновесия, встречающимися при работе со сплавами, узнают в рассмотренной диаграмме тот же самый тип, который получается в общем случае двух твердых тел, кристаллизующихся раздельно с образованием эвтектики.
Полностью смешивающиеся жидкости
607
Смесь Процентное изменение объема при смешении Изменение температуры, °C
я-Октан 1 я-Гексан / — 0,053 + 0,06
Бензол 1 Толуол J + 0,161 — 0,45
Этиловый спирт 1 Метиловый спирт/ . . . . + 0,004 — 0,10
Вода 1 Метиловый спирт/ . . . . — 2,98 + 7,85
Бензол 1 Этиловый спирт/ . . . 0,00 — 4,2
Первые три пары будут давать идеальные растворы вследствие из химического подобия, и два критерия подтверждают это предположение.
Но ие следует ожидать, чтобы последние две пары были идеальными, и это также подтверждается указанными критериями, за исключением случая бензол — этиловый спирт, где при смешении не происходит изменения объема. Это иллюстрирует тот факт, что ничтожно малое изменение объема или температуры для какой-либо отдельной смеси не является надежным критерием идеальности раствора. Причина этого ясна из рис. 115. Пунктирная линия представляет объем идеальной смеси компонентов А и В как функцию состава, а сплошная кривая соответствует случаю неиде-ального раствора. Ясно, что при
Рис. 115. Изменение объема, сопровождающее смешение двух полностью смешивающихся жидкостей.
смешении компонентов в соотношении, представленном точкой р, не произойдет никакого изменения объема, но для любого другого соотношения компонентов будет происходить изменение объема. Очевидно, что критерии 1) и 2) следует прилагать ле к одному, а к нескольким составам раствора. Без сомнения, критерий 3) будет наиболее надежным, но он не так удобен для применения, как два остальных. Первые три из указанных выше пар жидкостей имеют почти линейную кривую давления пара (р— х), тогда как две другие не обладают этим свойством. Из этого факта можно с уверенностью
608
ХИ. Равновесие при испарении и конденсации
заключить, что только первые три пары действительно образуют идеальные растворы.
Важно располагать хорошим средством проверки идеальности раствора, потому что состав пара очень трудно точно определить, и в то же время, если можно быть уверенным, что пар является смесью идеальных газов, то состав его легко вычисляется ☆ ). В тех случаях, когда отношение давлений пара двух компонентов в чистом состоянии (которое в дальнейшем мы будем называть для краткости „относительной летучестью”), близко к 1,00, незначительные ошибки при экспериментальном определении состава пара могут оказать большое влияние на расчеты по перегонке, и поэтому желательно располагать ВЫЧИСЛЕННЫМИ СОСТаВЗМИ Пара.
Наиболее важными уравнениями, применяющимися к случаю идеальных растворов со смесью идеальных газов в качестве паровой фазы, являются следующие (см. гл. IV):
Р=Рах+Рв^— х)> (177, гл. IV)
У —.Ра х 1—У Рв 1—х’ (178, гл. IV)
УР=Ра = Рах- (180, гл. IV)
(1 — j) р=рв=рв (1 — х). (181, гл. IV)
Уравнение (178, гл. IV) также можно записать в форме
^=1+(ааХ-1)л П)**)
И "-а-(Л1)Г ™
а уравнение (177, гл. IV) в форме
х = Р_. Рв (3) РА — РВ
Исключая х из уравнения (178, гл. IV) посредством уравнения (3),
имеем v = PAtP^£ti У Р(РА~РвУ ' 1
☆) Возможно, следует подчеркнуть, что идеальный раствор, подобно идеальному газу, представляет только предельный случай, к которому действительные растворы более или менее значительно приближаются. Для большого числа случаев приближение достаточно хорошо для практических расчетов. Следует также помнить, что в обычных уравнениях предполагается, что пар является идеальным газом, в то время как насыщенные пары даже при 1 атм могут отклоняться на несколько процентов от законов идеального газа.
&☆) Это уравнение, а также уравнение (178, гл. IV) включают отношение масс данного компонента, и поэтому концентрацию можно выразить или в мольных, или в весовых долях. Относительная летучесть а может иногда считаться постоянной, даже если раствор не идеальный и пар отклоняется от законов идеального газа.
Полностью смешивающиеся жидкости
609
и
РаРв
РА — (РА — Рв}У ‘
(5)
Эти уравнения графически представлены для случая постоянной температуры на рис. 116 и 117.
Кривая жидкости на рис. 116 является кривой, отвечающей уравнению (177, гл. IV). Кривая пара представляет график уравнения (4). Линии парциального давления представляют соответственно графики уравнений (180, гл. IV) и (181, гл. IV). Рис. 117 показывает
состав
Рис. 116. Диаграмма давление — состав для случая идеального раствора и идеального газа.
Рис. 117. Диаграмма состав пара — состав жидкости (у — х) для идеального раствора.
график уравнения (1). Все линии на рис. 116 очень легко могут быть найдены, если только известны рЛ и рв— давления пара компонентов в чистом состоянии. Для этого необходимо использовать только что приведенные уравнения или же чисто графические построения, которые для трех прямых линий не требуют объяснений. Так как, по уравнению (180, гл. IV),
Ра-^_ Ра.
У Р Р ’
то очевидно, что точки на кривой пара получаются путем деления отрезка на ординате х, образуемого линией, представляющей парциальное давление А, на соответствующий отрезок, образованный линией общего давления или кривой жидкости, т. е. у = АВ]СВ. Это определяет место точки пара D, которая должна быть при том же самом давлении, что и точка С на кривой жидкости. Эти две точки, соответствующие составам жидкости (х8) и пара (уЕ) при одинаковых давлении и температуре, должны представлять состав жидкой и 39 Химическая термодинамика
610
XII. Равновесие при испарении и конденсации
паровой фаз при равновесии. При этих же самых условиях любая горизонтальная линия (линия постоянного давления) между пределами Ра и Рв будет пересекать кривые жидкости и пара в точках, представляющих составы сосуществующих фаз в равновесии.
В тех случаях, когда нужно сделать лишь несколько расчетов, обычно проще пользоваться непосредственно уравнениями, чем употреблять их сначала для построения диаграммы р — х. Следующий пример иллюстрирует один из возможных типов вычислений:
Пример 2. Эквимольиая смесь бензола с толуолом (ее следует считать идеальным раствором) полностью испаряется при постоянной температуре 90° С. Чему будут равны давления в начале и в конпе процесса испарения, если давления пара бензола и толуола при 90° С соответственно равны 1008 мм и 404 мм рт. ст.?
По уравнению (177, гл. IV), /^ = 0,500X 10084-0,500X404 = 706 мм.
В конце испарения у = 0,500 и р2 дается уравнением (5):
_ 1008 X 404
Рг ~ 1008 — (1008 — 404) 0,500 ' ММ'
Практически перегонка проводится чаще при постоянном давлении, чем при постоянной температуре, и потому будут полезнее равновесные зависимости для постоянного давления. Не существует простых уравнений, подобных только что данным зависимостям, связывающих температуру и состав при постоянном давлении, но данные или соответствующие им графики легко получаются из уравнений (1) и (3) при приложении их к ряду температур между точками кипения двух чистых компонентов. Это показано в следующем примере.
Пример 3. Вычислить данные, по которым затем следует построить диаграммы t — х и у — х для системы бензол—толуол при давлении 1 атм^ если известны следующие данные по давлению пара чистых компонентов:
f,QC 70 80 90 100 ПО 120
Давление пара бензола, мм Hg 540 756 1008 1338 1740 2215
Давление пара толуола, мм Hg 206 287 404 557 741 990
Примем, что раствор идеальный и пар подчиняется законам идеальных газов.
При нанесении этих данных и определении точек пересечения кривых давления пара с горизонталью р = 760 находят точки кипения чистых компонентов; они будут соответственно равны 80,2 и 110,5° С. Затем между этими пределами произвольно выбирается ряд температур, с графиков считываются соответствующие давления пара, а х и у соответственна вычисляются из уравнений (3) и (1). Результаты приведены в следующей таблице:
Полностью смешивающиеся жидкости
611
t, °C мм ММ Рв Рт Х1) У1) у (из средней а)
80,2 760 288 2,64 1,00 1,00
82,0 815 307 2,65 0,892 0,957 0,953
85,0 877 340 2,58 0,782 0,903 0,900
90,0 1008 404 2,50 0,590 0,782 0,782
95,0 1150 475 2,42 0,422 0,640 0,645
100,0 1338 557 2,40 0,260 0,457 0,466
105,0 1530 644 2,38 0,131 0,264 0,273
110,5 1775 760 2,34 0,000 0,00
’) х vy — мольные доли бензола соответственно в жидкой фазе и в парах.
Те же самые данные можно очень легко получить с помощью графического построения, показанного на рис. 118. На диаграмме р—х проводят прямые линии от точки О до точек давле-
ния пара чистого толуола на ординате х = 0 при любой выбранной температуре. (Такой линией будет, например, линия OD.) Затем из этих точек проводят прямые линии до точек давлений пара чистого бензола на ординате х — 1 (напр., линия DE). Эти последние линии дают давление пара всех смесей при соответствующих температурах. Состав жидкости, кипящей при любой температуре, определяется опусканием перпендикуляра из точки пересечения горизонтали 760 мм с линией давления пара для этой температуры (АС для t = 95° С). Состав пара определяется из соотношения ЛВ/760 =
Обращаясь к приведенным в таблице данным, следует отметить, что Рв1рг> т. с. а изменяется только от 2,64 до 2,34. При использовании в уравнении (1) среднего значения а = 2,49 были получены значения у в графе 7; оказалось, что они близко совпадают со значениями в графе 6, основанными на переменной а. Такое применение уравнения (1) со средним а
Рис. 118. Определение данных по равновесию жидкость — пар для системы бензол — толуол при общем давлении в 1 атм.
дает очень удобную формулу для Данных при постоянном давлении, являющуюся достаточно точной для многих целей.
Неидеальные растворы. Идеальный раствор, как мы уже указывали раньше, представляет только предельный случай, к которому в большей или меньшей степени приближаются все реальные растворы, в зависимости от условий и от свойств системы. Бинарные растворы, заметно отклоняющиеся от идеального раствора, можно подразделить на четыре различных типа, согласно форме их изотер-39*
612
XII. Равновесие при испарении и конденсации
мической диаграммы р— х, что и показано на рис. 119, а, б, в и г й). На всех этих диаграммах верхняя кривая является кривой жидкости, т. е. точек кипения, а нижняя — кривой пара, т. е. точек росы. Типы I и II часто объеди-
5 г
' Рис. 119. Диаграммы давление — состав для неидеальных растворов.
проходить соответственно через максимум
няются вместе, но между ними существует определенное различие, заключающееся в том, что в случае I давление пара раствора превышает давление идеального раствора, а в случае II давление пара меньше, чем для идеального раствора (пунктирная линия представляет идеальный раствор). Тогда типы III и IV оказываются просто крайними случаями I и II, при которых отклонения от линейной зависимости для давления пара являются настолько значительными, что давление насыщенного пара неизбежно должно или минимум. Типичными
представителями этих четырех групп являются следующие системы при температурах, близких к нормальным температурам кипения:
I п III IV
Кислород—азот Хлороформ—этиловый эфир Вода—этиловый спирт Вода и некоторые кислоты
Этиловый спирт— этиловый эфир Хлороформ—бензол Сероуглерод—ацетон Хлороформ—ацетон
Бензол-циклогексан [227] представляет особенно интересный пример типа III, при котором два компонента имеют почти одинаковое давление пара, а отклонение от идеального раствора незначительно. Таким образом, мы имеем необычный случай максимума в давлении пара у почти идеального раствора87.
☆ ) Во всех случаях, кроме очень немногих, мы будем выражать состав системы содержанием компонента, имеющего более высокое давление пара г. е. более низкую точку кипения.
Полностью смешивающиеся жидкости
Соответствующие диаграммы температура—состав (t — х) и состав пара—состав жидкости (у — х)при постоянном давлении показаны на рис. 120, а, б, в, и на рис. 121. На обеих этих диаграммах типы I и И неразличимы друг от друга, а также и от идеального раствора. Тип III, который показывает максимум давления пара на диаграмме р — х, дает минимальную точку кипения на диаграмме t — х, а тип IV — наоборот.
Анализ как диаграмм р — х, так и диаграмм t — х показывает, что при максимуме или минимуме на любой кривой пар и жидкость совпадают по составу, и
поэтому растворы такого состава ведут себя при перегонке как чистое вещество, т. е. перегоняются, не-изменяя состава и сохраняя постоянной температуру кипения. Это, как мы увидим ниже (стр. 666), имеет очень важное значение для разделения жидкостей перегонкой. Смесь, которая соответствует максимуму или минимуму на кривой точек кипения, называется азеотропной смесью. Этот термин некоторыми авторами прилагается также к смеси, в которой су
ществуют две жидкие Рис. 120. Различные типы диаграмм темпера-фазы, потому что такая тУРа — состав.
смесь, подобно азеотроп-
ной в системе с одной жидкой фазой, будет перегоняться без изменения состава. Поскольку диагональ на диаграмме у — х представляет точки одинакового состава обеих фаз, кривые для типов III и IV будут
пересекать диагональ при составах, соответствующих максимуму или минимуму. Диагональ можно также представить линией, изображающей зависимость между составами жидкости и пара для смесей жидкостей, настолько сходных между собой, что они ведут себя как один компонент. Такими смесями являются стереоизомеры и большинство смесей, содержащих различные изотоны одного итого же элемента.
Влияние давления. До сих пор мы рассматривали только такие случаи, когда давление в системе было практически атмосферным. Для некоторых приложений представляет интерес кратко рассмотреть, что происходит при изменении давления. (Мы принимаем в качестве независимой переменной давление, а не температуру,
614
XII. Равновесие при испарении и конденсации
поскольку в большинстве случаев перегонка проводится при постоянном давлении.) Данные в этой области очень скудны, но рис. 122 представляет влияние давления на систему кислород—азот [62, 65] — одну из тех немногих систем, изученных совсем недавно, когда было опубликовано несколько исследований по углеводородным системам 88.
Основным результатом повышения давления является уменьшение расстояния между точками, отвечающими составам жидкой и паровой фаз. Это представляет общую тенден
цию, которую следует ожидать, потому что при возрастании давления (а следовательно, и температуры кипения) наблюдается постепенное приближение к критической области; как раз в этой
Рис. 122. Изобары для жидкой и паровой фаз системы кислород— азот.
(Давления даны в атмосферах.)
Рис. 121. Различные типы диаграмм у — х при постоянном давлении илн при постоинной • температуре.
области по аналогии с поведением чистого вещества можно рассчитывать найти критическую точку для смеси данного состава, и в этой точке обе фазы должны быть тождественны. Это явление, так же как и другие критические явления, будет более подробно рассмотрено в следующем разделе.
Другим важным результатом изменения давления является его влияние на состав азеотропной смеси. Поскольку последняя при перегонке ведет себя подобно чистому веществу, некоторые авторы придерживаются взгляда, что азеотропы представляют собой настоящие химические соединения двух компонентов. Практически не существует никаких определенных фактов, подтверждающих этот взгляд, а наиболее серьезным аргументом против него является то,
Полностью смешивающиеся жидкости
615
что азеотропный состав непрерывно изменяется по мере изменения давления или температуры. Табл. 20 показывает влияние давления на положение азеотропной точки в системе этиловый спирт — вода й).
Таблица 20
Влияние давления на состав азеотропной смеся
Давление, мм Hg
Температура кипения азеотропной смеси, °C
Мольный процент этилового спирта в азеотропной смеси
100
150
200
400
760
1100
1450
34,2
42,0
47,8
62,8
78,1
87,8
95,3
99,6
96,2
93,8
91,4
90,0
89,3
89,0
Критические явления. Критическая точка чистого вещества имеет
следующие три характерных признака.
1. Жидкая и паровая фазы тождественны.
2. Температура является максимальной температурой, при которой могут сосуществовать две фазы.
3. Давление является максимальным давлением, при котором могут сосуществовать две фазы.
В случае бинарных растворов эти три особенности уже не наблюдаются в одной точке, а обнаруживаются в трех различных точках. Это удоб
Рис. 123. Диаграмма давление — температура при постоянном составе для бинарного раствора в критической области.
нее показать на диаграмме давление— температура, подобной приведенной на рис. 123. Рисунок представляет поперечный разрез диаграм
мы давление — температура — состав
плоскостью постоянного состава. Она состоит из двух пограничных кривых — одной для жидкости и одной для пара того же состава, заключающих область, в которой могут сосуществовать две фазы, и встречающихся в критической точке С, где обе фазы становятся тождественными. Эта точка является точкой касания пограничной кривой и оги-
й) Данные взяты из Интернациональных критических таблиц.
616
XII. Равновесие при испарении и конденсации
Рис. 124. Кривые давление — температура для бинарной системы при постоянном составе.
бающей кривой АВ, которая связывает критические точки двух чистых компонентов. Точка С соответствует максимальной температуре, при которой пар данного состава может быть сконденсирован в жидкость, т. е. максимальной температуре, при которой могут сосуществовать две фазы в системе при данном общем составе. Эта точка, обычно называемая ,крикондентермической“ (критическая температура конденсации), соответствует признаку 2 критической точки чистого вещества. Точка М соответствует признаку 3. поскольку она отвечает максимальному давлению, при котором система данного состава может существовать в двух фазах ☆). Кривую DMC легче всего представить как кривую давления пара этой отдельной смеси. Подобным же образом, кривая ЕС С представляет геометрическое место точек росы для ряда давлений. Интересно отметить, что тогда как давление пара чистого вещества всегда возрастает с повышением температуры, в случае бинарного раствора давление может возрастать до максимума и затем, ' с дальнейшим ростом температуры, начать уменьшаться (это наблюдается около критической точки).
Диаграмма (рис. 123) дана для
одного состава. Для представления состава в качестве переменной можно применить систему из трех координат и получить трехмерную диаграмму, содержащую поверхность р — t — х. Для того чтобы представить, хотя бы частично, все три переменные на плоскости, можно применить следующий прием. Произведем большое количество сечений этой поверхности при постоянном составе и спроектируем все получающиеся кривые на одну плоскость, как показано иа рис. 124. Типичными системами, обладающими свойствами, иллюстрированными этой диаграммой, являются СО2 — SO2 [42] и этан — гептан [125]. Кривые 1 и 7 представляют кривые давления пара двух чистых компонентов, а остальные представляют кривые, типичные для смеси с ветвями для пара и жидкости. Точки, где ветвь пара одной кривой пересекает ветвь жидкости другой, представляют сосуществующие фазы при равновесии
1 и 7—кривые для чистых компонентов, 2— 6— для различных составов; С — соответствует критической точке; Cf — кри-кондентермической течке; Л — криконден-барической точке.
☆ ) Этой точке не присвоено названия, ио по аналогии с крикондентерми-ческой точкой ее можно было бы назвать »криконденбарической“.
Полностью смешивающиеся жидкости
617
(например, точка Д). Это ясно из того, что жидкая и паровая фазы при одинаковых давлении и температуре должны находиться в равновесии. Кривая СдСв является огибающей кривой, связывающей критические точки двух чистых компонентов, и представляет геометрическое место критических точек смесей. Она будет называться критической кривой. Форма этой кривой, показанная на рис. 124, будет одной из наиболее общих, но все же она не охватывает всех изученных случаев. Исследовались случаи, при которых критическая кривая обладала температурным максимумом и минимумом.
На рис. 125—127 приведены соответственно кривые р — х, t—х и у — х для системы того типа, который изображен на диаграмме р — t рис. 124. На диаграмме I (рис. 125) температура ниже критической температуры любого чистого компонента; на диаграммах II и Ш она выше критической температуры более летучего компонента, но ниже критической температуры другого компонента. Смеси, более богатые компонентом А, чем хг (диаграмма II) или х2 (диаграмма III), не могут образовывать двух фаз. На диаграмме I (рис. 126) критическое давление ниже критического давления любого компонента; на диаграмме II оно выше критического давления менее летучего компонента; на диаграмме III оно выше критических давлений обоих компонентов. Соответствующие кривые на диаграмме у — х при постоянном давлении показаны на рис. 127.
Очень интересное явление происходит в том случае, если изотермически сжимается смесь, состав которой лежит между составом критической точки и крикондентермической точки. На рис. 128, представляющем увеличенное изображение критической области рис. 123, линия АВ представляет изотермическое сжатие при постоянном общем составе, начиная от пара в точке А. При давлении, отвечающем точке D, достигается линия точек росы; если сжатие продолжается, количество жидкости сначала увеличивается, затем проходит через максимум и уменьшается и, наконец, исчезает совершенно, когда снова достигается линия точек росы (точка Е). Дальнейшее сжатие не будет вызывать разделения на две фазы. Это явление известно под названием обратной конденсации; впервые о нем сообщил Куэнен в 1892 г.
Изменение относительных количеств двух фаз легче представить на диаграмме р—х (рис. 129), поскольку любая вертикальная линия, проходящая через области двух фаз, рассекает горизонтальные соединительные линии на участки, длины которых пропорциональны количествам фаз (доказательство см. гл. XIII, стр. 671). Например, отношение HGfFH равняется отношению массы жидкости к массе пара. В точке D отношение равно нулю; то же самое наблюдается в точке Е. Между этими двумя точками оно, очевидно, проходит через максимум.
При любом составе слева от критической точки С наблюдаются явления конденсации обычного типа, т. е. жидкая фаза, появляю-
618
XII. Равновесие при испарении и конденсации
щаяся в точке росы, продолжает увеличиваться в своем количестве до тех пор, пока вся система не становится жидкой (в точке пузырьков). Это можно видеть, рассматривая процесс LN.
Рис. 125. Кривые давление —-состав в критической области.
Рис. 126. Кривые температура— состав в критической области.
Рнс. 127. Кривые у —х в критической области.
Рис. 128. Обратная конденсация.
Если относительное положение точек С и С' является таким, как это показано на рис. 130, то мы имеем особое положение: линия изотермического сжатия ADEB достигает теперь линии жидкости в точке D, и выделяющуюся фазу следовало бы считать паром й). Пока происходит сжатие, она будет возрастать в количе-
☆ ) В критической области нет резкого различия между жидкой и паровой фазами, а в гомогенной области выше критической точки различия иет совсем. В критической области может быть целесообразнее рассматривать «менее плотную» н «более плотную» фазы.
Полностью смешивающиеся жидкости
619
стве, проходя через максимум, а затем уменьшаться до нуля (в точке Е), когда снова пересекается линия жидкости. Это явление было предсказано Куэненом [139] и названо им „обратной конденсацией второго рода”, ио никогда не наблюдалось, хотя требуемое относительное положение точек С и С известно.
Подобные явления могут быть предсказаны для изобарного процесса при использовании диаграммы р — t или диаграммы t — х, за
Рис. 129. Обратная конденсация, показанная на диаграмме давление — состав.
С — критическая точка; О — криконден-термическая точка.
Рис. 130. Обратная конденсация второго рода.
С — критическая точка; С* — криконден-термическая точка.
исключением того, что в этом случае состав был бы промежуточ-ным между составами, отвечающими точкам С и М (рис. 123), и тип обратной конденсации зависел бы от относительного положения этих двух точек.
До последнего времени явление обратной конденсации представляло только теоретический интерес и не имело связи с какой-либо практической задачей. Это положение резко изменилось при развитии процессов извлечения нефти из скважин (связанных с использованием обратной конденсации). В некоторых глубоких скважинах, где преобладают высокие температуры, нефть существует главным образом в состоянии пара при давлениях порядка 100—200 атм. При понижении давления происходит обратная конденсация, потому что, хотя конденсация и обусловлена уменьшением давления и температуры, все же значительная часть ее происходит при изотермическом расширении. Остаточный газ после этого сжимается и возвращается в скважину для поддержания давления, так как если дать давлению снизиться, то нефть будет конденсироваться в скважине и поглощаться песками, из которых ее нельзя извлечь. Для дальнейших подробностей можно обратиться к статьям КатцаиКурата [122], Катца и Синглетерри [123] и Де Бака [54].
620
XII. Равновесие при испарении и конденсации
КОЛИЧЕСТВЕННОЕ ИССЛЕДОВАНИЕ НЕИДЕАЛЬНЫХ СИСТЕМ
Общие уравнения для фазовых равновесий в бинарной системе выведены в гл. IV. Теперь мы перейдем к применению некоторых из этих уравнений к системам таких типов, которые представляют интерес с точки зрения перегонки.
Законы Коновалова [330]. Сначала мы сделаем некоторые общие выводы, используя уравнения (138, гл. IV) и (139, гл.- IV) в следующих специальных формах, применимых к случаю постоянной температуры:
bv2dp = (y — x)^-dx, (6)
bV'dp = (х—у) dy, (7)
где Дг>] и ixv2 — соответственно сокращенная запись коэфициентов при dp в уравнениях (138, гл. IV) и (139, гл. IV)-
Разделив уравнение (6) на уравнение (7), получим
До2 _ d2ZLldx2 dx
Aih tfZy/dy2 dy • 17
Если состояние системы значительно удалено от критической области, то в качестве приближения можно написать
&v2 = vv, (9)
— vy. (10)
Из рассмотрения необходимых условий равновесия при постоянных давлении и температуре было показано [139], что
d2Z d-Z-^
или -ч—, 0. (11)
дх2 ду2 ' ' '
Из этого следует, что
^>0, (12)
dy '
т. е. возрастание содержания данного компонента в одной фазе всегда вызывает возрастание содержания его в другой.
Подобным же образом можно показать, что прибавление к системе такого компонента, который присутствует в паре в большем количестве, чем в жидкости (у/ х), будет вызывать возрастание давле-
ния. Это представляется общим правилом, за исключением области, близкой и критической, где нельзя ожидать, чтобы это было верно, так как уравнения (9) и (10) уже недействительны.
Из уравнений (6) и (7) следует, что
dp У — х d2ZL . „
dx Ди2 дх2 ' '
Количественное исследование неидеальных систем
621
и
dp x—y d*Zv . .
dy~ Avj ду2 • V*’/
Когда у = х, то
^ = ^ = 0. (15)
dx dy
Другими словами, во всех случаях, когда составы пара и жидкости одинаковы, на кривых р— х или р—у должны быть максимумы или минимумы (см. рис. 119), и наоборот, наличие экстремумов свидетельствует о тождестве состава фаз.
Эти три правила, известные как „законы Коновалова“, оказались полезными для установления общего направления кривых изотермических равновесий.
Уравнение Дюгема *). Это уравнение, некоторые формы которого были выведены в гл. IV, большей частью пишется в одной из следующих форм, применимых только к изотермическим системам, если пар является идеальным газом;
xrf In —[—(1—x)dlnpB = 0, (16)
у,Гх-т (167, гл- IV)
dy _у(1— у»)
Интегрирование любого ш этих уравнений дает соотношение, из которого можно вычислить составы пара, если известны составы жидкости как функции давления при постоянной температуре. Это является важным результатом, поскольку измерение давлений пара для ряда жидких смесей обычно значительно проще, чем определение состава равновесного пара.
Маргулес [164] предложил следующие решения уравнения (16):
Рв=Рв^-^ + л (18)
Эти уравнения также можно написать следующим образом:
1пУд= “2(1 -х)’ + |’(1 -х)’+ ..., (19)
1пТв=Ьхг+к^+.,.> (20)
й) Некоторые авторы предпочитают называть его уравнением Гиббса — Дюгема. Для этого имеются достаточные основания, поскольку оно является только частным случаем общих уравнений Гиббса.
622
XII. Равновесие при испарении и конденсации
где у— коэфициент активности. Пользуясь уравнением Дюгема, можно найти следующие соотношения между константами:
?2 = а2 4“ аз»
Р3= а3.
Константы а2 и а3 (или если желательно, то большее их число) могут быть найдены из р— х-данных. Завидский [265] применил уравнения (17) и (18), используя лишь две константы а2 и а3, которые были определены из наклона кривой р — х на концах ее, где х = 0 и 1, и получил вполне хорошее совпадение со своими соб-ственными экспериментальными измерениями. Уравнениями для этой цели служат:
(2”
и
?+? = !» [pA-(g)„,]-l»Pi. (22)
Леви [148] предложил два метода применения уравнений Маргулеса при вычислении составов пара по измерениям общего давления как функции состава жидкости. Использование лишь первой константы в рядах Маргулеса дает
1пул = а(1 —х)2, In ув = ах2.
Если раствор идеален, то 1пу = у—1. Используя это равенство в качестве приближения для растворов, которые незначительно отклоняются от идеального, получим следующее уравнение, эквивалентное уравнению Леви, но в несколько иной форме:
\/ .,—Раг< Ра^~х)^р
V У— рх-Гр\рА(\-х)+рвх}'
где Др — разность между действительным общим давлением и вычисленным общим давлением для идеального раствора.
Для растворов, которые значительно отклоняются от идеальных, Леви сохраняет две константы рядов Маргулеса и для определения их из конечных наклонов кривой общего давления пользуется методом Маргулеса, применяя скорее аналитический, чем графический метод. Таким образом, он легко получил
дх)х=л
^Р—Рв X
др\ =РА — Р dxj x=i 1 — х
Количественное исследование неидеальных систем
623
Для применения этих уравнений необходимы очень точные данные по общему давлению пара раствора в каждой из областей большого разведения и столь же точные данные по давлению пара чистых компонентов. Непосредственное определение разности давлений было бы желательно и в действительности оно очень важно для достижения требуемой точности89.
Карлсон и Колборн [40] произвели широкое изучение решений Ван-Лаара для уравнения Дюгема и нашли, что по многим бинарным системам они хорошо представляют данные и более удобны для применения, чем решения Маргулеса. Решениями Ван-Лаара являются
lgb-[ Лх~р’ (24)
L1 + В (1-х) ]
lgb = [ R(l — Х)р’ (25>
Ах J
откуда
Л = [1 + (1~g)1pB |2. (26)
B = lgv Г 1 _—gp—у, (27)
s 18 [. 1 (1 — Jf) 1g тв J ' '
где А и В—константы при данной температуре.
Если х = 0, то lgyA = X, lgyB = O и yst=1,0.
Если х=1, то lgYA = O, у = 1,0 и lgYe = B.
Так как,
по определению,
РУ Рах то для азеотропной смеси, для
Гл
и Ь />в(1-х)’ которой у = х,
р
^=Га И
ь = й
Следовательно, мы видим, что уравнения Ван-Лаара по данным определить равновесную кривую у — х, не нуждаясь в каких-нибудь других данных по составу.
Карлсон и Колборн предложили следующий метод нахождения у, х-данных на основании измерения общего давления.
можно вычислить константы для азеотропных смесей и
и В отсюда
Из определения коэфициентов активности можно написать, что
А
__ р — 1вРв<1—х)
*л Рах
v _ Р~ЧаРах Тв— Рв^-х) •
(28)
(29)
624
XII. Равновесие при испарении и конденсации
Подставляя в уравнение (28)ув = 1,0 (значение для х = 0), находят приближенные значения уд; нанося затем значения как функцию х и экстраполируя полученную кривую до х = 0, получают окончательное значение, которое, как мы уже показали, должно быть равно А. Подобное же применение уравнения (29) приводит к нахождению В. Таким образом, получают первое приближение к точным значениям и путем повторения процесса при использовании новых значений уд и ув, найденных из приближенных констант, получают следующий ряд величин. Последние обычно достаточно близки к точным величинам, но в некоторых случаях могут оказаться желательными и дальнейшие вычисления.
Кривые igy— х, полученные из уравнений Ван-Лаара или каким-либо иным методом интегрирования уравнения Дюгема, будут также полезны для сглаживания и интерполирования экспериментальных данных в этой области.
а/ Льюис и Морфри [156] получили составы пара по данным для составов жидкости путем приближенного ступенчатого интегрирования уравнения (167, гл. IV). Они допустили, что в узком интервале равновесное соотношение у — х может быть представлено одним из следующих уравнений:
х = а-\-Ьу,
При использовании линейной зависимости уравнение (167, гл. IV) легко интегрируется в пределах от хп уг, рх до х2, уг, р2, давая
(1—й —ft)lg?-=^l —alg^ = lg^2 (30)
1 — Уг Xi Pi и
fr X2 —^1
Уг~У\
Начиная от исходной точки хр уг, рг, где состав пара известен так же, как в точке, где х1=у1 (чистый компонент или постоянно кипящая смесь), у2 можно найти путем последовательного подбора при данных х2 и р2. Затем эта точка может быть использована в качестве исходной для следующей ступени и т. д. Этим путем можно охватить целый ряд составов, причем лучше начать с обоих концов и пользоваться обоими способами. Метод проверен на четырех обычных бинарных растворах органических жидкостей, и получено хорошее совпадение с экспериментально найденными составами пара. Метод этот является более трудоемким и, пожалуй, не пред
Количественное исследование неидеальных систем
625
ставляет преимуществ по сравнению с другими методами, более простыми для применения.
Эмпирические зависимости. В гл. IV мы получили общее уравнение (для случая, когда пар является идеальным газом)
У 1-у
(169, гл. IV)
Теперь допустим, что функцию тг можно связать с составом жидкости посредством степенного ряда
тг = а -|- bx -|- ex2 -|- dx3
Тогда
p = ft4-2cx4-3dx24- ...
дх 1 1 1
У ____ х е1>+ 2сх+ЗАс’Н--
1 —у 1 — х
(31)
Это будет полезным эмпирическим соотношением для алгебраического представления данных у — х. Для частного случая, когда с и d очень малы, уравнение (31) превращается в
У 1-У
(32)
1 —х
где а — эмпирическая константа. Для частного случая идеальных растворов
а = — Рв
В общем, величина я, определенная по уравнению (32), очень близка к константе как для изотермы, так и для изобары в случае растворов, которые незначительно отклоняются от идеальных. Например, Додж [62] •показал, что изобарные данные Доджа и Дунбара [65] для системы кислород — азот можно достаточно хорошо представить посредством уравнения (32) со следующими значениями я:
р, атм 0,50 1,00 5,00 10,00 15,00 20,00
о (отнесенная к мольной доле О2) 0,2027 0,2478 0,3798 0,4681 0,5292 0,5750
40 Химическая термодинамика
626
XII. Равновесие при испарении и конденсации
Выражая а как функцию давления с помощью эмпирического уравнения или графическим путем, получают простой способ увязки состава жидкости и пара при любом давлении в этих пределах, достаточно точный для технических целей.
Два следующих уравнения, повидимому, будут полезны для специальных случаев:
* V (33)[147]
1—> \1— xj ' J
и
1 РУу.^> = уР ЫРв . (34)г204]
SP(1— У)х dx Ра—Рв 1
-
% Уравнение (33) применяется для представления экспериментальных
данных у — х, но на нем нельзя основывать никаких теоретических выводов, потому что оно противоречит уравнению Дюгема. Уравнение (34) можно использовать для вычисления состава пара при данном давлении пара жидкости как функции концентрации. Авторы уравнения (34) считают, что оно в отдельных случаях позволяет осу-ществнть весьма точный расчет, но, согласно Льюису и Морфри, [ по крайней мере, в одном частном случае оно оказалось совершенно
неправильным.
t ' Приложения уравнения Дюгема й). Большая часть расчетов
[ по перегонке зависит от данных по равновесию жидкость — пар,
| и эти данные должны быть достаточно точны, если желают полу-
* чить результаты, заслуживающие доверия. Получение подобных
( данных трудно обеспечить, о чем свидетельствуют неточность и
I несогласованность результатов, приводимых в литературе, и по-
, этому важно располагать некоторыми способами для оценки их
совместимости. Зто является главной областью применения уравнения Дюгема.
Изотермические данные. Уравнение (16) можно легко преобразовать в уравнение
dp^dx _dpB/d(l—х)
Ра!х рвЦ1-х) ’ ( }
графическая интерпретация которого дана на рис. 131. На нем нанесены кривые парциальных давлений пара для системы этиловый спирт — хлороформ [226] при 55° С, и наклоны, требуемые уравнением (35), обозначены прямыми линиями. Так, если точка, где х = 0 и р = 0,
☆ ) Бблыная доля материала этого раздела основана на статьях Битти и Калингерта [13] и Карлсона и Колборна [40].
Количественное исследование неидеальных систем 6ZI
обозначена через О, а точка, где х=1 ир = О, обозначена через О', то получаем
-?А- = ОД, X
dPB d(\—x)
EF,
-^-z=O’D.
1 —x
Определяя наклон для этой системы при х = 0,50, найдем, что уравнение (35) удовлетворительно в пределах точности графического
определения наклона (около 1°/0). Таким образом, можно» заключить, что эти равновесные данные точны при условии, что пар можно принять за идеальный таз.
Приложение уравнения Дюгема в форме уравнения (35) включает допущение об идеальности газов, и поэтому всегда следует помнить, что несогласованность данных с уравнением не обязательно означает, что данные не точны. Если давление равно 1 атм или меньше этой величины, то следует ожидать совпадения в пределах нескольких процентов, если только не наблюдается ассоциации в парообразном состоянии, как для случая паров муравьиной или уксусной кислот.
Эти данные можно также проверить с помощью уравнения Дюгема в форме
Рис. 131. Проверка данных по равновесию жидкость — пар посредством уравнения Дюгема.
(Поданным для растворов этиловый спирт—хлороформ [226]).
--------rfy
(167, гл. IV)
и интегрированием его для определения р. Это производится графическим путем — нанесением (у — х)[у (1 —_у) как функции у и измерением 40*
628
XII. Равновесие при испарении и конденсации
Рис. 132. Использование уравнения Дюгема для проверки данных по равновесию жидкость— пар системы метанол — бензол при 40°С; точка р = 0, х = 0 в тексте обозначается 0 [146J.
площади под кривой. Скатчард и Реймонд [226] произвели эту проверку и получили очень хорошее совпадение между вычисленными и наблюденными величинами.
На рис. 132 показана проверка данных Ли [146] для системы метанол — бензол при 40°С. При х = 0,20 отношение наклонов
ВС 10 А = 1g = 0,1095 и отношение наклонов EF\O’D == 0,0830. По уравнению (35), эти два отношения наклонов должны быть равны. Поскольку эти отношения не совпадают и поскольку при таких низких давлениях эти два пара, в сущности, являются идеальными газами, приводится сделать вывод, что данные не очень точны.
Изобарные данные. Уравнение Дюгема выведено при допущении изотермических условий и строго приложимо только к изотермическим данным. Это обстоятельство должно налагать строгие ограничения на его применение, поскольку многие данные по равновесию жидкость — пар, получен
ные для использования при расчетах перегонки, являются изобарными. Если, однако, пользуются уравнением в следующей форме, выведенной в гл. IV,
dln^= — (151, гл.IV)
где аА\х и — коэфициенты активности у компонентов А и В, то
оно приложимо без значительной ошибки и к данным при постоянном давлении. Причину этого легко обнаружить, если написать
Количественное исследование неидеальных систем 629
что правильно в том случае, когда пар является идеальным газом и когда за стандартное состояние принято состояние чистого компонента в виде жидкости. Тогда как отдельные парциальные давления пара значительно изменяются с температурой, отношения их изменяются много меньше и
могут считаться практически постоянными в том сравнительно узком температурном интервале, с которым приходится иметь дело при изобарной перегонке. Битти и Калингерт [13] продемонстрировали справедливость этого допущения, показав, что изотермические данные по системе сероуглерод — ацетон при 35,2° С и изобарные данные при 760 леи (/ = 39 — 54° С) попадают на практически совпадающие кривые при нанесении их в виде коэфициентов активности в зависимости от мольной доли. Кривые также совпадают с требованиями, диктуемыми уравнением (151, гл. IV). Это свидетельствует о
Рис. 133. Зависимость коэфициентов активности, вычисленных по уравнению Ван-Лаара, от мольных долей [40].
том, что совпадение не
случайно.
Уравнение (151, гл. IV) можно также выразить в форме
</]87л 1 £]йТв dx х dx
(36)
и в этой форме оно может быть использовано для проверки изобарных данных с помощью отношения двух наклонов, т. е. аналогично использованию уравнения (35).
Типичные кривые активности легко можно найти из уравнений Ван-Лаара подбором значений А а В. Кривые, приведенные на рис. 133 (взяты из статьи Карлсона и Колборна) и основанные на значениях 4=1,13 и В = 0,49, очень хорошо воспроизводят данные по системе я-пропиловый спирт — вода при 1 атм. Диференцированием
630
XII. Равновесие при испарении и конденсации
уравнений Ван-Лаара получаем
/д1йтд\ =_ 2Л2
\ dx )х=й В ’
г <?igTB 1
р(1 —x)J(i-x)=o А ’
Г J'gTB.,1 =0
1.0(1 — х) J(l—Ж)=1
=q \ dx Jx=i
Эти уравнения полезны для указания направления кривых в конечных точках. Приближение горизонтальной касательной к значению у == 1,0 показывает, что закон Рауля, приложим к компонентам, мольная доля которых приближается к 1,0, а закон Генри требует, чтобы другой конец каждой кривой достигал конечного значения при конечном наклоне. Если действительные данные нанесены таким образом и расходятся с этими требованиями, то это служит качественным признаком неточности данных.
График у — х показывает, в какой степени любой бинарный раствор отличается от идеального, так как для идеального раствора обе кривые при у 1,0 должны быть горизонтальными линиями.
Из этих кривых, нанесенных по экспериментальным данным, можно сделать и другие качественные заключения. Например, наклоны линий Должны всегда обладать противоположным знаком при данном значении х; если наклон одной из кривых равен нулю (максимум или минимум), другая линия тоже должна иметь нулевой наклон при том же самом значении х (за исключением крайних точек); если для одного компонента значения у всегда больше единицы и не имеют максимума, то у для другого компонента должны быть также больше единицы. Следовательно, если экспериментальные данные нанесены таким образом, то часто сразу можно увидеть, что в некоторых областях данные неточны.
Пример 4. Проверить и оценить точность опубликованных данных по равновесию пар — жидкость для системы толуол — я-октан при 1 атм.
Температуры кипения для этой системы были определены в отдельных сериях опытов. Из графика этих данных можно определить температуры кипения, соответствующие различным равновесным составам. Затем необходимо иметь данные по давлению пара чистых компонентов в данном интервале кипения. Для расчета этих величин используем уравнения, выведенные Битти и Калингертом [13]:
толуол lgp = 7,7309 ---,
, 1941,4
я-октан lgp = 7,7503 ------—,
(р дано в миллиметрах ртутного столба и Т — в градусах Кельвина). Были получены две серии данных по составу жидкость — пар: одна, основанная
Количественное исследование неидеальных систем
631
на определении плотности, а другая — на определении показателей преломления. Для уменьшения количества подсчетов и уменьшения материала, подлежащего сведению в таблицы, в этом примере используются только данные, основанные на измерении плотности.
Таблица 21
Вычисленные результаты по проверке равновесных данных в системе толуол — н-октан
Мольная доля толуола Парциальное давление, мм Hg Коэфициент активности, f Логарифм коэфициента активности
жидкость (х) пар О') толуол октан толуол октан толуол октан
0,0970 0,1640 125 635 1,177 0,988 0,0708 —0,0052
0,2030 0,2970 226 534 1,084 1,005 0,0350 +0,0022
0,3000 0,4100 312 448 1,056 1,0.11 0,0278 0,0048
0,3460 0,4605 350 410 1,063 1,016 0,0265 0,0069
0,4075 0,5265 400 360 1,060 1,014 0,0253 0,0060
0,4800 0,5855 445 315 1,036 1,048 0,0154 0,0204
0,5270 0,6235 474 286 1,028 1,070 0,0120 0,0294
0,5710 0,6620 503 257 1,028 1,083 0,0120 0,0346
0,6145 0,6975 530 230 1,023 1,099 0,0099 0,0410
0,6630 0,7300 555 205 1,014 1,143 0,0060 0,0581
0,7215 0,7785 592 168 1,010 1,153 0,0043 0,0618
0,7570 0,8030 610 150 1,003 1,196 0,0013 0,0777
0,7945 0,8325 633 127 1,003 1,212 0,0013 0,0835
0,8235 0,8555 650 110 1,000 1,229 0,000 0,0896
0,8580 0,8825 671 89 1,000 1,249 0,000 0,0966
0,9075 0,9225 701 59 0,999 1,283 —0,0004 0,1082
0,9450 0,9550 726 34 0,999 1,251 —0,0004 0,0973
Парциальные давления и коэфициенты активности вычислялись затем прн допущении, что пары явлиются идеальным газом; полученные результаты сведены в табл. 21 и нанесены на рис. 134. Производя количественное сравнение наклонов посредством уравнения (36), найдем:
Мольная доля
0,25 0,50 0,75
Теоретическое отношение
/ 1—
наклонов =----------]
V х )
Действительное отношение наклонов
— 3,0 —1,0 —0,33
—0,40
632
XII. Равновесие при испарении и конденсации
Хотя наклоны не могут быть определены точно, но большие расхождения, обнаруживаемые этими числами, достаточны, чтобы показать, что экспериментальные данные не очень точны.
Рис. 134. Проверка данных по равновесию жидкость — пар с помощью уравнения Дюгема.
(По данным для сйстемы толуол — октан [28].)
Интегрируя уравнение (151,гл. IV),
Получение кривых коэфициента активности посредством графического интегрирования. Для исчерпывающего анализа изотермы нлн изобары для целого ряда составов можно графически проинтегрировать уравнение (151, гл. IV) и воспользоваться им для вывода ряда последовательных кривых у— х. Этот способ имеет некоторые преимущества по сравнению с интегрированием посредством таких уравнений как уравнения Маргулеса или ВанЛаара, так как он является строгим и не зависит от того, приложимо ли данное частное уравнение или нет.
Способ кратко можно охарактеризовать сле-
дующим образом, получаем
2
1
Мтл)2—HrJi
(37)
Принимаем в качестве нижнего предела х=1 (чистое А} и y=L Тогда уравнение (37) переходит в
*
1пТл=~
л=1
(38)
При другом пределе х = 0, согласно закону Генри, (ул)2 = Ад, и, следовательно, интеграл достигает постоянной величины, равной 1пЛд.
Частично смешивающиеся жидкости
633
Подобным же образом
X \п1в=~ J (39)
. . ..... ______^=0 ... __________.. .
и при х=1 интеграл достигает постоянного значения 1пйв.
С помощью этих уравнений любая кривая коэфициента активности может быть найдена из другой кривой посредством графического интегрирования. Например, примем, что кривая ув— х может быть временно фиксирована на основе экспериментальных данных. Наносим (1 —х)/х как функцию In ув, начиная с х= 1, где (1 — х)/х=0 и ув = &£— константе закона Генри. По мере приближения к другому пределу (х=0) величина (1—х)/х приближается к бесконечности, но интеграл, как показано выше, тем не менее приобретает постоянное конечное значение. Значение интеграла для любого данного х является площадью под этой кривой, определяемой многими методами.
Таким образом получается ряд кривых коэфициентов активности, которые должны совпадать с экспериментальными данными, если последние точны. Если же наблюдаются значительные отклонения, то это служит определенным признаком какой-то неточности в экспериментальных данных. При повторном определении положения одной из кривых коэфициента активности и повторном пересчете согласно описанному выше методу возможно с помощью нескольких подборов притти к термодинамически связанной паре кривых, которые лучше всего представляют данные в целом. Это один из способов сглаживания и согласования данных по равновесиям жидкость — пар при постоянной температуре или при постоянном давлении.
Попутно следует отметить, что испытание изобарных данных требует точных сведений по давлениям пара двух чистых компонентов во всем температурном интервале. Недостаток или отсутствие их делает невозможной проверку многих данных, имеющихся в литературе.
Для дальнейших примеров применения уравнения Дюгема в этой области можно обратиться к статье Битти и Калингерта [13].
ЧАСТИЧНО СМЕШИВАЮЩИЕСЯ ЖИДКОСТИ
К этому классу принадлежат многие пары жидкостей, имеющих промышленное значение, например анилин — вода, фурфурол — водам фенол — вода. Если к чистой жидкости А добавляют небольшое количество жидкости В, сохраняя температуру постоянной, то сначала В растворяется в А, образуя только одну жидкую фазу. Если добавление В продолжается, то достигается точка, при которой появляется другая жидкая фаза. Из правила фаз следует, что рассматриваемая
634
XII. Равновесие при испарении и конденсации
система в данном случае (прн наличии двух жидких и одной парообразной фаз) является унивариантной, и при фиксированной температуре состояние системы будет неизменным; другими словами, давление и состав всех трех фаз и свойства фаз фиксированы. Жидкие фазы будут насыщенными растворами одного компонента в другом, и дальнейшее добавление В приведет только к изменению относительного количества двух жидких фаз. Если общий состав жидкости в целом достигает состава, соответствующего насыщенному раствору А в В, то дальнейшее добавление В приведет к исчезновению той фазы, которая является раствором В в А.
Критическая температура растворения. Наблюдения, подобные только что описанным, можно производить и при других постоянных температурах, но состав двух насыщенных растворов будет изменяться с температурой. Во многих случаях по мере возрастания температуры обе жидкие фазы приближаются друг к другу по составу и, наконец, становятся идентичными, а выше этой температуры существует только одна жидкая фаза. Температура и состав, при которых это происходит, определяют „критическую точку", аналогичную той, при которой становятся идентичными жидкость и пар в однокомпонентной системе или в бинарной системе из полностью смешивающихся жидкостей. Такое поведение графически представлено на рис. 135, который показывает, ка
ким образом состав всех трех сосуществующих фаз изменяется с температурой. Так как имеется только одна степень свободы, то ясно, что давление не является независимой переменной, но определенным образом изменяется с температурой. Типичным примером системы, ведущей себя таким образом, является система фенол — вода [229]. Смеси гексана и метилового спирта дают подобную же диаграмму, за исключением того, что состав паровой фазы является промежуточным между составами двух жидких фаз. Смеси анилин—вода также дают диаграмму этого типа с верхней критической температурой растворения 90.
В некоторых системах, например в системе триэтиламин — вода, критическая точка для двух жидких фаз достигается при понижении температуры, как показано на рис. 136 [213]. Замечательно исключительно быстрое изменение растворимости обеих жидкостей при приближении к критической точке. Кривая пара является условной, поскольку экспериментальных данных для нее нет.
Критическая ' точка
Состав
Рис. 135. Диаграмма температура— состав для трехфазной системы (две жидкости и пар) с верхней критической температурой растворения для двух жидких фаз.
Частично смешивающиеся жидкости
635
Ввиду того, что известны системы как с верхней, так и с нижней критической температурой растворения, естественно рассчитывать на возможность обнаружить критические точки в одной данной системе, если наблюдения будут проведены в достаточно широком тем—- пературном интервале. Результатом была бы диаграмма, подобная показанной на рис. 137. Было открыто несколько систем с такими свойствами. Классическим примером является система никотин —
Рис. 136. Диаграмма температура—состав для трехфазной системы (две жидкости и пар) с нижней критической температурой растворения для двух жидких фаз.
Рис. 137. Диаграмма температура— состав для трехфазной системы (две жидкости и пар) с верхней и с нижней критическими температурами растворения для двух жидких фаз.
вода [115]. Возможно, что все системы показывали бы этот тип поведения, если бы не было того обстоятельства, что твердая фаза появляется раньше, чем достигается нижняя критическая температура, а критическая точка пар — жидкость достигается раньше верхней критической точки двух жидкостей.
На рис. 138 изображен тип диаграммы, даваемый смесями этиловый эфир—-вода [136], в которых раствор, богатый эфиром, становится идентичным с паром примерно при 200° С. Другим типом диаграммы, который следовало бы ожидать на основании общих принципов, будет диаграмма с нижней критической точкой жидкость— жидкость и верхней критической точкой жидкость — пар, показанная на рис. 139. .Этот тип диаграммы дают смеси этана с различными алифатическими спиртами, например этиловым и пропиловым.
Типичные диаграммы давление—состав. При перегонке систем, включающих частично смешивающиеся жидкости, интересуются
636
XII. Равновесие при испарении и конденсации
не только трехфазными равновесиями, но также н двухфазными. На рис. 140—142 показаны некоторые, полученные опытным путем, типичные диаграммы давление — состав (р — х) при постоянной температуре. На рис. 140, представляющем систему, подобную системе анилин — вода или фурфурол — вода (случай I), где давление насыщен
Состав
Рис. 138. Диаграмма температура — состав для трехфазвого равновесия в системе этиловый эфир — вода.
Критическая точка
жидкость-жидкость
Состав
Рис. 139. Диаграмма температура— состав для трехфазного равновесия в системе с нижней критической температурой растворения для жидких фаз и с иной критической точкой для жидкой и паровой фаз.
ного пара над равновесными растворами больше давления пара любого из компонентов, показаны следующие равновесия:
АВ — кривая давления пара ненасыщенной жидкости 2 (жидкость богата компонентом В).
АС — кривая пара в равновесии с жидкостью, состав которой меняется вдоль АВ. (Горизонтальная соединительная линия, например JK, связывает жидкую и паровую фазы в равновесии.)
ED— кривая давления пара ненасыщенной жидкости 1 (богатой А).
ЕС—кривая пара этого равновесия между паром и жидкостью 1 (HJ — соединительная линия, подобная линии JK}.
Точка В — раствор, богатый В и насыщенный А.
Точка D — раствор, богатый А и насыщенный В.
Все составы общей системы, лежащие между точками В и D, будут давать две жидкие фазы, имеющие составы, представленные точками В и D, и паровую фазу, имеющую нонвариантный состав точки С. Кривые BG и DF представляют равновесие между двумя насыщенными жидкостями при отсутствии паровой фазы. Наклон этих линий зависит от изменения объема при смешении компонентов. Если смешение сопровождается увеличением объема, то давление будет уменьшать смешиваемость, 'как показано на рис. 140, в противопо
Частично смешивающиеся жидкости
637
ложном же случае эффект будет обратный. Обычно эти двухфазные равновесия жидкость — жидкость не рассматриваются, поскольку они мало применяются при анализе перегонки. Влияние давления на сме-
Рис. 141. Диаграмма давление — состав для системы, содержащей две жидкие и одну паровую фазы (случай И).
Рис. 140. Диаграмма давление — состав для системы, содержащей две жидкие и одну паровую фазы (случай I).
шиваемость слишком мало, чтобы иметь какое-либо практическое значение.
Рис. 141 представляет собой случай II, когда давление насыщенного пара сосуществующих растворои яиляется промежуточным между
Рис. 142. Диаграмма давление — состав для системы, содержащей две жидкие и одну паровую фазы (случай III).
Рис. 143. Диаграмма температура — состав для системы, содержащей две жидкие и одну паровую фазы.
I,—жидкость 1; I, — жидкость 2.
давлением пара обоих чистых компонентов. АВ н СЕ—кривые давления пара ненасыщенных жидкостей, a AD и DE — кривые сосуществующих паровых фаз. Обе насыщенные жидкости, представленные
638
XII. Равновесие при испарении и конденсации
точками В и С, находятся в ствующего D.
На кривых давления пара нимумы для одной
равновесии с паром состава, соответ-
существуют также максимумы или ми-нз ненасыщенных жидких фаз. Случай максимума
Рис. 144. Соотношение между t—х-диа-граммами для полностью смешивающихся и для частично смешивающихся жидкостей.
(случай III) иллюстрируется на рис. 142. Такими свойствам^ обладает важная пара жидкостей: фенол — вода.
Постоянное давление. При решении проблем перегонки глав-
Рис. 145. у — х-кривые для систем из частично смешивающихся жидкостей.
ным образом приходится иметь дело с поведением систем при постоянном давлении, и поэтому кривые температура — состав при постоянном давлении более полезны, чем только что приведенные кривые. Однако лабораторные исследования равновесий обычно производятся при постоянной температуре, и диаграммы температура — состав должны затем строиться по данным для ряда диаграмм давление — состав. Диаграмма температура — состав для смесей, подобных анилину и воде, соответствующая диаграмме р— х (рис. 140), показана на рнс. 143. На этом рисунке отчетливо заметны различные области; всякие объяснения представляются излишними, поскольку диаграммы t — х и р—х подобны, причем одна является, в сущности, перевернутым изображением другой. Можно построить диаграммы t — х, соответствующие другим рассмотренным диаграммам р — х.
Связь между диаграммой рис. 143 и соответствующими диаграммами: 1) для полностью смешивающихся жидкостей (рис. 120, <5) и 2) для несмешивающихся жидкостей (рис. 114), очень интересна и показана на рис. 144. При наиболее низких температуре и давлении (р^диаграм
Частично смешивающиеся жидкости
639
ма приближается к диаграмме для пары несмешивающихся жидкостей, где обе жидкие фазы являются чистыми компонентами. По мере повышения температуры обе жидкости сближаются по составу между собой, и мы имеем типичную диаграмму для частично смешивающихся жидкостей. Если давление равно р4, то обе жидкости полностью смешиваются, и мы имеем ярко выраженный тип диаграммы с минимальной точкой кипения. При р3 линия постоянной температуры на диаграмме достигает критической температуры растворения, и обе жидкие фазы становятся идентичными.
Диаграммы у— х. Диаграммы состав пара — состав жидкостй, т. е. у — х, соответствующие диаграммам р— х, изображенным на рис. 140 (случай 1), 141 (случай II) и 142 (случай III), иллюстрируются на рис. 145 соответственно кривыми /, II и III. Общая форма этой диаграммы одинакова, независимо оттого, поддерживаются ли постоянными давление или температура.
Приложение законов простого раствора. Пусть А и В представляют два чистых компонента, а у и х — соответственно мольные доли А в паре и в жидкости. Допустим, что давление пара растворителя в любой из двух жидких фаз выражается законом Рауля (идеальный раствор), а для растворенного вещества — законом Генри. Эти допущения, конечно, являются строго правильными только в двух предельных случаях, когда х —►1 и х —>- 0; однако они практически справедливы и для сильно разведенных растворов. Тогда для раствора, богатого В, имеем:
PA = kAx> (40)
Рв = Рв(1— *)’
Р = Ра + Рв =
= ЬдХ Pg (1 х),
Р—Рв
^л — Рв ’
v z= Ра —= kAx
Р Р '
., kAp—Рв
У Р &А — Рв ’
_______kAx_____ kAx + pB(\ — х) ’
(41)
(42)
(43)
(44)
(45)
(46)
640 XII. Равновесие при испарении а конденсации
Подобно этому, для раствора, богатого А, можно вывести, что = + х>> (47) '=£f£,' ,48)
Поскольку парциальные давления А должны быть одинаковы над обоими равновесными между собой растворами и поскольку то же должно наблюдаться и для парциального давления пара компонента В, можно написать
*а=рЛ (50)
(51)
где индексы 1 и 2 относятся к насыщенным растворам, богатым соответственно А и В.
Приложение этих уравнений показано на следующем числовом примере.
Пример 5. При 110°С насыщенный раствор анилина в воде содержит 7,950/а анилина по весу, а насыщенный раствор воды в анилине содержит 88,05°/о анилина по весу. Давления пара чистого анилина и воды при 110° С соответственно равны 69,2 и 1073 мм. Построить диаграмму р — х —у для смеси при 110° С.
Мольные доли анилина в двух растворах равны
(7,95/93,1)
(7,95/93,1) + (93Л718) = 0’()162 (₽аствоР. богатый водой>-(88,05/93,1) „ ,
(88,05/93ЛН-(11,95/18)= °’587 (раствор’ богатый анилином).
По уравнению (50)
По уравнению (51) . 0,9838
*я=1073 ол1з=256°-
Общее давление для трехфазного равновесия вычисляется или по уравнению (42), или по уравнению (47).
По уравнению (42)
р = 2510 X 0,0162 4-1073 X 0,9838 = 1097.
Частично смешивающиеся жидкости
641
Линии р — х для обоих ненасыщенных растворов, согласно уравнениям (42) и (47), представляют прямые линии, соединяющие давление насыщенного пара чистого компонента с давлением насыщенного пара над сопряженными растворами. Значения у, вычисленные из уравнений (46) и (49), для принятых значений х приводятся ниже.
Раствор, богатый водой Раствор, богатый анилином
X 1 У X У
0,001 0,00234 0,587 0,0371
0,005 0,0116 0,650 0,0478
0,010 0,0231 0,700 0,0593
0,015 0,0344 0,800 0,0977
0,0162 0,0371 0,900 0,950 0,975 0,990 0,196 0,339 0,517 0,728
Чтобы дать диаграмму р—х—у для этой системы, эти результаты нанесены на рис. 146 й). Диаграмму i — х—у для системы при постоянном давлении можно получить нанесением ряда изотермических диаграмм и отсчетом величин при данном постоянном давлении.
Уравнение (46) можно также написать в форме
х + (рв/ЛЛ)(1-Х)’
и представить в такой же форме уравнение (49). Отношения pB/kA и ^в/Ра (аналогично относительной летучести а для идеальных растворов) не должны значительно меняться с температурой, и при использовании среднего значения можно вывести диаграмму у—х при постоянном давлении.
Этот метод получения равновесных данных для систем из частично смешивающихся жидкостей рекомендуется только в качестве первого приближения при отсутствии экспериментальных данных. Существуют серьезные сомнения относительно справедливости законов простого раствора даже для бесконечно разведенных растворов. Например, для случая фенол — вода, согласно данным Симса91, который обна-ружил постоянно кипящую смесь при 1 атм и 1,9О°/о (мольных) фенола в водном слое, отклонение от этих законов значительно.
< г
й) После того как эта задача была решена, появились данные Грисволда, Андреса, Арнета и Гарлэнда [95]. Эти исследователи дают составы сосуществующих фаз для раствора, богатого анилином, при 100° С. При сравнении их экспериментальных данных с вычисленными по иллюстрированному здесь методу, оказалось, что, несмотря на значительное отклонение от линейной зависимости р — х, вычисленные кривые у—р близко совпадают с экспериментальными.
41 Химическая термодинамика
Рис. 146. Диаграмма р — х—у для растворов анилин—вода при 110°С.
Рис. 147. Увеличенная диаграмма р — х—у для фазы, богатой водой. Система: анилин —вода при 110°С.
Тройные системы
643
ТРОЙНЫЕ СИСТЕМЫ
Рис. 148. Изображение состава тройной системы.
Ограничим рассмотрение случаем полностью смешивающихся жидкостей. Графический метод изображения, оказавшийся столь полезным для бинарных систем, применим и для этого случая, но он уже значительно сложнее. Если же следует рассмотреть больше трех компонентов, от него обычно следует отказаться совсем.
Состав трехкомпонентной системы удобно представить посредством равностороннего треугольника, как это показано на рис. 148. Это возможно благодаря тому свойству треугольника, что сумма перпендикуляров к трем сторонам, восстановленных из любой точки, равна высоте треугольника. Это легко показать нанесением эквидистантных линий, параллельных каждой из сторон треугольника. Эти линии, проведенные так, как это осуществлено на рис. 148, делят высоты на 10 рав-. ных частей. Если взять любую точку, например р, и опустить из нее перпендикуляры на стороны
треугольника, то увидим,
что отрезок pf равен двум единицам длины, отрезок ре — 3 единицам и отрезок pg—5 единицам, что в сумме дает 10. Тогда если вершины Л, В и С представляют соответственно 1ОО°/о компонентов А, В н С, то линии, параллельные противоположным сторонам, представляют 90°/о, 8О°/0 и т. д. компонента, отмеченного на соответствующей вершине. Следовательно, ab представляет все смеси, содержащие 9О°/о A, bd— все смеси, содержащие 10°/о С, а ас — все смеси, содержащие 1О°/о В. Сторона АВ будет характеризовать состав бинарной смеси А и В; то же справедливо для двух других сторон треугольника. Тогда точка р соответствует смеси, содержащей 20°/0 А, 50% В и ЗО°/о С.
Посредством перпендикуляра, восстановленного к плоскости этой треугольной диаграммы, можно представить другие параметры, как давление или температура, что и показано на рис. 149. Так как любая точка внутри этой призмы соответствует системе с тремя степенями свободы и так как однофазная трехкомпонентная система имеет четыре степени свободы, то одну из переменных следует
41*
644
XII. Равновесие при испарении и конденсации
Р= const*
Процент А
Рис. 149. Диаграмма температура—состав для тройной системы из полностью смешивающихся жидкостей.
считать фиксированной; в данном случае принимается постоянным давление. Если сосуществуют две фазы, при постоянном давлении будут две степени свободы, и поэтому мы будем иметь поверхность t— хА — хв для жидкой фазы и поверхность t—уА—ув для паровой фазы. Эти две поверхности соответственно показаны как DGFJEH и DKFMEL.
Составы любых двух сосуществующих фаз можно было бы представить прямыми (соединительными) линиями, связывающими эти две поверхности, но это не может служить какой-либо полезной цели и излишне усложнит диаграмму. Пересечения этих двух поверхностей с тремя вертикальными плоскостями, образующими грани призмы, дают знакомые линзообразные диаграммы t—х—у бинарных систем. Так, DHE представляет пересечение поверхности жидкости с передней вертикальной плоскостью призмы и является кривой I — х, т. е. кривой точек кипения бинарной смеси АВ. Подобно этому, DLE представляет пересечение с поверхностью пара и поэтому является кривой t—у, т. е. кривой точек росы для бинарной системы АВ.
Если мы рассматриваем любую плоскость, параллельную основанию призмы, например
плоскость QRS, то она будет пересекать две поверхности; линиями пересечения будут линии IF/V и UV соответственно для поверхности жидкости и пара. Эти две линии, которые спроектированы на основание (ab и cd), представляют составы фаз, которые сосуществуют при данных put, поскольку температура является постоянной вдоль любой горизонтальной плоскости. Обычно они не являются прямыми, но проводятся на фигуре как прямые линии просто для удобства. Чтобы найти составы обеих фаз, можно воспользоваться соединительными линиями, как показано посредством ef. По этой линии мы видим, что жидкость, состоящая примерно из 25°/0 А, 35°/0 В и 4О°/о С, при этом данном давлении и температуре находится
Тройные системы
646
в равновесии с паром, состоящим приблизительно из 65°/0 А, 2О°/0 В и 15°/0С.
При повороте трех вертикальных плоскостей призмы на 90° вокруг линий, соответствующих их основаниям, благодаря чему они будут лежать в плоскости основания, мы получим диаграмму, показанную на рис. 150; она удобна, поскольку показывает одно-
Рис. 150. Фазовая диаграмма для тройной системы.
временно фазовые зависимости как в трех бинарных системах, так и в тройной системе. Вся эта диаграмма в целом соответствует системе, находящейся при некотором постоянном давлении. Треугольная диаграмма в центре представляет горизонтальное сечение призмы и поэтому является плоскостью постоянной температуры и постоянного давления. Температура выбранная в этом случае, лежит выше точек кипения как А, так и В. Поэтому ей отвечает плоскость более высокого температурного уровня, чем плоскость, показанная на рис. 149 и лежащая выше точки кипения чистого А, но ниже точки кипения В.
Диаграмму у — х для тройных систем приданном р или t можно построить, как показано на рис. 151. Любая точка в треугольнике будет соответствовать некоторому составу жидкости (или пара), и будут существовать два ряда линий, один из которых состоит из линий, соединяющих все точки, отвечающие данному процентному содержанию компонента А в паре (или в жидкости), а другой — из
646
XII. Равновесие при испарении и конденсации
линий, отвечающих постоянному процентному содержанию компонента С в той же самой фазе. Во избежание путаницы на рисунке показано только по одной такой линии из каждого ряда. Пересечение в точке р должно соответствовать пару, содержащему 4О°/о А, 2О°/о В и 4О°/оС.
А
Процент В в жидкости
Рис. 151. у — х-диаграмма для тройных систем.
Равновесная жидкость, судя по координатам треугольника, оказывается состоящей из 2О°/0 А, 6О°/о В и 20°/0 С.
Грисволд и Динуидди [96] представили данные по составу пар— жидкость для тройной системы посредством двух диаграмм в обычных прямоугольных координатах. Такие диаграммы несколько легче для чтения, чем треугольные.
Рассмотренные нами примеры тройных смесей представляют случаи, в которых не образуется постоянно кипящих смесей. Такие азеотропные смеси хорошо известны в тройных системах, причем значительный промышленный интерес представляет система этиловый спирт — бензол — вода, которая усложняется также тем, что при 1 атм присутствуют две жидкие фазы. Рассмотрение трехкомпонентных систем с постоянно кипящими смесями или с частичной смешиваемостью жидкостей выходит за рамки этой книги.
Для тройных смесей, являющихся идеальными в жидкой фазе и пар которых можно считать идеальным газом, равновесные зависимости при постоянной температуре можно крайне просто выразить уравнениями, аналогичными уравнениям, использованным для бинар-
Системы из N-компонентов
647
ных систем. Так, если мы выразим все составы в мольных долях
компонентов А и В, то
Рл = ^л. <53>
Рв^Рв^в- (54)
Рс = Рс(1—ХЛ —хв)> (55)
Р = Раха+Рвхв + Рс^— ха~ хвЬ <56)
Уа=Р^’ <57>
Ув=~’ (58>
1—Ул~Ув=~ • <5Э)
Из этих уравнений можно легко вывести следующие уравнения:
Уа 1+(аЛС~ О-*л + (аВС—1)-«В ’
__
Ув ~~ 1 + (“АС — 1)^л + (аВС ~~ О *В ’ где
(60)
(61)
Эти уравнения можно также использовать в качестве хороших приближений для случая постоянного давления, поскольку отношение давлений пара чистых компонентов в тех относительно узких температурных интервалах, которые встречаются при перегонке, не претерпевает сколько-нибудь значительного изменения.
Рассмотрение неидеальных тройных систем выходит за рамки этой книги. Для частного случая тройной смеси кислород—азот — аргон можно обратиться к статье Хаузена [101]. Для более подробной трактовки тройных систем, как с математической, так и с графической точки зрения, можно обратиться к книге Куэнена [139].
СИСТЕМЫ ИЗ ^КОМПОНЕНТОВ
Для таких сложных систем графические методы не могут быть применены; поэтому следует прибегнуть к помощи чисто алгебраических соотношений. В общем случае существует зависимость в форме
Л = ? (Р. ХА> ХВ’ • • • XN-J- (62)
648
XII. Равновесие при испарении и конденсации
Поскольку имеется W степеней свободы (принимая две фазы), все, кроме одной из этих переменных, являются независимыми. Кроме того, функция будет зависеть от природы присутствующих компонентов. Если не сделать некоторых упрощающих допущений, то попытка дать продуктивную алгебраическую трактовку превратится в очень сложную задачу.
Идеальный раствор; паровая фаза представляет идеальный газ. Если можно принять, что жидкая фаза является идеальным раствором, тогда уравнение (62) можно значительно упростить, придав ему вид:
Л ~ г \Р> xi>- (63)
Другими словами, концентрация данного компонента в паре в данном случае зависит от его концентрации в жидкости (р и t фиксированы); на нее не будут влиять число и относительное содержание всех других компонентов в жидкости. Например, 10-мольная концентрация бензола в идеальном растворе будет давать определенное содержание бензола в паре при данных put, независимо от того, какими будут другие компоненты, и независимо от их относительных количеств. Если, однако, известно только давление или температура, то природа и относительные количества других компонентов должны входить в расчет. Для частного случая, в котором, кроме идеальности жидкого раствора, можно допустить, что пар является идеальным газом, функция ср (p,t, х{) становится очень простой. Таким образом, из ранее выведенных соотношений мы имеем
pi = pixi, (64)
у.р = р.х., (65)
p = '£pixi, (66)
y=Pi^L. (67)
Делением числителя и знаменателя правой части уравнения (67) на ре получаем
У ________________aiBxi____________ __ 'Wit (68)
' , ЛАВХА~1Г xB^TaCBxCJr -\~аНВхН ^3iBxi '
Относительные летучести а в этом случае относятся к компоненту В, но, конечно, можно выбрать любой из компонентов. (Отметим, что авв=1.) Совершенно аналогичными методами можно вывести, что
__ yi!aiB
^(Ус!асв) '
(69)
Системы из N-компонентов
649
Как и в случае бинарной смеси, эти уравнения являются строгими для постоянной температуры; но поскольку относительные летучести весьма незначительно изменяются с температурой, этими уравнениями можно пользоваться также и для случая постоянного давления с точностью, достаточной для расчетов по перегонке. При использовании уравнений (68) или (69) для определения а в случае постоянного давления необходимо знать температуру (потребуется только приблизительное ее значение). Температуру, полученную методом подбора, всегда можно проверить посредством соотношения
(70)
Пример 6. Идеальный жидкий раствор при 100° С и давлении в 2 атм содержит 20 мольных процентов толуола. Чему будет равна концентрация толуола в паре, находящемся в равновесии с этим раствором? Давление пара толуола при 100° С равно 557 мм рт. ст.
Из уравнения (65) получаем
Ут —
0,20 X 559 2X760
= 0,0735.
Отметим, что мы получили эту цифру, ничего не зная о других компонентах раствора. Однако их нельзя выбирать произвольно, потому что только определенные системы будут иметь указанные давление и температуру.
Если даны только давление и температура, то должен быть известен полный состав жидкой фазы, как показано нижеследующим примером.
Пример 7. Вычислить: i) состав пара, находящегося при атмосферном давлении в равновесии с жидкостью, которая состоит из 1О°/о н-гексана, 30% бензола, 40% w-гептана и 20% н-октана (все в мольных процентах); 2) равновесную температуру. Примем, что мы имеем идеальный раствор и идеальные газы и что давления пара чистых компонентов как функцию температуры можно представить уравнением igpMM = A — BjT°K. со следующими константами [13]:
А В
Гексан (компонент А) 7,7215 1654,6
Бензол (компонент В) 7,6559 1686,8
Гептан (компонент С) 7,5917 1750,0
Октан (компонент D) 7,7503 1941,4
Нормальные точки кипения лежат примерно от 69 до 126° С. Для решения методом подбора примем t = 100°C.
Затем из уравнений давления пара вычисляются следующие значения а, относящиеся к бензолу:
650
XII. Равновесие при испарении и конденсации
Вещество Давление пара, мм Hg Относительная летучесть
Гексан 1935 адз= 1,418
Бензол 1364 авв ~ I , 000
Гептан 798 асв = 0,585
Октан 352 «03 = 0,258
Нодставлии в уравнение (68), получаем
_________________1,418X0,100______________n . q[-
Уа — 1,418 X 0,100 + 0,300 + 0,585 X 0,400 + 0,258 X 0,200 ’ ’
№ = 0,412, ус = 0,322, №=0,071;
Ypixl = 19^5X0,100+1364X0,300+798X0,400+352X0,200=993. Следовательно, принятая температура является слишком высокой. Результаты еще нескольких подборов оказались следующими:
t°,C Давление пара
А В С D
90 1462 1023 592 253 741
95 1690 1186 689 299 860
Из графика '£pixi — t температура для 2ргхг = 760 определяется равной 90,9°С. При этой температуре значения а будут следующими:
в
аАВ= 1,425, авд = 1,000, acg = 0,577, aDB = 0,247;
с этими значениями а состав пара будет следующим:
№ = 0,197, № = 0,415, № = 0,320, № = 0,068.
Системы из N-компонентов
651
Различия между этими величинами и вычисленными на основании подобранной температуры 100° С хорошо укладываются в пределы ошибок, связанных с основными допущениями; из этого можно заключить, что для вычисления составов пара вполне достаточны даже очень приближенные температуры.
Неидеальный газ. Равновесное отношение К. Если пар нельзя считать идеальным газом, то обычно уравнение (63) пишут в форме:
= (71)
где — функции р, t, природы и состава раствора. Поэтому, хотя J( обычно называют „константой равновесия", но „равновесное отношение" было бы для нее более Подходящим термином. Обычно считают К не зависящей от состава, причем это равносильно допущению, что жидкая фаза является идеальным раствором. Это допущение, как мы уже показали, вносит очень большое упрощение.
Если пар является идеальным газом, то из уравнения (65) следует, что
(72)
По аналогии с этим уравнением Льюис и Люк [155] для случая, когда пар не является идеальным газом, пишут
, (73)
>р
где fp.— летучесть чистого компонента i при данной температуре и давлении пара чистого компонента, а / —летучесть чистого i в газообразном состоянии при температуре и общем давлении раствора.
Саудерс, Зельхаймер и Браун [237] пишут это уравнение в несколько другой форме, а именно:
K=f-± (74)
где/г—та же величина, что и f , но fL — летучесть компонента, существующего в виде жидкости при давлении и температуре раствора. Из уравнения для расчета изменения летучести с давлением (144, гл. Ill) при допущении, что объем жидкости не зависит от давления, легко получить
f RT (Р‘ Р)
m
Это отношение мало отличается от 1,00, пока давление меньше 100 атм, и поэтому уравнения (73) и (74), в сущности, эквивалентны.
Оба эти уравнения следует считать эмпирическими, не имеющими строго рационального основания. Попытку рационального вывода [68] можно сделать на базе общих уравнений для бинарной системы, выве
652
XII. Равновесие при испарении и конденсации
денных в гл. IV, что представляется желательным для выяснения некоторых ограничений этих уравнений. Из гл. IV имеем
I^v*dp »
(143, гл. IV)
Это — общее уравнение для двухфазного равновесия в бинарной системе. Если мы допустим, что обе фазы — жидкость и пар — являются идеаль-ными растворами, то ид и *&д нс зависяi oi состава и являются кциями только р и t. Допустим далее, что компонент А может существовать в чистом состоянии как жидкость или как пар при р и t раствора. (Это, конечно, невозможно для устойчивого равновесия. В любом идеальном бинарном растворе при давлении и температуре раствора более летучий компонент может существовать только в виде пара, а менее летучий компонент — только в виде жидкости.) Тогда можно написать
и поэтому величина
= v а , Va — v'l ,
^•°AdP
является по определению натуральным логарифмом летучести компонента А. Следовательно, уравнение (143, гл. IV) принимает следующий вид:
Этот вывод дан только для бинарного раствора, но можно распространить его на любой идеальный раствор, так как в таком растворе не получается различия от того, находится ли рассматриваемый компонент в другом индивидуальном компоненте или в сложной смеси.
В результате этого краткого обсуждения ясно, почему уравнение (74) представляет эмпирическую зависимость, которой следует пользоваться, отдавая себе отчет о пределах ее применения. Оно широко применялось, и выводы, основанные на нем, иногда производились без учета этих ограничений. С другой стороны, в настоящее время оно является единственным средством, имеющимся в распоряжении для вычисления равновесия в том случае, когда пар не является идеальным газом. С его помощью можно, по крайней мере, надеяться получить более точные результаты, чем с законом идеального газа, имея дело с системами при повышенных давлениях.
Системы из N-компонентов
653
При рассмотрении числового значения К. полезно помнить, что значение К для любого компонента точно зафиксировано и заранее определено в двух точках: К равно единице при давлении пара чистого компонента для рассматриваемой температуры и равно единице в критической точке для данной смеси. Эти два факта являются полезным руководством при рассмотрении тенденций в изменениях К-
Вычисление К- Равновесное отношение К легко определяется из экспериментальных данных; но такие данные еще очень скудны, и обычно бывает необходимо вычислять К из данных, имеющихся для чистых углеводородов и для немногих бинарных растворов с помощью уравнений, подобных уравнениям (73) и (74). Величины, найденные этим путем, широко использовались при расчетах, связанных с нефтяными углеводородами, и эти величины для нескольких обычных углеводородов можно получить из большого числа источников [30, 154, 230, 237]. Вследствие широкого применения этих данных представляется желательным более подробно исследовать методы, с помощью которых они были получены.
Эти величины находились двумя различными путями, причем каждый из них включает несколько сомнительные экстраполяции в нестабильные области. Рассмотрим, например, метод Льюиса и Кея, который основывается на уравнении (73). Если рассматриваемая температура выше критической, то величина/^, для любого чистого углеводорода не имеет никакого действительного физического значения, так как вещество, очевидно, не может иметь реального давления пара. Например, метан при 20°С принадлежит к этой категории. Подобным же образом, f не имеет действительного значения,. если температура ниже критической и давление выше давления пара, так как компонент при этих условиях не может существовать в виде газа. Бутан при 37,5° С и 20 атм может служить примером такого случая.
Льюис и Кей определили летучести для использования в уравнении (73) из обобщенного графика, на котором нанесены приведенные изотермы в координатах коэфициент летучести ☆) (отношение летучести к давлению) — приведенное давление. График Льюиса и Кея воспроизведен на рис. 152. Линия АВ представляет фазовую пограничную линию, т. е. она соединяет приведенное давление пара с приведенной температурой. В области выше АВ и выше т=1,00 чистые углеводороды устойчивы в виде газов, и летучесть легко вычисляется из р— v—Т-данных с помощью методов, рассмотренных в гл. VI. График в этой области в основном совпадает с рис. 34, который является обобщенной диаграммой коэфициента летучести для всех веществ. В области ниже АВ и т = 1,0 чистые углеводороды устойчивы только как жидкости, но график применяется в этой области
•£•) Этот термин может употребляться наравне с термином .коэфициент активности' для обозначения отношения / к р.
654
XII. Равновесие при испарении и конденсации
для нахождения f— летучести углеводорода в виде газа. Поэтому вместо нанесения коэфициента летучести жидкого углеводорода представляется более логичным вычислять значения f из уравнения (73) в виде
f =f - , JP ' Pt у ’
используя имеющиеся экспериментальные данные по растворам. Этот метод применялся для построения кривых рис. 152 в этой области. Он сводится к экстраполяции кривых, отвечающих области газовой
Рис. 152. Коэфициент летучести чистых углеводородов [154].
фазы, в которой газы являются неустойчивыми (в качестве основы для экстраполяции используются данные по растворам). Поскольку
х и у даются посредством экспериментальных данных ☆ ), значения
легко определяются из лилии АВ для
углеводородов, критические
температуры которых выше данной температуры. В случае углеводоро-
дов, критические температуры которых ниже данной температуры, как для СН4 при комнатной температуре, необходима экстраполяция кривой АВ в нестабильную область. Было предположено, что для экстраполяции можно применить уравнение (73), и Льюис и Кей исполь
зовали его для метана, применяя данные для раствора метан—пропан. Они пришли к выводу, что это будет удовлетворительным приближением при умеренных давлениях и для температур, не слишком превышающих критическую. Другим методом экстраполяции служил линейный график в координатах lgpt (или lg/p )—1/7'.
й) Данные Сэджа, Лэси и Шаафсма [224] по метану—пропану и Каммингса, Стона и Воланта [51] по н-пентану—н-гептану.
Системы из N-компонентов 655
<«
Вычисление К будет иллюстрировано несколькими примерами.
Пример 8. Вычислить К. для «-бутана при 50° С и 25 атм, используя метод Льюиса и Кея.
Из табл. III .Приложения':
критическая температура = 426° К, критическое давление = 36,0 атм.
я = 0,695.
оо
Из рис. 152 fplp = 0,52 и fp=0,52X25= 13,0.
Давление насыщенного пара бутана при 50° С [390] равно 4,9 атм,
4 9
:г = 4£ = 0,136. оО
Из рис. 152
fp.
= 0,9.
Р
Тогда
/₽;-4,4.
fp можно также найти непосредственно по графику летучестей жидких углеводородов при давлении их насыщенного пара, нанесенном как функция температуры. График приведен в книге Шервуда [230] и составлен Кеем.
Окончательно
fP. 4,4
к' ТзЛ”0’34
Пример 9. Вычислить Д' для метана при 38° С и 25 атм.
Этот пример отличается от предыдущего тем, что температура значительно, выше критической температуры метана, и поэтому для определения величины fp необходима значительная экстраполяция кривой давления пара выше критической точки.
Из табл. III .Приложения':
критическая температура = 191 °К, критическое давление = 45,8 атм.
т = 1,63, л = 0,546,
(по рис. 152) — 0,98,
Р
fP = 24,5.
Для получения значения fp давление пара СН4 можно экстраполировать на графике lg/>— 1/Г или, как предлагает Шервуд, можно нанести Ig/^ в зависимости от 1/т, получая прямую линию, которую можно экстраполиров’ать выше критической точки. Применяя последний способ, получаем
/р. = 190.
656
XII. Равновесге при испарении и конденсации
Тогда
~ 190 _78 К~ 215 -7Л
Метод, использованный для вычисления К Брауном и сотрудниками, существенно отличается от только что описанного. При температурах ниже критической летучесть пара при давлении насыщенного пара вычисляется по уже описанному методу или определяется из диаграммы коэфициентов летучести. Эта величина будет также летучестью жидкости при давлении пара, и из нее вычисляется летучесть жидкости при некотором другом давлении с помощью уравнения, связывающего изменение f с р, при допущении, что объем зависит от давления. Этот метод даст fL уравнения (74). fv вычисляется с помощью обычных методов для газа (до значений давления ниже давления насыщенного пара). В результате этих вычислений были найдены значения К для низших насыщенных углеводородов вплоть до пентана для т = 0,9 и 1,0 и до давлений примерно в 35 атм. Все эти значения К больше единицы, поскольку по вышеупомянутому методу К можно вычислить только в тех случаях, когда давление равно или меньше давления насыщенного пара. При давлении насыщенного пара К= 1 для всех случаев; для всех давлений, меньших давления пара, Значения /^<^1 получаются экстраполяцией
по графику 1gК—1gр с использованием в качестве основы кривых для т = 0,9 и 1,0, поскольку они практически охватывают весь интервал давлений (см. [237], рис. 1).
При температурах выше критической fv легко определяется с помощью общепринятых методов, но fL является летучестью жидкости при температуре выше критической, где жидкость не может существовать. Метод, использованный Брауном и сотрудниками для получения К в этой области, заключается в следующем. Значения К считываются с графика 1g К—1gр (только что рассмотренного вдоль изобар) и наносятся на график 1g К—т. Экстраполяция выше т=1,0 производится на основании разбросанных данных по теплотам растворения, которые дают возможность произвести вычисление наклонов посредством уравнения (207, гл. IV) и при помощи некоторых экспериментальных измерений К. Другими словами, в этой области К основано на данных по свойствам растворов, а не на данных по свойствам чистых веществ, как было сделано для области ниже т=1,0.
Эти методы имеют явные недостатки, но все же являются наилучшими из доступных в настоящее время. Представлялось желательным дать их краткое описание, чтобы пользующийся данными для К мог ощутить связанные с этим неточности и не строил бы на них необоснованных заключений.
Суммируем кратко неточности, связанные с вычислением К по уравнению (74):
1. Для всех значений 1 или давление будет меньше давления насыщенного пара при данной температуре, или температура будет
Системы из Ы-компонентм
657
выше критической. В обоих случаях термодинамически невозможно существование жидкой фазы для чистого компонента, и поэтому fL является эмпирической величиной.
Рис. 153. Константа фазового равновесия ЛГ для пропана. Критическая температура равна 97°С; критическое давление 42,0 атм [30].
2. Для всех значений К<^ 1 или давление будет больше давления пара, и поэтому не может существовать никакая устойчивая паровая фаза чистого компонента, или температура будет выше критической, и будет невозможна никакая жидкая фаза. Следовательно, или Д, или Д будут эмпирическими величинами.
42 Химическая термодинамика
658
XII. Равновесие при испарении и конденсации
Пользование диаграммами К. Диаграммы для низших насыщенных углеводородов вплоть до н-гептана, на которых нанесены изобары в координатах К—t, охватывающие широкий интервал температур и давлений, даны Брауном и Саудерсом [30]. Их график для пропана воспроизведен на рис. 153. Подобные диаграммы дают вычисленные значения К, и при пользовании ими следует отдавать себе отчет об их пределах, как это и было указано выше. Оказалось, что другие графики могут давать величины, значительно отличающиеся от этих значений К, особенно для метана и этана.
Применение значений К для вычисления состава любой фазы при равновесии жидкость — пар при данном составе другой фазы иллюстрируется следующим примером.
Пример 10. Вычислить давление и состав пара, равновесного с жидкостью, содержащей 5% СН4, 10°/о C2HS, 30% С3Н8, 25% пзо-С4Н10 и 30% н-С4Н10 (мольные проценты) и находящейся при 37,5° С.
Значения X взяты из диаграмм Брауна и Саудерса [30]. Так как X является функцией как р, так и t, а р неизвестно, то необходимо решение методом подбора. Критерием правильности найденного давления будет условие
Sy =
Мы также можем написать:
У/
= Sjfx=l.
KiXj ЪКх'
Преимущество этой формы уравнения для у заключается в том, что оно включает только отношения X и поэтому значительно не изменяется ни с р, ни с t. Следовательно, если хотят вычислить только состав пара, то требуется лишь приближенное значение р или t.
Подбор 1. Примем р=10,5 кг/см2.
Углеводород X Хх
Ci 8,5 0,425 0,320
С, 2,9 0,290 0,219
с3 1,15 0,345 0,260
изо-С4 0,57 0,143 0,108
н-С4 0,41 0,123 0,093
S#c = 1,326
Подбор 2. Примем р = 21,0 кг/см2.
У глеводород К Хх Хх ХХх~У
Ci 5,1 0,255 0,316
С, 1,66 0,166 0,206
- сз 0,72 0,216 0,267
изо-С4 0,36 0,090 0,111
я-С4 0,27 0,081 0,100
ZXx = 0,808
Системы из N-компонентов
659
Различия между составами пара для этих двух подборов едва ли существенны, и мы можем с уверенностью принять, что любой подбор, дающий ЕЛ-г с отклонением от единицы в пределах 20%, является удовлетворительным. Конечно, для получения давления необходим дальнейший подбор, но его можно свести к минимуму нанесением S/fx в зависимости от р. Этим путем мы находим, что если S/Cx=l, то />=15,4 кг'см2.
Метод совершенно одинаков и в том случае, если температура неизвестна, а давление определено.
Рис. 154. Константа фазового равновесия для метана и пропана в системе метан — пропан [221].
Если следует вычислить равновесный состав жидкости из состава пара, то метод остается тем же, за исключением того, что используются уравнения
х = £ (76)
и
У1К
2(У1Ю '
(77)
Экспериментальные равновесные отношения. Все значения К, которыми мы пользовались, вычислялись на основании определенных 42*
660
XII. Равновесие при испарении и конденсации
допущений и экстраполяций при наличии нескольких разбросанных экспериментальных точек, служащих для приближенной ориентировки. Экспериментальные данные в этой области очень отрывочны, но некоторыми исследователями сделано ценное начинание в области нефтяных углеводородов. Сэдж и Лэси [221] дали исчерпывающую сводку имеющихся в распоряжении данных по равновесным отношениям для метана в нескольких углеводородных смесях. На рис. 154, воспроизведенном из их статьи, показано влияние давления и температуры на К для метана н пропана в бинарной смеси метан — пропан. Отметим, что К—\ в критической точке системы и при давлении насыщенного пара пропана (при данной температуре). Например, кривая для пропана при 20° С начинается при К— 1 и р = 8,3 (приблизительно), т. е. при давлении насыщенного пара пропана. К уменьшается по мере возрастания давления, а затем увеличивается, достигая значения 1,0 примерно при 100 ат, т. е. при критическом давлении системы метан — пропан при этой температуре. При продолжении кривой выше К = 1 абсцисса дает значения К для метана. Совпадение величин, приведенных на рис. 154, с вычисленными Брауном и Саудерсом является вполне хорошим для пропана, но в случае метана имеется значительное расхождение.
Все вычисленные значения К основаны на допущении идеальности растворов, и некоторые данные, представленные Сэджем и Лэси, позволяют нам остановиться на этом допущении. В нижеследующей таблице даются значения равновесного отношения для метана в некоторых системах при 71 °C и 38 ат.
Система
Метан — пропан .... Метан — н-пентан . . . Метан — н-гексан . . .
Метан — циклогексан . . Метан — азот — гептан .
Метан — азот — толуол . Метан — азот — бензол .
Метан — чистая нефть . . .
К
3,89
6,30
7,08
9,63
6,86
12,4
14,6 (при 54,5°С; для 71°С значение будет больше)
Если бы эти растворы были идеальны, то значения К должны были бы быть одинаковы, между тем имеются значительные различия даже в случае систем, содержащих только насыщенные парафиновые углеводороды. С другой стороны, метан представляет крайний случай, и для высших углеводородов совпадение было бы лучше. В любом случае очевидно, что К является функцией не только р и t, но также природы и колйчества других компонентов. Сэдж и Лэси дали согласование значений К для метана в зависимости от молекулярного веса и химической природы присутствующих компонентов. Корреляция была с удовлетворительными результатами проверена на
Системы из N-компонентов
661
системе метан — сырая нефть, но при применении ее к системе метан— н-бутан получились худшие результаты.
Сэдж, Хикс и Лэси [219] представили пробную корреляцию значений К для некоторых легких углеводородов, включая гептан.
Браун и Саудерс также рассмотрели отклонения от закона идеальных растворов и показали, что наибольшие отклонения наблюдаются для углеводородов с низким молекулярным весом и когда данный компонент присутствует в относительно малом количестве. Они также дали график поправочных коэфициентов для различных углеводородов и для различной концентрации в жидкости.
Кей [125] сравнил свои измерения по системе этан—н-гептан со значениями К, данными Шервудом, и нашел прекрасное совпадение до тех пор, пока не было достигнуто приближение к критической области.
Другие признаки влияния природы раствора на значение К- показаны Сэджем и Лэси [222]. Данные по тройной системе метан— пропан—н-пентан при 37,5° С показали значительные изменения К для всех трех компонентов при 34, 68 и 102 атм по мере того, как раствор постепенно изменялся от 100-процентного пентана к 100-процентному пропану. Например, при 68 атм К для пентана изменяется от 0,08 до 0,50. Процентное изменение было меньше для пропана и метана. Этот эффект значительно слабее при более низких давлениях.
ГЛАВА XIII
ПРОЦЕССЫ ДЕСТИЛЛЯЦИИ
ОБЩИЕ ПРИНЦИПЫ
Возможность разделения растворов летучих веществ посредством различных процессов, объединяемых общим названием дестилляция, зависит от двух основных факторов: 1) различия в составах жидкой и парообразной фаз при равновесии и 2) различия в плотности этих фаз. Все операции, используемые в процессах дестилляции, включают соприкосновение между фазами (для осуществления определенных изменений в составе), за которым следует разделение фаз (обычно
Рис. 155. Элементарные процессы испарения и конденсации.
осуществляемое посредством силы тяжести). Второй фактор не связан с термодинамикой и не будет обсуждаться в дальнейшем, а первый фактор уже был довольно подробно рассмотрен в предыдущей главе. В данной главе мы разберем некоторые важные процессы, с которыми связываются эти два фактора.
Рассмотрим сначала некоторые общие принципы двух основных операций — испарения и конденсации, которые входят во все процессы дестилляции.
Интегральные испарение и конденсация. На диаграмме температура— состав для бинарной системы, показанной на рис. 155, точка А х1. При изобарном нагревании этой
отвечает жидкой смеси состава
жидкости она следует по пути АВ (постоянный состав). В точке J образуется первый пузырек пара, состав которого отвечает точке D. (Заметим, что при равновесии сосуществующие фазы должны иметь одинаковую температуру.) Если нагревание продолжается при пос-
тоянном составе и пар из системы не удаляется, то достигается точка, подобная точке Е, в которой система представляет смесь двух фаз (состава F и G). Если продолжается подвод тепла к системе (при постоянном давлении и при постоянном общем составе), то достигается точка Н, когда в системе остаются только следы жидкости (точка росы С). Выше точки Н начинается область перегре
Общие принципы
663
того пара. Если процесс испарения закончить раньше, чем будет достигнуто состояние системы, соответствующее точке Н, то такой процесс следует назвать частичным испарением, поскольку в системе будет присутствовать еще некоторое количество неиспарив-шейся жидкости. Если процесс проводится до состояния Н или дальше, то он считается полным испарением.
Процесс испарения, представленный линией JE, при котором всему получающемуся пару дают оставаться в соприкосновении со всей остающейся жидкостью, обычно называется однократным испарением. Его называют также интегральным испарением в отличие от диференциального или частичного испарения, которое будет рассмотрено ниже. Общим результатом будет разделение исходной жидкости состава хг на две фракции, одна из которых будет беднее компонентом А (состав х2), а другая богаче им (Ь)-
Процесс конденсации, противоположный только что описанному процессу, можно провести следующим образом. Начиная с точки В перегретая паровая смесь состава хг охлаждается при постоянном давлении; в точке И появляются следы жидкости. Если охлаждение продолжается, то количество жидкости непрерывно увеличивается, пока в точке J не останется лишь пузырек пара,- т. е. мы будем иметь только насыщенную жидкость. Дальнейшее охлаждение до точки А дает переохлажденную жидкость. Если состояние системы лежит между точками И и У, то охлаждение называется частичной конденсацией, поскольку остается несконденсированным некоторое количество пара.
Охлаждение до температуры, отвечающей точке J, или до еще более низких температур приводит к полной конденсации. Очевидно, что разделение компонентов достигается только при частичной, но не при полной конденсации.
Только что рассмотренный процесс конденсации, при котором весь полученный конденсат остается в соприкосновении с оставшимся паром и считается находящимся с ним в фазовом равновесии, будет называться интегральной конденсацией по аналогии с соответствующим процессом испарения. Максимально возможное обогащение пара более летучим компонентом, которого можно достигнуть с помощью такого процесса, соответствует для рассматриваемого случая составу yv
Сочетание этих двух процессов—испарения и конденсации — приводит к увеличению степени разделения жидкости состава х1 по сравнению с тем, что возможно при использовании любого из этих процессов в отдельности. Предположим, что жидкость испаряется до тех пор, пока не будет достигнуто состояние Е, а затем пар механически отделяется от жидкости и частично конденсируется (до состояния L). Тогда в системе будет присутствовать остаточный пар, представленный точкой D, и остаточная жидкость того же самого
664
XII!. Процессы дестиллщии
состава, что и исходная жидкость (точка J). Следовательно, общим результатом этого процесса будет разделение жидкости состава хг на две фракции — одну (/И) состава у, (жидкость после полной конденсации) и другую (F) состава х2. При многократном продолжении этого процесса над двумя жидкостями (отвечающими точкам F и М) с помощью соответствующего соединения остатков, которые, в свою очередь, подвергаются такому же процессу, можно разделить раствор А практически на чистые компоненты. Такой процесс очень трудоемок и никогда не используется на практике, поскольку тот же самый результат может быть достигнут гораздо более простым путем, но указанный принцип разделения используется во всех практических процессах.
Диференциальные испарение и конденсация. Испарение жидкости можно также провести следующим путем, который иллюстрируется рис. 155. Жидкость в состоянии J испаряется, и первая небольшая порция образующегося пара состава D сразу же удаляется из области соприкосновения с жидкостью, после чего целиком конденсируется. Оставшаяся жидкость теперь несколько менее богата более летучим компонентом; поэтому ее состав изобразится на линии жидкости точкой, расположенной немного левее точки J. Если испарение продолжается, причем каждую порцию вновь образовавшегося пара (теоретически бесконечно малую порцию) удаляют и конденсируют таким образом, чтобы остающаяся жидкость все время находилась в равновесии только с последней порцией выделяющегося пара, то точки, отвечающие составу неиспаряющейся жидкости, будут непрерывно перемещаться вдоль линии жидкости, а изменение состава пара будет изображаться соответствующими точками на кривой насыщенного пара. Этот процесс называется днференциальным испарением*); с его помощью можно получить небольшое количество (строго говоря, только бесконечно малое количество) чистого, менее летучего компонента, но невозможно получить даже самой малой доли более летучего компонента в чистом состоянии. При этом процессе принимают, что не происходит частичной конденсации, которая бы заставляла некоторое количество испарившегося материала возвращаться в перегонный куб в качестве орошающей жидкости.
Процесс, противоположный рассмотренному, а именно конденсация и непрерывное удаление от места соприкосновения с паром малых (теоретически бесконечно малых) порций образующейся жидкости, называется диференциальной или частичной конденсацией. Если начать процесс конденсации с пара в состоянии G, то диференциальная конденсация при полном ее проведении может дать небольшое количество чистого более летучего компонента, но наибольшее обогащение менее летучим компонентом будет соответствовать точке F.
&) Обычно этот процесс называют .простой перегонкой", но считается, что указанный выше термин имеет определенные преимущества.
Общие принципы
665
При сочетании этих двух процессов таким образом, чтобы конденсат непрерывно возвращался в перегонный куб в виде орошения, теоретически возможно произвести полное разделение жидкости состава х, на два чистых компонента. Такой процесс был бы очень неэкономичен из-за большого расхода тепла, поскольку все
тепло, затраченное на испарение, теряется при конденсации и конденсат нужно непрестанно вновь доводить до кипения.
Ректификация. Процессом перегонки, наиболее широко используемым в практике для разделения летучих жидкостей, является процесс ректификации, при котором в специальном контактном аппарате, известном под названием ректификационной колонны, поток пара не-
прерывно соприкасается с противоположно перемещающимся потоком жидкости. На рис. 156 схематически представлена простая ректификационная колонна. Поток жидкости Z.J, полученный из пара V} в конденсаторе, входит в верхнюю часть колонны, а поток пара V2, полученный в кубе, расположенном ниже колонны, входит в нижнюю часть ее, т. е. оба потока соприкасаются между собой по принципу противотока. Методы получения потоков и осуществления тесного соприкосновения между фазами могут изменяться, но это несущественно, поскольку иас интересует лишь принцип разделения. Рассмотрим какой-нибудь уровень в колонне (рис. 156); пусть пар V и жидкость L имеют составы, представленные соответственно точками Н и F на рис. 155. Пар Н ве находится в равно
весии с жидкостью F, с которой он соприкасается; рЙС jgg прин-равновесной жидкостью будет жидкость, соответствую- цип ректифика-щая точке С. Следовательно, имеется разность хими- пни. веского потенциала, и будет происходить массообмен
между паром и жидкостью. Это приводит к тому, что состав пара смещается по направлению к точке G, а состав жидкости — к точке С. Таким образом, пар становится богаче более летучим ком-
понентом, а в жидкости его содержание, наоборот, уменьшается. Кроме
того, следует отметить, что массообмен осуществляется как конденсацией, так и испарением. Другими словами, вместо того, чтобы
эти два процесса происходили раздельно в разных частях аппарата (что имеет место при фракционной перегонке), при ректификации они идут одновременно, что приводит к большой экономии тепла.
Рассмотренный процесс происходит также и на всех других уровнях колонны. Это приводит к тому, что восходящий пар постепенно обогащается более летучим компонентом, а стекающая жидкость становится беднее им. При установке в верхней части колонны конденсатора пар Vr конденсируется, и часть его удаляется в качестве дестил-лата, а остальная часть его возвращается в колонну в качестве орошающей жидкости А,, носящей название флегмы. Жидкость L2, покидающая
666
XIII. Процессы дестилляциа
ществующих при равновесии
сэ
5
Е о
100% В состав, проценты
100% А
Рис. 157. Разделение растворов с минимальной температурой кипения.
добно чистой жидкости.
нижнюю тарелку колонны, поступает в перегонный куб, где часть ее испаряется, образуя пар V2, а остаток удаляется. Для проведения непрерывного разделения бинарный раствор обычно подают в виде жидкости на некоторый промежуточный уровень колонны, а продукты удаляются соответственно из верхней и нижней частей колонны.
Разделение азеотропных смесей. Все процессы разделения растворов посредством перегонки зависят от различия в составе двух сосу-фаз. Если равновесная зависимость относится к типу растворов, имеющих максимум или минимум на кривой температуры кипения, тогда, как было показано в предыдущей главе, в этих точках максимума или минимума две фазы оказываются одинаковыми по составу, и дальнейшее разделение становится невозможным. На рис. 157, на котором представлена диаграмма температура — состав для раствора с минимумом температур кипения, хА представляет состав азео-' тропной смеси, которая при любом процессе перегонки ведет себя по-। состава xt можно разделить на чистый
компонент В и азеотропную смесь, но получение чистого компонента А при обычном процессе перегонки невозможно. Подобно этому, начав с раствора состава х2, можно разделить его на азеотропную смесь и чистый компонент А. Так как азеотропная смесь ведет себя подобно чистому компоненту, то диаграмму t—х можно считать состоящей как бы из двух диаграмм обычного типа, разделенных ординатой в точке хА.
Детали методов разделения азеотропных смесей выходят за рамки настоящей книги, но попутно можно отметить, что существует два общих способа, посредством которых можно провести разделение азеотропных смесей с помощью перегонки: 1) изменение давления при перегонке и, следовательно, изменение состава азеотропной смеси (см. гл. XII, раздел „Влияние давления“); 2) добавление третьего, а иногда и четвертого компонента, что приводит к изменению соотношения парциальных давлений насыщенного пара компонентов, благодаря чему разделение становится возможным. Последний метод обычно применяется для получения абсолютного спирта. Прибавление бензола к постоянно кипящей смеси этилового спирта и воды приводит к образованию тройной гетерогенной постоянно кипящей смеси (точка кипения равна 64,9°С при 1 атм), которую можно удалить из ректификационной колонны как дестиллат, причем абсолютный спирт остается в виде кубового остатка. Из дестиллата бензол извлекается посредством разбавления и механического разделения, а получающийся
Минимальная работа разделения
667
разбавленный спирт вновь концентрируется до состояния постоянно кипящей смеси в другой колонне ☆ ).
Азеотропная перегонка. Процесс разделения азеотропной смеси путем добавления нового компонента, образующего гетерогенную постоянно кипящую смесь, как в только что рассмотренном случае, обычно называется азеотропной перегонкой, хотя это и является неправильным применением термина, если ограничить термин „азеотропная смесь" гомогенной постоянно кипящей смесью. Подобный (по принципу) процесс приобрел в последние годы важное значение в связи с разделением растворов уксусная кислота — вода, причем тот же самый принцип приложим и в других случаях. Уксусная кислота и вода не образуют азеотропной смеси, но разгонка их трудна вследствие малой разницы между составом сосуществующих фаз. При добавлении третьего компонента, практически не смешивающегося с водой, дестиллатом при перегонке является не вода, а гетерогенная постоянно кипящая смесь воды и добавленного компонента, которая называется «улавливателем». Она кипит при более низкой температуре, чем вода, и поэтому значительно увеличивает конечную разность температур в системе. Например, если улавливателем является изопропиловый эфир, то точка кипения гетерогенной смеси с водой будет равна 61°С, и колонна в состоянии произвести разделение дестиллата и кубового остатка, кипящих соответственно при 61 и 118°С (уксусная кислота) вместо 100 и 118°С. Головной де-стиллат разделяется механически, а улавливатель возвращается в колонну. (Отметим сходство этого процесса с перегонкой с водяным паром.) Дальнейшие подробности, относящиеся к рассматриваемому процессу, можно найти в статье Отмера [183].
МИНИМАЛЬНАЯ РАБОТА РАЗДЕЛЕНИЯ
Осуществление разделения раствора посредством перегонки или другого какого-либо процесса требует затраты определенного количества энергии в виде теплоты или работы. В подавляющем большинстве процессов перегонки используется теплота, а не работа, но имеется несколько случаев, именно — низкотемпературное разделение газов и процессы повторного сжатия пара, когда для осуществления перегонки требуется затрата работы. С экономической точки зрения всегда желательно пользоваться возможно меньшим количеством теплоты или работы, и поэтому важно определить минимальное количество тепла или работы, которое должно быть затрачено для осуществления этого разделения. После того как эта величина будет найдена, ею можно будет пользоваться в качестве меры термодинамической эффективности любого действительного процесса.
Очевидно, что при перегонке потеря тепла происходит различными путями, например в конденсаторе с охлаждающей водой, с горячей
й) Более подробно см. статьи Кулей [45>], Гино и Кларка [97].
668
XIII. Процессы дестилляции
жидкостью, покидающей нижнюю часть колонны, и пр.; некоторое количество этого тепла можно сохранить с помощью соответствующих теплообменников. Это обстоятельство снова приводит нас к вопросу о том, чему равен наименьший расход тепла или работы на разделение при условии, что процесс проводится наиболее эффективно по сравнению со всеми возможными методами. В общем как теплота, так и работа зависят от характера процесса, и нельзя вычислить потребного минимума без детального рассмотрения процесса. Однако имеется один процесс, именно — обратимый изотермический, для которого работа измеряется изменением функции Р и поэтому зависит только от исходного и конечного состояний, а не от пути. ДЛ для процесса отвечает минимальной работе, необходимой для того, чтобы посредством изотермического процесса вызвать данное изменение.
Предположим, что требуется вычислить минимальную работу для разделения 1 моля жидкого питающего раствора состава xt на два раствора составов х2 и х3. Представим процесс перехода от исходного состояния системы к конечному состоянию посредством следующих обратимых изотермических стадий.
Стадия 1. Раздельное испарение двух компонентов из питающего раствора через полупроницаемые мембраны при равновесных давлениях и в соотношении, необходимом для образования первого продукта состава х2.
Стадия 2. Сжатие отделенных друг от друга паров до равновесных давлений компонентов над первым получающимся раствором (раствор состава х2).
Стадия 3. Конденсация паров в этот раствор через полупроницаемую мембрану.
Стадии 4, 5 и 6. Перевод двух компонентов из питающего раствора в раствор состава х3 (аналогично стадиям 1 —3).
Суммируя затем работы, связанные с проведением этих процессов, считая пары идеальными газами и пренебрегая объемом жидкости, получим на моль питающего раствора:
А ___________________7??1 Г Хг ^х* In d (-*•! -^-з) In ——
L х2 — х3 х2 — х3 pBi
I х3 (х2 Xl) |п рАз t 0 Х3) (х2 —-Гр РВз~1
Х3 рА^ Х2 Х3 pgJ ’
(1)
где индексы 1, 2 и 3 относятся соответственно к питающему раствору, первому и второму продукту, а индексы Ан В—к компонентам. Для частного случая разделения на два чистых компонента, уравнение (1) переходит в
А = — RT\ Х1 1п^-‘ + (1-х1)1П£В‘1 . (2)
L Ра, Рв, J
Уравнения (.1) и (2) можно подвергнуть дальнейшему упрощению.
Минимальная работа разделения
669
Для случая идеальных растворов, для которых
Pi=XiPi, (3)
уравнение (2) примет вид
A =RT [XjJnjq -|-(1 —Х1)1п(1 —де*)]. (4)£?)
Термодинамический к. п. д. действительного процесса перегонки обычно определяют расходом водяного пара, полагая, что минимальное количество водяного пара, требующегося для совершения минимальной работы, вычисляется согласно методам, рассмотренным в гл. Ш.
Пример 1. Вычислить минимальную работу, необходимую для того, чтобы эквимолярную смесь бензола и толуола разделить на два раствора, из которых один содержит 95% бензола, а другой — 95% толуола при температуре в 25°С. Найденную величину сравнить с работой, необходимой для полного разделения. Вычислить минимальное количество водяного пара, требуемое для полного разделения, если используется насыщенный водяной пар при давлении 3,5 ат и если наинизшая температура, при которой может быть осуществлена передача тепла, равна 20°С.
Примем, что бензол и толуол образуют идеальный раствор:
0,500, х2 = 0,950, х3 = 0,050, R = 1,987, Г=298,2°К.
Подстановка в уравнение (1) дает
А — —292 кал!моль питающего раствора.
Для разделения на чистые компоненты подстановка в уравнение (4) дает
Л = — 410.
Интересно отметить, что разделение на 99-процентные продукты увеличивает работу до 308 кал, т. е. наблюдается резкое увеличение расхода работы по мере приближения к чистым продуктам.
В гл. III было показано, что максимальная работа, получаемая в тепловой машине с помощью водяного пара, равна
А,акс.=« (Д//-W, где т—количество водяного пара (кг).
При использовании паровых таблиц Вукаловича получаем:
Д/7 =631,9 ккал/кг,
Д5 = 1,5871 ккал/кг-град,
4Ю ,
т = ,==.—о.. ,о_. =2,31 кг (на моль питающего раствора).
001,9 — 29о,2Х 1,58/1
2г) Это уравнение непосредственно следует из уравнения
А= &U— TAS,
поскольку для идеального раствора Д1/= 0. Строго говоря, оно приложимо к периодическому процессу, а соответствующим уравнением для непрерывного процесса будет
д=дя— гд$,
но разница между значениями, получаемыми с помощью этих уравнений, обычно ничтожно мала.
670
XIII. Процессы дестилляции
Так как имеется два моля питающего раствора на моль дестиллата, то минимальный расход водяного пара на моль дестиллата будет
2 X 2,31= 4,62 кг.
Для подобного расчета, примененного к определению термодинамической эффективности специального котла, можно рекомендовать статью Вебера [256].
ИНТЕГРАЛЬНОЕ (ОДНОКРАТНОЕ) ИСПАРЕНИЕ (КОНДЕНСАЦИЯ)
Рис. 158. Интегральное испарение (конденсация).
Этот тип процесса перегонки уже был рассмотрен качественно; следующей ступенью является вывод уравнений для расчета процес-сов. Ниже приведенный анализ будет ограничен случаем одной жидкой фазы. В этом разделе, как и в большинстве последующих разделов главы, мы будем пользоваться молями и мольными долями, однако следует отметить, что большинство получаемых зависимостей, если не все они, одинаково справедливы и для весовых единиц и для весовых долей.
Бинарная система. Элементарный процесс испарения и конденсации показан количество молей питающей
на рис. 158, на котором F означает
жидкости в случае испарения или количество молей питающего пара в случае конденсации. Процесс можно провести непрерывно или периодически. При непрерывном процессе поток пара может быть или параллельным потоку жидкости, или итти в противоположном ему направлении.
Из материальных балансов имеем
(5)
(6)
Испарившаяся доля будет
_ __ V___х — хр
~F x — v
(7)
Относительное количество двух фаз в конце процесса дается выражением
V^x—хр
L хр—у
Обычным допущением для периодического процесса является то, что получаемый пар находится в равновесии с остаточной жидкостью, т. е., что
Интегральное испарение и конденсация
671
где ®(х)— равновесное соотношение. При непрерывном процессе имеются два предельных случая: 1) равновесие между паром и остаточной жидкостью; 2) равновесие между паром и питающим раствором. В действительных случаях состав, вероятно, лежит между этими двумя пределами.
Уравнение (8) имеет простую, но часто очень полезную геометрическую интерпретацию. На рис. 155
х — хр___х2 — ___ЕЕ
xF—y Xi —у2 EG '
Другими словами, в любой точке двухфазной области, например в точке Е или L, перпендикуляр, проходящий через такую точку, делит горизонтальную соединительную линию на отрезки, длины которых пропорциональны количеству вещества в каждой фазе, причем отрезок, примыкающий к кривой пара, представляет жидкую фазу и, наоборот, отрезок, соединяющий данную точку с кривой жидкости, соответствует парообразной фазе (правило рычага).
Для случая интегральной конденсации легко показать, что соответствующими уравнениями являются:
и
„ L у —уР ~Е у — х
L __У—Ур
V Ур — х'
(9)
(Ю)
Многокомпонентная система. Уравнения для бинарной системы легко обобщить для обработки систем с любым числом компонентов. Материальный баланс дает, как и раньше,
F = L-^-V,
(5>
а баланс по какому-либо компоненту 1 дает
xFF = x1.Z-+y,-V. (11)
Исключая L, получаем
V xp. — xt
Е yt-Xi (12)
ИЛИ
ХР;~ Xl л- +*,- (13)
Используя равновесную зависимость в форме
y. = K{Xi, (71, гл. XII)
получаем
xFi (14)
672
XIII. Процессы десталляции
и так как Sjz=l, то
L (15) гг)
Для лучшего уяснения смысла уравнения (15) приводим следующую его форму, справедливую для случая системы, состоящей из трех компонентов—А, В и С:
_____ХрА______|_______^в______|-----^£.-------1 (16)
Сг+К1-Сг)/Ла]^Сг+((1-М/ЛвИ '
Для случая конденсации аналогичный вывод приводит к подобному же выражению. В этом случае лучше пользоваться соотношением
которое приводит к уравнению
где ур. — мольная доля любого компонента в питающем паре.
Отдельные члены суммы представляют собой составы сконденсированной жидкости. Для вычисления состава остаточного пара можно составить материальный баланс по каждому компоненту. Обобщенным уравнением баланса будет
/Р,.=.^и-Л)+-*Л- (18>
Для частного случая идеального раствора и идеального газа
(19)
и уравнение (15) переходит в следующее:
X Cz+(P/^)’(1-Cz)= 1 (20)
с аналогичным уравнением для конденсации.
Следует отметить, что уравнение (17) также приложимо и к тому случаю, когда способные конденсироваться пары смешиваются с некоторым неконденсирующимся газом, при условии, что растворимостью последнего в конденсате можно пренебречь.
£г) В уравнении (15) каждый член представляет состав остаточного пара. Другое уравневие, но одинаково пригодное для определения С, получается при использовании условия Sx/=1; в нем каждый член представляет состав конденсата.
Интегральное испарение и конденсация
673
Пример 2. Какая доля жидкой смеси, содержащей 10% (мольных) пропана, 65% н-бутана и 25% н-пентана, должна испариться в процессе однократного испарения при температуре 5°С и давлении в камере 600 мм рт. ст.?
Сделаем допущение, что жидкости будут идеальными растворами и что пар будет идеальным газом. Имеются следующие данные по давлению насыщенного пара при 4,5°С:
Пропан..................... 3800 мм рт. ст.
н-Бутан..................... 820 мм рт. ст.
н-Пентан...................190 мм рт. ст.
В данном случае приложимо и уравнение (20), но для нахождения + следует применить метод подбора. Пусть, например, Су = 0,500. Тогда, по уравнению (20),
_______0,100______।_______0,650______।______0,250________
0,5004-0,500 0,500 + |^Х0,500 0,500+|^Х 0,500
= 0,173 + 0,750 + 0,120= 1,043 ^ 1,00.
После нескольких подборов оказывается, что Су = 0,66, т. е. испаряется 66% (мольных) вещества. Следует отметить, что в этом расчете подразумевалась изотермичность процесса. В действительности однократное испарение более близко к адиабатному процессу. Условиям этой задачи полностью удовлетворило бы наличие жидкой смеси, достигающей дроссель-вентиля при несколько повышенной температуре, которую легко вычислить, исходя из того обстоятельства, что энтальпия во время процесса остается постоянной.
Пример 3. Дана жидкость, содержащая 6О°/о (мольных) бензола, 30% толуола и 1О°/о -иедаа-ксилола. Каким будет состав пара после испарения 50% (мольных) путем интегрального испарения, если конечное давление равно 1 атм?
Ниже приводятся данные по давлению насыщенного пара.
Давление насыщенного пара, мм Hg Температура, °C
60 70 80 90 100 110 120
Бензол .... 540 756 1008 1338 1740 2215
Толуол .... 139 206 287 404 557 741 990
ж-Ксилол . . . 51 78 116 168 238 330 449
Как и раньше, допустим, что жидкость является идеальным раствором, а пар—идеальным газом. Поэтому применимо уравнение (20); но теперь неизвестной величиной будет температура, и решение методом подбора должно быть произведено при различных температурах. Соответствующие давления насыщенного пара считываются с графика, построенного по вышеприведенным данным.
Приведем только окончательный результат подбора.
Пусть t — 95°С.
Давления насыщенного пара равны:
Бензол.................1150 мм рт. ст.
Толуол.................. 477 мм рт. ст.
Ксилол................. 199 мм рт. ст.
43 Химическая термодинамика
674
XIII. Процессы дестилляции
Мольные доли в паре равны:
Бензол..................0,725
Толуол .................0,233
Ксилол..................0,042
Состав жидкости легко определяется по материальному балансу.
Пример 4. Растворитель, содержащий 50% (объемных) бензола, 30% толуола и 20% ж-ксилола, полностью испаряется в потоке нагретого воздуха таким образом, что получающаяся смесь содержит пары растворителя в количестве 5,0 объемных процентов. Смесь сжимается до давления 3840 мм рт. ст. и охлаждается при постоянном давлении до 20°С. Сконденсированный растворитель и остаточный газ тщательно смешиваются при прохождении от конденсатора й ловушке, где конденсат выделяется и отсасывается так, что его можно считать находящимся в равновесии с остаточным паром. Принимая, что все смеси этих трех растворителей идеальны, вычислить процент возврата растворителя и состав регенерированного растворителя.
Давления насыщенного пара при 20°С равны:
Бензол............74 мм рт. ст.
Толуол............22 мм рт. ст.
ж-Ксилол.........6,4 мм рт. ст.
Плотности в граммах на миллилитр:
0,879 для Cg
0,866 для С7
0,866 для Cg
На 100 мл растворителя приходится:
50X 0,879 . _с,
—--------=0,563 моля Cg,
7о
30X 0,866 пооо „ —--------=0,282 моля С7,
20X 0,866 ._
—— = 0,163 моля С8; 10b
общее число молей = 1,008.
Мольные доли в исходном газе равны:
0,0500 = 0,0280 Cg,
^Х 0.0500 = 0,0140 С,, £1ЦХ 0,0500 = 0,0081 Cg.
Подставляя найденные данные в уравнение (17) и используя значения X/ = Pilp, получаем
0,0280 , 0,0140 , 0,0081 _.
+(74/3840) (1 — Се) * С* + (22/3840) (1 - Q Се+(6,4/3840) (1 — '
0,0280 , 0,0140 0,0081 _
+0,01927 (1 — Q + Zc + 0,00572 (1 — у + Zc -|- 0,001668 (1 — Q ~ b
Интегральное испарение и конденсация
675
Пусть
^=0,0400,
0,479 + 0,307 + 0,1945 = 0,9805, /
Се= 0,0390,-
0,487 + 0,315 + 0,199= 1,001.
0 0390
Следовательно, конденсируется-jj-QgQ-X 100 = 78°/0 растворителя. Мольный состав жидкости дается нижеприводимыми зиачениимн:
48,6®/о бензола,
31,5®/0 толуола,
19,9°/0 ксилола.
Состав пара можно найти из соотношения
В точке, отвечающей началу конденсации (точка росы), как для смеси паров, способных к конденсации, так и для смеси их с некон-денсируемыми газами, £с = 0; поэтому уравнение (17) переходит н уравнение
£?=’• (21>
или, для случая идеального раствора и идеальных газов, в уравнение
Эти два уравнения можно использонать для вычисления температуры или давления начала конденсации при данном значении одной из этих величин. Для определения давления расчет осуществляется непосредственно, но для расчета температуры необходимо прибегнуть к методу подбора.
В конце конденсации (в точке пузырьков) для смеси с неконден-сируемым газом действительно следующее соотношение:
(23) и для идеального газа
Последнее уравнение получается сразу, если принять 1 в уравнении, аналогичном уравнению (17) и представляющем сумму составов остаточного пара.
43*
676
XIII. Процессы дестилляции
Если исходить из питающей жидкости, то из уравнения (15), принимая Qv— 0, получаем уравнение линии кипения
2^=1.
ДИФЕРЕНЦИАЛЬНОЕ (ЧАСТИЧНОЕ) ИСПАРЕНИЕ (КОНДЕНСАЦИЯ)
Бинарные растворы. Так как в этом случае большое количество одной фазы в любой момент находится в равновесии только с незначительным количеством другой фазы, которая затем удаляется, то количественная обработка должна быть произведена с помощью анализа бесконечно малых. Как и раньше, будет рассматриваться только случай одной жидкой фазы.
Предположим, что в перегонном кубе находится L молей жидкости состава х, причем в некоторый момент испаряется dL молей, и пусть у обозначает состав пара. Материальный баланс по компоненту А для этого процесса выражается следующим уравнением:
Lx=ydL-j~(x— dx)(L — dL), (25)
или
— = (26)
L у—х‘
Интегрируя, получаем
L х1.
Г dL f dx
(27)
F Хр
1п^ = 1п^5У-=1п(1-^) = J (28)
XF
Интегрирование можно произвести для равновесных условий, если известна равновесная зависимость у — tp (х). Для разбавленных растворов можно принять закон Генри:
y = kx,
и тогда уравнение (28) переходит в
или
(29)
(30)
Используя зависимость для идеальных растворов в случае, когда паровая фаза является идеальным газом, именно, уравнение (1, гл. XII),
Диференциальное испарение и конденсация
677
легко можно привести уравнение (28) к форме
1п(1 — ' )=—ff. (31)
' v’ а — l*f(l—XL) 1 1 — XL
Это уравнение справедливо только для изотермического процесса, но оно* также может быть использовано и для изобарного процесса, если принять среднеарифметическое значение а, поскольку, как было показано в предыдущей главе, изменение а невелико.
Совершенно такой же прием в случае диференциальной конденсации приводит к уравнениям
yv
= (32)
М1-и=-^1п^ (33)
к 1 У F
И
1П (i _ у=—ц. in _ и , <34>
V Я_1 yF(l^yv) ур’ . >
которые аналогичны уравнениям (28), (29) и (31).
Может быть, здесь следует подчеркнуть, что эти уравнения применимы к предельному случаю, при котором а) перегонка проводится с бесконечно малой скоростью, т. е. равновесно, б) нет орошения жидкостью, после того как пар покидает кипящую жидкость, и в) нет увлечения жидкости паром. При любой действительной перегонке, осуществляемой в лаборатории или на заводе, удается лишь более или менее близко подойти к этому предельному условию.
Пример 5. Следует сконцентрировать посредством диференциального испарения при атмосферном давлении 100 кг 1-процентного (по весу) раствора фурфурола в воде. Какое количество раствора (по весу) следует перегнать для получения в дестиллате 99°/0 общего количества фурфурола? Каков будет состав этого дестиллата?
Равновесные данные для этой системы при 1 атм таковы:
Весовой процент Весовой процент
фурфурола в жидкости фурфурола в паре
0,20 1,5
0,40 3,0
0,60 4,4
0,80 5,8
1,00 7,0
При принятии зависимости y — kx
среднее значение k равно 7,32. В данном случае приложимо уравнение (29); тогда
хР= 0,0100,
0,00010 !
Xl~ i-cf-
678
ХШ. Процессы дестилляции
Подстановка в уравнение (29) дает
. г х 1 , 0,01
g(1 — 6,32 gl — Су’
1 — CV = 0,533, т. е. следует перегнать 46,7% раствора.
0 99
Концентрация фурфурола в дестиллате = X 100 = 2,12%.
Пример 6. Нанести кривые, показывающие зависимость составов кубового остатка, дестиллата и температуры кипения от количества отогнанного раствора (в весовых процентах), в результате диференциального испарения при 1 атм из 60-процентного (по весу) раствора бензола в толуоле.
Можно принять, что в этом случае применимо уравнение (31); будем пользоваться средним значением а = 2,50. Метод расчета будет продемонстрирован на примере.
Пусть xL — 0,400 А).
Подстановка соответствующих величин в уравнение (31) дает
Су = 0,61.
Состав дестиллата легко получается из материального баланса следующим образом:
Х/?=х£(1 —ty)-^xoty,
где xD — состав дестиллата. Тогда
_ Хр—xL(l —Су)
D tv
Дли случаи, когда xL = 0,400,
xD = 0,727.
Температура кипения любой жидкости в кубе определяется с помощью метода, описанного в примере 3, гл. XII.
Приняв ряд значений xL, получают данные для построения кривых; затем этн данные наносятся на рис. 159.
В тех случаях, когда не имеется простого алгебраического уравнения, выражающего равновесную зависимость, праные части уравнений (28) и (32) можно легко проинтегрировать; для этого строится график, причем на ось ординат наносятся значения \1(у— х), а на ось абсцисс х или у (в зависимости от того, что будет удобнее), после чего определяется площадь под кривой между данными пределами X или у.
Испарение илн конденсация систем, включающих две жидкие фазы, представляет значительный практический и теоретический интерес, но не рассматривается в этой книге.
Многокомпонентные системы. Примем, что равновесная зависимость имеет форму уравнения (71, гл.ХП), а именно:
У~К;Х{, и выберем в качестве стандартного вещества один компонент системы, например компонент В. Пусть раствор, подвергающийся
й) В этом примере х применяется для обозначения весовой доли.
Диференциальное испарение и конденсация
679
диференциальному испарению, содержит Lt молей некоторого компонента и LB молей стандартного компонента. Испарим бесконечно малое количество жидкости, причем получим пар, содержащий dLt
Рис. 159. Кривые диференциального испарения для системы бензол — толуол.
молей i и dLB молей В. При равновесии мы должны иметь
• Л_ Ув xi_ Кв*в ’ (35)
или dLt dLB _ KiJ± Kb Lb' (36)
или d\nLt = QdlnL.r Kb p (37)
Любое из этих уравнений можно написать для всех компонентов, за исключением В. В общем, как было показано в предыдущей главе, К янляется сложной функцией р, t и состава; но если мы примем, что жидкость будет идеальным раствором, то /С будет функцией только р и t. Отношение любых двух значений К очень мало меняется с р и i, и в качестве первого приближения можно проинтегрировать уравнение (37) при допущении, что отношение Ki/KB будет постоянным. Это допущение приводит к уравнению
le —= —. (381
g/7 Кв^Рв ' ’
680
XIII. Процессы дестилляции
Если пар является идеальным газом, то
Az Р£
Хв Рв'
Уравнения (38) и (39) можно объединить и записать в форме
(1 ty)xL (1 ^V^XLB
lg----------: a. lg -----------
хр. ‘В 6 Хрв
(39)
(40)
где а.в = К{}Кв или PijpB.
Материальный баланс отвечает уравнению
ХК—xLt (1 — Сv) -\-y£v,
(41)
где yt — мольная доля любого компонента в общем дестиллате. Должно существовать п—1 уравнений типа (40), п уравнений типа (41) и уравнение
Sxz.= l, (42)
т. е. всего 2л уравнений, любое из которых можно решить методом подбора для получения состава дестиллата и остатка при заданных составе питающей жидкости, давлении и доле испарившегося раствора.
Основным уравнением для конденсации, аналогичным уравнению (38) для испарения, будет уравнение
= (43)
А В П Св
ИЛИ
V, Vn
а^Рг=^Р~в- <44>
Используя тот же метод, который был применен длЬ расчета испа-
рения, получим
, (i-Qj'k , (i-QJ'Ko
«. 1g--------— lg----------
‘В 8 Ур. Угв
(45)
(46)
(47)
Пример 7. Пар, содержащий 300/0 (мольных) я-гексана, 300/0 «-гептана и 40% я-октана, частично конденсируется при атмосферном давлении. Принимая, что пар является идеальным газом и жидкость — идеальным раствором, сравнить состав остаточного пара для случая конденсации 8О°/о от общего числа молей, если конденсация будет: 1) интегральной и 2) диферен-цаальной.
Давление насыщенного пара можно вычислить по уравнению
1g/) (мм рт- ст.) = А — у .
Диференциальное испарение и конденсация
681
Используем следующие значения для констант [13]:
Углеводороды А В
Гексан 7,7215 1654,6
Гептан 7,5917 1750,0
Октан 7,7503 1941,4
Ниже приведен ряд давлений насыщенного пара смеси (мм рт. ст.):
£,°С Р(С6) /’(Q) Р(С8)
90 1462 592 253
95 1690 689 299
100 1935 798 352
105 2209 916 411
ПО 2526 1056 482
Для интегральной конденсации применимо уравнение (17), которое для рассматриваемого частного случая примет вид
УгА г Урв , Урс
^ + (Рд/р)(1-У + ^+(PbP)(1-Q + ^+(Ре/Р)(1-У = L
В рассматриваемом случае неизвестной переменной является конечная температура конденсации, поэтому рекомендуется решение методом подбора. При принятии £ = 95° С и подстановке соответствующих величин в последнее уравнение получаем
0,241 + 0,305 + 0,455 = 1,001.
Этот подбор настолько хорошо удовлетворяет уравнению, что дальнейшее уточнение нецелесообразно. Каждый член уравнения представляет мольную долю одного из компонентов в конденсате. Состав остаточного пара можно вычислить из уравнения (18), причем получаются следующие результаты:
Уу^ = мольная доля гексана в остаточном паре = 0,535,
Ууд = мольная доля гептана в остаточном паре =0,280, Уус = мольная доля октана в остаточном паре = 0,180.
Эти цифры можно незначительно исправить, если следует точно удовлетворить условию Sy у = 1. В случае диференциальной конденсации, — поскольку равновесная температура меняется на всем протяжении процесса, — следует выбрать среднюю температуру, при которой определяют а. В связи с этим обычно бывает полезно вычислять начальную и конечную температуры для всего процесса конденсации. Это можно легко осуществить с помощью урав
682
XIII. Процессы дестилляции
нений (22) и (24) при использовании метода подбора. Приближенные значения равны:
Начальная точка конденсации =108° С,
Конечная точка конденсации = 93° С.
Поскольку а с температурой значительно не изменяется, необходимо найти только ее приближенную величину, и для этой задачи можно принять температуру, равную 100° С.
ecB=S=o,44L
Прилагая к этому случаю уравнение (45), получим два уравнения: 1&УуА = 0.413 igyVB + 0,103, 1gУус = 2,27 lgyyB- 0,093.
Для решения по методу подбора принимаем у = 0,250.
Подстановка этого значения уув дает Vb
Ууа = Ъ,1\Т, Уус = 0,035, ^уу = 1,002.
Эта величина достаточно близка к единице для того, чтобы обойтись без дальнейшего уточнения подбором.
Следующая таблица суммирует результаты сравнения процессов:
Процесс Мольная доля компонента в остаточном паре
С6 С7 с8
Интегральная конденсация Диференциальная кондеи- 54 28 18
сация 72 25 3
Из этого можно заключить, что диференциальная конденсация дает значительно большее обогащение пара, чем интегральная.
Диферевциальвая коидеисация в присутствии ииертного газа. С самого начала примем, что мы имеем дело с идеальными жидкими растворами и идеальными газами и что инертный газ не растворим в конденсате в сколько-нибудь значительной степени. Основное уравнение совпадает с уравнением (44), и его легко превратить в уравнение
(1 — ^сУр)У у (1 — £еУр)Уу
ai В ------5-------- = lg ---V-------- >
В yF, V?B
(48)
Перегонка с водяным паром
683
в которому—мольная доля всего пара, способного конденсироваться в начале процесса, a — сконденсировавшаяся доля его. В этом случае уже не приложимо уравнение (47), но вместо него мы будем иметь
<«>
Это уравнение выводится следующим образом.
Для конечных количеств конденсата должно удовлетворяться следующее уравнение:
Sxz=l, (50)
а для фазового равновесия между идеальным раствором и идеальным газом — уравнение
XiPt=yvfp. (51)
Сочетание уравнений (50) и (51) дает уравнение (49).
Общий материальный баланс для пара, способного к конденсации, имеет вид
= (52)
Решая методом подбора уравнения (48), (49) и (52), можно вычислить сконденсировавшуюся долю пара и состав остаточного пара, если даны давление н конечная температура в конденсаторе. Средний состав конденсата получается с помощью материальных балансов для отдельных компонентов, что дает уравнение типа
У pi = (1 — W У и + *&Ур, (53)
где х{—средняя мольная доля данного компонента в общем конденсате.
ПЕРЕГОНКА С ВОДЯНЫМ ПАРОМ
Это название является общим термином, применяющимся к любому процессу испарения жидкости посредством введения в нее водяного пара, благодаря чему происходит непосредственное соприкосновение водяного пара и кипящей жидкости. Это позволяет проводить перегонку при пониженном давлении, поскольку водяной пар уравновешивает часть общего давления на систему, благодаря чему можно осуществлять перегонку при более низкой температуре. Тот же результат достигается при поддержании с помощью вакуум-насоса пониженного давления, но для случаев относительно нелетучих веществ, которые следует отгонять при достаточно низких температурах во избежание разложения, перегонку в вакууме трудно осуществить на практике. В некоторых случаях выгодна перегонка с водяным паром при пониженном давлении, другими словами, сочетание перегонки с водяным паром с перегонкой в вакууме.
684
ХП1. Процессы дестилляции
Поскольку дело касается общего принципа, вместо водяного пара можно пользоваться инертным газом, но водяной пар имеет некоторые практические преимущества, которые и привели к широкому применению его для этой цели.
Перегонка с водяным паром находит значительное применение при отделении жидкостей с низкой летучестью от нелетучего материала и при удалении растворителей и других летучих жидкостей из раствора в относительно нелетучих маслах или из твердых адсорбентов. В этой главе рассмотрение ограничено определением минимального расхода водяного пара для нескольких специальных случаев.
Случаи I. Жидкость, не смешивающаяся с водой; температура выше трехфазной точки кипения. Эта формулировка, относящаяся к точке кипения, предполагает, что тепло, необходимое для перегонки, поступает из другого теплового источника, а не непосредственно от острого водяного пара; обычно оно сообщается глухим паром, поступающим в змеевик или в рубашку. При использовании же перегретого водяного пара можно так же, как и в этом случае, перегонять при температуре, превышающей трехфазную точку кипения, и одновременно получать все тепло от острого пара, т. е. пара, вводимого непосредственно в жидкость, подлежащую перегонке.
Если рА и pw являются соответственно давлениями насыщенного пара жидкости, подвергающейся перегонке, и воды при данной температуре, тогда, согласно сформулированным условиям,
P'CPa+Pif’
и в кубе не будет присутствовать водяной слой.
Примем, что жидкость с помощью специальных устройств поддерживается при постоянной температуре и что водяной пар приходит в такое тесное соприкосновение с жидкостью, что удаляющаяся парообразная смесь находится в равновесии с жидкостью. Тогда будут справедливы следующие уравнения (при условии, что пар является идеальным газом и что жидкость ие содержит никакого растворенного вещества, заметно действующего на давление ее насыщенного пара):
УаР=Ра’ yw=\—yA=\-p-\
Разделив одно уравнение на другое, получаем
У а Ра __ Na У w Р~Ра Nw’
Тогда ту? NypMyy Mw P— рл ~™л NaMa рД
Это уравнение свидетельствует о том, что расход водяного пара, выраженный в килограммах водяного пара на килограмм перегоняе
(54)
(55)
(56)
(57)
Перегонка с водяным паром
685
мого вещества, будет уменьшаться с уменьшением давления, с повышением температуры и с увеличением молекулярного веса перегоняемой жидкости.
Пример 8. Следует перегнать ацетофенон при атмосферном давлении без образования водяного слоя, используя в качестве теплового источника только острый водяной пар. Водяной пар забирается от источника насыщенного пара при повышенном давлении и перед использованием расширяется до 1 атм. Чему должно быть равно давление водяного пара и минимальный расход его, если желательно поддерживать возможно низкое давление? При расчете тепловыми потерями следует пренебречь. Принять, что ацетофенон предварительно нагревается до температуры перегонки.
Будем считать, что ацетофенон перегоняется при постоянной равновесной температуре, определяемой условием, что тепло, отдаваемое водяным паром при охлаждении до этой температуры, будет равно теплоте парообразования. Наиболее низкая температура, при которой может происходить перегонка без образования водяного слоя, равна примерно 100° С. Примем, что эта температура равна температуре перегонки.
Давление насыщенного пара ацетофенона [188а]:
t = 202,4° С, р = 760 мм рт. ст.
t = 80,0° С, р = 10 мм рт. ст.
Температуры кипения воды при этих давлениях равны соответственно 100 и 11,3° С. При нанесении этих данных в виде линий Дюринга (см. раздел .Правило Дюринга', гл. VI) давление насыщенного пара при 100° С оказывается равным 25 мм.
По уравнению (57) имеем
18 xz 760 — 25
—— = —у, X-----=----— 4,4 кг водяного пара на килограмм ацетофенона.
/Пд 120 25
Скрытую теплоту парообразования ацетофенона при 100° С можно определить из графика функции Гильдебранда (см. гл. IX). Примем, что ацетофенон принадлежит к классу углеводородов. Это дает значение, равное 98 кал! г.
Один килограмм водяного пара при охлаждении от состояния насыщения до 100° С может доставлять 98/4,4 = 22,2 ккал.
Энтальпия насыщенного водяного пара при 1 атм равна 638,8 ккал/кг. Энтальпия, отвечающая расширившемуся водяному пару, равна 638,84-22,2 = = 661 ккал]кг.
Это соответствует насыщенному водяному пару при 8,2 кг/см2 (расширение при постоянной энтальпии). Следовательно, насыщенный водяной пар при этом давлении, расширяясь до 1 атм, будет достаточно перегрет для того, чтобы дать необходимую теплоту парообразования при охлаждении до 100° С.
Расход водяного пара по мере возрастания температуры перегонки будет уменьшаться, но по мере уменьшения расхода водяного пара очень быстро увеличивается необходимое давление водяного пара, так как энтальпия иасыщенного водяного пара очень мало изменяется с давлением. В действительности максимальная энтальпия насыщенного водяного пара равна 669,7 ккал/кг, причем эта величина соответствует давлению примерно в 30 кг]смг.
Случай II. Жидкость, не смешивающаяся с водой; температура соответствует трехфазной точке кипения. В том случае, если не обусловлен способ подачи тепла для перегонки (например, косвенно или же непосредственно с помощью использования перегретого водяного пара, как в примере 8), будут существовать две жидкие фазы,
686
XIII. Процессы дестилляции
и перегонка будет происходить в трехфазиой точке кипения, в которой
Р ~ Ра ~Ь Pw’
и уравнение (57) переходит в
ту?__Му? Pw
тА МА рА’
(58)
Для определения температуры перегонки можно нанести иа график рА-]~pw' в зависимости от температуры и по графику найти температуру, соответствующую общему давлению р. Очевидно, что для такой перегонки верхним пределом температуры будет точка кипения воды при данном давлении.
Уравнения (57) и (58) дают не общий расход водяного пара иа перегонку, а только то его количество, которое используется для переноса пара при перегонке. Кроме того, может еще потребоваться некоторое количество водяного пара для нагревания жидкости и испарения.
Влияние давления. Влияние давления зависит от пути, по которому PvjpA будет изменяться с температурой. Приблизительное представление о влиянии давления можно получить из уравнения Клаузиуса — Клапейрона и правила Трутона.
Из уравнения (46, гл. VI)
1п —= — (— — —. А И к Л Т2)
Применяя это к двум различным веществам, получаем
Lw~ LA(i И (Рдч __(Pg) е к \г, т,1
\РА/г \PaJi
(59)
Но если Tw и ТА приблизительно равны, то в качестве грубого приближения, основанного на правиле Трутона, можно считать
Поэтому
—^а-
Это указывает иа то, что, по крайней мере, для узких пределов изменения давления расход водяного пара, т. е. теплоносителя, практически не зависит от давления и что нельзя достигнуть больших преимуществ, изменяя давление. Экспериментальные данные показывают, что для жидкостей менее летучих, чем вода, pvlpA уменьшается с температурой, и поэтому увеличение давления будет до некоторой степени уменьшать количество острого пара, затрачиваемого на единицу дестиллата.
Перегонка с водяным паром
687
Случай III. Жидкость, частично смешивающаяся с водой. Этот случай существенно не отличается от случая несмешивающихся жидкостей; единственным различием является то, что давления насыщенного пара в уравнениях (57) и (58) не будут давлениями насыщенного пара чистых веществ, но должны определяться из фазовой диаграммы, подобной рис. 143, нли вычисляться по методам, указанным в гл. ХП, раздел .Применение простых законов растворения”. Если жидкость заметно смешивается с водой прн температуре конденсации, как, например, в случае анилина, то должны наблюдаться значительные потери, если только не применяются какие-нибудь меры для возвращения вещества, растворенного в водном слое. Это можно осуществить или посредством ректификации, или посредством экстрагирования соответствующей жидкостью.
Случай IV. Периодическая перегонка раствора летучей жидкости в нелетучей жидкости. Примем, что температура постоянна и превышает трехфазную точку кипения, благодаря чему отсутствует водяной слой. Для равновесного пара всегда имеет место соотношение
^ = Р^РА , (60)
<МА рА
где р — общее давление, а рА—парциальное давление летучего компонента А в паре, находящемся в равновесии с жидкостью.
Ввиду того, что
где х — мольная доля летучего компонента А в жидкости, В относится к нелетучему компоненту, a Ns — константа:
И
dNw=NB^^, (63)
или
= NBp f -^-dx----NB f -dx -. (64)
W B J PAd-^ BJ(l-x)2
По уравнению (64) расход водяного пара на перегонку между любыми данными пределами состава при постоянной температуре и общем давлении можно получить посредством графического интегрирования, если имеются данные по давлению насыщенного пара раствора как функции концентрации. Для частного случая идеального раствора
Ра = Рах-
688
XIII. Процессы дестилляции
Подставляя это значение рА в уравнение (64) и интегрируя, получаем
£. In х1(1~ *2) Р~Ра ( 1_________1 \
Л'в РА *2(1 —*1) Ра~ U— *2 1 —
(65)*)
Случай V. Непрерывная перегонка в противоточной колонне летучей жидкости из раствора в нелетучей жидкости. Из мате-
тел ,
“ "w я || А в в
Водяной f Освобожденный от
пар I | легких франций
раствор
Рис. 160. Диаграмма для случая V.
риального баланса колонны в целом (рис. 160) имеем
<66>
1 2 * 1
или от сечения СС до дна колонны:
^=^ВХуЕ^- (67)
Уравнение (67) можно написать в форме
1'=^.Х+Г>-^<в8)
Это уравнение выражает зависимость между составами двух потоков, проходящих через колонну. На диаграмме
К—X она является прямой линией ☆☆); будем называть ее рабочей линией. Другой зависимостью
между Y и X является соотношение фазового равновесия, которое при обычных обозначениях можно записать следующим образом:
Г=?(4
(69)
и которое должно удовлетворять конечным условиям, т. е.
АГ=О, К=0.
Кривая на диаграмме, отвечающая этой зависимости, будет называться кривой равновесия. Расход водяного пара в молях на моль растворителя В, т. е. Nw/Xs является величиной, обратной наклону рабочей линии [см. уравнение (68)]. Минимальный расход водяного пара при данных условиях будет соответствовать рабочей линии
й) Вывод этого уравнения несколько упрощается, если концентрацию выразить в единицах, используемых в случае V.
☆ ☆) Это обстоятельство и является причиной выбора этих единиц концентрации вместо «х и у.
... <}• 4 у,, 1
Перегонка с водяным паром 689
Ар X максимальным наклоном. Максимальный наклон определяется тем |!Г Обстоятельством, что ни одна рабочая линия не может пересечь /' линию равновесия, так как это противоречило бы второму закону, по- -Х ‘> скольку это требовало бы получения пара с более высоким содержанием 3;__растворенного веществ^, чем у равновесного пара. Следовательно,
, рабочая линия с максимальным наклоном будет линией, касатель-
ной к равновесной кривой. На рис. 161 показаны три такие, линии для обычного случая, когда водяной пар, подводимый к колонне, свободен от компонента, подлежащего перегонке, т. е. когда У2 = 0. Линия ВС является рабочей линией для минимального расхода водяного пара, если раствор следует довести до состава Х2. ОА является линией полного извлечения растворенного вещества из раствора, a DE — линией равновесия между входя
Рис. 161. Минимально^ количество водяного пара, требуемое для г непрерывной перегонки летучей жидкости из ее раствора в нелетучей жидкости в противоточной колонне.
щим раствором и выходящим паром. Во всех случаях состав выходящих паров дается пересечением рабочей линии с орди-
натой при Xt.
Частный случай идеального раствора. Для такого раствора
и поэтому
ур = хрл,
Y 1-1-г
и
х =
X 14-х’
а равновесная зависимость, выраженная уравнением (69), следующий вид:
у__ хРа
Р—(Ра~Р)х
н
Ур
Ра—Ч> — Ра)У'
принимает
(70)
(71)
Кривая равновесия выпукла к оси абсцисс (как* на рис. 161), если ’ РА^> р (обычный случай при перегонке),. и является асимптотой к ординате Х=р{{рА— р). Если рА<^р, то кривая выпукла к оси 44 Химическая термодинамика
♦
690
XIII. Процессы дестилляции
ординат, и асимптотой является абсцисса Y =рАЦр— рд). Если р — рА, то кривая равновесия будет прямой линией.
Пусть Xt будут координатами в точке касания рабочей линии и равновесной линии. Наклон рабочей линии в этой точке равен
Yt
xt-x2 •
Наклон равновесной линии равен
dY________________________________рдр
dX [p-(pA-p)Xt-p ’
(73)
причем эти два наклона равны, т. е.
v___ РаР
‘ [p-(PA~p)Xt]3'
(74)
Так как точка с координатами Yt, Xt лежит на кривой равновесия, то приложимо уравнение (70). Поэтому, исключая Yt из уравнений (70) и (74), получим
(75)
Подставляя Xt в уравнение (73) и замечая, что
1
NB (dY/dX) ’
имеем:
f = Р — ъУрХ2(рА—р)-[-Хг(РА--р)
\ / мин. Ра
\ Если Х2 = 0 (полное извлечение), то
(Nw\ = Р.,
\ Xg 1 мин. Ра '
(76)
(77)
Уравнения (76) и (77) применяются также к случаю, когда равновесие дается законом Генри в форме
УР = хкА.
Пример 9. Раствор бензола в минеральном масле для получения бензола должен подвергаться перегонке с водяным паром при атмосферном давлении. Сравнить минимальный расход пара, выраженный в килограммах водяного пара на литр получаемого бензола для: а) периодического процесса; б) непрерывного противоточного процесса.
Принять следующие допущения:
1. Богатый маслом раствор содержит 5% бензола по весу.
2. Остаток должен содержать 0,100/0 бензола по весу.
3. С помощью глухого пара поддерживается температура в 110° С.
4. Раствор масла можно считать идеальным и принять молекулярный вес масла равным 225.
5. Начальная температура раствора масла равна 20° С.
6. Теплоемкость масла равна 0,50 ккал!кг-град.
Перегонка с водяным паром
691
7. Водяной пар подается при барометрическом давлении, равном 1,75. атм. ‘Мольная доля бензола в всходном растворе
— - £
;__ ... Xj --.-78 -- = 0,132.
5 . 95
78^~225
Мольная доля бензола в остатке
х - 0Л0/78 _
г~ (0,10/78) + (99,9/225) ~0’00“88-
Рд — давление насыщенного пара бензола при 110° С = 1748 мм. Подставляя эти величины в уравнение (65), получаем
Nw __ 760 0,132 X 0,9971 .760 — 1748/ 1 1
NB ~~ 1748 lg 0,00288 X 0,868 1748 \ 0,9971 0,868/“
= 1,641 моля водяного пара на моль масла.
„ ’ 0,132 0,00288 П1._
На моль масла нолучим:^—=--------— =0,149 моля бензола,
0,868 0,997
0,149 X 78 х
—Qgylj—=13,2 мл бензола.
На литр полученного бензола потребуется
1,641 X 18 „
-----------= 2,24 кг острого водяного пара.
1 0,2?
Глухой пар необходим для нагревания загрузки в перегонном кубе до 110° С и для испарения бензола.
Средняя теплоемкость бензола в этом температурном интервале равна 0,45 ккал/кг-град. Скрытая теплота парообразования при 110° С, определенная по правилу Кистяковского ☆) или по Ватсону £?£•), равна 6750 кал!моль.
Расход тепла на моль масла равен
^225 X 0.50+ 78X0,45^ (110-20)1,8+ X6750= 11630кал.
Общий расход водяного пара при периодическом процессе равен 3,94 кг/л бензола. Для непрерывного процесса расход острого водяного пара дается уравнением (76). Так как
^ = 5® = 0,00289,
ТО
Nv L 760—2/760X0,00289(1748 — 760) +0,00289(1748 — 760) _
NB " 1748
= 0,383 моля водяного пара на мюль масла=0,222 кг/л бензола. Расход острого водяного пара непрерывной перегонки значительно меньше, чем для периодической. При отсутствии утилизации тепла расход глухого водяного пара будет тем же самым, но при непрерывном процессе в теплообменниках можно сохранить ббльшую долю тепла.
☆) Гл. IX, уравнение (34).
☆ Гл. IX, уравнение (36).
14*
692
XIII. Процессы дестилляции
Интересно отметить, что расход острого водяного пара для полного извлечения при непрерывном процессе равен
77^ X — 0,592 кг водяного пара иа литр бензола, тогда как при периодическом процессе ои был бы, согласно расчетному уравнению, бесконечно большим.
РЕКТИФИКАЦИЯ
Колонны, в которых обычно проводится ректификация, принадлежат к одному из двух типов: 1) насадочному и 2) тарельчатрму. Для ознакомления с конструкцией каждого из этих типов следует обратиться к учебникам по курсу процессов и аппаратов [287, 315а], но для этой книги существенным различием между этими двумя типами является то, что в насадочной колонне состав и температура двух противоположно направленных потоков жидкости и пара изменяются непрерывно, а в тарельчатой колонне они изменяются прерывно. Каждая тарелка тарельчатой колонны поддерживает слой жидкости, через который барботирует пар. Средние состав и температура жидкости на каждой тарелке отличаются на конечную величину от состава и температуры жидкостей на тарелках, расположенных выше или ниже данной. По мере увеличения числа тарелок или увеличения флегмового числа тарельчатая колонна в общем приближается по своему действию к насадочной, и в пределе, т. е. при бесконечно большом числе тарелок или при полном орошении, оба типа колонн, в сущности, становятся тождественными по принципу.
Ректификационная система состоит из соответствующей колонны, перегонного куба (называемого также кипятильником или испарителем), который служит для получения пара, питающего нижнюю часть колонны, и конденсатора для получения жидкости, орошающей верх колонны. Во всех случаях сама колонна практически представляет адиабатную систему (потери тепла в больших изолированных колоннах очень малы), и вся передача тепла происходит в кубе и в конденсаторе.
Ректификационная колонна может действовать периодически или непрерывно. В первом случае жидкость, подлежащая разделению, помещается в куб, и единственным питанием колонны служит пар из куба, который непрерывно изменяет свой состав. В непрерывно дев* ствующей колонне жидкость, подлежащая разделению, непрерывно подводится к промежуточной точке, называемой уровнем питания. Часть колонны, находящаяся выше этого уровня, называется укрепляющей нли ректификационной частью колонны (или просто верхней крлонной), а находящаяся ниже этого уровня называется исчерпываю щей частью, или отгонной, или просто нижней колонной. Из конденса тора выше колонны непрерывно отводится дестиллат, в которое сконцентрированы более летучие компоненты, а кубовый остаток богатый менее летучими компонентами, непрерывно забирается и
Ректификация
693
^перегонного куба. Можно также иметь раздельные непрерывно действующие укрепляющую и исчерпывающую колонны, причем главным различием между этими колоннами является то, что первая получает питание в виде пара, а вторая — в виде жидкости.
Применение термодинамики к анализу работы ректификационной колонны преследует две основные цели:
1. Определение теоретического числа тарелок в тарельчатой колонне, необходимых для заданных условий. Для насадочной колонны соответствующей величиной будет высота насадки, эквивалентная одной теоретической тарелке.
2. Определение минимально требуемой теплоты или работы.
Теперь мы перейдем к выводу количественных зависимостей, необходимых для осуществления этих целей, ограничив анализ бинарными системами, состоящими из полностью смешивающихся жидкостей.
Теоретическая или равновесная тарелка. Обычно теоретическую тарелку определяют как тарелку, содержащую слой жидкости, от которого поднимается однородный по составу пар, находящийся в фазовом равновесии с жидкостью, состав которой, в свою очередь, является средним из составов жидкости, покидающей тарелку, считая, что жидкость тщательно перемешивается. Это определение устанавливает стандарт, с которым следует сравнивать действительные тарелки. Отклонения действительных тарелок от идеальной обусловлены двумя главными причинами: 1) недостаточной длительностью контакта между паром и жидкостью, не позволяющей достигнуть значительного приближения к фазовому равновесию, и 2) неоднородным составом жидкости на тарелке в совокупности с тем, что поток жидкости не представляет среднего состава жидкости на тарелке.
К. п. д. тарелки. К. п. д. служит мерой для определения степени отклонения действительной тарелки от идеальной. Это отклонение определяется различными путями, причем наиболее обычным способом является определение среднего к. п. д. тарелки, который равен общему числу теоретических тарелок, необходимых для данного разделения при заданных условиях, деленному на действительное число тарелок, необходимых для осуществления рассматриваемого разделения. К. п. д. отдельной тарелки определяется уравнением
где _У„_1 — средний состав действительного пара, поднимающегося от (я—1) тарелки (тарелки ниже л-ной), уя— то же самое для к-ной тарелки, у*в—состав пара, находящегося в равновесии со средней жидкостью, стекающей с л-ной тарелки.
Числитель в уравнении (78) дает действительное обогащение пара данным компонентом, а знаменатель дает максимально возможное
694
XIII. Процессы дестилляции
обогащение Л). Уравнение (78) в этом виде определяет средний к. п. д. всей тарелки, но его также можно использовать для определения к. п. д. любой части площади тарелки; в этом случае он называется точечным или местным к. п. д. К. п. д. одной тарелки обычно является средней величиной, полученной путем суммирования н усреднения всех местных к. п. д.
Обсуждение различных факторов, влияющих на к. п. д. тарелк и, выходит за пределы этой книги, но желательно отметить, что тогда как местный к. п. д. никогда не превышает 1ОО°/о, остальные два к. п. д. могут превышать и часто действительно превышают эту величину. На первый взгляд это кажется нарушением второго закона; однако это обусловлено тбм, что в больших колоннах может создаваться значительный перепад концентраций в жидкости, благодаря чему жидкость на тарелке по среднему составу окажется значительно богаче легколетучим компонентом, чем переливающаяся жидкость. В результате, поднимающийся пар может иметь более богатый состав, чем состав, соответствующий фазовому равновесию с перетекающей жидкостью. Этот вопрос изучался несколькими исследователями; для ознакомления с подробностями следует обратиться к нх статьям [102, 134, 151]. '
Теоретическая эффективность насадочной колонны. Эта величина обычно выражается двумя способами: 1) высотой, эквивалентной теоретической тарелке (ВЭТТ), и 2) высотой, эквивалентной единице переноса массы (ВЭЕП). Первая величина равна длине секции колонны, которая осуществляет ту же самую степень обогащения, что и теоретическая тарелка. Формулируя это определение в несколько иной форме, можно сказать, что эта величина является высотой насадки, при которой средний пар, покидающий верхнюю часть колонны, находится в фазовом равновесии со средней жидкостью, покидающей нижнюю часть колонны. Физической интерпретации значения второй величины не существует. Математическое определение ее дается уравнением
ВЭЕП = р-----------, (79)
\Уу/(у* — У)
где Нр — общая высота насадки, a Vj и у2—конечные составы пара. Интеграл называется числом единиц переноса массы; он равен площади под кривой, выражающей зависимость 1/(у*—у) от у между данными пределами.
☆ ) Подобный же к. п. д. можно определить на основе состава жидкой фазы. Хотя в некоторых условиях он и может быть полезен, но он совершенно ие употребляется.
☆ ☆) Подобное уравнение можно написать, пользуясь и составами жидкости, но при перегонке обычно принимается, что пленка пара дает контролируемое сопротивление.
Ректификация
695
В общем случае число теоретических тарелок и число единиц переноса массы будут различны. Это обстоятельство в дальнейшем подвергнется количественному анализу (см. стр. 710). Хотя высота, эквивалентная теоретической тарелке, значительно чаще используется для
ч
Л'
Рис. 162. Схема ректификационной системы.
характеристики насадочной колонны, все же и применение величины, определяемой уравнением (79), покоится на надежной теоретической основе, поскольку она учитывает непрерывное, а не ступенчатое изменение.
Общие уравнения для адиабатной ректификационной колонны. Мы можем написать материальный и тепловой баланс для части системы выше уровня питания колонны, т. е. заключенной в пределах от АА' до ВВ' (рис. 162), следующим образом:
Vn — On-\-D (общий материальный баланс), (80)
Vny = Опх 4- Dxd (баланс компонента А), (81)
VnH=Onh-\-Dho-\- Qc (общий тепловой баланс). (82)
696
XIII. Процессы дестилляции
Для того чтобы сделать эти уравнения симметричными, напишем уравнение (82) в форме
VnH=Onh-\-Dh’D, (83)
где
(86)
(87)
(88)
(89)
Исключая Vn из уравнений (80) и (81), получаем
У ~ O_JLn Х П Л. П XD‘ (85)
- и । ~ п । ~
Из уравнений (80) и (81) можно исключить массы. Исключая V из уравнений (80) и (83), мы получаем два уравнения:
Oa__xD—y D у — х ' On, tt’p-H D ~~ H — h '
Отношение OnlD называется флегмовым числом.
Приравнивая уравнения (86) и (87), получаем
-Гр—У _h'D — H У—х H—h’
или h’D — H . H—h
h'D-h x + ~h'~-h Х°'
Уравнения (85) и (89) выражают соотношения между составами пара и жидкости на любом уровне колонны. Они совершенно общи, а потому свободны от каких-либо других допущений, кроме того, что колонна работает в установившемся состоянии и является адиабатной. Такое соотношение называется уравнением рабочей линии. Уравнение (89) можно написать в другой форме, именно:
(90)
Vc = °c + D- (91)
Сочетание этих уравнений с уравнением (84) дает
= AD + (zF + 1) (Яс - Ар)- (92)
Флегмовое число OC]D имеет особое значение и будет обозначаться буквой 7?. Если термин употребляется без пояснений, то под ним обычно подразумевается среднее флегмовое число. Зависимость между qc и флегмовым числом получается из уравнений (84) и (92);
Ректификация
697
? очевидно, что
9c = (^+1)(//c-Ad). (93}
Из уравнений (92) и (89)
> AHc-hpW-kH-^ + tH^h) ,_________H-h q
У~ (Hc-hD)R + (Hc-h) Х ' (tic—hD) R-]~(HC —h) D- ' >
Рабочая линия [уравнение (89) или (94)] фиксируется сразу, если известны состав дестиллата и флегмовое число. Можно ли нанести эту линию или нет, зависит от того, известны или нет энтальпии как функции состава при данном давлении.
Уравнения для укрепляющей части колонны можно получить аналогичным способом, составив баланс секций СС—DD'. Основными уравнениями баланса являются:
От = Ут-1-Г, (95)
Omx=Vmy+Wxw, (96)
Oj + QB=VmH+Whw. (97)
Поступая так же, как и раньше, легко получить следующие уравнения:
w (98)
V — X - ' om — w х Om-.w
У — xw _ _ x — xw (99)
77 — h’^ h—h’w ’
где Qb t * (100)
ИЛИ „ h'w — H 1 H~h (101)
hr -ф — h. X । h’w— h''w-
Уравнение (101) является уравнением рабочей линии для укрепляющей части колонны. Из общих балансов для всей системы получается следующий ряд уравнений:
F=D-\-W, (102)
Fzf=Dxd -}- Wxw, (103)
= Whw-\-Qc. (Ю4)
Уравнение (104) можно написать в форме
F/f = £)A'D4- Wh'w.
(105)
Символы zF и IF употребляются соответственно для обозначения состава и энтальпии питания, так как в общем случае питание может осуществляться или паром, или жидкостью, или смесью пара и
698
XIII. Процессы дестилляции
жидкости. Из уравнений (102) и (103) следует, что
D___гр —
W Xd — zP
и
F
W
Xq— x^y
XD— ZF
Из уравнений (102), (103) и (105) получим
(106)
(107)
(Ю8)
zf~~xw _Ip—h’w
XD— гР ^'d — Ip
Из уравнений (84), (92), (100), (104), (106) и (107) найдем, что
h’ <ХД~xw) 1Р—(гР — xw)(R-\-^)Hc~b-(zp — xw}hD ,iqq.
Ггч - 9 т, * ' '
Из уравнений (101) и (109) очевидно, что рабочая линия для укрепляющей части колонны фиксируется сразу, если даны флегмовое число, вид питания и составы дестиллата н кубового
Г" остатка. Из уравнений (98), (102), (106) и (107)
fy — xw} (xd — zp\ (ПО) D \У~Х J \ZF — Xw) ' /
Уравнение (110) является уравнением флегмового числа для исчерпывающей части, аиалогич-
2 |02 । \/г ным уравнению (86) для укрепляющей час-
-------г-----------ти. В случае отсутствия укрепляющей секции питательная жидкость входит в верхнюю ~ •----------------часть колонны, и обычный баланс приводит
Рис. 163. Соотношение к выражению
между потоками на различных уровнях.
Р __Ур—хУ
W ур~хр’
(1И)
в котором индекс F относится к верхней частя колонны. Отношение F/ W является флегмовым числом для десорбционной части колонны.
При рассмотрении любых двух уровней колонны между точками входа и выхода смеси из колонны (см. рис. 163) можно написать следующие три уравнения баланса:
+ (112)
1\У1 + °2Х2= ^yz-T-OjXj, (ИЗ)
K1/f1 + O2A2 = ^2 + OiV (И4)
Ректификация
699
Исключая Vj и V2, получаем ?
О2__ — + —
Oi— (Я2 - Л2) W-Л) + (Я2 - /А) (у2 - х2) • 1 ‘°’
В общем ясно, что О2 не равно Ог; другими словами, величина потока жидкости не является постоянной ни в одной секции колонны. Подобная же зависимость существует между Vy и V2.
Частные случаи. Большое количество общих уравнений можно крайне просто представить на диаграмме энтальпия—концентрация, но прежде чем рассматривать общие случаи, рассмотрим несколько частных, для которых можно значительно упростить общие уравнения.
Случай I. Теплота смешения и различия в теплоте, вызывающие изменение температуры, ничтожно малы. Это допущение приводит к уравнению
h = hD=hF = hw', (116) '
скрытая теплота парообразования при постоянном составе пара равна Н~ h = LAy + LB(\— у). (117)
Произведя подстановки этих величин в уравнение (94) и решая его относительно у, получим уравнение
__ U-м U? +1) ~ х + Твхр (1 I о.
У Lm{R+\)-Y(La-Lb](x-xd) ’ 1
в котором
= LAyc -|- LB (1 _ус). (119)
Если располагать данными по скрытым теплотам парообразования отдельных компонентов, то рабочие линии для укрепляющей секции легко можно найти по уравнению (118). Подобное же уравнение можно вывести для исчерпывающей секции колонны из уравнений (101) и (109).
Случай II. Одинаковые мольные скрытые теплоты. Это представляет дополнение к допущению в случае 1 и основывается на том обстоятельстве, что многиё пары сходных по своим свойствам жидкостей имеют приблизительно одинаковые мольные скрытые теплоты парообразования, как и требуется правилом Трутона. Подстановка ' в уравнение (118)
la=lb=l
приводит к уравнению
У = z?4-1 х /?-Н ‘ ^12°)
Также, поскольку
Ну=Н2
И
Н1 — h1= Н2 — Л2,
700 ~ XIII. Процессы дестилляции
уравнение (Й5) сводится к
О2 = О„ (121)
т. е. поток жидкости в колонне является постоянным (если считать на число молей). Из уравнения (112) также очевидно, что
=
Этот частный случай относят к постоянному потоку (выраженному
в молях).
- Уравнение рабочей линии (120) превращается теперь в уравнение
прямой линии с наклоном, равным и отрезком, отсекаемым на оси у, равным хо/(/?-[-1). Для получения столь же простого уравнения для исчерпывающей секции необходимо подробно рассмотреть состояние питания.
Влияние типа питания. Рассмотрим секцию тарельчатой колонны, расположенную на уровне питания, как показано на рнс. 164, и составим материальный и тепловой балансы по секции:
F^-On^Vm=Om+Vn, (122) ZFAf + (1-Z)F^+ Onhn+VmHm =
(Здесь I—часть питания, поступающая в виде жидкости.) Примем, что энтальпии насыщенного пара и жидкости постоянны, т. е. что
Нт = Н=Н т л И h =h—h. tn П ъ-
е
Тогда, исключив Vm и Vn из уравнений (122) и (123), получим
Om~On=F(?^+lӣ=g) ' (124)
= qF. (125)
Здесь q, как показывает уравнение, представляет количество тепла, требующееся для приведения одного моля питания в состояние насыщенного пара и деленное на мольную скрытую теплоту. Это становится более наглядным при рассмотрении нескольких частных случаев.
Ректификация 701
Случай I. Питанием служит перегретый пар: / = 0, Н—НР
q—отрицательно, а Н—НР представляет теплоту, которую следует отнять у пара для приведения его в состояние насыщения.
Случай 2. Питанием служит насыщенный пар:
(=0, Н=НР.
Следовательно,
q = 0.
Случай 3. Питанием служит смесь пара и жидкости:
H=HF, h = hp, q = l.
Случай 4- Питанием служит насыщенная жидкость-
Н=Нру h = hF, /=1, q=\.
Случай 5. Питанием служит жидкость при температуре ниже точки кипения:
Подстановка уравнения (125) в уравнение (98) дает
y — Oa-\-qF—V/X On-\-qF—WXw (126)
Если с помощью уравнений (106) и (107) исключить массы и обозначить отношение On]D через R, то это уравнение примет следующий вид:
У = /?4-Brf-£2 Х ~ х«г’ (127)
где
Е J
ZF — XW •
Уравнения (126) и (127) полезнее уравнения (98), поскольку поток . у в исчерпывающей секции непосредственно не дается, а его следует определить, исходя из потока в укрепляющей секции. Для условий ' *. питания в случае 4, которые являются, вероятно, одними Из наиболее щи часто встречающихся, уравнение (126) можно написать как
O„ + F W
On + Dxw-
702
XIJ1. Процессы дестилляции
Для случая постоянного мольного потока это уравнение можно также дать в следующей форме, включающей только отношения масс:
У = ^ГТХ~Т^Х^ <129>
Подобно аналогичному уравнению для секции укрепления, оно дает на диаграмме у — х прямую линию. Для одной укрепляющей колонны соответствующим уравнением является
<13°>
При использовании уравнений (106) и (107) и того обстоятельства, что для этого специального случая zF~xp, уравнение (129) можно дать в следующей общеупотребительной форме:
V__Rxf\~xD— (R + 1)XW r | <XF— xp)xW ( I □ < j
W-j-1) (X/r— Xw) (/?+ l)(Xf — Хцр)
Пересечение рабочих линий. Рабочие линии должны пересекаться, поскольку они имеют различный наклон; точка пересечения должна удовлетворять следующим условиям:
Ут=Уп=У/’ xm = xn = xj-
Исключая Оп из уравнений (86) и (126) и подставляя условия, отвечающие точке пересечения, получим
d(xd~ yj}=qF(xj— />)+ w(yj— х^- (132)
Сочетая это уравнение с уравнениями (102) и (103), получим
('33)
Прямая линия, выражаемая этим уравнением, является геометрическим местом точек пересечения двух рабочих линий. Она проходит через точку
и имеет наклон qf(q—1). Различные линии, соответствующие пяти рассмотренным случаям питания, приведены на диаграмме у — х рис. 165.
Использование фиктивного молекулярного веса. Так как
^А — ^АКА И
= /Ив).в,
Ректификация
703
то условие равенства мольных скрытых теплот будет удовлетворено, если
^в = ^а~- (134)
Рис. 165. Место пересечения рабочих линий.
7 _ < 0, наклон положительный, отрезок положительный (питание перегретым паром); i—q=Jit наклон равен нулю (питание насыщенным паром); 3 — q > 0 < 1, наклон отрицательный (питание жидкостью и паром); 4 — 9=1, наклон равен бесконечности (питание насыщенной жидкостью); 5 — 9>1, наклон положительный, отрезок отрицательный (питание переохлажденной жидкостью).
Можно удовлетворить этому условию, приписывая компоненту В фиктивный молекулярный вес, как это требуется уравнением (134). Фиктивная мольная доля А в растворе дается уравнением
„_______________ /1 ос \
Х — (тА1МА) + (тв1МвУ
Подставляя значение Мв из уравнения (134) и вводя весовую долю г, получаем
—<136>
Если для какой-нибудь системы мольные доли вычисляются по этому уравнению, то можно использовать упрощенные уравнения, применимые к случаю постоянного мольного потока. Однако следует отметить, что этот прием все же включает допущение о ничтожно малой теплоте растворения и о ничтожно малых различиях в теплоте, вызывающей изменение температуры, н поэтому не является законной заменой общих уравнений. Следует также отметить, что если
равновесная зависимость имеет форму, даваемую уравнением (1, гл. XII), она не зависит от- молекулярных весов и поэтому не изменяется при использовании фиктивного молекулярного веса.
Число теоретических тарелок. Определение числа теоретических тарелок является обычным применением уравнений, выведенных для ректификационных колонн. Однако следует отметить, что такое применение этих уравнений не является единственным. Существует определенная зависимость между состоянием и составом питания, составом обоих продуктов, флегмовым числом (или другим отношением масс) и числом теоретических тарелок; это соотношение можно использонать для выяснения любой из указанных величин, если даны остальные. Например, можно иметь уже действующую колонну и пожелать определить состав продукта, получаемого в данных условиях. Методы подсчета числа тарелок или любой другой переменной можно
704 XIII. Процессы дестилляции
подразделить на две группы: 1) графические методы и 2) аналитические методы. Графические методы, в свою очередь, можно подразделить на методы, основанные на применении диаграммы у—х, и методы, основанные на применении диаграммы энтальпия—концентрация (Н—х). Все эти методы будут давать одинаковые результаты при одинаковых основных допущениях; выбор между ними зависит главным образом от удобства или личного вкуса 92.
Айтору кажется,
Рис. 166. Схема секции тарельчатой колонны.
ите определение числа теоретических тарелок шшией точностью; об этом свидетельствует упоминания о дробном числе тарелок . Помня о целях нахождения числа теоретических тарелок, полезно предостеречь от необоснованных крайностей при осуществлении таких вычислений, так как большинство произведенных таким образом расчетов позднее требует принятия весьма приближенных к.п.д. тарелок, необходимых для получения числа фактических тарелок. Кроме того, приходится, например, учитывать то обстоятельство, что используемые равновесные данные часто бывают не слишком точны или же система ие адиабатна и т ц. Некоторое оправдание для рассмотрения дробного числа тарелок имеется в тех случаях, когда расчеты числа тарелок производятся исключительно для выявления влияния какого-либо параметра.
Общий принцип, положенный в основу определения числа тарелок, лучше всего 166, который представляет схематическое с
иллюстрируется рис;
изображение секции колонны, содержащей три тарелки. Различ-
ные составы жидкости и пара отмечены индексами для' обозиа чеиия тарелок, к которым они относятся. Примем, что известен состав хя+2 жидкости, стекающей на я —J— 1 тарелку. Тогда состав пара, поднимающегося от этой тарелки, можно вычислить из уравнения рабочей линии, поскольку такое уравнение связывает составы потоков,, проходящих на любом данном уровне. Если известен состав < пара уп+1, то состав жидкости, покидающей эту тарелку (хя+1), находится из уравнения фазового равновесия, так как определение i теоретической тарелки требует, чтобы оба потока находились в равно- ‘ -весии. По значению хя+1 из рабочей линии определяется уп, затем из кривой равновесия хп и т. д. Для того чтобы начать рассмотренный процесс расчета, мы должны иметь одни известный состав и значение флегмового числа, позволяющие фиксировать рабочую линию. В качестве одного из заданных условий задачи (в том случае,' если имеется одно неизвестное, можно применить метод подбора,
Ректификация
705
- результаты которого позднее можно проверить) обычно дается состав продукта хо; при полной конденсации он равен хс. При частичной — конденсации хс следует вычислять с помощью приведенных ранее уравнений по условиям, получаемым в конденсаторе.
Теперь обратимся к рассмотрению способа определения флегмового числа.
Флегмовое число. Уравнение (86) связывает флегмовое число на любом уровне укрепляющей секции с составом дестнллата и проходя-
Рис. 167. Изменение минимального флегмового числа OnjD с изменением уровня в колонне.
щих потоков. Разность (у— х) точно не фиксируется, но имеет совершенноопределенные пределы. Так как пар на всем протяжении колонны получается посредством испарения жидкостей, то ясно, что количество пара никогда не может превышать количества, соответствующего фазовому равновесию с жидкостью, из которой он получается; в противном случае происходило бы нарушение второго закона. Состав этой жидкости обычно должен быть менее богат легколетучим компонентом (хотя бы на ничтожно малую величину), чем состав жидкости, стекающей сверху (через которую проходит рассматриваемый пар); поэтому (у— х) имеет максимальное значение, соответствующее значению, принятому для фазового равновесия. Наоборот, стекающая жидкость получается прн конденсации пара, и предельный случай полной конденсации будет давать у = х\ следовательно, низшим пределом (у— х) будет нуль.
Максимальное значение (у — х) определяет минимальное значение OJD (флегмовое число) для любого уровня укрепляющей части [уравнение (86)]. На рис. 167 это значение нанесено для системы бензол— толуол, причем для а принималась средняя величина, равная 2,5, а содержание бензола в дестнллате принималось равным 99 мольным процентам. Результаты, представленные на фиг. 167, показывают, что минимальное флегмовое число для укрепляющей части медленно увеличивается по мере продвижения по направлению к низу колонны и будет максимальным на некотором уровне, выбираемом в качестве 45 Химическая термодинамика
706
XIII. Процессы дестилляции
уровня питания. Минимальное флегмовое число OmlD ниже уровня питания вычислялось по уравнению (ИО), причем принималось, что питание осуществляется насыщенной жидкостью. Для того чтобы сделать это значение сравнимым со значениями для укрепляющей части, из него вычиталось FfD, что давало значения OJD. Ход кривой типичен для большинства систем, но в некоторых случаях максимальное значение минимального орошения может наблюдаться на некотором более высоком уровне.
В адиабатной колонне количество орошения на различных уровнях не поддается регулированию, но может быть фиксирована максимальная величина, требующаяся для фазового равновесия на любом уровне. Эта максимальная величина различных минимумов является наименьшей возможной для рассматриваемого уровня (обычно уровня питания), но она не обязательно будет равна R(— OCID), потому что поток в общем случае не является стационарным. Для получения минимального OcjD из минимального OJD можно воспользоваться уравнением (115).
Предположим, что максимум минимумов равен 2,00 и приходится на уровне питания и что ОС)ОР, по уравнению (115), равно 1,20. Тогда найдем, что минимальное возможное значение R будет равно 2,00 X 1,20 = 2,40. Это значение называют „минимальным флегмовым числом**. При сочетании уравнений (86) и (115) можно получить уравнение для прямоговычисления минимального флегмового числа RM„a. Значение R, которое следует употреблять в уравнениях (94) и(118), должно быть больше этого минимума или в пределе равно ему. Для частного случая потока с постоянным мольным числом
Ос
D~~ D
и минимальное флегмовое число является постоянным, независимо от уровня в колонне. Величина R в уравнении (120) должна быть больше этого минимума. Если она равна минимуму, то данное разделение будет требовать бесконечно большого числа тарелок; если оно меньше минимума, то разделение невозможно. При увеличении отношения (выше минимума) число требующихся тарелок будет уменьшаться и достигать минимального значения в том случае, если поднимающийся пар полностью конденсируется, т. е. если R = oo. При введении этого значения в уравнение (94) уравнение рабочей линии превращается в
V = x, (137)
т. е. потоки жидкости и пара, проходящие через колонну, одинаковы по составу. Флегмовое число, которым следует пользоваться при установлении рабочих линий, будет лежать где-то между минимумом и бесконечностью. Его величина совершенно произвольна и может определяться только экономическими соображениями.
Ректификация 707
Графический метод Мак-Кэба — Тиле. Ступенчатый метод определения числа тарелок можно осуществить как аналитически (посредством двух уравнений, из которых одно является уравнением рабочей линии, а другое — уравнением линии равновесия), так и графически—по графику этих двух линий на диаграмме у — х. Частный случай потока с постоянным числом молей впервые был подробно рассмотрен Мак-Кэбом и Тиле [161]. Поскольку этот метод достаточно полно разобран в курсах процессов и аппаратов химической технологии, он излагаться не будет. Однако желательно указать, что тем же самым методом можно пользоваться как для общего случая, так и для частных случаев, в которых рабочие линии не являются прямыми. Линии легко наносятся в том случае, если имеются в распоряжении необходимые данные по энтальпии. Так как эти данные имеются лишь для очень немногих систем, а отклонение от линейности в подавляющем большинстве случаев невелико и может быть значительно уменьшено с помощью метода фиктивного молекулярного веса, то метод Мак-Кэба — Тиле практически является универсальным.
В случае жидкостей, очень близких но свойствам (например изомеров и изотопов) и поэтому требующих большого числа тарелок для разделения, графический метод становится очень трудоемким, и предпочтительнее будут некоторые алгебраические методы.
Алгебраические методы.. При выработке методов, описанных в нижеприводимом разделе, принималось, что жидкости являются идеальными растворами, а пар — идеальным газом; поэтому равновесная зависимость дается уравнением (1, гл. XII). Это представляет хорошее приближение для большинства систем, требующих большого числа тарелок. Для случая полного орошения получается следующее очень простое уравнение.
Начиная с нижней тарелки (№ 1) колонны, мы имеем
но так как для полного орошения
х2=уъ то
х., х.
Г^х; = аГ=7г или
Подобным образом
1 Л2
ИЛИ
Хг = а^
45*
708
XIII. Процессы дестилляции
тогда Х4 = а9Хи
или, обобщая для д-ной тарелки колонны, '^=ая~1. (138)
Л1
Если орошение поступает из конденсатора, в котором осуществляется полная конденсация, то
XD=Yn = aXn.
Подставляя в уравнение (138), получаем
или
1g а
(139)
Если Е—общий коэфициент фракционирования = XDIXV, то
IgE 1 —------1.
1g а
(140)
Если орошение поступает из дефлегматора, эквивалентного одной теоретической тарелке, то
п = ^-2. (141)
Iga ' >
Уравнение (139) можно также написать, как
Уп ___ап
1—Уп 1—V
что является уравнением для п равновесных тарелок, эквивалентным уравнению
V X
~— = а --------
1 —у 1 — X
для одной тарелки.
Для более общего случая, когда флегмовое число является конечной величиной, было разработано несколько методов. В тех случаях, когда равновесная кривая лежит относительно близко к диагонали и поэтому рабочие линии примерно параллельны равновесной кривой, можно показать, что число теоретических тарелок и число единиц переноса массы почти одинаковы; поэтому можно вычислить число тарелок по уравнениям, выведенным‘для переноса массы. Например, для случая равных скрытых теплот диференцирование уравнения (120) дает
(143)
Ректификация
709
При подстановке уравнений (143) и (120) в уравнение Л / > (' аУ
«(число единиц переноса массы) = J у*^у
получается
I dx
« = ---------1--------
Хр У* — х~ #-(XD— .И
(144)
(145)
с подобным же уравнением для исчерпывающей секции колонны. Это уравнение выведено Льюисом [152] и применялось им для расчетов по перегонке за много лет до введения понятия о единицах переноса
.массы. (Интегрирование он проводил графически). Додж и Хаф-ман [66] посредством уравнения (1, гл. XII), а также посредством закона Генри связали у* с х, причем они пользовались алгебраическим интегрированием, получая точные выражения для числа единиц переноса массы, которое прини малось равным числу тарелок. Для более подробного ознакомления следует обратиться к их статье.
Смокер [236] разработал метод аналитического определения числа тарелок, который луч-
Мольная доля в жидкости
Рис. 168. Метод Смокера для определения числа тарелок.
ше всего объяснить при помощи рис. 168. Основой его вычислений
служит перемещение начала координат осей у и х в точку пересечения рабочей линии с равновесной линией. Если уравнениями для рабочей и равновесной линий являются соответственно
у = тх -[- b
ах
У—1+(а-1)х’
(146)
(1, гл. XII)
и
то они при отнесении к новым осям принимают вид
у'=тх' (147)
и
+ (148)
где с = 1 -|- (а — 1) k.
710
XIII. Процессы дестилляции
k является координатой х при новом начале координат и легко определяется решением квадратного уравнения, получающегося исключением у из уравнений (146) и (1, гл. XII).
Начнем осуществление обычного ступенчатого расчета в точке г', х'о на рабочей линии, которая для случая укрепляющей секции будет представлять состав пара, покидающего верхнюю тарелку, если принять, что орошение поступает из конденсатора, в котором осуществляется полная конденсация. Состав жидкости на этой тарелке дается равновесным уравнением [перегруппировка уравнения (148)]:
c2v' х' = — г я —С(я —1)з»0 (149) •
но поскольку У'о = тх'О’
то тс2х'о (150)
я — тс (а — 1)х0
.Подобно этому, ( тс2х\ (151)
2 а — тс (а — 1) х[
или, сочетая уравнения (150) и (151), получаем
т2^х'о
я2 — тс (а — 1) (а-[-/яс2) х’о
(152)
Можно продолжать действовать этим способом; но сразу становится ясно, что уравнение (152) можно обобщить таким образом:
тп с2пх'п
х' =---------------------—— -----. (153)
° , Л.а.п — тпс2п , v '
я" — тс (я — 1)------т— х0
я — тс2 °
. I
Решая относительно п, получим следующее уравнение для числа тарелок в любой секции колонны:
lg ко U — Л1хй)/хл (1 — Мха)] lg(a/mc2) ’ (154)
где
Ж==тс(«-П
а — тс2
Если орошение полное, то /и=1, k—О; так как с— 1 и 41=1, то уравнение (154) превращается в
a^igW*„)
(155)
1g я
Ректификация
711
которое совпадает (включая в число тарелок и куб) с уравнением (139),
уже выведенным для этого частного случая.
Вариант метода Смокера можно основать и на случае полного орошения, при котором рабочей линией является диагональ у' = х'. Рабочую линию в общем случае можно превратить в диагональ; для
этого, во-первых, нужно перенести оси в точку пересечения, как и при методе Смокера, и затем повернуть их на угол 6, который равен 45° — <р, где w— угол, лежащий между рабочей линией и первоначальной осью х. С помощью хорошо известных соотношений
для поворота осей, х’, основанное на новых осях, можно связать с х; сочетание найденной з;висимости с уравнением (155) дает уравне-
ние для п, приложимое для общего случая. Вывод этого уравнения как интересное упражнение предоставляется изобретательности читателя.
Томсон и Битти[247]вывели упрощенные уравнения для вычисления количества теоретических тарелок для частного случая, в котором один из компонентов присутствует в питании в количестве, не превышающем 5°/0 (мольных). Их метод основан на допущении линейности рабочих и равновесных линий, и в нем используется ступенчатое суммирование, так же, как и в методе Смокера83.
Рис. 169. Зависимость между числом тарелок и числом единиц переноса массы.
Чис лэ тарелок как функция числа единиц переноса массы. Теперь мы в состоянии произвести количественное сравнение этих двух понятий. Иногда ошибочно утверждают, что эти два числа одинаковы во всех случаях, как бы велико ни было число единиц переноса массы. Соотношение между ними можно уяснить с помощью рис. 169. Между конечными составами пара уг и у2 имеется ряд отдельных ступеней, таких, например, как у*—уп, широко изменяющихся по составу. Каждая из них соответствует теоретической тарелке. С дру
гой стороны, единица переноса массы соответствует ступени, основанной на среднем из вертикальных отрезков у*—уп. Ясно, что число ступеней может быть или может не быть почти одинаковым в обоих случаях в зависимости от соотношения между рабочими и равновесными линиями. Если эти две линии параллельны, ясно, что оба эти числа должны быть равны. Это будет приблизительно правильно для идеального раствора близко кипящих компонентов, поскольку в таком случае равновесная линия никогда не будет сильно отклоняться от диагонали.
712
XIII. Процессы дестилляции
Пример 10. Вычислить а)число единиц переноса массы и б) число теоретических тарелок для следующих случаев:
1. Обогащение при полном орошении жидкого раствора н-гептана в метилциклогексане, содержащего 3,3 мольных процента н-гептана, до содержания 96 мольных процентов н-гептана. Примем идеальный раствор и а =1,07.
а) Положим, что можно применить к этому случаю уравнение (13) в статье Доджа и Хафмана [66], а именно:
2,303 / Xjy . , 1 —л./Л
•gVP+a|gj------- '
a — 1 хр 1 — xD)
Подставляя a =1,07, xD = 0,960 и xF— 0,033, получаем и =98.
б) К этому случаю приложимо также уравнение (139):
_96/4_
3,3/96,7
704,
1g 704
п- igi.OT -
98.
Результат одинаков по обоим методам и для любой системы этого типа; обе величины будут давать практически одинаковое число единиц переноса массы как при полном орошении, так и при любом конечном флегмовом числе.
2. Отгоняем легкие фракции из водного раствора, содержащего 5°/в (мольных) летучего компонента и подчиняющегося закону Генри, и доводим его до 0,01-процентного раствора при флегмовом числе, превышающем минимум в 1,2 раза. Примем константу закона Генри = 3,00.
а) Применим уравнение (12) из статьи Доджа и Хафмана, а именно:
2,303, Ахр-\-В П=~^Ахп+В’ где
k
A = k — 1—= (k—константа закона Генри),
Ке
По уравнению (111),
_ kxF — xw _ 3,00 X 0.050 — 0,00010
№)иин. — {k _ i j Хр — 2,00 X 0,050
(Яй)факт. = 1,20X1.50= 1,80,
А = 2,00—^ = 0,333,
l,Ov
„ 0,00010 - nnnn-cc
В — ort- = 0,0000556,
1,OV
и= 15,7.
Расход тепла на процессы перегонки
713
б) Применяя к этому случаю метод Смокера, можно леп:о найти, что lg(xF/xw)
1g (*И) ’
где т— наклон рабочей линии = RF/(RF—1), m-L§2_995 0,80“ 2’25,
п_ 1g (0,05/0,00010) 1g (3,0/2,25) ““ ’ '
В этом случае имеется заметное различие в числе единиц этих двух видов, и различие не связано с общим их числом. Например, посредством уменьшения флегмового числа эти величины можно значительно увеличить, но они не будут приближаться друг к другу м.
Рис.
170. Общая схема непрерывного процесса перегонки.
РАСХОД ТЕПЛА НА ПРОЦЕССЫ ПЕРЕГОНКИ
Практически любой непрерывный процесс перегонки можно для рассматриваемой задачи свести к общему результату, показанному на рис. 170. Питание, которым может быть или жидкость, или пар, или смесь жидкости и пара, разделяется на две части— пар V состава у (в некоторых частных случаях пар будет конденсироваться в жидкость, но для рассмотренного ниже доказательства не существенно, будет ли V жидкость или пар) и остаточную жидкость состава х.
Q представляет тепло, передаваемое окружающей среде или от нее, причем в первом Из общего материального баланса, баланса для компонента А и теплового баланса, получаем
случае оно считается отрицательным.
Fzp=Vy + Lx, Ffp=VH-\-Lh-[-( — Q).
(156)
(157)
(158)
Уравнение (158) можно дать и в форме, симметричной двум другим:
FIP = VH’-\-Lh, (159)
где
ЕЛИ
qv=H— Н'.
(160>
714
XIII. Процессы дестилляции
Исключение всех масс й) из уравнений (156), (157) и (159) дает
zP—х__Ip—h
у—х ~Н’ —h •
(161)
В общей форме это будет тем же самым уравнением, что и полученное выше, в гл. IX и в этой главе [см. уравнение (108)]. Из рас-суждений, рассмотренных в гл. IX, очевидно, что уравнение (161)
Рис. 171. Общий принцип нахождении расхода тепла, требуемого для процессов перегонки, по диаграмме энтальпия — концентрация.
можно представить на диаграмме энтальпия — концентрация в виде прямой линии, соединяющей точки, координатами которых являются
(zF, Ip),, (х, h) и {у, И’), а общее количество тепла на единицу пара непосредственно получается очень простым способом, иллюстрированным рис. 171. Прямая линия проводится по двум точкам h, х и IF, zF, после чего Н' определяется пересечением с ординатой при у. Н определяется сразу, если известны состояние пара и его состав.
Приведение передачи тепла к единице массы пара не является обязательным. Можно выбрать
любую другую массу и получить совершенно аналогичные зависимости.
Для аналитического определения требуемого расхода тепла уравнению (161) можно придать форму
?и=(Н-Л)-Х-^(/г-Л).
(162)
Развитием графического метода нахождения расхода тепла для процессов перегонки по диаграммам энтальпия — концентрация мы обязаны главным образом Полшону [191] и Савари [225]. Успех метода как средства строгого решения задачи зависит от точности данных по энтальпии для жидкой и паровой фаз рассматриваемой системы. Такие данные в настоящее время почти совершенно отсутствуют; поэтому метод представляет главным образом чисто теоретический интерес. В качестве первого приближения можно принять идеальность растворов, что делает изотермы линейными (см. гл. IX) и поэтому требует знания только свойств двух чистых компонентов. Любой из
й) Единицы массы несущественны. Килограммами можно пользоваться совершенно с тем же основанием, как и килограмм-молями.
Расход тепла на процессы перегонки
715
представленных ниже частных случаев без серьезного ущерба точности можно разобрать с помощью этой приближенной диаграммы.
В нижеследующих разделах будут кратко рассмотрены некоторые специальные случаи. Для более полной трактовки применения диа
грамм энтальпия — концентрация к задачам по перегонке и другим примыкающим областям см. книгу Бошняковича [24].
Непрерывная однократная перегонка. На диаграмме (рис. 172)
точка А представляет состояние охлажденная жидкость. Точка В представляет состояние кубового остатка, а точка С— состояние получаемого пара. Прямой линией, лежащей в основе всех построений на подобной диаграмме, в этом случае будет BAD. Расход тепла на единицу пара qv определяется непосредственно по диаграмме, как показано на ней, что находится в соответствии с уравнением (160).
Линии Л, ВС и EF—изотермы нли равновесные соединительные линии, связывающие составы двух насыщенных фаз, находящихся в равновесии. Они очень полезны при установлении возможных пределов, в которых могут изменяться определенные составы. Так, для за
питания; для питания служит пере-
Концентрация
Рис. 172. Определение расхода тепла на непрерывную однократную перегонку.
данных питания и кубового ос-
татка состав пара должен лежать где-нибудь между С и F, причем
первый предел соответствует равновесию между паром и кубовым остатком, а второй — равновесию между паром и питанием. При одно-
кратной перегонке обычно принимается, что кубовый остаток и пар находятся в равновесии; построение, показанное на рис. 172, основано на этом допущении. Если точка С находится справа от F, то можно сразу сказать, что представленный процесс неосуществим. Максимально возможный предел извлечения менее летучего компонента из питания дается точкой J, поскольку она обозначает состояние жидкости, находящейся в равновесии с полностью испарившимся питанием.
Дефлегмация. Построение для этого случая показано на рис. 173, на котором BAD является прямой линией, представляющей уравнения
Ур~—х Нр — h — qv=H’ — H.
(163)
(164)
716
XIII. Процессы дестилляции
Точка / соответствует максимально возможной концентрации легколетучего компонента в конденсаторе (полное орошение), и поэтому точка G дает максимально возможное обогащение пара. При данном
питающем и остаточном паре существует два предельных случая для состава конденсата: 1) параллельный поток с орошающей жидкостью, находящейся в
Концентрация
Рис. 173. Дефлегмация на диаграмме энтальпия — концентрация.
равновесии с остаточным паром (точка Е); 2) противоток с орошением, находящимся в равновесии с пита-1111П11 /'«•/\rrczo Т/Г О Г1 ГГТЛ ••»»<»
nnvm j^AVinu л j, ж до raoj ПСЛПЛ диаграммы видно, что для данного обогащения пара прямоточный конденсатор будет требовать значительно больше охлаждающей воды, чем противоточный конденсатор, поскольку для последнего случая точка D будет лежать на продолжении линии FA, а для первого — на продолжении линии, проведенной от Е до А.
Диференциальные испарение п конденсация. Уравнение для испарения можно
вывести непосредственно из уравнения (162) путем рас
смотрения количества пара dV, образующегося за бесконечно малый интервал времени, причем следует отметить, что питание и кубовый остаток должны отличаться по составу лишь на бесконечно малую величину. Эти условия сразу превращают уравнение (162) в уравнение
^=g=("-A)~(y-x)g.
(165)
Аналогичным уравнением для конденсации, легко получаемым с помощью подобного же способа, будет
-4v=-d^ = ^-h)-(y-x)d-^. (166)
Эти уравнения очень просто интерпретируются на диаграмме энтальпия— концентрация, что иллюстрировано для уравнения (165) (рис. 174). Наклон линии жидкости в точке С, т. е. dh!dx, равен
Расход тепла, на процессы перегонки
717
тангенсу угла ВС А, или
АВ dx у — х ' илн
АВ^-(у — х)£.
Из сопоставления этого уравнения с уравнением (165) сразу видно, что dQfdV = DB, как и показано на рис. 174.
Указанное построение дает только элементарное количество тепла, требуемое для испарения. Для конечного процесса следует
провести интегрирование. Так как
dQ=qvdV и
dV=Fdtv, то
(167) и
Су связано с х уравнением (28), a qv связано с х, как показано на рис. 174. Располагая рядом соответствующих значений и qv между указанными пределами, можно найти Q графически, определив интеграл как площадь под кривой ^v
Вышеизложенное главным образом имеет значение
Концентрация
Рис. 174. Диференциальное испарение на диаграмме энтальпия — концентрация.
иллюстрации принципов, а не практического метода нахождения расхода тепла для процесса фракционной перегонки. Расход тепла с достаточной точностью можно определить, допуская, что процесс является процессом однократного испарения между теми же самыми пределами состава питания и при том же среднем составе дестиллата.
Непрерывная перегонка в сочетании с дефлегмацией. Схематическое изображение этого процесса показано на рис. 175. Питательная жидкость состава хр непрерывно течет в перегонный куб, а кубовый остаток состава xw непрерывно отводится. Затем пар разделяется посредством дефлегмации на отогнанный продукт состава и на орошение состава х0. С помощью такого процесса можно получить, по существу, чистый компонент А в виде отогнанного продукта, если конденсация является диференциальной, но предельным
718
XIII. Процессы дестилляции
значением xw будет значение, соответствующее жидкости, находящейся в равновесии с паром состава yF = xF (см. рис. 176).
Пар к общему конденсатору
Рис. 175. Непрерывная перегонка, соединенная с дефлегмацией.
Три обычных кию баланса для системы в целом приводят к уравне- хр — xw hF — hw /1681 yD~xF H'-hF ’ ' '
в котором H' = HD + qc — qD, (169)
и к уравнениям qc~~ D
и qD D '
Уравнение (168) представляет прямую линию на диаграмме энтальпия— состав, связывающую точки (xF, hF), (xv, hw) и (yD, И'). Подобным образом те же самые балансы для одного конденсатора приводят к прямой линии между точками (yv, Hv), (х0, Ао) и (?D> ^")> где
^' = ^ + <7с-
(170)
Расход тепла на процессы перегонки
719
Из уравнений (169) и (170)
qD=H" — H'.
(171)
Рис. 176. Расход тепла на непрерывную перегонку в сочетании с дефлегмацией.
Графическое построение для нахождения qD показано на рис. 176. ALC и DEF являются прямыми, представляющими основу построения. yv принимается находящимся в фазовом равновесии с xw, которое может иметь любое значение между xF и составом, соответствующим точке G. Верхним пределом значения х0 является Уу, поскольку это будет требовать полной конденсации. Из диаграммы следует, что расход тепла при этом пределе будет приближаться к бесконечно большому значению (потому что линия DEF станет вертикальной). При изучении диаграммы также становится очевидным, что увеличение чистоты отогнанного продукта приводит к увеличению расхода тепла.
Ректификация. По уравнению (88) можно заметить, что любая рабочая линия в укрепляющей секции колонны на диаграмме энтальпия — концентрация является прямой линией, проходящей через точки (хд, h'D), (у, Н) и (х, h). Все рабочие линии для этой секции колонны (одной из таких линий является линия АВС на
рис. 177) будут проходить через точку С, положение которой зависит от флегмового числа, а следовательно, от количества тепла, отнимаемого в конденсаторе. [Последнее очевидно из уравнения (84).] Теплота qc, забираемая в конденсаторе на единицу количества погона,, определяется как показано на рис. 177.
Поскольку равновесная соединительная линия (в качестве двух примеров ее на рис. 177 показаны линии JK и FG) соответствует максимальному различию, которое может наблюдаться между паровой и жидкой фазами, находящимися в контакте, рабочие линии всегда должны иметь больший наклон, чем соединительные линии между теми же самыми точками. Если рабочие линии вертикальны (д/ = х), то из диаграммы сразу видно, что qc будет бесконечно большим, и мы имеем случай полного орошения. Если рабочие линии совпадают с соединительными линиями, то тогда они будут иметь наименьший
720
XI11. Процессы дестилляции
из всех возможных наклонов и поэтому будут пересекаться с ординатой xD в наинизшей из возможных точек, что означает минимальное количество тепла, забираемого из конденсатора. Это является случаем минимального орошения. Поскольку соединительные линии имеют различные наклоны, они будут пересекать ординату состава продуктов в различных точках, и минимальное орошение для секции
Рис. 177. Ректификация, изображенная иа диаграмме энтальпия — концентрация.
будет соответствовать наивысшей точке. Другими словами, минимальное орошение является максимумом минимумов, как и указывалось выше. Этот пункт иллюстрируется на рис. 178, который показывает три положения точки С, соответствующие пересечениям трех прямых линий, совпадающих с изотермами. Минимальное орошение определяется точкой С", где тепло должно удаляться, и по отношению к орошению она является максимумом для трех минимумов.
Если рабочая линия для любого уровня на диаграмме Н—х совпадает с изотермой, то из уравнений (84) и (88) получим
(?cU. = W-AD + (Я- А) . (172)
В подавляющем большинстве случаев минимум регулируется уровнем питания, и уравнение (172) переходит в
U. = Hf - hD + (HF - hF) . (173)
-VF~~XF
Расход тепла на процессы перегонки
721
Уравнение (108) показывает, что прямая линия связывает точку С, точку состояния питания N и точку М (см. рис. 177), координатами которой являются (х^,, Л'^). Уравнение (99) показывает, что все рабочие линии исчерпывающей колонны проходят через последнюю точку и поэтому она является осью для этих линий, аналогичной точке С
в ректификационной колонне. Отметим, что точка М фиксируется состоянием питания и местоположением точки С.
Величина qB представляет теплоту, затрачиваемую в кубе на единицу кубового остатка; она определяется на основе уравнения (100), как показано на рис. 177.
Из уравнений (102) — (104) можно вывести
х^ — hw— IF (174) zf — xd Ip—
где
h"D=hD + 4c-4D (175)
И
a —-B
Рис. 178. Минимальное орошение.
Основанное на этих уравнениях графическое построение для получения расхода тепла qD в кубе на единицу дестиллата показано на рис. 179. DAE—прямая линия, представляющая уравнение (174), а АВС—рабочая линия укрепляющей секции, положение которой зависит от величины сс—тепла, которое следует отнимать в кон денсаторе и которое, конечно, связано с флегмовым числом.
Минимальный расход тепла будет соответствовать минимальному флегмовому числу, которое, в свою очередь, определяется наинизшим возможным положением оси С. Из уравнений (173) —(175) легко получить следующую алгебраическую зависимость для вычисления минимального qD'.
= -IF-. (176)
•inn. уч- -X i t ум
Если неизвестно, какой уровень питания требует максимума различных возможных минимумов, то решение должно производиться методом подбора или же следует пользоваться графическим методом.
Если бы пограничные линии для насыщенных фаз были горизонтальными,—что наблюдается для случая потока с постоянным мольным числом, поскольку он предполагает равные скрытые теплоты, отсутствие 46 Химическая термодинамика
722
XIII. Процессы дестилляции
теплоты смешения и равные теплоты жидкостей, вызывающие изменение температуры,—то из диаграммы (рис. 179) ясно, что тепло, расходуемое в кубе, должно быть равно теплу, забираемому в конденсаторе (qc — qD), если питанием служит жидкость в точке кипения. Во всех остальных случаях эти две теплоты, как правило, не бывают равны.
Для получения или чистого компонента В при фракционирован
перегонке с конденсатором оро-
ной перегонке, или чистого А при
Рис. 179. Расход тепла на ректификацию.
шения в пределе будет требоваться бесконечно большое количество тепла; но при помощи процесса ректификации теоретически возможно полное разделение на два чистых компонента при затрате конечного количества тепла. Это можно обнаружить по рис. 179, на котором видно, что в пределе, когда xD — 1, qD будет все еще предельной величиной.
Определение числа тарелок. Хотя определение числа тарелок выходит за рамки определения расхода тепла, все же этот вопрос будет здесь рассмотрен, поскольку интересно показать, каким образом это можно осуществить по диаграмме энтальпия — состав с помощью ступенчатого метода, если имеется также в распоряже
нии равновесная кривая у — х, позволяющая определить положение соединительных линий в любой
точке. На рис. 177 точка F представляет состояние пара, покидающего
верхнюю тарелку колонны, если для получения орошения используется конденсатор, в котором осуществляется полная конденсация. Перемещаясь по соединительной линии, достигают точки G, которая соответствует составу жидкости на верхней тарелке, если она является теоретической тарелкой, как было определено выше. Прямая линия GC является рабочей линией, и поэтому точка J должна представлять состояние пара, поднимающегося с нижней тарелки. Продолжая это построение до тех пор, пока не будет достигнут уровень питания, получают число тарелок для укрепляющей секции. Число тарелок для исчерпывающей секции получают совершенно аналогичным способом, но рабочие линии проходят тогда через ось М.
Существует мало указаний на применение диаграммы энтальпия — концентрация для определения числа тарелок, поскольку пользоваться диаграммами у — х проще, и если не делать обычных упро-
Расход тепла на процессы перегонки
723
щающих допущений, то, в общем, получаются довольно правильные результаты. Различие между результатами, полученными по диаграмме Н — х и по диаграмме у — х с линейными рабочими линиями (метод Мак-Кэба — Тиле), обычно мало и не представляет практического значения, особенно если помнить, что на результаты часто сильнее влияют различия
в равновесных данных.
Из этого следует заключить, что основным применением диаграмм энтальпия — концентрация при перегонке является определение с их помощью количеств тепла и выяснение влияния определенных изменений, а не вычисление числа тарелок. В настоящее время главной помехой для практического использования этих диаграмм, даже для определения расходов тепла, является то, что имеется слишком мало данных для их построения.
Влияние потерь тепла. До сих пор мы считали колонну адиабатной, но интересно также рассмотреть влияние потерь тепла в колонне. Пусть qR и qE будут теплом, теряемым в окружающую среду укрепляющей и исчерпываю-
щие If Концентрация Xjj
Рис. 180. Влияние тепловых потерь колонны.
щей секциями колонны соответственно на единицу дестиллата и кубового остатка. Тогда уравнение теплового баланса для колонны
в целом принимает вид
где
FIP = Dh"D+Wtfw, (177)
4“ Яс 4“ 4ri (178)
Я в 4- Яе ’ (1'9)
На рис. 180 АВС является прямой линией, представляющей уравнение (177); на этой диаграмме указаны также и различные тепловые характеристики. При рассмотрении уровня выше верхней тарелки колонны оказывается, что qR = 0 и осью рабочих линий является Е. По мере движения книзу qR увеличивается до тех пор, пока не будет достигнут уровень питания, на котором qR достигает своего предельного значения, а ось рабочей линии — точки С. Другими словами, ось рабочей линии по мере движения к низу колонны сдви-46*
724
XIII. Процессы дестилляции
гается от £ к С. Из рис. 177 видно, что чем выше будет ось, тем меньшее количество тарелок окажется необходимым; поэтому фиксирование оси в точке С потребует меньше тарелок, чем в случае, когда ось сместится от С к Е. Из этого можно заключить, что для одинакового общего количества орошения или для одинакового общего количества тепла, отнимаемого в конденсаторе, соединенном с секцией укрепления, адиабатная колонна потребует меньше тарелок, чем колонна с тепловыми потерями. Формулируя это иначе, можно сказать, что лучше удалять общее количество тепла в конденсаторе, чем часть его
в конденсаторе, а остающуюся часть в колонне.
Подобные же рассуждения приложимы к исчерпывающей колонне, в которой по мере продвижения уровня равновесия от дна колонны к уровню питания ось рабочей линии сдвигается от D к А. Ось D рабочей линии требует наименьшего количества тарелок, и она будет фиксированной осью адиабатной колонны, потребляющей то же самое количество тепла, что и неадиабатная колонна. Отсюда можно заключить, что адиабатная отгонная колонна требует меньше тарелок (или при том же самом числе тарелок дает лучшее разделение), чем неадиабатная колонна при том же расходе тепла.
Подобные же выводы можно получить для случая колонн, работающих ниже температуры окружающей среды. В этом случае тепловым потоком будет тепло, попадающее в колонну извне.
Интересно также отметить, что если неадиабатная колонна, в которой тепло qc отбирается в конденсаторе, а тепло qR теряется в укрепляющей секции, изолируется затем для сведения потерь к минимуму, причем в то же самое время нет никакого увеличения qc, то ось фиксируется в Е, и поэтому разделение будет хуже, чем в неизолированной колонне. Для получения эффекта от изоляции следует увеличить охлаждение в дефлегматоре или же увеличить флегмовое число при использовании конденсатора,- в котором производится полная конденсация.
Приближенные методы определения расхода тепла. Методы, основанные на применении диаграммы энтальпия — концентрация, изящны и в то же время просты при условии, что в распоряжении имеется диаграмма. Данные, необходимые для построения такой диаграммы, обычно отсутствуют; даже если они доступны, то все же труд, потраченный на изготовление таких диаграмм, не оправдывается, если следует произвести только несколько вычислений. Диаграмма очень сильно упрощается, если принять линейную зависимость энтальпия—-концентрация для двух насыщенных фаз, поскольку мы в этом случае нуждаемся в термических данных только для чистых компонентов. Принимая средние теплоемкости постоянными, получим hA (мольная) = CpAMA(fA — /0), (180)
где /0 — нулевая температура. Если 1А является точкой кипения А, то значение hA получают на линии насыщенной жидко.ти.
Расход тепла на процессы перегонки
725
Для насыщенного пара
"а = Лл + Д4. (181)
Подобные уравнения применимы и для другого компонента. При использовании этих уравнений для определения местоположения пограничных линий для двух фаз можно затем найти qD посредством описанного выше графического построения или посредством уравнения (176).
Если жидкости сходны, можно пойти на дальнейшие упрощения и, принимая
И
/.д = Z.B= Z.5^), получить
НА = НВ=Н.
Подстановка в уравнение (176), соединенное с уравнением (86), приводит к уравнению
= (182)
для случая, когда питанием служит в основном жидкость, находящаяся при точке кипения. Уравнение имеет одинаковый вид, независимо от того, является ли R минимальным флегмовым числом или любой величиной, большей этого минимума. Если питание имеет температуру ниже точки кипения, то следует позаботиться о предварительном подогреве его.
Является крайне сомнительным, оправдывается ли использование действительной диаграммы энтальпия—концентрация хотя бы для какого-нибудь из немногих специальных случаев. Например, даже в случае смесей уксусная кислота — вода, когда разность скрытых теплот является необычно большой, различие при использовании диаграммы энтальпия — концентрация с прямыми и при использовании уравнения (182) получается незначительным и лежит в пределах точности, требующейся для большинства тепловых расчетов. Это показывает следующий пример.
Пример 11. Водный 50% раствор уксусной кислоты (мольные %%) следует разогнать при атмосферном давлении в непрерывно действующей колонне для получения дестиллата, содержащего 0,5% (мольных) СН3СООН, и кубового остатка, содержащего 99,0% (мольных) кислоты. Примем, что питание поступает при точке кипения, которая равна 104° С. Определить минимальное количество тепла, расходуемого в кубе на моль отогнанного продукта.
Использованы следующие данные:
й-) В действительности скрытые теплоты могут быть не равны! в этом случае рекомендуется пользоваться их средне-арифметическим значением.
726
XIII. Процессы дестилляции
Срсн соон= ^’522 кал/моль/град („Справочник инженера-химика“ Перри) (принимается постоянной для всего рассматриваемого интервала). £сн3соон — = 97,0 кал)г при нормальной точке кипения (118° С) = 5820 ккал/кг-моль). (Интернациональные критические таблицы).
£HsO==9700 ккал/кг-моль при 1 атм.
Пар, находящийся в равновесии с.50-процентным раствором, содержит 62,7°/tt (мольных) Н2О [48]. Все значения энтальпии будут даны в ккал/кг-моль выше нулевого уровня в 80° С.
ЛД(Л для воды) 18X20 = 360, hB = 60 X 0,522 X (118—80) = 1190, //• — 360 -J- 9700 — 10060, Нв = 1190 + 5820 = 7010.’
Рис. 181. Схема решения примера 11.
По рис. 181, основанному на построении, иллюстрированном на рис. 179, Уо (тепло, расходуемое в кубе на 1 кг-моль отогнанного продукта) = = 32 000 ккал. Это предполагает, что минимальный расход тепла определяется равновесием на уровне питания, и поэтому рабочая линия ца диаграмме совпадает с соединительной линией для уроиня питания.Тот же самый расчет можно произвести аналитически по уравнению (176), в котором различные величины имеют следующие значения (принимая линейность кривых насыщения):
Н=Нр—Н^ур-\-Нв (1 —ур} = =10060X0,6274-7010 X 0,373 = 8940,
й = hp = hAXp-\- hB (1 — хр) — 775, /1^=368,
/F= hF,
zf=xF= 0,500.
По уравнению (176)
^O = 31500.
По уравнению (182) 4d~ -|-1),
_ хп Ур ''мин. * ’
Ур— XF
_ (97005820\ /0,995—0,627
2 ДО,627 —0,500
+-1 = 30300.
Различие между двумя методами составляет только около 5°/0. Конечно, следует помнить о допущении, которое было сделано относительно хода линяй насыщения, но можно считать, что использование правильных линий не будет значительно изменять выводов.
Расход тепла на процессы перегонки 7Z1
К. п. д. ректификационной колонны. В начале этой главы были даны методы вычисления минимальной теоретической работы или расхода тепла для разделения растворов. Располагая этой величиной как стандартом, мы в состоянии исследовать эффективность любого рассмотренного процесса перегонки. В качестве примера вычислим термодинамический к. п. д. процесса ректификации в обычной адиабатной колонне.
Пример 12. Чему равен термодинамический к. п. д. процесса ректификации для проведения полного разделения, описанного в примере 1 при использовании водяного пара при 3,5 а»?
_ 1,00—0,715 «мин. — 0>715 _ 0,500 — ’ ’ £т = 7650 ккал)кг-моль.
По уравнению (182) qD = 7650(1 +1,33) = 17800 ккал!кг-моль дестиллата. Считая, что конденсат водяного пара должен удаляться при температуре насыщения, получаем:
17800 по „ расход водяного пара = = 5-= 28,2 кг, Оо1,У
термодинамический к. п. д. = X 100= 14,7°/о-ZO, Z
Из этого примера можно заключить, что с термодинамической точки зрения ректификационная колонна является очень неэффективным аппаратом. Конечно, большинство действительных колонн будет еще менее эффективно, чем эта колонна, вследствие необходимости использования орошения, сильно превышающего минимальное. В обычной адиабатной ректификационной колонне существует несколько причин, из-за которых возникают необратимые эффекты: 1) различия температур в кубе и конденсаторе, 2) падение давлении, обусловленное потоком жидкости, и 3) обмен веществом между фазами, происходящий не при фазовом равновесии. Эти необратимые ивлеиия и их влияние на термодинамический к. п. д. колонны были кратко рассмотрены в гл. X.
Снижение расхода тепла. В случае трудного разделения расход тепла может быть настолько большим, что стоимость водяного пара становится серьезной составляющей в общем расходе. Одним из возможных средств снижения расхода водяного пара является использование принципа многократного действия, который уже в течение долгого времени находит применение для единичной операции испарения. Этот принцип требует ряда кубов, каждый из которых работает при более низких давлении и температуре, чем предыдущий, благодаря чему пар из данного куба может конденсироваться в нагревающем змеевике следующего холодильника. Водяной пар будет подаваться только в кипятильник первого куба, имеющего наиболее высокое давление. Принцип правилен, но возможная экономия в большинстве случаев, очевидно, недостаточна для оправдания
й) См. пример 1.
728
XIII. Процессы дестилляции
увеличения первоначальной стоимости системы и больших производственных трудностей, и поэтому, насколько известно автору, в работе находится очень немного установок, работающих по принципу многократного действия. Использование этого общего принципа было предложено Калингертом [38] в 1925 г.; но недостатком его процесса является то, что орошение производится в дефлегматоре, и поэтому
в качестве нагревающей среды используется только получающийся пар.
Повидимому, этим путем можно достигнуть лишь небольшого выигрыша. Принципиально представляется более выгодным конденсировать сразу весь пар в кубе соседнего звена системы и затем разделять конденсат на орошение и продукт.
Схема повторного использования пара предложена Отмером [184] в качестве средства преодоления некоторых практических трудностей системы многократного действия. Одно из возможных применений системы показано на рис. 182. Колонна I— исчерпывающая колонна, работающая при повышенном давлении (порядка 3,5 кг/см2) с острым водяным паром. Пар, поступающий из верхней части колонны,
конденсируется в трубках кипятильника колонны II, которая рабо-
тает практически при атмосферном давлении; этот конденсат вводится в колонну II в качестве питания. Колонна II производит пол
ное разделение питания на два практически чистых компонента и совершенно не требует водяного пара. Действительно, в примере, данном Отмером (ректификация 2,5-процентного раствора ацетона в воде), вторая колонна производит избыток водяного пара, который можно отвести и использовать для каких-нибудь других целей.
Другим средством экономии тепла является использование теплового насоса или системы повторного сжатия пара, схематично показанной на рис. 183. В такой системе куб и конденсатор (который может быть общим, г. е. производящим как орошение, так и
Расход тепла на процессы перегонки
72Э
Рис.
183. Ректификационный процесс с использованием повторного сжатия пара.
дестиллат) соединяются в одну установку, и энергия для перегонки подводится в виде работы, а не в виде тепла. Для любого данного случая расход работы в компрессоре является определенной величиной и должен компенсироваться разностью энтальпий различных потоков вещества, входящих в систему и покидающих ее, плюс тепло, отбираемое в холодильнике и в охладителе или теряющееся путем излучения. Ясно, что эти различные количества энергии не будут уравновешиваться автоматически, т. е. должны применяться некоторые приспособления. Если работа слишком велика, то некоторое количество пара может проходить в конденсатор, вместо того,чтобы поступать в компрессор; если она слишком мала, то можно использовать добавочный нагрев водяным паром.
Из принципа теплового насоса, рассмотренного в гл. X, посвященной охлажде
нию, ясно, что количество работы зависит от разности температур, так как этим определяется количество потребного тепла. Например, в случае разделения растворов этиловый спирт — вода минимальная возможная разность составляет примерно 5°С; но приходится ее увеличивать еще на 5—10° для поддержания нужной скорости передачи тепла в куб. Как показывает следующий пример, это не очень благоприятный случай для применения данного принципа.
Пример 13. Произвести приближенное сравнение расхода энергии для процесса повторного сжатия пара (рис. 183) с расходом водяного пара для обычной ректификационной колонны при разделении водного раствора этилового спирта, содержащего 20°/о (мольных) последнего, на дестиллат, содержащий 86,0°/о С2Н5ОН, и кубовый остаток, который практически представляет собой чистую воду.
Примем, что расход тепла должен быть минимальным, т. е. что колонна должна работать с минимальным орошением.
По уравнению (86) для случая постоянного мольного орошения о __Уо ~У* К мин. ——----------------------------•
V* — х
730
XIII. Процессы дестилляции
Вследствие аномальной формы равновесной кривой для этой системы минимальное орошение, как показывает следующая таблица, не соответствует равновесию на уровне питания.
X д/* /?мин. х /?мин.
0,2000 0,5285 1,01 0,7900 0,8108 2,36
0,5000 0,652 1,37 0,8000 0,8175 2,43
0,7600 0,7905 2,28 ' 0,8100 0,8248 2,38
0,7800 0,8040 2,33 0,8200 0,8320 2,33
Равновесные данные представляют результаты Льюиса и Карея [153]. /?мин. принимается равным 2,43. По уравнению (182) ?/>= 9560 (2,43-|-1) = = 32800 ккалкг-моль отогнанного продукта. (9560 представляет среднюю из мольных скрытых теплот парообразования этилового спирта и воды при 1 атм.}
При вычислении работы сжатия примем, что получающийся пар является идеальным газом, что & (отношение теплоемкостей) = 1,16 и что разность температур в 10°С достаточна для передачи тепла в кубе. Это означает, что пары спирта должны конденсироваться при 110°С, когда давление его насыщенного пара равно 3,11 атм.
С помощью рис. 48 находим, что теоретическая работа сжатия иа 1 кг-моль пара равна ==: 1 квт-ч. Принимая к. п. д. равным 7О°/о и допуская сжатие всего пара, идущего из колонны, получаем
А = (ууу X 3,43 = 4,9 квт-ч.
В системе сжатия будет экономиться некоторое количество охлаждающей воды; но потребуется значительно большая поверхность нагрева в кубе, поскольку At для нагрева водяным паром будет равна 15,5°С, по сравнению с 10°С для нагрева паром, и коэфициент перехода в первом случае будет также больше.
Повторное сжатие пара будет выгодно только в тех случаях, ’ когда электрическая энергия очень дешева или когда разность температур, на преодоление которой следует затрачивать тепло, значительно меньше, чем в данном примере.
Отсутствие места не позволяет нам произвести анализ других систем с повторным использованием пара для сравнения их экономичности с экономичностью обычной ректификации. Это предлагается читателю в качестве интересной разносторонней задачи для самостоятельного решения 95.
ПРИМЕЧАНИЯ
1 При очень высоких давлениях, как показали недавно Кричевский, Большаков и Циклис [3331, газы тоже могут обладать ограниченной смешиваемостью, и, следовательно, смеси газов в этом случае будут расслаиваться, разделяясь на две фазы (см. Вступительную статью, разд. 3).
2 Правильнее было бы сказать .между теплотой и работой", так как опыты Румфорда были посвящены выяснению соотношения только между этими двумя величинами. См. также прим. 4.
® Необходимо указать на то, что молекулярно-кинетическое объяснение теплоты впервые дано М. В. Ломоносовым в его диссертации .Размышления о природе теплоты и холода" (1745—1746 гг.) [356].
В этой работе была доказана несостоятельность господствовавшей в то время теории, объяснявшей явления наличием в каждом теле особой субстан-п”и — теплорода, который перераспределяется между телами, вступившими ! юбмен, не изменяясь в своем количестве.
Если энергия характеризует состояние системы, то понятия теплота и ка могут быть отнесены только к процессу, так как всегда ассоциируются превращением, сущность которого состоит в передаче энергии.
Действительно, работа связана с взаимодействием между системой и внешней средой, возникающим в результате нарушения внешними силами равновесия в системе (в результате нарушения механического или других равновесий), т. е. представляет упорядоченную форму передачи энергии от системы к среде или, наоборот, — от среды к системе. Теплота, выделяемая или поглощаемая системой, является результатом нарушения термического равновесия между системой и внешней средой, т. е. представляет форму передачи энергии, обусловленную хаотическим („тепловым") движением частиц.
Теплота и работа являются единственно возможными формами передачи энергии от одного тела к другому. В справедливости этого определения [366, 367] легко убедиться из анализа уравнения (2); в соответствии с этим уравнением общее изменение энергии системы Д(7 вызвано двумя формами передачи энергии—теплотой (SQ) и работой (—S/4); других форм передачи энергии первое начало не предполагает.
6 Приведенное автором соотношение между энергией и массой (а не материей) U = с2т является строгим, но его нельзя интерпретировать как „возможность возникновения энергии из материи, и наоборот", как указано в тексте; указанный автором расчет может только привести к неправильному толкованию его. Соотношение это устанавливает, что изменение энергетического запаса системы всегда сопровождается соответствующим изменением массы. Эффект этот для обычных физических и химических процессов неуловимо мал, но в процессах, сопровождающихся много ббльшими энергетическими эффектами, такими, как превращения в атомных ядрах, эффект оказывается измеримым и подтверждается экспериментальными данными. Выделение большого количества энергии всегда сопровождается небольшим уменьшением массы.
732
Примечания
6 Следует сказать ,между протонами и нейтронами*. Современные представления об элементарных частицах и взаимодействии между ними изложены в обзорной статье Д. Д. Иваненко [301, 302]. См. также сборник «Мезон* [355].
1 С проблемой непосредственного превращения химической энергии топлива в электрическую энергию, при котором коэфициент полезного действия может быть доведен почти до 100е/ , можно познакомиться по монографии Давтяна |300]. В ней, наряду с изложением истории этой проблемы и теории топливного элемента, приводятся результаты теоретических и экспериментальных работ, проведенных автором в Энергетическом институте Академии наук СССР.
s В Советском Союзе работа по унификации научных терминов и буквенных обозначений проводится Всесоюзным комитетом стандартов при Совете Министров СССР и Комитетом технической терминологии Академии наук СССР, а по отдельным частным разделам также и другими организациями. По технической термодинамике по терминологии и обозначениям действуют стандарты ОСТ ВКС 6394 [360] и ГОСТ 3270-46 [296], охватывающие значительное число понятий и величин. Кроме того, в 1948 г. Комитетом технической терминологии выпущен проект «Терминологии технической термодинамики", охватывающий 186 понятий и включающий и обозначения их, который после широкого общественного обсуждения поступит как проект стандарта. В настоящее время заканчивается разработка проекта терминологии по физико-химическому анализу. (См. след, таблицу.)
Сравнение обозначений, применяемых различными авторами
Внутренняя энергия Энтальпия Свободная энергия Изобарноизотермический потенциал
Проект стандарта [389 и I, H(W) F O(Z)«)
Принято в данной книге и н F Z
Раковский [368,3691 и н F Z
Бродский 271 и н F ф
Путилов 367 и F
Колосовский 326 и н F ф
Капустинский 312 и н F (Л
У лих 393 и 1Г F G
Гетман и Даииэльс [294 Е И А F
Тейлор 384 и н А F
Льюис и Рендалл 351 Е . н А F
Гуггенгейм 299 Е н Е
Планк [364 и Е — ФГ
Ван-дер-Ваальс и Констамм 274 Е 1 $ с
Эйкен 403 и I F-o Fp
Партингтон 361 и И Е Z
Гиббс [87 е X ф г
Шоттки, Улих, Вагнер 228 и W Е G
Дюгем [70 и F ф>
Нернст 358 и А
*) В скобках указаны зшасные буквенные обозначения.
Примечания
733
» Если уравнение состояния имеет следующий вид р = (о, Т, N, 1\Гг,..., N, ...), т°, как показал Темкин [385], летучесть компонента газовой смеси определяется уравнением
ю Условия равновесия многокомпонентных двухфазных и многофазных систем рассмотрены в работах Сторонкина.
В первой работе [379] выведены диференциальные уравнения для сосуществующих фаз многокомпонентной двухфазной системы, являющиеся обобщением уравнений (138) и (139), и дано применение их к решению ряда вопросов термодинамической теории ^компонентных систем в общем виде и для особых случаев.
Во второй работе |380] найдены общие условия равновесия для многокомпонентных систем, в которых проходят химические реакции, и рассмотрено изменение давления насыщенного пара при изменении состава многокомпонентных двухфазных систем.
В третьей работе [381] выведены уравнения, характеризующие равновесие многокомпонентных многофазных систем, и на основании этих уравнений установлены свойства многофазных систем, имеющих экстремум давления и температуры.
Работы Сторонкина обобщены в монографии [381а].
и См. Вступительную статью, разд. 1.
12 Растворимости газов и газовых смесей в жидкостях под давлением посвящен ряд работ Кричевского и др. [333, 336, 337]. В частности, уравнение растворимости впервые было выведено Кричевским и Казарновским [337].
12а Следует отметить, что уравнение изобары растворимости впервые было предложено проф. Петербургского горного института Шредером [402].
126 Следует указать на обширное исследование Богуславского „Основы молекулярной физики и применение статистики к вычислению термодинамических потенциалов* [269], посвященное кинетическому толкованию и вычислению термодинамических функций.
13 Следует отметить, что отклонение поведения газа от закона Бойля впервые было установлено Ломоносовым, который в „Прибавлении к размышлениям об упругой силе воздуха* (1749) [356] установил, что „... плотности воздуха при очень больших сжатиях не пропорциональны упругостям его*.
1* См. Вступительную статью, разд. 3.
13 Существование ее впервые установлено Менделеевым [355].
16 Необходимо отметить, что уравнение, на котором основано понятие парахор, впервые установлено Бачинским |2б9].
11 Для вычисления критической температуры ароматических углеводородов (гомологов бензола), как показал Мамедов, можно воспользоваться приближенным экспериментальным уравнением [353а]:
Ткр = ( 2,0996— 0,00095 Тв,
где d — удельный вес жидкого углеводорода при 15,6/15,6° С.
Это уравнение может рекомендоваться для соединений с (Tnid3\ = — 500 -t- 750.
734
П римечания
Средняя погрешность расчета для 13 углеводородов оказалась равной
я = ± 1/ ——j = ± 4,6°; усредненная погрешность =
= • 100 = ±1,3%.
у tKp
18 Учению об уравнении состояния реального газа посвящена недавно вышедшая монография Вукаловича и Новикова [285]. В ней даны: сводка уравнений состояния, анализ важнейших уравнений, причем основное внимание уделено анализу уравнения Ван-дер-Ваальса, и общая теория уравнения состояния на основе современных представлений статистической физики.
19 В оригинале, очевидно, опечатка: .кроме газовой постоянной R*.
20 Для приближенных термодинамических расчетов при высоких давлениях и температурах Розен предложил уравнение [370].
pv = A-\-Bp-\-CT (83а)
или
z = a-\-bp -f-ct. (836)
Проверка этого уравнения на 6 газах и газовых смесях обнаружила достаточную точность его (погрешность расчета для п, тЭгЗ колеблется от 0,15 до 2,2%). В табл, ба приведены константы уравнения (836), а также указан интервал put, отвечающий отмеченной точности вычисления.
Таблица 6а
Константы уравнения Розена
Газ а й-103 с.КЯй) Область применения Средняя квадратичная ошибка, %
am °C
от до от до
n2 0,801 1,244 4,33 100 1000 20 200 1,15
n2 1,047 1,110 4,50 1000 6000 0 200 2,2
н. 0,990 0,721 3,76 100 1000 0 200 0,12
со 0,757 1,298 4,34 200 1000 С 200 1,0
Не 1,019 0,362 3,56 200 1000 0 200 0,25
N, + 3H2 0,973 0,818 3,81 100 1000 25 300 0,5
n24-3h, 0,969 0,862 3,74 200 1000 300 500 0,11
Воздух 0,767 1,207 4,32 200 1000 15 200 1,12
Воздух 0,788 1,193 4,28 1000 3000 0 50 0,97
Т 103
☆) Для идеального газа с откуда с = =866.
X / 0^2, X / о,х
Уравнение Розена полезно не только для вычисления (и интерполяции) объема, но и для расчета работы сжатия, что имеет существенное значение для нахождения работы цикла компрессора высокого давления (см. Вступи-
тельную статью, разд. 2).
Примечания
735
21 Парциальное давление компонента газовой смеси, т. е. часть общего давления р1г которая происходит от ударов молекул Z-того компонента о стенки сосуда, может быть определена и для реальных газовых смесей. Темкин [387] показал, что pi может быть выражено через летучесть компонента смеси с помощью уравнения
pi = xiRT fl ( T, xdp.
J v \ dp /
6
(104a)
Это уравнение в соответствии с соотношением (106, гл. IV) примет вид уравнения
Pi — xi^-dpt (1046}
о
которое позволяет вычислить парциальное давление через непосредственные опытные данные, определяющие объем как функцию давления и состава смеси при Т = const.
22 Распространенное в американской литературе мнение о большой точности этого метода (см. также [397]) не отвечает действительности. Так, уравнение Джиллиленда
Рт = хлРА + хвРв + ХАХВ (Wa + У5в)г, получающееся (для бинарных смесей) при сочетании уравнения (123, гл. V) с уравнениями (119, гл. V) и (121, гл. V), в применении к экспериментальным данным для смесей N2-|-H2 и СО-[-Н2 даег при высоких давлениях отрицательное значение последнего члена уравнения, хоти по Джиллиленду он должен быть всегда положительным.
23 Кричевский и Казарновский [338] предложили уравнение, содержащее одну константу
P~Pixi+p^x2+ax1x2(pi— рД. (123а)
В этом уравнении давления чистых компонентов р\ и ръ берутся при одном и том же объеме, равном объему смеси. Константа а зависит от температуры и определяется с помощью графика ax-ix-1 = z(pi—/>2). Казарновский показал [308], что для смесей, не содержащих полярных газов, константа а не зависит от температуры.
Марков [339] обобщил уравнение (123а) и распространил на тройные газовые с&еси.
Проверка уравнения Кричевского и Казарновского на обширном экспериментальном материале, охватывающем двойные и тройные газоиые смеси в широком интервале составов, давлений и температур, показала, чго расчет по этому уравнению дает вполне удовлетворительные результаты (средняя ошибка расчета р — v — 7-данных для изученных смесей меньше 0,5%). Существенным достоинством рассматриваемого уравнения является то обстоятельство, что для расчета по нему необходимо располагать только зависимостью р — v— Т для чистых газов.
Уравнение (123а) выведено на основании следующего допущения: при ностоинном общем объеме не происходит изменения энтропии при переходе небольшого количества одного из компонентов из идеального раствора в реальный того же состава, при условии, что оба раствора рассматриваются при равных мольных объемах.
736
П римечания
Розен и Темкин |385] показали, что уравнение Кричевского—Казарновского может быть выведено, исходя из теоретически обоснованного уравнения состояния со вторым вириальным коэфициентом
p = RT(l
? V \ V J
(1236)
Так как последнее уравнение справедливо только при умеренных давлениях, тогда как первое действительно и при высоких давлениях, то в области высоких давлений уравнение Кричевского — Казарновского должно рассматриваться как эмпирическое. Отметим также, что коэфициент а в общем случае должен зависеть от температуры.
24 Лишь в непосредственной близости к критической точке, т. е, при очень высоких давлениях, изобары в области жидкости становятся отличимы от нижней пограничной кривой.
25 Построение диаграммы — S, Н, ее анализ и примеры ее применения рассмотрены в Вступительной статье, разд. 4.
26 Для N2, О2, NH4, С2Н6 и GG12F2 диаграммы Igp — Н можно найти также в [293].
27 На протяжении всей книги автор пользуется таблицами Кинена н Кейеса [129]. При переводе эти таблицы везде были заменены таблицами Вукаловича [2&4], составленными на основании уравнения состояния Вукаловича (см. прим.28 * * * * * * * * * * * * * * * 44). Это привело, в частности, к замене табл. 7, 8 и 9 и рис. 28 и 29.
Сравнительный анализ таблиц термодинамических свойств водяного пара [275] показал, что таблицы Вукаловича полностью удовлетворяют существующим допускам и поэтому могут быть рекомендованы для широкого применения. Следует лишь отметить, что на S — //-диаграмме, приложенной к указанным таблицам, промежуточные изобары, нанесенные между изобарами 0,2 — 0,3; 0,3 — 0,4; 3 — 4 и 4 — 5 кг/см2, относятся соответственно к 0,24; 0,34; 3,4 и 4.4 кг!см2. При переиздании таблиц следовало бы нанести вместо указанных изобар изобары 0,25; 0,35; 3,5 и 4,5 кгфм2, а также весьма желательно было бы приложить к таблицам S— Т-днаграмму и нанести на ней (или на S —//-диаграмме) изохоры.
28 В табл. х7 воспроизводится небольшой раздел из табл. I таблиц Ву-
каловича.
В различных графах таблицы приводятся следующие величины:
Графа 1 — Температура в градусах Цельсия. Она охватывает пределы
от 0 до 374° (критическая температура равна 374,2° С) с интервалами в 1°.
Графа 2—То же в градусах Кельвина.
Графа 3—Давление насыщенного пара в кг/см2 или атм (при темпера-
туре, указанной в гр. 1 и 2).
Графа 4 — Удельный объем насыщенной жидкости (т. е. жидкости, находящейся в равновесии с паром в мГкг).
Графа 5—Удельный объем сухого насыщенного пара в м*1кг.
Графа 6 — Удельный вес сухого насыщенного пара в кг)м\
Графа 7 — Энтальпия насыщенной жидкости в ккал/кг (отсчитана от эн-
тальпии насыщенной воды при 0° G).
Графа 8—Энтальпия сухого насыщенного пара в ккал/кг.
Графа 9—Скрытая теплота парообразования ккал!кг, равная разности величин, указанных в графах 8 и 7.
Графа 10 — Энтропия насыщенной жидкости в ккал/кг-град (отсчитана от энтропии насыщенной воды при 0° С).
Графа II — Энтропия сухого насыщенного пара в ккал/кг-гра!.
Примечания
737
Значения свойств при температурах, отличных от указанных в таблице, можно получить с достаточной точностью посредством линейной интерполяции.
Таблица 7
Сухой насыщенный пар (по температурам)1
| Температура j Абсолютная темпера* тура Т,ок Давление р, кг,'см* Удельный объем Удельный нес сухого насыщенного пара V, кг{м* Энтальпия ! Теплота парообразования £, ккал{к? Энтропия
насыщенной жидкости м*1кг сухого насыщенного пара vn м*/кг j насыщенной жидкости Н>, ккал[кг сухого насыщенного пара /7", ккал; кг насыщенной жидкости 5', ккал[кг-град сухого насыщенного пара 3", ккал!кг>град
1 2 3 4 5 6 7 8 у 10 11
150 423,16 4,854 0,0010906 0,3924 2,548 150,9 655,5 504,6 0,4395 1,6320
151 424,16 4,985 0,0010917 0,3827 2,613 151,9 655,8 503,9 0,4419 1,6299
152 425,16 5,120 0,0010928 0,3732 2,679 153,0 656,1 503,1 0,4444 1,6277
153 426,16 5,257 0,0010939 0,3640 2,747 154,0 656,4 502,4 0,4468 1,6256
154 427,16 5,397 0,0010951 0,3551 2,816 155,0 656,6 501,6 0,4492 1,6235
Цифровой материал воспроизведен из таблиц Вукаловича.
В табл. 8 воспроизводится часть табл. II Вукаловича.
Графа I — Давление насыщенного пара в кг[см2 от 0,01 до 224 кг1смг (критическое давление равно 225,5 кг/см2).
Графа 2—Температура кипения в градусах Цельсия, соответствующая давлению, указанному в графе 1.
Графы 3—10 совпадают с графами 4—И табл. 7.
В других таблицах приводятся иногда энергия насыщенной жидкости, изменение энергнн при испарении и энергия насыщенного пара.
Таблица 8
Сухой насыщенный пар (но давлениям)1
Давление р, кг/см* Температура t, °C. Удельный объем Удельный вес сухого насыщенного пара 7”, кг/ И3 Энтальпия Теплота парообразования L* ккал^кг Энтропия
насыщенной жидкости м31кг сухого насыщенного пара Vм, мя;кг насыщенной жидкости /7\ ккал[кг сухого насыщенного пара /7", ккал/кг насыщенной жидкости S’, ккал[кг*град сухого насыщенного пара 3", ккал^кг-град
1 2 3 4 5 6 7 8 9 10
7,0 164,17 0,0011072 0,2778 3,600 165,6 659,4 493,8 0,4737 1,6029
7,2 165,31 0,0011086 0,2705 3,697 166,8 659,7 492,9 0,4764 1,6006
7,4 166,42 0,0011100 0,2636 3,794 167,9 660,0 492,1 0,4790 1.5984
7,6 167,51 0,0011114 0,2570 3,891 169,1 660,3 491,2 0,4815 1,5963
7,8 168,57 0,0011127 0,2508 3,988 170,2 660,5 490,3 0,4840 1,5942
1 Цифровой материал воспроизведен из таблиц Вукаловича.
47 Химическая термодинамика
738
Примечания
Энергия вычисляется из значений Н, р и » по уравнению
(У=Я-1(ро).
(6, гл. III)
В табл. 9 воспроизводится небольшая часть табл. III Вукаловича, которая
Т абляца9
Вода и перегретый пар <з (числа над ступенчатой линией относятся к воде)1
t 60 ат 6,5 ат 7,0 ат 7,5 ап
t = 158,3 = 0,3213 Н" = 657,8 S" = 1,6151 7. = 161,22 о- — 0,2979 Н" = 658,7 S«= 1,6088 ss-fs II II II II < % *• б'ЕцЧ V» = 0,2603 И" = 660,2 3» — 1,5974
V $ е | Н | S ° S V 1 " 1 *
150 0,0010906 150,9 0,4395 0,0010905 0,0011021 150,9 161,3 0,4395 0,4637 0,0010904 0,0011020 150,9 i6i,a 0,4394 0,4637 0,0010904 0,0011020 151,0 0,4394 161,3 0,4636
160 170 180 190 и,3232 0,3326 0,3416 0,3504 659,1 664,9 670,4 675,7 1,617b 1,6316 1,6439 1,6553
0,3058 0,3142 0,3224 664,2 669,7 675,0 1,6212 1,6337 1,6454 0,2827 0,2906 0,2983 663,2 669,0 674,4 1,6113 1,6242 1,6361 0,2626 0,2702 0,2776 662,2 1,6017 668,2 1,6152 673,7 1,6274
1 Цифровой материал воспроизведен из таблиц Вукаловича.
значительно обширнее двух других, так как в области перегретого пара имеются две независимые переменные вместо одной, что имело место в случае области насыщения.
В этой таблице указаны удельный объем, энтальпия и энтропия воды (числа над ступенчатой линией) и перегретого пара (числа под ступенчатой динией), а также удельный объем, энтальпия и энтропия насыщенного водяного пара и температура насыщения ts (все свойства даны в тех же единицах, что и в табл. I и II). В таблице даны значения свойств от 0 до 550° С (через каждые 10° С) и от 0,01 до 300 кг/см2 (с постепенно увеличивающимся интервалом от 0,01 до 10 кг/см2—в зависимости от величины давления).
Теплоемкость водяного пара (Ср и Cv) дается отдельно; она приведена соответственно в табл. IV и V в интервале 1—300 кг/см2 (1, 5, 10 и далее через каждые 10 кг/см2) и 100—540° С (через каждые 20°).
29 Все числовые значения будут взяты из таблиц Вукаловича или из диаграммы, сопровождающей эту таблицу.
30 См. Вступительную статью, разд. 4.
31 См. прим. ®.
32 Голубев и Кульчицкий [295] измерили теплоемкость 1/3-аз от оводородной смеси при ( = 25—150 и /> = 20 — 500 атм, причем экспериментальные данные вплоть до 200 атм совпали с уравнением (4).
33 Хотя имеющиеся таблицы и диаграммы для водяного пара охватывают область температур до 600° С и давлений до 300 атм, однако при высоких температурах и давлениях свойства пара большей частью найдены экстраполяцией экспериментальных данных. Между тем, в связи с широким внедрением в промышленность пара высоких параметров и в связи с повышением степени точности расчетов именно в этой области необходимы достоверные значения важнейших свойств водяного пара.
Примечания
739
В числе различных свойств особенно часто приходится пользоваться изобарными теплоемкостями. Так как в сверхкритической и особенно в критической области Ср велика и подвержена значительному изменению (см. стр. 283), то экстраполяция экспериментальных данных в эти области может привести к значительным ошибкам. В связи с этим необходимо указать на недавно разработанную во Всесоюзном теплотехническом институте новую методику определения теплоемкости водяного пара при высоких температурах и давлениях [391], позволяющую получить значения Ср, далеко превосходящие по точности ранее проведенные зарубежные исследования. Погрешность измерений оценивается авторами в ± 1,5°/0. Опыты производились при давлениях в 250, 275 и 300 атм и охватывали температурный интервал от 348 до 460° С.
34 В последнее время во Всесоюзном теплотехническом институте была разработана оригинальная методика определения объема водяного пара, превосходящая по точности методики, применявшиеся в других исследованиях [391а]. Были найдены удельные объемы в неизученных до сих пор критической и сверхкритической областях. Охвачены области о = 3,044 4*4,930 мл/г (6 изохор) до f = 413° С и /> = 300 атм.
35 Метод Деминга и Шуп, как показал Розен [371], может быть применен в том случае, когда в интервале (vb 7\ — v2, Т2) отклонение объемов от линейной зависимости
a = v-^22^L(r-r1)
>2 — >1
велико по сравнению с ошибками опыта. Это условие особенно существенно при вычислении Ср, так как в соответствии с уравнением (105, гл. III) в этом случае приходится производить двукратное диференцирование, которое дает результат, отличный от нуля только за счет искривления изобар ц=ср(Г).
Так, если считать мерой линейности изобар уравнение (83а, гл. V), то расчет показывает, что для водорода погрешность эксперимента примерно вдвое превышает отклонения от этого уравнения. Поэтому значения (Ср)нг, найденные по методу Деминга и Шуп, должны быть признаны неверными.
Практическая ценность функций Дна при соблюдении отмеченного выше условия ограничена дополнительным требованием, именно: необходимо, чтобы члены с производными (д&1дТ)р и (cajdT)p составляли лишь долю от общего значения правой части уравнений (23, гл. VI) и (25 гл. VI), так как только при этом условии ошибки, сделанные при нахождении (дЩдТ)р и (да!др)р, не войдут всей своей величиной в (dvvJ"),,. В то же время для ряда газов (например, для NH3 и СН4) указанные члены превосходят по абсолютной величине всю правую часть уравнений (23, гл. VI) и (25, гл. VI). В подобных случаях рекомендуется производить непосредственное диференцирование &Т.
36 См. Вступительную статью, разд. 2.
37 Так, например, были выведены уравнения для расчета летучих компонентов газовой смеси, подчиняющейся уравнению Ван-дер-Ваальса, Битти — Бриджмена, Бартлетта [331], Кричевского и Казарновского [3381 и др.
33 Уравнение Дюринга в отличие от уравнений (47, гл. VI) — (59, гл. VI) основывается на использовании метода сравнительного расчета. О теоретическом выводе его в сопоставлении с другими уравнениями, использующими этот метод, см. Вступительную статью, раздел 5.
39 Значительно более строгое по выводу и более точное по результатам уравнение, использующее тоже метод сравнительного расчета, предложено Киреевым. [3211.
47*
740
Примечания
Сопоставляя для двух веществ А и В уравнение Клаузиуса — Клапейрона,
представленное в форме dlnp. LA=T—^(p^A, (58а) dlnp„ LB=r-^p(pAv)B, (586)
относя их к одинаковой температуре и исключая ее из них, можно получить
V /А\ /о
(58в)
Величины I \ и I -%— 1 для двух каких-нибудь веществ, не слиш-ком различных по химическому характеру и по температурам кипения, оказываются пропорциональными при одинаковых температурах, т. е. отноше-
ние их
p4a\l )в
(58г)
можно принять не зависящим от температуры. Для большей ясности физического смысла этого допущения следует учесть, что в области низких давлений насыщенного пара, где можно принять pkv = RT, она равносильно допущению о пропорциональности теплот испарения сравниваемых веществ при одинаковых температурах, и
k = LaILb. (58д)
Интегрируя уравнение (58в), легко получить уравнение
lg Ра = k 1g Рв+С, (58е)
связывающее логарифмы давлений насыщенного пара двух веществ при равных температурах в линейной форме. Постоянная интегрирования С может быть определена, если известны давления пара обоих веществ прн какой-нибудь температуре, а постоянная k может быть определена или по соотношениям (58г) нлн (58д), или эмпирически по давлениям пара при какой-нибудь другой температуре. Применение этого уравнения подробнее рассматривается во Вступительной статье, разд. 5.
40 Вопросу теплоемкости насыщенных паров посвящен ряд работ Коло-совского с сотрудниками [326, 327|.
41 См. также стр. 440—446.
42 Колосовский и Гребенщиков [328] показали, что температурный коэфициент теплоты парообразования, взятый с обратным знаком, всегда больше разности изобарных теплоемкостей пара и жидкости, так как в отличие от уравнения (72) всегда
где
Д>0.
48 Исходя из подобия термодинамических параметров, Лукомский [349| предложил методику относительного расчета свойств чистых веществ на
Примечания
741
кривой равновесия жидкость — пар в области высоких давлений (т = 0,7 4- 1). Он построил обобщенные графики (на которых соответствующее свойство выражалось общей кривой) и на их основании нашел приближенные эмпирические уравнения. Точность расчетов за небольшим исключением составляет 6—20°/о- Для сходных веществ точность значительно повышается. Ниже приводятся расчетные уравнения (константы уравнений определены из соответствующих графиков).
Из графиков л = (т) и т 1g тс = ср (тс) найдено уравнение для расчета давления насыщенного пара
2,9 (1- -)
л =10 v х'. (76а)
По графикам рп (рж) = <р(т) и-(рж/ркр), (рп/ркр) = ? (т) получены уравнения для вычисления плотности пара и жидкости на кривой насыщения соответственно
£1 = 1--------------------1----------------- (766)
рж 0,26 (1 — т)*'* -4---------------7 4- 0,5
^3,8(1 —
И
— = 0,975 (1 — т) -f- 1,9 (1 — т)1/, 4- 1, Ркр
—=0,975(1—т)—1,9(1—г)'/»4-1. (76в)
Ркр
Из графика (£/£0) = <р(т), где Lo — теплота парообразования при 0,7т, найдено уравнение
у-=1,619(1 — т)0’4. 7-0
(76г)
Критические параметры (ркр и могут быть найдены или по указанным графикам, или вычислены с помощью уравнений
(76Д)
(76е)
полученных преобразованием уравнения л = ср(т).
44 Свойства перегретого водяного пара, приведенные в таблицах Вукаловича, найдены следующим путем.
Объем вычисляется по уравнению состояния
pv = RT [1 — — I v
-1
о2] ’
(76ж)
742
Примечания
в котором температурные функции А и В определяются уравнениями
т 2
bcxR
3+2mt
т 2
Сг!с\ \
Зт3 —4тг I
т 2 /
(76и)
где а и b — константы уравнения (42, гл. V), R — газовая постоянная и «j пи, и с2 — постоянные, вычисляемые по опытным данным [285]. Затем> пользуясь уравнением (76ж), по уравнениям (104, гл. 111) и (113, гл. III) были вычислены значения Ср и Cv с помощью значений внутренней энергии, найденных по уравнению (178, гл. III), и энтальпии, определенной по уравнению (6, гл. III). Наконец, по уравнению (86, гл. III) вычислялась энтропия. Необходимые для расчета значения СРа определялись по эмпирическому уравнению, заменившему величины, найденные из квантовой теории теплоемкости на основании оптических данных (анализ спектра водяного пара).
Таким образом, для расчета термодинамических свойств перегретого водяного пара понадобились данные по его сжимаемости и его теплоемкость при низком давлении. Результаты расчета оказались в хорошем согласии с опытными данными [285].
48 Для реальных газов показатель адиабаты не равен CpjCv (см. Вступительную статью, разд. 2): изменение его с температурой противоположно данным приводимой таблицы.
46 Обсуждение этого вопроса см. Вступительную статью, разд. 2.
47 При проектировании компрессоров этот вопрос решается в каждом конкретном случае путем расчета теплоотвода через рубашку.
48 См. Вступительную статью, разд. 2.
49 Для реальных газов вследствие необходимости учета в величине работы дополнительного члена это условие не будет соблюдаться; так, при высоком давлении оптимальное соотношение при двухступенчатом сжатии р2/Р1 — Рз1>!г приведет к тому, что А2 будет больше Ау
60 См. Вступительную статью, разд. 2.
и Практически эти эффекты учитываются коэфициентом подачи
Ф вплотн.е W'
где еплотн —коэфициент плотности (0,93—0,97), учитывающий утечки и ф —
, „ р2 j 7^2
коэфициент подогрева, равный ср —; соотношения 4> и таковы:
Р\
Рз Р1 1 2 3 4 5
ф 1 0,99 0,97 0,95 0,91
Примечания
743
» См. Вступительную статью, разд. 4.
«з См. Вступительную статью, разд. 4.
6-* Обычно считается критическим значением /?е = 2320.
и 1 пуаз =100 сантипуазам = 0,0102 Кг
56 Здесь желательно вспомнить, что, согласно термодинамическому определению, теплота является энергией в процессе перехода от одного места к другому с помощью одного из этих способов. Пока мы рассматриваем только энергию, содержащуюся в системе, мы не можем считать ее теплотой в обычном для термодинамики смысле слова. Однако нужно дать название энергии, содержащейся в теле и способной передаваться в виде теплоты; мы будем обозначать ее термином „термическая энергия*. См. прим. 4
57 Световое давление впервые было установлено и определено Лебедевым (344|.
68 Подробное изложение см. [315а].
69 Более подробную характеристику дифенила, дифенилоксида, даутерма и некоторых других жидких теплоносителей можно найти в книге Н. И. Гель-перина [288].
60 Рис. 68 построен по данным [171], несколько отличающимся от данных [293].
61 См. также [397].
62 Обширный экспериментальный материал по зависимости теплоемкости от температуры приведен в работах Курбатова [342, 343], посвященных изучению строения и свойств жидкостей. Исследовалось более 350 веществ и установлен ряд закономерностей. Так оказалось, что величина Ь/а в уравнении С = а-\-Ы для различных веществ примерно одинакова и для большинства изученных органических соединений близка к 0,0022; для жидкостей, получающихся сжижением трудно конденсируемых газов, эта величина близка к 0,0036; для заметно ионных молекул она падает до 0,0006.
63 Если для трехмерных кристаллов справедливо уравнение (31, гл. IX), то, как показал Тарасов [382, 383], для слоистых структур должен соблюдаться закон
С„ = аГ3, (31а)
а для линейно-полимеризованных молекул, т.е. для „одномерных* кристаллов,
С = АТ, (316)
где а и Л — константы.
Тем самым удается описать зависимость Ср от Т в области низких температур для многих веществ, не подчиняющихся закону Дебая, а та#же составить представление о структуре соединений.
64 Следует отметить, что для обычных жидкостей отклонение LrIT от 21 невелико. Исключения составляют ассоциированные вещества (вода, спирты, органические кислоты и др.).
® Использование графика у =/(т) было описано ранее Киреевым, предложившим и аналитическое выражение этой зависимости (доклад на IV Менделеевском съезде в Казани 16/VI 1926 г.) [316].
66 Эта работа Отмера как по выводу, так и по результатам полностью повторяет работу Киреева, опубликованную на 9 лет раньше (см. примечание зв), отличаясь от нее только большим числом примеров.
744
Примечания
87 Рассмотренный способ, независимо от Отмера, был предложен Гордоном [94].
88 Карапетьянц [314] предложил приближенный способ расчета, идея которого подсказана подобием кривых L = <f(T), приводящим к возможности их выпрямления вплоть до критической температуры после предварительного смещения в одну точку [(tKP — t) = О, L = 0]. (См. прим. 96.)
» Этот метод применим только к нормальным жидкостям, и большая ошибка для этилового спирта совершенно естественна.
ТО См. гл. XII.
И Изучению теплот испарения из растворов были посвящены некоторые работы Вревского [279—283] и Киреева [317]. Последним было показано, в частности, что парциальные теплоты испарения из раствора могут значи-тельно отличаться от теплот испарения из чистой жидкости.
72 Независимость теплового эффекта от пути перехода была установлена впервые русским академиком Гессом в 1840 г. и известна как закон Гесса.
ТО Речь идет, разумеется, об относительной ошибке.
то См. также [378].
то Влияние давления на теплоту реакции было определено Казарновским и Карапетьянцем для реакции синтеза аммиака [310]. Расчет, произведенный в интервале 250—500°С и 100—1000 атм, показал, что при высоких давлениях тепловой эффект реакции синтеза аммиака примерно на 20°/о превышает тепловой эффект при атмосферном давлении. Было также показано, что если исправить расчет на теплоту образования азотоводородноаммиачной смеси состава 0,2N2-|-0,6H2-)-0,2Nn3 из 7/з азотоводородной смеси и аммиака, полагая, что в условиях производства конечная смесь содержит примерно’ 20%NH3, то действие давления в значительной степени элиминируется.
Расчет, произведенный Казарновским, по более точным исходным дан-вж [309], в общем подтвердил результаты предыдущей работы.
то В русской литературе за единицу холодопроизводительности принята 1 ккал!кг.
я В связи с широким развитием в последние годы турбостроения можио' ожидать в ряде случаев возрождения воздушной холодильной машины в виде агрегата (состоящего из турбокомпрессора н воздушной турбины), для которого большие объемы циркулирующего воздуха являются не затруднением, а благоприятным фактором. Повышение индикаторного к. п. д., вызванное прямоточ-ностью движения воздуха в машине, и высокие значения ее механического к. п. д. могут обеспечить в крупных установках значительное повышение холодопроизводительности на 1 квт-ч затраченной энергии (см. [401]).
* 78 Все свойства заимствуем из книги С. Я. Герша [293] (см. диагр. XXIII).
то Возможность применения галоидбзамещенных углеводороде® в качестве рабочих агентов бинарных теплосиловых установок обсуждена в работе Гох-штейна [297].
80 Впервые идея Кельвина была развита Михельсоном (Москва, 1926) и доведена им до технического проекта [357].
81 Значения термодинамических констант воздуха, использованные в этом примере, заимствованы из книги Герша [293].
82 Можно также рекомендовать книги Герша [291, 292, 293], Фастовского' [394], Малкова и Павлова [353]. Исчерпывающая библиография по глубокому охлаждению и низким температурам составлена Малковым [352].
Примечания
745
83 См. Вступительную статью, разд. 6.
w Следует отметить, что при расчете равновесия важна не относительная, а абсолютная ошибка в величинах Д/7 и S.
ж Для дальнейших примеров применения приближения Нернста можно рекомендовать обратиться к книгам Перри [362], (188а], Тейлора [384], Нери-ста [358], Бродского [271], Раковского [368, 369], Капустинского [312], Герасимова и Крестовникова [290] и др.
86 Скорость изменения, как указывает автор па стр. 46, будет зависеть и от сопротивления процессу.
8т См. Вступительную статью, разд. 7.
88 На основании этих экспериментальных данных Лебедевым [344] были построены номограммы. Кроме общей номограммы, охватывающей диапазон давлений от 0, 5 до 20 атм при содержании азота (в жидкости и паре) от 0 до 100°'о, были построены 4 отдельные номограммы с охватом наиболее важных расчетных областей системы.
89 Обычное определение констант уравнений (17, гл. XII) и (18, гл. XII), осуществляемое либо по уравнениям (19, гл. XII) и (20, гл. XII) на основании найденных из опыта значений рА и рв, либо по уравнениям (21, гл. XII) и (22, гл. XII) на основании значений тангенсов углов наклона касательных (др[дх^х=я и {dpfdx^^Y не свободно от недостатков, так как первый способ-предполагает опытное определение парциальных давлений насыщенного пара, что требует сложного по аппаратуре и выполнению анализа парообразной фазы, а второй способ основан на недостаточно надежном графическом диференцировании путем проведения касательных в точках, в которых кривая обрывается.
Поэтому заслуживают рассмотрения способы, предложенные Литвиновым.
1. Сущность первого способа [345] заключается в следующем. Для получения значений {др]дх])х=о и [dp‘jdxx)x=i нужны лишь участки кривой, непосредственно прилегающие к ее концам, поэтому если действительную кривую р = ср (х) заменить некоторой другой р' — ср (х) и если последняя на. этих участках будет совпадать с первой, то получатся равенства:
\дх]х=о \дх Jх=о \дхJ х=1 V дх] Х=1'
Тогда для расчета потребуются четыре точки, две из которых будут лежать на концах кривой, т. е. отвечать давлению насыщенного пара чистых жидкостей, а две другие будут лежать близко к ее концам. В качестве последних рекомендуются точки х = 0,95 и х = 0,05, так как, с одной стороны, они достаточно близки к концам, чтобы дать хорошее совпадение двух кривых, а с другой— они не настолько близки к концам, чтобы привести к ухудшению результатов расчета вследствие того, что превышение одной точки над другой (например х—0,95 над х=1) может оказаться лежащим в пределах ошибок опыта. Существенным представляется и то обстоятельство, что вне интервала х = 0,95 ч- 0,05 на кривой р = ср(х) максимум или минимум наблюдается крайне редко. Из известных в настоящее времи нескольких десит-ков тысяч случаев образования азеотропов в бинарных смесих только в нескольких десятках случаев экстремум давлении насыщенного пара падает на интервалы х=1 ч-0,95 и х = 0,05ч-0.
Если же вне указанного интервала имеется экстремум давления насыщенного пара, то расчет необходимо ориентировать на значения х > 0,95« и х < 0,05.
746
Примечания
По точкам х=1, х = 0,95, х = 0,05 и х = 0 с помощью интерполяционной формулы Лагранжа Литвинов получил расчетное уравнение р’ — у М ’(третьей степени относительно х), подстановка в которое значений Рд, рв, рх=о,95 и рх=о,о5 позволяет найти р' — ф(х), затем вычислить производные, входящие в уравнения (21, гл. XII) и (22, гл. XII), а по ним константы уравнений (17, гл. XII) и (18, гл. XII) после чего легко найти парциальные давления и общие давления при любых концентрациях.
Проверка этого метода на пяти смесях, отличающихся характером кривых р=?(х), дала удовлетворительные результаты.
2. Если интерполяционная кривая дважды пересекает опытйую кривую, что получается, когда обе экспериментальные точки или точки перегиба лежат в интервале х — 0-5-1, то они не будут совпадать на интересующих нас участках. Хотя этот случай встречается очень редко, однако его также необходимо учесть, для чего интерполяционную кривую надо исследовать на максимум, минимум и точку перегиба. Если это исследование укажет на пересечение, то необходимо ввести еще одну опытную точку, расположив ее между точками, в которых происходит пересечение. Однако расчет по полученному в этом случае интерполяционному уравнению (четвертой степени относительно х) является громоздким; поэтому Литвинов рекомендует следующий способ [346]:
Если х близко к 1 или к 0, то рА или рв соответственно могут быть вычислены по закону Рауля. После того, как найдено общее давление, рв и рд определяются по уравнению (219, гл. IV); преобразовав полученные уравнения для х = 0,95 и х= 0,05, Литвинов получил уравнения, позволяющие производить вычисления констант уравнения (17, гл. XII) и (18, гл. XII) по Рл, Рв я двум значениям р.
Проверка этого метода в соответствии с привлечением закона Рауля помазала, что совпадение с экспериментальными данными тем лучше, чем ближе раствор к идеальному. Так как расчет парциальных и общего давлений (после того как определены значения констант уравнения Маргулеса) является трудоемким, то была построена номограмма, дающая отклонение от алгебраического решения на 1—1,5%.
Дополнительная проверка обоих способов расчета на семи смесих [347J подтвердила достаточную их точность.
90 Алексеев установил [267], что если разделить горизонтальные линии, -соединяющие точки растворов 1 и 2 (конноды), пополам и соединить точки деления, то получается прямая линия, пересечение которой с кривой растворимости дает критическую точку. Правило Алексеева является приближенным и соблюдается при выражении состава в весовых процентах.
91 Воспроизведено в ]250].
92 Утверждение автора, что выбор между диаграммами у — х и Н—х не имеет значения, является ошибочным. Диаграммой у— х можно пользоваться в тех случаях, когда компоненты ректифицируемой смеси имеют одинаковые или очень близкие между собой скрытые теплоты парообразования. В тех случаях, когда скрытые теплоты компонентов смеси значительно отличаются друг от друга, точный расчет возможен только по диаграмме Н—х„
93 Простой аналитический метод определения зависимости между числом теоретических тарелок и величиной флегмового числа для случая идеальной -бинарной смеси разработан Волковым и Жаворонковым [278].
Львов [350] предложил новый способ расчета ректификации многокомпонентных смесей, в основу которого положены следующие предпосылки: 1) любая пара компонентов при данных р и t ведет себя независимо от прочих компонентов и 2) в рабочих условиях ректификации (переменная температура) влияние прочих компонентов на поведение любой рассматриваемой пары может быть сведено к влиянию на изменение температуры по высоте
Примечания
747
колонны при данном давлении. Расчет ректификации сводится к расчету разделения бинарной смеси, в качестве которой выбирается i определяющая пара", которую труднее всего разогнать. Критерием такой пары являются наибольшие значения минимального флегмового числа. Все изложенные методы аналитического расчета являются лишь приближенными. Математически точное решение этой задачи впервые было дано Гельпериным [289].
94 За последние 10—15 лет в связи с широким развитием промышленности моторного топлива интересы исследователей обратились к усовершенствованию методов лабораторной ректификации.
Из многочисленных работ, посвященных выяснению влияния различных факторов на эффективность и производительность лабораторных колонок, заслуживают внимания работы Казанского и его сотрудников [303—307|.
Вопросу унификации методики определения погоноразделительной способности лабораторных ректификационных устройств посвящена статья Обо-ленцева и Фроста [359|. Авторы предложили: 1) формулу и номограмму для расчета числа теоретических тарелок, которым эквивалентна реальная погоноразделительная способность ректификационного устройства на данном рабочем режиме, и 2) метод определения к. п. д. лабораторных ректификационных устройств. Ими был также рассмотрен и продемонстрирован на графиках и примерах ряд зависимостей (между числом теоретических тарелок, относительной летучестью, концентрацией легкокипящего компонента в загрузке и дестяллата, количеством дестиллата), устанавливаемых предлагаемой формулой. В работе подчеркнута также необходимость унификации методики определения числа теоретических тарелок, а при опубликовании данных о ректификации— необходимость указывать не только число теоретических тарелок, но и примененные колонки и методику определения.
В своем обзоре Розенгарт |372] изложил основные сведения по теории ректификации и привел описание современных высокоэффективных лабораторных колонок. В статье приведена обширная библиография.
95 Более подробные сведения о расчете перегонки можно найти в книгах Трегубова [312], Гельперина [287], Фастовского [394], Федорова [395] и др.
96 Расчет, упомянутый в примечании 68, производится либо графически — по „выпрямленному графику" L = '?(tKp—t), либо аналитически — по уравнению
р— const = k [при (tKp— t) = (tKp — Г)], (44а)
отвечающему неизменности отношения теплот парообразования данного и стандартного вещества при температурах, равно удаленных от их критических температур.
Для осуществления расчета необходимо располагать зависимостью £' = <р(Г), величинами t'Kp и tKp и значением h при какой-либо одной температуре (например, LB). В качестве стандартного вещества необходимо выбрать воду.
Средняя ошибка расчета L вплоть до tKp для 50 веществ оказалась равной 2%; для СН4, С2Н4 и С3Н,ОН погрешность превысила 49/0.
Закономерное расположение прямых L = ср (Ткр — Г) в гомологическях рядах, или, что одно и то же, закономерное для гомологов изменение k уравнения (44а), позволяет оценить зависимость А от Г для соединения данного гомологического ряда при полном отсутствии экспериментальных данных.
ПРИЛОЖЕНИЕ
Таблица I
Эквиваленты давления
Мегабары или мегадины кг(см2 (ат) атм Столб Hg при 0°С Столб НаО при 15°С мм Hg фунт / / кв. / дюймы фунт / / кв. фут
см1 м дюймы м дюймы футы
1 1,0197 0,9869 0,7500 29,53 10,21 401,8 33,48 750,044 14,50 2088,0
0,9807 1 0,9678 0,7355 28,96 10,01 394,0 32,84 735,514 14,22 2047,17
1,0133 1,0333 1 0,76 29,92 10,34 407,2 33,93 760 14,70 2116,8
1,3333 1,3596 1,316 1 39,37 13,61 535,7 44,64 1000 19,34 2784,96
0,03386 0,03453 0,03342 0,02540 1 0,3456 13,61 1,134 25,399 0,4912 70,733
0,09768 0,09991 0,09670 0,07349 2,893 1 39,37 3,281 73,492 1,421 204,624
0,002489 0,002538 0,002456 0,001867 0,07349 0,02540 1 0,08333 1,867 0,03610 5,1984
0,02986 0,03045 0,02947 0,02240 0,8819 0,3048 12 1 22,40 0,4332 62,3808
0,06895 0,07031 0,06804 0,05171 2,036 0,7037 27,70 2,309 51,710 1 144
1 г/г.и2 — 980,597 дин/смг
1 дина/см»^ 0,001018 г/сл«
Таблиц* if
Эквиваленты энергии
Джоуль (107 эрг) кгм ккал квт-ч л. с.-ч. л X атм. кал Брит. тепл, единицы (BTU) Футофунты
1 0,10197 0,032390 0Д2778 0,063725 0,009869 0,239 0,0s9486 0,7376
9,80597 1 0,0023424 0,062724 0,033653 0,09678 2,3438 0,009296 7,233
4183 426,9 1 0,001162 0,001558 41,29 999,936 3,968 3,068-10»
3,6-106 3,671-10“ 859,98 1 1,341 35,528-10» 860,445-10» 3,415-10» 2,655-10“
2,6845-106 2,7375-10“ 641,7 0,7457 1 26,494-10» 641,659-10» 2,545-10» 1,98-10®
101,33 10,333 0,02422 0,042815 0,0*3774 1 24,218 0,09612 74,73
1054 107,5 0,25200 0,032928 0,033930 10,40 252 1 778,1
1,356 0,1383 0,033241 0,063766 0,0650505 0,01338 0,3241 0,001286 1
Таблица III
Критические константы и константы а и Ь уравнения Ван-дер-Ваальса1) (единицы для а и Ь: атм, мл, ° К, моли)
Вещество ТКР, °к ркр, атм vKp, мл! МО ль PkPVkP КГкр Константа Ван-дер-Ваальса2)
аХЮ-6 Ь
Азот 126,0 33,5 56 0,182 1,348 38,6
Аллилен 401
Аммиак 405,5 111,5 72,4 0,242 4,192 37,3
Api ин Гои?/ 48 /0,0 0,293 1,35 32,2
Ацетилен .... 309,1 61,7 113 0,275 4,398 51,37
Бензол 561,6 47,7 256 18,78 120,7
Бромистый иодо-
род 363 84 4,46 4,4
я-Бутан 426,0 36,0 250 14,32
Вода 647,3 218,2 56,8 0,234 5,454 30,6
Водород 33,2 12,8 64,3 0,302 0,245 26,6
Воздух 132,4 37,2 82,4 3) 0,282 1,338 36,34
93,02 4) 0,321
я-Гексан .... 507,9 25,9 367 24,83 176,5
Гелий 5,2 2,3 60,5 0,326 0,0334 23,2
я-Г ептан .... 540,0 26,8 427 30,90 206,6
Диизобутил . . . 549,9 24,5 482 35,05 230,1
Диметиламин . . 437,7 52,4 10,38 85,61
Диизопропил . . 500,5 30,6 357 23,24 167,7
Дифтортетрахлор-
этан 551,1
Дихлордифторме-
тан 384,6 39,6 221 0,277 10,62 99,53
Дихлорметан . . 489 51,4 13,22 97,59
Диэтиламин . . . 496,4 36,58 19,18 139,1
Закись азота . . . 309,6 71,7 98? , 3,975 44,2
Изобутан .... 407,1 37,0 249 12,72 105,3
Изопентан .... 460,9 32,92 308 18,33 143,5
Йодистый водород 424 82 6,226 53,0
Кислород .... 154,3 49,7 74,3 0,292 1,362 31,8
Криптон 210,6 54,2 105?5) 2,32? 40?
Ксенон 290 58 4,118 51
Метан 190,7 45,8 99,2 ‘ 0,290 2,264 42,78
Монометиламин . 430,0 73,6 7,137 59,9
Монопропиламин 496,9 46,76 15,01 109,0
Моноэтиламин . . 456,3 55,54 10,66 84,30
Неон 44,4 26,9 41 0,30 0,216 17,6
Озон 268? 92? 2,217 29,9
Окись азота". . . 179? 65? 1,40 28?
Окись углерода . 134,4 34,6 89,9 0,282 1,48 39,9
я-Октан 569,3 24,6 490,1 —— 37,42 237,3
я-Пентан .... 470,3 33,0 311 и и- 19,04 146,1
Пропан 369,9 42,01 194 — 9,249 90,23
Пропилен .... 364,8 45,0 182 - 8,396 83,12
Ртуть 1172 180 19,87 66,77
Продолжен и е
Вещество Ткр,° к Ркр> атм VKp, мл/моль PkPvkP ТКр Константа Ван-дер-Ваальса г)
аХ10"6 Ь
Сероводород . . Сернистый ангидрид Серный ангидрид Сероокись углерода Синильная кислота Тетрафторид кремния .... Трифторид бора . Трифтортрихлор-этан Углекислота . . . Фосген Фосфор Фосфористый водород Фтор Фтористый метил Хлор Хлористый водород Хлористый метил . Хлористый пропил Хлористый этил . Циан Циклогексан . . . Этан Этилен 373,5 430,3 491,4 378 458,6 272 260,8 460,7 304,3 456? 948,1 325 144 317,6 417 324,5 416,2 503,2 460,3 401 554,1 305,2 282,8 88,9 77,7 83,8 61 56,9 50 49,2 72,9 80 65 55 58,0 76 81,6 65,8 45,18 52,0 59,0 40,1 48,8 50,9 123 126 139 ' 96,1 124 137 309 137 134 0,262 0,210 0,281 0,276 0,267 0,294 4,461 6,767 8,201 6,664 10,49 4,204 3,931 3,604 31,95 4,617 1,179 4,940 6,499 3,67 7,48 16,31 11,57 6,016 21,48 5,419 4,48 43,1 56,8 60,2 63,6 82,55- 59,5 54,35- 42,8 121,4 51 26,1 56,1 56 40,8 64,96- 114,1 90,79 69,9 139,9 64,15 57,19
х) Большинство данных взято из циркуляра Бюро стандартов [36]. Данные по углеводородам заимствованы у Эдмистера [74].
8) Вычислены по критическим давлениям и температуре по уравнениям
_ 27 RSTKp.
0 — 64 Ркр ь=^, %Ркр /? = 82,06.
9) Критическая точка.
*) Крикондентермическая точка.
s) Указывает сомнительные значения.
ЛИТЕРАТУРА
А. Список авторов в порядке латинского алфавита
1. Adams L. Н., Chem. Rev., 19, 1—26 (1936).
2. Adams L. H., J. Am. Chem. Soc., 53, 3769—3813 (1931).
3. Akerlof G., J. Teare, J. Am. Chem. Soc.. 59. 1855 (1937).
♦4. Antoin C„ Compt. rend., 107, 681 (1888).
5. Aston J. G., Ind. Eng. Chem., 34, 514 (1942).
6. Bahlke W. H., W. B. Kay, Ind. Eng. Chem., 24, 291—301 (1932).
7. Bartlett E. P., H. L. Cuppies, T. H. Tremearne, J. Am. Chem. Soc., 50, 1275 (1928).
8. Bartlett E. P., H. C. Hetherington, H. M. Kvalnes, T. H. Tremearne, J. Am. Chem. Soc., 52, 1363, 1374 (1938).
9. Bean H. S., E. Buckingham, P. S. Murphy, Bur. Standards J. Research, 2, 562—658 (1929).
10. Beattie J. A., Phys. Rev., 31, 680—690 (1928); 34, 1615—1620 (1929); 36, 132—145 (1930).
11. Beattie J. A., Proc. Nat. Acad. Sci., 16, 14—19 (1930).
12. Beattie J. А., О. C. Bridgeman, Proc. Am. Acad. Arts. Sci., 63,229—308 (1928).
13. Beattie H. A., G. Calingaert, Ind. Eng. Chem., 26, 904 (1934).
14. Beattie J. A., C. Hadlock, N. Poffenberger, J. Chem. Phys., 3, 93—96 (1935).
15. Beattie J. A., S. Ikehara, Proc. Am. Acad. Arts. Sci., 64,127—176 (1930).
16. Beattie J. A., H. G. Ingersoll, W. H. Stockmayer, J. Am. Chem. Soc., 64, 548 (1942).
17. Beattie J. A., W. C. Kay, J. Kaminsky, J. Am. Chem. Soc., 59,1589—1590 (1937).
18. Beattie J. A., С. K. Lawrence, J. Am. Chem. Soc., 52, 6—14 (1930).
19. Beattie J. A., G. L. Simard, G. Su, J. Am. Chem. Soc., 61, 26—27 (1939).
20. Beattie J. A., W. H. Stockmayer, Reports on Progress in Physics, 7, 195 (1940).
-21. Bennewitz K-, W. Rossner, Z. physik. Chem., 39B, 126 (1938).
22. Benning A. F., R. C. Me Hamess, Ind. Eng. Chem., 32, 698—701 (1940).
23. Bichowsky F. R., F. D. Rossini, .The Thermochemistry of the Chemical Substances', Reinhold Publishing Corporation, New York (1936).
-24. Bosnjakov'c F., Technische Thermodynamic, Vols. 1 and 2, T. Steinkopf, Leipzig (1935).
25. Boston O. W., Chem. Met. Eng., 30, 56—59 (1924).
26. Branch G. E. K-, 7. Am. Chem. Soc., 37, 2316—2326 (1915).
27. Brinkworth J. H., Proc. Roy. Soc., A3, 124—133 (1926).
28. Bromiley E. C., Quiggle D., Ind. Eng. Chem., 25, 1136 (1933).
29. Brown G. G., Proc. 19th Annual Convention, Natural Gasoline Assoc. America, May (1940), pp. 54—74.
30. Brown G. G., M. Souders Tr., .The Science of Petroleum', pp. 1544—1579, Oxford University Press, New York (1938).
31. Brown G. G., M. Souders, R. L. Smith, Ind. Eng. Chem., 24, 513—515 (1932).
32. Bryant W. M. D„ Ind. Eng. Chem., 25, 820—823 (1933).
33. Buckingham E., Bur. Standards Sci. Paper, 57 (1907).
34. Buckingham E., Bur. Standards J. Research, 9, 61—79 (1932).
35. Buffington R. M„ W. K- Gilkey, Ind. Eng. Chem., 23, 254 (1931).
Литература
753
36. Bur. Standards Circ., 279; Tables of Thermodynamic Properties of Ammonia, 142 (1923).
37. Burgess G. K-. Bur. Standards J. Research, 1, 635 (1928).
38. Calingaert G., Chem, Met. Eng., 32, 362 (1925).
39. Calingaert G., D.S. Davis, Ind. Eng. Chem., 17, 1287—1289 (1925).
40. Carlson H. С., A. P. Colburn, Ind. Eng. Chem., 34, 581 (1942).
41. Carr A. R., D. W. Murphy, J. Am. Chem. Soc., 51, 116 (1929).
42. Caubet F., Z. phxsik. Chem., 40, 257—367 (1902).
43. Clark J. A., R. W. Miller, Chem. Met. Eng., 48, 74 (1941).
44. Clayton J. O., W. F. Giauque, J. Am. Chem. Soc., 54, 2610—2626(1932); 55, 5071—5073 (1933).
45. Cooley L. C., Chem. Met. Eng., 34, 725 (1927).
46. Cope J. Q., W.K. Lewis, H.C. Weber, Ind. Eng. Chem., 23,887—892 (1931).
47. Copson R. L., P. K- Frolich, Ind. Eng. Chem., 21,1116—1117 (1929).
48. Cornell L. W., R. E. Montonna, Ind. Eng. Chem., 25, 1331 (1933).
*49. Coulson E. A., E. F. G. Herington, J. Am. Chem. Soc., 69, 597 (1947).
50. Cox E. R., Ind. Eng. Chem., 15, 592 (1923).
51. Cumminngs L. W. T., Stones F. W„ Volante M. A., Ind. Eng. Chem., 25,728 (1932).
52. Dana L. J., Proc. Am. Acad. Arts. Sci., 60, 241—267 (1927).
53. Davis H. N., Meeh. Eng., 47, 107—108 (1925).
54. De Back E. E., Petroleum Eng., 10, 141 (1939).
55. Deming W. E., L. E. Shupe, J. Am. Chem. Soc., 52,1382—1389 (1930).
56. Deming W. E., L. E. Shupe, J. Am. Chem. Soc., 53,863—869 (1931).
57. Deming W. E., L. E. Shupe, Phys. Rev., 40,848—859 (1932).
58. Deming W. E., L. E. Shupe, Phys. Rev., 37,638—654 (1931).
59. Deming W. E., L. E. Shupe, Phys. Rev., 38, 2245—2264 (1931).
60. Dobratz C. J., Ind. Eng. Chem,, 33, 759—762 (1941).
61. Dodge B. F.,Ind. Eng. Chem., 24, 1353—1363(1932).
62. Dodge B. F., Chem, Met. Eng., 35, 622 (1928).
63. Dodge B. F., Ind. Eng. Chem., 22, 89—90 (1930).
64. Dodge B. F., Trans. Am. Inst. Chem. Eng., 34, 529—567 (1938).
65. Dodge B. F., A. K. Dunbar, J. Am. Chem, Soc., 49, 591 (1927).
66. Dodge B. F., J. R. Huffman, Ind.Eng.Chem., 29, 1434(1937).
67. Dodge B. F„ C. Housum, Trans. Am. Inst. Chem. Eng., 19, 117 (1927).
68. Dodge B. F., R. H. Newton, Ind. Eng. Chem., 29, 718 (1937).
69. Drew T. B., R. P. Generaux, Trans. Am. Inst. Chem. Eng., 32, 17—19 (1936).
70. Duhem P., Traiti ilementaire de micanique chimique fondle sur la Ther-modynamique, Paris, 1897—1899.
71. Eastman E. D., U. S. Bur. Mines Tech. Paper, 445 (1929).
72. Eastman E. D., Bur. Mines Inf. Circ., 6125 (1925).
72a. Eastman E. D„ Chem. Rev., 18, 257 (1936).
73. Edmister W. C., Ind. Eng. Chem., 28, 1112—1116 (1936).
74. Edmister W. C., Ind. Eng. Chem., 30, 352—358 (1938).
75. Edmister W. C., Ind. Eng. Chem., 32, 373 (1940).
76. Ellenwood F. O., N. Kulik, N. R. Gay, Cornell Univ. Eng. Expt. Sta. Bull.,
30 (1942).
77. Ewell R. H„ Ind. Eng. Chem., 32, 778 (1940).
78. Flock E. F., Bur. Standards J. Research, 5, 481—505 (1930).
79. Fiock E. F., D. C. Ginnungs, W. B. Holton, Bur. Standards J. Research, 6, 881—900 (1931).
80. Fluid Meters, Their Theory and Application, Part 1, 4th ed., American Society of Mechanical Engineers, New York (1937).
81. Fowler R.H., E.A.Guggenheim, Statistical Thermodynamics, Cambridge (1939).
82. Fugassi P„ C. L. Rudy, Jr., Ind. Eng. Chem., 30, 1029—1030 (1938).
83. Genereaux R. P„ Chem. Met. Eng., 44, 241—248 (1937).
84. Giauque W. F., J. Am. Chem. Soc., 49, 1864 (1927).
85. Giauque W. F., Ind. Eng. Chem., 28, 743—750 (1936).
48 Химическая термодинамика
754
Литература
86. Giauque W. F., J. W. Stout, J. Am. Chem. Soc., 58, 1144—1150 (1936).
87. Gibbs J. W., Trans. Connecticut Acad., 3, 108—248, 343—524 (1875—1878).
88. Gillespie L. J., Phys. Rev., 34, 352 (1929).
89. Gillespie L. J., J. Phys. Chem., 33, 354—360 (1929).
90. Gillespie L. J„ J. Math. Phys. Mass. Inst. Tech., 4, 84—96 (1925).
91. Gilliland E. R„ Ind. Eng. Chem., 28, 212—215 (1936).
92. Gilliland E. R., R. C. Gunness, V. O. Bowles, Ind. Eng. Chem., 28, 370—372 (1936).
93. Gordon A. R., J. Chem. Phys., 2, 65—72 (1934).
*94. Gordon D. H., Ind. Eng. Chem., 35, 851 (1943).
95. Griswold J., Andres D., Arnett E. F., Garland F. M_, Ind. Eng. Chem., 32, 878 (1940).
96. Griswold J., J. A. Dinwiddie, Ind. Eng. Chem., 34, 1188 (1942).
97. Guinot H., F. W. Clark, Trans. Inst. Chem. Enp,. 16. 189 (1938).
98. Haldane T. G. N„ J. Inst. Elec. Eng, 68, 666—675 (1930).
99. Haltenberger W., Jr., Ind. Eng. Chem., 31, 783—786 (1939).
100. Hausen* H., Forsch. Qebiete Ingenieurw., 2, 319—326 (1931).
101. Hausen H., Forsch. Qebiete Ingenieurw., 613,9—22 (1935).
102. Hausen H., Forsch. Qebiete Ingenieurw., 7, 177 (1936).
103. Havlicek J., L. Miskovsky, Helvetica Phys. Acta, 9,161—207 (1936).
104. Heck R. С. H., Meeh. Eng., January, p. 9—12 (1940).
* 105. Henglein F. A., Zts. f. Elektroch., 26, 431 (1920).
* 106. Henglein F. A., Zts. f. phys. Chem., 98,1 (1921).
* 107. Herz W., Zts. f. anorg. u.‘ allg. chem., 124, 56 (1922).
108. Hildebrand J. H., J. Am. Chem. Soc., 37. 970 (1915).
109. Hoge H. J., F. G. Brickwedde, Bur. Standards J. Research, 22, 351 (1939).
110. Holborne L., J. Otto, Z. Physik, 33, 1 (1925).
111. Horsley L. H„ Anal. Chem., 19, 508—600 (1947).
112. Horsley L. H„ E. C. Britton, H. S. Nutting, Anal. Chem., 19, 601—602(1947).
113. Horsley L. H„ H. S. Nutting, Anal. Chem., 19, 602—603 (1947).
114. Horsley L. H., Anal. Chem., 19, 603—609 (1947).
115. Hudson C. S., Z. physik. Chem., 47, 113—115 (1903).
116. Jackson D. H., Ind. Eng. Chem., 28, 522—526 (1936).
117. Jakob M„ Z. Ver. deut. Ing., 56, 1980—1988 (1912).
117a. Jessen, F. W., J. H. Lightfoot, Ind. Eng. Chem., 28, 840 (1936).
118. Jones G. W„ Lewis B., Friauf J. B., Perrott St. J., J. Am. Chem. Soc., 53, 869 (1931).
119. Just! E_, H. Liider, Forsch. Qebiete Ingenieurw., 6, 211 (1935).
120. Kalustian P„ Refrigerating Eng., 28, 188—193 (1934).
121. Kapitza P., Proc. Roy. Soc., A147, 189 (1934).
122. Katz D. L., F. Kurata, Ind. Eng. Chem., 32, 817 (1940).
123. Katz D. L„ C. D. Singleterry, A. J. M. E. Tech. Pub., 971 (1938).
124. Kay W. B„ Ind. Eng. Chem., 28, 1014—1019 (1936).
125. Kay W. B„ Ind. Eng. Chem., 30, 459—465 (1938).
126. Kearton W. J., Turbo-blowers and Compessors, Sir Isaac Pitman a. Sons, Ltd. London (1926).
127. Keenan J. H., Meeh. Eng„ 54, 195—204 (1932).
128. Keenan J. H., Meeh. Eng., 48, 144—160 (1926).
129. Keenan J. H., F. G. Keyes, Thermodynamic Properties of Steam, N.-Y, 1936.
130. Kelley К- K., High Temperature Specific Heat Equations for Inorganic
Substances, U.S. Bur. Mines.Bull, 371(1934); The Entropies of Inorganic Substances, U.S. Bur.Mines Bull., 350 (1932); U.S. Bur.Mines Bull.,393 (1936).
130a. Kelley К. K-, J- Am. Chem. Soc., 51, 180—187 (1929).
130b. Kelley К. K., Ind. Eng. Chem., 21, 353—354 (1929).
131. Keyes F. G., Proc. Nat. Acad. Sci., 3, 323—330 (1917).
132. Keyes F. G., L. B. Smith, H. T. Gerry, Proc. Am. Acad. Arts Sci., 70, 319—364 (1936).
Литература
755
133. Keyes F. G., R. L. Taylor, L. B. Smith, J. Math. Phys., 1, 211—242 (1922).
134. Kirschbaum E„ For sc h. Gebiete Ingenieurw., 8, 63 (1937).
135. Kirst W. E., W. M. Nagle, J. B. Castner, Trans. Am. Inst. Chem. Eng., 36, 371—390 (1940).
136. Klobble E. A., Z. physik. Chem., 24, 615 (1897).
137. Knoblauch O., W. Koch., Z. Ver. deut. Ing., 72, 1733—1739 (1928).
138. Knowlton J. W., F. D. Rossini, Bur. Standard. J. Research, 22, 415 (1939).
139. Kuenen J. P., Theorie der Verdampfung und Verflilssigungvon Gemischen, und der fraktionierten Destination, J. A. Burth., Leipzig (1906).
140. Kvalnes H. M„ V. L. Gaddy, J. Am. Chem. Soc., 53, 397—399 (1931).
141. Lane С. T., Rev. Set. Instruments, 12, 326—331 (1941).
142. Lappie С. E., Trans. Am, Inst. Chem. Eng., 39, 385—428 (1943).
143. Larson A. T„ J. Am. Chem. Soc., 46, 367—372 (1924).
144. Larson A. T„ R. L. Dodge, J. Am. Chem. Soc., 45, 2918—2930 (1923).
145. Lawless A. J., Heating, Piping and Air Conditioning, August and September 1940.
146. Lees S. C., J. Phys. Chem., 35, 3558 (1931).
147. Lehfeldt R. A., Phil. Mag. (5), 40, 397 (1895).
148. Levy R. M„ Ind. Eng. Chem., 33, 928 (1941).
149. Lewis G. N„ Proc. Am. Acad. Arts Sei., 37, 49 (1901).
150. Lewis G. N., R. E. Gibson, J. Am. Chem. Soc., 39, 2554 (1917).
151. Lewis W. K., Jr., Ind. Eng. Chem., 28, 399 (1936).
152. Lewis W. K., Ind. Eng. Chem., 14, 492—497 (1922).
153. Lewis W. K., J. S. Carey, Ind. Eng. Chem., 24, 882 (1932).
154. Lewis W. K., W. C. Kay, Oil Gas J., 32, No. 45, 40, 114 (1934).
155. Lewis W. К., C. D. Luke, Trans. A. S. M. E. (P. M. E. 54—8), 55—61 (1932).
156. Lewis W. K-, E. V. Murphree, J. Am. Chem. Soc., 46, 1—7 (1924).
157. Lewis W. K„ H. Weber, Ind. Eng. Chem., 14, 485 (1922).
158. Lobo W. E., Friend L., Skaperdas G., Ind. Eng. Chem., 34, 821 (1942).
159. Lotka A. J., Elements of Physical Biology, The Williams Wilkins Company, Baltimore (1925).
160. McAdams W. H., J. C. Morrell, Ind. Eng. Chem., 16, 375 (1924).
161. McCabe W. L., E. W. Thiele, Ind. Eng. Chem., 17, 605 (1925).
162. Macintire H. J., Chem. Met. Eng., 45, 682 (1938).
163. Mackey B. H„ N. W. Krase, Ind. Eng. Chem., 22, 1060 (1930).
164. Margules M., Sitzb. Akad. Wiss. Wien, Math. Naturw. Klasse, 104. 1243 (1895).
165. Maron S. H., D. Turnbull, Ind. Eng. Chem., 33, 408 (1941).
166. Maron S. H., D. Turnbull, Ind. Eng. Chem., 34 , 544 (1942).
167. Masson L, L. G. Doley, Proc. Roy. Soc. (London), 103A, 524 (1923).
168. Meissner H. P., E. M. Redding, Ind. Eng. Chem., 34, 521 (1942).
169. Meissner W., Ind. Eng. Chem., 33, 1440—1443 (1941).
170. Merkel F-, Z. Ver. deut. Ing., 72, 109 (1928); Zeit. ges. Khlte. Ind., 35, 130 (1938).
171. Millar R. W., J. D. Sullivan, Thermodynamic Properties of Oxygen and Nitrogen, Bur. Mines Tech. Paper, 424 (1928).
172. Mollier R„ Z. Ver. deut. Ing., 48, 271—275 (1904).
173. Molstad M. C., F. B. Varga, Chem. Met. Eng., 44, 143 (1937).
174. Morgen R. A., Ind. Eng. Chem., 34, 571—574 (1942).
175. Newton R. H„ Ind. Eng. Chem., 27, 302 (1935).
176. Newton R. H„ B. F. Dodge, Ind. Eng. Chem., 27, 577 (1935).
177. Newton R. H„ B. F. Dodge, J. Am. Chem. Soc., 56, 1287—1291 (1934).
177a. Oestrich, Forsch. Gebiete Ingenier, 7, 287—291 (1936).
178. Osborne N- S., Bur. Standards J. Research, 4, 609—629 (1930).
179. Osborne N. S., H. F. Stimson, E. F. Fiock, Bur. Standards J. Research, 5. 411—480 (1930).
49 Химическая термодинамика
756
Литература
180. Osborne N. S., H. F. Stimson, D. C. Ginnings, Meeh. Eng., 57, 162 (1935); Bur. Standards J. Research, 18, 389—447 (1937).
181. Othmer D. F., Ind. Eng. Chem., 32, 841 (1940).
182. Othmer D. F., Ind. Eng. Chem., 34, 1072 (1942).
183. Othmer D. F., Ind. Eng. Chem., 27, 250 (1935).
184. Othmer D. F., Ind. Eng. Chem., 28, 1435 (1936).
185. Page L., Introduction to Theoretical Physics., Chap. VIII, D. Van Nostrand Company, Inc., New York (1928).
186. Page R. T., Ind. Eng. Chem., Anna], ed., 7, 355—358 (1935).
187. Parks G. S„ Ind. Eng. Chem., 29, 845—846 (1937).
188. Parks G. S., Chem. Revs., 18, 325—334 (1936).
188a. Perry T. H., Chemical Engineer’s Handbook, 2-d ed., N.-Y, 1941.
189. Plank R., Z. Ver. deut. Ing., 60, 187—193 (1916).
190. Plank R., J. Kuprianoff, Z. ges. Kalts Ind., Beihefte 1, 9—65 (1929).
191. Ponchon M., Tech. Moderne, 13, 20—24, 55—58 (1921).
192. Prosen E. J. R„ F. D. Rossini, Bur. Standards J. Research, 27, 289 (1941);
519 (1941).
193. Ramsperger H. С., C. W. Porter, J. Am. Chem. Soc., 48, 1267—1273(1926);
50, 3036—3038 (1928).
194. Randall M., B. Longtin, Ind. Eng. Chem., 30, 1063—1067, 1188—1192, * 1311—1315 (1938).
195. Redlich О., P. W. Schutz, J. Am. Chem. Soc., 66, 1007 (1944).
196. Refrigerating Data Book. Third edition, American Society of Refrigerating * Engineers, New York (1936).
197. Rechenberg C., Einfache und fraktionierte Destination, in Theorie und Praxis, Lpz (1923).
198. Robinson H. M„ H. Bliss, Ind. Eng. Chem., 32, 395—398 (1940).
199. Roebuck J. R„ Proc. Am. Acad. /Irfs. S«., 60, 537—596 (1925); 46,287(1930).
200. Roebuck J. R„ Proc. Am. Acad. Arts. Sci., 64, 287—334 (1930).
201. Roebuck J, R., H. Osterberg, Phys. Rev., 48, 450 (1935).
202. Roehl E. J., Ind. Eng. Chem., 30, 1320—1322 (1938).
203. Roeser W. F.,H. T. Wensel.Bur. Standards J. Research, 14,247—282(1935).
204. Rosanoff M. A., C. W. Bacon, J. F. Schultze, J. Am. Chem. Soc., 36, 1993—2004(1914).
205. Rossini F. D., Bur. Standards J. Research, 6, 791 (1931).
206. Rossini F. D., Chem. Rev., 27, 1—15 (1940).
207. Rossini F. D„ Ind. Eng. Chem. 29, 1424 (1937).
208. Rossini F. D„ Bur. Standards J. Research, 6, 1—35 (1931).
209. Rossini F. D., Bur. Standards J. Research, 6, 37—49 (1931).
209a. Rossini F. D„ Bur. Stand. J. Res., 8, 119—139 (1932).
210. Rossini F. D„ Proc. Nat. Acad. Sci., 17, 343—347 (1931).
211. Rossini F. D„ Bur. Standards J. Research, 13, 189—202 (1934).
212. Rossini F. D., J. W. Knowlton, Bur. Standards J. Research, 17, 635 (1938).
213. Rothmund V., Z. physik. Chem., 26, 461 (1898).
214. Ruhemann M., J. Inst. Petroleum, 28, 215—239 (1942).
215. Ruhemann M., The Separation of Gases, Oxford, Clarendon Press, New York (1940).
216. Ruhemann M., B. Ruhemann, Low Themperature Physics, Cambridge University Press, London (1937).
217. Rumford В. T., An Inquire Concerning the Source of the Heat Which is Excited by Friction. Phil. Trans, 88, 80—102 (1798).
218. Sage В. H., H. D. Evans, W. N. Lacey, Ind. Eng. Chem., 31, 763 (1939).
219. Sage В. H., B. L. Hicks, W. N. Lacey, Статья, представленная на 8-м полугодовом собрании А. Р. J., май 24, 1938.
220. Sage В. Н, Е. R. Kennedy, W. N. Lacey, Ind. Eng. Chem., 28, 601 (1936).
221. Sage В. H., W. N. Lacey, Ind. Eng. Chem., 30, 1296 (1938).
Литература
757
222. Sage В. Н., W. N. Lacey, Статья, представленная на 22-м годичном собрании А. Р. J., ноябрь 7, 1941.
223. Sage В. Н., R. Н. Olds, W. N. Lacey, Перепечатка для 23-го годичного собрания А. Р. J., ноябрь 8, 1942.
224. Sage В. Н., J. G. Schaafsma, W. N. Lacey, Ind. Eng. Chem., 26, 1218—1224 (1934).
225. Savarit R„ Arts et metiers, pp. 65, 142, 178, 241, 266, 307 (1922).
226. Scatchard G., C. L. Raymond, J. Am. Chem, Soc., 60, 1278—1287 (1938).
227. Scatchard G., S. E. Wood, J. M. Machel, J. Phys. Chem., 43, 119—130 (1939).
228. Schottky W., H. Ulich, C. Wagner, Thermodynamik, Berlin, 1929.
229. Schreinemakers F. A„ Z. physik, Chem., 35, 549—579 (1900).
230. Sherwood T. K-, Absorption and Extraction. McGraw-Hill, Book Co., Inc., New York (1937).
231. Smith E. S„ Jr., Trans. A. S. M. E„ 52, HYD-52-7b, 89—109 (1930).
232. Smith L. B., J. A. Beattie, W. C. Kay, J. Am. Chem. Soc., 59, 1587—1589 (1937).
233. Smith L. B., F. G. Keyes, Proc. Am. Acad. Arts Sci., 69, 285—314 (1934).
234. Smith L. B., F. G. Keyes, H. T. Gerry, Proc. Am. Acad Arts Set., 69, 137—168 (1934).
235. Smith R. L., К. M. Watson, Ind. Eng. Chem., 29, 1408—1414 (1937).
236. Smoker E. H„ Trans. Am. Inst. Chem. Eng., 34, 165—172 (1938).
237. Souders M., Jr., C. W. Selheimer, G. G. Brown, Ind. Eng. Chem., 24, 517 (1932).
238. Spencer H. M., J. N. Flannagan, J. Am. Chem. Soc., 64, 250—253 (1942).
239. Spencer H. M., J. L. Justice, J. Am. Chem. Soc., 56, 2311—2312 (1934).
240. Sporn P., D. W. McLenegan, Heating, Piping and Air Condit., 402 (1935).
241. Stickney A. B., Refrigerating Eng., 24, 334—342 (1932).
•242. Temperature, Its Measurement, Methods Control in Science and Industry, Am. Inst, of Physics and N. B. S. Res. Coun. Reinholds Publishing Cor-porat 1941.
243. Thiele E. W., W. B. Kay, Ind. Eng. Chem., 25, 894—898 (1933).
244. Thomas C. L., G. Egolff, J. C. Morrell, Ind. Eng. Chem., 29, 1260—1267 (1937).
245. Thompson M. de K., Electrochem. Soc. Preprint., 82—8, Oct. 12, 1942.
*246. Thomson G. W„ Chem. Rev., 38, 1 (1946).
247. Thomson G. W., H. A. Beatty, Ind. Eng. Chem., 34, 1124 (1942).
248. Tunnell G„ J. Phys. Chem., 35, 2885—2913 (1931).
249. Van Nuys С. C„ Chem. Met. Eng., 28, 207—210, 255—256, 311—313. 359—362 (1923).
250. Walker W„ K. Lewis, W. H. McAdams, E. R. Gilliland, Principles of Chemical Engineering, 3rd ed., New York (1937).
251. Waterfill R. W„ Ind. Eng. Chem., 24, 616 (1932).
252. Watson К. M., Ind. Eng. Chem., 23, 360 (1931).
253. Watson К. M., Ind. Eng. Chem., 35, 398 (1943).
254. Watson K. M„ E. F. Nelson, Ind. Eng. Chem., 25, 880—887 (1933).
255. Watson К. M., R. L. Smith, Nat. Petroleum News, July 1936.
256. Weber H. C., Trans. Am. Inst. Chem. Eng., 34, 569 (1938).
257. Whitwell J. C„ Ind. Eng. Chem., 30, 1157—1161 (1938).
258. Wien W., F. Harms, Handbuch der Experimental Physik, vol. IX, Leipzig (1929); раздел Gasverflussigungund ihre thermodynamischen Grundlagen, p. 47—185.
259. Williams V. C., Trans. Am. Inst. Chem. Eng., 39, 93—111 (1943).
260. Wohl A., Z. Physik. Chem., 87, 1—39 (1914).
261. Work L. T., V. W. Haedrich, Ind. Eng. Chem., 31, 464—477 (1939).
262. York R., Ind. Eng. Chem., 32, 54—56 (1940).
263. York R., Ind. Eng. Chem., 34, 535—544(1942).
49*
758
Литература
264. Young S., Distillation Principles and Processes, New York (1922k
265. Zawidski J., Z. Physik. Chem., 35, 129 (1900).
266. Zetnansky M. W., Heat and Thermodynamics, 2d ed. McGraw-Hill Book, Inc., New York (1943).
Б. Список авторов в порядке русского алфавита
267. Алексеев В. Ф., Горный журнал, № 6 (1885).
268. Баранаев М. К., Журн. физ хим.., 9, 69 ( 937).
269. Бачинский А. И., Изе. физ. Ин-та, 2, 60 (1922).
270. Богуславский С. А., Научные известия, 1 (1922).
271. Бродский А. И., Физическая химия, т. I и И, Госхимиздат, 1948.
272. Бриджмен П. В., Физика высоких давлении, ОНТИ, 1935.
273. Бриджмен П. В., Новейшие работы в области высоких давлений, Гос-иноиздат, 1948.
274. Ван-дер-Ваальс И. Д., Констамм Ф., Курс термостатики, ч. I и II, Госхимиздат, 1936.
275. Варгафтик Н. Б., Известия ВТИ, № 9, 16 (1947).
276. Винер Р. Р., Термохимические расчеты, Госиноиздат, 1949.
277. Виноградов Г. В., Красильщиков А. И., Атлас номограмм по физической химии, Техтеорет, М. — Л., 1940.
278. Волков В. Л., Жаворонков Н. М., Хим. пром., № 9, 12—15 (1947).
279. Вревский М. С., ЖРФХО. ч. хим., 42, 1 (1910).
280. Вревский М. С., ЖРФХО, ч. хим., 42, 702 (1910).
281. Вревский М. С., ЖРФХО, ч. хим., 59, 69 (1927).
282. Вревский М. С., ЖРФХО, ч. хим., 61, 1875 (1929).
283. Вревский М. С., Фаерман Г. П., ЖРФХО, ч. хим., 61, 1897 (1929).
284. Вукалович М. П., Термодинамические свойства водяного пара (таблицы и диаграммы), Госэнергоиздат, 1946.
285. Вукалович М. П., Новиков Н. И., Уравнение состояния реального газа, Госэнергоиздат, 1948.
286. Гамбург, Д. Ю., Нефт. хоз. № 9, 50 (1947).
287. Гельперин Н. И., Дестилляция и ректификация, Госхимиздат, 1947.
288. Гелипарин Н. И., Выпарные аппараты, Госхимиздат, 1947.
289. Гельпарин Н. И., Кислород, № 4 (1947); № 8 (1948).
290. Герасимов Я. И., Крестовников А. И., Химическая термодинамика в цветной металлургии, ч. I—III, ОНТИ, 1933—1934.
291. Герш С. Я., Эффективные циклы глубокого охлаждения и новые принципы разделения воздуха, Машгиз, 1946.
292. Герш С. Я., Разделение газов методом глубокого охлаждения, Гос-топтехиздат, 1947.
293. Герш С. Я., Глубокое охлаждение. Советская наука, 1947.
294. Гетман Ф. и Даниельс Ф., Основы физической химии, Госхимиздат, 1941.
295. Голубев И. Ф., Кульчицкий Н. В., Журн. хим. пром., № 6, 36 (1938).
296. ГОСТ 3270 — 45. Основные расчетные понятия термодинамики.
297. Гохштейн Д. П., Журн. тех. физ., 9, 707 (1939).
298. Гребенщиков Б. Н., Журн. физ. хим., 6, 944 (1935).
299. Гуггенгейм Э. А., Современная термодинамика, Госхимиздат, 1941.
300. Давтян О. К., Проблема непосредственного превращения химической энергии топлива в электрическую, Изд-во АН СССР, 1947.
301. Иваненко Д. Д., Успехи физ. наук, 32,149 (1947).
302. Иваненко Д. Д., Успехи физ. наук, 32, 261 (1947).
303. Казанский Б. А., Коперина А. В., Батуев М. И., Доклады АН СССР, 56, 819 (1947).
304. Казанский Б. К., Либерман А. Л., Сергиенко С. Р., Тарасова Б. А., Платэ А. Ф., Журн. общ. хим., 12, 112 (1947).
Литература
759
305 Казанский Б. А., Либерман А. Л., Стерлигов О. Д., Журн. общ. хим. 13, 125(1943).
306. Казанский Б. А., Розенгарт М. И., Журн. общ. хим., 12, 246 (1942).
307. Казанский Б. А., Розенгарт М. И., Соловова О. П., Изв. АН СССР, отд. хим. наук, 97 (1947).
308. Казарновский Я. С., Журн. физ. хим., 14, 1640 (1940).
309. Казарновский Я. С., Журн. физ. хим., 18, 364 (1944).
310. Казарновский Я. С., Карапетьянц М. X., Журн. физ. хим., 15, 966—973 (1941).
311. Капица П. Л., Журн. тех. физ., 9, 2, 99 (1939).
312. Канустинский А. Ф., Термодинамика химических реакций и ее применение в неорганической химии, Изд. 2-е, ОНТИ, 1935.
313. Капустинский А. Ф., Ядимирский К. Б., Журн. физ. хим., 22, 1271 (1948).
314. Карапетьянц М. X., Хим. пром., № 6, 21 (1945).
315. Карапетьянц М. X., Нефт. хоз., № 10, 50 (1947).
315а. Касаткин А. Г., Основные процессы и аппараты химической технологии, Госхимиздат. 1948.
316. Киреев В. А., ЖРФХО, ч. хим., 61, 2331 (1929).
317. Киреев В. А., Журн. физ. хим., 1, 233 (1930).
318. Киреев В. А., Журн. физ. хим., 2, 233 (1931).
319. Киреев В. А., Журн. общ. хим., 3, 622 (1933).
320. Киреев В. А., Журн. физ. хим., 14, 1475 (1940).
321. Киреев В. А., Журн. физ. хим., 15, 451 (1941).
322. Киреев В. А., Сборник работ по физической химии, 181 (1947).
322а. Киреев В. А., Сборник работ по физической химии, 127 (1947).
323. Киреев В. А., Каплан С. И„ Злобин В. Н., Журн. пром, хим., 7, 133 (1934).
324. Кистяковский В., Уравнения и коэфициенты, определяющие свойства веществ при нормальном атмосферном давлении, и математический анализ химического равновесия, Пятигорск, 1922.
325. Кноблаух, Райш, Хаузен и Кох, Таблицы и диаграммы водяного пара, Госэнергоиздат, 1933.
326. Колосовский Н. А., Химическая термодинамика, Госхимиздат, 1932.
327. Колосовский Н. А., Термодинамические исследования, САОГИЗ, 1934.
328. Колосовский Н. А., Гребенщиков Б. Н., Журн. эксп. и теор. физ., 1, 179 (1931).
329. Колосовский Н. А., Меженин Н. С., Журн. общ. хим., 1 (63), 616 (1931).
330. Коновалов Д., Об упругости паров растворов, 1928.
331. Кричевский И. Р., Журн. физ. хим., 9, 659 (1937).
332. Кричевский И. Р., Журн. физ. хим., 14, 434 (1940).
333. Кричевский И. Р., Фазовые равновесия в растворах при высоких давлениях, Госхимиздат, 1946.
334. Кричевский И. Р., Большаков П. Е., Журн. физ. хим., 15, 184 (1941).
335. Кричевский И. Р., Гамбург Д. Ю., Журн. физ. хим., 17, 215 (1943).
336. Кричевский И. Р., Ильинская А. А., Журн. физ. хим., № 17, 225 (1943).
337. Кричевский И. Р., Казарновский Я. С., Журн. физ. хим., 6, 1320 (1935).
338. Кричевский И. Р., Казарновский Я. С., Журн. физ. хим.. 13, 378 (1939).
339. Кричевский И. Р., Марков В., Журн. физ. хим., 15 (1941).
340. Кричевский И. Р., Хазанова Н. Е., Журн. физ. хим., 13, 106 (1939).
341. Кричевский И. Р., Циклис Д. С., Журн. физ. хим., 17, 126 (1943).
342. Курбатов В. Я., Изв. технол. ин-та, 1, 1 (1927).
343. Курбатов В. Я., Труды ЛХТИ, 13, 3 (1947).
343а. Лебедев М. Е., Кислород, № 3 (1948).
344. Лебедев П. Н., Сэбр. соч., 1913.
345. Литвинов Н. Д., Журн. физ. хим., 13, 119(1939).
346. Литвинов Н. Д., Жчрн. физ. хим., 14, 552 (1940).
347. Литвинов Н. Д., Журн. физ. хим., 14, 782 (1940).
348. Лошаков Л. М., Труды МХТИ им. Менделеева, 7, 136 (1940).
760
Литература
349. Лукомский С. М., Журн. техн, физ., 12, 393 (1942).
350. Львов С. В., Хим. пром., № 6, 15 (1947).
351. Льюис Г. Н., Рендалл М., Химическая термодинамика, ОНТИ, 1936.
352. Малков М. П., Кислород, № 6 (1946).
353. Малков М. П., Павлов К. Ф., Справочник по глубокому охлаждению в технике, Гостехиздат, 1947.
353а. Мамедов А. М., Азерб. нефт. хоз., № 1—2, 27 (1947).
354. „Мезон", Сборн. статей под ред. И. Е. Тамма, Гостехиздат, 1947.
355. Менделеев Д. И., Основы химии, т. I, стр. 424, Госхимиздат, 1947.
356. Меныпуткин Б. Н., Труды М. В. Ломоносова по физике и химии. Изд-во АН СССР, 1936.
357. Михельсон В. А., Собр. соч., т. 1, стр. 319, 1930.
358. Нернст В., Теоретические и опытные основания нового теплового закона, ГИЗ, 1929,
359. Оболенцев Р. Д., Фрост А. В., Нефт. хоз., № 8, 35 (1947).
360. ОСТ ВКС 6394.
361. Партингтон Д. Р., Раковский А. В., Химическая термодинамика, ГОНТИ, 1932.
362. Перри Д. Г., Справочник инженера-химика, т. I, ОНТИ, 1937.
363. Пиатти Л., Рекуперация летучих растворителей, Госхимиздат, 1934.
364. Планк М., Термодинамика, Госиздат, 1925.
365. Попов М. М., Термометрия и калориметрия, Госхимтехиздат, 1934.
366. Путилов К. А., Изв. АН СССР, ч. хим., № 4 (1937).
367. Путилов К. А., Лекции по термодинамике, вып. 1—5, Всесоюзное хим. об-во им. Д. И. Менделеева, М., 1939.
368. Раковский А. В., Курс физической химии, Госхимиздат, 1939.
369. Раковский А. В., Введение в физическую химию, Госхимиздат, 1938.
370. Розен А. М., Журн. физ. хим., 19, 469 (1945).
370а. Розен А. М., Диссертация, Моск, ин-т хим. машиностр., Москва, 1941.
371. Розен А. М., Хим. пром., № 9 (1945).
372. Розенгарт М. И., Успехи хим., 17, 204 (1948).
373. Скляренко С. И., Баранаев М. К., Журн. физ. хим., 6, 1192 (1935).
374. Скляренко С. И., Баранаев М. К., Журн. физ. хим., 6, 1180 (1935).
375. Скляренко С. И., Баранаев М. К., Журн. физ. хим., 6, 1204 (1935).
376. Скляренко С. И., Баранаев М. К., Журн. физ. хим., 12, 271 (1938).
377. Скляренко С. И., Баранаев М. К., Межуева К. П., Журн. физ. хим.. 14,
839 (1940).
377а. Стэлл Д. Р., Таблицы давления паров индивидуальных веществ, Ино-издат, 1949.
378. Сыркин Я. К., Дяткина М. Н., Химическая связь и строение молекул, Госхимиздат, 1947.
379. Сторонкин А., Журн. физ. хим., 15, 50 (1941).
380. Сторонкин А., Журн. физ. хим., 15, 68(1941).
381. Сторонкин А., Журн. физ. хим., 15, 159 (1941).
381а. Сторонкин А., Об условиях термодинамического равновесия многокомпонентных систем, изд. Ленингр. Гос. Орд. Ленина университета' 1948.
382. Тарасов В. В., Докл. АН СССР, 46, 22 (1945).
383. Тарасов В. В., Докл. АН СССР, 46, 117 (1945).
384. Тейлор X. С., Физическая химия, Химтеорет, т. I, 1935; т. II, 1936.
385. Темкин М. И., Журн. физ. хим., 17, 269 (19(3).
386. Темкин М. И., Журн. физ. хим., 17, 414 (1943).
387. Темкин М. И., Журн. физ. хим., 19, 72 (1945).
388. Темкин М. И., Шварцман Л. А., Успехи хим., 17, № 2, 259 (1948).
389. Терминология технической термодинамики (проект), АН СССР, Комитет технической терминологии, 1948.
Литература
761
390. Тиличеев М. Д., Давление насыщенного пара углеводородов. Физико-химические свойства индивидуальных углеводородов, вып. П, Гостех-издат, 1947.
391. Тимрот Д. Л., Варгафтик Н. Б., Ривкин С. Л., Известия ВТИ, № 4, 391а. Тимрот Д. Л., Изветия ВТИ, № 1, 1 (1949).
392. Трегубов А. Н., Теория перегонки и ректификации, Гостоптехиздат, 1946.
393. У лих Г., Химическая термодинамика, Госхимтехиздат, 1933.
394. Фастовский В. Г., Разделение газовых смесей, Гостехиздат, 1947.
395. Федоров В. С., Испарение и ректификация, АЗГОН Т И, 1Д.9.
396. Фрост А. В., Тепловые и термодинамические свойства углеводородов. Физико-химические свойства индивидуальных углеводородов, вып. I, гл. 2, стр. 101—116, 132, Гостоптехиздат, 1945.
397. Хоуген О., Ватсон К., Физико-химические расчеты в технике, Госхимиздат, 1941.
398. Циклис Д. С., Диссертация, Гос. ин-т азота, Москва, 1943.
399. Циклис Д. С., Журн. физ. хим., 20, 181 (1946).
400. Циклис Д. С., Журн. физ хим., 21, 349 (1947).
401. Цыдзик В. Е., Бармин Е. Л. и Вейнберг Б. С., Холодильные машины и аппараты, Машгиз, 1946.
402. Шредер И. Ф., Горн, журн., № 11, 272 (1890).
403. Эйкен А., Курс химической физики, вып. I, II, III, Госхимтехиздат, 1933
ИМЕННОЙ УКАЗАТЕЛЬ
А
Адамс Л. Г. 175, 185, 752
Акерлоф Г. 459, 752
Алексеев В. Ф. 746, 758
Аллен 494
Амага Э. Г. И, 41, 214, 246, 248, 292
Андрес Д. 641, 754
Антуан С. 31, 752
Арнет Э. Ф. 641, 754
Астон Д. Г. 574, 752
Б
Балке В. Г. 238, 752
Баранаев М. К. 36, 758, 760
Бармин В. П. 761
Бартлетт Э. П. 213, 220, 248, 249, 253, 255, 257, 739, 752
Батуев М. И. 747, 758
Бачинский А. И. 733, 758
Бениевитц К. 437, 752
Беннинг А. Ф. 243, 752
Бернулли 370, 372, 386, 408
Бергело Д. 235, 236, 237
Бин Г. С. 388, 390, 752
Битти Г. А. 6, 9, 626, 629, 630, 633, 649, 681, 711, 757
Битти Д. А. 169, 240, 242, 243, 244, 249, 251, 255, 256, 257, 272', 274, 280, 289, 301, 357, 360, 739, 752, 757
Блисс Г. 272, 560, 733, 756
Богуславский С. А. 5, 733, 758
Больцман Л. 40, ПО, 129, 419
Большаков П. Е. 15, 16, 731, 733, 759
Бойль Р. 208, 210, 219, 220, 228, 230, 232, 234, 235, 241, 354
Бостон О. В. 383, 752
Боулс В. О. 577, 586, 754
Бошнякович Ф. 460, 715, 752
Бранч Д. Э. К. 752
Браун Г. Г. 218, 258, 259, 651, 653, 656, 657, 658, 660, 661, 752, 757
Браун Ф. 557, 578
Бриан В. М. Д. 434, 752
Бриджмен О. К. 9, 240. 242. 243. 244, 249, 251, 255, 256, 272, 274, 280, 289, 301, 360, 739, 752
Бриджмен П. В. 212, 758
Брикуидди Ф. Г. 31, 754
Бринкворт Д. Г. 273, 752
Бриттон Э. К. 754
Бродский А. И. 5, 732, 745, 758
Бромилей Э. К. 632, 752
Букингем Э. 110, 388, 390, 752 Бурджэс Г. К. 49, 50, 753 -Буффиягтон Р. М. 242, 752 Быховский Ф. Р. 471, 752 Бэкон К. В. 626, 755
В
Вагнер К. 732, 757 ‘
Ван-дер-Ваальс И. Д. 220, 228, 229, 230, 231, 232, 234, 236, 237, 239, 243, 247, 248, 249, 250, 272, 288, 289, 305, 323, 324, 353, 357, 732, 734, 739, 750, 751, 758
Ван-Лаар Д. Д. 623, 624, 629, 630, 632
Ван-Нуис К. К. 542, 544, 757
Варга Ф. Б. 390, 400, 755
Варгафтик Н. Б. 736, 739, 758, 761
Вант-Гофф Г. Я. 82, 193
Вагсон К. М. 221, 222, 258, 296, 297, 298, 300, 441, 444, 579, 691, 743, 744, 757, 761
Вебег Г. К. 218, 296, 441, 670, 755, 757
Вейнберг Б. С. 761
Вензель Г. Т. 50, 755
Вентури 366, 373, 374, 375, 380, 381, 385, 388, 389, 392, 394, 400, 404
Вильямс В. К. 450, 451, 757
Вин В. 550, 757
Вииер Р. Р. 432, 758
Виноградов Г. В. 27, 758
Волаит М. А. 654, 753
Волков Б. Л. 746, 758
Воль А. 237, 238, 272, 757
Именной указатель
76з
Ворингер. 28
Вревский М. С. 36, 744, 758
Вукалович М. П. 5, 105, 117, 119, 271, 276, 281, 282, 298, 308, 311, 315, 333, 350, 365, 422, 669, 734, 736, 737, 738, 741, 742, 758
Г
Гамбург Д. Ю. 9, 333, 758, 759
Ганнес Р. К. 577, 586, 754
Гарлэнд Ф. М. 641, 754
Гедрих В. В. 350, 757
Гейланд. 538
Гельмгольц Г. 102, 131
Гельперин Н. И. 743, 747, 758
Генглейн Ф. А. 23, 754
Генри В. 41, 154, 174, 175, 198, 577
586, 587, 630, 632, 633, 690, 709, 712
Герасимов Я. И. 745, 758
Герингтон Э. Ф. Д. 36, 753
Гетман Ф. 732, 758
Герц В. 22, 754
Герш С. Я. 20, 354, 358, 497, 743, 744,
758
Гесс Г. Г. 5, 744
Гиббс Д. В. 85, 134, 158, 159, 162,
181, 621, 732, 754
Гиббсон Р. Д. 439, 755
Гильдебранд Д. Г. 441, 444, 685, 754
Гипо Г. 667, 754
Голицын Б. Б. 5
Голубев И. Ф. 738, 758
Гольборн Л. 227, 754
Гордон Д. Г. 744, 754
Гохштейн Д. П. 744, 758
Гребенщиков Б. П. 740, 758, 759
Грисволд Д. 641, 646, 754
Гуггенгейм Э. А. 732, 753, 758
Гудзон К. С. 635, 754
Гудри 424
Гудинаф. 238
Гэдди В. Л. 292, 755
Гэй Н. Р. 280, 299, 753
Гюккель Э. 454
Д
Давтян О. К. 732, 758
Дальтои Д. 7, 246, 247, 253
Дана Л. Д. 242, 307, 753
Даниельс Ф. 732, 758
Де Бак Э. Э. 619, 753
Дебай П. 439, 454, 743
Девис Г. Н. 38, 238, 753
Девис Д. С. 306
Деминг В. Э. 242, 243, 251, 255, 256 280, 281, 290, 291, 739, 753
Джерри Г. Т. 239, 261, 304, 754, 757
Джилкей В.'К. 242, 752
Джили ленд Э. Р. 251, 577, 586, 735, 754, 757
Джиллеспай Л. Д. 173, 563, 564, 754
Джиннингс Д. К. 261, 572, 579, 753, 756
Джиок В. Ф. 109, НО, 207, 242, 572, 575, 753, 754
Джонс Г. В. 480, 754
Джоуль Д. П. 65, 68, 84, 85, 86, 97, 139, 210, 211, 225, 235, 261, 285, 286, 298, 299, 314, 351, 355, 356, 358, 360, 361, 362, 363, 524
Джэкоб М. 526, 533, 754
Джэксон Д. Г. 515, 754
Димок Д. Б. 560
Динуидди Д. А. 646, 754
Дитеричи 235, 236, 237
Добратц К. Д. 437, 753
Додж Б. Ф. 5, 7, 9, 218, 298, 362, 542, 543, 559, 560, 562, 564, 565, 573, 574, 586, 598, 614, 625, 651, 709, 712, 753, 755
Додж Р. Л. 562, 563, 755
Доллей Л. Г. 257, 755
Дрью Т. Б. 417, 753
Дунбар А. К. 543, 614, 625, 753
Дэви 68, 84, 185
Дюгем П. 154, 186, 190, 194, 621, 622, 623, 624, 626, 627, 628, 632, 633, 732, 753
Дюлонг. 438
Дюринг Е. 21, 22, 305, 459, 685
Дяткина М. Н. 744, 760
Ж
Жаворонков Н. М. 746, 758
Женеро Р. П. 407, 417, 753
3
Завидский Ж. 622, 758
Зельхаймер К. В. 651, 653, 656, 757
Земанский М. В. 110, 758
Злобин В. Н. 34, 759
И
Иваненко Д. Д. 732, 758
Ивелл Р. Г. 568, 753
Иессен Ф. В. 215, 754
764
Именной указатель
Икехара С. 255, 752
Ильинская А. А. 733, 759
Ингерсолл Г. Г. 292, 752
Иорк Р. 293, 333, 757
Истмен Э. Д. 434, 571, 598, 753
К
Казанский Б. А. 747, 758, 759
Казарновский Я. С. 733, 735, 736, 739, 744, 759
Калингерт Д. 306, 626, 629, 630, 633, 649 681 728 752
Каллендар 226
Калустиан П. 347, 754
Каминский Д. 242, 752
Капица П. Л. 527, 538, 754, 759
Каплан С. И. 34, 759
Капплс Г. Л. 213, 249, 752
Капустинский А. Ф. 5, 33, 732, 745, 759
Карапетьянц М. X. 744, 759
Карей Д. С. 755
Карлсон Г. К. 623, 626, 629, 753
Карно Л. Н. С. 85, 96, 97, 106, 107, 108, 111, 112, 115, 116, 121, 122, 124, 483, 484, 486, 487, 488, 489, 490, 492, 493, 495, 498, 502, 521, 522, 536, 543
Карр А. Р. 753
Касаткин А. Г. 692, 743, 759
Кастнер Д. Б. 424, 425, 755
Катц Д. Л. 619, 754
Квалнес Г. М. 213, 220, 292, 752, 755
Квиггл Д. 632, 752
Кей В. Б. 238, 258, 333, 616, 661, 752, 754, 757
Кей В. К. 242,653, 654, 655, 752, 755, 757 Кейес Ф. Г. 5, 9, 239, 240, 243, 261, 279, 280, 304, 306, 313, 315,736, 754, 755, 757
Келли К. К. 438, 440, 447, 572, 575, 754
Кельвин В, 85, 96, 97, 99, 102, 106, 107, 108, НО, 111, 521, 523 (см. также Томсон В.)
Кеннеди Э. Р. 286, 756
Кизон В. Г. 539
Кинен Д. Г. 5, 122, 123, 286, 315, 736, 754
Киреев В. А. 22, 24, 32, 33, 34, 739, 743, 744, 759
Кирст В. Э. 424, 425, 755
Киршбаум Э. 694, 755
Кистяковский В. А. 23, 441, 443, 579, 691, 759
Клапейрон 21, 30, 153, 187, 188, 189, 196, 209, 232, 263, 276, 302, 310, 311, 312, 314, 315, 316, 440, 441, 442, 443, 686, 740
Кларк Д. А. 539, 753
Кларк Ф. В. 667, 754
Клаузиус Р. 21, 30, 85, 95, 96, 97, 99, 102, 110, 114, 127, 153, 187, 188, 189, 196, 263, 302, 310, 311, 312, 314, 315, 316, 440, 441, 442, 443, 686.
Клейтон Д. О. 572, 753
Клоббл Э. А. 635, 755
Клод Ж. 527, 536, 538, 549
Кноблаух О 281 ?ЯЯ, 2Я4 7S5 750
Кноултон Д. В. 473,“ 575, 755, 756 ’ '
Кобэ Ф. 616, 753
Кокс Э. Р. 305, 306, 753
Колборн А. П. 623, 626, 629, 753
Колосовской Н. А. 5, 579, 740, 759
Коновалов Д. П. 5, 620, 621, 759
Констамм Ф. 732, 758
Коп Д. К. 218, 296, 753
Коперина А. В. 747, 758
Копсон Р. Л. 304, 753
Корнелл Л. В. 726, 753
Кох В. 281, 283, 284, 755, 759
Кразе Н. В. 280, 755
Красильщиков А. И. 27, 758
Крестовников А. И. 745, 758
Кричевский И. Р. 5, 9, 15, 16, 17, 731, 733, 735, 736, 739, 759
Кулей Л. К. 667, 752
Кулик Н. 280, 299, 753
Кульсон Э. А. 36, 753
Кульчицкий Н. В. 738, 758
Куприянов Ю. 324, 756
Курата Ф. 619, 754
Курбатов В. Я. 743, 759
Куэнен Д. П. 602, 617, 619, 620, 647, 755
Кэмиингс Л. В. Т. 654, 753
Кэртон В. Д. 320, 754
Л
Лайтфут Д. Г. 215, 754
Лан К. Т. 755
Лаппл К. Э. 412, 413, 755
Ларсон А. Т. 562, 563, 568, 755
Лебедев М. Е. 745, 759
Лебедев П. Н. 743, 759
Леви Р. М. 622, 755
Ледюк. 246
Лефел ьдт Р. А. 626, 755
Ле-Шателье А. 557
Ли С. К. 628, 755
Именной указатель
165
Либерман А. Л. 747, 758, 759
Линде К. 238, 278, 530, 538, 540, 546, 549
Литвинов П. Д. 745, 746, 759
Лобо В. Э. 410, 755
Ломоносов М. В. 731, 733
Лонгтен Б. 600, 756
Лоренц Р. 254
Лотка А. 94, 755
Лоулесс А. Д. 523, 755
Лоуренс К. К. 242, 752
Лошаков Л. М. 242, 760
Лугинин 5
Лукомский С. М. 740, 760
Львов С. В. 746, 760
Льюис Б. 480, 754
Льюис В. К. 218, 296, 441, 624, 626, 651, 653, 654, 655, 694, 709, 746, 753, 755, 757
Льюис Г. Н. 110, 146, 148, 149, 162, 289, 292, 294, 439, 598, 755, 760
Лэси В. Н. 259, 286, 301, 312, 654, 659, 660, 661, 756, 757
Людер Г. 434, 480, 754
Люк К. Д. 296, 651, 755
М
Майер Р. 84
Мак-Адамс В. Г. 441, 444, 746, 755, 757
Макинтайр Г. Д. 498, 505, 755
Маккей Б. Г. 280, 755
Мак-Кэб В. Л. 463, 707, 723, 755
Мак-Ленеган Д. В. 523, 757
>Максвелл К. 94, 95
Мак-Харнесс Р. К. 243, 752
Малков М. П. 744, 760
Мамедов А. М. 733, 734, 760
Маргулес М. 186, 621, 622, 623, 632, 746, 755
Марков В. 735, 759
Марон С. Г. 243, 300, 755
Массье. 102
Меженин Н. С. 579, 759
Межулева К. П. 36, 760
Мейсснер В. 443, 446, 755
Мейсснер Г. П. 222, 755
Менделеев Д. И. 733, 760
Меныпуткин Б. Н. 731, 733, 760
Меркель Ф. 460, 755
Миллар Р. В. 269, 273, 276, 315, 428, 450, 451, 466, 527, 528, 743, 755
Миллер Р. В. 539, 753
Мисковский Л. 272, 314, 754
Михельсон В. А. 744, 760
Милстед М. К. 390, 400, 755
Молье Р. 264, 268, 269, 270, 271, 496,
755
Монтониа Р. Э. 726, 753
Моргеи Р. А. 454, 462, 755
Моррел Д. 441, 444, 598, 755, 757
Морфри Э. В. 242, 624, 626
Мунтерс. 510, 512
Мурфи П. С. 389, 752
Мурфи Э. В. 753, 755
Мэкел Д. М. 612, 757
Мэссон Д. 257, 755
Н
Нельсон Э. Ф. 258, 297, 757
Нернст В. 304, 568, 580, 581, 732, 745, 760
Новиков Н. И. 734, 742, 758
Нуттинг Г. С. 754
Ньютон Р. Г. 216, 294, 295, 296, 362, 559, 565, 573, 651, 753
Нэгл В. М. 424, 425, 755
О
Оболенцев Р. Д. 747, 760
Олдс Р. Г. 301, 757
Осборн Н. С. 261, 314, 755, 756
Остерберг Г. 356, 756
Отмер Д. Ф. 24, 442, 444, 445, 667,
728, 743, 756
Отто Д. 227, 754
П
Павлов П. Ф. 744, 760
Паркс Г. С. 474, 561, 575, 586, 756
Партингтон Д. Р. 732, 760
Пельтье. 482
Перри Д. Г. 498, 511, 685, 726, 745,
756, 760
Перрот Г. С. Д. 480, 754
Пиатти Л. 26, 760
Пито. 96, 372, 373, 395, 396, 398
Планк М. 96, 99, 110, 569, 760
Планк Р. 284, 324, 756
Платен. 510, 512
Платэ А. Ф. 747, 758
Пойнтинг. 7, 200, 201
Понтон М. 460, 714, 756
Попов. 766
Портер К. В. 578, 756
Поффенбергер Н. 242, 752
766
Именной указатель
Прозен Э. Д. Р. 473, 756 Пти. 438 Пуазейль. 407 Пуассон. 9, 329 Путилов К. А. 5, 731, 760 Пэдж Л. 129, 474, 756 Пэдж Р. Т. 402, 756 Смит Л. Б. 239, 242, 261, 306, 754 755, 757 Смит Р. Л. 218, 258, 296, 297, 298, 300, 304, 752, 757 Смит Э. С. 387, 757 Смокер Э. Г. 709, 711, 713, 757 Соловова О. П. 747, 759 Спенсер Г. М. 434, 436, 757
Р Спорн П. 523, 757 Стаут Д. В. 242, 575, 754 Стенлей Г. М. 560
Райш О. Е. 283, 739 Раковский А. В. 5, 745, 760 ТЧ .4 Г» ОП'Т Г*<1МЗ<1П О. OUI Рамспергер Г. К. 578, 756 Рауль Ф. М. 153, 179, 191, 198, 577, 630, 639, 746 Реахенберг. 26, 756 Ребук Д. Р. 286, 361, 362, 756 Реддинг Э. М. 222, 755 Редлих О. 36, 756 Реймонд К. Л. 626, 627, 628, 757 Рейнольдс. 40, 366, 379, 380, 388. 396, 399, 407, 417 Рендалл М. 148, 289, 292,294, 598,600, 732, 756, 760 Ривкин С. Л. 739, 761 Робинсон Г. М. 272, 756 Розанов М. А. 626, 756 Розен А. М. 9, 10, 12, 13, 734, 736, 739 760 Розенгарт М. И. 747, 759, 760 Росснер В. 437, 752 Россини Ф. Д. 454, 471, 473, 474, 572, 574, 575, 586, 752, 755, 756 Ротмунд В. 634, 756 Руди К. Л. 437, 753 Руеманн Б. 550, 756 Руеманн М. 541, 556, 756 Румфорд. 68, 84, 731, 756 Рэзер В. Ф. 50, 756 Рэл Э. Д. 305, 756 Стерлигов О. .Д. 747, 759 Стефан. 40, 419 кАПКИИ А. О. UZU, UZ.1, Стимсон Г. Ф. 261, 314, 755, 756 Стон Ф. В. 654, 753 Сторонкин А. 733, 760 Стэлл Д. Р. 30, 757 Су Г. 242, 752 Сыркин Я. К. 744, 760 Сэдж Б. Г. 259, 286, 301, 312, 315, 654, 659, 660, 661, 756, 757 Сюлливан Д. Д. 269, 273, 276, 315 428, 450, 451, 467, 528, 537, 743. 755 Т Тарасов В. В. 743, 760 Тарасова Б. А. 747, 758 Тейлор Р. Л. 306, 755 Тейлор X. С. 732, 745, 760 Темкин М. И. 31, 32, 733, 735,736,760 Теннел Г. 148, 248, 757 Тиле Э. В. 258, 707, 723, 755, 757 Тиличеев М. Д. 31, 655, 761 Тимрот. 739, 761 Тир Д. 459, 752 Томас. 403, 404 Томас К. Л. 598, 757 Томсон В. 139, 210, 211, 225,235,261, 285, 286, 298, 304, 351, 355, 356, 357, 358, 360, 361, 362, 363, 524, 526,533 Томсон Г. В. 31, 711, 757
С Томсон М. К. 433, 757
Савари Р. 460, 714, 757 Саудерс М. 218, 651, 653, 656, 657, 658, 600, 601, 752, 757 Сегден С. 222 Сершенко С. Р. 747, 758 Симард Г. Л. 242, 752 Симс. 641 Синглетерри К. Д. 619, 754 Скаперда Г. Т. 410, 755 Скатчард Г. 612, 626, 627, 628, 757 Скляренко С. И. 36, 760 Торнбулл Д. 243, 300, 755 Трегубов А. И. 747, 761 Тримерн Т. Г. 213, 220, 249, 752 Трутон Ф. 22, 23, 440, 686, 699 У Уайт. 23 Уайтвелл Д. К. 395, 757 Уейнут. 410 Умов Н. А. 5
Именной указатель
767
Улих Г. 732, 757, 761 Уокер. 746, 757 Уорк Л. Т. 350, 757 Уотерфилд Р. В. 498, 757 Ууд С. Э. 612, 757 ц Циклис Д. С. 15, 17, 731, 759 Цыдзик В. Е. 744, 761 Ш
Ф Шаафсма Д Г. 315, 654, 757 Шарль. 208
Фаерман Г. П. 744, 758 Фаннинг. 41, 407, 410 Фарадей. 68 Фастовской В. Г. 744, 747, 761 Фаулер. 753 Федоров В. С. 747, 761 Фиок Э. Ф. 261, 314, 572, 579,752,755 Фланнаган Д. Н. 436, 757 Фрайнд Л. 410, 755 ФриуФ Д- В- 480, 754 Фролих П. К. 304, 753 Фрост А. В. 747, 760, 761 Фугасси П. 437, 753 Фуко. 426 Шварцман Л. А. 31, 32, 760 Шервуд Т. К. 653, 655, 661, 757 Шиллер Н. Н. 5 Шоттки В. 732, 757 Шрайнемакерс Ф. А. 634, 757 Шредер И. Ф. 733, 761 Штокмайер В. Г. 242, 257, 752 Шульце И. Ф. В. 626, 756 Шуп Л. Э. 242, 243, 251, 255, 256, 280, 281, 290, 291, 739, 753 Шутц П. В. 36, 756 Э Эванс Г. Д. 312, 756 Эглофф Г. 598, 757
X Эдмистер В. К. 272, 299, 300, 327, 435, 751, 753
Хавличек Ж. 272, 314, 754 Хазанова Н. Е. 8, 759 Хальтенбергер В. 459, 754 Хармс Ф. 550, 757 Хаузен Г. 283, 284, 647, 694, 754, 755 Хафман Д Р. 709, 712, 753 Хезерингтон Г. К. 213, 220, 752 Хек Р. К. 434, 754 Хикс Б. Л. 661, 756 Хог Г. Д. 51, 754 Холдан Т. Г. Н. 523, 754 Холтон В. Б. 572, 579, 753 Хорслей Л. Г. 754 Хосум К. 542, 753 Хоуген О. 743, 748, 761 Хэдлок К. 242, 752 Эйкен А. 732, 761 Эйлер Л. 155 Эйнштейн А. 437 Элленвуд Ф. О. 280, 299, 753 Эштрих. 364, 755 Ю Юел Д. Э. 560 Юнг С. 28, 307, 606, 758 Юсти Э. 438, 480, 754 Юстис Д. Л. 434, 757 Я Якоб М. 284 Яунмирской. 33, 759
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
А
Абсолютно-черное тело 420
Абсолютные, — давление 51
— нуль 109
— температура 78,106
— шкала температур 48
— энтропия 569
Абсорбционное охлаждение 508
Автотермическая реакция 589
Аддитивность, — давление 245, 247
—объемов 246, 248
— парахоров 222
— теплоемкостей 447
Адиабатные, — истечение 20, 283,404
— коэфициент полезного действия детандера 346
—• процесс 9, 11, 69, 324
— реакция, сравнение с изотермической 587
— —, пример расчета 588
Азеотропные смеси 613
----, влияние давления 614, 666
----, — температуры 35
----, — третьего компонента 36,666
-----, перегонка 667
----, разделение 666
----, состав 34—35
----, тройные 646
-----, условия образования 34
Активность 149, 175
—, влияние давления, температуры и состава 175
— компонента раствора 162, 172, 175, 180
—, коэфициент 204, 628
—, — газов 294, 654
—, — графики 628, 632
Алексеева правило прямой средней линии 746
Амага коэфициент сжимаемости 214
Амага — Ледюка закон аддитивности объемов 246
Атмосфера, нормальная (физическая) 36, 51 техническая 36, 51
Б
Бартлетта уравнение состояния 248
Бачинского—Сегдена правило 222
Бернулли теорема 310, 386, 408
Вертело уравнение состояния 236
Бинарные растворы (см. также Жидкость-пар, Равновесие, Растворы) -----, диференциальные испарение и конденсации 676
-----, интегральные испарения и конденсации 680
------, критические явления 615
------, уравнения для идеального раствора в обеих фазах 193
-----, — для равновесия идеального раствора и идеального газа 191
-----, — для фаз, состоящих из идеальных газов 188
Бинарный цикл 505
-----, пример расчета 560
Битти уравнение состояния 243
Битти — Бриджмена уравнение состояния 240
-----вычисление энтальпии с помощью его, пример 273, для смесей 248
----- таблица констант 242
Бойля закон 208
— критерий 227
— точка 219, 241
-----, вычисление с помощью приведенного уравнения состояния 207
Британская тепловая единица (BTV) 749
В
Вакуум 52
Вакуумные охлаждения 513 -----, пример расчета 515 Ван-дер-Ваальса уравнение состояния 228
— — — вычисление летучести, пример 288
— —• — вычисление объема, пример 233
П редметный указатель
769
Вин-дер-Ваальса вычисление работы, пример 323
-------- давление газовой смеси 247, 248
—-------- константы, вычисление 231
------------, таблица 749
—-------, приложимость его 233
Ван-Лаара решение уравнения Дюгема 622, 630
Вант-Гоффа «ящик равновесия» 82
Вентури измеритель 373 -----, потери давления 381
Вероятность и второй закон 93 Весовая доля 56
Вечный двигатель второго рода 97
Внутренняя энергия 86,103 (см. также
Энергия) ---------- влажного пара 302 ------ газовой смеси 168 ------двухфазной системы 302 ------, обозначения, применяемые различными авторами 732
----- перегретого пара 276
-----, пример расчета 276 ----- идеального газа 150 ---------- раствора 165 ------ уравнения 135
Водяной пар, аккумулятор 365
-----•, диаграмма, S — Н 269
-----, —, расчет свойств, примеры 270 -----, диаграмма S — Т 266 -----, расход при перегонке, пример 685
— —, таблицы свойств 736 -----, теплоемкость 280 Воздуха разделение 544 -----, процесс Линде 546 Воля уравнения состояния 237 Второй закон термодинамики 85, 91
-----—и вероятность 93 -----— и необратимые процессы 91 ----- и неупорядоченность 92 -----, история 85
--------, математическая формулировка 127
--------, нарушения в теплообменниках 427
--------, пример 428 --------, формулировка 95
Вторые производные, равенство нх 59
Вукаловича — Новикова уравнение состояния 741
Высота, эквивалентная единице переноса массы 694
—, эквивалентная теоретической тарелке 694
Г
Газовая постоянная 211
Газовые смеси, вычисление давления, пример 249
--------объема, пример 259
------, закон аддитивности давлений
245, 247, 253
----------объемов 246, 252
-----идеальные 244
--------, вычисление плотности, пример 246
------, коэфициенты сжимаемости 251
-----, критические давления и температура 258
— —, метод Джиллиленда для расчета сжимаемости 256
------, процессы разделения 540
-----, работа разделения 527
------, расслаивание 15, 54
— —, — в системе азот-аммиак 16
-----реальные 247
--------, вычисление давления, пример 249, 252, 256
-----, растворимость в жидкостях 206
-----, средний молекулярный вес 246
-----, уравнения состояния 245 (см. также Уравнение состояния)
Газы, дросселирование (см. Джоуля — Томсона эффект, Дросселлирова-ние)
— идеальные (см. Идеальные газы)
—, отклонение от идеальности 209
—, поток, измерение 383
—, влияние изменения состояния 393
—, максимальный расход 390
— , коэфициенты расширения 387
— , общие уравнения 383
— , расчетные уравнения 387
— , случай идеального газа 381
— поток в трубопроводах 409
—’ — максимальный 411
—, —, сравнение изотермического и адиабатного 412
—, растворимость в жидкостях под давлением 195, 733
— реальные (см. Реальные газы)
—, сжатие (см. Сжатие)
—, сжижение (см. Сжижение)
—, теплоемкость (см. Теплоемкость)
Генглейна уравнение 23
Генрн закон 174, 198, 630
Гесса закон 469
Гетерогенные реакции 584
Гиббса — Дюгема уравнение (см. Уравнение Дюгема)
Гомогенная газовая реакция 582
770
Предметный указатель
Гомогенная газовая реакции, пример расчета равновесного превращения 583
Графические — изображение работы 65
— методы вычисления термодинамических свойств 290
Гудинафа уравнение состояния 238
Д
Давление 51
— абсолютное 51
— барометрическое 52
— вакууметрическое 52
—, влииние на перегонку с водяным паром 686
—, — на равновесие пар — жидкость 614
—, — на растворимость 7, 195
—, — на теплоемкость 277
— ,— на энтальпию 296
— «внутреннее» 229
— единицы измерения 51, 36 .
— , закон аддитивности 247
— и напор 381
— критическое 222
— манометрическое 51
— насыщенного пара
— бензола 25
—-------, диаграмма Кокса 305
--------, графические расчеты 27
--------, линейная зависимость 1g р от 1 303
--------, методы сравнительного расчета 21, 739
--------, номограмма Киреева 29
--------, правило Дюринга 21, 22, 305
--------, различные уравнения 303
-----— сложных эфиров 26
--------смеси толуол-вода 604
--------, уравнение Антуана 31
--------, уравнение Ван-дер-Ваальса 305
------ —, уравнение Генглейна 23
--------.уравнение Киреева — Отмера 24, 739
—-------, уравнение Лукомского 740
--------, уравнение Нернста 305
— остаточное 363
— парциальное 190, 245
------компонента реальной смеси 248
— , пределы применения 52
— приведенное 216
— реальной газовой смеси 247
— сверхбарометрическое 51
Давление световое 419
— статическое, пример расчета 408
----, эквиваленты, таблицы 747
Дальтона закон аддитивности давлений 245
Двигатель, вечный второго рода 95
— солнечный 122
Движущая сила процессов 72
--------, связь с равновесием 75
---- химической реакции 552
Двухфазная область, вычисление свойств 301
Дебая — Гюккеля предельный закон 454
Дестиллат 665
Дестилляция (см. Испарение, Перегонка, Ректификации)
Детандер 320
— Кавицы 539
----, коэфициент полезного действия 346
Дефлегмации
— , расход тепла 715
Джиллилеида уравнение состояния 256
Джоуля закон 210
— опыт 65
----, связь с идеальным газом 210, 211
— эффект 363
----примера расчета 364
Джоуля — Томсона эффект (дросселирование) 210, 354
—-------, вычисление 354
----—, — аналитическим методом, примеры 360, 362
—-------, — по диаграмме S — Н 18
--------, примеры расчета 270, 358
--------, по коэфициенту активности 362
--------, изменение энтропии, пример 270
--------, изотермический (процесс)
352
----— интегральный 358
----—, качественные выводы 353
--------, коэфициент 139, 285, 355
------- —, кривая инверсии 356, 532
-------------, приведеннаи 358
--------, определение из него Ср. 286
— , получение низкой температуры 526
--------, поправка на кинетическую энергию 354
—-------, температура инверсии из
уравнения Битти 357
--------, точка инверсии 356
Диаграмма, давление — объем 65
Предметный указатель
771
Диаграмма, давление— состав (р—х)
—,------анилин — вода 642
—,------, идеальный раствор и иде-
альный газ 602
—,------, критическая область 618
—,------, неидеальные растворы 611
—, давление температуры для чистого вещества .262
—,------для бинарного раствора 615
— индикаторная 66
— Кокса 305
— констант фазового равновесия 658
--- для метана и пропана 659
— коэфициентов активности
— газов 296
-------- углеводородов 654
— работы сжатия 341
—, состав жидкости — состав пара
(у—Х)
—,-----------, идеальный раствор
609
—,-----------, критическая область
618
—,-----------, типичная 614
—,---------, тройная система 646
—,-----------, частично смешиваю-
щихся жидкостей 638
—, температура — состав (t—х)
—•------,бензол-толуол, пример 610
—,------, критическая область 618
—, — —, растворимые жидкости 613
—,------, тройные смеси 644
—,------, частично смешивающиеся
жидкости 634
— треугольная 643
---, состав жидкости — состав пара 646
---, температура — состав 634, 654
— энтальпия-состав (И—х) 658
--------для определения минималь-
ного флегмового числа 720
--------, построение 464
--------для определения числа тарелок 722
--------для растворов едкого натра
463
--------для смесей кислород-азот
466
--------, приложение к задачам смешения 460
--------, приложение к процессам перегонки 713
— энтропия-температура (S — Т) 265
-------- для водяного пара 266
--------для холодильного цикла сжа-
тия пара 495
-------- для углеводородов 310
--------для углекислоты (см. после стр. 748).
--------, расчет мощности одноступенчатого компрессора, пример 332
--------, расчет работы изотермического сжатия, пример 323
--------, расчет холодильной установки, примеры 501, 502, 530
—энтропия—энтальпия (Молье) (S—Н)
17, 264, 268
-------- для водяного пара 269
--------, процесс дросселирования 18
-------- истечения 20
--------фазовая для тронной смеси 645
Диафрагмовый измеритель 374
----, потеря давления 381
----, коэфициенты расхода для остроугольных измерителей 390
Дитеричи уравнение состояния 235
Диференциальное, испарение 664, 676
----, двухкомпонентные системы 676
----, многокомпонентные системы 678
----, расход тепла 716
— конденсация 664, 676
----в присутствии инертного газа 682
----, пример расчета 680
— уравнения для двухкомпонентных
двухфазных систем 181
---- однокомпонентной двухфазной системы 135
Дросселирование (см. Джоуля— Томсона эффект)
Дюгема уравнение (см. также Уравнение Дюгема) 186, 190, 194, 621
----. метод расчета Литвинова 745
----, применение к равновесиям при перегонке 625
----, проверка изобарных данных 628
Дюлонга и Пти правило 438
Дюринга правило 305
Е
Единицы давления 51, 748
— мощности 68
— работы 68
— теплоты 69
— энергии 749
— концентрации растворов 56
— переноса массы 694
тп
Предметный указатель
Ж
Жидкости, дросселирование 18
— истечение 375
— несмешивающиеся 604
— переохлажденные 260
—, полностью смешивающиеся 606
—, процессы дестилляции 662
—, теплоемкость 438
—, частично смешивающиеся 633
Жидкость-пар, равновесие, двухкомпонентные системы 604
--------в системе анилин-вода, пример 640
--------’ в системе метанол-бензол, 628
--------в системе толуол-н-октан, пример 630
--------, влияние давления 613
--------, исследование уравнения
Дюгема 632
--------, критические явления 615
---, многокомпонентные системы 648
----------, примеры расчета 649
--------неидеальные бинарные си-
стемы 620
----------------, законы Коновалова 620
----------------, уравнение Дюгема 621
----------------, эмпирические закономерности 625
---однокомпонентные системы 301
3
Закон (см. также Правила, Уравнение)
— Амага — Ледюка 246
— Бойля 208
— Генри J 74, 198, 630
— Гесса 469
— Дальтона 245
— Дебая — Гюккеля, предельный 454
— действующих масс 555
— Джоуля 210
— идеального газа 208
— Коновалова 620
— разбавленных растворов 196
— Рауля 179, 191, 198
— соответственных состояний 215, 234
— Стефана — Больцмана 419
— Шарля — Гей-Люссака 208
Задачи термодинамики в примеиении к химической технологии 45
И
Идеальные газы 208
Идеальные газы, вычисление свойств 149
-----, законы, отклонения от иих 211, 218
-----, основные уравнения 150, 321
-----,-----, пример расчета 150 -----------, поток 384 -----------, работа сжатия 322, 325 -----------,----------, пример расчета 323 -----------, равновесие 203
----- смеси 244
-----, уравнение состояния 149, 208
Идеальные растворы 178
-----, активность компонента 180
-----и идеальный газ, уравнения р — х — у 180
-----, критерий 606
-----, летучесть компонента 179
-----, общие уравнения для фазового равновесия 193
-----, перегонка с водяным паром 689
-----, равновесие 203
-----, уравнения для перегонки 608 -----, — для фазового равновесия 191, 193
-----, химический потенциал компонента 179
Изменения состояния (см. Процессы)
Измерение потока газов 383
--------, влияние изменения состояния 392
-----идеального газа 384
-----------, коэфициент расхода 377
-----------, — адиабатный 385
-----------, — значения 380
--------, уравнения для практического применения 387
--------, числовые примеры 398
Измеритель Вентури 373
— диафрагмовый 374
— потери давления 381
— сопловой 375
— термический 403
Изобара, критическая 265
—, равновесие в системе азот-кнсло-род 614
Изобарный (изобарно-изотермический) потенциал изменение, 131, 133, 732 -----, аддитивность значений A Z 567 —----, мера «чистой» работы 131
-----, образование органических соединений 568
-----, химическая реакция 558
-----, критерий равновесия 145, 160
----- раствора 164
-----, связь с параметрами состояния 141
Предметный указатель
773
Изобарный, стандартное изменение 251, 567
—Изолированная система 55
— —, изменение энергии 124
-----энтропии 123
Изотерма а — р 227
— углекислоты вблизи критической точки 230
Изотермические процессы -----, максимальная работа 132 -----, работа сжатии 11
-----, расширение идеального газа 322
-----, сжатие идеального газа 322
Изохоры, кривизна их 229
— приведенные 219
Изоэитальпы 256, 285
Инверсионная температура 356
Индикаторная диаграмма 66
Интегральная теплота испарения 449
Интегральное испарение (конденсация) 662
-----, двухкомпонентные системы 670
-----, многокомпонентная система 671
-----,-----, примеры расчета 673, 674
-----, углеводороды, пример 658, 673
-----, уравнения 670
Испарение диференциальное(см. Дифе-ренциальиое испарение)
—, изменение энергии, пример 105
— интегральное (см. Интегральное испарение)
—, процессы 599
— -, теплота (см. Парообразования, теплота)
Истечение 20, 375, 404
— влажного пара 20
— перегретого пара 26
-------, пример расчета 271 »
Истинная теплоемкость 70
К
Каллеидара, уравнение состояния 226
Калориметр дроссельный 359
Калория 69
Капицы детандер 538
Карно принцип 85, 97, 106
-----, доказательство Кельвина 107
— цикл 97, 111, 124
Каскадный процесс 524, 539
Кейеса уравнение состояния 239
Кельвина нагревательная машина 521
— температура 106
Кинетическая энергия 89
-----потока 367
50 Химияеская термодинамика
Кислород, низкотемпературное получение 547
Кистяковского уравнение 23, 441
— —, пример расчета 443
Киреева — Отмера уравнение 24, 739
Клаузиуса — Клапейрона уравнение21,
187, 189, 2эЗ, 302, 440
Клода процесс 536
-----, пример расчета 536
Кокса диаграмма 305
Компонент 53
Компрессоры, вредное пространство 333
—,------, влияние на объемный к. п. д.
335, 339
—, индикаторная диаграмма 345
—, классификация 317, 319
—, к. п. д. механический 344
—,---------объемный 324
—, минимальная работа на I ступень 329
— многоступенчатые 319, 335
-----, влияние вредного пространства 339
------, работа цикла 337, 341
—, необратимые эффек ы 344
—, объем, описываемый поршнем 333
— одноступенчатые 319, 328
-----, влияние вредного пространства 333
-----, идеальный цикл 328
-----, работа цикла 329, 11
— пароструйные 318, 347
-----, пример расчета 350
—, производительность, примеры 340, 492, 497
—, число ступеней 343
Конденсация 599
— диференциальная 664, 676
-----в присутствии инертного газа 682
----, тепловой эффект 716
— интегральная (однократная) 661,670
-----, двухкомпонентная система 670
-----, многокомпонентные системы 671
-----тепловой эффект 715
— обратная 616
—, сравнение диференциальной и интегральной 680
— частичная 663
Конноды (соединительные линии) 602
Коновалова законы 620
Константы (см. также Постоянная)
— взаимодействия 254
— закона Генри 198
— равновесия 201, 555
----- влияния давления 562
------- — температуры 204, 557
774
Предметный указатель
Константы, вычисление 557
-----, — косвенное 540
-----, —, метод Темкина и Шварцмана 31
-----, — по термическим дайиым 565, 571
-----, —, приближенные методы 527
-----, —, приближенный метод Нернста 580
— —, — синтеза муравьиной кислоты 577
-----, — синтеза этилбензола 579
-----, — синтеза этилового спирта 560, 574, 586
— уравнений состояния газовой смеси 254
— фазового равновесия (см. Равновесное отношение)
Концентрация, способы выражения ее 56
Коппа правило 438
Коэфициент активности 204, 294, 621, 628
—• летучести (см. Активность, коэфициент)
— отклонения 9
— подачи 344
— полезного действия детандера 346
-----компрессоров 324, 344
----- перегонки 669
-----ректификации при низкой температуре 542
------ ректификационной колонны 727
----- сопла 405
-----тарелки ректификационной колонны 727
------- тепловой машины 98, 106
----- теплообменников 431
-----термический 421
----- цикла Карно 483
— расхода, адиабатный 385
----- гидравлический 388
------ для остроугольных измерителей 390
— — средний 389
— расширения 387
—- —, график значений 389
— сжимаемости 212 (см. также Сжимаемости коэфициент)
— трения 407, 410
-----, член в уравнении жидкого потока 370
— фракционирования, общий 708
— характеристический 258
— холодильный 484
Кривые (см. также Линии)
— инверсии дроссель-эффекта 356
Кривые минимумов яа кривых »—л 235
— ортобарная 265
— плавления 262
— - пограничная 602
— равновесия 688
— сублимации 262
Критические, давление, вычисление
222, 740
—, — газовой смеси 258
— константы, таблица 525, 750
— объем, вычисление 222
— отношение давлений в потоке 391
— состояние 212
— температура 212
— — ароматических углеводородов j 733
— —, вычисление 221, 740
---- газовой смеси 258
----растворимости 634
— точка 13, 212. 262
----, критерий 228
— явления в бинарных смесях 615 )
Кричевского и Казарновского уравне-
ния состояния 735
Л
Летучесть 146
—, влияние давления и температуры 148
—, вычисление по графику коэфициента сжимаемости 292
—, — по уравнению состояния 288/ •:
301 z ,
—, — по функции а 292
— компонента смеси 162, 172
—-------, влияние температуры, дав-
ления и состава 177
— — — идеального раствора 179
.— коэфициент (см. Активность, коэфициент) ।
— и работа изотермического сжатия 330
—, приближенное уравнение 289
Ле-Шателье — Брауна принцип 557 1
Линде разделительная колоииа двой-
ной ректификации 549
— процесс 546
----, пример расчета 546
— цикл 530
----, пример .расчета 530
— уравнение состояния 238, 278
Линии (см. также Кривые)
— пограничные 262
— постоянного перегрева 266
— постоянной сухости пара 267
— постоянной энтальпии 266, 285
Предметный указатель
775
Ланни постоянной энтропии 263 — отношения количества тепла при
---ректификации 703
.--точек росы 267, 675
•- — рабочие 678, 696
' Ломоносова теория теплоты 731
М
Магнитная проницаемость 207
Магнитное силовое поле 207
— напряжение 207
Максвелла «демоны» 95
Макросостояние 52
Максимальная работа 131
Максимально полезная работа 134
Максимальный выход продуктов реакции 591
Маргулеса решения, уравнения Дюгема 621
Механический эквивалент теплоты 85
Микросостояние 52
Многокомпонентные системы, дифе-ренциальная перегонка и конденсация 678
-----, интегральное испарение и конденсация 671
— —, растворимость 602
Многоступенчатое сжатие 335
Молекулярный вес, средний 246
-----, фиктивный 702
Мольная доля 56
Мощность (см. также Работа) 67
— единицы измерения 68
— на сжатие, многоступенчатый компрессор, примеры 340, 342
— на накачивание 413
-----несжимаемые вещества 413
-----сжимаемые вещества 415
-----охлаждение 485, 501
Н
Нагревание 420
— . изменение энтропии 116
— водяным паром 422
— индукционными токами 426
— инфракрасным излучением 426
— органическими теплоносителями 423
— расплавленными солями 424
— ртутью 424
— с помощью холодильного цикла 521
— топочными газами 425
— электрическим током 425
Напор 372
50*
Напор общий 372
-----, пример расчета 373
— потенциальный (гравитационный) 372
— скоростной (динамический) 372
-----, пример расчета 373
— статический (напор давления) 372 Насыщенный водяной пар как теплоноситель 422
--------, свойства 736
-----:--, теплоемкость 308
Независимые переменные 57
-----—, замена 57
-----, уравнение изменения их 138
Необратимость в цикле паровой машины 125
—, причины 92
—, — в компрессорах 344
—, — при охлаждении 488
—, степень 78
Необратимые процессы 71
-----, изменение энтропии в них 114
-----, обращение 72
-----, примеры 71
-----, связь со вторым законом 72,92
Нернста приближенный метод 580
— тепловая теорема 568
.V-компонентные системы 647
О
Обобщенные силы, обратимые 205
-----, машина 98
— —, падение воды 81
-----, переход тепла 76
-----, процесс 76, 82
-----, — как стандарт для сравнения 79
-----, расширение газа 79
—, смешение газов 80
— химическая реакция 81, 133
Обратные, конденсация 617
— переход тепла 76
— цикл Карно 98, 111, 124
Объем влажного пара 302
—, вычисление по изобарной теплоемкости 283
—, — по энтальпии 284
—, — при высоком давлении 220
—, закон аддитивности 178, 248
—, извлекаемый из сосуда для хранения 223
— критический 222
—, определяемый но эффекту Джоуля-Томсона 286
— остаточный, а 227, 291
776
Предметный указатель
Объем приведенный 216
— реальной газовой смеси 248
Объемная доля 57
Объемный к. п. д. компрессора 324
Одновременно протекающие реакции 593
Однократное испарение (см. Интегральное испарение и конденсация)
Ортобарная кривая 17, 265
Осуществимость реакции 566
-----, пример расчета 579
Охлаждение 482
— абсорбционное 508
-----, аммиачная установка 511
-----, непрерывный процесс 510
-----, периодический цикл 509
-----с помощью «ледяного шара» 510
-----, система Шатен — МунтерсабЮ
— вакуумное 513
-----водяным паром 513
-----, расход водяного пара 516
-----, система центробежного сжатия 516
— единицы измерения 488
—, методы 482
— механическое 483
—, низкотемпературные процессы 524
—, получение твердой углекислоты 517
—, пример расчета 518
— при обратимом и необратимом расширении 364
— сжатием 494
— —, многоступенчатое сжатие и расширение 494
-----посредством расширения газа 489
-----—, сравнение с процессом расширения газа, пример 489
—, система Аллена 494
—, циклы 483, 488
—, — бинарный 505
—, —, расширение газа 489
—, —, сжатия пара 494
П
Пар влажный 261
—, вычисление свойств 20, 301
—, дросселирование 18
— водяной (см. Водяной пар)
— насыщенный 261
— —, вычисление свойств 302, 740
— перегретый 261
— водяной 736, 741
—, вычисление свойств 271
—, дросселирование 18
Пар, сухость (степень сухости) 267
Парахор 222
Парообразования, теплота 440
—, — ассоциированных веществ 441
—, —, влияние температуры 310, 441
—,— диференциальная, расчет 184, 189, 193, 441
—,------, — по методу Ватсона 441,
444
—,------, — по методу Карапетьян-
ца 744
—,------, — по методу Киреева —
Отмера 442, 444, 743
—.------. — по метолу Лукомскопо
740 ' • '
—,------, — по методу Мейсснера
443, 446
------, — по уравнению Клаузиуса— Клапейрона 306, 307
—,------, — с помощью функции
Гильдебранда 441, 444
—, — растворов 192, 437
Парциальное, давление 189, 246
Парциальные мольные величины 154
--------для бинарного раствора 156
--------, объемы 178
-------- теплоемкости 447
Пельтье эффект 482
Первый закон термодинамики 84
--------- справедливость 87
-------- история 84
--------математическая формулиров-
ка 87
-------- ограничения 91
----- — применение к потоку 370
Перегонка (см. также Испарение) 662
—, азеотропная смесь 667
— непрерывная в сочетании с дефлегмацией 716
— простая (однократная) 663
—, работа минимальная 667
—, —, пример расчета 669
—, расход водяного пара 685
—, расход тепла 699, 713
—,------на непрерывную однократ-
ную перегонку 714
—,------, приближенные методы вы-
числения 724
— с водяным паром 683, 685
-----,-----, примеры расчета 690
-----------, сравнение периодическо-
го и непрерывного процессов 690 г
—, способы 692
— фракционная 664
Пито трубка 375
-----, уравнения для нее 395
-----, —, пример расчета 397
Предметный указатель
ТП
Плавления кривая 262
.------гелия 264
—""теплота 446
Планка постулат 568
_2—Плотность насыщенных жидкости и пара, вычисление 740
Поверхностная энергии, 62 Повторное сжатие пара 729 Пограничная кривая 602 Пойнтинга соотношение 200 — эффект 7
Полупроницаемая мембрана 80, 668 Постоянная (см. также Константы) — газовая 211 — химическая условная 580 Постоянно испаряющиеся смеси 7 Постоянно кипящие смеси (см. Азеотропные смеси)
Постулат Планка 568
Потенциал изобарный (изобарно-изотермический) 133
—обозначения, применяемые различными авторами 732
— химический 158
Потенциальные, напор 372
— , энергия 88
Поток адиабатный, коэфициент расхода газа 385
—, баланс механической энергии 371 — в трубопроводах 406
— , газы 409
— максимальный 411
—, сравиеиие изотермического и адиабатного 412
— газовый, измерение 383
— значения коэфициента расхода 385, 388
— жидкостей в измерителях напора 375
— идеального газа 384
— , измерение, «фактор приближения» 377
— , измерители напора 373
— , --диафрагмовые 374
•-------термические 403
' —, коэфициент расхода 377
•—,-----расширения 387
— максимальный 411
—, максимальный расход и критическое отношение 390
—, мощность на накачивание 413
—, напор 372
—, общие уравнения энергии 339
—, потери механической энергии на расширение и сжатие 408
—-, примеры расчета 397
—, рабочие уравнения 396
Поток, разностное давление 376, 380
—, расход из хранилища при переменном напоре 382
—, скорость средняя 368
—, сопловый измеритель 375
—, теорема Бернулли 370, 408
—, «точка сжатого сечения» (vena contracta) 376, 380, 388
— , трение 371
— , трубка Пито 375
Правила (см. также Закон, Уравнение)
— Бачинского—Сегдена 222
— Дюлоига и Пти 438
— Дюринга 305
— Коппа 438
—прямой средней линии Алексеева 746
— рычага 671, 16
— Трутоиа 740, 23
— фаз 180, 600
-----, вывод 181
-----, применение к одиокомпоиент-ным системам 261, 267
-----, применение к двухкомпонентным системам 601
Приведенные, давление 216
— координаты 215
— объем 216
— температура 216
— уравнение состояния 234
«Пригодность» 121
— , пример расчета 123
Принципы Карно 85, 97, 106
— Ле-Шателье — Брауна 557
Простая перегонка (см. Интегральное испарение и конденсация)
Процессы, адиабатный 10, 11, 69, 324
— Гейланда 538
— изобарно-изотермический 133
— изобарный 321
— изотермический 323
— Клода 536
— Линде 530
— необратимые (самопроизвольные) 71
— обратимые 76
— охлаждения 482
— повторного использования пара 729
— политропные 327
— разделения газа, основные прин-
ципы 540
Псевдокритическая точка 258
Пуассона уравнение 325
Пуазейля уравнение 406—407
Работа «внутренняя» 64
— всасывания 67
— выхлопа 67
778
Предметный указатель
Работа, графическое изображение 65
—, единицы измерения 68
— изобарного процесса 321
— изобарно-изотермического процесса 134
— изотермического обратимого цикла 133
— индикаторная 66
— как форма передачи энергии 731
— максимальная 131
— максимально полезная 134
— механическая 64
— многоступенчатых компрессоров 319
— иа валу 367
— на охлаждение 485
----примеры расчета 485
— на перегонку, минимальная 667
----пример расчета 669
—, обусловленная давлением среды 65
— одноступенчатых компрессоров 328
--------, пример расчета 332, 335
— поверхностного натяжения 64
— разделения газов, минимальная 527
•----- —, пример расчета 528
— сжатия (расширения) 320
— — адиабатного 325
— —, вычисление из летучести 330
----газа, периодический процесс 321
-----, диаграмма 342
----из диаграммы энтропия — температуры 341
---- изотермического 322
--неидеальиого газа 10, 341
— сжижения, минимальная 527
----, пример расчета 528
— цикла 65, 329
----компрессора 11, 328, 337
— электрическая 64
Р
Рабочая линия 688, 696
----, пересечение 702, 703
— —-> уравнение 696
Равновесие 73, 153
— гетерогенное (см. Фазовое равновесие)
— гомогенное 161
—, жидкость-пар (см. Жидкость-пар, равновесие)
— и движущая сила 75
— истинное 73
— константа (см. Константа равновесия)
—, критерии 74, 145, 159
Равновесие, общие уравнения 153
— фазовое 160, 180, 185, 473
— химическое (см. Химическое равновесие)
— частичное 75
—, ящик Вант-Гоффа 82
Равновесное отношение (константа фазового равновесия) 651
—, влияние температуры 196
—, вычисление 653
----, примеры расчета 655, 658
—, диаграмма для некоторых углеводородов, 656, 657
—, значения для некоторых углево-дородных систем 660
Равновесное превращение 581
----, гомогенная газовая реакция 582
----,---------, примеры расчета 672
— —, одновременно протекающие реакции 595
----,---------, примеры расчета .598
----, реакция в газовой и жидкой фазах 584
----,----------------пример расчета 686
----, сравнение изотермической и адиабатной реакций 587.
— —,---------------пример рас-
чета 588
Разбавленные растворы 196
----, изменение точки кипения 199
----, понижение точки затвердевании 200
----, реакция в газовой и жидкой фазах 584
Растворимость, вычисление по теплоте плавления 446
— газов в жидкостях 195, 733
— газов взаимная 15
----, критическая точка 15
— жидкостей, взаимная 602
— — в сжатых газах 10
----, критическая точка 634
Растворы, активность компонента 163, 172, 175, 180
— бинарные (см. Бинарные растворы)
— газообразные 301
— идеальные (см. Идеальные растворы)
— истинные, сравнение с коллоидными 54
— концентрации 56
— , летучесть компонента 162
— неидеальные 611
— разбавленные 196
—, теплота парообразования 192, 456
—, уравнения, общие 154
Предметный указатель
779
'Растворы, активность для фазового равновесия 160
— энтальпия 165, 300, 301, 446
Расход жидкости из хранилища при
_ переменном напоре 382
— тепла (см. также Теплота)
— — при дефлегмации 715
---- при перегонке 713
---- при постоянном давлении 270
---- при ректификации 719
Расширение адиабатное и обратимое, пример 271
— джоулевское (эффект Джоуля) 363
— дроссельное (см. Джоуля — Томсона эффект)
—, изменение энтропии 114
—изоэнтальпийное, пример 270
—, коэфициент 387
— насыщенного пара 309
— свободное 351
—-, сравнение обратимого и необратимого 363
Рауля закон 179, 191, 198, 690
Реакции, автотермические 589
— адиабатные 7, 587
— гетерогенные 584
— гомогенные, газовые 582
—, жидкая фаза 584
—, изменение энтропии 116
— изотермические 587
----, сравнение с адиабатными 587
—, максимальный выход 591
—, направление 581
•г— обратимая 81, 133
----, проведение в гальваническом элементе 81, 217
—, одновременно протекающие 593
--------, количественные зависимости 595
—, осуществимость 566
—, теплота (см. Теплота реакции)
Реальные газы 211
----адиабатный процесс 10
----, изменение потока 392
----, работа сжатия 11, 341
----, расчет термодинамических свойств 9
----, согласие законов с опытом 257
----, теплоемкость 280
—'—, уравнения состояния 224
----, энтальпия 297
Рейнольдса критерий (число) 366, 380, 396
Ректификация 548, 665, 692
— адиабатная, воздуха 541
— , колонна 692
— , —, влияние потерь тепла 723
—, —, влияние типа питания 700
—, —, исчерпывающая часть 692
—, —•, коэфициент полезного действия 727
— , — Линде, двойная 549
— , — насадочная 694
----- неадиабатная 723
— , —, общие уравнения для адиабатной колонны 695
— , —, постоянный поток 700
— , —, укрепляющая часть 692
— , —, уравнения для частных случаев 699
— , —, флегмовое число 703
— , —, число тарелок 703 (см. также Теоретические тарелки)
—, коэфициент полезного действия при низкой температуре 542
— лабораторная 745
— обратимая 542
— при низких температурах 548
—, принцип многократного действия 727
—, процесс повторного использования пара 728
—, процесс повторного сжатия пара 724
— , расход тепла 719
—,----, влияние потерь 723
— , —• —, приближенные методы расчета 724
•-------—,-----, пример расчета 725
— ,---, снижение 727
термодинамический к. п. д. при низ-
кой температуре 542
пример расчета 542
Розена уравнение состояния 734
С
Свободная энергия 131, 732
----- и параметры состояния 141
-----, мера работы обратимого изотермического процесса 132, 668
Свойства, влияние предистории 61
—, вторые производные 59
—, вычисление по сжимаемости графическими методами 290, 291
—, — по эффекту Джоуля—Томсона 285
—, конечные изменения 60
— обобщенные 293
-----, аналитические выражения 301
------ на кривой равновесия жидкость-пар 740
—, основные уравнения 57
780
Предметный указатель
Свойства парциальные мольные 157
— перегретого пара, аналитический метод расчета 271
— смесей 295
—, совокупность 261, 313
—, —, совпадение друг с другом, пример 314
—, таблица 269, 270
—, —, примеры расчета 270
— экстенсивные 103, 261
----, общие уравнения 154
Сжатие адиабатное, изменение температуры 327
- —'—, работа 325
— водяного пара 422
— , вычисление работы 322
— изотермическое, работа 330
— многоступенчатое 335
----, число ступеней 343
— насыщенного пара 309
— неидеального газа 341
— пара 422
Сжижение водорода 532
— гелия 532
— газов двухступенчатым дросселированием 534
----, влияние предварительного охлаждения 533
----, максимальный выход из эффекта Джоуля — Томсона 530
----, машина для расширения (детандер) 535
----, каскадный метод 539
----натуральных 569
----, процесс Капицы 527, 538
----, процесс Клода 536
----, процесс Линде 530
Сжимаемости коэфициент Амага 214
---- азота 213
----, вычисление давления 213
----,------, пример расчета 214
।---,------летучести 292
----газов под давлением ниже
1 атм. 215
—---газовой смеси 251
-----------, закон аддитивности давлений 252
-----------, закон аддитивности объемов 251
----, обобщенные диаграммы 217, 218
----, примеры расчета давления и объема 220, 221
----, функция приведенных давления
и температуры 216
Системы, бивариантная 56
— гетерогенная 54
Системы, гомогенная 53
— двухкомпонентная 55
— двухфазная 301
—• изолированная 123
— нонвариантная 263
— однокомпонентная 54, 260
— трехкомпонентная 55, 643
— унивариантная 56, 634
Скорости, звуковая 391, 411
—• истечения
---по диаграмме S—Н 20, 272
— потока в круглых трубах, отношение средней к максимальной 397
— потока средняя 368
— процесса и термодинамика 45
— реакции 551
Скоростной (динамический) напор 372
Скрытая теплота 71
---парообразования (см. Парообразования теплота)
--- плавления 446
Смеси азеотропные (см. Азеотропные смеси)
— идеальные (см. также Газовые смесн)
— бинарные 187, 615, 670
— постоянно испаряющиеся 36
— тройные 643
Смешение, законы для газов 247
—, Процесс, изменение энтропии 116
— ,—, тепловые эффекты 448, 460
Соединительные линии (конноды) 602, 644
Сопла, расчет потока 404
— , истечение газов 404
— , коэфициент полезного действия 405
— , суживающиеся и расширяющиеся
Соответственных состояний закон 215, 234
Состояния 52
— , воспроизводимость 62
— , —, твердое тело 62
— критическое 211
— макроскопическое 52
— микроскопическое 52
— параметры (переменные) 56
— соответственные 215
— стабильные 62
— стандартные 62, 173, 572, 575
— уравнения (см. Уравнения состояния)
Сохранение энергии 66
Средний молекулярный вес смеси 246
Стандартные величины 63, 205, 557, 558
— вещество 734
Предметный указатель
781
Стандартные состояния 62, 173, 572, 575
— теплоты 205, 561, 557
— условия 63
---Степени свободы 56, 181
Степень расширения 384
— сжатия 327
------------пример расчета 335
— сухости (сухость) пара 267, 367
Стефана — Больцмана закон 420
Сублимации кривая 262
Т
Тарасова уравнения 743
Температура 47
— абсолютная (термодинамическая) 106
— , абсолютная шкала 106
— , абсолютный нуль 49, 109
— , влияние на теплоту реакции 475
— , — на константу химического равновесия 204
— горения, теоретическая 480
— , изменение при адиабатном дросселировании 360
— , — при адиабатном сжатии 327
— , измерение 48, 51
— инверсионная 356
— , качественное представление 47
— Кельвина, абсолютная 49
— кипения 302, 736
— — нормальная 222
растворителя 199
— , количественное определение 47
—,-, величина градуса 47
— • критическая 212, 221
---------- растворимости 634
— , международная шкала 49
— наивысшаи 49
— , нижний предел 49
— очень низкая, получение 109, 207
— по шкале идеального газа 108
— приведенная 216
— состав, диаграмма 613, 618, 634,
644
— характеристическая 439
— , эмпирическая шкала 47
— , энтропия, диаграмма 265 (см. также Диаграмма энтропия-температура)
Теоретические тарелки 693, 703
• и число единиц переноса массы
711
-------, коэфициент полезного действия 693
---, определение числа 704, 747
----,------, алгебраические методы 707
----,------, метод Волкова и Жаворонкова 746
----,------метод Гелытерина 747______
----, — —, метод Львова 746
----,------, метод Мак — Кэба — Ти-
ле 707
----,------, метод Смокера 709
----,------по диаграмме энтальпия-коицентрация 722
«Тепловая смерть» 127
Тепловые, источник 59
— машина 77
----, коэфициент полезного дейст- вия 98, 106
----, цикл 47
— насос 98, 487
— эффекты (см. Теплоты)
Теплоемкость 70
— , аддитивность 447
— азота 281
— азотоводородной смеси 280
— в критической точке 254
— , влияние давления 277
— водяного пара 281
— газов, влияние давления 277, 280, 300
---- и структура органических соединений 437
— —, отношение 325
— —, таблица 435
----, углеводородов 433
— единицы 71
— жидкостей 438, 743
— изобарная 70
----, вычисление 278
— изохорная 70
— истинная 70
— , квантовая теория 439
— мольная 70
— насыщенного пара 307
--------, примеры расчета 308, 311
— насыщенной жидкости 307
— , обобщенный график 300
— , пример расчета 299
— отрицательная 236
— парциальная мольная 447
— политропного процесса 12
— при низких температурах 440
— , разность Ср — Со 140, 278
— , расчет по эффекту Джоуля — Томсона 285
— соотношения 140
— средняя 71, 433
----, таблица 435
— твердых тел 438, 743
782
Предметный указатель
Теплоемкость, влияние температуры
— удельная 70
Теплоносители 420
— , вода 421
— , водяной пар 422
— органические 423
----- даутерм 423
— , расплавленная соль 424
—, ртуть 424
—, смесь NaNO2, NaNO3 и KNOS 424
—, топочные газы 425
Теплообменники 427 __ ,Т ГТ/Л ГТ ГГ/ЛИЛ tv sxvvzv'T'vv v> л , пилшпЦп&п 1 nvavonuju Дкянлопл
431
---------, пример расчета 431
—, нарушение в них второго закона, примеры 428
—, способ действия 383
Теплоотдатчик 76
Теплоприемник 76
Теплота 68
— , бесполезно теряемая 118 , примеры расчета 119, 120
—, вызывающая изменение энтропии 432
— единицы измерения 69
— изобарного процесса 104
---------, пример 105
— изобарного процесса изменения состояния 270
— изомеризации 472
— изотермического процесса 71
— изохорного процесса 276
— испарения (см. Парообразования теплота)
—, история 68, 84
— как форма передачи энергии 731
—, количественное выражение 69
— , механический эквивалент 85
— образования 472
— обратимой реакции 117
— парообразования (см. Парообразования теплота)
— , передача, способы 418
— плавления 446
— разбавления 73
— бесконечного 454
— растворения 448
-----дифференциальная 448
----- интегральная 448
-----, расчет по растворимости 196
— , расход иа перегонку 713
— , — на ректификацию 719
— «скрытая» 71 — смешения 460 , примеры расчета 454, 455, 462
Теплота фазового превращения 440
— физического процесса 432
— химических реакций 467
------- —, илияние давления 476
--------, влияние температуры 455
--------, вычисление из равновесных данных 473
--------общая 476
--------, связь со структурой 474
--------стандартная 205, 471, 557
-----, — суммирование 471
Теплотворность низшая 478
Терминология технической термодинамики 732
Термодинамика 45
—, задачи ее в применении к химическим процессам 45
— и скорость процесса 46
Точки Бойля 219, 234, 241
— замерзания, понижение для раз-' бавлениых растворов 200
— кипения, изменение для разбавленных растворов 199
-----, правило Дюринга 305
— —, смесь толуол-вода 604
— криконденбарнческая 616
— крикондентермическая 616
— критическая 13, 212, 262
-----, взаимная растворимость газон под давлением 640
------, растворимость жидкостей 634
— псевдокритическая 258
— росы (пузырьков)
-----, однокомпоненгная система 267
-----, смесь 675
— сжатого сечения 376, 380, 388
— тройная 263
Третий закон термодинамики 45, 440, 558
--------, приложение к химическому равновесию 31, 567
Тронные смеси 643
-----, графическое изображение состава 643
-----, диаграмма у — х 646
-----, диаграмма t — х 644
-----, неидеальные 648
------, уравнение для идеальной смеси 647
-----, фазовая диаграмма 645
Трутона правило 440,743
У
Удельная теплоемкость 70
Уеймута уравнение 410
Уравнение (см. также Законы, Правила)
Предметный указатель
783
Уравнение Бернулли (см. Бернулли теорема)
— Букингема 388
—• Ван-Лаара 623
— Дюгема 185, 621, 626, 627
— Женеро 407
закона действующих масс 203, 555
— Карапетьянца 747
— Киреева — Отмера 24, 739
— Кистяновского 23, 423
— Клаузиуса—Клапейрона 187, 188, 263, 302, 440, 730
— Мейсснера 443
— непрерывности потока 376
— Пойн1инга 200
— Пуазейля 406, 407
— Пуассона 325
— рабочей линии ректификационной колонны 696
— равновесия, общее 152
---для люэого числа компонентов
и фаз 158
Состояния Бартлетта 248
— Вертело 236
— Битти 243
— Битти — Бриджмена 240 (см. также Битти — Бриджмена — уравнение состояния)
— Бюро стандартов США для аммиака 238
— Ван-дер-Ваальса'228 (см. также Ван-дер-Ваальса уравнение состояния)
—, вириальная форма 241
— Воля 237
— Вукаловича — Новикова 741
— газовых смесей 254
— Гудинафа 238
— Джиллиленда 256
— Дитеричи 236
— Каллендара 226
— Кейеса 256, 239
— Клапейрона 209
— Кричевского и Казарновского 735
— Линде 238, 278
—, общая форма 224
—, определяющие объем 238
—, — предельные условия 226
— приведенные 234
— Розена 734
— со вторым вириальным коэфициен-том 736
— эмпирические 225
— Тарасова 743
— Уеймута 410
— У — х(перегонка), эмпирические 625
— Фаннинга 407, 410
— Шредера 733
Ф
Фаза 53, 54
Фазовое равновесие, диференциальные уравнения 181
-----идеальных растворов 133
— — многокомпонентных многофазных систем 185
-----, общ те уравнения 160
-----однокомпонентных систем, уравнение 186
-----раствора постоянного состава 187
----- теплоты реакции для него 473
-----, термодинамические методы определения 185
-----, фаза идеального газа 188
Факторы емкости 64
— интенсивности 64
Фаннинга уравнение 407, 410
Фиктивный молекулярный вес 702
Флегма 665
Флегмовое число 696, 698, 705
-----минимальное 705, 706
--------по диаграмме Н — х 720
Формулы (см. Законы, Правила, Уравнения)
Фракционная перегонка (см. Днферен-циация, испарение и конденсация) -----, кривые для раствора бензол-то-луол 679
Функции, Гильдебранда 441
— а (.остаточный объем”) 226, 739
— А («остаточный объем”) 226, 739
X
Характеристическая температура 439
Химическая постоянная, условная 580
Химический потенциал 158
-----, влияние давления, температуры и состава 175
-----, компонента идеального раствора 180
-----, связь с летучестью и активностью 162
-----, уравнения 170
Химическое равновесие 201, 551
-----, влияние давления 561
-----, влияние температуры на константу равновесия 204
-----,-----, примеры расчета 559, 561, 571, 581
-----, выход 581
------ гетерогенное 584
--------, примеры расчета 574, 571
-----гомогенное в газовой фазе 582
784
Предметный указатель
Химическое равновесие, идеальные газы 203
-----, идеальный раствор 203
— — и скорость реакции 551
-----константа (см. Константа равновесия)
Холодильные агенты 498, 744, 525
-----, сравнение их 499
-----, фреоны 500, 744
— коэфициент 484
-----, пример расчета 492
— оборудование, оценка его 498
— процессы 482
__е 1 1 ein ею .jviunvDtui UA1,
Холодопроизводительность 488
—примеры расчета 502
Ц
Цикл бинарный 505
— жидкого воздуха 533 '
— замкнутый 66
— Карно 98, 111, 124
-----обращенный 483, 486
— Клода 536
— компрессора 328
— Линде 530
— низкотемпературный 529
— охлаждения, идеальный 483
-----, реальный 488
— твердой углекислоты 517
— тепловой машины 77
Ч
Частично смешивающиеся жидкости 633
-----зависимость р — х—у 634
— правило прямой средней линии Алексеева 746
----- равновесия при перегонке 633
Число единиц переноса массы 695
----------, пример расчета 711
— компонентов 181
— Рейнольдса 371
— степеней свободы 181, 260, 600, 643
—• тарелок ректификационной колонны (см. Теоретические тарелки)
— флегмовое 696
Ш
Шарля— Гей-Люссака закон 208
Шкала температурная
— абсолютная 48
— величина градуса 47, 107
Шкала идеального газа 49
— Кельвина 106
— международная 49
—, установление 48
—, фиксированные точки 50
— термодинамическая 48
— эмпирическая 47
Шредера уравнение 733
Э
Эвтектика 606
Эжектор 347
— пароструйный 347
-----, расход водяного пара 348
—, теория 347
Эквивалентность массы н энергии 87
Эквиваленты давления, таблица 748
— энергии, таблица 749
Экстенсивные свойства 103, 154, 261
-----, общие уравнения 154
Энергетический баланс 87
Энергия 86
— внутренняя 86, 108 (см. также Внутренняя энергия)
— внутриядерная 89, 90
— единицы измерения 748
—, источники 92, 421.
— кинетическая 354
—, классификация 88
— лучистая 93, 419
— обесцененная 92, 118, 119
— поверхностная 93
—, превращение 90
— потенциальная 88
-----, обусловленная потоком (высотой) 367
— свободная 131, 732
— и параметры состояния 141
—, мера работы изотермического обратимого процесса 132
—, обозначения, применяемые различными авторами 101
— «тепловая» 89
— химическая 89
—, цепь превращений 90
—, эквивалентность массе <?7
— , эквиваленты таблицы 748
Энтальпия 101, 103
— влажного пара 302
—, влияние давления 297
—, влияние температуры, уравнения и таблицы 433, 434, 436
— газовой смеси 169
—, зависимость от параметров состояния 138
Предметный указатель
785
Энтальпия, изменение 104
—, концентрация, диаграмма (см. Диаграмма энтальпия—состав)
—, мера изобарного теплового эффекта 104
обобщённый график 297
—, обозначения, принятые различными авторами 732
— перегретого пара 271
—.------, пример расчета 273
—, связь 105
— растворов 165, 302, 447
-----, вычисление по давлению насыщенного пара или э. д. с. 459
Энтальпийные диаграммы (см. Диаграмма энтальпия — температура,
Энтропия — энтальпия)
Энтропия 102
— абсолютная 569
-----, метод расчета Киреева 33
-----, метод расчета Капустннского и Яцимирского 33
— влажного пара 302
— газовой смеси 169
—, изменение
—, —, изолированные системы 123
—, — при дросселировании 115
—, — при нагревании 116
—, — при необратимом процессе 114
—, — при переходе тепла, пример 115
Энтропия абсолютная при смешении, пример 116
— , — при химической реакции 33, 117
— и вероятность 129
— и обесцененная энергия 118
— и самопроизвольные процессы 111
— как функция, характеризующая второй закон 111
— калориметрическая 553, 569
—, количественное определение 111
—, общее определение 110
— перегретого пара, вычисление 286
— при абсолютном нуле (нулевая) 439, 569
— раствора 167
— смеси идеальных газов 167
—, температура, диаграмма (см. Диаграмма эн гропия — температура)
—, уравнения, связывающие с параметрами состояния 136
—, физический смысл 128
—, числовое значение 128
Эффект Джоуля 363
— Джоуля — Томсона (дросселирование) (см. Джоуля — Томсона эффект) — тепловые (см. Теплоты)
Я
«Ящик равновесия» Вант-Гоффа 82
ОГЛАВЛЕНИЕ
Стр.
Предисловие редактора............................................ 5
Вступительная статья............................................. 7
Из предисловия автора............................................37
Список важнейших обозначений.........•.......................... 39
Глава /. Определения и основные понятия..........................45
Глава II. Первый и второй законы термодинамики ................. 84
Глава III. Количественное выражение двух основных законов. Термодинамические функции........................................ 101'
Глава IV. Общие уравнения равновесия ...........................153
Глава V. Соотношения давление — объем — температура для газов . . 208
Глава VI. Термодинамические свойства газов и жидкостей..........260
Глава VII. Сжатие и расширение газов ...........................317
Глава VIII. Термодинамика потока................................366
Глава IX. Передача тепла и тепловые эффекты.....................418
Глава X. Охлаждение.............................................482
Глава XI. Равновесие химических реакций.........................551
. Глава XII. Равновесие при испарении и конденсации ............599
Глава XIII. Процессы дестилляции................................662
Примечания......................................................743
Приложения:.....................................................731
Литература .....................................................752
Именной указатель...............................................762
Предметный указатель............................................768