/












































































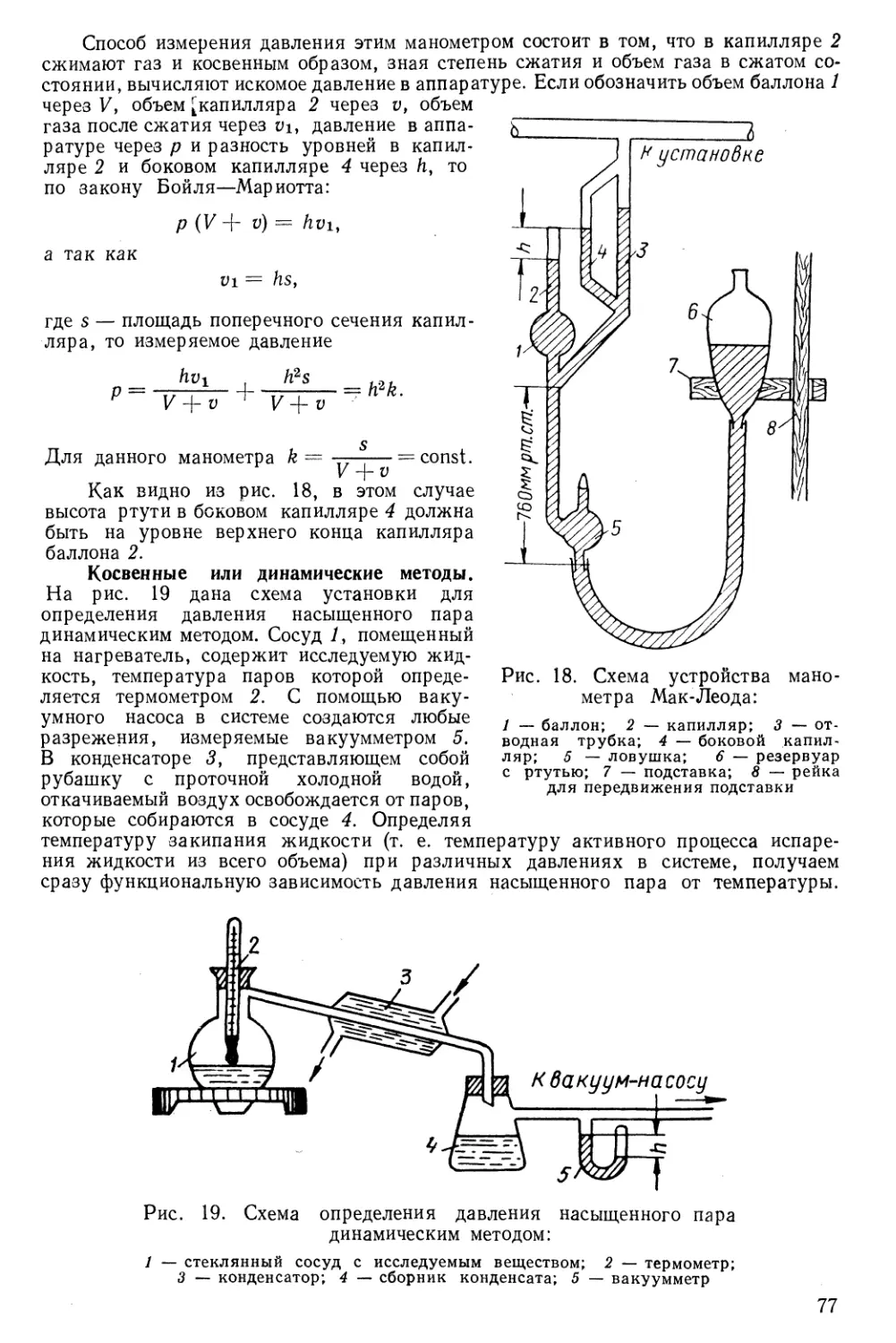




















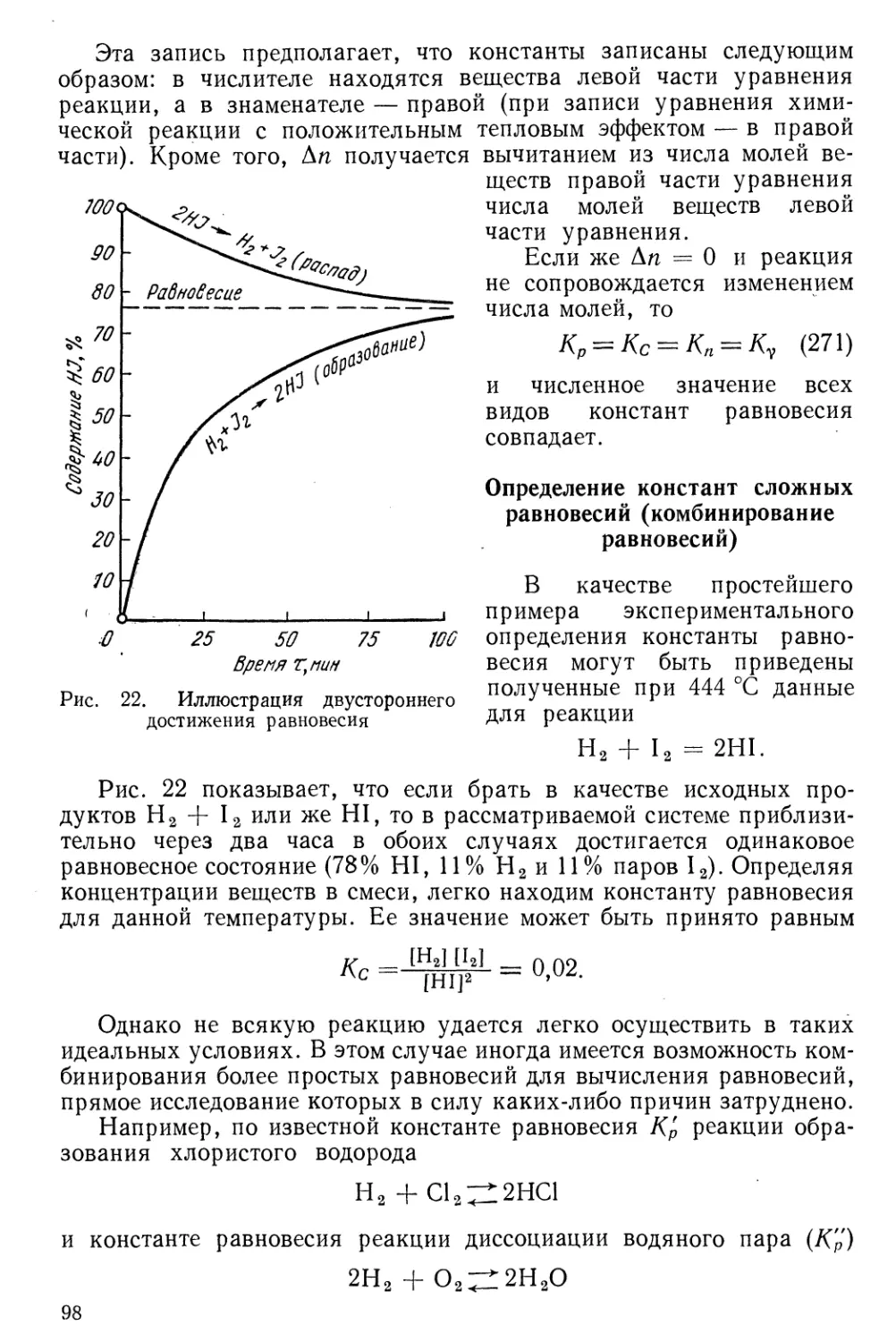


































































































































































Text
A. H. КРЕСТОВНИКОВ, В. Н. ВИГДОРОВИЧ
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
ИЗДАНИЕ ВТОРОЕ, ИСПРАВЛЕННОЕ И ДОПОЛНЕННОЕ
Допущено Министерством высшего и среднего
специального образования СССР в качестве учебного пособия
для студентов металлургических специальностей вузов
МОСКВА «МЕТАЛЛУРГИЯ» 1973
УДК 536.7
УДК 536.7
Химическая термодинамика. Крестовников А.
рович В. Н. Изд. 2-е, М., «Металлургия», 1973
А. Н., В и г д о-
с. 256
В книге изложены основные вопросы химической термодинамики
и ее приложение к учению о равновесии. Рассмотрены основы учения
о фазах и некоторые вопросы термодинамической теории растворов,
связанные с описанием фазовых равновесий и химического
взаимодействия компонентов.
Учебное пособие предназначено для студентов физико-химических
и металлургических специальностей, факультетов и высших учебных
заведений, а также может быть использовано широким кругом
специалистов: металлургами, материаловедами, химиками-технологами, фи-
зико-химиками.
Многие примеры и задачи подобраны для учащихся из области
материаловедения и металлургии. Ил. 118. Табл. 9. Список лит.:
80 назв.
Александр Николаевич Крестовников
Виленин Наумович Вигдорович
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
Редактор издательства В. П. Молокова Технический редактор И. А. Сперанская
Переплет художника Е. Н. Волкова
Сдано в набор 16/VIII 1972 г. Подписано в печать 5/П 1973 г. Т-03304
Формат 60x90Yie- Бумага типографская № 2. Печ. л. 16,0. Уч-изд. л. 17,09
Тираж 9 000 экз. Заказ 1928. Изд. №5523. Цена 81 коп.
Издательство Металлургия» 119034, Москва, 2-й Обыденский пер., 14
Ленинградская типография № 6 «Союзполиграфпрома при Государственном комитете
Совета Министров СССР по делам издательств, полиграфии и книжной торговли
193144, Ленинград, ул. Моисеенко, 10
к 3103-054
К 040(01 )-73 И 73
ОГЛАВЛЕНИЕ
Предисловие 6
Введение 7
Основные принятые обозначения 9
Часть первая
Термодинамика и учение о химическом равновесии
Глава I
Первое начало термодинамики
§ 1. Содержание первого начала '• 11
§ 2. Математическое выражение первого начала термодинамики 15
§ 3. Связь между единицами измерения энергии 17
§ 4. Термодинамические процессы 18
§ 5. Теплоемкость и формы ее выражения 29
Связь между Су и Ср (30). Эмпирические закономерности теплоемкостей
(30). Температурная зависимость теплоемкостей (32). Связь между
истинной и средней теплоемкостями (34). Опытное определение
теплоемкостей (35)
§ 6. Термохимия как раздел термодинамики 38
Связь между тепловыми эффектами реакции при постоянном объеме
и постоянном давлении (40). Закон Гесса (42). Тепловые эффекты,
сопровождающие некоторые химические реакции (44). Зависимость
теплового эффекта от температуры (уравнение Кирхгофа) (48)
Задачи , 52
Глава II
Второе начало термодинамики
§ 7. Содержание второго начала 58
§ 8. Необратимые и обратимые процессы 59
§ 9. Математическое выражение второго начала термодинамики 60
Коэффициент полезного действия тепловой машины (60). Цикл Карно
(61). Энтропия (63).
§ 10. Статистический характер второго начала термодинамики ...... 69
Энтропия и -термодинамическая вероятность состояния (69).
Антинаучное толкование второго начала термодинамики (71)
§11. Уравнение агрегатных превращений 72
§ 12. Опытное определение давления насыщенного пара 76
Задачи 79
Глава III
Характеристические (термодинамические) функции
§ 13. Внутренняя энергия 82
§ 14. Энтальпия 83
§15. Изохорно-изотермический потенциал (свободная энергия) 84
§ 16. Изобарно-изотермический (термодинамический) потенциал 85
§1 гу С\г' »»»»АиД"И ■■■tin II и I ——JM———^—Я—Л .
I/. Объединенные первое и второе начала термодинамики (уравнение
Гиббса—Гельмгольца) 87
§ 18. Направление протекания процессов и термодинамические условия
равновесия ""..... 88
§ 19. Химический потенциал и термодинамические характеристики
растворов 89
1* 3
Глава IV
Учение о химическом равновесии
§ 20. Закон действия масс и константа равновесия 93
Различные формы констант равновесия (96). Определение констант
сложных равновесий (комбинирование равновесий) (98)
§ 21. Приложение закона действия масс к гетерогенным равновесиям ... 99
§ 22. Термическая диссоциация 101
§ 23. Методы экспериментального определения констант равновесия .... 103
§ 24. Связь максимальной работы с константой равновесия (химическое
сродство) 105
§ 25. Зависимость константы равновесия от температуры 110
§ 26. Недостаточность первого и второго начал термодинамики для расчетов
химического сродства 114
Задачи 115
Глава V
Третье начало термодинамики
§ 27. Тепловая теорема Нернста 121
§ 28. Постулат Планка 126
§ 29. Расчет абсолютных значений энтропии 127
§ 30. Расчеты по равновесиям с помощью таблиц стандартных величин ... 130
Задачи 135
Глава VI
Методы приближенных термодинамических расчетов
§ 31. Теплоемкости 138
§ 32. Теплоты и энтропии агрегатных превращений 141
§ 33. Стандартные значения энтропии 144
§ 34. Теплоты образования 145
§ 35. Приближенные и ускоренные расчеты констант равновесия 148
Частьвторая
Учение о фазовом равновесии
Глава VII
Важнейшие понятия и определения
§ 36. Физико-химическая система и фаза 159
§ 37. Составная часть системы и компонент 160
§ 38. Термодинамическая степень свободы и правило фаз 162
§ 39. Геометрический строй диаграмм состояния 166
Глава VIII
Однокомпонентные и двухкомпонентные системы
§ 40. Однокомпонентные системы и аллотропия (полиморфизм) 167
§ 41. Двухкомпонентные системы 172
§ 42. Основы термического анализа 174
§ 43. Диаграмма состояния системы с простой эвтектикой 176
§ 44. Диаграммы состояния системы с химическим соединением ,. 185
Соединение с конгруэнтной точкой плавления (185). Соединение с ин-
конгруэнтной точкой плавления (187)
§ 45. Диаграмма состояния с неограниченной растворимостью компонентов
в жидком и твердом состоянии 192
§ 46. Диаграмма состояния с ограниченной растворимостью компонентов
в твердом состоянии * 199
4
& 47. Диаграмма состояния с ограниченной растворимостью (расслаиванием)
в жидком состоянии 206
§ 48. Темп (интенсивность) кристаллизации 209
§ 49. Давление насыщенного пара компонентов раствора 214
§ 50. О видоизменении диаграммы состояния под влиянием давления ... 216
Глава IX
Трехкомпонентные системы
§ 51. Диаграмма состояния с неограниченной растворимостью компонентов
в жидком и твердом состояниях 222
§ 52. Диаграмма состояния с неограниченной растворимостью в жидком
состоянии, полной нерастворимостью компонентов в твердом состоянии
и образованием тройной эвтектики при затвердевании 225
§ 53. Диаграмма состояния с ограниченной растворимостью компонентов в
твердом состоянии и образованием эвтектической смеси твердых
растворов • 229
§ 54. Диаграмма состояния тройной системы с химическим
взаимодействием двух компонентов 231
§ 55. Диаграммы растворимости 233
Глава X
Основы геометрической термодинамики
§ 56. Зависимость изобарного потенциала от температуры и давления . . . 240
§ 57. Зависимость изобарного потенциала от состава системы 241
§ 58. Об относительном расположении линий фазовых равновесий на
диаграммах состояния . 245
§ 59. Геометрическая термодинамика, химическая топология и физико-
химический анализ 250
Список литературы 254
ПРЕДИСЛОВИЕ
С химической термодинамикой знакомятся студенты всех физико-
химических и металлургических специальностей. Большей частью
химическая термодинамика включается в курсы физической химии
или материаловедения, а также в спецкурсы. Курсы, читаемые
в разных учебных заведениях, значительно различаются как по
своему объему, так и той принципиальной основе, на которой они
строятся. Такое положение вполне естественно, если учесть
огромное разнообразие практических приложений физической химии и
объектов рассмотрения в материаловедении. К сожалению, для
изучения классических положений и современных проблем химической
термодинамики перегрузка учебной программы учебных заведений
не позволяет выделять достаточное количество часов. Поэтому
учебный процесс требует наибольшей концентрации изучаемого
материала и тщательного его подбора для придания курсу
цельности.
При создании данного учебного Тюсобия были приняты во
внимание прежде всего те специфические задачи химической
термодинамики, которые представляют интерес для металлургов. В учебном
пособии изложены сведения, позволяющие использовать для
практических целей многочисленные и самые разнообразные данные по
физико-химическим константам, имеющиеся в справочной
литературе. Круг охватываемых вопросов включает в себя основные законы
термодинамики и термохимии, а также основные разделы учения
о фазовых равновесиях и термодинамической теории растворов.
При подготовке «Химической термодинамики» (избранные главы)
была поставлена задача удовлетворить запросы широкого круга
металлургов, которые пожелали бы самостоятельно освоить основы
химической термодинамики. Вместе с тем многие примеры и задачи
были подобраны аля учащихся, специализирующихся по
материаловедению и металлургии.
Порядок расположения материала и некоторые особенности его
изложения рассчитаны на последовательное чтение. Опыт работы
с первым изданием позволил сделать изложение еще более
компактным и за счет этого ввести во втором издании дополнительные
разделы. Однако, возможно, поставленная задача все еще решена не
до конца. В этом отношении большую помощь могут оказать
читатели (специалисты, преподаватели, студенты), которым авторы за
все замечания и пожелания будут благодарны.
ВВЕДЕНИЕ
В термодинамике рассматриваются взаимные превращения
теплоты и различных видов энергии. Она представляет собой
дисциплину, или скорее даже метод, который широко используется
физиками, химиками и исследователями в других областях науки
для установления внутренней связи между различными явлениями
природы и обобщения накопленного экспериментального материала.
Поскольку энергетические превращения сопутствуют всем
материальным изменениям и энергия характеризует меру движения материи,
а движение представляет собой неотъемлемое свойство материи и
основную форму ее существования, то область приложения
термодинамики охватывает огромное количество физических и химических
явлений.
Термодинамика является дедуктивной наукой. Она
рассматривает различные проблемы с помощью математического аппарата и
опирается при этом на три исходных положения — основные начала
(или законы) термодинамики, которые в свою очередь основаны на
многочисленных наблюдениях различных исследователей.
Первое начало термодинамики представляет собой закон
эквивалентности энергии. Оно дает возможность выражать различные
виды энергии некоторыми эквивалентными величинами.
Второе начало термодинамики является законом о направлении
процесса и дает возможность предсказать, произойдет ли при
данных условиях процесс в данном направлении.
Третье начало термодинамики — закон об абсолютном значении
функции, называемой энтропией. Оно позволяет производить
расчеты по химическим равновесиям, не воспроизводя эти равновесия
экспериментально.
Выбор исходных понятий (например, понятия о внутренней
энергии системы) при формулировке первого начала термодинамики,
статистический характер второго начала, а также некоторые
особенности третьего начала термодинамики приводят к ограниченной
применимости термодинамического метода. Полученные с помощью
этого метода результаты применимы лишь к материальным системам
с большой массой. Выводы термодинамики справедливы лишь при
макроскопическом рассмотрении описываемых явлений,
отвлеченном от атомно-молекулярной структуры вещества.
Термодинамика рассматривает лишь такие явления, которые
относятся к равновесиям или непрерывным последовательностям
равновесных состояний, т. е. к бесконечно медленным процессам.
Этой дисциплине предлагалось даже название «термостатика»,
хорошо отражающее сущность предмета. Однако этот термин не
привился.
В термодинамике важнейшими являются такие свойства, как
теплота, температура и их производные. Вместе с тем совершенно
не учитываются время и координаты пространства.
7
Как следствие этого ряд проблем физики и химии принципиально
не может быть разрешен термодинамическим методом. Таковы,
например, проблемы, связанные с изменением времени и касающиеся
вопросов кинетики или диффузии, которые сопряжены с
необходимостью представления о молекулярном, атомном или даже
внутриатомном строении вещества. Других ограничений для применения
термодинамики нет.
Указанный недостаток классической термодинамики, иногда
называемой феноменологической термодинамикой, восполняется
методами статистической термодинамики и термодинамики необратимых
процессов, взаимно дополняющими друг друга.
Уравнения термодинамики всегда связывают несколько величин,
которые характеризуют состояние системы и позволяют вычислить
некоторые из них на основании экспериментально полученных
значений для остальных. Эти соотношения, кроме того, можно
геометрически представить лишь в некотором абстрактном (не
физическом) пространстве. Вместе с тем математический аппарат
термодинамики прост и ограничивается использованием дифференцирования
и интегрирования.
Однако многие понятия термодинамики чрезвычайно абстрактны,
и в сочетании с кажущейся наглядностью выводов и трактовок это
составляет основные трудности для глубокого понимания
фундаментальных основ предмета и безошибочного их практического
использования.
Термодинамический метод применяется для решения самых
разнообразных проблем различных областей науки. Обычно при
рассмотрении содержания термодинамики и ее приложений выделяют
общую, техническую и химическую термодинамику. В общей
термодинамике излагаются основные начала термодинамики
и.непосредственно вытекающие из них следствия. В технической
термодинамике рассматривается применение тех же законов и их следствий
к тепловым двигателям. Наконец, содержание химической
термодинамики состоит в применении термодинамического метода к
изучению химических процессов. Она изучает превращения теплоты,
связанные с химическими реакциями и агрегатными превращениями.
При этом формулируются закономерности, позволяющие
определять направление и предел протекания этих процессов. Химическая
термодинамика оказывается плодотворной при решении вопроса
об устойчивости химических веществ, а также при отыскании
способов, предотвращающих образование нежелательных веществ, она
же позволяет указать рациональные значения температуры,
давления и прочих параметров для осуществления химических процессов,
определить пределы их протекания, а также полезна при решении
многих других металлургических и технологических задач.
С помощью термодинамики химическая наука за последнее время
продвинулась намного вперед, перешагнув за тесные рамки
эмпиризма. Теоретический анализ позволяет делать новые широкие
обобщения, а также дает возможность предвидеть основные
характеристики химических процессов.
8
ОСНОВНЫЕ ПРИНЯТЫЕ ОБОЗНАЧЕНИЯ
, D, G, R — участники химических реакций (в общем виде)
fr» d, g, r — стехиометрические коэффициенты
at — активная концентрация (активность) компонентов Л и В
в растворе А + В (ал и ав)
Ау — максимальная работа при постоянном объеме
А у — нормальное сродство при постоянном объеме
Ар — максимальная работа при постоянном давлении
А' — нормальное сродство при постоянном давлении
С — атомная или молекулярная истинная теплоемкость
с — удельная истинная теплоемкость
С, с — средняя теплоемкость (атомная, удельная) в интервале
температур от tx до t2, ° С
Сс — средняя теплоемкость (атомная, удельная) в интервале
температур от 0 до t, ° С
Су—теплоемкость при постоянном объеме
Ср — теплоемкость при постоянном давлении '
С — концентрация компонента В в сплаве А + В
С/ — мрлярная концентрация участника i реакции (Св, Cd, Cq^Cr)
F — изохорно-изотермический потенциал (свободная энергия)
А/7 — изменение изохорно-изотермического потенциала
F — число термодинамических степеней свободы (вариантность)
fi — коэффициент активности компонентов А и В в растворе А + В
if А и /б)
G — изобарно-изотермический (изобарный, термодинамический)
потенциал
AG — изменение изобарно-изотермического потенциала
т — масса вещества
Н — энтальпия
АЯ — изменение энтальпии
i — истинная химическая постоянная газообразного участника
реакции, константа уравнения давления пара (At — их
алгебраическая сумма)
V — условная (эмпирическая) химическая постоянная
газообразного участника реакции (At' — их алгебраическая сумма)
/ — константа интегрирования (в уравнении Габера)
k — постоянная Больцмана
Кс — константа равновесия, выраженная через концентрации
Кр — константа равновесия, выраженная через парциальные
давления
Кп — константа равновесия, выраженная через число молей
Ку — константа равновесия, выраженная через мольные доли
К — константа скорости химической реакции
k — число компонентов (к правилу фаз)
Ц/) — уравнение ликвидуса [L' (/) — его производная по
температуре]
N0 — число Авогадро
9
Ni — число частиц в данном микросостоянии (Nlt N2, Л"3,...)
п — число молей вещества
р — давление
pi — парциальное давление участников / реакции
(Рв, PD, PG и pR)
р — число составных частей (к правилу фаз)
г — число реакций
5 — энтропия
AS — изменение энтропии
Т — температура по шкале Кельвина (° К) [Т — t + 273 (точнее
Т = t+ 273,16)]
t — температура по шкале Цельсия (° С) [t = Т — 273 (точнее
t= T — 273,16)]
Qy — тепловой эффект при постоянном объеме в термодинамическом
обозначении
Qp — тепловой эффект при постоянном давлении в
термодинамическом обозначении
Qy — тепловой эффект при постоянном объеме в термохимическом
обозначении
Qp — тепловой эффект при постоянном давлении в термохимическом
обозначении
R — универсальная газовая постоянная
q — доля кристаллов в кристаллизующемся сплаве
U — внутренняя энергия
Л(/ — изменение внутренней энергии
V — объем
W — энергия смешения
Waa, Wbb, Wab — энергия взаимодействия одноименных и разноименных атомов
или молекул в растворе
xi — молярная концентрация компонента i в растворе или расплаве
(хл и хв)
Ui — молярная концентрация компонента i в твердом растворе
(УА и УВ)
у — число фаз
а, Ь, с — коэффициенты уравнения температурной зависимости средней
теплоемкости
а, р, у — коэффициенты уравнения температурной зависимости
истинной теплоемкости
ее (/) — уравнение солидуса [ее' (t) — его производная по
температуре]
6 — число пробегов химической реакции
у. А и к в — концентрация, % (атомные или молекулярные), компонентов
А и В
Ка и Kb — концентрация, % (по массе), компонентов А и В
[i — опытная молекулярная масса
[ii — химический потенциал компонента i в растворе
о (^ъ Ш» Ш>- • •)
jit. — нормальный химический потенциал компонента i в растворе
(\i°A и р°в)
Фт — приведенный потенциал при температуре Т, °К
АФ7 — изменение приведен-ного потенциала при температуре Г, ° К
Q — термодинамическая вероятность состояния системы
Часть первая
ТЕРМОДИНАМИКА И УЧЕНИЕ
О ХИМИЧЕСКОМ РАВНОВЕСИИ
Глава I
ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ
§ 1. СОДЕРЖАНИЕ ПЕРВОГО НАЧАЛА
Первое начало термодинамики представляет собой частный
случай закона сохранения материи и рассматривает сохранение энергии
при различных превращениях одних форм движения материи в
другие.
На заре развития термодинамики в ней в основном
рассматривались взаимопереходы теплоты и работы, поэтому прежде всего
было установлено сохранение энергии при переходе теплоты в
механическую работу и эквивалентность теплоты и работы. Позднее
была установлена эквивалентность теплоты многим видам энергии.
Первое начало рассматривается термодинамикой как постулат,
поскольку оно не может быть выведено или доказано какими-либо
логическими приемами. Содержание первого начала термодинамики
вытекает из обобщения многолетнего опыта, накопленного
человечеством в результате практической деятельности. В настоящее время
его справедливость признана всеми естествоиспытателями, поскольку
ни одно из следствий, к которым оно приводит, не находится в
противоречии с опытом. Исторически сложилось несколько
формулировок первого начала термодинамики, которые рассматривают
объективно существующий закон с различных сторон и
свидетельствуют о том, что исследователи приходили к его формулировке
разными путями.
Закон сохранения энергии в самом общем виде был впервые
сформулирован М. В. Ломоносовым (1748 г.), который в письме
к Эйлеру писал:- «Все перемены, в натуре случающиеся, такого
суть состояния, что сколько чего у одного тела отнимается, столько
же присовокупится к другому. Так, ежели где убудет несколько
материи, то умножится в другом месте». «Сей всеобщий естественный
закон простирается и в самые правила движения: ибо тело,
движущее своей силой другое, столько же оное у себя теряет, сколько
сообщает другому, которое у него движение получает».
В установлении современной формулировки первого начала
большую роль сыграли работы Г. И. Гесса, Майера, Джоуля и Гельм-
гольда. Остановимся на некоторых из этих формулировок.
Устанавливая зависимость между различными видами энергии,
Гельмгольц писал: «Энергия в природе не уничтожается и не
возникает вновь, а лишь переходит из одного вида в другой. Таким
образом, если в каком-либо процессе энергия одного вида исчезает, то
11
взамен ее появляется энергия в другой форме и в строго
эквивалентном количестве».
Поскольку самые различные виды энергии способны к взаимным
превращениям в строго эквивалентных количествах, то отсюда
вытекает формулировка Джоуля: «В любой изолированной системе
общий залас энергии сохраняется постоянным».
Так как все виды энергии являются формами движения материи,
взаимопревращение которых происходит всегда в одинаковых
соотношениях, то формулировка Майера выражает закон сохранения
энергии как закон неуничтожаемости движения. Ф. Энгельс,
подчеркивая эту мысль, писал: «Изменение формы движения является
всегда процессом, происходящим по меньшей мере между двумя
телами, из которых одно теряет определенное количество движения
такого-то качества (например теплоту), а другое получает
соответствующее количество движения такого-то другого качества
(механическое движение, электричество, химическое разложение).
Следовательно, количество и качество соответствуют здесь друг другу
взаимно и обоюдосторонне»*. Движение материи неуничтожимо как в
количественном, так и в качественном смысле. Движение материи
всегда сохраняет безграничную способность к качественным
превращениям из одной формы в другую.
Из указанных формулировок следует невозможность создания
такого механизма, который позволил бы получить работу, не
затрачивая на это соответствующего количества энергии. Иначе
говоря, вечный двигатель (перпетуум мобиле) первого рода
невозможен.
Можно дать еще много других различных формулировок первого
начала, которые логически связаны между собой и могут быть
выведены одна из другой. Однако для того, чтобы рассмотреть
математическую формулировку первого начала и имеющие для нас
наибольшее значение следствия, вытекающие из первого начала
термодинамики, необходимо познакомиться с таким
термодинамическим понятием, как внутренняя энергия.
Внутренняя энергия. Из разных форм энергии для
характеристики химических процессов особенно важна та энергия, которая
в скрытой форме заключена в каждом теле и имеет своей причиной
движение молекул, атомов и их составных частей. Эта энергия
называется внутренней энергией. Она равна полной энергии, за
вычетом кинетической и потенциальной энергии системы как целого.
Весь запас внутренней энергии складывается из энергии
поступательного, колебательного и вращательного движения всех
элементарных составных частей вещества, а также энергией их
взаимодействия, если оно имеет место **.
Количество внутренней энергии зависит от массы и природы
* К. Маркс, Ф. Энгельс. Сочинения. Изд. 2-е, т. 20. Госполитиздат,
1961, стр. 385.
** Классическая термодинамика не занимается изучением отдельных слагаемых
внутренней энергии. Это является предметом изучения статистической
термодинамики.
12
тела и от его состояния, т. е. от внешних условий, в которых
находится это тело. Основную роль при этом играют объем тела У,
давление Р и его температура Т. Эти три переменных параметра
однозначно определяют термодинамическое состояние тела и
изменяются в большей или меньшей степени при любом
физико-химическом процессе. Внутренняя энергия обычно обозначается
символом U.
Поскольку внутренняя энергия зависит еще от количества взятого
вещества, то для определенности условимся в дальнейшем относить
ее к молю вещества. В термодинамике для измерения внутренней
энергии так же, как в термохимии для измерения тепловых
эффектов химических реакций и превращений, общепринятым стало
применять тепловые единицы: калория (кал) или килокалория (ккал
или Кал). Калорией называется количество теплоты, необходимое
для нагревания 1 г воды на 1° С; килокалорией — количество
теплоты, необходимое для нагревания 1000 г воды на 1° С. Обе эти
величины относятся к интервалу температур от 14,5 до 15,5° С.
Международной системой единиц рекомендована для
предпочтительного использования энергетическая единица джоуль (дж)9 которая
определяется как работа силы в один ньютон (н) на пути в один
метр (м), а ньютон определяется как сила, придающая массе 1 кг
ускорение 1 м/сек2. Соотношение между упомянутыми единицами
следующее: 1 дж = 0,23901 кал или 1 кал = 4,1840 дж (см. табл. 1
на стр. 18).
При изменении состояния тела или системы изменяется
внутренняя энергия. Если в первоначальном состоянии 1 внутренняя
энергия была Ul9 а в конечном состоянии 2 она стала £/2, то величина
AU = U2 — Uг называется изменением внутрен ней
энергии. Термодинамика оперирует этим понятием и лишь
в редких случаях прибегает к рассмотрению абсолютной величины
внутренней энергии *.
Согласно созданной Эйнштейном теории относительности,
всякое изменение внутренней энергии AU системы должно изменять
ее массу Am на величину, которая получается делением энергии
на квадрат скорости света с, поскольку
AU = с2Ат. (1)
Но эти изменения массы в силу большой величины скорости света
(с = 3 X 1010 см1сек) незначительны для всех тех явлений, которые
рассматриваются химической термодинамикой. Поэтому даже при
химических реакциях с наибольшими изменениями внутренней
энергии обычное взвешивание не может доказать изменения общей
массы реагирующих веществ и остается справедливым закон
сохранения массы, сформулированный в работах М. В. Ломоносова и
Лавуазье.
* Определение количества энергии, отдаваемого или приобретаемого системой,
не представляет затруднений, но измерение полной энергии системы считалось до
последнего времени неосуществимым (для практических целей в таком измерении нет
необходимости и до настоящего времени).
13
Представим себе, что перевод вещества из состояния 1 в
состояние 2 связан с изменением внутренней энергии Д{/', а при обратном
переходе из состояния 2 в состояние 1 другим путем изменение
внутренней энергии составляет AU". Другими словами, мы при
этом получаем выигрыш или потерю энергии AU' — AU" y что
невозможно, поскольку является нарушением закона сохранения
энергии. Таким образом, изменение внутренней энергии системы
при любом процессе зависит не от пути этого процесса, а лишь от
начального и конечного состояний.
Это еще одна формулировка первого начала термодинамики.
Из нее следует, что каждому состоянию соответствует определенная
величина внутренней энергии независимо от того, каким путем это
состояние было достигнуто. В связи с £тим возможна другая
формулировка этого положения: внутренняя энергия есть однозначная
функция состояния.
Если обратиться к бесконечно малым процессам, которые
сопровождаются бесконечно малым изменением внутренней энергии
системы, то необходимо отметить, что величина dU обладает теми же
свойствами, что и AU: она не зависит от пути перехода системы из
одного состояния в другое. Дифференциалы, обладающие таким
свойством, в математике называются полными дифференциалами
функций. Свойством полного дифференциала является также то,
что он равняется сумме частных дифференциалов рассматриваемой
функции по переменным.
Например, если z является однозначной функцией переменных х и у, т. е.
* = / (ху),
то
dZ=(lk)ydX+{-w)Xdy'
где первое слагаемое представляет собой частный дифференциал функции z по
переменной х при постоянном значении у, а второе слагаемое — частный дифференциал
функции z по переменной у при постоянном значении х.
Легко видеть, что повторное дифференцирование ( -т~ ) по у и ( —г- \ по а:
приводит к одному и тому же результату
d2z d2z
дх ду ду дх '
так как результат двойного, так называемого перекрестного, дифференцирования не
зависит от порядка, в котором переменные принимают постоянное значение.
Следовательно, в выражении
dz = M dx + Ndy
признаком того, что z = f (х, у) есть однозначная функция состояния, a dz есхь
полный дифференциал этой функции, является равенство
/ дМ \ _ / dN \
\ ду )х ~\дх)у'
14
Тот факт, что полный дифференциал не зависит от пути перехода, представляют
в виде математического выражения
dz = О,
где z — однозначная функция состояния;
dz — полный дифференциал этой функции;
ф — интегрирование по замкнутому контуру.
Другими словами, если интеграл по замкнутому контуру от некоторой функции
равняется нулю, то подынтегральное выражение представляет собой полный
дифференциал этой функции.
Используя указанные математические понятия, можно кратко
в аналитической форме представить первое начало термодинамики
следующим образом:
2 1
J dU + J dU = О
1 2
или
<j)dt/ = 0. (2)
Из уравнений (1) и (2) следует, что внутренняя энергия является
однозначной, непрерывной и конечной функцией параметров,
определяющих состояние системы. Выражение (2) широко используется
в классической термодинамике, поскольку является наиболее общим
выражением одного из ее отправных положений — первого начала
термодинамики.
§ 2. МАТЕМАТИЧЕСКОЕ ВЫРАЖЕНИЕ
ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ
Рассмотрим изолированное вещество, для которого отсутствует
энергетический обмен с окружающей средой. Внутри такой системы
могут происходить лишь процессы, сопровождающиеся взаимными
превращениями различных видов энергии в строго определенных
соотношениях. Поскольку общий запас энергии этой системы —
постоянная величина
U = const,
то
dU = 0.
Изменение внутренней энергии может происходить за счет
подвода к системе некоторого количества теплоты из окружающего
пространства или отвода теплоты от системы. Подвод теплоты к системе
увеличивает запас ее внутренней энергии, а отвод, наоборот,
уменьшает.
Запас внутренней энергии системы может изменяться в
результате работы, совершаемой системой или производимой внешними
силами над системой. Если система осуществляет работу против
действия внешних сил, то ее внутренняя энергия уменьшается.
15
§
Если работа совершается внешними силами над системой, то запас
энергии системы увеличивается.
Условимся в дальнейшем пользоваться следующими
обозначениями. Теплота, получаемая системой извне, положительна (Q > 0);
теплота, отводимая от системы, отрицательна (Q <<0).
Положительной (А > 0) будем считать ту работу, которую совершает сама
система (работа расширения); отрицательной (А <0) условимся
считать ту работу, которую совершают внешние силы над системой
(работа сжатия). Соответственно dQ и dA характеризуют бесконечно
малые изменения, которые претерпевает система. Изменение
внутренней энергии будем считать положительным, если процесс
сопровождается приращением внутренней энергии системы.
Отрицательное значение будет соответствовать убыли внутренней энергии.
Теперь первое начало термодинамики можно представить в виде
уравнения
dQ=,dU + dA. (3)
Уравнение (3) соответствует следующему положению:
поглощаемое системой тепло расходуется и на увеличение внутренней энергии
системы и на совершение внешней работы — работы расширения
Все члены уравнения, а следовательно, и обе части уравнения должны
быть выражены в одних и тех же единицах.
Если рассматривать не дифференциально малые, а конечные
количества теплоты, внутренней энергии и работы, то
Q=U + A. (4)
Однако такая запись требует некоторых уточнений. Не следует
думать, что теплота, работа и внутренняя энергия оцениваются
одинаково. Хотя теплота и работа могут измеряться в одинаковых
единицах, эти величины значительно отличаются друг от друга по
своей физической сущности. Теплота и работа не являются формой
энергии, они представляют собой лишь форму перехода энергии от
одного тела к другому. Кроме того, теплота и работа являются
абсолютными величинами и в уравнении (4) связаны с разностной
величиной — внутренней энергией. Поэтому обычно для того, чтобы
более выпукло представить различие свойств энергии, теплоты и
работы, первое начало термодинамики в интегральной форме
записывается иначе:
Q = U2 - Ux + A (5)
или
Q = AU + А. (6)
Так как среди рассматриваемых величин только дифференциал
внутренней энергии обладает свойством полного дифференциала,
а теплота и работа зависят от пути перехода и их дифференциалы не
являются полными дифференциалами, то первое начало
термодинамики в дифференциальной форме иногда записывается следующим
образом:
8Q = dU + 8А. (7)
16
Необходимо заметить, что указанные формы математической
записи первого начала термодинамики справедливы лишь при
принятых условных обозначениях. Возможны иные условные
обозначения. Например, когда отрицательным считают увеличение, а не
уменьшение внутренней энергии. В этом случае форма записи должна
быть изменена:
Д[/ = А — Q. (8)
Однако физический смысл уравнений при этом полностью
сохраняется, но их использование всегда требует внимательного
отношения к принятым условным обозначениям.
Рассмотрение полученных уравнений позволяет сделать важные
заключения. Например, если имеет место процесс, при котором
U = const или Д(/ = О, Q = Л, т. е. вся подводимая теплота к
системе полностью расходуется на производство работы, а работа,
производимая над системой, полностью превращается в теплоту
и выделяется системой.
Обнаруживается также возможность использования внутренней
энергии. Если Q = 0, то —Д£/ = А. Другими словами, при
исключении теплового обмена системы с окружающей средой (адиабатная
система) работа расширения в системе может произойти только за
счет убыли запаса ее внутренней энергии. Кроме того, при А = О
имеем Q = Д[/, т. е. если система не может совершать работу
расширения, то вся подводимая к системе теплота расходуется только
на увеличение ее внутренней энергии.
§ 3. СВЯЗЬ МЕЖДУ ЕДИНИЦАМИ ИЗМЕРЕНИЯ ЭНЕРГИИ
Можно установить связь между единицами измерения теплоты
и единицами измерения механической работы. Эти величины
связаны через коэффициент пропорциональности, одинаковый для всех
видов процессов. Многочисленные и точные опыты позволили
вычислить этот коэффициент пропорциональности.
Оказалось, что механический эквивалент теплоты 1 кал =
= 0,427 кГм> а обратная ему величина (термический эквивалент
работы) 1 кГм = 1/0,427 кал = 2,34 кал.
Сущность экспериментального определения эквивалентов
различных видов энергии состоит в следующем. Каким-либо способом
затрачивается определенное количество энергии (механической,
электрической и т. д.) и производится точное определение
выделившейся при этом энергии другого вида (например, теплоты). При этом
устраняются все возможные энергетические потери.
Международной системой единиц установлена единая мера
энергии, теплоты и работы—джоуль. Выражая ее через основные
единицы этой системы и используя сокращенные обозначения (дж —
джоуль, н — ньютон, кг — килограмм, м — метр и сек — секунда),
получаем:
1 дж = (1 «).(1 м) - (J ^)НЫ1р|42)-(1 м).
17
Однако часто используются так называемые внесистемные единицы
энергии, теплоты и работы: калории (кал), килограммометры (кГ-м)
и литр-атмосферы (л-ат). Кроме того, применяются кратные
и дольные единицы джоулей и калорий. Наименование таких единиц
образуется с помощью приставок к простым наименованиям. В
зависимости от кратности и дольности могут использоваться
приставки: тера (1012), гига (109), мега (106), кило (103), гекто (102), дека
(10), деци (10-1), санти (10~2), милли (Ю~3), микро (10~6), нано
(10~9), пико (К)"12), фемто (К)"15) и атто (10~18).
В табл. 1 приведены переводные коэффициенты для различных
единиц измерения энергии.
Таблица 1. Соотношение между различными единицами энергии
Единицы
измерения энергии
1 дж
1 кал
1 кГм
1 л-ат,
дж
1
4,1840
9,80665
101,328
кал
0,23901
1
2,3438
24,218
кГм
0,101971
0,42684
1
10,383
л-ат
0,009866894
0,04129
0,09678
1
Примечание. 1 ккал = 1- К)3 кал; 1 л-ат = 1-103 см3-am и 1 дж = ЫО7 эре.
§ 4. ТЕРМОДИНАМИЧЕСКИЕ ПРОЦЕССЫ
Состояние системы определяется всей совокупностью ее свойств
и при изменении одного из них изменяются и все остальные. Так,
например, изменение давления влияет в той или иной степени на все
свойства системы: температуру, объем и др. Во избежание излишней
сложности ограничимся рассмотрением только таких свойств, как
давление, объем и температура, и только таких процессов, при
которых происходит изменение этих свойств. Что же касается
электрических, магнитных, оптических и т. п. свойств, то мы будем
предполагать их неизменными.
Если ограничиться изучением только давления, объема и
температуры системы и не принимать в расчет ее электрическое и
магнитное состояние, а также предположить, что система находится в
покое, то состояние такой системы определится совершенно
однозначно указанными тремя величинами: испытываемым давлением р,
занимаемым объемом V и температурой Т. Следовательно, давление,
объем и температура должны быть связаны между собой
зависимостью, которая в самом общем виде выражается уравнением:
, / (р, V, Т) = 0. (9)
Это уравнение носит название уравнения состояния
и представляет собой уравнение некоторой поверхности в
координатной системе трех взаимно перпендикулярных осей: р, V и Т
18
f(W)*o
(рис. 1). Эта поверхность называется термодинамической
поверхностью. Всякое состояние системы изображается
некоторой фигуративной (изобразительной) точкой, лежащей
на этой термодинамической поверхности. При изменении состояния
системы происходит перемещение фигуративной точки по
термодинамической поверхности. Обычно для удобства вместо
пространственного представления термодинамической поверхности пользуются
ее проекциями на координатные плоскости: давление — объем,
давление — температура, объем — температура.
Конкретные уравнения состояния определяются опытным путем.
Вид их в огромном большинстве случаев сложен. Мы ограничимся
в данном разделе
рассмотрением лишь уравнений
состояния так называемого
идеального газа, т. е.
воображаемого агрегатного
состояния вещества: оно
представляет собой
совокупность беспорядочно
движущихся атомов или молекул,
взаимодействие между
которыми осуществляется не
иначе, как через упругие
столкновения. Другими
словами, это состояние
характеризуется высокой
разреженностью вещества, при
которой среднее расстояние
между атомами и
молекулами в каждое данное мгновение весьма велико, а взаимодействие
между ними настолько мало, что, не допуская какой-либо
погрешности, его можно не принимать во внимание. Некоторые газы,
в частности водород или азот, при высоких температурах или
низких давлениях могут в известном приближении служить примером
этого гипотетического (идеального) состояния газа.
Рассмотрим конкретный вид уравнения состояния идеального
газа.
Если изменение состояния системы происходит при постоянном
значении давления, то такой термодинамический процесс
называется изобарическим или изопьезическим.
Изменяющиеся при этом параметры — объем и температура — связаны
между собой уравнением, которое явилось результатом
экспериментальных исследований Гей-Люссака:
(10)
Температура Т
Рис. 1. Пространственное
изображение термодинамической
поверхности
V = V0 (1 + at),
где
>*
V — объем газа при температуре t9 отмечаемой по шкале
Цельсия (°С);
V0 — объем газа при температуре 0° С;
а — коэффициент термического расширения.
19
Численное значение коэффициента а составляет величину
1 1
а
273,16
Для температуры tx
Vi = V0(l
273 '
к
273
(И)
(12)
изохора
а для температуры /.
V.
2 = V0(l +
273
(13)
Разделим уравнение
(12) на уравнение (13) и,
учитывая, что
Т = t + 273, (14)
где Т — абсолютная
температура,
отмечаемая по шкале
Кельвина (°К),
получим
Рис. 2. Линий важнейших термодинамических
процессов и их краткие характеристики
V*
-Tjr- ИЛИ
1 2
V2 =
Го
Следовательно:
т
const.
(15)
(16)
Полученное соотношение носит название закона Гей-Люссака
и его можно сформулировать следующим образом: объем данной
массы газа при постоянном давлении прямо пропорционален
абсолютной температуре. График этой зависимости в координатах
давление—объем представляет собой прямую линию, параллельную оси
абсцисс (рис. 2), и называется изобарой.
Термодинамический процесс, который происходит при постоянном
объеме, называется изохорическим или изопикническим.
Изменяющиеся при этом параметры — давление и температура — связаны
уравнением, которое получило название закона Шарля:
_ Тх
р2
т<
или
El
Р2_
То
Следовательно:
--- = const.
(17).
(18)
Таким образом, давление данной массы газа при постоянном
объеме прямо пропорционально абсолютной температуре. График
этой зависимости в координатах давление—объем представляет
20
собой прямую линию, параллельную оси ординат (рис. 2), и
называется изохорой.
Термодинамический процесс, который происходит при
постоянной температуре, называется изотермическим процессом.
Изменяющиеся при этом параметры — давление и объем — связаны
уравнением, экспериментально полученным Бойлем и Мариоттом:
~Г=~у- или PlVt = p,V2 = • • • (19)
Следовательно:
pV = const. (20)
Таким образом, при постоянной температуре объем данной
массы газа обратно пропорционален давлению. Учитывая, что
плотность газа есть величина обратная объему, т. е.
Р = -р-, (21)
указанное соотношение, известное под названием закона Бойля—
Мариотта, может быть сформулировано следующим образом: при
постоянной температуре плотность газа пропорциональна его
давлению. Зависимость объема и давления, при постоянной температуре
в координатах давление — объем представляет собой равностороннюю
гиперболу, асимптоты которой совпадают с осями координат, и носит
название изотермы (рис. 2).
Однако в действительности очень редко приходится встречать
термодинамические процессы, которые протекают таким образом,
что один из переменных параметров состояния (температура, объем
или давление) сохраняет постоянное значение. Чаще происходит
одновременное изменение всех параметров.
Поскольку известны зависимости между каждой парой из трех
переменных при постоянстве третьей, то имеется возможность найти
соотношение между всеми тремя переменными, характеризующими
состояние идеального газа.
Пусть в каком-то произвольном состоянии моля газа эти
переменные имеют значение р, V и Г, а в другом состоянии того же
количества газа переменные имеют значения ръ Vx и 7\. Перейдем
в две стадии из одного состояния в другое. Сначала изменим давление
от Р до р при постоянной температуре Т. При этом объем изменится
от У до V и, согласно закону Бойля—Мариотта:
pV = pxY. (22)
Затем изменим температуру от Г до Тг при постоянном давлении р1у
отчего объем изменится от V до Vly так как давлению рх и
температуре 7\ отвечает одно единственное значение объема Ух. Согласно
закону Гей-Люссака:
21
Исключая V из уравнений (22) и (23), находим, что
pV = P1V1
т тл '
(24)
Так как оба состояния (р, Vy Т и р1э Vly 7\) были выбраны
произвольно, то для любого состояния произведение давления на объем,
деленное на абсолютную температуру, есть величина постоянная:
-f- = R, (25)
где R — величина, которая не зависит от р, V и Ту но может
зависеть от количества молей данного газа. Для того чтобы исключить
влияние этого фактора, преобразуем уравнение (25) таким образом,
чтобы оно было справедливо для любого числа молей:
pV = nRTy (26)
где п — число молей газа, a R относится к 1 моль газа.
Это — одна из основных закономерностей, которым подчиняется
поведение идеального газа. Примечательно, что в ней отсутствуют
параметры, зависящие от природы газа. Эта зависимость получила
название основного объединенного уравнения состояния идеального
газа, или уравнения Менделеева —К лапейрона.
Входящая в уравнение постоянная R называется
универсальной газовой постоянной.
Один из важнейших газовых законов, открытый. Авогадро,
позволяет определить численную величину универсальной газовой
постоянной R. Закон Авогадро гласит: при одинаковом давлении
и одинаковой температуре одинаковые объемы различных газов
содержат одинаковое число молекул. Этому закону подчиняются
только идеальные газы. При нормальных условиях (О °С и
760 мм pm. cm.) объем одного моля идеального газа V0 = 22,415 л.
Число молекул, содержащихся в этом объеме, т. е. в одном моле,
называется постоянной Авогадро и составляет
N0 = 6,023-1023.
Численная величина R зависит, очевидно, от выбора единиц
измерения давления, объема и температуры. Будем измерять
давление атмосферами (760 мм рт. ст.)у объем-литрами (объем 1000 г
дистиллированной воды при 4° С) и температуру — градусами
абсолютной шкалы (°К). Поскольку при р0 = 1 am и Т0 = 273,16 °К
объем одного моля (п = 1) идеального газа VQ = 22,415 л, то
"^ = ^7з1б5 = °>08206 л-ат/(моль-град).
Используя соотношения различных единиц измерения энергии,
представленные в табл. 1, можно получить численное выражение
22
постоянной R в различных единицах измерения:
R = 0,08206-1,0133-109 = 8,314 дж/(моль-град);
R = 0,08206-24,214 = 1,987 кал/(моль-град);
R = 0,08206-103-760 = 62 320 мл-мм рт. ст./(моль-град) *.
Легко видеть, что R имеет размерность энергии, отнесенной
к одному молю и к одному градусу. Это обстоятельство позволяет
определить физический смысл газовой постоянной. Универсальная
газовая постоянная равна работе расширения, которую совершает
один моль идеального газа при изобарическом повышении его
температуры на один градус. Постоянная R есть та величина, на
которую разнятся молекулярные теплоемкости при р = const (Cp)
и при V = const (Cv)y т. е.
Cp-CV = R. (27)
Это уравнение получило название уравнения Майера.
Уравнение (25) pV = RT представляет собой уравнение
поверхности второго порядка, которое графически изображается в виде
гиперболического параболоида. Эта поверхность относится к числу
линейчатых поверхностей, т. е. к таким поверхностям, которые могут
быть образованы движущейся прямой. Прямые, укладывающиеся
на этой поверхности, распадаются на две группы, получившие в
нашем случае название изохор и изобар, так как они попадают в
сечение гиперболического параболоида pV = RT плоскостями при
V = const или р = const. Сечение плоскостью при Т = const
представляет собой разностороннюю гиперболу.
В термодинамике, кроме рассмотренных процессов при V =
= const, p = const и Г = const, используется представление о так
называемом адиабатическом процессе. Все точки кривой,
определяющей этот-процесс, лежат на поверхности pV = RT, но не
попадают в сечение плоскостью, параллельной какой-либо из
координатных плоскостей, поскольку ни один из параметров не сохраняет
постоянного значения. Характерной особенностью этого процесса
является отсутствие всякого теплового обмена с окружающей средой.
В этом случае уравнение первого начала термодинамики (3)
dQ = dU + dA вследствие отсутствия подвода какого-либо
количества теплоты к системе или его отвода от системы, т. е. при
dQ = 0, (28)
принимает следующий вид:
dU + dA = 0. (29)
* Поскольку в физической химии большинство величин относится к молю, то
часто размерность «моль» опускается. Кроме того, для приближенных расчетов,
принимая во внимание неточность газовых законов достаточно положить R =
= 0,082 л-am/град, или R = 2 кал/град.
23
Учитывая, что
dU = CydT * (30)
и
dA = pdV **, (31)
получаем
CvdT + pdV - 0. (32)
Подставляя значение p = RTIV из уравнения (26):
CvdT + RT -~ = 0. (33)
Разделив это уравнение на Т (позднее будет показано, что Т =£ 0),
получим
Cv~-\-R^-^ 0. (34)
Подставляя в это уравнение значение R из уравнения (27), получаем
Cv ^- + (Ср - Cv)^- = 0. (35)
Разделив уравнение (35) на Cv и обозначив
Су
получим
= Y, (36)
-yL + (Y-l)-yL=0. (37)
Интегрирование уравнения (37) дает
In Т + (у — 1) In V = const"'. (38)
Потенцируя уравнение (38), получаем
TVy~l = const". (39)
Подстановка вместо V его значения из уравнения (26) позволяет
преобразовать уравнение (39) к виду
ГУ"7 = const'. (40)
Наконец, подставляя вместо Т его значение из уравнения (26),
получаем
pVv = const. (41)
Уравнения (39), (40) и (41) представляют собой аналитическое
выражение адиабатического процесса. Эти уравнения
* Это уравнение является следствием того, что теплоемкость при постоянном
объеме Су представляет собой первую производную внутренней энергии по темпера-
(du\ и
туре, т. е. Су = \-ppj. ria доказательстве этого положения остановимся ниже, при
рассмотрении теплоемкостей.
** Это выражение будет также выведено несколько позже.
24
содержат величину у, которая получила название
адиабатического коэффициента и представляет собой
отношение молекулярной теплоемкости при постоянном давлении Ср к
молекулярной теплоемкости при постоянном объеме Cv. Согласно
молекулярно-кинетической теории, величины Ср ие Су кал/(моль-град)
идеального газа, а следовательно, и адиабатический коэффициент
определяются строением молекулы газа:
S cv . v
Одноатомный газ 5 3 1,66
Двухатомный газ 7 5 1,44
Трехатомный газ 9 7 1,28
Сопоставление уравнений, характеризующих процессы при р =
= const, V = const, T = const и dQ = О, показывает, что эти
процессы можно представить в обобщенном виде:
pVn = const. (42)
Придавая показателю степени п значения от 0 до оо, получаем
уравнения, характеризующие самые разнообразные возможные
термодинамические процессы. Процессы, отвечающие произвольному
значению пу получили название политропических процессов.
Наиболее часто используемые частные случаи политропических
процессов нам уже известны:
1) изобарический процесс (п = 0):
pV° = const,
р = const;
2) изотермический процесс (п = 1):
pV = const;
3) адиабатический процесс (п — у):
р]/у = const;
4) изохорический процесс (п = сю):
1
рУ00 — const, р °° V = const,
p°V = const, V = const.
На рис. 2 совместно представлено графическое изображение
этих процессов в виде проекции на координатную плоскость
давление—объем. График наглядно показывает, что по мере увеличения
показателя степени п усиливается крутизна линии, представляющей
данный процесс.
Величина площади, ограниченной этими линиями и осью абсцисс,
характеризует работу изменения объема при соответствующем
процессе. Следовательно, эта работа для каждого из рассмотренных
процессов будет различной.
Работа термодинамических процессов. В термодинамике для
иллюстрации различных положений широко используется
наглядная модель, называемая «цилиндром с поршнем». Цилиндр с одной
стороны ограничен дном, а с другой — подвижным поршнем, который
позволяет изменять объем содержащегося внутри цилиндра газа,
однако перемещение его не сопровождается трением о внутреннюю
сторону стенок.
Представим себе такой цилиндр, наполненный идеальным газом.
Газ оказывает давление на стенки сосуда и на внутреннюю
поверхность поршня. Величина давления газа р. Величина поверхности
поршня s. Следовательно, величина силы, стремящейся переместить
поршень вверх, определится как
F = ps. (43)
Если при этом поршень переместится на высоту dx, то
совершенная элементарная работа
dA = F dx = ps dx. (44)
Учитывая, что при этом произошло изменение объема газа:
dV = s dx, (45)
получаем в окончательном виде выражение для элементарной работы:
dA = р dV. (46)
Теперь выясним, к чему приводит интегрирование этого общего
выражения для различных частных случаев.
Изохорический процесс характеризуется
постоянством объема. Следовательно, V = const, dV = 0; поэтому
dA =0. (47)
Поскольку этот процесс протекает в замкнутом пространстве
(поршень, закрывающий цилиндр, неподвижно закреплен в одном
из положений), то расширение и связанная с ним работа отсутствуют.
В этом случае первое начало термодинамики
dQ == dU Л- dA
приобретает вид
dQ - dU (48)
или
Q - At/, (49)
т. е. убыль внутренней энергии системы обращается в теплоту,
выделяющуюся при протекании процесса, или, наоборот, вся
поглощенная теплота тратится на приращение внутренней энергии.
Рассмотренное положение позволило Джоулю опытным путем
показать, что если при постоянной температуре изменять объем или
давление идеального газа, то его внутренняя энергия остается
постоянной. Иначе говоря, внутренняя • энергия идеального газа
есть функция только его температуры.
26
В опытах Джоуля использовали два сосуда: из одного газ был
откачан, а другой содержал газ под давлением. Оба сосуда
помещали в два калориметра и соединяли трубкой с краном. Кран
открывали и давление в сосудах выравнивалось. После этого первый сосуд
приобретал приблизительно столько же теплоты, сколько терял
второй. Так как при этом никакой внешней работы не совершалось
(А = 0) и тепловой эффект в сумме приблизительно равен нулю
(Q = 0), то> согласно уравнению (49), Д[/ = 0, т. е. внутренняя
энергия газа не изменялась при увеличении его объема и
уменьшении давления.
Изобарический процесс характеризуется
постоянством давления. Следовательно, выражение dA = р dV после
интегрирования при условии р = const приобретает вид:
А = р (Va - V,). (50)
Работа, совершаемая при этом процессе (на внешнюю
поверхность поршня действует постоянное давление, например
атмосферное), пропорциональна изменению объема газа. Раскрывая скобки
и используя уравнение (25), приходим к другому выражению работы
изобарического процесса:
A = R(Tt-T1). (51)
Работа изобарического процесса пропорциональна изменению
температуры газа. При постоянном давлении подведение теплоты
к газу приводит к тому, что газ совершает положительную работу
(работу расширения), сопровождающуюся увеличением объема (см.
уравнение (50)] и повышением температуры [см. уравнение (51)].
Изотермический процесс характеризуется
постоянством температуры. Следовательно, dA = p dV, но, согласно
уравнению (25), р = RT/V, поэтому
<1А = Д£- dV. (52)
Интегрируя это уравнение и полагая Т = const, получаем
А = RT In -£-. (53)
По закону Бойля—Мариотта -гг" — \ поэтому
A = RT\n-^. (54)
Поскольку, согласно опытам Джоуля, при постоянной
температуре внутренняя энергия идеального газа остается постоянной, то
уравнение первого начала термодинамики (при Т = const и Д£/ = 0)
приобретает следующий вид:
Q = A. (55)
что при изотермическом процессе работа, совер-
гемой, полностью превращается в теплоту, которую
27
система выделяет в окружающее пространство, или, наоборот,
подводимая к системе теплота полностью расходуется на производство
работы расширения.
Адиабатический процесс характеризуется
выражением dQ = 0. Следовательно, уравнение первого начала
термодинамики в этом случае имеет вид dU + dA = 0
dA = —dU. (56)
Так как dU = Cv dTy то
dA = — Cv dT. (57)
Интегрирование дает следующую
зависимость:
А = -Су(Тг- Т2). (58)
или
п
^Изогперпа
чч \ Адиабата
\ \ I Изохора
*, ^!
Изобара х ■
Рис. 3. Графическое изображение
работы расширения идеального газа от
объема Vх До объема V2
Осуществляя подстановку
. piVi
Согласно этому уравнению,
работа адиабатического процесса
пропорциональна изменению
температуры газа, а также
пропорциональна его теплоемкости при
постоянном объеме. Последнее
обстоятельство позволяет сделать
заключение, что при адиабатном
расширении газа, имеющего более
сложное строение молекул, можно
получить большую работу.
Тг
получаем
R
А =
и Тс
R
Су
R
(PiVi — pzVz).
(59)
Так как R = CD — CVy то
Cv
A==c^c~v(Pivi-P*v^
c,
Используя адиабатический коэффициент у = -=г-
к новому выражению:
А =
1
7-1
(PiVi — р2^2).
приходим
(60)
Произведем замену: р^х = RTt и p%V2 = #Т2. Тогда
R
А
1
(7\-Г2).
(61)
Важно заметить, что работа расширения, величина
быть подсчитана по формулам (59)—(61), при адиаб;
совершается исключительно за счет убыли внутренн
28
стемы, а работа сжатия полностью расходуется на увеличение
внутренней энергии, поскольку отсутствует какой-либо иной
энергетический обмен между системой и окружающей средой.
На рис. 3 для сравнения представлено графическое изображение
работы расширения, совершаемой идеальным газом при изохори-
ческом, изобарическом, изотермическом и адиабатическом
процессах. Очевидно, наибольшей является работа при р = const, затем
при Т = const, далее при dQ = 0 и наименьшая — при V = const.
То же самое имеет место и для работы сжатия при возвращении газа
в исходное состояние с той лишь разницей, что ее величина будет
отрицательной.
§ 5. ТЕПЛОЕМКОСТЬ И ФОРМЫ ЕЕ ВЫРАЖЕНИЯ
Теплоемкость представляет собой одну из наиболее важных
характеристик вещества.
Из уравнения (3) первого начала термодинамики dQ = dU + dA
следует, что
dQ dU , dA _
dT ~ dT ' dT '
Величина
называется теплоемкостью. Теплоемкость показывает, какое
количество теплоты необходимо подвести к телу для заданного
повышения его температуры. В данном случае величина теплоемкости
определена как количество теплоты (дифференциально малое),
которое необходимо для дифференциально малого изменения
степени нагретости тела. Это так называемая истинная
теплоемкость.
Часто пользуются величиной средней теплоемкости
Легко видеть, что средняя теплоемкость равняется количеству
теплоты, необходимому для нагревания тела на 1°, причем t2 и t1 —
конечное и начальное значения температуры тела.
Если ^1 = 0 °С, a t2 принимает значение t> то уравнение (63)
упрощается:
С = -у- (65)
и величина С носит название средней теплоемкости в интервале
температур от 0 до ty °C.
В зависимости от того, к какому количеству вещества относится
рассматриваемая теплоемкость, различают удельную (на 1 г),
атомную (на 1 г-апгом) или молекулярную (на 1 моль вещества)
теплоемкости. Размерности этих теплоемкостей различны: удельной —
29
кал/{г-град); атомной — кал/(г-атом-град); молекулярной —
кал/(моль • град).
Поскольку величина Q зависит от пути процесса, то от него
также будет зависеть и величина теплоемкости. Чаще всего
используется теплоемкость при постоянном объеме Cv (изохорическая
теплоемкость) и теплоемкость при постоянном давлении Ср
(изобарическая теплоемкость), т. е.
Cv =
dU \ dQv
v~\dT
И
Здесь: уравнение (66) получено из уравнений (62) и (63) для
V = const (при этом dA = 0), а уравнение (67) получено
аналогично, но для р = const (здесь функция Н отличается от функции U
на величину совершаемой работы).
Связь между Ср ,и Cv
Как уже установлено уравнением (27), для моля идеального газа
связь между Ср и Cv выражается зависимостью
Cp-Cv = R. (68)
Для твердого состояния различие между Ср и Cv незначительно,
однако зависимость представляется более сложным уравнением *.
а2
ср—cv = T —— у0,
где а — изобарический коэффициент термического расширения;
к — изотермический коэффициент сжатия;
V0 — молярный объем при 0° К;
Т — температура.
При этом
а =
1 / dV
Vn \ дТ
ж\ m
И
■4(f), <70>
Эмпирические закономерности теплоемкостей
Из многочисленных эмпирических соотношений, предлагавшихся
для вычисления теплоемкостей твердых тел, наиболее часто для
приближенных расчетов используется правило Дюлонга—Пти,
а также правило Неймана—Коппа (известно под названием правила
аддитивности).
* Для вывода этого соотношения требуется привлечение второго начала
термодинамики; мы примем уравнение без доказательства.
30
Оба правила справедливы лишь в известном приближении и имеют
многие исключения.
Дюлонг и Пти сформулировали правило, согласно которому
атомная теплоемкость элементов в твердом состоянии, т. е.
произведение атомной массы элемента А на его удельную теплоемкость су
есть величина, приблизительно одинаковая для всех элементов и
в среднем
Ас = 6,2-г-6,4 кал1(г-атом-град). (71)
В формулировке этого правила не принято во внимание
значительное влияние температуры на теплоемкость, однако его принято
относить к небольшому интервалу комнатных температур.
Экспериментальные определения для кадмия, галлия, кальция, таллия,
свинца и индия дают следующие значения их атомных теплоемко-
стей при комнатной температуре, кал/(г-атом-град)\ 6,22; 6,23;
6,26; 6,29; 6,32 и 6,39 соответственно. Эти примеры подтверждают
правило Дюлонга и Пти. Исключения из этого правила составляют
такие элементы, как бор, углерод, кремний, сурьма и некоторые
другие.
Правило аддитивности, сформулированное Нейманом и Коппом,
позволяет приблизительно вычислять теплоемкость . химических
соединений. Согласно этому правилу, молекулярная теплоемкость
химических соединений в твердом состоянии, т. е. произведение
молекулярной массы на удельную теплоемкость, равна сумме
атомных теплоемкостей элементов, входящих в данное соединение.
Принимая во внимание правило Дюлонга и Пти для соединения,
состоящего из п атомов, можно написать
Мс = п (6,2-г-6,4) кал/(моль-град). (72)
Например, нас интересует теплоемкость сложного химического
соединения — криолита Na3AlF6 или AlF3-3NaF. Молекула этого
соединения состоит из десяти атомов. Следовательно, молекулярная
теплоемкость криолита будет приблизительно равна 6,3 -10 =
= 63 кал/(люль* град).
Экспериментально определенные значения теплоемкости криолита
при комнатной температуре составляют (по данным различных
авторов), кал/(моль-град): 51,60; 51,64 и 52,30. Эти значения
отличаются от рассчитанного значения 62 кал/(моль - град) на+16,8; +16,7
и +15,6% соответственно.
Подсчет величин молекулярных теплоемкостей для соединений
FeSi (6,3 X 2 = 12,6), Hg2Cl2 (6,3 х 4 = 25,2), Sb2S3 и Са2Те3
(6,3 X 5 = 31,5), а также для CaW04 [6,3 Хб = 37',8 кал/(моль - град)]
при сопоставлении с экспериментальными данными (11,4; 28,3;
29,5; 38,8 и 36,8 кал/(моль - град) дает следующие расхождения:
+ 10,5; —10,9; +6,8; —18,8 и +2,7% соответственно.
Для приближенных термодинамических расчетов часто такая
точность (до ±20%) оказывается достаточной.
31
Температурная зависимость теплоемкостей
Температура значительно влияет на величину теплоемкости и
это влияние различно в различных температурных интервалах.
Качественно характер изменения теплоемкости с изменением
температуры для металлов без превращений в твердом состоянии
представлен на рис. 4. По оси абсцисс отложена температура по шкале
Кельвина и нанесены точки, соответствующие комнатной
температуре (Ткомп)у температуре плавления (Тпл) и температуре кипения
(^кип)- По оси ординат отложены значения истинной теплоемкости С,
\
C=6,2+6,U
c*d*jrt+7T
С ^ Const
\
CF=5
CV=J
-I
-—Г
коми
Г
кип
Рис. 4. Схема изменения теплоемкости с температурой
относящейся к грамм-атому рассматриваемого металла. Учитывая
незначительное различие теплоемкостей Ср и Cv для твердого и
жидкого состояний, в этих случаях будем отвлекаться от этого
обстоятельства.
Для металлов в твердом состоянии в области низких температур
характерна зависимость теплоемкости от температуры,
изображаемая графически кривой, которая отвечает уравнению кубической
параболы. С понижением температуры теплоемкость быстро падает,
и при приближении температуры к абсолютному нулю теплоемкость
асимптотически стремится принять нулевое значение.
При повышении температуры теплоемкость резко возрастает.
Зависимость теплоемкости от температуры на этом участке
температур (от О °К) в большинстве случаев хорошо передается
полуэмпирическим уравнением Дебая:
С
v
Г'
(73)
в котором коэффициент пропорциональности а зависит от природы
рассматриваемого металла.
В области комнатных температур (Гкомн) теплоемкость
принимает значение, определяемое правилом Дюлонга и Пти.
32
Дальнейшее повышение температуры выше ТкОМН вплоть до Тпл
вызывает непрерывное увеличение теплоемкости до значений 7,0—
8,0 кал/(г-атом-град). Этот участок температур представляет
наибольший практический интерес. Зависимость теплоемкости от
температуры в этом интервале обычно принято выражать с помощью
эмпирических уравнений, имеющих вид степенных рядов:
С = а + РГ + уТ2 +••• (74)
Ввиду вполне достаточной точности часто ограничиваются тремя
членами степенного ряда, т. е. членом, содержащим Г2; тогда
уравнение принимает вид уравнения параболы:
С = а + РГ + уТ\ (75)
Иногда можно ограничиться двумя членами ряда, и тогда
зависимость выражается линейным уравнением:
С = а + р7\ (76)
Если при сравнительно низких температурах кривизна
увеличивается, а при высоких температурах зависимость теплоемкости
приобретает почти линейный характер, то, по предложению Келли,
используется уравнение вида
С = а + Р-71 + уТ~\. (77)
более точно описывающее указанную зависимость.
Уравнения зависимости теплоемкости от температуры могут
быть представлены через температуру как по шкале Кельвина
с = f (Т),
так и по шкале Цельсия
С = ф (*).
Поскольку Т °К и t °C отличаются друг от друга на постоянную
величину 273°, то уравнения в этом случае будут отличаться лишь
величиной коэффициентов.
Теплоемкость для жидкого состояния характеризуется
величиной, которая в большинстве случаев меньше величины теплоемкости
для твердого состояния. Применением точных калориметрических
измерений теплоемкостей было показано, что изменение
температуры жидкости вплоть до кипения практически не оказывает влия-
нвд**на величину теплоемкости, и она остается постоянной.
Поскольку большинство металлов в газообразном состоянии
является одноатомными, то их теплоемкости определяются
следующими величинами:
Cv = 3 и Ср = 5 кал/(г-атом-град).
3 А. Н. Крестовников 33
Связь между истинной
и средней теплоемкостями
Различают среднюю теплоемкость, относящуюся к изменению
температуры на некоторую конечную величину и равную
отношению количества сообщенной теплоты к соответствующему изменению
температуры:
С = • , , или С =
t% — * 1 t
и истинную теплоемкость, соответствующую бесконечно малому
изменению температуры:
dQ
С =
dt
Зависимость теплоемкости от температуры для интервала
комнатных, средних и высоких температур выражается эмпирическими
уравнениями степенного ряда. При этом для коэффициентов в
уравнениях истинной теплоемкости будем пользоваться буквами
греческого алфавита (а, р и у), а для коэффициентов уравнений средней
теплоемкости — буквами латинского алфавита (а, бис).
Выясним условия перехода от уравнений истинных теплоемкостей
к уравнениям средней теплоемкости. Например, если нам известно
уравнение истинной теплоемкости
С = а + $t + yt*9 *
а необходимо получить уравнение для средней теплоемкости в
интервале температур от tx до t2, то переход осуществляют следующим
образом. Известно, что
ё=Л-г и с= dQ
следовательно,
t2 — t\ d
dQ'=Cdt и Q = jCdt.
Тогда
5 =
Q
и
tt tx
1
t2 — ^i
« + 4- (h +12) + 4" Ci + 'A + й) • (78)
Таким образом, для перехода от уравнения истинной
теплоемкости к уравнению средней теплоемкости в интервале температур
от tx до t2 необходимо уменьшить коэффициенты уравнения
соответственно в один, два и три раза, вместо температуры в первой
степени подставить сумму (tx -f- t2), а вместо температуры в квадрате
подставить неполный квадрат этой суммы {t\ + t\t2 + t\).
34
Если от уравнения истинной теплоемкости
С = а + $t + yt*
необходимо перейти к уравнению средней теплоемкости для
интервала температур от 0 до t °C, то полученная зависимость (78)
упрощается при tx ~ О, a t2 принимает значение t. Следовательно,
имеем
t t
о о
В этом случае для перехода
от одного уравнения к другому
достаточно изменить величину
коэффициентов. Если уравнение
истинной теплоемкости имеет
вид
С = а + $t + yt\
а уравнение средней
теплоемкости имеет вид
C = a + bt + ct\
то связь коэффициентов a, р и у
с коэффициентами a, by с
выражается соотношениями:
а = ос, 6 = -|-; с *= -I. или а = а, р = 26; 7 = Зс. (80)
Отсюда видно, что а = а, 6 < р и с < у; следовательно, средняя
теплоемкость, определенная для интервала температур от 0 до t °C,
всегда будет меньше истинной теплоемкости при температуре L
Это легко показать графически. На рис. 5 зависимость истинной
теплоемкости представлена кривой АВ. Площадь, ограниченная
этой кривой и осью t представляет собой величину теплоты,
затрачиваемой на нагревание от 0 до температуры t °C. Средняя
теплоемкость в данном интервале температур не зависит от температуры и
изображается горизонтальной прямой, выбранной таким образом,
что расположенная под ней площадь тоже соответствует теплоте,
затрачиваемой на нагревание_от 0 до t °C. Выполнив эти построения,
убеждаемся в том, что с^> с.
Опытное определение теплоемкостей
Известно много различных способов определения теплоемкостей. Обычно
калориметрические опыты дают только средние теплоемкости, так как опытная разность
температур всегда конечна. Остановимся на более простом способе измерения
средней теплоемкости твердых и жидких тел методом смешения.
Ct В C'fJZL
Рис. 5. Пояснение соотношения между
средней С и истинной С теплоемкостью
Измерения производят с помощью калориметрической установки,
принципиальная схема которой изображена на рис. 6. Определенное количество вещества,
теплоемкость которого хотят исследовать, запаивают в ампулу/, из которой предварительно
откачивают воздух. Материалом ампулы может служить серебро, платина, кварц
и др. Далее ампулу с веществом подвешивают во внутреннем пространстве
электрической печи 2 и нагревают до необходимой температуры, величина которой контро-
лируется помещенной в печь термопарой 3.
I Когда установится постоянство температуры,
пережигают проволочку, удерживающую
ампулу, после чего она свободно падает в
калориметр, расположенный под печью. Главной
частью калориметра является тонкостенный
металлический стакан 4 с калориметрической
жидкостью (чаще всего для этой цели служит
вода). Стакан защищен от неравномерных потерь
теплоты медной оболочкой 7. Теплота вещества,
находящегося в ампуле, переходит к
калориметрической жидкости до тех пор, пока их
температуры не выравняются. Перемешивание
жидкости мешалкой 5 облегчает быстрое достижение
теплового равновесия в калориметре. Величину
подъема температуры определяют с помощью
очень точного термометра 6.
Количество теплоты, переданное образцом
калориметру, определяют следующим образом:
fci&SSS\\^\^
i.
Q = ы 2 "i
т
it
Рис. 6. Схематическое
изображение калориметрической
установки для определения теплоем-
костей твердых или жидких тел
при высоких температурах:
1 — ампула с исследуемым
веществом; 2 — печь; 3 — термопара;
4 — калориметрический стакан;
5 — мешалка; 6 — термометр; 7 —
оболочка калориметра
где At — изменение температуры калориметра
(рис. 7);
J] Ciitii — общая теплоемкость всех частей ка-
i
лориметра.
Далее определяют среднюю удельную
теплоемкость:
Q
m(t1 — t2)
или среднюю атомную (молекулярную)
теплоемкость:
Q
n(h-t2) '
С =
где т — масса образца (п — число грамм-атомов (молей) в образце); tx —
начальная температура и t2 — конечная (установившаяся) температура образца.
Описанный калориметр часто называют калориметром смешения.
Если в калориметре устанавливается температура тающего льда, т. е. 0° С, то
такой калориметр носит название ледяного калориметра. В этом фгучае
калориметрической «жидкостью» является смесь воды со льдом. При этом количество теплоты,
переданной калориметру, определяется прямым или косвенным измерением
уменьшения объема при таянии льда.
Иногда калориметрической жидкостью является массивный медный блок с
высверленной полостью для принятия ампулы. Благодаря хорошей теплопроводности
температура в таком блоке быстро выравнивается. Температуру измеряют с помощью
нескольких термопар, холодные спаи которых помещают в медном блоке, а горячие —
в печи. Таково принципиальное устройство медного калориметра.
При калориметрических определениях возникают ошибки и неточности,
обусловленные главным образом теплообменом прибора с внешней средой. Однако применяе-
36
мая методика определения изменения температуры в калориметре позволяет вводить
поправку на теплообмен.
Чаще пользуются графическим методом определения истинного изменения
температуры в калориметре. Для этой цели производят запись показаний температуры
как до сбрасывания ампулы в калориметр, так и в течение некоторого времени после
сбрасывания. Наблюдения заносят в таблицу, а затем переносят на график (рис. 7).
Ось абсцисс в данном случае является осью времени, а ось ординат — осью
температуры. Соединяя полученные точки на графике плавной линией, получаем кривую,
которая может быть разделена на три участка; abt be и cd; каждый из этих участков
соответствует определенному периоду опыта.
В предварительный период (ЛВ) фиксируется закон, по которому изменяется
температура калориметра вследствие теплового воздействия среды. На графике
наклон линии ah в этот период свидетельствует о том, что в данном случае калориметр
А ВС U
Время т
Рис. 7. Графический метод внесения поправки на теплообмен при
калориметрических испытаниях
медленно охлаждается (возможен также обратный случай, когда калориметр будет
медленно нагреваться). Сразу же после сбрасывания ампулы начинается следующий
главный период опыта (ВС). В этот период происходит очень быстрое изменение
температуры, и ее обычно приходится фиксировать через меньшие промежутки
времени. Самыми различными средствами стараются сократить продолжительность
главного периода опыта. Концом главного периода считают момент, когда опять
устанавливается определенный закон изменения температуры калориметра. На нашем
графике этот заключительный период опыта обозначен отрезком CD, а наклон
кривой cd соответствует тому случаю, когда калориметр охлаждается.
Обработка полученного графика сводится к следующему. Главный период опыта
делится на две равные части и через его середину восстанавливается перпендикуляр.
Далее кривые аЪ и cd продолжаются до пересечения с этим перпендикуляром.
Расстояние между полученными точками соответствует тому изменению температуры,
которое имело бы место, если бы удалось сократить до нуля продолжительность
установления теплового равновесия в калориметре, а следовательно, и устранить в этот
период влияние теплообмена калориметра с окружающей средой. Таким образом
определяется истинное значение изменения температуры.
Применяются также другие методы определения изменения температуры.
Получив значения средних теплоемкостей для различных интервалов температур,
приступают к определению уравнения, представляющего зависимость теплоемкости
от температуры. Допустим, что найденные значения для теплоемкостей равны ~Сг
С2 и С3 при соответствующих температурах нагрева ампулы перед сбрасыванием в
калориметр tu t2 и t3. Калориметр ледяной. Зависимости средней теплоемкости от
температуры необходимо представить уравнением степенного ряда
С= а+ bt+ ct*.
37
Подставив в это уравнение значения t, равные tlt t2n t3, получим соответственно
Это позволяет составить систему трех уравнений с тремя неизвестными:
Cl = a + btl + ct
ъ
C2^a+bt2+ ct\\ C3 = a+bts + ct\. (81)
Неизвестными являются коэффициенты уравнения степенного ряда a, b и с.
Решая систему уравнений (81), получаем конкретные значения этих коэффициентов.
Далее, используя соотношения (80), переходим от уравнения средней теплоемкости
к уравнению истинной теплоемкости.
Калориметрия смешения очень осложняется при измерении малых тепловых
эффектов (доли кал/г) или при измерении в области высоких температур (выше
2000° К). При низких температурах более чувствительным и надежным оказывается
метод обратной калориметрии.
Более же совершенными оказываются методы адиабатической калориметрии.
Они основаны на постепенном нагреве образца. Электрическим током с
контролируемым количеством подводимого джоулева тепла; при этом температура образца
повышается. При этом удается с достаточной точностью приблизиться к определению
истинной теплоемкости, особенно если уменьшать количество подводимого к образцу
тепла и интервал температуры нагрева образца. Теплопотери в окружающую среду
исключаются созданием адиабатических условий нагрева. Во время нагрева с
определенной скоростью образца, помещенного внутри обогреваемого стакана, температура
образца оказывается то незначительно выше, то ниже температуры обогреваемого
стакана. Через определенные промежутки времени их температуры сравниваются,
что соответствует адиабатическим (точнее квазиадиабатическим) условиям.
Истинная теплоемкость вычисляется при этом по формуле:
W
L~ dt '
т
dx
где W — мощность нагревательной спирали внутри образца;
т — масса образца;
dt
-j скорость изменения температуры образца.
Точность определения теплоемкости этим методом во многом зависит от
правильности поддержания заданных режимов калориметра: скорости нагрева, минимальной
разности температур при установлении адиабатных условий и т. д. Все это
предопределяет необходимость автоматизации измерений. Автоматические адиабатные
калориметры различаются по способу регулирования импульсов подачи электрической
мощности и способу учета поправок на несоблюдение адиабатности условий
измерения:
С = а+ $t+yt2.
§ 6. ТЕРМОХИМИЯ КАК РАЗДЕЛ ТЕРМОДИНАМИКИ
При протекании химических реакций происходит глубокое
изменение вещества, сопровождающееся или поглощением или выдече-
нием энергии. Этой энергией в большинстве случаев является
теплота. Значение тепловых эффектов, сопровождающих реакции,
позволяет сознательно | управлять ими, меняя соответствующим
образом внешние условия среды.
Термохимия представляет собой дисциплину, которая занимается
экспериментальным изучением тепловых эффектов химических
реакций. Возникновение термохимии произошло задолго до создания
термодинамики и ее приложения к химическим превращениям — хими-
38
ческой термодинамики. Сначала она представляла собой раздел
химии, занимающийся изучением тепловых эффектов химических
реакций. В настоящее время термохимия составляет обширный
раздел химической термодинамики, располагающий опытным
материалом по теплотам реакций и занимающийся их систематикой
и формулировкой общих закономерностей. В силу исторической
преемственности в термохимии удержались некоторые понятия и
обозначения, отличные от тех, которые впоследствии были введены в
термодинамику в период независимого развития термохимии и
термодинамики.
Особенно важно обратить внимание на то, что в термохимии при
обозначениях тепловых эффектов применяются знаки, обратные
тем, которыми мы пользовались до сих пор. Так, например, та
теплота, которая подводится к системе и ею поглощается, в
термохимии рассматривается как отрицательная, а теплота, выделяемая
системой, считается положительной. Поэтому при термохимическом
рассмотрении тепловых эффектов будем пользоваться обозначением Q
в отличие от О — обозначения, принятого в термодинамике. Таким
образом, между термохимическими и термодинамическими
обозначениями устанавливается следующее соотношение:
Q = —Q, или -Q = Q. (82)
Многообразие химических превращений по тепловому балансу
можно разделить на две группы: реакции, протекающие с
выделением теплоты, — экзотермические и реакции,
протекающие с поглощением теплоты, — эндотермические. К
экзотермическим реакциям относится большинство реакций образования
химических соединений из простых веществ, например:
С (графит) + 02 = С02 + 94,05 ккал;
С (алмаз) + 02 = С02 +94,50 ккал;
Pb + S (ромбич.) - PbS + 22,54 ккал;
Zn + 02 + Н2 = Zn (ОН)а + 153,5 ккал.
К эндотермическим реакциям относится большинство реакций
диссоциации химических соединений на простые вещества, например
2Н20 (жидк.) = 2Н2 + 02 — 136,64 ккал,
а также некоторые другие реакции:
С (алмаз) + 2S (ромбич.) = CS2 (жидк.) — 21,0 ккал,
2С (графит) + Н2 = С2Н2 — 54,19 ккал.
Существуют реакции, сопровождающиеся выделением или
поглощением незначительного количества тепла, а при некоторых
условиях тепловой эффект может стать почти нулевым, например
KI + 1а = К13 + 0,8 ккал.
39
Запись химических реакций с указанием теплового эффекта
реакции получила название термохимических уравнений. Так, для
реакции образования или распада озона имеем:
203 = 302 + 69 ккал.
Это термохимическое уравнение показывает, что при распаде
двух молей озона выделяется 69 ккал или что при образовании двух
молей озона из трех молей кислорода поглощается 69 ккал теплоты.
В реакциях образования данного соединения из простых веществ
тепловые эффекты обычно рассчитываются на один моль продукта
реакции, и коэффициенты подбираются с таким расчетом, чтобы
у формулы главного продукта, записанного в правой части
уравнения, коэффициент был равен единице. Например:
Zn (тв.) -f ~y 02 (газ) = ZnO (тв.) -f- 83,17 ккал.
На общее выделение или поглощение теплоты при той или иной
химической реакции заметное влияние оказывают такие физические
явления, как переход реагирующих- веществ из одного агрегатного
состояния в другое, поскольку все подобные переходы сами связаны
с поглощением или выделением энергии:
Н20 (тв.) = Н20 (жидк.) — 1,44 ккал,
С (алмаз) — С (графит) -|~ 0,45 ккал.
Поэтому термохимические уравнения должны содержать
указание на агрегатное состояние продуктов реакции, а для
кристаллических веществ — указание на их модификацию, если возможно их
существование в нескольких модификациях.
С термохимическими уравнениями можно производить
алгебраические действия: переносить члены из одной части уравнения в
другую, складывать уравнения, вычитать их одно из другого и т. д.
Связь между тепловыми эффектами реакции
при постоянном объеме и при постоянном давлении
Когда химическая реакция протекает при постоянном объеме,
то отсутствует работа системы против сил внешнего давления. В этом
случае А = 0 и, согласно первому началу термодинамики:
Q = и2— иг = At/.
Принимая во внимание термохимические обозначения, получаем
для реакции при V = const
Qv - —Qv - —(U2 - U±) - -AUy (83)
т. е. выделенная системой теплота (Qv или —Qv) равна убыли ее
внутренней энергии (—AU) при переходе из исходного состояния
с большим запасом энергии (иг) в состояние с меньшим запасом
энергии (U2).
40
Если химическая реакция протекает при постоянном давлении,
то системой будет производиться работа расширения. В этом случае
А = Р {V2 — Vx) и, согласно первому началу термодинамики:
Q = U2 - иг + р (V2 - Vt). (84)
Далее, принимая во внимание термохимические обозначения,
получаем для протекания реакции при р = const
QP = -QP = -(ия -uj-p (v2 - vj (85)
ИЛИ
QP = (Ut + PVt) - (Ua + PV2) = -(Я2 - Ях) = -ЛЯ. (86)
Сумма U + pVy обозначенная нами через Я, получила название
энтальпии. Она представляет собой термодинамическую
функцию, которая, подобно внутренней энергии, является функцией
состояния, а ее дифференциал — полным дифференциалом. Таким
образом, для этой функции
j>dH = 0. (87)
Следовательно, тепловой эффект химической реакции при
постоянном давлении (Qp или —Qp) равен убыли энтальпии (—АН) при
переходе из состояния с большей энтальпией (Нг) в состояние с
меньшей энтальпией (#2):
QP = -QP = -(H2-HJ = -AH. (88)
Подставляя в уравнение (85) выражения Q = —(£/2 — £Л),
AV = V2 — Viy получаем
QP - Qv — P&V. (89)
Величина AV = V2 — Vx представляет собой увеличение объема
вследствие того, что в результате реакции число молей газообразных
участников реакции возрастает на An = п2 — пх молей. Согласно
уравнению Менделеева—Клапейрона:
pAV = AnRT, (90)
т. е.
Qp=Qv — bnRT (91)
или, поскольку для обозначения тепловых эффектов часто
пользуются термодинамическими обозначениями через AU и АН:
АН - AU + AnRT. (92)
Таким образом, тепловой эффект при постоянном давлении (Qp)
отличается от теплового эффекта при постоянном объеме (Qv) на
величину работы, совершаемой за счет изменения числа молей
газообразных участников реакции (AnRT), когда реакция протекает при
41
p = const и может происходить изменение объема системы *.
Например, при реакции
СО + Н20 - С02 + Н2 + Q
не происходит изменения числа молей газообразных участников
реакции и An = 0. Следовательно, совершаемая при этом работа
равна нулю (А = 0) и Qp = Qv.
Реакция N2 + 3H2 = 2NH3 + Q сопровождается уменьшением
числа молей газообразных участников: An = 2 — 4 = —2.
Следовательно, реакция сопровождается уменьшением объема<#а
совершаемая при этом работа отрицательна (А <0). В этом случае Qp >
Наоборот, при реакции
Zn (тв) + H2S04 (жидк) = ZnS04 (жидк) + Н2 (газ) +~Q
возникает 1 моль газа (водорода). Следовательно, An = 1 — 0 =
= +1, происходит увеличение объема, а совершающаяся работа
расширения положительна (А > 0). -В этом случае Qp <CQy-
В системах, состоящих только из жидких или твердых веществ,
изменения объема очень малы. Поэтому для них обычно
пренебрегают различием между Qp и Qv и полагают, что
Закон Гесса
В основе термохимии лежит закон, согласно которому тепловой
эффект реакции не зависит от промежуточных стадий и определяется
лишь начальным и конечным состояниями системы. Этот закон был
открыт путем сопоставления опытных данных, а затем
экспериментально проверен Г. И. Гессом (1836—1840 гг.).
Формулировка этого закона следует непосредственно из первого
начала термодинамики. Действительно, анализируя равенства Qv =
— —AU и Qp = —А Я, приходим к выводу: тепловые эффекты
превращений, происходящих при V = const или при р = const,
не зависят от пути перехода от одного состояния к другому,
поскольку определяются соответствующим изменением функций
состояния U или Я.
Таким образом, если изданных исходных продуктов можно
различными путями получить заданные конечные продукты, то независимо
от вида промежуточных реакций суммарный тепловой эффект будет
одним и тем же. Разумеется, что вполне точным этот закон является
при том условии, что процессы протекают при постоянном объеме
или постоянном давлении.
* Для реакций с малыми величинами тепловых эффектов и большим изменением
числа молей газообразных участников реакции знаки Qp и Qv могут не совпадать.
42
Например (рис. 8), двуокись углерода можно получить, сжигая
углерод в кислороде непосредственно до двуокиси углерода С +
-f 02 = С03 + Qi или получая сначала сзкись углерода, а затем
сжигая ее до двуокиси:
C+l/202 = CO+Q2,
CO+l/202 = C02 + Q3.
Оба эти пути имеют одинаковое начальное состояние (С и 02)
и одинаковое конечное состояние (С02). Под одинаковостью
состояния подразумевается полное тождество по химическому составу,
Выделяется 94,05ккал
Рис. 8. Схематическое пояснение закона Гесса на примере
получения двуокиси углерода сжиганием углерода различными
путями
по условиям существования (температура, давление и пр.), по
агрегатному состоянию и кристаллической модификации, по степени
дисперсности и т. п.
По закону Гесса, тепловой эффект сжигания углерода до
двуокиси углерода должен быть равен суммарному тепловому эффекту
при сжигании его через окись углерода, т. е.
Qi = Q2 + Q3.
Закон Гесса имеет большое практическое значение. Например,
с его помощью можно установить тепловые эффекты реакций,
экспериментальное определение которых представляет большие трудности.
Для этих целей широко используются следствия из закона Гесса.
Первое следствие: тепловой эффект разложения
какого-либо химического соединения равен по абсолютной величине
и противоположен по знаку тепловому эффекту образования этого
соединения.
Второе следствие: если совершаются две реакции,
приводящие из различных начальных состояний к одинаковым
конечным, то разница между тепловыми эффектами представляет собой
тепловой эффект перехода из одного начального состояния в другое.
43
I p e т ь е следствие: если совершаются две реакции,
приводящие из одинаковых начальных состояний к различным
конечным, то разница между их тепловыми эффектами представляет
собой тепловой эффект перехода из одного конечного состояния
в другое.
Тепловые эффекты, сопровождающие некоторые
химические реакции
Теплота образования. Когда из простых веществ образуется
какое-либо соединение, то этот процесс сопровождается некоторым
тепловым эффектом. Количество теплоты при этом не одинаково для
различных соединений; кроме того, значения тепловых эффектов
существенно меняются с изменением внешних параметров системы
(например, температуры).
Теплотой образования принято называть тепловой эффект
реакции образования 1 моль данного соединения из простых веществ.
Стандартной теплотой образования называют теплоту образования
соединения, отнесенную к температуре 25 °С (298 °К) и 1 am
(760 мм рт. ст.). Если известна эта величина, то термохимический
расчет позволяет определить теплоту образования при других
значениях параметров состояния (см. стр. 48).
Теплота образования химического соединения из простых
веществ не определяет собой энергии связи между атомами в молекуле
данного соединения. Для этой цели могла бы служить теплота
образования соединения из атомов, значительно отличающаяся от теплоты
образования простых веществ. Однако для многих простых веществ
теплоты образования их из свободных атомов еще неизвестны.
Величины теплоты образования определяются или опытным путем
с помощью различных калориметров, или расчетным путем и
приводятся для различных соединений в таблицах стандартных
термохимических величин.
Практическое значение теплот образования заключается в том,
что, зная теплоты образования всех веществ, участвующих в той
или иной реакции, можно рассчитать тепловой эффект этой реакции.
Так, согласно закону Гесса, тепловой эффект реакции равен разности
между суммой теплот образования конечных продуктов и суммой
теплот образования исходных веществ с учетом коэффициентов при
формулах этих веществ в уравнении реакции. Это — четвертое
следствие из закона Гесса. При этом необходимо помнить,
что для тех реакций, в которых участвуют простые вещества, теплота
образования этих простых веществ условно принята равной нулю.
Например, если известно, что
1) S + 02 = S02 + 70,94 ктл\
2) 2Cu + S = Cu2S + 19,0 ккал:
3) 2Cu + VaO, = Cu20 + 39,89 ккал,
то имеется возможность определить тепловой эффект реакции:
2Cu20 + Cu,S = 6Cu + S02 + Q.
Подсчет по теплотам образования дает:
1-70,94 + 6-0— 1-19,0 — 2-39,8 - —27,66 ккал.
Полученный результат легко проверить. Для этого необходимо
из первого термохимического уравнения вычесть второе и удвоенное
третье уравнение.
Теплота сгорания. Для многих соединений не удается
осуществить реакцию образования их из простых веществ и, тем более,
нельзя измерить теплоту образования. В большинстве случаев,
однако, удается осуществить реакцию
полного сгорания. Определяемая при этом
теплота сгорания имеет не меньшее практическое
значение для термохимических расчетов,
чем теплота образования.
Теплотой сгорания называется тепловой
эффект реакции полного сгорания одного
моля данного соединения до образования
высших окислов. Поскольку с изменением
температуры и давления величина теплоты
сгорания может существенно меняться, то
для термохимических расчетов важно ввести
представление о стандартной теплоте
сгорания. Стандартной теплотой
сгорания называется теплота сгорания,
отнесенная к стандартным условиям, т. е.
25 °С (298 °К) и 1 am (760 мм рт. ст.).
Определение теплот сгорания обычно
производят в особом приборе —
калориметрической бомбе. На рис. 9
приводится принципиальная схема прибора,
с помощью которого ведутся определения
теплот сгорания. Для того чтобы горение шло энергично, в бомбу
вводят чистый кислород под высоким давлением. Так как
калориметрическая бомба должна выдерживать значительные давления,
то ее делают в виде толстостенного цилиндра / с навинчивающейся
крышкой 2. Материалом служит либо нержавеющая, либо
углеродистая сталь, платинируемая изнутри для предохранения от
разъедания. Отвешенное количество исследуемого вещества помещают
в чашку 4 и поджигают, пропуская электрический ток через
проволочную спираль 3. Бомбу помещают в калориметр и определяют
количество выделившейся при сгорании теплоты.
Практическое значение теплот сгорания состоит в том, что по ним
непосредственно можно определять тепловые эффекты реакций, так
как тепловой эффект реакции равен разности между суммой теплот
сгорания исходных веществ и суммой теплот сгорания конечных
продуктов с учетом коэффициентов при формулах этих веществ
в уравнении реакций. Это пятое следствие из закона
Гесса.
+
Рис. 9. Схематическое
изображение устройства
калориметрической
установки для определения
теплот сгорания веществ:
1 — толстостенный цилиндр;
2 — навинчивающаяся
крышка; 3 — проволочная
спираль; 4 — чашка
45
Например, необходимо определить теплоту образования метана
из элементов по реакции С + 2Н2 = СН4 + Q, если известно, что
1) С + 02 = С02 + 94,05 ккал,
2) Н2 + V202 = Н20 + 68,32 ккал,
3) СН4 + 202 - С02 + 2Н20 + 211,93 ккал.
Производим подсчет:
Q= 1-94,05 + 2 68,32— 1-211,93= + 18,76 ккал.
Для проверки полученного результата можно из суммы первого и
удвоенного второго уравнений вычесть ретье уравнение.
Теплота растворения. Процесс растворения в
огромном большинстве случаев представляет собой наложение двух
процессов: физического и химического, поскольку происходит
равномерное распределение частиц растворяемого вещества среди частиц
растворителя и в то же время эти частицы вступают во взаимодействие
с частицами растворителя.
Растворение газов всегда сопровождается выделением теплоты.
Жидкости растворяются большей частью с выделением теплоты.
Например, с выделением теплоты происходит растворение кислот
в воде, спиртов в воде, хлорноватистой кислоты в спирте, амидобен-
зола в ксилоле и др. Растворение сероуглерода в спирте и в эфире,
уксусной и синильной кислотах, в воде и других растворителях
сопровождается поглощением теплоты.
Растворение солей, как правило, сопровождается выделением
теплоты, если они способны вступать в химическое взаимодействие
с растворителями и образуют сольваты (гидраты — при растворении
в воде); так, например, растворяются в воде хлористый кальций,
йодистый барий, сульфаты натрия и меди и др. Соли же, не дающие
при растворении сольватов (гидратов), а также сами сольваты
растворяются с поглощением теплоты.
Знак теплового эффекта определяется, очевидно, тем, какой из
процессов преобладает: либо процесс разрушения кристаллической
решетки твердого вещества, сопровождающийся поглощением
теплоты, либо процесс сольватации (гидратации), связанный с
выделением теплоты.
Большое значение имеет количественное соотношение
растворенного вещества и растворителя. Теплота образования растворов
различного состава из одних и тех же компонентов может сильно
отличаться по величине; при увеличении разбавления теплота
образования раствора приближается к постоянной величине.
Теплотой растворения принято называть количество теплоты,
выделяемое или поглощаемое при растворении моля вещества в таком
количестве растворителя, при котором дальнейшее прибавление
растворителя уже не вызывает изменения температуры раствора.
Обычные табличные значения теплот растворения относятся к
растворению одного моля вещества в 200—400 моль растворителя (или более),
46
и это условие в случае образования водных растворов записывается
следующим образом:
НС1 + aq = НС1-а<7 + 18,5 ккал,
КОН + aq = КОН-aq + 13,0 ккал,
NH4OH + aq = NH4OH-a<7 — 6,0 ккал.
Зная теплоту растворения безводной соли и ее кристаллогидрата,
можно, например, определить теплоту образования
кристаллогидрата. Так, опытом установлено, что
Na2C03 + aq = Na2C03-aq + 6 ккал,
Na2C03- 10H2O + aq = Na2C03-a<7 — 16 ккал.
Следовательно, по закону Гесса, разность теплот обоих процессов
соответствует теплоте образования кристаллогидрата соды:
Na2C03 + 10Н2О = Na2CO3-10H2O + 22 ккал.
Теплота нейтрализации. Согласно теории электролитической
диссоциации, молекулы электролитов, т. е. солей, кислот и
оснований, обладают способностью диссоциировать в растворе на ионы,
причем сильные электролиты распадаются нацело:
КС1 - К++С1 -,
НС1 - Н+ + С1-,
NaOH = Na+ + OH-.
Вода почти не диссоциирует на ионы., поэтому нейтрализацию
едкого кали соляной кислотой НС1 + КОН = КС1 + Н20 можно
записать в ионной форме так:
h+ + ci- +к+ + он- = к+ + С1- + н2о
или иначе, принимая во внимание неизменность ионов калия и хлора,
стоящих в левой и правой частях уравнения:
н+ + он- - н2о.
По аналогии можно предположить, что вообще взаимная
нейтрализация сильных оснований и кислот состоит в нейтрализации ионов
водорода и гидроксила, а следовательно, тепловой эффект всех
подобных реакций должен быть одинаков. Действительно, точные
опыты показали, что количество теплоты, выделяющееся при
нейтрализации сильных оснований сильными кислотами, во всех
случаях практически одинаково и равно теплоте образования воды из
ионов:
LiOH + HC1 - LiCl + H20 + 13,7 ккал,
КОН + HN03 =■ KND3 + H20 + 13,7 ккал,
NaOH + HC1 = NaCl + H20 + 13,7 ккал,
Н+ +..ОН- = Н20 + 13,7 ккал.
47
Зависимость теплового эффекта
от температуры (уравнение Кирхгофа)
Очень часто бывает необходимо знать тепловые эффекты реакций
при высоких температурах, в то время как таблицы термохимических
величин содержат только стандартные тепловые эффекты,
относящиеся к температуре 25° С (298° К). Первое начало термодинамики
позволяет определить зависимость тепловых эффектов от
температуры и производить соответствующие пересчеты.
ЪВ и UD
при T+dT
Выделяется количество теплоты
Конечное
состояние
5
с: ^
Qo со
с:
а + аа
gG и гк
при T+dT
со4
4-
-о
С)
с:
«о
со
СО С}
ЪВ и йВ
при Т
Выделяется количество теплоты
+•
а
Начальное
состояние
Рис. 10. Схема к выводу уравнения зависимости теплового эффекта реакции
от температуры
Представим себе химическую реакцию
ЪВ + dD ■= gG + rR + Q,
В и D
G и R
Ь, d, g и г
Q
где .... в и и — начальные продукты;
- конечные продукты;
- стехиометрические коэффициенты;
- тепловой эффект реакции при какой-то
температуре Т.
При какой-то другой температуре Т + dT, незначительно
отличающейся от Г, тепловой эффект этой реакции Q + dQ мало
отличается от Q.
Проведем превращение двумя путями (рис. 10). Сначала реакцию
осуществим при температуре Т: при этом выделяется количество
теплоты Q; затем конечные продукты нагреем от Т до Т + dT\ при этом
придется затратить некоторое количество теплоты (gCG + rCR) dT.
Идя по другому пути, сначала начальные продукты нагреем от Т до
Т + dT и затратим на это количество теплоты (ЬСВ + dCD) dT\
затем осуществим реакцию при температуре Т + dT; при этом
выделится количество теплоты Q + dQ. Таким образом, оба пути при-
48
водят к одному и тому же состоянию; следовательно, если
описанные процессы протекали при постоянном объеме или при постоянном
давлении, то суммарные тепловые эффекты на обоих путях равны
друг другу:
Q - (gCG + rCR) dT^- (ЬСВ + dCD) dT + Q- dQ. (93)
Приведение подобных членов в уравнении (93) приводит его к виду
dQ - 1(ЬСВ + dCD) - (gCG + rCR)] dT (94)
или
-§r = (bCB + dCD) - (gCa + rCR). (95)
Обозначая сумму теплоемкостей начальных продуктов реакции
S Сн = ЬСВ + dCD, (96)
а суммарную теплоемкость конечных продуктов реакции
S Ск = gC0 + гС* (97)
получаем
~37г= 5jCh —ЦСК. (98)
Температурный коэффициент теплового эффекта реакции равен
изменению теплоемкости системы, происходящему в результате
данной реакции. Уравнение (98) представляет собой уравнение
Кирхгофа в дифференциальной форме.
Знак температурного коэффициента теплового эффекта
определяется разностью начальной и конечной теплоемкостей системы.
Если
1l ch >> 2j ck,
т. е. теплоемкость системы убывает, то
dQ
dT
>0.
и возрастание температуры сопровождается увеличением теплового
эффекта.
Если 2 Ся <> Е^к» т- е- теплоемкость системы возрастает, то
dQ
dT
и возрастание температуры сопровождается уменьшением теплового
эффекта. Наконец, возможно постоянство теплового эффекта реакции.
Это имеет место при
2j ч* = 2j Чо
когда не происходит изменения теплоемкости системы и
dQ _0
4 А. Н. Крестовников 49
Если тепловой эффект определен при V = const, то изменение
теплоемкости тоже должно быть определено при V = const. Точно
так же тепловому эффекту при р = const соответствует изменение
теплоемкости при р = const. Следовательно:
dQv _
(IX),-(IX), (99)
И
dQ
?- = (ScH)-(ScK). (юо)
Принимая во внимание, что Qv = —At/ и Qp = —АЯ, получаем:
■^- = (ScJy-(ScJK. (mi)
-^ = (IX)p-(IX)p- (102)
Уравнение Кирхгофа распространяется на агрегатные
превращения (в условиях их протекания без изменения давления). Для
обозначения тепловых эффектов этих превращений пользуются
термодинамическими обозначениями, которым приписываются
положительное значение, если они соответствуют поглощению теплоты, и
отрицательное — выделению теплоты. Таким образом, для процесса
испарения имеем:
Ц А"исп /■> /-> /1 П*ЧЧ
ftp — °пар ижидк> Ч1ио/
где ЛЯИСП — теплота испарения, а Спар и Сжидк — теплоемкости
пара и жидкости.
Для процесса плавления
dЛЯПЛ __ р р П04Л
где АЯПЛ — теплота плавления, а Сжидк и Ств — теплоемкости
в жидком и твердом состояниях.
Для возгонки
а А/твозг г> р /1ПЧ\
~tff —ипар °тв> \1KJO)
где АЯвОЗГ — теплота возгонки.
Для аллотропного перехода а —> (3 имеем:
—-^±i = C3-Ca, (106)
где АЯа->р — теплота аллотропного перехода, а Са и Ср —
теплоемкости различных полиморфных модификаций.
50
Перейдем к интегральной форме уравнения Кирхгофа, которая
чаще используется при практических расчетах. Для этого
интегрируем уравнение (98):
dQ=(ECH-ECK)d7\
I dQ= J (2cH-2cK)d7\
Qr0 г°
т
Qr-Qr,= Н2сн-1ХК
Окончательно получаем:
т
Qt = Qt0+ К£Сн-2с>Г. (107)
Это и есть уравнение Кирхгофа в интегральной форме, где QT
—тепловой эффект при температуре Т и QTo — тепловой эффект при
какой-то другой температуре Т0. Это может быть 0 °К, тогда
т
Qr = Qo + J (S с- — S с«)dT» (108>
0
или 298° К (25 °С), тогда
т
Qt = Q298 + J (S Си - 2 Ск) dT, (109)
298
или любая другая температура. Выражение (2 Сн — 2 Ск) является
функцией температуры, поскольку теплоемкость всех веществ
зависит от температуры. „Эта зависимость выражается уравнениями
степенного ряда:
Св = ав + $вТ + увТ\ (110)
CD = aD + pDT + yDT\ (111)
C0=aG + pGT +7^, (112)
CR = aR + faT + yRT*. (113)
Следовательно,
Qr = $T0 + \ (b(XB + d(XD ~ £aG — mR) dT +
To
f j ДОБ + dfo - rfG - rfo) T dT +
Го
Го
*
+ J ФУ в + <%> - ёЧа - Пи) T2 dT. (114)
51
Откуда, упрощая обозначения, получаем:
^ = $„ + ^1 + ^1 + ^1, (115)
где QT — тепловой эффект при температуре Т;
Q0 — гипотетический тепловой эффект при Т = О °К
(константа, определяемая по эмпирическим
данным для температурных зависимостей теплоем-
костей участников реакции);
Да, Ар, А у — алгебраические суммы соответствующих
коэффициентов в уравнениях степенного ряда для
температурных зависимостей теплоемкостей
участников реакции, умноженных на те
стехнеметрические коэффициенты, которые стоят перед
формулами этих веществ в уравнении химической
реакции.
Таким образом, зависимость теплового эффекта от температуры
мшкет быть выражена уравнением степенного ряда. В справочниках
имеются конкретные уравнения степенного ряда для тепловых
эффектов многих реакций. Уравнение (115) обычно называют
развернутым уравнением Кирхгофа.
В заключение следует сделать несколько замечаний. Во-первых,
необходимо помнить, что интегрировать уравнение (107) можно
лишь в том случае, если известны уравнения степенных рядов тепло-
емкостей, полученные для всего интервала температур от Т0 до Т.
Во-вторых, для того чтобы получить уравнение зависимости
теплового эффекта от температуры, недостаточно располагать только
уравнениями теплоемкости всех участников реакции — необходимо знать
также величину теплового эффекта при какой-либо температуре в
рассматриваемом интервале температур. В противном случае мы
вынуждены прибегать лишь к неопределенному интегрированию, в
результате которого в уравнении появляется постоянная
интегрирования.
Задачи
Задача 1. Определенное количество идеального газа, занимающего объем 10 л
при температуре 27° С и давлении 1 сап, адиабатно сжимается до объема 1 л. До
какого значения при этом поднимается его температура и чему будет равна затраченная
на сжатие работа, если у = Ср/Су = 1,66 (одноатомный газ) и если у = 1,4
(двухатомный газ).
Решение. Для адиабатного процесса, согласно уравнению (39), имеем:
Га=7\(-^М =(273+ 27) (-у-J =1371° К.
Работу определяем по уравнению (61) для п молей;
А = nCv{T1 — T2).
Так как, согласно уравнениям (26) и (51):
PiVi
52
R = 0,082 л-ат/(моль-град) и Су = 3 кал/(моль-град), то
Л = ~Ж~ Cy(Tl ~Г2) = "0,082-300 '3(3°° ~~ 13?1) = _13°6 КтЛ'
Для случая у— 1,4 при Су = 5 кал/(моль-град) имеем Т2 = 753° К и Л —
= —922 кал.
Сопоставление полученных результатов позволяет сделать заключение, что по
мере усложнения строения молекул газа уменьшается затрачиваемая на адиабатное
сжатие работа и происходит меньшее повышение температуры.
Задача 2. Определенное количество идеального газа занимает объем 5 л при
температуре 27° С. После адиабатного расширения газ занял объем 6,02 л при 5° С.
Требуется найти теплоемкости газа при постоянном давлении и при постоянном
объеме.
Решение. Прежде всего, согласно уравнению (39), определяем значение
адиабатного коэффициента:
v_ luTi — lgT2 1-14107
Далее, решая систему двух уравнений:
Р = 1,4107
и
R = Ср — Су = 1,987 кал/(моль-град),
получаем Су = 4,83^ 5 и Ср = 6,815^ 7 кал/(моль*град). Следовательно,
молекулы рассматриваемого идеального газа двухатомны.
Задача 3.3 г водорода находятся под давлением 5 am при температуре 0° С. После
расширения при постоянном давлении газ занял объем 15 л. Требуется определить
совершенную работу, количество полученной газом теплоты и изменение внутренней
энергии.
Решение. Величину работы определим по уравнениям (50) и (26):
А- Р (V2 - Vx),
RTX m RTX
Vi = n
p M
откуда
A = p(v*—M"'J^p-) = 5 (15—Г°'082-?-)= 41-5 л-ат-
Величину поглощенной теплоты, полагая Ср=7 кал/(моль• град), определим по
уравнениям:
Q = пСр (Г, - 7\) = -|- Ср (Та - Тг)
и
Т*
pV2 MpVt
nR R
откуда
Q
м
Ч^-гО'-Илда-"»)-»»""-
Изменение внутренней энергии определяем по уравнению (6):
1 QQ7
W = Q — A = 3536-41,5-i^-= 2536 кал.
UjUoZ
53
Задача 4. По известной температурной зависимости молекулярной теплоемкости
газообразного водорода при постоянном объеме Су = 4,850+ 0,700» 10"3 t
вычислить все остальные формы теплоемкости при температуре 1000° С.
Решение. Подставляем t = 1000° С и получаем
Су = 4,850 + 0,700- Ю-3-1000 = 5,550 кал/(моль-град).
Поскольку а = 4,850 и (3 = 0,700» 10"3, то, согласно соотношениям (83):
а = а = 4,850 и Ь = -|- = 0,350-10~3.
Поэтому средняя молекулярная теплоемкость по уравнению (82):
Ъу = 4,850 + 0,350- Ю"3-1000 = 5,200 кал /(моль -град).
Далее по уравнению (70):
Ср — Су -f # = 7,537 кал /(моль -град)
и
Ср = Су + R = 7,187 кал/'(моль-град).
Удельные теплоемкости получаются делением Су, Су, Ср и Ср на молекулярную
массу водорода М = 2:
Су = 2,775 кал/(г-град)\ Ср = 3,768 кал/(г-град);
Су = 2,600 кал/(г-град); Ср *= 3,093 кал/(г-град).
Задача 5. Истинная удельная теплоемкость железа при температуре 100° С
равна 0,1124 кал!(г-град); средняя удельная теплоемкость в интервале температур
0—100° С составляет 0,1089 кал. Требуется найти температурную зависимость
удельной теплоемкости железа.
Р е ш е н и е. На основании уравнений (78) и (79), исключая член уравнения
степенного ряда, содержащий квадрат температуры, составляем систему двух
уравнений:
С = а+ $t;
C = a + -^t,
откуда определяем значение коэффициентов аир:
0,1124 = а+ 0100, а= 0,1054;
0,1089 = а + -|- 100, р = 0,7-10~3.
Следовательно:
С = 0,1054 + 0,07- Ю-3* кал/(г-град).
Задача 6. Удельная теплоемкость углекислого газа при постоянном давлении
равна 0,217 кал/(г-град). Требуется определить отношение Ср/Су.
Решение. По удельной теплоемкости находим молекулярную теплоемкость:
Ср = срМ = 0,217'44 = 9,548 кал/(моль-град).
По уравнению (67) определяем теплоемкость при постоянном объеме:
Су = Ср — R = 9,548 — 1,987 = 7,561 кал/(моль-град).
Затем определяем адиабатический коэффициент:
ср 9,548
Су 7,561
1,263.
Задача 7. Истинная молекулярная теплоемкость двуокиси углерода (газ) при
постоянном давлении выражается уравнением
Ср = 7,00 + 0,0071Г — 0,186-10" &Г2.
54
Требуется вычислить количество теплоты, необходимое для нагревания 200 г газа от
температуры 27 до 227° С при постоянном давлении и при постоянном объеме.
Решение. По коэффициентам а = 7,00, Р = 0,0071 и у = —0,186-10~5
находим коэффициенты а= 7,00; Ъ = 0,00355 и с = —0,062» 105. Далее по
уравнению (78) находим среднюю теплоемкость для интервала температур от 27 (300° К)
до 227° С ( 500° К)
£р = 7,00 + 0,00355 (300 + 500) — 0,062-10"6 (3002 +
+ 300-500 + 5002) 9,536 кал/(моль-град)
и определяем
m — 200
QP = -~CP (tt - h) = -^-9,536 (227 - 27) = 8670 кал.
Затем по уравнению (68) находим теплоемкость при постоянном объеме:
gy = ср — R = 9,536 — 1,987 = 7,549 кал/(моль• град)
и определяем
т = 200
Qv=~Cv{t2- tx) = -^p 7,549 (227- 27) = 6863 кал.
Обратим внимание на то, что Qp> Qv, т. е. при постоянном объеме на нагрев
расходуется меньшее количество теплоты по сравнению с нагревом при постоянном
давлении.
Задача 8. Зная стандартные теплоты образования окиси свинца, воды (пар) и
двуокиси свинца, равные соответственно 52 700, 57 800 и 65 300 кал/моль, определить
тепловой эффект реакции восстановления водородом двуокиси свинца до окиси с
образованием воды (пар).
Решение. По четвертому следствию из закона Fecca:
~Qp = 52 700 + 57 800 — 65 300 = 45 200 кал/моль.
Для проверки суммируем уравнения:
Pb + Va 02 = РЬО + 52 700 кал
и
На + Va Оа = Н20 (пар) + 57 800 кал,
и вычитаем уравнение
РЬ + Оа = РЬ02 + 65 300 кал,
после чего получаем
РЬ 02 + На = РЬ О + НаО (пар) + 45 200 кал.
Задача 9. Стандартные теплоты образования воды (жидкой) и двуокиси углерода
при постоянном давлении соответственно равны 68 320 и 94050 кал/моль.
Стандартная теплота сгорания метана равна 211 930 кал/моль. Требуется найти теплоту
образования метана из простых веществ при постоянном давлении и при постоянном
объеме.
Решение. По пятому следствию из закона Гесса:
Qp = 2-68 320 + 94 050 — 211 930 = 18 760 кал/моль.
Для проверки суммируем удвоенное уравнение
Н2 + Va 02 = Н20 (жидк) + 68 320 кал
с уравнением
С + 02 = С02 + 94 050 кал
и вычитаем уравнение
СН4 + 202 = С02 + 2Н20 (жидк) + 211 930 кал,
после чего получаем
С + 2Н2 = СН4 + 18 760 кал.
Далее находим теплоту образования метана при постоянном объеме. Согласно
уравнению (91):
Qv=sQP+ &nRT = 18 760 + (1—2). 1,987-298 = 18 180 кал/мшь.
55
Задача 10. Воспользовавшись значениями стандартных тепдот образования:
Qpbs = ^2 ^00, QpbO = 52 450 и QSq2 — 70 940 кал/моль, определит^,., изменение
энтальпии и внутренней энергии для реакции 2PbS + 302 = 2РЮ +>2^>02.
Решение. По четвертому следствию из закона Гесса
~Q = 2-52 450 + 2- 70 940 — 2-22 300 = 202 180 кал!моль
или
АН = —Qp = —202 180 кал/моль.
По уравнению (92)
AU = АН — AnRT = — 202180 — (2 — 3) 1,987-298 = — 201588 кал/моль.
* Задача 11. На основании следующих данных:
4NH3 + 302 = 2N2 + 6Н20 + 362 400 кал, (I)
2Н2 + 02 = 2Н20 + 136 800 кал, (II)
NH3 + aq= NH3a? + 8400 кал (III)
подсчитать теплоту образования газообразного аммиака и водного раствора аммиака.
Решение. Умножаем уравнение (I) на +1/4, уравнение (II) на +3/4 и после
сложения результатов получаем
V2N2 + 3/2Н2 = NH3 + 12 000 кал. (IV)
*
Складывая уравнение (IV) с уравнением (III), получаем
V2 N2 + 3/2H2 + aq = NH3-aq + 20400 кал.
Задача 12. Молекулярная теплоемкость при постоянном давлении для
газообразного бромистого водорода зависит от температуры и меняется согласно уравнению
для газообразного брома
и для водорода
С^Вг = 6,5 + 0,00177,
СВг2 = 6,5 + 0,0064Г
с£г« =6,5 + 0,0017\
Требуется найти уравнение температурной зависимости теплоты образования
бромистого водорода и рассчитать значение теплового эффекта этого процесса при
температуре 1000° К, если при 320° С тепловой эффект равен 12 100 кал/моль.
Решение. Прежде всего определим необходимое значение алгебраической
суммы теплоемкостей веществ, участвующих в реакции:
1/2H2 + V2Br2 = HBr+Q,
т. е.
Е Сн - S Ск = г1*с*% + 1/2СВг* - C*Br = 0,0027.
Подставляем полученное значение в уравнение (111):
т т
Qt=QTq+ Г (2Сн — 2Ск) dT== 12 100+ I °>mTdT== П748 + 0,001Г2,
То т0
после чего подсчитываем тепловой эффект реакции образования газообразного
бромистого водорода при 1000° К:
#1000° к = 11 748 + 0,001 • 10002 = 12 748 кал/моль.
56
Задача 13. Молекулярная теплоемкость аммиака при постоянном давлении
задается следующим уравнением:
cnh3 = 804 + 0оз7Г + о,0551Г2,
а молекулярные теплоемкости водорода и азота при постоянном объеме задаются
уравнениями
С*2 = 4,92 + 0,0317Г + 0,0631Г2
и
С^2 = 4,66 + 0,0377\
Требуется определить теплоту образования аммиака при постоянном давлении при
температуре 700Q С, если ее значение при 20° С равно 11 890 кал1моль.
Решение. Предварительно необходимо, воспользовавшись уравнением (67),
перевести теплоемкости для азота и водорода из формы Су в форму Ср. Затем
определяем значение алгебраической суммы теплоемкостей веществ, принимающих
участие в реакции
V2N2 + 3/2Н2 = NH3 + Q,
т. е.
S Сн - Е Ск = Va Ф + 3/2С£2 - С?Нз = 5,41 + 0,03435Г - 0,054945Г2.
Полученное значение подставляем в уравнение (Ш):
973° К
Q9730 К == ^298° К + J [ 2j ^н ~" 2j ^к J dT ~
298° К
973° К
= 11 890 -f- I (5,41 + 0,03435Г - 0,064945Г2) dT ~- 14 280 кал/моль.
298° К
Задача 14. Молярная теплота испарения этилового спирта (М — 46) при 15° С
равна 6600 кал/моль. Средние удельные теплоемкости жидкого спирта и его паров
соответственно равны 0,530 и 0,360 кал/(г-град) в интервале температур от 0 до 789 С.
Требуется определить количество теплоты, необходимой для испарения 500 г спирта
при 60° С.
Решение. Воспользуемся уравнением (103):
т
АЯ60° С = АЯ15° С + ) (Спар — Сжидк) dT>
То
определим сначала удельную теплоту испарения:
60° С
А//60° С = АЯ15° С + J (Спар — Сжидк) dT =
15° С
6600
46
+ (0,360 — 0,530) (60 — 15) = 135,8 кал/г9
а затем количество теплоты, необходимой для испарения 500 г спирта:
Q = тАН = 500-135,8 = 67 910 кал.
Глав a~II.
ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
§ 7. СОДЕРЖАНИЕ ВТОРОГО НАЧАЛА
Первого начала термодинамики недостаточно для полного
описания термодинамических процессов. Оно позволяет точно найти энер-<
гетический баланс этих процессов, но не дает никаких указаний об
их направлении и о возможности их протекания. Общей
закономерностью, позволяющей находить направление и устанавливать
возможность или невозможность этих процессов, является второе
начало термодинамики. Кроме того, второе начало устанавливает те
условия, при которых превращение какого-либо запаса тепловой
энергии в полезную работу будет проходить наиболее полно.
Второе начало термодинамики, так же как и первое, является
результатом обобщения многолетнего человеческого опыта и,
следовательно, опирается на огромный накопленный экспериментальный
материал.
Исторически второе начало термодинамики было сформулировано
гораздо раньше первого, но со временем оно получало все новые и
новые толкования, а его формулировки становились все более
строгими. Впервые основное положение второго начала было дано
М. В. Ломоносовым (1747 г.): «. . . холодное тело В, погруженное
в теплое тело Л, не может воспринять большую степень теплоты *,
чем какую имеет Л». Первая математическая формулировка условий
превращения теплоты в полезную работу была сделана Сади Карно
(1824 г.). Он же вывел следствия, имеющие большое значение для
конструирования паровых машин. В работах немецкого физика
Клаузиуса (1850 г.) и английского физика Томсона (лорда Кельвина)
(1854 г.) были развиты идеи, которые вышли далеко за пределы
первоначально поставленной теплотехнической задачи. Несколько позже
Максвелл, Больцман и Гиббс установили связь второго начала с мо-
лекулярно-кинетическими представлениями. Это привело к
статистическому толкованию второго начала термодинамики.
Некоторые из формулировок второго начала наглядны и непо-^
средственно связаны с опытом, другие более абстрактны, но
являются более удобными для математического развития теории. По
Томсону: «Различные виды энергии стремятся переходить в теплоту,
а теплота, в свою очередь, стремится рассеяться, т. е. распределиться
между всеми телами наиболее равномерным образом». В этой
формулировке содержится представление о том, что в природе
происходит процесс рассеяния тепловой энергии, вследствие чего второе
начало термодинамики иногда называют законом рассеяния или
деградации тепловой энергии. По Клаузиусу: «Теплота никогда не
переходит с более холодного тела на более горячее, тогда как обратный
переход протекает самопроизвольно».
* Под степенью теплоты М. В. Ломоносов понимает нагретость тела, т. е.
температуру.
58
Подобно тому, как первое начало вводит функцию состояния —
внутреннюю, энергию, второе начало в форме, приданной ему
дальнейшими работами Кла/зиуса, 'вводит функцию состояния,
названную им энтропией.[Согласно второму началу, в то время как
внутренняя энергия изолированной системы остается неизменной, ее
энтропия при всех самопроизвольных процессах увеличивается^]
К вышесказанному необходимо также добавить, что содержание
второго начала иногда формулируется как невозможность создания
вечного двигателя (перепетуум мобиле) второго рода,
представляющего собой такую машину, которая заимствует тепло из резервуара
какой-либо температуры и превращает его в работу, охлаждая этот
резервуар и не производя больше никаких изменений в окружающих
телах.
Существует еще несколько логически связанных друг с другом
формулировок второго начала термодинамики, которые требуют
более подробного знакомства с понятием обратимых и необратимых
процессов, а также с понятием энтропии.
§ 8. НЕОБРАТИМЫЕ И ОБРАТИМЫЕ ПРОЦЕССЫ
Рассмотрение изменения функции, названной энтропией, привело
к разделению процессов на два класса. К первому классу относятся
так называемые необратимые процессы, в которых энтропия
увеличивается. Ко второму классу относятся обратимые процессы, при
которых энтропия остается неизменной. Остановимся подробнее на
характеристике этих процессов.
Необратимые процессы. Повседневный опыт показывает, что
существуют процессы, которые протекают самопроизвольно.
Наиболее яркими примерами таких процессов являются переход теплоты
от горячего тела к холодному, замерзание переохлажденной
жидкости, расширение газа в пустоту, взаимная диффузия газов или
жидкостей. Это все примеры одностороннего течения процессов. Они
всегда направлены в сторону приближения к равновесному
состоянию и прекращаются, когда это состояние достигнуто. При
теплопередаче равновесие определяется равенством температур, при
кристаллизации — равенством давлений во всем объеме, при диффузии —
равенством концентраций. Все эти примеры самопроизвольных
(спонтанных) процессов характеризуются существенным общим призна-
' ком: они сопровождаются превращением различных видов энергии
в теплоту, в теплота равномерно распределяется между всеми телами
системы. При этом подведение к системе того количества теплоты,
которое освободилось при процессе, не вызывает обратного течения
ни одного из названных процессов. Важно заметить, что косвенными
путями можно вернуть систему в первоначальное состояние, однако
при этом неизбежно придется произвести какие-либо энергетические
изменения в окружающей среде. В J противном случае необходимо
было бы признать возможность вечного двигателя второго рода.
Таковы характерные черты самопроизвольных или необратимых
термодинамических процессов, к которым относится огромное боль-
59
шинство реальных процессов, поскольку всегда имеется трение,
превращение электрической, световой и прочих видов энергии в
тепловую и т. д.
Обратимые процессы. Вполне обратимые процессы представляют
собой абстракцию и являются гипотетическими, но имеют большое
значение для теоретических исследований. Однако многие процессы
можно вести в таких условиях, чтобы их отклонение от обратимости
было как угодно малым. Характерной особенностью этих процессов,
таким образом, является то, что в каждой малой стадии процесса
состояние системы бесконечно близко к равновесному состоянию.
Следовательно, при изменении направления процессов,
проходящих последовательный ряд таких бесконечно близких состояний,
можно не только вернуть систему и окружающую ее среду в
первоначальное состояние, но и заставить их (систему и среду) обратно
пройти в точности те же изменения, что и при прямом процессе.
Примером обратимых процессов может служить адиабатическое
расширение или сжатие идеального газа. Однако этот процесс может
быть обратим лишь при условии полной теплоизолированности
системы и бесконечно медленного изменения объема и давления газа,
необходимого для быстрого выравнивания температуры.
Изотермическое расширение или сжатие идеального газа тоже может быть
обратимым процессом при условии немедленного теплообмена с
окружающей средой, необходимого для сохранения постоянства температуры.
Таким образом, и адиабатический, и изотермический процессы будут
обратимы при условии бесконечно медленного их протекания и
исключения трения. Следовательно, обратимые процессы являются
идеальными, предельными случаями реальных процессов.
§ 9. МАТЕМАТИЧЕСКОЕ ВЫРАЖЕНИЕ
ВТОРОГО НАЧАЛА ТЕРМОДИНАМИКИ
Коэффициент полезного действия тепловой машины
Для того чтобы прийти к математической формулировке второго
начала термодинамики, необходимо сначала познакомиться с
принципом действия тепловых машин. Работа таких машин
характеризуется коэффициентом полезного действия.
Если обозначить через Qx — количество теплоты, полученное
системой (машиной) от нагретого тела (нагревателя, теплоотдатчика),.
а через Q2 — количество теплоты, переданное системой более
холодному телу (теплоприемнику, холодильнику), то разность Qx — Q2
представляет собой, очевидно, количество теплоты, превращенное
в работу А (рис. 11):
A = Qi-Qa. (116)
Если тепловая машина работает без трения и тепловых потерь
в окружающую среду, то отношение
Qi — Q2, = JL — ц (117)
Qi Qi ' к }
является коэффициентом полезного действия,
Цикл Карно
Рассмотрим термодинамический процесс, состоящий из двух
изотермических (АВ и CD) и двух адиабатных (ВС и DA) обратимых
изменений состояния, представленных на рис. 12. Этот идеальный
цикл получил название цикла Карно.
Предположим, что первоначально система находится в
состоянии Л, ^характеризуемом объемом Уг и температурой 7\. Данным
значениям Vх и 7\ соответствует определенное значение давления Plf
которое определяется по уравнению Менделеева—Клапейрона (26).
Далее, систему подвергнем
изотермическому расширению (АВ)
при 7\ = const до объема 1/2>
после чего она приходит в со-
Нагреватель
Работа
A = Q1-Q2
Теплота
Теплота
1
J—<U
MQ.H.^.Q» I. II -Ь—.
«^р»
Холодильник
V, vt V2 V3
Рис. И. Схема перехода теплоты
в работу
Объем V
Рис. 12. Круговой процесс Карно
(идеальный термодинамический цикл)
стояние В, характеризуемое параметрами 1/2, Тг и р2. Переход из
состояния А в состояние В связан с производством работы, которая
совершается за счет подводимой к системе теплоты:
Q1 = RTX In
V,
V^
(П8)
Затем происходит адиабатное расширение системы (ВС) до
объема V3» когда система переходит в состояние С, характеризуемое
параметрами У3> Т2 и /?3- Этот переход системы из одного состояния
в другое связан с производством работы, которая совершается за
счет убыли внутренней энергии системы и составляет
AU - Cv (7\ - Т2).
(119)
Допустим далее, что система изотермически сжимается (CD) при
г2 = const до объема 1/4 и приходит в состояние!), характеризуемое
параметрами 1/4, Т2 и/?4. Работа внешних сил, затраченная на сжатие,
переходит в отдаваемое окружающей среде тепло, величина которого
составит
- Q2 = - RT2 ln:-£-. (120)
Наконец, пусть система адиабатно сжимается (DA) до
первоначального объема Vx и температуры 7\, т. е. приходит в первона-
61
чальное состояние А, которое при объеме Уг и температуре 7\ должно
обязательно иметь и давление рг. При этом затрачиваемая работа
расходуется на увеличение внутренней энергии системы:
At/ - Cv (7\ - Га); (121)
легко видеть, что это увеличение внутренней энергии при сжатии
в точности равно убыли внутренней энергии при расширении.
Складывая уравнения (118)—(121), получаем:
Q1-W-Q2 + W = Q1-Q2 = RT1\n^ +
+ Су(Т2-Тг)-ЯТ21п^ + CV(T1-T2) -
= /?Т'11п-^-—/?Г21п-^-. (122)
Поскольку для адиабаты ВС имеем
TiVT1 = r2VT\ (123)
а для адиабаты DA
TlVrl = T2Vr\ (124)
то, разделив уравнение (123) на уравнение (124), получаем
vT) =\У7) ' и ~K~~v7' {125)
Следовательно, уравнение (122) можно представить так:
Qi-Q2 = tf(A-Ta)ln-£-. (126)
Частное от деления этого уравнения на уравнение (118) дает
коэффициент полезного действия
ti= Ql~Q* = Tl~Ti , (127)
где Qx — теплота, получаемая от теплоотдатчика (нагревателя) с
температурой 7\, a Q2 — теплота, отдаваемая теплоприемнику
(холодильнику), имеющему температуру Т2.
Таким образом, коэффициент полезного действия тем больше, чем
ниже температура Т2 и чем больше разница между 7\ и Т2\ он мог бы
стать равным единице только в том случае, если бы температура 7\-
равнялась бы температуре абсолютного нуля.
Уравнение (127) можно представить в ином виде:
1_Ql=:1_^l или -§*- = -£-, (128)
41 * 1 41 * 1
или
Ql-^^O. (129)
1 1 1 2
62
Кроме того, можно показать, что коэффициент полезного действия
не зависит от рода вещества системы, а также, что коэффициент
полезного действия цикла Карно (г]), состоящего из обратимых
процессов, больше коэффициента полезного действия любого другого
кругового цикла (г)'), состоящего из необратимых процессов и
имеющего потери на трение:
Л >V- (130)
Высокое значение коэффициента полезного действия цикла Карно
является следствием не его специфической формы, составленной из
двух изотерм и двух адиабат, а обратимостью всех составляющих
его йроцессов. При этом подразумевается прежде всего обратимый
обмен теплотой (без потерь в окружающую среду) между
нагревателем, газом и холодильником и отсутствие потерь на трение
движущимися частями тепловой машины. Возможны и другие идеальные
циклы, в которых выполняются эти условия и коэффициент
полезного действия которых определяется также по формуле (127). Так,
цикл Эриксена составляется из двух изобар и двух изотерм, а цикл
Стирлинга из двух изохор и двух изотерм.
Заметим, что тепловые машины могут различаться по
техническому назначению. В рассмотренных случаях получали теплоту от
нагревателя, производили работу и отдавали неиспользованную
теплоту холодильнику. Такие устройства, если ставится задача
охлаждать более холодный резервуар, называют холодильными машинами.
Если же ставится задача
нагревать более нагретый
резервуар путем отнятия
теплоты от холодильника,
производством работы и
передачей неиспользованной
теплоты нагревателю, то такое
устройство называют
тепловым насосом.
Энтропия
Рассмотрим некоторый
произвольно взятый цикл
(рис. 13), состоящий только
из обратимых процессов.
Площадь, ограниченная
контуром этого цикла в координатах объем-давление, соответствует
величине теплоты, превращенной в работу. Разобьем этот контур
большим числом адиабат. Через точки пересечения адиабат с кривой цикла
проведем изотермы. При этом получаем малые циклы Карно, каждый
из которых состоит из двух изотерм и двух адиабат. Площади этих
циклов при достаточной близости адиабат между собой ничтожно
мало отличаются по площади от циклов, ограничиваемых адиабатами
и контуром цикла.
#
%
>**, \
\ V
Объем V
Рис. 13. Произвольный цикл, состоящий из
обратимых процессов
63
В предельном случае, когда число циклов Карно бесконечно
велико, сумма их площадей равна общей площади цикла.
Для каждого из бесконечно малых циклов, согласно
уравнению (129), справедливы равенства:
dQ[ dQ2 ^
?! Т2
ч и \\\ ш
dQY dQ2 dQx dQ2
—г, -*- = О, —v, -г = 0 и т. д. (131)
1 1 1 2 1 1 1 2
Суммируя эти равенства, получаем
dQ[ dQ2 dQ'l dQ2 dQl dQl
* f I ff rr i (fi
Tx T2 Tx T2 Тг Т.л
т. е. алгебраическая сумма приведенных теплот (так называют выра-
dQ Q\
жение ~y~ или-5м цикла равна нулю:
В пределе алгебраическая сумма N -=г- переходит в интеграл,
взятый по замкнутому контуру:
^- = 0. (134)
Поскольку интеграл по замкнутому контуру от некоторой
функции равен нулю, то подынтегральное выражение представляет собой
полный дифференциал этой функции, а сама функция представляет
собой функцию состояния системы. Эта функция получила название
энтропии и обозначается буквой 5 (Клаузиус, 1865 г.).
Следовательно:
<j)dS = 0, (135)
и
^-^dS (136)
для всех обратимых процессов.
Если будем рассматривать цикл, состоящий из необратимых
процессов, то, согласно неравенству (130), будем иметь
j) dS<0
(137)
и
dQ
Т
64
<dS. (138)
Таким образом, широко используемая математическая
формулировка второго начала термодинамики может быть представлена
или в интегральной форме:
§■^-^0 (139)
или в дифференциальной форме:
dQ^TdS, (140)
при этом знак равенства относится к обратимым (идеальным)
процессам, а- знак неравенства — к необратимым (самопроизвольным)
процессам.
Понятие энтропии с первого взгляда кажется несколько неясным
и трудно понимаемым. В то же время это понятие является
универсальным и высокоэффективным средством термодинамики при анализе
различных явлений. Попытаемся сделать понятие энтропии более
конкретным.
Прежде всего энтропия является функцией состояния.
Аналитическое выражение этого положения дается уравнением (136).
Следовательно, изменение этой функции не зависит от пути перехода и
зависит только от значения энтропии начального и конечного
состояний.
Очень часто понятие энтропии используется в качестве меры
деградации (рассеяния) энергии. Для того чтобы иллюстрировать
справедливость этого положения, построим наши рассуждения
следующим образом. Предположим, что имеются три тела (/, 77 и 77/),
температура которых 7\, Т2 и Т3, причем Тг > Т2 > Т3. Рассмотрим
соотношение величин работ, которые получаются, если вести
процессы между телами / и /77, // и ///, / и //. Если Q3 — теплота,
полученная телом ///, во всех случаях одинакова, то для разности
температур 7\ и Г3 имеем
A1 = Q1r\1 = Q1(l-TJT1)9
а для разности температур Г2 и Т3
A,- Q.r|, = Q2 (1 - 7УГ2).
Определим разность Аг — А2 = А, т. е. работу для разности
температур 7\ и Т%:
А = А - А, = Q, (1 - 7УЛ) - Q2 (1 - Ts/T2) = Q1- Q.TJ^ -
= Qi-Q» — Ta (Si-St) = Qi - Q, — Ts AS. (141)
Следовательно:
Qi - Q2 = A + T3AS, (142)
5 A. H. Крестовников 65
где А — работа, связанная с передачей теплоты от тела с
температурой 7\ к телу с температурой Г2;
Qi — Q2 — непревращенная в работу теплота;
Д5 — разность энтропии при температурах 7\ и Г2;
Т3 — какая-то температура, более низкая, чем 7\ и Т2-
Отсюда чем больше Д5, тем большая часть теплоты рассеивается
в окружающем пространстве, не превращаясь в полезную работу.
Следовательно, энтропия является мерой бесполезной теплоты или,
как еще иногда ее называют, мерой обесцененной энергии.
Большинство свойств систем, имеющих количественное
выражение, можно разделить на две группы. Свойства, которые при
соприкосновении систем выравниваются (температура, давление и другие),
называются интенсивными, а те свойства, которые суммируются
(объем, масса и другие), называются экстенсивными.
Величина энергии может быть представлена через произведение
двух величин, одна из которых выражает как бы напряженность
энергии (фактор интенсивности), а другая выражает то, на чем
данная напряженность может проявиться (фактор экстенсивности или
емкости). Например, приращение потенциальной энергии может быть
выражено произведением Fdh, т. е. произведением силы на
приращение высоты; приращение электрической энергии Edq, т. е.
произведением электродвижущей силы на количество переносимого
электричества; приращение поверхностной энергии ads, т. е.
произведением поверхностного натяжения на приращение поверхности.
Величины F, Еу а являются факторами интенсивности, a h, q, s —
факторами экстенсивности или емкости. С указанных позиций тепловая
энергия также может иметь фактор интенсивности (таковым является
температура) и фактор экстенсивности или емкости (таковым
является энтропия).
Как и всякое другое экстенсивное свойство, энтропия является
аддитивным свойством. Это означает, что энтропия системы равна
сумме энтропии составных частей. Энтропия пропорциональна массе.
Отсюда следует, что величина энтропии может относиться к
различному количеству вещества. Чаще всего эту величину относят к молю,
грамм-атому или грамму вещества.
Размерность энтропии легко определить из уравнения (136). Она
представляет собой единицы энергии, деленные на градус. Кроме
того, необходимо относить энтропию к определенному количеству
вещества. Единицами энергии в этом случае чаще всего являются
кал и ккал. Таким образом, чаще всего энтропия имеет следующую
размерность: кал/(г-град), кал/{г-атом-град) и кал/(моль-град). Для
второй и третьей формы размерности в последнее время стали
применять сокращенное обозначение э. е., расшифровываемое как
«энтропийная единица».
Размерность энтропии совпадает с размерностью теплоемкости.
Однако отсюда не следует аналогия их физического значения.
Теплоемкость характеризует количество теплоты, необходимой для
нагревания тела на 1°. Энтропия характеризует количество рассеянной
энергии, отнесенное к 1° данной температуры.
66
Проанализировав некоторые конкретные свойства энтропии,
рассмотрим способы вычисления энтропии.
Вычисление энтропии. Напомним, что если рассматриваемый
процесс сопровождается увеличением энтропии (AS !>0), то он
является самопроизвольным, если уменьшением (AS < 0), то —
несамопроизвольным.
Рассмотрим изменение энтропии в том случае, когда процесс
происходит при р = const. В этом случае
dQ - CpdT (143)
и, следовательно:
dS = -^- = -^. (144)
Интегрируя, получаем:
J «е- J ^
И
St,-STi=1 ^ (145)
Для взятия интеграла необходимо знать уравнение зависимости Ср
от температуры для всего интервала температур от 7\ до Т2.
В случае Ср = const, как это имеет место, например, для
идеальных газов, уравнение (145) упрощается:
5r,=Sr, + CplnA, (146)
или
AS = Cpln-b-. (147)
В уравнениях (145)—(147) под 7\ подразумевается любая
температура, меньшая чем 72. Забегая несколько вперед, скажем, что,
согласно третьему началу термодинамики, энтропия при абсолютном
нуле равна нулю; следовательно, при 7\ = 0 °К, когда Т2 становится
равным Ту имеем:
'т
ST=\5*-dT. (148)
о
Если процесс происходит при V = const, то
dQ - CvdT
и, следовательно:
dS = J^ = £vML. (149)
5*
67
После интегрирования получаем
Srt = STi+]'^f-t (150)
Ту
или
ST=\^f-. (151)
о
Если Cv = const, то
AS = C„ In-£*-• (152)
* 1
Если процесс происходит при Т = const, то, поскольку
dQ = dU + dA
и
получаем
Подставляя р = -=^-, имеем
dQ = TdS,
dU = 0,
ЛЛ - pdV,
TdS = pdK. (153)
RT
откуда
AS = flln-^. (156)
TdS^^dV, (154)
dS = R-^9 (155)
а после интегрирования
ЛЯ = /?1п.
Пользуясь законом Бойля—Мариотта {ргУг = /?2^2)> получаем
AS = #ln-^. (157)
Таким образом, изотермические превращения, происходящие без
изменения внутренней энергии системы, идут за счет затрачиваемой
при этом теплоты:
Q =/^ In £ = ЯГ In-£Ц (158)
ч Рг
количественно равной совершаемой при этом работе, определяемой
по уравнениям (53) и (54).
Если изотермическим превращением является изменение
агрегатного состояния: плавление, испарение, сублимация или аллотроп-
68
ный переход, то изменение энтропии, называемое в этом случае
энтропией превращения, определится следующим образом:
AS = -P-, (159)
где Q — тепловой эффект превращения.
Поскольку энтропия является функцией состояния, то изменение
энтропии можно определить, если известно значение параметров
начального и конечного состояний.
Допустим, что начальное состояние S± характеризуется
параметрами рх и 7\, а состояние 52 — параметрами р2 и Г2, если за
независимые переменные выбраны давление и температура. Раз
изменение энтропии не зависит от пути перехода, то удобнее осуществить
переход из состояния Sx в состояние S2 сначала при рг = const
(при этом температура меняется от Тг до Т2), а затем — при
Тг = const (при этом давление меняется от рг до р2).
Суммируя изменение энтропии по этим двум процессам, получаем
1 1 Р2
AS = Ср In 4^ + # In-^-. (160)
Точно так же при независимых переменных температуре и объеме
изменение энтропии составит:
AS = CKln-^+tfln-^. (161)
1 1 Vi
При независимых переменных давлении и объеме, подставляя
в уравнение (160) или (161) значения температур 7\ = —^- и 72
Р у
^-, а также учитывая, что Ср — Cv = R и Cp/Cv = y> полу-
__ * 2у2
чаем
AS^Cyln^. (162)
Таким образом, мы рассмотрели некоторые уравнения,
позволяющие вычислить энтропию или изменение энтропии по известным
изменениям параметров системы и по известным теплоемкостям. Однако
существуют и другие методы определения энтропии. Например, по
измерениям электродвижущих сил гальванических элементов или
по измерениям констант равновесия и температурным зависимостям
того и другого.
§ 10. СТАТИСТИЧЕСКИЙ ХАРАКТЕР
ВТОРОГО НАЧАЛА ТЕРМОДИНАМИКИ
Энтропия и термодинамическая вероятность состояния
Огромное значение для развития всего естествознания имело
установление молекулярно-кинетического представления о
механизме возрастания энтропии во всех реальных процессах.
69
Допустим, что имеется система, состоящая из нескольких
различных газов. Перед началом диффузии такую систему можно
рассматривать как упорядоченную. Когда диффузионное смешение газов
закончится и газы будут равномерно перемешаны, система становится
беспорядочной. Указанный процесс идет в природе самопроизвольно,
т. е. с увеличением энтропии или, как мы теперь видим, с
увеличением беспорядка. Иначе говоря, системы стремятся к более
беспорядочному состоянию. Следовательно, таких систем будет в природе
все больше и больше, и системы с большой энтропией, обладающие
большей беспорядочностью, более вероятны. Процессы,
соответствующие тому, чтобы в одной части системы сгруппировались
молекулы одного вида, а в другой части — другого вида, являются
настолько маловероятными, что практически могут считаться
невозможными.
Состояние вещества может быть описано двояким способом. Во-
первых, мы можем охарактеризовать макросостояние следующими
параметрами: давление, объем и температура. Эти статистические
величины и характеризуют средние свойства большого числа молекул
(атомов). Во-вторых, можно задать положение каждой молекулы
в пространстве, направление и скорость ее движения, т. е.
охарактеризовать микросостояние вещества. При этом одно и то же
макросостояние возможно при самых различных микросостояниях.
Термодинамической вероятностью
состояния системы называется число микросостояний, посредством
которых данное состояние может осуществиться. Обозначим
термодинамическую вероятность через Q. Применяя теорию вероятностей,
законы которой в сочетании с законами механики образуют
статистическую механику, можно, с одной стороны, определить
термодинамическую вероятность состояния, а с другой — связать
термодинамическую вероятность с энтропией.
Больцман, используя приемы статистической механики, показал,
что энтропия системы S может служить характеристикой
термодинамической вероятности данного состояния системы (Q). Связь между
ними выражается уравнением
5 = R In Q - kN0 In Q, (163)
где k — постоянная Больцмана, определяемая отношением
универсальной газовой постоянной к числу Авогадро, т. е.
Важно (не по формальным причинам) обратить внимание на то,
что формула (163) относится к числу замечательнейших
соотношений (всего их три), рожденных научной мыслью. Первое
соотношение—это формула Эйлера {еш — —1), которая связывает простым
соотношением величины, обладающие совершенно различными
свойствами (две трансцендентные величины и одну мнимую). Второе —
это формула Эйнштейна [AU = с2Дт (1), упомянутая на стр. 13],
содержащая только физические величины. Это обеспечивает возмож-
70
ность экспериментальных проверок, совершенно различающихся по
своей материальной сущности величин (движение — инерция,
энергия— масса и пространство — время). Третья—формула Больдмана
(163), связывающая физические величины (их численные
значения определяются выбором системы единиц) с математической
величиной (термодинамической вероятностью). Отсюда становится
ясным теоретическое значение формулы Больцмана, позволяющей дать
материалистическое толкование второго начала термодинамики и
понятия энтропии. Доказывается, далее, что термодинамическая
вероятность есть число комплексий *, посредством которых данное
пространственное распределение может осуществиться.
Следовательно, если молекул N0 и они распределяются так, что в каждом
из микросостояний окажется JV^, JV2, N3 и т. д. молекул, то
причем
JV0 = N± + N2 + N3 + • • • (166)
Анализ полученных соотношений приводит к следующему. Если
происходит увеличение энтропии, то это является следствием либо
увеличения числителя, либо уменьшения знаменателя в формуле (165).
Увеличение числителя соответствует увеличению числа молекул, и
если система замкнута, то это невозможно. Знаменатель будет иметь
минимум тогда, когда отдельные сомножители будут равны между
собой. Это соответствует совершенно равномерному распределению
молекул. В этом случае будем иметь максимальную энтропию.
Антинаучное толкование второго начала термодинамики. Так как
равномерное распределение частиц (по молекулярно-кинетической
теории) является наиболее беспорядочным, наиболее хаотичным, то
с увеличением энтропии растет хаос в системе. Отсюда и вытекает
одна из формулировок второго начала термодинамики, выдвинутая
Больцманом: «Мир стремится к хаосу», которая перекликается с
формулировкой Клаузиуса: «Энтропия мира стремится к максимуму».
Такие представления дали повод для различных идеалистических
толкований второго начала термодинамики, переплетающихся с
религиозными и мистическими настроениями, и послужили толчком
для создания «теории» тепловой смерти Вселенной. Возрастание
хаоса мира или энтропии мира, по мнению сторонников этой «теории»,
должно в конце концов привести к полному выравниванию
температуры во всей вселенной, что означало бы невозможность протекания
каких бы то ни было процессов и, следовательно, «тепловую смерть
* Комплексий представляют собой соединения, различающиеся друг от друга
только самими элементами и образующиеся с определенным чередованием. Число
комплексий С определяется как число возможных сочетаний из п элементов по k
элементам:
г = rt!
~ k\ {n—k)\ '
71
Вселенной». Эти антинаучные положения впоследствии были
разбиты, поскольку они основаны на ложном понимании Вселенной,
как чем-то ограниченной системы. В действительности Вселенная
безгранична, не имеет ни начала, ни конца как во времени, так и
в пространстве.
Рассматривая термодинамические законы применительно к
бесконечной вселенной, следует иметь в виду, что могут быть
отступления и протекать процессы менее вероятные, например с
уменьшением энтропии (в этом приходится все более убеждаться при
исследовании космических явлений). Термодинамический метод
исследования не может механически переноситься на ядерные реакции.
Метафизическая трактовка второго начала, вытекающая из
формулировок Клаузиуса и Больцмана, подвергалась критике с разных
точек зрения различными учеными: Смолуховским, Планком, Ван-
дер-Ваальсом, а с наиболее общей —философской точки зрения
концепция тепловой смерти была опровергнута Ф. Энгельсом в его труде
«Диалектика природы».
§ П. УРАВНЕНИЕ АГРЕГАТНЫХ ПРЕВРАЩЕНИЙ
Приложение второго начала термодинамики к агрегатным
превращениям позволяет вывести зависимость, которая получила
название уравнения Клаузиуса—Клапейрона. Для процессов
парообразования эта зависимость связывает производную давления
насыщенного пара по температуре, измерение объема и тепловой эффект. Для
процессов плавления и аллотропных превращений она связывает
производную температуры превращения по давлению, изменение
объема и тепловой эффект.
Выведем уравнение Клаузиуса—Клапейрона. Для этого
переведем вещество из одного агрегатного состояния в другое, оставляя
постоянным давление (р = const). Предположим также, что
температура тоже будет оставаться неизменной (это вполне вероятно),
т. е. превращение будет изотермическим (Т = const). При этом
переход вещества из одного состояния в другое сопровождается
совершением работы
А = р (V, - VJ. (167)
Проведение того же процесса при других температурах требует
другого давления, а следовательно, совершения иной работы. Для
выяснения влияния температуры продифференцируем уравнение (167)
по температуре (считая разность V2 — V\ не зависящей от
температуры):
dT - ^2 Vl) dT '
Умножив на Т (Т ф 0) левую и правую части уравнения, получаем:
72
Поскольку, согласно уравнению (127),
Qi - Q» Гх — Г2
или
в пределе имеем (при Qx — Q2 = dA; Qx = Q; 7\ — Т2 = dT и
7-1 = T),
dA _ dT
Q - T '
Q = T^r. (169)
Складывая уравнения (169) и (168), окончательно получаем
Q = 7 (V2 - V0-зг • (170)
В случае процесса сублимации это уравнение принимает вид:
<?«**, = Т (Vnap - VTB) -J-, (171)
в случае процесса испарения:
Ч?исп = •* (•'пар ^жидк) "^~ ' ( '
Здесь: Ссубл и QHcn — теплоты сублимации и испарения; Vnap —
объем данного количества вещества в парообразном состоянии; VTB и
^жидк — объем того же количества вещества в твердом и жидком
состоянии; dp/dT — температурный коэффициент изменения
давления насыщенного пара. Кривая сублимации исходит из начала
координат, где /? = 0 при Т = 0° К; кончается же в точке, давление и
температура которой соответствуют равновесию пара, жидкости и
кристаллов. В свою очередь из этой точки начинается кривая
испарения, кончающаяся в точке, давление и температура которой
соответствуют исчезновению различий между паром и жидкостью:
^пар = ^жидк и Л#исп = 0.
Обе кривые — возрастающие, обращены выпуклостью в сторону
оси температур и имеют одну общую точку.
В этой точке берет начало кривая плавления:
<2пл = Т (Ужидк - Утв) ■%■ , (173)
гДе Q™ — тепловой эффект, сопровождающий плавление. Наклон
кривой определится изменением объема при плавлении: если Ужидк —
— VTB > 0, то dp/dT >0 и функция будет возрастающей; если
^жидк—^тв <0> то dp/dT < 0 и функция" будет убывающей
(имеется в виду QnjI > 0). Заканчиваться кривая должна в точке
исчезновения различий между жидким и твердым состоянием
вещества (Ужидк = VTB и QnjI = 0)* однако, из-за затрудненности диф-
73
фузионных процессов при высоких давлениях построение кривых
вплоть до этой точки неосуществимо.
Если имеет место процесс аллотропного (полиморфного)
превращения, то он описывается уравнением:
Qa+fi = T(Vfi-Va)-$lr, (174)
где Qa->p —тепловой эффект аллотропного превращения и
{V$ — Va) — изменение объема данного количества вещества при
аллотропном превращении.
В уравнениях (173) и (174) величина dp/dT может быть определена
как величина, обратная коэффициенту, характеризующему
изменение температуры плавления или превращения а —> р с изменением
внешнего давления (т. е. -г—).
В процессах плавления и аллотропного превращения изменение
объема обычно невелико, в то время как при испарении и возгонке
разность объемов значительна вследствие того, что в газообразном
состоянии вещество занимает объем, во много раз превышающий
объем того же вещества в конденсированном состоянии. Последнее
обстоятельство позволяет сделать ряд упрощений. Во-первых, так
как при обычных условиях Ужидк и VTB во много раз меньше Упар, то
можно предположить, что Ужидк ^ 0 и Утв ^ 0. Тогда
Q = TVmp-^r. (175)
Во-вторых, допустим, что в этом случае
v = -^-
тогда
Q-^4-. (176)
Р dT
Произведем разделение переменных и интегрирование:
dp Q dT
R T2 '
(177)
In
J>2
Pi
ИЛИ
= £(£-£)• (178)
^t-l^P- (179)
Здесь допустили третье предположение, заключающееся в том,
что Q не зависит от температуры, т. е. Q = const. Это справедливо
лишь при интегрировании в небольшом интервале температур.
Часто уравнение (179) встречается в виде
lg Pl 4,5737^2 ' ^10 ;
74
где натуральный логарифм переведен в десятичный [для перевода
натуральных логарифмов в десятичные пользуются соотношением
In А = 2,303 lg Л, а универсальная газовая постоянная
представлена в калориях (R = 1,987 кал/(моль• град)].
Неопределенное интегрирование уравнения (177) дает
Q 1
1§Р = —
4,573 Т
+ const.
(181)
Отсюда видно, что если рассматривать lg р как функцию IIТ, то
эта зависимость является нелинейной. Используя построение этой
линейной зависимости по двум значениям давлений рг и р2 ПРИ соот-
1п760Ъ-
тр
Температура Т
*р
Рис. 14. Схема использования
зависимости логарифма давления (Р, мм pm.cm.)
от обратной температуры (Г, ° К) для
графического определения температуры
кипения (Гкип) и теплоты испарения
(Q ~- —R tg а) по известным значениям
Pi и Рг» соответствующим Гх и Г2
Рис. 15. Зависимость теплоты
испарения жидкости от температуры:
Т — тройная точка (равновесие
кристаллов, жидкости и пара); Т —
критическая температура (при Q = 0);
Q0 — гипотетическая теплота испарения
(при Т = 0° К)
ветствующих значениях температур Тг и Т2, можно определить
температуру кипения и теплоту испарения. Последняя определяется
по уравнению Q = —R tg а (рис. 14).
Если учесть уравнение зависимости теплового эффекта от
температуры (115)
Q = Qo +
Да гр , Ар
г +
Ду
(182)
1 " ' 2 * ' 3
графическое изображение которого дается на рис. 15, то после
подстановки этого значения Q в уравнение (177) получаем
Оо лгг А<* агг АР dT— Ау
d\np
dT — 42rdT
TdT.
(183)
RT2— xt ~* 2R ~* 3R
Интегрирование этого выражения по температуре дает уравнение,
которое получило название развернутого уравнения Клаузиуса—
Клапейрона:
\пр =
Qo 1
Аа
R Т
--^-1пТ—-^гТ —
А|5
R
2R
Ау
6#
Т2 + const. (184)
75
§ 12. ОПЫТНОЕ ОПРЕДЕЛЕНИЕ ДАВЛЕНИЯ
НАСЫЩЕННОГО ПАРА
Определение давления насыщенного пара осуществляется двумя методами:
статическим и динамическим. В первом случае измеряют давление над жидкостью
при определенной температуре, во-втором — температуру ее кипения при данном
давлении.
Прямые или статические методы. При средних температурах в простейшем
случае может быть использована барометрическая труба (рис. 16), помещенная в термо-
Q
8
5:
со
^ш
а
Рис. 16. Схема определения
давления насыщенного пара
статическим методом:
а — барометрическая трубка
без исследуемой жидкости; б —
трубка с исследуемой
жидкостью
з-э
Рис. 17. Схема метода определения
давления насыщенного пара при
температурах ниже комнатной:
1 — манометрическая трубка; 2 — стеклянный
сосуд с исследуемым веществом; 3 —
термостат; 4 — термометр
стат. Поверх ртути находится испытуемая жидкость. Разность уровней ртути с
соответствующей поправкой на плотность самой жидкости (р) дает давление насыщенного
пара при данной температуре:
h
р = 760 — h — мм рт. ст.
Так как ртуть обладает собственным давлением насыщенного пара, то иногда вместо
ртути используют легкоплавкие сплавы, а для высоких температур — олово в
кварцевой барометрической трубке.
При температурах ниже комнатных используется прибор, принципиальная схема
которого дана на рис. 17. Стеклянный сосуд 2 с исследуемой жидкостью снабжен
манометрической трубкой 1, изогнутой под прямыми углами. Сосуд помещается в
термостат 3, предназначенный для получения низких температур. Давление
насыщенного пара над жидкостью определяется разностью между атмосферным давлением,
отсчитываемым по барометру, и высотой ртути в трубке 1. Температура измеряется
термометром 4.
При сравнительно точных работах давление измеряется более совершенными
манометрами. Для этой цели наиболее распространен манометр Мак-Леода (рис. 18),
который состоит из баллона 2, снабженного в верхней части капилляром 1. К
отводной трубке 3, соединяющей манометр с аппаратурой, припаян боковой капилляр 4,
предназначенный для сравнения высот столбиков ртути в капиллярах 1 и 4. С
помощью ловушки 5, перекрывающей воздух, манометр соединен с резервуаром ртути 6,
установленным на подъемной подставке 7, передвигающейся по вертикальной рейке 8,
76
Способ измерения давления этим манометром состоит в том, что в капилляре 2
сжимают газ и косвенным образом, зная степень сжатия и объем газа в сжатом
состоянии, вычисляют искомое давление в аппаратуре. Если обозначить объем баллона 1
через V, объем [капилляра 2 через v, объем
газа после сжатия через vi, давление в
аппаратуре через р и разность уровней в
капилляре 2 и боковом капилляре 4 через /i, то
по закону Бой ля—Мариотта:
а так как
р (V + v) = hvi,
vi = hs,
где s — площадь поперечного сечения капил-
ляра, то измеряемое давление
hvx
V + v
+
h2s
V+v
нч.
Для данного манометра к
= const.
V + v
Как видно из рис. 18, в этом случае
высота ртути в боковом капилляре 4 должна
быть на уровне верхнего конца капилляра
баллона 2.
Косвенные или динамические методы.
На рис. 19 дана схема установки для
определения давления насыщенного пара
динамическим методом. Сосуд 1., помещенный
на нагреватель, содержит исследуемую
жидкость, температура паров которой
определяется термометром 2. С помощью
вакуумного насоса в системе создаются любые
разрежения, измеряемые вакуумметром 5.
В конденсаторе 3, представляющем собой
рубашку с проточной холодной водой,
откачиваемый воздух освобождается от паров,
которые собираются в сосуде 4. Определяя
температуру закипания жидкости (т. е. температуру активного процесса
испарения жидкости из всего объема) при различных давлениях в системе, получаем
сразу функциональную зависимость давления насыщенного пара от температуры.
Рис. 18. Схема устройства
манометра Мак-Леода:
J — баллон; 2 — капилляр; 3 —
отводная трубка; 4 — боковой
капилляр; 5 — ловушка; в — резервуар
с ртутью; 7 — подставка; 8 — рейка
для передвижения подставки
IF^S
Рис. 19. Схема определения давления насыщенного пара
динамическим методом:
J — стеклянный сосуд с исследуемым веществом; 2 — термометр;
3 — конденсатор; 4 — сборник конденсата; 5 — вакуумметр
77
Среди других динамических методов определения давления насыщенного пара
над твердыми металлами, которые характеризуются очень малыми величинами
давления насыщенного пара, следует назвать метод, основанный на измерении скорости
испарения через очень малое отверстие, а также метод взвешивания испаряющейся
нити.
Определение давления насыщенного пара
над разлагающимися и химически активными
сплавами и соединениями
Измерения давления насыщенного пара компонентов сплава или соединения
осложняются тем, что в паре могут присутствовать молекулы соединения, ассоциации
атомов и сами атомы компонентов, вследствие чего возникает необходимость
исследовать также химический и молекулярный состав пара. Кроме того, жидкие и твердые
вещества вследствие разложения меняют свой химический состав, и результаты
измерения должны быть отнесены не только к определенной температуре, но и к
определенной концентрации.
Большими возможностями обладает масс-спектрометрический метод, который
основан на определении отношения массы к заряду ионов, образующихся при
пересечении газового или парового потока вещества с пучком электронов. По
интенсивности ионных пучков рассчитывают давления компонентов газового (парового)
потока. Сплавы или соединения для этой цели испаряют из графитовых,
металлических или других тиглей через отверстие с диаметром от 0,3 до 3 мм (в методе Кнуд-
сена) или с открытой поверхности (в методе Лэнгмюра). Хотя масс-спектрометр
ический метод очень чувствителен, тем не менее абсолютные значения давлений с его
помощью измерить нельзя. Кроме того, в большинстве случаев взаимодействие
вещества с электронами приводит к изменению атомно-молекулярного состава газа или
пара.
Давления насыщенного пара компонентов газовой или паровой смеси могут
быть определены методом изоляции объема. Для этого часть сосуда из жаростойкого
стекла или кварца, имеющая определенный объем, после установления равновесия
отделяется с помощью перепайки от части, содержащей испаряемое вещество, а
затем образовавшийся в ней конденсат подвергается химическому анализу. Результаты
анализа служат исходными данными для расчета давлений пара компонентов по
уравнению Менделеева—Клапейрона (26).
Если пар над разлагающимся веществом имеет несложный состав, применимы
следующие методы: весовой, точки росы и оптической плотности.
При весовом методе используются рычажный механизм, аналитические весы и
ампулы с двухзонным нагревом. Разложение вещества в одной части ампулы и его
конденсация в другой части по показаниям весов (взвешивание не всегда производят
непрерывно, оно часто осуществляется через определенные отрезки времени)
позволяют определить количество перенесенного вещества, пропорциональное его летучести.
В методе точки росы вещество помещается в ампулу с отростком. Температура
отростка медленно понижается до появления конденсата, в то время как остальная
часть ампулы с веществом выдерживается при постоянной заданной температуре.
По зафиксированной температуре появления первых капель конденсата («росы»)
определяется давление насыщенного пара над веществом.
Измеряя оптическую плотность пара с помощью фотометрирования
монохроматического пучка света, также можно судить о давлении насыщенного пара над
веществом, поскольку эти две характеристики пропорциональны друг другу.
Все три перечисленных метода требуют предварительных экспериментов с
летучим компонентом. После калибровки прибора становится возможным вести измерение
давлений этого летучего компонента над разлагающимся веществом.
Следует упомянуть группу прямых статистических методов измерения давлений
насыщенного пара, использующих нуль-инструмент. Исследуемое вещество
помещают в ампулу, соединенную капилляром с стеклянным или кварцевым
нуль-инструментом (деформируемыми под действием давления спиралью или изогнутой
стенкой — «ложечкой»). Сначала нуль-инструмент градуируют по веществам с
известным давлением с применением противодавления инертного газа, т. е. определяют
градуировочную поправку к величине противодавления по степени деформации
нуль-инструмента.
78
В последнее время все более широкое применение получают методы,
использующие радиоактивные изотопы.
Задачи
Задача 1. Атомная энтропия свинца при стандартных условиях равна
15,49 кал/{г-атом-град).
Требуется определить энтропию твердого свинца при температуре плавления
327° С если температурная зависимость теплоемкости свинца выражается
уравнением,
Ср= 5,77+ 2,02-10"3 Т.
Решение. Для получения ответа задачи воспользуемся уравнением (145):
ST2=STl + J"f-dr;
600° К
_1
298° К
5б00° к = 5298о к + J -jr-(5,77 + 2,02.10-»r)dT =
= 15,49 + 5,77-2,303 Jg-|^_ +2,02.10-3(600 —298) =20,14 кал/(г-атом-град).
Задача 2. Требуется определить изменение энтропии при плавлении 1 кг меди,
если удельная теплота плавления меди 41,6 кал/г, а температура плавления 1083° С.
Решение. Поскольку плавление меди происходит изотермически,
воспользуемся уравнением (159):
AS = -jr = -4М- = 0,03068 кал/(г-град).
>.
Следовательно, изменение энтропии при плавлении 1 кг меди составит
30,68 кал/град.
Задача 3. Удельная теплота испарения воды при 100° С равна 539 кал/г,
удельная теплоемкость воды 1 кал/(г-град) и удельная теплоемкость пара при постоянном
давлении 0,477 кал/(г-град). Требуется найти изменение энтропии при превращении
100 г воды, взятой при 0° С, в пар при 120° С.
Решение. Искомое изменение энтропии складывается из трех частей:
изменение при нагреве от 0 до 100° С, изменение при испарении (изотермический
процесс) и изменение при нагреве^от 100 до 120° С.
Следовательно:
т т
1 КИП '2
dT , QHcn , Г п dT .
ST2~ST1- J Сжидк — + -f~ + J Спар"у
откуда
т
1 КИП
373° К 393° К
dT , 539 , Г Л А„ dT 373 . 539
л о Г dT , 539 , Г Л.__ dT 0 оло t 373 , 539 ,
AS= J — + -373-+ J °'477— ==2'303]SW + -37y +
273° К 373° К
39-i
+0,477.2,303\g-^-= 1,7817 кал/(г-град).
Полученное значение изменения энтропии является удельным; для превращения
100 г воды изменение энтропии составит 178,17 кал/град.
Задача 4. 12 г кислорода охлаждаются от 20 до —40е С. Одновременно давление
повышается от 1 до 60 am. Каково при этом изменение энтропии, если молекулярная
теплоемкость кислорода при постоянном давлении равна 6,97 кал/(моль-град).
79
Решение. Поскольку величина изменения энтропии при переходе из
состояния Pi = 1 am и ti = 20° С в состояние Рг = 60 am и ^ = —40° С не зависит от
пути перехода, то для облегчения решения задачи представим себе этот переход
состоящим из двух процессов: изобарического и изотермического. Тогда, согласно
уравнению (160), имеем
AS = Ш
М
12 / 233
(cp,n^ + «,n|L) =
32
(233 1 \
6,97-2,303 lg-293" + 4>573 \g~^ \ = -3,312 кал/град.
Задача 5. Температура превращения ромбической серы в моноклинную при
давлении 1 am равна 95,6° С, теплота превращения 3,12 кал/град. Требуется найти
разность объемов ромбической и моноклинной серы, если dT /dp = 0,0399 град/am.
Решение. Для определения разности объемов воспользуемся уравнением
(174), откуда
at/ Q dT 3,12.41,3-0,0399 ЛЛ10ПС ч/
А1/ = -Т ' Ж = Ш> = °'°1395 См3/г'
Для получения ответа в см3!г потребовалось воспользоваться соотношением единиц
измерения энергии: 1 кал =41,3 см3-ат.
Задача 6. Плотность жидкого и твердого висмута при температуре его
плавления (271е С) соответственно равна 10,005 и 9,637 г/см3. Атомная теплота плавления
висмута 2600 кал/г-атом. Требуется определить, при какой температуре висмут
плавится при давлении 50 am.
Решение. Предварительно определим значение коэффициента dT/dp из
уравнения (173):
544 ( ! 1—\
dT_ = Т(Ужтк-УТВ) = V Ю.005 ^TlJ_
dp Q 2600 ' грао/am.
Поскольку —г— < 0, то температура плавления понижается при увеличении
давления. Далее, воспользовавшись приближенным соотношением
dT _ AT
dp ~ Ар '
определяем температуру плавления висмута при давлении 50 am:
's = 'i + (P.- Pi) -^ = 271 + (50 - 1) (-0,004) = 270,8° С.
Задача 7. Удельный объем льда при 0° С равен 1,091 см31г, воды— 1,000 см3/г,
теплота плавления льда 80 кал!г. Требуется определить изменение температуры
плавления льда при изменении давления на 1 am и при какой температуре плавится лед
при давлении собственного пара (4,6 мм рт. ст.).
Решение. В соответствии с уравнением (173) легко найти значение
коэффициента изменения температуры плавления с давлением:
а = Т (W-ri.) = 273 ьооо-1;091 = _0>0075 град/ат_
dp Q 80-41,3
Далее можно определить температуру плавления льда после изменения
давления (4,6—760)/760 = —0,9938 am. Поскольку при 1 am температура плавления 0° С,
то при его уменьшении на 0,9938 am температура плавления станет равна
t = Ap-^~ = (_o,9938) (—0,075) = +0,0073° С.
80
Задача 8. Давления насыщенного пара этилового спирта при температурах 70
и 80° С соответственно равны 540,9 и 811,8 мм рт. ст. Требуется подсчитать
удельную теплоту испарения спирта.
Решение. Воспользуемся уравнением агрегатных превращений в
интегральной форме (180):
п__ 4,573ГхГ2 , р2 _ 4,573.343-353 811,8 _ 010
Ц~~ М(Т2-Тг) lg Pl "" 46(353-343) Ig 540,9 "" ZiZ кал,г'
Глава III
ХАРАКТЕРИСТИЧЕСКИЕ (ТЕРМОДИНАМИЧЕСКИЕ)
ФУНКЦИИ
Для того чтобы однозначно характеризовать свойства системы
состояние которой определяется какими-либо параметрами (р, V и Т)
при термодинамическом равновесии, недостаточно знать уравнение
состояния этой системы, связывающее давление, объем и
температуру. Для полной характеристики нужно знать еще уравнение,
которое позволяет определить энергию системы по данным параметрам
состояния. Анализ первого и второго начала термодинамики
показывает, что число уравнений, необходимых для полной характеристики
состояния системы, можно свести с двух до одного, поскольку все
необходимые для этого величины могут быть выражены через
некоторую функцию независимых переменных и ее производные.
Такие функции получили название характеристических функций.
Наиболее часто в качестве таких функций используются:
внутренняя энергия (£/), энтальпия (Я), свободная энергия (F) и
термодинамический потенциал (G) *.
Любое свойство системы может быть выражено через эти
характеристические функции (U, Я, F и G) и их производные в явной форме.
Однако использование той или иной функции зависит от конкретных
условий, которые диктуют нам выбор независимых переменных, а это
в свою очередь определяет выбор используемой характеристической
функции. Наиболее простые в математическом отношении выражения
для различных свойств системы получаются, если рассматривать U
как функцию от V и 5:
U = f (V, S); (185)
Я как функцию от р и 5:
F как функцию от V и Т:
и G как функцию от р и Т
H = f(p, S); (186)
F = f (V, T) (187)
G=/(p, Г). (188)
* Энтропия (S) также относится к числу характеристических функций. Она была
рассмотрена в предыдущих разделах (см. стр. 63—69).
6 А. Н. Крестовников 81
При применении функций U и Н одной из независимых
переменных является энтропия. Невозможность непосредственно измерять
энтропию представляет значительное неудобство при использовании
функций U и Я. В этом заключается одна из причин того, что чаще
пользуются функциями F и G при удобных для измерения
независимых переменных V и Т или Р и Т. Вместе с тем нельзя пользоваться
во всех случаях только одной из этих функций. Например, если мы
попытаемся использовать функцию F в случае, когда по условиям
опыта надо взять в качестве независимых переменных р и 7\ то
функция F не будет характеристической и даст выражение для ряда
свойств системы в неявной форме. Следовательно, для данного
случая необходимо воспользоваться функцией G.
§ 13. ВНУТРЕННЯЯ ЭНЕРГИЯ
В соответствии с уравнением (185) внутренняя энергия является
характеристической функцией при независимых переменных V я S.
Как и в случае всякой другой функции состояния, дифференциал
функции U является полным дифференциалом и, следовательно,
может быть представлен как сумма частных дифференциалов по
независимым переменным:
W=(-w)sdV + {-%r)vdS' («О)
Воспользовавшись уравнением (3) первого начала
термодинамики dQ = dU + dAy где dA = pdV, и уравнением (136) второго
начала термодинамики dQ = TdSy получаем
TdS = dU + pdV,
откуда
dU - TdS — pdV. (190)
Сравнивая уравнения (189) и (190), видим, что
{■ж)у = т <191>
И
(■W)s = -P- <192>
Таким образом, зная, что U = / {V, 5), легко получаем значения
недостающих для характеристики системы переменных Тир,
Причем становится очевидным, что температура является мерой
возрастания внутренней энергии системы с увеличением энтропии при
постоянном объеме, а давление — убыли внутренней энергии с
увеличением объема при постоянном значении энтропии. Направление
и кривизна кривых определяются по первой и второй производным
от соответствующей функции по параметру состояния (рис. 20). Ре-
82
n
S= const
Ц
V= const
П
T- const
F
V= const
-*~ V
a
n
S~ const
H
P= const
■*■«
T- const
■*^v
•*~p
Рис. 20. Схемы изменения характеристических функций при
соответствующих независимых параметров
Р- const
изменении
зультаты анализа зависимости внутренней энергии от объема и от
энтропии приведены на рис. 20, а.
Кроме того, изменение внутренней энергии системы при
постоянном объеме определяет величину теплового эффекта соотношением (83)
AU = Qv = — Qv.
§ 14. ЭНТАЛЬПИЯ
По уравнению (186) энтальпия является характеристической
функцией при независимых переменных р и S.
Следовательно:
""= (f-)sd"+ (-£),«*
(193)
Изменение энтальпии системы при постоянном давлении
определяет величину теплового эффекта соотношением (88):
A# = QP = -QP.
Из уравнения (3) первого начала термодинамики dQ = dU + dA,
где dQ = dH и dA — pdV при р = const, получаем dH = dU + pdV,
а после интегрирования:
2 2 2
\ dH = J dU + p J dVf
i i i
#2 - нг = u2 - и, + p (v, - vj,
или
Яа - ffx = (U% + pV2) - (Ux + PVX).
Отсюда следует, что связь между функциями Н и U определяется
соотношением:
Я = U + pV. (194)
6* 83
Дифференцируя полученное выражение, получаем:
dH = dU + pdV + Vdp.
Далее подставляем значение dU из уравнения (190):
dH = TdS — pdV + pdV + Vdp,
после чего получаем
dH = TdS + Vdp. (195)
Сравнивая уравнения (193) и (195), получаем, что
Легко видеть, что температура является мерой возрастания
энтальпии системы с увеличением энтропии при постоянном давлении,.
а объем — мерой возрастания энтальпии с увеличением давления
при постоянном значении энтропии (рис. 20, б).
Иногда энтальпию называют теплосодержанием. Кроме того, тот факт,
что HU = —Qy, а Д# = —Qp, послужил причиной возникновения терминологии,
согласно которой U и Н называют тепловыми функциями. Кроме того, U иногда
называют внутренней энергией системы при постоянном объеме, а Я — внутренней
энергией системы при постоянном давлении.
§ 15. ИЗОХОРНО-ИЗОТЕРМИЧЕСКИЙ ПОТЕНЦИАЛ
(СВОБОДНАЯ ЭНЕРГИЯ)
По уравнению (187) свободная энергия является
характеристической функцией при независимых переменных V и Т.
Следовательно:
dF=(-w)rdV+(-w)vdT- (198>
Согласно первому началу термодинамики: dQ = dU + dA или
dA — dQ — dU", откуда
dA = TdS — dU.
Интегрируя это уравнение при постоянном объеме и температуре,
получаем:
Av = Т (52 - Sx) - (U2 - U,)
или
Av = -l(U2 - TS2) - (U, - TSJ].
Если ввести обозначение
F = U — TS, (199)
то
Ау - -(Fa - Fx) = -AF, (200)
т. е. работа системы при постоянной температуре и постоянном объеме
равна убыли функции F в данном процессе. Из выражения (199)
следует, что при постоянной температуре и постоянном объеме в работу
84
не может перейти вся внутренняя энергия системы U. В работу
превращается только ее часть, остающаяся после вычета TS из U.
Ее обозначили F и назвали свободной энергией. Таким образом,
свободной энергией называется та часть внутренней энергии системы,
которая может быть превращена в работу при постоянной
температуре. Эта работа (Av) называется полезной и, если процесс протекает
обратимо, является максимальной. Величина полезной
максимальной работы Av не включает в себя работу изменения объема,
поскольку, кроме постоянства температуры, в выводе принималось
также постоянство объема системы. Эта работа может быть,
например, электрической, совершаемой гальваническим элементом.
Та часть внутренней энергии, которая при постоянной
температуре не превращается в работу (TS), называется связанной или
бесполезной энергией. Чем больше энтропия системы, тем больше ее
связанная энергия, поскольку энтропия представляет собой
связанную энергию, приходящуюся на 1 град.
Дифференцируя уравнение (199), получаем:
dF = dU — TdS — SdT.
Далее подставляем, согласно уравнению (190):
dU = TdS — pdV,
поэтому
dF = —pdV — SdT.
Сравнивая уравнения (198) и (201), находим, что
Таким образом, давление является мерой убыли свободной
энергии системы с увеличением объема при постоянной температуре,
а энтропия — мерой убыли свободной энергии с увеличением
температуры при постоянном объеме системы (рис. 20, в).
§ 16. ИЗОБАРНО-ИЗОТЕРМИЧЕСКИЙ
(ТЕРМОДИНАМИЧЕСКИЙ) ПОТЕНЦИАЛ
Термодинамический (изобарный) потенциал является
характеристической функцией при независимых переменных р и Т и,
следовательно:
Так же как энтальпия отличается от внутренней энергии на
величину pV, так и термодинамический потенциал отличается от
свободной энергии на pV (рис. 21), т. е.
G = F + pV = U — TS + pV. (205)
85
(201)
(202)
(203)
Проводя процесс при постоянном давлении и температуре, получаем
+(G2 - Gl) = +(F2 -FJ+p (Vt - V,),
или, обозначая
Ap = -(G2 - Gx) = -AG
и принимая во внимание, что
-(F2 - FJ =
получаем
AP = Av + p (V, - Vt),
■&F = Av,
(206)
где Ар — полная максимальная работа системы, равная убыли
термодинамического потенциала при постоянной температуре и
постоянном давлении. Она состоит из полезной максимальной работы
системы А у и работы сжатия р (У2 — VJ.
//
"^
TS
. TS .
■ 4j; <т% ^
^
f
£
як _
PV
Рис. 21. Схема соотношений
между основными
термодинамическими функциями
Дифференцируя уравнение (205), получаем
dG = dU — TdS — SdT + pdV + Vd/?. (207)
Далее подставляем dU = TdS — pdV согласно уравнению (190),
после чего получаем:
dG = Vdp — SdT. (208)
Сравнение уравнений (204) и (208) показывает, что
dG
т
(-£)
V
(209)
и
ас
аг
) =-s-
/р
(210)
Легко видеть, что объем системы является мерой возрастания
термодинамического потенциала системы с увеличением давления при
постоянной температуре, а энтропия — мерой убыли
термодинамического потенциала с увеличением температуры при постоянном
давлении (см. рис. 20, г).
Тот факт, что AF = —Av, a AG = —Ар, послужил причиной
возникновения терминологии, согласно которой F и G называют
рабочими функциями. При этом F иногда называют свободной энергией
системы при постоянном объеме, a G — свободной энергией системы
при постоянном давлении. Кроме того, F называют еще функцией
или свободной энергией Гельмгольца, a G — функцией, потенциалом
или свободной энергией Гиббса. В последнее время, по предложению
Академии наук СССР, получила распространение новая терминоло-
86
гия, согласно которой функция F носит название изохорно-изотер-
мического (или изохорного) потенциала, а функция G — изобарно-
изотермического (или изобарного) потенциала.
Заканчивая анализ характеристических (термодинамических)
функций и их производных, следует заметить, что, приравнивая
правые части формул (191) и (196), (192) и (202), (197) и (209), (203)
и (210), можно получить связь между частными производными
характеристических функций по различным параметрам, известные под
названием уравнений Максвелла:
\ dS )v~ \ dS )р' \ dV )s~~\ dV )т'
дН\ ( dG \ ( dF \ ( dG
dp
)s~- \ dp )т; \ dT)v~\ dTjp-
§ 17. ОБЪЕДИНЕННЫЕ ПЕРВОЕ И ВТОРОЕ НАЧАЛА
ТЕРМОДИНАМИКИ (УРАВНЕНИЕ ГИББСА—ГЕЛЬМГОЛЬЦА)
Согласно уравнению (199) и значению энтропии, которое дается
выражением (203), имеем:
Р = и + Т{ж)у <211)
Это и есть уравнение Гиббса—Гельмгольца для процессов,
протекающих при постоянном объеме. Напишем это уравнение для двух
состояний системы:
Рг = и1 + Т (Щ
, V
и
Р2==и2 + Т(
dF2
дТ Jv
Далее, вычитая одно из другого, получаем
дТ
Отсюда:
Лу = 0,+ Т(^-)Г; (212)
это другая форма записи уравнения (211).
Согласно уравнениям (205), (194) и значению энтропии, которое
дается уравнением (210), имеем
■AG = A# + r(-^L) . (213)
87
Это уравнение Гиббса—Гельмгольца для процессов,
протекающих при постоянном давлении. Повторяя рассуждения,
аналогичные тем, которые применялись для вывода уравнения (212),
получаем уравнение
AP = QP + T(1w-)P> (214)
которое является другой формой записи уравнения (213).
§ 18. НАПРАВЛЕНИЕ ПРОТЕКАНИЯ ПРОЦЕССОВ
И ТЕРМОДИНАМИЧЕСКИЕ УСЛОВИЯ РАВНОВЕСИЯ
Так как, согласно второму началу термодинамики, в системе
возможно только увеличение энтропии (при необратимых процессах),
но не уменьшение и, как крайний случай,— сохранение постоянства
энтропии (при обратимых процессах), то система находится в
устойчивом равновесии, если ее энтропия максимальна в данных условиях!
Следовательно, общим условием протекания процессов в
направлении устойчивого равновесия является условие
dS > 0, (215)
где знак равенства относится к равновесному состоянию, а знак
неравенства к самопроизвольным процессам.
Условию равновесия можно придать и другие формулировки.
Согласно уравнениям (201) и (208), имеем
dF = —p dV — S dT
и
dG = V dp — S dT.
Таким образом, учитывая выражение (215), при постоянной
температуре и постоянном объеме общим условием протекания
процессов в направлении достижения устойчивого равновесия будет условие
dF < 0; (216)
следовательно, при постоянной температуре и объеме в любой
системе самопроизвольно могут протекать только те процессы, которые
сопровождаются уменьшением F, причем пределом их протекания,
т. е. условием равновесия, является достижение некоторого
минимального для данных условий значения F.
Точно так же при постоянной температуре и постоянном давлении
общим условием протекания процессов и равновесия будет условие
dG<0; (217)
следовательно, в системах, находящихся при постоянной
температуре и постоянном давлении самопроизвольно могут протекать
только процессы, сопровождающиеся уменьшением G, причем
пределом их протекания, т. е. условием равновесия, служит некоторое
минимальное значение G для данных условий.
Аналогичные формулировки можно сделать в отношении всех
характеристических функций. Таким образом, при всех самопроиз-
88
вольных процессах происходит уменьшение характеристических
функций U, Я, F и G, а равновесие характеризуется их
минимальным для данных условий значением.
§ 19. ХИМИЧЕСКИЙ ПОТЕНЦИАЛ
И ТЕРМОДИНАМИЧЕСКИЕ ХАРАКТЕРИСТИКИ РАСТВОРОВ
В ходе химических процессов происходит не только изменение
таких параметров системы, как объем, давление и температура,
но также изменяется состав. В приложении к таким системам для
рассмотрения равновесия Гиббс предложил использовать
представление о химическом потенциале, который обозначается буквой \i
с соответствующими индексами.
Математически химические потенциалы представляют собой
частные производные характеристических функций (выбор их
определяется выбором независимых параметров системы) по числу молей
данного компонента системы при постоянных значениях
независимых параметров и количеств всех остальных компонентов:
Vll=(^L) ; (218)
li1=(^L) ; (219)
ri \ дпг JPts, п2, п3, . . . v ;
^=1^-) ; (220)
^1_" \~дгц)р, т, п2,п3, . . . ' '
Если ввести мольные концентрации (доли)
*/ = V2>/, (222)
то:
\ ОХг /V, S, х2, х3, . . .
^=(1г) ч '- <224)
\ axt IP, S, х2, х3, . . .
И-
1-('£)v.T.x,.xt,...'' (225)
^=Мг) т • (226)
\ 0Х1 /Р, Т, Х2, Х3, . . . V
Поскольку Uу Я, F и G являются функциями состояния, то при
nt = const имеем:
pl = (*L] =(™) =(*L) =(Д (227)
ri \dnijv,s \dnijp,s \дщ)у,т \dniJp9T K '
и при ^ = const имеем:
И* ==Л'ШГ)у9 s = (-dxjjp, s = \-dx-Jv, т = \-дх-)р, т> (228)
89
т. е. химический потенциал данного вещества / не зависит от выбора
независимых переменных, определяющих состояние системы.
Химический потенциал представляет собой фактор интенсивности
(степень напряженности) химической энергии данного вещества
в системе, поскольку химическую энергию, подобно любому другому
виду энергии, можно рассматривать как произведение фактора
интенсивности на фактор емкости. Фактором емкости в данном случае
будет число молей или концентрация данного вещества.
Следовательно, приращение химической энергии можно представить в виде
\it dni или \xi dxt. Кроме того, в состоянии равновесия не происходит
превращений химической энергии, т. е.
£ fi, dnt = 0 (229)
или
£ \it dxt = 0. (230)
Химический потенциал является мерой изменения
характеристической функции при соответствующих постоянных параметрах и
массах (концентрациях) всех веществ, за исключением массы
(концентрации) того компонента, количество которого изменяется в
системе. Следовательно, химический потенциал можно рассматривать
либо как соответствующий тепловой эффект (Qv = —AU или Qp =
= —Л#), либо как совершаемую работу (Av = —AF или Ар =
= —AG) при изменении массы (концентрации) данного вещества
в системе и заданном выборе переменных.
Представим себе, что несколько систем образуют одну общую
систему, причем фактор емкости этой новой системы равен сумме
факторов емкости ее составных частей, а факторы интенсивности
исходных систем одинаковы. Если же это не так, то в системе начнется
процесс, идущий в сторону выравнивания факторов интенсивности
за счет изменения факторов емкости. Следовательно, для равновесия
системы необходимо равенство химических потенциалов каждого
вещества во всех участках системы. Это положение широко
используется в химической термодинамике при изучении равновесий.
Представим себе раствор двух веществ А и В. Взаимодействие
между ними будем характеризовать с помощью энергий
взаимодействия разноименных (WAB) и одноименных (WAA и WBB) частиц
в рассматриваемом растворе. Для этого введем понятие «энергии
смешения», определяемое выражением
W = WAB— Waa + Wbb . (231)
Здесь WAB <0, WAA<^0 и WBB <0, поэтому W <0, если
взаимодействие разноименных частиц сопровождается большим
изменением энергии, чем взаимодействие одноименных частиц, и^>0,
если соотношение этих энергий обратное. Численное значение
энергии смешения, как правило, относится к 1 моль раствора.
Растворы, в которых энергия смешения не зависит от
температуры и концентрации раствора, принято называть регулярными.
Растворы относят к идеальным, если W = 0.
90
Энтальпия раствора двух рассматриваемых веществ может быть
записана в следующем виде:
Н = хлНл + хвНв + xAxBW, (232)
где #л и Нв — мольные энтальпии веществ, образующих раствор.
Слагаемое
Д#см - xAxBW (233)
получило название энтальпии (теплоты) смешения или избыточной
энтальпии (Л#изб) раствора; для соблюдения правила размерностей
оно отнесено к 1 моль раствора.
Оценим теперь энтропию смешения (ASCM), которую также
называют избыточной (в этом случае она обозначается AS^) или
конфигурационной (ASK0H<i>) энтропией раствора. Для этого воспользуемся
формулами (163)—(166). Тогда:
AS = R (In QAB — In QA — In QB), (234)
где
и
О - In {Na + Nb) ! /9^
iiAB ~ Ш NA\NB\ (235)
QA = Q^ = 1 , (236)
если предположить полностью неупорядоченную конфигурацию
взаимного расположения частиц А и В в растворе. Используя
приближение Стирлинга
In N1 ъ* (N In N) — Ny (237)
получаем
ASCM = — # (ха In *л + #в 1п хв). (238)
Следовательно, энтропия раствора может быть определена из
выражения
S = xASA + xBSB — R {xA In xA + хв In xB), (239)
где S.4 и SB — мольные энтропии веществ, входящих в раствор.
Изобарный потенциал раствора запишется в виде
■ G = хАНА — xATSA + *БЯ Б — xB7SB + зд^ +
+ RT (xA In xA + *Б In *в). (240)
Отсюда выводим выражение для химических потенциалов веществ,
образующих раствор:
^=Ы-) =^ + ^ln*4+0-*4)V (241)
\ А / р, I
И
^=-(-5Г-) =Й + ^1пдсв + (1-хв)2^. (242)
91
Здесь:
р'А = НА - TSA (243)
H = Hb~TSb- (2«)
величины, определяемые природой веществ А и В и температурой;
их называют нормальными химическими потенциалами. Вторые
слагаемые уравнений (241) и (242) определяются концентрацией и
температурой, а третьи — концентрацией и энергией смешения
соответственно. Преобразуем уравнения (241) и (242) к виду:
РА^\ь°А + ЯТ\паА (245)
Н = Н + ЯТ In aB, (246)
где
Яа = !аХа и aB = foKB\ (247)
/il=e-(1-^)*v и fB = e-V-xB)tw. (248)
Величины ал и ав получили название активных концентраций
(активностей), fA и fB — коэффициентов активности. Можно видеть, что
коэффициенты активности связаны с энергией смешения (а > х
и f < 1 при W < 0; а < х и / > 1 при № > 0).
Для идеальных растворов W = 0, / = 1 и а = х. Идеальные
растворы могут описываться с помощью уравнений для химических
потенциалов:
Va = Va + ЯГIn *л (249)
^Б = ^В + Я^ 1П *В- (250)
Реальные растворы принято описывать с помощью величин f
и W. При этом постулируется справедливость законов идеальных
растворов, а отклонение поведения реального раствора от идеального
характеризуется с помощью величины (кроме f и W)
AGCM = A#CM-7ASCM, (251
которая называется термодинамическим отклонением, изобарным
потенциалом смешения или избыточным изобарным потенциалом
(обозначения AGCM и AGH36 равнозначны).
Глава IV
УЧЕНИЕ О ХИМИЧЕСКОМ РАВНОВЕСИИ
Химическая система находится в равновесии, если ее состояние
при заданных внешних условиях (например, температуре и
давлении) во времени не изменяется.
Однако это односторонняя характеристика равновесия.
Различают три возможных вида равновесия: стабильное (устойчивое или
92
истинное), лабильное (неустойчивое) и метастабильное
(относительное) равновесие.
Стабильное равновесие характеризуется абсолютным минимумом
соответствующих характеристических функций. При таком
равновесии всякое достаточно малое воздействие вызывает какое-либо
малое изменение состояния системы, причем перемена направления
воздействия вызывает перемену направления изменения состояния
системы, а будучи выведенной из состояния равновесия, система
сама возвращается в него после прекращения воздействия на нее.
Таким образом, стабильное равновесие может быть достигнуто
как бы с разных сторон.
Лабильное равновесие характеризуется тем, что при любом
бесконечно малом воздействии система претерпевает конечное изменение,
в результате которого система приходит к стабильному
равновесию.
Метастабильное равновесие характеризуется относительным
минимумом соответствующих характеристических функций. В этом
случае воздействие на систему может привести к конечным
изменениям в направлении стабильного равновесия, однако некоторые
бесконечно малые воздействия вызывают настолько малые изменения
системы, что изменение направления воздействия обусловливает
перемену направления происходящего изменения, а следовательно,
система будет возвращаться в исходное состояние после прекращения
этих воздействий.
Таким образом, истинное химическое равновесие, помимо
неизменности со временем, должно отвечать минимальному значению
характеристических функций, которое только возможно при данных
выбранных независимых переменных.
Кроме того, равновесие не означает полного покоя. Напротив,
при равновесии в системе идут процессы, но в противоположных
направлениях, так что их результаты взаимно уничтожаются. С этой
точки зрения химическое равновесие является динамическим
(подвижным) равновесием.
§ 20. ЗАКОН ДЕЙСТВИЯ МАСС И КОНСТАНТА
РАВНОВЕСИЯ
Рассмотрим химическую реакцию в гомогенной газовой среде:
ЪВ + dD^lgG + rRy
где ft, d.t g и г представляют собой стехиометрические коэффициенты
соответствующих веществ Б, D, G и R. Между веществами В и D
существует сродство, т. е. стремление к взаимодействию. Оно же
имеется и у веществ G и R. Следовательно, реакция протекает в обоих
противоположных направлениях. В таких случаях при написании
реакции вместо знака равенства часто пользуются противоположно
направленными стрелками.
93
Для скоростей обеих противоположно направленных реакций
(vx и v%) можно составить следующие выражения *:
vi = kiCbBCdD (252)
и
V2 = ШСц, (253)
где ^и^2 — константы скоростей реакций, а Св, CD, CG и С# —
молярные концентрации, которые, будучи возведенными в
соответствующие степени (ими являются стехиометрические коэффициенты),
определяют скорости реакций vx и v2.
Если vx > v2, то молекул G и R будет образовываться больше,
чем молекул В и D (при этом концентрации СБ и CD уменьшаются,
а концентрации CG и Сд увеличиваются). Если v± <J и2> т° больше
будет образовываться молекул ВиО, анеОи /? (Св и CD
увеличиваются, а Сб и Q уменьшаются). Наконец, если vx = и2> т- е- °бе
реакции протекают с одинаковыми скоростями, то никаких видимых
изменений происходить не будет, так как в единицу времени будет
образовываться одинаковое число молекул В, D, G и R. При этом
устанавливается равновесие и, пользуясь выражениями для
скоростей прямой и обратной реакций, можно подойти к весьма важному
понятию о константе равновесия:
Vl = V2 = hC'sd = k2CgGCrR, (254)
откуда
* = £ = w-' (255)
i ^GUR
где К — константа равновесия, равная отношению констант
скоростей прямой и обратной реакций. Следовательно, для составления
выражения константы равновесия в числителе дроби пишется
произведение концентраций веществ левой части уравнения, в
знаменателе — правой части уравнения. Иногда поступают наоборот. Кроме
того, обычно уславливаются писать реакцию в экзотермическом или
эндотермическом направлении.
Численные значения концентраций всех веществ возводятся
в степени, равные числу участвующих в реакции частиц данного
вещества.
В каждом конкретном случае следует обращать внимание на то,
как написано уравнение химической реакции, для которого
составлена константа равновесия. Для одной и той же реакции в
зависимости от того, в каком направлении и для какого числа молей она
написана, константы равновесия имеют совершенно различные
численные значения. Например, при 450 °С имеет место следующее:
* Это следует из приложения теории вероятностей к рассмотрению скоростей
химических реакций, происходящих путем вероятностного столкновения частиц
(молекул, атомов или ионов), зависящего от их концентрации и долевого участия,
согласно материальному балансу по уравнению химической реакции.
94
1) 3/2H2 + V2N2 = NH3,
*i = ^Щщ = 0,00649 (J5JL)-1;
Л J*
2) 3H2 + N2 = 2NH3)
K2 = K\ = 0,421- io-4(J^L\-2;
3) NH3 = 3/2H2 + V2N2,
is 1 i с л i / -моль \
K3=_= 154,1 (—);
4) 2NH3 = 3H2 + N2,
/C4= -^ = 2,37-10* (-^M*.
Численное значение константы равновесия характеризует
состояние равновесия при данных условиях и не меняется с изменением
исходных концентраций реагирующих веществ. Величина константы
равновесия не зависит от того, какие из веществ, участвующих в
данной реакции, применяются в качестве исходных продуктов и в каких
соотношениях они введены в реакцию.
Константа равновесия связывает концентрации всех веществ,
участвующих в реакции, и нельзя изменить концентрацию ни одного
из них, чтобы это не повлекло за собой соответствующего изменения
концентраций всех остальных веществ, участвующих в реакции,
которое приводит к прежнему численному значению константы
равновесия при данных условиях. Таково содержание закона
действия масс.
Нами были приведены рассуждения, использующие кинетические
представления о достигаемом в системе равновесии. Теперь
рассмотрим термодинамический вывод1, закона действующих масс.
Рассмотрим в условиях равновесия гомогенную газовую
химическую реакцию
ЬВ + dD^gG + rR.
Обозначим через \iB, \iD, \iG и \iR химические потенциалы
реагирующих веществ при равновесии. Поскольку химический потенциал
является мерой изменения характеристических функций при
изменении количества компонента, то в состоянии равновесия [см. (229) ] *
Y1iiidni = biiB-i-diiD — giiG — riiR = 0, (256)
* Поскольку среди щ только одна переменная является независимой, то можно
уравнения (229) или (256) записать в следующем виде:
S № dni = £ у№* d& = °»
где Vi — стехиометрический коэффициент при компоненте /, а е—новая переменная,
которую как внутренний независимый параметр реакции принято называть числом
пробегов реакции. В некоторых случаях использование этого понятия оказывается
полезным при термодинамическом анализе химических реакций.
95
так как образование g молей вещества G и г молей вещества R
сопровождается расходом Ъ молей вещества В и d молей вещества D.
Учитывая уравнения (249) и (250), можно записать:
|i, = |i? + OThiC,. (257)
Тогда имеем
+ RT (b In CB + din CD — g In CG — r In CR) = 0, (258)
откуда
ln 7T$- = -W- M + Wd ~ №°c ~ 4). (259)
Правая часть этого уравнения зависит только от температуры
и при ее постоянстве для данной реакции есть величина постоянная.
Обозначая ее через In Kp, получаем:
pb pd
U£UD
ИЛИ
In -f-^ = In Kc, (260)
LGLR
pb pd
bGbR
Различные формы констант равновесия
Константа равновесия может быть выражена через молярные
концентрации веществ, участвующих в реакции. В этом случае
она обозначается Ксщ и связь между равновесными концентрациями
веществ Б, D, G и Ry как это было показано, можно представить
соотношением (2611.
Кроме того, константу равновесия можно" выразить через
парциальные давления веществ, участвующих в реакции:
РЬяРп
КР - ~тт- • (262)
Константу равновесия также можно представить через число
молей веществ, участвующих в реакции:
nbRndn
Кп=-§-^ (263)
nGnR
или через соответствующие мольные доли:
Ку = ?Т7-- (264)
Мольной долей называется отношение
(265)
я,
96
где nt — число молей вещества i в смеси, а %щ — общее число молей
всех веществ в смеси.
В общем случае константы равновесия /(с, Кр, Кп и Ку
различны по численной величине. Установим связь между ними. Для
установления связи между Кс и Кр воспользуемся уравнением
состояния идеальных газов:
pV = nRTy
откуда
л. = -
Pi = ^-RT= СДТ,
где Q — концентрация вещества i, выраженная в молях на литр.
Отсюда:
Jb nd pb pd
V ИвИ° Б D /Drp^b+d-g-r _ v D b+d-g-r
или
Kp = Kc(RT)~A\ (266)
где А/г = (g + r) — (b + d) = nK — nH — изменение числа молей
в результате реакции.
Так как для идеальных газов парциальные давления связаны
с общим давлением Робщ и мольной долей у£ данного вещества в смеси
равенством
Pi = Y/Робщ, (267)
то получаем:
К — РвР° — УвуС° (п \b+d-e-r —к in \bJrd-z-r
'V g r g г \"общ/ /^V \"общ/
pgPr VSY/?
ИЛИ
Kp = Ку (Р06Щ)"ЛД- (268)
Воспользовавшись соотношением
Пс
Pi — 7;Робщ — "vi Робщ1
получаем:
„ _ £вРЪ_ = ^D_ ( ^\Ь+" -'" = к ( "общ \ b+d'g-r-
или
^-'•(^""-'"("Г- (269)
Таким образом, если An =f= 0 и реакция сопровождается
изменением числа молей, то имеет место соотношение:
Кр = /Сс (^T)_An = Ка (Щ~АП = Kv (робщГАп. (270)
7 А. Н. Крестовников 97
90
80
70
- Рабнобесие
<%
СЛ
^*J>
'г f/>*
C/7Q&J
Эта запись предполагает, что константы записаны следующим
образом: в числителе находятся вещества левой части уравнения
реакции, а в знаменателе — правой (при записи уравнения
химической реакции с положительным тепловым эффектом — в правой
части). Кроме того, Дя получается вычитанием из числа молей
веществ правой части уравнения
числа молей веществ левой
части уравнения.
Если же Дя = 0 и реакция
не сопровождается изменением
числа молей, то
Кр = Кс = Кп = Ку (271)
и численное значение всех
видов констант равновесия
совпадает.
Определение констант сложных
равновесий (комбинирование
равновесий)
В качестве простейшего
примера экспериментального
определения константы
равновесия могут быть приведены
полученные при 444 °С данные
для реакции
Н2 + I2 = 2HI.
Рис. 22 показывает, что если брать в качестве исходных
продуктов Н2 + 12 или же HI, то в рассматриваемой системе
приблизительно через два часа в обоих случаях достигается одинаковое
равновесное состояние (78% HI, 11% Н2 и 11% паров 12). Определяя
концентрации веществ в смеси, легко находим константу равновесия
для данной температуры. Ее значение может быть принято равным
Кс =-°Ц1^1 = 0,02.
Однако не всякую реакцию удается легко осуществить в таких
идеальных условиях. В этом случае иногда имеется возможность
комбинирования более простых равновесий для вычисления равновесий,
прямое исследование которых в силу каких-либо причин затруднено.
Например, по известной константе равновесия Кр реакции
образования хлористого водорода
25 50 75 WG
Время TytiuH
Рис. 22. Иллюстрация двустороннего
достижения равновесия
Н2 + С1
2НС1
и константе равновесия реакции диссоциации водяного пара (Кр)
2Н2 + 02^Г2Н20
98
можно определить константу равновесия сложной химической
реакции (Ю:
4НС1 + 02^2С12 + 2Н20.
Так как
2
v' __ ^Н/С12 к" _ РН2Рр2
Ар — § ' ^Р — 2"
РНС1 %20
^HCI^Oo
и
^D =
то легко видеть, что
Р 2 2 '
Pci/h2o
Ю2 '
Точно так же, зная константу равновесия К'р для реакции
2Н20^2Н2 + 02
и константу равновесия К'р' для реакции
FeO^Fe+-i-02,
легко находим константу равновесия Кр для реакции
FeO + Н2 = Fe + Н20.
При этом
Ук
В каждом отдельном случае соотношение констант равновесия
определяется характером реакций, способом написания реакций
и их констант равновесия.
§ 21. ПРИЛОЖЕНИЕ ЗАКОНА ДЕЙСТВИЯ МАСС
К ГЕТЕРОГЕННЫМ РАВНОВЕСИЯМ
При выводе закона действия масс для гомогенных химических
реакций мы считали, что все компоненты реакции находятся в
газообразном состоянии. Если же в реакции участвуют жидкие или
твердые вещества, не образующие растворов друг с другом, то при
данной температуре парциальное давление этих конденсированных
участников реакции является величиной постоянной так же, как
является постоянным давление насыщенного пара данного вещества
при постоянной температуре. Эти постоянные величины можно
ввести в константу равновесия, которая в этом случае будет
определяться парциальным давлением только газообразных участников
реакции.
Применим это положение, например, к^реакции диссоциации
углекислого кальция, протекающей по уравнению:
СаОтв -f- С02 газ = СаС03 ТВ.
7* 99
В этом случае
РСаО^СО;
Кг
РСаСО,
но так как СаС03 и СаО находятся в конденсированном состоянии
И (При Т = COnst) рСаС03 = COHSt И /?СаО = Const, TO
следовательно, давление /?со2 должно быть для каждой данной
температуры величиной постоянной, не зависящей от количества
углекислого кальция и окиси кальция.
Можно аналогично поступать для любой другой реакции.
Например:
ZnO(TB) ~\- СО(Га3) ч—-^П(ЖИдК) + со2(
газ)*
кР
Рсо
Рсо2 '
или
CdO(TB) -|- С(ТВ) ^=^С(1(Жидк) ~Ь
СО.
1
Рсо
Аналогичный результат получим для всех реакций, в которых
только один из участников реакции находится в газообразном
состоянии. К этому случаю относится большинство реакций диссоциации
карбонатов и бикарбонатов, кристаллогидратов, аммиакатов, окислов
и сульфидов. Давление газообразного продукта диссоциации в этом
случае получило название давления (или упругости) диссоциации.
При равновесии, соответствующем данной температуре, давление
диссоциации в каждом отдельном случае является совершенно
определенной величиной.
Если рсо2 <СК\ т. е. не достигнуто равновесное давление С02,
то СаС03 будет диссоциировать; если рСо2 > К', то будет
происходить образование СаС03. В обоих случаях система приходит к
равновесному состоянию при рсо2 = Кр.
При изменении температуры изменяется давление диссоциации.
Характер этой зависимости позволяет сделать заключение об
устойчивости соединений.
Для примера рассмотрим давление диссоциации некоторых
кислородных соединений:
2Ag20^4Ag + 02, Kp=Po2\
2Cu0^2Cu + 02, К'Р = Ро2;
2Cu20 ^ ^Cu + 02, Кр = рош.
Температурная зависимость давления диссоциации схематически
представлена на рис. 23. Пересечение горизонтали, соответствующей
100
парциальному давлению кислорода в земной атмосфере (21%),
с соответствующими кривыми давления диссоциации дает
температуры, выше которых соединения будут легко диссоциировать, а ниже
диссоциация будет подавляться наличием парциального давления
кислорода воздуха. Анализ
графика приводит также к
заключению, что среди Ag20,
CuO и Cu20 наиболее
устойчивым является Си20 и
наименее устойчивым Ag20. Ниже
1030 °С устойчивыми являются
и окись меди и закись меди;
с повышением температуры от
1030 °С теряет устойчивость
окись меди CuO, а выше 1600° С
неустойчива и закись меди Си20.
Аналогичные рассуждения мо
гут быть проведены для оценки
поведения тех же соединений
в атмосфере чистого кислорода
при давлении 1 am (рис. 23).
1ат
МО МО
1030 1150.
Теппература?С
1600 то
Рис. 23. Температурная зависимость
давления диссоциации закиси- серебра,
окиси и закиси меди
§ 22. ТЕРМИЧЕСКАЯ ДИССОЦИАЦИЯ
Весьма многие вещества, будучи нагретыми до высокой
температуры, разлагаются на составные части, причем степень этого
разложения увеличивается с повышением температуры. Приведем
несколько примеров термической диссоциации, когда все участники
равновесия находятся в газообразном состоянии:
I2^2I, N204^2N02, PC15^PC13 + C12.
Численно состояние диссоциированного вещества характеризуют
степенью диссоциации а, за которую принимают отношение числа
распадающихся молекул к числу молекул до распада. Отношение
общего числа молекул после диссоциации к числу молекул до
диссоциации принято называть коэффициентом диссоциации /. Иногда
его называют изотоническим коэффициентом.
, Выясним связь между а и i.
Предположим, что первоначально имелось п молекул, тогда в
результате диссоциации распалось па молекул. Если каждая молекула
распадается на т молекул, то число вновь образовавшихся молекул
составит та д. Нераспавшихся молекул осталось п — па.
Следовательно, всего стало п—па-\-тпа молекул.
Отсюда:
п — /га + тип
п
- = 1 — а + am = 1 -f- а (m — 1),
или
а =
*•— 1
т
(272)
101
Таким образом, если а может принимать значения от 0
(отсутствие диссоциации) до 1 (полная диссоциация), то i принимает при
этом соответственно значения от 1 до т; причем т может принимать
различные значения в зависимости от вида реакции.
Например:
H2S^H2 + V2S2, m = 1,5;
S08;±SOa + V202, m= 1,5;
2N02^2NO + 02, m - 1,5
или
COCl^CO + Cl2, m = 2;
Na04^2NOa; m = 2 и т. д.
С другой стороны, значение коэффициента диссоциации
получается из соотношения:
i~, (273)
где М — теоретическая молекулярная масса вещества;
|л — опытная молекулярная масса того же вещества (она меньше
теоретической вследствие диссоциации вещества).
Следовательно, по аномалии молекулярной массы можно
определить коэффициент диссоциации. Это в свою очередь позволяет
определить степень диссоциации. Далее можно определить константу
равновесия реакции диссоциации. Связь константы равновесия и
степени диссоциации в каждом отдельном случае будет различной.
Наиболее простым случаем реакции диссоциации будет:
I. ^1 +1
п О О
п — па па па
Под уравнением химической реакции в первом ряду указано
число молей до диссоциации, во втором ряду — после диссоциации.
Отсюда если реакция имеет место в каком-то определенном объеме, то
„ папа а2
К = — -. п
п п — па 1 — а
В случае диссоциации водяного пара
2Н20 ^2Н2 + 02
п О О
1
п — па па -£-па
имеем общее число молекул после диссоциации:
2 ni = п — па + па + -у па = п ( 1 + -яг) •
102
Тогда парциальные давления Н20, Н2 и 02 составят:
п (1 — а) __ 1 — а
Рн2о ~~ Робщ 7 а \ ~ Робщ ^Г~ '
Ч'+т)
а
Рн2 — Робщ ^Г~ '
1
2
а
Ро2 ~ Робщ а
1 +"о"
У
откуда:
К„ =
Рн2Ро2 Робщ0? а3
Р „2
Рн20 2(l+-|-)(l-a)3 2
если робщ ~ 1 ат и а очень мало.
§ 23. МЕТОДЫ ЭКСПЕРИМЕНТАЛЬНОГО ОПРЕДЕЛЕНИЯ
КОНСТАНТ РАВНОВЕСИЯ
Огромное разнообразие самых различных реакций, а также
свойств реагирующих веществ обусловливает многообразие методов
исследования равновесия, каждый из которых должен учитывать
особенности изучаемого равновесия.
Для исследования газовых гомогенных равновесий необходимо
знать парциальные давления газов, находящихся в системе. Эти
давления лишь в редких случаях могут быть измерены
непосредственно, и их обычно вычисляют из наблюдений других различных
свойств, важнейшим из которых является плотность.
Наиболее удобным для этих целей оказался метод вытеснения
воздуха (рис. 24). В используемом приборе цилиндр 1 содержит
жидкость. Она доводится до кипения и тем самым поддерживается
определенная и постоянная температура в сосуде 2. Навеска
исследуемого вещества или нескольких веществ помещается в верху узкой
трубки сосуда 2 в ампуле 4 и поддерживается там палочкой 3.
Жидкость в цилиндре 1 кипятят до прекращения выделения пузырьков
воздуха из трубки 5. После этого подводят ее под эвдиометр 6
(газоизмерительная трубка) и, вынув палочку 3, сбрасывают ампулу в
сосуд 2. Ампула разбивается и вещества испаряются, диссоциируют
или происходит их взаимодействие. При этом вытесняется
определенный объем воздуха. Делают отсчет в эвдиометре 6 и по определяемым
величинам массы, объема, давления и температуры вычисляют
плотность вещества.
В различных специальных случаях прибор видоизменяется
сообразно требуемым условиям. Для окисляющихся веществ прибор
наполняется инертным газом, для легко разлагающихся веществ
применяется вакуум. При высоких температурах сосуды делаются
103
из фарфора, платины, иридия и нагреваются в электрической печи.
Кипящей жидкостью выбирают серу, нафталин, анилин и другие
вещества. Ампулу с веществом сбрасывают электромагнитом и
температуру измеряют термопарой.
Этот метод очень часто применяется для определения плотности
пара самых различных веществ. Кроме того, им широко пользуются
для исследования реакций
диссоциации.
В тех случаях, когда в газовой
системе имеется водород, его
парциальное давление может быть
определено методом полупроницаемых
перегородок (рис. 25). При этом систему i,
содержащую водород, помещают в
термостат 2, а внутрь этой системы
помещают платиновый, палладиевый или
иридиевый сосуд 3, из которого
выкачан воздух; сосуд соединен с маномет-
г
"^О QOOOQOOOO
/Г насосу
Рис. 24. Схема метода вытеснения
воздуха:
1 — цилиндр с кипящей жидкостью
(термостат); 2 — стеклянный сосуд для
испарения исследуемого вещества;
3 — устройство для удержания ампулы;
4 — ампула с исследуемым веществом;
5 — трубка для отвода избытка
воздуха; 6 — эвдиометр
I ^^
оооооооооо
Рис. 25. Схема метода полупроницаемых
перегородок:
1 — камера с исследуемыми веществами; 2 —
термостат; 3 — сосуд, стенки которого пропускают
водород; 4 — манометр
ром 4. При высокой температуре
водород диффундирует сквозь
материал стенок сосуда 3, а другие
газы не диффундируют. Диффузия продолжается до наступления
одинакового давления водорода в исследуемой системе 1 и внутри
сосуда 3. Это давление и есть парциальное давление водорода,
измеряемое манометром 4. Этим методом, например, можно
исследовать равновесие:
Н2О^Н2 +V202.
Если систему, находящуюся в равновесии при высокой
температуре, быстро охладить, то равновесие не успеет сдвинуться, и по
результатам анализа полученной смеси (иногда гетерогенной) можно
вычислить количество газов в момент равновесия. Это используется
в методе с последующей закалкой (рис. 26). Прибор представляет
собой кварцевую трубку 4, в которую помещается восстанавливаемое
вещество 3. Трубка помещается в термостат 2 и через выпускную
трубу 1 в нее поступает восстанавливающий газ, а через выпускную
104
трубку 5 отводятся продукты восстановления и оставшийся
восстанавливающий газ. Они поступают из зоны высокой температуры
печи в зону с более низкой температурой. При этом газ не успевает
перейти в состояние равновесия и сохраняет свой состав. Далее
его подвергают анализу.
Разновидности этого метода применяются для исследования как
гомогенных, так и гетерогенных равновесий. Этим методом, например,
Г
+
о о о о о о о
J
V
о о о о о о о о о
Рис. 26. Схема метода газового потока
с последующей закалкой:
1 — впускная трубка; 2 — термостат;
3 — исследуемое восстанавливаемое
вещество; 4 — кварцевая ампула; 5 —
выпускная трубка для газов
0 2 4 6 в /О /2
Спорость пролумамая газа,л/гшн
Рис. 27. Схема графического
определения константы равновесия
экстраполяцией к нулевой
скорости пропускания газа
можно определить константу равновесия реакции восстановления
окиси меди окисью углерода:
СиО + СО - Си + С02,
Кг
Рсо
Рсо.2
Однако, пропуская газ через трубку с восстанавливаемым
веществом с неодинаковой скоростью, приходят к различным значениям
константы равновесия. При повышенных скоростях в системе не
успевает установиться равновесие, а при слишком малых скоростях
не удается осуществить закалку газовой смеси. Поэтому
исследование производят при нескольких скоростях, а полученные результаты
экстраполируют к нулевой скорости пропускания газа (рис. 27).
Для определения константы равновесия применяются также
многие другие экспериментальные методы.
§ 24. СВЯЗЬ МАКСИМАЛЬНОЙ РАБОТЫ С КОНСТАНТОЙ
РАВНОВЕСИЯ (ХИМИЧЕСКОЕ СРОДСТВО)
Способность различных веществ взаимодействовать между собой
с образованием новых веществ была замечена давно и получила
наименование химического сродства. Однако далеко не сразу был
найден критерий, который мог бы служить мерилом этого сродства.
Решение вопроса осложнялось незнанием причин, вызывающих
химическое взаимодействие, в то время как опыты свидетельствуют
о существовании каких-то сил, приводящих к нему. Существование
105
таких сил бесспорно, каким бы образом ни объяснялась их природа *.
Решение проблемы было осуществлено с помощью термодинамики.
Было отвергнуто предположение о том, что скорость реакции
пропорциональна химическому сродству. Также была показана
несостоятельность гипотезы о равенстве теплового эффекта реакции
и химического сродства. Голландский физико-химик Вант-Гофф
предложил новую теорию химического сродства, которая, не
объясняя природы химического сродства, ограничивается указанием
способа его измерения и тем самым дает ответ на поставленную задачу
о количественной оценке химического сродства.
Вант-Гофф предложил в качестве мерила химического сродства
использовать максимальную работу, т. е. изменение свободной
энергии, если реакция протекает при V = const:
Av = —AF,
для изменения термодинамического потенциала, если реакция
протекает при р = const:
Ар - —AG.
Максимальная работа удовлетворяет всем требованиям,
предъявляемым к величине, характеризующей сродство: 1) она не зависит
от пути, по которому протекает реакция, а зависит лишь от свойства
и их начального и конечного состояний; 2) ее знак определяет
направление реакции, поскольку реакция протекает в направлении
совершения положительной максимальной работы; 3) она
представляет собой величину той энергии, которую нужно приложить к
системе, чтобы остановить реакцию, т. е. чтобы преодолеть силы
химического сродства; 4) кроме того, ее величина определяет величину
константы равновесия.
Остановимся на доказательстве последнего обстоятельства.
Изучение связи максимальной работы с константой равновесия требует
более конкретного рассмотрения проблемы химического сродства
отдельных реагентов друг к другу. Прежде всего следует найти
какую-либо возможность измерения максимальной работы
химической реакции. Для решения этой задачи с целью более наглядного
изложения часто рассматривают так называемый «ящик равновесия»,
или «ящик Вант-Гоффа» (рис. 28), который представляет собой
гипотетическую модель, позволяющую каждый исходный реагент в
отдельности сжимать в цилиндре с поршнем 1 и 2 от исходного
давления до равновесного. Затем через проницаемую (только для данного
вещества) перегородку реагент переводится в резервуар 5,
содержащий смесь всех реагентов, парциальные давления которых отвечают
равновесию, а получающиеся в результате реакции вещества через
*В настоящее время различают несколько видов межатомного взаимодействия,
каждый из которых, однако, обычно встречается лишь в сочетании с другими.
Основные среди них: гетерополярный (ионный), гомеополярный (ковалентный),
металлический, а также ван-дер-ваальсовский (остаточный) тип взаимодействия.
106
подобные же перегородки выводятся в цилиндры 4 и 5, где они
расширяются (каждое в отдельности) до неравновесных давлений. Коли^
чество веществ в резервуаре 3 очень велико, поэтому ввод или вывод
того или другого из них практически не вызывает изменения
давления или концентраций. Этот факт образно выражают словами,
говоря, что в ящике имеется «океан равновесия».
Суммируя все работы, совершенные изотермически и обратимо,
можно получить максимальную работу реакции.
Для того чтобы дать решение задачи в общем виде, рассмотрим
реакцию
ЬВ + dD^lgG + rR + Q,
где В, D, G и R— газообразные вещества, способные реагировать
между собой; b, d, g и г — стехиометрические коэффициенты
уравнения; Q — тепловой эффект
реакции при данной
температуре. Определим
максимальную работу этой
реакции.
Равновесные парциальные
давления веществ В, D, G
и R обозначим через pBi pD>
pG и pR. При этом положим,
что какое-то неравновесное
состояние системы этих ве-
7= const
1
ш
PD
рв
Рц
Рг.
Pr
I
R
ё
Рис. 28. «Ящик Вант-Гоффа»
ществ характеризуется парциальными давлениями, которые будем
обозначать р'в, pD, pG, p'R, и примем это состояние за исходное.
Исходные давления не равны равновесным, поэтому система
будет переходить в состояние равновесия. Если этот переход
осуществить изотермически и обратимо, то получим максимальную
работу. С помощью поршня / переводим Ъ молей вещества В от
давления рв до рв, после чего вводим его в резервуар 3. При этом
совершается работа:
Л = *ДГ1п—.
Рв
Далее с помощью поршня 2 переводим d молей вещества D от
давления рЬ до pD, а затем также вводим в резервуар 3. Работа
в этом случае составит:
Р'о
A« = dRT In
Pd
Выведем теперь из резервуара 3 с помощью поршня 4
образовавшиеся g молей вещества G, а затем переведем их от давления pG
до ph. Совершенная работа составит:
A3 = gRT In
Рд_
Pg
107
Отвод г молей вещества R из резервуара 3 с помощью поршня 5
с последующим переводом их от давления р% до р% сопровождается
работой:
Pr
Суммируем все работы, имевшие место при переходе к
равновесному состоянию:
Р_в_
Рв ' Ро
AP = — AG = bRT\n~ + dRTIn ^ +
+ gRTIn Pg- + rRT\n% = RTIn (/ffg(/d)" -RTIn4^•
Po P« (РоГЮ PqP«
Так как
ff r Ар
является константой равновесия, а
1п / >\е, Лг = b ln Рв + dlnpD — glnpG — г lnpR = 2j v lnp/
(как условно принято обозначать алгебраическую сумму логарифмов
неравновесных парциальных давлений, умноженных на
соответствующие стехиометрические коэффициенты), то
Ар - -AG = RT (Ц vlnp; - ln /Cp). (274)
Это уравнение применяется для реакций, протекающих при
постоянном давлении. Оперируя в выводе уравнения зависимости
максимальной работы от константы равновесия вместо парциальных
давлений концентрациями, получаем другую формулу, применяемую
для реакций, протекающих при постоянном объеме:
Av = -AF = RT (2 v In С\ — In Kc)• (275)
Если Ь + d = g + г, иными словами если реакция протекает
без изменения числа молекул, то Кр = /Сс и, следовательно, Лр =
= Ау. Однако в общем случае Ар =f= Ay.
Анализируя уравнения (274) и (275), получившие название
уравнений изотермы химической реакции
или уравнений изотермы Вант-Гоффа, нетрудно
заметить, что химическое сродство не есть неизменное свойство
вещества: оно меняется с изменением концентраций, парциальных
давлений и с изменением температуры.
Если максимальная работа реакций Ар = —AG или Ау = —AF
положительна, то
Ар = —AG > 0 и Av = —AF > О
и тогда
£ v In pi > 2 v ln Кр и 2 v In С\ > ln Kc •
108
Это означает, что
(РвУ (PrY P%Pr
(CB)b (СоУ c»Bcj
(СсУ(С*У >cbcR ■
Следовательно, система, находящаяся в неравновесном состоянии,
будет стремиться к равновесию; при этом концентрация и
парциальное давление веществ В и D будут уменьшаться, а для веществ G
и R эти величины будут увеличиваться; следовательно, реакция
пойдет в направлении
ЬВ + dD -* gG + rR + Q.
При этом чем больше максимальная работа, тем более система
удалена от состояния равновесия и тем больше химическое сродство
веществ В и D друг к другу.
Если максимальная работа реакции Ар = —AG или Av = —AF
отрицательна, то Ар = —AG <0 и Av = —AF <0:
Е vlnp/<ln/Cp и ^] v In Ct- << In /Cc,
откуда
(РвУ(РоУ ^рУр (Св)" (Со)" ^ СП
(РаУ(РнУ pPr (СоУКУ " « •
Следовательно, система, находящаяся в неравновесном состоянии,
будет переходить в равновесное; при этом концентрации и
парциальные давления веществ G и R будут уменьшаться, а для веществ В
и D эти величины будут увеличиваться. Таким образом, реакция
пойдет в обратном направлении:
ЬВ + dD <- gG + rR + Q.
При этом чем больше максимальная работа, тем дальше
находится система от равновесия и тем больше химическое сродство
веществ G и R друг к другу.
Если окажется, что максимальная работа реакции равна нулю,
то это означает, что:
Е v In pi = In Кр и Е v In C't = In Кс у
(РвУ (р'оУ _ к „ (СвУ (С'оУ
т. е. система уже находится в состоянии равновесия, и каждое из
возможных направлений протекания реакции равновероятно.
Таким образом, мы видим, что, находясь в различных
соотношениях, реагирующие вещества обладают различным сродством друг
109
к другу. Для того чтобы иметь возможность сравнивать сродство
различных веществ, было введено понятие нормального сродства.
К уравнению нормального сродства легко подойти, если положить
исходные концентрации или исходные парциальные давления всех
веществ равными единице. Тогда
А°р = — AG° = — RT In KP (276)
и
Лу = — AF° = — RT In Kc - (277)
о о
Следовательно, сравнивая Av или Ар различных веществ, мы
получаем характеристику степени их удаленности от состояния
равновесия при концентрации каждого из них С\ = 1 моль1л или
парциальном давлении р\ = 1 am.
§ 25. ЗАВИСИМОСТЬ КОНСТАНТЫ РАВНОВЕСИЯ ОТ ТЕМПЕРАТУРЫ
Полученные сведения о равновесии при постоянной температуре
можно дополнить. Найдем зависимость константы равновесия от
температуры. Дифференцируя уравнение изотермы реакции (274)
по температуре (с учетом, что р'в, ро, ph и р'# заданы и, следовательно,
от температуры не зависят), получаем
dAD dl±G ^, , _ dinKn
-2Г = ^^RZ>v\npL-R\nKp-RT—uA.
Подставляя полученные выражения для Ар и —~ в уравнение
Гиббса—Гельмгольца (213) и (214)
_ dAp
Ар ==: Чр ~г *■ $y '
получаем
RT^vlnp'i—RTlnKp^Qp+RT^vlnp'i —
d In Ко
-RT\nKp-RT*—^A,
откуда
- din Kn
Qp = RT* ^
dT '
Обычно это уравнение записывают в виде (уравнение Вант-Гоффа)
d In Ко Qd ~ ~
р Р (278)
dT ~ RT* '
или
dlnKp _ ДЯ
dT ~~ RT2 '
Для реакций, протекающих при постоянном объеме:
d\nKc Qy
df~~ ^RT*
(279)
(280)
ПО
или
Полученные уравнения устанавливают связь между изменением
константы равновесия с температурой и тепловым эффектом реакции.
Уравнения (278) и (279) относятся к условиям р = const и называются
уравнениями изобары химической реакции. Уравнения (280) и (281)
относятся к условиям V = const и называются уравнениями изо-
хоры химической реакции.
Все эти уравнения представлены в дифференциальной форме.
Интегрирование уравнений (278) и (280) при Q = const приводит
к выражению: __
dlnK = 2§*dT9
\пК = — -|-~ +const, (282)
где К = Кр (если Q = Qp) или К = Кс (если Q = Qv). Из
уравнения (282) видно, что логарифм константы равновесия (In К) линейно
зависит от обратного значения температуры (1/Т). При этом если
Q > 0, т. е. реакция является экзотермической, то прямая на
графике образует с положительным направлением оси абсцисс тупой
угол и с повышением температуры увеличивается константа
равновесия (в нашем написании). Если Q <0, т. е. реакция является
эндотермической, то указанный угол на графике оказывается острым
и увеличение константы равновесия происходит с понижением
температуры (рис. 29). Анализируя оба случая, видим, что повышение
температуры всегда смещает равновесие в эндотермическом
направлении.
Сделанный вывод представляет собой частный случай принципа
Ле-Шателье *, который позволяет в простейших случаях предвидеть
* Принцип Ле Шателье (иногда его называют правилом Ле Шателье) является
обобщением закономерностей химических и физических процессов, отличается в
результате широкого охвата рассматриваемых явлений многообразными и не всегда
достаточно строгими формулировками. Суть принципа хорошо передается некоторыми
его наименованиями: принцип подвижного равновесия, принцип смещения (или
сдвига) равновесия, принцип наименьшего принуждения, принцип химической
косности и др.
Впервые формулировки принципа даны в работах Даламбера и Гаусса (начало
XVIII века). Формулировка Ле Шателье (1884 г.) проверялась Эренфестом и
уточнялась Брауном (1887 г.). Имеются строгие формулировки принципа для конкретных
случаев и в общем виде. Например: индуцированное изменение параметров системы
Pi к Х( (факторы интенсивности и экстенсивности соответственно) вслед за
индуцирующим воздействием на систему регулированием параметра Р/ (фактор
интенсивности) уменьшает абсолютную величину изменения, зависящего от него и
сопряженного с ним по виду энергии параметра Xj (фактор экстенсивности), причем это
уменьшение тем больше, чем меньше индуцируемые изменения параметров. Так,
увеличение давления на идеальный газ (Р/ = Р) вызывает увеличение его температуры (Pi =
= Т) и энтропии (Xj = S). Вместе с тем уменьшается объем (Xj = V), чем
ослабляется результат внешнего воздействия, причем тем больше, чем меньше изменяется
температура и энтропия.
111
направление процесса. Согласно принципу Ле Шателье, изменение
внешних условий (температуры, давления) физико-химической
равновесной системы вызывает в ней реакции, противодействующие
производимому изменению.
Например, рассмотрим равновесия, применяемые при получении
двуокиси азота из азота и кислорода воздуха:
N2 + 02^!:2NO — 43,2 кал,
2NO + 02^T2N02 + 26,7 кал.
Согласно принципу Ле Шателье, первое равновесие при высоких
температурах будет сдвигаться в сторону образования N0, поскольку
*""k
1/7/Г
Рис. 29. Схема температурной
зависимости константы
равновесия для экзотермической
(Q> 0) и эндотермической
(Q< 0) реакции
система должна реагировать на повышение температуры процессом,
направленным на ее понижение, т. е. процессом отвода теплоты;
второе равновесие будет сдвинуто в сторону образования N02 при
понижении температуры, чтобы компенсировать отвод теплоты от
системы.
Попутно отметим возможность применения принципа Ле Шателье
для оценки влияния общего давления на химические равновесия.
Влияние давления можно рассмотреть на примере равновесия,
используемого при синтезе аммиака:
N2 + 3Ha^2NH8.
Из четырех молей газа получаются два моля газа, если реакция
протекает слева направо. При этом происходит сжатие.
Следовательно, повышение давления в системе вызывает процесс,
стремящийся уменьшить давление, т. е. способствует образованию
аммиака *.
* Влияние температуры и давления на сдвиг равновесия можно описать также
аналитически. Используя понятие числа пробегов химической реакции е (см.
примечание на стр. 95), можно получить: .
де \ ( дАН \ l / d2AG
и
,)в=(
\~др~)т = \
дг
дАЦ
дг
дг*
/ д2 AG \
\ дг2 ) т\
Так как (d2AG/ds2)T,p^> 0, то знак производных (dE/dT)p и (de/dp)x определяется
знаком (dAH/de)r,p и (dAU/dz)T,p соответственно. Кроме того, можно видеть, что
строгое соблюдение принципа Ле Шателье возможно лишь при постоянстве
соответствующих параметров.
112
Произведя интегрирование уравнений изобары и изохоры
химической реакции, можно получить уравнения, позволяющие вычислять
тепловой эффект химической реакции из измерений двух констант
равновесия при двух температурах:
dlnK = ^dT,
* 2 * 2
Если температурный интервал Г2—7\ не очень велик, то можно
пренебречь температурной зависимостью теплового эффекта реакции.
Тогда
1п*г,-1п*п = х(^~~тг)'
или
1пЙ;-1%г- <283>
Для решения конкретных задач это уравнение используется в
более удобном виде, учитывающем, что In А = 2,303 lg А и R = 1,987:
Ig^T- 4,673^7-, ' (284)
Если К = Кс> то Q = Qi/> и ес^и /С = /Ср, то Q = Qp.
Между тем тепловой эффект химической реакции в общем случае
зависит от температуры, и уравнения (283) и (284) верны лишь в
отдельных случаях. Учитывая зависимость теплового эффекта от
температуры [уравнение (115)]:
при интегрировании получаем
л 1 is Q dT
и Ш А = -jFr J¥ >
Да rp , Ар гр2 | Ay фз i \ dT
JrflnJC = J^(Qe + ^T + ^ + ^T» +
Г2
lnJC=-^+ Аа1пГ+^ + -^- + ■■ ■ + / (285)
ШЛ RT ^ \R ^ 2R ^ 2-3/? ' ^7' ^°0)
где / — константа интегрирования.
Это уравнение обычно называют развернутым уравнением
изохоры (или изобары) химической реакции.
Подставив выражение (285) в уравнение нормального сродства
А° = — RT In К,
8 А. Н. Крестовников 113
получаем
А° =Q0 — AaTlnT
АрГ2 АуТ*
2-3
#77.
(286)
Это уравнение получило название уравнения Габера.
Обращаем внимание на то обстоятельство, что Q = А° только
при температуре О °К, а при остальных температурах Q =f= A° и
тепловой эффект не может служить истинным критерием химического
сродства.
§ 26. НЕДОСТАТОЧНОСТЬ ПЕРВОГО И ВТОРОГО НАЧАЛ
ТЕРМОДИНАМИКИ ДЛЯ РАСЧЕТОВ ХИМИЧЕСКОГО СРОДСТВА
По уравнениям (285) и (286) можно вычислить любое равновесие,
а также нормальное сродство реакции. Однако для этого, помимо уже
известных нам термических величин, а именно теплового эффекта
- 0 реакции и теплоемкостей, необ-
">А k ^ , ходимо знать постоянную
интегрирования /. Попытка
определить ее посредством обычно
применяемого метода начальных
условий не дает положительных
результатов. Поэтому остается
возможным лишь опытное
определение ее величины
измерением равновесия хотя бы при
одной какой-то температуре.
Без этого невозможно
раскрыть ту неопределенность,
которую содержат в себе
уравнения (285) и (286) с
неизвестной постоянной интегрирования.
Неопределенность уравнения Габера (286) в этом случае наглядно
иллюстрируется рис. 30. Ход кривой
П - П Л- АаГ I- АРГ'2 J_ АУТЗ 4-
Чт — Vo п j 1 2 * 3 г ' ' *
ясен из температурных данных, поскольку уравнение не содержит
неизвестных величин. Когда Т = 0 °К, то Q = Q0, следовательно,
известна точка пересечения этой кривой с осью ординат. Кроме
того, величина коэффициентов Да, Д(5 и Ау вполне определенно
задает линию, выражающую рассматриваемую зависимость. Между
тем наличие константы / делает неопределенным положение кривой:
Apr2 Apr3
О Т
Рис. 30. Относительное положение
кривой Q = ЧГ (Г) и семейства кривых А =
= ф (Г) в области низких температур
A° = Q0 — AaTlnT —
2-3
#77.
Известно, что если Т = 0 °К, то А° = Q0 и, следовательно, эта
кривая приходит к оси ординат в точке, соответствующей Q0. Однако
всем возможным значениям / соответствуют определенное положение
и кривизна кривой, и имеется целое семейство кривых (см. рис. 30),
114
встречающихся при Т = О °К в одной точке. Константа / не может
быть выбрана произвольно, а должна иметь вполне определенное
значение, иначе химическое сродство вообще не может быть
однозначной функцией температуры. Следовательно, без постановки опыта
по изучению равновесия нет возможности выбрать какую-либо одну
кривую из их бесчисленного множества, каждая из которых
отличается величиной константы интегрирования.
Удалось измерить изменения физических свойств,
сопровождающие химические процессы: давления паров, теплоемкости, массы
и объемы реагирующих веществ, температуры и тепловые эффекты.
Однако разрешение основной задачи химической термодинамики —
определение химического сродства — натолкнулось на
недостаточность первого и второго начал термодинамики для расчетов сродства
на основании имеющихся измерений, так как для того, чтобы узнать
сродство при любой температуре, необходимо прибегнуть все же к
непосредственному измерению химического сродства или константы
равновесия, по крайней мере, для одной температуры. Иначе нельзя
найти величину постоянной интегрирования /.
Решение Ыернстом задачи вычисления химического сродства,
не прибегая к исследованию химического равновесия, по одним
только термическим данным (тепловым эффектам реакций и теплоем-
костям веществ, участвующих в ней), а также работы Планка
привели к введению новых теорем и постулатов, составляющих
содержание третьего начала термодинамики.
Задачи
Задача 1. После нагрева до 500° С 2 г фосгена при давлении 1 am занимают
объем 1,985 л. Требуется вычислить степень диссоциации фосгена при данных
условиях и парциальные давления всех газов.
Решение. Воспользовавшись уравнением Менделеева—Клапейрона (26)
и соотношением (273), получаем
pV-i-jJi-RT.
1,55.
откуда находим значение коэффициента диссоциации
. _ Мру __ 99» 1-1,985 _^
1 "~ mRT '~ 2-0,082.773
Далее для реакции диссоциации
СОС12 ^± СО + С12
с помощью выведенного ранее уравнения (272) при пг — 2 легко определить
значение степени диссоциации:
i — 1 1,55— 1 л _._ Гр-Л/
а = ——- = 2_{ = 0,55 = 55%.
Для нахождения парциальных давлений
Pi = Робщ -w-ч
8* 115
необходимо найти величину щ и 2/г/, т. е. число молей данного газа в смеси и
суммарное число молей. Поскольку
"со = ЛС12 = па
и
"сось^яО— а).
то
2 л/ = л (1 +а).
Обращаем внимание на то обстоятельство, что при подстановке в отношение
ni/Hni значений П( и V/it- числитель и знаменатель освобождаются от величины п —
исходного числа молей, т. е. определение парциальных давлений можно производить
исходя только из известного значения степени диссоциации:
а , 0,55 Л«--
Рсо = Рс\2 = Т+^ = l 'X55" = °'355 ат
и
1 — а 1 — 0,55 Л ООЛ
Рсос!2 = Робщ Т^ = 1 • t + о 55 = 0,290 «п.
Задача 2. При температуре 830° С и давлении 1 am степень диссоциации
сероводорода на серу и водород равна 8,7%. Найти константы равновесия этой реакции
диссоциации: Кр и Кс-
Решение. Запишем рассматриваемую химическую реакцию в
экзотермическом направлении:
2Н2+ S2= 2H2S,
тогда константы равновесия запишутся следующим образом:
Ph2Ps* „ ^Нг^Бг
PH2S CH2S
Найдем парциальные давления газов в смеси, для чего предварительно опре-
деляем пн = a, ns —-~-, яН8= 1 —а и £ щ = 1+ —; используя эти данные
и помня, что парциальные давления газов в смеси не зависят от исходного числа
молей, получаем
Рн2 — Робщ
a
i+4
a
Ps, = Р
оощ п
1+-о-
И
PH2S ~ Робщ
1- a
i+ a
2
Ко=„ РобЛ»3, > = „ .';°:0873..^ = 3,78-10-4 аот
Следовательно:
р ^ (1 — а)2 (2 + а) = (1 —0,087)2 (2 + 0,087)
и, согласно соотношению (266), при А/г = 2 — 3 = —1
Кс = Кр(RT)An = о^'.^'оз = 0,79-Ю-» моль/л.
116
Задача 3. При температуре 930° К константа равновесия реакции образования
водяного газа (СО + Н20:^!:С02 + Н2) равна единице. Смесь, состоящая из 20%
окиси углерода и 80% водяього пара, нагревается до 930° К. Требуется определить
состав смеси при равновесии.
Решение. Поскольку константа равновесия рассматриваемой реакции равна
единице, то способ ее написания безразличен. Кроме того, реакция идет без
изменения числа молей вещества, принимающих участие в реакции, поэтому справедливо
соотношение (271).
Обозначив количество двуокиси углерода и водорода через х (количество
участников реакции получается поровну), соответственно для окиси углерода и водяного
пара получаем (20 — х) и (80 — х). Подставив эти значения в формулу константы
равновесия, легко находим неизвестные равновесные концентрации:
[СО] [Н20] (20-х) (80-х) _,
АС = ,~п , гц . = 7S — 1 >
[С02] [Н2]
х2 = (20 -
х =
-*)(80-
= 16.
X2
— х),
откуда
Следовательно, состав равновесной смеси будет: 16% С02, 16% Н2,4% СО и 64% Н20.
Задача 4. Если нагреть 2,94 моль иода и 8,1 моль водорода в закрытом сосуде
до 444° С, то после достижения равновесия образуется 5,64 моль йодистого водорода.
Требуется определить, сколько образуется йодистого водорода, если исходить из
5,3 моль иода и 7,94 моль водорода.
Решение. Будем исходить из 2,94 моль иода и 8,1 моль водорода. Поскольку
по реакции образуется 5,64 моль йодистого водорода, то водорода остается 8,1 —
V~ = ^'^ моль, а иода 2,94 ~— = 0,12 моль. Поэтому константа равновесия
при температуре 444° С равна
к [Н2][12] _ 5,28-0,12
Обозначаем через х количество йодистого водорода, образующегося при той же
температуре из 5,3 моль иода и 7,94 моль водорода. Тогда
('"-т)(м-т)
fHdlbl
[HI]2 x
*= m.» =- -^ ^ = 0,02,
откуда получаем значения хг — 20,53 моль (отбрасываем как нереальное) и х2 ~
— 9,48 моль, которое принимаем за ответ задачи.
Задача 5. Требуется определить, сколько молей водорода надо добавить к
одному молю йодистого водорода в условиях предыдущей задачи, чтобы степень
диссоциации йодистого водорода стала равной 10%.
Решение. В условиях равновесия диссоциация 1 моль йодистого водорода
приводит к образованию — моль водорода и а/2 моль иода, в то время как остается
(1 —а) моль йодистого водорода (а — степень диссоциации). Добавление водорода
(обозначим это количество — х моль) увеличивает количество йодистого водорода и
уменьшает при этом количество иода, т. е. происходит подавление диссоциации, и
степень диссоциации становится равной а'. Следовательно, после достижения равно-
(а' \ а'
— -f х j моль, иода — моль и йодистого водорода (1 — а') моль.
По условиям задачи а' = 10%, поэтому
ОД , \ ОД
№+')
1Н,][1,] =\2 "7 2_=о02
[Hip (1 —оду2 ■А
откуда х = 0,274 моля, что и является ответом задачи.
117
Задача 6. Пользуясь условиями двух предыдущих задач, определить, сколько
молей водорода надо взять на каждый моль иода, чтобы перевести иод на 90%
в йодистый водород.
Решение. Очевидно, что чем больше взять водорода, тем полнее будет иод
переведен в йодистый водород. Если исходить из 1 моль иода, то после его
взаимодействия с водородом (обозначим вступающее во взаимодействие количество водорода
х моль) по условию задачи остается 1 —0,9= 0,1 моль, водорода остается (л: —
— 0,9) моль, а йодистого водорода образуется 1,8 моль.
По этим данным составляем уравнение
к== [H2][I2] __.(*-0,9)-ОД _ л ро
А [HI]2 (L8)2
откуда х = 1,548 моль. Полученный результат представляет собой ответ задачи.
Задача 7. Сульфогидрат аммония диссоциирует по уравнению
NH4HS(TB) ^ NH3 (газ) + H2S(ra3).
При 25° С равновесное давление продуктов диссоциации равно 500 мм рт. ст.
Требуется определить константу равновесия Кр и общее давление в состоянии
равновесия после того, как в сосуд, содержащий сульфогидрат аммония, введен аммиак
под давлением 300 мм рт. ст.
Решение. Поскольку в результате диссоциации образуется равное
количество молей NH3 и H2S, то и их парциальные давления pNH = Ph2s = """о" =
= 250 мм рт. ст.
Следовательно:
КР = Pnh3Ph2s = 250-250 = 6,25-10*.
После введения в рассматриваемую систему аммиака диссоциация сульфогидрата
аммония подавляется. Обозначив парциальное давление аммиака и сероводорода
соответственно (л: + 300) и х, составляем уравнение
КР = Pnh3Ph2s = (* + 30°) * = 6.25-104.
откуда после решения получаем два значения: xx<i 0, которое является нереальным,
и х2 = 141, 6 мм рт. ст. Теперь легко подсчитать общее давление:
Робщ = Pnh3 + Ph2s = (141>6 + 30°) + 141>6 = 583>2 ** Рт- сп-
Задача 8. При 713° К константа равновесия Кр реакции
Sb2S3 (тв) + ЗН2 (газ) <^ 2Sb(TB) + 3H2S(raa)
равна 0,429.
Требуется определить, чему равна мольная доля водорода в газовой фазе и будет ли
результат зависеть от общего давления.
Решение. Рассматриваемая реакция протекает без изменения числа молей
газообразных участников реакции. Поэтому давление не будет оказывать влияния на
равновесие.
Обозначив мольную долю водорода черех х и соответственно мольную долю
сероводорода через (1 —х), составляем уравнение
г>3 (1 д.\3
/Ср=-1^1 = - ^-1- = 0,429,
рн2 х
откуда получаем ответ зааачи: х= 0,5701.
Задача 9. Используя условия предыдущей задачи, определить, сколько молей
сурьмы образуется и сколько молей водорода израсходуется, если исходить из 1 моль
сульфида сурьмы.
118
Решение. Обозначим количество образовавшейся сурьмы через х моль\
тогда количество образовавшегося сероводорода составит 3/2 х моль, а количество
оставшегося водорода (1 — 3/2 л:) моль. Следовательно:
Рн * (3/2*)3
Кр = -^- = \ = 0,429.
р3Нз (1-3/2*)3
Отсюда х= 0,287 л*ол&— это количество образовавшейся сурьмы, а 3/2 х= 3/2X
Х0,287= 0,431 моль — количество израсходованного водорода.
Задача 10. Требуется определить, будет ли при температуре 444° С
происходить образование йодистого водорода в смеси газов, состоящей из водорода, иода и
йодистого водорода, если концентрации этих веществ имеют следующие значения,
моль! л:
н2 F2 hf
а) 2
б) 1,5
в) 1
5
0,25
2
10
5
10
Значение константы равновесия при указанной температуре К = 0,02.
Решение. Для получения ответа задачи следует использовать формулу (235),
согласно которой в случае «а» имеем
Av = - AF = RT (Jn 11Щ1^] ~ In Kc) =
= 1,987.717.2,303 (lg -^ lg 0,02^ = + 2289 кал/моль,
т. е. У] vln Ct > In Kc, и реакция пойдет в сторону диссоциации йодистого водо
рода.
В случае «б» имеем
Av = — bF= 1,987.717.2,303 (lg -^~^- - lg 0,02) =
= — 408 кал/моль,
т. е. ^ v In C( <C In Kc, и реакция пойдет в сторону диссоциации йодистого
водорода.
В случае «в» имеем:
Av = - AF = 1,987-717.2,303 (lg -|^- - lg 0,02 ) = 0,
т. е. 2V ^n ^i — ^п К с* и система находится в состоянии равновесия.
Задача 11. Требуется определить сродство окиси кальция к двуокиси углерода,
взятой при температуре 810° С и давлении 1 am, если давление диссоциации
углекислого кальция при указанной температуре равно 678 мм рт. ст.
Решение. Поскольку для реакции СаО(тв) + С02 = СаС03 /твч имеем
Кр = Рсо , воспользуемся формулой:
Ар = - AG = RT (In p'c02 - In pCOz) =
= 1,987.1083-2,303 (lg 1 — fe-|e|") = 244>7 кал1моль-
Задача 12. Требуется найти нормальное сродство для реакции 2СО + 02 ^1
>2С02 при температуре 1000° С, если степень диссоциации двуокиси углерода при
этой температуре и давлении 1 am равна 3- 10"3%.
119
Решение. Определяем парциальное давление газов в смеси:
а
— а _ ~2~
14-— 1 4- —
^2 ^2
_ 1 —а
РС02 — Робщ ^~ •
1+ ^
Далее, исключая из многочленов слагаемые, содержащие величину а в степени
выше второй, получаем
2
К — РѰа2 РОбЩ
Ар — о ^ о *
Рсо2 2
Наконец, по уравнению (276) определяем нормальное сродство
А°р = — RTlnКр = — 1,987-1273-2,303 lg Ь(3-10 &) = 80810 кал/моль.
Задача 13. Требуется подсчитать нормальное сродство реакции 2Ag + V2O2 —
= Ag20, если известно, что давление диссоциации закиси серебра при 25° С равно
5. 10"4 am.
решение. Нормальное сродство подсчитываем по уравнению (236),
учитывая, что Кр = Pq22[
A°n = — AG=^- 1,987.298-2,303lg(5-10 "4),/з = 9055 кал/моль.
Задача 14. Требуется показать, что тепловой эффект реакции 2NO^z^N2+02
не зависит от температуры, если известно, что зависимость константы равновесия от
4725 5
температуры определяется уравнением lg У К = 0,5441 ~—, а также найти
численное значение теплового эффекта этой реакции.
Решение. Воспользуемся уравнением (278). Для этой цели сначала определим
значение d In K/dT:
i/2 \gK= 0,5441 — 4725,5/Г,
In К = 2,303-0,5441.2 — 2,303-4725,5-2-1 /T
din К 2,303-4725,5-2
dT ~ Г2 '
после чего убеждаемся в независимости теплового эффекта от температуры и находим
его значение:
Q^RT2 ^~ = 1,987.2,303-4725,5-2 == 43 600 кал/моль.
Задача 15. Константа равновесия реакции Н2 + С12^±2НС1 может быть вы-
ражена уравнением lg К = 7^+ 0,440 lg T — 2,16. Требуется определить
тепловой эффект этой реакции при температуре 1000° К.
Решение. Сначала определим значение d In K/dT:
In/C= 2,3°^9586 +0,440 In T -2,16.2,303,
din К 2,303-9586 0,440
dT ~ T2 + T '
120
после чего по уравнению (278) определяем значения теплового эффекта при
температуре 1000° К:
- D-2dlnK , OQ7 l/wv2 /2,303-9586 . 0,440 \
Q = ^Г ~df~ ^ ' ' \ —Ш002— + "ТббО") = 44 700 кал1тль'
Задача 16. Константа равновесия реакции Н2 + I2^z^2HI при температуре
360° равна 0,015, а при температуре 444° С 0,02. Требуется сравнить среднее значение
теплового эффекта этой реакции в указанном интервале температур с нормальным
сродством при температуре 444° С.
Решение. По уравнению (284) находим значение теплового эффекта:
4,5737^0 КТг 4,573.717-633 , 0,02 010Л .
Q-T^Y;lg-K^-== 84 lg w = 312°кал1моль'
По уравнению (277) находим значение нормального сродства:
А° = — RT In К = —4,573-717 lg 0,02 = 5572 кал/моль.
Г л а в а V
ТРЕТЬЕ НАЧАЛО ТЕРМОДИНАМИКИ
§ 27. ТЕПЛОВАЯ ТЕОРЕМА HEPHCTA
Нернст высказал простое и довольно вероятное предположение
(1906 г.), что кривые Q = г|) (Т) и А° = ср (Т) не только встречаются
при Т = 0 °К, но и касаются друг друга. Говоря иными словами,
обе кривые вблизи абсолютного нуля имеют общую касательную,
т. е. касательные имеют одинаковый наклон к осям координат,
поэтому первые производные в точках касания, представляющие
собой тангенсы угла наклона, равны между собой. Это так
называемый постулат о касательной, который аналитически может быть
выражен следующим образом:
lim^=lim4^. (287)
Производные в формуле (287) представляют собой
dQ .. /AQ\ dA° ... /АЛ°\ /оооч
^ =aV5 \St ) И ИГ =llZ { W) ■ <288>
Следовательно, в формуле (287) приравниваются пределы при
Т —> 0 от пределов при А71—> 0. Это соответствует тому, что
касательные к кривым Q'= я|з (Т) и А° = ф (Т) еще до достижения Т =
=0 °К, т. е. в области, близкой к 0 °К, будут очень мало отличаться
друг от друга.
Кроме того, при очень низких температурах свойства твердых
тел перестают зависеть от температуры, а это требует, чтобы общая
касательная шла параллельно оси температур, т. е.
lim^ = lim^- = 0. (289)
121
Таково основное содержание высказанных Нернстом положений,
относящихся к свойствам конденсированных систем и получивших
название тепловой теоремы Нернста. Вначале эти положения были
высказаны в виде гипотезы, однако вскоре они бкгли подтверждены
экспериментально, например исследованием электродвижущих сил
гальванических элементов при комнатных температурах.
Таким образом, среди множества кривых правильной будет та,
которая горизонтально достигает оси ординат (рис. 31). Отсюда
вытекает целый ряд полезных
следствий.
Продифференцируем по
температуре уравнение зависимости
теплового эффекта от
температуры:
Qt = Qo + ^t +
учитывая, что Q = const:
dQ
dT
Рис. 31. Относительное положение
кривых "Q = ¥ (Г) и А0 = ф (Г) по
тепловой теореме Нернста
= Да + APT + ДуТ2 +
(290)
При Т = 0 °К все члены, содержащие 7\ становятся равными
нулю. Кроме того, согласно тепловой теореме Нернста, при Т = 0 °К
dQ
и
dT
= 0. Следовательно:
Да - 0. (291)
Теперь продифференцируем по температуре уравнение Габера:
A° = Q0 — AaTlnT
4£_72 — ^У-Тг
RTI,
учитывая, что Q0 = const и Да = 0:
dA° = —дрг— ^У-Т2-
dT
RTI.
(292)
При Т = 0 °К все члены, содержащие 7\ становятся равными
нулю. Кроме того, по тепловой теореме Нернста при Т = 0 °К и
dA Idt равно 0. Следовательно, поскольку R =/= 0, имеем
/ = 0. (293)
Доказав равенства Да = 0 и / = 0, можно упростить уравнение
Габера и прийти к формуле, не содержащей константы
интегрирования и точно определяющей ход кривой температурной зависимости
химического сродства:
A° = Q0- рГ2
7"з
•* • • •
(294)
122
Эта формула позволяет по тепловому эффекту и теплоемкостям
реагирующих веществ вычислить химическое сродство при любой
температуре, не прибегая к исследованию равновесия.
Ограничиваясь, как это обычно и делают, членами, содержащими температуру
не больше второй степени, получаем для случая V = const:
A; = -RTlnKc = Q0.v-A$T\
а для случая р = const:
А°р = - RT In Кр = Qo, р - AF2-
(295)
(296)
Впрочем для конденсированных систем различием между A°v
и Ар можно пренебречь. Тогда
A° = —RT\nK=Qo — APT2. (297)
Эти уравнения применимы для всех конденсированных систем
и позволяют распространить выводы, сделанные для температур
вблизи абсолютного нуля, на область более высоких температур.
Это справедливо, однако, только при условии, что кривые Q = -ф (Т)
и А0 = ф (Т) не имеют разрывов и перегибов, вплоть до температур
плавления или полиморфных превращений.
Далее, перепишем уравнение Гиббса—Гельмгольца (212) или
(214):
— г1А°
A° = Q+T^r
в следующем виде:
А° dT = QdT + T dA
или
А° dT — TdA° = QdT.
Разделим обе части уравнения на (—Т2), зная, что Т ф 0:
TdA° — A°dT Q dT
Интегрируем:
откуда
где
J-2
j2
= — J-~dT +const,
о
Л° = —Tj-^dT + constT,
о
const
A°-Qo
То
dA'
)
dT )т-*о
0.
(298)
123
Согласно уравнению Кирхгофа (108):
т
Qr = Qo + J(EC„-ECK)d7\
0
поэтому, подставляя значение QT в подынтегральное выражение
уравнения (298), получаем:
т т „
](7}ся-Ъск)аг
0
f2 \ * J fh
А° = -Т 1 %^ + Т 2 = -dT
о
или
£ cK)ат
A°=;-RT\nK =-'Q0 + T ) и -щ ОТ. (299)
о
Полученные формулы удобны в том отношении, что значение
интегралов находится непосредственно из таблиц, которые получили
название таблиц Митинг и содержат значения:
CpdT; }L-¥r~dT; T [J—14—dT (300)
для ряда температур многих веществ. Значение этих интегралов
находится графически с помощью планиметра или же методом
взвешивания площадей.
Если известно значение теплоемкостеи данного вещества при
различных температурах, то, нанося эти значения на график
(ордината — Ср, абсцисса — Т), получаем кривую, которая вместе с осью
абсцисс и ординатой, отвечающей требуемой температуре 7\
ограничивает площадь, соответствующую интегралу J Cp dT от 0 до
т
Т °К. Так находится величина J Cp dT. Поскольку теплоемкость
о
наиболее заметно меняется в области низких температур, то интеграл
будет определен тем точнее, чем ниже температуры, при которых
производились определения теплоемкостеи. Дальнейший ход кривой
определяется экстраполяцией по уравнению Дебая (73) или по
другим уравнениям.
Поступая точно так же, а именно: нанося на график найденные
значения интеграла J Cp dT от 0 до Т °К и разделенные на Т2
124
как функцию температуры и вычисляя полученные функции, находим
величину
Использование таблиц Митинг, содержащих значения
выражений (300), облегчает вычисление равновесий и в свое время широко
практиковалось.
Введение понятия характеристической температуры позволило
для всех веществ получить одну и ту же единственную кривую
теплоемкости, являющуюся функцией температуры. Это дало возможность
составить общие таблицы для определения выражений (300), так
называемые таблицы функций Планка—Эйнштейна и Дебая.
Применение тепловой теоремы
(третьего начала термодинамики) к газовым реакциям
В соответствии с тепловой теоремой Нернста неопределенная
константа интегрирования в уравнении, определяющем сродство
между жидкими или твердыми веществами, равна нулю.
Для газообразных систем эта константа интегрирования может
и не быть равной нулю. Однако Нернст показал, что с помощью
особого приема * на основании тепловой теоремы можно получить
и уравнение для расчета газовых реакций. Им было получено
уравнение
т
ЧКР = -j^r + т j (Sc-^£ck) dT + д г. (301)
о
где Qp — тепловой эффект реакции в твердой фазе при
Т = 0 °К;
(2 Сн — 2 Ск) — алгебраическая сумма теплоемкостей
твердых веществ, принимающих участие в
реакции; А/ называется константой
интегрирования уравнения газового равновесия и равна алгебраической сумме
констант интегрирования в уравнениях давления пара (181) или так
называемых истинных химических постоянных газообразных
участников реакции.
Истинные химические постоянные не зависят от агрегатного
состояния веществ и его кристаллической модификации; они
определяются экспериментально либо теоретически и содержатся в
специальных таблицах.
* С помощью проведения цикла, состоящего из обратимой конденсации исход-
веществ, реакции между ними в конденсированной фазе и последующей обрати-
ных
мой возгонки.
125
Ввиду отсутствия исчерпывающих данных по температурной
зависимости теплоемкостей веществ, особенно при низких и очень
высоких температурах, расчет по точным формулам не целесообразен.
Поэтому, а также с целью упрощения вычислений Нернстом было
предложено приближенное уравнение
ig^==-'A^+ij5AvigT+Ar' (зо2)
где Qp — тепловой эффект реакции при комнатной температуре;
Av — изменение числа молекул при данной реакции;
А*" — алгебраическая сумма условных (эмпирических)
химических постоянных газообразных участников реакции.
Величина условных химических постоянных определяется
методом подбора или по эмпирическим формулам и для различных
веществ оказывается различной. Кроме того, V может значительно
отличаться от i по величине, а также может иметь различный знак.
Специальные таблицы содержат значение /'. При отсутствии
значений условных химических постоянных их можно принять для
одноатомных газов равными 1,5, а для прочих — равными 3.
Так как
и
§ 28. ПОСТУЛАТ ПЛАНКА
<*Av dAF dAp dAG
_ :— И7ТИ tL rr;
dT dT ИЛ dT dT
dAF AC dAG AC
^-=-AS ИЛИ _ = -AS,
согласно (203) и (210), то соотношение (289) означает: вблизи
абсолютного нуля все реакции, протекающие в конденсированных
системах, не сопровождаются изменением энтропии. Планк расширил
это заключение, распространив его на энтропии реагирующих
веществ, и выдвинул постулат:
limS = 0 или S0°/c = 0. (303)
Наглядное подтверждение справедливости постулата Планка
находим в статистическом толковании энтропии как характеристики
термодинамической вероятности состояния (163). При постепенном
охлаждении величина термодинамической вероятности резко
уменьшается, и при Т = 0 термодинамическая вероятность достигнет
минимального значения, т. е. единицы. Значит данное
макросостояние — состояние идеального кристалла при абсолютном нуле —
может быть осуществлено единственным микросостоянием, и так как
Q = 1, то
S = k In Q = k In 1 = 0.
Следовательно, при абсолютном нуле энтропия правильно
образованного кристалла любого элемента или соединения в чистом состоя-
126
нии равна нулю, а при любом другом состоянии вещества энтропия
его больше нуля.
Логическое развитие постулата Планка приводит к важным
следствиям, которые представляют собой содержание третьего начала
термодинамики. Следует отметить, однако, что, несмотря на большое
значение третьего начала термодинамики, по своей общности оно
уступает первому и второму началам термодинамики. В отличие от
них оно не приводит к определению каких-либо фундаментальных
величин, подобных внутренней энергии или энтропии, а только
ограничивает значения одной из них. В силу этих причин третье начало
термодинамики часто называют тепловым законом, подчеркивая
тем самым его отличие от первого и второго начал термодинамики.
Из постулата следует, что ряд свойств конденсированных систем —
G, Я, F, U, Ср, Cv и др. —вблизи абсолютного нуля перестают
зависеть от температуры. Значит, еще не доходя до Т = 0 система
приходит в такое состояние, что достижение абсолютного нуля
становится принципиально невозможным. Так, в соответствии с
постулатом Планка:
lim С, = lim Су = 0 (304)
Г-» 0 Г-»0
и теплоемкость делается исчезающе малой при достаточном
приближении к абсолютному нулю (но еще до Т = 0), поэтому нельзя
отнятием теплоты достичь температуры 0 °К.
Наличие предельно низкой температуры было предсказано еще
М. В. Ломоносовым. Он указывал на существование «. . .наибольшей
и последней степени холода, состоящей в полном покое частиц. . . .»
Формулируя принцип недостижимости абсолютного нуля, часто
исходят, как и при формулировке первого и второго начал
термодинамики, из невозможности вечного двигателя (перпетуум мобиле)
третьего рода: нельзя построить машину, которая работала бы за
счет охлаждения тела до абсолютного нуля.
Отсюда следует, что коэффициент полезного действия тепловой
машины не может быть равным единице, так как это потребовало бы,
чтобы температура холодильника равнялась бы абсолютному нулю,
т. е. Т2 = 0 в уравнении (127).
С помощью адиабатического размагничивания парамагнитных
веществ было осуществлено охлаждение до 0,0034 °К (1948 г.).
§ 29. РАСЧЕТ АБСОЛЮТНЫХ ЗНАЧЕНИЙ ЭНТРОПИИ
Мы уже рассматривали вычисление изменения энтропии, которым
сопровождаются различные процессы. Были получены уравнения
(147), (152) и (158)—(162), которые позволяют осуществить эти
вычисления через изменение различных параметров системы. Сейчас
нам предстоит познакомиться с вычислением абсолютного значения
энтропии, поскольку, в соответствии с постулатом Планка,
необходимость выбора условного начала отсчета этой функции теперь
отпадает.
127
Определение абсолютного значения энтропии различных веществ
при тех или иных температурах возможно, если известны
теплоемкости этих веществ при всех температурах от абсолютного нуля
до интересующей нас температуры, а также тепловые эффекты и
температуры всех фазовых переходов, если они имеют место в этом
температурном интервале. В самом деле, пусть рассматриваемое
вещество при интересующей нас температуре Т находится в
кристаллическом состоянии и в той же модификации, что и при
абсолютном нуле. Тогда его энтропия при температуре Т определяется
равенством (148):
т
О
Если же в интервале температур от 0° К до Т вещество при
температуре Та->$ переходит из одной кристаллической модификации
в другую, то надо учесть возрастание энтропии при этом переходе,
которое, согласно уравнению (159), для данного случая составляет
где Д#а_р — тепловой эффект процесса.
Следовательно, в этом случае
ST= [CldT + ^^+ [ $dT,
О
Т ^а->р
а->0
где Ср — теплоемкость низкотемпературной модификации;
Ср — теплоемкость высокотемпературной модификации.
Если вещество при интересующей нас температуре находится
в жидком состоянии, то
СР ,т . ЛЯа->|3 , I _!£_
ST=: -f-dr+r-^p+ -f-dT +
0 а->Р Та
Т
лт-r С ГЖИДК
MlHL , 1£ dT,
пл т
1 пл
где Д#пл — теплота плавления;
Тпл — температура плавления;
£,жидк — теплоемкость вещества в жидком состоянии.
Наконец, если вещество находится в газообразном состоянии и
Qucu представляет собой теплоту испарения его при температуре
128
кипения Ткип, а С™3 — теплоемкость в газообразном состоянии, то
sT= J -^dT+-^f + j -f-dr +
0 Ta->$
+ ^7+ J -^r-dT + ~f^+ J -т-л-. (305)
'пл кип
Таким образом, можно определить абсолютное значение
энтропии веществ в различных состояниях при любых температурах. Если
Рис. 32. Схема графического метода Рис. 33. Схема графического^ метода
определения абсолютного значения эн- определения абсолютного значения
тропии по зависимости Ср/Т от Т энтропии по зависимости Ср от In Г
вещество имеет всего лишь одну кристаллическую модификацию,
то первые три члена уравнения (265) заменяются интегралом,
соответствующим энтропии твердого вещества при температуре его
плавления. Наоборот, при наличии большего числа твердых
модификаций число членов уравнения соответственно увеличится.
Хотя абсолютное значение энтропии можно найти и с помощью
теоретических уравнений для температурной зависимости теплоем-
костей и с помощью уравнений, составленных по опытным данным,
все же наиболее надежным является графический метод расчета;
он же представляется и наиболее удобным.
Для этой цели обычно строятся графики в координатах Ср/Т
и Т или же в координатах Ср и In T (рис. 32 и 33). Площадь, которая
ограничена кривой, осью абсцисс и крайней ординатой при
интересующей нас температуре (Т или In Т), соответствует энтропии при
данной температуре, за вычетом энтропии фазовых переходов. Эта
площадь оказывается разделенной на части, каждая из которых
отвечает величине соответствующих интегралов уравнения (305).
Определение абсолютного значения энтропии можно произвести
и другими методами. Один из них, например, основан на
использовании спектральных данных и данных о строении молекул. При этом
совершенно не используются допущения, принятые при термохими-
9 А. Н. Крестовников 129
ческом определении энтропии. Полное согласие между результатами
определения энтропии этими двумя методами указывает на
надежность обоих методов и, в частности, на справедливость постулата
Планка.
§ 30. РАСЧЕТЫ ПО РАВНОВЕСИЯМ С ПОМОЩЬЮ ТАБЛИЦ
СТАНДАРТНЫХ ВЕЛИЧИН
г
Существуют три вида таблиц стандартных величин: 1) таблицы
изменений энтальпии при образовании одного моля при стандартных
условиях; 2) таблицы изменений изобарного потенциала
образования одного моля вещества при стандартных условиях; 3) таблицы
энтропии, рассчитанных на 1 моль вещества при стандартных
условиях.
За стандартные условия принято считать температуру 25° С
(точнее 298,16 °К) и давление 1 am. Стандартные изменения
энтальпии, изобарного потенциала и энтропию принято обозначать
следующим образом: А#298> AG298 и S2%*
Вспоминая, что —АН = Qp, будем иметь в виду, что первый вид
таблиц представляет собой таблицы теплот образования веществ
при р = 1 am и t = 25 °С; второй вид таблиц (при условии —AG =
= Ар) представляет собой таблицы максимальных работ
образования вещества при р = 1 am, а третий вид таблиц позволяет
определить изменение энтропии реакции и характеризует величину
связанной энергии реакции при стандартных условиях.
Сравнение уравнений (213) и (210)
позволяет получить уравнение Гиббса—Гельмгольца в следующем
виде:
AG - АЯ- Г AS. (306)
Из этого уравнения, которое связывает AG — свободную энергию,
АН — полную энергию и TAS — связанную энергию при р = const,
следует, что при расчетах можно пользоваться только какими-либо
двумя видами таблиц, а по уравнению (306) вычислять данные
таблицы третьего вида.
Все подсчеты по таблицам стандартных величин основаны на
свойствах аддитивности AG, АН и S, поэтому они сводятся к простому
алгебраическому суммированию, аналогичному расчетам по закону
Гесса. Так, изменение энтальпии реакции АН равно алгебраической
сумме изменений энтальпий при образовании веществ — участников
реакции, причем с плюсом берутся продукты реакции, а с минусом —
исходные вещества. Это же относится и к AG и AS реакции. Следует
заметить, что АН и AG простых веществ равны нулю, поэтому они
в таблицах не помещены.
130
Таблицы стандартных величин позволяют определить AG, АН
и AS реакции только при стандартных условиях. Для нахождения
этих величин при любой температуре нужно знать математическую
зависимость их от температуры.
Согласно уравнению Кирхгофа (Ш), имеем:
т
Qr = Qo + J (Е С - Е ск) <я\
О
или
т
AHT = AHTo + \kCpdTy
где АС, = S Ск - 2 Са.
Если Г0 = 298 °К, а р = 1 am, то
т
АН°Т - А#о98 + j АСР dT. (307)
298
Согласно уравнению (145), имеем:
т
с сР
о
или, если Т0 ~ 298 °К и р = 1 am, то
S°r = S;98+ J^rfT. (308)
298]
Отсюда для изменения энтропии получаем:
т
С АС
A5r = AS2098+ \ -y^-dT. (309)
298
Если уравнение (307) или (309) подставить в уравнение
AG°T = AH°T — TAS°Ty
то получим:
т т
AG°T = А#2°98 + JACPdT-ТAS°29S-T §^dTy
29В 298
или
т т
AG°T = AH0m-TAS0m+ fJACpdT-T§^dT. (310)
298 298
Теплоемкости, которые помещены под знак интеграла, также
зависят от температуры, и это следует учитывать при использовании
уравнения (310) для расчетов.
9* 131
Согласно уравнениям нормального сродства (276) и (277):
а°р = — ^GT=:-Rт\nкp;
следовательно, при любой температуре Т
AGT
Ш J\p = dj. .
Для практических целей удобней пользоваться уравнением,
в котором натуральный логарифм переведен в десятичный, а
универсальная газовая постоянная выражена в калориях. Тогда
]gK AGr (ЗЦ)
s р 4.573Г v ;
Таким образом, получив значение AG° (величина нормального
сродства при температуре Т), по уравнению (311) легко находим
значение константы равновесия при данной температуре.
Аналогично для реакций, протекающих при V = const, получаем
№ = ^- (312)
о
Однако для определения AFt потребуется знание температурной
зависимости теплоемкостей реагирующих веществ при V = const.
Теория металлургических процессов основана на применении этих
расчетов к процессам получения металлов из руд. Результаты таких,
расчетов содействовали развитию новейших методов химической и
металлургической переработки.
Приведенная ниже табл. 2 составлена на основании наиболее
общепринятых значений стандартных величин. Полные таблицы
приводятся в термохимических и физико-химических справочниках
и обзорах и чаще содержат данные только по двум термодинамиче-
о о
ским функциям: Д#298 и ^298- Третью величину легко вычислить
ПО формуле AG298 = Д#298 — 298AS298-
о о
Термодинамические функции АНт и ASt довольно сложно
определить при высоких температурах экспериментально, а выше 1000 °К
эта задача в настоящее время практически невыполнима. Тем не менее
для газов имеются достаточно надежные методы статистического рас-
о о о о
чета термодинамических функций Ср, St, Нт и Gt для интервала
температур от 298 °К до нескольких тысяч градусов. При этом
используются молекулярные параметры: моменты инерции,
характеристические частоты колебаний молекул и др., определяемые в свою
очередь спектроскопическими методами. Для этих методов характерно
вести экспериментальные измерения, а затем и расчеты лишь для
интервалов температур. Однако это не вносит какие-либо
принципиальные затруднения, так как для расчета химических равновесий
необходимы не абсолютные значения термодинамических функций,
а их изменения в результате реакций.
132
Таблица 2. Стандартные термодинамические функции
Вещество
и состояние
Элементы
Н2 (газ)
Н (газ)
02 (газ)
03 (газ)
С, графит
(ТВ.)
С, алмаз
(ТВ.)
N2 (газ)
С12 (газ)
С1 (газ)
Вг2 (ж)
I, кристалл.
(ТВ.)
12 (газ)
S, ромб.
(тв.)
S2 (газ)
Li (тв.)'
Na (тв.)
К (тв.)
Mg (тв.)
Са (тв.)
А1 (тв.)
Си (тв.)
Zn (тв.)
Fe, а-форма
(тв.)
Мп (тв.)
Ag (тв.)
.о
оо 2
О СМ »<.
£ 2
ч §
1 «
<5»
оо 2
05 ^
о <м ^
^ 2
< §
1 *
и простые веществе
0
—51,8
0
—33,93
0
—0,450
0
0
—28,43
0
0
— 15,12
0
—29,64
0
0
0
0
0
0
0
0
0
0
0
0
—48,3
0
—38,85
0
—0,220
0
0
—25,04
0
0
—4,64
0
— 18,28
0
0
0
0
0
0
0
0
0
0
0
о.
•
«t
^
оо -^
о см а
со «
г
31,2
27,41
49,00
57,0
1,39
0,59
45,77
53,24
38,02
36,7
27,9
62,29
7,62
53,64
6,7
12,31
15,2
7,77
9,95
6,77
7,97
9,95
6,49
7,60
10,2
Вещество
и состояние
2S §
О СМ <^
^ 2
< 3
1 *
оо 2
а5 ^
О СМ ^
О 2
< §
1 *
о.
•
о
^ 5
00^
о см а
со «
Неорганические соединения
Н20 (газ)
Н20 (ж)
Н202 (ж)
ОН (газ)
НС1 (газ)
НВг (газ)
HI (газ)
HCN (газ)
H2S (газ)
NH3 (газ)
N20 (газ)
NO (газ)
N02 (газ)
N204 (газ)
СО (газ)
С02 (газ)
S02 (газ)
S03 (газ)
CS2 (газ)
СС14 (газ)
LiCl (тв.)
NaCl (тв.)
NaBr (тв.)
КВг (тв.)
Nal (тв.)
KI (тв.)
LiOH (тв.)
NaOH (тв.)
КОН (тв.)
MgO (тв.)
СаО (тв.)
А1а08 (тв.)
+ 57,800
+ 68,318
+44,87
—5,98
+22,03
+8,31
—6,50
—32,90
+4,80
+ 11,04
— 19,47
—21,53
—7,96
—2,24
+ 26,393
+94,030
+70,94
+94,44
—35,2
+25,6
+97,6
+98,3
■ПТИТ"
+86,8
+94,1
+69,2
+78,7
+ 115,3
+ 102,7
+ 102,6
+ 145,9
+ 151,7
+54,629
+56,661
+28,2
—4,85
+22,000
+ 12,50
—0,46
—28,51
+7,83
+3,94
—24,77
—20,10
—12,28
—23,44
+32,778
+94,242
+71,6
+87,0
— 17,6
+393,3 -
-16,5
г91,6
zUJL^
-97,6
-83,1
-90,5
-68,5
-77,5
-105,1
-90,8
-93,0
-138,1
-141,7
1-371,4
44,80
16,71
24,4
43,91
44,60
47,43
49,4
54,63
49,2
45,95
52,58
52,2
57,5
72,7
47,28
51,09
59,0
56,4
76,0
74,2
14,2
■1L1
19,1
17,8
21,0
23,5
25,0
13,7
12,4
23,0
6,4
9,0
12,4
Все сказанное привело к тому, что за последнее время получили
применение таблицы приведенных потенциалов («фи-потенциалов»)
и расчеты с их помощью. Уравнение
bG°T = — RTlnKD
для этой цели преобразовывают к виду:
R\nKt
Atf
a (g°t — н°0) д#;
о
ДЯГ
АФ; ^, (313)
где Д#о — изменение энтальпии в результате химической реакции
(тепловой эффект) при О °К. Величина
Фт = JLT-± (314)
называется приведенным потенциалом при температуре Т °К- Можно
видеть, что
* ^О П J> JtjQ
Ot=:St -т-±. (315)
т
Здесь вполне возможно определение абсолютного значения энтропии
St и изменения энтальпии при повышении температуры от О °К до
Т °К. Такие расчеты предварительно выполняются для химических
веществ по формулам:
Sr = )-£-dT+ ?>^p- (316)
и
1
Н°т - Hi = J С; dT -f S АЯ„ (317)
О
после чего результаты сводятся в таблицы. В формулах (316) и (317)
величины Д#,- и Ti представляют собой энтальпии (тепловые эффекты)
и температуры фазовых (аллотропных, полиморфных, агрегатных)
превращений. Для начала отсчета энтальпий применяется некоторая
температура; в данном случае О °К при 1 am, однако могут
использоваться и другие точки температурной шкалы (273,16 °К при 1 am
или 298,16 °К при 1 am). Состояние химических веществ при этих
условиях называют стандартным состоянием. Пересчет от одного
стандартного состояния к другому не сложен. Например, полагая
начало отсчета 0 и 298 °К, получаем функции Ф * и Ф **
соответственно (иногда вместо этих используются другие обозначения: Ф'
и Ф"). Функция Фт находится статистическими методами из моле-
кулярных параметров, а функция Фт рассчитывается по формуле
** _ *
^298—^0
фг =ФТ+ ™т и-. (318)
Разделять приведенные потенциалы на Ф * и Ф ** удобно, так как
они для сравнительно простых газов и достаточно полно изученных
твердых тел находятся с большой точностью, а Л#298, как правило,
определяется из калориметрических данных, часто менее точных.
Таким образом, всякое достаточно точное определение может быть
использовано для пересчетов с определением более точных значений
константы равновесия по формуле
о
АН
RlnKp = A<Z>** =?£. (319)
134
В табл. 3 приведены выдержки из таблиц приведенных
потенциалов для некоторых химических веществ.
Таблица 3. Таблицы термодинамических функций, рассчитанных
для практически используемых температур
Вещество и
стандартное состояние
5, (газ)
АЯ° = 53 200
А #°98 = 53 740
AG298 = 44 060
S2, (газ)
АЯ° = 29 935
АЯ°98 = 29 860
AG°98 = 18 430
х
о
273
298
1200
2000
273
298
1200
1500
5,684
5,658
5,093
5,085
7,600
7,760
8,900
8,960
*5?
о О
1
о К
1 449
1 591
6 361
10 417
1 949
2 141
9 888
12 570
ОО
О)
о см
1
—142
0
4770
8826
—192
0
7747
10 429
град)
^
О К Q
СО ^
39,684
40,086
47,547
50,138
53,96
54,510
66,300
68,300
град)
С)
*s
- <ч
34,380
34,750
42,247
44,929
46,826
47,330
58,060
59,920
град)
С)
"^
40,203
40,086
43,572
45,727
54,660
54,510
59,840
61,350
Примечание. Значения А// и AG даны в кал/моль,
Зная табличные значения приведенного потенциала Фг всех
веществ, участвующих в реакции, можно определить изменение при-
веденного потенциала АФ^ в результате реакции, а затем по
формуле (313) или (319) рассчитать константу равновесия реакции.
Задачи
Задача 1. Требуется найти стандартное изменение изобарного потенциала для
реакции 4НС1 + 02 = 2Н20 + 2С12, если известно, что для водяного пара —AG298 =
= 54 629 кал!моль, а для хлористого водорода — AG298 = 22 000 кал!моль.
Решение. Производим подсчет следующим образом:
AG298 (реакции) = 2 ^298 (Н2°) ~ 4 AG°m (HC1) -
= 2-(— 54 629) — 4 (— 22 000) = — 21 258 кал.
Задача 2. Требуется определить изменение изобарного потенциала реакции
образования метана из простых веществ при стандартных условиях, а также константу
равновесия Кр при температуре 250° С, если известны следующие данные: для
метана А#298 = —19 200 кал!моль\ для графита Sggg = ^^ кал!(г- атом-град); для
водорода 5298 — 31,2 кал!(моль град); для метана S°298 = 44,5 кал!(моль-град).
Решение. Определим сначала изменение изобарного потенциала для
реакции 2Н2+ С— СН4 при стандартных условиях:
АО°ш = А#;98 (СН4) - 298 [s°m (СН4) - S;98 (С) - 2S;98 (H2)] -
= — 19 200 — 298 (44,5 — 1,39 — 2-31,2) = — 13 452 кал!моль;
135
отсюда находим значение константы равновесия Кр:
luKt
ag:
13452
4.57371
КР
Рк
РснА
4,573-298
= 1,349.10-1°.
10.13;
Задача 3. Требуется найти для реакции НС1 + V402 = V2CI2 + V2H20
значение изменения изобарного потенциала при стандартных условиях и константу
равновесия Кр для температуры 25° С, если известно, что для хлористого водорода
—А^298 ~ 22 030 кал/моль и S298 — 44,6 кал/'(моль град); для паров воды ЛЯ298 ==
= —57 800 кал/моль и S298 = 44,80 кал/(моль-град); для хлора
= 53,24 кал/(г-атом-град); для кислорода S29s — 49,00 кал/(г-атом. град).
Решение. Вычисления производим аналогично предыдущей задаче:
4-АЯ;98(Н20)-ЛЯ;92(НС1)
°298
ли
298 (реакции)
— 298
У 5298 (С12) + "2~ 5298 (Н2°) _ 5298 (HC1) 4
1
^298 (^2)
(— -i-57800 + 22030^ — 298 (-—53,24 +
+4-44'8
- 44,6
49
■»)
4537 /сал.
г
4537
ig ** ~ 4^737 ~ 4,573 • 298 ~ 4'555'
/Сд =
Phc\PqZ
PcuPhIo
= 3,6.10"4.
Задача 4. Требуется определить состав равновесной смеси, полученной из
равных объемов воды и окиси углерода при температуре 600° К на основании следующих
данных:
для двуокиси углерода ЛЯ298 = —94 030 кал/моль и S298= 51,09 кал/(моль- град);
для окиси углерода ЛЯ298 = —26393 кал/моль и S298 = 47,28 кал/(моль, град);
для паров воды Л#298 =—57 800 кал/моль и S298 = 44,80 кал/(моль. град); для
водорода S298 = 31,20 кал/(моль- град), ЛС = 1,72 кал/(моль- град).
Решение. Согласно уравнению (270), получаем:
Т т
лс:
ля;
Т AS,
+j
ACpdT
1
ЛС;
dT
Т~~ "1J298 Л ^°298
298 298
и для реакции Н20 + СО = Н2 + С02 имеем:
AG°T = [ЛЯ2°98 (С02) - ЛЯ2°98 (СО) - ЛЯ^8 (Н20)] -
- т [s°m (co2) + s;98 (н2) - s;98 (н2о) - s;98 (со)] -
АС?Т (In -25Г+
— 600 (._
— 1,72-600 (2,303 lg
1
\ = (— 94 030 + 26 393 + 57 800)
298 ' Т
600 (51,09 4-31,20 — 47,28 — 44,80) —
298 ^ 600 /
4002 кал /моль.
136
Отсюда:
AGr 4002
lg Kp = С573Г = "" 4,573 • 600 = 2,532°'
Kp = 3,404 -lO"2.
Далее, обозначая содержание С02 и Н2 через х, для Н20 и СО будем иметь
(50 л;)2
(50 — х) и (50 — х). Тогда %— 3,404- 10'2 и х = 42,23%. Следовательно, рав-
новесная смесь будет содержать, %: 42,33 С02, 42,23 Н2, 7,77 СО и 7,77 Н20.
Задача 5. Требуется найти степень диссоциации а для реакции S2 = 2S при
Р= 0,001 am и Т= 1200° К.
Решение. В табл. 3 находим значения Ф* для S2 и S при 1200° К, равные
соответственно 58,060 и 42,247 кал/(моль-град).
CjTO ТО Я Я *
Дф* = 2-42,247 — 58,06 = 26,43 кал/(моль- град).
Теплота реакции S2 = 2S при 0° К равна разности теплот образования S2 и 2S из
ромбической серы (см. табл. 3):
АН°0 = 2 • 53 200 — 29 935 = 76 465 кал/моль.
По уравнению (313) получаем:
,о
* А^о 76 465
4,573 lg Кр = АФГ Л. = 26,43 - ^? = - 37,29;
lgKp = ~ 8,002.
Далее:
1 ы 1 Ра2 , 0,001а2 -ппо
IgKp = tefZTJ? = ]* 1-а2 = 9'"8'
откуда:
Кр= 9,95-10"9 и а = 0,0031.
Задача 6. Требуется рассчитать равновесие реакции
ZnS + СО = COS + Zn
при 1000° С, если значения функций Ф* для этой температуры известны, кал/моль:
22,97 для ZnS; 52,252 для СО; 63,32 для COS и 16,69 для Zn. Все участники реакции
находятся при 1000° С в газообразном состоянии, кроме соединения ZnS.
Стандартные теплоты образования равны, кал/моль:—48 500 для ZnS;—26 416 для СО
и 32 800 для COS. Стандартная теплота сублимации цинка —31 190 кал/г-атом.
Решение. Сначала находим
Лф1200° с = 63>32 + 16,69 — 22,97 — 52,252 = + 4,793 кал/моль;
затем
Д#°98 = — 48 500 — 26 416 + 31 190 + 32 800 = — 73 306 кал/моль.
Отсюда по уравнению (319) получаем:
4,573 lg Кр = АФ*Г - ^~- = 4,793 - ^- = - 52,79;
lgкр = lg — = _ 1153 и Ко = 2,95-10-12 am.
137
Глава VI
МЕТОДЫ ПРИБЛИЖЕННЫХ ТЕРМОДИНАМИЧЕСКИХ
РАСЧЕТОВ
Термодинамические и термохимические методы исследования
вооружили теоретиков и практиков мощным оружием научного
предвидения, поскольку с их помощью возможно, осуществление
теоретического анализа самых различных физико-химических процессов без
экспериментального их проведения.
Термодинамические методы расчета химических равновесий в
настоящее время широко используются в работе ученых, технологов-
проектировщиков, работников заводских лабораторий,
специалистами в области химии, физики, металлургии, теплотехники,
энергетики и многих других областях. Вместе с тем из-за огромной
диспропорции между числом известных и числом изученных веществ
работники указанных категорий постоянно испытывают затруднения,
связанные со сложностью и особенно с длительностью и
утомительностью проведения подобных расчетов. Кроме того, опубликованные
данные по термохимическим величинам являются весьма неполными,
некоторые данные вообще не могут считаться достаточно надежными
и их применение требует предварительной проверки.
Поэтому очень важно научиться оценивать неизвестные и
недостающие величинь! с достаточной точностью, а также уметь
пользоваться приближенными, так называемыми упрощенными и
сравнительными методами расчета. Эта задача не является очень трудной.
Тем не менее такая работа требует известного опыта и умения удачно
выбрать метод расчета в каждом отдельном случае, и, если это
необходимо, разработать новые методы.
§ 31. ТЕПЛОЕМКОСТИ
В различных областях металлургии знание теплоемкостей при
комнатной температуре и их температурных зависимостей является
весьма существенным, поскольку большинство расчетов включает
в себя в качестве первого и основного звена более или менее точное
определение теплоемкости. Вместе с тем экспериментальное
определение теплоемкостей является настолько трудоемким делом, что
невозможно рассчитывать на полное исследование всех веществ и
в широком интервале температур.
Для оценки теплоемкостей газов можно руководствоваться
следующими данными.
Одноатомные газы, к которым относятся многие пары металлов,
обладают постоянной теплоемкостью Су я^ 3 кал/(г-атом - град) или
Ср ^ 5 кал/(г-атом • град). Двухатомные газы при комнатных
температурах обычно имеют значение теплоемкости Ср =
= 7 кал/(моль-град). Максимальное возможное значение
теплоемкости Ср = 9 кал/(моль • град) достигается при высоких температурах,
хотя для некоторых газов, например для хлора и брома, Ср >
£> 7 кал/(моль-град) даже при комнатной температуре. Повышение
138
теплоемкости от 7 до 9 кал/(моль-град) происходит непрерывно и для
более легких молекул (до М = 40); уравнение
Ср = 6,7+ 1,0-10"3 Т
хорошо передает значения теплоемкостей в интервале температур
от 300 до 2300 °К. Для веществ, имеющих М > 100, в указанном
температурном интервале теплоемкость Ср = 9 кал/(моль • град) и
ее можно считать постоянной величиной. У соединений водорода
эти закономерности нарушаются.
Приближенное среднее значение теплоемкости многоатомных
газов можно получить, прибавляя к шести (или к пяти для
линейных молекул) учетверенное число межатомных связей в молекуле,
причем безотносительно к тому, являются ли связи простыми или
кратными. Таким образом, для А1203 в 1азообразном состоянии
С/г= 6 + 4-4 = 22 кал/(моль• град). Для С02 имеем Ср = 5 +
+ 4-2 = 13 кал/(моль-град). Подмечено, что чаще всего для легких
молекул Ср <С 13, а для тяжелых Ср >> 13 кал/(моль-град^.
Теплоемкости жидкостей не сильно отличаются от теплоемкостей
тех же веществ в твердом состоянии. Для многих неорганических
веществ теплоемкость в жидком состоянии находится в пределах
7—8 кал/(моль • град). Если опытных данных нет, то считают, что
Ср = 7,25 кал/(моль • град), или допускают аддитивность атомных
теплоемкостей элементов, составляющих рассматриваемую жидкость.
О температурной зависимости теплоемкости веществ в жидком
состоянии известно очень мало (принято считать ее постоянной
величиной^.
Теплоемкость при постоянном давлении твердых простых веществ
при комнатной температуре находится в пределах 6,2—
6,4 кал/(г-атом - град) (правило Дюлонга и Пти). Эта величина
возрастает с повышением температуры и при температуре первого
фазового превращения (или плавления при отсутствии аллотропии)
примерно одинакова для всех элементов и составляет 7,00—
—7,25 кал/(г-атом-град). Для получения теплоемкостей соединений
указанные значения должны быть умножены на число атомов в
молекуле (правило Неймана и Коппа). Если в левой и правой частях
уравнения химической реакции число атомов одинаково, то для
большинства реакций значение АС^ можно принять равным нулю.
Теплоемкости почти всех элементов известны, по крайней мере,
для комнатных температур. Известны они также и для многих
соединений. Поэтому в тех случаях, когда теплоемкости при высоких
температурах неизвестны, их можно вычислить, допуская линейное
возрастание теплоемкости с температурой. При помощи такого приема
можно получить значения, мало отличающиеся от истинных.
Пример. Теплоемкость окиси кадмия CdO при температуре 25° С (298° К) равна
Ср= 10,33 кал/(моль-град). Температура плавления CdO равна 1375° С (1648° К);
для этой температуры Ср = 2-7,25= 14,5 кал/(моль-град). На основании
имеющихся двух значений теплоемкости получаем:
10,33 = а + |3. 298, 14,5 = а + р. 1648,
откуда а = 9,41 и Р= 3,89- Ю-3, поэтому
Ср= 9,41 + 3,89- 1(Г37\
139
NS
*L
■—ч •-*
га ^
ч-*',—^
-э
га
■cop
1 |
i i
NN
-i >-t
СГ^н4
га и
%_• %_•
?л?л^^2 3 ^ o-~~~~^3 D 3^ h^w со П Mto МП'8 »
3 3 3 3
слслоо
га ^^.
*
н
ra
н
ra
н
ra
н
ra
О Z OO QOO fW£
ra
ra
со тз
CO H
H
CO
w
и-*ч
H
ra
я
fc»
H
ro
•-I
Co
w
>^-'
'h
ra
ra
*
CO
w
о
я
о
тз
о
S
со
Со
W
•-J
Со
W
га
^-чН Я
Ч ГО О
га • н
^ со
^ 5я
Со Я ро Со
oonnon
&OOQ
ТЗьа'
W
CO
W
*
CO
w
CO
w
•-J
Co
w
ro
Co >-j
Co "8*
W Я
^ H
ro -a
ч!^ ГО
Dd>>>>>>
CO ,—v О '—'M ^4/-s
H со w j^ H
•*—v ГО '""^ ^
r5 N^ н ^ ro w
О ГО &3 •
ro
ro
о 4^
J^i73^ tO»—СЛ
^°5ЪЪо4^Ъ
OlOlOl05^»-vlS^v)CDOOSOiCOWvj^OO»-v)0005050500
tO 0> СЛ -^ OO CD >—' ЮСПОО^^Ч^СЛСЛ*-1 CD -^ CD СЛ >—' О-^СЛОО-^
СЛСЛЧЮСО^-^^-^-ЧО СЛ О 00 tO tO CD 00 00 CD О О 00 00 О
OO
tO tO •"""* •—' tO •—* •—*
00 <J> (Л tO^jD ^< О* #* & OO 0Dpi
Ъо~ю~4*.Ъ> -^ о "ел"*-* оэЪо сл~ю сл
OO •—' tO 00 ОЭ CD 4^ О 00 00
•— •—. -ooo>—'oo^tooooooo»—'•—•—oo, ослаэаэаэо^оо to oooo
CT> OO
OO О
tOtOCD4^005-<100CDCDOOCDCDOCncDa>
СЛСЛ00005ЧЬЗО^ОООО^УЮ
CT> СЛ
OO О CD tO tO ►— tO -v] ^T OO CD CD СЛ tO
OO СЛ^ОЧОСП^СО •—' 00 СЛ-<J
OO >-*
0»-WWO f-OO) , СЛ, •—
4^ 05 tO 00 •—» OO»—'O
OCT>-<JOOOO ОЧО
СЛ СЛ
CT>
о
СЛ
о
I II I I I II I I II II II II I I II I I I w I I
00
to
о
ст>
о
00 О
СЛ »—'
4i» CD
сл а>
ст>
to
CD
CD
to
— 00
^ OO
to a>
OO
CD »—
о
OO
to
4^
to
СЛ
tO tO 00 00^— 4^
"сл ^ЪчЪч CD^
OO CT> О CD 4^ СЛ
4i»
о
to
СЛ
00
to
is
О CD CD
КЭ
03
ft)
в
ft)
о
H
a
о
?!
a»
Й
TO
о
О)
о
о
I
О!
Ю Ю Ю tO Oi СЛ
сл to
СЛ
юсл to ►—
СЛ
•— to^^tototototototototooototo»—o»-'
СЛСЛ СЛ СЛСЛСЛСЛ4^
м- tO »—' •— tO Ю tO »—' tO 4^ 00 СЛ
>« >* >* >* >*
СЛ СЛ СЛ СЛ СЛ
to to
CD CD
0O 0O
to
CD
0O
to to
CD -^>
0O 00
■^ 4^
О <l
О OO
4^ 00
О СЛ '
о ■
to to н
00 OOg
00
СЛ
Д CO
totototototototototototooo^
-<J-<J-<J-<JCDCDCDCDCDCD-<JCD Oa^
OOOOOOOOOOOOOOOOOOOOOOOO O^
СЛ.
00*
' KS tO
►o *—* •—* ►—* •—* ►—* Сл **"5
-^ 4^ 0O 0O 0O 00 -vj« J
■^ О О О О СЛК
OOOO О О Я
to*
vJtS
ООЯ
^ •—' ^ 00 00
to to to to to to to to to to to to to to to to to to
■^-^-^ ■<!-^-^-^ CD-4 CD CD-4 CD ^ CD CD-^ CD CD
000000000000000000000000001_-1^OOOOOOCOOO
' ' I I I I I I I I I I i ®S
to
СЛя CD CT> СЛ Oa
Os tOCDOOd
оя о од
to oo to to ►— to
СЛ -^ О О О О
о оо о о о о
о о о о
о •—* •
СЛ СЛ -^ •—* С7^
О О 00 О О
о о о о
to to to to to no to
CD CD CD CD CDa^ ->4
00 СЛ 00 OO 0ОЙ O0
■ I I I I I. I
►s ►-* ►— н
^я^
ooa
H-
о
A
a
о
о
н
a
о ft) X)
ft)
s
я
H
Co
я
03
о
s
s
X
H
5!
•o
p
»
X
<T
X
s
X
H
я
и
о
2
ж
о
о
н
ГР
"я?
я
Существует и другой простой метод определения температурной
зависимости теплоемкости соединения, если известно ее значение при
какой-либо температуре. В этом случае исходят из предположения,
что зависящая от температуры часть теплоемкости аддитивно
складывается из зависящих от температуры частей теплоемкостей
элементов, составляющих данное соединение. Важно только, чтобы
уравнения теплоемкости относились к одинаковым агрегатным состояниям.
Пример. Теплоемкость соединения NiSi при температуре 25° С (298° К) равна
17,38 кал/(моль.град). Теплоемкости составляющих это соединение элементов равны:
Ср = 5,90 + 2,05- 10"3 Т (для Ni),
Ср = 5,70 + 1,02. 10"3 Т — 1,06. 10бТ"2 (для Si).
Комбинируя эти уравнения, получаем для NiSi
Ср= 17,5+ 5,12. 10"3 Т — 1,06. 10бГ-2.
В табл. 4 приведены значения коэффициентов теплоемкостей
ряда веществ. Полные таблицы можно найти в термохимических и
физико-химических справочниках и обзорах. В табл. 4 приводятся
значения коэффициентов а, |3, у и уг уравнений:
Ср = а + РГ + уТ2 или Ср =± а + £Г + ^Т"2.
§ 32. ТЕПЛОТЫ И ЭНТРОПИИ АГРЕГАТНЫХ ПРЕВРАЩЕНИЙ
Величина теплот различных превращений в ряде случаев
оказывает еще более существенное влияние на результаты
термохимических расчетов, чем температурные изменения теплоемкостей.
Существующие методы оценки теплот превращений являются
довольно надежными.
Теплоту испарения вещества можно определить, пользуясь
правилом Пикте—Трутона. Это правило гласит, что энтальпия
испарения, деленная на абсолютную температуру испарения, является
приблизительно одинаковой для всех веществ:
Д5ИСП = -5М- =22 — 25 кал/(моль • град).
* исп
Это правило не оправдывается только для веществ с низкой
температурой кипения, а также в тех случаях, когда имеет место
ассоциация или диссоциация молекулы в газообразном состоянии,
что редко наблюдается для веществ, представляющих интерес для
металлургов.
Энтропия плавления веществ не является такой постоянной
величиной, как энтропия испарения. Однако, согласно правилу Комп-
тона—Ричардсона, энтропия плавления для большинства металлов
почти постоянна. Эта величина приблизительно равна 2,2 кал/(г-
атолсград) и относится только к истинным металлам,
характеризующимся кристаллическими решетками: гранецентрированной и объем-
ноцентрированной кубической и плотноупакованной
гексагональной. Для других типов решеток (например, типа алмаза) или
веществ с низкой температурой плавления (например, для азота и ар-
141
гона) энтропия плавления имеет большие значения. Энтропия
плавления является периодической функцией порядкового номера
элемента в таблице Д. И. Менделеева. Наибольшей энтропией плавления
в каждом периоде обладают элементы подгруппы фтора. Наименьшей
энтропией плавления среди металлов обладают щелочные и
щелочноземельные металлы. В подгруппе щелочных металлов наибольшую
энтропию плавления имеет литий. В подгруппе щелочноземельных
металлов энтропия плавления возрастает от бериллия к кальцию,
а затем уменьшается от кальция к барию. В подгруппе бора
максимум энтропии плавления приходится также на элемент четвертого
периода — галлий. В подгруппе углерода наблюдается уменьшение
энтропии плавления. в ряду кремний—германий—олово—свинец.
Для элементов подгруппы фтора энтропия плавления немного
возрастает с увеличением порядкового номера.
Для многих сплавов, имеющих в твердом состоянии структуру
неупорядоченного твердого раствора, энтропия плавления является
аддитивной и может быть вычислена по энтропиям плавления
металлов, входящих в состав сплава. Обычно значение энтропии плавления
в этом случае 2,2 кал/(г-атом-град). Для упорядоченных сплавов
в качестве приближенного значения можно принять величину
3,5 кал/(г-атом-град).
Труднее предсказать значения энтропии плавления химических
соединений. Можно лишь приближенно их оценить на основании
сравнения энтропии плавления соединений одного и того же
структурного типа.
Расчетные методы оценки теплот плавления химических
соединений непрерывно совершенствуются из-за крайней ограниченности
экспериментальных данных, особенно для химически активных,
тугоплавких и диссоциирующих при плавлении соединений.
Гипотеза о независимости плавления элементов в соединении или
аддитивность теплоты плавления соединения, как правило, не
подтверждаются экспериментом. Обычно расчет основывают на гипотезе
аддитивности энтропии плавления соединения:
АНА в = ASA ВТА в (320)
ASAmBn = mASA+nASBy (321)
где ASA и ASB — энтропии плавления элементов А и В
соответственно;
тип — стехиометрические коэффициенты в химической
формуле соединения АтВп.
В ряде случаев учитывают разупорядочение структуры при
плавлении:
&$лтвп = т ASA + п AS в + ф, (322)
где г|? — поправка, учитывающая разупорядочение структуры при
плавлении.
142
Значение г|) рассчитывается по формуле
ib = —kR(m + n)[ —— In —; — In —г— ) =
Y v ' } \m-\-ti mJrn[tnJrn m-\-nj
= 4,573£(m + n)(-2J-lg-4- + -4-lg—%-), (323)
где k — поправочный коэффициент, учитывающий степень разупо-
рядочения структуры при плавлении, и R — универсальная газовая
постоянная. В случае соединений, полностью сохраняющих при
плавлении свою структуру ближнего порядка (например, при
плавлении по типу «полупроводник—полупроводник»), имеем k^Q. В
случае радикального изменения структуры ближнего порядка
(например, при плавлении по типу «полупроводник—металл») имеем k ^ 1.
В реально наблюдаемых случаях этот коэффициент принимает
промежуточные значения.
Пример. Неорганические соединения InSb и AlSb плавятся с уменьшением
объема по типу «полупроводник—металл» (примем k~ 1); соединение Bi2Te3 —
с увеличением объема по типу «полупроводник—полупроводник» (примем k = 0).
В табл. 5 сравниваются расчетные и экспериментальные данные.
Можно видеть, что правдоподобность расчетов возрастает при
переходе от правил аддитивности теплот плавления к правилу
аддитивности энтропии плавления. Кроме того, введение поправки г|) явно
свидетельствует о сближении расчетных и эмпирических результатов.
Таблица 5. Сравнение результатов экспериментального АЯЭКСП и расчетного
(по правилу аддитивности) определения теплот плавления &Н
Соединение типа
Атвп
InSb
AlSb
Bi2Te3
Температура
плавления. °К
ТА
430
933
544
тв ,
904
904
723
*ЧА
798
•i
1353
859
Энтропия
плавления,
кал
(г-атом-град)
ASA
1,80
2,68
4,80
АЗВ
5,25
5,25
5,80
Поправка
Ф
2,76
2,76
0
Теплота плавления
А<дд
5,53
7,25
17,7
А"адд
5,60
10,6
23,2
АН
7,90
14,5
23,2
, кал/моль
ЛЯэксп
9,00 +
+ 0,60
19,9 +
+3,3
28,3 ±
+0,70
Поскольку используемые в расчете величины энтропии плавления
относятся к различным температурам, дальнейшее
совершенствование расчета возможно путем учета температурных зависимостей эн-
ропий плавления соединения и составляющих его элементов.
143
§ 33. СТАНДАРТНЫЕ ЗНАЧЕНИЯ ЭНТРОПИИ
Стандартные значения энтропии всех элементов, представляющих
интерес для металлургии, известны более или менее точно. Интересно
отметить, что энтропии простых веществ и ионов элементов, как
это было показано В. А. Киреевым и А. Ф. Капустинским, проявляют
периодичность в зависимости от порядкового номера.
Данные об энтропиях сплавов и интерметаллических
соединений в твердом состоянии очень немногочисленны. Поэтому для
приближенной оценки энтропии упорядоченных сплавов и соединений
можно пользоваться суммированием энтропии составляющих их
элементов. Для неупорядоченных сплавов производят такое же
вычисление, но в полученный результат вносят поправку, наибольшее
значение которой не превышает 1,4 кал/(г-атом-град).
Метод суммирования энтропии нельзя применять к
неорганическим соединениям. Для них был предложен ряд эмпирических
формул, которые позволяют производить расчеты для галогенидов,
окислов и сульфидов металлов.
Энтропия силикатов приблизительно равна сумме энтропии
составляющих окислов. Это относится также к другим двойным
окислам, например шпинелям. Более сложные закономерности были
установлены для карбидов (Кубашевский и Эванс).
Величины энтропии веществ, находящихся в газообразном
состоянии, подчиняются следующим закономерностям. Для
одноатомных газов с атомной массой выше 45 среднее значение стандартной
энтропии равно 41 кал!(моль • град)\ для газов с атомными массами от 4
до 45 величина стандартной энтропии составляет 30—
40 кал/(моль-град). Значения стандартных энтропии двухатомных
газов, имеющих молекулярную массу (М) в пределах от 20 до 300,
могут быть найдены по эмпирическому уравнению:
S298 = 52,7 + 0,043М — 240 ЛГ1.
Стандартные энтропии многих других газообразных соединений
выражаются уравнением:
S°m = 39,0 + 0,34М — 6,2-10~4 М2.
Приведенные уравнения позволяют производить подсчеты,
среднее отклонение которых составляет ±2 кал/(моль-град).
Стандартные энтропии веществ, находящихся при комнатных
температурах в жидком состоянии, могут быть успешно вычислены
путем прибавления энтропии плавления к стандартной энтропии
веществ в твердом состоянии или вычитания энтропии испарения из
энтропии вещества в газообразном состоянии.
Предлагались также приближенные формулы для оценки
молекулярных абсолютных стандартных энтропии соединений. Например,
для некоторых соединений хорошие результаты дает формула:
Sm - 9,15 (4,573 lg-^ + lg-^-) + 8,2.
144
Здесь М — молекулярная масса соединения;
Тпл — температура плавления соединения, °К;
р — плотность соединения, г! см3.
§ 34. ТЕПЛОТЫ ОБРАЗОВАНИЯ
Значение константы равновесия термохимическим методом
определяется через известные значения теплот образования соединений
и изменения энтропии. Мы уже рассмотрели методы оценки величины
энтропии, а что касается оценки величины теплот образования, то,
к сожалению, методы экспериментального изучения теплот
образования не очень надежны и данные имеются лишь для ограниченного
числа соединений; поэтому для их оценки желательно использовать
все возможные в каждом конкретном случае методы.
В стандартном состоянии энтальпия простых веществ условно
принимается равной нулю. Кроме того, поскольку теплоты
образования соединений мало изменяются с температурой, часто делается
допущение, что эти величины являются постоянными при всех
температурах, если только не происходит никаких изменений в агрегатном
состоянии веществ.
Требующая решения задача значительно облегчается, если
элементы, входящие в состав соединения, образуют несколько
соединений. Тогда можно воспользоваться закономерным изменением
теплоты образования при переходе от одного соединения к другому.
Так, например, если известна теплота образования одного
соединения, то теплота образования другого соединения может быть
получена из предположения, что она аддитивна, считая при этом
соединение состоящим из соединения с известной теплотой образования и
свободного элемента. Чаще всего получаемые таким образом
значения оказываются преуменьшенными. Однако использовать такой
прием представляется возможным далеко не всегда.
Пример. Натрий и сурьма образуют два соединения: Na3Sb и NaSb. Известно,
что для первого соединения теплота образования равна 47,2 ккал(моль\ тогда,
согласно Na3Sb + 2Sb = 3NaSb + А#298» получаем:
^298 ~ —^— ~ 1^,7 ккал/моль.
Существуют также определенные зависимости между теплотой
образования соединений металлов, имеющих одинаковый стехио-
метрический состав и одноименный анион, и логарифмом
порядкового номера катиона в Периодической системе Д. И. Менделеева.
Существующие закономерности позволяют оценивать теплоты
образования путем интерполирования и экстраполирования по
Периодической системе. Этот очень удобный метод, получивший название
правила термохимической логарифмики (рис. 34), был разработан
А. Ф. Капустинским.
М. X. Карапетьянцем было показано, что между различными
физико-химическими свойствами существует линейная зависимость.
Хотя строгого физико-химического обоснования этому факту в на-
Ш А. Н. Крестовников 145
стоящее время нет, справедливость такой гипотезы получила
подтверждение на большом экспериментальном материале. Практически
поступают следующим образом. По осям координат откладывают
экспериментальные
данные. Так как эти данные
определены с некоторой
погрешностью, прямая
линия оказывается
сглаженным выражением
зависимости; по ней-то и
производят интерполяцию или
экстраполяцию для
определения неизвестных
величин. Применение
описанного приема,
получившего название метода
сравнительного расчета,
тем надежнее, чем
однотипнее подобраны к
рассмотрению изученные и
изучаемые соединения.
Несомненно, заманчивым
является использование метода сравнительного расчета с переходом
к рассмотрению сразу нескольких свойств однотипных соединений.
Кроме того, значение метода сравнительного расчета для предсказа-
0,6 1,0 1,2 /,4 1,6 1,6
Логарифп поря д.код ого номера катиона
го
Рис. 34. Иллюстрация правила
термохимической логарифмики на примере кислородных
соединений и хлоридов элементов III группы
периодической системы элементов Менделеева
1
t
.1
§
1-
1^
*/#?
$700
&
I
50
'
-«с
<sfe
Пп^,
V
1
1
i
Ti
300
250
6
150 200 250 300 350 U00 200 250 300
Теплота образования[-АНМе 0 (а) и - ДНнео2 (б)3ккал/ноль]
Рис. 35. Иллюстрация применения метода сравнительного расчета для
определения теплот образования трихлорида \ ванадия (а) и тетрахлорида
гафния (б)
ния свойств соединений не уменьшается, если корреляционная
зависимость окажется криволинейной.
Примером использования метода сравнительного расчета может
служить определение теплот образования трихлорида ванадия и
тетрахлорида гафния (рис. 35). На^рис. 36 и 37 приведены другие
146
зависимости, которые могут быть использованы для сравнительных
расчетов.
В качестве эмпирической закономерности для определения
приближенных значений теплот образования можно использовать
зависимость теплоты образования от изменения объема при образовании
соединения, рассматривая эту зависимость в пределах соединения
данного структурного типа (рис. 36).
I
S
I
t
I
1
80
70
60
50
ЬО
30
20
10
О
AJCo
УрЬТе
ПС
Dh Qo
11 Dog
,
No СП
o/kL
SrSeo/
KF°>
°/°Cc
oVN
W
BaoJ
im
lr
■
W 20 30 40 50
Изменение обьепа,9/*
60
I
1
I
1
t
1
80
70
60
50
40
30
20
70
0
Ca1
/
/
Fe
Co?
pi
7
/tig
Attn
/zn
Cr
PSr
6Ca
i
/234
Нормальный потенциал^
Рис. 36. Связь теплового эффекта и относи- Рис. 37. Связь теплот образования
тельного изменения объема при образовании и нормальных потенциалов для
соединении структурного типа NaCl хлоридов МеС12 в водном растворе
Наблюдается также приближенно линейная зависимость между
теплотой образования соединения Д# и нормальным потенциалом
металла е0, входящего в состав этого соединения. Линейность более
строго выполняется для теплот образования соединений,
находящихся в растворе (рис. 37). При этом значение коэффициента k =
== dkHlds0 для солей различных кислот меняется в пределах от
25,5 до 17,8 и в среднем равно 21,2.
При использовании только одного из методов оценки нельзя
быть уверенным в правильности полученных данных. Следует для
определения одной и той же величины применять несколько методов,
и тогда путем сравнения найденных значений можно судить об их
надежности, причем различие величин не должно превышать 5—
10 ккал/моль.
10*
§ 35. ПРИБЛИЖЕННЫЕ И УСКОРЕННЫЕ РАСЧЕТЫ
КОНСТАНТ РАВНОВЕСИЯ
Уравнения (310) и (311) позволяют из термодинамических данных
вычислять изменение изобарного потенциала реакции, а
следовательно, и константы равновесия. Это имеет большое практическое
значение, так как экспериментальное изучение равновесий в
лаборатории длительно и требует значительных затрат. Кроме того, в ряде
случаев рассчитанные по термохимическим данным константы
равновесия более достоверны, чем экспериментальные, в силу целого ряда
причин.
Наибольшее значение имеет расчет равновесий в тех случаях,
где необходимо оценить возможность технического использования
какой-либо представляющей интерес, но мало изученной реакции.
Если хотя бы приблизительно можно предсказать зависимость
положения равновесия от внешних условий, то все равно расчет даст
указания на перспективы практического осуществления реакции.
Однако и для хорошо исследованных и применяемых в технике
реакций расчеты могут оказаться полезными для решения вопросов
усовершенствования производства. В зависимости от характера этих
практических вопросов в используемые методы расчета могут быть
внесены изменения, упрощающие и уточняющие расчеты.
Например, поскольку выше стандартной температуры (25° С)
теплоемкости изменяются с температурой значительно медленнее и
по более плавным кривым, чем в области низких температур, можно
получить точные результаты с помощью приблизительных сведений
о теплоемкостях. На этом основаны заслуживающие особого
внимания методы расчета, разработанные Улихом (1939 г.).
Приближения Улиха. Преобразованиям подвергается
уравнение (310):
т т
AG°T = АЯ2°98 - Т AS°29S + j ACP dT-T^^dT.
298 298
Теплоемкости, помещенные под знаком интеграла, зависят от
температуры, и это затрудняет использование уравнения (310) для
расчетов. Было предложено делать два возможных
последовательных приближения. Первое приближение во многих случаях дает
достаточно хорошую характеристику реакции при различных
температурах выше стандартной. Оно заключается в том, что АСР = 0,
т. е. реакция считается протекающей таким образом, что суммарная
теплоемкость системы не изменяется. Тогда уравнение (270)
принимает вид:
AG°T = AH°29s-TASl98. (324)
Второе приближение используется, когда нет возможности
воспользоваться температурной зависимостью теплоемкости. В этом
случае принимают АСР = а, где а выбирается на основании при-
148
близительной оценки теплоемкостей при комнатной температуре.
Тогда уравнение (310) преобразуется следующим образом:
AG"T = АНт — Т ASm -г а (Т — 298) - аТ In -^,
AG°T = АН°298 - Т ASm + аТ (1 - Щ- - In -^-) ,
или
AG г = АН;9& - Т AS°29S - дГ (In -^ + -^ - 1) . (325)
Обычно это уравнение записывают так:
AGt = AHm - Т ASlm - aTf (T), (326)
где
f(T) = \n-^ + ^—l. (327)
Значение функции / (Т) легко найти с помощью специальных
таблиц.
Расчет по Темкину — Шварцману. Удобный прием для вычислений
предложили М. И. Темкин и Л. А. Шварцман. Если АСР задано
степенным рядом
АСР = Да + Apr + АуТ\ (328)
то
АСт = АЯ2°98 - Т AS°2m - Т [Д а/0 (Т) + Щх (Т) + Ayf2 (T)], (329)
где функции /0 (Г), /х (Г) и /2 (Т) определяются для любой
температуры с помощью таблиц, составленных авторами. Приняты две
системы обозначения этих функций (и обе используются в равной
мере):
fo{T) = M0 = \n^ + ^-l (330)
и
f (Т) _ м _ тп 1 Я*п+1 298^ mn
Гп(1)-Мп- п{п+{) + {п+х)Т — • V6i)
(при п =£ 0).
Если уравнения теплоемкостей задаются наряду с уравнением
типа (328) степенным рядом
АСР = Да + Дрг + АугТ~\
то получаем
AG°T =. Д#2°98 - Т ASm - Т [Да/Ь (Т) + ДРД (Г) +
+ Дт/2(Л + ДТ1/-2(Г)1. (332)
149
<^4^4^4^4^СОСО(^СОЮ^ЮЮ^^^^ООООСОСОСОСОООООООС>Э^^^^С^
ОЧ^ЮОЧС^ЮОЧСЛЮОЧОТЮОЧСЛЮОЧСЛ^ООЧСЛЮОЧСЛЮОЧСЛЮОЧСЛЮОЧСЛЮОЧСЛЮОСОЧСЛО
OCOOWOCOOCOOCOOCOOCOOWOCOOCOOCOOCOOCOOCOOCOOCOOCOOCOOCOOCOOCOOW
05 05
о
о о
о
05 05 05 05 0 05
05
05
05
05
05
05
05
05
05
05
05
05
05
05 05
to
ЬОЮ^^^^ООООСОСОСОСООООООООО^^^^О^О^О^О^СЛСЛСЛОТ4^4^4^4^СОСОС^
ЮОЧС1ЮОЧС^ЮОЧС^ЮОЧСЛЮОЧСЛЮОЧ|СЛЬЭОЧСЛЮОЧСЛЬЭОЧСЛЮО^СЛЮОЧСЛЬЭО
050050050050050050 05 0050050050050050050050050050050050050050050050050
-j ел to to to ^i
05 О 05 СЛ О СО СО
ОО
4^
ОО
4^
ОО
4^
ОО
4^
ОО
4*
ОО
4^.
ОО
4^
ОО
4^
ОО
4^
ОО
4*>
ОО
4^
ОО
4^
ОО
4^
ОО
4*>
00
4^
СО
4^
ОО
4^
ОО
4*.
ОО
4*.
ОО
4^
ОО
4^
ОО
4^
ОО
4^
ОО
4^
00
4^
05 05
OOOOOOOOOOOOOOOOOOOOb-H'MMh-MMh-Mh-MMh-Mh-MH-MWh-bjbJtotowwtOWWWW^Oi
050505-^'<!-^I*^-^'^I,<IOOOOOOOOOOOOCOCOCOC£50000
О5-^00О>— ГО4^Сла500О*—С0СЛ05С0О00СЛ^ОК?СЛ00
0^00С0Ю4^СООСЛС0СЛО^С0ЮС0ОС0^-1О^О--^К>С0
a^ooa^a^wto-^-^to4^oo50o4^05Co^-oo4^4^oai05to
-Ч! *— 05 О) СЛ 4^ "Ч СО СЛ О
H-^b3WWWW4^^0l0505SlOOCDO^-tOWOiaia)OCOC00500
4^Чн-слсОООООЮООСООСТ|4^^-ь-о^-Ю050Ч01С£)а)СЛСТ)00
Cn05^0WWtO(»ai(»^05^(»^OWtOWOCON^WWOOO
СОСЛООО^СОООСЛСЛСЛ^^^10СЛ04^К)^-000104^СОСОСООО
ООООООООООООООООООООООООООООООООООООООООООООООООООООО
00 -^ -^ -<| ■<! ■<!
^- СО 00 *Ч СЛ 4^
4^ СО -vj tO СО 4^
►—• СО СЛ СЛ СЛ 4^
^^^О^С7>05а^а^а^СЛСЛСЛСЛСЛСЛ4^4^4^4^4^СОСОСОСОСОСОЮ^ЮЮ^^
<^^ОС>Э^СЛ4^ЮОСО^СЛ4^^000^СЛСО^СО^СЛСО^О^СЛСО^СО^СлСО^СО^СЛС^
к-сл^-<^^—сп^-4^со^-а^сх>слэслсх>с£>юооа^а^оо^сооооосо^ооспа^оо4^^
^СЛСОС>ЭСОЬОООО^СЛа^04^00<^СОЬО^ОСЛ^^СлОа^4^СОСОа^Ю^ЬО^СОСО
СЛ СЛ СЛ СО
ООООООООО О О О О О О О О <0>^^^p>^^^^^^p>^^p>^C^CDCDCDCDCDCDCD О О О О О О О О О О О О
^^^^^^^ЪоЪоЪо^ЪэЪоЪэЪэЪоТо'ьо'ю ьоюююЬю^"^"^^^"^ооооооЬоооооооЬЬооо
ООО^СЛ4^СОЮОСОС»^С7>4^СОЮ^ОСО^а^СЛ4^СОЬО^ООО^а^СЛ4^СОЮ^ОСОО^
^-00^4^000СО^ООЭ1ЮСОООа^ОЯСОЮСОС005а^4^(^^-^-СОСО
4^05^050500(XOOWCflOOCOWO^OW(»COCOWK-005WW^^^OWCOW^W(XC0005^4^^
4^- ОО
ооооооооооооооооо оооо оооо оооооооооооооооо о ооо оо о о о о о о
"со "со со "to "to "to То "to "to "to to "to ~to~^-'*>—~>—~н—~н—'L- ^"к-%-">—"к-"н-"о о"о о о о о о о о "о "о "о "о "о "о "о "о "о "о "о "о "о "о "о "о "о "о
СОЮ^СООО^О>СЛ4^СОЮ^ОСООО^О^СЛ4^СОСОЮ^ООСООО^^а^а^Сл4^4^СОС^
05CO^COOOO^<^4^4^tOCOtOtOtOCOW^^^C^^-tO^^OtOa^COb-(^
ЮЮь-С*305ЮСОООО)О^ЮСЛЧЧЮЧЧЧ^СЯСОЮ4^Ч4^Юа»С^»- CDOOhOOi^WvlOOCDCncDCOCO^tOCOOOCOCOCO
О О О CD
ОО
оооооооооооооооооо оо о о ооо о ооо оооооооооооо о ооо оооооооо
"со "со "w "со "со "со "со "со "со ЪоЪо "со "w^
05СЛСПСЛ4^4^4^СОСОЮЬО^^^ООСОСОСЮ00^^05СЛСЛ4^СОЮ
►—, ^(^^-004^^-^4^COa^^-^tOOOCOOOCOOOt00004^^tOCO^OO^-t04^4^^4^4^COtOCD^4^
00004^4^^СЛСЛОСОЮО^О^СО^СЛООС04^4^(^05СООО^-СО^-ООСОСОЮСОСОСЛО
СЛ
04WN30^0itOO^a\t0040lN30SOit004CntOONUit004CriN304ait004aiK)
О СО OjJO OWOWOWOOJOOJOOOOWOWOOOOOJOCO OJJ0 О CO О GO О^СО О СО О СО О СО
tOtOtOtOtOtOtOtOtOtO^-^->— ^->—' ^-4h— к-^-к-^-.— к-к-н— ^-b-^h— h— к-к-к-h— н-н— и- н- н- н- н-
^^н-н-н->^ООООсОЮЮЮОО(Х(^(»ЧЧЧЧ050)0505С^СД№СЛ^^^^МУООО:ЮЮ
юоч^юочслюочслюочсдюочсдьоочслюочслюо^слюочслюочсл
р op op op о 05 о 05 op op Q-рз OCTiooiOOiOOiOW op о Ст) op op op op о
Оо Ъо Ъо Ъо Ъо Ъо Ъо Ъо Ъо Ъо Ъо Ъо Ъо оо оо оо оо Ъо Ъо Ъо
ооооооооо ооооооооооо оо о ооооооооооооооооо
^^^~Ф^~^~Ф^~^~Ф^^~^"^~^~Ф^~^ СД СП СлЪлЪлЪл СЛ ОЗ Оз Оз Оз ОЗ ОЗ
OOOb-H-^WWWW^h^(^CT)05440000CDOOH-^-toC^4^4^CnCT)4CO(»CDOb-NDW4i.Cn
О^СЮЮО^ь-^О^СС^Ю^ОСаь-оЮЧ^ООю^О^С^ОООФОО^ОООЧФО^СлС^ОтО
owH-vjo54^i^^vjcD^cow^^o^wcowoooQo^ocoaiwoco^oooaiOooo5^ai
О Oi О) ^-ОЗ О CO-<J ОО to Ф- ^-СЛ Oi to О CO ОЗ О ОО О О to ОО ОЗ Ф-О Оз СО СО О ■<! Оз ОО О Оз Оз СО
~J~J"~Г-Г-Г*Г"^Г-Г*Г-J-Г-Г*Г-Г^РГ^Г^Г-Г-Г-Г-Г-Г^РРРРР Р Р Р.° РР о о о о
^^^--о^^^^^ГП^^^^Й000 о о о оЪ~со~со~с©Ъ~со~со~с©~соЪоЪоЪоЪоЪоЪо
£5£^^^Рч^№Г^ОТ£л4*'со^>Г0^00"^СЛСЛ^СОн- ОСООО-^ОЗСЛ4^КЗн-ОСр^СГЗСЛ^Ю
ОЗОзООООООь-,— co~WCO^CO^COOCOCOC©tOO^O>C00303COCoX*0^0
О'сон-соою^слоозооюослсл^ оооослоЪ^ооооо^юслосоюю^ооюсл-Зо^
w3 *^4 "-vj ю q-j
ооооооооооо о оо оооооооооооооооооооооооооо
СО СО СО СО СО СО ОоЪоЪоЪоЪоЪоЪоЪо^^^^^^^^ЪзЪзЪз Ъэ Ъэ Ъэ Ъэ Ъэ Ъэ Ъч ~СЛ Ъч Ъч ~СД Ъч Ъч ~СП ~4ь.
СП СЛ Ф- СО to О СО'00 ЧСЛ^СОЮОСОООЧСЛ4^00Юь- СООО-^ОзСЛСОЮ^-000-<103СЛ»4^Ю>—'ОсО
со оз ел»— о-^елю^-оооэсоюоо^Ф-сосоооелФ-'— соозелю^-^озсоюсоооелсоосоозелю
05^^СОСлЗЮСС05ь-ОЧ4^0С£|О^^ОЮО^№ОЧС1ЮЬоОСО(^СЛ^К)ООСОСОа1Ч0505
^-о о о о о о о о о о о о о о ооооооооооооооооооооооооо
о со со со со оо оо ооЪо оо оо -^Vj -v] ■^"•^"оз'оз ЪэЪэ as as ЪлЪч'сл "ел "ел "ел Ъг^^~4^~4^~Ф-^~Ф-ЪоЪоЪо"со
о^елсо^-со^ел4^юоооозФ-со>—i со-^озф-юосо-^озф-со^-ооо-^слф-ю^-ооо-^озф-
ооосо^со^сооо^-оососоо^о^о^-^о^оюа^оосоаз^-елОФ-о^^-елю^ь^о^со'—оо
004s.03-^ltOOO-Nl-^^-03tOCOtO-^'— ОООн-сон-ОСООООЗСООЧ^СЛС^ООЗ^^Ч"-1 -^ ОЗ -^ О
оо о оооо о о о о о оо оооооооооооооооооооооооооо
ф^ь^^^ф^^^й^Ф-Ф-Ф- Ф-"ф- ►£*■ Ф-"^"^^^"^^"4^"ф-"ф-1^"со"со"со"^
COCOCOCOCOCOtOtNOtOtOtOtOtO^-^-^-^-^-^-ОООООСОСОСОСОСООООООООО^^^^О^О^Оз
а^елФ-ю^-ооо^а^Ф-со^-ооо^елФ-юооо^елсо^-со^елсо^-со^^юсо^Ф-юсо^с^
WOOa5^OC0^O05WSWSC*5C3OWC0^N3WSO5000500WWW^05^C003C0W^O
о
/N
о
о
Се
о
I
со
о
I
га
о
I3
о
съ
о
S1
Оъ
/2 (T) и /_2 (Т) определяются из специально составленных таблиц,
содержащих эти значения для температур от 0 до 2000° С через
каждые 50° С (промежуточные значения находятся из той же таблицы
простым интерполированием).
В рассмотренном приеме расчеты равновесия облегчены за счет
предварительного табулирования всех температурных функций,
входящих в уравнение для определения логарифма константы
равновесия химической реакции.
Таблица температурных функций впервые была составлена
М. И. Темкиным и Л. А. Шварцманом. Затем она была
воспроизведена в целом ряде монографий и руководств по физической химии и
химической термодинамике.
Выше приводится табл. 6, дополненная Л. П. Владимировым:
искомые величины определены не только в функции от температуры
по шкале Цельсия, но и в функции от температуры по шкале
Кельвина, в силу чего интервалы между найденными значениями (ранее
50°) сузились до 25°, а также расширены пределы температуры:
от 200 до 2500 °К (вместо прежнего интервала от 0 до 2000° С).
Пример. Требуется определить значение константы равновесия реакции
СН4 + 202 == С02 + 2Н20 при температуре 1200° К.
Из справочных таблиц находим значения тепловых эффектов образования
участников реакции:
для метана
о
Д#298 = — 17 889 кал/моль,
для двуокиси углерода
Д#298 — — 94 030 кал/моль,
для паров воды
Д#90я = — 57 800 кал/моль,
298
после чего определяем значение А#298 Для реакции:
ДЯ298 = + 17 889—94 030 —2-57 800 = — 191741 кал.
Затем по таблицам находим абсолютные значения энтропии для всех участников
реакции:
для метана
о
S298 = 44,46 кал/(моль-град);
для кислорода
S298 = 49,00 кал/{моль-град)\
для двуокиси углерода
S298 = 51,09 кал/(моль-град);
для паров воды
S298 — 44,80 кал/(моль-град),
откуда изменение энтропии в результате реакции составит:
Д5298 == — 44,46 — 2-49,00 + 51,09 + 2-44,80 = —1,77 кал/град.
152
Поскольку известны температурные зависимости теплоемкостеи участников
реакции:
для СН4
Ср = 3,422 + 17,845- 10"3Г — 4,165-10-67"2
для 02
Ср = 8,643 + 0,202- 10"3Г — 1,03- ЮбГ-2,
ДЛЯ СО 2
Ср == 10,34 + 2,74- 10~3Т — 1,955.105Т"2,
для Н20
Ср = 7,219 + 2,374- 10"2Г — 0,267- KPT'*,
то можно рассмотреть численные значения коэффициентов теплоемкостеи:
Да = —3,422 — 2- 8,643 + 10,34 + 2-7,219 = +4,07,
ДР= (—17,845 — 2.0,202+2,740+2-2,374) 10"3 = —10,761- КГ3,
ДТ= +4,165. 10" 6
д?1 = (+2,06 — 1,955 — 2- 0,267). 105 = —0,429- 105.
Далее, так как температурных функций для Т — 1200° К в таблицах Темкина—
Шварцмана не содержится, находим их значения для двух ближайших температур:
Т= 1223,16° К (950° С) Т= 1173,16° К (900° С)
ЦТ 0,81756-Ю-з 0,85240-Ю-з
М0 . . ' 0,6552 0,6240
Мх 0,3498-Ю"3 0,3263.10»
М2 0,212Ы06 0,1925-10е
М_2 0,3216-Ю"5 0,3129-Ю-б
Полученные данные подставляем в уравнение (332), представляя его в следующем
виде:
1пКр =
AGT ЛЯ°98 AS°m
RT RT R
•--L [Да/0 (Т) + ДРД (Г) + Ду/2 (Т) + AYl/_2 (T)],
откуда
-0,81756-10-8 1,77 4,07-0,6552
lg К™ = 4^73 191,741 + 1Щ- 4^73~ +
, 10,761-0,3498 4,165-0,2121 , 0,429-0,3216 , „.„„,„
+ 4^73 4^73 + 4,573 = + 34,°342
и аналогично получаем:
lg#ii7s = + 35,50513.
Затем интерполяцией (если на АТ = 50° приходится Alg К = 1,47081, то для
AT = 27° имеем Alg/C = 0,794237) находим значение:
ig ^1200 = 34,710893,
Ki20o= Рсо*Рн>° = 0,5140-10»*
Рсн/о,
Л. П. Владимиров предложил два приема простого и быстрого
расчета химических равновесий. Первый из них автор назвал
приближенным, второй — практически точным.
153
Прием приближенного расчета по Л. П. Владимирову.
Уравнение изобары химической реакции, связывающее значение константы
равновесия с изменением энтальпии в результате реакции
d In Кр АН°Т
df ="етг>
после интегрирования дает
АН°Т
In Кр = -gjr- + const'
или
АН°Т
№=-~г +const. (335)
В то же время объединение первого и второго начала
термодинамики, получившее название уравнения Гиббса—Гельмгольца (306):
AG0T=zAH0T — TkS°T,
совместно с уравнением нормального сродства
AGr= —RT In KP
дает выражение
или
АН°Т AS°T
In Кр = -^р I ^~-
АН°Т AS°T
1§^-4,573Г+ 4,573 * <336)
Составление уравнений (335) и (336) раскрывает сущность
константы интегрирования в уравнении (335):
const = 0,21858 А5°г.
Если теперь принять, что вследствие незначительности изменения
величин теплоемкостеи с изменением температуры в результате
реакции не происходит изменения теплоемкости, т. е. АСР = 0,
то получаем приближенное уравнение:
IgKp = - 0,21858 (^р - Ь8ж) . (337)
Следовательно, для любой реакции имеет место простое выражение
\gKp = ~ + N, (338)
где М и N — численные функции от А#298 и A^gs, a именно:
М = — 0,21858 АЯ298, (339)
N= + 0,21858 AS298. (340)
154
Числовые значения этих функций для наиболее употребительных
газообразных, жидких и твердых веществ содержатся в таблицах
Л. П. Владимирова. Ниже приводится выдержка из этих таблиц
(табл. 7).
Таблица 7. Значения функций М и N к приближенному расчету
равновесий по Л. П. Владимирову
Вещество
и состояние
Н2 (газ)
Н (газ)
Не (газ)
N2 (газ)
02 (газ)
03 (газ)
S2 (газ)
S6 (газ)
S8 (газ)
Н20 (газ)
N20 (газ)
N0 (газ)
N02 (газ)
N204 (газ)
N205 (газ)
СО (газ)
С02 (газ)
СН4 (газ)
Вг2 (ж.)
Hg (ж.)
Н20 (ж.)
Н202 (ж.)
СС14 (ж.)
РС13 (ж.)
SiCl4 (ж.)
Ag (тв.)
А1 (тв.)
Значение
М
0
— 11322,444
0
0
0
—7541,010
—6382,536
—6072,152
—5921,332
+ 12633,218
—4295,097
—4721,328
—1739,897
—488,619
—131,148
+5774,009
+20557,886
+ 3910,178
0
0
+ 14932,730
+9777,645
+5545,375
+ 16590,222
+32784,000
0
0
функции
N
0
+ 5,1563
0
0
0
—3,6153
+8,5618
+ 10,1596
+ 10,5443
—2,3194
—3,8667
—0,6492
—3,1519
—15,5301
— 18,9334
+4,6861
+0,1524
—4,2142
0
0
—8,5204
—12,1950
—12,2033
—8,3891
— 11,7465
0
0
Вещество
и состояние
В (тв.)
Bi (тв.)
С, алмаз
(тв.)
С, графит
(тв.)
S, ромб.
(тв.)
S, монокл.
(тв.)
Sb (тв.)
Zn (тв.)
Zr (тв.)
СиО (тв.)
Си20 (тв.)
FeO (тв.)
Fe203 (тв.)
Fe304 (тв.)
MnS (тв.)
MoS3 (тв.)
CaS04 (тв.)
BiCl3 (тв.)
NaCl (тв.)
KF (тв.)
Na2Si03 (тв.)
ВаС03 (тв.)
Значение
М
0
0
—99,017
0
0
+ 16,374
0
0
0
+8415,330
+9289,650
+ 14054,694
+42623,100
+58339,002
+ 10273,260
+ 13438,298
+ 73333,590
+ 19803,348
+ 21492,971
+29401,196
—81136,896
+83606,786
функции
N
0
0
—0,1696
0
0
+0,350
0
0
0
—4,8245
—8,8998
—3,8475
— 14,2088
— 18,0339
+0,7585
—3,0142
— 19,6888
—10,4438
—4,7126
—5,1782
—16,4383
—12,7356
Для определения AM и A<V изучаемой реакции из
вспомогательной таблицы выписываются значения М и N для всех участников
реакции, которые затем алгебраически суммируются. Для конечных
продуктов реакции значения М и N берутся с тем знаком, который
приводится в таблице. Для исходных продуктов реакции М и N
берутся со знаком, противоположным тому, который приводится
в таблице.
Если в реакции участвуют по две, три и более молекулы
реагирующих веществ, значения М и N соответственно удваиваются,
утраиваются и т. д. В общем случае для химической реакции
ЬВ + dD = gG + rR + Q
определение ДМ и AAf производится следующим образом:
ДМ = gMQ + rMz — ЬМВ — dMD
155
и
AN = gNa + rNA — bNB — dND.
Пример. Требуется определить значение константы равновесия химической
реакции СН4 + С02 = 2СО + 2Н2 ПРИ температуре 1000° К-
Выписываем из табл. 7 значения М и N для всех участников реакции (кроме
простых веществ) и суммируем их:
для- метана М = +3910,178 и N = —4,2142;
для окиси углерода М = +20557,886 и N = +0,15235;
для окиси углерода 2М = +11548,018 и 2N = +9,37228.
Далее определяем AM и AN:
AM = 2М (СО) — М (СН4) — М (СОа) = 11548,018 —
— 3910,178 — 20557,886 = —12920,0,46;
AN = 2N (СО) — N (СН4) — W (С02) =
= 9,37228+ 4,2142 —0,15235= +13,43413.
Умножаем AM на \/Т и складываем с AAf:
lg кр = —12920,046. 0,001 + 13,43413 = +0,5141
и получаем:
Кр = рсо/?н2 = 3 26?
РснАРсо2
Прием практически точного расчета по Л. П. Владимирову.
Особенность этого приема заключается в том, что необходимые
для вычисления функции заранее вычисляются для простейших
соединений, образующихся из элементов. Это дает возможность
вычислять логарифм констант равновесия любых сложных
реакций путем алгебраического сложения соответствующих каждому
реагенту заранее вычисленных функций.
Такой прием основан на следующих соображениях. Логарифм
константы равновесия любой сложной газовой реакции может быть
представлен алгебраической суммой логарифмов парциальных
давлений всех участников реакции. Вместе с тем алгебраическая сумма
логарифмов парциальных давлений реагентов представляет собой
алгебраическую сумму логарифмов констант равновесия реакций
образования из элементов всех этих участников данной реакции.
Следовательно, вычисление логарифмов констант равновесия
сложных газовых реакций может быть практически сведено к
алгебраическому суммированию логарифмов констант равновесия реакций
образования из элементов всех участников реакции. Это приводит
к целесообразности заранее табулировать величины АЯ, AS и АСР.
Расчет значений логарифмов констант равновесия газовых
реакций описываемым методом производят по вытекающей из точной
формулы
т т
ACpdT
AGr = A#298-rAS298-r \ ™L——dT
156
упрощенной формуле
lg Kp = -Щ^ + A/ (AS) + Ас0М0 + Д^ +
+ Дс2М2 + Лс_2М_2, (341)
каждый член которой представляет алгебраическую сумму
соответствующих реагентов одной из вышеупомянутых функций (АЯ, AS
и Аср). Это позволяет производить расчет в более удобной форме.
Функции Af (АЯ) и Af (AS) в общем случае для химической
реакции ЬВ + dD = rR -\- gG -\- Q определяются с помощью
специально составленных таблиц (в табл. 8 приведена выдержка из
этих таблиц):
А/ (АЯ) = gf (AHG) + rf (АЯЛ) - bf (ПНв) - df (AtfD); (342)
A/ (AS) = gf (ASG) + rf (ДЯ*) - bf (AHB) - df (AHD), (343)
где f (АЯ) - —0.21858ДЯ298 и / (AS) - 0,21858AS298 Для
соответствующего реагента.
Точно так же
Ас0 - gc0 (G) + rc0 (R) - Ьс0 (В) - dc0 (£>), (344)
Aq =* gq (G) + rq (/?) — bcx (B) — dcx (D) (345)
и т. д., где Ас0 = 0,21858Да, или с0 = 0,21858а, и Асг = 0,21858Др,
или сг = 0,21858р, для соответствующего реагента, принимающего
участие в реакции.
Функции М0, Ml9 М2 и Л1_2 содержатся в специально
составленных таблицах (см. табл. 6).
П р и м е р. Требуется определить значение константы равновесия реакции
горения метана в кислороде СН4 + 20 2 = С02 + 2Н20 при температуре 950° С.
Из табл. 8 выписываем следующие значения:
сн4 о2 со2 но2
(АЯ) 3910,18 0 20557,89 12633,49
(AS) —4,2328 0 0,1419 —2,3231
с0 0,7480 1,8892 2,2600 1,5779
сг 3,9006-Ю"3 0,0442-Ю-3 0,5989 -10~3 0,5489-10"3
с2 —0,9104-10"6 — — —
с_2 — 0,2251 • 105 —0,4273-105 —0,0585-105
А/(АЯ) = +41914,68,
A/ (AS) = —0,27148,
Ас0 = 0,889620,
Дс1= —2,352139- 10" 3,
Ас2= —0,9104. 10" 6,
Ас_2= —0,093772- 105.
Далее по табл. 6 находим значения для Т = 1223° К:
1/Г= 0,81756. 10"3,
М0 = 0,6552,
Мг = 0,3498. 103,
Ма= 0,2121. 10е,
Л1_а= 0,3216- 10-*.
157
Таблица 8. Значения функций / (ЛЯ), / (AS) и / (АСр)
к практически точному расчету равновесий по J1. П. Владимирову
Вещество
и состояние
Ag (тв.)
А1 (тв.)
Bi (тв.)
Вг2 (газ)
С, алмаз (тв.)
С, графит (тв.)
С1 (газ)
С12 (газ)
S2 (газ)
S, ромб, (тв.)
S, монокл. (тв.)
02 (газ)
03 (газ)
Zn (тв.)
Zr (тв.)
А1203 (тв.)
ВаО (тв.)
СиО (тв.)
Си20 (тв.)
СО (газ)
С02 (газ)
СН4 (газ)
ОН (газ)
Н20 (газ)
НаО (ж.)
NiS (тв.)
AsCl3 (ж.)
SnCl4 (ж.)
NaCl (ж.)
NH4C1 (ж.)
SiF6 (газ)
AgN03 (тв.)
Na2Si03 (тв.)
/(ЛЯ)-Ю-»
0
0
0
1,65902
0,09902
0
—6,31696
0
—6,38254
0
—0,01637
0
—7,54100
0
0
83,05040
29,06114
8,41533
9,28965
5,77401
20,55789
3,91018
1,29618
12,63349
14,93273
4,06559
16,32793
27,84707
21,49297
16,38257
78,70066
6,42625
79,34454
/(AS)
0
0
0
4,9989
—0,1696
0
5,6000
0
8,5618
0
0,0350
0
—3,6153
0
0
—16,2853
—3,9130
—4,8245
—8,8398
—4,6861
0,1419
—4,2328
0,9414
—2,3231
—8,5204
—
—7,1213
— 12,4241
—4,7126
— 17,5236
—7,4798
—15,9364
—
Со
1,2525
1,0798
0,9814
1,8411
0,4765
0,8962
1,0863
1,6558
1,8754
0,7825
0,7781
1,8892
2,1552
1,1594
0,2312
5,9956
—
2,0262
3,2568
1,3661
2,2600
0,7480
1,4098
1,5779
—
2,0219
—
5,5891
2,4000
2,5792
5,0164
4,1159
6,8634
f (AS)
CfW*
0,2761
0,6470
1,1803
0,2129
0,6907
0,2230
—
0,5299
0,0656
1,3639
1,5213
0,0442
0,5377
0,5246
0,9049
0,6689
—
1,0492
1,2459
0,4571
0,5989
3,9006
0,1924
0,5489
—
0,7432
—
0,0437
0,8525
6,9946
0,5792
3,4973
2,0984
с2•10"
—
—
—0,0777
—
—
—0,2111
—
—
—
—
—
—
—
—
—
—
—
—
—0,1003
—
—0,9104
—
—
—
—
—
—
—
—
—
—
—
c_2.10-5
—0,0131
—
—
—
—0,3235
—0,4590
—
—
—
—
—
—0,2251
—0,2885
—
0,1224
—1,8514
—
—
—
—
—0,4273
—
0,6831
—0,0585
—
—
—
—0,4087
—
—
— 1,0317
—
— 1,4142
Полученные данные подставляем в уравнение (341):
\g кр = 41914,68. 0,81756. 10"« — 0,27148 + 0,889620-
• 0,6552 — 2,3521 • 0,3498 — 0,9104-0,2121 — 0,093772.
.0,3216= +34,58407,
откуда получаем
2
КР = Pc°sPH*° = 3,838-103.
Рсн/о,
Часть вторая
УЧЕНИЕ О ФАЗОВОМ РАВНОВЕСИИ
Изучение физико-химических процессов, происходящих в простых
и сложных химических системах, неразрывно связано с учением о
фазах. Учение о фазах, иначе называемое учением о равновесии
гетерогенных систем, представляет собой широкое обобщение
закономерностей, которым подчиняется обширный ряд процессов,
изучаемых физической химией и состоящих в изменении агрегатного
состояния или кристаллического строения веществ.
Основы этого учения были заложены Гиббсом, однако
только после трудов академика Н. С. Курнакова и его
последователей это учение перестало представлять только теоретический
интерес. Оно вошло в ряд прикладных наук: металлургию, галургию
и многие другие.
Глава VII.
ВАЖНЕЙШИЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ
§ 36. ФИЗИКО-ХИМИЧЕСКАЯ СИСТЕМА И ФАЗА
Физико-химические или термодинамические системы, обычно
представляющие собой вещество или смесь веществ, находящихся
в определенном изолированном объеме при данной температуре и
давлении, подразделяются на гомогенные и гетерогенные.
Гомогенной называется система, внутри которой нет
поверхностей раздела, отделяющих друг от друга части системы,
различающиеся по свойствам.
Гетерогенной называется система, внутри которой
имеются поверхности раздела, отделяющие друг от друга части
системы, различающиеся по свойствам.
Состояние физико-химической системы всегда характеризуется
определенными параметрами.
Параметрами системы называются величины,
характеризующие систему и поддающиеся изменению при непосредственном
воздействии на систему. Обычно параметрами являются объем,
температура, давление и соотношение компонентов в системе
(концентрация). Такие параметры, как напряженность магнитного и
159
электрического полей, освещенность и другие, хотя и являются
параметрами системы, но принимаются во внимание только в некоторых
специальных случаях.
Фазой называется совокупность гомогенных частей системы,
обладающих в равновесном состоянии одинаковыми
термодинамическими параметрами (или вся система, если она гомогенна).
Гомогенные части системы могут объединяться в фазу и при различии их
термодинамических свойств, но при условии возможности их
выравнивания путем непрерывного
изменения в процессе
достижения равновесия.
По агрегатному состоянию
фазы делятся на твердые,
жидкие и газообразные. Фазы,
находящиеся в твердом или
жидком состоянии, обычно
называют конденсированными
фазами, а физико-химические
системы, состоящие
исключительно из твердых и жидких
фаз, называют
конденсированными системами.
Наличие трех агрегатных
состояний воды при определенной
температуре и определенном
давлении представляет собой пример трехфазной физико-химической
системы (рис. 38, а). Другим примером гетерогенной
физико-химической системы может служить система хлористый натрий — вода,
в которой при определенных значениях параметров в равновесии
могут находиться четыре фазы: пар, насыщенный раствор хлористого
натрия в воде, лед и нерастворившиеся кристаллы соли (рис. 38, б).
а О
Рис. 38. [Пример гетерогенных физико-
химических систем—трехфазной (а),
четыре^'фазной (б):
/ — пар; 2 — лед; 3 — вода; 4 — насыщенный
раствор; 5 — кристаллы соли
§ 37. СОСТАВНАЯ ЧАСТЬ СИСТЕМЫ И КОМПОНЕНТ
Составными частями системы называются
химически индивидуальные вещества, составляющие данную систему
и способные к самостоятельному существованию, будучи
изолированными от других частей системы. Рассмотрим систему,
соответствующую химической реакции:
NH4C1 (тв.) 7Г NH3 (газ) + НС1 (газ).
Очевидно, что в данном случае имеются три составные части,
поскольку каждое из трех веществ может существовать отдельно как
самостоятельное химическое вещество (индивидуум). Отдельные
солевые ионы не могут являться составными частями системы.
Поясним сказанное примером. Рассмотрим систему хлористый
натрий — вода. Данная система имеет только две составные части:
NaCl и Н20, но не четыре (NaCl, H20, Na+ и С1~), хотя хлористый
натрий диссоциирует на ионы по уравнению NaCl 7^ Na+ + CI".
160
Ионы Na+ и С1~ не являются составными частями системы, так
как их нельзя из нее выделить в виде самостоятельных химических
веществ.
Понятие составная часть системы отличается от понятия
компонент системы. Для уяснения связи между этими двумя
понятиями рассмотрим систему, в которой имеет место реакция:
NH4C1 (tb.)^NH3 (газ) + НС1 (газ),
содержит две фазы, т. е. твердую и газообразную. Эта система,
изолированная от окружающей среды, удовлетворяет равновесному
соотношению
^NH8^HC1 _ js
^NH4C1
Рассмотрение данной системы заставляет прийти к выводу, что
хотя она состоит из трех составных частей — NH4C1, NH3 и НС1,
способных к самостоятельному существованию в изолированном
виде, но число компонентов в данном случае равно единице.
Действительно, твердая фаза состоит из NH4C1, но и газообразная
фаза состоит из того же компонента, ибо NH3 и НО находятся в
газообразной фазе в эквимолекулярных количествах (CNh:, = Сна).
Другими словами, если известна концентрация только одного
вещества из трех, то система тем самым будет полностью определена.
Если же извне в систему ввести избыток NH3 или НС1, т. е.
нарушить их эквимолекулярное соотношение, то система станет двух-
компонентной. Тогда компонентами будут NH4C1 и NH3 либо
NH4C1 и НС1 в зависимости от того, какое вещество вводится в
систему дополнительно.
Таким же образом может оказаться, что система, образованная
из трех солей и воды, имеет пять составных частей, если раствор
солей является насыщенным и все они выпали в осадок, но четыре
компонента (три соли и вода). Действительно, пусть имеем систему,
состоящую из NaCl, K2S04, КО и Н20. Очевидно, что в результате
обменной реакции образуется еще одна составная часть — Na2S04
(2NaO + K2S04^Na2S04 + 2 КО), которая способна к
самостоятельному существованию вне системы. Однако состав каждой
фазы может быть выражен посредством трех солей (NaCl, K2S04,
КО) и воды.
Рассматривая эти примеры, можно сделать заключение, что
число компонентов, из которых построена
данная система, равно числу составных
частей системы минус число возможных
реакций. Аналитически это выражается следующим образом:
* = Р — Л
где k — число компонентов;
р — число составных частей;
г — число реакций.
11 А. Н. Крестовников
161
Пример. Рассмотрим систему, состоящую из кислорода и водорода,
изолированную от окружающего пространства (рис. 39). Температура системы около
2000° С. При этой температуре идет реакция образования газообразной воды:
2Н2 + 02 —> 2Н20.
Таким образом, система имеет три составные части: Н2, 02 и Н20 (р = 3).
Поскольку в системе может протекать единственная реакция (г— 1), она является двух-
компонентной:
£=3—1 = 2.
После ознакомпления с методом расчета числа компонентов легче
перейти к определению самого понятия компонент.
Независимыми компонентами или компонентами
системы называются
индивидуальные вещества, наименьшее
число которых необходимо и
достаточно для образования всех
фаз данной системы. Под
индивидуальным веществом понимаются как простые
вещества, так и химические соединения.
§ 38. ТЕРМОДИНАМИЧЕСКАЯ СТЕПЕНЬ СВОБОДЫ
И ПРАВИЛО ФАЗ
Рис. 39. Пример
изолированной
системы (водород—
кислород)
Термодинамической степенью свободы
(сокращенно называемой просто степенью свободы
и обозначаемой буквой F) является произвольно
меняемый параметр системы. Причем это
изменение должно приводить к изменению числа
или природы фаз данной системы.
Число параметров состояния, способных
произвольно меняться без нарушения
фазового равновесия, называется числом
степеней свободы системы или ее
вариантностью.
• Допустим, что какая-либо однокомпонентная система
характеризуется определенными параметрами: температурой 7, давлением р
и объемом У; эти параметры связаны между собой уравнением
состояния: f (/?, У, Т) = 0. Тогда из трех перечисленных параметров два
будут являться независимыми параметрами, а один — зависимым.
Другими словами, мы задаемся произвольно двумя параметрами,
а система «сама» устанавливает третий параметр.
Если система состоит из одной только газообразной фазы, то
можно произвольно менять (оставляя систему в том же газообразном
состоянии) оба независимых параметра, например температуру и
давление. Это соответствует тому, что система имеет две степени
свободы, т. е. является двухвариантной.
Если в системе имеется равновесие жидкость^ пар, то, не
нарушая этого равновесия, можно менять только один параметр, так как
один из параметров уравнения состояния f (р, У, Т) = 0 становится
фиксированным, поскольку пар над жидкостью является насыщен -
162
ным. Другими словами, три параметра (температура 7, давление р
и объем V) связаны двумя уравнениями: уравнением состояния и
уравнением агрегатного превращения:
dp Q
dT TAV'
где Q — скрытая теплота превращения (в данном случае —
испарения), а А У — изменение объема при этом превращении.
Система в этом случае является одновариантной.
Число степеней свободы для каждой
системы определяется разностью между
числом параметров данной системы и числом
уравнений, связывающих эти параметры.
Перейдем теперь к рассмотрению основного соотношения учения
о фазах, называемого правилом фаз.
Рассмотрим какую-либо однокомпонентную двухфазную систему,
например систему вода^ пар, обе фазы которой находятся в
равновесии при некотором значении давления и температуры. Пусть одна
фаза (вода) имеет следующие удельные или мольные значения объема
энтропии, энтальпии и изобарного потенциала: Vl9 Sly Нг и Gx.
Другая фаза (водяной пар) характеризуется иными значениями тех
же величин: V2> S2, #2 и ^2-
Предположим, что к системе подведено такое количество теплоты
dQ, чтобы некоторое количество вещества перешло из одной фазы
в другую (из жидкой в парообразную).
Согласно второму началу термодинамики:
dQ = Т (S2 — Sx) dm, (346)
где dm — некоторое количество вещества.
Изменение энтальпии составит
dH = (Н2 — Нг) dm. (347)
При этом может совершиться работа, скажем, по подъему поршня,
если система была отделена от окружающей среды при помощи
цилиндра с поршнем:
dA = p (V2 — Vx) dm. (348)
Согласно первому началу термодинамики:
dQ = dU + dA. (349)
Поэтому имеем
Т (S2 — Si) dm - (Я2 — #i) dm + p (V2 — VJ dm, (350)
откуда, сокращая на dm и раскрывая скобки, получаем:
#i - TSX + pV1 = H2 - TS2 + pV2; (351)
но так как Я — TS + pV = G (где G — изобарный потенциал), то
полученное уравнение (351) можно написать следующим образом:
Gx = G2. (352)
11* 163
Таким образом, если две фазы системы, состоящей из одного
компонента, находятся в равновесии, то их удельные или мольные
изобарные потенциалы равны.
Число термодинамических степеней свободы рассмотренной
системы определяется следующим подсчетом: число параметров — три,
число связывающих эти параметры уравнений — два (уравнение
агрегатного превращения и равенство изобарных потенциалов двух
фаз). Следовательно, система является одновариантной.
Сделаем теперь следующий шаг и рассмотрим систему, которая
имеет у фаз и содержит k компонентов. Для упрощения рассуждений
предположим, что каждый компонент в каком-то количестве
находится в каждой фазе.
Состояние отдельной фазы описывается уравнением \\> (р, V, Т) =
= 0. Вводя вместо объема обратную ему величину — концентрацию
С = —, получаем уравнение состояния в такой форме ф (р, С, Т) =
= 0.
Так как наши рассуждения ведутся в пределах одной системы,
то давление (р) и температура (Т) для всех фаз одни и те же, т. е.
для всех фаз имеем два общих параметра (р и Т).
Если каждый компонент находится в каждой фазе, то для одного
компонента имеем, таким образом, у концентраций (С1У С2, С3, . . .
. . ., Су) во всех фазах, а для k компонентов имеем ky
концентраций. Итого для рассматриваемой системы имеем число параметров
2 + ky (два параметра р и Т и ky концентраций).
Подсчитаем теперь количество уравнений, связывающих эти
параметры. Для каждой фазы будем иметь уравнение состояния:
Ф (р, 7, С1У С2, С3, . . ., Ck) — 0. (353)
Всего таких уравнений у, т. е. столько же, сколько фаз.
Равновесие в многокомпонентной системе характеризуется равенством
химических потенциалов (i компонентов во всех фазах. Поэтому
для каждого компонента имеем еще дополнительно у — 1 уравнений.
Так, четыре фазы будут иметь три уравнения химических
потенциалов:
Конечно, первая фаза может соприкасаться и с четвертой фазой,
и тогда справедливо будет уравнение \1г = jx4; но это уравнение не
будет самостоятельным, так как оно следует из предыдущих
уравнений.
Для k компонентов получаем всего k (у — 1) таких уравнений.
Итак общее количество уравнений у + k (у — 1). Согласно
определению, данному в предыдущем разделе, число степеней свободы
системы (F) равно разности между числом параметров и числом
связывающих эти параметры уравнений:
F = (2 + ky)- [y + k(y-\)]. (355)
Раскрывая скобки, получаем:
F = k — у + 2, (356)
164
т. е. число степеней свободы равняется
разности между числом компонентов и числом
фаз плюс два.
Полученное уравнение представляет собой правило фаз в его
классической формулировке. В этом виде оно и применяется при
анализе большинства физико-химических систем.
Как видно из вывода этого уравнения, двойка появилась
вследствие принятого нами допущения, что на состояние равновесия могут
влиять только два параметра: температура и давление. Однако имеют
место случаи, когда на равновесие могут оказывать влияние и
другие параметры. В этих случаях двойка заменяется на
соответствующее число параметров /, способных оказывать влияние на
равновесие системы. Учитывая этот факт, можно записать правило фаз
в наиболее общем виде:
F = k-y + f. (357)
В металлических системах один из параметров, а именно давление,
в практических условиях изменяется в таких небольших пределах,
что это не оказывает существенного влияния на процессы и
превращения в сплавах. В связи с этим в применении к металлическим
сплавам правило фаз записывается уравнением
F = к —у + 1, (358)
в котором единица соответствует единственному параметру
(температуре), способному вызывать изменение фазового равновесия.
В некоторых случаях (например, при равенстве концентраций
какого-либо компонента в двух фазах) в рассматриваемой системе
возникают условия, позволяющие составить дополнительные
уравнения для характеристики системы. Дополнительные уравнения,
связывающие параметры системы, уменьшают вариантность системы
на столько единиц, на сколько увеличилось число уравнений,
характеризующих систему.
Отсутствие одного или нескольких компонентов в одной из фаз
не оказывает влияния на число степеней свободы, так как число
параметров, характеризующих систему, и число уравнений
изменяются на одно и то же число, а следовательно, разница между ними,
представляющая собой вариантность системы, остается без
изменения.
Например, определим вариантность двухкомпонентной системы
(соль + вода), состоящей из четырех фаз: соль ^ лед
^насыщенный раствор ^ пар (см. рис. 38, б). Число компонентов системы
к = 2. Число фаз, составляющих систему, у = 4. Параметры,
способные изменить состояние равновесия, — температура и давление,
т. е. / = 2. Подставляя найденные значения в уравнение (358),
получаем:
F^k — y + f = 2 — 4 + 2-0.
Двухкомпонентная система, состоящая из четырех фаз, имеет
ноль степеней свободы, т. е. является безвариантной системой. Это
165
означает, что четырехфазное равновесие в двухкомпонентнои системе
может осуществляться только при строго определенных значениях
температуры и давления.
В специальной литературе термины «безвариантность», «одно-
вариантность», «двухвариантность» и т. д. часто заменяют терминами
с приставками, заимствованными из латинского (ин-, моно- и бива-
риантность) или греческого (нон-, уни- и дивариантность) языков.
§ 39. ГЕОМЕТРИЧЕСКИЙ СТРОЙ ДИАГРАММ СОСТОЯНИЯ
Как было уже отмечено, состояние системы описывается
уравнениями вида ф (р, С, Т) = 0.
Численные значения параметров системы для каждого
конкретного случая определяют положение некоторой точки в трехмерной
системе координат С, /?, Т (рис. 40).
Каждая точка, взятая в этой
системе координат, характеризует
определенное состояние системы.
Например, точка а на рис. 43
соответствует состоянию системы
при параметрах р1У Сг и Тг.
Условились такую точку называть
изобразительной или
фигуративной точкой данной системы.
Однако на практике нет
необходимости всякий раз
использовать трехмерное пространство;
достаточно определить только два
параметра, так как третий
определится из уравнения ср (/?, С,
Т) — 0, т. е., иными словами,
для однозначного определения
состояния системы достаточно
определить положение
фигуративной точки в двухмерной системе
координат, например р и Т или С и Т. Такое графическое
изображение уравнения состояния системы носит название диаграммы
состояния.
Важно заметить, что, несмотря на широкое использование в
термодинамике уравнений состояния равновесных систем, мы не знаем
действительные уравнения состояния. Все попытки найти
математическое выражение уравнения состояния приводят к весьма
громоздким формулам, которые к тому же редко оправдываются в реальных
системах.
С этой точки зрения диаграммы состояния приобретают
огромный интерес. С их помощью на основании экспериментальных данных
мы получаем возможность наглядно изобразить в виде
геометрических образов действительное взаимоотношение
физико-химических параметров, определяющих состояние системы.
166
Рис. 40. Система координат для опре
деления состояния системы
Глава VIII.
ОДНОКОМПОНЕНТНЫЕ И ДВУХКОМПОНЕНТНЫЕ
СИСТЕМЫ
§ 40. ОДНОКОМПОНЕНТНЫЕ СИСТЕМЫ
И АЛЛОТРОПИЯ (ПОЛИМОРФИЗМ)
П{римером однокомпонентной системы может служить не только
любосе простое вещество, но и химическое соединение, обладающее
во всех агрегатных состояниях.
i
1
I
'в
а
37<4°С
195 am
%0075°С
0,006 am
Температура t, °C
Рис. 41. Схема диаграммы состояния
воды в координатах
давление—температура
строгго определенным составом
Важшо, чтобы систему составлял
определенный химический
индивидуум. На рис. 41
схематический, т. е. для большей
наглядности без соблюдения
масштабна, изображена диаграмма
состояния воды в области
невысокие давлений. Кривые OQ,
ОМ и ON делят диаграмму
состояния, изображенную на
координатах Р и t, на три
поля!. Эти поля представляют
собой совокупность точек,
каждая из которых обозначает
определенное агрегатное
состояние. Поле S соответствует
кристаллическому, поле L —
жидкому, а поле G — газообразному состоянию. Все точки,
принадлежащие тому или иному полю на диаграмме, отвечают
однофазному состоянию и, согласно правилу фаз (F = k — у + 2 =
= 1 — 1+2 = 2), обладают двумя степенями свободы. Это
означает, что одновременное изменение и давления и температуры
в пределах данного поля не вызывает появления других фаз.
Линия ON представляет собой границу между полем L и полем G.
Вследствие этого любая точка линии ON соответствует состоянию
равновесия между жидкостью и паром:
Поскольку в равновесии находятся две фазы (жидкость и пар),
то, согласно правилу фаз (F = k — у -\- 2 = 1 — 2 + 2=1),
система обладает одной степенью свободы. Это означает, что, не
нарушая фазового равновесия на линии ON, произвольно можно менять
только один параметр. Для того чтобы произвольное повышение
температуры не вызвало исчезновения жидкости, необходимо на
определенную величину повысить давление. Если же произвольно
понизить давление в системе, то для сохранения фазового равновесия
необходимо понизить и температуру [см. уравнения (170) и (172) на
стр. 73].
167
Легко видеть, что линия ON одновременно выражает зависимость
давления насыщенных паров от температуры. Кроме того, линию ON
можно трактовать как зависимость температуры кипения от внешнего
давления. В связи с этим линия ON получила название кривой
испарения.
В точке N кривая кипения обрывается, так как при температурах
выше 374° С никакое давление не может перевести пар в жидкость.
Эта точка диаграммы получила название критической точки
кипения, а ее параметры (для воды t = 374 °С и Р = 195 am) —
критических параметров.
Линия ОМ представляет собой сосокупность точек,
соответствующих двухфазному равновесию между твердым и жидким
состоянием:
Как и всякое другое двухфазное равновесие в однокомпонентной
системе, равновесие L^S обладает одной степенью свободы, что
соответствует одному произвольно меняемому параметру. Это
положение подробно разбиралось для случая равновесия пара и жидкости
[см. также уравнения (170) и (173) на стр. 73].
Линию ОМ можно назвать кривой плавления, так как
она изображает зависимость температуры плавления от внешнего
давления.
Легко видеть, что в случае воды увеличение давления
перемещает температуру плавления в сторону более низких температур.
Однако это связано с аномальным поведением воды: ее плотность
в твердом состоянии меньше плотности в жидком состоянии. Обычно
кривая плавления описывает увеличение температуры плавления
с повышением внешнего давления. Следует заметить, что давление
влияет на температуру плавления не так заметно, как на
температуру кипения; это находит свое отражение в том, что кривая
испарения всегда оказывается значительно более пологой, чем кривая
плавления. В большинстве случаев кривая плавления занимает почти
вертикальное положение. Экспериментально кривую плавления.уда-
лось проследить до очень высоких давлений, но при этом критическая
точка не была обнаружена.
Линия OQ является кривой сублимации. Точки этой
линии соответствуют состоянию равновесия между твердой и
газообразной фазами:
S^G.
Это равновесие также обладает только одной степенью свободы,
т. е. позволяет независимо изменять только один из параметров р
или t [см. уравнения (170) и (171) на стр. 73].
Кроме изображения совокупности точек, соответствующих
двухфазному равновесию S^G> линия OQ характеризует также
зависимость давления пара над твердым веществом от температуры. Вместе
с тем она показывает влияние внешнего давления на температуру
сублимации. Линия OQ на диаграмме состояния воды располагается
в области очень малых давлений, но, как можно заключить из на-
168
клона кривой, температура сублимации очень чувствительна к
величине внешнего давления. Зависимость температуры сублимации при
сверхнизких давлениях не изучалась, но теоретически можно
предположить, что кривая сублимации, если не образуются новые фазы,
имеет своим началом точку абсолютного нуля.
Линии ON, ОМ и OQ имеют общую точку О, называемую
тройной точкой. Она соответствует состоянию системы, в котором
пар, жидкость и кристаллы находятся в равновесии. По правилу
фаз число степеней свободы такой системы равно нулю, так как F —
= fe — 0 + 2 = 1—3 + 2 = 0.
Это означает, что сосуществование трех фаз может продолжаться
как угодно долго только при определенных и строго постоянных
значениях параметров. Для воды такими параметрами являются t =
= 0,0075° С и Р = 4,579 мм рт. ст. = 0,006 am.
Из нанесенных на диаграмму состояния линий мы еще не
рассмотрели линию ОУУ+ представляющую собой продолжение кривой
кипения. Физический смысл линии ONx заключается в том, что она
изображает зависимость давления насыщенного пара над
переохлажденной жидкостью. Ниже температуры тройной точки жидкость
является метастабильной фазой, поэтому, как видно из диаграммы
состояния, давление пара над ней выше, чем над твердой фазой,
стабильной при этих температурах.
Мы рассмотрели диаграмму состояния воды. Однако большинство
диаграмм состояния однокомпонентных систем, в том числе и
диаграмма состояния воды в области высоких давлений, оказываются
более сложными. Причина этого кроется в явлении полиморфизма,
свойственного очень многим веществам. Остановимся подробнее на
этом явлении.
Аллотропия (по-гречески — «другая форма») химических
элементов и полиморфизм (по-гречески — «многоформность»)
химических соединений представляют собой явления, связанные со
способностью веществ существовать в различных кристаллических
модификациях. Кристаллические модификации различаются по своим
физическим свойствам, несмотря на тождественность химических
свойств вещества, вследствие различного пространственного
расположения частиц вещества.
Всем известное простое вещество — сера может иметь две
кристаллические модификации: ромбическую и моноклинную. Однако
не следует думать, что существование обеих модификаций
равновероятно. Вероятность образования той или иной модификации
определяется условиями, в которых находится данное вещество, а в ряде
случаев — условиями охлаждения.
Предположив наличие двух модификаций а и р у некоторого
вещества Л, исследуем изменение давления паров над каждой из этих
модификаций с изменением температуры.
На диаграмме, приведенной на рис. 42, кривая ab описывает
изменение давления насыщенного пара а-модификации с
температурой; иными словами, это — кривая равновесия между твердым и
парообразным состоянием. То же самое относится к кривой cd, кото-
169
рая описывает изменение давления насыщенного пара р-модификации.
Как можно видеть из этой диаграммы, при температуре tl9 ниже
некоторой температуры t0, давление пара Р-модификации больше, чем
давление пара а-модификации, т. е. Р-модификация будет менее
устойчива, чем а-модификация и, следовательно, будет происходить
переход вещества из Р-модификации, если таковая еще имеется,
в а-модификацию; Р —* а.
Переход этот представляется как испарение р-модификации,
давление пара над которой высоко, и конденсация паров на
частицах а-модификации, давление
пара над которыми ниже.
По мере повышения
температуры разница в давлениях
пара а- и р-модификаций
становится все меньше, и в
некоторой точке О при температуре
tQ они становятся равными
(кривые пересекаются). Таким
образом, в состоянии,
описываемом точкой О,
термодинамически равновероятно
существование обеих модификаций.
Следовательно,точка О является
точкой взаимного перехода
а^—- р.
При дальнейшем повышении
температуры давление
насыщенного пара для а-модификации становится больше, чем для
Р-модификации, и потому при t2 > t0 неустойчивой становится а-модификация.
Однако необходимо оговориться, что неустойчивые модификации
могут существовать весьма продолжительное время, если рядом
нет более устойчивой модификации, которая в этом случае играет
роль «затравки».
Таким образом, в твердом состоянии ниже температуры t0
устойчива а-модификация, выше температуры t0 устойчива р-модификация.
Кривая bd изображает зависимость давления пара над
жидкостью, поэтому точка Ь соответствует температуре плавления
а-модификации fe), а точка d — Р-модификации (^
ПЛ
t0 tj
Температура t
-пл
^твер
Рис. 42. Схема равновесия пар
дое тело при наличии различных
кристаллических модификаций (энантиотроп-
ные вещества)
пл
Вещества, на диаграммах состояния которых имеются точки
перехода одной модификации в другую, получили название энантиотроп-
ных веществ. Примером являются сера, железо, титан, азотнокислый
аммоний и др.
Между тем для большой группы веществ не существует такой
температуры и такого давления, при которых можно наблюдать
переход одной кристаллической модификации в другую. Такими моно-
тропными веществами являются фосфор, углерод, карбонат
кальция, сернистая ртуть и др.
На рис. 43 представлена диаграмма состояния, на которую
нанесены кривые температурной зависимости давления пара для а-моди-
170
фикации (кривая ab) и р-модификации (кривая cd) монотропного
вещества. Как видно из диаграммы, эти кривые пересекаются при
температуре, лежащей выше температуры плавления вещества, когда
вещество утратило все признаки кристаллического состояния. Таким
образом, монотропные вещества имеют лишь мнимую точку перехода
существующих модификаций. При всех температурах ниже
температуры плавления устойчивой является а-модификация, а (5-модифи-
кация не имеет температурного интервала устойчивости.
Как правило, в момент образования какого-либо вещества,
имеющего несколько кристаллических модификаций, вначале образуются
1
&
«5;
^
I
d /
^
^ '
--""р |у
с У\
У
у
ОС
/ >L
ЛХ
yi I
1 1 ,
Температура t
ПЛ
Рис. 43. Схема равновесия пар
^[твердое тело при наличии различных
кристаллических модификаций (монотропные
вещества)
Температура t
Рис. 44. Схема диаграммы состояния
серы (параметры точек Л, Б, С иЯ
приведены в тексте)
менее устойчивые из них, которые затем уже переходят в более
устойчивые.
Диаграмма состояния серы представляет собой пример одноком-
понентной системы, в которой имеет место полиморфное превращение.
Рассмотрение этой диаграммы (рис. 44) позволяет выяснить условия
образования ромбической, моноклинной, жидкой и парообразной
фаз серы. Приведенные на диаграмме кривые характеризуют:
кривая DA —изменение давления пара над ромбической серой в
зависимости от температуры;
- изменение давления пара над моноклинной серой
в зависимости от температуры;
- зависимость давления пара над жидкой серой от
температуры;
- влияние давления на температуру перехода
кривая АС
кривая CF
кривая АВ
кривая СВ
кривая BE
о —► о
^ромб ■<— с-'монокл>
изменение температуры плавления моноклинной серы
в зависимости от давления;
изменение температуры плавления ромбической серы
в зависимости от давления.
171
Жидкую серу можно переохладить ниже точки плавления, а
ромбическую серу перегреть выше точки превращения в моноклинную
серу. Поэтому кривые DA, CF и BE могут быть продолжены внутрь
поля устойчивости моноклинной серы (ABC). Физический смысл
продолжения кривых:
АН — кривая давления пара над перегретой ромбической серой;
СН — кривая давления пара над переохлажденной жидкой
серой;
ВН — кривая плавления перегретой ромбической серы.
На этой же диаграмме
изображены тройные точки, отвечающие
нонвариантным равновесиям
следующих фаз:
А (95,5 °С, 0,004 ммрт. ст.) —
ромбическая сера ^ моноклинная
сера 7^ пар;
В (151 °С, 1288 am) —
ромбическая сера 7^ моноклинная сера 7^
7^ жидкая сера;
С (119,3 °С,0,02 мм рт. ст.)—
моноклинная сера 7^ жидкая
сера 7"* пар;
Я (112,8 °С, 0,01 ммрт.ст.)—
перегретая ромбическая сера 7"*
жидкая
/40
120
100
I
1
I
80
60
40
20
\ш
?
?
?
Е
7 V
Ш 1
Ш V
/ '
ш
ч
ш
L
€ переохлажденная
сера 7^ пар.
Пример более сложной
диа-
0
500
100 200 300 400
Температура ?, °С
Рис. 45. Диаграмма состояния висмута
граммы состояния однокомпонент-
ной системы показан на рис. 45.
При давлении l am висмут
существует лишь в одной аллотропной
форме (обозначена: /), имеющей
ромбоэдрическую кристаллическую решетку. Однако повышение
давления приводит к появлению еще семи аллотропных форм
(обозначены II—VIII). Штрихами указаны недостаточно изученные
границы фазовых областей.
§ 41. ДВУХКОМПОНЕНТНЫЕ СИСТЕМЫ
Двухкомпонентными называются физико-химические системы,
в состав которых входят только два компонента. Этими
компонентами могут являться как простые вещества, так и различные
химические соединения. Соотношение между компонентами способно
в значительной степени изменять свойства системы. Это означает,
что для однозначного определения состояния системы в данном
случае потребуются следующие параметры: Р, Т, Сх и С2, где
Сх и С2 — концентрации компонентов, составляющих систему.
В соответствии с этим уравнение состояния двухкомпонентной
системы имеет следующий вид:
Ф (/?, 7\ Съ С2) = 0.
172
Очевидно, что если определить три параметра, то четвертый
определится из уравнения состояния, и диаграмма состояния может
быть построена в трехосной системе координат. Удобней
воспользоваться для этой цели координатами р, С и Т.
При изучении температур плавления и температур фазовых
превращений некоторых двухкомпонентных систем, особенно в
случае различного рода металлических и шлаковых систем, без
существенной погрешности можно считать давление постоянным, а
следовательно, ограничиться построением
диаграммы в координатах С и Т.
На рис. 46 изображены
координатные оси, которые обычно
применяются для построения таких
диаграмм. Ось абсцисс,
представляющую собой ось концентраций,
изображают в виде отрезка, длину
которого принимают за 100%.
Концы этого отрезка будут
соответствовать чистым компонентам, т. е.
100% А и 100% S, если систему
составляют компоненты Ли В.
Любая точка, помещенная на отрезке,
делит его на части, которые
количественно характеризуют
химический состав сплава. Так, точка CL показывает, что в сплаве
содержится 60% В (отрезок СХА) и 40% А (отрезок С\5). Часто на ось
концентраций наносят только количество одного из компонентов,
так как количество другого легко находится по разности.
Необходимо обратить внимание, что ось концентраций может
быть отградуирована различным образом: % (по массе), % (ат.)
0^
•с
I
!
I
А
ой
I
I
I
\ ч
100А
08
80А 60А MA 20A
20В Ь08 60В 808
Концентрация С, %
0А
/008
Рис. 46. Оси координат,
применяемые при изображении диаграмм
состояния двухкомпонентных
систем
или % (мол.), доли (по массе), атомные или мольные доли и т. п.
Наиболее часто требуемый переход от % (по массе) к % (ат.)
и обратно может быть осуществлен по формулам:
*л =
ЮОЯд
^Л + ^в
м
А
КА
к
в
100
(359)
мв
и
Ал =
\оомл
100
к
- 1 4-
м
А
Мв
В
** + **= 100,
(360)
где кА и кв — % (ат.) и ХА и лв — % (по массе) компонентов А
и В с атомной или мольной массой Мл и Мв соответственно.
Ось ординат представляет собой ось температур. Принятыми
шкалами являются: международная, стоградусная или шкала
Цельсия (°С) и абсолютная, термодинамическая или шкала Кельвина
(°К). Однако применяются также и другие шкалы: Реомюра (°R),
173
Фаренгейта (°F) и Ренкина (°Ra). Соотношения между градусами
различных шкал следующие:
1 °С - 0,8 °R = 1,8 °F = 1 °К = 1,8 °Ra;
1° R - 1,25 °С - 2,25 °F - 1,25 °К = 2,25 °Ra;
1° F = 0,556 °C - 0,445t°R - 0,556 °K - 1 °Ra.
Любая точка на диаграмме состояния является носителем
определенного физического смысла. Так, фигуративная точка а (рис. 48)
показывает, что сплав состава Сг (40% А и 60% В) находится при
температуре t\. Однако на диаграмму состояния наносят только
точки, характеризующие процессы плавления или затвердевания,
фазовые переходы и т. п. Линии, соединяющие эти точки, носят
определенные названия. Так, линия, представляющая собой
совокупность точек начала кристаллизации, носит название
ликвидус (от латинского слова «жидкий»); выше ликвидуса сплавы
находятся в однофазном жидком состоянии. Линии, ниже которых
сплав находится в твердом состоянии, называют с о л и д у с (от
латинского слова «твердый»).
Для анализа и проверки правильности построения диаграмм
состояния широко применяется правило фаз, которое для данного
случая записывается в следующем виде:
F = k-y+ 1,
поскольку из двух переменных параметров один (а именно давление)
принимается постоянным.
Правило фаз позволяет заранее предсказать, что в двухкомпо-
нентной системе максимальное число равновесно сосуществующих
фаз равно трем (у = k — F + 1 = 2 — 0+1=3), так как
вариантность системы не может быть меньше нуля. Другими словами,
трехфазное равновесие в двухкомпонентной системе является
безвариантным равновесием.
Равновесие двух фаз в двухкомпонентной системе является одно-
вариантным (F — k — у + 1 = 2 — 2+1 = 1). Однофазное
равновесие такой системы обладает вариантностью, равной двум. Это
означает, что давление, которое мы принимаем за неизменный
параметр, тоже можно было бы изменить без нарушения фазового
равновесия.
§ 42. ОСНОВЫ ТЕРМИЧЕСКОГО АНАЛИЗА
Если при охлаждении физико-химической системы не происходит
никаких превращений, сопровождающихся выделением или
поглощением тепла, то температура будет непрерывно понижаться.
Графическое изображение результатов процесса в координатах время
охлаждения — температура даст в этом случае кривую, для которой
характерно непрерывное изменение угла наклона касательной к оси
абсцисс (кривая / на рис. 47).
Однако непрерывность в ходе кривой охлаждения нарушается,
если в системе имеет место какое-либо превращение,
сопровождающееся тепловым эффектом. При этом могут быть два случая: отво-
174
■*■*
димое тепло полностью возмещается выделяющимся теплом в
процессе данного превращения (безвариантное превращение) и
отводимое тепло возмещается только частично (одновариантное
превращение).
В первом случае, пока протекает превращение, несмотря на отвод
тепла, температура остается постоянной и на кривой охлаждения
появляется горизонтальный участок, отвечающий температуре
соответствующего превращения (кривая ///). Величина
горизонтального участка кривой определяется рядом факторов. При всех про-
.чих равных условиях величина площадки будет тем больше, чем
больше теплота превращения и
чем больше масса данной системы;
с увеличением скорости
охлаждения площадка будет уменьшаться.
Во втором случае температура
превращения не будет оставаться
постоянной и выделяющееся
тепло лишь несколько изменит
плавность охлаждения в области
температур, соответствующих
интервалу данного превращения
(кривая //).
Содержанием термического
метода анализа является
определение температур превращений на
основании изучения кривых
охлаждения (или нагревания) физико-
химических систем.
При построении диаграмм плавкости широко используются
кривые охлаждения, полученные переводом систем из области жидкого
состояния в область твердого состояния. Экзотермическим
превращением, температура которого в данном случае определяется,
является кристаллизация — переход из жидкого состояния в твердое.
При этом процессе вместо беспорядочного скопления хаотически
движущихся молекул жидкости, слабо между собою связанных *,
образуется упорядоченная кристаллическая структура с
фиксированным положением атомов в узлах пространственной
кристаллической решетки (атомы имеют лишь колебательные движения около
некоторого определенного положения). Таким образом, запас
внутренней энергии системы в твердом состоянии оказывается меньшим,
чем в жидком, в силу того, что в твердом состоянии отсутствует
хаотическое движение молекул. Этот избыток энергии жидкого
состояния по сравнению с твердым и отдается системой при
кристаллизации в виде теплоты кристаллизации.
Следует обратить внимание на вид кривых охлаждения. Если
в исследуемом веществе ,не происходит превращений, то кривые
Время
Рис. 47. Различные типы (/, //, ///)
кривых охлаждения (tu и tK —
температуры начала и конца превращения;
t
кр
температура кристаллизации)
* Исследования показывают, что и в жидкостях имеется известная
упорядоченность частиц в областях порядка 10—20 А (так называемый ближний порядок).
175
обращены выпуклостью к оси времени (закон охлаждения Ньютона),
т. е. по мере уменьшения перепада температур между веществом
и окружающей средой интенсивность охлаждения уменьшается.
При выделении теплоты за счет превращения в веществе кривизна
уменьшается и может стать обратной.
Нетрудно представить себе вид кривых нагревания; они
практически симметричны кривым охлаждения.
Помимо рассмотренного простого варианта термического
анализа применяется так называемый дифференциальный термический
анализ (метод ДТА — его сокращенное наименование в специальной
литературе). Если на рис. 47 сместить кривые // и /// влево до
совмещения их верхних участков с кривой /, то расхождение между
этими кривыми (по вертикали) будет соответствовать различию
температур одновременно охлаждаемых образцов, в одном из
которых превращений не происходит. Практически в методе ДТА
поступают следующим образом: измеряют разность температур двух
образцов (эталона и исследуемого вещества) и записывают ее во
времени одновременно с простой записью кривой охлаждения.
Дифференциальная запись позволяет с большей чувствительностью
фиксировать начало и конец превращений.
Широко практикуется также одновременная регистрация
нескольких свойств вещества при охлаждении и нагревании.
§ 43. ДИАГРАММА СОСТОЯНИЯ СИСТЕМЫ
С ПРОСТОЙ ЭВТЕКТИКОЙ
Допустим, что образующие систему два компонента в жидком
состоянии полностью растворимы друг в друге, а в твердом
состоянии совершенно нерастворимы; при затвердевании сплав
распадается на смесь кристаллов чистых компонентов. Такая система
характеризуется диаграммой состояния с простой эвтектикой
(рис. 48).
Отметим следующие элементы диаграммы: область, лежащая
выше aeby — однофазная область жидкого состояния L; области
аер и beq — двухфазные области L + А и L + В\ область ApqB —
область исключительно твердого двухфазного состояния А + Б,
причем слева от точки е структура сплава будет представлять собой
первичные кристаллы А и эвтектическую смесь (А + В), а справа
от точки е — первичные кристаллы В и эвтектическую смесь (Л -f В).
Необходимо помнить, что эвтектика не является самостоятельной
фазой, а лишь дисперсной смесью двух фаз — А я В.
Линия aeb — ликвидус диаграммы. Выше этой линии все сплавы
находятся в однородном жидком состоянии, ниже — в двухфазном
состоянии: жидкость и первичные кристаллы (или А или В). Таким
образом, переход фигуративной точки сплава через линию ликвидус
соответствует началу процесса кристаллизации, появлению первых
кристаллов твердой фазы.
Линия peg является солидусом диаграммы. При переходе
фигуративной точки через линию солидуса исчезают последние следы
жидкой фазы, и ниже ее сплав находится в твердом состоянии.
176
Точка еу соответствующая состоянию расплава, одновременно
насыщенного по отношению к кристаллам А и В, носит название
эвтектической точки. Состав, соответствующий точке в,
называется эвтектическим, а горизонталь, проведенная через эгу
точку, — эвтектической горизонталью.
Если мы желаем знать химический состав того или иного сплава
(например, сплава г), то, опуская перпендикуляр на ось концентра-
I
с:
5:
со
А+э6т(А+В)
г'
В+Эвт(А+В)
I
_б
е'{ ,е,
К о и и. ентр о ц и я
it
В
Рис. 48. Схема диаграммы состояния системы с простой эвтектикой
ций, получим точку г', которая показывает нам, сколько в сплаве
содержится компонента А (отрезок г'В) и компонента В (отрезок г'А).
Другими словами:
%Л-44-ШОо/0; %В= Г'А
,АВ
АВ
ЮОо/
или
%А
%в
г'В
г'А
С помощью диаграмм состояния могут решаться два типа задач:
определение количественного соотношения фаз в сплаве данного
химического состава (фазовый сплав) и определение химического
состава равновесных фаз в данном сплаве (состав фаз). Рассмотрим
примеры решения подобных задач для сплавов системы с простой
эвтектикой.
12 А. Н. Крестовников
177
Например, если состояние сплава описывается фигуративной
точкой k, то относительное количество жидкой и твердой фаз при
температуре tk будет
% L W % L k'B
__ == И ПИ -— —
%В kex ИЛИ %В kfe[ '
Приведенное соотношение получило название правила р ы'-
ч а г а. Горизонтальный отрезок ехЬ' является частью изотермы,
проходящей через точку k, и носит название конноды. Концы
конноды позволяют определить состав фаз (по оси концентраций),
а отрезки конноды — фазовый состав (по правилу рычага). Жидкость
при температуре tk имеет состав, определяемый точкой е1у т. е.
количества компонентов А я В в жидкости находятся в следующем
соотношении:
о/о Л e[B
Кристаллы, находящиеся в равновесии с этой жидкостью, имеют
состав: 0% А и 100% В (точка Ь' проектируется в точку В).
Легко видеть, что чем выше температура, тем жидкость сплава k'
богаче компонентом В.
Если обратимся к сплаву г, то обнаружим, что в двухфазной
области жидкость обогащается компонентом В не при повышении
температуры, а при понижении ее. Это связано с тем, что при
понижении температуры из сплава г выпадают кристаллы А (начиная
с температуры tri), а из сплава k выпадают кристаллы В (начиная
с температуры 4t).
Так как правило рычага широко используется для анализа
диаграмм состояния, покажем его справедливость на примере сплава k
(рис. 48).
Пусть относительное количество твердой фазы при состоянии
сплава, определяемом точкой k, равно х. Тогда относительное
количество жидкой фазы будет (1 — х)у так как весовое количество всего
сплава мы принимаем за единицу.
Количество компонента В в жидкой фазе равно е[А. Количество
компонента В в твердой фазе равно 100% (т. е. А В), так как в
интервале температур от tkt до te выпадают только кристаллы чистого
компонента В. Во всем сплаве количество компонента В равно kA,
Таким образом, можем написать равенство
е[А(\—х) +ABx = k'Al,
откуда
k'A — е[Л е[ ek
■у Л- •
~~ ЛВ— ехА ~~ e[B ~ e±b' '
1 , exk exbf — exk kb'
exb' ~~ exb' ~ exb'
178
Таким образом, для фигуративной точки k можно написать:
W
Относительное количество L 1 — х ехЬ' W
Относительное количество В х exk exk '
"или
%L __ W
o/0 В ~ exk #
Полученный результат соответствует правилу рычага.
Теперь рассмотрим процесс кристаллизации отдельного сплава,
например сплава г. Первую стадию этого процесса запишем таким
образом:
I у, I p
1_1__Л + 1(г1-е).
В дальнейшем при разборе диаграмм состояния мы всегда будем
применять такой способ записи. Читать ее следует так: из жидкости
в интервале температур от tfl до te выпадают кристаллы
компонента Л, а состав жидкости меняется от г до е.
Жидкость состава е оказывается в равновесии одновременно
с кристаллами А и кристаллами В, поэтому начинается их
совместная кристаллизация (вторая стадия):
1(е)-^эвт(Л + В).
Такую запись следует читать: при постоянной температуре te
из жидкости эвтектического состава е кристаллизуется эвтектика
(Д + В).
Первая стадия кристаллизации соответствует выпадению
первичных кристаллов и, согласно правилу фаз:
F = А — у + 1 = 2 — 2 + 1 = 1,
является одновариантным процессом. Переменным параметром здесь
является температура, или, иначе говоря, кристаллизация идет
в интервале температур.
Вторая стадия кристаллизации связана с одновременным
существованием трех фаз L, Л, В и, согласно правилу фаз:
F = fe — г/+1=2 — 3+1=0,
является безвариантным процессом, т. е. представляет собой
одновременную кристаллизацию из жидкости двух твердых фаз. Этот
процесс носит название эвтектической кристаллизации и
происходит при постоянной температуре.
На кривой охлаждения этого сплава (рис. 49) будем иметь точку
перегиба при температуре tri, так как процесс выделения из
жидкости первичных кристаллов сопровождается выделением тепла,
которое лишь частично возмещает отводимое тепло и сказывается только
в замедлении охлаждения. Далее, при температуре te кривая
охлаждения будет иметь горизонтальный участок, так как процесс одно-
12* 179
димое тепло
<3
с;
t
П
временного выпадания кристаллов А и В сопровождается
выделением такого количества тепла, которое полностью возмещает отво-
Охлаждение сплава ниже температуры
кристаллизации эвтектики (te) происходит по
закону, имевшему место выше
температуры trxy так как охлаждение
не сопровождается выделением тепла.
На рис. 50 представлены наиболее
характерные кривые охлаждения
ряда сплавов, по которым строится
диаграмма состояния с простой
эвтектикой.
Кривая А соответствует
охлаждению чистого компонента А. В
полном соответствии с правилом фаз
(равновесие двух фаз однокомпонент-
ной системы является
безвариантным равновесием) кристаллизация
чистого компонента происходит при
постоянной температуре, а именно
•е
L+A
L+A+B
О
Время
Рис. 49. Кривая охлаждения
сплава г (см. рис. 48)
при tJ
Кривые охлаждения I и II
соответствуют охлаждению доэвтек-
тических сплавов I и II. Кристаллизация этих сплавов протекает
в две стадии: сначала выпадают первичные кристаллы А (в сплаве /
начиная с температуры tl9 а в сплаве // — с температуры /2)» а за"
Q
Время
Концентрация
Рис. 50. Кривые охлаждения (слева) сплавов, образующих систему
с простой эвтектикой, и соответствующая диаграмма состояния (справа)
тем происходит эвтектическая кристаллизация. Так как условия
одновременного насыщения жидкости кристаллами А и В для
различных сплавов одни и те же, то эвтектическая кристаллизация
всех сплавов происходит при одной температуре, а именно te.
180
Кривая охлаждения /// соответствует охлаждению сплава чисто
эвтектического состава.
Интересно было бы выяснить структуру сплавов этой системы.
Для этой цели обратимся к рис. 51, на котором представлено
схематическое изображение микроструктуры пяти сплавов системы
с простой эвтектикой соответственно положению их на диаграмме
состояния рис. 50. Для большей наглядности кристаллы компонента А
в сплавах № 1 и 2 изображены с четко очерченными гранями (что
может свидетельствовать о малом поверхностном натяжении на
границе раздела кристалл—жидкость в момент кристаллизации),
а кристаллы В в сплавах № 4 и 5 изображены с округленными гра-
№1 №9 №3 №4- , N95
Рис. 51. Микроструктура сплавов системы с простой эвтектикой
(схема)
нями (что условно отвечает большому поверхностному натяжению
на их поверхности). Мелкая смесь кристалликов А и В в сплаве
№ 3, представляющая собой эвтектику, изображена с помощью
коротких разбросанных штрихов.
Впервые процесс эвтектической кристаллизации детально
исследовал А. А. Бочвар. Он показал, что в одних местах охлаждающегося
расплава при достижении жидкостью эвтектического состава и
температуры (соответствующей температуре кристаллизации
эвтектики) зарождаются кристаллы одного компонента, а в других
местах— другого компонента. Так, например, в каком-то объеме
охлаждающейся жидкости возникает кристаллический зародыш
компонента А. По мере роста зародыша компонента А (т. е. по мере
осаждения на нем атомов компонента А) окружающая этот кристалл
жидкость обогащается другим компонентом и, таким образом,
создаются благоприятные условия для зарождения
кристаллического зародыша компонента В. Как только рядом с кристаллом
компонента А появится кристаллик компонента 5, создадутся условия
для разразнивающей диффузии, т. е. условия для выхода каждого
из двух видов атомов, находящихся в жидкости (А и В), к своим
центрам кристаллизации. Дальнейшая кристаллизация вследствие
этого протекает со скоростью, превышающей скорость
кристаллизации компонентов А и В из жидкости того же состава. При этом
в процессе эвтектической кристаллизации одна фаза или один из
компонентов играет ведущую роль. Очевидно, что роль ведущего
будет играть тот компонент, который имеет большую скорость
кристаллизации.
181
Кривые фазового равновесия на диаграмме состояния системы
с простой эвтектикой (ае и еЬ на рис. 48 и 50) могут быть описаны
с помощью теории растворов.
Рассматривая компонент А как растворитель, а компонент В —
как растворяемое вещество, можно видеть, что кривая ае на рис. 48
и 50 является кривой, отображающей понижение температуры
плавления (или замерзания) компонента А при добавлении компонента В.
Эта кривая может быть описана из условия равенства химических
потенциалов жидкого раствора В и А (|Лл) и кристаллов А {\ьта)'*
\1а = \ьа- (361)
Далее, используя уравнения
Ра = (\Сдж + RTlnxA+(l- xAfWm (362)
и
^ = (^Ау\ (363)
находим понижение температуры плавления компонента А. Имея
Н% - TS% + RT InхА + (1— xa)2Wx = Ял - TSTj?, (364)
получаем
ДУ ^1А—1 щ^ Д5Л ^bi)>
где
ЬНА = т-Ил, ASA = AHA/TA = S%-ST?
и 7л—энтальпия, энтропия и температура плавления
компонента А. Соответственно отсюда при Wm = 0 (идеальный раствор),
ТАТ ^ Т\ и In хА ^ —хв при хА —> 1 (разбавленный раствор)
получаем
AT = TA - Т ~ ^ хв. (366)
Рассматривая компонент В как растворитель, а компонент Л как
растворенное вещество, для кривой eb на рис. 48,50 получаем:
ы __iB-i ___—__— + —_—, (^ь/)
где ДЯБ = Нв- НЪ\ ASB = АЯБ/ГБ - Sg — SbB и ГБ
-энтальпия, энтропия и температура плавления компонента В соответственно.
Предполагая, что раствор идеальный и разбавленный, получаем:
АТ = Тв-Т~-г^хА. (368)
Выражения (366) и (368) известны как формулы Вант-Гоффа
для понижения температуры плавления (или замерзания)
растворителя. Множители RT2jAHA и RT%/AHb определяются только
свойствами растворителя и не зависят от свойств растворяемого вещества;
182
они получили название криоскопических постоянных. Понижение
температуры растворителя пропорционально мольной доле
растворенного вещества при образовании им с растворителем идеальных
и разбавленных растворов. Однако, если W}K =f= 0, то это, согласно
формулам (365) и (367), скажется на величине понижения
температуры плавления; при Wm > 0 понижение температуры плавления
1-х ТТЛ/
будет меньше, чем для идеальных растворов, на величину д^ \уж,
а при Wm <0 — больше на ту же величину.
Рассмотренные аналитические выражения позволяют
энергетически охарактеризовать взаимодействие компонентов, если линии
ликвидус построены экспериментально. По известным значениям
АНА и Тл или АНВ и Тв можно рассчитать энергию смешения вдоль
ликвидуса [для этого преобразуем формулы (365) или (367)]:
AHA(l-^-\+RTlnxA
W* = L^y (369)
или
АН в (1 - -yA + RT in хв
W* = ^^ • (370)
Зная Wm, по формулам (247) и (248) могут быть рассчитаны
соответствующие активности и коэффициенты активности. При
1^ж = 0 появляется возможность определить термодинамические
характеристики компонентов. На это указывают приближенные
формулы (366) и (368). Кроме того, из формул (369) и (370) получаем:
in г __^Н{ТА-Т) _АНА 1 ЛЯЛ mn
ШХа ~~ WJT ~" R ' Т ~ЖХ { >
и
)nv -АНв(Тв-Т) _АНВ 1 АНВ П7сА
m хв _ ^-^ _______ ______ {ди)
Это — логарифмики Шредера—Ле-Шателье, которые показывают,
что для идеальных растворов кривые ликвидуса на диаграммах
состояния систем с простой эвтектикой могут быть спрямлены путем
представления их в координатах In xi =г-. Тогда тангенс угла
наклона прямой даст возможность определить энтальпию (скрытую
теплоту) плавления, а отрезок на оси ординат — температуру
плавления Чем. правые равенства в формулах (371) и (372)]. Расчеты
могут быть произведены графически и аналитически; при этом надо
иметь в виду, что In А == 2,303 lg Л, R = 1,987 и 2,303-1,987 =
= 4,573. Придадим формулам (371) и (372) расчетный вид:
где xt выражается в мольных долях, Д#, —в кал/моль (или кал/г-атом)
и 1\ — в градусах абсолютной шкалы.
183
Если на оси ординат откладывать lg х, а на оси абсцисс — 103/7\
то тангенс угла наклона прямой в этих координатах (tg а) будет
отрицательным, так как угол будет тупым (а > 90°). Энтальпия
плавления определится из произведения
AHt - —4,573 tg a, (374)
где Д#; будет иметь размерность ккал/моль (или ккал/г-атом).
Пример. На рис. 52 приведены две экспериментально изученные
диаграммы состояния: германий—антимонид алюминия (а) и крем-
1100
1500
1300
1/00
900
700 ^——
Ge 20 ЬО 60 80 AlSb Si 20 40 60 60 AlSb
Концентрация, % (пол.) Концентрация, % (мол.)
Рис. 52. Диаграммы состояния систем германий—антимонид алюминия (а)
и кремний—антимонид алюминия (б)
19*г.р
19U
1,68
1,82
^
а
X
N
ЧЧ
<р{\
..ТуЛ .
0,830
0,880
1000
Т(°Ю
1,UO
0600
0700
1000
т(°Ю
Рис. 53. Кривые ликвидуса диаграмм состояния германий—антимонид
алюминия (а) и кремний—антимонид алюминия (б) в координатах
1n*Ge(si)-10»/7TK)
ний—антимонид алюминия (б). Точки на диаграммах состояния
соответствуют результатам термического анализа сплавов. Энтальпии
плавления германия и кремния равны 8,2 и 11,6 ккал/г-атом
соответственно. Температуры плавления германия и кремния равны
943 °С (1216 °К) и 1420 °С (1693 °К) соответственно. На рис. 53
эти данные использованы для расчета кривой ликвидуса
компонентов германия и кремния (см. дополнительные построения на рис. 53).
Можно видеть, что в координатах lg xi — 103 Т рассчитанная
зависимость линейна и в обоих случаях совпадает с эксперименталь-
184
ными точками в области х{: —> 1 и Т —> Ть (разбавленные растворы).
Можно также видеть, что растворы германия с антимонидом
алюминия являются идеальными WjK = 0 и расчет совпадает с
экспериментом. Растворы кремния с антимонидом алюминия
характеризуются W}K << 0. Это означает, что разноименные частицы
взаимодействуют с большей энергией, чем одноименные
^AlSb^Si > "у! W^AlSb ^ AlSb + Wsi ^> Si J •
Кроме того, энергия смешения не остается постоянной и уменьшается
по абсолютной величине по мере увеличения температуры и
концентрации кремния.
у
лхву
§ 44. ДИАГРАММЫ СОСТОЯНИЯ СИСТЕМЫ
С ХИМИЧЕСКИМ СОЕДИНЕНИЕМ
Допустим, что компоненты данной системы в жидком состоянии
полностью растворимы друг в друге, а в твердом состоянии
совершенно нерастворимы и образуют химическое соединение, стойкое
при всех температурах вплоть до точки его плавления.
Два компонента могут вступать в химическое взаимодействие
друг с другом, давая химическое соединение
хА + уВ = АХВ,
Свойства соединения
будут совершенно отличными
от свойств образующих его
компонентов А и В.
Соединение с конгруэнтной
точкой плавления
Если химическое соединение
АхВу устойчиво при всех
температурах вплоть до
температуры его плавления, то в этом
случае говорят, что соединение
имеет конгруэнтную точку
плавления.
В простейшем случае
диаграмма состояния
рассматриваемой системы будет иметь
вид, изображенный на рис. 54.
Принцип построения этой диаграммы такой же, как и принцип
построения предыдущей диаграммы. Диаграмма представляет
собой как бы две диаграммы состояния с простой эвтектикой
А—АХВУ и АхВу — В. В ней имеются две эвтектические точки
#! и е2, соответствующие эвтектикам, которые соединение АхВу
образуют с компонентами А и В. Ликвидусом этой диаграммы
185
*ХВЦ
в
Концентрация
Рис. 54. Диаграмма состояния системы,
компоненты которой образуют устойчивое
химическое соединение
является линия ае1те2Ь, солидусом — эвтектические горизонтали
сех& и fe2q.
Наиболее замечательной точкой этой диаграммы является
точка т, для которой концентрации компонентов Л и В в системе
соответствуют химическому соединению АхВу. В этом случае
система ведет себя как однокомпонентная. На кривой охлаждения
имеется горизонтальная температурная остановка, как в случае за-
Время Концентрация
Рис. 55. Кривые охлаждения сплавов системы, в которой образуется
устойчивое химическое соединение, и соответствующая диаграмма состояния
стывания чистого компонента. Горизонтальный участок кривой будет
соответствовать температуре плавления химического соединения
(рис. 55).
Максимум, создаваемый кривой ликвидуса, может быть либо
острым, т. е. кривые сходятся под острым углом, образуя так
называемую сингулярную (особую) точку, или закругленным в той или
иной степени. Последнее обусловливается тем, что химическое
соединение АХВУ при плавлении диссоциирует по уравнению
АхВу = хА + уВ.
Наличие чистых Л и Б в сплаве понижает температуру
плавления АХВУ и переносит ее из точки т1 в точку т (рис. 54). По степени
закругления максимума можно судить о степени диссоциации
химического соединения при плавлении.
Соединение АхВу обычно имеет максимальную в системе
электропроводность, большую твердость и хрупкость. Температура его
плавления может быть выше температуры плавления чистых
компонентов. При всем этом не следует, однако, забывать, что
соединение АХВУ отнюдь не является самостоятельным третьим
компонентом, ибо состав его выражается химическим уравнением
реакции между А и В.
В ряде случаев в системе может образоваться несколько таких
соединений, и диаграмму состояния в этом случае можно представить
себе состоящей из нескольких диаграмм состояния с простой эвтек-
186
тикой. Примером таких двухкомпонентных систем могут служить
Си—Mg, Си—Се, Mg—Si, Mg—Sn, Mg—Pb, Mn—P, Bi—Fe, Ca—Mg
и др.
Рассмотрим примеры кристаллизации некоторых сплавов,
обозначенных на рис. 55. Стадии кристаллизации сплава / можно
записать так:
t —t
L-Л эът (А-\-AxBy).
Сплав // кристаллизуется только в одну стадию:
L-Л эвт (А+ АхВу).
Сплав /// кристаллизуется в две стадии:
L —Л АХВУ + L (3 - в1),
L Л эвт {А+ АХВи).
Наконец, сплав АХВУ кристаллизуется как чистый компонент:
L ^ АХВУ.
Соединение с инконгруэнтной точкой плавления
Рассмотрим систему, в которой два компонента в жидком
состоянии полностью растворимы друг в друге, а в твердом состоянии
нерастворимы и образуют химическое соединение, которое не может
быть расплавлено без разрушения. Этот тип диаграммы состояния
системы относится к случаю, когда химическое соединение,
образованное при взаимодействии компонентов, настолько неустойчиво
при высоких температурах, что до плавления оказывается нацело
диссоциированным по схеме:
АХВУ = хА + УВ.
Так как такое химическое соединение не может быть расплавлено
без полной диссоциации, то кривая ликвидус не имеет максимума,
и диаграмму этого типа иногда называют диаграммой состояния
со скрытым максимумом.
Диаграмму состояния со скрытым максимумом имеют многие
двухкомпонентные системы, причем часто в системе образуется
несколько неустойчивых химических соединений (например системы
Сг—С, Си—Ga и др.).
Рассмотрим простейший случай, когда в системе образуется одно
неустойчивое химическое соединение. Диаграмма состояния этого
типа представлена на рис. 56. Линия aecf представляет собой
ликвидус. Прямые beg и cmd — солидус. Прямая beg в то же время
является эвтектической горизонталью и соответствует температуре
187
кристаллизации эвтектики. Прямая fimd носит название перитекти-
ческой горизонтали или линии разрушения химического соединения
и соответствует температуре, при которой происходит разрушение
химического соединения при нагревании.
Важно отметить два следующих обстоятельства, которые
оказывают значительное влияние на характер превращений в сплавах,
составляющих систему с неустойчивым химическим соединением.
Эвтектическая жидкость (состава точек е и е') насыщена как
относительно кристаллов чистого
компонента Л, так и
относительно кристаллов
химического соединения АХВУ,
составы которых на оси
концентраций лежат по
разные стороны от состава
эвтектической жидкости.
Вследствие этого при охлаждении
жидкости происходит
одновременная кристаллизация
кристаллов А и кристаллов
АхВу-
Жидкость состава точки р
(или pf) также одновременно
насыщена и по отношению
к кристаллам чистого
компонента В, и по отношению
к кристаллам химического
лежат по одну сторону от
А
Рис,
А2В,
У
6
Концентрация
56. Диаграмма состояния с
неустойчивым химическим соединением
но составы их
соединения АхВуу
состава эвтектической жидкости. Это последнее обстоятельство
делает невозможным образование кристаллов одного типа без
растворения кристаллов другого типа. Например, образование
кристаллов АхВу из жидкости состава р происходит при
одновременном растворении в ней кристаллов компонента В.
Для уяснения сущности происходящих в этой системе процессов
рассмотрим ход кристаллизации некоторых сплавов этой системы,
кривые охлаждения которых представлены на рис. 57.
Состав сплава / соответствует эквивалентным количествам ком*
понентов А и В согласно реакции:
хА + уВ = АхВу.
Следовательно, в данном случае оба компонента нацело
связываются в химическое соединение, которое, однако, устойчиво лишь
ниже температуры tp. Выше этой температуры химическое
соединение разлагается на составляющие его компоненты А и В. Как
видно из диаграммы состояния, компонент В при температурах
выше tp существует в виде твердой фазы, находящейся в равновесии
с жидкостью, содержащей некоторое количество В. Другими словами,
жидкость сплава / выше температуры tp является насыщенной по
отношению к кристаллам В и выделение этих кристаллов из жидкости
188
будет первой стадией кристаллизации сплава 7. Затем при
постоянной температуре (согласно правилу фаз — это безвариантное
превращение, так как F = k — у + I =2 — 3+1 =0) произойдет
растворение этих кристаллов в жидкости и одновременно выделение
Так как в сплаве I компоненты А и В находятся в стехиометри-
ческом соотношении, то при температуре tp сплав нацело
превращается в кристаллы химического соединения АХВУ (без избытка
кристаллов В или жидкости).
Легко видеть, что если рассматривать сплавы, лежащие правее
ординаты АхВу1 т. е. сплавы, в которых имеется избыток компо-
Рис. 57. Кривые охлаждения сплавов с неустойчивым химическим
соединением и соответствующая диаграмма состояния
нента В против необходимого для образования АХВУ количества,
то компонент В не сможет войти целиком в состав этого соединения
(так как весь компонент А окажется связанным раньше, чем
израсходуется весь компонент В) и избыток В войдет в структуру твердого
из нее кристаллов химического соединения АхВу. Такой процесс
называют перитектическим.
Итак, имеем следующие стадии кристаллизации сплава /:
L—-^B + L(Pl-p);
B + L(p)^AxBy.
состояния в виде отдельной фазы. Таким образом, кристаллизация
сплава // идет по следующим стадиям:
L —-^ В + L (р2 — р), В + L (Р\ -> АхВу + В (избыток).
Очевидно также, что если концентрация сплава лежит левее
ординаты АхВуу то в сплаве после перитектического превращения
окажется в избытке жидкость, состав которой изображается
точкой р. Так как эта жидкость является насыщенной по отношению
к кристаллам химического соединения АхВуу то дальнейшее охлаж-
189
дение вызывает выпадение этих кристаллов. Однако в отличие от
перитектического превращения состав жидкости не восстанавливается
за счет кристаллов В и происходит обогащение жидкости
компонентом А. В результате этого жидкость достигает эвтектического
состава, при котором она одновременно насыщена и чистым
компонентом А и химическим соединением АхВу. Поэтому процесс
кристаллизации сплава /// заканчивается образованием эвтектики
(А + АхВу). Следовательно, имеем четыре стадии кристаллизации
сплава ///:
L XX В + L (р3 - р\ В + L (р) X АХВУ + L (избыток),
LXX АХВУ + L{p-e)%L(е)X эвт(АхВу + А).
Таким образом, в структуре сплава /// будем иметь первичные
кристаллы АХВУ, окруженные эвтектикой (АХВУ + А).
Как видно из диаграммы, те сплавы, фигуративные точки
которых лежат левее точки р, кристаллизуются как простые
эвтектические сплавы. Так, например, сплав IV кристаллизуется в две стадии:
L —X АХВУ + L (e1 -e)y L X эвт (АХВУ + А).
Нередки случаи, когда в двухкомпонентных системах образуется
несколько химических соединений, причем одни из них имеют
конгруэнтную, а другие — инконгруэнтную точку плавления.
Например, в системе MgCl2—Н20 образуются пять различных
кристаллогидратов, однако только один из них плавится конгруэнтно. В
системе Си—La образуются четыре химических соединения, из
которых только два плавятся конгруэнтно.
Сделаем несколько замечаний относительно природы химических
соединений, которые до сих пор обозначались АХВУ, где А и В —
компоненты соединения; х и у — стехиометрические коэффициенты,
целые числа.
Свойства этих соединений совершенно отличны от свойств
компонентов, входящих в состав соединения. Это связано с тем, что
компоненты вступают между собой в сильное химическое
взаимодействие и образуют кристаллы, обладающие своей особой
кристаллической решеткой.
Прежде всего соединениями типа АхВу могут быть соединения,
формулы которых отвечают следующим правилам валентности:
Mg2Si, Mg2Ge и Mg2Pb (здесь магний обладает валентностью +2,
а элементы IV группы Периодической системы элементов Д. И.
Менделеева — валентностью —4); Mg3P2, MgsAs2, Mg3Sb2 и Mg3Bi2
(магний с валентностью +2 соединяется с элементом V группы,
имеющим валентность —3) и др. Такие соединения, как правило,
образуют элементы — металлы с элементами переходного к
металлоидам типа.
Металлы образуют между собой так называемые
интерметаллические или «электронные» соединения. Соединения этого типа харак-
190
теризуются определенными значениями отношения валентных
электронов к числу атомов, называемыми электронными концентрациями.
Существуют соединения с электронными концентрациями: — (или
1,50), имеющими гранецентрированную кубическую решетку (3,
т. е. атомы расположены по вершинам куба и в центрах его граней,
21
например CuZn, Cu3Al, Cu5Sn и др.; -р^- (или ~1,62), имеющие
сложную кубическую решетку у, например Cu5Zn8, Cu9Al4, Cu3iSn8
7
и др., а также -j- (или 1,75), имеющие гексагональную решетку е,
например CuZn3, Cu5Al3, Cu3Sn и др.
Важную группу соединений образуют так называемые
полновалентные соединения с тетраэдрической координацией атомов. Они
обладают полупроводниковыми свойствами и являются изоэлектрон-
ными аналогами элементов IV группы Периодической системы
элементов Д. И. Менделеева (алмаз, кремний, германий, а-олово).
Структуры этих соединений являются производными от
структуры алмаза. Соединения P-SiC, GaP, GaAs, GaSb, InP, InAs, InSb,
BeS, a-ZnS, CuCl, CuBr и Cul имеют структуру кубической
цинковой обманки или сфалерита, а соединения a-SiC, A1N, GaN,
InN, BeO, ZnO, p-ZnS, CdS, CdSe, CdTe и Agl имеют структуру
гексагональной цинковой обманки или вюртцита. Эти соединения
образуются попарно элементами IV и IV, III и V, II и VI, I и VII
групп Периодической системы и поэтому обозначаются Л1УВ1У,
ЛШВУ, AuBYl и AlBvn. При этом электронная концентрация
оказывается равной 4, каждый атом окружен четырьмя ближайшими
соседями и образуются устойчивые 8-электронные связи, которым
приписывается преимущественно ковалентный тип связи с малой
долей ионной составляющей.
Однако многие соединения, которые обнаруживаются в сплавах,
не поддаются описанию с помощью простых правил.
Ликвидус химических соединений может быть рассчитан с
помощью формул теории растворов. Например, используя понятие
энергии смешения
w = wab-Waa~w™,
для соединения типа А В может быть получено выражение:
\пЩ\-х) = -Шж.±г+Щ^- 2(*-r°'5)2 W*, (375)
где АНАВ и ТАВ — энтальпия (теплота) и температура плавления
соединения АВ. Если линия ликвидуса построена экспериментально,
то формула (375) может быть преобразована для определения энергии
смешения вдоль ликвидуса:
Wm = - RT
2(х— 0,5)
ln4x(I-x) + |^j(-^-l)]. (376)
191
Здесь в формулах (375) и (376): х — атомная доля того
компонента, со стороны которого рассматривается ветвь ликвидуса
соединения.
Пример. Термодинамический анализ кривых ликвидуса (со
стороны компонентов Аш) для соединений AmBw показал, что
энергия смешения отрицательна и зависит от температуры, а
следовательно, и от концентрации. Поэтому исследованные системы
(Ga—Р, In—Р, Ga—As, In—As, Ga~Sb и In—Sb) не могут быть
отнесены к регулярным. Во всех системах по мере приближения
к эвтектической точке, которая расположена вблизи компонента Л111,
энергия смешения стремится к нулю. Сильное взаимодействие
разноименных атомов вблизи максимума постепенно уменьшается, и
раствор приближается к идеальному вблизи эвтектической точки.
Важно отметить, что при W)K = О формула (375) упрощается
и приобретает вид:
l„4*(l-*) = -^.-f+ ^, (377)
откуда следует, что в координатах In 4х (1 — х) — ЦТ ликвидус
соединения становится прямолинейным и по тангенсу угла наклона
прямой можно определить энтальпию (теплоту) плавления, а по
отрезку на ординате — температуру плавления соединения.
§ 45. ДИАГРАММА СОСТОЯНИЯ С НЕОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ КОМПОНЕНТОВ В ЖИДКОМ
И ТВЕРДОМ СОСТОЯНИИ
Переходя к изучению системы, в которой оба компонента во
всех соотношениях растворимы друг в друге как в жидком, так и
в твердом состоянии, необходимо прежде всего познакомиться
с новым понятием — твердый раствор.
Как известно, кристаллическое строение веществ
характеризуется вполне определенным пространственным расположением в
кристалле атомов, ионов и молекул. Поэтому, если мысленно соединить
линиями эти частицы, то получим некоторую решетчатую структуру,
называемую пространственной кристаллической решеткой (рис. 58).
Для каждого вещества характерен определенный тип
кристаллической решетки и определенное расстояние между частицами. Однако
это представление структуры в известной степени является
модельным и понятие «кристаллическая решетка» следует воспринимать
как некоторую условность.
Взаимное расположение и связь атомов обусловлены силами
взаимодействия между ними.
В случае ионной решетки (т. е. такой решетки, в узлах которой
располагаются ионы) связь осуществляется электростатическим
притяжением разноименных ионов, что схематически представлено
на рис. 59, а. Такого рода связь называется ионной,
электровалентной или гетеропол ярной' связью и
является характерной для многих неорганических веществ, солей.
192
Энергия связи подобных веществ достаточно велика. Они имеют
чаще всего высокую температуру плавления и значительную
механическую прочность.
В случае молекулярной решетки (т. е. такой решетки, в узлах
которой расположены молекулы) связь осуществляется силами
Ван-дер-Ваальса, т. е. притяжением взаимно поляризованных мо-
Рис. 58. Пространственная кристаллическая решетка
лекул (рис. 59, б). Чаще всего такое строение имеют органические
кристаллические вещества. Силы эти невелики, поэтому такие
вещества обычно весьма легкоплавки и обладают малой механической
прочностью.
Наконец, к третьему типу относятся вещества с атомной
решеткой, в узлах которой расположены отдельные атомы. Типичными
©
о б в
Рис. 59. Схематическое изображение различных видов химической связи:
а —- ионная связь; б — ван-дер-ваальсова связь; в — ковалентная связь
представителями этого класса веществ являются многие металлы.
Связь атомов такой структуры, обычно называемая атомной,
ковалентной или гомеополярной связью,
осуществляется взаимным притяжением атомов или ядер атомов к
обобщенным валентным электронам, которое уравновешивает силу
отталкивания между самими атомами. Для уяснения сущности
явления обобщения или, иначе говоря, коллективизации электронов
обратимся к схеме, изображенной на рис. 59, в. При сближении
атомов на расстояние, соизмеримое с величиной электронных орбит,
13
А. Н. Крестовников
193
a
б
Рис. 60. Кристаллическая решетка
простого вещества (А) (а) и
твердого раствора замещения В в А (б)
очевидно, возможен случай, когда электрон атома 2 окажется под
более сильным воздействием ядра атома /, чем под воздействием
ядра атома 2, и тогда электрон атома 2 опишет свой путь вокруг
ядра атома 1; затем электрон, попадая вновь под воздействие поля
ядра 2, опишет свой путь вокруг ядра атома 2 и т. д. В результате
такого движения электрон описывает вокруг обоих атомов путь
в виде восьмерки (так называемый «обменный эффект»). Подобным
же образом ведет себя и электрон атома 2. Таким образом,
произошла коллективизация двух
электронов, они стали общими для обоих
атомов.
При большом числе атомов и
электронов картина делается
значительно сложней.
Атомная или ковалентная связь,
осуществляемая коллективизацией
электронов, является весьма
прочной.
Однако прочность этого вида
связи в значительной степени
определяется природой вещества. Достаточно указать на такие
различные в химическом отношении вещества, как натрий и вольфрам.
Хотя тип связи у них одинаков, натрий плавится при температуре
97,7° С, а вольфрам — при температуре 3377 °С.
Рассмотрим вопрос о том, каким образом будут образовываться
кристаллические решетки при тесном соприкосновении одного
вещества с другим (например, при охлаждении расплава этих
веществ) .
Пусть имеем в сплаве компоненты Л и В. При некоторых
условиях часть узлов решетки вещества Л (рис. 60) может оказаться
занятой атомами (ионами, молекулами) вещества В. В результате
получаются кристаллы, имеющие тип кристаллической структуры,
подобной структуре вещества Л, но в состав которых входят, кроме
атомов Л, еще и атомы В (рис. 60). Такие кристаллы и носят
название твердых растворов.
Очевидно, что свойства твердого раствора определяются
относительным содержанием входящих в его состав компонентов. Однако
эти свойства отнюдь не являются средними по отношению к
свойствам компонентов Л и В. Зависимость большинства свойств твердых
растворов от их состава носит криволинейный, а не прямолинейный
характер.
Вопрос о том, решетка какого компонента лежит в основе твердого
раствора, определяется посредством рентгенографического анализа.
Каждой кристаллической структуре соответствует определенная
система линий на рентгенограмме.
Ренгтенограмма представляет собой фотографическую пленку,
на которой под воздействием рентгеновских лучей, прошедших через
кристаллы какого-либо вещества и отраженных атомными рядами
этих кристаллов, появляется ряд линий, размещение которых опре-
194
деляется расположением атомов в кристаллах исследуемого вещества,
а следовательно, определяется кристаллической структурой этого
вещества. Таким образом, если в нашем распоряжении имеются
рентгенограммы компонентов А и В и их твердого раствора (рис. 61),
то можно с полным основанием считать, что твердый раствор
образован на основании кристаллической решетки компонента Л,
поскольку характер линий и их относительное расположение на
рентгенограмме компонента А и твердого раствора сходны.
При этом, однако, изменяется расстояние между линиями
рентгенограммы (21 ф 21г), что говорит об изменении параметров
решетки при образовании
твердого раствора при ее
неизменном строении. Это
и понятно, ибо размеры
атомов (молекул, ионов)
обоих компонентов
неодинаковы.
Из сказанного должно
быть ясно, что
непрерывный ряд твердых
растворов могут образовывать
вещества, имеющие
одинаковый тип
кристаллической решетки и
довольно близкие по величине
межплоскостные
расстояния, а также более или
менее близкое атомное
строение (т. е. должны быть близко расположены в Периодической
системе элементов Д. И. Менделеева). Последнее условие
необходимо потому, что при растворении элементов, имеющих различную
валентность, сильно меняется электронная концентрация (т. е.
количество электронов на один атом), а это ведет к изменению самой
решетки; непрерывного ряда твердых растворов с одним и тем же
строением решетки в этом случае не получается.
Диаграмма состояния двухкомпонентной системы, в которой
образуется при застывании расплава непрерывный ряд твердых
растворов, показана на рис. 62 и бЗ.Такую диаграмму состояния имеют
многие двухкомпонентные сплавы. Так, неограниченная
растворимость компонентов наблюдается в системах Си—№ Fe—Ni Fe—Co
Mo—W, Ge—Si, Be-Sb и др. '
Линия tBabcdtA на рис. 62 представляет собой линию ликвидус
диаграммы; линия t^xbxcxdxtA — линию солидус. Область,
заключенная между линией солидус и линией ликвидус, соответствует
области двухфазного равновесия жидкости и кристаллов твердого
раствора.
Так, в сплаве / при температуре /а в равновесии будут
находиться жидкость состава b и кристаллы состава Ьи а при
температуре t8 — жидкость состава с и кристаллы состава сг. Относи-
16 195
Рис. 61. Пример рентгенографического
исследования решетки твердого раствора:
а — рентгенограмма компонента А\ б ~
рентгенограмма компонента В; в — рентгенограмма твердого
раствора
тельные количества жидкости и твердой фазы находятся по правилу
рычага. Так, например, при температуре /2 имеет место следующее
соотношение:
% а ~~ lb '
а при температуре t3 для того же сплава:
% L тс1
% а тс
Плавность хода кривой ликвидус с постепенным понижением
температуры плавления от tB (более тугоплавкий компонент) к tA
, , _». !__ . 6
Время Концентрация
Рис. 62. Диаграмма состояния и кривые охлаждения сплавов системы
с образованием непрерывного ряда твердых растворов
(менее тугоплавкий компонент) позволяет сделать вывод, что все
сплавы данной системы имеют однотипную кристаллическую
решетку. В связи с этим нет смысла различать понятия «твердый
раствор А в В» и «твердый раствор В в Л», поэтому все кристаллы
твердой фазы данной системы принято называть а-твердым раствором.
Затвердевание сплавов (сплав /) происходит в интервале
температур (от t± до Qy так как система при этом находится, согласно
правилу фаз, в состоянии одновариантного равновесия:
F = й —# + 1 = 2 —2 + 1 = 1.
При этом процесс кристаллизации сопровождается изменением
состава как жидкости (по линии ликвидус), так и выпадающих
кристаллов твердого раствора (по линии солидус).
Следуя принятой нами схеме записи процессов кристаллизации,
можно записать процесс кристаллизации сплава / так:
L ——+ а (а± — d±) -f L (a —■ d).
196
По мере изменения концентрации жидкой фазы с понижением
температуры количество этой фазы все уменьшается, а когда
ордината сплава пересекает линию солидус, то жидкая фаза, состав
которой в этот момент определяется точкой d, вовсе исчезает и сплав
целиком переходит в твердое состояние. Не следует забывать, что,
говоря о равновесии жидкой и твердой фаз при той или иной
температуре кристаллизационного интервала, мы имеем в виду средний
состав как всей выпавшей к этому моменту твердой фазы, так и
оставшейся жидкой. Но так как при каждой более низкой температуре
выпадающие новые порции твердой фазы отличаются по
относительному содержанию Л и В от ранее выпавших (в сторону уменьшения
в их составе атомов более тугоплавкого компонента В), то очевидно,
что в твердом состоянии кристаллы сплава не будут однородны по
химическому составу. Центральные области кристаллических зерен,
закристаллизовавшиеся раньше, будут обогащены более
тугоплавким компонентом. Однородность достигается диффузионным путем
в твердом состоянии.
Для описания кривых ликвидуса и солидуса системы с
неограниченной растворимостью компонентов в жидком и твердом
состоянии воспользуемся равенством химических потенциалов обоих
компонентов в жидкой и твердой фазах, находящихся в равновесии.
Введем обозначения: хА и хв — концентрации (мольные доли)
компонентов А и В соответственно в жидкой фазе; уА и ув — те же
концентрации- в твердой фазе; W}K и WTB — энергии смешения
компонентов в жидкой и твердой фазах соответственно.
Тогда:
= Н% — TS% + RT In xA + (1 ~ xAfWA\
tf = ^ + RT In yA + (1 - yA)2Wn -
= HTI - TSTI + RT In yA + (1 ~ у л) Vtb
^ = ц0ж + RT In xB + (1 - xB)2Wm =
= Я5 - TS2 + RT In xB + (1 - xB)2Wy\
\iT£ = \ioB + RT In у B + (1 - yBfwn =
- HBB - TSBB + RT In уB + (1 - yB)2WTB.
Приравнивая
Ж ТВ Ж ТВ
И'Л — \*>А, И \1В = \1В ,
получаем
T==J_ xBWm-y2BWTB+RMA
Ta Уа
и
т-JL *л^ж - У2л^тв + R АЯБ
Тв Ув
(378)
(379)
(380)
(381)
(382)
(383)
197
где
ASA =
Та
HZ-tf?, AHB
= S^ — S^, AS# :
Я ж
7в
Ятв
Б >
Эж
,4
STB
ТА и ТБ — энтальпии (теплоты), энтропии и температуры
плавления компонентов А и В соответственно.
*3
^
^
1
Ч)
С;
t:
й?
tA
tmax
L + oc
а
L
а
I
1
*л
ОС
L + CL
+ ■
Lrmn
L
•
и
6
Концентрация
В
Концентрация
В
Рис. 63. Диаграммы состояния систем с неограниченной растворимостью
компонентов в жидком и твердом состоянии:
а — с max; б — с min
Зависимость Т от х и у выразить в явном виде удается лишь для
случая Wm — WTB = О, когда жидкий и твердый растворы будут
идеальными. Тогда:
АНв / 1 1 \
ХА
1
в
АН
R
A /J 1
~\ Т Т
А
АНВ /J 1
R \R T
— р. ч
В<
И
1
АНВ ( 1
R \ Т Т
В,
Уа =
*"а/ 1
1
1-е R \т Т*<
АНВ ( 1 1_
# I т т
в,
(384)
(385)
Это — уравнения Зельца. Из них можно получить путем деления
друг на друга уравнения Ван-Jlaapi:
= ьнА ( 1 1
У А ~
Ш-^ =
R
ТЛ
)
(386)
и
1п JUL = Анв
Ув
R
\Т ТВ)'
(387)
198
Нетрудно видеть, что при уА = 1 и ув = 1 уравнения Ван-
Лаара переходят в логарифмику Шредера—Ле-Шателье [см.
формулы (371) и (372)].
Интересный анализ уравнений (382) и (383) был выполнен
Д. С. Каменецкой. Она показала, что при Wm =h WTB, W* <2RT
и W™ <^2RT ликвидус и солидус имеют общую экстремальную
точку с координатами:
ASA - -|/~Д^ __ (Д5л __ Д5б)
Хв-Ув- ASa _ ASb
(388)
и
7 = Г^ + юл ** =Г* + здд ** (389)
При этом, если №ж < №тв, то Т < Тл и Т > 7В, т. е. получается
диаграмма состояния с максимумом (рис. 63, а). Если №ж < WTB,
то 7 < ТА и 7 < ТБ, т. е. получается диаграмма состояния с
минимумом (рис. 63, б). ,
§ 46. ДИАГРАММА СОСТОЯНИЯ С ОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ КОМПОНЕНТОВ
В ТВЕРДОМ СОСТОЯНИИ
Диаграммы состояния, рассмотренные в §§ 43 и 44, хотя и
встречаются в металлургической и технологической практике, тем не
менее являются упрощенным и идеализированным изображением
большинства диаграмм состояния. Представить себе полное
отсутствие растворимости компонентов в твердом состоянии можно лишь
теоретически, практически же даже при очень значительном различии
атомов взаимодействующих веществ образование твердых растворов
(см. § 45) и некоторая растворимость должны иметь место.
Однако растворимость может быть настолько малой, что при
используемых для изучения диаграмм состояния методах она не может
быть определена.
Диаграмма состояния системы, в которой оба компонента во
всех соотношениях растворимы в жидкой фазе, а в твердом
состоянии обладают ограниченной растворимостью, представлена на
рис. 64. До некоторой концентрации (а или с) атомы компонента В
статически равномерно распределяются в решетке растворителя Л,
и структура сплава в твердом состоянии представляет собой
однородный твердый раствор В в Л (а-твердый раствор). Линия ас
является линией предельной растворимости для а-твердого раствора.
С другой стороны, если к компоненту В будем добавлять
компонент Л, то образуется однородный твердый раствор Л в В, который
назовем р-твердым раствором. Линия bd диаграммы представляет
собой линию предельной растворимости для Р-твердого раствора.
199
Итак, на диаграмме состояния область AtAacA соответствует
а-твердому раствору, область BtBbdB — р-твердому раствору.
Линия tAetB является ликвидусом, а линия tAaebtB — солидусом
диаграммы. Прямая aeb есть эвтектическая горизонталь. При
температуре te жидкость состава е является насыщенной по
отношению к а-твердому раствору состава а и по отношению к р-твердому
Время Концентрация
Рис. 64. Диаграмма состояния и кривые охлаждения сплавов системы
с ограниченной растворимостью компонентов в твердом состоянии
раствору состава 6, т. е. все сплавы, линия фигуративной точки
которых при охлаждении пересекает эвтектическую горизонталь,
будут иметь последней стадией кристаллизации образование
эвтектики, состоящей из мелкой смеси кристаллов а- и Р-твердых
растворов. При этом в доэвтектических сплавах эвтектической
кристаллизации будет предшествовать образование кристаллов а-твердого
раствора, а в заэвтектических — образование кристаллов Р-твердого
раствора.
Рассмотрим кристаллизацию некоторых сплавов. Процесс
кристаллизации сплава / ничем не отличается от кристаллизации
сплавов системы с образованием непрерывного ряда тведых растворов:
1(1__2)-Ла(Г-2').
Сплав // кристаллизуется в две стадии:
L —^ а (3' — а) + а (3 — е),
L(^a(a) + P(4).
В этом случае при эвтектической температуре жидкая фаза
становится насыщенной как по отношению к a-кристаллам, так и по отно-
200
шению к компоненту В, который, кристаллизуясь, растворяет в своей
решетке какое-то количество компонента Л, образуя твердый
раствор |3.
Кристаллизация сплава ///, очевидно, не отличается от
кристаллизации сплава /. Однако вместо кристаллов а-твердого раствора
образуются кристаллы (3-твердого раствора, по отношению к
которым оказывается насыщенной жидкость сплава ///:
L(4-5)—^|J(4'-5').
Рассматривая эту диаграмму, полагаем, что растворимость
компонентов в твердом состоянии с температурой не меняется (линии ас
и bd являются вертикальными прямыми). Однако в большинстве
случаев понижение температуры обычно приводит к некоторому
уменьшению растворимости (с понижением температуры
уменьшается предел растворимости как компонента В в решетке
компонента Л, так и компонента Л в решетке компонента В). Это
изменение описывается кривыми ас и bd на рис. 65, который представляет
собой обычный вид реальной диаграммы состояния с ограниченной
растворимостью в твердом состоянии.
Время Концентрация
Рис. 65. Диаграмма состояния и кривые охлаждения сплавов системы
с ограниченной растворимостью в твердом состоянии (в случае изменения
растворимости с температурой)
Рассмотрим процессы, протекающие при охлаждении сплавов
этой системы. Наибольший интерес для нас будут представлять
сплав / и сплав // (рис. 65). Первую стадию кристаллизации сплава /
можно записать так:
L —^ а (V — 2') + L (1 — 2).
201
В интервале температур от t2 до £4 происходит простое
охлаждение без каких-либо превращений, после чего
а (2')
t.— t
КОМИ
а(4 — с) + р(5 — d).
Атомы компонента J3, выходя из решетки ос-твердого раствора,
образуют решетку компонента В, которая содержит некоторое
количество атомов компонента Л, т. е. образуется Р-твердый раствор.
Концентрация
Рис. 66. Диаграмма состояния системы
с ограниченной растворимостью в твердом
состоянии в случае образования
компонентами устойчивого химического
соединения
Концентрация
Рис. 67. Диаграмма состояния системы
с ограниченной растворимостью в
твердом, состоянии в случае образования
компонентами неустойчивого
химического соединения
Сплав // кристаллизуется в три стадии:
L—^-i<x(3' — a) + L(3 — e), L~4> a (a + P(u),
t —t
, ч е коми . ч
a (a) ► a (a — c),
p {b) If^L0^ p ф - d).
Системы, имеющие рассмотренный тип диаграммы состояния,
очень многочисленны. Среди них можно назвать Zn—Cd, Си—Ag,
Сг—Ni, Sn—Pb, KN03—NaN03 и др.
Явление ограниченной растворимости в твердом состоянии может
иметь место и в системе с образованием устойчивого химического
соединения (рис. 66), и в системе с образованием неустойчивого
соединения (рис. 67). В этих системах образуются а-твердый раствор
на основе решетки компонента Л, Р-твердый раствор на основе
решетки компонента В и ^-твердый раствор на основе решетки
химического соединения АхВу, который часто называют
промежуточной фазой.
Сделаем ряд замечаний о промежуточных фазах.
Прежде всего следует обратить внимание на то, что различные
компоненты могут образовывать промежуточные фазы с различными
интервалами гомогенности по оси концентраций. Кроме того, одни
202
и те же компоненты могут образовывать в системе по нескольку
промежуточных фаз.
Отсутствие интервала гомогенности можно представить себе
лишь теоретически. Практически же он всегда может быть обнаружен,
если воспользоваться достаточно чувствительными методами
исследования.
Промежуточные фазы могут представлять собой твердые растворы
замещения (см. § 45) на основе соединения. В ряде случаев могут
встречаться твердые растворы вычитания. При этом твердый раствор
на основе решетки химического соединения образуется не путем
замены атома одного компонента в углах решетки на другой атом,
как в твердых растворах замещения, а путем изъятия атомов из
некоторого числа узлов решетки с оставлением этих узлов пустыми.
Так происходит растворение кислорода в закиси железа FeO (при
добавлении кислорода освобождаются узлы, которые занимали
атомы железа), титана или ванадия в карбидах титана или ванадия
соответственно (пустые места при этом образуются в узлах, ранее
занятых углеродом). Твердые растворы вычитания иногда
называются твердыми растворами с дефектной решеткой.
Промежуточные фазы могут быть образованы так, что атомы
металла располагаются в узлах одной из простых кристаллических
решеток (объемноцентрированной кубической, гранецентрированной
кубической или компактной гексагональной), а атомы металлоида
внедряются в пустоты этой решетки. Такие промежуточные фазы
называют твердыми растворами внедрения или фазами внедрения.
Карбиды и нитриды переходных металлов являются типичными
представителями фаз внедрения (WC, TaC, NbC, TiC, VC, TaN,
TiN, WN, ZrN, VN и др.). Для образования фаз внедрения
отношение атомных радиусов, как правило, должно удовлетворять
условию 'металлоида ^ ^»"^металла*
Промежуточные фазы, как уже отмечалось, образуются на основе
химических соединений компонентов. Состав этого соединения
устанавливается по результатам изучения зависимости физических свойств
от химического состава в пределах интервала гомогенности
промежуточной фазы. При пересечении ординаты химического
соединения наблюдается резкое изменение закономерности влияния состава
на рассматриваемое физическое свойство (на кривой
свойство—состав наблюдается сингулярная точка). Промежуточные
фазы с соединением в интервале гомогенности, на которые
приходятся сингулярные точки на кривых зависимости свойств от состава,
получили название д а л ь т о н и-д о в.
Если же в интервале гомогенности промежуточной фазы на
кривых зависимости свойств от состава не обнаруживается
сингулярных точек, то такие промежуточные фазы называются бертол -
л и д а м и и считаются твердыми растворами на основе мнимых
(не существующих при данных условиях) соединений.
Мы уже познакомились с особенностями превращения (см.
рис. 57), которое носит название перитектического. Рассмотрим
теперь другой случай перитектического превращения, отличающегося
203
от рассмотренного тем, что температура начала кристаллизации
в системе непрерывно повышается от одного компонента к другому
(рис. 68). Такой тип диаграммы состояния имеют системы Си—Со,
NaN03—AgN03 и др.
Наличие ограниченной растворимости компонентов в твердом
состоянии ведет в этом случае к тому, что жидкий раствор,
находящийся в равновесии с двумя насыщенными растворами, содержит
большее количество одного из компонентов, чем каждый из твердых
Рис. G8. Диаграмма состояния системы в случае ограниченной
растворимости компонентов при наличии перитектического превращения
растворов. Этим обстоятельством определяется ряд особенностей
в кристаллизации сплавов, относящихся к этому типу.
Рассмотрим кристаллизацию сплава /. От точки / до точки с
процесс не имеет никаких особенностей и сводится к образованию
кристаллов твердого раствора р, состав которых изменяется по
солидусу от точки /до точки d. При этом состав жидкости
изменяется по ликвидусу от точки / до точки а. В точке с сплав состоит
из жидкой фазы состава точки а. Однако в точке с жидкая фаза
насыщена также кристаллами сс-состава точки с, которые должны
выделиться в условиях некоторого переохлаждения. При переходе
системы из двухфазного состояния в трехфазное число степеней
свободы будет равно нулю:
F = fe — #+1=2 — 3+1=0,
следовательно, превращение должно происходить при постоянной
температуре. При этом происходит уже знакомое нам перитектическое
превращение, заключающееся в выделении кристаллов а-твердого
раствора путем взаимодействия ранее выделившихся кристаллов
204
Р-твердого раствора с жидкой фазой, т. е. кристаллы р будут
взаимодействовать с жидкой фазой и только при этом условии
образовывать кристаллы а. Это превращение в сплаве / будет протекать
до полного исчезновения кристаллов р и жидкой фазы. Дальнейшее
охлаждение этого сплава вызывает выделение кристаллов р-твердого
раствора в связи с уменьшением растворимости компонента В в а-
твердом растворе. Если охлаждение проводить достаточно медленно,
то состав а-твердого раствора должен изменяться по линии сс\
а состав Р-твердого раствора — по линии dd!'.
Таким образом, процесс кристаллизации сплава / можно
записать следующим образом:
L-^$(Y-d) + L(l-a)9 L(a) + $(d)^a(c)y
al£^0^a(c-c') + $(d-d').
В сплаве / относительное количество жидкой и твердой фаз
в начале перитектического превращения
%L _ cd
% Р ~~ ас
точно соответствует полному превращению жидкости и р-кристал-
лов в a-кристаллы при постоянной температуре.
Очевидно, что при кристаллизации сплава // мы будем иметь
иное соотношение:
% L _ fd ^ cd
% Р ~~ ~af ^ ас '
т. е. жидкость исчезает раньше, чем растворяются все р-кристаллы.
В связи с этим кристаллизация сплава // происходит следующим
образом:
L —^ Р (2' — d) + L (2 — d)t
L (а) -\- р (d) —> а (с) -\- р (избыток),
t —t
. ч п коми , /ч
а (с) ► а (с — с ),
p(d)Ji^sp(d_d').
В сплаве /// в отличие от предыдущих сплавов в итоге
перитектического превращения исчезает не жидкая фаза, а кристаллы р-
твердого раствора, так как, согласно правилу рычага:
%L _ bd cd
% Р ~" ~ab > ~ac '
В конце перитектического превращения, когда останутся две
фазы—жидкий раствор состава точки а и кристаллы а-твердого
раствора состава точки с, сплав будет кристаллизоваться путем,
обычным для кристаллизации твердых растворов. Из жидкости,
меняющей свой состав от а до В, будут выпадать кристаллы а,
205
изменяющие состав от с до В'. В интервале температур от t6 д, t7
происходит простое охлаждение, а начиная с температуры t7 до
комнатных температур происходит изменение растворимости, в
результате чего из а-кристаллов выделяются р-кристаллы. Итак, для
сплава /// имеем следующие стадии кристаллизации и
перекристаллизации в твердом состоянии:
LA"^p(3'-d) + L(3-fl),
L (a) -f- P (d) —+a(c)-\-L (остаток),
L(a — В)—~Ла(с — 6').
Затем следует простое охлаждение в интервале температур от t&
до t7 и, наконец,
Ч~ КОМН /\ I О / J/4
а > а(т — с ) -f- р (п — а).
»
Сплав IV будет кристаллизоваться как однофазный твердый
раствор:
1(4 —5)—"i-a (4' —5').
Легко представить себе окончательную структуру сплавов,
процесс кристаллизации которых был рассмотрен. Так, сплав IV будет
содержать при комнатной температуре только кристаллы а-твердого
раствора. Сплавы / и /// будут состоять из кристаллов а-твердого
раствора, причем сплав /// будет содержать большое количество
этих выделений. Наконец, структура сплава // будет представлять
собой конгломерат кристаллов а-твердого раствора, образовавшихся
в результате перитектического превращения, и кристаллов (З-тверддго
раствора, нерастворившихся при этом превращении.
§ 47. ДИАГРАММА СОСТОЯНИЯ С ОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ (РАССЛАИВАНИЕМ) В ЖИДКОМ
СОСТОЯНИИ
В системе, диаграмма состояния которой представлена на рис. 69,
в определенном интервале температур и концентраций (область
anckdmb диаграммы) однородность жидкой фазы нарушается.
Ниже линии anckdmb в системе вместо однородного взаимного
раствора образуются две несмешивающиеся жидкие фазы —
раствор А в В (состав его определяется точками линии kb) и раствор
В в А (состав ejo определяется точками линии Ы). Подобные
кривые на диаграммах состояния называются линиями
предельного насыщения или линиями расслаивания.
Поскольку в жидком состоянии нет какой-либо строгой
структуры, как для твердых тел (кристаллическая решетка), то
принципиально безразлично, какой из двух жидких компонентов (А
или В) считать растворителем. Принято обычно считать
растворителем тот компонент, который входит в данную жидкую фазу в
большем количестве.
206
I*
I
с
nf
qL _^j
L f
L+A/
e
:tu*
у— N_//
1 * \m y\
Xs ^
b
L+B
A + B
/_.
В
■
Концентрация
В
В сплавах, составы которых лежат правее или левее области
ограниченной растворимости, концентрации второго компонента
недостаточно, чтобы образовать вторую жидкую фазу. При
температуре выше tK система при любых концентрациях А и В остается
в состоянии однородного раствора (это объясняется происходящим
в данном случае увеличением взаимной растворимости с повышением
температуры).
Рассмотрим процесс кристаллизации сплава / (в области
расслаивания). При температуре t2j когда фигуративная точка этого сплава
окажется на линии akb, из
однородной жидкой фазы
начнут появляться капли второй
жидкой фазы состава точки d.
С дальнейшим понижением
температуры размеры этих
капель будут увеличиваться
и состав второй фазы будет
изменяться по линии kb от
точки d в сторону увеличения
в ней концентрации
компонента В до точки Ь. Состав
исходной жидкой фазы будет
при этом изменяться по
линии ka в сторону
уменьшения концентрации в ней
компонента В от точки с до
точки а.
Таким образом, в данном случае имеет место диффузионный
переход компонента В из первоначальной жидкой фазы во вновь
образовавшуюся и все возрастающее обогащение последней компонентом В.
При достижении некоторой температуры tM эта фаза становится
полностью насыщенной компонентом В (состав этой фазы
определяется точкой Ь) и начинается выделение компонента В (ставшего
при этой температуре избыточным) в твердую фазу, т. е. начинается
кристаллизация компонента В.
Жидкая фаза, из которой выделяется при tu компонент 5,
обедняется им и состав ее приближается к составу второй жидкой фазы
(состав точки а при температуре tM).
Согласно правилу фаз, это превращение, получившее название
монотектического превращения, протекает при постоянной
температуре:
F = k — y+1-2 — 3+1-0,
поскольку у представляет две жидкие фазы L (а) и L (Ь) и одну
твердую фазу В. Состав жидкости L (а) (точка а) при этом не
изменяется.
Следовательно, выделяющейся при кристаллизации компонента В
теплоты оказывается достаточно для того, чтобы компенсировать
тепловые потери системы при теплоотдаче в окружающую среду,
207
Рис. 69. Диаграмма состояния с
ограниченной растворимостью (расслаиванием) в
жидком состоянии
и температура системы не изменяется. Следовательно, процесс
кристаллизации компонента В из выделяющей его жидкой фазы
происходит аналогично процессу кристаллизации его из расплава чистого
компонента В с той, однако, разницей, что в конце этого процеса
жидкая фаза не исчезает, а только изменяет свой состав (от точки b
до точки а). Таким образом, по окончании монотектического процесса
система будет иметь только
две фазы: кристаллы
компонента В и жидкость
состава точки а и система будет
находиться уже в состоянии
одновариантного равновесия:
F = k — y+ 1 =
= 2 — 2+1=1.
Концентрация &
Рис. 70. Монотектическая диаграмма
состояния в случае образования компонентами
областей ограниченной растворимости
При дальнейшем
понижении температуры состав
жидкой фазы начнет
изменяться по линии ликвидуса
(от а до е) с выделением
в твердую* фазу
компонента В. Затем из жидкости
эвтектической жидкостью, проис-
состава еу которая является
ходит кристаллизация эвтектики (А + В).
Итак, кристаллизация сплава протекает следующим образом:
L
*м г , ч . г , , *. г ,,. *м
->L(c — a) + L(d — 6), L (Ь)-^ L (a) + В,
L—ZllL{a — е) + В, L (е) -Л эвт (A f В).
Окончательная структура сплава / будет состоять из первичных
кристаллов В и окружающей их эвтектики (А + В).
Кристаллизация сплава // будет отличаться от кристаллизации
сплава / тем, что она начнется с выделения при температуре tt
первичных кристаллов компонента В. Таким образом, в этом случае
будем иметь:
L
t«—t
L
**- fe
*B + L(p — b)9 Hp)-±B+L (a),
B + L(a — e)y L (e) -^ эвт (A + B).
Горизонталь atM на диаграмме носит название монотекти-
ческой горизонтали. Все сплавы, фигуративные точки
которых не попадают на эту горизонталь, кристаллизуются как
обычные эвтектические сплавы с выделением первичных кристаллов
компонента А или В и эвтектики (А + В). Кристаллизация такой
системы может происходить и с образованием твердых растворов
компонентов. В этом случае диаграмма принимает вид, показанный
на рис. 70.
208
Кристаллизация сплавов этой системы будет происходить
следующим образом:
сплав /:
L^L{g-a) + L{h-b),
t
М
L{b)-+L (a) + р(с),
£ _!^ £ (а - с) + р (с -/),
сплав //:
L(e)->3BT[a(d) + P(f)];
L-!iJ^p(9_c) +L(p-6),
t
L{b)-+L (a) + p (c),
L(e) ^эвт [a (d)+ ?(/)]•
Расслаивание в жидком состоянии наблюдается во многих
системах. Так, например, значительные области расслаивания имеются
на диаграммах состояния систем Си—РЬ, Си—Cr, A1—Те, Си—S,
Zn—Pb, Si—Na, Pb—Ga и др.
§ 48. ТЕМП (ИНТЕНСИВНОСТЬ) КРИСТАЛЛИЗАЦИИ
Рассмотрим в системе с неограниченной растворимостью в жидком
и твердом состояниях два сплава / и // (рис. 71, а), которые имеют
одинаковые интервалы кристаллизации. Сравнивая массу
кристаллов, образовавшуюся в этих сплавах при их охлаждении до середины
А
Концентрация
а
В
Время
Рис. 71. Сравнение количеств закристаллизовавшейся фазы в двух (/ и //)
сплавах'(а) и их кривых охлаждения (б) в системе с неограниченной
растворимостью в жидком и твердом состояниях
14 А. Н. Крестовников 209
интервала кристаллизации, можно видеть, что в сплаве / к этому
моменту больше половины массы сплава еще остается жидким,
а в сплаве // больше половины массы сплава обратилось в кристаллы.
Сплавы / и // различаются относительным расположением
ликвидуса и солидуса. Можно видеть, что различие в кристаллизации
сплавов / и // сказывается и на характере кривых охлаждения
(рис. 71, б).
Для характеристики прироста количества кристаллов при
прохождении сплавом интервала кристаллизации вводится понятие
темпа или
интенсивности кристаллизации (i).
Количественно это понятие может
быть выражено как производная
от доли твердой фазы q по
температуре /, взятой с обратным
знаком:
dq
dt
(390)
Безвариантные превращения,
например эвтектическая и пери-
тектическая кристаллизация,
характеризуются значением темпа
i = оо. Для одновариантных
превращений величина i будет
конечной.
Обозначим уравнение кривой
ликвидуса L (/), а уравнение
солидуса a (t). При любой температуре в интервале кристаллизации
сплава С от /н до tK можно найти соотношение масс жидкой и твердой
фаз (рис. 72). При температурах t-\-dt и t доля кристаллов составит
d'd" a'd'
q" = —»пг и q' = ,,, , соответственно. При изменении
температуры на dt изменение доли кристаллов составит:
Концентрация
Рис. 72. Схема к выводу основного
уравнения темпа кристаллизации
dq = q" — q' =
L(t +dt) — C
L(t) — C
L(t + dt) — a(t +dt) L (t)
Учитывая, что
L(t + dt) = L (t) + Lf (t) dt
a(t)
(391)
(392)
и
a(t + dt) =a (t) + a' (t) dt, (393)
где U (t) и a' (t) — производные уравнений ликвидуса и солидуса
по температуре, соответственно получаем:
С [U (t)-a'(t)]+L(t) a' (t) -L'{t)a'(t) ^
dq'- [L{t)-a(t)f
В итоге получаем основное уравнение темпа кристаллизации:
(394)
С [//(/) — а'(/)]+!(/) а'(/) — L' (t)at
[L{t)~a(t)f
(395)
210
Это уравнение в наиболее общем виде отображает связь темпа
кристаллизации с ходом кривых ликвидуса и солидуса диаграммы
состояния. Темповая функция i (С, t) изображается поверхностью,
характерной особенностью которой является ее линейчатый характер:
уравнение (395) линейно относительно С и линии пересечения
поверхности i (Ct t) изотермическими плоскостями являются прямыми.
Рассмотрим некоторые частные примеры.
В случае диаграммы состояния с простой эвтектикой вид
темповой функции существенно упрощается:
i = %$■, (396)
так как a (t) = 0; в случае прямолинейного ликвидуса
L (0 = — at + Ь (397)
для темповой функции получим:
' = <*=W • (398)
Вдоль ликвидуса в этом случае темповая функция будет иметь вид
1 = (b-atf = "С" ' (3")
так как при этом С = —at + b.
Можно видеть, что в системе с простой эвтектикой с понижением
температуры сплава в интервале кристаллизации происходит
монотонное уменьшение темпа кристаллизации. Он возрастает прямо
пропорционально содержанию второго компонента от нулевого
значения при его нулевой концентрации. С увеличением угла между
линией L (t) и положительным направлением оси температур темп
кристаллизации убывает. При увеличении интервала
кристаллизации темп также убывает.
Вдоль ликвидуса темп меняется следующим образом: он
возрастает по мере приближения к температуре плавления чистого
компонента.
Результаты расчетов для L (t) = —0,0(Ш -f 0,5 приведены на
рис. 73.
Если ликвидус диаграммы состояния имеет нелинейный вид,
темповая функция приобретает ряд особенностей. При L" (f) <0
(ликвидус обращен выпуклостью вниз) по мере понижения
температуры сплава будет происходить уменьшение темпа за счет
одновременного роста знаменателя L2 (f) и уменьшения числителя Z/ (t)y
тогда как в случае прямолинейного ликвидуса уменьшение темпа
происходит лишь за счет роста знаменателя L2 (t). Таким образом,
в случае выпуклого ликвидуса с понижением температуры
уменьшение темпа будет происходить более резко, чем в случае
прямолинейного ликвидуса.
При вогнутом ходе ликвидуса L" (t) > 0 рост знаменателя L2 (/)
сопровождается ростом числителя L' (t), что при определенных зна-
14* 211
чениях температуры начнет сказываться на характере изменения
темпа, нарушив монотонность его изменения. Часть изотемповых
линий трижды пересечет кривую ликвидуса.
О 0J 0,2 0,3 Ofi 0,5
Концентрация (доли)
Рис. 73. Изображение темповой
функции для случая
прямолинейного ликвидуса в системе с простой
эвтектикой:
/ — изотемповые линии на поле
диаграммы состояния: 7—5; 2 — 10;
3 — 15; 4 — 40; 5 — 25; 6 — 30; 7 —
35; 5 — 40; 9 — 45; 10 — 50; 11 — 100;
12 — 200; II — изменение темпа вдоль
ликвидуса, L (/) = —at -\- b, a = 0,001
и Ь = 0,5
1000
I
«ч
§ 600
I
%ьоо
I
400-
% J 00
I
^200
I
1 wo
Si
л
О 0,1 0,2 0,3 Ofi 0,5
Концентрация (доли)
Рис. 74. Изображение темповой
функции для случая
криволинейного солидуса в системе с простой
эвтектикой:
I — изотемповые линии на поле
диаграммы состояния: 7 — 10; 2 — 25;
3—50; 4 — 100; 5 — 200; 6 — 500;
темпа вдоль ликтзи-
116.65
дуса L = TeeJ^t ~ °Л
II — изменение
L =
На рис. 74 представлена темповая функция для кривой ликвидуса
диаграммы с простой эвтектикой:
116,65
L(t) =
116,5 + *
o,i.
(400)
Прослеживая изменение темпа вдоль ликвидуса, наблюдаем минимум
(i = 34,3-10"4, t = 417° С и С = 0,1).
Для того чтобы исследовать темповую функцию для диаграммы
состояния системы с неограниченной растворимостью в жидком и
212
твердом состоянии, представим кривую ликвидуса в виде:
___ fiJiO — M)
L(t)
а кривую солидуса в виде:
а (О
<*i + (1 — fli) bt9
(401)
= (hjl — bt)
а2 + 0 — <h)bt
(402)
где fli и а2
параметры, определяющие положение и форму
кривых, а Ь — масштабный множитель. Темповая
функция имеет вид:
i= b C[a1a2{\-btf]-a1a2{\-~btf
(а2-а1)(6/)(1-602
(403)
Эта функция терпит разрыв при t = 0, / = 1/4 и ^ = а2.
Уравнение проекции линии стока поверхности темповой функции получим
из dildt = 0. Тогда:
axa2 (1 — С) (1 — М)2 — С (б/)3 = 0. (404)
На рис. 75 представлена темповая функция для случая аг —
= 1/а2 = V2 и 6 = 1 -10"3. Штрихами нанесена проекция линии
/ООО
500
О
0,2 ОМ 0,6 0,8 1,0
О
0,2 0,Ь 0,0 0,0 1,0
Концентрация (доли)
Рис. 75. Изображение темповой функции для случая неограниченной растворимости
компонентов в жидком и твердом состоянии:
/ — изотемповые линии на поле диаграммы состояния: 1 — 26,7; 2 — 30; 5—40; 4— 60;
5 — 100; // — изменение темпа вдоль ликвидуса L (/) и солидуса а (/); ах = \/а2— х1г\
Ь~ ЬЮ-3 [см. уравнение (403)]
стока. Там же (рис. 75, б) можно проследить изменение темпа вдоль
кривой ликвидуса и кривой солидуса. Темп кристаллизации как
для начала, так и для конца интервала кристаллизации безгранично
возрастает по мере приближения к крайним ординатам, проходя
через минимум.
Решение аналогичных задач для ряда других случаев
представлено на рис. 76. Ликвидусу и солидусу диаграмм приданы
различные очертания; на поле диаграмм состояния нанесены проекции ли-
213
ний стока, а ниже показано изменение темпа вдоль ликвидуса и соли-
дуса. Сплавы, лежащие левее /', кристаллизуются с монотонно
уменьшающимся темпом. Сплавы, лежащие правее d\ наоборот,
кристаллизуются с повышением темпа. У сплавов, лежащих между /'
и d\ с понижением температуры темп кристаллизации сначала пони-
Концентрация (дола)
Рис. 76. Положение линии стока и изменение темпа кристаллизации вдоль
ликвидуса L (t) и солидуса a (t) для случаев аг = 1/а2 = г/5 (/); аг = V5;
«2 = V2 (//); fli=2Ha2=5 (///)
жается, а после встречи фигуративной точки с проекцией линии стока
темповой функции увеличивается. Имеются сплавы, у которых в
начале и в конце кристаллизации темп одинаков.
§ 49. ДАВЛЕНИЕ НАСЫЩЕННОГО ПАРА КОМПОНЕНТОВ
РАСТВОРА
До сих пор при рассмотрении двухкомпонентных диаграмм
состояния считалось, что давление насыщенного пара компонентов
постоянно или ничтожно мало. Однако оно может быть рассчитано
с привлечением теории растворов. Исследования показывают, что
давления паров компонентов могут быть значительными и могут
существенно меняться с концентрацией и температурой.
Запишем зависимости давления насыщенного пара компонентов А
и В от их концентраций и температуры. Сначала воспользуемся
законом Рауля [уравнения (405) и (406)] и активными
концентрациями [уравнения (247)], а затем перейдем к коэффициентам актив-
214
ности [уравнения (248)] и энергиям смешения [уравнения (231),
(233), (238) и (251)]. Получаем:
(405)
Ра = Рлал
и
и
(406)
(407)
(408)
Здесь ра и рв—давления насыщенного пара компонентов А и В
над раствором с концентрацией этих компонентов Ха и хв соответ-
о о
ственно (причем Хл + Хв = 1), рл и рв —давления насыщенного
пара чистых компонентов А и В. Далее.
Рв=Р&в\
Ра = Ра!аха
о
Рв = Рв1вхв •
Рв = Рв*ве А ' •
Учитывая, что
\прл = —
АН
исп А
RT
-f- const
и
1прБ = —
дя
исп Б
яг
const,
(409)
(410)
(4И)
(412)
получаем:
и
АН
Pa = const аХа^
рв = const д#д£
ЯГ
(413)
ЛЯ
исп В
AW
RT
(414)
где л/*исп л и А/^ИСп в — энтальпии испарения компонентов А
и В соответственно; const^ и const^ — постоянные в уравнениях
температурной зависимости давления насыщенного пара этих
компонентов. Зная W, а также р\ или АЯИСП л и const^, можно
рассчитать давление насыщенного пара компонента А над раствором при
любой температуре и концентрации. Давление насыщенного пара
компонента В можно рассчитать аналогично или по разности, так
как
Робщ = РА+РВ. (415)
Нетрудно заключить, что при W = 0 или / = 1, т. е. для
идеальных растворов, изотермы давления насыщенного пара компонентов
и общее давление линейно связаны с атомной (или мольной)
концентрацией.
Если одноименные атомы или молекулы в растворе
взаимодействуют друг с другом с большей энергией, чем разноименные (W > 0
215
или /> 1 —«положительное отклонение от идеальности»), то
давление насыщенного пара компонентов будет больше, чем в случае
идеального раствора. При W <0 или / <0 («отрицательное
отклонение от идеальности») давление насыщенного пара компонентов
над раствором будет меньше, чем в случае идеального раствора.
На рис. 77 схематически показано изменение давления
насыщенного пара компонентов и общего давления для различных случаев
взаимодействия компонентов.
Важно обратить внимание на то, что при равновесии фаз, т. е.
в двухфазных областях кристаллы — жидкость,
кристаллы—кристаллы и жидкость —жидкость, давления насыщенного пара компо-
Дбухфазная
область
А В А В А -4 В А В
Концентрация
Рис. 77. Зависимость давления насыщенного пара компонентов А (/) и В (2) и
общего давления Р0бщ (<?) при различных типах взаимодействия компонентов в сплавах:
а — идеальный раствор; б — положительное (W > 0 или / > 1) и в — отрицательное (W <
< 0 или / < 1) отклонение от идеальности соответственно; г — равновесие двух растворов
нентов над равновесными фазами одинаковы и в пределах
двухфазной области при данной температуре общее давление остается
постоянным.
Давление насыщенного пара компонентов над раствором следует
отличать от внешнего давления. В частном случае внешнее давление
можно называть «поршневым». Поршневое давление может либо
непосредственно передаваться раствору или сплаву, находящемуся
в конденсированном состоянии, либо посредством инертного или
нейтрального газа или жидкости. Однако паровая фаза под поршнем
может быть образована раствором или сплавом, тогда понятие о
давлении насыщенного пара и о внешнем, поршневом давлении
совпадают.
§ 50. О ВИДОИЗМЕНЕНИИ ДИАГРАММЫ СОСТОЯНИЯ
ПОД ВЛИЯНИЕМ ДАВЛЕНИЯ
С изменением внешнего давления вид диаграммы состояния
системы может весьма сильно изменяться.
Например, в системе анизол—уретан (рис. 78) при увеличении
давления происходит смещение температур плавления компонентов
и эвтектической точки в сторону больших концентраций уретана,
а затем и полное ее исчезновение.
216
Другим примером подобного явления может служить смещение
(в результате изменения внешнего давления) ликвидуса и
эвтектической точки в системе Al—Si (рис. 79). Сплав с 11,7% Si,
кристаллизующийся при обычном давлении (1 am), как эвтектический, т. е.
имеющий в структуре одну только эвтектику (а + (3), при давлении
в 20 000 am кристаллизуется как доэвтектический, выделяя
первичные кристаллы а, а затем эвтектику (а + |3).
Рассмотрим вопрос о возможности непрерывного перехода одного
типа диаграмм состояния в другой при изменении внешнего
давления. ,——_ |/4?0в
Анизол Уретан
Концентрация
Рис. 78. Влияние давления на
положение ликвидуса и
эвтектической точки в системе анизол—
уретан:
1 —\ am; 2—2000 am; 3—3000 am
Чч>
%ff60*
1
1 «'
^
L
^^*Z^^I
1 ^<^ /
S У 1
S Л I
/ 2 Ifi
J-/oc// L+fi \
\ \/\ /
(\11Л?
\Щ7о
577° \
OL+J3•
А1
Si
Концентрация S/, %
Рис. 79. Диаграмма состояния системы
алюминий—кремний при давлениях:
/ — 1 am; 2—20 000 am
Двухкомпонентные соединения при понижении температуры
могут образовываться по следующим трем наиболее общим схемам
взаимодействия фаз:
L + S->AxBy, (I)
Lx-\-L2->AxBy, (II)
1-М,. ("О
Выясним возможность перехода при изменении давления от
диаграмм состояния, отвечающих превращениям по схемам (I) и (II),
к диаграмме состояния, отвечающей превращению (III). Очевидно,
что в случаях (I) и (II) образование соединения происходит инкон-
груэнтно, а в случае (III) —конгруэнтно. Таким образом, все три
схемы образования соединений и отвечающие им типы диаграмм
существенно различны. В соответствии с этим они получили
собственные наименования: схема (I) — перитектическая, схема
(II) — синтектическая, схема (III) называется
превращением в дистектической точке.
Переход от схемы (I) к схеме (III) возможен при изменении
давления путем смещения фазовых областей в положение, при котором
состав жидкой фазы совпадает с составом химического соединения,
а твердая фаза 5 перестает принимать участие в превращении, ко-
217
торое претерпевает соединение (рис. 80, а). При дальнейшем
изменении давления видоизменение диаграммы состояния перетекти-
ческого типа завершается переходом в диаграмму с дистектической
точкой. Интерес представляет тип диаграммы состояния,
реализующийся при определенном давлении Р*. Точку, которая является
предельной, так как в нее вырождаются перитектическая, дистектиче-
ская и эвтектическая точки диаграммы, обозначим R.
Переход от схемы (II) к схеме (III) возможен при изменении
давления. При этом вырождается различие между жидкими фазами
вплоть до совпадения их составов с составом химического
соединения (рис. 80, б) или смещения фазовых областей в положение, при
Рис. 80. Схематическое изображение перехода от одного типа диаграммы
состояния двухкомпонентной системы к другому типу под влиянием
внешнего давления
котором состав одной из жидких фаз совпадает с составом
химического соединения, а другая жидкая фаза перестает принимать
участие в превращении, которое претерпевает соединение (рис. 80, в).
В первом случае (рис. 80, в) при дальнейшем изменении
давления видоизменение синтектического типа диаграммы состояния
завершается переходом в диаграмму с дистектической точкой. При
определенном давлении Р* имеет место тип диаграммы состояния,
который в данном случае содержит промежуточную точку (назовем
ее У), являющуюся предельной для синтектической и
дистектической точек. Поскольку соединение в этой точке при плавлении
диссоциирует (АхВу—^хА + уВ), то ветви ликвидуса этого
соединения плавно переходят одна в другую, т. е. имеют в этой точке общую
касательную, которая перпендикулярна ординате соединения.
Во втором случае (рис. 80, а) при дальнейшем изменении
давления видоизменение синтектического типа диаграммы состояния
завершается переходом в диаграмму монотектического типа с
дистектической точкой. При определенном давлении Р* имеет место новый
тип диаграммы состояния, который в данном случае содержит
промежуточную точку W, аналогичную уже рассмотренным точкам R
и У и являющуюся предельной для синтектической, дистектической
и монотектической точек. Однако горизонталь этой диаграммы,
заканчивающаяся в точке W', не является синтектической или
монотектической, так как кристаллизующаяся при этой температуре
жидкая фаза имеет состав, одинаковый с составом химического со-
'218
единения. Если ордината сплава пересекает эту горизонталь, то,
хотя образованию конгруэнтно плавящегося соединения и
предшествует процесс расслоения, одна из образующихся жидких фаз не
принимает участия в фазовом превращении при температуре
горизонтали. При этой температуре система ведет себя как однокомпо-
нентная, так как составы двух фаз (L + АХВУ) одинаковы.
Положение ликвидуса химического соединения (рис. 80, в), как
следствие диссоциации соединения (АхВу—> хА + уВ), таково, что
горизонталь нонвариантного превращения является ее
продолжением, т. е. касательной (см. стр. 249).
Контуры промежуточных диаграмм состояния, соответствующих
определенному давлению Р*, на схемах рис. 80 нанесены тонкими
линиями. Все точки этих диаграмм, в том числе и промежуточные
(R, V и W), удовлетворяют правилу фаз Гиббса, причем точки Ry V
и W являются точками трехфазного равновесия.
Такого рода переходные точки в двухкомпонентных системах
не могут отвечать четырехфазному равновесию, поскольку по
сравнению с прочими точками двухкомпонентных систем в данном случае
максимально допустимое число фаз ограничивается дополнительным
условием (уравнением) равенства составов жидкой и твердой фаз.
Пример. В системах галлий — мышьяк, индий — мышьяк,
галлий — фосфор и индий—фосфор при повышении давления
происходит переход от инконгруэнтного образования химических
соединений к конгруэнтному. Переход соответствует параметрам (Р*, am;
**, °С): 0,9 и 1243 для GaAs; 0,33 и 942 для InAs; 13—20 и 1510—
1525 для GaP; 60 и 1062 для InP.
Глава IX.
ТРЕХКОМПОНЕНТНЫЕ СИСТЕМЫ
В практике наряду с двухкомпонентными системами часто
приходится иметь дело с системами, составленными из трех
компонентов.
Уравнение состояния таких систем связывает между собой пять
параметров и имеет следующий вид:
/ (Сь С2, С3, Р, Т) = 0.
Отсюда следует, что уравнение состояния трехкомпонентной
системы графически не может быть изображено плоскостной
диаграммой. Однако, полагая давление практически неизменяемым, можно
построить пространственную диаграмму, изображающую связь
между концентрациями компонентов и температурой. Для этой цели
избирается пространственная система трех координат (рис. 81).
Две оси, расположенные, по чисто геометрическим соображениям,
под углом 60°, служат для изображения концентрации двух
компонентов. Ось, взаимно перпендикулярная к двум другим осям, слу-
219
жит для изображения температуры. Преимущество такой системы
координат состоит в том, что если угол между осями концентраций
равен 60° и масштаб их одинаков, то полученный треугольник
является равносторонним и позволяет достаточно просто определять
концентрацию третьего компонента. Этот треугольник, называемый
концентрационным, служит основанием для диаграмм трехкомпо-
нентных систем. Сами диаграммы при этом выглядят подобно
трехгранной прямоугольной призме.
Из геометрии известно, что любой равносторонний треугольник
обладает следующими свойствами: сумма перпендикуляров,
опущенных из любой точки внутри треугольника на его стороны, равна
его высоте, т. е. (рис. 82, а)
Ма + Mb + Мс = CD\
сумма отрезков прямых, параллельных сторонам треугольника и
проведенных через любую точку внутри треугольника, равна стороне
треугольника, т. е. (рис. 75, б)
Ma + Mb + Мс = АВ = ВС = АС
или Мах + МЬг + Мсг - АВ = ВС - АС.
Эти свойства равностороннего треугольника используют для
определения состава тройного сплава.
Если принять высоту треугольника ABC (рис. 82, а) за 100%
весового количества сплава, то величина перпендикуляра,
опущенного из точки, соответствующей данному сплаву, на сторону,
противолежащую какому-либо компоненту, будет выражать
процентное количество этого компонента. Так, для сплава, изображаемого
точкой М, имеем следующее содержание компонентов:
%А = Ма, %В = Mb и %С = Мс.
Сумма всех компонентов
%А + %В + %С = Ма+ Mb + Мс - 100%.
Если за 100% весового количества того или иного тройного сплава,
принять одну из сторон треугольника ABC (рис. 82, б), то полагая,
что составы, отвечающие чистым компонентам Л, В и С,
соответствуют вершинам треугольника, весовые проценты компонентов будут
определяться отрезками линий, параллельных сторонам
треугольника и противолежащих данным компонентам. Для сплава, состав
которого изображается точкой М, имеем:
%А = Ма9 %В - Mb и %С = Мс.
Если учесть, что Ма = Маг = аах= схВ, Mb = Mbx= bb±= cA>
Мс = Мсх = схс, то, принимая за 100% сторону АВ, можем написать:
%А =сгВ9 %В =сАп %C = ccv
Последний способ получил более широкое применение, так как
позволяет непосредственно отсчитать содержание того или иного
компонента на стороне треугольника по нанесенным на ней
делениям.
220
На концентрационный треугольник обычно наносится сетка с
определенной ценой одного деления. Такая сетка показана на рис. 83.
Если сплав изображается точкой х, то, проведя линии, параллель-
i
I
!
1
Рис. 81. Оси координат,
применяемые для
построения диаграмм состояния
трехкомпонентных систем
АО 10 20 30 40 50 60 70 80 90 ЮОВ
100 90 80 70 60 50 40 30 20 10 О
Рис. 83. Концентрационный треугольник
с координатной сеткой
Рис. 82. Иллюстрация свойств
равностороннего треугольника, применяемого для
выражения концентраций
трехкомпонентных систем
В А
Рис. 84. Изображение
трехкомпонентных сплавов с постоянным
содержанием компонента Л (а) и с постоянным
отношением содержания компонентов
А и С (б)
ные сторонам треугольника, находим состав сплава: А =20%,
В = 30% и С = 50%.
Разобрав геометрические основы концентрационного
треугольника, обратимся теперь к некоторым его свойствам. Важнейшими из
них являются следующие:
1. Вершины треугольника соответствуют чистым компонентам.
2. Точки, лежащие на сторонах треугольника, изображают
состав двухкомпонентных сплавов.
3. Точки, лежащие внутри треугольника, изображают состав
трехкомпонентных сплавов.
4. Точки, лежащие на прямой, параллельной стороне
треугольника, изображают сплавы с постоянным содержанием компонента,
противолежащего данной стороне (рис. 84, а).
221
5. Точки, лежащие на прямой, выходящей из вершины
треугольника, изображают сплавы с постоянным отношением содержаний
компонентов, изображаемых двумя другими вершинами (рис. 84, б).
§ 51. ДИАГРАММА СОСТОЯНИЯ С НЕОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ КОМПОНЕНТОВ
в жидком и твердом состояниях
Диаграмма состояния трехкомпонентных сплавов с
неограниченной растворимостью в твердом и жидком состояниях может иметь
место лишь в случае, если каждая из двойных систем обладает
неограниченной растворимостью
компонентов в обоих состояниях.
На рис. 85 представлена
пространственная диаграмма
подобной системы. Она состоит из
двух поверхностей. Поверхность
ликвидуса проходит через
соответствующие линии ликвидуса
двойных систем. Поверхность со-
лидуса проходит через
соответствующие линии солидуса
двойных систем.
Превращения, происходящие
при переходе таких сплавов из
жидкого в твердое состояние,
протекают подобно превращениям
в двойных сплавах,
кристаллизующихся с образованием
непрерывного ряда твердых растворов.
Точки поверхности ликвидуса
соответствуют температурам начала
кристаллизации тройного твердого
раствора при охлаждении. Точки
поверхности солидуса
соответствуют исчезновению последних
следов жидкости, т. е. концу
кристаллизации тройного твердого
Рис. 85. Пространственное
изображение диаграмм состояния тройной
системы с неограниченной растворимостью
компонентов в жидком и твердом
состояниях
раствора.
Согласно правилу фаз, процесс кристаллизации тройных
сплавов этой системы является двухвариантным процессом:
F = k — y + I =3 — 2+1-2.
Рассмотрим кристаллизацию сплава /. При некотором
переохлаждении из жидкого расплава состава точки х выпадают кристаллы
тройного твердого раствора состава точки а, лежащей на
поверхности солидус. Прямая, проведенная через точки а и tx (коннода),
лежит в плоскости, параллельной основанию диаграммы. При
каждой данной температуре в системе имеет место равновесие между
222
твердой и жидкой фазами, составы которых лежат на
противоположных концах конноды. При этом в процессе кристаллизации жидкая
фаза изменяет свой состав по поверхности ликвидус (по некоторой
кривой /х6), состав же твердой фазы меняется по поверхности соли-
дус (по кривой at2). Состав твердого раствора при изменении
температуры претерпевает независимое изменение, поскольку система
имеет две степени свободы, и второй степенью свободы является
состав твердого раствора. В силу
этого кривые txb и at2 имеют
двоякую кривизну, т. е. не лежат
в одной плоскости. Полное
затвердевание сплава заканчивается
в точке t2y лежащей на поверхности
солидус. Если кривые t-^b и at2
спроектировать на плоскость
основания диаграммы, то получим
кривые линии ахтх и хпЬх. При
этом линия ахтх изображает
изменение состава твердой фазы, а
линия хпЪх изображает изменение
состава жидкой фазы при
кристаллизации сплава /.
Символически процесс
кристаллизации сплава / можно
изобразить следующим образом:
L(x — Ьг) ——*■> а(аг — х).
Пространственная модель
диаграммы состояния, несмотря на
большую наглядность, для
практических целей почти не
употребляется ввиду сложности ее
изображения и искажения размеров
концентрационного треугольника
диаграммы и других ее элементов.
Поэтому в реальных случаях для того, чтобы составить
представление о диаграмме состояния, изображают серию изотермических и
политермических разрезов, совокупность которых позволяет судить
о характере превращений, претерпеваемых сплавами системы при
изменении их температуры.
Изотермическими разрезами диаграммы состояния называются
изображения диаграмм в сечении плоскостью, параллельной
плоскости концентрационного треугольника. Иногда эти разрезы
называют горизонтальными разрезами диаграммы.
На рис. 86 изображено пространственное расположение
изотермических разрезов, проведенных в области температур, лежащих
между температурами плавления чистых компонентов.
Изотермические разрезы при этих температурах являются оптимальными для
более или менее полной характеристики диаграммы. Изотермический
223
Рис. 86. Пространственное
расположение изотермических разрезов при t1
и t2 (при этом fc> ^i> tB и
разрез при температуре tx (при tB < tx > /с) представлен на рис. 87.
В разрезе имеются три области: область твердых растворов а,
область жидких растворов L и область гетерогенного равновесия
жидкого и твердого раствора L -\- а. Границы раздела областей
представляют собой сечение поверхностей солидус и ликвидус, а прямые
линии, проведенные в области равновесия двух фаз, являются конно-
дами.
Концы коннод изображают составы жидкой (на поверхности
ликвидус) и твердой (на поверхности солидус) фаз при температуре,
Рис. 87. Изотермические раз- Рис. 88. Пространственное располо-
резы при температурах tx (а) жение но литер мических разрезов
и t2 (б) (см. рис. 86)
соответствующей температуре данного изотермического разреза. На
том же рис. 87 изображен изотермический разрез при более низкой
температуре t2 (при tA < t2 < tB). Этот разрез отличается от
предыдущего более широкой областью твердого раствора.
Политермическими разрезами называют изображения диаграмм
в сечении плоскостью, перпендикулярной к плоскости
концентрационного треугольника. Иногда эти разрезы называют
вертикальными разрезами диаграммы.
Исходя из практических соображений, удобно строить
политермические разрезы, либо проходящие через вершину одного из
компонентов, либо проходящие параллельно одной из сторон
концентрационного треугольника.
Пространственное расположение этих основных видов
политермических разрезов представлено на рис. 88. Сплавы, попавшие в
разрез по линии CD, характеризуются одинаковым отношением содер-
224
жаний компонентов А и В. Сплавы, попавндие в разрез по линии MN,
параллельной АВ, содержат равное кошичество компонента С.
Плоскостное изображение этих разрезов дается на рис. 89.
Для полного представления о диаграмме состояния необходимо
построить несколько изотермических и несколько политермических
разрезов, количество которых зависит от степени сложности
интересующей нас диаграммы.
Важно заметить, что о составе равновесных фаз при
определенной температуре позволяют судить только» изотермические разрезы.
tr
ее
/77'
/77,
П
/7,
ОС
С
D
N
N
а
&
Рис. 89. Политермические разрезы по литии CD (а) и MN (б)
(см. рис. 88)
Линии политергушческих разрезов не являются линиями равновесия,
как это имеет место в двухкомпонентных (сплавах. По этим линиям
нельзя определять составы твердой и жидкой фаз, находящихся
в равновесии, так как составы растворов изменяются не в одной
плоскости. Политермические разрезы позволяют судить о
температурах начала или конца превращений сплавов, попадающих в
данный разрез.
§ 52. ДИАГРАММА СОСТОЯНИЯ С НЕОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ В ЖИДКОМ СОСТОЯНИИ, ПОЛНОЙ
НЕРАСТВОРИМОСТЬЮ КОМПОНЕНТОВ В ТВЕРДОМ
СОСТОЯНИИ И ОБРАЗОВАНИЕМ ТРОЙНОЙ ЭВТЕКТИКИ
ПРИ ЗАТВЕРДЕВАНИИ
Диаграмма состояния трехкомпонентных сплавов
рассматриваемого типа может иметь место лишь в случае, когда каждая пара
компонентов, составляющих систему, образует систему с простой
эвтектикой.
В этом случае можно логически наметилъ три возможные стадии
кристаллизационного процесса.
Так как насыщение жидкой фазы каждым содержащимся в ней
компонентом достигается при различной температуре и различной
15 А. Н. Крестовников 225
концентрации в зависимости от природы компонента, то, очевидно,
при понижении температуры жидкость вначале насыщается каким-то
одним из трех компонентов. Какой из компонентов будет
кристаллизоваться первым, зависит от положения сплава в системе, т. е. от
первоначальной концентрации этого компонента в сплаве. При
дальнейшем понижении температуры жидкость, обогащенная двумя
другими компонентами, становится насыщенной относительно какого-то
второго компонента (в зависимости
от его относительной
концентрации в сплаве), оставаясь при этом
насыщенной и относительно
первого компонента. Как только это
достигается, начинается
одновременное выделение из жидкости
двух компонентов, т. е. начинается
кристаллизация двойной
эвтектики. При этом, очевидно,
относительная концентрация третьего
компонента в жидкости
возрастает, так как два других
компонента все время из нее уходят
в твердую фазу. Кроме того,
с понижением температуры
растворимость этого компонента
должна уменьшаться.
Следовательно, при некоторой
температуре жидкость окажется
насыщенной одновременно
относительно всех трех компонентов, и
потому начинается их
одновременное выпадение в твердую фазу, т. е.
произойдет кристаллизация
тройной эвтектики. Это одновременое
насыщение жидкости по
отношению ко всем трем компонентам,
а следовательно, и эвтектическая
кристаллизация для всех сплавов системы происходят при одной
и той же температуре, как это и следует из правила фаз:
/7 = ^_^+1=3 — 4+1=0,
где у = 4, так как в равновесии находятся три твердые фазы (Л,
В и С) и жидкий раствор. Понижение температуры происходит
только после окончательного затвердевания последних капель
жидкости.
Рассмотрим поверхности различных стадии кристаллизации
сплавов, составляющих данную систему (рис. 90).
Совокупность трех поверхностей начала первичной
кристаллизации компонентов представляет собой поверхность ликвидус
диаграммы состояния. Эта поверхность проходит через ликвидус соот-
226
Рис. 90. Пространственное
изображение диаграммы состояния трехкомпо-
нентной системы, кристаллизующейся
с образованием тройной эвтектики
чистых компонентов
ветствующих двойных диаграмм компонентов, составляющих
данную тройную систему.
Поверхностью начала первичной кристаллизации компонента Л
является поверхность t^E-^EE^t^ Поверхностью начала первичной
кристаллизации компонента В является поверхность tBExEE%tB*
Поверхностью начала первичной кристаллизации компонента С
является поверхность ^ЕХЕЕ^С.
Поверхности начала кристаллизации двойных эвтектик
образуются перемещением прямой, один конец которой скользит по
ординате чистого компонента (Л, В
или С), а другой — по кривой
ЕгЕ, Е2Е или Е3Е. Поверхностью
начала кристаллизации двойной
эвтектики (Л + В) являются
поверхности А'ЪЕХЕА' и В'сЕ^ЕВ'.
Поверхностью начала
кристаллизации двойной эвтектики (А + С)
являются поверхности А'аЕъЕА'
и C'gE3EC. Поверхностью начала
кристаллизации двойной
эвтектики (В + С) являются
поверхности B'dE2EB' и c'fE2EC.
Поверхности начала кристал-^
лизации двойных эвтектик прохо-
Рис. 91. Проекция диаграммы
состояния системы, образующей тройную
эвтектику чистых компонентов, на
плоскость концентрационного
треугольника (см. рис. 83)
дят через соответствующие
эвтектические горизонтали двойных
диаграмм состояния. Линии
пересечения поверхностей начала
кристаллизации двойных эвтектик
ЕгЕ, Е2Е и Е3Е носят название
линий двойных эвтектик. Их общая точка называется точкой тройной
эвтектики. Проведенная через нее плоскость А'В'С, параллельная
плоскости концентрационного треугольника ABC, носит название
эвтектической плоскости или плоскости
кристаллизации тройной эвтектики. Плоскость А'В'С
является поверхностью солидус данной диаграммы состояния.
Как и предыдущая диаграмма состояния тройной системы,
диаграмма с тройной эвтектикой может быть представлена в виде серии
изотермических и политермических разрезов. Однако для
исчерпывающего представления о диаграмме в данном случае требуется
большее число разрезов, чем это необходимо для диаграммы с
неограниченной растворимостью.
Диаграмма может быть представлена в виде проекции на плоскость
концентрационного треугольника ABC. Такая проекция приведена
на рис. 91. Линии еге, е2е и е3е представляют собой проекции линий
пересечения поверхностей первичной кристаллизации компонентов
Л, В и С (т. е. линий двойных эвтектик ЕгЕ, Е2Е и Е3Е). Таким
образом, области Aexee3A\ Вгхег2В и Се2еегС есть проекции
поверхностей первичной кристаллизации компонентов^Л, В и С. Если фи-
15* 227
гуративная точка сплава лежит внутри этих поверхностей, то
кристаллизация начинается с выпадения первичных кристаллов
соответствующего компонента. Если фигуративная точка сплава лежит
на линиях ееъ ее2 или ее3, то кристаллизация сплава начинается сразу
с кристаллизации двойной эвтектики, а в структуре будут
отсутствовать первичные кристаллы.
Линии Аеу Be и Се есть проекции на плоскость треугольника
линий А'Е, В'Е и СЕ, которые являются линиями пересечения
поверхностей начала кристаллизации двойных эвтектик (А + В), (В + Е)
и (А + С) с поверхностью кристаллизации тройной эвтектики
(А + В + С). Если фигуративная точка сплава лежит на линиях
Ае, Be или Се, то в сплаве отсутствует процесс образования
эвтектики и вслед за первичными кристаллами кристаллизуется тройная
эвтектика А'ЪЕ^с.
Таким образом, области АеВ, ВеС и АеС (рис. 91) есть проекции
поверхностей (рис.90) А'ЬЕгсВЕА', B'dEJCEB и C'g Е3аА'ЕС/
которые являются поверхностями начала кристаллизации двойных
эвтектик (А + В), (В + С) и (А + С).
Наконец, точка е является проекцией точки тройной эвтектики Е.
Если фигуративная точка сплава лежит в этой точке, то жидкий
раствор является одновременно насыщенным, по отношению ко всем
трем компонентам. При охлаждении такого сплава кристаллизация
протекает в одну стадию с образованием тройной эвтектики.
На плоскость концентрационного треугольника обычно
проектируют изотермы, т. е. наносят проекции линий пересечения
изотермических плоскостей с поверхностью ликвидус. На рис. 91 нанесены
изотермы температур tl9 t2, t3 и t^ причем t± > t3 > t2 > t±. С
помощью изотерм, нанесенных на проекции диаграммы состояния,
легко можно определить температуру начала кристаллизации того
или иного сплава.
Рассмотрим кристаллизацию некоторых наиболее характерных
сплавов названной системы. Например, проследим кристаллизацию
сплава / (рис. 90—91).
На первой стадии кристаллизации
На этой стадии кристаллизации сплав имеет, согласно правилу
фаз, две степени свободы:
F = k — y+l=3 —2+1=2.
При этом независимыми переменными являются температура
(кристаллизация идет в интервале температур от tx до t3) и
концентрация сплава. Изменение этих величин не нарушает двухфазного
состояния сплава. На второй стадии
L —Л эвт (В + Q + L (t'a — Е).
Эта стадия характеризуется одной степенью свободы:
F = k — у+1=3 — 3+1 = 1
228
и тоже протекает в интервале температур. Состав жидкости при этом
изменяется по линии двойной эвтектики. Равновесие трех фаз обычно
принято для определенной температуры изображать с помощью так
называемого коннодного треугольника, вершины которого
соответствуют составу фаз, находящихся в равновесии. Положение
коннодного треугольника при температуре t3i т. е. в начале процесса
кристаллизации двойной эвтектики, представлено треугольником
Шг (рис. 90).
В конце процесса кристаллизации двойной эвтектики коннодный
треугольник лежит в плоскости кристаллизации тройной эвтектики
(треугольник В'ЕС на рис. 90). Третья стадия кристаллизации
сплава /:
Процесс кристаллизации тройной эвтектики, как указывалось
выше, протекает при постоянной температуре, так как число
степеней свободы равно нулю:
F = k — y+ I =3 — 4+1=0.
Полностью затвердевший сплав состоит из первичных
кристаллов В, двойной эвтектики (В + С) и тройной эвтектики (Л + В + С).
Кристаллизацию сплава // можно выразить следующим образом:
L-^li? эвт (В + С) + L2 (*a- £),
1(£)-^эвт(Л + В + С).
Сплав II не содержит первичных кристаллов и состоит только
из двойной и тройной эвтектики.
Кристаллизация сплава ///:
1-!с!*С + 1(*4 —£), L(E)^bbt(A + B + Q.
Этот сплав не содержит двойной эвтектики и состоит из
первичных кристаллов С, окруженных тройной эвтектикой (Л + В + С).
Сплав IV (сплав состава точки е) кристаллизуется в одну стадию:
L (Е) -^ эвт {А + В + С).
Сплав представляет собой равномерную мелкую смесь
кристаллов трех компонентов Л, В и С, т. е. тройную эвтектику (Л + В + С).
§ 53. ДИАГРАММА СОСТОЯНИЯ С ОГРАНИЧЕННОЙ
РАСТВОРИМОСТЬЮ КОМПОНЕНТОВ В ТВЕРДОМ СОСТОЯНИИ
И ОБРАЗОВАНИЕМ ЭВТЕКТИЧЕСКОЙ СМЕСИ
ТВЕРДЫХ РАСТВОРОВ
Рассмотрим диаграмму состояния системы, компоненты которой
образуют тройные твердые растворы, построенные на основании
кристаллической решетки какого-либо компонента, в которую
входят атомы двух других компонентов. Какой именно компонент яв-
229
ляется растворителем, зависит от положения фигуративной точки
сплава на концентрационном треугольнике, т. е. от соотношения
составляющих данный сплав компонентов. Таким образом, в
системе образуются три типа твеодых растворов —а, р и у,
построенных на основании решеток
компонентов А, В, С
(предполагается, конечно, что все
три компонента взаимно
растворимы до какого-то пре-
деладруг в друге). Диаграмма
состояния этой системы (рис.
92) будет иметь три области
однородных твердых
растворов:
а (область tAh'hPaaftA)y
Р (область tBb'bQcc't£)
и
у (область tcd'dRff'tc).
Рис. 92. Пространственное изображение
диаграммы состояния трехкомпонентной
системы с ограниченной взаимной
растворимостью компонентов в твердом
состоянии
Рис. 93. Проекция диаграммы
состояния системы с ограниченной
растворимостью компонентов в твердом
состоянии на плоскость концентрационного
треугольника (см. рис. 92)
За пределами этих областей, которые проектируются на плоскость
концентрационного треугольника в области hPa, bQc и dRf,
находятся уже смеси этих растворов с образованием двойных и тройных
эвтектик (рис. 93).
В связи с этим имеем следующие поверхности кристаллизации
(рис. 92):
tAExEE^tA — поверхность ликвидуса твердого раствора с
решеткой компонента А (а-раствор);
tBEiEEitB — поверхность ликвидуса твердого раствора (J;
230
tcE2EE3tc — поверхность ликвидуса твердого раствора у;
tAa'p'titA — поверхность солидуса твердого раствора а;
tBb'Qc'tB — поверхность солидуса твердого раствора р;
tcf'Rd'tc — поверхность солидуса твердого раствора у\
P'a'Eib'Q'ЕР — поверхность начала кристаллизации двойной
эвтектики (а + (3);
P'ER'f'E^h'P' — поверхность начала кристаллизации двойной
эвтектики (а + y);
Q'ER'd'E2c'Q — поверхность начала кристаллизации двойной
эвтектики ((3 + у)\
Р'а'ЕxbrQ'Р' — поверхность конца кристаллизации двойной
эвтектики (а + |3);
P'h!E3f R'P' — поверхность конца кристаллизации двойной
эвтектики (а + у)\
Q'cfE2d'RfQf — поверхность конца кристаллизации двойной
эвтектики ((3 + у);
P'Q'R' — плоскость кристаллизации тройной эвтектики
(а + р + 7).
Таким образом, сплавы, не лежащие внутри треугольника PQR',
заканчивают кристаллизацию вне плоскости кристаллизации
тройной эвтектики. Следовательно, тройную эвтектику будут
образовывать только те сплавы, фигуративные точки которых на
концентрационном треугольнике лежат внутри треугольника PQR.
Определение структуры сплава, образующейся в результате
кристаллизации, следует производить, исходя из рассмотрения
поверхностей кристаллизации, которые пересекает фигуративная
точка сплава при его охлаждении.
Структуры сплавов, фигуративные точки которых лежат в
областях AaPhA, BbQcB и CdRfC, соответственно представляют собой
кристаллы а, р и Y-
Сплавы областей aPeQb, cQeRd и fRePh соответственно будут
иметь структурной составляющей двойные эвтектики (а + Р),
(Р + у) и (а + 7).
При этом сплавы, попадающие внутрь треугольника PQR,
будут содержать еще тройную эвтектику.
Первичные кристаллы а, р и у содержат все сплавы, кроме тех,
которые попадают на линию кристаллизации двойных эвтектик:
ееъ ее2 и ее3.
Сплавы, лежащие на прямых Ре, Qe и Re, в своей структуре
содержат соответственно первичные кристаллы а, р, у и тройную
эвтектику (а + Р + у), но не содержат двойной эвтектики.
§ 54. ДИАГРАММА СОСТОЯНИЯ ТРОЙНОЙ СИСТЕМЫ
С ХИМИЧЕСКИМ ВЗАИМОДЕЙСТВИЕМ ДВУХ КОМПОНЕНТОВ
В рассматриваемом случае третий компонент химически не
взаимодействует с двумя остальными. Конечно, это только частный
случай, так как возможно образование нескольких двойных и тройных
химических соединений.
231
Диаграмма состояния тройного сплава, когда одна из пар
компонентов образует устойчивое химическое соединение, представлена
на рис. 94.
Как видно, эта диаграмма делится химическим соединением М
на две самостоятельные эвтектические диаграммы. В основании
первой из них лежит треугольник
АМС. Кристаллизация тройной
эвтектики (А + С + М)
происходит при температуре точки
Е в плоскости треугольника
АМС. В основании второй
части диаграммы лежит
треугольник АВМ, кристаллизация
сплавов этой части
заканчивается при температуре точки Е'
в плоскости треугольника
А2М2В2 образованием тройной
эвтектики (А + В + М).
С;
с
с __д_:*
Рис. 94. Пространственное изображение
диаграммы состояния трехкомпонентной
системы с образованием двойного
устойчивого химического соединения
Рис. 95. Проекция диаграммы
состояния системы с образованием двойного
устойчивого химического соединения М
на плоскость [концентрационного
треугольника
В данном случае химическое соединение М образуется в
результате химического взаимодействия компонентов В и С по
реакции
хВ + уС = ВХСУ.
Ликвидус этой диаграммы состоит из четырех поверхностей
начала кристаллизации:
tAEJiEbE'ExtA — поверхность начала кристаллизации
компонента Л;
tcE^EE^tc —поверхность начала кристаллизации
компонента С;
232
tBE2EfE1tB — поверхность начала кристаллизации
компонента В\
tME2E'EbEE3tM — поверхность начала кристаллизации
химического соединения М.
Диаграмма имеет следующие поверхности начала криеталлиза-
ции двойных эвтектик:
Е1Е,А2а2Е1 —двойной эвтектики (А + В)\
и Е1Е'В2Ь1Е1
Е^с^С^Е^ — двойной эвтектики (А + С);
и E^axAxEE^
Е2Е'В2Ь2Е2 — двойной эвтектики (В + М)\
и Е2Е'М2т3Е2
Е3ЕС1с2Е3 — двойной эвтектики (С + М)\
и Е3ЕМ1т1Е3
ЕъЕАгтгЕь^ ЕъМ1т2Еь, —двойной эвтектики (А + М).
EL z. Hi гл. 2CL3J-d к
и EbE'M2d3Eb
Солидус диаграммы представлен двумя плоскостями
кристаллизации тройных эвтектик А1С1М1 и А2М2В2.
Проекция этой диаграммы на плоскость концентрационного
треугольника ABC показана на рис. 95.
На этом заканчиваем рассмотрение диаграмм состояния тройных
систем. Рассмотренные виды тройных диаграмм отнюдь не
охватывают всех возможных случаев взаимодействия трех компонентов
и дают только метод подхода к их изучению.
§ 55. ДИАГРАММЫ РАСТВОРИМОСТИ
Выше были рассмотрены основные схемы кристаллизации
веществ из жидкого расплава, в котором они взаимно растворены друг
в друге. Этим схемам соответствуют определенные геометрические
модели (диаграммы состояния).
Естественно, что такие же схемы можно
применять в том случае, когда
вещества кристаллизуются не из
расплава, а из водного раствора. Например,
если охлаждать от комнатной
температуры раствор какой-либо соли в воде,
то диаграмма растворимости будет
иметь вид, показанный на рис. 96.
Здесь линия tAE является линией
Соль + L
Лед + соль
начала кристаллизации чистого льда, ,А . °£ ,рв
а линия tBE — линией предельной рас- ° а) Концентрат
творимости соли в воде. Когда фигура- „ пп _
тминяятпикя пягтпппя прпргрцрт тшнмт РИС< 96, ДИагРамма Раствори-
тивная точка раствора пересечет линию мости соли в воде
tAE, то появятся первые кристаллы
льда. Пр.и дальнейшем понижении температуры происходит
увеличение содержания соли в растворе. Наконец, при температуре tE
избыточная соль выпадает в твердую фазу и жидкая фаза
затвердевает в мелкокристаллическую смесь льда и соли.
233'
Очевидно, что эта диаграмма состояния является диаграммой
состояния эвтектического типа. Мелкокристаллическая смесь льда
и соли состава СЕ является эвтектической смесью, которая, как
видно из диаграммы (tE <J tB и tB = 0° С), кристаллизуется при
температуре ниже нуля. В практике этим явлением часто пользуются
для получения охладительных смесей (например, смеси поваренной
соли и снега).
Иногда эвтектику в системе вода—соль называют криогидратом,
а эвтектическую точку — криогидратной точкой.
Растворение солей или кислот в воде может сопровождаться
образованием химических соединений — гидратов, которые будут
кристаллизоваться как
самостоятельные компоненты. В этих
случаях диаграмма растворимости
будет иметь вид, аналогичный виду
диаграммы состояния двухкомпо-
нентной системы с химическим
соединением.
Если раствор заключает в себе
не одну, а две соли, то состав его
может быть выражен точкой на
концентрационном треугольнике,
одна из вершин которого
соответствует чистому растворителю, а две
другие — растворенным солям.
Температура раствора изображается по
оси, перпендикулярной к плоскости
концентрационного треугольника.
Растворимость солей при разных
температурах будет в этом случае
изображаться совокупностью поверхностей, аналогичных
поверхности ликвидуса в диаграммах состояния трехкомпонентных систем.
Растворимость солей может рассматриваться и в ином
направлении — не при замерзании, а при изотермическом испарении
(выпаривании) растворителя. Рассмотрим несколько таких диаграмм.
Пусть имеются две какие-то соли — Sx и S2, растворенные в воде
(рис. 97). Точка Q на оси абсцисс отвечает концентрации
насыщенного раствора соли Sx в воде в том случае, когда только одна эта соль
присутствует в растворе.
При добавлении в раствор некоторого количества соли S2
насыщающая концентрация соли Sx будет уменьшаться (ввиду общности
солевых ионов). При этом поступают следующим образом:
растворяют некоторое количество соли S2, а затем уже насыщают раствор
солью Sx. Таким образом получают ряд точек я, Ъ, с, d и т. д.;
отвечающих насыщающей концентрации соли Sx в растворе при
содержании различных количеств соли S2. Линия QA стье линия
насыщения раствора солью S2 в зависимости от содержания соли Sx.
Аналогичным образом получается линия РА, являющаяся
линией насыщения раствора соли S2 в зависимости от содержания
234
^
1
1
1
1
*
1
I
1
-
р
а'
3"
2"
1"
В
L + $2
Лт
<^г^—t
А
L
!/' \2'\3'
L + Sf + S2
\ с
\d
1
\С
\b L+Sr
» п , ,»г г, Jfr.
И20 Концентрация conuSt
Рис. 97. Изотермическая диаграмма
растворимости в воде двух солей
с одноименными ионами
соли Sx. Точка Л, принадлежащая одновременно двум линиям
насыщения, характеризует насыщение раствора обеими солями, которые
при испарении этого раствора будут одновременно выпадать из
раствора в твердую фазу. Эта точка носит название эвтонической
точки. Таким образом, имеем следующие области диаграммы:
H2OPAQ — область ненасыщенных растворов обеих солей (L);
РАВ — область смеси растворов, насыщенных солью S2,
с этой же солью в твердом состоянии (L + Sj);
ВАС—область смесей твердых солей Sx и S2 с раствором,
насыщенным обеими солями (L + Sx + S2).
Эта диаграмма относится к какой-то одной постоянной для
данного случая температуре tx. Рассмотрим на этой диаграмме процесс
выпаривания ненасыщенного раствора / при постоянной
температуре tv По мере испарения растворителя, т. е. по мере уменьшения
его количества, концентрация Si и S2 в растворе будет
увеличиваться. Следовательно, фигуративная точка раствора будет
перемещаться в сторону больших концентраций солей (точки 2, 3
и т. д.). Перемещение это происходит по прямой линии 1су так как
отношение концентраций обеих солей остается постоянным, т. е.
Sx 1 — V 2— 2' 3 — 3'
Таким образом, имеем:
L(l)XL(c)+nap.
При достижении фигуративной точкой линии насыщения раствора
солью Sx (точка с) начинается выпадение этой соли в твердую фазу,
т. е. уменьшение ее концентрации в жидкой фазе, и, следовательно,
увеличение концентрации соли S2. Так как раствор при этом остается
насыщенным по отношению к Sx, то фигуративная точка раствора
перемещается по линии с А. При этом имеем
L (с — А) —^ Sx + пар.
По достижении точки А раствор, оставаясь насыщенным
относительно соли Sx, насыщается и солью S2 и обе эти соли начинают
выпадать в твердую фазу в виде смеси (эвтоники); этот процесс
продолжается до удаления последних капель растворителя.
Изотермическая диаграмма растворимости несколько
усложняется, если соли вступают в химическое взаимодействие друг с
другом, т. е. образуют двойную соль. В этом случае двойная соль
обладает собственной ветвью растворимости на линии насыщения
раствора.
На рис. 98 изображена изотермическая диаграмма растворимости
солей SxhS2b воде, причем соли Si hS2 образуют в результате
взаимодействия двойную соль SxS2. Линия CD диаграммы является линией
насыщения раствора солью Sb линия АВ—солью S2, а линия
ВС — двойной солью SxS2. Точки В и С представляют собой эвтони-
ческие точки, в которых раствор является насыщенным по отноше-
235
нию к солям S2 и SxS2 (в точке В) или по отношению к солям Sjl
H_SiSa (в точке С).
В случае образования одной из солей устойчивого
кристаллогидрата изотермическая диаграмма растворимости будет подобна
представленной на рис. 99. На рисунке видно, что кристаллогидрат
тоже имеет собственную ветвь растворимости на линии насыщения
раствора.
Ряд изотермических диаграмм, построенных для различных
температур, можно объединить в одну политермическую диаграмму,
описывающую изменение состояния раствора в зависимости от тем-
W Концентрация S, И20 Концентрация S, SinH2°
Рис. 98. Изотермическая диаграмма Рис. 99. Изотермическая диаграмма рае-
растворимости в воде солей 5Х и 52 творимости в воде солей Si и 52 в случае
в случае образования двойной соли образования одной из солей устойчивого
S1-S2 гидрата S^n H20
пературы и концентрации солей. На рис. 100 показана такая
политермическая диаграмма растворимости солей MgCl2 и KCI в воде.
На плоскости APN' изображена диаграмма состояния системы
вода—соль MgCl2. Точка В является эвтектической точкой этой
системы. Отдельные ветви имеют следующее значение:
N'L' —выпадает гидрат MgCl2-2H20;
US' —выпадает гидрат MgCl2-4H20;
S'R' —выпадает гидрат MgCl2-6H20;
R'H' —выпадает гидрат MgCl2-8H20;
Н'Е'—выпадает гидрат MgCl2-10H2O;
Е'В —выпадает гидрат MgCl2-10H2O;
АВ — кристаллизуется лед.
На плоскости APQ изображена диаграмма состояния системы
вода—соль КС1. Эвтектической точкой этой системы является точка С.
Ветви диаграммы имеют следующее значение:
CQ — выпадают кристаллы КС1;
АС — кристаллизуется лед.
Поскольку точка А является точкой замерзания воды, то она
соответствует 0° С на оси температур.
Плоскость N'PQ представляет собой изотермическую диаграмму
растворимости при температуре кипения воды и носит название
236
границы кипения. Таким образом, точка Р соответствует
100° С на оси температур.
Точка D является эвтектической точкой системы вода —
MgCla-KCl.
Рассмотрим теперь значение поверхностей, совокупность
которых представляет собой политермическую диаграмму раствори-
Рис. 100.
Политермическая диаграмма
растворимости в воде содей
MgCl2 и КС!
НоО
мости. Каждая поверхность является границей насыщения раствора
по отношению к какой-либо твердой фазе.
Так, поверхность DFMKLNQCD соответствует насыщению по
отношению к КО; поверхность NLL'N'N' —MgCl2-2H20;
поверхность LL'S'SKL — MgCl2-4H20; поверхность SS'R'RS — MgCl2X
X6H20; поверхность RR'H'R—MgCl2-8H20; поверхность
HH'E'EH — MgCl2 • 10Н2О; поверхность EE'BDFE — MgCl2 X
Х12Н20; поверхность FEHRSKMF — MgCl2 -KC1 -6H20 и,
наконец, поверхность ABDCA соответствует кристаллизации из раствора
самого растворителя, т. е. выпадению кристаллов льда.
Сечение политермической диаграммы растворимости плоскостями,
перпендикулярными оси температур, позволяет получить
изотермические диаграммы растворимости для любой температуры.
237
Глава X.
ОСНОВЫ ГЕОМЕТРИЧЕСКОЙ ТЕРМОДИНАМИКИ
Диаграмма состояния является результатом экспериментального
исследования. Однако построение диаграммы состояния не является
результатом одной только эмпирики. Наряду с огромным
экспериментальным материалом, накопленным в этой области, большое
значение имеют различные теории, позволяющие частично или полностью
построить элементы диаграммы состояния. Созданные на
сегодняшний день методы теоретического изучения систем лишь отчасти
способны решить эту важную задачу и не позволяют надежно
контролировать правильность полученных экспериментальных данных. Эти
теории не позволяют также однозначно предвидеть появление новых
типов диаграмм состояния и характер перехода известных типов
диаграмм друг в друга при значительных изменениях давления.
Вместе с тем вопрос этот важен не только теоретически, но и
практически во всех тех областях, в которых надо считаться с особенностями
протекания процессов при высоких давлениях, что имеет место в
некоторых отраслях химической технологии, а также при образовании
минералов и горных пород. Еще большее значение приобретает этот
вопрос в связи с тем, что известные диаграммы состояния изучались
почти исключительно при атмосферном давлении, которое, как
известно, не представляет собой особой точки на шкале давлений.
Наиболее объективным методом, позволяющим контролировать
имеющиеся диаграммы состояния, а также методом предсказания
возможных типов диаграмм состояния является классический метод
геометрической термодинамики, иногда называемый методом
изобарного потенциала или методом
поверхностей изобарного потенциала. Этот метод
прост и точен, а главное его достоинство состоит в наглядности и
универсальности.
В основе метода геометрической термодинамики,
использующего геометрические построения и представление об изобарном
потенциале, лежит первое и второе начала термодинамики.
Согласно первому началу термодинамики, для всех однородных
(гомогенных) и неоднородных (гетерогенных) систем имеем:
dQ = dU + pdVy
где dQ — элементарный тепловой эффект;
dU — элементарное изменение внутренней энергии системы;
dA — элементарная работа, относящаяся к рассматриваемому
термодинамическому процессу.
Согласно второму началу термодинамики:
где dS представляет собой элементарное изменение энтропии,
являющейся мерой рассеянной системой энергии.
238
Объединяя уравнение с неравенством, получаем:
dU + pdV — TdS ^ 0.
Препятствием к этой математической операции может служитв
лишь случай, когда Т = 0. Однако этот случай исключается, пос-
скольку, согласно третьему началу термодинамики, абсолютный
нуль недостижим.
Если рассматривать процесс при р = const и 7 = const, то
левая часть полученного неравенства представляет собой элементарное
изменение изобарного потенциала. Изобарный потенциал G,
согласно определению, является функцией переменных р и Т:
G = f(p,T)
ИЛИ
G - U + pV — TS,
откуда при р = const иГ = const имеем:
dG = dU + pdV — TdS.
Анализируя приведенные выражения, приходим к выводу, что
dG^ 0.
Это неравенство представляет собой основное положение, на
которое опирается геометрическая термодинамика. Содержание этого
выражения сводится к следующему. Самопроизвольные
(естественные) процессы совершаются таким образом, что дифференциал
изобарного потенциала является отрицательной величиной. Это
означает убывание величины изобарного потенциала. После некоторых
изменений система приходит в состояние равновесия, которое
характеризуется минимальным при данных условиях (р = const,
Т = const) значением изобарного потенциала, и тогда изменение
изобарного потенциала прекращается (dG = 0). Это будет предельным
случаем любого самопроизвольного процесса. Другими словами,
все самопроизвольные процессы, приводящие системы к равновесию,
можно символически охарактеризовать следующим образом:
Изобарный потенциал G при неравновесном состоянии системы
может принимать любые значения, являющиеся большими, чем
определенные приданных р и Т значениях изобарного потенциалаGmin.
Неравновесные системы самопроизвольно переходят в равновесное
состояние и их изобарный потенциал принимает в конце концов
значение Gmln, чем обычно и характеризуется достижение равновесий
в системе.
Рассмотрим зависимость изобарного потенциала от температуры,
давления и состава.
239
§ 56. ЗАВИСИМОСТЬ ИЗОБАРНОГО ПОТЕНЦИАЛА
ОТ ТЕМПЕРАТУРЫ И ДАВЛЕНИЯ
Принимая во внимание, что изобарный потенциал является
функцией состояния, изменение которой при переходе системы из
одного состояния в другое не зависит от пути перехода (т. е.
дифференциал изобарного потенциала является полным дифференциалом
i и равняется сумме частных дифференицалов
по независимым переменным), можем
записать:
= (-£>+("
!
1
I
f
dG
ОТ
dT.
р
Температура Т
Рис. 101. Зависимость
изобарного потенциала от
температуры (р = const)
поскольку
С другой стороны, дифференцируя
нение
G = U + pV — TS
по всем переменным, получаем:
dG = dU + pdV + Vdp — TdS -
или
dG = VdP — SdT,
урав-
SdT
dU = TdS — pdV.
Сравнивая уравнения для dG, обнаруживаем, что
= -S.
(
dG
дТ
p
Так как энтропия 5 может иметь только положительные
значения (5 ^ 0), то с возрастанием температуры происходит
уменьшение изобарного потенциала G. Кроме того, поскольку с повышением
температуры происходит возрастание энтропии 5, т. е.
ж)р>°- <416>
то повторное дифференцирование дает:
дЮ) <о.
) р
(
ат2
(417)
Это позволяет сделать заключение, что кривая зависимости G от Т
обращена выпуклостью от оси абсцисс. Геометрическое изображение
функции G = / (Т) при р = const приводится на рис. 101. Все точки,
лежащие выше нанесенной на графике кривой, соответствуют при
данном давлении и соответствующих температурах неравновесным
состояниям системы. Точки, лежащие на кривой, отвечают
равновесным состояниям при соответствующих температурах.
Важно обратить внимание на следующее обстоятельство: каждая
фаза системы в общем случае обладает собственным изобарным
потенциалом. Энтропия различных фаз различна, следовательно,
различна и кривизна кривых G = f (Г), поэтому возможно пересечение
240
кривых при какой-то определенной температуре, как это показано
на рис. 102. При этой температуре система оказывается устойчивой,
т. е. находится в состоянии равновесия при наличии в ней обеих фаз.
Однако при всех остальных температурах минимальный изобарный
потенциал система приобретает только при ^ i
полном переходе одной из фаз, обладающей ^ *
большим изобарным потенциалом, в
другую, обладающую меньшим при данной
температуре изобарным потенциалом.
Аналогично тому, как это было сделано
для температурной зависимости изобарного
потенциала, анализируем его зависимость
от давления и обнаруживаем, что
(J°.\ =v
\ dp Jt
Так как объем всегда характеризуется
положительной величиной (V > 0), то с
возрастанием давления происходит увеличение
изобарного потенциала. Кроме того,
поскольку
aKN <0, . (418)
ТО
др
d2G
др2
<0
(419)
Температура Т
Рис. 102. Изменение
изобарного потенциала с
температурой (р = const):
L — для жидкого
состояния; 5 —для
кристаллического состояния
I
|
и, следовательно, кривая G — f (р)
обращена выпуклостью от оси абсцисс, как это
показано на рис. 103.
ДаблеяиеР
Рис. 103. Зависимость
изобарного потенциала от
давления (Т = const)
§ 57. ЗАВИСИМОСТЬ ИЗОБАРНОГО ПОТЕНЦИАЛА
ОТ СОСТАВА СИСТЕМЫ
Этот вопрос более сложен, чем предыдущие, и его легче
рассмотреть применительно к отдельным случаям. При этом для простоты
будем заниматься только двухкомпонентными системами.
Если компоненты совершенно нерастворимы друг в друге и во
всем интервале концентраций от Л до В образуется смесь
компонентов, то изобарный потенциал такой системы находится по правилу
аддитивности, как это показано на рис. 104. При этом изобарный
потенциал сплава (смеси) является линейной функцией
концентрации.
Если компоненты полностью растворимы друг в друге (случай
образования неограниченных жидких или твердых растворов), то
зависимость изобарного потенциала от концентрации должна
выражаться плавной непрерывной линией. Положение этой линии в
общем случае аналогично положению, приведенному на рис. 105,
16 А. Н. Крестовников 241
и характеризуется следующим. Кривая изобарного потенциала ветре
чает оси ординат в точках, соответствующих изобарным
потенциалам чистых компонентов. Это положение является очевидным и не
т ребует какого-либо доказательства.
Кривая изобарного потенциала выпукла к оси абсцисс на всем
своем протяжении, если растворы устойчивы при любом составе.
Это положение легко может быть доказано методом от противного.
Так как всякое нарушение однородности в системе приводит к
образованию* смеси, изобарный потенциал которой определяется по
правилу аддитивности, то очевидно, что при этом всегда будет
происходить увеличение изобарного потенциала системы.
Рис. 104. Изобарный
потенциал смеси компонентов,
не растворяющихся друг
в друге (р = const, Т =
= const)
Состав
Рис. 105. Изобарный
потенциал смеси компонентов,
неограниченно
растворяющихся /др^г в Друге (р =
= const, T = const)
Кривая изобарного потенциала касается крайних осей ординат.
Для доказательства этого положения необходимо рассмотреть
изменение тангенса угла наклона касательной к кривой изобарного
потенциала, который образуется между касательной и положительным
направлением оси составов. Это будет первая производная:
dG
= tg06.
Очевидно, что эта производная представляет собой также
химический потенциал одного из компонентов в растворе, т. е. работу
переноса единицы массы компонента из данной фазы в другую:
dG
dxi
= (X/
Величина химического потенциала для растворов определяется
выражением:
\ii = \JL°i + RT In xit
где \it — химический потенциал компонента /, концентрация
которого в растворе составляет xt\
242
fx/ — величина, определяемая температурой и природой
компонента i\
R — универсальная газовая постоянная;
Т — температура.
Анализ общего выражения, рассматриваемого по отношению к
компоненту В:
-Ц- = VB - рв + RT In хв, (420)
позволяет сделать следующие заключения.
Если хв —> 0, то In хв —» —оо,
следовательно, \iB —* —оо. Это означает, что
lg а __» —со и а —* 90°, т. е. касательная
Состав
Рис. 106. Изобарный
потенциал при равновесии чистого
компонента с раствором
(р = const, Т = const):
L — изобарный потенциал
расплава; Gd — изобарный потен-
циал кристаллов компонента В
к кривой изобарного потенциала раствора
при хв —> 0 стремится совпасть с осью
ординат чистого компонента А.
Аналогичное может быть доказано для ординаты
чистого компонента В.
Рассмотрим теперь некоторые
случаи представления фазовых равновесий.
Прежде всего нас будут интересовать
такие равновесия, как равновесие
раствора с одним из компонентов и
равновесие двух растворов.
На рис. 106 приводится пример
равновесия жидкого раствора (расплава) с
компонентом В в твердом состоянии. Состав
расплава, который является насыщенным
по отношению к кристаллам В, можно
найти, проведя касательную из точки,
соответствующей изобарному потенциалу
кристаллов 5, к кривой изобарного
потенциала расплава. Точка касания будет
отвечать искомому составу расплава (точка с).
Очевидно, что система кристаллы В —
расплав состава с будет обладать
наименьшим изобарным потенциалом (и,
следовательно, будет наиболее устойчивой),
поскольку все остальные линии (например,
для расплавов с' и с") расположены выше
касательной.
На рис. 107 дан пример равновесия
расплава и твердого раствора. Составы
равновесных при данных условиях
растворов, которые фиксируются определенным положением кривых
изобарного потенциала, определяются проведением к обеим кривым
общей касательной. Наличие такой касательной свидетельствует
о равенстве химических потенциалов компонентов в обеих фазах,
что соответствует равенству работ переноса единицы массы ком-
16* 243
Состаб
Рис. 107. Изобарный
потенциал при равновесии двух
растворов
(р = const, T — const; L —
изобарный потенциал расплава;
а — изобарный потенциал
твердого раствора
понента из одной фазы в другую, т. е. устанавливается равновесие.
Точки касания отвечают искомым составам равновесных растворов.
Это положение требует более строгого доказательства, поскольку
возможен случай равенства химических потенциалов компонентов
при равенстве углов наклона касательных, когда они не являются
продолжением друг друга. Мы ограничимся тем, что уже было
сказано по этому поводу.
Если в некоторой области концентраций раствор оказывается
неустойчивым и распадается на два других раствора, то в этом
случае на кривой изобарного потенциала появляются два минимума
Состав
Рис. 108. Изобарный потенциал
при равновесии двух твердых
растворов (р = const, T ~ const)
Состав
Рис. 109. Изобарный
потенциал системы, в которой
образуется химическое
соединение (p=const, Т = const)
и максимум (рис. 108). Проведение общей касательной к минимумам
кривой изобарного потенциала позволяет найти составы
равновесных друг другу растворов, которые образуются в области
неустойчивости однородного раствора.
При образовании в системе химического соединения ход кривых
изобарного потенциала при достижении ординаты этого соединения
ничем не отличается от хода тех же кривых при достижении
ординаты чистого компонента. Разумеется, это будет справедливо лишь
в том случае, когда химическое соединение оказывается устойчивым
и степень его диссоциации равна нулю (а = 0). При полной
диссоциации химического соединения (а = 1) кривые изобарного
потенциала не изменяют своего хода и аналогичны кривым изобарного
потенциала для однородного раствора. Оба указанных случая
иллюстрируются на рис. 109. Последовательное изменение возможного
положения кривых изобарного потенциала, которое происходит
в полном соответствии с отмеченными выше закономерностями, дает
возможность получить все точки диаграммы состояния.
Соответствующие геометрические построения выполнены на рис. 110 и 111
для диаграммы состояния с простой эвтектикой и для диаграммы
состояния с непрерывным рядом твердых растворов. Из этих
построений легко видеть общие принципы, используемые методом
геометрической термодинамики во всех остальных случаях.
244
Заканчивая рассмотрение основных положений и методов
применения геометрических построений с использованием представлений
об изобарном потенциале, следует указать на возможности, которые
открывает перед нами использование этого метода. Прежде всего
в нашем распоряжении оказывается метод анализа правильности
построения диаграмм состояния
предсказывать те или иные
варианты фазового равновесия
при различных условиях. При
этом сделанные выводы не
будут находиться в противоречии
с правилом фаз. Даже в тех
случаях, когда применение
правила фаз встречает затруднения,
возможно разрешить их.
§ 58. ОБ ОТНОСИТЕЛЬНОМ
РАСПОЛОЖЕНИИ ЛИНИЙ
ФАЗОВЫХ РАВНОВЕСИЙ
НА ДИАГРАММАХ СОСТОЯНИЯ
Метод геометрической
термодинамики может быть
использован для строгого
объективного доказательства
правильности (или неправильности)
относительного расположения
линий фазового равновесия на
диаграммах состояния.
На рис. 112 и 113
представлены диаграммы состояния
двухкомпонентных систем с
ограниченной растворимостью
компонентов в твердом состоянии и наличием стояниях
нонвариантного превращения — эвтектического
и перитектического. Там же изображено относительное расположение
линий изобарного потенциала для равновесных фаз при
температуре нонвариантного превращения (te и tP)9 при температуре
более высокой (/х) и при более низкой (tf2).
При температуре нонвариантного превращения в равновесии
находятся три фазы: расплав (L), твердый раствор В в А (а) и твердый
раствор А в В (Р). Такое равновесие характеризуется общей
касательной ко всем трем кривым изобарного потенциала (L, а и р).
Таковыми являются прямые аеЬ (рис. 112) и pab (рис. ИЗ).
При некотором перегреве (до tx) или переохлаждении (до t2)
наряду со стабильными фазовыми равновесиями вследствие задержки
нонвариантных превращений становятся возможными метастабиль-
ные фазовые равновесия. И стабильные, и метастабильные
равновесия характеризуются общими касательными к кривым изобарно-
245
Затем мы получаем возможность
А 6
Состад
Рис. ПО.
Построение диаграммы
состояния системы
с простой
эвтектикой
Состад'.
Рис. 111.
Построение диаграммы
состояния системы
с неограниченной
растворимостью
компонентов в
жидком и твердом со-
изотермического потенциала. Имеющиеся при этом равновесия
представлены в табл. 9, в которой приведены равновесные фазы, а в
скобках указывается точка, определяющая состав данной фазы.
Продолжения линий фазового равновесия в область их метаста-
бильности на представленных на рис. 112 и 113 диаграммах состоя-
Рис. 112. Применение метода геометри- Рис. 113. Применение метода геомет-
ческой термодинамики к анализу относи- рической термодинамики к анализу
тельного расположения линий фазового относительного расположения линий
равновесия на диаграмме состояния эвтек- фазового равновесия на диаграмме
тического типа состояния перитектического типа
ния проходят в двухфазных областях. Обращаясь к диаграммам
изобарного потенциала, видим, что это находит отражение в таком
относительном расположении общих касательных,
характеризующих фазовые равновесия, при котором касательные для стабильных
равновесий располагаются ниже касательных для метастабильных
равновесий.
Если представить себе такое относительное расположение
линий фазовых равновесий на диаграмме состояния, когда
продолжения этих линий в область их метастабильности лежат в однофазных
246
Таблица 9. Фазовые равновесия в системах эвтектического
[и перитектического типа
Диаграмма состояния эвтектического
типа (рис. 112)
температура
ti>te
*2 \ *е
фазовое равновесие
стабильное
L (/) + а (с)
L (g) + Р (k)
а (е) + Р (s)
метастабиль-
ное
a (d) + р (/г)
L (р) + а (т)
Z, («) + Р (г)
Диаграмма состояния перитектического
типа (рис. 113)
температура
h>tp
^2 ^ tp
фазовое равновесие
стабильное
L (с) + Р (/г)
L(r)+a(fe)
a (e) + Р (Л)
метастабиль-
ное
I (rf) + a (/)
a (e) + Р (g)
L (s) + р (m)
Рис. 114. Термодинамически невозможные варианты относительного
расположения кривых фазового равновесия на некоторых основных
типах диаграмм состояния
областях (рис. 114), то в этом случае в результате выполнения
геометрических построений общие касательные для метает аби л ьных
равновесий расположатся ниже, чем общие касательные для
стабильных равновесий. Поскольку такое их расположение
противоречит общепринятому представлению о величине изобарного
потенциала в стабильном и метает аб ильном состояниях, то следует
247
заключить, что исходное положение, приведшее к такому варианту,
невозможно.
В ряде случаев, однако, экстраполяция кривых в область
однофазного равновесия не является опровержением термодинамической
возможности такого варианта их относительного расположения.
Рис. 115. Некоторые примеры возможных случаев продолжения линии
фазового равновесия в однофазные области диаграмм состояния
Это относится к мнимым участкам кривых фазового равновесия после
их пересечения в различных экстремальных точках диаграмм
состояния (рис. 115).
Особое внимание заслуживают отдельные случаи, когда
относительное расположение линий фазового равновесия таково, что в
двухфазной области допускается экстраполяция лишь одной линии фазо-
\
\
—(
\
^
А*'"—"'* "
^7 ■
ч s l \
ч / I
\/ /
а
д
Рис. 116. Относительное расположение ветвей ликвидуса на диаграммах
состояния с химическим соединением
вого равновесия, в то время как другая линия при экстраполяции
совпадает с горизонталью нонвариантного превращения
(горизонталь является касательной к рассматриваемой кривой в точке их
пересечения). Таково расположение линий на диаграмме состояния
с так называемой промежуточной точкой (рис. 116,
а)—промежуточной между дистектической (рис. 116,6) и перитектической
(рис. 116, в) точками диаграмм состояния с химическим
соединением (см. также рис. 80, а). Соответствующее доказательство
методом геометрической термодинамики приводится на рис. 117.
Таким образом, рассмотренное правило об относительном
расположении кривых фазового равновесия имеет ограничения. Поэтому
248
в каждом отдельном случае следует анализировать
термодинамическую возможность имеющегося относительного расположения
кривых фазового равновесия, используя объективный и наглядный
метод геометрической термодинамики и избегая бессознательного
применения правила о невозможности продолжения кривых
равновесия в однофазных областях диаграмм состояния.
А В А „В
Состав Состав
Рис. 117. Применение метода геометрической термодинамики к анализу
относительного положения линий фазового равновесия на диаграммах
состояния с химическим соединением
Используя аналогичные приемы термодинамического
доказательства, можно установить закономерности относительного
расположения границ фазовых областей трехкомпонентных диаграмм
состояния.
На рис. 118 приводятся некоторые примеры возможного и
невозможного относительного расположения кривых фазового
равновесия на изотермических сечениях трехкомпонентных диаграмм
состояния. Продолжения линий, ограничивающих однофазную об-
219
ласть, могут лежать либо в различных двухфазных областях, либо
в одной трехфазной области. Невозможны случаи, когда
продолжения этих линий лежат в однофазной области или продолжение
одной линии попадает в двухфазную область, а другой — в
трехфазную.
Рис. 118. Пример возможного (а и б) и невозможного (в и г) относительного
расположения линий фазового равновесия на изотермических разрезах
диаграмм состояния трехкомпонентных систем
Все указанные закономерности относительного расположения
кривых на диаграммах состояния могут быть использованы для
проверки правильности полученных результатов экспериментального
изучения системы и для приближенного прогноза возможного
расположения смежных с изученными областями других фазовых
областей диаграмм состояния.
§ 59. ГЕОМЕТРИЧЕСКАЯ ТЕРМОДИНАМИКА
ХИМИЧЕСКАЯ ТОПОЛОГИЯ И ФИЗИКО-ХИМИЧЕСКИЙ АНАЛИЗ
Задача изучения или, как говорят, построения диаграмм
состояния в настоящее время решена лишь в незначительной части.
Построено около 20% диаграмм состояния двухкомпонентных систем,
около 4—5% —трехкомпонентных систем и т.д. Следовательно,
250
предстоит еще очень большая экспериментальная и теоретическая
работа.
Среди методов построения диаграмм состояния были рассмотрены
только основные экспериментальные (простой и диференциальный
термический анализ, стр. 174—176) и теоретические приемы (по
зависимостям изобарного потенциала от температуры и состава,
стр. 238—250). Практически же применяется значительно большее
число приемов. Почти любое физическое или химическое свойство
при рассмотрении его зависимости от температуры, состава и
других параметров состояния может быть использовано для
построения границ (поверхностей, линий) фазовых областей диаграмм
состояния. В формулировке общих закономерностей изменения свойств
в зависимости от состава и других параметров состояния, а также
в практическом использовании установленных закономерностей при
изучении фазовых областей и точек диаграмм состояния состоит
предмет научного метода, называемого
физико-химическим анализом.1
Физико-химический анализ как научный метод принципиально
отличается от другого подхода к изучению физико-химических
систем, при котором детально исследуются разнообразные свойства
различных химических объектов (элементов, соединений, фаз и их
смесей) по возможности в более чистом виде или преднамеренно
легированном состоянии с целью познания их природы и выявления
возможностей их практического использования.
Как известно, аналитические выражения, описывающие
фазовые равновесия, очень громоздки и лишь приближенно описывают
границы фазовых областей. Поэтому в учении о фазовых
равновесиях преобладает использование геометрических образов —
графических изображений диаграмм состояния. Это часто служит
основанием для того, чтобы именовать теорию и практику учения
о фазовых равновесиях геометрической термодинамикой2.
Результатом использования физико-химического анализа
является построение геометрических образов-графиков свойство —
состав, свойство-—температура и т. п., причем элементы этих
геометрических образов строго соответствуют фазовым областям
изучаемой физико-химической системы. Другими словами, каждому
участку или каждой области графического изображения зависимости
свойство — параметр состояния соответствует определенная
фазовая область рассматриваемого простого или сложного химического
объекта. В этом состоит важнейшая идея физико-химического
анализа, и это же послужило причиной относить его к так называемой
1 Знакомясь с физико-химическим анализом, полезно обратить внимание на
отличие термина «физико-химический анализ» от других внешне схожих терминов,
таких как «физическая химия» (дисциплина о наиболее общих закономерностях,
которым подчиняются химические превращения), «физико-химия» (комплекс знаний
по физике и химии) и, тем более, от термина «химическая физика» (дисциплина о
физических превращениях химических элементов).
2 Здесь термин «геометрическая термодинамика» трактуется более широко,
чем это было сделано на стр. 238
251
химической топологии. Здесь же заметим, что
практическое использование геометрических изображений для изучения
физико-химических (термодинамических) систем в отдельных
случаях дает основание считать физико-химический анализ и
геометрическую термодинамику синонимами1.
Основная идея физико-химического анализа распадается на два
взаимосвязанных принципа: принцип непрерывности и принцип
соответствия. Их сущность состоит в следующем.
Изучаемое свойство в зависимости от изменения одного или
нескольких параметров состояния (концентрация, температура,
давление и т. п.) обязательно меняется непрерывно, т. е. без разрывов,
без резких скачков. Это означает, что бесконечно малое изменение
параметра состояния меняет свойства системы тоже на бесконечно
малую величину.
Безусловно, непрерывность зависимостей свойство — параметр
состояния не исключает проявления на них экстремальных точек
и линий, какими являются максимумы и минимумы и которые
могут видоизменяться вслед за преобразованием координат, а также
специальных точек и линий. Среди специальных точек и линий
следует выделить встречающиеся на зависимостях свойство —
параметр состояния точки и линии перегиба (изменение знака кривизны)
и так называемые особые или сингулярные точки (узловые,
возврата, излома, прекращения и т.д.), а также соответствующие
складки; и те, и другие — инвариантные относительно
преобразования координат. Специальные и особые или сингулярные точки,
линии и складки соответствуют границам фазовых областей, а
участки линий и поверхностей, заключенные между ними,
соответствуют самим фазовым областям. В этом состоит используемый в
физико-химическом анализе принцип соответствия.
Конкретный вид зависимости свойство — параметр состояния
(положение точек и линий начала и прекращения, значения
производных, их знак и темп изменения) сложным образом
определяется и тем свойством, которое избрано для изучения системы, и
физико-химической природой изучаемой фазы или совокупности
фаз.
Важнейшие закономерности, сформулированные в результате
накопления экспериментальных данных в течение нескольких
десятилетий практического использования методов физико-химического
анализа, служат надежной основой для рационального изучения
физико-химических объектов и контроля правильности получаемых
результатов. До сих пор наблюдавшиеся противоречия служили
поводом для обнаружения методических упущений. Так, например,
обстояло дело с аномальной линейной зависимостью микротвердости
от концентрации в ряде полупроводниковых систем (Qe—Si,
InAs—InP и др.)- Эта зависимость в ранее изученных системах
имела характер плавной кривой с максимумом. Оказалось, что
причина аномалии крылась в неодинаковой хрупкости сплавов с раз-
1 Здесь термин «геометрическая термодинамика» трактуется более узко.
252
личной концентрацией. Учет хрупкого разрушения микротвердо-
стных отпечатков привел к исправлению «аномалии».
Иначе обстоит дело с некоторыми более поздними
экспериментами, которые сопряжены с более прецизионными измерениями
некоторых структурно чувствительных свойств. Так, например,
изучение изотерм вязкости и электропроводности растворов
InSb—QaSb показало наличие сингулярных точек (пиков) в
пределах достаточно надежно установленных областей гомогенности.
Другой пример, подтверждающий факт проявления
межмолекулярного и межатомного взаимодействия (при определенных составах
в пределах гомогенных областей) связан с наблюдением
сингулярных линий (складок) при измерении микротвердости по разрезам
с постоянным содержанием основы сплавов и переменным
соотношением концентраций легирующих добавок в системах: Си—Ni—Be
(складка приходится на разрез Си—NiBe), A1—Mg—Si (складка
приходится на разрез Al—Mg2Si) и др.
Характерен пример с "системой Qe—Si- Ее компоненты образуют
непрерывный ряд твердых растворов, однако зависимость ширины
запрещенной зоны от концентрации в этой системе имеет точку
излома и, следовательно, состоит из двух участков, а не из одного из-за
качественно отличающегося характера межзонных переходов в
электронной структуре атомов твердых растворов на основе Qe и Si
слева и справа от точки излома соответственно. «Нарушение»
принципа соответствия происходит из-за высокой структурной
чувствительности изучаемого свойства, в данном случае чувствительного
к электронной структуре атомов твердого раствора (гомогенной
фазовой области).
Поскольку каждый раз рассматривались равновесные состояния
сплавов, становится вполне очевидной необходимость пересмотра
и уточнения принципа непрерывности и главным образом принципа
соответствия, используемых в физико-химическом анализе. К тому же
вполне ясно, что современный физико-химический анализ пригоден
для решения значительно большего числа задач, чем их ставит
химическая термодинамика и в том числе геометрическая
термодинамика .
СПИСОК ЛИТЕРАТУРЫ
Учебники и учебные пособия по физической химии
Герасимов Я- И., ДревингВ. П., Еремин К. Н. и др. Курс
физической химии, т. I—II. М., «Химия», 1969—1970.
Даниэльс Ф., Альберти Р. Физическая химия. М., «Высшая школа»,
1967.
ЖуховицкийА. А.,Шварцман Л. А. Физическая химия. М., Метал-
лургиздат, 1963.
К и реев В. А. Краткий курс физической химии. М., Госхимиздат, 1962,
1963; М., «Химия», 1969.
К и р е е в В. А. Курс физической химии. М.—Л., Госхимиздат, 1956.
Киселева Е. В., Каретников Г. С, Кудряшов И. В. Сборник
примеров и задач по физической химии. М., «Высшая школа», 1970.
Мельви н—X ь ю з Э. А. Физическая химия, кн. 1—2. М., ИЛ, 1962.
Николаев Л. А., Тулупов В. А. Физическая химия. М., «Высшая
школа», 1967.
Учебники и учебные пособия по термодинамике и термохимии
А к о п я н А. А. Химическая термодинамика. М., «Высшая школа», 1963.
Владимиров Л. П. Термодинамические расчеты равновесий
металлургических реакций. М., «Металлургия», 1970.
ГовертонМ. Т. Термодинамика для инженеров. М., «Металлургия», 1966.
До дж Б. Ф. Химическая термодинамика. М., ИЛ. 1950.
КарапетьянцМ. X. Химическая термодинамика. М.—Л., Госхимиздат,
1953.
К и р е е в В. А. Методы практичзских расчетов в термодинамике химических
реакций. М., «Химия», 1970.
Крестовников А. Н., Вигдорович В. Н. Химическая
термодинамика. М., Металл у ргиздат, 1961, 1962.
КричевскийИ. Р. Понятия и основы термодинамики. М., «Химия», 1970.
КубашевскийО.,ЭвансЭ. Термохимия в металлургии. М., ИЛ, 1954.
Л е о н о в В. Ф. Термодинамика. М., «Высшая школа», 1968.
Мортимер К- Теплоты реакций и прочность связей. М., «Мир», 1964.
Мюнстер А. Химическая термодинамика. М., «Мир», 1971.
ПолторакО. М. Лекции по химической термодинамике. М., «Высшая школа»,
1971.
Пригожий И., Дефей Р. Химическая термодинамика. Новосибирск,
«Наука», 1965.
Путилов К- А., Термодинамика. М., «Наука», 1971.
Скуратов С. М., Колесов В. П., Воробьев А. Ф. Термохимия,
ч. I—II. М., изд. МГУ, 1964, 1966.
Ф е р м и Э. Термодинамика. Харьков, изд. ХГУ, 1969.
Э в е р т Д. Введение в химическую термодинамику. М., ИЛ, 1963.
Эллиот Д. Ф., Глейзер М., Рамакришна В. Термохимия
сталеплавильных процессов. М., «Металлургия», 1969.
254
Справочные издания по физической химии,
термодинамике и термохимии
БрицкеЭ. В., КапустинскийА. Ф., Веселовский Б. К- и др.
Термические константы неорганических веществ. М., изд. АН СССР, 1949.
ВерятинУ. Д.,МаширевВ. П., РябцевН. Г. и др.
Термодинамические свойства неорганических веществ. М., Атомиздат, 1965.
Войтович Р. Ф. Тугоплавкие соединения. Термодинамические
характеристики. Киев, «Наукова думка», 1971.
ВукаловичМ. П.,Кириллин В. А.,Ремизов С. А. и др.
Термодинамические свойства газов. М., Машгиз, 1953.
Герасимов Я. И.,Крестовников А. Н.,Шахов А. С. Химическая
термодинамика в цветной металлургии. В 4-х т. М., «Металлургия». 1960—1966
Т. I — 1960; т. II — 1961; т. III — 1963; т. IV — 1966.
КарапетьянцМ. X., КарапетьянцМ. Л. Основные
термодинамические константы неорганических и органических веществ., М., «Химия», 1968.
Термические константы веществ. Справочник в 10 вып., под ред. В. П. Глушко.
Вып. 1—4. М., изд. ВИНИТИ АН СССР, 1965—1970.
Термодинамические свойства индивидуальных веществ. Справочник в 2-х т., под
ред. В. П. Глушко. Изд. 22-е, т. I—И. М., изд. АН СССР, 1962.
Термодинамические свойства неорганических веществ. Справочник под ред.
А. П. Зефирова. М., Атомиздат, 1965.
У к кс К. Е., Блок Ф. Е. Термодинамические свойства 65-и элементов, их
окислов, галогенидов, карбидов и нитридов. М., «Металлургия», 1965.
Физико-химические свойства окислов. Справочник, под. ред. Г. В. Самсонова.
М., «Металлургия», 1969.
Физико-химические свойства элементов. Справочник, под ред. Г. В. Самсонова.
Киев, «Наукова думка», 1965.
F i с h t e R. Die thermodynamischen Eingenschften der Metall—chloride. Berlin,
1953.
Gmelin's Handbuch der anorganischen Chemie. Leipzig—Berlin, 1924.
Handbook of chemistry and physics. A ready-reference book of chemical and
phisical data. 37-th ed.—46-th ed., Cleveland, Ohio, 1955—1966.
К e 1 1 e у К. К. Bull. United Staties Bureau of Mines. Washington, 1934—1961.
Kubaschewski O., Evans E. Metallurgical thermochemistry. Lnd.,
1951; 2-d ed., 1955; 3-d ed., 1958.
Kubaschewski O., Evans W. U., Alcock С Metallurgical
thermochemistry, 4-d ed., Oxford, 1967.
Landolt—Bornstein. Physikalisch—chemische Tabellen, 5 Aufl., Bd. I—II. Berlin
1923; Erganzugsbande: I, 1927; 5 ( 2 Teile), 1931; II (3 Teile), 1935—1936.
L a n g e W. Die thermodynamischen Eigenschaften der Metalloxyde. Berlin,
1949.
M e 1 1 о r J. W. Comprehensive treatise on inorganic and theoretical chemistry.
Lnd. — N. Y. — Toronto, 1952, v. I—XVI.
ReidR.C,Sherwood Т. К. The properties of gases and liguids their
estimation and correlation. N. Y. — Toronto—Lnd., 1958.
R о s s i n i F. D., W a g m a n D. D., E v a n s W. H. a. o. Selected values of
chemical thermodinamic properties (Circular of the National Bureau of Standards 500).
Washington, 1952.
W a g m a n D. D., E v a n s W. Y., P a r k e r V. B. Selected values of chemical
thermodynamic properties. Washington, 1965—1969.
Учебные пособия по теории растворов
Вагнер К- Термодинамика сплавов. М., Металлургиздат, 1957.
ДаркенЛ. С.,ГурриР. В. Физическая химия металлов. М.,
Металлургиздат, I960.
Кириллин В. А., Шейндлин А. Е. Термодинамика растворов. М.,
Госэнергоиздат, 1956.
Ламсден Дж. Термодинамика сплавов. М., Металлургиздат, 1959.
С в е л и н Р. А. Термодинамика твердого состояния. М., «Металлургия», 1968.
Ш а х п а р о н о в М. И. Введение в теорию растворов. М., Гостехтеоретиздат,
1956.
255
Учебные пособия по фазовым равновесиям
Аносов В. Я. Краткое введение в физико-химический анализ. М., изд-во
АН СССР, 1959.
АносовВ. Я-, Погодине. А. Основные начала физико-химического
анализа. М., изд-во АН СССР, 1947.
Воловик Б. Е., Захаров М. В. Тройные и четверные системы. М.,
Металл ургиздат, 1948.
Древинг В. П., Калашников Я. А. Правило фаз. М., изд-во МГУ,
1964.
Захаров А. М. Диаграммы состояний двойных и тройных систем. М.,
«Металлургия», 1966.
Захаров А. М. Диаграммы состояний четверных систем. М., «Металлургия»,
1966.
Кур на ков Н. С Введение в физико-химический аназиз. М.—Л., изд.
АН СССР, 1947.
ПалатникЛ. С, Ландау А. И. Фазовые равновесия в
многокомпонентных системах. Харьков, изд. ХГУ, 1961.
ПерельманФ. М. Изображение химических систем с любым числом
компонентов. М., «Наука», 1965.
П е т р о в Д. А. Тройные системы. М., изд-во АН СССР, 1953.
Уббелоде А. Плавление и кристаллическая структура. М., «Мир», 1969.
ЧалмерсБ. Теория затвердевания. М., «Металлургия», 1968.
Эддисон У. Аллотропия химических элементов. М., «Мир», 1966.
Юм — РозериВ., Христиан Дж., Пирсон В. Диаграммы
равновесий металлических систем. М., Металлургиздат, 1956.
Справочные издания по растворам и фазовым равновесиям
Вол Е. А. Строение и свойства двойных металлических систем, т. 1—2. М.,
Физматгиз, 1959—1962.
Диаграммы состояния металлических систем. В 15-и вып. М., изд-во ВИНИТИ
АН СССР, 1956—1972. Вып. I — 1956; вып. II — 1959; вып. III — 1960; вып. IV —
1961; вып. V — 1962; вып. VI — 1962; вып. VII — 1963; вып. VIII — 1963; вып. IX —
1966; вып. X — 1966; вып. XI — 1968; вып. XII—1969; вып. XIII—1970; вып.
XIV—1971; вып. XV—1972.
Диаграммы состояния неметаллических систем. Окислы металлических
элементов и кремния. В 8-и вып. М., издание ВИНИТИ АН СССР, 1965—1972.
Вып. 1—1965; вып. И —1966; вып. III —1967; вып. IV—1968; вып. V —1969;
вып. VI —1970; вып. VII —1971; вып. VIII —1972.
Кауфман Л., Бернстейн X. Расчет диаграмм состояния с
помощью ЭВМ. М., «Мир», 1972.
Коган В. Б.,Фридман В. М. Справочник по равнозесию между жидкостью
и паром в бинарных и многокомпонентных системах. М., Госхимиздат, 1957.
Несмеянов Ан. Н. Давление пара химических элементов. М., изд-во АН
СССР, 1961.
Справочник плавкости солевых систем, ч. I. M., изд-во АН СССР, 1961.
Хансен М., Андерко К. Структура двойных сплавов, т. 1 —2. М.,
Металлургиздат, 1962.
Эллиот Р. Структура двойных сплавов. М., «Металлургия», 1969.
HaughtonJ. H., Prince A. Constitutional diagrams of alloys (a
bibliography). Lnd., 1956.
Kubaschewski O., Cattrell T. A. Thermochemical data of alloys.
Lnd., N. Y., 1956.
Shunk F. A. Constitution of binary alloys. McGraw-Hill. Lnd. — N. Y.;
1969.
SmithellsC. J. Metals reference book. Lnd., 1949; 2—d ed., Lnd., v. I, 1955; <
v. 2, 1957; 3—d ed., Lnd., v. 1—2, 1962; 4—d ed; Lnd., v. 1—3, 1967.