














































































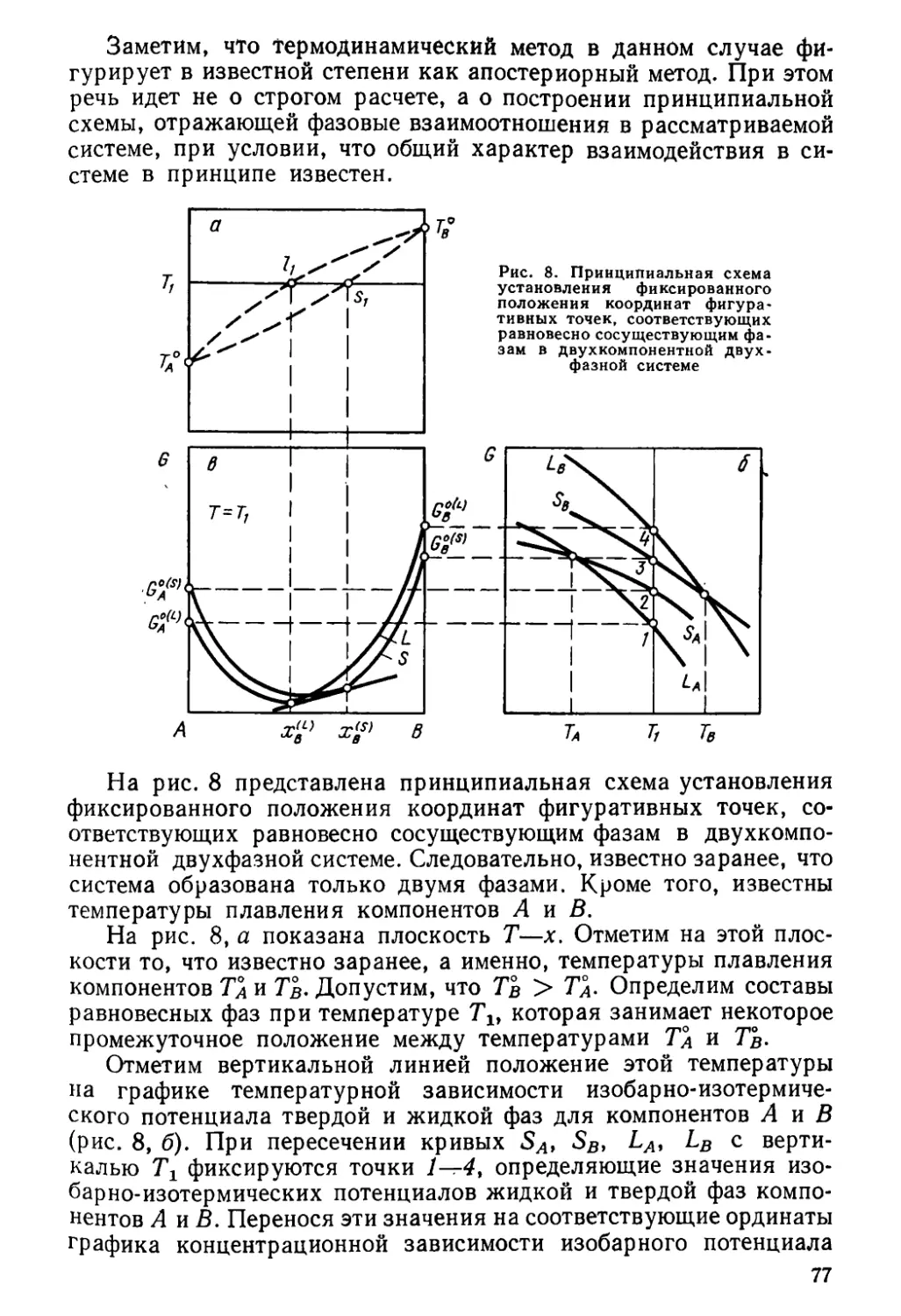



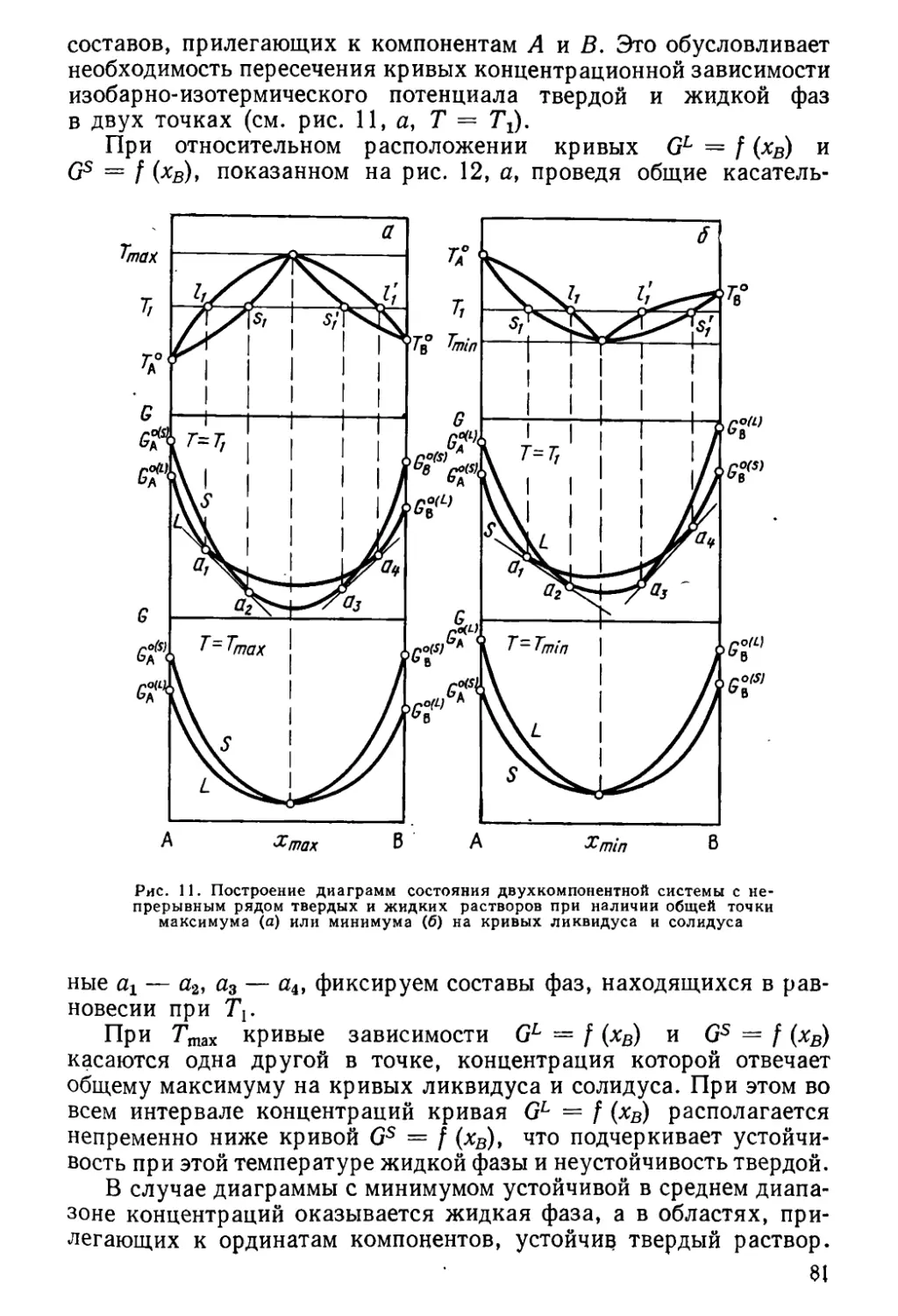






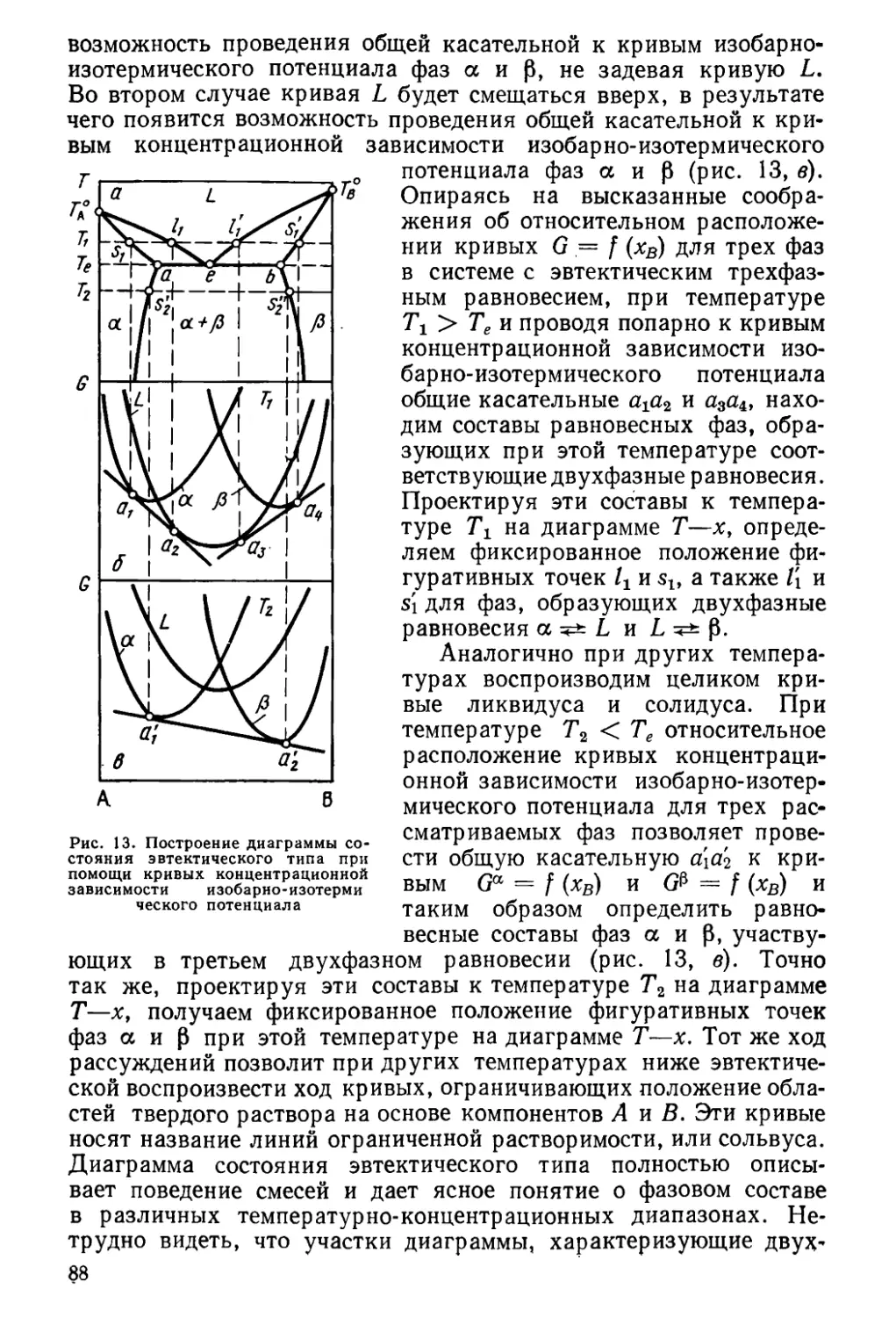











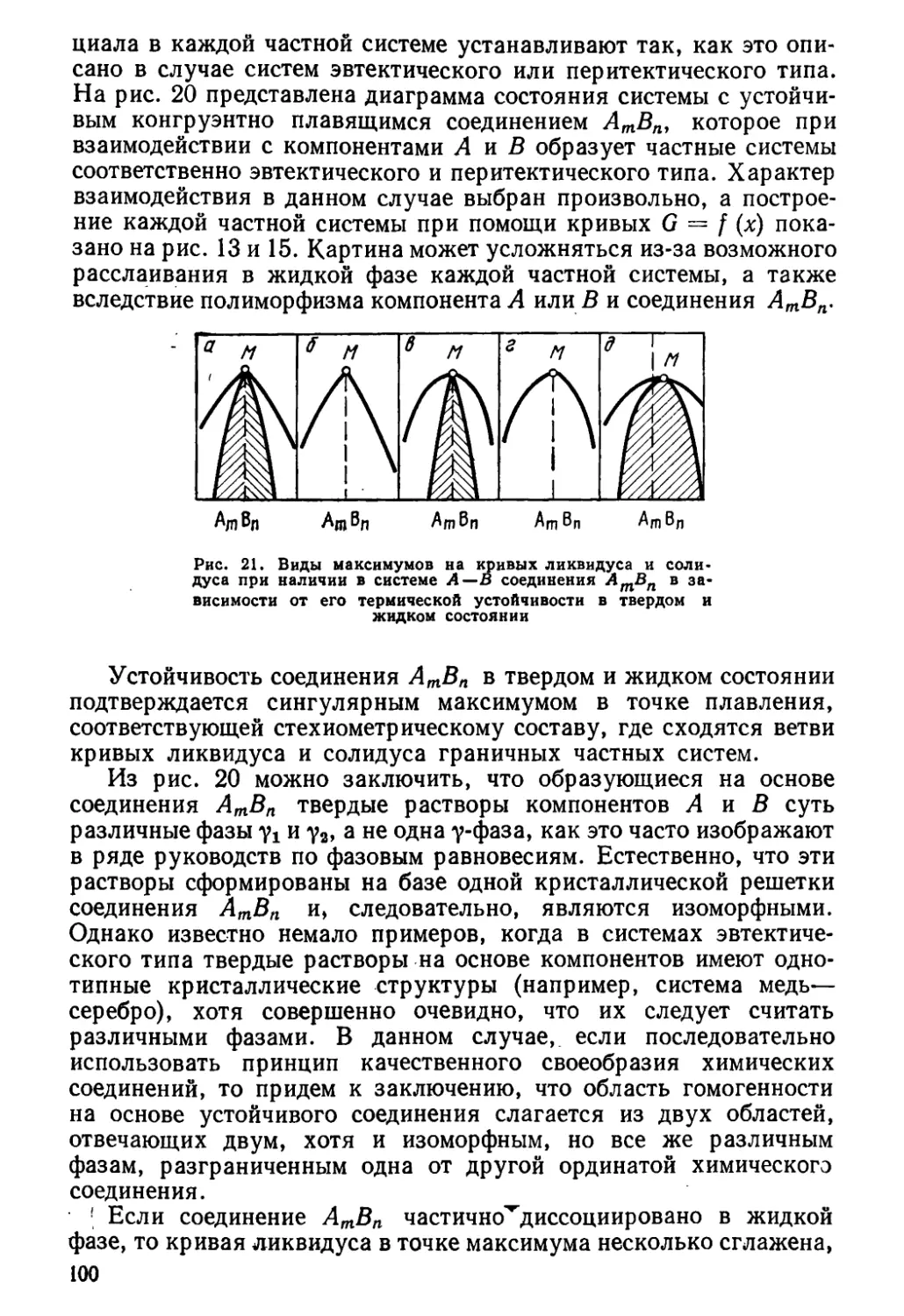






































































































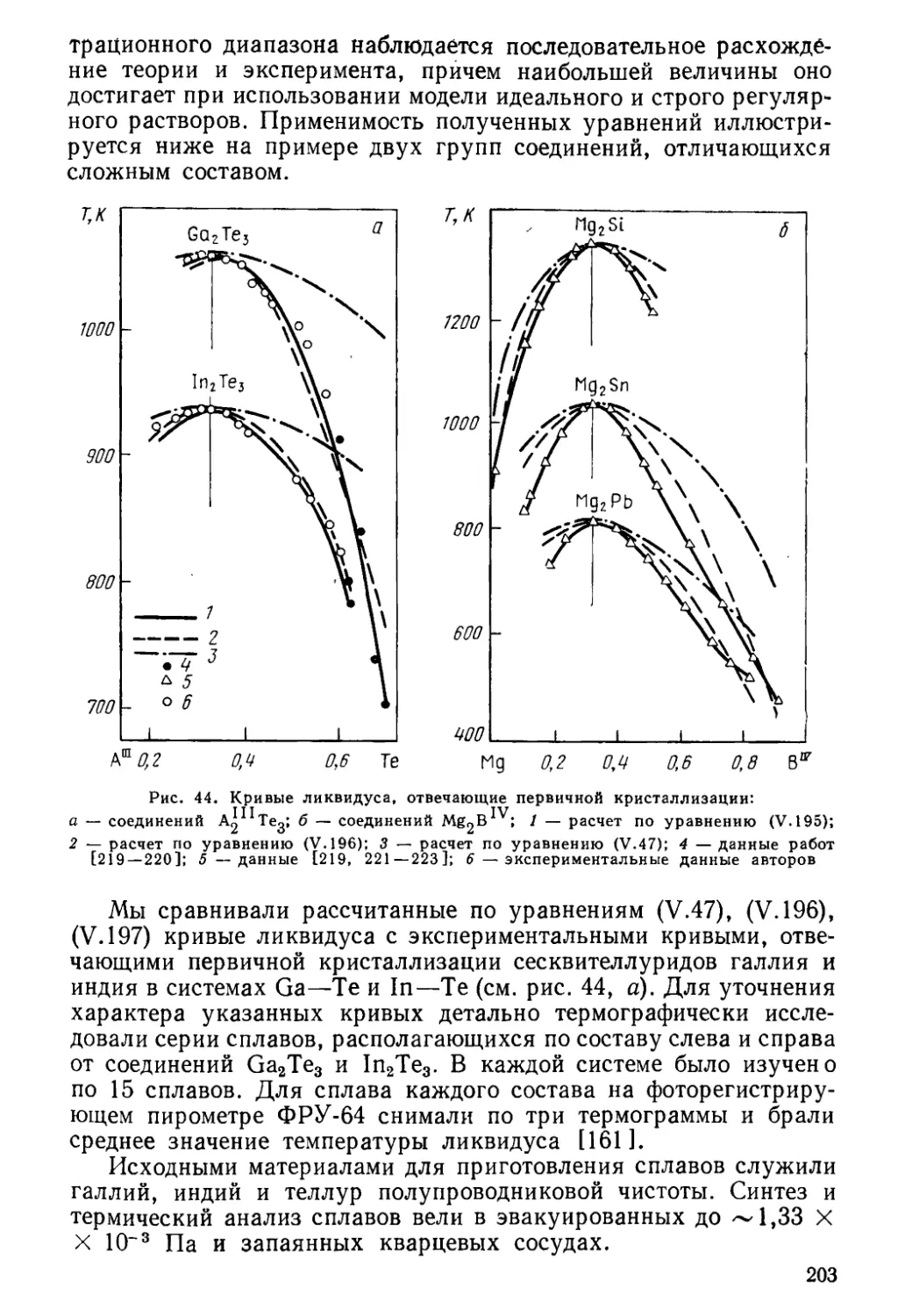






















































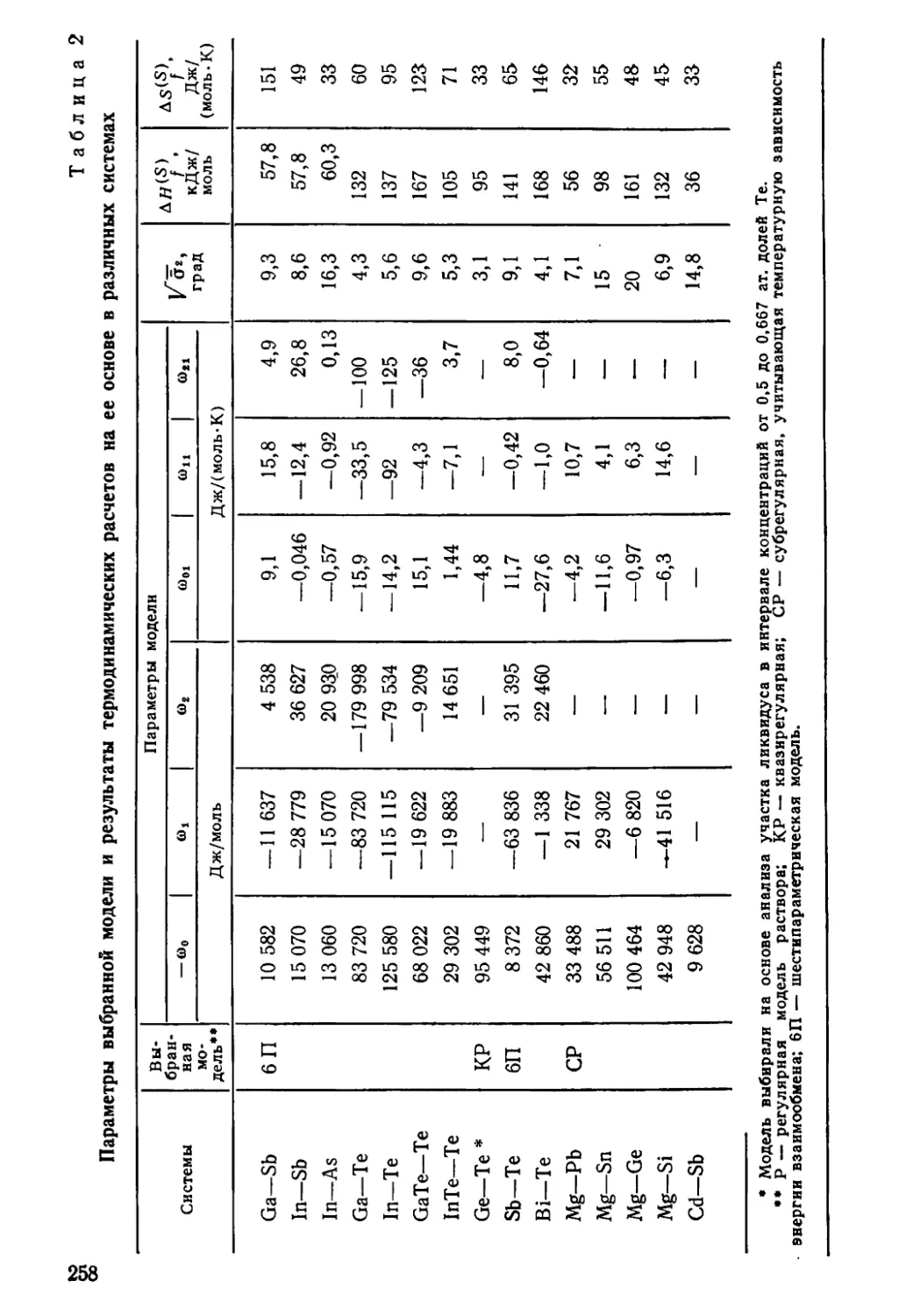
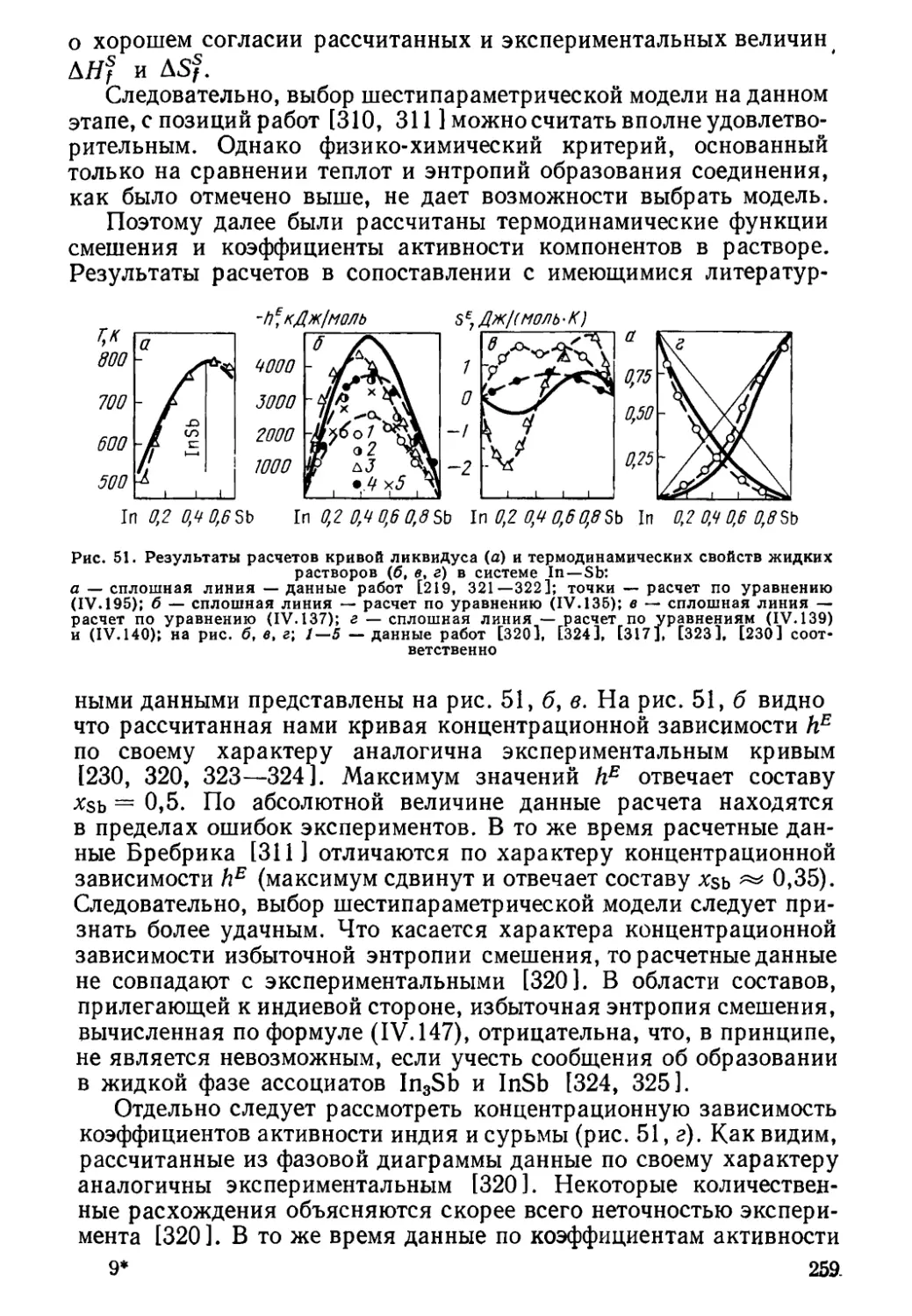





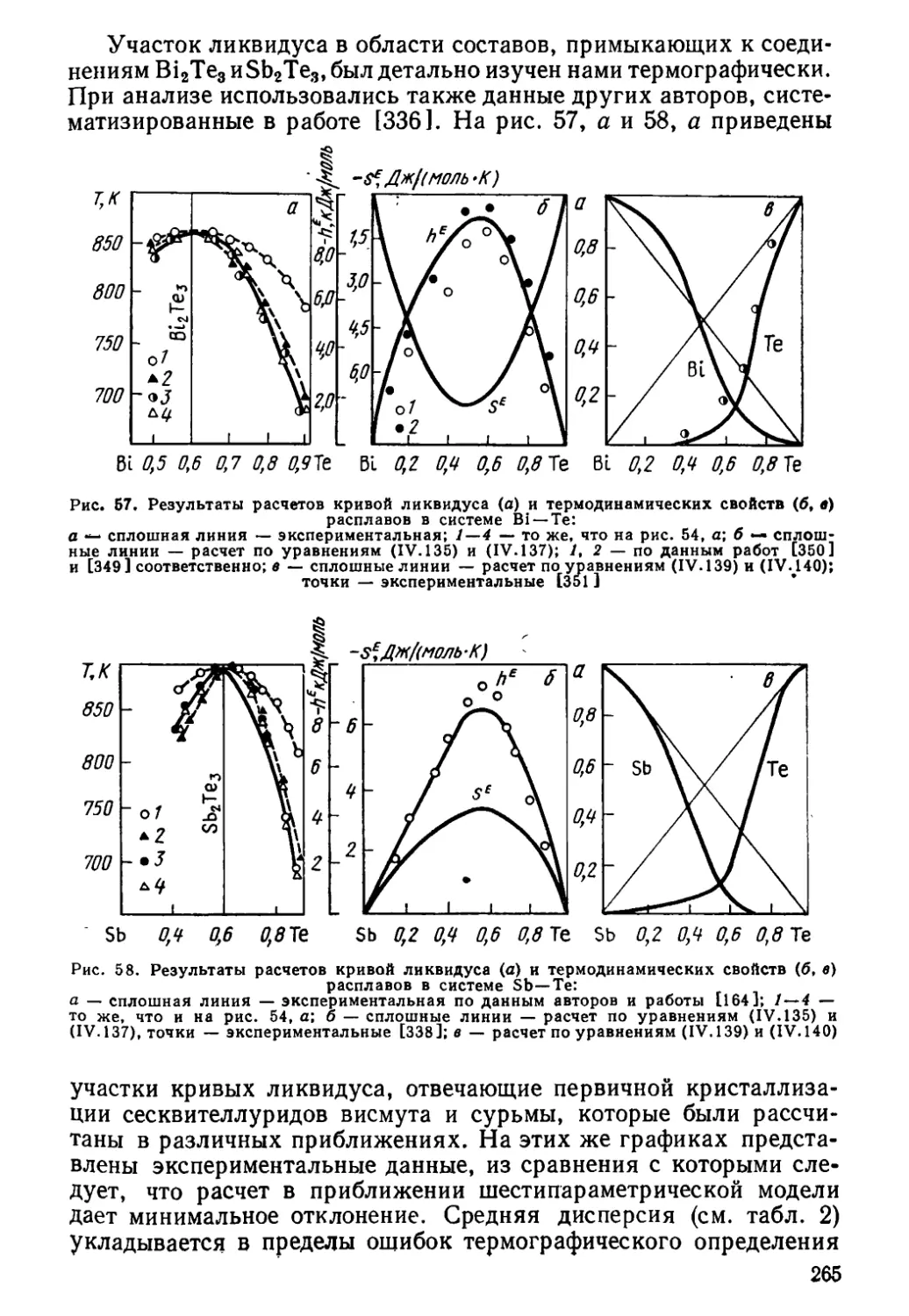


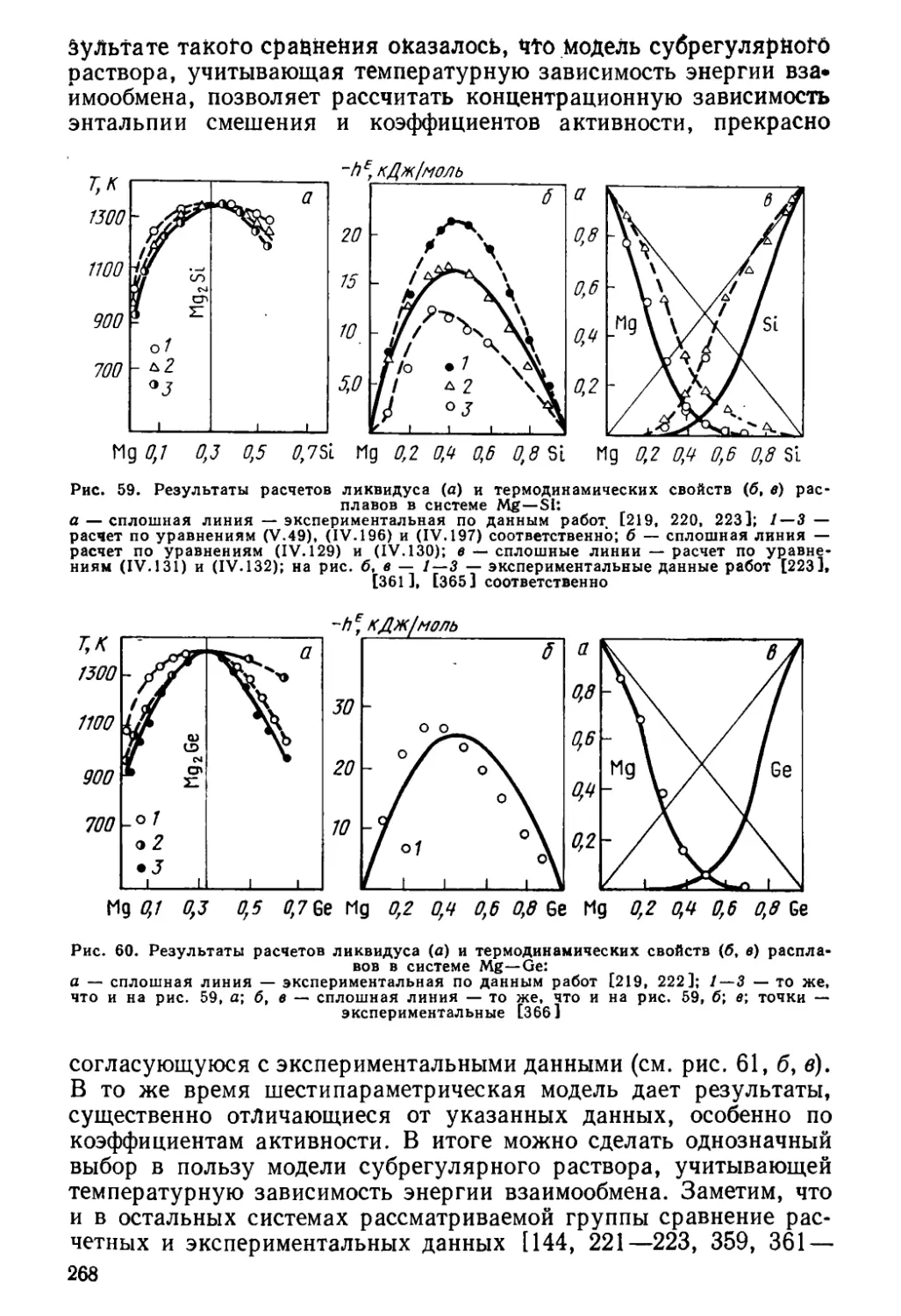














































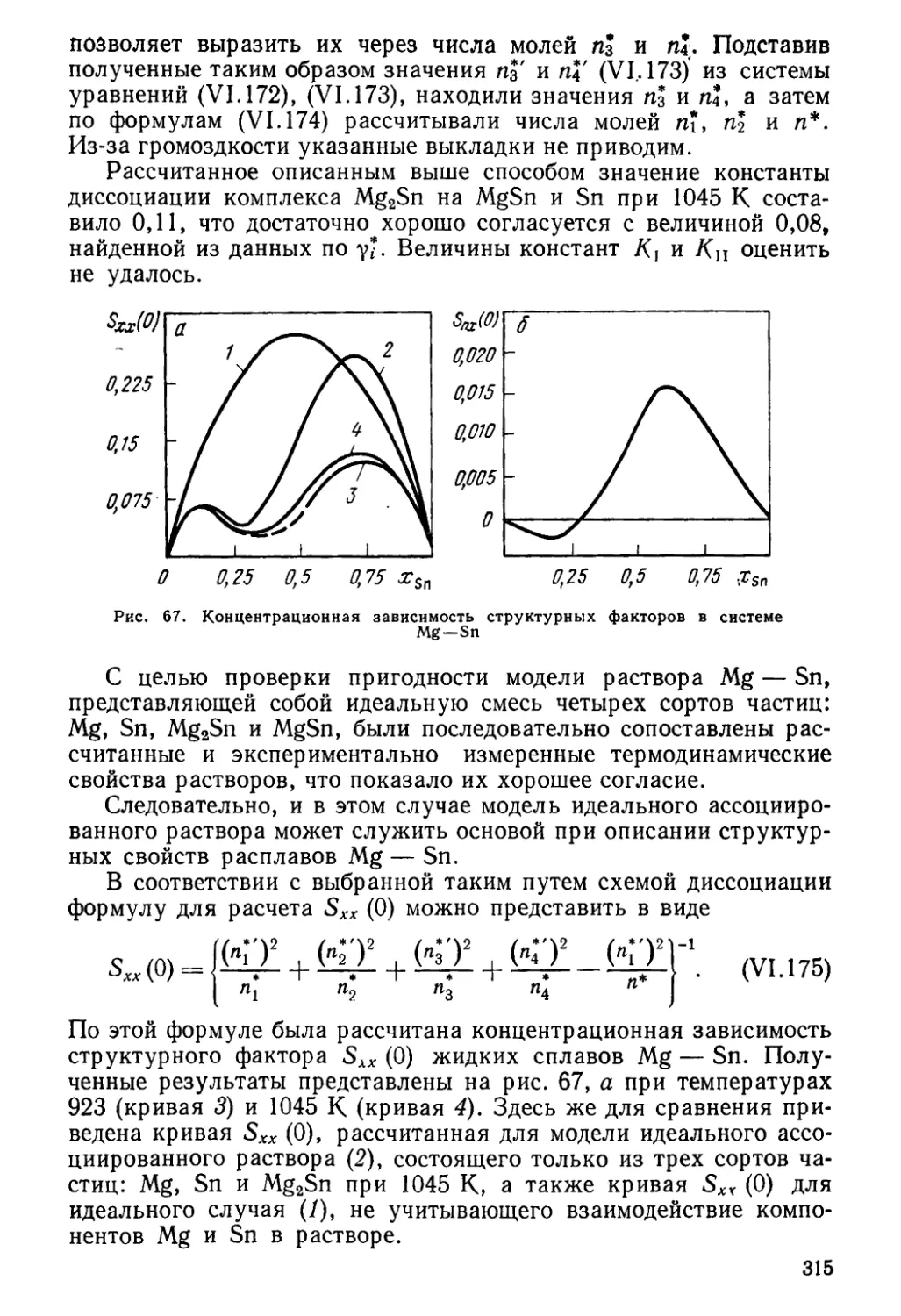





















Author: Глазов В.М. Павлова Л.М.
Tags: термодинамика энергетика химия физика физическая химия учебник по химии фазовые равновесия издательство металургия
Year: 1981




;
%t»
\ \\
f
;I
.
:
щ
r
$
1
.'!t1:-:..:;t
?!
.»
m
"'.;.
;.
1
Q
1>.:
::
t.t:ii
..
...
'"-
ti
'\\7i_\
Iii. :.'
&.
'('t\
""i(
'
.!'<:!
й
t.
. \:
.
. ...;,.
a
\
2.4'!'
_
,.
,
5
<
.
_
.
6,
r
'
: ;
:b
'" 1I
. -::<,'.
1. .' · 'tt ,
."",,
iF."
"
. .....
· ...
'\!-
_
.,
t...
_ t;
тt ,
:
_ -.
... .. ... ..;.
.
It:} .
. 5 J!
.
4
g--.
' .
"-t
..
..
..
B
:1S
1.
1 '
'"
' r SL
.&,.
.{$i
- ...
. ..
!
..
-&i.
'
' .. .
..... ."
!
.. . ..r-I -..,
.. _",. !:
:w.....
. .... '''.\
t
!>.
.
."'"
..:.i.....
w
::;,.
. -о
,i'"
4'"
........
'""...."" .
.
'.
'
CI
,
.
.
"
" 400
.....
""'.
. '-
:
'1'
!'
..: .
-. ,,
... ..
...... ... . ,." ..
.. ..' '...
... .;, · .,.
"'
. .....
, ,
.je'''' - ..
.
,
..:
"Jt..
..
....
:
. .t
... "
;."
.
... .. '-:.
. ""
'
4\.
.
..
.
,.....
.
.
r!.
l
f
i
t.
..,.
..
" '.
. <\ ... ..' -. !
. \ ..... Щ'\
.. -
-.
_
-';
.
:..:S
.
'R
..
"\ .......
..
<'
.--=t-;*
;I
-.:t
l
"',
.",;'t
-!. ....." '- ' ... ..... ....
...
..--=-
.
'i
1
:.i
"
'
il ,
o.
""""'"
:
\
i
'i
'
".
.
,.
"
'
$
..
........ -""""
.
...
»:;....,. . ... » _
..........-:
1t
.....
4 7
....... "'о.........:... . .o:
.:.......
.......
.r "5'''
.
!,.."
4j
J
' '1. ,
.
---.
ъ
., . i...
-::;
"'
t""
:i . . с:' " .
.
I
t
, .'
i' . I&
. "::."
"'
' >-
:. . ..
t ..'
'.'-
.. ·
.
..п.....
"
A
4..
W'''''
A.
-- ,
f
,,'.
. i-
.
.
._. '
"i
t
-
.
.... ':ос . .
.'
. ;о' ':'.
.:
.U:.
.. . ...
. -:. .
...
.:
t
.
.... . <. .....
...
.
.:А ,lL
...
4 .. '" -
'.
- ..
;'"",
... "::'..
' ... -.,
....
.
.;
,'.-
'...
" .. .' . - A
: "'. ... ';' .'
.
...... .., '"
:
ff '.
,,
..
..
. .
"'?h-
.
....... ' N-"
8'i
..,.. :
' ... . ""- "а.. .. ...
'
";'i
.
'"
'
E ":
. .
'"
.... " "о.
\1
.
...
.. .... '. .... ......, :"а.
g . '"
-.. . 1 ..
.
4....]o.
. ',- -
"'.:IS ." ......
"
" .' .
:
.
. tt1 .::
'
":V-
.
',
I
' ... '
y
.. '
:. :-m
" 1 1
"
."
'О.. ....
...
.........
.,..."\.....
..
. _t:
......
'C'
.........
,"'.'" .'
J.'
!O
,.
..
.
.-r"
..
..
· ......
-
., .'.
.
.
\.....
. ,'.
IIJ.
:
. ,,'"
:-
. Y".iI.......
)O
. "5
"
'.
t.'
_
-
.
h
'} . .g
..
. ::-
?-. "'-,,
;.
. :
..
.
,
.
::!.
"",' ..
I "
-:
..
"'\.
.:... '
', .' .
.. .
.
") .....
it.\
:.'
.
'
.
ё-
,"\. .
' Ь
"'" ". .:.'Т - .. м
"
:
Ъ. . _ .-.-.
,..
I
!" II
'J ," #i. t
. -'
:-.. . '.
J-'
:. "'" .
. ...:
.."'!.
. .... t;_
.
.....
'.
...
, r. '..
........ ,. .. .... .. ..
,
4
- .... . .... <>-
:h.- ..... , ".
.....
"'" . ..... '" '- '
11' ..
.......... .....
. $
-::.'
"
. '-.... а' .
. ' _ . .. \... , .
.
...,,
, . -
I
"',," ..
. -. .
t
", .
.
.' . -
_
... "': :-
.
.. " . _. ...
'.'...
<-
:;: . "'".,. . .
.
; ..А:
-$ :.
...... ::,'
·
- "!'
...
-
. .."....... --:-.. .
.... .;
... ....
-:'\ .
. .
;.'.
.,. _.-:.Эr
.,,
... ,!
,
:-.
..),
:.. "'... · -
i: " i:'
t'-
..,... . ·
- ,
.':'
"
.i.
"""'3
. .'
...
:
...,
48'
2- '.
;;
.k
..
-.......... .
."'".'"'.
.ц.. ' '
4:\
. .....
It: :
.
' ...... "" .
-
..t
Z
':. .:
....,..
. е:. '. .......
...,
. S,
.
.
.
'
S:i
..
. .
. :" :.
. 41 . " . . ..
.. .. '\,;
":' .
. '. .....::
'.. ,\'
.
"'
: -.
. .
- :' '.' , ;'_.: .....,... ....'-"."
....
....,
!'-.-
h'"
."t'"' ....
.....,,
. . . ".
. .
.
.
",. А 1ь..
:-
':-" ....
.' Y.
"'" ..
. . .'
<
..
. .
"-.
. ..".А...... .
I;'
.,.'
.
'
":. .:-.
'.'
'.
.
.
. ......
...
'.
. .....:.,
.00#
' .
1';' ..._'
.
.
. -...
.- ..... -
&.""
'
.' "
'
J"
:'" . . ..... .. ,
........ '"
.. .-"
. . ......
" J!__
.
---:.' ":'\ .... .. ," 'J.'
.
..... '.. 11.....
-','\. "-
,.. .
....
.
. . .
. .....
'..
.' ".. . .
t' .
.
........
.
,_.. '..
:- ........' ....'С" ...... .... А.......
.. ...
...:
"'
'
;.: А;- 1
. ,",'
..."..,.
...... .
..,:....
t"c.
. ..."':ti' 4.
;!r -... . ..
. . ': "
'r'
. '-( ",. .
.. .
\'" '.
.
'';'''-
........":"'...
_"1., IL
'
..
. '.-:..
-.
.
·
. I
.,
: :... . "-
. ..
.
.
.. .'
'.
.
'
."
..
;'..
'.. ; . .......
:.......
....
.;..
......... \ .". . . Iho'. _,
"'.....,.
....... ц ..-
...
'''''':..,,' -....
- .' -"..
:'(r>..... . ·
..... . t '
." .,.......
...,.. --
: .
-.. .. .... .
'
.: ... " . ... . ..
"",...,...... .". .
[
::
.'-i=-l.-w.'
";'
't'....... ..
.
;'i:;<i.
r
,
' :...... : . . . ...... i".,.:' '\.. . >
-;. ''':'
''h ......... ....'
;"\."'"
. -::
'
.... . . ....
.. '.
.).. -" .. .. .... .... .
. . .. . .:.: ;.
.
., .
. . '''\.,...
". .
: :.....
' .' ':;'l...
-
..
.... .
....
.
........, ..
.,.
.. ) "i
".""-"
..: .,..
. ...-..:
'
_::""4.
"(.'. "
'4I.
.'
;
"
.
.."
.
. -:-.... . .... ,,",. .. - . . '-- .
........
.: ...;., ,t
. '..i.. ."
','"
.
"'=' "*,.
..
.
.' .
'.' ," ..:
.. i
... "t
.'
. .
-. -(-
2
"'
.
. . <1:
... _... ..1.-..... ....4'... - t1
.
- . '"\,.' .
. .
..
..
- ""w ... --n # "'" J:.
....
.
.
..... 't ... .::.". .....:...
-
' .....
1.... ...... .
.'
. ...
-- . .
......
. .
.._ \ . _
. ..... '. J: ... _'
_
.
,
.
",,,.
,-...
.... ...
. ,.. .' . '. '....
..
"'" ..' . .!; . -. .'
.
- ',": .' .. , :
.... "'
- :-. - ....-. . .'... "'\.. . . -
. . $
.... '. '" . .... . +
.... . .... .... ........
.!. -':'
, .....
...,. " $,.... . -'"'- .,. -;...
--:
10\:'.. '" .,. ...... """"'
., ........
... ..",
"# ... 8'8'... . ..
.
.....
.
....t
.
..;;: ,..
'..
" '
,.,.'
'(.
.....
.
, \ '"
,
'.
..
- :-:;,", .(.
..-r-'
.
..
""'''''
''. I
. " ..
-........
- ...
.
..... ." ..... ...'-..::.
., ".
....
i
;
.
: 0"\". &,;.' '*' ...
.
-.' :- .....
"', .
.
....
4
'.: '\ '*
. s.
......;:.. "
. .' ...... '.
...: .,...
,. 'Ь:L '.. ..
.,
. ...
"
"'
:ъ. ,,
. - ..:-
.....
t
.
.
.
\:.... "" "":l.-:' ,,;. .
.'1\: " ' i9
.
I(
. '.о.
-' _:''-'1.
...
.., ,. 1
"
:
......
:
.
:' .
.,," ".:
" .,
.
..,;
."'......
..
;.
y
'$':
, .\ '.., .................."';..' .,... '.,
'. 1
'
. . . 'Jo- _
. .... - .,*
.
..,.,,-
. ....1. .
т . ....
.... ''У
. ...
......
.
....
.
...,
,'"
.;-
..,,:.
'.
\IL
.
..,... ':- 1 "
.
- ..' -- J
.tt' .
.. i
.
....
-. ......
.
'\;
.
. ........
·
. М ....... & . "'""\,oir.
.,
:..
.. ...", 'с
'
,... ' . '"'
-:- ..,..... :-
М! :- .
;
. -'
...'"
..
"k
1lf..i:s:.:c .:
..
. "..,
'
.'
.'
', :.....,. ....
.......
....
.
.,...
-'
..)O
.
.,
..""'"
'
';'
S""
"
": ".,"
.
..>c:.. 't't... ..
.
.. ;.... ....
............ ......,);.
...,.....::
.....- ...... .. . CI' .... .. ". ...
,. · -, rt{
> ..:
::.
.
'..k
"" .., .
. .'>.1.
., 4 ,,:' ,..' N - . . K
.:'
.
-...
.
,:!" q
'L-
' - ,,:t.'"!"
..
""':
'
' . ,':
.
*-
,
\ ..
,
..
.: :
'-"
.... ';" .
1.
.:.
.:.t..
- ....... .. .....,..\.. .
.... "....:
.. -
--::Jo. -
.
. ф.... ....:-
A:...
.. .......,.
"V
.
"L
. '-,
v'
,
. ..'
"f.!:'
..
., "'
. __,,-
.
,i.-::
....... '
'\' ........
..... .......
. .... .....
'о .
:
.
.-.....
'
...- '-'"
"
..
..
""'V
.... ........ ...
. . .....,.. . - ...
....
.........., .......",.
. "'"\ . _..," '"
......4 .:». , i'r-' ...t .
. - . __
. .....:::... . . '. ,
..:::: .... :.. ""1'.. ,
'.' . 't.
..:..""
""- ,'. :
........
. "'
.."' '"';"о
-.... ..
;::-...
"' ". _ -. ' .
.
. 't
.....
.
:.... . . ....... ...,
_
. '. ос....... ;..
:- а.-....... ," . . ... .
':'-
" ....
_.....
..
........h.r...
..
........ ..
"
.....
...:.... ""'--
_..
В. М. Глазов, Л. М. Павлова
ХИМИЧЕСКАЯ
ТЕРМОДИНАМИКА
И ФАЗОВЫЕ
РАВНОВЕСИЯ
(Двухкомпонентные металлические
и полупроводниковые системы)
Москва «МЕТАЛЛУРГИЯ» 1981
УДК 536.7:541.123.24
Рецензент — Проф. докт. хим. наук В. Б. Уфимцев
УДК 536.7: 541.123.24
Химическая термодинамика и фазовые равновесия. (Двухкомпонентные
металлические и полупроводниковые системы). Глазов В. М., Павлова Л. М.
М., Металлургия, 1981. 336 с.
Книга посвящена термодинамическому анализу диаграмм фазовых
равновесий. Даны трактовка, прогноз и расчеты диаграмм состояния двухкомпонентных
металлических и полупроводниковых систем. Раскрыты термодинамические
закономерности сплавообразования и поведения металлических и
полупроводниковых фаз в различных условиях. На основе различных модельных
представлений доведены до простых расчетных формул количественные описания фазовых
равновесий. Разработана проблема термической стабильности конгруэнтно
плавящихся промежуточных фаз в точке плавления, что дает возможность
управления степенью дефектности структуры при выращивании монокристаллов и
тонких слоев полупроводниковых соединений.
Предназначена для широкого круга научных работников в области
физической химии, металловедения и полупроводникового материаловедения, а также
может быть полезна аспирантам и студентам соответствующих специальностей.
Ил. 67. Табл. 4. Библиогр. список: 436 назв.
ИБ № 2055
ВАСИЛИЙ МИХАЙЛОВИЧ ГЛАЗОВ
ЛИДИЯ МИХАЙЛОВНА ПАВЛОВА
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА
И ФАЗОВЫЕ РАВНОВЕСИЯ
(Двухкомпонентные металлические
и полупроводниковые системы)
Редактор издательства Af. С, Архангельская
Художественный редактор А. И. Гофштейн
Технический редактор В. М. Курпяева
Корректоры: Л. М. Зинченко, Е. В. Якиманская
Переплет художника В, Н. Иванова
Сдано в набор'22.12.80. Подписано в печать 01.06.81. Т-21118. Формат
бумаги 60X90Vie. Бумага типографская № 1. Гарнитура литературная.
Печать высокая. Печ. л. 21. Усл. кр-отт. 21. Уч.-изд. л. 23,10. Тираж 2260 экз.
Заказ 422. Цена 3 р. 70 к. Изд. № 0404
Издательство «Металлургия», 119034, Москва, Г-34, 2-й Обыденский пер.* д. 14
Ленинградская типография МЪ 6 ордена Трудового Красного Знамени
Ленинградского объединения «Техническая книга» имени Евгении Соколовой
Союзполиграфпрома при Государственном комитете СССР по делам издательств,
полиграфии и книжной торговли. 193144, г. Ленинград, ул. Моисеенко, 10
п 31008—146
040(01)-81 100~~81 2603000000
© Издательство «Металлургия» 1981
ПРЕДИСЛОВИЕ
Книга посвящена проблеме, связанной с разработкой основ
теоретического анализа диаграмм фазовых равновесий. Хорошо
известно, какую важную роль играют диаграммы состояния в
металлургии и материаловедении, химической технологии,
галургии, нефтепереработке и т. д. Изучение диаграмм состояния
помогает выбрать оптимальные составы смесей и сплавов, разработать
режимы их термической обработки, определить эффективность
процессов разделения и глубокой очистки веществ при перегонке,
ректификации, зонной и направленной кристаллизации. Однако
обширная термодинамическая информация, которую можно
извлечь из диаграмм состояния, до последнего времени практически
не использовалась вследствие сравнительно слабого развития
обоснованного анализа фазовых равновесий.
Авторы предприняли первую попытку представить общую
картину термодинамического анализа диаграмм состояния и
показать широкие возможности этого подхода при выяснении
характера взаимодействия компонентов и строения фаз.
В книге обобщены исследования авторов, проводившиеся на
кафедре физической химии Московского института электронной
техники за последнее десятилетие, а также многочисленные
литературные данные.
При разработке проблемы авторы широко использовали
современную вычислительную технику, благодаря применению которой
стало возможным решение многих задач.
Авторы выражают надежду, что их труд сыграет определенную
роль в развитии затронутых вопросов и послужит началом
широкого применения термодинамического метода к анализу
равновесий в металлических, полупроводниковых, солевых, органических
и других системах.
Авторы искренне благодарят чл.-корр. АН СССР Я. И.
Герасимова и проф. В. Б. Лазарева, с которыми неоднократно
обсуждалась проблематика монографии, а также проф. В. Б. Уфимцева,
взявшего на себя труд по ее рецензированию. Кроме того, авторы
признательны своим коллегам по работе проф. Е. Б. Соколову и
проф. А. С. Пашинкину за полезную дискуссию, а также Л. И. Эта-
жовой и Л. А. Сергеевой за помощь в оформлении рукописи.
I*
Глава I
НЕКОТОРЫЕ ОБЩИЕ СООТНОШЕНИЯ
ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
1. СИСТЕМЫ, ИХ СВОЙСТВА И КЛАССИФИКАЦИЯ.
ТЕРМОДИНАМИЧЕСКИЕ ПАРАМЕТРЫ И УРАВНЕНИЕ
СОСТОЯНИЯ. ТЕРМОДИНАМИЧЕСКИЕ ПРОЦЕССЫ
Как известно, термодинамический метод является универсальным методом
изучения многих разнообразных процессов, позволяющим обобщать
наблюдаемые явления с целью установления взаимосвязей между ними.
Широкое применение этого метода к анализу процессов, связанных с
изменением химического состава в рассматриваемых материальных объектах,
привело к выделению особой термодинамической дисциплины, получившей
название химической термодинамики. Творцом химической термодинамики
следует по праву считать Гиббса [1].
Раскрытие глубоких изменений, сопровождающих перераспределение масс
реагирующих веществ, в свою очередь оказало исключительно сильное
воздействие на развитие термодинамического метода.
Таким образом, области применения термодинамического метода
необычайно широки и, следовательно, целесообразно сформулировать самые общие
представления, позволяющие абстрагироваться от специфики отдельных веществ
и рассматривать материальные тела и их взаимодействия как сферу приложения
термодинамического метода в целом.
Поэтому прежде чем перейти к изложению основ метода химической
термодинамики, необходимо сформулировать ряд основных понятий и определений,
строгость обозначения которых диктуется исключительной жесткостью
взаимосвязей, возникающих при конкретном рассмотрении различных материальных
объектов с помощью термодинамического метода.
Материальный объект (и даже его части) или определенную совокупность
материальных объектов, гипотетически выделенных нами из окружающей среды,
принято называть системой. Система, в которой происходит материальный
обмен между составляющими ее телами (массообмен или теплообмен), называется
термодинамической. Термодинамический метод применим лишь к системам,
состоящим из большого числа молекул, т. е. к макроскопическим системам.
Система имеет точные пространственные границы, отделяющие ее от
окружающей среды. Этими границами могут служить реальные физические
поверхности раздела или воображаемая математическая поверхность. Система может
быть однородной и неоднородной, причем в последнем случае она может быть
макро- и микронеоднородной (состоящей из отдельных однородных тел или
имеющей непрерывно изменяющуюся степень однородности, например градиент
концентрации).
Состав системы целесообразно с физико-химической точки зрения
выражать отношением числа молей каждого вещества к общему числу молей всех
составляющих систему веществе Если система содержит п± молей вещества A±t
п2 молей вещества А2 и т. д., то молярная доля, например, первого вещества
определится из выражения
i
Очевидно
£*' = !• (1.2)
Целесообразно вместо понятия «вещество» пользоваться понятием
«компонент». Под компонентами понимают вещества, составленные из атомов или
молекул одного сорта. Это определение предварительное и не учитывает воз-
4
можности протекания химических реакций. Понятие «материальный объект»
целесообразно заменить понятием «фаза», под которой понимается любое
однородное тело, отличающееся от ближайшего окружения резким изменением свойств
на границах. Это определение — также предварительное. Строгое
термодинамическое обоснование его будет дано после изложения основ термодинамического
метода.
В зависимости от степени изолированности систем от окружающей среды
различают изолированные, закрытые и открытые системы.
Системы, которые совершенно не взаимодействуют с окружающей средой,
называются изолированными. Системы, которые не обмениваются
с окружающей средой частицами (атомами, молекулами, ионами), образующих
их веществ, но,взаимодействуют с ней иным путем (например, посредством
теплообмена, механической работы, излучения), называют закрытыми.
Системы, которые обмениваются с окружающей средой частицами всех
составляющих веществ или части веществ, называются открытыми.
Состояние системы может быть определено совокупностью ее свойств. Все
величины, характеризующие какое-либо макроскопическое свойство
рассматриваемой системы, называются термодинамическими
параметрами. Различают внешние и внутренние параметры.
Те макроскопические величины, которые определяются взаимоотношением
внешних тел по отношению к данной системе, называются внешними параметрами,
а те, которые определяются взаимодействием и состоянием частей, составляющих
данную систему, внутренними. Первоначальный выбор независимых
термодинамических параметров может быть любым, но если он уже сделан, то его нельзя
произвольно менять в процессе решения задачи, и поэтому все переходы от
одной совокупности независимых параметров к другой нужно проводить в
соответствии с математическими правилами.
Независимые термодинамические параметры чаще всего поддаются
непосредственному измерению (например, температура, давление, мольный или
удельный объем, концентрация и т. д.).
Различают интенсивные параметры (или факторы интенсивности) и
экстенсивные (или факторы емкости). Интенсивными называют параметры, значения
которых не зависят от массы. Это все молярные и удельные свойства,
температура, давление и т. д. Интенсивные свойства могут иметь одно и то же значение
во всей системе или изменяться от точки* к точке. Величины их не аддитивны.
Экстенсивными называются такие свойства, величины которых пропорциональны
массе. Они обладают аддитивностью. Это объем системы, ее масса или число
молей и т. д. Интенсивные свойства — это специфические свойства системы в
данном состоянии. Поэтому в качестве независимых термодинамических параметров
используют обычно интенсивные свойства.
Если ограничиться изучением термодинамических свойств простой системы,
т. е. не принимать во внимание влияние внешних силовых полей (например,
гравитационного, электростатического или магнитного) и, кроме того,
предположить, что система находится в покое, то состояние ее совершенно однозначно
определится тремя величинами: объемом V, давлением р и абсолютной
температурой Т. Следовательно, объем, давление и температура должны быть
связаны между собой некоторой зависимостью, которая в самом общем виде
выражается уравнением
/(р, V, Т)=0. (1.3)
Уравнения, связывающие между собой параметры состояния, называются
уравнениями состояния. Соотношение (1.3) — термическое уравнение
состояния.
При переходе системы из одного состояния в другое изменение ее свойств
не зависит от способа перехода, а определяется лишь начальным и конечным
состоянием системы, т. е. термодинамическими параметрами в этих двух
состояниях. Таким образом, изменение состояния системы можно характеризовать,
рассматривая лишь ее исходное и конечное состояния.
5
Глава I
НЕКОТОРЫЕ ОБЩИЕ СООТНОШЕНИЯ
ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ
1. СИСТЕМЫ, ИХ СВОЙСТВА И КЛАССИФИКАЦИЯ.
ТЕРМОДИНАМИЧЕСКИЕ ПАРАМЕТРЫ И УРАВНЕНИЕ
СОСТОЯНИЯ. ТЕРМОДИНАМИЧЕСКИЕ ПРОЦЕССЫ
Как известно, термодинамический метод является универсальным методом
изучения многих разнообразных процессов, позволяющим обобщать
наблюдаемые явления с целью установления взаимосвязей между ними.
Широкое применение этого метода к анализу процессов, связанных с
изменением химического состава в рассматриваемых материальных объектах,
привело к выделению особой термодинамической дисциплины, получившей
название химической термодинамики. Творцом химической термодинамики
следует по праву считать Гиббса [1].
Раскрытие глубоких изменений, сопровождающих перераспределение масс
реагирующих веществ, в свою очередь оказало исключительно сильное
воздействие на развитие термодинамического метода.
Таким образом, области применения термодинамического метода
необычайно широки и, следовательно, целесообразно сформулировать самые общие
представления, позволяющие абстрагироваться от специфики отдельных веществ
и рассматривать материальные тела и их взаимодействия как сферу приложения
термодинамического метода в целом.
Поэтому прежде чем перейти к изложению основ метода химической
термодинамики, необходимо сформулировать ряд основных понятий и определений,
строгость обозначения которых диктуется исключительной жесткостью
взаимосвязей, возникающих при конкретном рассмотрении различных материальных
объектов с помощью термодинамического метода.
Материальный объект (и даже его части) или определенную совокупность
материальных объектов, гипотетически выделенных нами из окружающей среды,
принято называть системой. Система, в которой происходит материальный
обмен между составляющими ее телами (массообмен или теплообмен), называется
термодинамической. Термодинамический метод применим лишь к системам,
состоящим из большого числа молекул, т. е. к макроскопическим системам.
Система имеет точные пространственные границы, отделяющие ее от
окружающей среды. Этими границами могут служить реальные физические
поверхности раздела или воображаемая математическая поверхность. Система может
быть однородной и неоднородной, причем в последнем случае она может быть
макро- и микронеоднородной (состоящей из отдельных однородных тел или
имеющей непрерывно изменяющуюся степень однородности, например градиент
концентрации).
Состав системы целесообразно с физико-химической точки зрения
выражать отношением числа молей каждого вещества к общему числу молей всех
составляющих систему веществ. Если система содержит tii молей вещества А^
п2 молей вещества А2 и т. д., то молярная доля, например, первого вещества
определится из выражения
*i = "i/5>- (1.1)
i
Очевидно
2*1 = 1. (1.2)
i
Целесообразно вместо понятия «вещество» пользоваться понятием
«компонент». Под компонентами понимают вещества, составленные из атомов или
молекул одного сорта. Это определение предварительное и не учитывает воз-
4
можности протекания химических реакций. Понятие «материальный объект»
целесообразно заменить понятием «фаза», под которой понимается любое
однородное тело, отличающееся от ближайшего окружения резким изменением свойств
на границах. Это определение — также предварительное. Строгое
термодинамическое обоснование его будет дано после изложения основ термодинамического
метода.
В зависимости от степени изолированности систем от окружающей среды
различают изолированные, закрытые и открытые системы.
Системы, которые совершенно не взаимодействуют с окружающей средой,
называются изолированными. Системы, которые не обмениваются
с окружающей средой частицами (атомами, молекулами, ионами), образующих
их веществ, но'взаимодействуют с ней иным путем (например, посредством
теплообмена, механической работы, излучения), называют закрытыми.
Системы, которые обмениваются с окружающей средой частицами всех
составляющих веществ или части веществ, называются открытыми.
Состояние системы может быть определено совокупностью ее свойств. Все
величины, характеризующие какое-либо макроскопическое свойство
рассматриваемой системы, называются термодинамическими
параметрами. Различают внешние и внутренние параметры.
Те макроскопические величины, которые определяются взаимоотношением
внешних тел по отношению к данной системе, называются внешними параметрами,
а те, которые определяются взаимодействием и состоянием частей, составляющих
данную систему, внутренними. Первоначальный выбор независимых
термодинамических параметров может быть любым, но если он уже сделан, то его нельзя
произвольно менять в процессе решения задачи, и поэтому все переходы от
одной совокупности независимых параметров к другой нужно проводить в
соответствии с математическими правилами.
Независимые термодинамические параметры чаще всего поддаются
непосредственному измерению (например, температура, давление, мольный или
удельный объем, концентрация и т. д.).
Различают интенсивные параметры (или факторы интенсивности) и
экстенсивные (или факторы емкости). Интенсивными называют параметры, значения
которых не зависят от массы. Это все молярные и удельные свойства,
температура, давление и т. д. Интенсивные свойства могут иметь одно и то же значение
во всей системе или изменяться от точки* к точке. Величины их не аддитивны.
Экстенсивными называются такие свойства, величины которых пропорциональны
массе. Они обладают аддитивностью. Это объем системы, ее масса или число
молей и т. д. Интенсивные свойства — это специфические свойства системы в
данном состоянии. Поэтому в качестве независимых термодинамических параметров
используют обычно интенсивные свойства.
Если ограничиться изучением термодинамических свойств простой системы,
т. е. не принимать во внимание влияние внешних силовых полей (например,
гравитационного, электростатического или магнитного) и, кроме того,
предположить, что система находится в покое, то состояние ее совершенно однозначно
определится тремя величинами: объемом V, давлением р и абсолютной
температурой Т. Следовательно, объем, давление и температура должны быть
связаны между собой некоторой зависимостью, которая в самом общем виде
выражается уравнением
/ (/>, V, T) = 0. (1.3)
Уравнения, связывающие между собой параметры состояния, называются
уравнениями состояния. Соотношение (1.3) — термическое уравнение
состояния.
При переходе системы из одного состояния в другое изменение ее свойств
не зависит от способа перехода, а определяется лишь начальным и конечным
состоянием системы, т. е. термодинамическими параметрами в этих двух
состояниях. Таким образом, изменение состояния системы можно характеризовать,
рассматривая лишь ее исходное и конечное состояния.
5
С математической точки зрения, утверждение независимости изменения свойств -
от пути эквивалентно тому, что бесконечно малое изменение свойства является
полным дифференциалом.
Согласно теореме математического анализа, интеграл от полного
дифференциала не зависит от пути интегрирования. Справедливо обратное
утверждение; если изменение какой-либо величины не зависит от пути превращения, то
эта величина является свойством системы. В качестве примера простой системы
может служить идеальный газ. Для описания поведения газа необходимы лишь
два из трех параметров состояния, так как третий параметр определится из
термического уравнения состояния.
Комбинируя законы Бойля—Мариотта и Гей-Люссака—Шарля, получим
уравнение Клапейрона:
pV=nRT, (1.4)
где R — универсальная газовая постоянная; п — число молей.
Смесь идеальных газов также подчиняется уравнению состояния вида (1.4).
В этом случае п равно общему числу молей смеси, а общее давление р согласно
закону Дальтона,— сумме парциальных давлений газов, составляющих смесь, т. е.
Р = РА + Рв + РС+..., (1.5)
где рА, рв, рс — парциальные давления составляющих смеси.
Введем предварительно одно из самых важных понятий термодинамики —
«термодинамическое равновесие». Если состояние системы не изменяется с
течением времени, т. е. ее параметры сохраняют постоянное значение, то такая
система находится в состоянии равновесия. Следует подчеркнуть, что при
термодинамическом равновесии постоянное значение имеют только параметры,
характеризующие данную систему в целом. Понятие «абсолютного» равновесия или
«равновесия с учетом всех мыслимых изменений» не имеет физического смысла.
Равновесные процессы характеризуются тем, что между свойствами системы,
претерпевающей равновесное изменение состояния, в течение всего процесса
существует определенная зависимость, описываемая уравнением состояния
типа (1.3), тогда как при неравновесных процессах само понятие давления,
температуры и других параметров, определяющих состояние системы, часто теряет
всякий смысл, так как в различных точках в один и тот же момент эти величины
могут обладать совершенно различными численными значениями.
Термодинамические пооцессы могут быть также обратимыми и необратимыми.
Понятие обратимых и необратимых изменений было введено Планком в 1879 г. Г21.
Обратимым является равновесный процесс, если при проведении его в
обратном направлении система приходит в начальное состояние и во внешней среде
при этом не происходит никаких изменений. Те процессы, которые не
удовлетворяют этому условию, называются необратимыми. К их числу, следовательно,
относятся все неравновесные процессы, а также равновесные процессы,
допускающие обращение, но с изменением окружающей среды.
Понятие «обратимый процесс» является более общим, чем понятие
«равновесный процесс». Действительно, при совершении равновесного процесса
конкретной системой необязательно, чтобы в сопряженных системах процессы также
были равновесными.
Если термодинамическая система осуществляет процесс в прямом и
обратном направлениях и в результате последнего возвращается в исходное состояние,
то считают, что такая система совершила циклический, или круговой, процесс.
В зависимости от условий протекания в термодинамике различают
изотермический, изобарический, изохорический и адиабатический процессы.
2. ПАРЦИАЛЬНЫЕ МОЛЬНЫЕ ВЕЛИЧИНЫ И ИХ СВОЙСТВА
Рассмотрим парциальные мольные величину, играющие ^исключительно
важную роль в термодинамике однородных систем, состоящих из"двух или
большего числа компонентов.
Положим, что Y является каким-либо экстенсивным свойством данной
однородной системы, содержащей л/ молей вещества Л/, зависящим от температуры,
б
давления и количеств различных составных частей системы, т. е. можно
записать
Y = f(T,p,nltn2t...tnk). (1.6)
Тогда величина #/, определяемая соотношением
У1 = (дУ/д1ц)т.р.п1+г (1-7)
называется парциальной мольной величиной вещества Л/.
В отличие от молярных парциальные мольные величины могут быть
положительными и отрицательными, так как молярные величины характеризуют
свойства системы, а парциальные мольные величины — изменения свойств.
Выведем некоторые уравнения, связывающие парциальные мольные
величины. Поскольку любое экстенсивное свойство является однородной функцией
первого порядка от независимых переменных nv п2* ...» п& то, согласно теореме
Эйлера, можно записать
2 "C'fry-"'») « , {(Г, р, пи „.... пк). (LB)
i
С учетом выражения (1.7) тождество (1.8) можно представить в следующем
виде:
г=2>г,. (1-9>
i
В уравнении (1.9) отражена связь между экстенсивным свойством и
парциальными мольными величинами составляющих систему веществ. Из него
следует, что значение экстенсивного свойства может быть определено, если
известны количества и парциальные мольные свойства отдельных веществ.
Продифференцируем тождество (1.9) по п,- (/ = 1, 2, ..., А), при постоянных
р и Т.
С учетом соотношения (1.7) для каждого / получим
2>/Шд"/)р.Г = 0 (1.10)
i
или ^ni(d9jldni)p,T=-Q< (I.11)
i
Отсюда следует, что значения парциальных мольных величин не являются
независимыми. Для системы, состоящей из двух веществ, уравнение (1.10)
принимает вид:
Ч (дуг/дп^р, т + п2 {dy2}dnt)p% т = 0. (1.12)
Уравнения (1.10) и (1ч 12) описывают зависимость между приращением
какой-либо величины yi и других величин этого рода в случае бесконечно малого
изменения состава системы при постоянных давлении и температуре.
Соотношения (1.9) и (1.10) называются в химической термодинамике
парциальными молярными условиями. В соответствии с теоремой Эйлера
соотношение (1.11) характеризует парциальные мольные величины как однородные
функции нулевого порядка, т. е. для всех i
Si (7\ P. /«i, /"2 lnk) = Si (Т, р. пь п2 nk). (I.13)
Это значит, что если размеры системы увеличиваются в / раз, то значения
парциальных мольных величин остаются неизменными. Таким образом,
парциальные мольные величины являются интенсивными параметрами, поскольку
они зависят не от общего количества отдельных частей, а только от состава, т. е.
от относительного количества составных частей. Поэтому если к данной
однородной системе прибавить одновременно такое количество отдельных веществ,
что относительный состав системы не изменится, то и парциальные мольные
величины останутся прежними. Парциальные мольные величины, как и всякие
7
интенсивные свойства, определяются давлением, температурой и относительным
количеством составных частей или составом системы.
Если речь идет об одном моле системы, то уравнения (1.9) и (1.10) можно
записать в следующей форме:
У^^хт. (1.14)
i
%*i№/dxi)=0. (1.15)
i
Применяя уравнения (1.15) к бинарной системе, получим, выбрав в
качестве независимой переменной х2,
*i (dyildx2)Pt т + х2 (ду2/дх2)р% т = 0 (1.16)
или фуфх2)р, т1(Шдх2)р% т = — х21х±. (1.17)
Из соотношения (1.17) следует, что производные (дух/дх2) и (ду2/дх2) всегда
имеют обратные знаки; при х± = х2 = 0,5 наклоны кривых уг == / (x2)t у2 = / (**)
одинаковы и отличаются только по знаку; если одна кривая имеет максимум,
то другая кривая при том же составе — минимум.
Поведение парциальных мольных величин при бесконечном разбавлении
можно также рассмотреть с помощью уравнения (1.17). Положим, например,
что х2-> 0. Тогда могут возникнуть две ситуации: (dy1/dx2)Pf r может или
стремиться к нулю, или оставаться конечной величиной. В последнем случае
производная (ду2/дх2)р т должна стремиться к бесконечности, так как ее произведение
на стремящееся к нулю отношение х2/х± остается конечным. Другими словами,
если х2= 0, то кривая Ух становится горизонтальной, а кривая у2 —
вертикальной.
Полученные в данном разделе соотношения будут нами широко
использованы и при решении конкретных задач, связанных с анализом фазовых
равновесий в двухкомпонентных системах.
3. НУЛЕВОЙ ПРИНЦИП.
ПЕРВОЕ И ВТОРОЕ НАЧАЛА ТЕРМОДИНАМИКИ
Принято считать, что системы находятся в термическом равновесии, если
между ними при отсутствии теплоизоляции не происходит теплообмена. Вполне
очевидно, что при соприкосновении в различной степени нагретых тел с течением
времени устанавливается одинаковая температура. Это заключение, сделанное
с открытием термометра, является одним из четырех постулатов термодинамики
и называется законом термического равновесия (нулевой принцип).
В современной термодинамике этот закон сформулирован Фаулером
следующим образом: если система 1 находится в состоянии термического
равновесия по отдельности с системами 2 и 3, то эти две последние системы находятся
в состоянии термического равновесия между собой.
Рассмотрим три простые системы, способные обмениваться теплотой.
Запишем условие термического равновесия системы 1 с системой 2 в виде
уравнения:
/Ч(рМД; р2, V2)=0. (I.18)
Аналогично для систем 1 и 3 справедливо
f^ipW1; рз, 1/*)=0. (1.19)
Далее, согласно нулевому принципу в равновесии должны находиться
также системы 2 и 3, т. е.
/23 (р\ у*. рзэ уз) = 0> (1.20)
Поскольку все переменные оказываются взаимосвязанными,
соотношение (1.20) должно выводиться из выражений (1.18) и (1.19). Математически это
8
возможно, если рассматриваемые переменные будут присутствовать в
комбинации как некоторые функции, например р1 и V1 должны входить в виде функции
х (р1* V1)', Р2 и V2 — в виде функции у (р2, V2); а /?3 и Vs — в виде функции
z (р3, V3). Тогда уравнения (1.18)—(1.20) можно представить в виде
/12(*,*/)=0; (1.21)
/13(#>2)=0; (1.22)
Z23 (*, г) = 0. (1.23)
Теперь принципиально возможно исключить одну из трех переменных в
каких-либо двух из этих уравнений. Например, уравнения (1.21) и (1.22) можно
решить относительно у и приравнять результаты.
Если аналогично использовать другую пару уравнений, то в конечном итоге
можно записать равенство трех функций:
х (р\ V1) = у (р\ V2) = г (р3, V9). (1.24)
Это дает основание считать, что существует некоторое свойство системы
/ (р, V) такого рода, что из условий t1 = t2 и t1 = Z3 следует t2! = Р. Это
свойство называется эмпирической температурой. Таким образом, термический
принцип равновесия ведет к аналитическому критерию термического
равновесия, определяемого равенством температур. Приведенный закон позволяет
установить, является температура двух тел одинаковой или разной и по этому
признаку судить о направленности теплового потока или об его отсутствии. •
Опыты Румфорда, Дарвина, Гей-Люссака, Майера, Джоуля-Томсона [3]
в той или иной мере с неопровержимой убедительностью свидетельствуют об
эквивалентности тепла и работы. Принципиальное значение имеют работы
Майера, который по сути первый сформулировал первое начало термодинамики,
дав совершенно правильное толкование знаменитому опыту Гей-Люссака. Майер
вычислил также механический эквивалент теплоты для кругового процесса.
Систематические, прецизионно поставленные опыты Джоуля,
последовавшие за открытием Майера, полностью доказали постоянство механического
эквивалента тепла для системы, совершающей круговой процесс. На основе этого
можно сделать следующее заключение: в замкнутой системе сумма всех видов
энергии постоянна; при их взаимопревращениях энергия не теряется и не
создается вновь. Так формулируется закон сохранения энергии, являющийся одним
из важнейших всеобщих законов природы. Этот закон был назван Клаузиусом
первым началом термодинамики.
Из принципа эквивалентности вытекает невозможность создания вечного
двигателя, т. е. такого двигателя, который непрерывно бы производил работу
без всяких затрат.
В общей форме принцип эквивалентности можно записать:
j)bQ = j)&A, (I.25)
где 6Q и ЬА — бесконечно малые теплота и работа. Знак ф обозначает
интегрирование по замкнутому контуру.
Рассмотрим важнейшие следствия из принципа эквивалентности тепла и
работы. Представим себе изолированную систему. Для изолированной системы
внутренняя энергия постоянна, т. е. U = const, а следовательно, dU = 0.
Теперь представим, что мы сообщаем системе какое-то количество тепла,
т. е. повышаем температуру. Если объем при этом остается постоянным, то
никакой работы, связанной с расширением, не производится, и все сообщаемое
системе тепло пойдет на изменение внутренней энергии. Если при нагревании
будет изменяться и объем, то подводимое тепло будет расходоваться не только
на изменение внутренней энергии, но и на совершение внешней работы, т. е.
работы расширения. Следовательно,
$Q=dt/+64 (I.26)
9
или в интегральной форме
Q=MJ+A. (1.27)
Условимся тепло, подводимое к системе, считать положительным, а тепло,
отданное системой, отрицательным. Тогда уравнение (1.27) можно представить
в виде
Q = (tfn-tfl) + i*, (I-28)
т. е. сообщаемая теплота идет на повышение запаса внутренней энергии от U\
до Uл и на совершение внешней работы. Уравнение (1.28) — математическое
выражение первого начала термодинамики.
Для случая, когда единственным видом работы, который совершает система,
является работа расширения, уравнение (1.28) принимает вид
vn
Q = Uu-Ui+ J pdV. (1.29)
vl
Отсюда при p — const
Qp = Uu~-Ui + p(Vu-Vi) (1.30)
или
• Qp = tfn-tfi = Atf, (1.31)
dQp^dH. ' (1.32)
Таким образом, теплота, поглощаемая в процессе, протекающем при
постоянном давлении, равна изменению энтальпии системы, если единственным
видом работы является работа расширения.
Согласно первому закону термодинамики, при переходе одной формы
энергии в другую полная энергия изолированной системы сохраняется. Но несмотря
на то, что закон сохранения энергии отражает основной, неизменный принцип
всех природных явлений, он не определяет, какие из них на самом деле могут
совершаться и какие нет. Ответ на этот вопрос дает второй закон или второе
начало термодинамики. В фундаментальном труде французского инженера Карно
[4] «Размышления о движущей силе огня и о машинах, способных развивать
эту силу» (1824 г.) предпринята первая, еще весьма несовершенная попытка
сформулировать то, что в настоящее время называется вторым началом
термодинамики. В своей работе Карно поставил три основных вопроса: 1) необходимое
условие для преобразования тепла в работу; 2) условие, при котором
трансформация тепла в работу может достичь максимального эффекта; 3) зависит ли
коэффициент полезного действия тепловой машины, дающей максимальный эффект,
от природы рабочего вещества. Карно отмечает, что к. п. д. всех обратимых
тепловых машин одинаковы и зависят не от рода работающего тела, а только от
предельных температур, между которыми эти машины работают. Идеи Карно
математически развил Клапейрон (1834 г.), который также впервые предложил
графическое изображение цикла Карно, состоящего из двух изотерм и двух
адиабат идеального газа.
Дальнейшее формирование второй закон получил в трудах Клаузиуса
(1850 г.) и Томсона (1851 г.) [41.
Клаузиус формулировал второе начало термодинамики следующим
образом: теплота не может переходить самопроизвольно от более холодного тела
к более теплому. Позднее выражение «самопроизвольно» Клаузиус заменил
другим — «без компенсации», т. е. без каких-либо изменений
термодинамического состояния рабочего тела или других участвующих в процессе тел. Такая
формулировка второго закона термодинамики именуется постулатом Клаузиуса.
Справедливость постулата Клаузиуса в его первой формулировке представляется
самоочевидной и подтверждается совокупностью 'опытных данных, связанных
в первую очередь с нашими наблюдениями. Этот постулат Клаузиуса надо по-
Ю
нимать в широком аспекте. Как неоднократно указывал Клаузиус, это
положение ни в коем случае не должно просто означать, что тепло непосредственно не
переходит от более холодного тела к более теплому (последнее само собой
понятно и следует уже из определения температуры). Настоящий смысл
положения Клаузиуса заключается в том, что тепло вообще никаким способом с
помощью какого бы то ни было процесса не может Сыть перенесено с более
холодного тела на более теплое без того, чтобы не произошли другие изменения
(«компенсация»). Только пользуясь этим более широким толкованием положения
Клаузиуса, как отмечает Планк 12 J, можно, исходя из него, делать заключения
относительно каких угодно природных процессов. Планк формулирует второе
начало термодинамики так: невозможно построить периодически действующую
машину, вся деятельность которой сводится к поднятию тяжести и охлаждению
теплового резервуара.
Согласно Планку, необратимость природных процессов в целом может быть
выражена следующим образом: процессы, которые никаким способом нельзя
полностью обратить, называются необратимыми; все остальные процессы —
обратимы. Например, невозможно каким бы то ни было способом полностью
обратить процесс, при котором тепло возникает благодаря трению, т. е. если
падающий груз, будучи соединен с соответствующим механизмом, превратит
раооту в тепло, произведя трение в воде или ртути при помощи колеса с
лопастями, то совершенно невозможно провести процесс в обратном направлении так,
чтооы груз и вся система заняли бы точно прежнее положение, так как часть
энергии в виде тепла неооратимо рассеется в окружающей среде.
Обратимые процессы протекают бесконечно медленно. Процессы, идущие
с измеримой быстротой, не могут быть ооратимыми, потому чго благодаря их
скорости внутреннее распределение энергии не успевает в точности следовать
за изменением внешних условий. Это влечет за собой некоторое удаление от
теплового равновесия, которое неизбежно связано с факторами необратимости.
Поскольку обратимый процесс складывается из совокупности ряда состояний
равновесия, которое не требует изменений в окружающем пространстве, он может
пройти как в прямом, так и в обратном направлениях.
Таким образом, обратимые процессы, как и вообще всякие процессы,
осуществимые в идеальных условиях, хотя и невозможны в действительности, тем
не менее имеют некоторую реальность в том смысле, что они не противоречат
законам природы, и если их нельзя воспроизвести в полном виде, то можно
воссоздать приблизительно и притом с любой степенью приближения. Очевидно,
что все реальные процессы отличаются один от другого по степени необратимости.
В термодинамике количественной мерой степени необратимости процессов
является введенная Клаузиусом новая функция состояния, названная им
энтропией.
Согласно Клаузиусу, степень необратимости процесса целесообразно
определять не с помощью количества переданного тепла Q, а с помощью величины
приведенного тепла Q/7\ где Т — температура по абсолютной
термодинамической шкале [5]. Значение Q/T в необратимом процессе и является возрастанием
энтропии системы. Если мы обозначим энтропию в начале процесса через Slf
а в конце его через 52, то
S2-S1=Q/T. (I.33)
Физический смысл энтропии достаточно сложен. Выражая изменение энтропии
в необратимом процессе как разность энтропии в конце и в начале процесса,
мы тем самым уже подразумеваем, что энтропия является свойством системы,
т. е. функцией параметров состояния, поэтому изменение энтропии не зависит
от характера протекания процесса, а определяется только параметрами его
начала и конца.
Рассматривая энтропию как функцию состояния и включая в число
параметров состояния состав, будем иметь
dS = (dS/дГ W dT + (dS/dp)T, ndp + £ (dSjdn^ Рч ч dnt. (1.34)
11
Энтропия является экстенсивным свойством, обладающим аддитивностью.
Математическое доказательство существования энтропии хорошо
прослеживается в методе Клаузиуса.
Из последовательного рассмотрения цикла Карно [4] следует, что к. п. д. У)
обратимой машины можно представить в виде
Ц = (Qi - Q2VQ1 = (71! - T2)/Tv (1.35)
где Qi — количество тепла, передаваемое рабочему телу нагревателем, Q2 —
количество тепла, отдаваемое рабочим телом холодильнику, 7\ и Т2 —
максимальная и минимальная температуры теплообмена рабочего тела с источником
теплоты соответственно.
На основании уравнения (1.35) для любого обратимого цикла имеем
(f> (&Q06P/T) = 0.
Так как интеграл по замкнутому контуру от 6Q/T равен нулю и,
следовательно, изменение этой величины не зависит от формы пути, то 6Q/71 есть полный
дифференциал некоторой функции параметров состояния 5, которую, как
отмечалось, Клаузиус назвал энтропией.
Следовательно, для обратимого процесса имеем
dS= 6Q/T или 6Q= TdS (I.36)
Соотношение (1.36) является основным для толкования второго начала
термодинамики и для развития термодинамики вообще.
Как известно [5], во всех случаях к. п. д. необратимой машины Карно т|'
меньше к. п. д. обратимой машины т), т. е.
Ч'<Л- - (1-37)
Тогда на основании зависимости (1.35) с учетом выражения (1.37) приходим
к заключению, что приведенная теплота нагревателя в необратимой машине
Карно меньше приведенной теплоты холодильника. Поэтому
$Wh«*p/T)<0. (I.38)
В дифференциальной форме в общем виде получим
TdS>6Q, (I.39)
где знак неравенства соответствует необратимым изменениям, а знак равенства
обратимым.
Выражение (1.39) представляет собой полную математическую
формулировку второго начала термодинамики в дифференциальном виде.
Из рассмотрения аналитического выражения второго начала термодинамики
в виде (1.39) следует, что энтропия может служить критерием направленности
термодинамического процесса.
Исходя из уравнения первого закона термодинамики (1.26) и
соотношения (1.39), в общем случае получаем
TdS>dU+6A. (I.40)
Выражение (1.40) является объединенным уравнением первого и второго
начал термодинамики. Отсюда следует, что при необратимом процессе работа,
совершаемая системой, всегда меньше, чем при обратимом процессе в данных
условиях. Поэтому работа обратимого процесса — максимальная. Всякое
вмешательство необратимости в ходе процесса сказывается на уменьшении работы
системы.
12
4. СВЯЗЬ ЭНТРОПИИ С РАЗЛИЧНЫМИ ПАРАМЕТРАМИ
И НЕКОТОРЫЕ СООТНОШЕНИЯ
МЕЖДУ ПРОИЗВОДНЫМИ ФУНКЦИЯМИ
Для решения задач, связанных с использованием принципа энтропии,
целесообразно рассмотреть ее зависимость от других термодинамических величин..
Из уравнения (1.40) для обратимого процесса будем иметь
dS^jdU+^pdV. (I.41)
или, выражая внутреннюю энергию через энтальпию, можно записать
dS=ldH-±VdP> (I.42)
где все дифференциалы суть полные дифференциалы, так как S, (У, Я, V и р
являются функциями состояния. Следовательно, \1Т представляет собой
интегрирующий множитель.
Выражения (1.41) и (1.42) можно рассматривать как объединенные формулы
для обоих начал термодинамики, описывающие все обратимые изменения
состояния, т. е. эти уравнения относятся к такому бесконечно малому изменению
состояния, которое переводит систему из состояния равновесия в бесконечно
близкое состояние, являющееся также состоянием равновесия. Для необратимых
изменений эти уравнения неверны.
Из математического анализа следует, что полный дифференциал должен
удовлетворять соотношениям взаимности. В случае простой системы мы имеем
лишь две независимые переменные и одно соотношение взаимности. Из
сравнения выражения для полного дифференциала внутренней энергии при
переменных Т и V и уравнения первого начала термодинамики
6Q = [ (dUldV)T + p]dV + (dUIdT)v dT. (1:43)
Подставив значение 6Q из выражения (1.43) в уравнение второго начала
термодинамики, получим
dS = Т-1 [(dU/dV)T + p] dV + Г"1 (dU/dT)v dT. (1.44)
щг [T-i (dUldT)y] = ^r [T-i (dU/dV)T + рЩ (1-45)
После дифференцирования и несложных преобразований получим
(dU/dV)T = Т (др/дТ)у — р. (1.46)
Соотношение (1.46) характеризует зависимость внутренней энергии от объема
в изотермическом процессе.
Подставив уравнение (1.46) в выражение (1.44), окончательно получим
(dS/dV)T=(dp/dT)v (I.47)
или (с учетом соотношения между частными производными параметров
уравнения состояния)
(dS/dV)T = (dp/dT)v = - (dV/dT)P!(dV/dp)T. (I.48)
Сравнивая выражение для полного дифференциала энтальпии простой
системы при переменных Тирс уравнением первого начала термодинамики, найдем
6Q= (dHjdT)pdT+ [(дН/др)т- V] dp. (I.49)
Используя выражения (1.49) и (1.42), получим
dS = T-i [(дН/др)т - V] dp + Т'* (дН/дТ)р dT. (I.50)
13
Соотношение взаимности в этом случае можно записать следующим образом
^[Г-* (дН/дТ)Р]т = ± [Т-1 (дН/др)т - V/T]p. (I.51)
После дифференцирования
(дН/др)т = -Т (dV/dT)p + V. (1.52)
Это соотношение характеризует зависимость энтальпии от давления в
изотермическом процессе.
Подставив выражение (1.52) в уравнение (1.50), получим
(dS/dp)T = -(dV/dT)p. (I.53)
Выражения (1.46) и (1.52) являются термодинамическими уравнениями
состояния, так как производные (dU!dV)r и (дН/др)т налагают
термодинамическое условие, которому должно удовлетворять любое эмпирическое
уравнение состояния.
Уравнения (1.46) и (1.53) позволяют, используя данные о термических
свойствах вещества, находить калорические величины — внутреннюю энергию и
энтальпию, и, наоборот, по известным калорическим величинам вычислять
термические свойства вещества.
Наконец, найдем зависимость энтропии от температуры.
Из определения истинной теплоемкости
C = 6Q/dT. (I.54)
Подставив в выражение (1.54) значение 6Q из (1.36), имеем
С = TdS/dT. (I.55)
Это выражение справедливо для любого бесконечно малого изменения
температуры в системе независимо от того, каким путем оно происходит. Обычно
нагревание проводят либо при постоянном давлении, либо при постоянном
объеме.
В соответствии с уравнением (1.55) получим:
при р = const
dS = Cpd\nT (I.56)
или
(dS/dT)p = Cp/T; (I.57)
при V = const
dS = Cvd\nT (1.58)
или
(dS/dT)v=:Cv/T. (Ь59)
Продифференцировав уравнения (1.57) и (1.59) соответственно по р и по V
с учетом того, что
Cv = (dU/dT)v и Ср = (дН/дТ)р, (1.60)
получим:
d*S/6Tdp = Г-i (дСр/др)т = Г"* (д*Н/дТдр); (1.61)
d*S/dTdV = Г-* (dCy/dV)т = Г"* (д*ЩдТдУ). (1.62)
На основании уравнений (1.53) и (1.61),
(дСр/др)т = — Т (dW/dT*)p. (1.63)
Согласно выражению (1.62) с учетом соотношения (1.47), найдем
(дСу1дУ)т = Т (д*р/дТ*)у. (1.64)
14
В простейших случаях при постоянных объеме и давлении интегрирование
соотношений (1.56) и (1.58) приводит к следующим выражениям для энтропии:
S (р,Т) в j (Ср/Т) dT + const; (Ь65)
5 (V, Т) = [ (Су/Г) dT + const. (1.66)
Если мы хотим найти увеличение энтропии вещества, температура которого
повышается при постоянном давлении или объеме от Т\ до Гц, то, очевидно,
необходимо знать характер температурной зависимости теплоемкости.
Разность энтропии двух состояний вычисляется особенно просто, если
обратимый переход из одного состояния в другое происходит при постоянной
температуре. Это имеет место, например, при агрегатных превращениях. Тогда на
основании уравнения второго начала термодинамики
SII-SI=^' (I67>
где q — скрытая теплота фазового превращения, отнесенная к одному молю
вещества; о — молярная энтропия; верхний индекс Л означает
индивидуальное вещество.
Формулу (1.67) можно использовать для вычисления изменения энтропии
при обратимом плавлении тел, испарении и т. д. Здесь тоже надо различать
случаи, когда V = const и /?= const. При постоянном объеме не совершается
никакой работы, и скрытая теплота равна увеличению внутренней энергии Ди°,
а при постоянном давлении она равна увеличению тепловой функции Гиббса A/i°:
gv = Au°; qp = Ah°. (1.68)
Рассматривая изотермический переход вещества из одного агрегатного
состояния в другое, например жидкости в пар (кипение), отметим, что
равновесие имеет место при давлении пара, равном внешнему приложенному давлению.
В этом случае (dS/dV)т тождественно равно As°/Aa°, где As° и Да°
увеличение энтропии и объема в расчете на 1 моль испарившегося вещества. Тогда,
согласно выражению (1.47), можно записать
dp/dT = As°/Au°, (I.69)
или, учитывая соотношения (1.67) и (1.68) для фазового перехода жидкость—
пар,
dp/dT = Ah°v [T°B(v'M -i>°<^)]-\ (1.70)
где Ah°v — скрытая теплота испарения; t>°(G) и v°^L) — мольные объемы пара и
жидкости при температуре кипения (индексы G и L соответствуют газообразной
и жидкой фазам); Т° — температура кипения чистого вещества.
Выражение (1.70) — известное уравнение Клапейрона—Клаузиуса. Оно
является, по существу, прямым следствием второго начала термодинамики.
При переходе жидкости в пар объем пара во много раз превышает объем
жидкости, в связи с чем значением v°^ в формуле (1.70) можно пренебречь.
Тогда
dp/dT ~Ah°v(vG)Tl)-i. (I.71)
Выражение (1.69) носит общий характер и может быть использовано для любых
фазовых переходов.
15
Для процесса плавления уравнение Клапейрона—Клаузиуса имеет особое
значение, поскольку оно характеризует наклон кривой равновесия кристаллы—
жидкость. В этой связи его целесообразно представить в следующем виде:
dTldp = Т'т (0в<« - v^)/Ah'm. (1.72)
В этом уравнении производная dTldp имеет смысл барического коэффициента
кривой плавления вещества и характеризует изменение температуры плавления
в зависимости от давления, &h°m — теплота плавления.
Учитывая взаимосвязь между объемом и плотностью вещества,
уравнение (1.72) можно записать в виде
dTldp = (d<*> -d<L>) (AS^^dW)-1. (1.73)
где d — плотность индивидуального вещества (индекс S соответствует твердой
фазе); Asm — энтропия плавления.
Важно подчеркнуть, что для испарения и для плавления скрытая теплота,
а следовательно, и энтропия фазового превращения всегда положительна, тогда
как разность удельных объемов в случае плавления может быть и
положительной, и отрицательной. Последнее наблюдается у воды, однако не является
исключением", как это отмечено в ряде работ [6] и др., и характеризует довольно
распространенное явление в природе, особенно присущее полупроводникам,
отличающимся рыхлой упаковкой в твердом состоянии (см. [7]).
Из уравнений (1.72), (1.73) непосредственно вытекает закономерность,
согласно которой при y°(L) — i>0(S) < 0 или d(S) — d^ < 0 производная dp/dT
отрицательна, и, следовательно, при повышении давления температура
плавления убывает. В случае, когда v°^ — а*5)> 0 или d^ — d^ > 0,
температура плавления возрастает, так как производная dp/dT положительна.
5. ХАРАКТЕРИСТИЧЕСКИЕ ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ
И УРАВНЕНИЕ ГИББСА—ГЕЛЬМГОЛЬЦА
Непосредственное применение обоих начал термодинамики дает возможность
решать разнообразные конкретные задачи. Метод воображаемых обратимых
циклов, который во всех случаях приводит к правильному решению задачи,
нельзя считать совершенным, поскольку он требует чисто искусственных
построений и обходных путей при решении конкретных задач. Поэтому широкое
распространение получил другой, более простой метод — метод
термодинамических (характеристических) функций, который по праву можно назвать
методом Гиббса.
Понятие характеристических функций в термодинамику было впервые
введено Массье (1869 г.). Гиббс дал этим функциям несколько иное
определение и последовательно применил их к решению ряда вопросов химической
термодинамики.
Характеристическими называются функции состояния системы, через
производные которых наиболее просто и притом в явном виде могут быть выражены
все термодинамические свойства системы. При этом под термодинамическими
свойствами понимаются такие физические свойства, которые зависят только от
температуры, давления (или объема) и состава. Термин «характеристические
функции» не совсем удачен, поскольку он создает впечатление, что
характеристичность есть свойство функции. В действительности же, характеристичность
функции есть следствие выбора независимых переменных (параметров
состояния).
Необходимость введения характеристических функций была обоснована
Гиббсом следующим образом. Объединенное уравнение двух начал
термодинамики для обратимых процессов (1.41) связывает пять переменных: 7\ 5, (/, р
и Vy определяющих состояние системы, находящейся в равновесии. Каждая
из указанных величин может рассматриваться и как параметр состояния, и как
16
функция состояния. Для определения состояния системы ограничимся пока
случаем двух независимых переменных. Тогда остаются три неизвестных
параметра, для определения которых, помимо уравнения (1.41), необходимо иметь
еще два уравнения, связывающих те же самые величины между собой.
Вторым уравнением может быть термическое уравнение состояния, а третье
уравнение можно построить, учитывая соотношения между параметрами или
функциями состояния, вытекающие из конкретных свойств рассматриваемой
системы. Это уравнение, приводящее к решению задачи, содержит новую
функцию, которая и называется термодинамической функцией.
Среди термодинамических функций, к числу которых может относиться
любая функция состояния, и число которых, вообще говоря, неограниченно велико,
следует выделить характеристические функции, которые, как уже указывалось,
обладают тем свойством, что при определенном выборе параметров состояния
частные производные характеристической функции по ним равны одному из
параметров состояния. Очевидно, что число характеристических функций
невелико.
Гиббс вводит четыре функции, которые и называют, собственно,
характеристическими: внутреннюю энергию U, энтальпию Я, изохорно-изотермический F
и изобарно-изотермический G потенциалы.
Систематика Гиббса следует непосредственно из объединенного уравнения
первого и второго начал термодинамики (1.41), которое целесообразно представить
в виде
dU = TdS — pdV. (I.74)
Рассмотрим последовательно вывод характеристических функций и зададим
значения частных производных. Согласно уравнению (1.74), внутренняя энергия
является функцией независимых параметров S и V. Поскольку внутренняя
энергия есть функция состояния, ее дифференциал является полным и, следовательно,
dU = (dU/dS)v dS + (dU/dV)sdV. (1.75)
Уравнение (1.74) должно тождественно удовлетворяться при любых
значениях dS и dV. Это возможно только в одном случае, если
(dU/dS)v = Т\ (1.76)
(dU/dV)s = -p. (1.77)
Следовательно, согласно определению, внутренняя энергия является
характеристической функцией при переменных S и V.
При другом же выборе переменных это сделать невозможно. Если данную
термодинамическую функцию рассматривать как функцию таких переменных,
для которых она не является характеристической, то другие сопряженные
термодинамические величины невозможно выразить в явном виде с помощью этой
функции.
Вывод остальных характеристических функций (Я, F и G) основан на
преобразовании Лежандра, которое состоит в том, что одновременно с изменением
независимых переменных, стоящих в правой части равенства (1.74), меняется
и функция, стоящая под знаком дифференциала в левой части этого равенства.
Вычитая d (TS) из обеих частей уравнения (1.74), после несложных
преобразований получим
d(U — TS) = -SdT — p dV. (1.78)
Уравнение (1.78) есть полный дифференциал функции
F= U—TS. (1.79)
Эта^ функция введена впервые Массье, а Гельмгольц назвал ее свободной
энергией. Поэтому она называется свободной энергией Гельмгольца, или свободной
энергией при постоянном объеме. По современной терминологии, ее называют
изохорно-изотермическим потенциалом. Из уравнения (1.78) следует, что F
17
является функцией Т и V. Записав выражение для полного дифференциала как
суммы частных производных, получим
dF = (dF/dT)v dT + (dF/dV)T dV. (K80)
Сравнивая выражения (1.78) и (1.80), будем иметь
(dF/dT)v = — S; (I.81)
(dF/dV)T = — p. (I.82)
Следовательно, свободная энергия Гельмгольца является
характеристической функцией. Используя преобразование Лежандра, перейдем к независимым
переменным S и р. Прибавив d (pV) к обеим частям уравнения (1.74), после
некоторых преобразований найдем
d(U + pV) = TdS + Vdp. (I.83)
Полученное выражение есть полный дифференциал энтальпии Н = U + pV
или тепловой функции Гиббса, которая, следовательно, является
характеристической функцией двух независимых переменных S и р. Учитывая, что Н — это
функция состояния, можно записать
dH = (dH/dS)p dS + (dH/dp)s dp. (1.84)
Сравнивая выражения (1.83) и (1.84), получим *
(dH/dS)p = T; (1.85)
F*(dH/dp)s = V. (1.86)
Вычитая из обеих частей уравнения (1.74) дифференциал d (TS — pV) и
переходя к независимым переменным Т и р, будем иметь
d(U — TS+ pV)=—SdT+ Vdp. (1.87)
Таким образом, полученное выражение — полный дифференциал новой
функции G:
G = U— TS+ pV= F+ pV. rt.88)
Из уравнения (1.87) следует, что G — это функция температуры и
давления. Ее называют свободной энергией Гиббса или, по современной терминологии,
изобарно-изотермическим потенциалом. Функция G является
характеристической при независимых переменных Т и р. Ее также ввел Массье.
Однако именно Гиббс раскрыл ее глубочайший смысл и широко
использовал в развитии химической термодинамики. Собственно поэтому функция G и
получила название свободной энергии Гиббса.
Учитывая, что свободная энергия Гиббса есть функция состояния, можно
записать
dG = (dG/dT)p dT + (dG/dp)T dp. (1.89)
Сравнивая выражение (1.87) и (1.89), получим
(dG/dT)p = -S; (1.90)
(dG/dp)T = V. (1.91)
Больше преобразований уравнения (1.74), аналогичных приведенным,
сделать нельзя. В итоге имеем четыре характеристические функции U, Я, F, G.
Беря частные производные этих функций по соответствующим параметрам,
получим восемь выражений: (1.76) и (L77); (1.81) и (1.82); (1.85) и (1.86); (1.90)
и (1.91), которые имеют большое значение и смысл при решении различных
физико-химических задач. Ими мы в дальнейшем и будем пользоваться. Как уже
отмечалось, термодинамических функций множество и в принципе роль
термодинамической функции ""может играть любой параметр состояния, например,
энтропия или объем. Однако параметры"состояния, взятые в виде таких
произвольных функций, характеризуются сложными соотношениями между частными
производными* Это нетрудно показать, используя ту же последовательность
18
рассуждений, что и при выводе Четырех характеристических функций. Ё
некоторых же работах [9j энтропию причисляют к характеристическим функциям.
Представляет интерес рассмотрение вторых производных
термодинамических потенциалов по сопряженным независимым переменным. Таким способом
могут быть получены различные соотношения между термодинамическими
параметрами системы.
Из указанных уравнений получим:
d2U/dS dV = (dT/dV)s = - (dp/dS)v; (1.92)
d2F/dT dV = (dS/dV)T = (dp/dT)v\ (1.93)
d2H/dS dp = (dT/dp)s = (dV/dS)p; (1.94)
ЗЮ/дТдр = — (dS/dp)T = (dVldT)p. (1.95)
Соотношения (1.92)—(1.95) являются следствиями объединенного уравне"
ния первого и второго начал термодинамики и считаются основными
дифференциальными уравнениями. Впервые они были получены Максвеллом.
Продифференцируем уравнение (1.90) по Т при р= const:
(дЮ/дТ% = — (dS/dT)p. (I.96)
С учетом выражения (1.57) будем иметь
(дЮ/дТ*)р = — Ср/Т. (1.97)
Продифференцировав (1.97) по р при Г= const и поменяв в левой части
уравнения порядок дифференцирования, получим
l(d*jdT*) (dG/dp)T)p = - Г-1 (дСр/др)т. (1.98)
Учитывая равенство (1.91), на основании выражения (1.98) приходим к
соотношению, аналогичному (1.63). Зная уравнение состояния и значение Ср при
одном давлении, можно, пользуясь полученным уравнением, вычислить Ср при
других давлениях.
Аналогично из выражений (1.81) и (1.59) получим
(d*F/dT*)v = — Су IT. (1.99)
Продифференцировав уравнение (1.99) по V при Т= const и, изменив в левой
части порядок дифференцирования, с учетом соотношения (1.82) получим
выражение, аналогичное (1.64).
Рассмотрим, какой физический смысл имеют приращения
характеристических функций при некоторых процессах. Необходимо отметить, что работа
системы складывается из работы вследствие увеличения объема и работы
вследствие внутренних сил, не сопровождающейся изменением объема. Однако
уравнение (1.74) учитывает только один вид работы — работу по преодолению
внешнего давления, поэтому его следует обобщить. Обозначим все другие виды работы
как произведение обобщенной силы на изменение обобщенной координаты
%Х(с1У{ = дА*тах^ (1.Ю0)
i
где ЬАJjax — сумма элементарных полезных работ (кроме работы расширения).
19
С учетом тождества (1.100) запишем выражения для дифференциалов
характеристических функций (1.74), (1.78), (1.83), (1.87) в следующем виде:
dU = TdS-pdV -^ XidYa (1-Ю1)
i*
dF = —SdT — pdV — J] XidYi; (1.102)
i
dH ^TdS + Vdp-^XtdYr, (1.103)
i
dG= -SdT+Vdp—% XidYi. (1.104)
i
При постоянных V и всех обобщенных координатах dY'; в соответствии с
выражением (1.101) имеем
dU = TdS = bQv,Yit (I.105)
т. е. приращение внутренней энергии равно количеству теплоты, полученной
системой.
При постоянной энтропии (dS = 0) из равенства (1.101) следует, что
- (dU)s = PdV+ 6A'max = 6Лтах. (1Ю6)
т. е. при этом условии уменьшение внутренней энергии равно количеству
максимальной работы, произведенной системой.
При постоянном давлении (dp = 0) и постоянных обобщенных
координатах (dYi = 0) в соответствии с выражением (1.103) получим
dH = TdS = dQp,Yi, (I.107)
т. е. при этих условиях приращение энтальпии равно количеству теплоты,
поглощенному системой.
При постоянных энтропии (dS = 0) и давлении (dp = 0)
-<*Я = ЦХг^=6Л;ах. (1.108)
i
Следовательно, при этих условиях уменьшение энтальпии равно
максимальной работе системы, не связанной с изменением объема.
Физический смысл свободной энергии Гельмгольца легко выяснить,
вычислив работу в изотермическом процессе.
На основании выражения (1.102) при постоянной температуре (dT = 0)
имеем
-{dF)T=pdV + bA*max=bAm!iX, (I.109)
т. е. в изотермическом обратимом процессе максимальная работа, совершаемая
системой, равна убыли свободной энергии. При постоянных температуре (dT = 0)
и объеме (dV = 0)
-№.у = »С <1Л1°)
Таким образом, при изохорно-изотермическом процессе убыль свободной
энергии Гельмгольца равна полезной максимальной работе, которая не
включает в себя работу, связанную с изменением объема.
Если изотермический процесс необратим, то мы получаем работы меньше,
чем возможно, так как часть ее не используется, теряясь при необратимом
процессе.
20
Г1ри постоянных температуре и давлении (dT= 0 и dp = 0), согласно
выражению (1.104),
-(<к?)Р,г=бл;ах. (Liu)
Следовательно, уменьшение свободной энергии Гиббса определяется
максимальной работой, не связанной с изменением объема при изобарно-изотерми-
ческом процессе.
Для справедливости уравнений (1.109)—(1.111) температура системы
необязательно должна оставаться постоянной на протяжении всего обратимого
процесса, необходимо только, чтобы температура источника, от которого система
получает или которому отдает тепло, равнялась температуре системы в ее
начальном (конечном) состоянии, как впервые указал Гиббс [1].
Анализируя связь между изохорно-изотермическим потенциалом и
внутренней энергией, а также соответственно между изобарно-изотермическим
потенциалом и энтальпией, можно получить важные соотношения, имеющие
значение в практике термодинамических расчетов.
Связь между свободной энергией Гельмгольца и внутренней энергией
задается уравнением (1.79), которое, однако, не позволяет судить собственно о связи
между указанными величинами, поскольку, кроме F и U, в него входит энтропия S,
которая сама является термодинамической функцией состояния.
Учитывая, однако, соотношение (1.81) и подставляя его в выражение (1.79),
получаем
F = U + T(dF/dT)v. (I.112)
Выражение (1.112) называется уравнением Гиббса-Гельмгольца и имеет
важное значение в изотермических процессах, отличающихся тем, что их
течение сопровождается производством работы практически без изменения объема
(таких, как работа гальванического элемента, пластическая деформация,
различные процессы в растворах, работа в электрическом и магнитном полях, и т. д.).
Аналогично можно прийти к соотношению между изобарно-изотермическим
потенциалом и энтальпией:
G = H — TS. (I.113)
Однако из этого выражения не видна явная связь между G и Я, поскольку
в него входит энтропия «S. Заменяя энтропию в равенстве (1.113) ее значением
из уравнения (1.90), получим
G = Я + Т (dGldT)p. (I.114)
Это также уравнение Гиббса—Гельмгольца, которое имеет важнейшее
значение при анализе изобарно-изотермических процессов (в частности в
термохимии).
Рассмотренные свойства функций F и G дают возможность установить связь
между максимальной работой процесса, протекающего равновесно, и теплотой
того же процесса, но протекающего неравновесно, и получить еще один вариант
уравнения Гиббса—Гельмгольца [10].
При расчетах интегральных значений характеристических функций
получают не абсолютные значения, а их разности в любых двух состояниях системы.
При этом одно из состояний системы фиксируют, принимая за стандартное,
которому приписывают произвольное, но удобное значение характеристической
функции.
Выведем общую формулу для расчета характеристической функции G (р, Т)
при различных давлениях и температурах. Сначала найдем зависимость
величины G (р> Т) от давления при Т = const. Эта задача легко решается
математически путем изотермического интегрирования выражения (1.91). Запишем
получающееся при этом уравнение:
G(P2> T2) -0(pv Тш) = J Vdp. (1.115)
21
Интегрируя выражение (1.97), с учетом соотношения (1.92) находим зависимость
функции G (р, Т) от температуры при постоянном давлении р±:
т т
а ^2
O^.rj-G^.rj^-Ste^ro^i-yj-J^J Ср(Рт Т) dT> (М16>
Учитывая выражение (1.115), окончательно будем иметь
G (д. Тл) =G(Pi, Тг) - S (pit Т±) (Г, - Tt) +
т т
' 2 »
+ J Vdp- J dT J Cp (Pj,' T)dT. (1.117)
3i h
Аналогично можно получить уравнение для расчета характеристической
функции F в зависимости от объема и температуры:
F &2> Т2) = F (V19 Tt) - 5 (Vv Tx) (Tj- Tt) -
2 2 2
-jpdV- I dT J 3L*ZilI)d7\ (1.118)
Глава II
УЧЕНИЕ О ХИМИЧЕСКИХ ПОТЕНЦИАЛАХ
И ОБЩАЯ ТЕОРИЯ ТЕРМОДИНАМИЧЕСКОГО
РАВНОВЕСИЯ
1. ФУНДАМЕНТАЛЬНЫЕ ТЕОРЕМЫ И УРАВНЕНИЯ.
УРАВНЕНИЕ ГИББСА—ДЮГЕМА
В гл. I отмечено, что термодинамические системы могут быть открытыми и
закрытыми. Оба начала термодинамики получены для закрытых систем. При
переходе от простых систем к сложным, образованным многими компонентами,
возможность обмениваться массами веществ с внешней средой, а также между
отдельными частями системы приводит к заключению об изменении состава,
который, следовательно, должен быть включен в число параметров состояния
системы.
Между открытыми и закрытыми системами в термодинамическом отношении
нет никакого различия. Гиббс очень остроумно вышел из затруднения при
рассмотрении сложных систем, полагая данную закрытую систему как совокупность
фаз, каждая из которых может рассматриваться как открытая [1]. Открытые
и закрытые системы различаются главным образом условиями своего
существования и, следовательно, возможностями изменения своих составов, поскольку
составы открытых и закрытых систем подчинены различным условиям
материальной изоляции [11].
22
Учитывая, что главным объектом нашего рассмотрения являются
гетерогенные двухкомпонентные системы, обобщим уравнения для
характеристических термодинамических функций на случай открытых систем, включив при этом
состав в число основных параметров состояния. Рассмотрим основные
термодинамические теоремы для открытых систем.
Первая теорема. При любых приращениях параметров состояния
термодинамической системы, если независимыми переменными являются энтропия,
объем и число молей компонентов, справедливо следующее соотношение для
полного дифференциала внутренней энергии:
k
dU = TdS-pdV + J] (Wldnt)s9v,ntdnt. (II.1)
Для доказательства справедливости данного соотношения учтем выражение
для полного дифференциала внутренней энергии сложной системы:
А
dU = (dUfdT)VtndT + (dU/dV)TtndV+Jl (dUfdnt)Ttvtnjdni. (II.la)
Когда открытая система подвергается такому изменению, при котором ее
состав остается постоянным, то ее состояние меняется точно так же, как это
происходило бы в случае однородной закрытой системы. Для такого случая
в соответствии с уравнениями (1.76) и (1.77) можно записать:
(dU/dS)Vin = T; (II.2)
(dU/9V)s9n = -p- (".3)
Подставляя значения частных производных из выражений Ш.2) и (Н.З)
в уравнение (11.1а), получим соотношение (ИЛ), что и требовалось доказать.
Величина производной (dU/dtii)sf vf n.- была названа Гиббсом химическим
потенциалом i'-того компонента:
to=(dUfdnt)s9vtnr (П.4)
Введение понятия «химический потенциал» явилось особенно ценным для
химической термодинамики, так как он играет основную роль в определении
химических и фазовых равновесий.
Чтобы нагляднее представить себе смысл химического потенциала, будем
рассматривать каждый вид энергии в уравнении (ИЛ) как произведение двух
величин: обобщенной силы (фактора интенсивности) и обобщенной координаты
(фактора емкости). Все производные внутренней энергии по независимым
параметрам при условии постоянства остальных параметров являются обобщенными
силами. В связи с этим производную (dU/dni)stv,nf также можно считать
обобщенной силой или фактором интенсивности, определяющим процесс
перераспределения числа молей компонентов в системе. Таким образом, химический
потенциал при перераспределении масс компонентов играет ту же роль, что и
давление при изменении объема или температура в процессе теплообмена. Можно
говорить о химическом потенциале компонента в каждой точке системы точно
так же, как говорят о его концентрации или о мольной доле.
Можно получить так же, как и в случае с d(J, выражения для полных
дифференциалов функций F, Я и G. Полный дифференциал функции F при
переменных Г, V и л/ будет
k
dF = (dF/dT)VtndT + (dF/dV)TtnmdV+ £ (0F/d/t,)r, к, „,<*"/. (II.5)
23
Здесь и далее индекс п означает постоянство содержания всех компонентов
в системе, a nj — постоянство всех, кроме t-того. Приняв во внимание
соотношения (1.81) и (1.82) при условии фиксирования всех пп получим
k
dF = — SdT-pdV + £ (dF/dn^T^v.rtfdrii, (11.6)
t=l
Рассматривая Н как функцию переменных S, р и я/, имеем
k
dH = (dH/dS)Py n dS + (дН/др)3, ndp + 2 (дН/dtiih, Pt njdni. (II.7)
или с учетом соотношений (1.85) и (1.86)
к
dH=TdS + Vdp + £ (dH/dni)sшР9 njdfii. (11.8)
Полный дифференциал (? как функция 7\ р и я/ запишется следующим
образом:
k
dG = (dG/dT)PjndT + (dG/dp)Ttndp+ £ №дгц)р%т%п1*П1. (II.9)
i=i
или, приняв во внимание выражения (1.90) и (1.91) при условии постоянства
всех П[у
k
dG= -SdT + Vdp+Y, (дв/дп^т, р, nj dnt. (II. 10)
1=1
Отметим, что постоянство энтропии при изменении массы t-того компонента
возможно только при специально подобранном режиме температур, и
уравнение (II.4) оказывается практически непригодным. Поэтому на практике
химический потенциал всегда определяют в виде производных от других функций.
Рассмотрим возможность выражения химического потенциала через другие
характеристические функции.
Вторая теорема. Частные производные всех характеристических функций
по массе i-того компонента при постоянстве остальных соответствующих
параметров состояния равны между собой.
Докажем, что
R = (dU/dni)St у, nj = (дН/дпс)3> Pf nj = (dF/dni)Tf v, n}- =
= №dni)T9P9ni. (П.11)
Продифференцируем выражения для характеристических функций Я, F и G
и в получившиеся результаты подставим значение dU из уравнения (II.1).
Сопоставляя полученные данные с выражениями (II.6), (II.8) и (11.10), убеждаемся
в справедливости соотношения (11.11). Окончательно имеем
k
dU = TdS-pdV+y£l \ndnr, (11.12)
t=i
к
dH = TdS + V dp+Yi Pi dns, (П.13)
i=\
k
dF = — SdT-pdV+ J] ludna (11.14)
t=i
k
dG^ — SdT + Vdp+^ptdrii. (11.15)
i=l
24
Наибольшее практическое распространение получило выражение
химического потенциала через свободную энергию Гиббса:
to = (dQ/dnt)ptT9nr (П-16)
поскольку процессы, связанные с изменением состава системы, изучаются
большей частью при постоянных температуре и давлении. Из уравнения (11.16) видно,
что химический потенциал (-того компонента является парциальной мольной
величиной свободной энергии Гиббса.
Дифференциальные уравнения (11.12)—(11.15) Гиббс назвал
фундаментальными, чтобы подчеркнуть, что они выражают связь между характеристическими
функциями и их переменными и, следовательно, дают исчерпывающую
термодинамическую характеристику смеси, состоящей из к компонентов.
Фундаментальные уравнения эквивалентны одно другому. Поэтому для полного описания
термодинамических свойств системы сложного состава необходимо иметь только
одно какое-то фундаментальное уравнение.
Легко заметить, что термодинамические потенциалы ((/, Я, F и G) являются
линейными однородными функциями соответствующих экстенсивных величин
(S, Vt пх, /i2, •••> я*)- Применяя к ним теорему Эйлера об однородных функциях,
получим интегральные выражения:
U = TS-pV + 11Л+ |*л + ... + №*; (П.17)
Н = TS + \x,xnt + jv*2 + ... + iiknk; (11.18)
F = — pV + №! + \i2n2+ ... + nknk\ (11.19)
k
G = IV4+ [ляяя + ... + Wk = Ц Rrt/- (11.20)
Эти формулы можно также получить интегрированием уравнений (11.12)—
(11.15) при условии постоянства всех интенсивных свойств 7\ /?, \iv fi2, ..., [ik-
Так как интенсивные величины характеризуют состояние системы,
интегрирование при указанном условии имеет смысл увеличения массы смеси без
изменения ее состояния.
Согласно изложенному термодинамические свойства системы полностью
определены, если, например, известна функция G (7\ р, nlt n2, ..., п£. Из
выражения (11.20) следует, что термодинамические свойства системы однозначно
определены, если химические потенциалы jxf- всех к компонентов системы
известны как функции независимых переменных 7\ р, п1у я2> •••» nk- Эти k
химических потенциалов связаны между собой и с другими термодинамическими
параметрами соотношениями, которые легко получить, пользуясь
фундаментальными уравнениями (11.12)—(11.15). Так как G — функция состояния, a dG—
полный дифференциал, то для любой пары компонентов справедливо так
называемое соотношение взаимности:
(ф;,/^)г.Р, п1Ф( = №/*»/)г. p. niH- (»-21)
Используя теорему Коши, на основании выражений (11.16), (1.90) и (1.91)
(причем последние два с учетом всех щ = const), легко убедиться, что
(дц/дТ)р9 п - - (dS/dni)Tf Pt n. = _s,; (II.22)
(дщ1др)т> п = (дУ/дпс)т tPt П] = vi% (II.23)
где S; и vt — парциальные мольные энтропия и объем компонента i соответственно.
Если продифференцировать уравнение (11.17) и учесть выражение (11.12),
то получим еще одно фундаментальное уравнение, выведенное Гиббсом:
k
SdT — Vdp+J^riidiii^O. (II.24)
t=i
25
Это уравнение необычно в том отношении, что независимыми переменными
его являются интенсивные параметры (температура, давление, химические
потенциалы \ilt №» ••->№)' а в качестве коэффициентов использованы экстенсивные
величины (5, V$ tit).
Для изменений, происходящих при постоянных температуре и давлении,
на основании уравнения (IL24) можно записать
к
2] л* 4^=0. (11.25)
Соотношение (11.25) носит название уравнения Гиббса—Дюгема, Оно
связывает между собой изменения химических потенциалов компонентов в изобарно-
изотермическом процессе и имеет большое значение в термодинамике растворов.
Уравнение (II.25) позволяет рассчитать для изобарно-изотермических
процессов одну из величин ф/ (например, ф^, если известны остальные.
При постоянных Тир полный дифференциал йуц будет
k
d\H = £ (dto/dnflT, p, n... dnj. (II.26)
/=-1 '
Подставив выражение (11.26) в уравнение (11.25), получим
Ц 2 nt (dto/dnflr. p. i, dni = 0. (11.27)
* i
Так как последнее уравнение справедливо для любых значений dnj, должно
выполняться условие
£ Щ Шдпдт, Р, я = 0, (11.28)
что совпадает с выражением (1.10), если рассматривать pi/ как парциальную
мольную величину.
Кроме того, соотношение взаимности (11.21) позволяет вместо
соотношения (11.28) записать
Е«|№Й)г.м , = 0. (И.29)
i '
Следует отметить, что общее описание парциальных мольных .величин,
приведенное в гл. I, полностью приложимо к химическим потенциалам.
Используя уравнение (11.29), для двойной смеси можно записать
Ч (diijdttj) + п2 (dfii/дяа) =0 (II .30)
(здесь и далее индексы у частных производных химических потенциалов
опущены).
Выберем мольную долю второго компонента в качестве независимой
переменной. Тогда
dni/dn2 = x1/n-diii/dx2,
отсюда
d^i/dx2 = п/х^д^/дп^
Подставив выражение (11.31) в уравнение (11.30), найдем
дуч/д** = — п/х2 • дцх1дп^
Аналогично
дц2/дх2 =n/xvdiL2/dn2\
д\к21дх2 = — ntx2-d\i2ldni.
26
(11.81)
(11.32)
(11.33)
(11.34)
(11.35)
На основании выражения (11.28), используя соотношения (11.31) и (11.34),
легко убедиться, что
*i (d\ii/dx2) + х2 (д[12/дх2) = 0. (11.36)
Очевидно, что молярную свободную энергию Гиббса g с учетом
уравнения (11.20) можно выразить как
k
i
С помощью соотношений (11.37) и (11.36) легко убедиться, что для двойной
системы справедливо также
0g/d*a = И* — Pi- (11.38)
В общем случае для fe-компонентной смеси
Я?/д*,=|1,-!1*. (П-39)
где под k подразумевается компонент, мольная доля которого изменяется
зависимо, т. е. находится из уравнения связи (1.2).
Дифференцируя выражения (11.38) по х2у получим
d2gldx\ = d]i2/dx2 — дрх/дх2. (I I.40)
Подставив в это выражение значение д\лх1дх2 из соотношения (11.36), найдем
d'g/dxl = Эр2/дх2 (1 + xjxx). (11.41)
или d[i2/dx2 = xld2g/dx22. (II.42)
Аналогично d\ijdx2 = — х2 d2g/dxl- (11.43)
Уравнение (11.23) для индивидуального вещества при Т = const можно
представить в виде
d\i° = v°dp. (11.44)
Подставляя значение v° для одного моля из уравнения (1.4) в выражение
(11.44), получим
d[i° = RT(dp/p). (II.45)
Интегрируя полученное уравнение при Т= const, найдем выражение для
химического потенциала идеального газа:
ц° (Г, р) = р,°* (Г) + RT Inр, (II.46)
где \i°* (T) — постоянная интегрирования, величина которой зависит от
природы газа и температуры.
Из уравнения (11.46) видно, что \i°* (T) представляет собой химический
потенциал газа при данной температуре, когда давление его равно единице.
По определению смесь газов в объеме V при температуре Т называется
идеальной, когда ее свободная энергия F равна сумме свободных энергий, которые
имели бы отдельные составляющие, если бы каждая из них занимала тот же
самый объем при той же самой температуре:
F=» £F(7\ V, n,)= 5>,/J, (11.47)
/ /
где f°. — молярная свободная энергия Гельмгольца (индекс i относится к
составляющей газовой смеси).
27
Приняв Vi = 1 на основании выражения (1.118) легко убедиться, что для
одного моля t-той составляющей смеси идеальных газов
/; = /;* (Г) -RT In V], (II.48)
где f°* (T) — постоянная интегрирования, не зависящая от объема и
являющаяся функцией температуры.
После подстановки выражения (11.48) в равенство (11.47), получим
F= J\n{ [ /Г(Г)-ЯГ1п(У/я,)]. (П.40)
Дифференцирование выражения (11.49) по Л/ приводит к следующему
соотношению для химического потенциала составляющей смеси идеальных газов:
щ = (dF/dm)Tt V, nj = /Г (Т) + RT + RT In (щ/V). (II.50)
Химический потенциал составляющей газовой смеси можно выразить также
через парциальное давление. Смесь идеальных газов подчиняется тому же
уравнению состояния, что и чистые газы:
pt = niRTIV, (11.51)
т. е. парциальное давление компонента i, входящего в состав газовой смеси, равно
давлению, которое создавалось бы щ молями чистого компонента в том же
объеме и при той же температуре.
Приняв во внимание уравнение (11.51), на основании выражения (11.50)
можно записать
W— |*;(Г) + /гПпр,. (11.52)
В этом уравнении [if (Т) = /?* (Г) + RT (1 — In RT) зависит только от
температуры и совпадает с определяемой из выражения (11.46) функцией \i°* (T)
для чистого компонента i.
Если вещество i является единственным компонентом системы, то рь равно
общему давлению и выражения (11.52) и (11.46) совпадают.
Заменив pi в соотношении (11.52) на /?*,-, получим
lH = \i0i(T,p) + RT\nxif (II.53)
где jij (Т. р) = р] (Т) + RT lnp, (II.54)
т. е. fit- (Т, р) — химический потенциал чистого газа при температуре Т и
давлении р.
Запишем для любого реального газа уравнение, аналогичное
выражению (11.46):
^ = И°*(П + #Г1пф(Г, Р), (Н.55)
где ц°* (Т) — та же функция, что и для идеального газа; ф — фугитивность
или летучесть, возникающая из-за существования в реальном газе
межмолекулярных взаимодействий.
Функция ф зависит от температуры и от давления. Ввел эту функцию
Льюис (1901 г.).
Форма уравнения (11.55) имеет то преимущество, что химический потенциал
реального газа с учетом фугитивности можно записать точно так же, как и для
идеального газа, только давление заменяется на фугитивность. Отметим, что ф
подобно р интенсивная переменная. При достаточно низких давлениях все газы
близки к идеальным и фугитивность становится равной давлению (закон Гиббса—
Дальтона), т. е.
Нт(ф/р)= 1. (11.56)
28
По аналогии с выражением (11.52) для смеси реальных газов запишем
^ = Ц*(Г) + ЯГ1пфЬ (11.57)
где ц* — та же функция, что и для идеальных газов, а все эффекты
межмолекулярного взаимодействия включены в фугитивность ф/, являющуюся, таким
образом, функцией температуры и парциальных давлений, или, что то же самое,
функцией от Т, р, rti, ла, ..., л*.
Уравнение (11.57) определяет функцию ф/. Это уравнение, как и ранее,
сохраняет общую форму уравнения для смеси идеальных газов* но парциальные
давления в нем заменены на фугитивности.
Как и для чистого газа, фугитивности компонентов в смеси можно рассчитать
по уравнению
р
RT In (Ф,/р,) = lim [(*,_ *")*>. (11.58)
р*
где верхний индекс «id» относится к значению величины t^ в идеальной смеси*
Для расчета фугитивности одного из компонентов смеси при данной
температуре необходимо знать только его парциальный молярный объем как функцию
давления. При этом можно использовать непосредственно определяемые
значения vi или уравнение состояния.
Распространяя уравнение (11.53) на реальные смеси, запишем
lit = fi) (Г, р) + RT In ah (II.59)
где fij (7\ р) — та же функция, что и в уравнении (11.53), а все эффекты,
связанные с межмолекулярным взаимодействием, включаются, таким образом,
в функцию й( (7\ р, nv ..., пь), названную Льюисом активностью компонента i.
При уменьшении общего давления активность каждого компонента
стремится к его мольной доле. Следует отметить, что а/ подобно xi — интенсивная
величина.
Введем понятие коэффициента активности
yi = at/xif (II.60)
обладающего следующим свойством:
limvt = l. (И.61)
В смеси идеальных газов все коэффициенты активности равны единице.
Значения (1 — Yt) или 1п V/ можно, следовательно, использовать как меру
отклонений от законов идеальных газов.
Связь между фугитивностью и коэффициентом активности устанавливается
соотношением
yi = 4tlPt* (И.62)
вытекающим из выражений (11.54), (11.59) и (11.57). Коэффициенты активности
можно рассчитывать тем же способом, что и летучести. На основании (11.62)
уравнение (11.58) можно записать
р
RT \пу(=Пт [(vt—vftdp. (11.63)
Приведенные выражения являются общими не только для газовых смесей,
но и для смесей вообще, включая конденсированные фазы, однако в каждом
конкретном случае должно быть правильно выбрано стандартное состояние.
29
Учение о химических потенциалах лежит в основе общей термодинамической
теории гетерогенных равновесий и составляет суть подхода Гиббса к
рассмотрению этой проблемы в самом общем виде. Преимущества подхода на основе
химических потенциалов по сравнению с другими методами неоднократно
обсуждались в литературе и мы на них не останавливаемся. Отметим только, что данный
метод всегда дает правильное решение, как будет показано при рассмотрении
некоторых вопросов.
2. ПРИНЦИП РАВНОВЕСИЯ ГИББСА
Одной из главных проблем термодинамики является необходимость
конкретной формулировки условий термодинамического равновесия для
различных систем. Эта задача может быть решена чисто математическим путем на
основе метода Гиббса, который дал общую формулировку принципа равновесия
материальной системы, изолированной от всех внешних влияний, в двух
совершенно эквивалентных формах.
Хотя Гиббс не остановился на обосновании условий равновесия, в
действительности они являются следствием обоих начал термодинамики в форме
(1.41) и (11.12).
Метод толкования термодинамического равновесия, данный самим Гиббсом,
весьма лаконичен и поэтому достаточно труден для понимания.
Большая заслуга дальнейшего развития метода Гиббса принадлежит Ван-
дер-Ваальсу [12]. Последовательное и строгое изложение и творческое
развитие идей Гиббса и Ван-дер-В аальса дано в классической работе А. В. Сторон-
кина [13], чьим методом мы и будем пользоваться при изложении основ принципа
равновесия Гиббса и критериев устойчивости термодинамических систем.
Пусть рассматриваемая ^-компонентная система полностью изолирована
от внешней среды, т. е. подчинена следующим условиям:
U ss= const
V = const;
«!= const; (11.64)
л2 = const;
tik = const.
Тогда из выражения (1.40) следует
TdS^O. (11.65)
Если изолированная система находится в неравновесном состоянии, то,
согласно второму началу термодинамики, при подходе к равновесию с учетом
Т> 0 ее энтропия будет самопроизвольно возрастать. Возрастание энтропии
изолированной неравновесной системы возможно лишь благодаря необратимому
перераспределению энергии и веществ между ее различными частями. Когда
обмен теплом и веществами между частями изолированной системы станет
обратимым, энтропия системы примет постоянное значение в строгом
соответствии с заданными значениями энергии, объема системы и масс веществ. В итоге
при учете, что Т ф 0, будем иметь
dS = 0. (11.66)
На основании изложенного можно сформулировать следующее
фундаментальное положение, называемое принципом равновесия Гиббса: изолированная
система находится в состоянии равновесия, если ее энтропия при всех
возможных изменениях, совместимых с условиями изоляции, остается постоянной или
уменьшается, т. е.
(*s)u.v,»ui,1,....nJk<0. (H.67)
30
В выражении (11.67) знак равенства справедлив при протекании в системе
обратимых процессов, а знак неравенства — необратимых. В случае
изолированных систем последние могут носить только флуктуационный характер.
Сформулированное свойство энтропии носит вероятностный характер,
однако возможность самопроизвольного уменьшения энтропии изолированной
макроскопической системы, как показал Больцман, весьма маловероятна [14].
Сама идея этого заключения принадлежит Гиббсу, который сформулировал, что
невозможность нескомпенсированного уменьшения энтропии сводится,
по-видимому, к невероятности. Это и было отмечено Больцманом.
В соотношении (11.67) отражен тот факт, что энтропия изолированной
системы в состоянии равновесия имеет условный максимум. Максимальное
значение энтропии изолированной системы определяется заданными значениями
энергии, объема системы и масс, а следовательно, и числа молей компонентов.
Из объединенного уравнения первого и второго начал термодинамики при
условии постоянства объема и масс компонентов получим
{dU<TdS)v,4, *»..., ч- (П.68)
Поскольку Т — величина положительная, то согласно выражению (11.68)
внутренняя энергия и энтропия в случае равновесных процессов всегда
изменяются симбатно. Если V ф const, то справедливо выражение (1.41), т. е. при
таком условии энергия и энтропия не обязательно изменяются симбатно.
Рассмотрим систему при заданных значениях объема и общего числа молей
компонентов, в которой протекает неравновесный процесс. Энтропия такой системы
будет самопроизвольно возрастать. Чтобы воспрепятствовать этому, необходимо
соответствующим образом отводить от системы теплоту. При поддержании
энтропии постоянной внутренняя энергия системы уменьшается до тех пор, пока
не будет достигнуто состояние равновесия. Следовательно, при приближении
системы к состоянию равновесия внутренняя энергия системы уменьшается,
если энтропия и объем системы, а также число молей компонентов
поддерживаются постоянными. Таким образом, для состояния равновесия внутренняя
энергия системы имеет условный минимум.
На основе представления об изменении внутренней энергии принцип
равновесия можно сформулировать так: система находится в состоянии
равновесия, если ее внутренняя энергия при всех процессах, не нарушающих условия
постоянства энтропии, объема и общих количеств компонентов, остается
постоянной или возрастает. Иначе1
(»)3,v,»i, «,,.•■.»* >0. (П.69)
Отметим, что первая формулировка принципа равновесия предусматривает
полную изоляцию системы, включая и условие адиабатичности. Вторая
формулировка предполагает, что между системой и окружающей средой происходит
теплообмен.
В связи с различными внешними условиями принцип равновесия Гиббса
можно выразить при помощи других термодинамических потенциалов [13]:
№)s9ptnlt «„..., «л>0, (11.70)
l*F)T9v9nl9n%r..9nk>0. (11,71)
(^)г,р,пь^,...,па>0. (11.72)
Рассмотренные формулировки принципа равновесия Гиббса эквивалентны
в том смысле, что из одной формулировки при известных допущениях следуют
все остальные. Однако они не тождественны, поскольку их содержание и при-
v l В выражениях (11.67), (11.69) и далее даны вариации энтропии и
внутренней энергии, а не дифференциалы, поскольку при этом речь идет о
виртуальных изменениях, составляющих более обширный класс изменений [13].
31
менимость подчинены различным условиям существования систем [13].
Условие применимости конкретной формулировки принципа равновесия Гиббса
выражается в закреплении определенного набора термодинамических
параметров.
3. УСЛОВИЯ ФАЗОВОГО РАВНОВЕСИЯ.
ДЕЙСТВИТЕЛЬНЫЕ И ВОЗМОЖНЫЕ КОМПОНЕНТЫ
Рассмотрим условия равновесия фаз. В этой связи необходимо дать более
строгое определение понятиям «фаза», «компонент» и некоторым другим,
которые были введены нами предварительно в гл. I и которыми мы до сих пор
пользовались в тех случаях, когда не требовалось их строгого толкования.
Компонентами Гиббс называл индивидуальные вещества, приращения
концентраций которых независимы и выражают все возможные изменения в
составе рассматриваемой системы. При этом под индивидуальными веществами
понимаются простые вещества и химические соединения.
По Гиббсу, все компоненты делятся на действительные и возможные. Под
действительными компонентами следует понимать такие вещества, которые
присутствуют хотя бы в исчезающе малых количествах во всех гомогенных частях
или фазах гетерогенной системы. Под возможными компонентами
подразумеваются вещества, которые входят в состав некоторых фаз, но в данной фазе
отсутствуют.
Если в системе отсутствуют обратимые химические реакции, то все вещества,
образующие систему, являются компонентами, поскольку их концентрации могут
изменяться независимо, т. е. в этом случае' понятие «компонент» совпадает с
понятием «вещество» и число компонентов равно числу веществ, содержащихся
в системе.
Если в системе протекают обратимые химические реакции, то концентрации
лишь части веществ, входящих в состав системы, могут изменяться независимо.
Это объясняется тем, что благодаря протеканию обратимых химических реакций
существуют количественные связи между концентрациями веществ. Такие связи
описываются термодинамическими уравнениями, число которых равно числу
независимо протекающих обратимых реакций. Поэтому в случае систем с
химическими превращениями число независимых компонентов будет всегда меньше
числа веществ, входящих в состав системы, и равно разности между числом
сортов частиц, самостоятельно существующих в системе, и числом независимо
протекающих обратимых реакций.
Не следует смешивать число компонентов с числом исходных веществ,
смешением которых была получена рассматриваемая система. Число компонентов
может быть и больше, и меньше числа исходных веществ в зависимости от
химических особенностей системы [13].
Из сказанного следует, что выбор компонентов, как подчеркивает
А. В. Сторонкин, произволен и неоднозначен. На вопрос, из каких компонентов
состоит система, в которой протекают обратимые химические реакции4, нельзя
дать однозначный ответ. Число же компонентов — совершенно определенное.
Необходимо также отметить, что число компонентов зависит и от условий,
при которых существует изучаемая система. Изменяя условия, можно
возбуждать или подавлять химические реакции в системе и таким образом уменьшать
или увеличивать число связей, накладываемых на изменение концентраций
веществ.
Понятие «фаза» было введено Гиббсом в скрытой форме в связи с выводом
правила фаз. Большая заслуга в уточнении этого важнейшего понятия в теории
гетерогенных равновесий принадлежит Ван-дер-Ваальсу и А. В. Сторон-
кину [12, 13].
При выводе правила фаз Гиббс предполагал, что каждой фазе отвечает свое
фундаментальное уравнение типа (11.24) и, следовательно, существует столько
независимых уравнений этого вида, сколько фаз имеется в рассматриваемой
системе. Физический смысл этого заключения состоит в том, что каждая фаза
обладает своим видом зависимости термодинамических свойств от параметров
состояния, что и отличает ее от других фаз, кроме того, фаза гомогенна и имеет
32
макроскопические размеры. Последнее связано с тем, что уравнение (11.24)
справедливо при условиях, что фаза находится в равновесии внутри себя,
внешнее поле или отсутствует, или оно постоянно и можно пренебречь
поверхностными явлениями. Таким образом, основная суть понятия «фаза» отражена в
уравнении типа (11.24), а поверхность раздела между фазами является лишь внешним
признаком фазы.
Уравнение, которое выражает свойственную данной фазе зависимость
термодинамических свойств от параметров состояния, Ван-дер-Ваальс назвал
уравнением фазы.
В соответствии с современными представлениями, основанными на идеях
Гиббса, под фазой• следует понимать совокупность телесных комплексов,
термодинамические свойства которых одинаково зависят от параметров состояния,
т. е. описываются одним и тем же уравнением фазы, в качестве которого можно
взять любое из фундаментальных уравнений Гиббса. При этом под телесными
комплексами подразумеваются макроскопические гомогенные части,
образованные индивидуальными веществами, либо молекулярными или атомными
смесями веществ и отделенные одна от другой поверхностями раздела [13].
Системы, состоящие из нескольких фаз, являются гетерогенными системами.
При подходе к термодинамическому анализу гетерогенных равновесий
возникает проблема, связанная с оценкой возможного числа равновесно
сосуществующих фаз и числа параметров данной системы, которые можно изменить
произвольно, не нарушая равновесия. Гиббс рассмотрел общие закономерности,
которым подчиняются равновесные системы, состоящие из любого числа фаз и
любого числа компонентов, и вывел закон, определяющий вариантность
равновесной гетерогенной системы. Этот закон вошел в учение о гетерогенных
равновесиях под названием правила фаз.
Положим, что система состоит из т веществ и содержит г фаз при
температуре Т и давлении р.
Если в системе, состоящей из m веществ и г фаз, протекает / независимых
реакций, то число независимых переменных (вариантность или число степеней
свободы системы)
/ = (т — /) — г+ 2 или /= £ — г+ 2, (11.73)
где к — число компонентов.
В зависимости от числа степеней свободы принято различать системы нонва-
риантные (/= 0), моновариантные (/= 1), дивариантные (/= 2) и
поливариантные (/> 3). Согласно равенству (11.73), максимальное число сосуществующих
фаз наблюдается в нонвариантных системах (&+ 2). Так, в случае однокомпо-
нентных систем максимальное число сосуществующих фаз равно трем, а в
случае бинарных систем — четырем. В нонвариантных системах невозможны
никакие изменения системы без уменьшения числа фаз.
Формула (11.73) является математическим выражением правила фаз: число
степеней свободы гетерогенной системы равно числу компонентов минус число
фаз плюс два.
Общеизвестный вывод выражения (11.73) предполагает, что каждый
компонент присутствует в каждой фазе, т. е. является действительным. Однако если
допустить, что один из компонентов (например, i) является возможным, т. е.
отсутствует в одной из фаз (например, а), то это не отразится на числе степеней
свободы. Действительно, при этом в выражении (11.73) исчезает одно из
связывающих уравнений, например
|*? = И?. (И.74)
но одновременно мы должны ввести условие отсутствия / в фазе а, т. е.
*? = 0. (П.75)
При введении каждого из условий отсутствия компонента, подобных
равенству (11.75), мы теряем одно из условий (11.74). Общее число условий при этом
остается неизменным и правило фаз (11.73) сохраняет свою форму.
2 Глазов В. М. и др.
33
Из уравнения Гиббса следует, что цифра Два появилась вследствие
принятого допущения о том, что на состояние системы влияют только два параметра:
температура и давление. Однако бывают случаи, когда влияют и другие
параметры. В этом случае правило фаз в общем виде записывается так:
f=k-r+N,
где N — соответствующее число параметров.
Для многих конденсированных систем давление меняется столь
незначительно, что не оказывает влияния на процессы и превращения в данной системе.
Тогда правило фаз формулируется следующим образом:
/=*-г+ 1.
В некоторых случаях (например, при равенстве концентраций какого-либо
компонента в двух фазах) возникают условия, позволяющие составить
дополнительные уравнения для характеристики системы. Появление этих
дополнительных уравнений, связывающих параметры системы, вызывает уменьшение
вариантности системы на столько единиц, на сколько увеличилось число уравнений,
характеризующих систему.
Вариантность / определяет число степеней свободы системы без учета масс
фаз. Если же в число переменных включают и массы фаз, то общее число
независимых переменных определяет полную вариантность F системы: F= /+ г.
Использование этой величины позволяет полнее описать объемные, поверх-
костные и объемно-поверхностные фазовые процессы.
Дальнейшее развитие путей приложения правила фаз к системам
различного типа содержится в работах Сторонкина и его школы (см., например, [11,
15] и др.).
Следуя методу Гиббса, рассмотрим условия равновесия фаз. Положим, что
собственно химические реакции между компонентами в рассматриваемом
случае исключены. Тогда все химические превращения сводятся к перемещению
компонентов из одной части системы в другую. Следовательно, условия
равновесия относятся только к распределению компонентов между отдельными фазами
системы. Для нахождения этих условий допустим, что система состоит из фаз,
содержащих к компонентов. Рассмотрим все возможные (виртуальные)
изменения фаз с теми ограничениями, при которых записано выражение критерия (11.69),
т. е. при постоянстве общей энтропии системы, общего объема и общего числа
молей.
Запишем в развернутом виде эти условия следующим образом:
ф<5 = Sa + Sp + ...+'Sr - Cs = 0;
фу = Va + Vp + ... + V - С v = 0;
Ф1 = n? + n? + ... + n\ — Cx = 0; (11.76)
<*k = n% + nl+...+nrk-Ck = 0.
где C$, Cyy Cly ..., Ck—постоянные.
Уравнения связи (11.76) накладывают ограничения на изменения параметров
рассматриваемой гетерогенной системы.
Согласно общему условию (11.69), принцип равновесия Гиббса для
^-компонентной r-фазной системы запишется в виде
(£«/Л ^0. (Н.77)
34
Для нахождения условий, при которых внутренняя энергия системы
принимает условный минимум, используем метод Лагранжа. Составим следующую
функцию:
Ф = U + ЯяФз + Лу% + >i<I>i + ... + h®k> (II.78)
где Я§, Ху, Kv ...Дд — постоянные множители.
Как известно, функция Ф будет иметь безусловный минимум, когда
внутренняя энергия системы достигнет условного минимума. Следовательно, частные
производные функции Ф по параметрам состояния системы, указанным в
уравнениях (11.76), для состояния равновесия равны нулю.
Согласно уравнениям (11.76) и (11.78) с учетом соотношений (1.76), (1.77)
и (II.4), находим:
дФ/dSf = Г/ + ls = 0;
дФ/dV* = — р*+ Яу = 0: (11.79)
дФ/дп{ = ц{ + К. = 0.
На основании результата решения системы уравнений (11.79) для
рассматриваемого случая условия равновесия ^-компонентной г-фазной системы можно
записать следующим образом:
Т^ = Т^ = = ТГл
(11.80)
Ра=
ц? =
tf=
рр=..
■И? —
■rf--
.~f;
• =tf;
■•-Й-
Первые два соотношения в условии (11.80) являются уравнениями
термического и механического равновесия системы, а остальные характеризуют
условия равновесного распределения каждого компонента по всей системе. Таким
образом, при равновесном распределении компонента в закрытой системе его
химический потенциал имеет одинаковое значение во всех фазах, составляющих
данную систему.
Уравнения равновесия (11.80) выведены при условии отсутствия или
постоянства внешнего поля, а также при условии пренебрежения влиянием
поверхностей раздела между частями системы на ее термодинамические свойства.
При выводе также предполагалось, что все компоненты содержатся во всех
фазах. Это предположение не вносит никаких ограничений в условия
равновесия (11.80).
Детализируем формулировки условий фазового равновесия в гетерогенной
системе в связи с делением компонентов на действительные и возможные.
Представим себе систему с произвольным числом фаз при некоторых заданных
значениях температуры и давления.
Рассмотрим процесс перехода некоторого количества йпг действительного
компонента из а-фазы в (3-фазу. Суммарное изменение изобарно-изотермического
потенциала, которое согласно принципу равновесия (11.72) должно быть больше
или равно нулю, можно записать таким образом:
<6G)p, Т. »„ „. = (вО/ав,)« г. „,. „.dnf+idG/dn^ «„„.«foS^O
(11.81)
или, согласно выражению (11.16), учитывая, что изменение числа молей dn?
и dray численно равны, но противоположны по знаку, в другой форме
(A-tf)dni^0. (II.82)
2* 35
Так как drtx по определению — величина положительная, из выражения (11.82)
имеем
И?-^0. (П.83)
Поскольку первый компонент для системы в целом и, в частности, для р-фазы
является действительным компонентом, т. е. хотя бы и в малом количестве, но
заведомо имеется в этой фазе, то наряду с только что проанализированным
процессом мы вправе рассмотреть и обратный процесс, а именно перенос dn^ молей
из Р- в а-фазу. В соответствии с этим теперь условие равновесия (11.81) приводит
к соотношению
»*? —мЗ^о. (IL84)
Итак, если рассматриваемый компонент присутствует в обеих фазах, то
условие равновесия приводит к зависимостям (11.83) и (11.84), которые
справедливы только при знаке равенства, тогда как при знаке неравенства они взаимно
исключаются. Отбрасывая на этом основании знак неравенства, мы убеждаемся,
что при термодинамическом равновесии химический потенциал любого
компонента одинаков во всех фазах, для которых этот компонент является
действительным, т. е. мы приходим, таким образом, к условиям равновесия в виде (11.80).
Изложенные соображения приводят к существенно иному выводу
относительно возможных компонентов. Если, например, первый компонент присутствует
только в а-фазе, а в других фазах отсутствует, то можно себе представить
процесс перехода этого компонента из а-фазы в любую другую, что приводит к
соотношению (11.83). Однако этому процессу нельзя противопоставить обратный
процесс перемещения этого компонента из какой-либо фазы, где нет даже его следов,
в а-фазу. В этом случае соотношение (11.84) не применимо, и поэтому мы не
можем игнорировать знак неравенства.
Следовательно, на основании термодинамики можно утверждать лишь, что
химический потенциал возможного компонента в той фазе, где этот компонент
присутствует, меньше химического потенциала (или в крайнем случае равен ему)
того же компонента в других фазах, где при равновесии нет даже его следов.
Иначе говоря, если для какого-либо компонента отсутствует равенство
химических потенциалов в разных фазах, то при термодинамическом равновесии такой
компонент сосредоточивается в той фазе (или в тех фазах), где его химический
потенциал минимален [16].
Таким образом, вопрос об игнорировании существования возможных
компонентов выходит за рамки термодинамики и решен нами с привлечением моле-
кулярно-кинетических представлений [17].
4. УСЛОВИЕ ХИМИЧЕСКОГО РАВНОВЕСИЯ.
ЗАКОН ДЕЙСТВУЮЩИХ МАСС
Для систем, в которых между компонентами протекают химические
реакции, общее состояние термодинамического равновесия следует конкретизировать
следующим образом. Химическое равновесие — это неизменяющееся во времени
состояние системы, в состав которой входят компоненты, способные реагировать
между собой химически, и продукты реакции. Термодинамически можно
установить условия равновесия в такой системе и показать, как на нее влияет
изменение параметров состояния (температуры и давления).
Пользуясь методом Гиббса, рассмотрим систему, в которой проходит
реакция:
Mi + ъА2^±укАк + v;+1^+1, (11.85)
где v. и v'. — стехиометрические коэффициенты исходных веществ и продуктов
реакции соответственно; Л, — символ i-того вещества.
36
Стехиометрическое уравнение такой реакции в общем виде следующее:
J\viA£^0. (II.86)
i
По условию продукты реакции берут с положительным знаком, а исходные
вещества — с отрицательным. В данном случае рассматриваемую
^-компонентную г-фазную систему можно получить смешением (k— 1) веществ Лх, Л2, ...,
Ak-v поскольку, по предположению, первые два вступают в обратимую
реакцию (11.85) и образуют два других вещества.
Пусть п*, л£, ..., л£_х — число молей смешиваемых веществ. Эти величины
постоянны, так как изучаемая система — закрытая. После установления
химического равновесия система будет состоять из (&+ 1) веществ Аъ Л2, ..., Л^+1,
число молей которых равно соответственно nv л2, •••» nk+1. Очевидно, что для
веществ, не принимающих участие в реакции (Л3, Л4, ..., Л^х), величины С/
и щ одинаковы.
Запишем теперь уравнения связи для рассматриваемой системы. При
составлении уравнений связи для веществ Ах и Л2 учтем, что равновесные значения
числа молей этих веществ (пг и п2) не равны их значениям в момент смешения (п[
и п'2), так как часть веществ Аг и Л2 расходуется на образование веществ Л$
и Ak+i в соответствии с уравнением химической реакции (11.85). Тогда
уравнения связи для рассматриваемой системы будут иметь вид:
Ф5 = £ S/-Cs = 0;
г
av= £ vi — Су= о;
^1 = 1 *{+ (vi/vOt n{-Cx =0; (11.87)
r
фз= I] ^-C3 = 0;
ф*-1 = £ «i-i"c*-i =0'
Первые слагаемые в выражениях для Фх и Ф2 представляют собой общие
числа молей непрореагировавших веществ Л2 и Л2, а вторые слагаемые —
количество веществ Лх и Л2, израсходованное на образование веществ Ak и Л^+А.
Вполне очевидно, что при отсутствии в системе химических реакций
уравнения (11.87) переходят в выражения (11.76). Отсюда следует, что условия
равновесия (11.80), выведенные для систем без химических превращений, справедливы
и для систем с химическими превращениями. Однако в первом случае они
необходимы и достаточны, а во втором они лишь необходимы.
Для установления дополнительных условий равновесия закрытых систем,
в которых протекают химические реакции, примем во внимание, что изменения
числа молей веществ, принимающих участие в обратимой реакции, в
рассматриваемом случае взаимно связаны, согласно уравнению (11.85):
I4h|4h|e«il:ler|i+il=svi:v2sv*:v*+i' (IL88>
где в прямых скобках — абсолютные значения приращений числа молей.
37
Дифференцирование функции Ф, составленной согласно выражению (11.78)
с помощью уравнений связи (11.87), по числу молей (например, вещества А±
в /-той фазе) и приравнивание результатов дифференцирования нулю с учетом
соотношения (11.88) приводят к окончательному результату:
Vil*{ - *А = чЫ + v'k+A+v (11.89)
Уравнение (11.89) выражает условие химического равновесия в /-той фазе.
Во всех других фазах, в которых протекает данная реакция, условие
химического равновесия выражается аналогично, поскольку согласно уравнениям (11.80)
химические потенциалы веществ имеют одинаковые значения во всех фазах.
В общем случае условие равновесия для собственно химической реакции
вида (11.85) примет вид
2>/И/=0, (П.90)
где суммирование проводится по всем участникам реакции. Таким образом»
в общем виде условие гетерогенного равновесия, согласно Гиббсу, определяется
уравнениями (11.80) и (П.90).
Из условия (11.90) легко получить основной закон химического равновесия—
закон действия масс, если химические потенциалы ji/ в явной форме выразить
через концентрации или парциальные давления компонентов. Впервые закон
действующих масс был сформулирован Гульдбергом и Вааге (1867 г.) на основе
кинетического рассмотрения идеальных* газов. Такой подход приемлем только
для простейших реакций. В общем же случае закон действующих масс вытекает
из термодинамических соображений.
Пусть в однородной системе, состоящей из идеальных газов, протекает
химическая реакция (11.85). Подставив значение химического потенциала из
уравнения (11.52) в условие химического равновесия (11.90), получим
Е^<Г>+Е^*Г1пр,=0. (П.91)
i i
откуда 2>>p,=-£v,|i;(r)/l?'7\ (II.92)
i i
т.е. левая часть уравнения (11.92) — функция только температуры f (T).
Заменим сумму логарифмов pi логарифмом произведения /?,, а / (Т) логарифмом
некоторой функции К1^ (Т). Тогда уравнение (11.92) можно переписать в виде
1пП(рУ<) = 1п^(Г)._ (11.93)
где pi — парциальные давления составляющих системы в момент равновесия.
Величина К*£* выраженная через равновесные парциальные давления в
идеальной газовой смеси, есть функция только температуры и не зависит от
суммарного давления и парциальных давлений веществ в исходной смеси, т. е. от
относительных исходных количеств веществ. Отметим, что К1* для равновесной смеси
реальных газов зависит от давления. Величина К1* называется константой
химического равновесия или просто константой равновесия, а уравнение (11.93)
выражает закон действующих масс в применении к идеальным газам.
Очевидно, что значение константы равновесия реакции — не произвольное.
Отсюда можно заключить, что выбор стандартного состояния для начала отсчета
химического потенциала зависит от природы реагирующих веществ в том
случае, когда имеется возможность химического взаимодействия между
составляющими смеси. Поэтому представляет определенный интерес другой вывод закона
действующих масс, который не требует привлечения стандартных состояний.
38
Следуя Гуггенгейму [18], рассмотрим два набора определенных значений
парциальных давлений р., обозначенных соответственно через р\ и р'\. Тогда
для каждого t-того вещества имеем, независимо от какого бы то ни было выбора
стандартного состояния
F/-K- = #rin(p';/p;.). (п.94)
Предположим теперь, что набор величин р|, а также р\ отвечает каждый
состоянию химического равновесия при той же самой температуре. Тогда,
согласно условию химического равновесия (11.90), для реакции общего вида (11.85)
можем записать
vi^, + v2*4 + • • • = vV - + v'2\l', + . • .;
Al A2
Вычитая одно уравнение из другого и принимая во внимание
выражение (11.94), после несложных преобразований окончательно получим
=(\)W--/(4)V,(4)V-
Так как подобное состояние должно быть справедливо для любого другого
набора равновесных значений р,- при той же самой температуре, отсюда следует,
что равенство (11.95) эквивалентно выражению (11.93).
Константа равновесия связывает парциальные давления всех веществ,
участвующих в реакции, и нельзя изменить парциальное давление ни одного из них,
чтобы это не повлекло за собой соответствующего изменения парциальных
давлений всех участников реакции и не привело бы к прежнему численному
значению константы равновесия при данных условиях. Таково содержание закона
действующих масс.
Формулировку закона действующих масс, соответствующую уравнению
(11.93), можно сохранить также и для реальных газов, если парциальные
давления участников реакции заменить их фугитивностями. Тогда
П (фУ') =4>i 1(Р2 2. • . |ф?'Ф2§ . . • =*ф(Г). С"'96)
Приняв во внимание, что \i* (T) в уравнениях (11.52) и (11.57) одна и та же,
получим
*"(Г) = *Ф(Г)- О1'97)
Величина /Сф (Т) не зависит от давления.
Не повторяя аналогичных рассуждений, укажем только, что для реального
раствора
v7 V
П (а>) = «i Ч \ . .1*11Ф . . .=Ка(Т,р), (11.98)
i
причем
К1*(Т,р) = Ка(Т.р)- (П.99)
39
Формулы для константы химического равновесия (11.93) и (П.96) можно
применять и в том случае, когда в реакции, кроме газов, участвуют твердые или
жидкие вещества (гетерогенные реакции). Тогда в правую часть формулы будут
входить парциальные давления или летучести паров конденсированных веществ.
Если в реакции участвуют жидкие или твердые вещества, не образующие между
собой растворов, то при данной температуре парциальное давление этих
конденсированных компонентов реакции является величиной постоянной, зависит только
от температуры и не зависит от парциальных давлений других газов или паров,
имеющихся в системе. Поэтому для удобства расчетов в выражениях (11.93)
и (IL96) вводят давления паров конденсированных веществ в константу
равновесия, которая в этом случае будет определяться парциальным давлением только
газообразных участников реакции.
Механизм гетерогенной реакции (например, протекает она в газовой фазе
или на поверхности раздела фаз) не влияет на вывод закона действующих масс
в гетерогенных системах, поскольку термодинамика позволяет судить о
равновесии процесса лишь по исходному и конечному состояниям системы.
5. КРИТЕРИИ УСТОЙЧИВОСТИ ФАЗ И ГЕТЕРОГЕННЫХ
СИСТЕМ
Общие условия равновесия термодинамических систем заключаются в том,
что в состоянии равновесия энтропия и термодинамические потенциалы имеют
экстремальные значения. Так, для систем с постоянными энтропией, объемом и
числом молей компонентов условием равновесия является минимум внутренней
энергии, что выражается двумя соотношениями б1/=0и62£/>0. При выводе
конкретных условий равновесия достаточно лишь первого соотношения (dU = 0).
Однако равенство нулю первой вариации является лишь необходимым условием
экстремума и не обеспечивает минимального значения внутренней энергии.
Достаточным условием минимума внутренней энергии является положительное
значение ее второй вариации, которое и обеспечивает устойчивость равновесия.
Термодинамические неравенства, выражающие условия устойчивости,
наряду с принципом равновесия составляют основу химической термодинамики
гетерогенных систем. Мы приводим гиббсовский вывод критериев устойчивости
фаз, систематизированный главным образом в работах А. В. Сторонкина [13].
Критерий устойчивости данной фазы относительно образования внутри
нее новых фаз.
Внутренняя энергия гетерогенной системы, в которой наряду с г старыми
фазами имеется s новых фаз, равна
г s
С/= 2 [/'+ Ц Ц1- (11.100)
i=l /=1
Здесь и далее индекс i соответствует старым фазам, а индекс / — новым.
Предположим, что система получена смешением Cv С2» ..., Ck-t молей
веществ Ах, А2, ..., Ak-i и что в системе протекает обратимая реакция вида (IL85).
Принцип равновесия для рассматриваемой системы представим в виде
£ «(/< + J «//"L Vt с t с > 0. (11.101)
Запишем уравнения связи, выражающие условия, при которых внутренняя
энергия системы имеет условный минимум:
Ф5 = 2S'+ S S''-Cs = 0; (11.102)
i=i /=i
40
Ф1= £ *! + £ »{ +(viM) ( £ *J+ £ ni) -Ci =0;
Ф2= £ «2 + £ *J + (v2/v^) I £nj + £ «i) -C2 = 0;
/=1 /=1 V/-i /=1 /
ф3= 5>з+Г4-с3 = о;
/=1 /=i
} (11.104)
/=1 /-1
На основании выражения (11.12) с учетом условий (11.80) соотношение (11.101)
можно представить в следующем виде:
[£«/{+г £es'-p£ev'+iAi £««{+...+
/=1
/=1
£ ц+11
/=1
>0.
Согласно уравнениям (11.102)—-(11.104), справедливо:
J] 6S/=-£ 6S>;
1=1 /=i
£ 6У< = - £ 6К';
/«1 /=1
£ M = -£ M-(Vi/v;) (£ *!,'+£ 6^; |
f-=l /=I \t=l /=1 / I
£ 6n< = -£ Ц- (VvJ)( £ К + £ 6лП ;
£&1! = -£Ц;
(11.105)
(11.106)
(11.107)
i=i
/=i
Efiii = -l6"U
/=1
/=i
(11.108)
На основании уравнений (11.88), (11.89) и (11.106)—(.11.108) выражение
/П.105) сводится к следующему виду:
£ (6t// - T6\S' + pbVl - \1гЬп{ - ... -
£=1
-^+16«i+o]svCi CftV
(11.109)
41
Учитывая независимость изменения экстенсивных величин UJ\ 5;, W, n{, ...,
njUl» относящихся к данной новой фазе, от изменений таких же величин,
относящихся к другим новым фазам, на основании выражения (11.109) имеем
ЫЛ — T6Si + pbVf — \kYbn[ - ... — ^+1 вп{+1 > 0. (11.11 0)
Выражение (11.110), взятое со знаком равенства, дает дополнительное
условие равновесия относительно образования новых фаз. Если новые фазы в
начальный момент их образования рассматривать как гомогенные, то в результате
суммирования левой части соотношения (11.110) для новых фаз
макроскопического размера найдем
(//-TS' + pV/-|i1n{-...-|ift+1nj;+1>0. (11.111)
Это выражение, являющееся критерием устойчивости фаз относительно
образования внутри них новых фаз, имеет большую практическую ценность.
Уравнение (11.111) нетрудно преобразовать к виду:
(у1 + и*) — и*0>0. (11.112)
Левая часть условия (11.112) выражает приращение внутренней энергии§
связанное с образованием новой макроскопической фазы в состоянии UJ и
изменение состояния старой от Ul0 до U1.
В условии (11.111) не учтена работа образования поверхности раздела между
старой и новой фазами и поэтому оно является лишь достаточным критерием
устойчивости фаз относительно конечных изменений состояний.
Если условие (11.111) выполняется и поверхностное натяжение на границе
раздела между старой и новой фазами положительно, т. е. для образования
диспергированной фазы необходимо затратить работу и, следовательно, более
строгое условие (11.110) тем более выполняется, то рассматриваемая фаза устойчива
относительно образования новых фаз (диспергированных и недиспергированных).
В случае отрицательного поверхностного натяжения макроскопическая фаза
будет неустойчивой и использование выражения (11.111) не имеет смысла.
Однако критерий устойчивости (11.111) не выполняется для метастабиль-
ных фаз (пересыщенный раствор или переохлажденный расплав и т. д.), которые
неустойчивы по отношению к образованию из них других макроскопических фаз,
когда поверхностные явления можно не учитывать. Поэтому он не является
необходимым. Если в метастабильную фазу внести зародыши новой, более
устойчивой фазы, то процесс ее роста до макроскопических размеров происходит
самопроизвольно, а работа ее образования будет отрицательна.
Следовательно, достаточный критерий устойчивости фаз относительно
образования в них новых фаз можно сформулировать следующим образом: фаза
устойчива относительно конечных (прерывных) изменений состояния, если
работа образования новой фазы макроскопических размеров положительна [13].
Таким образом, достаточный критерий устойчивости позволяет отличать
стабильные состояния от метастабильных. Стабильные и метастабильные
состояния устойчивы относительно образования диспергированных фаз. Поэтому
условие (11.110) справедливо для стабильных и метастабильных состояний и
является необходимым и достаточным условием устойчивости фаз относительно
прерывных (конечных) изменений состояния.
Критерий устойчивости фаз относительно бесконечно
малых изменений состояния (флуктуационных процессов)
Фазы, неустойчивые по отношению к бесконечно малым изменениям,
являются абсолютно неустойчивыми и, следовательно, не могут существовать.
Поэтому критерий устойчивости фаз по отношению к бесконечно малым
изменениям позволяет отличать реализуемые состояния подвижного равновесия от
нереализуемых.
42
На основании интегрального критерия устойчивости (11.111) при любых
отклонениях от равновесия фазы со средой должно выполняться неравенство
ДГ Д5 —ДрДИ + А|х1Д/11+... + Д^ЛяЛ>0, (11.113)
где Д — точное приращение соответствующей величины при переходе из
исследуемого состояния в примыкающее (бесконечно близкое).
Полученное Гиббсом неравенство (11.113) является необходимым и
достаточным критерием устойчивости двух бесконечно близких состояний фазы
одного относительно другого.
С помощью неравенства (11.113) можно установить качественную связь
между изменениями сопряженных термодинамических параметров. Левая часть
неравенства (11.113) содержит (fc + 2) слагаемых. Частные условия устойчивости
получим, фиксируя значения (£ + 1) параметров по одному из каждого
слагаемого, кроме того, которое рассматривается, причем из фиксированных (k + 1)
параметров, по крайней мере, один должен быть экстенсивным.
Выполнение первого условия фиксирования необходимо для того, чтобы при
постоянстве (k + 1) параметров в неравенстве (11.113) имелось только одно
отличное от нуля слагаемое, а второе условие исключает из рассмотрения такие
случаи, когда при постоянстве (k-\- 1) интенсивных параметров невозможны
изменения состояния фазы и когда, следовательно, неравенство (11.113) теряет
смысл. Тем самым мы включаем массу фазы в число переменных.
Фиксируя в соответствии с изложенными условиями значения (&+ 1)
параметров, из неравенства (11.113) получаем:
(ДГ/Д5)а>0; (11.114)
(Др/ДУ)а<0; (11.115)
(Д^/Дл/ЬХ), (11.116)
где индекс а указывает способ фиксирования параметров, который определяется
характером рассматриваемого процесса изменения состояния фазы.
Неравенства (11.114)—(11.116) являются необходимыми и достаточными
условиями термической, механической и химической устойчивости фаз
относительно бесконечно малых изменений состояния. Согласно этим неравенствам,
температура и энтропия, а также химические потенциалы и сопряженные с ними
числа молей компонентов изменяются всегда симбатно, давление же и объем
изменяются всегда антибатно.
Как известно, если производная любого интенсивного параметра по
соответствующему экстенсивному положительна, то положительно и отношение
полных приращений, которое, однако, может быть положительным и тогда, когда
производная равна нулю. Поэтому достаточный критерий устойчивости фаз
относительно бесконечно малых изменений состояния можно представить в виде.
dTdS — dpdV + dii1dnl + ...+d\ikdnk>Q; (II.117)
(dT/dS)a>0; (11.118)
(dp/dV)a<0; (Н.119)
№Л)в>о. (ПЛ20)
Способ фиксирования параметров аналогичен использованному при
получении выражений (11.114)—(П.116). Если в неравенствах (11.117)—(IIЛ20)
ввести знак равенства, то получим необходимые условия устойчивости. На гра"
нице устойчивости одна из производных (11.118) или (11.119) обращается в нуль,
тогда как обратные им величины в нуль не обращаются. Достаточный критерий
Устойчивости (11.113) можно переписать в другой форме, которая полезна при
решении некоторых конкретных задач:
«2£/>0. ' (11.121)
43
• Установим достаточный критерий устойчивости (11.120) через вторые
производные термодинамических потенциалов в общем виде, используя
последовательность рассуждений, примененную в работе Мюнстера [19].
Введем в неравенство (11.113) обобщенные координаты X и Y и перепишем
его в виде
^Д^Д^Х). (11.122)
Внутренняя энергия как функция обобщенных координат У имеет вид
U = U(YltY2 Ym).
т т
Отсюда dU= X ^ldYi)i^idyi = YXldY*-
i=\ i=l
(II.123)
(11.124)
На основании выражения (11.124), используя преобразование Лежандра,
получим общее уравнение для термодинамических потенциалов:
(№k=dU-'£d(ХМ) = f,XtdYt - 2Xt dYt - J]Yt dXt =
1=1 £=1 1=1 1=1
k m
= -^r,dX,+ £ XjdYh (11.125)
r=l /=fe+l
(11.126)
или в интегральной форме
ЧГЛ = ЧГЛ(Х/.К/),
где i = 1, 2, ..., k; j = k-\- 1, ..., т.
Выразим теперь неравенство (11.122) через вторые производные
термодинамического потенциала Wk- Для этого нужно AYS (s— 1, 2, ..., k) и ДХ/ (t =
= k + 1, ..., m) представить как функции Х[ и У/. Так как в неравенстве (11,122)
ограничивались только вторым порядком, то получим линейную систему
уравнений:
k m
AYS = Y (dYsldXi) AXi + £ (dYs/dYj) ДГ,;
f=i /=*-H
£ m
ДХ, = ^ (<ЭХ,/аХ,) AX, + £ {dXtlWflbYt.
t=l
(11.127)
На основании выражения (11.125) имеем
Отсюда
ак./дХ/^—д^/дХ.д*,;
ах//ау/ = а»ул/дУг/дУг/.
Из уравнения (11.125) вытекает следующее соотношение Максвелла:
д»У/аХ,дК/ = - (dYsf&Yf) = (dXt/dXi). (II.131)
44
(11.128)
(11.129)
(11.130)
Если подставить выражения (11.127) с учетом соотношений (IT. 129)—(11.131)
в неравенство (11.122), то
k tn k k
2 ays {±Х;+ £ bXt лгу = £ £ (ак5/ах.) дх? +
/em m ft
+ £ £ (^,/аК/)АГ,АХ/+ £ S(dX//dX/)AX,AK/ +
t=i /=ft+i /=ft-fl t=i
m m
(11.132)
В результате первоначальное выражение распадается на две квадратичные
формы, первая из которых зависит только от экстенсивных, а вторая — только
от интенсивных параметров.
Неравенство (11.132) в общем случае выполняется тогда, когда первая
квадратичная форма определена положительной, а вторая — отрицательной. Если
исключить вариации, которые изменяют лишь массу фазы, приняв Ym— const,
то получим достаточные условия устойчивости относительно бесконечно малых
(непрерывных) изменений состояния в форме, пригодной для всех
термодинамических потенциалов:
lb"Vk(Xl9...,Xk)]Yj<0; (П.133)
lWk(Yk+L...,Ym-i)]Ytn9xi>0. (П.134)
Учитывая знак квадратичных форм, эквивалентная, а практически более
важная формулировка может быть представлена в следующем виде:
для главных миноров нечетного порядка
ld*¥k/dXtdXs\<0; (II.135)
для главных миноров четного порядка
\д*Ук/дХсдХ8\>0; (11.136)
для всех главных миноров
\d*ykidYjdYt\>0. (II.137)
Условия в форме (11.133) и (11.134) впервые даны Шоттки [20].
При условии постоянства величин 7\ р и ^]п;для термодинамического по-
i
тенциала Гиббса получим следующее соотношение, эквивалентное
выражению (11.134):
№(хг **-i)]P,7>0. (H.138)
или для всех главных миноров
S Г \d*g/dXidxA>09 (*\/ = l,2 ft —l). (11.139)
Для бинарной системы выражение (11.139) сводится к условию:
(dVfo?)p.r>0. (II.140)
С учетом выражений (П.39) и (11.43) критерий устойчивости можно переписать
и виде
id\i2ldx2)PtT>0. * (II.141)
45
В общем случае критерий устойчивости требует, чтобы поверхность
термодинамического потенциала Гиббса, отвечающая устойчивым относительно нет
прерывных изменений состояниям фазы, была выпукла относительно
координатных осей концентраций.
Обычные формулировки условий устойчивости одновременно являются и
ограничениями на величины термодинамических потенциалов [см., например,
неравенства (11.121) и (11.139)]. Из условий устойчивости были выведены также
ограничения на парциальные мольные термодинамические функции смешения
[21]. Эти ограничения носят дифференциальный характер. В работе [22] на
основе строгих соотношений для коэффициентов активностей компонентов или
активностей, вытекающих из условий устойчивости относительно непрерывных
изменений состояния [23], без каких-либо допущений получены ограничения на
свободную энергию образования раствора в интегральной форме.
Необходимые критерии устойчивости
гетерогенных систем относительно
непрерывных изменений состояния
До сих пор были рассмотрены критерии устойчивости однородных систем
и отдельных фаз гетерогенных систем. Покажем теперь, что гетерогенная система
в целом будет находиться в состоянии устойчивого равновесия, если критерии
устойчивости относительно непрерывных изменений состояния выполнены для
каждой из составляющих фаз. Ограничимся рассмотрением однокомпонентной
системы, состоящей из двух фаз а и Р, имеющих макроскопические размеры.
Предположим, что при бесконечно малом изменении состояния системы число
молей фаз па и nQ остается неизменным, т. е.
л/ = const (/ = а, р). (11.142)
Рассмотрим возможные возмущения А£/ при соблюдении следующих
дополнительных условий:
Ф$ = nasa + /tM - Cs = 0;
фу = nava + rfiv& — Cv = 0;
фп =ла + /гр —Сл =0,
(11.143)
Здесь s и v — молярные энтропия и объем, определяемые по уравнениям:
s = S/n; v = V/n. (II.144)
На основании уравнений (11.143) с учетом условия (11.142) найдем
na6sa + /грб5Р = 0; 1
na6va + nhv* = Q; (II.145)
6ла = 6>£ = 0. J
При равновесии для всех возможных возмущений вариации первого порядка
равны нулю. В результате получаем условие устойчивости в следующем виде:
«Ч/-4-
^•M4^w+^.W]>'
(11.146)
46
Используя равенство U = паиа + пРи? (и— молярная внутренняя
энергия) и соотношения (11.145), запишем неравенство (11.146) в развернутом виде:
62{/=-±-п«*>2
1 dV
i aV l aV
1 дЧ
](Ssa)2 +
/ i d2ua i aV \ . „ 21
(11.147)
Так как условие (11.147) должно быть справедливым для любых значений па
и tv , то, подставив па = пг = 1, получим критерии устойчивости в явном виде:
aV/as<a»2 + dV/ds<P)2>o;
PiP/do(ot)2 + д*иР/до<®2 > 0;
Лв
+
dW
*<«)»т 5s<P>2 \ dv™2^ dvW*
dV*
Л*
*V
i2„p
asaau'
Д +
d*«'
(11.148)
(11.149)
>
>o.
(11.150)
Для устойчивости всей системы в целом необходимым условием является
устойчивость отдельных фаз. Следовательно, согласно достаточному критерию
устойчивости фаз относительно непрерывных изменений состояния, должны
выполняться неравенства:
dsW
dV70s<a>2>O; dVVds<P>2>0;
д*иа/до<а)2 > 0; d WP>2 > 0;
Ла
dV
dsi№ dvi№
dsadva
d\* \2
>0;
ds*dv{
>0.
(11.151)
(11.152)
Очевидно, что из неравенств (11.151) следует неравенства (11.148) и (11.149),
а из неравенств (11.151) и (11.152) вытекает также и неравенство (11.150).
Для теории гетерогенных систем существенный интерес представляет связь
между изменениями сопряженных интенсивных и SKereHwHBHbix параметров,
отнесенных не к отдельным фазам, а ко всей гетерогенной системе в целом. Эта
связь, как показано Сторонкиным, устанавливается необходимым условием
устойчивости гетерогенных систем [13]:
dT dSg-dp dV* + d\xx dnf + ... + d\ik dn%> 0.
(11.153)
Неравенство (11.153) устанавливает ограничение на изменения интенсивных
параметров (7\ р, \iv ..., fifc)> относящихся в силу условия равновесия фаз как
к отдельным фазам, так и к гетерогенной системе в целом, и экстенсивных
параметров (Sg, yet net ##м n|), относящихся лишь к гетерогенлой системе в целом.
47
Согласно условию (11.153), для сопряженных термодинамических
параметров (кроме давления и объема) справедливо
(дХ£/дУ()а> 0, (11.154)
где Yf — экстенсивное свойство гетерогенной системы.
Для давления и объема справедливо следующее неравенство:
(dp/dVe)a<0. (II.155)
Из выражений (11.154) и (11.155) следует, что сопряженные интенсивные и
экстенсивные параметры гетерогенной системы всегда изменяются симбатно,
за исключением давления и объема, изменяющихся в противоположных
направлениях. Это положение справедливо, если система находится в состоянии
устойчивого равновесия, если протекающие в системе процессы вызывают изменения
состояния фаз, и, наконец, если при этом в каждой паре сопряженных
параметров, за исключением той, которая изменяется, один параметр постоянен.
Неравенства (11.153)—(11.155) являются необходимыми, но не достаточными
условиями устойчивости относительно непрерывных изменений состояния,
поскольку может представиться такой случай, когда эти неравенства будут
выполнены, а состояние гетерогенной системы будет неустойчивым. Однако, если
гетерогенная система находится в состоянии устойчивого равновесия и если
протекающие в ней фазовые процессы вызывают изменение состояния фаз, то
условие (11.153) и его следствия (11.154) и (11.155) обязательно выполняются.
Частным, но важным случаем условия (11.154) является неравенство
(д^/дп?) g>0. (ПЛ56)
Неравенство (11.156) справедливо для гетерогенных систем, у которых число
компонентов больше числа фаз (k > г).
При переходе к молярным долям неравенство (11.156) можно представить
в виде
<**«W.f «f_,M.-W"S Л-,1>1>0- 0МИ)
Согласно выражению (11.157), химический потенциал и общая молярная
доля данного компонента гетерогенной системы изменяются симбатно при
условии постоянства температуры, давления и отношения общих молярных долей
остальных компонентов. По физическому смыслу наложенных условий
неравенство (11.157) имеет более общее значение, чем неравенство (11.156), так как
в нем условие постоянства числа молей компонентов заменено менее жестким
ограничением — условием постоянства отношений молярных долей. Очевидно,
если выполняется неравенство (11.157), то, как следствие, соблюдается и
аналогичное неравенство (11.156).
В заключение отметим, что расчеты, связанные с использованием уравнений
кривых моновариантных равновесий, обычно требуют привлечения тех или иных
модельных представлений (отсутствие растворимости, идеальный или
регулярный раствор), что, несомненно, снижает ценность результатов. Применение
условий устойчивости при анализе уравнений фазовых равновесий позволяет
без каких либо допущений в некоторых случаях выявить интервал возможных
значений параметров состояния (см. например, работу Тойкки и Сусарева [23],
в которой показано применение условий устойчивости для выявления
концентрационной области расположения эвтектик и точек минимума температуры
кристаллизации в системах твердое тело—жидкость).
46
6. ПРИНЦИП СМЕЩЕНИЯ РАВНОВЕСИЯ
(ПРИНЦИП ГИББСА—ЛЕ-ШАТЕЛЬЕ)
Вопрос о направленности течения процесса при внешнем
воздействии на систему является одним из наиболее кардинальных
в химической термодинамике, так как на его основе в конечном
итоге решается проблема наиболее целесообразного варианта
проведения того или иного процесса.
Простейшая формулировка принципа смещения равновесия
была дана Ле-Шателье (1884 г.)- Суть ее сводится к тому, что
система, находящаяся в равновесии, реагирует на внешнее
воздействие таким образом, чтобы уменьшить это воздействие. Такая
формулировка, по существу, охватывает принцип стабильности.
В простейших случаях принцип Ле-Шателье эквивалентен
условиям стабильности и практически дублирует принцип
наименьшего принуждения, сформулированный еще в работах Даламбера
и Гаусса. Однако ограниченность и неточность данной
формулировки с позиций химической термодинамики может в ряде случаев
привести к совершенно неправильным заключениям.
Браун (1887 г.), опираясь на условия устойчивости равновесия,
попытался дать более строгое обоснование принципа Ле-Шателье.
Однако практически не улучшил существа дела.
Имеется мнение, что всеобъемлющая формулировка этого
принципа, чрезвычайно сложна. Поэтому И. Пригожий и Р. Де-
фэй [24 ] считают более логичным обсуждать возникающие в связи
с этим проблемы в терминах соответствующей теоремы модерации,
которая дает однозначный ответ и которую можно вывести,
опираясь на понятие химического сродства по Де-Донде [25]. В этом
смысле можно считать, что в неравенстве Де-Донде (A'v > 0)
отражена наиболее общая формулировка принципа Ле-Шателье [24].
Анализ процессов, послуживших основанием для вывода
принципа Ле-Шателье—Брауна, позволил П. С. Эпштейну [26]
сделать заключение, что они относятся к двум совершенно
различным типам процессов смещения равновесия и, следовательно, сам
принцип должен распадаться, по крайней мере, на два различных,
не связанных между собой правила. При этом процессы,
связанные с превращением и переходом веществ (химические и фазовые
процессы), принадлежат к одной группе, а процессы, связанные
с участием так называемых вторичных сил — к другой.
Детальный анализ обеих групп процессов содержится в
монографии А. В. Сторонкина [13]. Последовательное рассмотрение
смещения химических и фазовых равновесий приводит к
следующим выводам.
Математически принцип смещения химического равновесия
в самой общей форме можно представить в виде следующих
неравенств:
(д\пК/дХ{)х.ф.*^0у если ДГ^О, . (II.158)
где /С — константа равновесия реакции типа (11.85).
49
При постоянных давлении и напряженности внешних силовых
полей (электрического и магнитного) справедливо
(d\nK/dT)p,xt^Ot если Д#°^0, (11.159)
где Д#° — стандартная теплота реакции при постоянных Тир.
Неравенства (11.158) и (11.159) справедливы и для гомогенных,
и для гетерогенных химических реакций.
Смысл неравенств (11.158) заключается в том, что изменение
обобщенной силы Xt (кроме Xt = Т) при постоянстве остальных
сил вызывает смещение химического равновесия в таком
направлении, при котором изменение сопряженной обобщенной
координаты ДУ7 препятствует изменению обобщенной силы.
Неравенства (11.159) обусловливают то, что с изменением температуры
при постоянных остальных обобщенных силах химическая
реакция всегда смещается в таком направлении, при котором
развиваемый реакцией тепловой эффект препятствует изменению
температуры (принцип смещения подвижного равновесия Вант-
Гоффа). Учитывая противоположный характер направленности
давления и объема, давление в качестве обобщенной силы берут
со знаком минус (Xt = —р). Исходя из этого для давления (при
постоянстве остальных обобщенных сил) неравенства (11.158)
принимают вид:
(dln/C/dp)r.x,saO, если ДУ°^=0. (11.160)
Смысл неравенств (11.160) сводится к тому, что при повышении
давления химическое равновесие сдвигается в сторону
уменьшения объема системы и наоборот.
В случае фазовых равновесий константу равновесия К можно
рассматривать как коэффициент распределения /-того вещества
между равновесными фазами. В этом случае справедливы
следующие неравенства, характеризующие смещения равновесного
распределения отдельных веществ между сосуществующими фазами
под воздействием внешних сил:
(dfoKi/dXjh^. SO, если Д#,а0; (11.161)
(д In Ki/dT)Pt xk S 0, если Мг\ S 0, (II. 162)
где Д*/,; — стандартные парциальные мольные величины.
По своему физическому смыслу неравенства для фазовых
равновесий (11.161) и (11.162) совершенно аналогичны неравенствам
(11.158) и (11.159) для химических равновесий.
Поэтому можно дать общую формулировку принципа смещения
равновесия, связанного с превращением и переходом веществ
в результате химических или фазовых взаимодействий. При
изменении обобщенной силы Xt и постоянстве остальных обобщенных
сил химическое и фазовое равновесия смещаются в том
направлении, при котором сопряженная обобщенная координата Yt
испытывает приращение, противодействующее изменению обобщенной
50
силы [131. В такой формулировке принцип смещения справедлив
для истинного динамического равновесия и неприменим к так
называемым «замороженным» или «ложным» равновесиям. Кроме
того, он справедлив только в случае воздействия одной какой-
нибудь силы при фиксировании всех остальных и при учете
возможных изменений знаков тепловых, объемных и других эффектов,
сопровождающих рассматриваемые процессы.
Рассмотрение процессов, протекающих при участии так
называемых вторичных сил, под которыми следует понимать силы,
индуцируемые под действием первичных (непосредственно
первоначально воздействующих на систему) сил, впервые приведено
в работах Гиббса [1 ] и связано с условиями устойчивости системы
относительно непрерывных изменений состояния.
Если следовать логике рассуждений Гиббса 11] и выражать
массы компонентов, как это принято в данной работе, через число
молей, то можно получить следующее неравенство:
{d\kkldnk)Ht v, Й1,.... н_х > (d\Lk/dnk)T, v\ цг..., H_t > 0. (11.163)
Неравенство (11.163) выражает так называемый сокращенный
[21 ] принцип Ле-Шателье—Брауна относительно параметров \ik
и nk для гомогенных процессов, протекающих с участием
вторичных сил.
В литературе часто приводится формулировка принципа Ле-
Шателье—-Брауна на основе вывода Эренфеста [27], согласно
которой внешнее воздействие, выводящее гомогенную систему из
равновесия, стимулирует в ней процессы, стремящиеся ослабить
результат этого воздействия. Подчеркивая недостаточность этой
формулировки из-за отсутствия указаний относительно условий
проведения процесса смещения равновесия, А. В. Сторонкин
отмечает [13], что она существенно проигрывает в общности
формулировке Гиббса, данной в виде (11.163).
Заслуга Гиббса в установлении принципа смещения
равновесия была показана впервые Дюгемом [28], а не А. И. Русановым
и М. М. Шульцем, как это ошибочно отмечается в работах [13,
29]. Роль Гиббса в развитии принципа смещения была хорошо
известна самому Ле-Шателье, который по данному поводу
остроумно заметил, что между решением частных задач отдельными
исследователями и общим методом решения Гиббса такая же
разница, как между решением задач на экстремум древними греками
и дифференциальным исчислением. Однако в первом случае общий
метод был создан до появления частных решений, тогда как во
втором он был разработан на основе обобщения частных
решений (см. [30]).
Из приведенного высказывания Ле-Шателье следует не только
сам факт его осведомленности о роли Гиббса в решении
рассматриваемой проблемы, но также и четкое понимание соразмерности
степени общности собственной и гиббсовской формулировок.
51
В связи с изложенным, а также учитывая, что более общая
формулировка была дана Гиббсом по крайней мере на десять лет
раньше, чем вышла работа Ле-Шателье, принцип смещения
равновесия с полным основанием следует называть принципом Гиббса—
Ле Шателье.
Последовательный анализ неравенства Гиббса позволил
авторам работ [13, 29] получить обобщенное выражение принципа
смещения равновесных гомогенных процессов с участием
вторичных сил в виде
{dYjdX^ > (д¥{/дХ{)а> > > (dYt/dXda^ > {dYjdX^
(11.164)
где % = Yly X2j . . ., Xi_Xi Хиъ . . ., Xt\
a2 = Yly Y2, X3, ..., X;_1} Xi+V ..., Xt\ fl/_1=/1, ..., Y(_Ly
Yi+x> . . ., Yt_lt Xt\ at=Y1% . . ., Yi_x, Yi+L, . . ., Yt.
Из рассмотрения неравенства (11.164) следует, что по мере
замены условий постоянства обобщенных сил условиями
постоянства соответствующих сопряженных параметров производная
dYjdXi уменьшается, поскольку при этом возрастает число
вторичных сил, противодействующих влиянию первичной силы
на состояние равновесия. Действующие вторичные силы являются,
очевидно, функциями первичной силы X,, поскольку все
производные в цепочке (II, 164) описывают моновариантные процессы
смещения равновесия.
Таким образом, при моновариантном процессе смещения
равновесия интенсивность воздействия первичной силы на
сопряженный параметр уменьшается по мере сокращения числа
фиксированных других обобщенных сил среди (t — 1) фиксированных
параметров, где t — число независимых параметров.
Учитывая, что принцип смещения равновесия с участием
вторичных сил является прямым следствием критериев
устойчивости, которые, как было отмечено, приложимы к гетерогенной
системе, то, очевидно, смещение равновесия этой системы
целесообразно рассмотреть по аналогии с гомогенными системами
на указанной основе.
Нетрудно показать, что термодинамический потенциал
Гиббса Gg, относящийся к гетерогенной системе в целом,
обладает теми же свойствами по отношению к переменным Sg, Vg
и nf гетерогенной системы, что и рассмотренная Гиббсом
соответствующая функция для гомогенной системы. Следовательно,
вывод неравенств, характеризующих принцип смещения равновесия
гетерогенных систем с участием вторичных сил, вполне
аналогичен выводу для случая гомогенных систем.
52
При этом, однако, в случае гетерогенной системы необходимо
учесть требование, согласно которому фиксированные величины
следует выбирать таким образом, чтобы рассматриваемая
гетерогенная система была моновариантной [13]. В соответствии с
правилом фаз Гиббса это требование выполнимо при условии, что число
фаз меньше числа компонентов гетерогенной системы.
С учетом этого требования по аналогии с гомогенной системой
можно представить принцип смещения равновесия гетерогенной
системы с участием вторичных сил в виде следующей цепочки
неравенств:
(dYf/dXdaf > (dYf/dXt)^ > >
> [midXt)aU > (dYfJdXt)af9 (11.165)
где параметры af, af и т. д. расшифровываются точно так же, как
и в случае неравенств (11.164), но относятся к гетерогенной системе
в целом (на что указывает индекс «£»).
Фигурирующие в неравенствах (11.165) обобщенные
координаты Y\g) имеют характер брутто-величин, также относящихся
ко всей гетерогенной системе в целом. В соответствии с
ограничительным условием, согласно которому число фаз меньше числа
компонентов, цепочки неравенств (11.165) короче цепочки
неравенств (11.164).
Рассмотренные случаи сдвига равновесия иллюстрируют
достаточно сложный характер содержания принципа Гиббса—Ле-
Шателье, однако они дают основание для конкретного анализа
различных систем. В случае гетерогенных фазовых равновесий
возможна еще большая детализация данного принципа и, в
частности, интерес представляет рассмотрение принципа смещения
вдоль линии фазового равновесия в двухкомпонентных системах.
7. СМЕЩЕНИЕ ВДОЛЬ ЛИНИИ РАВНОВЕСИЯ.
ОБОБЩЕННОЕ ДИФФЕРЕНЦИАЛЬНОЕ УРАВНЕНИЕ
ВАН-ДЕР-ВААЛЬСА
Смещение вдоль линии равновесия фаз в гетерогенной системе
можно представить в различной форме. Частными выражениями
принципа смещения вдоль линии фазового равновесия являются
законы Гиббса—Коновалова [1, 31], законы Вревского [32]
и т. п. В наиболее общей форме рассматриваемый принцип дан
Ван-дер-Ваальсом [12], который получил дифференциальное
уравнение двухфазного равновесия в двухкомпонентной системе. Это
Уравнение впоследствии было обобщено А. В. Сторонкиным на
многокомпонентные системы [13, 33].
Уравнение Ван-дер-Ваальса в сочетании с условиями
устойчивости, выведенными Гиббсом, позволяет дать исчерпывающую
характеристику термодинамических свойств двухфазных систем
53
[33]. На его основе возможны рассмотрение и анализ диаграмм
состояния, в связи с чем остановимся более подробно на
обосновании этого уравнения.
Простой и наглядный вывод уравнения Ван-дер-Ваальса можно
дать из фундаментальных уравнений Гиббса для первой и второй
фаз:
s«dT ~vadp + dp? + х? {drf - rfjif) = 0; (11.166)
sp AT -vbdp + d\x\ + x\ (dfif - d|if) - 0. (II. 167)
Вычитая первое уравнение из второго и учитывая, что, согласно
выражению (11.39),
^2 - Hi = №/дх2)Рщ Ту (11.168)
получим
(sp - sa) dT - (ир - va) dp + (4 - 4) d (dg/dx2)Pt т = 0.
(11.169)
Распространяя рассмотрение на многокомпонентные
двухфазные системы А. В. Сторонкин получил аналогичное соотношение
в виде
№ - va) dp = (sp - sa) dT + *i (*? - *?) d (d*/e*,)p. г. ;/вМ.
(11.170)
Согласно выражениям (11.169) и (11.170), дифференциал
d (dgldXi)Pt т, xi=hj может быть отнесен как к первой, так и ко
второй фазам. В соответствии с этим дифференцирование при записи
указанного дифференциала в развернутом виде должно
проводиться по переменным состояния или первой, или второй фаз.
Так как частные производные (dg/dxi)PtT,x:4:i для каждой фазы
имеют свой вид функциональной зависимости от параметров
состояния фазы, то из формул (11.169) и (11.170) вытекают два
независимых дифференциальных уравнения. Условия равновесия
многокомпонентной двухфазной системы будут выражаться
следующей системой дифференциальных уравнений:
(0э _ „«) dp = (sp _ sa) dT + T (*? - x?) d (dg/dxtf;
fe-1
(op - va) dp = (sp - sa) dT + Б (x? - x?) d (dgfdxtf; (11.171)
d (dg/дх,)*, т. x.t+i = d (dg/dxt)l т. xl+t.
54
В последних уравнениях i принимает значение от 1 до (k — 1),
кроме i = sy где s — номер лишнего уравнения. Нетрудно
показать [10, 13, 33], что если уравнение
d (dg/dxt)l Tt Х1Ф& = d (dg/dxfp, г. Х}ф5 (IIЛ72)
является лишним, то оно должно вытекать из уравнений системы
(11.171).
Раскрывая дифференциалы d (dg/dx() последние (k—2)
уравнения системы (11.172) можно записать в развернутом виде
следующим образом:
[{dsjdxif - (ds!dxt)*\ dT - [(dv/дх^ -
- (dv/dxi)*] dp-D (dgfdxt)* + D (dg/dxtf = 0, (11.173)
где D(dg/dxt) = ^(d'g/dxidx,)dxi (11.174)
— дифференциал по составу.
Используя представления о парциальных мольных величинах
(см. гл. 1), можно записать
(ds/dx^T, р, х.ф1 = Si - sk. (II.175)
Отсюда (ds/dxtf - (ds/dx()a = (§? - s?) - (& - S?) =
= [bR\*+*>-Mi*+to)/T. (11.176)
Здесь Ай[а">Р) и А/Ца"*Р) — парциальная мольная теплота перехода
1-того и fe-того компонентов из а- в |5- фазу.
Аналогично нетрудно убедиться, что
(dv/dxUT.^x.+^Vt-Vb. (II.177)
Далее (dv/dxtf - {dv/dxt)a = М)^ - AtJJT p\ (H. 178)
где
(11.179)
ду(а->р) — приращение парциального молярного объема /-того
компонента при переходе из а- в р-фазу.
55
С учетом соотношений (11.173)—(11.179) систему
дифференциальных уравнений (11.171) можно записать следующим образом:
*-1
р - vat - I! (4 - х?) (do/ftr,)"] dp =
= р - sa - *Г (*? - xf) (ds/dxt)«] dT +
+ Б S (4 - х?) [<%(<>*,• ах,)] <**?;
i=i /=i
|> _ у« _ *|! (jc? _ х?) (&«р] Ф =
= Ь - sa - *|! («? - xf) (Os/дхА dT
+
1=1 J
+ SI (4 -*?) iSfg/idXidxi)] dxf, (11.180)
[(Ай{^Р) - Ай^р))/Л dr + (Aitf**P) - Av^V) dp -
- d (З^)*5 + d (ag/a^)a = o,
где I = 1, ..., s— 1, s+1, ...,£—1.
Два первых уравнения системы (11.180) названы Сторонки-
ным обобщенными дифференциальными уравнениями Ван-дер-
Ваальса, а последние (k — 2) уравнения — дополнительными
условиями равновесия. Два первых уравнения являются более
общими и позволяют лучше раскрыть термодинамические
закономерности гетерогенной системы по сравнению с дополнительными
условиями. Из первых двух уравнений системы (11.180) следует, что
для однокомпонентной двухфазной системы
(ф - xf*) dp = (sP - s<*) dT, (11.181)
т. е. получаем известное уравнение Клапейрона—Клаузиуса.
Для двухкомпонентной двухфазной системы находим уравнение,
выведенное впервые Ван-дер-Ваальсом:
\xfi _ rj* — (*Р — ха) (dv/дх)] dp = [sP - sa — (*P — x*)x
X (ds/дх)] dT + (d2g/dx2) (*P - x*) dx. (11.182)
Производные dv/дх, dsldxy d2g/dx2 и дифференциал dx должны
относиться или к а-, или к Р-фазе.
Физический смысл множителей Да и As при dp и dT
соответственно в обобщенном уравнении Ван-дер-Ваальса заключается
в том, что они характеризуют изменение объема и энтропии
двухфазной системы при изобарно-изотермическом образовании одного
моля р-фазы из бесконечно большого количества а-фазы, т. е.
56
являются характеристиками процесса, несовместимого с
условиями равновесия между фазами [12—13, 33].
В работе [34] дана другая наглядная интерпретация
физического смысла коэффициента As, связанная с равновесным
превращением а-фазы в р-фазу. Согласно полученному авторами этой
работы выражению, As определяется следующими
экспериментально измеримыми величинами: дифференциальным тепловым
эффектом равновесного изобарического образования р-фазы в
системе, содержащей первоначально только один моль а-фазы,
температурным эффектом фазового процесса и молярной
теплоемкостью а-фазы.
Физический смысл коэффициентов при dxj в первых двух
уравнениях системы (11.180) сводится к разности фазовых эффектов
для /-того и &-того компонентов, характеризующих влияние изо-
барно-изотермического изменения состава фазы в результате
фазового процесса (при движении фигуративной точки состава по
конноде) на химические потенциалы компонентов в данной фазе.
Производные химических потенциалов по составу характеризуют
эффекты растворения и выделения при фазовом процессе.
Фазовые эффекты являются мерами эффектов растворения и
выделения (или, как отмечается в работе А. В. Сторонкина [13] «всали-
вания и высаливания») при изменении состава фаз в результате
фазовых процессов (по коннодам).
Очевидно, тепловые эффекты, связанные с фазовыми переходами
а —> р и Р —> а, можно представить как
q{*+fi) = т&а+М\ q{^d) = Ts^^\ (11.183)
где <7(а->Р) и q{^a) — дифференциальная мольная теплота
образования р-фазы из a-фазы и наоборот, соответственно.
Эти величины по абсолютным значениям не равны между собой,
так же как и соответствующие рассматриваемым переходам
объемные изменения и(а_>Р) и а(Р_>а), поскольку они отвечают процессам,
имеющим одно и то же исходное, но различные конечные
состояния, и, следовательно, не являющихся обратными по отношению
одно к другому [13].
Обычно в расчетах уравнение Ван-дер-Ваальса может быть
использовано, если на основе модельных представлений известен
вид зависимости изобарно-изотермического потенциала фазы от
переменных состояния. Его применяют и в том случае, когда
коэффициенты уравнения (11.182) разлагаются в ряд в окрестности
некоторой точки диаграммы состояния и расчеты проводятся по
локальным уравнениям различного приближения [35—38].
Если двухфазная многокомпонентная система удовлетворяет
лишь условиям частичного равновесия (вследствие того что в
присутствующих фазах имеется неодинаковое количество веществ
или по каким-то причинам не реализуется какое-либо условие
межфазового равновесия), то применение обобщенного дифферен-
57
циального уравнения Ван-дер-Ваальса встречает некоторые
затруднения. В работе А. Н. Мариничева и А. В. Сторонкина [39]
получено модифицированное обобщенное дифференциальное
уравнение Ван-дер-Ваальса, позволяющее описывать гетерогенные
системы, находящиеся в частичном равновесии, а также состоящие,
например, из двух частей, разделенных полупроницаемой
мембраной, т. е. уравнение, пригодное для описания систем^самого
различного типа. Предлагаемое в работе [39] уравнение при
условии полного термодинамического равновесия и присутствия
в сосуществующих фазах одинакового количества веществ
переходит в обобщенное дифференциальное уравнение Ван-дер-Ваальса.
8. ОБ АДЕКВАТНОСТИ РАЗЛИЧНЫХ ПОДХОДОВ
К ВЫВОДУ ПРИНЦИПА СМЕЩЕНИЯ ВДОЛЬ ЛИНИИ
РАВНОВЕСИЯ
При выводе принципа смещенного равновесия в термодинамике
явно обозначались два различных подхода.
Один подход, получивший развитие в работах Гиббса [1],
Планка [2], Ван-дер-Ваальса [12], а также А. В. Сторонкина
[33] основывается на принципе равновесия Гиббса в приложении
к гетерогенной системе с неизменяющимися массами компонентов.
Другой подход к выводу принципа смещенного равновесия
содержится в работах Шоттки [20], Пригожина и Дефэя [24].
Этот подход базируется на понятии функции химического
сродства или аналогичной ей, которая не предполагает строгой
материальной изоляции системы и, следовательно, может быть
применена к открытым системам (т. е. возможен обмен массами
компонентов с внешней средой). В. К. Семенченко [40], хотя и не
указывает на использование понятия функции сродства, однако по
логике вывода пользуется им в неявной форме.
Ван-дер-Ваальс [12] и А. В. Сторонкин [33], применяя
методологию Гиббса, получил дифференциальное уравнение
двухфазного равновесия в двухкомпонентной и поликомпонентной
системах.
Аналогичные по содержанию, но отличающиеся по форме
уравнения представлены в работах [20, 24, 40]. Они существенно
отличаются по виду от уравнения Ван-дер-Ваальса, поэтому
возникает вопрос о взаимоотношении двух указанных подходов к
выводу принципа смещенного равновесия.
В работе В. К. Семенченко [40] отмечается, что
математическая формулировка принципа смещенного равновесия сводится
к нахождению условий равновесия, т. е. к варьированию одного
из термодинамических потенциалов при постоянстве всех
термодинамических сил и последующему дифференцированию по этим
силам (т. е. к смещению равновесия), тогда как выведенные
Ван-дер-Ваальсом условия смещенного равновесия в форме (11.182)
содержат двукратное дифференцирование по температуре и дав-
58
лению. На основании этого сделано заключение [40], что
уравнение (11.182), а также его обобщения на любое число сил и
компонентов неверны.
Взаимоотношение двух указанных подходов к выводу
принципа смещения вдоль линии равновесия рассмотрено ниже на
основе нашей работы [41 ]. Покажем, что несмотря на внешние
различия результаты выводов принципа смещенного равновесия при
использовании двух указанных подходов на самом деле адекватны.
Возьмем для наглядности двухкомпонентную систему.
При втором подходе, согласно Пригожину и Дефэю [24],
выражения для дифференциалов сродства можно представить
в виде системы уравнений;
dA[ = djif - d|if, dA2 = dp? - d\&. (11.184)
Поскольку \ii = / (7\ p, x9) для полного дифференциала
химического потенциала с учетом соотношений (11.22) и (11.23)
можно записать
d[it = —St dT + бi dp -f (d\ii/dx2)Py T dx2. (II. 185)
После подстановки уравнения (11.185) в выражение (11.184)
с учетом (11.168), и имея в виду уравнение Гиббса—Дюгема вида
(11.36) и равенство сродства нулю вдоль линии равновесия,
получим
Д*ГР) dT - А £>|°~Р) dp - х% (-ЦТ d$ +
\ (м2 /р,Т
+ *(Ш,.т** = °'> С"-186)
Д*ГР) dT - ДбГ*> dp + {\- х2") (%)apT dx% -
-o-^(4s-):.r^»°.
где &s\a^] = S? - gf; Av\a^} = б? - vf. (11.187)
Умножая первое уравнение системы (11.186) на х[9 а второе на х!2
можно исключить переменные dx\ или dx%. В результате будем
иметь
(4 Ы{ГЬ) + 4 Ai#~P)) dT - (4 Av[a^ + 4 A^P)) dp -
-№-x§)(d2g/dx!)adxZ = 0;
(x? AS}**» + *? Д4°~Э)) dT - (xf A^"* + (II. 188)
+ x? Д t»^P)) dp - (*? - 4) (d2g/dxlf dx% = 0.
Как видим, уравнения (11.188) также содержат в себе полное
термодинамическое описание двухкомпонентных двухфазных
систем.
59
В работе В. К. Семенченко [40] на основании использования
аналогичного подхода было получено следующее уравнениег:
(m? &sx + mf As2) dT — (mf Д 61 -f m§ Д 62) dp +
+ (m\n% - m?mf) фа dca = 0, (II. 189)
где m — число молей i-того компонента;
ca = mf/m?\ Фа = (dlH/dmx)lTtma. (II. 190)
Уравнение (II. 189) также дает полное термодинамическое
описание двухфазного равновесия в двухкомпонентной системе. Для
доказательства адекватности двух рассмотренных подходов к
выводу принципа смещенного равновесия вначале покажем, что
уравнение (11.189) эквивалентно первому уравнению системы
(11.188). Перейдем от переменных состава в виде чисел молей
к переменным состава в виде мольных долей.
Выразим производную (d^dm^T через (д\1г/дх^, г- По
аналогии с выражением (11.32) для рассматриваемого случая будем
иметь
Ф\ц1дтх)1 т = (дрг/дхд* [m?7(m? + m2a)2]. (II.191)
Подставим равенство (11.191) в выражение (11.189):
(mf Asi + ml As2) dT — (m? Avx + m\ Av2) dp +
4- (д\Ы/дхх)% тT2 (m?*2a - wP2xf) d (xx/x2)* = 0- (II. 192)
Разделив равенство (11.192) на (mf + mf) и приняв во
внимание, что
d(xlfx2)a = — (l/xia))2dx?, (II.193)
найдем
(х? A5i + 4 As2) dT - (jcf Л61 + 4 Аб2) dp -
- (l/x2a) (0^/3*0?,r (*?*" - *!!*?) dx2a = 0. (IIЛ94)
На основании выражения (11.168) получим
(&8/Э4)Р.т = (3ni/&fi),.г - (Зцз/ахОр.г. (И. 195)
Из уравнения Гиббса—Дюгема
(д\12/дхг)р, т = [- (1 - х2)/х2] • (а^/5^, г. (И. 196)
1 В уравнении (11.189) и его производных сохранена символика,
приведенная в работе [40].
60
После подстановки равенства (11.196) в выражение (11.195)
получим
№Л)р,г= -х2 (d2g/dxl)PtT (11.197;
или (dni/d*i)p,т = х2(d2g/dxl)Ptт. (П. 198)
Подставим соотношение (11.198) в выражение (11.194) и с
учетом того, что х{х% — х$х% = х% — л|, придем к первому
уравнению системы (11.188). В этом пункте можно констатировать
идентичность подходов и конечных результатов работ [20, 24, 40].
Чтобы привести уравнение (11.188) к уравнению Ван-дер-Ваальса
(11.182), преобразуем множители при dT и dp.
С учетом значений Д$<а^Р> и Д$<а-^> из выражения (11.187),
принимая во внимание соотношение (1.14), можно множитель
при dT первого уравнения системы (11.188) представить в виде
(4 Asia"P) + 4 Д*ГР)) = s* - jrfs? - *f *?• (И. 199)
Прибавим к выражению (11.199) и вычтем из него
sa=xasa + x?sa. (II.200)
После несложных преобразований получим
(4 д*ГЭ) + 4 л^Р)) = sp - $а - (4 - *?) (sa - sf).
(11.201)
Согласно уравнению (11.175),
(s? - sa) = (ds/d*2)?. г- (Н.202)
Следовательно, окончательно множитель при dT в первом
уравнении системы (11.188) примет вид
(4 Mi**** + 4 Д«^Ю) = sp - sa - (4 - x2a) (ds/dx2)aPt т.
(11.203)
Нетрудно видеть, что полученное значение множителя при dT,
согласно выражению (11.203), отвечает соответствующему
множителю в уравнении Ван-дер-Ваальса (11.182). Аналогично
преобразуется и множитель (л;РДб{а^>л| Дб<а-^>) при dp в уравнении
(11.188). В итоге оказывается, что уравнение (11.189) сводится
целиком к уравнению Ван-дер-Ваальса (11.188). Учитывая
доказанную эквивалентность уравнений (11.188) и (11.189), можно
заключить, что наиболее отличное по форме уравнение Семенченко
(11.189) также сводится к уравнению Ван-дер-Ваальса (11.182).
Отсюда следует, что несмотря на отмеченные различия в подходах
при выводе принципа смещенного равновесия в двух указанных
группах работ, получаемые конечные результаты вполне
адекватны. Это может служить подтверждением того, что между
открытыми и закрытыми системами в термодинамическом отношении
нет никакого различия. Они различаются лишь в возможностях
61
изменения своих составов, которые для открытых и' закрытых
систем подчинены различным условиям материальной изоляции
[11, 13].
Однако, если сравнивать уравнения в форме (11.188) с
уравнениями Ван-дер-Ваальса, особенно применительно к
многокомпонентным системам, то, безусловно, следует отдать предпочтение
последним.
В отношении многокомпонентных двухфазных систем метод
Ван-дер-Ваальса и метод, основанный на анализе сродства, также
приводят к одному и тому же результату, однако метод Ван-дер-
Ваальса в форме, развитой А. В. Сторонкиным [13, 33], отличается
изяществом и простотой при использовании. Покажем это на
примере трехкомпонентных двухфазных систем.
Дифференциалы химических потенциалов в случае трехком-
понентной системы будут иметь вид:
d\it = — Si dT + vtdp -f (д\зц1дх^р% T, xt dx1 + (д\1,-/дх2)р, т, Xl dx2y
(11.204)
а для дифференциалов сродства можно записать
d4=d|i?-;d|i?. (II.205)
Вдоль линий равновесного сосуществования двух фаз
сродство равно нулю, и эти линии удовлетворяют уравнениям вида
(11.205), если положить в них правые части равными нулю.
Таким образом, для описания условий равновесия в трехкомпонент-
ной двухфазной системе в дифференциальном виде будем иметь
следующую систему уравнений:
dtf - d|if = As[a^ dT - Д v[a^ dp + (дц/дхг)* _ « dx? +
P» * t л2
+ №\ldx2)a a dxt - (dpildxt£ . «dx\ -
P» J • *1 P> l • *2
-(дцх/дхд* _ хфЬ
Pt * > *i
d$ _ d\$ = As^p) dT - At#~w dp + (11.206)
+ (бца/агО" _ xa dx? + (d^dXit « dxt -
Pt * t *2 "' ' 1
-(dtoldxtf _ *dx\-(dv.2idx2f T jdxb
p, i t *2 P' * 1
d$ - d(i§ = ASpw dT - Attf"P) ф + (дць/дхд* т x* dx? 4-
P, J f *2
+ Шдх2)1 r> x«dx? - (faldxtf T> ^ dxl - (dii3/dx2fp r ^ dxl
62
Полученные соотношения можно упростить, используя
молярную свободную энергию Гиббса g.
Согласно равенству (11.39),
(dg/dxx) = \лг- \i3\ (dg/dx2) = \i2- (x3. (11.207)
На основании уравнений (11.207) и соотношения Гиббса—
Дюгема в виде
xi d^i + *2 d\i2 + (1 — *i — х2) d\iu = 0 (11.208)
нетрудно убедиться, что
(д\лх1дхх)р, т.х, = (1 - хг) (d2g/dx\)Pi Tt Хг - х2 (d2g/dxx дх2)р> т\
(d\ii/dx2)Pi т, Хх = (1 - *i) (d2g/dxx dx2)Pf т — х2 (d2g/dx22)Pt Тш Xl\
(d\i2/dxx)p, т,х2 = (1 — х2) (d2g/dxx дх2)р, т — хх (d2g/dx2)Pt ТвХш.
(11.209)
(d\i2/dx2)Pt т,Х1 = (1 — *2) (d2g/dx22)Pi TtXl — хх (d2g/dxx dx2)Pt T\
{d\xzidxx)Pt т,х2 = —xi {d2g/dx2)Pt TtXt - x2 {d2gldx\dx2)Pt т\
(д[хз/дх2)р, T,Xl = —*2 (d2g/dx%)Pt T,Xl — *i (d2g/dxx dx2)Pt r.
Подставив выражения (П.209) в соотношения (11.206), получим
следующую систему уравнений:
ф? - фр = Л*Г Р) dr - А иГР> dp -f 1(1 - xf ) (<fg/<*2)a -
-x? (d2g/dXldx2)a] dx? + [(1 - x?) (d2g!dXl dx2)a -
-~x? (d2g/dxT) Axl - [(1 - x\) {d2gldx\f -xlx
x {d2gidXldx2f\dx\- [(1 -xf)(aV^a.v2)p -
—*§ {d2gldx\f\ dxl = 0; (11.210)
d^2a- d|*| = A s2°"P)d7 - A u2°~P) dp + [(1 - x2a)(^g/ах, dx2)a -
-tf №ajc?)e] dxf + [(1 - x%) (ifg/dxir - *f (SPg/дхг дх2)а) х
x ^ _ [(i _ 4) {d2gldxxdx2Y - x^ifgldx2)*] d£ -
- [(i - x\) {d2gidx22f - jff (a2g/^ a*2)p] d4 = 0;
dtf - d|if = A4a"P) dr - A i^p) dp + [x? {d2gldx\)a - *2a x
x (cfg/дхг dx2)a] dx? + u? (a2^ <fr2)a - *2a (a2g/a^)a] x
x d*? + [*? (a2g/^)p + 4 (d2g/dXl dx2f\ dx\ +
+ [4 (cfr/un <*2)P + 4 (cfg/dx2)*] dx\ = 0.
63
Вычитая третье уравнение системы (11.210) из первого и из
второго уравнений, получим
(Д5<а^> - Д5^ю) dT - (Aoj^P' - Avia^) dp +
+ (д^/дх2)* dx{a) + (d2g/dXl dx2)a dx? - {fgldffl dxt -
— (d2gldxx dx2f dx\ = 0; (11.211)
(Д$»Ю _ AgJ**») dT - (A6^w - Aof*w) dp +
+ (tfg/dXi dx2f dxf + (d?g/dxl)a dx? - (afyud dx2f dx\ -
— (cPg/d$*d& = 0. (11.212)
Умножив уравнение (11.211) на х\, а уравнение (11.212) на х$
и сложив с третьим уравнением системы (11.210), окончательно
найдем
[4№***> - ASS"^) + 4.(A^P) - Asi°*w) + ASi^w]dT-
— U? (Aeia^> - де^>) + 4 (де^Р) - A6ia^>) +
+ AvJTwl ^ + (*? - *?) l- №*! <*2)a +
+ (4 - x?) [(d2g/dx22)a - (d2g/dxx dx2)a] dx? = 0. (11.213)
Как видим, для получения окончательного результата на
основе использования функции сродства уже для трехкомпонентной
системы требуются довольно сложные расчеты. В уравнении
(11.213) также дано полное термодинамическое описание
двухфазной трехкомпонентной системы.
Между тем для получения уравнения, содержащего
аналогичную информацию, — мы можем использовать обобщенное
уравнение Ван-дер-Ваальса—Сторонкина. Действительно, положив число
компонентов, равное трем, на основании уравнений (11.180) будем
иметь
[SP _ s« _ (4 _ х«) (ds/dxx)a - (x$ - x2a) (ds/dx2)a] dT -
_ [^ _ t,« _ (xf _ x«) (dv/dxx)a - (xf - jc?) (dv/dx2)a]dp +
+ (*f - *?)T(dfy0*Da - №/дхгдх№ dx?+ (4 - x%) x
x l(#g/d4)a - (d2g/d.*1/d*2)a] ж# = o. (11.2H)
Уравнения (11.213) и (11.214) вполне адекватны, поскольку
уравнение (11.213) целиком сводится к соответствующему
уравнению Ван-дер-Ваальса—Сторонкина (11.214). Докажем это так же,
64
как это было сделано для двухкомпонентной системы и преобразуем
множители при dT и dp. Тогда
Л (я? -«?) - 4 (si - в?) + 4 tep ~ *?) -
X (<fe/A*i)a - (4 - *2a) (ds/dx2)a. (11.215)
Так же можно преобразовать множитель и при dp. На
основании уравнений (11.213), (11.215) и аналогичного (11.215)
выражения для множителя при dp получим окончательно выражение
(11.214), что и требовалось доказать.
Таким образом, можно сделать вывод, что метод, развитый
Ван-дер-Ваальсом и А. В. Сторонкиным [12, 13, 33], вполне
корректен. Это удобный и универсальный метод, дающий реальные
возможности для анализа гетерогенных равновесий.
Глава III
ФАЗОВЫЕ РАВНОВЕСИЯ
В ДВУХКОМПОНЕНТНЫХ СИСТЕМАХ
1. ОБЩИЕ ПРЕДСТАВЛЕНИЯ О ДИАГРАММАХ СОСТОЯНИЯ
При анализе равновесия фаз в гетерогенных системах
конечной целью является установление строгих взаимосвязей между
параметрами, характеризующими состояние системы. Зная
зависимость между параметрами состояния, можно не только
определить состояние равновесия гетерогенной системы, но и
предсказать характер фазовых превращений при изменении температуры,
давления и концентрации в определенном направлении.
Как известно, начиная с работ Гиббса в термодинамике
гетерогенных систем, наряду с аналитическим широко используют
геометрический метод. Графическое толкование гетерогенных
процессов и равновесий, основанное на строгих закономерностях,
отличается большой наглядностью и позволяет охватить предмет
исследования в целом. Это позволило выделить самостоятельный
раздел термодинамики, получивший название геометрической
термодинамики [42—44].
Графическое толкование изменения характеристических
термодинамических функций в зависимости от параметров состояния и
установление на этой основе закономерных графических
соотношений между ними лежит в основе учения о диаграммах
состояния гетерогенных систем. Таким образом, диаграммой фазового
равновесия, или диаграммой состояния, называется графическое
изображение соотношений между параметрами состояния. Каждая
точка на диаграмме состояния, именуемая фигуративной точкой,
3 Глазов Е. М. и др. 65
oпpeдeляet численные значений параметров, характеризующих
данное состояние системы. Как известно, установить вид
уравнений состояния (особенно для конденсированных систем) трудно,
кроме того, без дополнительных данных их недостаточно для
полной характеристики термодинамического состояния системы
[13]. Поэтому экспериментальное и теоретическое построение
диаграмм состояния является главным, если не единственным
путем в решении проблемы описания гетерогенных равновесий
при анализе реальных систем. Диаграмма состояния служит
рабочим инструментом для технологов при определении
направленности процессов, связанных с фазовыми переходами, выборе
режимов термической обработки материалов, оптимальных составов
сплавов для определенного целевого назначения, и т. п. Большое
значение имеют диаграммы состояния в таких важнейших областях,
как материаловедение, галургия, нефтепереработка, химическая
технология и т. д. Определяя роль диаграммы состояния, Н. С. Кур-
наков [45] отмечал, что все детали процесса химического
взаимодействия, например, появление новых фаз и определенных
соединений, образование жидких и твердых растворов, находят точное
и определенное отражение в том геометрическом комплексе
поверхностей, линий и точек, который образует химическую
диаграмму.
Вскрывая общие связи в сочетании отдельных элементов
диаграммы, можно придать языку химической диаграммы стройную
систему, благодаря которой ее чтение и использование при
изучении химического взаимодействия между компонентами
утрачивают кажущуюся сложность [46].
Диаграмма состояния аккумулирует в себе также обширную
термодинамическую информацию, извлечение которой возможно
на основе сочетания графического и аналитического методов
химической термодинамики. В отличие от любых других диаграмм,
изображающих зависимость между какими-либо величинами с
помощью кривых или поверхностей, в диаграммах состояния каждая
точка независимо от места ее расположения имеет физический
смысл, так как отражает определенное состояние системы [44].
Диаграмма состояния дает ответ на вопрос о том, сколько
и какие конкретно фазы образуют систему при данных значениях
параметров состояния.
В соответствии с выбором температуры, давления и
концентрации как основных параметров состояния гетерогенной системы,
мы однозначно приходим к выбору свободной энергии Гиббса
в качестве основной термодинамической функции, ее
характеризующей.
Если рассматривать однокомпонентную систему, то, очевидно,
свободная энергия Гиббса будет функцией только двух параметров
состояния —температуры и давления:
G = ф (7\ р).
66
(III.1)
Полагая равновесие двухфазным (например, равновесие в точке
плавления), в числе прочих имеем условие равновесия
G<s) = G<L>. (HI.2)
На основании равенства (III. 1) получим 4
С^ = Фв(Г, р); G5=(f(7\ р). (1Н.З
Решая совместно уравнения (III.3) с учетом выражения (III.2),
найдем
Ф (Г, р) = 0. (III.4)
Уравнение (III.4) представляет цилиндрическую поверхность,
на которой лежит кривая аг — а2 пересечения поверхностей G<L)
Рис. 1. Поверхность изобарно-изотермического
потенциала однокомпонентной системы при равновесии
твердой и жидкой фаз (а) и при возможных
моновариантных равновесиях в однокомпонентной системе
и трехфазном нонвариантном равновесии (б)
Рис. 2. Диаграмма
состояния однокомпонентной
системы
и G<S) (рис. 1). При пересечении с плоскостью Т—р эта
поверхность образует линию а\—#2, положение которой в системе
координат G—Т—р определяется как
Ф (Г, р) = 0; G = 0. (III.5)
Положение той же линии в координатах Т—р можно
представить уравнением
Р = / (Т).
(III.6)
Следовательно, линия а\—а^ является одновременно
графическим изображением уравнения (II 1.6) и проекцией линии
пересечения поверхностей G(S) и G(L) на плоскость р—7\ Ее можно
построить, исходя из диаграммы изобарно-изотермического
потенциала.
3* 67
Для трех фаз однокомпонентной системы получаем (рис. 1, б),
пересечение трех поверхностей для изобарных потенциалов
соответственно жидкой, твердой и паровой фаз, следы которого
проектируются на плоскость р—Т.
Если теперь представить отдельно результат указанного
построения (рис. 2), то полученная диаграмма, которую собственно
и называют диаграммой состояния, будет характеризовать
фазовые взаимоотношения в однокомпонентной системе при различных
значениях параметров состояния. Вполне очевидно, что данная
диаграмма, хотя и не дает точного представления о характере
изменения самой функции Гиббса при изменении состояния
системы, однако ее связь с изобарно-изотермическим потенциалом
отчетливо прослеживается на основе описанных построений.
Таким образом, диаграмма состояния передает связь между
параметрами состояния (температурой и давлением) при
одновременной связи их с изменением функции состояния,
характеризующей фазы, образующие систему и их взаимные переходы.
Областям существования фаз L, G и S на диаграмме состояния,
т. е. однофазным областям, отвечают соответствующие поверхности
на диаграмме изобарно-изотермического потенциала (рис. 1, б),
линиям совместного существования попарно твердой и паровой
а\—а жидкой и паровой а—к' жидкой и твердой фаз а—а2
отвечают линии пересечения поверхностей (3s и GG, GL и GG, GL и (3s
соответственно. Точке совместного существования трех фаз L,
S и G (иначе тройной точке) отвечает общая точка трех
поверхностей Gs, GL и GG на рис. 1, б, для которой
Gs=,GL = G(l. (III.7)
При переходе к многокомпонентным системам в качестве
параметров состояния, помимо температуры и давления, фигурируют
также соответствующие концентрации. При этом картина хотя
и усложняется, но не меняется принципиально. Точно так же,
как и в случае однокомпонентной системы, в более сложных
системах можно перейти от пространственной диаграммы,
характеризующей зависимость изобарно-изотермического потенциала от
каких-либо двух параметров состояния (приняв остальные
постоянными), к плоской диаграмме состояния в координатах Т—х,
р—х и р—Т (х— концентрация одного из компонентов).
Принципиальная схема построения и перехода к диаграмме Т—х
для двухкомпонентной двухфазной системы при постоянном
давлении дана в работах Д. А. Петрова [46], В. П. Зломанова [47]
и др.
Двухкомпонентная система, в принципе, должна изображаться
в трехмерном пространстве при использовании в качестве
переменных температуры, давления и концентрации одного из
компонентов, поскольку концентрация второго компонента
определяется из уравнения связи вида (1.2).
68
Пространственная фигура может быть последовательно
воспроизведена с помощью соответствующих плоских сечений,
получаемых описанным способом на основе анализа зависимости изобарно-
изотермического потенциала от соответствующих двух
параметров состояния при постоянном третьем. Следовательно, для
использования геометрического метода важно знать общий
характер зависимости изобарно-изотермического потенциала от
температуры, давления и от концентрации отдельно.
2. ЗАВИСИМОСТЬ ИЗОБАРНО-ИЗОТЕРМИЧЕСКОГО ПОТЕНЦИАЛА
ОТ ТЕМПЕРАТУРЫ, ДАВЛЕНИЯ И КОНЦЕНТРАЦИИ
Общий характер зависимости изобарно-изотермического
потенциала от температуры и давления определяется непосредственно
значениями соответствующих производных из уравнений (1.90)
и (1.91). Из этих выражений следует, что изобарно-изотермический
потенциал с ростом температуры убывает и мерой уменьшения его
при нагревании системы служит энтропия при постоянных
давлении и концентрации. Увеличение давления приводит к
возрастанию свободной энергии Гиббса, а мерой роста ее в этом
случае является объем при постоянных температуре и
концентрации. Кривизна кривой температурной зависимости изобарно--
изотермического потенциала определяется знаком второй
производной. Из выражения (1.90) имеем
(дЮ1дТ\, х. = - (dS/dT)p, v (III.8)
Учитывая, что при нагревании системы энтропия всегда
возрастает, получим
(а2о/аг2)р,^<о. (ш.9)
Следовательно, кривая температурной зависимости изобарно-
изотермического потенциала обращена вогнутостью к оси абсцисс.
Точно так же, дифференцируя выражение (1.91), найдем
(дЮ/др*)т, Х[ = (dV/dp)T, Xr (III. 10)
Учитывая, что с ростом давления объем всегда уменьшается
и, следовательно, производная (dV/dp)T,x. отрицательна, получим
(d*G/dp*)TtXi<0. (III.ll)
Следовательно, зависимость изобарно-изотермического
потенциала от давления также обращена вогнутостью к оси абсцисс.
Данные, представленные на рис. 3, иллюстрируют изменение
свободной энергии Гиббса в зависимости от соответствующего
параметра состояния при неизменных остальных параметрах.
Особое значение в теории диаграмм состояния имеет температурная
зависимость G, поскольку графическое толкование этой зависи-
69
мости для совокупности фаз данной системы дает
непосредственное и наглядное представление о фазовых переходах.
Если снова обратиться к однокомпонентной системе, то
анализ относительного расположения кривых изобарно-изотермиче-
ского потенциала для твердой и жидкой фаз дает, во-первых,
отчетливое представление о температуре плавления, а, во-вторых,
графически наглядно и совершенно однозначно иллюстрирует
условие равновесия в форме (III.2).
В самом деле, при температурах ниже температуры плавления
в соответствии с критерием устойчивости стабильна твердая фаза
и, следовательно, ее изобарно-изотермический потенциал
располагается в области более низких значений по сравнению с жидкой
Рис. 3. Зависимость изобар-
но-изотермического
потенциала:
а — от температуры, р =
= const; б — от давления,
^* Г = const
фазой. При нагревании выше температуры плавления
наблюдается обратная картина и, следовательно, в момент плавления
кривые GL = f (Т) и Gs = / (Т) должны пересекаться (рис. 4).
Обоснованный выше характер кривых зависимости изобарно-
изотермического потенциала от температуры доказан совершенно
однозначно по знаку второй производной (II 1.9). Тем не менее
в целом ряде учебников и монографий по материаловедению
кривые G = f (Т) или F = f (T) часто представляют неверно:
выпуклостью к оси абсцисс. Учитывая широкую распространенность
этой ошибки, И. И. Новиков особо обсудил этот вопрос в своей
работе [48], хотя правильное толкование характера
температурной зависимости свободных энергий Гиббса и Гельмгольца
содержится во многих руководствах по химической термодинамике
и фазовым равновесиям (см. например, [45, 49—50]).
Проиллюстрируем фиксирование температур двух- и
трехфазного равновесий с помощью кривых температурной зависимости
изобарно-изотермического потенциала. На рис. 5, а приведена
чаграмма состояния однокомпонентной системы. В соответствии
условием равновесия в тройной точке, описываемой выражением
(III.7) при учете относительной стабильности фаз в конкретных
температурных диапазонах для давления р*, отвечающего
равновесному давлению сосуществования трех фаз, получим картину,
показанную на рис. 5, б. Для давлений р{ и рп соответственно
ниже и выше р* относительное расположение кривых
температурной зависимости изобарно-изотермического потенциала фаз S,
L и G показано на рис. 5, в и г.
70
Согласнб условиям монобарйантных равновесий S +± О, S +* L
и L+±G кривые изобарно-изотермического потенциала
пересекаются одна с другой при температурах, отвечающих
температурам фазовых переходов при давлениях /?, и рп соответственно.
Такая картина вытекает из рассмотрения трехмерной зависимости
изобарно-изотермического потенциала от параметров состояния,
> I
Рис. 4. Относительное расположение
кривых температурной зависимости изобарно-
изотермического потенциала твердой и
жидкой фаз для однокомпонентной
системы
Рис. 5. Определение фиксированного
положения температур двух- и трехфазного
равновесий в однокомпонентной системе
о которой уже указывалось, и еще раз подчеркивает органическую
взаимосвязь диаграммы состояния с диаграммой
изобарно-изотермического потенциала.
Рассмотрим концентрационную зависимость свободной
энергии Гиббса. Общий характер этой зависимости вытекает
непосредственно из критерия устойчивости относительно непрерывного
изменения состояния сжггемы. Физическая картина устойчивости
данного состояния фазы относительно примыкающих состояний
описана в общем виде в гл. II. Чтобы подойти к обоснованию
общего характера концентрационной зависимости свободной
энергии Гиббса, необходимо конкретизировать выражение для
критерия устойчивости.
Из анализа критерия устойчивости фаз относительно
образования внутри них новых фаз в форме аналогичной
(Н.111) с учетом уравнения (11.20) вытекает условие устойчивости
71
данного состояния относительно всех возможных примыкающих
состояний в виде неравенства
— \ik (nl — n'k) > О,
где одним штрихом обозначено состояние, анализируемое на
устойчивость, а двумя штрихами — примыкающее состояние. При
постоянных температуре и давлении это неравенство приобретает вид:
(AG- S |i'/A/n)>0.
В то же время
ДО~Е|*;Дл<+1/2!(62С)р,г +
(III.12)
(III.13)
Подставляя уравнение (III. 13) в выражение (III. 12), получим
соотношение, аналогичное (11.138),
(S2G)p,r>0.
(III.14)
Рассматривая изобарно-изотермйческий потенциал как функ*
цию молярных долей компонентов, можно неравенство (III. 14)
представить в следующей форме:
S Ъ фЮ1дх1 dxj) &xt 6xf
I Ц Ц (дЮ/дх; дх}) bxt Ьх, I > 0. (III.15)
L *—l /—l 1p*t
Из неравенства (III.15), согласно теореме Сильвестра, вытекает
следующее выражение:
Сц G12 • • • Git k_x
G2i G22 • • • G2, k-l
Д*.х =
>o.
(III.16)
I Gk-i. i Gft-i, 2 • ♦ • G*-i, k_x |
Отсюда для тройной системы получим неравенство
I Он G121
IG21 G221
А2 =
>0;
для двойной системы
Ai = Gii > 0.
В выражениях (III. 16)—(III. 18)
Gtj^&GldXidXj.
72
(III.17)
(111.18)
(III.19)
Учитывая произвольную нумерацию компонентов, можно
записать
Git > 0. (111.20)
Исходя из сказанного и, в частности, опираясь на критерий
устойчивости в форме (III. 15), можно сделать общее заключение
о том, что поверхность изобарно-изотермического потенциала,
отвечающая устойчивым относительно непрерывных изменений
состояниям, должна быть выпуклой относительно осей
концентраций.
На основе сделанного
заключения, фиксируя параметры состояния
Тир для двухкомпонентной
гомогенной системы, можно представить
общий характер концентрационной
зависимости
изобарно-изотермического потенциала (рис'. 6).
Обоснование вида этой важнейшей
зависимости будет законченным, если мы
конкретизируем характер ее
кривизны и объясним, как она
соприкасается с ординатами компонентов
А и В.
Пусть точки С и D (рис. 6)
отвечают двум состояниям,
являющимся примыкающими, и
отличаются одна от другой по
концентрации на величину Ах. Проведем касательную ахаг к кривой
G = f (х) в точке С. Тогда величина отрезка DK может быть
определена из уравнения
Рис. 6. К обоснованию характера
концентрационной зависимости
изобарно-изотермического потенциала
гомогенной фазы в
двухкомпонентной системе при Т = const, p =
= const
DK = (G + AG) - [G + (dG/ддг) Ax);
Щ = 1/2!. (d2G/dx2) (Ax)2 ^ .
(111.21)
(111.22)
Согласно условию устойчивости (III. 15), величина отрезка DK
должна быть положительной. Отсюда следует вывод, что кривая
концентрационной зависимости изобарно-изотермического
потенциала в окрестности точки С должна быть обращена выпуклостью
к оси составов, если точка С отвечает устойчивому относительно
непрерывных изменений состоянию. Сделанное заключение
находится в полном согласии с общим выводом, вытекающим из
критерия устойчивости в форме (III. 15).
При этом важно подчеркнуть, что оба из примыкающих
состояний являются устойчивыми. Аналогичное заключение можно
сделать на основании представления об изменении
изобарно-изотермического потенциала при нарушении однородности фазы. В самом
деле, если бы компоненты данной системы не растворялись один
в другом, а образовывали механическую смесь, то изобарно-
73
изотермический потенциал этой смеси определялся бы по правилу
аддитивности. В случае образования этими же компонентами
однородной устойчивой фазы (что задается по условию) всякое
нарушение однородности будет приводить к повышению изобарно-изо-
термического потенциала. Следовательно, вся кривая должна быть
обращена выпуклостью к оси составов.
О сопряжении рассматриваемой кривой с ординатами А и В
можно судить по изменению угла наклона касательной к
зависимости G. = f(x) по мере приближения к чистым компонентам.
Согласно определению, данному в гл. II [уравнение (11.39)],
нетрудно заключить, что тангенс угла наклона касательной к
кривой изобарно-изотермического потенциала (см. рис. 6) есть
разность химических потенциалов между вторым и первым
компонентами в фазе данного состава, т. е.
tg a = (dG/dxB)Pi т = ив- цА = Д(1. (111.23)
Тогда из выражения для химического потенциала раствора
в простейшем случае
Дц = Дц0 + RT In (xB/xA). (III.24)
При хв —>0; \п(хв/хА)—> — оо; А[х —♦ — оо. (III.25)
Отсюда на основании выражений (II 1.24) и (II 1.23)
tga-> — оо; a->90° (III.26)
Это значит, что кривая концентрационной зависимости
изобарно-изотермического потенциала по мере приближения к
ординате А стремится совпасть с ней, а в момент достижения касается
ординаты в точке, отвечающей значению G для чистого
компонента А при данных значениях давления и температуры.
Применяя аналогичные рассуждения к компоненту Б, приходим
к такому же заключению.
Таким образом, общий характер концентрационной
зависимости свободной энергии Гиббса установлен совершенно
однозначно. Эта зависимость в сочетании с температурной
зависимостью G (см. рис. 3) позволяет прогнозировать возможные варианты
характера фазового равновесия в двухкомпонентных системах.
Взаимосвязь тангенса угла наклона касательной к кривой
концентрационной зависимости изобарно-изотермического
потенциала с химическими потенциалами компонентов в форме (III.23),
дает основание для геометрического толкования условия
равновесия фаз в двухкомпонентной системе.
Покажем, что, проведя общую касательную к кривым
концентрационной зависимости изобарно-изотермического потенциала,
можно установить составы фаз, находящихся в равновесии.
Рассмотрим двухкомпонентную систему А—5, в которой при неко-
74
торой температуре Тг в равновесии находятсягжидкая L и твердая S
фазы. Очевидно, каждая из фаз характеризуется собственной
кривой зависимости свободной энергии Гиббса от состава (рис. 7).
Условием проведения общей касательной к двум кривым является,
во-первых, равенство угловых коэффициентов в точках касания,
а во-вторых, принадлежность точек касания одной и той же
прямой. Учитывая характер
рассматриваемой фундаментальной
зависимости, первое условие можно
записать в виде
(dG/dxB)LPtT = (dG/dxBfPtT.
(111.27)
Это условие является
необходимым, но недостаточным,так как
касательные к двум кривым
могут иметь одинаковый наклон
на разных уровнях, т. е. точки
касания могут не принадлежать
одной и той же прямой. Поэтому
помимо равенства угловых
коэффициентов необходимо записать
условие принадлежности точек
касания к одной и той же прямой. Очевидно, таким условием
является (рис. 7)
GL = GS- (4 - хк) (dG/dxB)p, т. (111.28)
После некоторых преобразований с учетом выражения (III.27)
получим
GL - xLB (dG/dxB)LPt т = Gs - х| (dG/dxBfPt T. (111.29)
Отсюда, принимая во внимание соотношения (11.37) и (11.38),
находим
(Ш.ЗО)
Выбрав в качестве независимой переменной концентрацию
компонента А и повторяя ход приведенных рассуждений, можем
записать
Рис. 7. К обоснованию двухфазного
равновесия в двухкомпонентной
системе (Г = Tt) при помощи кривых
концентрационной зависимости изо-
барно-изотермических потенциалов
• твердой и жидкой фаз
L S
\*>А = \*>А'
Gh-xLA(dG/dxA)LPtT
Gs-.xsA(dG/dxAfp,T
или \ib = \ir
(111.31)
(111.32)
В выражениях (111.30) и (III.32) отражено выполнение условий
равновесия твердой и жидкой фаз при данных температуре и
давлении. Следовательно, проведение общей касательной к кривым
концентрационной зависимости свободной энергии Гиббса является
75
Геометрической интерпретацией условия фазового равновесия й
позволяет однозначно фиксировать составы сосуществующих фаз.
Установленное геометрическое условие равновесия широко
используют на практике при установлении возможных видов равновесия
в двухкомпонентных системах. Отрезок общей касательной,
заключенный между концентрациями равновесных фаз, является конно-
дой, а ее проекция на плоскость Т — х преобразуется в отрезок
прямой, соединяющий координаты фигуративных точек,
символизирующих сосуществующие при данных условиях фазы.
Если рассматривать зависимость изобарно-изотермического
потенциала в двухкомпонентной системе для каждой фазы отдельно
(одновременно от состава и от температуры), то, очевидно, эта
зависимость будет представлять собой поверхность в виде желоба,
обращенного выпуклостью к плоскости Т—х. Отметим, что линии
пересечения этого желоба с ограничивающими плоскостями G—Т
для компонентов А и В являются кривыми температурной
зависимости изобарно-изотермического потенциала этих компонентов,
аналогичными показанным на рис. 3.
При рассмотрении двух кривых концентрационной зависимости
изобарно-изотермического потенциала в изотермическом сечении
имеем картину, показанную на рис. 7. Проводя общую
касательную, с учетом того, что значения свободной энергии Гиббса внутри
интервала концентрации между сосуществующими фазами всюду
ниже изобарно-изотермического потенциала каждой из фаз,
заключаем, что в указанном интервале при данных значениях
температуры и давления устойчивой является смесь равновесных
фаз, и изобарный потенциал смеси определяется по правилу
аддитивности. Мы уже отмечали, что в конденсированных системах роль
давления сравнительно невелика и в известных пределах ею можно
пренебречь. В этом случае для двухкомпонентных систем в
качестве параметров состояния, определяющих характер фазовой
диаграммы на плоскости, остаются температура и концентрация.
Фиксируя один из этих параметров, получаем возможность
установить четкую зависимость изобарно-изотермического потенциала
от другого параметра. Анализ относительного расположения этих
зависимостей для различных фаз позволит установить характер
фазового равновесия в системе.
3. ТЕРМОДИНАМИЧЕСКИЙ ВЫВОД ОСНОВНЫХ ТИПОВ ДИАГРАММ
СОСТОЯНИЯ ДВУХКОМПОНЕНТНЫХ СИСТЕМ
ДВУХФАЗНОЕ РАВНОВЕСИЕ
Принцип установления характера фазового равновесия на
основе приведенных выше температурной и концентрационной
зависимостей изобарно-изотермического потенциала сводится
к установлению фиксированного положения фигуративных точек
с координатами равновесно сосуществующих фаз на
плоскости Т—х.
76
Заметим, что термодинамический метод в данном случае
фигурирует в известной степени как апостериорный метод. При этом
речь идет не о строгом расчете, а о построении принципиальной
схемы, отражающей фазовые взаимоотношения в рассматриваемой
системе, при условии, что общий характер взаимодействия в
системе в принципе известен.
'в
Рис. 8. Принципиальная схема
установления фиксированного
положения координат
фигуративных точек, соответствующих
равновесно сосуществующим
фазам в двухкомпонентной
двухфазной системе
На рис. 8 представлена принципиальная схема установления
фиксированного положения координат фигуративных точек,
соответствующих равновесно сосуществующим фазам в
двухкомпонентной двухфазной системе. Следовательно, известно заранее, что
система образована только двумя фазами. Кроме того, известны
температуры плавления компонентов Л и В.
На рис. 8, а показана плоскость Т—х, Отметим на этой
плоскости то, что известно заранее, а именно, температуры плавления
компонентов Та и Т°в. Допустим, что Т°в > Т°А. Определим составы
равновесных фаз при температуре 7\, которая занимает некоторое
промежуточное положение между температурами Та и Тв.
Отметим вертикальной линией положение этой температуры
на графике температурной зависимости изобарно-изотермиче-
ского потенциала твердой и жидкой фаз для компонентов А и В
(рис. 8, б). При пересечении кривых SAf SBt LM LB с
вертикалью 7\ фиксируются точки 1^4, определяющие значения изо-
барно-изотермических потенциалов жидкой и твердой фаз
компонентов А я В. Перенося эти значения на соответствующие ординаты
графика концентрационной зависимости изобарного потенциала
77
(рис. 8, в), мы тем самым определяем крайние точки G\{L) и G°a{S\
а также G°BiS) и Gb{L) этой зависимости.
Далее, учитывая общий характер концентрационной
зависимости изобарно-изотермического потенциала, согласно рис. 6,
соединяем соответствующими кривыми попарно точки G°a{L)
и G°b{L\ а также G°a{S) и G°b{S). В итоге имеем две пересекающиеся
кривые концентрационной зависимости изобарно-изотермического
потенциала для жидкой и твердой фаз соответственно, проводя
к которым общую касательную, получаем составы хв и х% жидкой
Рис. 9. Диаграмма состояния двухкомпо-
нентной системы с непрерывным рядом
твердых и жидких растворов
Рис. 10. Характер концентрационной за-,
висимости изобарно-изотермического
потенциала в двухкомпонентной системе при
распаде твердого раствора или расслоении
расплава
ъ
G
ft
(У.
а1/^~^
I ,CXT + OC2
V*2
'1 \
1 \ 1
1 1
1 \
i /
i /
i
i i
«2
*<*,
Х<хг В
и твердой фаз, находящихся в равновесии при температуре 7\-
Чтобы получить соответствующие сопряженные точки на диаграмме
состояния Т—ху необходимо эти составы спроектировать на
горизонталь 7\, обозначенную на рис. 8, а. Таким образом,
фиксированные положения фигуративных точек 1г и sx символизируют
состояние сопряженных жидкой и твердой фаз, находящихся
в равновесии при температуре 7\.
Аналогично приведенному для любой другой температуры в
интервале между Та и Т°в можно найти соответствующую пару
сопряженных точек. Соединяя общими кривыми все lt и st и
замыкая их в точках Та и Тв, получим простейший вариант фазовой
диаграммы, характеризующей равновесие только двух фаз
(рис. 9). Если равновесными фазами являются жидкая (L) и
твердая (а) фазы, то данная диаграмма состояния характеризует
равновесие системы, в которой образуются непрерывные ряды
твердых и жидких растворов. Аналогичная картина характерна и для
системы жидкость—пар.
Кривую, соединяющую все точки 1Ь и отвечающую
температурам начала выделения твердой фазы из жидкой (кристаллиза-
78
ции) при охлаждении, принято называть кривой ликвидуса.
Соответственно кривую, объединяющую все точки S/ на рис. 8, а
и отвечающую температурам конца процесса преобразования
жидкой фазы в твердую при охлаждении или началу выделения
жидкой фазы из данной твердой (плавлению) при нагревании,
называют кривой солидуса.
Приведенные формулировки для рассматриваемой системы
являются совершенно строгими. Однако система, представленная
на рис. 8 или 9, простейшая и характеризуется единственной
ветвью ликвидуса и единственной ветвью солидуса. Применительно
к более сложным системам, которые будут рассмотрены ниже,
указанные формулировки носят частный характер, поскольку ничего
не говорят о границах кривых. Между тем эти границы
определяются пределами применимости соответствующего частного
уравнения фазы. В этой связи в работе [13] дано строгое
определение кривых ликвидуса. Под этими кривыми понимают ветви
диаграммы температур начала кристаллизации, которым отвечают
различные частные уравнения фаз. Такое определение тесно
увязано со строгим термодинамическим определением понятия фазы
и является общим.
Рассмотренный вариант фазовой диаграммы (рис. 9) включает
типичное двухфазное равновесие, которое характерно абсолютно
для всех других систем и фигурирует в них в качестве
обязательного элемента. В дальнейшем мы будем этот случай фазового
равновесия связывать с переходом из жидкого состояния в твердое
или наоборот, поскольку речь пойдет о конденсированных
системах. Тем не менее рассуждения о формировании или
термодинамическом конструировании различных типов диаграмм фазового
равновесия могут быть распространены на любые другие виды
фазовых равновесий.
Если же говорить о конденсированных системах, то, очевидно,
что помимо описанного важнейшего вида равновесия между
жидкой и твердой фазами возможно существование равновесия
между двумя жидкими либо между двумя твердыми фазами в
случае разрыва растворимости. Заметим, что путь установления
фиксированного положения координат равновесных фаз на
диаграмме Т—х остается тем же самым, что и в случае равновесия
между жидкой и твердой фазами.
На рис. 10 схематически показано, как фиксируются
координаты равновесных фаз аг и сс2, которые могут быть соответственно
жидкими или твердыми. При этом в связи с неустойчивостью
однородного раствора в интервале концентраций xai—xaz на
кривой зависимости свободной энергии Гиббса от состава появляется
максимум и одновременно два минимума. И в данном случае
основной прием определения составов равновесных фаз при
температуре 7\ сводится к проведению общей касательной к кривой
концентрационной зависимости изобарно-изотермического
потенциала, Это графическое выполнение условия термодинамического
79
равновесия гетерогенной системы в форме равенства химических
потенциалов данного компонента в каждой из сосуществующих
фаз.
Таким образом, нами рассмотрены основные виды двухфазных
равновесий в гетерогенной конденсированной системе. Если
рассматривать варианты фазовых равновесий, которые можно
термодинамически сконструировать в двухкомпонентнои системе, то
их в принципе может быть достаточно много, особенно при условии,
что один или оба компонента имеют по несколько полиморфных
превращений. Такие варианты подробно рассмотрены в работе
[46] и принципиально ничего нового не дают по сравнению с
рассмотренными видами двухфазных равновесий.
Рассмотрение фазовой диаграммы типа приведенной на рис. 9
показывает, что добавление компонента В к компоненту А
приводит к увеличению температуры плавления последнего, тогда как
добавление А к В, наоборот, снижает его температуру плавления.
Причины того или иного характера изменения температуры
плавления при добавлении одного компонента к другому
заключаются в особенностях межатомного взаимодействия на
электронном уровне. Однако если ясна направленность взаимного влияния
компонентов на температуру их плавления, и известно, что
равновесие в системе двухфазное, то можно совершенно четко, пользуясь
описанным методом, показать характер фазовой диаграммы.
Для термодинамики гетерогенных систем, помимо
рассмотренного, представляют интерес два других случая взаимного влияния
компонентов при добавлении одного к другому на температуру
плавления, а именно:
а) температуры плавления снижаются;
б) температуры плавления повышаются.
Если выполняются условия (а) или (б), то кривые ликвидуса
и солидуса на соответствующей диаграмме состояния имеют
общую точку: в первом случае минимума, а во втором — максимума.
Вид фазовых диаграмм этого типа показан на рис. 11, а и б.
Термодинамический вывод этих диаграмм с помощью кривых
концентрационной зависимости изобарно-изотермического потенциала
аналогичен приведенному выше, однако в интервале температур между
точками максимума (или минимума) и соответственно ближайшей
температурой плавления одного из компонентов изотерма 7\
показывает наличие двух разных двухфазных равновесий по
концентрации левее и правее точки экстремума. Каждая пара
сопряженных фаз при данной температуре характеризуется собственным
условием фазового равновесия и, следовательно, может быть
интерпретирована графически при помощи соответствующих кривых
концентрированной зависимости изобарно-изотермического
потенциала, к которым приведена общая касательная.
При этом необходимо иметь в виду, что в случае диаграммы
с максимумом твердая фаза устойчива в каком-то среднем
диапазоне концентраций, тогда как жидкая фаза устойчива в областях
80
составов, прилегающих к компонентам А и В. Это обусловливает
необходимость пересечения кривых концентрационной зависимости
изобарно-изотермического потенциала твердой и жидкой фаз
в двух точках (см. рис. 11, а, Т = Тг).
При относительном расположении кривых GL = / (хв) и
qs = f (хв), показанном на рис. 12, а, проведя общие касатель-
А Я max В A %min В
Рис. 11. Построение диаграмм состояния двухкомпонентной системы с
непрерывным рядом твердых и жидких растворов при наличии общей точки
максимума (а) или минимума (б) на кривых ликвидуса и солидуса
ные ах — а2, а3 — а4> фиксируем составы фаз, находящихся в
равновесии при Т{.
При Ттах кривые зависимости GL = f (хв) и G5 = / (хв)
касаются одна другой в точке, концентрация которой отвечает
общему максимуму на кривых ликвидуса и солидуса. При этом во
всем интервале концентраций кривая GL — f (xB) располагается
непременно ниже кривой G5 = / (хв), что подчеркивает
устойчивость при этой температуре жидкой фазы и неустойчивость твердой.
В случае диаграммы с минимумом устойчивой в среднем
диапазоне концентраций оказывается жидкая фаза, а в областях,
прилегающих к ординатам компонентов, устойчив твердый раствор.
$1
Это также обусловливает пересечение кривых GL = f (хв) и Gs =
= f(xB) в двух точках (см. рис. 11, б, Т = 7\). Проводя точно
так же две общие касательные ах—а2 и а3—а4 к кривым
концентрационной зависимости свободной энергии Гиббса твердой и жидкой
фаз, фиксируем составы равновесных фаз, определяемые концами
коннод sx— 1Х и s[ — /j.
При температуре минимума на кривых ликвидуса и солидуса
кривые GL = / (jcb) и (3s = f (xB) касаются одна другой в точке,
1 J V 1
\ \! V j
ш<
1 1 1
, 1 / ,1
/ \J I
К ' /
1 ;
1
^ В
В
Рис. 12. К обоснованию трехфазного эвтектического (а) и пе-
ритектического (б) равновесия в двухкомпонентной системе
при помощи кривых концентрационной зависимости изобарно-
иэотермического потенциала
отвечающей по концентрации составу минимума, при
непременном относительно более низком расположении последней кривой
во всем интервале концентраций. В точках максимума и минимума
составы жидкой и твердой фаз одинаковы, т. е.
xLB^xl (Ш-33)
Выражение (II 1.33) является дополнительным условием,
снижающим вариантность системы в данной точке на единицу.
Поэтому в точке максимума или минимума для изобарического
сечения системы равновесие должно быть нонвариантным и сплавы,
отвечающие составу экстремальных точек на рис. 11, ведут себя
как однокомпонентные системы.
ТРЕХФАЗНОЕ РАВНОВЕСИЕ
Применив правило фаз Гиббса к двухкомпонентной системе
при постоянном давлении увидим, что максимальное число фаз,
одновременно участвующих в равновесии, равно трем. Это огра-
8.2
ничивает число возможных трехфазных равновесий по следующим
соображениям. При любом фазовом превращении в г-фазной
системе число превращающихся фаз может быть равно 1,2, ...,
г—1, а число образующихся — соответственно г—1, г—2, ..., 2,1.
Очевидно, в трехфазной системе может быть максимум две
превращающиеся фазы, чему отвечает одна образующаяся фаза.
Соответственно и образующихся фаз может быть максимум две, чему
соответствует только одна превращающаяся фаза. В двухкомпонент-
ной системе в простейшем случае возможными фазами являются
твердые растворы аир, образующиеся на основе компонентов А
и В соответственно, а также жидкий раствор X. Тогда, очевидно,
в соответствии с изложенным относительно числа и характера
возможных трехфазных равновесий в двухкомпонентной системе
можем записать
L^a + Р; (111.34)
L + р^>а. (111.35)
Фазовые равновесия (Ш.34)и (III.35) называют соответственно
эвтектическим и перитектическим. В соответствии с нонвариант-
ным характером трехфазного равновесия в двухкомпонентной
изобарической системе равновесия (III.34) и (III.35)
наблюдаются при постоянной температуре, соответственно эвтектической Те
или перитектической Тр. Концентрационные пределы этих
превращений ограничены пределами областей гомогенности на основе
компонентов А и В при эвтектической температуре либо
протяженностью устойчивого состояния жидкой фазы, с одной стороны,
и протяженностью области твердого раствора на основе одного
из компонентов, с другой стороны, при перитектической
температуре.
Чтобы зафиксировать положение эвтектической либо
перитектической горизонтали на диаграмме фазового равновесия в
координатах Т—х, можно воспользоваться тем же приемом
геометрической термодинамики, что и в случае установления
двухфазного равновесия.
Из общего условия фазового равновесия для эвтектического
и перитектического превращений в системе А—В имеем
\1л = |А = м; \ib = |ig = |ii (HI.36)
В соответствии с выражениями (11.80) и (III.29), при учете
равенства (II 1.27) можем записать
Ga - хав (dG/dxB)p, т = Gp - х% (dG/dxB)P. т =
= GL-xb(dG/dxB)P.T\ (Ш.37)
Ga - х\ (dG/dxA)Pt т = Gp - A (dG/dxA)P, т =
= GL - xLA (dG/dxA)PtT. (Ш.38)
83
Выражения (111.37) и (111.38) представляют собой развернутую
форму условий фазового равновесия (III.36) и, кроме того, как
было показано при рассмотрении двухфазного равновесия,
являются условиями проведения общей касательной к кривым
концентрационной зависимости изобар но-изотермического потенциала
сосуществующих фаз. Следовательно, геометрической
интерпретацией условия трехфазного равновесия в двухкомпонентной системе
является общая касательная de'b' или p'db' к кривым G = f (xB)
для соответствующих трех фаз. При этом фиксируются совершенно
строго составы равновесных фаз хв, х% и хв, а проектирование
этих составов на диаграмму Т—х к температурам Те или Тр
(рис. 12) позволяет установить фиксированное положение
эвтектической (aeb рис. 12, а) либо перитектической (pab, рис. 12, б)
горизонтали. Заметим, что уже из выражений (III.34) и (III.35)
вытекает совершенно четкое положение эвтектической и
перитектической горизонталей относительно температур плавления
компонентов А и В для конденсированных систем. В самом деле,
поскольку превращающейся фазой при эвтектическом превращении
в процессе охлаждения является жидкость, а образуются при этом
две твердые фазы аир, можно заключить, что левая ветвь кривой
Ga = f (хв) будет всюду ниже левой ветви кривой* G<L> = / (хв),
а это означает, что
GSa<Gla. (III.39)
Точно так же приходим к выводу, что правая ветвь кривой 0^ =
— f (хв) должна быть всюду ниже правой ветви кривой GL =
= / (хв)- Откуда следует
G|<Gi (HI.40)
Из сопоставления с кривыми температурной зависимости
изобарно-изотермического потенциала для однокомпонентной
системы (см. рис. 4) на основании выражений (III.39) и (III.40)
будем иметь
Та>Т,\ П>Те. (111.41)
В соответствии с этим результатом наклоны правых и левых
ветвей на кривых концентрационной зависимости
изобарно-изотермического потенциала должны быть выбраны не случайно,
а вполне определенно. Обе ветви кривой для жидкости — наиболее
крутые. Левая ветвь для фазы а идет круче, чем для фазы р,
поскольку обе ветви должны прийти в общую точку, отвечающую
значению изобарно-изотермического потенциала компонента А
при эвтектической температуре, т. е. в точку G°aS). Аналогично
правая ветвь для фазы р круче, чем для фазы а, поскольку обе
ветви должны встретиться в общей точке G°bS) при данной
температуре. Таким образом, можно вполне обоснованно установить
фиксированное положение кривых зависимости G = / (хв) для
84
всех трех фаз, опираясь на значения изобарно-изотермических
потенциалов твердой и жидкой фаз компонентов А и В при
рассматриваемой температуре. Относительное расположение этих
потенциалов на ординатах компонентов устанавливается из
графика G = f (T) при учете соотношений (III.41).
Рассуждая аналогично о перитектическом превращении при
положении точки р, как показано на рис. 12, б, получим
Gla<GSa\ (III.42)
Gfl>G|. (III.43)
откуда следует (см. рис. 4), что
Га<Тр<Гв. (111.44)
Кривые концентрационной зависимости изобарно-изотерми-
ческого потенциала при температуре трехфазного равновесия
расположены в общем случае на разных уровнях, однако ко всем
трем кривым можно провести единую касательную. Как мы видели,
для установления условий трехфазного равновесия не требуется
привлекать никакие дополнительные представления о характере
двухфазных равновесий и о том, какой из компонентов является
растворителем, а какой растворенным веществом. Важно только
правильно зафиксировать параметры и четко представлять,
концентрация какого компонента является независимой переменной,
а какого — зависимой.
Следовательно, приведенное доказательство условия
трехфазного равновесия в двухкомпонентной системе носит общий
характер и из него не вытекает никаких дополнительных следствий и
ограничений на значения изобарно-изотермических потенциалов
сосуществующих фаз. Поэтому утверждение, что для
рассматриваемых нонвариантных равновесий условия (III.37) и (III.38),
вытекающие непосредственно из условий равновесия Гиббса и
являющиеся одновременно его геометрической интерпретацией,
оказываются необходимыми, но не достаточными [51 ], лишено,
на наш взгляд, должных оснований. Заключения авторов работы
[51 ] о том, что при трехфазном равновесии в двухкомпонентной
системе производные от свободных энергий сосуществующих фаз
по концентрации не только равны, но и обращаются в нуль, а сами
значения свободных энергий одинаковы, сделаны на основании
рассмотрения двухфазных равновесий в ликвидусно-солидусной
области для каждой из твердых фаз и в области распада твердых
растворов. При этом авторы допускают погрешность, приравнивая
для области распада производные по концентрациям разных
компонентов, что эквивалентно утверждению о равенстве разностей
химических потенциалов компонентов В и А в фазе а и А и В
в фазе р. В этом пункте вывод авторов работы [51 ] вступает в
противоречие с фундаментальными условиями равновесия фаз,
полученными Гиббсом на совершенно строгой основе без каких-либо
85
модельных представлений и экстраполяции линий фазового
равновесия. Заключение же авторов о равенстве изобарных потенциалов
при трехфазном нонвариантном равновесии в двухкомпонентной
системе, во-первых, ставит это равновесие в совершенно
исключительное положение по сравнению, например, с двухфазным, для
чего в принципе нет каких-либо оснований, поскольку двухфазное
равновесие также рассматривается при данной фиксированной
температуре и, следовательно, вариантность системы не должна
играть роли. Во-вторых, из этого заключения вытекает вывод
о равенстве химических потенциалов обоих компонентов между
собой. Такое равенство, хотя и не опровергается в принципе
методами термодинамики, однако, совершенно очевидно, что оно
может быть физически реализовано лишь при исключительном
сочетании характеристик компонентов и фаз и, следовательно,
представляет собой частный случай равновесия фаз.
Рассмотренные выше эвтектическое и перитектическое
трехфазные равновесия в двухкомпонентной системе носят общий
характер, поскольку характеризуют принципиальную
возможность течения процесса превращения одной фазы с образованием
двух других либо превращения двух фаз, сопровождающегося
образованием третьей фазы. Однако от того, в каком агрегатном
состоянии (даже для конденсированных систем) находятся
взаимодействующие фазы, возможны и другие виды трехфазных
равновесий в двухкомпонентной системе.
В случае, если все три фазы — твердые (что осуществимо при
наличии у одного из компонентов хотя бы одного полиморфного
превращения), возможны трехфазные равновесия, аналогичные
эвтектическому и перитектическому, которые называют
соответственно эвтектоидным и перитектоидным. Их можно представить
следующим образом:
Y^a + P; (IIU5)
Y+a^p. (III.46)
При наличии в определенном температурно-концентрационном
диапазоне двух жидких и одной твердой фазы возможны
трехфазные равновесия, получившие название монотектического и синтек-
тического. Им соответствуют следующие фазовые реакции:
Lx^L2 + a; (III.47)
Ъ + Ццьа. (Ш.48)
Нетрудно видеть, что по своей природе моногектическое
трехфазное равновесие (III.47) аналогично эвтектическому (III.34)
либо эвтектоидному (III.45), а синтектическое (III.48) аналогично
перитектическому (III.35) либо перитектоидному (III.46). Вполне
очевидно, что агрегатное состояние фаз не накладывает никаких
дополнительных ограничений на условия трехфазного равновесия
и поэтому термодинамическое обоснование трехфазных равновесий
86
(III.45)—(III.48) в принципе ничем не отличается от приведенного
выше обоснования эвтектического и перитектического равновесий
(см» рис. 12).
Сочетание различных видов двух- и трехфазных равновесий
приводит к разным фазовым соотношениям в системе и,
следовательно, к разным видам диаграмм состояния. Возможные
видоизменения характера кривых моновариантного равновесия при
сочетании различных фазовых областей в пределах одной
диаграммы фазового равновесия весьма многообразны. Базируясь на
вышеописанных принципах обоснования двухфазных и
трехфазных равновесий при помощи кривых концентрационной
зависимости изобарно-изотермического потенциала, рассмотрим вывод
некоторых основных вариантов диаграмм состояния двухкомпо-
нентных систем.
ОБОСНОВАНИЕ ОСНОВНЫХ ТИПОВ ДИАГРАММ СОСТОЯНИЯ
ДВУХКОМПОНЕНТНЫХ СИСТЕМ
Рассмотрим систему с трехфазным эвтектическим равновесием.
На основании изложенного выше имеем исходное положение,
согласно которому взаимное добавление компонентов А и В
приводит к понижению их температур плавления. Нанесем на
диаграмму состояния Т—х положение эвтектической горизонтали
aeb и температуры плавления компонентов А и В (рис. 13, а).
Покажем, как будет изменяться относительное расположение
кривых концентрационной зависимости изобарно-изотермического
потенциала трех фаз, сосуществующих при эвтектической
температуре в случае повышения или понижения температуры
относительно Те.
Анализируя физико-химическую сущность трехфазного
эвтектического равновесия, нетрудно заключить, что оно является
сочетанием трех двухфазных равновесий L <± a, L^p иа^±р.
Этот вывод вытекает из рассмотрения общей касательной к кривым
концентрационной зависимости изобарно-изотермического
потенциала трех рассматриваемых фаз при эвтектической температуре.
В самом деле, отрезок касательной ае' (см. рис. 12) указывает
на первое из трех указанных двухфазных равновесий, отрезок
е'Ь' — на второе и, наконец, db' на третье. Следовательно,
эвтектическую горизонталь можно рассматривать как совокупность
трех коннод,символизирующихналичиетрех указанных двухфазных
равновесий.
Учитывая относительное расположение температур плавления и
эвтектической горизонтали aeb, приходим к заключению, что при
повышении температуры будут стабилизироваться первые два
Двухфазных равновесия, а при понижении — третье. При этом
кривая концентрационной зависимости изобарно-изотермического
потенциала жидкой фазы в первом случае будет смещаться вниз,
так что минимум ее расположится ниже минимума, по крайней
мере, одной из кривых двух других фаз (рис. 13, б). Это исключает
возможность проведения общей касательной к кривым изобарно-
изотермического потенциала фаз а и р, не задевая кривую L.
Во втором случае кривая L будет смещаться вверх, в результате
чего появится возможность проведения общей касательной к
кривым концентрационной зависимости изобарно-изотермического
потенциала фаз аир (рис. 13, в).
Опираясь на высказанные
соображения об относительном
расположении кривых G = f (хв) для трех фаз
в системе с эвтектическим
трехфазным равновесием, при температуре
7\ > Те и проводя попарно к кривым
концентрационной зависимости
изобарно-изотермического потенциала
общие касательные а±а2 и а3а4,
находим составы равновесных фаз,
образующих при этой температуре
соответствующие двухфазные равновесия.
Проектируя эти составы к
температуре 7\ на диаграмме Т—х,
определяем фиксированное положение
фигуративных точек 1Х и slf а также 1[ и
si для фаз, образующих двухфазные
равновесия a^±L и L +± р.
Аналогично при других
температурах воспроизводим целиком
кривые ликвидуса и солидуса. При
температуре Т2 < Те относительное
расположение кривых
концентрационной зависимости
изобарно-изотермического потенциала для трех
рассматриваемых фаз позволяет
провести общую касательную a[af2 к
кривым Ga = f (хв) и G& = / (хв) и
таким образом определить
равновесные составы фаз аир,
участвующих в третьем двухфазном равновесии (рис. 13, в). Точно
так же, проектируя эти составы к температуре Т2 на диаграмме
Т—х, получаем фиксированное положение фигуративных точек
фаз а и р при этой температуре на диаграмме Т—х. Тот же ход
рассуждений позволит при других температурах ниже
эвтектической воспроизвести ход кривых, ограничивающих положение
областей твердого раствора на основе компонентов А и В. Эти кривые
носят название линий ограниченной растворимости, или сольвуса.
Диаграмма состояния эвтектического типа полностью
описывает поведение смесей и дает ясное понятие о фазовом составе
в различных температурно-концентрационных диапазонах.
Нетрудно видеть, что участки диаграммы, характеризующие двух-
88
Рис. 13. Построение диаграммы
состояния эвтектического типа при
помощи кривых концентрационной
зависимости изобарно-изотерми
ческого потенциала
фазное равновесие между твердыми растворами ha основе А и В,
с одной стороны, и жидкостью — с другой, представляют собой
части диаграммы с непрерывным рядом твердых и жидких
растворов (см. рис. 9), а участок, ограниченный линиями сольвуса, можно
трактовать как срезанный купол расслоения, показанный на
рис. 10. Подробное описание переходов из одной фазовой области
в другую, а также процесса кристаллизации или плавления сплавов
различного состава не входит в задачи нашей работы. Такое
описание содержится во многих руководствах по гетерогенным
равновесиям [46, 52—59]. Сам же механизм эвтектической
кристаллизации, как сложное явление формирования из жидкости двух
кристаллических фаз описан вставшей классической работе А. А. Боч-
вара [60] и трактуется им как последовательное выделение
ведущей и ведомой фаз в соответствии с сопровождающим
кристаллизацию изменением состава окружающего раствора и пересыщением
его в пользу то одной, то другой фазы.
Особый интерес представляет сочетание линий солидуса и
сольвуса. На рис. 13 показано, что это сочетание осуществляется
в точке максимальной протяженности твердых растворов на
основе компонентов Л и В. В литературе такие точки названы
точками предельного насыщения.
Заметим, однако, что термодинамика не накладывает никаких
ограничений на направленность кривых солидуса и сольвуса,
поэтому максимальная протяженность областей гомогенности
необязательно должна наблюдаться именно при эвтектической
температуре. В принципе максимум растворимости может
располагаться и выше и ниже эвтектической температуры. Тем не менее
представленная картина отражает достаточно распространенный
случай, когда каждая кривая моновариантного равновесия
меняется так, как показано на рис. 13, а, и поэтому естественно, что
максимальная растворимость отвечает точке их встречи.
Рассматриваемый случай, хотя и достаточно распространенный,
все же следует рассматривать как частный. Максимальная
протяженность области гомогенности на основе одного из компонентов
ниже линии эвтектического (или другого трехфазного равновесия)
встречается довольно редко. Значительно более распространен
случай максимальной растворимости выше температуры
трехфазного равновесия, получивший название ретроградного солидуса.
Вид участка фазовой диаграммы с ретроградным солидусом при
трехфазном эвтектическом равновесии показан на рис. 14. Как
видим, максимальная протяженность области твердого раствора на
основе компонента А, отмеченная на кривой солидуса буквой т,
отвечает температуре Тт, которая выше температуры трехфазного
эвтектического равновесия. Необычность представленной картины
заключается в том, что если рассматривать процесс охлаждения
из жидкости, например, сплава состава хъ то оказывается, что
после полного затвердевания жидкости состава 1Х при температуре
7\ в точке sx он в процессе дальнейшего охлаждения плавится при
89
более низкой температуре в точке s2 с выделением жидкости сд*
става /2, сопряженной с s2 при температуре Т2.
Поскольку плавление вещества в процессе охлаждения
физически трудно объяснить, в литературе даже встречались
утверждения, что существование ретроградного солидуса невозможно (см.
например работу [61 ]). Однако еще
в 1908 г. Ван-Лаар [62]
термодинамически обосновал возможность
существования ретроградного
солидуса. В дальнейшем проблема
ретроградного плавления обсуждалась
в работах [63—66].
Экспериментальные исследования, особенно систем
с участием германия и кремния,
показали [67], что у большинства из
них максимум протяженности твер-
\qo(q дого раствора на основе указанных
веществ расположен выше
температуры трехфазного равновесия и,
следовательно, солидус является
ретроградным. Учитывая большой интерес,
который представляет ретроградное
плавление для теории и практики, мы
рассмотрим его термодинамическое
обоснование и возможности
аналитического описания (в гл. V). Здесь же
отметим только, что с позиций
геометрической термодинамики
принципиальная возможность существования
ретроградного солидуса никаких
возражений не вызывает. На рис. 14, був
показано положение кривых
концентрационной зависимости изобарно-
изотермического потенциала твердой
и жидкой фаз при температурах Тт и
Т2 (причем Тт > Т2 > Те).
Как видим, положение
фигуративных точек равновесных фаз на
диаграмме Т — х фиксируется совершенно однозначно путем
проведения общих касательных ахаг и а\сС2 к кривым G - / (хв)
фаз, находящихся в равновесии при каждой температуре.
Понижение температуры от Тт до Г2 лишь смещает соответствующие
кривые в область более высоких значений
изобарно-изотермического потенциала в связи с общим ходом его температурной
зависимости и увеличивает интервал концентраций между точками
касания к кривым G<* = f (хв) и GL = / (хв). Однако видимых
причин смещения кривых изобарно-изотермического потенциала
вдоль оси концентраций на основании только геометрической
90
Рис. 14. Ретроградный сслидус
в системе эвтектического типа и
относительное расположение кри-
вых концентрационной
зависимости изобарно-изотермического
потенциала твердой и жидкой фаз
при различных температурах
термодинамики указать нельзя. Следует отметить, что
ретроградный характер солидуса возможен не только при
эвтектическом, но и при других возможных трехфазных равновесиях
в двухкомпонентной системе, так как характер трехфазного
равновесия в принципе не связан с сущностью рассматриваемого
явления. Важно только, чтобы область двухфазного равновесия
между твердым и жидким растворами была нисходящей.
Если в системе имеется трехфазное перитектическое
превращение, то можно конкретно судить об его расположении по
температуре относительно точек плавления компонентов А и В, что
отмечено выше неравенством (III.44). Вполне естественно, что
соотношение между температурами плавления компонентов может
быть и обратным, однако температура трехфазного перитектиче-
ского превращения также будет занимать промежуточное
положение между температурами плавления А и В.
Следовательно, рассматривая вопрос о фазовых соотношениях
в двухкомпонентной системе при трехфазном перитектическом
превращении, мы можем нанести на диаграмму Т—х положение
перитектической горизонтали и обозначить температуры
плавления компонентов А и В согласно соотношению (II 1.44). Положение
перитектической горизонтали обосновано при помощи кривых
концентрационной зависимости изобарно-изотермического
потенциала на рис. 12, б. Считая картину, представленную на рис. 12, б,
исходной, покажем, как будет изменяться относительное
расположение кривых концентрационной зависимости
изобарно-изотермического потенциала каждой из фаз, формирующих перитектическое
равновесие в случае повышения и понижения температуры
относительно перитектической горизонтали.
Анализ физико-химической сущности трехфазного перитекти-
ческого равновесия дает основание считать, что оно так же, как и
эвтектическое трехфазное равновесие, представляет собой
сочетание тех же трех двухфазных равновесий L =** a, L+* $ и а+± $,
однако в данном случае промежуточное по составу положение
занимает а-фаза, тогда как в случае эвтектического превращения
это положение занимала жидкая фаза. Естественно, что роль ос-
фазы при перитектическом превращении существенно иная,
поскольку жидкая фаза в случае эвтектического превращения
является превращающейся, а а-фаза — образующейся согласно
соотношениям (III.34) и (III.35). Поэтому при охлаждении ниже
температуры перитектического превращения а-фаза
стабилизируется, тогда как жидкая фаза при охлаждении ниже температуры
эвтектики исчезает. Это, по существу, обусловливает характер
смещения концентрационной зависимости кривой
изобарно-изотермического потенциала а-фазы при изменении температуры
относительно перитектической горизонтали.
Учитывая сказанное, перитектическую горизонталь раЪ можно
рассматривать как совокупность трех коннод pb, pa и ab,
что отчетливо видно по наличию общей касательной к кри-
91
вым изобарно-изотермического потенциала всех трех фаз на
рис. 12, б.
Можно сделать заключение, что с ростом температуры выше Тр
будет стабилизироваться двухфазное равновесие L +* р, тогда
как при понижении температуры ниже Гр в соответствующих
концентрационных диапазонах стабильными будут два других
двухфазных равновесия L+±a и а+*ф.
На диаграмме
изобарно-изотермический потенциал — состав это будет
выражаться в относительном
смещении кривых концентрационной
зависимости G для каждой из
рассматриваемых фаз. При этом, так как а-фаза
является образующейся при
охлаждении, кривая
изобарно-изотермического потенциала этой фазы в случае
понижения температуры, например
до Т2 (рис. 15, а), сместится вниз,
так что минимум этой кривой
окажется ниже минимума, по крайней
мере, одной из кривых
концентрационной зависимости G для двух других
фаз (рис. 15, б). При повышении
температуры, например до 7\ (рис. 15, а),
кривая Ga = f (хв) сместится вверх
так, как это показано на рис. 15, е.
Учитывая изложенные соображения
об относительном расположении
кривых G = f (хв) для трех фаз,
составляющих перитектическое равновесие,
при повышении и понижении
температуры следует в каждом случае для
определения составов равновесных
фаз проводить возможные общие
касательные к кривым
концентрационной зависимости изобарно-изотерми-
ческих потенциалов соответствующих
фаз. Из анализа рис. 15, в при 7\ > Тр можно заключить, что
в интервале концентраций между точками касания а{ и а^
стабильным будет двухфазное равновесие L ч* Р, поскольку изобарно-
изотермический потенциал смеси двух этих фаз, определяемый
для каждого состава положением на общей касательной а\—а£,
всюду ниже изобарно-изотермического потенциала отдельно
каждой из фаз. Проектируя составы, отвечающие точкам касания
а\ и й2 к температуре 7\ на диаграмме Т—х, получим
фиксированное положение конноды 1^, определяющей сопряженные точки на
кривых ликвидуса и солидуса при данной температуре.
Аналогично при других температурах в интервале Тр—Тв можно во-
92
Рис. 15. Построение диаграммы
состояния перитектического типа при
помощи кривых концентрационной
зависимости
изобарно-изотермического потенциала
спроизвести целиком кривые ликвидуса и солидуса,
ограничивающие область двухфазного равновесия L *± р.
При температуре Г2 < Тр относительное расположение кривых
концентрационной зависимости изобарно-изотермического
потенциала таково (рис. 15, б), что к ним можно провести две общие
касательные ах—а2 и а3—а4. Это обусловлено смещением
промежуточной кривой G(a> вниз в связи со стабилизацией a-фазы при
охлаждении. Очевидно, положение общей касательной ах—а2
ограничивает интервал концентраций /2—s2, в пределах которого
стабильной является смесь фаз а и L при данной температуре.
Положение касательной а3—а4 ограничивает при Г2 интервал
устойчивости двухфазного равновесия a :«=►: р. Проектируя общую
касательную а3—а4 к температуре Т2 на диаграмме Т—х> получим
положение конноды s'z—s'2, определяющей составы равновесных
фаз аир при данной температуре. Изменяя температуру в пределах
между Тр и ТА> можно описанным путем воспроизвести кривые
ликвидуса и солидуса, ограничивающие интервал устойчивости
двухфазного равновесия a +± L, и соответствующие участки кривых
сольвуса, ограничивающие положение области двухфазного
равновесия a =*=* р.
При температуре Т2 в интервале концентраций между
точками аг и аъ кривая концентрационной зависимости изобарно-
изотермического потенциала a-фазы для каждого состава
определяет наинизшее возможное значение этой функции, что и
обусловливает стабильное однофазное состояние в этом
концентрационном пределе при данной температуре. Ниже температуры
плавления компонента А кривая Ga = / (хв) окажется всюду ниже
кривой GL = f (xB), что в конечном итоге обусловит возможность
определения области единственного стабильного в этом
температурном интервале двухфазного равновесия а ^± р. В результате
описанных построений на плоскости Т—х воспроизводится диаграмма
состояния перитектического типа (рис. 15, а), которая наряду
с эвтектической является важнейшей типовой диаграммой,
поскольку иллюстрирует одно из главных типовых сочетаний
трехфазного равновесия с соответствующими двухфазными.
Завершая рассмотрение эвтектической и перитектической
диаграмм состояния, заметим, что при построении этих диаграмм
с помощью методов геометрической термодинамики мы в обоих
случаях предполагали содержание двух кристаллических а- и
Р-фаз, отличающихся собственным строением, свойствами и,
следовательно, характеризующихся самостоятельными
уравнениями фаз. Поэтому в отличие от А. Б. Млодзеевского [43],
который при обосновании этих диаграмм использует только одну
кривую концентрационной зависимости изобарно-изотермического
потенциала для твердой фазы, аналогичную приведенной на
рис. 10, мы сочли более строгим вывод с использованием двух
самостоятельных кривых Ga = f (xB) и G$ = f (xB). При этом
конечный результат оказывается одним и тем же.
93
Если не учитывать агрегатного состояния фаз, то эвтектическое
и перитектическое равновесия в принципе полностью охватывают
возможные сочетания трехфазного равновесия с двухфазным.
Поэтому рассмотрение возможных вариантов равновесия с учетом
иного агрегатного состояния фаз в двухкомпонентной системе
с позиций геометрической термодинамики практически ничего
Рис. 16. Фазовые равновесия в окрестности трехфазного монотектиче-
ского (а) и синтектического (б) равновесия с соответствующим
относительным расположением кривых концентрационной зависимости изобарно-
изотермического потенциала при температурах выше и ниже
температуры нонвариантного превращения
принципиально нового не содержит. Это положение наглядно
иллюстрируется на примере трехфазных равновесий с участием
двух жидких фаз. В соответствии с соотношениями (III.47) и
(II 1.48) одна или две жидкие фазы могут играть роль
превращающихся. При этом образующимися фазами могут быть либо жидкая
в сочетании с твердой, либо одна твердая. В первом случае, как
отмечалось выше, трехфазное равновесие ■— монотектическое, а во
втором — синтектическое. Наличие двух жидких фаз в
двухкомпонентной системе сопряжено с разрывом растворимости в
определенном концентрационном диапазоне и появлением двухфазного
равновесия L± +* L2, которое характеризуется на диаграмме фазо-
94
вого равновесия Т—х куполом расслаивания, показанным на
рис. 10. Сочетание этого купола с горизонталью трехфазного
равновесия возможно двояким путем. В первом случае, когда
превращающейся является одна жидкая фаза, а образующимися твердая
и другая жидкая фазы, формируется монотектическое трехфазное
равновесие (рис. 16, а), а во втором случае, когда
превращающимися являются две жидкие фазы, а образующейся — твердая,
формируется трехфазное синтектическое равновесие. В случае
монотектического равновесия жидкая фаза Ьг занимает
промежуточное положение по составу между твердым раствором а (или р)
и жидкой фазой L2. При рассмотрении варианта системы, когда
температура плавления компонента А выше, чем у компонента В,
очевидно, что температура трехфазного монотектического
равновесия Тм занимает промежуточное положение между Та и Тв.
Рассматривая горизонталь монотектического трехфазного
равновесия апгп по аналогии с описанными выше эвтектическим
равновесием как сочетание трех коннод, получим картину
относительного расположения кривых концентрационной зависимости изо-
барно-изотермического потенциала трех равновесных фаз,
аналогичную показанной на рис. 12, а. Следовательно, увеличение
температуры выше монотектической, должно приводить к
стабилизации двух двухфазных равновесий L =*±= а и Lx +=* L2. Понижение
температуры ниже монотектической горизонтали должно
способствовать стабилизации равновесия а :+* L2. Изменение
относительного расположения кривых концентрационной зависимости изо-
барно-изотермического потенциала трех рассматриваемых фаз
будет в принципе аналогично показанным на рис. 13, б, в для
системы эвтектического типа. Однако принимая во внимание
расслоение в жидкости и учитывая, что при определенном
перегреве две жидкие фазы перейдут в одну, вместо двух кривых
концентрационной зависимости G, использованных для эвтектической
системы, в данном случае достаточно одной кривой для жидкой
фазы — типа показанной на рис. 10. При этом относительные
перемещения соответствующих участков этой кривой и кривой
Ga = / (хв) при изменении температуры полностью аналогичны
наблюдаемым в эвтектической системе.
Таким образом, с позиций геометрической термодинамики
монотектическое трехфазное равновесие аналогично эвтектическому.
Рассмотрим точно так же синтектическое трехфазное
равновесие, когда две жидкие фазы Lx и L2 являются превращающимися,
а твердый раствор a—образующийся. Приходим к заключению,
что, во-первых, горизонталь трехфазного синтектического
равновесия pdq расположена выше температуры плавления наиболее
тугоплавкого компонента (например, Л), а во-вторых, нагрев выше
температуры синтектического трехфазного равновесия Ts
приводит к стабилизации двухфазного равновесия Lx =*=* L2, а
охлаждение ниже 7^ стабилизирует двухфазные равновесия Ьг +* а и
L2^± a.
95
При этом относительное расположение кривой Ga = / (хв)
и соответствующих участков единой кривой GL = / (хв) при
интектической температуре аналогично показанному на рис. 12, б,
а выше и ниже ее качественно воспроизводит картину,
представленную на рис. 15, б, в для перитектической системы. Таким
образом, синтектическое трехфазное равновесие с позиций
геометрической термодинамики является аналогом перитектического.
Отличие заключается только в агрегатном состоянии одной из
трех фаз, участвующих в равновесии.
Рис. 17. Диаграмма фазовых равнове- Рис. 18. Диаграмма фазовых
равновесий -двухкомпонентной системы при сий двухкомпонентной системы при
наличии монотектического и перитек- синтектическом и эвтектическом трех-
тического трехфазных равновесий фазных равновесиях
В итоге общая картина фазового равновесия в окрестностях
синтектической горизонтали pdq может быть представлена так,
как показано на рис. 16, б.
Заметим, что фиксированное положение линий моновариантного
равновесия в системах, представленных на рис. 16,
устанавливается в принципе так же, как в случае систем эвтектического и
перитектического типа, при помощи кривых концентрационной
зависимости изобарно-изотермического потенциала при различных
температурах.
Общая картина относительного расположения кривых Ga —
— f (хв) и GL = / (хв) при температурах выше и ниж:е монотекти-
ческой (синтектической) горизонтали показана на рис. 16.
Возможные варианты фазовых диаграмм при наличии в системе
купола расслаивания и трехфазных монотектического и синтекти-
ческого равновесий показаны на рис. 17 и 18.
Рис. 17 иллюстрирует наличие в системе двух трехфазных
равновесий — монотектического и перитектического. Возможен
вариант, когда вместо перитектического равновесия будет
эвтектическое. То же самое относится и к системе, представленной на
рис. 18, т. е. вместо эвтектического равновесия возможно пери-
тектическое.
96
Как видим, появление в системе дополнительно еще только
одной жидкой фазы существенно усложняет общую картину
фазового равновесия в двухкомпонентной системе. Очевидно,
образование промежуточных твердых фаз в двухкомпонентной системе
также должно отразиться на диаграмме состояния. Как правило,
промежуточные твердые фазы формируются на основе определенных
химических соединений [68 ], которые могут плавиться конгруэнтно
либо распадаться в результате перитектического превращения.
При изучении систем с промежуточными фазами возникает
вопрос о концентрационной зависимости изобарно-изотермического
потенциала этих фаз. Рассмотрение этого вопроса в работах [42—
44, 50, 53] не всегда строго термодинамически выдержано и
поэтому вносит известную неопределенность в толкование
проблемы. Прежде всего необходимо оперировать термодинамически
строго обоснованным понятием фазы. Существование в системе
определенных химических соединений требует уточнения
принадлежности растворов на их основе к одной или разным фазам.
Мы будем строго придерживаться изложения этого вопроса,
данного в работе [13], считая его последовательно
термодинамически выдержанным.
Как известно, природа фаз определяется особенностями
межмолекулярного взаимодействия, которое в первую очередь
обусловлено сортом частиц, их образующих. От природы частиц,
образующих данную фазу, зависят величина и характер сил обменного
взаимодействия, приводящих к формированию определенных
химических связей. Если растворы и фазы различаются родом
образующих их частиц, то их химические составы (речь идет об
истинных составах) качественно различны и термодинамические
характеристики этих фаз описываются разными фундаментальными
уравнениями [13]. Это очень важное заключение приводит к
выводу, что такие растворы даже в пределах одной гомогенной
системы следует рассматривать как самостоятельные фазы. Различие
между зависимостями свойств растворов, имеющих качественно
иные химические составы, от параметров состояния должно
проявляться, если не в виде самих функций, то по крайней мере
в значениях постоянных величин, фигурирующих в уравнениях
этих функций и отражающих специфику межчастичного
взаимодействия. В случае растворов или фаз переменного состава данному
качественному составу, или, иначе говоря, данному набору частиц
по сорту отвечает конечный интервал количественных составов
в данной системе, в пределах которого только и существует строго
определенный вид зависимости термодинамических и иных свойств
от параметров состояния. Положение о том, что характер
зависимости свойств от параметров состояния определяется качественным
химическим составом, весьма существенно и названо Сторонкиным
[13] принципом качественного своеобразия определенных
химических соединений. По существу этот принцип дает возможность
четко определить принадлежность растворов на основе определен-
4 Глазов В. М. в др. 97
ных химических соединений к самостоятельным фазам и,
следовательно, более строго использовать геометрическую термодинамику
при установлении общего характера фазового равновесия в системе
с промежуточными фазами.
Итак, если в системе образуется устойчивое недиссоциирующее
химическое соединение АтВп9 то, очевидно, растворы,
образованные добавлением компонента А к АтВп и Б к АтВп, следует
относить к различным фазам независимо от агрегатного состояния
системы. Здесь мы подходим к важному выводу о том, что в
результате взаимодействия между компонентами А и В образовалось
химическое соединение АтВп, отличающееся собственной (как
правило, совершенно иной по сравнению с А и В) кристаллической
структурой, если речь, идет о твердом состоянии, и своей особой
структурой ближнего порядка, если речь идет о жидком состоянии.
На основании этого устойчивое химическое соединение в двух-
компонентной системе следует рассматривать как самостоятельный
компонент, расчленяющий данную систему на самостоятельные
подсистемы.
Заметим, что этот важный вывод лежит в основе всего
дальнейшего рассмотрения фазовых соотношений в системах с
промежуточным фазами на основе определенных химических соединений.
Замечательно, что к этому заключению пришел еще Д. И.
Менделеев [69], рассматривавший возможность образования
определенных химических соединений в растворах. Он подчеркивал, что
«растворы разбиты, расчленены определенными соединениями,
если среди растворов они находятся».
На необходимость рассматривать определенные химические
соединения в качестве компонентов указывал также Н. М. Витторф
[53]. Следует отметить, что последовательное использование этого
положения с учетом сказанного выше о принадлежности растворов
на основе соединения к самостоятельным фазам коренным образом
изменяет подход к установлению фазовых соотношений в системе
при помощи кривых концентрационной зависимости изобарно-
изотермического потенциала фаз. Использование в качестве
параметра состояния брутто-состава сплавов в пределах каждой
частной системы требует учета и внесения корректив.
В свете изложенного рассмотрим жидкий раствор, в котором
образуется определенное недиссоциирующее соединение АтВп.
Так как это соединение рассматривается как компонент, то при
каждой данной температуре оно характеризуется самостоятельным
значением изобар но-изотермического потенциала. Следовательно,
растворы А + АтВп и АтВп + В являются самостоятельными
фазами. На этом основании общий характер изменения изобарно-
изотермического потенциала от брутто-состава жидкого раствора
определится кривой, показанной на рис. 19. Из рис. 19 следует, что
каждая частная система, имея свою жидкую фазу, характеризуется
собственной кривой зависимости G = f (x), которая всюду
обращена выпуклостью к оси составов. При этом, очевидно, отпадает
98
необходимость обсуждения вопроса о возможности проведения
общей касательной ахаг к двум указанным кривым, как это сделано
в работе [53], поскольку они принадлежат по существу разным
системам и только для наглядности и характеристики системы
А—В в целом изменение изобар но-изотермического потенциала
дано в функции брутто-состава, т. е. в зависимости от мольной
доли компонента В.
Даже незначительная диссоциация соединения АтВп в корне
изменяет положение дел. Качественный молекулярный состав
Рис. 19. Концентрационная
зависимость изобарно-изотермического
потенциала в системе с устойчивым недис-
социированным соединением
в жидкой фазе
т п
Т\
тА
L /
rfa e bY,
К L
1 г'хШГг \n\
1 л*г' Ш \fl\
H<*Vi \
тй°
АщВп
Рис. 20. Диаграмма состояния системы
с конгруэнтно плавящимся ыедиссо-
циирующнм соединением
слева от ординаты соединения перестает в принципе отличаться от
состава справа от нее, и в итоге имеется одна жидкая фаза. Так
как речь идет об одной фазе, концентрационная зависимость ее
изобарно-изотермического потенциала должна описываться кривой
типа, показанной на рис. 6.
Указанные рассуждения относятся и к системам в твердом
состоянии, поскольку принципиальной разности между жидкими
и твердыми растворами нет. Рассмотрим два принципиально
различных типа диаграмм состояния с химическими соединениями:
1) устойчивыми вплоть до температуры плавления и переходящими
в жидкую фазу того же состава, т. е. плавящимися конгруэнтно;
2) участвующими в трехфазном перитектическом превращении
в качестве промежуточной фазы, т. е. плавящимися инконгруэнтно.
Конгруэнтно плавящиеся соединения при отсутствии
диссоциации в твердом и жидком состоянии разделяют двойную систему
А—В на соответствующее число подсистем, в пределах которых
фазовое равновесие может быть представлено одним из описанных
выше видов простых диаграмм состояния. При этом фиксированное
положение линий фазовой диаграммы при помощи кривых кон-
центрацион ной зависимости изобарно-изотермического потен-
4* QQ
циала в каждой частной системе устанавливают так, как это
описано в случае систем эвтектического или перитектического типа.
На рис. 20 представлена диаграмма состояния системы с
устойчивым конгруэнтно плавящимся соединением АтВПУ которое при
взаимодействии с компонентами А и В образует частные системы
соответственно эвтектического и перитектического типа. Характер
взаимодействия в данном случае выбран произвольно, а
построение каждой частной системы при помощи кривых G = f (х)
показано на рис. 13 и 15. Картина может усложняться из-за возможного
расслаивания в жидкой фазе каждой частной системы, а также
вследствие полиморфизма компонента А или В и соединения АтВп.
А/пВл АщВп АтВп АтВп АтВл
Рис. 21. Виды максимумов на кривых ликвидуса и соли-
дуса при наличии в системе Л—В соединения АтВп в
зависимости от его термической устойчивости в твердом и
жидком состоянии
Устойчивость соединения АтВп в твердом и жидком состоянии
подтверждается сингулярным максимумом в точке плавления,
соответствующей стехиометрическому составу, где сходятся ветви
кривых ликвидуса и солидуса граничных частных систем.
Из рис. 20 можно заключить, что образующиеся на основе
соединения АтВп твердые растворы компонентов А и В суть
различные фазы Yi и у2, а не одна у-фаза, как это часто изображают
в ряде руководств по фазовым равновесиям. Естественно, что эти
растворы сформированы на базе одной кристаллической решетки
соединения АтВп и, следовательно, являются изоморфными.
Однако известно немало примеров, когда в системах
эвтектического типа твердые растворы на основе компонентов имеют
однотипные кристаллические структуры (например, система медь—
серебро), хотя совершенно очевидно, что их следует считать
различными фазами. В данном случае, если последовательно
использовать принцип качественного своеобразия химических
соединений, то придем к заключению, что область гомогенности
на основе устойчивого соединения слагается из двух областей,
отвечающих двум, хотя и изоморфным, но все же различным
фазам, разграниченным одна от другой ординатой химического
соединения.
' Если соединение АтВп частично^диссоциировано в жидкой
фазе, то кривая ликвидуса в точке максимума несколько сглажена,
100
причем степень диссоциации прямо связана с радиусом кривизны
ликвидуса. При этом соединение АтВп уже не делит жидкий
раствор на две различные фазы в рамках единой гомогенной
системы и в результате во всей системе А—В имеется только одна
жидкая фаза. Это следует учитывать при соответствующих
построениях с помощью геометрической термодинамики. Если же
соединение АтВп диссоциирует не только в жидком, но и в твердом
состоянии, сглаженный максимум будет также и на кривой солидуса,
причем, как и в случае жидкого состояния, величина радиуса
кривизны будет характеризовать степень диссоциации
соединения. При этом следует иметь в виду, что при существенной
диссоциации соединения максимум на диаграмме состояния может
быть сдвинут относительно стехиометрического состава. Вполне
очевидно, что на основе частично диссоциированного в твердом
состоянии соединения формируется только одна, а не две фазы,
как в случае недиссоциированного соединения. На рис. 21, а—д
показаны виды максимумов М на кривых ликвидуса и солидуса,
характеризующие различные варианты сочетаний этих кривых
в случае наличия или отсутствия диссоциации соединения АтВп
в твердом и жидком состоянии. Однако независимо от вида
максимума общий характер фазового равновесия в системе с
промежуточной фазой определяется характером ее взаимодействия с
компонентами в твердом и жидком состоянии, а следовательно, и видом
возникающих трехфазных равновесий.
Если химическое соединение образуется по перитектической
реакции, общий принцип обоснования фазовых соотношений
в окрестности перитектической горизонтали рМЬ не отличается
от простой перитектической системы, которая была рассмотрена
ранее (см. рис. 15). Однако из-за образования твердого раствора
на основе компонента А и соответствующего двухфазного
равновесия а^± L возникает проблема сочетания его со
стабилизирующимся при охлаждении ниже температуры перитектической
горизонтали двухфазным равновесием с участием фазы на основе
соединения АтВп. Возникающие при этом трехфазные равновесия могут
быть различными. На рис. 22 показана одна из возможных
диаграмм состояния с инконгруэнтно плавящимся соединением АтВп.
Рассматривая возможность обоснования такой диаграммы при
помощи кривых зависимости изобарно-изотермического
потенциала от концентрации, необходимо иметь в виду, что на основе
соединения образуется либо одна, либо две фазы, а следовательно,
и при геометрических построениях число кривых G = f (x) должно
соответствовать числу фаз. Иными словами, при
термодинамическом обосновании систем типа показанной на рис. 22, следует
учитывать принцип качественного своеобразия определенных
химических соединений.
Заметим, что формирование в процессе перитектического
превращения устойчивого соединения АтВп стехиометрического
состава дает основание считать, что ниже перитектической гори-
101
зонтали оно играет роль компонента со всеми вытекающими отсюда
последствиями. В частности, при температуре перитектической
горизонтали равновесие фаз можно обосновать, проведя общую
касательную ага2 через точку Ga в , отвечающую значению изо-
барно-изотермического потенциала соединения АтВПУ к кривым
концентрационной зависимости его для жидкой фазы L и твердой
фазы р так, как это показано
на рис. 23. Далее, проектируя
составы, отвечающие точкам
касания, к температуре Тр на
диаграмме Т—х, получим фик-
Рис. 22. Диаграмма состояния
системы с инконгруэнтно плавящимся
Рис. 23. Изображение трехфазного пе-
ритектического равновесия в двухком-
понентной системе с инконгруэнтно
плавящимся соединением ЛтВп
методом геометрической термодинамики
сированные положения фаз, находящихся при этой
температурке в равновесии совместно с соединением АтВп.
Рассмотренный на рис. 23 вариант образования определенного соединения
непосредственно в процессе перитектической реакции характерен
именно для данного соотношения между температурами
плавления компонентов и трехфазного равновесия. Естественно,
что это соотношение может быть и иным, однако в
принципе ход рассуждений будет таким же. Соединение АтВп может
сочетаться с перитектической горизонталью в любой точке и
в пределе может совпадать с точкой р. В этом случае плавление
соединения АтВп трактуется как конгруэнтное в переходной
точке, а трехфазное равновесие — как равновесие между жидким
и твердым соединением АтВп и фазой р [70]. Однако такое
совпадение физически едва ли достижимо, точнее вероятность его весьма
мала и поэтому реально соединение будет плавиться конгруэнтно
либо инконгруэнтно, в связи с чем горизонталь трехфазного
равновесия будет соответственно эвтектической либо
перитектической.
102
Используя метод построения концентрационной зависимости
изобарно-изотермического потенциала фаз в двухкомпонентной
системе, можно прогнозировать возможность экстраполяции линий
моновариантного равновесия и при этом судить о правильности
их относительного расположения. Суть принципа использования
метода геометрической термодинамики заключается в том [50 ], что
при проведении общих касательных к системе кривых G = / (х),
характеризующих метастабильное и соответственно стабильное
состояние, эти кривые должны располагаться в области более
низких значений изобарно-изотермического потенциала.
Заканчивая рассмотрение общих принципов построения
возможных типов диаграмм фазового равновесия двухкомпонентных
систем, отметим, что представленные выше примеры далеко не
исчерпывают всего многообразия случаев сочетания одно-, двух-
и трехфазных равновесий и, следовательно, возможных диаграмм
состояния. В частности, нами не затронуты системы,
отличающиеся неполной взаимной растворимостью компонентов, но
имеющие достаточно широкие области твердых растворов,
характеризующихся общей точкой на кривых ликвидуса и солидуса, т. е.
включающие элемент диаграмм состояния, представленных на
рис. И. Не рассмотрены системы с эвтектоидными либо перитек-
тоидными трехфазными равновесиями, а также многие другие
случаи. Это обусловлено тем, что предметом дальнейшего анализа
на основе термодинамики растворов будет главным образом
двухфазное равновесие между твердыми и жидкими растворами. Кроме
того, полный перечень возможных видов равновесия и невозможен
пока в принципе, поскольку этот вопрос до настоящего времени
не решен до конца.
Касаясь роли давления в процессе формирования новых
сочетаний линий моновариантных равновесий на диаграмме
Т—х, отметим обнаруженный одним из авторов и
детализированный в работах [71—72] случай фазовых взаимоотношений при
изобарическом сечении, отвечающем давлению в тройной точке
одного из компонентов.
При наложении внешнего давления возможны и другие
принципиально новые типы диаграмм состояния простейших двойных
систем.
В работе [73] при рассмотрении возможностей сочетания
областей двухфазного равновесия в двухкомпонентной системе
отмечается, что, помимо горизонтального касания, возможно и боковое,
что приводит к принципиально новым видам диаграмм состояния.
При рассмотрении основных типов диаграмм состояния
двухкомпонентных систем мы во всех случаях исходили из факта
образования твердых растворов на основе. компонентов и
определенных химических соединений. Между тем принципиальной
возможности существования однокомпонентных фаз в
многокомпонентной гетерогенной системе термодинамика не отвергает.
Данное утверждение базируется на представлениях Гиббса о дей-
103
ствительных и возможных компонентах, рассмотренных в
предыдущей главе.
На это со всей определенностью указал К. А. Путилов [16],
несмотря на то, что попытка обоснования отрицания абсолютной
нерастворимости термодинамическим путем принадлежит такому
авторитетному ученому, как М. Планк [2].
б
м
\МАтВп
N
АтВр + В
В А АтВп
В А
Щ Рис. 24. Диаграммы состояния с^пренебрежимо^малыми областями
растворимости в твердом состоянии
Таким образом, доказательство существования абсолютной
нерастворимости по существу сводится к оценке реальности
существования возможных компонентов, причем подход к решению
вопроса должен быть не термодинамическим, а базироваться
на молекулярно-кинетических представлениях.
A xLJ АтВп х£
Рис. 25. Построение диаграмм двухфазных равновесий в системах с пренебрежимо
малыми областями растворимости в твердом состоянии:
а — смесь двух твердых компонентов; б — равновесие твердого компонента А и жидкого
раствора; е —» равновесие химического соединения и жидкого раствора
Подобные соображения, связанные с молекулярно-кинетиче-
скими представлениями о тепловом движении атомов и диффузии,
позволили одному из авторов совместно с И. И. Новиковым [17]
сделать заключение о принципиальной невозможности
существования возможных компонентов в гетерофазной многокомпонентной
системе. Отсюда вытекает важнейшее следствие о принципиальной
104
невозможности диаграммы состояния с так называемой «простой
эвтектикой» и ее модификаций (рис. 24).
Обоснование типов диаграмм состояния, показанных на этом
рисунке, с помощью метода геометрической термодинамики,
данное в ряде работ (см. [42—44, 50]), следует считать условным или
приближенным (рис. 25). Точнее можно сформулировать так: если
бы однокомпонентные фазы в гетерогенных системах, образованных
двумя и более компонентами, существовали (в данном случае
компонентами считаются также недиссоциирующие химические
соединения), то доказательства, приведенные в работах [42—44,
50 и др. ], были бы абсолютно строгими. Если же полагать
ничтожно малыми изменения изобарно-изотермического потенциала
компонента при незначительной растворимости, то в выводах на
основе геометрической термодинамики следует учитывать только
температурную зависимость G для такого компонента.
Г л а в а IV
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕРМОДИНАМИЧЕСКОЙ
ТЕОРИИ РАСТВОРОВ КОНДЕНСИРОВАННЫХ
СИСТЕМ
1. ОБОБЩЕННОЕ ПОНЯТИЕ РАСТВОРА.
ТЕРМОДИНАМИЧЕСКАЯ КЛАССИФИКАЦИЯ РАСТВОРОВ
Растворами называют такие системы переменного состава,
в которых одно вещество сравнительно равномерно распределено
в среде другого или других веществ. Мы употребили выражение
«сравнительно равномерно», поскольку полное однородное
распределение встречается крайне редко. Раствор в общем случае может
иметь любое агрегатное состояние: твердое, жидкое и газообразное,
т. е. различают газовые, жидкие и твердые растворы. Жидкие
растворы делят на две группы: растворы неэлектролитов и
растворы электролитов. В случае газообразной системы более принято
говорить о газовой смеси, а не о газовом растворе. Твердые
растворы менее обычны, чем жидкие, но с термодинамической точки
зрения они аналогичны.
Процесс растворения отнюдь не представляет собой простого
распределения молекул или ионов, поскольку в большинстве
случаев он связан с проявлением различных взаимодействий между
разно- и^одноименными частицами. Это обусловливает природу
раствора и степень отклонения от идеальных закономерностей.
Подчеркнем, что неправильно сравнивать состояние
растворенного вещества, даже в очень разбавленном жидком растворе,
с состоянием его молекул в газе. Об этом весьма наглядно
свидетельствует хотя бы то, что теплота растворения твердого вещества
105
обычно очень близка к теплоте плавления и значительно
отличается от теплоты сублимации.
Простейшие составные части раствора, которые могут быть
выделены в чистом виде и смешением которых можно получить
растворы любого возможного состава, называются компонентами
раствора.
С термодинамической точки зрения все компоненты раствора
равноценны, поэтому деление их на растворитель и растворенное
вещество условно, особенно для систем, представляющих собой
непрерывные ряды растворов. Обычно растворителем называют
компонент, присутствующий в растворе в значительно большем
количестве по сравнению с другими компонентами, либо
компонент, который в чистом виде при данных условиях имеет такое же
агрегатное состояние, в котором находится раствор (если другие
компоненты в чистом виде имеют иное агрегатное состояние).
Условимся в дальнейшем растворитель обозначать индексом 1,
а индексы 2, 3, ... относить к различным растворенным веществам.
Экстенсивные свойства растворов определяются давлением,
температурой и количеством каждой составной части, а
интенсивные — давлением, температурой и относительным количеством
различных составных частей или составом.
Для измерения концентраций в термодинамике растворов
наиболее употребителен способ, основанный на представлении
о мольных долях [см. уравнения (1.1) и (1.2)].
В теории растворов рассматривают два основных класса:
идеальные и неидеальные (реальные) растворы. Идеальные
растворы подразделяют на бесконечно разбавленные, в которых
мольные доли всех растворенных веществ близки к нулю, и
совершенные растворы, которые остаются идеальными при любых
концентрациях. Термодинамическая классификация растворов
основана на характере уравнений для химических потенциалов
компонентов раствора.
Так, идеальный — это такой раствор, в котором для каждого
компонента справедливо выражение
ц \d (7\ р, х) = \х] (7\ р) + RT In xu (IV. 1)
где \х°с (Т, р) — химический потенциал t-того вещества в
стандартном состоянии. Член RT In xt соответствует изменению
химического потенциала в результате смешения (при образовании
идеального раствора).
Заметим, что, несмотря на сходство в записи выражений (11.53)
и (IV. 1), зависимость химического потенциала от приложенного
давления в этих двух случаях совершенно различна.
По аналогии с уравнением (IV. 1) зависимость
химических потенциалов компонентов неидеального раствора от
106
концентрации можно записать, согласно работе [80], в виде
ц, (7\ р, х) = М: (Г, р) + ЯГ lnx;Yi, (IV.2)
где Y/ — коэффициент активности.
Стандартное значение химического потенциала \х° (Г, р) — это
его значение при активности (x^i = at)9 равной единице.
Есть два способа нормировки стандартного состояния:
1. Симметричный — для растворов, в которых оба компонента
выступают как равноправные, за стандартное состояние каждого
из компонентов принимают состояние чистого компонента. Именно
этот способ используют при расчете функций смешения на
основании экспериментальных данных.
2. Несимметричный — когда в качестве стандартного состояния
компонентов выбирают их состояние в предельно разбавленном
растворе, т. е. близкое к идеальному. Сущность его заключается
в следующем:
а) для вещества 1, являющегося растворителем, в качестве
стандартного состояния выбирают его состояние в чистом виде;
б) для растворенных веществ за стандартное состояние
принимают их состояние в предельно разбавленном (хг-+ 1, х{ —> 0)
растворе, т. е.
(л? (Г, р) = lim (|xf — /?Г lnjff) (i = 2, 3 k).
При выборе стандартного состояния подразумевают
соответствующую нормировку коэффициентов активности.
Для первого случая
limT, = l, (/ = 1, 2, .... *)J (IV.3)
Для второго случая
lim v? = l; limY? = l, (i = 2, 3, . . ., k). (IV.4)
*Х-И Х.-+0
Общее условие идеальности, как легко установить, сравнив
соотношения (IV. 1) и (IV.2), заключается в том, что для
идеального раствора во всей области концентраций
7/ (Т9 Р, *i, . . ., хк_г) = 1 (1 = 1,2,..., АО-
Преимущество использования коэффициентов активности
состоит в том, что они позволяют в уравнениях для неидеального
раствора сохранить формальное сходство с уравнениями для
идеальных растворов. Коэффициент активности растворителя в
противоположность коэффициентам активности растворенных компонентов
из чисто арифметических соображений не является наиболее
подходящей функцией для измерения отклонений от идеальности.
Поэтому для растворителя удобнее вместо коэффициента
активности использовать другой поправочный фактор, называемый
107
Осмотическим коэффициентом Вьеррума и Гуггенгейма [18],
который вводят следующим образом:
|ii = |i;(7\ p) + ORT\nxl9 (IV.5)
где Ф —► 1 при ^-+1 и x2tз...—*0. Сопоставляя это выражение
с уравнением (IV.2), найдем, что
ф—1 «ЬуИпх!. (IV.6)
При использовании осмотического коэффициента формальное
сходство с уравнением для идеальных растворов отчасти теряется,
но зато коэффициент Ф намного больше чувствителен к небольшим
отклонениям от идеальности, чем коэффициент активности.
2. ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ СМЕШЕНИЯ.
ОСНОВНЫЕ СВОЙСТВА И ЗАКОНЫ ИДЕАЛЬНЫХ
РАСТВОРОВ
Прежде чем перейти к изложению основных свойств идеальных
растворов, дадим определение функции смешения.
Пусть у — мольное значение некоторой аддитивной
термодинамической функции (U, #, F, G, S, V и т. д.). Мольную функцию
смешения обозначим символом ум. По определению
Ум=У(хи . . ., *1-ь Р, Т) - S xtytip, T) (IV.7)
или уы =у(хи • • ., х*-ь V, Т) - S .ф!(У, Г), (IV.8)
где у] — мольное значение термодинамической функции для
чистого /-того компонента.
Из определения следует, что функции смешения учитывают
изменения термодинамических функций раствора при образовании
его из чистых компонентов. С помощью этих функций можно
описать термодинамические свойства раствора в широкой области
концентраций.
Согласно выражениям (IV.7) и (IV.8), для чистых компонентов
функции смешения равны нулю.
Парциальная мольная функция смешения определяется
разностью
9" = У{-Уи (IV.9)
где, согласно уравнению (1.7),
8i = (ду/дп,)р, т. пц.{.
108
Так как на основании выражения (1.9) любая аддитивная
мольная величина связана с парциальными величинами соотношением
y=»E*fc, (iv.io)
то функцию смешения, учитывая уравнение (IV.9), можно
представить в виде
к к к к
у" = Е хт - S x,yi = Е *, (д{ - у]) = Е *<Р"- (iv.ii)
i=l i=l t=l t=l
Таким образом, соотношения, определяющие связь между
функциями смешения ум и y*t совершенно аналогичны
соотношениям, устанавливающим зависимость между аддитивной величиной
термодинамической функции и парциальными мольными
значениями этой функции. Основываясь на этой связи, нетрудно
убедиться, что для бинарной системы справедливо
tf = gu-x2(d£/dxlP.T, (IV. 12)
vS = gu^x1(dgu/dx%)PtT. (IV. 13)
Аналогичным образом
ЙГ = /zM - х2 (dhu/dx$p, г, Й2 = hu + xx (dhM/dx2)Pt Ty (IV. 14)
sf = sM - х2 {dsuldx2)Pt г, 52м - sM + хг (dsM/dx2)Pt т. (IV. 15)
Пользуясь основным исходным выражением для определения
идеального раствора (IV. 1), можно вывести все другие свойства,
которыми должен обладать идеальный раствор.
На основании соотношений (IV.1) и (IV.9), можно записать
ц? <w> = tf - р] = RTlnxt. (IV. 16)
Отсюда, приняв во внимание выражение (IV.ll), изобарно-изотер-
мический потенциал смешения можно представить в следующем
виде:
gM <"> = £ x,|i? (W) = ЯГ Е *, In*. (IV.17)
i=i f=i
Такая простая форма уравнения для свободной энергии
смешения характерна только для идеальных растворов.
Соответствующая этому уравнению энтальпия смешения равна
нулю. Действительно, на основании связи между изобар
но-изотермическим потенциалом и энтальпией можно записать
Лм = - T*{d(g«/T)/dT]p, x± Хк (IV.18)
или после подстановки выражения (IV. 17)
ьг <"> = -т2 S хй [а(и? т1т)1ат\Р.х, = о. (iv.i9)
f=l '
109
Объем системы при образовании идеального раствора также не
изменяется, т. е. для идеальных растворов объем смешения равен
нулю.
Это следует из взаимосвязи между объемом и изобар
но-изотермическим потенциалом, а также из уравнения (IV. 17)
о- <«) = (дё« <">/ф)г, Жк = i; .v, (фм <">/а»)г,,, = о.
(IV.20)
Из выражений (IV. 19) и (IV.20) также следует, что внутренняя
энергия смешения, равная
uu = hM-pvM, (IV.21)
также должна быть равна нулю для идеального раствора.
Наконец, дифференцируя выражение (IV. 17) по температуре,
найдем энтропию смешения при образовании идеального раствора:
k
sm ad) e _ №м dd)/dT) _R 2 ^ in^.. (iv,22)
Все перечисленные выше свойства соблюдаются для идеального
раствора одновременно. Так, смесь, образующаяся без теплового
эффекта и без изменения объема, еще нельзя считать идеальной.
Необходимо, чтобы и энтропия смешения удовлетворяла
соотношению (IV.22).
На основании выражений (1.114), (11.16), (1.7), (11.23) и
уравнения для химического потенциала компонента идеального раствора
легко убедиться, что в идеальном растворе парциальные мольные
энтальпии htd и парциальные мольные объемы v\ld) компонентов
зависят только от Т и р:
v\d = (dtf/dp)T.x - [ф°/ (Л р)/др]т = v].
Для средней мольной энтальпии и среднего мольного объема
идеального раствора на основании равенства (IV. 10) и
приведенных выше соотношений можно записать
к к
hid = S xth\d - t xth], (IV.23)
vid = £ xtv\d = Б xfflt. (IV.24)
Отсюда следует, что в этом случае h\d и v\d равны соответственно
мольной энтальпии Щ и мольному объему v] чистого компонента i.
Значения h и v зависят от состава раствора, поскольку в общем
случае у\ ф у).
ПО
Для идеальных совершенных растворов формулы (IV.23) и
(IV.24) справедливы во всем интервале концентраций от xt = 1
до xt = 0.
В то же время в растворе, который становится идеальным
лишь при достаточном разбавлении, последние уравнения точно
выполняются только при х19 близком к единице. В предельно
разбавленном идеальном растворе для растворителя
но для растворенных веществ в общем случае
Л? <">*/& vf^^vl
где верхний индекс* означает принадлежность данной величины
к предельно разбавленному раствору.
Это различие между идеальным (совершенным) и идеальным
разбавленным раствором является следствием того, что энтальпия
и объем смешения, которые равны нулю для идеального раствора,
не обязательно должны быть равны нулю для идеального
разбавленного раствора. Рассмотрим предельно разбавленный двойной
раствор. Энтальпия его компонентов до смешения равна
Н = tiih\ + n2h*2, }
тогда как энтальпия раствора определяется равенством
H = nlh\ + n2fit{id).
Отсюда нетрудно убедиться, что
h«=x2{RVid)-hl).
Таким образом, образование предельно разбавленного раствора
из чистых компонентов при постоянных Тир сопровождается
тепловым эффектом, величина которого определяется последним
уравнением. Аналогично и объем смешения
if-*(0?(M)-<&
где образование разбавленного идеального раствора в общем
случае сопровождается сжатием или расширением. Но несмотря на
это, средняя мольная энтальпия и средний мольный объем в
некотором интервале концентраций (отвечающем образованию
идеального раствора) линейно зависят от мольной доли.
Учитывая, что при несимметричном способе нормировки для
двойного разбавленного идеального раствора
8 = *iMi + *2И<2 -f - RT (xx In xi + х2 In x2),
имеем s = (xisl + x2s*{ld)) — R (xx In xx + x2 In x2).
Отсюда видим, что энтропия смешения двух чистых компонентов
при образовании разбавленного идеального раствора не равна так
называемой «идеальной» энтропии смешения и отличается от нее
ill
на величину хг (§Z(id) — s°2). «Идеальная» же энтропия смешения
относится к процессу смешения чистого компонента 1 с
гипотетическим веществом, мольная энтропия которого равна sj.
Из законов термодинамики трудно определить условия, при
которых растворы становятся идеальными. Экспериментально
установлено, что все достаточно разбавленные растворы, в которых
мольные доли всех растворенных веществ достаточно близки
к нулю, ведут себя как идеальные. К такому же выводу приводит
и статистическая термодинамика [81 ]. Концентрация, при которой
в данном растворе начинают обнаруживаться заметные отклонения
от идеальности, сильно зависит от природы образующих его
веществ. Растворы, для которых отклонение от идеальности нельзя
заметить на протяжении всего интервала концентраций,
исключительно редки и встречаются только тогда, когда растворитель и
растворенное вещество обладают сходным химическим строением
и имеют близкие термодинамические характеристики в чистом
состоянии (температуры кипения, энергии испарения, давления
паров при заданной температуре и т. д.).
Идеальный раствор является удобным стандартом для
сравнения с любым реальным раствором. Законы поведения идеальных
растворов являются предельными законами, соблюдающимися
тем точнее, чем больше сходство между компонентами.
Используя для химического потенциала растворенного
компонента / в паре уравнение (IL57), на основании условия фазового
равновесия можно записать
fi7 = \i°i(7\ p) + RT\nxi=VL°i{G) (T) + RTInФ|f (IV.25)
где ф; — фугитивность компонента i в^насыщенном паре над
раствором.
Отсюда находим
Vt^kiXi. (IV.26)
Здесь
£ = ехр [[£ (Г, р) - |ife (G) (T)]/RT). (IV.27)
Если допустить, что паровая фаза является смесью идеальных
газов, то соотношение (IV.26) примет вид
р" = *Л, (IV.28)
где pi — парциальное давление насыщенного пара компонента i
над раствором.
Формулы (IV.26) и (IV.28) выражают закон Генри для
идеального раствора: парциальное давление пара растворенного вещества
пропорционально его мольной доле. Величина kt называется
коэффициентом Генри. В идеальном (совершенном) растворе
ki = pi, где р°с — давление насыщенного пара вещества /.
112
Из уравнения (IV.27) следует, что величина kt является
функцией только р и Т и не зависит от концентрации. Можно также
показать, что при обычных давлениях на значение kL давление
практически не влияет. Зависимость kt от температуры, согласно
выражению (IV.27), можно определить по формуле
dlnkj _ 1 d[^(L)(r,p)/T] г d^]^ (T)JT]
dT ~ R dT R dT ~
~ RT* ==: RT2 ' (IV./yj
где Ah°it v — мольная теплота испарения компонента i из
идеального раствора в равновесный с ним пар.
Другим более узким определением идеального раствора может
служить утверждение, что все растворенные компоненты
подчиняются закону Генри [см. формулу (IV.26) или (IV.28)].
Исходя из закона Генри, пользуясь соотношением Гиббса—
Дюгема, можно вывести закон Рауля:
В случае идеального поведения газовой смеси над раствором
закон Рауля приобретает вид:
РС = 'РЛ, (IV.30)
или несколько в иной форме:
(pI-pi)/pI=E*<. (iv.3i)
I
Это уравнение показывает, что относительное понижение
парциального давления пара растворителя равно сумме мольных
долей растворенных веществ.
Общее давление пара раствора определяется законом Дальтона.
Отсюда, учитывая равенство (IV.30), для двухкомпонентного
раствора получим:
Р = Pi (1 - х2) + plx2 = р\ + (р\ - р\) ,v2, (IV.32)
так что общее давление пара идеального бинарного раствора
является также линейной функцией мольной доли.
Выполнение условий (IV.30) или (IV.31) при нескольких
различных концентрациях означает, что химические потенциалы
компонентов имеют форму (IV. 1) и следовательно, раствор является
идеальным. Таким образом, изучениеТдавления пара позволяет
судить об идеальности данного раствора.
Составы идеального раствора и его насыщенного пара в общем
случае различны, т. e.~*<L>^= х^.
Условие x{L) = x{G) при всех концентрациях соблюдается
только при равенстве давлений насыщенного пара обоих чистых
113
компонентов. Характер зависимости я<°> от #(L> может сильно
изменяться с температурой.
Константа равновесия реакции, протекающей в растворителе,
который не принимает в ней участия, определяется уравнением
(11.93). Если сделать подстановку в закон действия масс (11.93)
из закона Генри (IV.28), которому подчиняется в идеальном
растворе каждый компонент, то получим
('v' 'v' \
л;, ; ~ < со$&- = & (т.. л. (iv.33)
где Кх {Ту р) — константа для данных растворителя, температуры
и давления.
При больших разведениях константу равновесия (IV.33) можно
выразить через мольные концентрации или моляльности [24].
Для выяснения зависимости Kxd (T, р) от температуры на
основании уравнения изобары Вант-Гоффа [10], а также соотношений
(IV.33) и (IV.29) получаем
\d\nKxd(T, Р)] _ АЯо<0> VI У(Щу_ АЯ°<а> . _т ^
[ дТ Jp~ RT* Zj RT* "~ RT* Ka — Lyb),
(IV.34)
где Д//° — теплота реакции при постоянном давлении в идеальном
растворе.
Подобным же образом для зависимости Klxd (7\ р) от давления,
используя выражения (IV.33), (IV.27) и (11.23), найдем
\d\nKid(T,p) 1 V v'0' AV° «*> lmm . ~ п\тоъ
[ д-р \t = -L-RT= RT^{a^U S)' (IVl35)
i
где AV° — изменение объема реакции, протекающей в идеальном
растворе.
Рассмотрим две фазы аир, каждая из которых является
идеальным раствором. Общее условие химического равновесия
для данной системы в отношении компонента i будет
|i? = |if. (IV.36)
Далее в соответствии с выражением (IV.25), принимая во внимание
равенство (IV.26), получим
x?/x? = k?/$ = lCi(p, Г), (IV.37)
где Ki (р> Т) — коэффициент распределения растворенного
компонента i между двумя фазами аир.
Формула (IV.37) выражает закон распределения Нернста для
идеальных растворов, согласно которому отношение мольных
долей компонента в двух идеальных растворах, находящихся
114
в равновесии между собой, зависит только от температуры и
давления и не зависит от состава растворов.
Сравнивая формулы (IV.29) и (IV.37), получаем зависимость
Ki от температуры:
г dinKito, т) 1 /ainff \ / a in ^ \ _ ан]^^
[ дТ Jp- \ дТ )р \ дТ )р- RT* >
(IV.38)
где АЛ1(а">Р) — стандартная теплота перехода компонента i из
а- в р-фазу, отнесенная к одному молю.
3. ЗАКОНЫ НЕИДЕАЛЬНЫХ РАСТВОРОВ.
ИЗБЫТОЧНЫЕ ТЕРМОДИНАМИЧЕСКИЕ ФУНКЦИИ.
КЛАССИФИКАЦИЯ ОТКЛОНЕНИЙ ОТ ИДЕАЛЬНОСТИ
Как уже отмечалось, коэффициент активности компонента
в неидеальном растворе является функцией температуры, давления
и состава раствора. На основании уравнения Гиббса—Гельмгольца
(1.114), а также соотношений (11.16) и (1.7) имеем
№-]..,"*• (iv-39»
Отсюда легко убедиться, что коэффициент активности t-того
компонента в неидеальном растворе зависит от температуры:
(д In yVdT)Pt x. = ± \д/дТ [(М, - ^)/Т)\р, Х( =
= -(й,-й;)/ЯГ2, (IV.40)
где Щ — парциальная мольная энтальпия t-того компонента
в стандартном состоянии.
Аналогично на основании соотношений (11.23) и (IV.2) можно
получить
(д\пу</др)т = (vi - v))/RT9 (IV.41
где v] — парциальный мольный объем f-того компонента в
стандартном состоянии.
Если в уравнение Гиббса—Дюгема (11.25) подставим
выражение (IV.2) и при этом воспользуемся оператором D для
обозначения изменений от состава при постоянных температуре и давлении,
то для одного моля смеси получим
Е XlD Ы* т - RT { Е xtD (Inxt) + Е xtD (Inу,)) = 0. (IV.42)
Однако первое выражение в правой части равенства (IV.42)
тождественно равно нулю. Отсюда как следствие должна
равняться нулю и вторая сумма:
£x,D(lnY,)p,r==0. (IV.43)
i=i
115
Состояние бинарного раствора можно определить переменными
Т, р, л:2, что позволяет записать уравнение (IV.43) в форме:
(1 - х2) (д In уг/дх2)Ру т + х2(д In уъ/дх2)р% т = 0. (IV.44)
Тогда In ух = — J (x2/xx)dlny2. (IV.45)
о
Отсюда следует, что для вычисления коэффициентов
активности компонентов бинарного раствора, например первого
компонента, в некоторой области составов раствора, начиная с х2 = 0,
достаточно знать коэффициенты активности второго компонента.
Это особенно важно в тех случаях, когда какой-либо из
компонентов раствора обладает измеримым давлением пара.
Интегрирование уравнения (IV.45) проводят обычным
графическим способом. Вблизи значения х2 = 1 вычисление
коэффициента активности первого компонента затруднено, так как
подынтегральная функция при этом пределе стремится к
бесконечности. Чтобы уменьшить ошибку расчета, желательно
располагать данными по коэффициентам активности второго
компонента для составов, предельно разбавленных относительно
первого компонента, когда точность экспериментального определения
величин, входящих в формулу (IV.45), значительно ниже. Поэтому
для оставшейся области концентрации, примыкающей к чистому
второму компоненту, пользуются различными способами
интерполяции, коэффициентов активности. Один из таких способов
заключается в разложении логарифма коэффициента активности
в ряд по степеням концентрации:
lnY2 = ao*i + ai*iH .
Вблизи х2 = 1 ряд быстро сходится и можно ограничиться двумя
его первыми членами. Тогда значения а0 и аг легко определить,
построив график функции:
In 72/^1 = ^0 + ^X1.
Полученные таким образом значения а0 и аг используют при
интегрировании уравнения (IV.45) для построения оставшейся
части кривой In y2 как функции x2/xv
Вводя осмотический коэффициент растворителя, определяемый
уравнением (IV.5), для изменений состава при постоянных
температуре и давлении в соответствии с выражением (IV.43) можно
записать
—хг In (l/^)DO + (1 - Ф) 2 dxt + E xtD In y/ = 0. (IV.46)
t=»2 t=2
116
Это уравнение выведено Бьеррумом. В частности, для бинарного
раствора уравнение (IV.46) можно записать в виде
(1 - х2) In (1 - х2) DO + (1 - Ф) dx2 + x2D In у2 = О (IV. 47)
или в интегральной форме:
х% х%
—lnY2 = J [(1 — ^/x8]ln(l -x2)DQ> + j (1 —0)d\nxt.
о о
(IV.48)
Таким образом, коэффициент активности растворенного
вещества в данном неидеальном бинарном растворе можно
вычислить, если осмотический коэффициент растворителя известен для
всех растворов, более разбавленных, чем рассматриваемый
раствор, при тех же самых температуре и давлении.
Исходя из уравнения (IV.2), для давления пара
растворенного компонента неидеального раствора вместо соотношения
(IV.28) получим
Pi=btxiyi. (IV.49)
При симметричном способе нормировки kt = pj. С помощью
выражений (IV.49), (IV.29) и (IV.40) легко показать, что
/ дInpt \ _ / rd\nkt \ . / din у, \ _ &hi, v(T)
\ дТ )Pt х. ~ \ дТ )Рч х; ^\дТ )р, Х}~~ RT* '
(IV.50)
где Д/i/.V (Т) —мольная теплота испарения /-того вещества из
данного раствора.
Подобным же образом на основании формул (IV.49), (IV.27),
(11.23) и (IV.41) при симметричном способе нормировки получим
д\пр \ v]{L) uf —u°.(L) v\
(~V" )тЛх. = JrT + ~LW = Wr • <IV-51)
Численные значения kt и yt зависят от выбора способа
нормировки, но произведение ktyi равно отношению pjxi и не зависит
от выбора стандартного состояния.
Формулы для давления пара растворителя совершенно
аналогичны тем, которые были выведены для растворенных компонентов.
В данном случае они следующие:
Р! = Р?*17ь (IV.52)
(д In pJffDp, Xt = А/и, vlRT*, (IV.53)
(д In Pl/dp)T, Xl = vf/RT (a = L, S). (IV.54)
117
При несимметричном способе нормировки для бинарного
неидеального раствора можно записать:
Pi=Pi*iYi. .(IV.55)
p2 = k2x2y2. (IV.56)
В рассматриваемом случае предельным законом для
растворителя будет закон Рауля (IV.30), а для растворенного вещества
закон Генри (IV.28).
Сравнивая выражения (IV.55) и (IV.30) или (IV.56) и (IV.28),
видим, что при несимметричном способе нормировки для любого
компонента величина коэффициента активности выражается
отношением действительного давления пара к идеальному:
Vi = Pi/Pi' = ft/Pi*i; V2 = P2/p2d =* Pdk2x2. (IV.57)
В симметричном способе нормировки, согласно формуле (IV.3),
из уравнения давления пара (IV.49) следует, что в этом случае
(IV.58)
при *i =1 р\ = ku
при х2 = 1 pi = k2j
т. е.
Pi = Pi^iYb Р2 = P2X2Y2. (IV.59)
Отсюда 7i = ft/Pi* = Pil{pi*i)l 72 = PdP? = pdiplx*)* (IV.60)
Таким образом, преимущество симметричного способа
нормировки заключается в том, что в нем все компоненты
рассматриваются одним и тем же способом. Коэффициенты активности
(IV.60) имеют простой смысл. Они равны отношению
наблюдаемого парциального давления к тому, которое наблюдалось бы,
если раствор был идеальным. Таким образом, при использовании
симметричного способа нормировки коэффициенты активности
служат непосредственной мерой отклонения раствора от
идеальности.
Подставив в уравнение (IV.44) значения коэффициентов
активности из соотношений (IV.59), получим
(1 -x2)(dlnPl/dx2)Tt p + x2(d\np2/dx2)TtP = 0. (IV.61)
В соответствии с выражениями (IV.54), (IV.51) парциальные
давления компонентов зависят от общего давления, но при
обычных давлениях возможные изменения pt с давлением ничтожно
малы в связи с малыми значениями vt.
Поэтому формула (IV.61) практически сводится к уравнению
Дюгема — Маргулеса:
(1 - х2) (д In pJdx2)T + х2 (д In р2/дх2)т = 0, (IV.62)
в котором постоянной при дифференцировании остается только
температура.
118
Это уравнение, связывающее парциальные давления,
справедливо при любых отклонениях от идеальности в растворе. При его
выводе допускалось лишь, что газообразная фаза является
идеальной газовой смесью и что парциальные мольные объемы
компонентов в растворе пренебрежимо малы.
Допустим, что рх и рг определены экспериментально. Тогда
уравнение (IV.62) приводит к простому критерию, с помощью
которого можно оценить точность результатов эксперимента.
Уравнение (IV.62) можно переписать в виде
Pl/*1 Р2/*2 '
При графической интерпретации полученного результата
можно заключить, что др2/дх2 есть наклон касательной к кривой
р2 = f (*г) в точке ху а р2/х2 есть наклон прямой, соединяющей
точку х с началом координат. К таким же заключениям можно
прийти, рассматривая кривую рх = / (х^.
Из уравнений (IV.30) и (IV.57) следует, что
71=71=Р1/(рвЛ) (IV.64)
и
72/72 =k2/p°2. (IV.65)
Поскольку 7? -► 1 при xt -> 0, последнее уравнение означает, что
lim 72 = k2/pl (IV.66)
Поэтому выражению (IV.65) можно придать вид
72/72* = Hm 72 (IV.67)
или 72/72 = Hm 721. (IV.68)
С помощью этих уравнений можно легко переходить от одного
способа нормировки к другому. Если, например, мы определили
72 как функцию состава, то формула (IV.67) позволяет вычислить
величину 7*-
Условие равновесия для химической реакции, протекающей
в растворителе, который не принимает в ней участия,
определяется уравнением (11.90). Подставив в это равенство значение jit-
из уравнения (1V.2), легко убедиться, что для закона действия
масс в неидеальных растворах справедливо
In П (хфр = - £ v,tf (Г, p)IRT = In Ka (7\ р) (IV.69)
i i
ИЛИ П {Xwfi = (xftfl (Х'2УУ*/(Х1У1Г' (Af2V2)V. = Ka (T, p),
(IV.70)
119
где константа равновесия Ка (7\ р) для данных растворителя,
температуры и давления постоянна. Равным образом выражение
(IV.70) можно переписать в виде
(X{f> (4)V(*l)V' (^)V.'. = Ка (Т, р) (Y.)V' (Y2)V.'./(yDV' Ы*» = К,
(IV.71)
где величина К'х зависит от концентрации.
Зависимость Ка (Т, р) от температуры и давления для
неидеального раствора, согласно соотношениям (IV.69), (IV.39)
и (11.23), имеет вид:
Г д1пКа(Т, р) 1 АЯ° <«). г д\пКа(Т, р) 1 АУ°(а)
L дТ \Р- RT* ' [ др }т~ RT
(IV.72)
В соответствии с выражением (IV.71)
На основе этого уравнения при учете соотношений (IV.40) и
(IV.72) получим
(д In К'х/дТ)р = Д#°7#Г2, (IV.74)
где Д#а — теплота реакции в данном растворе.
Аналогичным образом нетрудно убедиться, что
(д In Кх/др)т = -AVVRT, (IV.75)
где AFa —изменение объема при протекании реакции в данном
растворе.
На основании соотношений (11.90) и (IV.2) при равновесии двух
неидеальных фаз будем иметь
ц? _ $=,*; <a> - и; <» + яг in iw/^i =
= ЯГ In Kt (T, p) + RT In [(хф)"/(х1Ъ)»] = 0. (IV.76)
В уравнении (IV.76) введено обозначение
р] «*> _ ц; <» = ЯГ In /С, (Г, р). (IV.77)
ft ft
Отсюда Ki(T, р)=-^> (!V.78)
где коэффициент распределения Kt (7\ р) для данной пары
растворителей является функцией только температуры и давления.
Уравнение (IV.78) представляет собой общую форму закона
распределения Нернста [82].
Если одна из фаз является предельно разбавленным
идеальным раствором, а другая —нет, то уравнение (IV.78) позволяет
рассчитать коэффициент активности у{ в неидеальной фазе.
120
По аналогий с предыдущим имеем
[d In К< (Г, р)/дТ]р = (h](Э) - tit W)/RT2 = Mi] &^IRT\
(IV.79)
[д In Kt (7\ р)/3р]г = -&о\(0"Р)//?Г (IV.80)
где A/i] (а_>р> — стандартное изменение энтальпии, a AuJ(а_>>р) —
стандартное изменение объема при переносе 1 моля компонента i
из раствора а в раствор р. Величины A/t](a"*w и А^(а_>р)
являются функциями только от Т и р и не зависят от состава фаз.
Из соотношений (IV.78), (IV.40) и (IV.79) следует также, что
[а1п(//К)]^ (ft? - hf)/RT2 - Ahl^/RT2. (IV.81)
Знания активности и коэффициента активности каждого
компонента недостаточно для выяснения причин отклонений реальных
растворов от идеального поведения. Поэтому в термодинамике
растворов широко пользуются так называемыми избыточными
величинами термодинамических функций.
Ранее на примере идеальных растворов было введено понятие
о термодинамических функциях смешения. Определение этих
функций легко распространяется и на неидеальные растворы.
Разность между термодинамической функцией смешения уы
для реального раствора и значением этой функции для идеального
раствора уы(ш) при тех же Т и р называется избыточной
термодинамической функцией. Избыточные термодинамические
функции обозначают символом уЕ (Е — первая буква английского
слова excess — избыточный). Таким образом, согласно
определению,
ув=уи^^цы)ф (IV.82)
Величина уЕ представляет собой избыток (положительный или
отрицательный) данного термодинамического свойства раствора
по отношению к тому же свойству идеального раствора
сравнения, составленного из тех же компонентов. Для чистых
компонентов, очевидно, уЕ = 0.
Изобарно-изотермический потенциал при смешении
компонентов, приводящем к образованию неидеального раствора, вместо
выражения (IV. 17), можно найти из следующего уравнения:
к k
Мы = Ъ xt\if = RTix( \nxiyi. (IV.83)
i—\ i= 1
При выводе этого уравнения, как и других избыточных
термодинамических функций, используют симметричный способ
нормировки. Взаимные соотношения между избыточными
термодинамическими функциями, выраженными в различных системах
сравнения, рассмотрены в работе [83].
121
Сравнивая выражения (IV. 17) и (IV.83), получаем
k
gE = gM- g" <w> « RT 2 Xi In yh (IV.84)
где gE — избыточный изобарно-изотермический потенциал. На
основании уравнения (IV.84) при учете соотношений (IV. 12)
и (IV. 13) для двух компонентного раствора можно записать
(if = RT In vi = gE - x2 [dgEldx2)T^
4 = RT\ny2 = gE + xl{dgEldx2)TtP.
Аналогичные выражения справедливы и для других избыточных
термодинамических величин: -
Sf = sE - х2 {dsE/dx2)PtT, s$=*sE + xx (dsE/dx2)p, T\ (IV.86)
Я? = hE - х2 (dhE'/dx2)Pt г, fif - hE + хх (dhE/dx2)p, T. (IV.87)
Избыточные термодинамические функции легко рассчитать из
общетермодинамических соотношений. На основании
соотношения (1.90) избыточную энтропию определяют дифференцированием
уравнения (IV.84) по температуре:
к к
s* = —ЯГ 2 xf (д In yi/dT)Pt, - Я Е * In v/. (IV.88)
Избыточную энтальпию определяют, подставляя соотношения
(IV.84) и (IV.88) в уравнение (1.113):
hE = -ЯГ2 £ Xj [д In ?//<ЗГ)р. Ж|. (IV.89)
Для расчета избыточного объема дифференцируют уравнение
(IV.84) по давлению
к
vE^RT1£i xl (д In yJdp)T, Xr (IV.90)
*=i *
Избыточную внутреннюю энергию определяют подстановкой
соотношений (IV.89) и (IV.90) в уравнение иЕ = hE —pvE\
UE = _RT ^Г g Xj(d ln yi/dT)pt ^ + р g x. {д 1П у1/др)т> ^ J .
(IV.91)
При обработке экспериментальных данных часто встречаются
случаи, когда конденсированные растворы не образуют
однофазную систему во всем исследуемом диапазоне концентраций. Однако
чтобы исключить влияние различия в агрегатном состоянии
чистых компонентов на термодинамические функции растворов,
обычно за стандарт принимают состояние чистых компонентов
в том же агрегатном состоянии, что и раствор, и при той же
температуре. Это позволяет легко сравнивать термодинамические
122
функции растворов между собой и проводить общий анализ
свойств растворов, сопоставляя их с соответствующими
величинами для идеальных смесей.
Найденные опытным путем значения активностей
пересчитывают на новое стандартное состояние следующим образом.
Пусть экспериментально определена активность компонента Л
в жидком растворе при температуре Г* (которая находится ниже
температуры плавления А) по
отношению к чистому твердому
веществу Л. Состояние, в котором
находится переохлажденный
жидкий компонент А при температуре
опыта Т* будем считать новым
Стандартным состоянием.
Рассмотрев процесс перехода вещества А
между тремя фазами:
переохлажденным жидким компонентом Л,
жидким раствором Л—В и
твердым чистым веществом Л, получим
следующее соотношение для
пересчета значений активности Л на
новое стандартное состояние [84 ]:
аА = Аа\. (IV.91a)
Здесь
Л = ехр(1/ЯГ) -Д/4,л(1-
тт,А
-Т*/Гт,А)- j Ac°PtAdT +
Т*
+ Г \(Ас;гА/Т)с1т\,
Т* )
аА представляет собой активность компонента Л в растворе
относительно переохлажденной жидкости, аА — активность
компонента Л в растворе по отношению к чистому твердому веществу,
кс°Рг а — разность теплоемкостей компонента Л в твердом и жидком
состояниях в расчете на 1 моль. Активность второго компонента В
находят обычным путем по уравнению (IV.45) для всех
концентраций раствора.
Если экспериментальным путем определяется активность более
легкоплавкого компонента В (рис. 26), то расчет ведут в такой
последовательности: 1) интегрированием уравнения Гиббса —
Дюгема определяют активность аА\ 2) по уравнению (IV.91a)
пересчитывают активность вещества Л на новое стандартное
состояние — переохлажденный жидкий компонент Л; 3) если это
можно сделать с достаточной точностью, то экстраполируют функ-
Рис. 26. Диаграмма фазовых
равновесий и активности компонентов двойного
сплава с гетерогенной областью:
аА и аВ ~~ активности компонентов А
и В в сплаве по отношению к чистым
жидким веществам Л и В при
температуре Т*, а* — активность
компонента А по отношению к чистому твердому
12а
ции аА и ав на гетерогенную область и твердые растворы
компонента В в веществе А.
Избыточные термодинамические функции тесно связаны с
экспериментально измеряемыми величинами, такими как давление
пара над раствором, теплота смешения при постоянном давлении
и др.
Сравнивая избыточные термодинамические свойства реальных
растворов, можно судить о межмолекулярном взаимодействии
в них.
В этом смысле по характеру отклонений от идеальности
раствора можно судить о природе растворЬв и классифицировать
их. При этом различают два предельных случая:
1. Регулярные растворы, для которых
Лв»Г|а*|, (IV.92)
и, следовательно,
gE&hE. (IV.93)
В этом случае отклонения от идеальности обусловлены главным
образом теплотой смешения.
2. Атермальные растворы, для которых
hE<£T\sE\, (IV.94)
и, следовательно,
gE « - TsE. (IV.95)
В этом случае отклонения от идеальности в значительной мере
обусловлены энтропийными изменениями.
4. КОНЦЕПЦИЯ РЕГУЛЯРНОГО РАСТВОРА.
РЕШЕТОЧНАЯ МОДЕЛЬ РАСТВОРА.
СТРОГО РЕГУЛЯРНЫЙ РАСТВОР И ЕГО МОДИФИКАЦИИ
Анализ фазовых равновесий на строго термодинамической
основе, как было показано в гл. II и III, возможен при
использовании двух, в своей основе связанных подходов. Однако оба
метода, будучи абсолютно строгими, не позволяют рассматривать
конкретные системы, так как дают только качественную картину
фазовых соотношений. Для перехода к численным решениям
требуется привлечение модельных представлений о характере
межмолекулярного взаимодействия в растворах, позволяющих
получить конкретную форму выражения термодинамических
функций и вывести соотношения между параметрами состояния
рассматриваемой системы.
Привлечение определенных моделей позволяет не только
рассчитать определенные свойства и сравнивать их с экспери-
124
ментом, но также высказать важные суждения по поводу
определенных отклонений рассматриваемой системы от модели, что
тоже дает весьма ценную информацию относительно физико-
химической природы фаз, образующих эту систему.
Существенную роль в развитии термодинамического описания
фазовых равновесий, начиная от работ Бирона [85] и Ван-Лаара
[86, 87], сыграла модель регулярного раствора и ее последующие
модификации. Эта модель основана на представлении о
независимости энергии системы от характера атомного распределения
и учитывает только различие в энергиях взаимодействия
разноименных и одноименных атомов, являющихся ближайшими
соседями.
Сам термин «регулярный раствор» впервые был введен Гиль-
дебрандом [88—90], по определению которого регулярный
раствор характеризуется нулевыми значениями избыточного объема
и избыточной энтропии смешения, а концентрационная
зависимость энтропии смешения определяется так же, как и у идеального
раствора. Метод Ван-Лаара получил дальнейшее развитие в
работах Карлсона и Колбэрна [91], Скэтчарда [92], Гильдебранда
[89—90, 93—94], Герцфельда и Гейтлера [95] и других авторов.
В работах Скэтчарда [92], Гильдебранда и Вуда [96] было
получено выражение для избыточной внутренней энергии одного
моля жидкого раствора, на основании которого коэффициенты
активности регулярной смеси можно представить в виде
RT In Yi = v\<S>\ (Ьг - б2)2, RT In y2 = %Ф? (6i - 82)2. (IV.96)
где Ф/ и 8/ — объемная доля и параметр растворимости,
введенный Гильдебрандом, соответственно.
Перепишем систему (IV.96) в форме, удобной для вычислений,
допуская, что v\ = v\:
#Г In yi = *>*!, (IV.97)
[ RT\ny2=<i>xl (IV.98)
где ш — параметр (энергия взаимообмена), который
рассчитывают из опытных данных.
Отметим свойство регулярного раствора, вытекающее из его
определения. Условие sE = 0, характеризующее регулярные
растворы, означает, что:
In7i =/0/Л. \ny2 = f(l/T). (IV.99)
Действительно, sE равно нулю при любых значениях чисел молей
компонентов, т. е.
dsE/dn1=^0\ dsE/dn2 = 0. (IV. 100)
В соответствии с выражением (1.90) на основании соотношений
(IV. 100) можно записать
д2ВЕ/дп±дТ = 0; d2gE/dn2dT = 0, (IV.101)
125
Отсюда, принимая во внимание уравнение (IV.84) и соотношение
Гиббса —Дюгема, нетрудно убедиться, что
dRTlnyJdT^O; (IV Л 02)
dRTlny*fdT = Q, (IV. 103)
что эквивалентно выражениям (IV.99)..H, наоборот, соблюдение
условий (IV.99) означает, что sE = 0.
. Таким образом, в регулярных растворах логарифм
коэффициента активности обратно пропорционален абсолютной
температуре.
Из уравнения (IV.84) и соотношений (IV.97), (IV.98) вытекает,
что избыточный термодинамический потенциал при образовании
регулярного раствора, а следовательно, и его энтальпия
определяются относительно простым выражением:
gE&hB^WbXt. (IV. 104)
Так как избыточная энтропия регулярного раствора принята
равной нулю, то в соответствии с уравнением (1.90) из выражения
(IV. 104) вытекает, что параметр со не должен зависеть от
температуры.
Поведение некоторых реальных растворов приблизительно
удовлетворяет уравнению параболы (IV. 104). Однако
параболическая форма для hE или gE не может служить доказательством
регулярности раствора.
Теория регулярного раствора внутренне противоречива. Как
уже отмечалось, энтропия регулярного раствора должна быть
равна энтропии идеального раствора, и отклонения от идеальности
обусловлены только тем, что энтальпия смешения не равна нулю.
В основе этого допущения лежит предположение о независимости
величины изменения энтропии при образовании смеси от
теплового эффекта, которое может строго выполняться только для
идеальных смесей, где /iM равна нулю. Конечные значения hM как
положительные, так и отрицательные оказывают влияние на
величину энтропии смешения.
Величина sE даже в тех случаях, когда она составляет
незначительную часть от sM<M>, играет существенную роль для
многих свойств смесей [97].
Используя соотношение Гиббса —Дюгема (IV.43), нетрудно
убедиться, что в интервале концентраций, где коэффициент
активности растворителя определяется уравнением вида (IV.97),
коэффициент активности растворенного вещества равен
lnv2 = ow? + A (IV. 105)
где J — постоянная интегрирования, ос — постоянный параметр.
Очевидно, что система уравнений
In vi = axi In уз = а*? + J (IV. 106)
не идентична системе уравнений (IV.97) и (IV.98).
126
Из сопоставления выражений (IV.97), (IV.98) и (IV. 106)
следует, что если уравнения вида (IV.97) и (IV.98) справедливы для
всей концентрационной области от хх = 0 до хг = 1, то постоянная
интегрирования в (IV. 106), очевидно, должна равняться нулю.
Однако это заключение не согласуется с экспериментальными
данными для многих реальных систем [98].
По мнению А. Г. Лесника [99], класс регулярных растворов
не так широк, как предполагалось при введении понятия о
регулярных растворах, но такие растворы встречаются чаще, чем
можно было ожидать из общих соображений.
Теория строго регулярных растворов относится к числу
наиболее простых решеточных теорий растворов. Она достаточно
строго обоснована с помощью статистической термодинамики
[81, 100, 101 ], тогда как ее модификации (субрегулярная,
квазирегулярная и другие модели) являются эмпирическими, поскольку
в них без должного физического обоснования вводятся поправки
на температуру, концентрацию и пр. Подробное изложение
теории приводится также в монографиях [102—104].
Основной параметр в теории строго регулярных растворов —
энергия взаимообмена (<о12) реакции образования единичной
связи 1—2 —определяется соотношением
«12 = «12 - 1 < 2 («и + и22) (IV. 107)
и характеризует различие в энергиях взаимодействия одинаковых
молекул и молекул разного рода.
Величина 2а>12 характеризует изменение потенциальной
энергии системы при замене пар 1—1 и 2—2 на две пары 1—2:
(1 — 1) + (2 — 2) = 2 (1 — 2), (IV. 108)
т. е. в процессе, который и называется квазихимической
реакцией.
Приближенный метод оценки величины конфигурационного
интеграла с помощью приемов комбинаторики на основе
предположения о «независимости пар» молекул (см. подробнее в
работах [102—104]) приводит к выражению
Xi*/[(tfi - Х1Я) (N2 - Х12)] = ехр (-йн/ИГ), (IV. 109)
где Ыг и N2 — числа частиц первого и второго сорта
соответственно; Х12 —среднее число пар 1—2 в растворе.
Эту зависимость называют квазихимической формулой или
уравнением квазихимического равновесия вследствие ее
формального сходства с выражением для константы равновесия
квазихимической реакции (IV. 108). Уравнение квазихимического
равновесия играет в теории строго регулярных растворов важную роль.
Оно устанавливает связь между средним числом пар 1—2 в
растворе и энергией взаимообмена.
Из соотношения (IV. 109) вытекает, что при ©12 = 0, очевидно,
Х12 = N1N2/(N1 + Af2), т. е. любое распределение молекул в
растворе равновероятно, иначе говоря, молекулы распределены
127
хаотически. При <о12 Ф 0 имеются отклонения от хаотического
распределения, т. е. «эффект упорядочения». Если ю12 < О» то
Х12 > N1N2/(Nl + ЛГ2), если ©1Я > 0, то Х12 < NM^ + N2).
Таким образом, ©^характеризует структуру раствора.
Для удобства вычислений термодинамических функций в
теории строго регулярных растворов вводят два параметра г\ и р,
которые определяют с помощью следующих соотношений:
г) = ехр ЫкП X* = 2^^/К^ + N2) (р + 1)]. (IV. 110)
Молярную свободную энергию смешения строго регулярного
раствора можно рассчитать согласно соотношению [103]
р «gM = N (Л1дГ + х2$) = RT{(1 - х2) In (1 - х2) +
+ х2 Ых2 + (-J-) [(1 - *2) In ф + 1 - 2*а)/(1 - х2) (1 + р) +
+ х2 In ф - 1 + 2х2)/х, (1 + Р)1} • (IV. 111)
где 2 — координационное число.
Избыточная свободная энергия равна
Коэффициенты активности компонентов, как это следует из
выражения (IV. 112), равны
*-НЖТ- <IV-"4>
Принимая во внимание соотношение Гиббса — Гельмгольца,
изменение энтальпии при образовании раствора из чистых
компонентов можно оценить следующим образом:
h\ c~u™= zco12X12 = [2z/( 1 + Р)] х2 (1 - х2) Afa13. (IV. 115)
Если энергия взаимообмена ю12 = 0, то, согласно
соотношениям (IV. ПО), г) = 1 и р = 1. Подставляя Р = 1 в формулы
(IV. 113) и (IV. 114), получаем Ух=\ и у2 = 1, т. е. раствор
в данном случае идеальный.
С помощью выражения sE = (hM —gE)/T, учитывая формулы
(IV. 112) и (IV. 115), можем найти выражение для энтропии строго
регулярного раствора.
Нетрудно убедиться, что функции gE/RT, hu/RT>
определяемые соотношениями (IV. 112) и (IV. 115), при заданном значении х2
зависят только от безразмерного энергетического параметра
128
®12/kT, который называют приведенной энергией взаимообмена
единичной связи 1—2. Формула (IV. 111) для изменения свободной
энергии gM и вытекающие из нее соотношения (IV. 113) и (IV. 114)
для коэффициентов активности рассматриваются в теории строго
регулярных растворов как «первое приближение» к описанию
реального поведения растворов.
Наряду с «первым приближением» в теории рассматривается
еще более. грубое «нулевое приближение», которое получают,
разлагая при небольших значениях ю12 параметр (} в ряд по
степени cb12/kT с точностью до членов первого порядка относительно
й12/ЛГ.
В нулевом приближении будем иметь
g* = RT[(l -*2)1п(1 -*з) + *21п*21 + *а(1 —xJNz&Ut
(IV. 116)
gE = frM = Х2(! _x2)Nzu12, (IV.117)
RTlnyi^zN&utfi, (IV. 118)
RT 1пу2 = гЫ&12#, (IV. 119)
sE = 0. (IV. 120)
Из формул (IV. 113)—(IV. 115) и (IV. 117)—(IV. 119) вытекает,
что при положительных значениях ю12 (образование пар 1—2
энергетически невыгодно) наблюдается эндотермический эффект
смешения и положительные отклонения от идеального поведения
раствора; при ю12 < 0 тепловой эффект смешения экзотермичен,
отклонения от идеального поведения отрицательные. Если ш12 >
> 0, то наблюдается тенденция систем к расслаиванию.
Все рассмотренные выше уравнения могут быть выведены при
условии, что энергия взаимообмена зависит от температуры [106].
В практике расчетов эта зависимость, как правило, принимается
линейной. Однако в работе Растоги [107] при допущении
постоянства коэффициента объемного расширения [108] было показано,
что зависимость <о12 = / (Т) является экспоненциальной, что
подтверждается экспериментальными данными [109, ПО]. Теория
конформальных растворов [111] также приводит к
экспоненциальной зависимости <о12 от температуры [112].
Допущение о линейной зависимости ©12 от Т никак не
противоречит теоретически полученному выводу об экспоненциальной
зависимости й12 от температуры, поскольку в небольшом
температурном диапазоне экспоненциальную кривую всегда можно
аппроксимировать прямой линией. Однако необходимо помнить,
что допущение о линейной температурной зависимости энергии
взаимообмена есть не более, чем приближение, и что более строгой
является экспоненциальная зависимость ю12 от Т.
В работе ЕсинаШЗ] показано, что уравнения первого
приближения строго регулярных растворов переходят к виду уравне-
5 Глазов В. М. и др. 129
ний нулевого приближения не только при малых абсолютных
значениях со12, но и при любых больших | ю121, если со12 < 0 (сильно
упорядоченные системы), а концентрация одного из компонентов
невелика. В этих случаях соответствующие параметры имеют
иногда иной физический смысл, чем в уравнениях регулярных
растворов. Кроме того, условные энергии взаимообмена в
противоположность регулярным растворам неодинаковы в формулах для
коэффициентов активности и для теплоты смешения.
«Эффект упорядочения» обычно не определяет полностью
величину избыточной энтропии смешения sE, которую можно выразить
в виде суммы тепловой (изохорной), объемной и конфигурационной
частей. В свою очередь тепловой вклад в энтропию может быть
дифференцирован с выделением колебательной, электронной,
а иногда и парамагнитной составляющих.
При достижении высоких температур можно допустить
независимость тепловой части избыточной энтропии от температуры,
что эквивалентно линейной зависимости энергии взаимообмена
от температуры.
С учетом сказанного, в нулевом приближении
квазирегулярных растворов получим следующие выражения для свойств
раствора [114]:
/г£ = ^2ш0^2, (IV. 121)
s* = Ыгщххххгу (IV. 122)
gE = Nz (ш0 - ЩьТ) хгх2> (IV. 123)
где ш0 и ©oi — параметры, не зависящие от температуры.
Для первого приближения квазихимической модели учет
температурной зависимости энергии взаимообмена приводит к
формулам:
hE = ЫгщХ1х2 [1 _ 2(сэ0 — &01T)/kT}9 (IV. 124)
gE = Nz К - ©оЛ *Л {1 - [(©о - ©и T)lkT\ хгх2\. (IV. 125)
Отклонение растворов от идеального поведения проявляется
в заметной асимметрии концентрационной зависимости
термодинамических свойств. Эта асимметрия обусловлена тем, что энергия
смешения при образовании раствора не полностью определяется
заданием наиболее вероятного числа парХ12 разносортных атомов
и соответствующих им энергий межчастичного взаимодействия щ-г
Эти энергии — не строго постоянные, а зависят от температуры
и состава раствора.
Наиболее простые соотношения можно вывести в
предположении, что энергия взаимообмена 012 линейно меняется с составом.
Несмотря на то, что последнее предположение строго обосновать
не удается, им широко пользуются, и в ряде случаев теоретические
выводы согласуются с опытными данными.
130
Харди [115] сделал попытку учесть зависимость энергии
взаимообмена от состава раствора с помощью модели
субрегулярного раствора.
В этой модели предполагается, что энергия взаимообмена
линейно зависит от состава, а энтропия смешения совпадает
с идеальной. В приближении модели субрегулярного раствора
формула нулевого приближения квазихимической теории для gE
имеет вид
gE = hE^zNx^{ _я2)(<»о_Й1я2). (IV.126)
Из соотношений (IV.85) и (IV. 126) нетрудно убедиться, что
в приближении модели субрегулярного раствора коэффициенты
активности компонентов раствора определяются выражениями:
RT In vi = Nzxl \{Щ - Щ) + 2*2©i], (IV. 127)
RT\ny2 = Nz(l- x2f (ю0 + 2*2%). (IV. 128)
Применение модели субрегулярного раствора при
термодинамическом анализе фазовых диаграмм бинарных систем
рассмотрено в работе [116].
В соответствии с результатами работ [114, 117, 118],
соотношение (IV. 126) удовлетворительно описывает изотермы
термодинамических свойств ряда силикатных расплавов, сульфидных
и металлических растворов, характеризующихся не только
асимметричными кривыми gE и hE, но и знакопеременными
отклонениями от закона Рауля. Однако это выражение не отражает
зависимости gE от температуры.
Предположения о линейной зависимости энергии смешения
от температуры и состава приводят к уравнениям [114, 119, 120]:
gE = Nzx2 (1 - *2) [щ - Й01Г -f" х2 (ю, - ffiuT)b (IV. 129)
sE = Nzx2(l -x2)&0i + Nzx&(l-X2)&iu (IV. 130)
RT In V! = Nzxl [®o - ©i - (®oi - ©n T)\ + 2Nzx\ (&г - ffin7),
(IV.131)
RT In V2 = Nz (1 - x2f [©o - й01Т + 2*2 (йх - ©u7)]. (IV. 132)
Ввиду того что применение моделей строго регулярного и
субрегулярного растворов, рассмотренных выше, ограничено,
предпринимаются попытки их модифицирования. Цель этих попыток —
расширить пределы применимости моделей для расчета фазовых
равновесий, для термодинамического анализа экспериментально
построенных диаграмм состояния, а также термодинамических
свойств. Одна из таких довольно удачных попыток была
предпринята Шарки и др. [121], которые предлагают усложнить
модель субрегулярного раствора, принимая во внимание не только
двойные, но и тройные группы атомов.
5* 131
Результирующая энтальпия смешения, полученная в этой
работе, имеет вид
hM « ахх\х2 + «2*1*2 — аз*?*!» (IV. 133)
где Од = 2&l2/zRT, (IV. 134)
ах и а2 — параметры, учитывающие структурные и объемные
эффекты при смешении, а также конфигурационную энтальпию
смешения. Параметр а3 в принципе должен зависеть от состава,
поскольку радиальное распределение атомов меняется с составом,
однако при расчетах его принимают постоянным.
Из анализа выражения (IV. 133) легко убедиться, что при
аг = <ха и а3 = О имеем приближение регулярного раствора;
при ах Ф а2 и а3 = 0 — субрегулярную модель, а при ах = а2
и —а3 < О — квазихимическое приближение.
Полученный в работе [121] результат целесообразно
модифицировать с учетом линейного характера температурной
зависимости энергии взаимообмена. Представим соотношение (IV. 133)
в более удобном виде, совпадающем по форме записи с
приближением Маргулеса:
/lM « Х\Х2 (о)0 + <д\Х2 + (02Х2)> (IV. 135)
Аналогичное разложение в ряд может быть применено и для
избыточной мольной энтропии смешения:
*£ = *1*2 Г <*пЛ*П. (IV. 136)
Ограничиваясь тремя первыми членами разложения, получим
уравнение
sE = хгх2 (щ\ + соцлсг + 0)21*2). (IV. 137)
Отсюда, с учетом выражений (IV. 135) и (IV. 137), можно записать
для избыточной свободной энергии
gE = хгх2 [со0 - ©oi^ + (о)! - <опТ) х2 + (<о3 - со21Г)х\\. (IV. 138)
Используя уравнение (IV.85), в рассматриваемом
приближении выразим коэффициенты активности компонентов:
RT In Yl = 4 [Щ — (0! — (С001 — 0)ц) 71] +
+ х\[2 ((ох - (опГ) + (о)2 - со21Г)(3*2 - 2)], (IV. 139)
RT In у2 = *? [о)0 - щхТ + 2 (о)! - сопТ) х2 +
+ 3(<©2-©2iT)xf]. (IV. 140)
Принципиальным недостатком теории строго регулярных
растворов является допущение о том, что изменение объема при
смешении равно нулю. Следствия, вытекающие из данного допущения,
в рамках квазихимического метода расчета термодинамических
132
функций были тщательно проанализированы в работе И. Р. Кри-
чевского [122]. Полученные там формулы отличаются от
уравнений нулевого приближения теории строго регулярных растворов
дополнительным членом (р — pTj v, где v — объем смеси.
Позднее были предложены другие методы расчета
термодинамических функций растворов, исходящие из межмолекулярных
сил, которые позволяют учесть и влияние «объемного» эффекта.
И. Пригожий и др. [123—125], используя методы теории
«свободного объема» жидкостей, разработанные Леннард-Джонсом
и Девонширом [126, 127] в приложении к растворам, выдвинули
теорию «свободного объема» растворов. Другой интересный подход
к этой проблеме был предложен Лонжет-Хиггинсом [111 J. В
развитой им конформальной теории растворов модель жидкого
состояния вообще не используется, но введены довольно жесткие
ограничения относительно межмолекулярного взаимодействия.
Молекулы «конформального» раствора должны быть близки по
форме, размерам и межмолекулярному взаимодействию. На
основании теории конформальных растворов коэффициенты активности
бинарного раствора определяются выражением:
RTlnyt = tfEMdl2$ (IV. 141)
которое весьма похоже на уравнение для коэффициентов
активности компонентов в нулевом приближении теории строго
регулярных растворов. В уравнении (IV. 141) параметры Es и d12
рассчитывают из экспериментальных данных по теплотам
испарения и критическим параметрам соответственно. Сравнительный
анализ моделей конформального и регулярного растворов дан
в обзоре [128].
В заключение отметим, что в реальном растворе форма и
размеры молекул компонентов могут заметно отличаться одни от
других, что часто служит причиной отклонения свойств растворов
от идеальных.
В первых работах Флори [129—131], Хаггинса [132—133]
и других исследователей основное внимание было уделено
изучению наиболее простого случая, когда избыточная энергия
смешения раствора равна нулю, и, следовательно, избыточная энтропия
является единственной причиной отклонения от идеального
поведения (атермические растворы).
За последние годы наметились большие успехи в
аналитическом представлении термодинамических свойств растворов,
благодаря появлению новых корреляционных уравнений Вильсона,
Ренона — Праузница, UNIQUAC и др. [134]. Эти уравнения
в большей мере, чем ранее существовавшие, основаны на
молекулярных представлениях, хотя ни одно из них нельзя считать
результатом последовательной обработки той или иной
молекулярной модели раствора. Поэтому параметры этих уравнений также
находят по существу эмпирическим путем.
133
5. ФОРМАЛЬНО-МАТЕМАТИЧЕСКОЕ ОПИСАНИЕ
ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ РЕАЛЬНЫХ СИСТЕМ
Поскольку явные зависимости термодинамических свойств
от параметров состояния не могут быть получены с достаточной
точностью, часто приходится использовать их формальное
описание. Рассмотрим некоторые широко применяемые
интерполяционные формулы.
Любое интегральное избыточное термодинамическое свойство
уЕ или парциальное избыточное термодинамическое свойство
t-того компонента yf в бинарной системе А — В можно
представить в виде простых рядов по составу, предложенных Маргуле»
сом [1351:
УЕ = хгх~2 (<7о + ?i*2 + q*4 H )> (IV. 142)
yf = х\ (а0 + ахх2 + а2х\ Н ), (IV. 143)
V* = х\ (bQ + Ьгх2 + Ъ2х\ +•••). (IV. 144)
где у может быть свободной энергией Гиббса, энтальпией,
энтропией и т. д.
Коэффициенты этих рядов не являются независимыми. Связь
между ними определяется соотношениями:
я* = (п + 1) (qn - qn+i)> bn = (n+ \)qn,
ап = Ъп- bn+1 (n + Щп + 2) (IV. 145)
Если принять, что все коэффициенты, кроме q0, a0i b0 равны
нулю, то получим уравнения регулярного раствора (IV.97) и
(IV.98). Ограничиваясь рассмотрением только первых двух
членов разложения, придем к модели субрегулярного раствора.
Использование соотношений Маргулеса неудобно из-за того,
что во всех трех уравнениях присутствуют разные коэффициенты.
Кроме того, замена в выражениях (IV. 142)—(IV. 144) переменной
х2 на хг также приводит к новому набору коэффициентов. Другой
недостаток состоит в том, что в концентрационном диапазоне от
х — 0 до х = 1 ряды Маргулеса в сильной степени
неортогональны. Например, для рядов Маргулеса, включающих члены
шестого порядка, степень неортогональности Р = 0,9931. В случае
неортогональных полиномов значения первых коэффициентов
сильно зависят от степени ряда, т. е. при увеличении степени
ряда требуется пересчитывать все ранее найденные значения
коэффициентов. Отсюда следует вывод, что численные значения
коэффициентов неортогональных полиномов не могут заключать
в себе определенного физического смысла. Это делает невозможным
сопоставление свойств различных систем путем сравнения
численных значений коэффициентов.
К недостаткам уравнений Маргулеса следует отнести и то, что
в области малых значений х при больших п величина множителя #"
134
становится пренебрежимо малой. Поэтому коэффициеОТ an
должен приобрести очень большое значение, чтобы вклад слагаемого
апхп стал заметным. В этой связи при Р > 0,99 коэффициенты
в уравнениях Маргулеса должны рассчитываться со значительно
большей точностью, чем исходные данные [136]. В работе [137]
показано, что при машинном расчете для уравнения Маргулеса,
содержащего 19 членов, недостаточно даже двойной точности.
Отмеченных недостатков можно избежать, представив
концентрационную зависимость термодинамических свойств в виде
ортогональных полиномов [138].
Хотя для аппроксимации зависимостей термодинамических
свойств от состава в бинарных системах можно использовать
различные ортогональные функции, число коэффициентов,
необходимое для получения желаемой точности согласования
экспериментальных и теоретических кривых, в значительной степени
зависит от выбора того или иного полинома. Поскольку в
соответствии с положениями квазихимической теории зависимости
избыточных термодинамических свойств от состава описываются
алгебраическими, а не тригонометрическими или
трансцендентными уравнениями, то следует отдать предпочтение
алгебраическим ортогональным полиномам. При этом выбираемые
ортогональные функции должны приводить к соблюдению закона
Рауля и обеспечивать конечные значения и плавный характер
зависимости о = / (х) в предельно разбавленных растворах [138].
Кроме того, так как многие термодинамические свойства должны
удовлетворять соотношению Гиббса — Дюгема, то при выборе
ортогональных полиномов следует исходить из простоты
дифференцирования и интегрирования. Желательно также, чтобы при
сохранении первого или двух первых слагаемых ортогональных
функций получались выражения для регулярного и
субрегулярного растворов соответственно.
Перечисленным условиям удовлетворяют полиномы Лежандра,
которые являются строго ортогональными в интервале
концентраций 0 < х <: 1. Первые пять функций Лежандра в этом
концентрационном диапазоне имеют вид:
P*(x) = L
Р±(х) = 2х— 1,
Р2(х) = б*2 — 6х+ 1. (IV. 146)
Р3 (х) = 20*8 — 30*2 + 12*.— 1,
р4 (Х) = 70*4 — 140*3 + 90х2 — 20х + 1.
135
Общая рекуррентная формула Для полинома Рп (х) запишется
следующим образом [134]:
(2/г— 1) (2дс— 1) 0 я—1
*.<*)-■
Рп-1
•Р*-л(х).
(IV. 147)
Графическое изображение пяти полиномов Лежандра показано
на рис. 27, на котором видно, что в концентрационном диапазоне
О <: х < 1 значения всех полиномов не превышают ±1. В отличие
от функций х, х2, г5, ..., которые приобретают максимальное
значение при х = 1 и равны нулю при х = 0, полиномы Лежандра
имеют экстремумы при различных
значениях х. Кроме того, начиная
с функции Р3 (х) каждый из
последующих полиномов Лежандра имеет
на один экстремум больше, чем
предыдущий. Поэтому разница в
согласовании экспериментальных и
теоретических кривых не увеличивается вблизи
составов х = 1 или х = 0, как при
использовании простых степенных
рядов. Рис. 28 также наглядно
показывает отсутствие взаимосвязи между ко-
вэффициентами полиномов Лежандра,
первых пяти ортогональных по- Т^К K3K Каждая фуНКЦИЯ Рп (Х) Ха-
ЛИНконцентр*цийРо <"н<е?вале растеризуется различным видом
функциональной зависимости. Выражения
для избыточной свободной энергии
смешения раствора и коэффициентов активности компонентов с
помощью ортогональных полиномов Лежандра в общем виде можно
представить следующим образом:
я'
Pi
1,0
0,2
О
-0,2
-0,6
-1,0
А Р"
L X ^^ ^г ^^
\/ Рг
у ' I
У А
\чп\
Ы//\
i I
О 0,2 0,1 0,6 0,8 X
п=0
п'
RT\nyx=xl% апРп{х2\
л=0
(IV. 148)
/?ПП72=^Ь ЬпРп{х2).
В этих уравнениях связь между коэффициентами qn, an и Ь,г
определяется соотношениями:
an = (n+l)qn + (2n+l) Е%Я4*(-1)\ (IV. 149)
k=n'-n
bn = (n+\)qn + (2n+l) Б qn+k,
(IV. 150)
136
?„ = an/(tt+l) + (2tt+l) g" (re + fe)^|
w A+1)
(IV.151)
?n=M«+i)+(2n+i) 2 (n+y^+t+D' <IV-152>
»,.«,+(2«+1)Т-^ЙЙ- 0V.1S9
Ограничиваясь первыми тремя слагаемыми, с учетом выражений
(IV. 146), (IV. 149) и (IV. 150) соотношения (IV. 148) можно
переписать в виде
qE = хгх2 [<7„ + Яг (2*2 - 1) + Л (6x1 - 6*2 + 1)], (IV. 154)
RT\ny1=x22[q0 + ql(4x2-3) + q2(l8x22-24x2 + 7)], (IV. 165)
Д7 In 72 = х\ [<7o + <7i (4х2"1) + ?2 (184-12х2+1)]- .
(IV. 156)
Выражения для коэффициентов активности компонентов,
записанные в виде (IV. 155) и (IV. 156), уже не являются ортогональными
функциями. Поэтому чтобы иметь строго ортогональные
полиномы во всех трех уравнениях, следует сохранить три набора
различных коэффициентов.
Представление термодинамических функций в виде
ортогональных полиномов Лежандра особенно целесообразно в системах
с сильным химическим взаимодействием, характеризующихся
сложным характером зависимостей термодинамических свойств.
Однако как показано на примере бинарных систем Sn — Hg,
Sn — Cd и Sn — Zn [138], эти полиномы полезно также
использовать для сравнительного описания поведения
термодинамических свойств и в более простых системах, где согласование
расчетных и экспериментальных кривых достигается с учетом
пяти или меньшего числа коэффициентов, которым в этом случае
можно приписать определенный физический смысл, опираясь на
молекулярно-статистические представления.
В работах [139—140] для описания избыточных интегральных
функций растворов было предложено использовать
тригонометрический ряд Фурье для синусов:
уЕ = £?„ sin (шт*), (IV. 157)
который ортогонален в концентрационном интервале 0 < х <: 1.
Основное возражение против такого представления состоит в том,
что аппроксимация термодинамических свойств
тригонометрическими функциями на некоторых концентрационных участках
137
не удовлетворяет термодинамическим требованиям [138].
Выражения для избыточных парциальных мольных величин вида
п'
У\ = 5? Яп fsln (nnx2) — ппх2 cos (ппх2)],
п=1
полученные на основании уравнения (IV. 157), также не являются
ортогональными функциями.
Термодинамические свойства растворов можно представить
и с помощью тригонометрического ряда Фурье для косинуса
в виде выражений [138]:
в?
91
= *i Ub + S an cos (nnx2) t (IV. 158)
= х\ \а0 ~\ г С — а0 — ^ ап (sin (nnx2)/nn +
+ л:^ cos (ппх2)) |, (IV. 159)
у* = х2 С — %*2 -S fln sin (плх2)/пл . (IV. 160)
где С — постоянная интегрирования.
Здесь функции (IV. 159) и (IV. 160) также не являются
ортогональными. Кроме того, в простых системах при одной и той же
точности согласования с экспериментальными данными
аппроксимация термодинамических свойств тригонометрическими рядами
требует учета большего числа членов по сравнению с простыми
рядами или полиномами Лежандра.
Избыточные термодинамические свойства растворов можно
успешно описать хорошо изученными полиномами Чебышева.
В этом случае выражения, аналогичные (IV. 148), примут вид:
п' п'
уЕ = хгх2^ qnTn(x), У\=&Ъ апТ1{х),
9i = x\t ЬпТп(х), (IV. 161)
где первые три полинома Чебышева, преобразованные для
интервала 0 < х < 1, определяются соотношениями:
Г0(х)=1, 7\(*) = 2*-1, Г2(*) = 8*2-8*+1. (IV. 162)
Общая рекуррентная формула для полинома Тп имеет вид:
Тп (х) = 2 (2* - 1) Г».* (х) - Тпл (х). (IV. 163)
138
Коэффициенты 6 соотношениях (IV. 161) связаны между собой
следующим образом:
*,, = (*+ !)?*+ Sf (-\)k(2n + 2&k)qn,k (IV.164)
(6 = 1/2, если п = 0, 6=1, если п > 0),
Ья=(л+1)?я + £ "(2/1 + 28*) W- (IV.165)
/г=1
Умножая полиномы Чебышева на множитель (4х — 4д:2)~1/4, можно
привести их к строго ортогональному виду для интервала 0 <
< х < 1, но эта процедура приводит к большим математическим
сложностям и не дает никаких преимуществ по сравнению с
использованием полиномов Лежандра.
Для представления концентрационной зависимости
термодинамических свойств используют также уравнения Редлиха —Ки-
стера [140]:
yE = x1x*T>q,(l -2*2)М, (IV. 166)
Ух = 4 I Я1 ((2/ - 1 - 2/*2] (1 - 2х2)'-2} (1V.167)
У* = х\ Е qt [(1 - 2jx2) (l - 2х2Г2]. (IV. 168)
/=i
Для уравнения (IV. 166) с семью слагаемыми наибольшее значение
параметра неортогональности Р равно 0,9669, что существенно
лучше по сравнению с величиной 0,993Ь для уравнения
Маргулеса.
Полиномы Лагерра и Эрмита ортогональны в диапазоне
0 <: х < оо и —оо < х < оо соответственно и поэтому непригодны
для аппроксимации термодинамических функций бинарных
систем.
Вильяме [139] предложил метод группирования полиномов
в Zn (x) — функции и представил избыточные свойства, а не
параметр со в виде ряда по Zn (x). Преимущество такой
аппроксимации заключается в том, что она согласуется с квадратичной
зависимостью Даркена для разбавленных растворов. К числу
недостатков следует отнести лишь приблизительную
ортогональность Z-функций и отсутствие рекуррентной формулы для членов
с п > 6.
Остальные ортогональные полиномы, не нашедшие здесь
отражения, как правило, непригодны для простого представления
термодинамических свойств.
139
Крупковский [141] предложил для учета концентрационной
асимметрии избыточной свободной энергии смешения ввести
коэффициент асимметрии kf не зависящий от температуры, по
уравнению
^"fl-^r-1*0-*^' (IV'169)
или, в случае противоположного характера асимметрии, по
формуле
^(^г-**10"*0*"1' (IV,170)
и соответственно для коэффициентов активности компонентов
In Yl = со' (Г) {(1 - x2)k - [k/(k - 1)] (1 - х*)*"1 + 1/(* - 1)Ь
(IV.171)
lnY2 = c0'(T)(l-;t2)ft (IV. 172)
или
1пу1 = (о/(Л(1-.^, (IV.173)
In Va = со'(Л {(1 -х^-Щк - 1)](1 -*0*-i+ l/(ft- 1)},
(IV. 174)
где со' (Г) = а/71г, а и г — константы, характеризующие данный
раствор, со' (Г) — приведенное значение энергии взаимообмена,
равное со (T)/RT. Легко убедиться, что при k = 2 формулы Круп-
ковского переходят в симметричные соотношения регулярных
растворов.
Соотношения (IV. 171)—(IV. 174) удовлетворяют уравнению
Гиббса — Дюгема. Выбор той или иной системы уравнений
зависит от характера симметрии, свойственной данной системе [142].
Параметры со и £ находят из экспериментальных данных. При
практических расчетах обычно принимают, что ю (Т) линейно
зависит от температуры, следовательно,
со' (Г) = ш (T)/RT = а/Т ± Ь. (IV. 175)
В случае положительных отклонений от закона Рауля
соотношение (IV. 172) применимо к компоненту с меньшим атомным
радиусом [143]. Из экспериментальных данных получено, что
для положительных отклонений от закона Рауля показатель k,
как правило, находится в области значений от 1 до 2, однако
указывается [144], что при отрицательных отклонениях от закона
Рауля k > 2. Подробный анализ функций fx^x^ и /2 =
= jc* — Ш(к — 1) 1 4"1 + \l(k— 1) для значений 0, 1 <: k < 3 дан
в работе [145]. Установлено также, что коэффициент асимметрии
тем ближе к 1, чем больше асимметрия таких термодинамических
свойств, как энтальпия смешения или избыточная свободная
энергия.
140
Хотя метод Крупковского и позволяет учесть незначительную
асимметрию концентрационной зависимости термодинамических
свойств, он непригоден для систем со знакопеременными
отклонениями от закона Рауля [117]. Существенный недостаток этого
метода заключается еще и в том, что в рассматриваемом
приближении выражения для коэффициентов активности компонентов
представлены в виде произведения двух функций:
У1(х, T) = f(T)f(x)t
одна из которых характеризует зависимость от температуры,
а другая — от концентрации. Следствием такого формального
представления термодинамических свойств является то, что
при Т = const интегральные и парциальные мольные величины
строго пропорциональны одни другим, а избыточные величины
не зависят от температуры [146].
При анализе данных по фазовому равновесию наиболее часто
используют вариант линейной температурной аппроксимации
параметра со. В этом приближении рассмотренные выше полиномы
по форме записи остаются такими же, но допускается, что каждый
из входящих в них параметров линейно связан с температурой.
В соответствии с изложенным выражения для коэффициентов
активности компонентов бинарного раствора в приближении
различных полиномов можно представить в следующем виде:
в приближении рядов Маргулеса
RT In vi = xl [ft - ft - faoi - fti) T] + x\ [2 (ft - qnT) +
+ (ft - ЯпТ) (3*2 - 2) + • • •], (IV.176)
/?TlnY2 = ^too-fti7, + 2(9l-ft1r)x2 + 3(ft-ft171)xi+...];
в приближении полиномов Лежандра
RT lnvi = *1 too - ЯохТ + (ft - qnT) (4x2 - 3) -f
+ (ft - ftiT) (18x1 - 24*2 + 7) + •.. ],
RT In Y2 = x\ [ft - ЯохТ + (ft - ftiT) (4*2 - 1) +
+ (ft - qnT)(l8xi- \2x2 + 1) + ...]; (IV.177)
в приближении полиномов Редлиха — Кистера
RT In yi = xl [ft - qoiT + (ft - ftiT) (3 - 4*2) +
+ (ft - fti^) (5 - 6*2) (1 - 2x2) +...],
RT In y2 = x\ [ft - ftiT + (ft - qnT) (1 - 4x2) +
+ (ft - qnT) (1 - 6x2) (1 - 2xt) +...]; (IV. 178)
141
в приближении тригонометрических рядов Фурье для синусов:
RT In yi = £ Як [sin (кпхъ) — knxt cos (knx2)] —
k=\
—T S Qk [sin (knx2) — knx2 cos (knx2)],
k=l (IV. 179)
RT In Ya = S 9* lsin (fowfe) + *ЛЛ:1cos (*^^a)l ~~
—71 X!: q'k [sin (Ллаго) -f- u^i cos (&л#2)]-
В работе [147] предложена модель линейно-температурной
аппроксимации, согласно которой в уравнениях, связывающих
коэффициенты активности компонентов с избыточными
парциальными мольными свойствами растворов,
|if = RT In yi = hE - Tsf, (IVЛ 80)
(if = RT In 72 = й? - Tsb (IV. 181)
Величины /if и sf не зависят от температуры и произвольным
образом зависят от состава. Используя для представления
концентрационной зависимости избыточных парциальных мольных
свойств гиперболические функции, выражения для коэффициентов
активности компонентов можно записать следующим образом
[1471:
RT In yi = axl {1 + sh С (х2 — *ь) — С{1 — *2)chC (*2 — x0)\ —
—Tbx\ {1 + sh D (x2 — x0) — D (1 — *2) ch D (*2 — x0)b
(IV. 182)
Я7 In Y2 = a (1 - x2f { 1 + sh С (*a - *0) + Cx,chC(;c2 - *0)} —
—Tb(l - x2f \l + shD(x2- x0) -\- Dx2chD(x2- x0)}.
где С и D — параметры, не зависящие от температуры и состава;
*2 = хо — координата точки, относительно которой ведется
рассмотрение и которая может иметь любое значение от 0 до 1.
В работах [140, 148] допускается более сложная по сравнению
с линейной зависимость параметра со от температуры.
Способ, предложенный в работе [140], основан на разложении
в ряд Тейлора по температуре функции gE (T, х).
Ограничиваясь первыми тремя членами разложения, с учетом
общеизвестного термодинамического соотношения
g£ = hE- TsE
и выражений (1.90) и (1.97), найдем
gE(T, x) = hE -TsE-cE (Т-в-Т\п^-), (IV.183)
142
где 0 — температура, при которой проводят разложение, значения
hEy sE и сЕр берут при Т = 0.
Соотношение для gE (7\ х) можно представить также с помощью
одного из рассмотренных выше полиномов, например полиномов
Редлиха — Кистера, в виде
k
gE (Г, Х) = Xlx2 Б ю, (Т) (1 - 2х2)/-Ч (IV. 184)
Сравнивая выражения (IV. 183) и (IV. 184), можно записать
к
gE(T, *) = *i*22 [hf-Tsf -cf (r-e-Tln-J-)] X
X (1 -2^)М. (IV. 185)
Авторы работы [148] получили выражения для коэффициентов
активности компонентов в виде двойного ряда Тейлора по составу
и температуре, представив величину х2 (d\i2/dx2)Tt р следующим
образом:
N М
х2 (д^дх^г, Р = Б Б (W (Т - Т0)т (хг - Хо)", (IV. 186)
л=0 т=0
где х0, Т0 — координаты точки, вокруг которой производится
разложение, (Jm„ — коэффициенты разложения. Записав (х2 —х0)п
в уравнении (IV. 186) в виде бинома Ньютона
(х2 -х0)п = S cqn{-\)qхЪхГя (IV.187)
<7=0
и поделив обе части уравнения (IV. 186) на x2i получим
N М
(dii2!dx2)T, р = (1 /х2) Б Б Рт„ (Т - Го)'" (~ 1)" х% +
п=0 пг—0
N М /1-1
+ Б ГР««(Г-7о)жЕс£(-1)'4х5_в'1 (IV.188)
л—О т=0 <7=0
Проинтегрировав уравнение (IV. 188) по х2, получим
ъ(х9 r) = inx2E S Рт«(г^г0)т(-1Г4 +
W М ( л-1 " )
+ Б Б Рт„ (Г ^ Г0)т Б 4 {-\)qxlxTql{n -q)\+ J2,
n=l m=Q {<7=0 )
(IV. 189)
где J2 — постоянная интегрирования.
В соответствии с законом Рауля,
lim ц2 (х, Т) = |х5 (*, Т) + ЯГ In x2. (IVЛ90)
143
Из сравнения выражений (IV. 189) и (IV. 190) следует
ЯГ=£ £ РтЛГ-ТоП-!)"*?, (IV.191)
h = \il(x, Л-ES Рт„(7' - Г0)т £ 4(-1)<4'(" - Я)-
(IV. 192)
Учитывая уравнения (IV. 191) и (IV. 192), выражение (IV. 189)
можно записать в виде
N М
(i2 (х, Т) = & (Т) + RT In ж2 + Б £ pmn (Г - Го)"1 х
х Г £ с« (-1/4 {хГя - 1)/(п - <7)1 • (IV. 193)
U=0 - J
Используя соотношение Гиббса — Дюгема и уравнение (IV. 186)»
получим
(1 - х2) (д^/дх^т, р = £ £ (-1)" pmn (г - То)" (хг - х0)».
я=0 пг—0
(IV. 194)
Проделав те же преобразования, получим аналогичные выражения
для цх (*, Т):
ц(х, T) = ii\(T) + RT\n(l -х2) + £ £ ^„(-ХПТ-ТоГх
/г=1 m=0
X { "£ (-1)' cl (1 - xo)" [(1 - х2Г" - 1 ]/(n -<?)}, (IV. 195)
где RT = £ £ pm„ (Г - To)" (1 - *„)"• (IV. 196)
n=0 m=0
С учетом формул (IV. 193) и (IV. 195) выражение для избыточной
свободной энергии смешения можно записать в виде:
М N ( п-\
g£= £ £ Ртп(Г-Г0Г (-1)"(1 -Х2) £ (-1)40 -ХоУХ
X 1(1 - **)""' - 1 ]/(п - q) + *2 £ (-1)М4(*Г< - 1)/(« - q)\
(IV.197)
144
Применяя соотношение (1.90) к избыточным величинам, легко
убедиться, что
s£=SS Р««(Т - То)"1-1 m( (-1)"(1 -х2) S (-1)«сяп х
X (1 - *оП(1 - х2)п-д - 1]/(п - q) + *2 2J (-\)qcUl X
х(*Г'-0/(1-<7)}- (IV. 198)
Из выражения (1.113) в приложении к избыточным величинам
и соотношений (IVJ97) и (IV. 198) получим
М-Х N (
ftE=SS [|W + (m + 1)(W.„Л (Т- То)"(-1)" (1 -Хг) X
m=0 л=1 I
х £ (-1)М0 -*<Л(1 -^Г'- 1 ]/(«-?) +
<7=0
+*2 Е (-1)М4(*Г<- i)/(n-<7)+ £ fWr-ТоГ х
<7=0 П=1
х f(-i)n(i-Vi;(-i)'4(i -*л(1-*«г*- п/(л-9)+
+ *2|(-1Г(М(Г- 0/(л-9)]}. (IV.199)
Для нескольких простых случаев из соотношений (IV. 193)
и (IV. 195) можно получить выражения для химических
потенциалов компонентов, которые соответствуют известным моделям
растворов [148]. Отсюда следует, что между интерполяционными
формулами и теоретическими уравнениями имеется прямая связь
и можно предполагать, что члены более высоких порядков вызваны
отклонениями от теоретических представлений.
6. КОНЦЕПЦИЯ АССОЦИИРОВАННЫХ РАСТВОРОВ
Как уже отмечалось, в регулярных растворах теплота смешения
по порядку величины близка к тепловой энергии RT
(~2500 Дж/моль при обычных температурах). Однако имеется
обширный класс реальных растворов, в которых энергия
взаимодействия между молекулами на порядок и даже больше может
превышать тепловую, в результате чего в растворе образуются
относительно устойчивые молекулярные конфигурации
различного сорта. Образование таких конфигураций приводит к измене-
145
нию вращательного и колебательного спектров составляющих
молекул. В общем случае в растворе различают молекулы двух
видов:
1) молекулы или мономеры, на вращения которых не
повлияли соседние молекулы;
2) ассоциативные комплексы — любые сравнительно
непрочные соединения между молекулами одного или разного рода,
вращательное и колебательное состояния которых в результате
взаимодействия изменились (ниже для краткости используем
термин «ассоциат»),
В 1908 г. Долежалек [1511 высказал соображение о том, что
все неидеальные растворы подчиняются закону Рауля, а
наблюдаемые отклонения от идеальных закономерностей являются
кажущимися и связаны с неучетом сольватации, приводящей
к образованию различных комплексов. По существу, в работе
Долежалека речь шла об ассоциатах различного рода,
усложняющих молекулярный состав раствора.
Растворы, в которых наряду с ван-дер-ваальсовыми
взаимодействиями имеются специфические направленные
взаимодействия, называют ассоциированными растворами. При
теоретическом рассмотрении их равновесных свойств возможны два
подхода [149].
Основной из применяемых в настоящее время методов
базируется на теории ассоциативных равновесий, в которой раствор
рассматривается как смесь ассоциатов разного рода и мономеров,
а равновесие между ними определяется законом действующих
масс. Другой подход основан на решеточной модели раствора,
образованного мономерными молекулами, между которыми
существуют сильные направленные взаимодействия.
Области применения двух указанных методов описания
ассоциированных систем различны [150]. Решеточная теория,
разработанная в наиболее общем виде в трудах Баркера, имеет
преимущества при исследовании систем с направленными
взаимодействиями, не приводящими к образованию определенных
соединений (в частности, теория позволяет описывать системы с
ассоциацией в циклы и разветвленные цепи различного размера и
строения).
При изучении систем, в которых неаддитивность
взаимодействий играет существенную роль (наиболее распространенный
случай), необходимо обращение к теории ассоциативных
равновесий. Она исходит из определенной модели реакции ассоциации,
например, реакции типа^
тА + пВ^АтВп
или реакции последовательной ассоциации:
Aul + Ax^Ah
146
приводящей к образованию ассоциатов различных размеров (i —
= 1,2,..., оо). Основное уравнение теории представляет закон
действующих масс, а основные параметры — константы
равновесия реакций ассоциации и энтальпии ассоциации.
В ассоциированном растворе, который по определению состоит
из мономеров и ассоциатов, не рассматривают те взаимодействия
между различными видами молекул, которые достаточно сильны,
чтобы привести к ассоциации. Отсюда следует, что взаимодействия
между ассоциатами и мономерами — ван-дер-ваальсовские и
поэтому ассоциированные смеси по свойствам не должны сильно
отличаться от простых растворов неполярных молекул. Иными
словами, в ассоциированном растворе большую часть отклонений
от идеальности объясняют взаимодействиями, приводящими к
образованию самих ассоциатов.
В зависимости от степени приближения смесь ассоциатов
и мономеров считают идеальной, регулярной, атермальной и т. д.
[102, 151—154].
Идеальный ассоциированный раствор (идеальная смесь
мономеров и ассоциатов при всех концентрациях) — простейшая
модель, в которой пренебрегают различием в энергиях ван-дер-
ваальсовых взаимодействий между молекулярными формами
различного сорта и разницей в размерах этих форм.
Стекки [1551 обобщил концепцию идеальных ассоциированных
растворов, предположив, что различия в ван-дер-ваальсовых
взаимодействиях между частицами можно учесть с помощью
коэффициентов активности молек/лярных форм в приближении
модели регулярного раствора. Следовательно, такая модель
представляет собой регулярную смесь мономеров и ассоциатов и
называется регулярным ассоциированным раствором.
В настоящее время эту теорию широко используют при анализе
фазовых равновесий в двойных и квазидвойных системах,
включающих полупроводниковые соединения, где присутствие
ассоциатов в жидкой фазе весьма вероятно [156].
Химические потенциалы компонентов
ассоциированного раствора
Следуя Пригожину [24], рассмотрим ассоциированный
раствор компонентов А и В. В общем случае можно записать, что
бинарный раствор А — В включает молекулярные индивиды типа
A,, Bt9 AkBt (г, /, ky I = 1, 2, 3, ...). Если общие числа молей А
и В в растворе равны пА и пв, а числа молей различных
фактически присутствующих в растворе молекулярных форм равны паг
n>Bt> nAkBr то (в расчете на мономерные единицы)
пА-Ъ гпаг + Е S knAkBly (IV.200)
Г к I
пв = Ц tnB. + Б S 1пАв (IV.201)
147
Химические потенциалы присутствующих в растворе
молекулярных форм определяются выражением
ра = (dG/dna)Pt г. Прфа (а = Ar; В,; AkB<) (IV.202)
Измеряемые на опыте химические потенциалы компонентов А и В
равны:
рА = (dG/dnA)p, Tt Пв- рв = (dG/dnB)Pt т. пА. (IV.203)
Ассоциаты в растворе находятся в равновесии между собой
и с мономерами А1 и Bv Возможные реакции между этими
частицами можно представить уравнениями:
А, = rAx; Bt = tB±; AkBt = kAx + IBX. (IV.204)
При равновесии должны соблюдаться условия:
\*>аг = r\iAl\ [*>Bt = t)*>Bx\ \^Акв1 = k\iA± + 1рвг (I V.205)
Запишем полный дифференциал G при постоянных Тир для
ассоциированной системы и будем рассматривать ее как смесь
различных ассоциатов.
Тогда
dG = Е \iAr dnr + Е Vb{ dnt + E E Рал. dnA^ (IV.206)
Используя условия (IV.205), на основании выражения (IV.206)
получим
dG = \iA Е rdnAf + \ib, E t dnBt + \iA, E I] & ^л.в, +
+Ц^Е Е /d/Ц^,. (IV.207)
Согласно уравнениям (IV.200) и (IV.201),
dnA — Е г ^плг + Е Е * ^п^в., ^в =
г т k I * 1
= Е * *ta, + Е Е / ^Л. (IV.208)
Отсюда можно записать
dG = \iAl dnA + PBtdriB. (IV.209)
Для любой двойной системы с постоянными Тир
dG = \iAdnA + \iBdnB. (IV.210)
Уравнения (IV.209) и (IV.210) должны быть тождественны
при любых значениях dnA и dnBf что возможно при условии
M = Mn JAB = (*Bi. (IV.211)
Таким образом, измеряемые на опыте химические потенциалы
компонентов |лА и \iB равны химическим потенциалам мономерных
молекул. Соотношения (IV.211) — общие, они справедливы для
любой системы, в которой протекают реакции ассоциации,
независимо от типа этих реакций и степени отклонения ассоцииро-
148
ванной смеси от идеальной. Они применимы не только к жидким
системам, но и к ассоциированным газам.
На основании выражений (IV.211) и закона действующих масс,
используя общеизвестные термодинамические соотношения, можно
получить формулы для расчета термодинамических функций
смешения ассоциированных растворов.
Активности компонентов регулярного
ассоциированного раствора
Покажем теперь, каким образом можно рассчитать
коэффициенты активности регулярного ассоциированного раствора.
Приняв во внимание условие (IV.211), запишем уравнение
Гиббса—Дюгема при постоянных Тир для бинарной смеси,
в которой происходит ассоциация молекул, в виде
nAd\iA2t + nBdiiBl=0 (IV.212)
Подставив в формулу (IV.212) выражения (IV.200) и (IV.201),
получим:
£ rnA d\iA< + Ti 2] ЬпАф, d\iA< -f E tnBi d\iB, +
r r * k I m 1 t t l
+ S I>^fl/<W==0 (IV.213)
k I * L x
Отсюда, приняв во внимание соотношения (IV.205), найдем
Ц tiArd\iAr+ Е nBid\LBt+ S S ЧВ/^Л =0. (IV.214)
Подставив в уравнение (IV.214) выражение для химических
потенциалов молекулярных форм в развернутом виде, легко
убедиться, что
S *Ard In yAr + 2 *Btd In yBf + S S *^B,d In y^, = o,
(IV.215)
где уагу yst и yAkBl — коэффициенты активности
присутствующих в растворе молекулярных индивидов различного рода.
Отсюда видно, что уравнение (IV.215) по форме совпадает
с уравнением Гиббса—Дюгема для смеси, состоящей из веществ Ап
Bt и AkBh
Известно, что коэффициент активности компонента i
регулярного n-компонентного раствора определяется уравнением [156]
RT In yt - t Щ1$ +SS xhxj К- + ©« - (o,y), (IV.216)
где co,;- — энергия взаимообмена, характеризующая парные
взаимодействия частиц в растворе.
Соотношение (IV.216) удовлетворяет уравнению (IV.215) и
используется для определения коэффициентов активности
молекулярных индивидов в регулярном ассоциированном растворе.
149
Глава V
ТЕРМОДИНАМИЧЕСКИЙ АНАЛИЗ ФАЗОВЫХ
ДИАГРАММ В ПРИБЛИЖЕНИИ РАЗЛИЧНЫХ
ТЕОРИЙ РАСТВОРОВ
1. АНАЛИЗ КРИВЫХ ФАЗОВЫХ РАВНОВЕСИЙ
В ДВУХКОМПОНЕНТНЫХ СИСТЕМАХ
НА ОСНОВЕ ТЕОРИИ ИДЕАЛЬНЫХ РАСТВОРОВ
ЛОГАРИФМИКА ШРЕДЕРА. ЭНТРОПИЯ ПЛАВЛЕНИЯ
И ФОРМА КРИВОЙ ЛИКВИДУСА. РАСЧЕТ ЭВТЕКТИЧЕСКОЙ
ТОЧКИ
Логарифмика Шредера известна как математическое
выражение кривой ликвидуса диаграмм состояния с простой эвтектикой.
Прежде чем конкретизировать вид уравнения кривой
ликвидуса в приближении теории идеальных растворов, целесообразно
использовать общетермодинамический вывод, основанный на
привлечении дифференциального уравнения Ван-дер-Ваальса — Сто-
ронкина [12—13].
Рассмотрим двухкомпонентную двухфазную систему при
постоянном давлении, в которой компоненты образуют непрерывный
ряд идеальных жидких растворов и имеют пренебрежимо малую
растворимость в твердой фазе. Схематический вид такой
диаграммы состояния представлен на рис. 24, а. Выберем Т и х2
в качестве независимых переменных, определяющих состояние
системы.
Запишем теперь дифференциальное уравнение
Ван-дер-Ваальса для равновесия между идеальным жидким раствором и
твердым компонентом 1. Учитывая, что в этом случае xf — 1,
xf = 0, дифференциальное уравнение Ван-дер-Ваальса можно
представить в форме
/ дТ \ь 4{#*1д*1)р.т /vn
\ д*ш )р ~~ s[<5> - sL + х£ {ds/dx2)LPtт* КУА)
где х\ — мольная доля компонента В в насыщенном растворе.
Принимая во внимание, что жидкая фаза идеальна, с помощью
соотношений
^-* <L>* + *<% ' (V.2)
(d2g/dxt)LPt т = (dii2/dx2)LPi T/( 1 - *2L), (V.3)
соотношение (V. 1.) легко преобразовать к виду
[Afcl. m (T)/R] d (1/Г) = -d In xf, (V.4)
где AAjf m (T) — теплота плавления первого компонента при
температуре 7\
150
Интегрируя уравнение (V.4) от температуры плавления
первого компонента до некоторой температуры 7\ получим
т1,т ГТи m "I
RT\nx^-Ah\tfn(l -T/Tlm)+ j J Ac0Ptla\nT\dT9
т L г J
(V.5)
где Ac°Pt i — разность теплоемкостей первого компонента в
твердом и жидком состояниях.
Конкретный вид уравнения (V.5) зависит от способа
получения. Известны еще по крайней мере две другие формы
уравнений кривых ликвидуса в бинарных простых эвтектических
системах, полученные методом цикла и интегрированием функции
Планка G/T.
В работе [157] доказана эквивалентность всех трех форм и
показано, что порядок интегрирования в интеграле, стоящем в
правой части выражения (V.5), изменять нельзя.
Таким образом, уравнение (V.5) определяет кривую первичной
кристаллизации первого компонента (левая ветвь ликвидуса на
рис. 24, а), если раствор идеален и твердые растворы не
образуются. Разность теплоемкостей в твердом и жидком состояниях
Дер, 1 в общем случае зависит от температуры. При AcPi i = О
-lnxf = (Ah°lt JR) (l/T - l/T°u m). (V.6)
Аналогичное соотношение можно записать для кривой
первичной кристаллизации второго компонента (правая ветвь ликвидуса
на рис. 24, а)
-\n4 = (bth;jR)(l/T-l/Tl.m)- (V.7)
Уравнения вида (V.6) и (V.7) в такой форме были предложены
И. Ф. Шредером [158—159] для определения растворимости
малорастворимых веществ (предельно разбавленные растворы).
Логарифмикой Шредера широко пользуются при расчетах теп-
лот плавлений индивидуальных веществ на основании данных по
фазовому равновесию, в системах эвтектического типа при
условии подтверждения идеальности раствора каким-либо косвенным
методом.
Если отложить логарифм мольной доли растворенного
вещества в зависимости от 1/Г, то при соблюдении условий (V.6) или
(V.7) график окажется прямой линией, наклон которой дает
нам значение теплоты плавления растворителя, а точка,
соответствующая на графике мольной доле xi: = 1, температуру
плавленая /-го компонента.
151
В тех случаях, когда Ah], m (T) зависит от температуры,
полезнее строить график In x\L) в функции от 1п(Т/Т°{,т). Чтобы
проиллюстрировать это положение, перепишем выражение (V.4)
в виде
(д In 4/д In 7% = Ahefi m (T)/RT. (V.8)
Продифференцировав это выражение по In 7\ получим
[d2\nx^/(d\nTf]p^AcPt tlR-bh], m(T)/RT. (V.9)
Если зависимость In х\ от In T линейная, то вторая
производная, определяемая уравнением (V.9), равна нулю. В точке
плавления А/г°,т = 77, т As;,m- Следовательно, с достаточно
хорошим приближением можно принять, что
Asit m « AcPt i,
Ah], m (T) - Ahl m - Ac°Pt t (Ti. M-T) = As], mT. (V.10)
Подставив выражение (V.10) в соотношение (V.8) и интегрируя
от 77, m до Ту получим
lnjrf-»(As:tm/R)In(7,/rJiW) (V.11)
В соответствии с этим выражением график функции In x\L)
от In (T/T°it m) также будет прямой линией, тангенс угла наклона
которой определит величину энтропии плавления
растворителя As?, m.
Необходимо, разумеется, помнить, что уравнения вида (V.6)
или (V.7) применимы только к идеальным растворам и даже для
них являются приближенными.
Используя соотношение (V.6), покажем также, что форма
кривой кристаллизации вблизи ординат чистых компонентов
позволяет при отсутствии калориметрических данных распознавать
соединения с низкой энтропией плавления.
Перепишем уравнение (V.6) в виде
T = Ahlm/(Asittn-R\nx$). (V.12)
Двойное дифференцирование этого уравнения по xt приводит к
выражению
13)
Знак этой производной, определяющий кривизну линии
ликвидуса, зависит от знака выражения:
2Я-Д*\ж + Я1пд^ (V.14)
15?
Очевидно, что в области, прилегающей к началу кривой, т. е.
при x\L) ->- 1,
(д2Т/дх% < 0, если As), м > 2R; (V. 15)
(д2Т/дх$р>0, если As], m<2R. (V. 16)
Оба случая схематически изображены на рис. 28. Первому из них
соответствует энтропия плавления растворителя в точке
плавления больше, чем 2R, второму — меньше.
Представляет интерес проанализировать, например,
уравнение (V.6) при л£ ->- 0. В этом случае In (1 — х\) можно при-
т
т°
'т,А
Рис. 28. Виды кривых ликвидуса:
вблизи ординат чистых
компонентов:
а — ASj m > 2R; б — ASj m <
<2R
Л хв А хв
ближенно заменить просто —х£. Тогда соотношение (V.6)
преобразуется к виду:
Т-Т\.т = - {ЯТ\9 mT/Ahl M) xf; (V. 17)
или приближенно
Т-Т\9М = -(RTl m/AtCu m)4<0. (V. 18)
Соотношение (V.18) выражает известный закон понижения
температуры замерзания Вант-Гоффа.
В работе [160] сделана попытка ответить с помощью
термодинамики на вопрос, почему температура начала затвердевания двух-
компонентного сплава отличается от температуры
кристаллизации чистого компонента. Исходя из условий термодинамического
равновесия показано, что причина такой разницы — влияние
второго компонента на давление пара, а следовательно, на
химический потенциал первого компонента. По мнению авторов
работы [160], эта интерпретация является более общим правилом,
относящимся и к идеальным, и к неидеальным растворам, к
системам из компонентов, практически не растворяющихся в
твердом состоянии, и к системам, обладающим заметной
растворимостью в твердом состоянии.
СИСТЕМЫ С НЕПРЕРЫВНЫМ РЯДОМ ТВЕРДЫХ РАСТВОРОВ.
УРАВНЕНИЯ КРИВЫХ ЛИКВИДУСА И СОЛИДУСА
Теория идеальных растворов в приложении к жидкому и
твердому растворам дает возможность описать только диаграмму
состояния типа «сигара», схематически показанную на рис. 9.
153
Пусть компоненты 1 и 2 образуют идеальные растворы й жйД*
ком и в твердом состоянии. Пусть Т и х2 — независимые
переменные.
Легко убедиться, принимая во внимание соотношения (V.2),
(V.3) и (IV. 1), что для рассматриваемого случая
дифференциальное уравнение Ван-дер-Ваальса примет вид:
Преобразуем соотношения (V.19) и (V.20) к виду, удобному
для интегрирования:
(*f*?A/i°i, m - x$xfAhl, m + x[Ah\, m)aT =
= _ (*f _ 4) RT2 d In x£, (V.21)
(xfxfAh'Um-x\;xfAhlm-{-
+ xfbhl,m)dT= - (4 - 4) RT2 d In xl. (V.22)
Вычитая (V.21) из (V.22), получим
(xf - *XL) Ahl, m dj = _ (^ _ xf) RT2 (d In *f - d In *2L). (V.23)
Учитывая, что
Al —л1 = л2 —л2 »
выражение (V.23) можно привести к виду:
din (4/4) KmW (V94,
d(l/T) ~~ R ' \*-*V
Аналогично можно также получить
dln(*f/«f) А/^^Г)
d (1/71) - Я ■ (V^
В выражениях (V.24) и (V.25) под величиной Ah], m (T)
следует понимать теплоту плавления /-того компонента при
данной температуре Т. Интегрируя уравнение (V.24) от Т\,т до Т
при условии, что АЛг, m не зависит от температуры, получим
4/4 = ехр - 1(Д/£. m/tf) (1/rJ. m - 1/Г)]. (V.26)
Аналогичное уравнение получим на основании выражения (V.25)
4/xi = ехр - [(Ahl, mlR) (UK m - 1/7% (V.27)
Перепишем его в форме
(1 - *f)/(1 - 4) - ехр -[(Aft°lf J/?) (1/т;в m - 1/Г)]. (V.28)
154
Введя обозначения
%г = (Ah°u JR) (1/т\9 т - 1/Г), • (V.29)
_ Я2 = (A/i;, m/#) (1 /г;§ т - 1/7), (V.30)
и решая совместно уравнения (V.26) и (V.28), получим для линии
ликвидуса
х\ = (е-и - l)/(e'Xt - ^) (V.31)
и для линии солидуса
х$ = (*-** - l)/(e" (*1+м - 1). (V.32)
Соотношения (V.31) и (V.32) были получены Ван-Лааром и
могут быть применены, в частности, для расчета кривых
ликвидуса и солидуса в идеальных системах, образующих непрерывные
ряды твердых и жидких растворов.
Особенно простым является случай, когда температуры
плавления и теплоты плавления обоих компонентов почти одинаковы.
При этих условиях после решения уравнений (V.26) и (V.28)
получим
На основании выражений (V.29) и (V.30) легко убедиться, что
последнее равенство возможно, если Т = Т°т> т. е. в этом случае
кривые ликвидуса и солидуса совпадают, вырождаясь в одну
горизонтальную прямую. Н. С. Курнаков [451 указывает, что
примерами систем с такого рода диаграммой состояния служат
системы, компоненты которых являются левыми и правыми
оптическими изомерами.
Уравнение (V.26) можно получить и более простым путем.
Химический потенциал компонента i в идеальном растворе
определяется уравнением (IV. 1).
При равновесии будем иметь
И(L) (Г, р) - ^(5) (Г, р) = RT In xf/x[, j
Левая часть уравнений (V.33) представляет собой изменение
мольной величины свободной энергии Гиббса при плавлении
компонента.
Для выбранной нами температуры Т в соответствии с
выражениями (1.113) и (11.37) это изменение равно
р)^ _ ц° <s> = Agif m = Ah]t m (T) - TAs], m (T). (V.33a)
В точке плавления компонента / величина Ag°(i m равна нулю.
Отсюда, принимая во внимание соотношение
As)t m=Ah°i9 mlT°it m>
155
и допуская, что величины А/Й, т и As", m незначительно изменяются
с температурой, можно записать приближенное равенство:
|iiiL) - |i*(S) » Aftl. « (1 - TIT], M). (V.336)
Подстановка этого выражения в уравнения (V.33) приводит к
формулам (V.26) и (V.28).
ДИФФЕРЕНЦИАЛЬНОЕ УРАВНЕНИЕ ЛИКВИДУСА В СИСТЕМЕ
С КОНГРУЭНТНО ПЛАВЯЩИМСЯ СОЕДИНЕНИЕМ.
ИНТЕГРИРОВАНИЕ УРАВНЕНИЯ В СЛУЧАЕ ИДЕАЛЬНОГО
РАСТВОРА
Практически важным представляется рассмотрение
равновесия двух фаз в системе с конгруэнтно плавящимся химическим
соединением типа АтВП9 отличающимся пренебрежимо малой
областью гомогенности. Пригожий и Дефэй рассмотрели [24] этот
случай на основе функции химического сродства. Возможность
конкретного решения задачи с привлечением модели регулярных
растворов была рассмотрена нами [1611. При этом наметился
подход к получению интегральных форм уравнения кривой ликвидуса
в системе с конгруэнтно плавящимся соединением типа АтВп.
Покажем возможность вывода дифференциального уравнения
кривой ликвидуса в указанной системе на основе принципа
смещенного равновесия в форме, данной Ван-дер-Ваальсом — Сто-
ронкиным, и дадим его решение в приближении модели
идеального раствора.
Рассмотрим двухкомпонентную двухфазную систему с
соединением АтВп при постоянном давлении. Диаграмма состояния
такой системы представлена на рис. 20. Пусть Т и х2 (мольная
доля компонента В) — независимые переменные, определяющие
состояние данной системы. Для рассматриваемого случая
дифференциальное уравнение Ван-дер-Ваальса можно записать в виде
(9T\L (4-4)W*2)£r ,Vo44
Учитывая, что дифференциальная мольная теплота
образования твердой фазы из жидкой определяется выражением (см. [13])
$«-*> = [ss -sL-(x$- 4) (ds/dx2)Lp, т) Т, (V.34a)
а также, принимая во внимание уравнение
xfх[ — 4*? = *f - 4 (V.346)
и соотношение (V.3), преобразуем уравнение (V.34) к виду:
;a-»s)
,2 --——)\aZ)nrIL
(V.35)
156
Поскольку в рассматриваемом случае в равновесии с жидким
раствором находится твердая фаза, представляющая собой
конгруэнтно плавящееся соединение с узкой областью гомогенности,
то можно считать, что ее состав практически постоянен и для
данного температурного интервала совпадает с составом соединения.
Следовательно,
*2»л/(т + л), (V.36)
где тип — стехиометрические коэффициенты.
Перепишем (V.35) с учетом выражения (V.36):
(V.37)
Учитывая, что один моль фазы, отвечающий составу
соединения, соответствует образованию [\/(т + п)] аналитических
молей соединения АтВп, величину (т + п) q(L->s> можно
рассматривать как дифференциальную мольную теплоту кристаллизации
этого соединения из насыщенного раствора, т. е.
(m + n)«7(L"S)=^X (V.38)
С учетом этого выражения уравнение (V.37) запишем в виде
(©; - - [-"■ (* - т^г) (Ж. г] /«ад- <v-*>
Это уравнение в дифференциальной форме выражает
функциональную зависимость температуры ликвидуса конгруэнтно
плавящегося соединения от состава, т. е. представляет собой
аналитическое выражение кривой ликвидуса, отвечающего первичной
кристаллизации соединения АтВп. Кроме того, уравнение (V.39)
полностью совпадает с выражением, полученным Пригожиным
и Дефэем [24], Отсюда следует важный вывод о том, что два
различных по своей природе подхода к анализу гетерогенного
равновесия дают один и тот же результат. Это дополнительный
аргумент в пользу плодотворности использования принципа
смещенного равновесия в форме, предложенной в работах [12, 131.
Он же свидетельствует о необоснованности утверждения [40]
о несостоятельности уравнения Ван-дер-Ваальса. -
Анализ уравнения (V.39) прежде всего показывает, что если
жидкость имеет тот же состав, что и находящееся с нею в
равновесии соединение, т. е.
4/(1 — х£) = п/т,
а производная от химического потенциала (d\i2/dx2)pt t не есть
бесконечность (степень диссоциации соединения в жидкости не равна
157
нулю), то (дТ/дх2)р = 0, т. е. на кривой ликвидуса температура
проходит через экстремум.
Если ограничиться рассмотрением устойчивых состояний, для
которых, согласно формуле (11.141),
(d\i2/dx2)Pt т > О,
то легко убедиться, что экстремум является максимумом. В самом
деле, во всех известных до сих пор случаях величина последней
дифференциальной мольной теплоты кристаллизации Ял'^ <0
и в соответствии с уравнением (V.39)
(дТ/дх2)р>0 при д£/(1 -x\)<njm\
(дТ/дх2)р<0 при 4/(1 — х%)>п/п%.
При подходе к интегрированию уравнения (V.39) необходимо
использовать модель, которая позволила бы конкретизировать
запись отдельных величин, в него входящих. Процесс
кристаллизации соединения с узкой областью гомогенности при некоторой
температуре Т можно представить в виде
m (Л)£ас + «4 ** (AmBnf. (V.40)
Изменение изобарно-изотермического потенциала процесса
(V.40) определится из выражения
- AG = гщьа + WLb - \i°a{X (V.41)
или, раскрывая значения химических потенциалов, получим
— AG = — Ш* + mRT In уг + nRT In y2. (V.42)
Используя уравнение Гиббса—Гельмгольца и учитывая
соотношение (V.42), можно записать выражение для теплового эффекта
Я{а^в\ входящего в уравнение (V.39) в общем случае в следующем
виде:
&Х = Я11Х8) + RT* lm <a ln Ъ/*ПР.х + п (д In y2/dT)Pt x].
(V.43)
В общем случае
(dlbJdxtfi. т = RT/x* + RT (д In y2ldx2)Lp> T. (V.44)
Решим задачу на основе модели идеального раствора.
Феноменологически дифференциальную мольную теплоту
кристаллизации соединения АтВп из насыщенного раствора можно
представить в виде двух слагаемых: теплоты сегрегации и теплоты
кристаллизации соединения при данной температуре:
fiftS - - АС. Атвп (Т) + «Л (V.44a)
где qE — теплота сегрегации.
158
Для идеального раствора величина qE равна нулю, й,
следовательно, величина qjt^ равна теплоте кристаллизации
соединения при данной температуре.
Кроме того, в соответствии с выражением (V.44) в идеальном
растворе
(dv*/dx2)f; = RT/xJi. (V.45)
Подставив это соотношение в уравнение (V.39), получим
(AC, AmsjRT2) dT = (л/4 - m/( 1 - *£)) dxl (V.46)
Отсюда, считая теплоту плавления соединения в рассматриваемом
интервале температур постоянной, получим при интегрировании
уравнения (V.39) от точки, отвечающей составу конгруэнтно
плавящегося соединения, в которой х% = nl(m + п) и Т = Т°т> Атвп,
до точки с координатами х£ и Т
Это уравнение можно записать также в другом виде, удобном
для графического представления:
да:
гвп I 1 1 \ .
\Г Гт'Атвп)
= -1пГ(«±^!!(1 _^Г(4)П]- (V-48)
График функции ln[(x2)n(l — х^)"1] = /(1/ТЛИк) представляет
собой прямую линию, по тангенсу угла наклона которой можно
определить теплоту плавления соединения АтВп.
В работе [162] на основе дифференциального уравнения Ван-
дер-Ваальса рассмотрены локальные закономерности в ходе
кривых, характеризующих равновесие в бинарных системах с
идеальной фазой при изобарических условиях. Допуская, что жидкий
раствор идеален (при этом твердый раствор может и не быть
идеальной фазой), и принимая во внимание равенство (V.346),
дифференциальное уравнение Ван-дер-Ваальса легко привести к виду:
(bs{L^S)/RT) dT + (xilx\ _ x?Ix[) dx\ = 0, (V.49)
где As<L_>s> < 0 — дифференциальная мольная энтропия
кристаллизации идеальной фазы.
В точках х\ = х\ = 0 и х\ — х\ = 1 величины xf 1х\
обращаются в неопределенность вида 0/0.
159
Пользуясь правилом Лопиталя, находим предельные значения
функций (д£/д£ — xflx[) в точках xf = х[ = 0 и jcf = xf- = 1:
Для точек Xi = 0 и хг = 1 уравнение (V.49), согласно
равенствам (V.50) и (V.51), преобразуется к виду:
-[Л/12. mlR (Tl, mf\ (dT/dxsX *-о = (дх£/д4)Р. *-о - 1, (V.52)
-[AAl. «/Л (Tl. m)2] (flr/dxf),.^ = 1 - (ctf/d*f),.,,_!. (V.53)
В силу очевидных неравенств АЛт, * > 0 и R > 0 из
уравнений (V.52) и (V.53) следует взаимосвязь между знаками
производных от Т и величины (dx\ldx\)Ptx в точках, отвечающих чистым
компонентам двухфазных бинарных систем твердая фаза —
идеальный расплав:
1) если (<97/fof)p.,1=oA0, то 1 Д (dxt/dxf)PtXl=0
2) если (дТ/дх?)РвХ1=г/\09 то (dx^dxf)PiXt^ AU
где знак Д в каждом случае выражает одно и то же отношение
(больше, равно, меньше).
Соотношения (V.52) и (V.53) характеризуют связь между
предельным ходом линий солидуса и линий состав идеальной фазы —
состав неидеальной фазы.
Согласно третьему закону Гиббса—Коновалова [1, 31], в
бинарных системах равновесные величины xf и л£ при условии
постоянства температуры и давления изменяются всегда сим-
батно. Поэтому производные (дТ1дх§)р могут быть заменены на
{дТ1дх[)р.
В результате такой замены получим аналогичные
закономерности в ходе линий составов сосуществующих фаз и линий
ликвидуса.
2. ВЗАИМОСВЯЗЬ РАЗЛИЧНЫХ ТИПОВ ДИАГРАММ
ФАЗОВЫХ РАВНОВЕСИЙ С ХАРАКТЕРОМ
МЕЖМОЛЕКУЛЯРНОГО ВЗАИМОДЕЙСТВИЯ
Исходя из известных выражений для термодинамического
потенциала, типы диаграмм состояния можно исследовать
аналитически, а также с помощью методов геометрической
термодинамики.
160
Для вывода уравнений кривых моновариантных равновесий
в двухкомпонентных двухфазных системах используют три
подхода.
Наиболее распространенный способ основан на условиях
равновесия Гиббса, которые записывают в виде равенства
химических потенциалов компонентов:
Н*=|*?. |i2=|i§. (V.54)
Второй способ основан на уравнениях, представляющих собой
другую форму записи условий фазового равновесия через
термодинамические потенциалы фаз:
G06 - А (дС/дх2)« = Сэ - х§ (dG/dx2f,
Ga + (I - х?) (dG/dx2)a = G* + (1 - 4) (dG/дх*?. ( ' )
При x2 = xa = x& на основании первого соотношения (V.55)
можно записать
G«-G$ = х2 [(dG/dx2)<* - (dG/dx2f].
Принимая во внимание выражение (III.27), придем к выражению,
которое лежит в основе третьего подхода:
-G« = G*. (V.56)
Теория идеальных растворов в применении к жидкому и
твердому растворам дает возможность описать только диаграмму
состояния типа «сигары».
Основные типы простейших диаграмм состояния, кроме пери-
тектического, можно получить, принимая для жидкого состояния
модель идеальных, а для твердого — неидеальных
растворов.Однако идеальные растворы даже в жидком состоянии
встречаются крайне редко, поэтому наибольшее значение имеет анализ
диаграмм состояния, использующий модель неидеальных
растворов как для твердого, так и для жидкого состояния.
Определенную роль в развитии этой проблемы сыграла теория регулярных
растворов, применение которой и в настоящее время в ряде
случаев весьма плодотворно.
Применение теории регулярных растворов к
сосуществующим фазам дало возможность впервые с единой точки зрения
описать все простейшие типы диаграмм состояния.
Использование теории регулярных растворов при
теоретическом изучении равновесия фаз началось с работ Беккера [163],
который рассмотрел частный случай равновесия фаз — распад
твердого раствора, идущего без фазового перехода (т. е. когда
кристаллические решетки обоих компонентов одинаковы).
Недостатком беккеровского приближения является допущение
симметричного характера кривой распада, что редко наблюдается
в реальных системах. Однако существенно здесь то, что, поль-
6 Глазов В. М. и др. 161
эуясь уравнением Беккера, по экспериментальным данным можно
оценить роль межмолекулярного взаимодействия в поведении
раствора.
Пинес [164, 165] обобщил метод Беккера, рассмотрев случай
распада (расслоения) растворов, идущего с фазовым переходом.
Полученные Пинесом результаты, касающиеся связи
характера межмолекулярного взаимодействия с видом диаграммы
равновесия, получили дальнейшее развитие и уточнение при помощи
геометрического метода в работах Д. С. Каменецкой [166—168].
Используя второй подход, получим уравнения кривых
расслаивания и моновариантных равновесий, отвечающих первичной
кристаллизации компонентов, в приближении модели регулярного
раствора.
Величину молярной свободной энергии Гиббса для двухкомпо-
нентного регулярного раствора при р = const можно записать
следующим образом:
g (Г, х2) = (1 - х2) у; (Т) + xz[il (T) +
+ RT [(1 - х2) In (1 - х2) + х2 In x2] + ft<*> (*), (V.57)
где |iHT)=«-rse,. (V.58)
Дифференцирование уравнения (V.57) по х2 приводит к
выражениям следующего вида:
(dg/dx2)T = RT In Ml - x2)\ + (dh* (x2)/dx2)r -
-V*(T) + v*(T), (V.59)
(d2g/dxl)T = RT/[x2 (1 - x2)] + {d'hE (x2)/dx*)T. (V.60)
Уравнение бинодали (расслоение в жидкой фазе) или спино-
дали (распад твердого раствора) легко получить, приравняв
производную (V.60) нулю.
Аналогично тому, как это сделано в работе [169],
представим энтальпию смешения раствора в виде тригонометрического
ряда Фурье для синусов:
п
/i<£>(*2) = S aks\nknx2. (V.61)
Такая форма записи удовлетворяет условию: hE = О при х = О
и х = 1 и допускает произвольную зависимость параметра
межмолекулярного взаимодействия от концентрации. Поэтому на
основе выражения (V.61) удается провести более общий анализ
процесса расслоения или распада, чем в приближении строго
регулярной модели, согласно которой данный процесс возможен лишь
при положительных значениях ш, а кривая распада симметрична.
Используя соотношения (V.60) и (V.61), уравнение для спино-
дали можно записать в виде
п
RT = (1 — х2) х2п% Ц k2ak sin knx2.
k=\
162
Критическая точка (Т = Тк и х = xk) является максимумом
спинодали. При Т <Tk твердый раствор распадается.
На рис. 29 изображены кривые распада, соответствующие
энтальпиям смешения (V.61) при k = 1, 2, 3 с положительными
и отрицательными значениями ak. На этом рисунке видно, что
для регулярных растворов характер кривой распада зависит от
величины отношения параметра межмолекулярного
взаимодействия к значению RT. Обобщая изложенное, можно сказать,
например, что фактически все системы с положительной теплотой
смешения (hE > 0) должны иметь предел несмешиваемости в твер-
Рис. 29. Области расслаивания,
соответствующие функциям
избыточной энтальпии
п
~ 2- ak s*n ^лх для значений * =
= 1, 2 и 3:
верхний график — положительные
значения с^, нижний —
отрицательные; значения RT^/a^. a —
2,47; б — 7.85; в — 13,54; г —
(—2,47); д — (—7,85); е — (—22,21)
дой фазе. Экспериментально это не подтверждается. Причина
этого несоответствия с теорией заключается в следующем. Если hE
недостаточно велика, то величина Tk настолько низка, что
скорость процесса распада в твердом состоянии будет бесконечно
медленной; таким образом, как правило, образуется твердый
раствор, который фактически является метастабильным. Вследствие
малых скоростей диффузии большая часть диаграмм состояния,
вероятно, неточна в области низких температур.
Для простоты взаимосвязь между характером
межмолекулярного взаимодействия и видом фазового равновесия рассмотрим
в рамках модели строго регулярного раствора.
Подставив выражения (V.57) и (V.59) в (V.55) с учетом
соотношений (V.58) и (IV. 104) и пренебрегая зависимостью теплоты
плавления от температуры, получим формулы, связывающие
равновесную температуру с концентрациями сосуществующих фаз в
приближении модели строго регулярного раствора:
Т =
Т =
4«-*40-4L))/0-*f)l '
(1 -xtf<»L-(l-x$y<*s + Ah02fm
&*2tm-R\n(xk/4)
6*
(V.62)
(V.63)
163
При этом для каждой фазы в случае отсутствия в ней расслоения
должны выполняться соответственно условия устойчивости
(11.140):
d4)TiP 4 (1-4)
дЖ =_2<о*+ *Г >0. (V.64)
94] т., 4(^-4)
При (oL < 0 и cos < 0 эти неравенства выполняются при любой
температуре. Если же (oL > 0 или cos > 0, то написанные
неравенства при некоторой температуре и концентрации переходят
в равенство; при дальнейшем понижении температуры величины
(d2g/dxl)r, p или (d2g/dxl)f, P становятся отрицательными в
некотором интервале концентраций, т. е. на кривой gL (xL) или
gs (Xs) появляется максимум.
Если бы зависимость Т от xL и Xs можно было выразить в
явной форме Т (xL) или Т (xs)r то было бы легко рассчитать кривые
равновесия и определить вид этих кривых в зависимости от
перечисленных констант. Однако решить эти уравнения в явной форме
не удается, но некоторые частные случаи диаграмм могут быть
рассмотрены аналитически.
Случай I. В обеих фазах связь однотипных и разноименных
атомов одинакова, т. е. o>L = a)s = 0 и в чистых компонентах
происходит фазовое превращение с тепловым эффектом,
например плавление. В этом случае получается диаграмма с
непрерывным рядом твердых растворов типа «сигары». Ниже будет
показано, что «сигара» получается и при других значениях coL и cos.
В данном случае можно решить систему уравнений (V.62) и (V.63)
в явной форме. При этом получается уравнение ликвидуса (V.31)
и солидуса (V.32).
Случаи II и III. В интервале температур, в котором протекает
фазовое превращение, нет расслоения или распада. В этом
случае кривые равновесия имеют общую точку (максимум или
минимум) (см. рис. 11).
Если в уравнениях (V.62) и (V.63) положить xL = xs = xe, то
получится
д ° 1 / (AS1. rnf - (Asl, m - As2. m) К, m ~
»1, m
X, —
X,m-4B< mVK-^)]
(V.65)
те = т\,т+ш\7ш xi = Tim + ^-=^-(i-xey-. (v.66)
©«■>-
m
Asj
X2 —
, m "
Tl.
-As2>
m +
m
Ы1-
As°2
m
164
При этом если о/" > (os, то Те > T]t т, т. е. температура точек
равных концентраций выше точек плавления чистых
компонентов. Получается диаграмма состояния с максимумом. Если coL <
< G)s, то Те < 77, т, получается диаграмма состояния с
минимумом.
Таким образом, если в твердом растворе тенденция к
объединению разноименных атомов выражена сильнее, чем в жидком, то
получается диаграмма состояния с максимумом, в противном
случае — с минимумом. На основании соотношений (V.65) и (V.66)
можно также заключить, что диаграмма состояния с максимумом
или минимумом прямо свидетельствует о неидеальном поведении
смеси, поскольку для идеальных растворов такой тип диаграммы
принципиально невозможен [170—173].
Опираясь на уравнение (V.65), А. А. Костарев [174] уточнил
условия образования диаграмм состояния с непрерывным рядом
твердых растворов. Используя условия существования
экстремума на кривых ликвидуса и солидуса, он пришел к выводу, что
в случае, когда экстремум находится за пределами реально
существующего интервала концентраций (0—1) или вообще
отсутствует, получается диаграмма состояния типа «сигары». Анализ
при указанных условиях приводит к следующим неравенствам:
As?* m — (As°lt m — AsL w) X
v ГЛс° Asl, mA4, м(Г1. m-T2, m)
L or — or .
x€^l\ xe<0. (V.67)
Последовательное решение неравенств (V.67) приводит к
необходимому и достаточному условию реализации
сигарообразных диаграмм [174]:
Asl, m (Tl, m - T°lt m) < со5 - o)L <: bslt m(T°2, m - T°u m). (V.68)
Величину As°it m можно вычислить из диаграмм состояния по
наклону кривых в точках j£=*f = 0Hjt£=*f=l:
4S;..-Rn.„|m[(g)'-/(g)s]r_r.J/(T;.„-r,„,
" *4—«П.-{|»[(^)1/(^Лм,..}/(П..-П-».
Ширина сигары зависит от величины As?, m, чем она меньше, тем
уже сигара.
Аналогично [174—175] на основании выражения (V.65) и
условий существования экстремума в интервале 0 < хе < 1 по-
165
<0;
лучают неравенства, выполнение которых обусловливает
формирование диаграммы состояния с минимумом
со5 - oL > As^, w (f2t m - T\9 m) (V.69)
и с максимумом
о5 - coL < ASolt m (г;. т - 7-;, «). (vjo)
Случай IV. При очень больших энергиях смешения в твердой
фазе взаимная растворимость компонентов практически
отсутствует. При этом уравнения (V.62) и (V.63) приобретают
наиболее простой вид и характеризуют
кривые ликвидуса в системе
эвтектического типа.
а. Уравнение первичной кристалли»
зации первого компонента
(V.71)
б. Уравнение первичной
кристаллизации второго компонента
Рис. 30. Кривые ликвидуса при л^ = U, 1 = z т •
различных значениях энергии Д$2 m — R In Х<>
гмршрнна »
(V.72)
смешения
При (oL = 0 уравнения (V.71) и (V.72) совпадают с уравнениями
линий ликвидуса Шредера—JIe-Шателье. Таким образом, члены
(a|co)l и (1 —xk)2 &L характеризуют отклонение поведения
раствора от идеального. На рис. 30 схематически показано влияние
coL на ход кривых растворимости.
В точке х = 0 или х = 1 все три кривые имеют общую
касательную, наклон которой определяется величиной As°, m.
Из анализа кривых растворимости, которые получаются при
различных по знаку значениях o)L, вытекает, что при со1- = 0
закон Рауля выполняется для большего интервала концентраций,
чем при coL <0 или 0L >0, т. е. при a>L — 0, как и можно было
ожидать, раствор наиболее близок к идеальному.
Отметим, что среди эвтектических диаграмм можно встретить
все три типа линий ликвидуса (coL =0, coL >0 и со'- <0), но
для большинства эвтектик, по-видимому, характерно все же
положительное значение (oL.
Как отмечается в работе [174], анализ диаграмм состояния
эвтектического или перитектического типа более сложен, чем
анализ диаграмм состояния с непрерывным рядом твердых
растворов, но и здесь можно дать качественное и даже
полуколичественное описание условий реализации того или иного типа равновесия.
166
Так, используя грубое приближение Гильдебранда [93],
согласно которому диаграммы состояния с эвтектикой и
перитектикой рассматриваются как результат наложения равновесий с
непрерывным рядом твердых растворов на кривые распада, для
простой эвтектики следует добавить к (V.69) еще достаточное
условие распада твердой фазы [174]:
со5>2£Г;,т> (V.73)
где 77, т — температура плавления более легкоплавкого
компонента.
Для простой перитектики, поскольку жидкая фаза в отличие
от твердой не распадается, o)s > (oL, и неравенство (V.68)
переходит в условие [174]:
О < 0)S - C0L < As°2t m (Tl9 m - Т\9 m), (V.74)
дополненное условием распада твердой фазы [83]:
os>i?(r;,w + r;,m). (V.75)
Теория регулярных растворов не позволяет дать
количественный критерий образования промежуточных фаз и формирования
соответствующих диаграмм состояния в двухкомпонентных
системах. Однако в рамках формализма регулярных растворов
можно рассмотреть относительное расположение кривых
ликвидуса в системах с конгруэнтно плавящимися соединениями.
Проанализируем уравнение кривой ликвидуса конгруэнтно
плавящегося соединения AmBn (V.39) в приближении
регулярного раствора.
Для регулярного раствора
Тогда уравнение (V.44) преобразуется к виду
(dii2/dx2)LPiT = RT/xk - 2coL (l - x2L).
После подстановки этого выражения в уравнение (V.39) получим
(f)L^-lrir[± ^МИ?- 2(0^(1 -*Ы /#*/>.
\dxjp \т {_xlJ[xl \ "U4Am»n
(V.76)
Уравнение (V.76) описывает в дифференциальной форме
равновесие первичных кристаллов АтВп с регулярным жидким раствором.
Для регулярного раствора теплотой qE в уравнении (V.44a)
можно пренебречь. Тогда приближенной формой
соотношения (V.76) в обсуждаемом случае является выражение
(®:-«',U--£r)[f-'"i<i-rf>]K
(V.77)
167
При анализе этого выражения необходимо рассмотреть два
случая, соответствующих положительным и отрицательным откло^
нениям от закона Рауля.
При отрицательных отклонениях от закона Рауля
коэффициенты активности меньше единицы, следовательно, coL <0.
Тогда из уравнения (V.77) вытекает, что при o>L < 0 для любых
значений х\ соблюдается неравенство
\dT/dx2\L>\dT/dx2\id^.
Отсюда можно заключить, что если раствор характеризуется
отрицательными отклонениями от закона Рауля, то максимум
конгруэнтно плавящегося
соединения — более острый, чем в
идеальном растворе.
При положительных
отклонениях от закона Рауля
коэффициенты активности больше
единицы и G)L >0. В этом случае
для всех значений х\
\dT/dx2\L<\dT/dx2\i<HL\
т. е. максимум соединения
становится более пологим, чем в
идеальном растворе. При очень больших
положительных значениях coL
может возникнуть область
несмешиваемости. Фазовая диаграмма при
этом будет иметь вид,
представленный на рис. 31, который
соответствует системе с нижней
критической температурой смешения [24].
Рассмотрим возможность решения уравнений (V.62) и (V.63)
с помощью геометрического метода Розебома, который заключается
в исследовании различий в степени кривизны функций свободных
энергий для сосуществующих фаз.
Согласно работе [167], сначала исследуем геометрическим
методом Розебома поверхности свободной энергии сосуществующих
фаз, определяемые соотношениями:
gLiT,xL)-(l-4)lhw+*M{L)-
-r[(i-*2LH(i)+4-4<L)] +
+ RT [41п4 + (1 - 4) In (1 - 4)] + "L4 (1 - 4), (V.78)
gs(T,4) = (i-4)A°1(S)+4/l;(S)-
-r[(i-*fR(S>+4S)4(S,l +
+ RT ц i _ xi) in (l - 4)+4 in 4\ + <*s*l (i - *f )• (v.79)
168
Рис. 31. Диаграмма состояния с
ограниченной растворимостью в жидкой
фазе при наличии нижней критической
точки
Эти уравнения представляют в координатном пространстве g,
Т, х некоторые желобообразные поверхности gL и g^, касающиеся
плоскостей х = О и х = 1. Уравнения линий касания получим,
положив в формулах (V.78) и (V.79) значение*, равным нулю или
единице.
При х\ = xf = 0 (чистый компонент 1)
gL(T,0) = hl<L)-TsVL) = ^L>(T);
2s (Г, 0) = h\{S) - Ts\(S) = и!(S) (Г) (V.80)
при х\ — *f = 1 (чистый компонент 2)
gs (Г, 1) = hiiS) - 7^(S) = ^(S) (T). (V.81)
При исследовании характера фазовых равновесий в системе
можно воспользоваться сечениями поверхностей энергии Гиббса gL
и gs плоскостями заданных постоянных температур. При этом
форма и взаимное положение проекций линий пересечения на
плоскость (g, х)у как было показано в гл. III, определят
концентрацию сосуществующих фаз. Исследуем при помощи первых и
вторых производных g по х и радиуса кривизны р особенности
поведения указанных проекций.
При заданных условиях (Т = const, p = const)
(dg/dxfo т = (1 - 2xL) <*L + RT In [xL/( 1 - xL)\ -
-[i0i(L) + ^(L). (V.82)
Отсюда следует, что при xL -> 0 (dg/dx)pfr -> —°°, т. е.
кривая g'" = f (x1) (для удобства обозначим ее gx) касается оси х = 0
и направлена вниз; при xL ->■ 1 (dg/dx)lp\ т ->• оо, т. е. кривая gi
касается ординаты л; = 1 и направлена вверх. Корни уравнения
(l~2xL)coL + ^ln[xV(l-^)]-fi;(L) + ^(L)=0 (V.83)
дадут те значения х , при которых кривая gx имеет максимум
или минимум. Для решения этого уравнения геометрическим
способом его целесообразно представить в виде
(1 - 2xL) <*L/RT + <£ = In [(1 - xL)lx% (V.84)
где U = WL)-v\{L))lRT. (V.85)
Левая часть уравнения (V.84) в функции от х представляет
собой прямую, наклон которой зависит от знака и величины
энергии смешения, а также от температуры. Обозначим эту
прямую I1L, а ординаты точек прямой Оп. Правая часть уравне-
169
ния (V.84) в функции от х представляет собой кривую, которую
обозначим Ку а ординаты ее точек Оц*
Рассмотрим различные возможные положения прямой Я
относительно кривой /С.
Случай I. coL <<0.
В этом случае наклон прямой положителен, и прямая
пересекает кривую К в одной точке (см. рис. 32). Следовательно,
кривая gx имеет одну экстремальную точку.
При coL <0 производная (d2g/dx2)p,T >0 [см. (V.64)], т. е.
кривая gx имеет минимум (см. рис. 6). Величина {d gldx )Pt т равна
Рис. 32. Геометрическое решение Рис. 33. Геометрическое решение
уравнения (V.84) при ш^ < 0 уравнения (V.84) при 0 < со^ <
< 2RT
разности наклонов прямой UL и касательной к кривой /С. При
coL <0 эта разность положительна. Положение минимума (xmin)>
определяемое точкой пересечения Я с /С, меняется с
температурой: при ст >0 *mln < 1/2; при Ст <0 xmln > 1/2; при ст = О
Случай 2.
При положительной энергии смешения наклон прямой Я
отрицателен. При этом возможны три варианта.
А. При небольших coL прямая IJL пересекает кривую К в
одной точке (рис. 33), в которой, согласно формуле (V.64),
производная (d2g/dx2)pt т > 0.
Следовательно, при небольших положительных значениях coL,
когда выполняется условие (V.64), кривая gx имеет один минимум,
положение которого определяется точкой пересечения Я с К.
При понижении температуры положение минимума смещается от
*mln > 1/2 ДО *mln < 1/2.
170
Б. При некоторой критической температуре Тк прямая UL
касается кривой /С. Тогда
Гл = 2*л(1-**)©//?.
(V.86)
На основании этого выражения легко убедиться, что состав,
отвечающий критической точке, равен 0,5. Подставив значение
xL = 0,5 в выражение (V.84), найдем, что стк = 0, т. е. при
критической температуре в соответствии с уравнением (V.85)
химические потенциалы компонентов
одинаковы.
В. При Т <Tk прямая TlL
пересекает кривую К в трех точках
(рис. 34), причем Ст >0. В двух
крайних точках выполняется
условие (V.64), а в средней
Ж.,~-~+
RT
4(i-4)
<о.
(V.87)
Рис. 34. Геометрическое решение
уравнения (V.84) при ©L > 2RT
Следовательно, на кривой gx
имеются два минимума, а между ними
один максимум (см. рис. 10). При
температурах ниже критической
система расслаивается. Если к кривой
К провести две касательные а%(и и
аъа±% параллельные прямой Я , то
в интервале концентраций между точками касания х'в и хв
выполняется условие (V.87). Абсциссы точек касания
соответствуют точкам перегиба. Вне этого интервала соблюдается условие
(V.64). Итак, кривая gLx может быть двух видов в зависимости от
величины со :
1) при coL <0 и при всех значениях coL, удовлетворяющих
условию 0 < (oL <CRT/[2xL(l — xL)]y кривая gx имеет один
минимум, так как наименьшее значение правой части
неравенства равно 2RТу то его можно записать в виде: coL <<
< 2RT;
2) если в некотором интервале концентраций, включающем
А - &L>RT
концентрацию х = 0,5, выполняется неравенство L j— y
[2х (I х )]
т. е., если coL >2#7\ то кривая gx имеет два минимума, а
между ними один максимум (случай расслоения).
171
Для более полной характеристики кривой gx можно еще
определить ее радиус кривизны:
т (1/1 + [(1-2**) (*>L/RT) +4 + In [xL/(i -xL)\f }3
P """ -2<x>L + RT/[xL{\+xL)]
(V.88)
Если coL >0, то при T = Tk pL -> oo, при T > Tk pL >0
и при Г < 7\ в некотором интервале концентраций pL < 0. При
со*- < 0 всегда pL > 0, т. е. кривая обращена во всех точках
выпуклостью вниз. При прочих равных условиях, чем больше
энергия смешения, тем больше радиус кривизны. Чтобы выяснить
зависимость pL от температуры, положим xL = 1/2. При этом
р^ в W 1+(с£)213/(- 2coL + ART). (V.89)
С повышением температуры ст уменьшается [см. формулу (V.85) ]
и pL тоже уменьшается.
Аналогично можно исследовать поверхность gs = f (Xs, Т).
Зная форму кривых свободной энергии сосуществующих фаз,
можно при помощи метода Розебома [176—177] рассмотреть
различные типы диаграмм состояния.
Условие формирования диаграмм состояния с непрерывным
рядом твердых растворов.
a) coL = (os = 0, т. е. взаимодействие одноименных и
разноименных атомов в обеих фазах одинаково. При этом прямые П
и IIs параллельны оси х и каждая пересекает кривую К в одной
точке. Уравнение (V.84) в этом случае приобретает вид:
для жидкой фазы: cf = In [(l — xL)/xL], (V.90)
для твердой фазы: cf = In [(l — я5)/*5]. (V.91)
Оба уравнения могут быть решены аналитически. Кривые gx
gt в этом случае имеют по одному минимуму. Расстояние между
минимумами (*пнп — хт\ п) зависит от разности ст —ст.
Расстояние между точками касания xL и Xs общей касательной к
кривым gx и gx также зависит от этой разности: чем меньше ст — cf >
тем ближе значения хьих . Если принять, что ст больше ст, то
прямая IJL окажется выше прямой Я5 (см. рис. 35, а). При
понижении температуры точки Xmm и хтт перемещаются влево,
причем все время Xmin <#min. Если теперь построить диаграмму
как геометрическое место проекций на плоскость (л:, Т) точек
касания xL и xs общей касательной к кривым gx и gx, то получится
диаграмма состояния с непрерывным рядом твердых растворов
типа сигары (см. рис. 9).
172
го о о ^^ г
б) со = со <0; Т\%тФ Г2, т. При этом условии прямые П
и П параллельны между собой, а наклон их положителен (см.
рис. 35, б). При понижении температуры, как и в предыдущем
случае, значение *тт все время меньше х^т. Изменение xL и *s
(концентраций сосуществующих фаз) происходит, главным
образом, из-за различного характера перемещения кривых gx и gx
с температурой, и в итоге также получается диаграмма состояния
с непрерывным рядом твердых растворов типа сигары.
Рис. 35. Геометрическое решение уравнения (V.84) при:
<os = 0; б — &L — ©s < 0; в — 0 < &L = (0S = 2RTb <2RT
в) 0 < со*- = cos = 2RTk < 2RT; Т* < Т. Как уже
отмечалось, при положительной энергии смешения прямые UL и /7s
имеют отрицательный наклон. При небольших значениях со
каждая из этих прямых пересечет кривую К только в одной точке
(см. рис. 35, в). При понижении температуры х^\п <4ш и xL <
<* . В итоге получается сигара. При дальнейшем понижении
температуры прямая /7s коснется кривой К при некоторой
температуре ТКу а при еще более низких температурах пересечет
кривую К в трех точках, что соответствует образованию на
кривой gx двух минимумов и, следовательно, распаду твердой фазы.
На основании соотношений (V.62) и (V.63) для
рассматриваемых случаев уравнение сигары может быть записано в виде
„._ co[(4)2-(*f)2]+A/.;,m (v92)
или
Г-
^,m-Kln[(l-4)/0-*f)]
*> [Q-4)2-О-*f)2]+ */.;,„
A%.m-*K*2L)/(*f)
(V.93)
173
При отрицательных энергиях смешения сигара получается
более узкой, чем при 0 = 0. При 0 >0 характер кривых может
быть более сложным, особенно при больших значениях 0.
L S T S
г) 0 <0 <2/?7\ В этом случае положения хт\п и хтщ
близки одно к другому (рис. 36, а). Радиус кривизны pL кривой gx
меньше радиуса кривизны gx согласно выражениям (V.88) и
(V.89). В этом случае, как было показано ранее, получается диа-
Рис. 36. Геометрическое решение уравнения (V.84) для диаграммы
состояния с непрерывным рядом твердых растворов при наличии
общей точки на кривых ликвидуса и солидуса:
а — минимума; б — максимума
грамма состояния с точкой равных концентраций с минимумом
(см. рис. 11, б).
д) 05 < 0L <C2RT. В этом случае pL >os. Положения
минимумов кривых gx и gx близки, так как при Т^>Т\,т разность
ст — ст мала, и прямые П и П пересекают К в близких точках
(рис. 36, б). Таким образом, при таком соотношении энергий
смешения обеих фаз на диаграмме состояния получается точка
равных концентраций с максимумом.
Далее, используя аналогичные построения, можно получить
критерии формирования других видов диаграмм состояния.
При условии 0L <2#T, a 0s >2#Г (значения температур
плавления чистых компонентов отличаются мало) получается
диаграмма состояния с ограниченной растворимостью в твердой фазе
с эвтектической точкой.
При 0L <2RTy a 0s >2RT{Tl,m > Т°г,т) получается
диаграмма состояний с ограниченной растворимостью в твердой фазе
с перитектической точкой.
174
При G)L >2#7\ a)s <^2RT должно иметь место расслоение
в жидкой фазе и неограниченная растворимость в твердой фазе.
При со*->2#7\ (os >>2RT (растворимость в обеих
фазах
пред-
ограничена) получается диаграмма фазовых равновесий,
ставленная на рис. 17.
Таким образом, используя метод Розебома и применяя
геометрические построения, Д. С. Каменецкая последовательно
рассмотрела возникновение раз-
Т I I личных типов фазовых диаграмм
в зависимости от величины и
знака энергии смешения в
сосуществующих фазах при учете
Рис. 37. Схема построения линии
температур равных значений изобарно-
изотермических потенциалов твердой
и жидкой фаз проектированием точки М
на изотерму Тх в точку М'
Рис. 38. Кривые равных значений G'
для гипотетических систем, в которых
жидкие растворы идеальны, а твердые
регулярны с hE — а2 sin 2nx:
а — высокотемпературная область; б —
низкотемпературная область
в отдельных случаях соотношения между температурами плавления
чистых компонентов. Из работы Д. С. Каменецкой следует, что
связь -между типом фазовой диаграммы и характером
межмолекулярного взаимодействия (величина и знак со), описываемого
в рамках регулярного приближения, не однозначна. В пользу
такого заключения говорят также результаты термодинамического
доказательства, данного в работе Оонка и Спренкелса [169] на
основе анализа кривых равных значений изобарно-изотермиче-
ского потенциала фаз.
В соответствии с работами [169, 178] покажем это. Используя
соотношения (V.56), (IV.7), (IV. 17) и (IV.84), запишем
выражение для линии равных значений изобарно-изотермических потен-
175
циалов твердой и жидкой фаз Т0 (х) (см. рис. 37) в общем виде:
Г ,ух _ Р ~*2) Г!, ,»Ч m +^2, m4 m + AftE (*) ,V q44
(1 - x2) As1§ m + x2As2_ m - As£ (x)
где AhE = /i£ <« (*) - hE <s> (A As£ = sE <« (x) - sE <*> (*).
(V.95)
Ограничиваясь рассмотрением линий Т0 (х) в рамках модели
регулярных растворов соотношение (V.94) для удобства анализа
можно переписать в виде
Т /И Р -*2> Г1. тЦ. т + *2Г2. mA4. m ,
У о V**/ — о 7—^ " \ *~
Asl, m+MAs2, m-Asl,m)
+ —о ^!W . (V.96)
Asl, m + MAs2, m-Asl, m)
Для случая, когда при всех значениях х соблюдается условие
AhE (х) = hE {L) (x) — hE (S) (x) = 0 (обе сосуществующие фазы
идеальные растворы или обнаруживают одинаковые отклонения от
идеальных), второе слагаемое в правой части уравнения (V.96)
исчезает. В этой ситуации кривая Т0 (х) описывается первым
слагаемым в формуле (V.96) и называется нулевой линией. Как
правило, разность Asl, m — Asi|m мала по сравнению с
величиной As°i, m и нулевая линия незначительно отличается от прямой,
соединяющей температуры плавления чистых компонентов. Это
отклонение определяется отношением Aft (x) к Asi, m. Таким
образом, характер кривой AhE (x) непосредственно отражается на
форме кривой равных значений изобарно-изотермических
потенциалов фаз.
В качестве иллюстрации на рис. 38 показано последовательное
изменение характера кривых равных значений G сосуществующих
фаз в модельных двухкомпонентных двухфазных системах в
зависимости от величины разности между температурами плавления
чистых компонентов (7^, т — Т\у т). При расчетах линий Т0 (х)
допускалось, что жидкая фаза представляет собой идеальный
раствор, а твердая—регулярный, характеризующийся теплотой
смешения, определяемой по формуле hE (5) = а2 sin 2лх с
положительным значением сь^ и областью расслаивания. Кроме того,
предполагалось, что Asi,m = As°2,m.
Согласно результатам расчетов, проведенных в работах [169,
178], кривые равных значений изобарно-изотермических
потенциалов сосуществующих фаз адекватно отображают тип фазовой
диаграммы, иными словами, максимумы, минимумы и точки
перегиба на кривых моновариантных равновесий точно совпадают
с соответствующими особенностями линий Т0 (х).
176
В соответствии с этим положением кривые равных значений G,
представленные на рис. 38, а, б, будут соответствовать следующим
типам диаграмм состояний:
а) кривая 4 отвечает случаю, когда Т\ч т = T\t m. На
соответствующей диаграмме состояния должны быть два экстремума:
максимум и минимум;
б) кривые /—3 отвечают случаю, когда T\t m — T\t m > О,
и иллюстрируют влияние повышения температуры плавления
второго компонента TZ, m, в результате которого положение
экстремумов сдвигается к координатам чистых компонентов. На
кривой 3 экстремумы еще присутствуют, соответствующий тип
фазовых равновесий будет такой же, как и в предыдущем случае. На
кривой / экстремумы отсутствуют, производная (dT0 (x)/dx)p >0
при всех значениях х. Этот случай соответствует типу диаграмм
фазовых равновесий с непрерывным рядом твердых растворов. На
кривой 2 наблюдается случай, когда экстремумы находятся в
точках х = 0 и х = 1 (производная (dT0 (x)ldx)p ~ 0), где кривые
ликвидуса и солидуса соприкасаются. Эта кривая отвечает
граничному варианту, когда тип диаграммы изменяется от случая (а)
к сигаре.
в) кривые 5—7 отвечают случаю, когда разность Tl, m —
— T\,m <0, и иллюстрируют влияние на форму линий Т0 (х)
понижения температуры плавления второго компонента Т£, т, в
результате которого положения экстремумов на кривых Т0 (х)
сближаются.
На кривой 7 экстремумы отсутствуют, при всех
значениях х производная (dT0 (x)/dx)p < 0, а соответствующий тип
диаграммы будет «сигарой». Кривая 6 отражает промежуточный
случай, когда положения экстремумов совпадают, в результате на
линии Т0 (х) наблюдается точка перегиба. Наличие этой точки
отвечает случаю, когда кривые ликвидуса и солидуса на диаграмме
фазовых равновесий также имеют общую точку перегиба. При этом
изменяется тип диаграммы фазовых равновесий от вида (а) к
сигаре.
Рис. 38, б отвечает диапазону температур, когда в твердой фазе
происходит расслаивание, и, следовательно, кривые равных
значений изобарно-изотермических потенциалов сосуществующих фаз
пересекаются со спинодалью. В этом случае пересечение кривой
равных значений G, характеризующейся отсутствием экстремумов
(кривая 9), со спинодалью приводит к образованию диаграмм
фазовых равновесий перитектического типа. При пересечении
кривых Т0 (х), имеющих максимум и минимум, возможно несколько
вариантов:
1. Минимум линии Т0 (х) находится внутри области
расслаивания, а максимум за ее пределами (кривая 10). Этому
соответствует диаграмма фазовых равновесий с наличием эвтектики и
максимума на кривых ликвидуса и солидуса в определенном
интервале концентраций.
177
2. Оба экстремума линии Т0 (х) (кривые 3—6 на рис. 38, а)
находятся внутри области расслаивания; этому соответствует
диаграмма фазовых равновесий перитектического типа.
3. Кривая равных значений G касается спинодали, а максимум
и минимум расположены за пределами области несмешиваемости
(кривая S); этому соответствует диаграмма фазовых равновесий
с наличием перитектики и максимума и минимума на кривых
ликвидуса и солидуса в определенном интервале концентраций.
Заметим, что кривая 2 при пересечении со спинодалью
приводит к промежуточному случаю, соответствующему переходу от
диаграммы фазовых равновесий перитектического типа к диаграмме
вида (3), тогда как кривая 6 не отражает промежуточный случай.
Пользуясь изложенным выше методом, Оонк и Спренкелс
[169] проанализировали взаимосвязь различных типов фазовых
равновесий с характером межмолекулярного взаимодействия
в рамках модели регулярных растворов, исключив из
рассмотрения случаи полиморфизма и расслоения в жидкой фазе.
Для описания концентрационной зависимости разности теплот
смешения жидких и твердых растворов использовались
выражения вида:
Aft* (х) = S Да* sin knx, (V.97)
где выбор величины п основывался на результатах анализа
реальных систем.
Результаты, полученные в работе [169], свидетельствуют о том,
что один и тот же тип диаграмм фазовых равновесий может быть
связан с различным характером межмолекулярного
взаимодействия и, наоборот, один и тот же характер межмолекулярного
взаимодействия может привести к возникновению разных типов
фазовых равновесий.
Магер, Лукас и Петцов [179—180] на основе теории регулярных
растворов рассмотрели условия, при которых изменяется тип
фазовой диаграммы. Они использовали экспериментальный
материал для 420 систем. Энергетическим параметром сравнения
служила величина RTM = R {T\t m + Tlt m)/2. Кауфман и Берн-
стейн [181 ] на основе модели идеальных и регулярных растворов
разработали методы расчета различных фазовых диаграмм с
помощью ЭВМ.
В работе [168] Д. С. Каменецкая проанализировала критерии
образования диаграмм состояния различного типа в зависимости
от соотношения основных величин, определяющих главные
особенности диаграмм состояния при различных давлениях: co(L),
<d<s>, vE <l> и ve<sK
В заключение отметим, что все попытки прогнозировать
возможные типы диаграмм состояния на основе модели регулярных
растворов в силу объективной ограниченности этой модели сле-
178
дует рассматривать как чисто качественные. При этом наиболее
последовательный подход к данному вопросу, осуществленный
в работах Д. С. Каменецкой, по существу, мало прибавляет к
качественным выводам, получаемым на основе метода Розебома. Но
качественный прогноз эволюции видов фазовых равновесий такой
подход все же дает.
Однако даже качественные прогнозы ограничиваются самым
главным допущением, согласно которому эволюция диаграмм
происходит при сочетании купола расслоения в твердой фазе с
двухфазным равновесием между жидкой и твердой фазами, хотя такой
подход в принципе ниоткуда не вытекает. При этом
предполагается изоморфность структур компонентов, что не может
считаться строгим условием из-за своей ограниченности.
3. АНАЛИЗ КРИВЫХ ЛИКВИДУСА И СОЛИДУСА
В СИСТЕМАХ С НЕОГРАНИЧЕННОЙ
И ОГРАНИЧЕННОЙ РАСТВОРИМОСТЬЮ
Термодинамический анализ фазовых равновесий раскрывает
возможности для выяснения характера межмолекулярного
взаимодействия, а следовательно, и раскрытия физико-химической
природы фаз. Важную информацию в этом отношении дает оценка
величины и знака энергии взаимообмена между компонентами,
позволяющая судить о преимущественном характере тех или иных
парных взаимодействий в растворе, что непосредственно приводит
к выводам о тенденциях к образованию соединений либо
расслаиванию. Оценка же температурно-концентрационной зависимости со
помогает выявить направленность структурных изменений при
варьировании важнейших параметров состояния. На основе
анализа диаграмм состояния можно извлечь информацию о
термодинамических функциях, характеризующих образование тех или
иных фаз, либо, напротив, располагая соответствующими
термодинамическими данными, можно рассчитать отдельные элементы
фазовых диаграмм или оценить достоверность имеющихся.
В общем случае при выражении через коэффициенты
активности и термодинамические характеристики плавления
компонентов уравнения линий моновариантного равновесия в двухкомпо-
нентной системе можно представить в следующем виде:
RT In (1 - x2)Ly[ - RT\n(l - x2fyf = AsJ, W(T - т\9 m)
(V.98)
или
RT In x\y\ - RT In xfyl = Д%, «(71 - Tl9 m). (V.99)
В системах с неограниченной растворимостью компонентов
в твердом и в жидком состоянии эти уравнения справедливы во
всем интервале концентраций. В системах с ограниченной
растворимостью в твердом состоянии (эвтектического, либо перитектиче-
ского типа) эти уравнения строго применимы только к раствори-
179
телю, поскольку в данном случае стандартное состояние
компонента 5, растворенного в Л, не эквивалентно состоянию
чистого В. Исключение могут составлять системы с ограниченной
растворимостью, образованные изоморфными компонентами.
Конкретный вид уравнений (V.98) и (V.99) зависит от
выбранной модели, в приближении которой проводится анализ
межмолекулярного взаимодействия. Так, используя для коэффициентов
активности компонентов выражения (IV. 139) и (IV. 140), получим
следующие соотношения, характеризующие двухфазное
равновесие S4±L и связывающие температуру с концентрациями
сосуществующих фаз:
<т + (4°>o)L - (4%Г - (4)2 0 -24) <tf +
+ (^)2(I-2*f)o.f + (4)3(34-2)<o|--
(*?)3(3*f-.2)<*
(V.100)
Ч т+(*Ki)L - (*H0S - (4)2 0 - 24) < +
+ (*f f (1 - 2xi) fflfx + (4)a (34 - 2) < -
-(*?)'(a*f-2K-rin [(i-4)/(i-4)l
a*;, « + (4)2 «tf - (4 )2 •?+24 (4)2 * -
- 24 (4)2 cof + 3 (44)2 ^ - 3 (xfxfy «of
As;,m+(4)2<1-(4)4s1 + 2(4)24<x- * { '
- 2 (*f)2 4<4+з (44)2 < - з (44)2 «4 -
-r in (4/4)
На основании этих соотношений при определенных условиях
легко получить уравнения линий моновариантного равновесия
5 *=* L в приближении других простых моделей растворов, в
частности, субрегулярной, квазирегулярной, строго регулярной,
идеальной и др.
В бинарных системах наиболее широко используют метод
анализа межмолекулярного взаимодействия в приближении теорий
строго регулярных и квазирегулярных растворов [см.
уравнения (V.62) и (V.63) ]. В этом случае для систем с непрерывным
рядом твердых растворов параметры (nL и cos можно выразить в
явном виде:
пт {(1 -4)2 ш [(1 -4)/0 -4)1 - (4)2'" {414))
(1-4)2(4)2-0-4)2(4)2
(1-4)2м1т(т-т\,т)/т1т-(4)2ы1.т(т-Т1Ж.т
(i-4)2(4)2-(i-4)2(4)2
(V.102)
180
со5 = ©о — ®oiT =
rt ((i -4)2m [(i -4)/(i-4)] - (4У i" (*f/4)i
(4)20-*!)2-(*1Ж)2
(l-4)2<m(7--^.m)/r;,w-(4)2Aft;,CT(r-r;tCT)/r;|OT
(V.103)
Для систем с незначительной областью твердых растворов
при анализе межмолекулярного взаимодействия обычно
обращаются к уравнениям типа (V.71) и (V.72). Тогда для точки
эвтектического состава получим
ТУ? 1П [(1 -4W)/4w\ ~ К т (Г\.т-Т.) -
coL = ~Ау(7У~Гг) . (V.104)
(1-2*£(.,)
Часто при расчетах значения экспериментальных точек в
координатах температура — состав подставляют непосредственно
в уравнения (V.102)—(V.104), с помощью которых и определяют
параметры энергии взаимообмена. При этом естественный разброс
экспериментальных данных приводит к изменению величины
энергии смешения, вызывая сомнения в пригодности той или иной
модели раствора. Влияние ошибок в исходных экспериментальных
данных на величину <dl и <os проанализировано в работе Фостера
и Вудса [182]. Правильный расчет параметров coL и (0s основан
на подборе значений, которые приводят к наилучшему
совпадению теоретически рассчитанных кривых ликвидуса и солидуса
с экспериментально найденными. Критерием расхождения может
служить величина а, определяемая из соотношения [183]
а2=<Тлик + асол, (V.105)
где
м
О'лик = 2т' V лик, расч * лик, эксп/ /Ai, (V.IUOJ
N
^сол == 2j \' сол, расч — Л сол, эксп/ /*». (V.IU/J
k=\
181
Здесь М и N — число экспериментальных точек. Операцию
поиска проводят до тех пор, пока не будет найдено минимальное
отклонение а.
Наибольшее допустимое отклонение атах в каждом
конкретном случае устанавливают в достаточной мере произвольно,
исходя из точности экспериментальных данных. Как показал
анализ, выполненный в работе [183], условию
a<<W (V.108)
могут удовлетворять одновременно несколько моделей растворов.
При этом для разных моделей значения подбираемых параметров,
определяющих температурную и концентрационную зависимость
энергии взаимообмена, могут сильно различаться. Вследствие
этого при больших ошибках эксперимента расчет таких
термодинамических свойств сплавов, как энтропия и энтальпия смешения,
основанный только на анализе фазовых диаграмм, приводит к
ненадежным результатам. Однако получаемые значения GE сплавов
и коэффициенты активностей компонентов хорошо согласуются
с экспериментом.
В общем случае решение задачи оценки параметров,
определяющих температурно-концентрационную зависимость энергии
взаимообмена, возможно лишь с привлечением ЭВМ.
Для частного случая, когда энергии взаимообмена в жидкой
и твердой фазах равны, уравнения (V.62) и (V.63) можно
преобразовать к виду
К4)2- {4У\ © - q; = in[(l - *f)/(i - 4)]9
[(l - 4У - 0 - 4)2] *—(& = in (4/4), (V.109)
где Q; = (Ah), JR) (1 - Т/Т], m), (V.109a)
со' — приведенная энергия взаимообмена.
В работе [184] предложен простой прием решения системы
уравнений (V.109) с помощью некоторой универсальной функции,
который позволяет быстро рассчитать диаграмму состояния при
различных значениях энергий взаимообмена и, следовательно,
определить из условия наилучшего прохождения рассчитанной
кривой через экспериментальные точки энергии взаимообмена
в фазах. С этой целью уравнения (V.109) преобразуют к виду
C(xL)-C(^) = Q;-Qb D{xL)-D(xs) = Q[, (V.110)
где функции
С (г) = (1 — 2г) со' + In [г/(1 — 2) ],
D(z) =22со' + In (1 —2) (V.lll)
табулированы в весьма широких пределах.
Используя соотношения (V.98) и (V.99) и выражения для
коэффициентов активностей компонентов, записанные в виде (IV.113)
и (IV. 114), можно проанализировать кривые фазовых равновесий
182
в приближении квазихимической модели. Такой анализ
проводили для систем Si (Ge) — металл в работе [185]. Полученный
результат дает удовлетворительное согласие с
экспериментальными данными.
Джордан предложил [186] метод анализа межмолекулярного
взаимодействия в бинарных системах А—В в приближении модели
регулярного ассоциированного раствора типа А + В + В2.
Для данного случая активности компонентов бинарного
раствора определяются [187] уравнениями:
ЯА = У Л* А = У At*At = йАг> (V- J * 2)
аВ = УВХВ = ув^В./ув^Вг = ^Bt/aBr (V. 1 13)
Коэффициенты активностей мономеров Аг и Вх можно рассчитать
по методу [187]:
RT\пуАх^со*4, КГ1пУвг ~©**л, (V. 114)
где со* — эмпирический параметр, учитывающий взаимодействие
между структурными единицами раствора.
Чтобы найти выражения для xAt и хВх через экспериментально
определяемые величины, рассмотрим равновесие реакции димери-
зации
2В1+±В2,
для которого справедливо
Ка(Р, T) = (X2BJXBt)(y%JyB,).
Во избежание громоздкости обозначений здесь и в дальнейшем
опустим индексацию при обозначении константы равновесия.
Условия сохранения масс компонентов для рассматриваемого
раствора можно записать в виде
xAt=xA(l+xB2), (V.115)
ХВг = ХВ ~ ХВш (1 + ХА). (V. 1 16)
Подставив эти уравнения в выражение для константы
диссоциации димера, получим
К = (уВ1/увш) [хв - xBi (1 + хА)\2хв%. (V. 117)
С помощью уравнения Гиббса—Дюгема, записанного в виде
хАА 1п У Ах + хвА In yBt + xBid In уВш = О,
и уравнений (V.114)—(V. 116) найдем зависимость для In ув2-
Интегрируя от хв = 1 до хв и считая смесь частиц Вг и В2
в чистом компоненте В идеальной, получим
RTltiyBt = 2v>*x2A. (V.118)
183
После подстановки этого выражения и второго уравнения из
системы (V.114) в (V. 117), получим
К = [хв-хВш(2 + хА)]*/хВш.
Отсюда легко убедиться, что
х* = (1 - *л) (QT - 1)/(1 + хА) (Q* + 1), (V. 119)
где Q* = y^l+4(l-jfj)//C. (V.120)
Подставив выражение (V.119) в (V.115) и (V. 116), получим
xAt = 2хл (хА + Q*)/(l + xA) (Q* + 1), (V. 121)
xBt = 2xB/(l+Q% (V.122)
Окончательно, подставив соотношения (V.121) и (V.122) вместе
с уравнениями (V.114) в формулы (V.112) и (V.113), получаем
выражения для коэффициентов активности компонентов
регулярного ассоциированного раствора типа А + В + В2:
RTlnyA = aXB + RT\n[2(xA + Q*)/(l+xA)(Q*+l)l (V.123)
/?rin^ = a)*4 + ^ln[(l+Q°)/(l+Q*)], (V.124)
где Q° = К1 + 4//С. (V.125)
Для полностью диссоциированной смеси хВг -^ 0 и /С->-°о.
При этом Q становится равным единице, а уравнения (V.123) и
(V.124) переходит в выражения для коэффициентов активности
регулярного раствора.
Используя формулы (V.98), (V.39), (V.123) и (V.124), можно
проанализировать кривые фазовых равновесий в бинарной
системе А—В в приближении модели регулярного
ассоциированного раствора А + В + 52. Так, в случае малой взаимной
растворимости в твердой фазе соотношения (V.98) и (V.99) примут
вид:
«• (*&)' +Ч.
— Я In (2 (Q* + 1 —*|{)/[(2 —*fe) (Q* + Oil
*'0-*д)2+Чт
(V.126)
(V.127)
Выражение (V.126) было использовано в работе [1861 для
анализа линии ликвидуса, отвечающей первичной
кристаллизации висмута в системе Bi—Ga. Полученный результат хорошо
согласуется с экспериментальными данными.
Следствием данного частного рассмотрения является
заключение о неправомерности привлечения модели регулярного
раствора для термодинамического анализа диаграмм фазовых равно-
№
Ёесий в системах, растворы которых могут содержать ассоциаты
одного из компонентов.
Как отмечается в работе [186], располагая изотермическими
данными о Y/> нетрудно рассчитать условные значения энергии
взаимообмена по уравнению
со = /?Г1п^/(1 -*iY>
(V.128)
где со — условное значение со.
В то же время на основании, скажем, выражения (V.123)
имеем
со =
RT
= со* +
2(*A + Q*)
+ 4 1П (1+xa)(Q* + 1)
(V.129)
а) При хл = О
принимает вид
это соотношение
со (хА = 0) = со* +
• + RT In [2Q7(1 + Q°)L (V.130) А —
Отсюда при отсутствии ассоциатов
в растворе (tf-^oo, Q° = 1) будем
иметь: со = со* = со, т. е. регулярный
раствор. В случае сильной ассоциации
одного из компонентов (Q°-^«>, К-*
со
со* + RT In 2.
Рис. 39. Концентрационная
зависимость условной энергии
взаимообмена при различных
степенях димеризации
компонента Б в растворе (Г = const)
0) при хА = 0 получим
(V.131)
б) При хл = 1 с помощью правила Лопиталя на основании
выражения (V.129) найдем
& - со* + RT/K. (V.132)
Как и в первом случае, при К -> <*>, S = со* = со. в) Для
/С > 0, очевидно, характерно некоторое промежуточное
положение энергии взаимообмена между двумя рассмотренными
крайними случаями. Графически описанную картину
концентрационной зависимости со при различных степенях димеризации
компонента В в растворе можно иллюстрировать схемой,
представленной на рис. 39. Приведенные выше уравнения широко применяют
для анализа характера межмолекулярного взаимодействия в
приближении модели регулярного раствора и различных ее
модификаций не только в бинарных, но и в квазибинарных системах. Так,
в работе [183] проведен систематический термодинамический
анализ кривых фазовых равновесий в квазибинарных системах с
непрерывным рядом твердых растворов, образованных
соединениями АШВУ, в приближении модели квазирегулярных рас-
1S5
творов и ее частных случаев. По нашему мнению, такой подход
допускает практически полное отсутствие диссоциации
соединений в растворах.
При анализе межмолекулярного взаимодействия в
квазибинарных системах типа АтВп — CpDq возможны и другие способы
описания кривых фазовых равновесий. Остановимся на некоторых
из них.
Предположим, что соединения-компоненты практически не
диссоциируют в твердом растворе и существенно диссоциируют
в жидком. Получим общие термодинамические уравнения,
позволяющие рассчитать кривые фазовых равновесий в этом случае.
При р = const для твердого раствора можно записать
^тв„(Г)==^(т\(Л + ^1п^та„(1-4А), (V.133)
|xSA(T) = ii°clVg(T) + RTlnyScpDqxScpDq, (V.134)
а для жидкого раствора, учитывая диссоциацию
соединений-компонентов,
VAmBn(T)=miiLAi(T) + n^(T), (V.135)
rify(Т) = PA W + «А Ю- <У*i36>
При равновесии будет справедливо:
\1Атвп = \1Атвп = т\к\ + /i|i^, (V. 137)
|*сЛ = |1сЛ = РА + <п&19 (V. 138)
где химический потенциал мономеров определяется уравнением
rf = \i]iL) (Т) + RT InTWf (i = Au Bu Cu Dx). (V. 139)
Поскольку, согласно принятой модели, соединения-компоненты
почти полностью диссоциируют в жидкой фазе, то в выражении
(V.139) под величинами yt и х( следует понимать коэффициенты
активности и аналитические мольные доли веществ Л, 5, С и D
в растворе. При этом жидкую фазу можно рассматривать с
хорошим приближением как четырехкомпонентный раствор.
Поэтому далее индекс 1 у мономерных форм будет опущен.
В точках чистых компонентов на основании выражений (V.137)
и (V.138) будем иметь
■4Й. - Ы(Х - тАШ + п^\ (V.140)
К%-»с%-р6аг+№>1Ь\ (V.H1)
где верхний индекс * указывает на принадлежность чистому
соединению.
186
На основании формулы (V.33a), учитывая зависимость ДА?, m
и As°it m от температуры, можно записать
VAmBn = НАЛ» + Д«т, АтВп(Тт. АтВп - Т) -
- ДсПт^.л» ~Г-Г1п(г;,л я/Г)], (V.142)
где Дс£ — разница в теплоемкостях твердого и переохлажденного
жидкого соединения.
Подставив соотношение (V.142) в (V.140) и пренебрегая
разницей в теплоемкостях Ас°р, получим
й%= m^(L) + п\1*в(L) - As;. Атвп(Тт. Атвп - T). (V.143)
Объединив это выражение с уравнением (V.137), получим
окончательно
Inуа в (1 - Хс d ) = mIn *u?A*A
+ nln f—-^-—] + Asm,A mBn(T°m, AmBn-T)/RT. (V.144)
\Ув n/("i + n) I
Аналогично из формулы (V.138) найдем
+
1п7?р0Др? = р1п -
?£*с
Yc(L)P/(P + <7)
+ 9ln L<^ + (?)] + AV cnDfl(r;. C„D„ - Г)/Я7\ (V.145)
-p-f* ""-p"?
При m = n = p = q=l уравнения (V.144) и (V.145)
приобретают вид:
1П ySAB(l -XSCD) = 1П [уЬАук/уАа)увШ] +
+ ln(4^4) + As°mMB \Tm, AB — T)/RT; (V.146)
In YCD*CD = In [7^0/тС (LVd{L)] +
+ ln(4^4) + As°m,Co (^m, CD — r)//?7\ (V.147)
Уравнения, аналогичные по виду (V.144)—(V.147), для
тройных и, как частный случай, квазибинарных.систем типа АтВп —
— CqBr были получены Джорданом в работе [188].
Соотношения (V.144)—(V.147) также широко применяют для
анализа фазовых равновесий в квазибинарных системах.
Вид уравнений (V.144)—(V.147) в каждом конкретном случае
определяется выбором модели раствора, в приближении которой
проводят анализ межмолекулярного взаимодействия.
187
Анализ изотерм, принадлежащих поверхности ликвидуса
тройных систем Ga—As—Zn и Ga—Р—Zn, включая и квазибинарные
разрезы GaAs1—Zn и GaP—Zn, на основе соотношений вида (V.146)
в регулярном приближении был проведен в работе [189].
Базируясь на методе Гуггенгейма [190], Стринфеллоу и
Грин [185] предложили метод оценки коэффициентов активностей
компонентов трехкомпонентной смеси в квазихимическом
приближении. Результаты [185] могут быть использованы для анализа
равновесий с помощью уравнений (V.146) и (V.147) в
квазибинарных системах типа АС—ВС. В этом случае жидкий раствор следует
считать трехкомпонентным.
Для оценки коэффициентов активности t-того компонента
k-компонентной смеси в квазихимическом приближении можно
использовать метод, описанный в [191].
В работе [192] рассмотрен метод анализа межмолекулярного
взаимодействия в квазибинарной системе АС—ВС для случая,
когда в твердой фазе соединения не диссоциируют, а жидкая фаза
представляет собой регулярную смесь типа А + В + С + АС +
+ ВС.
4. ТЕРМОДИНАМИЧЕСКОЕ ОБОСНОВАНИЕ
РЕТРОГРАДНОГО СОЛИДУСА.
РАСЧЕТ РЕТРОГРАДНОГО СОЛИДУСА
В РАЗЛИЧНЫХ ПРИБЛИЖЕНИЯХ
Подавляющее большинство двойных систем с участием
германия и кремния, а также ряда других полупроводников отличается
ретроградным солидусом или так называемой отрицательной
растворимостью. Сущность рассматриваемого явления, как было
показано выше (см. гл. III), заключается в том, что максимальная
протяженность области гомогенности на основе, например,
компонента А располагается выше температуры нонвариантного
превращения в двухкомпонентной системе (см. рис. 14).
Как отмечалось выше, на первый взгляд, ретроградный
характер солидуса представляется необычным явлением, поскольку
процесс плавления совершается при отводе тепла, т. е. при
охлаждении сплава. Возможно, в противоположность обычному
ретроградное плавление сопровождается выделением, а не
поглощением тепла. Однако вопрос этот до настоящего времени не изучен.
Между тем, еще в 1908 г. Ван-Лаар, используя модель,
подобную модели регулярных растворов, теоретически рассчитал
несколько бинарных фазовых диаграмм и показал возможность
существования диаграмм состояния с отрицательной
растворимостью [62, 66]. С тех пор возможность ретроградной растворимости
была практически забыта на долгое время. Вероятно, это в
какой-то мере можно объяснить тем, что Ван-Лаар не выделил
особую точку максимальной растворимости на кривой ретроградного
солидуса, а также отсутствием экспериментального материала
188
вплоть до 1926 г. Поэтому когда в работах [193—194] был
экспериментально обнаружен ретроградный характер солидуса
в системе цинк—кадмий со стороны цинка, авторы отнеслись
к этому явлению как к невозможному и пытались объяснить его
аллотропным превращением в цинке. Только в 1924 г. и позже
Рауб с сотрудниками [195—197] обнаружил значительное число
диаграмм состояния с ретроградной растворимостью. В 1948 г.
Мейеринг [65] проанализировал ретроградное явление в общем
виде, использовав дифференциальное уравнение Ван-дер-Ваальса,
и получил приближенную формулу, позволяющую предсказывать
ретроградный характер солидуса в бинарных металлических
системах на основе данных о составе сосуществующих фаз при
температуре эвтектического превращения.
В соответствии с работой [65] для двухфазной двухкомпонент-
ной системы при постоянном давлении запишем дифференциальное
уравнение Ван-дер-Ваальса в виде
Wr ¥1Щ7Т • (УЛ48)
Принимая во внимание, что по условию устойчивости величина
производной (d2g/dx2)^t т всегда положительна, из уравнения
(V.148) приходим к выводу, что в точке максимальной
растворимости на кривой ретроградного солидуса должно выполняться
условие
(dxfdT)f = 0, если (ds/dxfPt т - (ss—sL)/(xs—xL) = 0. (V. 149)
Рассмотрим бинарную металлическую систему, компоненты
которой имеет одну и ту же энтропию плавления s°it m.
Запишем выражения для энтропии твердой и жидкой фаз,
принимая энтропию смешения идеальной и учитывая правило
Ричардса, согласно которому энтропия плавления металла
определяется соотношением
As], m = bRy
где b может принимать значения в интервале от 1 до 2,7. Тогда
справедливо
^ = (1-4)5;(5Ч44(5)-/?[(1-4)1п(1-4)+41п4],
(V.150)
— Я [(1—4) In (1-4)+ 4 In4L (V.151)
Подставив выражения (V.150) и (V.151) в (V.149), после
несложных преобразований получим формулу Мейеринга:
lgxf = lgx2L _ 0,4346/4 + l(l — 4)/4] X
х lg(l-4)-[(l-4)/4]lg(l-4). (V.152)
189
ig*|
С помощью уравнения (V.152) Мейеринг построил график,
представленный, на рис. 40, который позволяет предсказывать
знак производной (дТ1дх)% а следовательно, и характер солидуса
в бинарных системах. На этом графике заштрихованные площади
ограничивают область сосуществующих составов Xs и xLy где
указанная производная \дТ/дх\^, ^ оо. При этом верхняя
заштрихованная площадь соответствует значениям ft от 1 до 1,2,
а нижняя — от 2,3 до 2,7. Очевидно, что для сопряженных
значений Xs и xLy попадающих
в область ниже
заштрихованной полосы, величина
производной (dTldx)sp —
положительная, и, наоборот, выше этой
полосы — отрицательная.
Согласно Мейерингу, для
классификации кривых
солидуса по ретроградному или
нормальному типу с помощью
графика, изображенного на рис.
40, достаточно располагать
данными по составу жидкой и
твердой фаз только при темпера-
туре эвтектического превраще-
tfQ ния. В самом деле, если
производная (dTldx)sp при температуре
эвтектики имеет отрицательный
знак, то при более высоких
температурах она ни в одной
из точек не примет значения,
равного оо, поскольку минимум Xs на кривой солидуса не
наблюдается. В этом случае вид кривой солидуса будет нормальным.
В то же время, если при температуре эвтектики (дТ/дх)р
положительна, то при повышении температуры солидус будет иметь
вертикальную касательную при условии, что он проходит через
точку плавления чистого компонента. Здесь мы имеем дело с
кривой ретроградного солидуса. В этом случае возможны два
исключения; первое связано с аллотропным превращением в чистом
компоненте, второе — с расслоением в жидкой фазе; на их
анализе мы подробно не останавливаемся.
В результате проведенного анализа Мейеринг пришел к
выводу, что ретроградный солидус возможен в бинарных системах,
где растворимость в твердом состоянии незначительна, а
температура эвтектического превращения существенно ниже
температуры плавления чистого твердого растворителя.
К сожалению, работа Мейеринга долгое время оставалась
в тени, и вопрос о ретроградном солидусе в соответствующей
учебной и монографической литературе не нашел отражения.
190
Рис. 40. К термодинамическому
обоснованию ретроградного солидуса по
Мейерингу [298]
За последние 10—15 лет положение дел резко изменилось в связи
с массовым обнаружением ретроградного солидуса в двойных
системах с участием полупроводников [67]. Появились работы,
в которых возможность рассматриваемого явления обосновывалась
вновь [64, 198—200]. Заметим, что подход Мейеринга строг
только до известного предела. Конечное же уравнение (V.152)
является приближенным вследствие ограниченности правила Ри-
чардса, что было отмечено одним из авторов в работе [201 ] при
анализе периодической зависимости энтропии плавления простых
тел от порядкового номера элемента в таблице Д. И. Менделеева.
Тем не менее, качественные выводы Мейеринга можно считать
вполне обоснованными, поскольку те максимальные отклонения
от правила Ричардса, которые наблюдаются в действительности,
не могут изменить принципиально выводов, сделанных на основе
уравнения (V.152) и графика на рис. 40.
Причина ретроградной растворимости становится более
понятной при определении положения линии солидуса по известной
кривой ликвидуса [64, 199—200].
Для упрощения трактовки термодинамических причин
отрицательной растворимости авторы работ [64, 199—200] допускают,
что твердая фаза ведет себя как регулярный раствор, а жидкая
фаза, находящаяся в равновесии с твердой выше эвтектической
температуры — как идеальный раствор. Разность химических
потенциалов растворенного компонента 2 в насыщенном
разбавленном твердом растворе и чистого компонента 2 определяется
соотношением
^ <*> _ ^ <s> = RT In V2 <S)*2 (S). (V. 153)
В то же время
H2*<s>_H°2<s> = /if (S> -Tif<s>. (V.154)
Из соотношений (V.153) и (V.154) находим
RT In V2 (S)*2(S) = hi(S) - TSf (S). (V. 155)
Так как по условию рассмотрения твердый разбавленный
раствор является регулярным, то по определению
s^S) = -R\nx2<S). (V.156)
Отсюда ^rlnV2MS) = /if (S>. (V.157)
Поскольку RT In 72(S) = osAS) 2 «* <os.
то в рассматриваемом случае
/if (S) «=> const,
т. е. не зависит от состава.
191
Так как разбавленный твердый раствор находится в равновесии
идеальным жидким раствором, то можно записать
^ <*> + RT In 4 {S) + ft?{S) = ill(L) + RT In 4,
или In (xl <S>/*2L) = (Дц2 - ft* (S))/tf Г,
(V.158)
(V.159)
цессе плавления второго
Атомная доля
Рис. 41. Кривые солидуса в системе
Ge—Me, рассчитанные для
различных значений h*z (S) (Дж/моль~1);
те
где А|Ш2 — изменение мольной свободной энергии Гиббса в про-
компонента при температуре 7\
Учтя формулу (V.33a),
соотношение (V.159) можно записать в виде:
ln(*2MS7*2L) = lntf2 =
= (Mlm-h^S))/RT-As2tjR.
(V.160)
Здесь и далее индексы Тир при
обозначении коэффициента
распределения Kt (T,p) опущены.
Для получения кривой солидуса
/ (xf, Т) = 0 необходимо исключить
из уравнения (V.160) величину х\.
Если принять во внимание, что
в разбавленном твердом растворе
х2 ((( 1, а жидкая фаза идеальна, то
для х^ можно воспользоваться
уравнением Шредера в форме (V.6).
Решая совместно уравнения (V. 160)
и (V.6), можно рассчитать кривую
солидуса,. если известны такие
термодинамические параметры, как температуры и теплоты плавления
обоих компонентов, а также парциальная мольная теплота смешения
/tf <s> растворенного компонента в твердом растворе. Как
показывает опыт, тенденция к ретроградной растворимости
непосредственно связана с величиной ftf <s). Тогда если величина Щ <5>
относительно низкая, то кривая солидуса не обнаруживает
ретроградного характера, но с ее увеличением тенденция к ретроградной
растворимости растет. Причиной того, что ретроградная растворимость
не наблюдается у большинства металлических систем, является
слишком низкое значение RE <s). Фактические значения /if*5),
необходимые для появления ретроградности, весьма большие.
Используя соотношения (V.160) и (V.5) и задаваясь произвольно
различными значениями для величин ftf <5\ Тармонд и Стразерс
[199] построили четыре кривые солидуса в системе с участием
германия, где в качестве растворенного второго компонента
было выбрано произвольное вещество с теплотой плавления
14,6 кДж/моль и температурой плавления 1160 К. Эти результаты
приведены на рис. 41.
192
/
6
— 92; 2 — 114; 3 — 46; 4 — 23
— кривая ликвидуса, рассчитан
ная по уравнению (V.162)
Из рисунка видно, что кривые солидуса, соответствующие
более высоким значениям Щ <5>, проявляют тенденцию к
ретроградной растворимости.
Высокое значение /zf <S) связано с низкой растворимостью
компонента в твердой фазе. В результате этого явление ретро*
градной растворимости наблюдается, как правило, только в си*
стемах с низкой растворимостью. Большинство систем, в которых
растворителями служат германий и кремний, являются системами
такого типа. Для приведенного выше примера было необходимо
сделать предположение, что твердый и жидкий растворы ведут
себя соответственно регулярно и идеально. Эти ограничения,
однако, не противоречат общему выводу о том, что ретроградная
растворимость непосредственно связана с большим значением
величины Щ <s>.
Авторы работы [199] проанализировали также явление
ретроградной растворимости с помощью другого термодинамического
параметра — коэффициента распределения /Q. Записав выражение
для коэффициента распределения KZ при температуре плавления
чистого растворителя в виде
In Kl = (Д/£ т - hi {S))/RT°h m - A*;, JR (V.161)
и подставив его в формулу (V.160), они получили следующее
уравнение:
In /С2 = (Т[, Jt) In Kl + (As2. JR) (Tl. m/T - 1), (V. 162)
где 77, m — температура плавления чистого растворителя.
Коэффициент распределения определяется экспериментально.
Анализ систем с участием германия для различных значений
показывает [199], что чем меньше величина этого параметра,
тем более ярко выражена ретроградная растворимость, причем
максимум растворимости с увеличением Kl сдвигается к более
низким температурам.
По данным работы [199], для осуществления ретроградного
солидуса величина К°2 должна быть меньше 0,1, а значение Щ (S>
больше 23148 кДж/г-моль.
Причину, обусловливающую ретроградность, Тармонд в
работе [199] объясняет следующим образом. В системах с
ретроградным солидусом, как показывает опыт, температура эвтектики
обычно довольно низкая, а ее состав в сильной мере сдвинут
к растворенному компоненту М. Вследствие этого химический
потенциал растворенного вещества М в жидком растворе должен
проходить через максимум. Это легко уяснить, если за меру
химического потенциала выбрать изменение парциального давления М
вдоль кривой ликвидуса. В точке плавления чистого растворителя,
например германия, парциальное давление М равно нулю. По
мере добавления М температура ликвидуса медленно падает, в то
время как концентрация увеличивается очень резко;
следовательно, парциальное давление М тоже растет. В области больших
' Глазов В. М. и др. 193
концентраций вещества М состав ликвидуса изменяется с
температурой медленно, поэтому преобладающим становится
температурный фактор, и парциальное давление М начинает падать.
Таким образом, химический потенциал или парциальное
давление растворенного вещества М вдоль кривой ликвидуса должны
проходить через максимум. Поскольку при равновесии химические
потенциалы компонентов в сосуществующих фазах равны, то
на кривой солидуса в рассматриваемом случае должен
наблюдаться максимум растворимости.
Так как твердый раствор разбавленный, то в соответствии
с законом Генри концентрация растворенного вещества М будет
прямо пропорциональна его давлению пара при постоянной
температуре. При этом если константа Генри не зависит от
температуры, то максимум растворимости на кривой ретроградного
солидуса будет иметь место при температуре, соответствующей
максимуму давления вещества М над насыщенным жидким
раствором. Поскольку в реальном случае наблюдается температурная
зависимость, максимум растворимости М должен наблюдаться
при более высоких температурах, чем максимум его парциального
давления.
Общий способ установления ретроградной растворимости
твердых растворов на основе зависимости логарифма равновесного
коэффициента распределения от логарифма мольной доли
растворенного вещества (примеси) дан в работе А. Н. Киргинцева [202].
Для двухкомпонентной системы, состоящей из твердой и
жидкой фаз, при постоянном давлении справедливы
дифференциальные уравнения Ван-дер-Ваальса (см. гл. II) в форме
(dT\L *?-*2 (&g\L ,v - ъ
*L\S - ±lA f^\s . (V164)
/ dT \s
\ dx2 )p
В соответствии с видом диаграммы состояния с ретроградным
солидусом (см. рис. 14) из уравнений (V.163) и (V.164), полагая
справедливость условий устойчивости
(d2g/dxl)Lp,T>0 и (d2g/dxi)lr>0,
находим, что в интервале между температурой плавления
растворителя и точкой максимума растворимости m справедливо
ss-sL-(xl- 4) (dsldx2)Lp, г > 0,1
в» -^ _ (4-4) (ds/dx2)sp, т > 0; J (V- ЬЬ)
194
в точке т
ss-sL- (*f - 4) (ds/dx2)Lp, г > О
s* _ s^ _ (xs _ 4) (№2)s r = о; Г ^V- bb>
в интервале между точками mud
ss _ sL - (*f _ 4) (ds/d*2)£, r > 0
s* _ s^ _ (xf _4)(ds/dx2)sp, T<0.1 {W'b)
Разделив уравнение (V.163) на (V.164) и учитывая, что,
согласно формуле (11.43),
(d2g/dxl)p, г = — (д[ц/дх2)р, Т/х2,
после несложных преобразований можно получить следующее
выражение для равновесного коэффициента распределения:
<д\пКЛ ss-sL- (*f-4) fo/^2)* г (^i/^2)p, г j
д 1п *2 J р§ т ss - sL - (*f - 4) (дз/дь)^ т (d\ijdx2)spt T
(V.168)
Сравнивая это соотношение с условиями (V.166) и (V.167), видим,
что в точке m
(д In Kjd In х%)Рш т = - 1, (V. 169)
а в интервале между температурой нонвариантного равновесия
и точкой m (рис. 14)
(д In К2/д In 4%, т < - 1. (V. 170)
Таким образом, если зависимость логарифма коэффициента
распределения от мольной доли примеси отвечает условиям
(V.169) и (V.170), то кривая солидуса имеет ретроградный характер
и, наоборот, если эти условия не выполняются, то кривая
солидуса имеет обычный вид. На рис. 42 показана зависимость lg K2
от lgA^ для системы KCI—SrCl2 (SrCl2 — примесь) по данным
работы [203].
На рис. 42 видно, что при lg х\ = —0,6 осуществляется
условие (V.169), т. е. в системе KCI—SrCl2 имеется ретроградная
растворимость.
Существуют два термодинамических подхода к оценке величины
растворимости легирующих элементов в полупроводниках.
Первый основан только на рассмотрении гетерогенного равновесия
твердой и жидкой фаз; второй учитывает также ионизацию
атомов легирующего компонента при образовании раствора. С
помощью первого подхода удается дать качественное описание
кривых солидуса, при этом для описания поведения твердой фазы
обычно используют модель регулярного раствора, а для
описания жидкой фазы модель идеального [199] либо регулярного
раствора [204].
7* 195
Так, авторы работы [199] провели расчет солидуса в двойных
системах Ge—Sb, Gey—Си и Si—Си, допуская, что жидкие растворы
в этих системах идеальны, а твердые — регулярны. В работе
[205] показано, что жидкие растворы в двойных германиевых и
кремниевых системах с различными элементами /В, IIIB, IVB
и VB подгрупп периодической системы лучше описываются с
помощью теории регулярных растворов. Учитывая такое поведение
жидких сплавов и рассмотрев условия гетерогенного равновесия
твердой и жидкой фаз в приближении теории регулярных
растворов, В. М. Козловская и Р. Н. Рубинштейн [206] предложили
для расчета кривой солидуса
использовать уравнение следующего вида:
ln/C2-(A/i2.m-o)S)//?r +
+ (coL/RT)(l - 4У - As2> JR.
(V.171)
Недостаточность такого подхода либо
его модификаций типа, указанного
в работе [204], для систем с
участием полупроводников очевидна,
так как при их легировании
элементами донорного и акцепторного
типа существенную роль играют
процессы ионизации в твердой фазе, на
что обратил внимание Раисе [207—
209]. В этой связи нами была
рассмотрена возможность расчета линии солидуса в
полупроводниках с учетом степени ионизации атомов примеси в твердом
растворе на их основе [210]. При этом поведение доли неионизиро-
ванных атомов в твердой фазе мы рассматривали в приближении
регулярных растворов.
Рассматривая гетерогенное равновесие при образовании
твердого раствора на основе полупроводника, можно записать
Рис. 42. Иллюстрация
применимости условий (V.169) и (V.170) для
установления ретроградного
солидуса на примере системы KCl—SrCl2
JL:
:js^jf + et
(V.172)
где J n J — неионизованное состояние легирующего
компонента в твердой и жидкой фазах соответственно, /f и е —
ионизованное состояние легирующего компонента и носитель заряда.
Согласно условию гетерогенного равновесия, химические
потенциалы растворенного вещества в сосуществующих фазах
одинаковы, т. е. \*k = fif.
В случае образования реального раствора
H2L = ^^> + RT In 4V2,
196
(V.173)
Отсюда по условию равновесия
£(L) + RT In xU\ = pi(S> + RT In xl iS)y2 (S)
или
xl <s> = 4L)yiL)hi(S) exp [(^ <L) - ^ ^)/RT] x
X exp [(^(S) - ^ (S))/W1. (V.174)
Допустим, что разницей между значениями \il(S) и \il(S) можно
пренебречь.
С учетом известного допущения (V.33a)
*; <s> = (4Ly»/y; <s>) exp [as;. m (r;. m - тут. <у.т)
С другой стороны, согласно работе [24], химический
потенциал растворенного компонента равен химическому потенциалу
нейтральной формы, т. е.
,*(S) + RT In xl <S)Y2 iS) = W(S)). + W In (xl <*>)„ (yl <*>)*,
(V.176)
где индекс п означает принадлежность к нейтральному состоянию.
Считая стандартным состоянием нейтральных атомов и
растворенного компонента одно и то же стандартное состояние, например,
состояние чистого твердого компонента, на основании формулы
(V.176) можно записать
у; <« в (,; («)я (v; («у,; (S) = Fb (t; <s>)n) (ул77)
где F„ — доля неионизованных атомов легирующего компонента
в твердом растворе.
При высоком уровне легирования величину Fn можно найти
с помощью функции распределения Ферми—Дирака
Fn = {1 + 1/2 exp \(Ef - Ea)lkT]}-\ (V. 178)
Fn = {1 + 1/2 exp [(Ed - Ef)lkT]\'\ (V.179)
где Еа и Ed — энергии ионизации акцепторов и доноров
соответственно, отсчитываемые, так же как и уровень Ферми £),
от верхнего края валентной зоны.
С учетом выражения (V.177) уравнение (V.175) можно
представить в виде
4 iS)/*2 = Т2/Ы iS))n Fn exp [(As;, JRT) (f2t m - T)]. (V. 180)
В приближении регулярных растворов для рассматриваемого
случая можно записать
^=ехр[(со^Г)(1-4)2],
(7;<5>)„ = ехр((со5/ЯГ)[1 -(*2*(5>Ш, (V.181)
где (os — энергия смешения атомов полупроводника и
нейтральных атомов легирующего компонента в твердой фазе.
197
Подставив эти выражения в уравнение (V.180), получим
К2 = х\ <S>/4L) = F-1 ехр {(Д4. JRT) (Т°а. т-Т) +
+ (<oL/RT) (1 - 4)2 - (<о5/*Г) [1 - (4 <s>)„]2). (V.182)
Учитывая незначительную растворимость легирующего элемента
в полупроводнике, можно пренебречь величиной (x*2{S))n. Тогда
К2 = Fnl ехр [(Д5°2, JRT) (Tl m - Т) +
+ (со*7ЯГ) (1 - 4)2 - <*S/RT] • (V. 183)
Допустим, что поведение нейтральных атомов в твердой фазе
описывается строго в рамках модели регулярных растворов. В этом
случае при-температуре плавления полупроводника будем иметь
Kl = (F°n)-1 ехр [(As°2, jRTl, m) (Т°2, m - Т°и m) +
+ со° {L)/RTU m - <s>s/RTl, Л (V. 184)
где верхний индекс ° при Fn и со указывает на принадлежность
этих величин к температуре плавления полупроводника T\t m.
Решим уравнение (V.184) относительно cos:
cos = bs2t m (Tl, m - К m) + co°(L) - RT°U m \n(FnKl). (V. 185)
Подставляя полученное значение cos в формулу (V.183), получим
уравнение кривой солидуса:
х\ <5> = x^Fn1 ехр [(Asl, JRT) (Т°1ш m - Т) +
+ (coL//?7) (I -X\Y + (Т\. JT) In (FX) - со0 <L>/RT]. (V.186)
Величину F°n можно найти, полагая, что при температуре
плавления полупроводник является собственным и, следовательно,
уровень Ферми проходит практически посередине запрещенной
зоны, т. е.
F°n = {1 + 2"1 ехр l(-Ea + HE/2)/kT]}-\ (V.187)
F0n = {1 + 2"1 ехр \{Ed - ЬЕ12)1кТ\\~\ (V. 188)
где А£ — ширина запрещенной зоны полупроводника при
температуре его плавления.
Таким образом, зная положение уровня Ферми в твердой фазе,
коэффициент распределения легирующего компонента при
температуре плавления полупроводника, изменение ширины
запрещенной зоны с температурой, энтропию плавления легирующего
компонента и его потенциал ионизации, а также ход кривой
ликвидуса в исследуемой системе можно с помощью уравнения (V.186)
рассчитать кривую солидуса.
Пользуясь описанным методом, мы произвели расчет кривых
солидуса в двойных системах германия с алюминием, галлием,
индием, мышьяком и сурьмой. При вычислениях принимали во
198
внимание температурную зависимость энергии смешения
компонентов в расплаве. Сведения о ходе кривых ликвидуса, а также
необходимые для расчетов данные заимствовали из работ [67,
200]. Положение уровня Ферми определяли из графика Блэкмора
[211 ]. Полученные результаты приведены на рис. 43 в
сопоставлении, с экспериментальными данными. Из приведенного рисунка
следует, что результаты расчетов достаточно хорошо согласуются
с экспериментом. Наблюдающиеся незначительные расхождения
можно объяснить недостаточно строгим выбором стандартного
состояния для легирующего компонента.
Рис. 43. Кривые солидуса
для некоторых двойных
систем германия с
легирующими элементами донорного и
акцепторного типа:
Nд а — концентрация
доноров или акцепторов;
кривые — расчетные по
уравнению (V.186); точки —
экспериментальные данные [67]
Впервые последовательно подход к оценке величины
растворимости с учетом ионизации атомов легирующих компонентов был
осуществлен в работе Леховека [212]. Если допустить, что
энтальпия, необходимая для перевода одного моля нейтральной
легирующей добавки из твердого раствора в жидкий сплав, линейно
зависит от температуры, а поведение доли нейтральных атомов
в твердой фазе идеально, т. е.
Ы(5))*=1, (V.189)
то на основании соотношений (V.174) и (V.177) придем к
выражению
(Х; &)jxt = (7f/e) [F°nKtf/yl iL>fi-m/T, (V. 190)
где G = exp [(AsS, m —a)/R], которое совпадает с формулой
Леховека.
Описанный выше подход к оценке положения солидуса в
системах с участием полупроводника в качестве основы позволяет
решать и обратную задачу, т. е. устанавливать положение уровня
Ферми при данной концентрации легирующей добавки при
заданной температуре. При этом в качестве первичных используют
199
ZK
1100 \-
Данные о характере гетерогенного равновесия полупроводник-
примесь. Такая возможность была нами рассмотрена в работе
[213]. При сопоставлении полученных результатов для германия
с графиком Блэкмора получено вполне удовлетворительное
согласие.
5. АНАЛИЗ КРИВОЙ ЛИКВИДУСА КОНГРУЭНТНО
ПЛАВЯЩЕГОСЯ СОЕДИНЕНИЯ АтВп В РАЗЛИЧНЫХ
ПРИБЛИЖЕНИЯХ НЕИДЕАЛЬНЫХ РАСТВОРОВ
ИНТЕГРАЛЬНАЯ ФОРМА УРАВНЕНИЯ КРИВОЙ ЛИКВИДУСА
В РАЗЛИЧНЫХ ПРИБЛИЖЕНИЯХ МОДЕЛИ РЕГУЛЯРНОГО
РАСТВОРА
На основе дифференциального уравнения Ван-дер-Ваальса
нами было получено общее термодинамическое выражение (V.39),
которое описывает кривую ликвидуса конгруэнтно плавящегося
соединения АтВп в дифференциальной форме. Как отмечалось,
это выражение адекватно уравнению Пригожина и Дефэя [24],
полученному на основе функции химического сродства. Этот
результат подчеркивает общность подхода на основе уравнения
Ван-дер-Ваальса и дает основание считать выражение (V.39)
вполне строгим.
Общее решение этого уравнения может послужить основой
для использования конкретных моделей растворов при
термодинамическом описании кривых ликвидуса в системах
рассматриваемого типа. При решении этого уравнения в идеальном
приближении наметился общий подход к получению интегральных форм
уравнения ликвидуса в системе с конгруэнтно плавящимися
соединениями, связанный с привлечением конкретных моделей
[214]. В данном случае дадим общее решение уравнения (V.39)
и покажем, как на его основе можно получить уравнения для
различных приближений теории регулярных растворов.
Подставим в уравнение (V.39) выражение производной
химического потенциала по концентрации (d\i2/dx2)p, t- В результате
после разделения переменных найдем
- (qjt& IT2) dT = - [mR/{ 1 - *£)] dx\ +
+ (/i/?/*2)d*2 - [mRx&l(l—4)] d lnyt + nRdlnyt (V.191)
Принимая во внимание выражение (VI.43), для коэффициентов
активности можем записать
(1 —4) d In7i + *2 d In72 = 0. (V.192)
После подстановки d In y2 из выражения (V.192) в уравнение
(V.191) получим
q^X d (ЦТ) = mRd In (l - x\) + nRd Inx\ +
+ mRd In y[ + nRd In y*. (V. 193)
200
Полагая, что Цлтвп не зависит от температуры, и интегрируя
уравнение (V.193) в пределах от Т = T°mfA в , х% = п (т + п)
до Г, *£■, получим
^Lln^(1"^ +Шп ,(L)Y2*2 1-
[ Уг{L) mftm + п) ^ у2 <L> п/(т + п) J
= As^. Атвп (Т-Т°т, Атвп), (V. 194)
где индекс «штрих» относится к составу соединения. Уравнение
(V.194) является общей интегральной формой уравнения
ликвидуса, отвечающей первичной кристаллизации фазы на основе
конгруэнтно плавящегося соединения АтВп.
До этого вывод и решение уравнения (V.39) —
термодинамически строгие, если не считать допущения о независимости
Яа*в от температуры. Вполне очевидно, что уравнение (V.194)
может служить основой для вывода модельных форм уравнений
кривой ликвидуса. Для этого нужно выразить коэффициенты
активности в соответствующем приближении и подставить их
в выражение (V.194).
Используя уравнения (IV.139) и (IV.140), на основе (V.194)
получим уравнение кривой ликвидуса конгруэнтно плавящегося
соединения АтВПУ включающее наиболее часто употребляемые
частные случаи других преобразований (модели субрегулярного,
строго и квазирегулярного растворов и др.) ив соответствии с
результатами, полученными в работе [114], позволяющее с
достаточно хорошим приближением прогнозировать термодинамические
свойства растворов, в частности, теплоту смешения:
Т = {со£ [пг (4)2 + л (1 - *2L)2 - mn/(m + n)] +
+ (of [m (4)2 (2*2 - 1) + Ъгх\ (l - x\f - mn2/(m + nf] +
+ 0)2 [m (4)3 (3x£ ~ 2) + 3n (4)2 (1 - x\Y - mnV(m + nf] +
+ AC^m^Mcooi[-m/2/(m + n) + m(4)2 + n(l-x2L)2] +
+ (ou [- mnV(m + nf + m (4)2 (2x2L - 0 + Ъгх\ (l - x\f] +
+ ©{2 [— mn3/(m + nf + m (*2 )3 (34 - 2) + 3n (*2 )2 x
X (1 _ x^f] - R In [(m + «)m+7mY] -
- R In [(1 - *2T (4)4 + As;, л^Г1. (V.195)
Единое уравнение кривой ликвидуса в системах
рассматриваемого типа имеет смысл при частичной диссоциации соединения
АтВп. В противоположном случае, согласно работе [13], раствор
201
делится ординатой соединения на две фазовые области, а левая
и правая ветви ликвидуса должны описываться самостоятельными
уравнениями.
В приближении модели субрегулярного раствора, допускающей
температурную зависимость энергии взаимообмена (со2 = 0t
w2i = 0)» на основе уравнения (V.194) получим
АС Атвп + ©о N4)2 + п 0 - х^)2-тп/(т + п)] +
Т + «[■ [т (*£)а (24 - 1) + 2я4 (1 - 4)2 ~ "*"7(™ + *)2] (V1%)
Asm,AmBn-<l4m"l(m + n)-m4-n([-4L))2]-
— ©ft (mn2/(m + л)2 — m (*£)2 (2х£ — l) — 2ш£ X
Х (1 _ 4)2] _ R ln [(m + л)т+п (, _ ^Ljrn (^)"/(тт„")]
Это выражение аналогично уравнению, полученному нами
[161] прямым интегрированием дифференциального уравнения
кривой ликвидуса соединения Ат Вп с использованием указанного
приближения. Заметим, что при отсутствии температурно-кон-
центрационной зависимости параметра межмолекулярного
взаимодействия со выражение (V.196) преобразуется к виду [161]
r_ M°mtAmBn + ^(m + n)№-n/(m + n)Y
При т = п = 1, т. е. в простейшем случае соединения А В,
уравнение (V.197) переходит в известное уравнение, полученное в
работах Виланда [215], а также Нугаре и Потье [216]:
_, А^^ + <(24-1)72
Г=л4мв-«Ч^0-4)Г (УЛ98)
Аналогичный (V.I98) результат получен Г. М. Кузнецовым и
М. П. Леоновым [217] при рассмотрении процесса плавления
одного моля кристаллов соединения АтВп с пренебрежимо малой
областью гомогенности.
Заметим, что попытка [218] решить уравнение (V.39)
приближенным методом интегрирования (метод вариации произвольной
постоянной) с использованием модели регулярного раствора
привела к результату, не совпадающему с уравнением Виланда,
вследствие чего полученное в работе [218] выражение нельзя
считать приемлемым.
В заключение отметим, что нами были апробированы
практически все описанные выше модели раствора на многих группах
конгруэнтно плавящихся химических соединений. При этом
оказалось, что в сравнительно небольшом интервале концентраций
относительно стехиометрического состава большинство моделей
дает удовлетворительное соответствие расчетных и
экспериментально найденных кривых ликвидуса. При расширении концен-
202
трационного диапазона наблюдается последовательное
расхождение теории и эксперимента, причем наибольшей величины оно
достигает при использовании модели идеального и строго
регулярного растворов. Применимость полученных уравнений
иллюстрируется ниже на примере двух групп соединений, отличающихся
сложным составом.
|_1 I I 1 4/7/71 1 I | | |
^0,2 0,Ц 0,6 Те Мд ОЛ 0,Ц 0,6 0,8 Ъш
Рис. 44. Кривые ликвидуса, отвечающие первичной кристаллизации:
а — соединений А^Те^ б — соединений Mg2BIV; / — расчет по уравнению (V.195);
2 — расчет по уравнению (V.196); 3 — расчет по уравнению (V.47); 4 — данные работ
[219 — 220]; 5 — данные [219, 221—223]; 6 — экспериментальные данные авторов
Мы сравнивали рассчитанные по уравнениям (V.47), (V.196),
(V.197) кривые ликвидуса с экспериментальными кривыми,
отвечающими первичной кристаллизации сесквителлуридов галлия и
индия в системах Ga—Те и In—Те (см. рис. 44, а). Для уточнения
характера указанных кривых детально термографически
исследовали серии сплавов, располагающихся по составу слева и справа
от соединений Ga2Te3 и 1п2Те3. В каждой системе было изучено
по 15 сплавов. Для сплава каждого состава на фоторегистриру-
ющем пирометре ФРУ-64 снимали по три термограммы и брали
среднее значение температуры ликвидуса [161].
Исходными материалами для приготовления сплавов служили
галлий, индий и теллур полупроводниковой чистоты. Синтез и
термический анализ сплавов вели в эвакуированных до ~1,33 X
X 10"3 Па и запаянных кварцевых сосудах.
203
Расчеты кривых ликвидуса, отвечающих первичной кристаЛЛи*
зации соединений Ga2Te3 и 1п2Те3 по уравнениям (V.47), (V.196)
и (V.197), были выполнены с привлечением ЭВМ.
Из сравнения всех расчетных и экспериментальных данных
нетрудно заключить, что они достаточно хорошо согласуются
между собой в сравнительно узком концентрационном диапазоне.
При расширении концентрационного диапазона наблюдаются
расхождения между теорией и экспериментом в первую очередь при
использовании модели идеального, а затем строго регулярного
растворов. Модель субрегулярного раствора описывает
экспериментальные данные во всем интервале концентраций.
Совершенно аналогичная картина наблюдается в случае
соединений Mg2BIV (где BIV— Si, Ge, Sn, Pb). Результаты расчетов
по уравнениям (V.47), (V.196) и (V.197) в сопоставлении с
экспериментальными данными работ [220—223 ] приведены на рис. 44, б.
Из анализа полученных результатов расчета вытекает, что
модель субрегулярного раствора наилучшим образом описывает
экспериментальные данные по кривым ликвидуса [220—223].
Однако соответствующие расчеты функций смешения не всегда
дают удовлетворительное согласие с экспериментом.
ИНТЕГРАЛЬНОЕ ТЕРМОДИНАМИЧЕСКОЕ УРАВНЕНИЕ КРИВОЙ
ЛИКВИДУСА КОНГРУЭНТНО ПЛАВЯЩЕГОСЯ СОЕДИНЕНИЯ
(УРАВНЕНИЕ БРЕБРИКА)
Важную роль в установлении истинного строения раствора
играют термодинамические соотношения, описывающие кривые
ликвидуса с помощью экспериментально измеряемых
термодинамических свойств растворов и самих химических соединений.
Такие соотношения были получены в работах Бребрика [147,
224].
Используя результаты этих работ, получим уравнение кривой
ликвидуса, устанавливающее непосредственную взаимосвязь
между термодинамическими свойствами жидкой и твердой фаз.
Рассмотрим конгруэнтно плавящееся соединение АтВПу
находящееся в равновесии с жидкой двухкомпонентной фазой
состава х\. Допустим, что соединение имеет очень незначительную
область гомогенности, близкую к х2 = п/(т + /г).
При равновесии выполняется равенство химических
потенциалов каждого из компонентов в сосуществующих фазах [см.
уравнение (V.54)].
Эти равенства определяют составы равновесных
сосуществующих твердой и жидкой фаз при любой температуре.
Умножив первое уравнение из (V.54) на т, а второе на п
и сложив их, получим
m\ii + n\i2 = mfxf + fl[*f. (V.199)
204
Запишем выражения для химических потенциалов компонентов
в жидкой фазе в развернутом виде:
vi = \h Ш + ЯГ In vf (1 - 4)l (V.200)
|Х2 = ^2(L) + #Т In 72*2. (V.201)
Подставив эти выражения в уравнение (V.199), получим
mRT In [vf (1 — *£)] + nRT In (?£*£) -
= m (|if - i*I(L)) + n(|if - ,12(L)). (V.202)
Учитывая, что по условию рассмотрения область гомогенности
соединения близка к х2 = nl(m + n), правую часть соотношения
(V.202) можно рассматривать как свободную энергию
образования твердого соединения состава АтВп из жидких компонентов А
и В.
Тогда соотношение (V.202) примет вид
mRT In [vf (1 - 4) + nRT In [yffi] = AGf (V.203)
где
AGf = A#f-TASf,
AGf, AHf и AS/ — свободная энергия, энтальпия и энтропия
образования твердого соединения из жидких компонентов
соответственно. Покажем далее, что уравнение (V.203), по существу,
идентично интегральной форме полученного нами ранее
уравнения кривой ликвидуса конгруэнтно плавящегося соединения
АтВп типа (V.194).
Свободная энергия смешения при образовании жидкого
раствора стехиометрического состава определится следующим
образом:
А^(L) = mRT In [y[{L) ml(m + n)] + nRT In [y2 {L) n/(m + n)].
(V.204)
Очевидно, что величины AGf и AG/(L) связаны между собой
соотношением:
AGf==AG;(L)-AG;,^B„, (V.205)
где AGm, л в — изменение изобарно-изотермического
потенциала реакции (V.40) для процесса плавления. При температуре
плавления соединения Т°т,Атв , как уже отмечалось [см.
уравнение (V.33a)], значение AGm, лтв равно нулю, и, следовательно,
AGf = AG;<L). (V.206)
205
При любой другой температуре, принимая во внимание
формулу (У.ЗЗа) и пренебрегая разницей в теплоемкостях исходных
веществ и продуктов реакции (V.40), на основании соотношения
(V.205) можно записать
AG? = ДО;(L) + Ыгт, Атвп [(Г/г;. л1Пвп) - 1]. (V.207)
Отсюда, подставив выражения (V.207) и (V.206) в (V.203),
получим уравнение ликвидуса вида (V.194).
В соответствии с выводом работы [224], используя уравнение
Кирхгоффа, представим зависимость энтальпии и энтропии
образования соединения АтВп от температуры в виде степенного
ряда. С точностью до второго члена разложения будем иметь
АЯ? - Л/* {S) + а (Г - 7V АтВп) + Ь(Г2_ I AmBn)l% (V.208)
AS? = ASf {S) + aln(T/T°Mt лтв) + Ь(Т - Т°т, А^ (V.209)
где а и Ь — постоянные, индекс • указывает, что данная
величина относится к температуре плавления соединения. На
основании выражения (V.206), приняв во внимание, что в точке
плавления соединения
ДО;. Атвп = АС, АтВп - Т°т, AmBnAsm, AmBn = О, (V.210)
можно записать
mRTl.AmBn\n[yTiL)(l-x?L))]+nRrm,AmBn\n[yfwx'2^} =
= AHf <s> - Т°т, AmBnAST (s> + A*;. AmBn - T°m, AmBAS°m, ,тв„.
(V.211)
Подставив в формулу (V.211) соотношения (IV.180) и (IV.181),
получим из членов, не содержащих Vm,A B , выражение
AHf <s> = mh? EL + nhf EL - tim, л», (V.212)
mrrv
из членов при Тт, лтв — соотношение
ASf {S) = m'sT EL + nsf EL - AS;, AmBn -
-Rln[(l-£iLT(x2iL)H (V.213)
Уравнения (V.212) и (V.213) являются дополнительными
уравнениями, устанавливающими связь между
термодинамическими свойствами твердой и жидкой фаз.
206
Подставив в уравнение (V.203) соотношения (IV.180), (IV.181),
(V.208), (V.209), (V.212) и (V.213), получим уравнение кривой
ликвидуса конгруэнтно плавящегося соединения в окончательном
виде:
Ыг
m
АтВп + » (*f * - *f ") + « (*»" ~ *f ") +
+ а(Т°т.Атвп-Т) + аТ1п(т/Гт,АтВп) +
+ Ь(Тт А В -TYI2
+ /?in{[(i-^<L>)/(i-4)]mKa)/4n
Это уравнение можно решить методом последовательных
приближений. В отличие от ранее полученных соотношений (V.195)—
(V.198) уравнение (V.214)—более общее, поскольку при его
выводе не была использована конкретная модель для описания
поведения жидкой фазы. Уравнение (V.214) следует всегда
использовать вместе с уравнениями (V.212) и (V.213).
Выражение (V.214) можно упростить, если представить
величину AGf в виде линейной функции от температуры следующим
образом:
AGf = AHf -TA~sf + b (Г), (V.215)
где б (Т) — линейно-температурный член, вносящий небольшой
вклад в AGf; AHf и ASf — средние значения энтальпии и
энтропии образования соединения в определенном интервале
температур.
Допустим, что среднее значение энтальпии образования
соединения связано с ее значением при температуре плавления
соотношением
A/ff = A#f (5) + A, (V.216)
где А — поправочный член.
Подставив последнее выражение в уравнение (V.215), легко
убедиться, что
А^> = Д5Г^ + (А/г;,лтв„) +
+ Ь(Гт,Атвп)/Т°т,АтВп. (V.217)
Подставив соотношения (V.215)— (V.217) в формулу (V.203) и
учтя выражения (IV. 180), (IV.181), (V.212) и (V.213), получим
207
более простое уравнение для кривой ликвидуса конгруэнтно
плавящегося соединения АтВп в неявной форме:
Л/4. Атвп - А - в (Т) + m («fL - n?EL) +
T = +n(n!L-h!EL)
+ m (*«• - sf^) + n (s$L - sfEL) +
+ r m f[(i-4w)/(i -4L))]m(4(L)/4L))n)
Это уравнение можно строго решить относительно температуры,
если пренебречь величинами б (Г) и б (Тт,Атвп)- Получаемое
при этом выражение аналогично уравнению (V.214) и отличается
от него лишь поправочным членом А к величине теплоты
плавления соединения АтВп. Однако предпосылки, положенные в
основу вывода этих уравнений, различны.
Установим, что при каких условиях в общем случае допустимо
пренебрежение членом б (Г) в уравнении (V.218). Конечно,
это отчасти зависит от точности расчета и области рассматриваемых
температур.
Сопоставляя выражения (V.218) и (V.214), получим
а [Т - Гя. AmBn + T In (Г,Гп, AmBn)] + Ь(Т- Гт, Л-А)»/2 =
= Д (1 - Т/Т'т, АтВп) + б (Г) - ТЬ (Гт. Атвп)/П. *твп. (V.219)
Таким образом, сравнивая выражение
ДА;, Атвп 4- т (hfL - h?EL) + n (hEL - hfEL) +
+ Д(1-Г/Т;,Лтв„) (V.220)
с выражением
АС. лпвп + т {REL - hTEL) + п (hEL - h?EL) +
+ Ь(1-Т/Гт,АтВп) + Ъ(Т)-
-ЩГт,АйА)/Гя.лтвя, (V.221)
можно оценить величину возможной ошибки.
УРАВНЕНИЕ КРИВОЙ ЛИКВИДУСА КОНГРУЭНТНО
ПЛАВЯЩЕГОСЯ СОЕДИНЕНИЯ АВ В ПРИБЛИЖЕНИИ
МОДЕЛИ РЕГУЛЯРНОГО АССОЦИИРОВАННОГО РАСТВОРА
Температурно-концентрационный характер зависимости
термодинамических свойств в системах с конгруэнтно плавящимся
соединением часто свидетельствует в пользу существования не-
распавшихся молекул соединения в жидкой фазе. Поэтому
представляет определенный интерес описание кривых фазовых
равновесий в приближении модели регулярного ассоциированного
раствора, состоящего из мономеров и ассоциатов,
208
Рассмотрим бинарную систему А—В, в которой образуется
конгруэнтно плавящееся соединение АВ. Предположим, что жидкий
насыщенный раствор представляет собой регулярную смесь
трех сортов частиц: мономеров AL и BL и ассоциатов АВ.
Чтобы рассчитать кривую ликвидуса первичной
кристаллизации соединения АВ по уравнению вида (V.194), записанного для
случая т = п = 1,
RT In laicfraub] = Д/й, лв - Т Asmm, ABy (V.222)
необходимо знать коэффициенты активностей компонентов.
Согласно работе [187], в рассматриваемом случае имеем
а а = аАх = уахХах, ав = aBl = увххвх. (V.223)
На основании формулы (IV.216) коэффициенты активности
частиц Аи Вг и А В трехкомпонентной регулярной смеси
определяются уравнениями:
RT 1П yAt = xb^At-Вг + XAB®At-AB +
+ xBixab (<*Мх-лв — ®bx-ab + ®ax-bx)\ (V.224)
RT In yBt = хлво^-лв + А^-в +
+ xAixAB (<*bx-ab + ®ax-bx — ®ax-ab)\ (V.225)
#Г 1П УАВ = Xax«>Ax-AB + X%t<0Bl-AB +
+ ^i^B» (©Л.-ЛВ + <*BX-AB — ®AX-Bt). (V.226)
Чтобы рассчитать коэффициенты активности мономеров Аг и Вг
и ассоциатов АВ по формулам (V.224)—(V.226), необходимо знать
мольные доли хАх9 хв19 хав и параметры межмолекулярного
взаимодействия со/;.
Запишем закон' действующих масс для реакции ассоциации
А + В = ЛВ в виде уравнения
К = (хАххВх/хав) (УахУвх/Уав). (V.227)
В рассматриваемом случае условия сохранения масс компонентов
будут иметь вид
fh — пАх + пав, пч = пВх + плв. (V .228)
Используя определения мольных долей компонентов и
молекулярных индивидов, соотношения (V.228) легко преобразовать к виду
хах = ха— хвхАв\ (V.229)
хВх = хв — хахав. (V.230)
Представив выражение (V.227) в виде
In К = In уАх + In yBl — In уав + In (xAxxbJxab), (V.231)
209
с помощью уравнений (V.224)—(V.226) и соотношений (V.229),
(V.230), нетрудно убедиться, что
In К = In {[хАхв (1 + *Ав)2 — хав)/хав] +
+ (vAi-bJRT) [1 - хАХв (1 + хав)2] -
- Ы^ав/RT) [1 - хв (1 + хлв)2] -
- (Пвглв/RT) [1 - ды (1 + Wl. (V.232)
К сожалению, уравнение (V.232) является трансцендентным
относительно Хав и не имеет решения в явном виде, если со,-; Ф 0.
Однако принципиально при использовании ЭВМ на основании
формулы (V.252) можно рассчитать зависимость хАВ от мольной
доли одного из компонентов, если параметры К и о^ известны.
Далее, подставив значения хАв в уравнения (V.229) и (V.230),
можно оценить мольные доли мономеров при заданных составах.
Подставляя значения xAl, xBt и хАв Для данного состава раствора
хА и хв, из выражений (V.224) и (V.225) сначала можно
рассчитать коэффициенты активности мономерных форм In yAt и In yBl,
а затем с помощью уравнений (V.223) — коэффициенты активности
компонентов А и В. Однако, как правило, параметры К и щ^
неизвестны. Поэтому для расчета величин аА и ав регулярного
ассоциированного раствора используют приближенный метод,
предложенный Джорданом [187].
По этому методу, разложив соотношения (V.224) и (V.225)
в ряд Тейлора по хв и хА при хв = 0 и хА = 0 соответственно.;
полагая при этом, что со^-лв = <ubx-ab> и принимая во
внимание выражение (V.232), получим следующие приближенные
уравнения для коэффициентов активности мономерных форм Аг
и Вг:
RT In yAl « (1/[(1 + Ki)2<*Ai-ab] +
+ Ktl[{\ + Ki)<*Ax-Bt\] x% + • • • = <**xh + • • •, (V.233)
RT In yBt « {1/[(1 + Kif <*Ax-ab\ +
+ Ki/[(l + Ki) cd^-bJ} x*a+..-= ©Va + • • •, (V.234)
где
со* = 1/[(1 + Kif vAi-ab\ + Ki№ + Ki) cd^-bJ, (V.235)
Ki = K exp [(со^-в, - VArABVRT]. (V.236)
На основании выражений (V.236) и (V.235) легко убедиться,
что в случае полной ассоциации (/(/ = /( = 0)
«>* = (oAt {Bi)-ab, (V.237)
а в случае полной диссоциации (/С; = К = оо)
<*>* = соЛ-в. (V.238)
210
Используя уравнение Гиббса—Дюгема в виде
хАх d In yAl + xBl d In yBl + xAB d In yAB = 0
соотношения (V.229), (V.230), (V.233), (V.234) и условие
нормировки улв —* 1 при хв = 0,5, получим для In yAB
RT In уАВ = (<о*/2) (1 — 4хАхв). (V.239)
Подставив выражения (V.233), (V.234) и (V.239) в уравнение
(V.227) с учетом соотношений (V.229) и (V.230), получим
1п/С = (со*/2#Т) + In [хАхв (1 + хАву - хАВ] - In xAB. (V.240)
Уравнение (V.240) можно легко решить относительно хАВ в виде
хАВ = (1 - />)/(!+Р). (V.241)
Здесь Р = Vl-4xAxBl{\+K') =
= |Л-4*^(1-<za), (V.242)
где /(' — кажущаяся константа диссоциации комплекса А В,
связанная с истинным значением константы К уравнением
Кх = К ехр [—ю#/2ДГ]. (V.243)
В общем случае (дс^ =#= *в) Я есть доля мономерных атомов А
и В в растворе, т. е.
Я = ("л, + лВ1)/(лл + пв). (V.244)
В то же время Кх = а2/(1 — а2), где а — степень диссоциации
комплекса АВ в точке, отвечающей составу соединения хв = 0,5,
зависит от температуры.
На основании выражения (V.242) нетрудно убедиться, что
в точке хв = 0,5
Р = а°.
Если пАх = пА и nBi = nB, то Р = а° = 1, т. е. соединение Л В
полностью диссоциировано в жидкой фазе. После подстановки
уравнения (V.241) в соотношения (V.229), (V.230) получим
xAl = (хА - х5 + Р)/(1 + Р), (V.245)
xBl = (хв - *л + Р)/(1 + Я). (V.246)
Наконец, объединив уравнения (V.245), (V.246), (V.233) и (V.234)
с уравнениями (V.223), получим следующие выражения для
активностей компонентов бинарного раствора, в котором
происходит взаимодействие, приводящее к образованию ассоциатов
типа АВ:
аА = yAlxAi = [(хА - хв + Р)1{\ + Р)\ ехр (<o*x%/RT), (V.247)
ав - yBlxBl = [(хв -хА + Р)1{\ + Р)] ехр (<o*x\/RT). (V.248)
Выражения (V.247) и (V.248) удовлетворяют уравнению
Гиббса—Дюгема для двухкомпонентной системы.
211
Подставив эти выражения в уравнение (V.222), после
несложных алгебраических преобразований придем к уравнению кривой
ликвидуса первичной кристаллизации конгруэнтно плавящегося
соединения АВ в приближении теории регулярного
ассоциированного раствора, полученному Джорданом [187]:
т &КпшАВ + Ы (хв -0,5)»
где S (хв) = [(1 + Р)/(\ + a°)f/(4xAxB). (V.250)
Рассмотрим два предельных случая.
1. Случай полной диссоциации соединения А В в жидкой фазе,
когда а° = 1, Я = 1.
Тогда выражение (V.250) преобразуется к виду
S(xB) = \/(4хАхв),
и выполняется равенство (V.238). Следовательно, в этом случае
справедливо уравнение (V.198).
2. Случай полной ассоциации соединения в жидкой фазе.
Это значит, что жидкий раствор состоит либо из мономеров Аг
и ассоциатов АВ, либо из мономеров Вг и ассоциатов АВ. В
данном случае на основании соотношений (V.229), (V.241) и (V.230):
1 + Р = 2хв при хв > хА,
1 + Р = 2хА при хв < хА.
(V.251)
Подставив эти выражения в уравнения (V.249) и (V.250), придем
к следующему результату:
^ 1п (xbIxa) + Лг>т, ЛВ
Таким образом, в этом случае фазовая диаграмма распадается
как бы на две самостоятельные диаграммы А—А В и В—Л В, а
соединение А В ведет себя как растворитель.
В данном приближении уравнение кривой ликвидуса (V.249)
конгруэнтно плавящегося соединения АВ содержит два параметра
со* и а°, которые по условию рассмотрения не зависят от
концентрации.
Чтобы упростить расчеты, обычно допускают, что параметры со*
и а° не зависят также и от температуры. Значения этих параметров
для небольших величин а° можно оценить графически.
212
Вычитая уравнение (V.253), записанное для полностью
ассоциированного раствора, из уравнения (V.249), где а0 Ф 0,
получим для значений хв >>хА
2"1 In [S (хв) (хА/хв)] = [(о* - ®B-ab)/RT] (хв - 0,5)2. (V.254)
Принимая во внимание формулы (V.250) и (V.242) и введя
обозначение (хв — 0,5) = Ах, соотношение (V.254) можно
преобразовать к виду:
In (1 + а°) + [К - Пв-лвУЯТ] (А*)2 =
= In {[1 + 2 Ах |/1 + (а°/2Дх)2 — а02]/^ + 2 Ах)}. (V.255)
Отсюда легко убедиться, что для а° < 1 (например, а° <: 0,2)
при значениях 2Дх >> а° правую часть с достаточно надежным
приближением можно приравнять нулю, за исключением области,
прилегающей к составу х = 0,5. Кроме того, для небольших
значений а° справедливо In (1 + а°) я^ а°.
Таким образом,
ао + [((0* _ и^укг] (Ах)2 ^ 0. (V.256)
Выразив из соотношений (V.253) и (V.256) значения ыв_АВ
и приравняв их между собой, получим следующую приближенную
формулу:
ыв-ав = [— RT In (хА/хв) + Т As°m, ав -
-Л/4, лв}/2 (Ах)2 = со* + RTa*/(Ax)2. (V.257)
Отсюда следует, что функция <дВ_АВ от аргумента Л* должна
представлять собой гиперболу с постоянным предельным значением
со* для больших значений Ах. Кроме того, зависимость (ов_АВ
в координатах от Т/(Ах)2 есть прямая линия, по тангенсу угла
наклона которой можно оценить величину параметра а°, а по
величине отрезка, отсекаемого на ординате, значение
параметра со*.
Метод анализа кривых ликвидуса конгруэнтно плавящихся
соединений А В на основе теории регулярных ассоциированных
растворов был использован в работах [187, 193] для систем
А11 — BY\ A1Y — BVI, в которых предполагается сильная
ассоциация в жидкой фазе.
В работе Осамуры и Мураками [225] на примере пяти
бинарных систем AUBY рассмотрен промежуточный случай — модель
«регулярного частично ассоциированного раствора» в
предположении, что мольная доля ассоциатов в жидкой фазе относительно
мала, так что с достаточно хорошим приближением выполняются
соотношения (1 + хАВ)2 & 1, xAlxBl *& хАхв.
213
В соответствии с выводом работы [2251 с учетом принятых
допущений на основе выражений (V.223)—(V.225) и формулы для
константы равновесия реакции
аАв1(аАхаВ1) = С0 ехр [ЛЯ° {L)/RT] (V.258)
будем иметь
Хав = ХаХвРав (1+2 bxtijiBlRTh (V.259)
где A#0(L) —теплота образования комплекса А В в жидкой
фазе, С0 — постоянная, связанная с его энтропией образования:
Рав = С0 ехр [(АН°(L) - а\в + <*ax-bx)\IRT (V.260)
(Пав и (Пав равны соответственно функциям: (ь)ах-ав — g>bx-ab)/2
и (тх-Ав + <ьвх-ав)12, Ах = 0,5 — хг.
При условии, что величина выражения (A#°L + солв —
— (fiAt-Bt) значительно больше значения соля, после
подстановки уравнения (V.259) в формулу для избыточной свободной
энергии жидкой регулярной смеси, состоящей из мономеров Аи Вг
и комплексов А В вида
f =8 ==хахвх®ах-вх + хА1Хав®ах-ав~г
+ Хв*А#йвх-Ав + хАВ АЯ°(L) (V.261)
с помощью известных термодинамических соотношений получим
следующие выражения для коэффициентов активности
компонентов:
RT 1П у А = Х^Аг-В, + (ДЯ° (L) + (й+АВ -
- <*ах-вх)Р°ав [1 — (1 — 4A.v) oTab/RT], (V.262)
RT In yB = x2AaAi_Bi + (Д#°(t) + G)^ -
- со^-в.) Рлв [ 1 + (1 + 4 Ах) afo/RT], (V.263)
где второе слагаемое учитывает вклад за счет ассоциатов.
В рассматриваемом приближении [225]
О) = G>Al-Bt + (АЯ° (L) + Ы+АВ ~ <йАх-Вх) Р°АВ(1+ ^~АВ Ах/RT).
(V.264)
В этом уравнении множитель (1 -f- 4a>ABAx/RT) зависит от
концентрации. Поэтому для случаев, когда со^в Ф 0, параметр со
зависит от состава.
Значение параметра со в приближении модели регулярного
частично ассоциированного раствора можно рассчитать на
основании данных по фазовым равновесиям следующим образом.
214
Для двух точек ликвидуса, отвечающего первичной
кристаллизации соединения А В, принадлежащих одной и той же изотерме,
на основании выражения (V.264) можно записать
y^RTfa —(x)u)/(Axl — Ахп) =
= 4 (Д#°(L) + <о\в - «>Al_Bl) <*>АВС0 exp (A/RT), (V.265)
где А = АЯ°(L) + <ьАх_Вг - со^, (V.266)
нижние индексы I и II означают растворы, лежащие слева и
справа от состава соединения соответственно.
Отсюда построив график зависимости In у = / (1/71), можно
определить значения двух функций: А и [4 (A#0(L) + cdJb —
— WAt-Bi) (uabC0]. С помощью графика функции г = со —
— yAx/RT в зависимости от exp (AIRT) можно оценить еще две
величины: cu^-Bi и (AHC(L) + <ь\в— сол,-^) Со- В итоге из
данных по фазовым равновесиям в бинарной системе будем иметь
четыре уравнения с пятью неизвестными параметрами: С0,
(OAt-Bn <ОЛ,-ИВ, <*>Bt-AB И A//°<L).
Наиболее вероятное значение образующихся в растворе
комплексов А В при данной температуре рассчитывают из условия
минимума свободной энергии раствора gM. Поэтому в качестве
пятого уравнения можно воспользоваться выражением
dgM/dxAi + dgM/dxBl = dgM/dxABy (V.267)
из которого, принимая во внимание соотношение gM = gM <w> +
+ gE и формулу (V.261), нетрудно оценить параметр С0.
Как будет рассмотрено далее, параметры взаимодействия между
мономерными молекулами в жидкой фазе можно рассчитать также
другим способом, например, используя данные об электроотри-
цательностях и параметрах растворимости чистых компонентов.
Теплоту образования бинарных комплексов АВ в жидкой фазе
можно рассчитать по уравнению [225]
A#°w= const /,Д (V.268)
где Д. — степень ионности; D — фактор дегибридизации.
Расчеты [225] параметров межмолекулярного взаимодействия
на основе данных по фазовым равновесиям, а также по формуле
(V.268) и из данных по электроотрицательностям компонентов
хорошо согласуются между собой.
6. УРАВНЕНИЕ КРИВОЙ ЛИКВИДУСА
И ТЕРМОДИНАМИЧЕСКИЙ АНАЛИЗ СИСТЕМ
С ИНКОНГРУЭНТНО ПЛАВЯЩИМСЯ СОЕДИНЕНИЕМ
Как было показано выше, методы термодинамики растворов
дают возможность аналитически описать кривую ликвидуса в
системах с конгруэнтно плавящимися соединениями. Однако во
многих системах образуются химические соединения, плавящиеся
инконгруэнтно [219, 220].
215
На рис. 45 схематически показана диаграмма состояния
двойной системы с каскадом перитектических превращений, в
результате которых образуются химические соединения различного
состава. Из этого рисунка следует, что в общем случае для таких
систем процесс инконгруэнтного плавления можно представить
в виде следующей перитектической реакции:
AmBsn+±Lp + AkBf, (V.269)
где Lp — расплав состава р, насыщенный одновременно
относите тельно кристаллов АтВп и AkBt.
В случае пренебрежения ролью
давления, что типично для
конденсированных систем, трехфазное
равновесие (V.269), как показано
в гл. III, нонвариантное.
Строго термодинамическое опи-
сайие кривой ликвидуса в системе
с каскадом инконгруэнтно
плавящихся соединений в литературе
отсутствует.
Отдельные участки этой
кривой, отвечающих первичной
кристаллизации одного из соединений
с узкой областью гомогенности,
в принципе можно описать с
помощью уравнения Бребрика
(V.214). В приложении к инкон-
груэнтным соединениям оно
остается таким же строгим, как и для
конгруэнтно плавящихся
соединений, поскольку в него не
входят термодинамические
характеристики, характеризующие собственно процесс плавления
соединения. Однако для термодинамического анализа кривой ликвидуса
в системе типа показанной на рис. 45 уравнение Бребрика
ненаглядно, так как оно описывает не только физически
реализуемый ликвидус (например, ер рис. 45), но также и гипотетический
участок рМ. Следовательно, уравнение Бребрика нечувствительно
к переходу через перитектическую горизонталь и не выявляет
физико-химической сущности инконгруэнтного плавления,
приводящего к появлению совершенно нового вида двухфазного
равновесия, включающего твердую фазу иного состава и природы,
чем соединение АтВп.
Подход к решению задачи на основе уравнения Виланда
(V.198) требует знания таких характеристик, как теплота и
температура плавления соединения. Понятно, что в случае
инконгруэнтного плавления эти характеристики физически
нереальны. Попытка [226] преодолеть эту трудность путем расчета
21Q
L
Р'л
^1>ч /
/м
1 '/'^
\\j(-+AmBn
I e
\\<х
J а+АтВп
)
>
АтВп*
+АкВг
у
> 1
\а&+А 1
Ат7^/7
AKBt
5
Рис. 45. Принципиальная схема
диаграммы состояния с инконгруэнтно
AkBi
гипотетических величин на основе экспериментально определяем
мой энтальпии перитектического превращения, на наш взгляд,
неудачна, поскольку полученный результат включает в себя член
[тп/(т + п) ] coL, который представляет собой теплоту смешения
при образовании физически несуществующего жидкого
регулярного раствора состава п/(т + п). Кроме того, авторы [226]
допускают равенство энергий смешения компонентов для указанного
несуществующего раствора и для раствора состава /?, что, конечно,
необоснованно. По этим причинам результат работы [226] нельзя
считать достаточно корректным с физико-химической точки
зрения.
В связи с этим нами была сделана попытка [227] получить
единое уравнение кривой ликвидуса в системе с каскадом пери-
тектических превращений, которое должно учитывать физико-
химическую сущность инконгруэнтного плавления для каждого
соединения.
Допустим, что протяженность областей твердых растворов
на основе соединений и компонента В пренебрежимо мала и ее
можно не принимать в расчет. Запишем перитектическую реакцию
(V.269) с учетом соотношений масс образующихся фаз:
AmBsn ^ х (1 - р) A L + хрВ^г + yAkBf, (V.270)
где, согласно материальному балансу,
х = [m/(l —p)]+kln — p(m + n) 1/(1 — р) X
X[p(k + I) — I], (V.271)
у= [mp — n(l—p)Vlp(k + l)—l]. (V.272)
Тогда изменение изобарно-изотермического потенциала при
перитектической реакции (V.270) можно представить в виде
AGP ^x(l-p)iiLA + xpiib + yv,°A$\ - ti°A{X = 0. (V.273)
Здесь верхний индекс «°» относится к чистому веществу.
Подставляя в формулу (V.273) развернутое выражение для
химических потенциалов \з^ и используя модель регулярного раствора
для описания жидкой фазы, после несложных преобразований
получим
AGp=ax(l-p)|iVL)+^(L) +
+ *(1 -p)RTp\n(l -p) + XpRTplnp + xp(l -p№ +
+ ^Й-^3Я = 0, (V.274)
где coL — энергия смешения компонентов в жидкой фазе.
Для удобства дальнейших расчетов прибавим к выражению
(V.274) и вычтем из него величину гц1°вЬ). Далее учтем, что
W + /l*№) - f4(i> = -AGf iAiBj). (V.275)
217
Принимая во внимание соотношения (V.275) и (V.272), получим
AGP = -AGf {AfnBn) + У AGf ИЛ) + х (1 - р) RTp In (1 - р) +
+ х/?#Гр lnp + jcp(l — р) aL = 0. (V.276)
В этом выражении AGf — свободные энергии образования
твердых соединений АтВп и AkBt из жидких компонентов А и В.
Раскрывая выражения для AG. и AG/, согласно формуле
(1.113), на основе уравнения (V.276) получим
АЯ„ - Тр ASP = — AHf (лтвЛ) + Тр ASf (лтвЛ) +
+ у bHfM) - уТр ASf ИЛ) + х (1 - р) RTP In (1 - р) +
+ xpRTp In /? + xp (1 - /?) <oL = 0. (V.277)
Отсюда, объединяя члены, не содержащие 7^, найдем следующее
выражение для энтальпии рассматриваемой перитектической
реакции:
АНР = -&Н*(Атвя) + у AHf {AkBl) + *р (1 - р) <Л (V.278)
Аналогично из членов при Тр получим выражение для энтропии
перитектической реакции:
ASp = -ASsf{AmBn) + yASf{AkBl)-
— jc/? (1 — р) In (1 — р) — xpR In p. (V.279)
Решая уравнения (V.278) и (V.279) относительно Д#^лтвл)
ASf^mart) и подставляя результаты в уравнение Бребрика
(V.203), получим следующее аналитическое выражение кривой
ликвидуса инконгруэнтно плавящегося соединения в
приближении теории регулярных растворов:
-ДЯр + у Mf(AkBi) + oL [xp (1 -р) -
Т~ -Д5Р + у^sf{AkBl)-ЯШЦ1-р?^у»] + ' (V*280)
+ R\n[(l-xLB)m(x^]
где xB — концентрация жидкого раствора, соответствующая
ликвидусу при данной температуре.
В данном случае мы пренебрегаем температурной
зависимостью энтальпии и энтропии образования соединения АтВПу
полагая ее незначительной.
218
Это уравнение отличается тем, что в него заложены параметры
перитектического превращения /?, АНР, ASpy а также
термодинамические характеристики образования фазы, кристаллизующейся
первично выше температуры перитектической реакции
образования соединения АтВп. Следовательно, при переходе через пери-
тектическую горизонталь в область более высоких температур
вид уравнения (V.280) изменится за счет именно этих
характеристик.
Когда выше Тр кристаллизуется инконгруэнтно плавящееся
соединение AkBt (рис. 45), кривая ликвидуса рр' будет
описываться уравнением, аналогичным (V.280), однако вместо р> АНР
и ASP в нем будут фигурировать параметры следующего
перитектического превращения, т. е. р\ АН'Р и ASP соответственно, а
вместо AHf(AkBt) и ASf(AkBt) — параметры образования р-фазы.
Соответствующим образом изменяются параметры х и уу
определяемые из уравнений (V.271) и (V.272).
Если соединение AkBt плавится конгруэнтно, то, очевидно,
параметры перитектического превращения в уравнении (V.280)
будут отсутствовать, и в этом случае
MfiA Ю-©ЧА (xLBf+ I(1 -4)21
Т = 'У* , \l, lm > (V-281)
т. е. получаем уравнение Бребрика, в котором коэффициенты
активности записаны в приближении теории регулярных
растворов.
Наконец, если устойчив твердый раствор на основе
компонента В и растворимость незначительна, то, очевидно, k = О,
/ = 1, и тогда
Т = [АС, в + <*L (1 - 4)7(Л4, в-R In xLB). (V.282)
Нетрудно заметить, что это уравнение эквивалентно
уравнению Каменецкой (V.72).
Предлагаемый подход к описанию кривой ликвидуса в системе
с инконгруэнтным соединением позволяет по диаграмме
состояния, построенной экспериментально, судить о таких важных
термодинамических параметрах, как АНру ASpf Atff^s,) и
AS/(V*,)-
Проведенный анализ носит общий характер и не зависит от
выбора модели раствора. Модель может быть любой, вплоть до
формально-математической, использующей ряды любой
протяженности. Соответствующие видоизменения уравнения (V.280)
б удут чисто математическими.
219
В заключение проиллюстрируем рассмотренный метод на
примере системы Аи—РЬ, в которой имеется «каскад» из трех пери-
тектических реакций (рис. 46), отвечающих образованию
соединений AuPb3, АиРЬ2 и Аи2РЬ [219—220].
Для этой системы с помощью уравнения (V.280) были рассчи-
таны]участки кривой ликвидуса еръ р^р2 и р2Рз» соответствующие
первичной кристаллизации
указанных соединений. Результаты
расчета показаны сплошной
линией. Пунктиром отмечены
результаты расчетов по уравнению
Бребрика, а точками — данные
термического анализа [228]. Как
видно на рис. 46, согласие
расчетных и экспериментальных
данных хорошее.
По диаграмме состояния были
рассчитаны значения энтальпий
перитектических реакций, а также
термодинамические параметры
образования соединений из жидких
компонентов. Результаты
расчетов представлены в табл. 1.
В последнем столбце помещены
значения свободной энергии
образования соединений Au2Pb и
AuPb2 из жидких компонентов
при соответствующей Тр из
работы [230 ]. Как видно из сравнения
с рассчитанными в нашей работе величинами AGf, согласие
достаточно хорошее, что указывает на приемлемый выбор модели
и корректность общего подхода.
Таблица 1
Термодинамические свойства инконгруэнтно плавящихся соединений
и параметры перитектических реакций в системе Аи—РЬ
т,к
700
600
500V
400
О. \
см \
^
<
<+
| /
k
\ \
1 _ I
VO CL
_!_. . i_
0,15 0,30 0,45 0У6 0,15 0,9
Атомная доля РЬ
Рис. 46. Сопоставление рассчитанных
по уравнению (V.280) и
экспериментальных [228] кривых ликвидуса в
системе золото—свинец
Соединение
Au2Pb
AuPb2
AuPb3
Ьч
691
527
490
к
X
. ч
СХ К
1 0,43
0,72
0,78
ч
s
5
<
32,6
23,9 *
67,4
< 3-
i ^
1 х
48,6
38,9
88,7
ад)
Л
« ч
со S
11
1 П
37,7
37,7
136,0
JQ
ч
1 *ч
1 к
22,6
17,5
22,2
30]
«N
ч
СО*—S
1 ^
■ X
30,6
20,5
* Звездочкой отмечено значение АЯо для AuPb2, полученное
экспериментально [229—2301
220
7. РАЗЛИЧНЫЕ МЕТОДЫ ОЦЕНКИ ЭНЕРГИИ ВЗАИМООБМЕНА
НА ОСНОВЕ ЭКСПЕРИМЕНТАЛЬНЫХ ДАННЫХ
Из рассмотренных выше подходов к расчету линий фазовых
диаграмм в системах с различным характером взаимодействия
компонентов следует, что практически при любом выборе модели
раствора принципиально важную роль играет правильная оценка
параметра, характеризующего роль обменных сил между атомами
и трактуемого как энергия взаимообмена.
Чаще всего при исследовании и анализе экспериментально
построенных диаграмм состояния строят температурно-концен-
трационную зависимость о вдоль кривой ликвидуса, используя
приближение регулярного раствора [231—238]. Смысл таких
построений сводится в конечном итоге лишь к качественному
суждению о характере межмолекулярного взаимодействия в
системе и о степени отклонения ее от регулярности в том или ином
интервале концентраций. Для более строгих оценок и надежных
количественных выводов следует использовать комбинированный
подход, связанный с привлечением дополнительных методов
оценки со.
Анализ возможных взаимосвязей энергии взаимообмена с
различными физико-химическими свойствами показывает, что
величину coy,- можно оценить из экспериментальных данных по тепло-
там смешения, давлению пара, растворимости, составу и
температуре азеотропной и эвтектической точек, критической
температуре расслаивания, избыточным объемам и т. д.
Отметим, что большинство описанных методов оценки со
требует дополнительных данных, помимо основного измеряемого
свойства. Исключение составляют методы, основанные на
измерении теплот смешения и давления пара.
По тепло там смешения»
Теплоты смешения достаточно надежно определяют
калориметрическим методом [239], который, несмотря на трудоемкость,
широко распространен. По формуле (IV. 104)
(о = Л7ад. (V.283)
Следовательно, для определения энергии взаимообмена достаточно
одного калориметрического измерения теплоты смешения.
По давлению пара
Для термодинамических исследований пользуются тензиметри-
ческими измерениями [240]. Если известен молекулярный состав
паровой фазы, тензиметрические данные позволяют достаточно
надежно оценить коэффициент активности, а следовательно, и
221
энергию взаимообмена. Из соотношений (IV.97), (IV.98) и (IV.59)
имеем
/7i = РЛ exp (cox! RT), (V.284)
Р2 = РЛ exp M/RT). (V.285)
Отсюда
Величину PixJp^Xx иногда называют делящим множителем.
Из уравнения (V.286) можно заключить, что для определения
энергии взаимообмена достаточно экспериментально измерить рг
и р2 Для какого-либо одного состава, за исключением х = 0,5.
В ряде случаев такое определение энергии взаимообмена хорошо
согласуется с расчетными данными на основе диаграммы фазового
равновесия [241 —243].
На основе данных об электроотрицательностях
и атомных объемах компонентов
В работе [244] предложен метод оценки параметра
межмолекулярного взаимодействия со;7 в двойных и квазидвойных
системах, основанный на использовании электроотрицательностей %L
атомных объемов (vl) и параметров растворимости (6,)
компонентов, образующих исследуемую систему. Такой подход дает
возможность оценивать со/м опираясь на величины, которые для
простых веществ определены и табулированы. Выведем формулу для
расчета со;/, включающую указанные параметры. Для расчетов
используем выражение Скэтчарда [92]. Для простых жидкостей,
у которых пар близок к идеальному, величину энергий
взаимодействия ии можно отождествить с энергией испарения M°it у.
Тогда можно записать
&Ui,v = CuVh (V.287)
где сц —платность когезионной энергии.
Согласно Полингу [245], энергию взаимодействия
разноименных полярных частиц можно найти из уравнения
"12 - ("ii"22)1/2 + 3 • Ю4 (Xi ~ Х2)2- (V.288)
Учитывая формулу (V.287), из уравнения (V.288) получим
сп = (спс22)1/2 + 3 • 104£ (х, - Х2)7№)1/2, (V.289)
где k —константа, значение которой определяется разницей
в постоянных экранирования растворов различной природы и
взаимной ориентацией взаимодействующих частиц, поскольку
222
вклад в энергию связи из-за разности электроотрицательностеи
определяется в основном кулоновскими силами.
После подстановки уравнения (V.289) в уравнение Скэтчарда
[92] найдем
- 6 • 10Ak (Xl _ X2)V№)1/2] Ф1Ф2. (V.290)
Примем сц = 6} (V.291)
(обычно размерность параметра [б] = Дж1/2-м~5/2). Подставляя
значение hM из выражения (V.290) в (V.283) при учете (V.291),
получим
со = [v\v2l{xlV\ + x2v2)] [(8i - б2)2 -
-6.104A(xi~X2)2/№)1/2]. (V.292)
Для составов, примыкающих к какому-либо из компонентов, т. е.
для относительно малых значений xt соотношение (V.292) можно
представить в виде
(d = i,; (8i - б2)2 — 6104^ {vjv))112 (xi - X2)2. (V.293)
Величину константы k в уравнениях (V.292) и (V.293) теоретически
рассчитать невозможно. Поэтому обычно ее находят, сопоставляя
выражения (V.292) или (V.293) со значениями со, оцененными из
других данных, например, по давлению пара или фазовому
равновесию.
Параметр растворимости bt рассчитывают обычно с помощью
выражения
6, = (ДЛ;,у-ДГ)/с£. (V.294)
При подходе к оценке ДЛ°, у с использованием тензометриче-
ских данных и уравнения Клапейрона—Клаузиуса следует,
однако, проявлять большую осторожность и пользоваться
значениями теплот фазовых переходов и уравнениями температурной
зависимости давления пара простых веществ, тщательно
отобранными при помощи ЭВМ путем согласования данных на основе
закона Гесса и периодического закона Менделеева (см. [246—248 ]).
С помощью уравнения (V.292) в работе [244] были оценены
значения энергии взаимообмена в ряде бинарных систем с
участием германия и кремния, которые сопоставлены с результатами
определения этой величины на основе анализа фазовых диаграмм.
Из сравнения полученных данных следует, что описанный метод
оценки параметра со на основе данных по электроотрицательностям
и атомным объемам дает достаточно надежные результаты.
Рассмотрим применение данной методики на квазибинарных
системах. Такая задача была рассмотрена нами на примере
систем Ge— InP и Ge—GaP [249].
223
Ё работах [250—2511 показано, что характер фазового
равновесия в квазибинарных системах Qe—In(Ga)P описывается
диаграммами состояния эвтектического типа с незначительной
растворимостью в твердом состоянии.
Анализ кривых ликвидуса в приближении регулярных
растворов [250], а также расчеты [252] степени диссоциации фосфидов
вдоль кривых ликвидуса в рассматриваемых системах
показывают, что соединения существенно диссоциированы в жидкой
фазе. Это дает основание для рассмотрения характера
межмолекулярного взаимодействия в системах Qe—In (Ga) P при помощи
описанной выше методики (основанной на использовании данных
об электроотрицательностях и атомных объемах компонентов
с привлечением теории регулярных растворов), распространенной
на трехкомпонентные системы.
На основании анализа парных взаимодействий в тройной
системе А—В—С можно записать следующие соотношения,
связывающие температуру ликвидуса, концентрацию в тройной
системе и величины энергий взаимообмена в соответствующих
двойных системах:
RT In КАВ (Т) = (aABxB + <дАСхс) (хв + хс) + (солвхл + совсхс) х
X (хА + хс) — (овсхвхс — (оАсхАхс + RT In (хАхв), (V.295)
RT In Кс (Т) = Кв - (ов (Л) с) хв М) +
+ <ол (В) с (хс - хА iB)) + RT In (xA iB)/xc). (V.296)
В этих формулах
КАВ(Т)=уАувхАхв, (V.297)
Кс (Т) = уА {в)ХА (в)/усхс (V.298)
Уравнения (V.295) и (V.296) записаны при условии хА +
+ хв + хс = 1 и отвечают областям первичной
кристаллизации А В и С соответственно. Численные значения величин
RT In Кав (Т) и RT In Кс (Т) определяются из
экспериментальных данных по фазовым диаграммам соответствующих двухком-
понентных систем с помощью следующих уравнений:
RT In КАВ (Т) = (олб (Л + 4) + RT In (xAxB), (V.299)
RT In Кс (Т) = (ол {в) с (хс - хА {В)) + RT In (xA {В)/хс). (V.300)
Эти уравнения записаны при условии, что хА + хв = 1 и хА (В) +
+ хс=1.
Учитывая, что в данном конкретном случае рассматриваются
квазибинарные системы типа С—(АВ), можно записать
хА=хв = х. (V.301)
С учетом этого условия уравнения (V.295) и (V.296) можно
представить в виде
224
2агх (х — 1) + а2 - —2#Г In x, (V.302)
я3* + а4 = RT In [(1 — 2*)/*]. • (V.303)
В этих уравнениях значения коэффициентов а19 а2, а3 и а4
рассчитывают на основе данных по энергиям смешения в
соответствующих двойных системах при помощи следующих уравнений:
fli = 2 (солс + <овс) — солв; (V.304)
а2 = ®АС + щс - RT In Кля (Л 5 (V.305)
а* = ®ав — «5с — 3о)ж> (V.306)
а4 = олс - #Г In Кс (Г). (V.307)
Значения энергии смешения, характеризующие парные
взаимодействия в двойных системах Ge—In, (Ga), (Р) и In (Ga)—Р,
были рассчитаны нами с помощью уравнения (V.292).
Необходимые для расчетов значения входящих в уравнение (V.292)
величин были взяты из работ [87, 245].
При расчетах были использованы двойные диаграммы
состояния, представленные в работах [67, 205, 253, 254]. На основе
данных по энергиям смешения, рассчитанных с помощью
уравнения (V.292), были вычислены значения коэффициентов аи а2,
а3 и а4, входящих в уравнения кривых ликвидуса (V.302) и (V.303).
Уравнения (V.302), (V.303) решали относительно х для
заданной температуры ликвидуса графическим способом. Для перехода
к мольным долям при каждой данной температуре в
квазибинарной системе С—А В (А В—InP и GaP, С—Ge) использовали
соотношение хАВ = xl{\ —х).
Результаты расчетов представлены на рис. 47 и 48 в
сопоставлении с экспериментальными данными работ [250—251].
Из рис. 47 следует, что результаты расчетов по кривым
ликвидуса в системе Ge—GaP достаточно близки к экспериментальным
данным, полученным методом термического анализа [250].
В системе Ge—InP картина несколько сложнее. Результаты
расчета линии ликвидуса, отвечающей первичной кристаллизации
твердого раствора на основе фосфида индия, хорошо согласуются
с данными работы [250], которые резко отличаются от данных
работы [251]. В случае линии ликвидуса, отвечающей первичной
кристаллизации твердого раствора на основе германия,
наблюдается обратная картина, хотя из-за ограниченности данных в этом
температурно-концентрационном диапазоне сделанный вывод
нельзя считать строгим.
Учитывая, что в обеих рассматриваемых работах чистота
исходных материалов, условия эксперимента и использованная
аппаратура были практически одинаковыми, объяснить
наблюдаемые расхождения можно только различными степенями
переохлаждения, достигаемыми в соответствующих концентрационных
8 Глазов В М. и др#
225
интервалах, что может быть обусловлено различиями в тепловых
режимах охлаждения. В данном случае термодинамический расчет,
выполненный с использованием опорных данных по атомным
объемам и электроотрицательностям компонентов, помогает
разобраться в существе дела и наметить равновесный вариант
диаграммы состояния, показанный на рис. 48 жирными линиями.
Наблюдаемые расхождения между расчетными и
экспериментальными данными можно объяснить тем, что фосфиды галлия и
индия (особенно второй) диссоциированы в растворах не пол-
т.к
1373
1273 V
1173
1073 Г
г*
Г L
Г OL+P
I I
• /
*2
°5
ill
0,4 0,6 0,8 GaP
Рис. 47. Диаграмма состояния системы
Ge—GaP:
/ — расчет по уравнениям (V.292),
(V.295); 2 — экспериментальные
данные авторов
Ge 0,2 Ofi 0,6 0,8 InP
Рис. 48. Диаграмма состояния системы
Ge—InP:
1 — расчет по уравнениям (V.292),
(V.295) и (V.296); 2 — данные работы
[251]; 3 — экспериментальные данные
авторов
ностью. Об этом свидетельствует определенная корреляция между
величинами расхождений и степенью диссоциации, оцененной
приближенно вдоль кривых ликвидуса систем Ge—In(Ga)P [252].
По вязкости
В работе [255] показана возможность оценки энергии
смешения в жидкой фазе со*- на основе данных по вязкости бинарного
или квазибинарного раствора. В соответствии с работой [256],
коэффициент диффузии в растворе связан с коэффициентом
активности следующим соотношением:
Dt=D\(\+d\rL4*ild\nx\\
(V.308)
где D] — коэффициент самодиффузии i-того компонента.
Допуская, по Мелвин—Хьюзу [257, с. 704], применимость закона
Стокса, в соответствии с уравнением (V.308), будем иметь
1/л «= (1/л") (1 + a inyj-/a injtf).
(V.309)
226
Используя уравнение А. И. Бачинского [258] и суммируя
величины Ctr$Xi (где с-ь — константа в уравнении А. И.
Бачинского, т]* — текучесть, т. е. величина, обратная вязкости), а также
допуская, что сх = с2 = с, в случае идеального раствора можем
записать
l/y\id=4/4i + 4/ib> (V.310)
где % и т]2 — текучести чистых компонентов.
Подставляя это выражение в формулу (V.309), найдем
1М = (х^/г\1 + 4/г\2)(1+д1пу^/д\п4У (V.311)
В простейшем случае для модели строго регулярного раствора
в соответствии с формулой (IV.97) уравнение (V.311) можно
переписать в виде
1/П = W/t|? + 4/t|S) [l -2{xlxjfi>L)/RT]- (V.312)
Используя это выражение, можно оценить значение энергии
смешения coL на основе экспериментальных данных по вязкости.
В работе [255] это показано на примере ряда сульфидных систем.
В заключение отметим, что при выводе уравнения (V.311) был
сделан ряд допущений, среди которых наименее строгим является
выражение коэффициентов диффузии через вязкость. При более
строгом подходе необходимо использовать более точные
соотношения (см. работы [259, 260]).
В работе О. А. Есина и др. [261—263] указана возможность
оценки энергии смешения из значений мольных объемов и
коэффициента поверхностного натяжения.
Из рентгенографических данных
Методика определения параметра межмолекулярного
взаимодействия в твердых растворах полупроводников с изовалентным
замещением из рентгенографических данных предложена и
проиллюстрирована на примере квазибинарных систем,
образованных соединениями ANBB~N, в работах В. Т. Бублика [264].
Фурье-компоненты флуктуации концентрации связаны в
макроскопической теории с термодинамическим потенциалом
твердого раствора следующим образом [265—266]:
\/<\bx+\2>=(l/RT){d2g/dx2 + $x) при J->0, (V.313)
где q — волновой вектор, ф** — вторая производная
термодинамического потенциала, обусловленного упругими силами, по
концентрации.
Измеряя интенсивность диффузного рассеяния в симметрич-
-> -►
ных относительно узла точках обратного пространства (+q, —q),
можно выделить часть рассеяния, обусловленную статистиче-
8* 227
скими смещениями, и построить зависимость <|Ал:->|2> =
= / (q2). Экстраполяция полученной прямой к \q\ = 0 дает [264]
со (0) = /гГ Г 1/< | Ajc-> I2 > — (1/^1^2)] при q-+ 0, (V.314)
где со (0) — Фурье-компонента энергии смешения.
Отсюда, на основании соотношений (V.313), (V.314), (IV.93)
и (IV. 104), легко установить связь:
d2gEldx\ = со (0) - cpL- (V.315)
Если кристалл с кубической решеткой считать упруго
изотропным континуумом, то с некоторым приближением можно
использовать уравнение [264]
q& = [2/С (1 - 2<т)/3 (1 - а)] [(3/а) (da/ax)]2, (V.316)
где а — параметр решетки; о — коэффициент Пуассона; К =
= (1/3) (Gn + 2G12); Gty — модуль упругости раствора.
Используя для функции gE приближение строго регулярного
раствора, получим соотношение, связывающее величину
параметра межмолекулярного взаимодействия с величинами,
устанавливаемыми из рентгенографических данных в виде
ю = [-ю(0) + фУ/2. (V.317)
Однако здесь следует учитывать, что использование приближения
регулярного раствора для сплавов, характеризующихся заметной
деформацией кристалла, не является надежным приближением.
Для строго регулярных растворов должна, по-видимому,
наблюдаться связь:
со = со (0)/2. (V.318)
Поэтому возможность трактовки, по Бублику, величины со как
аддитивной суммы двух составляющих (упругой и «химической»)
следует признать весьма грубым приближением. При более
сложной картине поведения реального раствора значение
параметра со с рентгенографическими параметрами будет связано,
по-видимому, более сложной зависимостью.
8. РАСЧЕТ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ СПЛАВОВ
ПО ДАННЫМ О ФАЗОВЫХ РАВНОВЕСИЯХ
Между термодинамическими свойствами сплавов и диаграммами
состояния имеется тесная связь. В гл. III было показано, что если
известны концентрационные зависимости свободных энергий
сплавов в широком интервале температур, то с их помощью можно
построить диаграмму состояния. Однако в связи с тем, что фазовые
диаграммы большинства бинарных систем хорошо изучены,
целесообразнее их использовать для вычисления некоторых
термодинамических параметров, например теплот плавления компонен-
228
тов, теплот и энтропии образования сплавов, химических
потенциалов или коэффициентов активностей компонентов и др.
Известны многочисленные примеры использования диаграмм
фазовых состояний сплавов для определения их
термодинамических свойств [267—271].
Сформулированы некоторые закономерности. Например,
сплавы, имеющие диаграмму состояний эвтектического типа либо
диаграмму с областью расслаивания в твердом или жидком
состоянии, имеют, как правило, положительные энтропии и теплоты
образования; отрицательные теплоты образования характерны
для сплавов с промежуточными фазами [272].
Эти выводы, однако, не имеют строгого доказательства. Но,
как показано Г. Ф. Ворониным [273] на основе введенного им
нового класса термодинамических функций (парциальных
мольных функций гетерогенных смесей), в ряде случаев удается
обосновать подобные корреляционные связи.
В соответствии с этой работой сформулируем следующие
правила:
1. Если в системе без промежуточных фаз область первичного
твердого раствора на основе одного из компонентов пренебрежимо
мала, то твердый раствор на основе второго компонента имеет
положительную теплоту и энтропию образования, т. е.
Л«(Р)^0, 5м(Р)^0. (V.319)
Это правило применимо к сплавам с любым типом диаграмм
состояний, имеющим в твердой фазе область расслаивания,
примыкающую к чистому компоненту.
2. Для области составов, отвечающих первичной
кристаллизации 1-го компонента, будем иметь
ЯГ {L) + ДйГ, „ 3* О, *Г (L) + A*. m 5* О, (V.320)
где/z"*^ и S\ —парциальные мольные свойства образования
жидкого сплава, принадлежащие кривой ликвидуса при
симметричном способе нормировки.
3. В системах с вырожденной эвтектикой можно указать
предельные значения интегральных функций жидкого сплава:
Л-^-О-дг^ДА;,,», sM(L)^-(l-x2)As;,m, (V.321)
где индекс 1 принадлежит более тугоплавкому компоненту.
Так как величина (Ам (L) —TsM(L)) должна быть всегда
отрицательной, то при температурах, лежащих выше температуры
плавления 1-го компонента, для энтропии образования сплава
можно записать более «жесткое» условие:
sm (D ^ _ (1 _ Х2) дЛ;в т/т\ (V.322)
Если интегральные функции образования сплава
положительны, то неравенства (V.321) и (V.322) выполняются всегда,
229
если же они отрицательны, то эти неравенства (V.321) и (V.322)
ограничивают их величину.
Указанные неравенства можно обосновать и для систем с
незначительной областью твердых растворов вблизи 1-го
компонента, у которых эвтектическая точка по составу несколько
удалена от второго компонента, поскольку продолжение ветви
первичной кристаллизации первого компонента за эвтектическую
температуру приводит диаграмму состояний к только что
рассмотренному виду.
4. В системе с приблизительно центральным положением
эвтектической точки на оси концентраций с незначительной областью
растворимости в твердом состоянии для обоих компонентов имеем
s« w ^ _ (l _ х2) Ahl m/7\ sM (L) ^ - х2 Д/£ JT. J ( ' '
Как отмечено в работе [273], неравенствам (V.323) должны
удовлетворять и термодинамические свойства системы с областью
расслаивания в жидкой фазе и с перитектически плавящимся
соединением.
Общий недостаток всех известных решений задачи
извлечения термодинамических свойств из данных только по фазовому
равновесию заключается в применении тех или иных
предположений о характере температурно-концентрационной зависимости
термодинамических функций раствора. Наиболее распространено
приближение теории регулярных растворов. Однако, как
свидетельствует эксперимент, многие сплавы, как правило, имеют
значительные избыточные энтропии образования.
Метод расчета термодинамических свойств в двухкомпонент-
ных двухфазных системах, разработанный авторами работ [140,
148, 274—277], не требует каких-либо предварительных сведений
о природе раствора. В основе метода, разработанного Хискесом
и Тиллером [275—277], лежит формальное представление
химических потенциалов компонентов в виде двойного ряда Тейлора
по температуре и концентрации с произвольно выбранным числом
членов [см. уравнения (IV.193) и (IV. 194)]. Задача расчета
заключается в определении величины коэффициентов при каждом
члене ряда.
Чтобы нагляднее представить ограничения, налагаемые
уравнениями (IV. 191) и (IV. 196) на коэффициенты рт/г, запишем
разность (Т— Т0)т в виде бинома Ньютона:
т
(т - т0)т = Е <4 (-i)m-/ тг'т1.
Подставив (Т — Т0)т в формулу (IV. 196), получим
М М N
Е т1 Е £ (1 -х0г$,ппс1т(-1)т-'тг1 =RT.
1=0 m=l n=0
230
(V.324)
(V.325)
Так как уравнение (IV. 196), а следовательно, и (V.325) должны
выполняться при любых значениях 77 коэффициенты при
одинаковых степенях Т в правой и левой части выражения (V.325)
должны быть равны. Таким образом, получаем систему из (М + 1)
уравнений вида:
£■ £ / « 1 m 1 ( Я ПРИ /=1,
SSM^UHr'C1- л * /safcl (V.326)
m=/ n=0 { U При / 7^ 1,
где / = 0, 1, 2, .... М.
Аналогично из уравнения (IV. 191) имеем
м N [ R пои / = 1
2 1П-1Гт-'*п47Г'= о * , ' (V.327)
где I = О, 1, 2, ..., М.
Таким образом, ограничения, налагаемые уравнениями (IV. 191)
и (IV. 196) на коэффициенты pLi, приводят к 2 (М + 1) N
линейным уравнениям, содержащим (М + 1) (N + 1) неизвестных
параметров $lmn*
Недостающие (М + 1) (Af — 1) уравнения можно получить
из условий фазового равновесия.
Для каждого значения температуры равновесного
сосуществования фаз аир имеем два уравнения фазовых равновесий, что
определяет минимально необходимое для выбранной модели число
экспериментальных данных, так как для определенности задачи
требуется, чтобы число неизвестных не превосходило числа
уравнений.
Подставив выражения для химических потенциалов
компонентов (IV. 193) и (IV. 195) в условия фазового равновесия и введя
обозначения:
"-1
/{(т, п) - (-1)» (Т - Т0Г ^ -^V 40 - *о)Ч(1 - 4Г-1],
(V.328)
<7=0
л~1
fUm, n) = (T-r0)ffl2-eV^K4r?-l]. (V.329)
9=0
231
йолучим для каждого значения Т дополнительные ураЁНений
вида:
N м г э 1
|n=l m=0 ( xi)
(V.330)
N М / Р \
2 2 о?<т'п) &. - $(т'«) pw=*г 1п НИ+ й(Г)-
п=1 т=0
(V.331)
где gl (Т) = ^ (Р> (Г, р) - ^(а) (Г, р),
^2(Г) = ^(Р)(Т, р)-^<°>(7\ р).
Используя широко известное соотношение для изменения
химического потенциала чистого вещества при фазовом переходе
вида (V.142), имеем
г1. rn Tl, m i о .
gi(T) = As\,m(T-T\,m)+ j AcPtldT-T J ^i]d7\
(V.332)
g3(T) = Asl,m(T-r2im)+ J Ac'p,2dT-T j -^ df,
(V.333)
где Acp, t — разность теплоемкостей чистого компонента i в а-
и fi-фазах при температуре 7\
Соотношения (V.332) и (V.333) справедливы только для систем
с непрерывным рядом твердых растворов и для некоторых систем
эвтектического типа.
Если компоненты образуют кристаллические фазы с различной
структурой, то вместо этих соотношений следует использовать
Другие.
В качестве примера рассмотрим систему эвтектического типа,
в которой твердая фаза на основе компонента 1 имеет структуру,
отличную от структуры твердой фазы на основе компонента 2.
Тогда в качестве стандартного состояния компонента 1 в 0-фазе
следует выбрать реальное состояние компонента 1, а в качестве
стандартного состояния компонента 2 — его состояние в
бесконечно разбавленном р-растворе. И наоборот, стандартным
состоянием компонента 2 в |5'-фазе будет состояние чистого второго ком-
232
понента, а компонента 1 — его состояние в бесконечно
разбавленном р'-растворе. Поэтому в этом случае приходится полагать, что
И (L) (7\ Р) - pi*(П (7\ р) = А + ВТ + СТ\п 7\ (V.334)
|Л2(L) (Т, Р) - \i?(Р) (7\ р) = D + £Г + FT In Г, (V.335)
где Af Bf Cf D9 E, F — коэффициенты, определяемые из условий
химического равновесия вместе с Pmn.
Вид выражений (V.334) и (V.335) выбран по аналогии с
обычным выражением для \iiiL) — [х!<5). Таким образом, решение
системы, составленной из уравнений вида (IV, 191), (IV. 196),
(V.330) и (V.331) с учетом (V.332) (V.333) или (V.334) и (V.335),
дает возможность рассчитать параметры Ртя, и следовательно,
химические потенциалы компонентов, на основании которых затем
можно вычислить все другие термодинамические свойства сплавов
исследуемой системы.
Изложенный метод Хискеса и Тиллера можно несколько
обобщить, записав исходное уравнение (IV. 186) в виде
*2 Ш\(х, Т)/дх2]т, , = Е XJ PL (Т - ТоГ (*2 - *о)п> (V.336)
т=0 я—.0
где М1 зависит от природы рассматриваемой фазы /, a Nlm> кроме
того, еще и от значения т. Это дает возможность использовать
любой допустимый набор членов разложения, а также
предполагать различные модели раствора для разных фаз.
В этом случае, разумеется, число неизвестных уже не равно
2 (N + I) (M + 1), кроме того, среди уравнений вида (V.326)
и (V.327) могут появиться линейно зависимые, а число ограничений
сократится.
За исключением небольшого числа простых случаев,
коэффициенты Pmn не имеют физического смысла. Рассмотрим два
примера. Координаты начальной точки разложения примем
равными х0 — Т0 = 0, 1 — х0 = 1.
А. Строго регулярный раствор.
М = 1, Л^о = 2, Nx = 0.
Для нахождения соотношений, связывающих между собой
коэффициенты Ртл» используем уравнения (V.326) и (V.327).
Из выражения (V.326) следует:
. при / = 0 р00 + Рог + Рог = 0, при / = 1 р10 = R.
Из уравнения (V.327):
при / = 0 роо = 0, при / = 1 р10 = R.
Отсюда получим окончательно
Роо = 0, рхо = R, pox = -ft* (V.337)
233
Подставив в формулу (IV. 195) значения М = 1, N0 = 2,NX =
= 0, xQ = Т0 = 0, 1 — х0 = 1 при учете соотношений (V.337),
получим выражение для химического потенциала 1-го
компонента в виде
А (7\ х) = \i[{i) (T) + RT In (I - Л'О +
1 Wm п-1
+ Т.- 1: pL(--inrr2H-i)4[(i-*Dn-?-n/(n-?) =
m=0 я=1 <7=0
= ц!(0 (Г) + «Г In (1 - xi) + fe2/2. (V.338)
Аналогично на основании выражений (IV. 193) и (V.337)
нетрудно убедиться, что
ц«(7\ x) = li°2w(T) + RT\nxl2 +
1 Nm n-\
+ I- !■ Р^Г1 £ (-1)4 lW,-,) - U/(n - q) =
m=0 n=l <7=0
= ^(0 (Г) + RT In jc| -h Po- (1 — 4)2 /2. (V.339)
Полученные выражения (V.338) и (V.339) аналогичны
уравнениям для химических потенциалов компонентов (IV.97) и (IV.98)
для строго регулярного раствора.
Б. Квазирегулярный раствор:
М = 1, ЛГ0 = 2, #! = 2.
Легко убедиться, что в этом случае уравнения (V.326) и (V.327)
для / = 0 аналогичны (V.337). Из соотношений (V.326) и (V.327)
при I == 1 следует
Рю + Pn + Р12 = #> Poo + Poi + P02 = 0»
Poo = 0, P10 - fl, (V.340)
откуда p01 = -poa, pu = -pia. (V.341)
Так как соотношения, связывающие рп с р12, и р01 с р02,
одинаковы, то выражения для химических потенциалов в этом
случае можно получить, добавив к выражениям (V.338) и (V.339)
соответственно члены Т$12х\12 и Тр12 (1 —х2)2/2,
|i{ = ii[{i) (Г) + RT In (1 - xi) + (4° 2/2) (p02 -f Р12Г), (V.342)
^ = ^ «> (T) + RT In 4 + [(1 - 4)2/2] (P02 + Pi27> (V.343)
Эти уравнения аналогичны выражениям для химических
потенциалов компонентов в приближении модели квазирегулярного
раствора.
Уравнения (V.330) и (V.331) являются обычными линейными
уравнениями относительно коэффициентов р«л и, в принципе,
234
для их решения достаточно иметь число уравнений, равное числу
неизвестных [следует иметь в виду, что на ^1тп накладываются
связи, задаваемые уравнениями (V.326) и (V.327)].
Однако, чтобы исследовать характер температурной
зависимости термодинамических функций в широком интервале
температур и иметь возможность надежно экстраполировать
полученные зависимости за пределы данного интервала, а также чтобы
получить значения расчетных параметров, согласующиеся со
всеми имеющимися экспериментальными данными, необходимо
использовать как можно больше экспериментальных точек во
всем температурном диапазоне сосуществования фаз. В этом
случае число уравнений вида (V.330) и (V.331) может превышать
число неизвестных и решение находят одним из методов
математической статистики.
В работе Броувера и Оонка [140] разработан метод,
позволяющий на основании экспериментально построенной Т—х
диаграммы рассчитать такие избыточные термодинамические
функции смешения, как энтропия и энтальпия, а также избыточную
теплоемкость растворов.
Метод Броувера и Оонка так же, как и предыдущий, основан
на линейной модели регрессии, вытекающей из условий
термодинамического равновесия фаз. Однако в отличие от способа Хи-
скеса и Тиллера в данной модели избыточные термодинамические
функции получают разложением функции gE (Г, х) в ряд
Тейлора с использованием ортогональных полиномов Редлиха—
Кистера.
В соответствии с работой [140], на основании выражения
(IV.85), используя для избыточной свободной энергии раствора
соотношение (IV. 184), для системы эвтектического типа будем
иметь
Hf (Г, х) = х\ £ ©, (7) {[2/ - 1 - 2/х,1 (1 - 2х2)'-2}, (V.344)
\£ (Т, х) = (1 - х2)2 £ со,- (Г) [(1 - 2jx2) (1 - 2х'2у-Ц, (V.345)
где штрихом отмечены параметры, относящиеся к раствору с
концентрацией Х2 > Х2 (е) .
Здесь могут представиться три варианта:
1) эвтектика со стороны первого компонента, в качестве
функции регрессии может служить уравнение (V.345); 2) эвтектика
со стороны второго компонента; в качестве функции регрессии
может служить уравнение (V.344); 3) эвтектика приблизительно
в центре; уравнение регрессии получают, вычитая соотношение
(V.344) из выражения (V.345).
235
В последнем случае, полагая, например, k = 2, получим
следующую функцию регрессии [140]:
Hf(7\ x')-|if(7\ x) = {Af-Tsf + cf[T-e-
- Г In (Г/6)]} [(1 - х f - х2] + {Af - 7sf +
+ с* [Т - 0 - Т In (Г/9)]}'[(1 - /)2 (1 - 4/) - х2 (3 - 4х)],
(V.346)
где hf, sf, cf — константы в выражении для температурной
зависимости параметра со,- [см. уравнение (IV.185)]. Для кривой
расслаивания справедливо:
nf (*i) ~ И-f (*п) - ЯГ In (1 - *„)/(1 - хг)9
[i2 (х{) - uf (*н) = RT In (*„/*,), (V.347)
где нижние индексы I и II означают сосуществующие растворы.
Для кривой расслаивания функцию регрессии в общем виде
можно записать следующим образом:
/ (х, Т) = [^ (хп) - \if (хц)]г - Ы (хг) - |if (хг)]т. (V.348)
Для систем с непрерывным рядом твердых растворов функция
регрессии имеет вид выражения (IV. 185).
Экспериментальные значения химических потенциалов
оценивают из данных по фазовым равновесиям с привлечением
формулы
lif (S) (Г, xf) = RT In tf/xf) - Asl, m (T - 71, m) +
+ A4f, [Г - Tl. m - T In (T/tf. m)] + |*f (L) (Г, xf)- (V.349)
Из этого уравнения видно, что для системы с непрерывным
рядом твердых растворов химические потенциалы компонентов
в какой-либо из фаз можно рассчитать при условии, если известны
избыточные термодинамические функции для другой фазы.
На основе согласия экспериментальных и рассчитанных
величин gE или (if находят оценки параметров модели,
описывающей избыточные термодинамические свойства в изучаемой
системе.
Расчет изотерм термодинамических свойств сплавов
рассмотрен Г. Ф. Ворониным [274]. В соответствии с этой работой [274]
покажем, как можно рассчитать парциальные и интегральные
236
свойства сплавов из фазовой диаграммы с простой эвтектикой.
Схематический вид диаграмм представлен на рис. 49, где хе и
Те — координаты эвтектической точки, 7\ и Т2 — температуры
соответствующих ветвей кривых ликвидуса.* За стандартное
состояние для раствора принято состояние чистых жидких
компонентов. Допускается, что температурная зависимость теплот и
энтропии смешения жидких сплавов столь незначительна, что
ею можно пренебречь.
Вдоль всей кривой ликвидуса расплав находится в
равновесии либо с кристаллами первого компонента, либо с кристаллами
второго компонента, поэтому величины
$ для составов и температур кривой
ликвидуса равны изменению
химических потенциалов соответствующих
компонентов в процессе их
неравновесного затвердевания.
Основываясь на этом, найдем
выражения для парциальных мольных
изобарно-изотермических потенциалов
смешения жидких (пересыщенных)
твердых растворов при температуре Те.
Согласно формуле (V.33a):
принимаем во внимание выражение
\il = hi-Tsh (V.350)
которое получается путем
дифференцирования (1.113) по щ при постоянных Т и р и учитывая, что
по условию рассмотрения ftf и s" не зависят от температуры,
нетрудно убедиться, что
Рис. 49. Схема к расчету
термодинамических свойств сплавов
из фазовой диаграммы по
методу Воронина [274]
^Г (*2, Те) = |1Г (*2, ТХ) - $ (Х2) (Те - 7\), (V.351)
Ц2 (*2, Те) = $ (х2} Т2) - §? (Х2) (Те - Т2). (V.352)
Отсюда, подставив уравнения вида (V.33a), получим
rf (*2, Те) = As;, г (Тг - Т°т% х) - $ (х2) (Те - 7\), (V.353)
V* (*2, Те) = Asm§ 2 (Т2 - Т°т, 2) - Й (х2) (Те - Т2). (V.354)
Равенства (V.353) и (V.354) справедливы в различных областях
составов. Первое при хе > х2 > 0, а второе при 1 > х2 > хе.
Однако границы использования этих соотношений можно
расширить, экстраполируя известные участки кривой ликвидуса за
эвтектическую температуру до температуры Тп. Таким образом,
вблизи точки хе% Те оба уравнения (V.353) и (V.354) выполняются
одновременно. Точность экстраполяции кривой ликвидуса
падает по мере удаления от состава хе, поэтому участки диаграммы,
примыкающие к чистым компонентам, в расчетах не используют.
237
Уравнения (V.353) и (V.354) содержат четыре неизвестные
функции: sT, S2, \i\ (x2y Те), ^2 (*2, Те). Дифференцируя выражения
(V.353) и (V.354) по составу и используя соотношение (1.16) для
парциальных мольных энтропии смешения, легко исключить
последние из системы и получить выражения, связывающие между
собой величины \iy (x2, Те), \х% (х2у Те) и их производные по составу.
Далее нетрудно, пользуясь соотношениями (IV. 12) и (IV. 13),
заменить \i? (x2, Те) и \l$ (лг2, Те) на величину gM (x2y Те). Согласно
работе [274], получающееся в результате этих преобразований
уравнение имеет вид
р,уч <*У(*2, Te) ■ fl/rJ^M(*2, Те)
- (^M£;rg)I2=J+F {X2) [gU {X2>Te) -gM (x<>Te)]=°>
(V.355)
где коэффициенты
P (*•) = Тг- Т%, (V.356)
n/„ \ _ (T2 — Te) dTx {T1 — Te) dT2 /у qc7\
f/v>»_ Г ! (T*-T*) dTi . _! (Л-Ге) dr2] v 5g
и величины
gM (х9, Тъ) = (1-хв) (Те - П., О Л4,! + .ve (Г* - П,, 2) Л4,2,
(V.359)
[dgM (*2, Te)/dx2]e = (Г, - Г;, 2) As°m, 2 - (Г, - Hi. l) As^, X
(V.360)
полностью определяются формой кривой ликвидуса и свойствами
чистых компонентов, уравнения (V.359) и (V.360) — начальные
условия интегрирования.
Помимо этих условий, интегральная кривая задана и на
границах интервала (0,1)
£м (0, Те) = #м (1, Те) = 0, - (dg«/dx2)Xt=Q = (dgIA/dx2)x^1 = оо.
(V.361)
Рассмотренный способ оценки термодинамических свойств
проиллюстрирован в работе [274] на примере системы Bi—Cd
с использованием метода «избранных» точек [278]. Рассчитанные
функции смешения сплавов Bi—Cd, как показано в работе [274],
удовлетворительно согласуются с экспериментальными данными.
В работе Прокса [279] предложен метод расчета избыточных
термодинамических функций смешения растворов gE и sE на
основе изобарических данных по фазовым равновесиям в бинарной
системе и теплотам смешения. Рассмотрим два случая.
238
1. Расчет избыточной энтропии растворов в системах
эвтектического типа с пренебрежимо малой взаимной растворимостью
компонентов.
В рассматриваемом случае условия фазового равновесия для
компонентов будут иметь вид:
V& (*, Т) = ^(S) (7), ^ (*\ Т) = ii°BiS) (Г),
где штрихом обозначены параметры, относящиеся к раствору
с концентрацией хв>Хв(е).
Отсюда принимая во внимание формулу (V.350), для
растворов с концентрацией Хв < хв (е) можно записать
sLa(T) = [RLa(*, T)-h°A{S)(T)]/T + sAiS)(T). .
Значение парциальной мольной энтропии компонента А при
температуре Тэ, для которой имеются экспериментальные данные
по теплотам смешения, можно оценить по формуле
SA (Г) = [hLA (X, Т) - h°/S) (T)]/T + SA{S) (T) +
тэ
+ J [cLp.a(x, T)!T]dT, (V.362)
т
где cPiA—парциальная мольная теплоемкость компонента А
в растворе.
Отсюда парциальную мольную энтропию смешения компонента
А при температуре Тэ можно определить из уравнения
sZtL)(x, r) = sLA(x, Г)-^>(Г) =
Тэ
= [hLA (х, Т) - tiA(S) (Т)]/Т + j [cLp, А (х, T)IT] dT -
т
t°m,k Г'
- j K^(T)/T]dT- J [cp'L2/T]dT-Ah°m,A(rm.A)/T;n,A.
(V.363)
После несложных преобразований это соотношение приводится
к виду [279]:
sj? (L) (х, Г) = [hEA(t> (х, Т) + А/С. А (Т)УТ +
Тэ T°m, A
+ J Up, (i' (х, Т)/Т] dT + j [Ас;, л (Т)/Т\ dT -
т т
-bh°m,A(rm,A)trm.A. (V.364)
239
Аналогично можно рассчитать парциальную мольную энтропию
смешения компонента В для растворов с концентрацией Хв >
> *в (е) по уравнению
-М tf.) (/> Г) _ [RE (Z.) (/> т) + д^ в (Г)]/Г +
Гэ т°т, В
+ j [<£ V (Т)/Т\ dT+ j [Ас°р, в (Г)/Л dr-ДС. в (Г;. B)lfm, в-
Т Т
(V.365)
Значения s%{L){x, Тэ) и 5л (/) (*, Г3) для составов хв<.хв{е)
и *£ > хв (е) рассчитывают по уравнению Гиббса—Дюгема
соответственно:
sA (х)
-М (L) / грэ\ -М (L) / \ f /1 \ -1 t-M (L) / ч
5В (л:, Т ) = 5В (*е) — J (1 — *в) *В ^Л О),
М Ы
tf (L) (*. Г) - tf (L) (*) - j (1 - ^в)"1 *в ^ <L) (x).
*B (*e)
Избыточную энтропию бинарного раствора А—В при температуре
Тэ можно определить по уравнению
sE ™ (х9 Г) = (1- xLB) tf {L) (х, Г) + xLBs% (L) (*, П +
+ R [(1 - *в) In (1 - 4) + *в In *£]. (V.366)
2. Расчет избыточной энтропии растворов в системах
эвтектического типа при взаимной растворимости компонентов.
В этом случае условия фазовых равновесий для
компонентов А я В примут вид:
tf (Г, /) = tf (Т, Xs), и& (Г, *'<L>) = р.! (Г, *'<*>)
или с учетом соотношений (V.350) и (IV.2) в другой форме записи:
fij?(L) (х, Т) - Т&{L) (х, Т) = hAiS) (Г) - Ts^(S) (Г) + #Г In <&
йв (L) (*, Г) - Г^(L) (*, Г) = h°B{S) (T) - TsB{S) (T) + RT In а|.
Аналогично тому, как это было сделано в первом случае, для
системы с взаимной растворимостью компонентов в твердом
состоянии можно получить уравнения, позволяющие рассчитать
парциальные мольные энтропии смешения компонентов А и В
в жидком растворе, которые по форме записи будут отличаться
от соотношений вида (V.364) или (V.365) лишь дополнительными
слагаемыми —R In (l — хв) и —Rln хв соответственно. Даль-
240
нейшая обработка результатов с целью оценки величины sE{L)
в данном случае аналогична той, которая была рассмотрена в
первом варианте.
Для расчета величины gE (L) (Г9) в обоих случаях можно
воспользоваться известным термодинамическим соотношением:
' gE = hE-TsE.
В системах с конгруэнтно и инконгруэнтно плавящимися
химическими соединениями на основе фазовой диаграммы можно
оценить значения термодинамических функций образования
промежуточных фаз.
Из уравнений (V.202) кривой ликвидуса соединения АтВп
вытекает, что, располагая данными о коэффициентах активности
компонентов в насыщенных растворах соответствующего состава,
можно рассчитать термодинамические параметры образования
соединения A#f и ASf.
При отсутствии данных о термодинамических свойствах
сплавов данной системы, располагая достаточно надежными данными
по фазовому равновесию, значения yt можно оценить с хорошим
приближением из условия наилучшего совпадения теоретически
рассчитанной кривой ликвидуса с экспериментально
построенной. Далее, пренебрегая зависимостью A//f и ASf от
температуры в исследуемом температурном диапазоне, совместным
решением системы линейных уравнений вида (V.202), например, по
методу наименьших квадратов, можно рассчитать средние
значения теплоты и энтропии образования исследуемого соединения. Так,
например, используя для анализа кривой ликвидуса конгруэнтно
плавящегося соединения выражение вида (V.195), теплоту и
энтропию образования твердого соединения из жидких
компонентов можно рассчитать с помощью следующих выражений,
включающих частные случаи и других более простых моделей
раствора:
AHf = [тп/(т 4- п)\ щ + [тп2/(т + nf\ coi -(-
+ [тпг/(т + nf] щ - AC, Атвп> (V.367)
ASf = \mn/(m + n)} co01 + [jnn2/(m + л)2]-а>ц +
+ [mn3/(m + nf] co21 - As°m, Л||Л| + R In mmnn/(m + n)m+n.
(V.368)
В заключение покажем, какие термодинамические
характеристики сплавов можно рассчитать, построив зависимость
М*0н.в«/(1/Гя«)<
241
Из условия химического равновесия имеем jxf = pf, или,
в раскрытом виде,
^ <s> - ц; ™ = д£. ,(Т - г;. ,•) = RT In [*ftf/*fVf ]• (V.369)
При незначительной растворимости в твердой фазе активносхь
af можно принять равной единице. В этом случае в уравнении
(V.369) удается разделить энтропийную и тепловую составляющие.
Подставляя выражение вида (V.350) в уравнение (V.369),
получим
In (*f)Нас = - (AR\S+L)/RT) + (1/Л) (Asls^>). (V.370)
Здесь Айр£) = AC. i + ftf {L\ (V.371)
AsrL) = AS;,, + 5f(L), (V.372)
где Ah\s^L) и As!5"*10 — предельные парциальные мольные
теплота и энтропия растворения /-того компонента.
Из уравнения (V.370) следует, что если
не зависят от температуры, то предельную мольную теплоту
растворения f-того вещества можно определить по тангенсу угла
наклона прямой In (л£)нас = / (1/^hk), а из отрезка, отсекаемого
на оси ординат при 1/ГЛИк = 0 вычислить AlP^L). Далее, зная
теплоту и энтропию плавления /-того компонента, по формулам
(V.371) и (V.372) легко рассчитать парциальную мольную
энтальпию смешения и избыточную парциальную мольную энтропию
смешения /-того вещества в жидкой фазе.
Как было установлено авторами работы [280],
прямолинейная зависимость In (jcf)Hac от 1/Глик наблюдается только при
низких концентрациях х\, т. е. при очень больших разбавлениях
(<2ат %). Это означает, что допущение о независимости A/z[s~>L)
и AsJs->L) от температуры не является строгим. Поэтому данный
способ позволяет рассчитывать величины Afi|s_>z') и As|s_> )
только при значительных разведениях раствора (#нас-^0).
Как показано Малиновским [281 ], величины парциальных
мольных энтальпий и энтропии компонентов можно оценить
также из наклона кривых ликвидуса в эвтектической точке
следующим образом.
Для системы эвтектического типа с пренебрежимо малой
взаимной растворимостью компонентов имеем
^(^(5)/Т)р = ^(^/Г)р. (V.373)
Раскрыв полные дифференциалы в обоих частях уравнения (V.373),
получим
RT2 (д In atldxt)p% т = bh?+L) (дТ/дхс)р. (V.374)
242
Записав выражение вида (V.374) для каждого из компонентов,
после несложных преобразований придем к уравнению
= RT2 [хг (д In аЦдхг) - х2 (д In а2/дх2)]р, г,
где kt = (дТ/дх^р.
Принимая во внимание соотношение Гиббса—Дюгема, для
эвтектической точки будем иметь
(*! ЬЯ[8+1)кг)е = (х2 Mt^L)k2)e. (V.375)
Используя формулу Ah\s^L) = TAs\s^L)t аналогичное
выражение можно записать для парциальных мольных энтропии
растворения компонентов в точке эвтектики:
(хг As[s*L)kx)e = (дг2 A4s^L)k2)e. (V.376)
С помощью выражения
Ah^L) = (я, Ah[s+L))e + (x2 Ahis-*L))e, (V.377)
где Ahls^L) — интегральная мольная теплота растворения
эвтектического сплава, получим еще одно соотношение, которое можно
применять при практических расчетах (см. работу [281]).
На основании соотношений (V.375) и (V.377) нетрудно
убедиться, что
(№[s-*L))e = [M*i*i + *i*2>] btis+L); (V.378)
(Aft^L))e = [hl{x2kx + x2k2)\ bh{es+L). (V.379)
При температуре эвтектики
Ti,m
Ак1т(Ге)=^АН],т{т1т)- j Acp}s+L)dT. (V.380)
Принимая во внимание соотношения (V.380) и (V.371), с помощью
(V.378)—(V.379) получим выражения, позволяющие
рассчитывать парциальные мольные теплоты смешения компонентов в точке
эвтектики из наклона кривых ликвидуса и свойств чистых
компонентов, вида:
(ftf (L))e = {kiH&h + xfa)\ Ah?->L) -
Л.т
-Mh.AT\.m)+ J Acp^L)dT;
Te (V.381)
(fif №>), = [k\!(x2k, + x2k2)] Ah?-*L) -
T2, m
-Ah°2,m(f2,m)+ j Ac°PTL)dT.
Te
243
Аналогично тому, как это было сделано выше, при учете (V.372)
и выражения dAs]t т = &c°p^j*L) d In 7\ получим следующие
уравнения для расчета парциальных мольных энтропии смешения
компонентов в эвтектической точке из наклона кривых ликвидуса
и свойств чистых компонентов:
(Sl (L))e - [fc/(*l*l + ХгШ &S{eS*L) -
Tl, m
-bsUm(T\.m)+ j AcPTL)dT,
r2, m
-AsJ.«(^fm)+ J bcp%+L)dT.
9. ОЦЕНКА СТРУКТУРНОГО ФАКТОРА
В РАСПЛАВАХ БИНАРНЫХ СИСТЕМ НА ОСНОВЕ
АНАЛИЗА ДАННЫХ ПО ФАЗОВЫМ РАВНОВЕСИЯМ
На основе анализа фазовых диаграмм можно получить также
информацию о структуре раствора. В работах Марча и Бхатии
[282—285] выведены уравнения, связывающие форму (тангенс
угла наклона) кривых моновариантных равновесий со
структурным фактором Sxx (0) в длинноволновом пределе, обусловленным
флуктуациями концентрации в жидком сплаве и твердом растворе.
Для твердых растворов рассмотрение более правильно проводить
в терминах «ближнего порядка».
На основании формулы (11.188) при р = const наклоны
касательных к кривым ликвидуса и солидуса, отвечающим первичной
кристаллизации компонентов, после умножения числителя и
знаменателя на Т можно представить в следующем виде:
\ дх2 )р
т(4-*!)№/д4)ьР.т
(V.382)
и
\ Эх, )р - (£ М^Ц + XL ЩЗ+D) ' С-000>
где M\s+L) = tii —hi.
В соответствии с выражениями (V.39) и (V.3) для касательной
к кривой ликвидуса, отвечающего первичной кристаллизации
соединения ApBq, имеем
(dTldx2)Lp = Т [п (1 _ х^) - mxk\ (d2g/dx2)Lp, rfflftf- (V.384)
244
Бхатиа и Торнтон [286] ввели представления о структурных
факторах Snn (q)9 Snx (q) и Sxx (q), соответствующих корреляциям
плотность—плотность (пп)у плотность—концентрация (пх) и
концентрация—концентрация (хх), где п—плотность заданного
числа частиц.
В длинноволновом пределе (q ->■ 0) структурные факторы
Snn (0), SnX (0) и Sxx (0) жидких смесей приобретают простой
физический смысл:
SM(0)-((An)»)M, (V.385)
S„x(0) = <AttAx)> (V.386)
Sxx(0) = n((Axf), (V.387)
где ((An)2) —среднеквадратичная флуктуация заданного числа
частиц, {(Ал:)2) —среднеквадратичная флуктуация концентрации,
не связанная с сортностью частиц, (An Ал:) — корреляция между
этими двумя флуктуациями, величины Ал: и Дп рассчитывают по
формулам:
Ах = гг1 [хАпг — (1 —х) Ап2]; (V.388)
Ап = Ап± + An2. (V.389)
Основываясь на принципах статистической термодинамики,
Бхатиа и Торнтон получили уравнения, связывающие корреляцию
концентрация—концентрация со второй производной изобарно-
изотермического потенциала смеси по концентрации [286 L
Обозначим среднее число атомов двух сортов в данном
макроскопическом объеме У системы, находящейся в термическом
равновесии с окружающей средой через пг и п2, а отклонения числа
частиц от средних значений через Д^ и Дп2.
Тогда вероятность таких отклонений определится из
уравнения
W = W0 exp Г— 2 Fa Дл, An^kT], (V.390)
где W0 — нормировочный множитель; k — постоянная Больц-
мана,
Ft! = Fjt = (PF/dtii dnfa v = (д^/дп^у,Tt п.ф. =
^idpjldnfar.n^. (V.391)
Нетрудно убедиться, что
(д^/дп^т, Уч п{ф. = (dpi/dnfa p, Щф] + VtVj/фт, (V.392)
где рг — изотермическая сжимаемость раствора при постоянном
составе [108]. Используя формулу (V.392), можно записать
£ Fi} Ащ Ап; = S (d\ii/dnJ)T9Ptai . Ащ Anf +
if i\
+ {vx A/ii + v2 Ап2)2/фг. (V.393)
245
С помощью соотношения Гиббса—Дюгема и выражений (V.388)
и (V.389) уравнение (V.393) преобразуется к виду [286]:
£ Ftj Д/г. Anj = ИМ2)] [А/г + п Дхб]2 + В (Ал:)2, (V.394)
I. /
где
б = (vx - и2)/[(1 -x)v± + xv2] = (n/V) (бх —б,); (V.395)
В « (л>1) (дрг/дпдт, р, я = (cfc/dx2)/". (V.396)
Так как функция № дает распределение типа гауссовского, то
среднеквадратичные флуктуации соответствующих величин
будут определяться выражением
{AnjAni) = kT/Fij. (V.397)
Подставив выражение (V.394) в (V.390), с учетом равенства (V.397)
получим следующие формулы для структурных факторов Sxx,
Snn и Snx в длинноволновом пределе:
Sxx (0) = RT/(d*G/dx*)TiP = (1 - xt)l(d In ajdxdp, r, (V.398)
Snn (0) = (n/V) kT$r + S2S„ (0), (V.399)
Snx(0) = -VSxx(0). (V.400)
Подставив соотношение (V.398) в уравнения (V.382)—(V.384),
получим выражения, связывающие форму кривых
моновариантного равновесия со структурным фактором Sxx (0) в системах
эвтектического типа [уравнения (V.401) и (V.402)] и в системе
с конгруэнтно плавящимся соединением [уравнение (V.403) ]
(JL\L = Rt>(4-4) (V 401)
/iL\s = *r(4-*f) (V402)
lp S*x (0) (х\ Дй}*-»« + x\ AHf^L>) '
т2[я-
5^(0)
(■|L^.^-^+g^. (V.403)
<7
С помощью соотношения (V.398) можно рассчитать
структурный фактор Sxx (0) в рамках различных моделей растворов,
используя соответствующие экспериментальные данные.
Встречающиеся в литературе способы оценки Sxx (0) требуют
информации о термодинамических свойствах растворов, которая для
большинства систем отсутствует [287, 288]. Однако число систем,
где изучены фазовые равновесия, довольно велико.
Авторы разработали метод расчета структурного фактора
Sxx (0) на основе анализа данных по фазовым равновесиям в
бинарных системах.
246
Изменение Sxx (0)] с концентрацией в приближении ряда
простых моделей растворов, в частности, идеальной, регулярной,
конформальной и модели Флори, обсуждалось в работах [284,
289—290].
Как отмечалось ранее (см. гл. IV), в'реальных системах для
описания концентрационной зависимости термодинамических
свойств смесей обычно применяют формально-математический
подход, базирующийся на использовании либо простых степенных
рядов Маргулеса, либо различных ортогональных полиномов,
среди которых часто применяют полиномы Редлиха—Кистера,
Лежандра, тригонометрические ряды Фурье. Используя
соотношения (V.398).,(IV.176)—(IV.179) и допуская, что коэффициенты
указанных полиномов зависят от температуры по уравнению,
предложенному в работе [140]:
</ = <7, - Tqn + qj2 (Т - 0 - Г In (Г/9)),
где 9 — начальная температура разложения, отвечающая
экспериментальной температуре в какой-либо точке, для структурного
фактора Sxx (0) можно записать следующие выражения:
а) в приближении рядов Маргулеса:
S«(0) = *!*Jl - (2Xlx2/RT) [q0 - q01T + q02 (Т—в—Т ln-J-) +
+ (3* - 1) (ft - qnT + ?i2 (T - 0 - T In -J-) ) +
+ 3*2(2x2 - 1) (<72 - q21T + q22 (т - 0 - Г In-J-))] J"1;
(V.404)
б) в приближении полиномов Редлиха—Кистера:
Sxx(0)=^xlx2^l-(2x1x2/RT) [q0~q0lT + q02(T-Q-T\n^-) +
+ 3(l-2x2)(qi-qiJ + q1%(T-Q-Tln^-)) +
+ (5-24jc1x2)((72-(?21T + ?ft(r-0^rin^))]j'1; (V.405)
в) в приближении полиномов Лежандра:
S«(0) = x^l-Px&JRT) [q0 - q0lT + q02 (Т—в—Т ln-J-) +
+ 3(2x%-l)(q1-quT + qJM(j'-e-Tln^)) +
+ (7 - 36*л) (ft - qnT + qu (т - 0 - Лп-у-))]}"1; (V.406)
247
г) в приближении тригонометрического ряда Фурье для
синусов:
Sxx (0) - х±х2 1 - (xxx2IRT) I [qk - Tqkl -f
+ qk2 (T-Q—T In (779))] (6n)2 (1 — x2) sin (£л*2) } . (V.407)
Приравнивая нулю значения соответствующих параметров,
на основании соотношений (V.404)—(V.406) можно прийти к
выражению для структурного фактора, совпадающему по форме
записи с уравнением, полученным в приближении регулярного
раствора [290]:
5"ГО-[1-2(%Г)«ЛГ (V-408)
В идеальном растворе со = 0 и, следовательно,
S£(°)=*i** (V.409)
Как будет показано далее, оценки постоянных, составляющих
параметр межмолекулярного взаимодействия со, можно получить
методом наименьших квадратов путем согласования с данными
по фазовым равновесиям.
Выбрав таким образом модель раствора, с помощью одной из
формул (V.404)—(V.407) легко рассчитать концентрационную
зависимость структурного фактора Sxx (0) в исследуемой системе,
а затем, если известны изотермическая сжимаемость и плотность
смесей, можно оценить и структурные факторы Snn (0) и Snx (0)
[286],
Таким образом, анализ фазовых равновесий позволяет
получить полезную информацию о структуре растворов. Получаемые
при анализе структурные факторы связаны с парциальными
структурными факторами Фабера и Займана [291 ], по которым
можно судить о функциях радиального распределения,
10. ВЫБОР МОДЕЛИ РАСТВОРА НА ОСНОВЕ
СОГЛАСОВАНИЯ С ЭКСПЕРИМЕНТАЛЬНЫМИ ДАННЫМИ
ПО ФАЗОВЫМ РАВНОВЕСИЯМ
Математическая задача выбора модели раствора на основе
сравнения остаточных дисперсий часто возникает при
моделировании фазовых равновесий, прогнозировании термодинамических
свойств, исследованиях характера межмолекулярного
взаимодействия в неидеальных системах и т. п.
Анализ, проведенный авторами работ [287, 288], показал, что
число систем с изученным фазовым равновесием значительно
превышает число систем, для которых известны термодинамические
свойства фаз. Поэтому попытки определить параметры на основе
248
экспериментальных данных по фазовым равновесиям
представляют существенный интерес.
При термодинамическом анализе фазовых диаграмм широко
используют модели идеального, регулярного, субрегулярного
растворов, квазихимическое приближение и ряд других
подходов. Среди критериев, позволяющих выбрать лучшее согласие
рассчитанных для различных моделей кривых фазовых
превращений с экспериментальными данными, следует выделить два
основных:
1) математический критерий Фишера (F-критерий], который
выявляет лучшее согласие со статистической точки зрения [292—294 ];
2) физико-химический критерий, заключающийся в расчете
не только кривых фазовых превращений, но и термодинамических
характеристик растворов и соединений (например, химических
потенциалов компонентов, энтальпии и избыточной энтропии
смешения растворов, общего давления пара и парциальных давлений
паров компонентов, термодинамических функций образования
соединений и т. д.), в результате чего удается установить, для
какой модели согласие между расчетными и экспериментальными
данными наилучшее.
Вопрос об определении параметров сводится к экстремальной
задаче минимизации некоторой функции — критерия,
позволяющего сравнивать экспериментально измеренные и рассчитанные
по модели величины.
Выбор критерия, как правило, существенно отражается на
оценках погрешностей физико-химических констант,
фигурирующих в исходной термодинамической или математической модели.
Так как всякий эксперимент связан с появлением случайных
ошибок, то для выбора модели возможно использование различных
методов математической статистики, например максимального
правдоподобия, наименьших квадратов, байесовского метода,
чебышевской аппроксимации (метод минимакса) и др. [292,
294—301].
В случае, если измерения независимы одно от другого и
известен закон распределения, наиболее обоснованный вид критерия
дает принцип максимума правдоподобия. Так, если ошибка
распределена по нормальному закону, приходим к минимизации
суммы квадратов невязок (методы наименьших квадратов) [296—
298]. В случае распределения Лапласа приходим к минимизации
просто суммы модулей невязок. В последние годы число работ,
в которых экспериментальные данные обрабатываются методом
наименьших квадратов (МНК) растет. Метод наименьших квадра-
->
тов реализует поиск таких параметров 6, для которых
«взвешенная сумма» квадратов невязок имеет минимум:
minSQ = rain £ (Ifi-lcXМ'М^ -хХ (V.410)
в, xi 6, х(
249
где х* — вектор, состоящий из k измеренных в /-том эксперименте
величин, включающих случайные ошибки; хь — вектор этих же
величин, свободных от случайных ошибок; Mt —
ковариационная матрица измеряемых величин размера kxk, индекс «Г»
означает операцию транспонирования. При этом должны быть учтены
функциональные связи между истинными значениями измеряемых
переменных при каждом наблюдении, которые включают
параметры, подлежащие оценке. Предполагается, что матрица Mt —
диагональная, а элементы ее представляют собой дисперсии
измеряемых переменных.
Существующие методы оценки статистически обоснованы только
для случаев, когда используемая модель является линейной
функцией относительно неизвестных параметров [298, 301 ].
Условия, при которых оценки, полученные МНК, имеют
смысл наилучших линейных оценок параметров [296, 302] (а если
закон распределения невязок близок к нормальному, то и смысл
оценок наиболее вероятных значений [303]), следующие:
1) экспериментальные ошибки в определении независимых
(контролируемых) переменных отсутствуют; 2)
экспериментальные ошибки измерения независимой переменной (функции отклика)
имеют распределение Гаусса с нулевым средним значением;
3) результаты наблюдений не зависят одно от другого для
заданной экспериментальной точки и для различных точек; 4) модель
для описания функции отклика адекватна, т. е. позволяет
воспроизвести истинные значения переменной; 5) уравнения связи ли-
->
нейны относительно подбираемых параметров 0.
Для бинарных равновесных трехфазных конденсированных
систем измеряемыми величинами являются давление /?,
температура Т, мольные доли компонентов в жидкой xLt твердой Xs и
газообразной хУ фазах. Сточки зрения математической статистики
в данном случае не могут быть строго выделены независимые
переменные и отклики, поскольку все величины определяются
экспериментальной, следовательно, включают ошибку измерений.
В то же время, в соответствии с правилом фаз Гиббса, для полного
описания трехфазной бинарной системы достаточно задать лишь
одну переменную. Следовательно, с термодинамических позиций
только одна из измеряемых величин, характеризующих
трехфазное равновесие в бинарных системах, может считаться
независимой. В конденсированных двухфазных системах, где давление
мало влияет на равновесие, также только одно измеряемое
свойство из набора Т, xL или Xs может рассматриваться как
независимое. В бинарных двухфазных системах жидкость—пар две
величины можно рассматривать как независимые.
Таким образом, при оценке параметров модели раствора на
основе согласования с полным набором данных по фазовым
равновесиям в бинарной двухфазной системе требуются два
уравнения-ограничения.
250
В качестве независимой переменной выбирают мольную долю
компонента в жидкой фазе, определяемую с наибольшей
точностью.
На практике требование детерминированности
контролируемых переменных часто не выполняется. Если неопределенность
при фиксации независимых факторов невелика, то использование
обычных методов регрессионного анализа не приводит к
существенным погрешностям. В противном случае значения оценок
параметров могут заметно отличаться от действительных величин.
В частности, оценки, полученные методом наименьших квадратов
без учета ошибок в определении контролируемых переменных,
могут оказаться либо сильно смещенными, либо просто
несостоятельными [302].
Критерий оптимальности оценок параметров вида (V.410)
используют, в принципе, и в том случае, когда модель нелинейна
относительно искомых параметров (см. например, работы [292,
297, 293]. В этих ситуациях исходную задачу нелинейной
оптимизации с помощью простых многократных преобразований часто
пытаются свести к линейному регрессионному анализу. Этого
удается достичь, например, применяя экспоненциальные или
логарифмические функции.-
Формальное использование таких манипуляций из-за ошибок
в измерении в переменных, как правило, приводит к тому, что
величины параметров, найденные упрощенным способом, очень
сильно отличаются от значений, полученных при решении
исходной задачи. При нормальных ошибках измерений параметры,
полученные с помощью преобразований, могут иметь даже
неправильные знаки [292]. Наиболее часто используемый прием
линеаризации путем разложения в ряд Тейлора может привести из-за
ошибок, возникающих при ограничении ряда, также к
существенному отличию решений от наилучших оценок. В принципе,
возможен поиск параметров МНК и без предварительной
линеаризации [305—307].
Для практической обработки результатов экспериментов
важно знать, какой метод позволяет наиболее эффективно
осуществить поиск экстремума SQ [306].
Перепишем подлежащую минимизацию функцию (V.410) через
введенные описанным выше образом понятия зависимых и
независимых переменных. С этой целью сначала с помощью двух
уравнений связи следует представить зависимые переменные
системы через независимые в явной форме:
F = /(*> 0); Z = ?(x, 6), * (V.411)
где Y и Z — векторы истинных значений зависимых перемен-
ных Y и Z; Y = { Yi\^=\ и Z = {Z,-}£U; x — представляет собой
вектор размерности N (k — 2); (k — 2) — общее число незави-
251
симых переменных, которое в рассматриваемом случае равно
двум или единице.
Каждый набор (k — 2) элементов из х можно представить
в виде вектора xh соответствующего t-тому эксперименту.
Предположение, что ошибки измеряемых переменных некор-
релированы, т. е., что матрица Ml1 диагональна, позволяет
записать критерий (V.410) в виде
N (
min SQ = min J Q_ (yt - Y])2 + -L (Z j - Zf) +
+ 24-W-^)i (V-412)
ft G*; i
где a2^—дисперсии ошибок в определении независимых пере-
менных; оу. и а| — дисперсии ошибок в определении откликов.
Встречающиеся алгоритмы поиска минимума критерия (V.412),
как правило, основаны на линеаризации уравнений связи путем
разложения их в ряд Тейлора по параметрам и истинным
значениям независимых переменных и на переходе от задачи с
ограничениями к задаче без ограничений [308].
В связи с поиском минимума функционала вида (V.412)
возникают трудности, к которым относятся: определение начальных
приближений для параметров, численный расчет производных по
параметрам и особенно проблема сходимости, которая играет
решающую роль при квазилинеаризации [293].
Обычно при использовании хороших начальных оценок
параметров и истинных значений измеряемых переменных сходимость
достигается очень быстро. При плохих начальных оценках
сходимость может быть очень медленной или ее вообще нет.
В ряде работ [294, 309] на основе принципа максимального
правдоподобия рассмотрен способ оценки параметров модели,
который также является общим, так как учитывает ошибки в
определении всех величин, но отличается от рассмотренного выше тем,
что не требует разделения переменных на зависимые и независимые.
Если измерения независимы и модель способна описать
экспериментальные данные, а невязки &i распределены нормально
с некоторой дисперсией о], то в этом случае критерий
оптимальности оценок имеет вид:
N 2
min SQ = min V % = 1ТМ~Ч, (V.413)
Г f ft °i
где е, = f (x% V)=y]- у и (V.414)
М — диагональная матрица с элементами о*.
252
В соответствии с принципом максимального правдоподобия
в общем виде дисперсии невязок величин, характеризующих
равновесие в двухфазных бинарных системах, определяются
соотношением:
К сожалению, встречающиеся в литературе способы оценки
параметров моделей гетерогенных равновесий учитывают не все,
а зачастую совсем не учитывают ошибки в определении
измеряемых переменных и поэтому не приводят к наилучшим
статистическим оценкам параметров [111, 140, 147, 148, 275—277, 310—
312]. Одна из основных целей выбора модели раствора состоит
в получении выражения, которым можно было бы пользоваться
для достоверного прогнозирования различных
термодинамических свойств, фазовых равновесий и т. д. Степень достоверности
предсказаний будет зависеть от степени достоверности самих
экспериментальных данных и модели. Поэтому метод оценки
параметров должен также предусматривать и наличие меры
надежности прогнозирования. Эта задача тесно связана с оценкой
неопределенностей в значениях определяемых параметров.
Применение метода максимального правдоподобия удобно
в практическом отношении, поскольку для этого не требуется
знать абсолютные значения дисперсий, которые, как правило,
неизвестны, а достаточно располагать статистическим весом Wt:
Wi = o2/o2c или W£=l/al (V.416)
где ot — обычно принимают равной инструментальной ошибке
при измерении соответствующей переменной; а2 — дисперсия
наблюдаемых переменных с одинаковым весом.
Оцениваемые параметры модели не зависят от величины а2 и
могут быть рассчитаны без нее. Однако для анализа ошибок в
значениях параметров, вычисляемых по МНК, необходимо
располагать величиной а2, которую можно оценить при помощи
соотношения
s2 = £(a2) = s/(W-p). (V.417)
Общие формулы для вычисления различных характеристик
точности модели приведены в работе [292] и др.
Совместное распределение вероятностей оценок параметров
в нелинейном случае обычно является очень сложной функцией,
которую не только трудно вычислить, но и очень неудобно
использовать, даже если бы она была известна. Поэтому на практике
не стремятся к точному подсчету этого распределения или каких-
либо его моментов, а характеризуют ошибки оценок условными
253
способами [297]. Практическое использование доверительных
интервалов при оценке параметров нелинейных моделей
рассмотрено в работе [313].
Если для описания некоего набора данных* используют две
различные модели, то уравнение (V.417) приводит к двум
различным оценкам s2. Если при этом окажется, что величина s2 для
второй модели меньше, чем для первой модели, то вторая модель
лучше описывает экспериментальные данные. Однако чтобы
проверить, является ли это улучшение статистически значимым,
следует воспользоваться F-распределением (критерием Фишера),
которому подчиняется величина
F(v, |i) = sX (V.418)
равная отношению большей величины дисперсии к меньшей, если
считать распределение ошибок нормальным, где v и \i — степени
свободы для первой и второй моделей соответственно. Если F >
> Fi-a (v, |ы), то следует считать, что вторая модель лучше
первой. Если F < Ft_a (v, (x), то ни одной из моделей нельзя отдать
предпочтение. Здесь величина а характеризует статистическую
надежность при выборе модели.
В работе [314] задачу выбора из различных гипотез одной,
которая наилучшим образом отражает экспериментальные данные,
предлагается решать на основе критерия отношения вероятностей,
который требует минимального объема информации, основан на
вычислении простых выражений и обладает рядом оптимальных
свойств.
Вопрос о статистической проверке гипотез для случая, когда
модель выбирают на основе согласования с несколькими наборами
однотипных данных, рассмотрен в работе [294].
Для контроля адекватности модели двумя наиболее
существенными характеристиками являются доверительный интервал в
каждой точке измерения и случайность остаточных отклонений [292].
Если измеренное значение лежит вне соответствующего
доверительного интервала, то это может быть вызвано следующими
причинами: 1) дисперсии зависимых и независимых переменных
заданы слишком малыми; 2) модель неадекватна.
Наиболее надежное заключение об адекватности модели
делается на основании изучения остаточных отклонений измеренных
значений от вычисленных. Если модель адекватна, то эти
отклонения оказываются чисто случайными величинами. Наоборот, если
в их распределении имеется определенная закономерность, то
модель выбрана неверно. Оценки параметров 0Ь 62, ... вр по
МНК для различных вариантов вектора измеряемых величин не
совпадают; кроме того, различают также и меры их точности. В
качестве откликов при оценке параметров моделей бинарного
равновесия жидкость—кристалл используют величины Т и уг В
работах [294, 315] дан сравнительный анализ влияния выбора
определенной функции отклика на точность оценки параметров при ана-
254
лизе данных по равновесию жидкость—пар. Наиболее важный
вывод, вытекающий из результатов работы, заключается в том,
что в бинарных двухфазных системах метод максимального
правдоподобия, в принципе, позволяет находить надежные оценки
параметров при использовании данных только по двум переменным,
например, р—xL или Т—xL.
Включение дополнительной информации, вопреки мнению
авторов работ [111, 316], не улучшает точности оценок
параметров, но обеспечивает возможность проверки взаимной
согласованности экспериментальных данных [315].
В работах Бребрика [147, 310—312] метод МНК использован
при выборе модели раствора для систем с конгруэнтно
плавящимися соединениями, который позволяет количественно согласовать
данные по фазовому равновесию с термодинамическими свойствами
соответствующих соединений и растворов.
Отличительной особенностью метода Бребрика является
использование дополнительных уравнений, связывающих
термодинамические характеристики раствора со свойствами конгруэнтно
плавящегося соединения в точке плавления, с помощью которых
можно зафиксировать ряд искомых параметров разложения.
Данный метод был широко апробирован на примере систем
группы Л111—Bv [147, 310, 312]. Значения параметров моделей
с помощью МНК находим путем минимизации функционала вида:
[N -11/2
Д Vh экой - Th расч)2/(ЛГ -/>)], (V.419)
где р —число подбираемых параметров; # —число
экспериментальных точек.
Было проанализировано семь моделей раствора: идеальная,
строго регулярная, квазирегулярная, линейно-температурная
аппроксимация, модель, учитывающая линейную зависимость
энергии взаимообмена от температуры и концентрации, с
использованием рядов Маргулеса и квазихимическое приближение.
При этом в каждом частном случае параметры модели находили
из условия (V.419) двумя путями. В первом варианте ни на один
параметр модели не накладывалось ограничение. Во втором
варианте на основании формул (V.212) и (V.213) при учете
конкретных выражений для избыточных парциальных мольных
термодинамических свойств раствора составляли дополнительные
уравнения связи, которые либо строго фиксировали значения отдельных
параметров, либо накладывали определенные ограничения на их
изменения. " •
Наилучшее совпадение расчетного и экспериментального
ликвидуса не является достаточным критерием адекватности модели
и раствора.
Этот вывод наглядно иллюстрируется результатами [147, 310].
Пять моделей раствора, а именно: строго регулярная, квазирегу-
255
лярная, линейно-температурная аппроксимация, модель,
построенная с использованием рядов Маргулеса и квазихимическое
приближение приводят к среднеквадратичным отклонениям, лежащим
в пределах ошибок эксперимента. Однако значения
термодинамических характеристик A#f и ASf, рассчитанные для строго
регулярного, квазирегулярного и квазихимического растворов,
неудовлетворительно согласуются с экспериментальными
значениями. На основании этого можно заключить, что перечисленные
модели раствора не адекватны для описания свойств систем AmBv.
Кроме того, рассчитанные с помощью этих моделей
термодинамические свойства систем также свидетельствуют в пользу данного
вывода, хотя во многих работах утверждается обратное [317].
Что касается линейно-температурной аппроксимации и модели
с использованием рядов Маргулеса с точностью до членов третьего
порядка, то они обе приводят к удовлетворительному согласию
расчетной кривой моновариантного равновесия и
термодинамических характеристик соединения, причем в последнем случае это
согласие даже несколько лучше.
В работе [311 ] была предпринята попытка расширить
концентрационный диапазон применимости единой модели раствора,
опираясь на эвтектические точки, которые автор считает наиболее
надежно установленными.
В работе [3111 также проанализировано влияние ошибок
в определении термодинамических свойств соединения на значения
параметров модели раствора.
В работе [312] проиллюстрирован выбор модели двух-
компонентного раствора с привлечением методики расчета
[318, 319].
Выбор модели, при котором сопоставляются все свойства [312,
318, 319 ] (кривые ликвидуса и солидуса, термодинамические
свойства растворов и т. д.), лишен практического смысла, потому что
не остается параметров, которые можно было бы описать на основе
данной модели, по крайней мере при тех же условиях. Однако
практически такой выбор все же полезен, так как позволяет
оценивать термодинамические свойства растворов при других
значениях параметров состояния, а также рассчитывать иные
кривые моновариантного равновесия, например, кривые соль-
вуса. Кроме того, используя периодический закон Д. И.
Менделеева, можно достаточно уверенно подходить к расчетам в
системах, образованных аналогами соответственно при «анионном» или
«катионном» замещении компонентов, пользуясь уже только
имеющимися надежными данными по фазовым равновесиям.
Если термодинамические характеристики растворов и
соединений отсутствуют, модель можно выбирать, последовательно
рассчитывая и сопоставляя параметры, характеризующие энергию
взаимообмена на основе различных экспериментальных данных
(вязкость, плотность, электроотрицательность и т. д.).
256
В случае совпадения полученных на основе различных данных
значений параметров энергии взаимообмена можно с известным
приближением считать соответствующую модель обоснованной и на
ее основе рассчитывать термодинамические характеристики
растворов и соединений.
Вся система не всегда может быть описана в рамках одной
модели. Особенно это относится к системам с промежуточными
фазами различной физико-химической природы. Тогда при выборе
модели следует ограничивать температурно-концентрационные
пределы ее применимости. -hElix -х )L
Проиллюстрируем на ряде систем 2з \ '"
возможности расчета термодинамиче- ' |
ских параметров растворов на
основе фазовых диаграмм для систем,
включающих полупроводниковые
соединения различных групп
простого и сложного составов.
Системы Alu—Bv ' о 0,2 0,4 0,6 0,8 1,0
Рассмотрим ряд систем Л111—5V, Рис. 50. Зависимость функции
В КОТОРЫХ Образуются ШИРОКО ИЗ- /i^/(*In*Sb)L от состава в системе
г г j л. ~ In —Sb при 900 К по данным ра-
вестные полупроводниковые фазы бот [зго]
типа AlllBv. Модель
субрегулярного раствора в этих системах неадекватна эксперименту. Так,
например, характер поведения функции hu/(XiXt)t рассчитанной,
по данным работы [320] и построенной графически на рис. 50,
наглядно свидетельствует о том, что растворы In—Sb не
субрегулярные. К этому заключению мы пришли на основе
сопоставления рассчитанных с помощью формул (IV. 137) и (IV. 138)
избыточных термодинамических функций смешения с
экспериментальными данными работы [320]. Из этого сопоставления вытекает
вывод о нелинейном характере концентрационной зависимости
энергии взаимообмена в системе индий—сурьма. Поэтому была
предпринята попытка применить шестипараметрическую модель,
предусматривающую квадратичный характер указанной
зависимости. Расчет параметров модели с использованием уравнения (V. 196)
вели путем минимизации суммы квадратов невязок (V.419) методом
прямого поиска. Значения найденных таким путем параметров
приведены в табл. 2. На рис. 51, а показана рассчитанная по
уравнению (V.196) кривая ликвидуса в сопоставлении с
экспериментальными данными [321, 322]. Как видно на рисунке, согласие
достаточно удовлетворительное.
Рассчитанные на основе данных по температурам ликвидуса
при помощи выражений (V.367) и (V.368) значения
термодинамических параметров образования твердого антимонида индия из
жидких индия и сурьмы представлены в табл. 2. Из сопоставления
этих значений с результатами работ [229, 311] следует вывод
У Глазов В. М. и др. 257
Таблица 2
Параметры выбранной модели и результаты термодинамических расчетов на ее основе в различных системах
Системы
Ga—Sb
In-Sb
In—As
Ga—Те
In—Те
GaTe—Те
InTe—Те
Ge—Те *
Sb-Te
Bi—Те
Mg—Pb
Mg—Sn
Mg—Ge
Mg—Si
Cd-Sb
бранная
модель**
6П
KP
6П
СР
— ©о
10 582
15 070
13 060
83 720
125 580
68 022
29 302
95 449
8 372
42 860
33 488
56 511
100 464
42 948
9 628
«»
Дж/моль
— 11 637
—28 779
— 15 070
—83 720
—115 115
—19 622
—19 883
—
—63 836
— 1 338
21 767
29 302
—6 820
—41 516
—
Параметры модели
©2
4 538
36 627
20 93_0
— 179 998
—79 534
—9 209
14 651
—
31 395
22 460
—
—
—
—
—
©01
9,1
—0,046
—0,57
— 15,9
— 14,2
15,1
1,44
—4,8
11,7
—27,6
-4,2
— 11,6
—0,97
-6,3
—
*>»
©81
Цж/(моль-К)
15,8
— 12,4
—0,92
—33,5
—92
-4,3
-7,1
—
—0,42
-1,0
10,7
4,1
6,3
14,6
—
4,9
26,8
0,13
— 100
— 125
—36
3,7
—
8,0
—0,64
—
—
—
—
—
Van,
град
9,3
8,6
16,3
4,3
5,6
9,6
5,3
3,1
9,1
4,1
7,1
15
20
6,9
14,8
кДж/
моль
57,8
57,8
60,3
132
137
167
105
95
141
168
56
98
161
132
36
Дж/
(моль-К)
151
49
33
60
95
123
71
33
65
146
32
55
48
45
33
* Модель выбирали на основе анализа участка ликвидуса в интервале концентраций от 0,5 до 0,667 ат. долей Те.
• * р —. регулярная модель раствора; КР — квазирегулярная; СР — субрегулярная, учитывающая температурную зависимость
энергии взаимообмена; 6П — шестипараметрическая модель.
о хорошем согласии рассчитанных и экспериментальных величин
ДЯ? и AS?.
Следовательно, выбор шестипараметрической модели на данном
этапе, с позиций работ [310, 311] можно считать вполне
удовлетворительным. Однако физико-химический критерий, основанный
только на сравнении теплот и энтропии образования соединения,
как было отмечено выше, не дает возможности выбрать модель.
Поэтому далее были рассчитаны термодинамические функции
смешения и коэффициенты активности компонентов в растворе.
Результаты расчетов в сопоставлении с имеющимися литератур-
1п 0,2 0,Ч0,6Ъ\> 1к\ 0,2 0,4 0,6 0,8 ЪЬ In 0,2 0,4 0,6 0,8 Sb In 0,2 0,4 0,6 0,ff$b
Рис. 51. Результаты расчетов кривой ликвидуса (а) и термодинамических свойств жидких
растворов (б, в, г) в системе In—Sb:
а — сплошная линия — данные работ [219, 321—322]; точки — расчет по уравнению
(IV. 195); б — сплошная линия — расчет по уравнению (IV. 135); в — сплошная линия —
расчет по уравнению (IV. 137); г — сплошная линия — расчет по уравнениям (IV. 139)
и (IV.140); на рис. б, в% г\ 1—5 —данные работ [320]. [324], [317], [323], [230]
соответственно
ными данными представлены на рис. 51, б, в. На рис. 51, б видно
что рассчитанная нами кривая концентрационной зависимости hE
по своему характеру аналогична экспериментальным кривым
[230, 320, 323—324]. Максимум значений hE отвечает составу
#sb = 0,5. По абсолютной величине данные расчета находятся
в пределах ошибок экспериментов. В то же время расчетные
данные Бребрика [311] отличаются по характеру концентрационной
зависимости hE (максимум сдвинут и отвечает составу xSb ^ 0,35).
Следовательно, выбор шестипараметрической модели следует
признать более удачным. Что касается характера концентрационной
зависимости избыточной энтропии смешения, то расчетные данные
не совпадают с экспериментальными [320]. В области составов,
прилегающей к индиевой стороне, избыточная энтропия смешения,
вычисленная по формуле (IV. 147), отрицательна, что, в принципе,
не является невозможным, если учесть сообщения об образовании
в жидкой фазе ассоциатов In3Sb и InSb [324, 325].
Отдельно следует рассмотреть концентрационную зависимость
коэффициентов активности индия и сурьмы (рис. 51, г). Как видим,
рассчитанные из фазовой диаграммы данные по своему характеру
аналогичны экспериментальным [320]. Некоторые
количественные расхождения объясняются скорее всего неточностью
эксперимента [320]. В то же время данные по коэффициентам активности
9* 259.
индия и сурьмы, рассчитанные по шестипараметрической модели,
также согласуются с рядом других расчетных данных [326].
На основании анализа табл. 2 и рис. 51 можно сделать
заключение о достаточной обоснованности выбора
шестипараметрической модели для описания физико-химической природы растворов
системы In—Sb и о возможности на ее основе рассчитать
термодинамические свойства этих растворов, опираясь на надежные
данные по фазовой диаграмме.
Опираясь на выбранную описанным путем модель раствора и
данные детальных термографических исследований кривых
ликвидуса в области составов, прилегающих к соответствующим соеди-
-^,Дж[моль
woo
-£Дж11моль-К)
Лп 0,2 0/4 0,6 0,8 ks In 0,2 0,4 0,6 0,8 As
In 0,2 0,4 0,6 0,8As
Рис. 52. Результаты расчетов кривой ликвидуса (а) и термодинамических свойств (б, в)
жидких растворов в системе In —As:
а — по данным работ [219, 321—322], точки на кривой — расчет по уравнению (V.195);
б — расчет по уравнениям (IV. 135) и (IV. 137); в — расчет по уравнениям (IV. 139) и
(IV.140)
нениям [321 ], мы предприняли расчеты термодинамических
свойств жидких растворов In—As и Ga—Sb. Результаты расчетов
представлены в табл. 2 и на рис. 52 и 53. На рис. 52, б видно, что
избыточная энтропия смешения мала по абсолютной величине, что
качественно согласуется с заключением авторов работы [327] о
регулярности поведения растворов In—As вблизи температуры
плавления арсенида индия. Во всяком случае, можно однозначно
сформулировать, что энергия взаимообмена практически не
зависит от температуры и является сложной (скорее всего,
квадратичной) функцией концентрации.
Представленные на рис. 53, б концентрационные зависимости
избыточных теплот и энтропии смешения растворов Ga—Sb
достаточно хорошо коррелируют с имеющимися экспериментальными
данными по своему характеру, но хуже согласуются по
абсолютной величине. Данные по теплотам смешения [328—330]
отличаются от приведенных на рисунке на величину порядка
2 кДж/моль. Избыточная энтропия положительна и отличается от
данных работы [331 ] не более чем на 2,5 Дж/(моль« К).
Рассчитанные на основе полученных оценок параметров значения
коэффициентов активности (рис. 53, в) занимают промежуточное
положение между экспериментальными результатами работ [331—333].
260
Значения теплот и энтропии образования твердых арсенида индия
и антимонида галлия из жидких компонентов, рассчитанные в
выбранном приближении, заметно отличаются от литературных
данных [229, 316—317, 326], которые, в свою очередь, плохо
согласуются между собой. Величина дисперсии при сравнении
расчетного и экспериментального ликвидуса в системе In—As достаточно
велика (см. табл. 2), тогда как в системе Ga—Sb она находится
в пределах ошибок при термографическом исследовании.
-^Дж/моль зЕ7Дж1(моль-К)
OGgl 0,2 07Ц 0,6 0,8ЪЪ Ga 0,1 0,4 0,6 0,8 Sb Ga 0,2 0,4 0,6 0,8 Sb
Рис. 53. Расчеты кривой ликвидуса (а) и термодинамических свойств (б, в) жидких
растворов в системе Ga — Sb:
а — сплошная линия — по данным работ [219, 321—322], точки на кривой — расчет
по уравнению (V.195); б — сплошные линии — расчет по уравнениям (IV.135) и (IV. 137);
1,2 — данные работ [331], [328] соответственно; в — сплошные линии — расчет по
уравнениям (IV.139) и (IV.140); 1—4 —данные работ [332], [330], [333], [329]
соответственно
Системы Aul— BWI
В качестве представителей этой группы нами рассмотрены
двойные системы галлия и индия с теллуром. По имеющимся
данным [219, 220], в этих системах образуются устойчивые
конгруэнтно плавящиеся моно- и сесквителлуриды.
Учитывая сложный характер фазового равновесия в системах
Ga—Те и In—Те, связанный с наличием областей расслаивания
и инконгруэнтно плавящихся соединений Ga3Te2, GaTe3, In9Te7,
1п3Теб и 1п2Теб, целесообразно выбирать модель раствора на основе
монотеллуридов, охватывающих по первичной кристаллизации
наибольший температурно-концентрационный диапазон. В этой
связи нами были последовательно рассмотрены модели идеального,
строгорегулярного, субрегулярного раствора, шестипараметриче-
ская модель. Результаты расчетов ликвидуса при сопоставлении
с экспериментальными данными, приведенные на рис. 54, а и 55, а,
показывают, что наименьшее расхождение между теорией и
экспериментом наблюдается в случае шестипараметрической модели
(см. также табл. 2). Достаточно хорошее согласие с
экспериментом дает также расчет по субрегуляторной модели, учитывающей
температурную зависимость энергии взаимообмена.
261
Расчеты теплот и энтропии образования монотеллур ид ов галлия
и индия по шестипараметрической модели дают хорошее согласие
с данными работ [229, 334—336]. Рассчитанные
термодинамические свойства были сопоставлены с экспериментальными данными.
In 0,3 0,4 0,5 It In 0,2 0,4 0,6 0,8 Те In 0,2 0,4 0,6 0,8 Те
Рис. 54. Результаты расчетов кривой ликвидуса (а) и термодинамических свойств (б, в)
жидких растворов в системе In —Те:
а — сплошная линия — данные работ [219, 220]; 1—4 — расчет по уравнениям (V.47),
(V.198), (V.196), (V.195) соответственно; б — сплошные линии — расчет по уравнениям
(IV. 135) и (IV.137); /, 2 — данные работ [338], [339] соответственно; в — сплошные
линии — расчет по уравнениям (IV. 139) и (IV. 140), точки — по данным работы [339]
| -в^Дж-моль^К"1
GQ 0,4 0,5 0,6Те • Ga 0,2 0,4 0,6 0,8Те Ga 0,2 0,4 0,6 0,8Те
Рис. 55. Результаты, расчетов кривой ликвидуса (а) и термодинамических свойств (б,- в)
жидких растворов в системе Ga —Те:
а — сплошная линия —данные работ [219, 220]; 1—4 — то же, что и на рис. 54, а;
б.—сплошные линии — расчет по уравнениям (1У.135) и (IV.137); точки — данные
работы [337]; в — сплошная линия — расчет по уравнениям (IV. 139) и (IV. 140), точки —
данные работы [339]
На рис. 54, б и 55, б приведены концентрационные зависимости
избыточных теплот и энтропии смешения при образовании жидких
растворов в сопоставлении с экспериментальными данными работ
[337—339].
Из рис. 54, б следует, что по своему характеру расчетные
данные аналогичны результатам работы [338 ], но отличаются от при-
262
веденных в работе [339] положением максимума. Из-за
существования относительно устойчивого соединения 1п2Те3 (см. работу
[340]) расчетные данные и результаты работы [338] следует
признать более надежными. Что касается некоторых количественных
расхождений, то, учитывая значительные ошибки при
экспериментальных оценках теплот смешения, можно считать, что они
относительно невелики. Из рис. 55, б следует, что рассчитанные нами
по формуле (VI. 145) теплоты смешения достаточно хорошо
согласуются с данными Кастанета и Бергмана [337 ] по общему
характеру концентрационной зависимости и по абсолютной величине.
На рис. 54, в и 55, в показаны концентрационные зависимости
коэффициентов активности компонентов в рассматриваемых
системах, вычисленные в избранном приближении по формулам (IV. 149)
и (IV. 150).
Сопоставление этих кривых с данными Пределя с соавторами
1339], полученными на основе измерений упругости пара,
показывает заметное расхождение. Однако, учитывая картину,
представленную на рис. 54, б и 55, б, данные работы [339 ] следует
признать недостаточно надежными, что может быть, кроме того,
обусловлено неучетом сложного состава паровой фазы при оценке
коэффициентов активности.
Пользуясь выбранной моделью раствора, мы произвели
соответствующие расчеты для частных систем GaTe—Те и InTe—Те.
Переход к частным системам обусловлен относительной
стабильностью монотеллуридов галлия и индия в жидкой фазе [340,
341 ]. Параметры энергии взаимообмена, а также
термодинамические функции образования твердых сесквителлуридов из жидких
теллура и монотеллуридов представлены в табл. 2. Сопоставление
энтальпий образования сесквителлуридов галлия и индия с
данными работ [229, 334, 342, 343] показывает хорошее согласие,
что свидетельствует в пользу правильности выбора шестипара-
метрической модели.
Системы Л1У—5VI
В наших исследованиях данная группа была представлена
системой Ge—Те. По имеющимся сведениям [219—220], в этой
системе имеется одно конгруэнтно плавящееся соединение и
предполагается существование инконгруэнтного соединения GeTe2
[344]. Монотеллурид германия отличается односторонним
характером области первичной кристаллизации [345], настолько, что
в работе [70 ] это соединение предполагали плавящимся в
переходной точке. На самом же деле соединение плавится конгруэнтно,
однако левая ветвь ликвидуса, соответствующая первичной
кристаллизации монотеллурида германия, чрезвычайно мала. Анализ
правой ветви ликвидуса в различных приближениях показывает
(рис. 56, а), что в концентрационных пределах от 0,5 до 0,67
атомной доли теллура ликвидус хорошо описывается при помощи всех
263
моделей, кроме идеальной. Величина дисперсии не превышает
обычных значений ошибки при термическом анализе. Однако при
*те > 0,67 во всех случаях наблюдаются резкие расхождения
между расчетом и экспериментом, что может быть обусловлено
существованием дителлурида германия, либо какими-нибудь
другими причинами. Во всяком случае, можно только на основании
проведенных расчетов сделать однозначный вывод о
необходимости уточнения характера фазового равновесия в районе хТе = 0,67.
Из сопоставления термодинамических функций образования моно-
Ge 0,5 0,6 0,7 0,8Те ,Ge 0,2 0,4 0,6 0,6 Те Ge 0,2 0,4 0,6 0,8Те
Рис. 56. Результаты расчетов кривой ликвидуса (а) и термодинамических свойств (б, в)
расплавов в системе Ge—Те:
а — сплошная линия — данные работ [219, 220, 345] (1—4 — то же, что и на рис, 54, а);
б, в — расчет на основе уравнений (IV.97) и (IV.98)
теллурида германия, рассчитанных в различных приближениях,
с экспериментальными данными работ [229, 346—347] следует
вывод о том, что квазирегулярная модель — наилучшая и,
следовательно, может быть использована для описания
термодинамических свойств растворов в этой системе. Результаты расчетов
избыточных термодинамических функций и коэффициентов
активности германия и теллура для квазирегулярной модели в
зависимости от состава приведены на рис. 56, б и 56, в соответственно.
Отсутствие литературных данных не позволяет сравнить расчет
с экспериментом и делает результат, представленный на рис. 56,
пока единственным.
Попытки описания других систем Alw— BVI успехом не
увенчались. Одной из возможных причин этого может быть сложная
схема диссоциации соответствующих соединений.
Системы Лу— BVI
В качестве представителей этого класса нами рассмотрены
системы Bi—Те и Sb—Те, в которых образуются сесквителлуриды
соответственно висмута и сурьмы, получившие широкое
практическое распространение как термоэлектрические материалы.
264
Участок ликвидуса в области составов, примыкающих к
соединениям Bi2Te3 HSb2Te3, был детально изучен нами термографически.
При анализе использовались также данные других авторов,
систематизированные в работе [336]. На рис. 57, а и 58, а приведены
',«
850
800
750
700
-&&
ь-
CNJ
СО
"о/
*2
-«J
_|
а
"**§?*
\\
*\ Ц
V
V
1 1— 1
w
щ
BL 0,5 0,6 0,7 0,8 0,9Те BL 0rZ 0,4 0,6 0,8 Те BL 0,2 0,4 0,6 0,8 Те
Рис. 57. Результаты расчетов кривой ликвидуса (а) и термодинамических свойств (б, в)
расплавов в системе Bi — Те:
а — сплошная линия — экспериментальная; 1—4 — то же, что на рис. 54, а; б *-
сплошные линии — расчет по уравнениям (IV. 135) и (IV. 137); /, 2 — по данным работ [350]
и [349] соответственно; в — сплошные линии — расчет по уравнениям (IV.139) и (IV.140);
точки — экспериментальные [351 ]
^ -8*ДК/(М0ЛЬ-К)
LK
850
800
750
700
&ж
~~ /ж
"о/ £
*z <°
-•j
*<*
i
*T"T !
\v
V
W
а
0,8
0,6
W
о,г
- Sb Vs.
X
1 ~|
6/\
/ ы
Х^ 1 \
Sb 0,4 0,6 0,8Те Sb 0,2 0,4 0,6 0,8 Те Sb 0,2 0,4 0,6 0,8 Те
Рис. 58. Результаты расчетов кривой ликвидуса (а) н термодинамических свойств (б, в)
расплавов в системе Sb—Те:
а — сплошная линия — экспериментальная по данным авторов и работы [164]; 1—4 —
то же, что и на рис. 54, а: б — сплошные линии — расчет по уравнениям (IV.135) и
(IV. 137), точки — экспериментальные [338]; в — расчет по уравнениям (IV. 139) и (IV. 140)
участки кривых ликвидуса, отвечающие первичной
кристаллизации сесквителлуридов висмута и сурьмы, которые были
рассчитаны в различных приближениях. На этих же графиках
представлены экспериментальные данные, из сравнения с которыми
следует, что расчет в приближении шестипараметрической модели
дает минимальное отклонение. Средняя дисперсия (см. табл. 2)
укладывается в пределы ошибок термографического определения
265
кривых ликвидуса. Значения найденных при этом параметров
энергии взаимообмена представлены в табл. 2, где приведены
также рассчитанные по температурам ликвидуса с помощью
уравнений (V.367) и (V.368) значения энтальпий и энтропии
образования сесквителлуридов висмута и сурьмы. Из сопоставления этих
величин с полученными термодинамическими расчетами на основе
стандартных термодинамических функций, приведенных в
справочнике [348], можно заключить, что расхождения укладываются
в пределы ошибок при оценке рассматриваемых величин. Таким
образом, согласно работе [317], шестипараметрическую модель
можно считать приемлемой для описания фазового равновесия и
термодинамических свойств сплавов в системах Bi (Sb)—Те. Однако
для надежного заключения о правильности выбираемой модели
мы считаем целесообразной увязку рассчитываемых на ее основе
термодинамических свойств растворов в широком интервале
концентраций с экспериментальными данными, если они имеются.
На рис. 57, б представлена рассчитанная нами кривая
концентрационной зависимости теплоты смешения в системе Bi—Те,
которая практически идеально согласуется - с
экспериментальными данными Микавы с соавторами [349], а также Блачника и
Эннинга [350]. Результаты расчетов коэффициентов активности
висмута и теллура на основе фазовой диаграммы в шестипараме-
трической модели приведены на рис. 57, в, где представлены для
сравнения имеющиеся экспериментальные данные Бребрика,
полученные на основе результатов измерений давления пара [351 ]
с использованием уравнения температурной зависимости давления
пара чистого теллура [239]. Как видим, результаты расчетов
вполне удовлетворительно согласуются с экспериментом. Расчет
избыточной энтропии смешения (рис. 57, б) показывает, что она
отрицательна и по абсолютной величине относительно невелика.
Расчеты избыточных термодинамических функций смешения
в системе Sb—Те представлены на рис. 58, б в сопоставлении с
экспериментальными данными работы [338]. Видно, что расчет
прекрасно согласуется с экспериментом, в связи с чем выбор модели
следует в данном случае признать удачным и результаты расчетов
коэффициентов активности сурьмы и теллура, представленные на
рис. 58, б, можно рекомендовать как достаточно надежные.
Избыточная энтропия смешения в данной системе весьма незначительна
по абсолютной величине, но положительна по знаку, что указывает
на преобладание эффекта роста энтропии при смешении по
сравнению с уменьшением в результате упорядочения, обусловленного
химическим взаимодействием. В рассмотренной выше системе Bi—
Те картина обратная. На первый взгляд, это можно расценивать
как некоторое несоответствие, особенно, если учесть относительно
более высокую термическую стабильность теллурида сурьмы по
сравнению с теллуридом висмута [352—353]. Однако следует
учитывать суммарный эффект взаимодействия в жидкой фазе, который
в системе Bi—Те оказывается более выраженным из-за явного
266
пройблеййя МонотеллурйДа бисмуга как химического йнди&ида
в жидкой фазе [352], несмотря на то, что это соединение плавится
инконгруэнтно [336], чего в случае системы Sb—Те не
наблюдается [352].
Подводя некоторый итог анализу фазового равновесия и
термодинамических свойств сплавов в системах Bi (Sb)—Те, можно
отметить, что по шестипараметрической модели рассмотренные
системы описываются вполне удовлетворительно, и, следовательно,
ее можно рекомендовать для использования в данном случае и
для аналогичных систем с селеном.
Системы An—Blv
Представителями этой группы в наших исследованиях служили
двойные системы магния с кремнием, германием, оловом и
свинцом, в которых образуются конгруэнтно плавящиеся соединения
Mg2BIv.
Подход к выбору модели в этих системах такой же, как и в
предыдущих случаях. На рис. 59—62 представлены участки кривых
ликвидуса, рассчитанные в различных приближениях. Здесь
же приведены экспериментальные кривые, заимствованные из
работ [219—223, 322]. Анализ показывает, что модель
субрегулярного раствора, учитывающая температурную зависимость энергии
взаимообмена, и шестипараметрическая модель практически
одинаково хорошо описывают кривые ликвидуса, отвечающие
первичной кристаллизации соединений Mg2BIV (см. также рис. 44, б).
Повышенные значения дисперсии (см. табл. 2) в системах
Mg—Sn и Mg—Ge обусловлены, по нашему мнению, применением
неточных значений теплот плавления соответствующих
соединений, заимствованных из работ [354—355].
Расчет термодинамических параметров образования
соединений Mg2BIV на основе данных по температурам ликвидуса с
привлечением двух указанных моделей дает мало отличающиеся
результаты. Сравнение расчетных значений энтальпий и энтропии
образования соединений Mg2BIV с экспериментальными данными
работ [335, 356—359] показывает, что они хорошо согласуются
между собой. Поэтому для выбора модели, как и в случае
соединений ЛШБУ, удачное описание кривых ликвидуса и достаточно
хорошая оценка термодинамических функций образования
соединений Mg2BIV оказываются недостаточными.
Для окончательного решения вопроса о наиболее подходящей
модели раствора в рассматриваемых системах мы сопоставили
избыточные термодинамические функции смешения и
коэффициенты активности компонентов, рассчитанные в двух
приближениях, с экспериментальными данными. В качестве опорной в
данном случае была избрана система Mg—Sn, расплавы которой
наиболее подробно изучены в работах [144, 222, 360—363], причем
данные разных авторов хорошо согласуются между собой. В ре-
267
зультатё такого срайнейия оказалось, Wo модель субрегулярйоГО
раствора, учитывающая температурную зависимость энергии вза»
имообмена, позволяет рассчитать концентрационную зависимость
энтальпии смешения и коэффициентов активности, прекрасно
-"*,
70 \
15
10 \
м\
кДж/моль
6
/^
/iO
' ^\\
'——i— 1 i i ч
а
0,8
0,6
0,4
0,2
- \\N
Zjl
в J
ХА
Mg tf,7 0,3 0,5 0,7Si Mg 0,2 ОМ 0,0 0,8 Si Mg 0,2 0,4 0,6 0,8 Si
Рис. 59. Результаты расчетов ликвидуса (а) и термодинамических свойств (б, в)
расплавов в системе Mg—SI:
а — сплошная линия — экспериментальная по данным работ [219, 220, 223]; 7—3 —
расчет по уравнениям (V.49), (IV.196) и (IV. 197) соответственно; б — сплошная линия —
расчет по уравнениям (IV. 129) и (IV. 130); в — сплошные линии — расчет по
уравнениям (IV. 131) и (IV. 132); на рис. б, в — 1—3 — экспериментальные данные работ [223],
[361], [365] соответственно
-ЬЕ,кДж/моль
/,*
1300
1100
900
700
1М
о2
• J
CD
CD
СП
21
L
-^^ а
>& ^ч>
\
1 1
а
0,8
0,6
о,ч
0,2
Мд\
/ « -»
6/f
X /Ge
^un 1 \
Мд 0,1 0,3 0,5 0,7to Мд 0,2 0,4 0,6 0,8 Ge Мд 0,2 0,4 0,6 0,8 Ge
Рис. 60. Результаты расчетов ликвидуса (а) и термодинамических свойств (б, в)
расплавов в системе Mg—Ge:
а — сплошная линия — экспериментальная по данным работ [219, 222]; 1—3 — то же,
что и на рис. 59, а; б, в — сплошная линия — то же, что и на рис. 59, б; в; точки —
экспериментальные [366]
согласующуюся с экспериментальными данными (см. рис. 61, б, в).
В то же время шестипараметрическая модель дает результаты,
существенно отличающиеся от указанных данных, особенно по
коэффициентам активности. В итоге можно сделать однозначный
выбор в пользу модели субрегулярного раствора, учитывающей
температурную зависимость энергии взаимообмена. Заметим, что
и в остальных системах рассматриваемой группы сравнение
расчетных и экспериментальных данных [144, 221—223, 359, 361 —
268
362, 364—367] оказывается в пользу модели субрегулярного
раствора, хотя экспериментальные данные в системах Mg—Si и Mg—Ge
менее убедительны по сравнению с системой Mg—Sn. Возвращаясь
к первому этапу выбора модели, связанному с оценкой параметров
-в^Дж/шль-К)
Щ0,2 0УЧ 0,6 %8Sn Щ 0,2 0,4 076 0,8 Sn Ид 0,2 0,4 0,6 0,8Ъг\
Рис. 61. Результаты расчетов ликвидуса (а) и термодинамических свойств (б, в)
расплавов в системе Mg—Sn:
а — сплошная линия — экспериментальная по данным работ [219, 222]; 1—3 — то же,
что и на рис. 59, а\ б, в — сплошная линия — то же, что и на рис. 59, б, в; 1—3 —
экспериментальные данные работ [361], [360], [363] соответственно
Рис. 62. Результаты расчетов ликвидуса (а) и термодинамических (б, в) свойств
расплавов в системе Mg—Pb:
а — сплошная линия — экспериментальная, по данным работы [221], /—3 —то же,
что и на рис. 59, а; б, в — сплошная линия ~ то же, что и на рис. 59, б, в; 1—4 —
экспериментальные данные работ [363], [365], [359], [364] соответственно
модели и термодинамических функций образования соединений
Mg2BIV, мы отдаем предпочтение соответствующим величинам,
рассчитанным по модели субрегулярного раствора (см. табл. 2).
Рассчитанные в данном приближении экспериментальные
кривые концентрационной зависимости избыточной энтропии
смешения во всех рассмотренных системах имеют сложный характер,
однако его толкование едва ли целесообразно, если учесть, что
269
по абсолютной'величине избыточные энтропии смешения очень
малы [менее 1 Дж/(моль-К)1.
Однако на*основе данных|по давлению пара в работе [361],
значения избыточной энтропии смешения получаются
отрицательными по знаку и значительными по абсолютной величине [от —1,7
до —7,5 Дж/(моль-К) в точке максимума]. Учитывая сложный
характер испарения сплавов в рассматриваемых системах, сделать
однозначное заключение в пользу данных работы [361 ]
затруднительно.
В то же время, опираясь на собственные расчеты и учитывая
данные работ [144, 356, 360, 362—365], можно даже в принципе
пренебречь избыточной энтропией смешения и при расчетах
ограничиться субрегулярной моделью.
Таким образом, на основе проведенного анализа можно
заключить, что для описания фазового равновесия и термодинамических
свойств сплавов в системах Mg—Blv следует рекомендовать
модель субрегулярного раствора.
Системы Au—Bv
В данной группе нами подробно рассмотрена система Cd— Sb.
Интерес к этой системе обусловлен возможностью получения
полупроводникового моноантимонида кадмия, который широко
применяют в электронной технике [368]. Из рассмотрения фазовой
диаграммы системы кадмий—сурьма следует, что, помимо
стабильного моноантимонида кадмия, возможно образование мета-
стабильных фаз более сложного состава, причем возможность
реализации метастабильных равновесий существенно зависит от
кинетики процесса кристаллизации. Поэтому подходить к анализу
данной системы при опоре на равновесную диаграмму следует
с осторожностью, учитывая возможность смещения истинных
температур фазовых переходов из-за легкости отхода от равновесного
течения этого процесса.
Последовательный расчет кривой ликвидуса, отвечающего
первичной кристаллизации моноантимонида кадмия с
использованием различных моделей, показывает (рис. 63, а), что модели
субрегулярного раствора и шестипараметрическая наиболее близки
к имеющимся экспериментальным данным. Модель строго
регулярного раствора показывает отклонение от экспериментального
ликвидуса в среднем на 14,3 К. Расчеты термодинамических
параметров образования антимонида кадмия из компонентов во всех
приближениях, кроме идеального, дают практически не
отличающиеся между собой величины, которые идеально согласуются с
данными Хальтгрена [359] и близки к приведенным в справочнике
Глушко [229]. Следовательно, на первом этапе анализа в данном
случае трудно отдать предпочтение какой-либо из моделей,
поскольку величина дисперсии по ликвидусу при учете ненадежности
экспериментальных данных не может играть существенной роли.
270
Расчеты избыточных термодинамических функций смешения и
коэффициентов активности кадмия и сурьмы показывают, что
наилучшее согласие с экспериментальными данными работ [369—375]
дает модель строго регулярного раствора. Таким образом,
создается противоречивая ситуация. С одной стороны, модель строго
регулярного раствора наилучшим образом описывает
термодинамические свойства расплавов системы кадмий—сурьма во всем
диапазоне концентраций, а с другой, — она хуже описывает
экспериментальный ликвидус по сравнению с шестипараметрической и
-Г1Е7кД?к1моль
Са 0,2 0,4 0,6 Sb Cd 0,2 0,4 0,6 0,8 Sb Cd 0,2 0,4 0,6 0,8 Sb
Рис. 63. Результаты расчетов кривой ликвидуса (а) и термодинамических свойств (б, в)
расплавов в системе Cd —Sb:
а — сплошная линия — экспериментальная по данным работ [219, 3681; 1—4 — то же,
что и на рис. 54, с; б т- сплошная линия — расчет по уравнению (IV. 104); в —
сплошная линия — расчет по уравнениям (IV.97) и (IV.98). На рис. б, в — 1 — 7 —
экспериментальные данные работ [369], [372], [373], [374], [370], [371], [375] соответственно
субрегулярной моделями. В связи с этим нами дополнительно
исследовано давление пара над расплавами различного состава
с использованием метода точек кипения. Полученные результаты
прекрасно согласовались с результатами более
высокотемпературных измерений давления пара, выполненных в работе [375] с
использованием метода потока. Данные наших измерений и
результаты работы [376] были совместно обработаны на ЭВМ, и на основе
полученных результатов рассчитаны коэффициенты активности,
которые хорошо согласовались с экспериментальными данными,
полученными методом э. д. с. [370]. Следовательно, результаты
исследований термодинамических свойств расплавов кадмий—
сурьма следует признать более надежными, нежели кривая
ликвидуса, построенная методом термического анализа.
На этом основании можно сделать выбор в пользу модели
строго регулярного раствора и использовать ее для надежного
расчета диаграммы фазового равновесия.
Для большей надежности использования строго регулярной
модели целесообразно использовать прием, связанный с оценкой
энергии взаимообмена на основе различных экспериментальных
данных. Нами были использованы объединенные данные по давле-
271
нию пара, теплотам смешения [373, 377] и данные, полученные
методом э. д. с. [370].
Во всех трех случаях были получены значения энергии
взаимообмена, малоотличающиеся между собой (в среднем около
14,6 кДж/моль). Рассчитанный на основе этих данных ликвидус,
отвечающий первичной кристаллизации моноантимонида кадмия,
показан на рис. 63, а. Рассчитанные в строго регулярном
приближении избыточные термодинамические функции смешения и
коэффициенты активности в сопоставлении с данными работ [369—
371, 373, 374, 377] и наших собственных исследований по
давлению пара приведены на рис. 63, б, в.
Проведенные расчеты нельзя рассматривать как окончательный
выбор модели раствора в рассмотренных системах. Авторы отдают
себе отчет в том, что предпринятая ими попытка расчета
термодинамических свойств растворов на основе данных по фазовым
равновесиям в системах с конгруэнтно плавящимися соединениями
требует дальнейшего уточнения, поскольку расчеты показали,
что выбор модели на основе данных по фазовым равновесиям
представляет собой многоэкстремальную задачу, успешное решение
которой в большой мере зависит от выбора метода минимизации,
вида функционала и других исследований.
Г л а в а VI
ТЕРМОДИНАМИЧЕСКИЕ ИССЛЕДОВАНИЯ
ДИССОЦИАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ
1. ФОРМА И ПОЛОЖЕНИЕ МАКСИМУМА
НА КРИВЫХ ЛИКВИДУСА И СОЛИДУСА
Громадный экспериментальный материал, накопленный
школой Н. С. Курнакова, позволил ему высказать положение,
согласно которому форма (радиус кривизны) и положение максимума
кривых плавкости тесно связаны со степенью диссоциации
химического соединения на образующие его компоненты [45, 378].
Совершенно недиссоциирующему при плавлении соединению
отвечают острые (сингулярные) максимумы кривых ликвидуса и соли-
дуса (радиусы кривизны кривых в этой точке равны нулю) (см.
рис. 21, а, б). Если соединение диссоциирует лишь в жидкой фазе,
кривая ликвидуса приобретает плавный максимум, а кривая соли-
дуса сохраняет острый (рис. 21, в, г). Если диссоциация проходит
в той и другой фазах, оба максимума плавные (рис. 21, д). С
увеличением степени диссоциации соединения в одной из фаз возрастает
радиус кривизны соответствующей кривой.
Школой Н. С. Курнакова было экспериментально показано, что
для некоторых диссоциирующих соединений состав максимума не
272
совпадает с его химической формулой, в связи с чем соединения
были подразделены на дальтониды и бертоллиды [45].
Отмеченная выше связь формы и положения максимума кривых
ликвидуса и солидуса со степенью диссоциации химического
соединения термодинамически обсуждалась в работе [20].
На этом основании Крегер [198] предлагает аппроксимировать
кривые ликвидуса и солидуса соединений, диссоциированных в
различной степени, конкретными математическими выражениями
(гипербола при частичной диссоциации и парабола при полной
диссоциации).
Несмотря на большое значение, которое имеет ход кривых
моновариантного равновесия вблизи состава соединения для
теории физико-химического анализа, до недавнего времени ему не
уделялось достаточного внимания. По существу имеется
единственная обобщающая работа О. А. Есина [379], в которой
предпринята попытка восполнить этот пробел.
Рассмотрим вопрос, вытекает ли форма максимума на кривых
ликвидуса и солидуса из общих термодинамических
положений или для этого необходимо привлечь дополнительные
данные.
Гиббс показал [1], что пологий максимум кривых
температура — состав вытекает из общих термодинамических положений,
в то время как существование острого максимума не доказано
термодинамически, и базируется на тех же экспериментальных
результатах, что и теория предельно разбавленных растворов.
Несмотря на это, вопрос о возможности термодинамического
обоснования существования сингулярного максимума продолжал
время от времени обсуждаться в литературе.
Так, в 1889—1892 гг. между Ле-Шателье и Розебомом [380—
382] состоялась дискуссия о форме максимума для СаС12-6Н20
на диаграмме плавкости системы СаС12—Н20. В итоге, авторы
сошлись на том, что точность эксперимента недостаточна для
решения вопроса. Кроме того, Ле-Шателье правильно указал, что
общие термодинамические положения не могут дать однозначного
ответа на этот вопрос.
Вид диаграммы состояния с образованием соединения типа
АтВп может быть получен с помощью геометрического метода на
основе рассмотрения кривых изобарно-изотермического
потенциала. При этом, как подробно обсуждалось ранее (см. п. 3, гл. III),
априори следует допустить наличие или полное отсутствие
диссоциации соединения в растворе, в зависимости от чего вид кривой
изобарно-изотермического потенциала будет тем или другим (см.
рис. 6 и 19). Поэтому, как справедливо отмечено в работе [383],
такой подход также не дает строгого термодинамического решения
вопроса о форме кривой максимума соединений. Вид кривой
изобарно-изотермического потенциала при отсутствии диссоциации
соединения на компоненты длительное время служил предметом
острой научной дискуссии (см. работы [53, 384, 386, 387]).
273
Ван-дер-Ваальс и Констамм [12], полемизируя с Планком
[2], писали: «О составе определенных фаз термодинамика может
сказать столь же мало, как мало она может ответить на вопрос,
какие химические вещества существуют и в каких фазах они могут
существовать». И далее: «Из наших выводов никоим образом не
вытекает, что всякое вещество способно находиться в равновесии
с другими веществами и соответственно получаться обратимым
путем из других веществ».
Таким образом, существование сингулярного, или острого,
максимума не вытекает непосредственно из общих
термодинамических положений, но и не отрицается ими. В качестве
предельного случая допускают существование совершенно недиссоцииро-
ванного на компоненты соединения (степень диссоциации а° равна
нулю).
Анализ двойных диаграмм состояния с конгруэнтно
плавящимися химическими соединениями показывает, что в ряде случаев
максимум кривой плавкости отклоняется от стехиометрического
состава. Соединения с заметным отклонением от стехиометрии
были названы Н. С. Курнаковым бертоллидами. При этом было
отмечено, что бертоллиды представляют собой промежуточные
фазы переменного состава на основе диссоциированного
химического соединения [45]. Позднее с учетом данных о непрерывном
переходе от дальтонида к бертоллиду было показано, что
принципиального различия между ними нет и имеющиеся различия в
характере диаграмм плавкости — следствие особенностей
межфазового (гетерогенного) и внутрифазового (химического) равновесия
[379, 388]. В ряде работ [20, 389, 390] отмечена связь формы и
положения максимума на диаграмме плавкости со степенью
диссоциации соединения. Однако аналитическая зависимость в явном
виде между величинами отклонения от стехиометрии и степени
(либо константы) диссоциации в указанных работах не выведена.
Большой шаг вперед в количественном решении данной задачи
представляют работы А. Б. Млодзеевского [391—392] и О. А. Еси-
на [379, 393]. Исходя из теории идеальных ассоциированных
растворов, А. Б. Млодзеевский получил количественную связь между
сдвигом максимума и степенью диссоциации. О. А. Есин уточнил
решение А. Б. Млодзеевского и обобщил его на случай реальных
систем [379, 393].
Исходя из условий фазового равновесия (11.80), для двухком-
понентной двухфазной системы можно записать
И* — }4 = [Ав — [Ал- (VI. 1)
Введя коэффициенты активностей соответствующих
молекулярных форм улху Увг и уав, можно записать химический
потенциал /-того компонента и закон действующих масс в виде
\1в = \1в (7\ р) + RT In [(хв - и)1{\ - и)] yBl (VI.2)
274
и аналогичного соотношения для компонента А:
(\-Хв- и) (хв - и)/и (1 - и) = Куав/уа1Ув1 = К\ (VI.3)
где и—число молей АВУ приходящееся на один брутто-моль
фазы.
На основании формул (VI.2) и (VI.3) любую из разностей (VI. 1)
можно представить в виде
VB - JM = \L°B -\L°A + RT 1П [(XB - Uflu (1 — И) /С'] +
+ RT In (vAjyBi). (VIA)
В точке максимума
где б — отклонение максимума на кривой плавкости от стехио-
метрического состава.
Подставляя последние соотношения в формулу (VI.4) и
принимая во внимание равенство (VI. 1), находим
лГкУ» VK'^+tf +28J/Jt'w+1 = expf *Д~М
V K{L) |//('(S) +462 +26(/^(S) +1 ' Р\2#Гтах j'
(VI.5)
где qA = №S) + RT In vi) - (^(S) + Я Г In Yl),
Яв - (^(L) + ЯГ In ^) - (^<L) + RT In tft). (VLb)
В соответствии с выражением (IV.78)
КаУаМ^К'а, KBykJySBt = KB, . (VI.7
/Сг* — кажущийся коэффициент распределения *-того компонента.
Принимая во внимание соотношения (VI.7), из выражения
(VI.5) получаем окончательный результат:
l/^ Kg^+g + 26/gT^ = / ^Г
У /С (L) J/*' <s) + 4б2 + 26 VK {S) + 1 У Кв
Из этого уравнения в предположении, что изменения
коэффициентов активности малы по сравнению с K'isK K'{L\ Ка и Kb
непосредственно следует, что б только тогда отлична от нуля,
когда соединение диссоциирует в обеих фазах (K'{S) > 0 и /C(L) >
>>0) или коэффициенты распределения компонентов между
фазами не равны (вчастности, К'аФКв)- Но эти особенности как раз
и характерны для бертоллидов и промежуточных между ними и
дальтонидами соединений.
Наоборот, если, например, Ка = #в, и правая часть уравнения
(VI.8) равна единице, то левая часть может стать равной ей при
любых значениях констант диссоциации (и в частности, K'{S) и
K'^L)) только тогда, когда 6 = 0, так как K'{L) всегда больше K'{S)-
275
Другими словами, равенства коэффициентов распределения
компонентов вполне достаточно, чтобы при любых степенях
диссоциации соединения в обеих фазах сдвиг максимума отсутствовал.
Наконец, если хотя бы одна из констант диссоциации равна
нулю, например в твердой фазе, то при любых коэффициентах
распределения, для того чтобы не вступить в противоречие с
уравнением (VI.8), нужно положить 6 = 0. Следовательно, чтобы
максимум не сдвигался, достаточно того, чтобы соединение
совершенно не диссоциировало хотя бы в твердой фазе.
Взаимосвязь величины сдвига максимума с константой
диссоциации была получена О. А. Есиным [379, 393] несколько иным
путем в виде
a-xa-itf— *Ьл*л-Хв) (VI9)
2[2{l-KA) + xLAi(KA-KB)] ^ }
Из анализа этого соотношения следует, что состав максимума
будет совпадать с формулой химического соединения тогда, когда
либо оно не диссоциировано в твердой фазе, либо коэффициенты
распределения между фазами одинаковы. Выполнение одного из
этих условий обеспечивает отсутствие сдвига.
Сдвиг будет наблюдаться при одновременном соблюдении двух
условий: диссоциации соединения в обеих фазах и неравенства
коэффициентов распределения. Абсолютное значение сдвига тем
больше, чем сильнее диссоциация соединения в жидкой, а
следовательно, и в твердой фазе и чем больше разность коэффициентов
распределения компонентов между фазами (Кл—Кв)- Максимум,
как следует из уравнения (VI.9), сдвигается (от 0,5) в сторону
компонента, коэффициент распределения которого больше.
Эти выводы легко обобщить на случай образования любого
соединения А с В сложного состава [379].
Позднее возможность отклонения максимума кривой плавкости
от стехиометрического состава соединения была доказана Цер-
нике [394] на основе модели «диссоциации» с использованием
теории понижения точки замерзания. В работах Ходкинсона [395,
396] приведено аналогичное объяснение этого явления на основе
молекулярной модели разупорядочения.
2. ТЕРМОДИНАМИЧЕСКОЕ ОБОСНОВАНИЕ РАЗЛИЧНЫХ
ПУТЕЙ ОЦЕНКИ ПАРАМЕТРОВ ДИССОЦИАЦИИ
КОНГРУЭНТНО ПЛАВЯЩИХСЯ СОЕДИНЕНИЙ
В литературе имеется ряд сообщений, в которых предприняты
попытки математически обосновать взаимосвязь ликвидуса со
степенью диссоциации конгруэнтно плавящегося соединения.
Так, по инициативе Н. С. Курнакова, Н. В. Липин попытался
[397, 398] математически описать кривую плавкости
диссоциирующих соединений. Пользуясь формально математическим мето-
276
дом, Н. В. Липин вывел уравнение кривой плаб кости бинарногб
соединения, в котором степень диссоциации представлена как
функция температуры и параметра диссоциации (К) в неявном
виде, в связи с чем для практических расчетов это уравнение
оказалось непригодным. Упомянутая задача в явном виде впервые
была решена Н. И. Степановым [399], который получил уравнение
кривой плавкости бинарного соединения, диссоциированного
в жидкой фазе, исходя из анализа поверхности выделения
двойного соединения в тройной системе.
Однако применение уравнения Н. И. Степанова для расчета
константы диссоциации химического соединения из вида диаграммы
плавкости требует знания двух гипотетических величин: теплоты и
температуры плавления соединения при отсутствии диссоциации,
которые нельзя определить экспериментально. Это уравнение,
по-видимому, можно использовать в случае очень малой
диссоциации.
Ван-дер-Ваальс и Констамм [12], исследуя зависимость
наклона касательных к кривым фазовых равновесий от степени
диссоциации, использовали полученное ими уравнение (11.182)
в форме
Для того чтобы обойти трудности, связанные с непосредствен-
ным анализом реальных систем, авторы разбили величину
термодинамического потенциала g на две части. Одна из частей
справедлива для весьма разреженного состояния фазы, а другая является
поправкой на «конденсированность». В результате получается
выражение
\д#~ )р, т = ^Т [ 1-х —и + Т^П ""
(2x-l)*u(x-u)(l-x-u) I ,VT « n
(х — w)2(l— x — и)2ф J> VVi-11;
где Ф = и (x — и) + и (1 — x — и) + (х — и) (1 — х — и).
Если теперь обратиться к точке диаграммы, где брутто-составы
фаз одинаковы, т. е. там, где xL = xs, то согласно уравнению
(VI. 10), производная (дТ/дх)р может не быть равной нулю только
при условии, что вторая производная (d2g/dx2)pf т бесконечна. Как
видно из соотношения (VI. 11), это условие возможно в том случае,
когда либо 1 — xL — uL = 0, либо xL — uL == 0. Но эти условия
означают, что в точке максимума соединение А В совершенно не
диссоциировано на компоненты. Истинное число молей его равно
аналитическим мольным долям компонентов uL = 1 —xL = xL = V2.
По мнению Ван-дер-Ваальса и Констамма, до тех пор, пока
выкристаллизовывающееся соединение диссоциировано хотя бы
в малейшей степени, (дТ/дх)р = 0 для xL = xs и максимум кривой
277
ликвидуса пологий. Отсюда не следует, однако, что ход кривой
Т = f (х) всегда останется одинаковым независимо от степени
диссоциации соединения и что для совершенно недиссоциирован-
ного соединения эта кривая скачкообразно достигает резкой
вершины. Наоборот, из хода кривой Т = f (x) вблизи максимума
можно сделать заключение о степени диссоциации.
Для иллюстрации непрерывности перехода от плавного
максимума к острому с падением степени диссоциации Ван-дер-Ваальс
и Констамм на основе выражения (VI. 11) рассмотрели частный
пример соединения, степень диссоциации которого равна нулю
в твердой фазе и очень мала в жидкой. Полученное авторами
уравнение для касательной в точке, близкой к максимуму, имеет вид:
(-q(s->D/RT2) (pridx)L = 3/2 - (5а0/4 Axt), (VI. 12)
где и0—число продиссоциировавших молей соединения АВ;
AxL = 1/2 — xLt причем AxL < 1, а и0 < kxL.
Из соотношения (VI. 12) видно, что для малодиссоциированных
соединений производная в точке, не слишком далекой от максимума,
тем больше, чем меньше степень диссоциации. В пределе, когда
ио = О, уравнение (VI. 12) дает выражение, отличное от обычного,
вытекающего из теории предельно разбавленных растворов, что,
видимо, связано с рядом допущений, сделанных при его выводе.
Вследствие этого данное уравнение имеет приближенный
характер, а его практическое применение затруднено [379].
Шоттки, Улих и Вагнер [20] исследовали формы кривых
ликвидуса и солидуса и их взаимную связь также в предположении
малой степени диссоциации соединения в терминах упорядочения
и разупорядочения. Однако степень или константа диссоциации
соединения не входят в явном виде в их уравнения, а сделанные
формально математические упрощения не позволяют точно отнести
разбираемый ими случай к какому-либо из обычных классов
растворов (предельно разбавленных, идеальных, совершенных).
Только в случае предельно разбавленного жидкого раствора
компонентов в химическом соединении, диссоциация которого
в расплаве весьма мала и полностью отсутствует в твердой фазе,
авторы работы [20] дают весьма простое уравнение вида
]A(a0L)2 + 4(A/)2 - «oL = -g^L) A77(RT2,„), (VI. 13)
где a0 — степень диссоциации соединения АВ при температуре
плавления.
Это уравнение хорошо передает непрерывность изменения
формы максимума кривой ликвидуса с ростом степени диссоциации.
Так, если а0 = 0, то
Д77(2 AxL) = -Rfl AB/q{S+L), (VI. 14)
что идентично соответствующему закону предельно разбавленных
растворов. Двойка у AxL является результатом различия масшта-
278
бов на оси составов: абсциссы в уравнении (VI. 14) уменьшены
вдвое по сравнению с общепринятыми.
Далее, если а£ Ф 0, a Aa:l ->- 0, то для состава, отвечающего
формуле соединения
lim | ДГ/Ах |L->0 = dT/dxA = 0,
и максимум пологий.
Наконец, для заданного малого AxL понижение AT тем меньше,
чем больше а£. Иначе говоря, максимум тем более полог, чем
выше степень диссоциации соединения.
О. А. Есин [379] исследовал зависимость наклона касательных
к кривым фазовых равновесий от степени диссоциации, используя
модель идеального ассоциированного раствора.
Пусть компоненты А и В в двухкомпонентной двухфазной
системе способны образовать одно и то же химическое соединение
А В в обеих фазах в соответствии со схемой:
^нас ~Г "нас •<— At)
\\l itL _ t (VI.15)
■Днас ~г -^нас <— А£> •
В соответствии с выводом О. А. Есина [379], ограничимся
рассмотрением одной половины диаграммы состояния, где хв и х%
меньше или равны половине. Из элементарных соображений
следует, что
1„(2_а)хв т
ах.
в
ХАВ- !_(1_а)^ •
(VIЛ 6)
Отсюда величина, обратная константе диссоциации соединения
А В9 примет вид:
1 xLAn \—oll l — О — «L)*«
KL *А*Вх *L l-(2-aL)*£ '
Используя уравнения (VI.16) и (VI.17), для идеального
ассоциированного раствора находим
^b\l __ RT \ aL , *в(1-*в) v
X
дхв )р,т «Ч \l-(l-aL)^ l-(l-«^
Из этого соотношения следует, что для a > 0 величина (дцв1дхв)р, т
конечна, а для a = 0 бесконечно велика.
279
Уравнения (VI. 17) и (VI.18) в сочетании с производными для
кривых солидуса:
/ дТ у (4-4)(дЫд*в)1тТ д,т ,Qv
и ликвидуса:
дают все необходимое для анализа формы максимумов в
зависимости от степени диссоциации химического соединения, если
пренебречь изменением <7<а+Р) с составом в рассматриваемой области.
В частности, отсюда легко получить все основные типы
максимумов, указанные Н. С. Курнаковым [45] для химических
соединений [379].
Рассмотренный метод, основанный на исследовании наклона
касательных к кривым плавкости в точках максимума, неудобен
тем, что не позволяет проследить непрерывность перехода
пологого максимума в острый с падением степени диссоциации.
Действительно, наклон касательной в точке максимума для любых а > О
всегда равен нулю, кроме единственного случая, когда а = 0.
Рассмотрение же наклона касательных в точках, близких к
максимуму при допущении незначительной диссоциации соединения
[12], удобно лишь для предельно разбавленных растворов [12, 20].
Кроме того, анализ с использованием модели идеальных
ассоциированных растворов, допускающий любую степень диссоциации
соединения А В осложнено громоздкостью исходных уравнений.
В этом отношении большой интерес представляют работы
А. Б. Млодзеевского [44, 391—392], который дал выражение для
радиуса кривизны максимума как функции констант диссоциации
химического соединения типа А В в обеих фазах. Радиус кривизны
максимума различен для каждого значения а и становится равным
нулю только при а = 0.
С помощью геометрического метода А. Б. Млодзеевский вывел
уравнение связи между формами кривых изобарно-изотермических
потенциалов и кривых плавкости. С этой целью он рассмотрел
кривые G в весьма малом интервале температур Т°т — 7\ что
"позволило ему разложить величины GL и Gs в ряд и ограничиться
первым членом разложения.
Принимая в первом приближении кривые G вблизи максимума
за параболы, А. Б. Млодзеевский показал [44], что кривые
ликвидуса и солидуса вблизи максимума будут также
параболами. В итоге А. Б. Млодзеевский [391, 392] получил уравнения,
связывающие радиус кривизны ликвидуса и солидуса с константой
диссоциации соединения ЛВ и величиной отклонения максимума
от стехиометрии.
259
Однако, как справедливо указыбаёт 6. А. Ёсин [379], вывод,
сделанный А. Б. Млодзеевским, не лишен
формально-математических (термодинамически не конкретизированных) допущений.
Это заключается в том, что параболичность кривых Т = f (*,)
обосновывается А. Б. Млодзеевским законами предельно
разбавленных растворов, а полученная форма кривых
термодинамического потенциала, равно как и применяемое уравнение для G,
относятся к идеальным ассоциированным растворам.
О. А. Есин [379, 393] получил уравнения А. Б. Млодзеевского
обычным термодинамическим путем, не прибегая к
дополнительным допущениям о формах кривых вблизи максимума, и обобщил
их для случая реальных систем, введя коэффициенты активности.
Очевидно, что радиус кривизны в точке максимума кривой
плавкости обратно пропорционален второй производной от
температуры по составу:
Ртах = [(д2Т/дх%)р, „ах]"1. (VI.21)
Таким образом, вся задача сводится к нахождению этой
второй производной.
Используя условия равновесия твердой и жидкой фаз в двух-
компонентной системе в форме (11.182), данной Ван-дер-Ваальсом
и Констаммом при р = const с учетом выражения вида (V.34a)
и аналогичного ему соотношения для дифференциальной мольной
теплоты образования жидкой фазы из твердой, имеем
( дТ \S _ т(4-4)№7**в)1т ,VT 99ч
\^)Р ~ W^ ' (VL23)
Деля одно выражение на другое, с учетом выполнения,
согласно работе [13], в точке максимума равенства
qis+Q = —q{L+s)y (VI.24)
находим
д4 L №<«,/
(VI.25)
Рассматривая в уравнениях (VI.22) и (VI.23) наклоны касатель
ных как функции х%, хв, Т и (d2g/dx|)p, т и допуская постоянство
qi<x->$)f получим в точке максимума для солидуса
( д*Т \S Гтах / d*g
(VI.26)
281
и аналогично для линии ликвидуса
д*Т \l Tmm ( d*g \L
1 д*Т у
[ *4 ),
(VI. 27)
! pt max
Отсюда в соответствии с формулами (VI.21) и (VI.25) радиусы
кривизны примут вид:
0s _ (*ш/*№~*М) т28)
Ртах— — 7v2T7TJys FT^TTTTys /^2^/^2.. \L V vvi.^o;
7-тах (^4)Lx [ (*У*4)£.х ~ №<*!)Lx] '
■^гаах
(VI. 29)
Далее, чтобы получить явную зависимость радиуса кривизны
от константы диссоциации соединения Есин [379, 393]
воспользовался соотношением вида (V.3).
Таким образом, как и во всех предыдущих случаях, вся суть
задачи сводилась к раскрытию выражения (d\iB/dxB)PfT.
Дифференцируя уравнение (VI.2) по хв и используя (VI.3)
для отыскания производной и по хВу находим (d\iB/dxB)PiT. Так
как состав в точке максимума в общем случае не совпадает с
формулой химического соединения, то подставляем вместо хв
величину 1/2 + б, где сдвиг максимума 6 не превышает 1/2. В
результате после несложных преобразований получим
(J*-\ _ 4RT if K' + i ,
l^max^1"462) У К'+** +
КГ (1-26) (d\nyABtBtt AJdxB)muX
+ V\ + iMK')V\ + (№lK') [К' + ЮГНМ VK' + 462 + 26 ] +
\ а /шах
где
дхв L„ дхв
В частном случае идеального ассоциированного раствора
уравнение (VI.30) примет вид:
/J!£_\s - W л Г ^+> .... **Г 1 з
\ a^/max_1_46a ^ ^ + 4б2 "1-46* х« ' tV1"^
где х = К(# + 46а)/(# + 1).
282
Радиус кривизны максимума, например, ликвидуса на основании
формул (VI.29) и (VI.92), можно найти из выражения
Если же соединение А В совершенно недиссоциировано в твердой
фазе (Ks = 0), то при учете в точке максимума равенств (VI.24) и
q{S+L) = (щ + пу1 Д/4, АВ (VI.34)
(VI.33) переходит в уравнение
p"*-=~~SSTZV^ТГ = -1ЩТ^ (VI-35)
так как в этом случае, как было показано, 8 = 0.
Уравнения (VI.33) и (VI.35) позволяют не только вывести все
возможные сочетания острого и пологого максимумов, но и
показать непрерывное изменение кривизны максимума со степенью
диссоциации для идеальных ассоциированных растворов. Из
уравнения (VI.35) непосредственно видно, что чем больше константа
диссоциар(и K{L\ тем больше радиус кривизны максимума, тем он
более пологий. При полном отсутствии диссоциации (KL = 0)
радиус кривизны равен нулю и максимум острый.
То же вытекает и из более сложного уравнения (VI.33). Чтобы
представить зависимость р от К в явном виде из этого уравнения,
необходимо знать взаимосвязь б с /С. Для этого воспользуемся
выражением (VI.9).
В рассматриваемом приближении идеального
ассоциированного раствора энтальпию одного брутто-моля какой-либо из фаз
можно найти из соотношения
h = (xB- и) h°B + (1 - хв - и) КА + uh°ABi (VI.36)
где h°Bi h°Ay h°AB —энтальпии компонентов Л, В и соединения АВ
при заданных р и Т и данном агрегатном состоянии.
Из выражения для константы диссоциации соединения и
формулы (VI.36) нетрудно убедиться, что
<j^L) = (1/2) Ahm, AB + S (АС, А - АС. в) -
■ —(1/2)AH°Wxs + (1/2)MI0^kl, (VI.37)
где АЯ°—мольная теплота диссоциации соединения А В
при-заданных р и Т в идеальном ассоциированном растворе.
Из совокупности уравнений (VI.33) и (VI.37) можно судить,об
изменении р с К*
Следуя работе [393], проанализируем эти уравнения.
Так как по условию рассмотрения р принято отрицательным и
величина q(s-*L> >0, то из уравнения (VI.33) следует, что KL >
£> Ks. Иначе говоря, в точке максимума диссоциация в жидкой
283
фазе больше, чем в твердой. Так как значения обеих констант
меняются в одном и том же направлении ( от 0 до оо), это означает, что
KL растет быстрее, чем Ks-. Из неравенства KL > Ks следует
также, что коэффициенты распределения Ка и Kb будут
правильными дробями, равными в предельном случае нулю или единице.
Легко показать, что знаменатель формулы (VI.9) в этом случае
всегда положителен и, следовательно, знак сдвига определяется
только знаком разности (Ка — Кв). Абсолютная величина б
монотонно растет с xlax и К от нуля до значений
, с, _J_ 1
Отсюда следует, что с ростом KL величины V{KL + 462)/(/CL + 1)
и V (Ks + 462)/(/Cs+ 1) возрастают от нуля до единицы, причем
первая быстрее, чем вторая. Таким образом, второй множитель
уравнения (IV.33) непрерывно увеличивается с /(L.
Ввиду того что величина (1 —4б2) уменьшается медленнее
(от 1 до 0), чем растет значениеxL (от 0 до 1), первый множитель
уравнения (VI.33) увеличивается вместе с /CL. Что касается
третьего множителя, то его изменение с к1, согласна формуле
(VI.37), примет вид *
_^A#°(s)J^f_. (VI.38)
В этом выражении теплота диссоциации всегда положительна,
в то время как разность теплот плавления компонентов может
иметь любой знак. Поскольку, однако, производная от 6 меньше
1/2, а от х5 меньше единицы и обе они достаточно малы, то знак
всего выражения (VI.38) определяется Д#°(1>, т. е.
положительный. Следовательно, абсолютное значение q<s+L) также
увеличивается вместе с KL. В самом неблагоприятном случае, когда
соединение практически целиком диссоциировано в обеих фазах и
максимум почти лежит на ординате первого компонента, коэффициенты
распределения сближаются, dbldKL ->• 0, а производная от Xs
стремится к I. Тогда приращение (dq(s*L)ldKL) весьма незначительно
превышает величину 1/2 (ДЛт, а + А/4, в — ДАт, лв). Так как
при этих условиях теплота плавления соединения А В мало
отличается от суммы теплот плавления компонентов, то знак
выражения (VI.38) остается по-прежнему положительным. Таким
образом, все три множителя выражения (VI.33), а следовательно, и | р |
монотонно возрастают с KL. При KL = 0, р^ах = 0 и максимум
острый. Следовательно, положение, выдвинутое Н. С. Курнако-
вым, получает термодинамическое обоснование, по крайней мере,
для идеальных ассоциированных растворов.
284
Однако значение р зависит не только от /С, но и от других
величин, характеризующих природу веществ А, В и АВ. «Пока не
будет доказано, — пишет О. А. Есин, — что изменение этих величин
при переходе от одного соединения к другому влияет на величину
р так же, как и изменение К, нельзя безоговорочно сравнивать
степень диссоциации различных соединений, пользуясь лишь
кривизной максимума» [393].
В самом деле, даже в простейшем случае соединений,
диссоциированных лишь в жидкой фазе, на основании формулы
(VI.35) видно, что абсолютное значение радиуса кривизны
максимума может быть различным при одной и той же константе
диссоциации (KL) в зависимости от величины Ah°m, ab- Анализ
уравнения (VI.30), применимого для реальных растворов
любой концентрации, показывает, что отдельные предельные
закономерности, справедливые для идеальных ассоциированных
растворов, могут быть получены для реальных растворов в
предположении, что выражения (д In улв, Ахвх1дхв)ты и (д In ув/дхв)тах
конечны.
Эти предположения, необходимые для перехода к острому
максимуму, вполне оправданны и вытекают из термодинамического
определения коэффициентов активности, а именно из того, что
при переходе к предельно разбавленным растворам частиц Аг
и Вг в соединении А В эти коэффициенты стремятся к единице.
Но для того чтобы вывести все основные виды диаграмм
состояния двухкомпонентных двухфазных систем, образующих
химическое соединение, и, в частности, показать непрерывность
перехода пологого максимума в острый с ростом диссоциации,
нужно допустить, что члены уравнения (VI.30), содержащие
коэффициенты активности, меньше по абсолютной величине и
медленнее меняются с изменением степени диссоциации, чем первое
слагаемое. Обсуждение надежности этих допущений лежит, однако,
за пределами чисто термодинамического исследования и может
быть проведено на основе эксперимента или молекулярно-кинети-
ческой теории.
Позднее в работах [400, 401 ] метод оценки степени
диссоциации конгруэнтно плавящихся соединений, основанный на анализе
кривизны ликвидуса, был обобщен авторами на случай, когда
в системе образуется сложное соединение АтВп.
Рассмотрим диаграмму состояния А—В с конгруэнтно
плавящимся соединением АтВп с узкой областью гомогенности и
практически недиссоциирующим в твердой фазе. Допустим, что в
системе имеются лишь следующие равновесия:
AmBsn z± AmBLn ^ т (Л)^ас + п (B)L. (VI.39)
Как было показано в работе [379], на основе теории
идеального ассоциированного раствора, при осо = 0 величина
производной (d2g/dx%)max бесконечно велика. Учитывая это, на основании
285
формул (VI.29), (VI.34), легко убедиться, что при отсутствии
диссоциации в твердой фазе радиус кривизны ликвидуса можно найти
из соотношения
<£ М°т' АтВп 1 /VT 40}
р*« - (т + П) Гт. лтвп (aV«4)J. т * { }
Для рассматриваемого случая
Н* = V* + RT In \(хв - /ш)/[1 - и (m + /г - 1)]}. (VI.41)
Отсюда, принимая во внимание формулу (V.3), легко убедиться,
что
d*g \ __ 1 — и (т + д — 1) — (^и/^д) [1 -*Б (от + я— 1)]
d*| J г~ (*в-/ш)[1-и(т + л-1)1(1-.хв)
(VI.42)
Допустим, что в точке, отвечающей составу соединения, число
молей ПАтвп, находящихся в расплаве, максимально, т.е.
справедливо условие
(ди/дхв)ша = 0. (VI.43)
Учитывая это, получаем
D /max
Из выражений (VI.44) и (VI.40) для точки максимума найдем
"™*-1^ТГ+—Ш Aho > (VI.45)
* пг п
где уменьшаемое представляет собой число аналитических молей
соединения, приходящихся на один брутто-моль фазы стехио-
метрического состава, а вычитаемое — число продиссоциировав-
ших молей соединения АтВп.
Отсюда, используя определение а, находим для точки
максимума
. aL (m + n)> ^Т2,Атвп ^ .
^.лтвп
При т = п = 1 выражение (VI.46) переходит в уравнение вида
(VI.35) для бинарного соединения.
Методы оценки степени диссоциации соединений,
предложенные в работах А. Б. Млодзеевского и О. А. Есина, были впервые
применены Н. Н. Пацуковой [383] для расчета ао ряда двойных
солей. Для определения радиуса кривизны в точке максимума
был использован интерполяционный полином Лагранжа.
286
В работах [376, 402—404] был разработан метод оценки
степени диссоциации соединения в жидкой фазе на основании
данных о температурах кристаллизации или давлении пара
растворов, расположенных вблизи состава соединения.
Согласно работе [403], выберем в качестве растворителя
соединение ЛВ, диссоциирующее на атомы Л и 5. Рассмотрим
добавление к нему вещества Л, принимая его за второй компонент.
Нетрудно убедиться, что
Хав = Щ(1 -а)/л, (VI.47)
хАх = (ami + m2)/nt (VI.48)
xBt = amxln, (VI.49)
где п — общее число частиц в смеси, п — (1 — а) тг + щ;
mt — аналитическая концентрация /-того вещества, выраженная
в моляльностях.
На основании формулы (VI.47), выражения Гиббса—Дю-
гема midpii + /n2d[i2 = 0 и условия химического равновесия \iAb =
= \iAt + \1вх при т1 = const можно записать следующую
систему уравнений:
\ от2 Jmt I от2 Атх \ от2 J тх
(VI.50)
/ д\пхАВ\ гд in (дщ -f щ2) 1 / dlna\ ■ р/ д\пп\ = Q
\ дт2 )тх L дт2 ]тх \ дт2 /mt*~\ дт2 ) тх
Для решения этой системы необходимо дополнительное,
четвертое уравнение. В качестве такого уравнения в работе [403]
было предложено соотношение вида
л i (д In П11дт2)тх — т\ {да1дт2)тх = 1, (VI. 51)
где пг = атх + /я2.
На основании формул (VI.50) и (VI.51), получим
/ 01п*цЛ ^ щ g
V дт2 )тх (2ат1 + т2)(т1 + т2)* к # ;
В приближении модели идеального ассоциированного
раствора справедливо
RT \ дт2 /тх ~~* \ дт2 )тх ~~ \ дпГ2 )тх 9
где р! — парциальное давление пара соединения над раствором.
Отсюда, подставив выражение (VI.53) в (VI.52), получим
a e _ "§5Г [ (щ + т2) (дInPl/dm2)mi + l\- (VL54)
287
В то же ЁрёМй, используя Данные о температуре
кристаллизации растворов вблизи состава соединения, химический
потенциал растворителя можно представить в виде [403]
t*i = V* (Т, р) - (Д/4. x/rm9 0 Д7\ (VI.55)
/ 0|*i\ ML, i / дАТ \ *Т / д№т г \
\ дт2 )т1 ~ ~7^7" \&ч')т1 ~ Т°тЛ\ дт2 )m% > (VI.56)
где производная (дД/4, \/дт2)т1 характеризует изменение Ahm, \
вследствие подавления диссоциации растворителя. Допуская, что
эта производная будет небольшой при малых значениях т2,
можно приближенно записать [403]
(дц1/дт2)т1« - (Л/4, х/Гт, х) (дЬТ/дт2)т1. (VI.57)
Принимая во внимание выражение для криоскопической
постоянной [24, 131]
%-R(rm.i)2/Ah0milmu
соотношение (VI.57) можно переписать в виде
(d|ii/ftn2)mi = - (RT°mt Х/Вктх) (dAT/dm2)mt. (VI.58)
Совместное решение уравнений (VI.58) и (VI.52) при учете (VI.53)
приводит к выражению
а = "2^7[(т1 + т%)(дИТ/дтш)т1 ~ l\ (VL59)
или в другой форме записи (см. [405]):
Г\ J m я о I
. МГПш Ав (Гт9 Ав) х2 (1 - х2) | дТ/дх2 |; - 1 J * <VL6°)
Как показано в работе [403], на основании равенства
Щ (d^JdmlYmt = — (d\X2ldm2)mty
где индекс «°» указывает на принадлежность к точке плавления
соединения; с помощью выражений (VI.53) при учете (VI.52)
или (VI.58) и соотношения Гиббса—Дюгема получаются
следующие уравнения, позволяющие рассчитать степень диссоциации
соединения в точке плавления из данных по фазовому равновесию
или давлению насыщенного пара соединения А В:
"•— ^(a-in^;/ (VL61)
а° " 1^Ш]Щ^ = ~ 2m, (#f/dnQ-m ' (VL62)
После несложных преобразований легко убедиться, что
соотношения (VI.62) и (VI.35) эквивалентны.
288
а ==
I— 2х9
2х2
Кроме того, автор работы [403] обобщил соотношения (VI.61)
и (VI.62) на случай, когда соединение S диссоциирует по сложной
схеме, а именно:
v^S^v^ + v^S,
И it
vAa vBb
где а и b — число частиц, которые в свою очередь образуются
из молекул А и В соответственно.
В результате получаются выражения:
«о = -abvABv J\vA (avA + bvB) m\ №^\ 1, (VI.63)
«o = — QkabvABvB/[vA (avA + bvB) mx {d2T/dml)°mi]. (VI.64)
В работе [406] были проанализированы следствия,
вытекающие из включения в расчетные формулы для aL коэффициентов
активности частиц, образующих ассоциированный раствор,
которые оценивались в приближении регулярных растворов. В
результате автор пришел к заключению, что величина ошибки,
возникающей из-за пренебрежения неидеальным поведением смеси
частиц А1у Вг и АВ, практически близка к нулю, если aL = 0 ч-
-^-0,01 и составляет ~4—5% для aL = 0,1.
Джордан [187] для описания кривой ликвидуса первичной
кристаллизации соединения А В в приближении регулярного
ассоциированного раствора получил уравнение вида (V.249),
включающее два неизвестных параметра: степень диссоциации
соединения а0 и функцию энергий обменного взаимодействия частиц о*
в растворе. Так как каждой температуре ликвидуса отвечает свое
значение а°, использование упомянутого уравнения в форме,
предложенной автором, для расчета параметров диссоциации
соединения на основе анализа кривых ликвидуса не представляется
возможным.
[3. ОЦЕНКА СТЕПЕНИ ДИССОЦИАЦИИ ХИМИЧЕСКИХ
СОЕДИНЕНИЙ В РАЗЛИЧНЫХ ПРИБЛИЖЕНИЯХ
[[ТЕРМОДИНАМИКИ РАСТВОРОВ
Количественная оценка степени диссоциации, с помощью
подхода, в основе которого лежит идея Н. С. Курнакова о
взаимосвязи кривизны ликвидуса со степенью диссоциации конгруэнтно
плавящегося соединения, может быть достигнута двумя
путями:
1. Детальное экспериментальное исследование кривой
ликвидуса, отвечающего первичной кристаллизации соединения АтВПУ
с последующей обработкой этих данных на ЭВМ с целью
формальной аппроксимации при помощи различных полиномов. Такой путь
Ю Глазов В. М. и др. 289
можно назвать формально математическим, поскольку описание
ликвидуса с помощью полиномов не связано с особенностями
характера межмолекулярного взаимодействия в системе. В то же
время оцениваемая формально математически кривизна ликвидуса
может считаться экспериментальной, поскольку в основе оценки
лежат экспериментальные данные, а точность оценки определяется
степенью надежности экспериментальной методики и числом
экспериментальных точек на кривой.
2. Модельное описание кривых ликвидуса для различных
моделей термодинамики растворов и установления на основе
общетермодинамических соотношений конкретных выражений
для параметров диссоциации в соответствии с принимаемой
моделью.
Ограниченность первого подхода определяется необходимостью
детального экспериментального исследования кривой ликвидуса,
особенно в окрестности соединения АтВпу что не всегда возможно.
Ограниченность второго подхода связана с определенной
неадекватностью принимаемой модели и рассматриваемого
раствора.
Оценка степени диссоциации, если известен радиус кривизны,
затруднений не вызывает. Для модельного подхода целесообразно
вывести и проанализировать выражения для параметров
диссоциации. Отметим, что этот подход был развит нами в работах
[321, 400, 401, 407—411 ] и подкреплен сравнением с
экспериментальной оценкой радиуса кривизны на ряде конкретных систем,
включающих соединения типа АВ и АтВп. Рассмотрим оценку
параметров диссоциации химических конгруэнтно плавящихся
соединений в обобщенном виде, а затем дадим некоторые
иллюстрации на примере ряда систем, включающих полупроводниковые
соединения различного типа.
Из сравнения уравнений (VI.40) и (VI.46) следует, что степень
диссоциации соединения АтВа в жидкой фазе связана со второй
производной изобарно-изотермического потенциала по
концентрации следующим выражением:
. = [(m + »)i^.yn1t (VI65)
На основании формул (IV.140) и (V.3) для второй производной
изобарно-изотермического потенциала в приближении шестипара-
метрической модели будем иметь
[й)Р. Г ^^sf -21щ~ °*т+к " WuT) {3хв ~1)+
+ Зхв (ш, - <*иТ) (2хв - 1)]. (VI.66)
290
Для .точки максимума, когда хв = п/(т + п), получим
( d*g V _ (т + п)* f r 2m/i
\ д*в Л. г ~ ™~ I m> т * ~~ (« + ")2
X [щ - ©„rS, лтвл + •%=£ («I - о)цГт, лтв„) +
+ 3(Д~? (юг - (оЛ- л^«)]}• (VL67)
Подставляя выражение (VI.67) в (VI.65), получим для степени
диссоциации соединения АтВп в рамках шестипараметрической
модели:
«О = RTm. AmBn [RTm% АтВп - 2 (т™п)Ш [®0 ~ «Л. ^mBn +
+ 2/1 — ТП / - ^-,с * | 3/1 (ft — ftl) , фо - ч 1 ) ""*
1М^Г (^ - V"7*»' Am*n) + (т + я)« (<°* - С021Гт', ЛтВл) J J .
(VI.68)
На основании этого уравнения можно прийти к уравнениям для
степени диссоциации в моделях субрегулярного,
квазирегулярного и строго регулярного растворов.
Так, в модели субрегулярного раствора, учитывающей
температурную зависимость энергии взаимообмена, предполагается
ее линейная зависимость от концентрации и, следовательно,
о)2 = 0; со21 = 0. (VI.69)
В соответствии с этим для данной модели
<*0 = RT°mt AmBn {RTm, AmBn - {*™п)г [*>0 ~ ©Ol^m, AmBn +
В модели квазирегулярного раствора допускается независимость
энергии взаимообмена от концентрации и, следовательно, помимо
условия (VI.69),
о)! = 0; (ои = 0. (VI.71)
Тогда из уравнения (VI.68) получим
«0L = RTm. AmBn [RT°mt AmBn - (m2^n)a (©0 - ЩгТщ, AmBn)] * •
(VI.72)
Наконец, в модели строго регулярного раствора отсутствует также
и температурная зависимость энергии взаимообмена (со01 = 0)
и, следовательно,
4 = RT'm. лтВп [*К, AmBn - ffifa] -1. (VI.73)
10* 291
Для соединения типа АВ уравнения (VI.68), (VI.70), (VI.72)
и (VI.73) принимают вид:
^ ^m,^(«+2co01+(D11)-(2(o0 + a>1) • ^iJV
«о = То {2R , ч_ - (VI.75)
' т, ЛВ (z/< *Г ©oi) ^0
а£ = 2RT°mt AB/(2RT°m, ав - ©»). (VI.76)
Таким образом, в простейшем случае соединения АВ для шести-
параметрической модели и модели субрегулярного раствора
степень диссоциации описывается одним и тем же уравнением, что
определяется известной концентрационной симметричностью
соединений этого типа.
В. А. Ротенберг с соавторами [218] получил уравнение,
близкое по виду к (VI.76), дифференцированием уравнения вида
(V.77). Полученное уравнение отличается от (VI.76) отсутствием
двойки в числителе. Данное расхождение связано с
использованием уравнения Млодзеевского, в которое входит
дифференциальная мольная теплота образования моля фазы, равная по величине
половине теплоты плавления соединения АВ> что не было
принято во внимание авторами работы [218 ], тогда как в нашем случае
это учтено.
Говоря об оценке степени диссоциации соединений типа АтВп
и АВ с помощью уравнений (VI.73) и (VI.76), уместно отметить
следующее. Эти формулы записаны в приближении модели строго
регулярного раствора, которая, как отмечалось выше (см.,
например, рис. 43), предполагает полную диссоциацию соединения.
В итоге налицо противоречие. С одной стороны, применяемая
модель допускает полную диссоциацию, с другой, — по этой модели
рассчитывается степень диссоциации, т. е. предполагается
частичная диссоциация. Чтобы разрешить это противоречие, обратимся
вновь к получению цепочки уравнений (VI.68)—(VI.76). Эти
уравнения были выведены на основе использования шестипараметри-
ческой модели, включающей параметры со0, co0i, (*>i, <ои, со2 и (о21>
связанные с характером межатомного взаимодействия, но не
имеющие ясного физического смысла, как например, энергия
взаимообмена в модели строго регулярного раствора. Поэтому
если говорить о приложении модели строго регулярного раствора
к системам с частично диссоциированными соединениями, то
очевидно, что это можно сделать лишь формально, поскольку
физический смысл, который вкладывается в понятие энергии
взаимообмена, и предполагает наличие лишь слабых сил
межмолекулярного взаимодействия (типа ван-дер-ваальсовых), в данном
случае утрачивается. В этой связи со0 следует рассматривать как
один из формальных параметров, который не имеет физического
292
смысла энергии смешения в модели строго регулярного раствора,
применяемой к системам без промежуточных фаз (см. также
работу [2531).
Иными словами, модель строго регулярного раствора в
понимании ее как физической модели строения раствора в принципе
не может быть применена к системам с частично
диссоциированными конгруэнтно плавящимися соединениями. Поэтому
приложение этой модели к указанным системам носит вполне
формальный характер, а параметр со0 включает в себя все виды
взаимодействий в растворе как слабых, так и сильных. Следовательно,
по отношению к системам с химическими соединениями можно
говорить лишь о математическом выражении кривых фазовых
равновесий, а не об описании строения растворов в данном
приближении.
В соответствии с изложенным уравнение Виланда и его
аналоги для соединений АтВп, если исключить случай полной
диссоциации, дают возможность определить некий эффективный
параметр со0, который можно использовать в соответствующих
расчетах.
Таким образом, отмеченное выше противоречие разрешается
на указанной основе. Это было важно отметить, потому что в
некоторых работах [412—413] применение формального аппарата
теории регулярных растворов к системам с частично
диссоциированными соединениями понимается в буквальном смысле, т. е.
расценивается как использование строгой физической модели.
Кроме того, авторы работ [412, 413], рассматривая проблему
диссоциации в терминах теории упорядочения и разупорядоче-
ния, имеющих в основном структурный смысл, отождествляют
степень разупорядочения со степенью диссоциации, что,
естественно, неверно, так как расплавы частично диссоциированных
соединений могут отличаться самой различной степенью
структурного разупорядочения, не связанной со степенью диссоциации.
Рассмотрим [411] возможность оценки степени диссоциации
соединений АтВп с привлечением одного из наиболее формальных
подходов к описанию поведения растворов, развитого в работах
Крупковского [141, 142].
По Крупковскому, на основании выражений (V.3) и (IV. 172)
можем записать
(d2g/dx2B)p> т - RT/[xB(l -хв)\ - Ы (Г) (1 - хв)к-\ (VI.77)
Для точки максимума выражение (VI.77) преобразуется к виду
/ d*g\ (m + n)kRT0mAmBn-kco(T)mk-ln
\ д*в )р,т (m + n)k-2mn
(VI .78)
293
Подставляя выражение (VI.78) в (VI.65), получим
(m + n)kRT° , R
a0L —-*- ' тп k . • (VI.79)
Аналогично для противоположного вида асимметрии кривой
ликвидуса имеем
(т + n)k+l RT°m , „
«oL = яг^ тп к 1 2 • (VI.80)
(т + n)fe+1 RTmt АтВп - Ы (Т) nk~W }
Уравнения (VI.68)—(VI.76), а также (VI.79) и (VI.80)
выведены для случая, когда максимум на кривой плавкости отвечает
стехиометрическому составу. Однако, как отмечалось при
рассмотрении классификации соединений, по Н. С. Курнакову,
при значительных степенях диссоциации максимум на кривой
плавкости заметно сдвигается (см. рис. 21, д). Выше мы показали
возможность учета сдвига максимума при оценке параметров
диссоциации в общем виде. Проиллюстрируем эту возможность
при использовании модельных представлений. Для простоты
ограничимся моделью строго регулярного раствора в системе
с конгруэнтно плавящимся соединением типа АВ [414]. Имеем
(dG/dc)Pt т = (\1°в- |М) + RT In [с/(\ - с)] + (1 - 2с) <о0, (VI.81)
где с = х% = х%. (VI.82)
Из уравнений (VI.81) и (VI.82), учитывая, что
с =1/2 + б, (VI.83)
получим б = -^ *А . % V7ч ?2—L . (VI.84)
Если разности химических потенциалов в числителе уравнения
(VI.84) выразить через соответствующие коэффициенты
распределения компонентов А и В между твердой и жидкой фазами,
то получим
8=«^. (VI.85)
Если указанные разности выразить через теплоты и температуры
плавления компонентов, то найдем
g _ А*ш, Л [(TmJTm. а)-1]~ АС, В [(^шах/У°«, в) ~ *] (щ ggx
2К-«о)
Уравнения (VI.84)—(VI.86) характеризуют величину
отклонения от стехиометрии для модели строго регулярного раствора
соединений типа АВ. В этих выражениях трудно определить оо£.
Однако переходя к оценке степени диссоциации, этот параметр
можно исключить.
294
Запишем выражение для отношения радиусов кривизны
ликвидуса и солидуса в точке максимума вида:
Ртах ... (KL + 462)(KS+\)
ГПЫХ
0C44«W+!) • (VI'87)
Из этого уравнения получим
Ks+l I \ KL + l pm
(VI.88)
max
Выражение для радиуса кривизны кривой ликвидуса с учетом
уравнения (VI.88) приобретет вид
Ртах" 4RT2 V 46) !_/pS /L U/3 ' (V1W>
^^'тах 1 \гтах/^тах/
Выразим радиус кривизны ликвидуса через параметр со0 при
наличии сдвига максимума относительно стехиометрического
состава. Для этого необходимо записать выражения для второй
производной изобарно-изотермического потенциала по
концентрации в принятом приближении.
На основании формулы (V.3) для строго регулярного раствора
найдем
(d2g/dxl)°Pt т = RTnf'x/\c{l - с)] - 2g& (VI.90)
или, подставляя значение с из выражения (VI.83),
(d2g/dx2B)°*T = RTmax/{\ - 4б2) - 2©5. (VI.91)
Подставляя эту формулу в уравнение (VI.29), получим
j^ 2*Гтах-со*(1-4б*)
Р-» 2Гшах К-<)[ЭДГШ1Х-<(1-4*«)]^ (У1-У>
max
Разделив выражение (VI.28) на (VI.29) при учете уравнения
(VI.91), будем иметь
(PLA1/2 2*Гтах-<(1-4б*)
\^j ~ мгтИ1Х-^(1-4#) (VL93)
или, вычитая из единицы левую и правую части уравнения (VI.93),
1 _ Pmax ^ V ° 9±±1 > . (VI 94)
(.Ртах J 2*Тв-«*(1-4в»Г (VLy4)
295
Сравнивая уравнения (VI.89) и (VI.92) и учитывая соотношение
(VI.94), можно показать, что
[(** + 4б2)/(^ + I)]1/2 = 2RTJMX/[2RTma - <4 (1 - 4б2)]
(VI.95)
или, приняв
\2RTmJ[2RTmax -o)0L(l - 4б2)]}2 = Af, (VI.96)
на основании формулы (VI.95) найдем выражение для расчета
степени диссоциации соединения АВ с учетом сдвига максимума
относительно стехиометрии строго регулярного раствора:
о£ = -|/"(M_462)/(1 -462). (VI.97)
В полученном выражении учтено также поведение твердой фазы,
поскольку, во-первых, в исходное уравнение сдвига максимума
входила формальная энергия взаимообмена в твердом состоянии
(со£), а, во-вторых, сам сдвиг максимума б предполагает
диссоциацию в твердом состоянии.
Отметим также, что полученные для строго регулярных
растворов выражения для параметров диссоциации соединений А В
при сдвиге максимума иллюстрируют в принципе общий подход
к решению вопроса о диссоциации соединений любой сложности
с привлечением других моделей вплоть до шестипараметрической.
Использование того или иного аппарата для расчета определяется
обоснованным выбором модели раствора. Привлечение моделей
квазирегулярной, субрегулярной и шестипараметрической при
одновременном учете сдвига максимума приводит лишь к
постепенному усложнению формул, однако принципиально ничего
нового не дает. Поэтому сдвиг максимума на кривой плавкости
конгруэнтно плавящегося соединения учитывается нами только
в приближении строго регулярных растворов для соединений А В
и в других приближениях для более сложных соединений здесь
не рассматривается.
Отдельно остановимся на расчете степени диссоциации
конгруэнтно плавящегося соединения АтВп вдоль кривой ликвидуса
в приближении модели регулярного ассоциированного раствора.
В работе [415] был предложен метод оценки степени
диссоциации конгруэнтно плавящегося бинарного соединения АВ,
обладающего узкой областью гомогенности, вдоль кривой
ликвидуса для регулярного ассоциированного раствора [187] с
привлечением термодинамических данных по э. д. с.
В данной работе предпринята попытка обобщить этот способ
на случай образования сложного соединения АтВп и показать
принципиальную возможность использования других
термодинамических данных для решения указанной задачи.
296
Рассмотрим бинарную систему А—В с конгруэнтно
плавящимся соединением сложного состава типа АтВп> обладающим
узкой областью гомогенности [менее 1% (ат.)].
Предположим, что в жидкой фазе соединение АтВп
диссоциирует по схеме реакции (VI.39), константа равновесия которой
определяется уравнением:
К = (Jtf Д / *Атвя) (АЛ / УАтвп). (V1.98)
Таким образом, жидкий раствор можно представить как
молекулярную смесь трех сортов частиц: мономеров Аг и Вх и ассо-
циатов АтВп. Для описания поведения такой смеси воспользуемся
моделью регулярного ассоциированного раствора.
Согласно работе [24], для рассматриваемого случая
справедливы соотношения (V.223). Найдем выражения для
коэффициентов активности компонентов аА и ав.
Условия сохранения масс компонентов для рассматриваемого
случая можно записать в виде
nA = nAl + mnAmBn; (VI.99)
пв = пВх + ппАтВп. (VI. 100)
Используя определения мольных долей компонентов и
молекулярных форм соотношения (VI.99) и (VI. 100) легко преобразовать
к виду:
*AX = ха - [т - хА (т + п - 1)] хА в\
г / , 1м (VI.101)
xBl = *в - [п - хв (т + п - 1)] хАтВп.
В приближении модели регулярного раствора коэффициенты
активности тройной смеси Аг + Вх + АтВп принципиально можно
рассчитать по уравнению вида (IV.216). Однако чтобы
воспользоваться этим уравнением, необходимо знать параметры со* ^ / (*>,
которые неизвестны.
Для расчета коэффициентов активности мономерных форм
молекулярной смеси Ах + Вх + АтВп воспользуемся
приближением вида (V.233) и (V.234). Для нахождения коэффициента
активности уА в воспользуемся соотношением Гиббса — Дюгема
вида
xAld In yAl + xBl d In yBl + xAmBnd In yAmBn -0. (VI. 102)
Подставив в это уравнение соотношения (VI. 101), (V.233) и (V.234),
после несложных преобразований получим
(2(0W) [-хв (т + п) + п] dxB = -d In yAmBfl. (VI. 103)
Интегрируя это уравнение в пределах от хв = п/(т + п) до хв
и используя условие нормировки Ултвп = 1 при хв = п/(т + п)у
получим следующее выражение для коэффициента активности
297
7л в в приближении модели регулярного ассоциированного
раствора:
*т 1п УЛтВп = V**Km + n)] [п -(т + п) хв\\ (VIЛ 04)
С помощью уравнений (V.233), (VI. 101), (V.233) и (V.234) получим
следующие выражения для активностей компонентов А и В:
а а - ялх = {*а - [т -хА(т-\-п- 1)] *Атв^ exp (со*4//?г),
(VI. 105)
<*в = авг = {хв - [п - хв (т + п - 1)] хАтВп} exp (<»*x2A/RT).
(VI. 106)
На основании формулы (V.203), допустив независимость A#f
и ASf от температуры, что, как известно, справедливо для
достаточно широкого температурного диапазона, приходим к
следующему равенству:
<*A<*% = aJPa£, (VI. 107)
где а\ и a'i — активности компонента / в растворах,
принадлежащих ликвидусу и лежащих слева и справа от состава соединения
хв = nl(m + п) на одной и той же изотерме. Условимся величины,
характеризующие раствор, находящиеся справа от состава
соединения, отмечать штрихом. *
В соответствии с выводами работы [13] для случая, когда
соединение АтВп незначительно диссоциировано в жидкой фазе,
регулярные ассоциированные растворы, лежащие слева и справа
от состава соединения, должны характеризоваться различными
значениями параметра со*. Тогда, объединив уравнения (VI. 105),
(VI.106) и (VL107), получим
[хА^[т^хА(т-\-п^1)]хАтВп}тх
Х{Хв — [п — хв(т + п—\)] хАтВп)п ехр [со* (тх% + nx2A)/RT] =
= [хА- [т - Х']?{т + п- \)]х'Атвп}тХ
X [х'в — [п-х'в(т + п- \)]хАтВп)пх
Xexp[u'(mx'£ + nx'l)/RT]. (VI.108)
Это уравнение содержит четыре неизвестные величины *Атвп.
х'лтв , со* и <о*'. Для их определения необходимо иметь систему
из 4 уравнений. Так как в рассматриваемом случае (р = const)
константу равновесия реакции (VI.39) следует рассматривать
только как функцию температуры, на основании соотношений
(VI.98) и (V.223) имеем
^ла%/аАтвп = а'Ата'вп/аАтВп. (VI.109)
298
Отсюда, учитывая равенство (VI. 107), найдем
ХАтВпУАтВп = ^АтВпУАтВп. (VI. 1 10>
Подставив сюда выражение (VI. 104), получим второе уравнение
х^вп ехр {(m+*n)RT [п-(т + п) xBf\ =
-xWn™p{im + n)RT U-{m + n)x'BV). (VI.111>
Два других уравнения можно получить, привлекая экспери-
ментальные термодинамические данные. В частности, можно*
использовать данные по давлению насыщенного пара вдоль линии'
трехфазного равновесия GLSa в . Для этого случая
дополнительные уравнения будут иметь следующий вид:
Р = Р*л{хл — [т-хА{т + п- 1)] хАтВп) х exp [(-jjjr) хв] +
+ Р°в [хв - [п - хв (т + п - 1)] *ИтВл} ехр [("|г) ха] >
(VI.112>
/>' = Pa {x'a- [m-x'A(m + n- 1)]x^J ехр [(-^г).^2] +
(VI. 113)
где ри/?' — общее давление насыщенного пара над насыщенными
жидкими растворами при данной температуре,
располагающимися по составу слева и справа от соединения АтВп соответственно.
В случае использования данных по теплотам смешения
компонентов на основании формулы (IV.89) при учете соотношений
(V.223), (V.233), (V.234) и (VI.101) для тройного регулярного-
ассоциированного раствора будем иметь
r,,rv [ (т + «) (*А + Х%) Х А В
hE <« = (0* \xAXB - , V n jfS +
, [n-(m + n)xBf ) . *Атвп
f (т + пЩт + п-1)хАтРп+1] f Ч"ая' (m + n-1)*,^+1 >
(VI. 114).
ft _со (Vb- (m + n_1)<leA+1 +
-U [n-(m + n)x'B]2 | , *лтв„
(m + n)Um + n-l)x'A B + 1] T""' (m + n-l)x'A „ +1'
(VI. 115)
299»
тде AHf — теплота образования жидкого АтВп из жидких
компонентов. Если имеются данные по э. д. с, можно записать
—zFE = RT\naiy (VI.116)
—zFE' = RT In a,, (VI.117)
где at — активность t-того вещества, определяемая по одному
из уравнений (VI.105) или (VI.106).
При наличии надежных данных по температурной зависимости
AGf для оценки мольной доли ассоциатов и параметра со* можно
воспользоваться системой только двух уравнений, одно из которых
будет иметь вид
{хА -[т-хА(т+п- 1)] хАтВп)т X
х{хв-{п-хв{т + п- \)]хАтВп)пу:
X exp [w* {mx% + nx2A)/RT] = exp (AGflRT\ (VI. 118)
а вторым может служить любое из уравнений (VI. 112) —(VI.117).
Аналогичная система будет справедлива и для другой ветви
.ликвидуса. Далее величину степени диссоциации соединения
АтВп можно рассчитать по формулам:
для левой ветви ликвидуса
Ып)[(т + п-1)хАтВп+1] ' (VLUy>
для правой ветви ликвидуса
—х'а в + (хл/т) \(т + п — 0 х'л в + Ч
а' = тп К П т" J. (VI. 120)
(хА/т)[(т + п- 1) *Атвп+ 1\
Рассмотренные выше способы оценки параметров диссоциации
в различных приближениях термодинамики растворов можно
использовать в тех случаях, когда на основе предварительного
анализа выбрана модель раствора в рассматриваемой системе.
Для систем, включающих полупроводниковые фазы
различного состава, выбор модели раствора был нами достаточно
надежно обоснован ранее (см. гл. 5, п. 9). Опираясь на полученные
результаты и используя уравнения для соответствующих моделей,
мы оценивали параметры диссоциации полупроводниковых
соединений различных групп. Результаты такой оценки
представлены в табл. 3.
Полученные данные целесообразно сопоставить с
экспериментальными результатами в тех случаях, когда кривая ликвидуса
подробно и достаточно надежно исследована.
Оценка степени диссоциации конгруэнтно плавящегося
соединения по кривизне ликвидуса требует аналитического
представления экспериментальной зависимости Тлик = f (хв), для чего,
300
Таблица £
Параметры диссоциации полупроводниковых соединений
различных групп, определенные экспериментально
и оцененные методами термодинамики растворов
Соединение
Mg2Pb
Mg2Sn
Mg2Ge
Mg2Si
CdSb
CaSb
InSb
InAs
GaTe
InTe
Ga2Te3
In2Te3
GeTe*
SnTe
PbTe
Sb2Te3
Bi2Te3
Степень
аппроксимирующего
полинома
Чебышева
5
6
7
8
3
4
5
4
4
6
7
7
—
5
10
3
6
VT*.
град
4,2
3,2
1,8
1,5
2,0
1,9
2,7
1,8
2,8
4,0
2,7
2,2
—
3,9
3,7
2,1
1,0
-Р9Ю'
12,44
8,87
7,27
10,93
29,04
12,0
12,97
7,11
5,48
2,089
12,15
16,89
—
3,960
4,908
14,20
40,42
«I
0,24
0,18
0,17
0,26
0,32
0,16
0,14
0,13
0,16
0,04
0,13
0,14
—
0,07
0,08
0,20
0,43
аРасч
0
0,28
0,26
0,33
0,30
0,47
0,34
0,52
0,51
0,17
0,11
0,21
0,18
0,16
—
—
0,20
0,57
Модель
раствора
СР
Р
6П
СР
6П
КР
6П
* Анализ ликвидуса в системе Ge—Те проводили в интервале концентраций от
50 до 67% (ат). Те
как правило, используют различные полиномы. Случайные-
ошибки, которые особенно сказываются при нахождении второй
производной (д2Тлш/дхв)таХу определяют целесообразность
нахождения зависимости, точно описывающей экспериментальные
данные. В этой связи следует проводить сглаживающую
аппроксимацию.
Аппроксимация экспериментальных зависимостей рядами
Тейлора предполагает разложение в ряд вблизи какой-либо
выбранной точки, поэтому используется лишь при условии сверхбыстрой
сходимости ряда. Применение интерполяционных многочленов,
таких как полиномы Лагранжа, Ньютона, также не всегда
оправдано вследствие случайных ошибок эксперимента.
Использование в качестве аппроксимирующей функции
степенного ряда
7 = я0 + Д1Х + я,л:2Н \-апхп (VI. 121}
предполагает для нахождения коэффициентов разложения решение
системы линейных уравнений [416]. Однако для вычисления
коэффициентов многочленов высокой степени (практически п > 2)
в наиболее важном случае измерений одинаковой точности эта
30 Г*
система оказывается практически малопригодной ввиду большой
потери точности при ее решении, которая сказывается тем больше,
чем выше степень многочлена. С повышением степени многочлена
весь расчет приходится повторять сначала.
От указанных недостатков свободна система ортогональных
полиномов Чебышева р0 (х)9 рг (х), р2(х), ... на множестве точек
■Ль х2, ..., xN с весом Wt >0. При этом аппроксимирующий
многочлен имеет вид
Т = soPo(x) + sxPl(x) + - ■ ■ + snPn(x). (VI.122)
Параметры этого выражения вычисляют по формуле
N
Sj=^ , (/ = 0, 1, ..., N), (VI.123)
которые практически не зависят от степени искомого полинома.
Применение в качестве аппроксимирующих уравнений
полиномов Чебышева не позволило получить удовлетворительные
данные по кривизне ликвидуса в точке плавления соединения из-за
смещения максимума аппроксимирующей кривой от точки х =
= nl(m + п).
Алгоритм, предложенный Пеком [417—418], позволяет
аппроксимировать экспериментальную зависимость, с помощью
полиномов Чебышева, причем аппроксимирующая кривая проходит
через k заранее фиксированных точек.
С помощью алгоритма Пека можно также перейти от
полиномиального разложения (VI. 122) к форме простого степенного
ряда с коэффициентами с0, си ..., сп> что позволяет находить
кривизну ликвидуса в аналитической форме и тем самым повышает
точность дифференцирования.
Применение преобразования координат х' = х — х01 где х0 =
— п/(ш + п) — состав соединения, дает возможность представить
полином в виде ряда
Т= ^„(^* (VI.124)
откуда вторая производная (д2Т/дх2)хв=п/(т+п) определится из
выражения
(д*Т/дх*)х=п/{т+п) = 1/р = 2с2. (VI.125)
Изложенный метод аппроксимации апробирован нами на
шестнадцати соединениях. При этом фиксировалась точка плавления
соединения хв = п/(т + л), Т = Т°т, лтвп, которая, как правило,
является табличной величиной. Максимум аппроксимирующей
кривой во всех шестнадцати случаях совпал с максимумом криво»
302
ликвидуса. Оптимальную степень полинома определяли из уело*
вия стабилизации дисперсии с использованием критерия Фишера.
Полученные значения радиусов кривизны ликвидуса и степени
диссоциации соединений приведены в табл. 3 в сопоставлении
с величинами, рассчитанными методами термодинамики растворов
на основании выбранных моделей. Данные таблицы показывают
вполне удовлетворительную сходимость расчетных и
экспериментальных данных.
Заметим, однако, что в случае соединений А111 Ву это
расхождение оказывается наибольшим по сравнению с остальными
рассмотренными группами соединений. Максимальное
расхождение наблюдается у антимонида и арсенида галлия. Отмеченное
несоответствие расчетов и экспериментов, хотя и не
принципиально для выбора модели раствора (если учесть последовательную
увязку всех термодинамических свойств растворов In—Sb),
но все же требует дополнительного рассмотрения.
4. ДИССОЦИАЦИЯ БИНАРНОГО СОЕДИНЕНИЯ
В КВАЗИБИНАРНЫХ РАСТВОРАХ ТРЕХКОМПОНЕНТНЫХ
СИСТЕМ
Выше мы рассмотрели поведение соединений в бинарных
растворах. Представляет определенный интерес проанализировать
поведение соединений при их растворении в других веществах,
т. е. рассмотреть более сложные, так называемые квазибинарные
системы, с тем, чтобы проследить за изменением термической
стабильности соединений не только от температуры, но и от степени
разбавления раствора. Такая задача часто встречается в практике
легирования полупроводников и металлов, в связи с чем ее общее
рассмотрение, данное нами в [405], имеет определенное
практическое значение.
Рассмотрим простейшую квазибинарную систему АВ—С,
в которой конгруэнтно плавящееся соединение диссоциирует на
составляющие атомы. Пусть раствор, составленный из молекул
Аъ Ви АВ, С, ведет себя как идеальная смесь частиц. Такое
допущение не слишком грубое, поскольку практически вся
неидеальность учитывается актом химического взаимодействия,
приводящего к образованию соединения АВ в растворе. Тогда,
согласно работе [24], можно записать
v;;B + RT\nx'ABy'A; = l,-j:RTlnxAB, (VI.126)
где х'ав — аналитическая мольная доля соединения в растворе;
7лв — коэффициент активности А В; \к°аВ и \fAB — стандартные
химические потенциалы вещества АВ и молекулярной формы А В
соответственно.
Из этого выражения
хав1{*'авУав) = exp [(^'fl - vAB)/RT]. (VI.127)
303
Изберем способ нормировки, согласно которому у'лв -*• 1 при
х'ав -* 1. Тогда можно записать
*\в = «р!(^в- VAB)/RT] (VI.128)
Сравнивая формулы (VI.127) и (VI. 128), получим
*лв = **лв*'лвУлв- (VI-129)
Здесь и далее верхний индекс * означает принадлежность к
чистому веществу А В.
Выразим х*лв через степень диссоциации соединения АВ.
Согласно определению,
*лв = "лв/(»лв + "'л + 'Ы (VI.130)
Используя уравнения материального баланса для числа частиц
в растворе при диссоциации соединения А В со степенью а0 и при
учете выражения (VI. 130), получим
*^ = 0-«•)/(!+*')." (VI.131)
Согласно определению понятия степени диссоциации
соединения А В в растворе, можем записать
а = Па/ПАВ = ХА (В)/(ХАВ + *А (В))' (VI.132)
Далее из равенства хА + хв + хАВ + хс = 1, принимая во
внимание, что хс = х'су'с* а также при учете соотношений (VI. 129)
и (VI. 131), получим
*л -х'в = (1/2) [ 1 ~ *4bVhb (1 - « )/(J + «') - *cYcl- (VI-133)
Подставляя в уравнение (VI. 132) значения ха (В) из формулы
(VI. 133) и хАВ из (VI. 129), при учете соотношения (VI. 131)
получим окончательное выражение для степени диссоциации бинарного
соединения в квазибинарном растворе:
аш ^'-'cVc-'лв^ (VI 134)
1-ХсУс + ХАвУАв(1-а)/{1+а)
Это уравнение — общетермодинамическое и его дальнейшее
конкретное использование определяется выбором модели раствора,
от которого зависит выражение для коэффициентов активности ус
и улв-
Рассмотрим возможность использования уравнения (VI. 134)
на примере системы GaSb — Ge, которая типична для
исследуемого случая. Необходимые для расчета сведения о кривых
моновариантного равновесия по данным исследования квазибинарной
диаграммы состояния GaSb — Ge [421 ] представлены в табл. 4.
304
Таблица 4
Значения степени диссоциации GaSb вдоль кривой
ликвидуса диаграммы состояния GaSb — Ge
и исходные данные для их расчета
&
</Э
СО
- О
X
1,00
0,80
0,70
0,62
0,60
0,48
*
*«
X
ч
ь.
985
952
926
905
918
968
XI
с/Э
СО
- О
а
1,00
0,72
0,56
0,46
0,44
0,36
си
- о
а
0,00
0,14
0,22
0,29
0,31
0,40
о
в
0,34
0,33
0,32
0,31
0,31
0,34
.о
С/)
СО
к О
в \\ н
0,34
0,40
0,45
0,48
0,49
0,52
0,37
0,28
0,21
0,16
0,10
*
*«
X
t ч
ь*
1014
1048
1066
1092
1115
х>
00
СО
- О
а
0,28
0,21
0,16
0,08
0,06
си
- а
а
0,49
0,57
0,61
0,67
0,73
о
a
0,35
0,36
0,37
0,38
0,38
8
0,57
0,62
0,68
0,73
0,82
Значения степени диссоциации чистого антимонида галлия
при температуре Т находили из уравнения
In Кт = —ДЯ° {L)/RT + AS° (L)/#, (VI. 135)
где Кт — константа диссоциации чистого GaSb, Д#° и AS° —
стандартные энтальпия и энтропия реакции диссоциации GaSb
соответственно.
Величину A#0(L) рассматриваемой реакции определяли по
уравнению
Atf°(L)=2/i£(L)/(l-4L))' (VI.136)
где hEiL\ согласно формуле (V.212), равна
Лв<« = (АЯГ + АСл|),/2.
Значение A#/S) рассчитывали обычным способом из
стандартной теплоты образования GaSb [229]. Величину а0 принимали
равной 0,34 (см. табл. 3). Далее на основе уравнения (VI. 135)
вычисляли значение AS0(L). Полученные таким образом величины
A#°<L> и AS°<L> реакции диссоциации GaSb в жидкой фазе
оказались равными 19 590 Дж/моль и 3 Дж/(моль-К) соответственно.
Значения коэффициентов активности германия вдоль кривой
ликвидуса в рассматриваемой системе определяли из условия
фазового равновесия [см. формулу (V.142)], допуская отсутствие
разницы в теплоемкостях между твердым и переохлажденным
жидким германием и пренебрегая растворимостью антимонида
галлия в твердом германии. Указанные допущения не являются
грубыми, поскольку величина Acp.Ge, как правило, весьма
незначительна, а величина растворимости GaSb в Ge, по данным
[422], не превышает 1,5% (мол.).
11 Глазов В. М. я др. 305
4*CaSb
Активность антимонида галлия была рассчитана с помощью
уравнения Гиббса — Дюгема в предположении пренебрежимо
малой температурной зависимости активности германия.
Результаты оценки аЬе и abasb указанным способом приведены в табл. 4.
Подставив в уравнение (VI. 134) значения аЬе, abasb и а°, мы
рассчитали величины степени диссоциации антимонида галлия вдоль
кривой ликвидуса в системе GaSb — Ge (табл. 4). Из анализа
полученных данных следует, что по мере роста температуры и
степени разбавления антимонид галлия диссоциирует все в большей
степени. Наглядно поведение рассматриваемого соединения в его
растворах с германием
иллюстрируется на рис. 64, где представлена
температур но-концентрационная
(вдоль кривой ликвидуса)
зависимость степени диссоциации
антимонида галлия в системе GaSb—Ge.
Видно, чтов растворах, содержащих
до—36—40% (мол.) GaSb, антимонид
галлия диссоциирован весьма
существенно, что в полной мере
объясняет поведение легированных
галлием и сурьмой германиевых
растворов при вытягивании
монокристаллов германия методом Чох-
ральского, когда галлий и сурьма
захватываются по отдельности в
соответствии с их индивидуальными
коэффициентами распределения (см. работы [67, 423]). При
этом экспериментально построенная диаграмма состояния не
отражает равновесия между твердой и жидкой фазами,
поскольку линии, соединяющие составы жидкой и твердой фаз
при каждой данной температуре, не являются коннодами.
Сделанное заключение хорошо согласуется с данными
физико-химического анализа в рассматриваемой [424] и аналогичных системах
с привлечением метода вязкости [425].
Значительная диссоциация антимонида галлия в жидких
растворах на основе германия также получила прямое
подтверждение в опытах по рентгенографическому исследованию быстро-
охлажденных (со скоростью более 106 К/с) сплавов [426—428].
Расчеты, выполненные нами на других системах Ge — AluBv,
показали в общем картину, аналогичную представленной
на рис. 64.
Полученные результаты в совокупности с экспериментальными
данными [423—428 ] позволяют сделать заключение об отсутствии
равновесия между жидкой и твердой фазами подобно тому, как
это наблюдается в двойных системах, поскольку процесс
диссоциации чистого соединения, происходящий всегда в растворе,
усиливается вследствие эффекта разбавления.
306
Рис. 64. Изменение степени
диссоциации антимонида галлия вдоль
кривой ликвидуса в системе Ge—
GaSb
5. ОЦЕНКА СТРУКТУРНОГО ФАКТОРА В ЖИДКИХ
РАСТВОРАХ ПО КРИВИЗНЕ ЛИКВИДУСА В ДВОЙНЫХ
СИСТЕМАХ С КОНГРУЭНТНО ПЛАВЯЩИМИСЯ
СОЕДИНЕНИЯМИ
Системы с конгруэнтно плавящимся соединением
характеризуются содержанием в жидкой фазе не только атомов
составляющих компонентов, но и химических комплексов различного
состава. В этом случае, как было показано в работе [429],
величину среднеквадратичной флуктуации концентрации Sxx (0) можно
рассчитать, располагая сведениями только о константах
равновесий реакций, протекающих в растворе.
В соответствии с законом сохранения масс для одного брутто-
моля раствора компонентов Л и Б, содержащего, кроме отдельных
атомов А и В, также и химические комплексы Ат.Вп.> где / равно
3, 4, ..., k (k — общее число молекулярных форм в растворе),
справедливо:
k
п\= 1 -х2—Ът1пг (VI.137)
nl = x2- Ъщп), (VI.138)
k k
п =* Г. п) = 1 - £ (т.- + п: - \)п). (VI.139)
/=1 /-3
Здесь п* — число молей /-той молекулярной формы, п* — общее
число молей в одном брутто-моле раствора.
Согласно этим формулам,
^' = -1- £ту/г*\ (VI.140)
/=з
k
%' = 1-!."/"/'> (VI.141)
/=з
k
п' = — £ (my + nf - 1) п]\ (VI. 142)
/=з
где штрихом обозначена первая производная по концентрации.
Принимая во внимание выражения (VI. 137) и (VI. 138),
свободную энергию жидкой смеси можно представить в виде
k k
ё^Ъ я*/|1/ = (1 — *2) hi + *2H* — S п) дО/ +
/=1 /=3
+ RT £, п) \п(п]/п) + £ п]А,, (VI. 143)
11* 307
где
AGy = — RT In Кj = miV\ + np\ - \i°}; (VI. 144)
Aj = RTlnyr,
Kj — константа диссоциации /-того комплекса.
Продифференцировав уравнение (VI. 143) по концентрации
с учетом выражений (VI. 139)—(VI. 141), соотношения (IV.43)
и уравнения для константы диссоциации /-того комплекса
\ 1У1/ ^ v 2У2/ ; g/c (VI. 145)
л/Т//я
получим следующее выражение для структурного фактора смеси:
s-w-«■/(■&),.- £4
Vя/ ) fn*48
j«!7
/J П]
После дифференцирования уравнения (VI. 143) по давлению найдем
v = (dg/dp)T, к, = (1 — х2) v\ + x2t>2 —
k k
- Е «J AV/ + £ "/ (&fydp)r, * (VI. 147)
/=3 /—1
где AVj = (dAGy/dp)r. ,.
Продифференцировав уравнение (VI. 147) по концентрации и по
давлению, получим выражения для функций:
б = — (\/v) (dv/dx2)T, p, (VI.148)
Q = RTfa!v (VI. 149)
(Рт- — изотермический коэффициент сжимаемости), с
использованием которых по формулам (V.399) и (V.400) можно оценить
структурные факторы Snn (0) и Snx (0).
Расчет равновесного числа молей молекулярных форм,
присутствующих в растворе, а следовательно, и концентрационной
зависимости структурного фактора Sxx (0) связан, согласно
равенству (VI. 146), с использованием определенных приближений
для Yi> например идеального, регулярного или конформного
раствора, модели Флори [289—290].
Обычно применяют простейшее приближение идеального
ассоциированного раствора, когда все у,- = 1> поскольку наибольшие
силы межмолекулярного взаимодействия учитываются при этом
допущением о существовании химических комплексов.
Рассмотрим сначала наиболее простой случай, когда в смеси
образуются комплексы лишь одного вида.
308
Обозначим через п\, п2 и п| число молей А, В а комплексов
АтВп соответственно. Тогда уравнения (VI. 137)—(VI. 142)
приобретут вид:
п\ = 1 — х2 — mnl; п\ = х2 — тц; п = 1 — (т + п — 1) п*3,
(VI. 150)
«1 =—1— mnl'; п\ =1— nnl ; п = —{т-\-п—\)щ .
(VI.151)
Полагая Yi = Тг = 7л в . = 1, на основании формул (VI. 144)—
(VI. 147), получим 1429'] '
Sxx(0) = \iyifln) - (п'У/п'У, (VI.152)
6--т^)г.Р.*Г (i-^K+^-^V (VL153)
= -??-{(l -x2)vyTl) + x2v^2)+[^(nlAVj)]Tx}, (VI.154)
гдеЛ^-ЯГ^.
I 5p Jr,,, + кг ^ + „; + „; n* ) > (VI.155)
и = (1 — x2) v[ + ад — «3 6У;. (VI.156)
Принимая во внимание, что константа диссоциации химического
комплекса Ат.Вп не зависит от концентрации, с помощью
выражения
(пГ/iO111/ {пЦпр
Kf = - Чт^ — (VL157)
41п
и соотношений (VI. 150)—(VI. 151) можно убедиться, что
„♦' [{т + п)х2-п]ппъ
™3 = • * * . ; 7~о о * * * 2 * * ♦ * * ♦ ' (V1.10SJ
n\n2n3 (m + Л ~~ I) — т Л Л2Л3 ~~ П Л П1П3 ~~ Л Л1П2
Таким образом, опираясь только на значение К- комплекса Ат.Вп.,
решив совместно систему уравнений (VI. 150) и уравнение (VI. 157),
можно рассчитать истинные числа молей молекулярных форм
в растворе, а с помощью выражений (VI. 158) и (VI. 151) величины
п*\ Затем по формуле (VI. 152) можно определить структурный
фактор Sxx(0).
309,
Используя данные по плотности чистых компонентов и
соединения АтВпу по формуле (VI. 156) можно оценить величину AVjy
которая характеризует изменение объема при диссоциации
комплекса АтВп, Из уравнения (VIЛ54), привлекая данные о
сжимаемости чистых компонентов и соединения АтВПУ можно
рассчитать производную (d&Vj/dp)TiX. Подставляя полученные таким
образом значения ДУу и (dAVjldp)T%x в уравнения (VI. 153) и
(VI. 154), можно рассчитать изменение параметров б и 0 во всем
концентрационном диапазоне, а следовательно, и структурные
факторы Snn (0) и Snx (0).
От структурных факторов Sxyy Snn и Snx в длинноволновом
пределе легко перейти к парциальным структурным факторам
Фабера [291 ], используя следующие соотношения:
flii = в + [1/(1 - x2f\ - 26/(1 - х2) + 62S,, - х2/(\ - х2)у
а22 = 0 + [1/4 + 2Ь/х2 + б2] Sxx - (1 - х2)/х2у (VI. 159)
<h% = 9 + {б2 + (1 - 2л:2)б/[(1 - х2)х2] - 1/[(1 - х2)х2\\ Sxx+ 1,
где aif (q) = 1 + (n/V) J [*/у (г) - 1 ] *&<' d*r;
n — общее число частиц в объеме v\ gif (r) — парные функции
распределения.
Проиллюстрируем далее возможность расчета
концентрационной зависимости структурного фактора Sxx (0) на основе анализа
данных по кривизне ликвидуса в двойных системах.
Рассмотрим диаграмму состояния А — В с конгруэнтно
плавящимся соединением АтВп с узкой областью гомогенности и
практически не диссоциирующим в твердой фазе. Допустим, что
в системе имеются лишь равновесия вида (VI.39). Константу
(VI. 157) для процесса диссоциации АтВп по реакции (VI.39)
можно записать в виде
Kf = mmnnatf+n/{[\ + a0(m + n- l)f+л"1 (1 -a0)}. (VI. 160)
На основании выражения (VI. 160) с учетом (VI.46) легко
рассчитать константу диссоциации сложного соединения АтВп,
если известны радиус кривизны кривой ликвидуса при Тт, лтвпУ
а также теплота и температура плавления соединения. Значение /С/
при какой-либо другой температуре можно оценить, пользуясь
методикой, предложенной нами в п. 5 гл. VI. Этот способ был
использован авторами для расчета при помощи уравнения (VI. 152)
концентрационной зависимости структурного фактора Sxx (0) в
системе Cd — Sb при температурах 723, 823 и 903 К.
По имеющимся данным [219, 368], в этой системе образуется
одно устойчивое конгруэнтно плавящееся соединение моноанти-
монид кадмия и возможно образование метастабильных фаз более
310
сложного состава. Была рассмотрена следующая схема
диссоциации антимонида кадмия:
CdSbs^=;CdSbL^:CdL + SbL. (VI.161)
Возможность реализации равновесий с образованием более
сложных молекул не учитывалась.
Результаты оценки радиуса кривизны кривой ликвидуса
в точке плавления соединения CdSb, приведенные в табл. 3, были
получены на основе термографических исследований диаграммы
состояния Cd — Sb в области, прилегающей к составу CdSb в
интервале концентраций от 40 до ~57% (мол.) Sb 15 сплавов.
Константы диссоциации антимонида кадмия при температурах
823 и 903 К оценивали с помощью соотношения (VI. 135).
Полученное значение степени диссоциации CdSb в точке плавления
использовали для расчета термодинамических параметров
реакции диссоциации жидкого антимонида кадмия (VI. 161). Значение
теплоты образования твердого антимонида кадмия из жидких
компонентов A#f заимствовалось из работы [430]. Рассчитанные
энтальпия и энтропия диссоциации жидкого CdSb равнялись
12,3 кДж/моль и —1,2 Дж/(мольК) соответственно. Значения
константы и степени диссоциации антимонида кадмия равны
0,1439; 0,35 и 0,1687; 0,38 соответственно при температурах 823
и 903 К.
Чтобы проверить применимость модели идеального
ассоциированного раствора для описания поведения жидкой фазы в системе
Cd — Sb, последовательно сопоставляли интегральные и
парциальные мольные термодинамические свойства растворов,
рассчитанные для указанной модели, с экспериментальными
данными [359, 370, 374, 431, 432], что показало в итоге хорошее их
согласие.
В соответствии с этим был сделан вывод [433], что эта модель
может служить достаточно надежной основой описания
структурных особенностей расплавов в системе Cd — Sb.
Значения констант диссоциации жидкого антимонида кадмия,
полученные нами, были использованы для расчета с
использованием модели идеального ассоциированного раствора
концентрационной зависимости структурного фактора Sxx (0) жидких
сплавов в системе Cd — Sb по формуле
Sxx (0) - К )7"i + (n2J/n2 + (п;вУ1пАВ - \tCJIn... (VIЛ 62)
с учетом выражений (VI.150), (VI.151) и (VI.158).
На рис. 65 представлены рассчитанные на основании данных
по температурам ликвидуса вблизи моноантимонида кадмия
концентрационные зависимости флуктуации концентрации Sxx (0)
для жидких сплавов Cd — Sb при температурах 729, 823 и 903 К.
Видно, что изменение структурного фактора при всех указанных
температурах представляет собой симметричные кривые с двумя
311
Максимумами одинаковой высоты, между которыми находится
минимум. По мере увеличения концентрации сурьмы от нуля
значения структурного фактора возрастают, но находятся ниже
идеального. Максимальное значение Sxx (0) приходится на
концентрации ~0,25 и ~0,75 атомных долей сурьмы, минимальное
значение — на состав соединения. С ростом температуры глубина
минимума уменьшается.
Сравнение зависимостей, представленных на рис. 65, с
характером изменения избыточной энтропии смешения (рис. 66) поз-
8в,Дж/(моль-К)
0,25 0,5 Ot75 Sb
Атомная доля
Рис. 65. Концентрационная
зависимость флуктуации концентрации в жид*
ких сплавах Cd—Sb при различных
температурах. К:
1 — 729; 2 — 823; 3 — 903; 4 —
идеальный раствор
0,25 0,5 0,75
Атомная доля
Рис. 66. Избыточная интегральная
энтропия смешения расплавов при
температуре, К:
1 — 729; 2 — 823; 3 — 903
воляет сделать вывод, что основной вклад в избыточную энтропию
расплава Cd — Sb связан с изменениями структуры, т. е. носит
конфигурационный характер, тогда как другие вклады, такие
как колебательный, электронный и т. п. играют, по-видимому,
гораздо менее существенную роль.
Избыточная энтропия раствора в системах, подобных
рассмотренной, определяется (см. работу [102]) уменьшением числа
возможных способов ориентации мономерных частиц при
связывании их в комплекс и сопровождающих этот процесс уменьшением
числа независимых частиц в системе (отрицательный вклад),
а также — появлением третьей разновидности частиц
(положительный вклад). Очевидно, в случае системы Cd — Sb роль
последнего фактора становится превалирующей и избыточная энтропия
при формировании рассматриваемых растворов оказывается
положительной.
Если в системе содержится сложное соединение типа АтВп,
оно может диссоциировать различными путями. В каждом
конкретном варианте выбор схемы диссоциации требует специального
рассмотрения. Такой случай был проанализирован нами на
примере системы Mg — Sn [434], в которой образуется одно
конгруэнтно плавящееся соединение состава Mg2Sn.
312
Были рассмотрены следующие две схемы диссоциации Mg2Sn
в жидкой фазе:
Mg2Sn ^ 2Mg + Sn; Mg2Sn ^ MgSn + Mg. (VIЛ 63)
MgSn ^ Mg + Sn. (VI. 164)
В первом варианте допускалось, что жидкий раствор представляет
собой идеальную смесь только трех сортов частиц: мономеров Mg
и Sn и комплексов Mg2Sn. При выборе такой схемы диссоциации
константу диссоциации соединения Mg2Sn в точке плавления
можно рассчитать с помощью предложенной нами ранее формулы
(VI.46).
Экспериментальная оценка радиуса кривизны кривой
ликвидуса в точке плавления Mg2Sn была нами проведена на основе
детального термографического исследования 18 сплавов в
интервале концентраций от 20 до 50% (ат.) Sn (см. табл. 3).
Однако из сопоставления рассчитанных значений активностей
Mg и Sn с экспериментальными данными [4351 следует, что модель
раствора Mg — Sn, представляющая собой идеальную смесь трех
сортов частиц: мономеров Mg и Sn и комплексов Mg2Sn не
адекватна эксперименту.
Поэтому мы предприняли попытку рассмотреть более сложный
механизм диссоциации молекул Mg2Sn в жидкой фазе по схеме
(VI. 163). При этом допускалось, что жидкий раствор представляет
собой идеальную смесь четырех сортов частиц: Mg, Sn, Mg2Sn
и MgSn. Обозначим их числа молей в растворе соответственно
через rtu /i2, nt и гй.
Коэффициенты активности Mg и Sn можно представить в виде
_ *1 = Д1^П + *11*1*2 + 2/Ч*Г*2 /ут jgg4
71 *i ^„ + ^„«2+2^1*1**2 ' '
*2 * 1*11 + #11*1*2 + 2К\*\ *2 /лгт , ~Cv
72 = ^Г=* кк л-к S + kS* ' (VI. 166)
где x* — истинная мольная доля i-той молекулярной формы
в растворе; хъ х2 — аналитические мольные доли компонентов
Mg и Sn в расплаве соответственно:
Ki=*x№/xl; (VI. 167)
/Сп=*Г2*2/*з. (VI.168)
На основании формул (VI. 165) и (VI. 166) в пределе будем иметь
при х\ ->- 0, Хъ -> 1:
Y?-*i/0+*i). (VL169)
при х* -+ 1, х\ -+ 0:
$ - КгКпККгКц + КХ + *„). (VI. 170)
313
Таким образом, располагая данными о предельных значениях
коэффициентов активностей компонентов при достаточном
разбавлении у*, можно оценить величины констант образования
частиц Mg2Sn и MgSn из Mg и Sn. При расчетах значения у*9
заимствованные из работ [144, 360, 366], принимали равными
0,0078 и 0,0043 при 923 К и 0,0104 и 0,00080 при 1045 К для Mg
и Sn соответственно. Рассчитанные таким образом значения
констант К\ и Ки составляли 0,0134; 0,00085 и 0,0079; 0,00046 при
температурах 1045 и 923 К соответственно.
Основной недостаток такой методики оценки констант
диссоциации присутствующих в растворе молекулярных форм
заключается, во-первых, в том, что коэффициенты активностей
компонентов в разбавленных растворах измеряют с достаточно большой
погрешностью, и, во-вторых, в том, что величину у* одного из
компонентов устанавливают, как правило, не экспериментально,
а находят интегрированием по уравнению Гиббса — Дюгема,
что, несомненно, связано с включением дополнительных ошибок
в определении у*.
В этой связи нами была предпринята попытка рассчитать
константы диссоциации рассмотренных реакций при Т°т, Mg2sn,
опираясь на данные пор и активности Mg в точке плавления
соединения, и сопоставить их с полученными ранее результатами.
Для точки плавления соединения, согласно выражению (VI.40),
имеем
—ASm> Mg2Sn/(9/?T'mf MgoSnPmax) =
= (3 - ЗЛ - В)/[(2 - ЗЛ) (1 - Л)], (VI.171)
где А = 2п*3+п1 (VI.172)
В = 2лГ + лГ. (VI. 173)
Величину Л рассчитывали из данных работы [366] по активности
Mg в точке плавления соединения по формуле
А = (хг — ах)1{\ — аг).
Подставляя значение А в уравнение (VI. 171), находили
значение В.
Числа молей молекулярных форм в растворе, отвечающем
составу соединения при Т°т> Mg2sn, необходимые для расчета
констант К\ и /(п, находились совместным решением системы
уравнений (VI. 172) и (VI. 173). С этой целью, используя уравнения
закона действующих масс для реакций диссоциации (VI. 164),
записывали систему уравнений, решение которой относительно
производных nV и пХ с учетом уравнений материального
баланса:
п{ = Х\ — 2/Хз — nl, п*2 = х2 —• Пз —гц, п* = 1 — 2п% — /ij.
(VIЛ 74)
п9 = 1 - 2п* - пХ
314
позволяет выразить их через числа молей nl и nl. Подставив
полученные таким образом значения п\' и пХ (VI, 173) из системы
уравнений (VI. 172), (VI. 173), находили значения nl и /г*, а затем
по формулам (VI. 174) рассчитывали числа молей п*, nl и я*.
Из-за громоздкости указанные выкладки не приводим.
Рассчитанное описанным выше способом значение константы
диссоциации комплекса Mg2Sn на MgSn и Sn при 1045 К
составило 0,11, что достаточно хорошо согласуется с величиной 0,08,
найденной из данных по у*. Величины констант /(, и Ки оценить
не удалось.
SnxiO)
0,020
0,015
0,010
0,005
0
6
/~\
I _ i _ ±_
0,25 0,5 0,75 XSn
0,25 0,5 0,75 ;rSn
Рис, 67. Концентрационная зависимость структурных факторов в системе
Mg—Sn
С целью проверки пригодности модели раствора Mg — Sn,
представляющей собой идеальную смесь четырех сортов частиц:
Mg, Sn, Mg2Sn и MgSn, были последовательно сопоставлены
рассчитанные и экспериментально измеренные термодинамические
свойства растворов, что показало их хорошее согласие.
Следовательно, и в этом случае модель идеального
ассоциированного раствора может служить основой при описании
структурных свойств расплавов Mg — Sn.
В соответствии с выбранной таким путем схемой диссоциации
формулу для расчета Sxx (0) можно представить в виде
Sxx(0)
* \ *
+
(«Г)2 , «у к'у
2Ы
+
(VIЛ 75)
По этой формуле была рассчитана концентрационная зависимость
структурного фактора Sxx (0) жидких сплавов Mg — Sn.
Полученные результаты представлены на рис. 67, а при температурах
923 (кривая 3) и 1045 К (кривая 4). Здесь же для сравнения
приведена кривая Sxx (0), рассчитанная для модели идеального
ассоциированного раствора (2), состоящего только из трех сортов
частиц: Mg, Sn и Mg2Sn при 1045 К, а также кривая Sxx (0) для
идеального случая (/), не учитывающего взаимодействие
компонентов Mg и Sn в растворе.
315
Из рисунка видно, что зависимость структурного фактора
Sxx (0) жидких сплавов Mg — Sn от концентрации при 923 и
1045 К представляют собой асимметричные кривые с двумя
максимумами различной высоты, между которыми находится минимум,
отвечающий составу химического соединения Mg2Sn. С ростом
температуры высота максимума со стороны Sn в рассматриваемом
температурном интервале от 923 до 1045 К увеличивается, а со
стороны Mg почти не меняется. Сопоставление зависимости
структурного фактора Sxx (0) от состава в системе Mg — Sn с
изменением избыточной энтропии смешения показывает, что она
определяется не только конфигурационной частью. Очевидно,
существенную роль играют также непозиционные вклады.
С использованием данных по плотности [436] была сделана
попытка рассчитать концентрационную зависимость структурного
фактора S,u (0) жидких сплавов Mg — Sn по формуле (V.400)
с учетом выражения (VI. 148) при Т = 1073 К. Полученная
кривая Snx (0) (рис. 67, б) характеризует изменение корреляции
плотность — концентрация в зависимости от состава расплавов
Mg — Sn при содержании в нем частиц четырех сортов.
* ♦ *
В заключение нам хотелось бы подвести некоторые итоги и наметить пути
Дальнейшего поиска в этой интересной области физической химии. Прежде всего
следует подчеркнуть, что в деле термодинамического анализа фазовых диаграмм
двухкомпонентных систем достигнут определенный прогресс. Речь идет в первую
очередь об использовании диаграмм фазовых равновесий для извлечения
заложенной в них информации о термодинамических свойствах и строении фаз.
Наибольший успех в этом отношении достигнут при исследовании жидких растворов
в широком диапазоне составов. Этому способствовало использование
фундаментальных идей, заложенных в трудах Гиббса и Ван-дер-Ваальса. Наиболее
плодотворным при решении целого ряда задач оказалось использование
дифференциального уравнения Ван-дер-Ваальса, описывающего двухфазное равновесие в двух-
компонентной системе. В данном случае нам представляется целесообразным еще
раз подчеркнуть фундаментальный характер указанного уравнения, особенно
в связи с тем, что выведенные нами на его основе дифференциальные уравнения
кривых моновариантного равновесия в двухкомпонентных системах (кривые
ликвидуса и солидуса) полностью адекватны аналогичным уравнениям,
полученным И. Пригожиным на основе представлений о функции химического сродства по
де Донде.
Использование строгих термодинамических дифференциальных уравнений
кривых моновариантного равновесия для получения информации о
термодинамических свойствах и строении фаз конкретных систем с использованием фазовых
диаграмм требует либо привлечения дополнительной термодинамической
информации, либо введения модельных представлений. В первом случае задачу удается
выдержать термодинамически строго до конца.
Помимо рассмотренных в монографии, в последний год появились работы,
успешно решающие задачу этого плана [437, 438] *.
Во втором случае речь идет о решении обратной задачи, которая относится
к классу математически некорректных [439]. В отношении задачи, связанной
* См. также Дегтярев С. А. Развитие методов расчета термодинамческих
свойств двойных сплавов с использованием диаграмм состояний. Автореф.
канд. дис. М., 1981, МГУ им. М. В. Ломоносова.
316
с определением термодинамических свойств только на основе экспериментально
построенной фазовой диаграммы, мы встречаемся с различными источниками
неопределенности, среди которых в первуто очередь следует выделить:
а) незнание истинной модели раствора; б) наличие ошибок в определении
температур и составов фигуративных точек кривых моновариантного равновесия;
в) неоднозначность оценки параметров модели раствора вследствие наличия
значительных, а порой и непреодолимых математических трудностей расчетного
характера.
Поэтому в литературе иногда встречаются достаточно пессимистические
высказывания относительно возможностей успешного решения таких задач [440].
Однако, как отмечают А. Н. Тихонов и В. Я- Арсенин, уже сейчас разработаны
достаточно надежные методы решения некорректных задач по приближенным
данным, устойчивые к малым изменениям последних [439].
Поэтому любые попытки развития путей оценки термодинамических свойств
из данных по фазовой диаграмме представляют несомненный интерес. Авторы
отдают себе отчет, что выполненные ими расчеты и рассмотренный вариант возможности
оценок термодинамических свойств растворов по фазовой диаграмме не исчерпал
всех возможных источников улучшения результатов и поэтому обладает
ограниченной предсказательной силой. Однако уже сейчас путем последовательного
системного анализа, связанного с постепенным усложнением модели при
одновременном подборе хорошего алгоритма, удается получать устойчивые оценки
параметров и достаточно надежные значения активностей компонентов, интегральных
свободных энергий Гиббса и теплот смешения в широких концентрационных
диапазонах. Этого пока нельзя сказать в отношении энтропии смешений, хотя эта
величина даже при использовании экспериментальных данных оценивается крайне
ненадежно. В свете изложенного параметры моделей, представленные в табл. 2,
могут быть в принципе улучшены, а вместе с этим соответственно улучшены и
рассчитанные на их основе другие величины.
Среди проблемных задач, поставленных и частично решенных в данной
монографии, нам хотелось бы выделить проблему диссоциации конгруэнтно
плавящихся соединений в связи с тем, что внесение ясности в вопрос о термической
устойчивости соединения имеет первостепенное практическое значение в двух
аспектах: а) при разработке технологии получения полупроводниковых
соединений с заранее заданной степенью дефектности их структуры и б) при разработке
процесса сложного легирования полупроводников элементами донорного и
акцепторного типов в связи с анализом реакции донорно-акцепторного
взаимодействия, которая по сути эквивалентна реакции диссоциации соответствующего
донорно-акцепторного комплекса.
Указанные вопросы требуют дальнейшего развития и могут успешно решаться
на основе методов и результатов, изложенных в настоящей книге.
Отметим также, что, наверное, впервые в столь полной форме дано
систематическое рассмотрение природы и методов расчета ретроградного солидуса.
Тем не менее решение этого вопроса требует дальнейшего усовершенствования,
в первую очередь на основе более рационального выбора стандартного состояния
растворенного вещества.
Среди перспективных расчетных методов при подходе к оценкам
термодинамических и структурных свойств решением прямой задачи на основе фазовой
диаграммы вкупе с другими термодинамическими свойствами целесообразно выделить
метод сплайн-функций [441, 442], который был успешно применен к некоторым
металлическим и полупроводниковым системам в работах Г. Ф. Воронина и
С. А. Дегтярева [443].
Этот метод нам представляется особенно прогрессивным при подходе к
оценкам радиуса кривизны ликвидуса в точке плавления конгруэнтно плавящегоя
соединения.
Таким образом, даже беглый перечень проблемных вопросов, связанных
с основной темой монографии, и возможных путей их решения, показывает, что
требуются дальнейшие усилия многих исследователей для успешного и полного
решения главной задачи, касающейся развития наших представлений о строении
и свойствах фаз различной физико-химической природы на основе данных о
фазовых равновесиях.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Гиббс Дж. В. Термодинамические работы. Пер. с англ./Под ред. В. К. Се-
менченко. М.—Л., Гостехиздат, 1950. 493 с. с ил.
2. Планк М. Термодинамика. Пер. с нем. А. Н. Фрумкина. Л.—М.: Госиздат,
1925. 312 с. с ил.
3. Кричевский Я. Р. Понятия и основы термодинамики. 2-е изд., пересмотр,
и доп. М.: Химия, 1970. 440 с, с ил.
4. Второе начало термодинамики. [Сборник работ]. Сади Карно — В. Том-
сон Кельвин — Р. Клаузиус — Л. Больцман. — М. Смолуховский/Под ред.
и с предисл. Л. К. Тимирязева. М.—Л.: Гостехиздат, 1934. 311 с.
5. Ферми Э. Термодинамика. Пер. с англ. Б. А. Вайсмана. Харьков: изд.
Харьковского гос. ун-та, 1969. 140 с.
6. Карякин Я. Я., Быстрое К- Я., Киреев Я. С. Краткий справочник по
физике. М.: Высшая школа, 1969. 598 с.
7. Глазов В. M.t Чижевская С. Я., Глаголева Я. Я. Жидкие
полупроводники. М.: Наука, 1967. 244 с.
8. Радушкевич JI. В. Курс термодинамики. М., Просвещение, 1971. 288 с.
9. Киреев В. А. Краткий курс физической химии, изд. 4-е М.: Химия, 1970.
639 с.
10. Курс физической химии. Изд. 2-е/Под общ. ред. Я. И. Герасимова. М.:
Химия, Т. 1. 1969. 592 с, т. 2, 1973. 624 с.
11. Сторонкин Л. В. — Вестн. ЛГУ, 1956, № 16, с. 74—84.
12. Ван-дер-Ваальс Я. Д., Констамм Ф. Курс термостатики. Термические
равновесия материальных систем. Пер. с нем. Б. Я. Гордона./Под ред. А. В. Ра-
ковского. М.: ОНТИ, 1936, ч. I, 452 с; ч. II. 439 с.
13. Сторонкин А. В. Термодинамика гетерогенных систем, ч. 1—2. Л.: Изд-во
ЛГУ, 1967. 448 с.
14. Больцман JI. Лекции по теории газов. Пер. с нем./Под ред. Б. И. Давыдова.
М.: Гостехиздат, 1956. 557 с.
15. Сторонкин А. В., Жаров В. Т., Мариничев А. Я. — ЖФХ, 1976, т. 50.
№ 12, с. 3048—3057.
16. Путилов К- А. Термодинамика. М.: Наука, 1971. 375 с.
17. Глазов В. М., Новиков И. Я. — ЖФХ, 1974, т. 48, № 5, с. 1134—1137.
18. Гуггенгейм Е. А. Современная термодинамика, изложенная по методу Гиббса.
Пер. с англ./Под ред. С. А. Щукарева. М.—Л.: Госхимиздат, 1941. 188 с.
19. Мюнстер А. Химическая термодинамика. Пер. с нем. Е. П. Агеева./Под
ред. Я. И. Герасимова. М.: Мир, 1971. 296 с.
20. Schottky №., Ulick Я., Wagner С. Thermodynamic Berlin, J. Springer, 1973,
S. 619.
21. Сторонкин А. В., Белоусов В. Я.—ЖФХ, 1965, т. 39, № 1, с. 174—177.
22. Тойкка А. М.у Сусарев М. Я. — В кн.: Химическая термодинамика и
термохимия. М.: Наука, 1979, с. 156—159.
23. Тойкка А. М.9 Сусарев М. Я. — ЖПХ, 1978, № 12, с. 2704—2709.
24. Пригожин Я., Дефэй Р. Химическая термодинамика. Пер. с англ./Под ред.
В. М. Михайлова. Новосибирск: Наука, 1966. 510 с.
25. DeDonder Tn., Van Rysselberghe P. Thermodynamic Theory of Affinity. A book
of principles. Stanford: Univ. Press; London: H. Milford; Oxford: Univ. Press,
1936. 142 p.
26. Эпштейн П. С. Курс термодинамики. Пер. с англ. Н. М. Лозинской,
Н. А. Толстого. М.—Л.: Гостехиздат, 1948. 419 с.
27. Эренфест Я. — Журн. рус. физ. об-ва, 1909, т. 41, с. 347.
28. Duhem P. Le potentiel thermodynamique. Et ses applications a la mecanique
chimique et a Г etude des phenomenes cecetriques. Paris: Hermann, 1886.
29. Русанов А. Я., Шульц M. M. — Вестник ЛГУ, т. 15, № 4, Серия физики
и химии, 1960, № 1, с. 60—65.
30. Франкфурт У. Я., Френк А. М. Джозайя Виллард Гиббс. М.: Наука,
1964. 280 с.
31. Konowalow D, — Wied. Annalen, 1881, Bd 14, S. 34—52.
318
32. Вревский М. С. Работы по теории растворов. М.—Л.: Изд-во АН СССР,
1953. 334 с.
33. Сторонкин А. Об условиях термодинамического равновесия
многокомпонентных систем. Л.: Изд-во ЛГУ, 1948. 122 с.
34. Жаров В. Я., Коробов С. В. — В кн.: Химическая термодинамика и
термохимия. М.: Наука, 1979, с. 126—130.
35. Жаров В. Т., Сторонкин Л. В. — ЖФХ, 1975, т. 49, № 12, с. 3048—3052.
36. Молочко В. А.у Федоров Я. Я., Курдюмов Г. М. — Труды по химии и хим.
технолог. Горьковского гос. ун-та, 1969, вып. 3, с. 40—44.
37. Сторонкин А. В. —ЖФХ, 1966, т. 40, №8, с. 1673—1679.
38. Hayes Л., Chipman J. — Trans. AIME, 1939, v. 135, p. 85—132.
39. Мариничев А. Я., Сторонкин А. В. — В кн.: Химическая термодинамика
и термохимия. М.: Наука, 1979, с. 16—19.
40. Семенченко В. /(. — ЖФХ, 1975, т. 49, № 1, с. 247—249.
41. Глазов В. Af., Павлова Л. М. — ДАН СССР, 1978, т. 240, № 1, с. 104—107.
42. Млодзеевский А. Б. Термодинамика и теория фаз. Введение в учение о
состоянии вещества с точки зрения термодинамики. М.—Л.: Госиздат, 1922.
172 с.
43. Млодзеевский А. Б. Теория фаз с применением к твердым и жидким
состояниям. М.—Л.: ОНТИ, 1937. 122 с.
44. Млодзеевский А. Б. Геометрическая термодинамика. М.: Изд-во МГУ, 1956.
92 с.
45. Курнаков Я. С. Введение в физико-химический анализ. 3-е доп. изд. Л.:
ОНТИ, 1936. 194 с.
46. Петров Д. А. Тройные системы. М.: Изд-во АН СССР, 1953. 316 с.
47. Зломанов В. П. Р—Т—х диаграммы двухкомпонентных систем. М.: Изд-во
МГУ, 1980. 152 с, ил.
48. Новиков Я. Я. — ФММ, 1958, т. 6, № 4, с. 768.
49. Бочвар А. А. Металловедение. М.: Металлургиздат, 1956. 496 с, ил.
50. Крестовников А. Я., Вигдорович В. Я. Химическая термодинамика.
2-е изд. М.: Металлургия, 1973. 256 с.
51. Огородников В. В., Огородникова Л. Л. — ЖФХ, 1975, т. 49, № 1, с. 30—34.
52. Аносов В. #., Погодин С. Л. Основные начала физико-химического анализа.
М.—Л.: Изд-во АН СССР, 1947. 877 с.
53. Витторф Я. М. Теория сплавов в применении к металлическим системам.
Спб.: 1909. 434 с.
54. Древинг В. Я., Калашников Я- А. Правило фаз с изложением основ
термодинамики. 2-е изд. М.: Изд-во МГУ, 1964. 456 с.
55. Захаров М. В., Румянцев М. В., Туркин В. Д. Диаграммы состояния
двойных и тройных металлических систем. М.: Металлургиздат, 1940. 227 с, ил.
56. Райнз Ф. Диаграммы фазового равновесия в металлургии. Пер. с англ.
А. Г. Спектора./Под ред. Б. Г. Лифшица. М.: Металлургиздат, I960.
376 с. с ил.
57. Тамман Г. Руководство по гетерогенным равновесиям. Пер. с нем. Н. П. Луж-
ной, В. С. Егорова./Под ред. А. Г. Бергмана. Л.: ОНТИ, 1935. 328 с.
58. Финдлей Л. Правило фаз и его применение. Пер. с англ. В. В. Нестеровой/
Под ред. и с добавлениями А. В. Раковского. М.: ГОНТИ, 1932. 304 с.
59. Юм-Розери В., Христиан Дж., Пирсон В. Диаграммы равновесия
металлических систем. Пер. с англ. Т. Н. Кадыковой, Т. В. Краснопевцевой,
М. П. Равдель./Под ред. Я. П. Селисского. М.: Металлургиздат, 1956.
399 с, ил.
60. Бочвар А. А. Исследование механизма и кинетики кристаллизации сплавов
эвтектического типа. М.—Л.: ОНТИ, 1935. 82 с.
61. Захаров Л. М. Диаграммы состояний двойных и тройных систем. М.:
Металлургия, 1964. 300 с. с ил.
62. Van Laar У. /.—Z. Phys. Chem., 1908, Bd 63, H. 2, S. 216—253.
63. Кузнецов Г. М.у Кузнецова С. /С. — Неорг. матер., 1966, т. 2, № 4, с. 643—
648.
64. Свелин Р. Л. Термодинамика твердого состояния. Пер. с англ. И. Г. Ряб-
цева, В. П, Мамирева. М,: Металлургия, 1968. 316 с.
319
65. Meejering J. L. — Philips Res. Rept., 1948, v. 3, № 4, p. 281—302.
66. Van Laar J. J. — Z. Phys. Chem. (1908B) Bd 64, S. 257—297.
67. Глазов В. М., Земское В. С. Физико-химические основы легирования
полупроводников. М.: Наука, 1967. 372 с.
68. Юм-Розери В. Введение в физическое металловедение. Пер. с англ. В. М.
Глазова, С. И. Горина. М.: Металлургия, 1965. 204 с.
69. Менделеев Д. Я. Избр. соч., т. 3. Л.: ОНТИ. 1934, с. 210.
70. Новиков И. Я.— ДАН СССР, 1955, т. 100, № 6, с. 1119—1121.
71. Крестовников А. Я., Вигдорович В. Я. — ЖФХ, 1957, т. 31, № 6, с. 1345—
1351.
72. Крестовников Л. Я., Вигдорович Л. Я. — В кн.: Чистые металлы и
полупроводники. М.: Металлургиздат, 1959, с. 93.
73. Аптекарь И. Л., Исаева Л. Г. — ДАН СССР, 1975, т. 224, № 1,с. 120—123.
74. Лифшиц Б. Г. Металлография. М.: Металлургия, 1971, с. 150.
75. Лебедев Т. А. Некоторые вопросы общей теории сплавов. Л.: Лениздат,
1951, с. 27.
76. Prince A. Alloy Phase Equilibria. Amsterdam, 1966, p. 74.
77. Глазов В. М., Вигдорович В. Я. Микротвердость металлов и
полупроводников. 2-е изд. М.: Металлургия, 1969. 248 с.
78. Глазов В. М., Глаголева Я. Я., Корольков Г. А. — Изв. АН СССР. ОТН,
1957, № 8, с. 89—94.
79. Петров Д. Л., Буханова А. Л.—ЖФХ, 1954, т. 28, № 1, с. 161—172,
80. Льюис Дж., М. Рендалл. Химическая термодинамика. Пер. с англ. А. Н. Лук-
ницкого./Под ред. П. А. Ребиндера с доп. О. Редлиха. Л.: ОНТИ, 1936.
532 с.
81. Фаулер Р.> Гуггенгейм Э. Статистическая термодинамика. Пер. с англ.
И. И. Рогачева, Т. С. Рубинштейн. М.: ИЛ, 1949. 612 с.
82. Нернст В. Теоретическая химия с точки зрения закона Авогадро и
термодинамики. Спб., 1904. 620 с.
83. Hala Е. —Collect. Czech. Chem. Communics, 1979, v. 44, № 8, p. 2455—
2459.
84. Евсеев М. Л., Воронин Г. Ф. Термодинамика и структура жидких
металлических сплавов. М.: Изд-во МГУ, 1966. 131 с.
85. Бирон Е. В. — ЖРФХО, 1909, т. 41, с. 569.
86. Van Laar J. У. — Z. phys. Chem., 1910, Bd 72, № 6, S. 723—751.
87. Van Laar /. J. — Z. Phys. Chem., 1929, Bd 185, S. 35.
88. Hildebrand J. Я. — Proc. Nat. Acad. Sci., 1927, v. 13, p. 267.
89. Hildebrand J. Я. — J. Amer. Chem. Soc, 1929, № 51, p. 66—75.
90. Hildebrand J. Я., Prausnitz J. M., Scott R. L. Regular and Related
Solutions. N. Y., Van Nostrand Reinhold, 1970, p. 228.
91. Carlson Я. CColburnA. P. — Ind. Eng. Chem., 1942, v. 34, № 5, p. 581—
589.
92. Scatchard G. — Chem. Rev., 1931, v. 8, №2, p. 321—333.
93. Hildebrand J. Я., Scott R. L. Regular Solutions. Englewood (N.Y.):
Prentice-Hall, 1962, 180 p.
94. Гильдебранд Д. Г. Растворимость неэлектролитов. Пер. со 2-го англ. изд.
Б. Я. Герчикова, Б. Г. Зискинда./Под ред. М. И. Темкина. М.: ГОНТИ,
1938. 167 с.
95. Herzfeld К. F., Heitler W.—Z. Electrochem., 1925, Bd 31, S. 536—539.
96. Hildebrand J. Я., WoodS. E. —J. Chem. Phys., 1933, v. 1, № 12, p. 817—
822
97. Киреев В. Л. — ЖФХ, 1940, т. 14, № И, с. 1456—1468.
98. Darken L. S. — Trans. Metall. Soc. AIME. 1967, v. 239, № 1, p. 80—89.
99. Лесник А. Г. Модели межатомного взаимодействия в статистической теории
сплавов. М.: Физматгиз, 1962. 100 с.
100. Guggenheim E. A. Mixtures, Oxford, 1952, 270 p.
101. Rushbrooke G. S. Introduction to Statistical Mechanics. Oxford, The
Clarendon Press, 1949. 334 p.
102. Смирнова Н. А. Метод статистической термодинамики в физической химии.
М.: Высшая школа, 1973. 480 с.
320
103. Шахпаронов Mt Я. Введение в молекулярную теорию растворов. М.: Гос-
техиздат, 1956. 507 с.
104. Prigogine I. The Moleculare Theory of Solutions. Amsterdam,
North-Holland Publishing Company, 1957, 448 p.
105. Киттель Ч. Статистическая термодинамика. Пер. с англ. О. А. Ольхова.
Под ред. С. П. Капицы. М.: Наука, 1977. 336 с.
106. Guggenheim Е. А. — Trans. Faraday Soc, 1948, v. 44, p. 1007—1012.
107. Rastogi R. P.— J. Indian Chem. Soc, 1962, v. 39, JSfe 3, p. 215—219.
108. Колосовский H. А. Химическая термодинамика. Л.: Госхимиздат, 1932.
446 с.
109. Rastogi R. P., Nigam R. K. — Trans. Faraday Soc, 1959, v. 55, p. 2005—
2012.
110. Rastogi R. P., Nigam R. /0 — Proc Natl. Inst. Sci. India, I960, v. 26A,
p. 184—194.
111. Longuet-Higgins Я. С — Proc Roy. Soc, London, 1951, v. 205A, p. 247.
112. Rowlinson J. S. — Proc. Roy. Soc, London, 1952, v. 214A, p. 192—206.
ИЗ. Есин О. А. — Труды Ин-та металлургии УФ АН СССР, 1971, вып. №25,
с. 15—20.
114. Срывалин Я. Т., Есин О. Л., Ватолин Я. А. и др. — ЖФХ, 1968, т. 42,
№ 3, с. 717—722.
115. Hardy Я. /е. —Acta Metallurgica, 1953, v. 1, № 12, p. 202—209.
116. Teh-cheng Tien, Hsi-hsiung Chu, Acta Met. Sinica, 1965, v. 8, № 1,
p. 38—50.
117. Срывалин И. Г., Есин О. А. — Научн. докл. высшей школы. Сер.
металлургия, 1959, № 1, с 5—10.
118. Срывалин И. Т., Есин Т. Л., Ватолин Я. Л. — В кн.: Физическая химия
металлургических расплавов. Свердловск: Наука, 1969, вып. 18, с 3—44.
119. Rao М. 1/., HiskesR., Tiller W. Л. —Acta Metallurgica, 1973, v. 21, № 6,
p. 733—740.
120. RaoM. V., Tiller W. A. — Mater. Sci. and Eng., 1973, v. 11, №2, p. 61—68.
121. Sharkey R. jL., Pool M. /., Hoch Af. — Met. Trans., 1971, v. 2, № 11,
p. 3039—3049.
122. Кричевский Я. Р. — ЖФХ, 1944, т. 18, N° 3-4, с. 187—193.
123. Prigogine /., Garikian /. — Physica, 1950, v. 16, p. 239—248.
124. Prigogine /., Mathot V. — J. Chem. Phys., 1952, v. 20, p. 49—57.
125. Prigogine I., Bellemans A. — Discussions Faraday Soc, 1953, v. 15, p. 80—93.
126. Lennard-Jones J. E., Devoshire F. — Proc Roy. Soc, London, 1937, v. A163,
p. 53-70.
127. Lennard-J ones J. £., Devonshire F. — Proc Roy. Soc, London, 1938, v. A165,
p. 1—11.
128. Buchowski Я. Colloq. Nat. Centre Nat. Rech. Sci, 1965; Sept, 1966, p. 161—177.
129. Flory P. J. — J. Chem. Phys., 1941, v. 9, p. 660—661.
130. Flory P. /. — J. Chem. Phys., 1942, v. 10, p. 51—61.
131. Flory P. /. Principles of Polymer Chemistry: Cornell U. P., Ithaca (N. Y.),
1953, 672 pp. with ill.
132. Huggins M. L., Ann. N. Y. — Acad. Sci., 1942, v. 43, p. 1—32.
133. Huggins M. L. —J. Chem. Phys., 1941, v. 9, p. 440.
134. Kemeny S. — Kern, kozl., 1979, v. 51, M 3—4, p. 311—344.
135. Margules M. — Sitzber. Akad. Wiss. Wien, Mathemat. Natur. W., 1895,
AB/11, a Bd 104, S. 1243—1278.
136. Eliezer J., Howald R. A. — U. S. Dep. Commerce Nat. Bur. Stand. Spec.
Pull., 1978, № 496, p. 846—906.
137. Bale C. W.y Pelton Л. D. — Canad. Met. Quart., 1975, v. 14, № 3, p. 213—
219.
138. Bale C. W.y Pelton Л. D. — Met. Trans., 1974, v. 5, p. 2323—2337.
139. Williams 0. — Trans. Met. Soc AIME, 1969, v. 245, p. 2565—2570.
140. Brouwer N., Oonk J. Л. /. — Z. physik. Chem. Neue Folge, 1977, Bd 105,
H 3/4 S 113 123
141. Kmpkowski Л. — Bull. Acad. Polan Sci. et Lett,, 1950, ser. A, № 1, p. 15.
321
142. Krupkowski Л. —Bull. Acad. Polan Sci, ser. sei techn., 1959, v. 7, № 5,
p. 333—340.
143. Ptak W. — Arch. Hutnictwa, 1968, v. 13, № 3, p. 251—272.
144. Mozer Z., Fitzner K. — Thermodyn, Nucl. Mater., Vienna, 1975, v. 2,
p. 379—390.
145. Krupkowski Л., Fitzner K- — Arch. Hutnictwa, 1975, v. 20, №4,
p. 529—537.
146. Knobloch G. — Kristall uns Technik, 1975, Bd 10, № 6, S. 605—616.
147. Brebrick R. F. — Met. Trans., 1971, v. 2, № 6, p. 1657—1662.
148. Hiskes Д., Tiller W. Л. — Mater. Sci. Eng., 1968, v. 2, № 6, p. 320—330.
149. Смирнова H. A. — В кн.: Химия и термодинамика растворов. Вып. 2.
Л.: Изд-во ЛГУ, 1968, с. 3—42.
150. Смирнова Н. А. — Теорет. и эксперим. химия, 1974, т. 10, вып. 6, с. 781—
786.
151. Dolezalek F. — Z. Phys. Chem., 1908, Bd 64, S. 727—747.
152. Hildebrand J. tf., Scott R. L. The Solubility of Nonelectrolytes. 3-d ed.
New York: Dover Publishing Inst., 1964, 488 p.
153. KehiaianH. 1/., Sosnowska-Kehiaian /C. — Bull. Acad. Polan Sci, ser. sci
Chem., 1964, v. 11, № 10, p. 591—596.
154. Sarolea-Mathot L. — Trans. Faraday Soc, 1953, v. 49, p. 8-^20.
155. Stecki J. — Roczniki Chem., 1959, v. 33, p. 255—257.
156. Jordan A, S. — J. Electrochem., 1972, v. 119, JSfe 1, p. 123—127.
157. Malinovsky M., Safarikova A. Zb. pr. Chemicko-technol. fak. SVST 1975—
1976. Bratislava, 1978, p. 225—230.
158. Schroder I. F.—Z. phys. Chem., 1893, Bd 11, № 4, p. 449—465.
159. Шредер И. Ф. — Горный журнал, 1890, № 11, с. 272.
160. Tyrkiel E. — Metalloznawstwo i obrob. ciepl., 1978, № 34, str. 7—11.
161. Глазов В. M., Павлова Л. М.у Москвинова Н. Л. —ДАН СССР, 1975,
т. 225, Я° 5, с. 1096—1099.
162. Горбунов А. И. — Теорет. и эксперимент, химия, 1969, т. 5, № 6, с. 805—
163. Becker R.—Z. fur Metallkunde, 1937, Bd 29, № 8, S. 245—249.
164. Пинес Б. Я. — ЖЭТФ, 1943, т. 13, № 11—12, с. 411—417.
165. Пинес Б. Я- Очерки по металлофизике. Харьков: Изд-во Харьковского
гос. ун-та, 1961, 412 с, ил.
166. Данилов В. #., Каменецкая Д. С. — ЖФХ, 1948, т. 22, № 1, с. 69—79.
167. Каменецкая Д. С. — ЖФХ, 1948, т. 22, №1, с. 81—89.
168. Каменецкая Д. С. — ЖФХ, 1964, т. 38, № 1, с. 73—79.
169. Oonk Н. А. У., SprenkelsA. — Rec. trav. Chim., 1969, v. 88, № 11, p. 1313—
1331.
170. Каменецкая Д. С. — В кн.: Проблемы металловедения и физики металлов.
М.: Металлургиздат, 1949, с. 113—121.
171. Каменецкая Д. С. — ЖНХ, 1958, т. 3, №3, с. 607—610.
172. Глазов В. М., Крестовников А. И., Менделевич А. Ю. — ЖФХ, 1969,
т. 43, № 12, с. 3067—3069.
173. Глазов В. М., Крестовников Л. Я., Менделевич А. Ю. — В кн.:
Теоретические и экспериментальные методы исследования диаграмм состояния
металлических систем. М.: Наука, 1969, с. 116—119.
174. Костарев Л. Л.—ЖФХ, 1977, т. 51, №4, с. 929—931.
175. Bennet В. W., Shiftet G. У., Aaronson H. J. — Calphad, 1978, v. 2, № 3,
p. 281—284.
176. Roozeboom H. W. B.—Z. phys. Chem., 1893, Bd 12, № 3, S. 359—389.
177. Roozeboom H. W. B.—Z. phys. Chem., 1899, Bd 30, № 3, S. 385—412.
178. Oonk H. A. J. — Rec. trav. Chim., 1968, v. 87, № 12, p. 1345—1358.
179. Mager 7\, Lukas H. L„ Petrov G. — Z. Metallkunde, 1972, Bd 63, H. 10,
S. 638—646.
180. Mager Г., Petzow G. —Z. Metallkunde, 1972, Bd 63, H. 11, S. 702—709.
181. Кауфман Л., Бернстейн X. Расчет диаграмм состояния с помощью ЭВМ.
Пер. с англ. А. Л. Удовского, Г. И. Хохловой, Д. Б. Чернова./Под ред.
И. Л. Аптекаря, А. Я. Шеплева. М.: Мир, 1972. 328 с,
322
182. Foster L. M., Woods J. F. — J. Electrochem. Soc, 1971, v. 118, № 7,
p. 1175—1183.
183. Brebrick R. F., Panlener R. J. — J. Electrochem. Soc, 1974, v. 121, №. 7,
p. 932—942.
184. Романенко В, Я., Иванов-Омский В. Я. — ДАН СССР, 1959, т. 129,
No з с 553 555
185. StrinfeilowG. В., Green Р. Е. — J. Phys. Chem. Solids, 1969, v. 30, p. 1779—
1791.
186. Jordan A. 5. — Met. Trans., 1976, v. 78, p. 191—202.
187. Jordan A. S. — Met. Trans., 1970, v. 1, p. 239—249.
188. Jordan A. S. — Met. Trans., 1971, v. 2, p. 1959—1963.
189. Jordan A. S. — Met. Trans., 1971, v. 2, p. 1965—1970.
190. Guggenheim £. Л. — Trans. Faraday Soc, 1937, v. 33, p. 151—159.
191. Хубер Д. — В кн.: Процессы роста и синтеза полупроводниковых
кристаллов и пленок. Ч. 2. Новосибирск: Наука, 1975, с 212—218.
192. Laugier А. — Rev. Phys. Appl., 1973, v. 8, № 3, p. 259—270.
193. Jenkins C. Я. M. — J. Inst, of Met., London, 1926, v. 36, № 1, p. 63—97.
194. Stockdale D. — J. Inst, oi Met., London, 1930, v. 44, № 1, p. 75—80.
195. Raub E. — Z. fur Metallforschung, 1947, Bd 2, S. 119—120.
196. Raub £., Engel A. — Z. fur Metallforschung, 1946, Bd 1, S. 76—81.
197. Raub £., von Polaczek-Wittek A.—Z Am Metallkunde, 1942, Bd 34, S. 93—96.
198. Крегер Ф. Химия несовершенных кристаллов. Пер. с англ. В. П. Злома-
нова, В. А. Левицкого, Б. А. Поповкина./Под ред. О. М. Полторака.
М.: Мир, 1959. 656 с.
199. Thurmond С. £., Struthers J. D. — J. Phys. Chem., 1953, v. 57, p. 831—
835
200. Trumbore P. A. — Bell. Syst. Techn. J., 1960, v. 39, № 1, p. 205—234.
201. Глазов В. М. — ЖФХ, 1972, т. 46, Mb 3, с 606—611.
202. Киргинцев А. Я. — Изв. АН СССР. Сер. хим., 1971, № 6, с. 1313—1314.
203. Киргинцев А. Я., Исаенко В. А., Косяков В. Я. — Изв. АН СССР. Сер.
хим., 1970, Ня 2, с. 439—441.
204. Кузнецов Г. М. Исследования в области материаловедения контактов
металл—полупроводник. Автореф. дис на соискание учен, степени докт.
техн. наук. М., 1972 (Московский институт стали и сплавов).
205. Thurmond С. D., Kowalchik М. — Bell. Syst. Techn. J., 1960, v. 39, № 1,
p. 169—204.
206. Козловская В. M.t Рубинштейн Р. Я. — ФТТ, 1961, т. 3, с. 3354—3362.
207. Reiss Я.—J. Chem. Phys., 1953, v. 21, № 7, p. 1209—1217.
208. ReissЯ., Fuller C. S. —J. of Metals AIME Trans., 1956, v. 8, № 206,
p. 276—282.
209. Reiss Я., Fuller C. S., Morin F. — Bell Tel. Techn. J., 1956, v. 35, № 3,
p. 536—636.
210. Глазов В. М., Павлова Л. М. — ЖФХ, 1978, т. 52, № 4, с. 854—857.
211. Blakemore J. S. — Proc Phys. Soc, 1958, v. 71, p. 692—697.
212. Lehovec A".—J. Phys. Chem. Solids, 1962, v. 23, p. 695—709.
213. Глазов В. AT, Павлова Л. М. — Докл. АН СССР, 1977, т. 232, № 3, с. 607—
610.
214. Павлова Л. AL, Глазов В. М. — ЖФХ, 1978, т. 52, № И, с 2774—2778.
215. Vieland L. J. — Acta Metallurgica, 1963, v. 11, p. 137—142.
216. Nougaret P., Potier A. — J. Chem. Phys. et phys-chem. biol., 1969, v. 66,
№ 4, p. 764—768.
217. Кузнецов Г. Af., Леонов М. Я., Лукьянов А. С. — ДАН СССР, 1975, т. 223,
N° 1, с. 124—126.
218. Ротенберг В. Л., Кузнецов Г. М. — ЖФХ, 1974, т. 48, № 2, с 464—466.
219. Хансен М., Андерко К- Структуры двойных сплавов. Пер. с англ ./Под
ред. И. И. Новикова, И. Л. Рогельберга. М.: Металлургиздат, 1962.
220. Эллиот Р. П. Структуры двойных сплавов. Пер. с англ./Под ред. И. И.
Новикова, И. Л. Рогельберга. М.: Металлургия, 1970.
221. Eldridge J. Af., Miller £., Komarek К. L. — Trans. Met. Soc AIME, 1965,
v. 233, № 7, p. 1303—1308.
323
522. Eldrldge J. M., Miller £., Komarek K. L. — Trans. Met. Soc. AIME, 1966
v. 236, N° 1, p. 114—121.
223. GeffkenR.y Miller E. — Trans. Met. Soc. AIME, 1968, v. 242, № 11; p. 2320—
2328.
224. Brebrick R. F. — Met. Trans., 1976, v. 7A, N 10, p. 1609—1610.
225. OsamuraK., Murakami Y. — J. Phys. Chem. Solids, 1975, v. 36, № 9, p. 931—
937
226. Корпев В. В., Дутчак Я. Я., Коренчук Н. М. — ЖФХ, 1977, т. 51, № Ю,
с. 2563—2567.
227. Павлова Л. М.у Глазов В. М. — ДАН СССР, 1978, т. 241, № 6, с. 1371—
1374.
228. Legendre В., Souleau С. — Bull. Soc. Chim. Trans., 1973, № 7—8, p. 2202—
2206.
229. Термические константы веществ./Под ред. В. П. Глушко и др. М.: ВИНИТИ,
1972, вып. VI, с. 184.
230. Hultgren #., Desai, Pr. D.y Hawkins D. J. et al. — Selected values of the
thermodynamic properties of binary alloys. Amer. Soc. Metalls: Metals Park,
Ohio, 1973, 1435 p.
231. Глазов В. М.у Довлетов K-, Малкова А. С. — Электрон, техника. Сер.
матер., 1975, № 9, с. 74—78.
232. Глазов В. Af., Довлетов /С., Малкова А. С. — Электрон, техника. Сер.
матер., 1975, № 10, с. 64—68.
233. Глазов В. Af., Лебедева Л. В., Павлова Л. М. — Электрон, техника. Сер.
матер., 1973, № 7, с. 49—55.
234. Глазов В. Af., Павлова Л. М. — Неорг. матер., 1977, т. 13, № 1, с. 15—17.
235. Глазов В. М.у Павлова Л. Af., Акопян Р. А. — ЖФХ, 1978, т. 52, № 7,
с. 1811—1812.
236. Глазов В. М.у Павлова Л, М.у Грязева Н. Л. — Неорг. матер., 1975, т. И,
№ 3, с. 418—423.
237. Глазов В. М.у Павлова Л. Af., Евдокимов А. В. — ЖФХ, 1976, т. 50,
№ 10, с. 2489—2493.
238. Глазов В. Af., Павлова Л. М.у Лебедева Л. В. — В кн.:
Термодинамические свойства металлических сплавов. Баку: Изд-во ЭЛМ, 1975, с. 372—375.
239. Попов М. Af. Термометрия и калориметрия. М.: Изд-во МГУ, 1954. 942 с.
240. Новоселова А. В.у Пашинкин А. С. Давление пара летучих халькогенидов
металлов. М.: Наука, 1978. 112 с, ил.
241. Глазов В. М.у Бурханов А. С, Салеева Н. Af. —ЖФХ, 1975, т. 49, № 7,
с. 1658—1661.
242. Глазов В. М.у Коренчук Н. Af. — ЖФХ, 1971, т. 45, № 8, с. 2107—2208.
243. Глазов В. Af., Пашинкин А. С, Бурханов А. С. — ЖФХ, 1978, т. 52,
Но 5, с. 1136—1138.
244. Strinfellow G. Б.—Mat. Res. Bull., 1971, v. 6, p. 371—380.
245. PaulingL. C. The Nature of the Chemical Bond 3-d ed., Ithaca, New York:
Cornell University Press, 1960. 644 p.
246. Герасимов Я- Я., Глазов В. Af., Лазарев В. Б. и др. — ДАН СССР, 1977,
т. 235, Кя 4, с. 846—849.
247. Герасимов Я- Я., Глазов В. М.у Лазарев В. Б., Жаров В. В. — ДАН
СССР, 1978, т. 242, № 4, с. 868—871.
248. Герасимов Я. И.у Глазов В. М.у Лазарев В. 5., Жаров В. В. — ЖФХ,
1979, т. 53, № б, с. 1361—1368.
249. Глазов В. М.у Павлова Л. М.у Передерий Л. И. — Неорг. матер., 1980,
т. 16, № 4, с. 595—598.
250. Глазов В. М.у Павлова Л. М.у Передерий Л. И. — Неорг. матер., 1977,
т. 13, № 2, с. 209—213.
251. Угай А, А.у Соколов Л. И.у Гончаров Е. Г. — В кн.: Полупроводниковые
материалы и их применение. Воронеж: Изд-во ВГУ, 1974, с. 71—76.
252. Павлова Л. Af., Передерий Л. И. — В кн.: Проблемы микроэлектроники.
М.: МИЭТ, 1978, вып. 38, с. 96—101.
253. Thurmond С. D. — J. Phys. Chem. Solids, 1965, v. 26, JSfe 5, p. 785—802.
324
254. Boomgard Л, Schol К. — Philips Res. Kept., 1957, v. 12, p. 127—140.
255. Бармин Л. #., Есин О. Л., Добровинский Я. Е. — ЖФХ, 1970, т. 44,
№ 10, с. 2560—2563.
256. Герцрикен Я., Дехтяр Я. Я. Диффузия в металлах и сплавах в твердой
фазе. М.: Физматгиз, 1960. 182 с.
257. Мелвин-Хьюз Э. А. Физическая химия. Пер. с англ. Е. Н. Еремина,
О. М. Полторака, Ю. В. Филиппова./Под ред. Я. И. Герасимова, т. 2,
М.: ИЛ, 1962. 679 с.
258. Бачинский А. И. Избранные труды. М.: Изд-во АН СССР, 1960, с. 51.
259. Глазов В. М., Белоусов В. У., Щеликов О. Д. — ЖФХ, 1977, т. 51, № 9,
с. 2288—2292.
260. Глазов В. М., Щеликов О. Д.у Белоусов В. У. — Физ. и техн.
полупроводников, 1976, т. 10, № 10, с. 1998—2001.
261. Есин О. Л.— ЖФХ, 1969, т. 43, № 1, с. 228—231.
262. Ухов В. Ф., Есин О. А., Ватолин Н. Л., Дубинин Э. Л. — Труды
института металлургии УФАН СССР, 1969, вып. 18, с. 87—103.
263. Ухов В. Ф., Есин О. А., Ватолин Н. Л., Дубинин Э. Л. — В кн.:
Термодинамические и термохимические константы. М.: Наука, 1970, с. 114—119.
264. Бублик В. Т., Горелик С. С, Зайцев А. Л. Твердые растворы
элементарных полупроводников и полупроводниковых соединений. Научн. труды
МИСиС, 83. М.: Металлургия, 1974, с. 48—60.
265. Кривоглаз М. Л., Смирнов А. Л. Теория упорядочивающихся сплавов. М.:
Физматгиз, 1958. 388 с.
266. Кривоглаз М. Л. Теория рассеяния рентгеновских лучей и тепловых
нейтронов реальными кристаллами. М.: Наука, 1967. 336 с.
267. Вагнер К. Термодинамика сплавов. Пер. с англ. А. Г. Спектора./Под ред.
А. А. Жуховицкого. М.: Металлургиздат, 1957. 180 с.
268. Ламсден Дж. Термодинамика сплавов. Пер. с англ. В. Н. Вигдоровича
и В. М. Глазова./Под ред. Н. Н. Сироты. М.: Металлургиздат, 1959. 440 с.
269. Hauffe /(., Wagner C.—Z. Electrochem, 1940, Bd 46, № 3, S. 160—170.
270. Shottky W. F., Bever M. B. — Acta Metallurgica, 1958, v. 6, № 5, p. 320—
326.
271. Wagner C. — Acta Metallurgica, 1958, v. 6, № 5, p. 309—319.
272. Sauerwald F., Brand P., Mem W. — Z. fur Metallkunde, 1966, Bd 57, № 2,
s. 103—108.
273. Воронин Г. Ф. — В кн.: Современные проблемы физической химии. М.:
Изд-во МГУ, 1976, т. 9, с. 29—31.
274. Воронин Г. Ф. — ДАН СССР, 1971, т. 196, № 1, с. 133—135.
275. Hiskes R. — Generation of chemical potential by analysis of phase diagrams.
Stanford, Calif.: Stanford Univ., 1968, 264 p. (From Dis. Abst. В 1969, v. 29,
№ 11, p. 4198).
276. Hiskes R., Tiller W. Л. — Mater. Sci Eng., 1969, v. 4, № 2—3, 163—
172.
277. HiskesR., Tiller W. A. — Mater. Sci. Eng., 1969, v. 4, № 2—3, p. 173—184.
278. Ланцош. К- Практические методы прикладного анализа. Справочное
руководство. Пер. с англ. М. 3. Кайнера./Под ред. А. М. Лопшица. М.:
Физматгиз, 1961. 517 с.
279. Proks J. — Chem. zvesti, 1978, v. 32, №4, p. 433—440.
280. Kleppa O. /., Weil J. Л. — J. Amer. Chem. Soc, 1951, v. 73, p. 4848—
4850.
281. Malinowsky M. — Chem. zvesti, 1971, v. 25, №2, p. 92—96.
282. Bhatia A. 5., March N. H. — Physics Letters, 1972, v. 41A, № 5, p. 397—399.
283. Bhatia А. В., March. N. H. — Physics Letters, 1975, v. 51A, № 7, p. 401—
402.
284. Bhatia А. В., March N. H. —J. Phys. Metal Phys., 1975, v. 5, p. 1100—
1106.
285. Bhatia A. B.y March N. H. — Phys. Chem. Liquids, 1975, v. 4, p. 279—283.
286. Bhatia А. В., Thornton D. E. — Phys. Rev. В., 1970, v. 2, № 8, p. 3004—
3012.
325
287. Ansara /. — Int. Met. Rev., 1979, v. 24, No. 1, p. 20—53.
288. Bennet L. Я., Kahan D. J.9 Carter J. С — Mater. Sci. and Eng., 1976,
v. 24, № 1, p. 1—17.
289. Bhatia А. В., Hargrove W. Я., March N. H. — J. Phys. Solid State Phys.,
1973, v. 6, p. 621—630.
290. Bhatia A. B. — In: Liquid Metals, Ed. by R. Evans and D. A. Greenwood.
Conference Ser. Number 30, The Institute of Physics Bristol and London,
1976, p. 21—28.
291. Faber T. E. Introduction to the Theory of Liquid Metals, Cambridge U. P.,
London, 1972, 587 pp. with ill.
292. Хартман K-, Лецкий Э., Шефер Б. Планирование эксперимента в
исследовании технологических процессов. Пер. с нем. Г. А. Фомина и Н. С. Лец-
кой./Под ред. Э. К. Лецкого. М.: Мир, 1977. 552 с.
293. Бард Й. Нелинейное оценивание параметров./Пер. с англ. В. С. Дуженко
и Е. С. Фоминой./Под ред. В. Г. Горского. М.: Статистика, 1979. 336 с.
294. Bjorn V. Model Fitting and Discrimination for Vapour-Liquid Equilibrium
Calculation. A critical Evolution of Liquid Mixtures Models, Goteborg, 1978,
108 p.
295. Denting W. E. Statistical Adjustement of Data N. Y., Wiley, 1943.
296. JIuhhuk /О. В. Метод наименьших квадратов и основы математико-стати-
стической теории обработки наблюдений. Изд. 2-е. М.: Физматгиз, 1962.
349 с.
297. Клепиков Н. Я., Соколов С. Н. Анализ и планирование экспериментов
методом максимума правдоподобия. М.: Физматгиз, 1964. 184 с.
298. Рао С. Р. Линейные статистические методы и их применение. Пер. с англ.
А. М. Кагана и др./Под ред. акад. Ю. В. Линника. М.: Наука, 1968. 546 с.
299. Gulglion С, Domenech 5., Enjalbert M. — J. phys. chim. et phys. chim. biol.,
1977, v. 74, No. 9, p. 867—874.
300. Clifford A. A. Multivariate Error Analysis. N. Y., Halsted Press Wiley,
1973.
301. Дрейпер Н. P., Смит Г. Прикладной регрессионный анализ./Пер. с англ.
и под ред. Ю. П. Адлера и В. Г. Горского. М.: Статистика, 1973. 392 с.
302. Федоров В. В. Теория оптимального эксперимента. М.: Наука, 1971,312 с, ил.
303. Белеванцев В. И., Малкова В. И., Пещевицкиц Б. И. — Изв. Сиб. отд.
АН СССР, 1977, № 14, сер. хим., вып. 6, с. 35—40.
304. Mezaki #., Draper N. R., Johnson R. A. Ind. anl Eng. Chem. Fundam.,
1973, v. 12, № 2, p. 251—254.
305. Химмельблау Д. Прикладное нелинейное программирование. Пер. с англ.
И. Н. Быховской и Б. Т. Вавилова. Под ред. М. Л. Быховского. М.: Мир,
1975. 534 с, ил.
306. Reklaitis J. V., Phillips D. Т. — AIIE Trans., 1976, № 3, p. 235—256.
307. Powell P. #., MacDonald J. R. —Computer J., 1972, v. 15, p. 148—154.
308. Anderson T. F., Abrams D. S., Grens E. A. — AJChE Journal, 1978,
v. 24, № 1, p. 20—28.
309. Sutton T. L., MacGregor J. F. — Can. Journal Chem. Eng., 1977, v. 55,
N° 5, p. 602—608.
310. Brebrick R. Л — Met. Trans., 1971, v. 2, № 12, p. 3377—3383.
311. Brebrick R. F. — Met. Trans., 1977, v. 8A, № 3, p. 403—414.
312. Rao M. V.t Tiller W. A. — J. Phys. Chem. Sol., 1970, v. 31, p. 191—198.
313. Damert K. — Chem. Techn. (DDR), 1976, Bd 28, № 2, S. 74—76.
314. Nowak M., Flork W. Proceedings of the 5th symposium Computers in
chemical Engineering, 1977, The High Tatras, CSSR, с 111—114.
315. Van Ness Я. С, Pedersen F., Rasmussen P. — AJChE Journal, 1978, v. 24,
№ 6, p. 1055—1063.
316. Lukas H. L., Henig E. Th.> Zimmermann B. — CALPHAD, 1977, t. 1, № 3,
p. 225—236.
317. Panish M. В., Ilegems M, — In: Progress in Solid State Chemistry, v.7,
chapt 2, H. Reiss and J. O. McCaldin, eds. Oxford, Pergamon Press, 1972,
p. 39—83.
326
318. Rao M. V., Tiller W. A. — J. Mater. Sci., 1972, v. 7, p. 14—18.
319. Rao M. 1Л, Tiller W. A. — J. Mater. Sci. and Eng., 1974, v. 14, p. 47—54.
320. Hoshino Я., Nakamura Y.> Shimoji M.t Niwa K. — Ber. Bunsenge Phys.
Chem., 1965, Bd 69, № 2, S. 114—118.
321. Глазов В. M., Павлова Л. М. — ЖФХ, 1976, т. 50, № И, с. 2764—2768.
322. Шанк Ф. А. Структуры двойных сплавов. Справочное издание. Пер. с англ./
Под ред. И. И. Новикова, И. Л. Рогельберга, М.: Металлургия, 1973.
323. Itagaki /С-, Yагата А. — Res. Inst. Miner. Dressing Metall, Tohoku Univ.
Sendai, 1975, v. 39, № 8, p. 880—887.
324. Predel В., OehmeG. — Z. fur Metallkunde, 1976, Bd 67, № 12, S. 826—834.
325. Глазов В. M. — Изв. АН СССР. ОТН, 1960, N° 5, с. 190—194.
326. Горбов С. Я. Термодинамика полупроводниковых соединений AnI^v. Итоги
науки и техники. Сер. Химическая термодинамика и равновесия./Под ред.
Я. И. Герасимова. Т. 3. М.: Изд-во ВИНИТИ АН СССР, 1975. 151 с.
327. Каратаев В. В., Мильвидский М. Г., Немцова Г. А. — Неорг. матер.,
1975. т. 11, № 5, с. 830—834.
328. Predel В., SteinD. W. — J. Less-Common Metals, 1971, v. 24, № 4, p. 391—
403.
329. Gambino N., Bros /.-P. — J. Chem. Thermodyn., 1975, v. 7, p. 443—451.
330. Chao Peng-nien, Mo Chin-chi. — Acta Met. Sinica, 1965, v. 8, p. 32.
331. Bergman C, Laffite M., Maggianu У. — High Temperature High Pressures,
1974, v. 6, № 1, p. 53—60.
332. Герасименко Л. Я., Кириченко И. В., Ложкин Л. Я., Морачевский А. Г.—
В кн.: Защитные металлические и оксидные покрытия, коррозия металлов
и исследование в области электрохимии. М.—Л., Наука, 1965.
333. Данилин В. Я., Яценко С. Я. — В кн.: Труды Института химии Уральск,
филиала АН СССР, Свердловск, 1970, вып. 20, с. 142—145.
334. Вечер А. Л., Мечковский Л. Л., Скоропанов А. С. — Неорг. матер., 1974,
т. 10, № 12, с. 2140—2143.
335. Кубашевский О., Эванс Е. Термохимия в металлургии. Пер. с англ.
К. А. Новосельцева./Под ред. А. Ф. Капустинекого. М.: ИЛ, 1954. 424 с.
336. Полупроводниковые халькогениды и сплавы на их основе./Абрикосов Н. X,.
Банкина В. Ф., Порецкая Л. В., Скуднова Е. В., Чижевская С. Н./М.:
Наука, 1975. 220 с. с ил.
337. Castanet R., Bergman С. — J. Chem. Thermodyn., 1977, v. 9, p. 1127—1132.
338. Maekawa Г., Yokokawa Г., Niwa /C. — J. Chem. Thermodyn., 1972, v. 4,
p. 153—157.
339. Predel В., Piehl /., Pool M. J. — Z. fur Metallkunde, 1975, Bd 66, H. 5,
S. 268—274.
340. Глазов В. Af., Чижевская С. Я. — ДАН СССР, 1962, т. 145, № 1, с. 115—
118.
341. Глазов В. М., Чижевская С. Я. — ЖНХ, 1962, т. 7, № 8, с. 1933—1937.
342. Аббасов А. С, Никольская А. В., Герасимов Я- Я., Васильев В. Я. — ДАН
СССР, 1964, т. 156, № 6, с. 1399—1401.
343. Напп Я., Burow F. — Angew Chemie, 1956, Bd 68, № 11, S. 382.
344. Родионов А. В., Евсеев А. М. — В кн.: Термодинамические и
термохимические константы. М.: Наука, 1970. с. 67—73.
345. Абрикосов Я. X., Шелимова Л. Е. Полупроводниковые материалы на
основе соединений AlvBvl. M.: Наука, 1975. 197 с.
346. Садыков К. В., Семенкович С. А. — В кн.: Химическая связь в
полупроводниках и термодинамика. Минск: Наука и техника, 1966, с. 153—157.
347. Bergman С, Castanet R. — Ber. Bunseng., 1976, Bd 80, № 8, S. 774—775.
348. Карапетьянц М. X., Карапетьянц М. Л. Основные термодинамические
константы неорганических и органических веществ. М.: Химия, 1968.
471 с.
349. Maekawa Г., Yokokawa Т., Niwa К— J. Chem. Thermodyn., 1971, v. 3,
№ 1, p. 143—146.
350. Blachnik R.y Enninga E. — Thermochim. Acta, 1974, v. 9, p. 83—86.
351. Brebrick R. F. — J. Phys. Chem. Solids, 1969, v. 30, p. 719—731.
327
352. Глазов В. M.t Крестовников А. Я., Глаголева Я. Я. — Неорг. матер.,
1966, т. 2, № 3, с. 468—475.
353. Порецкая Л. В., Абрикосов Я. Л\, Глазов В. М. — ЖНХ, 1963, т. 8,
№ 5, с. 1196—1198.
354. Мелех Б. Т., Семенкович С. А. — Неорг. матер., 1967, т. 3, № 7, с. 1265—
1266.
355. Beardmor P., Howlett В. W.y Lichter В. D. — Trans. Met. Soc. AIME, 1966,
v. 236, № 1, p. 102—108.
356. Еременко В. Я., Лукашенко Г. Я. — Неорг. матер., 1965, т. 1, № 8,
с. 1296—1297.
357. Киреев В. А. Методы практических расчетов в термодинамике химических
реакций. 2-е изд. М.: Химия, 1975. 536 с.
358. Самсонов Г. В., Перминов В. М. Магниды. Киев: Наукова думка, 1971,
344 с, ил.
359. Selected Values of Thermodynamic Properties of Metals and Alloys. [Hult-
gren R., Orr R. L., Anderson P. D., Kelly К. К.] N. Y.: John Wiley and
Sons., 1963.
360. Еременко В. Я., Лукашенко Г. М. — Укр. хим. журн., 1963, т. 29, № 9,
с. 896—900.
361. Eldridge J. AT, Miller £., Kotnarek К. L. — Trans. Met. Soc. AIME, 1967,
v. 239, № 6, p. 775—781.
362. Nauak A. /f.f Oelsen W. — Trans. Indian Inst. Metals., 1971, v. 24, № 6,
p. '66—73.
363. Sharma R. A. — J. Chem. Thermodyn., 1970, v. 2, № 3, p. 373—389.
364. Лантратов М. Ф. — ЖНХ, 1959, т. 4, с. 1415—1419.
365. Срывалин И. Г., Есин О. Л., Лепинских Б. М. — ЖФХ, 1964, т. 38,
№ 5, с. 1166—1172.
366. Eldridge J. М., Miller £\, Komarek К. L. — Trans. Met. Soc. AIME, 1966,
v. 236, № 8, p. 1094—1098.
367. Knappwost A.—Z. phys. Chemie (Frankfurt), 1959, Bd 21, S. 358—375.
368. Полупроводниковые соединения группы AllBv. [Лазарев В. Б.,
Шевченко В. Я., Гринберг Я. X., Соболев В. В.]. М.: Наука, 1978, 256 с, ил.
369. Elliott J. F., Chipman J. — Trans. Faraday Soc, 1951, v. 47, p. 138—148.
370. Geffken 7?., Kotnarek K. L., Miller E. — Trans. Met. Soc. AIME, 1967, v. 239,
p. 1151—1160.
371. Masse J. D. G., Orr R. L., Hultgren R. — Trans. Met. Soc. AIME, 1966,
v. 236, № 8, p. 1202—1204.
372. Scheil £., Lukas Я. L. — Z. Metallkunde, 1961, Bd 52, S. 417—422.
373. Seltz Я., De Witt B. J. — J. Amer. Chem. Soc, 1938, v. 60, p. 1305—1308.
374. Wittig F. £\, Gehring E. — Ber. Bunsenges. Phys. Chem., 1966, Bd 70,
№ 7, S. 717—723.
375. BakerE. Я. — Trans. Inst. Mining and Met., sect., C, 1969,, v. 78C, p. 83—86.
376. BrayfordJ. R., WyattP. A. H. — Trans. Faraday Soc, 1956, v. 53, p. 642—
646.
377. Scheil E.—Z. Metallkunde, 1942, Bd 34, S. 242—246.
378. Курнаков Я. С, Жемчужный С. Ф. — ЖРФХО, 1912, т. 44, с. 1964—
1983.
379. Есин О. А. — Изв. сектора физ.-хим. анализа, 1949, т. 17, с. 38—63.
380. U Chatelier — Compt. rend., 1889, v. 108, p. 566—567; 801—803.
381. Roozeboom Я. W. B. — Z. phys. Chem., 1889, Bd 4, № 1, S. 31—65.
382. Roozeboom H. W. B. — Z. phys. Chem., 1892, Bd 10, № 4, S. 477—593.
383. Пацукова Н. Н. Исследование в области термодинамики двойных солей.
Канд. дис, М., ИОНХ им. Н. С. Курнакова, 1953.
384. Ruer R. — Z. phys. Chem., 1907, Bd 59, № 1, S. 1—16.
385. Van Laar J. J. —Z. phys. Chem., 1909, Bd 60, H. 2, S. 197—220.
386. Млодзеевский А. Б. — Изв. Ин-та физ.-хим. анализа, 1928, т. 4, № 2,
с. 247—271.
387. Шишкин Я. Я.—ЖОХ, 1940, т. 10, с 12—13.
388. Агеев Я. В., Макаров Е. С. — ЖОХ, 1943, т. 13, с. 242—248.
389. Иванов О. С, — Изв. АН СССР. Отд. хим. наук, 1944, № 4, с. 192—200,
328
390. Сирота Я. Я. — ДАН СССР, 1944, т. 44, с. 331— 335.
391. Млодзеевский Л. Б. — Изв. Ин-та физ.-хим. анализа, 1933, т. 6, N° 1$
с. 13—18.
392. Млодзеевский Л. Б. — Изв. сектора физ-хим. анализа, 1943, т. 16, № lj
с. 13-18.
393. Есин О. Л. — Изв. сектора физ.-хим. анализа, 1949, т. 19, с. 151—154.
394 Zernike J. — Chemical Phase Theory. A comprehensive treatise on the
deduction, the applications and the limitation of the phase rule Deventer, Klu-
ver, 1957. 494 p.
395. Hodgkinson R. J, — J. Electronics, 1956, v. 1, № 6, p. 612—624.
396. Hodgkinson R. J. — J. Electronics, 1956, v. 2, № 2, p. 201—203.
397. Липин Я. В, —Изв. Ин-та физ.-хим. анализа, 1928, т. 4, с. 49—58.
398. Липин Я. В. — Изв. Ин-та физ.-хим. анализа, 1928, т. 4, с. 59—64.
399. Степанов Я. Я. — Изв. Ин-та физ.-хим. анализа, 1928, т. 4, № 1, с. 327—
330.
400. Глазов В. M.t Павлова Л. М. —ДАН СССР, 1975, т. 225. № б, с. 1347—
1350.
401. Глазов В. M.t Павлова Л. М. — Неорг. матер., 1977, т. 13, № 2, с. 217—
221.
402. Mauntford G. Л., Wyatt P. А. Я. — Trans. Faraday Soc, 1966, v. 62,
p. 3201—3216.
403. Wyatt P. Л. Я. — Trans. Faraday Soc, 1956, v. 52, p. 806—815.
404. Wyatt P. Л. Я. — Trans. Faraday Soc, 1960, v. 56, № 448, p. 490—497.
405. Глазов В. M.t Павлова Л. М. — ЖФХ, 1977, т. 51, № 11, с 2783—2787.
406. Dawber У. G., Wyatt Р. Л. Я. — J. Chem. Soc, 1958, v. 52, p. 3636—3638.
407. Глазов В. М., Павлова Л. М. —ДАН СССР, 1974, т. 218, № 3, с 600—
603.
408. Глазов В. М., Павлова Л. М. Фазовые равновесия гетерогенных систем.
Теория растворов в приложении к анализу фазовых равновесий. М.: Изд-во
МИЭТ, 1975. 156 с.
409. Глазов В. М., Павлова Л. М. — Неорг. матер., 1976, т. 12, № 12, с 2114—
2119.
410. Глазов В. М., Павлова Л. М. — Неорг. матер., 1978, т. 14, № 5, с. 824—
826.
411. Павлова Л. M.t Шевченко В. #., Маренкин С. Ф. и др. — ДАН СССР, 1948,
т. 238, № 1, с. 108—111.
412. Дутчак Я- Я., Корпев В. В., Коренчук Я. М. —ЖФХ, 1977, т. 51, № 10,
с. 2568—2572.
413. Корпев В. Д., Коренчук Я. M.t Дутчак Я- Я. —ЖФХ, 1977, т. 51, № 1,
с. 16—20.
414. Глазов В. М., Павлова Л. М. — ЖФХ, 1977, т. 51, № 4, с 821—825.
415. Schneider M.y Guillaume Л С. — J. Phys. Chem. Solids, 1974, v. 35, № 4,
p. 471—478.
416. Румшиский Л. 3. Математическая обработка результатов эксперимента.
М.: Наука, 1971, 192 с.
417. Агеев М. Я., Алик В. П., Марков Ю. Я. Библиотека алгоритмов. М.:
Советское Радио, 1976. с. 44.
418. Peck Т. Е. L. —Soc Ind. Appl. Math. Rev., 1962, v. 4, № 2, p. 135—141.
419. Регель А. Р., Глазов В. М. Периодический закон и физические свойства
электронных расплавов. М.: Наука, 1978. 309 с.
420. Глазов В. ЛЬ, Павлова Л. М.—Термическая диссоциация
полупроводниковых соединений. М.: МИЭТ, 1980. 79 с
421. Глазов В. М., Чижевская С. Я. — ДАН СССР, 1959, т. 129, № 4, с. 869—
872.
422. Глазов В. M.t Петров Д. Л., Чижевская С. Я. —Изв. АН СССР. ОТН,
1959, № 4, с. 153—155.
423. Глазов В. М., Земское В. С, Журкин Б. Г. и др. — Труды ин-та
металлургии им. А. А. Байкова АН СССР, 1963, вып. 14, с. 108—119.
424. Глазов В. М. — ЖНХ, 1961, т. 6, № 4, с. 933—936.
425. Глазов В. М. — Изв. АН СССР. ОТН, 1962, № 1, с 89—93.
329
426. Велов А. Ф., Акопян Р. Л., Глазов В. М., Потемкин А. 0. — ДАН СССР,
1978, т. 238, №5, с. 1128—1131.
427. Глазов В. М. — В кн.: Проблемы металловедения цветных сплавов. М.:
Наука, 1978, с. 181—192.
428. Глазов В. М., Павлова Л. М., Евдокимов А. В. — ДАН СССР, 1977, т. 232,
Mb 2, с. 371—374.
429. Bhatia Л. В., Hargrove W. Я., Thornton D. E. — Phys. Rev., 1974, v. 9,
№2, p. 435—444.
430. Zabdyr L. — Arch. Hutn., 1979, v. 24, № 1, p. 95—101.
431. Ptak W., Zabdyr L. — Arch. Hutn., 1975, v. 20, №3, p. 379—387.
432. Малкова А. С, Зеленцова Я. Л., Павлова Л. Af. — В кн.:
Термодинамические свойства металлических расплавов. Алма-Ата: Наука, Каз. ССР,
1979, т. 2, с. 62—64.
433. Павлова Л. М., Глазов В. М. — ДАН СССР, 1980, т. 254, №5, с. 1162—
1166.
434. Павлова Л. М., Глазов В. М., Поярков /С. Б. — ДАН СССР, 1980, т. 255,
№ 5, с. 1173—1177.
435. Eldridge J. M., Miller £., Komarek tf. L. — Trans. Met. Soc. AIME, 1967,
v. 239, p. 775—781.
436. Еременко В. Я., Иващенко Ю. Я., Хиля Г. П. — Изв. АН СССР. Металлы,
1977, № 3, с. 38—40.
437. Воронин Г. Ф., Дегтярев С. А. — ДАН СССР, 1980, т. 254, № 5, с. 1146—
1149.
438. Pelton Л. D. — Вег. Bunsenges. Phys. Chem., 1980, Bd 84, S. 212—218.
439. Тихонов Н. А., Арсенин В. Я- Методы решения некорректных задач. М.:
Наука, 1979. 286 с.
440. Налимов В. В., Голикова Т. И. Логические основания планирования
эксперимента. 2-е изд. М.: Металлургия, 1981. 152 с.
441. Форсайт Дж., Малькольм М. М., Моулер К- Машинные методы
математических вычислений: Пер. с англ. М.: Мир, 1980. 280 с.
442. Завьялов Ю. С, Квасов Б. И., Мирошниченко В. Л. Методы сплайн-функций.
М.: Наука, 1980. 352 с.
443. Дегтярев С. Л., Воронин Г. Ф.— В кн.: Термодинамика металлических
систем. Ч. 1. Алма-Ата; Наука, 1979, с. 38—43.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аддитивность 5, 12, 74, 108, 109, 226, 228
Адекватность 254 — 257, 313
Активность 18, 107, 121, 148, 149, 183,
211, 242, 298, 300, 306, 314
— коэффициент 29, 107, 108, 115 — 121,
125—133, 136, 140, 142, 148, 149, 179 —
184, 188, 200, 201, 209, 210, 214, 221,
226, 259, 260, 263, 264, 266, 268, 271,
274, 275, 281, 285, 297, 305, 314
Акцептор 197
Алгоритм 302
Анализ регрессионный линейный 251
— термический 203, 220, 225, 263, 271
— физико-химический 306
Ассоциат 146, 147, 185, 208, 211—214,
259, 300
Ассоциация 149, 210, 212, 213
Бертоллиды 273—275
Больцмана постоянная 245
Бачинского уравнение 227
Бьеррума уравнение 117
Бребрика уравнение 204, 216 — 219
Ван-дер-Ваальса уравнение
дифференциальное 54, 150, 154, 156, 157, 159, 189,
194, 200
обобщенное 53—65
Вант-Гоффа уравнение изобары 114
Вектор волновой 227
Вероятность 245, 250, 253
Вес статистический 253, 304
Вещество растворенное 106, 107, 111 — 113,
151, 191 — 197
Взаимодействие межмолекулярное 146, 147,
162, 179, 180—186, 209, 215, 218, 222,
224, 227, 228, 291, 308
Виланда уравнение 216, 293
Вязкость 226, 256, 306
Газ идеальный 6, 112, ИЗ
Газовая постоянная 6
Гаусса распределение 250
Гельмгольца энергия свободная 17
— — — молярная 27
— работа максимальная 20
Генри закон 112, 113, 118, 194
Гесса закон 223
Гиббса —Гельмгольца уравнение 16, 21,
115, 158
Гиббса—Дюгема уравнение 26, 59, 60,
63, 113, 123, 126, 140, 142, 149, 211, 287
Гиббса метод термодинамический 4, 30, 77
характеристических функций 16
— правило фаз 33, 34, 82
— принцип равновесия 30, 35, 58
— уравнение дифференциальное
фундаментальное 24, 25, 54
— условия равновесия 30 — 32, 40, 153,
161, 196, 231, 239, 240, 274, 281, 305
— энергия свободная 16, 17, 66, 69 — 79,
109, ПО, 121, 134, 168, 228, 307
максимальная работа 21
молярная 27, 62, 155, 162, 192
образование соединений 205, 218
смешения 109, 215
избыточная 122, 128, 132, 136,
140, 142, 214, 235
Давление 112, 113, 193, 194, 221, 223,
249, 250, 263, 266, 270, 271, 287, 288,
296, 297, 299, 308
Дальтона закон 6, 113
Дальтониды 273—275
Диаграмма состояния фазовая 53, 58, 59,
65—68, 72, 80, 103, 104, 131, 150, 155,
156, 160, 179, 182, 188, 194, 212, 219 —
224, 228, 229, 237, 241, 249, 257, 260,
266, 270 — 274, 279, 303—306
основные типы 65—105
перитектического типа 92, 93, 96,
161, 166, 167, 174, 177—179
плоская 68
пространственная 68
с непрерывным рядом твердых
и жидких растворов 78, 81, 82, 89, 153,
161, 164 — 167, 172 — 174, 177, 179, 185
областью расслаивания 229
химическими соединениями
99 — 102, 261, 263, 267, 285
— характер межмолекулярного
взаимодействия 160—179
— — — эвтектического типа 88—90, 93,
96, 166, 167. 174, 179, 224, 229, 237—240
Дисперсия 252 — 254, 303
Диссоциация 185, 201, 219, 226, 264,
283, 286, 296, 297, 303—306, 310—315
— константа 183, 211, 275, 276, 282 —
285, 308—315
— степень 101, 158, 211, 226, 273 — 278,
282, 285 — 290, 292 — 296, 300—304, 306
Доля мольная 4, 106, 150, 151, 160, 186,
194, 195, 209, 210, 213, 225, 250, 251,
297, 313
— объемная 125, 223
Донор 197
Закон действующих масс 36, 38, 114, 119,
149, 209, 274, 314
— периодический 223, 256
— понижения температуры замерзания 153
— сохранения массы вещества 183, 209,
297, 307
Замещение анионное 256
— изовалентное 227
— катионное 256
Изомер оптический 155
Изотерма 188, 215
Интеграл конфигурационный 127
Интервал доверительный 254
Ионизация 195 — 197, 199
— энергия 197, 198
Каменецкой уравнение 219
Каннода 57, 91, 95, 306
Карно цикл 10, 12
Кирхгоффа уравнение 206
Клаузиуса—Клапейрона уравнение 15, 16,
56 223
Комплекс 211, 214, 215, 230—234, 255,
307 312 315
Компонент 4, 27, 32, 33, 66, 86, 104, 150,
151, 171, 180, 186
Континуум упруго-изотропный 228
Коши теорема 25
Коэффициент барический 16
— кажущийся 275
— объемного расширения 129
— осмотический 108, 116, 117
— поверхностного натяжения 227
— полезного действия 10, 12
— расширения 50, 114, 120, 192—195,
198, 275, 284, 306
Кривые ликвидуса 150, 151, 157, 160,
179, 181, 190, 191, 201—204, 209, 214 — ,
216, 219, 226, 229, 237, 238, 241—244,
246, 256, 257, 260, 266, 267, 305
— — радиус кривизны 280, 282, 295,
302, 303
уравнение 163, 202, 212, 215, 217,
218
— моновариантного равновесия 96, 103,
161, 176, 244, 246, 256
331
— плавкости 275, 276, 280, 294, 296
— равных значений изобарно-изотерми*
ческого потенциала 175, 177
— расслаивания 161 — 163, 167, 236
— солидуса 89, 154, 179, 181, 190—195,
198, 244, 256, 273
радиус кривизны 245
расчет 198
уравнение 153, 163, 196
— сольвуса 88, 89, 256
— термодинамического изобарно-изотер-
мического потенциала 280в 281
Кристаллизация 151, 152, 157 — 159, 184,
203, 204, 209, 211—216, 219, 220, 224,
225, 229, 230, 237, 261, 263, 265, 267,
270
Критерий оптимальности 249—252
— полезного действия 12
— устойчивости 40, 53, 189, 194
• гетерогенных систем 46, 48
гемогенной фазы 40—47, 51,52, 71, 72
— физико-химический 249
Крупковского метод 139
Лагранжа метод неопределенных
множителей 35
Лагранжа полином 286, 301
Лапласа распределение 249
Легирование 195, 199, 306
Лежандра полином 135, 136, 137 — 141
Ликвидуса поверхность 188
Линеаризация 251, 252
Максвелла уравнение 19, 44
Маргулеса ряд 247, 255, 256
— уравнение 118, 139
Матрица 25, 252
Машина тепловая 10, 12
Метод вариации произвольной постоянной
202
— геометрический 65, 162, 169, 280
— геометрической термодинамики 105, 160
— «избранных точек» 238
— максимального правдоподобия 255
— математической статистики 235, 249,
250
— наименьших квадратов 241, 248—250,
253, 255
— потока 271
— точек кипения 271
Множитель интегрирующий 13
Мономер 146, 148, 183, 208, 210, 212,
214, 215, 312, 315
Невязка 249, 250, 257
Независимые переменные 150, 154, 156
Нернста закон распределения 114, 120
Нормировка, способы 107, 118, 304
— условие 211, 297
Ньютона бином 143, 230
— полином 301
Область гомогенности 156—158, 188, 202—
205, 216, 285, 297
Объем атомный 222, 224, 226
— мольный парциальный 25
средний 110, 111
— молярный 46, 110, 227
«*- «свободный», теория 132
— смешения ПО, 111
избыточный 122, 221
Оптимизация нелинейная 251
— среднеквадратичное 256
Ошибка случайная 249, 256
Параметры интенсивные 5, 47, 48
— модели 255, 257
— независимые 5
— термодинамические 5, 48, 192, 193,
228, 257, 311
332
— экстенсивные 5, 7, 12, 25, 42, 43, 47, 48
Переход фазовый 161, 270
Плавление 90, 97, 99, 155, 188, 192, 205f
206, 219, 263
Плотность 245, 248, 256, 310
Полином ортогональный 39, 134, 235, 247
Полупроводник 188, 191, 195—200
Постоянная криоскопическая 288
Порядок ближний 244
Потенциал изобарно-изотермический (см.
Гиббса свободная энергия) 158, 205,
217, 237, 245, 273, 290, 296
кривые, относительное расположе-,
ние 80—82, 85, 87, 88, 91—96
поверхность 46, 67, 73
— термодинамический 17, 19, 46, 160,
227, 277
— — избыточный 126
— химический 23, 48, 59, 131, 157, 161,
186, 191, 230—237
в растворах 106—113, 147—149
газа идеального 27, 28
реального 28, 29
связь с термодинамическими
потенциалами 24
Превращение аллотропное 180, 189
— монотектическое 94, 96
— нонвариантное 188, 189
— перитектическое 91, 93, 101, 213, 216,
217, 219
— полиморфное 80, 100
— синтектическое 94, 96
— фазовое 164
— эвтектическое 87, 88, 91, 189
Предел длинноволновый 244—246, 310
Приближение квазихимическое 130, 188,
249, 255
— «нулевое» 191
Процесс изотермический 6, 41
— необратимый 6, 11 — 13
— обратимый 6, 11, 12
— равновесный 270
Пуассона критерий 228
Работа 12
— максимальная 12, 20, 21
— полезная 19
— расширения 8, 19
Равновесие 150, 157, 159, 160, 164, 194,
204, 237, 273, 274
— гетерогенное 20, 32, 33, 65, 89, 195
— двухфазное 53, 66, 70, 71, 76—82,
84—89, 94, 101, 103, 180, 216
— жидкость—кристалл 254
— жидкость—пар 255
— квазихимическое уравнение 127
— константа 38, 39, 49, 50, 114, 120, 127,
183, 297, 298, 307
— метастабильное 270
— механическое 35
— моновариантное 48, 67, 71, 89, 179,
180, 273, 304
уравнение линии 162, 179, 180, 186
— монотектическое 86, 94—96
— нонвариантное 67, 83, 86, 195, 216
— перитектическое 83 — 86, 91, 94, 102
— перитектоидное 103
— принцип смещения 9, 17, 50, 51, 53, 156,
— синтектическое 94—96
— термическое 8, 35, 36, 245
— трехфазное 67, 70, 71, 82, 87, 89, 94,
96, 101 — 103, 216, 250
— фазовое 32, 65, 80, 103, 112, 124, 125.
150, 151, 156, 161, 178, 179, 182, 184-
187, 208, 223, 224, 230, 238. 241, 244,
246, 248, 253, 256, 261, 264, 266, 267, 270
условия 32—37, 83. 85, 161
— — геометрическая интерпретация 74—
76, 84
— химическое 36, 38, 114, 119, 242, 287
— эвтектическое 83 — 87, 89, 91, 94
•— эвтектоидное 103
Разбавление, степень 303, 306
Разрез квазибинарный 188, 306
Распределение 246, 253, 254
— нормальное 252
Рассеяние диффузное 227
Расслаивание 100, 129, 164, 171, 175,
177, 179, 190, 221, 229, 230
— купол 78, 89
Раствор:
ассоциированный 146, 213
— идеальный 281, 282—285, 315
— регулярный 147, 149, 183, 184, 208,
210—213
атермальный 124, 133
бинарный (двухкомпонентный) ИЗ, 116 —
118, 122, 141, 149, 179.186,226, 240,
256, 297, 300, 303
идеальный 106 — 115, 150 — 156, 158, 159,
161, 165, 191, 192, 195, 196, 226, 278
квазибинарный 304
конденсированный 122
конформальный 129, 132
модель 48, 124, 127, 142, 146, 156. 158
161, 180—184, 187, 188, 195, 201 —
204, 213, 214, 219—221, 231, 233,
246—250, 252—255, 257—263, 266—
271, 280, 287, 290—293, 297, 298, 308
насыщенный 299
неидеальный 106, 107, 115, 117—121,
161, 165, 200
определение 105
предельно-бес конечное разбавление 106,
107, 111, 112, 120, 135, 150—153,
233, 314
реальный 106, 112, 121, 124, 158, 228, 285
регулярный 124—127, 130—133, 135,
145, 147. 149, 157, 160—163, 167,
175—178, 184, 185, 188. 191, 224,
226, 228, 230, 233, 234, 248, 260
структура 241, 248
твердый 23, 90, 100, 103, 161. 178, 180,
J85, 191, 192, 225. 236, 237, 244
— изоморфный 100
— термодинамическая классификация
105, 106
— четырехкомпонентный 186
Растворение 105, 151 — 153, 180, 190, 193,
194
— теплота 105, 242, 243
Растворимость 151, 179, 180, 184, 188—
195, 197—199, 221, 224, 230, 239, 240,
242
— параметры 215, 222, 223
Растворитель 106, 107, 111, 114, 126,
151-153, 180, 193, 194
Рауля закон 113, 118, 140—143, 146, 168
Реакция квазихимическая 127
Редлиха—Кистера полином 235
ряд 139, 141, 143, 247
Решетка кристаллическая кубическая 228
параметры 228
Ричардса правило 189, 191
Розебома метод 168, 172, 175, 179
Свойства избыточные 134
интегральные 138, 311
парциальные мольные 255, 311
— термодинамические 108, 121, 138, 182,
201, 204, 206, 208, 228—230, 233, 236,
241, 246—249, 253 — 257, 260 — 267, 270,
271
— экстенсивные 106
Сильвестра теорема 72
Система:
бинарная (двухкомпонентная) 53, 58,
59, 65, 68, 72. 80. 109, 150, 156, 159,
160, 179, 180, 183—191. 194, 198,
208, 211, 215, 222—225, 228, 238,
244, 246
вариантность 33, 34, 82
двухфазная 53—57, 148. 150, 156, 160,
189. 230. 250
дивариантная 33
гетерогенная 33—35
гомогенная 51, 52, 97, 101
закрытая 5, 22, 23. 37
идеальная 154
изолированная 5, 9. 10
квазибинарная 222—227, 303
конденсированная 34, 66, 79, 80, 216, 250
макроскопическая 4
многокомпонентная 54, 57, 58, 62, 68, 103
моновариантная 33, 53
неидеальная 248
нонвариантная 33
однокомпонентная 33, 66, 67
однофазная 122
открытая 5, 22, 23, 58, 61
перитектического типа 179
с конгруэнтно плавящимся соединением
246
— непрерывным рядом твердых
растворов 180, 181, 185, 232, 236
состояние 150, 156
— параметры 66, 68, 85, 97, 98, 179
228. 256
— стандартное 107, 117, 123, 197, 199,
229, 232, 237
— уравнение 5, 6, 14, 17, 66
— устойчивое 158
термодинамическая 4, 22
трехкомпонентная (тройная) 62, 72, 188,
224, 306
трехфазная 250
эвтектического типа 150, 151, 179, 232,
235, 242
Смесь Л-компонентная 188
— молекулярная 297
— тройная 297
Соединение химическое, плавящееся инкон-
груэнтно 99 — 102
конгруэнтно 99. 101, 156, 157
диссоциирующее 99—101, 278
недиссоциирующее 98, 99, 105
Солидус 189, 190
— радиус кривизны 245
— ретроградный 89, 91, 188—194
Сольватация 146
Степень ионности 215
— свободы, число 33, 34
— структурного разупорядочения 293
Структура 179, 311
Тейлора ряд 210, 230, 235, 251, 252, 301
Текучесть 226
Температура:
кристаллизации 287. 288
критическая 171, 221
ликвидуса 203, 225, 237, 257, 266, 267,
311
плавления 80. 151. 155, 180. 192, 193,
207, 216, 260, 294, 310
термодинамическая абсолютная 11, 126
эмпирическая 9
Теплоемкость 14, 57, 151, 187, 206, 232,
235, 239, 305
Теплота:
диссоциации 283, 284
дифференциальная мольная 57
испарения 15, 113, 117
кристаллизации, дифференциальная
молярная 157—159
ззз
образования 213, 229, 241, 259, 261,
262, 266, 267, 300, 305
парциальная мольная 55, 192
плавления 16, 106, 150, 151, 154, 155,
190, 192, 205—207, 216, 228, 229,
242, 267, 277, 289, 292, 294, 310
приведенная 12
реакции 114, 120
сегрегации 158
смешения 124, 145, 163, 201, 217, 221,
237, 259—263, 266, 268
стандартная 205
фазового превращения 15, 16
Термодинамика, второй закон 8, 10—12
— геометрическая 65, 93, 95, 96, 101, 103
— нулевой принцип 8
— первый закон 8—10, 12
Точка:
азеотропная 221
замерзания 150
критическая 163, 171
переходная 102, 263
плавления 151, 193, 302, 313, 314
тройная 103
фигуративная 65, 76, 88
эвтектическая 181, 221, 230, 237, 242 —
246, 256
Упругость, модуль 228
Фаза 5, 32, 33, 66, 87, 97, 98, 100, 101
— кристаллическая 232
— однокомпонентная 103, 105
— промежуточная 241
Фактор структурный 244—246, 307—312,
316
Ферми—Дирака распределение 197
Ферми-уровень 197, 199
Фибера—Зеймана структурный фактор 248
Функция термодинамическая 16, 17, 179,
235, 266
смешения 107—112, 121, 238
избыточная 121, 124, 264
— характеристическая 16, 23
Фурье ряд 138, 141, 247, 248
Цикл обратимый 12
Число координационное 128
Чохральского метод 306
Эвтектика 48, 189, 190, 191, 193, 229, 235
— вырожденная 229
— температура 89, 91, 237
Эйлера теорема 7, 25
Электроотрицательность 215, 222 — 224, 226,
250
Энергия взаимообмена (смешения) 125—
131, 140, 166, 170, 172, 174, 175, 179,
181, 182, 185, 197, 199, 202, 203, 215,
221—226, 256, 257, 260—263, 266—
268, 271, 291—293
— внутренняя 8—10, 15, 17, 19, 20, 23,
40,'42, 44, 47
— связи 223
Энтальпия 10, 17, 20
— испарения 223
— мольная 110
парциальная ПО, 115
средняя ПО
— образования 205—207, 218
— перитектического превращения 217—
220
— растворения 242
— смешения (избыточная) 109, 122, 126,
132, 140, 162, 163, 182, 242
— стандартная 305
— сублимации 223
Энтропия 11, 12, 130, 134, 184
— изолированной системы 31
— как критерий направленности
процесса 12
— кристаллизации 157, 158
— молярная 46
образования 214, 229, 259, 262, 266
— парциальная мольная 25, ПО
— плавления 16, 150, 189, 191, 198, 242
— растворения 292
— связь с различными параметрами 190
— смешения 110, 111, 125 — 131, 182,
189, 237 — 240, 242, 244, 259, 260, 262,
266, 267, 269, 270
— избыточная 122, 123, 242, 259, 260
— стандартная 305
— фазового превращения 16
Эффект тепловой ПО, 111, 158
— фазовый 57
— упорядочения 126, 130, 131
— эндотермический 129
ОГЛАВЛЕНИЕ
ПРЕДИСЛОВИЕ 3
ГЛАВА I. НЕКОТОРЫЕ ОБЩИЕ СООТНОШЕНИЯ
ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ 4
1. Системы, их свойства и классификация.
Термодинамические параметры и уравнение состояния.
Термодинамические процессы 4
2. Парциальные мольные величины и их свойства б
3. Нулевой принцип. Первое и второе начала
термодинамики 8
4. Связь энтропии с различными параметрами и
некоторые соотношения между производными функциями 13
5. Характеристические термодинамические функции и
уравнение Гиббса—Гельмгольца 16
ГЛАВА II. УЧЕНИЕ О ХИМИЧЕСКИХ ПОТЕНЦИАЛАХ
И ОБЩАЯ ТЕОРИЯ ТЕРМОДИНАМИЧЕСКОГО
РАВНОВЕСИЯ 22
1. Фундаментальные теоремы и уравнения. Уравнение
Гиббса—Дюгема 22
2. Принцип равновесия Гиббса 30
3. Условия фазового равновесия. Действительные и
возможные компоненты 32
4. Условие химического равновесия. Закон
действующих масс 36
5. Критерии устойчивости фаз и гетерогенных систем 40
6. Принцип смещения равновесия (принцип Гиббса—
Ле-Шателье) 49
7. Смещение вдоль линии равновесия. Обобщенное
дифференциальное уравнение Ван-дер-Ваальса 53
8. Об адекватности различных подходов к выводу
принципа смещения вдоль линии равновесия ... 58
ГЛАВА III. ФАЗОВЫЕ РАВНОВЕСИЯ В
ДВУХКОМПОНБИТНЫХ СИСТЕМАХ 65
1. Общие представления о диаграммах состояния 65
2. Зависимость изобарно-изотермического потенциала
от температуры, давления и концентрации .... 69
3. Термодинамический вывод основных типов диаграмм
состояния двухкомпонентных систем 76
ГЛАВА IV. ОСНОВНЫЕ ПОЛОЖЕНИЯ
ТЕРМОДИНАМИЧЕСКОЙ ТЕОРИИ РАСТВОРОВ
КОНДЕНСИРОВАННЫХ СИСТЕМ 105
1.. Обобщенное понятие раствора.
Термодинамическая классификация растворов 105
2. Термодинамические функции смешения. Основные
свойства и законы идеальных растворов 108
3. Законы неидеальных растворов. Избыточные
термодинамические функции. Классификация отклонения
от идеальности 115
4. Концепция регулярного раствора. Решеточная
модель раствора. Строго регулярный раствор и его
модификации 124
5. Формально-математическое описание
термодинамических свойств реальных систем 134
6. Концепция ассоциированных растворов 145
335
ГЛАВА V. ТЕРМОДИНАМИЧЕСКИЙ АНАЛИЗ ФАЗОВЫХ
ДИАГРАММ В ПРИБЛИЖЕНИИ РАЗЛИЧНЫХ
ТЕОРИЙ РАСТВОРОВ 150
1. Анализ кривых фазовых равновесий в двухком-
понентных системах на основе теории идеальных
растворов 150
2. Взаимосвязь различных типов диаграмм фазовых
равновесий с характером межмолекулярного
взаимодействия 160
3. Анализ кривых ликвидуса и солидуса в системах
с неограниченной и ограниченной растворимостью 179
4. Термодинамическое обоснование ретроградного
солидуса. Расчет ретроградного солидуса в
различных приближениях 188
5. Анализ кривой ликвидуса конгруэнтно
плавящегося соединения АтВл в различных
приближениях неидеальных растворов 200
6. Уравнение кривой ликвидуса и
термодинамический анализ систем с инконгруэнтно плавящимся
соединением 215
7. Различные методы оценки энергии взаимообмена
на основе экспериментальных данных 221
8. Расчет термодинамических свойств сплавов по
данным о фазовых равновесиях 228
9. Оценка структурного фактора в расплавах
бинарных систем на основе анализа данных по
фазовым равновесиям 244
10. Выбор модели раствора на основе согласования
с экспериментальными данными по фазовым
равновесиям 248
ГЛАВА VI. ТЕРМОДИНАМИЧЕСКИЕ ИССЛЕДОВАНИЯ
ДИССОЦИАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ ... 272
1. Форма и положение максимума на кривых
ликвидуса и солидуса 272
2. Термодинамическое обоснование различных путей
оценки параметров диссоциации конгруэнтно
плавящихся соединений 276
3. Оценка степени диссоциации химических
соединений в различных приближениях термодинамики
растворов 289
4. Диссоциация бинарного соединения в
квазибинарных растворах трехкомпонентных систем 303
5. Оценка структурного фактора в жидких растворах
по кривизне ликвидуса в двойных системах с
конгруэнтно плавящимися соединениями 318
БИБЛИОГРАФИЧЕСКИЙ СПИСОК 330
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ 331