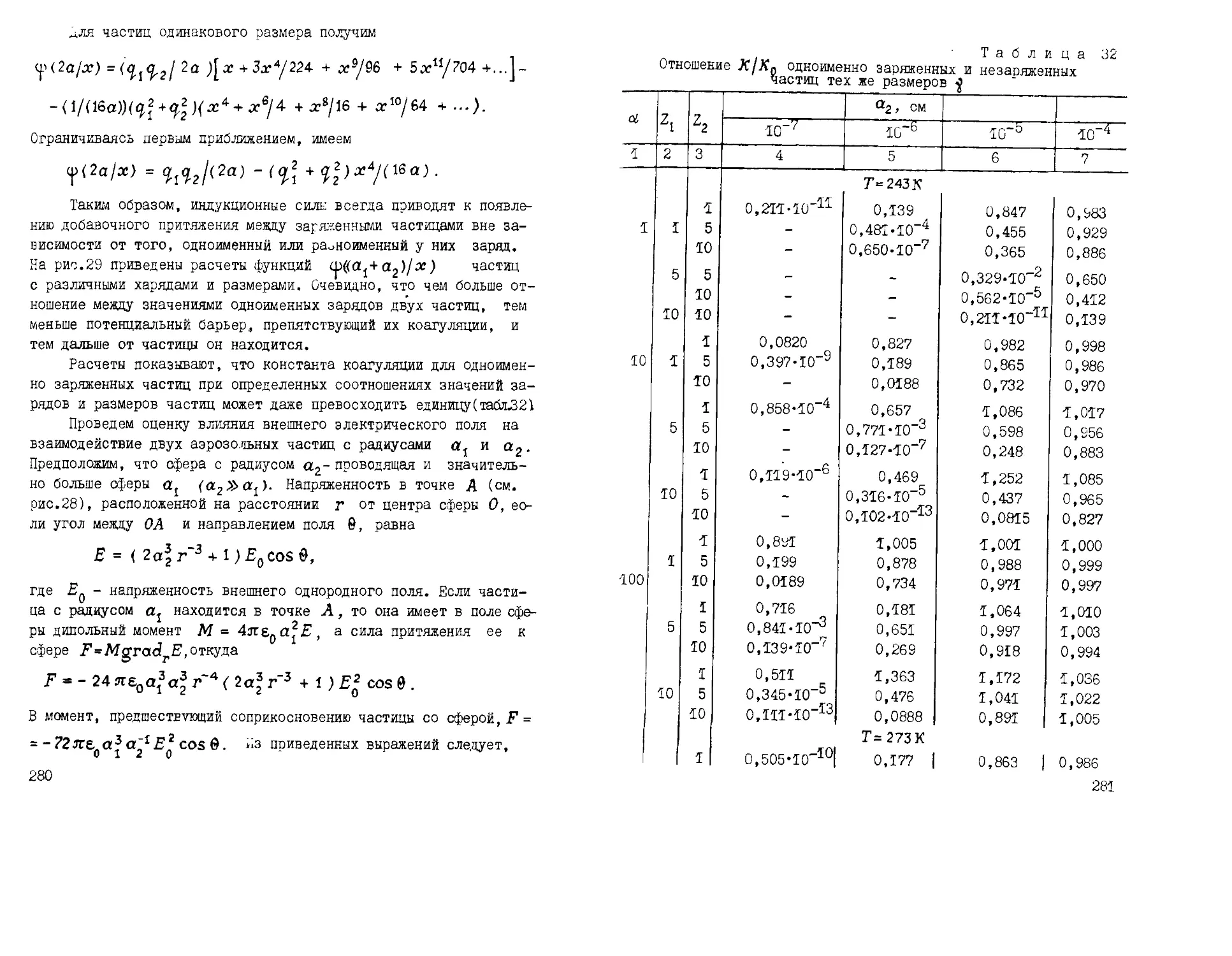
/
















































































































































































Author: Ивлев Л.С.
Tags: метеорология климатология термодинамика энергетика химический состав аэрозолей аэрозоли структура аэрозоли
Year: 1982




Text
ЛЕНИНГРАДСКИЙ ОРДЕНА ЛЕНИНА
И ОРДЕНА ТРУДОВОГО КРАСНОГО ЗНАМЕНИ
ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ имени А. А. ЖДАНОВА
Л. С. Ивлев
ХИМИЧЕСКИЙ СОСТАВ
И СТРУКТУРА
АТМОСФЕРНЫХ АЭРОЗОЛЕЙ
ЛЕНИНГРАД
ИЗДАТЕЛЬСТВО ЛЕНИНГРАДСКОГО УНИВЕРСИТЕТА
1982
Печатается по постановлению
Редакционно-издательского совета
Ленинградского университета
УДК 551.510+536.7
У. в л е в Л.С. Химический состав и структура атмосферных
аэрозолей. -Ji.: Изд-во Ленингр. ун-та, 1982. Ил. - 36,
табл. - 33, Оиблиогр. - 241 назв. -368 с.
В монографии рассмотрены основные источники аэрозоль-
аэрозольного вещества, механизмы распределения стоков и трансформа-
трансформации аэрозольных частиц в воздушной среде. Представлен обзор
экспериментальных данных о структуре и химическом составе
аэрозолей в чистой и загрязненной атмосфере и предложены
аэрозольные модели атмосферы.
Книга предназначена для специалистов в области метеоро-
метеорологии, физики и химии атмосферы, атмосферной фотохимии, гео-
геохимии, а также для аспирантов и студентов старших курсов фи-
физических и географических факультетов университетов.
Рецензенты:
акад. И.В.Соколов-Петрянов , канд.хим.наук Б.И.Огород-
Б.И.Огородников (Научно-исслед. физ.-хим. ин-т им.Л.Я.Карпова),
канд. физ.-мат. наук|А.В.Федынский| (Центр, аэролог,
обсерв.), Сев.-Зап. территориальное управление по гид-
гидрометеорологии и контролю природной среды
„ 1903040000 - 180 й,„,
И 076@2) - 82 ЬА~*г
Издательство Ленинград-
Ленинградского университета, •
1982 г. *>«
Г»
j чуб*.
йГ. (>c/»i-' .:.(,:••
г. Огрддоэдк
рата
Б 3 Е к 2 Н Л 2
Увеличение общей загрязненности атмосферы и связанные с
этим глобальные изменения ноосферы и биосхеры, в частности
изменения климата Земли [15, IS], вызвали повышенным интерес к
физико-химическим свойствам и структуре атмосферных аэрозолей.
Аэрозоли, существенно влияя на изменения радиационного режима
атмосферы и ^зовых переходов в ней [19, Зс], являются важней-
важнейшей компонентой загрязненной атмосферы. Количественные оценки
роли аэрозолей в процессах, вызывающих изменения энергетическо-
энергетического баланса в системе атмосфера - Земля, затруднены как в силу
сложности корректного решения подобной задачи, так и в связи с
большой неопределенностью наших сведений об атмосферных аэрозо-
аэрозолях и чрезвычайно высокой изменчивостью их структуры.
Определенные сведения о структуре атмосферных аэрозолей
требуются для решения теоретических [29, 81 ] и прикладных [Ю,
35, 56, 74] задач физики атмосферы и других областей науки и
техники. В большинстве случаев при этом необходимо знание толь-
только отдельных характеристик аэрозолей: счетной или массовой кон-
концентрации, индикатрисы рассеяния, коэффициентов ослабления или
поглощения света и т.д. Непосредственные измерения этих харак-
характеристик не всегда возможны, да и сами характеристики сильно
варьируют по величине в зависимости от различных внешних усло-
условий. Поэтому в расчетах часто используются модельные представ-
представления об атмосферных аэрозолях или их характеристиках, которые
позволяют по имеющимся у исследователя исходным данным полу-
получить требуемые параметры и характеристики.
3
Пространственная структура аэрозолей заметно изменяется во
времени в результате процессов генерации, выведения из атмосфе-
атмосферы и распространения в ней аэрозольных частиц. Но в действитель-
действительности такое разделение условно, поскольку все три типа процес-
процессов взаимосвязаны. Если при рассмотрении аэрозолей ограничивать-
ограничиваться определенным диапазоном размеров исследуемых частиц, необхо-
необходимо помнить, что эти же процессы приводят к переходу частиц из
одного диапазона размеров в другой и оказываются неразделимыми.
Рассматривая физическую картину существования аэрозолей во всем
диапазоне их размеров, следует выделить четвертый тип процес-
процессов - процессы трансформации спектра размеров аэрозольных час-
частиц.
Задачу построения физической картины формирования аэро-
аэрозольной структуры можно разделить на два этапа: 1) формирование
пространственной глобальной структуры аэрозолей (массовой кон-
концентрации частиц) в результате действия первых трех типов про-
процессов; 2) формирование локальной структуры аэрозолей в резуль-
результате действия всех факторов. Полученные на втором этапе оезуль-
таты молено использовать для уточнения характеристик процессов
первых типов.
Процессы генерации аэрозольных частиц в основном определя-
определяют их физико-химические свойства и концентрацию в атмосфере.
Эти процессы очень мало исследованы даже для наземных источни-
источников, а точность определения мощности различных источников не
превышает порядка измеряемой величины.
В результате длительного существования аэрозольных частиц,
их переноса, перемешивания и взаимодействия особенности физико-
химических свойств частиц от различных источников нивелируются,
и возникает тип аэрозолей, который называют фоновым. В разных
слоях атмосферы характеристики фоновых аэрозолей претерпевают
существенные изменения.
Процессы распространения и выведения аэрозолей из атмосфе-
атмосферы определяются как метеорологическими (геофизическими) факто-
факторами, так и свойствами самих аэрозольных частиц. К процессам,
которые определяются метеорологическими факторами, относятся
упорядоченный конвективный и адвективный переносы частиц и в
значительной степени их перемешивание в результате турбулент-
4
ной диффузии. (Для очень крупных'частиц нельзя пренебрегать
влиянием на диффузию массы частиц.) Индивидуальными свойствами
частиц определяются в первую очередь такие процессы их выведе-
выведения из атмосферы, как седиментация и осаадение на препятствиях,
В процессе распространения или выведения частиц из атмос-
атмосферы масса и состав последних трансформируются в результате ко-
агуляционного и конденсационного роста, а также гетерогенных
реакций. Коагуляция, конденсация и гетерогенные реакции форми-
формируют спектр размеров частиц, определяя вид функции распределе-
распределения частиц по размерам [21, 34]. Эти процессы вместе с процес-
процессами генерации обусловливают величину комплексного показателя
преломления вещества аэрозольных частиц и их форму, т.е. опре-
определяют оптические свойства частиц [17].
Строгое математическое решение проблем, связанных с опи-
описанием всех процессов, определяющих формирование аэрозольной
структуры, и построением полной аэрозольной модели, в настоя-
настоящее время невозможно как из-за недостаточности знаний их ха-
характеристик, реализующихся в атмосфере, так и из-за чисто ма-
математических трудностей. На современном уровне знаний об атмос-
атмосферных аэрозолях и при технических возможностях XX века пред-
представляется наиболее разумным создание аэрозольных моделей с ис-
использованием при этом всех имеющихся экспериментальных и теоре-
теоретических результатов. Поэтому необходимо обобщить и проанали-
проанализировать имеющиеся данные, объяснить основные закономерности и
свойства аэрозольной структуры атмосферы. Несомненный интерес
представляет лабораторное и натурное моделирование основных
процессов, которые определяют химический состав, структуру ат-
атмосферных аэрозолей и их физико-химические свойства.
По механизмам образования вещества диспергированной фазы
аэрозолей, в значительной степени определяющим их физико-хими-
физико-химические свойства, можно выделить три основных типа процессов:
1) конденсационно-коагуляционные, в том числе гетерогенная нук-
леация, 2) химические реакции и 3) диспергирование аэрозоль-
аэрозольного вещества. Для аэрозолей конденсационного происхождения
спектр размеров частиц начинается от размепов, определяемых не-
несколькими атомами или молекулами, т.е. от I нм. Максимум рас-
распределения таких частиц определяется интенсивностью процесса
генерации аэрозольного вещества, степенью его пересыщения в
среде и временем жизни частиц, а большинстве случаев этот мак-
максимум находится в диапазоне при г = 0,05+0,30 мкм. Частицы
конденсационного происхождения в атмосфере далеко не всегда
становятся ядрами конденсации. Их счетная концентрация в при-
с о о
земном слое атмосферы колеблется от 10 до 10 см . (Величины
счетной концентрации, выходящие за границы указанных значении,
наблюдаются только з условиях сильного загрязнения воздуха.)
Массовая концентрация частиц конденсационного происхождения ред-
редко превышает 50 мкг/м . Конденсационные аэрозоли образуются так-
также в результате газофазных химических реакций окислов азота,
серы, органических соединений между собой и с другими атмосфер-
атмосферными газами. Однако при наличии в атмосфере аэрозольных частиц
с газофазными реакциями успешно конкурируют гетерогенные хими-
химические реакции. Образующиеся в результате этих реакций частицы
могут существенно отличаться характеристиками от аэрозолей чис-
чисто конденсационного происхождения. Их модальные радиусы будут
несколько больше: 0,15-0,80 мкм, счетная концентрация в призем-
А *5 —.*3
ном слое от 10 до 10*" см , а массовая может достигать несколь-
нескольких миллиграмм на кубический метр воздуха.
Частицы дисперсионного происхождения, образующиеся в ре-
результате разрушения поверхности или всего объема сплошных твер-
твердых или жидких тел, имеют максимум распределения по размерам
обычно в облает/, г2-1 мкм. Правда, в случае диспергирования
растворов может происходить испарение растворителя и тогда мак-
максимум распределения частиц по размерам может сдвигаться в об-
область г 4 1,0 мкм. Такой процесс происходит, например, при
диспергировании морской воды и скоростях ветра около 7 м/с и
более. Верхний предел размеров частиц дисперсионного происхож-
происхождения определяется возможностью длительного их существования в
атмосфере (более часа) и находится в области г 4 10 мкм.Счет-
мкм.Счетная концентрация частиц дисперсионного происхождения может из-
изменяться от их полного отсутствия до 10 - 10 см . При счет-
счетных концентрациях таких частиц > 104 см массовая концентра-
концентрация достигает нескольких миллиграммов в одном кубометре возду-
воздуха, на несколько порядков превышая массовую концентрацию кон-
конденсационных аэрозолей.
дисперсионные аэрозоли, исключая аэрозоли, образующиеся
при разбрызгивании воды на морской поверхности, менее гигроско-
гигроскопичны, чем конденсационные. Однако значительная доля более мел-
мелких частиц конденсационного происхождения при длительном сов-
совместном существовании захватывается крупными частицами диспер-
дисперсионных аэрозолей: образуются частицы смешанных аэрозолей [Ж)],
спектр размеров которых подобен спектру размеров аэрозолей дис-
дисперсионного происхождения.
Так как физические и физико-химические свойства частиц аэ-
аэрозолей в первую очередь определяются их размерами, то класси-
классификация аэрозольных частиц по этому параметру является важней-
тей. Рациональной классификацией является разбиение частиц на
три большие группы: г < ОД; ОД 4- г 4 1,0; г > 1 мкм. В
флзике аэрозолей эти группы соответственно называются мелкодис-
мелкодисперсной, среднедисперсной и грубодисперсной фракциями. В метео-
метеорологии первая группа называется частицами Айткена, или ядра-
ядрами конденсации, - по способу обнаружения частиц с помощью ре-
регистрации капель, образующихся при сильном пересыщении воздуш-
воздушной среды, в которой частицы находятся [НО], впервые применен-
примененному дж. Айткеном. Зторая группа называется оолыдими частица-
частицами, или частицами Ми [34]. Рассеяние видимого света на этих
частицах хорошо описывается с помощью теории, развитой Г.Ми.
Другим фактором, определившим выделение частиц довольно узкой
области размеров в отдельную группу, является соизмеримость
длины свободного пробега молекул воздуха в приземном слое с
порядком размеров этих частиц.
Счетная концентрация гигантских частиц обычно очень мала
(W < 1 cm~j), но зато их массовая концентрация может опреде-
определять общую массовую концентрацию аэрозолей.
3 табл.1 приведены классификация частиц атмосферных аэро-
аэрозолей, их роль в основных метеорологических явлениях и соотно-
соотношение между количеством частиц и их массой на основании данных
Х.Кяге, полученных для относительно чистого воздуха в призем-
приземном слое на континенте [110J -
Аэрозоли, в значительной' степени определяющие многие ат-
атмосферные процессы и явления, представляют собой важнейший объ-
объект исследований практически во всех областях науки об атмос-
I
О I
Q) Щ
ЕЗ c\3
О W—
s к а>
tf Fh К
\э о
si
1°
a> a>
О.Я
о ш
о
со
сб
ч>" ¦
Я Ч>
S К
О "
р,
ш
в
о сб
я
СцЯ
Е- О
а> о
S су
со
Сб ft
?13 с
о
о
м
о
I
и
о
с-
I
сг>
со
СП
I
о
СП
СП
СП
I
СП
З1
о
са
о
вв
<х>
Сб
О.
а>
я
О
о со
и о
о п
а-
3)
со
ш
S
I
5
о
и
CD
f О
о в1
f О
ок
S
ы
о
S • О.
о сб н
о о к
д о ч>
Е<св Ч
О S CD
о I
S!
о о
о
с
о
сб со
К S
со
я
S
о,
8
о
о ш
©р.
О Я
S
о
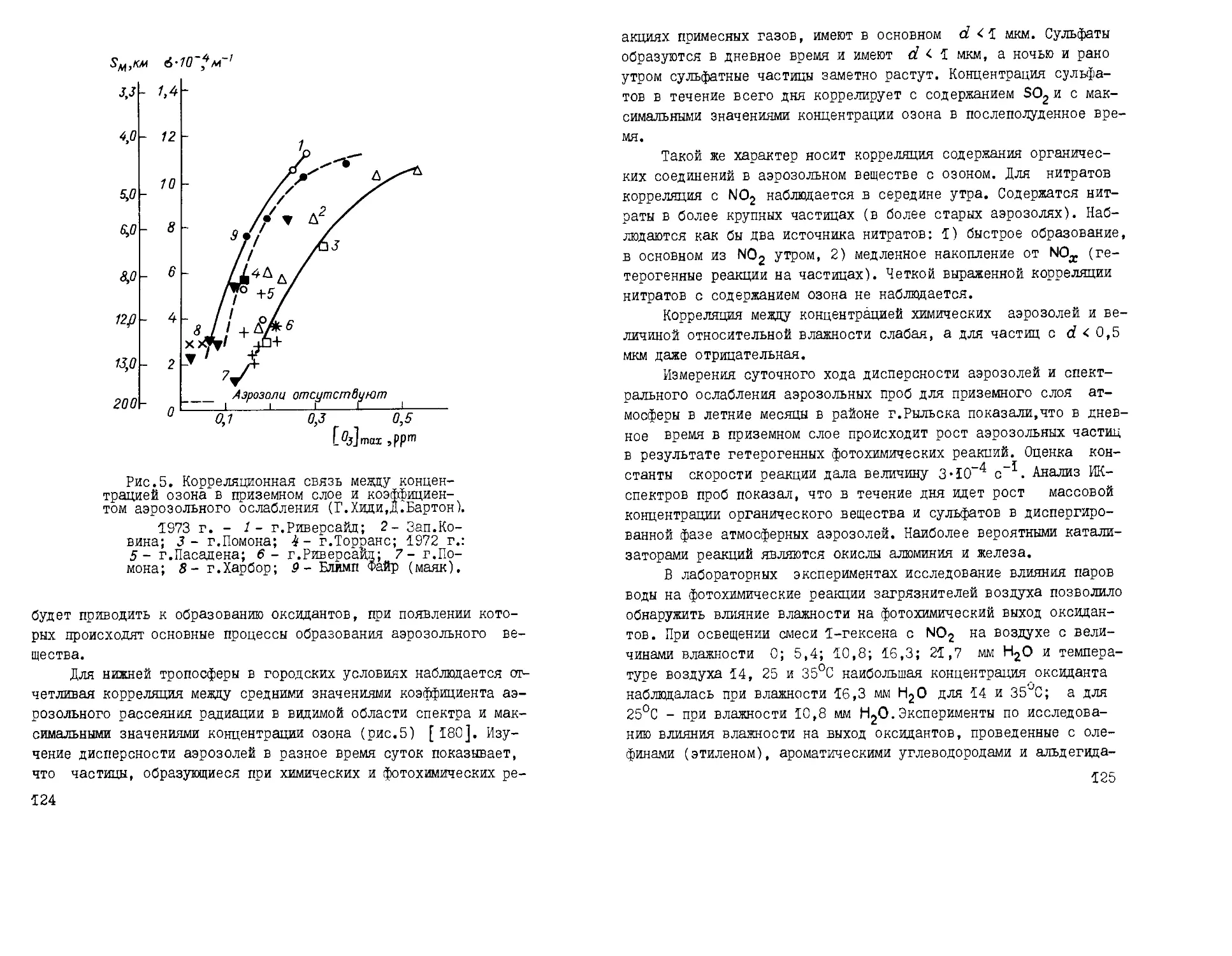
^ере.' Наиболее изучена роль аэрозолей в процессах облако- и
осадкообразования [34, 70].
3 последние годы выполнено огромное количество исследова-
исследований по оптике атмосферных аэрозолей, результаты которых частич-
частично обобщены в ряде монографий [29, 36, 77]. Однако эти резуль-
результаты приводят к выводу о том, что без априорных знаний микро-
микроструктуры аэрозолей оптические измерения не дают полностью од-
однозначного ответа о состоянии аэрозолей в момент измерений.Дос-
таточно определенно доказано, что в атмосферных оптических яв-
явлениях в видимой области спектра важную роль играют не толь-
только большие частицы, но и гигантские.
Относительно мало исследований о роли аэрозолей в явлени-
явлениях атмосферного электричества. Здесь два момента определяют
роль аэрозолей: 1) мелкодисперсные частицы (ионы) в значитель-
значительной степени обеспечивают проводимость воздуха, а среднедисперс-
ные и грубодисперсные, захватывая эти ионы, резко понижают про-
проводимость воздуха; 2) аэрозольные частицы с г^0,1 мкм, полу-
получившие в результате различных физических процессов заряд опре-
определенного знака, могут довольно длительное время сохранять его
и, следовательно, способствовать процессам разделения зарядов
в пространстве.
В химии атмосферы роль аэрозолей определяется тем, что по-
последние являются конечными продуктами различных химических и
ротохимических реакций; средой, в которой проходят реакции;
катализаторами ряда химических реакций, которые в чистой газо-
газовой среде при атмосферных условиях не вдут [105, 128]. Аэро-
Аэрозольные частицы, являющиеся конечными продуктами химических ре-
реакций в нижней атмосфере, легко выводятся из атмосферы облака-
облаками и осадками, а в верхней атмосфере могут являться важным фак-
фактором в изменении радиационного режима атмосферы.
Из сказанного видно, что, как уже было отмечено, из-за
большой неопределенности наших знаний о ряде физических и хи-
химических характеристик атмосферных аэрозолей, а также из-за
очень больших вариаций этих характеристик, многие утверждения
о роли аэрозолей в различных физико-химических процессах в ат-
атмосфере пока не имеют точных количественных оценок.
Задача, стоявшая перед автором этой монографии, заключа-
заключалась в создании модели атмосферных аэрозолей для решения урав-
уравнений переноса излучения в видимой и инфракрасной областях
спектра в земной атмосфере при различных метеорологических ус-
условиях.
Автор начал с анализа и обобщения экспериментальных дан-
данных о микроструктуре атмосферных аэрозолей и создания на их ос-
основе аэрозольных моделей. Этот путь оказался чрезвычайно труд-
трудным. Модели получались сложными, а главное, не всегда соответ-
соответствовали модельным оптическим характеристикам и общепринятым
представлениям, так как микрофизическая аппаратура не всегда
позволяет измерять те параметры аэрозолей,которые ответственны
за соответствующие оптические характеристики (неполный спектр
размеров частиц, испарение и другие изменения микроструктурам
характеристик до момента их измерения и т.д.), переход к опти-
оптическим характеристикам аэрозолей делался некорректно из-за не-
незнания показателя преломления вещества частиц, пренебрежения
их несферичностью и т.д., неправомерно сравнивались результаты
локальных микроструктурных измерений аэрозолей с их интеграль-
интегральными оптичеокими свойствами.
Эти обстоятельства заставили автора углубленно заняться
проблемой соответствия оптических и микроструктурных характе-
характеристик атмосферных аэрозолей. Это было тем более важно, что
развитие дистанционных методов зондирования атмосферы, особен-
особенно лазерного зондирования за последние 10 лет поставило перед
исследователями задачу определения микроструктурных характе-
характеристик аэрозолей по оптическим данным. Решение обратной зада-
задачи оптики аэрозолей вызвало необходимость получения априорной
информации о микроструктурных характеристиках аэрозолей. На
этой основе у автора возник тесный творческий контакт с коллек-
коллективом сотрудников Института оптики атмосферы СО АН СССР. Учас-
Участие автора и руководимого им коллектива в Комплексном энерге-
энергетическом эксперименте (КЭНЭКС), а также в Атлантическом тропи-
тропическом эксперименте (АТЭП-74) в семидесятых годах позволило на-
накопить оольшой экспериментальный материал по микроструктурным
характеристикам атмосферных аэрозолей. В результате при подго-
подготовке собранного автором материала к публикации оказалось,что
10
весь материал не укладывался в рамках традиционных схем. Поэто-
Поэтому автор решил разделить свою работу на микрофизическую и опти-
оптическую части. Это было оправдано и тем, что ряд вопросов, рас-
рассматриваемых в настоящей монографии, не находит еще большого
интереса у специалистов в области оптики атмосферы, в то вре-
время как большинство специалистов в области изучения аэрозолей
как объекта метеорологических исследований далеки от вопросов
гптики аэрозолей.
Однако даже в значительно более узкой по тематике моногра-
монографии автору пришлось затронуть вопросы, в которых он не облада-
обладает достаточным опытом собственных исследований, поэтому он в
ряде разделов ограничился обзором соответствующей литературы.
Естественно, что монография в определенной степени - обобщаю-
обобщающий проблему обзор. Экспериментальный материал, использованный
в это? работе, получен сотрудниками лаборатории физики аэрозо-
аэрозолей Ленинградского государственного университета С.Д.Андреевым,
Ь..1.«мохойским, В.М.Жуковым, а. В. Решетниковым, А.Ю.Семовой,
В.К.Соломатиным, научным сотрудником Главной геофизической об-
обсерватории им. А.Л.Воейкова В.И.Ивановым, научным сотрудником
Ленинградского гидрометеорологического института Е.Г.Головиной,
а также дипломантами Н.О. Грлгоровым, М.И.Коноваловым и други-
другими. Ряд экспериментальных исследований был совместно выполнен
со старшим научным сотрудником НИФХИ им. Л.Я.Карпова Б.И.Ого-
родниковым, с руководителем лаборатории ЦАО А.В.Федынским. В
написании раздела о коагуляции аэрозолей большую помощь оказа-
оказала старший научный сотрудник Главной геофизической обсервато-
обсерватории им. А.И.Воейкова Ю.А.Довгалюк и старший преподаватель Нов-
Новгородского политехнического института О.А.Одинцов.
Фотохимические исследования проводились совместно с науч-
научными сотрудниками Ленинградского гидрометеорологического инсти-
института и Научно-исследовательского института аналитической химии
Б.Г.Сиротой, В.Н.Колбенковым, С.Н.Хворостовским и другими. Ав-
Автор глубоко признателен всем названным коллегам. Искреннюю бла-
благодарность автор выражает также профессору Ленинградского гид-
гидрометеорологического института В.Г.Морачевскому, который вни-
внимательно прочитал первоначальный вариант монографии, указал на
ряд ошибок и неточностей в тексте.
Автор надеется, что эта работа принесет определенную поль-
пользу тем, кто интересуется вопросами химии и структуры атмосфер-
атмосферных аэрозолей. И
Глава I
ИСТОЧНИКИ АТМОСФЕРНЫХ АЭРОЗОЛЕЙ
И ПРОСТРАНСТВЕННО-ВРЕМЕННАЯ СТРУКТУРА
ИХ ХИМИЧЕСКОГО СОСТАВА
Содержание аэрозолей в атмосфере обусловлено не только
вкладом источников разной мощности (интенсивности поступления
аэрозолей), а также такими факторами, как место образования аэ-
аэрозолей в атмосфере, характеристики воздушных потоков, облаков
и осадков, но и скоростью выведения'этих аэрозолей из атмосфе-
атмосферы, которая, в свою очередь, зависит от спектра размеров и хи-
химического состава генерируемых частиц. Кроме того, источники
аэрозольных частиц определяют (по крайней мере вблизи от них)
концентрацию, дисперсность и химический состав аэрозолей в ат-
атмосфере.
Физико-химические свойства аэрозолей существенно зависят
от того, как распределены химические соединения в частицах раз-
разных размеров. В частности, оптические свойства характеризуются
в первую очередь химическим составом больших частиц.
Поскольку основная масса аэрозольных частиц содержится в
нижних слоях атмосферы, то и такие важные климатообразующие
факторы, как влияние на оптическую прозрачность атмосферы и по-
поглощение коротковолновой солнечной радиации, определяются свой-
свойствами аэрозолей нижних слоев, являются их наиболее нестабиль-
нестабильной и изменчивой частью. Поэтому представляет интерес устано-
установить, можно ли обнаружить определенные закономерности в хими-
химическом составе аэрозолей в нижних слоях атмосферы и какова роль
12
местных источников в формировании глобального аэрозольного ;..:-та.
Очевидно, что если аэрозольные частицы местного происховд,;.^:?
способны долго существовать и переходить в более высокие слои
атмосферы, то это приведет к росту концентрации аэрозольного фо-
фона. Следовательно, в этом случае можно говорить о росте влияния
аэрозолей местного происхождения и, в частности, антропогенных
аэрозолей на климат Земли. Для протяженных и постоянно действую-
действующих источников, таких, как почвы и морская поверхность, глобаль-
глобальный характер химического состава частиц и влияния их на радиа-
радиацию очевиден.
Для проблем, связанных с облако- и осадкообразованием осо-
особый интерес представляет задача определения пространственно-вре-
пространственно-временной структуры ядер конденсации и кристаллизации. В частнос-
частности, необходимо знание концентрации и дисперсности частиц мор-
морской соли и сульфатов.
§ 1.1. Глобальные оценки мощности источников аэрозолей
Полной и достаточно точной инвентаризации всех источников
аэрозольных частиц до настоящего времени не проведено. Оценки
мощности большинства источников выполнены с точностью до поряд-
порядка измеряемой величины.
Основными источниками аэрозолей являются почвы, растения,
поверхность морей и океанов, вулканы, метеоритные потоки, лес-
лесные пожары, химические и фотохимические реакции в атмосфере, хо-
хозяйственная деятельность человека. По оценкам С.Дэвиса мощность
природных источников аэрозолей составляет 2,31'10^ т/год, из
которых 1,10'1Сг т/год приходится на солевые частицы из океана
[74]. (Последние существуют в атмосфере не более нескольких дней,
так как быстро вымываются осадками.) Мощность антропогенных ис-
источников аэрозолей оценена им в 2,96'Ю т/год, т.е. несколько
больше 10% от общей мощности источников.
Химические и Фотохимические реакции в атмосфере ответст-
ответственны за появление мелкодисперсной фракции вторичных аэрозолей.
Аэрозольные частицы образуются из продуктов реакций сернистого
газа, органических соединений, аммиака, сероводорода, окислов
13
азота и некоторых других газов с окислителем типа озона, а так-
также с водяным паром и солевыми аэрозольными частицами.
верхний и нижний пределы количества углеводородов, выбра-
выбрасываемых в атмосферу,, составляют @,72+2)-10 т/год. Около
1,8-Ю7 т/год поступает в атмосферу от стационарных источников
США. Содержание очень мелких частиц ( г < 2 мкм) составля-
составляет примерно 22% от общего количества, что равно 4-10 т/год.
По Е.ГольдОергу эта величина для земного шара в целом равна
I.Z'IO7 т/год. 3 Отчете Национальной администрации по контролю
загрязнения воздуха в США приведен общий выброс органических
веществ, равный 2,9«107 т/год. Можно предполагать, что 75$ это-
этого количества не метан, а более сложные соединения, которые,
возможно, переходят в аэрозольное состояние (вторичные аэрозо-
аэрозоли). Ь Отчете Комиссии по исследованию критических проблем
окружающей среды приведено значение 9-10 т органических соеди-
соединений,что является, вероятно, верхним пределом [74].
Весьма эффективным источником мелкодисперсных фракций ор-
органических аэрозолей является растительность. Согласно Ф.Венту,
биосфера ежегодно выделяет в атмосферу около 0,2-10 т терпенопо
добных или слабо окисленных углеводородов, что создает естествен-
естественный фон ~ 3-6 мкг/м3 [237 ].
В последние годы активно обсуждается гипотеза об образова-
образовании органических веществ и соединений серы из морских растений
и поверхностной пленки океана. По оценке Дж- Довелока в атмос-
атмосферу выделяется 10 т/год диметилсульфида ((CH^^S)морскими
водорослями, листвой и почвой. Д. Хитчкок подверг критике эту
оценку. По его данным диметилсульфида выделяется не больше
B+5)-106 т/год. Следовательно, более важен учет выделения
сероводорода анаэробными бактериями [181].
Мощность различных источников серы определяется следующи-
следующими цифрами: океан вносит 130, процессы сжигания - 150, вулка-
вулканы - 2 Мт/год. Мощность процессов гниения и выделения серы бак-
бактериями (в виде H2S) точно не установлена. Общая интенсивность
выделения серы, приведенной к [SO4], 550 Мт/год. Интенсив-
Интенсивность процессов вымывания сульфатных аэрозолей осадками оцене-
оценена такими цифрами: над океаном - 217, над сушей - 258 Мт/год.
14
Остальные 75 Мт/год должны удаляться из атмосферы с помощью дру-
других процессов - сухого выпадения и растворения.
Источниками выделения в атмосферу сернистого газа, кроме
вулканов, являются промышленные предприятия A0 т/год), а се-
сероводорода - в основном растительность. Оценка выделяемого рас-
растительностью и вулканами сероводорода весьма неопределенна.
Кояно предполагать, что порядок величины выделяемой массы тот
г.е, что и для сернистого газа. В перерасчете на [S04]" газо-
газовые источники дают ~ 4-10 т/год.
Еде менее определенны оценки мощности источников окислов
7 Р
азота и аммиака: 10 - 10 т/год. Причем лишь часть упомянутых
газов (.около 15+30%') идет на образование аэрозольных частиц.
Е.Гольдберг оценил скорость образования сульфатов в ат-
атмосфере вследствие сгорания ископаемого топлива, сравнив содер-
содержание сульфатов в ледниках в настоящее время и в те периоды,
когда влияние человека отсутствовало, как l,3*10s т/год. Ж.Пе-
терсон и Х.Юнге дают величину 2>108 т/год [74].
Объем промышленного выброса сернистого газа и окислов азо-
азота за последние 20 лет в связи с переходом на сжигание газов
вместо угля заметно увеличился. Причем большое количество этих
газов окисляется до ангидридов кислот и растворяется в облач-
облачных каплях. (Попытки определить источники серы по измерениям
изотопа34S не дали обнадеживающих результатов из-за сильной
перемешанности сернистых соединений от разных источников.) Вре-
Время жизни SO2 в тропосфере составляет от одного часа до нес-
нескольких дней, а сульфатных частиц - около одной-двух недель.
Переход S02 в SO^ происходит неполностью, что подтверждается
нелинейностью отношения [SO2]/ [S04]~2. Превращение его в SO*
происходит в процессе переноса S02 на большие расстояния. О
слабой растворимости SO2 в воде и медленном его переходе пос-
после этого в сульфаты свидетельствует тот факт, что концентрация
SOj с высотой изменяется значительно медленнее, чем концентра-
концентрация водяного пара. Большинство измерений [SO^2] свидетельст-
свидетельствует о континентальном происхождении сульфатов. Например, от-
отношение [S04] /[Na] в атмосфере Северной Атлантики изменяет-
изменяется от 0,54 до 20, в то время как для морской воды это отноше-
отношение равно 0,25. Так как значительная часть соединений серы ан-
15
тропогенного ироисховдения,
то наблвдается заметное уменьшение
ее
ее концентрации летом и увеличение зимой, когда возрастает ин-
интенсивность выбросов сернистых соединений различными теплоэнер-
теплоэнергетическими объектами. В приземном слое и в тропосфере на высо-
высотах от 50 до 3000 м обнаруживается слабая зависимость концент-
концентрации сернистых соединений от высоты, а в нижележащем слое
концентрация выше при устойчивом состоянии атмосферы. Указан-
Указанные закономерности хорошо объясняются особенностями распростра-
распространения выбросов сернистых соединений промышленного происхожде-
происхождения. Оценка времени жизни S02 вблизи источников равна ^ —Юч,
причем концентрация сульфатов редко превосходит концентрацию
S02.
Разные
няются
оценки времени жизни SO2 в нижней атмосфере объяс-
различными условиями окисления сернистого газа и превра-
щения его в аэрозольные частицы.
Окислы азота и группа аммония
ропогенное происхождение:
N0
nh1|
имеют в основном ант-
гениие nFv«^~^ U0x - 3-Ю7, NH+ - 13-Ю7 т/год;
inu активно участвует в образовании фотохимических, аэрозолей,
N02 растворяется в воде и в результате реакции окисления обра-
HN03. Вертикальный градиент HNO, такой же, как у Н20,
зует
тогда как содержание
медленнее
большая часть NH3
._„_г NH3 с высотой уменьшается значительно
Это можно считать доводом в пользу утверждения, что
образует сульфат аммония. Локальное отноше-
- от оощей массы возникающих аэрозолей. Двуокись серы оказы-
оказывает влияние на фотообразование органических аэрозолей: малые
концентрации S02 подавляют образование аэрозолей, а большие
усиливают. Параллельно с образованием органических аэрозолей
возникает серная кислота. Большая часть органического вещества
в фотохимических аэрозолях, составляющая 25-30% от общей массы,
относится к моно- и дикарбоновьм кислотам.
Образование аэрозольных частиц из сернистых соединений и
окислов азота в загрязненной атмосфере по Х.Юнге происходит тре-
тремя основными способами.
1) Фотохимическое окисление и
гетерогенные газовые реакции проис-
происходят в засушливых районах и высоких олоях тропосферы. По
jp.Коксу скорость конверсии сернистого газа составляет 0,03%
от S02» окисляемого за 1 ч в чистом воздухе фотохимическим пу-
путем [146]. Ж.Брикар показал, что если концентрация S02 в воз-
воздухе сильно возрастает, то достигается уровень насыщения, выше
которого образование частиц не зависит от концентрации S02 [125].
Смесь кислорода, азота и следов сернистого газа при корот-
коротковолновом облучении не образует аэрозольные частицы, тогда как
з присутствии NO2 и N^0 пно происходит. Ж.Брикар считает, что
1рямое фотоокисление $02 не дает достаточного числа ядер кон-
хенсации, и для образования аэрозольных частиц необходимо при-
о©
- г
ние
fNO,l/f NHJ для воздушного бассейна над городом состав- |,уТствие N02.
L 3J/L 3J .,.,.,/, _ „„„ттво mMiYnriTCJt Сходные а
ляет 0,91 - 1,4, т.е.
на
NH4N0
в этом случае приходится
ми,
I0I5J? от всей массы аэрозолей для частиц сг<5 мкм (пример-
(примерно 10-14 мкг/м3).
Скорость образования нитратных загрязнений оценивалась
Ж.Петерсоном и Х.Юнге по составу продуктов сгорания ископаемо-
ископаемого топлива как 3,5*10 т/год. Примерно такие же величины при-
приводятся в работах [11, 74, 76].
Количество образующихся вторичных аэрозолей и их химичесН
кий состав известны далеко не полностью. Исследования фотохи-
фотохимических аэрозолей показали, что они состоят в основном
окислов металлов, солей нитратов и сульфатов
и воды. Сульфаты и нитраты, входящие в органические аэрозоли
образуются при фотоокислении S02 и N02 и составляют примерно
Сходные фотохимические реакции происходят также с терпена-
выделяющимися из растительных веществ. Представляет инте-
из
азотной кислоты
рее исследование возможности окисления гидрокарбонатов до угле-
углерода. Если такого рода реакции в атмосфере происходят, то ста-
становится понятной корреляция между содержанием сульфатов в ат-
атмосфере и поглощением солнечной радиации.
2) Каталитическое окисление в
рисутствии тяжелых металлов в
большой степени зависит от присутствия подходящих катализато-
катализаторов (ионов тяжелых металлов) и может быть достаточно быстрым
сильно загрязненном воздухе. При определенных значениях рН
процесс прекращается. Реакция проходит как в сухом воздухе,
ак и в облачных каплях.
|2 1922 V[7
16
а: ¦ четче ч y
3) Реакция аммония с двуокисью
серы в присутствии жидкой воды
(реакция облачных капель) зависит от по-
поступления NHj. Если значение рН поддерживается достаточно вы-
высоким, например вследствие поступления NH3 из газовой фазы, то
реакция может продолжаться.Аму.иак и аммиачные соединения явля-
являются, в свою очередь, продуктами процессов сгорания и распада
органического вещества (в особенности животного происхождения).
Механизм образования сульфата аммония эффективен только в при-
присутствии жидкой воды, т.е. в атмосферных слоях, где существуют
облака и туманы. Модельные расчеты показывают, что скорость
окисления в облачных каплях равна 12$ SO2 в I ч. Измерения с
самолетов содержания частиц (NH4JS04 показывают, что макси-
максимум концентрации сульфатных частиц часто наблюдается под ниж-
нижней границей облака. Чаотицы оульфата аммония могут оставаться
взвешенными в воздухе после испарения капель облаков и туманов.
Начальные ядра сульфата аммония имеют радиус примерно
З-iO"8 см, такие ядра переходят в капли с размером 1СГ6 ом.Так
как время жизни сульфатных частиц больше времени жизни газовых
компонентов, из которых они образуются, то этот механизм может
существенно влиять на глобальное содержание аэрозолей.
Кроме серной и азотной кислот и их солей в атмосфере мо-
могут образовываться также соляная кислота и ее соли. Этот про-
процесс происходит в результате освобождения с поверхности моря
хлора в газообразной фазе. Количество хлора, выделяемого в ат-
атмосферу, не рассчитывалось.
Не упоминается в литературе возможность фотохимических и
химических реакций образования атомов углерода. Такой процесс
может происходить в результате неполного сгорания углеводоро-
углеводородов (их обугливание). Предполагается, что этот процесс не мо-
может происходить в атмосфере, так как для него потребуется вы-
высокая температура (около 700°С). Однако присутствие сильных
катализаторов (солей и окислов различных металлов), а также
сильных окислителей (возбужденных атомов кислорода, озона,раз-
озона,разных кислот).коротковолнового солнечного освещения, вероятно,
может приводить к возникновению реакции окисления углеводоро-
углеводородов и образованию смол и сажи.
18
Морская поверхность вносит в глобальное массовое содержа-
содержанке атмосферных аэрозолей не менее 10-20$ частиц. Е.Эрикоон оце-
оценивает общее количество соли, поступающей в атмосферу, более
чем в 10^ т/год. Большинство частиц быстро возвращается в оке-
океан и только 10 т оседает на суше. Химический состав этих час-
частиц соответствует примерно химическому составу сухого остатка
морской воды: NaCl- 78; MgCl2- И; CaS04,Na2S04, K2S04- 11%.
Роль морских солей в создании глобальной картины атмосфер-
атмосферных аэрозолей довольно ограничена. В частности, по вертикали
они распространяются только до высоты 1-2 км, так как активно
участвуют в образовании облачных капель. Б.Мейсон полагает,что
одна десятая часть ядер конденсации облачных частиц состоит из
морской соли, а остальная представляет собой смесь продуктов
природных и антропогенных пожаров [70]. Данные по концентрации
(коэффициенту обогащения [X]/[Na]) в морских аэрозолях та-
таких элементов, как Fe, Cu, Zn,$r, показывают, что аэрозоль-
аэрозольное вещество не может образовываться только из поверхностной
пленки морской воды. Коэффициент обогащения сильно зависит от
размера частиц. Например, отношение содержания органических ве-
веществ к содержанию Na растет с уменьшением размеров частиц.
Для всех перечисленных компонентов, а также для С, Р, NOj на-
наблюдается повышенное содержание этих элементов .в микрослое
A00^150 мкм).
Часть органического вещества морского происхождения выно-
выносится в атмосферу в ооставе мороких солевых частиц. Содержание
углерода в аэрозольных частицах морской соли изменяется от 0Д5
до 0,47 мкгДг с отношением [C]/[NaCl], равным 0,01-0,03 при
концентрациях морской соли больше 9 мкг/м3. При уменьшении кон-
концентрации соли до 2,5 мкг/м3 отношение [C]/[NaCl] возрастает
до 0,19, что подтверждает гипотезу о выделении органических ве-
веществ высыхающими морокими водороолями. Исследования состава и
структуры мороких аэрозолей В.Гнддингоом и М.Бейкер показа-
показали, что частицы дымки покрыты органической пленкой, состоящей,
вероятно, из омыленных жирных кислот и белков, а не жирных кис-
кислот, как предполагалось ранее. Концентрация органического ве-
вещества дымки такова, что образование монослоя в результате прос-
простой адсорбции из воздуха маловероятно. Следует предположить,
19
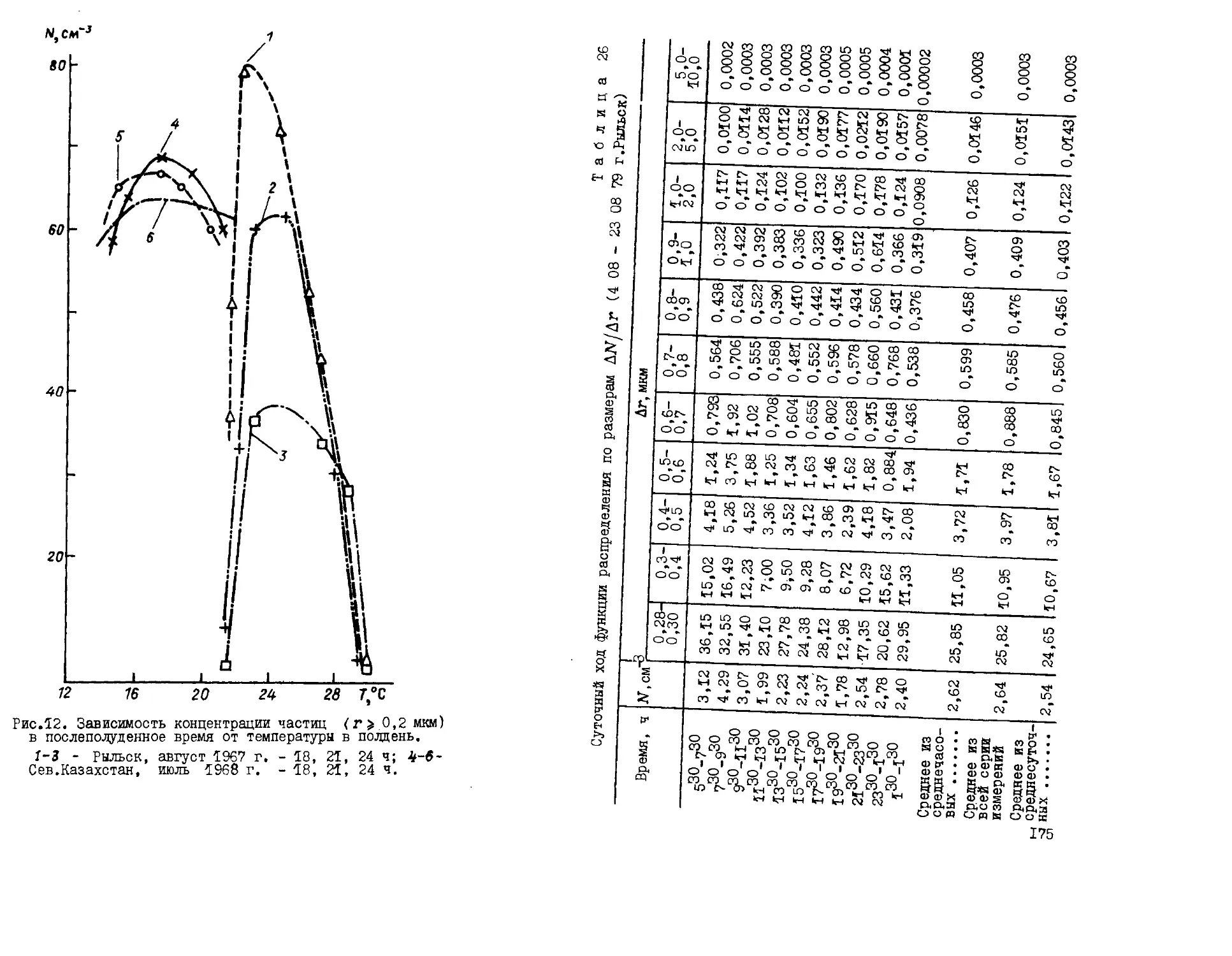
},мкг\м
что эти частицы образуются при диспергировании поверхностного
слоя океана.
до настоящего времени неясны все механизмы выделения аэро-
аэрозольного вещества океаном. Так, В.С.Савенко, основываясь на ки-
кинетической теории конденсированных состояний Я.И.Френкеля, рас-
рассмотрел возможность переноса ионов из воды в газовую среду. По
его мнению, такой перенос реален
и мохет быть одним из механизмов
выделения в атмосферу ряда ионов
[83], участвующих в образовании
атмосферных аэрозолей. Р.Лемлих
полагает, что растворяемое ве-
вещество разделяется внутри жид-
жидкости и удаляется путем адсорб-
адсорбции на поверхности пузырьков
[193]. В.Д.Корж показал, что ин-
интенсивность испарения электроли-
электролитов из морской воды определяет-
определяется их относительной концентраци-
концентрацией [83]. Явление фракционирова-
фракционирования обусловлено различием по-
поверхностной и объемной концент-
концентраций солей в морской воде.Фрак-
воде.Фракционирование морских солей воз-
возможно при замерзании морской во-
воды. Образующиеся при этом крис-
кристаллы льда состоят из чистой во-
воды, а растворы солей, обладаю-
обладающие разной растворимостью, вы-
вытесняются в ячейки между крис-
кристаллами. Соли из перенасыщенно-
перенасыщенного раствора выделяются в такой
последовательности: углекислый
кальций (СаСО3), сульфаты, в
основном в виде мирабилита Na2SO4 • 10 Н20, и после всех хло-
хлористый натрий (NaCl). Так как выветривание солей изо льда мо-
может происходить в процессе их вымерзания, то будет наблюдаться
20
V,MJC
Рис.1. Зависимость кон-
концентрации хлоридов в призем-
приземном слое от скорости ветра
на побережье Черного A),
Японского B), Балтийского
E), Баренцева D) морей.
обогащение аэрозолей частицами, содержащими [Са]+2 и
Процесс выноса морских солей в атмосферу при срыве водных
капель и пузырей с гребней волн подробно обсуждался в работах
[9, 110]. Следует отметить, что особенно интенсивное образова-
образование частиц, содержащих морские соли, происходит в прибрежной зо-
зоне как в результате прибойного разбрызгивания морской воды, так
и выделения солей с поверхности песка и камней при их обсыхании.
,1нтересные данные о зависимости концентрации морских аэрозолей
от скорости ветра для разных водных бассейнов приведены в моно-
монографии О.П.Петренчук (рис.1) [76].
Вулканы при извержениях выбрасывают в атмосферу колоссаль-
колоссальное количество дыма и пепла: до 75-Ю6 м3 ( "* 2,0*10 т вещест-
вещества). Часть этих частиц вместе с газами вулканического происхож-
происхождения может подниматься в стратосферу, на высоту более 20 км.
Самые мелкие частицы сохраняются в стратосфере несколько лет.
Несмотря на эпизодичность вулканических извержений, можно оце-
оценить их среднегодовую мощность величиной порядка (I*-4)-ICr т.
Кроме того, следует учесть, что сернистый газ, выбрасываемый в
стратосферу, вступает в фотохимические .реакции и также образу-
образует аэрозольные частицы. Для приближенной оценки можно предполо-
предположить, что в стратосферу его попадает •» 10 т/год. По ходу ак-
аккумуляции монтмориллонита в осадочных морских породах Е.Гольд-
берг оценил выброс продуктов вулканических извержений в атмос-
атмосферу величиной I,5-10 т/год [74]. Наименьшее значение равно
2,5-107 т/год.
Роль вулканов в общем загрязнении атмосферы иногда сущест-
существенно преувеличивается. Хотя очевидно, что наиоолее мощные из-
извержения вулканов действительно сильно загрязняют верхние слои
атмосферы, но для нижних слоев вулканы как загрязнители имеют
лишь локальное значение. Р.Кейдлом были сделаны оценки средне-
среднегодового вулканического загрязнения соответственно тропосферы
и стратосферы некоторыми соединениями: НС1 - 7,5*10 и2,8*10 ;
HF - 2.8.104 и 1,4-Ю3; S02- 7,5'Ю6 и 2,8-Ю5 т/год. По мне-
мнению Р.Кейдла, фреоны дают хлора и фтора значительно больше.Оче-
больше.Очевидно, роль паров НС1 и HF в образовании глобального фона аэ-
аэрозолей ничтожна так же, как и роль S02 вулканического проис-
происхождения в тропосфере [105].
21
Вулканическая пыль, судя по некоторым данным, может даже
в тропосфере присутствовать достаточно длительное время. По
крайней мере в ледниковых отложениях Антарктиды обнаружена вул-
вулканическая зола, которая была перенесена на расстояние не менее
4000 км, причем возраст исследованных отложений состазлял от
1,8 до 16 млн. лет.
Ф.Фольцем приведена оценка общего количества вулканическо-
вулканического вещества в стратосфере в период после извержения вулкана
Агунг - 1,3*10 т. Инжекция вулканической пыли и SO2 происхо-
происходила в основном в северном полушарии в зоне широт 40-58°. Че-
Через два месяца после извержения вулкана наблюдались аэрозоль-
аэрозольные слои в глобальном масштабе на высоте 15 (в Арктике) и 20
(на экваторе) км. Время пребывания этих вулканических продук-
продуктов в атмосфере составляло примерно один год.
Относительно недавнее извержение вулкана Этна позволило
оценить роль вулканических загрязнений в районе, где высок уро-
уровень антропогенных загрязнений. Анализ проб, взятых в июне
1973 г. с помощью сканирующего электронного микроскопа и рент-
генофлуоресцентным методом показал, что загрязнение атмосферы
элементами Cd, Hg.Cu, Zn - одного порядка, элементом Sc -
значительно больше, а элементом РЬ - значительно меньше, чем
в результате человеческой деятельности.
Химический анализ дыма и пыли для разных вулканов показы-
показывает преимущественное содержание соединений кремния - 60-80,
сульфатов - 30-Ю, кальцитов - 3-10, соединений алюминия -0-20;
железа - 1-10$. Химический состав продуктов вулканических вы-
выделений для разных вулканов различен (табл.2, 3). Е.Мроз, ис-
исследовавший с помощью забора проб импактором и нуклепор-фильт-
рами продукты деятельности вулкана Химаеу (Исландия), отмечает
обогащенное содержание в аэрозолях таких элементов, как Вг ,
Se.Sb, In [208]. Анализ проб частиц, взятых из эруптивных га-
газов вблизи вулкана Гекла, позволил предположить, что основная
масса частиц состоит из осколков лавы, но присутствуют и такие
кристаллические соединения, как сульфаты кальция и аммония, а
также мелкие сферические включения.
Р.Кейдлом было замечено, что частицы, собранные из газов
вулкана Килауэа до и после извержений, имели намного большее
22
Таблица 2
Коэффициенты обогащения EF различных элементов
в аэрозольном веществе при извержениях вулканов
относительно их содержания в земной коре
Эле
мент
К
Na
Mg
Mn
Sc
La
V
Co
Hf
Cr
J
Cs
Zn
Pb
Cu
Cd
s
As
Sb
Se
Cl
Br
Hg
Вулкан
Изал-
ко
0,056
0,19
0,30
0,026
_
_
_
_
_
_
_
_
_
_
_
_
-
Хиыа-
еу
-
-
-
0,90
0,40
_
3,3
0,40
_
_
53
_
_
115
_
_
-
Кила-
Килауэа
_
-
-
-
-
-
_
-
-
-
-
_
_
_
_
_
200
17000
_
_
35000
Май-
он
_
0,040
0,27
-
-
-
-
-
-
-
-
_
_
-
60
_
-
_
-
Аре-
нал
0,030
0,080
0,13
-
-
-
-
-
-
-
-
-
-
_
_
-
13
-
-
-
25
_
-
Этна
8,9
15,0
-
5,9
0,30
-
2,4
4,4
-
2,8
-
20
580
320
240
1900
22000
800
660
180000
31000
36000
13000
Толба-
чикский
-
1,9
-
-
1,6
4,1
-
4,0
16,8
32,0
30,8
1620
950
-
2200
-
-
2780
11000
-
28,5
230
1740
Авгус-
Августин
0,43
1,3
0,78
0,78
1,5-
0,69
1,6
0,75
-
7,5
-
-
4,3
20
13
233
-
173
84
-
17+8000
1060
5000
Примечание,
щения EF = 1.
Для Al.Si.Fe коэффициент обога-
23
Таблица 3
Содержание отдельных элементов в воздухе вблизи
фумарол (август 1974 г., усредненное значение) и при
извержении вулкана Толбачикский (северный прорыв,
август 1976 г.), мкг/м3
Элемент
Fe
Na
Са
К
Вг
Си
С1
Sr
Sc
Th
Hf
Yb
Та
Cd
As
Mo
Hg
CS
Lu
Sm
Se
Sb
Co
Zn
Rb
AS
La
Аи
Долина
гейзеров
180,0
25,0
11,0
12,0
5,5
5,5
6,3
2,0
0,023
0,030
0,020
0,047
2,7
1,4
1,1
0,73
0,21
0,041
0,0095
0,50
0,17
0,12
0,12
0,30
0,070
0,050
0,010
0,022
Фумаролы
на влк Ключев
екая Сопка
30,0
3,2
1,5
1,5
0,68
1,4
0,70
2,3
0,023
0,020
0,008
0,015
0,26
0,22
0,38
0,36
0,054
0,010
0,00042
0,38
0,13
0,050
0,025
0,30
0,10
0,050
0,0085
0,0063
на влк Безы
мянный
50,0
17,0
2,7
5,0
2.4
1,9
1,0
2,0
0,017
0,063
0,011
0,024
1,90
0,90
0,80
0,71
0,24
0,015
0,0010
0,23
0,090
0,050
0,025
0,30
0,10
0,050
0,0085
0,060
Влк Толба-
Толбачикский
120,0
12,0
1,0
1,0
0,50
1,4
2,7
2,5
0,14
0,050
0,032
0,029
1,0
< 0,50
< 1,0
< 0,20
0,25
0,068
0,00046
< С, ю
0,39
0,32
0,56
0,13
0,062
0,052
-
-
24
содержание растворимых солей, чем частицы, собранные из газов,
выделяющихся из лопающихся верхушек фонтанчиков лавы. Возмож-
Возможное объяснение этого явления заключается в том, что при извер-
извержении сернистый газ смешивается при высокой температуре с кис-
кислородом воздуха и окисляется до трехокиси серы, которая перехо-
переходит при наличии влаги в кислоту и растворяет соли металлов.
Г.Мак-Дональд подсчитал, что масса газов, выделившихся при из-
извержениях того же вулкана, составила ~ 0,5% от массы извержен-
изверженной лавы, причем на сернистый газ приходится всего 10$ от обще-
общего количества изверженного газа [59, 105, 209].
Таким образом, вулканические продукты представляют собой:
1) частицы тонко измельченной лавы и продукты истирания стенок
кратера; 2) капли серной кислоты с растворенными кристалличес-
кристаллическими веществами, часть из которых, возможно, является продук-
продуктом сублимации магмы; 3) сернистый газ и сероводород, не успев-
успевшие окислиться до выброса в атмосферу.
Почвы и пустыни. Вдали от морских и промышленных районов
химический состав аэрозольных частиц практически полностью оп-
определяют почвы. Косвенные оценки позволяют предполагать, что в
приземном слое почва генерирует около 50% всех аэрозольных час-
частиц (по массе). Абсолютная продукция почвенных частиц составля-
ет не менее A*5)-10 т/год [19J.Однако химический состав
этих частиц не идентичен составу почвы, так как минералы и дру-
другие почвенные продукты не одинаково диспергируются, выветрива-
выветриваются и поднимаются в атмосферу. Основные компоненты пыли - сое-
соединения кремния, глиноземы, карбонаты и кальциты, окислы желе-
железа. Количество органических соединений в аэрозолях почвенного
происхождения сравнительно невелико и не превосходит 10%.
Оценивая количество частиц горных пород и почвенных час-
частиц, поступающих в атмосферу вследствие естественных процессов,
Е.Гольдберг экстраполировал скорость накопления материалов вы-
выветривания в ледниках и морских осадочных породах для расчета
глобального среднего выброса частиц и получил величины в пре-
пределах A+5)'10 т/год. Г.Робинсон и Е.Робинс оценили средний
выброс частиц равным 2*10 т/год, приняв при расчетах, что кон-
концентрация атмосферной пыли - величина постоянная. Ж.Петерсон и
Х.Ккге при экстраполяции количества естественной и образовавшей-
25
ся в результате сельскохозяйственных работ пыли, поднятой вет-
ветром над США, получили близкую величину - 2,5-10 т/год [74].
Наиболее вероятно, что эти значения сильно занижены, и их мож-
можно принять как величину вклада почвенных аэрозолей в фоновую
глобальную концентрацию атмосферных аэрозолей.
Некоторые оценки по мощности и распространению аэрозолей
почвенного происхождения позволяют делать наблюдения с косми-
космических спутников Земли [26]. Однако неточность определения дио-
перснооти пыли приводит к значительным ошибкам в таких оценках.
По оценкам О.П.Петренчук из почвы поступает в атмосферу
9,6-Ю8 т/год аэрозольного вещества [76].
Чаоть пыли, осевшей на земную поверхнооть, может попадать
в атмосферу вторично. Эффективность вторичного ветрового захва-
захвата пылевых частиц не менее 10$ [69].
Биологичеокие частицы (пыльца, споры, грибки, микробы,
вирусы) имеют диаметр в ооновном более 1 мкм. Эти чаотицы пе-
переносятся на большие расстояния и присутствуют даже в страто-
стратосфере. Например, споры грибков находили над Карибским морем на
расстоянии не менее 1000 км от источника, пыльца обнаружива-
обнаруживалась на расстоянии 2500 км от возможного источника, а морокие
оактерии - на раостояниях до 130 км от побережья в глубь мате-
материка. При заборах проб частиц в ловушки на высотах 11-22 км
были собраны споры ряда плесневых грибков [99, 105] .
С помощью аэрозольного опектрометра А.Гётц и О.Прейнинг
собрали пробы чаотиц естественных аэрозолей с размерами 0,09
мкм < г < 0,5 мкм (западные районы США). При повторном про-
просмотре проб через неоколько дней обнаружилось уменьшение раз-
размеров и числа мельчайших частиц [105]. По-видимому, это связа-
связано с медленным иопарением летучего органического вещества пос-
после их осаждения на подложку, поэтому можно предполагать, что в
атмосферных аэрозолях роль органической фракции больше, чем
следует из оценок массы органичеокого вещества в пробах.
Натурные олтичеокие эксперименты по наблюдению процесса
образования аэрозолей из органических веществ проводились
В.Н.Капуотиным и Ю.С.любовцевой [52].
Значительное выделение аэрозольного вещеотва наблюдается
при высыхании морских водорослей: после полудня на морском по-
26
;ережье в результате этого процесса концентрация ядер Айткена
возрастает до 10 см~3 и более.
Р.Янике [184] утверждает, что интенсивность образования ор-
органических аэрозоле;., того же порядка величина, что и интенсив-
интенсивность образования почвенных, мороких и сульфатных аэрозолей.
Зремя жизни частиц с г < 0,01 мкм меньше суток, а для частиц
0,01 < г @,1 мкм - около 80 сут. По его оценкам четверть
всех ядер Айткена состоит из органического вещества.
Продукты сгорания растительности. Из этих веществ наибо-
наиболее существенны продукты сгорания, образованные в результате
лесных пожаров и сжигания отходов лесоразработок - главным об-
образом, сажа, которая может поглощать заметную долю падающей на
Землю солнечной радиации, и пепел. Оценка вклада этого источни-
источника в общее содержание аэрозолей весьма неточна. Так, экстрапо-
экстраполяция данных по вкладу твердых продуктов сгорания лесов в США
на весь земной шар ооставила 3*10 т/год. Очевидно, что оценка
занижена. Например, Р.Фленом подочитано, что только в результа-
результате сгорания кустарников в саваннах Африки в атмосферу поступа-
поступает частиц 8-107 т/год [99]. Х.Нойбергер подсчитал, что при сте-
степном пожаре на площади 0,5 га образуется в среднем около
мелких частиц [105]. По оценке [74], полученной также глобаль-
глобальной экстраполяцией данных о лесных пожарах в США, доля массы
продуктов сгорания растительности по всему земному шару состав-
составляет 1,5-10 т/год.
Многочисленные данные свидетельствуют, что частицы, обра-
образующиеся при леоных пожарах, не только существенно увеличива-
увеличивают общее содержание аэрозольных частиц в глобальном масштабе,
но и заметно влияют на погоду в местном и региональном масшта-
масштабах, в частности, они увеличивают концентрацию активных ядер
конденсации, задерживая процесс коалесценции при образовании
ДОВДЯ.
Промышленность и транспорт вносят относительно небольшой
зклад в общее содержание аэрозолей и примесных газов в атмос-
атмосфере, но большая доля промышленных загрязнений является весь-
весьма токсичной. Наиболее токсичны соединения ртути, овинца, раз-
различные органические соединения. Большинство газовых примесей
Участвует в процессе образования вторичных аэрозолей.
27
Удельное выделение вредных веществ
или получении 1 т
Таблица 4
Вещество
Пыль
Сернистый
газ
Окислы азота
Углеводоро-
Углеводороды и альде-
альдегиды
3,4-бензпи-
рен
Прочие ор-
органические
вещества
Источник
Тепловозы
ТЭЦ
Автотранспорт ..
Авиация
Прочие
Очистные устрой-
устройства
ТЭЦ
Печи
Автотранспорт ..
Авиация
Воздухонагрева-
Воздухонагреватели
Котельные теп-
тепловозы
Прочие
Автотранспорт ..
Прочие
Автотранспорт ..
Котельные
Автотранспорт ..
Авиация
Прочие
Топливо
Уголь
120
6,0
21
8,2
8,2
3,3
0,03
Мазут
5,4
11,0
7,2
1.1
7-1О
Газ
Коксо-
Коксовый
2,8
1Д
0,83
0,83
Л-
0-0,5
2--I0
0*1,0
Домен-
Доменный
1,0
0,36
0,30
0,18
Основные источники промышленных аэрозольных и газовых за-
загрязнений атмосферы связаны с четырьмя отраслями хозяйственное
деятельности человека: энергетикой, транспортом, химической
28
п атмосферу при расходовании
продукта, кг/т
природ-
природный
1 1 1 1 1
5,0
3,7
:
1,6
ii i i i i i
Дизель-
Дизельное
2,0*5,0
2,0*5,0
1,0+10
0,5
0*80
-
0,1*1,0
10~3
до 100
0,1*4.5
Бензин,
керосин
0+4,0
0,5*5,0
0,02+50
0*50
-
0*20
10*2103
1,0*15,0
0,5+20
Мусор
2,0+15,0
1,0
-
0,05+1,0
0,05+30
0,7+50
Другие
Цемент
320-350
—
—
-
-
продукты
Цветные
металлы
1,0+10
0+1,0
-
-
-
промышленностью и металлургией. В глобальном масштабе наиболь-
;шя интенсивность загрязняющих атмосферу выбросов приходится
на тепловые электростанции, нефтехимические предприятия, ме-
металлургические комбинаты и автотранспорт. Удельное выделение
29
некоторых примесей в результате деятельности человека приведе-
приведено в табл.4. Наибольшее количество особо токсичных компонентов
выбрасывают в атмосферу автотранспорт и некоторые химические
предприятия.
Уже отмечалось, что атмосферные загрязнения по всему зем-
земному шару распределены весьма неравномерно. Это определяется не-
неравномерностью распределения источников загрязнений, малым вре-
временем жизни большинства загрязняющих примесей, особенностями
атмосферной циркуляции и атмосферными процессами выведения за-
загрязнений из атмосферы.
Наибольшее количество источников антропогенных загрязне-
загрязнений сосредоточено в промышленно развитых районах в средних ши-
широтах северного полушария. Особенно интенсивно загрязняется ат-
атмосфера Западной Европы (Рурский промышленник ра:.он, Бельгия,
Нидерланды и южная часть Англии). Загрязняющие компоненты,имею-
компоненты,имеющие малое время жизни, опасны только для самих районов с ин-
интенсивными источниками загрязнение. з настоящее время практи-
практически во всех таких районах существует постоянно действующая
служба контроля уровня загрязненности атмосферы. Одно из пер-
первых мероприятий, применяемых в борьбе с опасным уровнем атмос-
атмосферной загрязненности - увеличение высоты труб заводов и теп-
тепловых электростанций - приводит к увеличению времени жизни вы-
выбрасываемых загрязнений, а следовательно, к увеличению загряз-
загрязненности над более обширными территориями Земли, например, ат-
атмосферы скандинавских стран. Неравномерности распределения за-
загрязнений в атмосфере способствует отсутствие перемешивания
воздушных масс северного и южного полушарий; преимущественное
направление воздушных масс, определяемое широтой местности,
сезоном года, рельефом и близостью обширных водных бассейнов;
пространственная неравномерность в образовании облачности и вы-
выпадении осадков. Вследствие указанных причин для отдельных в
основном долгоживущих компонентов, таких, как аэрозоли, SO2 ,
NO^.CO, ЮН3,фреоны, существуют проблемы глобального и ло-
локального загрязнений, а для других, главным образом короткожи-
вущих, таких, как органические соединения и соединения ртути
и свинца, - проблема локального загрязнения.
В случае промышленных аэрозолей наибольшая доля выбрасы-
выбрасываемых в атмосферу частиц приходится на продукты сгорания.
30
;;о л.Грину и В.Лейну [27] состав аэрозольных частиц из дымовых
труб следующий: сажа - 48-27; смола - 1; зола - 51-62$ (пред-
(представляет собой главным образом соединения кремния и кальция).
Общее количество аэрозолей промышленного происхождения со-
составляет по разным оценкам, от 5-10 до 45/? по массе от всех аэ-
аэрозолей, т.е. примерно E-10)*108 т/год. Верхние оценки пред-
представляются завышенными. Размеры аэрозольных частиц промышлен-
промышленного происхождения лежат в наиболее оптически активном диапазо-
диапазоне 3,1 мкм 4 г & 1 мкм, а вещество частиц промышленного про-
происхождения содержит компоненты, сильно поглощающие солнечную
радиацию: сажу, соединения железа, смолы и токсичные вещества
(окислы свинца, соединения ртути, органические вещества); 96%
всей массы ртути в атмосфере присутствует в газообразном сое-
стоянии. Основными антропогенными источниками выбросов ртути
являются химические заводы, тепловые электростанции, печи, ав-
автомобили, самолеты, сточные воды. Наибольшую опасность для жи-
живых организмов представляют органические соединения ртути.Один
из важнейших процессов образования таких соединений - усвоение
ртути бактериями в сточных водах с последующим иопарением
ее в атмосферу. Наиболее высокие концентрации ртути наблюдалжь
вблизи естественных источников - гейзеров - 28 мкг/м3при сред-
средних значениях 0,2+0,9 мкг/мэ. В сельских районах США наблюда-
наблюдались концентрации ртути от 0 до 0,015 мкт/м3, а в городах -
до 1 мкг/м3. Около магистралей концентрация ее не превышает
0,04 мкг/м3.
Особое внимание в работах, посвященных исследованию атмос-
атмосферных загрязнений, уделяется загрязнениям, создаваемым автомо-
автомобильным транспортом. В выхлопных газах автомобилей обычно со-
содержится очень большое количество частиц диаметром 0,02-0,06
им и небольшое - крупных частиц. В среднем общая масса этих
твердых частиц составляет около 0,08$ от массы потребляемого
двигателем горючего, причем около половины этого вещества пред-
представлено соединениями свинца, в основном в виде его окислов.
Эти соединения присутствуют в атмосфере в газообразном и аэро-
аэрозольном состоянии, причем наибольшую опасность для здоровья
представляют газообразные соединения свинца. Наблюдается тес-
тесная корреляция между интенсивностью автомобильного движения и
концентрацией свинца.
31
Оценки содержания свинца в ледниковых отложениях на Кав
зе (ледник Алибек) за 1893-1933 и 1940-1973 гг. показали, ч
для первого периода концентрация свинца составила 0,8, а д.
второго - 8 мкг/л [28]. (Свинец и его соединения как катализ*
торы фотохимических реакций играют определенную роль в образ!
вании смога.)
Автотранспорт является также важнейшим антропогенным ис^
никоМ' выброса органических соединений. По некоторым оценкам
вклад автотранспорта в общее загрязнение атмосферы органичеы
веществом составляет 50-60$ [31]. Состав органических газовш
аэрозольных примесей в атмосфере по основным группам вещест
распределен следующим образом: непредельные углеводороды - 27,
предельные углеводороды - 32,2; ароматические углеводороды -
альдегиды и кетоны - 2,2; прочие органические соединения
34,4$.
Состав углеводородов в отработанных газах бензиновых и д
зельных двигателей изучен достаточно подробно. К идентифициро-
идентифицированным органическим соединениям принадлежат альдегиды, такие
как формальдегид, ацетальдегид и акролеин. Были идентифициров!
ны также кетоны, спирты, эпокси-соединения, органические нит-
нитраты и нитриты. К последним относится пероксиацетилнитрат -
RCO3NO2(HAH), играющий важную роль в образовании смога типа
лос-анжелесского. Вероятно, это смешанные ангидриды органичес-
органических перкислот (метил, этил, пропил, фенил) с азотной кислотой.
Так как основным источником выбросов углеводородов и свш
ца является автотранспорт, между содержанием этих компонентов i
приземном слое наблюдается высокая степень корреляции. Следова
тельно, можно контролировать уровень загрязненности атмосферы
углеводородами путем относительно простого измерения содержа-
содержания свинца в атмосфере. Мероприятия по очистке промышленных вы
бросов и оздоровлению атмосферы привели к понижению уровня за-
загрязненности атмосферы в 1971 г., но затем снова стал наблюдал
ся общий рост их содержания, особенно трех компонентов: 1 рас-
растворимых сульфатов, 2) нерастворимых нитратов, 3) органических
веществ, в частности полициклических углеводородов типа бензо-
пиренов, в состав которых входят Pb, Fe, Zn.Mn, Ni.Cr, Cd .
32
В глобальной картине атмосферного загрязнения весьма за-
заметна неравномерность загрязнения атмосферы северного и южного
полушарии. lio Е.Робинсону 93$ SO2 и 95% N02 антропогенного про-
происхождения выделяются в атмосферу в северном полушарии. Причем
количество антропогенной серы по [74] - 0,73*10 т/год - сос-
составляет примерно треть от общего количества выделяемой в атмос-
iepy серы B,20-10° т/год).
Значительно большие интенсивности глобальных источников
аэрозоле»; приводятся в монографии [76]. О.П.Петренчук исходит
;:з следующих положений: 1) баланс примесей, поступающих в ат-
мосзру и выводимых из нее в течение года, в глобальном масшта-
масштабе замыкается; 2) количество осадков на всем земном шаре сос-
составляет 577-10 км3, а средняя минерализация - 6,2 мг/л; 3)вювд
облаков в общее содержание растворимых примесей в атмосфере при-
принимается равным 50$; такое количество растворимых примесей со-
содержится в подоблачном слое атмосферы; 4) основные компоненты
растворимых примесей берутся в следующем соотношении: морские
соли - 23, частицы почвы - 26, соединения серы - Ы%. При этих
предположениях общая мощность источников аэрозолей равна при-
примерно 1,2*10 т/год, без учета недолгоживущей грубодисперсной
фракции аэрозолей в нижнем (до 250 м) слое атмосферы. О.П.Пет-
О.П.Петренчук получила уточненные по сравнению с Э. Эриксоном оценки
вклада источников морских солей по данным ионного стока хлори-
хлоридов в океан: A,23*1,5)*10 т/год, примерно совпадающую со зна-
значением 1,3-10 т/год (по первому варианту оценок глобальных ис-
источников ).
Эти оценки, как и более ранние, весьма приближенны. В част-
частности, они могут измениться при учете процессов образования аэ-
аэрозолей вследствие химических реакций в атмосфере, различного
времени жизни растворимых примесей (например, механизм много-г
кратного обводнения и высыхания аэрозольных частиц), механизма
вторичного подъема частиц в воздух, ненадежности экстраполяции
данных по северному полушарию на южное, возможности протекания
химических реакций образования сульфатов в пробах осадков и об-
облачной воды и т.д. Однако можно утверждать, что большинство сде-
сделанных ранее оценок характеризуют нижнюю границу интенсивности
источников аэрозольных частиц, тогда как оценки по [76] ближе
у •¦еальной величине.
3 1V22
33
Следует отметить, что сдвиг верхней границы размеров аэро-
аэрозольных частиц в ту или другую сторону при оценках интенсив-
интенсивности источников существенно влияет на оценку глобальной мощ-
мощности источников. Особенно важно, что соотношение между интен-
сивностями разных источников также сильно изменится, так как
дисперсность частиц от разных источников различна. Это обстоя-
обстоятельство является главным при возникновении противоречий в оцен-
оценке роли разных источников аэрозолей.
Таким образом, общее количество аэрозольной материи, выде-
Q ТО
ляющейся в земную атмосферу, составляет примерно 2*10 -1,2-10
т/год. В монографии М.Е.Берлянда приводится значение 2,3«10
т/год [llj. Табл.5 иллюстрирует оценки мощности разных источни-
источников аэрозолей.
Следует отметить, что доля аэрозольных частиц, возникаю-
возникающих в результате превращений газовых примесей в атмосфере,весь-
атмосфере,весьма значительна. Более того, для аэрозолей антропогенного проис-
происхождения количество аэрозольных частиц, образующихся из газа,
в несколько раз превышает количество частиц, непосредственно
выбрасываемых в атмосферу. По мнению М.Е.Берлянда, запыленность
атмосферы можно рассматривать как конечный результат превраще-
превращения газовых примесей и некоторый итоговый показатель загрязне-
загрязнения атмосферы [11].
§ 1.2. Источники и химический состав
стратосферных аэрозолей
Вклад наземных источников в общее содержание стратосфер-
стратосферных аэрозолей невелик, если не считать эпизодических изверже-
извержений вулканов. В остальных случаях тропосферные аэрозоли могут
проникать в стратосферу только в результате конвективного подъ-
подъема частиц в экваториальной зоне. Большое количество аэрозоль-
аэрозольных частиц образуется в стратосфере в результате химических и
фотохимических реакций из окислов азота и серы. Главным источ-
источником антропогенных аэрозолей в нижней стратосфере и верхней
тропосфере являются продукты сгорания авиационного топлива.
Общая масса этих продуктов в нестоящее время составляет A+5)х
34
со
о
со
со
k-t
О
o
5+2
о
CO
со
00
кН
о
кН
О
кЧ
3
It
5! О Н
a w о
m
Pi
CD
a i
о
LO
5
«
и
CD
0 8 % S
О К (=ч со
в од ч
со о
о о о
о о о о
О СО О tO
И
О Я
а о
и ^
<о п Ч
д о со
Ж Ж Р
&8 р
я
а
со
о
а
го
со
со
3
р.
8
cq
k-t
о
S
о
о
о*
о
<*о о
с\г ^
+ +
LO LO
о
О)
СМ
О (
00 ;
си
о
о
seed к
CD CD Я
QP, Н Я
са
р.
CD
РдР.
U <о
23
3 К Ч
ОО !>
о со о
CD V
CfS ^ -— —-
OS a) \D pq
а
К
ани
ени
о ее
О lo
0р кН кН
LO CV о"
о о о
О !> LO
+ + ч-
о о о
^] СО LO
«.н
Z
о «м as
sj о
со S
и\
CD СО О
со
К
я
S
Si
аз
О Д
W
0) <D
О Ф
CD Ен
Е^—'
?§§
о
S
СО'
о
о
о
кН
О)
m
хЮ6 т/год, причем объем такого рода загрязнений непрерывно рас-
растет. Количество пылевой материи внеземного происхождения срав-
сравнительно' невелико. В атмосферу ежесуточно вторгается 100-200млн.
метеоритов, что эквивалентно примерно A+5)-10 т космическо-
космического вещества. В ряде работ приведены данные о количественном со-
содержании элементов в метеоритах [34, 59, 93]. Среднее содержа-
содержание их по массе таково: [О] - 33, [Fe]- 29, [Si]- 17, [Mg]-14,
[S] - 2,1, [Ni]- 1,7, [Ca]- 1,4, [Al] - 1, [Na]- 0,7%. Kpo-
ме них в метеоритах присутствуют в достаточно заметных количес-
количествах Сг, Mn,Ti, К, Р, Со. По данным Г.Ньюкирка, количество
пылевой материи внеземного происхождения в стратосфере состав-
составляет примерно 105? от общего содержания аэрозольных частиц в
стратосфере [210]. Некоторое представление о массе пылевой ма-
материи внеземного происхождения можно получить из данных по от-
отложениям на дне океана и во льдах Антарктиды и Гренландии. Ус-
Установлено, что микроскопические шарики (довольно обычная соста-
составляющая отложений) на дне океана имеют космическое (метеорит-
(метеоритное )происхождение.Они являются типичной составляющей глобеги-
рянового ила и обнаруживаются в нем до глубины 3 м, что соот-
соответствует возрасту осадочных пород не меньше 1-3 млн. лет. На
I кг осадочных пород приходится около 1000 таких шариков. По
химическому составу они сходны с метеоритами и отличаются от
земных минералов. Так как для вещества космического происхож-
происхождения считается характерным высокое содержание никеля B,5%),
то это позволяет определить массу метеоритного вещества, падаю-
падающего на Землю. Примерная величина аккреции метеоритного веще-
вещества составляет около 5*10 т/год для всей Земли, или 3«10~
г/см3 с, причем почти вся масса метеоритного вещества прихо-
приходится на микрометеориты.
Исследования в Гренландии и Антарктиде показали, что чер-
черные блестящие намагниченные шарики, вероятно, имеющие метеорит-
метеоритное происхождение, обнаруживаются во льдах на глубинах, кото-
которые соответствуют возрасту льда (а следовательно, и возрасту
шариков), - около 750 лет. Это исключает возможность их инду-
индустриального происхождения. Форма и характеристики поверхности
таких частичек несовместимы с предположением вулканического их
происхождения. Изучение таких частиц в арктических льдах в нас-
36
тоящее время систематически используется для получения информа-
информации о флуктуациях годового притока микрометеоритов в далеком
прошлом и в настоящее время. В соответствии с одной из программ
на протяжении целого года одновременно собирались выпавшие за
день осадки на двух арктических станциях - в центральной Аляс-
Аляске и на о.Корнуолл (Канадский Арктический архипелаг). На обеих
станциях частота выпадения намагниченных частиц с диаметром
больше 3 мкм, состоящих главным образом из железа,составляла
примерно 1 част./(см -сут). Согласованность данных, полученных
на обеих станциях, а также их совпадение с результатами подоб-
подобных же исследований, проведенных в пустынных районах Калифор-
Калифорнии и Нью-Мексико (США), свидетельствует об их метеоритном про-
происхождении. Химический анализ частиц, собранных в Арктике, по-
показывает, что это преимущественно магнетиты и что их химичес-
химический состав сходен с химическим составом метеоритов [59, 92] .
Поток метеоритного вещества по этим данным больше самого верх-
верхнего предела значений принятых оценок среднегодового потока
частиц в атмосферу Земли. Сходные результаты получают из анали-
анализа пылевых отложений в сфагнуме в разных отдаленных от промыш-
промышленных центров районах земного шара. Это позволяет предполагать,
что поток пылевой материи в стратооферу составляет около 10
т/год. Такие же значения потока получаются и по измерениям со-
содержания пыли в верхней атмосфере [45, 49, 59, 210].
Согласно имеющимся данным важнейшим элементом вещества
стратосферных аэрозолей является сера в виде кислоты, сульфа-
сульфата и персульфата аммония, поэтому возникает необходимость ус-
установить источник выбросов серы, обеспечивающий наличие оерно-
кислотных и сульфатных аэрозолей над обоими полюсами. Сущест-
Существующие гипотезы о влиянии вулканических извержений, переносе
сернистого газа из тропосферы и т.д. оказываются недостаточно
удовлетворительными. В полярных районах сказывается влияние та-
такого источника серы, как морской лед, но едва ли он является
достаточно мощным. Поэтому Дж.Ловелоком было предположено,что
большую роль в выносе серы в атмосферу играет диметилсульфид,
содержащийся в морской воде. Другим возможным источником суль-
сульфатных частиц в стратосфере может быть карбонил сульфида (хи-
(химически инертный в тропосфере), попадающий в нижнюю стратосфе-
37
ру в результате диффузии и конвективных потоков в районе эк-
экватора.
Прямые измерения структуры стратосферных аэрозолей начали
интенсивно проводиться в конце 50-х - начале 60-х годов. Наибо-
Наиболее важным результатом на первом этапе исследований явилось об-
обнаружение Х,?яге стратосферного аэрозольного слоя на высотах
17-20 км. Им было установлено, что основными компонентами ве-
вещества стратосферных аэрозольных частиц являются сульфат и пер-
персульфат аммония [2, 5, 19, 45, 110], количество серы составля-
составляет 8-14?? от общей массы аэрозольных проб и наблюдается присут-
присутствие Si и Fe. С.Моссопом было обнаружено, что почти в каж-
каждой сульфатной частице содержатся нерастворимые в воде вещест-
вещества, главным образом силикаты. Р.Кейдл предположил, что в состав
стратосферных аэрозолей входят продукты лесных пожаров [105].
Ю.Шедловски были проведены оценки вклада метеоритных осадков
[50]. Кроме того, группой Х.Юнге было установлено, что количест
ва [NH4] недостаточно, чтобы считать стратосферные аэрозоли
состоящими из сульфатов одного аммония. Более того, он предпо-
предположил, что аммоний адсорбируется аэрозольным веществом уже пос-
после экспозиции пробы. Это было позднее подтверждено работами
группы Р.Кейдла. В частности, было обнаружено,что в стратосфер-
стратосферном слое доля аммония не превышает 105?, но возрастает с высотой
в средних широтах, начиная с 20 км, а на экваторе - с 27 км.
Группой Е.Бигга было установлено наличие в стратосферном аэро-
аэрозольном слое серной кислоты и преимущественное присутствие суль
фата аммония ниже и выше стратосферного аэрозольного слоя [117,
118]. Следует отметить, что группой Р.Кейдла было выявлено, что
изменение отношения концентрации серной кислоты к сульфату ам-
аммония обратно пропорционально размеру частицы. Эмпирически бы-
было показано, что наиболее вероятное соотношение серной кислоты
и воды в каплях сернокислотных стратосферных аэрозолей 3:1.Сог-
3:1.Согласно результатам их работ капли серной кислоты являются оонов-
ным компонентом стратосферных аэрозолей [105, 110, 117, 118,
224].
Анализы частиц, собранных на ИХБ (бумажные) и полистироло-
полистироловые фильтры во время полетов самолетов на высоте около 18 км
над тропическими районами Центральной и средними широтами Се-
Северной Америки, показали достаточно близкие результаты. На по-
тшстироловых фильтрах было обнаружено малое количество нитратов,
либо они отсутствовали. Содержание кремния на тех же фильтрах
было намного выше, чем на фильтрах ИХБ. Ионы аммония отсутство-
отсутствовали в стратосферных пробах, отобранных над тропиками, но при-
присутствовали в пробах, отобранных в средних широтах на фильтрах
обоих типов. Концентрация хлора была высокой на фильтрах ИХБ,
но очень низкой - на полистироловых. Отношение [Cl]/[Br] для
проб на полистироловых фильтрах равно 12-19 (для морской воды -
300). В пробах, взятых на фильтры ИХБ, обнаружено очень высокое
соединение иона [ NO3], который, очевидно, присутствует в стра-
стратосфере в виде HNOj [105], что соответствует и данным спект-
спектральных наблюдений. В составе вещества стратосферных аэрозолей
кроме сульфата аммония (NH4JS04 удалось обнаружить также
(NH4JS,Og и две формы нитрозилсерной кислоты: NOHSO, к NOHS 0?.
Результаты экспериментальных исследований 70-х годов нес-
несколько противоречат данным более ранних исследований группы
Х.шге (табл.6): получены более высокие массовые концентрации
аэрозольного вещества - примерно в 10-30 раз, кроме того, наб-
наблюдаются отличия по химическому составу. В первую очередь сле-
следует отметить более высокое содержание Si и Fe. 3 исследова-
исследованиях советских ученых при определении химического состава аэро-
аэрозольных частиц особое внимание обращалось на содержание Fe, Ni,
Al.Ca, Si, Mg. в частности, было обнаружено высокое содержа-
содержание Fe для высоты 30-31 км - примерно 0,01 - 0,05 мкг/м3. Вы-
Высокое отношение концентрации Ni к Fe свидетельствует о кос-
космическом происхождении вещества частиц, в состав которых вхо-
входят эти элементы. Можно предполагать, что основными химичес-
химическими соединениями, содержащими Fe в стратосфере, являются окио-
лы и гидроокислы железа. Другим существенным результатом этих
исследований является обнаружение высокой доли гигантских час-
частиц в общей концентрации аэрозолей, что объясняет высокие мас-
массовые концентрации аэрозольного вещества.
Специальных исследований содержания в стратосфере аэро-
аэрозольных компонентов, поглощающих солнечную радиацию, не прово-
проводилось. Однако нагрев стратосферы в экваториальных областях на
6-7 С сразу после извержения вулкана Агунг и на 2-3°С в после-
38
39
Таблица 6
Химический состав стратосферных аэрозолей, мкг/м3
Компо-
Компонент
S04
Si
(базальт)
NH4
NO3
NO2
Na
Cl
Br
Fe
Mg
Al
Ca
Ni
Стратосферные аэрозоли
по Х.шге
0,0068
0,00011
•
•
•
•
•
.
0,00043
0,00010
по Р.Кейдлу
0,11+0,36
0+0,19
0+0,043
0+3,4
•
0+0,05
0+0,07
0,0006+0,007
•
•
по Г.Ходи
0,01+4,0
0+0,7
0,05
0+0,01
О.обб
0
0 '
0,001+0,05
o.oi
0,002+0,09
о.сй
0+0,003
0,002
•
по Л.С.йвлеву
0,002+0,05
0,08+0,12
•
•
•
•
•
•
0,015+0,068
0,012+0,024
0,025+0,067
0,0012+0,0036
Примечание. Здесь и в табл. 7, 8, 14, 18, 24
точки означают, что анализы на компонент не проводились.
дующие годы свидетельствует о большом содержании в этих
слоях сажи и окислов железа, что согласуется с данными о повы-
повышении содержания Fe в стратосферных аэрозолях.
Наличие значительной доли минеральной составляющей в стра-
стратосфере может быть связано как с большим вкладом вулканическо-
вулканического пепла в состав аэрозолей, так и с вкладом продуктов распа-
распада метеоритов. Модно согласиться с оценкой Г.Ньюкирка доли про-
продуктов космического происхождения, составляющей 10-20$, если
предположить, что годовое выпадение космической материи на
40
земную поверхность больше 105 т, а время жизни стратосферных
аэрозольных частиц около года.
Весьма интересным и важным фактом является обнаружение в
составе аэрозолей значительной доли нитратов, что свидетельст-
свидетельствует о фотохимических процессах образования стратосферных аэро-
аэрозолей не только из сернистого газа и других соединений серы,но
и из окислов азота.
Аэрозольное отношение смеси по массе для стратосферы, ве-
вероятно, колеблется от 10~и до 10"9. Весьма высокие значения
отношения смеси E«10~9) были получены в дрейфовых полетах на'
высоте 30-31 км летом 1968 г. [45].
§ 1.3. Химический состав аэрозолей
в приземном и нижних слоях атмосферы
Фоновые аэрозоли. Анализ химического состава аэрозолей в
нижних слоях атмосферы показывает его сильную зависимость от
локальных источников аэрозолей.
Оценка тенденции роста глобальных загрязнений, в том чис-
числе роста глобального содержания аэрозолей в атмосфере Земли,
требует проведения фоновых измерений, т.е. измерений в таких
районах земного шара, где отсутствуют антропогенные источники
аэрозолей, а также нет влияния других мощных локальных источ-
источников, например пустынь. Общепризнаны в качестве фоновых стан-
станций наблюдения за составом атмосферы обсерватория "Мауна-Лоа"
(Гавайские острова) и обсерваторий в Антарктиде. В названных
пунктах земного шара наблюдается определенное увеличение кон-
концентрации NO2 (важный фактор в образовании фотохимических аэ-
аэрозолей) .
Х.Юнге считает, что измерения концентрации морских аэро-
аэрозолей в Южном полушарии дают также фоновые величины. В Совет-
Советском Союзе фоновые измерения можно проводить на севере Сибири
и на Дальнем Востоке, а также в горах Памира и Тянь-Шаня. Наи-
Наибольшее количеотво фоновых измерений проведено в обсерватории
"Мауна-Лоа" на высоте 3,4 км. По данным измерений концентраций
N02, SO2 и аэрозолей в 1970 г. были обнаружены выоокая кор-
корреляция содержания SO2 и аэрозолей, а также слабая корреляция
41
концентраций этих компонентов с содержанием РЬ, т.е. можно по
латать, что источник выбросов исследуемых веществ находится ни
же обсерватории; [N02] не коррелирует с содержанием SO2 и наи
более вероятно, что источником NOj (Для района обсерватории
"Мауна-Лоа") являются вулканы. Обнаружено, что содержание Na
на высоте 3,4 км составляет всего 1% от содержания его на уров
не моря. Это подтверждает мнение, что время жизни этого злемен
та в аэрозольном состоянии очень невелико. Изменение общей кон
центрации аэрозолей вверх по склону горы, на которой расположе
на обсерватория, составляет три порядка, а при удалении от бе-
берега она изменяется по закону \aW'- l/lgl. Наблвдается слабое
влияние аэропорта Хило на уровень концентрации некоторых компс
нентов.
Особый интерес представляют результаты измерений концент-
концентраций разных элементов в Антарктиде с целью оценки влияния pas
личных источников на глобальное загрязнение. По значению коэф-
коэффициента обогащения, определенного следующим образом:
_ [Хатм]/[А1атм]
CX1/IAI]
все элементы можно разбить на три грудин.
В первую группу входят элементы с EF = 0,8f22: K.Ca.Sc
V, Mn, Fe, La,Sm, Eu.Th, которые квалифицируются как аэрозш
ные компоненты естественного, в основном почвенного, происхож-
происхождения. Во вторую группу входят элементы с EF = 4,4f7,0: Mg,
Сг, Са, С1, которые попадают в атмосферу в результате вулка-
вулканических извержений, из океана и частично в результате антрош
генных загрязнений.
Элементы третьей группы с EF = 49f43000 - Na.Cu.Pb,
Zn, Se, Вг, Sb - того же происхождения, что и элементы вто
рой [105, 216]. Элементы J, Вг и С1 присутствуют в атмосфе
ре в аэрозольном и газообразном состояниях. Время жиэни этих
элементов в Антарктиде значительно больше, чем на Гавайских а
ровах, причем фоновые концентрации иода и брома ничтожны, ад)
хлора концентрации различаются почти на порядок (для станций
"Мак-Мурдо" и "Южный полюс").
42
Результаты измерений концентрации Pb, Na, Mgr, Са, К,
Al, Se, Th, Fe, Zn, Вг станцией "Амундсен-Скотт" показали, что для
2п, Си, Sb, Se. Вг, Pb основными источниками являются высоко-
высокотемпературные.
Фоновые аэрозоли в Антарктиде в основном морского происхож-
происхождения * частично принеоенные из верхней атмосферы, причем соеди-
соединения серы вулканического происхождения. Антропогенный вклад в
содержание фоновых аэрозолей в этом районе очень мал.
Измерения фоновых концентраций SO^2 над морской поверх-
поверхностью в разных точках земного шара дают близкие значения т-
= Ч\2 мкг/м3 при отсутствии локальных источников на расстоянии
более 500 км. Над разными районами суши фоновые концентрации SO^
заметно различаются. Например, более высокие фоновые концентра-
концентрации наблюдаются на восточном побережье Северной Америки: ~ 5
мкг/м3, где главными компонентами аэрозолей являются H2SO4 и
NH4HSO4 ;часто наблвдается присутствие значительной доли(NH^jSCj,.
К фоновым можно отнести также эпизодические измерения сос-
состава аэрозолей в глубинных районах южной Америки, где исследо-
исследовались концентрации Pb,S,Ca,Fe, ионов К,Cl для двух фрак-
фракций: tf <1 и tf >1 мкм. Соединений РЬ обнаружено очень мало, а
соединения S- находились в основном во фракции с d < 1 мкм. Аэ-
Аэрозолей морского происхождения зарегистрировано значительно мень-
меньше, чем над континентальными районами северного полушания. Ана-
Анализ соотношения [S]/[K] не позволил выявить каких-либо законо-
закономерностей.
Анализ состава и структуры фоновых аэрозолей позволил Э.Ме-
сарошу выделить четыре основных типа аэрозольных частиц - ядер
конденсации: морскую соль, сульфат аммония, серную кислоту,смзсь
хлористого натрия и сульфата аммония. Причем по его наблюдени-
наблюдениям ядер NaCL над континентом очень мало, и наблвдаются в основ-
основном гигантские частицы [96, 203].
Фоновые измерения органического компонента атмосферных аэ-
аэрозолей единичны. Так, анализ аэрозольных проб, взятых на запад-
западном побережье Ирландии в 50 и от уреза воды на высоте 31 и, по-
показал, что для воздушных маос, приходящих из Атлантики, общая
массовая концентрация аэрозольного вещества составляет 8,6 -
4,8 мкг/м3, а количество органичеокого вещества доотигает 105?
от общей величины.
43
Было выделено пять фракций органического вещества, соотно-
соотношение между массами которых зависело от направления и скорости
ветра: 1) В - основные соединения ~ 8а; 2) А - кислоты, фе-
фенолы - 27%; 3) N - нейтральные вещества - 64%; 4) полярные и
ароматические вещества; 5) алифатическая фракция. Две последние
фракции по массе не превышали 1% от всего количества органичес-
органического материала.
В континентальных воздушных массах фоновое содержание ор-
органического вещества, очевидно, изменяется в более широких
пределах. Весьма сильно изменяются также значения фоновых кон-
концентраций пылевых частиц, содержащих такие элементы, как Si, At,
Са. Причем наблюдается заметный сезонный ход концентраций этих
элементов в атмосфере. Важный компонент вещества фоновых аэро-
аэрозолей - соединения железа. В пылевых частицах основными типами
таких соединений являются окись железа Fe2O3 и гидроокиси
П F"e2Oj -m Н2О.В океанических аэрозолях 5sFe присутствует в ви
де аморфных окислов в высокодисперсных частицах либо агрегиро-
агрегирован к более крупным, а значительная часть 55 Fe содержится в ви-
виде химичеоки более активных гидроокислов. Фоновые концентрации
Fe изменяются от 0,01 до 0,2 мкг/м3.
Городские аэрозоли. Обстоятельных исследований требует хи-
химический состав аэрозолей в крупных промышленных районах, замет-
заметно отличающихся от естественных аэрозолей целым рядом характе-
характеристик: концентрацией, дисперсностью, микроструктурой частиц и
их химическим составом. Своеобразный тип сложных индустриальных
атмосферных загрязнений представляет собой смог.
Обычно различают два типа смогов: образующийся при сжига-
сжигании угля и фотохимический. Оба типа смогов обязательно содержат
три класса вторичных аэрозолей, образующихся при фотохимических
и темновых реакциях: продукты трансформации окислов азота, дву-
двуокиси серы и органических загрязнений, в частности фотополимера
Смог первого типа (лондонский) состоит в основном из сме-
смеси угольного дыма и тумана. В этом смоге присутствуют довольно
крупные частицы сажи и капли раствора серной кислоты (продукт
окисления сернистого газа). Для него характерно высокое содер-
содержание канцерогенных соединений (полициклических углеводородов,
образующихся при неполном сгорании угля), в частности 3,4-бенз-
44
пирена. Концентрация серной кислоты в мощных смогах достигает
нескольких миллиграммов на кубометр воздуха, а 3,4-бензпирена -
нескольких десятых микрограммов на кубометр воздуха.
готохимический смог (лосанджелесский) образуется в воздухе,
загрязненном парами органических соединений и окислов азота,
при освещении его солнечным светом. Частицы фотохимического смо-
смога значительно более мелкодисперсны, чем в смоге первого типа.
р.Кейдл и его сотрудники обнаружили, что большая часть веществ,
собранных в Лос-Анджелесе при сильном смоге, состояла из капель,
часть которых содержала темнокоричневые клейкие водонераствори-
мые органические соединения. При испарении капель на их поверх-
поверхности образуется пленка, препятствующая быстрому испарению да-
даже при влажности около 30% [105].
Из минеральных веществ в смогах обнаружены сульфаты аммо-
аммония и кальция, а также фториды. Доля капель в фотохимическом
смоге по оценке К.Уитби составляет от 15 до 50% от общего чис-
числа частиц [24].
В воздухе больших городов практически всегда присутствуют
компоненты, характерные для смогов.
Содержание некоторых элементов в воздухе Северной Америки
представлено в табл.7 [105, 110, 180]. Следует отметить, что
уровень концентрации SO2 в приземном слое над тем же районом
за период 1960-1970-х гг. понизился на 50%, в основном из-за
увеличения высоты дымовых труб. Однако концентрация сульфатов
в городах осталась постоянной, а в селах увеличилась. Прямые
измерения показывают, что 40% сульфатных аэрозолей содержат сер-
серную кислоту. Результаты непрерывных наблюдений содержания и хи-
химического состава аэрозолей в течение двух лет в Сент-Луисе при
заборе проб с разделением на фракции: d > 2,4 и d < 2,4 мкм
показывают хорошую корреляцию между концентрацией серы в мел-
мелких частицах и всей массой мелких частиц. Во фракции с d <
< 2,4 мкм содержится около 90% сульфатного вещества, что сос-
составляет примерно 35% от всей массы этой фракции.
Результаты измерений водорастворимых компонентов аэрозоль-
аэрозольного вещества на содержание ионов Н+, NH+, SO"?, N07 на по-
оережье Балтийского моря и в районе Гётеборга позволили обна-
обнаружить два типа аэрозолей: "черные" пробы,содержащие сульфаты,
45
46
a
X
3
г?
с?
(A
1—'
с
N
О
.о
1°
1?
<
в}
О
(Л
ород
P-,
о
C\J
о
ю
о
ьч
о
1
о
кН
о
о
кН
о
о
со
о
C\J
о
кН
о
о
CNJ
о
о
со
со
ш
о
о
•=ч
о
кН
ю
in
о
кЧ
< ¦>
C\J
8
о
о
со
о
о
со
о
1>
о
ог
кЧ
о
о
to
кЧ
ати ..
а
Г)
ю
о
1
о
о
?5
со
о
о
кЧ
о
кЧ
о
in
кЧ
о
а
S
о
кЧ
о
о
а.
о
о
со
о
с >
со
о
о
in
о
о
кН
о
со
о
со
о
со
о
ю
о.
о
Виндз
о
1Л
кЧ
о
о
1
8
о
о
кЧ
О
о
кЧ
О
О
со
о
о
кН
-Сити
о
Канза
О
ю
о
ю
•
о
со
о
о
ю
t4
о
о
кЧ
а
Нью-Й
00
со
о
C\i
СТ)
о
о
ю
in
о
о
СП
m
8
о
ю
о
о
С\}
о
~„
кЧ
см
о
со
ю
*
о
Фреон
C\J
о
CSi
to
о
кЧ
м
кЧ
о
со
кЧ
о
о
о
ю
о
о
СО
кЧ
to
о
СТ)
СП
о
о
кЧ
ог
св
Помон
й
о
о
кЧ
о
о
1
о
кЧ
о
о
ог
о
о
ог
о
C\J
ю
ь для
полии
со о
Средн
метр
сажу, марганец и металлы, связанные с приходом континентальных
в03дулных масс из Западной Европы, и "белые" пробы всех осталь-
остальных приходящих воздушных масс. Максимальное содержание аниона
[S0A] соответствует максимальному содержанию катиона [N04]
и минимальному [N03]. Основная масса сульфатов в виде (NH4),S04,
( NH4 KH<S04J, NH4HS04 находится в высокодисперсной фракции.
Анализ воздуха в Париже обнаружил присутствие в нем азотис-
азотистой и серной кислот, аммиака, серистого газа, сероводорода, пеп-
пепла, сажи, дегтя, а также пылевых частиц, содержащих кремний,
окислы железа и цинка, барий, мышьяк и растительную пыльцу [31].
При построении модели аэрозолей интерес представляет опре-
определение влияния индустриальных источников загрязнений на загряз-
загрязненность атмосферы вдали от них. Такого рода исследования были
проведены по программе комплексного энергетического эксперимен-
эксперимента в районе Запорожья, а затем в районе Тбилиси и Алма-Ата
(табл.8). Влияние городов обнаруживалось на расстояниях в нес-
несколько километров и на высотах не менее 3 км [41]. Были иссле-
исследованы аэрозольные слои (дымовые купола) над этими городами.
Компоненты антропогенных и естественных аэрозолей, содержащие
железо, так же, как и сажа, весьма эффективно поглощают солнеч-
солнечную радиацию. Возможно, зто способствует возникновению инверси-
инверсионных слоев в атмосфере, особенно в промышленных районах, что
ведет, в свою очередь, к еще большему накоплению аэрозольных и
газовых загрязнений. Измерения химического состава аэрозолей в
Запорожье, Рустави и Алма-Ате показали высокое содержание саже-
сажевых частщ в их атмосфере: от 10 до 35$ по массе от общего со-
содержания органических веществ. В центре Ленинграда содержание
сажевых частиц в отдельных измерениях достигало 30-40$ от обще-
общего содержания аэрозольных частиц (по массе). Не обнаружено вы-
высокого содержания аниона [ S04 ]. Во всех названных городах
оно в основном не превышало 5 мкг/м3. (Следует отметить, что
данные были получены путем химического анализа фильтров, на ко-
которых могло не остаться легкоиспаряющейся серной кислоты^ Зна-
Значения массовых концентраций Fe,Al,Mg,Mn в отдельных про-
пробах сильно изменялись, что свидетельствует о присутствии в го-
городском воздухе гигантских частиц, содержащих химические сое-
соединения этих элементов. Время жизни таких частиц в атмосфере
должно быть весьма непродолжительным.
47
Таблица'
Элементный состав аэрозолей над городами СССР
по измерениям в осенне-летние месяцы 1968-1974 гг., мкг/м3
Компо-
Компонент
Fe
AL
In
са
Mg
A\n
Ni
Си
Pb
Ag
О
Ti
[so4]
[N03l
Город
Ленин-
Ленинград
B0 проб)
2,0
0,7
2,5
2,0
0,10
0,02
0,03
0,2
_
10,0
2,0
Алма-Ата
B5 проб)
3,0
0,2
0,5
2,5
1,2
0,08
0,07
0,2
0,2
-
-
-
5,0
Зало-
C0 проб)
15,0
7,0
1.5
7.0
2,5
0,5
0,10
0,5
•
0,03
0,10
15,0
3,0
Рустави
E2 пробы)
35,0
50,0
5,0
85,0
25,0
•
-
2,5
•
-
-
5,0
10,0
Рыльск
A5 проб)
7.0
0,5
2,0
2,0
0,5
0,1
0,3
0,3
0,15
0,06
0,05
8,0
-
Томе
C5 пр
0,45
0,30
0,42
1,80
0,21
0,02
о,т
0,4С
0.0Е
-
-
1,0
Примечани
для каждого города.
е. Измерения проводились в 1-3 пункт»
Континентальные аэрозоли. Существование значительного к<
личества аэрозолей в атмосфере обусловлено действием пыльш
бурь, которые заносят пыль далеко в океан и даже переносят i
из Африки через Атлантику [19, 221]. Именно это обстоятельств
вызывает очень сильную изменчивость поля аэрозолей над Атланз
ческим океаном .Массовая концентрация пылевых частиц диаметра
более 0,5 мкм изменяется от 10 мкг/м3 вблизи побережья Саха-
Сахары до 10 мкг/м3 над Антарктидой. Общая массовая концентрат
пыли в пустыне может достигать 1 мг/м3, а вдали от источник)
30-300 мкг/м3. Процентное содержание пород в почвах пустынь
примерно следующее: монтмориллонита - 35, каолинита - 20, ил-
48
лита - 20, кварца - 5, нитрата калия - 5, лимонита и других
соединений - 5$, [229].
В монографии Х.Юнге [110] приведен химический состав пыли
над пустыней Сахарой: SiO2- 37+55, Al2Oj- 0+20, Fe2O3-I*16,
СаС03- 6+22, Mn3O4- 2+4; MgO- 0,4+3$. Имеются небольшие ко-
количества К, Na, Cu, H2S04, HCl. Следует отметить, что 58$
частиц имеют d 4 0,5; 24$ - d = 0,5+1,0 и 4$ - d > 1 мкм.Пыль
сильно обогащена окислами железа и марганца по сравнению с грун-
грунтом - источником пыли. Вероятно, при образовании мелкой пыли
процессы физического и химического разделения действуют селек-
селективно, однако механизм фракционирования не изучен.
Элементный анализ сахарской пыли, осевшей на иллюминаторах
самолета ИЛ-18 при полетах над Атлантическим океаном в период
АТЭП - 74, показал следующий массовый состав: Si - 18,7+27, Al-
31+33, Fe - 14+15, Са - 5+6, Mg- 4+4,8$. (Повышенное содер-
содержание Al,возможно, вызвано эрозией поверхности корпуса самоле-
самолета.) В переводе на химические соединения получаем SiO2- 20+30,
Al203- 29+37, Fe2O3- 10+10,7, CttCOj- 6,4+7,6,MgO-3,3+4,Q$.
Основными минеральными компонентами сахарской пыли являют-
являются кварц - 50, слюда - 18, известковый пшат - 18$.
Для пыли из западной Гвинеи характерно высокое содержание
диатемии (основной компонент СаСО3). Относительно высокий про-
процент - 3,5% от общей массы составляют рудные минералы.
Неоднократно отмечались случаи выпадения в Европе пыли
пустынного проиоховдения, имеющей красный цвет. Этот цвет об-
обусловлен присутствием в ней значительного количества окислов
железа, в основном Fe2O3. Так, из облака, пришедшего из южно-
южного Марокко и западной Сахары, над Пиренеями выпала пыль без дож-
дождя. Интенсивность осадка составляла от 0,4 до 8,0 г/м , а об-
общая масса осевшей пыли была оценена в 700 т. В веществе пыли
были определены следующие элементы (в порядке убывания по мас-
массе): Са, Fe, Al, Mg, Na, Si, К. Половина всей массы пыли
имела размеры в интервале d - 7,3+80 мкм и состояла из слюды,
кварца, карбонатов, более мелкие частицы - из филлитов. Пере-
Перенос сахарской пыли на большие расстояния неоднократно наблю-
наблюдался и на снимках .полученных с помощью космических спутников
Земли.
4.1922
49
Р.Рекс и Е.Голъдберг, исследовавшие содержание кварца в ''\
морских глинах в восточной части Тихого океана, обнаружили за- j
метную зависимость концентрации частиц кварца от широты, на ко-
которой взята проба. Очевидно, что кварц и другие минералы пере- ':
носятся в атмосфере из засушливых районов далеко в Тихий океан
[74].
Примерно такой же состав пыли обнаружен и над горами, пуо-
тынями, полупустынями, степями, пашнями. Следует добавить, что
в составе пыли могут наблвдаться в относительно больших коли-
количествах также соединения магния, натрия и калия. '
Максимум распределения частиц почвенного происхождения по!
размерам, как правило, находится в субмикронной области разме-
размеров, но весьма вероятны также максимумы в области размеров
1 4 г 4 5 мкм. Частицы почвенного происхождения с радиусом
меньше 0,1 мкм могут образовываться, в частности, при кристал-
кристаллизации на поверхности почвы солей, растворенных в грунтовых
водах. По С.Тумею мощность генерации таких частиц составляет
Ю4 - Ю5 част/(см2-с) [233]. .'
Особенно интенсивное выделение мелких частиц в атмосферу
происходит на солончаковых почвах. По измерениям, проведенным
экспедицией КЭНЭКС-70 в пустыне Кара-Кум, вклад солончаков как
источника аэрозолей в общее (массовое) содержание частиц в пр«
земном слое не превышает 20-30$ для районов пустынь и полупус*
тынь [77], внося основной вклад в счетную концентрацию частиц.
Исследование пыльных бурь на юго-западе Северной Америк!
1972-1975 гг. с помощью самолетных и наземных измерений дада
величину вертикального потока пыли в атмосферу от 0,25*10 д<
2,2-Ю~° г/(см2-с).При этом ее массовая концентрация на высоте
1 и изменялась от 0,1 до 0,5 мг/м3. В приземном слое в распр*
делении частиц по объему наблюдались две моды: для частиц с
г = 1+30 мкм и г = 30+100 мкм. В тропосфере вторая мода исчв
зает [122]. Чрезвычайно важно, что с поверхности почвы могу?
генерироваться и более мелкие частицы. Например, результата
нейтронного-активационного анализа проб, взятых над Аравийскш
морем четырехкаскадным импактором с абсолютным фильтром, поМ
зали, что в аэрозолях присутствует большое количество частиц'
с d <: ОД мкм, содержащих Mn.Cr, Fe. Это свидетельствует с
почвенном происхождении таких частиц.
50
Большой цикл исследований содержания элементов в неоргани-
неорганических аэрозолях был выполнен в 1967-1975 гг. сотрудниками ла-
лаборатории физики аэрозолей Ленинградского университета. Иссле-
Исследовались пробы, взятые в приземном слое атмосферы в различных
климатических зонах Советского Союза: в горах (Кавказ, Тянь-
Шань), степи (Северный Казахстан), пустыне (Кара-Кумы), лесах
(вблизи Томска), лесостепи (около Рыльска), на морском побе-
побережье (Крым), над морем (Каспийское море, Атлантический океан),
над промышленными районами (Ленинград, Запорожье). Измерения
проводились в летне-осенний период. Некоторые результаты изме-
измерений представлены в табл.8, 9. Полученные данные обнаружили
весьма сильную пространственно-временную изменчивость химичес-
химического состава аэрозолей. Существует, например, суточный ход со-
содержания химических компонентов, обусловленный суточными вари-
вариациями интенсивности возникновения аэрозоля, а также конвектив-
конвективных и адвективных процессов в" атмосфере. Для протяженных ис-
источников аэрозольных частиц (почвы, морская поверхность) аэро-
аэрозоли оказываются сильно перемешанными уже в нижних слоях атмос-
атмосферы (см. табл.8).
Измерения химического состава аэрозолей в приземном слое
на большом удалении от антропогенных источников в западной Аф-
Африке (Берег Слоновой Кости) в 10 и 100 км от берега Гвинейско-
Гвинейского залива с ноября 1974 г. по август 1975 г. позволили обнару-
обнаружить максимальную массовую концентрацию аэрозолей в январе и
июле, достигавшую 3 мкг/м3, и минимальную, равную 0,5 мкг/м3,-
в конце сухого сезона; 30$ аэрозольного вещества было морско-
морского и континентального происхождения, а 70$ аэрозолей образова-
образовалось при окислении SO2 в атмосфере. Очевидно, что эти резуль-
результаты соответствуют данным фоновых измерений. Измерения содер-
содержания соединений серы в аэрозольном веществе в десяти пунктах
п-ва Флорида показывают, что сульфаты присутствуют практичес-
практически в диапазоне 0,25 4 d 4 4 мкм, причем в основном в облас-
области d <2 мкм. Главным компонентом аэрозольного вещества явля-
является сульфат аммония (NH4J S04. Обнаруживается также серная
кислота H2SO4. Непрерывные измерения с 1972 г. в Альпах (го-
(гора Банк, высота 1780 м) также свидетельствуют о том, что в
чистом воздухе главными компонентами аэрозольного вещества яв-
51
Содержание химических элементов в аэрозолях
Таблица 9
Место и время
измерений
Пойма р.Сейм (г.Рыльск
1967 г., июль-август
Эльбрус, Я = 3100 м
1958 г., август
31
август
Ерминтау (Сев. Казах-
Казахстан)
1968 г., август
Шувалово (пригород
Ленинграда)
1968 г., апрель-май
Пустыня Kapa-KjM
(г.Репетек)
1970 г., октябрь
Пос.Анката (Уральская
область)
1971 г. .июль-август
0,3+2,0 0,5+0,5
1,0+1,0
1,2+1,0
1,2+0,8
0,6+0,6
0,50+0,50
0,02+0,010
0,10+0,05 С,02+0,02
0,30+0,20
0,07+0,01
0,10+0,20
1,0+2,0
5,0+5,0
2,1+1,0
1,5+1,5
1,0+2,0
ляются ионы [1ЧНд]+и [S04] [221]. Концентрация аэрозолей гор
ного происхождения очень низка, а их характеристики весьма пос
тоянны. Содержание РЬ и Zn составляет 10-20 нг/м3. При мор-
морских воздушных массах присутствуют ионы Na и С1. Поэтому мож-
можно сделать вывод, что химический состав аэрозолей определяет-
определяется происхождением воздушных масс (тропические или полярные), а
массовая концентрация - тем, откуда приходят воздушные массн
в район измерений - с континента или с моря.
Вывод об однородности химического состава аэрозолей в нвя
них слоях атмосферы следует также из спектроскопических изме-
измерений сухих остатков дождевых проб и проб аэрозолей, соСранни
Ф.Фольцем в тропической зоне (Панама, Пуэрто-Рико), в умерен-
умеренных (Бедфордшир, Англия; штат Массачуэетс,США, Майнц, Централ
ная Европа), в субарктичеокой зоне (Аляска) и арктических (за
падное побережье Гренландии) широтах.
52
приземного слоя, мкг/м3
0
0
0
0
0
0
Ni
,50+0
,06+0
,5+0,
,35+0
,03+0
,02+0
,40
ДО
5
,35
,04
,20
Al
0,3+0
1,5+1
,3
,5
15,0+10,0
3,0+2
0,4+0
0,2+0
.5
,5
.3
2
2
4
6
1
3
Са
,5+2,0
,0±1,5
,0+4,0
,0+6,0
,7+3,5
,5+5,0
0
0
4
Си
,3+0,3
, 15+0,10
,5+4,5
0,3+0,3
0,03
0,03
Сг
од+од
0,05+0,10
0,2+0,2
0,03+0,03
0,01
0,01
2,0+2
0,2+0
0,4+0
,0
,15
,4
0,10+0,10
0,1+0
од+о
,5
,2
0 химическом составе приземных аэрозолей можно судить так-
также по химическому составу осадков. Наибольшее количество работ
по этому вопросу посвящено исследованиям соотношения количест-
количества сульфатов, карбонатов и нитратов в осадках. Это соотношение
определяется процессами в системе СО2- вода - растворенное ве-
вещество аэрозольных частиц. Все компоненты нейтрализуют друг
друга, и система приходит в равновесие с СО2 в атмосфере. Ми-
Минерализация осадков, кроме места выпадения их, зависит от вре-
времени года и интенсивности осадков. Например, минерализация
осадков в районе Фрунзе составляет 26,2 - 44,4 мг/л. Основной
минеральный компонент СаСО3. Минимальная минерализация осад-
осадков наблвдается в летние месяцы. Интенсивные осадки имеют ма-
малые величины отношений [NO3]/[S04], [ N(-Kl/[ NH4l •
0.П.Петренчук отмечает устойчивый химический состав осад-
осадков из фронтальных облаков. Анализ синоптичеоких условий дает
53
J
основание сделать вывод, что сера имеет континентальное проис-.
хождение. А.Струмплер получил, например, следующие концентра-
концентрации некоторых элементов в осадках для западных районов Север- ;
ной Америки (шт.Небраска): Ag - 8,4-iO, Al- 3,5-iO, Cd -
3,l-10~\ Си- 4,4-iCT3, Mn- 5,2-iO, Pb - 4,8'ICT3, Zn -
1-iO мг/л. По его мнению, Ag и Cd - явно антропогенного про-
происхождения, А1 и Мп - из почв. В осадках над Японией обнару-
обнаруживается большая доля частиц почвы из пустынь и полупустынь се-
северного Китая и Монголии, которые служат ядрами снежных крис-
кристаллов. Значительное количество частиц, идентифицированных как,
частицы оь-кварца, ot-кристобалита и пироксены, - вулканичес-
вулканического происхождения. Отдельные ядра отождествляются с минерала-
минералами глины и хлористого натрия, причем содержание последнего про
вытает содержание глин примерно в три раза.
Аномальный химический состав аэрозолей в приземном слое
может наблвдаться при действии мощных естественных источников:
пылении открытых почв, активной деятельности фумарол и гейзе-
гейзеров в вулканических зонах.
В 1974, 1976 и 1980 гг. лабораторией физики аэрозолей Ле-
Ленинградского университета были проведены измерения химическсго
состава аэрозолей на Камчатке в районе Ключевской Сопки, вул-
вулканов Толбачикский, Безымянный и в долине гейзеров. Некоторн*
данные об элементарном составе вулканических аэрозолей пред-
представлены в табл.3 и 10.* Следует отметить очень высокую кон-
концентрацию Fe, Вт, Сг, Си и особенно редкоземельных злементСЯ
Cl, Sr, Та. Определенной закономерности в суточном ходе со-
содержания различных элементов в аэрозольном веществе не обна-
обнаруживается.
Аэрозоли над морской поверхностью. Концентрация солевых
частиц над океаном может достигать 100см~3,но в среднем она
составляет ~ 1 см. Максимум в распределении по размерам Щ1
ходится на солевые частицы с диаметром около 0,3 мкм. Частила
морского происхождения могут проникать далеко в глубь суши.
* Нейтронно-активационный элементный анализ аэрозольных
проб проводился в Институте ядерной физики АН УзССР под руко-
руководством Р.А.Кулматова.
54
Например, различие между концентрацией солевых частиц над океа-
океаном и на расстоянии 1500 км от берега, наблвдавшееся Р.Кейдлом,
было незначительным. Чаще наблюдается резкое уменьшение содер-
содержания морских компонентов (Cl, Na) уже на первых километрах в
глубь суши. Например, на побережье Черного моря NaCl преобла-
преобладает в аэрозольном веществе уже в 2 км от берега, а на побе-
побережье Азовского моря в непосредственной близости от моря основ-
основными компонентами являются НСО3~, Са2+, 0|", что объясняется
сильным влиянием континентальных источников аэрозолей [76]. Да-
Даже в центральных районах Тихого и Атлантического океанов обна-
обнаруживается высокое содержание Fe, Al,Mn, присутствие кото-
которых характерно для аэрозолей континентального происхождения РЬ,
V - антропогенного происхождения и Си, происхождение которо-
которого неясно.
Поверхностная пленка океана толщиной 100 - 150 мкм может
быть эффективным источником не только органических аэрозолей,
но и частиц, содержащих ряд элементов, не характерных для мор-
морской воды. Расчеты отношения величины EF ([X]/[Na]) для аэ-
аэрозольной пробы, взятой в воздухе на высоте 20 м (о.Оаху), к
EF для проб поверхностной пленки воды показали, что для Na,
К,Са, Mg эта величина не превышает двух, а для' Сг, Си, Fe,
Zn - больше 2000-20000.
Частицы морских солей в основном очень крупные. Содержа-
Содержание их сильно зависит от относительной влажности. В сухое вре-
время года содержание Fe,Se, Au, Eu в аэрозолях увеличивается в
два-четыре раза по сравнению с периодом дождей, что свидетель-
свидетельствует о сильном пылении почвы в приморских районах. Измерения
содержания V над восточной частью Тихого океана и Гавайскими
островами показали, что максимальное содержание V в аэрозолях
0,8 нг/м3, а среднее - 0,1 нг/мэ, что не может быть объяснено
только наличием морских аэрозолей. Основными источниками выде-
выделения в атмосферу ванадия могут быть выветривание горных пород,
эрозия почв, вулканические извержения и хозяйственная деятель-
деятельность человека. Поэтому можно сделать вывод, что частицы кон-
континентального происхождения участвуют в образовании морских аз-
розолей, причем примерно 2/3 выделяемого ванадия антропогенно-
антропогенного происхождения.
55
Таблица 10
Суточный ход содержания элементов (август
Время, ч
1815
О15
315
?25
1215
1515
1815
2115
2315
О15
Элемент
Си
4,1
9,7
зд
3,9
3,3
7,7
0,59
0,3
0,3
3,3
Вг
2,4
2,6
2,1
1,9
2,2
2,0
2,0
2,2
2,5
2,7
Сг
5,1
3,3
0,5
1,7
2.7
4,8
0,9
3,0
2,9
1.6
С1
2,8
0,7
0,7
0,7
0,77
0.7
1,0
1.4
0,7
0,7
Та
2,3
1.6
1.6
2,2
1.7
2,0
1.8
1.6
1.7
3,4
Cd
0,77
од
0,61
1,0
1,1
0,18
1.5
2,1
1,3
0,57
As
2,6
1.1
0.1
0,22
0,83
од
0,69
1.7
0,1
0,1
Mo
1,2
0,7
2,2
0,17
0,44
0,18
0,90
0,06
1,0
0,67
1974 г., долина гейзеров), мкг/мэ
Sm
0,064
0,21
0,29
0,08
0,097
0,17
1.2
0,40
0,08
0,08
HS
0,30
0,25
0,31
0,12
0,32
0,22
0,25
0,43
0,097
0,19
Yb
0,02
0,01
0,01
0,019
0,031
0,01
0,024
0,01
0,054
0,02
Th
0,04
0,072
0,06
0,12
0,04
0,04
0,04
0,074
0,093
0,063
Sc
0,012
0,012
0,003
0,054
0,006
0,003
0,011
0,0082
0.0И
0,007
Au
0,040
0,055
0,0047
0,050
0,46
0,030
0,021
0,018
0,016
0,012
Lu
0,00030
0,00054
0,00070
0,00010
0,00070
0,00042
0,00010
0,00044
0,0075
0,00010
Исследования вклада в структуру морских аэрозолей вещест-
вещества иного происхождения с помощью анализа данных по коэффициен- -
там обогащения для разных элементов относительно Na и построе-
построение уравнений регрессии для Mg, Ca,K, Sr, Cl приводят также
к выводу, что в морских аэрозолях высоко содержание вещества
континентального происхождения [19].
Среднее отношение [Ca]/[Na] в морской воде равно пример-
примерно 0,038. (В дистиллятах морской воды, взятой в Черном и Сре-
Средиземном морях, а также в Тихом океане, значение этого отноше-
отношения меняется от 0,091 до 0,143.) В морских аэрозолях по Х.Юнге
это отношение [Ca]/[Na] составляет 0,096, а по данным работы
[76] оно изменяется от 0Д4 до 0,28. Этот факт частично можно
объяснить фракционированием ионов при переходе их в аэрозоль-
аэрозольное состояние. Однако Е.Гофман и Р.Дьюс не обнаружили значи-
значительного фракционирования ионов [183]. Полученное ими отноше-
отношение [Ca]/[Na] равнялось 0,045 для морских аэрозолей и изменя-
изменялось до 0,114 при ветрах с континента. Следует отметить, что
обогащение аэрозолей соединениями, содержащими Са, может проис-
дить в результате разрушения кораллов. Например, самолетные из-
измерения концентрации аэрозольных частиц с помощью счетчика Пол-
Поллака, проведенные над Австралией, показали, что в морском воз-
воздухе ниже самого низкого уровня инверсии она равняется 220 см ,
а вблизи рифов возрастает до 1590 см.
Радиоактивный анализ 19 элементов для проб, взятых в се-
северной Атлантике, позволил проверить по соотношению пяти ред-
редкоземельных элементов в почве, сланцах и хондритных метеоритах
различные гипотезы о происхождении вещества морских аэрозолей.
Наилучшее соответствие наблвдалось для соотношения элементов в
почвах.
Анализ проб, взятых 8 - 26 У1 71 г., выявил сильное влия-
влияние адвекции воздушных масс с континента на содержание и коэф-
коэффициенты обогащения для Mg, Са и К относительно Na, причем
наблюдалась сильная зависимость их от скорости ветра. Исследо-
Исследования фракционирования ионов [Na]+ и [S04]npn распылении
морской воды в лабораторных условиях показали отсутствие это-
этого процесса. Однако при натурных исследованиях над северной Ат-
56
57
лантикой значение отношения [S04]/[Na]+B морских аэрозоладЗ
оказалось в три раза больше, чем в воде, причем 50$ избыточных!
ионов [S04] и 90$ ионов [ NH4] содержалось в частицах с d <¦¦«
< 0,45 мкм, что, возможно, свидетельствует о газофазных реакцщ!
ях образования сульфатов над морем [166]. ;i
Образование сульфатов над морской поверхностью происходят]
в результате окисления диметилсульфвда, сернистого газа и се1
водорода, источниками которых является морская биомасса, так,
например, большое количество диметилсульфвда ввделяется в а1
феру при гниении морских водорослей. В пользу газофазного и
низма окисления сернистых соединений свидетельствуют эксперт
ментальные данные о преимущественном содержании сульфатов и оси
ной кислоты в мелкодисперсной фракции морских аэрозолей. В 1
частности, в частицах с d < 0,5 мкм обнаруживаются значитель-1
ные концентрации SO2,
(NH4JS04,
H2SO4 , а в частицах с
24 4
< 0,1 мкм доля сульфатов в аэрозольном веществе превышает ^
Однако вряд ли в приводном слое происходитгазофазное фото
химическое окисление. Более вероятны ион-молекулярные реакци
а также каталитическое и фотокаталитическое окисление серниоиЦ
соединений в мелких водных каплях ( SO2 + Н20 + 03 + катаяиза|
тор). Это предположение подтверждается экспериментально наблян
даемой зависимостью концентрации сульфатов в приводном слсЦ'
атмосферы от относительной влажности. Препятствием для значи-|
тельного роста сульфатных частиц, вероятно, является повышен»!
кислотности среды при образовании серной кислоты и сульфатов.-'¦
В приводном слое атмосферы наблюдается также возникнове-$
ние окислов азота и нитратных соединений в результате окисле-4
ния аммиака, который не прореагировал с сернистными соедине- 1
ниями.
Количество органического вещества в морских аэрозолях
превышает 10 мкг/м3, но оно также содержится в основном в су<
микронной фракции. Большинство органических соединений прису
ствуют в морском воздухе как в газообразном, так и в аэрозод
ном соотоянии. В частности, концентрация формальдегида дося|
ет 5,4 нг/м3, а концентрация л-алканов (С10*С28) изменяете
от 1 до 25 нг/м3.
58
Измерения на о.Сан-Николас в 130 км от Лос-Анжелеса пока-
показывают, что содержание Si в аэрозолях составляет - 20,морской
соли - И, сульфатов, нитратов и соединений аммония - больше
25$. Отношение [Cl] /[Na] = 2,4, т.е. выше, чем для морской
no;
зяачительно отли-
воды. Содержание Са, Br. Pi, S04 ,
чается от содержания этих компонентов в городских аэрозолях.
В аэрозольных пробах, взятых А.Месарош на научно-исследо-
научно-исследовательском судне "Профессор Визе" в ноябре 1971 - феврале 1977гг.
в 600 километрах от материка, были обнаружены в основном части-
частицы сульфата аммония, хлорвда натрия и оерной кислоты [202].При-
[202].Причем большинство частиц было не сферической, а вытянутой формы.
Вместе с тем методом колец Лизеганга в импакторных пробах,вэя-
тых на о.Куба, были обнаружены в большом количестве частицы
NaCl.
Особое внимание в ряде исследований обращалось на коэффи-
коэффициенты обогащения для Cl,J, Br. Было обнаружено уменьшение
отношения [Cl]/[Na] в аэрозолях по сравнению с морской водой.
Потери хлора для Пуэрто-Рико - 0,22 мкг/м3 A3$ от общего содер-
содержания); в заливе Сан-Франциско - 1,60 мкг/м3 E4$); 90? потерь
наблюдается в шестом и седьмом каскадах импактора (мелкие час-
частицы) и менее 20$ - в первых двух каскадах. Установлена линей-
линейная связь между величиной потерь хлора и содержанием NO2 в воз-
воздухе: 0,06 мк-моль/м3С1 соответствует 1 мк>моль/м3 NO2, из че-
чего следует, что главной причиной потерь'хлора является реакция
HNO3 + NaCl —> HCl + NaNO3- М.Петерсон и Р.Скорер анали-
анализировали пробы осадков с тремя градациями отношения [Cl]/[Na]=
= R : В < 1,5; R > 2,16 и 1,5 < Р < 2,16 (последний случай
соответствует F «1,8 для морской воды) для проверки гипоте-
гипотезы Кауэра о химических и фотохимических реакциях с участием
NaCl. Однако окончательного вывода о причинах несоответствия Л
третьей градации для большинства проб сделать невозможно из-за
больших ошибок измерений [212].
Недостаток хлоридов в морских частицах малого размера на-
наблюдался также в пробах, взятых вблизи Лос-Анжелеса в 100 и
от берега на высотах 8 и 30 м, что объясняется образованием
кислой среды в "старых" аэрозолях: (А) + NO2 -* С12+ Н1Ч03 .
В пользу вывода о заметном ввделении хлора из атмосферных аэро-
59
Таблица IT'
оахарской пыли над трогшчеокой Атлантикой
Состав (процентный по массе) компонентов
Материал
Иллит
Мусковит
Кварц
Кальцит
Каолинит
Плагиоклаз
Хлорит
Монтморилонит
Микроклин
Всего:
есто
изме-
ения
1
2
3
I
2
3
1
2
3
1
2
3
1
2
3
1
2
3
1
2
3
1
2
3
1
2
3
SiO2
26,15
31,38
31,38
20
13
13
~
2,74
3,88
2,74
3,37
3,03
3,03
1,06
1,06
1,46
1,53
1,53
1,53
1Д5
0,86
0,86
56,0
54,74
54,0
тю2
0,005
0,006
0,006
":
-
0,025
0,036
0,025
;
0,001
0,001
0,002
":
-
0,031
0,043
0,03
]
А12О3
13,0
15,6
15,6
-
2,39
3,38
2,39
1,02
„ 0,92
0,92
¦ 0,83
0,83
1Д4
0,59
0,59
0,59
0,53
0,40
0,40
18,36
21,72
21,04
(имически!
СО2
-
-
. 4,39
2,41
3,3
-
-
-
-
-
4,39
2,41
3,30
Примечание: 1- о.Сал, 2 - о.Барбадос, 3 -г.Май-
компонент
Fe2O
1,25
1,5
1,5
-
_
-
0,006
0,008
0,006
0,004
0,003
0,003
0,076
0,076
од
0,025
0,025
0,025
-
-
-
1,36
1,61
1,64
FeO
_
-
-
-
_
-
_
-
-
_
_
-
0,75
0,75
1,03
_
—
-
_
-
-
0,75
0,75
1,03
MnO
_
-
:
-
_
-
_
_
-
_
_
-
0,02
0,02
0,03
_
_
-
_
_
-
0,02
0,02
0,03
MgO
2,2
2,64
2,64
0,004
0,002
0,003
_
-
-
0,005
' 0,004
0,004
0,79
0,79
1,1
0,096
0,096
0,096
_
-
-
3,09
3,53
3,84
CaO
0,10
0,12
0,12
5,59
3,07
4,19
-
-
-
0,04 .
0,036
0,036
-
-
-
0,05
0,05
0,05
-
-
-
5,78
3,28
4,39
Na2O
0,15
0,18
0,18
-
_
-
0,0096
0,013
0,0096
0,55
0,49
0,49
_
-
-
0,003
0,003
0,003
—
_
-
0,71
0,69
0,69
K2O
4,15
4,98
4,48
-
-
-
0,02
0,03
0,02
0,018
0,01
0,01
-
-
-
0,001
0,001
0,001
0,32
0,24
0,24
4,51
5,26
4,75
ами. Количество связанной воды от 4,8 до 5,8$.
61
60
золей овидетельотвугат и результаты химического анализа ооадков
и морокой воды. В чаотнооти, отношение [нсоз]/[с*] в осадках
в деоятки раз выше, чем в морской воде.
Для J и Вг установлено обогащение оодержания в морской
воздухе, причем наблвдаетоя заметное обогащение аэрозолей J:
на первых ступенях каокадного импактсра в 100, а на фильтре -
в 14000 раз, т.е. аэрозольные чаотицы, оодержащие йод, в ос- •¦
новном мелкие. Лабораторные экоперименты показали,что в естест—.
венных уоловиях обогащение аэрозолей иодом идет в 20 раз ин-
интенсивнее. Содержание Вг в воздухе коррелирует о содержанием
РЪ. Ооновным источником, по крайней мере газообразного брома,
является морская соль. Газообразный бром плохо раотворяется и
оущеотвует в атмосфере неоколько недель. Его содержание над ;/
о.Оаху в Тихом океане ооставляет 50 нг/м3. В аэрозольном оос-
тоянии брома примерно в четыре-десять раз меньше, а время жиз-
жизни его в семь раз больше. Так как в аэрозольных чаотицах раз-
разных размеров наблвдается увеличение оодержания брома в оамнх
мелких и самых крупных, то можно оделать вывод, что существую»
два иоточника появления аэрозольного брома и часть его из аэрсь
золей улетучиваетоя. •'
Разделение Вг и J происходит при переходе этих злемен- ;
тов в аэрозольное ооотояние. Измерения отношения [Cl]/[Na] '
для разных фракций частиц также подтверждает переход части [С1]
в газообразное состояние.
Большой экспериментальный материал по химичеокому ооотаву
аэрозолей в приводном олое Атлантичеокого океана был получен
в период проведения научного эксперимента АТЭП-74. Влияние са-
хароких пылевых бурь наблвдалооь вплоть до полуоотрова Флорида
Для иллюотрации приведен процентный веоовой оостав пылевнх чао>
тиц, по данным Дж.Просперо, для о-вов Сал и Барбадос, г.Майами
(табл. И) [226].
Как видно из табл.11, для всех пунктов маооовая доля ос--;
новных химичеоких соединений, характерных для пыли, в общем
содержании аэрозолей составляет 65-70$.
Измерения аэрозолей в этом районе проводились на научно-
исследовательокйх судах "Паооат" и "Профессор Визе" лаборато-
лабораторией физики аэрозолей Ленинградского универоитета. Некоторые
62 ;.'
данные по элементному составу аэрозолей в приводном олое по из-
измерениям на этих судах приведены в табл.12, 13. "Професоор Ви-
Визе" находился в период наблвдений западнее Дакара на пути про-
прохождения пылевых масс из Сахары, а "Паооат" - вблизи экватора,
где сказалось влияние оахарских пылевых вынооов.
Анализ аэрозольных проб, взятых на "Профеосоре Визе", дает
возможность сделать вывод, что даже в приводном олое атмосферы
значительна доля аэрозольных частиц неморокого происхождения.
Об этом свидетельствует относительно высокое оодержание в про-
пробах таких элементов, как Al, Fe, Са . Однако соотношение эле-
элементов в пробах, взятых на "Профессоре Визе", оущеотвенно от-
отличается от ооотношения элементов в самолетных пробах, получен-
получениях в том же районе Атлантики. Можно определенно оказать, что
аэрозольные частицы, содержащие Fe, Al, РЪ не опуокалиоь из
тропосферы в приводный слой в районе нахождения "Профессора Ви-
Визе". Обратная картина наблвдаетоя в пробах, взятых на "Пасоате"
(~0° ш., 10° з.д.). Содержание и соотношение между элементами
в самолетных пробах и пробах, взятых на "Пасоате", очень оход-
ны. Это позволяет предположить, что в районе нахождения "Паооа-
та" выпадают чаотицы из пылевого облака, идущего из Африки, в
частности из Сахары, что объяоняетоя оущеотвованием определен-
определенного воздушного течения, оносящегс пылевое облако в нижней его
части к югу, в район нахождения "Паооата".
Для оптических характеристик аэрозолей очень важно содер-
содержание влаги в чаотицах. Экоперимент по оценке оодержания влаги
в атмосферных аэрозолях морокого происхождения был выполнен о
помощью нефелометра, имеющего обогреваемую подводящую трубку
[216]. Нагревание этой трубки до 150°С уменьшало раосеивающую
способность аэрозольных частиц для морокого воздуха на 505?.При
измерениях аэрозолей над о.Оаху оигнал уменьшался на 65-75$, а
при измерениях в верхней тропоофере всего лишь на 20$. Это сви-
свидетельствует о том, что морские аэрозоли не попадают в верхнего
тропосферу. Повышенная гигроокопичнооть частиц над оотровом,
по-видимому, обуоловлена приоутотвием в них оульфатов и, воз-
возможно, серной кислоты.
По наблюдениям А.Вудкока в ряде олучаев образование мор-
морских туманов пелноотью объяоняетоя роотом солевых чаотиц при
63
Таблица 12
Содержание элементов в приводном слое' Атлантического океана
вблизи экватора (НИС "Профессор Визе", АТЭП-74), мкг/м3
Время взятия
пробы (дата.ч;
24
27
13
31
1
07; 12
07; 12
08; 12
14
08; 12
09; 09
12
18
Na
3
I
2
7
5
8
1
3
,0
,7
,0
,3
,2
,0
,8
.5
К
0,70
0,45
0,50
1,8
1,3
2,1
0,40
1Д
[sq.1
0,40
1,2
0,40
1,0
0,50
1,8
1,5
1,9
Fe
5,0
1,8
6,1
13,1
7.1
0,46
0,46
3,1
Ni
0,20
0,15
0,73
0,70
0,09
0,21
0,20
0,20
Элемент
Al
1,5
2,0
1,2
7,2
1,3
0,49
1,4
0,47
Са
0,80
2,5
0,46
1,2
0,71
12,0
10,0
12,0
Pb
_
-
0,036
0,10
0,10
-
-
-
Mn
0,040
0,050
0,067
0,18
0,11
0,26
0Д6
0,10
Mgr
1
1
1
3
2
4
2
4
.0
,3
,3
,8
,1
.3
,7
,1
Cu
0,050
0,060
0,050
0,13
0,070
0,90
0,50
0,90
zn
1
2
0
0
0
o,
-
,3
,3
,98
,30
,16
30
Cr
0,050
-
-
-
-
0,16
-
-
in
В
Таблица 13
Содержание элементов в приводном слое Атлантического океана
вблизи экватора (НИСП "Пассат", АТЭП-74), мкг/м3
Время взятия
пробы (дата, ч
24 06; 09
15
25 06; 09
30 06; 15
4 07; 09
6 07; 12
7 07; 09
8 07; 15
9 07; 12
15 07; 09
12
29 07; 09
30 07; 12
6 08; 09
12
10 08; 06
о 12 08; 09
Элемент
Fe
0,60
0,80
0,72
0,60
1,2
44,0
23,0
9,2
0,60
12,0
20,0
17,0
0,50
6,3
10,0
8,0
28,0
Ni
-
-
-
-
-
-
-
-
0,21
-
0,19
-
-
-
0,20
-
Mg
0,18
0,18
-
17,0
0,24
25,0
21,0
9,1
-
3,7
26,0
13,0
5.1
9,3
8,9
3,5
23,0
Си
0,18
3,0
0,18
6,6
0,12
10,0
9,0
2,0
0,12
0,35
3,2
0,41
0,27
-
-
0,15
6,1
Al
0,60
0,78
0,30
22,0
2,4
22,0
26,0
8,5
2,4
8,3
29,0
15,0
31,0
4,1
10,4
8,0
24,0
Mn
0,036
0,054
0,018
4,6
0,060
7.2
5,6
-
0,0060
0,15
4,4
0,20
0,20
2,0
1.8
0,10
2,6
_
_
_
260
190
80,0
—
26,0
28,0
20,0
6,0
11,0
110
12,0
195
Pb
_
_
_
_
_
_
_
-
_
0,18
_
0,14
-
_
-
0,10
-
Zn
_
_
_
_
_
_
_
_
3,8
_
_
_¦
_
_
_
-
Сг
_
_
_
...
_
_
0,030
_
_
_
-
66
я
<u
s
ЗЯТИЯ
о
а
?н
с
-1
to
н
В1
аГ
Й
я и
4>
¦—
я
о
с,о
(Г
с
1
со
из
С*}
«¦
о
о
г>
СО
8
о
о
о
S3
о
О5
со
о
о
ьч
о
со
о
со
кЧ
1
1
1
о
fcrl
C\i
C\i
о
кЧ
С-
кЧ
О
кН
кЧ
О
со
кЧ
1
1
1
О
О5
C\i
О
г-
о
C\i
о
ID
О
о
IV
05
о
СП
о
кН
1
'
О
CNJ
О
о
¦"*
о
о
krl
c\i
кч
о
о*
1
1
О
о
кЧ
СО
ID
C\i
О
О
со
о
ю
я
СП
о
«.о
с\г
о
со
1С
СП
о
о
со
кН
СО
кЧ
О
СО
C\J
со
кЧ
о
кЧ
О
О
О
О
0-
кН
О
СП
о
о
го
1
1
о
кЧ
кЧ
из
О
ю
о
со
о
из
CV
о
о
1
1
о
г-
о
кЧ
О
из
СО
о
Cxi
о
из
о
8
О5
о
«о
1
1
1
о
м
о
со
КЧ
о
о
кЧ
О
"=Г
ID
О
1
Г)
о
о
о
о
о
-**
р,
8
о
со
кЧ
090
о
о
о
о
ID
СО
ID
О
О5
О
C\J
кЧ
1
1
О
ID
Ю
О
со
кН
О
со
о
О5
о
со
кЧ
CV
к-Н
1
С\!
кН
и->
кН
О
О5
о
Cxi
о
о
чч
CV
о
со
из
о
о
о~>
8
СП
о
со
и
1
•4е
см
о
о
о
со
-*?
о
S
о
о
ю
,080
о
о
ю
со
кЧ
о
<о
со
CD
к-Н
1
1
1
о
CV
со
о
О5
CV
о
из
CV
1
о
(О
из
со
кЧ
О5
о
со
кЧ
1
о
со
о
1
о
сп
кг!
со
кН
о
ю
CV
я
о
¦"*
о
из
о
о
со
кЧ
8
со
о
05
C\J
о
ID
О
!>
О5
О
а>
о
г-
ID
О
о
СО
ю
о
о
ю
•4е
о
О5
о
О5
о
ю
см
увеличении относительной влажности воздуха, если предположить,
что диапазон размеров эффективных ядер конденсации 0 4 - 4 мкм
[240].
§ 1.4. Химический состав тропосферных аэрозолей
Особенности поведения аэрозолей разной химической природы
обусловливают различие их химического состава на разных высо-
высотах. Вертикальная структура химического состава аэрозолей в тро-
тропосфере характеризуется, как правило, слабым убыванием массовой
концентрации большинства элементов, входящих в состав неоргани-
неорганических аэрозолей. Для отдельных элементов, как, например, Fe
это убывание может даже отсутствовать. ' '
Наиболее сильно уменьшается с высотой концентрация Si что
по-видимому, связано с относительно большими размерами частиц '
содержащих кремний. Очень наглядной иллюстрацией разнообразия
вертикальных профилей содержания элементов являются данные
представленные на рис.2, а также некоторые результаты самолет-
самолетных исследований атмосферных аэрозолей, проведенных по програм-
программе комплексного энергетического эксперимента (табл.14). Эти дан-
данные показывают, что химический состав континентальных.аэрозо-
континентальных.аэрозолей в нижних слоях атмосферы может заметно изменяться, сохра-
сохраняя некоторые характерные особенности, т.е. можно с большой
уверенностью говорить о достаточно хорошей перемешанности тро-
тропосферных аэрозолей. Характерный химический состав аэрозолей в
нижних слоях тропосферы для европейской территории Советского
Союза представлен в табл.15.
Для высот больше 5 км практически уже не наблюдается за-
зависимость химического состава аэрозолей от локальных источни-
источников, это подтверждают И.Блиффорд о сотрудниками, которые изу-
изучали вертикальную структуру некоторых химических элементов
главным образом для оценки роли почвенных иоточников аэрозо-
аэрозолей) в трех точках земного шара о сильно различающимися при-
природными условиями: в Тихом океане' в 250 км от Санта-Барбары
в Долине смерти (шт.Калифорния) и в дельте р.Ориноко. Всюду
отмечалось значительное увеличение концентрации Si и уменьше-
67
Н,км
0,5
1,0
0,5 1,0
т,отн.ед.
Рис.2. Вертикальные профили относительного содержания
отдельных элементов в аэрозолях (Д.Жиллет).
а - г.Скотсблаф, шт.Небраска; б - побережье
Тихого океана; в - Долина Смерти, шт.Калифорния.
70
Таблица 15
Химический состав и концентрация аэрозолей в слое
2оО-1000 м над европейской территорией Советского Союза
(в среднем за год) [96], мкг/м3
Компонент
S02
Cl"
N0"
NHj
Na+
K +
Mg+
са+г
Всего
Район
Север
A3 проб)
0,29
0,58
0,05
0,07
0,29
0,10
0,03
0,21
1,62
Северо-запад
E3 пробы)
3,76
2,33
0,18
0,065
1,46
1,28
0,55
1,74
11,95
Юго-запад
(95 проб)
6,96
2,26
1,44
1,92
1,88
1,11
0,56
1,50
17,63
Юго-восток
(-16 проб)
3,44
1,89
0,68
0,84
1,39
0,96
0,41
1,79
11,40
женная слоистая структура содержания элементов, характерных
для сахарской пыли, причем на больших высотах обнаруживается
относительное обогащение Fe.Al в пылевом слое и Ca.Mg под
пылевым слоем (рис.3).
Следовательно, использовать понятие "морские аэрозоли"
применительно к химическому составу имеет смысл лишь при отсут-
отсутствии вблизи мощных источников аэрозолей с другим химическим
составом и для нижних слоев атмосферы ( Н 4 2-3 км).
Хотя основным компонентом морских аэрозолей являются-хло-
являются-хлориды, однако содержание сульфатов и органических компонентов
может быть также весьма существенным. Определение концентрации
Mg, Ni, Co, La, Cr, V, Ва , Sr в нижней тропосфере над мор-
морской поверхностью, показало, что соотношение концентраций этих
элементов соответствует тем же соотношениям в горных породах,
для Pb, Sn и Zn концентрации на порядок выше и свидетельст-
71
6,0
2,0
-^— Х- ¦¦» f>
Са
О1 1 1 1 1 1 I I'll
6,0
2,0
-I I 1 1 I I i
6,0
4.0
2,0
-
- /
-У
1
X
вует об их антропогенном происхождении.В последние годы отмече-
отмечено значительное увеличение концентраций металлов в тропосфере,
особенно РЪ. Обнаружено равномерное распределение по высоте Мп
и Cd, концентрация которых не зависит от времени,как и SOj» и
хорошая корреляция между содержанием этих трех компонентов; РЬ
коррелирует с СО и наблюдается в основном в нижних слоях ат-
атмосферы.
Сильное влияние на содержание аэрозолей в тропосфере ока-
оказывают метеорологические условия. Наблюдается увеличенное со-
содержание металлов в районе инверсий: для Fe - в облаках над
и под инверсией, для Ag - под температурной инверсией и в об-
облаках (в 10-100 раз); постоянная концентрация элементов в об-
облачном слое и уменьшение ее под облаком.
Наиболее хорошо изучен химический состав минеральной оос-
тавляющей аэрозолей над континентами, определяемой химическим
составом почв (табл.16). Кроме компонентов, указанных в таблДЗ,
наблюдаются относительно большие содержания Си, Ва, Ni, Se,
Сг, Zn.
Для оценки влияния аэрозолей на климат чрезвычайно важно
знание содержания в атмосфере сажевых частиц, но таких сведе-
сведений очень мало. По измерениям, проведенным в Швеции, отношение
количества углерода к количеству неорганического вещества изме-
изменяется от 0,28 до 1,03. Такое высокое содержание углерода ха-
характерно для загрязненных районов земного шара. Можно предпо-
предполагать, что общее содержание органических веществ приблизитель-
приблизительно в два раза больше содержания сажи, т.е. в нижних слоях ат-
атмосферы фоновая концентрация сажевых частиц может достигать ве-
величины: ОД - 5 мкг/м3. Эта сажа входит, вероятно, в соотав
смешанных аэрозольных частиц.
Некоторые данные о загрязнении тропосферы промышленными
выбросами были получены при полетах в районе Сланцев и Киришей
(табл.17). Можно отметить относительно низкую общую массовую
Рис.3. Абсолютные концентрации содержания элементов в тро-
тропосферных аэрозолях над тропической Атлантикой (западнее Да-
Дакара) в период проведения АТЭП-74.
а - 4 августа; 6-13 августа; в - 9 сентября.
73
о я:
и и
я я я
нон
К Ен
' OW
он
О
о
S.
<U I
и :
со
со
I Ен
ч S
rog
со
ОУЭ1ПОООО СО О
С- С\! СО Q 00 кЧ кЧ 0000
СОУЭСМОООС^ ОО
^?)СМ1ПСПСМкчО ф-^г
из из о О
кнОУЭСОСОСООО О
СМкЧООООООО О
?' см1 см1 А1 А1 А1 4' 3' Л1 ' ' Л1
N W О
СОСОкЧСМкНкНО» •
о о
_
in о
изсмсосм^оо ¦=*
СМкчОООООООкЧО
+1 +1 +1 +1 +1 +1 +1 +1 +1 +1 +1 I
кн^оооотиз-^соФсмиз
см m см \р • »оо • ^* • •
• » « О О -ООсОкЧ
Г^ ¦"* кЧ ^
Таблица 17'
in оо со из со с^ с^
со см о со
из
CM ID
О ID
С\!
S
о
fP У
С\!
т* U3
из о
С\!
С\!
00 С\!
О CD Ф
ID О
о о. '
из
(Л 1П
о in см ¦# о
• ' ' ' ' ^г ^ ' ' '
и со ^< о о
см
С\! ¦*
1П С\!
О^УЭ Ю 00 О N
из I
— . - . CNJ кЧ О
С\!С\!ЮкчОкчОкЧ
СО О О С\! О О
СО
О О О О СП
1Пизюоог^изос-с-и
изоюспизюсо-^'кн-^'кчо
СП00^С\!С\!С\!1г1ОО •• • •
С\! О О О
.^ г-> О) <0 Я
|«оа
Содержание элементов над промышленными центрами
Ленинградской области, мкг/м
¦*"% _— /» ¦ ш л *т fn
^ЛвМбНТ
Fe
Zn
Pb
К
Br
Си
Cr
As
Se
Sb
Co
Ag
Sr
Cd
Аи
Mo
Hg
Та
Се
Yb
Lu
Th
Hf
Cs
Sc
Sm
La
1,5
2,2
Высота,
4,*
Кириш
5,0
<0,6
<0,2
<5,0
<0,7
3,0
< 0,5
0,2
0,09
< O.I
< 0,15
-
1,0
0,30
0,13
0,020
< 0,040
0,80
0,010
< 0,005
0,0004
-
0,016
i 0,02
0,0050
-
<0,OI
3,0
< 0,6
<0,l
< 5,0
< 0,6
< 0,05
0,80
< 0,1
0,05
0,1
0,05
-
1.0
< 0,10
0,0011
0,10
< 0,040
0,60
0,40
0,007
-
-
-
< 0,02
0,0030
0,030
< 0,01
2,5
< 0,6
од
5,0
0,7
1,2
0,10
< 0,1
< 0,05
ОД
0,16
-
1,0
< 0,10
0,0012
0,13
< 0,040
0,28
0,013
0,006
0,0005
0,010
0,0070
< 0,02
0,0025
0,035
< 0,01
км
1,5
2,5
4,0
Сланцы
2,7
<0,3
0,15
< 10
0,40
0,25
0,12
< 0,05
< 0,02
-
0,05
< 0,005
-
< ОДО
-
0,010
-
•
•
< 0,005
0,0004
0,005
0,010
< 0,01
0,0030
-
< 0,005
2,0
<0,3
0,20
<10
0,30
ОДО
0,20
<0,05
<0,02
-
0,05
-
-
<одо
-
0,050
-
•
< 0,005
-
-
-
<0,01
0,0020
0.05
<0,005
0,5
<0,3
0,050
< 10
0,25
0,07
0,08
<0,05
<0,02
-
0,03
-
-
<одо
-
0,005
-
•
•
<0,005
-
-
-
< 0,01
-
-
< 0,005
75
концентрацию аэрозолей над этими промышленными районами и внсо-
кую для отдельных антропогенного происхождения элементов: V, Se,
As.Sb.
Аномально высокие концентрации аэрозолей в тропосфере наб-
наблюдаются во время пыльных бурь и вблизи действующих вулканов.
По наблюдениям Дж.Просперо основная масса пыли переносится че-
через Атлантический океан в направлении Центральной Америки в
слое от 1,5 до 7,7 км. Средняя концентрация этой пыли в районе
о.Барбадос составляет 61 мкг/м3, а в нижних слоях тропосферы -
22 мкг/м3. Общая масса пыли, переносимой через меридиан, пере-
пересекающий о.Барбадос, около 25-27млн. т. Неоднократно наблюдал-
ся подъем бурями минеральной пыли пустынь экваториальной Афри-
Африки до высоты 5-7 км. Температура запыленного слоя может быть
на 5-6°С выше температуры пограничного слоя, что создает оиль-
нуго инверсию в нижнем атмосферном слое. Облако пыли пересекает
Атлантику за 5 - 7 сут.
Максимум образования пылевых облаков в экваториальной Аф-
Африке наблюдается в июне - августе. В связи с сильными засухами
в начале 70-х годов наблюдалась тенденция к усилению ветровой
эрозии. По оценкам В.Грингела и Р.Мюлейзена, сделанным по из-
измерениям электропроводности воздуха, сахарская пыль переносит-
переносится над северной Атлантикой в слое воздуха 1,2 - 3 км, где мас-
массовая концентрация пылевых частиц достигает 1500 мкг/м3. Это
составляет 2,5 - 4 г для воздушного столба с площадью основа-
основания 1 м2, что соответствует экспериментальным данным, по осажде-
осаждению пыли на горизонтальные площадки. Такого же порядка величи-
величины были полученн при самолетных и судовых измерениях концентра-
концентрации аэрозолей в период АТЭП-74. Существенное отличие этих оце-
оценок от оценок Дж.Просперо, вероятно, вызвано различием в спо-
способах отбора проб. 3 пробоотборнике, используемом Дж.Просперо,
при скоростях ветра v > 2 м/с происходит заметное уменьшение
эффективности захвата грубодисперсных частиц, которне состав-
составляют основную массу пылевой материи.
Л.Радке с помощью измерений с самолета в окрестности вул-
вулкана Бейкер (Аляска) были построенн изолинии концентраций хи-
химических соединений и полученн оценки интенсивности выделения
вулканических продуктов: H2S - 0,35 - 1,3, аэрозолей - 38 г/с
76
Скорость увеличения числа частиц вулканичеокого происхождения
на высоте 3,7 км составила 1,9-10 с , а на высоте 11,3 км -
2,6-Ю с , что подтверждает мнение о существенной роли вул-
вулканических извержений только для больших высот.
§1.5. Химический состав аэрозольных частиц разных
размеров
Как уже отмечалось ранее, вопрос имеет принципиальное зна-
значение для физико-химических процессов трансформации аэрозолей
и выведения их из атмосферы, а также для атмосферной оптики.
Однако пока имеются только отрывочные качественные данные, хо-
хотя различие в химическом составе частиц разных фракций было за-
замечено давно. Причины этого различия в общем понятны. Они об-
условленн в первую очередь характером распределения по разме-
размерам аэрозольных частиц от разных источников, избирательностью
процессов удаления их из атмосферы к физико-химическим свойст-
свойствам аэрозольных частиц, а следовательно,я возрастом последних.
В частности, химжчеокий состав мелкодисперсной фракции будет
определяться в основном веществом частиц, образующихся из га-
зовнх примесей. Самые крупнне частицы, очевидно, образуются в
результате механического разрушения пород. Х.Юнге приводит дш-
ные по содержанию [S04], [NH4]+ и [Ct ]" в больших и ги-
гигантских частицах [110]. Содержание первнх двух типов ионов
изменяется приблизительно одинаково, что свидетельствует о су-
существовании этих ионов в атмосфере в основном в виде частиц
сульфата аммония с максимумом распределения их по размерам в
области г <1,0 мкм. Для хлоридов максимум распределения,оче-
распределения,очевидно, сдвинут в сторону более крупных частиц (табл.18).
Часто максимум распределения частиц определенного хими-
химического состава по массе приходится на гигантские. Измерения
аэрозолей многокаскадным импактором Андерсена в районе г.Оса-
г.Осака показали, что Al, Ca, Sc, Ti , La, Се, Sm, Vb, Lu.Hf
Та, ТЬ находятся главным образом в частицах о d > 3,0 мкм;
Se, As , Cd, Sb имеют максимум в распределении в области
d < 1,0 мкм, a Na.Cl, К, V, Сг, Mn, Fe , Co, Zn , Вг, Pb, Ag,
77
Таблица 18
Относительное содержание (по массе) ионов в частицах
разных размеров
Ионы
so;
NH +
NOJ
ci-
со;2
са+г
Na+
к+
Ядра
Айткена
По X.
39,5
23,7
4,0
2,6
•
5,3
•
•
Большие
частицы
Юнге
33,8
24,6
7,7
6,2
.
3,1
•
•
г» L. Т 0 мРгм
1 % 1 |U MXuu
По
П.Ф.Свистову
38,0
3,2
6,9
10,5
3,2
15,0
7,7
9,3
Гигантские
частицы
По
Х.Юнге
28,6
9,5
14,3
14,3
•
9,5
•
•
по
П.Ф.Свистову
61,3
2,7
1.1
6,7
0,5
6,9
5,6
9,4
Примечание. Автор данной монографии предполага-
предполагает, что доля всех указанных ионов в растворимом веществе аэро-
аэрозолей ооставляет 93-95$, а доля ионов по Х.Юнге - 75-80$.
Sc, Ba, W имеют два максимума в распределении, которые чаще
всего наблвдаются в интервале d = 1+3 мкм. Аналогичные изме-
измерения импакто'ром Андерсена в Нагоя также выявили различную хи-
химическую природу частиц, имеющих разные размеры. Модальный ди-
диаметр в распределении частиц по масое <^qi~ 0,5 мкм обуслов-
обусловлен наличием частиц сульфата аммония и характерен для измере-
измерений в зимние месяцы, а модальный диаметр tf « 40 мкм, наблю-
наблюдавшийся особенно отчетливо летом,- частиц нитратов.
Над городами США Сг, Cd, Mg имеют максимум распределе-
распределения для частиц с d > 1,5 мкм, а РЬ и Fe - для частиц с d <
< 1,5 мкм. По измерениям в районе Бостона малые размеры име-
имеют частицы, содержащие Zn.Se.Sb, Вг ; элементы Al.Se, Fe,
Th содержатся в наиболее крупных частицах, а Со, Mn CI
наблюдаются в составе частиц промежуточных размеров. Характер-
Характерно бимодальное распределение для частиц, содержащих РЬ и Вг.
Элементы Вг, J, РЬ наблюдаются главным образом в частицах,
78
уловленных за импактором на фильтр; 50$ РЬ и Вг обнаруживает-
обнаруживается в частицах с d < 0,2 мкм, почти весь Вг и J содержится в
частицах с г < 0,25 мкм, а С1 в частицах с г > 1 мкм. Р.Хейн-
дрикс обнаружил, что ряд оксифильных элементов принадлежит ве-
веществу частиц с of < 1,0 мкм, а С1, Вг, J, К, V, Mn, Zn, Se, As,
Jr\ , Hg - веществу еще более мелких, субмикронных частиц,
что связано с недостаточной степенью старения большинства аэро-
аэрозольных частиц для достижения ими динамического равновесия с
воздушной средой.
В большинотве исследований особое внимание уделяется дис-
дисперсности трех важнейших компонентов облачных ядер конденсации:
сульфатов, хлоридов и нитратов. Дж.Лодж обнаружил, что сульфат
аммония практически весь содержится в частицах, имеющих диапа-
диапазон размеров 0,08 мкм < г < 0,8 мкм. Лабораторные эксперимен-
эксперименты показали то же самое, несмотря на то что реакция образова-
образования сульфата аммония происходит главным образом на твердых' час-
частицах. Электронная дифракция позволила идентифицировать части-
частицы (NH4JS04h NaNO3. Почти все частицы, содержащие Na, CI,
NOJ, имеют размеры больше 1 мкм. Сходные результаты получены
в исследованиях Э.Месароша, А.Месарош, Г.Георгии. Интересные
результаты по величине отношения [S]/[Cl] для частиц разных
размеров получены для тропосферных аэрозолей: для частиц с раз-
размерами г- 0,2 мкм зто отношение 4:1, для г- 2 мкм - 4 :10
и для г - 4 мкм - 13 : 10. Максимум содержания серы приходил-
приходился на довольно узкий интервал размеров вблизи г— 0,2 мкм, а
максимум содержания хлоридов на интервал г = 2+3 мкм. Промежу-
Промежуточный максимум в распределении частиц по массе в области око-
около 1 мкм состоит из частиц другой природы.
Измерения Э.Месароша и А.Месарош в свободной атмосфере
показали, что максимум в распределении сульфатных чаотиц сдви-
сдвинут в область ядер Айткена. Экспериментально установлено, что
Al сконцентрирован в больших частицах, в гигантских частицах
наблюдается повышенное содержание Mg и Na, в частицах Айтке-
Айткена заметна повышенная концентрация V i 01. П.Ф.Свистовым бы-
были предложены модели химического состава растворимого вещест-
вещества аэрозолей для двух диапазонов размеров частиц: для грубо-
дисперсной фракции с г > 1 мкм с основными ионами Na+, К + Са**
79
CO , SO2 и для среднедисперсной фракции с г<1 мкм с ос-
основными ионами Н+, NH+, S02, ОН".
Полученные с помощью двухкаскадного фильтра результаты по
содержанию поглощающих коротковолновую радиацию компонентов ч
разных фракциях аэрозолей свидетельствуют о том, что основная
доля сажи содержится в больших частицах и ядрах Айткена, т.е. в
тех фракциях, где заключена основная доля частиц серной кисло-
кислоты и сульфатов. Существует хорошая корреляция между данными по
поглощению коротковолновой радиации в атмосферной толще и со-
содержанием в ней аниона SO^2. Можно предполагать, что частицы
сажи присутствуют в каплях серной кислоты. Наличие мельчайших
частиц сажи на поверхнооти капель серной кислоты может препят-
препятствовать испарению и диссоциации молекул серной кислоты.
Г л а в а 2 .
ХИМИЧЕСКИЕ ПРОЦЕССЫ ОБРАЗОВАНИЯ И РОСТА АЭРОЗОЛЬНЫХ ЧАСТИЦ
(фотохимические и гетерогенные реакции)
Сильная изменчивость аэрозольного компонента атмосферы об-
обусловлена, в частности, химическими процессами образования лег-
коконденсирующихоя продуктов и химическими реакциями на аэро-
аэрозольных чаотицах. Реакции образования диспергированной фазы
атмосферных аэрозолей происходят между разными реагентами:
1) в газовой фазе, 2) в водных каплях, 3) на поверхности твер-
твердых аэрозольных частиц, являющихся катализаторами. В газовой
фазе основными процессами образования легкоконденсирующихся
продуктов являются процессы окисления SOj до SO3 и NQ^flo N20.
с последующим образованием серной и азотной кислот; окисление
и полимеризация органических соединений в основном непредель-
непредельных углеводородов (особенно олефинов). Эти процессы идут в
присутствии сильных окислителей (озона, атомарного и синглет-
ного кислорода,различных радикалов) и при облучении смеси реа-
реагентов коротковолновой солнечной радиацией (фотохимические про-
процессы). Реакции окисления в растворах и на аэрозольных части-
частицах происходят как на свету, так и в темноте (с меньшими око-
ростями).
Некоторые газы растворяются в каплях с обратимой гидрата-
гидратацией и диссоциацией (NH3 и С02) и участвуют в процессах об-
образования диспергированной фазы аэрозолей лишь косвенным обра-
образом, другие участвуют в необратимых превращениях и реакциях с
растворенными окислами серы (S02) и азота (КЮд.).
При каталитических реакциях скорость превращения газового
компонента А в вещество диспергированной фазы аэрозолей опреде-
определяется в значительной степени не скоростями каталитических ре-,
акций в растворах или на поверхности твердых частиц, а интенсив-
интенсивностью захвата компонента А аэрозольными частицами: в случае
водных растворов - скоростью переноса молекул газа в капле и
растворимостью газа в воде; в случае твердых частиц - скоростью
переноса молекул газа к поверхности частицы, скоростью абсорб-
абсорбции на поверхности частиц и скоростью диффузионного переноса
молекул в порах частицы.
Если скорооть абсорбции на поверхности частиц высока, то
определяющую роль играет скорость диффузионного переноса моле-
молекул А в среде В к частице. При радиусе частицы порядка или
меньше средней длшш овободного пробега молекул газа справедли-
справедливо следующее выражение для диффузионного потока молекул А на
частицу:
где Яя- средняя длина свободного пробега молекул А в газе В;
оЬд - коэффициент прилипания; Лд - концентрация диффундирую-
диффундирующего газа А; гл - радиус абсорбирующей частицы; | - параметр,
зависящий от молекулярных масс пгА,тв и диаметров молекул ве-
вещества и окружающего газа, изменяющийся от 0,02 до 3,2. Знаки
плюс и минус указывают направление движения молекул относитель-
относительно поверхности частиц. При Х/га -» оо скорооть диффузионного
переноса пропорциональна площади частицы. Для частиц с радиу-
радиусом, значительно превышающим среднюю длину пробега молекул га-
газа, имеем
' <п -л. W I + сА/Г, \, . B.2)
где с - коэффициент скольжения, приблизительно равный едини-
единице для большинства газов; ВА - коэффициент диффузии пара ве-
вещества А в среде В. В этом случае при Л/га -* скорость
диффузионного переноса пропорциональна радиуоу частицы. Из
этих формул легко оценить роль атмосферных аэрозольных частиц
82
разных размеров в каталитических реакциях. Если не рассматри-
рассматривать случаи реакций в облаках и в очень редко встречающихся гру-
бодисперсннх образованиях, то можно считать, что процессы хими-
химических превращений газов происходят на частицах, имеющих г4 =
= 0,5 мкм.
Скорость захвата компонента А из воздуха на твердых по-
пористых аэрозольных частицах при малой скорости адсорбции моле-
молекул и газа на поверхности частицы по сравнению со скороотью диф-
диффузии внутрь частицы должна будет определяться уравнением вида
Jl=~*AVro4/<3a>I B#2)
где S - общая площадь поперечного сечения пор на наружной по-
поверхности частицы; a - гидравлический радиус пор; г^ - попра-
поправочный коэффициент по Е.Тиле, дается выражением
где Kj. - константа скорости реакции в расчете на 1 см2 поверх-
поверхности пор частицы.
Стационарное распределение концентрации молекул А в чао-
тице л. при реакции первого рода определяется уравнением
(l/r2)D?d<r2dnA/dr)/dr -КлпА'О B.4)
с граничными условиями Лд- л , г = г0 и dnA/dr'O, r*0,
где г - радиальное направление от центра частицы, ZL - коэффи-
коэффициент диффузии молекул А в порах, КА - константа скорости ре-
реакции на поверхности частицы, выраженная числом молекул 1 моль
в I см3 за 1 с. Реакция лимитируется диффузией D~ и при малых
значениях 5 протекает внутри частицы, а при больших значени-
значениях С, когда скорость реакции намного выше скорости диффузии
в порах, определяющую роль играет реакция на поверхности час-
частицы.
Закупорка пор в аэрозольных частицах смолистыми и маоля-
нистыми органическими веществами снижает способность частиц к
их росту в результате химических превращений газообразных про-
продуктов. Сведения о пористости атмосферных аэрозольных чаотиц
весьма ограничены. Например, методом БЭТ (адсорбционный метод
83
С.Брунауэра, П.Эммета и Е.Теллера) было установлено, что ве-
величина адсорбирующей поверхности для частиц, взятых в воздухе
над Питтсбургом, изменялась в зависимости от сезона от 1,98 до
3,05 м /г. Это меньше, чем у специально приготовленных порош-
порошков активированного угля или силикагеля A00+200 иг/т), но на-
намного больше поверхности сферических частиц с тем же распреде-
распределением по размерам.
Можно считать, что для процессов трансформации аэрозолей
гетерогенные процессы играют первостепенную роль. В пользу это-
этого утверждения свидетельствуют экспериментальные данные о нали-
наличии корреляции между количеством твердых аэрозольных частиц в
атмосфере и скоростью превращения окислов серы и азота в суль-
сульфаты, нитраты и соответствующие кислоты. В частности, существо-
существование капель в атмосфере практически возможно лишь в присутст-
присутствии ядер конденсации,что предполагает прохождение гетероген-
гетерогенных каталитических реакций на ядре.
Гомогенные газофазные реакции образования легкоконденси-
рующихся продуктов могут играть существенную роль, когда кон-
концентрация аэрозольных частиц в атмосфере невысока и вещество
этих частиц не обладает заметными каталитическими свойствами.
Наибольшее значение из газофазных реакций имеют фотохимические.
На первой стадии фотохимической реакции происходит образование
электронно-возбужденных молекул реагента при облучении их све-
светом (первичный фотофизический процесс). На следующей стадии
электронно-возбужденная молекула может либо вступить в реакцию
с другими молекулами, либо передать часть энергии возбуждения
другим молекулам (вторичный фотофизический процесс) и перейти
в состояние с более низким уровнем возбуждения и с большим вре-
временем жизни. В новом возбужденном состоянии молекула также мо-
может вступать в химические реакции, которые практически невоз-
невозможны для невозбужденной молекулы .Из нефотохимических газофаз-
газофазных реакций образования аэрозолей наиболее важными являются
ион-молекулярные реакции, происходящие в основном в высоких
слоях атмосферы. В атмосфере практически не наблюдается дейст-
действия какого-то только одного процесса перехода газовых компонен-
компонентов в аэрозольное состояние. Обычно действует одновременно не-
I
Аэрозоли
i
Сухое
осажденищ
Конденсирую-
Конденсирующийся газ
о
S
Он
84
сколько различных процессов с участием нескольких примесей и
аэрозольных частиц разного химического состава.
Вое перечисленные процессы в конечном итоге приводят к из-
изменению содержания в атмосфере не только аэрозольных частиц,
но и ряда примесей, в частности к выведению из атмосферы окис-
окислов серы и азота. Этот механизм выведения примеси из атмосферы
можно изобразить с помощью схемы, представленной на рис.4.
§ 2.1. Химические реакции соединений серы
В земную атмосферу сера в газообразном состоянии поступа-
поступает примерно в количестве 200 - 300 млн. т/год в виде сернисто-
сернистого газа SO2, сероводорода H2S, сульфидов, в основном диметил-
сульфида (CH3JS и карбонилсульфида (CSO). Предполагается,что
сероводород быстро окисляется до сернистого ггча, после чего
идет дальнейшее окисление SO2 с образованием сернокислотных
и сульфатных аэрозолей. Делались попытки построить модели кон-
конверсии серы с учетом сероводорода, однако исследований деталь-
детального механизма конверсии H2S в атмосфере не проводилось. Так-
Также практически не исследовался механизм конверсии сульфидов.
Окисление сернистого газа в атмосфере происходит с помощью всех
упомянутых ранее процессов: 1) реакций в водных растворах,
2) газофазных реакций, 3) реакций на поверхности аэрозольных
частиц. Оценить роль каждого процесса в окислении SO2 доста-
достаточно сложно. Однако можно утвердцать, что гетерогенные реак-
реакции окисления SO2 на твердых пылевых частицах играют важную
роль в загрязненной городской атмосфере, где велико содержание
в аэрозольном веществе элементов, являющихся катализаторами ре-
реакций окисления. Реакции в водных каплях необходимо привле-
привлекать для рассмотрения процессов конверсии SO2 в тропосфере.
Газофазные реакции окисления SO2 могут объяснить процесс об-
образования аэрозольного слоя в стратосфере. Оценки скоростей ре-
реакции по всем трем механизмам у разных исследователей сильно
различаются. Существуют серьезные сомнения в важности роли фо-
фотохимических газофазных реакций в нижней атмосфере. В то же
86
время регрессионный анализ показывает, что наблюдаются значи-
значимые корреляционные связи между концентрацией сульфатов и интен-
интенсивностью солнечной радиации в диапазоне длин волн Я=0,29*0,50
мкм при разных концентрациях SO2-
Гетерогенные реакции окисления сернистого газа. Установле-
Установлено, что для того чтобы скорости гетерогенных реакций были дос-
достаточно большими, необходимо выполнение по крайней мере одного
из следующих условий: I) присутствие в водном растворе компо-
компонента, нейтрализующего кислоту, 2) наличие реакционно-активных
окисей металлов, 3) наличие каталитических частиц. По Х.Коеру
сульфаты образуются всегда при конденсации водяного пара в ат-
атмосфере. Катализаторами реакций окисления могут быть ионы Мп,
Си, Fe, концентрация которых должна быть ~ I мг/л. Присутст-
Присутствие NM3 способствует окислению SO2. Процесс окисления этого
типа рассматривался в работе Х.Юнге и Т.Райана [186], которые
исследовали темновое окисление SO г в воде в присутствии хло-
хлористого железа FeCl2 и аммиака NH3. Роль NH3 сводилась к
улучшению растворимости SO2. Ван-ден-Хевель и Дж.Мейсон наблю-
наблюдали реакции окисления SO2 в водных каплях в присутствии NH3
без ионов металлов. Конечным продуктом был сульфат аммония. По
В.Скотту и П.Хоббсу в незапыленной облачной атмосфере может ид-
идти реакция окисления SO2 в системе SO2 — Н2О — СО2— NHj.
Механизм окисления SO2 в водных каплях, содержащих MnSO4.
может быть на основании кинетических исследований представлен
следующим образом:
SO, + Мл
.+ 2
/V\nSO?2,
2 Мл SO*2,
MnSOt2 + H2° ==* Мл+2+ HSO" +H+,
3 К7 *
HSO" + Н
87
В системе MnS04 (I мкг/м3) - Н20 (/= 95$) - SO2 A0 млн)
за 24 ч образуется 35 мкг/м3 H2SO4.
Возможно также некаталитическое окисление SO2 кислородом
в водных каплях. Для облаков с водностью 1 г/м3 при рН= 3+6
скорость выведения SO2 из атмосферы ~ A + 0Д5)-10~5 ч.
Окисление SO2 в водных каплях может идти также при растворе-
растворении в воде озона и органических загрязнителей. В экспериментах
С.Пенкетта было показано, что скорость окисления SO2 при пар-
парциальном давлении БОг G млн~^ ) в присутствии озона E млн )
может достигать 13$ ч , если происходит совместная адсорбция
SO2 и озона водяными каплями.
Установлен факт ускорения конверсии S02 на поверхности
окислов металлов по сравнению с гомогенным фотоокислением в га-
газовой фазе. II.Уроне исследовал реакции окисления SO2 в присут-
присутствия различных окислов: А1203, СаО, СгО, Fez03, PbO, V203.
Скорости реакций в присутствии этих окислов оказались значи-
значительными, причем реакция окисления шла даже в темноте. Экспери-
Экспериментально наблюдались высокие скорости окисления SOj без осве-
освещения системы в присутствии Fe304, А12О3, РЪО , Р*20,,а так-
также в присутствии угольной пыли (возможно, сажи). Окисление
при этом может достигать 30$ ч , превышая в шесть раз ско-
скорость гомогенного окисления в газовой фазе.
На важность гетерогенных каталитических реакций в атмос-
атмосфере косвенно указывает наличие корреляции между количеством
выбросов твердых частиц в атмосферу и скоростью превращения
SO2 в серную кислоту и сульфаты.
Таким образом, можно сделать вывод о том, что гетероген-
гетерогенные процессы для преобразований газообразных соединений серы в
соединения, существующие в жидкой и твердой фазах, играют ве-
ведущую роль для условий загрязненной атмосферы. В чаотности,
окисление SO2 до серного газа также происходит значительно
быстрей в присутствии аэрозольных частиц. Реакция образования
серной кислоты H2SO4 из серного газа S03 очень быстрая и,
очевидно, легче происходит в присутствии диспергированной фаза
Газофазные реакции окисления SO2- Для газофазного меха-
механизма конверсии SO2 в атмосфере очень важен процесс фотохи-
фотохимического окисления сернистого газа. Особенности фотохимичес-
фотохимического поведения молекул SO2 в атмосфере в значительной степени
обусловлены тем, что фотодиссоциация S02 на SO и 0 энергети-
энергетически невозможна вплоть до А, = 218 нм (при поглощении овета
с Л < 210 нм происходит диссоциация SO2). При облучении
SOg светом с Я } 240 нм образуются электронно-возбужденные
молекулы, которые флуоресцируют из трех состояний. Пары SO2
дают несколько систем полос поглощения в ультрафиолетовой об-
области. Первая полоса между 390 и 260 нм довольно слаба, имеет
хорошо выраженную тонкую структуру и, по-видимому, не связана
с возбуждением флуоресценции. Вторая' простирается от 244 до
200 нм и совпадает от Я = 210 нм со спектром возбуждения флу-
флуоресценции. Этот спектр плохо поддается анализу, особенно в
длинноволновой части.
Спектр флуоресценции при возбуждении монохроматическим све-
светом с А = 210 нм (искровая Линия Zn) имеет сравнительно про-
простую структуру, в которой периодически повторяются колебатель-
колебательные частоты 1370, 1150 и 520 см . Эти частоты соответствуют
трем основным частотам невозбужденной молекулы SO2, наблюдав-
наблюдавшимся в спектре комбинационного рассеяния сернистого газа.Окис-
газа.Окисление SO2 фотохимическим путем определяется реакциями сернис-
сернистого газа в фотовозбужценном состоянии S02 CВг). Это утверж-
утверждение основано на исследовании флуоресценции и фосфоресценции
фотовозбужденного сернистого газа и факте замедления процесса
фотоокиоления сернистого газа при добавлении биацетила, являю-
являющегося сильным тушителем ^SO2 ; 3SO2 представляет собой би-
радикал с неспаренным электроном в $р-орбите серы. Его реак-
реакционная способность исследовалась в ряде работ [113, 126, 135,
143]. Установлено, что в процессе фотоокисления SO2 в призем-
приземном слое атмосферы может вступать в реакции с Н', НО2, НСО3,
RO, RCOO2. ROOO, RO2. O<lD), ОCР), Ог A&р), 02A?+),
03, NO3,N2O5,OH (R- радикал СН3, C^k и т.д.). Б результате
наблюдения реакций неизлучающей возбужденной молекулы S02 с
молекулами СО и C2F4 высказано предположение, что фотовозбуж-
фотовозбужденный атом кислорода в молекуле SO2 может переходить к этим
молекулам, образуя SO-радикал. Присутствие радикала SO при
фотоокислении сернистого газа может повлиять на скорость реак-
реакции фотоокисления.
89
Наиболее высокие скорости тушения 'SO2 наблюдались в
столкновительных реакциях с молекулами 03, углеводородов и
Н2О. Образуется 3SC>2 при возбуждении его излучением в облас-
области длин волн 400 - 340 нм, а также в результате интеркомбинаци-
интеркомбинационных переходов при столкновительных процессах сернистого газа
в фотовозбужденном синглетном состоянии * SOj.
Образование 1ЭОг происходит при возбуддении облучением в
области длин волн 340 - 240 нм. Скорость генерации ^Ог значи-
значительно выше, чем скорость генерации 3 SO3, поэтому важно оце-
оценить скорость перехода * SO2 в
основными реакциями образования
новительыые реакции:
3,
оценки показывают,что
из
являются столк-
ISO, + SO,
"ea
3so2
so
2.
:so,
м
3 SO, + M ,
где М - третья молекула ( N2,O2, Н2О, Аг, Не, Хе, СО2, О3, N0 ).
Значительная часть *S02 тушится по реакции
+ М
Отношение констант скоростей ^ss/(^sS + K9SL 0,09. Расче~
ты общей скорости генерации 3SO2 в атмоофере при поглощении
солнечного света дают величины от нескольких десятых процента
до нескольких процентов за час (в зависимости от внсоты солнца
и концентрации газовых молекул М).
Фотохимическое окисление было обнаружено экспериментально
при освещении сернистого газа излучением с Я = 313 нм. Ф.Блэ-
кетом предложена следующая схема фотоокисления сернистого газа:
SO2 + h-9 (Я < 218 нм)
SO + О,
so.,
so4 + о2
so3 + о3
90
Однако экспериментально при процессе фотоокисления SO2 не ре-
регистрировались S04 и О3.
Т.Халлом квантовый выход реакции окисления SO2 был опре-
определен величиной порядка ф~10 [169]. Им предложена схема
реакции
3so2+ so2
so3
soCl") — SO3
B'5)
Необходимость присутствия окислителя не доказана и не получено
прямых спектроскопических доказательств образования SO4. B03-
моано, что при реакции фотоокисления сернистого газа может об-
образовываться метастабильный димер - OjS - 0 - SO или реакци-
реакционно-активный тетраксид S04. однако по П.Лейтону более вероят-
вероятно возникновение бирадикала
О
о
s - о -о
По схеме Ф.Блэкета возможна реакция с сохранением спина
электрона
+ 0Л31~) — SO*.
Возбужденный в синглетное состояние SO4 тушится
so;
S04
+ м
+ о,
— so4
so3 +
B.6)
В этом случае константа реакции с О2 при малых концентрациях
реагентов будет зависеть от общего давления смеси. Считая, что
концентрации ^§Ог и 3SC>2 ПРИ фотолизе чистого SO2 постоян-
постоянны, получим величину квантового выхода Ф, не зависящую от кон-
концентрации SO2. что подтверждается экспериментальными исследо-
исследованиями Р.Кокса [135]. По Р.Коксу при возбуждении SOj излуче-
излучением с Я = 285 нм тушение *S02 при столкновениях приводит
в 8$ случаев к инверсии спина и в 92$ к релаксации в основное
состояние, т.е. ^8$/(^8х+ Kgi) = 0,08. Наблюдается слабая
зависимость этого отношения от длины волны излучения: увеличе-
91
чение <Р до ОД при А = 302 нм. Экспериментально определенная
Р.Коксом величина квантового выхода окисления SO2 в S03 при
освещении 500-ваттной ртутной лампой среднего давления оказа-
оказалась равной Cp<SO2—- SO3) = @,38 + ОДО).1О. В этом
случае отношение К6 /(Kg + Кд) = 1/26, т.е. только 1/26 дезак-
тивационных столкновений 3S02 с S02 при комнатной темпера-
температуре приводит к химической реакции. Эти данные хорошо укладыва-
укладываются в схему фотолиза чистого SO2, предложенную Р.Коксом.Роль
кислорода по Р.Коксу заключается в его участии в реакции,
SOCI") + 02 + M —- SO,
м
B.7)
В присутствии малых количеств кислорода 0г G,2 гПа) при пар-
цианальном давлении SO2, равном 38,0 гПа, квантовый выход ока-
оказался равным 5*10 , что свидетельствует о заметном вкладе ре-
реакции окисления SO-радикалов. При облучении солнечным светом
величина квантового выхода оказалась на порядок меньше: Ф —
= 3«10~4. Оценки квантового выхода, сделанные П.Лейтоном, да-
дали величины в пределах от 3>Ю~3 до 0,3. Для концентрации SO2
в пределах 0,2 - 0,6 млн в присутствии водяного пара Фм
е20,3,а при концентрациях SO 2 - 16,9 гПа и 02 - 14,1 гПа в от-
отсутствие влаги Ф «*¦ 0,036. Однако нельзя сделать определенный
вывод о том, что уменьшение Ф связано с отсутствием Н20 и вы-
высокой концентрацией S02 и 02. Т.Халл полагает, что реакция фо-
фотоокисления первого порядка по SO2 и не зависит от концент-
концентрации О2. Им был получен квантовый выход, равный 0,01 при
D(SO2)= 74,5+306,0 гПа и р@2)= 66,5+255 гПа и освещении
солнечным светом ( 9 = 20°).
Для низких концентраций SO2 более реальна Ф = 0,003.
Такая же величина квантового выхода была получена в работе[44]
для парциального давления S02 в кювете от 7 до 166 гПа и от-
отношений парциального давления SO2 к давлению воздуха от 0,04
до 14,5 при облучении газовой смеси ртутной лампой ДРШ-1000 и
лампой сверхвысокого давления СВД-Т20. Продукты реакции наблю-
наблюдались как в объеме кюветы, так и на стенках. В ходе экспери-
эксперимента велись наблюдения за концентрациями реагентов и получаю-
получающихся продуктов реакции с помощью методов КРС, УФ- и ИК-спект-
роскопии, а также по рассеянному излучению (табл.19).
92
Таблица 19
Кинетика образования аэрозолей при фотоокислении
сернистого газа (ДРШ-1000 включается на 20 с шагом 10 с)
f, с
1
го
50
80
110
140
170
200
230
260
290
320
350
380
410
440
470
500
530
560
590
620
650
680
710
1
"о
йр
(9-9СП
Л, нм
350
Psof
Рн о~
700
32,
40
2(гПа)
2
0,85
0,71
0,61
0,52
0,47
0,44
0,43
0,43
0,42
0,44
0,43
0,44
0,46
0,46
0,47
0,48
0,50
0,48
0,51
0,50
0,55
0,55
0,55
0,56
3
1,0
0,93
0,81
0,82
0,76
0,68
0,64
0,58
0,54
0,51
0,53
0,46
0..44
0,43
0,42
0,42
0,43
0,42 (
0,43
0,43
0,43
0,45
0,46
0,45
350
Раа,'
рн = ,
г(гПа
4
0,92
0,80
0,69
0,62
0,57
0,54
0,53
0,52
0,52
0,54
0,56
0,56
0,59
0,61
0,61
0,63
0,63
0,63
0,64
0,66
0,67
0,67
0,69
0,68
700
40,
32
5
0,99
0,96
0,89
0,87
0,81
0,71
0,70
0,69
0,64
0,62
0,59
0,58
0,56
0,55
0,55
0,55
0,54
0,54
0,54
0,55
0,56
0,56
0,60
0,58
350
PsoT 5
п 0= /
700
г(гПа)
6
0,90
0,82
0,79
0,71
0,68
0,67
0,65
0,63
0,65
0,66
0,66
0,66
0,67
0,68
0,69
0,70
0,73
0,72
0,73
0,74
0,75
0,74
0,76
0,76
7
0,98
0,94
0,93
0,89
0,86
0,83
0,80
0,76
0,74
0,70
0,68
0,67
0,66
0,63
0,62
0,62
0,61
0,60
0,61
0,61
0,60
0,61
0,63
0,64
488
Psotm
=13
=40?гПа)
8
1,6
-
0,88
-
1.0
-
0,94
-
1.0
-
1,0
-
-
1,25
-
1,86
-
1,20
-
-
-
-
-
93
Продолжение табл. 19
1
740
770
800
830
860
890
920
950
980
1010
1040
1070
1100
ИЗО
1160
Ц90
1220
1250
1280
1310
1340
1370
1400
2
0,55
0,56
0,59
0,59
0.S0
0,64
0,64
0,66
0,68
0,69
0,70
0,72
0,73
0,74
0,75
0,76
0,77
0,80
0,82
0,82
0,88
0,88
0,89
3
0,46
0,47
0,49
0,51
0,52
0,54
0,56
0,59
0,58
0,60
0,64
0,64
0,68
0,69
0,71
0,73
0,74
0,76
0,78
0,83
0,84
0,85
0,85
4
0,71
0,72
0,72
0,73
0,75
0,73
0,75
0,77
0,77
0,78
0,78
0,82
0,85
0,86
0,84
0,89
0,89
0,89
0,90
0,92
0,93
0,95
0,96
5
0,61
0,63
0,63
0,67
0,69
0,71
0,72
0,75
0,77
0,79
0,81
0,86
0,84
0,83
0,82
0,85
0,95
0,95
0,95
0,98
0,98
0,98
0,98
6
0,76
0,77
0,76
0,74
0,78
0,80
0,80
0,80
0,81
0,81
0,81
0,83
0,83
0,86
0,85
0,86
0,87
0,87
0,89
0,89
0,89
0,88
0,89
7
0,64
0,65
0,66
0,67
0,69
0,72
0,73
0,74
0,75
0,76
0,77
0,79
0,81
0,81
0,82
0,83
0,84
0,85
0,85
0,86
0,86
0,87
0,86
8
210
-
1,40
-
-
-
-
-
-
-
1,20
-
-
-
-
-
-
-
-
1,25
-
-
_
Примечание. Общее давление в кювете во всех экс-
экспериментах р0 =767 + 1 гПа.
При освещении смеси SO2 и О2 излучением с длиной волны
Я = 315 нм и изменением относительного содержания SO2 от 100
до 60% величина квантового выхода для чистого SO2 получилась
равной 5-10" и увеличилась до 0,22 с повышением отнооительно-
го содержания О2 до 40$. Фотометрические наблвдения концент-
концентрации SO2 при начальных парциальных давлениях от 26,6 до
133 гПа в присутствии кислорода от 79,8 до 545,0 гПа в случае
использования излучения с
94 ¦
Я, = 313 нм дали Ф = 1,7.10, а
при интегральном освещении в области Я. = 280+420 нм Ф5*
=• 2-10"^, причем не наблвдалось зависимости величины квантово-
квантового выхода от концентраций кислорода, сернистого газа, а также
N2, CO2 и инертных газов. Различные эксперименты дают оценки
величины Ф от 10 до 10. Например, для смеси SO2 и О2 при
освещении ее на А = 313 нм получена величина Ф= G,2 + 1,4)х
хНГ3.
Низкие значения величины квантового выхода для гомогенно-
гомогенного окисления SO2 порядка 1*10"' приводятся в работе 1112],
причем такие же величины квантового выхода получены в результа-
результате исследований фотоокисления SO2 в чистой и городской атмос-
атмосфере. Возможные объяснения больших вариаций величины квантово-
квантового выхода фотоокисления SO2 предложены Р.Коксом: 1) каталити-
каталитическая роль стенок кюветы, 2) разогрев газа при высоких давле-
давлениях за счет поглощения излучения. (При давлении SO2 примерно
900 гПа 505? излучения поглощается в миллиметровом слое у окна
кюветы.)
Весьма сильно различаютоя оценки скоростей реакции фото-
фотоокисления SO2- Т.Халлом приводится величина скорости фотоокис-
фотоокисления при парциальных давлениях для SO2 - 74,5+306,0 гПа, О2-
66,5+266 гПа и освещении солнечным светом, равная 0,05$ -ч .
По Е.Герхардту и Х.Джонстону реакция фотоокисления Первого по-
порядка по S02 и скорость конверсии So2 в SO3 в лабораторных
условиях при освещении в диапазоне длин волн Л = 365+295 нм
К = 0,68#>ч~*, а для натурных условий освещения А = 0,1%-ч~^
[162]. Максимальная скорость прямого фотоокисления S02 -К =
= 2,45ё-ч~^, а более быстрые скорооти конверсии SO2 наблюдают-
наблюдаются в присутствии двуокиси азота. Увеличение скорости исчезнове-
исчезновения SO2 наблюдается также при повышении величины относитель-
относительной влажности. Оценки скорости исчезновения SO2 по наблюдению
за концентрацией изотопа 3^S при облучении солнечным светом
SO2 (-p(SO2)= 12+19 млн и 50$ относительной влажности) да-
дают скорость исчезновения SO2 в полуденное время 0,6$-ч , а
в послеполуденные часы скорость конверсии So2 уменьшается до
0,12.4-*.
Можно предполагать, что широкий разброс данных в скорости
окисления SOg и в величине квантового выхода связан, в част-
95
SO3 +
S03
ности, с тем, что существует несколько механизмов реакций окис-
окисления SO2 при освещении и в присутствии различных газовнх ком-
компонентов. Реакции B.5) и B.7) являются не единственными в про-
процессе фотообразования SO3. К сожалению, мало известно о химии
радикалов S04 и SO. В лабораторных экспериментах необходимо
учитывать реакции со стенками камеры. В чистом SO2 важнейшей
реакцией, очевидно, является реакция B.5), причем низкая кван-
квантовая эффективность обусловлена большой эффективностью деакти-
деактивации 3SO2 в S02 при столкновительных реакциях. Радикал SO
в основном реагирует со стенками кюветы и теряется. С ростом
концентрации кислорода квантовый выход фотоокисления SO2 дол-
должен расти. Однако при ультрафиолетовом облучены; SO3 может на-
наблюдаться фотолиз
C44 нм) S02 + 0(гР),
03 + h< C00 нм) SO + O2CI)
и в присутствии О2 это должно вести к появлению озона 03,что
уменьшит квантовый выход фотоокисления S02. Анализ возможных
фотохимических реакций в системе SO2 и 02 был проведен Ж.Буфа-
лини, который показал, что если реакции прямого окисления SO2
молекулярным и атомарным кислородом существенны, то скорости
образования озона и SO, должны быть пропорциональны. Это не
было обнаружено ни в одном эксперименте [113]. Р.Кейдл и П.Ма-
гилл установили, что окисление SO2 озоном пренебрежимо мало:
в смеси с относительными содержаниями S02 от 1 до 10$ при 1%
Н2О и очень высоком содержании О3 A%) за 24 ч прореагирова-
прореагировало меньше 0,1$ S02. Реакция SO2 с 03 при комнатной темпера-
температуре, вероятно, не идет.
Окисление S02 атомарным кислородом ОCР) в присутствии
третьей частицы в атмосферных условиях также не может играть
существенной роли, так как значительно быстрей идет реакция
атомарного и молекулярного кислорода с образованием озона. Ана-
Анализ всех возможных реакций кислорода с S02 показывает, что
для атмосферных условий, возможно, важной реакцией является
SO, + М
S04 + М ,
B.8)
96
а для лабораторных условий, когда отношение концентрации SO2 к
концентрации 02 достаточно велико
S03
B.9)
Возможен другой путь окисления SOj :
3SO2 + O2CZ- ) -
SO* ,
so
3SO2
SO* + M S04 + M ,
so4 + o2
so3
В этом случае снова встает вопрос о наблюдаемости S04 и 03.
Экспериментально получаемая контанта реакции, если идут приве-
приведенные реакции, будет зависеть от концентрации смеси. Для кон-
концентрации SO2, равной 0,2 млн , и величине относительной влаж-
влажности Ъ0% было получено, что не больше 1/1400 молекул 02 O&g)
реагирует с S02 (реакция B.8)). Реакция B.9) является экзо-
экзотермической реакцией второго порядка ( Ш = -2,61«Ю~^9 Дж) с
низким активационным порогом, однако скорость этой реакции ма-
мала.
При определенных условиях из смеси газов S02 , Oj. H20
могут образовываться молекулы серной кислоты. Присутствие мо-
молекул HjO для фотохимических реакций важно как источник ради-
радикалов ОН и ООН.Исследования скоростей реакции SO2 с радика-
радикалом ОН были проведены Р.Коксом [135]. Известна реакция гидро-
ксила ОН с сернистым газом с образованием HSO3
SO2 + ОН — HSO3 ( йН = -5,7Л09Дж), B.10)
имеющая скорость порядка 0,23 - 1,4#.ч~*. Химия HSOj практи-
практически не изучена. Этот радикал имеет неспаренный электрон в
сере, который взаимодействует с молекулярным кислородом,а так-
также, возможно, с гидроксилом
—^ H2SO4 + М,
7.1922
hso3 + он + м
hso3+ о2
HSO, .
5
97
Дальнейшие реакции радикала HSO5 могут приводить к образова-
образованию пермоносерной, пердисерной и серной кислот в условиях го-
горячего факела и повышенного содержания озона.
Реакции радикала ООН с SO г следующие :
ООН + SO2
ООН + SO,
он + so3
* HO2SO2
(ДН = - 1,22.10^9Дж),
( Ш = - Т,74-10~19Дж).
Скорость первой из этих реакций порядка О.ВБ^'Ч, а для вто-
второй значительно выше. Неоднократно рассматривалась возможность
газофазной нефотохимической реакции при атмосферных условиях
в системе NH3-SO2-H2O. В частности, учитывалась реакция об-
образования в стратосфере сульфита аммония NH4S03. Взаимодейст-
Взаимодействие SOj с NH3 в присутствии, молекул воды при I — 24°С идет
по реакции: 2NH3+ SO2 + НгО з=& (NH4JSO3, а при I * 14°С
и низких влажностях: 2NH3+ SO2 + НгО 5= (NH4JS2O5. Экспе-
Экспериментальные данные по фотохимии SO2 - N0^ системы достаточно
разноречивы. Например, для концентраций N02, равной 1-2 млн~^
и S02, равной 10-20 млн , при облучении с Л = 295*365 нм
никакого влияния NO2 на скорость реакции окисления SO2 обна-
обнаружено не было; NO2 играет роль третьей частицы в реакции
so2 + о + м
so3
м
Возможно, концентрация NO2 влияет на ход реакции
S04 + NO
SO3
NO2.
А.Альтшулер и Ж.Буфалини считают это обстоятельство несущест-
несущественным в образовании SO3 [1I3]. Однако если SO3 образуется
больше по реакции S04 + SO2 -* 2SO3, чем по реакции B.7), то
NO2 будет способствовать уменьшению скорости образования SO3.
А.Альтшулер и Ж.Буфалини подтвердили, что более эффективна
убыль S03 или деактивация S02 в реакциях с N0 .
Дж.Калвертом с сотрудниками исследовались реакции двуоки-
двуокиси серы с окислами азота
SO2 + NO3
SO3+ NO2
SO,
SO3 + 2К1О2(или N2O4),
S02 +
NO,
SO3 + NO .
Оценки скоростей первых двух реакций привели к выводу о второ-
степенности этих процессов в атмосфере, загрязненной N0^ и
углеводородами. При приблизительно равных концентрациях К102 и
S02 двуокись азота участвует в конверсии S02 как источник
атомарного кислорода. При Я, = 404,7 нм происходит фотолиз
N0,
N02
NO(X2Jt) + 0CР).
•Если NO2 B атмосфере мало, то главную роль играет цепочка ре-
реакций : 0CР) + 02 —- 03 — 03+ NO —NO2 + O2.Добавление N0^
к низким концентрациям S02 может приводить к различным резуль-
результатам. В присутствии N0 может происходить фотолиз SO2 с об-
образованием SO, но N0 может термически окислиться. Прямое окис-
окисление сернистого газа двуокисью азота может происходить в тем-
темноте. Эта реакция наблюдалась при высоких концентрациях S02»
N02 и отсутствии кислорода. В присутствии кислорода в темноте
сернистый газ практически не реагирует с N0 и N02. Очевидно,
что эффективность реакции атомарного кислорода с сернистым га-
газом (реакция B,10)) должна расти с увеличением отношения
[SO2]V [ ЫОЛ] при достаточно высокой скорости образования ато-
атомарного кислорода в реакциях окислов азота, в частности с озо-
озоном. Убыль S02 в присутствии N02 и 03 наблюдалась эксперимен-
экспериментально. Возможна следующая реакция окисления S02 с окислами
азота:
- S0$ + N02 .
N03 или N2O5
S02
Исследование реакции S02 с N02 при приблизительно одинаковых
концентрациях обоих реагентов показало, что в результате реак-
реакции образуется белый твердый порошок с отношением содержания
j как 1:1,
Работы группы Ж.Брикара подтверждают схему окисления
сернистого газа атомарным кислородом. Прямое фотоокисление S02
при концентрациях, близких к реальным A0~2 млн), не играет
значительной роли в образовании аэрозолей. В частности, не за-
S02 к
98
99
мечено образования частиц с г > Ю мкм при освещении с Л >
> 300 нм смесей SO2, O3, N2 и водяного пара. Ж.Брикар с
сотрудниками приходит к выводу, что для образования аэрозолей
из сернистого газа необходимо наличие еще одного или несколь-
нескольких газовых загрязнителей. Специальными опытами было показано,
что роль такого загрязнителя-активатора может играть, например,
NO2 при концентрациях 0,5-2 млн , являющийся фактически ис-
источником атомарного кислорода при фотодиссоциации (квантовый
выход при Я 4 366 нм примерно равен единице). Вероятно, в ра-
работах, подтверждающих схему Ф.Блэкета, реагирующие газы не бы-
были в достаточной степени очищены от примесей [123, 125].
Достаточно полный обзор реакций SO2 с углеводородами и
другими органическими соединениями сделан в работах [ИЗ, 126,
153]. Наблюдается образование сульфиновой <С4Н10502),дисульфи-
новой и дисульфоновой кислот в виде бесцветного или светложел-
того дыма. Аэрозоли возникают в смеси SO2 с ацетоном, этаном,
пропаном, л-бутаном, изобутаном и п -пентаном в отсутствие ¦
кислорода О2 при освещении с Я > 230 нм. Для SO2 - это реак-
реакция первого порядка с квантовым выходом, увеличивающимся с чис-
числом атомов углерода в соответствующих предельных углеводородах
от 0,006 для метана до 0,26 для пентана. В случае олефинов
квантовый выход ниже: от 0,02 до 0,05. По другим измерениям ве-
величины квантовых выходов несколько меньше: от 0,0Т для метана
и 0,07 для изобутана.
Продукты взаимодействия для углеводородов с сернистым га-
газом с помощью хромотографа были идентифицированы как сульфоно-
вая кислота и тиосульфонат и наблюдалось образование радика-
радикала HSO2-
Имеющиеся данные свидетельствуют о сильном взаимодействии
я-электронов олефинов с электрофильной молекулой SO2 C В^)•
Г.Бадкок с коллегами рассматривает два альтернативных механиз-
механизма реакции
3so2
3SO,
RH
RH
RSO2H ,
SO,H + R.
Предпочтение отдается второму механизму. Олефины частично пре-
препятствуют образованию аэрозолей при фотоокислении SO2 , если
100
концентрации SO2 и углеводородов малы. Так как возбужденный S02
более эффективно реагирует с кислородом, чем с углеводородами,
то для реакций SO2 с углеводородами большее значение имеют ре-
реакции SO2 в невозбукденном состоянии с органическими свободны-
свободными радикалами. Этот механизм окисления SO2 через перекисный
бирадикал вдет при низких концентрациях реагентов в системе
SO2- O2- RH [135]:
SO2 + 03 + RCH = HCR
-SO3 + 2RHCO
h2so4.
Предельные углеводороды и высшие олефины могут образовывать со-
сополимеры с SOj, например, сополимер 2-пентана с SO2, сополи-
сополимер циклогексана с S02- В продуктах реакции SO2 с углеводоро-
углеводородами обнаружены карбонильные соединения и окисленные органичес-
органические функциональные группы (альдегиды, кетоньг, органические кис-
кислоты). Слабые карбонильные полосы в области 5,7 мкм обнаружены
по ИК-спектрам в реакциях SO2 с л-бутаном, изобутаном и нео-
пентаном. Однако в большинстве исследований аэрозолей не обна-
обнаружено, а в качестве конечных продуктов указываются альдегиды
и кетоны, которые могут образоваться в ходе реакций озона с уг-
углеводородами.
Общий вывод сводится к тому, что реакции смеси SO2 - RH
не играют большой роли в окислении SO2 и образовании аэрозо-
аэрозолей. Несмотря на то, что SO2 не способствует образованию и
росту ядер конденсации для органических аэрозолей, однако, если
в смеси присутствует NH3, то наблюдается довольно интенсивное
образование аэрозолей, особенно при наличии в системе циклогек-
циклогексана и сс-пинена. Преимущественным химическим соединением в
аэрозольном веществе в этом случае является сульфат аммония. Су-
Существенное увеличение скорости реакции в смеси олефинов и NO2
наблюдается при добавлении SO2. Такой же эффект наблюдается
для ацетиленов и ароматических углеводородов, но для последних
эффект увеличения скорости образования продуктов реакции выра-
выражен слабей. А.Альтшулер отмечает, что эффект усиливается, если
в реакции участвует несколько ароматических соединений, напри-
например толуен и ксилен - вещества, характерные для автомобильных
выхлопов. Основными конечными продуктами реакций такого рода
смесей являются озон, альдегиды, кетоны, СО , органические нит-
101
раты и в небольшом количестве эпоксиды, алкоголи, зстеры, перо-
ксиды. В реакциях с участием низших углеводородов, в частности
олефинов, образуется серная кислота и нитритные вещества [ИЗ,
126, 153].
Количественных оценок влияния добавления SO2 на скорости
рассматриваемых реакций практически нет, так же, как и оценки
скоростей окисления SO2. Скорость реакции окисления SO2 увели-
увеличивается с уменьшением концентрации SO2, однако количество об-
образующегося сульфата пропорционально концентрации SO2 и толь-
только 15-35Й переходит в сульфаты. Добавление S02 в систему RH +
+ NO2 подавляет образование повышенной концентрации оксидантов,
а В.Вильсон и К.Леви утверждают,что в сухом воздухе добавление
SO, в диапазоне концентраций 0-0,75 млн"'1- в систему 1-бутен
D млн ) -N0A млн ) уменьшает скорости конверсии [126, ISO].
Присутствие озона в смеси SO2, N0^ и некоторых углеводоро-
углеводородов может приводить к образованию серной, сульфиновой и сульфо-
новой кислот даже в темноте (например, из 1,5-гексадиена, цик-
лопентана, циклогексана), и скорости реакций сильно зависят от
концентрации озона. При освещении образуются те же продукты.
Практический интерес представляют исследования влияния ве-
величины относительной влажности на скорость реакции и выход ко-
конечной продукции для смеси SOj-NO^—RH . Спектральные дан-
данные об аэрозолях, образующихся в системе 2-метил-2бутен + NO2+
+ SO2 при величинах относительной влажности от 10 до 50%, пока-
показывают, что для меньших влажностей наблюдается относительно га-
лая концентрация серной кислоты, значительно большая концентра-
концентрация нитратных соединений и более четкая идентификация химичес-
химических соединений, чем в случае 50-процентной влажности. Однако
существенного влияния увеличения влажности на скорость фото-
фотоокисления SO2 в большинстве исследований не обнаружено. По
измерению рассеяния от аэрозолей было установлено, что увели-
увеличение влажности приводит к уменьшению коэффициента рассеяния.
Вероятно, это следствие разбавления образующейся в результате
реакции кислоты и выпадения капель на стенки. Увеличение влаж-
влажности усиливает ингибиторный эффект малых концентраций SO2
B,0 млн )на образование аэрозолей.Возможен'также рост частиц
в результате коагуляции и конденсации. Влияние влажности на
скорости реакций существенно зависит от того, какое органичес-
органическое соединение участвует в реакции, причем наблюдается законо-
закономерность замедления реакции окисления в сухой атмоофере. Силь-
Сильная зависимость максимального числа образующихся аэрозольных
частиц Nmax от концентрации SO2 и величины относительной вла-
влажности / выражается эмпирической формулой
^пиис* C<SO2>/2<H2O).
При добавлении СО в систему RH- NO^-SOj— H20 сущест-
существенным моментом является окисление СО гидроксильным радикалом
с образованием гидропероксидрадикала как промежуточного продук-
продукта реакции. Численные расчеты показывают, что при этом скорость
исчезновения RH уменьшается незначительно (при начальной кон-
концентрации СО, равной 100 млн), но увеличивается скорость та-
таких конечных продуктов, как пероксиацетил, нитрат, озон, фор-
формальдегид.
Таким образом, учитывая состав атмосферы для газофазного
окисления SO2 с последующим образованием сернокислотннх аэро-
аэрозолей, из возможных реакций можно ввделить следующие:
з$О2
М — SO3 + Нг0 -* H2SO4 ,
см
3-
SO2 + ООН —> SO3 + ОН -^ H?SO4 ,
К » (9± 1,8) ¦ 10~16 смъ- иояехул-*- с'1 при 300 К,
3so2 + он + м —> hoso2 —> h2so4 ,
К= 2Ю'асм3- молекул-*-с'1 при [ М ] е 2,2 Ю1'см'3,
SO2 + 03 + RCH = HCR
— S03 + 2RHCO + НгО — H2SO4,
К = 6,6-Ю'17 см3, молекул'1, с'1 при концентрации
олефина о, 1 млр8 '%
S02 + R00 S03 + R0 + Н20 > H2SO4 ,
К~9-Ю~10 см3- молекул'1.с'1 для СН3ОО .
103
А. Биренцвайг и др. [119] еще больше ограничивает количество
реакций окисления SO2. Они полагают, что окисление SO2 в ат-
атмосфере производится радикалами НО2, ОН СН3О3, а озон идет
на образование радикала ОН.
§ 2.2. Химические реакции соединений азота
Существенную роль в процессах образования аэрозольного ве-
вещества в атмосфере играют соединения азота. В предыдущем разде-
разделе отмечалась роль NH3 в процессах окисления SO2 и образова-
образования сульфата аммония. Роль окислов азота в процессе окисления
сернистого газа менее определенна. Значительно важней, что в
результате химических и фотохимических превращений с участием
окислов азота, кроме азотной кислоты и ее солей в качестве про-
промежуточных продуктов, образуются некоторые реакционноспособные
молекулы и радикалы, которые, в свою очередь, участвуют в соз-
создании аэрозольного вещества. Некоторые из образующихся молекул
обладают токсичными свойствами, например пероксиацетилнитрат
(ПАН). Кроме того, окислы азота активно взаимодействуют с озо-
озоном, уменьшая его концентрацию в атмосфере.
Для окислов азота NO2 и N?0 важна возможность фотодиссо-
фотодиссоциации при облучении их коротковолновой солнечной радиацией, до-
достигающей нижней стратосферы и даже тропосферы:
N2O + Л-Э < Л 4 337 нм) -*¦ Ы2 + OAi>) , B.И)
с квантовым выходом около единицы;
N2O + Л-5 < Л < 250 нм ) — N0 + N < * $ ) ,
210 нм)
N2
0AS),
2
N02 + ho ( A -v 398 нм) -*• NO(X2jt) + 0CP)
с квантовым выходом, равным единице. Свет с Я > 398 нм также
может приводить к дисооциации молекулы N02,вероятно, воледст-
вие влияния вращательной энергии на этот процесс. Константа ско-
скорости реакции B.11) равна 3-Ю0 с. Образующийся атомарный
104
кислород реагирует с N2O и дает молекулы N0. Однако скорость
фотодиссоциации N20 в тропосфере при освещении солнечным ове-
том меньше 1'Ю* с. В этом случае вклад образующихся 0('D)
и N0 по реакции B.1!) в фотохимические тропосферные процеосы
незначителен. Фотопроцессы для окиси азота N0, а также высших
окислов N03 и N2O5 изучены слабо. Происходят они при более
коротковолновом излучении, чем для N20 и N02, и не играют су-
существенной роли в химических и фотохимических реакциях образо-
образования аэрозолей.
Высшие окислы являются промежуточными продуктами реакции
низших кислот. Молекула N03 появляется в результате реакции
озона с двуокисью азота. Эта реакция протекает довольно медлен-
медленно (К = З+7-iO^1'7 см3/(мс-лекул«с)). В дальнейшем молекула N03
участвует в двух быстрых темновых реакциях с другими окислами
азота и в реакциях с органическими соединениями:
N03 + NO - 2N02,
N03
N02
Реакции N03 с углеводородами могут играть важную роль в фото-
фотохимических процессах образования смога. Так, константа реакции
с 2,3-диметил-2-оутаном К = 101 см3/(молекул-с); для реакции
с этиленом - К = 9,3-10~16 см3/(молекул-с) [171, 215].
Молекула N2O5, образующаяся в основном в результате тем-
новой реакции окислов азота, вступает в реакции с первичными
загрязнителями (гетерогенный процесс) и с основными газовыми
компонентами воздуха. Большинство реакций медленные. Важней-
Важнейшей реакцией для образования, аэрозольного вещества является
взаимодействие N2O5 и конденсированной на аэрозольных части-
частицах воды с образованием азотной кислоты и в дальнейшем нитра-
нитратов. Наиболее реакционноспособной молекулой из окислов азота
является окись азота N0 - с химической точки зрения свободный
радикал, имеющий нечетное число неспаренных р-электронов. В
олучае реакций с радикалами N0 образует нейтральные молекулы,
а в случае реакций с нейтральными молекулами - новые радикалы.
Существенны следующие гомогенные реакции с кислородом:
2N0 + О,
2N0,
NO + Ог
NO
O3 —
N02 + О ,
N02
2 + O2 .
Предполагается, что в этих реакциях принимают участие молекулы
СО,Н, Н02, ОН и формальдегид. Реакция N0 с 03 важна для
стратооферного озонного слоя. Молекула N02, поглощая квант све-
света, диссоциирует с образованием N0, но поскольку процесс фото-
фотоокисления N0 в N02 идет быстрее, то концентрация N02 при
освещении медленных газовых смесей увеличивается. Окись азота
вместе с двуокисью азота реагирует с молекулами воды:
N0
Н20 -> 2HNO2
Константа скорости этой реакции при комнатной температуре
1-1СГ4 м /(молекул-с). В нижних слоях атмосферы эта реакция
может иметь продолжение:
HNO, + ho
N0 + ОН
и служить источником радикалов ОН, которые играют важную роль
в атмосферной фотохимии. Особенно важна эта цепь реакций в
процессах трансформации при наличии солнечной радиации [165,
169, 171].
Молекула КЮ2, несмотря на свою радикальную природу (у нее
нечетное количество р-электронов), при комнатной температуре
практически не вступает в реакции с 02,^, С02, СО и други-
другими нейтральными молекулами и некоторыми радикалами. Поэтому
роль двуокиси азота в фотохимии атмосферы состоит в ее транс-
трансформации на свету в N0 и 0('Р) и создании тем самым возмож-
возможности для появления различных промежуточных и стабильных про-
продуктов в реакциях N0 и 0CР) с другими молекулами.
Из гетерогенных реакций окислов азота исследованы следую-
следующие реакции в водных растворах;
+ н2о
2H+ + 2
— 2H+ +
2N0
2,
NO + NO2 + H2O -
2NO2+H20 ==> H+ + NO" + HONO,
106
HONO
NO2 + O3
2NO2 + H.
HNOS04 н
3HNO2 5
ONO2 + h
+ OH
(BH2
2so4
1- H20
1,0 -
"
0)
5==
HNO3 +
- H +
н2о ¦
HNOSO,
HNO2 •
2NO +
+ N0"
t- N02 ,
+ o2,
\ + MN03'
+ h2so4,
H20,
+ ROH .
В работе [129] обсуждается гетерогенная реакция N02 с
частицами морской соли NaCl.B результате этой реакции образу-
образуется хлористый нитрозил NOCI, обладающий сильной коррозионной
способностью.
§ 2.3. Окисление и полимеризация органических соединений
Среди множества органических соединений, присутствующих в
атмосфере, с точки зрения фотохимии наибольший интерес предста-
представляют такие соединения, которые фотодиссоциируют при облучении
коротковолновым солнечным светом с образованием активных ради-
радикальных продуктов. К таким органическим веществам относятся
альдегиды и кетоны.
Фотодиссоциации альдегидов. Фотодиссоциация формальдеги-
формальдегида, который составляет 40% от общего количества альдегидов в
загрязненной атмосфере, идет при облучении с Я 4370 нм с об-
образованием атомов водорода и формильных радикалов:
I—- н + нсо i
НСНО + ho 1
н2 + со
II
Сумма квантовых выходов обеих реакций равна единице, при-
причем для Л = 313 нм квантовые выходы примерно равны. В силу
малой активности молекулярного водорода и окиси кислорода су-
существенную роль играет только первая реакция. Формильные
107
радикалы реагируют с молекулярным кислородом:
н + о2 + м —> но2 + м .
нсо
но2 + со ,
образуя молекулы НО2, которые играют важную роль в образова-
образовании фотохимических аэрозолей.
Фотолиз высших альдегидов дает формильные радикалы (НСО)
в реакции разложения первого типа при облучении с Я < 340 нм:
RCHO +
R + НСО.
Например, фотодиссоциация ацетальдегида при облучении с Я =
= 313 нм:
сн3сно
сн3 + нсо.
Метильные радикалы инициируют следующие реакции:
сн,
сн3ог н
сн3о
м
N0
сн3о2
м ,
СН3О + N0
2,
но2 + нсно,
в ходе которых происходит образование Н0г и конверсия N0 в
N02. Продукты фотодиссоциации альдегидов могут вступать в даль-
дальнейшие реакции с органическими загрязнителями воздуха. Фотовоз-
Фотовозбуждение алифатических альдегидов при облучении с Я i 340 нм
на воздухе приводит к реакциям образования промежуточных про-
продуктов с алифатическими и ароматическими углеводородами. Вве-
Введение окислов азота в состав реагентов увеличивает скорости
реакции примерно в 10 раз [165, •ТбЭ, 171, 215].
Важность фотолиза альдегидов для образования фотохимичес-
фотохимического смога подтверждена количественными данными по скорости об-
образования HOj при фотолизе НСНО, полученными Дж.Калвертом с
сотрудниками.
Фотодиссоциация кетонов. Исследование продуктов фотодис-
фотодиссоциации кетонов показало, что они могут быть как в молекуляр-
молекулярной, так и в радикальной форме. Например, диссоциация ацетона
происходит по следующим альтернативным механизмам:
108
СК.СОСН, +
з з
I >
—
| ,.
СН,СО + СН,
+ со
I
II
Первичный квантовый выход фотодиссоциации ф = 0,9, причем
<Pi/(<Pi +Ф2)= 0,07 при возбуждении А = 313 нм.
Реакции радикальных продуктов фотолиза. Важность учаотия
радикалов в фотохимических процессах в загрязненном воздухе
впервые была высказана П.Лейтоном и подтверждена работами по
математическому моделированию процессов в смоговых камерах. Ос-
Основную роль среди этих реакций играют реакции с углеводородами
и окислами азота. В присутствии кислорода реакции алкильных и
формальных радикалов с углеводородами маловероятны. С большей
вероятностью идут реакции с кислородом, в результате которых
образуются пероксиальные, включая НО^, и пероксиациальные ра-
радикалы:
R+ О,
RO2.
RCO
RCOO
II
О
Образовавшиеся пероксирадикалы могут реагировать с атмосферны-
атмосферными загрязнителями.
Механизм и кинетика реакций Н02 с углеводородами иссле-
исследованы мало. На основании исследования продуктов реакции С?Н4
С3Н6 и г - С4Н4 с Но2 можно думать, что первым шагом этих
реакций является присоединение Н02 к. двойной связи:
=сн2
снз
С
ООН
с
н
I
— с - н
н
I
с
I
ООН
— с — н
Для объяснения образования конечных продуктов предполагается
изомеризация и распад промежуточного продукта:
сн3 н
сн,
н
сн3- с-с-н-сн2-с- сн2о
сон
НСНО
ООН ОН
Сложности в изучении таких реакций усугубляются трудно учиты-
учитываемым вкладом процессов на стенках реакционного сосуда.
Пероксирадикалы реагируют с окислами азота по следующим
схемам:
ROO' + NO—-NO2
R0',
RCO"
RCOO" + NO — NO,
О О
RCOO" + NO2—> RCOONO2 ,
о о
RCOO' + N0 +(О2)—- R00' + N0
С02
Появляющиеся в результате этих реакций радикалы могут вступать
в реакции:
R0" + 02 — RA)CHO + НО" ,
R0" + N02 —> RONO2 ,
RO' + NO —> RONO ,
RCO"
II
О
CO,
RO*
Продолжением рассмотренных реакций может быть цепь реакций
RCO -^ RCO0'-^-RCO"-^-R*-^- RO' ^* RO* •^>HO' ^* ОН .
II II 2 z
О О
Таким образом, за время жизни R- и RCO"- радикалов боль-
большое количество молекул NO перейдут в N02, которая, поглощая
коротковолновое солнечное излучение ( А. = 295+430 нм), дает
начало реакциям, приводящим к образованию фотохимических аэро-
аэрозолей [130, 171, 215].
Вторым важным следствием этой цепи реакций является обра-
образование гидроксильного радикала НО2. Когда концентрация N0
«О
мала, пероксиацильные и пероксиалкильные радикалы могут взаимо-
взаимодействовать с NO2, образуя пероксиацетилнитраты (ПАН) и алкил-
пернитраты <ROON02), весьма токсичные компоненты фотохимическо-
фотохимического смога:
N0. -
СН,СОО
CK.C00NO, (ПАН).
о о
Образование токсичных компонентов ПАН и ППН (пероксипропил-
нитрат) наблюдалось экспериментально при освещении смеси кето-
нов с ЬЮ2. ППН образуется из дизтилкетона,вероятно,через сле-
следующие реакции:
О
II
сн3сн2с*
о
я
СН3СН2СОО
о
II
—*¦ сн3сн2соо*
о
ch3ch2coono2 .
Исследование в модельных условиях фотоокисления азометана пока-
показало, что реакция свободных метилпероксиальных радикалов с N0
дает N02 и СН3<3 :
СН3ОО* + N0
СН3О*
N02.
Выводы о роли основных классов свободных радикалов в фотохими-
фотохимических реакциях в загрязненной атмосфере сводятся к следующему.
Алкильные радикалы (R) быстро присоеди-
присоединяют 02 с образованием пероксиалкильных радикалов ( R00).
В дальнейших реакциях участвуют в основном радикалы R00 .
Ацильные радикалы <RCO), как и алкильные,
быстро присоединяют кислород, образуя пероксиацильные радикалы
"^°°, которые и включаются в механизм образования фотохимичес-
О
кого смога.
Пероксиалкильные радикалы( R00)
реагируют с N0, ускоряя его конверсию в N02.
_ / RCOO\
Пероксиацильные радикалы! и 1мо-
гут реагировать с N0, а при малых количествах N0 с NO2 > об-
образуя стабильные пероксиацитилнитраты (ПАН).
Ацилатные радикалы (*Ч° \ получаются в
T>e3Y.n>iaie реакции NO и пероксиацильных радикалов; нестабиль-
нестабильны и распадаются с образованием алкильных радикалов и СО2.
Алкоксильные радикалы^ИО), включая
ОН, получаются в результате реакций пероксиальных радикалов с
N0 и озона с олефинами. Гидроксильный радикал ОН образуется
также в результате превращений неорганических соединений:
H0NO + Л-3 < Я < 400 нм) - ОН + N0 ,
Н20 > 2 ОН
Н,0 + Л* ( Л < 370 нм ) +
20Н.
Это единственный радикал, обнаруженный экспериментально в натур-
натурных условиях в концентрации л- 10 см . Высокую реакционную
способность ОН-радикалов иллюстрирует их реакция с типичным
насыщенным углеводородом - пропаном:
н2о
с3н7
(А"= 5-Ю3 л-моль-с).
Радикал С3Н? реагирует с молекулярным кислородом, образуя пе-
роксирадикал, который способен переводить N0 в N02. Реакции
ОН-радикала с другими углеводородами также приводят к образо-
образованию перекисных радикалов. Схематически это можно представить
следующим образом:
ОН + олефины-* R00 + альдегиды {К - 2,5*10 мольбе),
R0O+H20(K=8*10 моль• с ),
3 1
1
~3
)
2
{К = 3,8*Ю~3 мольбе).
ОН + ароматические углеводороды.
ОН + парафины -*¦ R00 + Н20
Результаты исследований по кинетике реакций ОН-радикала
свидетельствуют о высоких скоростях таких реакций. В частности,
это подтверждают исследования кинетики реакций ОН-радикала с
ароматическими углеводородами: толуолом, ксиленом (диметилбен-
зол) и триметилбензолом.
§ 2.4. Реакции кислорода и озона
Почти все химические процессы образования аэрозольного ве-
вещества в атмосфере связаны с реакциями окисления. Роль невоз-
бужденного молекулярного кислорода в этих реакциях незначитель-
незначительна. Более существенна роль в такого рода реакциях атомарного
И2
кислорода, озона и возбужденного молекулярного кислорода. Про-
Процессы образования аэрозольного вещества ведут к выведению кис-
кислорода и озона из газовой фазы и, следовательно, изменяют ес-
естественный ход химических реакций в атмосфере и, возможно, ве-
величину концентрации атомарного кислорода и озона. Рассмотрим
последовательно важнейшие состояния кислорода в атмосфере и воз-
возможные реакции с их участием.
Атомарный кислород 0CР) образуется в
основном при фотодиссоциации окиси азота:
N02 + ho < А < 43Онм) — N0 +0CЯ) (К= 0,266 мин).
Атомарный кислород в возбуж-
возбужденном состоянии О (ID) может образовываться с
большим квантовым выходом в загрязненной атмосфере в результа-
результате фотолиза 03 светом с Л < 308 нм. Другой возможный путь их
появления - фотолиз NO2. Квантовая эффективность этой реакции
при Я = 213,8 нм близка к единице: O(l?>2)/ N2 > 0,97.
Дезактивация O(Ii?2) в тропосфере происходит в основном в ре-
результате физического тушения при столкновениях с N2,O2,Xe,
СО,СО2 с переводом атомов O0D2) в невозбужденное состояние
ОCР). Существенным является и химическое тушение синглетно-
возбужденного атомарного кислорода, играющее важную роль в фо-
фотохимических реакциях.
Озон Од возникает в результате быстрой реакции ато-
атомарного кислорода с молекулярным:
М -* 0г
М
(К«2-10~13 моль-с).
02 + М * 0г
Возможен и другой путь образования озона - в результате реак-
реакции кислорода и бирадикалов типа [146]
RCHOO' + О,
RCHO'
О3 .
В реакциях образования фотохимических аэрозолей может прини-
принимать участие синглетный кислород O2(I^g.) и O.OZg).Известны
следующие процессы, приводящие к образованию O2A2g):a) пря-
пряфб
мое фотовозбуждение:
8 192Z
?g) I
757.8 им
M » 1262^9 км
o2Oi+)
О2 + М +
M ;
б) перенос энергии в газовой или гетерогенной фазе от ор-
органических примесей или от NOg* и SO2* •
в) фотолиз озона: О3 + Ло * *31° НЕ (
г) экзотермические химические pearaniH:NO ¦
д) гидролиз пероксиацетилнитрата:
2ОН
Н20
02 •
3^ 3^
0ONO2 О*
Эти процессы имеют следующие скорости образования: прямое
фотовоэбувдение - 9,2-Ю" млн -ч ; фотолиз озона 0,1 млн -ч,
перенос энергии - 80 млн «ч , хемивозбувдение - 1140 млн~^ч~?
Тушение возбужденного синглетного кислорода O2(^&g) может про-
происходить либо при переносе энергии на подходящий тушитель, об-
обладающий ниэкорасположенным уровнем энергии ( < 7880 см),ли-
см),либо через образование в момент столкновения комплекса с перено-
переносом заряда. Последние представления были развиты Е.Огрызло с
сотрудниками на основании наблвдавшейся корреляции между кон-
константами тушения в газовой фазе и потенциалами ионизации туше-
тушения. Эффективными тушителями O2<IAg)no механизму образования
комплекса с переносом заряда являются амины и серосодержащие
соединения.
Реакции атомарного кислорода ОCР). Первичные реакции с
органическими соединениями схематически можно представить сле-
следующим образом:
олефины + 0(ъР) —- R + RCO,
ароматические углеводороды + О(зр)—> R + ОН,
парафины + О (Щ —> R + ОН ,
альдегиды + О CР) —> RCO + ОН .
114
Считается, что взаимодействие с ОCР), а не с озоном яв-
является первым шагом в цепи реакций фотоокисления органических
веществ. Величины скоростей реакций с олефинами и ароматически-
ароматическими углеводородами указывают на электрофильный характер взаимо-
взаимодействия, что проявляется в присоединении кислорода к двойной
связи. Например, для реакций О(*Р)с цис-2-пентеном предлагает-
предлагается следующий механизм.
Присоединение ОCР) к двойной овязи с образованием триплет-
ного бирадикала
СН,
, С|НВ
с=с
I I
н н
ОCР)
Время жизни бирадикала оказывается достаточно большим для
того, чтобы произошла изомеризация. Образовавшиеся цис- и транс-
трансизомеры либо распадаются, либо стабилизируются после отвода
энергии при столкновениях.
В результате внутримолекулярной миграции атома кислорода
может образовываться возбужденный метил-2-пропилкетон
СН,
с2н
2н5
w
~К -[
сн3
ссн2с2
¦}•<
распад
сн,ссн2с2н5
"h
н2с2н5
И "О" Н
о
Возбужденный кетон распадается или стабилизируется.
Внутримолекулярная миграция алкильной группы приводит к
образованию возбужденного 2-метилбутанола, который может рас-
распадаться или стабилизироваться:
СН,
о* н
[нсснс.н,!*-—
1 II J I ¦
распад
СН,
.НССНС.Нс
II 2 5
0 о
Механизм реакции О{3Р) с ароматическими углеводородами
изучался в основном Р.Цветановичем [139]. В результате реакций
ОCР) с бензолом и толуолом были обнаружены устойчивые продук-
t:i: вода, окись углерода, фенол (из CgHg) или крезол (из
C7Hg)n налет на стенках кюветы, представляющий собой полимер-
115
il [природу и
ное вещество. Полимерный продукт имеет алифатическую.;^ этого
содержит функциональные группы СНО, ОН.СО.Образовя»Ифатичес_
продукта представляет интерес, потому что сходные ''¦ГМн0ГИе из
кие соединения были обнаружены в натурных услозиях. ;| и ве_
этих соединений содержат пять или шесть атомоз углэррх 'ика_
роятно, образуются в результате присоединения
лов типа ОCР) или ОН к ароматическому кольцу, поЬЩ j^HCOgfl"H_
дует раскрытие кольца, реакции с О2 и т.д. Налриме^,,
нение ОCР) к бензольному кольцу:
О* :
Раскрытие кольца может происходить по схеме I
-Н /Н:
— СН == СН-СН=СН-СН=С :
-н 01 '
4 0" ; Нелетучие
Последующие реакции ненасыщенного бирадикала дают
продукты (полимеры). Может быть предложена альтерня'.^
ма: » /Ч^-ОК
ивная схе-
JT\
:,;рно
1+со
, тушение
Реакции атомарного кислорода О ('I»). Химическое^ r об
синглетно-возбужденного атомарного кислорода привод^ присут_
зованию реакционноспособных радикалов. Так, пары вол
r\n n \ треимущест-
ствующие в атмосфере .удаляют 9% атомов Ш1 D2>
венно в результате реакции 2ОA?>2) + Нг - 2ОН' реакция
Следовательно, при высокой концентрации озока ц
может быть важным источником гидроксильных радикя.?то3
¦ j inp е в ращ в—
модействии с предельными углеводородами вероятность,
ния OAD9) в ОCР) низка. Однако с высокой эффя,-'
* . .11.66 важная
осуществляются химические реакции двух типов: наибо* г-С-
иэ них реакция включения 0{:D2) в С-Н -связх (но , „
связи) с образованием колр^чтельно-возбужденнкх "го'г
тов с избытком энергии в (9,7+11,0)-10
не
буж
Дж.
гог.
В г,й
спирты распадаются в зависимости от внешнего давления и числа
степеней свободы образовавшейся молекулы. В частности, в газо-
газофазной реакции O(!D) с циклобутаном образуется "горячий" ко-
колебательно-возбужденный циклобутанол в основном электронном со-
состоянии, мономолекулярно распадающийся на этан, ацетальдегид
(константа скорости реакции 1,2-10И с). Время жизни циклобу-
танола 8,3-10 ^ с, и его дезактивации происходит практически
при каждом соударении.
Второй этап реакции OC^D) - отрыв атома водорода от предель-
предельного углеводорода с образованием гидроксила ОН и алькильного ра-
радикала (?. Вероятность этих реакций в два-три раза ниже; O(Z>)
с готовностью вступает в реакцию с фторированными хлоралканамк,
содержание которых в тропосфере в виде аэрозолей значительно.
Так, константы скорости для CFCl 3, CF2Cl2, CHF2Cl CF2CICF2CI
равны 3,5-Ю11; 3.2-1011; 2Д-1011; и 2.0-1011 л-мол-с со-
соответственно. Основными продуктами в реакции с CFC1, и CF2Cl2
являются COFCI и COF2. Вероятно, одновременно образуется и мо-
молекулярный хлор. Взаимодействие 0(^0) с фторированными хлор-
алканами в тропосфере несущественно, поскольку содержание O^D)
невелико. Так как в стратосфере концентрация О(Ф) значительно
выше, то эта реакция может иметь значение, особенно если учесть,
что образующийся в результате ее молекулярный хлор под жест-
жестким ультрафиолетом в стратосфере диссоциирует и может вызвать
цепную реакцию
ci2
o2-*cio+o-ci+o2.
Реакции озона. Сравнение констант скоростей реакций озона
с различными газовыми компонентами атмосферы показывает, что
наиболее важными для процессов трансформации газов в аэрозоль-
аэрозольное вещество реакциями являются реакции озона с окислами азота
N0 и N02 и с олефинами (табл.20). Когда концентрация N0 в ат-
атмосфере невелика (для фотохимических смогов это наступает пос-
после полудня), определяющими становятся реакции озона с олефи-
олефинами. Эмпирические скорости образования аэрозолей из разных
олефинов следующие? пропилен - B,7 + 0,Э)-108, 1-бутен -
D,6 + 1,4)-108, 1-гексен - A,6 + 0,5).107, 1-октен -
D,9 + 1,8)-107, 1-децен - A,0 + 0,5)-10~36, 1-дедецен -
117
Таблица 2С
Скорости реакций озона с окислами азота и некоторыми
органическими соединениями при комнатной температуре
Химическое соединение
Константа скорости
^11
Относительная
скорость
Окись азота
Двуокись азота .,
Углекислый газ ..
Этилен
Пропилен
Изобутен
1-бутен
Тетраметилэтилен
Триметилэтилен ..
Ацетилен
Ацетальдегид
Метан
Толуол
Цис-2-бутен
Транс-2-бутен ...
1-гексен
1-пентен
Изобутилен
2-гексен
A,1 + 0,2)-104
19 + 3
0,6-Ю
0,93 +.0,09
7,5 + 0,6
8,2
6,1
48
10
906
296
52
20
0,72
7,2 • 10'
8,1
113,1
-3
1-3
«2,8 + 0,6
2,6 + 1,2
2,9 f 2,1
2,2 + 3,8
1,0
0,8 + 1,1
0,8 * 2,0
2,9 + 1,9
A,1 + 0,3)-10 35, щклогексен - D,9 + 1,8)-10 37 мкг-моль
х м~3«с .В реальной атмосфере скорости реакции олефинов с озо-
озоном могут сильно отличаться от приведенных.
Обнаруживается сильная зависимость скоростей реакций от
концентрации реагентов, температуры и присутствия посторонних
примесей, в частности кислорода и двуокиси серы. Например, по-
повышение температуры от 225,2 до 363,1 К приводит к 2-17-крат-
2-17-кратному увеличению константы скоростей реакции с алканами в зави-
зависимости от их типа.
118
В присутствии кислорода константы скоростей реакции пони-
понижаются. По-видимому, кислород захватывает свободные радикалы,
образующиеся в первичном фотохимическом акте реакции ,и предот-
предотвращает их взаимодействие с озоном. При наличии примеси SO2 в
смеси олефинов с озоном эффективно образуются сернокислотные
аэрозоли. Механизм этой реакции до конца не установлен. Темпе-
Температурная зависимость константы скорости и влияние кислорода на
скорость реакции обнаружены для взаимодействия озона с диенами.
Озон эффективно взаимодействует со сложными углеводорода-
углеводородами (>Cj0) - монотерпенами, которые в довольно больших коли-
количествах выделяются лесной растительностью. Константы скоростей
реакции для различных терпенов колеблются от 1,6*'1О " до 1,бх
xICT моль-с . Механизм реакции озона с углеводородами изу-
изучен недостаточно. Предполагается, что первичным продуктом ре-
реакции является озонид или мольозонид - продукт присоединения
озона к молекуле углеводорода, который распадается на биради-
кал (или, возможно, цвиттерион) и карбонильное соединение:
R, R,,, R. ,0 R.
\
ш
С = С
С
\
У° Ч/У*/
IV
IV
(мольозонид)
(бирадикал)
Дальнейшими превращениями бирадикала объясняют большой набор
продуктов этих реакций. Например, в случае пропилена обнаруже-
обнаружены формальдегид, вода, метанол, метан, СО и СО2,которые могут
возникнуть в ходе превращений
^ сн3он + со
сн3-сн-о-о
н2о
сн4
сн2
со2
о
Первичный озонид может трансформироваться путем отрыва атома
водорода. Например, для случая 1-бутена возможна следующая схе-
схема превращений:
119
,0-0
сн3сн-снсн2он
•0-0
сн2сн2снсн2он
/ООН
сн3сн2снсно
сн3сн3сн2сч
¦ ООН
• сн2
Некоторые из этих промежуточных соединений (например, кетогид-
ропероксид транс-2-бутена) обладают высокой стабильностью. Од-
Однако, как правило, эти продукты нестабильны и при распаде дают
альдегиды, кетоны, СН2,СО и большие количества атомарного ки-
кислорода.
Хемилашнесценцию в реакции озона с олефинами, зависящую
от структуры олефина , связывают с распадом диоксиэтанов, обра-
образующихся при отрыве атома водорода.
Гетерогенные процессы распада молекул озона изучались в
работах [128, 156]. Основная трудность оценки роли аэрозолей
в выведении озона из стратосферы заключается в отсутствии дан-
данных по эффективности каталитического действия вещества аэро-
аэрозольных частиц на скорость распада молекулы озона, т.е. отно-
отношения числа активных столкновений молекул озона с веществом к
общему числу столкновений ($ ). В лабораторных исследованиях
получены значения о, изменяющиеся для различных веществ от
10 до 101 (табл.21). Численные оценки изменения концентра-
концентраций некоторых газов в стратосфере в результате гетерогенных ре-
реакций озона и атомарного кислорода были выполнены Крутценом и
др. с использованием модельных представлений о структуре стра-
стратосферных аэрозолей (табл.22) [128].
Реакции синглетного кислорода О^Дд^.Характерными яв-
являются следующие:
а) присоединение к олефинам, имеющим избыточную электрон-
электронную плотность на двойной связи с образованием 1,2-диокси-
этанов:
120
Таблица 21
Зависимость доли активных столкновений J и времени
жизни х для систем озон - А12О3 и других компонентов от
температуры
Параметр
i
т, мин
Iga -102, см-мин
Т,К
295
2.3-I0
3 + 0,2
3 + 0,3
2 + 0,3
3 + 0,3
5 + 0,5
250
5.4-10
2,2 + 0,2
2 + 0,3
2 + 0,3
2 + 0,3
2 + 0,3
209,5
I.5-I0
1,5 + 0,1
5 + 0,5
2 + 0,3
4 + 0,5
2 + 0,3
Компо-
Компонент
А12О3
Алюмо-
си.-игат
MgO
ПО2
\
II
О-о
R»~ Riv
б) окисление олефинов, содержащих алкильный атом Н до'
гидроперекисей
R R
ООН
в) 1,4-циклоприсоединение к диеновым углеводородам с обра-
образованием циклических перекисей:
г) взаимодействие с восстановителями - фенолами, серосо-
серосодержащими соединениями и аминами.
121
Таблица 22
Влияние гетерогенных реакций 03 и О на концентрацию
активных газов в стратосфере на высоте 20 км
Клмпп
лишни
нент
н
ОН
но2
но3
N0
N02
СН4
н2о2
со
N20
0
°3
Концентрация молекул в '
Азрозоли
отсутствуют
9,8-Ю
4,4-10 5
2,4-Ю7
6,2-Ю9
1,7-Ю9
2,6-109г
2,3-Ю12
ЗД-108
9,8-Ю10
4.6-1011
2,2-Ю6
3,9-Ю12
J/f = Ю мкм *
9,5-Ю
4,3-Ю5
2.2-107
5,8-Ю9
2,0-Ю9
2,6-Ю9
2,3-Ю12
2,8-Ю8
9.9-1010
4,6-Ю11
1,9-Ю6
3,4-ТО12
[ см3
$1? = 1 мкм
5,8-Ю
2,2-Ю5
4,0-Ю6
6.8-107
8,2-Ю9
9,1-Ю7
2,3-Ю12
9Д-106
1,2-Ю11
4,5-10П
1,8'104
2,9-Ю10
Наиболее хорошо изучены реакции первого типа. Показано,
что они могут идти через образование промежуточных продуктов -
пероксиранов:
с о:
V
к
\
или цвиттерионов
Ч
с
с— с
*IV
Лш
ш
Квантовомеханический расчет позволил установить, что про-
промежуточным продуктом в реакции 02<*До.) с этиленом и пропиле-
пропиленом является соответствующий пероксиран, а в реакции с винила-
мином и 2,3-дигидропираном - цвиттерион. Взаимодействие
с 2-метилбутеном-2 и 1,2-диметилциклогексаном в основ-
О2( A
122
ном происходит в соответствии в реакцией второго типа. В газо-
газофазной реакции кислорода O2(IAg) с олефинами различия в кон-
константах скорости обусловлены почти полностью различиями в энер-
энергиях активации. Как для линейных олефинов, так и для цикличес-
циклических пентенов введение метильной группы уменьшает энергию актива-
активации на 6,3 кДж. Введение второй группы в цис-положение по отно-
отношению к первой приводит к дополнительному падению энергии на
3,25 кДж. Для ряда циклогексана энергия активации более сильно
зависит от метального замещения. Снижение энергии активации,
как принято считать, связано с электрофильным характером син-
глетного кислорода. Энергия активации в растворе примерно на
12,6 кДж меньше энергии активации в газовой фазе. Этим объясня-
объясняется увеличение константы скорости в растворе на два порядка.
Константы скоростей реакции O2(*Ag) с олефинами соизмеримы с
константами скоростей реакций озона с ними и на пять порядков
меньше, чем соответствующие константы для реакций с ОCр)
(см. табл.20). Так как концентрация ОCр) в атмосфере порядка
10 млн , а 02(Мя.) - порядка 10 млн , то окисление оле-
олефинов синглетным кислородом 02<1Лд.) не должно давать значи-
значительного вклада в расход олефинов в загрязненной атмосфере. Од-
Однако при прямом гидролизе загрязнителей воздуха, например ПАНа,
концентрация О2<1Дл-),а значит, и его вклад в реакцию может
значительно возрасти. К такому же результату может привести из-
избыточное содержание в загрязненной атмосфере других органичес-
органических соединений (бензол, нафталин, бензальдегид), способных эф-
эффективно и многократно фотогенерировать по механизму переноса
энергии.
§ 2.5. Реакции образования аэрозолей в реальной атмосфере
В реальной атмосфере практически всегда присутствуют окис-
окислы серы и азота, углеводороды, водяной пар, твердые и жидкие
аэрозольные частицы. Особенности протекания химических и фото-
фотохимических реакций в несильно загрязненной атмосфере, очевид-
очевидно, в значительной степени будут определяться радиационным ре-
режимом и влажностью воздуха. Наличие коротковолновой радиации
Т23
Sm,km 6-10~?M
-¦*...-/
3,3
4,0
5,0
6,0
8ft
12fl
13,0
200
I 1,4
- 12
10
- 8
_ 6
- 4
- 2
0
Аэрозоли отсутствуют
II I Г
0,1
0,3
0,5
Рис.5. Корреляционная связь между концен-
концентрацией озона в приземном слое и коэффициен-
коэффициентом аэрозольного ослабления (Г.ХидиД.Бартон).
1973 г. - 1 - г.Риверсайд; 2- Зап.Ко-
вина; 3- г.Помона; 4- г.Торранс; 1972 г.:
5 - г.Пасадена; 6 - г.Риверсайд; 7-г.По-
7-г.Помона; 8- г.Харбор; 9- Блимп Файр (маяк).
будет приводить к образованию оксидантов, при появлении кото-
которых происходят основные процессы образования аэрозольного ве-
вещества.
Для нижней тропосферы в городских условиях наблюдается от-
отчетливая корреляция меаду средними значениями коэффициента аэ-
аэрозольного рассеяния радиации в видимой области спектра и мак-
максимальными значениями концентрации озона (рис.5) [180]. Изу-
Изучение дисперсности аэрозолей в разное время суток показывает,
что частицы, образующиеся при химических и фотохимических ре-
124
акциях примесных газов, имеют в основном d <1 мкм. Сульфаты
образуются в дневное время и имеют d < 1 мкм, а ночью и рано
утром сульфатные частицы заметно растут. Концентрация сульфа-
сульфатов в течение всего дня коррелирует с содержанием SO2 и с мак-
максимальными значениями концентрации озона в послеполуденное вре-
время.
Такой же характер носит корреляция содержания органичес-
органических соединений в аэрозольном веществе с озоном. Для нитратов
корреляция с NO2 наблюдается в середине утра. Содержатся нит-
нитраты в более крупных частицах (в более старых аэрозолях). Наб-
Наблюдаются как бы два источника нитратов: 1) быстрое образование,
в основном из NO2 утром, 2) медленное накопление от NOj. (ге-
(гетерогенные реакции на частицах). Четкой выраженной корреляции
нитратов с содержанием озона не наблвдается.
Корреляция меаду концентрацией химических аэрозолей и ве-
величиной относительной влажности слабая, а для частиц с d < 0,5
мкм даже отрицательная.
Измерения суточного хода дисперсности аэрозолей и спект-
спектрального ослабления аэрозольных проб для приземного слоя ат-
атмосферы в летние месяцы в районе г.Рыльска показали,что в днев-
дневное время в приземном слое происходит рост аэрозольных частиц
в результате гетерогенных фотохимических реакций. Оценка кон-
константы скорости реакции дала величину 3-Ю с. Анализ ИК-
спектров проб показал, что в течение дня идет рост массовой
концентрации органического вещества и сульфатов в диспергиро-
диспергированной фазе атмосферных аэрозолей. Наиболее вероятными катали-
катализаторами реакций являются окислы алюминия и железа.
В лабораторных экспериментах исследование влияния паров
воды на фотохимические реакции загрязнителей воздуха позволило
обнаружить влияние влажности на фотохимический выход оксидан-
оксидантов. При освещении смеси 1-гексена с NO2 на воздухе с вели-
величинами влажности 0; 5,4; 10,8; 16,3; 21,7 мм Н2О и темпера-
температуре воздуха 14, 25 и 35°С наибольшая концентрация оксиданта
наблюдалась при влажности 16,3 мм Н2О для 14 и 35'JC; а для
25°С - при влажности 10,8 мм h^O.Эксперименты по исследова-
исследованию влияния влажности на выход оксидантов, проведенные с оле-
финами (этиленом), ароматическими углеводородами и альдегида-
125
ми, подтвердили влияние влажности на этот процесс, но опреде-
определенные выводы по механизм влияния из этих экспериментов сде-
сделать нельзя.
Ж.Буффалини и А.Альтшулер, исследовав влияние паров воды
на фотоокисление этилена в системе этилен - NO^, а затем на
фотоокисление тетраметилэтилена, л-бутана, бутена-1 и метил-
этилена, не обнаружили влияния воды на скорость фотореакции
[ИЗ ]. Аналогичный результат получен для смеси этилена с N0^-
Вообще объяснить механизм увеличения реакционной способности
компонентов системы при добавлении паров воды сложно. Малове-
Маловероятно, чтобы молекулы воды принимали участие в свободно-ради-
свободно-радикальном механизме фотоокисления углеводородов. Возможно, влия-
влияние влажности есть результат воздействия стенок кюветы на фо-
фотохимические реакции. Значительная часть свободных радикалов,
образующихся при фотоокислении углеводородов, может гибнуть на
стенках кюветы, а введение водяных паров может дезактивировать
реакционные центры стенок, увеличивая тем самым время жизни
свободных радикалов. Другое объяснение влияния влажности на
ход фотохимических реакций заключается в связывании водой окио-
лов азота в реакциях типа
N2O5 + Н20 — 2HNO3
NO+NO2+H2O — 2HN0,
(Л* = 1,0.103моль.с),
(К = гД-Ю'^1
Так как лабораторные исследования образования аэрозолей
из сернистого газа показывают отчетливую зависимость интенсив-
интенсивности образования частиц от влажности, то следует сделать вы-
вывод, что в натурных условиях механизм образования аэрозолей
несколько иной. В частности, высокая относительная влажность
может приводить к закупорке пор в аэрозольных частицах, играв-
игравших роль катализаторов в реакции окисления двуокиси серы.
Экспериментами в смоговых камерах было обнаружено, что
-126
двуокись серы влияет на процессы образования фотохимических
аэрозолей из олефинов: введение малых концентраций S02 подав-
подавляет процессы образования аэрозолей, а большие концентрации SCL
способствуют образованию аэрозолей. Из сказанного можно сде-
сделать вывод, что процессы фотохимического и химического образо-
образования аэрозолей могут протекать различным образом не только из-
за различных соотношений между реагентами, но и из-за влия-
влияния на химические реакции физико-химических процессов: кон-
конденсации, адсорбции и коагуляции.
Глава 3
СТОКИ И ПЕРЕНОС АЭРОЗОЛЕЙ В АТМОСФЕРЕ
Экспериментальные измерения показывают, что соотношение
между аэрозолями различного происхождения сильно изменяется в
пространстве и времени. Это соотношение определяется не только
мощностью ближайших источников аэрозолей, но и временем суще-
существования аэрозольных частиц различного происхождения, которое,
в свою очередь, зависит от физико-химических свойств частиц,
места их нахождения и метеорологических процессов в атмос-
атмосфере.
Время жизни микропримесей в атмосфере. Для решения проб-
проблемы загрязнения воздуха, особенно аэрозольного, представляют
интерес по крайней мере три временные характеристики. Первой
является общее время жизни х микропримеси, второй - величина,
характеризующая время химических и физико-химических превраще-
превращений исходных веществ % t третьей - должна быть величина, опре-
определяющая время существования частиц в исследуемом локальном
объеме пространства тд . При процессах, определяющих величину
тх у происходит трансформация вещества без его удаления из ат-
атмосферы (химические реакции, конденсация, коагуляция, которая
в конечном итоге влияет на скорость удаления вещества из атмо-
атмосферы). Эти процессы рассмотрены в других главах. Третья вели-
величина тд - определяется как процессами полного удаления ве-
вещества из атмосферы, так и процессами его переноса в другой
объем пространства (диффузия, адвекция, конвекция). Последние
не всегда способствуют выведению вещества из атмосферы, но
128
обычно способствуют выравниванию относительной концентрации ве-
лества в разных точках пространства.
Экспериментальные измерения времени жизни аэрозолей и ис-
исходных газообразных продуктов показывают, что эта величина мо-
может меняться в очень широких пределах. Так, если в стратосфере
время существования наиболее мелких частиц около трех лет, то
в тропосфере оно гораздо меньше - от нескольких дней до нес-
нескольких месяцев, в зависимости от физико-химических свойств и
характеристик внешней среды. Например, время жизни сернистого
газа и аэрозольных частиц, образующихся из продуктов реакций
сернистого газа с другими веществами, не более пяти дней. По
мнению Е.С.Селезневой, время жизни сульфатов в атмосфере про-
промышленного района значительно меньше: для случая с осадками оно
составляет 0,88 - 0,73 сут, без осадков - 3,2 сут. (Расчеты вы-
выполнены методом Х.Юнге.) По экспериментальным данным Е.С.Селез-
Е.С.Селезневой, время жизни сульфатов, образующихся над промышленным
центром, для осадков составляет 3-4 и 15-20 ч без осадков.
При таких малых временах жизни перенос сульфатов происхо-
происходит не более чем на 300 - 350 км от города. Вместе с тем спут-
спутниковые наблюдения показывают, что сульфатные аэрозоли распро-
распространяются и на тысячи километров от источника [195]. Время
жизни других гигроскопических аэрозольных частиц несколько боль-
больше, но примерно того же порядка ( 4 12 дней). К этой величине
близко и время жизни всех аэрозолей промышленного происхожде-
происхождения. Например, в работе [19] оно предполагается равным 30 сут.
Время жизни аэрозольных частиц малогигроскопических и нераст-
нерастворимых с г < 5 мкм в тропосфере может быть примерно два-че-
два-четыре месяца. Самые большие аэрозольные частицы с г > 10 мкм
существуют меньше суток. Однако если аэрозольные частицы попа-
попадают в безоблачные слои атмосферы, они могут существовать и
более длительное время. Наблюдения Р.Янике и А.Шютца особеннос-
особенностей переноса пыли из пустыни Сахары при северо-восточном пас-
пассате показали, что основная масса пыли переносится выше уров-
уровня пассатной инверсии в слоях с низкой относительной влаж-
влажностью. При этом могут переноситься даже очень крупные части-
частицы, вплоть до частиц размером около 100 мкм. Сухие выпадения
лнлевых масс происходят за сотни и тысячи километров от источ-
1922
129
ника, причем очень часто на противоположных от источника скло-
склонах гор, например лёссовая пыль в Памире, пыль из пустыни Саха-
Сахары во французских Пиринеях. Часть осевшей на землю пыли (~1($)
может вторично попасть в атмосферу. Очевидно, что вторичное
поступление аэрозолей возможно и с поверхности океана. Общее
содержание аэрозолей разного происхождения Mj, г-см - мож-
можно определить как функцию интенсивности образования этих час-
частиц (мощности источника) 7j (т/год), времени их жизни t,- (или
скорости стока R{). Для стационарного случая Mi=rili и !•«/?..
Такого рода оценка справедлива как для всей атмосферы, так и
для локальных объемов. Экспериментальное определение xi в боль-
большинстве случаев очень затруднено, а часто и невозможно из-за
сильной пространственной неоднородности концентрации исследуе-
исследуемого вещества.
Существует несколько основных механизмов стока аэрозолей
из атмосферы: седиментация, вымывание аэрозольных частиц обла-
облаками, туманами, осадками, сухое осаждение на препятствиях. Уда-
Удаление из атмосферы газовых примесей, образующих аэрозольные
частицы, также регулирует концентрацию аэрозолей в атмосфере.
Большинство газовых примесей особенно активно удаляется из ат-
атмосферы в результате адсорбции на водяных каплях и увлажнен-
увлажненных поверхностях.
Однако в большинстве случаев процессы выведения частиц из
атмосферы представляют собой совместное действие диффузии пе-
переноса частиц и собственно механизмов стока.
§ 3.1. Диффузия и перенос аэрозолей в атмосфере
Перенос примеси в атмосфере происходит как в результате
конвективных и адвективных движений воздушных масс, так и в
результате турбулентной и молекулярной диффузий. Характер пе-
переноса вещества в вертикальном и горизонтальном направлениях
воздушными потоками и турбулентной диффузией существенно раз-
различен. Скорости вертикальных восходящих и нисходящих токов
воздушных масс имеют порядок величины»от нескольких десятков
до нескольких десятков сантиметров в секунду и лишь в очень
130
-едких случаях достигают величин в несколько десятков метров в
секунду. Эти токи по масштабу не превышают нескольких километ-
километров. Скорости горизонтальных движений могут быть значительно
больше: ст десятых до сотен метров в секунду, а масштабы пере-
переведения достигают сотен и тысяч километров.
Такая же ситуация наблюдается и с турбулентными пульсация-
пульсациями воздушных масс, обусловливающими диффузию примеси: масштабы
турбулентных пульсаций в горизонтальных направлениях значитель-
значительно превосходят масштабы пульсаций в вертикальном направлении.
Турбулентная диффузия способствует выравниванию концентра-
концентраций аэрозолей N<x,y) в горизонтальных направлениях и выравни-
выравниванию величины аэрозольного отношения смеси а-тл\т в верти-
вертикальном направлении.
3 случае броуновской (молекулярной) диффузии, вызванной
тепловым движением молекул, принципиальной разницы между вер-
вертикальным и горизонтальным направлениями нет. Из общей тео-
опи случайных процессов молекулярная диффузия должна описывать-
описываться дифференциальным уравнением Планка - Фоккера
дУ дАУ/ d2$W
di дх ох2
где W(x,t) - вероятность состояния частицы; А - характеризу-
характеризует среднюю скорость систематического процесса; У - случайные
изменения переменной х ; х - должна быть непрерывно меняющей-
меняющейся величиной, т.е. вероятность больших отклонений за малое
время достаточно быстро стремится к нулю. В частном случае А =
= v, У = В = (х* -x2)/Bi ). Влияние броуновской диффузии на вырав-
выравнивание концентрации частиц в пространстве ограничивается воз-
воздействием на частицы размером меньше -1,0 мкм и очень небольши-
небольшими перемещениями их за конечный промежуток времени.
...еханизм турбулентной диффузии в атмосфере усложняется
тем, что беспорядочные пульсации скорости воздушных потоков
иг.сгат частотный спектр от сотых долей секунды до суток и более,
а масштабы турбулентных вихрей - от нескольких миллиметров до
сотен километров. Облако примеси для пульсаций большого мас-
масштаба, превышающего его размеры, ведет себя как одна частица.
Характеристики перемещения облака описываются в неподвижной
131
(эйлеровой) системе координат уравнением диффузии Фикка вида
дх
di
дх/ дуГаду] ' dzVzdz '¦ C*2)
В то же время частицы внутри облака смещаются относительно друг
друга (в лагранжевой системе координат) вследствие случайных
пульсаций скоростей воздушных потоков меньших масштабов - по-
порядка величины расстояния между частицами. Эти пульсации увели-
увеличивают расстояние ."°нду частицами. По мере расширения системы
аэрозольных частиц (струи или облака) возрастает и масштаб
пульсаций, участвующих в лагранжевой диффузии.
При лагранжевом описании турбулентности вдали от подсти-
подстилающей поверхности можно предполагать, что турбулентность изо-
изотропна, т.е. нет существенной разницы между горизонтальными и
вертикальной составляющими пульсаций скоростей воздушных пото-
потоков. Очевидным недостатком описания турбулентной диффузии при-
примеси с помощью уравнения C.2) является пренебрежение зависи-
зависимостью коэффициента перемешивания от масштаба турбулентных пуль-
пульсаций. Кроме того, использование этого уравнения предполагает,
что диффузия вещества происходит в направлении уменьшения его
концентрации. Мгновенное значение потока этого вещества пропор-
пропорционально градиенту ее концентрации, поэтому максимальная ско-
скорость распространения диффундирующих частиц должна быть равна
бесконечности. В общем случае это уравнение для описания тур-
турбулентной диффузии в атмосфере не всегда пригодно. Однако для
стационарных и квазистационарных задач его использование дос-
достаточно оправдано.
Статистический подход к вопросу распространения примеси в
атмосфере, учитывающий временное изменение масштаба турбулент-
турбулентных пульсаций, был начат работами Дж.Тейлора. Им было получено
выражение
'*. -, _ . . ..
C.3)
где у<1) - расстояние, на которое частица увлечена турбулент-
турбулентными пульсациями v' от начала координат за время I ; R(%) =
~'2 -
= v'(l)v'(t
132
гР
одноточечная нормированная лагранже-
ва автокорреляционная функция. Обобщение этого результата для
трехмерного случая было сделано Г.Бэтчелором. При больших i
автокорреляционная функция Р достаточно быстро стремится к ну-
нулю и частица "забывает" свое начальное движение, что позволяет
вернуться к описанию диффузии с помощью уравнения C.2). Одна-
Однако для горизонтальных направлений (большие вихри) необходимо
очень большое время диффузии, чтобы можно было пользоваться
этим уравнением. О.Сеттоном была предложена диффузионная мо-
модель, в которой лагранжева корреляционная функция R(t,) зависит
от турбулентной энергии vr\ вязкости ¦? и времени ?. Приме-
Применимость схемы О.Сеттона в дальнейшем была расширена введением
дополнительных эмпирических параметров.
Строго говоря, необходимо либо отказаться от использова-
использования: закона Фикка для диффузии аэрозольных частиц, либо придать
ему такой вид, который соответствовал бы распространению диф-
диффундирующих частиц с конечной скоростью.
Пусть частицы в каждый момент 0, 1, 2, ... могут находить-
находиться в любой из точек трехмерного пространства с координатами х =
- И , у~}1, 2 = kl ( г, } , к = 0, +1, +2, ...), т.е. в уз-
узловых точках кубической решетки с шагом I, причем за промежу-
промежуток времени от пх до (n+l)z любая такая частица может с
равной вероятностью 1/6 перейти в любую из шести соседних уз-
узловых точек. Обозначив число частиц в узле (il, jl, kl ) в мо-
момент времени пх через J^i,it]c,n, легко перейти к рекуррентно-
ку соотношению
Ni
C.4)
Зсли изменения Nij^n за пространственный или времен-
временной :таги относительно малы (в атмосфере это справедливо в слу-
случае среднемасштабной диффузии), легко перейти от C.4) к не-
непрерывному случая. Концентрация частиц в точке (х,у,г) в
момент времени i определится соотношением
•133
Тогда можно написать:
с<х,у, г, Ьт) = t\c<x+l,y,z,t) + c(x-l,y, z,t) +
y+l,z,i) + c<x,y-l,z,i) + c(X,y,2+l,l) + c(x,y, z-l, ?)].
для потока частиц между двумя соседними узлами имеет мес-
место соотношение
I4 дс
C.5)
бт Ьх
(с точностью до членов высшего порядка малости). Зто выражение
для потока частиц между двумя соседними узлами (точками прост-
пространства, отстоящими друг от друга на расстоянии Z) предполага-
предполагает равномерное движение частиц в течение промежутка времени со
скоростью vx = I /x .
Если реальное движение частиц между двумя соседними узла-
узлами является более сложным, то предлагаемую схему движения час-
частиц можно рассматривать как усреднение сложного движения. Под
потоком частиц в этом случае понимается его значение, усреднен-
усредненное по интервалу времени х .
Аналогично C.5) можно написать выражения для потока и меж-
между любыми другими точкаг.ш пространства, разделенными интерва-
интервалом I. Поэтому через единицу площади с ортом внешней нормали
П в единицу времени проходит число частиц, равное
1 Г г* дс Л z* дс Л z4 дс
ТЛ-Тг7хСО&(ПХ)-
Z*
6т ду
-
6т
Величина потока в этом случае равна
Z2
< I > = -
VC
C.6)
где символ < I > - среднее значение потокг. частиц за промежу-
промежуток времени х . Классический закон Фикка получается из C.6)
посредством соотношения
134
I = Urn < I >
т-»о
C.7)
а фикковское дифференциальное уравнение диффузии получается с
помощью C.7) и уравнения непрерывности для мгновенного значе-
значения потока частиц:
дс
5Т
C.8)
В случае, когда применимость классического закона Фикка,
выражаемого соотношением C.7), становится неоправданной, со-
соотношение C.8) должно быть усреднено по интервалу времени г.
При этом получается
С
-V\
Используя соотношение C.6) из C.8), получаем
(ЗЛО)
Выражение (ЗЛО) есть уравнение баланса примеси в точке
г за промежуток времени т, если каждая частица этой примеси
за это время может переходить из произвольной точки простран-
пространства в любую другую точку, удаленную от нее на расстояние I ,
т.е. на сферу, описанную радиусом Z с центром в этой точке.
Его можно получить, если все члены правой части равенства
(ЗЛО) разложить в ряд Тейлора по I, сохранив первые неисчеза-
ющие члены этого разложения, содержащие производные по прост-
пространственным координатам.
Рассматриваемая схема не полностью соответствует реальный
движениям частиц в турбулентном потоке. При развитой турбулент-
турбулентности в атмосфере спектр скорости частиц простирается от нуля
до некоторого максимального значения практически непрерывным
образом. Однако такое сложное движение частиц всегда можно за-
заменить некоторым усреднением - эффективным движением, соответ-
соответствующим рассмотренной схеме. В этом случае скорость г?х=1/х
135
будет иметь смысл максимальной скорости распространения диффун-
диффундирующих частиц.
Соответствующее дифференциальное уравнение,описывающее та-
такое эффективное движение частиц, можно получить с помощью раз-
разложения левой части уравнения (ЗЛО) в ряд Тейлора по парамет-
параметру I. Разложение следует производить до членов второго поряд-
порядка малости по т, включительно, так как правая часть C.10)
фактически представляет собой тейлоровское разложение функции
C(r,i) по параметру I с точностью до членов ОA2), в то вре-
время как величины I и х в рассматриваемой схеме имеют один по-
порядок малости (их отношение равно гг и представляет собой ко-
конечную величину). Получим
Ы
2 Ы2
бт
Если I не зависит от пространственных координат, то
\72с =
дгс и> дс
1 - 2
1Г2 Ы
(З.Я)
где u)=i/x, v=irx/y/3. Здесь:/ имеет смысл максимальной
скорости изотропной диффузии в трехмерном пространстве, а а> -
частота перехода частицы из одной эффективной турбулентной не-
неоднородности, занимающей объем пространства с линейным разме-
размером I, в другую аналогичную неоднородность.
Уравнение (З.И) описывает диффузию частиц, обусловленную
однородной и изотропной турбулентностью. Требование монохрома-
монохроматичности турбулентности в этом случае подменяется условием за-
замены действительной картины движений на эффективную.
Согласно некоторым наблюдениям в низкочастотной части спе-
спектра турбулентности существуют вихри, периоды которых состав-
составляют минуты и даже десятки минут и которые в первом приближе-
приближении можно считать монохроматическими, однородными и изотропны-
изотропными. В общем случае они не могут покрывать всего пространства,
занимаемого турбулентной средой, сплошным образом. Тогда диф-
диффундирующее облако должно принять "отростковую структуру". Мел-
Мелкомасштабная область турбулентности будет обеспечивать квази-
квазинепрерывное заполнение турбулентной среды диффундирующей при-
136
кесью. Для одномерного случая такого рода уравнения предлага-
предлагались в работах Е.С.Лялина и А.Слонина.
Конечно, уравнение C.11) описывает турбулентную диффузию
в слишком идеализированном вцце. Например, в пограничном слое
атмосферы диффузия никогда не является изотропной. Обобщение
на случай неизотропной диффузии не представляет труда, если
придать параметрам гг и <о соответствующий тензорный харак-
характер. Случай неоднородной и и неизотропной диффузии представля-
представляет наибольший интерес. Однако, если считать параметры гг и о>
функциями координат, соответствующее уравнение решить не прос-
просто даже на ЭВМ.
В предыдущем изложении предполагалось, что среднее тече-
течение (например, ветер) отсутствует. При наличии такого течения,
направленного, скажем, вдоль оси х и имеющего скорость и, урав-
уравнение C.11) в его левой части следует дополнить членом и 4е-.
В более общем случае кроме ветра имеют место седиментация при-
примеси, а также общий подъем или опускание воздушных масс в ре-
результате конвективных потоков (± w). Если скорости воздушных
потоков и седиментации частиц можно считать величинами, не за-
зависящими от пространственных координат и времени, то удобно
уравнение диффузии C.11) записать в системе координат, движу-
движущейся вместе с ветром и одновременно участвующей в вертикаль-
вертикальном движении со скоростью (-v$±w). Для перехода к неподвиж-
неподвижной системе координат, жестко связанной с поверхностью Земли,
в окончательном выражении достаточно воспользоваться формула-
формулами перехода х = Х- ut, i/=Y, z = Z + (v$±ur)t, где x,y,z -
подвижные координаты, X,Y,Z - неподвижные и предполагается,
что ось X направлена вдоль скорости ветра, а ось Z - верти-
вертикально вверх.
Вымывание примеси можно учесть введением закона экспонен-
экспоненциального убывания концентрации диффундирующих частиц, имеюще-
имеющего ВИД
где
c(v,t) является решением уравнения (З.И).
137
Если рассматриваемые эффекты имели место только в проме-
промежутке времени от момента it до момента ?2, то
^) di,
C.13)
где фильтрующий оператор Z (?~?j) Z аг-1^) представляет
собой произведение функций Хевисайда, определяемых как
!0, если х & О,
1, если х > О
и не относится к функции с (r,t).
Нетрудно видеть, что функция сл(г,г) удовлетворяет урав-
уравнению
Однако нет необходимости заниматься решением уравнения C.14)
потому, что с a <r,i) выражаетоя через c(r,t) посредством от-
отношения C.12) и C.13), а функция С(г,1) находится как реше-
решение уравнения C.11).
При диффузии в пограничном слое атмосферы в качестве крае-
краевых условий должны быть взяты условия на подстилающей поверх-
поверхности (z =0) и на бесконечнооти (|г| -»«>). При |г|.-* оо
концентрация должна обращаться в нуль. При z = 0 краевое ус-
условие должно отражать характер взаимодействия диффундирующих
частиц с подстилающей поверхностью. Если примесь полностью по-
поглощается этой поверхностью, то в качестве краевого условия
2=0 должно быть взято однородное условие Дирихле. Если при-
примесь полностью отражается от подстилающей поверхности, то ус-
условие должно иметь вид однородного условия Неймана.
В промежуточном случае, т.е. когда примесь частично отра-
отражается и частично поглощается на поверхности z = 0, условие
имеет вид неоднородного смешанного условия Дирихле - Неймана.
В практических задачах этим условием пользоваться не приходит-
приходится, потому что, как правило, характер взаимодействия диффунди-
диффундирующих частиц с подстилающей поверхностью точно неизвестен.По-
138
этому целесообразно, например, считать подстилающую поверх-
поверхность идеально отражающей диффундирующие частицы, т.е. считать
поток примеси через поверхность z = 0 равным нулю (вся примесь
з воздухе). В этом случае задачу для полупространства z »• О
удобно заменить задачей для безграничного пространства соглас-
согласно методу изображения, хорошо известному из электростатики.
В качестве начальных условий в общем случае должны быть
заданы как начальное значение функции c(r,i), так и скорость
изменения этой функции в момент времени i =0, т.е. значение
~г . Однако в некоторых случаях задача может быть постав-
поставит г = о
лена и без начальных условий. Это имеет место в задачах о рас-
распространении граничного режима, если этот режим действует дли-
длительное время.
Разнообразие метеорологических условий, сильно влияющих
на турбулентную диффузию загрязняющих примесей, в ряде случаев
ставит под сомнение возможность применения теоретических схем
турбулентной диффузии для реальной атмосферы. Поэтому большое
значение имеют накопление статистического материала по распре-
распределению примесей в реальной атмосфере и создание различного ро-
рода эмпирических формул [233]. При наличии направленных движе-
движений воздушных масс основная масса аэрозольных частиц и других
примесей переносится этими движениями (адвекцией и конвекцией}
Зосходящие потоки могут перевернуть вертикальный профиль кон-
концентрации аэрозолей и привести к возрастанию концентрации час-
частиц с высотой, особенно в отсутствие постоянно действующих на-
наземных источников частиц. Такие случаи наблюдаются при измере-
измерениях над водной поверхностью (море или большое озеро) в вечер-
вечернее время, а иногда и в дневное, когда отсутствует адвекция.
Горизонтальный перенос аэрозолей так же, как и турбулент-
турбулентная д-лад-узия, способствует уравниванию концентраций аэрозолей
в различных точках атмосферы. В случае необходимости решения
задачи переноса аэрозолей от источника загрязнений при ветре
горизонтальную координату берут по направлению ветра и решают
"зунерную задачу. Как уже говорилось ранее, перенос аэрозолей
адвекцией играет существенную роль даже в приземном слое. В
стратосфере этот механизм определяет распространение аэрозо-
аэрозоли по всему земному шару.
139
§ 3.2. Сухое осаадение аэрозольных частиц
Седиментация. Скорость оседания аэрозольных частиц в ат-
атмосфере под действием силы тяжести зависит не только от разме-
размеров частицы, но от удельной плотности и плотности упаковки ве-
вещества частицы, а также от высоты слоя атмосферы, в котором
рассматривается этот процесс.
Расчет установившейся скорости осаждения сферических час-
частиц с размерами менее 20 мкм сравнительно прост. Рассматривает-
Рассматривается режим движения частиц с числом Маха намного меньшим единицы.
Причем для всех чисел Кнудсена число Рейнольдса для потока с
частицей также меньше единицы ( Re„,= rv$ /-9 < 1 ). В этом слу-
случае скорость седиментации определяется выражением ir$ = Bmg,
в котором подвижность частицы В согласно эмпирическим соотно-
соотношениям равна
10'2W~1
10 10г Ю3 vs,cm/c
В =
[1,257+ Q400 езср A,10 Af/г )]¦*?[ ,
где %• - средняя длина свободного пробега молекул; п - вяз-
вязкость среды. Соответственно интенсивность потери частиц вслед-
вследствие седиментации
*•-¦*(?)¦*¦•(?)¦
где А* 1.3-106 см2 для
у = 1 г/см3 при I = 20°С.
Таким образом, скорости падения частиц, а следовательно,
интенсивности их выведения из разных слоев атмосферы и время
их пребывания в этих слоях зависят от довольно большого числа
параметров среды и самих частиц, из которых наиболее сущест-
существенными являются размеры последних и плотность воздуха. На
рис.6 приведены скорости падения сферических аэрозольных час-
частиц на разных высотах (кривые 1-к, 8-12) и в приземном слое
(кривые 5-€), а также время их падения с высоты 80 км (кри-
(кривые 8-12'), где могут-образовываться частицы после сгора-
сгорания метеоритов, в предположении, что конвективные потоки от-
отсутствуют. Сравнение их со скоростями конвективных движений
воздушных масс, наблюдаемых в атмосфере @,5+20 см/с), пока-
140
ЗОТсит
10'" -
10
Рис.6. Скорости падения и время седиментацион-
ного пребывания частиц разных размеров в атмосфере.
/ -4-1?B)для частиц с о = 0,2; i; i; 1;
г/смз и г= 0,1; 0,1; 1,0; 2J0 мкм соответст-
соответственно; 5,6 - для частиц с о = 1 г/см3 в зави-
зависимости от г; 7 - ьх - скорость восходящего по-
потока на разных высотах, уравновешивающая ско-
скорость седиментации и 8-12 - ir<(z) для час-
частиц с р=2 г/смз и г= 0,2; 0,5; 1; 5; 10 мкм
соответственно;8'-12'- время пребывания частиц,
падающих из космоса с высоты 80 км.
зывает, что седиментацией для частиц с г < 20 мкм почти всег-
всегда можно пренебречь для аэрозолей приземного слоя и даже для
тропосферы. Для частиц с г < 5 мкм до высот 30 км и г< 1 мкм
до высоты 50 км скорости седиментации сравнимы со скоростями
конвективных потоков, а частицы с г 4- 0,5 мкм могут находить-
находиться длительное время взвешенными в атмосфере на высотах ~ 70 -
80 км. Для частиц с рыхлой структурой и несферической формой
скорость падения может быть значительно меньше, чем для соот-
соответствующих частиц сферической формы.
Для приземного слоя атмосферы наибольший интерес представ-
представляют данные по скорости падения водных, сажевых, кварцевых час-
частиц, а также частиц окислов железа, имеющих наибольшую плот-
плотность (из относительно распространенных в атмосфере аэрозоль-
аэрозольных частиц) (табл.23).
Большой интерес представляют результаты исследований тра-
траекторий движения малых частиц в атмосфере [200], показывающие,
что в ряде случаев частицы опускаются по спирали. Это сущест-
существенно уменьшает их скорость падения и увеличивает время пребы-
пребывания в атмосфере.
Сухое осаждение частиц. Вблизи нижней границы атмосферы
частицы удаляются из атмосферы с помощью трех механизмов: се-
седиментации на поверхность объекта, диффузионного осаждения на
различных телах и инерционного осаждения на препятствиях. Поч-
Почти во всех случаях все три процесса действуют одновременно. Пе-
Перенос вещества из турбулентного газа к ограничивающей подсти-
подстилающей поверхности в упрощенном виде можно представить осу-
осуществляющимся через турбулентный пограничный слой в сторону
вязкого или ламинарного подслоя, прилегающего к граничной по-
поверхности. Лимитирующим фактором в переносе частиц к поверх-
поверхности будет молекулярная диффузионная способность переносимых
веществ [230]. (Коэффициент турбулентной диффузии в атмосфере
изменяется от 10 до 10 смг/с.) Когда поверхность достаточ-
достаточно шероховата, вязкий подслой приобретает турбулентноподобный
характер, заметно возрастает сопротивление у поверхности, вы-
вызывая увеличение передачи импульса.
Полагают, что вязкий характер диффузионного слоя сохраня-
сохраняется в случае шероховатости поверхности, и на расстоянии нес-
142
Таблица 23
Скорости падения сферических частиц в приземном слое
атмосферы, см/с
г
1
0
0
0
0
0
0
I
1
2
2
3
4,
5
7,
ю,
,мкм
,01
05
,1
3
5
7
0
5
0
5
0
0
0
5
0
1
1
2
1
4
6
1
2
5
7
0
0
0
0
1
Вода
,85.
,67-
.91-
,58.
,31-
,99-
,35-
,92-
,06«
,82-
,111
Д96
,306
,750
,20
10~Ь
10~4
10
10
10~3
ю-3
10
10~2
10~2
Ю
3
3
5
2
7
1
2
5
9
0
0
0
0
1
2
Вещество
Сажа
,42-
,09-
,38-
,92-
,97-
,29-
,50-
,40-
,32-
,145
,205
,363
,566
,29
22
10~5
ю-4
10
10
10
10~2
ю-2
10
10~2
Кремнезем
4
7
4
1
1
3
7
0
0
0,
0
0,
2,
3,
5,0-10~5
,51'10~4
,86-10~4
.27.10-3
Д6-10
,89>10~2
64»10~2
88«10~2
137
211
300
529
826
02
24
9
8
1
7
2
3
6
Гематит
,25-Ю-5
,35-ТО-4
,46-Ю
,54-Ю-3
,16-10
,50-10-2
,75'Ю
0,149
0
0
0
0
1
3,
6,
,253
,391
,555
980
53
75
00
кольких микрон от поверхности скорость ооаждения определяют мо-
молекулярные процессы. Грубые оценки скоростей осаждения можно
получить из соотношения
C.15)
- коэф-
F - напряжение на поверхности; и* - скорость свободного те-
течения; ^ - плотность газа. Коэффициент пропорциональности в
143
где $с = */D^; -0 - кинематическая вязкость газа;
фициент молекулярной диффузии вещества оь ;
этом соотношении зависит от характера поверхности. Для плоские
поверхностей с шероховатым травяным покрытием А.Чемберлен по-
получил некоторые значения ir_, которые оказывались всегда выше
или равны vs, причем наименьшая скорость осаждения характер-
характерна для частиц с радиусом ОД мкм [105].
Следует заметить, что наличие механизма седиментации мо-
может увеличить скорость осавдения ve на объект. К этому приве-
п(т),см
10
V
\\
е
дет также наличие электроста-
электростатического заряда на частице
или объекте. Инерционное осаа-
дение с учетом эффекта зацеп-
зацепления играет ведущую роль при
относительных скоростях дви-
движения частицы порядка метров
в секунду и более.
Весьма интенсивным погло-
поглотителем аэрозольных частиц
является растительность и дру-
другие шероховатые объекты [225],
Например, при измерениях со-
содержания аэрозолей в призем-
приземном слое в Запорожье в двух
близко расположенных точках
было получено, что концентра-
концентрация аэрозольных частиц в пар-
парке была примерно в пять раз
меньше, чем в соседней точке,
расположенной вне парка (po.7i
В то же время растительность
является источником аэрозоль-
аэрозольных частиц, частично и тех,
которые были ранее ею захва-
захвачены.
Эксперименты по иссле-
исследованию вымывания пыльцы амб-
амброзии и ядер Айткена хвойными и широколиственными лесами бы-
были проведены Х.Нойбургером и другими. Густой хвойный лес уда-
144
.\
ю-'
10
г, мкм
Рис.7. Концентрации частиц
разных размеров вблизи промыш-
промышленного источника.
1 - на открытой площад-
площадке- 2 - в молодом парке в
200 м от нее (г.Запорожье,
июль, 1972 г.).
лял более 80% пыльцы и до 34$ ядер Айткена (субмикронная фрак-
фракция). Широколиственные деревья были способны удалять примерно
s два раза меньшее число субмикронных частиц. Г.Ланге, Ж.Росин-
ский и Ц.Нагамото провели серию экспериментов с деревьями и
одиночными иглами хвойных в аэродинамической трубе. Для отдель-
отдельных игл и листьев заряд до восьми единиц не оказывал существен-
существенного влияния на эффективность захвата пылинок с d~ 2 мкм при
скоростях ветра 1,2-1,6 м/с. Средняя эффективность захвата час-
частиц иглами кедра и ели оказалась около 6%. При скорости ветра
выше 2 м/с наблюдалось сдувание пылинок [226]. Эффективность
захвата частиц при скорости ветра 0,2-0,5 м/с. изменяется от 1
до 10$. Сдувание пылинок прекращается после нескольких часов
эксперимента.
Картина выведения аэрозольных частиц из нижних слоев ат-
атмосферы лесной .растительностью усложняется еще тем, что воз-
воздушный поток, содержащий аэрозоли, частично обтекает лесной
или парковый массивы. Скорость воздушного потока внутри леса
уменьшается. Меняется также режим турбулентного перемешивания.
Летом 1968 г. автором в районе Рыльска был проведен экспери-
эксперимент с одновременным измерением вертикальных профилей счетной
концентрации аэрозолей и турбулентных пульсаций воздушной мас-
массы внутри соснового леса. Обработка и анализ полученных резуль-
результатов не позволили сделать однозначных выводов о зависимости
между измеряемыми параметрами. Сравнение же данных по концент-
концентрации аэрозолей внутри соснового леса с данными, полученными
в пойме р.Сейм на расстоянии 1 км, показало, что наиболее за-
заметное уменьшение концентрации аэрозолей в лесу наблюдается в
предполуденное время (в три-пять раз), тогда как в вечерние ча-
часы в отдельных случаях значения счетной концентрации аэрозолей
в лесу были даже больше, чем в пойме реки.
Эффективным поглотителем аэрозольных частиц является вод-
водная поверхность, в частности поверхность океана.
Большинство теоретических и экспериментальных оценок ин-
интенсивности сухого и влажного вымывания аэрозолей из атмоофе-
рн было получено при изучении вымывания из атмосферы радиоак-
радиоактивных продуктов. Средние скорости вымывания различных радио-
радиоактивных продуктов в нижней атмосфере колеблются в пределах от
10.1922
145
нескольких десятков до нескольких сантиметров в секунду. Напри-
Например, скорость сухого осаждения короткоживущих продуктов распа-
распада радона оказалась для зимы - 0,8 см/с, а для лета - 4,0 см/с.
Таким образом, скорость сухого вымывания определяется ве-
величиной коэффициента турбулентного обмена, скоростью седимента-
седиментации частиц и характером подстилающей поверхности, на которой
осаждаются аэрозольные частицы. Осаждением аэрозольных частиц
на очень больших твердых частицах, выпадающих из атмосферы,мож-
атмосферы,можно пренебречь. Вклад сухого осаждения в общую скорость удале-
удаления аэрозолей из атмосферы в умеренных широтах составляет при-
примерно 10-20%. В районах с малым количеством осадков этот вклад
значительно больше. Следует отметить, что конденсационный и ко-
агуляционнни рост частиц увеличивает их массу и размеры ж, сле-
следовательно, скорость осаждения.
§
3.3. Вымывание аэрозолей облаками и осадками
Важнейшим из процессов удаления как аэрозольных частиц,
так и газовых примесей из атмосферы является очищение воздуха
облаками и осадками. Этот процесс достаточно сложен и его эф-
эффективность определяется эффективностью переноса примесей к мес-
месту очищения (облакам и туманам), внутриобластным очищением эле-
элементами облака (вымывание облаками), подоблачным очищением осад
нами (вымывание осадками [116, 141]).
Вымывание в облаках и подоблачное вымывание определяются
в основном следующими параметрами: 1) размером и концентрацией
частиц в атмосфере, 2) размером и концентрацией облачных и до»
девых капель, дейотвувдих на коллекторы, 3) запасом жидкой во-
воды в облаке при непрерывной конденсации, 4) значением рН и ха
ническим составом облачной и дождевой воды, 5) степенью раство
римости газов и частиц в водных каплях.
Внутриоблачное вымывание с трудом поддается оценке. Мини-
Минимальная скорость удаления будет определяться суммой скоростей
зародышеобразовашш и потерь из-за соударений. Оценка первой
скорости дает ~ 0,1 см/с и выше. Скорость удаления из-за со-
соударений определяется выражением
146
C.16)
где Ep<rt,r.) - эффективность соударений частиц разных разме-
размеров. Она может рассчитываться, если известен спектр облачных
капель и эффективность соударений частиц с каплями. Подоблач-
Подоблачное вымывание рассчитывается проще (гравитационная коагуляция).
для значений гл/гк « 1 при радиусах капель гк, превышаю-
превышаю50 мкм, и г.%, 0,5 мкм
(Stfc + 0,25J
с учетом эффекта зацепления.
4
Для более мелких частиц
р
Re.
Теоретические расчеты эффективности захвата частиц капля-
каплями были выполнены С.Гринфельдом, а также рядом других исследо-
исследователей [116, 141]. Например, М.Дана провел вычисления коэф-
коэффициента вымывания для монодисперсных и полидисперсных аэрозо-
аэрозолей в диапазоне радиусов частиц от 0,001 до 10 мкм [141]. В
большинстве теоретических работ достаточно полно исследованы
случаи броуновского осаждения частиц на облачных и дождевых
каплях и инерционного захвата частиц при средних числах Вэйноль-
дса. Особое внимание уделяется учету эффектов "зацепления" и
силы тяжести в области минимума вымывания аэрозольных частиц.
Вымывание всех частиц определяется двумя процессами: захватом
частиц облачными каплями (ot>) и удалением облачных капель
($). Меньший по величине коэффициент определяет скорость уда-
удаления частицы из атмосферы: для г 4 0,01 мкм &&& и вымыва-
вымывание определяется удалением облачных частиц из атмосферы; для
наиболее крупных частиц оь 4. & и более важным становится учет
процесса захвата частиц облачными каплями. Модельные экспери-
эксперименты по определению коэффициентов захвата частиц каплями, а
также телами разных форм, близких к встречающимся в атмосфере
(ледяные частицы, снежинки, нити и иглы растений), позволили
оценить влияние несферичности на величину коэффициента захвата.
147
Причем экспериментально исследовалось также влияние испарения
капли на величину коэффициента захвата частиц, и было показано,
что ледяные кристаллы, растущие в смешанных облаках, обладают
большей способностью собирать инородные частицы, чем капли
воды.
Основные закономерности влажного вымывания аэрозольных час-
частиц четко сформулированы в работе [21]: 1) вымывающее дейстше
жидкокапельных облаков на тонкодисперсный аэрозольный компо-
компонент пропорционально сумме поверхностей всех капель (T^-JVr^);
2) вымывающее действие жидкокапельных облаков по отношению к
самым мелким аэрозольным частицам пропорционально сумме радиу-
радиусов всех капель (т~* ~NrK); 3) вымывающее действие жидких
облаков и осадков на очень мелкие аэрозольные частицы обратно
пропорционально поверхности этих частиц (т ~ г^2 ) ; 4) ми-
минимум влажного вымывания наблюдается для частиц с размерами,
для которых влияние броуновской диффузии на осаждение уже дос-
достаточно мало, а влияние зацепления еще недостаточно эффективна
В области минимума вымывания сечение захвата на несколько по-
порядков меньше геометрического. Экспериментально область миниму-
минимума влажного вымывания была обнаружена автором при измерениях
концентрации и дисперсности аэрозольных частиц в приземном слэе
в районе Рыльска и в Северо-Западном Казахстане в 1967-1968 гг.
В последние годы были выполнены модельные эксперименты по
определению коэффициента захвата аэрозольных частиц падающими
водными каплями. Так, Л.Ванг и Х.Пруппахер экспериментально оп-
определили эффективность захвата аэрозольных частиц из JnAl3
с удельной плотностью 1,32 г/см и радиусом около 0,25 мкм вод-
водными каплями (в диапазоне 150-2500 мкм), падающими с высоты
35 м при счетной концентрации модельных частиц в камере 10^-
V —Я
-10 см . Экспериментальная зависимость коэффициента захвата
ап
от размера капель в отсутствие электрических зарядов на
каплях совпадает с теоретической. С увеличением радиуса капли
от 150 мкм наблюдается уменьшение Ег„ до 2*10 при 250 мкм
с последующим увеличением до 10~2 при гк = 525 мкм и затем
монотонное уменьшение ?гр вплоть до гк ? 1000 мкм. Такая за-
зависимость коэффициента захвата частиц каплей объясняется тем,
что захват происходит на тыловой части капли и обусловлен осо—
148
бенностями турбулизации среды за падающей каплей, зависящими от
числа Рейнольдса для капли. Наличие заряда на капле увеличивает
коэффициент захвата. Аналогичные исследования на частицах AgCl
в диапазоне 0,15 мкм 4 г 4 0,45 мкм для водных капель ра-
радиусом от 0,6 до 2,6 мм показали, что для капель с гк = 0,62;
0,82 и 0,98 мм наблюдается минимум коэффициента захвата ?г„
аэрозольных частиц с га - 0,25 мкм при отсутствии электричес-
электрического заряда на капле. Это объясняется особенностями захвата
частиц передней и задней областями капли. При изменении разме-
размера капли от 0,62 до 2,60 мм наблюдается почти линейное убыва-
убывание коэффициента захвата Е^ для аэрозольных частиц с га =
= 0,15; 0,25 и 0,36 мкм. Зависимость Ег от величины поверх-
поверхностного заряда капли для незаряженных аэрозольных частиц име-
имеет симметричный характер относительно знака заряда. При опреде-
определенных величинах плотности поверхностного заряда наблюдается
максимум эффективности захвата частиц, обусловленный появлени-
появлением индуцированного заряда на аэрозольной частице. (Для капель
меньших размеров максимум наблюдается при меньших значениях
поверхностного заряда.) Дальнейшее увеличение поверхностного
заряда выше предельных значений приводит к частичному отеканию
заряда с капель на частицы и уменьшению коэффициента захвата.
Определение эффективности вымывания аэрозольных частиц дождевы-
дождевыми каплями в подоблачном слое с помощью одновременного измере-
измерения дочерних продуктов распада радона Rn в дожде, облаках и
у поверхности земли, выполненное В.Хиксом, дало величину ин-
интегрального коэффициента эффективности вымывания ?"_ * 10 -
Экспериментальные наблюдения показывают, что вымывание
частиц осадками в сильной степени зависит от типа осадков.
К.Итагаки и СДоенума, Е.Рейтер и Х.Георгии установили, что
снежные хлопья и падающие ледяные кристаллы являются более эф-
эффективными "мусорщиками", чем капли дождя [93, 110]. Х.Геор-
Х.Георгии считает, что высокая очищающая способность плавающих снеж-
снежных хлопьев обусловлена их большой поверхностью. Тщательное
исследование вымывания аэрозолей снежинками было проведено И.
Кагоно и др. [200]. Изучение сухого остатка снежинок различной
конфигурации под оптическим и электронным микроскопами позво-
149
Таблица 24
Вымывание химических элементов
осадками, мкг/мэ
Тип осадков, мес-
место наблвдений
Дождь, Ерментау,
30 УШ S8
Снегопад,Ленинград
3-4 1У 69
Номер
пробы
1
2
3
4
5
S
(Контр.]
1
2
3
5
6
(Контр.)
Эле
Mg
1,10
2,4
1,0
1,2
0,6
0,03
0
2,0
1.S
0
1,5
0,039
Мп
2,е
0,6
0,3
0
0
0,005
0,01
0,06
0.0S
0,05
0,02
0,017
РЬ
•
0
0
0
0
0
0,47
0,75
0
•
0,011
Fe
19,5
2,0
3,3
2,7
1,9
0,07
2,4
8,7
1.67
2,67
10,0
0,029
лило выявить, что кристаллы разной формы захватывают аэрозоль-
аэрозольные частицы примерно одинаково. Количество захваченных частиц
с г, < 0,2 мкм для одного кристалла порядка нескольких тысяч,
а поверхностная плотность частиц примерно 2,7*10° см . Иссле-
Исследование расположения и состава частиц в кристалле позволяет ут-
утверждать, что захват частиц происходит в основном во время па-
падения снежинок, а не в облаке. Определяемый из эксперимента но-,
эффициент захвата получается выше теоретических оценок. Было
также обнаружено, что коэффициент захвата увеличивался при
уменьшении интенсивности снегопада.
Сходные результаты были получены в измерениях вымывания
аэрозолей приземного слоя осадками различных типов в работах
автора. Был выявлен селективный характер вымывания как для
разных фракций частиц (рис.8), так и для различных химических
150
мент
Ni
1,1
0,3
0,3
0
0
0
0,11
0,03
0,03
0
0
0
Al
1,0
11,5
5,3
4,0
1,6
0,14
0,10
1,15
0,53
0,40
0,16
0,14
Са
15,0
3,5
0
0
0
0,4
15,0
0,35
0
0
0
0
Си
4,4
3,3
1,9
2,5
1.2
0
0,43
0,33
0,19
0,25
0,12
0
0
0
0
0
0
0
0
0
0
0
0
0
Zn
4,8
5,8
0
2,8
0
0
0,48
0,58
0
0,28
0
0
Сг
_
-
_
—
-
-
0
0
0,24
0,32
0,64
0
соединений (табл.24) [43, 45]. В частности, было обнаружено
слабое вымывание аэрозольных частиц, содержащих элементы Fe и
А1. Очевидно, что это связано не только со слабой гигроскопич-
гигроскопичностью частиц, в состав вещетва которых входят эти элементы,
но и с различным химическим составом частиц разных размеров.
Наблюдающееся некоторое увеличение содержания этих элементов
во время дождя свидетельствует о существовании тропосферных сло-
слоев аэрозолей, содержащих в своем составе окислы этих металлов.
Вымывание аэрозольных частиц облаками и туманами весьма
эффективно в первую очередь для гигроскопических и смешанных
частиц, поэтому химический состав проб в облаках и туманах не
вполне соответствует химическому составу всех атмосферных аэ-
аэрозолей. Подтверждением неполного соответствия химического со-
состава частиц являются данные о составе сухих аэрозолей туманов
и облаков, полученные Л.Куроива, Г.Ямамото и Т.Отаке.
151
й Ну
0,9
0,7
0,5
0,3
0,1
-
с
1
У
г
1
_?
\
X
X
. л
Y
\
\
i
{
г
X
X
/
\
V
\
_<
с /
\ /
J
(
A
i
<*<
A/2
tit
: •¦¦ &
1/ \
i
/
/ <
/yS j
1
1
1 1
1 1
J ;
P
4
T
f
b
3
4
0,02 0,05 0,1 0,2 0,5 1,0
5,0 10 г,мкм
Рис.8. Отношение концентрации'частиц разных размеров после
осадков к концентрации до их выпадения.
1 - через 2 ч после дождя (г.Рыльок, 17 08 67 г.);
2 - во время доадя (г.Ерментаз, 30 07 68 г.); 3- че-
через 10 мин после доадя (там же); ? - во время дож-
дождя (г.Рыльск. 21 07 69 г.): 5 - через 30 мин после
дождя (г.Рыльск, 21 07 69 г.).
Так как при выпадении осадков дождевые капли и снежинки
дополнительно захватывают аэрозольные частицы из подоблачного
слоя, то химический состав осадков несколько отличается от сос-
состава облачной воды. Большой материал по этим вопросам собран и
проанализирован в монографии Х.Юнге. Результаты исследований
химического состава облачной воды, полученные за последние го-
годы сотрудниками Главной геофизической обсерватории им. А.И.Во-
А.И.Воейкова в различных районах Европейской территории Советского
Союза на высотах 250-1000 м, позволяют сделать вывод о том,
что содержание нерастворимых компонентов в большинстве случаев
составляет 70-80$ от общей концентрации аэрозолей. В отдельных
пробах содержание нерастворимых веществ в подынверсионных сло-
152
ях достигало 300 - 600 мкг/м3. Вывод о том, что в северных рай-
районах концентрация нерастворимой части аэрозолей обычно не пре-
превышала 0,5 мкг/м3 и составляла 20 - 30$ от общего количества
аэрозолей, весьма интересен, но может быть не вполне точным
ввиду возможного неучета некоторых компонентов, например Fe.At.
Существенным для оценки интенсивности очищения воздуха яв-
является анализ состава осадков, ведущийся в настоящее время в
больших масштабах в разных странах. В частности, отмечается
большая концентрация некоторых металлов (особенно свинца) в
осадках, увеличение содержания кислоты в осадках над Скандина-
Скандинавией и другими частями Европы [74, 110]. Большой материал по
химическому составу осадков был проанализирован Е.С.Селезневой
с сотрудниками для территории Советского Союза. Из месячных и
годовых карт содержания различных химических компонентов в осад-
осадках следует, что эти компоненты распределяются в основном зо-
зонально, причем количество минеральных компонентов увеличивает-
увеличивается с севера на юг. В наиболее чистых осадках на севере в лес-
лесной зоне средняя минерализация 8-15 мг/л. К югу она возраста-
возрастает до 35 - 40 мг/л в районах Средней Азии. Минерализация об-
обусловлена в первую очередь сульфатами и гидрокарбонатами. Со-
Содержание хлоридов в осадках быстро уменьшается с удалением от
морских побережий в глубь континента.Оценки количества выпада-
выпадающих с осадками минеральных веществ дают 10 т/год мине-
минеральных солей [76].Отметим,что эта величина значительно прево-
превосходит приведенные ранее оценки мощности источников аэрозольно-
аэрозольного вещества.Следует также учесть,что значительная доля аэро-
аэрозольных частиц не вымывается осадками.
Е.С.Селезневой и ее сотрудниками было показано, что основ-
основной вклад в минерализацию облачной воды вносят сульфаты, а не
хлориды. Доля морских еэрозолей, вымываемых облаками, лишь 15-
20%, а 80-85$ примесей в составе имеют континентальное проис-
происхождение [96].
Измерения в северных районах интересны, в частности, не
только для оценки удельного веса аэрозолей различного происхож-
происхождения, но и для оценки интенсивности из глобального выведения
из атмосферы. В этой связи важны результаты анализа химическо-
химического состава льдов Гренландии, Арктики и Антарктиды. Для них ха-
153
рактерно присутствие остатков минерального вещества, принесен-
принесенного из более низких широт [19]. Необходимо учесть при этом,
что время жизни Са+2 примерно в пять-десять раз больше SO^2,
и, следовательно, в атмосфере высоких шрот содержание этих ксм-
понентов должно быть одного порядка. Практически почти нет дан-
данных, особенно для высоких широт, по содержанию в осадках (а
следовательно, и в аэрозолях) соединений Fe, Al и Si, в то
время как исследования последних лет указывают на весьма боль-
большой удельный вес компонентов, содержащих эти элементы. Очень
мало известно и об интенсивности выведения из атмосферы углеро-
углерода. Можно предполагать, что он удаляется почти также интенсив-
интенсивно, как сульфаты. Содержание углерода в осадках над Швецией
составляет для дождей 1,7 - 3,4, а для снега 0,8 - 1,9 мг/л.
Это свидетельствует о том, что основная доля углерода в аэро-
аэрозольных частицах, выводимых из атмосферы осадками, растительно-
растительного происхождения. Возможно, большая часть сажи быстро коагули-
коагулирует до гигантских размеров вблизи источников и не участвует в
образовании глобального аэрозольного фона.
Глава 4
РАСПРЕДЕЛЕНИЕ АЭРОЗОЛЕЙ В АТМОСФЕРЕ
Одной из важнейших характеристик аэрозольной частицы яв-
является ее размер, во многом ответственный за ее физические
свойства. Определение размера отдельной аэрозольной чаотвды,
вообще говоря, задача нетривиальная, особенно, если частица
имеет неправильную форму. Еще более оложно определение соответ-
соответствующей этому понятию характеристики для аэрозольной системы.
Сложность задачи заключается не только в трудности эксперимен-
экспериментального определения размеров и формы всех частиц системы в
каком-то объеме пространства, но и во введении такой характе-
характеристики аэродисперсной системы^как функции распределения час-
частиц по характеристикам, адекватно отражающим данную систему.
§ 4.1. Функция распределения частиц по размерам
Диспергированную фазу аэродисперсной системы можно пред-
представить как множество N векторов, имеющих т компонентов, со-
соответствующих тем характеристикам аэрозольной частицы, кото-
которые для нас определяют ее однозначно (размеры, форма, химичес-
химический состав частиц, электрический заряд и т.д.). Это множест-
множество векторов будет определять микроструктурное состояние дис-
диспергированной фазы в любой момент времени в виде точки в об-
общем фазовом пространстве. Если число дисперсных систем в обоб-
обобщенном фазовом пространстве достаточно велико, то в это прост-
пространство можно ввести непрерывную функцию о , равную плотнос-
155
ти систем, т.е. плотности точек, изображающих системн. Устано-
Установив точность определения компонентов вектора, отвечающего аэро-
аэрозольной частице, можно задать размеры некоторого объема в обоб-
обобщенном фазовом пространстве, в котором все изображающие точки
систем, попавших в него, будут физически неразличимы. Набор и
точность измерения переменных определят уровень описания сис-
системы. Совокупность систем, для которых можно ввести непрерыв-
непрерывную функцию у составит статистический ансамбль. Отдельная
система ансамбля - его индивидуальная реализация. Предел.от-
Предел.отношения функции р^ к количеству всех дисперсных систем при
беспредельном увеличении этого количества будет представлять
многочастичную плотность вероятности того, что каждая частица
дисперсной системы находится в соответствующем бесконечно ма-
малом объеме фазового пространства, будет определять коллектив N
частиц в целом. Такая N-частичная функция по эксперименталь-
экспериментальным данным для атмосферных аэрозолей практически вряд ли может
быть построена, да и содержит она значительно больше информа-
информации, чем это необходимо для описания определенных физических
свойств коллектива частиц в целом, Поэтому для упрощения ана-
анализа применяют так называемое сокращенное описание, в частнос-
частности унарную функцию распределения.
Унарная функция распределения получается из N -частичной
при предположении, что пространства изменения переменных всех
частиц совпадают. Интегрируя N-частичную функцию по перемен-
переменным всех частиц кроме f-й, получим плотность вероятности то-
того , что параметры : -й частицы будут находиться в бесконечно
малой окрестности переменной точки обобщенного фазового про-
пространства при произвольных значениях параметров других частиц.
Чтобы получить плотность вероятности для произвольной одной
частицы коллектива, т.е. получить унарную функцию, следует про-
провести усреднение по всем частицам коллектива. Соответствующее
выражение для унарной функции через N-частичную будет иметь
вид
Унарная функция распределения частиц по размерам часто называ-
называется спектром размеров частиц. В общем случае спектр размеров
¦156
частиц изменяется как во времени,так и в пространстве. Поэтому
среднее по статистическому ансамблю может характеризовать пове-
поведение отдельной реализации в момент I, если события, происхо-
происходящие в большинстве реализаций ансамбля, происходили за конеч-
конечный промежуток (l-Ai, i ) в отдельной реализации. Среднее
по ансамблю будет приближенно равно среднему для произвольной
реализации по промежутку времени порядка М , причем отклоне-
отклонение среднего по ансамблю от среднего по времени будет тем мень-
меньше, чем больше изображающих точек сгруппируется в бесконечно
малом промежутке фазового пространства. Этого можно добиться,
увеличивая промежутки времени &i (At должно быть значительно
меньше характерного времени изменения унарной функции распреде-
распределения) или огрубляя условие различимости отдельных реализаций
ансамбля с помощью усреднения всех или какой-нибудь части пере-
переменных, характеризующих коллектив частиц отдельной реализации,
например укрупняя интервалы деления частиц по размерам. Увели-
Увеличение минимально различимого объема фазового пространства долж-
должно быть существенно меньше размеров неоднородностей этой харак-
характеристики, а это определяется возможностью зафиксировать с по-
помощью физических измерений минимально различимый интервал раз-
размеров частиц Аг(г) и видом унарной функции распределения
(спектра размеров частиц).
При изучении физических процессов трансформации аэрозолей
в атмосфере имеют дело с унарной функцией
<q<x,y,z,r,t)= N(x,y,z,t)y<x,y,z,r,t). D.2)
По определению функция (l/N)<q(х,у,z,r,t)drdxdydz пред-
ставляет собой вероятность попадания произвольной частицы
диспергированной фазы с радиусом (г, r+dr) в момент време-
времени t в достаточно малый объем dxdydz, в котором расположе-
расположена точка с радиус-вектором o(x,y,z).
На основании сказанного величины М, йг
ограничены сверху: М «(dlny/dl)'1; Дг «<(Hn<p
(ДУI/3 _ (Да:ДуД2I/з « (Дгпср)'1. Функция y<x,y,z,
г, t) имеет вероятностный характер. Для однородной системы из
N частиц функция <q(r,i)]N характеризует средневзвешенную
157
вероятность попадания частиц в интервал размеров ( г, r + dr)
и единичный интервал пространства. Поскольку -ттСССС <f(r,l)*
«drdxdydz = I, то г3 °
<p(r,i) dr = nil),
D.3)
т.е. <fCr,?) - нормирована на концентрацию частиц. Отождест-
Отождествлять функцию (f (r,i) с фактической концентрацией частиц в
заданном единичном объеме в заданном интервале размеров можно
лишь для объемов, в которых среднее число частиц, определяемое
выражением Ф(г, I )dr, достаточно велико, а флуктуациями их
числа, обусловленными вероятностным характером функции <f(r,i),
можно пренебречь. Если система пространственно неоднородна, то
переход к реальным концентрациям более сложен. Для достаточно
большого числа частиц необходимо рассматривать достаточно боль-
большие конечные интервалы: AV=AxAyAz и Ьг. Аз условия <f(x,
у, 2, г, i) AVAr » 1 имеем, что AVAr » 1 / <р (г, х, у,
z, I) и, следовательно, AV и Аг не могут быть взяты малыми
одновременно. С уменьшением объема AV увеличивается интервал .
Аг, и наоборот.
Требование представительности статистической выборки на-
накладывает серьезные ограничения на интервалы А г , AV: AV
должно быть много больше характерного объема неоднородности
системы, а Аг должно быть достаточно малым, чтобы функция <р
не менялась существенно. Практически необходимо усреднить функ-
функцию ю по некоторому пространственному масштабу, обеспечиваю-
обеспечивающему достаточное количество частиц.
В унарной функции ш (х, у, 2, г, i ) переменная г может
быть заменена по соображениям удобства на другую: У«4яг*/3-
при рассмотрении коагуляциэнных процессов с жидкими частица-
частицами, т = 4згрг3/3 - при рассмотрении процессов загрязнения
атмосферы различными источниками, 5 = 4яг2 - при изучении оп-
оптических свойств аэродисперсных систем.
Эмпирическая зависимость унарной функции распределения от
времени и пространства устанавливается по измерениям унарной
функции от А1 и iV, которые могут бнть разными для различ-
158
ных моментов времени и точек пространства. Нормировка унарной
функции С *°<р (х, у,2, г, i)dr=n(x,y,z,t) на концентрацию
частиц имеет преимущества перед другими по следувщим причинам:
I) любой другой способ нормировки не позволяет изучать времен-
временные изменения системы, 2) вид функции практически не зависит от
точности определения счетной концентрации частиц во всем диапа-
диапазоне изменения размеров частиц.
Вместе с тем нормировка на концентрацию частиц несколько
затрудняет сравнительный анализ изменения вида спектра размеров
частиц, так как концентрации аэрозольных частиц в атмосфере мо-
могут изменяться на неоколько порядков. В этом случае использует-
используется нормировка на одинаковую концентрацию. Нормировка на едини-
единицу для полного спектра размеров нереальна из-за эксперименталь-
экспериментальных трудностей определения в первую очередь rmln. Кроме того,
в области rmln происходят наиболее сильные изменения фунции
распределения, а следовательно, условия, накладнваемне на АУ
ш AT, должны быть очень жесткими. Ограничения функции со сто-
стороны rmin определяются либо возможностями экспериментальных ме-
методов измерения размеров частиц, либо ролью частиц разных раз-
размеров в рассматриваемых физических процессах. Например, при по-
получении спектров частиц с помощью оптического микроскопа г . ?.
mm
%, 0,20 - 0,25 мкм, а при рассмотрении оптических явлений в ат-
атмосферных аэрозолях гт5л?, 0,04 мкм.
Описание спектра размеров аэрозольных частиц в виде фор-
формулы, зависящей только от г, предполагает, что длительная трш-
сформация функции распределения частиц по размерам в результа-
результате различных физических процессов в атмосфере приводит к дина-
динамическому равновесию содержания частиц во всех интервалах спек-
спектра и созданию стабильного распределения частиц по размерам.
По-видимому, такое стабильное распределение не единственно и
оно зависит от каких-то внешних параметров среды, которые нуж-
нужно определить, но во всяком случае оущеотвует ограниченное ко-
количество функций распределения частиц по размерам. Теоретичес-
Теоретические исследования существования автомодельных решений уравнения
коагуляции показали, что для некоторых частных случаев действи-
действительно имеются автомодельные решения и спектр размеров частиц
"забывает" свое первоначальное соотояние. Однако в области
159
"х. оста" распределения условие многократности столкновений мо-
&ет оказаться недостаточным и автомодельного распределения для
7* > j\p может не существовать.
Формулы, приводимые разными авторами, могут быть разбиты
на эмпирические, подбираемые в соответствии с конкретными экс-
экспериментальными данными; универсальные четырехпараметрические
эмпирические формулы, описывающие почти все получающиеся в экс-
эксперименте распределения; теоретические формулы, выведенные на
основе определенных физических представлений о закономерностях
образования и трансформации аэрозолей.
К первой группе формул, описывающих распределение частиц
по размерам, в частности, относится формула Юнге
П(Г)
L
dr
где С - коэффициент, определяемый концентрацией частиц и усло-
условиями нормировки, а $ - показатель степени, изменяющийся от
трех до шести для различных условий измерения. Она приближенно
описывает спектр размеров частиц в диапазоне изменения радиуоа
от ОД до 1,0 мкм. Широкое употребление формулы Юнге для опиоа-
ния экспериментальных данных связано не только с простотой оп-
определения показателя степени в этой формуле, но и с тем, что
приближенный расчет аэрозольных коэффициентов ослабления и рас-
рассеяния радиации для юнговских распределений весьма прост и по-
позволяет легко проводить сравнения спектрального хода этих ко-
коэффициентов с формулой Ангстрема для спектрального хода ослаб-
ослабления радиации в атмосфере в видимой области спектра. Правомер-
Правомерность этих операций оправдана тем, что в атмосфере в большинст-
большинстве случаев для видимой области спектра оптические характеристи-
характеристики аэрозолей определяются в основном частицами, имеющими раз-
размеры от 0,1 до 1,0 мкм.
Теоретические обоснования применимости степенных функций
распределения частиц по размерам для атмосферных аэрозолей мож-
можно найти в работах С.Фридлендера, В.И.Смирнова и др. [87-89,
159].
Большое количество эмпирических формул было предложено
для аэрозолей дисперсионного происхождения. Чаоть из этих фор-
формул может в некоторых случаях достаточно удовлетворительно опи-
160
сывать экспериментальные данные по атмосферным аэрозолям.
Нормально-логарифмическое распределение было одним из пер-
первых использовано для описания спектра размеров аэрозольных час-
частиц, функция распределения имеет следующий вид:
где о?г - геометрический средний диаметр частиц; &г - геомет-
геометрическая стандартная девиация: lad = Т. (nlgd)/N ;
lge> - ^Y,<nlgd -nlgdrJ/N' ;
lgdm = nlgdr + 2>303 (nm/2) lg*er ;
o?; = (i/lg(yr y^T) JVeocp [- (ig d - Ig dr )/2 lg\ ] 6 lg d;
площадь поперечного сечения частиц -
масса -
К эмпирическим формулам относится формула Хргиана - Мазина
П(г) - Аг2ехр(-Ъг), D.5)
которая хорошо описывает результаты измерений дисперсности в
аэрозолях конденсационного происхождения.
Среди универсальных эмпирических формул широкое примене-
применение нашло модифицированное гамма-распределение, использованное
с определенными рекомендациями Д.Дейрменджаном в расчетах мат-
матриц рассеяния для шести моделей распределения частиц атмосфер-
атмосферных аэрозолей по размерам
п<г) =
D.6)
и. 1»?.г
1РЛ
Легко показать, что предлагаемая четырехпараметрическая
функция распределения случайных величин является обобщением
большинства известных эмпирических и теоретических законов ста-
статистического распределения случайных величин. При определенных
значениях параметров она преобразуется з нормальный закон Гаус-
Гаусса-Лапласа, законы Максвелла, Пирсона и другие.
Модифицированная Г-кфункция для расчета оптических харак-
характеристик атмосферных аэрозолей была предложена Ж.Де-Луюзи с со-
соавторами в виде п(гу = Аг*ехр(-г).
В.И.Смирнов для описания спектра размеров аэрозольных час-
частиц использовал функцию
п(г) = а г
,-*,
D.7)
для которой достаточно просто определяются моменты р-то поряд-
порядка
М(р)
.и интегральное распределение
N(r) = j b(i~k)/s f((k-i)/s) %((k-s-l)/s ; b/rs) .
Эта функция, в частности, может достаточно хорошо заменять
функцию Юнге п(т) = - С г'4 при значениях к = 4, s = 1 в диа-
диапазоне изменения радиуса от 1,4«10 до 12 мкм.
Следует отметить, что интегральная функция распределения
n(r)dr используется многими исследователями в
Лпт
силу того, что ряд методов измерения спектра размеров частиц
позволяет измерять именно эту функцию. Например, для нормаль-
нормально-логарифмического распределения интегральная функция распре-
распределения при m = 3 имеет вид
Г(г) = [erf(z) + 1 ],
где z - (lnr-lnrr)/[VTbnerr-TOlner/^J'rr- средний
геометрический радиус, <уг - стандарт геометрического откло-
отклонения.
162
Как уже говорилось ранее,вид функции распределения частии
по размерам определятеся методом (выбор At , ДУ и Дг ) .местом
и временем измерения {x,ytz,i) > основными характеристиками
источников аэрозолей (начальная функция, возраст и химический
состав частиц).
§ 4.2. Методы измерения концентрации
и распределения частиц по размерам
Методы измерения аэрозольной структуры в атмосфере можно
разделить на дистанционные, в основном оптические, и прямые -
микрофизические. Практически все микрофизические методы связа-
связаны с предварительным забором пробы воздуха с последующими опе-
операциями,либо регистрации каких-то физических характеристик этой
среды, обусловленных присутствием аэрозольных частиц (оптичес-
(оптических, электрических и т.д.), либо осаадения аэрозольных частиц
на подложку. Уже при аспирации пробы воздуха в прибор происхо-
происходит определенное искажение спектра размеров частиц, особенно в
грубодисперсной фракции, вследствие их инерционности: увеличе-
увеличение концентрации гигантских частиц при линейной скорости забо-
забора пробы, меньшей, чем скорость набегающего потока (в случае
совпадения направления обеих скоростей), уменьшение их концент-
концентрации при линейной скорости забора пробы, большей скорости на-
набегающего потока, или при несовпадении направлений обеих ско-
скоростей. Учет искажения спектра из-за аспирационного эффекта не
представляет принципиальных трудностей.
В самом приборе искажение естественной структуры аэрозоль-
аэрозольных частиц может происходить в результате фазовых переходов ве-
вещества частиц и среды, а также под влиянием процессов, проис-
происходящих при осаждении частиц на препятствие. Особенно сложна
задача эффективного осаждения на подложку мелкодисперсной фрак-
фракции. В приборах без осаждения частиц на подложку основные труд-
трудности связаны с техническими возможностями регистрации наибо-
наиболее мелких частиц и неоднозначностью интерпретации регистриру-
регистрируемых сигналов. В атмосферных исследованиях пользуются практи-
практически всеми названными методами.
Т63
Основные приборы, используемые для изучения пространствен-
пространственно-временной структуры аэрозолей: готоэлектрические счетчики,
фильтры, импакторы и счетчики ядер конденсации. Эпизодически ис-
используются нефелометры, масс-спектрометр аэрозольных частиц
Гетца, седиментометры, электростатические счетчики, &, - радио-
радиометры и еще некоторые приборы с узкоспециальными задачами.Эти
приборы не являются универсальными, дающими достаточно полную
характеристику структуры аэрозольных частиц. Каждый тип прибо-
прибора измеряет какую-то характеристику аэрозолей для определенно-
определенного диапазона размеров частиц или для частиц, имеющих определен-
определенные физико-химические свойства, например способность служить
ядрами конденсации. Следовательно, при анализе и обобщении ре-
результатов экспериментальных исследований необходимо учитывать
особенности применявшихся методов измерений и находить логичное
объяснение кажущимся противоречиям в результатах измерений аэ-
аэрозольной структуры различными приборами и методами. Следует
также помнить, что практически все эти приборы не являются аб-
абсолютными. В ряде случаев невозможна даже надежная тарировка
зтих приборов из-за чрезвычайного разнообразия природы атмос-
ферннх аэрозолей и особенностей проведения эксперимента в атмо-
атмосфере, например при ракетных измерениях.
Особенности и характерные недостатки основных используе-
используемых приборов следующие: 1) фотоэлектрический счетчик измеряет
рассеянную радиацию от частицы, приблизительно пропорциональ-
пропорциональную квадрату радиуса частицы, диапазон измеряемых частиц огра-
ограничен минимальным радиусом - примерно ОД мкм; 2) фильтр прак-
практически улавливает все аэрозольные частицы, но часть этих час-
частиц может испариться или раствориться при обработке фильтра,
по размерам частиц ограничений практически нет, измерение
очень низких концентраций частиц при ограниченном времени за-
забора пробы затруднено; 3) импактор селективен по отношению к
частицам разных размеров, при соударении частиц с препятстви-
препятствием возможно разрушение частиц, возможно испарение частиц и их
коагуляция на подложке, очень сложна обработка проб под элект-
электронным микроскопом; 4) счетчик ядер конденсации измеряет кон-
концентрацию частиц, которые перерастают при пересыщении в
капли.
164
1'э сказанного следует, что счетные концентрации частиц,
полученные этими приборами, существенно различны. Различаться
может также и вид кривой распределения частиц по размерам. По-
Поэтому при приведении данных измерений концентрации частиц, по-
полученных первыми тремя типами приборов, указывают минимальный
размер частиц, определяемый в указанных измерениях, а для дан-
данных, полученных с помощью счетчика ядер конденсации, указыва-
указывают, что измерялись ядра конденсации.
Большинство методов измерения спектра размеров частиц не
позволяет проводить тонкую градацию частиц по размерам. Напри-
Например, у фотоэлектрических счетчиков это связано с особенностями
оптических свойств отдельных частиц в области г~Я . Например,
Дж.Роэен использует аэростатный фотоэлектрический счетчик для
измерения только двух (эпизодически трех) градаций размеров
частиц: d %¦ 0,3 и d 4 0,5 мкм [223]. Фотоэлектрические счет-
счетчики, используемые для измерения спектра размеров частиц в бо-
более широком интервале, имеют в основном около десяти градаций,
а в отдельных случаях до 30 - 60, что явно недостаточно для
точного описания функции распределения. Анализ фильтровых и
импакторных проб аэрозолей с помощью оптического и электронно-
электронного микроскопов показывает, что в реальных спектрах наблюдают- .
ся заметные отклонения от гладких кривых распределения. В слу-
случае серийных обработок аэрозольных проб количество градаций
при оптической и электронной микроскопии также ограничено из-
за методических и экспериментальных трудностей. Использование
каскадных импакторов тоже предполагает резкое ограничение ко-
количества градаций по размерам до пяти - шести фракций. Счетчи-
Счетчики спектра размеров частиц по электрической подвижности [195]
z индукционные счетчики также дают ограниченное число града-
градаций, кроме того, они предполагают определенные зависимости меж-
между размером частиц и их зарядом, которые могут не выполняться.
Практически непрерывные кривые распределения получают с по-
помощью метода диффузионных батарей и спектрометра Гетца. Од-
Однако эти методы имеют очень серьезные ограничения по примене-
применению для исследования атмосферных аэрозолей.
На данных, полученных разными методами, сильно сказыва-
сказывается влияние пространственной неоднородности функции распре-
165
деления частиц по размерам. Флуктуация экспериментальной функ-
функции распределения будут проявляться при уменьшении объема ис-
исследуемых аэрозолей и времени регистрации спектра размеров.Экс-
размеров.Экспериментальную проверку влияния времени измерения на вид функ-
функции распределения провели Т.Градель и Дж.Франей прибором, по-
позволяющим измерять спектр размеров г от 0,29 до 4,94 мкм за
10,8 с. Были обнаружены весьма заметные флуктуации функции рас-
распределения, растущие с увеличением размера частиц, что подтвер-
подтверждает высказанные ранее соображения о влиянии &1 и AV на
§ 4.3. Глобальные типы распределения
В глобальном масштабе выделяют три общих типа распределе-
распределения частиц по размерам в тропосфере: фоновый, океанический и
континентальный. Идеализированные кривые, демонстрирующие су-
существенные черты этих трех типов распределения, изображены на
рис.9. Фоновый тип распределения аэрозолей по размерам предпо-
предполагается репрезентативным для средней и верхней тропосферы.Кри-
тропосферы.Кривые а и «Г соответствуют случаям существования и отсутствия
непрерывной генерации мелкодисперсной фракции, причем фоновая
счетная концентрация всех аэрозольных частиц для очень чисто-
чистого воздуха предполагается равной 700 см~^.
Самый нижний слой воздуха над океанами, высотой около
2 км, содержит морские частицы. Океанический тип распределе-
распределения частиц по размерам отличается от фонового в интервале ра-
радиусов от 0,5 до 20 мкм, в котором концентрация частиц океани-
океанического типа увеличивается по сравнению с фоновой за счет час-
частиц, возникающих из морских брызг. Общая концентрация частиц
морского происхождения невелика - меньше 10 см .
ё нижней тропосфере над сушей предполагается репрезента-
репрезентативным третий тип распределения частиц по размерам - континен-
континентальный. Счетная концентрация всех частиц в этом случае воз-
оастает до 104 см в сельской местности и до 3*104 см~3 в не-
со
больших городах. В крупных городах она превышает 10 см .Рас-
.Распределение частиц с г < 0,1 мкм считается неопределенным.
166
dlgr
,см
-з
Эти типы распределений - слишком грубое приближение к дей-
действительности. Распределение аэрозольных частиц по размерам в
большей степени зависит от происхождения этих частиц, их хими-
химической природы и различных метеорологических (особенно микрофи-
микрофизических) характеристик среды, чем от концентрации частиц, хо-
хотя в общем случае и справед-
справедливо утверждение, что увели-
увеличение счетной концентрации
приводит вследствие усиле-
усиления коагуляции к перестрой-
перестройке спектра размеров частиц.
Затем коагуляционные эффекты
между всеми частицами, их
конденсационный рост и уда-
удаление из атмосферы (седимен-
(седиментация, вымывание и инерцион-
инерционное осаждение) приводят к
трансформации спектра разме-
размеров частиц. Оценки действия
этих факторов на частицы раз-
разных размеров были сделаны
Х.Юнге, Д.Хайди [178, 180],
И.Блиффордом и др. [122, 178,
180, 211].
На рисЛ0 изображено из-
изменение постоянной скорости
процессов выведения частиц в
зависимости от размеров час-
частиц по И.Блиффорду. Исполь-
Используя для начальной микрострук-
микроструктуры распределение Х.Юнге,
И.Блиффорд рассчитал измене-
10
10г г.мкм
Рис.9. Глобальные типы рас-
распределения частиц по размерам
(Х.Юнге).
I - фоновый тип; 2 - конти-
континентальный; 3 - океаничес-
океанический; а,О - возможные вариа-
вариации при непрерывном образо-
образовании частиц. Стрелка пока-
показывает влияние загрязнения
на положение максимума рас-т
пределения.
ние спектра размеров частиц
со временем под воздействием
названных факторов. Распреде-
Распределение имеет тенденцию со временем сужаться как со стороны ма-
малых, так и больших размеров. При этом модальный радиус переме-
1В7
щается в область больших значений до г — 0,1+0,2 мкм. Такие
условия могут реализоваться, например, в сухой чистой тропосфе-
тропосфере. Анализ наблюдаемых распределений, как правило, приводит к
аппроксимации его степенной
функцией Х.Юнге. Величина по-
показателя степени характеризу-
характеризует скорость убывания концент-
концентрации частиц с увеличением их
размеров. Этой формулой наи-
наиболее удовлетворительно опи-
описывается распределение частиц
по размерам в интервале г от
0,2 до 0,8 - 1 мкм, т.е. в об-
области таких размеров частиц,
которые определяют рассеива-
рассеивание свойства аэрозолей в ви-
видимой области спектра. Пока-
Показатель степени, равный четы-
четырем, характерен для централь-
центральной Европы, Англии и Северной
Америки. Примерно для трети
измеренных распределений час-
частиц по размерам в приземном
слое "атмосферы в районе Рыль-
ска получено юнговское рас-
распределение с 0=4. Для
степных районов Прикавказья
с помощью импакторных измере-
измерений получены распределения,
которые достаточно удовлетво-
ю-*
W'6
10-е
W4
ю-2
а
-
-
h * if
- /J
- //
, /
3
i i
i v
i i i
i
i
i
i
J
-Si
Iff*
10~2
10
-6
г, см
Рис.10. Скорости выведения
и гиперации частиц разных раз-
размеров из единичного объема ат-
атмосферы под влиянием различных
процессов.
/ - седиментация; 2 - кон-
конденсация; 3,3а - захват час-
частицами (коагуляция )\к,ка-
осадки.
рительно описываются форму-
формулой Х.Юнге с ^ = 3,5+4.
Теоретически юнговское распределение для тропосферы и
стратосферы получил С.Фридлендер, исходя из условий динамичес-
динамического равновесия процессов ухода и прихода частиц разных разме-
размеров вследствие процессов генерации, коагуляции и седиментации
[159]. Экспериментально наблюдаются распределения частиц по
168
размерам и концентрации, соответствующие фоновым морским аэро-
аэрозолям при счетной концентрации около 700 см и массовой кон-
концентрации 20 мкг/м3, которые хорошо описываются степенной за-
еисимостью с -0 = 4 для г Ь 0,2 мкм.Отклонения от этой кар-
картины объясняются существованием источников аэрозольных частиц
больших размеров с разными модами и, кроме того, эксперименталь-
экспериментально установленным фактом исключительно сильного влияния влажнос-
влажности на изменение спектра размеров частиц. То что частицам раз-
разного происхождения свойственны специфические распределения,час-
распределения,часто очень далекие от юнговского, показано, например, в работах
;,..Хайди [178, 180].
Анализ функции распределения аэрозольных частиц по разме-
оам можно использовать для оценки вклада различных механизмов
в образование атмосферных аэрозолей. Так, процессы дробления
при образовании дисперсных аэрозолей (пыли) приводят к нормаль-
нормально-логарифмическому распределению с максимумом плотности рас-
распределения в основном в области т Ъ. 1,0 мкм. Конденсационный
или фотохимический процесс образования аэрозолей из веществ,
имеющих очень низкую упругость насыщения пара, также приводит
:: образованию одномодальных распределений, но с максимумом плот-
плотности распределения частиц по размерам в области г 4 0,1+0,3
мкм. При действии различных процессов генерации аэрозолей бу-
будут образовываться распределения с несколькими относительными
максимумами функции распределения частиц по размерам. Коагуля-
ционный механизм роста частиц и механизм седиментации ведут к
размыванию относительных максимумов и образованию распределе-
распределения типа Юнге. Следовательно, многомодовые распределения мо-
могут сохраняться, если мала эффективность коагуляции, либо ма-
мало время существования аэрозолей после их образования.
интересно отметить, что распределения с несколькими мак-
максимумами наблюдались Р.Фенном (рис.11), а также другими иссле-
исследователями в приземном слое воздуха при невысоких значениях
счетной концентрации аэрозолей и в случае присутствия вблизи
места измерений нескольких источников аэрозолей. Обобщая ре-
результаты 329 серий измерений микроструктуры аэрозолей на вы-
высотах от 0 до 9 км, сделанный над сушей (Северная Америка) и
Тихим океаном, И.Блиффорд и Д.Жилетт [122] пришли к выводу,
169
о,г
Рис.11. Функция распределения частиц по размерам, полу-
полученным с помощью аэрозольного спектрометра Р.фенном в при-
приземном слое (а) и тропосфере (о ).
что микроструктура глобальных аэрозолей может быть охарактери-
охарактеризована как композиция трех перекрывающихся нормально-логариф-
нормально-логарифмических распределений для частиц следующих классов:а) субмик-
субмикронная фракция; б) большие частицы, определяющие рассеяния
света; в) гигантские частицы ( г > 1 мкм), являющиеся продук-
продуктами эрозии почвы, биологических процессов или разбрызгивания
и последующего испарения капель морской воды (над океаном).
К аналогичному выводу пришли К.Уитби и другие исследова-
исследователи, измерявшие распределение аэрозольных частиц по размерам,
начиная с частиц г = 0,015 мкм с помощью электростатическо-
электростатического счетчика. Особенно убедительно независимость поведения трех
фракций аэрозолей проявляется при анализе функций распределе-
распределения частиц по массе или объему. В большинстве случаев масса
фракции гигантских частиц значительно превосходит массу двух
170
(больших
d <* 0,17+
4= 8,75
других фракций. 3 таких кривых распределения частиц по размерам
отчетливо наблюдается минимум в области г = 1+2 мкм. Однако мо-
может наблюдаться и непрерывный рост кривой dV/dr с увеличением
размеров частиц. Подобного рода кривые были получены автором
при измерениях в приземном слое атмосферы в Северо-Западном Ка-
Казахстане, когда в атмосфере присутствовали частицы дисперсион-
дисперсионного происхождения, в том числе и субмикронных размеров.
На основе статистической обработки данных по функции рас-
пргделения аэрозольных частиц по размерам в разных точках США
К.Уитби получил следующие данные по структуре аэрозолей:среднее
отношение массы грубодисперсной фракции к массе тонкодисперсной
примерно равно двум; для чистого воздуха - при d< 1 мкм т =
= 4 мкг/м3, а при d > 1 мкм т = 9 мкг/м3; для средних усло-
условий т = 50 мкг/м3 при d < I мкм, а при d > 1 мкм т = 120
мкг/м3; средний геометрический диаметр ядер Айткена d& —
¦* 0,017 мкм; e>s= 1,74; для аккумулятивной фракции
аэрозольных частиц) - sv <** 2,05, df-0,34 мкм при
+0,80 мкм; для грубодисперсной фракции ffv « 2,33,
мкм при d - 3,5+25 мкм.
Экспериментальные данные, полученные автором и сотрудни-
сотрудниками его лаборатории путем забора аэрозольных проб на разных
высотах в атмосфере, позволили предложить некоторые типовые
модели распределения аэрозольных частиц по размерам (табл.25),
которые имеют характерные особенности: наличие диапазона 1,54
& г 4 5 мкм, где плотность распределения выше, чем следует
из распределения Юнге; пониженное содержание частиц с г 4
? 0,3 мкм по сравнению с данными оптических измерений.
Следует отметить, что при обработке проб, взятых на фильт-
фильтры ФПП-15, терялись частицы органического происхождения. Так
как микроструктурные характеристики в этом случае показывают
малую чувствительность спектра частиц (в основном минеральных)
к величине влажности, то можно предполагать, что при увеличе-
увеличении влажности растут в первую очередь частицы органическо-
органического происхождения.
171
Таблица 25
Экспериментальные модели распределения аэрозольных частиц
по размерам (uV/Дг) (нормировка ^ Дг
V — ДгИООсм3)
_
7*, МКМ
0,04
0,05
0,07
ОДО
0,15
0,20
0,25
0,30
0,40
0,50
0,70
0,80
0,90
1,00
1/50
2,00
2,50
3,00
5,00
10,00
30
150000
80000
30000
10000
3000
2500
700
200
141
70
17,5
8,00
1,50
0,50
0,50
0,30
0,0010
0,0001
0,00000
0,00000
Модель
23-30
100000
60000
25000
10600
5000
2800
700
300
144
80
7.0
5,60
5,60
4,50
3,00
3,00
0,60
0,07
0,00001
0,00001
слоя атмосферы, км
15-25
88000
75000
52000
20000
4500
1250
700
300
131
112
25,0
17,0
12,0
8,50
1,60
0,20
0,06
0,015
0,00003
0,00001
S-15
150000
90000
15000
5600
4000
1900
1000
150
110
85
20,5
19,0
17,0
15,0
4,00
2,00
0,40
ОДО
0,00007
0,005
1-9
150000
90000
30000
12000
5500
2800
700
300
90
45
27,0
23,0
23,0
27,0
3,30
3,00
1,40
0,40
0,040
0,005
0-1
93300
37000
9300
4000
2200
1070
530
400
220
70
12,7
8,80
6,80
5,50
2,50
1,05
0,63
0,31
0,075
0,00145
§ 4.4. Распределение аэрозолей
в приземном слое атмосферы
Подробный анализ данных по концентрации аэрозолей в при-
приземном слое атмосферы приведен в работах [19, 24, 38, 110].
Работы последних лет не добавили существенно новых результатов
к уже имеющимся. Отметим лишь, что вариации концентрации час-
частиц в приземном слое особенно велики: для ядер конденсации от
W4 до 101 см~3, для больших аэрозольных частиц - от 10° до
10~к см и определяются как свойствами подстилающей поверхнос-
поверхности и других источников, так и метеорологическими характеристи-
характеристиками; влажностью, временем суток, скоростью ветра. В последние
годы выяснилось, что перестройка структуры аэрозолей происходит
особенно сильно в приземном слое от 0 до 500 м. Здесь наиболее
отчетливо проявляется влияние подстилающей поверхности, сезона
года и времени суток на концентрацию и вертикальный профиль кон-
концентрации аэрозолей. В частности, представляют интерес кривые
зависимости счетной концентрации аэрозолей, имеющие колоколооб-
разный вид, в послеполуденное время от температуры воздуха в
полдень (рис.12). Очевидно, что такая зависимость определяется
взаимодействием процессов генерации аэрозолей почвой (нагрева-
(нагревание почвы) и процесса уноса аэрозолей в более высокие слои ат-
атмосферы (конвективный подъем воздушных масс). Эти процессы мо-
могут приводить к появлению различных типов вертикальных профи-
профилей концентрации аэрозолей в нижних слоях атмосферы, в том чис-
числе может наблюдаться возрастание концентрации аэрозолей с вы-
высотой.
Большой цикл исследований структуры аэрозолей в приземном
слое проводился в районе Рыльска в осенне-летние периоды, начи-
начиная с 1967 г. В измерениях летом 1979 г. кроме продолжения про-
программы исследований структуры аэрозолей с помощью забора проб
на фильтры были проведены сравнения различных методов определе-
определения структуры аэрозолей. Аэрозольные пробы в приземном слое
брались на фильтры типа ФПП при одновременных измерениях дис-
дисперсности аэрозолей с помощью фотоэлектрического счетчика
АЗ-4М. Более 700 спектров, полученных с помощью фотоэлектричес-
фотоэлектрического счетчика в период со 2 по 23 августа, были подвергнуты
статистической обработке с целью выявления среднего суточного
хода функции распределения частиц по размерам, а также средних
функций распределения за каждые сутки и за весь период измере-
измерений. Полученные результаты приведены в табл.26 и 27. Обработка
проводилась без приведения к нормированным значениям счетной
концентрации и выбрасывания данных, которые представлялись
аномальными. Несмотря на это, все средние Функции распределе-
173
172
80\
60 \
Z0\
12
16
20
28
т;с
Рис. 12. Зависимость концентрации частиц (г> 0,2 мкм)
в послеполуденное время от температуры в полдень.
1-3 - Рыльск, август 1967 г. - 18, 21, 24 ч; *-«-
Сев.Казахстан, июль 1968 г. - 18, 21, 24 ч.
H trl М М м in
о м м о о о о о" о о* d
oooooo
CO O
П.О.Я до т
ОО cq Ocq 6B
К O5
4 <J
со а
о
О.
ф
о
в
в
Еч
3
ф
CD
Н
Ф
ft
176
I
оо
со со
о см ы см
П ГО О1 О) 0J П
оооооо
I
ОО
О
ОО
кНСМ
) О N И N О| ы и ¦» uj ш 1ч ч, - ..¦„ ..........
эооооооооооо ооооооо
зооооооооооо ооооооо
оооооооооооо ооооооо
о* о" о" о" о о о* о" о* о о о ооооооо
05 СМ кН
ш „ Ч1 05 СО -
8 8 8 88S
О
з^исо^юсоосбп
ч, vH СМ СМ н и СМ И Н ОН
оооо ооооооо
оооооооооооо ооооооо
ississssEsas. 5 s.a.g.tsa
о* о" о* о* о* о* о* о* о* о о о ооооооо
¦si1 СМ О
о см n
„ „ . -. _ ,- ¦*
со cd со см кг1 ¦* со
"-> Ч1 ¦si1 Ю VC СМ •*
о о о о о
ЮЮ?>-ЮЮ1^05с
мсмю^^юч1^ , ., .
эоооооооо ооооооо
со en
о*о*
о-со
ОО
COLOCNicd^CD^kHlOtDt^ t-^v-jv^^-.».- n,^.
СОМ0)СМ(ЛЧ(ЮФЧ|(БЮ^^101СОЧ|ММ|
оооооооооооооооо о"о
ю to со cd
кН кН СО СО
К со ю ю
СМ кН
О- Oj
Ч1 Ю
ЭООООкНООООООО ОООООО
ОО
Q0O
см со
ев
I
СГ)^ЮОЮО^ООСОС?)иОСООЮ
(ОСМО-СМЮ1Псои>СОСМС?)СООСМ
ЮЮЮ05?>.С?)СОкч^ООСПС^ Ю О
OOo'o"cMOO*kHOkrtOOOkrlo'oOO
о ю ю !>¦ Q 1>
о см о со см см
оо^ oinoooino
COCDkHCQ^CMCOtOCOCOCOtr)
CD?>-CDOOC0CMC005t?)kHC0
s
Ч1 Щ 1> щ wj nj ^i- ^ щ -- —
QCMkHCMOOCOCM^CMOCMOCVCOtO
CM CO О M
О N (О О
ю см
СО СО CO krt О
Ю СО СМ СМ О
CDCMOIO CVCMINCM
мсмюмсоимч1
со to
COO
CnCDCNmCOOOCDkrtCO
СМ кН М
со" о со <J>
о со со
CO Ю Ю Ю Ю
" t?) Ч1 О И
СО О N N NN
¦^i1 О Ю О О О
СПСОЮСОСПСПСООЮкнР^^кЧ Ш О Ч1 О fflM
СМСОкН^СОкНСМСМксНСМкНСЧ) кНкНкНкНСМ^С
«?«y«PEgSS8SSss
СО CD к1 СО
¦si1 кН СО О-
CM CM CV кН СМ
01 °> g S n°,
' СО^
сососососососососососососо
ооооооооооооо
¦^•ЮС^СОСПОкНСМСО^ЮСОС-
УМНМНИИи
ния частиц по размерам, полученные с помощью общего усреднения
данных, усреднения среднесуточных и двухчасовых средних, оказа-
оказались весьма сходными (см. табл.26). Анализ полученных данных
подтвердил, что основными процессами, определяющими средний су-
суточный ход функции распределения, являются механизмы генерации
частиц, конвективного подъема и диффузии, а затем седиментации
частиц. При усреднении не проявилось влияния конденсационного
механизма на изменение дисперсности частиц.
На графике зависимости появления относительных максимумов
для частиц разных размеров от времени обнаруживаются определен-
определенные закономерности (рис.13): после восхода солнца в утренние ча-
часы относительные максимумы наблюдаются практически для частиц
всех размеров - генерация дисперсионных (пылевых) частиц; в ве-
вечерние часы максимумы начинаются с наиболее крупных частиц и
закономерно передвигаются в сторону более мелких - седимента-
седиментация частиц из более высоких слоев атмосферы. Есть еще одна
ветвь суточного перемещения максимумов, которая начинается в
момент восхода солнца с размера порядка г = 0,1 мкм и перед-
передвигается до частиц с размерами г = 0,7+0;8 мкм. На рис.13 она
соответствует прямой. Если предположить, что в приземном слое
наблюдается рост каких-то частиц, то изменение массы этих час-
частиц во времени описывается экспонентой с константой скорости
К = 3-Ю с.
Возникает вопрос, почему в ранее проведенных измерениях не
было обнаружено проявления гетерогенного процесса роста аэро-
аэрозольных частиц в суточном ходе функции распределения частиц по
размерам? Основная причина - в отсутствии достаточно длинных
рядов наблюдений для маломеняющихся воздушных масс.Наблюдения
1979 г. проводились в весьма специфических условиях. В предут-
предутренние часы в месте проведения измерений (пойма р.Сейм) еже-
ежедневно происходило интенсивное вымывание аэрозолей каплями ту-
тумана и процессы генерации и трансформации частиц каждый раз на-
начинались заново во всем приземном слое атмосферы. Адвективных
потоков воздушных масс в нижнем слое атмосферы в период наблю-
наблюдений практически не наблюдалось.
В наблюдениях предыдущих лет проводились только фильтро-
фильтровые заборы аэрозолей. Как известно, очень большой вклад в филь-
12 .тг
177
тровые измерения, особенно в массовую концентрацию, вносят ги-
гигантские частицы дисперсионного происхождения (пыль) о г «
гг 10 мкм. В фотоэлектрическом счетчике эти частицы на входе
отсекались, что и позволило более отчетливо увидеть вклад фото-
фотохимических аэрозолей в распределение частиц по размерам.
Так как время нахождения аэрозольных частиц в приземном
слое сравнительно мало: от нескольких часов до нескольких су-
суток, а процессы генерации частиц очень разнообразны, то и спе-
спектр размеров аэрозольных частиц здесь наиболее широк: от сотых
микрона до нескольких десятков микрон, причем распределение чао-
твд по размерам сильно зависит от местннх источников и быстро
меняется в течение суток.
Один из наиболее дискутируемых вопросов в научной литера-
литературе по структуре атмосферных аэрозолей - соотношение медцу
морскими и континентальными аэрозолями. Если лет 20 назад пре-
преобладающим было мнение о достаточно четком разделении типов
морских и континентальных аэрозолей, то в последние годы эта
точка зрения подвергается серьезным сомнениям и вызывает возра-
возражения. Многочисленные исследования микроструктуры аэрозолей в
приземном слое атмосферы свидетельствуют о том, что морской тип
распределения аэрозолей по- размерам в значительной степени оп-
определяется физическими условиями образования спектра, в первую
очередь высокой относительной влажностью воздуха над морской
поверхностью, а не особым химическим составом аэрозольных час-
частиц. Кроме того, специфические аэрозольные частицы морского
происхождения существуют только в небольшом диапазоне высот нац
морской поверхностью( z 4 1,5+2 км) и проникают всего на
несколько километров в глубь суши. Например, измерения на по-
побережье Атлантики в районе Бреста показали, что аэрозоли мор-
морского, континентального и городского происхождения сильно пере-
перемешаны. При ветре, дующем с моря, показатель степени юнговско-
го распределения был следующим: -Р**-0 - 1 =1,8 для диапазона
размеров 0,3-4,0 мкм, 2,7 - для области 4,0-40 мкм и 4,9 - для
г > 40 мкм. Для случая, когда аэрозоли можно было назвать чис-
чисто городскими, о* = 3,5 в диапазоне 0,3-4,0 мкм, 2,9 - для
г = 4,0-40 мкм и 6,0 - для г> 40 мкм. Распределение час-
частиц по массам достаточно хорошо описывалось тремя нормально-ло—
179
гарифмическими распределениями. Измерения на Кубе показали, что
98% спектров описываются нормально-логарифмическими распределе-
распределениями для области г - 0,005+5 мкм с максимумом распределения
в области 0,01 - 0,05 мкм.
В работах группы сотрудников Главной геофизической обсер-
обсерватории им.Воейкова при измерениях аэрозолей на побережье Чер-
Черного и Азовского морей с помощью трехкаскадного импактора было
получено юнговское распределение для частиц 1 мкм^г^Т2 мкм
с ¦?*, меняющимся от 1,4 до 4,2 при среднем значении -9\
Л^М — 0,75 см . Экстра-
Экстра= 2,5 и счетной концентрации ^>1МКМ ,
поляция этой величины на интервал d ¦>• 0,5 мкм дает величину по-
порядка 5-10 см. При измерениях счетной концентрации на о.Сан-
Николас в 130 км к западо-гого-западу от Лос-Анджелеса были по-
получены значения 20 - 100 см , а при измерении концентрации
3
ы а , р
Айткена - в среднем 2000 см. Средняя масса аэрозольных
/3 11$
здер Айткен рд
частиц оказалась равной 29,8 мкг/м3, причем лишь 11$ массы аэ-
аэрозольного вещества соответствовало морской соли.
В измерениях, проведенных над морской поверхностью при
удалении от Токио до о-ва Огасаваре, наблюдалось сильное умень-
уменьшение концентрации крупных частиц, что не могло быть объяснено
только седиментацией аэрозолей. Исследовалось изменение спект-
спектра размеров частиц при распространении их с суши над океаном
на расстоянии до 1000 км к югу от Токио по подвижности атмос-
атмосферных ионов для диапазона 3 - 80 нм и фотоэлектрическим счет-
счетчиком для частиц с г > 0,1 мкм. Было обнаружено, что наибо-
наиболее медленно изменения происходят в диапазоне размеров 0,01
мкм 4 г ^0,1 мкм, причем наблюдается уменьшение счетной
концентрации и смещение максимума распределения частиц по раз-
размерам в сторону малых частиц [207].
Максимум в распределении частиц по размерам для естествен-
естественных аэрозолей был обнаружен в области г — 3*4'10 см. Па-
Параллельные измерения дисперсности радиоактивных аэрозолей по-
показали, что размер радиуса максимума распределения для них ока-
оказался в полтора раза больше, чем для естественных аэрозолей.
Измерения над океанской поверхностью вдали от берегов
весьма противоречивы и выявляют лишь отсутствие однородного
типа распределения, характерного для морских аэрозолей. В цен-
180
тральной Атлантике летом 1974 г. было получено юнговское распре-
распределение с -i * = 3 для диапазона размеров частиц с 1-20 мкм.
А.Г.Лактионовым наблюдалось эффективное вымывание гигантских
аэрозольных частиц в тропической зоне Атлантики, нарушающее
типичный вид морского распределения. А.Месарош отмечает слож-
сложный вид функции распределения аэрозольных частиц на расстоянии
600 км от берега. Аппроксимация спектра юнговским распределени-
распределением дает значения показателя степени ¦?*, равным пяти для час-
частиц с г< 0,5 мкм, \>* =14-2 для 0,5 4 г 4 1,5 мкм, а для
13$ спектров ¦?*= 3 при г ^ 1,5 мкм.
Большой наблюдательный материал по дисперсности аэрозолей
в районе тропической Атлантики, полученный в период АТЭП-74,
позволил провести статистический анализ результатов измерений
для разных слоев атмосферы и выявить основные закономерности
дифференциальной функции распределения частиц по размерам. Наи-
Наиболее типичные функции распределения представлены в табл.28.
Для всех них характерно наличие заметных отклонений от гладких
функций распределения, типа степенных или нормально-логарифми-
нормально-логарифмических в области гигантских частиц.
Сравнение функций распределения частиц по размерам, полу-
полученных в приводном слое в тропической и северной Атлантике, с
функциями распределения частиц по размерам, полученными в рай-
районах, удаленных от океана (Прибалтика, Сибирь), показывают,что
важнейшим условием наблюдения океанического типа распределения
частиц по размерам является условие высокой относительной влаж-
влажности воздуха в течение длительного {промежутка времени.
Можно достаточно уверенно предполагать, что для естествен-
естественных приземных аэрозолей в континентальных районах основным ис-
источником частиц является почва, кроме районов с густой расти-
растительностью в период интенсивного выделения растительностью уг-
углеводородов класса терпенов.
А.Г.Лактионов обнаружил, что значительная доля спектров
аэрозольных частиц в приземном слое атмосферы не описывается
формулой Юнге, а имеет выпуклый и вогнутый характер (в лога-
логарифмическом масштабе). Установлена отрицательная корреляция
181
Распределение аэрозольных частщ
(см~3)
над
Таблица 28
тропической Атлантикой в период проведения АТЭП-74
Г, мкм
0,05-0,1
0,1-0,15
0,15-0,2
0,2-0,25
0,25-0,3
0,3-0,4
0,4-0,5
0,5-0,75
0,75-1,0
1-1,5
1,5-2,0
2-2,5
2,5-3,0
3-4
4-6
>6
N<>i,0)
"oft*
НИСП "Пасса!
20 07
36,2
35,3
27,9
17,0
4,90
11,7
6,40
4,50
1,70
0,080
0,125
0,062
0,016
0,008
0,003
0,0001
0,293
150,9
29 07
123,0
67,2
31,0
14,6
5,40
3,03
1,75
1,40
0,300
0,168
0.0455
0,0164
0,0033
0,0020
0,0005
0,0001
0,236
247,9
" 0° ш., 12° з.д.
30 07
98,4
52,2
16,4
10,2
6,00
10,2
3,20
4,60
1,60
0,032
0,058
0,025
0,020
0,030
0,0080
0,0004
0,173
203,0
31 07
48,0
63,7
14,2 .
4,43
1,20
0,93
2,50
0,133
0,235
0,133
0,019
0,0035
0,0032
0,015
0,0045
0,0005
0,179
135,5
10 08
20,4
84,0
36,2
13,2
8,20
15,8
6,00
10,0
3,60
0,230
0,890
0,!160
0,080
0,150
0,040
0,0020
1,55
198,0
Примечание: Самолетные измерения проводились
между мелкими и крупными (гигантскими) частицами. Причем
спектр размеров гигантских чаотиц слабо изменялся с изменени-
изменением величины относительной влажности, т.е. основная доля ги-
гигантских частиц - твердые негигроскопические частицы .Некото-
.Некоторая корреляция мелщу содержанием чаотиц в разных областях
спектра наблюдается для спектров дымок.
НИС "Пр
12 07
53,0
46,4
28,1
15,0
7,60
4,20
2,35
2,10
0,960
0,170
0,110
0,0410
0,0240
0,0300
0,0850
0,0098
0,470
160,2
оф.
ъ
76
82
31
13
8
5
3
3
0
0
0,
о,
о,
о,
о,
о,
1,
Визе"
0 з.д
20 07
,5
,0
,5
,7
до
.70
,25
40
380
410
305
120
0850
0520
0730
0203
07
205,6
1ОС
.
47
42
19
14
6
7
4
1
0
0
0
о,
о,
о,
о,
о.
о,
С Ш
30 07
,3
,0
,7
,6
,50
,20
Д5
,90
870
310
213
072
051
120
0810
0240
871
145,1
85
300
,0
176,0
78
39
4
2
0
0
0
о
1
о,
9,
о.
о,
о.
1,
.5
.7
.46
.36
,714
,803
,683
166
16
0469
0999
148
0375
0061
66
389,4
Самолетные
1500
49
.0
102,0
65
32
1
1
0
0
.0
0
0,
0,
о.
0.
0,
о.
0,
.0
,0
.71
.51
,189
,257
357
081
571
0435
220
0366
0406
00207
995
253,2
3000
36
67
43
31
4
3
1
2
1
0
2
0,
1,
0,
о,
о,
4,
,0
.0
.5
.0
,45
,84
,06
,52
,62
,482
17
166
16
145
172
0062
3
195,3
измерения
27
64
67
44
И
7
2
4
1
0
1,
0,
0,
0,
о.
i
1,
4500
.0
.0
.0
f **
.5
f W
,86
.46
.04
16
31
249
08
0761
145
oeeV^
0445
0019
69
231,0
12 07 (первые пять столбцов) и 30 07 (последушцие).
Следует отметить, что в работах по нахоадению корреляцион-
корреляционных связей между разными фракциями спектра размеров аэрозоль-
аэрозольных частщ часто делаются неверные выводы из отрицательной кор-
корреляции или отсутствия корреляции. Отрицательная корреляция меж-
между двумя частицами спектра обычно свидетельствует о сильном
182
183
Продолжение табл. 28
западнее г.Дакар (высоте
6400
34,0
31,5
26,0
12,3
1,46
3,25
1,65
1,13
0,891
0,463
0,324
0,0491
0Д44
0,0318
0,0328
0,00425
1,05
113,2
600
56,0
68,1
31,0
25,4
7,80
5,10
4,06
4,47
4,25
3,56
2Д6
0,466
0,117
0,0122
0,0080
0,0002
212,5
1500
83,0
85,3
102,0
85,0
21,7
17,0
7,31
10,6
5,62
4,78
2,79
0,591
0,179
0,0253
0,0080
0,0040
425,9
, м)
1800
75,0
68,0
137,0
220,0
28,4
22,10
16,5
7,29
2,83
2,47
1,38
0,337
ОД 66
ОД 01
0,0140
0,0070
581,6
2000
54,0
31,5
46,0
102,0
25 Д
14,0
7,23
8,01
2,60
1,59
0,418
0,0774
0,0140
0,005
-
-
292,5
3000
47,0
25Д
28,2
44,2
18,2
13,1
21,3
12,7
5,77
3,92
1,56
0,347
0,089
ОД 46
0,053
0,0050
221,7
6000
12,4-
8,50
6,00
5,25
4,80
3,50
2,36
2Д0
0,916
0,736
0,485
0,287
0,133
-
-
-
47,5
взаимодействии между этими частями спектра, а положительная кор-
корреляция - о слабом взаимодействии и общей причине появления
частиц. Следовательно, отсутствие корреляции не может толковать-
толковаться однозначно как признак полной независимости разных частей
спектра.
Прямые измерения мелкодисперсной фракции приземных аэрозо-
аэрозолей показали, что максимум распределения частиц по размерам на-
находится обычно в области г- C+6)«10-~2 мкм, а минимум около
г - 2,5-Ю мкм.
Исследования особенностей структуры аэрозолей почвенного
происхождения проводились в пустынях Средней Азии, степях и по-
полупустынях Казахстана и Южного Урала. Для частиц с г > 0,25мкм
были получены величины счетных концентраций JV- 3,2+1,7 см~^.
При аппроксимации гистограмм распределения частиц по размерам
формулой Юнге показатель степени оказался изменяющимся от 1,2
ло 5 при среднем значении 5* =2,7.
Заметно отличающиеся от приведенных данные по счетной кон-
концентрации были получены в работах автора по измерениям в пус-
пустыне Кара-Кум и в степях Северо-Западного Казахстана [19, 41,
77]. Для данных, полученных в пустыне Кара-Кум, наблюдается ус-
устойчивость спектрального распределения частиц по размерам для
леей тропосферы и в разное время суток. Возможно, это объясня-
объяснялось тем, что измерения проводились поздней осенью, когда не
наблвдалось сильных изменений характеристик воздушных масс и
конвективные процессы в атмосфере были ослаблены. В данных по
Кара-Кумам в приземном слое весьма заметно проявился суточный
ход счетной концентрации аэрозолей для воех уровней высот -
от 2 до 15 м - с максимумом в вечерние часы и минимумом в ут-
утренние, в то время как спектральная плотность распределения час-
частиц по размерам оказалась практически не зависящей от времени
суток. Только в вечерние часы наблюдалось небольшое относитель-
относительное увеличение концентрации гигантских аэрозольных частиц. При
описании распределения частиц по размерам двухмодалышм распре-
распределением dtf/tfr = а^ехр (SrOi /г) + а2г'6 ехр (-6 г02/г ) оба
модальных радиуса оказались очень устойчивыми: ? =0,04+
+0.06 мкм, ?и = 8+12 мкм. В измерениях в районе Южного'Ура-
Южного'Урала не наблюдалось такого четко выраженного суточного хода счет-
счетной концентрации, как в пустыне Кара-Кум, т.е. механизмы, вызы-
вызывающие генерацию частиц, не имели явно выраженного суточного
хода. Однако функция плотности распределения по размерам имела
достаточно четко выраженный суточный ход: в первой половине дня
184
185
12
16
РисЛ4. Суточный ход содержания четырех фракций частиц
на высотах 0,5 и 2 м по трехсуточной серии наблюдений (пос.
Анката, 16-18 07 71 г.).
1- г = 0,2+0,5; 2- г = 0,5+1,25; J- г = 1,25+2,2;
4 - г » 2,2 мкм.
наблвдалось более сильное падение относительной концентрации
частиц с ростом их размеров при г«= 0,20+0,5 мкм, увеличение
К вечеру фракций гигантских частиц с г > 2 мкм и уменьшение
относительной доли частиц с г = 0,4+1,5 мкм. Причем суточный
ход функции распределения частвд по размерам для разных высот
был несколько различен, отставая по времени с высотой. Это ил-
иллюстрируется рисЛ4, где приведены результаты измерений по су-
суточному ходу четырех фракций частиц на высоте 0,5 и 2 м.
Изменения спектра размеров аэрозольных частиц в районе Ш-
ного Урала в течение суток происходило не только в приземном
186
-ц
10"
слое, но и во всей толще атмосфе-
атмосферы аналогично изменениям спектра
размеров частиц в приземном слое.
В Северной Америке измере-
измерения аэрозолей почвенного проис-
происхождения проводились в пустыне
Мохава, где функция распределе-
распределения частиц по размерам оказалась
бимодальной с максимумом при
dm- 0,3+0,4 мкм и «^4,0 мкм.
Зто удовлетворительно согласует-
согласуется с многочисленными результата-
результатами измерений структуры приземных
аэрозолей естественного проиохож-
дения, проведенными автором в раз-
различных районах Советского Союза.
Следует отметить сходный суточный
ход счетной концентрации частиц,
полученный в пустыне Мохава К.Уит-
би (рис.15) ж др. и в наших рабо-
работах. Измерения концентрации час-
частиц, поднимаемых в атмосферу в
результате ветровой эрозии почв
в шт.Небраока, были предприняты
Д.Еиллетом, И.Блиффордом и'Ч.Фен-
стером. Кроме функции распределе-
распределения, которая для г = 1+6 мкм имела вид dN/dXnr — сг~^, а
для г = 0,3+1 мкм была еще более пологой, проводились гради-
градиентные измерения счетной концентрации (на z = 1,5 и 6 м) и
скорости ветра (на z =1,5; 3,0; 6,0 м). Самолетные измере-
измерения показали, что влияние почвенных частиц на структуру аэро-
аэрозолей сказывается вплоть до высоты 11,9 км.
Несмотря на достаточно большое разнообразие спектров раз-
размеров частиц почвенного происхождения, в первую очередь аридно-
аридного, для них можно отметить и определенные закономерности (рис.1Б\
Обращает на себя внимание заметное отклонение функции распреде-
распределения от степенной (юнговской) в диапазоне г>1,0 мкм. Это
187
10'
10'
<1,мкм
Рис.15. Суточный ход дис-
дисперсности аэрозольных час-
частиц по К.Уитби ( 28-29
08 69).
1 - 15;ч; 2 - 01 ч;
3 - 04 ч.
dlgd
100
50
0,01
0,1
1.0
10
100
cL,mkm
Рис.17. Объемное распределение частиц по'размерам
(К.Уитби) в разных пунктах США.
особенно отчетливо проявляется на функциях объемного или мас-
массового распределений частиц по размерам (рис.17). Многомодаль-
ность распределений указывает на наличие нескольких источников
аэрозольных частиц и на плохое слипание частиц этого типа при
столкновениях.
188
§ 4.5. Распределение городских аэрозолей
Распределения частиц по размерам в аэрозолях антропоген-
антропогенного происхоадения исключительно разнообразны. Хозяйственная
деятельность человека способствует в первую очередь возникно-
возникновению аэрозолей фотохимического происхождения, которые в на-
начальный момент имеют очень небольшие размеры (порядка десятков
189
нанометров), а затем в результате гетерогенных реакций и коагу-
ляционного роста, достигают нескольких десятых микрометра.Сред-
микрометра.Средний модальный радиус этих частиц около 0,2 мкм. Другой тип аэ-
аэрозолей антропогенного происхоадения дисперсионный. У таких
аэрозолей распределение частиц по размерам хорошо описывается
нормально-логарифмической функцией с наиболее вероятным (модаль-
(модальным) радиусом г = 2+3 мкм.
Наличие мощных источников аэрозольных частиц со специфичес-
специфическими свойствами обусловливает особенности распределения частиц
по размерам. Причем эти распределения практически всегда дале-
ки от описываемых формулой Юнге. Например, в Запорожье даже в
относительно слабо загрязненные дни наблюдается увеличенное со-
содержание частиц с г <=¦ 3+20 мкм. Величина этого вторичного
максимума в распределении частиц по размерил определяется как
местом измерений, так и временем суток (рис.1С). Поздним утром
и днем достаточно устойчиво наблюдаются максимумы функции рас-
распределения частиц по размерам в диапазоне г « 3*10 мкм, веро-
вероятно, в основном дисперсионного происхоадения (с поверхнооти
городских сооружений и дорог, строительная пыль). Во второй
половине дня пыль поднимается и в нижние слои тропосферы. В ве-
вечерние часы начинается опускание наиболее грубодисперсных фраи-
ций аэрозолей к поверхности земли и появляется максимум в функ-
функции распределения в области г * 20*50 мкм B2+23 ч). В более
поздние часы максимум смещается в сторону все более мелких час-
частиц и перед восходом солнца он находится в области r= S+12MKM.
Большинство гигантских частиц по электронно-микроскопи-
электронно-микроскопическим снимкам представляют собой,сложные рыхлые конгломераты
из более мелких частиц, что свидетельствует о повышенной спо-
способности частиц промышленного происхождения к коагуляционному
росту. Часто наблюдаются цепочки из частиц, характерные для
различных дымов, электронно-микроскопический анализ показыва-
показывает также повышенное по сравнению с естественными аэрозолями
содержание мелкодисперсной фракции. Наиболее мелкие частицы с
г = 0,02+0,05 мкм имеют почти правильную сферическую форму.
Плотность вещества этих частиц весьма различна: наблюдаются
как частицы с большой удельной плотностью (окислы металлов),
так и малой (сажи). Так как большинство рыхлых конгломератов
190
«о
¦о
t
IT
ео
i
1 1
1 1
-
с
. о
*!--
i
о
т
^ Е-* CdO
Й о и> tr
..8
со
а « i
В1 К
B'S
о о>
S ""
-м w«,s
00 ы
P-.
191
содержит частицы приблизительно одинаковой плотности, то можно
предполагать, что происходит быстрый коагуляционный рост таких
частиц вблизи их источника. В пробах, взятых при повышенной
влажности, наблюдались сферические конгломераты с разной плот-
плотностью вещества - гигроскопические частицы типа сульфатных.
Особенности микроструктуры промышленных аэрозолей, напри-
например в районе Рустави, обусловлены в основном особенностями их
источников - высокой концентрацией этих частиц в приземном слое
атмосферы и метеорологическими условиями этого района. Основны-
Основными генераторами аэрозольных частиц района г.Рустави являются
трубы промышленных предприятий, выбрасывающие специфические аэ-
аэрозольные частицы и газы, которые в дальнейшем в процессе фото-
фотохимических реакций частично переходят в твердое и жидкое аэро-
аэрозольное состояние. Некоторое количество аэрозольных частиц об-
образуется в результате подъема пылевых частиц с поверхности (ок-
(окружающие горы, городские территории, промышленные стройки), а
также из выхлопных автомобильных газов. Так, в период измере-
измерений (декабрь 1972 г.) при безветренной погоде и сравнительно
небольшом количестве транспорта в районе Рустави главным источ-
источником аэрозолей являлись выбросы из труб промышленных предпри-
предприятий. Так как в утренние и вечерние часы, а в некоторые дни и
в дневное время наблюдались инверсии на высотах несколько со-
сотен метров, то создались условия, благоприятствующие усиленно-
усиленному загрязнению приземного слоя атмосферы промышленными аэрозо-
аэрозолями, в силу чего наблюдалась высокая концентрация частиц в
течение всего периода измерений. Причем аэрозоли благодаря ин-
инверсии распространялись почти равномерно по всей площади про-
промышленного района. Особенно высокая концентрация наблюдалась
вблизи главных промышленных предприятий города: в основном час-
частицы грубодисперсной фракции с г %, 10 мкм. Для частиц мень-
меньших размеров счетные концентрации были приблизительно одинако-
одинаковы почти во всех точках наблюдений, различаясь не более чем в
два-три раза.
Сухая погода и уменьшенная по сравнению с летними месяца-
месяцами интенсивность солнечной радиации не способствовали появле-
появлению смоговых явлений. В течение всего периода наблюдений смог
не возникал даже при очень высокой концентрации аэрозольных
192
частад. Указанные аэрозоли по свойствам были ближе к пылям, чем,
например,аэрозоли в Запорожье и Алма-Ате. Это отчетливо прояви-
проявилось при электронно-микроскопическом анализе аэрозольных проб:
для руставских проб типично присутствие большого числа частиц,
имеющих плотную однородную структуру и неправильную форму с ост-
острыми гранями и углами. Такого рода частицы характерны для раз-
размельченных горных пород. Эти частицы сравнительно редко образо-
зьшали конгломераты. Эффективность коагуляции таких частиц не-
невысока, по крайней мере при тех влажностях (/ 4 70$), которые
наблюдались в период проведения измерений.
В тропосфере над городом была зарегистрирована весьма ма-
малая концентрация частиц с г > 4 мкм: не наблюдалось конгломе-
конгломератов частиц, дисперсность их была почти постоянна. В распреде-
распределении аэрозолей довольно отчетливо проявлялось несколько нало-
наложенных друг на друга одномодальных распределений частиц по раз-
размерам, соответствующих разным источникам.
Аэрозоли приземного слоя атмосферы в разных районах Ленин-
Ленинграда по своей микроструктуре очень разнообразны. Однако для
большинства районов города можно отметить низкие концентрации
частиц с размерами больше 5 мкм ( N г > 5 МК1И ? Ю~3 см),
особенно в удаленных от центра районах. Одной из причин относи-
относительной чистоты воздушного бассейна Ленинграда является посто-
постоянное Еымывание аэрозольных частиц облаками и осадками, а так-
:ке вынос загрязненного воздуха за пределы города адвективными
воздушными потоками,в первую очередь ветрами,дуюшими с моря.
Исследования дисперсности аэрозолей в Алма-Ате проводи-
проводились в апреле 1972 г. и в ноябре-декабре 1973 г. Для аэрозоль-
аэрозольных частит! воздушного бассейна этого города характерно быст-
быстрое нарастание концентрации мелких частиц в течение суток и
уход большой части аэрозолей в ночное время вниз по долине
Р.Алма-Атинки. Вследствие этого аэрозольные частицы в городе
не успевают "состариться" и они достаточно мелкодисперснн.Боль-
мелкодисперснн.Большинство частиц, явно фотохимического происхождения, образова-
образовались из продуктов выхлопов автомобилей и других двигателей и
представляют собой конгломераты мелких сфер приблизительно од-
одного диаметра ( d =* 0,03 мкм). Типичные функции распределения
аэрозолей по размерам для Алма-Аты представлены в табл.29.
13.1Щ
193
Таблица 29
Распределение аэрозольных частиц в тропосфере над Алма-Атой
по самолетным измерениям ДЛ/ (,см-з) 9 ноября 1973 г.
г, мкм
0,02-0,05
0,05-0,10
0,10-0,15
0,15-0,20
0,20-0,25
0,25-0,37
500-
-800
1250
4816
950
1850
162
230
43,5
98,0
17,4
19,0
8,44
7,98
800-
-1200
980
2300
760
1400
120
200
48,0
73,0
19,7
19,1
S.06
7,01
1200-
-1600
865
1350
580
1020
105
176
45,0
83,0
19,0
18,5
6,73
5,16
1700-
-2100
810
1180
530
750
88,0
135
37,0
44,0
18,7
17,9
11,1
4,27
Я, км
2200-
-2500
560
950
400
680
8S,0
95,0
33,5
29,0
17,9
16,9
7,42
6,64
2800-
-3200
420
670
340
545
78,0
87,0
27,0
2S,0
12,4
11,8
6,54
5,82
3200-
-3600
380
490
2S0
400
74,0
81,0
31,0
23,0
14,8
10,8
9,84
5,94
3R00-
-4000
270
350
200
310
57,0
65,0
20,5
22,0
11,0
И,8
7,80
5,24
4400-
-4800
160
210
120
145
43,0
51,5
15,7
24,0
7,46
15,6
4,80
5,01
5200-
-5500
95,0
82,0
76,0
64,0
38,0
40,0
13,8
14,3
7,80
8,01
3,85
3,57
0,37-0,62
0,62-0,88
0,88-1,25
1,25-1,88
1,88-2,5
> 2,5
7,31
8,88
4,58
2,00
0,71
0,59
0,30
0,23
0,13
0,064
0,076
0,028
2444
4816
8,65
8,15
3,22
2,90
0,58
0,60
0,32
0,22
0,067
0,060
0,028
0,039
1940
4010
6,14
6,24
4,31
3,17
0,78
0,90
0,29
0,30
0,059
0,090
0,031
0,042
1632
2662
9
5
5
,78
,04
,86
9
7
4
,75
,52
,85
2,56
0,74
1,20
0,38
0,39
0,094
0,120
0,038
0,034
1522
2140
7,30
0,81
1,28
0,40
0,47
0,084
0,170
0,045
0,056
1064
1794
8,08
7,92
5,34
3,36
0,91
0,93
0,42
0,31
0,115
0,156
0,039
0,056
899
1358
8,27
7,90
4,81
3,88
0,87
0,94
0,35
0,37
0,100
0,102
0,062
0,054
764
1024
Продолжение
7,35
8,40
4,52
3,78
1,14
1Д2
0,64
0,59
0,180
0,200
0,062
0,064
580
778
6,91
8,40
4,14
3,54
1,18
1,23
0,80
0,44
0,215
0,220
0,078
0,048
364
465
табл.29
7,39
8,86
4,04
6,21
0,93
1,12
0,67
0,68
0,170
0,200
0,095
0,034
248
229
Примечание. Каждая первая строка (начиная со второго столбца) соответствует
измерениям в 6 ч 00 мин - 6 ч 40 мин утра, вторая- - в 11 ч 20 мин -11 ч 40 мин.
Наибольшее количество измерений функции распределения го-
городских аэрозолей проведено в США [105, 179, 234, 239, 240] с
помощью миннесотской системы анализа аэрозолей [195].
Основной прибор этой системы - счетчик размеров частиц по
электрической подвижности, позволяющий регистрировать частицы
с d } 0,0030 мкм. Измерения известного в научной литературе
"коричневого денверского облака" показали, что доля по массе
субмикронных частиц в облаках изменяется от 1 до 73$, причем
четко различаются две фракции частиц: с г
)
0,01 (вблизи
шоссе с интенсивным автомобильным движением) и г - 0,2 мкм.
02
Коричневый цвет облако имело из-за особенностей распределения
частиц по размерам. К.Уитби и др. показали, что могут существо-
существовать фракции с максимумами распределения d^0* ®'^ и ^оз ^
Ч 1 мкм. Проведенные вблизи автомагистрали (на расстоянии ЭОм)
измерения аэрозолей в шт.Калифорния в диапазоне размеров 0,003-
40 мкм позволили обнаружить максимум в распределении d^. —
==¦ 0,02 мкм (в области d > 0,015 мкм). Удельный объем этих
частиц в среднем был AVj - 17,1 мкм • см , а счетная концент-
концентрация ANj = B*3)-10 см, причем при уменьшении влажности
обе величины возрастают.
Измерения в Миннеаполисе также обнаружили бимодальный ха-
характер распределения городских аэрозолей: первый максимум рас-
распределения, приходящийся на d - 1,0 мкм, - конденсационного
происхождения, второй - около d — 5,0 мкм - дисперсионного.
Х.§литер, измерявший концентрацию и размеры частиц авто-
автоматическим счетчиком ядер конденсации в восьми километрах от
лопенгагена#обнаружил, что в большинстве измерений средний раз-
размер частиц уменьшается с увеличением общей концентрации частиц
приблизительно по закону NoS[ii ¦=*¦ \Jr . По теоретическим и экс-
экспериментальна'.; данным значения модального диаметра равны ОД -
0,2 мкм. Однако в Лос-Анджелесе, например, величина модального
радиуса аэрозольных частиц равна около 0,5 мкм. По мнению
С.Ьридлэндера, причина несоответствия - в отсутствии надежных
данных о скорости окисления SO2 а капельках [158].
§ 4.6. Распределение аэрозолей в тропосфере
Вертикальный профиль аэрозолей в тропосфере исследовался
глазным образом с помощью самолетных заборников проб и нефело-
нефелометров. На основе данных большинства измерений обнаруживается
слоистость вертикальной структуры тропосферных аэрозолей. Осо-
Особенно отчетливо проявляется слоистость вертикального профиля
Н,км
Рис.19. Вертикальные профили счетной концентрации аэрозоль-
аэрозольных частиц с г •? 0,2 мкм по самолетным измерениям A971 г.).
а - северо-западный Казахстан - 16 июля: 1 - 10 ч 40 мин;
2 - 12 ч; 3 - 23 ч; h - 17 июля, 1 ч 10 мин; 6 - там же,
5-12 июля; 6-18 июля: в - Черное море: 7-31 июля;
8-3 августа.
концентрации больших и гигантских частиц (рис.19-21). Сравни-
Сравнительно небольшое количество измерений вертикального профиля
счетной концентрации ядер Айткена не дает возможности сделать
зквод о его слоистости, однако недавно выполненные Дж.Розеном
аэростатные измерения концентрации таких ядер показывают, что
вертикальный профиль их не описывается экспонентом и что для
немногочисленных данных относительно частиц г < 0,1 мкм -
ядер конденсации (ядер Айткена) - характерно прежде всего су-
197
го
го
АО О
ю
-10 -5 0 5
Т,°С
Рис.20. Вертикальные профили счетной концентрации частиц
с г*0,2 ми.' над промышленными районами.
а - г.Запорожье, 22 августа 1972 г.: / - 02 ч; 2-08 ч;
6 - г.Алма-Ата - г.Узун-Агач, 29 ноября 1973 г.: 3 -
г.Алма-Ата, 7 ч; к - там же 10 ч 30 мин; 5 - г.Узун-Агач,
7 ч 30 мин; 6 - там же 11 ч: в - г.Тбилиси - г.Рустави:
7,8 - с декабря 1972 г., 10 ч и 14 ч 30 мин соответст-
соответственно; г.Красноводск: 9-9 декабря 1972 г., 9 ч: про-
профили температуры, полученные одновременно {7-9).
шественное расхождение результатов прямых измерений (счетчика-
.'.и ядер) и по косвенным методикам. Концентрация ядер изменяет-
изменяется от 10 до 100 част/см3.
Анализ высот наиболее вероятного появления аэрозольных
слоев показывает, что эти высоты практически всегда располага-
располагаются г?ч:':е или ниаз высот инверсии и изотермии, где резко ос-
ослаблена турбулентность и отсутствует конвективный перенос воз-
воздушных масс, т.е. подынверсионные аэрозольные слои образуются '
в результате подъ;ма аэрозольного вещества из приземного слоя
атмосферы, а налгнверсионные слои - в результате опускания
аэрозолей вулканического происхождения и аэрозолей, образую-
193
:".хся ? результате фотохимических реакций и при работе авиа-
¦¦.:с?пг:х дг-игателей. В тропосфере высоты наиболее вероятного пс~
<г?глняя аэрозольнчх слоев обьчно соответствуют высотам наиболее
веослтнего появления облачности. 3 средних хиротах такими высо-
высотами являются уровни: 2-2,5; 6-7 и 10-12 ил. Следует отметить,
что существует резкий переход от приземных аэрозолей к тропо-
тропосферным примерно на высоте
S0C-1CCO м, причем выше Цк*
500 к особенно сильно умень- ' f
дается содержание мелкодис-
мелкодисперсной фракции и больших аэ-
аэрозольных частиц.
Сотрудниками лаборато-
лаборатории физики аэрозолей Ленин-
Ленинградского университета с по-
помощью самолетных зондировок
были получены данные по вер-
вертикальным профилям счетной
концентрации частиц для раз-
разных климатических зон в
новноы для частиц с Г i-
i- 0,2 мкм. Особый интерес
представляют вертикальные
профили счетной концентрации
больших частиц, полученные в послеполуденные часы над Каспий-
Каспийским морем (август, 1973 г.). Наблюдалось явное возрастание
счетной концентрации частиц с высотой вплоть до потолка зонди-
зондирования F*7 км). Такой профиль, по-видимому, объясняется на-
наличием развитой конвекции и отсутствием источника аэрозолей в
месте зондирования, когда вклад морских аэрозолей пренебрежи-
пренебрежимо мал по сравнению с аэрозолями почвенного происхождения.
Вертикальная структура счетной концентрации атмосферных
аэрозолей до высоты 30 км исследовалась с помощью аэростатных
аэрозольных приборов: фильтров, фотоэлектрических счетчиков и
счетчиков ядер конденсации. Примеры вертикальных профилей аоро-
золей, полученных с помощью аэростатного импактора, представ-
представлены на рис.22.
ы, см1
Рис.21. Вертикальные профили
концентрации частиц с г» 0,2 мкм
над тропической Атлантикой (за-
(западнее Дакара) в период проведе-
проведения АТЭП-74.
а - 4 июля: 5 - 4 августа
1S74 г.
!:¦
ного цвета, тогда как первые каскады собирали красную пыль. Зна-
Значительная доля частиц была составлена из вещества от разных ис-
источников, и большое количество частиц пыли из Сахары было пок-
покрыто оболочкой, которую они, возможно, получили, попав в какой-
либо из циклов конденсации, идущих в облаке.
В исследованиях, проведенных с борта самолета ИЛ-12 в экс-
экспедиции АТЭП-74, были получены сходные результаты, однако в
большинстве проб максимум в распределении частиц по размерам
приходился на частицы с г4 0,5 мкм. Вертикальные профили счет-
счетной концентрации пылевых частиц свидетельствуют о том, что оо-
новная масса аэрозольного вещества заключена в слое 1,5 - 7 км
с максимумом на высотах 3+5 ж, причем счетная концентрация час-
частиц радиусом 0,85-1,0 мкм изменяется примерно в 20 раз: от
2,8 см в пылевом слое до 0,12 см вне слоя. Интересным фак-
фактом является выявленная в этой экспедиции необычная зависи-
зависимость спектра размеров аэрозольных частиц от высоты: увеличе-
увеличение с высотой доли грубодисперсной фракции аэрозолей (~ 1 мкм)
Это свидетельствует о сравнительно недавнем появлении аэрозоль-
аэрозольных частиц (несколько часов, суток), о довольно высокой величи-
величине скорости восходящих потоков (превосходящей скорость падения
гигантских аэрозольных частиц) и, возможно, о значительной ро-
роли коагуляции в изменении спектра аэрозольных частиц (вероятно,
развитая турбулентность внутри пылевого слоя резко увеличивает
скорость коагуляции). Влияние седиментации сказывается на фор-
формировании профиля частиц с г> 1 мкм. Повышенная концентрация
этих частиц наблюдается вблизи нижней границы пылевого слоя.
Спектр размеров частиц сахарской пыли имеет многомодальный ха-
характер.
§ 4.7. Распределение аэрозолей в стратосфере
3 последние годы получено большое количество принципиаль-
принципиально важного и нового материала по вертикальной структуре стра-
стратосферных аэрозолей. Это связано в первую очередь с развитием
методов лидарного зондирования, а также с большим комплексом
исследований по программе изучения ант шогенного воздействия
202
на климат, в частности с работами Е.Бигга с сотрудниками и груп-
группы исследователей из университета шт.Вайоминг в США.
Анализ экспериментальных исследований, выполненных сотруд-
сотрудниками Ленинградского университета, показывает, что наблюдает-
наблюдается весьма сложный и изменчивый характер вертикального профиля
счетной концентрации аэрозолей (рис.23) с относительно высокой
частотой наблюдения аэрозольных слоев под и над слоями инверсии
и изотермий: 10-13; 17-18; 22-23,5 км. Центры аэрозольных сло-
слоев чащег всего располагаются на высотах 13,5; 16,5; 19,2; 21,8;
24 км. Величины коэффициентов аэрозольного ослабления на этих
высотах в видимой области спектра достигают 0,01 - 0,03 км,
а счетной концентрации для частиц с а > 0,2 мкм - 0,1 - 1,0
см~3. Замечено неполное соответствие вертикального профиля аэ-
аэрозолей по оптическим и микрофизическим измерениям счетной кон-
концентрации, что свидетельствует об изменчивости спектра разме-
размеров и состава аэрозольных частиц с высотой. Слой Юнге по дан-
данным большей части измерений (в первую очередь летних) расслаи-
расслаивается на два, что соответствует существованию на этих широтах
двух тропопауз (полярной и тропической). При измерениях в зим-
зимнее время и поздней осенью имеет место уменьшение мощности аэ-
аэрозольных слоев и их "размазывание".
исследования с помощью аэрозольного радиозонда, проведен-
проведенные сотрудниками университета шт.Вайоминг, позволили получить
обширный массив данных по вертикальным профилям счетной кон-
концентрации аэрозолей в тропосфере и особенно з стратосфере.Двух-
канальным счетчиком частиц аэрозольного зонда измерялась кон-
концентрация двух фракций частиц диаметром болое 0,3 и 0,5 мкм
при заборе воздуха с помощью насоса производительностью 0,75
л/мин. Для устранения загрязнения от оболочки радиозонда счет-
счетчик был подвешен на расстоянии 90 м от оболочки. Лзмерения про-
производились как при подъеме, так и при парашютном спуске аэро-
аэрозольного зонда.
Определение концентрации аэрозолей в каждой точке произво-
производилось путем усреднения данных регистрации 500 отдельных час-
частиц. Вертикальный профиль суммарной концентрации частиц диа-
диаметром больше 0,3 мкм имел разрешение по высоте 250 м, ошибка
измерений концентрации составляла около 5%. В некоторых случа-
203
Рис.23. Результаты аэростатных импакторных измерений частщ
1-М - 1966 г.; 5-7 - 1971 г.
Н.км
10 N.cm'
с 0,2 мкм (г.Рыльск).
(июль - август).
ях осуществлялся совместный запуск аэрозольного зонда с импак-
тором, что дало возможность определить химический состав час-
частиц, которые в нижней стратосфере представляли собой преиму-
преимущественно капли серной кислоты и сульфатные частицы. Поэтому
при калибровке аэрозольного зонда принимался показатель прелом-
преломления непоглощающих частиц, равный 1,45.
Главная цель стратосферных аэрозольных исследований в вы-
выяснении, каков источник фоновых стратосферных аэрозолей (обра-
(образуются они вследствие реакций между газами или путем коагуля-
коагуляции более мелких частиц, какие изменения могут быть в этой свя-
связи вызваны появлением продуктов выхлопа двигателей сверхзвуко-
сверхзвуковых самолетов; какова изменчивость глобального распределения
аэрозолей).
Полученные результаты указывают на большую изменчивость
содержания аэрозолей в тропосфере и нижней стратосфере, но весь-
весьма высокую их стабильность на высотах более 20 км, что позволя-
позволяет предполагать существование устойчивого естественного страто-
стратосферного фона аэрозолей. Основной вклад в изменчивость общего
содержания аэрозолей в стратосфере вносит слой, расположенный
между местной тропопаузой и уровнем 15 км, поэтому наблвдаетоя
четкая корреляция между высотой тропопаузы и общим содержанием
аэрозолей в стратосфере (зона изменчивости содержания аэрозо-
аэрозолей почти исчезает летом и достигает максимума зимой). Измере-
Измерения показали, что определяемая по двум фракциям дисперсность
аэрозолей почти не изменяется в стратосфере: отношение концен-
концентрации частиц двух фракций (с диаметром более 0,3 и 0,5 мкм)
составляет четыре - пять, что соответствует показателю степе-
степени в формуле Юнге, равному ** = 3.
Максимум счетной концентрации располагается на высотах
22-23 км в экваториальном поясе, снижаясь параллельно тропо-
тропопаузе до 17-18 км в полярных районах. В слое максимума отноше-
отношение смеси для аэрозолей составляет 8-10 част/мг воздуха. Это
эквивалентно отношению смеси по массе, равному примерно
1,3*10 г/г. На уровне тропопаузы отношение смеси может в за-
зависимости от времени года уменьшаться примерно в десять раз.
Максимум концентрации частиц расположен на 4 км ниже максиму-
максимума аэрозольного отношения смеси, причем максимальная концент-
206
пация частиц с d > 0,3 мкм составляет 0,5 - 1,5 см 3. Эти ре-
iv-ьтаты хорошо согласуются с данными оптических измерений.
Выше 50 км микрофизические методы исследования вертикаль-
вертикальной структуры аэрозолей применялись для их изучения в зоне се-
себрястых облаков [150, 151, 176]. Счетная концентрация частиц
на высотах, соответствующих высотам серебристых облаков G7 -
г5 км), изменяется от 10~4 до 10~3 см~3. Этот высотный аэрозоль-
аэрозольный слой, вероятно, очень неустойчив и может образовываться в
сезультате двух механизмов: 1) торможения и сгорания микромете-
микрометеоритов, 2) подъема аэрозольных частиц конвективными потоками
из более низких слоев атмосферы. Скорость восходящих потоков в
этом случае должна достигать 50 см/с и более. По В.Уэббу такая
скорость вполне реальна. Так как восходящие потоки вызваны ра-
радиационным нагревом всей толщины атмосферы в дневное время, то
очевидно, что максимальные значения счетной концентрации аэро-
аэрозолей на высоте серебристых облаков должны наблюдаться в ве-
вечернее время. Это соответствует известным экспериментальным
данным. Размеры частиц на таких высотах практически не могут
превышать 1-1,5 мкм из-эа быстрого увеличения скорости паде-
падения частиц с увеличением их размеров. Судя по оптическим дан-
данным, особенно по наблюдениям с космических кораблей, существу-
существуют слои аэрозолей на высотах 30 и 45 км, которые также связаны
с особенностями температурной стратификации и располагаются
вблизи зон изотермии [19].
Вертикальная структура аэрозолей в высоких слоях атмосфе-
атмосферы исследовалась также с помощью ракетных импакторов и фото-
фотоэлектрических счетчиков сотрудниками Центральный аэрологичес-
аэрологической обсерватории совместно с исследователями из Ленинградского
и Новосибирского университетов. Полученные данные подтверждают
сложность вертикального профиля счетной концентрации аэрозолей
для высот больше 30 км. Однако об определенных высотах преиму-
преимущественного появления слоев по данным прямых измерений гово-
говорить трудно. Отмечена изменчивость дисперсности частиц как
с высотой, так и от запуска к запуску на одной высоте (рис.24).
Первый модальный радиус находится в области r0J = 0,03*0,06мкм.
Большинство частиц представляют собой плотные сферы.
207
Л V
Wi
, отн.
.еЗ.
10
10
10
1О'г
JL
W
1
г, мкм
Рис.24.Распределение аэрозольных частиц по размерам в высших
слоях атмосферы по ракетным импакторным измерениям.
/ - 52 км; 2 - 54+60 км; 3 - 65+73 км: Ч - 78+83 км; 5 -
95 км (сентябрь 1980 г., В.М.Жуков, Л.С.Ивлев).
В стратосфере, где процесс коагуляции затруднен из-за .та-
.талой концентрации частш^Дж.Фрэндом было обнаружено бимодальное
-аспределение, а распределение Х.Юнге приближенно выполняется
глхъ для частиц с г » 0,4 мкм. Первый максимум распределения
частиц по размерам находится в области ядер Айткена, а второй-
в диапазоне г = 0,3 f 0,4 мкм. По Х.Юнге, а также дж.Ьрэнду
это - сульфатные частицы, образовавшиеся из сернистого газа,
заброшенного в атмосферу при извержениях вулканов. СЛлоссоп, ис-
исследовавший структуру аналогичных частиц, взятых с помощью са-
самолетного импактора, обнаружил, что частицы имеют несульфатное
ядро, а сульфаты образуют лишь оболочку, что не противоречит
гипотезе Х.Юнге. Однако расчеты З.Мартелла показывают,что час-
частицы, обнаруженные Дж.Фрэндом и С.Моссопом, не обязательно дол-
должны иметь фотохимическое происхождение, а могут образовываться
в результате коагуляции в тропосфере. Другое объяснение образо-
образования этого максимума по Е.Мартеллу - ошибка, возникающая при
обработке пробы, вследствие коагуляции частиц на подложке. Бо-
Более поздние исследования указывают на возможность образования
части из OCS.
По результатам исследований автора распределение Юнга при-
приближенно выполняется для частиц с г ? 0,2 мкм. Наблюдаются
распределения, аналогичные полученным Дж.Фрэндом, однако они
характерны всегда для тонких слоев, которые чередуются со слоя-
слоями, где выполняется распределение Х.Юнге. Выше 22 км часто об-
обнаруживается резкое уменьшение доли крупнодисперсной фракции -
показатель степени изменяется от четырех к пяти. Подобное рас-
распределение было определено по ракетным пробам в зоне серебрис-
серебристых облаков [ 49].
Измерения дисперсности стратосферных аэрозолей, выполнен-
выполненные Е.Ьиггом с помощью аэростатных подъемов аэрозольных импак-
торов, показывает, что в верхней тропосфере и стратосфере час-
часто наблюдаются многомодовые распределения частиц по размерам,
причем в стратосфере спектр частиц значительно уже, чем в тро-
тропосфере. Этот факт может свидетельствовать о большом времени
жизни стратосферных частиц (типичные спектры аэрозольных час-
частиц на разных высотах представлены на рис.25).
К1922
2С9
dN
N.omH.ed..
0,01
0,5 г,мкм
210
1].Еиггом отмечается сезонная изменчивость распределения
частиц по размерам для разных диапазонов высот. Доля крупных
частиц в стратосфере заметно возрастает весной (для южного по-
луцария в сентябре-ноябре) и имеет минимум осенью и летом. Наи-
Наиболее стабильна для разных сезонов концентрация частиц с г >
>С,23 мкм.
Злектронно-микроскопические снимки аэрозольных проб пока-
показывают, что стратосферные аэрозольные частицы очень часто пред-
представляют собой сложные агрегаты мельчайших частиц различного
химического состава. Средняя функция распределения частиц по
размерам в нижней стратосфере по данным 23-х осенне-летних аэ-
аэростатных полетов, совершенных в средней полосе европейской тер-
территории Советского Союза, представлена в табл.30. Характерно
увеличенное по сравнению с данными Х.Юнге содержание гигант-
гигантских аэрозольных частиц с радиусом от 1,35 до 2,7 мкм. Частиц
с г Ь- 4 мкм практически не наблюдается. Концентрация мельчай-
мельчайших, субмикронных частиц, начиная с высоты 20 км и до высоты
25-27 км, уменьшается очень мало, что подтверждает гипотезу об-
образования мелких частиц непосредственно в нижней стратосфере.
Измерения с помощью счетчиков ядер Айткена [214] также подтвер-
подтверждают эту гипотезу.
При анализе данных по дисперсности атмосферных аэрозолей
необходимо всегда помнить, что практически не существует уни-
универсальных приборов, дающих надежные сведения о распределении
частиц по размерам в достаточно широком диапазоне. Поэтому
всегда представляют наибольший интерес данные, полученные од-
одновременно разными методами. Такого рода данные были получены
в советско-американском эксперименте по изучению стратосфер-
стратосферных аэрозолей, проведенном в Рыльске в 1975 г. Результаты из-
измерений фотоэлектрическим счетчиком, импактором и фильтром по-
показали значительные расхождения в величинах счетной концентра-
Рлс.25. Распределения аэрозольных частиц в стратосфере
по аэростатным измерениям.
а - по Дж.Фрэнд (Г); по С.Моссопу B); по Г.Ферри,
Х.ЛеммуE;- ?_ по данным измерений электрической
подвижности ионов A8 II 74); 6 - по импакторным
измерениям Е.Бигга.
211
212
ю
С\2
со
о
i
^н
<я
а
а
>>
и
СЯ
о
с-
о
о
to
«н
о
со
о
о
а.
о ююю
(М<?>(МСО
МООО
« *> к ¦>
ю
о
о
о
о
о
о
1
1
О
О
О Ю<О
ст> сосхгоо
О СЧНОО I
о оЪоо
О00С
о шг>с
СО "*ООО
ктН
*
о
о
i i i i i i i Т
oooooooo
I I I I
ЛСОСООООО
СО1Н1кЧкНк
С0*ОСОкН1Н1
оюююопинооо
ЮШММсООООООООСООЮ
OOOOOOOOOOOCONCW
I I
co^ocoojoo
нсосг^»
окнсо?>с\г^н
ОЩ^СМ^ИОООО •
ОПЮ«ОООООО
гаюоэошоюою
ОООЫкнОЛЛгЮ
о
0
-юооо
1OQOOH
I I I I I k
оюо
I I I I I kHOiCOino
схгсоюсооюоюою I I i I -
OOOOOOOOOOHNNCO^
, А
^о о,
О S
0 s
:н X
Л s
**J о
1 a
I ft
ю е
к:
со
Ш Ен
И
>§<
Ш S
с
о х
к я
СО ?Ц
а
о о
Сч Ен
О К
К СЯ
0 с
са В
а.
а «'
<г> я
щ О
1 р<
О Ей
О Ц
Е-1 S
S °
о со
о сч
со t^
Я ей
Е-1
СЯ
сч
CD О
ft a
со
со
s ь м:
о со
s ч ft
со о
а о, >>
к
я s
.,:,: частиц разных размеров. Наиболее серьезное расхождение в"
результатах измерений наблюдается для высот, соответствующих
слою Юнге. Концентрация частиц по данным импактора оказалась
значительно более низкой, чем по данным фотоэлектрического счет-
счетчика. При анализа фильтровых аэрозольных проб под электронным
микроскопом были обнаружены круглые светлые пятна на пленке с
довольно узким распределением их по размерам. Верхняя граница
размеров этих светлых пятен приходится на d = 0 4*0 5 мкм
а максимум на ^= 0,03 мим. Предпшгоженже, что этж п'устоты в
пленке образовались в результате наличия в ней капель серной
кислоты, позволяет удовлетворительно увязать с данными ' полу-
полученными разными приборами.
Сравнительный анализ данных измерения концентрации и дис-
дисперсности аэрозолей, полученных всеми тремя методами, был про-
проведен и для других слоев атмосферы. При этом предполагалось
что распределения частиц по размерам в области г > 0 1 мкм'на
всех высотах подобны распределению Юнге, но с разными'показате-
разными'показателями степени. Данные, полученные с помощью фильтра и шпактора
нормировались по показаниям фотоэлектрического счетчика в об-'
ласти г = 0,15+0,25 мкм.
Различие в данных, полученных тремя методами, вызвано дву-
двумя обстоятельствами: 1) наличием в атмосфере частиц, содержа-
содержащих в себе большую долю жидкой фазн (капли, растворы и т п )-
) отклонением ввда реального распределения от распределения'
-tare. В частности, для высот 0,5 - 3 км при измерениях в Рыль-
ске следует предположить заметное отклонение от степенного за-
ксна распределения в области г = 0,45+0,50 мкм. Этих частиц
в атмосфере заметно больше, чем следует из распределения Юнге.
счетчиГ ТДЫ СПраВедливы' если ^тать фотоэлектрический
счетчик прибором, дающим абсолютные данные. Однако такой фото-
фотоэлектрический счетчик, кроме очевидного недостатка - измерения
концентращш частиц только в двух диапазо - ия
ные результаты по измерению концентрации частиц, если частицы
Ферические или имеют разные показатели преломления. вГ
ZZ'Z С1Ш>НЫе расховденй* в Данных измерений фото!
злек рическим счетчиком, импактором « фильтром, наблвдакшеся
высотах 3,0 - 7,0 км, соответствуют присутствию в этих сло-
213
ях большого количества несферических частиц и частиц, имеющих
красновато-бурый оттенок. Если последние считать частицами оки-
окислов железа, которые имеют показатель преломления больше 2,4
в видимой области спектра, то очевидно, что данные фотоэлектри-
фотоэлектрического счетчика в этих слоях атмосферы также нельзя считать
достаточно надежными.
§ 4.8. Глобальная пространственно-временная
структура аэрозолей
Глобальная пространственно-временная структура атмосфер-
атмосферных аэрозолей получена в значительной степени в результате оп-
оптических измерений прозрачности атмосферы.
Относительно простые измерения оптической толщи атмосферы
в видимой области спектра позволяют вести временной и простран-
пространственный мониторинг содержания аэрозолей во всем столбе атмос-
атмосферы. Анализ имеющихся экспериментальных данных по оптической
толще атмосферы на длине волны Л = 0,5 мкм показывает, что
для незамутненных районов земного шара обнаруживается достаточ-
достаточно четкая широтная зависимость величины оптической толщи х .
В районе Северного и Южного полюсов т = 0,05, а на экваторе
т =0,3. Значения оптической толщи, полученные при измерениях
над морокой поверхностью, несколько ниже, чем при измерениях в
континентальных условиях.
Большой интерес представляют длинные ряды наблюдений про-
прозрачности атмосферы на фоновых станциях, в частности в обсерва-
обсерватории "Мауна-JIoa". Они показывают, что для фоновой концентра-
концентрации аэрозолей в атмосфере выше уровня "Мауна-Лоа" ( z * 4000 ы)
вклад стратосферных аэрозолей является основным, а вариации в
содержании фоновых аэрозолей обусловлены сезонными процессами
образования и ухода аэрозолей из атмосферы, вулканическими из-
извержениями. Для сезонного хода замутненности атмосферы харак-
характерно наличие максимума в октябре-ноябре и минимума в апреле-
мае. После сильной замутненности атмосферы, вызванной вулкани-
вулканическими извержениями начала шестидесятых годов, наблюдается
ее постепенное снижение. Измерения на других фоновых станциях,
214
расположенных на различных широтах, показывают, что максимум
замутненности атмосферы наблюдается в начале лета, а минимум в
октябре-декабре месяце. Мутность атмосферы постоянно испытыва-
испытывает значительные колебания. Пб данным актинометрических наблюде-
наблюдений имело место резкое увеличение мутности в атмосфере в 1963г.,
вызванное извержением вулкана Агунг в марте 1963 г., хотя тен-
тенденция к увеличению мутности наблюдалась уже в период 1960 -
1962 гг., и, возможно, связана с многочисленными ядерными взрн-
вами, проведенными в это время.
Сезонный ход мутности атмосферы объясняется развивающими-
развивающимися весной турбулентными движениями воздушных масс. Данные пос-
последних лет в основном повторяют сезонный ход, полученный Анг-
Ангстремом на основе многочисленных измерений в 30-х годах.
Прогнозы угрожающего роста глобальных загрязнений атмосфе-
атмосферы, обусловленных хозяйственной деятельностью человека, делают
необходимым проведение критического анализа имеющихся данных
наблюдений т. Тенденция увеличения содержания углекислого га-
газа способствует усилению парникового эффекта атмосферы, и, сле-
следовательно, потеплению климата. Поэтому наблюдающееся за пос-
последние десятилетня некоторое похолодание климата побудило выс-
высказать предположение, что оно обусловлено компенсирующим эффек-
эффектом роста планетарного альбедо, вызванного увеличением запылен-
запыленности.
Однако разнообразные данные противоречат выводу о постоян-
постоянное росте запыленности. Так, в период 1964-1970 гг. общая тен-
де;.-,ия к прояснению атмосферы после извержения Агунга неодно-
неоднократно нарушалась. В это время имели место многочисленные слу-
случаи отдельных помутнений атмосферы с последующими быстрыми про-
прояснениями. В экваториальной зоне в этот период произошло нес-
несколько крупных извержений вулканов: Таал A965 г.), Фернандина
A968 г.) и др., продукты извержения которых были заброшены
непосредственно в стратосферу. Частичное загрязнение верхней
тропосферы и стратосферы вызваны испытаниями атомных и водород-
водородных бомб в Китае на поверхности Земли и в атмосфере. В Арктике
в этот период произошли извержения вулканов Традент A963 г.),
Суртсей A963 г.), Ридаут A966 г.) и Гекла A970 г.). Вулка-
Вулканическая пыль и газы при извержениях арктических вулканов рас-
ггространялись лишь в атмосфере северного полушария, Вместе с
тем значительная часть продуктов извержений в экваториальной зо-
зоне поступала в атмосферу умеренных шрот северного полушария.
При изпержениях вулканов кроме пепла и сернистых газов в стра-
стратосферу обычно попадает значительное количество водяного пара,
который может там длительно существовать. Особенно это сущест-
существенно при извержениях подводных и прибрежных вулканов.
Таким образом, в последние годы в атмосферу северного по-
лугаркя поступило значительно большее количество не только про-
продуктов деятельности человека, но и вулканических выбросов. Вов-
Вовлечение этих продуктов в сезонную циркуляцию стратосферных воз-
воздушных масс проявилось в средних широтах в значительном увели-
увеличении осенне-зимнего максимума замутненное™.
Х.Эльзассер приводит следующие противоречащие выводу о рос-
росте глобальной загрязненности факты: 1) мутность атмосферы в вос-
восточной части США (вне городов) в период 1962-1966 гг.была срав-
сравнима с мутностью в Вашингтоне в 1903-1907 гг.; 2) электропро-
электропроводность воздуха в южной части Тихого океана оставалась неиз-
неизменной с 1908 г.; 3) уточненные данные измерений мутности в
"Мауна-Лоа" показали, что минимальные значения мутности не из-
изменялись в период 1958-1962 гг., а к 1970 г. вернулись к "до-
агунговскому" уровню; 4) измерения фактора мутности ранним ле-
летом в Антарктиде не обнаружили какой-либо тенденции роста с
1949-1952 по 1966 гг.; 5) не выявляют заметной тенденции изме-
изменения прозрачности атмосферы данные наблвдений в обсерватории
Ловелла, начатые в 1955 г.; 6) анализ результатов измерений пря-
прямой солнечной радиации на длине волны 0,39 мкм, проведенных в
Смитсонианской обсерватории за 1902-1950 гг., не выявил тенден-
тенденции к уменьшению прозрачности атмосферы; 7) наблюдения во мно-
многих городских районах, где до 40-х годов имела место сильная
замутненность атмосферы, обнаружили к 1970 г. существенное
уменьшение фактора мутности (соответственно запыленности);
8) исследования ядер конденсации не обнаружили заметного антро-
антропогенного влияния; 9) геоморфологические данные свидетельству-
свидетельствуют об увеличении содержания пыли в атмосфере за последние нес-
несколько тысячелетий с 26 до 720 мкг/м3, что намного превышает
антропогенные эмиссии.
216
Особую роль в выявлении глобальной тенденции изменения
прозрачности атмосферы сыграл анализ данных аэрозольных и акти-
нометрических измерений в обсерватории "Мауна-Лоа", которые
удовлетворяют критериям "фоновой станции".
Данные измерений прямой солнечной радиации на Мауна-Лоа за
период I958-I97I гг. не выявляют какой-либо тенденции, которая
может быть связана с влиянием деятельности человека. А.Дайер,
проанализировавший вековой ход замутненности атмосферы по дан-
данным наблюдений прямой солнечной радиации в СССР A961-1968 гг.),
Японии A951-1972 гг.), США A958-1971 гг.) и Австралии A953-
1972 гг.), в качестве характеристики замутненности использовал
"пылевой индекс" D = cos 0 1л ( Т/1) ( © - зенитный угол
Солнца; 1,1- потоки прямой солнечной радиации, измеренной при
одинаковом зенитном угле Солнца @) в "исходном" и рассматри-
рассматриваемом году соответственно). За исходные I приняты доагунгов-
ские 1961-1962 гг. Анализ хода пылевого индекса показывает,что
вс всех рассмотренных случаях происходило постепенное повышение
прозрачности атмосферы после извержения вулкана Агунг A963 г.)
до доагунговского уровня (исключение составляют лишь данные для
Японии за 1971-1972 гг.), т.е. никакой заметной вековой тенден-
тенденции глобального повышения замутненности атмосферы не наблюдает-
наблюдается. Следует подчеркнуть, что для надежных выводов о тенденциях
глобальной замутненности атмосферы необходимы длительные высо-
высокоточные измерения (точность измерений должна быть около 1%).
Измерения вертикальных профилей коэффициентов прозрачнос-
прозрачности атмосферы показывают, что прозрачность всей толщи атмосферы
определяется, в первую очередь, прозрачностью ее нижних слоев.
В некоторых случаях на нижние 2-3 км атмосферы приходится до
80-905? ослабления солнечной радиации всей толщей атмосферы, по-
поэтому для выводов об общем изменении ее запыленности можно ис-
использовать лишь данные станций, где наблюдается стабильный сос-
состав нижних слоев атмосферы и по возможности отсутствуют источ-
источники аэрозолей на земной поверхности.
Большой массив данных о вертикальной структуре аэрозолей
получен с помощью прожекторного зондирования. В частности, од-
одна из первых оптических моделей структуры атмосферных аэрозо-
аэрозолей была построена Л.Элтерманом по данным прожекторного зонди-
217
Измерения с пилотируемых космических кораблей позволили1
оценить глобальную пространственно-временную изменчивость поля
стратосферных аэрозолей. Основные выводы этих измерений хорошо
согласуются с данными аэростатных и самолетных измерений стра-
стратосферных аэрозолей. Особо следует отметить обнаружение при из-
измерениях с космических кораблей аэрозольного слоя на высотах
40-50 км, возможно, возникшего в результате попадания на эти вы-
высоты вулканических газов и водяного пара. Анализ возможностей
решения обратных задач атмосферной оптики применительно к изме-
измерениям из космоса показывает, что для корректного решения тако-
такого рода задач необходимо задание точной априорной информации,
в первую очередь о комплексном показателе преломления вещест-
вещества аэрозольных частиц.
Эффективным методом наблюдения вертикальной структуры ко-
коэффициента рассеяния атмосферных аэрозолей является сумеречный
метод, требующий, однако, высокой чистоты нижних слоев атмосфе-
атмосферы и отсутствия облачных образований. По этой причине количест-
количество измерений структуры аэрозолей этим методом относительно не-
невелико. Интересно отметить, что проведенные О.Б.Васильевым ле-
летом 1971 г. единичные спектральные измерения сумерек в Ленин-
Ленинграде показали существование отчетливо выраженного аэрозольно-
аэрозольного слоя на высотах около 45 км, что хорошо согласуется с данны-
данными измерений с космических кораблей. Разрешение этого метода го
высоте ~ 4 км. Более тонкую структуру вертикального профиля
аэрозолей оптическими методами получают при аэростатных зонди-
зондированиях атмосферы со спектральными и актинометрическими прибо-
приборами. Эти измерения показывают ярко выраженную слоистую струк-
структуру стратосферных аэрозолей по крайней мере до высоты 30 км.
Большинство исследователей связывают наличие аэрозольных слоев
с температурной стратификацией атмосферы. Это положение верно
в общем случае. Однако следует предположить, что в ряде случа-
случаев определенная температурная стратификация атмосферы, в свою
очередь, формируется в результате существования аэрозольных
слоев, которые могут образовываться не только в результате вер-
вертикальных движений воздушных масс, но и вследствие генерации
частиц в атмосфере, действия электрических сил и т.д.
218
Быстрая изменчивость вертикальной структуры стратосферных
аэгозолей может быть обусловлена различными факторами: турбу-
турбулентностью,конденсацией,коагуляцией и адвекцией воздушных масс.
Наиболее правдоподобным выглядит последний фактор,который пред-
предполагает существование горизонтальной неоднородности стратосфер-
стратосферных аэрозолей.
Зесьма интересен факт сильного уменьшения коэффициента об-
обратного рассеяния радиации на высотах, соответствующих повышен-
повышенным значениям относительной влажности. Повышенная относительная
влажность воздуха может приводить к изменению микроструктуры аэ-
оозолей: образованию более компактных частиц, изменению их по-
показателя преломления, ускоренному выпадению частиц из слоя,что
вызывает уменьшение коэффициента обратного рассеяния, а также
изменению хода фотохимических реакций образования аэрозольных
частиц.
Параллельные измерения структуры аэрозолей с помощью лида-
ра и фотоэлектрического счетчика выявили заметное расхождение в
содержании аэрозолей, полученном этими методами, в том числе на
высотах 24-28 км. Возможно, что заниженные значения содержания
аэрозолей в стратосфере, измеренные лазерным методом, могут по-
получаться и из-за неудовлетворительности методики обработки ла-
лазерных измерений.
Очевидно, что одной из главных проблем при моностатичес-
моностатическом лазерном (лидарном) зондировании атмосферных аэрозолей яв-
является нахождение соответствия между коэффициентом обратного
рассеяния радиации аэрозолями ся и коэффициентом аэрозольно-
аэрозольного рассеяния бр или счетной концентрацией аэрозолей JVa. Ко-
Коэффициент обратного рассеяния б"я чрезвычайно чувствителен к
реальной части показателя преломления, что делает интерпрета-
интерпретацию данных лидарного зондирования весьма неоднозначной. Поэто-
Поэтому ряд данных по вертикальному профилю атмосферных аэрозолей,
полученных с помощью лидарного зондирования, можно считать лишь
качественными характеристиками содержания аэрозолей в атмосфе-
атмосфере. Зыесте с тем это обстоятельство позволяет использовать ли-
дарнос зондирование для определения не только количественного
содержания аэрозолей в атмосфере, но и для определения некото-
некоторых х;!мических компонентов в области резонанса обратного рас-
рассеяния.
219
Наиболее ценным качеством лидарного зондирования, deccnopi
но, является возможность картирования распределения аэрозолей в
атмосфере и его динамики на достаточно больших расстояниях(даль
ность действия лдцеров определяется их техническими данными и
условиями измерений). Полученные с помощью лидарного зондирова-
зондирования данные по вертикальному профилю аэрозолей позволили сущест-
существенно уточнить картину формирования вертикальной структуры аэ-
аэрозолей и модели вертикальных профилей аэрозолей.
Слои повышенной замутненности воздуха имеют достаточно
большую протяженность в горизонтальных направлениях (по Е.Биг-
гу не менее ЗОС км), причем существуют высоты преимущественно-
преимущественного появления слоев, характерные для разных широт. Отчетливо вы-
вырисовывается широтный ход слоя Юнге. Возможно, что для тропо-
тропосферных слоев также существует широтный ход, однако отсутству-
отсутствуют данные для малых широт.
Следует также отметить сильную зависимость характера все-
всего вертикального профиля аэрозоля от широты местности. Очень
вероятно, что в верхней тропосфере, а может быть и в стратосфе-
стратосфере, в летний период аэрозольные слои, особенно на низких и сред-
средних широтах, тлеют структуру, сходную с облачной, что обуслов-
обусловлено ячеистой структурой конвективных токов, причем в этом слу-
случае нет резкой качественной разницы между образованием аэрозоль-
аэрозольного слоя и облака. Результаты микроструктурных исследований
вертикального профиля аэрозолей с помощью самолетных зондиро-
зондировок позволяют сделать вывод, что вертикальная структура аэро-
аэрозолей и микроструктура частиц в тропосфере в зависимости от
места проведения зондирования имеют чрезвычайную изменчивость.
Зависимость аэрозольной структуры от мест зондирования наблю-
наблюдается вплоть до высоты 8-9 км.
Летальный анализ данных наблюдений пространственно-времен-
пространственно-временных вариаций глобального поля концентрации стратосферных аэро-
аэрозолей по результатам зондирований на десяти станциях, располо-
расположенных в диапазоне широт от 85 с.ш. до 90° ю.ш., показали, что,
кроме слоя Юнге наблюдаются тонкие слои аэрозолей большой го-
горизонтальной протяженности, которые связаны либо с переносом
воздуха через разрывы в тропосфере умеренных широт, либо с вул-
вулканической активностью.
анные аэростатных аэрозольных зондирований, выполненных
я -озеном с сотрудниками на дрейфующей арктической станции
'¦о0 с.::.) в декабре 1971 и 1972 гг., на двух антарктических
станциях ";..ак кердо" G8° ю.ш.) и "Кжный полюс (90° ю.ш.) - в
январе 1S72 и 1S73 гг. позволили получить сведения об особен-
особенностях распределения аэрозолей диаметром более 0,3 мкм в поляр-
полярной стсатосфере. Обнаружено, что средние эффективные размеры
частиц в верхней тропосфере меньше, чем в стратосфере.
Несмотря на весьма сильную изменчивость аэрозольной струк-
структуры как во времени, так и в пространстве, накопленный экспери-
экспериментальный материал позволяет говорить о глобальном характере
изменения структуры аэрозолей в зависимости от широты места из-
измерений и времени года.
К числу наиболее существенных особенностей глобального рас-
распределения стратосферных аэрозолей принадлежат: годовой ход,
наиболее заметно проявляющийся в умеренных широтах северного
полушария, и подобие вертикальных профилей концентрации аэрозо-
аэрозолей в районах Северного и Южного полюсов (даже в разное время
года), указывающее на общность механизмов образования аэрозо-
аэрозолей. По-видимому, в полярных областях в отличие от более низ-
низких широт годовой ход концентрации аэрозолей выражен не очень
сильно.
Обращает на себя внимание также отчетливая корреляция меж-
между общим содержанием аэрозолей в стратосфере и высотой тропо-
тропопаузы. Это связано, по-видимому, с тем, что основной зоной из-
изменчивости аэрозолей является нижняя стратосфера. Практическое
отсутствие вариаций содержания аэрозолей в слое ~ 20 км указы-
указывает на то, что продолжительность существования аэрозолей, за-
заброшенных на эти высоты вулканическими извержениями или иным
способом, должна быть очень большой, тогда как ниже 20 км она
гораздо короче и в среднем составляет около шести месяцев.
Стабильность аэрозолей в верхней атмосфере является, по-
видимому, глобальной. Так, вертикальные профили концентрации
аэрозолей, полученные в Арктике с интервалом в один год, не об-
наруниваит изменений на высотах более 15 км,но указывают на со-
содержанке аэрозолей в более низких слоях, аналогичное наблюдав-
наблюдавшемуся в ...арами (шт.Вайоминг, СЮ.)-.
220
221
По данным измерений на "Кжном полюсе" с интервалом в один
год практически не наблвдалось изменений концентраций аэрозоле.
на всех высотах, что позволяет рассматривать эти данные как тя
пичную характеристику фоновых аэрозолей. Результаты наблвденм!
на нескольких станциях в северном полушарии дают возможное!!
сделать вывод о наличии четко выраженного минимума аэрозольно-
аэрозольного отношения смеси в слое 15-22 км в умеренных широтах, кото.
рый может быть обусловлен наличием стока аэрозолей на уровне
тропопаузной "ловушки". Поле концентраций аэрозолей подобной
лю концентрации радиоактивных компонентов. Хотя долгопериодны!
изменения содержания аэрозолей малы, на высотах > 20 км все
же наблвдается заметный годовой ход с амплитудой 10
для всей толщи стратооферы. То, что максимум содержания аэро-
аэрозольных частиц в стратосфере приходится на зимние месяцы, вер<
ятно, связано с отсутствием переноса стратосферных частиц втр
посферу в этот сезон. Выше 20 км практически никаких сезонных
вариаций содержания аэрозолей не наблюдается - здесь их конце!
трация наиболее стабильна. Кроме сезонных вариаций стратосфер-
стратосферных аэрозолей наблюдается еще более долгопериодные,' природа
которых неясна.
Имеющиеся данные свидетельствуют об уменьшении концентра
ции частиц стратосферных аэрозолей диаметром более 0,3 мкм в
четыре-пять раз в течение десятилетнего периода со времени иа
вержения вулкана Агунг A963 г.), но на эту тенденцию налагав
ся флуктуации, достигающие 300%.
Результаты измерений структуры аэрозолей в тропосфере 1
стратосфере можно свести к следующим.
1. Вертикальные профили счетной концентрации имеют всеЩ
слоистую структуру, которая в значительной степени связана 0
профилем температуры: а) аэрозольные слои находятся вблизи JJ
вней, где затруднен турбулентный обмен (инверсии, изотермми)}
б) аэрозольные слои могут приводить к непосредственному натр
ву окружающего воздуха.
2. Спектр размеров и природа частиц тропосферных аэрозо-
аэрозолей в значительной степени определяются наземными источникам!
особенно пылением почв. Почти всегда наблюдаются распределил
с несколькими модами.
3. Профиль счетной концентрации частиц в стратосфере име-
имеет четко выраженный максимум над тропопаузой. Обнаруживается
определенный широтный ход высоты аэрозольного слоя: для поляр-
полярных широт высота слоя около 15 км, а для тропиков - выше 20 км.
Вариации счетной концентрации больших частиц в слое очень за-
заметны и в значительной степени связаны с вариациями вулканичес-
вулканической активности. Отмечен также сезонный ход содержания частиц в
атмосфере. 0 вариациях счетной концентрации ядра Айткена доста-
достаточно определенно сказать трудно., но имеющиеся данные позволя-
позволяют сделать вывод, что в нижней стратосфере эти вариации значи-
значительно меньше, чем для больших частиц. Изменения счетной кон-
концентрации гигантских частиц, вероятно, достигают трех порядков
величины. Связи этих вариаций с вулканической деятельностью не
обнаруживается.
4. Распределение частиц по размерам многомодальное. Можно
выделить три основные моды, соответствующие частицам ядра Айт-
Айткена ( г ^ 0,1 мкм), большим (ОД < г 4 1,0 мкм) и гигант-
гигантским частицам ( г > 1,0 мкм).
222
нию работы его образования. Равновесие такого комплекса с па-
паром неустойчивое, так как термодинамический потенциал проходит
в этой точке через максимум (метастабилъное состояние). Для та-
такого комплекса, находящегося в неустойчивом равновесии с имев-
имевшимся пересыщенным паром, суммарная работа образования по Дж.
Гиббсу равна
E.3)
*¦-?•
где S - поверхность образующейся фазы. Используя уравнение
Дж.Кельвина, описывающее зависимость давления насыщенного пара
над каплей от ее радиуса, выражение E.3) можно написать в ви-
виде
ДФ= 16зст2в2 /Cfk2T2 In2/),
где /- пересыщение пара; т- масса молекулы пара; р - плот-
плотность жидкости (при условии пренебрежения величиной плотности
пара).
Из теории флуктуации следует, что вероятность возникнове-
возникновения зародыша новой фазы в единице объема за единицу времени оп-
ределяется выражением
™
E#4)
полученным впервые М.Фольмером и А.Вебером, где К - кинетичес-^
кий множитель, N - счетная концентрация зародышей [235]. Для
стационарного случая по Р.Беккеру и В.Дёрингу множитель К ра-
равен
= V
Здесь ?• - число молекул в зародыше; i - поток молекул кон-
конденсирующегося газа на единицу площади поверхности конденсиро-
конденсированной фазы; Cj - число молекул в 1 см3 пара; NA - число Аво-»;
гадро; оь - коэффициент конденсации; М - молекулярная масса
пара.
Я.И.Френкелем был сделан более строгий и простой вывод
уравнения для скорости образования зародышей и было показа-
показано, что
226
m
ехр1-йФ/(кТ)] , E<4)
где Z - коэффициент Зельдовича [100]. Уравнение Беккера - Дё-
ринга отличается от уравнения Френкеля множителем. Так как та-
такие физические характеристики зародыша, как плотность вещества
и величина поверхностного натяжения, зависят от размера зароды-
зародыша, то делались неоднократные попытки учесть влияние этих фак-
факторов на величину работы образования зародышей и скорости их
образования [30, 109].
Рядом исследователей были сделаны попытки учесть зависи-
зависимость термодинамических параметров от величины зародыша, а при
вычислении работы по образованию зародыша - также физические
условия протекания процесса.
В работах Л.М.Щербакова предполагается, что для объектов
малой величины избыток свободной энергии связан не только с его
поверхностью, но относится к объекту в целом. Для достаточно
малых капель отступление от аддитивного распределения термоди-
термодинамических потенциалов (в частности, свободной энергии) требу-
требует добавления в выражение для работы образования зародыша но-
новых членов, зависящих от размера объекта, учитывающих капилляр-
капиллярный эффект. При учете капиллярных эффектов второго рода выра-
выражение для работы образования зародышей сферической формы при-
приобретает вид
E.6)
а для скорости их образования -
\20Sexp[-4/(kT)]. E.4")
Б.В.Дерягиным и Л.М.Щербаковым была учтена внутренняя структу-
структура зародыша: распределение плотности в зародыше от центра к
границе, наличие поверхностного слоя и т.д. Оказалось, что
критический размер зародыша в этом случае определяется толщи-
толщиной поверхностного слоя.
Используя представления квантовой статистики,Дж.Лоте и
Г.Паунд указали на то, что при расчете работы образования за-
227
родыша необходимо учитывать добавочные члены, связанные с изме-
изменением свободной энергии Гиббса вследствие отделения группы мо-
молекул от большого комплекса, активизации поступательной степе-
степени свободы, активизации вращательной степени свободы. Получен-
Полученный ими поправочный коэффициент в выражении для скорости обра-
образования зародышей весьма велик. Например, для водяного пара
при Г = 300 К этот коэффициент равен Ю17 [197].
Однако в работах Х.Рейсса [70, 218, 219] на основе ста-
статистической механики эти выводы Дж.Лоте и Г.Паунда опроверга-
опровергаются.
Х.Рейссом отмечается, что применение обычных термодинами-
термодинамических величин недопустимо в тех случаях, когда размеры зароды-
зародыша становятся соизмеримыми с толщиной переходного слоя между
жидкостью и паром, т.е. для зародышей, содержащих не более двух
десятков молекул. Сформулированная им статистически-механичес-
статистически-механическая трактовка конденсации не требует, чтобы зародыши были жид- "
кими каплями сферической формы и нет необходимости подробно
рассматривать равновесие фаз между зародышем и газовой фазой.
X.Рейсе разделяет пространство, занимаемое паром, на ячейки
объемом -r-g'v, где v - объем молекулы, a g' - некоторое
положительное число, больше или меньше единицы. Количество яче-
ячеек, содержащих молекулы, выражается формулой
n<g} = ? ехр [- {kT(g'-g -
W(g)}l(kT)], E.7)
р [
где N - общее число молекул; W(g) - усредненная по времени
энергия взаимодействия для ячеек с g молекулами, число кото-
которых усредняется по выбранному контуру. Экстремальное зна-
значение n<g> определяется из условия dn(g)/dg=0, т.е.
- кТ In (g/gr) = dW(g)/dg . E.8)
Когда g мало, W(g) = 0 ж nJnax(g>) = n(g') . Численная
шготнооть молекул в этой наиболее вероятной ячейке g j(g'v) -
= N/V равняется средней плотности всего объема пара. Когда
g велико, W(g) - конечно, и условие E.8) определяет ми-
минимальное значение n(g) = n(g*). Ячейки с g>g* имеют
бо'лъшую вероятность приобрести молекулы, чем их потерять, а
228
для ячеек с g<g* характерно обратное. По Х.Рейссу скорость
образования ядер, т.е. комплексов с g = g* в предположении
устойчивого распределения ячеек:
E.4"
где W(g*) = [kT(g'-g* + g*ln(g*Jg') + W<g*)] -обра-
-обратимая работа, затрачиваемая на образование зародыша. Экспери-
Экспериментальная проверка теории гомогенной нуклеации показала, что
она хорошо описывает кинетику фазовых переходов неполярных ве-
веществ, таких, как бензол, л-гексан, л-пентан и пр., а для
полярных веществ, способных к ассоциации (вода,метиловый спирт),
обнаружено значительное расхождение с теорией. Так, для воды
теория и эксперимент дают расхождение в 104 раз. Если учесть
вращательно-поступательный парадокс, то расхождение может сос-
составить 10 раз. Это послужило причиной для создания теории
нуклеации водяного пара, основанной исключительно на молекуляр-
молекулярных представлениях: 1) кластеры имеют определенную структуру;
2) внутренние свойства молекул, образующих кластер, изменяются
незначительно; 3) время жизни кластеров доотаточно велико,что-
велико,чтобы определить их колебательный спектр; 4) ассоциация молекул
воды обусловлена водородной связью; 5) углы между водородными
связями образуют приблизительно тетраэдр. Такая модель позво-
позволяет с помощью аппарата статистической термодинамики рассчи-
рассчитать изменение свободной энергии образования кластеров, потен-
потенциальную энергию межмолекулярного взаимодействия в кластере,
а также скорость нуклеации. Для замкнутой системы объема V
из N(gy невзаимодействующих кластеров размером g при посто-
постоянной температуре Т сумма по состояниям Q определяется как
Q = [ Ztg")] * /N<g) I , а свободная энергия системы ф =
= - kTln Q. Из последних двух соотношений энергия образова-
образования одного кластера равна:
E.9)
Статистическая сумма Z(g) для единичного кластера записыва-
229
вается в виде Z<g> = ZnZBf>ZK0/i exp[-EnЦкТ)\, где Zn - посту-
поступательный, ZSp - вращательный, ZKOrt - колебательный вклады
в статистическую сумму. Экспоненциальный член учитывает межмо-
лекулярное взаимодействие в кластере ( Еп - полная потенциаль-
потенциальная энергия кластера). Тогда выражение для свободной энергии
образования кластера имеет вид
»(?)• E-и>
Из этого выражения следует, что структурам кластеров из
20, 35, 47, 57 молекул соответствуют локальные минимумы изме-
изменения свободной энергии, что обеспечивает их преимущественное
существование по сравнению с кластерами других размеров. Рас-
Рассчитанные моды колебаний для кластерной додекаэдрической струк-
структуры из 20 молекул расположены в диапазоне частот 40-230 см~*,
а для кластеров меньших размеров - в интервале частот 60 -
700 см . Молекулярная модель сравнивалась с эксперименталь- ,
ними данными по измерению скорости гомогенной нуклеации в тем-
температурном интервале 267-272 К и дала удовлетворительное сог-
согласие [140, 168].
Эта модель, трактующая кластер как структуру с определен-
определенной геометрией, позволяет отказаться от применения термодина-
термодинамических параметров макросистемы к микросистеме из нескольких
молекул.
Столкновения молекул пара могут приводить к образованию
комплекса необязательно в условиях пересыщения. В частности,
полярные молекулы воды могут существовать в ассоциированном
состоянии в виде димеров и более сложных комплексов. Образова-
Образование и разрушение димеров можно рассматривать как равновесную
химическую реакцию: Н2<Э + Н2О т— <Н2ОJ. Константу равнове-
равновесия этой реакции Кс можно представить следующим образом:
*с= C((H2OJ)/C*(H2O) =
где ^((Н2ОJ) и <?(Н?О)- статистические суммы мономеров и
димеров ? энергия водороной вязи Выиснне суммы
2 ??
димеров, ?н - энергия водородной связи. Вычисленные суммы
для условий t = 20°С, р =1000 гПа,
<?((Н2ОJ) и
230
е (н2О) = 13 гПА дает величину С((Н2ОJ) = 0,О1С(НгО) при
?н = 4,2-10~20 Дж. При уменьшении ?н до 3,3-Ю0 Мл вели-
величина С((ИгО)г) уменьшается до 0,001 С(НгО). Температурная за-
зависимость концентрации димеров выражается следующим образом:
р((Н2ОJ) = (А/Г1.7>рг<Н20)ехр[-(Ь?н-с)/Г],
где А,Ъ,С - эмлирически подбираемые коэффициенты. Расчет кон-
концентрации связанных молекул воды проводился также с использова-
использованием теории ассоциации, согласно которой в газе, помимо сил
Ван-дер-Ваальса,на расстояниях, много меньших радиуса действия
этих сил, действуют ассоциативные силы. Реальный газ представ-
представляет собой смесь групп связанных молекул с разным количеством
молекул в этих группах и находящихся в термодинамическом равно-
равновесии. Концентрация таких групп в реальном газе находится из
уравнения состояния газа через концентрацию мономеров: я-. -
= с~пе\Уе~*, где rig^NgjN - молярные доли; Ng - количест-
количество групп из g . молекул; N - общее число молекул; V -. объем
газа; Cg - температурный коэффициент, для g}3 Cg^ikgjT*)*
*exp(-Eg/(kT)); для димеров сг = (*2/гг;вхр(-/2 ЦкТ)).
Коэффициент kg зависит от физико-химических свойств газа.
Оценки концентрации димеров, сделанные по приведенным формулам
для обычных атмосферных условий, дают С((Н2ОJ)^ ЮС(Н2О).
Однако теория ассоциаций более оправдана в области сравнитель-
сравнительно высоких температур (t i- 50°С) и давлений.
Оптические проявления димеров воды обнаружены в инфракрас-
инфракрасной области спектра, где наблюдается смещение частота валент-
валентных колебаний гидроксильной группы в сторону низких частот,
увеличение полуширины этих полос, а также рост их интенсивнос-
интенсивности. В реальной атмосфере димерный компонент далеко не исчерпы-
исчерпывает перечень возможных кластерных структур водяного пара.Кро-
ме того, в атмосфере гомогенная конденсация наблюдается при
конденсации продуктов горения (микрометеорное вещество, вулка-
вулканические извержения, некоторые антропогенные источники) и в
результате химических реакций с углеводородами.
На начальной стадии конденсации вероятность возникновения
комплексов из большего числа молекул значительно возрастает.
Моделирование процессов конденсации паров йодистого серебра,
231
хлористого натрия и металлического серебра с помощью вычисли-
вычислительных машин показало, что вероятность прилипания (коэффици-
(коэффициент аккомодации) молекул пара к малым зародышам новой фазы
(размерами 10-15 молекул) на несколько порядков ниже макроскопи-
макроскопического коэффициента конденсации. Следовательно, в процессе об-
образования конденсационных аэрозолей основную роль играет коагу-
коагуляция малых зародышей, поэтому получающиеся частицы должны ха-
характеризоваться достаточно широким распределением по размерен
и высокой счетной концентрацией [95].
В экспериментах Б.И.Старо с сотрудниками помимо аэрозолей '
с такими характеристиками наблюдались также аэрозоли с узким
распределением частиц по размерам и низкой счетной концентра-
концентрацией и аэрозоли с бимодальным распределением частиц по разме-
размерам. Образование этих типов аэрозолей объясняется наличием ге-
гетерогенной конденоации на нелетучих примесных ядрах, имеющих
размеры в несколько молекул, и зависимостью коэффициента кон-
конденсации от размера кластеров. Эта зависимость приводит к силь-
сильному увеличению длительности инкубационного периода гомогенной
конденсации, что может воспрепятствовать образованию частиц по
гомогенному механизму и способствовать.гетерогенной конденса-
конденсации. Вследствие одновременного действия обоих механизмов раз- ¦;
меры гомогенно образованных частиц оказываются существенно мень-
меньшими и аэрозольные частицы приобретают бимодальное распределе-
распределение по размерам с первым максимумом распределения в области
г. = 1+7,5 нм и вторым в области г2тах= 20+27,5 нм. :
Большой цикл исследований равновесного и неравновесного
образований кластеров водяного пара с помощью численных расче- )
тов на ЭВМ был проведен Э.Пурингом и Е.Баклом. В частности,ими .}
определен размер кластера, обладающего макроскопическими свой- '¦¦
ствами A22 молекулы Н2О). Константа равновесия для димера
водяного пара при 272 К по их расчетах равна 4*10 см
[95]. .¦;
Полярные молекулы воды могут образовывать относительно ус-.'
тойчивые комплексы с включениями некоторых инородных молекул
(H2S, SO2, СО2 , N2O , СН4, Аг, Кг, Хе , Rn и т.п. не имею-
имеющих сродства с водой), так называемые водные клатраты или га- '
зовые гидраты. Элементарная ячейка водного клатрата построена ..
Из 2ь молекул воды, связанных в 12 пятиугольников, образующих
додекаэдры с молекулами воды в 20 вершинах. Так как пентагональ-
ные твердые тела не могут быть плотно упакованы в пространстве,
то регулярная кристаллическая решетка из пентагональных додека-
додекаэдров должна включать также другие координационные многогранни-
многогранники. При рассмотрении водных клатратов используются два типа
комбинаций многогранников: первый тип с додекаэдрами и тетра-
додекаэдрами, второй тип с гексадодекаэдрами и додекаэдрами.
Более крупные многогранники имеют в центре полости, в которые
включены молекулы-гостьи. Эти молекулы-гостьи могут быть одно-
одного сорта в полостях обоих типов, а также, возможно, что боль-
большие и малые полости заняты молекулами разных сортов. Такие клат-
тзаты называют двойными, или смешанными. Водные клатраты сущест-
существуют в твердой фазе, имеющей поверхность, способную присоеди-
присоединить молекулы воды с помощью водородных связей, причем вероят-
вероятность ухода этих молекул с поверхности клатратов ниже, чем с
поверхности льда или жидкой воды. Вследствие этого водные клат-
ратк могут быть активными зародышевыми центрами. Размеры самых
маленьких клатратов (додекаэдров) порядка 1,0-1,2 нм, а двой-
двойные клатраты могут быть значительно больших размеров.
Между молекулами включенного вещества и окружающими вод-
водными молекулами решетки клатрата не существует ни водородных,
ни ковалентных, ни ионных связей, а слой молекул воды клатрат-
ной решетки изолирует включенные молекулы от окружающей среды,
вследствие чего они не могут участвовать в химических реакциях
с молекулами вне клатрата. Одной из характерных особенностей
клатратов является их стабильность при температуре выше 0°С.
Например, газовый гидрат двуокиси серы существует при темпера-
температуре 7°с. Термодинамические измерения и расчеты теплоты обра-
образования гидратов газов показываю, что между компонентами сое-
соединения действуют только ван-дер-ваальсовы силы.
исследования Б.И.Стыро и его сотрудников кинетической ус-
устойчивости атмосферных частиц, содержащих продукты распада ра-
радона, легко образующего водные клатраты в элементарной атмос-
|ерной ячейке, приводят к возможности допущения стабильного су-
i-рстзования в атмосфере соединений молекул реальных газов и
парсв ь группах клатратного типа размерами ~ 10 см.
232
233
при этом повышается кинетическая устойчивость системы "
ферный воздух". :1
Наличие заряда у зародыша способствует возникновению ус-
устойчивых комплексов молекул - заряженных кластеров и капель.
Общее изменение свободной энергии системы с зарядом а при об-'
разовании зародышевых капелек из пара равно
| -<ра) + 4яг2<5 + cf/dtr).
f/dtr). EЛ1)
Предполагая, что dsjdr- 0, из условия равновесия капли и па-, г
ра получим .\
E.12)
( RTf/M)lnf = 26/r - <
In/ приобретает максимальное значение при r = r* = f
Если радиус г больше г*, капля будет расти с уменьшени- ;
ем свободной энергии, т.е. капля будет устойчивым зародышем, i
Так как давление насыщенного пара над заряженной каплей меньше, :
чем над незаряженными каплями того же размера, то такие капли ;¦
могут существовать в атмосфере при относительной влажности мень- *
ше 100%. Так, для капель с единичным электрическим зарядом ;,-
равновесный радиус при влажности 40% равняется тг = 3,6*10 сц |
а при 80% - Tj = 3,Э-10~° см. В реальной атмосфере такие кап- л
ли будут моментально захватываться более крупными частицами. . .'*
Приведенные термодинамические формулы не учитывают поляри- |
зации капли ионом. Для полярных молекул воды поверхностная знер- ]
гия капли, возможно, будет изменяться не только от величины за- '
ряда, но и его полярности. Этим объясняют более низкую сте-
пень пересыщения для капель с отрицательным зарядом по сравне-
нию с положительно заряженными (см., например,[81]).
Легкие ионы не могут вырасти в капли в результате последо- |
вательного присоединения к ним молекул водяного пара, более вв- |
роятен захват иона молекулярным комплексом или зародышем, не-
недостаточно большим, чтобы иметь отчетливо выраженный двойной , |
электрический слой, характерный для большой капли воды. Сило-
Силовое поле молекул зародыша (атом кислорода наружу) будет стре- Ц
миться ориентировать молекулы пара относительно поверхности за-|
родыша,благоприятствуя развитию водородных связей и поверхност-:
ной структуры. Захват отрицательного иона увеличивает сило-
234
'!
поле поверхности зародыша и способствует адсорбции поляр-
808 молекул воды, а для положительно заряженного зародыша эф-
Г^шность захвата молекул уменьшается. В отсутствие пересы-
ия пара в атмосфере могут существовать устойчивые заряжен-
Ш6Н кластеры. Они были экспериментально обнаружены относитель-
НЫе едавно вначале с помощью измерений ракетными масс-спект-
1° еТрами на высотах 60-85 км, а позднее в аэростатных полетах
высотах 27-32 км. Исходными ионами при образовании заряжен-
Н«* водных кластеров являются ионы O+.NOta сами кластеры мо-
™, быть типа NO+(H2O)m и Н+(Н2О)П. Обнаружены кластеры,сс-
/ержащие до шести и более молекул воды. В лабораторных услови-
условияхна масс-спектрометре были получены заряженные водные класте-
рн со степенью ассоциации до восьми
§ 5.2. Гомогенная гетеромолекулярная конденсация
Развитая Х.Рейссом и др. на основе представлений статисти-
статистической механики теория гомогенной конденсации достаточно прос-
просто обобщается на случай систем из двух и более химических ком-
компонентов [170, 189, 190, 217, 230].
Изменение свободной энергии при конденсации бинарной сис-
системы А (воды) и В (другое конденсирующееся вещество) равно
согласно Х.Рейссу
EЛЗ)
где ис - химические потенциалы двухкомпонентной смеси, когда
газ и жидкость находятся в равновесии над плоской поверхностью
смеси; и - реальные химические потенциалы в пересыщенной ат-
атмосфере; S - площадь поверхности капли, зависящая от концент-
концентраций Пд и пв ; бдВ - поверхностное натяжение капли, завися-
зависящее от концентрации вещества В в растворе; х = п 1(п, + пд) .
* Большой комплекс исследований условий образования заря-
заряженных водных кластеров с различной степенью ассоциации выпол-
выполнен на кафедре физики атмосферы Ленинградского университета
Е.Г.Авдиевым.
235
Для бинарной системы условия образования капли критического
диуса г* следующие:
длФ \ =0)
что приводит к формуле для критического значения свободной
гии капли
E.14) :
Равновесное число капель критического размера и критического ¦
состава х* пропорционально произведению концентрации молекул
(М К) ( АФ*/(кТ)) щи ий ф 4#г
М. + К) еэср (- АФ*/(кТ)), площади критической сферы
и потоку конденсирующегося газа на единицу площади fig =N.kTx,
хB51т.вкТ)~*12. Окончательно выражение скорости образования
зародышей (зародыщ/(см -с)) с учетом, что -Л^**-^» имеет вид-
E.15)'
Основными химическими соединениями, участвующими в гомо-j
генной гетеромолекулярной конденсации в атмосфере могут быть.:
(при определенных условиях) серная и азотная кислоты. Киангои;;
и др. были созданы вспомогательные таблицы для расчетов скорое
ти ядрообразования бинарных и тернарных систем компонентов •
Н20, H2S04, HNOj [189]. В частности, ими вычислена "актив*
ность" а, определяемая как парциальное давление р газа в ед!
ницах равновесного давления пара над жидкой фазой при поотоян-j
ной температуре Г для разных систем. Например, для системы^
Н2О - H2SO4 : аB5°С) = р{ H2SO4 )/A,33-10)Па, а для систем*]
Н2О-НГЧО3: а<2О"С) = р ( HN03)/F0-103 )Па . (При гетеромолв|
кулярной конденсации активность а всегда меньше или равна ед|
нице, а относительная влажность предполагается меньше 100$.) j
Зависимости скорости ядрообразования от влажности и вапя^
чины активности очень сильные. Для HNO3 при величине "¦!
a(HNO3)<0,i (^(HNOj^l.O'lO17 см скорость ядрообразовда
ния очень мала при достаточно высокой относительной влажностй|
воздуха. Для серной кислоты заметное ядрообразование наблвда^
ся даже при a(H2SO4) <= 0,001 (для I = 25°С соответствуем
236 1
давлению паров H2SO4 р = 1,33-Т0~7Па и п(Н?0А)*3-107 см 31
При уменьшении относительной влажности воздуха до величин мень-
ше 20;»' гетеромолекулярное ядрообразование в системе HNO3-H2O
практически прекращается, а для системы H2SO4-H2O резко
уменьшается.
3 тернарной системе H2SO4-HNO3 - Н2О условия ядрооб-
оазования менее жесткие,чем в бинарных системах, в частности
возможна конденсация HNO3 при значительно меньшем парциальном
давлении.
"екоторые характеристики процесса гетеромолекулярной гомо-
гомогенной конденсации в системе Н2О - H2S04 для условий страто-
стратосферного аэрозольного слоя Юнге приведены в табл.31. 0 реаль-
реальном вкладе этого процесса в образование аэрозольного слоя мож-
можно говорить лишь для скорости генерации больше •J-IO см • с .
Анализ результатов численного моделирования образования заро-
дытей капель серной кислоты показывает, что устойчивый серно-
сернокислотный слой на высотах 17-22 км может существовать, если ско-
рость генерации молекул SO3 будет больше 10 см • с , а кон-
концентрация молекул водяного пара больше 8*10 см . Такие ус-
условия легко реализуются при сильных вулканических извержениях,
когда в стратосферу забрасываются сотни тысяч тонн подяного па-
пара и сернистого газа, и затем происходит окисление SO2 до SO3.
В случае, когда сильных вулканических извержений длительное
время нет, диффузионный поток сернистого газа из тропосферы в
нижнюю стратосферу не обеспечивает необходимой скорости генера-
генерации молекул SO3, так как S02 окисляется в тропосфере. Тогда,
вероятно, существенным фактором становится диффузионный поток
химически более устойчивого карбонилсульфида OCS, который окис-
окисляется уже в стратосфере. Наиболее энергетически выгодное со-
соотношение масс серной кислоты и воды в зародышевой капле раст-
раствора для температуры слоя Юнге Г = 218 К примерно равно 23:27.
Температура замерзания такого раствора 230 К, а существование
переохлажденной жидкой кашш раствора возможно при температуре
на 30-40 ниже точки замерзания,1причем добавление в раствор
10% HNO3 понижает точки замерзания до 223 К. Таким образом,
существование жидких капель раствора серной кислоты в нижней
стратосфере вполне объяснимо. Начальные размеры зародышей око-
237
Таблица 31
Характеристики процесса гетеромолекулярногв
для разных концентраций паров серно!
N( НгО),
см
8,75-Ю12
8,75-1012
8,75-Ю12
8,75-Ю12
8,75-Ю13
8,75-Ю13
8,75 «Ю14
JV, см-3
Н2О
H2SO4
5-Ю5
5-Ю6
5-107
5-Ю8
5-Ю6
5-Ю5
5-
I05
дж/мг
0,08002
0,07952
0,07923
0,7936
0,08004
0,08017
0,08014
Л*(Н20)
..„ J
132 •
34
13 j
9 1
14 1
21 \
4 ,'i
ло F+10)•10 см, однако они быстро укрупняются вследствие ДА
фузионного потока молекул серной кислоты (или SO3) и водяного!
пара. Очевидно, что фактором, лимитирующим их размеры, будеЖ
концентрация молекул серной кислоты в окружающей воздушной щ
де. Для стационарного случая :!
di
dNa
dr
dr =0.
см, о"*
При J(H2SO4)= г.О-Ю^ см~°. c~l общая поверхность аэрозо
частиц должна быть примерно 7,2-10~8 см^см3, что соответзд
обшей концентрации примерно в несколько частиц в кубичем
сантиметре при среднем геометрическом радиусе их равном 0,1
§ 5.3. Адсорбция и набухание
Гетерогенный механизм конденсации в реальных атмосфер
условиях оказывается практически всегда более эффективна!, i
гомогенный. Для больших ядер конденсации начальная стадия Ц
рогенной конденсации - абсорбция и адсорбция молекул пара -jj
238
гомогенного образования зародышей
кислоты и воды при Г = 218 К
п*
Н2О
H2S04
77
25
11
7
6
•11
2
Г* см
1,3.10~7
8,'б-10~8
6,45-10~8
5,7 -Ю"8
5,8 -10~8
6,9 -10"8
6,4 -10~8
5,56-10~19
2.52-109
1,49-Ю9
9,31-100
1.12.109
1,69-10~19
7.21-100
см'3с
ю-72
9,9-Ю7
3.7.101
3,5-10
6,7-Ю
'5,8-10~15
3,5-10°
исходит при парциальном давлении пара значительно меньшем,чем
давление насыщения. Растворение в конденсате вещества частицы
согласно закону Рауля приводит к снижению давления насыщения
пара над поверхностью частицы.
Для частиц с шероховатостями и порами упругость насыщения
над вогнутыми поверхностями может быть значительно ниже упру-
упругости насыщения над плоской поверхностью. Вследствие этого кон-
конденсация пара будет приводить к заполнению вогнутых поверхнос-
поверхностей и микрокапилляров внутри частиц при значениях относитель-
относительной влажности (или парциального давления других конденсатов),
далеких от насыщения [ 64 ]. Для некоторых веществ процесс мик-
микрокапиллярной конденсации сопровождается заметным увеличением
объема частиц без изменения их формы, так называемым набухани-
набуханием [100J. Механизм адсорбции весьма детально был исследован в
классических работах С.Брунауэра, Я. де-Бура, Я.И.Френкеля и
других.
Весьма наглядная трактовка механизма адсорбции содержится
в монографии Я. де-Бура [16].
Молекулы газа, сталкиваясь с поверхностью, какое-то время
контактируют с ней. При этом происходит обмен энергией. В слу-
случае зеркального отражения обмена энергией не происходит. Если
239
время пребывания молекулы газа на поверхности намного превыше- \
ет период колебаний молекул поверхности, то происходит полный
обмен энергией, и молекула газа приобретает ту же температуру,
что и поверхность. Характеристикой взаимодействия молекулы га-
газа с поверхностью является коэффициент аккомодации а = (TJ -
- Тх)/( Т2 - Т1 ), где Tj - температура газа, 72 - темпера-
температура поверхности и Т2Г - температура молекул газа, испаряющих-
испаряющихся с поверхности. Для случая зеркального отражения а = 0, a -
при полном обмене энергией а = 1. Увеличение шероховатости по-
поверхности приводит к увеличению коэффициента аккомодации, а кро-
кроме того, к увеличению времени адсорбции молекул газа т. Время
адсорбции определяется в соответствии с теоретическим уравне- >
нием, предложенным Я.И.Френкелем: т = xoeQ /(RT) , где т —'
период колебания адсорбированной молекулы в направлении, пер- ¦
пендикулярном к поверхности (имеет порядок 10"^ с), Q - теп-.
лота адсорбции, выделяющаяся при переходе молекулы из газооб-
газообразного состояния в адсорбированное. Энергия физической ад-
адсорбции, при которой связь между поверхностью и адсорбирован- '<
ной молекулой осуществляется силами Ван-дер-Ваальса, имеет по-'
рядок 1-3-1СГ20 ?к и может достигать 1,3*10~19 Дж. (При пооя
леднем значении Q величина т равна примерно 100 с при комна**
ной температуре.) ;
Число адсорбированных молекул на единицу площади можно в»
разить, как 5-пх, где л - число молекул, ударяющихся о ejsM
ничную поверхность за 1 с. Для водяного пара Q %. 6,5*10"* Дж:
в зависимости от вещества поверхности, а т ? 5-10 с для
t - 110°С. Например, при относительной влажности 60% и темпера
туре 20°С не менее 20/2 поверхности будет покрыто молекулами вЛ
ды. Для ряда газообразных веществ, особенно органических, дол^
поверхности, покрытой адсорбированными молекулами, может бытЙ
очень большой даже при очень низких парциальных давлениях эищ
газов вследствие больших значений т. |
Если не все ударяющиеся молекулы адсорбируются, а
рая их доля ql отражается, то 6 = л<1- а)г. Подставляя
ражения для л и х в формулу для числа адсорбированиях на
¦ ницу площади молекул, получаем
240
E.16)
уравнение адсорбции при допущениях И.Ленгмюра: 1) значение теп-
теплоты адсорбции всех молекул, ударяющихся о свободную поверх-
поверхность, одна и та же и не зависит от присутствия других адсор-
бирозанных молекул; 2) молекула, ударяющаяся об адсорбирован-
адсорбированную молекулу, немедленно возвращается в газовую фазу, приобре-
приобретает вид б = волт/(ео + лт), или через степень покрытия:;
9«<г/б? = (ят/у0)/<1 + пх/ео) = кпр/{1+кпр), где кп =
= (JV/ }/2яМЯГ)т / бг0 . По этой модели адсорбированные молеку-
молекулы остаются на своих местах в течение всего времени адсорбции
х, что не всегда соответствует реальной картине адсорбции. .
Если молекулы, ударяющиеся о слой уже адсорбированных мо-
молекул, испытывают достаточно сильное притяжение, то могут вы-
выполняться условия для адсорбции более чем одного слоя, т.е.
для полимолекулярной адсорбции. Уравнение БЭТ, описывающее по-
полимолекулярную адсорбцию, было выведено С.Брунауэром, П.Эмме-
том и Е.Теллером. Ими предполагается, что в процессе образова-
образования слоя адсорбированные молекулы его не оказывают влияния
друг на друга, а энергия адсорбции в пределах каждого из слоев
постоянна. Выражение для е в этом случае
в =
EЛ6-)
где а=(бу/2лМЯТ ) /(Nz), к = x/xj, т-время адсорбции мо-
молекул, непосредственно связанных с поверхностью; т^ - время
адсорбции молекул, адсорбированных на первом и последующих сло-
слоях. Выражение для объема адсорбированных молекул будет следую-
Wm: V=kpVm/(<i-p)[l+(k-l)pl<l]. Здесь Vm- объем
адсорбированного газа, когда вся поверхность адсорбента покры-
покрыта монослоем. При рассмотрении отдельной молекулы первого ад-
адсорбционного слоя трудно представить, что молекула второго слон,
расположенная на изолированной молекуле, связана настолько про-
прочно, что теплота адсорбции имеет величину порядка теплоты ис-
испарения. Только в том случае, когда молекулы первого слоя име-
ят плотную двумерную упаковку (конденсированы), можно ожидать,
«.Mi 241
что теплота адсорбции следующих слоев молекул близка по вели-
величине к теплоте испарения жидкости.
Уравнение, описывающее адсорбцию в термодинамических веля
чинах, было выведено Дж.Гиббсом [22 "\ и может быть представлено
в виде
где с - концентрация молекул в трехмерной (газовой) фазе;
(дг/дс )s - частная производная от поверхностной энергии по
трехмерной концентрации при постоянной величине поверхности 5,
Полагая адсорбированную фазу двумерным газом, можно написать
уравнение Ван-дер-Ваальса для адсорбированного газа, которое''
может качественно описывать явления адсорбции и двумерной ков*
денсации, когда силы притяжения между молекулами поверхности i
адсорбента становятся достаточно большими. Уравнения Ван-дер^-
Ваальса в виде, предложенном М.Фольмером, записывается следу»
щим образом:
FiA- Ь2> = ЯТ. E.18
Здесь F - двумерное давление (сила на единицу длины поверх-
поверхности); Ь2 ~ поправочный член для двумерного газа, аналогич-
аналогичный члену в уравнении Ван-дер-Ваальса ( Ъг - N/c); А - площад
поверхности на 1 моль двумерного газа. Учет поправочного член
обусловленного ван-дер-ваальсовыми силами притяжения, приводя
к более сложному уравнению состояния двумерного газа
( F + а2/А2)(А + Ь2 ) = ЯТ, E.18'
где а2=(яЛГг/Dс?4)НЗ/4)о12?, * -поляризуемость, d - Д1
метр молекулы. .г
Поведение большинства адсорбируемых на поверхности молей
кул плохо описывается уравнением Ван-дер-Ваальса для двумерж
"Cto газа в силу их анизотропнооти. Для изолированных адоорбир|
ванных молекул их ориентация относительно поверхности буде* jj
ределяться поляризуемостью молекулы, в то время как в случА
группы молекул силы Ван-дер-Ваальса будут стремиться распоя
жить длинные молекулы параллельно друг другу, а плоские - °Й
242 '<
ентировать параллельно их плоскости. Взаимная ориентация моле-
молекул сама по себе приводит к увеличению критической температуры
двумерного состояния и благоприятствует конденсации. Однако вза-
взаимная параллельная ориентация диполей уменьшает взаимное при-
притяжение молекул и препятствует конденсации. Дипольный момент
у адсорбированных молекул может наводиться адсорбентом.
У адсорбентов с сильно развитой пористой поверхностью ад-
адсорбированные молекулы в углублениях окружены значительно боль-
большим числом поверхностных атомов, чем при адсорбции на плоской
поверхности. Вследствие аддитивности сил Ван-дер-Ваальса тепло-
теплота адсорбции в этом случае будет значительно больше, чем для
ровной поверхности. Адсорбция в капиллярах может привести к то-
тому, что теплота адсорбции молекул даже в последующих слоях ад-
адсорбированных молекул будет выше теплоты конденсации адсорбата.
Теплота адсорбции последующих слоев молекул может несколь-
несколько увеличиваться,если молекулы нижележащего слоя поляризованы
аакиы образом,что возможна индуцированная поляризация в следую-
следующих слоях.Заметных изменений размеров частиц при такой адсорб-
адсорбции не наблюдается:происходит заполнение ми кр о капилляров и ми-
кро^ероховатостей поверхности частиц и увеличение диаметра ча-
частиц на несколько десятых нанометра.Существеннее изменение фи-
физических и химических свойств поверхности частиц,которые могут
сказываться на оптических свойствах последних и электрических
явлениях, а также на протекании различных химических и фотохи-
фотохимических реакций.
Двумер'" ¦ давление, создаваемое адсорбированными молекула-
молекулами, может .иимать весьма большие значения, поэтому давление
на стенки адсорбента может приводить к структурным изменениям
адсорбента, особенно, когда имеются капилляры, в которых про-
происходит адс^-'мия. Капилляры могут расшириться, увеличивая про-
пространство, ютупное для адсорбата. Двумерное давление достига-
достигает особенно больших значений, когда происходит двумерная кон-
конденсация. Наблюдается набухание адсорбента, при котором объем
системы может увеличиваться в несколько раз (даже в сотни раз).
многие вещества набухают при адсорбции воды: силикагель, жела-
желатина, шерсть, крахмал, древесный уголь, различные полимеры.
243
Я.И.Френкель рассматривал набухание как процесс осмотичес-
осмотической природы, вызванный не потенциальными, а кинетическими си-
силами, т.е. тепловым движением молекул наполнителя, и связанный
не с уменьшением энергии, а с увеличением энтропии.Поверхность
полимера играет в процессе набухания ту же роль, что и полупро-
полупроницаемая мембрана в случае обычного осмоса. Проникновение на-
наполнителя может иметь место при условии, что между молекулами
обоего сорта существует определенное притяжение (вещество ад-
адсорбента смачивается). Следует отметить, что при отсутствии на-
наполнителя изменение объема полимерного вещества адсорбента бы-
было бы связано с проявлением упругих сал, соответствующих изме-
изменению 'взаимной потенциальной энергии молекул. В присутствии на-
наполнителя эти силы не имеют значения и упругое напряжение, выз-
вызванное набуханием, определяется относительно малым модулем уп-
упругости.
Аля неполимеров этот процесс может происходить подобным
же образом, если межмолекулярные силы взаимодействия вещества
адсорбента по порядку величины близки к силе взаимодействия
между молекулами адсорбента и адсорбата. Происходит как бы час-
частичное растворение адсорбента в адсорбате.
А
§ 5.4. Гетерогенная конденсация
Атмосферные ядра, на которых конденсируется водяной пар,
могут быть различной природы и обладать разными физико-химичес-
физико-химическими свойствами. Различают три основннх типа ядер: гигроскопи-
гигроскопические, негигроскопические и смешанные. Они могут быть смачи-
смачиваемыми и несмачиваемыми. Вещеотво смешанных ядер состоит из
гигроскопического и негигроскопического материалов. Очевидно,
что на начальной стадии конденоационного роста смешанные ядра
будут вести себя как гигроскопические. Процесс гетерегенного
конденсационного роста частиц можно разделить на три стадии:
1) повышение влажности среды до величины, когда происходит по-
полимолекулярная адсорбция и конденсация в микрокапиллярах и на
шероховатостях поверхности частиц, 2) обводнение ядра до прев-
превращения его в каплю насыщенного раствора, 3) рост капли раство-
244
оа. Если капелька формируется на растворимом ядре, то равновес-
равновесная упругость пара у ее поверхности уменьшается на величину, за-
зависящую от природы и концентрации растворяемого вещества. Для
негигроскопических ядер процесс гетерогенной конденсации закан-
заканчивается обводнением ядра, после чего рост может идти только
при влажности, превышающей влажность насыщения. Для смешанных
ядер третья стадия конденсационного роста частиц быстро прекра-
прекращается из-за недостатка растворимого вещества.
Выражение для равновесной упругости пара у поверхности ка-
капельки раствора впервые было выведено Х.Кёлером. По Б.Дж.Мей-
сону [70] оно имеет следующий вид:
imM/(W
где р'г - упругость пара у поверхности частицы с радиусом г;
Ря> ~ упругость пара над плоской поверхностью; оБ - плот-
плотность воды; Og - плотность раствора, 6' - поверхностное натя-
натяжение раствора; m - масса растворяемого вещества в граммах;
М - молекулярная масса воды; i - коэффициент Вант-Гоффа, за-
зависящий от химической природы вещества частицы и степени дис-
диссоциации (т.е. от концентрации) растворяемого вещества; W -
обратимая работа, затраченная на образовансе капли раствора.
Весьма важны для процесса конденсации поверхностные свойства
частиц: смачиваемость, электрическая структура и т.д. Учет этих
свойств приводит к изменению величины работы образования капли.
Рассмотрим твердое сферическое ядро радиусом гя, которое
в результате конденсации молекул водяного пара растет и стано-
становится зародышем с радиусом гг> гя . В качестве меры смачивае-
смачиваемости используем угол смачивания <^>, заключенный между поверх-
поверхностью сконденсированной фазы и подстилающей поверхностью. Обо-
Обозначим газообразную, жидкую и твердую фазы воды индексами л, 6,
т, а величины удельной поверхности энергии между фазами епв,
6вт>етп и граничные поверхности Sp6 , SgT,STn. Для сфе-
сферического ядра SeT = SnT = 4згг| .
Одним из эффективных механизмов конденсации водяного па-
пара является конденсация на шероховатых поверхностях, происходя-
происходящая при относительной влажнооти, значительно меньшей 100$ [61,
245
64]. По М.Фольмеру работа образования зародышевой капли Л*
есть разность мевду работой, затраченной на образование поверх-
поверхности зародыша Ая, и работой, полученной при создании зароды-
зародыша Аи, т.е.
E.20)
из условия равновесия поверхностных сил:
получаем
7
3cos<? " 2г*/гз
E.20)
Для абсолютно смачиваемого ядра
°
= 0°, а для абсолютно не-
р p
смачиваемого <р = 180°. Более строгий вывод величины работы
образования капли на ядре приводится, например, в монографии
А.И.Русанова [82].
Из формулы E.20) видно, что работа образования капли на
полностью несмачиваемом ядре (<р = 180°) по сравнению со сма-
смачиваемым существенно увеличивается.
Скорость гетерогенного образования капель с участием водо-
нерастворимых частиц согласно Н.Флетчеру описывается выражени-
выражением E.4) для гомогенного образования капель
1
где гя - радиус аэрозольной частицы; К= с'а>(&вп$3/(
53 - площадь поверхности кристаллического зародыша на водоне- .
растворимом субстрате; с' - число отдельных молекул водяного
пара, адсорбированных единицей площади поверхности; и) - час- /
тота столкновений молекул воды на единицу площади поверхности ,.;
конденсационной фазы. Вблизи 0°С кинетический фактор изменяет- >"
ся от 10 ^ до ТО в зависимости от адсорбирующей эффективное- ;
ти частиц относительно водяного пара. Изменение свободной энер-;
гии при образовании зародыша на нерастворимой частице Щ
где х=гя/г3;
246
f(m,X)= i + ±
f mx2{[(x-m)/U]-l};
U = (l + xz- 2mx)ll2 и m = cos0 . При полной смачиваемости
частицы 9 = 0, m = l, r3 = гя, 4Ф3 = 0, и критическое пере-
пересыщение получается из уравнения Кельвина. Размеры капель на
ядрах конденсации определяются не только физико-химическими
свойствами ядра и величиной относительной влажности, но и тем,
как меняется влажность воздуха во времени [86, 96]. Если за
начало отсчета первой стадии роста частиц принять момент дости-
достижения концентрацией водяных паров в среде равновесного значе-
значения над плоской поверхностью насыщенного раствора вещества яд-
ядра сн, то т = (сн/рН2М/(рЯ:Г»(б/гс) , где C -скорость
увеличения концентрации водяных паров в среде (гЛсм^с)); гс -
начальный радиус ядра. Оценка времени осуществления второй ста-
стадии т, требует знания количества влаги тп, которое может 5°-
глотить частица до полного ее растворения, т.е. знания радиуса
полностью обводненного ядра г . Этот радиус определяется как
ттп~г ffi^c'» где ^с " коэффициент, учитывающий раствори-
растворимость вещества частицы и изменение плотности. По Н.А.Фуксу
для частиц с радиусом в диапазоне 10 - 10 см
[103] формула
dr
dx
-Oct.
Дс
применима
E.21)
где « - четвертая часть средней абсолютной скорости молекул
пара; ai. - коэффициент конденсации; ов - плотность воды;раз-
ность концентрации Дс = бт, откуда т2 = у'2о rc(|/Jb^-l)/(-9«^),
Изменение поверхностных свойств капли может как замедлить,
так и ускорить протекание первых двух стадий: 1) уменьшение е
ускоряет процесс, 2) увеличение поверхностного сопротивления
проникновению молекул воды в объем ядра ( сь ) замедляет про-
процесс. Время протекания третьей стадии определяется с помощью
ранее приведенной формулы Н.А.Фукса, в которой Дс = ^(т^ +
- гъ
)• Расчеты т1( *г2
показыва-
247
ют, что т3 всегда значительно меньше времен первых двух ста- *?
дий роста капли (доли секунды), тогда как х1 и т2- величины
одного порядка (сотни секунд). Для движущихся воздушных масс '
такая значительная задержка в установлении равновесного разме-
размера капли может существенно влиять на пространственную структу-
структуру аэрозолей.
§ 5.5. Эмпирические формулы роста гигроскопических
и смешанных аэрозольных частиц при увеличении влажности ';
Наиболее простым и относительно изученным типом завж-й
симости микроструктуры аэрозолей от величины относительной вла-
влажности является случай, относящийся к росту гигроскопических'1;
частиц при увеличении влажности. Классическим примером роста .-'¦
гигроскопических аэрозольных частиц при изменении влажности мо'
гут служить морские аэрозоли, диспергированная фаза которых ос-»:
стоит в основном из NaCl [188] и сульфатов. j
Неоднократно проводились расчеты размеров капель растворе
различных гигроскопических веществ для разных величин относи-*:
тельной влажности, которые показывают удовлетворительное согла-2
сие с экспериментальными измерениями равновесных радиусов чао- ;
тиц из одного вещества [23, 111, 154, 196]. Например, расчет-'•;
ные величины относительной влажности, при которых происходит П9ч|
реход частиц в капельное состояние, следующие: NaCl - 78, ¦<
(NH4JSO4 - 73, NH4Cl-86, MgS04- 85, NaNO3- 78$ [46]..
Однако в реальных условиях, в частности в смогах, практи-^
чески нет частиц, состоящих из одного химического соединения, j
поэтому эмпирические формулы могут точнее описывать рост реаль^
ных аэрозольных частиц, чем теоретические. В последние годы 6irf
ли разработаны методы, позволяющие вводить в основное уравнешк!
термодинамического равновесия между сферой с жидкой оболочкой^
окружающим сферу влажным воздухом эмпирические поправки, раошй
ряющие возможности оценок массы, размера, средней плотности чщ
тиц и среднего эффективного показателя преломления вещества 1
как функций относительной влажности [133, 173, 187, 227]. ^
В общем виде эмпирическая формула термодинамического рав-
равновесия сферы с окружающим влажным воздухом записывается фор-
формулой:
ъа
ер[2втв<1+У)/<рв/?в7»], E.22)
где aga - водная активность жидкости вещества частицы для
плоской поверхности и авр - для поверхности части при давле-
давлении водяного пара р. и температуре Т; X - представляет со-
собой поправочную функцию срг1^-рг1ръ)ръ., где с изменяется
от 0,002 до 0,008 в диапазоне изменения температуры от +30 до
-30°С, У - эмпирическая поправка в экспоненциальном выражении,
ави/ава = ав +z ( 2 - эмпирическая поправка, изменяется от
0,004 A-яв) до 0,01 (I-aB) в диапазоне температур +30 -
-30°С). Основную роль в этой эмпирической теории играет поня-
понятие коэффициента возрастания массы для описания водной актив-
активности. Причем рассматриваются четыре различных случая обводнён-
обводнённых частиц: 1) частица, состоящая из одного растворимого в во-
воде соединения (NaCl, (NH4JSO4, MgrCl2 и т.д.)); 2) частица,со-
частица,состоящая из смеси растворимых веществ; 3) частица, состоящая из
нерастворимой и растворимой частей, покрытая водной оболочкой
при идеализированных условиях; 4) частица, покрытая водной обо-
оболочкой в реальных условиях.
Для первого простейшего случая возможно четыре эквивалент-
эквивалентных определения ' ав, содержащих разные эмпирические коэффици-
коэффициенты: ав = лв/(лв + ?лс); ав =[лв/(лв + лс)] а; ав=$влв/(лв+лс);
ав = ехр<-Ф-?лс)/лв , где лв - число молей воды;- лс - чис-
число молей сухого растворяемого вещества; фд -кажущийся осмо-
осмотический коэффициент; $э - коэффициент активности воды; ф -
фактический осмотический коэффициент; ? - фактор Вант-Гоффа;
¦? - число молей ионов из одного моля сухого растворимого ве-
вещества (коэффициент диссоциации). Коэффициенты г и Ф для раз-
разных химических соединений табулированы. Описание ав через от-
отношение массы твердого вещества тпс к массе конденсированной
воды производится с помощью двух эмпирических параметров: г> =
= Ф-?МВ jMc и M-C=i-WB/Mc - соответственно экспоненциально-
248
249
го и линейного коэффициентов возрастания массы. Выражения ав
как функции концентрации раствора тс/те через параметры п
(/) и а=<1 -рлг/т)
и ис имеют вид ав
В /
с/е с
пв) и ав=<1 -р.слгс/тв)-».
и ис д ав р^сс/в) в рс/в
Величины а. и /гг /тс экспериментально измеряются, что позволя-
позволяет табулировать параметры rjc и |ХС, которые зависят от ад оп-
определенным образом для каждой соли.
При заданных температуре Г и давлении р величина ав
прямо пропорциональна количеству воды, приходящейся на единицу
массы сухого растворимого вещества. Причем при бесконечном раз-
разбавлении ав = 1, фактический осмотический коэффициент Ф =1
и фактор Вант-Гоффа \ = -0. Тогда 7с = J?c^ae=1>; Ус SWae=I)
/
П.Винклером использовалось несколько иное аппроксимацион-
ное выражение для ав: аъ= i-cbmcjmB, предполагающее, что
& постоянно и определяется как oi=iMB/Mc подобно линей-
линейному коэффициенту возрастания массы Мс.
Для случая смеси растворов предполагается, что каждая .
соль в растворе ведет себя независимо от присутствия других со-
солей. Тогда все выражения имеют такой же вид, но используются
усредненные величинн -о, Ф, i, Мс, цс , у.с , а тс, пс представ-
представляют суммы тс•, п.; по всем компонентам раствора.
При описании идеализированного покрытия водной оболочкой
неполностью растворимой частицы предполагается, что можно пре-
пренебречь внутрифазовыми явлениями между растворимым и нераство-
нерастворимым веществом частиц и присутствием поверхностно-активных ве-
веществ. В этом случае в выражениях для ав вместо тс вводит-
вводится величина т0, равная сумме масс растворимого и нерастворимо- ч
го вещества частиц.
Для описания реального обводнения аэрозольной частицы в .;
выражения для п и и вводят поправочный эмпирической козффици-з
ент $. Величина поверхностного натяжения раствора частицы мо- ,
жет быть выражена в виде следующей эмпирической формулы: Сс я '
= бгв(Г) + Ьтс1тв, где 6В(Т) -поверхностное натяжение :
чистой воды при температуре Т, а Ъ примерно равна 0,02 Н/м ;
(для NaCt Ъ = 0,0276 Н/м, для <NH4JSO4 Ь = 0,0164 Н/м). ,
Зависимость бв от температуры имеет вид бв(Т) = 6Гв(Гд) + ';
+ а(Т0-Т), гдебв(Г0= 273, 16 К) = 0,0756 Н/м, а =1,53 X
250
10-5 н/(м-град) в диапазоне изменения температур от -10 до
+-.С°С. Зависимость ffc от наличия поверхностно-активных ве-
веществ выражается фактором $е. Предполагается, что объем части-
частицы с конденсированной водой равен сумме объемов вещества и кон-
конденсированной воды. Тогда очевидно, что
3- E'23)
Окончательное выражение для f=pr/pB приобретает вид
Это уравнение справедливо, когда ав ^ agn , т.е. для величи-
величины a , при которой достигается полное покрытие частицы водной
оболочкой. Для оценки этой величины можно использовать соотно-
соотношение ma(aBi)/m0 =apB/p0, причем для практических целей
достаточно считать ct =1. Порог водной активности соответст-
соответствует наибольшей водной активности, при которой линейный коэф-
коэффициент возрастания массы и достигает относительного максиму-
максимума, т.е. момента, когда растворимое вещество частицы полностью
растворилось.
При более низких величинах влажности f и соответственно
величины ав справедливо уравнение адсорбции БЭТ, которое Г.Хе-
нел приводит к виду mB/m0= uaB/(l-aB), подразумевая,что
У =ав, а и - линейный коэффициент возрастания массы. Для
переходного режима &ъ{~ &&el^ ав ^ аъ1 вводится поправка
на кривизну (для частиц меньше 0,1 мкм) S(aB)=S(aBf)aB/aBj .
При значениях относительной влажности, близких к значениям уп-
упругости насыщения водяного пара, в уравнение термодинамическо-
термодинамического равновесия для частицы вводится поправка в показатель экс-
экспоненты:
Р
т0
251
Размеры аэрозольных частиц сферической формы определяются'
из полученных формул с помощью выражения ¦•
E.25)'*
в предположении справедливости равенства J = аа и пренебреже-"*
ния влиянием кривизны поверхности частицы, т.е. для /4 0,90—j
0,95 и rQ » 0,1 мкм. 1
С учетом влияния кривизны поверхности частицы и в предпо->а
ложении /=йв имеем
-ar
+ ft u/л ^
Применимость этой формулы значительно шире: для 0,70 <
< 0,99 и Г„ > 0,04 мкм.
Простое эмпирическое выражение для зависимости г
предложено Ф.Кастеном:
E.26)
Оно неоднократно использовалось для расчетов метеорологическо)
дальности видимости при разной влажности. Показатель степени
6 относительно слабо зависит от размера частицы, относив
влажности и природы частиц. Для разных моделей аэрозолей Г.
нелом были получены значения е в диапазоне 0,18-0,285
В работе [196] в качестве эмпирической формулы для о:
ления равновесного радиуса частиц г* при относительной
ности f, имеющих радиус сухого ядра rrf, предложено вы;
r = oi г ^, где <» и ^ определяются природой вещества
величиной влажности. Проверка этой формулы для 19 элек
(NaCl, NH4NO3, CaCl2, <NH4JS04 и т.д.) в диапазоне :
ности 81-99,5% показала, что ошибки расчетов радиуса час
меньше 5%.
Для многокомпонентных систем экспериментально было пом
но существование как положительных, так и отрицательных откл*
нений от правила аддитивности [23, 196].
252
§ 5.6. Образование ледяных частиц в атмосфере
Оазовые переходы воды в атмосфере происходят в основном
при отрицательных температурах.Прямое образование льда,т.е. ге-
могенная кристаллизация водяного пара,в реальной атмосфере яв-
явление, вероятно, невозможное. Даже при температурах до -20°С
значительная часть облаков состоит либо из переохлажденных вод-
водных капель, либо из одновременно сосуществующих водных капель
и ледяных кристаллов. Это свидетельствует не только о гетеро-
гетерогенном характере образования ледяных частиц в атмосфере, но и
о том, что только часть аэрозольных частиц служит активными яд-
ядрами образования ледяных кристаллов. При понижении температуры
воздуха концентрация активных ядер увеличивается по экспоненте:
л=лоеэср(?ДГ>, где ? = 0,6 град, ДГ=273К-Г, причем
пространственные вариации активных ядер конденсации относитель-
относительно невелики [105]. Различные частицы способствуют образованию
льда при разных температурах. Известно, что возможна активиза-
активизация ядер при низких (-40°С) температурах и пониженной влажнос-
влажности (но не менее 50$).
Гетерогенное образование льда может осуществляться различ-
различными способами:
1) сублимацией молекул водяного пара на тввердой частице
с образованием ледяного зародыша;
2) конденсацией водяного пара на ядре с последующим замер-
замерзанием;
3) нуклеацией при иммерисионном замерзании, когда активное
ядро полностью погружено в каплю;
4) контактной нуклеацией при столкновении переохлажденной
капли с ядром.
Степень влияния различных примесей и частиц на температу-
температуру замерзания капель определяет их льдообразующую активность.
Наибольшее значение температуры, при которой образуется ледяная
фаза - предельная или пороговая температура активизации веществ.
Было обнаружено, что льдообразующая активность всех веществ име-
имеет две пороговые температуры, причем более высокая соответству-
соответствует пересыщению паров воды относительно водной поверхности, а
253
другая - относительно льда. Экспериментально также установлено!
что основным процессом гетерогенного образования ледяных чао- ¦'
тип, является двухстадийный процесс: конденсация - замерзание
[73, 95].
Наиболее сложной проблемой оказалась задача выявления ос-
основных требований, обусловливающих активное льдообразование на
частице. Так как скорость гетерогенного образования льда опре-
определяется энергией, необходимой для образования ледяного зароды-
зародыша размером больше критического, то любая частица будет способ-
способствовать изменению фазового состояния путем предоставления ус-
устойчивой поверхности, на которой может расти зародыш, снижая
тем самым величину энергии образования устойчивого ядра [105]. '
Следовательно, для сублимационного образования ледяной частицы
необходимо условие водонерастворимости твердого ядра. Для ядра
замерзания прямой зависимости пороговой температуры кристалли-
кристаллизации от величины растворимости вещества ядра не наблюдается.
В.Я.Никандров показал, что образование льда на поверхности рао-
творимой подложки имеет место при достижении температур, cootv
ветствующих образованию эвтектики, т.е. смеси ледяных кристал-
кристаллов и кристалликов подложки без нарушения структур.
По некоторым данным вокруг растворенных гидрофильных мо*
лекул наблюдается тенденция упрочнения молекулярной структуры
воды, причем образовавшийся вокруг молекулы "айсберг", может
иметь льдоподобную структуру и служить "молекулярным ядром за-
замерзания" .
Очевидной характеристикой, влияющей на льдообраэующую ак-
активность частицы, является ее размер. При сублимации льдообра-»
зующая активность частиц падает с уменьшением размера частиц
для г 4 ОД мкм, а при замерзании это явление наблюдается дл
частиц с г = 0,03 мкм. Но определяющими льдообраэующую акти|
ность частиц свойствами являются свойства их поверхности,в чей'
стности характеристики межфазной границы раздела,соответствую:
щей тп = COS 6,@- угол смачивания) при гетерогенной кондено*
ции. По Н.флетчеру для сублимации тп = (еп-б"л )/ влп , а для
замерзания щ=(е8-еЛ)/5Лв , где 6п,ел,
с а - удельные Я
верхностные энергии на границах раздела частица-пар, лед, вод!
соответственно, а СЛГ1, вла - на границах раздела лед-пар,л«1
жидкость. Для неактивных ядер m = -1,а для высокоэффективных
m = +1. Скорости льдообразования по Н.Флетчеру будут опреде-
определяться: для процесса непосредственной кристаллизации на сфери-
сферическом ядре формулой
E.27)
где
ТО26 см, с,
4яблпгз2/3'
254
фактор, зависящий от свойств ядра; а* = гя/г3, а г3 =
= 2Мвблп/[рлЙГAп-|^)]; Для двухстадийного процесса конденса-
конденсация-замерзание
<р*/(кТ)]ехр[-^(т,хI^Ф31(кТ)), E.28)
где К2 - ТО см . с , ДФ* - приблизительно равно энергии
активизации, необходимой для самодиффуэии молекул в жидкости;
г3 = BеЛВМв/рА )JtTo ((Lc - 1И)/Т) dT, где Lc - скрытая
теплота сублимации, 1И - скрытая теплота испарения, TQ= 273K.
Следовательно, при льдообразовании необходимо, чтобы свог
бодная энергия, связанная с границей раздела между частицей и
льдом, была по возможности малой.
Это условие определяется химической и кристаллографичес-
кристаллографической природой льдообразующего вещества. Так как решетка льда
скреплена водородными связями, желательно, чтобы на поверхнос-
поверхности зародыша имелись водородные связи с той же прочностью и по-
полярностью, что и между молекулами во льду, и чтобы молекулы, об-
образующие водородные связи, имели вращательную симметрию, пото-
потому что такие молекулы обязательно будут направлять свои Н-свя-
зывающие группы к поверхности (например, водородные связи орга-
органических веществ и гидроксильных групп каолинита и других сили-
силикатов ).
Кристаллографическое сходство льдообраэующего вещества
предполагает набор атомов в решетке, близкий по геометрическо-
геометрическому расположению молекул воды в какой-либо плоскости кристалла
льда с низким индекоом, близкие длины связей и углы связей,
обеспечивающие атомное соответствие на границе раздела подлож-
подложки и льда. Благодаря этому возможно эпитаксиальное или ориентн-
255
рованное нарастание льда на подложке. Если имеются различия.то
решетки льда либо подложки могут упруго деформироваться для со-
соответствующего сцепления с другой решеткой, что предполагает
относительно низкий модуль упругости второго рода для подложки.
Большое кристаллографическое различие будет приводить к возник-
возникновению на границе раздела фаз дислокаций, что будет повышать
удельную поверхностную энергию бл. В этом случае пограничная
зона между ледяным зародышем и поверхностью субстрата может
быть представлена состоящей из отдельных участков хорошего крио-
таллографического соответствия, ограниченных линейными дислока-
дислокациями.
Хотя кристалло1рафическая природа органических веществ и
льда различна, однако В.Гартеном и Р.Хэдом были выделены четы-
четыре типа кристаллических структур органических веществ с льдопо-
добными пространственными схемами водородных связей, способных
повысить температуру замерзания капель: I) кристаллическая струк-
структура, в которой водородные связи, находящиеся на одинаковой
глубине от поверхности, расположены так, что они с определен-
определенной периодичностью D:1, 1:1) повторяют расположение Я-свя-
Я-связей в кристаллах льда; 2) структуры, в которых схема Я -свя-
-связей гексагональна и совпадает с кристаллической структурой льда
на базальной поверхности; 3) небольшие кластеры Я-связей в
структуре, находящиеся на расстоянии, близком к расстоянию меж-
между атомами кислорода в кристалле льда;'4) произвольно располо-
расположенные Н-связи, концентрация которых оптимальна.
Вместе с тем на основании большого числа опытов Л.Лодж и
Ф.Парунго пришли к выводу, что льдообразующая активность орга-
органических веществ может быть предсказана без учета их кристалло-
кристаллографической природы, а только на основе их молекулярной струк- •
туры.
Как уже отмечалось,существуют разные способы инициирования
образования ледяной фазы.Если аэрозольная частица окружена вла-
влажным воздухом,то в этом случае происходит адсорбция молекул во-
воды из окружающей газовой фазы на активных центрах сухой поверх-,
ности частицы, образующая двумерные участки воды, которые при
более высоком давлении пара растут, превращаясь в многослойные
трехмерные структуры. Чем крупнее эти структуры, тем выше заро-
256
дышеобразующая способность частиц. Для превращения этих скоп-
скоплений в лед требуется среда, насыщенная относительно воды,если
температура среды выше критической. Причем для каждого вещест-
вещества характерна своя температура льдообразования, которая опре-
определяется химической и кристаллографической природой льдообразу-
ющих частиц, а также наличием активных центров на их поверхнос-
поверхности - гидрофильных центров на преимущественно гидрофобной по-
поверхности - и дефектов поверхности (дислокаций).
Когда аэрозольная частица окружена переохлажденной каплей,
после захвата ее этой каплей или после конденсации водяного па-
пара на ней молекулы воды обладают уже определенным структурным
расположением - скоплением молекул, которое тем отчетливей вы-
выпажено, чем ниже температура. Стабилизация скоплений молекул вс-
—Т Т ТО
ды ( t ~ 10 *10 с) может происходить также путем адсорб-
адсорбции молекул воды активными центрами поверхности частицы.
Г.Эдварде и Л.Ивенс на основе экспериментальных исследова-
исследований предположили,что образование льда в переохлажденной воде за-
зависит от характера развития участков адсорбированного мономоле-
мономолекулярного слоя воды на твердой поверхности частиц. Мономолеку-
Мономолекулярный слой будет относительно беспорядочным с большой свобо-
свободой перемещения молекул воды вследствие поверхностной диффузии
при температуре выше характеристической, если поверхность час-
частицы имеет умеренное сродство к воде и в основном гидрофобна.
Ниже этой температуры при условии кристаллографического подо-
подобия твердая поверхность способна постепенно преобразовывать бес-
беспорядочный мономолекулярный слой в кристаллическое льдоподоб-
ное состояние BМ-лед). Это льдоподобное состояние стабилизи-
стабилизируется преимущественно латеральными (боковыми) водородными свя-
связями и закрепляется на поверхности ее активными центрами. Даль-
Дальнейшая нуклеация льда происходит лишь с очень небольшим переох-
переохлаждением или без него. Если твердая поверхность имеет сильное
сродство к воде из-за наличия полярных групп, избыточного чис-
числа водородных связей или сильно гидратированных катионов, то ад-
адсорбированный слой может приобрести крепко спаянную структуру,
которая непригодна для образования 2М-льда. В этом случае
.олжен образоваться второй адсорбированный, достаточно неупоря-
неупорядоченный слой, чтобы произошел переход в 2М-лед.
П тг 257
Молекулярная модель образования льда на поверхности с ис-
использованием представлений о кластерах была разработана Р.Плам-
мером, Б.Хейлом и др. [140, 168]. Предполагалось, что молеку-
молекулы воды связываются водородными связями, кластеры имеют хорошо
выраженную структуру, внутренние свойства молекул влияют на об-
образование кластеров, время жизни кластеров достаточно велико,
чтобы можно было охарактеризовать их колебательный спектр,клас-
спектр,кластеры на поверхности и кластеры в газовой среде ведут себя, как
смесь невзаимодействующих между собой идеальных газов, структу-
структура кластера на льдоподобной поверхности не сильно изменяется
связью с поверхностью. Свободная энергия формирования кластера
с уменьшением температуры возрастает, а скорость нуклеацин
уменьшается, так как уменьшается поток молекул к поверхности
кластера. Эффективность льдообразования, вероятно, определяет-
определяется возможностями возникновения кластеров в первом адсорбирован-
адсорбированном слое благодаря определенному расположению дентров адсорбции,
Для образования кластера энергетически благоприятно условие
примерного равенства теплоты адсорбции отдельной изолированной
молекулы энергии отдельной водородной связи. Тогда вокруг пер-
первой адсорбированной молекулы образуется кластер из пяти молекул
имеющий льдоподобную структуру. Вместе с тем А.Цетльмейер пола-
полагает, что для эффективного льдообразования необходимо, чтобы
адсорбированная молекула воды в первом монослое не имела связи
с другими молекулами (площадь миграции ~1,2«10~14 см2). При ре-
регулярной адсорбции молекул на всей поверхности адсорбента рост
кластеров замедляется, так как молекулы воды прежде чем адсор-
адсорбироваться, должны быть ориентированы определенным образом к
адсорбированной пленке. Более выгодна неравномерная адсорбция
около специфических точек в виде островков полислоев. А.Цеттдь-
мейер считает, что активные центры поверхности распространяют
свое силовое поле в глубину адсорбированных слоев, причем наб-
наблюдается усиление взаимодействия в следующих за первым моносло-
монослоем слоях молекул. Из изотерм адсорбции воды на различных повер!
ностях км были получены низкие значения энергии адсорбции и вн«
сокие - энтропии, что свидетельствует о высокой подвижности мо-
молекул адсорбированной воды в первом слое. Форма изотерм адсорб-
адсорбции при температуре выше 6,5°С для AgJ показывает, что вна-
258
чале формируется жидкая фаза, а при некотором давлении наблюда-
наблюдается фазовый переход в кристаллическое состояние. По А.Цеттль-
мейеру для активного льдообразования необходимо, чтобы теплота
адсорбции была меньше теплоты конденсации; теплота смачивания
должна возрастать при увеличении заполненности монослоя. Опти-
Оптимальным условием ядрообразования является также наличие парных
соседствующих гвдроксилов.
Расчеты формирования кластера на поверхности показывают,
что если энергия взаимодействия кластер - поверхность меньше
2,0-Ю0 Дж, то структура однослойного кластера менее стабиль-
стабильна, чем многослойная структура.Следовательно, для гидрофобной
поверхности многослойные кластеры в силу большей устойчивости
будут преобладать, и их рост перпендикулярен поверхности. Моле-
Молекуле воды над базисной плоскостью льда энергетически выгодно
ориентироваться одним протоном к поверхностной молекуле так,
чтобы строилась правильная решетка гексагонального льда. Энер-
Энергия связи для такой конфигурации получается равной 5,5*10 Дж.
Если оба протона направлены к поверхности, то энергия связи
уменьшается до 4,7*10 Дж. Во льду отсутствуют изолированные
места сильной связи, выделенные областями с отсутствием связи.
Вся поверхность притягивает молекулы, которые находятся в соо-
тоянии физической адсорбции и могут продвигаться к местам бо-
более сильной связи, преодолевая энергетический барьер 1,11 х
х ТО дж. И, следовательно, основную роль в образовании клас-
кластера на поверхности или роста кристалла льда по Р.Пламмеру иг-
играет поверхностная диффузия.
Так как каждое ядро в капле имеет свою характеристическую
температуру замерзания, то температура замерзания капли опреде-
определяется частицей, обладающей наивысшей характеристической темпе-
температурой (сингулярная гипотеза). Учитывая, что концентрация
льдообразующих ядер л =поехр($йТ), и вероятность содержа-
содержания в капле объемом V по крайней мере одного эффективного яд-
ядра при 9=-AT определяется как 1-е~п(в)У, процесс замер-
замерзания можно описать соотношением
In,
E.29)
259
откуда видно, что число замерзших ядер не зависит от скорости
охлаждения и постоянно, если переохлаждение постоянно.
Ж.Дюфур и Р.Дефей, предполагая, что при данной температу-
температуре все ледяные зародыши, образовавшиеся в совокупности водяных
капель одинакового размера, имеют равную вероятность достичь
размера критического зародыша в результате случайных флуктуа-
флуктуации отдельных молекул в воде и что природа присутствующих пос-
посторонних частиц не играет существенной роли (статистическая ги-
гипотеза), получили выражение такого же вида [148].
Экспериментальные данные показывают необходимость предпо-
предположения наличия совокупности ядер с собственными характеристи-
характеристическими диапазонами температур замерзания, причем вероятность
льдообразования за счет каждого ядра является функцией темпера-
температуры переохлажденной воды. Это качественно согласуется с меха-
механизмом замерзания Г.Эдвардса и Л.Ивенса.
Инициирование льдообразования при контакте частицы с вод-
водной каплей более эффективно, чем в случае ее погружения в кап-
каплю. Н.Флетчер полагает, что это объясняется частичной раствори-
растворимостью вещества,причем вода избирательно вытравливает наиболее
активные центры на поверхности частицы.
Однако согласно Л.Ивенсу, более высокую эффективность кон-
контактного замерзания капель можно объяснить адсорбционной мо-
моделью, предположив, что частица, погруженная в переохлажденную
воду, может иметь прочно адсорбированный слой, который неприго-
непригоден для превращения в 2М-лед. В начальный период контакта,про-
контакта,продолжительность которого равна периоду вращения молекул воды,
может образоваться разупорядоченный адсорбированный слой, кото-
который легче трансформируется в 2М-лед.
Определенную роль в процессе льдообразования играет также
электрическая природа поверхности частицы и наличие примесей
на поверхности и в переохлажденной воде. Для льдообразования
энергетически благоприятна совокупность узлов атомной решетки,
содержащая приблизительно равные количества положительно и от-
отрицательно заряженных частиц, так как в этом случае диполи во-
воды будут иметь произвольную ориентацию. Экспериментально было
показано, что эффективность нуклеации наибольшая вблизи изоэле-
ктрической точки. Кроме того, на эффективность льдообразования
з.чияет различная поляризуемость ионов подложки, благодаря кото-
do;": на поверхности образуется двойной электрической слой, что
сказывается на характере взаимодействия молекул воды с подлож-
подложкой, а следовательно, на величине поверхностной энергии под-
ленки и условиях ее смачивания. Попадая в поверхностный слой,
отделившиеся от кристалла, ионы понижают энергию поверхностно-
поверхностного натяжения, чем способствуют замерзанию капли.
влияние примесей на эффективность льдообразования зависит
ст концентрации и типа примесей. Известно, что ионы вследствие
их размеров и сильного радиального электрического поля могут
разрушать структуру воды вне адсорбционного слоя путем разрыва
водородных связей и образования гидратных оболочек, препятствуя
этим образованию льда. Вместе с тем примеси в решетке подлож-
подложки могут улучшать кристаллографическое соответствие ее решетке
льда, одновременно повышая эффективность льдообразования. Неко-
Некоторые примеси способствуют возникновению активных центров, в
частности в результате реакций на поверхности субстрата. Напри-
Например, добавление 0,6$KNO3 к AgJ увеличивает интенсивность ад-
адсорбции воды в десятки раз. Активные центры образуются также при
загрязнении подложки молекулами поверхностно-активных веществ,
а также гидрофобной поверхности гидрофильными примесями. Однако
адсорбция поверхностных газов и паров, например таких, как SO2,
NH3> NO2, активными центрами на поверхности льдообразующих час-
частиц заметно уменьшает их эффективность. "Отравляют" активные
центры также растворенные в капле вещества, особенно различные
соли. 3 отсутствие химических или других взаимодействий при кон-
концентрациях растворимого вещества, меньших 10~3 моль/л, влияние
их на э&Ьективность льдообразования не наблюдается.
Увеличение льдообразующей эффективности некоторых веществ
при облучении их ультрафиолетовым светом или гамма-лучами свя-
связано о возникновением новых активных центров.
260
261
Глава 6
КОАЛИЦИОННЫЙ И КОВДЕНСАЦИОННО-КОАГУЛЩИОНШй
МЕХАНИЗМ РОСТА ЧАСТИЦ
Коагуляция аэрозольных частиц в атмосфере является важней-
важнейшим физическим механизмом формирования спектра размеров атмос-
атмосферных аэрозолей. Определение роли коагуляции в процессах тран-
трансформации аэрозолей затруднено тем, что практически всегда в
атмосфере действуют одновременно несколько механизмов роста час-
частиц и их удаления иэ атмосферы. Взаимодействие этих процессов
не аддитивное. Кроме того, и сам механизм коагуляции весьма
сложен. Различные физические процессы эффективно способствуют
или препятствуют коагуляции аэрозольных частиц разных размеров.
Основные из них следующие.
Случайные флуктуации плотности и средней скорости молекул
воздуха приводят мелкие частицы (г < I мкм) в нерегулярное
(броуновское) движение, в результате которого происходят взаим-
взаимные столкновения частиц и их (броуновская) коагуляция [180].
Броуновское движение частиц приводит их к броуновской диф-
диффузии и способствует осаждению частиц на более крупные объекты.
Этот процесс является основной причиной сухого вымывания мелко-
мелкодисперсной фракции аэрозолей. Коагуляция в результате броунов-
броуновской диффузии возможна на крупные частицы атмосферной пыли,об-
пыли,облачные частицы и частицы осадков, движущиеся под действием си-
лн тяжести. В случае падающих с относительно большой скоро-
скоростью частиц мелкие частицы коагулируют в результате одновремен-
одновременного действия броуновской диффузии и конвективного переноса
воздушным потоком, обтекающим крупные частицы. Это случай кон-
262
вективной броуновской диффузии. Безынерционная неброуновская
( г < 1 мкм) частица должна обойти крупную вместе с обтекающим
эе воздухом. Однако вследствие конечных размеров частиц нор-
нормальная составляющая скорости воздуха на расстоянии радиуса ма-
малой частицы от поверхности большой не будет равна нулю и воз-
возможно "зацепление" мелкой частицы за крупную. Так как сила тя-
тяжести, действующая на падающие частицы, направлена претив кон-
конвективного переноса мелких частиц воздухом, обтекающим крупную
частицу, то эффективность конвективной броуновской коагуляции
несколько уменьшается. Это влияние седиментации осаждающихся
частиц по величине одного порядка с влиянием эффекта зацепле-
зацепления. Поскольку гидродинамическое поле крупной частицы неодно-
неоднородно, на движение мелких частиц в этом поле всегда будет ока-
оказывать влияние их инерция.
Возможны два режима движения под влиянием инерции в неод-
неоднородном поле среды: докритический, когда поведение частиц ана-
аналогично поведению воздушных частиц, обтекающих поверхность
крупной частицы, и сверхкритический, когда влияние сил инерции
столь велико, что траектории мелких частиц пересекают поверх-
поверхность больших. При сверхкритическом режиме наблюдается еще один
механизм коагуляции - инерционное осавдение. Эти три процесса
действуют совместно при явлениях гравитационной коагуляции па-
падающих частиц сравнимых размеров. На характер протекания коагу-
ляционных процессов в этих условиях оказывает влияние эффект
гидродинамического взаимодействия частиц. Силы гидродинамичес-
гидродинамического взаимодействия при малых скоростях движения (малых числах
Рейнольдса) препятствуют сближению частиц, причем они неограни-
неограниченно возрастают при их сближении, что приводит к гидродинами-
гидродинамическому запрету коагуляции [21]. Коагуляцию аэрозольных частиц
в некоторой степени усиливает турбулентность среды. Если внут-
внутренний (колмогоровский) масштаб турбулентности меньше или срав-
сравним с размерами частиц, то наблюдается случай турбулентного
блуждания частиц, аналогичный броуновскому. Это приводит к не-
непосредственной турбулентной коагуляции частиц. Внутренний мас-
масштаб турбулентности в атмосфере примерно равен или больше 1 мм,
следовательно, непосредственная турбулентная коагуляция имеет
место для очень крупных частиц, например хлопьев снега. В ос-
263
тальных случаях турбулентность будет усиливать коагуляционнче
процессы интенсивного перемешивания частиц на расстояниях,боль-
расстояниях,больших внутреннего масштаба турбулентности. Следовательно, харак-
характеристики турбулентности среды будут входить во внешние гра-
граничные условия. Кроме того, турбулентность приводит к появле-
появлению дополнительной регулярной неоднородности гидродинамическо-
гидродинамического поля на расстояниях между частицами, меньших внутреннего
масштаба турбулентности.
Турбулентные пульсации могут способствовать перебросу час-
частицы из области дальнйдействующих сил взаимного отталкивания в
области, где действуют силы взаимного притяжения частиц. Приро-
Природа этих сил может быть различной. В среде, температура которой
неоднородна, при характерных размерах неоднородностей, меньших
или сравнимых с размерами частиц, появляется сила, пропорцио-
пропорциональная градиенту температуры и действующая в направлении ее
уменьшения. Это явление, называемое термофорезом, вызвано тем,
что импульсы, получаемые частицей от более теплых областей сре-
среды, не скомпенсированы импульсами от более холодных областей.
Явление осаждения частиц на более холодную поверхность - тер-
термопреципитация - может наблюдаться при попадании аэрозольных
частиц в облачные системы.
В среде с неоднородностями парциального давления, сравни-
сравнимыми с размерами частиц, возникает сила, пропорциональная гра-
градиенту парциального давления, действующая в направлении умень-
уменьшения парциального давления (диффузиофорез). Для броуновских
частиц аналогичные явления называются термодиффузией и бародиф-
фузией.
Электрические силы взаимодействия частиц ответственны за
электростатическую коагуляцию. Эти силы могут возникнуть как
под действием внешнего электрического поля, так и из-за элект-
электрического заряда самих частиц. Внешнее электрическое поле мо-
может создать наведенный дипольный момент, причем все ориентиро-
ориентированные диполи будут взаимно притягиваться. Силы притяжения бу-
будут действовать между частицами, разноименно заряженными, ней-
нейтральными проводящими частицами с зарядом любого знака. Индук-
Индукционные (зеркальные) силы всегда способствуют коагуляции.
264
Силы взаимодействия возникают также в случае испарения или
конденсации частицы в среде. Из-за испарения частица и окружаю-
окружающая среда должны охлаждаться, в результате чего возникают тер-
мофоретические силы притяжения. Концентрационное поле испаряю-
испаряющегося вещества (водяной пар) в силу диффузиофореза действует
на другие частицы от испаряющейся частицы. Наличие воздушной
среды приводит к возникновению градиента парциального давления
воздуха, приблизительно равного по величине градиенту парциаль-
парциального давления испаряющегося вещества. Кроме того, в среде воз-
возникает гидродинамическое течение смеси от испаряющейся части-
частицы - стефановское течение, которое уносит аэрозольные частицы
от испаряющейся частицы. (Все эти силы называют силами фаси.)
В зависимости от конкретных условий силы Фаси могут как препят-
препятствовать, так и способствовать коагуляции частиц. Для атмосфер-
атмосферных аэрозолей не все из перечисленных эффектов играют одинако-
одинаково важную роль. Для роста аэрозольных частиц в свободной атмос-
атмосфере из коагуляционных механизмов важны: броуновская коагуля-
коагуляция, турбулентное перемешивание, электростатическая коагуляция.
Все остальные процессы существенны при вымывании аэрозольных
частиц из атмосферы и для процессов в облачных системах.
§ 6.1. Уравнение коагуляции
Трансформация спектра аэрозольных частиц в атмосфере опи-
описывается уравнением коагуляции. Уравнение коагуляции в дискрет-
дискретной форме для коллоидных систем было выведено М.Смолуховским,
а для непрерывных переменных - Х.Мюллером, получившим интегро-
дифференциальное уравнение, вопросам обоснования и пределам
применимости которого посвящено большое количество работ, в
частности группы теоретиков из Института экспериментальной ме-
метеорологии [21].
В строгой постановке уравнение коагуляции выводится с ис-
юльзованием понятия N -частичной функции распределения. Фено-
Феноменологическое рассмотрение процесса коагуляции может быть сде-
сделано исходя из понятия одночастичной функции распределения при
введении ряда допущений и предположений:
265
1. Не нарушая общности вывода, можно предположить однород-
однородный случай распределения N частиц в достаточно большом объе-
объеме V, такой, что можно пренебречь влиянием стенок, т.е.
)
,z,r,
где (j>(x,y, z, r, i ) - плотность распределения вероятности
нахождения частиц в соответствующем интервале пространства (х,
у, г, x+dx, y + dy, z + dz) и размеров (г, r+dr), норми-
нормированная следующим образом:
\\\ \ ^^х'1/'г'г^^х^У^2^г ~ * >
a <p<x,y,z, r,t) - функция, характеризующая количество
частиц в единичном интервале размеров, приходящееся на единич-
единичный объем. Используя определение концентрации частиц nil) -
= N/Ls и условие однородности, получаем n<t)=V°y(r,i)dr.
2. Предполагается, что расстояние между частицами намного
больше их собственных размеров, т.е. V=V^ 4яг3/3 « L3
(или параметр упаковки V/l3 « 1 ). Это условие можно запи-
записать и так:
\ ^nrz(o(r,i)dr « 1.
Jo ° '
3. Характерное время мевду столкновениями т = N/(dN/di)
много больше времени взаимодействия частиц, т.е. I ез « т,
что фактически означает возможность одновременного столкнове-
столкновения только двух частиц. (Для случая гидродинамического взаимо-
взаимодействия это условие должно быть усилено.)
4. Предполагается, что каждый акт столкновения частиц соп-
сопровождается их слипанием (быстрая коагуляция).
5. Предполагается, что в объеме осуществляется хорошая пе-
ремешиваемость частиц.
Для простоты рассматривается однородный случай в достаточ-
достаточно большом объеме, так что можно пренебречь влиянием стенок:
<р<г. I) = <? <х,у, z,r, I) = Nif (х, у, z, r, t ), т.е. не
учитывать локальные изменения, вызванные каким-либо актом коа-
коагуляции при анализе следующего акта. Это предположение ведет к
возможности рассмотрения процесса как марковского и вследствие
266
этого позволяет не учитывать предысторию процесса при построе-
построении уравнения коагуляции.
Используя предположения, что столкновения парные и процесс
марковский, напишем уравнение баланса с помощью двухчастичной
функции to (r.,r.,t), характеризующей количество пар частиц с
размерами т. и г_ в единичном интервале размеров в единичном
элементе пространства. Изменение функции (f(r,t) за время &i
обусловлено образованием частиц размером г из более мелких и
исчезновением частиц размером г в результате коагуляции их с
другими частицами. На уровне физической строгости можно запи-
записать
Здесь функция W характеризует частоту столкновения частиц
двух размеров ( т^ иг или г и rt), а множитель 1/2 перед
первым членом обуоловлен тождественностью частиц одинакового
размера и необходимостью исключить повторение одинаковых ситу-
ситуаций при интегрировании по г иг. Функции i^ и иг предпо-
предполагаются симметричными. Так как в силу хорошего перемешивания
до попадания в зону взаимодействия частицы распределены незави-
независимо, <p(r,r2,i)= <p (r,i ) f(r2,t) и уравнение F.1)при-
F.1)примет вид
Вищ функции и/ зависит от физических процессов, определяющих
коагуляцию. Для малой вероятности столкновения функцию w мож-
можно определить через отношение рассматриваемого промежутка вре-
времени At к х- времени жизни частиц в единичном объеме. (Вре-
(Время жизни частицы г в единичном объеме есть отношение этого
единичного к очищаемому ими вследствие коагуляции объему час-
частиц г» в единицу времени.) Тогда
267
j
Заменяя A(f/At на ду/dl и обозначая частоту парных столк-
столкновений в единице объема \jx(ryr2) через ЛГ(г1(гг) (кон-
(константу коагуляции), получим уравнение коагуляции в виде
F.2)
Следует отметить, что функция вероятности столкновения для час-
частиц, распределенных в рассматриваемом объеме случайно, для ко-
конечного At более точно выражается как ( At/x )[ 1 - At/г ] *
* (<p(rj,?) <p (r2,i) rfr-j dr2-\). Полученное выражение фор-
формально легко обобщается на неоднородный случай и на случай не-
непрерывно растущих частиц „
' 2 ^^K(rvri^<x,y,z,r1,i)^<x,y,z,r2,f)S(r-r1-r2)dr1cir2 -
- y<x,y,z,r, i)§ K(r,rt)t?(x,y,z,rvi) drt,
где и - скорость перемещения частиц, г - скорость непрерыв-
непрерывного роста (испарения). Проинтегрировав первый член в правой
части по г^, получим уравнение коагуляции
d<?(x,y,z,r,i) d[rv<x,y,z,r,t)]
T + дх»[иухуггЦ + LJL^1
F.3)
= |f Л"fr-rj,rt)tp(ar,j/,z,r-rI(i)cp(ar,^/,г,rj,i;rfrj -
268
6.2. Броуновская коагуляция
Физическая модель броуновской коагуляции частиц разработа-
разработана и обоснована в работах А.Эйнштейна и М.Смолуховского, П.Лан-
жевена и С.Чандрасекара [21, 89, 104]. Представляя броуновс-
броуновское блуждание частиц как марковский процесс, можно показать,
что это блуждение хорошо описывается уравнением А.Н.Колмогоро-
А.Н.Колмогорова
dW = _ dAW
дх
F.4)
di дх дх2
где предполагается, что A(t,x) = Urn ( i/т) i (х^-х) W(t, x,
t + т, х^) dx = Um[( xt -x) I x] - средняя скорость слу-
случайного процесса; 2*f(i, x.) = Urn \(i/x)(x.-xJ]W(t, X,
i + x x,)dx = Итп\( x,~х)г /х| - характеристика случайных изме-
* t-»oL * i
нений х, а также 1гт[(д:1-д:K/-г]= 0 ; W(l0,x0; i,x) -
вероятность перехода системы из состояния х0 при I в состоя-
ние х при t . Более частный вид этого уравнения
di ~ дх +
F.5)
- уравнение Эйнштейна - Планка - Фоккера, или уравнение План-
Планка - Фоккера. Уравнения F.4) и F.5) легко обобщаются на слу-
случай нескольких обобщенных координат.
Нетрудно видеть, что уравнение Планка - Фоккера имеет
смысл уравнения неразрывности для плотности вероятности W (г,
I), так как оно эквивалентно уравнению dW/dl + diu(AW +
+ y^W) = 0, где JW+tfvW- плотность потока вероятности.
В частном случае броуновского блуждания, когда А=и и
f = D (коэффициент диффузии), уравнение Планка - Фоккера бу-
будет иметь вид
-~ - - diu-(uW)
at
DAW.
F.6)
пункция
V -«W l.r) . [teBrt-
269
Удовлетворяет уравнению Планка - Фоккера ( гг-, и j - составляю-
составляющие обобщенных координат и скоростей).
Если справедливы предположения, что: 1) движения частиц
за последовательные промежутки времени i независимы, 2) сред-
средняя энергия поступательного движения частиц вдоль каждой оси
равна 1/B кТ), 3) частицы движутся независимо друг от друга,
то для очень малых частиц справедливы выведенные А.Эйнштейном
уравнения
~х2 = 2Dt , D = кТВ , ^-^
где В - подвижность частицы.
Бесконечное иррегулярное блуждание не слишком малых час-
частиц в газообразной среде поддерживается не просто случайным ха-
характером отдельных столкновений молекул среды с аэрозольной час-
частицей, а молекулярными флуктуациями полного импульса, переда-
передаваемого частице средой в разные промежутки времени, так как
время взаимодействия одной молекулы газовой среды с не слишком
малой частицей тг ~ Zu существенно превышает время между
двумя последовательными столкновениями отдельных молекул с этой
частицей тс ~ \j( пгиаг) (I- 10см - характерный радиус взаимо-
взаимодействия молекулы среды с частицей; пГа 10 см , и - 5х
хЮ см/с - тепловая скорость молекул газа). Об отдельных стол-
столкновениях молекул с частицей имеет смысл говорить, если а 4
4- Ю~6 см. Поэтому для дисперсных частиц вводят понятия кажу-
кажущейся длины свободного пробега Я}- и времени релаксации час-
частицы х=Втп. Обобщение выражения А.Эйнштейна для квадрата сме-
смещения частицы будет иметь вид
j дг|2 = т^тгъ { C* - 1 + eccp (-$>t)\, F.8)
где C » i /(Вт), или для смещения по дт
~~хг = 2D[i -т { 1-еар(-?/т)|] .
Полный импульс, передаваемый молекулами газа частице, мо-
может быть записан в виде двух слагаемых: регулярного и флуктуа-
цаонного. Если взаимодействие частиц с окружающей средой сла-
слабое, т.е. ^а^^аРг» та ^ Рг/Ла (>na,Va , ла - масса,объем
270
и концентрация частиц; р - плотность среды), то флуктуацион-
ная часть импульса не зависит от регулярной скорости частицы и
распределена по гауссовому закону. В этом случае можно напи-
написать уравнение движения отдельной частицы в виде уравнения
Ланжевена
+ F*
F.9)
где f. и F* - регулярная и флуктуационная составляющие си-
силы, действующей на частицу со стороны среды.
Предположим, что т* - время корреляции различных моле-
молекулярных флуктуации значительно меньше тр - времени релакса-
релаксации регулярного поля частицы. Тогда, усредняя уравнение Лан-
жевена по временам порядка Тж, получим немарковское уравне-
уравнение
где dv - общее приращение скорости частицы, a dvm~ случай-
случайное приращение скорости частицы за время di, $т - коэффици-
коэффициент регулярного трения, отнесенный к массе частицы, интеграль-
интегральный член, обусловленный влиянием инерционности среды, приводит
к немарковости процесса (вязкое последействие). Усреднение по
т порядка C^ приводит к уравнению F.9), описывающему бро-
броуновское блуждание как марковский процесс.
Выражения для ^ и В в общем случае зависят от чиола Кнуд-
сена, т.е. от отношения длины свободного пробега молекул газа
к радиусу частицы. В результате броуновского блуждания незави-
независимых частиц всегда существует вероятность их столкновения и
затем коагуляции. Обычно задача броуновской коагуляции оводит-
ся к задаче броуновской диффузии частиц к поглощащей сфере.
Кроме функции W(r,i) вводится функция W*<r,t), соответ-
соответствующая вероятности достижения частицей некоторой границы за
время t, удовлетворяющая уравнению
М* F.10)
при и = 0 и условиям W*(r,0)= 0 для точек, не лежащих на
границе, W* (г,0 ) =0 для точек, лежащих на границе.
271
Для монодисперсных частиц с радиусами а соприкосновение
произойдет в момент, когда расстояние между центрами этих час-
частиц будет равно 2а, поэтому задача броуновской диффузии час-
частицы с радиусом г к поглощающей сфере с таким же радиусом эк-
эквивалентна задаче расчета вероятности попадания частицы WBr,
i ) на границу сферы с радиусом 2а; W*(r,l) по Н.А.Фуксу вы-
выражается следующим образом:
W\r,«>= -^-
Если концентрация частиц пA), то количество частиц, попавших
на сферу 2а за время @,i), будет
Ш) = SnaDn(t)( t
F.11)
Величину 83iaD*K0 называют константой коагуляции. Процесс
можно считать стационарным при t » 4az/(nD) . Отказываясь от
неподвижности поглощающей сферы с учетом независимости смеще-
смещений частиц относительно друг друга, получаем, что ?>,_ = 2?, + 7?_,
и, следовательно, ежесекундно с каждой частицей сталкивается
2К n(i) частиц, а общее число столкновений в секунду K0n(tJ=
= - dn(t) Idl. Решение задачи о вероятности достижения части-
частицей сферы в более общем случае и ф 0 получается из уравнения
F.10) при соответствующих начальных и граничных условиях. Для
диффузии частиц с концентрацией n(i) это уравнение будет иметь
вид
дп<1)
di
+ div ыпA) - DAn(i),
или с учетом внешних сил
+ diu (u +
= D&n(i)
F.12)
F.13)
Последнее уравнение обычно называют уравнением Смолуховского,
или уравнением конвективной диффузии в безынерционном прибли-
приближении, описывающем диффузионное расплывание неоднородностей
концентрации частиц с неограниченной скоростью распространения
возмущения. Применение уравнений типа F.12) к броуновскому дви-
272
яению частиц справедливо лишь для интервалов времени I, боль-
больших времени релаксации частиц ? > 1/р, или для расстояний,
больших, чем кажущаяся длина свободного пробега А:-. Следова-
Следовательно, для случая, когда размер поглощающей сферы сравним с
/Ц , необходимо вводить поправку.
Эта поправка приближенно находится по методу граничной
сферы: поглощающая сфера окружается концентрической сферой ра-
радиусом . 2а +S, внутри которой поток частиц определяется с по-
иощью газокинетической теории и приравнивается диффузионному
потоку на границе внешней сферы [89]. Величина 8 определяет-
определяется как среднее расстояние, на котором окажутся отлетающие от
поверхности поглощающей сферы частицы, пролетевшие расстояние
Уточненное выражение # дается формулой
f
Ф(х)= 1?[^
где
J этом случае предполагается, что частица проходит путь 8 с
некоторой вероятностью рассеяния dx/Я^. Учет зависимости
свободного пробега от скорости частицы дает поправку не бо-
более 1/J.
В работе [21] обращается внимание на важность учета гид-
гидродинамического взаимодействия частиц на близких расстояниях
1^, что должно уменьшать константу броуновской коагуляции.
Сднако примерно на этих же расстояниях существенны и силы ко-
короткого, но сального их взаимодействия. Это может полностью
компенсировать влияние сил гидродинамического взаимодействия
частиц.
В общем случае броуновской коагуляции не каждое соприкос-
соприкосновение частиц приводит к их слипанию или слиянию. Следователь-
Следовательно, в окрестности поверхности сферы концентрация частиц пл
не обязательно равна нулю. Вдали от сферы концентрация частиц
;*вка n(i) = no0 . В гидродинамическом приближении условия
'8. I92Z
У73
для концентрации частиц должны задаваться на некотором расстоя-
расстоянии от сферы; 82 > у$~^В. Если 52 « 1% , то используются
экстраполяцивнные граничные условия кэдп/д8=п-пл при о" =
= 0 или г ' а^ + аг , где Л:э подбирается таким образом, что-
чтобы правильно определялся поток на поверхность 6" = 0, причем
кэ ~Vd/&- пРи Учете сил короткого взаимодействия гранич-
граничное условие имеет вид кэ дп/dS ж л-ла или 6"= If или
г = ctj + if + аг .
Решение задачи броуновской диффузии определяется величи-
величиной критерия подобия Ре « ??,12/^ (число Пекле) и отноше-
отношением числа Ре к числу Re (число Рейнольдса), называемому
числом Прандтля: Рг = Ре/Ре = C/D.
Константы броуновской коагуляции для частиц разных разме-
размеров приведены в монографии Н.А.Фукса [104]. Существенное уско-
ускорение коагуляции атмосферных аэрозольных частиц может наблюдать-
наблюдаться в результате сил взаимодействия частиц и при наличии внеш-
внешних сил.
Исследование броуновской коагуляции методом Монте-Карло
было осуществлено автором совместно с М.Н.Иошпе. При решении
задачи использовалась модель пространства с периодической струк-
структурой из элементарных кубических ячеек. В центре последних на-
находятся поглощающие сферы, к которым диффундируют случайным об-
образом распределенные в ячейке (в соответствии с законом равно-
равномерного распределения) частиц. В качестве проекции пути, про-
проходимого за i, берется величина
где п - случайная величина с нормальной плотностью распреде-
распределения /(п ) «= A/у/2атГ) е? '2 , что дает_значение среднего
квадрата смещения частицы вдоль оси ас: хг «• 2D \l - т [ I ~
- еаер<-i/t )U; т = 5т - время релаксации. При t»x
выражение для х? переходит в F.7), а при t «г, разлагая
экспоненту в ряд, получаем ~oc$ =31р2?2,где 1ог - средний ква-
квадрат скорости движения частицы.
Предполагается, что во всех ячейках диффузия протекает
одинаково, т.е. если частица покинула элементарную ячейку с
ребром а, то другая частица влетела в нее. Размер элементар-
274
ной ячейки должен значительно превосходить радиус поглощающей
сферы, а алгоритм броуновского блуждания частиц по элементар-
элементарной ячейке должен быть таким, чтобы не пропустить первого со-
соприкосновения частицы с поглощающей сферой. Как показали машин-
машинные эксперименты, объем элементарной ячейки должен быть на
лесть порядков больше объема поглощающей сферы, а путь между
двумя фиксациями: 1~\*Тг/10. Длина кажущегося свободного про-
пробега частиц для мелкодисперсной фракции оказывается больше
VJr/10, поэтому необходимо для определения момента соприкос-
соприкосновения частиц решать систему уравнений
X-
х2
¦*1
X2
У_
Уг
Z'
III
-И
г ж
где xi,y1,z. и х2'Уг'г2 ~ начальные и конечные координа-
координаты частицы за один шаг.
Пересечение траектории частицы с поверхностью сферы долж-
должно сыть действительным (рис.26). Для уменьшения числа шагов в
описании броуновского блуждания частиц элементарная ячейка бы-
была разделена на зоны по расстоянию от поглощающей сферы так,
что частицы, находящиеся в дальней от поглощающей сферы зоне,
могут проходить в среднем бо'льшие расстояния, чем частицы в
ближней зоне. В случае неудачного выбора средних путей частицы
[ d11 в зонах элементарной ячейки константа .коагуляции для
данной концентрации будет заниженной. Изменяя значения {<?Д и
проведя расчет константы коагуляции,можно наблюдать максимум К
в случае, когда \d { близки к оптимальным. После этого можно
оценивать влияние границ элементарной ячейки на константу коа-
коагуляции. На рис.27 для примера приведены результаты расчета
зависимости константы коагуляции от размеров элементарной ячей-
ячейки (а-п~У3) для частицы с г = 5-20"^ см. (Для участка АС
влияние границ ячейки можно не учитывать.)
Сравнение результатов расчета констант броуновской коагу-
коагуляции для частиц разных размеров с результатами расчетов, вы-
выполненных методом граничной сферы, а также на основе решения
сферической задачи Милна показывает хорошее согласие значений
275
К-Ю*cm3-с
8,5
6,5
2.5
0,0? 0,05 0,1
Рис.26. Модель броуновского блуж-
блуждания частиц в элементарной ячейке.
а - истинное (Л и фиктивное
B) столкновения частиц с погло-
поглощающей сферой; 5 - разделение
элементарной ячейки на зоны.
0,2
а,с/л
Рис.27. Влияние гра-
границ элементарной ячейки
на значение константы
коагуляции для частицы
с г = 5-Ю см.
констант для всего диапазона размеров частиц. Различие не пре-
превосходит 4$>. Метод Монте-Карло позволяет проводить расчет кон-
констант коагуляции и для значительно более сложных дисперсных
систем, когда не существует надежных способов расчета этих ве-
величин.
§ 6.3. Электростатическая коагуляция
Наибольшее значение для коагуляции твердых аэрозольных час-
тиц представляют электрические силы их взаимодействия.
Интегрирование уравнения F.12) с граничными условиями
п(г, 0)=л0 при г-»оо; л (г,0 )=0 при г = 2а приводит к сле-
следующему выражению для константы коагуляции монодисперсных час-
частиц:
К
Г
F.13)
276
Рис.28. Схема электростатического взаимодейст-
взаимодействия двух сфер.
где ср(г)= \ F(r)dr - потенциал силы F(r).
Отношение константы коагуляции с учетом сил взаимодействш
частиц К и без их учета Кп будет равно
или при замене х = 2а/г
для частиц размеров
K<ai,ctt) ,
и а2 имеем
W
При замене x*(al+a2)/r выражение для J имеет вид
еэсР t *P <^«i + aaV
Когда частицы притягиваются друг к другу (ш < 0 ), име-
имеем ¦J > 1 и, следовательно, увеличение константы коагуляции,
при оттаскивании частиц друг от друга (ср > 0 > наблюдается ее
уменьшение.
Сила взаимодействия двух заряженных сферических частиц с
радиусами cfj и а^, являющимися проводниками на расстоянии г
277
(рис.28), в отсутствие электростатического поля определяется
следующим выражением:
З(г2-а?)* + FЛ4)
-efJ -1/(г2-ар2 -1Дг2-а22J|.
В этом выражении первый член соответствует кулоновской си-
силе взаимодействия, а выражение в фигурных скобках - индукцион-
индукционной силе взаимодействия наведенных зарядов первого зеркального
отображения с зарядом о =eZ., с зарядом о и друг с дру-
другом. Выражение F.14) можно представить в виде ряда
~7S K }
В частности, для частиц с равным радиусом
лучаем
по-
по-(a3[r5)«i2
3a2jr2 + 4a4/r
5a6/r6
Отметим, что в работе [104] приведена формула силы взаимо-
взаимодействия двух проводящих частиц, отличающаяся от вышеприведен-
вышеприведенной численным коэффициентом в первом ряде (вместо 6 стоит коэф-
коэффициент 15).
Выражение для потенциала электростатической силы Ф<Т) =
= \ F (Г)<?г при замене x = (ai+a.)Jr получим в следую-
следующем виде:
или после подстановки выражения для электростатической силы
278
Юa\a\Ba*
4] 2**1 [ *4/2
2«*>*"/
г
a4*8/
...]J. F.15)
¦ / МГ
-5,2
f I
i
600 700
Is
Рис.29. Зависимости сил электростатического взаимодейст-
взаимодействия сфер разных размеров и зарядов.
/- Zj.f, 22=1; 2-Zj-I, Z2«10; 5 - Z,-I , Z^
-100; 4 - Zj-Ю, Z2-l ; 5-Z^lO, Z,-10. a-
= I0 см , a2 « 10см .
279
частиц одинакового размера получим
в )[х+3x4/224- + Х9/Ж
Ограничиваясь первым приближением, имеем
Таким образом, индукционные силы всегда приводят к появле-
появлению добавочного притяжения между заряженными частицами вне за-
зависимости от того, одноименный или равноименный у них заряд.
На рис.29 приведены расчеты функций (Mai+a2)/x) частиц
с различными харядами и размерами. Очевидно, что чем больше от-
отношение между значениями одноименных зарядов двух частиц, тем
меньше потенциальный барьер, препятствующий их коагуляции, и
тем дальше от частицы он находится.
Расчеты показывают, что константа коагуляции для одноимен-
одноименно заряженных частиц при определенных соотношениях значений за-
зарядов и размеров частиц может даже превосходить единицу (табл.3 2 \
Проведем оценку влияния внешнего электрического поля на
взаимодействие двух аэрозольных частиц с радиусами а^ и а2.
Предположим, что сфера с радиусом аг- проводящая и значитель-
значительно больше сферы аг (a2»ai). Напряженность в точке А (см.
оис.28), расположенной на расстоянии г от центра сферы О, ес-
если угол между О А и направлением поля б, равна
где к _ напряженность внешнего однородного поля. Если части-
частица с радиусом а. находится в точке А , то она имеет в поле сфе-
сферы дипольный момент М = 4леоа*Е, а сила притяжения ее к
сфере F=MgradrE, откуда
\ 3 cos 0 .
( 2а\ г'3 + i) E
F = - 24яеоа?а*
В момент, предшествующий соприкосновению частицы со сферой, F =
= -72же. a3a"x?2cos0 ^з приведенных выражений следует,
0 12 0
280
Отношение К/Ко одноименно заряженных
частиц тех же размеров ¦
Таблица 32
и незаряженных
а
1
1
10
100
f
zl
2
1
5
10
1
5
10
1
5
10
ZZ
3
I
5
10
5
10
10
1
5
TO
1
5
10
1
5
10
1
5
10
i
5
10
1
5
10
1
1СГ7
4
0,2И-10~И
-
-
_
-
-
0,0820
0,397-Ю"9
-
0,858-Ю
-
-
0,119-Ю
-
-
0.8У1
0,199
0,0189
0,716
0,841-10"^
0Д39-10
0,511
0,345-Ю
0Д11-Ю3
0,505-Ю0
O-Z, СМ
1G
5
7=243 К
0,139
0,481-10
0,650-Ю
_
-
-
0,827
0,189
0,0188
0,657
0,771-ТО
0Д27-10~7
0,469
0,316-Ю
0Д02-103
1,005
0,878
0,734
0,181
0,651
0,269
1,363
0,476
0,0888
Г=273К
0,177
!G~b
6
0,847
0,455
0,365
0,329-10~2
0,562-Ю
0,211-ТО1
0,982
0,865
0,732
1,086
0,598
0,248
1,252
0,437
0,0815
1,001
0,988
0,971
1,064
0,997
0,918
1,172
1,041
0,891
0,863
1СГ4
7
0,983
0,929
0,886
0,650
0,412
0,139
0,998
0,986
0,970
1,017
0,956
0,883
1,085
0,965
0,827
1,000
0,999
0,997
1,010
1,003
0,994
1,036
1,022
1,005
0,986
281
Продолжение табл.32
I
2
1
10
100
5
10
1
5
10
1
5
10
1
5
3
5
10
ь
10
10
1
5
10
1
5
10
1
5
10
1
5
10
1
5
10
I
5
10
1
5
10
5
10
4
_
-
_
-
-
0,112
0,499-Ю"8
-
0,276-Ю
-
-
0,828-10"^
-
-
0,909
0,244
0,0311
0,777
0,203-10~2
0,118-Ю
0,594
0,159-Ю
0,457-Ю2
0,29 -10"9
-
-
-
-
5
0,157-Ю
0,450-10"^
Г=273К
-
_
-
0,846
0,232
0,0310
0,714
0,186-ТО
0,108-10"^
0,545
0,146-Ю
0,419-10~12
1,005
0,891
0,761
1,176
0,692
0,318
1,362
0,533
0,122
Г=293К
0,203
0,301-Ю
0,135-Ю
-
-
6
0,499
0,310
0,676 «КГ*
0,244-Ю-4
0,505-Ю0
0,984
0,879
0,759
1,083
0,637
0,293
1,251
0,490
0,112
1,001
0,989
0,974
1,059
0,9997
0,929
1,161
1,044
0,909
0,872
0,525
0,338
0,0100
0,548-Ю
7
0,937
0,898
0,683
0,458
0,177
0,998
0,987
0,974
1,015
0,961
0,896
1,078
0,970
0,846
1,000
0,999
0,997
1,009
1,003
0,995
1,033
1,020
1,005
0,987
0,941
0,904
0,702
0,485
282
1
2
10
1
3
10
I
5
10
4
-
0,133
о.гот-ю
-
0
0
0
5
-
,856
,259
,407
Продолжение
6
0.291-10-9
0,985
0,887
0,774
табл.32
7
о,2Оа
0,999
0,988
0,975
10
100
5
10
1
5
10
1
5
10
1
5
10
1
5
10
1
5
10
1
5
10
0,525-10"
,-5
0,241-10
0,919
0,272
0,0408
0,813
0,329-10
0,383-Ю
0,664 а
0,369-10 "
,-2
0,356-Ю1
,-И
Г=293К
0,746
0,302-10
0,350-10"
0,591
0,339-10
0,327-10'
1,005
0,898
0,776
1,176
0,716
0,349
1,361
0,567
0,145
,-2
Г4
1,081
0,659
0,321
1,249
0,521
0,133
1,001
0,990
0,976
1,056
1,001
0,934
1,155
1,045
0,919
1,014
0,964
0,902
1,073
0,973
0,856
1,000
0,999
0,998
1,008
1,002
0,995
1,031
1,019
1,005
что сила притяжения частицы пропорциональна квадрату напряжен-
напряженности внешнего электрического поля и проявляется на сравнитель-
сравнительно близких к сфере расстояниях ( ~ г). Если сфера аг явля-
является диэлектриком, то следует пользоваться выражением для Е,
учитывающим диэлектрическую проницаемость вещества сферы ег
8 .
В случае, если обе сферические частицы проводящие, имеют
произвольные размеры (аг, аг) и находятся в электрическом по-
поле Ео, то на них возбуждаются электрические диполи с моментом
283
М = Е0<хг. Сила взаимодействуя двух таких диполей с радиусами
а1 и аг без учета индукционных сил, вызванных взаимной поля-
поляризацией частиц, определяется выражением
потенциал электростатической силы взаимодействия
с[ЧГ) = - E*aer~3{2coszB - sin20).
В этом случае
Значение ^ становится больше единицы при увеличении отно-
отношения радиусов частиц, а также при высоких напряженностях элек-
электрического поля, например при Е — 10 В/си, что соответству-
соответствует условиям грозовых облаков, $ » 1.
Потенциал сил взаимодействия при учете вторичных индукци-
индукционных сил, вызванных взаимной поляризацией частиц (с радиуса-
радиусами а), представляется в виде сложного тригонометрического ряда.
r~6
то в выражение для
Если учесть только члены ряда , р
вводится дополнительный член: SE^a^ г , где множитель о"
выбирается так, чтобы при 0 = 0 потенциалы ш(г,а), вычис-
вычисленные по точной формуле и по формуле с дополнительным членом,
совпали.
Знак потенциала сшг; определяется знаком 2 cos2 0 -
~sm20.TaK, если 1<?2 в <2, то ш<г)<0, а если ^?20>2,
то <1)(г)>0. Область около поглощающей сферы оказывается раз-
разделенной на зоны притяжения и отталкивания частиц. Зоны притя-
притяжения около полюсов сферы соответствуют углам 9, равным 0 и
ЗС, Этим объясняется возникновение в атмосфере длинных цепочек
сфер.
В задаче взаимодействия двух заряженных проводящих сфер с
радиусами а^ и а. и зарядами а и а , находящихся на рас-
расстоянии г друг от друга в однородном электрическом поле ? ,
направление которого составляет угол 6 с прямой, соединяющей
центры сфер, для силы, действующей на сферу радиусом а в нап-
направлении оси, соединяющем центры, получено выражение
2t4
В этом выражении ?0 - диэлектрическая проницаемость воздуха,
a F, F , F. , F., FK , F. , F~ - табулируемые коэффициенты.
Выражение для напряженности поля в точке А сферы радиусом а^
для (см. рис.28) случая, когда поле параллельно оси, соединяю-
соединяющей центры сфер:
где fj, Е2>Еъ " коэффициенты, значения которых для разных
сфер могут быть рассчитаны на ЭВМ. Общие закономерности для си-
силы взаимодействия частиц и величины потенциала (±>(г) те же,что
и в описанных частных случаях.
Представляет интерес рассмотреть возможность накопления
заряда аэрозольной частицей. Предположим, что твердая проводя-
проводящая частица сферической формы радиусом а^ находится в непод-
неподвижном воздухе и внешние поля отсутствуют. Частица заряжается
в результате оседания на ней монодисперсных сферических частиц
радиусом а у движущихся в направлении на первую частицу с пос-
постоянной скоростью V. Условие оседания частиц выражается нера-
неравенством mv2j2 > (<?2/Dя?0))( cf,il(ai + a2)), из которого
видно, что в отсутствие внешнего электрического поля предель-
предельный заряд частицы имеет квадратичную зависимость от относитель-
относительной скорости движения частиц. Так, если v = 0,25 см/с, то
для af = 6«10~4 см, а, = З'Ю см и q7= l,?-10~15 Кл пре-
предельный заряд <11^^2' а для СКОРОСТИ V=0,b см/с уже
О.. 4 5а.. без учета силы зеркального отображения, которая не-
несколько повысит эту величину.
Наличие внешнего электрического поля существенно усложня-
усложняет проведение этих оценок. Если внешнее поле однородно, то час-
частицы движутся только под действием внешнего поля, и сопротивле-
сопротивление воздуха определяется формулой Стокса. Поместив начало коор-
координат в центр частицы радиусом <2j, за текущую координату при-
примем г = $ а,, где а - безразмерный радиус-вектор, причем
направление движения частиц радиусом а2 противоположно нап-
направлению a .
Уравнение движения частицы радиусом а> и массой т запи-
285
сывается в виде
Так как dvjdl = (dv/dr)(drfdi) =vdv/dr= (v/ajdv/da ,
то (mvjai)dvlda=YiiTi, где J^.T- - сумма сил, действующих
на частицу. Эта сумма состоит из электрических и аэродинамичес-
аэродинамических сил (сила тяжести не учитывается). Здесь Fj - сила куло-
новского взаимодействия двух заряженных сфер с учетом сил зер-
зеркального отображения
где а, = - <^г <fl; @ = o^a'1; F2- сила взаимодействия заря-
заряда а2 с диполем частицы радиусом а., дипольный момент кото-
которой M1=-aI3E0, F2=-2<^2ЕОа; Е„-напряженность внешне-
внешнего электрического поля в предположении, что Е параллельно оси
a ; Fj - сила взаимодействия заряда <?, с диполем частицы,
имеющей радиус а2 и дипольный момент ^2 = ~а2^о* ^3 =
= -2аЁ.а3а1 aj3 ; F4 - сила взаимодействия диполей рассматри-
рассматриваемых частщ; F4 = -6 а\ Е» а~гa- F5=-6«^a.v - сила со-
сопротивления воздуха, где п - вязкость воздуха; F6 - сила вне-
внешнего электрического поля, действующего на свободную частицу
радиусом аг, F6 = ^20
Начальное (предельное) условие имеет вид
аюо
При исследовании решений этого уравнения необходимо найти та-
такое значение ц,^а, при котором V(a0) = 0 и v'(ao)= 0. Эти
условия необходимы и достаточны для определения значения пре-
предельного заряда о (г).
Решение уравнения с указанными условиями можно получить
численными методами. Начальные условия могут быть заменены ус-
условием v , = V. ¦ Уравнение решается с помощью метода
Рунге-Кутта.
1/\з приведенных уравнений видно, что возможность коагуля-
коагуляции частщ существенно возрастает, если движение последних от-
286
носительно друг друга таково, что сближает их расстояние, при
котором индукционные силы взаимодействия становятся достаточ-
достаточными для взаимного захвата. Одна из возможностей создания та-
такого движения - турбулентные пульсации воздуха, которые могут
перебрасывать одноименно заряженные частицы через потенциаль-
потенциальный барьер сил отталкивания. Следует отметить, что в силу отно-
относительно больших масштабов турбулентных пульсаций вероятность
выбрасывания частицы из потенциальной ямы сильного взаимодейст-
взаимодействия двух частиц, очевидно, исключительно мала.
Другая возможность коагуляции частиц заключается в разли-
различии скоростей падения частиц разных размеров. Но в этом случае
должна существовать очень большая разница в радиусах частщ
(дождевая капля и аэрозольная частица в подоблачном слое).
Таким образом, электростатические заряды на аэрозольных
частицах практически всегда способствуют увеличению скорости
коагуляции. Причем существует достаточно много механизмов на-
накопления электростатического заряда на частицах, в том числе пу-
путем коагуляции одноименно заряженных аэрозольных частщ.
Диффузионный механизм зарядки и .коагуляции заряженных час-
частиц обсуждался в работах [14, 21, 104], в частности экраниров-
экранировка дальнодействующих сил электростатического взаимодействия по-
подвижными ионами на расстояниях дебаевского радиуса (г„ = (Ц?*
*rijT)/2 где ц, - заряд иона; л. - концентрация ионов, Т -
температура в энергетических единицах ) и возможность использо-
использования уравнения стационарной диффузии ионов для описания про-
процессов диффузионной зарядки частиц. Условие пршиенимости ста-
стационарного уравнения: i^iZ*~пЯъ « 1 или fjf'^nr^R-M
для #= 10 см,где \ - время подстройки ионной концентрация,
а t, - время изменения величины заряда. Для стационарности
процесса необходимо, чтобы *2*V B o6lJ4HHX условиях в ат-
атмосфере как экранировкой дальнодействующих сил подвижными ио-
ионами, так и нестационарностьв диффузионной зарядки частщ мож-
можно заведомо пренебречь. Кногочастичные процессы дают вклад толь-
только в не зависящий от времени член, который может быть опущен
при 1~1г»( fB
М = (ig3 у1 +(
где
g3
верхние индексы при
g3 ? j
означают: к - коагуляцию, д - дебаевское взаимодействие.
287
t о.4. Решения уравнения коагуляции
Зопрос о существовании автомодельных решений для броунов-
броуновской коагуляции бил рассмотрен О.Н.Тодесом. »1м было получено
уравнение, которому удовлетворяло решение, зависящее от пере-
переменной, составленной из комбинации объема частиц и времени ко-
коагуляции. О.Н.Тодес постулировал существование автомодельного
решения из предположения, что система "забывает" начальные ус-
условия после нескольких актов коагуляции.
С.Фридлендером постулируется гипотеза о самосохранении рас-
распределения при достаточно больших временах жизни коагулирующих
аэрозольных частиц. Такое распределение принимает вид
(Г, i ) =
цн?)>
F.16)
где г)(г) - функция только размера, <D(i) - функция только вре-
времени [159]. Справедливость этой гипотезы была продемонстрирова-
продемонстрирована для броуновской коагуляции в стоксовом и в свободно-молеку-
свободно-молекулярном режимах.
А.А.Душников постулировал гипотезу о подобии распределе-
распределений в процессе коагуляции первоначально монодисперсных аэро-
аэрозольных частиц с одинаковой удельной массой. Аналитически бы-
было показано, что самосохранение должно достигаться, когда за-
зависимость коэффициентов коагуляции от размера частиц однород-
однородна, т.е. K(AV, Ли)=Я"К(У,и),гле V и U - объемы коагули-
коагулирующих частиц, и показатель однородности достаг чно мал. Само-
Самосохранение строго выполняется при А = 0, а при ЛфО лишь
приблизительно.
В более общей постановке вопрос о существовании решений
уравнения коагуляции формулируется следующим образом: какие
характеристики распределения частиц по размерам перестают за-
зависеть от начальных данных? Для уравнения коагуляции
= | СK(V-U%U)y(U,l)<?(V-U,i)dU - 4{{V,l)^ K(V,U)cp(U,t)dU,
если K(V,U) неоднородно, возможно только преобразование ти-
типа умножения
288
и <р
на постоянное число, т.е. изменение
масштаба времени коагуляции. Это позволяет решать уравнение ко-
коагуляции для нормированных распределений. Если K(V, U) _ од-
однородная функция порядка и, то допускаются преобразования не
только t, но и V: V = Ах, 1 = ^х. Тогда уравнение коагуля-
коагуляции примет вид
F.17)
дх
где & и «р - параметры преобразования функции распределения;
ядра выбираются из соображений нормировки; ^ и Л - свободные
параметры. Если наложить на параметры условие oi,^-5AI+M'= i ,
то уравнение F.17) формально сведется к первоначальному веду.
Однако при таком преобразовании могут измениться начальные ус-
условия (вид начальной функции if>(V, 0)). Для 6"-функции сох-
сохраняется полное подобие уравнения коагуляции, тогда как для ос-
остальных классов начальных функций подобие не имеет места и уни-
универсальность решения может следовать только из асимптотических
свойств решения уравнения. Это показывается с помощью теории
размерностей.
Если основными единицами измерения являются длина L, вре-
время Г, число частиц л,то размерность остальных величин сле-
следующая:
-T; [Ъ] =
у, - параметр однородности ядра, Ъг{(Х,и) = $(х,у), гЦХ,у)-
комбинация только ос и у для однородного случая размерности
Ь ' ' Mi - моменты функции начального распределения. Из че-
четырех первых величин можно образовать три безразмерных комби-
комбинации: tp =( VJN*^ ; l = (N0IV0)x ; r^ibNj-PIVj-W,
а остальные параметры представляются через комбинацию двух, взя-
взятых в качестве основных: Х; *У~^ (N IV )<i+i)M- 2 ; 1=0,
г о
X , С.
Решение уравнения записывается через безразмерную функцию
в виде
19. 1921
289
или
, ае),
если число моментов, определяющих начальное распределение ко-
конечно, так как моменты высших порядков определяются через мо-
моменты более низких порядков. Для § функции с|>=шE,т), тог-
тогда как для остальных начальных функций необходимо проанализи-
проанализировать влияние ае. Если начальное распределение не меняет без-
безразмерных параметров, т.е. Ф{х,т,эе)= оь^(Хл,^х, ае ), то,
выбирая ¦Jet , * = 1, получим
где ot(x) - функция времени, а ф- функция ае и |. Для слу-
случая j3 = fio= const и экспоненциального начального распределе-
распределения решение не зависит от моментов распределения больше перво-
первого. Асимптота решения <р(ге,т) = (\-хг)е~^~х^х , откуда
видно, что |«A-т)а», а универсальная функция Ф(|)= е~^ .
Уравнение для ф(|) будет иметь вид
В случае А , имеющей однородность порядка и., решение пред-
представляют в виде Ф(х, т) = <у.(т)Ф<|). Для возрастающего
ядра &*х+у асимптота распределения
у(х,т) = A-х)Bлего)А12х~1/2 ежр [ -хA-гJ/Bе0)],
где 6jj - дисперсия начального распределения.
Универсальная переменная выбирается в виде |, = х (I -
-тJ/Bб_),а функция Ф<!) = ?~3/ге~& . В этом случае у(х,
х) = <&(Х) ф(|,), где a(T) = {l-xLjt-V2Bei))-2. Ап-
проксиыационнсе выражение для ядра с параметром однородности
и, меняющимся в пределах 0-1, имеет вид
где | %
В.И.Смирновым методом производящей функции было найдено
полное асимптотическое решение уравнения стационарной коагуля-
коагуляции аэрозолей
290
1{тп)+В(гп)-
См
+ И X
применимое в интервале размеров частиц, в котором источник и
сток частиц практически не действует [88]. Коэффициент К(т,тпл
имеет довольно общий вид
где ols + ?s = Л. = const ; 0 ^ ots ; § <¦ 1 ; Л <¦ 1 ; min(eis ,^s
=-O,Aj- коэффициенты; n - произвольное число; Ф^О - произ-
произвольная функция.
Так как всегда возможна замена искомой функции g(m) =
- f(m)iP(m), то достаточно рассмотреть ядро K(mvm) =
показатель однородности К. В частном случае K(mi,m)=(mf*' +
+ mJ1) , где ff - целое число, четное при выборе знака ми-
минус, и.5 = Я, Предполагается, что функция источника достаточ-
достаточно быстро убывает с ростом m: I(m) ^ const езср(-т>г/т0),где
лг0 - характеристика ширины спектра источника.
Функция В(тп) отлична от нуля лишь для /п^М,при M»mD.
Для решения уравнения используется производящая функция Р
комплексного переменного z, определяемая в области \z\<l ин-
интегралом J?(z)=^c'g4m)zntfm.06o3Ha4HB Inz =-и, вводят функ-
функцию Q(u) = Р(е"и) = \ ?г(тп)ё~итdm , являющуюся преобра-
зованием Лапласа функций g(m) ж A/ 2ari) \ t m Q (u)eamdu,
где р>0 - вещественное число. Точка z -1 -'особая точка
функции F(z), а и = 0 - особая точка Q(u), поэтому интег-
интегрирование g(m) производится по контуру, обходящему особую точ-
точку, a R(z) вблизи z = 1 представляется в виде ряда. При ин-
интегрировании по контуру учитывается быстрое убывание множите-
множителя еитп при и<0, малость интеграла по исчезающе малому ра-
радиусу вокруг и = 0 и однозначность функции Q(u) на плоскос-
плоскости ц с разрезом от 0 до - оо.
Асимптотика функций g(m) и J(m) для mo<Lm<kM, где
М - нижняя граница действия функции стока, имеет вид
291
'1 g(m>,
где аг5 = |Я-2а5|а
Эти формулы применимы для описания асимптотического поведения
спектра масс частиц при стационарной коагуляции в интервале
масс, где достаточно узкий источник не влияет на спектр, а сток
еще не проявляется. Спектр g(m) является степенной функцией
масс. Если iv(m) - степенная функция т, то и спектр f(m)
также степенная функция. Показатель степени спектра g<m) за-
зависит только от показателя однородности относительно масс.Этот
спектр пропорционален корню квадратному из интенсивности источ-
источников. При Л , не слишком близком к единице, значения Л срав-
сравнимы только для частиц сравнимых масс, и спектр определяется
коагуляцией последних.
Для броуновской коагуляции в области 2«10 < г <
< 6.1СГТ мкм, когда 1^
стационарный асимптотический спектр масс выражается формулой
Ss(m) ft 0,161 (r^Pj(kT)) V2 тп*!2,
лов
где Р= jomI(m)dm- поток массы в систему от источника.Срав-
источника.Сравнение со случаем K=const показывает, что форма спектра в
обоих случаях одинакова.
Для гравитационной коагуляции, когда Кг-(Зла/2)Cпр/
/4) ml'3m2' , выражение для асимптотического спектра масс
имеет вид
Соответствующая полученным спектрам модельная функция рас-
распределения частиц по размерам записывается в виде
<рБ(Г) =0,235
. <рг(Г)= 0,573 р'1
!r-9/2 г«<г< -
292
Подобные результаты были получены ранее С.Фридлендером,
использовавшим в решении уравнения коагуляции теорию размернос-
размерностей, а также Дж.Клеттом, получившим решение задачи о стационар-
стационарной коагуляции для частного случая: K=rrvm^. Более широкие
возможности получения решения уравнения коагуляции представля-
представляют численные методы, не накладывающие жестких ограничений на
вид начальной функции и ядра К. Однако в этом случае возникают
трудности, связанные с большим объемом расчетов.
Моделирование эволюции распределения частиц по размерам
во времени производится с помощью дискретного аналога уравне-
уравнения коагуляции:
*' FЛ9)
где х = 2C6I'3(лсА3Г3/(р^т1)) - безразмерное время. Для
замыкания бесконечной системы используется быстрая сходимость
бесконечных рядов при }+к больше некоторого числа. Реально
с помощью ЭЦВМ решается не более 1000 уравнений такого вида,
что позволяет исследовать коагуляцию на сравнительно небольшом
начальном этапе, когда средний размер коагулирующих частиц пре-
превышает размер первичных частиц не более чем в несколько раз.
Устранение этой трудности в ряде работ достигается неравномер-
неравномерным разбиением частиц по массам, например рядом с усредненны-
усредненными массами" 2плг1,либо на начальном участке берется натураль-
натуральный ряд пт,, который, начиная с п= 5, заменяется рядом 2птп1
[Э4] . Данный метод использовался для исследования образования
самосохраняющегося распределения аэрозольных частиц по разме-
размерам при коагуляции в свободно-молекулярном режиме, т.е. для ус-
условий приземного слоя до Г 4 10 см. Асимптотический закон из-
изменения концентрации со временем N~ т~6/5, выведенный ранее
аналитически, устанавливался уже при т«10. Форма распределе-
распределения частиц по размерам оказывалась весьма близкой к нормально-
логарифмической.
Исследование влияния потенциала лондоновского притяжения
между двумя диэлектрическими сферами, учитываемого формулой Га-
макера путем введения в К поправочного множителя М,для неор-
неорганических веществ примерно равного М =2,2, а для органичес-
293
ких М = 1,39 (при одинаковых диаметрах коагулирующих сфер),
показало, что нарушения ;.ормы зависимости Net) и f(m) наблю-
наблюдаются при т > 10 и сохраняются еще при т » 1000. Наличие
меямолекулярных сил взаимодействия приводит к увеличению поли-
полидисперсности системы (функция Фридлендера <J>(^) расширяется).
§ 6.5. Влияние влажности на микроструктуру
атмосферных аэрозолей.
Морфология частиц
Важнейшим фактором изменчивости атмосферных аэрозолей,оче-
аэрозолей,очевидно, является присутствие в атмосфере водяного пара, содержа-
содержание которого варьируется от 10 до 10 г/г. Взаимодействие
аэрозолей и водяного пара может носить весьма сложный характер,
исследование этого процесса затрудняется тем, что не всегда
можно отделить эффекты, влияющие на структуру атмосферных аэ-
аэрозолей, в частности на их дисперсность, и действующие в ряде
случаев на аэрозоли одновременно с влажностью, от эффекта пря-
прямого влияния изменения влажности. Например, известно, что мно-
многие метеорологические и физические параметры атмосферы, в том
числе и относительная влажность воздуха, имеют суточный ход.
Дисперсность атмосферных аэрозолей тоже имеет отчетливо выра-
выраженный суточный ход. В результате может наблюдаться хорошая
корреляционная связь между дисперсностью аэрозолей (например,
концентрацией какой-то определенной фракции) и значением отно-
относительной влажности. Из физических соображений очевидно, что
в этом случае определяющими факторами будут суточный ход гене-
генерации частиц и конвекция воздушных масс. Кроме того, любой ис-
исследуемый объем атмосферы представляет собой открытую систему,
воздушная масса в которой может изменить свойства под внешним
воздействием или полностью обновиться.
Несмотря на сказанное, влияние влажности на дисперсность
атмосферных аэрозолей можно считать установленным фактом .При-
.Причем увеличение влажности даже при значениях относительной влаж-
влажности, далеких от величины насыщения, приводит к заметному рос-
росту аэрозольных частиц. Установлено, что в первую очередь уве-
294
личивается фракция радиусом ОД-0,3 мкм. Увеличение размеров
частиц от г = ОД до г = 0,2+0,25 мкм не мояет быть объясне-
объяснено в рамках классической теории конденсационного роста капель
растворов. В то же время грубодисперсные аэрозольные частицы
практически не увеличиваются. Это позволило сделать предположе-
предположение о разной природе и различных физико-химических свойствах
мелко- и грубодисперсной фракций атмосферных аэрозолей. Дейст-
Действительно (см. гл.1), наблюдается существенное различие в хими-
химическом составе мелко- и грубодисперсной фракций аэрозолей в
приземном слое атмосферы. В более высоких слоях разные фракции
имеют примерно одинаковый химический состав. Кроме того, в верх-
верхней тропосфере и более высоких слоях атмосферы почти полностью
отсутствуют частицы из органического вещества, однако поведе-
поведение частиц в зависимости от относительной влажности весьма схо-
сходно с тем, что наблюдается в приземном слое. В частности, появ-
появление перламутровых и серебристых облаков всегда связано с не-
некоторым повышением влажности в соответствующих слоях, которая,
однако, не достигает величины, необходимой для конденсационно-
конденсационного роста частиц.
Более детальное исследование влияния абсолютной и относи-
относительной влажности на содержание аэрозолей выявило, что сущест-
существуют разные типы такой зависимости: 1) увеличение концентрации
атмосферных аэрозолей с увеличением абсолютной влажности (в ос*-
новном по данным оптических измерений); 2) рост мелкодисперс-
мелкодисперсной фракции при увеличении относительной влажности в области
значений, далеких от наснщенияBО5»</* 70$); 3 )ув«шчение раз-
размеров аэрозольных частиц согласно расчетам гетерогенной кон-
концентрации на гигроскопических ядрах (&0% < f <. 100$);
4) уменьшение содержания гигантских частиц в атмосфере при от-
относительной влажности, блшзкой к насыщению; 5) аномальный ход
изменения концентрации аэрозольных частиц в зависимости от от-
относительной влажности. Кроме того, при оптических измерениях
случаи постепенного роста частиц с увеличением относительной
влажности от 50%', что можно интерпретировать как рост частиц
вследствие адсорбции водяного пара и частичного растворения ве-
вещества частиц.
295
Аномальный ход изменения концентрации аэрозольных частиц
в зависимости от относительной влажности Г.В.РозенОергом и его
сотрудниками объясняется конденсацией органических паров, выде-
выделяемых растительностью, на уже имеющихся ядрах и пассивацией
частицы поверхностно-активными веществами (ПАВ), входящими в
состав паров. По их мнению, в пользу конденсационного фотохими-
фотохимического механизма роста аэрозольных частиц свидетельствуют вы-
высокие скорости увеличения счетной концентрации в диапазоне г =
= 0,25+0,3 мкм до 4 част (см"^-ч), не достижимые при коагуляци-
онноы росте частиц для наблюдаемых начальных концентраций.Ско-
концентраций.Скорость конденсационного роста вычислялась по формуле
Jir2drjdt =
где Ъ - параметр, примерно равный единице, константа а зави-
зависит от молекулярной массы и степени пересыщения конденсируемых
паров, неизвестная для рассматриваемого случая; Я^ - длина
свободного пробега молекул. Выражение для изменения количест-
количества частиц в рассматриваемом диапазоне размеров в предположении
постоянства суммарной концентрации частиц для размеров выше
критического (а^— = 0,09 мкм) получено в виде
где Пр - начальная концентрация частиц с начальным радиусом
Тр . Удовлетворительное согласие расчетных данных с экспери-
экспериментальными получено при ai/si - 0,06. Для объяснения зависи-
зависимостей экспериментальных данных эффектом пассивации необходи-
необходимо предположить, что отношение объемной плотности ПАВ в орга-
органических парах, участвующих в процессе конденсации, к поверх-
поверхностной плотности молекул ПАВ при плотной мономолекулярной упа-
упаковке равно примерно 0,005. Наблюдались также другие типы за-
зависимости концентрации частиц от относительной влажности в том
же районе наблюдений (о.Саарема, июль-август 1973 г.). В част-
частности, описывались случаи, когда, несмотря на постоянство воз-
воздушной массы, аэрозольные частицы оказываются практически не-
нечувствительны к влажности в очень широком диапазоне ее измене-
изменения. Полагают, что такое поведение аэрозолей объясняется нали-
наличием значительной доли нерастворимых ядер конденсации, которые
296
ниже точки росы образуют сухую мглу и начинают активно участво-
участвовать в конденсационных процессах только по достижении насыще-
насыщения. При таком составе конденсационных ядер процесс помутнения
атмосферы мало зависит от относительной влажности и определяет-
определяется преимущественно механизмами пространственного переноса аэро-
аэрозолей. Присутствием таких частиц объясняются, например, резуль-
результаты измерений в районе пос.Карадаг, где вклад несферических
частиц в общее рассеяние аэрозолями достигал 80%.
Следует отметить, что сотрудниками Ленинградского универ-
университета в аэрозольных пробах, взятых в указанном районе, были
обнаружены в большом количестве гигантские частицы, имеющие
;орму палочек и идентифицируемые как натролиты или шпаты. Эти
частицы практически нерастворимы в воде и их присутствие в воз-
воздухе позволяет объяснить наблюдавшиеся ранее поляризационные
эйекты в рассеянии света аэрозолями.
Очевидно, что данные о зависимости структуры и содержания
аэрозолей в атмосфере от абсолютной влажности можно объяснить
опосредованной связью с относительной влажностью, свойственной
каждой воздушной массе. Однако возможна и прямая зависимость
концентрации и размеров аэрозольных частиц от величины абсолют-
абсолютной влажности, если аэрозоли образуются в результате реакций
окислов азота и серы с водяным паром в присутствии катализато-
катализаторов: образование растворов серной и азотной кислот в условиях
невысокой относительной влажности. Прекрасной иллюстраци-
иллюстрацией влияния внешних условий воздушной среды на формирование мик-
микроструктуры частицы являются результаты морфологического ана-
анализа аэрозольных проб, взятых с помощью самолетного фильтрово-
фильтрового заборника на разных высотах над Беринговым морем в марте
1973 г. [42], Эти данные представлены в табл.33. Видно, что
чем меньше высота, а следовательно, выше относительная влаж-
влажность, тем больше доля частиц сферической формы. Причем в ре-
:;у.'Л>тате более длительного существования частиц в средней тро-
тропосфере доля плотных сфер здесь значительно выше, чем в ниж-
нижней тропосфере, где частицы относительно'Ыолодые". Низкая
влажность в верхней тропосфере ведет к увеличению доли рыхлых
'¦астиц.
297
Таблица
Содержание частиц разных форм, %
Число
проб
4
10
3
я,
м
1000
4000
9000
1
18
41
12
2
65
30
40
Типы
3
_
2,0
-
4
8,0
2,5
15
частиц
5
10
9,0
6
7,5
3,7
13
7
7
7
_
,0
,0
1
3
4
8
,5
,8
,0
Примечание. 1- плотные сферы, 2 - неплотные сфе-
сферы, 3 - частицы с оболочкой из очень мелких частиц (с "шубой"),
4 - плотные несферические, 5 - рыхлые несферические, 6 - цепоч-
цепочки из более мелких частиц, 7 - рыхлые с плотными ядрами, 8 -
кристаллы и частицы с высохшей оболочкой.
Все указанные типы частиц были обнаружены не только на оп-
определенных высотах в атмосфере, но и в приземном слое при конк-
конкретных условиях (табл. 33-35). Так, в чистом сухом воздухе
(вблизи пос. Чолпон-Ата, оз.Иссык-Куль) наблюдались частицы,
сходные с частицами, которые считаются типичными для верхних
слоев атмосферы, а также отдельные частицы субмикронных раз-
размеров правильной сферической формы.
В более пыльном и влажном воздухе (вблизи пос.Тамги, оз.
Иссык-Куль) наблюдались частицы, сходные с частицами, обнару-
обнаруженными в сульфатном слое ?/нге, а также отдельные плотные час-
частицы неправильных форм и рыхлые агломераты, состоящие из ве-
вещества разной плотности и, вероятно, разного происхождения. В
приземном слое (вблизи Рыльска) большинство частиц имело фор-
форму близкую к сферической, однако эти частицы были составлены
из еще более мелких'частиц.
Однородными являются в основном частицы, состоящие, веро-
вероятно, из гигроскопических кристаллообразующих соединений (NaCl,
(NH4JS04 • NH4HS04 и т.п.). Для проб, взятых в приземном
слс-о (вблизи Томска), характерно присутствие агломератов оваль-
овальной формы из частиц невысокой плотности, а также частиц непра-
неправильной формы, например звездочек, по-видимому, фотохимичес-
фотохимической природы.
298
Я
•О
03
ц
о
и
а,
а
О:
О я)
аз о
1
о
X
со
Я
а.
"i
в*
аз
Si
ft
аз
g
о
.
1
8
в1
со
о
о
-=!•
о
О
' 1
и
см
кЧ
W
см
о
кЧ
1
СМ
о
is
48,
о
СО
СМ
кЧ
.29,
со
¦¦*
кЧ
Ю
со
C4J
со
LT3
СО
•ч1
кЧ
СО
О
кЧ
о
ю
о
о
ю
42,
о
•ч1
ю
к-1
О
СО
ю
см
со
Q
со
о
-ч-
СО
го
кЧ
СО
Я
СО
с-
см
ю
кч
СО
О
ю
кЧ
О
кЧ
О
кЧ
СО
со
г-
СО
-ч-
см
со
кЧ
о
см
со
СО
*?
СО
со
кч
m
У
кЧ
СО
о
о
см
о
in
кЧ
О
кЧ
•Ч1
СО
со
о
LT3
1
1
•ч1
см
см
со
кЧ
кЧ
Ю
LT3
О
LT3
СО
о
см
о
о
С\!
О
со
см
со
кч
LT3
о
о
со
кч
с^
1
о
кч
о
см
СО
СО
кЧ
см
см
со
о
о
со
о
(О
см
о
•ч*
СО
LO
•я-
со
о
со
со
1
1
со
кЧ
кЧ
см
см
-ч1
СО
кЧ
СО
см
ю
со
о
о
со
о
ю
М
см
кч
со
о
СО
кЧ
1
CD
о
о
кЧ
CD
СО
•ч1
кч
СО
кч
CD
О
О
5-0
со
о
о
со
кЧ
СМ
со
о
СО
о
СО
кч
о
кч
-ч-
о
со
.-ч1
СО
кч
г-
см
S
о
о
•Ч1
о
о
ю
СО
со
СО
о
СО
о
со
см
1
с-
о
СО
кч
о
СО
со
кЧ
-ч-
со
о
LT3
о
о
UJ
о
со
кч
о
м
со
о
со
о
1
СО
о
кЧ
•Ч1
о
о
о
о
кЧ
ю
1>
о
СО
о
со
о
кЧ
о
кЧ
о
см
о
см
о
LT3
кН
о
о
о
кч
ю
см
со
со
СО
S
см
со
о
см
со
S
ю
са
(Ц
о
Щ
* S S s
й в) Ч «
сг о 5
»!§!
аз а „
s s ^ S
е аз аз о
s * 1
со о
299
а, л
Таблица 35
Морфологическая структура аэрозольных частиц в тропосфере
(Рыльск, август 1975 г.)
Я, км
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
0,5
0,5
2,8
г,*
0,8
14
08
08
14
0,8
08
14
08
08
14
08
08
14
08
08
14
08
08
14
08
08
14
08
08
14
08
г, мкм
0,02-0,05
0,05-0,10
0,10-0,15
0,15-0,25
0,25-0,35
0,35-0,45
0,45-0,55
0,55-0,65
0,65-0,75
Тип частицы
1
1,2
7,2
15,7
5,5
1,9
6,0
12,8
10,1
20,5
6,0
-
48,2
5,4
-
9,6
10,7
-
-
-
-
-
-
-
-
-
-
-
2
1,6
5,6
-
14,4
4,5
57,5
22,2
2,9
6,8
26,7
8,6
11,0
9,7
20,0
9,6
-
12,3
15,1
_
13,9
-
_
24,2
-
-
-
-
3
52,5
42,2
38,0
18,4
16,5
31,8
8,9
6,3
-
7,5
8,9
10,8
8,6
12,3
10,1
-
7,0
-
19,4
-
-
-
6Д
9,3
-
-
-
4
44,7
45,0
15,0
55,7
61,0
47,0
45,6
55,4
15,3
42,2
35,1
16,2
29,0
18,7
3,4
14,3
36,9
1,9
6,4
30,5
-
50,0
30,3
-
-
-
1,2
5
_
-
-
6,0
4,8
-
8,3
11,7
2,9
12,1
37,7
26,4
46,2
31,6
14,2
75,0
33,3
27,2
74,2
55,6
18,3
50,0
39,4
5,7
100,0
-
3,3
6
_
-
7,2
-
11,3
18,5
2,2
13,6
27,6
5,5
9,7
26,2
17,4
-
-
10,5
-
_
-
20,0
-
-
-
-
-
-
Шг)
X
24,7
12,5
12,5
20,4
21,6
27,4
18,2
22,0
10,5
20,2
24,4
21,4
9,4
10,8
8,1
2,8
4,0
9,2
3,1
2,5
6,3
0,8
2,3
0,3
-
1,3
300
Продолжение табл.35
0,5
0,5
2,8
и,5
;,5
2,3
2
08
14
08
08
и
08
0
0
з
,75-0,
,02-0,
85
85
4
-
-
-
5,
3,
9,
8
6
I
5
13
7
8
-
-
-
,7
,9
,0
6
-
-
21,
14,
14,
3
I
1
7
42
44
35
-
-
-
,9
,5
,0
8
2,
14
19
26
0
7
8
8
9
-
-
-
1,6
10,1
7,1
10
-
-
0,5
§ 6.6. Конденсационно-коагуляционный механизм
роста частиц
В работах автора была выдвинута гипотеза о влиянии влаж-
влажности на скорость коагуляционного, а также конденсационно-ко-
агуляшонного роста частиц. Это влияние может быть обусловле-
обусловлено двумя факторами, возникающими при увеличении относительной
влажности воздушной среды: I) ростом эффективности слипания
коагулирующих частиц при больших значениях относительной вла-
влажности, 2) появлением дополнительных сил взаимодействия час-
частиц (вероятно, электростатических).
Первое предположение, казалось бы, противоречит выводам,
сделанным в монографии Н.А.Фукса [104] о слабой зависимости кон-
константы коагуляции от эффективности соприкосновения частиц. В
уравнении коагуляции полагается, что каждый акт встречи частиц
приводит к взаимной коагуляции. В реальной атмосфере при пос-
постоянном действии на две столкнувшиеся частицы различных сил, в
частности сил, создаваемых воздушными потоками, при малой шю-
дади соприкосновения твердых частиц величина силы слипания час-
частиц играет немаловажную роль. Так как при соприкосновении час-
частиц образуются капилляры, в которых может происходить конденса-
конденсация водяного пара при значениях относительной влажности, дале-
далеких от насыщения, то слипание частиц усиливается, и они стано-
становятся единым целым.
Расчет сил сцепления таких частиц показывает, что при по-
зышении влажности происходит их увеличение почти на два поряд-
порядка. Этот эффект особенно существен для частиц с размерами по-
301
рядка десятых и сотых микрометра и значительно меньше для более
крупных частиц,Кроме того, концентрация более крупных частщ
резко падает <dNjdr ~ - Сг'А) и, следовательно, резко уменьша-
уменьшается вероятность их столкновения.
Известно общее выражение для молекулярной силы взаимодей-
взаимодействия двух сухих частщ с одинаковым радиусом: F=3iroBe13 -
- ®jj), где rQ - радиус частщ; в.3 - свободная энергия еди-
единицы поверхности раздела одной частицы и промежуточной фазы,
отделяющей ее от другой частицы; бц - свободная энергия едини-
единицы поверхности раздела двух частщ.
Рио.ЗО. Схемы капиллярной конденсации.
а - две сферы; $ - конус и плоская поверхность.
Рассмотрим две сферические частицы с одинаковым радиусом,
между которыми находится некоторое количество воды (рис.30).
В силу малых размеров частиц не будем учитывать влияние грави-
гравитационного поля, искажающего кривизну поверхности воды.
Сила капиллярного взаимодействия двух сферических частиц
состоит из двух слагаемых. Первое - сила сжатия, возникающая в
связи с тем, что благодаря кривизне менисков по обе стороны по-
поверхности воды существует перепад давлений, определяемый пер-
первым законом Лапласа. Эта сила выражается формулой
F.20)
где 6 - поверхностное натяжение воды; S - проекция площади
граница вода - частица на плоскость, перпендикулярную направле-
направлению силы: Г[ i г2 - радиусы кривизны. Имеем S-=SKr* sin.2<p ,
где (о - угол, зависящий от количества воды, находящийся между
частицами. Второе слагаемое силы капиллярного взаимодействия оп-
определяет стягивающее действие поверхности воды. Оно представля-
представляет собой проекцию вектора поверхностного натяжения воды, дейст-
действующего по периметру смачивания на ось, соединяющую центры час-
частиц:
где 0 - краевой угол скачивания; I - длина периметра соприко-
соприкосновения воды с частицей. Это слагаемое играет существенную
роль при больших количествах воды (т.е. при больших <р).
Таким образом, общая сила капиллярного взаимодействия час-
частиц определяется следующим выражением:
-r~1) + 2sin<<p + 0)].
Радиусы кривизны, вычисленные в предположении, что
дуга окружности, описывается как
' -<I-cos<p)(l-sm«p+0))/cos(<p+e)]> F.22)
гг = ro(I-cos<p)/cos<(p + 0). F.23)
Подставляя F.22) и F.23) в F.21), получим
F = 2jte>r0cosf cos(-| + 0)[l - -i*g(f - |-)sin<p-sin2|] F.24)
Объем воды, заключенный между частицами, вычисляется по форму-
ле
302
V - 2яго2[{ I-cos<f)/cosz((p+8)]jcos2(| +0) + sin2 | -
-(y-<p-0)(sin<<p+9)-sine)/cos(<p + 0)}.
Полученные выражения позволяют рассмотреть влияние краево-
краевого угла смачивания 0 и количества воды в контакте частиц V
на силу капиллярного взаимодействия. Сила капиллярного взаимо-
взаимодействия исчезает при 0=я/2-<р/2. При ср/2 + 0 < JT/2 между
303
частицами действует сила притяжения, а при <р/2 + в > я/2 -
сила отталкивания, т.е. с увеличением угла 9 сила притяжения
уменьшается и становится равной нулю при 90=я/2, а при даль-
дальнейшем увеличении 9 наступает отталкивание частиц. С увеличе-
увеличением количества воды между сферами сила притяжения их уменьша-
уменьшается.
В случае полного смачивания сила капиллярного взаимодейст-
взаимодействия частиц определяется равенством
F = 6ЛГ0 sin2<f [ го(г? - г) + 2 ],
где rt = ro[sin<f -(I-cos<f)< I - sin<f)jcosy]; r2 = rQ( 1 -
- cos<P)/cos(p, а объем воды, заключенной между частицами, в
этом случае определяется выражением
V- 2orro3cos'2<f <I-coS<pJ[ I -(f -<p)tgcp].
Например, при <р = 45°, V =0,25 г3 см сила капиллярного
взаимодействия частиц F = 2,8-Ю rQH .
Из частиц несферической формы рассмотрим конусообразную
частицу. Сила капиллярного взаимодействия такой частицы (в мес-
месте контакта вершины конуса) с плоскостью также состоит из двух
слагаемых:
где б" - поверхностное натяжение; г. и г. - радиусы кривиз-
кривизны, ot - угол конуса; г. - радиус окружности смачивания, ко-
конуса; rt=r [!-(!-cosot)/(itgot(l +sin*)
т.е. имеем
Объем воды, заключенный между частицами и плоскостью, выража-
выражается равенством
V= mr2|[(rj + r2J +r22](l + sin*) + (r*j2){l
-(rt + r2)r2( sin otcosot + a +jr/2)|.
304
Механизм капиллярной конденсации между двумя соприкасаю-
соприкасающимися частицами сводится к следующему. Предполагая наличие
адсорбированной пленки молекулы воды на частицах, получим в
начальный момент конденсации в месте соприкосновения частиц
поверхность с максимальной кривизной. Когда давление шара над
этой поверхностью достигнет значения р = р езср [ - 1,5 * •
х 1О"ю(()<(р)/г0], пар станет насыщенным. Конденсация пара приведет
к продвижению жидкости в более широкую часть рассматриваемого
пространства. При этом кривизна поверхности уменьшится. Отноше-
Отношение p/ps станет равным единице при угле накопления <р= 53°10'.
Этому углу соответствует объем жидкой прослойки V^ = 0,4 r^.
Указанный механизм хорошо действует для частиц с г ? ОД мкм.
Для частиц больших размеров влажность, при которой происходит
коагуляционное слипание, близка к насыщению, и главную роль в
росте частиц начинает играть конденсация на поверхности части-
частицы. При испарении жидкой прослойки силы сцепления между части-
частицами уменьшаются и система частиц может рассыпаться. Гиотере-
зис вызван тем, что разъединение частиц происходит не сразу
после испарения воды, а значительно позже.
Увеличение влажности воздушной среды сопровождается раз- .
личными электрическими явлениями в среде и на поверхности аэ-
аэрозольных частиц (ориентированная адсорбция молекул воды на
поверхности частиц, зарядка и перезарядка частиц, поляризация
зарядов на частице и т.д.) Экспериментально установлено, что
увеличение относительной влажности ведет к накоплению отрица-
отрицательного заряда на аэрозольных частицах и положительного заря-
заряда на легких ионах.
Вследствие всех этих процессов происходит возникновение
индуцированных зарядов на увлажненных частицах. Они могут по-
появиться за счет дипольного момента молекул воды или разделения
заряда при растворении гигроскопического вещества (солей, кис-
кислот и т.п.). Роль электрического заряда частиц в их коагуляции
качественно можно оценить, представив, что размеры коагулирую-
коагулирующих сфер определяются изменяющимся радиусом взаимного притяже-
чия частиц, а подвижность частиц остается примерно прежней.Тог-
прежней.Тогда константа коагуляции частиц К- (г^, г-) увеличится во
305
столько раз, во сколько увеличился радиус сильного взаимодейст-
взаимодействия коагулирующих сфер (ш (г ) > к Т ) . Этот коэффициент уве-
увеличения эффективной константы коагуляции может быть порядка
10-10*" в зависимости от влажности и свойств аэрозольных час-
частиц. Причем наиболее заметно электростатический эффект увеличе-
увеличения скорости коагуляции должен проявляться в полидисперсных
системах. Как было показано ранее, при сильно различающихся за-
зарядах и размерах частиц наблюдается взаимное притяжение однои-
одноименно заряженных частиц. Наличие электрических зарядов на час-
частицах приводит также к улучшению условий их слипания друг с
другом.
В общем случае механизм капиллярной конденсации в прост-
пространстве между двумя соприкасающимися частицами сводится к сле-
следующему. В предположении наличяя адсорбированной пленки моле-
молекул воды на частицах произвольной формы после их сближения в
зазоре между ними образуется пленка жидкости с искривленной по-
поверхностью.
Пусть потенциал Гиббса молекул в фазе пара ifu, а в жид-
жидкой фазе ?ж. Если манжета между частицами образуется в ре-
результате конденсации молекул пара, то общее изменение свобод-
свободной энергии будет: ( Уж- Уп )п + eS, где S -поверхность
манжеты; е - поверхностное натяжение на границе раздела жид-
жидкость - пар. Поверхностная энергия манжеты, несущей заряд у,,
во внешней среде с диэлектрической постоянной е. выражается
следующим образом:
где ? - диэлектрическая постоянная жидкости. Тогда
VM - объем манжеты; А^ - число Авогадро; рж - плотность
жидкости; М - масса моля вещества жидкости.
Разность потенциалов Гиббса выражается формулой A'if =
= к Т In (pip*). гДе р - парциальное давление пара над искрив-
искривленной поверхностью манжеты, a ps - парциальное давление на-
насыщения.
306
Условие равновесия системы: д"У/д5=0, откуда оладует
Р М f * + _?_ i($S4S),dVrw
Ps ^Рж*^" fy/dS 8яе0е Л \ ^5
В дальнейшем величину Л//(Л^рж А7> обозначим через f.
Так как d^/dS = (к1 + к2), где •^Г1*г. и Jt = г.
радиусы кривизны нормальных взаимно перпендикулярных сечений
манжеты, то
Л6.25)
fs = - Л
Для жидкой манжеты! д(\
ла F.27) приобретает вид
lnPs = -
форму-
формуF.26)
В начальный момент образования перемычки (из адсорбирован-
адсорбированных молекул пара) величина к^ +
Г2м
ф
Г1м
^при
^ + кг = гГ| - тfj » 0 , так как
заряда формула F.28) соответству-
соответству2м 1м
ет формуле Томсона).
Когда давление пара над зтой поверхностью достигает зна-
значения
начинается конденсация пара в манжете, которая приводит к про-
продвижению жидкости в более широкую часть рассматриваемого прост-
пространства. При этом кривизна поверхности уменьшается ж увеличива-
увеличивается угол наполнения «р. Для того чтобы пар продолжал конден-
конденсироваться, необходимо увеличение давления р.
Для сферических частиц, находящихся друг от друга на рас-
расстоянии Л, величины *j и к2 зависят не только от их радиу-
радиусов Qj, а г и параметра манжеты - угла наполнения ср, но и от
расстояния между сферами А .
Для заряженных частиц зависимость угла наполнения to от
отношения давления пара над частицами к давлению насыщения бу-
будет иметь несколько иной характер - наполнение манжеты конден-
конденсированной жидкостью будет происходить при меньших значениях
307
pip.. Влияние заряда особенно сильно сказывается в области ма-
малых значений p/ps- От параметров манжеты зависит также капил-
капиллярная сила взаимодействия двух частиц.
На каждой из них экспериментально было установлено, что
при сближении двух капель под действием электрических сил в за-
зазоре образуется локальное конусообразное искривление. При сбли-
сближении вершин конусов должен наступить момент, когда потенциал
окажется достаточным для электрического пробоя прослойки возду-
воздуха между ними. Есть доказательства возможности появления раз-
разряда при сближении капель перед слиянием. Однако вопрос о ха-
характере разряда - искровой или коронный - не решен.Время разря-
разряда 10-10 с. Это время намного порядков меньше времени, ко-
которое требуется для стекания заряда с капли, если учитывать
электропроводность воды. Очевидно, что стекание заряда обеспе-
обеспечивается не электропроводностью воды, а электропроводностью ио-
ионизированного воздуха в прослойке между каплями. Процесс обра-
образования перемычки занимает время порядка 10 с.
Если заряженные аэрозольные частицы покрыты слоем адсорби-
адсорбированной воды, толщина которого определяется влажностью окружа-
окружающего воздуха, то представляется вероятным образование такой
же перемычки при сближении двух аэрозольных частиц. В отличие
от капель в последнем случае не происходит слияния, а перемыч-
перемычка становится поверхностью отрицательной кривизны, над которой
понижено давление водяного пара.
При слабом взаимодействии молекул твердых частиц, являю-
являющихся составными частями более крупной частицы, по сравнению с
взаимодействием молекул этих твердых частиц и молекул конденса-
конденсата (воды, серной кислоты, органических веществ) может наблю-
наблюдаться конденсационный рост частицы, внешне напоминающий набуха-
набухание. Конденсат проникает в микрокапилляры между мелкими части-
частицами внутрь более крупной при относительной влажности, дале-
далекой от насыщения. Причем чем мельче частицы, покрывающие свер-
сверху более крупную частицу (каплю),и чем плотнее их упаковка,
тем ниже относительная влажность, при которой происходит кон-
конденсация. Рост крупной частицы определяется концентрацией и
дисперсностью мелких частиц в воздухе и относительной влаж-
влажностью. Очевидно, что мелкие частицы не должны растворяться в
308
конденсате. Идеальным материалом для такого процесса могут слу-
служить мельчайшие частицы сажи при условии их смачиваемости. (Сма-
(Смачиваемость частиц сажи достигается присутствием молекул серной
кислоты.) Весьма вероятно, что этот процесс реализуется при
образовании промышленных загрязнений, особенно смогов.
Подобного рода процессы должны протекать и в свободной ат-
атмосфере. Например, этим механизмом можно объяснить довольно быс-
быстрое вымывание после лесных пожаров сажевых частиц, играющих
весьма важную роль в поглощении солнечной радиации, из нижних
слоев атмосферы. Несколько слабее обсуждаемый процесс сказыва-
сказывается на таких аэрозольных частицах почвенного происхождения,как
частицы кварца и окислов железа. Однако даже и для них в естес-
естественных условиях наблюдался заметный коагуляционный рост при
высокой концентрации аэрозолей и повышенной влажности воздуха.
Особенно отчетливая картина коагуляционного роста плохо смачи-
смачиваемых частиц в облаке сахарской пыли, несущемся над Атлантичес-
Атлантическим океаном, была обнаружена при зондированиях такого типа об-
облаков в экспедиции АТЭП-74. Оказалось, что в центре облака в
большинстве случаев наблюдается коагуляционное укрупнение час-
частиц. Причем степень их укрупнения хорошо коррелирует с повыше-
повышением относительной влажности. Сильное влияние влажности на
структуру аэрозольных частиц подтверждается результатами анали-
анализов проб, взятых в приземном слое атмосферы (на южном и север-
северном берегах оз.Иссык-Куль).
Лабораторный эксперимент, моделировавший процесс роста са-
сажевых частиц, также подтвердил сильное влияние влажности и при-
присутствия паров серной кислоты на скорость коагуляционного рос-
роста частиц сажи. Эффективные коэффициенты коагуляции в лабора-
лабораторном эксперименте значительно превышали теоретические.
Все рассмотренные эффекты существенно усложняют кинетичес-
кинетическое уравнение коагуляции полидисперсной системы с учетом кон-
конденсационного процесса, и вряд ли можно достаточно точно ввес-
ввести поправки на увеличение радиуса взаимодействия частиц и сил
сцепления. Однако очевидно, что влияние этих поправок значи-
значительно и в реальной картине трансформации атмосферных аэрозо-
аэрозолей они играют большую роль.
309
Конденсационно-коагуляционную гипотезу о трансформации
спектра атмосферных аэрозолей подтвердили результаты эксперимен-
экспериментов по влиянию влажности на рост аэрозольных частиц и изменение
электрических характеристик воздушной среды, проведенные в боль-
большой камере туманов Главной геофизической обсерватории им.А.И.Во-
им.А.И.Воейкова.
В процессе опытов определялись интегральная и спектральная
концентрация аэрозолей, их морфологический состав и полярные
электропроводности воздуха.
Перед увлажнением воздуха проводились фоновые измерения
его температуры, влажности и электропроводности, а также изуча-
изучалась структура аэрозолей. Увлажнение воздуха в камере произво-
производилось путем смачивания стенок и пола камеры, либо испарением
воды при нагревании ее в открытом сосуде. В каждом из опытов
удавалось провести комплексные измерения всех характеристик
только при одном значении влажности. При втором способе увлаж-
увлажнения воздуха измерения в каждом опыте проводились при измене-
изменении относительной влажности от 30 до 90$ с шагом 10$. Все при-
приборы были установлены на уровне 1,5-2,0 м от пола камеры. Аэро-
Аэрозольные измерения проводились на двух уровнях - 2 и 6 м.
Для выяснения связи между электрическими и структурными
характеристиками аэрозолей в замкнутом объеме было проведено
три серии опытов: фоновые при ,/ = 30$, в увлажненной камере
при двух способах увлажнения.
Во время опытов в камере без увлажнения обычно держались
температура ~ 22°С (февраль 1974 г.), относительная влажность
~ 30$. Положительная электропроводность в камере составляла
C,5-г5,5)-10~15 си/и, отрицательная C,3-5,0)-10~'15 см/м, что
на порядок ниже средних значений электропроводности в атмос-
атмосфере.
Исследования структуры аэрозолей показали четко выражен-
выраженное изменение ее с высотой. Примеры типичных распределений аэ-
аэрозольных частиц по размерам в камере приведены в табл.36 (про-
(пробы 2-5). Наблюдается заметное отклонение от юнговского распре-
распределения для гигантских частиц. Морфологический анализ проб
показал, что для них характерно наличие относительно большого
количества рыхлых частиц как правильной сферической, так и
310
Таблица 36
Абсолютные концентрации аэрозольных частиц разных размеров
при увлажнении в камере туманов, сы~3
d,
0
0
i
i
2
2
3
В
мкм
0,05
ОД
0,2
0,3
0,4
0,5
,6-0,
,8-1,
,0-1,
,5-2,
,0-2,
,5-3,
,0
с е
8
0
5
0
5
0
г о
2
12
60
32
20
7
12
15
9
18
3
2
3
1
197
,5
,0
,0
,5
,0
.5
,5
,5
,0
,0
,0
,5
,0
,0
3
6,0
45,5
61,1
39,0
25,0
14,0
10,5
7,5
6,0
1,0
0,30
_
-
215,9
]
4
17
56
71
45
36
23
22
10
3
I
0,
о,
о,
28S
ioMep
,5
,0
,5
,5
,0
,0
,3
,0
,5
,5
40
50
60
,0
5
114
81
75
23
13
9
14
1
2
1
0,
0,
о,
336
пробы
,5
,0
,0
,5
,0
,0
,0
,6
,8
,8
20
15
10
,6
10
33 ,'6
45,6
49,5
31,8
18,0
14,0
9,0
1.8
0,6
0,6
0,15
ОДО
0,50
205,2
И
48
179
149
51
14
7
.11
2
0
0
0,
о,
о,
463
,0
,0
,0
,0
,4
.2
,2
Д
,6
,4
27
20
16
,5
18
18
66
69
38
25
10
20
8
8
1
0,
0,
о,
242
,4
,0
,2
.0
,6
,8
,8
,8
,4
,2
21
21
10
Д
26
0
62
55
21
18
26
18
3
12
4
0,
0,
1,
225
,8
,4
,6
,6
,0
.4
,0
.6
,8
,0
80
20
00
,2
неправильной формы. Таких частиц рыхлых коагулянтов в пробах
было от 55 до 65/2 по счетной концентрации для всего исследуе-
исследуемого диапазона размеров частиц @,005 & г (, 5 мкм).
При первом способе увлажнения в результате баллоэлектри-
ческих эффектов абсолютные значения полярных проводимостей
очень резко менялись от опыта к опыту. Электронно-микроскопи-
Электронно-микроскопические снимки аэрозольных проб выявили наличие в них частиц,
которые можно было трактовать как высохшие капельки. Исследо-
Исследования морфологии частиц (пробы 10,11) показали, что в увлаж-
увлажненной камере доля рыхлых сферических частиц увеличивается, а
доля частиц неправильной формы и цепочек уменьшается. Наблюда-
Наблюдаются заметный рост концентрации частиц с d 4 0,4 мкм, умень-
уменьшение концентрации частиц с 0,4 4 d i 0,6 мкм и примерное
постоянство концентрации частиц с d > 0,6 мкм. Концентрация
крупных частиц с d > 3 мкм уменьшается. Последние результаты
противоречат как коагуляционной гипотезе, так и ранее получен-
полученным данным о вымывании аэрозолей в приземном слое атмосферы.
Они могут быть объяснены процессами, связанными с особенностя-
особенностями увлажнения камеры. Увеличение мелкодисперсной фракции с
d & 0,4 мкм наиболее вероятно связано с разбрызгиванием и вы-
высыханием капель воды, а уменьшение концентрации частиц с
d > 3 мкм - с вымыванием этих частиц. Таким образом, данный
способ увлажнения камеры вносил в измеряемые характеристики
значительные изменения, поэтому от него пришлось отказаться.
312
30 40 SO 60 70 80 Г, %
Рис.31. Изменение полярных проводимостей
" * А.., коэффициент?
ральной концент!
в опытах I и П.
воздуха Л+, Л... t коэффициента униполярности
А+/Л_, интегральной концентрации влажности
При увлажнении воздуха испарением воды путем подогрева в
каждом отдельном опыте удавалось получать значения всех измеря-
измеряемых величин при относительной влажности от 30 до 90$.На рис.33
показаны изменения полярных проводимостей, коэффициентов уни-
униполярности А+,А_, Л+/Л_ и интегральной концентрации аэро-
аэрозольных частиц с увеличением относительной влажности. В первом
случае как Л+, так и А_ уменьшаются с увеличением относи-
относительной влажности до 6052. Однако, как показывает ход A+/A_f
отрицательная проводимость уменьшается гораздо сильнее, чем по-
положительная. Затем до J - 80% наблюдается незначительный рост
проводимостей. Ход полярных проводимостей хорошо коррелирует с
ходом интегральной концентрации аэрозолей, которая имеет мак-
максимум при относительной влажности 60%, а затем падает. Во вто-
втором случае интегральная концентрация аэрозольных частиц прак-
тическа не менялась,А+ и А_так же,как А+/А_,изменяются не-
незначительно, причем А+/Д_ имеет обратный ход, т.е. А+умень-
иается сильнее, чем А_. Следовательно» уменьшение полярных
проводимостей происходило в основном в результате осаждения
ионов на аэрозольных частицах. Влияние на проводимость эффек-
эффекта перестройки спектра ионов по подвижности незначительно.
Структурные измерения показали, что наблюдался переход
мелкодисперсной фракции аэрозолей с d < 0,5 мкм во фракцию с
d ">. 0,5 мкм (см. табл.36). Причем поскольку в начальном сос-
состоянии отмечалось заметное отклонение от юнговского распреде-
распределения и счетная концентрация частиц была заметно выше, чем в
чистой атмосфере, радиус частиц увеличивался в довольно широ-
широком диапазоне. Эти результаты можно объяснить только с точки
зрения коагуляционной гипотезы.
Опыты однозначно показали, что увеличение относительной
влажности от 30 до 60-705? приводит к уменьшению полярных прово-
Дгмостей воздуха и увеличению интегральной поверхности частиц,
при бо'льших значениях влажности наблюдается рост полярных про-
водимостей и уменьшение интегральной поверхности аэрозолей. Ука-
Указанный характер изменения поверхности частиц подтверждает пред-
предположение о том, что наличие заряда на них облегчает адсорбцию
водяного пара, в результате чего наблюдается рост интегральной
поверхности аэрозоля.
313
Глава 7
АЭРОЗОЛЬНЫЕ МОДЕЛИ АТМОСФЕРЫ
Чрезвычайное многообразие процессов, регулирующих содер-
содержание и структуру аэрозольных частиц в атмосфере, и весьма из-
изменчивая их природа приводит к значительным пространственно-
временным вариациям аэрозольного компонента атмосферы.Поэтому
описание структуры атмосферных аэрозолей проще строить, исполь-
используя набор моделей для характерных условий.
Для решения многих теоретических и прикладных задач тре-
требуются весьма ограниченные сведения о структуре или характерис-
характеристиках аэрозолей - необходима только "частная" аэрозольная мо-
модель, например модель вертикального профиля коэффициентов аэ-
аэрозольного ослабления радиации для нескольких длин волн.
Пространственные масштабы аэрозольных моделей также раз-
различны. Например, для расчета переноса радиации в атмосфере не-
необходимы глобальные модели, а для задач, связанных с предотвра-
предотвращением вредных последствий аэрозольных загрязнений, - локаль-
локальные прогностические модели. В последнем случае необходимо зна-
знание динамики изменения аэрозольных характеристик в атмосфере.
Чаще всего наиболее реально получить какие-либо прямые
или косвенные характеристики, несущие информацию о структуре
аэрозолей. По этим, а также некоторым данным об условиях фор-
формирования аэрозольной структуры требуется восстановить дру-
другие аэрозольные параметры.
Полные аэрозольные модели могут быть построены на основе
знания процессов поступления аэрозольных частиц в атмосферу и
трансформации их в период пребывания и выведения из нее.
Ранее были рассмотрены основные процессы, регулирующие аэ-
аэрозольную структуру атмосферы: коагуляция, конденсация, диффу-
диффузия и седиментация частиц, конвекция и адвекция воздушных масс,
вымывание частиц облаками и осадками, осавдение на различных
препятствиях, Фотохимические и нефотохимические реакции образо-
образования аэрозольного вещества. Роль этих процессов в различных ус-
условиях существенно меняется. Для каждого из процессов можно на-
написать выражение потока частиц в секунда на единичный объем
воздуха и уравнение баланса всех потоков для стационарного слу-
случая или же уравнение изменения содержания частиц во времени для
нестационарного случая. Однако получающиеся интегродифференци-
альные уравнения в общем виде не решаются, даже если известны
все параметры различных процессов в атмосфере и граничные ус-
условия. Необходимы определенные упрощения этих уравнений и пред-
предположения о характеристиках процессов, регулирующих структуру
аэрозолей.
Существующие аэрозольные модели для разных слоев и для
всей атмосферы созданы в предположении стационарности процес-
процессов поступления и выведения аэрозолей из атмосферы, а также
равномерной распределенности частиц в горизонтальных направле-
направлениях. Обычно предполагается, что все аэрозольные частицы пред-
представляют собой сферы с равномерно перемешанным веществом. Коа-
Коагуляция приводит к образованию сфер с объемом, равным объему
коагулировавших частиц. Даже такая упрощенная одномерная зада-
задача вертикального распределения аэрозолей в атмосфере чрезвычай-
чрезвычайно сложна, особенно из-за недостаточного знания констант раз-
различных процессов. Эту задачу можно разбить на три: 1) созда-
создание моделей химического состава аэрозолей на разных высотах,
2) создание моделей вертикального профиля счетной или массовой
концентрации аэрозолей, 3) создание моделей распределения час-
частиц по размерам на разных высотах. Такой подход фактически пред-
предполагает дополнительные упрощения: слабую зависимость спектра
оазмеров частиц от химического состава и счетной концентрации
314
315
и слабую зависимость процессов выведения аэрозолей из атмосфе-
атмосферы от их химического состава.
Способ решения одномерной задачи вертикального распределе-
распределения аэрозольных частиц, заключающийся в разделении атмосферы на
ряд слоев, для которых уравнения, описывающие поведение аэро-
аэрозольных частиц, упрощаются в результате оценки роли различных
процессов, предлагался автором в его ранних работах. Некоторые
наиболее общие представления о закономерностях вертикальной
структуры аэрозолей этот метод позволяет получить, по крайней
мере для верхних слоев атмосферы. 'В отдельных случаях задача
построения вертикального профиля аэрозолей еще более упрощает-
упрощается, если аэрозольные частицы определенного типа не взаимодей-
взаимодействуют с другими частицами.
Задачу определения глобальной структуры атмосферных аэро-
аэрозолей можно сформулировать несколько по-иному. Если предполо-
предположить, что рассматривается задача распределения массы дисперги-
диспергированного вещества в атмосфере, то коагуляция и конденсация вли-
влияют на ее распределение лишь через изменение характеристик про-
процессов седиментации и вымывания частиц из атмосферы. (Водный
компонент аэрозолей в этой задаче не учитывается.) Одномерная
задача определения вертикального профиля становится достаточно
простой. Проблема заключается в определении коэффициентов тур-
турбулентной диффузии и функции распределения частиц по размерам
для генерируемых аэрозолей. Последняя с течением времени будет
изменяться в зависимости от начальной концентрации частиц,
времени и относительной влажности. Эти изменения можно модели-
моделировать с помощью численных расчетов на ЭВМ. Существенным обсто-
обстоятельством, облегчающим решение задач пространственной структу-
структуры аэрозолей, является резкое различие в физических условиях
существования частиц в тропосфере и стратосфере.
Наличие большого статистического экспериментального мате-
материала позволяет строить "частные" статистические и прогности-
прогностические аэрозольные модели. В связи с развитием дистанционных
методов определения аэрозольного компонента атмосферы, в пер-
первую очередь лидарных измерений, частные статистические аэро-
аэрозольные модели находят в настоящее время все большее примене-
применение. Прогностические модели создаются главным образом для опре-
316
деленных районов Земли и позволяют получить некоторые характе-
характеристики аэрозолей в приземном слое атмосферы [10, 58, 91].
Эмпирический подход к построению аэрозольных моделей осно-
основан на их создании по экспериментальным данным с учетом физи-
физической картины формирования структуры аэрозолей в атмосфере.
3 отличие от чисто статистического подхода такой путь построе-
построения моделей позволяет более точно передать характерные особен-
особенности аэрозольной структуры и использовать все имеющиеся экс-
экспериментальные данные, полученные разными методами.
С помощью микрофизических методов в основном определяются
счетная и массовая концентрации аэрозолей, а оптическими мето-
методами - коэффициенты ослабления, рассеяния и обратного рассея-
рассеяния. Оптических измерений проведено значительно больше, однако
переход от результатов этих измерений к основным аэрозольным
характеристикам весьма сложен и неоднозначен.
Итак, существуют три основных направления построения моде-
моделей атмосферных аэрозолей: 1) статистический набор и анализ ре-
результатов экспериментальных измерений какой-либо аэрозольной
характеристики, 2) создание физической картины формирования аэ-
аэрозольной структуры с использованием различных упрощений,
3) комплексный подход к созданию аэрозольной структуры на ос-
основе экспериментальных данных и теоретических представлений о
физике ее формирования.
Аэрозольный фактор в различных расчетах может быть учтен
с помощью использования определенных модельных представлений об
атмосферных аэрозолях, которые в зависимости от поставленных
задач, наших априорных знаний и мощности используемого для рас-
расчетов вычислительного аппарата могут быть более или менее слож-
сложными и приближенными к реальной картине.
Основными качествами аэрозольных моделей должны быть:дос-
быть:достоверность, простота, физичность и ее универсальность. В иде-
идеальном случае аэрозольные модели должны получаться одна из дру-
другой при разных уровнях точности, физической и математической
строгости описания аэрозольных характеристик. Универсальная
модель получается естественным путем, если можно решить систе-
систему уравнений, описывающих достаточно точно процессы возникно-
возникновения, трансформации и вымывания из атмосферы аэрозольных час-
317
тиц. Определенные попытки в этом направлении делались рядом
специалистов. Однако трудности, возникающие на этом пути, до
конца не преодолены, в частности из-за недостаточности экспери-
экспериментальных данных о физико-химических процессах в атмосферных
аэрозолях. В то же время имеющиеся экспериментальные данные о
их структуре позволяют строить эмпирические аэрозольные модели.
§ 7.1. Основные модельные характеристики
атмосферных аэрозолей
Наиболее важным характеристикам аэрозольных моделей явля-
являются: пространственная структура поля концентрации аэрозолей
т (г,х,у) или N(riX.y) распределение частиц по разме-
размерам л (г); химический состав частиц, определяющий в значитель-
значительной степени физико-химические свойства аэрозолей, в частности
плотность вещества частиц о ; форма частиц; для оптических рас-
расчетов - комплексный показатель преломления т = п -iэе ; изме-
изменение указанных характеристик во времени и в зависимости от ме-
метеорологических факторов.Одно из первых допущений .используемых
при создании аэрозольных моделей, - это предположение,что все
аэрозольные частицы имеют сферическую форму.Дальнейшими упроще-
упрощениями модельных представлений являются предположения равно-
равномерной распределенности химических соединеиий в аэрозолях оп-
определенного типа и в веществе аэрозольных частиц разных раз-
размеров, о горизонтальной однородности структуры атмосферных аэ-
аэрозолей на достаточно больших расстояниях (масштаб однороднос-
однородности может изменяться от нескольких сотен метров до тысяч кило-
километров).
Трудности, связанные с изменением распределения частиц по
размерам во времени и пространстве, в какой-то мере можно обой-
обойти, вводя набор разных типов аэрозолей, имеющих определенные
или закономерно изменяющиеся характеристики распределения. В
частности,удобно ввести обозначение N(r, x,y, z ) = N(x,y,
z)rii(T), где N(x,y,z) - счетная концентрация аэрозольных
частиц всех размеров, а щ<г)- плотность распределения частиц
по размерам,
318
= N(x,u,z),
= 1.
^ведя предположение независимого поведения разных типов аэро-
аэрозолей, получим
0
где индекс г определяет набор типов аэрозолей, которые исполь-
используются в построении данной модели; Я0 и i° - широта и долго-
долгота места применимости модели; i - время; /. - метеорологичес-
метеорологические факторы, определяющие изменчивость аэрозольной структуры.
Каадому индексу i соответствуют определенные значения
зависящие не только от химического состава постоянных компонен-
компонентов вещества частиц, но и от относительной влажности f0. В бо-
более общем виде наблвдается зависимость т не только от относи-
относительной влажности, но и от парциальных давлений других конден-
конденсирующихся компонентов атмосферного воздуха ( H2SO4, HNO3 ,
органических веществ). Важно также определить зависимость функ-
функции распределения частиц по размерам от высоты и относительной
влажности.
Частные модели отдельных аэрозольных характеристик даны,
например, в работах [ 79, 80, 211],
Вертикальный профиль концентрации. В большинстве работ ос-
основной вид зависимости концентрации аэрозольных частиц от высо-
высоты выражается формулой типа
Ni(z> -
G.2)
где zoi - высота образования или накопления аэрозолей; Н( -
полутолщина слоя, на которой, концентрация частиц уменьшается в
е раз; Я^ зависит от природы аэрозолей и условий их существо-
существования (температурной стратификации, турбулентного режима атмос-
атмосферы, скорости вымывания частиц). В более общем виде
для больших разностей высот необходимо вводить экспонен-
экспоненциальный множитель, учитывающий влияние изменения плотности
319
воздуха на концентрацию аэрозольных частиц. (Основные механиз-
механизмы переноса частиц связаны с переносом воздушных масс, а не ин-
индивидуальных аэрозольных частиц.) Однако в силу малой толщи
аэрозольных слоев (малых Hrf, изменчивости их структуры и не-
недостаточности наших знаний ос этой структуре обычно не вводит-
вводится дополнительный множитель в формулу для вертикальной струк-
структуры слоя.
Выражения для Яг или cp.(z) могут быть различными в зави-
зависимости от того, какое дифференциальное уравнение наиболее точ-
точно описывает физические процессы переноса аэрозольных частиц в
атмосфере в рассматриваемом случае. В решении дифференциально-
дифференциального уравнения Dfr^ + (ir$± w)N = 0, учитывающего лишь турбу-
турбулентную диффузию D, скорость седиментации vs и восходящих
(нисходящих) воздушных потоков w, полуширина слоя Н{= (vs +
+ \a)ID, а в решении уравнения В^Ч -ot,N = O, учитывающего
лишь диффузию и вымывание частиц из атмосферы *, Я^ получа-
получается равным (D/ <*) */2, где ct - коэффициент вымывания час-
частиц. Если основными факторами, определяющими вертикальную струк-
структуру аэрозоли, являются диффузия и коагуляция C > то уравне-
уравнение, описывающее вертикальный профиль аэрозолей, приводится к
виду
6D
Решение будет
N<z) =
Следует отметить, что эти формулы получают при очень боль-
них упрощениях: постоянстве коэффициентов диффузии, вымывания,
коагуляции, скоростей седиментации и воздушных потоков, а так-
также при предположениях, что частицы не очень сильно отличаются
по своим физико-химическим свойствам и что процессы распреде-
распределения частиц в атмосфере стационары. Очевидно, что все эти
предположения и упрощения далеки от реальности. Однако такие
упрощенные выражения могут использоваться для создания прибли-
приближенных моделей вертикального профиля аэрозольных частиц и про-
320
верки роли различных фактрров в формировании вертикальной струк-
структуры аэрозолей. Предполагая экспоненциальное уменьшение кон-
концентрации частиц, можно определить Hi иэ экспериментальных
данных по значениям .У(О) и NB^. Тогда Hi = z1/(lnN0-lnN1)
и N<Z) = | }
В гл.4 обсуждались экспериментальные данные по вертикаль-
вертикальному профилю счетной концентрации частиц. Отмечалось, что поч-
почти всегда наблюдаются слои с повышенной концентрацией вблизи
изотермий и инверсий (см. рис.20). Такого рода слоистые профи-
профили можно получить либо решая уравнения переноса аэрозолей с
восходящими и нисходящими потоками, либо поместив в определен-
определенные слои атмосферы источники аэрозольных частиц (химические ре-
реакции). Для описания таких слоев удобно использовать выражение
G.3)
где z0 - высота максимума аэрозольного слоя, смещающегося под
влиянием происходящих в атмосфере физических процессов, а па-
параметр Л определяется интенсивностью выбывания частиц из ат-
атмосферы (к = 5+12).
Функция распределения частиц по размерам. Большинство экс-
экспериментаторов отдают предпочтение описанию функции распределе-
распределения по формуле Юнге как наиболее простой и позволяющей быстро
делать различного рода оценки. Но формула Юнге не дает реаль-
реальной картины распределения частиц по размерам для г 4 г» 4
4 ОД мкм. Кроме того, практически всегда (а для -9^4 обя-
обязательно) необходимо ограничивать диапазон изменения размеров
частиц сверху. Обычно принимается J^ax = 10 мкм ['110]. Однако
измерения показывают, что в атмосфере витают частицы и больших
размеров. При описании их распределения по размерам формулой
Юнге необходимо увеличить показатель степени -9 до 5-6 [84].
В теоретических расчетах оптических характеристик аэрозо-
аэрозолей распределение частиц по размерам наиболее часто онисывает-
ся гамма-распределением, аналитические свойства которого хоро-
хорошо изучены.
Экспоненциальный спад кривой $ -распределения в области
р> rQ не отвечает имеющимся представлениям о динамике пере-
21.
321
хода в эту область более мелких частиц (коагуляционный и кон-
конденсационный рост частиц) и их удаления из атмосферы (вымыва-
(вымывание, осаждение на препятствиях, оседание), которые больше оп-
оправдывают степенной характер уменьшения концентрации частиц с
размером, наблвдаемый экспериментально [38, 87]. Физическим
представлениям не отвечает также степенной характер уменьшения
концентрации частиц с юс размером в субмикронной области, так
как, по крайней мере для конденсационного возникновения частиц
наблюдается экспоненциальная зависимость числа возникающих чао
тиц от размеров зародыша [l].
Условиям физической обоснованности удовлетворяет аналити-
аналитическое выражение D.7) для распределения частиц по размерам,
предлагающееся в работе [87]. Для нормированного распределена
D.7) коэффициент а выражается следующим образом:
Априорное исследование пределов применимости функции D.7
для описания распределения частиц по размерам может существен-
существенно ограничить область поисков параметров этого распределения
для любого реального случая. Остановимся на физических факто-
факторах ограничений. Если мы будем рассматривать распределение ча
тиц по размерам в области г > rQ , то для распределения, ха-
характеризующегося одним источником, необходимо dmjdr- А,г"ъ
< ei 0), иначе область, где dmjdr имеет максимум, будет
характеризовать новый источник частиц. Следовательно, на нисш
дающей ветви распределения вдали от максимума ¦? ^3+е . Боле<
того, если имеется постоянный стационарный поток вещества, not
тупающий из мелкодисперсной фракции в крупнодисперсную (беско-
(бесконечно долго действующий), то должно выполняться условие
г dr - Br~ei ( e.. i. 0 ) ; тогда -о ^ 4+е, (вели-
V
0
чины ? и ?j определяются характером вымывания крупнодисперФ
ной фракции из атмосферы). Верхний предел * по имеющимся эко
перименвальным данным не превышает 10-12, даже для неустанови
шегося процесса. Нижний предел модального радиуса аэрозольных
частиц можно положить равным 0,01-0,02 мкм, так как время су
ществования частиц меньших размеров в атмосферных условиях ни
тожно мало. Верхний предел не превышает 10 мкм даже в условия
пыльных бурь.
322
гтах ^ 5°I//J го
из математических свойств функции D.7) следует, что не
имеет смысла пользоваться значениями параметра тга»^.>25. Тогда
fe?L * °'02' а- *me< 50 и« следовательно, .
5°I//J го ' а rmin * 5O'^S *o • ПРОСМОТР значений функ-
функций, являющихся интегралами нормированной функции, от х i до
хж для разных 5 показывает, что при s < 0,5 значения интег-
интегралов, не превышают 0,7 для любых реальных зстал, т.е. функция
D.7) при s < 0,5 не может описать сколько-нибудь удовлетво-
удовлетворительно любые экспериментальные данные (больше трети частиц ле-
лежит вне области, описываемой функцией распределения). Следова-
Следовательно, реально нижняя граница s соответствует 5^п= 0,5.При
этом * = 6+7, а т = 3+3,5. Для s>i значения -9, которые
можно использовать для описания реальных распределений, возрас-
возрастают. Максимальное значение 5, которое можно использовать для
описания распределения частиц по размерам, Sj^^ 4 5. При тако-
такого рода распределениях диапазон изменения размеров всех частиц
гтох" 2'5го* гтт~ °>4 Д™ 5 = 5. Для s < I диапазон раз-
размеров частиц, описываемых аналитической функцией, наоборот,зна-
наоборот,значительно шире: при s = 0,5 г^^ 2500г , а г1гЛпаг 4«10~4г..
Таким образом, область значений т^т-1, необходимая для опи-
описания реальных распределений, ограничена: 0,5 4 wij 4 5, as
изменяется в диапазоне 0,5 4. s 4 2.
Подбор функции п(г) проводится по экспериментальным дан-
данным как дифференциальных, так и интегральных счетчиков частиц.
Экспериментальные дифференциальные кривые распределения частиц
по размерам могут быть существенно искажены по отношению к ре-
реальным распределениям в силу ряда технических и методических
причин. Интегральные (кумулятивные) кривые распределения надеж-
5,00
rn(r)dr для •?-функ-
•?-функций определяются выражениями
JVr« tfoj[(jns+f)/s; brs],
Nr = Nol[(ms-l)/s;mros/rs] .
Для нахождения параметров функции распределения использу-
используются также измерения характеристик, отражающих второй и третий
моменты распределения функции п(г): коэффициент аэрозольного
323
ослабления (рассеяния) радиации б, км (б-МB)) и массо-
массовая концентрация частиц р, мкм/м (р - МC)). Это, вообще гово-
говоря, весьма проблематично, так как 6 является функцией не толь-
только относительного размера частиц о = 2тсг/Х , но и комплексно-
комплексного показателя преломления вещества аэрозольных частиц т, а мас-
массовая концентрация частиц р определяется в основном присутст-
присутствием в атмосфере гигантских частиц, содержание которых может
сильно флуктуировать и не подчиняться найденным оптимальным фун-
функциям распределения частиц по размерам.
При создании модели функции распределения частиц по разме-
размерам представляют интерес закономерности изменения этой функции
от времени и от параметров среды. На первом этапе разумно пред-
предположить, что два процесса влияют на функцию распределения час-
частиц по размерам: коагуляция как фактор, определяющий изменение
п(г) во времени, и конденсация как фактор, определяющий влия-
влияние среды на изменение п(г)
n<r,t,f) = f(no<r),r<i,fo)).
G.4)
Наиболее простое предположение - стационарная коагуляция изме-
изменяет функцию распределения: сдвигает модальный радиус частиц,
не меняя общего вида функции. Тогда можно оценить изменение rfl
от времени. По М.Смолуховскому для монодисперсных аэрозолей
1
G.5)
Для полидисперсных аэрозолей с одномодальным распределением мож-
можно предположить, что вид зависимости изменения общей концентра-
концентрации частиц от времени примерно такой же. Вместо К^ необходимо
использовать эффективный коэффициент коагуляции, зависящий от
модального радиуса rQ(t) и коэффициента диффузии D :
Nil)
отсюда выражение для
G,5')
G.6)
324
где Cj - поправочный множитель, определяемый отношением реаль-
реального коэффициента диффузии к коэффициенту броуновской диффузии,
а также увеличением частоты столкновения частиц за счет других
факторов, например несферичности частиц и сил электростатичес-
электростатического взаимодействия.'
Столь простое выражение изменения rQ(i) связано с пред-
предположениями о том, что i достаточно велико (l^Zr^J{SkTClN()))t
константа коагуляции слабо зависит от размера частицы, а вид
функции п(г) таков, что ro<t)/r0(O) = ij(i)/r3<0> (масса
аэрозольного вещества постоянна).
Наибольшие ошибки при использовании выражения G.6) будут
в случае, когда начальная функция распределения не яЕ\дется од-
номодовой, а концентрация частиц вблизи второй моды не очень
сильно отличается от концентрации частиц вблизи первого макси-
максимума распределения. В этом случае частицы вблизи максимума г¦
будут преимущественно коагулировать с частицами вблизи второго
максимума г , и первый максимум исчезнет.
Предложенная концепция коагуляционного роста частиц позво-
позволяет провести грубые оценки общей концентрации и модального ра-
радиуса аэрозольных частиц при разных мощностях источника (раз-
(разной массовой концентрации аэрозолей) в зависимости от времени
их существования. Так, за половину суток концентрация частиц
уменьшится до (9,0*5,0)»104 см, а за месяц до 15С-5-ТОО см~^.
Модальный радиус таких частиц за 12 ч достигнет ~ 0,03 мкм при
тп — 10 мкг/м и 0,05 мкм при т. — 50 мкг/м°, а за месяц -
С,12 мкм в первом случае и 0,20 мкм во втором. Лишь при очень
большой интенсивности загрязнения атмосферы может наблюдаться
переход модального радиуса в область значений, больших единице
(примерно за одни сутки).
В реальной атмосфере, особенно в дневное 'время суток, оче-
очевидно, будет возникать аэрозольное вещество в результате хими-
химических реакций и диспергирования вещества с разными поверхнос-
поверхностями. В этом случае решение уравнения коагуляции существенно
усложняется. Однако прямые микрофлзические исследования аэро-
аэрозолей в нижних слоях атмосферы свидетельствуют о том, что гомо-
гомогенное образование диспергированной фазы аэрозолей несуществен-
несущественно влияет на их структуру при наличии в этих слоях пылевых час-
325
тиц. Наблюдается в основном рост частиц в результате гетероген-
гетерогенных реакций. Например, при измерениях в приземном слое в райо-
районе Рыльска наблюдалось увеличение размеров отдельных частиц в
течение дня с г - ОД до г* 0,7+0,8 мкм, по-видимому, в ре-
результате фотокаталитических реакций. В предположении реакции
первого рода константа скорости реакции оказывается порядка
10~4 с . Однако неизвестно в настоящее время, на какой доле
пылевых частиц вдет гетерогенная реакция образования аэрозоль-
аэрозольного вещества.
Генерация дисперсионных аэрозолей в атмосферу наблюдается
с момента восхода солнца и до полудня и зависит от ряда факто-
факторов, в первую очередь от типа подстилающей поверхности. Замет-
Заметное изменение функции распределения частиц этого типа будет про-
происходить в вечерние и ночные часы в результате седиментации
крупных частиц.
В реальной атмосфере изменение размеров частиц при конден-
конденсационном росте происходит значительно быстрей, чем в случае
коагуляции. При этом деформируется также и функция распределе-
распределения частиц по размерам. Относительное изменение размеров частиц
с ростом влажности зависит как от их радиуса, так и от их хими-
химического состава.
В ряде случаев можно считать, что в первом приближении от-
отношение г jr. не меняется, тогда функция п(г/гв) не изменится.
Для этого необходимо, чтобы выполнялось соотношение вида
G.7)
где г - размер сухой частицы, а гу - размер частицы при
f Н ФК ф
влажности f. Например, Ф.Кастен предлагает формулу
G.7')
где б0 изменяется от 0,18 до 0,255 в зависимости от типа аэро-
аэрозолей. В атмосфере наблюдается обычно влияние влажности не толь-
только на конденсационный рост аэрозольных частиц со смешанным
химическим составом, но и на фотохимические процессы образова-
образования и роста частиц, а также на их коагуляцию. Кроме того, изме-
изменение влажности часто коррелирует с движениями воздушных масс,
326
не обязательно содержащих аэрозоли одного типа и одинаковой кон-
концентрации. В силу этого для модальных представлений можно огра-
ограничиться использованием эмпирической формулы G.7'), по кото-
которой определяется изменение ro(fo)-
Изменение г. влечет за собой изменение наклона ниспадаю-
ниспадающей ветви кривой распределения в фиксированном диапазоне разме-
размеров частиц, т.е. экспериментально определяемой \>экс » несколь-
несколько меньшей значения -9 в формуле D.7). Разница между этими ве-
величинами будет тем значительней, чем ближе г. к измеряемому
диапазону размеров частиц. При конденсационном росте частиц,
описываемом формулой G.7'), не изменяется выражение функции
распределения вида D.7). Тогда функцию распределения частиц
по размерам, изменяющуюся в зависимости от коагуляции и конден-
конденсации, можно записать в виде
n(r,i,fB) =
еэср
.G.8)
Химический состав частиц. За достаточно длительное вре-
время существования аэрозольных частиц в результате коагуляции и
конденсации их вещество частично перемешивается. Наиболее пол-
полное перемешивание происходит в мелкодисперсной фракции и вда-
вдали от мощных источников частиц определенного химического сос-
состава. Поэтому можно ожидать, что в тропосфере частицы доволь-
довольно однородны по химическому составу и наиболее близки к моде-
модели, созданной по данным о мощности и химическом составе основ-
основных источников аэрозолей. Химический состав тропосферных аэро-
аэрозолей не должен полностью соответствовать этим данным, так как
не все аэрозольное вещество попадает в тропосферу и время жиз-
жизни частиц разного происхождения (а значит, и разной дисперснос-
дисперсности) неодинаково. Можно считать, что в верхнюю тропосферу не по-
попадают морские соли. Первоначальным продуктом конверсии сер-
сернистого газа в стратосфере является серная кислота. Однако при-
присутствие твердых аэрозольных частиц и окислов азота приводит
к определенному изменению состава стратосферных аэрозолей.
327
В нижней тропосфере и приземном слое атмосферы химический
состав частиц определяется в первую очередь ближайшими мощными
их источниками.Основными глобальными типами аэрозолей в ниж-
нижних слоях атмосферы будут: 1) морские, 2) континентальные (дис-
(дисперсионные), 3) городские (дисперсионного происхождения), 4)фо-
4)фотохимические (конденсационные).
Следует отметить, что так как аэрозоли разного химическо-
химического состава имею.т разную дисперсность, то даже при их перемеши-
перемешивании разные фракции будут иметь существенно разный химический
состав. В частности, в грубодисперсной фракции обнаруживается
значительной большая доля силикатов и других минералов, а вмел
кодисперсной преобладают продукты фотохимических реакций.
Комплексный показатель преломления вещества диспергирован-
диспергированной фазы. Комплексный показатель преломления вещества аэрозоль-
аэрозольных частиц получают разными способами. Например, Ф.Фольц опре-
определял in =n -Ы для проб аэрозолей, взятых в разных райо-
районах земного шара, с помощью известного метода КВг-техники.
Этот показатель определяется также из измерений оптических ха-
характеристик атмосферных аэрозолей, т.е. решается обратная зада
ча оптики аэрозолей при некоторых допущениях о виде функции рас
пределения частиц по размерам. Оценки показателя преломления
вещества аэрозольных частиц делались по данным об оптических
константах его главных составляющих [34, 47, 48].
Основным фактором изменения комплексного показателя пре-
преломления для разных типов аэрозолей является относительная вла
жность. Влияние влажности на изменение показателя преломления
определяется увеличением содержания воды в веществе частиц.
Предполагая, что вода не покрывает частицу сплошной оболоч-
оболочкой, а равномерно распределяется в ее веществе* можно использо
вать соотношение для определения реальной чаоти показателя пре
ломления вещества смешанного состава
Л ^~1 Л
71 = у ^i^i >
где с^ - объемная концентрация i-го компонента смеси. Исполь
зование такого же соотношения для мнимой части показателя пре-
преломления менее оправдано.
328
Определение доли воды в веществе частиц для разных влаж-
ностей и типов аэрозолей можно задать таблично по эксперимен-
экспериментальным данным , либо используя формулы типа G.7'):
(предполагается, что относительная доля воды в веществе каждой
частицы при данной влажности одинакова). В общем случае послед-
последние равенства несправедливы по следующей причине: структура оу-
хой частицы может быть рыхлой и первоначальная конденсация при-
приводит к уплотнению частицы и заполнению микрокапиллярных проме-
промежутков конденоирующейоя воды. Однако при влажностях больше 70-
80% эти процессы должны закончиться и дальнейший рост частиц
будет прояоходить приблизительно по уравнению G.7 ).
Следует отметить, что комплексный показатель преломления
вещеотва для сухих чаотиц может быть меньше эффективного пока-
показателя преломления из-за их рыхлости.
§ 7.2. Статистические оптичеокие модели
атмосферных аэрозолей
Большой цикл исследований оптических характеристик аэро-
аэрозолей в приземном слое атмосферы позволил Г.В.Розенбергу и
Г.И.Горчакову предложить классификацию типов оптической пого-
погоды [80]. Было выделено четыре основных типа: дымка, туманная
дымка, дымка с изморосью, туман. Для каждого типа оптической
погоды были определены основные особеннооти компонент матрицы
рассеяния в видимой области спектра. В дальнейшем было показа-
показано, что опектральный ход коэффициентов аэрозольного ослабления
радиации в видимой и ближней ИК-областях спектра также сущест-
существенно определяется типом оптической погоды. Эти выводы были
сделаны на основании статистической обработки результатов наб-
наблюдений оптических свойств атмосферных аэрозолей в приземном
слое в районе Звенигорода для условий достаточно чистой атмос-
атмосферы.
329
Дальнейшее развитие экспериментальных исследований опти-
оптических характеристик аэрозолей привело к появлению нескольких
отличающихся друг от друга точек зрения на создание оптических
моделей аэрозолей приземного слоя атмосферы: 1) важнейшим фак-
фактором формирования оптических свойств аэрозолей является тип
воздушной массы, в которой производятся оптические измерения;
2) существует тесная корреляция между коэффициентом ослабления
яри г =0,55 мкм и оптическими характеристиками аэрозолей в
широкой области спектра; 3) оптические свойства аэрозолей оп-
определяются в основном относительной влажностью.
Первый подход к моделированию оптических характеристик аэ-
аэрозолей, фактически к их прогнозированию, требует учета пред-
предыстории образования аэрозолей и набора большого статистическо-
статистического материала по юс оптическим характеристикам для различных воз-
воздушных масс. Схема прогноза оптических характеристик аэрозолей
получается сложной и недостаточно определенной. Концепция,выд-
Концепция,выдвинутая Г.В.Розенбергом и его сотрудниками, основана на утверж-
утверждении, что оптические овойства аэрозолей определяются в основ-
основном концентрацией мелкодисперсной фракции, изменчивость содер-
содержания которой определяет значение коэффициента аэрозольного ос-
ослабления при Я =0,55 мкм или метеорологическую дальность ви-
видимости в горизонтальном направлении.
Результаты исследований [19, 37, 40-42] позволяют сделать
вывод, что вклад грубодисперсной фракции в оптические характе-
характеристики атмосферных аэрозолей значителен. Но поскольку процес-
процессы формирования аэрозольной структуры довольно длительны и пос-
постоянны, они приводят к относительно стабильным распределениям
частиц как по размерам, так и по химическому составу, характер-
характерным для определенного слоя атмосферы. Наиболее сильным и быст-
быстродействующим фактором изменения аэрозольной структуры являет-
является относительная влажность воздуха, ответственная за содержа-
содержание воды в составе аэрозольных частиц (рис.32). Специфический
Рио.32. Влияние относительной влажности на комплексный
показатель преломления синтетической модели атмосферных аэ-
аэрозолей.
о - реальная часть, 6 - мнимая часть: 1 - / < 40%;
2 -/ = 40+65%; 3 - f = 65*75%: 4 - / = 75+82$;
S — J' = 82+92%.
330
n .
0,4 1
331
спектральный ход показателя преломления воды и изменение спект
ра размеров частиц при их конденсационном росте определяют в
значительной степени вариации спектрального хода коэффициентов
аэрозольного рассеяния, поглощения и ослабления радиации, а
также другие оптические свойства атмосферных аэрозолей.
Конечно, все упомянутые подходы являются слишком упрощен-
упрощенными. Наиболее оптимальным является сочетание двух последних
уточненных и дополненных концепций с учетом фактора истории об-
образования аэрозольной структуры.
Прогнозирование оптических свойств приземного слоя атмос-
атмосферы. Известно, что метеорологическая дальность видимости SM
в дьшках при фиксированной температуре Г коррелирует с абсолта
ной влажностью е, а коэф|ициент асимметрии рассеянного свето-
светового потока Ка. - с метеорологической дальностью видимости $м.
По величине Кл выделено десять классов нормированных индикат-
индикатрис, представленных в табличной форме
d&
Л1
я/2
Используя вероятностные зависимости, можно по значениям темпе-
температуры и абсолютной влажности восстанавливать значение 5М или
коэффициента аэрозольного ослабления для А =0,55 мкм и ожи-
ожидаемый класс атмосферных индикатрис рассеяния для данного мес-
места наблюдений. Так, плотность распределения 5М в приземном
слое атмосферы хорошо описывается эмпирическим выражением
где параметры S=aSM, SM= S/<q-l), C(SM) = ?/((<?-1 J
S, Ц ,a - определяются из экспериментальных данных для
наблюдения, причем для дымок
места
3,6 J^ + 600
° ~ S}. + 600
2,6
$1 +600
Для коэффициента асимметрии плотность распределения описывает-
описывается нормально-логарифмическим распределением типа
332
[- (InА1, -
где Ь - стандартное отклонение 1пА"а. Для индикатрис пологого
типа
Ъ = ( 0,9ЛГа2^ - 1 )/Км , К3„ = 18 S~°>43 - 0,4 .
Вероятность появления индикатрисы класса,характеризуемого зна-
значением Ка от ЛГа1 до
рал
для данной s , получим, взяв интег-
интег= f
- <P((lnKn -
где Ф(аг) = у2/я i eocp (-i2j2)dt - удвоенная функция Лап-
Лапласа. Зная К- можно вычислить среднюю индикатрису рессеяния
J{Q) для заданного значения метеорологической видимости SM.
Для проведения большинства расчетов переноса излучения
пользуются не табличными данными ijF), а аналитическими вы-
выражениями индикатрис рассеяния. Существует несколько аппрокои-
мационных выражений для индикатрис рассеяния в атмосфере в ви-
виде полиномов и рядов с экспоненциальными членами. В частности,
в астрофизике пользуются индикатрисой Хеньи - Гринстейна:
где g - некоторый параметр. Это выражение очень удобно в том
отношении, что весьма просто разлагается по полиномам Лежандра:
Кроме того, формула Хеньи - Гринстейна довольно хорошо аппрок-
аппроксимирует реальные индикатрисы рассеяния.
Вертикальные профили спектральных коэффициентов аэрозоль-
аэрозольного рассеяния, обратного рассеяния и ослабления радиации. Ре-
Результаты спектральных аэростатных исследований, проведенных ка-
333
федрой физики атмосферы Ленинградского университета, в сочета-
сочетании с данными других советских исследователей привели к созда-
созданию модели аэрозольного ослабления до высоты 100 км (аэрозоль-
(аэрозольная модель ЛГУ-67). Коэффициенты аэрозольного ослабления в сц
тосфере по этой модели значительно превосходят те же коэффици-
енты в модели Л.Злтермана A964 г.), полученные по данным ми»
рофизических измерений Юнге. Однако наибольший массив данных i
0 10 20 30 40 50 60 70
Н,км
Рис.ЗЗ. Вертикальные профили аэрозоль-
аэрозольного коэффициента ослабления для Л =0,55 мкм.
/ - по Г.В.Розенбергу, В.Н.Терешковой;
2,3 - по Л.Элтерману (модели 1Э64 и соот-
соответственно 1968 гг.); 4- модель ЛГУ,1968г.;
5 - модель ИФА, 1980 г.; € - модель Л.С.Ив-
лева, 1973 г.
вертикальной структуре оптических характеристик атмосферных i
розолей получен с помощью лазерного зондирования атмосферы. ,
зерные измерения содержания аэрозолей в стратосфере позволил
обнаружить отчетливо проявляющиеся изменения вертикального щ
филя стратосферных аэрозолей в зависимости от изменения шире
334
ярко выраженную слоистость аэрозолей, быструю изменчивость вер-
вертикального профиля во времени, резкое уменьшение аэрозольных ко-
коэффициентов обратного рассеяния радиации на высотах более 25 км.
Вариацжи вертикальных профилей стратосферных аэрозолей в-
зависимости от широты связаны с изменением температурной стра-
стратификации атмосферы и ослаблением конвекции в высоких широтах,
поэтому, чем выше широта, тем менее резко выражен стратосферный
слой аэрозолей и тем ниже он расположен. Характерные вертикаль-
вертикальные профили аэрозолей по .оптическим измерения» представлены на
рис.33.
§ 7.3. Блочная аэрозольная модель
По месту возникновения аэрозолей разобьем их на следую- .
щие типы: 1) наземного происхождения; 2) тропосферного проис-
происхождения; 3) стратосферы и высоких слоев атмосферы. В качестве
функций распределения для описания спектра размеров частиц возь-
возьмем л<г) = ar'6exp(-6r0jr), так как численные эксперимен-
эксперименты показывают, что в области •**>** экспериментальные данные
достаточно хорошо описываются зтой функцией с помощью варьиро-
варьирования Гр- Вертикальный профиль аэрозолей для наземных источ-
источников будем описывать экспоненциальной функцией, а для внутри-
атмосферных - функцией N(z) = bz*eocpfAz/zo), причем Н-
г 2г^/к. Комплексные показатели преломления задаются таблично.
Зависимости п(Г) и Яг (Я) от относительной влажности описывают-
описываются эмпирическими формулами, рассмотренными в § 7.1.
Аэрозоли наземного происхождения. Можно выделить четыре
типа аэрозолей наземного происхождения: 1) конденсационные (про-
(продукты сгорания и фотохимических реакций, конденсации веществ с
малой упругостью паров и т.п.); 2) дисперсионные (продукты вы-
выветривания и разрушения почв, горных пород и т.п.); 3) морские;
4) промышленные, в основном грубодисперсные (золы и пыли), так
как мелкодисперсные входят в первый тип аэрозолей»
Конденсационные аэрозоли. Модаль-
Модальный радиус rQ лежит в области 0,02-0,10 мкм. Общая концентра-
концентрация частиц приблизительно соответствует общей концентрации час-
частиц, наблюдаемой в приземном слое атмосферы, т.е. порядка
10 -10 см . Для частиц с г ^ 0,1 мкм концентрации около
200 см"? для частиц с г * 0,15 мкм - 100-50 см, для час-
335
тиц с г %. 0,22 мкм - 5-20 см . Основная масса этих частиц
хорошо смачиваема, они растут при изменении относительной влаж-
влажности, начиная с 10-20%, и легко становятся ядрами конденсации
при образовании облаков и туманов. 3 силу этих причин Я не
должно превышать 2 км, т.е.
01
<O)e*p (-
иредполагаем, что распределение частиц по размерам из-за отно-
относительно небольшого времени существования их не меняется с вы-
высотой
ч-1 --6
О,ЗаГEРге»р(-0C/г),
Го = 0,05 мкм,
Х
= 6.
о
Химический состав конденсационных аэрозолей примерно оп-
определяется следующими значениями концентрации различных компо-
компонентов в процентах по массе: [$О4]~2- 25; [NH4]+1- 8; ^з*"
10; Ж)^1 -2; SiO2 - 15; F^O,- 4; Ге+3(соли) - 1;А12О3-
5; С - 1; Са+г- 10; СГ1- 8; Na+I- 8; K+I- 1; Mg+2- l;
HjO (связанная) - 5; Cu.Mn и др. - 1%.
Показатель преломления вещества частиц конденсационных аэ-
аэрозолей, рассчитанный по методике, изложенной в работех [47,
48j для«разных значений относительной влажности приводится в
табл.37. В области длин волн 0,4-0,9 мкм спектрального хода
Л (А) практически не наблвдается. В этой области длин волн ве-
величина «(Я) может заметно варьироваться, не влияя существен-
существенно на значения л(А), так как концентрация сажи - основного
поглотителя коротковолновой радиации - составляет всегда нич-
ничтожную долю от общей концентрации и сильно изменяется в зави-
зависимости от присутствия источников.
Дисперсионные аэрозоли. Модальный ра
диус г0 лежит в области от 0,5 до 10 мкм. Точнее можно ска-
сказать, что существует несколько модальных радиусов для частиц
разной природы. Экспериментальные данные показывают, что
главным модальным радиусом частиц этого типа следует считать
г0 = 1,5+2,0 мкм (наиболее мелкодисперсная часть дисперсион-
дисперсионных аэрозолей ведет себя как конденсационные аэрозоли и вхо-
336
СГ) CD
22 1922
8
1 1
СПГ__
~ «==¦-
— 4
i
- 1
J
Л
//
/
- >
/
- !
VI
\ 1
/ у
/
1 1
Комплексный
Л.мкм
0.5
0,7
1.0
1,6
2,0
2.4
2,8
3.0
4,0
4.6
5.4
6,0
7.0
8.5
9.0
10,0
11,0
В I3.o
to 15,0
Таблица 38
показатель преломления вещества аэрозолей дисперсионного происхоздения
40^
п
1,67
1,65
1,65
1,64'
1,644
1,64
1,640
1,608
1,646
1,637
1,612
1,567
1,515
1,611
1,186
1,650
2,140
1,750
1,790
1,770
эе
0,0065
0.005
0,005
0,0051
0,0076
0.010
0,014
0,044
0,076
0,018
0,018
0,018
0,045
0,434
0,600
1,240
0,126
0,118
0,296
0,315
п
1,54
1,54
1,539
1,538
1,532
1,524
1,507
1,45
1,528
1,52
1,485
1,420
1,300
1,439
1,050
1,178
2,185
1,777
1,820
1,750
0,0065
0,004
0,003
0,003
0,0035
0,004
0,003
0,061
0,101
0,014
0,016
0,013
0,046
0,404
0,571
0,973
0,267
0,130
0,345
0,345
S = 65+75#
п
1,467
1,467
1,466
1,464
1,458
1,448
1,428
1,337
1,457
1,461
1,435
1,392
1,297
1,400
1,162
1,222
1,782
1,547
1,597
1,587
эе
0,0050
0,003
0,0025
0,0025
0,0025
0,0025
0,0025
0,090
0,177
0,011
0,016
0,012
0,085
0,285
0,334
0,611
0,154
0,117
0,268
0,367
/ =
п
1,382
1,382
1,381
1,379
1,372
1,361
1,337
1,227
1,412
1,392
1,370
1,333
1,262
1,342
1,229
1,262
1,487
1,352
1,362
1,395
75*85#
0,0025
0,0017
0,0015
0,0015
0,0015
0,0015
0,0015
0.108
0,203
0,010
0,015
0,008
0,072
0,135
0,199
0,300
0,090
0,102
0,237
0.349
S =
Й
1.342
1,341
1.340
1,338
1,330
1,316
1,285
1,133
1,412
1,378
1,350
1,322
,272
,326
,252
,268
.284
,212
,212
.322
85+95#
эе
0,0008
0,0007
0,0006
0,0006
0,0006
0.0006
0,0006
0,147
0,281
0,008
0,010
0,007 ,
0,102
0,056
0,073
0,115
0,061
0.II2
0,271
0.4II
Промышленные пылевые аэрозоли
(грубодисперсные) - весьма изменчивый тип аэрозолей. Модальный
радиус наиболее вероятен в области rQ = 3+4 мкм. Общая концен-
концентрация достигает нескольких частиц в 1 см . Источник следует
считать приподнятым на 0,5 км. Аэрозоли выводятся из атмосферы
весьма интенсивно, Я 't 1 км, в воздухе наиболее крупных и
сильно загрязненных городов Я достигает 2 км.
Химический состав вещества аэрозолей сильно меняется. Су-
Существенно, что в составе частиц высока доля сильных поглотите-
поглотителей коротковолновой радиации. Показатель преломления для ве-
вещества частиц промышленных аэрозолей по экспериментальным из-
измерениям в Ленинграде представлен на рис.34-
Морские аэрозоли. Микроструктура и концент-
концентрация морских аэрозолей сильно зависит от солености морской во-
воды, температурного режима приводного слоя и степени волнения
морской поверхности, а также формируется в результате химичес-
химических реакций в воздухе. Наиболее вероятным модальным радиусом
частиц из морских солей следует полагать г. =0,2 мкм. Части-
Частицы, образующиеся в результате химических реакций, в основном
сульфатные с модальным радиусом г = 0,05 мкм. Морские аэро-
аэрозоли весьма гигроскопичны и вымывание их из атмосферы вдет
весьма интенсивно. Вертикальный профиль этих частиц соответ-
соответствует экспоненте с Н - 0,5+1 км. Химический состав морских
аэрозолей рассматривался в гл.1. Основными компонентами вещест-
вещества этих частиц являются хлористый натрий и сульфаты, причем
мелкодисперсная фракция состоит преимущественно из сульфатов,
среднедисперсная - из хлористого натрия. Доля последнего уве-
увеличивается с увеличением размеров частиц. Оба компонента явля-
являются весьма гигроскопичными соединениями, поэтому наблюдается
сильная зависимость комплексного показателя преломления от от-
относительной влажности, наиболее просто рассматриваемая по фор-
формуле Ф.Кастена.
Аэрозоли тропосферного происхождения. В тропосфере аэро-
аэрозольные частицы ооразуются в результате фотохимических реак-
реакций, однако условно можно отнести к аэрозолям тропосферного
происхождения также частицы дисперсионного происхождения, по-
попавшие в атмосферу в результате мощных выбросов (например,вул-
340
канических извержений), конвективных и адвективных потоков воз-
воздушных масс, сильно загрязненных пылью (пылепесчаных бурь).
Следует также выделить тип аэрозолей, возникших на мелкодиспер-
мелкодисперсных ядрах конденсации, длительное время существовавших в ат-
атмосфере (например, продукты вулканических извержений, попавшие
в тропопаузу).
Конденсационные (фотохимичес-
уие) аэрозоли. Высота образования этих аэрозолей мо-
может изменяться от 1,5 до 4 км, что определяется температурной
статификацией атмосферы и высотой выброса сернистого газа,
окислов азота и других газовых компонентов, участвующих в фо-
фотохимических реакциях. По имеющимся экспериментальным данным
13 А,мкм
Рис.35. Комплексный показатель преломления сульфата аммония
рассчитанный по Крамерсу-Кронигу.
[19, 34] за z0 можно принять высоту 3 км. Значение Я колеб-
колеблется от 0,5 до 1,5 км. Модальный размер образующихся частиц
~ 0,08-0,15 мкм. Исходя из имеющихся экспериментальных данных,
NQ (z = 3 км) =100 см.
Основными химическими компонентами тропосферных конденса-
конденсационных аэрозолей являются сульфаты и капли серной кислоты.
Спектральный ход комплексного показателя преломления сульфата
341
аммония представлен на рис.35, данные по изменению размеров
сульултных частиц в зависимо., .и от влажности представлены в ра-
работах [70, 110, 173], что позволяет также вычислить изменение
показателя преломления при увеличении относительной влажности
воздуха.
дисперсионные аэрозоли. Этот тип свя-
связан в основном с эрозией почв и характерен для летних перио-
периодов времени. Высота и мощность слоя дисперсионных тропосферных
аэрозолей не имеют определенных характеристик, они должны оп-
определяться местными условиями.
Тропопаузные конденсационные
аэрозоли. 3 области тропопаузы весьма часто накаплива-
накапливаются аэрозоли конденсационного происхождения, ложно предпола-
предполагать, что это сульфатные частицы, но более "старые", чем в тро-
тропосфере. Поэтому модальный радиус может быть сдвинут в сторону
более крупных частиц, т.е. г - 0Д0 мкм. Общая счетная кон-
концентрация тропопаузных аэрозолей может сильно меняться в пре-
предельных случаях, достигая 10^ см. Экспериментальные данные
свидетельствуют о малой величине Я —0,5 км [37, 39, 84]. По-
Показатель преломления для этих частиц соответствует показателю
преломления сульфатных частиц.
Аэрозоли стратосферы и высоких слоев атмосферы. Главными
типами аэрозолей в стратосфере и в более высоких слоях атмосфе-
атмосферы считаются сульфатные аэрозоли слоя Юнге и аэрозоли космичес-
космического происхождения, i/южно, конечно, выделить в отдельные типы
и аэрозоли, попавшие в стратосферу из тропопаузы, аэрозоли -
продукты сгорания космических кораблей и продукты работы раз-
различного рода двигателей. Однако глобальную картину аэрозолей в
верхней атмосфере определяют только первые два типа.
Сульфатный аэрозольный слой в
стратосфере. В настоящее время характеристики этого
слоя изучены достаточно основательно. Вертикальная структура
Рис.36. Вертикальные профили (модельные) счетной концентрации
частиц для атмосферных аэрозолей.
/ - общая концентрация Na(z); 2 - N(z) для частиц с г^
^ ОД мкм; 3a.-tf(z) для частиц с 7*5- 0,15 мкм;3<Г- усреднен-
усредненный профиль по измерениям для Рыльска в августе 1975 г. \Ча-
- N(z)jxm частиц с г* 0,25 мкм; кд - то же для Рыльска,
август 1975 г.; 5-N(z) для частиц с г2. 0,5 мкм.
342
/
г-
//#%х ti,
слоя наиболее удовлетворительно описывается при zQ= 17 км и
Н = 3 км. Распределение по размерам имеет две моды т^= 0,03
и Го2 = О ДО мкм. Счетная концентрация -N4O,15 = 3.5 см ,
а в максимуме сульфатного слоя No~ W см . В качестве пока-
показателя преломления для расчетов можно использовать данные по
показателю преломления сульфата аммония.
Аэрозоли космического проис-
происхождения. Если предполагать, что перенос частиц в верх-
верхней атмосфере осуществляется вследствие их падения, то для час-
частиц одного размера вертикальный профиль определяется из усло-
условий постоянства потока частиц, т.е. ^j.,consivs = const. Расче-
Расчеты скорости гравитационного падения частиц показывают, что ха-
характер изменения скорости падения vs с высотой неэкспоненциа-
неэкспоненциален. Приближенно до уровня 30-25 км его можно, например, пред-
представить в виде линейной зависимости
» = иЬ"аBо>
Тогда
Вертикальные токи в верхней атмосфере могут приводить так-
также к образованию спорадических аэрозольных слоев.
Характер изменения распределения частиц по размерам косми-
космической пыли с высотой достаточно сложен, так как концентрация
мельчайших частиц, образовавшихся при сгорании микрометеоритов,
недостаточно высока для быстрого коагуляционного роста до раз-
размеров 0,03-0,1 мкм. Время существования субмикронных частиц в
высоких слоях атмосферы весьма длительно. Скорости падения час-
частиц разных размеров сильно различаются.
Очевидно, что должно наблюдаться смещение модального ра-
радиуса частиц с уменьшением высоты. Имеющихся данных пока недос-
недостаточно, чтобы построить удовлетворительную модель распределе-
распределения этих частиц по размерам (рис.36).
Показатель преломления вещества частиц космического проис-
происхождения наиболее близок к модели показателя преломления ве-
вещества частиц дисперсионного происхождения.
Новые экспериментальные данные свидетельствуют о большом
вкладе в вещество частиц верхней атмосферы окиси алюминия, яв-
являющейся продуктом антропогенного происхождения.
344
Указатель литературы
1. А м е л и н А.Г. Теоретические основы образования ту-
тумана при конденсации пара. Л., 1972. 303 с.
2. Пространственно-временные вариации оптических свойств
атмосферы, обусловленные взаимодействием атмосферного аэрозоля
с полем влажности /Андреев С.Д., И в л е в Л.С., С п а-
ж а к и н а Н.К., Я н ч е н к о Е.Л. - В кн.: Физика мезо-
сферы и мезосферных облаков. Метеорологические иооледования. М.,
1975, вып.22, с.34-49.
3. Артемов И.С. Влияние формы частиц на величину
константы скорости коагуляции аэрозолей. - Коллоидный журн.,
1947, т.9, * 4, с.225-230.
4. Асатуров М.Л. Конденсационный рост стратосфер-
стратосферного сульфатного аэрозоля. - Труды Гл. геофиз. обсерв., 1976,
вып.365, с.3-13.
5. Атмосферный аэрозоль и его влияние на пере-
перенос излучения: К итогам советско-американского аэрозольного экс-
эксперимента / Под ред. К.Я.Кондратьева. Л., 1978. 120 о.
6. Б а р р е П. Кинетика гетерогенных процессов. М.,1976.
399 с.
7. Батчер С.Чарлсон Р. Введение в химию ат-
атмосферы. М., 1977. 270 с.
8. Бейкер М..Емиленко А.С., Розен-
б е р г Г.Р. Радиационное модулирование состояния аэрозоля бе-
бегущими облаками. - Изв. АН СССР. Физика атмосферы и океана,
1978, т.14, * 8, с.855-865.
345
9. Беляев Л. И. О выносе морских солей в атмосферу
и о ходе его. - Труды Морск. гвдрофиз. ин-та АН СССР, 1955,
вып.7, с.49-63.
10. Б е р л я н д ini.E. Исследование атмосферных диффузий
и загрязнение атмосферы на современном этапе. - Метеорология и
климатология, 1974, т.2, с.250-330.
11. Берлянд ivi.E. Современные проблемы атмосферной
диффузии и загрязнения атмосферы. Л., 1975. 448 с.
12'. Озон и аэрозоли в нижней стратосфере /Бобров-
/Бобровский А.П., Иванов В.И., И в л е в Л.С, Сиро-
т а В.Г. - В кн.: Современное состояние исследований озоносфе-
ры в СССР. М., 1980, с.168-176.
13. Богданов B.C. Исследование аэрозолей, образую-
образующихся в радиационно-химических реакциях. - Журн. физ. химии,
1961, т.34, вып.6, с.613-617.
14. Борзилов В.А., С е д у н ов Ю.С., Сте-
Степанов А.С. Силы индукции между ионами воздуха и каплей. -
Изв. АН СССР. Физика атмосферы и океана, 1973, т.9, J§ 3, с.264-
271.
15. Б у д ы к о М.И. Изменения климата. Л., 1974. 230 с.
16. Б у р д в Я. Динамический характер адсорбции. М.,
1962. 290 с.
17. Б ы з о в а Н.Л. Рассеяние примеси в пограничном ела
атмосферы. М., 1974. 191 с.
18. Бинникова Т.В., Довгалюк Ю.А. К реше^
нию кинетического j-равнения коагуляции. - Труды Гл. гесфиз.об'
серв., 1974, вып.290, с.3-10.
19. Влияние аэрозоля на перенос излучения: возможные кли-
климатические последствия /Кондр-атьев К.Я., В а с и л 1
е в О.Б., И в л е в Л.С..Никольский Г.А., С м о к
т и й О.И. Л., 1973. 266 с.
20. 3 о л о щ у к В.М. Введение в гидродинамику грубодис
персных аэрозолей. Л., 1971. 208 с.
21. В о л о щ у к В.М., С е д у н о в tO.C. Процессы ко«
гуляции в дисперсных системах. Л., 1975. 320 с.
22. Г и б б с Дж.В. Термодинамические работы, т., 1950.
421 с.
346
23. Глушков В.Е., Ершов В.А., С а л о в В.А.
Хонденсационный рост крупных капель растворов. - Физика аэро-
аэродисперсных систем, 1976, вып.14, с.7.
24. Грабовский Р.И. Атмосферные ядра конденса-
конденсации. Л., 1956. 164 с.
1 25. Григоров Н.О., И в л е в Л.С. Эксперименталь-
Экспериментальное исследование процессов перехода твердых частиц в капельное
состояние. - Вестн. Ленингр. ун-та, 1971, Л 16, с.48-56.
26. Григорьев А.А., Липатов В.Б. Пыльные
бури по данным космических исследований. Л., 1974. 30 с.
27. Г р и н X., Л е й н В. Аэрозоли - пыли, дымы и тума-
туманы. Л., 1969. 427 с.
28. Д а в и т а я Ф.Ф. 0 возможности влияния запыленнос-
запыленности атмосферы на уменьшение ледников и потепление климата. - Изв.
АН СССР. Серия географическая, 1965, J* 2, с.3-23.
29. Дейрменджан Д. Рассеяние электромагнитного
излучения сферическими полвдисперсными частицами. М., 1975.
164 с.
30. Д е р я г и н Б.В. .Прохоров А.В. Уточненная
теория гомогенной конденсации и ее сравнение с опытом. - Докл.
АН СССР, 1972, т.207, № 6, с.1311-1313.
31. Д е т р и Ж. Атмосфера должна быть чистой. М., 1973.
379 с.
32. Дмитриев М.Т..Китросский Н.А. Неко-
Некоторые физико-химические процессы в воздухе, происходящие в ус-
условиях загрязнения атмосферы и океана. - Изв. АН СССР. Физика
атмосферы и океана, 1972, т.8, J* 1, с.103-107.
33. Дмоховский В.И., Иванов В.А., И в -
лев Л.С. Вертикальная структура аэрозоля в районе промышлен-
промышленного центра Запорожья. - Труды Гл. гесфиз. обсерв., 1974,
вып.332, с.73-81.
34. Довгалюк L.A., И в л е в Л.С. Физика водных
и других атмосферных аэрозолей. Л., 1977. 255 с.
35. Захаров В.М., К о с т к о 0.К* Метеорологичес-
Метеорологическая лазерная локация. Л., 1977. 222 с.
36. 3 у е в В.Е. Распространение видимых и инфракрасных
золн в атмосфере. М., 1970. 496 с.
347
37. 3 у е в В.Е., И в л е в Л.С, Кондрать-
е в К.Я. Новые результаты исследований атмосферного аэрозо-
аэрозоля. - Изв. АН СССР. Физика атмосферы и океана, 1973, т.9, В 4,
с. 371-385.
38. И в л е в Л.С. Аэрозольная модель атмосферы. - В кн.:
Проблемы физики атмосферы, 1969, вып.8, с.125-160.
39. И в л е в Л.С. Аэростатные исследования структуры ат
мосферного аэрозоля. - В кн.: Проблемы физики атмосферы, 1972,
вып.10, с.92-103.
40. И в л е в Л.С.Формирование спектра размеров аэро-
аэрозольных частиц в приземном слое атмосферы. - Докл. АН СССР,
1973, т.210, « 2, с.335-338.
41. Результаты исследований атмосферного аэрозоля /Ив -
лев Л.С, Андреев С.Д., Головина Е.Г. и др,-
В кн.: Загрязнение атмосферы, Вильнюс, 1976, № 3, с.131-145.
42. Аэрозольные исследования в экспедиции "Беринг" / И в-
л е в Л.С. .Дмоховский В.И. .Иванов В.А., С о-
л о м а т и н В.К. - Труды Гл. гесфиз. обсерв., 1975,вып.363,
с.37-43.
43. Вымывание аэрозоля в приземном слое атмосферы / И в -
лев Л.С., Иония В.А., С е м о в а . А.Ю., С п а ж а к и-
н а Н.К. - Труды Гл. гесфиз. обсерв., 1973, вып.293,с.161-172
44. И в л е в Л.С. и др. Лабораторные оптические иссле-
исследования образования диспергированной фазы из сернистого газа. -
В кн.: Материалы Всесоюз. совещ. по распространению оптическо-
оптического излучения в дисперсной среде. 1978, Обнинск. М., 1978,
с.200-201.
45. И в л в в Л.С, Огородников Б.И., С е -
м о в а А.Ю. Определение содержания некоторых химических эле-
элементов в стратосферном аэрозоле. - Труды ин-та пг:1кл. гесфиз.,
1976, вып.19, с.26-33.
46. И в л е в Л.С, Одинцов О.А. 0 влиянии влаж-
влажности на коагуляцию аэрозольных частиц. - Труды Гл. гесфиз.об-
гесфиз.обсерв. , 1974, вып. 290, с.30-37.
47. И в л е в Л.С, Попова СИ. Влияние влажности
на значения оптических констант вещества атмосферного аэрозо-
аэрозоля. - Изв. вузов. Физика, 1974, № 5, с.11-15.
348
48. И в л е в Л.С, Попова СИ. Комплексный пока-
показатель преломления диспергированной фазы атмосферного аэро-
аэрозоля. - Изв. АН СССР. Физика атмосферы и океана, 1973, т.9,
И 8, с. 1034-1043.
49. И в л е в Л.С, Федынский А.В. Исследование
структуры аэрозоля в верхней атмосфере. - В кн.: Метеорологи-
Метеорологические исследования. М., 1976, И 22, с.17-25.
50. Исследования по ядерной метеорологии и хи-
химии атмосферы / Под ред. Е.П.Махонько, Е.С.Селезневой. Л.,196а
299 с.
51. К а л в е р т Дж., П и т т с Дж. Фотохимия. М. ,196d
663 с.
52. Капустин В.Н., Любовцева Ю.С. 0 наб-
лвдении природных фотохимических дымок. - Изв. АН СССР. Физика
атмосферы и океана, 1976, т.12, И 6, с.620-626.
53. К а р о л ь И.Л.-Высотные самолеты и стратосфера. Л.,
1974. 41 с.
54. К а р о л ь И.Л. Радиоактивные изотопы и глобальный
перенос в атмосфере. Л., 1972. 380 с.
55. К а т ц м. Некоторые аспекты физической и химической
природы атмосферных загрязнений. - В кн.:. Загрязнение атмосфер-
атмосферного воздуха. Женева, 1962, с.105-170.
56. К а ч у р и н Л.Г. Физические основы воздействия на
атмосферные процессы. Л., 1978. 455 с.
57. К а ч у р и н Л.Г., Морачевский В.Г. Кине-
Кинетика фазовых переходов воды в атмосфере. Л., 1965. 145 с.
58. К е д д и А.В.К. Прогностическое моделирование за-
загрязнения воздуха: исследования в срорт -Вэлли. - В кн.: Мони-
Мониторинг состояния окружающей природной среды. Л., 1977, с.154-
173.
59. К е й д л Р.Д. Твердые частицы в атмосфере. М., 196&
284 с.
60. Кондратьев К.Я. .Бартенева О.Д. и
др. Аэрозоль в АТЭП и его радиационные свойства. -.Труды Гл.
геофиз. обсерв., 1977, вып.381, с.3-68.
61. К р ы с т а н о в Л. Избранные труды по физике атмос-
атмосферы. Л., 1968. 214 с.
349
62. Л е в и н Л.М. Исследования по физике грубодисперс-
ных аэрозолей. М., 1961. 215 с.
63. Л е в и ч В.Г. Физико-химическая гидродинамика. М.,
1959. 699 с.
64. Л е в к о в Л. Конденсация на шероховатых поверхнос-
поверхностях. - Изв. АН СССР. Физика атмосферы и океана, 1970, т.6, * (
с.790-797.
65. Леонтович М.А. Введение в термодинамику. М.
Л., 1951. 199 с.
66. Лушников А.А., С у т у г и н А.Г. Современ-
Современное состояние теории гомогенной нуклеации. - Успехи химии,
1976, т.45, вып.З, с.385-415.
67. Л я п и н Е.С. 0 турбулентном перемешивании воздуха
в атмосфере. - Метеорология и гидрология, 1948, * 5, с.13-23.
68. Мак-Ивен М., Ф и л л и п с Л. Химия атмоофе
ры. М., 1978. 375 с.
69. М а х о н ь к о К.П. Вторичное поступление в атмоа|
ру шли, осевшей на землю. - Изв. АН СССР, шизика атмосферы
океана, 1979, т.15, № 5, с.568-570.
70. М е й с о н Ь.Дж. Физика облаков. Л., 1961. 541 с.
71. Метеорология и атомная энергия / Под pej
Д.Слэйда. Л., 1971. 648 с.
72. М о н и н А.С., Я г л о м A.M. Статистическая гид]
механика, ч.1. 1965, 639 с; ч.2, 1967. 720 с.
73. Морачевский В.Г., Дубрович Н.А.
Льдообразующие свойства модифицированных ядер конденсации. -1
ды Гл. геофиз. обсерв., 1975, вып.356, с.142-147.
74. Непреднамеренные воздействия на кл»
мат / Под ред. Ы.И.Будыко. Л., 1974. 260 с.
75. 11 е р о в СП., X р г и а н А.Х. Современные проб.
мы атмосферного озона. Л., 1980. 287 с.
76. Петренчук О.П. Экспериментальные исследова
ния атмооферного аэрозоля. Л., 1979. 264 с.
77. П о л н ы й радиационный эксперимент / Под ред
К.Я.Кондратьева, Н.Е.Тер-Маркарянц. Ji., 1976. 238 с.
78. П р и с т л и С.Х.Ь. Турбулентный перенос в призек
ном слое атмосферы. Л., 1964. 122 с.
350
79. Радиационные характеристики атмосферы и
земной поверхности / Под ред. К.Я.Кондратьева. Л., 1969. 345 с.
80. Розенберг Г.В. Свойства атмосферного аэрозо-
аэрозоля по данным оптического исследования. - Изв. АН СССР. Физика
атмосферы и океана, 1967, т.З, № 9, с.936-949.
81.Русанов А.И. К термодинамике нуклеации на заря-
заряженных центрах. - Докл. АН СССР, 1978, т.238, J* 4, с.831-834.
82. Русанов А.К. Фазовые равновесия и поверхност-
поверхностные явления. Л., 1967. 388 с.
83. Савенко B.C., Корж В.Д. К вопросу о перено-
переносе солей морской воды в атмосферу при физическом испарении. -
Океанология, 1972, т.12, J* 5, с.936-940.
84. Результаты исследований естественных аэрозолей е
нижней тропосфере (до 550 м над уровнем моря1) /С арки-
сов С.Л., Степанова СИ., Степанов Г.В.,
1 о ргу ан и З.Г. - Труды Всес.оюз. высокогорн. геофиз.
ин-та, 1973, вып.24, с.95-106.
35.Свистов П.Ф. Исследование состава и некоторых
физико-химических свойств атмосферных аэрозолей над равными рай-
районами. Автореф. канд. дис, Л., 1967. 18 с.
86'. С е д у н о в Ю.С. Физика образования жвдкокапельной
фазы в атмосфере. Л., 1972. 207 с.
87. С м и р н о в В.И. Об аппроксимации эмпирических рас-
распределений облачных капель и других аэрозольных частиц. - Изв.
АН СССР. Физика атмосферы и океана, 1973, т.9, J* 1, с.54-65.
88. С м и р н о в В.И. Решения семейства уравнений ста-
стационарной коагуляции и модель спектра размеров частиц атмосфер-
атмосферного аэрозоля. - Изв. АН СССР. Физика атмосферн и океана, 1977,
т.13, « 3, с.274-286.
89. С м и р н о в В.И. Скорость коагуляционного и конден-
конденсационного роста частиц аэрозолей. - Труды Центр, аэролог, об-
обсерв., 1969, вып.91. 119 с.
90. Современное состояние исследований озоно-
сферы в СССР / Под ред. А.Х.Хргиана, Г.А.Кокина. М., 1980.
268 с.
91. Сонькин Л.Р. Вопросы прогнозирования фонового
загрязнения воздуха в городах. - Труды Гл. геофиз. обсерв.,
1974, вып.314, с.73-79.
351
У2. С т ы р о Б.И. Вопросы ядерной метеорологии. Виль-
Вильнюс, 1959. 418. с.
93. С т ы р о Б.И. Самоочищение атмосферы от радиоактив-
радиоактивных загрязнений. Л., 1968. 288 с.
94. С у т у г и н А.Г. .Гримберг А.Н. Самосохра-
Самосохраняющееся распределение аэрозольных частиц по размерам при коа-
коагуляции в свободно молекулярном режиме. - Изв. АН СССР. Физика
атмосферы и океана, 1975, т.И, И 9, с.956-959.
95. Т е з и с ы докладов УШ Международной конференции по
нуклеации / Под ред. И.И.Гайворонского. Л., 1973. 164 с.
96. Труды УШ Всесоюзной конференции по физике облаков
и активным воздействиям / Под ред. И.И.Гайворонокого, В.П.Ломи-
надзе. Л., 1970. 536 с.
97. У э б б В. Структура стратосферы и мезосферы. М.,
1969. 258 с.
98. Ф е д о р о в Е.К. Взаимодействие общества и природы.
Л., 1972. 87 с.
99. Ф е т т В. Атмосферная пыль. М., 1961. 336 с.
100. Френкель Я. И. Статистическая теория конденса-
конденсации (ассоциации) и полимеризации. - Журя. эксп. и теор. физики,
1939, т.9, И 2, с.199-207.
101. Френкель Я.И. Теория явлений атмосферного элек-
электричества. М. ;Л., 1949. 155 с.
102. Фреймундт Т.Н. Об асимптотике решений уравне-
уравнения коагуляции. - Труды Ин-та экспер. метеорол., 1974, вып.8
D6), с.102-107.
103. Фукс И.А. Испарение и рост капель в газообразной
среде. М., 1958. 91 с.
104. Фукс Н.А. Механика аэрозолей. М., 1955. 351 с.
¦105. Химия нижней атмосферы / Под ред. С.Расула. М.,
1976. 408 с.
106. Шишкин Н.С. Облака, осадки и грозовое электри-
электричество. М.; Л., 1954. 401 с.
107. Шляпинтох В.Я., Иванов В.Б. Тушение син-
глетного кислорода. - Успехи химии, 1976, т.45, вып.2,с.202-223.
108. Шмаус А.,Виганд А. Атмосфера как коллоид.
Л., 1933. 43 с.
352
109. Щербаков Л.М., Рыков В.И. 0 теплоте ис-
испарения малых капель. - Коллоидный нурн., 1961, т.23, вып.2,
с.221-227.
110. Юнге X. Химический состав и радиоактивность ат-
атмосферы. М., 1965. 423 с.
111.Abel И..Winkle г P.,Junge С. Stu-
Studies of size distributions and growth with humidity of natural
aerosol particles. - Final. Sci. Kept., 1969, AFCBL-69-O2O5,
112. Allen E.H., II с Q u i g g B.C., С a d 1 e B.C.
Фае photooxidation of gaseous sulfur dioxide in air. - Chemo-
sphere, 1972, vol.1, p.25.
115. Altshuller A.P., Bufallini I.I.
Photochemical aspects of air pollution. - Environ. Soi.Teohnol.,
1971, vol.5, M 1, p.39-63.
114. Atkinson fi.,*inlayson B.J.,
Pitts J.K. Fhotoionization mass spectrometer studies of
gas phase ozone - olefin reactions. - J. Amer. Chem.Soc. ,1973,
vol.95, N 23, p.7592-7599.
115. Atmospheric pollution / Ed. li.li.Benarie.
Paris. Elsevier, 1976. Ь<+9 p.
116. Beard K.V. Experimental and numerical collision
efficiencies for submicron particles scavenged by small rain-
raindrops. - J. Atm. Sci., 1974, vol.31, N 6, p.1595-1602.
117. Bigg S.K., Survey C.S. Sources of atmosphe-
atmospheric particles over Australia. - Atmos. Environ., 1978, vol.12,
N 8, p.1643-1655-
118. Bigg K.K., ? v i z Z., Thomps on W.J.
Electron microscope photographs of extraterrestrial particles.-
Tellus, 1971, vol.23, M 3, р.248-2ЬО.
119. Birenzvige A.. 11 о h а е n V.A. Gas to
particle conversion of sulfur dioxide reaction products at low
relfcive humidities. ASBC-SUNY Publ., N 363, 1975. 97 p.
120. Blanchard C.C. Surface active organic ma-
material on airborne salt particles. - In: Froc. Intern. Conf.
Cloud Phys. Toronto, Canada, 1968. 7 p.
121. Blifford J.H. Tropospheric aerosols. - J.
Geoph. Res., 1970, vol.75, N 15, p.3099-
23.
353
-122. Blifford J.H., G i 1 1 e t e D.A. The in-
influence of air origin on the chemical composition and size dis-
distribution of tropospheric aerosols. - Atmos. Environ., 1972,
vol.6, N ?, p.463-430.
123* formation and evolution of nuclei of condensation
that appear in the air initially free of aerosol / В г i -
c.ard J.,Billard F., С а Ь а п е It., U a d e 1 a i-
n e G. - J. Geoph. Нее., 9Ь8, vol.73, H 14, p.4487-4496.
124. Formation and properties of neutral ultrafine partic-
particles and small ions conditioned by gaseous impurities of the
air /Bricard J., С a b a n e M.,Madelalne G.,
V i g 1 a D. - J. Colloid. Interface Sci., 1972, vol.39i H 1,
p.42-58.
125. Boulaud D., Bricard J., U a d e 1 a i-
n e G. Aerosol growth kinetics during S02 oxidation. - Atmos.
Environ., 1978, vol.12, H 1-3, p.1?1-1?7.
126. Bufalini M. Oxidation of sulfur dioxide in
polluted atmospheres. - ihnviron. Sci. Technol., 1971, vol. 5,
S 8, p. 685-700.
127. Cadi e fi.D. Composition of the stratospheric sul-
fate layer. - Trans. Amer. Geoph. Union, 1972, vol.53i H 9,
p. 812-820.
128. С a d 1 e H.D. ,Crutzen P., Ъ h h a 1 t D.
Heterogeneous chemical reactions in the stratosphere.-J.Geoph.
fies., 1975, vol.80, H 24, p.3381-3386.
129. С a d 1 e R.D., Bobbins B.C. Kinetics of at-
atmospheric chemical reactions involving aerosol. - Disc. Farad.
Soc, 19bO, H 30, p.155-161.
130. С a 1 v e г t J.G., Demerjian K.L..K err
J.A. Computer simulation of the chemistry of a simple analogue
to the sunlight-irradiated auto-exhaust polluted atmosphere. -
Environ. Lett., 1973, vol.4, N 2, p.123-140.
131< Castleman A.*. Hucleation processes and aero-
aerosol chemistry. - Space Sci. Rev., 197*. vol.15, И 5,p.457-589-
132. CIAP Monographs, vol.3. Draft. U.S. Department of
transportation. Washington, D.C., 1974, p.118-199.
354
133* Cooper V.A., Knight Ch.A. Heterogeneous
nucleation by small liquid particles. - J. Aerosol Sci., 1975,
vol.6, S 4, p.315.
134. Cox B.A., Sandalls F.J. The photo-oxida-
photo-oxidation of hydrogen sulphide and dimethyl sulphide in air. -Atmos.
Environ., 197*, vol.8, H 12, p.1269-1289.
135. Cox B.A. Some experimental observations of aero-
aerosol formation in the photooxidation of sulfur dioxide. - J.
Aerosol. Sci., 1973, vol.4, p.473.
136. Crutzen P.J. A review of upper atmospheric
photochemistry. - Canad. J. Chem., 197*. vol.52, S 8, p.1569-
1581.
137• Crutzen F.J. The possible importance of CSO
for the sulfate layer of the stratosphere. - Geophys. Bes.
Lett., 1976, vol.3, M 2, p.73-76.
138. Cunningham P. Т., Johnaon S.,
Tang B.T. Variations in chemistry of airborne particulate
material with particle size and time. - Environ. Sci.Technol.,
197*, vol.8, Я 2, p.131-135.
139. Cvetanovic B.J. Excited state chemistry in
the stratosphere. - Canad. J. Chem., 1974, vol.52, N 8,p.1452.
140. Theory of nucleation of water. I. Properties of some
clath-rate-like cluster structures /Dace Ы., L u n d L.H.,
F 1 u m m e r P.L.M., К a s s n e r J.L., Hale B.H. - J.
Colloid. Interface Sci., 1972, vol.39, И 1( p.65-78.
141. D a n a M.T., H a 1 e s M.J. Statistical aspects
of the washout of polydisperse aerosols. - Atmos. Environ.,
1976, vol.10, И 1, p.45-50.
142. Davies CD. Size distribution of atmospheric
particles. - J. Aerosol bci., 1974, vol.5, M 3, p.293-3OO.
143. Davis D.D., Klauber G. Atmospheric gss
phase oxidation mechanisms for the molecule S02« - Int.J.Chem.
Kinet., 1975, vol.7, X 1, p.5*3-556.
144. D e 1 a n e у А.С, Pollock W.H., S h e d -
1 о v s к у J.P. Tropospheric aerosol» the relative contribu-
contribution of marine and continental components. - J.Geoph.Bes. ,1973»
К 27, р.Ь219-Ь225.
355
145. D e L u i s i J.J., Blifford J.H., T а к a-
m i n e J.A. Models of tropospheric serosol size distribution
derived from measurement at three locations. - J. Geoph. Res.,
1972, vol. 77. В 24, p.4529-4538.
146. Dodge M.C., Bufalini J.J. Photochemical
smog and ozone formation. - Adv. Chem., 1972( -Ы 113, P»232.
147. Doyle G.J. Self-nucleation in sulfuric acid -
water system. - J. Chem. Phys., 19Ы, vol.35, N 6, p.795»
148. Dufour J., D e f а у H. Thermodynamics of
clouds. New York; London, 1963. 255 p.
149. Dunham S.B. Detection of photochemical oxida-
oxidation of sulfur dioxide by condensation nuclei techniques. -
Nature, 1960, vol. 188, N 4744, p. 51.
150. barlow N.H. Electron microscope studies of
particles on sampling surfaces recovered from space with soun-
sounding rockets. - J. Geoph. Res., 1968, vol.73, N 13, p.4363«
151. I s г 1 о » И.Н., Ferry u.V.,Blanchard
II.В. Examination of surfaces exposed to a noctilucent cloud.
Aug.1, 1968. - J. ueoph. Res., 1970, vol.75, й 33, p.6736.
152. Nitrogen - sulfur compounds in stratospheric aerosols
/ F a r 1 о w N.H.,Snetsinger K.G., HayesD.M.,
L e m N.Y. - J. Geoph. Res., 1978, vol.83, S 12, p.6207-6211.
153. Finlayson B.J., Pitts J.N. Photoche-
Photochemistry of the polluted troposphere. SPX is now included with
U0x and HC in homogeneous and heterogeneous atmospheric che-
chemistry. - Sci., 197b, vol.192, N 4235, p.111-119.
154. Fitzgerald J.W. Approximation formulas for
the equilibrium size of an aerosol particle as a function - of
its dry size and composition and the ambient relative humidi-
humidity. - J. Appl. Meteorol., 1975, vol.14, N 6, p.1044-1049.
155. Photochemical smog systems; heterogeneous decay of
ozone /fox D.L., Kamens R., Jeffries H.E.,
S h a i г F.H. - Sci., 1976, vol.191, N 4231, p.1057-1058.
156. Freiberg J. Effects of relative humidity and
temperature on ironcatalyzed oxidation of SO2 in atmospheric
aerosols. -Environ. Sci. Technol., 1974, vol.8, К 8,p.731-734.
356
157. Freiberg J. The resction of sulfur dioxide
with ozone in water and its possible atmospheric significan-
significance. - Atmos. Environ., 1978, vol.12, И 12, p.2523.
1^8. F г i e d 1 ander S.K. Chemical element balan-
balances and identification of air pollution sources. - Environ.
Sci. Technol., 1973, vol.3, И 2, p.235-240.
159. Friedlander S.&. Theoretical considera-
consideration for the particle size spectrum of the stratospheric aero-
aerosol. - J. Ueteorol., 1961, vol.18, N 5, p.753-759.
160. Friend J.F. Trace material composition of the
lower stratosphere. - In» CIAP Proc. Survey Conf., Febr.15-16,
1972, p.71-79/
161. Gelbard F., Seinfeld J.H. The general
dynamica equation for aerosols. Theory and application to
aerosol formation and growth. - J. Colloid Interface Sci.,1979,
vol.68, H 2, p.363-383.
162. Gerhardt E.fi., Johnstone H.F. Pho-
Photochemical oxidation of sulfur dioxide in air. - Ind.iiag.Chem,,
1955, vol.47, N 8, p.972-976.
163. Goetz A., Pueschel R. Basic mechanisms
of photochemical aerosol formation. - Atmos. Environ., 1967,
vol.1, N 3, p.287.
164. G г a e d e 1 Т.Е. Sulfur dioxide, sulfate aerosol,
and urban ozone. - Geophys. fies. Lett., 197&» vol.3, N 3»P>1a1-
184.
165. Graham R.A., Johnston H. Kinetics of
the gas - phase reaction between ozone and nitrogen dioxide.-
J. Chem. Phys., 1974, vol.60, N11, p.4628.
166. Gravenhorst G. The sulphate component in
aerosol samples over North Atlantic. - "Meteor/ Forschungser-
gebn., 1975, Bd 10, H.1, S. 22-31.
167. Giisten H.,Penzhorn R.D. Photoohemi-
sche Reaktionen atmospharischer Schadstoffe. - Naturwiss.Rdsch.,
Bd 27, H.2, S.56-68.
168. Hale B.N., F 1 u m m e r P.L.M. On nucleation
phenomens. I. A molecular model. - J. Atmos. Sci., 1974, vol.
31, N 6, p.1615-1621.
357
169. Hall T.C. Photochemical studies of $0? and S02-
Jfcesis, Univ. Calif., Los Angeles, 1953. 248 p.
170. H a m i 1 1 P. The time dependent growth of H20-
HoSO^ aerosols by heteromolecular condensation. - J. Aerosol
Sci., 1975, vol.6, N 6, p.475-482.
171. Hampson fi.T., G а г v i n D. Chemical kine-
kinetic and photochemical data for modelling atmospherio chemist-
chemistry. U.S. Department of commerce, Nat. Bureau Stand. Те с tin.
Note, 1975, N 866. 114 p.
172. Handbook of aerosols. Washington.1949.205 p.
173* H a n e 1 G. The properties of atmospheric aarosol
particles as functions of the relative humidity at thermody-
nsmic. -Adv. Geophys., 1976, vol.19, p. 73-188.
173. Harder A.B. Formation of sulfate in the stra-
stratosphere through the gas phase oxidation of sulfur dioxide. -
J. Geoph. fies., 1975, vol.30, N 24, p.3399-3401.
175. H e с h t T.A., Seinfeld I.H.,Dodge U.C.
further development of a generalized kinetic mechanism for
photochemical smog. - Environ. Sci. Tecnnol., 1974,vol.8,N 4,
p. 327-339-
176. H emenway C.L.,Soberman B.K. Studi-
Studies of micrometeorites obtained from a recoverable sounding
rocket* - Astron. J., 1962, vol.67, N 2, p.116; 1962, N 3,
p. 121.
177. H i d у G.M. The dynamics of aerosols in the lower
troposphere in assessment of airborne particles. - In: Froc.
3rd Univ. Rochester Conf. Environ. Toxicology, C.C.Thomas.
Springfield, 1972, p.81-115.
178. H i d у G.M. e.a. Summary of the California aero-
aerosol characterization experiment. - J. Air Pollut. Cont.Assoo.,
1975, vol.25, N 11, p.1106-1114.
179. H i d у G.M., Brock J.fi. The dynemics of aero-
colloidal systems. New York, 19?0. 148 p.
180. H i d у G.M., Burton C.S. Atmospheric aero-
aerosol formation by chemical reactions. - Intern. J. Chem.Kinet.,
1975, vol.7, N 7, p.509-541.
181. Hitchcock D.H. Dimethylsulfide emission to
the global atmosphere. - Chemosphere, 1975, vol.4, N 3,p.i37-
138.
358
182. Hofmann D.J. Stratospheric aerosol determina-
determination. - Canad. J. Chem., 1974, vol.52, N 8, p.1519-1526.
183. Hoffman J.L., D u с e fi.A., H о f f m a n E.J.
Trace metals in the Hawaiian marine atmosphere. - J.Geoph.fies.,
1972, vol.77, N 27, p.5322-5329.
184. Jaenicke fi. The role of organio material in
atmospheric serosols. - Pageoph., 1978, vol.116, N 2-З1 p.282-
292.
185* J u n g e C.S. Important problems of global pollu-
pollution. - In: Proc. Intern. Conf. Structure, Composition and Ge-
General Circulation of Upper ana Lower Atm., Jen. 14-25, 1974t
Univ. of Melbourne, Australia, 1975i N 1, p.1-16.
186. J u n g e C.E., H у a n T.G. Study of sulfur di-
dioxide oxidation in solution and its role in atmospheric che-
chemistry. - Quart. J. Hoy. Ueteorol. Soc, 1958, vol.84, N 359i
p.46-55.
187. К a s t e n P. Visibility forecast in the phase of
pre-condensation. - Tellus, 1969, vol.21, В 5. p.631-635.
188. Keith C.H., A r о n s A.B. The growth of sea-
salt particles by condensation of atmospheric water vapour. -
J. Ueteorol., 1954', vol.11, N 2, p.173-184.
189. К i a n g C.S. e.a. Ternary nucleation applied to
gas-to-particle conversion. - J. Aerosol Sci., 1975, vol. 6,
N. 6, p.465-478.
190. К i a n g C.S. e.a. Heteromolecular nucleation theo-
theory applied to gas-to-p*rticle conversion. - Atmos. Environ.,
1973i vol.7, N 12, p.1279-1283.
191. Lamb H.H. Volcanic dust in the atmosphere,-Philo-
soph. Trans. Hoy. Soo., 1970, vol.266, N 1178, p.425-533.
192. L a z r u s A.L., Gandrud В., С a d 1 e fi.D.
Nitric acid vapor in the stratosphere. - J. Appl. lieteorol.,
-1972, vol.11, N 2, p.389-392.
193. Lemlich fi. Absubble processeat fosm fractiona-
tion and bubble fractionation. - J. Geoph. Res., 1972, vol.77»
N 27, p.5204-5210.
194. Liu В.У.Н., V h i t b у K.T., P u i D.T.H. A por-
portable electrical analyzer for size distribution measurement of
cE9
submicrcn aerosols. - J. Air Pollut. Cont. Assoc., 1974, vol.
24, и 11, p. 1067-Ю72.
195. Loo B.W. e.a. Large-scale measurement of airborne
particulate sulfur. - Atmos. Environ., 1978, vol.12, H 1-3,
p.759-771.
196. Low E.D.H. A theoretical study of nineteen con-
condensation nuclei. - J. Rech. Atmos., 1969. И 4, p.65-78.
197. Lothe J., Pound G.M. On the statistical
mechanics cf nucleation theory. - J. Chem. Phys., 1966, vol.
45, H 2, p.630-634.
198. Lundgren L.A., F a u 1 u в H.J. The mass
distribution of large atmospheric particles. - J. Air Pollut.
Gontr. Assoc., 1975. vol.25, И 12, p.1227-1231.
199. U a n t о n U.J. On the motion of в small particle
in the atmosphere. - Boundary-Layer lieteorol., 1974,vol.6,H 3-4,
p.487-504.
200. M a g с n о С. e.a. A measurement of scavenging ef-
effect of falling snow crystals on the aerosol concentration. -
J. Meteorol. Soc. Japan, 1974. ser.2, vol.52, p.407-416.
201. U с M u г г у Р.Н., Jfriedlander S.K.
Aerosol formation in reacting gases: relation of surface area
tc rate cf gas-to-particle conversion. - J. Colloid.Interface
Sci., 1978, vol.64, H 2, p.248-257.
202. Ueszaros E. On the formation of atmospherio
sulphate particulate in the winter months. - J. Aerosol Sci.,
1974, vol.5, N 5, p.483-485.
203. Ueszaros E. Some data on the chemical composi-
composition of atmospheric particles in the submicroscopic range of
sizes. - Id»3aras, 1966, vcl.70, H 5, p.257-261.
204. Ueszaros E..Varhelyi G. On the oon-
centration, size distribution and residence time of sulfate par-
particles in the lower troposphere. - Idtt;jaras, 1975, vol.79, H 5,
p. 267-273.
205. Miranda H.A., F e n n E. Stratospheric aero-
aerosol sizes. - Geophys. Res. Lett., 1974, vol.1, И 5, p.201-203.
206. Uisaki M., Ikegami M., К a n a z a -
w a I. Deformation of the size distribution of aerosol partic-
360
les dispersing from land to ocean. - J. lieteorol. Soc. Japan,
1975, vcl.53,, N 2, p.111-120.
207. 11 с h a t a P.C., A 1 о f s D.J. Insoluble conden-
condensation nuolei. - J. Atmos. Sci., 1975. vol.32, И 1, p.116-122.
208. Jt г о z E.J., Roller W.H. Composition of at-
atmospheric particulate matter from the eruption of Heimaey,
Iceland. - Science, 1975, vol.190, N 4213, p.461-464.
209. Hair P.V.H., V о h r a E.G. Growth of aqueous
sulphuric acid droplet as a function of relative humidity. -
J. Aerosol Sci., 1975, vol.6, N 3-4, p.265-271.
210. Hewkirk G. Meteoric dust in the stratosphere
determined by optical scattering techniques. - Smiths.Contrib.
Astrophys., 19ь7, Н 11, p.349.
211. Particulate models: Their validity and
application / Ed. J.H.Bliff ord. NCAR-TN-PROC-68, Aug. 1971,
1972. 294 p.
212. Paterson 11. P., Scorer R.S. The chemist-
chemistry of sea salt aerosol and its measurement. - Mature, 1975»
.vol.254, H 5500, p.491-495.
213. P i 1 a t M.J., Charlson R.J. Theoretical
and optical studies of humidity effects on the size distribu-
distribution of a hydroscopic aerosol. - J. Rech. Atmos., 1966,vol.2,
H 273, p. 165-170.
214. Podzimek I.,Sedlacek W.A., H a -
b e r 1 I.B. Aitken measurements in the lower stratosphere.-
Tellus, 1977, vol.29, H 2, p.116-127.
215. P г у d e L.I. Chemistry of the air environment.
Cummlngs Publ. Co., Menlo Park, Calif., 1973- 169 p.
216. Pueschel R.t., Bodhaine B.A., M e n-
d о n с a B.G. The proportion of volatile aerosols on the
Island of Hawaii. - J. Appl. Meteorol., 1973, vol.12, N 2,
p.308-315-
217. Re is 8 H. The kinetics of phase transition in
binary system. - J. Chem. Phys., 1950, vcl.18, И 6,p.840-848.
218. R e i s s H. The statistical mechanical theory of
irreversible condensation. - J. Chem. Phys., 1952,vol.20, N 8,
p.1216-1227.
361
219. Reiss H., Marvin D.C., Heist E.H.
The use of nucleation «nd growth a* a tool in chemical physics.
- J. Colloid. Interface Sci., 1977, vol.58, N 1, p.125-141.
220. Eeitsr R. .Sladkovic R., PU t z 1 K.
Die wichtigsten cheaischen Bestandteile dee Aerosols Sber Mit-
teleuropa unter Reinluftbedingungen in 1800 m. SeehOhe. - Me-
teorol. Bdsch., 1975, Bd 2в, Н.2, Б.37-55.
221. E о d h e H. Measurements of sulfur in the free
atmosphere over Sweden, 1969-1970. - J. Geoph. Bee. ,1972,vol.
77, N 24, p.4494-4499.
222. Rose W.I., S t о i b e r R.?. Xhe 1966 eruption
of Iz&lco volcano, ?1 Salvador. - J. Geoph. Res., 1969, vol.
74, К 12, p.3119-3130.
223. Rosen J.M. The boiling point of stratospheric
aerosols. - J. Appl. Meteorol., 1971, vol.10, N 6,p. 1044-1046.
224. Rosinski J. Sole of aerosol particles in
formation of precipitation. - Kev. Geoph. Брасе Fhys.» 1974,
vol.12, N 1, p.129-134.
225. Ho g in ti J.,Nagamoto C.I. Particle
deposition on and re—entrainment from coniferous trees. Ft.I.
- Kclloid. 2., 1965, Bd 204, Н.2, Б.111-119-
226. Б a v о i e D.L., Frospero J.M. Aerosol con-
concentration statistics for the northern tropical Atlantic. - J.
Geoph. Res., 1977t vol.82, H 37t p.5954-5964.
227. Seinfeld J.H. ,Ramabhadran Т.Е.
Atmospheric aerosol growth by heterogeneous condensation. -
Atmos. Environ., 1975, vol.9, N 12, p.1091-1097.
228. Shipley Б.Х. e. a. The evalution of lidar ex-
experiment to obtain meteorological and quality measurements
from a space shuttle platform.-Final Rept. on NASA Grant NSG.
Madison, Wisconsin, 1975* 60 p.
229. S 1 i n n W.G.H., S h e n S.F. Anisotropic brow-
nian diffusion and precipitation scavenging of submicron par-
particles. - J. Geoph. Ses., 1970, vol.75. N 11, p.2267-2270.
230. Stauffer D,, M о h n e n T.A., К i a ng C.B.
Hetsromolecular condensation theory applied to particle growth.
- J. Aerosol Sci., 1973, vol.4, N 6, p.461-471.
362
231. lakahashi X. Electric charge of small par-'
tides. - J. Atmos. Sci., 1972, vol.29, К 5, p.921-928.
232. Turbulent diffusion in environmental pollu-
pollution. - Adv. Geoph., 1974, VO1.18A. 462 p.; 18B. 389 p,
233. T w о m e у S. Atmospheric aerosols. &l*evier,197e.
681 p.
234. V 0 h r a K.G., S u b b в R.H.C., V a • u d e -
van K.N. Role of natural ionization in the formation of
condensation nuclei in the atmospheric air. - Meteorol. Sci.
Japan J,, 1968, vol.74, N 4, p.374.
235. V о 1 m e r M. Kinetik der Phasenbildung. - 2.
Phy*. Chem., 1929. Bd 25, Б.555.
236. Walter H. Coagulation and size distribution
of condensation aerosols. - Aerosol. Sci., 1973, vol.4, p.1-15.
237. Went F.W. Organic matter in the atmosphere and
its possible relation to petroleum formation. - Froc.Hat.Acad.
Bci. U.S., 19Ь0, vol.46, К 2, p.212.
238. W h i t b у K.I. e.a. Characterization of Califor-
California aerosols. I. Size distribution of freeway aerosol*. - At-
moe. Environ., 1975. vol.9, N 3, p.463-482.
239- W h i t b у K.I., Liu B.T.H., H u в а г R.B.
The aerosol size distribution of Los Angeles smog. - J.Colloid.
Interface Sci., 1972, vol.39, N 1, p.177-204.
240. Woodcock A.H. Marine fog droplets and salt
nuclei. Ft.I. - J. Atmos. Sci., 1978, vol.35, H 4, p.657-664.
241. 2immels I. The critioal condition* characte-
characterizing the formation of some heterogeneous nucleation systems.
- J. Colloid. Interface Sci., 1978, vol.65, N 1, p.59-66.
Оглавление
Введение 3
Глава I. Источники атмосферных аэрозолей и пространст-
пространственно-временная структура их химического сос-
состава 12
§1.1. Глобальные оценки мощности источников
аэрозолей 13
§ 1.2. Источники и химический состав страто-
стратосферных аэрозолей 34
§ 1.3. Химический состав аэрозолей в приземном
и нижних слоях атмосферы 41
§ 1.4. Химический состав тропосферных аэрозолей 67
§ 1.5. Химический состав аэрозольных частиц раз-
разных размеров '. 77
Глава 2. Химические процессы образования и роста аэро-
аэрозольных частиц (фотохимические и гетерогенные
реакции) 81
§ 2.1. Химические реакции соединений серы .... 86
§ 2.2. Химические реакции соединений азота ... 104
§ 2.3. Окисление и полимеризация органических
соединений 107
§ 2.4. Реакция кислорода и озона 112
§ 2.5. Реакция образования аэрозолей в реаль-
реальной атмосфере 123
Глава 3. Стоки и перенос аэрозолей в атмосфере 128
§ 3.1. Диффузия и перенос аэрозолей в атмосфере 130
364
§ 3.2. Сухое осаждение аэрозольных частиц ... 140
§ 3.3. Вымывание аэрозолей облаками и осадка-
осадками 146
Глава 4. Распределение аэрозолей в атмосфере 155
§ 4.1. Функция распределения частиц по разме-
размерам "
§ 4.2. Методы измерения концентрации и распре-
распределения частиц по размерам 163
§ 4.3. Глобальные типы распределения 166
§ 4.4. Распределение аэрозолей в приземном
слое атмосферы 172
§ 4.5. Распределение городских аэрозолей .... 189
§ 4.6. Распределение аэрозолей в тропосфере.. 197
§ 4.7. Распределение аэрозолей в стратосфере 202
§ 4.8. Глобальная пространственно-временная
структура аэрозолей 214
Глава 5. Конденсационный рост частиц 224
§ 5.1. Гомогенная конденсация "
§ 5.2. Гомогенная гетеромолекулярная конденса-
конденсация 235
§ 5.3. Адсорбция и набухание 238
§ 5.4. Гетерогенная конденсация 244
§ 5.5. Эмпирические формулы роста гигроскопи-
гигроскопических и смешанных аэрозольных частиц при уве-
увеличении влажности 248
§ 5.6. Образование ледяных частиц в атмосфере 253
Глава 6. Коагуляционный и конденсационно-коагуляционный
механизмы роста частиц 262
§ 6.1. Уравнение коагуляции 265
§ 6.2. Броуновская коагуляция 269
§ 6.3. Электростатическая коагуляция 276
§ 6.4. Решения уравнения коагуляции 288
§ 6.5. Влияние влажности на микроструктуру ат-
атмосферных аэрозолей. Морфология частиц 294
§ 6.6. Конденсационно-коагуляционный механизм
роста частиц 301
365
Глава 7. Аэрозольные модели 314
§ 7.1. Основные модельные характеристики ат-
атмосферных аэрозолей 318
§ 7.2. Статистические оптические модели ат-
атмосферных аэрозолей 329
§ 7.3. Елочная аэрозольная модель 335
Указатель литературы 345
ИБ * 1578
Лев Семенович Ивлев
ХИМИЧЕСКИЙ СОСТАВ И СТРУКТУРА
АТМОСФЕРНЫХ АЭРОЗОЛЕЙ
Редактор Г.А.Григеяч Художественный редактор А.Г.Голубев
Технический редактор Л.А.Топорина
Корректоры С.К.Школьникова, М.В.Зазулина
Подписано в печать 12.10.82. М-26637 Формат 60x81 I/I6.
Бумага тип. * 2. Печать офсетная. Уч.-изд.л. 18,60.
Усл.печ.л. 21,62. Усл. кр.-отт. 22,19. Заказ 1922.
Тирах 896 экз. Цена 2 р. 80 к.
издательство ЛГУ им. А.А .Жданова
I99I64. Ленинград, B-I64, Университетская наб., 7/9
Ордена Трудового Краоного Знамени
Первая типография издательства "Наука"
199034, Ленинград, В-34,9 линия,12