/






















































































































































































Text
РУКОВОДСТВО по НЕОРГАНИЧЕСКОМУ СИНТЕЗУ Редактор ЕБрауэр В шести томах
Москва «Мир» 19 8 6
РУКОВОДСТВО по НЕОРГАНИЧЕСКОМУ СИНТЕЗУ
Редактор Г. Брауэр Том 6
Перевод с немецкого канд. хим. наук И. А. Добрыниной, канд. хим. наук В. Н. Постнова и канд. хим. наук С. И. Трояиова
Мос ква «Мир» 1986
ББК 24.1
Р85
УДК 542
Handbuch der Praparativen Anorganischen Chemie
in drei Banden
Drifter Band
Unter Mitarbeit von
G. Brauer, W. P. Fehlhammer,
TT , „ n O. Glemser, H.-J. Grube, K. Gustav,
Herausgegeben von Georg Brauer w д Herrmann> s Herzog, H Lux,
H. Mflller, K- Ofele, E. Schill,
R. Scholder, H. Schwarz,
E. SchwarzmanH; K. Schwochau, A. Simon, J. Strahle
Dritte, umgearbeitete Auflage
Ferdinand Enke Verlag Stuttgart 1981
Шольдер P., Шварц X., Шилль Э., Фельхаммер В. П., Херманн В. А., Офеле К., Брауэр Г.
Р85 Руководство по неорганическому синтезу: В 6-ти т. Т. 6. Пер. с нем./Под ред. Г. Брауэра. — М.: Мир, 1986.— 360 с., ил.
В книге коллектива авторов из ФРГ, представляющей, по существу, энциклопедию неорганического синтеза, приведены методики получения более 3000 препаратов. Книга выходит в 6-ти томах. 6-й том содержит описание методик синтеза особых групп соединений н представляет собой перевод последней части (ч. 1П) третьего тома оригинального издания.
Предназначена для специалистов в самых различных областях науки и техники, а также для преподавателей и студентов химических вузов.
ББК 24.1
540
Редакция литературы по химии
© 1954, 3. Auflage, 1981, Ferdinand Enke Verlag
© перевод на русский язык, «Мир», 1986.
Часть III. Особые группы соединений
Глава 31. ГИДРОКСОСОЛИ
Р. Шольдер (переработано X. Шварцем) (Р. Scholder, Н. Schwarz)
Перевод канд. хим. наук Н. А. Добрыниной
Введение
Гидроксосолями называют группу комплексных соединений, в которых комплексный аниои состоит из центрального атома металла, координирующего в соответствии со своим обычным координационным числом гидроксид-ионы, являющиеся лигандами. Катионами в гидроксосолях служат щелочные металлы (как правило, натрий), щелочноземельные металлы, барий и стронций, иногда кальций. Реакцией двойного обмена можно получить немногочисленные гидроксосоли тяжелых металлов.
Гидроксосоли обычно получают из водных растворов с высокой концентрацией щелочи. Это значительно затрудняет синтез, а чистота веществ часто бывает неудовлетворительной. Чистые препараты иногда можно получить взаимодействием стехиометрических количеств компонентов при относительно низких температурах (50—140°С) [1]. При более высокой температуре гидроксосоли переходят в оксосоли (последние как раз и получают реакцией гидроксида металла со щелочью).
Во многих случаях оксометаллаты получают только из соответствующих оксидов [2]. В отдельных случаях удается получить оксогидроксосоли.
Образование комплексного аниона гидроксометаллата происходит согласно равновесию по схеме
М(ОН)Х Мх+ + хОН-, Мх+ + z/OH" [М(ОН^]<У-*>-
При этом действуют сильным основанием на плохо растворимый гидроксид металла, проявляющий «амфотерные» свойства и реагирующий как кислота.
К настоящему времени известны следующие металлы, способные образовывать гидроксосоли (расположены по степеням окисления):
M(II): Be, Mg, Sn, Pb, Сг, Мп, Fe, Со, Ni, Cii, Zn, Cd
М(П1): Al, Ga, In, Bi, Cr, Mn, Fe ’
M(IV): Sn, Pb, Ir, Pt
M(V): Sb
При получении кристаллических гидроксометаллатов щелочных металлов решающим является положение равновесия, приведенного выше. Большинство гидроксосолеи, и прежде всего гидроксосоли щелочных металлов, быстро разлагаются в воде и растворах разбавленных щелочей на соответствующие компоненты. Только гидроксосоли Sn(IV), Pt(IV) и Sb(V) образуют прозрачные водные растворы при комнатной температуре, тогда как гидроксометаллаты других элементов устойчивы лишь в виде осадков в растворах с высокой концентрацией щелочей. Гидроксометаллаты щелочноземельных металлов иногда бывают относительно устойчивы, главным образом из-за их низкой растворимости в разбавленных растворах. Повышение температуры приводит к смещению равновесия в сторону образования отдельных компонентов. Изучение
1870 Глава 31. Гидроксосоли
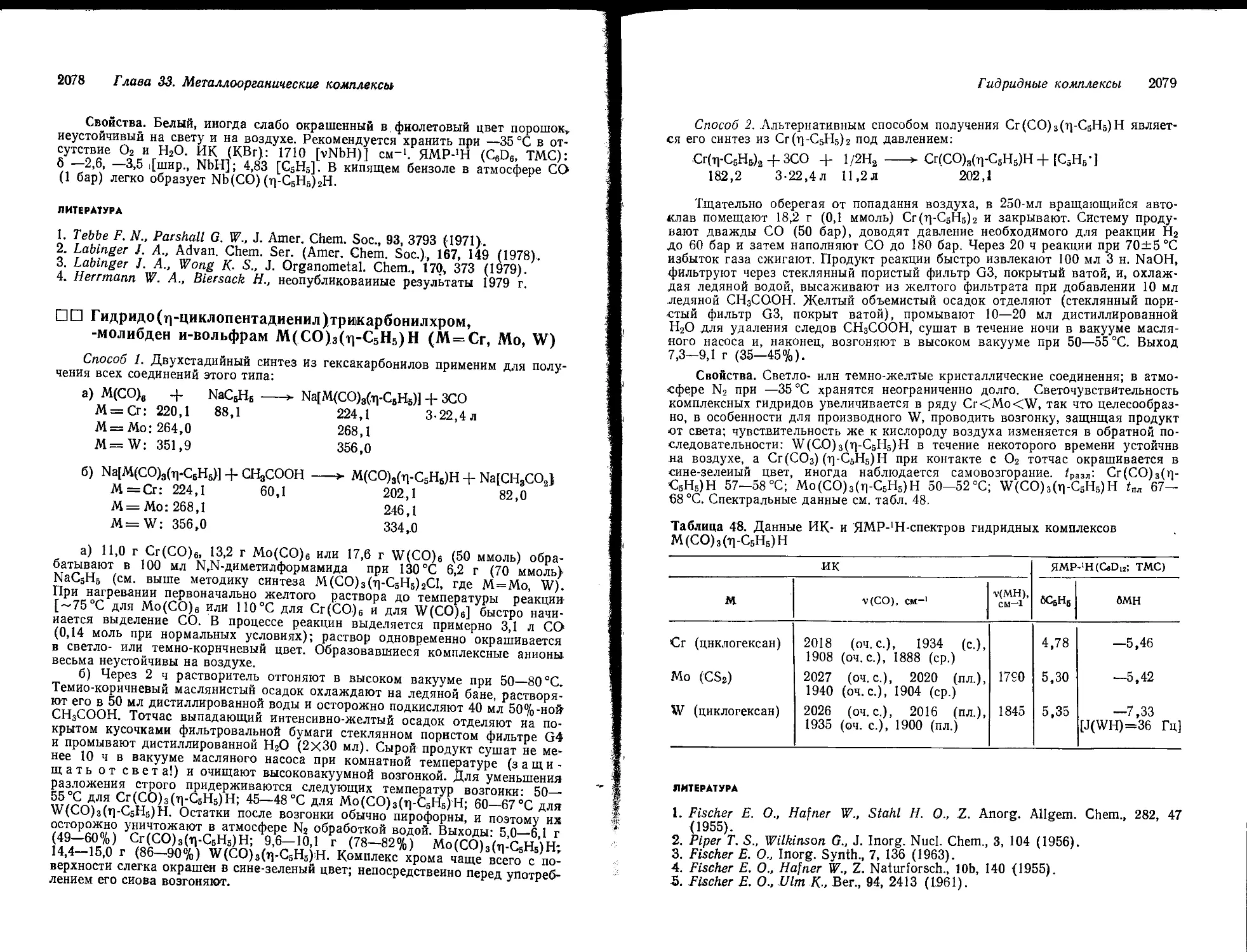
систем оксид металла — NaOH—Н2О при постоянной температуре позволило выявить ряд неожиданных индивидуальных особенностей (см. рис. 458). Так, с повышением концентрации щелочи растворимость гидроксосоли возрастает до максимума, а затем резко падает. Восходящей ветви кривой растворимости отвечает донная фаза состава М(ОН)т или МО„, а нисходящей ветви — гидроксометаллат натрия, растворимость которого с дальнейшим увеличением концентрации NaOH быстро падает. Из экспериментального факта — максимума растворимости гидроксосоли при концентрации NaOH более 30% (Юн.) —
Рис. 458. Растворимость в системе оксид металла — оксид натрия — вода в зависимости от концентрации NaOH.
следует, что гидроксосоли надо получать из растворов щелочей высокой концентрации (45—50%-ных), причем не только с целью получения высокого выхода. Гидроксометаллаты щелочноземельных металлов могут быть получены и из разбавленных растворов.
Микрокристаллические осадки обычно освобождают от захваченного маточного раствора путем высушивания их на пористой керамической пластинке. Для гидроксосолей этот метод неэффективен, и лучше применять встряхивание с изоамиловым спиртом (для гидроксометаллатов щелочных металлов) или обработку безводным метанолом с добавлением небольшого количества NaOH (для солей щелочноземельных металлов).
Как и галогенидные комплексы, гидроксосоли некоторых металлов, проявляющих степени окисления 2 и 3, образуют разнотипные соединения в зависимости от концентрации щелочи и температуры. Из малоустойчивых гидроксометаллатов щелочных металлов чаще всего получают натриевые соли; соли калия очень хорошо растворимы, и из концентрированных растворов щелочи выделяются вместе с твердым КОН.
Гидроксосоли бария и стронция выделяются в виде плохорастворимых веществ при добавлении простых солей Ва2+ или Sr2+ к растворам гидроксидов или оксидов соответствующих металлов в NaOH. В простейшем случае гидроксосоль можно получить одновременным добавлением по каплям концентрированного раствора перхлората металла и соли щелочного металла, взятых в стехиометрическом соотношении, к более или менее концентрированному горячему водному раствору NaOH. Надо отметить, что в концентрированном водном растворе NaOH растворимость NaC10< гораздо выше, чем NaCl.
Свободные гидроксокислоты, которые можно рассматривать как высшие гидраты оксидов металлов, не известны, за исключением лишь Нг[Р1(ОН)6]. Комплексный характеп гидроксометаллатов иногда проявляется в окраске растворов их солей. Правильность представлений о составе гидроксосолей обычно подтверждают результатами термического обезвоживания, способностью к образованию смешанных галогеногидроксосоедииений, проявлением изоморфизма, а также данными рентгенографических исследований. Несмотря на то что гидроксосолей известно довольно много, сведения об их структуре
Применение концентрированных щелочей 1871
ограничены. Причем имеющиеся данные для простейших гидроксосолей не позволяют однозначно судить о структуре гидроксоаииоиа и о координационном числе центрального иона металла. В литературе описаны случаи, когда гидроксоанионы трактуются как отдельные образования, а также случаи, когда гидроксоанионы скорее можно отнести к смеси соответствующих гидроксидов. Примерами первых являются: M2‘[Pt(OH)6] (M‘ = Li, Na) [3];
M2"[Cu(OH)6] (M" = Sr, Ba) [4J; Na[Zn(OHJ3] [5]; Ca[Zn(OH)3k-2H2O [6], а примерами вторых — Мн[8п(ОН)б] (Mn = Ca, Mg, Мп, Fe, Со, Ni, Си, Zn, Cd) [7]; М“[1г(ОН)6]’ (M'‘=Zn, Cd) [8]; M"[Pb(OH)6] (M“=Ca, Co) [9]; Fe[Ge(OH)6] [10]; M‘[Sb(OH)6] (M‘ = Na, Ag) [11], M2‘[Sn(OH)6] (M‘=Na, K); K2[Pt(OH)6] [12]; Ka[Ir(OH)6] [13]. Найдено, что целый ряд щелочноземельных гидроксометаллатов(III) типа M3II[MIII(OH)6]2 (М“ = Са, М1П=А1, Сг [14]; M" = Sr, М‘“=А1, Ga, Sc, In, Yb, Lu, Cr, Fe [14, 15]; M“ = Ba, Min=Al, Sc, In [16]) обладают структурой граната [17].
ЛИТЕРАТУРА
1. Scholder R., Schwochow F„ Angew. Chem., 78, 1102 (1966).
2. Scholder R„ Klemm IF., Angew. Chem., 66, 461 (1954); Scholder R„ Angew. Chem., 70, 583 (1958).
3. Tromel M„ Lupprich E„ Z. Anorg. Allgem. Chem., 414, 160 (1975).
4. Dubler E., Korber P„ Oswald H. R., Acta Cryst., B, 29, 1929 (1973^.
5. Schnering H. G. von, Naturwissenschaften, 48, 665 (1961); Angew. Chem., 77„ 1090 (1965); Habilitationsschrift, Universitat Munster 1963.
6. Liebau F„ Amel-Zadeh A., Kristall und Technik, 7, 221 (1972).
7. Strunz H., Contag B., Acta Cryst., 13, 601 (1960); Morgenstern-Badarau J., Billiet Poix P„ Michel A., C. R. Acad. Sci. Paris, 260, 3668 (1965).
8. Sarkozy R. F„ Chamberland B. L., J. Solid State Chem., 10, 145 (1974).
9. Levy-Clement C„ Morgenstern-Badarau I., C. R. Acad. Sci. Paris, Ser. C 270, 316 (1970).
10. Strunz H., Giglio M., Naturwissenschaften, 46, 489 (1959).
11. Schrewelius N., Z. Anorg. Allgem. CJiem., 238, 241 (1938).
12. Bjorling С. O., Arkiv Kemi, Mineral. Geol., 15 B, No 2 (1941).
13. Chamberland B. L., J. Less-Соттоц Metals, 44, 239 (1976).
14. Schwarz H., Z. Naturforsch., 22 b, 554 (1967).
15. Ito J., Frondell C„ Amer. Mineral., 52, 1105 (1967); Ito J., Amer. Mineral., 53, 1663 (1968); Ito J., Mater. Res. Bull., 3, 495 (1968).
16. Kwestroo IF., van Gerven H. C. A., van Hal H. A. M„ Mater. Res. Bull., 12, 161 (1977); Ahmed A. H. M., Glasser L. S. D., Acta Cryst., В 25, 2169 (1969).
17. Brandenberger E„ Schweiz. Mineralog. Petrogr. Mitt., 13, 569 (1933); Weiss R„ Grand jean D„ Acta Cryst., 17, 1329 (1964); Ducros P„ Durif-Varambon A., Bertaut E. F„ Delapalme A., Solid State Commun., 1, 85 (1963); Cohen-Addad C., Ducros P„ Durif A., Bertaut E. F„ Delapalme A., J. Physique, 25, 478 (1964).
Применение концентрированных щелочей
Исходные вещества. Обычно используют продажный препарат NaOH в виде гранул (пластинок) с содержанием 97—98% NaOH или твердый КОН со средним содержанием 85% (остальное вода).
Посуда. Используемая для синтеза стеклянная посуда должна быть устойчива к действию концентрированных щелочей (50%) при комнатной температуре в течение нескольких дней, а также в процессе синтеза гидроксосолей при комнатной температуре не должна разрушаться, иначе возможно загрязнение препарата. Горячие концентрированные щелочи быстро разрушают лю
1872 Глава 31. Гидроксосоли
бое стекло. Для таких опытов нужна специальная посуда из серебра, которая устойчива к действию кипящих щелочных растворов. Можно также использовать посуду из чистого никеля, что, однако, оказывается не дешевле, чем работать с серебряной посудой.
Фильтры. Обычно применяют иенские стеклянные пористые фильтры (G3 или G4, лучше всего № 17), однако от частого контакта с горячими щелочными растворами фильтры разрушаются и быстро приходят в негодность. При фильтровании горячих щелочных растворов рекомендуется обертывать нутч-фильтры (тигли с фильтрующим дном) полоской ткани, чтобы избежать возможного растрескивания. Если же используют горячую щелочь с концентрацией выше 50%, то нутч-фильтр обертывают жестяной манжеткой, предварительно смазанной глицерином. Пористые пластинки из известных искусственных материалов устойчивы к действию концентрированных щелочей, однако возможность их применения пока еще ие изучена.
Приготовление NaOH, не содержащего карбонатов. Сначала из продажного NaOH готовят в серебряной колбе 50%-ный раствор. Чтобы предотвратить осаждение избытка NaOH на дне колбы, ее все время встряхивают. Кипятить раствор с обратным холодильником нежелательно, так как горячая щелочь может выплеснуться через форштос холодильника. Приготовленный 50%-ный раствор щелочи оставляют медленно охлаждаться (в течение 2— 3 дней) при комнатной температуре, причем исключают контакт с СОг, закрыв колбу специальной защитной трубкой с коленом и шариком, заполненным 50%-ным раствором КОН*. В случае образования осадка Na2CO3 раствор щелочи фильтруют через стеклянный пористый фильтр 17G4 в отсутствие COj. Если полученный раствор прозрачен, то он практически не содержит карбонатов. Чтобы быстро избавиться от карбонатов, к 100 мл горячего 50%-ио-го раствора NaOH добавляют 1—2 г Ва(ОН)2-8Н2О и после охлаждения смеси до комнатной температуры сразу же ее фильтруют. ВаСО3 практически нерастворим в 50%-ном NaOH при комнатной температуре.
Разбавленные растворы NaOH готовят разбавлением 50%-ного раствора прокипяченной и охлажденной в отсутствие СО2 водой. Для растворов с концентрацией NaOH выше 50% отделение Na2CO3 проводят при 40—60 °C, так как при более низкой температуре кристаллизуется NaOH. Для получения такой высококонцентрированной щелочи из 50%-ного раствора NaOH отгоняют (без доступа СО2) в мерный цилиндр рассчитанный объем воды. Для исключения СО2 вся посуда, в которой находится щелочь, а также обратный холодильник и стеклянный нутч-фильтр, содержащие щелочные растворы, закрывают резиновыми пробками с защитными трубками, заполненными КОН.
Температуры кипения 20—70%-пых растворов NaOH и КОН определены Герлахом [2] (см. также [3]):
Концентрация NaOH (КОН): г/100гН2О % 25 20 42,8 30 53,8 35 66,7 40 81,8 45 100 50 122,5 55 150 60 233,3 70
U, °C: NaOH 108 116 121,5 128 134,5 142,5 150,5 160 180,5
кон 106 113 118 124,5 133 143 160,5 177,5 228
* В дальнейшем такую трубку будем называть просто «защитной трубкой». — Прим, перев.
Гидроксобериллаты 1873
О плотности и вязкости 12—72%-ных растворов NaOH в интервале 10— 70 °C см. данные Крингса [4].
Меры предосторожности. Для предохранения глаз от разъедающего действия щелочей необходимо работать в плотно прилегающих защитных очках. Если концентрированная щелочь попала под ноготь, то нужно как следует промыть палец под струей воды, а затем ополоснуть в разбавленной уксусной кислоте. При работе с серебряной посудой надо учитывать ее большую теплопроводность по сравнению со стеклянной.
ЛИТЕРАТУРА
1. Scholder R., Chem. Fabrik, 11, 541 (1938).
2. Gerlach G. Th., Z. Analyt. Chem., 26, 463 (1887).
3. Antropoff A. von, Sommer W., Z. Physik. Chemie, 123, 192 (1926).
4. Krings W., Z. Anorg. Allgem. Chem., 255; 294 (1948).
Тетрагидроксобериллат натрия Na2[Be(OH)4]
BeO + 2NaOH + H2O -----> Na2[Be(OH)4l
25,0 80,0 18,0 123,0
Растворы гидроксобериллата натрия, насыщенные при высокой температуре, обладают большой вязкостью, в значительной мере склонны к пересыщению и. часто не кристаллизуются даже при многодневном стоянии при комнатной температуре. Кристаллизацию Na2[Be(OH)4l вызывают внесением затравки (см. далее). К 160 г 56%-иого раствора NaOH добавляют 15 г ВеО и кипятят смесь в течение часа с обратным холодильником. Полученный раствор фильтруют при температуре не ниже 80 °C через пористый фильтр G4, затем фильтрат охлаждают до комнатной температуры и вносят в прозрачный раствор затравку. Через 12—24 ч отсасывают осадок через фильтр G3, один раз промывают 50%-ным раствором NaOH, распределяют вещество тонким слоем на керамической пластинке и сушат в вакууме над смесью КОН — силикагель. Выход —15 г (—20% в расчете на ВеО).
Кристаллы для затравки, а) Несколько миллилитров пересыщенного раствора гидроксобериллата натрия выдерживают при 0°С. Через несколько дней выпадают хорошо образованные кристаллы, имеющие форму брусочков, которые используют в качестве затравки.
б) К 1 мл пересыщенного раствора гидроксобериллата натрия добавляют при комнатной температуре гранулу твердого NaOH и оставляют раствор стоять в течение —8 ч. На поверхности гранулы образуются мелкие кристаллы, которые используют в качестве затравки.
Свойства. Бесцветные столбики. По способу с затравкой (а) получается более чистое вещество (столбики величиной I—2 мм), а по способу (б) — мелкие кристаллы, иногда плохо образованные, величиной 0,2—0,3 мм, чистота которых невысока из-за адсорбции или захватывания маточного раствора. Захваченный NaOH можно частично удалить, обрабатывая кристаллы абсолютным спиртом, содержащим 12—16% NaOH. (Растворимость равна 3—4 г Na2[Be(OH)4] в 100 мл спирта при 0°С.)
Взаимодействием осадка Na2[Be(OH)4] с раствором NaOH меньшей концентрации получают Na[Be(OH)3],
Тетрагидроксобериллат стронция Sr[Be(OH)i]
Ве(С1О4)2 + Sr(OH)2-8H2O -> Sr[Be(OH)4] -f- Sr(C104)2 + 16Н2О
207,9 531,5 164,7 286,5 288,2
2—1261
1874 Глава 31. Гидроксосоли
50 г Sr(OH)2-8H2O растворяют при 80—90°С в 500 мл Н2О и выпавший при 60 °C осадок SrCO3 отфильтровывают. К фильтрату при 60 °C добавляют по каплям при перемешивании раствор 10,4 г Ве(С1О4)2 в 15 мл Н2О и оставляют раствор иа ночь в сушильном шкафу при 60“С. Выпавшие кристаллы фильтруют через стеклянный пористый фильтр G4, промывают 25 мл абсолютного метанола и затем 5—7 г вещества дважды настаивают некоторое время со 100 мл абсолютного метанола. После фильтрования вещество промывают небольшим количеством абсолютного метанола и абсолютного эфира и быстро высушивают в вакууме над смесью КОН — силикагель.
Свойства. Кристаллы в виде брусочков. Устойчивы к действию абсолютного метанола. Медленно разлагаются водой.
ЛИТЕРАТУРА
1. Scholder R., Hund Н., Schwarz Н., Z. Anorg. Allgem. Chem., 361, 284 (1968).
Тетрагидроксомагнезиат натрия Na2[Mgf(OH)i]
Mg(OH)2 + 2NaOH -----> Na2[Mg(OH)4l
58,3 80,0 138,3
Na2[Mg(OH)4] получают из высококонцентрированных, по меньшей мере 61%-ных, растворов NaOH при их взаимодействии с твердым Mg(OH)2. Отгоняя 180 мл воды из 500 мл 50%-кого раствора NaOH, налитого в серебряную колбу, получают щелочь с концентрацией 65%. К горячему (100 °C) раствору щелочи добавляют 6 г Mg(OH)2. Для приготовления Mg(OH)2 прокаленный при 500 °C MgO кипятят с обратным холодильником в серебряной колбе с серебряной мешалкой в течение 20 ч при 100 °C, строго следя за отсутствием СО2 в системе. Затем, не прерывая нагревания, смесь из колбы пересасывают через серебряную трубку и полиэтиленовый шланг в тигель с фильтрующим дном 17G3 и фильтруют прн 110 °C на глицериновой бане. Вещество отсасывают по возможности досуха, затем отжимают на нагретой до 100 °C керамической пластинке и оставляют на ней же на 5 ч в вакуум-эксикаторе при 100°С. Таким образом получают 8—10 г относительно сухого, ио сильно загрязненного NaOH продукта. Для удаления остаточного NaOH 3 г измельченного продукта встряхивают 30—45 мин (в отсутствие влаги и СО2) со свеже-перегнанным изоамиловым спиртом (берут фракцию, собранную при 127— 129°C), затем фильтруют смесь через стеклянный фильтр 11G3 и промывают осадок 50 мл изоамилового спирта и эфира. Вещество окончательно высушивают в эксикаторе в течение нескольких часов на силикагеле, причем одновременно удаляются остатки эфира.
Свойства. Бесцветные микрокристаллические гексагональные листочки. При действии Н2О образуется кристаллический Mg(OH)2 (бруцит). При действии щелочных растворов метанола и этанола (8—15% NaOH) вещество разлагается даже при 0°С, изоамиловый спирт только при очень продолжительном действии вызывает отщепление NaOH. Рентгенограммы порошка Na2[Mg(OH)4] аналогичны рентгенограммам Na2[Co(OH)4] и Na2[Ni(OH)4) (высокотемпературная форма).
ЛИТЕРАТУРА
1. Scholder R., Keller С., Z. Anorg. Allgem. Chem., 317, 113 (1962).
2. Scholder R., Giesler E„ Z. Anorg. Allgem. Chem., 316, 237 (1962).
Гидроксостаннаты(П) 1875
Гексагидроксомагнезиат бария Ba2[Mg(OH)6]
Mg(C104)2 + Ва(С1О4)2 + 6NaOH----> Ba2[Mg(OH)6) + 6NaC104
223,2 672,5 240,0 401,0 734,6
К 500 мл 48%-ного раствора NaOH в серебряной колбе добавляют 4 г твердого Mg(ClO4)2 и такое количество почти насыщенного раствора Ва(С1О4)2, чтобы молярное отношение Mg(C104)2: Ва(С1О4)2 было равно 1 ; (3,5—4,0). Для этого потребуется 21,1—24,1 г перхлората бария. После 24 ч кипячения раствора с обратным холодильником раствор фильтруют через стеклянный пористый фильтр G3, промывают 100 мл метанола и 30 мл эфира, а затем встряхивают остаток в течение 2 ч с 250 мл метанола. Вновь фильтруют раствор, промывают метанолом и эфиром и сушат в вакуум-эксикаторе над силикагелем.
Свойства. Кристаллы — бесцветные призмы, часто плохо образованные. При действии воды (0°С) быстро и полностью разлагается. Комплекс изо-структурен Ba2j[Zn(OH)6] и Ваа[Си(ОН)б].
ЛИТЕРАТУРА
1. Schnering Н. G. von, Angew. Chem., 77, 1090 (1965).
2. Dubler E„ Korber P., Oswald H. R., Acta Cryst., B29, 1929 (1973).
3. Scholder R., Keller C„ Z. Anorg. Allgem. Chem., 317, 113 (1962).
Тригидроксостаннат(П) натрия Na[Sn(OH)3]
SnCL-2H.O -+-2NaOH ---> Sn(OH)2 + 2NaCl + 2H2O
225,6 80,0 152,7 116,9 36,0
Sn(OH)2 + NaOH ---> Na[Sn(OH)g]
152,7 40,0 192,7
Для получения Sn(OH)2 к молочного цвета раствору 25 г SnCl2-2H2O в 1,5 л воды добавляют при комнатной температуре небольшой избыток 10%-ного аммиака и разбавляют смесь до объема 2 л. После отстаивания слегка мутную жидкость фильтруют, добавляют 2 л воды и снова фильтруют, повторяя этот прием 2—3 раза. Осадок Sn(OH)2 сначала фильтруют через пористый фильтр 25G3, а затем (осторожно!) под слабым вакуумом на воронке Бюхнера и отмывают водой на фильтре от ионов С1-. В полученном в виде пасты осадке определяют содержание Sn(OH)2, прокаливая пробу осадка до образования SnO2. Выход Sn(OH)2 —85%, содержание Sn(OH)2 в пасте —50%. В конической стеклянной длинногорлой колбе на 150 мл растворяют 35 г NaOH в 23 мл воды. В раствор, имеющий температуру 50—60 °C, не обращая внимания на неполностью растворившийся NaOH, вносят пасту SnJOH)2 и колбу немедленно закрывают резиновой пробкой с защитной трубкой, заполненной раствором пирогаллола. После кратковременного встряхивания Sn(OH)2 растворяется в —50%-ной щелочи, причем выделяется небольшое количество темного SnO. Теплый раствор фильтруют через стеклянный пористый фильтр 17G4. Из прозрачного фильтрата, охлажденного до 0°С без доступа воздуха, через несколько часов кристаллизуется соль. Перед фильтрованием соли стеклянный тигель с фильтрующим дном 17G2 нагревают до ° С. Для удаления захваченной осадком щелочи кристаллы распределяют на холодной (0 °C) керамической пластинке и выдерживают в пустом вакуум-эксикаторе при 0—3°С. Выход ~6 г (33% в расчете на SnCl2-2H2O).
Свойства. Бесцветные, чуть заостренные на концах столбики, частично
2'
1876 Глава 31. Гидроксосоли
объединенные в агрегаты. Na[Sn(OH)3], помещенный в закрытый сосуд при О °C, через некоторое время разлагается и темнеет, чувствителен к действию влаги и кислорода воздуха. Препарат, счищенный с керамической пластинки, содержит 0,1—0,2 моля NaOH. В процессе выделения и высушивания препарата небольшая часть его переходит из Sn(II) в Sn(IV).
Кристаллическая структура орторомбическая (пространственная группа Р212121, а= 14,45 А, 6=16,79 А, с=5,89 А). Согласно [2], Na[Sn(OH)3] за 24 ч стояния под маточным раствором количественно переходит в изоморфную, ио более бедную водой бесцветную форму Na[Sn(OH)2,5O2,s]=Na4[Sn(OH)3]2' [Sn2O(OH)4], имеющую моноклинную кристаллическую структуру с а= = 17,35 А, 6=8,23 А, с=22,20 А, 3=101°. Под действием рентгеновских лучей оба эти соединения разлагаются за 3—4 ч.
ЛИТЕРАТУРА
1. Scholder R., Patsch R. Z. Anorg. Allgem. Chem., 215, 176 (1933); Scholder R., Krauss К, неопубликованные данные.
2. Schnering H. G. von, Angew. Chem., 77, 1090 (1965); частное сообщение.
Оксогидроксостаннат(П) бария Ba[Sn2O(OH)4]
2Na[Sn(OH)3] + Ba(OH)2-8H2O --> Ba[SnO(OH)4] + 2NaOH + 9H2O
385,4 315,5 458,8 80,0 162,1
Весь полученный из 2,5 г SnCl2-2H2O гидроксид Sn(OH)2 в виде пасты (см. выше синтез Nai[Sn(OH)3]) вносят в раствор 60 г NaOH в 50 мл НгО при 50 °C. Раствор охлаждают до 30 °C, добавляют горячий раствор 1 г Ва(ОН)2-8Н2О в 2 мл воды, оставляют стоять ~1 ч при этой температуре (без доступа воздуха) и отфильтровывают от темного SnO и выпавшего карбоната. Фильтрат нагревают до 65°C, добавляют горячий (95°C), предварительно отфильтрованный раствор 9 г Ва(ОН)2-8Н2О, после чего за несколько минут выделяется зеленовато-желтый осадок Ba[Sn2O(OH)4]. Осадок промывают декантацией, фильтруют, промывают на фильтре 50%-ным раствором NaOH, затем 50 мл 2%-ного раствора Ва(ОН)2-8Н2О в метаноле и, наконец, чистым метанолом при 0°С. Осадок высушивают на керамической пластинке над силикагелем. Выход 5 г (—20% в расчете на SnCl2-2H2O).
Свойства. Желтоватый микрокристаллический порошок. Разлагается водой. Содержит примесь ВаСО3. Исследования строения препарата показали, что он отщепляет 1 молекулу воды и переходит в Ba[Sn0(OH)2], структура которого включает цепи [SnO(OH)]. Возможно, что желтая окраска обусловлена полимерной структурой, так как анион [Sn2O(OH)]2~ бесцветен (ср. с Na[Sn(OH)3]).
ЛИТЕРАТУРА
1. Scholder R„ Putsch R., Z. Anorg. Allgem. Chem., 216, 176 (1933); Scholder R., Krauss К, неопубликованные данные.
2. Schnering H. G. von, частное сообщение (1976).
Тетрагидроксоманганат(П) натрия Na2[Mn(OH)4]
Мп(ОН)2 4- 2NaOH ---> Na2[Mn(OH)4]
89,0 80,0 169,0
Из 700 г NaOH и 700 мл воды готовят раствор NaOH и фильтруют его. Полученный раствор и 14 г микрокристаллического Мп(ОН)2 (синтез
Гидроксоферраты(П) . \87Т
Мп(ОН)2 см. в работе [1]) кипятят в серебряной колбе с обратным холодиль-ником без доступа воздуха в течение 1,5 ч. При этом основная часть Мп(ОН)»-растворяется. Раствор фильтруют при 125 °C через стеклянный фильтр 17G3. Фильтрат переливают в коническую колбу, снабженную защитной трубкой с пирогаллолом, охлаждают до 75 °C и некоторые время выдерживают при этой температуре. Прозрачный, слегка коричневый раствор быстро кристаллизуется. Осадок фильтруют, быстро промывают 50%-ным раствором NaOH при 70°C и высушивают на керамической пластинке без каких-либо осушающих средств в эксикаторе, заполненном азотом.
Свойства. Рассыпчатый желтый, местами коричневатый кристаллический порошок, чувствительный к действию О2 и Н2О. Вследствие незначительного окисления препарата кислородом воздуха он всегда содержит марганец в высших степенях окисления (Мп : активный кислород = 1 : 0,01—0,05).
ЛИТЕРАТУРА*
1. Scholder R., Kolb A., Z. Anorg. Allgem. Chem., 264, 209 (1951).
Тетрагидроксоферрат(П) натрия Ма2[Ре(ОН)4]
Fe + 2NaOH + 2H2O -----> Na2[Fe(OH)4J + H2
55,9 80,0 36,0 169,9 2,0
8 г восстановленного железа помещают в круглодонную серебряную колбу и приливают 350 мл 50%-ного раствора NaOH. К колбе на резиновых пробках присоединяют обратный холодильник (с защитной трубкой со ще-. лочью) и серебряную трубку, через которую пропускают очищенный азот. Для удаления воздуха смесь в колбе кипятят в течение 2,5 ч в токе N2. Полученный синий раство’р охлаждают до 120 °C и фильтруют на стеклянном фильтре через слой восстановленного железа при строгом отсутствии воздуха, собирая, фильтрат в склянку со 100 мл 50%-ного раствора NaOH (в атмосфере азота).; Раствор охлаждают в токе N2 в течение 12 ч, отфильтровывают от серо-зеленых частиц, промывают 5О°/о-ным раствором NaOH и высушивают иа керамической пластинке в эксикаторе с азотом. Выход 4 г (—16% в расчете на железо).
Свойства. Серо-зеленый микрокристаллический порошок, крайне чувствительный к влаге и кислороду воздуха. Под микроскопом наряду с полиэдрами Na2[Fe(OH)4] хорошо заметны бесцветные скошенные пластинки Na4[Fe(OH)7]-2Н2О (ср. ниже синтез гидроксоферратов(III) бария).
ЛИТЕРАТУРА
1. Scholder R„ Angew. Chem., 49, 255 (1936).
Тетрагидроксокобальтат(П) натрия Na2[Co(OH)4]
Со(ОН)2 + 2NaOH ----> Na2[Co(OH)4)
92,9 80,0 172,9
Исходные вещества: микрокристаллический Со(ОН)2 (см. далее) и порошок NaOH (см. далее) — смешивают в сухой камере, заполненной сухим N2, в молярном соотношении Со(ОН)2: NaOH=l : 2,01. Например, берут 1,018 г Со(ОН)2 (99,28%) и 0,879 г NaOH (99,5%). Смесь светло-розового цвета запаивают (в атмосфере азота) в маленькой стеклянной ампуле. Для
1878 Глава 31. Гидроксосоли
полного удаления кислорода рекомендуется ампулу с содержимым перед тем, как запаять, несколько раз вакуумировать и заполнить чистым азотом. После выдерживания ампулы в течение 14 ч при 120 °C взаимодействие компонентов приводит к образованию слабоспекшегося красно-фиолетового вещества, местами окрашенного в коричневый цвет, вероятно, из-за гидролиза. Для предотвращения окисления препарата в процессе его получения все манипуляции в сухой камере надо производить возможно быстрее.
Микрокристаллический Со(ОИ)2. К 400 мл кипящего 50%-ного раствора NaOH в серебряной колбе добавляют по каплям (в отсутствие СОг) раствор 82 г СоСЬ-бНзО в 103 мл HjO, после чего смесь кипятят еще полчаса. После охлаждения до комнатной температуры осадок фильтруют через вороику с фильтрующим дном G4 и промывают 80 мл 50%-ного раствора NaOH. Влажный осадок взбалтывают с 1 л горячей воды, содержащей сульфат гидразина, затем фильтруют, промывают еще 2 л горячей воды (с добавкой сульфата гидразина), потом ацетоном, эфиром и сушат в эксикаторе над силикагелем.
Порошок NaOH. Для удаления небольшого количества воды (1—2°/о) из NaOH и для последующего его распыления берут продажный препарат (пластинки или гранулы NaOH) и сначала переплавляют его в атмосфере N2. Лучше всего для этого использовать большие серебряные лодочки, в которые помещают около 20 г NaOH (желательно из только что вскрытой банки, чтобы уменьшить вероятность присутствия карбонатов). Лодочки с NaOH ставят в стеклянную трубку, которую помещают в трубчатую печь с температурой 500 °C. Вне зоны нагрева в трубку с обоих концов ставят две большие фарфоровые лодочки с Р«Ою для поглощения выделяющейся при плавлении воды. Полного удаления воды добиваются прокаливанием 15—20 г NaOH в течение 6 ч при 500 °C в токе чистого азота. Застывший расплав извлекают из серебряной лодочки в сухой камере с N2 и затем тонко измельчают его в агатовой ступке или (лучше) в герметичной шаровой мельнице с агатовыми шарами. Растертый в порошок NaOH (1—5 г) рекомендуется хранить до дальнейшего употребления в плотно закрытом бюксе. Анализ полученного порошка щелочи показывает, что содержание NaOH достигает ~99,5%.
Свойства. Микрокристаллический красно-фиолетовый порошок, чувствительный к действию О2 и НгО. По описанной методике получают низкотемпературную модификацию, тогда как из раствора при 155—160°С выделяется коричнево-фиолетовая высокотемпературная модификация. Рентгенограммы порошка обеих модификаций сходны с таковыми для форм Na2[Ni(OH)4] и Naa[Mg(OH)4].
Получение аналогов. Способом, аналогичным описанному для Na2[Co (ОН)«], можно получать очень чистые гидроксометаллаты двухвалентных металлов, например, соединения типа Na^[M"(OH)4] —путем нагревания тонко измельченных гидроксидов, взятых в стехиометрических количествах, в токе чистого азота. Так, низкотемпературную форму NagfMgfOH^] получают при 65 °C, а высокотемпературную— при 100 °C; Na2[Ni(OH)4] —при 140 °C; Na2[Cu(OH)4] — при 75 °C; Na2fZn(OH)4] — при 55 °t. Только лишь легко окисляющиеся гидроксометаллаты Наг[Со(ОН)4] и Na2[Mn(OH)4] (130°С) рекомендуется получать взаимодействием компонентов в замкнутой системе в атмосфере чистого N2, например в маленьких ампулах, запаянных в атмосфере N2. Обычно 15 ч бывает достаточно для завершения реакции. Метод годится также для получения гидроксоферратов(Ш) бария и натрия (см. ниже).
ЛИТЕРАТУРА
1. Scholder R., Schwochow F., Angew. Chem., 78, 1102 (1966).
2. Scholder R„ Weber H., Z. Anorg. Allgem. Chem., 216, 159 (1933).
3. Scholder R„ Giesler E„ Z. Anorg. Allgem. Chem., 316, 237 (1962).
4. Scholder R„ Keller C., Z. Anorg. Allgem. Chem., 317, 113 (1962).
Гидроксоникколаты 1879*
Гексагидроксоникколат(П) стронция Sr2[Ni(OH)6]
Ni(C104)2 + 2Sr(C104)2 + 6NaOH -> Sr2[Ni(OH)e] + 6NaC104
257,6 573,0 240,0 336,0 734,0
250 г NaOH растворяют в 455 мл H2O в серебряной колбе, добавляют 8 г SrfOHJj-SHjO, кипятят с обратным холодильником непродолжительное время и оставляют иа сутки стоять. Выделившийся SrCO3 отфильтровывают. В кипящий раствор полученной щелочи прибавляют по каплям раствор 35 мл Ni(C104)2 (7 г) и Sr(C104)2 (31 г) (молярное соотношение 1 :4, получение см. ниже) и кипятят реакционную смесь 12 ч с обратным холодильником без доступа СО2. Осадок 5гз[№(ОН)б] отфильтровывают от горячего раствора через пористый фильтр 17G4 (в отсутствие СО2), промывают 35%-ным раствором NaOH при комнатной температуре, а затем абсолютным метанолом. Осадок встряхивают в течение 8 ч с абсолютным метаиолом, фильтруют и промывают метаиолом и эфиром. При высушивании вещества в вакуум-эксикаторе иад силикагелем в течение нескольких часов удаляются следы эфира.
Приготовление раствора Ni(ClOt)2+Sr(ClOt)t. К 6,46 г NiCl2-6H2O и 15,97 г БгСОз добавляют 25 мл воды и постепенно прибавляют 24 мл 70%-иой НС1О4 (d 1,67). Полученный раствор, содержащий некоторый избыток НС1О4, упаривают для удаления НС1 до тех пор, пока не появится густой «туман» НС1О4, после чего смесь разбавляют 35 мл воды.
Свойства. Серо-зеленый мелкокристаллический порошок, не содержащий кристаллов строго определенной формы. Соединение не взаимодействует с по-лунасыщенным раствором Sr(OH)2 (0,35 г SrO на 100 мл НгО). Постепенно разлагается водой.
ЛИТЕРАТУРА
1. Scholder R., Giesler Е., Z. Anorg. Allgem. Chem., 316, 237 (1962).
Тетрагидроксокупрат(П) натрия Na2[Cu(OH)4]
CuO + H2O4-2NaOH-------> Na2[Cu(OH)4]
79,5 18,0 80,0 177,6
Неочищенный (сырой) продукт. В теплый раствор щелочи (без карбонатов), содержащий 500 г NaOH в 330 мл Н2О, насыпают 15 г чистого СиО и растворяют оксид непродолжительным кипячением с обратным холодильником. Темно-синий раствор при 110 °C осторожно разбавляют, вливая через обратный холодильник 140 мл воды, и фильтруют раствор при 100 °C в предварительно нагретую коническую колбу от незначительного нерастворившегося количества СиО. Фильтрат в конической колбе закрывают защитной трубкой с 50%-ным раствором КОН и оставляют кристаллизоваться в течение 6 дней в сушильном шкафу при 75 °C. Выпавший осадок промывают иа фильтре небольшим количеством 45—50%-ного раствора NaOH при комнатной температуре и высушивают иа керамической пластинке над H2SO4. Выход 13 г (~39% в расчете на СиО).
Если фильтрат после отделения СиО быстро охладить в смеси льда с NaCl, то вещество выделяется быстро в виде очень тонких светло-синих табличек, которые трудно освободить от избытка NaOH. Выход 20 г.
Чистое вещество. Для удаления значительных количеств захваченной щелочи темно-синие кристаллы после промывания 50%-ным раствором NaOH вносят (при комнатной температуре) в 150 мл 40%-ного NaOH, встряхивают 1 ч последовательно со следующими смесями: 150 мл СН3ОН-|-22,5 г NaOH
1880 Глава 31. Гидроксосоли
(18°С); 150 мл СНзОН+15 г NaOH (0°C); 150 мл СН3ОН + 1,5 г NaOH (—10 °C). После декантации кристаллы дважды настаивают с чистым СНзОН (—10 °C), фильтруют и промывают СНзОН (—15°C). Кристаллы сушат иа керамической пластинке над силикагелем в эксикаторе возможно малого размера.
Свойства. Твердые темно-синие кристаллы. Чистое вещество крайне чувствительно к влаге и на воздухе быстро становится темно-коричневым. Кристаллическая структура орторомбическая (пространственная группа P2i2t2i, а=6,75 А, 5 = 6,78 А, с=9,02 А).
ЛИТЕРАТУРА
1. Scholder R., Felsenstein R-, Apel R., Z. Anorg. Allgem. Chem., 216, 138 (1933).
2. Schnering H. G. von, Angew. Chem., 77, 1090 (1965).
Гексагидроксокупрат(П) бария Ba2[Cu(OH)6]
Na2[Cu(OH)4l + 2Ba(OH)2-8H2O ---> Ba2[Cu(OH)el + 2NaOH + 16H2O
177,6 631,0 440,3 80,0 288,2
К 250 мл 50%-иого раствора NaOH (без карбонатов) при 5 °C прибавляют раствор 10 г CuBr2 в 25 мл Н2О, нагревают смесь на водяной бане до 70 °C, отфильтровывают раствор от небольшого количества СиО и кипятят раствор с обратным холодильником при 130 °C. Готовят раствор 30 г Ва(ОН)2-8Н2О в 50 мл Н2О, фильтруют его горячим через складчатый фильтр н добавляют к первоначальному раствору. Выделившийся осадок сразу же отфильтровывают, переносят в коническую колбу, охлаждают до 0°С, в течение 5 мин встряхивают со 100 мл СНзОН при —10 °C, фильтруют (0°С) и промывают ацетоном и безводным эфиром. Удаляют эфир длительным выдерживанием в вакуум-эксикаторе. Выход 13 г (~66% в расчете на CuBr2).
Свойства. Светло-синий микрокристаллический порошок. Чистый препарат разлагается водой. Кристаллическая структура моноклинная (пространственная группа P2i/c; а=6,030 А, 6 = 6,440 А, с=10,115 А, (3=124,3°). Структура состоит из изолированных искаженных октаэдров [Сп(ОН)б]4- и похожа на сильно искаженную кубическую структуру KsPtCU.
Препарат изоструктурен Sr2[Cu(OH)6] (а=5,786 А, 6=6,154 А, с= =9,744 А, (3=124,15°).
ЛИТЕРАТУРА
1. Scholder R„ Felsenstein R., Apel R., Z. Anorg. Allgem. Chem., 216, 138 (1933).
2. Dubler E., Korber H. R., Oswald H. R., Acta Cryst., B29, 1929 (1973).
Гидроксоцинкаты натрия
В системе ZnO—Na2O—Н2О в зависимости от концентрации NaOH существуют 4 различных твердых гидроксоцинката: Na[Zn(OH)3]-ЗНгО; Na[Zn(OH)3]; Na2[Zn(OH)4]-2Н2О и Na2[Zn(OH)4],
Тригидроксоциикат натрия Na[Zn(OH)3]
ZnO + NaOH + Н2О ------> Na[Zn(OH)3]
81,4 40,0 18,0 139,4
Гидроксоцинкаты 1881
а) Неочищенный (сырой) продукт, получение кристаллов для затравки. К горячему раствору 185 г NaOH в 100 мл Н2О добавляют 105 г ZnO и кипятят смесь 1/2 ч с обратным холодильником. Затем смеси дают остыть до 100 °C и через холодильник добавляют понемногу 85 мл воды. Горячий раствор фильтруют и тотчас же охлаждают до 15 °C. Если в течение дня раствор не кристаллизуется, то в нескольких миллилитрах кипящего раствора гидроксо-цинката растворяют одну гранулу NaOH, охлаждают раствор в смеси льда с NaCl и выделившийся при большей концентрации NaOH тетрагидроксоции-кат используют в качестве затравки, помещая его в 5 мл охлажденного первоначального раствора. Потирая стеклянной палочкой о стеики пробирки с раствором, вызывают кристаллизацию тригидроксоцинката. Полученные таким образом кристаллы вносят как затравку в общую массу раствора, причем в течение часа выделяется тригидроксоцинкат. Осадок фильтруют, промывают 50%-ной щелочью и сушат в пустом эксикаторе на керамической пластинке. Выход 50—60 г (—28—33% в расчете на ZnO).
б) Получение чистого препарата. Кипятят раствор 60 г ZnO в 250 мл 51%-ного NaOH, фильтруют при 40°C и охлаждают до 15°C. Вызывают кристаллизацию вещества, внося затравку. Через 12 ч фильтруют раствор и далее делают все так же, как указано в п. «а». Выход 40 г (—40% в расчете на ZnO). 10 г влажного препарата встряхивают 2 ч со 150 мл щелочного раствора СН3ОН (100 мл СНзОН+15 г NaOH), затем фильтруют, промывают сначала фильтратом (СНзОН), а потом ацетоном и сушат над силикагелем. При таком способе высушивания полностью удаляется NaOH, и результаты анализа вещества на ZnO, Na2O и Н2О хорошо совпадают с теорией.
Свойства. Бесцветный микрокристаллический порошок (мелкие палочки). Быстро разлагается водой, а 10%-ным раствором NaOH в СН3ОН — в течение 1 ч. Устойчив в 15%-ном растворе NaOH в СНзОН при 18 °C. Препарат, полученный по способу (а), содержит примесь NaOH в количестве 0,1 моль на 1 моль гидроксоцинката. Кристаллическая структура тетрагональная (пространственная группа P42/mbc, а= 10,86 А, с=5,35 А).
Тетрагидроксоцинкат натрия Na2[Zn(OH)4]
ZnO + 2NaOH + Н2О ------> Na2[Zn(OH)4l
81,4 80,0 18,0 179,4
Готовят прозрачный раствор 195 г NaOH в 140 мл Н2О (с исключением СО2), растворяют в нем при кипении 56 г ZnO и фильтруют при 90 °C. Через 1 ч выделяются кристаллы, которые промывают 50%-иым раствором NaOH и сушат на керамической пластинке в пустом эксикаторе, распределив вещество возможно более тонким слоем. Выход —100 г (—81% в расчете на ZnO).
Свойства. Кристаллы — бесцветные тонкие микрокристаллические пластинки. Препарат содержит около 0,2 моль NaOH на 1 моль гидроксоцинката, причем щелочь удалить невозможно.
ЛИТЕРАТУРА
1. Scholder R„ Weber Н„ Z. Anorg. Allgem. Chem., 215, 355 (1933).
2. Scholder R„ Hendrich G., Z. Anorg. Allgem. Chem., 241, 76 (1939).
3. Schnering H. G. von, Naturwissenschaften, 48, 665 (1961); Angew Chem, 77, 1090 (1965).
Гексагидроксокадмиат бария Ba2[Cd(OH)6]
Cd(OH)2 + 2Ba(OH)2 • 8H2O ---> Ba2[Cd(OH)6] + 16H2O
146,4 631,0 489,1 288,2
1882 Глава 31. Гидроксосоли
В серебряную колбу помещают 250—400 г NaOH в 400 мл воды и 30 г Ва(ОН)2-8Н2О и перемешивают некоторое время при 90—100°С. Раствор фильтруют горячим, снова доводят фильтрат до кипения и добавляют раствор 5 г ацетата кадмия в 10 мл Н2О. После этого смесь кипятят еще 3 ч с обратным холодильником при постоянном перемешивании. Первоначально выпавший хлопьевидный осадок Cd(OH)2 переходит при кипячении в кристаллизующийся Вай[С<1(ОН)б], который отфильтровывают от горячего раствора, промывают 40%-ным раствором NaOH и сушат на керамической пластинке в пустом эксикаторе.
Свойства. Бесцветный микрокристаллический порошок. Препарат загрязнен NaOH, содержащимся в количестве 0,1—0,2 моль на 1 моль соли.
ЛИТЕРАТУРА
1. Scholder R., Staufenbiel Е., Z. Anorg. Allgem. Chem., 247, 259 (1941).
Алюминаты щелочных металлов
Как показано в работе [1], в растворах гидроксоалюминатов нет полианионов и существуют лишь мономеры состава [А14(ОН)4]“, тогда как твердые гидроксо- и оксогидроксоалюминаты натрия имеют сложный состав. В системе А120з—Na2O—Н20 из растворов с близкими значениями концентраций NaOH и А12О3 кристаллизуются (в зависимости от температуры) 3 различные алюмината натрия:
Гептагидроксоалюминат(тетра) натрия Na4<[Al(OH)7] -ЗН2О
Оксогидроксоалюминат (моно) натрия I 3Na2O • А12О3 • 2,67Н2О=Na6[Al6O4 (ОН) ы] Оксогидроксоалюминат (моно) натрия II 2Na2O • А12О3 • 2,5Н2О=Na4[Al4O3 (ОН) ю]
Из раствора алюмината калия всегда выделяется оксогидроксоалюминат калия К3О • А12О3 • ЗН2О — К2[А12О (ОН) 3].
В связи с этим интересен тот факт, что в системе ВаО—А12О3—Н2О, для которой получены структуры соединений (кроме структуры гидрограната ЗВаО'А12О3-6Н2О=Ва2([А1(ОН)б]2=Ва3|[А12О3(ОН)12]), вообще ие существует простых анионов, а присутствуют соединения:
2ВаО • А12О3 5Н2О= Ва2[А12 (ОН) 10]
а-ВаО А12О3 • 2Н2О=а-Ва ГАЮ (ОН) 2] 2
у-ВаО • А12О3- 2Н2О=у-Ва [А1О (ОН) 2] 2
а-ВаО • А12О3 • 4Н2О=а-Ва^[А14 (ОН) i3]
Гептагидроксоалюминат натрия Na4[Al(OH)7]«3H2O
AI(OH)3 + 4NaOH + ЗН2О ----► Na4[AI(OH),]-3H2O
78,0 160,0 54,0 2 92,0
В серебряной колбе растворяют при кипячении 300 г NaOH и 50 г А1(ОН)3 в 200 мл воды. Доводят температуру смеси до 60—65 °C и перемешивают смесь в течение 3 ч, затем фильтруют осадок через пористый стеклянный фильтр G2, быстро отжимают на керамической пластинке и сушат 3 дня в вакуум-эксикаторе над Р4Ою. Из маточного раствора вскоре выделяется вторая порция кристаллов, которые больше по размеру и лучше образованы. Их выделяют и сушат так же, как и кристаллы первой порцни. Вторую порцию кристаллов благодаря их величине легко очистить от захваченной веществом щелочи, поэтому из второй порции получается более чистый препарат.
Свойства. Длинные призматические оптически двуосные кристаллы, проявляющие двойное лучепреломление. Получить чистое вещество удается лишь
Алюминаты 1883
из раствора щелочи высокой концентрации (по меньшей мере 50%), причем это не так просто, как описано выше. Пока еще не известно, как промывать кристаллы, не вызывая одновременно их разложения. Даже 20%-ный раствор NaOH в СН3ОН или в абсолютном С2Н6ОН вызывает быстрое разложение вещества. Поэтому препараты содержат от 0,2 до 0,4 моль NaOH на 1 моль алюмината.
Оксогидроксоалюминат натрия I Na4[Al6O4(OH)i6] 6Al(OH)g + 6NaOH ---> Na6[AleO4(OH)16] + 4H2O
468,0 240,0 635,9 72,1
Прозрачный раствор алюмината, полученный кипячением 130 г NaOH и 50 г А1(ОН)3 в 100 мл воды, охлаждают до 40°C и выдерживают при этой температуре, непрерывно перемешивая, в течение 12—15 ч. Образовавшийся осадок фильтруют через пористый стеклянный фильтр G3 и промывают последовательно 30 мл холодного 30%-ного раствора NaOH, 30 мл 10%-ного раствора NaOH в метаноле, 30 мл метанола (с добавлением 10 капель предыдущего промывающего раствора), дважды промывают 50 мл этанола и 50 мл эфира. Препарат сушат в течение суток в вакуум-эксикаторе на керамической пластинке над Р4О10.
Свойства. Бесцветный сильно гигроскопичный микрокристаллический порошок. Кристаллы оптически одноосные. Полностью удалить щелочь очень трудно. По результатам анализа отношение Al: Na близко к 1: (1,02—1,04).
Оксогидроксоалюминат натрия II Ыа4[А14Оз(ОН)ю]
5А1(ОН)3 + 4NaOH ----> Na4[Al4O3(OH)10] + ЗН2О
312,0 160,0 418,0 54,0
Готовят кипящий раствор алюмината I (как указано выше), доводят температуру смеси до 100 °C и перемешивают при этой температуре в течение 8 ч. Выделившийся осадок сразу же фильтруют через пористый стеклянный фильтр G3, промывают последовательно 50 мл 25% -ного раствора NaOH в метаноле, 50 мл абсолютного метанола, 100 мл абсолютного этанола, 50 мл эфира и сушат в вакуум-эксикаторе на керамической пластинке над Na2O.
Свойства. Кристаллы в форме тонких табличек. Оптически одноосны. Полученное вещество достаточно высокой чистоты.
Оксогидроксоалюминат калия К2[А12О(ОН)6]
2А1(ОН)3 + 2КОН ------> К2[А12О(ОН)6] + Н2О
156,0 112,0 250,2 18,0
120 г КОН и 30 г А1(ОН)3 растворяют при кипячении в 100 мл воды, оставляют раствор несколько часов стоять при комнатной температуре, фильтруют, вносят затравку кристаллов (см. ниже) и встряхивают 24 ч. Микрокристаллический осадок промывают небольшим объемом 50%-ного раствора КОН, затем 150 мл 5%-ного раствора КОН в метаноле и, наконец, ацетоном. Вещество сушат в вакууме над силикагелем. Выход ~5 г ( — 10% в расчете на А1(ОН)3).
Без затравки кристаллизация затягивается и наступает лишь через несколько дней.
Для получения кристаллов затравки готовят раствор 20 г КОН и 5 г А1(ОН)3 в 10 мл воды. Раствор фильтруют при комнатной температуре, а затем встряхиванием в течение 12 ч добиваются обильной кристаллизации алюмината калия, служащего в качестве кристаллов-зародышей. Получить таким
1884 Глава 31. Гидроксосоли
же образом основную массу вещества нельзя, так как зародышевые кристаллы очень Хрупкие и их невозможно освободить от избыточного КОН.
Свойства. Бесцветный микрокристаллический порошок, может быть полу-.чен в'очень чистом виде.
ЛИТЕРАТУРА
1. lahr К. F„ Pernoil I., Bunsengesellsch., 69, 221, 226 (1965).
2. Fricke R., Jucaitis P., Z. Anorg. Allgem. Chem., 191, 129 (1930).
3. Jucaitis P., Z. Anorg. Allgem. Chem., 220, 257 (1934).
4. Scholder R., Kleeberg W., Schroder M., Naturforschung und Medizin in Deutschland 1939—1946 (FIAT-Review), Band 25, Anorg. Chemie, Teil III, 141.
5. Scholder R., Lieber IF., Dissertation W., Lieber, T. H. Karlsruhe 1955.
6. Thilo E., Gessner W., Z. Anorg. Allgem. Chem., 337, 238 (1965).
7. Gessner W., Z. Anorg. Allgem. Chem., 337, 254 (1965).
8. Ahmed A. H. M., Glasser L. S. D., Acta Cryst., В 25, 2169 (1969).
9. Ahmed A. H. M., Glasser L. S. D., Acta Cryst., В 26, 867 (1970).
10. Ahmed A. H. M„ Glasser L. S. D., Acta Cryst., В26, 1686 (1979). .11. Glasser L. S. D., Goivanoli R., Acta Cryst., В 28, 760 (1972).
12. Glasser L. S. D., Giovanoli R., Acta Cryst., В 28, 519 (1972).
Гексагидроксохромат(Ш) натрия Ыаз[Сг(ОН)6]
Cr(C104)s + 6NaOH ----> Na3[Cr(OH)6] + 3NaC104
350,3 240,0 223,0 367,3
Продажный препарат СггОз-лНзО содержит обычно большую примесь карбоната, поэтому при получении №з[Сг(ОН)б] лучше всего использовать водный раствор Сг(С1О4)3, приготовленный из CrjOa-nHjO. Образующийся при реакции со щелочью NaClO4 достаточно растворим в конц. NaOH. Берут необходимое количество продажного гидрата CrjOs-nHjO, отвечающее 3 г Сг2О3 (содержание Сг2О3 в гидрате определяют заранее), добавляют рассчитанное количество 20—25 %-ной НС1О4, упаривают до объема —25 мл, фильтруют раствор, вливают его в 300 мл 51%-ного раствора щелочи (без карбонатов) и кипятят —30 мин с обратным холодильником. Смесь охлаждают до 120 °C и фильтруют в склянку, предварительно подогретую до 95 °C. Темно-зеленый фильтрат переливают в серебряную колбу, закрытую защитной трубкой с 50%-ным раствором КОН, и ставят на 4 ч в сушильный шкаф при 85°C. Выделившийся осадок промывают на фильтре 2 раза небольшим количеством 40%-ного раствора NaOH (18 °C) н тотчас же встряхивают его 30 мин с 80 мл 5%-ного раствора NaOH в метаноле (18 °C). Вновь фильтруют смесь и промывают осадок несколько раз такими же щелочными растворами метанола и, наконец, избытком ацетона, который удаляется при длительном выдерживании вещества в вакуум-эксикаторе над силикагелем. Выход 5—6 г (57—68% в расчете на Сг2О3).
При охлаждении фильтрата после выделения Nag[Cr(OH)6] из него выпадают разнообразные агрегаты пластинок гепта- и октагидроксохроматов(Ш).
Свойства. Микрокристаллический зеленый порошок, состоящий из хорошо образованных полиэдров. В холодной воде сначала образует прозрачный раствор, из которого через некоторое время начинает постепенно выпадать хлопьевидный Сг2Оз-пН2О. Препарат получается очень чистым (Cr:Na = = 1 :2,99—3,02).
Гидроксохроматы 1885
ЛИТЕРАТУРА
1. Scholder R., Pdtsch R., Z. Anorg. Allgem. Chem., 220, 411 (1934).
Гексагидроксохромат(Ш) стронция Sr3[Cr(OH)6]2
(3SrO)-Cr2O3 + 6H2O ----> Sr3[Cr(OH)6]2
462,8 108,1 570,9
Твердофазной реакцией SrO (или SrCO3) с Cr2O3 в атмосфере чистого азота получают 3SrO-Cr2O3. Несколько граммов 3SrO-Cr2O3 помещают в платиновую пробирку-палец, которую заключают в автоклав (бомбу), налив туда 10-кратиый избыток воды. Бомбу выдерживают 2 сут при 150—200 °C. После высушивания над Р4О10 получают практически чистый Sr3[Cr(OH)6]2.
Соединение можно получить, добавляя по каплям раствор Sr(C104)2+ +Сг(С1О4)3 (молярное соотношение 3:2) к избытку кипящего разбавленного NaOH. Конечная концентрация NaOH должна быть около 2 н.
Свойства. Микрокристаллический зеленый порошок. Структура типа граната («гидрогранат», кубический; пространственная группа Ia3d, а— 13,143 А).
ЛИТЕРАТУРА
1. Schwarz Н., Z. Naturforsch., 22b, 554 (1967).
Гидроксоферраты(Ш) натрия
При окислении раствора Naa[Fe(OH)4] в 50%-ном NaOH кислородом воздуха получают соответственно: при 20—25 °C — октагидроксоферрат(Ш) натрия, при 50—60°С — гептагидроксоферрат(Ш), при 100—130°С — оливково-зеленый оксоферрат(III) и в кипящей 55—60%-ной щелочи — красный оксоферрат (III) NaFeO2. Безводный октагидроксоферрат(Ш) натрия получают твердофазной реакцией из Fe2O3-nH2O и NaOH.
Гептагидроксоферрат(Ш) натрия Na4[Fe(OH)7]«2H2O
Способ 1
2Na2[Fe(OH)4l + 4NaOH+1/2Оа4-5Н2О----> 2Na4[Fe(OH)7]-2H2O
339,7 160,0 16,0 90,0 605,8
Сначала получают раствор Na2[Fe(OH)4] в 50%-ной щелочи (NaOH) (см. выше) при температуре 120 °C, который фильтруют в коническую колбу со 100 мл 50%-ного раствора NaOH. В течение 12 ч в раствор Na2[Fe(OH)4] при 60°C пропускают через две промывалки с 50%-ным раствором NaOH сильный ток кислорода. Исходный зеленовато-синий раствор при этом постепенно обесцвечивается, и из него выделяются кристаллы. Осадок отфильтровывают, быстро промывают 50%-ным раствором NaOH и сушат в тонком слое на керамической пластинке в вакуум-эксикаторе над силикагелем.
Можно рекомендовать окисление феррата (II) бромом. Для этого колбу закрывают резиновой пробкой с отверстиями для защитной трубки со щелочью и для капельной воронки, из которой при 50—60 °C добавляют (при сильном встряхивании) раствор 2—3 мл Вг2 в 10 мл ССЦ до обесцвечивания раствора в колбе. Избытка брома допускать нельзя. Смесь оставляют стоять ‘2 ч при той же температуре, а затем обрабатывают осадок, как указано выше в способе с кислородом в качестве окислителя.
1886 Глава 31. Гидроксосоли
Способ 2
Fe(OH)s + 4NaOH + 2На0 -----> Na4[Fe(OH),]-2H2O
106,9 160,0 36,0 302,9
К раствору 10 г FeCl3-6H2O в 3—4 л Н2О по каплям добавляют небольшой избыток 10%-иого раствора NaOH при комнатной температуре, в результате чего выделяется Ре2Оз-пН2О. Осадок промывают декантацией, фильтруют и тщательно промывают на фильтре. Отвешенное количество Fe2O3-nH2O помещают в серебряную коническую колбу, добавляют такое же количество твердой NaOH, а затем 300 мл 50%-иого раствора NaOH. Колбу закрывают пробкой с защитной трубкой, заполненной 50%-ным раствором КОН, и ставят иа несколько дней в сушильный шкаф при 70 °C. При стоянии Ре2Оз-пН2О полностью переходит в почти бесцветный микрокристаллический осадок Na4Fe(OH)7]-2Н2О, плохо растворимый в концентрированных щелочах. О дальнейшей обработке препарата см. выше (способ 1).
Свойства. Почти бесцветный кристаллический порошок (скошенные пластинки, частично собранные в агрегаты). Очень чувствителен к действию влаги. Мгновенно разлагается водой и метанолом с образованием Fe2O3-nH2O. Разлагается уже в 30%-ном растворе NaOH при 18 °C.
Октагидроксоферрат(Ш) натрия NasFe(OH)3-5H2O
При окислении Na2[Fe(OH)4] с помощью О2 или при превращении осадка Fe2O3-nH2O (ср. с предыдущей методикой, способы 1 и 2) при комнатной температуре наряду с гептагидроксоферратом выделяется также и бесцветный октагидроксоферрат(Ш). Последний фильтруют через пористый стеклянный фильтр G4, промывают 50%-ным раствором NaOH и сушат в тонком слое на керамической пластинке в пустом эксикаторе.
Свойства. Почти бесцветный кристаллический порошок (тонкие иглы). При получении вещества обработкой свежеосажденного Fe2O3-nH2O превращение неполное. Препарат обезвоживают в токе азота при 90 °C до Na5[Fe(OH)8].
Безводный октагидроксоферрат(Ш) натрия Na^FefOHJsJ
В разбавленный раствор FeCl3 -6Н2О пропускают при комнатной температуре ток NH3, в результате чего выделяется осадок Fe2O3-nH2O, который многократно отмывают на стеклянном фильтре водой до отрицательной реакции на ион С1“. Влажный осадок высушивают до тех пор, пока он не начнет распыляться (Fe2O3-8H2O). Полученный таким образом Ре2Оз-8Н2О и растертую в порошок твердую щелочь (как и в случае получения Na2[Co(OH)4]) смешивают (в отсутствие СО2) в соотношении Fe: Na= 1 :5,00. При растирании навесок обоих веществ в агатовой ступке (в сухой камере с азотом) сначала образуется темно-коричневая паста, которая через 10 мин застывает, так что требуется интенсивное размешивание. Смесь помещают в серебряную лодочку и нагревают в течение 12 ч в токе N2 при 75 °C. Затем смесь снова растирают в порошок и выдерживают в тех же условиях еще 18 ч.
Свойства. Микрокристаллический белый, чуть желтоватый порошок.
ЛИТЕРАТУРА
1. Scholder R., Angew. Chem., 49, 255 (1936).
2. Scholder R., Kyri H„ Dissertation H. Kyri, T. H. Karlsruhe 1950.
3. Scholder R., Herrmann F., Dissertation Herrmann F., T. H. Karlsruhe 1967. 4. Scholder R., Schwochow F„ Angew. Chem., 78, 1102 (1966).
Гидроксоферраты(Ш) 1887
Гидроксоферраты(Ш) бария
Гидроксоферрат(Ш) бария получают добавлением по каплям раствора Fe(C104)3+Ba(C104)2 [молярное соотношение 1 : (2,5—3,0)] к горячему раствору NaOH. При использовании исходного 25—39%-ного раствора щелочи образуется гексагидроксосоль Ba3[Fe(OH)3]2, а при исходном 42%-ном растворе — гептагидроксосоль Ba2[Fe(OH)7] -0,5Н2О.
Аналогичным образом получают гексагидроксоферрат(Ш) стронция Sr3[Fe(OH)6]2 из 5%-ного раствора NaOH и гептагидроксоферрат(Ш) стронция Sr2[Fe(OH)7]-3H2O из 20%-ного раствора NaOH.
Смесь Fe(C104)3+Ba(C104)2 (молярное отношение 1:3), необходимую для получения гидроксоферрата(Ш) бария, готовят растворением 3,5 г РегО3 в смеси 35 мл 70%-ной НС1О4 и 25 мл концентрированной соляной кислоты. Для удаления НС1 смесь упаривают до появления густого дыма НС1О4. Этот раствор взбалтывают со взвесью 26 г ВаСО3 в 125 мл воды, а затем фильтруют.
Гексагидроксоферрат(Ш) бария Ba3[Fe(OH)e]2
В серебряную колбу наливают 180 мл 50%-ного раствора NaOH (без карбонатов) и разбавляют его 140 мл воды (без СО2) до 33%. Раствор щелочи нагревают (без доступа СО2) до кипения с обратным холодильником и прибавляют к нему приготовленную заранее (см. выше) смесь Fe(C104)3+ +Ва(С1О4)г. Выпадает Ba3[Fe(OH)3]2 в виде белого осадка. Смесь кипятят с осадком еще 1 ч, охлаждают до комнатной температуры, фильтруют через пористый стеклянный фильтр G3 и промывают 33%-ным раствором NaOH. Выделенный осадок встряхивают несколько минут с 200 мл абсолютного СН3ОН, затем фильтруют через пористый стеклянный фильтр G4, промывают абсолютным СН3ОН и абсолютным эфиром. Препарат сушат в вакуум-эксикаторе над Р4О10.
Препарат можно получить твердофазной реакцией БегОэ-пНгО с NaOH в соответствующем соотношении при 90 °C (см. выше синтез Naa[Co(OH)4]).
Свойства. Белые или слегка желтоватые шестиугольные пластинки. Водой разлагается с образованием РегОз-пНгО. Устойчив в абсолютном метаноле.
Гептагидроксоферрат(Ш) бария Ва2[Ре(ОН)7]«0,5НгО
Получение аналогично синтезу Ва3[Ре(ОН)б]г- Вместо 33%-ного раствора NaOH можно использовать 400 мл 50%-ного раствора NaOH.
Свойства. Белые, слегка желтоватые шестиугольные пластинки. Разлагается водой с образованием Fe2O3-nH2O. Длительное действие абсолютного метанола приводит к образованию коричневого раствора.
ЛИТЕРАТУРА
1. Scholder R. и. Mitarb., Dissertationen Т. Н. Karlsruhe: Zeiss W. 1953; Kreutz M. 1957; Muller P. 1966; Herrmann F. 1967.
2. Scholder R., Kreutz M„ Liebigs Ann. Chem., 653, 1 (1962).
Тексагидроксостаннат(1У) натрия Na2[Sn(OH)6]
Sn(OH)4 + 2NaOH ---> Na2[Sn(OH),]
186,7 80,0 266,7
Раствор SnCl4 в сильно разбавленной HC1 нейтрализуют NaOH (без карбонатов) по метилоранжу. Отфильтрованный и отмытый водой до отрицательной реакции на ион С1~ осадок гидрата SnO2-nH2O вносят порциями в избыток концентрированного горячего ( — 100’С) раствора NaOH, в котором он
1888 Глава 31. Гидроксосоли
полностью растворяется. Через некоторое время выделяется кристаллический гексагидроксостаннат. Кашицу кристаллов фильтруют (в отсутствие СОг), промывают 30%-ным раствором щелочи и несколько раз спиртом и эфиром.
Свойства. Бесцветный кристаллический порошок (тонкие шестиугольные пластинки); легко растворяется в Н2О, причем растворимость уменьшается с повышением температуры. Препарат всегда содержит небольшое количество адсорбированной щелочи. Чувствителен к действию СО?. Кристаллическая структура ромбоэдрическая (пространственная группа R3, а=5,84 A, a=61,2J; структура типа Cdl2).
ЛИТЕРАТУРА
I. Zocher Н., Z. Anorg. Allgem. Chem., 112, 1 (1920).
2. Reiff R., Toussaint S. M., Z. Anorg. Allgem. Chem., 241, 372 (1939).
3. Bjdrling C. 0., Arkiv Kemi, Mineral. Geol., 15 B, № 2 (1941).
Гексагидроксоплюмбат(1¥) натрия Маг[РЬ(ОН)6]
Способ 1. Электрохимическое получение [1, 2]
РЮ + Н2О + 2NaOH + ЗОН- — 2е -------> Na2[Pb(OH)e]
223,2 18,0 80,0 34,0 355,2
18,5 г желтого РЬО растворяют в 300 мл кипящего 13 н. (37%) раствора NaOH, фильтруют горячий раствор через пористый стеклянный фильтр G4 и охлаждают без доступа СО2. Из таких растворов гидроксоплюмбатов(П) при длительном стоянии постепенно выделяется кристаллический оксид РЬО. Поэтому лучше всего раствор сразу же после охлаждения до комнатной температуры подвергнуть электролизу. В данном случае раствор сначала декантируют от выделившегося РЬО.
Электролитической ячейкой служит прямоугольная стеклянная кювета на 300 мл, плотно закрывающаяся резиновой пластинкой. В соответствующих прорезях в крышке кюветы укрепляют трубку для ввода газа, термометр, мешалку с затвором (для предотвращения доступа воздуха), впаянный в стеклянную трубку проводник, подводящий ток к аноду, и керамическую диафрагму для отделения катодного пространства. Используют электроды из белой платины размером 5x5 см2. Электролиз проводят при комнатной температуре и плотности тока 0,02—0,03 А/см2. Находящийся в анодном пространстве сильнощелочной раствор гндроксоплюмбата(П) необходимо интенсивно перемешивать. В катодном пространстве скапливается концентрированный раствор NaOH. Гексагидроксоплюмбат(ГУ) натрия выделяется в виде белого «дождя» кристаллов. Осадку дают осесть, сливают прозрачный раствор, заливают кашицу кристаллов абсолютным спиртом, смывают кристаллы с жидкостью в меньшнй сосуд и настаивают с абсолютным спиртом до отрицательной реакции на щелочь жидкости над кристаллами. Чисто белые кристаллы при высушивании в вакууме становятся желтоватыми.
Способ 2. Получение химическим путем из РЬ(СН3СОО)4 [3]
Pb(CHsCOO)4 + 6NaOH -----> Na2[Pb(OH)8] + 4CH3COONa
443,5 240,0 355,2 328,3
В длинногорлую круглодонную стеклянную колбу на 1 л, снабженную мешалкой с затвором, капельной воронкой и защитной трубкой с 30%-ным раствором КОН, наливают 200 мл 30%-ного раствора NaOH (без карбонатов). Чтобы в кончик капельной воронки не попадали брызги щелочи, на него наде-
Гидроксоплюмбаты 1889*
вают стеклянную насадку диаметром 15 мм, выступающую ниже конца воронт ки на — 2 см.
При сильном перемешивании к раствору щелочи по каплям добавляют предварительно отфильтрованный желтоватый раствор 50 г РЬ(СН3СОО)4. (см. т. 3) в 200 мл СНС13 (растворитель высушен над КзСО3 с последующим добавлением 1 мл СН3СООН). В месте попадания капель раствора. РЬ(СНзСОО)< в щелочь образуется коричневый РЬО2-лНгО, который быстро, растворяется. Через некоторое время начинается выделение Na2[Pb(OH)6]. После того как весь раствор РЬ(СН3СОО)4 в СНС1з добавлен, смесь перемешивают до тех пор, пока суспензия не станет чисто белой. Осадку дают осесть, в течение часа, фильтруют и промывают один раз 30%-ным раствором NaOH' в метаноле, 2 раза чистым метанолом и, наконец, 3 раза эфиром. Все операции проводят в отсутствие СО2 и влаги. Препарат сушат на керамической пластинке в вакуум-эксикаторе над Р4Ою. При быстром проведении синтеза удается получить чисто белый препарат. Выход 33 г [—82% в расчете на> РЬ(СН3СОО)4].
Способ 3. Получение химическим путем из РЬО2 [3]
РЬОа + 2НаО + 2NaOH-------> Na2[Pb(OH),]
239,2 36,0 80,0 355,2
В круглодонную серебряную колбу на 1 л, снабженную плотно подогнанной серебряной трубкой и обратным холодильником с защитной трубкой: (с 30%-ным раствором КОН), помещают 175 г РЪО2, 80 г гранулированного. NaOH и 500 мл 50%-ного раствора NaOH (без карбонатов). Смесь нагревают прн перемешивании до кипения в течение 3—4 ч, при этом РЬО2 почти полностью переходит в желтый Na2PbO3. Лишь незначительная часть РЬО осаждается на дне колбы. Смесь охлаждают при перемешивании и добавляют' 300 мл воды. Суспензию переносят в круглодонную колбу на 3 л, приливают еще 700 мл воды при температуре ниже 50 °C и оставляют стоять на ночь. Прн стоянии желтый Na2PbO3 переходит в бесцветный Na2[Pb(OH)e]. После-добавления следующих 800 мл воды и взбалтывания Na2[Pb(OH)6] растворяется. Раствор фильтруют, к бледно-желтому фильтрату прибавляют 500 г-NaOH, которые растворяют, взбалтывая смесь, и оставляют раствор охлаждаться до комнатной температуры. Через несколько часов раствор фильтруют, через пористый стеклянный фильтр G3 и обрабатывают далее, как указано: в описании способа 2. Выход 240 г (—92% в расчете на РЬО2).
Если соль, полученная способом 2 или 3, имеет желтоватый оттенок, ее-следует очистить. Для этого 25 г Na2[Pb(OH)3] вносят небольшими порциями при перемешивании в 200 мл 15%-ного раствора NaOH, следя за тем, чтобы, перед внесением каждой порции предыдущая порция растворилась полностью.-. Раствор фильтруют от небольшого осадка, добавляют в еще горячий фильтрат 50 г NaOH и сильно встряхивают раствор, пока все не растворится. После охлаждения раствору дают постоять 1—2 ч, отфильтровывают чистые белые-кристаллы и промывают и высушивают их, как было указано в описании способа 2. Получается препарат высокой чистоты.
Свойства. Кристаллы — бесцветные гексагональные полиэдры. Устойчив в 22%-ном растворе NaOH до 18 °C. Вещество чувствительно к действию влаги, причем, поглощая воду, изменяет окраску. Кристаллическая структура-ромбоэдрическая, изоморфная Na2[Sn(OH)6], Пространственная группа R3;.. а=5,87 А, а=61,5°; d 3,975 (рассчит.), 3,94 (определено пнкнометрически).
ЛИТЕРАТУРА
1. Grube G., Z. Electrochemie, 28, 273 (1922).
2. Simon A., Z. Anorg. Allgem. Chem., 177, 109 (1929).
3. Scholder R„ Kindler H„ Dissertation H. Kindler, T. H. Karlsruhe 1952..
3—1361
•1890 Глава 31. Гидроксосоли
•Октагидроксоплюмбаг( IV) бария Ва2[РЬ(ОН)8]
Na2[Pb(OH)J + 2ВаС12 -2Н2О + 2NaOH ---->
355,2 488,6 80,0
----> Ba3[Pb(OH)8] + 4NaCl + 4НаО
617,9 233,8 72,1
В серебряную круглодоиную колбу на 500 мл вносят 200 мл 30%-ного >;раствора NaOH (без карбонатов), 15 г ВаС12-2Н2О и 9 г Na2[Pb(OH)6], закрывают колбу резиновой пробкой с обратным холодильником, снабженным защитной трубкой, наполненной 30%-ным раствором КОН. Смесь нагревают 1—2 и до кипения, изредка встряхивая колбу, затем смесь фильтруют через пористый стеклянный фильтр G4 и промывают последовательно 3 раза 30%-ным раствором NaOH, 3—5 раз метанолом и 3 раза эфиром. Осадок высушивают в вакуум-эксикаторе над P-iOio.
Свойства. Белый рассыпчатый порошок. Под действием влаги воздуха постепенно приобретает коричневую окраску.
ЛИТЕРАТУРА
I. Scholder R., Kindler Н„ Dissertation Н. Kindler, Т. Н. Karlsruhe 1952.
Глава 32. ИЗО- И ГЕТЕРОПОЛИСОЕДИНЕНИЯ
Э. Шилль* (Е. Schill)
Перевод канд. хим. наук С. И. Троянова
Изополисоединения
Методики синтеза изополисоедннений бора, фосфора и кремния помещены в» соответствующие главы, посвященные синтезу соединений этих элементов.
Обзоры по изо- и гетерополисоединениям**: Evans Н. Т., in: Dunitz J. D.,. Ibers J. A. (eds.), Perspectives in structural chemistry, vol. IV, Wiley, New York,. 1971; Weakley T. J. R„ In: Structure and bonding, vol. 18, Springer, Berlin — Heidelberg — New York, 1974; Tytko К. H., Glemser O., in: Emeleus H. J.,. Sharpe A. G. (eds.), Advances in inorganic chemistry and radiochemistry, vol. 19, Academic Press, New York, 1976; Tsigdinos G. A., in: Methodicum Chimicum, Bd. 8, Thieme, Stuttgart, 1974.
Трихромат калия КгСгзОю
Соль образуется из водного раствора К2СГ2О7 в присутствии избытка* СгО3.
20,4 г К2СГ2О7, 52,1 г СгО3 и 52,1 г воды нагревают до тех пор, пока все не растворится. К раствору снова добавляют воду, восполняя ее потери на испарение, и охлаждают до комнатной температуры. Кристаллы отфильтровывают на стеклянном пористом фильтре и высушивают путем просасывания-воздуха, не содержащего пыли. Выход 21 г.
Свойства. М 394,2. Темно-красные призматические кристаллы, не содержащие кристаллизационной воды. Растворение в воде сопровождается разложением соли.
Тетрахромат калия K2Cr40i3
Получение ведут так же, как трихромата калия, но со следующими количествами реагентов: 12,6 г К2СГ2О7, 57,4 г СгО3 и 42,3 г воды. Выход 21 г.
Свойства. М 494,2. Темно-красные или коричнево-красные таблитчатые г кристаллы. Растворение в воде сопровождается разложением соли.
ЛИТЕРАТУРА
1. Jager Е., Kruss G., Вег., 22, 2040 (1889).
2. Gmelins Handbuch der Anorganischen Chemie, 8. Aufl., Chrom. System-Nr. 52 B, Verl. Chemie, Weinheim, 1962, S. 509, 581, 582.
* Во втором издании «Руководства» на немецком языке эта глава была? написана Б. Грюттнером (В. Gruttner) и Г. Яндером (G. Jander).
** На русском языке можно рекомендовать следующую статью: Спи-цып В. И., Торченкова Е. А., Казанский Л. П. — Итоги науки и техники, сер. «Неорганическая химия», ВИНИТИ, 1984, т. 10. — Прим, перев.
3*
«1892 Глава 32. Изо- и гетерополисоединения
.Диванадат натрия Na4V2O7»15H2O
100 мл 1,1 М раствора Na3VO4 (полученного растворением V2Oa в рассчитанном количестве едкого натра, не содержащем карбонатов) подкисляют путем добавления по каплям и при энергичном перемешивании 25 мл 4,4 н. НС1О4. Для ускорения установления равновесия некоторое время нагревают на водяной бане, пока оранжевый раствор не обесцветится. После этого раствор концентрируют в вакууме при 25—30 °C. Выпавшие кристаллы отфильтровывают и промывают небольшим количеством воды.
Свойства. Бесцветные кристаллы, растворяющиеся в воде.
-ЛИТЕРАТУРА
1. Jander G., Jahr К.. F., Z. Anorg. Allgem. Chem., 211, 53 (1933).
Триванадат калия KV3Oe
3KVO3 + 2H+-----> KVSO9 + 2K+ + H2O
Эту соль получают из раствора монованадата калия (KVO3) при добав-.лении уксусной кислоты в количестве 1,4 моль на 1 моль ванадата.
В стакане, помещенном в водяную баню при 75 °C, растворяют 7 г моно-ванадата калия в 25 мл воды. К горячему раствору, имеющему концентрацию по ванадию ~2 М, при сильном перемешивании прибавляют из бюретки 70 мл 1 М раствора СН8СООН, причем носик бюретки должен быть погружен в раствор. Скорость прибавления раствора кислоты составляет 1 мл/мин.
Раствор красного цвета выдерживают в горячей бане и затем в течение ~15 ч охлаждают до комнатной температуры. Для кристаллизации прозрачный раствор охлаждают до 0 °C. Кристаллы отсасывают, промывают неболь-,шим количеством ледяной воды, а затем ацетоном.
Свойства. М 319,9. Оранжево-красные кристаллы в форме ромбов или .шестиугольных пластинок, а иногда в виде более крупных бипирамид.
Если исходить из монованадата аммония, таким же образом можно получить триванадат аммония NH4V3O8.
литература
1. Jahr К- F„ Jander Q„ Z. Anorg. AHgem. Chem., 220, 204 (1934).
:2. Kelmers A. D., J. Inorg. Nucl. Chem., 21, 45 (1961).
3. Evans H. T„ Block S., Inorg. Chem., 5, 1809 (1966).
Тексаниобат калия KsNbeOig^nHaO
В серебряном тигле нагревают Nb2Oa и КОН (молярное соотношение ~ 1 :20) до образования прозрачного расплава. Плав измельчают и растворяют в воде. Раствор отделяют декантацией от нерастворимого остатка и концентрируют в вакууме над конц. H2SO4 до выпадения кристаллов. Кристаллы промывают небольшим количеством воды и высушивают на воздухе.
Na8Nb6Oi9"nH2O осаждают из водных растворов калиевой соли действием NaOH. Белый мелкокристаллический осадок отсасывают, промывают водой, спиртом, эфиром и высушивают.
Свойства. Хорошо кристаллизующиеся соли; содержание воды зависит от условий получения. Большие прозрачные кристаллы калиевой соли выветриваются при хранении в «сухих» условиях, но сохраняют способность хорошо
Изополисоединения 1893
растворяться в воде. Натриевая соль образует различные кристаллические формы в зависимости от условий осаждения. Обе соли устойчивы в водных растворах при pH 13.
Na7HNbeOi9-nH2O получают путем двукратной перекристаллизации Na8Nb60i9 из водного раствора. Соответствующую калиевую соль лучше всего получать добавлением спирта к раствору, содержащему 10 масс.% или более K8Nb6O19.
Свойства. Na7HNb6Oi9-nH2O кристаллизуется в виде длинных иголок. Соответствующая калиевая соль легко теряет кристаллизационную воду, давая гидраты с меньшим содержанием воды.
Обе соли устойчивы в водных растворах только в области pH 9—13.
Метаниобат калия КбЫЬ6С>18«пН2О
К 2—4%-ному водному раствору K3Nb60i9 при охлаждении льдом и при энергичном перемешивании добавляют по каплям равный объем метанола. При этом образуется гидратированный метаниобат калия в виде аморфного хлопьевидного осадка. Его отфильтровывают, промывают 50%-ным метанолом и высушивают в мягких условиях.
Свойства. Чисто-белый порошок, хорошо растворимый в воде. Содержание воды колеблется в зависимости от условий получения. В водных растворах метаниобат устойчив лишь при pH ~8.
ЛИТЕРАТУРА
1. Jander G., Ertel D., J. Inorg. Nucl. Chem., 14, 77 (1960).
2. Спицын В. И., Лапицкий А. В. — Ж. прикл. хим., 1953, т. 26, с. 117.
Гексатанталат калия КзТа6О19- 16Н2О
В серебряном тигле нагревают 25 г КОН и 5 г Та2О5 до образования прозрачного расплава. Застывший плав выщелачивают небольшим количеством горячей воды. Раствор фильтруют, собирая фильтрат в полиэтиленовый стакан, с тем чтобы исключить возможность загрязнения силикатами. Неочищенный продукт получают осаждением этанолом. Три раза производят перекристаллизацию из раствора в разб. КОН, осаждая соль добавлением этанола.
Свойства. Кристаллы длиной до 1 см в виде шестигранных призм с несколько притупленными ребрами. Кристаллы растворяются в воде, давая раствор с сильнощелочной реакцией.
При хранении в очень «сухих» условиях происходит выветривание кристаллов. Содержание кристаллизационной воды колеблется.
ЛИТЕРАТУРА
1. Jander G., Schulz Н„ Z. Anorg. Allgem. Chem., 144, 233 (1925).
2. Jander G., Ertel D., J. Inorg. Nucl. Chem., 3, 139 (1956).
3. Nelson W. H., Tobias R. S., Inorg. Chem., 3, 985 (1963).
(mo-
Гексатанталат натрия Na8Ta6Oi9»«H2O
Размельченный плав, образовавшийся при сплавлении Ta2Os с NaOH (_
лярное отношение 1:5), выщелачивают 10-кратным (по массе) количеством холодной воды. Остаток растворяют в воде при 80 °C. Раствор упаривают при
1894 Глава 32. Изо- и гетерополисоединения
Свойства. Мелкие пластинчатые кристаллы (п=33). 1%-ный водный раствор имеет pH 8,58.
ЛИТЕРАТУРА
1. Спицын В. И., Шаврова В. Н. — Ж. общ. хим., 1956, т. 26, с. 1258.
Октамолибдат аммония (NH4)4Mo8O26’rtH2O (п=4, 5)
2(NH4)eMo7Oa4+ 10MoOs ----> 3(NH4)4Mo8O2e
24,7 г продажного препарата гептамолибдата аммония (NHiJeMorOjiX Х4Н2О растворяют в 1500 мл воды и раствор нагревают до кипения. При перемешивании медленно приливают суспензию 14,4 г МоОз в 500 мл воды, поддерживая температуру 80—90 °C. Перемешивают еще 3—4 ч, пока весь МоО3 не растворится. При необходимости раствор профильтровывают. Прозрачный раствор упаривают в ротационном испарителе до объема 300—400 мл. Раствор оставляют при комнатной температуре на 2—3 дня. Выпавшие кристаллы отфильтровывают, промывают небольшим количеством ледяной воды и высушивают на воздухе. Выход 34 г.
Свойства. Большие бесцветные кристаллы, выветривающиеся на воздухе.
ЛИТЕРАТУРА
1. Aveston J., Anacker Е. W., Johnson J. S., Inorg. Chem., 3, 735 (1964).
2. Lingqvist I., Arkiv Kemi, 2, 325 (1950).
3. Tytko К. H„ Schdnfeld B„ Z. Naturforsch., 30b, 471 (1975).
Полиоктамолибдат аммония (NH4)en(Mo8O27)„-4nH2O
Соль выкристаллизовывается при долгом стоянии (несколько недель) насыщенного или 0,20—0,25 М раствора (NH4)eMo7O24-4H2O.
Свойства. Бесцветные игольчатые кристаллы, плохо растворимые в воде. Соответствующие натриевую и калиевую соли таким путем получить не удается.
ЛИТЕРАТУРА
1. Boschen J., Buss В., Krebs В., Acta Cryst., В 30, 48 (1974).
2. Glemser О., Wagner Q., Krebs В., Tytko К- H., Angew. Chem., 82, 639 (1970).
3. Tytko К. H., Schdnfeld B., Z. Naturforsch., 30b, 471 (1975).
12-Вольфрамат натрия Naio(H2Wi2042) ’27H2O
(Паравольфрамат натрия В)
Способ 1 [1—5]. 20 г Na2WO4-2H2O растворяют при нагревании в 40 мл воды и нейтрализуют 2 н. НС1, используя лакмус в качестве индикатора. Для этого необходимо взять ~23,5 мл раствора НС1 (или 1,2 моль НС1 на 1 моль Na2WC>4). Раствор должен иметь pH 6,8. Для кристаллизации раствор прн комнатной температуре выдерживают в вакуумном эксикаторе с H2SO4*.
Способ 2 [6, 7]. К 1,5 М. раствору Na2WO4-2H2O при энергичном переме
* Использование вакуумных эксикаторов с конц. H2SO4 запрещено действующими в настоящее время правилами техники безопасности. — Прим, перев.
Изополисоединения 1895
шивании прибавляют порциями из бюретки ~5 и. HNO3. Образующийся студенистый осадок геля WO3 при перемешивании должен раствориться. Прибавление кислоты продолжают до достижения pH 6. Раствор отфильтровывают от коллоидных частиц на мембранном фильтре № 4 и оставляют для кристаллизации прн комнатной температуре. Кристаллы отфильтровывают, тщательно промывают водой и высушивают на стеклянном пористом фильтре путем про-сасывания воздуха. В течение нескольких дней кристаллы выдерживают на воздухе между листами фильтровальной бумаги.
Свойства. Большие прозрачные или молочно-белые, триклинные кристаллы. литература
1. Rosenheim A., Z. Anorg. Allgem. Chem., 96, 160 (1976).
2. Scheibler C„ J. Prakt. Chem., 83, 284 (1861).
3. Jander G„ Kriierke U., Z. Anorg. Allgem. Chem., 265, 244 (1951).
4. Aveston J., Inorg. Chem., 3, 981 (1964).
5. Ke pert D. L., Progr. Inorg. Chem., 4, 199 (1962).
6. Saddington K.., Cahn R. W., J. Chem. Soc. (London), 1950, 3526.
7. Glemser 0., Holznagel W., Holtje W., Schwarzmann E., Z. Naturforsch., 20b, 725 (1965).
Мета-12-вольфрамат натрия Na6(H2W12O10)-21H2O
(Метавольфрамат натрия)
Способ 1. Раствор 20 г Na2WO4-2H2O в 200 мл воды нагревают до кипения и в него вносят порциями избыток желтой вольфрамовой кислоты WO3-xH2O. Суспензию кипятят ~2 ч, причем кроме избытка вольфрамовой кислоты в осадок выпадают белые нерастворимые продукты. После кипячения и фильтрования раствора его pH равен ~3. Раствор упаривают на водяной бане и ставят для кристаллизации в эксикатор с H2SO4.
Способ 2. В трехгорлой колбе с KPG-мешалкой, обратным холодильником и капельной воронкой нагревают до кипения раствор 40 г паравольфрамата натрия в 600 мл воды. В кипящий раствор прибавляют по каплям водную суспензию WO3-xH2O. При падении каждой капли в прозрачный раствор происходит появление желтой мути, которая за счет образования метавольфрамата почти (фазу исчезает. Скорость падения капель устанавливают таким образом, чтобы перед падением следующей капли раствор уже был совершенно прозрачным. Окончание реакции узнают по появлению неисчезающего желтого окрашивания и помутнения раствора. Добавляют еще некоторый избыток WO3-xH2O и смесь кипятят ~2 ч. После этого отфильтрованная проба раствора не должна давать осадка при прибавлении нескольких капель разбавленной (1:1) соляной кислоты. Реакционную смесь фильтруют через стеклянный пористый фильтр G3 и затем через мембранный фильтр № 4. Раствор упаривают на водяной бане до сиропообразной консистенции, охлаждают в бане со льдом и вызывают кристаллизацию путем трения стеклянной палочкой о стенку. Выпавшую соль отфильтровывают и высушивают в потоке воздуха.
Свойства. Бесцветные бипирамидальные тетрагональные кристаллы. Легко выветриваются, при выдерживании над H2SO4 теряют почти всю кристаллизационную воду. Хорошо растворяются в воде.
ЛИТЕРАТУРА
1. Scheibler С., J. Prakt. Chem., 83, 301 (1861).
2. Jander G., Kruerke U., Z. Anorg. Allgem. Chem., 265, 244 (1951).
3. Glemser O., Holznagel W., Holtje W., Schwarzmann E., Z. Naturforsch., 20b, 725 (1965).
1896 Глава 32. Изо- и гетерополисоединения
12-Вольфрамовая кислота HeClhW^OtopnHzO
(Метавольфрамовая кислота)
Свободную кислоту можно выделить из раствора NaafHaWwOw) по методу Дрекселя (см. ниже в разд. «Гетерополисоедииения. Общие методы получения»). Через слой эфирата пропускают ток сухого воздуха; твердый продукт отжимают на пористой глиняной пластинке. Получают относительно хорошо сохраняющийся препарат, растворимый без остатка в воде. Однако некоторые из препаратов уже через несколько дней превращаются в желтую вольфрамовую кислоту.
Лучших результатов можно достичь, получая кислоту путем ионного обмена из кристаллического метавольфрамата натрия. 20 г. NastlbW^CUo) 'ХН2О растворяют в 50 мл воды. Далее получают прозрачный элюат, ие подверженный гидролизу при упаривании. Выход 18 г.
Свойства. Крупные кристаллы в виде октаэдров, иногда образуются также ромбоэдры или бипирамиды. На воздухе выветриваются. Хорошо растворяются в воде. Разбавленные растворы кислоты могут длительное время храниться на холоду; при нагревании наступает коагуляция. Концентрированные растворы часто коагулируют уже при незначительном нагревании.
ЛИТЕРАТУРА
1. Rosenheim A., Kohn F., Z. Anorg. Allgem. Chem., 69, 253 (1911).
2. Schott G., Harzdorf C„ Z. Anorg. Allgem. Chem., 288, 15 (1956).
3. Glemser 0., Holznagel W., Holtje W., Schwarzmann E., Z. Naturforsch., 20b, 725 (1965).
12-Ниобоманганат(1У) натрия Nai2[Mn(Nb60i9)2] »50H2O
Сначала получают раствор комплексоната марганца (II) из 5 ммоль М.п(СН3СОО)2-4Н2О, 5 ммоль динатриевой соли EDTA, 10 ммоль NaOH и 20 мл воды. Этот раствор добавляют по каплям в кипящий раствор гекса-ниобата натрия (13 г соли в 300 мл воды). Прибавляют 3% H2O2, пока ие прекратится усиление оранжевого окрашивания раствора. При охлаждении раствора образуются светло-оранжевые кристаллы, которые перекристаллизовывают из 0,2 М раствора ацетата натрия. Повторная обработка остатка пероксидом водорода улучшает выход. Перекристаллизацию ведут из теплого 0,2 М раствора ацетата натрия. Кристаллы промывают водно-спиртовым раствором (4:1) и сохраняют над насыщенным раствором Na2SOr ЮН2О.
Свойства. Кристаллы оранжевого цвета, выветривающиеся при выдерживании на воздухе.
ЛИТЕРАТУРА
1. Flynn С. М., Stucky G. D., Inorg. Chem., 8, 332 (1969).
10-Ванадат дикалия-димарганца K2Mn2Vio028’12H20
К раствору 20,7 г монованадата калия KVO3 в 500 мл воды добавляют 14 г ацетата калия и затем при интенсивном перемешивании по каплям 9 мл ледяной уксусной кислоты. К полученному раствору приливают при перемешивании раствор 6,8 г MnSO4-H2O в 20 мл воды. Упаривают в ротационном испарителе до объема 300 мл, дают остыть и отфильтровывают выпавшие кристаллы.
Свойства. Л4 1361,67. Золотисто-желтые игольчатые кристаллы.
Изополисоединения 1897
ЛИТЕРАТУРА
1. Radau С., Liebigs Ann. Chem., 251, 114 (1889).
2. Flynn С. M„ Pope M. T„ J. Amer. Chem. Soc., 92, 85 (1970).
10-Ванадат дикалия-дицинка КгХпгУюОгвИбНгО
Раствор 20,7 г монованадата калия KVO3 в 500 мл воды подкисляют разб. СНзСООН до pH 5. Прибавляют раствор 6,6 г Zn(CH3COO)2-2H2O в 100 мл воды и полученный раствор упаривают в ротационном испарителе до объема 300 мл. Дают остыть и отфильтровывают выпавшие кристаллы.
Свойства. М 1454,6. Крупные кристаллы оранжевого цвета.
ЛИТЕРАТУРА
1. Radau С., Liebigs Ann. Chem., 251, 114 (1889).
2. Evans H. T., Inorg. Chem., 5, 967 (1966).
Г етерополисоединения
Общие методы получения
Гетерополисоединения можно получить путем смешивания раствора растворимого молибдата, ванадата или вольфрамата с раствором, содержащим гетероатомы. После смешивания раствор подкисляют до значения pH, при котором данное гетерополисоединение устойчиво, а затем нагревают. При охлаждении или иногда после упаривания раствора получают соль гетерополикислоты.
Если соли таким простым способом не получаются или не могут быть выделены в достаточно чистом виде, то их выделяют из растворов соответствующих кислот средней концентрации путем насыщения хлоридами металлов или добавлением рассчитанных количеств карбонатов металлов. Последним способом соли получаются в более чистом виде и с лучшим выходом, однако при добавлении карбоната следует соблюдать определенную осторожность, поскольку его избыток вызывает, как правило, разложение гетерополианиона.
Гетерополикислоты получают следующими способами: 1) путем экстракции диэтиловым эфиром подкисленного водного раствора гетерополисоединения (метод Дрекселя); 2) путем ионного обмена из гетерополисоли.
Способ 1. Метод Дрекселя. В делительной воронке к возможно более концентрированному (иногда сиропообразной консистенции) раствору натриевой соли гетерополнкислоты прибавляют 1/3 объема диэтилового эфира и энергично встряхивают, чтобы насытить раствор эфиром. Затем небольшими порциями, каждый раз сильно встряхивая, добавляют охлажденную льдом конц. НС1 (37%), которая не должна содержать железа. При этом жидкость не должна нагреваться; при необходимости делительную воронку охлаждают водой. Получающаяся свободная кислота дает с эфиром продукт присоединения, который в виде тяжелых маслянистых капель оседает на дно и образует третий слой. После отстаивания и осветления этот слой сливают в колбочку. Если при добавлении новой порции соляной кислоты на границе эфира и раствора больше не образуется маслянистых капель, это свидетельствует об окончании реакции. К полученной маслянистой жидкости прибавляют примерно равный объем воды н путем пропускания тока чистого сухого воздуха через раствор удаляют эфир. Оставшийся прозрачный водный раствор кислоты помещают в вакуум-
1898 Глава 32. Изо- и гетерополисоединения
ный эксикатор над кони. H2SO«* до начала кристаллизации, а затем для поглощения оставшегося количества НС1 раствор выдерживают над твердым КОН. В случае 12-вольфрамоборной кислоты кристаллизацию ведут в эксикаторе с Р2О5, что предотвращает возможность разложения гетерополикислоты за счет улетучивания борной кислоты. Следует указать на то, что во время встряхивания и осаждения третьего слоя необходимо прибавлять соляную кислоту, поскольку она расходуется при образовании продукта присоединения эфира. Применение серной или азотной кислоты не рекомендуется, так как эти кислоты в дальнейшем удаляются не так легко, как соляная кислота.
Способ 2. Ионный обмен. Преимущество этого способа состоит в высокой степени чистоты конечного продукта. Исходным веществом служит хорошо растворимая в воде гетерополисоль, очищенная многократной перекристаллизацией. В связи с отчетливо выраженной кислотной функцией гетерополикислот и с их склонностью подвергаться действию восстанавливающих агентов рекомендуется использовать в работе катнонообменники, содержащие сульфогруппы (например, пермутит RS, дауэкс 50W), проявляющие только сильнокислотные функции и практически не способные служить восстановителями. Выбор конкретных условий работы зависит от устойчивости, качества и количества получаемой гетерополикислоты. Руководствуются следующими ориентировочными правилами: обменная емкость обычно составляет 2 мг-экв./см3 ионообменной смолы (насыпной объем). Целесообразно работать с возможно более концентрированными исходными растворами, которые медленно (~2— 5 мл/мин) пропускают через колонку. Растворы свободных кислот упаривают в ротационном испарителе до небольшого объема и прн необходимости помещают в эксикатор для кристаллизации.
Этот метод нельзя использовать для получения гетерополикислот, имеющих в водном растворе сильнокислую реакцию. Кроме того, некоторые затруднения возникают, если в растворе присутствуют посторонние соли (NaCl, NaNOg и т. д.), поскольку при прохождении через ионообменную колонку образуются НС1, HNO3 и т. д. Слишком сильнокислая среда при упаривании элюата препятствует кристаллизации свободной гетерополикислоты.
На преимущества этого метода было указано выше. Недостаток его состоит в том, что необходимо исходить из чистых кристаллических солей щелочных металлов, которые часто могут быть получены лишь косвенным путем через стадию свободной кислоты, выделяемой по методу Дрекселя. Значительное время требуется на стадии концентрирования обычно сильно разбавленных растворов гетерополикислот, полученных путем ионного обмена.
ЛИТЕРАТУРА
1. Drechsel Е., Вег., 20, 1452 (1887).
2. Rosenheim A., Jaenicke J., Z. Anorg. Allgem. Chem., 101, 224 (1917).
3. Baker L. C. W., Loev B., McCutcheon T. P., J. Amer. Chem. Soc., 72, 2374 (1950).
4. Element R., Z. Anorg. Allgem. Chem., 260, 267 (1949).
5. Hein F., Lilie H., Z. Anorg. Allgem. Chem., 270, 45 (1952).
6. Copaux H., Ann. Chim. Phys., (8), 17, 217 (1909).
12-Молибдофосфорная кислота Нз(РМ01204о)»пНгО (n = 5—29)
H3PO4 -|- 12MoOs ---> H8(PMoi2O40)
Способ 1 [1]. К кипящему раствору 6,3 г 25%-ной фосфорной кислоты в 100 мл воды небольшими порциями прибавляют 35 г МоОз. Кипячение продолжают еще 2—2,5 ч. Раствор отфильтровывают от нерастворившегося остат
* См. предыдущее примечание. — Прим, перев.
Гвтерополисоединения 1899
ка и с целью очистки продукта встряхивают с эфиром. (При этом добавлять кислоту не надо.) К эфирату добавляют воду в количестве 1/5 его объема. Эфир удаляют в ротационном испарителе. В том случае, если водный раствор имеет зеленоватый оттенок, к нему прибавляют несколько капель 3%-ного Н2О2. Для кристаллизации кислоты раствор помещают в холодильник или в вакуумный эксикатор.
Способ 2 [2]. 50 г молибдата натрия Na2MoO4-2H2O растворяют в 150 мл воды. Раствор пропускают через катионообменник (дауэкс 50WX4) и к элюату прибавляют при перемешивании 2 г 85%-ной фосфорной кислоты и 5 мл 3%-ного НгО2. Раствор упаривают в ротационном испарителе до начала кристаллизации; для кристаллизации оставляют при комнатной температуре. Очистку кислоты производят через эфират, как указано при описании способа 1. Можно также сначала слить растворы молибдата натрия и фосфорной кислоты, а затем пропустить через ионообменник. Однако при этом молиб-дофосфорная кислота может разложиться.
Свойства. Желтые хорошо образованные октаэдрические кристаллы, хорошо растворяющиеся в воде.
ЛИТЕРАТУРА
1. Rosenheim A., Jaenicke J., Z. Anorg. Allgem. Chem., 101, 248 (1917). 2. Пат. США 3446575 (1969).
12-Молибдофосфат аммония (МН4)3(РМо12О40) «пН2О
2,5 г (NH4)H2PO4 растворяют в 50 мл воды и прибавляют 50 мл коиц. HNO3. К этому раствору при перемешивании приливают раствор 50 г (NH4)eMo7O24-4H2O в 150 мл воды. Сразу же выделяется осадок; его отфильтровывают, промывают горячей водой, к которой добавлено несколько капель конц. HNO3, и высушивают на воздухе.
Свойства. Желтая мелкокристаллическая соль, плохо растворяющаяся в воде.
ЛИТЕРАТУРА
1. Kehrmann F„ Z. Anorg. Allgem. Chem., 7, 417 (1894).
12-Молибдофосфат бария Ваз(РМо1204о)2«пН20
К насыщенному раствору 12-молибдофосфорной кислоты прибавляют избыток насыщенного раствора ВаС12. Сразу же в виде крупных кристаллов выделяется бариевая соль. Ее отфильтровывают, промывают небольшим количеством холодной воды и дважды перекристаллизовывают из горячей воды, подкисленной несколькими каплями конц. HNO3.
Свойства. Лимонно-желтые октаэдрические кристаллы, растворяющиеся в воде.
ЛИТЕРАТУРА
1. Kehrmann F„ Z. Anorg. Allgem. Chem., 7, 417 (1894).
12-Молибдокремневая кислота H4(SiMoi204o)-nH2O (n=5—29)
Способ 1. 50 г Na2MoO4-2H2O растворяют при нагревании до 60 °C в 200 мл воды. При энергичном перемешивании прибавляют по каплям сначала 34 мл 25%-ной НС1, а затем раствор 5 г растворимого иатрневого стекла (d 1,37) в 50 мл воды. Продолжая перемешивать, прибавляют по каплям
1900 Глава 32. Изо- и гетерополисоединения
60 мл конц. НС1. Если после охлаждения выпадает кремневая кислота, ее отфильтровывают на стеклянном пористом фильтре. Гетерополикислоту экстрагируют из раствора эфиром. Для этого раствор охлаждают в бане со льдом, прибавляют 100 мл эфира и 100 мл ледяной конц. НС1 порциями по 20 мл. Раствор перемешивают в делительной воронке, производя круговые движения с тем, чтобы избежать образования эмульсии. Эфират 12-молибдокремневой кислоты осаждается в виде маслянистых капель, образующих третий слой. Его отделяют, прибавляют воды (1/5 объема эфирата), после чего эфир удаляют в ротационном испарителе. Водный раствор помещают для кристаллизации в эксикатор с Р4Ою- Если раствор окрашен в зеленый цвет, к нему добавляют несколько капель бромной воды.
Способ 2. Раствор 30 г (МН4)бМо7О24-4Н2О в 100 .мл воды прибавляют к раствору 2,6 г Na2SiC>3 в 100 мл 0,1 н. NaOH. После фильтрования раствор пропускают через сильнокислотный катиоиообменник в Н+-форме. Элюат упаривают в ротационном испарителе до начала кристаллизации. Очистку гетерополнкислоты через эфират проводят, как указано выше (способ 1).
Свойства. Желтые хорошо образованные октаэдрические кристаллы, хорошо растворяющиеся в воде. Соли щелочных, щелочноземельных металлов н Sd-переходных элементов лучше всего синтезировать путем взаимодействия карбоната металла с 12-молибдокремневой кислотой.
ЛИТЕРАТУРА
1. North Е. О., Haney W-, Inorg. Synth., 1, 127 (1939).
2. Strickland J. H. D„ J. Amer. Chem. Soc., 74, 862 (1952).
3. Пат. США 3446575 (1969).
12-Молибдоцерат(1У) аммония (NH;)8(CeMo12O;2)-8FLO
7 г нитрата аммония-церия(ГУ) (МН4)2Се(МОз)б растворяют в 140 мл воды. Этот раствор добавляют по каплям при перемешивании к кипящему раствору 30 г (NH4)eMo7O24-4H2O в 100 мл воды. После остывания отфильтровывают выпавший темно-желтый кристаллический осадок. Его промывают 1%-ным раствором нитрата аммония, затем метанолом и высушивают на воздухе. Выход 15 г.
Свойства. М 2251,81. Темно-желтые кристаллы, плохо растворяющиеся в воде.
12-Молибдоцерат(1У) аммония-водорода
(N Н4)6Н2( СеМо12О42) • ЮН2О
Приготавливают насыщенный при 65 °C раствор (NH4)s(CeMo12O42)-8Н2О в 1 н. H2SO4. После фильтрования раствор охлаждают и к нему прибавляют насыщенный раствор нитрата аммония. Выпавшие кристаллы отфильтровывают, отмывают от сульфат-ионов насыщенным раствором нитрата аммония, затем промывают метанолом и высушивают на воздухе.
Свойства. Светло-желтые кристаллы, плохо растворяющиеся в холодной, несколько лучше в горячей воде.
12-Молибдоцериевая(1У) кислота Нз(СеМо12О42) «пН2О
Кислоту можно получить методом ионного обмена, но не по Дрекселю (через эфират). Приготавливают раствор кислой соли или суспензию аммониевой соли в воде (10 мг соли/1 мл воды), которые пропускают через ионо-
Гетерополисоединения 1901*
обменник (например, амберлит IR 200, дауэкс 50WX4). При упаривании раствора 12-молибдоцериевой(1У) кислоты в ротационном испарителе получают-застывшее в виде стеклообразной массы желтое вещество.
ЛИТЕРАТУРА
1. Baker С. W„ Gallagher G. A., McCutcheon Т. Р., J. Amer. Chem. Soc., 75,.. 2493 (1953).
2. Шахова 3. Ф., Гаврилова С. А. — Ж. неорг. хим., 1958, т. 3, с. 1370.
12-Молибдоарсенат(¥) калия K3(AsMoi2O40)«nH2O
30 г (NH4)6Mo7O24-4H2O нагревают в фарфоровой чашке с раствором1.. КОН (9 г КОН в 20 мл воды) до полного удаления аммиака. После охлаждения раствор разбавляют 50 мл воды, охлаждают льдом и при перемешивании вливают в охлажденную льдом конц. HNO3. При этом раствор остается прозрачным. После прибавления рассчитанного количества As2O5 (1,84 г 3As20sX Х5Н2О), растворенного в 50 мл воды, раствор окрашивается в желтый цвет. При нагревании до 60—70°C начинается выпадение желтого осадка. По. охлаждении осадок отфильтровывают, промывают холодной водой и высушивают на воздухе.
Свойства. Мелкокристаллический порошок. Плохо растворяется в холодной, несколько лучше в горячей воде.
ЛИТЕРАТУРА
1. Pufahl О., Dissert. Univers. Leipzig, 1888.
б-Молибдохромат(Ш) аммония (МН4)з(СгМо6О24Н6)-пН2О (n=4—7)
К нагретому до кипения раствору 12 г NH4Cr(SO4)2-12Н2О в 50 мл воды прибавляют по каплям при перемешивании раствор 30 г (NH4)вМо7О24-4Н2О' в ПО мл воды. При этом цвет раствора медленно меняется от зеленого до-сиреневого. pH раствора поддерживают при этом на уровне 4,5 путем добавления разб. H2SO4. При охлаждении выкристаллизовывается соль розового-цвета.
Свойства. М 1197,7 (7Н2О). Розовые четырехугольные пластинки или чешуйки. Хорошо растворяется в горячей воде.
ЛИТЕРАТУРА
1. Hall R. D., J. Amer. Chem. Soc., 29, 692 (1907).
2. Rosenheim A., Schwer H., Z. Anorg. Allgem. Chem., 89, 226 (1914).
б-Молибдохромовая(Ш) кислота Нз(СгМо6О24Н6)«пН2О
Насыщенный при 60 °C раствор аммониевой соли пропускают через колонку с сильнокислотным катионообменником. Элюат упаривают досуха в ротационном испарителе.
Свойства. Зеленый мелкокристаллический порошок, растворяющийся» в воде. Можно получить разнообразные соли трехосновной кислоты. При-pH 6,5 гетерополианион полностью разрушается. Раствор кислоты окрашен? в розовый пвет.
0902 Глава 32. Изо- и гетерополисоединения
ЛИТЕРАТУРА
1. Baker L. С. W., J. Amer. Chem. Soc., 77, 2136 (1955).
»6-Молибдохромат(1П) натрия Маз(СгМовО24Нб)’8Н2О
Раствор 145 г Na2MoO4-2H2O в 300 мл воды подкисляют прибавлением кони.. HNO3 до pH 4,5. К этому раствору при перемешивании прибавляют раствор 40 г Сг(МО3)з'9Н2О в 40 мл воды и нагревают до кипения. При этом цвет раствора постепенно меняется от фиолетового до розового. Раствор кипятят в течение ~1 мин н фильтруют через стеклянный пористый фильтр. Затем его оставляют на 6 дней стоять при комнатной температуре. Выпавшие кристаллы отфильтровывают, несколько раз промывают холодной водой и высушивают иа воздухе. Выход 78 г.
Свойства. М 1230,6. Крупные розовые кристаллы, постепенно выветривающиеся на воздухе, что сопровождается образованием пентагидрата в виде порошка розового цвета.
ЛИТЕРАТУРА
1. Tsigdinos G. A., Dissert. Univers. Boston, 1961.
*6-Молибдородат(1П) аммония (NH4)3(RhMoGO24Hf,)-7H2O
В раствор 65 г (NH4)eMo7024-4H20 в 260 мл воды прибавляют раствор 13 г Na3RhCl6-12H2O в 35 мл воды. Смесь нагревают на водяной бане, пока цвет раствора из красного не перейдет в желтый. Охлаждают до 15 °C. Выпавшие желтые кристаллы отфильтровывают, несколько раз промывают ледяной -.водой и высушивают на воздухе.
Свойства. М 1248,8. Желтые кристаллы, хорошо растворяющиеся в горячей воде. Свободную кислоту можно получить из аммониевой соли путем -ионного обмена.
.ЛИТЕРАТУРА
1. Tsigdinos G., Diss. Univers. Boston, 1961; Diss. Abstr., 22, 732 (1961).
*6-Молибдоникколат(П) аммония (NH^^NiMogC^HgpSHaO
Раствор 22 г (NH4)6Mo7024-4H20 в 200 мл воды нагревают до кипения. К нему при перемешивании добавляют по каплям раствор 2,6 г NiSO4-6H2O в 150 мл воды. Цвет раствора при этом меняется из зеленого через сине-зеленый в желто-зеленый. Раствор кипятят в течение 10 мин, а затем фильтруют. К фильтрату прибавляют 10 г NH4C1 и дают остыть. Выпавшую соль отфильтровывают, промывают водой и высушивают на воздухе.
Свойства. М 1186,4. Соль голубого цвета. Умеренно растворяется в холодной, несколько лучше в горячей воде. Свободную кислоту можно получить путем ионного обмена.
.литература
'1. Matijevic Е., Kerker М„ Beyer Н„ Theubert F., Inorg. Chem., 2, 581 (1963).
Гетерополисоединения 1903s
6-Молибдоиодат натрия Na5(IMo6O24)«nH2O (n=26, 34)
2№зНаЮв + 12MoOs + 2Na2COs -----> 2Nas(IMoeO24) + 2CO2 + 2H2O
В 300 мл воды вносят 20 г Na3H2IOe, 59 г МоО3 и нагревают. Через некоторое время прибавляют 7 г Ыа2СОз. Когда все растворится, раствор сильно* упаривают. При этом выпадают хорошо образованные белые ромбоэдрические-и, кроме того, многогранные несколько желтоватые призматические кристаллы.
Свойства. Ромбоэдрические кристаллы, легко выветриваются на воздухе, становясь при этом чисто белыми и непрозрачными. На формульную единицу приходится 34 молекулы воды. Соль хорошо растворяется в воде.
Блестящие желтоватые асимметричные призмы не выветриваются на воздухе. Содержание воды соответствует 26Н2О. Вещество растворяется в воде.
6-Молибдоиюдная кислота Н5(1Мо6О24) «пН2О
25 г натриевой соли растворяют в 100 мл воды. Раствор пропускают через сильнокислотный катионообменник. При упаривании прозрачного и бесцветного элюата наблюдается выделение газа, что свидетельствует о (по крайней-мере частичном) разложении. Образуется светло-желтое кристаллическое вещество. Выход 25 г.
Свойства. Светло-желтое кристаллическое вещество, хорошо растворяющееся в теплой воде.
ЛИТЕРАТУРА
1. Blomstrand С. W., Z. Anorg. All gem; Chem., 1, 10 (1892).
6-Молибдоалюминат натрия Na3(AlMo6O24H6)«llH2O
Раствор 132 г Na2MoO4-2H2O в 400 мл воды подкисляют до pH 4,5 добавлением по каплям и при перемешивании конц. HNO3. К этому раствору прибавляют раствор 32,5 г A1(NO3)3-9H2O в 60 мл воды. Нагревают до кипения,, еще горячий раствор фильтруют и оставляют при комнатной температуре на 2—3 недели, выпавшие кристаллы отфильтровывают, несколько раз промывают небольшими порциями холодной воды и высушивают на воздухе. Выход 60 г.
Свойства. М 1259,6. Крупные белые кристаллы, выветривающиеся на воздухе с образованием пентагидрата. Хорошо растворяется в воде. Свободную кислоту получают путем ионного обмена; устойчивы лишь разбавленные растворы кислоты.
(NH4)3(A1Mo6024H6).7H20 получают аналогичным образом.
литература
1. Tsigdinos G., Diss. Univers. Boston 1961.
2. Baker L. C. W., J. Amer. Chem. Soc., 77, 2136 (1955).
3. Hall R. D„ J. Amer. Chem. Soc., 29, 692 (1907).
б-Молибдоферрат(Ш) аммония (NH4)3(FeMo6O24H6).nH2O (n=5—10)
В Двухлитровом стакане растворяют 30 г гептамолибдата аммония, (NH4) бМо7О24 • 4Н2О в 600 мл воды. Раствор нагревают до 45 °C и затем нагревание прекращают. К раствору при энергичном перемешивании прибавляют’
Й 904 Глава 32. Изо- и гетерополисоединения
по каплям раствор 9 г NH4Fe(SO4)2-12Н2О. Каждая следующая капля должна попадать в раствор лишь после растворения желтого осадка, образовавшегося при падении предыдущей капли. Раствор при этом не должен охлаждаться ниже 40 °C. Раствор фильтруют через складчатый фильтр и оставляют на 1— .2 дня при комнатной температуре. Выпавшие кристаллы отфильтровывают и .промывают небольшим количеством холодной воды.
Свойства. Белые кристаллы. Водные растворы соли разлагаются при на-.греванни выше 45 °C. Свободная кислота неустойчива, ее можно получить лолько в виде очень разбавленных растворов.
«ЛИТЕРАТУРА
1. Rosenheim A., Schwer Н„ Z. Anorg. Allgem. Chem., 89, 224 (1914).
2. Baker L. C. W„ Forster Q., Tan W., Scholnick F., McCutcheon T. H., J. Amer. Chem. Soc., 77, 2136 (1955).
<6-Молибдокобальтат(1П) натрия Ма3(СоМовО24Нб)’8Н2О
Раствор 121 г Ма2МоОг2НгО в 200 мл воды подкисляют до pH 4,5 путем добавления по каплям при интенсивном перемешивании конц. HNO3. Затем прибавляют по каплям при перемешивании раствор 23 г CoSC>4-7H2O -в 40 мл воды. Образующийся осадок снова переводят в раствор, добавляя немного конц. HNO3. После этого приливают раствор 30 г Na2S20e в 35 мл воды и смесь нагревают. При этом может образоваться синий осадок, растворяющийся, однако, при дальнейшем нагревании. Цвет раствора медленно меняется от красного до зеленого. Раствор кипятят до прекращения выделения ^кислорода, фильтруют в горячем состоянии через стеклянный пористый фильтр и оставляют на 2—3 недели при комнатной температуре. Выпавшие кристаллы -отфильтровывают, промывают несколькими небольшими порциями холодной воды и высушивают на воздухе. Выход 39 г.
Свойства. М 1227,53, Зеленовато-синие кристаллы, теряющие воду при выветривании на воздухе и переходящие при этом в зеленый порошок. Водный раствор соответствующей гетерополикислоты получают путем ионного ^обмена.
ЛИТЕРАТУРА
1. Friedheim С., Keller F., Вег., 39, 4306 (1906). '2. Tsigdinos G. A., Diss. Univers. Boston, 1961.
18-Молибдодифосфат аммония (NH4)6(P2Mo18O62) • 14Н2О
К раствору 100 г Na2MoO4-2H2O в 400 мл воды прибавляют при перемешивании 15 мл 85 %-ной Н3РО4 и 80 мл конц. НС1. Образовавшийся желтый 'раствор кипятят с обратным холодильником в течение 8 ч. Если за счет вос-- становления гетерополнсоединения раствор окрасится в зеленый цвет, прибавляют несколько миллилитров 3%-ного Н2О2. Затем при перемешивании в раствор вносят твердый NH4CI до насыщения и приливают еще 200 мл насыщенного раствора NH4C1. При стоянии из раствора выпадают желтые кристаллы 18-молибдодифосфата аммония.
Кристаллы отфильтровывают, растворяют в 100 мл воды. К раствору прибавляют 400 мл диоксана, что вызывает выпадение аммониевой соли. Ее отфильтровывают, промывают сначала смесью диоксана с водой (3:1), потом .диоксаном и эфиром. Выход 25 г.
Гетерополисоединения 1905
Свойства. Желтые кристаллы, хорошо растворяющиеся в воде. Соль можно также рассматривать как димерный 9-молибдофосфат аммония. литература
1. Rosenheim A., Traube A., Z. Anorg. Allgem. Chem., 91, 75 (1915).
2. Wu H„ J. Biol. Chem., 43, 183 (1920).
18-Молибдодифосфорная кислота НеСРгМо^Овг)«ПНгО
Свободную шестиосновную кислоту получают из аммониевой соли методом Дрекселя (экстракцией эфиром) или путем ионного обмена.
ЛИТЕРАТУРА
1. Wu Н., J. Biol. Chem., 43, 189 (1920).
2. Tsigdinos G., Diss. Univers. Boston, 1961.
18-Моли1бдодиарсенат(¥) натрия NaG( As2Mo18OG2)-23^0
Водный раствор Na2HAsO4-7H2O нагревают до кипения и прибавляют порциями МоО3. Некоторое время суспензию кипятят. Затем фильтруют и темно-желтый фильтрат упаривают в ротационном испарителе. Натриевая соль выпадает в виде желтых кристаллов.
Свойства. Кристаллы — желтые моноклинные призмы, растворимые в воде. Можно рассматривать как димерный 9-молнбдофосфат натрия.
ЛИТЕРАТУРА
1. Rosenheim A., Traube A., Z. Anorg. Allgem. Chem., 91, 92 (1915).
18-Молибдодимышьяковая(¥) кислота He(As2Moi8O62)«25H2O
Способ 1. Из раствора натриевой соли путем экстракции эфиром в присутствии соляной кислоты (по Дрекселю).
Способ 2. Из раствора натриевой соли путем ионного обмена на ионооб-Менннке в Н+-форме с последующим упариванием водного раствора кислоты в ротационном испарителе.
Свойства. Темно-красные триклйннЫё Кристаллы, легко разлагающиеся С отщеплением воды. Хорошо растворяется в воде.
ЛИТЕРАТУРА
1. Rosenheim A., Traube А., Т. Anorg. Allgem. Chem., 91, 75 (1915).
2. Pujahl О., Diss. Univers. Leipzig, 1888.
9-Молибдоманганат(1¥) аммония (NH4)e(MnMo9O32)-6H2O
Способ 1 [1]. 30 г гептамолибдата аммония (NH4)eMo7O24-4H2O растворяют в 300 мл воды. Этот раствор подкисляют серной кислотой до pH 4,5— 4,0, после чего прибавляют свежеприготовленный раствор 2,9 г КМпО4 в 200 мл воды. Перемешивая раствор, его нагревают до 70—80 °C. Медленно 4—1361
Г906 Глава 32. Изо- и гетерополисоединения
по каплям (1 капля за 5 с) прибавляют 3%-ный Н2О2 до тех пор, пока фиолетовая окраска перманганата не перейдёт в красно-коричневую. Раствор фильтруют горячим. Ставят на ночь в холодильник и отфильтровывают выпавшие кристаллы. Их промывают небольшим количеством ледяной воды и высушивают на воздухе. Выход 25 г.
Способ 2 |[2, 3]. Производят окисление Мп2+ в азотнокислом растворе молибдата действием пероксодисульфата.
Свойства. М 1646,7. Блестящие ромбоэдрические кристаллы с цветом от оранжево-красного до коричнево-красного. Хорошо растворяется в горячей воде.
ЛИТЕРАТУРА
1. Friedheim С., Samuelson М., Z. Anorg. Allgem. Chem., 24, 67 (1900),.
2. Hall R. D„ J. Amer. Chem. Soc., 29, 692 (1907).
3. Schaal R., Souehay P., Anal. Chim. Acta, 3, 114 (1949).
9-Молибдомарганцевая(1¥) кислота Н6(МпМо9Оз2) «пНгО
Свободную кислоту получают путем иоииого обмена с последующим упариванием раствора в ротационном испарителе.
Свойства. Темио-коричиевый мелкокристаллический порошок. При взаимодействии с карбонатами металлов могут быть получены разнообразные соли. При этом значение pH не должно превышать 4,5.
9-Молибдоникколат(1¥) аммония (NH4)6(NiMo9O32)«6,5H2O
Раствор, содержащий NiSO4 и (NH4)2S2O8 в молярном соотношении 1 : 1, прибавляют по каплям к рассчитанному количеству 10%-ного раствора гептамолибдата аммония, нагретого до 95 °C. При интенсивном перемешивании раствор кипятят в течение 5 мин, быстро фильтруют и охлаждают. Соль содержит небольшую примесь катионно-связанного никеля. Для получения натриевой соли, свободной от таких примесей, концентрированный раствор аммониевой соли пропускают через катионообменник в №+-форме.
Свойства. М 1659,3. Темно-красные кристаллы, сходные по структуре с 9-молибдоманганатом аммония.
ЛИТЕРАТУРА
1. Hall R. D., J. Amer. Chem. Soc., 29, 692 (1907).
2. Baker L. C. W., Weakley T. J. R„ J. Inorg. Nucl. Chem., 28, 447 (1966).
1О-Молибдодикобальтат(III) аммония
(N H4) g (C02M010O30) *10 H2O
75 г (МН4)бМо7О24-4Н2О растворяют в 225 мл воды. К этому раствору прибавляют раствор 15,5 г Со(СНзСОО)г-4Н2О в 400 мл воды. В полученный красный раствор вносят 30 г активированного угля в виде зерен и 100 мл 18%-кого Н2О2. Смесь нагревают до кипения и кипятят до прекращения выделения кислорода. Образовавшийся темно-зеленый раствор отфильтровывают в горячем состоянии от активированного угля. Фильтрат помещают в холодильник при 0—3°С. Выпавшие темно-зеленые кристаллы отфильтровывают, промывают небольшим количеством ледяной воды и высушивают на воздухе. Выход 45 г.
Гетерополисоединения 1907
Свойства. М 1959,3- Темно-зеленые кристаллы, хорошо раствориющиеся в воде. Соль может быть разделена на оптические изомеры.
ЛИТЕРАТУРА
1. Friedheim С., Keller F., Вег., 39, 4306 (1906).
2. Tsigdinos G. A., Diss. Univers. Boston, 1961.
3. Ama T„ Hdaka J., Shimura Y., Bull. Chem. Soc. Japan, 43, 2654 (1970).
11-Молибдо-1-ванадофосфорная кислота
H4(PMouVO40)*nH2O (na; 32—33)
6,1 г NaVO3 растворяют при нагревании в 100 мл воды. К этому раствору прибавляют раствор 8,9 г Na2HPO4-2H2O в 100 мл воды. По охлаждении смесь подкисляют добавлением 5 мл конц. H2SO4, причем цвет раствора становится красным. К полученному раствору прибавляют раствор 133 г Na2MoO4-2H2G в 200 мл воды и затем при интенсивном перемешивании добавляют по каплям 85 мл конц. H2SO4. При этом раствор окрашивается в светло-красный цвет.
Охлажденный во льду раствор встряхивают с 400 мл эфира. Эфират гетерополикислоты образует средний слой. Его отделяют и путем пропускания воздуха из него удаляют эфир. Остаток представляет собой вещество оранжевого цвета. Его растворяют в 50 мл воды и раствор помещают в эксикатор с H2SO4 для кристаллизации. Выпавшие кристаллы отфильтровывают, промывают небольшим количеством воды и высушивают на воздухе.
Свойства. М 2256,8 (32Н2О). Кристаллы оранжевого цвета, хорошо растворяющиеся в воде.
ЛИТЕРАТУРА
1. Tsigdinos G. A., Hallada С. J., Inorg. Chem., 7, 437 (1968).
10-Молибдо-2-ванадофосфорная кислота
H5(PMoi0V2O40).nH2O 34—35)
Получение этой кислоты аналогично получению H4(PMonV04o). Берут следующие количества реагентов: 24,4 г NaVO3 в 100 мл воды, 7,1 г Na2HPO4-2H2O в 100 мл воды, 5 мл конц. H2SO4, 121 г Na2MoO4-2H2O в 200 мл воды, 85 мл конц. H2SO4, 500 мл эфира. Эфират гетерополикислоты образует в этом случае нижний слой. Его дальнейшую обработку ведут так же, как это описано выше. Выход 35 г.
Свойства. М 2247,9 (34Н2О). Большие кристаллы красного цвета.
9-Молибдо-З-ванадофосфорная кислота
Н6(РМо9УзО40).пН2О (па; 34—35)
Получение ведут аналогично синтезу предыдущего препарата. Берут следующие количества реагентов: 36,6 г NaVO3 в 200 мл воды, 7,1 г Na2HPO4X Х2Н2О в 100 мл воды, 5 мл конц. H2SO4, 54,5 г Na2MoO4-2H2O в 150 мл воды, 85 мл конц. H2SO4, 400 мл эфира. Эфират гетерополикислоты образует средний слой. Выход 7 г.
Свойства. Л4 2203,9 (34Н2О). Кристаллы красного цвета. Кислота разлагается в сильнокислом растворе, ее нельзя получить на катионообмеиниках. Этим можно объяснить невысокий выход.
4»
1908 Глава 32. Изо- и гетерополисоединения
ЛИТЕРАТУРА
1. Tsigdinos G. A., Hallada С. J., Inorg. Chem., 7, 438 (1968).
12-Ванадофосфат аммония (NH4)7(PV12O36)’nH2O
К раствору 28 г NH4VO3 в 300 мл воды прибавляют по каплям при интенсивном перемешивании 4,8 г Н3РО4 (d 1,7). Раствор нагревают до 60 °C и приливают 50 мл 1 и. НС1. Выпавший V2Os отфильтровывают. Из фильтрата, окрашенного в темно-красный цвет, выпадают фиолетово-красные кристаллы. Их отфильтровывают и промывают небольшим количеством холодной воды.
Свойства. Фиолетово-красные кристаллы, растворяющиеся в воде с образованием раствора, окрашенного в темно-красный цвет. При нагревании раствора соединение разрушается и осаждается V2O5.
ЛИТЕРАТУРА
1. Rosenheim A., Pieck М., Z. Anorg. Allgem. Chem., 98, 223 (1916).
12-Ванадофосфат серебра Ag7(PVi2O36)«nH2O
30,5 г NaVOs растворяют при нагревании в 200 мл воды, а 18,7 г Na2HPO4-2H2O растворяют в 80 мл теплой воды. Оба раствора сливают вместе и при перемешивании добавляют по каплям 26,2 мл конц. HNO3. Образовавшийся темно-красный раствор разбавляют водой до объема 330 мл.
42,5 г AgNO3 растворяют в 660 мл воды и раствор при интенсивном перемешивании прибавляют по каплям к раствору ванадата-фосфата. Выпавшую серебряную соль отфильтровывают и высушивают в вакуумном эксикаторе над Р4О10. Выход 33 г.
Свойства. Коричиево-черные кристаллы, плохо растворяющиеся в воде.
ЛИТЕРАТУРА
1. Jander G„ Jahr К. F„ Witzmann Н„ Z. Anorg. Allgem. Chem., 217, 75 (1934).
13-Ванадоманганат(1¥) калия K7(MnV13O38)«18H2O
Способ 1. 23,6 г V2Os и 18,0 г К2СО3 вносят в 1000 мл воды и затем при перемешивании нагревают до 60—80 °C. Для ускорения растворения V2O5. добавляют 1—2 мл 30%-ного Н2О2. После того как все количество V2OS растворится, избыток Н2О2 разлагают кипячением и горячий раствор фильтруют. Горячий фильтрат подкисляют до pH 5—4, добавляя по каплям и при энергичном перемешивании 1 н. HNO3. К этому раствору, нагретому до 60—70°C, медленно прибавляют по каплям, интенсивно перемешивая, два раствора: свежеприготовленный раствор перманганата калия (3,2 г КМпО4 в 200 мл воды} и 100 мл 1%-ного Н2О2.
После прибавления растворов перемешивают еще в течение 15 мин, а затем упаривают раствор в ротационном испарителе до объема 400—500 мл, еще горячим фильтруют и оставляют фильтрат на ночь в холодильнике. Выпавшие кристаллы отфильтровывают, промывают небольшим количеством ледяной воды и высушивают на воздухе. Выход 15 г.
Способ 2 [1]. 35,9 г KVO3 растворяют в 1000 мл воды. К раствору добавляют по каплям 20 мл I н. HNO3. После этого прибавляют раствор 3,4 г MnSO4-H2O в 20 мл воды и 10,8 г K2S2O8, причем раствор перемешивают и
Гетерополисоединения 19091
поддерживают при температуре —80 °C. Перемешивание и нагревание продолжают еще в течение 5—7 ч, пока раствор не упарится до объема —300 мл. Горячий раствор фильтруют, затем к фильтрату прибавляют 40 мл 1 М раствора ацетата калия. Оставляют на ночь. Выпавшие кристаллы отфильтровывают, промывают небольшим количеством ледяной воды и высушивают иа воздухе.
Беря в качестве реагентов соли натрия или аммония, можно аналогичным образом получить 13-ванадоманганаты(1У) натрия или аммония.
Свойства. М 1923,15. Блестящие октаэдрические кристаллы с цветом от оранжево-красного до коричнево-красного. Хорошо растворяются в горячей воде.
ЛИТЕРАТУРА
1. Flynn С. М., Pope М. Т., J. Amer. Chem. Soc., 92, 85 (1970).
12-Вольфрамокремневая кислота H4(SiW1204o)"7H20
1000 г NajWOi^HjO растворяют в 2000 мл воды. К раствору прибавляют 75 г натриевого растворимого стекла (d 1,37). Раствор нагревают до кипения и добавляют по каплям (со скоростью 1 капля/с) 60 мл 24%-ной соляной, кислоты. Если при этом образуется осадок кремневой кислоты, его отфильтровывают на стеклянном пористом фильтре. Раствор охлаждают, прибавляют при охлаждении 400 мл ледяной концентрированной соляной кислоты и встряхивают в делительной воронке с эфиром. Эфират 12-вольфрамокремневой кислоты образует маслянистые капли, опускающиеся вниз. Для очистки эфират растворяют в 1000 мл 3 и. НС1 и полученный раствор снова встряхивают с эфиром. От основного количества эфира эфиратиый слой освобождают путем продувки чистым сухим воздухом, после чего начинают образовываться кристаллы. В заключение прибавляют воду в количестве, соответствующем половине объема имеющегося раствора, и удаляют остатки эфира просасыванием очищенного воздуха. Для кристаллизации кислоты раствор помещают в вакуумный эксикатор.
Свойства. М 2038,4. Блестящие бесцветные кристаллы, имеющие форму октаэдров. Хорошо растворяется в воде.
ЛИТЕРАТУРА
1. North Е. О., Inorg. Synth., 1, 130 (1939).
12-Вольфрамосиликат калия K4(SiW12O40)«nH2O (п=9, 18)
Готовят водный раствор кислоты (1 масс. ч. кислоты на 3—4 масс. ч. воды) и определяют его концентрацию путем выпаривания и прокаливания аликвотной части. При нагревании прибавляют небольшими порциями рассчитанное количество К2СО3. Прозрачный раствор, реакция которого должна быть еще кислой, упаривают на водяной бане или в ротационном испарителе до */г—’А первоначального объема. При охлаждении калиевая соль K^SiWijO») X XnHjO осаждается сначала в виде гексагональных призматических кристаллов, потом выделяются также ромбические кристаллы. Перекристаллизацию производят из горячей воды.
Свойства. Бесцветные гексагональные призматические кристаллы (18Н2О); легко выветриваются. Хорошо растворяются в горячей воде и несколько хуже
1910 Глава 32. Изо- и гетерополисоединения
в холодной. Ромбические кристаллы (9Н2О) представляют собой, по-видимому, соль «изокислоты». Они выветриваются не так быстро.
ЛИТЕРАТУРА
1. Rosenheim A., Jaenicke J., Z. Anorg. Allgem. Chem., 101, 243 (1917).
12-Вольфрамофосфорная кислота H3(PW1204o)*nH20 (n = 5—29)
Кислоту получают по методу Дрекселя путем извлечения эфиром из раствора натриевой соли Na3(PWi204o)-пНгО, содержащего конц. НС1. Обычно получают кристаллы, окрашенные в желтый или зеленоватый цвет. Если используемую натриевую соль предварительно 1—2 раза перекристаллизовать, то кислота выделяется в виде прозрачных бесцветных кристаллов.
Кислота также хорошо получается методом ионного обмена. При этом используют насыщенный раствор натриевой соли. Бесцветный элюат сначала концентрируют в ротационном испарителе, а кристаллизацию ведут в вакуумном эксикаторе иад конц. H2SO4*.
Свойства. Большие блестящие кристаллы, имеющие форму октаэдров. Хорошо растворимы в воде. Окрашенные кристаллы неустойчивы и часто уже через несколько часов превращаются в порошок. Напротив, бесцветные кристаллы могут сохраняться в течение месяцев.
ЛИТЕРАТУРА
1. Rosenheim A., Jaenicke J., 7.. Anorg. Allgem. Chem., 101, 251 (1917).
12-Вольфрамофосфат натрия Na3(PWi204o)«nH20 (п^ЗО)
12Na2WO4+Na2HPO4+23HCl—>Nas(PW1204o)+23NaCl
Раствор 50 г Na2WO4-2H2O и 25 г Na2HPO4-12Н2О в 80 мл воды упаривают при —80 °C до начала кристаллизации, т. е. до образования тонкой кристаллической корочки. Затем при перемешивании прибавляют 75 мл 24%-ной НС1 (d 1,12). Образующийся осадок снова полностью растворяется. Упаривают на водяной бане до начала кристаллизации. Перекристаллизовывают из воды.
Свойства. Большие бесцветные столбчатые кристаллы, иногда с зеленоватым оттенком. Растворимы в воде.
ЛИТЕРАТУРА
1. Rosenheim A., Jaenicke J., Z. Anorg. Allgem. Chem., 101, 251 (1917).
12-Вольфрамоборная кислота H5(BWi204o)»nH2O
Способ 1 [1]. Из Na2WO4-2H2O сначала получают натриевую соль Nas(BWi204o). При этом берут большой избыток борной кислоты, с тем чтобы нейтрализовать щелочную реакцию раствора Na2WO4, так что раствор становится кислым.
Из раствора натриевой соли кислоту выделяют путем добавления эфира и серной кислоты по методу Дрекселя.
* См. предыдущее примечание. — Прим, перев.
Гетерополисоединения 1911
100 г Na2WO4-2H2O и 150 г Н3ВО3 растворяют в 400—500 мл кипящей воды. Раствор кипятят, пока из пробы раствора при добавлении разб. НС1 не перестанет выпадать вольфрамовая кислота. После охлаждения отделяют отсасыванием раствор от кристаллической массы, состоящей из борной кислоты и полибората натрия. В раствор вносят дополнительно 70 г Н3ВО3 и упаривают, нагревая открытым пламенем горелки. Образующуюся при охлаждении кристаллическую массу отсасывают и промывают 33%-иой H2SO4. Маточный раствор, содержащий натриевую соль Na5(BWi2O40)-иН2О, обрабатывают по методу Дрекселя, встряхивая с 2—3-кратным объемом 33 %-ной серной кислоты и эфиром.
Способ 2 [2]. 100 г Na2WO4-2H2O растворяют в 200 мл воды, 1,56 г Н3ВО3 растворяют в 20 мл горячей воды. Оба раствора сливают вместе в трех-гордой колбе на 500 мл, снабженной обратным холодильником, мешалкой и капельной воронкой. При нагревании до 90 °C и перемешивании медленно, по каплям прибавляют 81,2 мл 30%-ной H2SO4. Перемешивание продолжают еще в течение 3 ч. Если при охлаждении образуется осадок, его отфильтровывают на стеклянном пористом фильтре G4.
Из этого раствора получают эфират, для чего готовят и затем охлаждают льдом следующие растворы. Раствор Г. 50 мл диэтилового эфира, который предварительно встряхивают с 75 %-ной H2SO4. Раствор 2: 80 мл 75 %-ной H2SO4, насыщенной диэтиловым эфиром.
Раствор гетерополисоединения и раствор 1 встряхивают в делительной воронке. После разделения фаз медленно, по каплям прибавляют раствор 2. Когда добавлена уже половина раствора 2, начинается осаждение маслянистого эфирата в виде третьей, тяжелой фазы, причем водный слой мутнеет. После добавления всего количества кислоты водная фаза снова становится прозрачной, а на границе масло/водный раствор появляется осадок сульфата натрия.
Отдельные фракции эфирата собирают вместе, добавляют воды (Vs их объема) и удаляют эфир в ротационном испарителе.
Для удаления ионов SO42- прибавляют 1,7 г ВаСОз. После фильтрования через стеклянный пористый фильтр G4 раствор упаривают в ротационном испарителе до начала кристаллизации. Кристаллическую массу высушивают в токе азота. При этом часто происходит разложение с образованием WO3-xH2O.
Свойства. Выделяющиеся кристаллы имеют форму октаэдров или гексагональных игл. Прозрачные октаэдрические кристаллы довольно устойчивы при хранении, хотя они при этом приобретают жирный блеск и желтоватый оттенок. Гексагональные игольчатые кристаллы менее устойчивы и скорее мутнеют, окрашиваясь в желтый цвет.
ЛИТЕРАТУРА
1. Rosenheim A., Jaenicke J., Z. Anorg. Allgem. Chem., 77, 244 (1912).
2. Lange G„ Diss. Univers. Hohenheim, 1969.
12-Вольфрамоборат натрия Na5(BW1204o)«nH20 (n«58)
Кристаллическую натриевую соль лучше всего получать из свободной кислоты путем ее взаимодействия с рассчитанным количеством Na2CO3. К раствору 42 г кристаллической гетерополикислоты прибавляют 3 г безводного Na2CO3. Упаривают сначала на водяной бане, а затем в эксикаторе над конц. H2SO4.
Свойства. Хорошо образованные октаэдрические кристаллы белого цвета.
1912 Глава 32. Изо- и гетерополисоединения
ЛИТЕРАТУРА
1. Rosenheim A., Schwer Н., Z. Anorg. Allgem. Chem., 89, 236 (1914).
18-Вольфрамодифосфат аммония (NHOe^WisOeO'MIbO
1 моль Na2WO4-2H2O растворяют в горячей воде и к раствору прибавляют 4 моль 85%-пой фосфорной кислоты. Желтый раствор при перемешивании кипятят с обратным холодильником в течение 3—5 ч. Для устранения образующихся продуктов восстановления в конце нагревания прибавляют несколько капель конц. HNO3.
Во время охлаждения вносят твердый NH4C1 в таком количестве, чтобы произошло выпадение гетерополисоли и раствор при этом обесцветился. Перекристаллизацию ведут из горячей воды.
Свойства. Лимоино-желтые или зеленовато-желтые кристаллы. Растворяются в воде. Свободную кислоту можно получить по методу Дрекселя (см. в начале разд. «Гетерополисоедииения»).
ЛИТЕРАТУРА
1. Kehrmann F., Z. Anorg. Allgem. Chem., 1, 432 (1892); Вег., 20, 1808 (1887).
2. Jander G., Banthien H., Z. Anorg. Allgem. Chem., 229, 142 (1936).
18-Вольфрамодиарсенат(¥) аммония (МН^^АвгХУ^Овг^ННгО
Гетерополисоедииение мышьяка получают так же, как соответствующее соединение фосфора.
ЛИТЕРАТУРА
1. Kehrmann F„ Z. Anorg. Allgem. Chem., 22, 290 (1900).
2. Rosenheim A., Jaenicke J., Z. Anorg. Allgem. Chem., 101, 270 (1917).
Глава 33. МЕТАЛЛООРГАНИЧЕСКИЕ
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
В. П. Фельхаммер, В. А. Херманн, К. Офеле (W. Р. Fehlhammer, W. A. Herrmann, К- Ofele)
Перевод канд. хим. наук В. Н. Постнова
Предварительные замечания
В связи с бурным развитием металлоорганической химии мы попытались в этой главе (в продолжение статьи Ф. Зееля «О карбонильных и нитрозильных комплексах:», приведенной во втором немецком издании данной книги) собрать воедино удобные методики получения довольно значительного числа наиболее употребительных металлоорганических соединений. Хотя мы стремились охватить по возможности больший спектр различных практически важных классов соединений, специалист заметит, что при отборе методик предпочтение отдано карбонильным комплексам металлов. По-видимому, это справедливо, и не только потому, что карбонилы металлов благодаря пионерским исследованиям В. Хибера и его школы заняли достойное место в современной химии комплексов, но прежде всего потому, что именно этот класс соединений наиболее широко используется в качестве исходных веществ в металлооргани-. ческой химии. Все отобранные препараты, по нашему мнению, являются типовыми и (или) наиболее часто используются как исходные вещества в металлоорганических синтезах. Для нахождения других типов металлоорганических соединений, ие вошедших в данную главу, рекомендуем обратиться к допол-. нительной литературе и прежде всего к книгам [1—3].
Методики синтезов в данном разделе составлены таким образом, что при строгом их соблюдении препараты получаются аналитически и спектрально чистыми. Там, где это целесообразно, наряду с указанием обычных физических характеристик (цвет, устойчивость на воздухе, /пл, /кип, растворимость) приведены спектральные данные, которые можно использовать при оценке чистоты получаемых соединений. Практически во всех случаях чистота получаемых образцов достаточна для их дальнейшего использования в синтезах. Величины загрузок (~1—60 г) препаративно оправданны и в отдельных случаях зависят также от доступности и стоимости исходных соединений. Мы сознательно не включили в книгу методики получения некоторых дешевых и легкодоступных реактивов, таких, например, как Fe(CO)s, Ni(CO)4*. Большинство приведенных методик многократно проверены нами на воспроизводимость, в том числе и при изменении загрузок.
ЛИТЕРАТУРА
1. King R. В., Organometallic Syntheses (R. В. King, J. J. Fisch, eds.), Academic Press, New York, London, 1965. Подборка хорошо воспроизводимых методик получения ряда металлоорганических соединений, в особенности, карбонилов металлов и их производных, а также циклопентадиенильных комплексов.
2. Inorganic Syntheses, McGraw-Hill, New York, 1939... Серия издается с 1939 г. Хорошо отработанные прописи синтезов ряда типичных комплексов
* Эти, а также некоторые другие исходные реактивы, как правило, имеются в продаже.
1914 Глава 33. Металлоорганические комплексы
переходных металлов. Предметный и формульный указатели в 10-м и 15-м томах. До сих пор вышло 19 томов.
3. Dub. М. (ed.), Organometallic Compounds 2. Aufl., Springer, Berlin, Heidelberg, New York, 1966. 1-е дополнение ко 2-му изданию: Springer, 1975. Весьма ценный сборник, охватывающий широкий ряд металлоорганических соединений, за исключением простых карбонилов металлов и нитрозильных комплексов. Для каждого соединения приводятся методы получения, физические характеристики и типичные реакции, в которые эти соедииеиия вступают. Кроме того, приведены ссылки на оригинальные источники и патенты.
Общая техника работы
Как правило, синтезы проводятся с использованием трубок Шленка, что стало обычным во всех металлоорганических лабораториях как у иас в стране, так и за рубежом. В качестве инертного газа используют азот, предварительно очищенный пропусканием последовательно через BTS-катализатор* и активированные молекулярные сита (5 А)**. Сведения об общей технике работы в лаборатории можно найти в ч. I данной книги (см. г. 1).
Препараты, разлагающиеся на воздухе, или комплексы, синтез которых непременно следует проводить в атмосфере инертного газа из-за неустойчивости иа воздухе исходных материалов или промежуточных стадий, помечены знаком □□.В таких случаях все стеклянные сосуды перед их употреблением многократно последовательно откачивают в вакууме масляного насоса и продувают азотом. Для нагревания колб используют электрические нагреватели (до 500 °C)***. Переливание жидких продуктов или растворителей проводят только в противотоке азота.
Все соединения, помеченные знаком □, как правило, не подвержены действию кислорода воздуха в кристаллическом состоянии. Их получение, одиако, следует вести в атмосфере инертного газа из-за неустойчивости в растворах, что иногда сказывается на общем выходе.
Препараты, не требующие применения атмосферы инертного газа, никак специально не обозначены.
Металлоорганические комплексы можно хранить в течение продолжительного времени в плотно закрытых трубках Шленка в атмосфере инертного газа и в холодильнике.
В работах с неустойчивыми на воздухе и (или) легко гидролизуемыми соединениями с особой тщательностью следует готовить используемые растворители, очистка которых и абсолютирование подробно описаны во многих руководствах****. Для абсолютирования наиболее употребительных растворителей используют двухлитровые колбы и осушители, указанные в табл. 45. Особенно аккуратно следует обращаться со сплавами Na/K (самовозгораются на воздухе!). Довольно стабильны и более удобны в употреблении ставшие недавно коммерчески доступными сплавы Na/Pb; однако они дороже и не так эффективны.
Очень удобна для абсолютирования и насыщения растворителя инертным газом аппаратура, приведенная на рис. 459. Растворитель, помещенный в двухлитровую колбу, в течение длительного времени (обычно несколько дней)
* Фирма-изготовитель Badische Anilin-u. Sodafabrik (6700 Ludwigshafen).
** О высушивании с помощью молекулярных сит см. брошюру «Тгоскпеп in Labor» («Лабораторные осушители») фирмы Е. Merck (6100 Darmstadt).
*** фирма-поставщик EGA-Chemie (7924 Steinheim/Albuch).
**** См. т. 1, ч. I данной книги; Bunge W. — В кн.: Methoden der organischen Chemie (Houben, Weyl-Miiller), 4 Aufl., Bd. 1/2, S. 765, Thieme, Stuttgart 1959.
Общая техника работы 1915
Таблица 45. Наиболее употребительные растворители и используемые осушители
Растворитель Квалификация чистоты иа торговом препарате Осушитель
Ацетон X. ч. Granusic Ва, молекулярные сита 3 А6
Ацетонитрил для анализа 1) Granusic Аа; г) поташ
Бензол х. ч. Сплав Na/K
Хлороформ для сиит. Granusic Ва
Циклогексан для синт. К
Эфир диэтиловый х. ч. Сплав Na/K
1,4-Диоксаи для синт. К
М,Ц-диметилформамид для синт. Молекулярные сита 4 А6 (хранить в посуде из темного стекла)
Диметиловый эфир диэтиленгликоля (диглим) чистый, 98% Молекулярные сита 4 А6
Диметиловый эфир этиленгликоля (моноглим) чистый, 99% Молекулярные сита 4 А6
Этанол 99,6 %1 X. ч. 1) Na; 2) диэтиловый эфир фталевой кислоты
я-Гексан, н-гептан X. ч. Сплав Na/K
Метанол X. ч. Mg (метилат магиия)
Метиленхлорид X. ч. Granusic Ва; сплав Na/Pb
н-Октан для сиит. N|a
н-Пентаи х. ч. Сплав Na/K
Тетрагидрофураи для сиит.” Предварительная осушка перегонкой иад LiAlH,; последующая очистка многодневным кипячением иад сплавом Na/K
Толуол для синт. Na
а Фирма-поставщик Т. J. Baker Chemicals (6080 Gross-Gerau).
б Фирма-поставщик Е. Merck (6100 Darmstadt 1).
вНастоятельно рекомендуется применение растворителя именно этой квалификации чистоты, так как иначе абсолютирование потребует значительной затраты времени! Использование безводного и тщательно обескислороженного тетрагндрофурана особенно необходимо в случае синтезов т]-циклопентадненильных комплексов металлов MCn-CsHsh (где M=V, Cr, Мп, Col, чрезвычайно чувствительных к влаге и кислороду воздуха.
кипятят с обратным холодильником над одним из осушителей, указанных в табл. 45. Затем двухходовой кран 5 с тефлоновым штуцером между двухлитровой колбой 7 и приемником 11 перекрывают и растворитель в слабом токе инертного газа (N2) перегоняют через отвод 4 и собирают в приемнике 11. Для освобождения от растворенного кислорода этот процесс перегонки в атмосфере азота многократно повторяют. Когда желаемая степень осушки достигнута, двухходовой кран 5 надлежащим образом перекрывают и растворитель через отвод 9 в противотоке азота сливают в приемник, предварительно заполненный азотом. К растворителю, подвергаемому абсолютированию над Na, К или сплавом Na/K, целесообразно добавить на кончике шпателя неболь-
1916 Глава 33. Металлоорганические комплексы
шое количество бензофенона, который в отсутствие следов кислорода или воды при действии щелочных металлов образует темно-сииий кетил-радикал Шленка и тем самым указывает на окончание процесса абсолютирования и полное отсутствие кислорода в растворителе. В основе всех методик синтеза лежат приемы работы и приборы, хорошо известные и часто используемые в лабо-
Рис. 459. Аппаратура циркулирующего действия для абсолютирования и обезгаживания растворителей.
/ — одноходовой кран (4 мм); 2 — нормальный шлнф 14,5/23; 3 — металлический змеевиковый холодильник; 4 — трубка, по которой поднимаются пары растворителя; 5 — двухходовой тефлоновый кран (4 мм); 6 — одноходовой край (4 мм); 7 — 2-лнтро-вая колба; 8 — нормальный шлиф 29/32 ; 9 — сливной капилляр (1,0 мм); 10 — нормальный шлнф 29/32; 11 — емкость для сбора растворителя в процессе кипения.
раториях органической химии* (например, двухлитровая трехгорлая колба с обратным холодильником Димрота, капельной воронкой и мешалкой; перегонка при нормальном давлении и вакуумная перегонка; кристаллизация, экстракция в аппарате Сокслета), и поэтому мы не будем их подробно описывать. Вся обычная лабораторная посуда должна быть приспособлена для работы в атмосфере инертного газа (одно- и двухходовые краны с оливами, ртутные затворы).
Высушивание исходных соединений или продуктов реакции проводят по необходимости в вакууме масляного насоса или в высоком вакууме.
Точки плавления соединений определяют в заплавленных капиллярах (скорость плавления около 2—3 °С/мин).
* Ср., например, Gatterman-Wieland. Die Praxis des organischen Chemikers, Teil I (Allgemeine Arbeitsanweisungen), 42. Auflage, de Gruyter, Berlin New York 1972.
Общая техника работы 1917
В большинстве случаев в качестве адсорбента для колоночной хроматографии применяют кизельгель 60 (размер частиц 0,063—0,200 мм)*. Все продажные носители для хроматографии перед использованием в течение нескольких часов обезгаживают в высоком вакууме и продувают азотом, этот процесс несколько раз повторяют. При обезгаживании при комнатной температуре активность носителя II—III степени. Более эффективное разделение неполярными элюентами (активность носителя I степени) достигается, если
Зв мм
Рис. 460. Грушевидный сублиматор.
а —собственно сосуд Для возгонки; б — палец сублиматора, / — керн 14,5/23; 2 —керн 14,5/23; 3 — одноходовой кран (6 мм) с оливой для надевания шланга; 4 — муфта, нормальный шлиф 45/40; 5 — керн, нормальный шлнф 45/40.
обезгаживать сорбент ~10 ч при 150 °C. Если нет специального примечания, то колоночную хроматографию проводят в колонках с рубашкой при охлаждении водой и небольшом избыточном давлении азота. Элюат далее упаривают в вакууме водоструйного насоса.
Сублимацию всегда проводят только в аппаратуре, специально приспособленной для работы в отсутствие воздуха.
Грушевидный сублиматор (с круглодоиной колбой, рис. 460) преимущественно используют для легко возгоняющихся соединений (до —100°C), величина загрузки порядка 10 г. В таком сублиматоре можно возгонять, например, следующие соединения, синтез которых описан в данной книге: Mn(n-C5H5)(CO)3, M(n-C5H5)H(CO)3 [M=Cr, Mo, W], МпХ(СО)5 [Х=С1, Вг, I], Fe(o-C3F7)I(CO)4 или Mn(n-C4H4N) (СО)з-
Если сырой продукт слишком мелкокристаллический, его следует покрыть стеклянной ватой, так как иначе сублимат в процессе возгонки будет загрязняться и потребуется повторная возгонка. Очень часто, особенно при очистке циклопентадиенильных комплексов, на кончике пальца сублиматора в самом начале возгонки осаждаются маслянистые вещества (в большинстве случаев это в основном дициклопентадиен) и возогнавшийся затем продукт может упасть снова в сырую массу; во избежание этого до начала возгонки основного продукта необходимо удалить образующийся маслянистый налет кусочком ваты.
* Фирма-поставщик Е. Merck (6100 Darmstadt 1); Artikel — Nr. 7734.
1918 Глава 33. Металлоорганические комплексы
Целесообразно так вести возгонку, чтобы хорошо высушенный продукт реакции нагревался в высоком вакууме до требуемой температуры возгонки по возможности медленнее, что позволит количественно удалить легколетучие маслянистые загрязнения.
Обычно приводимые в тексте температуры возгонки относятся к температурам иагрева бань (баии с силиконовым маслом) и, если речь не идет о верхней границе иагрева, являются приблизительными, поскольку очень сильно зависят от природы сырого продукта.
Так, например, если чистый Mn^-CsHs) (СО)3 непрерывно возгоняется уже при температуре ~35°С, то сильно загрязненные препараты, в особенности маслообразные или покрытые коркой очень твердые образцы, начинают возгоняться лишь при 70 °C или даже выше собственной точки плавления Mn^-CsHj) (СО)3 (77°С) за время от 3 до 30 ч.
Довольно часто предварительное высушивание сырых продуктов в вакууме при высокой температуре ведет к их цементированию, а это в свою очередь приводит к трудностям при извлечении их из реакционных колб. Кроме того, продукт реакции возгоняется лишь при температуре, более высокой, чем обычно. В таких случаях рекомендуется перед возгонкой механически раз:-мельчить сырой продукт (в атмосфере азота, если потребуется). Гораздо про-ще в процессе высушивания время от времени проверить шпателем консистенцию сырого продукта, затем перенести его в сублимационный аппарат еще в слегка влажном состоянии и уже после этого избавиться от остатков влаги. При больших загрузках (скажем, в колбах на 2 л) может происходить настолько сильное затвердевание [например, как при получении V(t]-CsH5)2, Cr(r]-C5H5)2, Мп(С5Н5)2, Со(т]-С5Н5)2], что существует большая опасность пробить стенку колбы при извлечении продукта и тем самым «испортить» вещества, крайне чувствительные к кислороду воздуха. При нагревании аппаратуры для возгонки обязательно надо применять термостатированные масляные бани (магнитная мешалка с контактным приводом). Электроплитки непригодны для этого, так как не позволяют с достаточной степенью точности контролировать температуру нагрева*, что может привести к нежелательному перегреву.
Извлечение сублимата (рис. 460, б) в случае чувствительных к кислороду воздуха соединений [например, Сг(т]-С5Н5)Н(СО)з] осуществляют через специально предусмотренный крестообразный шлиф. Для этого с одной стороны (например, 1) присоединяют предварительно заполненную азотом трубку Шленка, затем уже охлажденный прибор поворачивают на 90°, «палец» с сублиматом в атмосфере азота слегка вытягивают из муфты и сверху (2) плоским шпателем счищают возогнанное вещество с поверхности «пальца» вниз в приготовленную для этого трубку Шленка. Если количество применяемого для возгонки сырого продукта достаточно велико, весь процесс необходимо повте-рить несколько раз. О конце возгонки свидетельствует отсутствие сублимата при превышении заданного интервала температуры возгонки. Необходимо иметь в виду, что во многих случаях остатки после возгонки приведенных в данной главе соединений переходных металлов пирофорны. Уничтожать их следует осторожно, в вытяжном шкафу, аккуратно смывая водой в атмосфере азота. При больших загрузках, естественно, следует быть особенно осторожным (и, в частности, например, когда имеют дело с остатками после возгонки сандвичевых комплексов М(т]-С5Н5)2, где M=V, Сг, Мп, Со).
То же относится и к работам с удлиненной аппаратурой для возгонки (рис. 461), которую используют преимущественно при больших загрузках
* Недавно появившиеся в продаже регулируемые нагревательные приборы «Herastat-Heizhauben» (фирма-изготовитель Heraeus-Wittmann) также не подходят для нагрева аппаратуры для возгонки. При температурах свыше 200 °C применяют графитовые бани [например, при получении Mg(C5H5)2].
Общая техника работы 1919
сырого продукта (10—40 г), когда температура возгонки порядка 100°C. Эти приборы особенно пригодны для возгонки бисциклопентадиенильных комплексов Mfn-CsHsh (где M=V, Сг, Мп, Со). В большинстве случаев даже ие требуется использовать предусмотренный в приборе холодильник, так как из-за довольно высоких температур возгонки соединений последние начинают
Рис. 461. Аппаратура удлиненной конструкции для возгонки.
1 — одноходовой кран (6 мм); 2 — нормальный шлиф 45/50; 3 — нормальный шлиф 29/32 ; 4 — рубашка холодильника для охлаждения водой; 5 — граница поверхности масляной бани.
Рис. 462. Аппаратура удлиненной конструкции с погружаемой лампой для фотолиза (стекло «дюран 50», толщина стенок 2 мм, все удлиненные части расположены концентрически одна в другой).
1 — место для ртутной лампы; 2 — ввод средства для внутреннего охлаждения; 3 — нормальный шлиф 45/50; 4 — шлифованный кран 14,5 (4 мм);
5 — якорь магнитной мешалки (32 мм); 6 — внешнее охлаждение; 7 — пористая стеклянная пластинка G2; 3 — муфта, нормальный шлиф 14,5/23. служит для заполнения и для присоединения газометра, ртутного затвора н масляного счетчика пузырьков; 9 — трубка для подвода газа в реакционный раствор, снабженная винтовым краном 10.
кристаллизоваться на холодных стенках прибора уже на небольшом расстоянии от уровня масла в бане (5, рис. 461). Сублимат, как правило, очень прочно удерживается на стекле. В таких случаях помогает быстрое охлаждение зоны с сублиматом сухим льдом или (в особых случаях) кратковременное погружение в жидкий азот. Продукт затем счищают со стенок прочным стальным шпателем с загнутым концом в трубку Шленка, присоединяемую к аппа
1920 Глава 33. Металлоорганические комплексы
рату для возгонки через муфту 3 (НШ 29/32). Если при удалении сублимата требуется инертная атмосфера (N2), то аппарат для возгонки, естественно, следует расположить горизонтально.
Препаративные фотореакции с карбонилами металлов и их производными в зависимости от величин загрузок, интенсивности света, а также от условий эксперимента (например, термостабильности продуктов реакции, необходимости непрерывно подводить газ и т. п.) проводят в специальной аппаратуре, так называемом фотореакторе с погруженной лампой или реакторе типа Falling Film (т. е. фотореакторе с подвижным слоем)*. На рис. 462 представлена схема погружаемой лампы — весьма удобной и широко используемой в практике аппаратуры, рассчитанной на ртутную лампу высокого давления средней мощности** и имеющей следующие характеристики: 1) полезный объем составляет примерно 230 мл; 2) возможно как внешнее, так л внутреннее охлаждение до —80°C в течение длительного времени (2—3 суток); для более низких температур рекомендуется заменить муфту 3 с нормальным шлифом 45/50 на переходник подходящего размера с уплотняющей прокладкой; 3) фотоли-зуемый раствор интенсивно перемешивается посредством 32-мм якоря магнитной мешалки 5; 4) обеспечивается постоянное продувание реакционной смеси инертным или реакционным газом. Впаянная на нижнем конце ввода пористая стеклянная пластина 7 обеспечивает равномерный ввод газа. В качестве впускного вентиля 10 применяют игольчатый вентиль с тефлоновым уплотнением.
В так называемых фотореакторах типа Falling Film раствор, подвергаемый фотолизу, постоянно перекачивается из буферного сосуда в собственно реакционный сосуд с помощью тефлоновой турбинки. Основное достоинство данного способа заключается в том, что очень тонкий слой раствора, окружающий насадку, в которую вставляют лампу, непрерывно сменяется. Это особенно важно в тех случаях, когда в процессе фотолиза выделяются твердые частицы продуктов разложения. В обычных фотореакторах с погруженной лампой твердые частицы оседают иа насадке и значительно снижают эффективность фотолиза. Особенно надежной аппаратурой, пригодной, в частности, для проведения фотопревращений металлоорганических соединений в больших объемах, является представленный на рис. 463 фотореактор А 9356 фирмы NORMAG по Майеру. В качестве источника света используется мощная ртутная лампа (0,7 кВт, TQ 718, Original Hanau или 0,45 кВт Hanovia).
Примеры использования указанных фотореакторов см. ниже [см. получение V2(n-C5H5) (СО)5, Mn(n-C5HS) (СО)2[С(С6Н5)2], Cr(CO)2(CS2)h-C6H3(CH3)3], Cr(T]-C6Hs) (СО) (NO) (CS)]. Еще два примера фотоаппаратуры приведены ниже при описании методик получения [Mn(n-CSHS) (CO)2(n2-C8Hi4)1 и [VH(PF3)6J.
Тем не менее подчеркнем, что фотопревращения, в которых желаемый продукт быстро выпадает из раствора или выкристаллизовывается, не следует проводить в описанных здесь приборах [см., например, получение Fe2(CO)g]. В таких случаях рекомендуется проводить фотолиз в обычных трубках Шленка на солнечном свету. Этот очень простой и удобный метод позволяет получать чистые кристаллические продукты.
Синтезы многих карбонилов металлов и их производных требуют применения высокого давления (вплоть до 600 бар). Различают три типа автокла-вов***:
а) Вращающиеся автоклавы. Автоклав лежит горизонтально на ведущих роликах в закрепленном кожухе-печке и в процессе реакции с помощью мотора приводится во вращение вокруг собственной оси. В центре днища авто-
* Фирма-изготовитель О. Fritz (NORMAG), (6238 Hofheim/Taunus).
** Philips HPK 125 W или Original Напои Quarzlampen-GmbH TQ 150.
*** Фирма-изготовитель, например, Ernst Haage (Miilheim/Ruhr).
Общая техника работы 1921
2
Рис. 463. Аппаратура для фотолиза в падающем слое (типа Falling Film). / — трубка для подвешивания УФ-лампы (TQ 718 Hanau на 500, 600 илн 700 Вт нли лампа высокого давления Hanovla на 450 Вт); 2— стеклянная трубка (4 мм), укрепленная прочно на стенке трубки 2 н служащая для ввода слабого тока Аг н постоянного выдувания Оа, тем самым предохраняющая металлические части лампы от коррозии; 3 — холодильник с двойной рубашкой; 4 — вакуумная рубашка; 5 — место, где в потоке жидкости протекает фотохимическая реакция; 6 — рубашка холодильника (вода или раствор-охлаждающей солн); 7 — внешняя вакуумная рубашка; 8 — трубка, отводящая реакционный раствор; 9 — капиллярный кран для отбора проб; 10 — стеклянный игольчатый вентиль для регулирования циркуляции реакционного раствора; 11— буферный, промежуточный сосуд для реакционного раствора (например, на 1000 мл); /2 —спускной кран из тефлона; 13 — вращающаяся тефлоновая турбинка с вмонтированным магнитом, служащая для перекачивания жидкости н приводимая во вращение от внешнего вращающегося магнита; 14, 15 — стеклянный игольчатый вентиль; 16 — ввод газа; 17 — муфта (нормальный шлиф 29/32), служащая для присоединения газовой бюретки или ртутного затвора (для сброса избытка газа).
клава сделано углубление ~2 см, в которое вставляют кончик термопары. Термопара закреплена прочно, как и кожух, т. е. не вращается вместе с автоклавом, а свободно проворачивается в отверстии в днище автоклава. Измеряемая температура носит название «температуры рубашки», т. е. является внешней температурой.
5—1361
.1922 Глава 33. Металлоорганические комплексы
б) Качающиеся автоклавы. Автоклав прочно закрепляется в подвижном .нагревательном кожухе, который с помощью эксцентрично расположенного .мотора встряхивается во время реакции. Реакционная масса в автоклаве таким способом равномерно перемешивается. Измерение температуры произво-.дится термопарой, конец которой вставлен внутрь автоклава в специально приспособленную для этого трубку. Регистрируется таким образом не температура рубашки, а внутренняя температура в автоклаве. На это отличие следует обращать внимание, особенно в случае твердофазных реакций, так как в зависимости от толщины стенок автоклава существует определенный температурный градиент вплоть до 80 °C, который следует учитывать при определении периода нагревания реакционной смеси. В некоторых реакциях, таких, как, например, синтез Мп^-СзНз) (СО)3 из Mn(C5Hs)2, быстрый нагрев качающегося автоклава до конечной температуры реакции, как правило, приводит к разложению реакционной массы, поскольку исходное соединение начинает слишком быстро и экзотермично взаимодействовать с СО. Когда твердофазную реакцию во вращающемся автоклаве с внешним измерением температуры по техническим причинам необходимо провести в качающемся автоклаве с внутренним контролем температуры, следует учесть, что при конечной температуре 100 °C период нагревания должен быть увеличен по меньшей мере на 5 ч, а для температуры 200 °C — не менее чем на 8 ч.
в) Автоклавы с мешалками. Для хорошего перемешивания больших •объемов жидкости (свыше 1 л) используют обычно применяемые мешалки (в большинстве случаев магнитные, с постоянным магнитом), которыми снаб-жены, как правило, все вертикально закрепленные автоклавы на 1 или 2 л. Мешалки с помощью клинообразного ремня соединены с регулируемым мотором. Втулку мешалки необходимо охлаждать водой, особенно когда реакции проводят при повышенных температурах.
Автоклавы емкостью свыше 250 мл по соображениям безопасности долж-.ны быть снабжены предохранительными клапанами, которые в случае превы-.шения максимально допустимого давления открываются и обеспечивают сброс избыточного давления. Клапаны закрываются, когда будет снова достигнуто предельно допустимое давление. Небольшие автоклавы (100—250 мл) снабжают особыми разрывающимися шайбами, которые лопаются в случае превышения предельного давления, и обеспечивают тем самым сброс всего газа из автоклава. Разрывающиеся предохранительные шайбы необходимо заменять по крайней мере после каждого четвертого эксперимента. Для проведения автоклавных реакций, в которых используется высокое давление СО, рекомендуется применять автоклавы из хромоникелемолибденовой стали исключительно с медной внутренней поверхностью.
Насколько это возможно, кроме методик, требующих применения высокого давления, приведены также варианты с использованием обычного давле->ния. Это облегчит работу в тех случаях, когда нет в наличии специально оборудованной автоклавной лаборатории. Когда приведены «другие методы получения», чаще всего речь идет о методах, требующих больших затрат, или вариантах, идущих с малым выходом, недостаточно хорошо отработанных.
Следует еще раз указать на высокую токсичность карбонилов металлов или их производных. Все эксперименты необходимо проводить только в хорошо действующем вытяжном шкафу!
Названия всех металлоорганических соединений даны в соответствии с современной номенклатурой IUPAC [International Union of Pure and Applied Chemistry, Nomenclature of Inorganic Chemistry, Second Edition, Butterworth, London, 1970], причем применена повсеместно теперь используемая гапто-(т])-номенклатура [см.: Cotton F. A., J. Amer. Chem. Soc., 90, 6230 (1968)]. Если циклический лиганд координирован всеми своими центрами с атомом металла, цифровой индекс не указывают, например: бис(т)-циклопентадиенил)железо вместо бис (т]5-циклопентадиенил)железо. Названия-синонимы приведены толь-жо в тех случаях, когда речь идет об общепризнанных тривиальных или сокра-
Общая техника работы 192&-
щениых названиях. Стехиометрия уравнений реакций указана только тогда,, когда приведены побочные продукты, либо доказаны в данном конкретном случае, либо подразумеваются по аналогии.
Материал, изложенный в данной главе, есть плод усилий многочисленных, научных коллективов, предоставивших нам частично еще не опубликованные материалы. Особую благодарность мы выражаем д-ру X. Г. Альту (Н. G. Alt),, профессорам X. Бруииеру (Н. Brunner), В. Беку (W. Beck), X. Беренсу (Н. Behrens), Ф, Боиати (F. Bonati) и Ф. Кальдераццо (F. Calderazzo), д-ру Н. Г. Коинели (N. G. Connelly), профессорам П. Дикснё (Р. Dixneuf),. Дж. Эллермаиу (J. ЕПегшапп), Э. О. Фишеру (Е. О. Fischer), К- Флориани (С. Floriani), П. Хаймбаху (Р. Heimbach), М. Херберхольду (М. Herberhold), и X. Ф. Хербериху (Н. F. Herberich), д-ру Б. Ф. I. Джонсону (В. F. G. Johnson), д-ру К- Йонасу (К. Jonas), профессору X. Ф. Кляйну (Н. F. Klein), Д-ру Ф. Р. Крайслю (F. R. Kreissl), профессорам Т. Круку (Th. Kruck), Э. Линднеру (Е. Lindner) и П. Мэйтлису (Р. Maitlis), М. Мушиолю (М. Muschiol), . д-ру И. Планку (J. Plank), Б. Райтеру (В. Reiter), профессору Б. X. Робинсону (В. Н. Robinson), д-ру С. Д. Робинсону (S.D. Robinson), профессору Д. Сел-лману (D. Seltmann), д-ру Дж. Спенсеру (J. Spencer), профессорам О. Штель-церу (О. Stelzer), В. Штромайеру (W. Strohmeier) и Р. С. Тобиасу (R. S. Tobias), д-ру И. Вахтеру (J. Wachter), профессорам X. Вернеру (Н. Werner),. Г. Вильке (G. Wilke) и Г. Уилкинсону (G. Wilkinson).
Исходные соединения
□ Натриевый порошок
Обычный продажный натрий (палочки, кусочки) из-за малой поверхности реагирует в большинстве случаев достаточно медленно. Натриевая же пыль, (порошок) существенно более реакционноспособна. В двухлитровую толстостенную высокогорлую колбу, снабженную краном для ввода азота, помещают 45—50 г натрия в виде палочек или кусочков, заливают 1 л сухого ксилола и закрывают часовым стеклом. Содержимое колбы нагревают на плитке*. Когда кусочки натрия начнут распадаться от легкого на них нажатия стеклянной ’ палочкой (~98°С), нагревание выключают. Температура внутри колбы в этих условиях поднимается еще примерно до 105 °C. При этой температуре вставляют в колбу штангу ультрамешалки** таким образом, чтобы затягивающие отверстия находились приблизительно на 2 см ниже поверхности ксилола в; колбе, одновременно открывают кран ввода азота. При постоянном продувании азота начинают механическое размельчение*** расплавленного натрия (длительность включения мешалки 15±3 с). Образовавшуюся суспензию натрия серо-желтого цвета тотчас фильтруют еще в горячем состоянии через большой стеклянный пористый фильтр (G4). При этом следят, чтобы натрий не отсасывался досуха, иначе произойдет его сплавление. Когда последнюю > порцию натриевого порошка поместят на фильтр, его промывают небольшим количеством сухого холодного ксилола, затем промывают петролейным эфиром (fK„n 40—60°C), собирая фильтрат в другой приемник, и, наконец, промывают полученный порошок натрия петролейным эфиром многократно порциями по 50—100 мл, осторожно перемешивая оплавленной стеклянной палоч--
* Колбонагреватель Pilz-Heizmantel.
** Фирма-изготовитель IKA-Werk (7813 Staufen).
*** Внимание: критическим моментом получения натриевого порошка' является включение ультрамешалки. Если ксилола взято слишком много, в первые секунды может произойти выбрасывание верхнего слоя ксилола из-; колбы. Наготове держать огнетушитель! Работать в перчатках!
5‘
4924 Глава 33. Металлоорганические комплексы
кой. После этого высушивают в вакууме масляного насоса в течение нескольких часов. Первоначально отделенный ксилольиый фильтрат можно использовать повторно.
ОС Циклопентадиенил натрий NaC5H5
Циклопентадиеиилнатрий чаще всего используется в синтезах циклопен-тадиеиильных комплексов металлов. Его получают из такой слабой кислоты, как циклопентадиеи (рКа=15,5), обработкой либо Na (способ 1), либо NaH (способ 2). Оба способа в равной степени позволяют получить достаточно чистый продукт.
Способ 1
С6Н„ + Na ----> NaC5H5 + 1/2Н,
66,1 23,0 88,1
В двухлитровую трехгорлую колбу, снабженную обратным холодильником и капельной воронкой, помещают 23,0 г (1,0 моль) порошкообразного натрия в 500 мл тетрагидрофурана. При охлаждении льдом прикапывают 95 мл (76,4 г; 1,16 моль) свежеперегиаиного циклопентадиена* с такой скоростью, чтобы выделение газа было не слишком сильным. Для полного растворения Na требуется 3—4 ч. Когда раствор начнет окрашиваться в бледно-розовый цвет, охлаждение снимают. Реакция считается законченной, если раствор прозрачен и не осталось больше порошка Na. Растворитель удаляют в вакууме водоструйного иасоса при 30—40 °C, а остаток высушивают в высоком вакууме. Должен получиться сухой порошок. Отгонка растворителя при нормальном давлении нежелательна, так как продукт при этом образуется затвердевшим и трудно извлекается из колбы! Выход практически количественный.
Точно таким же образом можно получить KCsHs из К и CsH6 в бензоле [2].
Способ 2
C5He + NaH ----> NaC5Hs + Н2
66,1 24,0 88,1
К охлаждаемой льдом суспензии 24,0 г (1,0 моль) NaH в 500 мл тетра-тидрофурана (ТГФ) при интенсивном перемешивании магнитной мешалкой прикапывают 86 мл (69,1 г; 1,05 моль) свежеперегнанного циклопентадиена* в течение ~75 мин. Наблюдается интенсивное выделение газа, и смесь вскоре приобретает розовый оттенок; в конце реакции цвет углубляется до краснофиолетового (см. свойства). Растворитель при 30—40°C удаляют в вакууме водоструйного насоса и остаток высушивают в высоком вакууме. Выход количественный. Для реакций NaCsHs с соединениями металлов во многих случаях
* Мономерный циклопентадиен получают термическим расщеплением продажного дициклопентадиена. Для этого последний (/кип 170 °C) прикапывают в горячий декалин («140 °C) и непрерывно отгоняют мономерный продукт расщепления (1Кпп 34—41 °C), используя длинный (60 см) дефлегматор, обмотанный асбестовым шнуром, в охлаждаемый льдом приемник с CaClj. Обычно С5Н6 тотчас используют в синтезах, однако его можно хранить без заметной димеризации в течение многих недель при температуре сухого льда. Аналогичное верно и для метилциклопентадиена, дающего точно по такой же методике NaC5H4CH3.
Исходные соединения 1925
можно использовать непосредственно тетрагидрофурановый раствор, так что выделение чистого продукта оказывается излишним.
Другие способы. Взаимодействие Na (соответственно Li, К, Rb, Cs, Са, Sr) с циклопентадиеном в жидком аммиаке [1, 3]. Таким же способом можно получить LiCsHs, NaCsHs, КС5Н5, RbCsHs, CsC5H6, Ca(CsHs)2 или Sr(C5H5)2.
Свойства. Вещество белого цвета, очень чувствительное к влаге и кислороду воздуха; растворы в тетрагидрофуране в отсутствие любых загрязнений, в особенности следов кислорода и воды, бесцветны. /разл >300 °C. Реагирует с водой: NaCsHs+TUO-^NaOH+CsHs. Практически бесцветные растворы NaC5Hs могут быть получены лишь при тщательном исключении малейших следов кислорода. Для этого требуется также обескислороживать используемый циклопентадиен. Этого можно достигнуть прямой перегонкой свежепере-гианного циклопеитадиена в суспензию Na — ТГФ, соответственно NaH — ТГФ, в атмосфере азота. Во многих случаях это сложно осуществить. Обычно большие количества мономерного С5Н6 хранят над СаС12 в атмосфере азота в сосуде Дьюара с сухим льдом. Непосредственно перед употреблением необходимое количество циклопентадиеиа перекондеисируют в вакууме масляного иасоса или высоком вакууме в другую колбу, охлажденную жидким азотом и заполненную N2. Полученный таким образом дистиллат при небольшом избыточном давлении N2 оттаивает, и его тотчас же помещают в градуированную капельную воронку прибора для синтеза NaCsHs. Следует заметить, что для большинства циклопентадиенильиых комплексов металлов, описываемых в данной главе, использование розового или розово-фиолетового растворов NaCsHs в ТГФ никоим образом ие влияет на выход получаемых продуктов.
Если необходимо получить абсолютно сухой белый NaCsHs, то требуется (особенно для больших количеств, 100—300 г) многодневное откачивание остатков растворителя в высоком вакууме при 140 °C.
ЛИТЕРАТУРА
1. Ziegler К., Froitzheimer-KUhlhorn Н„ Hafner К., Вег., 89, 434 (1956).
2. Thiele К., Вег., 34, 68 (1901).
3. Fiescher Е. О., Hafner W., Stahl Н. О., Z. Anorg. Allgem. Chem., 282, 47 (1955); Fraenkel G., Carter R. E., McLachlan A., Richards J. H., J. Amer. Chem. Soc. 82, 5846 (1960).
Циклопентадиенилталлий TIC5H5
T12SO4 + 2C5H6 + 2KOH --> 2T1C5H5 + 2H2O + K2SO4
504,8 2-66,1 2-56,1 2-269,5 2-18,0 174,3
Растворяют 48,0 г (95 ммоль) T12SO4 и 20,2 г (0,36 моль) КОН в 400 мл Н2О (для ускорения подогревают), при комнатной температуре добавляют 13,2 г (0,2 моль) циклопентадиена и перемешивают смесь 10 мин. Выпавший осадок желтого цвета отделяют, промывают водой и минимальным количеством охлажденного СН3ОН и высушивают в высоком вакууме. Выход 46,5 г (91% на взятый T12SO4). Полученный продукт достаточно чистый для дальнейшего использования его в металлоорганических синтезах. Аиалитическн чистый препарат получают возгонкой в высоком вакууме при 80—100 °C. Циклопентадиенилталлий получается при этом в виде красивых желтых иголочек длиной до 1 см. Выход 30,7—34,8 г (60—68%).
Аналогично, исходя из метилциклопеитадиена, может быть получен метил-циклопентадиенилталлий [3]. Алкильные производные T1(ti-C5H4R) чувствительны к кислороду воздуха [4].
1926 Глава 33. Металлоорганические комплексы
Свойства. Светло-желтое кристаллическое вещество, устойчивое к действию влаги и кислорода воздуха, /ра3л >230 °C. Растворяется в тетрагидрофуране, хуже растворяется в метаноле, не растворяется в большинстве менее полярных органических растворителей. Структура: в кристалле зигзагообразные цепочки из атомов Т1 и фрагментов С5Н5 [5]. В парах имеет структуру полусандвича, симметрия [6].
ЛИТЕРАТУРА
1. Meister Н., Angew. Chem., 69, 533 (1957).
2. Chemische Werke Huis AG (Erf. H. Meister), D. P. 942989 (9 Mai 1956);
C. A. 52, 16249 (1958).
3. Reynolds L. T., Wilkinson G., J. Inorg. Nucl. Chem., 9, 86 (1959).
4. Katz T. L, Mrowca J. J., J. Amer. Chem. Soc., 89, 1105 (1967).
5. Frasson E„ Menegus F., Panattoni C., Nature, 199, 1087 (1963).
6. Tyler J. К, Cox A. P„ Sheridan J., Nature, 183, 1182 (1959).
□ □ Дициклопентадиенилмагний Mg(C5H5)2
Для получения Mg(CsH5)2 можно пользоваться двумя способами. Цикло-пентадиенилмагнийбромид, получаемый реакцией переметаллирования из MgBr(C2H5) и циклопентадиена, при нагревании в высоком вакууме до 210— 230 °C превращается в Mg(C5H5)2, который в этих условиях непрерывно возгоняется (способ 1). Особой простотой отличается прямое взаимодействие Mg и С5Н6 в газовой фазе (способ 2).
Способ 1
Mg + C2H5Br ----> MgBr(C2H6)
24,3 109,0 133,3
MgBr(C2H5) + С6Н6 ---> MgBr(C6H6) + С2Н6
133,3 66,1 169,3
дт
2MgBr(C5H5) ----> Mg(C6H6)2 + MgBr2
2-169,3 154,5 184,1
В двугорлую колбу на 1 л с обратным холодильником и капельной воронкой помещают 14,6 г (0,60 моль) Mg-стружки и 500 мл эфира. При интенсивном перемешивании прикапывают 48,1 мл (70,8 г; 0,65 моль) С2Н5Вг; для начала реакции в большинстве случаев требуются умеренное подогревание и добавка нескольких кристалликов иода на кончике шпателя. К получившемуся темно-серому раствору MgBr(C2H5) прикапывают 57,5 мл (46,3 г; 0,70 моль) свежеперегнанного циклопентадиена. Смесь нагревают с обратным холодильником 15 ч. После упаривания раствора в вакууме водоструйного насоса остаток (серого цвета) сушат в высоком вакууме. Затем продукт переносят в аппаратуру для возгонки, где при температуре 210—230 °C в высоком вакууме происходит термическое превращение MgBr(CsH5) в Mg(C5H5)2. Образующийся Mg(CsH5)2 тотчас же возгоняется и собирается в виде бесцветных кристаллов на конце пальца сублиматора, охлаждаемого водой. Твердый остаток за время возгонки (не менее 15 ч) становится настолько плотным, что требуется нагревание до —250 °C, чтобы использовать последние остатки MgBr(C5H5) для перевода их в дициклопентадиенилмагний. Выход 14,4—18,1 г (31—39%).
Способ 2
Mg + 2С5Н6 -----> Mg(C6H5)2 + H2
24,3 2-66,1 154,5
Исходные соединения 1927
Через трубку для сожжения (диаметром 3 см, длиной 60 см) при тщательном предохранении от малейших следов воздуха и воды пропускают ток Не и свежеперегнанного циклопентадиена над порошком магния, нагретого до 500—600 °C. Реакция начинается сразу и протекает в дальнейшем спокойно, пока весь циклопентадиен не прореагирует. Образующийся продукт оседает за зоной нагрева на холодных стенках трубки в виде бесцветных пушистых кристаллов. Время реакции около 4—5 ч. Выход практически количественный.
Свойства. Бесцветные кристаллы, при доступе воздуха тотчас происходит разложение, окраска становится коричневой; /пл 176—178 °C. В расплавленном состоянии не разлагается вплоть до 300 °C и выше. Растворим в бензоле и эфире. С водой реагирует энергично, образуя Mg(OH)2 и С5Н6.
ЛИТЕРАТУРА
1. Wilkinson G., Cotton F. A., Birmingham J. M., J. Inorg. Nucl. Chem., 2, 95 (1956).
2. Fraenkel G., Carter R. E., McLachlan A., Richards J. H„ J. Amer. Chem. Soc., 82, 5846 (1960).
3. Barber W. A., J. Inorg. Nucl. Chem., 4, 343 (1957).
4. Barber W. A., Inorg. Synth., 6, 11 (1960).
T]Циклопентадиенил (тр и-н-бутил) олово
Sn^^QHs) (H-CiHgJs
Sn^'-CsHs) (н-С4Н9)з — ценный, очень мягкий и доступный в больших количествах циклопентадиенилирующий реагент, широко используемый в химии металлоорганических комплексов. Он очень просто получается при взаимодействии NaC5H5 с продажным хлоридом три-н-бутилолова:
NaC5H6 + SnCl(H-C4H9)3 --> Snfr^-Q^) (н-С4Н9)3 + NaCl
88,1 325,5 355,1 58,4
88,1 г (1,0 моль) порошкообразного NaCsH5 (см. выше) суспендируют примерно в 1,4 л абсолютного, свободного от тиофена бензола и обрабатывают в четыре порции 299,4 г (247,7 мл; 0,92 моль) 8пС1(«-С4Н9)з. Перемешивают слегка разогревающуюся при этом суспензию еще ~2 суток при комнатной температуре и затем дают осесть не вступившему в реакцию NaC5H5 и NaCl. Получившийся светло-желтый раствор осторожно фильтруют через большой стеклянный пористый фильтр G3, покрытый дополнительно слоем стеклянной ваты. В том случае, если промывают коричневую студнеобразную массу на фильтре, нельзя фильтровать под давлением, иначе поры фильтра очень скоро забьются. Гораздо лучше, если реакционную смесь до фильтрования в несколько порций отцентрифугировать*. Остатки на фильтре, соответственно остатки после центрифугирования, многократно промывают бензолом (общий объем до 150 мл). Фильтрат, соответственно центрифугат, упаривают в роторном испарителе (вакуум водоструйного насоса, температура бани 30—50 °C) и остающееся желтое масло подвергают вакуумной перегонке. Зп^'-СдНд) (н-С4Н9)3 перегоняется при 102—104°C (1,5-10~2 мм рт. ст.) в виде бледно-желтого масла. Выход 222—248 г (68—76%). Величины загрузок можно варьировать без существенного изменения выходов.
Свойства. Бледно-желтое своеобразно пахнущее масло, неограниченно долго сохраняется в атмосфере сухого азота. На воздухе происходит быстрое
* Очень удобны лабораторные центрифуги JU II фирмы Heraeus-Christ. При их использовании фильтрование оказывается излишним.
1928 Глава 33. Металлоорганические комплексы
осмоление и изменение цвета. /кип 102—104°C (1,5-10-2 мм рт. ст.); 92— 94°C (4-Ю-3 мм рт. ст.). Спектр ЯМР-’Н (CS2, 24 °C, ТМС): о 5,91 (С5Н5); 7(Н—119Sn) 20,0 Гц; J(H—117Sn) 19,1 Гц.
Аналогичные соединения. Другие алкил (т]‘-циклопентадиенил) станнаны (например, Sn(T]‘-C5H5) (СН3)3, Sn(r]‘-C5H5)2(CH3)2, Sn(T]‘-CsH5)3CH3, Sn(rp-CsHs^H-CiHgb и т. д.) могут быть получены по аналогичной методике при соответствующем изменении молярного соотношения используемых реагентов. Соединения серии SnR^Tp-CsHs^-,,, как правило, легко разделяются перегонкой.
ЛИТЕРАТУРА
1. Fritz Н. Р., Kreiter С. G., J. Organometal. Chem., 1, 323 (1964).
Карбонилы металлов
□ □ Гексакарбонилванадий V(CO)6
V(CO)6 синтезируют лучше всего карбонилированием безводного VC!3 при высоком давлении с применением смеси Mg/Zn в качестве восстановителя и с последующим подкислением первоначально образующегося иона [V(CO)e]_
a) 2VC13 + 12СО + 4Mg------> Mg[V(CO)6]2 + 3MgCl2
2-157,3 12-22,4л 4-24,3 462,2 3-95,2
б) Mg[V(CO)6]2 + 2НС1 --> 2[VH(CO)J + MgCl2
462,2 2-36,5 2-220,0 95,2
в) 2[VH(CO)6] -> 2V(CO)3 + H2
2-220,0 2-219,0 22,4л
Во вращающийся автоклав емкостью 500 мл помещают 9,90 г (63 ммоль) сухого VC13, 220 г безводного и насыщенного азотом пиридина и — 16 г Mg/Zn-порошка (1 :2,7), который предварительно активирован 2 г иода. Доводят давление СО в системе до Ро=135 бар и вращают автоклав 12 ч прн 135 °C. При нагревании давление растет до ~200 бар, в конце реакции давление падает до —160 бар. После охлаждения автоклава и сжигания остаточного СО реакционную смесь смывают абсолютным пиридином в атмосфере Ь12 в колбу для декантации и оставляют на 24 ч. Затем после удаления жидкости остаток отфильтровывают на холоду при 1 мм рт. ст. Коричневый твердый продукт обрабатывают последовательно 200 мл Н2О и 400 мл эфира и при охлаждении льдом и встряхивании подкисляют водно-эфирный раствор 4 н. соляной кислотой, предварительно насыщенной азотом и взятой в количестве не менее 300 мл.
Все последующие операции проводят осторожно, по возможности в темноте и непременно в атмосфере азота. Как можно быстрее отделяют эфирный слой, промывают его разбавленной соляной кислотой, затем водой и сушат —20 ч над MgSO4. При удалении эфира получают сначала красно-коричневую жидкость, еще выделяющую водород. После окончательного удаления растворителя образуется твердое вещество. Его подвергают возгонке при 40—50 °C и давлении 10-3 мм рт. ст. Еще влажный V(CO)6 сушат в атмосфере N2 над Р2О5 и повторно сублимируют. Выход —2 г (—15%).
Свойства. Сине-зеленые, летучие, очень чувствительные к кислороду воздуха парамагнитные кристаллы. В атмосфере N2 разлагаются при 60—70 °C (в запаянном капилляре). В высоком вакууме возгоняется уже при 40°C. Растворяются в инертных органических растворителях, цвет раствора оранже
Карбонилы металлов 1929
во-желтый. С основаниями Льюиса идет оквслительно-восстановительное диспропорционирование. ИК (нуйол): 1976 [v(CO)] см-1; р,= 1,81 магнетона Бора (90—300 К). Кристаллическая структура описана в работах [2, 3].
ЛИТЕРАТУРА
1. Ercoli R., Calderazzo F„ Alberola A., J. Amer. Chem. Soc., 82, 2966 (1960). 2. Calderazzo F., Cini R., Ercoli R., Chem. Ind. (London), 1960, 934.
3. Calderazzo F., Cini R., Corradini P., Ercoli R., Natta G., Chem. Ind. (London), 1960, 500.
□ □ Гексакарбонилванадат(—I) [бис{ди(2-метокси-этиловый)эфир}]натрия [Na(C6Hi4O3)2] [V(CO)6]
Для синтеза гексакарбонилванадатов(—I) пригодна вышеописанная методика карбонилирования при высоком давлении с использованием в качестве восстановителя Na:
Fe(CO)g
VC1S + 4Na + 6 СО + 2 C6H14O3 ------->
157,3 4-23,0 6-22,4л 2-134,2
----> [Na(C6H14O3)3] [V(CO)J + 3 NaCl 510,3 3-58,4
В качающемся автоклаве (емкость 1 л, предельное давление 300 бар), предварительно заполненном аргоном, помещают 31,5 г (0,2 моль) безводного VCI3, 27,5 г (1,2 моль) натриевого порошка, 400 мл абсолютного диглима и 2 мл Fe(CO)s. Дважды продувают автоклав СО (последовательно доводят давление до 20—30 бар и сбрасывают его), доводят давление СО до 200 бар и нагревают в течение 48 ч при 160 °C, интенсивно встряхивая автоклав. За время реакции давление падает иа 45—50 бар. Реакционную смесь (черного цвета) фильтруют через пористый стеклянный фильтр G4 (диаметр 3 см), дополнительно заполненный стекловолокном, остаток на фильтре промывают диглимом дважды порциями по 100 мл. Фильтрат (желтого цвета) встряхивают в делительной воронке с 1 л гексана и отделяют нижний слой, в котором содержится желаемый продукт. Далее к нему добавляют смесь эфира и пентана по 200 мл каждого, и гексакарбонилванадат выпадает в виде желтых кристаллов. Продукт отфильтровывают, несколько раз промывают пентаном порциями по 50—100 мл и сушат в вакууме. Выход 56—60 г (55—59%).
Свойства. Желтые кристаллы, неустойчивые на свету, достаточно стабильны на воздухе, /Пл 173—176 °C (с разл.). Вещество растворяется в воде, умеренно растворяется в полярных растворителях типа тетрагидрофурана, ие растворяется в неполярных растворителях — бензоле, гексане. ИК (диглим): v(CO) 1850 см-L
Аналогичные соединения. По аналогичной методике из NbCls, соответственно TaClg, с использованием в качестве восстановителя сплава Na/K получают гексакарбонилметаллатные комплексы [К(С6Н14О3)3][М)(СО)б1 и [К(С6Н14О3)3][Та(СО)в] [1, 3].
литература
1. Werner R. Р. М., Podall Н. Е., Chem. Ind. (London), 1961, 144.
2. King R. B„ Organometallic Syntheses, v. 1, p. 82, Academic Press, New York, London 1965.
3. Ellis J. E., Davison A., Inorg. Synth., 16, 68 (1976).
1930 Глава 33. Металлоорганические комплексы
□ □ Гексакарбонилхром Сг(СО)6
Способ синтеза Сг(СО)в, приводящий к наибольшему выходу, заключается в восстановительном карбонилировании безводного СгС1з при высоком давлении в присутствии А1(С2Н5)з (способ 1); однако работа с триэтилалюминием требует повышенной осторожности и высокой квалификации. Более удобным в выполнении, но дающим меньшие выходы конечного продукта является способ карбонилирования безводного СгС13 при высоком давлении в присутствии смеси А1+А1С1з (способ 2).
Способ 1 [1]
А1(С2Н5)з
CrCl3 + 6 СО --------► Сг(СО)6
158,4 6-22,4 л 220,1
В трехгорлую колбу на 1 л, снабженную обратным холодильником, капельной воронкой и эффективной мешалкой, медленно прикапывают 270 мл (1,98 моль) А1(С2Н5)з к 200 мл диэтилового эфира. Реакцию ведут в атмосфере N2. Если происходит очень сильное разогревание, колбу обдувают холодным воздухом из фена. Остатки А1(С2Н5)3 в капельной воронке смывают в колбу эфиром (90 мл). Охлажденный раствор также в атмосфере N2 медленно вливают в качающийся автоклав на 1 л, в котором уже помещены 50,0 г (0,32 моль) безводного СгС13*. СгС13 перед использованием сушат —50 ч при 110 °C в высоком вакууме. Автоклав закрывают и продувают СО. Затем доводят давление СО до 150 бар и включают автоклав. Нагрев регулируют таким образом, чтобы температура в автоклаве за 1 ч поднялась до 90 °C; рабочая температура в автоклаве НО—115 °C. В течение последующих 5 ч дважды регулируют давление СО в автоклаве, каждый раз доводя его до — 200 бар. Еще через 15 ч реакция заканчивается. Охлажденное содержимое автоклава в атмосфере азота выливают через капельную воронку в трехгорлую колбу на 2 л. [Осторожно! Возможно самовоспламенение на воздухе!} Выкристаллизовавшийся в автоклаве продукт с помощью шпателя переносят в ту же колбу.
В отверстиях крышки автоклава может кристаллизоваться значительное количество Сг(СО)6. Его лучше всего удалить с помощью деревянного шпателя подходящего размера и также поместить в колбу.
Колбу затем снабжают эффективной мешалкой, обратным холодильником и капельной воронкой. Осторожным добавлением смеси 40 мл СН3ОН и 40 мл бензола, затем 800 мл 15—18%-ной НС1 (сначала со скоростью 1 капля за 10 с) разлагают оставшийся А1(С2Н5)3, сильно перемешивая содержимое колбы (на это уходит —5 ч). Не рекомендуется НС1 вливать непосредственно в раствор, в котором есть остатки А1(С2Н5)3; лучше, если корочка, образовавшаяся на стенке колбы в месте падения капли НС1, постепенно переходит в раствор.
Верхний керн, вставленный в обратный холодильник и соединяющий его с ртутным затвором, плотно не укрепляют, для того чтобы он мог послужить выпускным клапаном и легко приподняться в случае неожиданного повышения давления в колбе.
Автоклав также споласкивают разбавленной (1:1) соляной кислотой (100 мл), которую вливают в реакционную колбу по окончании гидролиза. Затем убирают обратный холодильник, на его место ставят насадку Кляйзена и прямой холодильник с приемником на 1 л, а два других горла колбы закры
* СгС13 используют продажный или получают его по прописи, приведенной в данной книге. Если есть сомнения в чистоте препарата, то перед высушиванием его кипятят с 2 н. НС1 и промывают водой.
Карбонилы металлов 1931
вают пробками. Чтобы избежать потерь, приемную колбу снабжают шариковым холодильником и, кроме того, охлаждают ее льдом. Затем содержимое колбы нагревают до кипения и Сг(СО)6 за 2—3 ч перегоняют с паром в виде бесцветных кристаллов. Получившийся продукт отсасывают на воронке Бюхнера, 2—5 раз промывают небольшим количеством СН3ОН и сушат на воздухе. Выход 60—65 г (86—94%).
Способ 2 [2]
А1+А1С13
СгС13 + 6 СО ---------► Сг(СО)6
158,4 6-22,4 л 220,1
Все операции проводят в атмосфере азота. В трубку Шленка помещают 46,3 г (0,29 моль) безводного СгС13, 46,3 г (1,72 моль) порошка А1, 40,0 г (0,30 моль) абсолютно безводного А1С13, примерно 5 г сплава Деварда и смесь хорошо растирают стеклянным пестиком. Затем переносят ее в стеклянный автоклавный «палец», предварительно также продутый азотом, добавляют для лучшего перемешивания во время реакции 10 стеклянных шариков и помещают «палец» в качающийся или вращающийся автоклав на 1 л. Добавляют еще ~250 мл бензола* и тотчас закрывают. Систему однократно продувают СО, доводя давление до 100 бар; для реакции необходимо давление СО порядка 260 бар. В течение 2 ч доводят температуру до 140 °C и поддерживают ее 50—60 ч. Максимальвое рабочее давление составляет приблизительно 460 бар (при необходимости в процессе реакции регулируют давление СО в системе). По окончании автоклав охлаждают и сжигают избыток СО. После продувания системы азотом автоклав открывают. Получившуюся кашицеобразную массу черного цвета переносят в трехгорлую колбу на 2 л с мешалкой, обратным холодильником и капельной воровкой на 250 мл. При интенсивном перемешивании и охлаждении льдом гидролизуют ледяной водой оставшийся А1С13. Когда при добавлении воды уже не будет заметно никакой реакции, капельную воронку заменяют газовводной трубкой от паровика и всю массу подвергают перегонке с паром (см. ч. 1т. 1). Сначала отгоняют бензол, затем, когда в холодильнике появляется кристаллическая корочка, приемник заменяют. Продукт отделяют на воронке Бюхнера, промывают небольшим количеством СН3ОН и отсасывают досуха. Далее переносят его в грушевидный сублиматор и возгоняют в высоком вакууме при 60—80 °C. Выход —50 г (78%).
После сжигания избытка СО реакционную смесь можно обработать иначе: образовавшуюся массу вытряхивают на круглый или складчатый фильтр. Оставшийся в автоклаве продукт смывают бензолом и помещают его в колбу на 1 л вместе с основной массой, предварительно высушенной на воздухе. Основное количество бензола отгоняют в вакууме водоструйного насоса при нагревании не выше 40°C (температура водяной бани). При этом Сг(СО)6 еще в заметной степени не улетучивается. Затем сырой продукт малыми порциями возгоняют при 10 мм рт. ст. и 70—80 °C на масляной бане. Сублимат загрязнен дифенилом, избавиться от которого можно перекристаллизацией из СС14. Выход колеблется в пределах 70—79%.
Свойства. Бесцветные, легко возгоняющиеся, устойчивые на воздухе кристаллы, /пл 154—155°C. Сг(СО)6 не растворяется в воде, ограниченно растворяется в органических растворителях. Растворы на свету и на воздухе разлагаются. ИК (газовая фаза): 2000,1 [v(CO)], 441 (v(CrC)] см-' ЯМР-
* Применяемый абсолютный бензол не должен содержать тиофена. Абсолютное исключение влаги имеет решающее значение для успешного синтеза Сг(СО)6 при высоком давлении.
1932 Глава 33. Металлоорганические комплексы
13С (CDC13, ТМС): б 212,1 [СО]. Давление пара: Igp (ммрт.ст.) = = 11,475—3622,9/7" в пределах 46,1 и 138,4 °C. Кристаллическая структура: см. [3]; d(Cr—С) = 1,916 А.
ЛИТЕРАТУРА
1. Podall Н. Е., Dunn J. Н., Shapiro И., J. Amer. Chem. Soc., 82, 1325 (1960); Vorschrift des Anorganisch-Chemischen Laboratoriums der Technischen Uni-versitat Mflnchen.
2. Fischer E. O., Hafner W., Ofele K„ Chem. Ber., 92, 3050 (1959).
3. Whitaker A., Jefferey J. W., Acta Crystallogr., 23, 977 (1967).
□ □ Пентакарбонилхромат(—П) натрия Na2[Cr(CO)5]
Восстановление Cr(CO)6 металлическим Na в жидком аммиаке впервые описал Беренс с сотр. [1]. Способ синтеза, приведенный ниже [2], очень похож на метод, модифицированный Эллисом с сотр. [3].
Сг(СО)„ + 2 Na-> Na2[Cr(CO)5] + СО
220,1 2-23,0 238,0 22,4 л
Одногорлую и двугорлую, предварительно градуированную, колбы Шленка емкостью по 500 мл каждая соединяют друг с другом с помощью холодильника, который можно охлаждать ацетоном с сухим льдом (рис. 464).
Ртутный затвор
Рис. 464. Установка для получения Na2[Cr(CO)5], / — реакционная колба; 2 —двугорлая колба с аммиаком; 3 — охлаждающая баня: ацетон/СО>.
Все части аппаратуры предварительно нагревают в высоком вакууме (используя фен). В атмосфере инертного газа (Аг или N2) в одногорлую колбу / добавляют 4,2 г (0,18 моль) Na в виде крупных кусков и 11,0 г (50 ммоль) тонко измельченного Сг(СО)в. В двугорлую колбу 2, которая служит для очистки и высушивания NH3, добавляют 1 г Na, охлаждают до —78 °C ацетоном с сухим льдом (3), конденсируют 350—400 мл NH3 и затем вместо газовводной трубки ставят стеклянную пробку. Во время конденсации холодильник заполняют охлаждающей смесью сухого льда с ацетоном. Охлаждающую баню 3 убирают из-под колбы с аммиаком 2 и помещают под реакционную колбу 7. Для переконденсации аммиака в реакционную колбу при осторожном
Карбонилы металлов 1933
перемешивании* нагревают двугорлую колбу 2 на ацетоновой бане, температуру которой предварительно доводят до —20 °C, и в аппаратуре создают небольшой вакуум. Чтобы температуру бани поддерживать при —20 °C и тем: самым создать непрерывную переконденсацию аммиака, используют регулируемую электроплитку. Затем аммиак в реакционной колбе 1 доводят до кипения (используя ту же ацетоновую баню), непрерывно перемешивая*. При этом змеевик, охлаждаемый ацетоном с сухим льдом, служит в качестве обратного холодильника. Время от времени, а также в моменты переконденсации в системе создают вакуум для избавления от выделяющегося моноксида углерода. Когда цвет аммиачного раствора изменится от синего до оранжево-желтого (3—7 ч), смесь нагревают еще 0,5 ч. После этого охлаждают до —45-ь -5—50 °C и оставляют иа некоторое время без перемешивания, чтобы дать-осесть NaNH2 и побочно образовавшемуся ацетилендиолату натрия (ПазСгОз). За это время заранее охлаждают приемную колбу на 500 мл до —78 °C (ацетон— сухой лед), а также охлаждают до —45ч—50°C стеклянный пористый фильтр (G3, 3X30 см) с рубашкой для охлаждения, предварительно прогрев-их в высоком вакууме, чтобы избавиться от малейших следов влаги.
Холодильник-змеевик снимают с реакционной колбы 1, тщательно следя за тем, чтобы в аммиачный раствор не попадала конденсирующая влага. Через тонкую стальную иглу (диаметром 1,5 мм), изолированную от возможного* нагревания и вставленную в резиновую пробку, при небольшом давлении передавливают на стеклянный пористый фильтр G3 сначала только отстоявшийся прозрачный оранжевый раствор и фильтруют его. Оставшуюся темно-коричневую суспензию (100—200 мл) за один прием сразу переносят на фильтр. Иглу с фильтра удаляют и споласкивают ее, а также реакционную колбу водным спиртом, чтобы не допустить высыхания NasCaOa- Для ускорения дальнейшего фильтрования рекомендуется проводить его при небольшом избыточном давлении (до ~2 бар). Следят также за тем, чтобы осадок иа фильтре не стал сухим (во избежание взрыва и воспламенения!). Сразу после фильтрования приемную колбу удаляют, а фильтр тотчас же заливают водным спиртом. Фильтрат максимально упаривают в вакууме водоструйного насоса. Остатки NH3 удаляют в высоком вакууме при комнатной температуре, для чего требуется несколько часов. Сухой продукт представляет собой очень пушистый светло-желтый порошок, достаточно чистый для его использования в дальнейших синтезах.
Для очистки продукт растворяют в 300—400 мл тетрагидрофурана и фильтруют. Чтобы мелкий осадок не забивал фильтр (G3), последний покрывают слоем (~3 см) промытого и прокаленного песка. Раствор вначале капает слегка мутным, пока на песке не образуется слой осадка. Поэтому первую порцию мутного фильтрата повторно фильтруют через тот же слой. Для ускорения процесса фильтрования применяют небольшое избыточное давление (~2 бар). Прозрачный раствор в зависимости от концентрации имеет цвет от желтого до оранжево-желтого. При удалении растворителя выпадают тонкие светло-желтые иголочки, при высушивании в высоком вакууме образуется желтый порошок. Выход 7,3 г (61%).
Особенно чистый продукт получается, если профильтрованный тетрагнд-рофурановый раствор упаривают до начала кристаллизации Na2[Cr(СО)5] и разбавляют 200 мл абсолютного эфира. Выпавший осадок отфильтровывают, промывают холодным эфиром (2x50 мл) и высушивают в высоком вакууме. Выход 6,6 г (55%).
Свойства. Желтое пирофорное вещество, стабильное в твердом состоянии; в предварительно прокаленной трубке Шленка в отсутствие малейших следов
* Так как тефлоновая оболочка обычно используемого якоря магнитной мешалки реагирует с Na+NH3 (жидк.), то применяют якорь, запаянный в. стеклянную трубку.
1934 Глава 33. Металлоорганические комплекса
воздуха сохраняется свыше месяца. Раствор в тетрагидрофуране также устойчив и может долго храниться. ИК (ТГФ): 1819 (с.), 1768 (с., шир.) [v(CO)J см-1; (нуйол): 1925 (сл.), 1802 (с.), 1770 (с.), 1644 (с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Behrens Н., Weber R., Z. Anorg. Allgem. Chem., 291, 122 (1957); Lindner E., Behrens H., Uhlig D., Z. Naturforsch, 28b, 276 (1973).
’2. Mayr А., неопубликованные результаты.
3. Ellis J. E., Hentges S. G., Kalina D. G., Hagen G. P., J. Organometal. Chem., 97, 79 (1975).
Декакарбонилдихромат(—I), -молибдат(—I)
и -вольфрамат(—I) натрия Na2[M2(CO)I0] (M=Cr, Mo, W)
2,2'-Дипиридил
2M(CO)6 + 2Na------------------->- Na2[M2(CO)10] + 2CO
M = Cr: 2-220,1 2-23,0 M=Cr: 430,1 2-22,4л
M = Mo: 2-264,0 M=Mo: 518,0
M = W: 2-351,9 M=W: 693,8
К суспензии 2,5 г (0,11 моль) порошка Na в 150 мл свежеперегнанного ^абсолютного тетрагидрофурана, помещенной в трубку Шленка емкостью 250 мл, снабженную ртутным затвором, добавляют —50 мг 2,2'-дипиридила и 0,05 моль гексакарбонила металла [11,0 г Сг(СО)6, 13,2 г Мо(СО)в или 17,6 г W(CO)6]. Смесь перемешивают* до тех пор, пока не прекратится выделение газа (Мо-производное: 20—30 ч; Сг- и W-производные: 40—60 ч). Для контроля протекания реакции целесообразно собирать выделяющийся газ (—1,2 л) в газометре. Реакционную смесь фильтруют через стеклянный фильтр G4 (диаметр 3 см), покрытый А12О3 и кусочками фильтровальной бумаги (толщиной соответственно 2 и 1 см), и красно-коричневый фильтрат упаривают в вакууме до объема —50 мл. Затем добавляют —100 мл 1,4-диоксана и оставляют стоять при 0°С в течение 1 ч. Выкристаллизовавшийся диоксано-:вый аддукт отфильтровывают на пористом стеклянном фильтре G3 и промывают диоксаном до тех пор, пока фильтрат не станет почти бесцветным. Чистый продукт получают после промывания эфиром и высушивания в высоком вакууме при 50°C до постоянной массы (—6 ч). Выход —90%.
Свойства. Микрокристаллическое соединение от светло- до темно-желтого цвета, чрезвычайно неустойчиво на воздухе (пирофорно). Растворяется в воде, тетрагидрофуране, пиридине, ацетоне, ацетонитриле, не растворяется .в диоксане, эфире и всех неполярных растворителях. ИК (CH3CN): М=Сг: 1983,5 (оч. сл.), 1917 (ср.), 1890 (оч. с.), 1793,5 (ср.) [v(CO)] см-1. М=Мо: 1983,5 (оч. сл.), 1937 (ср.), 1895 (оч. с.), 1795 (ср.) [v(CO)] см-1. M=W: 1978 (оч. сл.), 1943 (ср.), 1893,5 (оч. с.), 1794,5 (ср.) [v(CO)] см-1. Кристаллическая структура: см. [2].
ЛИТЕРАТУРА
1. Lindner Е„ Behrens Н„ Birkle S., J. Organometal. Chem., 15, 165 (1968). .2. Handy L. B„ Ruff J. K„ Dahl L. F„ J. Amer. Chem. Soc., 92, 7312 (1970).
* Нельзя использовать якорь магнитной мешалки в тефлоновой оболочке.
Карбонилы металлов 1935
□ □ Гексакарбонилмолибден Мо(СО)6
Мо(СО)6 получают восстановительным карбонилированием МоС15 при высоком давлении в присутствии сплава Деварда в качестве акцептора галогена-[ср. метод получения для W (СО)6].
Си-А1 МоС16 + 6СО ------► Мо(СО)6
273,2 6-22,4л 264,0
В стеклянный вкладыш на 750 мл, который подходит к качающемуся или-вращающемуся автоклаву иа 1 л, помещают 45,0 г MoClg и ~65 г сплава?. Деварда, смешанного приблизительно с таким же количеством по объему ко-
Рис. 465. Перегонка с паром гекса-карбонилмолибдена.
/ — колба для перегонки (2 л); 2 — охлаждаемый льдом приемник (колба на 2 л): 3 — холодильник (длина 60 см, внутренний диаметр 12—14 мм); 4 — подвижный стеклянный поршень с газонепроницаемым уплотнением в месте ввода (навинчивающийся колпачок), его широкий нижиий конец имеет зазор в 1—2 мм; 5 — к ртутному затвору; 6 — нормальный шлиф 29/32.
лец Рашига или стеклянных шариков. После добавления 200 мл абсолютного эфира помещают вкладыш в автоклав и нагревают при 100 °C в течение 24 ч при начальном давлении СО около 150 бар. Затем охлаждают, сжигают остатки СО, открывают автоклав и эфирный раствор переносят в двухлитровую-колбу 1 (рис. 465). К колбе присоединяют отводящий холодильник 3, снабженный насадкой для перегонки с паром, а также хорошо уплотненным стеклянным поршнем 4, который может перемещаться вверх и вниз внутри холодильника; во время перегонки поршень укреплен выше уровня отводной трубки. В качестве приемника служит колба 2, которую охлаждают льдом. Добавив 500 мл —20% НС1, разлагают сначала избыток сплава Деварда, затем' при низкой температуре отгоняют эфир и до перегонки с паром удаляют этот дистиллат из приемной колбы 2. В колбу 1 добавляют 500 мл Н2О, нагревают на плитке до кипения и перегоняют с паром Мо(СО)6. Кристаллизующийся в холодильнике карбонил время от времени стеклянным поршнем проталкивают в приемную колбу 2, чтобы избежать забивания холодильника. В отогнанном эфире также содержится небольшое количество Мо(СО)6. Его можно-извлечь, удалив растворитель в вакууме при возможно низкой температуре. Весь собранный продукт отфильтровывают на воронке Бюхнера, промывают небольшим количеством (—5 мл) холодного метанола от смолистых примесей' и отсасывают досуха. Возгонкой в вакууме масляного насоса при 25—40 °C получают —35 г (80%) белого кристаллического Мо(СО)6.
Свойства. Бесцветные, пушистые, очень устойчивые на воздухе кристаллы.
1ПЛ 150—151 °C (с разл.). Не растворяется в Н2О, умеренно растворяется в*
I i
1936 Глава 33. Металлоорганические комплексы
•органических растворителях. Растворы неустойчивы иа свету, легко окисляются на воздухе. ИК (газ): 2002,6 [v(CO)], 368 [v(Mo—С)] см-1; (и-гексаи): 1989,5 (оч. с.), 1957 (сл.) [v(CO)J см-1. Давление пара: lgp(MM рт. ст.) = «=11,174—3561,3/Г (от 55 до 145°C). Структура: см. [3, 4]; d(Mo—С) = «=2,063 А (электронографически).
'ЛИТЕРАТУРА
1. Arbeitsvorschriften der Anorganisch-chemischen Laboratorien der Techni-schen Universitat Munchen und der Universitaten Erlangen—Niirnberg und Regensburg; Hurd D. T., Inorg. Synth., 5, 135 (1957).
2. Brockway L. O., Ewens R. V. G., Lister M. W., Trans. Faraday Soc., 34, 1350 (1938).
3. Arnesen S. P., Seip H. P., Acta Chem. Stand., 20, 2711 (1966).
Гексакарбонилвольфрам W(CO)6
W(CO)6 синтезируют восстановительным карбонилированием WCU в присутствии в качестве акцепторов галогена порошка Си (способ 1) или сплава Деварда (способ 2).
•Способ 1
Си
TVC1, + 6СО ----->- W(CO)e
396,6 6-22,4л 351,9
Во вращающийся автоклав на 250 мл помещают стеклянный сосуд, в котором находятся 28,5 г (72 ммоль) WC16, 33 г (0,53 моль) тонкого медного порошка и 70 мл петролейного эфира (/кап 60—80 °C). Создают в системе давление СО ~300 бар и вращают автоклав при 140 °C. Через 48 ч необходимо дополнительно подкачать СО, через 72 ч реакция заканчивается. Содержимое автоклава переносят в грушевидный сублиматор с охлаждаемым «паль-цем> и после осторожного удаления растворителя в вакууме водоструйного насоса при комнатной температуре проводят высоковакуумную возгонку сырого продукта при 40—60 °C. При этом во избежание загрязнения при возгонке сырой продукт прикрывают стеклянной ватой. Выход —23 г (91%).
Способ 2
Си-А1
WC1, + 6СО -----► W(CO)e
396,6 6.22,4 л 351,9
Во вращающемся автоклаве на 500 мл нагревают при начальном давлении СО 200 бар и температуре 200 °C в течение 30—45 ч смесь 30,0 й (76 ммоль) WC16, 40 г (избыток) сплава Деварда и 200 мл абсолютного эфира. Реакционную смесь извлекают затем из автоклава водой и подвергают перегонке с паром (см. рис. 465). Сырой продукт на стеклянном пористом фильтре промывают небольшим количеством охлажденного льдом ацетона (2 мл) и отсасывают досуха. Возгонкой в вакууме при —50 °C получают —23 г (86%) бесцветного кристаллического W(CO)S.
Свойства. Бесцветные, пушистые, устойчивые иа воздухе кристаллы. /пл 169—170°C (с разл.). Не растворяется в Н2О, умеренно растворяется в органических растворителях. Растворы неустойчивы иа свету, легко окисляются на воздухе. ИК (газ): 1997,5 [v(CO)J, 374 [v(W—С)] см-1 ЯМР-13С
Карбонилы металлов 1937
(CDC13, внутр, стандарт ТМС): б 192,1 [СО]. Давление пара: IgpMMpT. ст.= = 10,947—3640,4/Г (от 65,6 до 137°C). Кристаллическая структура: см. [3, 4]; d(W—С) =2,058 А (электронографически).
ЛИТЕРАТУРА
1. Arbeitsvorscriften des Anorganisch-chemischen Laboratoriums der TU Mfln-chen sowie der Universitaten Erlangen—Niirnberg, Regensburg und Tubingen; Hurd D. T„ Inorg. Synth, 5, 135 (1957).
2. Brockway L. O, Ewens R. V. G„ Lister M. W., Trans. Faraday Soc, 34, 1350 (1938).
3. Arnesen S. P„ Seip H. P„ Acta Chem. Scand, 20, 2711 (1966).
□ □ Декакарбонилдимарганец Мп2(СО)ю
Декакарбонилдимарганец получают карбонилированием при высоком давлении солей Мп(П), например МпС12 или Мп(СН3СО2)2. Наилучших выходов достигают, если в качестве восстановителей используют алюминийалкилы (способ 1). Если нет в распоряжении автоклава высокого давления, то для получения Mn2(CO)io можно использовать Mn^-CHjCshU) (СО)з, проводя восстановительное карбонилирование при атмосферном давлении (способ 2). Последний способ, к сожалению, протекает с невысоким выходом (<20%), тем не менее вполне приемлем из-за доступности исходных материалов.
Способ 1
A1(C2Hj)2
2Мп(СН3СОА+ 10СО 3 Мп2(СО)10
•~«5UU Odp
2-173,0 10-22,4л 390,0
Соблюдать осторожность; А1 (С2Н5)3 способен самовоспламеняться!
В трехгорлой колбе на 1 л, снабженной обратным холодильником, высокоэффективной мешалкой и капельной воронкой, суспендируют 34,6 г (0,20 моль) безводного ацетата Мп(II) в 450 мл диизопропилового эфира. К этой смеси в атмосфере N2 медленно прикапывают 120 мл (0,88 моль) три-зтйлалюминия в течение —2,5 ч; если реакция слишком бурная, охлаждают колбу холодным воздухом (феном). Охлажденную смесь осторожно в атмосфере N2 переносят в качающийся автоклав на 1 л. Колбу и капельную воронку споласкивают в автоклав 40 мл абсолютного диизопропилового эфира. Автоклав быстро закрывают и продувают СО (50 бар при этом достаточно). Затем давление СО доводят до 200 бар, запускают автоклав и медленно нагревают до 80—85 °C. Когда реакция начнется, что видно по понижению давления, автоклав нагревают до 100 °C и выдерживают при этой температуре 20 ч. После охлаждения содержимое автоклава выливают (атмосфера N2f) через воронку в 2-литровую трехгорлую колбу; твердый осадок переносят в колбу шпателем. Колбу снабжают обратным холодильником, эффективной мешалкой и капельной воронкой. Затем при соблюдении таких же предосторожностей, как и в синтезе Сг(СО)6 (см. выше), по каплям добавляют смесь 400 мл Н2О и 100 мл конц. НС1, чтобы гидролизовать избыток А1(С2Н5)3. Далее, как и в случае Сг(СО)3, перегоняют Мп2(СО)10 с паром до тех пор, пока в холодильнике не будет больше образовываться желтый кристаллический Мп2(СО) ю. Дистиллат охлаждают почти до замерзания, чтобы было удобнее отделить Мп2(СО)10 от совместно отогнавшегося растворителя. Mn2(CO)to собирают на фильтре, промывают небольшим количеством холодного СН3ОН и сушат на воздухе. Выход —15 г (—38%).
6—1361
1938 Глава 33. Металлоорганические комплексы
Способ 2
Na гМп^-СНзСЛ) (С03) + 4С0 -> Мп2(СО)10
1 оар 2-218,1 4-22,4 л 390,0
Получение Мп2(СО)ю из Мп(т]-СН3С5Н4) (СО)3 при атмосферном давлении проводят в 4-литровой трехгорлой колбе, которая снабжена: а) краном для ввода N2, б) трубкой для ввода газа (СО), которая на несколько сантиметров погружена в растворитель; в) капельной воронкой на 250 мл с выравнивателем давления и воздушным (!) холодильником (оба с помощью переходов укреплены на одном боковом горле колбы); г) эффективной мешалкой (средний шлиф). После того как аппаратура продута N2, в колбу помещают 100 г (4,35 моль) Na кусочками и 500 мл сухого диглима. Смесь нагревают до кипения (161—164 °C) и диспергируют мешалкой расплавленный натрий. Затем прерывают ток N2 и вводят СО. Через 5—10 мин начинают медленно прикапывать 212 мл (300 г, 1,38 моль) Мп(т]-СН3С3Н4) (СО)3, время прибавления ~4 ч. Прн этом смесь необходимо нагревать до слабого кипения. Ток СО не должен быть слишком сильным. После прибавления карбонила смесь кипятят еще 1 ч и охлаждают до комнатной температуры. В реакционную смесь осторожно в течение 2 ч добавляют 400 мл изопропилового спирта, продолжая пропускание СО, после чего порциями (!) подкисляют образовавшуюся коричневую массу 350 мл 85 %-ной Н3РО4. Эта реакция весьма зкзотермична, поэтому необходимо эффективное охлаждение колбы сухим льдом. После завершения добавления кислоты в колбе образуется кашицеобразная масса, которая наряду с Mn2(CO)io содержит избыток фосфата натрия. Кристаллы (—500 г) отфильтровывают и для удаления фосфатов промывают горячей водой. Промывные воды должны стать почти бесцветными. Остаток желтого цвета, в основном Мп2(СО)ю, весит около 50 г. Для очистки его промывают еще несколькими миллилитрами изопропилового спирта, после чего ои становится более светлым. Окончательную очистку проводят высоковакуумиой возгонкой при 70—80 °C.
Все остатки от фильтрования объединяют и оставляют стоять иа 2 недели в прикрытой колбе Эрленмейера при комнатной температуре. За это время выпадает довольно значительное количество Мп2(СО)ю, который для препаративных целей является достаточно чистым (/пл 150—152°C). Общий выход 44-53 г (16—20%).
Свойства. Золотисто-желтые, в течение долгого времени стабильные на воздухе светочувствительные кристаллы. /пл 153—155 °C. Умеренно возгоняется в высоком вакууме уже при комнатной температуре. Растворяется практически во всех органических растворителях. Растворы на воздухе неустойчивы. ИК (СС14): 2044 (ср.), 2012 (с.), 1981 (ср.) [v(CO)] см-1. Кристаллическая структура: см. [3], симметрия Д4<1; d(Mn—Мп) =2,923 А.
ЛИТЕРАТУРА
1. Podall И. Е., Dunn J. Н., Shapiro И., J. Amer. Chem. Soc., 82, 1325 (1960). 2. King R. В., Stokes J. C., Korenowski T. F„ J. Organometal. Chem., 11, 641 (1968).
3. Dahl L. F„ Rundle R. E„ Acta Crystallogr., 16, 419 (1963).
□ □ Тетрахлороалюминат гексакарбонилмарганца(1) [Мп(СО)в][А1СЦ]
MnCi(CO) 5 + AiCi3 + CO --► [Mn (CO)6J [AiCl4]
230,4 133,3 22,4л 391,8
Карбонилы металлов 1939
Во вращающийся автоклав на 50 мл помещают хорошо растертую смесь 0,6 г (2,6 ммоль) МпС1(СО)5 и 0,7 г (5,2 ммоль) чистого свежевозогнанного А1С1з- После создания давления СО до 300 бар автоклав запускают и в течение 20 ч греют при 100 °C. Светло-серый порошкообразный продукт реакции переносят из автоклава на стеклянный пористый фильтр G4 с помощью эфира порциями до тех пор, пока фильтрат не станет прозрачным. Затем смывают продукт с фильтра 5—10 мл свежеабсолютированного тетрагидрофурана, высаживают добавлением к фильтрату диметилового эфира этиленгликоля, отфильтровывают выпавшие кристаллы и промывают их иа фильтре диэтиловым эфиром. Для окончательной очистки переосаждение повторяют еще 2—3 раза и высушивают продукт в высоком вакууме. Выход 0,63 г (62%).
Свойства. Бесцветные кристаллы, растворимые в тетрагидрофуране; не растворяются в диметиловом эфире этиленгликоля. В присутствии влаги соединение разлагается тотчас до МпН(СО)5 и СО2. При нагревании до 130— 140 °C также происходит разложение. ИК (нуйол): 2101 (с.) [v(CO)] см-1.
Диалоги. Получение менее чувствительного к гидролизу [Re(CO)6][AlC14] проводят аналогичным образом. Этилензамещенные производные [М(С2Н4)Я-(СО)6_J[PF6] (M=Mn, n=l; M=Re, п=1, 2) получают из МС1(СО)5, Aids и С2Н4 при 50—220 бар. Ацетонитрильное производное [Мп(МССН3)з(СО)3][РРб] синтезируют из МпВг(СО)5 в кипящем CH3CN с последующим высаживанием при действии NH4PF6. Синтезы других катионных комплексов [Мп(СО)5Ц + с фосфор- и азотсодержащими лигандами описаны в работах [6, 7].
ЛИТЕРАТУРА
1. Fischer Е. О., Fichtel К. Ofele К, Вег., 95, 249 (1962) ([Мп(СО)6] [А1СЦ]). 2. Hieber W„ Kruck Th., Angew. Chem., 73, 580 (1961) ([Re(CO)6][AlC14]). 3. Fischer E. O., Ofele K., Angew. Chem., 73, 581 (1961) ([MnC2H4(CO)5][AlC14]). 4. Fischer E. O., Ofele K-, Angew. Chem., 74, 76 (1962) ([Re(C2H4)2(CO)4][PF6]). 5. Reimann R. H., Singleton E„ J. Organomet. Chem., 59, C24 (1973) ([Мп(МССНз)з(СО)з][РР6]).
6. Kruck Th., Hofler M., Ber„ 96, 3035 (1963) ([Mn(CO)sL][PF6]).
7. Drew D„ Darensbourg D. J., Inorg. Chem., 14, 1579 (1975) ([M(CO)6L][PFe] (M=Mn, Re).
□ □ Декакарбонилдирений Re2(CO)10
Re2(CO)i0 можно получать простым и удобным способом из РегО? (способ 1), причем СО одновременно является восстановителем и лигандом. В принципе так же протекает образование и из KReO4 (способ 2).
Способ 1
Re2O, + 17СО -----> Re2(CO)10 + 7СО2
484,4 17-22,4 л 652,5 7-22,4 л
В абсолютно сухой вращающийся автоклав на 250 мл помещают 25,0 г (51,6 ммоль) Re2O7, тщательно избегая доступа воздуха и влаги, и, создав давление СО около 50 бар, удаляют остатки посторонних газов. Затем давление в автоклаве поднимают до 300 бар и медленно доводят внешний нагрев до 250 °C. Через 18 ч автоклав охлаждают до комнатной температуры и сжигают избыточный СО. Для удаления Re2O7 беловато-серый сырой продукт (порошкообразный или в виде корочек) промывают на фильтре водой до тех пор, пока фильтрат не станет нейтральным. Остаток подвергают возгонке в высоком вакууме при 66—75 °C; Re2(CO)I0 образуется в виде белых кристаллов. Выход составляет свыше 30 г (89%), если использовать очень чистый геп-6*
1940 Глава 33. Металлоорганические комплексы
токсид. Если исходный продукт загрязнен перреиатом, выход существенно снижается и не может быть заметно улучшен даже в более жестких условиях (400 бар, 270°C).
В случае больших загрузок (48 г Re2O7) необходимо в процессе реакции еще раз подкачать дополнительно СО (время реакции 40 ч). Полученный продукт в этом случае перекристаллизовывают из 750 мл кипящего ацетона. Выход 80—85%.
Способ 2
2Re + 7Н2О2 + 2КОН -------> 2KReO4 + 8Н2О
2-186,2 7-34,0 2-56,1 2-289,3 8-18,0
2KReO4 + 17СО ----> Re2(CO)j0 + 7СО2 + К2О
2-289,3 17-22,4л 652,5 7-22,4 л 94,2
18,6 г (0,1 моль) порошка Re суспендируют в небольшом количестве водь в круглодоиной колбе на 250 мл, снабженной капельной воронкой и обратным холодильником. При перемешивании магнитной мешалкой прикапывают 60 мл 30%-ного Н2О2 и затем кипятят 2 ч. После охлаждения фильтруют и нейтрализуют (охлаждая льдом) насыщенным раствором КОН по фенолфталеину. Выпавший KReO< отсасывают и высушивают (растворимость при 0°С: 10 г/л). Из маточника после упаривания можно выделить еще некоторое количество соли.
В шаровой мельнице получают тонкую смесь 10,0 г (35 ммоль) KReO4 и 50,0 г порошка Си. Реакционную смесь помещают во вращающийся автоклав на 250 мл, который однократно при давлении ~ 100 бар продувают СО, и создают рабочее давление СО около 320 бар. Затем автоклав нагревают в течение 24 ч при 300 °C. После охлаждения, сжигания избытка СО и продувания азотом автоклав открывают. Продукт реакции переносят в трубку Шленка. Тщательно споласкивая автоклав петролейным эфиром (/кип 40— 60 °C) и удаляя растворитель, получают еще некоторое количество сырого продукта, которое объединяют с основной массой. Для очистки проводят высоковакуумную возгонку при 80—100°C. Выход ~7,3 г (64%).
Свойства. Бесцветные листочки, устойчивые иа воздухе. /Пл 177 °C; при высокой температуре происходит разрушение с выделением металлического Re. Не растворяется в воде, умеренно растворяется в алифатических углеводородах, хорошо растворяется в тетрагидрофуране и метиленхлориде. Re2(CO)]0 можно возгонять при атмосферном давлении в токе СО при 140 °C. ИК (циклогексан): 2070 (ср.), 2014 (с.), 2003 (с.), 1976 (ср.) [v(CO)l см-1. Кристаллическая структура: см. [5], симметрия D^, d(Re—Re) =3,01 А, изоморфен Мп2(СО)ю-
ЛИТЕРАТУРА
1. Hieber W., Fuchs Н., Z. Anorg. Allgem. Chem., 248, 256 (1941).
2. Hileman J. C., Huggins D. K., Kaesz H. D., Inorg. Chem., 1, 933 (1962).
3. Fischer E. O., Scherzer К.., неопубликованные результаты.
4. Scherzer K„ Dissertation, Technische Univers. Mflnchen, 1979.
5. Dahl L. F., Ishishi E., Rundle R. E., J. Chem. Phys., 26, 1750 (1957).
Эннеакарбонилдижелезо Fe2(CO)9
Fe2(CO)2 образуется фотохимически из Fe(CO)s:
2Fe(CO)5 ---> Fe2(CO)9-|-CO
2-195,9 363,8 22,4л
Карбонилы металлов 1941
Смесь 24,0 мл (35,0 г, 0,18 моль) продажного Fe(CO)5 и 150 мл ледяной уксусной кислоты при интенсивном перемешивании магнитной мешалкой облучают в трубке Шлейка ртутной лампой высокого давления мощностью 150 Вт. Если выделяющийся Fe2(CO)9 осядет на стенках трубки Шленка, era счищают в смесь, чтобы скорость реакции не уменьшалась. Выпавшие золотистые кристаллы отфильтровывают, отмывают водой от уксусной кислоты, затем промывают спиртом, далее эфиром и наконец 1 ч высушивают в высоком вакууме. Выход 28,2 г (86%).
Наиболее простым способом получения Fe2(CO)9 является облучение реакционной смеси (см. выше) обычным солнечным светом до постоянной массы выпавшего продукта. В зависимости от интенсивности света и длительности облучения (не менее 10 ч при ярком солнечном свете) выход составляет до 95%.
Свойства. Блестящие, золотисто-желтые листочки, очень устойчивые в отсутствие влаги на воздухе, d 2,085 (18°C). Не растворяется в алифатических углеводородах, бензоле и эфире, очень мало растворяется в метаноле. Постепенно разлагается в тетрагидрофуране и галогеноуглеводородах. В атмосфере инертного газа при 35 °C хранится без разложения. В присутствии органических растворителей при ~60°С начинается разложение с образованием Fe(CO)s и Fe3(CO)i2 без выделения СО; в твердом состоянии в интервале от 100 до 120°C разложение протекает по схеме 3Fe2(CO)9->3Fe(CO)5+Fe3(CO) 12; Fe3(CO)i2-»-3Fe+12CO. ИК (КВг): 2080 (ср.), 2034 (с.), 1828 (с.) [v(CO)J см-1. Кристаллическая структура: см. [3]; d(Fe—Fe) =2,46 А.
ЛИТЕРАТУРА
1. Speyer Е„ Wolf Н., Вег., 60, 1424 (1927).
2. Keeley D. F., Johnson R. Е., J. Inorg. Nucl. Chem., 11, 33 (1959). 3. Powell H. M., Ewens R. V. G., J. Chem. Soc. (London), 1939, 286.
□ Додекакарбонилтрижелезо Fe3(CO)i2
Fe3(CO)i2 получают из Fe(CO)s переводом сначала в анионный гидридный комплекс [FeH(CO)4]_ и последующим его окислением:
Fe(CO)5 + 2NaOH ----> Na[FeH(CO)4] + NaHCO3
195,9 2-40,0 191,9 84,0
3Na[FeH(CO)4l 4- ЗМпО, -> Fe3(CO)12 4- 3«МпО» 4- 3NaOH
3-191,9 3-86,9 503,7 3-70,9 3-40,0
В двугорлой колбе на 2 л, снабженной обратным холодильником и капельной воронкой, помещают раствор 52,0 мл (75,0 г, 0,38 моль) Fe(CO)j в 200 мл СН3ОН и 56,0 г (1,40 моль) NaOH в 150 мл Н2О и полученную смесь перемешивают 0,5 ч магнитной мешалкой при комнатной температуре. К полученному коричневому раствору Na[FeH(CO)4] также при комнатной температуре добавляют насыщенный водный раствор NH4C1(~44 г NH4Cl в 150 мл Н2О).
Для окисления карбонилферрат-аниоиа при охлаждении льдом и перемешивании порциями прибавляют также заранее охлажденную суспензию МпО2, полученную из 85,0 г (0,54 моль) КМпО4 (см. ниже). Во время экзотермической реакции смесь принимает темно-красную окраску. Для полноты реакции перемешивают еще 1 ч при комнатной температуре. Непрореагировавший МпО2 затем разлагают осторожным прикапыванием раствора 50,0 г (0,18 моль) FeSO4-7H2O в 250 мл 10%-ной H2SO4. Через 20 мин добавляют 200 мл 50%-ной H2SO4. Еще через 20 мин перемешивания из раствора, окра-
1942 Глава 33. Металлоорганические комплексы
шейного от красноватого до зеленого цвета, выпадает черный осадок Fe3(CO) 12. Продукт отделяют на стеклянном нутч-фильтре, несколько раз промывают горячей конц. H2SO4 порциями по 50 мл, затем дважды по 100 мл Н2О, дважды по 20 мл 95%-ного С2Н5ОН и наконец многократно петролей-ным эфиром порциями по 50 мл, затем некоторое время сушат в высоком вакууме. Выход 39—49 г (60—75%). Такой продукт вполне пригоден для большинства препаративных целей. Возгонкой Рез(СО)12 (60 °C, высокий вакуум) с учетом значительных потерь (вплоть до 80%) можно получить спектрально чистый препарат.
Получение диоксида марганца. В широкогорлую 2-литровую колбу Эрлен-мейера помещают раствор 85,0 г (0,54 моль) КМпО< в 380 мл Н2О. После добавления приблизительно 10 мл 95%-ного С2Н5ОН -дожидаются сначала восстановительной реакции, для чего иногда требуется осторожное нагревание смеси на водяной бане. (Осторожно! Сильно экзотермическая реакция!) Когда реакция начнется, порциями при взбалтывании прибавляют ПО мл 95%-ного С2Н5ОН и оставляют стоять до тех пор, пока темно-фиолетовая окраска перманганата не перейдет в коричневую. Прежде чем применять суспензию МпО2 для окисления Na[FeH(CO)4], ее охлаждают льдом.
Свойства. Черные, умеренно устойчивые на воздухе кристаллы, в высоком вакууме возгоняются при температуре с60 °C. При длительном нагревании свыше 60 °C соединение разлагается с выделением металлического железа. Плохо растворяется в органических растворителях; раствор, окрашенный в зеленый цвет, при доступе воздуха вскоре разлагается с выделением оксида Fe(III). ИК (C2CI4): 2047 (оч. с.), 2024 (ср.), 1865 (сл.), 1834 (сл.) [v(CO)J; (н-гексан): 2046 (оч. с.), 2023 (ср.) [v(CO)] см-1. Кристаллическая структура: см. [4]; d(Fe—Fe)=2,69 соотв. 2,55 А.
ЛИТЕРАТУРА
1. Hieber W., Z. Anorg. Allgem. Chem., 204, 165 (1932).
2. Hieber W., Brendel G., Z. Anorg. Allgem. Chem., 289, 324 (1957).
3. King J?. B., Stone F. G. A., Inorg. Synth., 7, 193 (1963).
4. Wei С. H„ Dahl L. F., J. Amer. Chem. Soc., 88, 1821 (1966).
□ □ Тетракарбонилферрат(—II) натрия Na2[Fe(CO)4]
Fe(CO)s 4- 2Na --> Na2[Fe(CO)4] + CO
195,9 2-23,0 213,9 22,4 л
В двугорлую колбу на 2 л, предварительно прогретую и снабженную ртутным затвором и краном для ввода N2, в атмосфере азота помещают 4,6 г (0,2 моль) порошкообразного Na (см. выше его получение), 1,7 г (0,01 моль) бензофенона Ph2CO и 1,2 г свежеперегнанного (над Na+Ph2CO) тетрагидрофурана. Затем через стальную иглу (диаметром 1,5 мм) в реакционную колбу порциями вводят Fe(CO)s, помещенный предварительно в трубку Шленка в количестве (13,4 мл, 0,1 моль), достаточном для исчезновения синей окраски кетального радикала: сначала через некоторое время после добавления Fe(CO)s синяя окраска восстанавливается, тогда снова добавляют порцию Fe(CO)5, и так несколько раз. Избыток бензофенонкетила подавляет побочную реакцию Fe(CO)s+ [Fe(CO)4]2-->[Fe2(CO)8]2-+CO. Реакционная смесь поэтому должна всегда после каждого добавления Fe(CO)5 сохранять слабую голубую окраску. После добавления —10 мл Fe(CO)s начинает выпадать бесцветный Na2[Fe(CO)4]. Синяя окраска по мере течения реакции возобновляется все медленнее, так что добавление последних порций Fe(CO)5 может затянуться в самом неблагоприятном случае (в зависимости от степени измельчения Na) на 2—3 дня. По окончании реакции для более полного выпадения продукта добавляют 600 мл тщательно высушенного н-гексана или пентана,
Карбонилы металлов 1943
оставляют на некоторое время стоять и затем декантируют избыток раствора (цвет коричневый). Кристаллический продукт промывают еще одной порцией н-гексана (500 мл) и затем суспендируют в 500 мл «-гексана. Суспензию переносят на стеклянный пористый фильтр (G3) и фильтруют при небольшом избыточном давлении. Высушенные в высоком вакууме кристаллы образуют бесцветный порошок. Выход 18,8 г (88%).
Другие способы получения. 1) Из Fe(CO)s и металлического Na в жидком NH3 [2]. 2) Из Fe3(CO)12 и Na в жидком NHS [3—5]. 3) Аналитически чистый Ka[Fe(CO)4] (1ПЛ 270—273°C, с разл.) может быть получен из Fe(CO)j и K[BH(erop-C4H9)3] («K-Selectrid>, Aldrich Chemical Со.) с выходом 95— 100% по способу, описанному в работе [6].
Свойства. Белый, пирофорный микрокристаллический порошок, который в отсутствие воздуха и в темноте хранится практически неограниченное время. Растворяется в воде, диметнлформамиде, N-метилпирролидоне и гексаметаполе, хуже растворяется в тетрагидрофуране. ИК (нуйол): 1761 (оч. с.) [v(CO)l см-1. Na2[Fe(CO)4] • 1,5 (диоксан) [реактив Кольмана, фирма-поставщик Alfa Ventron] оказался ценным реактивом для органических и металлоорганических синтезов. Кристаллическая и молекулярная структура тетрагональная, пространственная группа симметрии Р42/т (а= 10,690 А, с= = 12,293 А); сильно искаженная тетраэдрическая молекула с взаимодействиями Na+.. .О-, Na+.. .С- и Na+.. .Fe- [7].
ЛИТЕРАТУРА
1. Collman J. Р., Finke R. G., Cawse J. N., Brauman J. I., J. Amer. Chem. Soc, 99, 2515 (1977).
2. Behrens H., Weber R„ Z. Anorg. Allgem. Chem., 281, 190 (1955).
3. Hieber W., Vohler 0., Z. Anorg. Allgem. Chem., 294, 219 (1958).
4. Bayer E., Brune H. A., Hock K. L., Angew. Chem., 74, 872 (1962).
5. King R. B., Stone F. G. A., Inorg. Synth., 7, 196 (1963).
6. Gladysz J. A., Tam. W„ J. Org. Chem., 43, 2279 (1978).
7. Chin H. B., Bau R., J. Amer. Chem. Soc., 98, 2434 (1976).
□ □ Октакарбонилдиферрат(—I) натрия Na2[Fe2(CO)8]
hv
' 2Fe(CO)6 4- 2Na ----->- Na2[Fe2(CO)8] + 2CO
2-195,9 2-23,0 381,8 2-22,4л
В трубку Шленка на 200 мл помещают раствор 4,00 мл (29,8 ммоль} Fe(CO)5 в 175 мл тетрагидрофурана и добавляют 145 г 1%-ной амальгамы Na (63,0 ммоль Na). При интенсивном перемешивании облучают смесь мощной ртутной лампой высокого давления (например, Hanau TQ718). Во время облучения трубку Шленка держат при комнатной температуре (охлаждающая рубашка). Через —1,5 ч реакция заканчивается (выделение газа 600—700 мл). Темный красно-корнчневый раствор вместе с выпавшими оранжевыми кристаллами декантируют с амальгамы и отгоняют в вакууме растворитель досуха. Красно-коричневый кристаллический остаток растворяют в 10—20 мл ацетона, фильтруют через стеклянный пористый фильтр G4 и снова отгоняют растворитель в вакууме. Сырой продукт смесью тетрагидрофуран — эфир 1 :1 (20 мл) переносят на стеклянный пористый фильтр G3 и промывают на фильтре эфиром порциями по 10 мл до тех пор, пока он еще будет окрашиваться в светло-розовый цвет. Затем кристаллы высушивают —24 ч в высоком вакууме при комнатной температуре. Выход 5,1 г (90%). Рекомендуется для лучшей сохранности Na-соль перевести в К(С2Н5)4-соль обработкой водным раствором [N(C2H5)4]I.
1944 Глава 33. Металлоорганические комплексы
Свойства. Чрезвычайно неустойчивое на воздухе, желто-коричневое, микрокристаллическое вещество. Хорошо растворяется в воде, ацетоне и ацетонитриле с красно-коричневым окрашиванием; умеренно растворяется в тетрагидрофуране, не растворяется в эфире и неполярных растворителях. Желтую Na-соль не удается без разложения полностью освободить от растворителя.
Чистые соли получают из водных растворов осаждением объемистыми катионами, например N(C2H5)4+, и перекристаллизацией из смеси ацетонитрил— эфир. ИК-спектр [М(С2Н5)4]2[Ре2(СО)8] (нуйол): 1920 (с., шир.), 1852 (с., шир.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Ruff J. К., Inorg. Chem., 7, 1818 (1968).
2. Hieber W., Brendel G., Z. Anorg. Allgem. Chem., 289, 324 (1957).
3. Farmery K„ Kilner M., Greafrex R., Greenwood N. N., J. Chem. Soc. (London) A, 1969, 2339.
Додекакарбонилтрирутений Ru3(CO)12
Способ 1
6Ru(C6H,O2)s + 24CO + 9H2-------> 2Ru3(CO)12 + 18C5H8O2
6-398,4 24-22,4л 9-22,4л 2-639,3 18-100,1
(C5HgO2 = ацетилацетон)
Ацетилацетонат натрия рекомендуется готовить непосредственно перед употреблением: 2,3 г NaOH в виде 40%-ного водного раствора перемешивают с 5,75 г (57,4 ммоль) ацетилацетоиа [пентандион-2,4]. Белую массу, образующуюся в процессе экзотермической реакции, охлаждают и применяют для последующего получения Ru3(CO)i2 без дополнительной обработки.
В 1-литровый качающийся автоклав загружают в указанвом порядке 5,0 г (19,1 ммоль) RuC13-3H2O, 7,0 г (57,4 ммоль) ацетилацетоната натрия (см. выше) и 140 мл СН3ОН, плотно закрывают и создают сначала рабочее давление Н2 40 бар, а затем СО 120 бар. Карбонилирование проводят при 145°C (внутренняя температура). Через 4 ч автоклав охлаждают до комнатной температуры и отфильтровывают оранжево-желтый кристаллический продукт. Дополнительно Ru3(CO)i2 выделяют при выпаривании маточника в вакууме водоструйного иасоса и экстракцией остатка в аппарате Сокслета кипящим петролейным эфиром (1кип 60—80°C). Обе фракции объединяют и очищают перекристаллизацией из горячего петролейного эфира (/кип 60—80°C), а также ацетона. Выход 2,0—2,8 г (50—70%).
Способ 2
со Zn, со
RuCl3-ЗН2О ----> RuCl^CO)* ----->- Ru8(CO)12
261,5 639,3
В двугорлой колбе на 500 мл, снабженной эффективным обратным холодильником и трубкой для ввода газа, кипятят раствор 7,00 г (26,8 ммоль) высушенного в вакууме RuCl3-3H2O (37—39% Ru) в 120 мл абсолютного 2-этоксиэтанола (моноэтиловый эфир этиленгликоля). Одновременно через раствор продувают интенсивный ток СО. В течение 6 ч первоначально красный цвет раствора постепенно переходит в лимонно-желтый, при этом обра-ауется карбонилхлоридный комплекс рутения RuClv(CO)z, строение которого не уточнено. Приблизительно через 8 ч раствор охлаждают до комнатной тем
Карбонилы металлов 1945
пературы, продолжая пропускать СО. Раствор можно оставить на ночь, сохранив слабый ток СО. Помутнение раствора практически не сказывается на выходе.
Далее к раствору добавляют 100 мл абсолютного этанола и 8,0 г (0,122 моль, избыток) порошка Zn. Применяемый Zn (30 меш, —140 отв./см2) предварительно очищают ~2 н. НС1, промывают Н2О и ацетоном и отсасывают досуха.
Затем раствор нагревают при 85 °C, интенсивно перемешивая и пропуская ток СО. Через 8—9 ч охлаждают, дают отстояться и декантируют оранжевожелтую суспензию от избытка Zn. Оранжево-красные кристаллы Ки3(СО)и отфильтровывают, промывают СН3ОН и высушивают в вакууме. По этой методике получают достаточно чистый Ru3(CO)i2. Еще некоторое количество продукта можно извлечь из остатков, используя смывной метанольный раствор и очищая Ru3(CO)i2, как это описано выше. Общий выход сырого продукта ~3,8 г.
Препарат перекристаллизовывают из горячего толуола с небольшим количеством н-гексана. От нерастворимых загрязнений (по-видимому, это кар-бонилгидрндный рутениевый кластер) избавляются, фильтруя раствор еще горячим. Выход 2,8—3,4 г (50—60%).
Другие способы. Карбонилирование при нормальном давлении ацетата гексакис (р,-ацетато)трисакваоксотрирутения (III) [Ки3О(О2ССН3)б(Н2О)3]-О2ССН3 в н-пропаноле при 80 °C. Выход 59% [4].
Свойства. Кристаллическое вещество, оранжевого цвета, довольно устойчивое на воздухе и на свету. Растворяется в большинстве органических растворителей, особенно хорошо в ацетоне. Не растворяется в воде. ИК (н-гек-сан): 2060 (с.), 2030 (с.), 2010 (ср.) [v(CO)] см-1. Кристаллическая структура: см. [5, 6]; d(Ru—Ru) =2,848 А.
ЛИТЕРАТУРА
1. Braca G., Sbrana G„ Pino Р., Chim. Ind. (Milano), 46, 206 (1964).
2. Johnson В. F. G„ Lewis J., Inorg. Synth., 13, 92 (1972).
3. Mantovani A., Cenini S„ Inorg. Synth., 16, 47 (1976).
4. James B. R„ Rempel G. L., Teo W. K, Inorg. Synth., 16, 45 (1976).
5. Corey E. R., Dahl L. F., J. Amer. Chem. Soc, 83, 2203 (1961).
6. Mason R„ Rae A. I. M., J. Chem. Soc. (London) A, 1968, 778.
Додекакарбонилтриосмий Os3(CO)i2
3OsO4 + 24CO ------> Os3(CO)12 + 12CO2
3-254,2 24-22,4л 906,7 12-22,4л
Смесь 4.0 г (15,7 ммоль) OsO4 и 90 мл С2Н5ОН, помещенную во вращающийся автоклав на 200 мл, обрабатывают СО в течение 12 ч прн 165 °C (внешняя температура) и начальном давлении 80 бар. После охлаждения автоклава льдом желтый кристаллический продукт отфильтровывают, промывают небольшим количеством ацетона и сушат в вакууме масляного насоса. Полученный препарат для большинства целей достаточно чист. Необходимую очистку проводят высоковакуумной возгонкой при 130 °C или перекристаллизацией из горячего толуола. Выход 3,3—3,8 г (70—80%). Для больших загрузок (до 25 г OsO4) можно использовать 1-литровый вращающийся автоклав.
Осторожно! Из-за чрезвычайно высокой токсичности OsO4 все операции необходимо проводить в хорошо действующем вытяжном шкафу!
Другие способы. Карбонилирование при высоком давлении галогенидов или оксидгалогенидов осмия в присутствии порошков Си или Ag. Выход около 90% [2].
4946 Глава 33. Металлоорганические комплексы
Свойства. Желтое кристаллическое вещество, легко возгоняется, на воздухе неустойчиво. Растворяется только в очень полярных органических растворителях. ИК («-гексан): 2070 (с.), 2036 (с.), 2015 (ср.), 2003 (ср.) (v(CO)] см-1. Кристаллическая структура; см. [3]; d(Os—Os) =2,88 А.
ЛИТЕРАТУРА
1. Johnson В. F. G., Lewis J., Inorg. Synth., 13, 92 (1972).
2. Hieber W., Stallman H„ Z. Elektrochem., 49, 288 (1943).
3. Corey E. R„ Dahl L. F., Inorg. Chem., 1, 521 (1962).
□ Октакарбонилдикобальт Co2(CO)8
Co2(CO)8 может быть получен с хорошими выходами карбонилированием прн высоком давлении ацетата кобальта(П) (способ 1), карбоната кобальта (II) (способ 2) или активированного металлического кобальта (способ 3).
Способ 1
2Со(СН3СО2)а-4Н2О8(СН3СО)аО-[-8СО + 2На--------->
2-249,1 8-102,1 2-22,4 л 2-22,4л
---> Соа(СО)8 + 20СН3СОаН 342,0 20-60,1
Во вращающийся или качающийся автоклав на 500 мл (предельное давление до 500 бар)* помещают 124,5 г (0,50 моль) Со(СН3СО2)2-4Н2О и 209 г (2,05 моль) ангидрида уксусной кислоты и удаляют посторонние газы из автоклава, создав давление Н2 около 100 бар и затем сжигая избыток водорода. Реакцию начинают при давлении Н2 60 бар и давлении СО 240 бар. Автоклав •нагревают до 165—175°C (внутренняя температура), и уже при 130°С интенсивно начинается реакция. Давление в автоклаве поднимается до —400 бар. Через 15 ч систему охлаждают до комнатной температуры и сжигают избыток газа в автоклаве. Содержимое переносят в 2-литровую колбу Бюхнера (в атмосфере аргона), разбавляют при перемешивании равным объемом воды « отфильтровывают выпавший кристаллический (цвет от оранжево-желтого до красного) осадок, хорошо промывают его водой на фильтре и несколько часов сушат в вакууме водоструйного насоса. Выход 47—53 г (55—62%). Продукт для превращения его в Со(т]-С5Н8) (СО)2 достаточно чистый. Аналитически чистый препарат получают осторожной высоковакуумной возгонкой при 30 °C [при более высокой температуре происходит образование Со4(СО)12].
Способ 2
2СоСО3 + 2Н2 + 8СО -------> Со2(СО)8 + 2НаО + 2СОа
2-118,9 2-22,4л 8-22,4л 342,0 2-18,0 2-22,4л
В качающийся автоклав на 500 мл помещают 15,0 г (0,126 моль) СоСО3 ( — 46% Со) и 0,05 г Со2(СО)8 и заливают 150 мл петролейного эфира (/кип 40—60 °C) (в атмосфере N2). Систему трижды продувают СО под давлением 50 бар, затем создают рабочее давление 280 бар Н2+СО (140 бар Н2 и 140 бар СО). Автоклав запускают, медленно нагревают до 150—160°C (внут
* Для небольших загрузок («0,15 моль Со(СН3СО2)2-4Н2О) преимущественно используют вращающийся автоклав на 250 мл, условия реакции те же (выход до 90%). При проведении синтеза по способу 1 рекомендуется восполнять потерю давления СО в автоклаве до первоначального примерно через 3 ч после начала реакции.
Карбонилы металлов 1947
ренняя температура) и выдерживают при этой температуре 3—4 ч. В процессе нагревания при 120 °C в автоклаве достигается максимальное давление около 360 бар. В конце реакции давление составляет около 300 бар. После охлаждения автоклава избыточный газ осторожно сжигают.
Внимание! В продукте реакции находятся следы летучего высокотоксичного СоН(СО)4! Прозрачный темный раствор фильтруют через складчатый фильтр в колбу Эрленмейера {в вытяжном шкафу! СоН(СО)<!] и оставляют кристаллизоваться при —10 °C. Образуются длинные, хорошо выраженные кристаллы, которые после сливания маточного раствора некоторое время высушивают в высоком вакууме. Выход 16,1—19,1 г (75—89%).
При быстром охлаждении до низкой температуры (ниже —20 °C) образуется мелкокристаллический или даже порошкообразный продукт, чрезвычайно неустойчивый иа воздухе (образование пирофорного Со4(СО)121).
Способ 3
2Со + 8СО --------> С02(СО)8
2-58,9 8-22,4л 342,0
Порошок Со, используемый в реакции, предварительно активируют водородом. Шланг от вентиля водородного баллона сначала соединяют с промывной склянкой, ничем не заполненной. Затем присоединяют промывную склянку с конц. H2SO4. Далее газовый шланг присоединяют к кварцевой трубке, закрепленной горизонтально, с колпачком на конце. Аппаратуру замыкают ртутным затвором, конец которого через шланг выведен в вытяжной шкаф. В кварцевую трубку помещают 58,9 г (1,0 моль) Со. Затем ток Н2 регулируют таким образом, чтобы пузырьки газа можно было считать. Через 30 мин воздух из системы будет вытеснен. Теперь осторожно нагревают трубку до 250—300 °C и при этой температуре продувают Н2 в течение 6 ч. По окончании восстановления систему охлаждают н заменяют ток Н2 током N2. Активированный порошок Со в токе N2 переносят в трубку Шленка, также предварительно заполненную азотом.
Во вращающийся автоклав на 250 мл в атмосфере N2 помещают 20,0 г (0,34 моль) активированного порошка Со. После однократного продувания системы СО при —100 бар в автоклаве создают рабочее давление 450 бар, нагревают до 130 °C и при этой температуре выдерживают 3 дия. Затем автоклав охлаждают, сжигают избыток СО и однократно продувают N2. В атмосфере азота образовавшиеся оранжевые (или цвета охры) кристаллы извлекают шпателем из автоклава и возгоняют в высоком вакууме при 25—30 °C. Препарат хранят в атмосфере азота в темноте. Выход 40 г (69%).
Свойства. Мелкие кристаллы оранжево-красного цвета, при длительном хранении на воздухе разлагаются; крупные кристаллы — красного цвета. В темноте и инертной атмосфере при —35 °C сохраняются неограниченно долго. При нагревании свыше 50 °C происходит быстрое разрушение с образованием Со4(СО)12. ИК («-гексан): 2071 (с.), 2069 (с.-оч. с.), 2059 (ср.), 2044 (оч. с.), 2042 (оч. с.), 2031 (с.), 2023 (с.-оч. с.), 1867 (ср.), 1858 (ср.) [v(CO)] см-1. Кристаллическая структура: см. [7]; d(Co—Со) =2,524 А; два мостиковых СО-лиганда, в растворе диссоциация и равновесие с немостиковой структурой.
ЛИТЕРАТУРА
1. Szabo Р., Marko L., Bor G„ Chem. Techn. Berlin, 13, 549 (1961).
2. Wender I., Sternberg H. W„ Metlin S„ Orchin M„ Inorg. Synth., 5, 190 (1957).
3. Mond L„ Hirtz H„ Cowap M. D„ Z. Anorg. Chem., 68, 207 (1910).
1948 Глава 33. Металлоорганические комплексы
4. Hieber W., Schulten Н„ Marin R., Z. Anorg. Allgem. Chem., 240, 261 (1939).
5. Blanchard A. A., Gilmont P., J. Amer. Chem. Soc., 62, 1192 (1940).
6. Wender 1., Greenfield H. W., Orchin M., J. Amer. Chem. Soc., 73, 2656 (1951).
7. Sumner G. G., Klug H. P., Alexander L. E., Acta Crystallogr., 17, 732 (1964).
□ □ Додекакарбонилтетракобальт Co4(CO)i2
3Co2(CO)8 + 2Co(C6H7O2)2 + 2Ha --> 2Co4(CO)12 + 4C5H8O2
3-342,0 2-257,1 2-22,4л 2-571,9 4-100,1
(C5H8O2 = ацетилацетон)
В качающийся автоклав на 500 мл помещают 34,2 г (0,1 моль) Co2(CO)g, 17,0 г (66 ммоль) ацетилацетоната Со(П) и 150 мл «-гептана. Реакционную смесь качают (или перемешивают*) 2 ч при 30 °C. Затем давление Н2 в системе доводят до 60 бар и выдерживают при 30 °C еще 8 ч. По окончании реакции избыток Н2 сжигают и содержимое автоклава фильтруют в атмосфере N2. Продукт реакции в виде черного кристаллического порошка несколько раз промывают охлажденным до —70 °C «-пентаном. Затем уже достаточно чистый продукт сушат в вакуум-эксикаторе. При очистке возгонкой в высоком вакууме при 80—90°C заметны потери (образование пирофорного Со!). Перекристаллизация возможна только для малых порций, так как соединение обладает плохой растворимостью.
Другие способы. Термолиз Сог(СО)8 в инертной атмосфере; получение больших количеств чистого Co4(CO)i2 этим методом неудобно [3].
Свойства. Черные кристаллы, чувствительные к кислороду воздуха. Вещество плохо растворяется в неполярных органических растворителях, в тетрагидрофуране разлагается. ИК («-гексан): 2103 (сл.), 2063 (оч. с.), 2055 (оч. с.), 2038 (ср.), 2027 (ср.), 2018 (пл.), -1990 (сл.), 1898 (оч. сл.), 1867 (ср.), 1831 (сл.) [v(CO)] см-1. Кристаллическая структура: см. [4]; Со«-тетраэдр, 4 (Со— Со) =2,49 А.
ЛИТЕРАТУРА
1. Ercoli R., Chini Р., Massi-Mauri М., Chim. Ind. (Milano), 41, 132 (1959).
2. King R. В., Organometallic Syntheses, v. 1, p. 103, Academic Press, New York, London, 1965.
3. Mond L., Hirtz H., Cowap M. D., J. Chem. Soc., 1910, 798; Z. Anorg. Chem., 68, 207 (1910).
4. Wei С. H„ Dahl L. F., J. Amer. Chem. Soc., 88, 1821 (1966).
□ □ Тетракарбонилкобальтат(—I) таллия TI[Co(CO)4]
Co2(CO)8 + 2T1 -----► 2T1[Co(CO)4]
342,0 2-204,4 2-375,4
4,0 г (11,7 ммоль) Co2(CO)g растворяют в 50 мл толуола и 3 ч перемешивают с 6,0 г (29,4 ммоль) Т1-стружки. Цвет раствора изменяется от темно
* Поскольку гидрирование идет при относительно низком давлении, реакцию можно проводить в лабораторном автоклаве на 200 мл с малыми загрузками, используя перемешнванне магнитной мешалкой.
Карбонилы металлов 1949
красного до желто-коричневого. Его фильтруют (стеклянный пористый фильтр G3), упаривают до половины объема и высаживают желтый Т1[Со(СО)4] добавлением 100 мл петролейного эфира. Осадок отфильтровывают и многократно промывают на фильтре петролейным эфиром для удаления следов Т1[Со(СО)4]3. Выход 7,0 г (80%).
Другие способы получения. Из Na [Со (СО) 4] и T1NO3 с 51%-ным выходом, а также из Hg[Co(CO)4]2 и металлического Т1 с выходом 57% [3].
Свойства. Мелкие кристаллы желтого цвета. tn„ 140—142 °C. Растворяется в полярных органических растворителях, не растворяется в углеводородах. Растворы очень неустойчивы на воздухе. ИК (СН2С12): 2043 (с.), 1972 (ср.), 1937 (оч. с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1 Pedersen S. Е., Robinson П7. R., Schussler D. Р., J. Organometal. Chem., 43, С44 (1972).
2. Schussler D. P„ Robinson W. R., Edgell W. F., Inorg. Chem., 13, 153 (1974).
3. Burlitch J. M., Theyson T. W., J. Chem. Soc. Dalton, 1974, 828.
□ □ Бис(тетракарбонилкобальтат) тетракис(пиридин)магния [Mg(C5H5N)4] [Со(СО)4]г
Получение основано на восстановительном расщеплении связи М—М в Со2(СО)в-
Mg-Hg
Со2(СО)8 4-Mg + 4C5H6N ----->- [Mg(C5H5N)4] [Со(СО)4]2
342,0 24,3 4-79,1 682,7
В сухой колбе на 250 мл готовят 1%-ную амальгаму путем интенсивного перемешивания 50 г Hg и 0,5 г (20,6 ммоль) Mg-порошка. После охлаждения до комнатной температуры добавляют раствор 3,40 г (9,9 ммоль) Со2(СО)в в 40 мл бензола и 5,0 г (63,2 ммоль) пиридина. Колбу неплотно закрывают и содержимое перемешивают 20 ч. Реакционная смесь изменяет цвет от красно-коричневого до темно-желтого. Раствор фильтруют через стеклянный пористый фильтр (G3), покрытый ватой, промывают бензолом, фильтрат упаривают в вакууме масляного насоса до 20—30 мл и разбавляют 100 мл н-пентаиа. Выпавший желтый порошок отфильтровывают на стеклянном пористом фильтре (G3), перекристаллизовывают из горячего бензола и сушат в высоком вакууме. Выход 4,8—5,8 г (70—85%).
Свойства. Желтое кристаллическое вещество, чувствительное к кислороду я влаге воздуха, при нагревании до ~ 150 °C разлагается, не плавясь. Растворяется в большинстве органических растворителей. В растворе тетрагидрофурана практически не диссоциирует. ИК (бензол): 2016 (ср.), 1939 (с.), 1764 (с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. McVicker G. V., Inorg. Synth., 16, 58 (1976).
2. Slaugh L. H„ Mullineaux R. D., J. Organometal, Chem., 13, 469 (1968).
3. McVicker G. V., Matya R. S„ Chem. Commun., 1972, 972.
1950 Глава 33. Металлоорганические комплексы
□ Додекакарбонилтетрародий Rh4(CO)i2
Rh4(CO)12 получают карбонилированием [RhCl(СО)2]2 при нормальном давлении (способ 1) или при высоком давлении (способ 2) в условиях, исключающих образование Rhs(C0)i6-
Способ 1
2[RhCl(CO)2]2 + 4СО ---> Rh4(CO)12
2-388,8 4-22,4л 747,7
В трехгорлую колбу на 500 мл, предварительно прогретую в высоком вакууме, снабженную обратным холодильником и трубкой для ввода газа, помещают 700 мг (1,8 ммоль) [RhCl(CO)2]2 и 2,0 г (23,8. ммоль) NaHCO3> высушенного при 100 °C.
Систему дважды попеременно вакуумируют и продувают током СО (высушенным над Р4О10), затем добавляют 300 мл абсолютного гексана. При перемешивании магнитной мешалкой в течение 2 ч через раствор продувают слабый ток СО. Затем добавляют к раствору 1 каплю (—0,02 мл) Н2О и еще 2 ч продувают ток СО. Избыток Н2О нежелателен, так как иначе образовался бы Rh6(CO)16 и выделился металлический Rh. После добавления воды цвет раствора быстро изменяется до темно-красного. По окончании карбонилирования раствор фильтруют, фильтрат упаривают в вакууме водоструйного насоса до —70 мл и кристаллизуют при температуре сухого льда. Темно-красные кристаллы перекристаллизовывают при той же температуре из смеси толуол +гексан (1:1) и затем сушат в высоком вакууме. Выход аналитически чистого Rh4(CO)i2 колеблется между 40 и 65%, однако, как правило, состав* ляет 55—60% (—400 мг).
Способ 2
Си
2[RhCl(CO)2]2 + 4СО --->- Rh4(CO)12
2-388,8 4-22,4л 747,7
Смесь 4,0 г (10,3 ммоль) [RhCl(CO)2]2 и 120 г (избыток) тонкой медной, проволоки помещают в стеклянный автоклавный вкладыш и заливают 150 мл. «-гексана. Реакционную смесь выдерживают в течение 4 дней в качающемся однолитровом автоклаве прн давлении СО 200 бар. Затем темно-красный раствор сливают с Cu-проволоки и охлаждают до —10 °C. Выход Rh4(CO)i2, составляет 2,0 г (52%). Перекристаллизацией из горячего гексана получают аналитически чистый препарат.
Другие способы. Карбонилирование при высоком давлении безводного-RhCl3 при 50—80 °C в присутствии металлических Си, Cd или Zn [3].
Свойства. Кирпично-красные устойчивые на воздухе кристаллы. /пл 76 °C,, свыше 150 °C разлагается. Плохо растворяется в алифатических растворителях, умеренно растворяется в бензоле и эфире. Разлагается при обработке концентрированными кислотами и щелочами, в последнем случае с образованием металлического Rh. При нагревании до 100 °C в атмосфере N2 разлагается до Rh6(СО) 16. ИК (гексан): 2075 (с.), 2070 (с.), 2062 (пл.), 2044 (ср.), 2016 (сл.), 1978 (сл.), 1885 (с.) [v(CO)] см-1. При загрязнении Rh6(CO)15, появляется дополнительно полоса 1811 см-1. Кристаллическая структура: см. [4]; d(Rh—Rh) =2,73 А.
ЛИТЕРАТУРА
1. Cattermole Р. Е„ Osborne A. G., Inorg. Synth., 17, 115 (1977).
2. Chaston S. H. H., Stone Р. G. A., J. Chem. Soc. (London) A, 1969, 500.
3. Hieber W., Lagally H., Z. Anorg. Allgem. Chem., 251, 96 (1943).
4. Wei С. H„ Wilkes G. R„ Dahl L. F„ J. Amer. Chem. Soc., 89, 4792 (1967).
Карбонилы металлов 1951
□ Гексадекакарбонилгексародий Rh6(CO)i6
Rh6(CO)I6 можно получить с хорошими выходами либо при нормальном давлении из тетраацетатодиродия (II) (способ 1), либо карбонилированием RhCl3-3H2O при высоком давлении (способ 2).
Способ 1
3Rha(CH3COa)4 + 22СО + 6Н2О-----> Rh6(CO)ie + 6СОа + 12СН8СОаН
3-442,0 22-22,4л 6-18,0 1065,6 6-22,4л 12-60,0
В двугорлую колбу на 250 мл, снабженную обратным холодильником и трубкой для ввода газа, помещают 3,0 г (6,8 ммоль) тетраацетатоднродия(П) [3], 150 мл пропанола и 6,0 мл 40%-ной HBF4 в Н2О. Пропуская слабый ток СО и перемешивая магнитной мешалкой, смесь нагревают 20 ч при 75—80 °C (температура водяной бани). При более высокой температуре выходы резко падают из-за значительного разложения в этих условиях. В процессе реакции Выпадает мелкий фиолетово-коричневый кристаллический осадок Rh6(CO)i6. По окончании реакции оранжево-коричневую суспензию охлаждают в атмосфере СО до комнатной температуры, отфильтровывают на стеклянном фильтре, промывают многократно малыми порциями охлажденного метанола и, наконец, сушат в высоком вакууме при ~50°С. Выход 2,1 г (87%).
Способ 2
6RhCl8 • ЗН2О + 25СО --> Rh6(CO)le + 9СОС1а + 18Н2О
6-263,3 25-22,4л 1065,6 9-98,9 18-18,0
В 1-литровый качающийся автоклав помещают 3,0 г (11,4 ммоль RhCl3-3H2O и 400 мл 90%-ного C2HsOH, создают давление СО 40 бар и нагревают при 60 °C в течение 4 дней. Далее охлаждают до комнатной температуры, сжигают избыток СО и смывают метанолом черный кристаллический продукт на воронку Бюхнера. После отсасывания и промывания горячим гексаном получают черные кристаллы. Высушенный в высоком вакууме препарат является аналитически чистым. Выход 1,8 г (89%).
Другие способы. Карбонилирование безводного RhCl3 при высоком давлении в присутствии металлических Си, Cd или Zn при 80—230 °C [4].
Свойства. Фиолетово-коричневые, блестящие, устойчивые на воздухе кристаллы; крупные кристаллы кажутся черными. Соединение разлагается при температуре свыше 200 °C. Не растворяется в алифатических растворителях, плохо растворяется в других обычных растворителях. Комплекс устойчив к действию кислот и щелочей. При нагревании выше —220 °C на воздухе образует металлическое зеркало. ИК (нуйол): 2075 (с.), 2025 (ср.), 1800 (с.) [v(CO)] см-1. Кристаллическая структура: см. [5], Rhe-октаэдр; d(Rh—Rh) = =2,776 А; 4 СО-группы, находящиеся над плоскостями родиевого октаэдра, образуют мостик между тремя атомами родия; по 2 концевые СО-группы у каждого Rh.
литература
1. James В. R„ Rempel G. L„ Тео W. К., Inorg. Synth., 16, 49 (1976).
2. Chaston S. H. H„ Stone F. G. A., J. Chem. Soc. (London) A, 1969, 500.
3. Rempel G. L., Legzdins P., Smith H„ Wilkinson G., Inorg. Synth., 13, 90 (1972).
4. Hieber W„ Legally H., Z. Anorg. Allgem. Chem., 251, 96 (1943).
•5. Corey E. R., Dahl L. F., Beck W., J. Amer. Chem. Soc., 85, 1202 (1963).
1952 Глава 33. Металлоорганические комплексы
Ароматические сандвичевые комплексы
□ □ Бис(т1-циклопентадиенил)ванадий ¥(т)-С5Н5)2 (Ванадоцен)
VC1S + 3NaC5H5 --> V(r]-C5H5)2 + 3NaCl + [C5H5-J
157,3 3-88,1 181,1 3-58,4
В трехгорлую колбу на 1 л, снабженную обратным холодильником и эффективной мешалкой, помещают 66,0 г (0,75 моль) NaC5H5 в 500 мл тетрагидрофурана и порциями постепенно прибавляют 42,5 г (0,27 моль) безводного VC13 с такой скоростью, чтобы растворитель чуть-чуть кипел. Всегда наготове следует иметь ледяную баню, так как реакция неожиданно может разойтись! (При небольших загрузках можно интенсивно перемешивать магнитной мешалкой.) Для завершения реакции смесь нагревают еще 2 ч. После охлаждения до комнатной температуры растворитель упаривают в вакууме водоструйного насоса, остаток высушивают в высоком вакууме и, наконец, выделяют продукт высоковакуумной возгонкой при 100—150 °C. Выход 22,5—26,9 г (46— 55%).
Другие способы. 1) Взаимодействие VC13 с цнклопентадиенилмагнийбро-мидом (молярное соотношение 1 :5) в кипящем эфире. Выход 33—40% [2]. 2) Взаимодействие УС12-2ТГФ (нз VCl3+Zn) с NaC3H5 (или его замещенными производными) в тетрагидрофуране. Выход 60—68% [3]. 3) Очень чистый V(r|-C5H5)2 можно получить в количестве 100—250 г (выход 78—86%) нэ УС13+ТГФ+1лА1Н4+ЫаС5Н5 [4].
Свойства. Фиолетово-черные, очень неустойчивые на воздухе кристаллы, /пл 167—168 °C. Очень хорошо растворяется в тетрагидрофуране, в растворе которого, как правило, и применяется в дальнейших синтезах. Кристаллическая структура: см. [5].
ЛИТЕРАТУРА
1. Wilkinson G„ Cotton F. A., Birmingham J. M., J. Inorg. Nucl. Chem., 2, 95 (1956).
2. Fischer E. 0., Hafner W., Z. Naturforsch., 9b, 503 (1954).
3. Kohler F. H., Prossdorf W., Z. Naturforsch., 32b, 1026 (1977).
4. Handlir К, Holecek J., Klikorka J., Z. Chem., 19, 265 (1979).
5. ITeiss E„ Fischer E. 0., Z. Anorg. Allgem. Chem., 278, 219 (1955).
О (т|-Циклогептатриенил) (ц-циклопентадиенил) ванадий
V(T]-C6H6) (СО)4 + С7Н8---> V(ryC7H7) (П-С5Н5) + 4СО + 1/2Щ
228,1 92,1 207,2 4-22,4л 11,2л
Смесь 4,56 г (20 ммоль) Vfrj-CsHs) (СО)4 и 60 мл (избыток) продажного циклогептатриена в течение 9 ч кипятят с обратным холодильником. После отгонки не вошедшего в реакцию циклогептатриена в вакууме масляного насоса при ~30°С образуется черный остаток, который после промывания ~10 мл охлажденного льдом эфира очищают возгонкой в высоком вакууме. Первоначально возгоняющиеся при 50—80 °C маслянистые загрязнения выбрасывают; основной продукт возгоняется при 100 °C. Выход 1,6—2,4 г (40— 60%).
Свойства. Пурпурные кристаллы. В течение короткого времени комплекс устойчив иа воздухе. Плохо растворяется в обычных органических раствори
Ароматические сандвичевые комплексы 1953»
телях. Растворы чрезвычайно чувствительны к следам кислорода. Парамагнитен (р.= 1,69 магнетона Бора). Кристаллическая структура: см. [3]; оба п овязанных кольца расположены симметрично и параллельно друг другу.
ЛИТЕРАТУРА
1. King R. В., Stone F. G. A., J. Amer. Chem. Soc., 81, 5263 (1959).
2. King R. В., Organometallic Syntheses, v. 1, p. 140, Academic Press, New York... London, 1965.
3. Engebretson G., Rundle R. E., J. Amer. Chem. Soc., 85, 481 (1963).
□ □ Бис(т]-циклопентадиенил)хром Cr(Ti-C5H5)2
CrCl3 + 3NaC6H5 --> Cr(n-C6H6)2 + 3NaCl+[C5H6.]
158,4 3-88,1 182,2 3-58,4
В 2-литровую трехгорлую колбу, снабженную обратным холодильником и эффективной мешалкой, помещают раствор 88,1 г (1,0 моль) NaCsHs в 600 мл тетрагндрофурана и при интенсивном перемешивании порциями (примерно по 8 г) прибавляют 47,5 г (0,3 моль) безводного СгС13. Поскольку реакция экзотермическая, прн необходимости применяют охлаждение ледяной водой. Для завершения реакции смесь кипятят еще 2 ч. После охлаждения до комнатной» температуры полученную суспензию (зеленого или в идеальном случае краснофиолетового цвета) упаривают в вакууме масляного насоса, а затем в высоком вакууме избавляются от летучих побочных продуктов до получения совершенно сухого порошка. Остаток подвергают высоковакуумной возгонке при 130—150 °C и получают продукт в виде ярко-красных кристаллов. Выход. 26,8—31,7 г (49—58%).
Другие способы. 1) Взаимодействие СгС13 с MgBr(CsHs) (молярное отношение 1:4) в кипящем эфире. Выход 60—70% [2]. 2) В препаративном отношении менее предпочтительным (выход ~30%) является взаимодействие-паров циклопента диен а с Сг(СО)6 в трубке для сожжения [3, 4]. 3) Термическое расщепление '[Cr(NH3)s] (CsHs)3 также препаративно менее удобно (выход <10%) [5]. 4) Особенно чистый продукт получают при взаимодействии небольших количеств (1—2 г) СгС12-ТГФ (из Сг+НС1) и NaCsHs (или замещенных производных, например NaC5H4CH3) в тетрагидрофуране. Выход 75— 83% [6]. 5) Очень чистый Cr^-CsHsJj в количествах 100—250 г (с выходом до 86%) получают из смеси CrCl3+Tr®+LiAlH4+NaC6Hs [7].
Свойства. Ярко-красные, чрезвычайно неустойчивые на воздухе кристаллы. <Пл 173—174 °C. Очень хорошо растворяется в тетрагидрофуране. Взаимодействием с точно дозируемым количеством кислорода получают тетрамерный,: также неустойчивый на воздухе окснд [Сг(С5Н5)О]4 [8]. Кристаллическая структура: см. [9].
ЛИТЕРАТУРА
1. Wilkinson G., Cotton F. A., Birmingham J. M., J. Inorg. Nucl. Chem., 2, 95» (1956).
2. Fischer E. O., Hafner W., Stahl H. O., "L. Anorg. Allgem. Chem., 282, 47'
3. Birmingham J. M„ Seyferth D., Wilkinson G„ J. Amer. Chem. Soc., 76, 4179* (1954).
4. Wilkinson G., J. Amer. Chem. Soc., 76, 209 (1954).
5. Fischer E. O., Hafner W„ Z. Naturforsch., 8b, 444 (1953).
7—1361
4954 Глава 33. Металлоорганические комплексы
-6. Kohler F. Н„ Prossdorf Г., Z. Naturforsch., 32b, 1026 (1977).
7. Handlir К-, Holecek J., Klikorka J., Z. Chem., 19, 265 (1979).
8. Fischer E. 0., Ulm K-, Fritz H. P., Chem. Ber., 93, 2167 (1960). '9. Weiss E., Fischer E. O., Z. Anorg. Allgem. Chem., 284, 69 (1956).
□ □ Дибензолхром Сг(т]-С6Нб)2
Дибензолхром получают по восстановительному методу Фишера — Хаф-яера взаимодействием безводного СгС13 и бензола в присутствии мезитилена Ф качестве катализатора.
а) ЗСгС18 4- 2А1 AlClg 4“ 6С8Н8 * 3[Сг(т]-С8Н8)21 [AICI4]
3-158,4 2-27,0 133,3 6-78,1 3-377,0
б) г^п-СвН^] [А1С14] 4- NaaS2O4 4- 12КОН ---->
2-377,0 174,1 12-56,1
----> 2Cr(r]-CeHe)2 4- Na2SO8 4- K2SO8 4- 2К[А1(ОН)4] 4- 8КС1 4- 2Н2О 2-208,2 126,0 158,3 2-134,1 8-74,6 2-18,0
а) В двугорлую колбу на 250 мл, предварительно прогретую при —300 °C, -снабженную обратным холодильником, при тщательной защите от кислорода воздуха помещают 12,7 г (80 ммоль) порошкообразного СгС13*, 1,75 г (65 ммоль) тонкого порошка А1 и 30,0 г (0,23 моль) свежевозогнаниого А1С13. После добавления 80 мл абсолютного бензола и 10 капель (—0,25 мл) абсолютного мезитилена смесь кипятят в течение 30—35 ч при перемешивании магнитной мешалкой.
б) За это время в 4-литровой трехгорлой колбе, снабженной эффективной мешалкой, готовят смесь ПО г КОН, 650 мл дистиллированной Н2О, 250 мл СНзОН и 1000 мл С6Нв и в атмосфере N2 прибавляют 70,0 г (0,40 моль) Na2S2O4.
Содержимое (желто-зеленого цвета) реакционной (двугорлой) колбы предварительно охлаждают и при интенсивном перемешивании вливают в заранее приготовленную, охлаждаемую льдом смесь в трехгорлой колбе. Остающиеся твердые частички на стенках с помощью шпателя также переносят в колбу. После 2-часового интенсивного перемешивания бензольную фазу отделяют, сушат КОН и упаривают в роторном испарителе. Остаток подвергают высоковакуумной возгонке при 150—170 °C. Выход 13,3—15,3 г (80—92%).
Если необходимо получить продукт, свободный от мезитилена, то комплексообразование проводят в запаянной трубке при 140 °C. В ампулу иа 60 моль помещают 5,0 г (32 ммоль) порошка СгС13,0,7 г (26 ммоль) порошка А1, 12,0 г (90 ммоль) свежевозогнаниого А1С13 и 24 мл абсолютного бензола. В вакууме отгоняют около 1/4 бензола и запаивают ампулу (в вакууме) при охлаждении ее до —190°C. После нагревания до комнатной температуры -смесь хорошо встряхивают, помещают в предохранительный железный патрон и при медленном вращении в течение 6 ч нагревают при 140 °C. Рекомендуется поместить защитный патрон в сушильном шкафу и приспособить его так, что-бы можно было вращать вокруг собственной оси системой приводов, используя для этого воздушное отверстие в сушильном шкафу. Последующую обработку содержимого ампулы после ее охлаждения до комнатной температуры ^проводят так, как это описано выше в п. «б».
* Безводный СгС13 кипятят с 2 н. НС1, хорошо промывают и высушивают.
<См. получение СгС13.)
Ароматические сандвичевые комплексы 1955»
Свойства. Черно-коричневые неустойчивые на воздухе кристаллы.
284—285 °C. Выше — 300 °C разлагается при обычном давлении, выше 250 °C — в высоком вакууме. Непрерывно возгоняется в высоком вакууме при 160 °C. Кристаллическая структура: см. [5].
ЛИТЕРАТУРА
1. Fischer Е. О., Hafner W., Z. Naturforsch., 10b, 665 (1955).
2. Fischer E. 0., Seeholzer J., Z. Anorg. Allgem. Chem., 312, 244 (1961).
3. Fischer E. O., Inorg. Synth., 6, 132 (1960).
4. Fischer E. O., Inorg. Synth., 7, 136 (1963).
5. Weiss E., Fischer E. O., Z. Anorg. Allgem. Chem., 284, 69 (1956).
□ □ Бис(циклопентадиенил)марганец Mn(C5H5)2
MnCl2 + 2NaC8H5 ----> Mn(C6H5)a + 2NaCl
125,8 2-88,1 185,1 2-58,4
а) Для приготовления безводного MnCh продажный MnC12-4H2O в течение нескольких дней сушат в сушильном шкафу при 180 °C. Затем тонко* измельчают в шаровой мельнице, 10 ч кипятят с SOC12 и, наконец, высушивают не менее 10 ч в высоком вакууме при 180 °C.
б) В 2-литровую двугорлую колбу, снабженную обратным холодильником и магнитной мешалкой*, помещают свежеприготовленный раствор 88,1 г (1,0 моль) NaCsHj в 500 мл тетрагидрофурана и порциями добавляют 62,9 г (0,5 моль) безводного МпС12 [см. п. «а»]. После 12-часового кипячения растворитель отгоняют в вакууме масляного насоса в охлаждаемую ловушку, а темно-коричневый остаток высушивают в высоком вакууме до порошкообразного состояния. Высоковакуумной возгонкой при температуре не выше 150 °C получают темно-коричневый кристаллический Мп(С5Н5)2. Выход 60— 70 г (65—72%). Успех синтеза определяется абсолютной безводностью исходных материалов и тщательным предохранением от доступа кислорода воздуха.
Свойства. Чрезвычайно неустойчивые на воздухе и в присутствии следов влаги кристаллы янтарного цвета, которые прн 158 °C переходят в порошкообразную модификацию красно-коричневого цвета, уже не пригодную для дальнейшего получения Мп(т]-С5Н5) (СО)3 (синтез см. ниже). 1пл 172—173 °C. На воздухе воспламеняется, особенно порошкообразный. Взаимодействует с FeCl2 с образованием ферроцена. Темно-коричневая модификация (пространственная группа симметрии Рпа 2t) имеет зигзагообразную полимерную структуру нз фрагментов Mn(C5Hs) с дополнительными мостиковыми С5Н5-лнган-дами [3].
ЛИТЕРАТУРА
1. Wilkinson G., Cotton F. A., Birmingham J. M„ J. Inorg. Nucl. Chem., 2, 95 (1956).
2. Arbeitsvorschrift des Anorganisch-Chemischen Institute der Universitat Regensburg.
3. Blinder W„ Weiss E., Z. Naturforsch., 33b, 1235 (1978).
* Чтобы смесь хорошо перемешивалась, используют длинный (32 мм У якорь магнитной мешалки. Для больших загрузок используют высокоэффективную мешалку с тефлоновым пропеллером.
7*
3956 Глава 33. Металлоорганические комплексы
Жис(т]-циклопентадиенил)железо Ре(т]-С5Н5)2
«(Ферроцен)
Синтез осуществляется в две стадии:
2FeCl3 + Fe ----> 3FeCl2
2-162,2 55,8 3-126,8
FeCla + 2C6He + 2(C2H6)2NH --> Fe(n-C6H6) + 2[(CaH6)NH2]Cl
126,8 2-66,1 2-73,1 186,0 2-108,6
16,2 г (0,1 моль) безводного FeCl3 (см. получение FeCl3) восстанавливают <в суспензии в 100 мл тетрагидрофурана кипячением в’ течение 5 ч в присутствии 2,79 г (50 ммоль) Fe-порошка. FeCl2 образуется в виде суспензии серо-•коричневого цвета. Растворитель досуха отгоняют в вакууме водоструйного насоса. При охлаждении льдом и интенсивном перемешивании добавляют к образовавшемуся остатку смесь 19,8 г (0,3 моль) циклопентадиена и 43,9 с (62,3 мл, 0,6 моль) диэтиламииа. Через 8 ч перемешивания при комнатной температуре упаривают содержимое в вакууме водоструйного насоса. Остаток обрабатывают петролейным эфиром (tKHn 40—60 °C) в аппарате Сокслета. Экстракт охлаждают до —35 °C; через несколько часов выпадает основное количество продукта в виде длинных темно-желтых кристаллов. Еще некоторое количество Fe(r]-C5H5)2 можно получить испарением маточного раствора. Общий выход 24,6—26,5 г (66—71%). Препарат можно очистить высоковакуумной возгонкой при 80—100 °C, однако для последующих синтезов такая очи-стка не является необходимой.
Другие способы. 1) Действие паров CsHe на нагретый Fe-порошок [3]. '2) Взаимодействие FeCl2 с MgBr(C5H5) [4].
Свойства. Совершенно устойчивые на воздухе ромбические или игольчатые кристаллы от темно-желтого до оранжевого цвета. /пл 173—174 °C. Термически устойчив при температурах свыше 500 °C. Растворяется в болыпин--стве органических растворителей. При действии сильных окислителей типа HNO3 окисляется до ферроцеиий-катиона Fe(r]-C5H5)2+; Fe(T|-C5H5)2-^-[Fe(r]-'CsHs)2]+ (£о=—0,3 В). Кристаллическая структура: см. [51; d(Fe—С) = =2,045 А.
ЛИТЕРАТУРА
1. Birmingham J. М., Seyferth D., Wilkinson G., J. Amer. Chem. Soc., 76, 4179 (1954).
2. Wilkinson G„ Org. Synth., 36, 34 (1956).
3. Miller S. A. et al., J. Chem. Soc. (London), 1952, 632.
4. Kealy T. J., Pauson P. L., Nature, 168, 1039 (1951).
• 5. Dunitz J. D„ Orgel L. E., Rich A., Acta Crystallogr., 9, 373 (1956).
Ферроцен как представитель ароматических углеводородов
Циклопентадиенильные группы в ферроцене и его гомологах рутеноцене •и осмоцене обладают ароматическим характером. Конкурентное ацилирование подтверждает большую реакционную способность ферроцена по сравнению с бензолом. Скорость ацилирования ферроцена превышает скорость ацилирования полусандвичевых комплексов Mn(T]-RCsH.() (СО)3 [R=H, СН3]. Другие типичные реакции электрофильного замещения доказывают тесное сходство этих комплексных систем с «классическими» ароматическими соединениями. В качестве примеров ниже рассмотрены ацилирование ферроцена по Фриде-.лю — Крафтсу и формилироваиие по Вильсмейеру.
Ароматические сандвичевые комплексы 1957
О 1,1'-Диацетилферроцен Ее(т|-С5Н4СОСНз)2
Специальным подбором условий реакции можно провести практически моноацилирование; последующее ацилирование идет преимущественно гетеро-аннулярно, так как ацильный заместитель вследствие своего акцепторного характера (—I/—М) значительно понижает электронную плотность в замещенном циклопентадиенильном кольце. Электроноакцепторный характер ацильного заместителя передается через атом металла на незамещенный лиганд, однако приводит лишь к незначительному ослаблению ароматичности незамещенного циклопентадиенильного кольца.
Ниже приводится пример преимущественного получения 1,Г-диацетилфер-роцена:
А1С13
Fefrj-CsHs^ 4" CHgCOCl >
186,0 78,5
----> Fe^-Cs^CCCHgJa (главный продукт) Ре(т]-С5Н4СОСН3) (т]-С5Н5) ф-270,1 228,1
+ Fe[T]-C6H8(COCH3)2J (П-СВН5) 270,1
Раствор 30,0 г (0,16 моль) ферроцена в 100 мл абсолютного CH2CI2 в атмосфере N3 приливают в течение 15 мин к интенсивно перемешиваемой смеси 53,0 г (0,40 моль) А1С13, 32 мл (0,45 моль) ацетилхлорида и 200 мл абсолютного CH2CI2, при этом наблюдается постоянное выделение НС1 и появление фиолетовой окраски раствора. После 2-часового перемешивания реакционную смесь разлагают ледяной водой, отфильтровывают от выпавшего А1(ОН)3 и многократно промывают СНС13. Органический слой отделяют, водный несколько раз экстрагируют СНС13. Объединенные органические вытяжки промывают водой до нейтральной реакции. Темно-красный раствор упаривают до объема —200 мл и разбавляют 100 мл циклогексана. При постепенном охлаждении до —35°C выпадают 24,4 г (56%) 1,1'-диацетилферроцена в виде рубиново-красных кристаллов, которые еще раз перекристаллизовывают.
Маточные растворы при необходимости можно подвергнуть хроматографической очистке на колонке в условиях, приводимых в оригинальной литературе. Получают еще 2,6 г 1,1'- и 1,1 г 1,2-диацетилферроцена, 1,9 г ацетилферроцеиа « 0,1 г ферроцена.
Свойства. 1,1'-Диацетилферроцен: рубиново-красные устойчивые на воздухе кристаллы. 1пл 130—131 °C. Плохо растворяется в эфире, циклогексане и воде, хорошо растворяется в хлороформе, тетрахлориде углерода и этилацетате.
По реакции Вильсмейера получают с хорошими выходами соответствующие альдегиды лишь для активированных ароматических соединений, таких, как эфиры фенолов или диалкнланилины. Бензол не формилируется в этих условиях. М. Розенблюм [1] применил реакцию Вильсмейера для ферроцена и тем самым подтвердил высокую реакционную способность последнего. Этим способом получают исключительно ферроценилальдегид; последующее форми-лированне вследствие сильной дезактивации формильной группой не происходит.
ЛИТЕРАТУРА
1. Rosenblum М., Woodward R. В., J. Amer. Chem. Soc., 80, 5443 (1958).
2. Broadhead G. D„ Osgerby J. M., Pauson P. L„ J. Chem. Soc. (London), 1958, 650.
1958 Глава 33. Металлоорганические комплексы
□ Формилферроцен Fe(r]-C5H5)(r]-C5H4CHO)
РОС13
Fe(T]-C6H6)a + C6H5N(CH3)CHO -->
186,0 135,2
--> Fe(n-C5H5) (т]-С6Н4СНО) +C6H3NHCH,
214,1 107,2
Смешивают 4,4 г (33 ммоль) N-метилформанилида н 5,0 г (33 ммоль) РОС13, 1 ч выдерживают при комнатной температуре (при этом образуется кристаллическая масса) и добавляют 20 мл хлорбензола. В атмосфере N2 за 15 мин при перемешивании добавляют небольшими порциями 2,99 г (16 ммоль) ферроцена и оставляют смесь стоять в закрытой колбе в течение —70 ч. После добавления ледяной воды смесь оставляют стоять еще 2 ч для завершения гидролиза комплекса и затем экстрагируют хлороформом.
Хлороформные вытяжки сушат безводным Na2SO4 и отгоняют растворитель в вакууме водоструйного насоса. Выделение ферроценилальдегида проводят хроматографически на А120з бензолом. Ферроценилальдегид отделяется от непрореагировавшего ферроцена, избытка формилирующего агента и прочих загрязнений на хроматографической колонке в виде красной полосы. После перекристаллизации из 25%-ного этанола или петролейного эфира (1ки» 40—60 °C) получают 2,56 г (75%) ферроценилальдегида.
Свойства. Коричнево-красные, устойчивые на воздухе кристаллы. tna 121—122 °C. Соединение перегоняется с паром н может быть очищено возгонкой в высоком вакууме при 70—80 °C.
ЛИТЕРАТУРА
1. Schlogl К- Monatsh. Chem., 88, 601 (1957).
2. Jutz Ch., Tetrahedron Lett., 1959, 1.
3. Broadhead G. D., Osgerby J. M., Pauson P. L., J. Chem. Soc. (London), 1958, 650.
Гексафторофосфат бис(т)-циклопентадиенил)железа(1П) [Fe(r)-C5H5)2] [PF6]
Тетрафтороборат (трацетилциклопентадиенил) (трцик-лопентадиенил)железа(Ш) [Ре(т]-С5Н4СОСН3)(т]-С5Н5)] [BF4]
Ферроцен и его производные легко окисляются различными методами с образованием солей ферроцения. В случае ферроцена или алкилферроцена это удается сделать действием конц. H2SO4; другим удобным в применении окислителем является AgBFi. Имеется исчерпывающий подбор литературы по окислению ферроцена и его производных [6].
I. Получение [Fe (n-C5H6)2] [PFJ
2Fe(n-C5H5)2 + 3HaSO4 + 2NH4PF, -->
2-186,0 3-98,1 2-163,0
---> 2[Fe(n-C5Hs)2] [PF6] + SO2 + 2NH4HSO4 + 2HaO 2-331,0 64,1 2-115,1 2-18,0
2,0 г (10,7 ммоль) ферроцена перемешивают в течение 15 ч при комнатной температуре с 20,0 мл (0,37 моль) конц. H2SO4. Темно-синюю реакционную смесь разбавляют водой до 200 мл, охлаждают, фильтруют и к фильтрату прибавляют концентрированный водный раствор 1,8 г (11,0 ммоль) NH4PFe.
Ароматические сандвичевые комплексы 1959
Выпавший осадок отделяют на пористом стеклянном фильтре (G3), промывают водой до нейтральной реакции и отсасывают досуха. Затем растворяют в 50 мл ацетона, добавляют 25 мл метанола, фильтруют и после промывания (3x25 мл эфира) высушивают в вакууме. Выход 3,1—3,3 г (87—92%).
Свойства. Темно-синие кристаллы, проявляют дихроизм. Хорошо растворяется в ацетоне, ацетоннтрнле н нитрометане, умеренно растворяется в ме-тиленхлориде и эфире. 210 °C (с разл.).
II. а) Получение AgBF4 (см. также том 1)
Ag2CO3 + 2HF -----> 2AgF -f- Н2О + СО; AgF + BF3 -----> AgBF4
275,5 2-20,0 2-126,9 18,0 22,4л 126,9 67,8 194,7
К 136 г (0,80 моль) AgNO3, растворенным в 400 мл Н2О, при интенсивном перемешивании и защите от прямого света добавляют раствор 38 г NaOH в 1200 мл Н2О, предварительно насытив его СО2. Выпавший Ag2CO3 после декантации помещают еще во влажном состоянии в тефлоновый стаканчик. Интенсивно перемешивая, порциями прибавляют 47 г 40%-ной HF (0,94 моль) и после прекращения выделения газа нагревают смесь на водяной бане в течение 30—40 мнн, пока масса не составит примерно 115 г. После охлаждения Добавляют 100 мл абсолютного метанола, хорошо перемешивают н декантируют с осадка. Эту операцию повторяют еще дважды. Остаток трижды взбалтывают с эфиром (по 100 мл), декантируют н еще во влажном состоянии переносят образовавшийся светло-коричневый продукт в колбу емкостью 250 мл и высушивают в вакууме, причем в конце нагревают до 60—70 °C. Из •объединенных метанольно-эфирных вытяжек после декантации получают еще некоторое количество желтоватого продукта, который промывают декантацией дважды по 25 мл метанола и трижды по 50 мл эфира н высушивают в вакууме. Общий выход составляет 90 г (89%). Соль гигроскопична и чувствительна к свету.
Последующие операции проводят, избегая влаги воздуха.
Суспендируют 90 г (0,71 моль) AgF в 180—200 мл нитрометана и при перемешивании пропускают через раствор ток газообразного BF3 (20 мл/мин) до тех пор, пока AgF полностью не растворится (80 мин). Избыток BF3 вытесняют током N2, не давая растворителю отгоняться. Темный раствор фильтруют в непрозрачную колбу и растворитель удаляют в вакууме; колбу при этом часто встряхивают, чтобы не образовалась прочная корочка соли. Через 15 ч высушивания в вакууме продукт размельчают под слоем пентана в атмосфере N2, защищая от света. Полученную соль сохраняют в темноте под слоем пентана. Выход 117 г (85%). AgBF4 бесцветен, гигроскопичен и светочувствителен. Хорошо растворяется в воде, эфире, толуоле, умеренно растворяется в бензоле и циклогексане, не растворяется в насыщенных углеводородах.
б) Получение i[Fe(T]-C6H4COCH3) (т)-С5Н5)] [BF4]
Fe(n-C5H4COCH8)(T)-CliHs) + AgBF4---->
228,1 194,7
----> [Fefo-C^COCH,) (т]-С8Н5)] [BF4] + Ag 314,9 107,9
Раствор 2,1 г (9,25 ммоль) ацетилферроцена в 50 мл эфира медленно прикапывают к раствору 1,80 г (9,25 ммоль) AgBF4 в 100 мл эфира. Черный осадок ацетилферроцениевой соли и мелкодисперсного Ag собирают на стеклянном пористом фильтре (G4) и промывают эфиром (5x30 мл). Ферроце-ниевую соль с фильтра смывают ацетоном, высаживают эфиром, отфильтровывают и сушат в вакууме. Выход 2,62 г (90%).
Свойства. Темно-синне дихроичные кристаллы, чувствительные к влаге. Растворяется в ацетоне, не растворяется в ТГФ, СН3ОН и эфире. Растворы в ацетоне быстро разрушаются. 110—111 °C (с разл.).
1960 Глава 33. Металлоорганические комплексы
ЛИТЕРАТУРА
1. Nesmeyanov A. N„ Materikova R. В., Lyatifov I; R., Kurbanov Т. Kh., Kochetkova N. S., J. Organometal. Chem., 145, 241 (1978).
2. Kemmit A. D. W., Sharpe D. W. A., J. Chem. Soc., 1961, 2496.
3. Olah G. A., Quinn H. W., J. Inorg. Nucl. Chem., 14, 295 (1960).
4. Laborvoschrift G. A. Wright, Department of Chemistry, The University Sheffield S3 7HF England.
5. Carty P., Dove M. F. A., J. Organometal. Chem., 28, 125 (1971).
6. Gmelins Handbuch der Anorganischen Chemie, Erganzungswerk zur 8. Auflage, Springer, Berlin usw., Bd. 14 (1974), Bd. 49 (1977), Bd. 50 (1978).
□ □ Гексафторофосфат (т]-циклопентадиенил)(т|-бен-зол)железа(П)[Ее(т)-С5Н5)('п-С6Нб)] [PFe]
Aicia+Ai
Fe(n-C5HS)2 + C6H6 + NH4PFe --------> [Fe(n-C5H6) (п-СвН6)] [PFe]
186,0 78,1 163,0 344,0
В трехгорлую колбу в атмосфере N2 помещают 7,5 г (40 ммоль) ферроцена, 27 г безводного А1С1з, 2 г Al-порошка и 100 мл абсолютного бензола, не содержащего тиофена; смесь перемешивают и оставляют на ночь кипеть на. плитке (минимальный нагрев). Цвет реакционной смеси изменяется от желто-зелеиого до красно-коричневого. Если используют недостаточно сухой бензол,, вначале наблюдается выделение HCL
Смесь охлаждают на ледяной бане до комнатной температуры и гидролизуют 200—250 мл ледяной воды. Поскольку эта реакция сильно экзотермическая, воду сначала добавляют по каплям, при необходимости колбу охлаждают на ледяной бане. Затем воду прибавляют быстрее, но интенсивно перемешивают. Избыток А1 отделяют на воронке Бюхнера. В фильтрате образуются две фазы, которые разделяют в делительной воронке. Верхнюю органическую фракцию от темно-желтого до коричневого цвета выбрасывают, нижнюю водную фракцию зеленого цвета многократно промывают петролейным эфиром до тех пор, пока он не станет практически бесцветным. Очищенную таким образом водную фазу переносят в стеклянный стакан н постепенно, интенсивно перемешивая, добавляют водный раствор 6,5 г (40 ммоль) NHiPFs. Тотчас же выпадает светло-зеленый пушистый, объемистый осадок, который оставляют на ночь. Раствор становится прозрачным, желто-зеленым, образовавшийся осадок очень мелкий и может забивать поры фильтра. Поэтому фильтрование проводят под давлением. Полученный продукт сушат над Р40ю. Выход неочищенной соли 8,0 г (58%).
Для очистки продукт растворяют в кипящем ацетоне и высаживают эфиром, потери при этом бывают значительными. Выход (чистого продукта) 6,0 г (44%).
Свойства. Желто-зеленая соль, устойчивая на воздухе, на свету чернеет, растворяется в полярных растворителях. При действии Li[AlH<] образуется нейтральный комплекс Fe(T]-C5Hs) (т15-СбН7). ЯМР-‘Н (ацетон, ТМС): 6 4,58 [синглет, CsHs]; 5,82 [синглет, С6Нв].
ЛИТЕРАТУРА
1. Khand I. U., Pauson Р. L., Wattz W. Е., J. Chem. Soc. (London) С, 1968, 2257.
2. Nesmeyanov A. N., Vol'kenau N. A., Bolesova I. N„ Tetrahedron Lett., 1963, 1725.
3. Green M. L. H., Pratt L., Wilkinson G., J. Chem. Soc. (London), 1960, 989.
Ароматические сандвичевые комплексы 1961
□ □ Бис(т]-циклопентадиенил)рутений Ru^-CsHs^
(Рутеноцен)
RuC18 + 3NaC5H5 ----> Rufij-CsHsJj 4- 3NaCl [C5H5*]
207,4 3-88,1 231,3 3-58,4
При слабом кипении в течение 80 ч нагревают в 200 мл тетрагидрофурана смесь 10,43 г (50 ммоль) RuCl3 и 17,6 г (0,20 моль) NaCsHs. Растворитель отгоняют досуха в вакууме водоструйного насоса н остаток подвергают высоковакуумной возгонке при 120—130 °C. Сублимат хроматографируют на колонке с А12О3 [активность I степени, колонка 20x2 см, при ( + 10)4-( + 15) °C] бензолом; Ru(t]-CsHs)2 элюируется в виде светло-желтой зоны. После удаления растворителя и повторной возгонки получают 5,6 г (48%) аналитически чистого желтого Ru(t]-C6H5)2-
Точно так же из OsCl< получают бис (туциклопентадненил) осмий, осмоцен, Os(t]-C5Hs)2 с выходом 23% [1].
Свойства. Устойчивые на воздухе светло-желтые ромбические кристаллы, /пл 199—200°C. Кристаллическая структура: см. [3]; d(Ru—С) =2,21 А.
ЛИТЕРАТУРА
1. Fischer Е. О., Grubert Н., Вег., 92, 2302 (1959).
2. Bublitz D. Е., McEwen W. Е., Kleinberg J., Org. Synth., 41, 96 (1961).
3. Hardgrave G. L., Templedon D. H., Acta Crystallogr., 12, 28 (1959).
□□ Бис(т]-циклопентадиенил)кобальт Со(т]-С5Н5)2
{Кобальтоцен)
СоС12 + 2NaC5H5 ----> Co(i]-C5H5)2 + 2NaCl
129,8 2-88,1 189,1 2-58,4
а) Безводный СоС12 получают из продажного СоС12-6Н2О 15-часовым -кипячением в SOC12 и 5-часовым высушиванием в высоком вакууме при 150 °C. Полностью безводный продукт чисто синего цвета.
б) В 1-литровую двугорлую колбу, снабженную обратным холодильником, помещают раствор 44,0 г (0,5 моль) NaCsH5 в 300 мл тетрагидрофурана, добавляют порциями при перемешивании 32,5 г (0,25 моль) СоС12 и нагревают '2 ч с обратным холодильником. Для хорошего перемешивания применяют длинный (32 мм) якорь магнитной мешалки или используют высокоэффективную мешалку (для больших загрузок). Затем при комнатной температуре отгоняют растворитель и сушат в высоком вакууме. Черный комкообразный остаток подвергают высоковакуумной возгонке при 100—130 °C. Выход Со(т]-С5Н5)2 составляет 36—40 г (76—85%).
Другие способы. 1) Реакция [Co(NH3)6]C12 с NaC5H5 (выход ~90%) [2]. 2) Взаимодействие Co(SCN)2 с KCSHS в жидком NH3 (выход 70%) [3].
Свойства. Кристаллическое соединение, очень неустойчивое на воздухе, •от пурпурного до черного цвета. Растворимо в бензоле и тетрагидрофуране. Очень легко окисляется с образованием устойчивого катиона [Co(t]-CsHs)2]+. Кристаллическая структура: см. [4].
ЛИТЕРАТУРА
1. Wilkinson G., Cotton F. A. et al., J. Inorg. Nucl. Chem., 2, 95 (1956)
2. Cordes J. F„ Ber., 95, 3084 (1962).
3. Fischer E. 0., lira R„ Z. Naturfirsch., 8b, 327 (1953).
4. Pfab W., Fischer E. 0., Z. Anorg. Allgem. Chem., 274, 316 (1953). ’’
1962 Глава 33. Металлоорганические комплексы
Гексафторофосфат бис(т)-циклопентадиенил)кобальта(П1} [Со(г)-С5Н5)2] [PF6]
(Гексафторофосфат кобальтоцения)
HgOg+Og
Cofn-^H^+NHiPF.-----------> [Co(n-C5H5)2] [PFJ
189,1 163,0 334,1
В 100-мл трехгорлой колбе растворяют 1,0 г (5,3 ммоль) Co^-CgHg)» в 30 мл тетрагидрофурана. При перемешивании добавляют несколько капель 35%-ного Н2О2 и в течение 30 мин продувают через раствор воздух, используя водоструйный насос. Раствор при этом из фиолетового .становится темно-коричневым. После добавления к нему водного раствора 0,87 г (5,3 ммоль) NH4PFe тотчас выпадает коричневатый осадок. Для полноты осаждения раствор оставляют на ночь. Осадок отделяют на стеклянном фильтре, промывают небольшим количеством охлажденного тетрагидрофурана и затем небольшим количеством ледяной воды. После перекристаллизации из воды сушат в высоком вакууме над силикагелем. Выход 1,4 г (79%).
Свойства. Ярко-желтое кристаллическое соединение, растворимое в сильнополярных органических растворителях; водные растворы обладают электропроводностью. Соединение устойчиво на воздухе и не разрушается даже концентрированными кислотами (HNO3, H2SO4). ИК (нуйол): 830 (оч. с.) [v(PF6)J см-'. ЯМР-'Н (D2O, ТМСвиешн, 25°C): 6 5,90 (синглет, С5Н5).
ЛИТЕРАТУРА
1. Adams Р. М., Raynor J. В., Advanced Practical Inorganic Chemistry, Wiley» London — New York, 1965, p. 112.
2. Werner H., частное сообщение.
□ □ Бис(т)-ци1клопентадиенил)никель Ni(T]-C5H5)2 (Никелоцен)
Синтез проводят в две стадии по «аминному методу». В качестве основания для депротонирования слабокислого цнклопентаднена используют диэтил-амин. Циклопентадиеннд натрия как переносчик цнклопентадненильного аниона в этом методе не применяют.
Ni 4- Вг2 ---► NiBr2
58,7 159,8 218,5
NiBra + 2C6He + 2(C2H6)2NH -> Ni(T)-C5H5)2 + 2[(C2H5)2NH2]Br
218,5 2-66,1 2-73,1 188,9 2-154,1
В 1-литровую четырехгорлую колбу, снабженную эффективной мешалкой» капельной воронкой, термометром и обратным холодильником, помещают 500 мл диметилового эфира этиленгликоля и 29,4 г (0,5 моль) Ni-порошка. При интенсивном перемешивании прикапывают 27,3 мл (80,8 г, 0,5 моль) брома с такой скоростью, чтобы температура не превышала 40 °C (охлаждают льдом). Когда сильно экзотермическое образование эфирата NiBr2 закончится, смесь высушивают в высоком вакууме. Желто-коричневый остаток осторожно-при охлаждении льдом и перемешивании обрабатывают 400 мл диэтиламииа (суспензия синего цвета). Затем по каплям прн комнатной температуре добавляют 98 мл (79,0 г, 1,20 моль) свежеперегнанного цнклопентаднена; примерно» через 30 мнн первоначально синий цвет смеси изменяется на зеленый. Перемешивают еще 12 ч прн комнатной температуре и растворитель упаривают в ва
Ароматические сандвичевые комплексы 1963
кууме водоструйного насоса при слабом нагревании. Остаток от интенсивнозеленого до коричнево-зеленого цвета обрабатывают 600 мл кипящего к-гек-сана в аппарате Сокслета, уже в процессе экстракции выделяются длинные темно-зеленые кристаллы Ni(r]-C5Hs)2. Гексановый раствор охлаждают льдом, фильтруют через пористый стеклянный фильтр (G4) и кристаллы сушат в высоком вакууме при комнатной температуре. Фильтрат упаривают до половины объема, охлаждают до —35 °C и выделяют еще около 10 г Nitn-CsHs)^ Общий выход 70—77 г (74—81%). Препарат достаточно чистый для его дальнейшего применения в синтезах. Высоковакуумной возгонкой при 90—110°С получают совершенно чистый продукт.
1,1- Диметилникелоцен Ni(T]-C5H4CH3)2 лучше всего получать из NiBr2 и Na[C5H4CH3], Выход 35—41%. 36—38 °C [4].
Другие способы. Взаимодействие NiCl2-6H2O с С5Нб+КОН в среде диметилового эфира этиленгликоля и днметнлсульфоксида. Выход 55—57% [5].
Свойства. Темно-зеленые с металлическим блеском иголочки или листочки. inn 173—174 °C. В твердом состоянии заметно разлагается на воздухе, особенно в мелкокристаллическом состоянии. В атмосфере инертного газа при —30 °C хранится неограниченно долго. Не растворяется в воде, хорошо растворяется в большинстве органических растворителей, за исключением алифатических углеводородов. Растворы на воздухе очень неустойчивы. Кристаллическая структура: см. [6, 7]; d(Ni—С) =2,18 А.
ЛИТЕРАТУРА
1. King R. В., Organometallic Synthesis, v. 1, р. 71, Academic Press, New York, London, 1965.
2. Fischer E. O., lira R., Z. Naturforsch, 8b, 217 (1953).
3. Wilkinson G., Pauson P. L„ Birmingham J. M„ Cotton F. A., J. Amer. Chem. Soc., 75, 1011 (1953).
4. Reynolds L. T., Wilkinson G„ J. Inorg. Nucl. Chem., 9, 86 (1959).
5. Jolly W. L„ Chazan D. J., Inorg. Synth., 11, 122 (1968).
6. Pfab W„ Fischer E. O., Z. Anorg. Allgem. Chem., 274, 317 (1953).
7. Ронова И. А., Алексеев H. B. — Ж. структ. хим., 1966, т. 7, с. 886.
П Тетрафтороборат трис(т]-циклопентадиенил)диникеля(П)
[Ni2(n-C5H5)3] [BF4]
2Nih-C5H5)24-HBF4 -----> [Ni2(T]-C5H5)3] [BF4] + С6Н6
2-188,9 87,8 399,5 66,1
В 100-мл трубке Шленка в атмосфере инертного газа помещают 1,13 г {6 ммоль) никелоцена в 10 мл пропионового ангидрида и при интенсивном перемешивании добавляют по каплям из пипетки ~0,6 мл точно 50 %-ной водной HBF4 (~3 ммоль)*. Необходимо следить, чтобы реакционная смесь не слишком разогревалась, для этого ее охлаждают на ледяной бане. Первоначально зеленый раствор становится темно-коричневым. После добавления кислоты перемешивают еще ~ 10 мни и, интенсивно перемешивая, медленно разбавляют 50 мл эфира. Выпадающий черно-фиолетовый мелкокристаллический осадок отфильтровывают на пористом стеклянном фильтре (G3) и многократно промывают эфиром. Фильтрат имеет слабо-зеленую окраску*. Выход 1,05 г (88%).
Аналогичным образом получают и замещенные трехслойные комплексы [Ni2(C5H4R)3] [BF4] (R=CH3, грет-С4Н9).
* Более разбавленная 38%-ная HBF4 ие дает желаемого продукта. С другой стороны, избыток кислоты ведет к разрушению соединения, фильтрат в «онце вместо слабо-зеленой имеет коричневую окраску.
1964 Глава 33. Металлоорганические комплексы
Свойства. Черно-фиолетовое, мелкокристаллическое соединение, в твердом состоянии умеренно устойчиво на воздухе. tnjI выше 130 °C (с разл.). Не растворяется в неполярных растворителях, без разложения растворяется только в пропионовом ангидриде и нитрометане. Такие растворители, как вода, диметилсульфоксид, ацетонитрил, тетрагндрофуран и ацетон, разрушают соединение. ИК-(КВг): 3120 (с.), 1421 (ср.), 1400 (с.), 1285 (сл.), 1109 (с.), 1080, 1035 (оч. с.), 1002 (с.), 890, 870 (сл.), 805 (с.), 772, 536 (ср.), 524 (с.) см-1. ЯМР-‘Н (CD3NO2, ТМС): 6 4,70 [синглет, 5Н]; б 5,4 [синглет, ЮН].
ЛИТЕРАТУРА
1. Werner Н., Salzer A., Synth. React. Inorg. Metal-Org. Chem., 2, 239 (1972); Angew Chem., 84, 949 (1972).
Ароматические карбонилы металлов
□ □ Бис(т]-циклопентадиенил)дикарбонилтитан
Ti(n-C5H5)2(CO)2
Ti(r]-C5H5)2(CO)2 удобнее всего получать из легкодоступного TiCl2(r]-С5Н5)2 карбонилированием СО при нормальном давлении в присутствии активированного алюминия. Приведенная здесь пропись пригодна для получения как для меньших, так и для больших порций (вплоть до двухкратной) Т1 (Т]-С5Н5) 2 (СО) 2.
HgC12
3TiCl2(ii-C5H6)2 4- 2Al + 6СО -> 3Ti(n-C5H5)2(CO)2 4.2AlCls
3-249,0 2-27,0 6-22,4л 3-234,1 2-133,3
В 2-литровую двугорлую колбу, снабженную эффективным обратным холодильником и трубкой для ввода газа, соединенной через редуктор с баллоном СО, помещают раствор 24,9 г (0,10 моль) TiChfn-CjHeh в 300 мл тетрагидрофурана, тщательно защищая от доступа кислорода и влаги воздуха. Затем в течение 30 мин насыщают раствор сухим СО н прибавляют 10 г (избыток) тонкого порошка А1 или Al-бронзы. Для активирования А1, служащего в качестве восстановителя и акцептора галогена, добавляют еще на кончике шпателя HgCl2, продолжая пропускать слабый ток СО. Реакция за 10 ч протекает количественно, если при интенсивном перемешивании* либо постоянно продувают слабый ток СО, либо создают н сохраняют в системе постоянное избыточное давление СО с помощью 2-литровой стеклянной емкости. После окончания реакции суспензию фильтруют через колонку с A12O3 (Woelm-Pharma, активность I степени, 50x3 см), охлаждаемую водой. Промывают до тех пор тетрагидрофураном, пока фильтрат не перестанет окрашиваться в светло-красный цвет. Растворитель удаляют в вакууме водоструйного насоса, остаток обрабатывают петролейным эфиром (^кип 40—60 °C) в аппарате Сокслета. Выход 18,7—19,9 г (80—85%).
Другие способы. Восстановительное карбонилирование TiOl-CsHs^BHi) под действием СО прн повышенном давлении (>100 бар) или при нормальном давлении в присутствии трнэтиламина [2, 3].
Свойства. Красно-коричневые кристаллы, возгоняются в высоком вакууме при —80 °C с сильным разложением. Разлагаются при температуре >90 °C. В твердом состоянии и в растворах вещество разрушается от влаги и кисло
* Крупный якорь магнитной мешалкн, например, 32 мм.
Ароматические карбонилы металлов 1965»
рода воздуха. Растворяется во всех полярных органических растворителях (особенно хорошо в тетрагидрофуране), плохо растворяется в петролейном» эфире. В галогенсодержащих растворителях (типа СНС1з) очень быстро разлагается. ИК («-гептан): 1975 (с.), 1897 (с.) [v(CO)] см-1. ЯМР-'Н (C6D2): 6 4,58 [синглет, С5Н5]. Кристаллическая структура: см. [4].
ЛИТЕРАТУРА
1. Demerseman В., Bouquet G„ Btgorgne М„ J. Organometal. Chem., 101, С24' (1975).
2. Murray J. G., J. Amer. Chem. Soc., 83, 1287 (1961).
3. Fachinetti G„ Fochi G„ Floriani C., J. Chem. Soc. Chem. Commun., 1976,. 230.
4. Atwood J. L„ Stone К- E., Alt H. G., Hrncir D. C„ Rausch M. D., J. Organometal. Chem., 132, 367 (1977).
□ (т]-Циклопентадиенил)тетракарбонилванадий
V(T)-C5H5)(CO)4
V(t]-CsH6) (CO) 4 получают карбонилированием V(i]-CsHs)2. Высоковакуумный способ 1 дает гораздо лучшие выходы, однако очистку сырого продукта-лучше проводить хроматографнчески, как это рекомендовано по способу 2.* (карбонилирование при нормальном давлении).
Способ 1
на
V(n-C5H6)2 + 4СО ----> V(t]-C5H5) (СО)4 + [С5Н5-]
181,1 4-22,4л 228,1
Во вращающийся автоклав на 250 мл помещают, тщательно предохраняя1 от влаги и кислорода воздуха, 27,2 г (0,15 моль) возогнанного V(t]-C5H5)2». Для удаления посторонних газов систему дважды продувают Н2 при НО бар. Реакцию проводят при давлении Н2 100 бар и далее СО до 320 бар. При медленном нагревании до необходимой температуры 140 °C рабочее давление в системе повышается до 470 бар. Реакция начинается уже при —75 °C, что заметно по небольшому падению давления. Через 5 ч реакции при 140 °C давление, как правило, составляет около 400 бар, через 20 ч — около 320 бар-(140 °C).
Через 15—20 ч автоклав охлаждают, сжигают избыток газа. Оранжевый-или оранжево-коричневый сырой продукт очищают высоковакуумной возгонкой при 70—100 °C. Возгоняющееся попутно желто-коричневое масло (в основном дициклопентадиен) при необходимости осторожно счищают кусочком-ваты. Остаток после возгонки чаще всего пирофорен, его уничтожают обработкой водой в атмосфере N2. Выход 29,1—31,1 г (85—91%).
В случае если сублимат еще влажный или не вполне чистого оранжевого-цвета, его подвергают повторной возгонке; возможна также хроматографическая очистка на колонке с SiO2 смесью пентан — бензол (10:1) небольших порций продукта (до 3 г), однако это требуется не всегда. Затвердевшие в сублимате захваченные маслянистые загрязнения легко удаляют промыванием-пентаном.
Способ 2
V(T]-C5H5)a + CO --> V(n-C5H6)2(CO)
181,1 22,4 л 209,1
V(n-C6H5)2(CO) + Na + ЗСО -----> V(t]-C5H5) (СО)4 + NaC5H6
209,1 23,0 3-22,4л 228,1 88,1
1966 Глава 33. Металлоорганические комплексы
Раствор 8,65 г (47,5 ммоль) возогнанного V(t]-CsHs)2 в 200 мл тетрагидрофурана перемешивают в течение 2 ч при комнатной температуре в атмосфере СО н добавляют затем 1,15 г (50 ммоль) Na-порошка. Раствор еще 12 ч перемешивают путем встряхивания в атмосфере СО. После удаления растворителя в вакууме водоструйного насоса остаток возгоняют в высоком вакууме при ~80°С. Выход сырого продукта 9,64 г (89%). Для последующей очистки от V(t]-C5H5)2 сырой продукт хроматографируют на колонке с кизельгелем смесью пентан — бензол (10:1). V(t]-C5H5) (СО)4 элюируется в виде оранжевой полосы. Выход 3,25 г (30%).
Другие способы. 1) Обработка в течение 7 ч при давлении СО 60 бар и температуре 117±6°С тетрагидрофуранового раствора сырого Vfn-CsHsJj (из VCh+NaCsHs). Выход23% [5].2) Взаимодействие [Ь1а(диглим)2] [V(CO)e] с HgCl(C5H5). Выход 78% [6].
Свойства. Светло-красные блестящие кристаллы, чувствительные к свету и кислороду воздуха, имеющие типичный карбонильный запах. 138 °C. При
длительном нагревании свыше 120 °C разлагается с выделением СО. Растворяется практически во всех органических растворителях. На свету превращается в V2(t]-C5H5)2(CO)s. ИК (гексан): 2031 (с.), 1931 (оч. с.), 1901 (пл.) [v(CO)] см-1. ЯМР-'Н (C6D6, ТМС): б 4,30 [синглет, С5Н5]. Кристаллическая •структура: см. [7].
ЛИТЕРАТУРА
1. Fischer Е. О.. Hafner W., Z. Naturforsch, 9b, 503 (1954).
2. Fischer E. О., Vigoureux S., Ber., 91, 2205 (1958).
3. Vigoureux S„ Dissertation, Technische Hochcshule Miinchen, 1958.
4. Fachinetti G., Del Nero S., Floriani C., J. Chem. Soc. Dalton, 1976, 1046.
"5. Hing R. B., Organometallic Syntheses, v. 1, p. 105—108, Academic Press, New York, London, 1965.
6. Werner R. P. M., Filbey A. H., Manastyrskyj, S. A., Inorg. Chem., 3, 298 (1964).
7. Wilford J. B„ Whitla A., Powell H. M., J. Organometal. Chem., 8, 495 (1967).
П (т)-Ацетилциклопентадиенил)тетракарбонилванадий
V(n-C5H4COCH3)(CO)4
Ацилирование V(t]-C5H5) (CO)4 по Фриделю — Крафтсу протекает так же легко, как и его прототипа ферроцена (см. выше). В качестве примера ниже приводится синтез У(г]-С5Н4СОСНз) (СО)4:
А1С13
V(n-C6HS) (СОД + СН3СОС1 ----> V(t]-C5H4COCH3)(CO)4 + HC1
228,1 78,5 270,1 36,5
В 500-мл колбу, снабженную обратным холодильником, помещают раствор 10,00 г (43,8 ммоль) VCq-CsHs) (СО)4 в 250 мл СН2С12 и к нему при интенсивном перемешивании прибавляют последовательно 11,70 г (87,7 ммоль) порошка А1С13 и 5,15 г (4,68 мл, 65,6 ммоль) СН3СОС1. Раствор, цвет которого быстро изменяется от оранжевого до темно-красного с одновременным выделением НС1, интенсивно кипятят с обратным холодильником в течение 7 ч. Затем реакционную смесь гидролизуют осторожным прибавлением 125 мл 2 н. НС1, хорошо перемешивают и дают слоям разделиться. Верхний водный слой декантируют, промывают нижний органический слой 100 мл Н2О, обрабатывают ~10 г К2СОз и сушат безводным Na2SO4. После фильтрования метилеи-
Ароматические карбонилы металлов 196Г
хлорид отгоняют в роторном испарителе, а оранжевый порошкообразный или» мелкокристаллический остаток возгоняют в высоком вакууме при 80—90 °C. Кристаллический красный сублимат хроматографируют бензолом на кизель-геле 60 (Merck 7734, активность I степени, колонка 60X2 см) для очистки от небольших количеств примеси VCq-CsHs) (СО) 4. Сначала элюируется ярко-желтая зона V(n-CSH5)(CO)4, затем темно-красная зона ацетильного производного. После отделения от исходного можно ускорить смывание с колонки,., добавив 20 об.% эфира. Растворитель удаляют в вакууме водоструйного насоса, а остаток сушат в высоком вакууме. Выход 6,65 г [56% в расчете на-V(ri-C5H5) (СО)4].
Свойства. Оранжевый или оранжево-красный кристаллический порошок (из эфира). В твердом состоянии выдерживает краткую экспозицию на воздухе. Разлагается при плавлении около 98 °C. Плохо растворяется в пентане,., хорошо растворяется в бензоле, очень хорошо растворяется в полярных растворителях, например СН2С12 или тетрагидрофуране. ИК (эфир): 2028 (с.), 1936 (оч. с.) [v(feO)]; (КВг): 1683 (с.) [v(C=O)J см-’.
ЛИТЕРАТУРА
1. Fischer Е. О., Plesske К., Вег., 93, 1006 (1960).
□ □ Ди-ц-карбонилтрикарбонилбис[(т]-циклопентадие-нил)ванадий] (V—V) V2(n-C5H5)2(CO)5
Способ 1
V(T]-CsH5)(CO)4 + 2Na ---> Na2[V(T]-CsH5) (СО),] + СО
228,1 2-23,0 246,0 22,4 л
Na2[V(T]-CsH5) (СО),] + 2Н+--> «V(r]-C5H5)H2(CO)a» + 2Na+
246,0 202,1 2-23,0
2«V(t]-C5H5)H2(CO),» --> V2(t]-C5H5)2(CO)5 + 2H2 + СО
2-202,1 372,1 2-22,4 л 22,4л
В двугорлой колбе на 500 мл, в дно которой впаяна вводная трубка с краном, готовят амальгаму натрия из 11,5 г (0,5 моль) Na и 75 мл (1000 г) Hg. Затем добавляют 9,12 г (40 ммоль) V(t]-C5H5) (СО)4 в 350 мл тетрагидрофурана. Суспензию интенсивво перемешивают 45 мин. При этом красно-оранжевый-цвет постепенно исчезает и одновременно выпадает светло-желтый осадок. Амальгаму натрия сливают через трубку в дне колбы, а растворитель отгоняют в вакууме водоструйного насоса досуха. К оставшейся в колбе желтой соли добавляют 100 мл 2 и. НС1. Наблюдаются интенсивное выделение газа и-образование пушистого зеленого осадка, который экстрагируют 700 мл петро-лейиого эфира. Растворитель упаривают примерно до 200 мл и хроматографируют темно-зеленый раствор на кизельгеле (Merck 7734, активность I степени, колонка 60x3 см) при 10—15°С смесью пентан — бензол (10:1). Сначала элюируется желто-коричневая зона V^-CsHs) (СО)4. Далее бензолом выделяют зеленую зону V2(ti-C5Hs)2(CO)5. После отгонки растворителя перекристаллизовывают остаток из смеси петролейный эфир — эфир (10:1) и сушат в> высоком вакууме. Выход 5,88 г (79%).
Способ 2
hv
2V(T)-C,HS) (СО)4 ---> V2(r]-C5H5)2(CO)5 + 3CO
2-228,1 372,1 3-22,4л
<1968 Глава 33. Металлоорганические комплексы
В охлаждаемом водой фотореакторе проточного типа с тефлоновой тур-'бинкой с подвижным слоем жидкости* облучают ртутной лампой высокого давления мощностью 0,7 кВт раствор 6,00 г (26,3 ммоль) VCn-CsHs) (СО) 4 'В 1,2 л тетрагидрофурана в течение 120 мин. Первоначально оранжево-желтый раствор уже через несколько минут начинает окрашиваться в зеленый цвет с выделением газа. Во время фотолиза вся аппаратура заполнена N2. Темио-зеленый раствор упаривают в роторном испарителе, а остающийся темнозеленый кристаллический осадок очищают хроматографически, как указано «в описании способа 1. После перекристаллизации из смеси петролейный эфир — эфир (10:1) при температуре от —35 до —78 °C получают 3,96 г (81%) темно-зеленых блестящих иголочек или ромбиков V2(t)-C3H5)2(CO)5, которые сушат в высоком вакууме.
Свойства. Темно-зеленые, неустойчивые иа воздухе кристаллы. 99 °C (с разл.). Растворяется во всех органических растворителях. Растворы легко •окисляются. ИК (нуйол): 2009 (ср.), 1955 (оч. с.), 1910 (оч. с.), 1871 (ср.), 1832 (оч. с.) [v(CO)] см-1. ЯМР-'Н (C6D6, ТМС): S 4,57 [синглет, С5Н5]. Структура: см. [3]; d(V—V) =2,462 А.
ЛИТЕРАТУРА
1. Fischer Е. О., Schneider R. J. I., Angew. Chem., 79, 537 (1967). "2. Herrmann W. A., Plank J., Ber., 112, 392 (1979).
3. Cotton F. A., Kruczynski L., Frenz B. A., J. Organometal. Chem., 160, 93 (1978).
Трикарбонил (трциклогептатриенил) ванадий V(t]-C7H7)(CO)3
V(CO)e + C7H8 ---> V(T]-C,H7) (CO), + V2H2 + 3CO
219,0 92,1 226,1 11,2 л 3-22,4л
Раствор 8,76 г (40 ммоль) V(CO)6 и 18,4 г (0,20 моль) циклогептатриена в 300 мл н-гексана кипятят с обратным холодильником, предохраняя от влаги «и кислорода воздуха. За время реакции выделяется около 2,7 л СО. Раствор (зеленого цвета) фильтруют еще теплым через пористый стеклянный фильтр (G3) с наполнителем для отделения от выпавшего коричневого аморфного лродукта разложения, упаривают в вакууме водоструйного насоса до ~30 мл и, постепенно охлаждая до —78 °C, оставляют кристаллизоваться. Выпавшие "блестящие кристаллы повторно перекристаллизовывают из н-гексана. Выход 1,9 г (21%).
Свойства. Темно-зелеиое кристаллическое вещество, не очень устойчивое «а воздухе. Быстро возгоняется в высоком вакууме при 60 °C. 1ПЛ 134—137 °C 4с разл.). Не растворяется в воде, растворяется в большинстве органических растворителей, например СН3ОН, эфире, ацетоне, СНС13, СС14 или петролейном эфире. Растворы зеленого цвета, очень легко окисляются. ИК (н-гептаи): 1975, 1915 [v(CO)J; (КВг): 1950, 1875 [v(CO)] см->.
ЛИТЕРАТУРА
1. Werner R. Р. М., Manastyrskyj S. A., J. Amer. Chem. Soc., 83, 2023 (1961).
* Объем буферной емкости 1,1 л. Фотореактор типа А9356, фирма-постав-щик Otto Fritz (NORMAG, Hofheim, Taunus).
Ароматические карбонилы металлов 1969
□ □ Гексафторофосфат тетракарбонил(т]-мезитилен)ванадия [V(CO)4{t)-C6H3(CH3)3}][PF6]
Комплексы типа [V(T)-ароматический углеводород) (СО) 4]+Х~ получают термическим замещением карбонильной группы в V(CO)g. Ниже описываемый способ пригоден для получения также и других комплексов с ароматическими лигандами, например с бензолом, толуолом и ксилолом. Промежуточно образующийся гексакарбонилванадатный комплекс может быть выделен и представляет собой термически весьма устойчивое соединение.
a) 2V(CO)6 + С6Н3(СН3)3 -> [V(CO)4{t1-C«Hs(CHs)s}] [V(CO)e] +2СО
2-219,0 120,2 502,2 2-22,4л
б) [V(CO)4h-CeH3(CH3)3}] [V(CO)6] + NH4PF6 ->
502,2 163,0
----> [V(CO)4{T]-C6H3(CH3)3}1 [PFJ + [NH4] [V(CO)6] 428,1 237,0
В 250-мл колбу, защищенную от света и изолированную от воздуха, помещают 3,04 г (13,9 ммоль) V(CO)6 и 75 мл мезитилена (избыток). Создав небольшое избыточное давление N2 ( — 10 мм рт. ст.), нагревают при 35 °C в течение 48 ч. Выделяющийся СО примерно через каждые 10 ч откачивают. Цвет раствора за время реакции изменяется от оранжевого до коричневозеленого. По окончании реакции смесь фильтруют, твердый осадок многократно промывают петролейным эфиром (ЦНп 40—60 °C) и сушат в высоком вакууме. Высушенный комплекс [У(СО)4{т]-С6Н3)з}] [V(CO)6] воспламеняется при контакте с кислородом воздуха. В фильтрате остается не вошедший в реакцию V(CO)6, который можно извлечь.
Остатки на фильтре (сырой продукт) извлекают 80—100 мл тетрагидрофурана. После фильтрования к красно-коричневому раствору при интенсивном перемешивании добавляют раствор 800 мг (4,9 ммоль) NH4PF6 в 40 мл тетрагидрофурана. Тотчас выпадает осадок красного цвета, который после промывания тетрагидрофураном высушивают в высоком вакууме. Выход 0,75 г (25% в пересчете на взятое количество V(CO)6).
Свойства. Микрокристаллическое вещество, красного цвета, в твердом состоянии устойчивое на воздухе. Разлагается при 102—104 °C. Не растворяется в петролейном эфире, плохо растворяется в тетрагидрофуране и эфире, умеренно растворимо в ацетоне. Прн комнатной температуре быстро взаимодействует с пиридином с выделением ~2—3 моль СО. ПК (ацетон): 2063 (оч. с.), 2015 (оч. сл.), 1979 (оч. с.) (v(CO)] см-1.
ЛИТЕРАТУРА
1. Calderazzo F., Inorg. Chem., 3, 1207 (1964).
□ □ Т етракарбонил (rj-циклопентадиенил) ниобий
Nb(n-C5H5)(CO)4
Единственный до сих пор известный способ, позволяющий получать большие количества Nb^-CjHs) (СОЦ с воспроизводимыми выходами, заключается в восстановительном карбонилировании при высоком давлении либо ниобоцен-дихлорида (способ 1), либо тетрахлоро (грциклопентадиеннл) ниобия (способ 2) в присутствии (А1+Си) -системы в качестве акцептора галогена. Ниже приводимая методика позволяет за 1 прием получить свыше 15 г ЦЬ(ц-C5Hj)(CO)4, а также пригодна как для больших, так и для меньших загрузок. 8—1361
1970 Глава 33. Металлоорганические комплексы
Способ 1
NbCl2(t]-CsH6)2 + 3Na + 4СО------> Nb(n-CSHS) (СО)4
294,0 3-23,0 4-22,4 л 270,0
В сухой вращающийся автоклав на 1 л помещают в атмосфере аргона смесь 10,9 г (0,474 моль) мелкого порошка Na, ~10 г порошка Си (размер частиц 0,04 мм) и 5 г Al-пыли в 600 мл бензола и добавляют 40,0 г (0,136 моль) NbCl2(T]-CsHs)2. Систему продувают СО при 100 бар и затем создают рабочее давление СО 330 бар (насыщение на холоду). При интенсивном перемешивании доводят температуру внутри автоклава за 3—5 ч до 135±3°С и перемешивают в этих условиях еще 125 ч. Максимальное давление в автоклаве составляет 470 бар. Затем автоклав охлаждают до комнатной температуры (за ~8 ч конечное давление 300 бар при 25°C), сжигают избыток газа и переносят содержимое (коричнево-красного цвета) в колбу емкостью 1000 мл. После фильтрования через фильтр G3, покрытый стеклянной ватой*, растворитель отгоняют в роторном испарителе (коричневое стекло!) до объема 100 мл и пропускают остаток через колонку с кизельгелем (0,063— 0,200, активность II степени, Merck 7734; 40X3 см), охлаждая ее водой и защищая от света; для фильтрования через колонку используют растворитель — бензол, причем промывают бензолом до тех пор, пока цвет раствора из темно-красного не перейдет в светло-желтый. Фильтрат (~700 мл) упаривают в роторном испарителе при температуре бани не выше 30 °C. Остаток, загрязненный небольшим количеством дициклопентадиена, возгоняют в высоком вакууме при 65—100°C (температура бани), охлаждая приемник проточной водой, и перекристаллизовывают из эфира с пентаном (от —35 до —78 °C). После высушивания в вакууме препарат получают аналитически чистым. Выход 19,6 г (53%). В автоклаве емкостью 500 мл синтез Nb(r]-C5HS)(CO)4 проводят при следующих условиях: 10,0 г (34,0 ммоль) NbCl2(T]-C5Hs)2, 2,70 г (0,118 моль) порошка Na, ~3 г порошка Си, —2 г Al-порошка, 180 мл бензола, начальное давление СО 330 бар, время реакции 50 ч, температура реакции 135±3°С (внутри автоклава). Ход синтеза, а также обработка и выделение продукта аналогичны приведенным выше. Выход 4,8—5,1 г (52— 55%).
Способ 2
NbCl4(T]-CsHs)+4Na + 4СО --------> Nb(n-CSHS) (СО)4
299,8 4-23,0 4-22,4 л 270,0
Карбонилирование проводят во вращающемся автоклаве на 1 л по методике, аналогичной вышеприведенной [60,0 г (0,20 моль) NbCl^-CsHs); 18,4 г (0,80 моль) Na-порошка; —14 г Си-порошка; —8 г А1-порошка; 600 мл тетрагидрофурана; исходное давление СО 330 бар; температура внутри автоклава 135±3°С; время реакции 130 ч, рабочее давление 450 бар; давление в конце реакции после охлаждения автоклава 280 бар]. Обработку и выделение продукта проводят так же, как указано в описании способа 1 (фильтрование коричнево-красного раствора через колонку с кизельгелем и последующая возгонка при 60—100°C). Выход 47,5—50,8 г (88—94%).
Другие способы. Окислительное циклопентадиенилирование гексакарбо-нилниобата(—I) согласно уравнению
[Nb(CO)J- + HgCl^-CgHs) ----> Nbh-CSHS) (СО)4 + 2СО + Hg + СГ
* Осторожно при очистке фильтра: остаток содержит следы непрореагировавшего Na!
Ароматические карбонилы металлов 1971
Выходы колеблются от 10 до 50%. Применимость этого способа существенно ограничена довольно затруднительным синтезом [3, 4] промежуточного гекса-карбонилниобата(—I) с низким выходом.
Способ получения исходя из NbCl5[NbCls/Na/NaCsH5/Tr0] характерен нестабильными выходами ( — 18%) и пригоден лишь для получения небольших количеств, не свыше 1—1,5 г Nb(T]-C5Hs) (СО)4 [1, 5].
Свойства. Блестящие красные игольчатые или ромбические кристаллы, выдерживают кратковременную экспозицию на воздухе. /пл 145—148 °C (без разл.). Вещество растворяется в большинстве органических растворителей, особенно хорошо в бензоле, эфире, метиленхлориде. Растворы очень неустойчивы на свету и воздухе. Из растворов в я-гексане на свету (рассеянный дневной свет или кратковременное облучение прямым солнечным светом) выпадает кристаллический, темный, почти черный Nb3(T]-C5H5)3(CO)7 [6].
Nb(T]-CsHs) (СО)4 термически довольно устойчив, можно кипятить целый день в бензоле без всякого изменения. ИК (СбН6): 2028 (с.), 1914 (оч. с.) [V(СО)] см-1. Кристаллическая структура: см. [7].
Меченные по углероду и по кислороду производные Nb(T]-C5H5)(CO)3(13CO) и Nb(t]-C5H5)(CO)3(13C1SO) получают по схеме
hv *с»о
Nb(r!-CSHS) (СО)4 -> Nb(n-C5HS) (СО)3(ТГФ) --->
---> Nb(t]-C8H8) (СО)3(*С*О)
(Степень обогащения —40% *С*О.)
ЛИТЕРАТУРА
1. Herrmann W. A., Biersack Н., Вег., 112, 3942 (1979).
2. Herrmann W. A., Biersack Н„ J. Organometal. Chem., 191, 397 (1980).
3. Werner R. P. M., Filbey A. H., Manastyrskyj S. A., Inorg. Chem., 3, 298 (1964).
4. Ellis J. E. Davison A., Inorg. Synth., 16, 68 (1976).
5. Анисимов К. H., Колобова Н. Е., Пасынский А. А. — Изв. АН СССР, сер. хим., 1969, с. 2238.
6. Herrmann W. A., Ziegler М. L., Weidenhammer К., Biersack Н., Angew, Chem., 91, 1026 (1979).
7. Bernal I., Creswick M., Herrmann W. A., J. Organometal. Chem. (1981).
□ □ Гексакарбонил-ц3(т]3Ст)2О)-карбонилтрис[(т]-циклопента-диенил)ниобий] (3Nb—Nb) КЬ3(т)-С5Н5)з(СО)7
При фотолизе NbCn-CsHs) (СО)4 в гексановом растворе тотчас выпадает кристаллический NbsCq-CsHs^CO)?, который наряду с шестью классическими терминальными СО-группами содержит одну терминальную и в то же времи дважды мостиковую карбонильную группу (так называемое «полутройное связывание»),
3Nb(n-CsHs) (СО)4 Nb3(n-CSHS)3(CO)7 + 5СО
3-270,0 670,1 5-22,4 л
Раствор 2,70 г (10 ммоль) возогнанного Nb(T]-C5H5) (СО)4 в 250 мл я-гек-сана помещают в термостатированную трубку Шленка из стекла дюран, снабженную ртутным затвором, и при +18 °C облучают солнечным светом (используют, например, криостат НААКЕ КТ 33, хладагент СН3ОН). В зависи-8*
1972 Глава 33. Металлоорганические комплексы
мости от интенсивности света время облучения составляет 15—80 ч. В этих условиях из раствора выпадают темные, почти черные кристаллы с металлическим блеском, временами их счищают со стенок реакционного сосуда. Фотолиз можно также проводить традиционным методом с использованием аппаратуры с погружаемой лампой, ртутной лампы высокого давления (например, TQ 150, Original Hanau). Однако в этом случае не всегда сразу получают кристаллический продукт. Кристаллы многократно промывают н-гексаном и сушат в высоком вакууме. Выход 1,45—1,83 г (65—82%).
Свойства. Темные, почти черные игольчатые кристаллы, в течение короткого времени стабильны на воздухе, не возгоняются. Разлагаются выше 100 °C, частично образуя исходный ЬЛДтрСзНз) (СО)4. Вещество умеренно растворяется в эфире, очень хорошо растворяется в метиленхлориде и тетрагидрофуране (растворы коричневого цвета, очень неустойчивые на воздухе). Соединение не хроматографируется. ЯМР-!Н (ацетон-de, +33 °C, ТМС): б 5,66 [с., С5Н5, 5Н]; 5,52 [с., С5Н5; ЮН]. ЯМР-13С (CDC13, +33°C, 15% обогащения 13СО, ТМС): б 95,26; 94,40 [С5Н5]; ~247 (шир.) [СО]. ИК (циклогексан): 1993 (сл.), 1973 (оч. с.), 1952 (сл.), 1934 (cpj, 1907 (с.) [v(CO)] см-1; (КВг): 1982 (сл.-оч. с.), 1961 (оч. с.), 1946 (ср.), 1915 (с.), 1889 и 1850 (с.-оч. с.), 1838 (с.), 1330 (сл.-ср.) [v(CO)] см-1; другие полосы (КВг) в области 3500—700 см-1: 3115 (сл., шир.), 1424 (сл., шир.), 1065 (сл.), 1011 (сл.), 1003 (сл.), 996 (сл.), 845 (сл.-ср.), 833 (сл.-ср.), 810 (ср.), 803 (ср.), 791 (сл.-ср.) см-1. Отнесение частот валентных колебаний на основании данных для Nb3(T]-C5H5)3(I3CO)7 (15% 13СО). Кристаллы моноклинные (пространственная группа симметрии Cs2ft-P2i/c; а= 17,17 А; 6 = 7,752 А; с= 18,76 А; 0=117,57°).
ЛИТЕРАТУРА
1. Herrmann W. A., Ziegler М. L., Weidenhammer К-, Biersack Н., Angew. Chem., 91, 1026 (1979).
Комплексы типа М(тр ароматический лиганд)(СО)з (M=Cr, Mo, W)
Комплексы переходных металлов наряду с ферроценовыми производными представляют, пожалуй, наибольшие возможности для варьирования органического лиганда. Самым простым способом получения их является нагревание соответствующего карбонила металла с ароматическим соединением. Оптимальная температура таких реакций (идущих с отщеплением СО-групп) равна 120—150 °C, поэтому необходимо использовать соответственно высококипя-щие органические растворители. Лучшими оказываются такие донорные растворители, как 2-метоксиэтиловый эфир, ди-ц-бутиловый эфир, диоксан и тетрагидрофуран, а также очень часто и их смеси. Для получения термически неустойчивых соединений, в первую очередь соединений Мо и W, или комплексов с очень реакционноспособными ароматическими лигандами следует применять реакцию обмена лигандов в замещенных металлкарбоиилах ML3(CO)3, где L — донорный лигаид со слабой обратной связью. Реакции замещения L протекают в таком случае гораздо быстрее, чем замена СО-групп. Обмен лигандов можно также значительно ускорить добавкой кислот Льюиса, которые образуют с отщепляющимся лигандом прочный аддукт. Для этих трех методов получения комплексов типа М(т]-ароматический лиганд) (СО) 3 далее будет дано лишь по одному примеру. Полный обзор литературы по этим комплексам для М=Сг можно найти в книге [1]. Кроме того, опубликованы подробные обзорные статьи [2—4] о получении и химических свойствах этих металлоорганических соединений.
Комплексы М(^-ароматический лиганд) 1973
ЛИТЕРАТУРА
1. Gmelins Handbuch der Anorganischen Chemie, Erganzungswerk zur 8. Auf-lage, Verlag Chemie, Weinheim, Bd. 3, 1971, S. 181—289.
2. SUverthorn W. E„ Adv. Organometal. Chem., 13, 47 (1975).
3 Sneeden R. P. A., Organochromium Compounds, Academic Press, New York 1975.
4. Semmelhack M. F„ New Applications of Organometallic Reagents in Organic Syntheses, Elsevir, Amsterdam 1976, p. 361—395.
□ Трикарбонил(т]-хлорбензол)хром Сг(СО)з(т]-С6Н5С1)
Cr(CO)6 + C6H5Cl Cr(CO)3(T]-C6HsCl) + 3CO
220,1 112,6 248,6 3-22,4л
В реакционную колбу на 50 мл с проволочной мешалкой (см. рис. 479) и ртутным затвором помещают 3,0 г (13,6 ммоль) Cr(CO)s, 6,0 мл (58,8 ммоль) чистого перегнанного С6Н5С1, 6,0 мл диглима (см. т. 1, ч. I) и 6,0 мл диоксана. Смесь перемешивают магнитной мешалкой, нагревая на бане при температуре ~ 145 °C в течение ~20 ч. За это время должно выделиться 650—700 мл СО. После охлаждения растворитель отгоняют в вакууме досуха и сублимируют остатки Сг(СО)6. Затем остаток растворяют в 5—10 мл бензола, добавляют 15 г кизельгеля и снова отгоняют в вакууме растворитель. Сорбированный на кизельгеле продукт переносят на колонку (80x3 см) с дезактивированным (5% Н2О) кизельгелем и последовательно элюируют 50 мл гексана, 750 мл смеси бензол — гексан (1:2), 100 мл смеси гексан — бензол (1:1) и 100 мл бензола. Комплекс Cr^-CsHgCl) (СО)3 проявляется на колонке в виде темножелтой полосы, которая идет вслед за бледно-желтой полосой, содержащей следы Сг(г]-С6Н6) (СО)з. Элюат отгоняют в вакууме досуха, остаток перекристаллизовывают из смеси эфир — гексан и кристаллический продукт после промывания 3x5 мл гексана высушивают в вакууме. Выход 2,2 г (65%).
Свойства. Темно-желтые, устойчивые на воздухе кристаллы, легко растворяются в бензоле и эфире, труднее в гексане. /пл 100,5—103,0 °C. Вещество возгоняется в высоком вакууме, частично разлагаясь, при температуре свыше 80 °C. ПК (С6Н12): 1992 (с.), 1929 (оч. с.) [v(CO)J см-1.
Аналоги. По способу, подобному приведенному, можно получать аналогичные комплексы хрома с бензолом и многими другими производными бензола, конденсированными ароматическими углеводородами, с пиридином и его производными.
ЛИТЕРАТУРА
1. Nicholls В., Whiting М. С., J. Chem. Soc. (London), 1959, 551.
2. Mahaffy C. A. L„ Pauson P. L., Inorg. Synth., 19, 154 (1979).
3. Silverthorne W. E., Adv. Organometal. Chem., 13, 70f. (1975).
4. Biedermann H.-G., Ofele K, Schuhbauer N., Taitelbaum J., Angew. Chem., 87, 634 (1975).
□ □ Трикарбонил(т]-толуол)молибден Мо(СО)з(г)-С6Н5СНз)
Mo(NCCHs)s(CO)s + C6HSCH3 ---> Mo(CO)3(r]-CcH5CH3) + 3NCCH3
303,1 92,1 272,1 3-41,1
В колбе на 250 мл, снабженной краном для ввода N2, обратным холодильником и ртутным затвором, кипятят при перемешивании магнитной ме
1974 Глава 33. Металлоорганические комплексы
шалкой 3,6 г (11,9 ммоль) Mo(NCCH3)3(CO)3 в 150 мл абсолютного толуола в течение 3 ч. После охлаждения избыток толуола отгоняют в вакууме. Желто-зеленый остаток извлекают эфиром, фильтруют (стеклянный пористый фильтр G4) и фильтрат упаривают в вакууме до минимального объема. После добавления гексана оставляют на холоду, выпавшие кристаллы отфильтровывают, промывают небольшим количеством гексана и сушат в вакууме. Выход 2,4 г (74%).
Свойства. Желтые, устойчивые в течение нескольких часов на воздухе кристаллы. Вещество легко растворяется в бензоле и эфире, хуже в гексане; растворы неустойчивы на воздухе. /пл 127—128 °C. В высоком вакууме возгоняется лишь при ~ 100°C, но с сильным разложением. ИК (гексан): 1987 (с.), 1913 (с.) [v(CO)] см-1.
Аналоги. Этим способом могут быть получены соответствующие комплексы вольфрама с бензолом и его производными. Аналогично получают Сг(т]-С4Н5М)(СО)з [C4H5N=пиррол] и Сг(т]-С4Н4Те)(СО)3 [С4Н4Те=теллурофен]. Для получения стирол-, иодбензол- и триптицентрикарбонилхрома вместо Cr(NCCH3)3(CO)3 можно использовать Сг(ИН3)з(СО)3.
ЛИТЕРАТУРА
1. King R. В., Fronzaglia A., Inorg. Chem., 5, 1837 (1966).
2. Ofele К, Dotzauer Е., J. Organometal. Chem., 42, C87 (1972).
3. Moser G. A., Rausch M. D., Synth. React. Inorg. Metal.-Org. Chem., 4, 37 (1974).
□ Трикарбонил(т]-тиофен)хром Cr(CO)3(T]-C4H4S)
Cr(CO)a(y-CH3C6H4N)3 + C4H4S + 3BF3O(CH3)2 -->
415,4 84,1 3-113,9
----► Cr(CO)3(n-C4H4S) + 3y-CH3C5H4NBFs + 3O(CH3)2 220,2 3-160,9 3-46,1]
1,35 г (3,25 ммоль) аран-трикарбонилтрис(у-пиколин)хрома* и 13 мл (0,164 моль) безводного тиофена нагревают в течение 5 мин при 40 °C в колбе на 100 мл, снабженной краном для ввода N2 и ртутным затвором. Затем, продолжая перемешивание, быстро прибавляют 1,00 мл (11,0 ммоль) свежепере-гнанного BF3-O(CH3)2 и еще перемешивают 15 мин. К реакционной смеси (красного цвета) добавляют 30 мл эфира, охлаждают до 0 °C и вливают 10 мл Н2О. Перемешивают 10 мин и отделяют эфирную фазу. Водный слой экстрагируют эфиром порциями по 10 мл до тех пор, пока экстракт не станет бесцветным. Объединенные эфирные вытяжки промывают Н2О (2X10 мл), сушат Na2SO4, отфильтровывают осушитель и отгоняют в вакууме досуха. Для удаления побочно образующегося пентакарбонил (у-пиколин) хрома остаток несколько раз промывают (по 3—5 мл) гексаном и перекристаллизовывают из смеси эфир — гексан. Выход 0,50 г (70%).
Свойства. Красные, в течение нескольких часов устойчивые на воздухе кристаллы. Вещество легко растворяется в бензоле и эфире, хуже в гексане, разлагается при ~ 145 °C. Возгоняется в высоком вакууме при ~90°С. ИК (С6Н12): 1985 (с.), 1914 (с.), 1898 (с.) [v(CO)] см"1.
* Это исходное соединение лучше всего получать по способу 26, описанному ниже в методике синтеза гран-трис(ацетонитрил)трикарбонилхрома Cr (NCCH3) 3(СО) 3.
Комплексы М(х\-ароматический лиганд) 1975
Аналоги. Этим методом можно получить соответствующие комплексы селенофена и различных производных тиофена. Исходя из Mo(CsH5N)3(CO)3 получают комплексы Мо(т]-С6Н5Х) (СО)3 (Х=С1, Вт, OR, COR, СН=СЙ3 и др.), которые нельзя получить другим методом. С замещенными инданами по этому способу в условиях кинетически контролируемой реакции получают преимущественно термодинамически неустойчивые изомеры.
ЛИТЕРАТУРА
1. Ofele К-, Вег., 99, 1732 (1966).
2. Novi М., Guanti G., Dell’Erba С., J. Heterocycl. Chem., 12, 1055 (1975).
3. Nesmeyanov A. N., Rybinskaya M. I., J. Organometal. Chem., 102, 185 (1975).
4. Gracey D. E. F., Jackson W. R., Jennings W. B., Mitchell T. R. B., J. Chem. Soc. (London) B, 1969, 1204.
Трикарбонил(гексаметилборазин)хром Сг(СО)з[т)-В3М3(СН3)6]
a) 3BC13 + 3[CH3NH3]C1 ---► C13B3N3(CH3)3 + 9HC1
3-117,2 3-67,5 225,9 9-36,5
6) C13B3N3(CH3)3 + 3CH3MgI ---> B3N3(CH3)„ + 3Mg(Cl)I
225,9 3-166,2 164,7 3-186,7
в) Cr(CO)3(CH3CN)3 + B3N3(CH3)„ --> Cr(CO)3[n-B3N3(CH3)e] + 3CH3CN
259,2 164,7 300,7 3-41,1
a) □ В-трихлор-М-триметилборазин [1, 2]. Реакцию проводят в трехгорлой колбе на 2 л, снабженной эффективной мешалкой, обратным холодильником и трубкой для ввода газа, доходящей почти до дна колбы. Обратный холодильник снабжают дополнительным обратным холодильником (с охлаждением ацетоном с сухим льдом) и ртутным затвором. Перез газовводной трубкой помещают промывную склянку с 90 мл (1,1 моль) ВС13. В колбу помещают 67,5 г (1 моль) i[CH3NH3]Cl и 1 л хлорбензола и нагревают смесь при интенсивном перемешивании до кипения. Слабым током N2 выдувают ВС13 из промывной склянки в реакционную колбу. Смесь кипятят до тех пор, пока не перестанет выделяться НС1. Затем фильтруют и растворитель отгоняют в вакууме. Твердый бесцветный остаток очищают возгонкой в высоком вакууме (1ПЛ 160—164°C). Выход 71,3 г (95%).
б) □ Гексаметилборазин [2, 3]. Реакцию проводят в трехгорлой колбе на 250 мл, снабженной эффективной мешалкой, капельной воронкой с обратным холодильником и ртутным затвором. После заполнения прибора N2 в колбу помещают 9 г (0,37 моль) Mg-стружки, 25 г (0,11 моль) свежевозогнаниого Cl3Br3N3(CH3)3 и 75 мл эфира. Затем в течение нескольких часов при перемешивании добавляют 51 г (0,36 моль) СН31 в 25 мл эфира. Смесь кипятят несколько часов, отгоняют эфир и сублимируют остаток в высоком вакууме. С., 97 °C. Выход 13,3 г (73%).
в) □□ Трикарбонил(гексаметилборазин)хром [4, 5]. К двугорлой колбе емкостью 250 мл, снабженной краном для ввода N2 и находящейся в термостатированной при 30 °C масляной бане, присоединяют последовательно две трубки Шленка. Первую охлаждают ледяной водой, вторую помещают в сосуд Дьюара, охлаждаемый смесью метанол — сухой лед до —20 °C. В колбу помещают 1,3 г (5,0 ммоль) Cr(CO)3(CH3CN)3, 4,1 г (24,9 ммоль) B3N3(CH3)e и 80 мл диоксана. При интенсивном перемешивании отгоняют в вакууме водоструйного насоса ~50 мл диоксана в обе трубки Шленка и снова добавляют
1976 Глава 33. Металлоорганические комплексы
в колбу примерно такое же количество диоксана. Эту процедуру повторяют до тех пор, пока в ИК-спектре реакционного раствора не будут проявляться только полосы поглощения СО-групп комплекса Cr(CO)3[T]-B3N3(CH3)6]. Образовавшийся оранжевый раствор фильтруют (стеклянный пористый фильтр G4) и растворитель удаляют в вакууме. Игольчатые оранжевые кристаллы перекристаллизовывают из смеси бензол — гексан при комнатной температуре и сушат в высоком вакууме. Выход 1,36 г (90%).
Другие способы. Из Сг(СО)6 и гексаметилборазина фотохимической реакцией [6].
Свойства. Оранжево-желтые кристаллы в форме иголочек или палочек, возгоняются в высоком вакууме и хранятся несколько дней на воздухе.
141 °C (с разл.). Вещество хорошо растворяется в полярных органических растворителях, умеренно в циклогексане и практически не растворяется в пентане или гексане. В хлорированных углеводородах постепенно разлагается. ИК (циклогексан); 1963, 1867 [v(CO)J см-1; (КВг): 1374 (v(BN)] см-1.
Аналоги. Таким же способом получают В,В,В-триэтильное производное Cr(CO)3[T]-(C2H5)3B3N3(CH3)3], которое при действии расплавленного B3N3(C2H5)6 превращается в трикарбонил(гексаэтилборазин)хром. Для получения смешанных алкилзамещеиных комплексов Cr(CO)3(R3B3N3R3') (где R,R' = CH3, H-C3H7, Ц30-С3Н7) также целесообразно гран-Сг(СН3СИ)3(СО)3 смешать с 10-кратным избытком соответствующего гексаалкилборазина без растворителя и нагреть до температуры плавления боразина [7].
ЛИТЕРАТУРА
1. Ryschkewitsch G. Е„ Harris J. J., Sister Н. Н., J. Amer. Chem. Soc., 80, 4515 (1958).
2. Prinz R„ Dissertation Technische Hochschule Mflnchen, 1967.
3. Wiberg E., Hertwig K., Z. Anorg. Allgem. Chem., 255, 141 (1947).
4. Prinz R., Werner H., Angew. Chem., 79, 63 (1967).
5. Werner H., Prinz R., Deckelmann E., Ber., 102, 95 (1969).
6. Deckelmann K., Werner H., Helv. Chim. Acta, 53, 139 (1970).
7. Scotti M„ Werner H„ Helv. Chim. Acta, 57, 1234 (1974).
□ □ Бис[трикарбонил(п-Циклопентадиенил)молибден] (Mo-Mo) [Mo(n-C5H5)(CO)3]2
Наиболее удобным методом синтеза [Мо(т]-С5Н5) (СО)3]2 является взаимодействие Мо(СО)6 с дициклопентадиеном при нагревании:
2Мо(СО)6 + С10Н12----> [Мо(п-С5Н5)(СО)3]2+6СО + Н2
2-264,0 132,2 490,1 6-22,4л 22,4л
Реакцию проводят в атмосфере N2 в 500-мл колбе, снабженной обратным холодильником. В качестве такового используют трубку с водяной рубашкой, чтобы легко возгоняющийся Мо(СО)3 возвращать из верхней части холодильника в реакционную смесь. Холодильник Димрота для этого непригоден.
Смесь 26,4 г (0,10 моль) Мо(СО)6 и 130 мл (127 г; 0,96 моль) дициклопентадиена* до тех пор нагревают при 135—145°C (температура бани), эффективно перемешивая, пока на холодные части аппаратуры не перестанет сублимироваться Мо(СО)6 из горячей реакционной смеси. (Легко возгоняющийся
* Используют дициклопентадиен марки «для синтеза». Применение технического дициклопентадиена приводит к получению менее чистого продукта, обработка которого затруднена вследствие загрязнения вязкой полимерной примесью.
Комплексы М(г\-аро магический лиганд)
1977
Мо(СО)6 счищают в процессе реакции стеклянной палочкой или металлическим шпателем снова в реакционную смесь.) Затем смесь охлаждают до комнатной температуры. Если реакция прошла до конца, образуется красный кристаллический продукт. (Если же реакция не закончилась или температура реакции была недостаточной, то в основном кристаллизуется белый Мо(СО)6, в этом случае смесь нагревают еще. Если реакционную смесь нагревали слишком высоко, то после охлаждения выпадает коричневатая масса, состоящая в основном из полимеров дициклопентадиена.)
Красио-фиолетовые кристаллы отделяют и для удаления дициклопентадиена и других органических загрязнений многократно промывают петролей-ным эфиром. Не вступивший в реакцию и не отмытый петролейным эфиром Мо(СО)6 удаляют возгонкой (—12 ч, 50°C, высокий вакуум, сублиматор). Полученный теперь продукт для большинства синтезов является достаточно чистым. Аналитически чистый образец получают из профильтрованного раствора в СНС13, который для полноты осаждения разбавляют (20% по объему) пентаном и охлаждают до —35 °C. Выход (после кристаллизации) 14,7— 18,4 г (60—75%).
Другие способы. Окисление воздухом Mo(Tq-CsH5)H(CO)3 в эфирном растворе. Этот метод используют, когда в наличии есть Мо(т]-С5Н5)Н(СО)3. Можно использовать также и сырой продукт, получаемый по следующей схеме: Mo(CO)6-^-Na[Mo(T]-CsH5) (СО)3]->Мо(т]-С5Н5)Н(СО)з (методику см. ниже), не выделяя сам карбонилгидрид. Выход до 80% |[4, 5].
Свойства. Красно-фиолетовые кристаллы, на воздухе устойчивы, <пл 215— 217 °C. При —150 °C вещество возгоняется в высоком вакууме с сильным разложением. Почти не растворяется в петролейном эфире, хорошо растворяется в бензоле и метиленхлориде. Растворы, а также загрязненные препараты заметно неустойчивы на воздухе. ИК (КВг): 1957 (оч. с.), 1926 (оч. с.), 1904 (оч. с.), 1891 (оч, с.) [v(CO)l см“!. Кристаллическая структура: см. [6], d(Mo—Мо) =3,222 А.
литература
1. Wilkinson G., J. Amer. Chem. Soc., 76, 209 (1954).
2. Hayter R. G„ Inorg. Chem., 2, 1031 (1963).
3. King R. B„ Organometallic Syntheses, v. 1, p. 109 ff., Academic Press, New York 1965.
4. Piper T. S., Wilkinson G„ J. Inorg. Nucl. Chem., 3, 104 (1956).
5. King R. B., Stone F. G. A., Inorg. Synth., 7, 107 (1963).
6. Wilson F. C., Shoemaker D. P., J. Chem. Phys., 27, 809 (1957).
□ □ Бис[дикарбонил(т]-циклопентадиенил)молибден] (Mo—Mo) [Mo(n-C5H5)(CO)2]2
[Mo(T]-C5H5) (CO)3]2 -> [Mo(t]-C5H5) (CO)2J2 + 2CO
490,1 434,1 2-22,4л
Реакцию проводят в атмосфере N2 в одногорлой колбе, снабженной обратным холодильником. Пропуская слабый ток N2, кипятят в течение 24 ч при перемешивании магнитной мешалкой 11,0 г (22,4 ммоль) [Мо(т]-C5H5)(CO)3]2 в 100 мл толуола. Охлаждают до комнатной температуры и растворитель отгоняют в вакууме масляного насоса (25—35°C). Сухой остаток экстрагируют в аппарате Сокслета кипящим СН2С12 (150 мл). Для одновременного фильтрования экстракта дополнительно вставляют насадку, заполненную SiO2, на которую насаживают гильзу с реакционным остатком, заполненную стеклянной ватой. После окончания экстракции к раствору СН2С12 добавляют еще —70 мл эфира и оставляют в холодильной камере при —35 °C.
1978 Глава 33. Металлоорганические комплексы
Выпавшие красно-коричневые кристаллы отделяют и промывают пентаном. Из маточника после упаривания и добавления эфира получают еще дополнительно некоторое количество продукта. Общий выход 6,9 г (71%).
Другие способы. Облучение раствора М(т]-С5Н3) (СН3) (СО)3 [М=Сг, Мо, W] в пентане. Скорость реакции уменьшается в ряду Cr, Mo, W [2].
Свойства. Кристаллы красно-коричневого цвета, очень устойчивы на воздухе, (пл 215—217°C (с разл.). Вещество плохо растворяется в пентане и эфире, хорошо в бензоле и метиленхлориде. Растворы на воздухе неустойчивы. ИК (КВг): 1900 (с.), 1850 (с.) [v(CO)] см-1. Чистоту образца лучше всего определять по данным спектра ЯМР-'Н (С5Н5, 6 5,21; CDC13). Структура: см. [3], d(Mo—Мо) =2,448 А; асимметричные карбонильные мостиковые лиганды.
ЛИТЕРАТУРА
1. Curtis М. D., Klingler К. J., J. Organometal. Chem., 161, 23 (1978).
2. Alt И. G., J. Organometal. Chem., 124, 167 (1977).
3. Klingler R. J., Butler W. M., Curtis M. D., J. Amer. Chem. Soc., 100, 5034 (1978).
Тетрафтороборат трикарбонил (т]-циклогептатриенил)-молибдена(О) [Мо(СО)3(т)-С7Н7)] [BF4]
Комплекс [Мо(СО)з(т)-С7Н7)] [BF4] очень легко получается по реакции гидридного отщепления из соответствующего олефинового комплекса Мо(СО)з(т]6-С7Н8) при действии на него тетрафторобората трифенилметильного катиона.
Мо(СО)3(пб-С7Н8) + [(C„HS)3C] [BFJ->[Мо(СО)3(п-С7Н7)] [BFJ + (С6Н5)3СН
272,1 330,1 357,9 244,3
К раствору 27,2 г (0,10 моль) Мо(СО)3(т]6-С7Н8) в 150 мл CH2CI2 добавляют 36,3 г (0,11 моль) [(СбН3)3С] [BF4] и перемешивают 30 мин. Образовавшийся осадок отфильтровывают (G3), промывают (5x50 мл) CH2CI2 и высушивают в вакууме. Выход 30,4—32,2 г. (85—90%).
Свойства. Микрокристаллическое, устойчивое на воздухе вещество от оранжевого до коричневого цвета. Медленно темнеет при нагревании выше ~ 140 °C, не плавится до 270 °C. Умеренно растворяется только в очень полярных органических растворителях, например ацетоне. ИК (КВг): 2030, 1990, 1950 [v(CO)] см-1.
ЛИТЕРАТУРА
1. Dauben Н. I., Ноппеп L. R., J. Amer. Chem. Soc., 80, 5570 (1958).
2. King R. B„ Organometallic Syntheses, v. 1, p. 141, Academic Press, New York, London, 1965.
□ □ Трикарбонил (т]-циклопентадиенил)марганец
Мп(т)-С5Н5)(СО)3
(Цимантрен*)
Самым лучшим способом синтеза Мп(т]-С3Н5) (СО)3 по чистоте и выходу сырого продукта является карбонилирование дициклопентадиенида марганца при высоком давлении (способ 1). Использование очень неустойчивого на воз
* Как и все синонимические названия комплексов сандвичевого и полу-сандвичевого типа, название «цимантрен» используют лишь для упрощенного обозначения органических производных: «цимантренил-...».
Комплексы М(т\-ароматический лиганд) 1979
духе Мп(С5Н5)2 можно избежать, если применять в качестве исходного дихлоро (дипиридин) марганец (способ 2). Последний образует Мп(т]-С5Н5) (СО)3 по реакции восстановительного карбонилирования при высоком давлении в присутствии СбНв (способ 2).
Способ 1 [1—4]
Mn(CsHs)2 + 3CO --> Мп^-СвНвИСОЬНЧОДИ
185,1 3-22,4 л 2и4,1
При абсолютном исключении воздуха и паров воды во вращающийся автоклав на 500 мл помещают 92,5 г (0,5 моль) возогнанного Мп(С3Н5)2. После продувания системы СО (50 бар) создают рабочее давление СО 250 бар. Затем медленно внешнюю температуру доводят до 105±5°С. Для успешного протекания экзотермической реакции, быстро начинающейся при 65 °C, особое значение имеет точное соблюдение рабочей температуры. Через 20 ч автоклав охлаждают до комнатной температуры. Желтый продукт, обычно в виде очень твердой кристаллической массы, подвергают высоковакуумной возгонке при 60—120 °C. Использование чистого кристаллического исходного продукта позволяет получать Mn^-CsHs) (СО)3 с выходом 78,5—86,7 г (77—85%). Превышение температуры реакции (>150 °C) приводит к черным порошкообразным продуктам разложения. Если реакция прошла не полностью, при открывании автоклава может воспламениться непрореагировавший Мп(С5Н5)2; необходимо при переносе сырого продукта в аппаратуру для возгонки работать в атмосфере N2.
Малые загрузки [~25 г Мп(С5Н5)2] проводят во вращающемся автоклаве на 250 мл (начальное давление СО 300 бар; температура 105±5°С, максимальное рабочее давление ~390 бар); выход практически не изменяется. Можно использовать качающийся автоклав с внутренним измерением температуры.
Способ 2 [5, 6]
н2 2MnCl2(CsHsN)2 + Mg + 2CSH„ + 6СО ---->
2-284,0 24,3 2-66,1 6-22,4л
----> 2Мп(п-С6Н5) (СО)3 + 2[C5HsNH]C1 + MgCl2 + 2C6H6N 2-204,1 2-115,6 95,2 2-79,1
а) Дихлоро(дипиридии)марганец. К кипящему раствору 150 г (0,76 моль) МпС12-4Н2О в 1,5 л 96%-ного С2Н5ОН по каплям прибавляют 125 мл (123 г; 1,55 моль) пиридина. Охлаждают до комнатной температуры, осадок отделяют, промывают эфиром и сушат при 120°С на воздухе. Выход ~190 г (90%).
б) Трикарбоиил(т]-циклопеитадиенил)марганец. В качающийся автоклав на 1 л помещают 80 г (1,21 моль) свежеперегнанного циклопентадиена, 500 мл М,Н-диметилформамида, 156 г (0,55 моль) МпСЬ-гСбНзП (см. п. «а») и 40 г (1,67 моль) Mg-стружки. Автоклав плотно закрывают и дважды продувают Н2 при давлении 80 бар (насыщение реакционной смеси водородом имеет решающее значение для успеха синтеза!) и затем при начальном давлении 70 бар Нг+120 бар СО автоклав нагревают до 170—180 °C (внутренняя температура). Выдерживают при этой температуре 12 ч. Во время реакции максимальное рабочее давление составляет ~390 бар.
Охлажденную реакционную смесь переносят в аппаратуру для перегонки с паром (трехгорлая колба на 2 л) и сначала гидролизуют добавлением по каплям ~200 мл Н2О. В результате интенсивной перегонки с паром в приемнике собирается Мп(т]-С3Н5) (СО)3 в виде масла золотистого цвета, которое при охлаждении кристаллизуется. Продукт для полноты кристаллизации охлаждают ледяной водой, кристаллы отделяют, высушивают и возгоняют в
1980 Глава 33. Металлоорганические комплексы
высоком вакууме при 50—90 °C. Выход 48—58 г (43—52%, считая на
MnCl2-2CsHsN).
Свойства. Желтое, легколетучее и совершенно устойчивое на воздухе соединение, обладающее камфарным запахом. t„„ 77 °C. Большие кристаллы сильно преломляют свет. Хорошо растворяется в большинстве органических растворителей, не растворяется в воде. ИК (С6Н6): 2026 (оч. с.), 1935 (оч. с.) [v(CO)] см-1. ЯМР-’Н (ацетон-de, ТМС): б 4,96 [синглет, С5Н5]. ЯМР-13С (CDC13, ТМС): б 83,1 [С5Н5]; б 224,9 [СО]. Кристаллическая структура: см. [7]. (^-симметрия фрагмента Мп(СО)3".
ЛИТЕРАТУРА
1. Fischer Е. О., Jira R., Z. Naturforsch., 9b, 618 (1954).
2. Brown J. E., Shapiro H., пат. США 2818417 (31.12.1957); С. A. 52, 8535 (1958).
3. Brown J. E„ DeWitt E. G„ Shapiro H„ пат. США 3127351 (31.3.1964); С. A. 61, 5434 (1964).
4. Fischer E. 0., Plesske K, Ber., 91, 2719 (1958).
5. Cordes J. F., Neubauer D., Z. Naturforsch., 17b, 791 (1962).
6. King R. B., Organometallic Syntheses, v. 1, p. 112, Academic Press, New York, London, 1965.
7. Berndt A. F., Marsh R. E., Acta Crystallogr., 16, 118 (1963).
□ Трикарбонил (т)-пентабромциклопентадиенил)марганец
Мп(т)-С5Вг5)(СО)з
(Пербромцимантрен)
МпВг(СО)5 + CsBr4Na --> Mn(T]-C5Br5) (СО)3 + N2 + 2СО
274,9 407,7 598,6 22,4л 2-22,4л
2,75 г (10 ммоль) МпВг(СО)5 и 4,08 г (10 ммоль) тетрабромдиазоцикло-пентадиена* перемешивают в 150 мл бензола в течение 15 ч при 55 °C (внутренняя температура), а затем для окончания реакции кипятят еще 10 мин. Раствор упаривают до объема ~30 мл и хроматографируют на колонке с кизель-гелем (Merck 7734, 0,063—0,200 мм, активность II—III степени, 25x3 см, +15 °C) бензолом. Коричнево-желтую бензольную фракцию испаряют в вакууме водоструйного насоса, остаток высушивают и очищают возгонкой в высоком вакууме. При 40 °C возгоняется около 30—50 мг Mn2(CO)1(), а затем при 65°C —4,55 г (76%) Mn(f]-CsBrs) (СО)3.
Свойства. Устойчивые на воздухе прямоугольные пластиночки (при кристаллизации из петролейного эфира). /пл 117—117,5 °C. Пербромцимантрен неустойчив на свету и хранится в темноте. ПК (гексан): 2044 (с.-оч. с.), 1983 (оч. с.) [v(CO)] см"3. ЯМР-13С (CDC13, ТМС, 30°C): б 220,90 [СО]; б 86,50 [С5Вг5].
ЛИТЕРАТУРА
1. Herrmann W. A., Huber М., J. Organometal. Chem., 140, 55 (1977).
2. Cram D. J., Partos R. D., J. Amer. Chem. Soc., 85, 1273 (1963).
* Получение см. в работах [1, 2].
Комплексы М(г\-ароматический лиганд)
1981
□ Трикарбонил(т]-пирролил)марганец Mn(T]-C4H4N)(CO)3 (Азацимантрен)
Мп2(СО)10 + 2C4H5N ---> 2Mn(n-C4H4N) (С0)3 ф- Н2 ф- 4С0
390,0 2-67,1 2-205,1 22,4 л 4-22,4 л
7,8 г (20 ммоль) Mn2(CO)io и 15 мл (14,4 г, 0,22 моль) свежеперегнанного пиррола кипятят в 50 мл октана в течение 20 ч. После охлаждения до комнатной температуры реакционную смесь (от темно-желтого до оранжевого цвета) хроматографируют на колонке (50x2,5 см, от 10 до 15°C) с кислым А12О3 (Woelm-Pharma) петролейным эфиром. Предварительно упаривать растворитель нецелесообразно, так как значительная часть продукта может при этом теряться. С помощью петролейного эфира избавляются от октана, затем бензолом элюируют светло-желтую полосу непрореагировавшего Мп2(СО)ю (~600 мг, 8%). Продукт реакции смывают ацетоном в виде темно-желтой полосы. После отгонки растворителя в вакууме водоструйного насоса желтый маслянистый осадок сушат в вакууме масляного насоса, перекристаллизовывают из смеси петролейный эфир — эфир при —78 °C и, наконец, возгоняют в высоком вакууме при 35—38 °C. Выход 4,0—5,0 г (49—61%).
Свойства. Темно-желтые, сильно преломляющие свет, устойчивые на воздухе иголочки (кристаллизация из смеси эфир — петролейный эфир), не очень устойчивые на свету. /пл 41 °C. Вещество умеренно растворяется в алифатических углеводородах, хорошо растворяется в бензоле, эфире, метиленхлориде, ацетоне. ПК (циклогексан): 2032 (с.), 1974 (оч. с.), 1964 (оч. с.) ,[v(CO)] см-1. ЯМР-'Н (CDCI3, ТМС): б 6,07 фа-Н], 5,21 ф0-Н] (псевдотриплеты). ЯМР-13С (CD2C12, ТМС, +25 °C): б 223,57 [СО]; б 107,57 фа-С]; б 87,48 [Р-С].
ЛИТЕРАТУРА
1. Joshi К- К-, Pauson Р. L., Proc. Chem. Soc. (London), 1962, 326.
2. Joshi К. K„ Pauson P. L., Quazi A. R., Stubbs W. H., J. Organometal. Chem., 1, 471 (1964).
3. King R. B., Efraty A., J. Organometal. Chem., 20, 264 (1969).
□ Трикарбонил(т]-циклопентадиенил)рений Re(T)-C5H5)(CO)3
Поскольку бис(т)-циклопентадиенил)рений неизвестен, полусандвичевый комплекс Re^-CsHs) (СО)3 в противоположность своему аналогу цимантрену Mn(T]-CsHs) (СО)з получают из карбонилсодержащего исходного продукта введением в него циклопентадиенильного лиганда. Существуют два способа: 1) прямое взаимодействие Re2(CO)10 с дициклопентадиеном, идущее с расщеплением связи Me—Me; выход Re^-CsHs) (СО)3 составляет 63—73%; 2) взаимодействие ReBr(CO)5 с Т1С5Н5; Re^-CsHs) (СО)3 образуется с более высоким выходом.
Карбонилирование Re(r]-C5H5)2H при высоком давлении (выход 16% в пересчете на Re-металл) не является препаративным методом получения [4].
Способ 1 [1]
Re2(CO)10 ф- С10Н12 ->- 2Re('q-C5H5) (СО)3 ф- 4СО ф- Н2
652,5 132,2 2-335,3 4-22,4л 22,4л
6,53 г (10 ммоль) Re2(CO)10 кипятят с обратным холодильником в течение 6 ч в 40 мл дициклопентадиена. Коричневый раствор разбавляют 30 мл пентана и охлаждают смесь сухим льдом. Выпадающие белые кристаллы отфильтровывают и возгоняют в высоком вакууме при 50—60 °C. Выход 4 2— 4,9 г (63—73%).
1982 Глава 33. Металлоорганические комплексы
Способ 2 [2, 3]
ReBr(CO)e + Т1С5Н6 ----> Re(n-C5H6) (С0)3 + Т1Вг
406,2 269,5 353,3 284,2
Смесь 8,12 г (20 ммоль) ReBr(CO)s и 5,93 г (22 ммоль) TIC5H5 в 100 мл тетрагидрофурана кипятят с обратным холодильником 16 ч. После фильтрования и испарения растворителя в вакууме водоструйного насоса получают беловатый порошкообразный остаток, который подвергают возгонке в высоком вакууме при 50—60 °C. Выход чистого Re(T]-CsH5) (СО)з 5,8—6,0 г (87—90%).
Свойства. Бесцветные, устойчивые на воздухе кристаллы. tn]l 110—112 °C. Растворимы во всех органических растворителях, растворы устойчивы иа воздухе. ИК (СС14): 2041 (оч. с.), 1939 (оч. с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Green М. L. Н., Wilkinson G., J. Chem. Soc. (London), 1958, 4314.
2. Fischer E. O., Fellmann W., J. Organometal. Chem., 1, 191 (1963).
3. Несмеянов A. H., Анисимов К- H., Колобова Н. Е., Барышников Л. И.—* Изв. АН СССР, сер. хим., 1963, 193; С. А. 58, 12588 (1963).
4. Pruett R. L., Moorehouse Е. L„ Chem. Ind. (London), 1958, 980.
□ Трикарбонил (т]-галогенциклопентадиенил)рений Re(T]-C5H4X)(CO)3 (Х = С1, Вг, I)
Моногалогенозамещенные производные трикарбонил (трциклопеитади-енил) рения получают простым способом циклопентадиенилирования соответствующих пентакарбонилгалогенидов рения диазоциклопентадиеиом.
ReX(CO)6 + C6H4N2 --------> Re(n-CSH5X) (СО)3 + 2СО + N2
361,7 (Х = Cl) 92,1 369,8 (Х = Cl) 2-22,4л 22,4л
406,2 (Х = Вг) 414,2 (X = Вг)
453,2 (Х=1) 461,2(Х = 1)
Трикарбоиил(т]-хлорциклопентадиенил)рений Ие(т]-С5Н4С1)(СО)3
2,89 г (8 ммоль) ReCl(CO)s обрабатывают 15 ммоль диазоциклопентадиеиа в 60 мл бензола при 90 °C (температура бани) в течение 3 ч. Реакционная смесь становится коричневой, выделяется газ. После охлаждения до комнатной температуры растворитель отгоняют в вакууме водоструйного насоса досуха. Черный порошкообразный осадок обрабатывают (2x40 мл) петролейным эфиром (/кип 40—60°C). После отгонки растворителя (в вакууме водоструйного насоса) продукт перегоняют в высоком вакууме при 105 °C (воздушная баня). Желтый дистиллат застывает при комнатной температуре: бледно-желтое твердое вещество перекристаллизовывают из пентана. Выход 2,40 г (81%).
Трикарбоиил(т]-бромциклопентадиенил)рений Re(T]-CsH4Br) (СО)з
4,06 г (10 ммоль) ReBr(CO)s нагревают с 25 ммоль диазоциклопентадиена в 120 мл бензола в течение 12 ч. Смесь из желтой становится коричневой, выделяется газ. После охлаждения до комнатной температуры растворитель отгоняют досуха, черно-коричневый порошкообразный остаток многократно, порциями по 30 мл, встряхивают с петролейным эфиром (/Ип 40—60 °C), желтый экстракт упаривают до объема ~50 мл и охлаждают при (—35)—
Комплексы М (^-ароматический лиганд) 1983
(—78) °C. Беловатые, сантиметровой длины, еще немного клейкие игольчатые кристаллы очищают перегонкой в высоком вакууме (температура воздушной бани 90—110°C). Дистиллат устойчив на воздухе и застывает, образуя бесцветную твердую, крупнокристаллическую массу. Выход 3,56 г (86%).
Трикарбоиил(т]-иодциклопеитадиеиил)рений Re(T]-C5H4l)(CO)s
Реакцию проводят аналогично описанным выше [4,53 г (10 ммоль) ReI(CO)5 и 18 моль диазоциклопентадиена; время реакции 15 ч]. Сырой продукт экстрагируют (4x25 мл) петролейным эфиром (/кип 40—60°C), перегоняют в высоком вакууме при 130—145 °C (воздушная баня) и перекристаллизовывают из пентана (—35°C). Выход 3,69 г (80%).
Другие способы. Реакция непрямого (в несколько стадий) галогенирования Re(т]-С5Н5) (СО)з. Выход 38% (Х=С1), 88% (Х=Вг), 85% (Х=1) [2, 3].
Свойства. Устойчивые, легко возгоняющиеся кристаллы белого цвета, очень хорошо растворяются в большинстве используемых органических растворителей. /пл 38,5—39,5°С (Х = С1); 58—59°С (Х=Вг); 67—68°С (Х=1). Спектральные данные см. табл. 46.
Таблица 46. Спектральные данные для Re(T]-C5H4X) (СО)3
X ик см-’ (гексан) ЯМР-’На 6(CDC13, тмс, 33’С) ЯМР-«С 6(CDC13, ТМС, 32 °C)
v(CO) а-Н Р-н со С5Н4Х
С1 2031 (оч. с.), 1952 (оч. с.) 5,52 5,22 192,96 104,31, 83,14, 83,08
Вг 2029 (оч. с.), 1951 (оч. с.) 5,55 5,23 193,02 84,25, 86,07, 84,49
I 2029 (оч. с.), 1952 (оч. с.) 5,57 5,23 193,12 84,29, 87,12, 84,91
а Все сигналы проявляются как псевдотриплеты.
ЛИТЕРАТУРА
1. Herrmann W. А., Вег., 111, 2458 (1978).
2. Несмеянов А. Н., Анисимов К. Н., Макаров Ю. В., Колобова Н. Е. — Изв. АН СССР, сер. хим., 1969, 357; С. А. 70, 115 308 (1969).
3. Несмеянов А. Н., Колобова Н. Е., Макаров Ю. В., Анисимов К- Н. — Изв. АН СССР, сер. хим., 1969, 1992; С. А. 72, 21 761 (1970).
□ Бис[дикарбонил(т]-циклопентадиенил)железо] (Fe—Fe) [Fe(r]-C5H5)(CO)2]2
2Fe(CO)6 + C10H12 --> [Fe(T]-CsHs) (CO)2]2 + 6CO + H2
2-195,9 , u 132,2 353,9 6-22,4л 22,4л
В двухлитровой трехгорлой колбе, снабженной обратным холодильником и эффективной мешалкой, нагревают до кипения в течение 20 ч смесь 900 г (6,8 моль) дициклопентадиена и 200 мл (292 г, 1,49 моль) Fe(CO)s. Температуру масляной бани держат в интервале 140—160 °C. Применять электрическую плитку не рекомендуется ввиду возможности локального перегрева реакционной массы (может образоваться ферроцен!).
1984 Глава 33. Металлоорганические комплексы
При охлаждении коричневой реакционной смеси до О °C продукт выпадает в кристаллической форме (цвет от красно-коричневого до черного). Кристаллы отделяют, промывают несколько раз порциями по 50 мл петролейным эфиром и сушат в высоком вакууме. Выход 221—235 г (84—89%).
Получаемый таким образом кристаллический продукт аналитически не совсем чистый, однако он вполне пригоден для дальнейшего использования в синтезе. Аналитически чистый препарат получают после перекристаллизации из смеси метиленхлорид — эфир.
Другие способы. Взаимодействие Fe(CO)5 с циклопентадиеном в автоклаве. Выход 78% {3].
Свойства. Темно-коричневые, устойчивые на воздухе кристаллы, возгоняются в высоком вакууме при 140—150°C (с частичным разложением). /пл 194°C (с разл.). Вещество не растворяется в воде, плохо растворяется в али-?>атических углеводородах, хорошо растворяется в полярных растворителях хлороформ, тетрагидрофуран, ацетон). Темно-коричневые растворы постепенно разлагаются при доступе воздуха с выделением оксида Fe(III). ПК (КВг): 1955 (оч. с.), 1940 (оч. с.), 1756 (оч. с.) [v(СО)] см-1. Спектр ЯМР-’Н (CDC13, ТМС): 6 4,79 [синглет, С5Н5]. Кристаллическая структура: см. [4—7].
ЛИТЕРАТУРА
1. Piper Т. S., Cotton F. A., Wilkinson G., }. Inorg. Nucl. Chem., 1, 165 (1955).
2. King R. B., Stone F. G. A., Inorg. Synth., 7, 110 (1963).
3. Hallam B. F., Pauson P. L., J. Chem. Soc. (London), 1956, 3030.
4. Mills O. S., Acta Crystallogr., 11, 620 (1958).
5. Bryan R. F., Greene P. T„ J. Chem. Soc. (London), A 1970, 3064.
6. Bryan R. F., Greene P. T., Newlands Al. J., Field D. S., J. Chem. Soc. (London), A 1970, 3068.
7. Campbell 1. L. C., Stephens F. S., J. Chem. Soc. (Dalton), 1975, 226.
□ Тетракис[карбонил(г)-циклопентадиенил)железо] (6Fe—Fe) [Fe(n-C5H5)CO]4
2[Fe(n-C5H5) (CO)2]2 -> [Fe(n-C5HS)CO]4 + 4CO
2-353,9 595,8 4-22,4л
Способ 1 [1]
28,4 г (80 ммоль) [Fe^-CsHs) (СО)2]2 помещают в трубку Шленка емкостью 500 мл, снабженную обратным холодильником, и в течение 12 дней кипятят в 260 мл ксилола при непрерывном перемешивании магнитной мешалкой в атмосфере N2. Получившуюся темно-зеленую смесь охлаждают до комнатной температуры, с помощью шприца в атмосфере N2 (давление N2) переносят мелкодисперсный осадок (черного цвета) непосредственно в насадку для экстракции и промывают его ксилолом' (3X20 мл) и 20 мл бензола. Далее экстрагируют в течение ~2 недель в аппарате Сокслета 250 мл эфира, восполняя по мере необходимости потерю растворителя. После охлаждения до комнатной температуры отфильтровывают выпавшие черные, хорошо сформировавшиеся кристаллы, промывают их эфиром, пентаном и сушат в высоком вакууме. Выход 4,5—5,0 г (19—21%).
Способ 2 [2, 3]. Вариант этого синтеза, дающий существенно большие выходы (54%), заключается в кипячении [Fe^-CsHs) (СО)2]2 в ксилоле в присутствии трифенилфосфина (~1 : 1,2).
Комплексы ЛГ(т]-ароматический лиганд)
1985
Свойства. Устойчивые на воздухе, черные, октаэдрические кристаллы. /пл 220°C (с разл., с образованием ферроцена). Вещество хорошо растворяется в СН2С12 и нитрометане, растворяется в ацетоне и эфире, плохо — в спиртах, не растворяется в воде. Растворы комплекса устойчивы на воздухе. Галогены и соли серебра типа Ag[SbF6] окисляют нейтральный комплекс до катиона [Fe(n-C5H5)CO]4+.
ИК (КВг): 1620 [v(CO)] см-1. ЯМР-'Н (СНС13, ТМС): б 4,84 [С5Н5]. Кристаллическая и молекулярная структура: орторомбическая, пространственная группа симметрии РЬса (а=9,580 А, Ь= 15,118 А, с=28,635 А). Молекула имеет структуру кубана симметрии Та: 4 лиганда СО расположены над плоскостями тетраэдра из 4 атомов железа с 6Fe—Fe-связями [4].
ЛИТЕРАТУРА
1. King К. В., Inorg. Chem., 5, 2227 (1966).
2. White A. J., J. Organometal. Chem., 168, 197 (1979).
3. White A. J., Cunningham A. J., J. Chem. Educ., 56, 317 (1980).
4. Neuman M. A., Trinh-Toan, Dahl L. F., J. Amer. Chem. Soc., 94, 3383 (1972).
□ □ Гексафторофосфат трикарбонил(т)-циклопентадиенил)-железа(П) [Fe(i!-C5H5)(CO)3] [PFe]
a) Na(K) [Fe(n-C5H6) (CO)2J + C1CO2C2H5 ->
200,0 108,5
-----> Fe(r)-C5HS) (CO)2CO2C2HS + Na(K)Cl 250,0 58,4
6) Fe(T]-CsHs) (CO)2CO2C2HS + HC1 -> [Fe(n-C5H5) (CO)3]C1 + C2H5OH
250,0 22,4л 240,4 46,1
в) [Fe(n-CSHS) (CO)3]C1 + NHaPF,, -> [Fe(n-C5H5)(CO)3 ] [PFJ + NH4C1
240,4 163,0 350,0 53,5
К охлаждаемому льдом раствору 40 ммоль [Fe(T]-C5Hs) (СО)2]~ в 400 мл тетрагидрофурана (синтезированному из 7,08 г [Fe(r]-C5H5) (СО)2]2 и амальгамы натрия, полученной по методике, приведенной ниже) медленно прибавляют раствор 40 ммоль (4,34 г, 3,84 мл) этилового эфира хлормуравьиной кислоты в 20 мл тетрагидрофурана и перемешивают 1 ч при комнатной температуре. Затем отгоняют растворитель в вакууме водоструйного насоса и экстрагируют остаток, предварительно высушив его в высоком вакууме, бензолом (5Х Х20 мл). Экстракт фильтруют через пористый фильтр (G3), покрытый листочком фильтровальной бумаги, пропускают через раствор слабый ток НС1 и декантируют растворитель с образовавшегося осадка. Все последующие операции проводят в атмосфере инертного газа. Осадок многократно промывают эфиром (по 20 мл), сушат в высоком вакууме и растворяют в небольшом количестве воды. Из предварительно профильтрованного водного раствора высаживают продукт, добавив водный раствор 6,5 г (40 ммоль) NH4PF6. Осадок отфильтровывают, сушат и переосаждают из ацетона эфиром. Выход 5,5 г (40%).
Другие способы. Взаимодействием FeBr(f]-C5H5) (СО)2, А1Вг3 и СО во вращающемся автоклаве (16 ч, 60 °C, 240 бар) получают [Fe^-CsHs) (СО)з]-[А1Вг4]; затем гидролизуют и комплексный катион высаживают в виде гексафторофосфата при добавлении NH4PF6. Выход 37% [2].
9—1361
1986
Глава 33. Металлоорганические комплексы
Свойства. Устойчивые на воздухе оранжевые кристаллы, /пл >150 °C (с разл.). При 250 °C происходит быстрое разложение и образование ферроцена.
ИК (нуйол): 2120 (оч. с.), 2068 (оч. с.) [v(CO)]; 835 (оч. с.) [v(PFe)] см-1. ЯМР-]Н (ацетон-de, ТМС): б 6,14 [синглет, С8Н8].
ЛИТЕРАТУРА
1. Busetto L., Angelici R. J., Inorg. Chim. Acta, 2, 391 (1968).
2. Fischer E. 0.. Fichtel K„ Ber., 94, 1200 (1961).
□ □ Бис [дикарбонил (т]-циклопентадиенил) рутений] (Ru—Ru) [Ru(t)-C5H5)(CO)2]2
Способ 1 [1, 2]
co
a) 2RuCls ---->- [RuCl2(CO)8]2
[2-207,4 512,0
6) [RuCl2(CO)3]2 + 2NaC8H8 ---> [Ru(i]-C8H5) (CO)2)2
512,0 2-88,1 444,4
a) 5 г (~20 ммоль) RuC18- (1— 3)H2O нагревают с обратным холодильником в колбе на 250 мл в 50 мл сульфинилхлорида SOC12 в течение суток. Далее избыток сульфинилхлорида сливают, сухой остаток растворяют в ~500 мл метанола и в течение 24 ч нагревают при 65 °C в автоклаве иа 1 л при давлении СО 10 бар. Бледно-зеленый раствор упаривают в роторном испарителе, остатки СН3ОН удаляют в высоком вакууме. Полученный таким путем [RuCl2(CO)3]2 (4,1 г, 80%) используют без дальнейшей очистки.
б) 4,1 г (8 ммоль) [РиС12(СО)з]2 (см. ниже также методику его синтеза) растворяют в —200 мл абсолютного, насыщенного N2 тетрагидро фурана и прибавляют в течение 1 ч к раствору NaC8H8 (полученному из 3 г Na и 8 мл С8Н6 в 120 мл тетрагидрофурана; см. выше методику синтеза NaC8H5). Цвет реакционной смеси вскоре изменяется от красного до оранжевого. Оставляют кипеть на ночь с обратным холодильником, затем охлаждают до комнатной температуры и отгоняют растворитель в роторном испарителе. Твердый осадок коричневого цвета обрабатывают бензолом (5x20 мл), экстракт фильтруют и растворитель отгоняют. Сырой продукт оранжевого цвета очищают хроматографированием на А12О3 (нейтральный, активность I степени) бензолом или, в случае мелких загрузок, возгонкой в высоком вакууме при 140 °C. Выход 1,8 г (50%).
Способ 2 [3]
a) Rus(CO)12 + ЗС8Н„ ----> 3Ru(t]-CsH8) (Н) (СО)2 + 6СО
639,3 3-66,1 3-223,2 6-22,4л
б) 2Ru(r]-C8H8) (Н) (СО)2 + 1/2Оя --> [Ru(n-C8H6) (СО)2]2 + Н2О
2-223,2 11,2 л 444,4 18,0
В колбе емкостью 500 мл кипятят с обратным холодильником раствор 1,0 г (1,6 ммоль) Ru3(CO)i2 в 200 мл н-гептана и избыток циклопентадиена (3 мл) в течение 3—4 ч в инертной атмосфере. В конце реакции получают светло-желтый раствор [Ru(r]-CsHs) (Н) (СО)2], через который в течение — 10 мин продувают воздух. Образуется коричневая суспензия. После упаривания в роторном испарителе остаток извлекают —5 мл СН2С12 и хроматографируют на А12О3 (активность I степени, колонка 20 см) смесью СН2С12 — петролейный эфир (1 : 1). Выход 640—750 мг (60—70%).
Комплексы М(у\-ароматический лиганд) 1987
Свойства. Оранжевое, устойчивое на воздухе твердое соединение, 1Ы 185 °C. Хорошо растворяется в бензоле, ацетоне, CH2CI2 и СНС1,; не растворяется в петролейном эфире.
ИК (КВг): 1957 (с.), 1940 (с.), 1773 (с.), 1761 (ср.) [v(CO)l см-'. ЯМР-'Н (C6D6, ТМС): 6 4,17 [синглет, С5Н5].
ЛИТЕРАТУРА
1. Blackmore Т., Bruce М. I., Stone F. G. A., J. Chem. Soc. (London), А 1968, 2158.
2. Fischer Е. О., Vogler A., Z. Naturforsch., A 17b, 421 (1962).
3. Humphries А. Р., Knox S. A. R„ J. Chem. Soc. (Dalton), 1975, 1710.
□ □ Гексафторофосфат трикарбонил (т]-циклопентадиенил)-рутения(П) [Ки(т]-С5Н5)(СО)з] [PF6]
a) RuC1(t]-C5H5) (CO)a + Aid, + CO--> [Ru(i]-CeHe) (CO) J [A1C1J
257,6 133,3 22,4л 419,0
6) [Ru(r]-C5H5) (CO),] [А1СЦ] + NH4PF„ ->
419,0 163,0
----> [Ru(ri-C8H8) (CO),] [PF,] + NH4[A1C14] 395,2 186,8
К раствору 0,45 г (1,8 ммоль) RuC1(t]-CsHs) (СО)2 в 40 мл бензола, нагретому до 40 °C, прибавляют 0,95 г (7 ммоль) А1С1, и в течение —2 ч через реакционную смесь пропускают интенсивный ток СО. Пипеткой удаляют бензол, вязкий остаток высушивают, растворяют в —10 мл ледяной воды и фильтруют в насыщенный водный раствор NH4PF6 (0,3 г). Тотчас выпадающий белый осадок отфильтровывают, промывают 5 мл Н2О, высушивают и переосаждают из ацетона эфиром. Выход 425 мг (60%).
Свойства. Белые, устойчивые на воздухе кристаллы, хорошо растворимые в ацетоне, ацетонитриле, мало растворимые в воде, эфире или бензоле. ИК (КВг): 2137 (оч. с.), 2075 (оч. с.) [v(СО)] см-'.
ЛИТЕРАТУРА
1. Kruse А. Е„ Angelici R. J., J. Organometal. Chem., 24, 231 (1970).
2. Blackmore T., Cotton J. D„ Bruce M. I., Stone F. O. A., J. Chem. Soc. (London), A 1968, 2931.
□ □ Дикарбонил(ц-циклопентадиенил)кобальт
Со(т]-С5Н5)(СО)2
Для получения Со (t)-CsH5) (СО) 2 используют два принципиально различных способа. Способ 1—замещение циклопентадиенильного лиганда в Со(т]-С5Н5)2 (кобальтоцен) под действием СО при высоком давлении. По другому способу (способ 2) вводят СзН5-лиганд путем частичного замещения СО-груп-пы и расщепления связи металл—металл в Со2(СО)8. Если имеют в распоряжении Со2(СО)8, то способ получения Со(г]-С5Н5) (СО)2 без применения высокого давления является наиболее удобным.
Идущее с малым выходом взаимодействие СО и Со(т]-С5Н5)2 при атмосферном давлении ввиду доступности Со2(СО)а уже не имеет препаративного значения (выход <12%) [6].
9*
1988 Глава 33. Металлоорганические комплексы
Способ 1 [1—3]
Со(т]-С6Н6)2 + 2С0 ------> Co(T]-CsH5) (С0)2 + [СеН6-]
189,1 2-22,4л 180,1
Во вращающийся или качающийся автоклав на 500 мл (предельное давление не меньше 250 бар) помещают в инертной атмосфере раствор 47,3 г (0,25 моль) возогнанного Со(г]-С5Н3)2 (синтез см. выше) в 250 мл тетрагидрофурана. Систему дважды продувают СО при 50 бар и создают рабочее давление СО 100 бар. Через 10 ч нагревания при 130 °C (внутренняя температура) избыток СО сжигают, красно-коричневую реакционную смесь упаривают в вакууме водоструйного насоса при температуре не выше 30 °C и оставшееся красное масло перегоняют в вакууме (/кип 29—31 °C при 1 мм рт. ст., охлаждаемый льдом приемник). Выход 10,8—12,2 г (24—27%).
Способ 2 [4, 5]
Со2(СО)8 + 2С5Нв ----> 2Со(п-С6Н6)(СО)2+4СО Д- Н2
342,0 2-66,1 2-180,1 4-22,4л 22,4л
Смесь 34,2 г (0,1 моль) Со2(СО)3 (синтез см. выше) и 50 мл свежепере-гнаиного циклопентадиена кипятят в 300 мл метиленхлорида. Сильное выделение газа приводит вначале к вспениванию, целесообразно поэтому реакцию проводить в колбе емкостью 1 л. Через 30 ч растворитель отгоняют при комнатной температуре в вакууме водоструйного насоса, а остаток перегоняют (/кип 29—31 °C при 1 мм рт. ст.) в охлаждаемый льдом приемник.
Дистиллат загрязнен (до 10%) дициклопентадиеном, который удаляют хроматографически. Применяют охлаждаемую водой колонку с кизельгелем (Merck; 0,063—0,200 мм, 100x2 см); продукт очищают порциями по 5 мл, элюент — петролейный эфир. Дициклопентадиен отделяют в виде светло-желтой зоны, идущей впереди растянутой темно-красной полосы Co^-CsHs) (СО)2. Растворитель удаляют в вакууме водоструйного насоса при комнатной температуре. Общий выход очищенного продукта 11,7—13,7 г (32—38%).
Свойства. Темно-красная, подвижная, устойчивая на воздухе и на свету, чрезвычайно токсичная жидкость с характерным неприятным карбонильным запахом. /пл —22°С; /кип 139—140°С (710 мм рт. ст.), 75°С (22 мм рт. ст.), 64°C (12 мм рт. ст.), 38°C (2 мм рт. ст.), 29—31 °C (1 мм рт. ст.). Растворяется во всех применяемых органических растворителях. Со(г]-С5Н5) (СО)2 можно при —35 °C в темноте хранить месяцами, если хорошо защищать от доступа кислорода воздуха.
ИК (CS2): 2028 (с.-оч. с.), 1967 (оч. с.) [v(CO)] см-1; (циклогексан): 2036 (оч. с.), 1974 (оч. с.) [v(CO)J см-1. ЯМР-'Н (CS2, ТМС): 6 5,00 [синглет, С5Н5].
ЛИТЕРАТУРА
1. Fischer Е. О., Jira R., Z. Naturforsch., 10b, 355 (1955).
2. King R. В., Stone F. G. A., Inorg. Synth., 7, 112 (1963).
3. King R. B., Organometallic. Syntheses, v. 1, p. 115—118, Academic Press, New York, London, 1965.
4. Piper T. S., Cotton F. A., Wilkinson G„ J. Inorg. Nucl. Chem., 1, 165 (1955).
5. Rausch M. D„ Genetti R. A., J. Org. Chem., 35, 3888 (1970).
6. King R. B„ J. Amer. Chem. Soc, 84, 4705 (1962).
Комплексы М(у\-ароматический лиганд)
1989
□ □ Дикарбонил (т]-циклопентадиенил)родий Rh(n-C5H5)(CO)2
[RhCl(CO)2]2 + 2TIC5H5 —> 2Rh(T!-C5H5) (СО)2 + 2Т1С1
388,8 2-269,5 2-224,0 2-239,8
Суспензию 7,78 г (20 ммоль) [RhCl(CO)2]2 (синтез см. ниже в этой главе) и 16,2 г (60 ммоль) TIC5H5 в 300 мл петролейного эфира перемешивают 15 ч при комнатной температуре в инертной атмосфере и в темноте. После фильтрования (стеклянный пористый фильтр G3, вата) интенсивно-желтый раствор упаривают в вакууме водоструйного насоса и переконденсируют затем при 45 °C в высоком вакууме оранжево-желтое масло, обычно загрязненное коричнево-зелеными продуктами разложения, в трубку Шленка, охлаждаемую жидким азотом. Конденсат растворяют в 30 мл петролейного эфира, фильтруют через стеклянный пористый фильтр G3, покрытый слоем кизельге-ля (2 см), освобождаясь от белых, малорастворимых побочных продуктов, и растворитель удаляют в вакууме масляного насоса. Выход 7,1—7,6 г (80— 85%).
Свойства. Желтое, подвижное, неприятно пахнущее масло; чувствительно к свету и кислороду воздуха. L,., —11 °C, /кип —240 °C. Очень хорошо растворяется во всех органических растворителях. ИК (пленка): 2051 (оч. с.), 1987 (оч. с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Fischer Е. О., Bittier К, Z. Naturforsch., 16 b, 225 (1961).
2. Knight J., Mays M. J., J. Chem. Soc. (London), A 1970, 654.
3. Herrmann W. A., Kriiger C., Goddard R., Bernal I., J. Organometal. Chem., 140, 73 (1977).
□ □ ц-Карбонилбис[карбонил(ц-циклопентадиенил)родий]
(Rh—Rh) Rh2(r)-C5H5)2(CO)3
2Rh(r]-C6H6) (CO)2 -> Rh2(n-C5H6)2(CO)3+CO
2-224,0 420,0 22,4 л
4,0 г (17,9 ммоль) Rh(T]-C5H5) (CO) 2 кипятят в течение 5 дней в 70 мл бензола, временами продувая через систему N2. Уже при достижении температуры кипения наблюдается постепенное выделение газа и изменение цвета от светло-оранжевого до светло-красного. Сырой маслянистый остаток, полученный после упаривания растворителя в вакууме водоструйного насоса, хроматографируют на кизельгеле 60 (активность II—III степени, колонка 30x2 см, 10—15°С). Смесью пентан — бензол (5:1) элюируют оранжево-желтую зону непрореагировавшего Rh(r]-C5H5) (СО)2, который получают в виде оранжевожелтого масла после отгонки растворителя; выход 0,82 г (22%). Бензолом выделяют Rh2(r]-C5H5)2(CO)3 в виде быстро сдвигаемой темно-красной полосы. После отгонки растворителя получают 2,52 г (67%) биядерного комплекса родия.
Свойства. Интенсивно-красные, устойчивые на воздухе, прозрачные кристаллы (кристаллизация из смеси эфир — СН2С12; 20:1). /пл 139°C. Растворяется во всех органических растворителях. ИК (КВг): 1967 (оч. с.), 1816 (оч. с.) [v(CO)] см-1; (пентан): 1989 (оч. с.), 1841 (с.) [v(CO)] см-1. Кристаллическая структура: см. [3], d(Rh—Rh) =2,681 А.
1990 Глава 33. Металлоорганические комплексы
ЛИТЕРАТУРА
1. Fischer Е. О., Bittier К., Z. Naturforsch., 16 b, 835 (1961).
2. Herrmann W. A., Kruger C., Goddard R., Bernal 1., J. Organometal. Chem., 140,73 (1977).
3. Mills 0. S., Nice J. P., J. Organometal. Chem., 10, 337 (1967).
□ □ Дикарбонил (т]-пентаметилциклопентади1енил)родий Rh[n-C5(CH3)5](CO)2
Полностью метилированное производное Rh^-CsHs) (СО)2 может быть получено карбонилированием при нормальном давлении [Rh (р-С1) С1(т)-С5(СН3)3}]2 в присутствии Zn в качестве восстановителя:
[Rh(p,-Cl)Cl {n-CS(CH3)5}]2 + 4СО + 2Zn ---->
618,1 4-22,4л 2-65,4
----> 2Rh[r1-Ce(CH3)6] (СО)2 + 2ZnCla 2-294,2 2-136,3
В колбе на 500 мл, снабженной эффективным обратным холодильником и краном для ввода азота, готовят в атмосфере СО суспензию 3,00 г (4,9 ммоль) [Rh(p,-Cl)Cl{r]-C5(CH3)5}]2 (получение см. ниже в этой главе) в 270 мл СН3ОН и ~2 г (избыток) по возможности очень мелкой Zn-пыли. При интенсивном перемешивании через кипящую суспензию продувают сильный ток СО. Через —10 ч охлаждают до комнатной температуры, фильтруют (стеклянный пористый фильтр G3) и растворитель осторожно (30—35 °C) отгоняют в вакууме водоструйного насоса. Остаток очищают высоковакуумной возгонкой при 60°C (10-3 мм рт. ст.), а также колоночной хроматографией иа кизельгеле (Merck 7734; активность II—III степени, водяное охлаждение; 25x1,5 см) или на флоризиле. Продукт хорошо отделяется смесью пентан — бензол (3:1) в виде быстро смещаемой оранжево-красной полосы от незначительных количеств биядерного комплекса Rh2[rj-C3(CH3)5]2(p.-CO)2, представляющего собой сине-фиолетовую полосу. После отгонки растворителя в вакууме водоструйного насоса (<35°С) родиевый комплекс перекристаллизовывают из смеси пентан — эфир при (—35) — (—78) °C и сушат в высоком вакууме (30 мин). Для повышения выхода рекомендуется остаток после возгонки (см. выше) снова (если потребуется, то и многократно) подвергнуть восстановительному карбонилированию в тех же условиях. Общий выход для предложенных здесь загрузок колеблется от 1,5 до 1,8 г (52—63%). Возгонку можно не делать, если сырой продукт экстрагируют пентаном (~60 мл) и хроматографируют.
Получение аналогичного иридиевого комплекса 1ф]-С5(СН3)8](СО)2 проводят взаимодействием [Ir(p-Cl)Cl{r]-C5(CH3)s}]2 с Fe3(CO)12 в кипящем бензоле [2].
Свойства. Оранжевые, довольно устойчивые на воздухе кристаллы. Область плавления и разложения 101—107 °C. Очень хорошо растворяется во всех органических растворителях. Растворы также устойчивы на воздухе.
ЯМР-‘Н (CDC13, ТМС): 6(СН3) 2,05 (дублет, VRh-H=0,4 Гц). ЯМР-13С (CD3NO2, ТМС): 6(С3Н5) 102,8 (VRh-c=3,5 Гц); 6(СО) 196,6 ('JRh_c= = 83,2 Гц); 6(СН3) 11,2. ИК (КВг): 2000 (оч. с.), 1950 (оч. с.) [v(CO)l см-'; (CH3NO2): 2010 (оч. с.), 1953 (оч. с.) [v(CO)] см-1.
Термическая обработка (например, возгонка при 80—85 °C, 10—20 мм St. ст.) приводит к образованию биядерного комплекса Rh2[T|-C5(CH3)5]2(|i-CO)2. 'ействием сильных кислот и последующим депротонированием получают Rh2(n-C5 (СН3)5]2(ц-СО)(СО)2 [3].
Комплексы М(у\-ароматический лиганд) 1991
литература
1. Kang J. W., Maitlis Р. М„ J. Organometal. Chem., 26, 393 (1971).
2 Kang J. W„ Moseley K-, Maitlies P. M„ J. Amer. Chem. Soc., 91, 5970 (1969).
3. Plank J., Riedel D„ Herrmann W. A., Angew. Chem., 92, 961 (1980).
□ □ (ц-Карбонил) бис [карбонил (rj-пентаметилциклопентадие-нил)родий] (Rh—Rh) КЬ2[т]-С5(СНз)5]2(ц-СО)(СО)2
Для получения желаемого биядерного комплекса исходный моноядерный комплекс КЬ[г]-С5(СНз)5] (СО)2 протонированием HBF4 переводят в катионный ц-гидридный комплекс i[Rh2{T]-C5(CH3)5}2(p.-H) (ц-СО) (СО)2] [BF4], При действии NaOCH3 происходит депротонирование с количественным образованием нейтрального биядерного комплекса Rh2[r]-C5(CH3)5]2(p-CO) (СО)2. Образование связи металл—металл, происходящее при действии кислоты, весьма важно в препаративном отношении, поскольку исходный комплекс не превращается в биядерный ни термически, ни при действии света.
. a) 2Rh[rj-C6(CH3)6] (СО)2 + HBF4 ---->
2-294,2 87,8
-----> [Rh2{r1.C6(CH3)6}2(|i-H) (jx-CO) (CO)J [BF4] + CO
648,1 22,4 л
6) [Rh2{T]-C5(CH3)5}2(p.-H) (|X-CO) (CO)2] [BF4] + NaOCH3 ->
648,1 54,0
---> Rh2[n-C8(CH3)6]2(|x-CO) (CO)2 + NaBF4 -R CH3OH 560,3 109,8 32,0
а) К раствору 294 мг (1,0 ммоль) Rh[r]-C8(CH3)8] (CO)2 в 20 мл эфира при комнатной температуре и интенсивном перемешивании прикапывают ~0,5 мл (избыток) 54%-ной HBF4 в эфире. В конце прибавления кислоты наблюдается выделение газа и образование биядерного р-гидридного комплекса. Через 10 мин интенсивного перемешивания темно-коричневый пушистый осадок отфильтровывают (стеклянный пористый фильтр G3) от светло-желтого реакционного раствора, отмывают эфиром (4x5 мл) от кислоты, хорошо перемешивая осадок, и сушат 3 ч в высоком вакууме. Выход 314 мг (97%).
Темно-коричневый порошок в атмосфере N2 хранится при комнатной температуре; при доступе воздуха происходит быстрое депротонирование.
б) 314 мг (0,48 ммоль) солеобразного продукта протонирования по способу (а) заливают при комнатной температуре насыщенным на холоду раствором NaOCH3—СН3ОН (30 мг NaOCH3 в 10 мл СН3ОН), тщательно защищая от доступа воздуха и света. Депротонирование происходит мгновенно, и нейтральный биядерный комплекс Rh2[r]-Cs(CH3)5]2(p.-CO) (СО)2 выпадает в виде карминово-красного мелкокристаллического продукта, плохо растворимого в метаноле. Продукт тотчас фильтруют (стеклянный пористый фильтр G3, защита от света и воздуха), по возможности быстро промывают метанолом (3—4 мл) и сушат 4 ч в высоком вакууме (+10°C). Полученный таким образом продукт аналитически чист, однако может быть перекристаллизован из смеси тетрагидрофуран — эфир (10:1, —35 °C) в темноте. Выход 258 мг (96%).
Свойства. Карминово-красный порошок или красно-фиолетовые кристаллы, неустойчивые на свету, однако их можно длительное время хранить на воздухе при комнатной температуре без заметного разложения, если защищать от опта. При нагревании в запаянном капилляре при 141—143 °C происходит
1992 Глава 33. Металлоорганические комплексы
выделение газа и образование темно-синего КЬ2[г]-С5(СНз)5]2(р.-СО)2. Хорошо растворяется в тетрагидрофуране и нитрометане, образуя темно-красный раствор; растворяется в метиленхлориде и бензоле, не растворяется в пентане. Все растворы на свету изменяют окраску на темно-синюю вследствие диспропорционирования на Rh[^-C5(CH3)5] (СО)2 и Rha[iq-C5(CH3)5]2(p-CO)2.
ИК (ТГФ): 1952 (оч. с.), 1791 (оч. с.) [v(CO)] см-1; (КВг): 1947 (оч. с.), 1794 (оч. с.) [v(CO)J см-1. ЯМР-‘Н (ТГФ-ds, —10°С, ТМС): 6(СН3) 1,91 (синглет) .
Соединение можно хранить очень долго (не неограниченно) при температурах ниже —30 °C, тщательно предохраняя от света и доступа кислорода воздуха.
ЛИТЕРАТУРА
1. Plank J., Riedel D„ Herrmann W. A., Angew. Chem., 92, 961 (1980).
2. Plank J., Dissertation, Univers. Regensburg, 1980.
□ □ Бис[( ц-карбонил)(т]-пентаметилциклопентадиенил)родий] (Rh—Rh) Rh2[n-C5(CH3)5]2(p-CO)2
Указанный комплекс образуется при термическом диспропорционировании комплекса КЬ2[г]-С5(СНз)5]2(1-1-СО) (СО)2:
3Rh2[T]-C6(CH3)5]2(p-CO) (СО)2 -->
3-560,3
----> 2Rh2[r]-Cs(CH3)6]2(p,-CO)2 + 2Rh[t]-C6(CH3)6] (СО)2 + СО 2-532,3 2-294,2 22,4;л
280 мг (0,5 ммоль) Rh2[T]-Cs(CH3)5]2(p.-CO) (СО)2 (см. предыдущую методику) растворяют в 50 мл тетрагидрофурана и 3 ч кипятят с обратным холодильником. Раствор из темно-красного быстро становится темно-синим. Растворитель отгоняют в вакууме водоструйного насоса, фиолетовый остаток хроматографируют на SiO2 (Merck 7734; активность II—III степени, охлаждение водой; колонка 30X1,8 см). Бензолом элюируют оранжевую полосу РЬ[г]-С5(СНз)3] (СО)2, а эфиром — темно-синюю полосу КЬ2[т]-С5(СНз)5]2(р--СО)2. Оба комплекса после отгонки растворителя водоструйным насосом перекристаллизовывают из пентана соответственно из смеси эфир — пентан. Выход 87 мг (89%) Rh[,r)-C5(CH3)s] (СО)2; 142 мг (80%) Rh.<[T]-C5(CH3)6]2(n-СО)2; выходы определяют по вышеприведенному уравнению реакции.
Другие способы. Фотолиз КЬ2[г]-С3(СНз)5]2(р.-СО) (СО)2 при 0 °C в тетрагидрофуране. Выход —96% (по вышеприведенному уравнению) [2].
Нагревание твердого КЬ[г]-С5(СНз)5] (СО)2 (80—85 °C; 10—20 мм рт. ст.) с 30%-ным выходом [3].
Кипячение КЬ[г]-С5(СНз)5] (СО)2 и (СНз)зСМО в ацетоне с выходом 80— 90% [1].
Свойства. Сине-фиолетовые блестящие кристаллы, устойчивые на воздухе и на свету. Растворяется в СН2С1г, тетрагидрофуране и нитрометане, почти не растворяется в пентане. В кристаллическом состоянии соединение устойчиво вплоть до ~ 168 °C.
ИК (СН2С12): 1726 (оч. с.) [v(CO)] см"1; (КВг): 1734 (оч. с.) [v(CO)1 см-1. ЯМР-‘Н (CDC1, ТМС): б(СН3) 1,65 (псевдосинглет).
ЛИТЕРАТУРА
1. Herrmann W. A., Plank J., неопубликованные результаты 1978—1980 гг.
2. Plank Dissertation, Univers. Regensburg, 1980.
3. Nutton A.. Maitlis P. M., J. Organometal. Chem., 166, C21 (1979).
Комплексы М(г\-ароматический лиганд)
1993
□ [ц-Диоксид серы (8)]бис[карбонил(т]-циклопентадиенил)-родий] (Rh—Rh) Rh2(T)-C5H5)2(CO)2(p-SO2)
Образуется практически с количественным выходом по реакции замены мостикового СО-лиганда на SO2 при действии диоксида серы на раствор комплекса p,-CO[Rh(r]-CsH5) (СО)]2 в эфире при комнатной температуре:
Rh2(r]-C5H5)2(CO)2(p.-CO) + SO2-> Rh2(r1-C5H5)2(CO)2(li-SO2) + COj
420,0 22,4 л 456,1 22,4 л
Раствор 105 мг (0,25 ммоль) pi-COfRtRq-CgHs) (СО)]2 в 20 мл эфира насыщают при комнатной температуре сухим SO2. При этом часть ЗО2-комплекса выпадает из раствора в виде карминово-красных кристаллов. Перемешивают 2 ч, повторяют пропускание SO2 и оставляют стоять еще на 5 ч. После отгонки растворителя в вакууме водоструйного насоса кристаллический остаток хроматографируют на кизельгеле-60 (Merck 7734; активность II—III степени; охлаждение водой; колонка 20X1 см). Смесью СН2С12—ацетон (5:1) элюируют ярко-красную, быстро идущую полосу 5О2-комплекса. После перекристаллизации из смеси ацетон — эфир (10:1; —25 °C) препарат аналитически чистый. Выход НО мг (97%).
Свойства. Кирпично-красные игольчатые кристаллы, устойчивые на воздухе как в твердом виде, так и в растворах. Очень хорошо растворяется в СН2С12, диметилформамиде и нитрометане; раствор темно-красного цвета; хорошо растворяется в ацетоне, хуже в тетрагидрофуране, не растворяется в эфире и алифатических углеводородах. При нагревании в запаянном капилляре выше 150 °C наблюдается постепенное просачивание в запаянном месте, а при 203—205 °C происходит бурное разложение со значительным увеличением объема.
ИК (СН2С12): 2040 (ср. плечо), 2008 (оч. с.) [v(CO)] см-1; (КВг): 2000 (оч. с.) [v(CO)J, 1207 (с.) [v(SO)], 1056 (с.) [v(SO)], 502 (с.) [6(SO)] см-‘. ЯМР-'Н (CDCI3, +29 °C, ТМС): б(С5Н5) 5,65 (псевдотриплет), 2/(Rh, Н) = = 0,44 Гц. ЯМР-13С (CD2C12; +29°С; ТМС: обогащение 13СО 47%; 22,63 МГц): 6 91,66 (С3Н5, дублет); ‘/(Rh, С) =3,5 Гц; 6 189,22 (СО, дублет); ‘/(Rh, С) = = 83 Гц.
Молекулярная структура: транс-конфигурация фрагментов (r]-C5H5)Rh(CO), которые связаны связью металл—металл и мостиковым SO2-фрагментом с координацией по атому серы.
Аналоги
В биядерных [ц-диоксид серы (S)]-комплексах Pd2Cl2(p-SO2) (p-DPM) [где DPM=(C6H5)2PCH2P(C6H5)2], [IrH(CO)2P(C6H5)3]2(p.-SO2), [IrH(CO)2-P(C6H5)3]2(p.-SO2) и [Fe(r]-C5H5) (CO)2]2(p.-SO2) атом серы с двумя атомами кислорода и атомами металла находится в тетраэдрическом окружении [2; обзорная статья по 502-комплексам].
ЛИТЕРАТУРА
1. Herrmann W. A., Plank Ziegler М. L., Balbach В., Вег. (1981).
2. Mingos D. М. Р., Trans. Metal. Chem., 3, 1 (1978).
□ □ Дикарбонил(р-циклопентадиенил)иридий
1г(л-С5Н5)(СО)2
[1гС1(СО)3]2 + 2Т1С5Н5 -> 21г(т]-С5Н5) (СО)2 + 2СО + 2Т1С1
623,4 2-269,5 2-313,3 2-22,4л 2-239,8
1994 Глава 33. Металлоорганические комплексы
Суспензию 2,0 г (3,21 ммоль) [1гС1(СО)3]2 (синтез описан в работе [2]) и 5,4 г (20 ммоль) возогнанного Т1СзН5 кипятят в 200 мл н-гексана в течение 30 ч с обратным холодильником. Желтый раствор фильтруют (стеклянный пористый фильтр G3, вата), избавляясь от коричневой суспензии, растворитель отгоняют в вакууме водоструйного насоса, а остаток для очистки от примесей пропускают через колонку с кизельгелем (0,063—0,200; активность II—III степени, 15x2 см) в смеси гексан — бензол (3:1). После упаривания светло-желтого раствора в вакууме водоструйного насоса получают 1г(г]-С5Н5) (СО)2, который для удаления остатков бензола сушат в вакууме масляного насоса. Выход до 96%.
Свойства. Золотисто-желтое масло, некоторое время устойчивое на воздухе, с характерным карбонильным запахом. Перегоняется при 120—140 °C (10~3 мм рт. ст.). При длительном воздействии кислорода воздуха разрушается. Очень хорошо растворяется во всех органических растворителях. Дипольный момент р,(25 °C) =3,75±0,07 дебай (в циклогексане). ИК (CgH6): 2037 (оч. с.), 1957 (оч. с.) [v(CO)] см-1. ЯМР-'Н (CgDg, ТМС): б 4,19 [синглет, С8Н5].
ЛИТЕРАТУРА
1. Fischer Е. О., Brenner К. S., Z. Naturforsch., 17b, 774 (1962).
2. Ginsberg А. Р„ Koepke J. W-, Sprinkle C. R., Inorg. Synth., 19, 19 (1979).
□ □ Бис[р,-карбонил(т]-циклопентадиенил) никель] (Ni—Ni) [Ni(r)-C5H5)CO]2
Комплексы Ni2(r]-CsH5)2(CO)2 и №3(т]-С5Нз)з(СО)2 получают совместным диспропорционированием бис (грциклопентадиенил)никеля и тетракарбонилни-келя:
Ni(n-CSHS)2 + Ni(CO)4 ---> 2[Ni(n-CsHs)CO]2
188,9 170,8 303,6
Осторожно! Ni(CO)4 вследствие его высокой летучести чрезвычайно ядовит. Все операции необходимо проводить в хорошо действующем вытяжном шкафу!
В колбе на 250 мл, снабженной обратным холодильником, кипятят в течение 8 ч 15,1 г (80 ммоль) Ni(r]-C5Hs)2 (получение см. выше) в 120 мл бензола и 30 мл (39,3 г; 0,23 моль) Ni(CO)4. Растворитель и избыток Ni(CO)4 удаляют в вакууме водоструйного насоса, а осадок (оливкового цвета с красноватым оттенком) хроматографируют на нейтральном Д120з (активность I степени, 10—15 °C; 20x3 см) для отделения от совместно получающегося комплекса Ni3(r]-CsH5)3(CO)2. Колонку промывают эфиром до тех пор, пока элюат не будет окрашиваться в розовый цвет. Отогнав растворитель в роторном испарителе, получают 12,6 г (52%) красного мелкокристаллического [Ni(r]-C5HS)CO]2, достаточно чистого для препаративных целей. Аналитически чистый продукт получают либо высоковакуумной возгонкой при 105— ПО °C, либо перекристаллизацией из смеси эфир — петролейный эфир (2:1).
Свойства. Темно-зеленые блестящие кристаллы, в проходящем свете от темно-коричневого до пурпурно-красного цвета, устойчивые на воздухе. Темнокрасные растворы в бензоле, эфире или ацетоне неустойчивы на воздухе, вызывают раздражение кожи. /пл 139°C (с разл.). ИК (КВг): 1880 (оч. с.), 1830 (оч. с.) [v(CO)] см-1. Дипольный момент р(20°C) =0±0,38 дебай (в бензоле).
Олефиновые, ацетиленовые и аллильные комплексы 199 5
ЛИТЕРАТУРА
1. Fischer Е. О., Palm С., Вег., 91, 1725 (1958).
2. Tilney-Bassett J. F„ J. Chem. Soc. (London), 1961, 577.
Олефиновые, ацетиленовые и аллильные комплексы
□□ Трис(т]-аллил)хром Сг(г)-С3Н5)3
a) C3H5CI + Mg —-> Mg(r]1-C3HS)C1
76,5 24,3 100,8
б) CrCIs + SMg^^CaH^Cl ---> Cr(n-C3H5)3 + 3MgCl2
158,4 3-100,8 175,2 3-95,2
Mg(r]>-C3H5)C1
Аппаратура: трехгорлая круглодонная колба иа 1 л; мешалка; обратный холодильник с ртутным затвором, заполненным маслом для счета пузырьков; капельная воронка.
В реакционную колбу, охлаждаемую на ледяной бане, помещают 26,7 г (1,1 моль) Mg-стружки и 150 мл сухого эфира. Затем прикапывают 76,5 г (1 моль) C3HsCl в 150 мл эфира с такой скоростью, чтобы как можно меньше пузырьков газа улетучивалось через ртутиый затвор; содержимое колбы интенсивно перемешивают. Реакция начинается сразу и заканчивается примерно через 10 ч. Аллильный реактив Гриньяра представляет собой белый кристаллический порошок, тонкую суспензию в эфире, и в дальнейшем используется в таком виде или в виде разбавленного эфирного раствора (например, 0,5 М).
Сг(т]-С3 Н5)3
В трехгорлую колбу на 1 л, снабженную эффективной мешалкой, капельной воронкой и низкотемпературным термометром, помещают 15 г (94,6 ммоль) возогнанного* СгС13, вакуумируют и заполняют аргоном. После добавления сухого эфира** суспензию охлаждают до —30 °C и при интенсивном перемешивании в течение 2 ч добавляют 624 мл 0,5 М раствора аллил-магнийхлорида (312 ммоль) в эфире. Температура не должна подниматься выше —25 °C. Из интенсивно-красного раствора выпадает светлый осадок. По окончании прибавления перемешивают еще в течение 12 ч при —20 °C и образовавшийся темно-красный раствор фильтруют через фильтр с охлаждающей рубашкой при —20 °C под аргоном для отделения от солей Mg. Растворитель отгоняют при —30 °C в высоком вакууме и остаток извлекают пеитаиом, предварительно охлажденным до —10 °C. Фильтруют еще раз и промывают пентаном до тех пор, пока раствор не станет прозрачным. Сг(г]-СзН5)з вымораживают при —78 °C и сушат в высоком вакууме при —40 °C. Выход 8,3 г (50%).
Свойства. Крупные кристаллы, самовозгорающиеся при доступе воздуха н разлагающиеся выше —10°С. Соединение хранят при —78 °C (сухой лед).
* Используют возогнанный СгС13 (Е. Merck, Дармштадт) или получают его по методике, приведенной в данном руководстве.
** Эфир и пентан сушат предварительно CaCh, а затем в атмосфере Аг над K/Na-сплавом в специальной аппаратуре (см. рис. 459).
1996 Глава 33. Металлоорганические комплексы
ЛИТЕРАТУРА
1. Kharasch М. S., Fuchs С. F., J. Org. Chem., 9, 364 (1944).
2. Oberkirch W., Dissertation, T. H. Aachen, 1963.
3. Heimbach P., частное сообщение.
□ □ (т]4-Бицикло[2.2.1] гептадиен-2,5)тетракарбонилхром, (т]4-бицикло [2.2.1] гептадиен-2,5)тетракарбонилмолибден М(т]4-С7Н8)(СО)4 (М=Сг, Мо)
М(СО)6 + С7Н8 ---------> М(г]4-С7Н8) (СО)4 + 2СО .
М=Сг: 220,1 92,14 256,2 2-22,4л
М=Мо: 264,0 300,1
СГ(т14-С7Н8)(СО)4
В трубке Шленка на 250 мл, снабженной обратным холодильником и магнитной мешалкой, помещают 20,0 г (91 ммоль) Сг(СО)в, 32 мл норборнадиена (С7Н8), 120 мл метилциклогексана и кипятят смесь в течение 36 ч. Сублимирующийся в холодильнике Сг(СО)6 время от времени возвращают в трубку Шленка. По окончании реакции растворитель удаляют в высоком вакууме при 40 °C и затем сублимируют не вошедший в реакцию Сг(СО)6 (25 °C, 10-3 мм рт. ст.); при 80 °C и 10-3ммрт. ст. возгоняют продукт реакции. Выход 6,8 г (29%).
Свойства. Желтые, неустойчивые на воздухе иглы (1ПЛ 92—93 °C), растворимые в полярных органических растворителях, например СН2С12. ПК (СНС13): 2030 (оч. с.), 1950 (пл.), 1935 (оч. с.), 1897 (оч. с.) [v(CO)] см-1. ЯМР-'Н (СС14, ТМС): б 4,42 (триплет, 4 Н), 3,73 (мультиплет, 2 Н), 1,30 (триплет, 2 Н).
Мо(г]4-С7Н8)(СО)4
Аналогично описанному выше смесь 15,0 г (57 ммоль) Мо(СО)6 и 20,9 г (227 ммоль) норборнадиена в 100 мл метилциклогексана кипятят 24 ч. Образовавшуюся желеобразную массу охлаждают, фильтруют через стеклянную вату и промывают (3X15 мл) петролейным эфиром. Фильтрат упаривают до половины объема и охлаждают до —78 °C. Выпавшие кристаллы отделяют на фильтре с охлаждающей рубашкой и сушат в вакууме масляного насоса. Затем в аппаратуре для возгонки сублимируют не вошедший в реакцию Мо(СО)6 (комнатная температура, 10~3 мм рт. ст.). Продукт реакции возгоняют при температуре до 80 °C и 10-3 мм рт. ст. При более высокой температуре идет разложение. Выход 9,0 г (53%).
Свойства. Светло-желтые, неустойчивые на воздухе кристаллы (1ПЛ 77— 78 °C), даже в атмосфере азота постепенно темнеют. Растворяется в полярных органических растворителях (тетрагидрофуран или СН2С12). ПК (СНС13): 2045 (с.), 1955 (оч. с., шир.), 1892 (оч. с., шир.), <[v(CO)] см-1, ЯМР-'Н (СС14): б 4,85 (триплет, 4 Н), 3,78 (мультиплет, 2 Н), 1,33 (триплет, 2 Н).
ЛИТЕРАТУРА
1. Bennet М. A., Pratt L„ Wilkinson G., J. Chem. Soc. (London), 1961, 2037. 2. Pettit p„ J. Amer. Chem. Soc., 81, 1266 (1959).
3. King R. B., Organometallic. Syntheses, v. 1, p. 122fL, Academic Press, New York, London. 1965.
Олефиновые, ацетиленовые и аллильные комплексы 1997
□ □ Трикарбонил(т]6-циклогептатриен)молибден
Мо(СО)з(т)6-СтН8)
Мо(СО)6 + С7Н8 --> Мо(СО)3(г]б-С7Н8) + ЗСО
264,0 92,1 272,1 3-22,4л
(С7Н8 = циклогептатриен)
Смесь 52,8 г (0,20 моль) Мо(СО)6, 400 мл метилциклогексана и 100 мл чистого циклогептатриена кипятят в течение 16 ч в колбе Шленка на 1 л, снабженной магнитной мешалкой и обратным холодильником. Красный раствор охлаждают до комнатной температуры и фильтруют. Фильтрат охлаждают до —78 °C и выпавшие красные кристаллы отделяют на стеклянном фильтре с охлаждающей рубашкой. Избыток гексакарбонила и другие летучие загрязнения удаляют длительным (8 ч) нагреванием в вакууме масляного насоса при 25—35 °C. Выход 21—41 г (40—75%).
Свойства. Оранжево-красные кристаллы (/пл 100—102 °C), растворимы в большинстве органических растворителей. ИК (CH2CI2): 1985 (оч. с.), 1914 (оч. с.), 1881 (оч. с.) [v(CO)] см-1. ЯМР-‘Н (CS2, ТМС): б 6,04, 4,88, 3,56, 2,56.
Сг(СО)3(г]6-С7Н8) получают тем же способом, однако выход, как правило, существенно ниже (!5—20%). Большая часть Сг(СО)6 не участвует в реакции, возгоняясь и оседая на стенках холодильника. Избежать этого можно, применив специально сконструированную аппаратуру («карбонильная груша» Офеле; см. рис. 479) и подобрав соответствующие растворители. Выходы удается повысить до 80% и более.
Красные кристаллы (/пл 128—130 °C) растворимы в большинстве органических растворителей. ИК (СНС13): 1981 (оч. с.), 1915 (с.), 1884 (с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. King R. В., Organometallic Syntheses, v. 1, р. 123, Academic Press, New York, London, 1965.
2. King R. B., J. Organometal. Chem., 8, 139 (1967).
□ □ (г]1-Аллил)пентакарбонилмарганец Mn('r)1-C3H5)(CO)5
a) Mn2(CO)10 + 2Na ---> 2Na[Mn(CO)5]
390,0 2-23,0 2218,0
6) Na[Mn(CO)5] + C3H5Br --> Mn(n1-C3H5) (CO)5 + NaBr
218,0 121,0 236,1 102,9
О получении тетрагидрофуранового раствора Na[Mn(CO)s] из 0,46 г (20 ммоль) Na в 46 г Hg, 3,90 г (10 ммоль) Мп2(СО)ю и 100 мл тетрагидрофурана см. ниже методику синтеза МпН(СО)5.
Раствор Na,[Mn(CO)s] декантируют от ртути в трубку Шленка и при перемешивании добавляют 2,4 г (20 ммоль) аллилбромида. Раствор окрашивается в светло-желтый цвет, и выпадает осадок NaBr. Растворитель отгоняют, остаток экстрагируют петролейным эфиром (3x20 мл). Экстракт фильтруют, петролейный эфир удаляют в вакууме. Дальнейшую очистку проводят хроматографированием (нейтральный А12О3, активность I степени, петролейный эфир) или перегонкой. В последнем случае образуется заметное количество Мп(г]-С3Н5) (СО)4. Выход 3,4 г (72%).
1998 Глава 33. Металлоорганические комплексы
Свойства. Желтое масло (/кип 48°С/2 мм рт. ст.), на воздухе быстро разлагается. При нагревании происходит отщепление СО и образование Мп(т1-СзН5) (СО)4. ИК (гексаи): 2110 (сл.), 2079 (ср.), 2024 (оч. с.), 2004 (ср.) [v(CO)] см-1; (пленка): 1620 (сл.) [v(C=C)] см-1. ЯМР-’Н (CDC13, ТМС): 6 1,85 (дублет, СН2, 2 Н); 4,77 (мультиплет, =СН2; 2 Н); 6,15 (мультиплет, = СН, 1 Н).
ЛИТЕРАТУРА
1. McClellan W. R., Hoehn Н. Н., Cripps Н. N., Muetterties Е. L., Howk В. W., J. Amer. Chem. Soc., 83, 1601 (1961).
□ (т]-Аллил)тетракарбонилмарганец Mti(t]-C3H5)(CO)4
Мп(т]1-С3Н8) (СО)5 -> Мп(т]-С3Н5) (СО)4 + СО
236,1 208,1 22,4 л
2,36 г (10 ммоль) Мп('П1-С3Н5) (СО)з помещают без растворителя в трубку Шленка, соединенную через насадку с ртутным затвором, и в течение 3 ч нагревают при 85°C (масляная баня). За протеканием реакции следят по объему выделяющегося СО. По окончании реакции продукт очищают хроматографически (нейтральный А12О3, активность I степени, петролейный эфир) или перегонкой (£Мп 66°C при 16 мм рт. ст.). Выход 1,73 г (83%).
Свойства. Бледно-желтые кристаллы (/пл 55—56°C), на воздухе медленно разлагаются, растворимы в большинстве органических растворителей. ИК (пленка): 2066 (с.), 1965 (оч. с., шир.) i[v(CO)] см-1; 1501 (сл.) [v(CCC)] см-1. ЯМР-'Н (ацетон-de, ТМС): 6 1,80 (дублет, СН2-анти, 2 Н], 7(НН) = = 11,2 Гц; 6 2,80 [дублет, СН2-с«н, 2 Н], J (НН) =7,5 Гц; 6 4,95 [мультиплет, CH, 1 Н]. ЯМР-13С и динамику см. [2].
ЛИТЕРАТУРА
1. McClellan W. R., Hoehn Н. Н., Cripps Н. N., Muetterties Е. L., Howk В. W., J. Amer. Chem. Soc., 83, 1601 (1961).
2. Oudeman A., Sorensen T. S., J. Organometal. Chem., 156, 259 (1978).
□ □ т]6-Циклогептатриен(г]-циклопентадиенил)марганец
Мп(т)-С5Н5)(г]6-С7Н8)
Mn(n-CSH5)(CO)3 + C7H3----> Mn(T)-C5Hs) W’-C.Hg) + 3CO
204,1 92,1 212,1 3-22,4л
В 200 мл бензола растворяют 3,0 г (33 ммоль) свежеперегнаниого цикло-гептатриена и 2,04 г (10 ммоль) Mn(r]-C5Hs) (СО)3 и в течение 8 ч облучают ртутной лампой высокого давления на 500 Вт (стекло дюран; охлаждаемая водой «аппаратура с погружаемой лампой»)*. Происходит выделение газа и
* Время реакции определяется интенсивностью облучения. Полноту прохождения реакции определяют ИК-спектроскопически по наличию или отсутствию исходного Mn(r|-C5HS) (СО)3. Замещение карбонильных лигандов проходит значительно быстрее при использовании фотореакторов для тонкого слоя типа Falling Film [изготовитель — фирма Otto Fritz GmbH (6238 Hofheim/Tau-nus)]. «Аппаратура с погружаемой лампой» есть в продаже [изготовитель — фирма Otto Fritz GmbH (6238 Hofheim/Taunus); фирма H. Mangels, Labor-und Destillationstechnik (1503 Bornheim/Roisdorf)]; cm. [3], а также выше текст к рис. 463 и сл.
Олефиновые, ацетиленовые и аллильные комплексы 1999
изменение цвета раствора от светло-желтого до темно-красного. Затем раствор для очистки от коричневого нерастворимого осадка пропускают через колонку (10X3 см) с кизельгелем или кизельгуром, растворитель отгоняют в вакууме водоструйного насоса и остаток сушат в высоком вакууме. Образующийся темно-красный порошок возгоняют в высоком вакууме при 60— 70°C. Выход 1,85 г (87%). Чистый сублимат не должен давать в ИК-спектре никаких полос поглощения СО в области 1800—2100 см-1.
Свойства. Термически устойчивые, но неустойчивые на воздухе игольчатые кристаллы от темно-красного до черно-красного цвета. ЯМР-*Н (толуол-d8, ТМС): 6 3,62 [синглет, С5Н5, 5 Н]; 5,58; 4,40; 2,60; 2,40; 0,04 [мультиплет, С7Н8, 8 Н].
ЛИТЕРАТУРА
1. Pauson Р. L., Segal J. A., J. Organometal. Chem., 63, С13 (1973).
2. Pauson Р. L., Segal J. A., J. Chem. Soc., Dalton, 1975, 2387.
3. Herrmann W. A., Schweizer I., Z. Naturforsch., 33b, 1128 (1978).
□ □ Дикарбонил (п2-циклооктен) (^-циклопентадиенил)-марганец Мп(т]-С5Н5)(СО)2(т12-С8Н14)
Мп(г]-С5Н5) (СО)3 + С8Н14 -> Мп(т]-С8Н5) (СО)2С8Н14 + СО
204,1 110,2 286,3 22,4л
Аппаратуру для УФ-облучения общей полезной емкостью 500 мл и с боковым отводом для взятия проб (ср. рис. 466) обертывают алюминиевой
Рис. 466. Аппаратура для облучения УФ-светом по Штромейеру (W. Stroh-meier).
1 — нормальный шлиф 24/29 для взятия проб; 2 — нормальный шлиф 45/40 ; 3 — тефлоновый игольчатый кран; 4 — нормальный шлиф 14/23; 5 —магнит.
фольгой и помещают в стакан на 3 л с холодной водой. Затем загружают в инертной атмосфере 400 мл обескислороженного я-гексана, 3,6 г (17 ммоль) Mn(r]-CSHS) (СО)3, 25 мл (0,17 моль) циклооктена и в течение ~5 ч облучают при перемешивании (лабораторная погружаемая лампа, Original Hanau, тип
2000 Глава 33. Металлоорганические комплексы
TQ 150/Z3)*. Далее фильтруют через тонкий (2 см) слой целлюлозы, а растворитель и избыток циклооктена отгоняют в высоком вакууме при комнатной температуре.
Оставшийся желто-коричневый продукт переносят в аппаратуру для возгонки в парах растворителя (рис. 467) и при комнатной температуре возгоняют сначала в высоком вакууме не вошедший в реакцию Мп(т]-С6Й5) (СО)3. Затем Мп(г]-С5Н5) (СО)2С8Н14 возгоняют таким же образом в высоком вакууме
Рис. 467. Аппаратура для возгонки при температуре горячих паров растворителя.
1 — сосуд для возгонки с нормальным шлифом 45/40; 2 — круглодонная колба (до 250 мм) с нормальным шлифом 29/32 (обеспечивает образование горячих паров растворителя);
3 — игольчатый вентиль (фирмы-изготовители; Laborcenfer GmBH. О. Fritz GmBH; Schott и Gen); 4 — обратный холодильник с нормальным шлифом 29/32; 5—подвод охлаждающей воды.
при 65 °C. Используемая здесь аппаратура, нагрев в которой осуществляется парами тетрагидрофурана (рис. 467), исключает опасность локального перегрева. Выход 2,8—3,0 г (58—62%).
Свойства. Желтый порошок, устойчив на воздухе, не гидролизуется. /пл 110—111 °C. Растворяется во всех органических растворителях. ИК (н-гек-сан): 1964 (с.), 1905 (с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Fischer Е. О., Herberhold М., in: Essays in Coordination Chemistry, Birkhauser Verlag Basel, 1965.
2. Butler I. S., Coville N. J., Fenster A. E., Inorg. Synth., 16, 53 (1976).
* В ИК-спектре (в н-гексане) обнаруживают лишь весьма незначительное количество исходного комплекса (2028 (с.-оч. с.), 1947 (оч. с.) (v(CO)] см-1).
Олефиновые, ацетиленовые и аллильные комплексы 2001
□ □ Трикарбонил (т]-циклобутадиен) железо
Fe(CO)3(r)-C4H4)
Ре(СО)з(г]-С4Н4) лучше всего получают по реакции а,Р-дигалоген-элими-нирования из цис-3,4-дихлорциклобутена-1 в присутствии Fe2(CO)9
C4H4CI2 + Fe2(CO)9 ---•> Fe(CO)3(r]-C4H4)
123,0 363,8 192,0
В двугорлую колбу на 500 мл, снабженную обратным холодильником с ртутным затвором и краном для ввода азота, помещают 20,0 г (15,4 мл, 0,163 моль) цис-3,4-дихлорциклобутена-1*, 125 мл абсолютного бензола, 25,0 г (68,7 ммоль) Fe2(CO)9 и нагревают до 50—55 °C (температура масляной бани). При этом начинается интенсивное выделение газа, которое примерно через 15 мин полностью прекращается. Fe2(CO)9 добавляют порциями по 8 г с интервалами 15 мин до тех пор, пока выделение СО не прекратится полностью. Для этого потребуется около 140 г (~0,38 моль) Fe2(CO)9. Время превращения цис-2,3-дихлорциклобутена-1 в циклобутадиеновый комплекс ~6 ч.
Реакционную смесь фильтруют через стеклянный пористый фильтр (G3), покрытый ватой, а остаток на фильтре промывают петролейным эфиром до тех пор, пока растворитель не перестанет окрашиваться. Объединенный фильтрат испаряют в вакууме водоструйного насоса при температуре не выше 25 °C. Маслообразный остаток подвергают далее фракционной перегонке (колонка Вигре 20 см); бензол и Fe(CO)s при 20—30°C (16 мм рт. ст.) отделяют от Ре(СО)з(г|-С4Н4), перегоняющегося при 44°C (3,5 мм рт. ст.). Необходимо тщательно контролировать давление при перегонке. Продукт собирают в приемник, охлаждаемый льдом, где он постепенно закристаллизовывается в виде желтых игл. Температура масляной бани: 35—40°C [Fe(CO)s и бензол] и 65— 68 °C [Fe(CO)3(r]-C4H4)]. Тщательный контроль температуры и давления устраняет образование Fe3(CO)i2 во время перегонки.
В тех редких случаях, когда дистиллат в конце перегонки загрязнен зеленым Fe3(CO)i2, используют хроматографическую очистку на А12О3 бензолом (охлаждаемая водой колонка, защита от прямого света). Описываемую здесь методику можно применять и для больших загрузок, по меньшей мере для трехкратного количества реагентов. Выход 13,8—14,4 г (45—46%, считая на C4H4CI2).
Свойства. Бледно-желтое масло, неустойчивое на свету и на воздухе (/Кип 47 °C при 3 мм рт. ст.) или соответственно желтые призмы (/Пл 28—32 °C). Растворяется во всех органических растворителях, образуя очень чувствительные к окислению растворы. В отсутствие следов О2, влаги и в темноте при —30°C хранится в течение многих месяцев без разложения. ИК (пленка): 2055, 1985 [v(CO)] см-1. ЯМР-‘Н (СС14, ТМС): 6 4,00 [синглет, С4Н4].
ЛИТЕРАТУРА
1. Amiet R. G., Reeves Р. С., Pettit R„ Chem. Commun., 1967, 1208.
2. Emmerson G. F„ Watts L., Pettit R., J. Amer. Chem. Soc., 87, 131 (1965).
3. Pettit R„ Henery J.. Org. Synth., 50, 21 (1970).
4. Pettit R., Emerson G. F., Advan. Organometal. Chem., 1, 143 (1964).
5. Pettit R., Henery J., Org. Synth., 50, 36 (1970).
* Фирма Fluka-Feinchemikalien (Buchs/Schweiz) поставляет этот препарат; получение см. [5].
10—1361
2002 Глава 33. Металлоорганические комплексы
□ т]-Бутадиен(трикарбонил)железо Fe(r]-C4H6)(CO)S
Fe(CO)6 + С4Н6 ---> Fe(i]-C4H6) (СО), + 2СО
195,9 54,1 194,0 2-22,4л
Примерно 55 мл (37,7 г, 0,70 моль) бутадиена перекондеисируют из стального баллона в охлажденную до —78 °C ловушку (ацетон+сухой лед) и затем быстро переносят в охлажденный до той же температуры вращающийся автоклав на 250 мл, в котором находится несколько кусков сухого льда. Затем тотчас прибавляют 50 мл (73,0 г, 0,37 моль) Fe(CO)s, закрывают автоклав и в течение 12 ч реакционную смесь нагревают при 140±5°С. После охлаждения автоклава до комнатной температуры и стравливания избыточного давления продукт реакции, представляющий собой жидкость оранжевого цвета, фракционируют. Сначала в вакууме водоструйного насоса при 35 °C отгоняют 40 г непрореагировавшего Fe(CO)5 (1кш 40°C при 55 мм рт. ст.). Основной продукт перегоняется в виде оранжевой жидкости в вакууме масляного насоса (температура бани —70°C). Повторной перегонкой при тех же условиях получают аналитически чистый препарат. Выход —11 г (15%, считая иа взятый Fe(CO)5).
Свойства. Устойчивая на воздухе жидкость оранжевого цвета. /пл 19 °C, <кип 47—49°C (0,1 мм рт. ст.). Смешивается со всеми органическими растворителями, не растворяется в воде. ПК (С2С14): 2053 (оч. с.), 1985 (оч. с.), 1975 (с.-оч. с.) [v(CO)] см-1. Кристаллическая структура: см. [4]; d(C—С) = = 1,45 А (соответственно 1,46 А).
ЛИТЕРАТУРА
1. Reihlen Н., Gruhl A., Hessling G. V., Pfrengle О., Liebigs Ann. Chem., 482, 161 (1930).
2. Hallam В. F„ Pauson P. L., J. Chem. Soc. (London), 1958, 642.
3. King R. B., Organometallic Syntheses, v. 1, p. 128, Academic Press, New York, London, 1965.
4. Mills O. S., Robinson G., Acta Crystallogr., 16, 758 (1963).
□ Бис(т)-бутадиен)карбонилжелезо Fe(r)-C4H6)2CO
В противоположность термическому декарбонилированию Fe(CO)s в присутствии бутадиена, приводящему к получению трбутадиен (трикарбонил) железа, фотохимическое замещение идет дальше с образованием бис(ц-бута-диеи) карбонилжелеза.
ftv
Fe(CO)5 + 2C4Hj --> Fe(r]-C4H6)2CO 4CO
195,9 2-54,1 192,0 4-22,4л
В сосуд иа 400 мл с погружаемой лампой (рис. 462), снабженный термометром и магнитной мешалкой и приспособленный для внешнего и внутреннего охлаждения, помещают в атмосфере аргона раствор 10,0 г (51 ммоль) Fe(CO)s и 54,1 г (1,0 моль) бутадиена в 300 мл пентана и присоединяют прибор к газовой бюретке. Охлаждающая жидкость протекает сначала через внешнюю, а затем через внутреннюю охлаждающие рубашки.
Раствор охлаждают до —35 °C и при перемешивании облучают ртутной лампой высокого давления (Philips HPK125W). Газовыделение начинается сразу и заканчивается примерно через 48 ч. Уже в процессе облучения происходит кристаллизация образующегося бис (г]-бутадиен) карбонилжелеза, который оседает на внешней стенке вследствие градиента температур. Таким образом внутренняя стенка охлаждающей рубашки остается прозрачной.
Олефиновые, ацетиленовые и аллильные комплексы 2003
При нагревании до 20 °C продукт снова растворяется. Для отделения от нерастворимых продуктов разложения суспензию фильтруют, при охлаждении фильтрата до —78 °C снова выпадают кристаллы Fefq-CJdehCO. Их отделяют от маточника и высушивают в вакууме. Выход 8,2—8,6 г (85—90%).
Свойства. При перекристаллизации из пентана (—78 °C) получают оран-жево-красные, тетрагональные, сантиметровой длины кристаллы ((Пл 130— 135 °C, с разл.). Соединение устойчиво на воздухе при комнатной температуре. ИК (н-гексаи): 1984,5 [v(CO)] см-1. ЯМР-*Н (C6De, ТМС): 6 4,27 (мультиплет, 2 Н), 1,0 (мультиплет, 2 Н); —0,35 (мультиплет, 2 Н).
ЛИТЕРАТУРА
1. Koerner von Gustorf Е., Buchkremer J., Pfajfer Z., Grevels F.-W., Angew. Chem., 83, 249 (1971).
□ □ Трикарбонил (г]4-циклогептатриен)железо
Fe(CO)3(r]4-C7H8)
Fe(CO)6 + C7H8 --> Ре(СО)3(г]4-С7Н8) + 2CO
195,9 92,1 232,0 2-22,4 л
В колбу на 250 мл, снабженную обратным холодильником с ртутным затвором, помещают 25,0 мл (36,8 г, 0,19 моль) Fe(CO)g и 25 мл циклогепта-триена в 70—80 мл этилциклогексана* и смесь кипятят 24 ч (выделение СО). Растворитель при слабом нагревании удаляют в вакууме масляного насоса. Остаток очищают перегонкой в высоком вакууме при ~60°С. Выход —8,7 г (20%).
Свойства. Жидкость красного цвета, не очень устойчива на воздухе. (пл — 5°C, (кип 70°C (0,4 мм рт. ст.). Растворяется во всех органических растворителях. ИК (пленка): 2050 (оч. с.), 1975 (оч. с.) [v(CO)] см-1, дипольный момент р,=2,43±0,04 дебай.
ЛИТЕРАТУРА
1. Burton R., Pratt L„ Wilkinson G., J. Chem. Soc. (London), 1961, 594. 2. Dauben H. J., Bertelli D. J., J. Amer. Chem. Soc., 83, 497 (1961).
□ □ т]-Аллил(трикарбонил)хлорожелезо FeCl(r)-C3H5)(CO)3
Fe2(CO)e + C3HSC1 --> FeCl(r1-C8Hs) (CO), + Fe(CO)6 + CO
363,8 76,5 216,4 195,9 22,4л
Получение проводят по прописи, предложенной для синтеза описываемого ниже FeBr(r)-C3H5) (СО)з [36,4 г (0,10 моль) Fe2(CO)9; 7,65 г (0,1 моль) C3H5CI, 50 мл гексана, 40°C, время реакции 120 мин]. После окончания реакции раствор фильтруют и до отгонки Fe(CO)s на несколько минут оставляют стоять на воздухе, а затем обрабатывают, как это описано ниже. Если окисление на воздух® не делают, то после упаривания получают красную, очень неустойчивую на воздухе смолу, из которой высоковакуумиой сублимацией полчают кроме основного соединения еще некоторое количество темно-красно
* В качестве растворителя используют также я-октан (1кип 136 °C) или КСИЛОЛ ((кип -138 °C).
10*
2004 Глава 33. Металлоорганические комплексы
го пирофорного кристаллического соединения, подробнее не исследованного. Выход РеС1(г]-СзН5) (СО)3 5,4 г (25%).
Свойства. Желтые, не очень устойчивые на воздухе иголочки. /пл 88— 89 °C (с разл.). Растворяется во всех органических растворителях. ИК (КВг): 2091 (оч. с.), 2013 (оч. с.), 1985 (оч. сл., плечо) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Murdoch Н. D., Weiss Е„ Helv. Chim. Acta, 45, 1927 (1962).
□ □ ц-Аллил (трикарбонил) броможелезо
FeBr(n-C3H5)(CO)3
Fe2(CO)9 + C3H5Br --> FeBr(r]-C3H5)(CO)3 + Fe(CO)5 + CO
363,9 121,0 260,9 195,9 22,4 л
Суспензию 36,4 г (0,10 моль) Fe2(CO)9 в 50 мл гексана и 12,1 г (0,10 моль) 1-бромпропена-2(аллилбромида) нагревают при перемешивании до 40 °C. Через 90 мин образуется темно-красный раствор, который фильтруют для удаления FeBr2, образовавшегося вследствие частичного разрушения. После удаления Fe(CO)s и растворителя в вакууме водоструйного насоса при 20—30 °C остаток оставляют кристаллизоваться при 0 °C. Продукт образуется в виде желто-коричиевых призм (4,0 г). Дальнейшим упариванием маточника получают еще 6,1 г кристаллов. Для очистки продукт перекристаллизовывают из петролейного эфира (tKIln 40—60°C). Выход 9,9 г (38%).
Свойства. Желто-коричневые призмы, не очень устойчивые на воздухе, /пл 86—87 °C (с разл.). Растворимы во всех обычных органических растворителях. ИК (КВг): 2095 (оч. с.), 2082 (сл., плечо), 2020 (оч. с.), 1991 (сл., плечо) ,[v(СО)] см-1.
ЛИТЕРАТУРА
1. Murdoch Н. D„ Weiss Е., Helv. Chim. Acta, 45, 1927 (1962).
□ □ т]-Аллил (трикарбонил) иодожелезо
FeI(r]-C3H5)(CO)3
Fel (грСзНд) (СО)з удобно получать взаимодействием аллилиодида либо е Fe(CO)s (способ 1), либо с Fe2(CO)9 (способ 2).
Способ 1
Fe(CO)5 + С3Н61 —> Fel(n-C3HS) (СО)3 + 2СО
195,9 168,0 307,8 2-22,4л
Смесь — 19,6 г (13,4 мл, 0,10 моль) Fe(CO)3 и 11,8 г (70 ммоль) 1-иод-пропена-2 (аллилиодида) перемешивают в течение 50 ч при 43—46 °C (внутренняя температура) и затем в вакууме масляного насоса доводят досуха. Образовавшийся коричневый мелкокристаллический продукт очищают возгонкой в высоком вакууме при —50°C. Выход —15,5 г (72%, считая на аллил-иодид).
Способ 2
Fe2(CO)9 + С3Н61 ---> FeI(r]-C3H5) (СО)3 + Fe(CO)6 + СО
363,8 168,0 307,9 195,9 22,4л
Олефиновые, ацетиленовые и аллильные комплексы 2005
Получают по методике, аналогичной методике синтеза FeBr(T]-C3H5) (СО)3 [36,4 г (0,10 моль) Fe2(CO)9, 50 мл гексана, 16,8 г (0,10 моль) С3Н31, 40°С, время реакции 45 мин]. Выход 20,6 г (67%).
Другие способы. Замена галогена в FeCl(r]-C3Hs) (СО)3 при действии Nal; выход 42% [2].
Свойства. Не очень устойчивое на воздухе кристаллическое соединение темно-коричневого цвета. /пл 98 °C (с разл.). Растворяется в большинстве используемых органических растворителей. ИК (КВг): 2078 (оч. с.), 2015 (оч. с.), 1982 (ср., плечо) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Plowman R. A., Stone F. G. A., Z. Naturforsch., Tell В, 17, 575 (1962).
2. Murdoch И. D., Weiss Е., Helv. Chim. Acta, 45, 1927 (1962).
□ □ Бис[т]-аллил(трикарбонил)железо] (Fe—Fe) [Fe(TC3H5)(CO)3]2
Для синтеза ,[Fe(r]-C3H5) (CO)3]2 бромокомплекс FeBr(r]-C3H5) (CO)3 no аналогии с реакцией Гриньяра обработкой Zn превращают сначала в «Fe(ZnBr) (r)-C3Hs) (СО) з», который при действии другой порции FeBr(rj-С3Н5) (СО)3 дает конечный продукт со связью металл—металл:
a) FeBr(r]-C6H6) (СО)3 + Zn -> {Fe(ZnBr) (т]-С3Н6) (СО)3}
260,9 65,4 326,3
б) {Fe(ZnBr) (г]-С3Н5) (СО)3} + FeBr(n-C3H5) (СО)3 ->
326,3 260,9
—[Fe(n-C3H5) (СО)3]2 + ZnBr2 361,8 225,4
Суспензию 5,0 г (19,2 ммоль) FeBr(r]-C3H5) (СО)3 и 2,94 г (45 ммоль) цинковой пыли интенсивно перемешивают в 150 мл эфира при комнатной температуре. Вскоре раствор приобретает темно-красный оттенок, а через 2 ч становится желто-коричневым. Избыток Zn отделяют, а к фильтрату добавляют еще 5,0 г (19,2 ммоль) FeBr(r|-C3H5) (СО)3. Раствор сразу становится красным. Через 30 мин перемешивания фильтруют, фильтрат упаривают в вакууме водоструйного насоса до консистенции масла и остаток при интенсивном перемешивании экстрагируют петролейный эфиром (1КИП 40—60 °C) тремя порциями по 100 мл. Объединенный экстракт упаривают до объема 150 мл и оставляют кристаллизоваться при температуре сухого льда. Приблизительно через 1 ч выпавшие кристаллы отделяют и несколько часов сушат при —78 °C в высоком вакууме. Выход 3,0 г (43%).
Свойства. Темно-красные, довольно устойчивые на воздухе кристаллы. Медленно разлагаются в растворе пентана при комнатной температуре. При действии Н2 идет образование Fe(r]2-C3H6) (СО) 4. При действии бутадиена-1,3 идет вытеснение аллильного лигаида и образование Fe(r]-C4H6) (СО)3. Растворимы в большинстве органических растворителей. Растворы чувствительны к окислению. В растворах происходит частичная диссоциация с образованием радикала [Fe(r)-C3H5) (СО)3]‘ . ИК (КВг): 2020, 1965 Jv(СО)] см-1; (гексаи): 2015, 1969 [v(CO)] см-1.
2006 Глава 33. Металлоорганические комплексы
ЛИТЕРАТУРА
1. Murdoch Н. D., Lucken Е. А. С., Helv. Chim. Acta, 47, 1517 (1964).
2. Putnik Ch. F„ Welter J. J., Stucky G. D., D’Aniello M. J., Sosinsky B. A., Kirner J. F„ Muetterties E. L., J. Amer. Chem. Soc., 100, 4107 (1978).
3. Muetterties E. L„ Sosinsky B. A., Zamaraev К- I., J. Amer. Chem. Soc., 97, 5299 (1975).
4. Несмеянов A. H., Крицкая И. И., Устынюк Ю. А., Федин Э. И. — ДАН СССР, 1967, т. 176, с. 341.
Тетрафтороборат трикарбонил(т]5-циклогептадиенил)железа [Fe(CO)3(T]5-C7H9)][BF4]
Fe(CO)3(Ti‘-C7H8)+HBF4 ----> [Fe(CO)3(n5-C7II9)l [BF4]
232,0 87,8 319,8
В полиэтиленовом стакане растворяют 23,2 г (0,1 моль) Ре(СО)з(т14-С7Нз) (его синтез приведен выше) в 150 мл пропионового ангидрида. Раствор охлаждают при перемешивании магнитной мешалкой на ледяной бане. При добавлении по каплям 22,1 г (0,12 моль) 48%-ной HBF< выпадает желтый осадок. Смесь перемешивают еще 20 мин, осадок отделяют, промывают СН2С12 (5x50 мл) и сушат в высоком вакууме. Выход ~28,8 г (~90%).
Свойства. Устойчивый на воздухе порошок желтого цвета. Не растворяется в большинстве органических растворителей, за исключением ацетона. ИК (КВг): 2110, 2053, 1980 [v(CO)] см-1.
ЛИТЕРАТУРА
1. Dauben Н. J., Bertelli D. J., J. Amer. Chem. Soc., 83, 497 (1961).
2. Burton R., Pratt L., Wilkinson G., J. Chem. Soc. (London), 1961, 594.
3. King R. B., Organometallic Syntheses, v. 1, p. 142, Academic Press, New York, London, 1965.
Дикарбонил (т]5-ци1клогептадиенил) иодожелезо FeI(CO)2(ri5-C7H9)
[FetCOW-C,^)] [BF4] + Nal ------> FeI(n-C7H9) (CO)2 + CO + NaBF4
319,8 149,9 331,9 22,4л 109,8
В колбе на 500 мл перемешивают при комнатной температуре 16,0 г (50 ммоль) [Fe(CO)3(r]6-C7H9)] [BF4] (см. полученный выше препарат) и 34,5 г (0,23 ммоль) Nal* в 250 мл ацетона. Происходит выделение СО и окрашивание раствора в коричнево-красныи цвет. Через 4 ч реакция заканчивается. Растворитель отгоняют в вакууме водоструйного насоса, а красно-коричневый остаток экстрагируют СН2С12 (3x100 мл). После фильтрования раствор СН2С12 снова упаривают в вакууме водоструйного насоса досуха, а получившийся кристаллический остаток промывают петролейным эфиром (/КИп 40— 60°С; 3x50 мл) и сушат в высоком вакууме. Выход 7,5—12,5 г (45—75%).
Свойства. Устойчивое на воздухе, красно-коричневое кристаллическое вещество. /„л 86—89°C (с разл.). Не растворяется в неполярных органических растворителях (например, в петролейном эфире), хорошо растворяется в таких полярных растворителях, как СН2С12 или ацетон. ПК (СН2С12): 2033, 1960 [v(CO)J см-1.
* Вместо Nal можно использовать эквимолярное количество Lil или KL
Олефиновые, ацетиленовые и аллильные комплексы 2007
ЛИТЕРАТУРА
1. Dauben Н. J., Bertelli D. J., J. Amer. Chem. Soc., 83, 497 (1961).
2. King R. B., Organometallic Syntheses, v. 1, p. 142, Academic Press, New York, London, 1965.
□ □ (т]-Аллил)трикарбонилкобальт Со(т)-С3Н5)(СО)з
a) Co2(CO)8 + 2Na ---> 2Na[Co(CO)4]
342,0 2-23,0 2-194,0
6) Na[Co(CO)4] + C3H5Br '-> Co(r]-C3H5) (CO), + CO + NaBr
194,0 121,0 184,0 22,4 л 102,9
В трубке Шленка, под которую подставляют жестяную баню, приготавливают 1%-ную амальгаму натрия из 460 г Hg и 4,6 г (0,2 моль) Na. Осторожно! Реакция сильно экзотермична и должна быть проведена в вытяжном шкафу (пары Hg)!
К полученной жидкой амальгаме прибавляют раствор 34,2 г (0,1 моль) Со2(СО)8 в 150 мл тетрагидрофурана и оставляют перемешивать на ночь. Затем образовавшийся раствор декантируют в двугорлую колбу на 500 мл, снабженную краном для ввода азота, ртутным затвором и капельной воронкой. Оставшуюся амальгаму затем разлагают осторожно изопропиловым спиртом.
При перемешивании медленно прикапывают раствор 24,2 г (0,2 моль) аллилбромида в 50 мл тетрагидрофурана. Раствор вспенивается (выделение СО), через 2 ч реакция заканчивается. Красный раствор упаривают в вакууме досуха, а остаток экстрагируют петролейным эфиром (3x20 мл). Экстракт фильтруют, растворитель отгоняют в вакууме, остаток хроматографируют иа А12О, (активность I степени, 40x1 см.) Возможна также очистка перегонкой масла, оставшегося после упаривания растворителя [2]. Выход 26,7 г (73%).
Свойства. Оранжевое масло. /пл —33 °C. На воздухе разлагается. ИК (н-гексан): 2060 (с.), 1970 (оч. с.) [v(CO)] см-' ЯМР-*Н (CDC13, ТМС): 6 4,80 [мультиплет, CH, 1 Н]; 2,93 [дублет, СН2-с«н, 2 Н], /(НН) =6 Гц; 2,15 [дублет, СНг-анти, 2 Н], /(НН) = 10 Гц.
ЛИТЕРАТУРА
1. McClellan W. R., Hoehn Н. Н„ Cripps Н. N., Muetterties Е. L., Howk В. W., J. Amer. Chem. Soc., 83, 1601 (1961).
2. Heck R. F., Breslow D. S., J. Amer. Chem. Soc., 83, 1097 (1961).
□ □ Гексакарбонил(ц,т]-дифенилацетилен)дикобальт (Co—Co) Co2(CO)6(C6H5C^CC6H5)
Co2(CO)8 + CeH6C==CCeH6 -> Co2(CO)e(CeH5C=CCeH6) + 2CO
342,0 178,2 464,1 2-22,4 л
Раствор 6,84 г (20 ммоль) Со2(СО)8 и 3,40 г (19 ммоль) дифенилацетиле-на в 50 мл петролейного эфира перемешивают 12 ч при комнатной температуре. Затем растворитель отгоняют в токе N2, а остаток растворяют в 250 мл СН3ОН, при необходимости нагревая метанольный раствор почти до кипения, и фильтруют еще горячим. При медленном охлаждении до комнатной температуры из раствора выпадают темные, пурпурно-красные иголочки или листочки. Выход 7,1—7,6 г [76—82%, считая иа Со2(СО)8]. Для очистки рекомендуется высоковакуумная возгонка при 90—100 °C либо повторная кристаллизация из СН3ОН.
2008 Глава 33. Металлоорганические комплексы
Свойства. Пурпурно-красные, почти черные кристаллы, не очень устойчивые на воздухе, 1ПЛ 109—НО °C. Хорошо растворимы почти во всех полярных органических растворителях. ИК (КВг): 2093, 2050, 2040 [v(CO)] см-1. Дипольный момент ц=2,0 дебай. Кристаллическая структура: см. [2].
ЛИТЕРАТУРА
1. Greenfield И., Sternberg Н. U7., Friedel R. A., Wotiz !. Н., Markby R., Wen-der I., J. Amer. Chem. Soc., 78, 120 (1956).
2. Sly W. G„ J. Amer. Chem. Soc., 81, 18 (1959).
□ р,-Дихлоротетракис(т]2-циклооктен) диродий
[RhCl(n2-C8H14)2]2
2RhCl3-3H2O 4- 4C8H14 ---> [RhCl(T]2-C8H14).2],
2-263,3 4-110,2 717,5
В трубке Шленка растворяют 2,0 г (7,6 ммоль) RhCl3-3H2O* в 35 мл смеси этанол — вода (5: 1), предварительно насыщенной N2, и прибавляют 4 мл циклооктена. Далее нагревают на термостатированной масляной бане при 35 °C в течение 1—2 дней, отфильтровывают желтый осадок, промывают небольшим количеством смеси этанол — вода (5:1) и сушат в высоком вакууме. Перекристаллизация из смеси CH2CI2 — петролейный эфир (1 : 1). Выход 1,5— 2,0 г (55—73%)- Выход повышают почти до 80%, «'ли к слегка упаренному маточнику добавляют кристаллики для затравки и перемешивают несколько дней.
Свойства. Желтые ромбические кристаллы, /пл >150 °C (с разл.), в течение нескольких недель устойчивы под азотом. Вещество плохо растворяется в бензоле и СНС13. Растворы очень неустойчивы. ПК (КВг): 1555 [v(C = C)], 1458, 1439, 1350 см"1.
ЛИТЕРАТУРА ,
1. Winkhaus G., Singer Н., Вег., 99, 3602 (1966).
2. van der Ent A., Onderdelinden A. L., Inorg. Synth., 14, 92 (1973).
□ Бис[ц-хлоро-{1,2:5,6-т]-( циклооктадиен-1,5)} родий]
[RhCl(n4-C8Hi2)]2
2RhCl3-3H2O+ 2C8H12 4- 2C2H5OH + 2Na2CO8- 10H2O ----->
2-263,3 2-108,2 2-46,1 2-286,2
--->- [RhCl(r)4-C8H12)]2 + 2CH3CHO + 4NaCl 4- 2CO2 4. 28H2O 493,1 2-44,0 4-58,4 2-22,4л 28-18,0
В колбу емкостью 250 мл, снабженную обратным холодильником и ртутным затвором, помещают в указанной последовательности: 4,00 г (15,2 ммоль) RhC13-3H2O ( — 38% Rh фирмы Degussa), 4,40 г (15,4 ммоль) Na2CO3-ЮН2О, 40 мл 80%-ного С2Н5ОН н 6,0 мл (5,29 г, 49 ммоль) циклооктадиена-1,5. Смесь кипятят с обратным холодильником 18 ч, при этом выпадает оранжевожелтый порошок. После охлаждения до комнатной температуры осадок отде
* Гидрат хлорида родия (III) RhCl3-xH2O с содержанием Rh -38% поставляет фирма Degussa (Hanau).
Олефиновые, ацетиленовые и аллильные комплексы
009
ляют и промывают до тех пор 80%-ным этанолом, пока фильтрат не даст отрицательную пробу на хлорид-ион. После высушивания в высоком вакууме получают аналитически чистый препарат. Выход 3,34 г (89%).
Свойства. Оранжево-желтый, устойчивый на воздухе порошок. Разлагается при 250 °C. Растворяется в полярных органических растворителях, например в СН2С12. Не растворяется в пентане. Из смеси СН2С1г—(СгН6)2О кристаллизуется в виде оранжевых призм. ИК (нуйол): 998, 964, 819 см-1 (характерные полосы поглощения). ЯМР-’Н (CDC13, ТМС): 6 4,3 [винильиые Н], б 2,6—1,7 [аллильные Н].
ЛИТЕРАТУРА
1. Chatt J., Venanzi L. М., Nature, 177, 852 (1956).
2. Chatt J., Venanzi L. M., J. Chem. Soc. (London), 1957, 4735. j
3. Giordano G., Crabtree R. H., Inorg. Synth., 19, 218 (1979).
□ □ Бис [ 1,2:5,6-т]-циклооктадиен-1,5)] никель
N1(T]4-C8H12)2
Ni(CsH7O2)2 4- A1(C2H5)3 2C8H12 --->
256,9 114,2 2-108,2
>- Ni(Tq4-C8I II2)2 -J- A1(C5H7O2)2C2H6 4- C2He 4- C2H4
275,1 254,3 30,1 28,1
[Ni(C5H7O2)2 — ацетилацетонат никеля
а) Никель(II)ацетилацетонат [Ni(acac)2] выпускают фирмы Merck-Schu-chardt (Мюнхен) и Konigswarter und Ebel (Хаген). Его можно также синтезировать из свежеосажденного гидроксида никеля Ni(OH)2 и ацетилацетона [5] или электрохимическим путем из металлического Ni и ацетилацетона в водном изопропаноле [6].
Успех синтеза бис (циклооктадиен)никеля в значительной степени определяется чистотой Ni(acac)2. Поэтому необходимо его дважды перекристаллизовать в атмосфере аргона из толуола или ксилола. Остатки растворителя удаляют в вакууме (10-4 мм рт. ст.). Циклооктадиен-1,5 (COD) выпускают химические заводы Hills, Marl и фирма Е. Merck (Дармштадт). COD очищают перегонкой в вакууме (^кип —45 °C при 13 мм рт. ст.) над АЦОСгНв) (С2Н5)2. Бензол обычным образом высушивают и затем перегоняют в атмосфере аргона над небольшим количеством NafAl^Hsh].
б) В стеклянный прибор (рис. 468) в атмосфере Аг помещают 77,1 г (0,3 моль) чистейшего ацетилацетоната никеля в 250 мл бензола и 172,8 г (1,6 моль) циклооктаднена-1,5. После охлаждения до 0°С* сифонируют 7 г (0,13 моль) жидкого бутадиена-1,3, который препятствует выпадению металлического Ni. При интенсивном перемешивании прикапывают за 3—4 ч раствор 37,6 г (0,33 моль) триэтил алюминия в 100 мл бензола. Первоначально зеленый раствор медленно становится оранжево-красным. Вскоре выпадают светло-желтые кристаллы. Смесь оставляют стоять —15 ч, например на ночь при 20 °C, для завершения кристаллизации.
На стеклянном пористом фильтре G3 кристаллическую массу отделяют,
* Охлаждение осуществляют непрямым способом, например путем прокачки алифатических углеводородов или метилциклогексана. Нельзя использовать спирт или ацетон! Взрывоопасно!
2010 Глава 33. Металлоорганические комплексы
промывают холодным бензолом (2X75 мл) и холодным эфиром (2x75 мл) и сушат в вакууме (10-4 мм рт. ст.). Выход 73,5 г (89%).
При необходимости, например когда из-за выделившегося Ni продукт темного цвета, его перекристаллизовывают из толуола. Для этого осадок растворяют при комнатной температуре в 3 л толуола, фильтруют и оставляют в холодильнике для кристаллизации. Вторую порцию более мелких кристаллов получают упариванием маточника до половины первоначального объема и повторным охлаждением.
Другие способы. Восстановление NiCl2 калием в присутствии циклооктадиена-1,5 с выходом ~40% [3].
Электрохимическое восстановление ацетилацетоната никеля (II) в присутствии циклооктадиена-1,5 с выходом —65% [4].
Рис. 468. Стандартная аппаратура для получения [NiBr^-CsHsJJa-
1 — трехгорлая колба на 1 л; 2 — капельная воронка на 250 мл с регулированием прикапывания жидкости с помощью игольчатого крана-шпииделя, сиабжеиная насадкой для подвода инертного газа; 3 — эффективная вакуумируемая мешалка, приводимая во вращение от внешнего вращающегося магнита; 4— штуцер со спиртовым термометром (от —90 °C до +30 °C); 5 — трехходовой кран.
Свойства. Лимонно-желтый кристаллический порошок или крупные желтые кристаллы (из бензола). На воздухе вещество разлагается в течение нескольких минут. tn]t (под Аг) 142°C (с разл.). Хорошо растворяется в бензоле, толуоле и тетрагидрофуране (при нагревании свыше 60 °C происходит разложение). Почти не растворяется в эфире и алифатических углеводородах. ЯМР-'Н (CSD6, внешний стандарт ТМС): 6 1,38 [СН2], 3,64 [СН].
ЛИТЕРАТУРА
1. Bogdanovic В., Kroner М., Wilke G„ Liebigs Ann. Chem., 699, 1 (1966).
2. Schunn R. A., Inorg. Synth., 15, 5 (1974).
3. Otsuka S., Rossi M., J. Chem. Soc. (London), A1968, 2630.
4. Lehmkuhl H., Leuchte W., Eisenbach W., Liebigs Ann. Chem., 1973, 692.
5. Gach F„ Monatshefte Chem., 21, 98 (1900).
6. Lehmkuhl H„ Eisenbach W., Liebigs Ann. Chem., 1975, 672.
Олефиновые, ацетиленовые и аллильные комплексы 2011
□ □ Бис[|х-бромо(т]-аллил)ни1кель] [NiBr(T]-C3H5)]2
2Ni(r]4-C8H12)2 + 2СзН5Вг -> [NiBr(r)-C8Hs)]2 + 4С8Н12
2-275,1 2-121,0 359,4 4-108,2
а) Подготовка реагентов. Аллилбромид выпускает фирма Merck-Schuch-hard (Мюнхен). Непосредственно перед применением его перегоняют еще раз под аргоном (/Кип 70°C при 760 мм рт. ст.).
б) В аппаратуре, изображенной на рис. 468, суспендируют 82,0 г (0,3 моль) бис (циклооктадиен-1,5) никеля (см. вышеописанный препарат) в 300 мл эфира*. При интенсивном перемешивании добавляют по каплям при (—25) — (—10) °C за 3—4 ч раствор 40 г (0,33 моль) аллилбромида (см. п. «а») в 50 мл
Рис. 469. Стеклянная воронка для фильтрования с охлаждающей рубашкой.
'.Г
эфира. Желтые кристаллы Ni(T]4-C3Hi2)2 постепенно растворяются, а раствор становится темно-красным. Для окончания реакции перемешивают еще 0,5— 1 ч при 0 °C и фильтруют раствор через стеклянный пористый фильтр G3 с охлаждающей рубашкой (рис. 469) для отделения от металлического Ni. Фильтрат охлаждают до —80 °C и оставляют кристаллизоваться на несколько часов.
Другие способы. Из бис(т]-аллилникеля) и НВг в эфире с выходом 88% [1] или из Ni(CO)4 и аллилбромида с выходом ~10% [3].
Свойства. Крупные красные кристаллы, при +20 “С устойчивы в течение нескольких часов. Хранят при температуре от —30 до —20 °C.
ЛИТЕРАТУРА
1. Wilke G. et al., Angew. Chem., 78, 157 (1966).
2. Heimbach P., частное сообщение.
3. Fischer E. O., Burger G., Z. Naturforsch., 16b, 77 (1961).
* Вместо эфира можно использовать в качестве растворителя толуол или диметиловый эфир диэтиленгликоля (диглим). Все используемые растворители обычными методами предварительно высушивают и затем в атмосфере At перегоняют над K/Na-сплавом.
2012 Глава 33. Металлоорганические комплексы
□ □ Бис[|х-метил(т]-аллил)никель] [М1(т]-С3Н5)СНз]2
□ □ Бис(т]-аллил)никель Ni(Ti-C3H5)2
□ □ rj-Аллил (метил) (трифенилфосфин)никель
Ni(n-C3H5)(CH3)[P(C6H5)3]
а) [№Вг(П-С3Н5)]2 + 2Mg(CH3)Cl ----> [Ni(n-C3H5) СН3]2 + 2MgBrCl
359,4 2-74,8 229,6 2-139,7
б) [Nifo-CgH^CH^ ---> Ni(n-C3H5)2 + Ni + 2[СН3-]
229,6 140,9 58,7 2-15,0
в) [N1(t)-C8Hs) СНд]2 + 2Р(СвН5)3 -> 2Ni(r1-C3H6)(CH3)[P(CeH6)3]
229,6 2-262,3 2-377,1
Подготовка реактивов. Диэтиловый эфир и пентан предварительно сушат над СаС12 и затем перегоняют над Na/K-сплавом или над Na[Al(C2H5)4].
а) [М(п-С3Н5)СН3]2 [1, 2]
В пятигорлую колбу (рис. 470) в атмосфере Аг помещают 10,1 г (28 ммоль) ГМ1Вг(п-СзН5)]2 (получение см. выше) и при перемешивании растворяют (частично) в 100 мл эфира [см. примечание о подготовке реактивов!]. Далее постепенно охлаждают до-------100 °C. Для этого реакционную колбу, сифон и
R
Рис. 470. Пятигорлые сдвоенные колбы для получения [№(т]-С3Н5)СНз]2-
1 — калиброванная воронка с охлаждающей рубашкой; 2 — высокоэффективная мешалка С внешним магнитным приводом; 3 — пористый стеклянный фильтр G3; 5—6—7 — мост, связывающий обе колбы; 8, 9 — хвостовые краны; J0 — баня для охлаждения (пентан, жидкий азот, —1S0 °C).
Олефиновые, ацетиленовые и аллильные комплексы 2013
воронку для фильтрования погружают в баню с охлаждающей смесью (например, с метилциклогексаном), которую охлаждают с помощью жидкого азота. Поступление охладителя регулируют автоматически с помощью системы, состоящей из емкости с охладителем и клапанного реле [например, криостат фирмы Gebr. Haake (Зап. Берлин)]. Когда температура в охлаждающей емкости (бане) превышает заданную, автоматически включается подводящий вентиль и жидкий N2 попадает в баню для охлаждения. Таким образом, любую желаемую температуру вплоть до —130 °C регулируют в течение дли тельного периода с точностью ±5 °C.
В градуированную капельную воронку 1 помещают 112 мл (56 ммоль) 0,5 М эфирного раствора Mg(CH3)Cl и через охлаждающую рубашку пропускают раствор метанола примерно при —60 °C. В течение 2 ч раствор реактива Гриньяра прикапывают в колбу, где он тотчас реагирует с соединением Ni. Образуется темно-фиолетовый раствор. Далее в течение 3 ч реакционную смесь выдерживают при —100 °C.
В процессе реакции кран 6 (рис. 470) открывают полностью для выравнивания давления между реакционной колбой и частью для фильтрования. После окончания реакции кран 6 закрывают и открывают кран 8. Давлением аргона определенную порцию реакционной смеси передавливают на фильтр 3 и кран 8 снова закрывают. В процессе фильтрования кран 9 держат открытым, чтобы избыток аргона удалялся из приемника. Продукт реакции находится в фильтрате, а побочно образующиеся неорганические соединения остаются на фильтре. Остаток на фильтре (темно-серого цвета) промывают 50 мл эфира и выбрасывают. Затем снимают с прибора, капельную воронку, вакуумную мешалку и насадку с термометром и фильтрат упаривают в вакууме (10-4 мм рт. ст.) при —90 °C досуха.
Красно-фиолетовый остаток при —100 °C извлекают 250 мл пентана (см. приготовление растворителей) и повторно фильтруют через стеклянный пористый фильтр (G3) с охлаждающей рубашкой. Пентановый раствор, свободный от галогенидов, является достаточно чистым для дальнейшего использования. При охлаждении до —130°С из раствора выпадают кристаллы бис[граллил(метил)никеля]. Выход 5,3 г (82%).
Аналогичным образом получают несколько более термоустойчивый комплекс бис[т]3-1,3-диметилаллил(метил)никель] с выходом 94% [3].
Свойства. Темно-фиолетовые кристаллы [М1(т)-СзН5)СН3]2 хранятся лишь при —78 °C. ЯМР-’Н (толуол-de, ТМС, —70 °C): б 5,33 [мультиплет, СН]; 2,76 [дублет, СН2-син, 7(НН-син)=7 Гц]; 2,25 [дублет, СН2-анти, j(HH)-днтц=13 Гц); —1,50 [синглет, Ni-CH3]; —1,88 [синглет, Ni-CH3], Кристаллическая и молекулярная структура бис[ц-метил-(1,3-диметил-т]-аллнл)никеля] моноклинная, пространственная группа симметрии Р2]/п; а=9,9594(9) А, & = = 9,3895(7) А, с=14,8434(8) А, [3=95,112(5)° [4]. 6
6) Ni(n-C3H5)2 [5]
Свежеприготовленные 100 мл раствора 4,59 г (20 ммоль) [М1(т]-СзН5)СНз]2 в эфире нагревают при постоянном перемешивании в течение 30 мин от —78 до —50 °C. При этом выпадает черный осадок металлического никеля, а темно-фиолетовый цвет раствора [М1(г]-СзН5)СНз]2 исчезает. Дают осадку осесть и фильтруют получившийся желтый раствор через стеклянный пористый фильтр (G3) с охлаждающей рубашкой. При —78 °C и давлении 10-4ммрт. ст. полностью удаляют растворитель. На последней стадии отгонки растворителя выпадают желтые кристаллы, которые возгоняют при —35 °C в вакууме (10-4 мм рт. ст.). Выход 2,68 г (95%).
Другие способы. Из NiBr2 и аллилмагнийхлорида в эфире с выходом — 80% [6], а также из бис[т)-аллил(галогено)никеля] по реакции диспропорционирования, например в NH3 или Н2О с выходом 40—45% [7].
2014 Глава 33. Металлоорганические комплексы
Свойства. Соединение цвета охры; /пл +1 °C, на воздухе возгорается и горит сильно коптящим пламенем. ИК: 1485 [v(CCC)] см-1. ЯМР-’Н (толуол-de, ТМС): транс-изомер 6 4,98 [мультиплет, СН]; 3,91 [дублет, СН2-сан, J(H-H-cuh) =7,5 Гц]; 1,75 [дублет, СН2-антн, /(Н-Н-анта) = 14 Гц]; ijuc-изо-мер б 5,00 [мультиплет, СН]; 3,68 [дублет, СН2-сан]; 2,18 [дублет, СН2-анта].
в) Ni(n-C3H5)(CH3)[P(C6H6)3] [1, 2]
К 10 мл 0,2 М раствора [Ni(r]-C8H6)CH3]2 в эфире при —78°С прикапывают пипеткой раствор 1,05 г (4 ммоль) Р(С6Н8)3 в 50 мл эфира. К концу добавления цвет раствора из темно-фиолетового становится желтым. Через 15 мин температуру смеси повышают до —10 °C. При этой температуре растворитель отгоняют в вакууме (10_2 мм рт. ст.) досуха. 'Остается осадок цвета охры. Его перекристаллизовывают из 100 мл пентана и сушат в вакууме (IO-2 мм рт. ст.) при 0°С. Выход 1,12 г (74%).
Свойства. Кристаллы цвета охры. В бензольном растворе соединение разлагается уже при 40°C. ЯМР-'Н (C8D8, ТМС): б 5,00 [мультиплет, СН]; 3,61; 2,88; 2,71; 1,91 [4 дублета, СН2]; 0,27 ,[Ni-CH8]; 7,55—7,06 [мультиплет, С.Н5].
ЛИТЕРАТУРА
1. Bogdanovii В., Bonnemann Н., Wilke G., Angew. Chem., 78, 591 (1966).
2. Heimbach P., частное сообщение.
3. Schenkluhn H., Dissertation, Univers. Bochum, 1971.
4. Kruger C., Sekutowski J. C., Berke H., Hoffmann R., в печати.
5. Bogdanovic В., Bonnemann H., Wilke G., Angew. Chem., 79, 817 (1967).
6. Wilke G„ Bogdanovic B., Angew. Chem., 73, 756 (1961).
7. Birkenstock U„ Bonnemann H„ Bogdanovic B., Walter D., Wilke G., Advan. Chem. Ser„ 70, 250 (1967).
□ □ (1,2:5,6:9,1 ^-т\-транс, транс, гранс-циклододекатриен)-никель №1(т]6-транс,транс,трансЛ,5,
Ni(C6H7O2)2 + 2А1(С2Н5)2(ОС2Н5) + транс, транс, транс-С12Н1Я ——>
256,9 2-130,2 162,3
----► С2Нв + С2Н4 + 'НЦг\в-транс,транс,транс-1,5,9-С12Н18) 4-
30,1 28,1 221 ,0
+ 2А1(С2Н5) (ОС2Н5) (С5Н7О2) 2-200,2
а) Приготовление реагентов, транс, транс, транс-1,5,9-Циклододекатриен (CDT) является продажным препаратом (например, Chemische Werke Huis). Его также легко можно получить из бутадиена-1,3 [2]. транс,транс,транс-1,5,9-CDT очищают двойной перекристаллизацией из спирта (/пл 34 °C) и перегонкой под Аг (/кип 111 °C при 20 мм рт. ст.). Эфир предварительно сушат, как обычно, а затем перегоняют в атмосфере аргона над K/Na-сплавом.
б) В аппаратуру, изображенную на рис. 468, в атмосфере Аг помещают 51,3 г (0,2 моль) ацетилацетоната никеля (II) [получение см. выше в методике-синтеза Ni(-q4-C4HI2)2, п. «а»], 70 г (0,42 моль) транс,транс,транс-1,5,9-цикло-додекатриена и 500 мл эфира [см. п. «а»]. Колбу охлаждают до 0°С (см. примечание к синтезу бис[1,2:5,6-т)-(циклооктадиен-1,5)]никеля) и при интенсивном перемешивании в течение 3 ч прикапывают 53,3 г (0,41 моль) этоксиди-этилалюминия в 60 мл эфира. Образование эфирного раствора алюминийорга-
Олефиновые, ацетиленовые и аллильные комплексы 2015
нического соединения идет с выделением тепла, поэтому всегда алюминийор-ганическое соединение вносят в растворитель, а не наоборот.
Первоначально зеленая реакционная смесь при добавлении А1-комплекса становится темно-коричневой. Ее перемешивают 17 ч при 0 °C. Затем раствор через погруженный в охладительную смесь сифон со стеклянным пористым фильтром (G2) передавливают в емкость на 1 л, ампулу или трубку Шлейка, охлажденную предварительно до —78 °C, избавляясь тем самым от кристаллов А1(С2Н5) (ОС2Н5) (С5Н7О2). Выпавшие моментально при охлаждении красные кристаллы отделяют от растворителя. Остаток эфира полностью отгоняют
Рис. 471. Высоковакуумная возгонка Ni (ц6-т,т,т-1,5,9-С12Н]8).
/ — ампула для возгонки; 2 — баня с водой; 3 — охлаждение водой; 4 — подвод инертного газа.
при 20°C (10-’ мм рт. ст.), а затем при 20°C (10-4 мм рт. ст.) отгоняют избыток циклододекатриена.
Комплекс очищают возгонкой (рис. 471). Для этого верхнюю часть ампулы 1 несколько раз обвивают охлаждающим шлангом 3. Нижнюю часть, в которой находится весь твердый продукт, погружают в водяную баню 2, нагретую до 50 °C. При 10~4 мм рт. ст. никелевый комплекс возгоняется и оседает на охлажденной части ампулы в виде темно-красных с металлическим блеском кристаллов. Возгонку проводят ~24 ч. Темный остаток на дне ампулы растворяют или суспендируют в эфире и отсасывают. После повторного промывания дна ампулы сублимат как можно быстрее растворяют при 20 °C в 500 мл эфира и кристаллизуют комплекс при —78 °C в виде красноватых игл. Маточник снова отсасывают (сифонируют) и продукт сушат при 10~4мм рт. ст. Выход 33,1 г (70%).
Другие способы. Электрохимическим путем из ацетилацетоната Ni(II) и циклододекатриена с выходом 21% [3].
Свойства. Красное кристаллическое соединение, 1пл ~90°С (с разл.), разлагающееся уже в присутствии каталитических количеств О2. Необходимо предохранять от попадания следов смазки. Растворы, например в тетрагидрофуране или эфире, термически менее устойчивы, чем сами кристаллы. При комнатной температуре и даже при —40 °C из раствора начинает выпадать металлический Ni. Растворы или суспензии можно сохранить лишь при температуре ниже —80 °C.
ЛИТЕРАТУРА
1. Bogdanovic В., Kroner М., Wilke G., Liebigs Ann. Chem., 699, 1 (1966).
2. Breil H„ Heimbach P., Kroner M., Muller H., Wilke G., Makromol. Chem., 69, 18 (1963).
3. Lehmkuhl H., Leuchte W., Eisenbach W., Liebigs Ann. Chem., 1973, 692.
Дихлоро(1,2:5,6-т]-циклооктадиен)палладий PdCh^-CgH^)
PdCl2 + C8H12 --> PdCl2(ri4-C8H12)
177,3 108,2 285,5
2016 Глава 33. Металлоорганические комплексы
Растворяют при нагревании 2,0 г (11,3 ммоль) PdCl2 в 5 мл конц. НС1. Охлажденный раствор разбавляют 150 мл 96%-ного этанола, фильтруют и при перемешивании добавляют 3,0 мл (24 ммоль) циклооктадиен!а-1,5. Тотчас выпадает желтый осадок, его через несколько минут отделяют, промывают эфиром (3X30 мл) и 2 ч сушат в высоком вакууме. Выход 3,0 г (93%).
Свойства. Желтый, устойчивый на воздухе порошок (<пл>210оС с разл.) или крупные желтые кристаллы (при перекристаллизации из кипящего СН2С12). Растворим в СНС13 и нитробензоле.
ЯМР-'Н (СНС13, ТМС): 6 6,32 [ = СН—]; 2,69 [-СН2-].
ЛИТЕРАТУРА
1. Drew D., Doyle J. R., Inorg. Synth., 13, 52 (1972).
□ □ Бис[1,2:5,6-т)-(циклооктадиен-1,5)] палладий
Pd('q4-C8Hi2)2
a) Li2C8H8 PdCI2(rpC8II12) -|- C8H12 --► Pd(^4-C8H12)2 -j- 2LiCl -|- CSHS
118,0 285,5 108,2 322,8 2-42,4 104,2
6) Pd(n4-C8H12)2 + 3CH,CH=CH2 =?=>: Pd(r|2-C3HB)3 + 2CsH12
322,8 3-42,1 232,6 2-108,2
Прибор состоит из двугорлой круглодонной колбы на 250 мл, снабженной краном для ввода Ns, магнитной мешалкой с якорем в тефлоновой оболочке и капельной воронкой с выравнивателем давления. Колбу охлаждают в бане (смесь изопропиловый спирт — сухой лед) до —50 °C, продувают сухим N2 и в противотоке N2 загружают в нее 5,7 г (20 ммоль) тонкоразмельченного-PdCl2(r]4-C8His) (см. полученный выше препарат) и 30 мл циклооктадиена-1,5*. С помощью шприца отбирают и помещают в капельную воронку точное количество свежеприготовленного раствора LisC8H3 (20 ммоль) в эфире [получение см. ниже методику синтеза Р1(г)4-С8Н12)2]и в течение 1 ч прикапывают к интенсивно перемешиваемой суспензии в колбе. Далее реакционную смесь нагревают до —35 °C, вместо N2 систему заполняют пропиленом и интенсивно перемешивают еще 1 ч. В течение этого времени суспензия становится почти черной.
Тем временем готовят аппаратуру для фильтрования при низкой температуре (рис. 472), на дно фильтровальной воронки насыпают слой 5 см А120з. (2). Заполняют аппаратуру пропиленом и устанавливают температуру верхней рубашки охлаждения —35 °C, нижней бани —40 °C. Затем реакционную смесь из колбы быстро выливают на слой А12О8, фильтруют и фильтрат упаривают при —30 °C, следя за тем, чтобы получившийся желтый раствор1 ни в коем случае не прикасался со стеклянными частями, температура которых выше —25 °C. Раствор упаривают до тех пор, пока не выпадет твердое соединение серого цвета. Избыток растворителя декантируют, а остаток промывают эфиром (2x10 мл, —40°C). Затем сырой продукт извлекают 70 мл жидкого-пропилена, который конденсируют при —65 °C в трубку Шленка 1. Получившийся раствор еще раз фильтруют через слой 1 см А12О3 в атмосфере N2 в точно такой же аппаратуре (рис. 472), но меньшего размера, охлажденной до —70 °C. Сорбент промывают 20 мл холодного пентана, к которому добавляют 2 капли циклооктадиена-1,5. Затем трубку Шленка с бесцветным фильтратом ставят в баню, охлажденную до —40 °C, и отгоняют пропилен. В трубке Шленка остаются бледно-кремовое твердое соединение и немного пентана,
* Эфир и циклооктадиен-1,5 должны быть абсолютно сухими и не содержать кислорода и непосредственно перед употреблением перегнаны в атмосфере N2 или Аг.
Олефиновые, ацетиленовые и аллильные комплексы 2017
который осторожно удаляют пипеткой. Получившийся продукт сушат при;
—30°C (0,01 мм рт. ст.). Выход 2,5—3,5 г (39—54%).
Другие способы. Из Pd-паров и циклоокта диена-1,5 в метилциклогексане_-при —120 °C с выходом 20% [3].
Рис. 472. Аппаратура для фильтрования при низкой температуре.
/ — трубка Шленка (—150 мл); 2 —слой Л120з иа поверхности стеклянного фильтра.
Свойства. Кремоватый порошок или бесцветные кристаллы, термически/ существенно более лабильные, чем соответствующие соединения Ni или Pt; хранятся при температуре не выше —20 °C. Вещество плохо растворяется в; некоординирующихся растворителях. Олефины, третичные фосфины и изонитрилы замещают оба COD-лиганда. ИК (нуйол, при низкой температуре):; 1335 (с.), 1316 (сл.), 1245 (ср.), 1238 (ср.), 1189 (ср.), 1161 (сл.) см-'.
ЛИТЕРАТУРА
1. Green М., Howard J. А. К.. Spenser J. L., Stone F. G. A., J. Chem. Soc. Dalton, 1977, 271.
2. Spenser J. L., частное сообщение.
3. Atkins R. M., Mackenzie R., Timms P. L., Turney T. W., J. C. S. Chem. Com-mun„ 1975, 764.
□ Моногидрат трихлоро(г]-этилен)платината(П) калия
K[PtCI3(n-C2H4)].H2O
(Соль Цейзе)
KJPtcm + Сгн4 ----> K[PtCl3(n-C2H4)] H2O + KCl
415,1 22,4л 386,6 74,6
Аппаратура. Двугорлая колба на 250 мл с краном для ввода N2, ртутный* затвор, магнитная мешалка и трубка для ввода газа.
11-1361
2018 Глава 33. Металлоорганические комплексы
В атмосфере Ns растворяют 4,15 г (10 ммоль) K.2[PtCl4] и 45 мг (0,2 ммоль) SnCls-2H2O в 50 мл соляной кислоты, не содержащей кислорода (3,5 моль; 15 мл дымящей НС1+35 мл Н2О; 20 мин продувают слабым током N2). Через перемешиваемый раствор в течение — 4 ч продувают слабый ток этилена (1—2 пузырька в 1 с). Цвет раствора при этом постепенно'изменяется от темно-красного до желтого. Раствор фильтруют через стеклянный пористый фильтр (G3). (Ни в коем случае не применять фильтровальную бумагу! Влажный продукт не трогать металлическим шпателем, иначе происходит разложение с выделением металлической Pt.) Затем охлаждают 30 мин при —20 °C. Выпавшие желтые иголочки (1,6 г) быстро отфильтровывают, промывают небольшим количеством ледяной воды с этанолом (1:1) и сушат в эксикаторе над конц. H2SO< и КОН. Оставшийся после фильтрования раствор испаряют в роторном испарителе (температура бани 40—50 °C) досуха, остаток растворяют в 15 мл метанола, раствор фильтруют и после отгонки растворителя получают дополнительно 1,6 г продукта в виде желтоватого мелкокристаллического порошка. Общий выход 3,2 г (83%).
Свойства. Длинные желтые игольчатые кристаллы. При ~75°С вещество начинает терять кристаллизационную воду и при 180 °C разлагается с выделением С2Н4. Устойчиво на воздухе, хорошо растворяется в воде, 1—5 М НС1, СН3ОН, С2Н5ОН и ацетоне; растворы постепенно разлагаются. Не растворяется в эфире и алифатических углеводородах. ИК (нуйол): 339 (оч. с.), 331 (оч. с.), 310 (с.) [v(Pt—Cl)] см-’. ЯМР-’Н (ацетон-de, ТМС): 6 4,26, 2Z(Pt—Н) — = 64 Гц. ЯМР-’3С (ацетон-de, ТМС): 6 67,2, ’/(Pt—С) = 194 Гц. Кристаллическая структура моноклинная (пространственная группа симметрии Р21/с; а= = 11,212 А, 6=8,424 А, с=9,696 А, Р= 107,52°). Квадратно-плоскостная координация с перпендикулярно ориентированными этиленовыми лигандами [3, 4].
ЛИТЕРАТУРА
1. Hartley F. R., Organometal. Chem. Rev., A6, 129 (1970).
2. Chock P. B., Halpern J., Paulik F. E., Inorg. Synth., 14, 90 (1973).
3. Jarvis J. A. J., Kilbourn В. T., Owston P. G., Acta Crystallogr., B27, 366 (1971).
4. Love R. A., Roetzle T. F„ Williams G. J. B., Andrews L. C., Bau R„ Inorg. Chem., 14, 2653 (1975).
□ т]-Этиленбис(трифенилфосфин)платина Р1(т)-С2Н4)[Р(С6Н5)з]2
NaBH4
Pt(Ti2-O2)[P(CeH6),]2.CeHe + C2H4 -> Pt(n-C2H4) [P(C6H5)3]2
829,8 22,4л 747,7
Аппаратура. Двугорлая колба на 250 мл с краном для ввода N2, магнитная мешалка, трубка для ввода газа и капельная воронка с выравнивателем давления.
В 100 мл этанола суспендируют 3,32 г (4 ммоль) Pt(O2) [Р(С6Н6)з]2-С6Нв (получение см. ниже в этой главе) и пропускают через раствор этилен в течение 15 мин. Затем очень медленно при перемешивании прикапывают 30 мл 0,1 М этанольного раствора NaBH4, не прекращая продувать этилен (~1 пузырек в 1 с). Через несколько минут образуется желтый прозрачный раствор. В конце прибавления постепенно начинает выпадать светлый осадок. Перемешивают еще 30 мин в атмосфере этилена, выпавший осадок отделяют на стеклянном пористом фильтре (G3), промывают 30 мл Н2О, 20 мл этанола, пентаном и высушивают. Выход 2,84 г (95%).
Другие способы получения. Исходя из Pt(COs) [Р(С6Н6)3]2 [2].
Олефиновые, ацетиленовые и аллильные комплексы 2019"
Свойства. Кремовый порошок, tnx 117—125 °C (с разл.). На воздухе некоторое время, а в атмосфере Ns вещество совершенно устойчиво. Растворяется в бензоле и хлороформе, не растворяется в алифатических углеводородах,, спиртах и Н2О. В CS2 образуется Pt(r)2-CS2) [Р(СвН6)з]2- ЯМР-'Н (CDC13„ ТМС): 6 2,15, 7(СН) = 146,5 Гц; V(PtH)=62 Гц. ЯМР-13С (CD2C12, ТМС): б 39,6, Z(Pt—С) = 194 Гц. Кристаллическая структура моноклинная (пространственная группа симметрии Р2,/а; а—16,46 А, 6=10,85 А, с= 17,85 А, 0 = = 100,5°), тригонально-плоскостная координация вокруг атома Pt, d(C—С) = = 1,434 А [3].
ЛИТЕРАТУРА
1. Cook С. D., Jauhal G. S„ J. Amer. Chem. Soc., 90, 1464 (1968).
2. Blake D. M., Roundhill D. M., Inorg. Synth., 18, 122 (1978).
3. Cheng P. T., Nyburg S. C„ Can. J. Chem., 50, 912 (1972).
□ т]-Дифенилацетиленбис(трифенилфосфи1н)платина
Pt(C6H5C=CC6H5)[P(C6H5)3]2
2PtCl2[P(CeH5)3]2 + 2CeH5C=CCeH5 + 3N2H4 --->
2-790,6 2-178,2 3-32,0
---> 2Pt(CeH5feCCeH5) [P(CeH5)sl2 + N2 + 2[N2H6]C1s.
2-897,9 22,4л 2-105,0
В двугорлой колбе на 250 мл с краном для ввода N2 суспендируют 0,95 г (1,2 ммоль) Р1С12[Р(С6Н5)з]2 (его синтез приведен в этой главе ниже) в 50 мл 96%-ного этанола, предварительно насыщенного N2, и при перемешивании прикапывают 0,8 мл 85%-ного гидразина. Через 5 мин к получившемуся желтому раствору добавляют 1,0 г (5,6 ммоль) дифенилацетилена в 30 мл этанола и нагревают 15 мин с обратным холодильником, при этом постепенно выпадает хлопьевидный осадок. Реакционную смесь медленно охлаждают до-комнатной температуры и выпавший мелкий светло-желтый кристаллический" осадок отделяют на стеклянном фильтре. Эту операцию и все последующие выполняют на воздухе. Кристаллы промывают 20 мл СН3ОН—Н2О (9:1), 10 мл эфира и сушат в высоком вакууме. Выход 980 мг (91%).
Свойства. Микрокристаллический устойчивый на воздухе порошок кремового цвета, /пя 161—164°C (с разл.), растворяется в СН2С12 и бензоле, не растворяется в спиртах и эфире. ИК (КВг): 1740 (ср.-с.) [v(C=C)] см-1. Кристаллическая структура триклинная (пространственная группа симметрии Р1; а=И,32 А, 6=15,83 А, с= 13,34 А, а=112°51', 0=113О26', у=83°40', приблизительно тригонально-плоскостная координация Pt, «цис-конфигурация» ацетиленового лиганда с <£С—СзС 140° [2].
ЛИТЕРАТУРА
1. Blake D. М.., Roundhill D. М„ Inorg. Synth., 18, 122 (1978).
2. Glanville J. О., Stewart J. M„ Grim S. O., J. Organometal. Chem., 7, P9 (1967).
Дихлоро [1,2:5,6-т]-(циклооктадиен-1,5)] платина PtCl2(n4-C8H12)
K2[PtCl4] + C8H12 -► PtCl2(r)4-C8H12) + 2KCI
415,1 108,2 374,2 2-74,6
11
2020 Глава 33. Металлоорганические комплексы
Растворяют 2,5 г (6 ммоль) K.2[PtCU] в 40 мл Н2О, фильтрует и добавляют к фильтрату 60 мл ледяной уксусной кислоты и 2,5 мл (20 ммоль) циклооктадиена-1,5. Интенсивно перемешивают и нагревают до 90° (водяная баня). Через —30 мин первоначально темно-красный раствор становится темножелтым, и выделяется мелкокристаллический осадок. Раствор в вакууме доводят до объема 30 мл и на фильтре отделяют осадок в виде светло-желтых иголочек. Его промывают 50; мл воды, этанолом и эфиром и сушат 1 ч при 100 °C. Выход 2,0 г (90%).
Другие способы получения. Из H2[PtCle] и циклооктадиена-1,5 с выходом «0% [4].
Свойства. Белые, устойчивые на воздухе кристаллы, 1ПЛ 250—280 °C (с разл.). Вещество легко растворяется в CH2CI2, СНС13 и нитрометане. ЯМР-'Н (СНС1з, ТМС): б 5,62 [=СН—], J (Pt—И) =65 Гц; б 2,71 [—СН2— ].
ЛИТЕРАТУРА
1. Chatt J., Vallarino L. М., Venanzi L. M., J. Chem. Soc., 1957, 2496.
2. Clark H. C., Manzer L. E., J. Organometal. Chem., 59, 411 (1973).
3. McDermott J. X., White J. F., Whitesides G. M., J. Amer. Chem. Soc., 98, 6521 (1976).
4. Drew D„ Doyle J. R., Inorg. Synth., 13, 48 (1972).
□ □ Бис [ 1,2:5,6-т]-(циклооктадиен-1,5)] платина
Pt(n4-C8H12)2
Этот комплекс, чрезвычайно важный для развития химии платины (0), был впервые получен в 1967 г. Мюллером и Гёзером [3] из Р1(изо-СзН7)2СОО и циклооктадиена (COD) с выходом 9%. Ниже приведены два метода синтеза, в которых легкодоступный комплекс PtC^COD восстанавливается в присутствии COD с помощью Li2C8H8 или кобальтоцена.
Способ 1 [1, 2]
a) 2Li С8Н8 >- Li2C8H8
2-6,9 104,2 118,0
б) Li2C8H8 + PtCl2(n<C8H12) + C8H12 ----> Pt(^-C8H12)2 + 2LiCl + С8Н8
118,0 374,2 108,2 411,5 2-42,4 104,2
Приготовление реагентов. Эфир, циклооктадиен-1,5 и циклооктатетраен-1,3,5,7 должны быть абсолютно сухими и свободными от О2 и непосредственно перед использованием перегнаны еще раз в атмосфере N2 или Ar. PtC12(r]4-С8Н12) (см. предыдущую методику) необходимо измельчить как можно тоньше чтобы обеспечить максимальное его использование, несмотря на плохую растворимость.
a) Li2C3H8. В двугорлой круглодонной колбе на 250 мл, снабженной магнитной мешалкой и краном для ввода N2, охлаждают до 0°С 150 мл абсолютно сухого, свободного от О2 эфира (см. выше). Затем помещают 0,7 г (0,1 моль) Li-проволоки или кусочков Li и 4,2 г (40 ммоль) циклооктатетраена (см. выше) и перемешивают 16 ч. После того как осядет образовавшийся в небольшом количестве осадок Li2C8H8, определяют молярность гидролизом и титрованием кислотой определенной порции образовавшегося раствора. Насыщенный эфирный раствор Li2C8H8 имеет молярность —0,24 М.
б) Pt(ri4-C8Hi2)2. Двугорлую колбу на 1 л, снабженную краном для ввода N2, капельной воронкой с выравнивателем давления и магнитной мешалкой, помещают в охлажденную до —40 °C баню. Далее помещают 7,4 г (20 ммоль)
Олефиновые, ацетиленовые и аллильные комплексы 2021
Р1С12(т]4-С8Н|2) и 30 мл свободного от О2 циклооктадиена-1,5 (см. приготовление реагентов) и закрывают горло полиэтиленовой пробкой. Свежеприготовленный раствор Li2C8H8 (20 ммоль) с помощью шприца переносят в капельную воронку и в течение 45 мин прикапывают в интенсивно перемешиваемую реакционную смесь, температуру которой поддерживают при —40 °C. Затем нагревают до +10 °C и в вакууме (0,01 мм рт. ст.) отгоняют летучие компоненты. Колбу заполняют Na, твердый остаток счищают со стенок колбы и размельчают с помощью шпателя. Атмосферу N2 заменяют на атмосферу этилена и добавляют 800 мл петролейного эфира, насыщенного этиленом и охлажденного до 0 °C. Через 10 мин интенсивного перемешивания образовавшийся раствор легким избыточным давлением этилена передавливают через слой 2 см А12О3 (активность II степени) в колбу на 2 л. Фильтрование ни в коем случае нельзя проводить с подсосом! Остаток после экстракции еще раз обрабатывают 100 мл петролейного эфира, насыщенного этиленом, и точно так же пропускают через А12О3. К объединенному фильтрату добавляют 1 мл циклооктадиена и упаривают растворитель при пониженном давлении до объема ~15мл. Первоначально бесцветный раствор быстро становится коричневым. Снова создают в системе атмосферу N2, осторожно пипеткой удаляют маточник и кристаллический продукт промывают несколькими порциями петролейного эфира. Уже достаточно чистый, но слегка окрашенный в коричневый цвет продукт в течение 15 мин сушат в вакууме (0,01 мм рт. ст.) при комнатной температуре. Выход 6,0 Г (42—73%).
Для дополнительной очистки сырой продукт растворяют в большом объеме (~200 мл/г) петролейного эфира, пропускают через слой 5 см А12О3 (активность II степени), упаривают растворитель до начала помутнения и охлаждением до —78 °C снова кристаллизуют.
Способ 2 [5]
PtCls(r|4-C8H12) 4- 2Со(П-С5Н5)2 + С8Н12 ->
374,2 2-189,1 108,2
----> Р1(т]4-С8Н12)2 + 2[Со(п-С6Н5)2]С1 411,5 2-224,6
При —78°С к 10 мл циклооктадиена-1,5 и 10 мл СН2С12 прибавляют 1,0 г (2,67 ммоль) порошка PtCl2(tq4-C8Hi2) [см. методику синтеза предыдущего препарата] и 1,01 г (5,34 ммоль) Со(т1-С5Н5)2 [получение см. выше в этой главе]. В течение 15 мин при перемешивании доводят температуру смеси до —30 °C, а затем, продолжая перемешивание, в течение 1ч — до —20 °C. Приливают к смеси 100 мл эфира (20°C), при комнатной температуре фильтруют через слой «А!2О3 для хроматографии» (нейтральный, обезгаженный, дезактивированный 7% воды слой: 5 см высоты и 1 см в диаметре) в приемник, предварительно охлажденный до —78°C. Сорбент промывают эфиром (4x50 мл). Фильтрат для полноты кристаллизации выдерживают 24 ч при —78 °C. Маточник сливают, продукт сушат в высоком вакууме (10~2—10-3 мм рт. ст., начинают при —78°C и заканчивают при 20°C). Выход 770—885 мг (70—78%).
Из маточника после упаривания досуха, экстракции эфиром, пропускания через А12О3 и вымораживания при —78 °C получают еще 45—65 мг (4—6%) чистого продукта.
Свойства. Белый кристаллический комплекс Pt (COD) 2, в заметной степени на воздухе не разлагается, однако не следует его долго держать открытым на воздухе или помещать в вакуум, так как легко идет потеря олефиновых лигандов. /пл 127—128°C (с разл.). Не растворяется в воде, растворяется в органических растворителях. Комплекс в эфирном растворе значительно устойчивее, чем в СН2С12. Оба лиганда COD легко могут быть замещены на другие олефины, ацетилены, фосфины или изонитрилы. ЯМР-'Н (C6D6): б 4,20 [муль
2022 Глава 33. Металлоорганические комплексы
типлет, 8Н, =СН—], /(Pt—Н) =55 Гц; 6 2,19 [мультиплет, 16Н, СН2]. ЯМР-13С (CeDe): б 73,3 [С=С], J(Pt—С) = 143 Гц; б 33,3 [СН2], J(Pt—С) = = 15 Гц. Кристаллическая структура триклинная (пространственная группа симметрии РТ; а= 10,605 А, 6=9,370 А, с=7,382 А, а= 110,33°, ₽ = 84,43°, у= = 107,38°) [4].
ЛИТЕРАТУРА
1. Green М., Howard J. А. К., Spenser J. L., Stone F. G. A., J. C. S. Chem. Commun., 1975, 3; J. Chem. Soc. Dalton, 1977, 271.
2. Spenser J. L„ частное сообщение.
3. Muller, Goser, Angew. Chem., 79, 380 (1967).
4. Howard J. A. K-, неопубликованные данные.
5. Herberich G. E„ Hessner B., Z. Naturforsch., 34b, 638 (1979).
Алкильные и ацетильные комплексы
□ □ Бис(т]-циклопентадиенил)диметилтитан
Ti(7i-C5H5)2(CH3)2
TiCl2(T)-C5H5)2 + 2LiCH3 -> Ti(n-C5II5)2(CH3)2 + 2LiCl
249,0 2-22,0 208,2 2-42,4
В 200 мл эфира суспендируют 12,5 г (50 ммоль) TiCl2(r]-C6H5)2, охлаждают до —78 °C и при хорошем перемешивании добавляют точно стехиометрическое количество (100 ммоль) LiCH3 в эфире. При этом тщательно защищают от дневного света, который может инициировать автокаталитическое разрушение продукта! В течение 30 мин доводят температуру до 0 °C, окраска реакционной смеси при этом изменяется от оранжевой до желтой. Как только красный цвет исчезнет, растворитель отгоняют в вакууме водоструйного насоса, а сухой желтый остаток тотчас экстрагируют петролейный эфиром (tKIln 40—60 °C) до тех пор, пока остаток не станет белым. Экстракт фильтруют через стеклянный фильтр, покрытый кусочками фильтровальной бумаги, фильтрат упаривают до объема —100 мл и для кристаллизации ставят на сухой лед на ночь. Оранжево-желтые иголочки после декантации маточника короткое время сушат в высоком вакууме. Выход 9,9 г (95%).
Свойства. Оранжево-желтое кристаллическое соединение. Устойчивость его непредсказуема: иногда не изменяется в течение 1—2 дней на воздухе, а иногда за то же время при —30 °C и в атмосфере Аг разлагается, по-видимому, автокаталитически, образуя продукт черного цвета, который подробно не исследован. Поэтому рекомендуется хранить полученное соединение при —78 °C в атмосфере Аг. Ti(T]-C5H6)2(CH3)2 неустойчив на свету, не возгоняется, хорошо растворяется во всех обычных органических растворителях. Спонтанно разлагается при нагревании свыше 100 °C. В присутствии СО фотохимически легко идет образование ТЦСО^трСзВДг.
ЛИТЕРАТУРА
1. Piper Т. S., Wilkinson G., J. Inorg. Nucl. Chem., 3, 104 (1956).
2. Clauss К., Bestian H., Justus Liebigs Ann. Chem., 654, 8 (1962).
3. Alt H. G., Di Sanzo F. D., Rausch M. D., Uden P. C., J. Organometal. Chem., 107, 257 (1976).
4. Rausch M. D., Boon W. H., Alt H. G„ J. Organometal. Chem., 141, 299 (1977).
Алкильные и ацетильные комплексы 2023
□ □ Бис(т]5-инденил)диметилтитан, -цирконий, -гафний М(т]5-С9Н7)2(СН3)2 (M = Ti, Zr, Hf)
Поскольку исходные соединения М(rp-Cgll?)2С12 (M=Ti, Zr, Hf), из которых действием LiCH3 прямо могут быть получены соответствующие диалкиль-ные производные, как правило, не являются продажными реактивами, рекомендуется синтезировать М(д6-С9Н7)2(СН3)2 (M=Ti, Zr, Hf) без выделения промежуточных стадий. Этот способ описан в литературе, иапример, для получения М(т]-С5Н5)2(СН3)2 |[здесь: инден вместо циклопентадиена в качестве исходного соединения; см. также несколько ниже синтез М(т^-СдН?) (СН3) (СО3)].
а) 2С9Н8 2-116,2 + 2LiR > 2LiC9H7 + 2RH 2-22,0 (R = CH3) 2-122,1 2-22,4л 2-57,1 (R = h-C4H8) —78 °C
б) 2LiC9H7 2-122,1 + МС14 >- MCI2(r|5-C9H7)2 + 2LiCl 189,7(M=Ti) 348,8(M=Ti) 2-42,4 233,0(M=Zr) 392,1 (M = Zr) 320,3 (M = Hf) 479,4 (M=Hf)
—20 °с
в) МС12(г|5-С9117)2 + 2LiCH3 -> М(П5-С9Н7)2(СН3)2 + 2LIC1
348,8(M=Ti) 2-22,0 308,3 (M=Ti) 2-42,4
392,1 (M=Zn) 351,6 (M = Zr)
479,4 (M=Hf) 438,9 (M=Hf)
Используют точные стехиометрические количества соответствующих реагентов, иначе на последней стадии образуются маслообразные продукты, которые очень плохо элюируются пентаном или петролейным эфиром. Выходы 60—75%.
Свойства. Все бис(т]6-инденил)диметилметаллы (Ti, Zr, Hf) неустойчивы на воздухе. В холодильнике в атмосфере N2 сохраняются длительное время. Ваксообразные светло-желтые Zn- и Hf-производные возгоняются в высоком вакууме при 100—НО °C. Грам Ю5°С (M=Ti), 107 °C (M=Zr), 120 °C (М= =Hf). Молекулярная структура: см. [2].
ЛИТЕРАТУРА
1. Samuel Е„ Setton R., J. Organometal. Chem., 4, 156 (1965).
2. Atwood J. L., Hunter E. W., Hrncir D. C., Samuel E„ Alt H., Rausch M. D„ Inorg. Chem., 14, 1757 (1975).
3. Samuel E„ Rausch M. D., J. Amer. Chem. Soc., 95, 6263 (1973).
□ □ 1,1-Бис('г]-циклопентадиенил)-2,(3,4,5-тетрафенилметаллы
М(Т]-С5Н5)2[С4(С6Н5)4] (M = Ti, Zr, Hf)
Из известных методов синтеза металлоциклов M(ri-C6H5)2[C4(C6H5)4] [M=Ti, Zr, Hf] фотохимический метод является наиболее предпочтительным,
2024 Глава 33. Металлоорганические комплексы
поскольку дает наибольшие выходы, а также позволяет получить производные гафния.
М(г!-С6Н6)2(СН3)2 + 2СВН5С-СС6Н5 ->
208,2(M=Ti) 2.178,2
251,5(M = Zr)
338,8 (M=Hf)
СеН5 CeH6
+ побочные органические продукты
I I СеН6 СеН6
534,5 (М = Ti)
577,8(M=Zr)
664,7(M = Hf)
В обычном приборе для проведения фотохимических синтезов в 300 мл пентана растворяют 10 ммоль исходного соединения [2,08 г Т1(т1-С6Н6)2(СНз)2, 2,52 г 7г(т]-С5Н5)2(СНз)2 или 3,38 г Н1(т1-С6Н6)2(СНз)2] и 5,34 г (30 ммоль) дифенилацетилена. При интенсивном перемешивании облучают в течение 10 ч ртутной лампой высокого давления (Hanovia L, 450 Вт). На стенках сосуда выделяются кристаллы металлоциклов, как правило, аналитически чистые. При охлаждении реакционного раствора до 0 °C в течение 2—4 ч выпадают еще кристаллы. После декантации раствора и промывания небольшим количеством пентана кристаллы сушат в высоком вакууме. Целесообразно проводить синтезы с загрузками не более 10 ммоль, так как при более высоких концентрациях выход сильно снижается из-за уменьшения светопропускаемо-сти растворов. Выход: 30% (M=Ti); 40% (M=Zr); 48% (M=Hf).
Свойства. M = Ti: темно-зеленые кристаллы, устойчивые на воздухе в течение 1—2 дней; /пл 165 °C (на воздухе); умеренно растворяется в пентане. M=Zr: оранжевые кристаллы, устойчивые на воздухе в течение 1—2 дней; /пл 149 °C (на воздухе); умеренно растворяется в пентане. М=Ш: желтые кристаллы, устойчивые на воздухе в течение ~6 дней; /пл 202°C (на воздухе); умеренно растворяется в пентане.
ЛИТЕРАТУРА
1. Alt Н„ Rausch М. D„ J. Amer. Chem. Soc., 96, 5936 (1974).
2. Rausch M. D., Boon U". H., Alt H. G., J. Organometal. Chem., 141, 299 (1977).
□ □ Пентаметилтантал Ta(CH3)5
a) 2TaCl6 -f- 3Zn(CH3)2 -► 2TaCl2(CH3)3 + 3ZnCl2
2-358,2 3-95,5 2-297,0 3-136,0
6) TaCl2(CH3)3 + 2LiCH3 -> Ta(CH3)5 %- 2LiCl
297,0 2-22,0 256,1 2-42,4
Осторожно! Диметилцинк загорается на воздухе и бурно реагирует с водой. Технику безопасной работы с такими соединениями см. в статье [7].
Алкильные и ацетильные комплексы 2025
Твердый пентаметилтантал чрезвычайно пирофорен и может неожиданно взорваться! Его получение и работу с ним следует проводить только со специальным защитным экраном!
Прибор состоит из трехгорлой колбы на 250 мл с краном для ввода N2 и магнитной мешалки. В одно горло колбы вставляют доходящую почти до дна стеклянную трубку, на конце которой впаян стеклянный пористый фильтр G3. Другой конец трубки вставляют в колбу Шленка на 250 мл, точная масса которой заранее известна. Два других горла колбы закрывают соответственно резиновой пробкой (Septum, см. также т. 1, ч. I данного руководства) и ртутным затвором. Весь прибор основательно продувают очищенным Ns, а затем помещают 18,25 г (0,05 моль) свежевозогнанного ТаС15 и 180 мл сухого обескислороженного 2-метилбутана. При интенсивном перемешивании к этой смеси с помощью дозирующего шприца через резиновую пробку добавляют 5,5 мл (0,08 моль) Zn(CH3)2. Тотчас выпадает белый осадок ZnCIs. Перемешивают при комнатной температуре 4 ч, фильтруют желто-зеленый раствор во вторую колбу, растворитель упаривают, а зеленоватый остаток [ТаС12(СН3)3] высушивают при 0 °C в вакууме масляного насоса. Колбу взвешивают, продукт растворяют (10 мл/г) в сухом, насыщенном N2 эфире. Эфирный раствор охлаждают до •—70 °C и добавляют 2 г-экв. 1 М эфирного раствора метилли-тия. При перемешивании смеси доводят температуру до —20 °C, при этом выпадает осадок LiCl. Эфир удаляют в вакууме при —20 °C и реакционную массу грязно-зеленого цвета экстрагируют охлажденным до •—20 °C 2-метил-бутаном [5 мл/г ТаС12(СН3)3]. Для этого ~5 мин перемешивают и при —20°C отфильтровывают от нерастворимого LiCI. (Осторожно! Концентрированные растворы Та(СН3)5 в алифатических углеводородах на воздухе самовоспламеняются!) После отгонки растворителя в вакууме при —20 °C получают Та(СН3)5 в виде зеленых кристаллов. Выход 7,7—9,0 г (60—70%).
Свойства. Очень летучее твердое вещество зеленого цвета, хранится в течение длительного времени лишь при температуре ниже —40 °C в атмосфере инертного газа. ЯМР-'Н (толуол d8, —10°C): 6 = 0,82. Термохимические свойства см. [5]. Фотоэлектронные спектры см. [6].
ЛИТЕРАТУРА
1. Galyer L., Wilkinson G., частное сообщение.
2. Juvinall G. L., J. Amer. Chem. Soc., 86, 4202 (1964).
3. Fowles G. W. A., Rice D. A., Wilkins J. D„ J. Chem. Soc, Dalton. 1973, 961.
4. Schrock R. R„ Meakin P., J. Amer. Chem. Soc., 96, 5288 (1974).
5. Adedeji F. A., Connor J. A., Skinner H. A., Galyer L., Wilkinson G., J. Chem. Soc. Chem. Commun, 1976, 159.
6. Galyer L., Wilkinson G., Lloyd D. R., J. Chem. Soc. Chem. Commun., 1975, 497.
7. Harney D. W„ Meisters A., Mole T., Aust. J. Chem., 27, 1639 (1974).
□ □ Трикарбонил (р-циклопентадиенил)метилмолибден и -вольфрам М(т]-С5Н5)(СНз)(СО)3 (М = Мо, W)
Получение проводят в две стадии:
a) M(CO)e + NaC6H5 —Na[M(n-C5H5) (СО)3] 4- ЗСО
М=Мо: 264,0 88,1 268,1 3-22,4 л
M=W: 351,9 356,0
2026 Глава 33. Металлоорганические комплексы
б) Na[M(T]-C5H5) (СО).] + CHSI -> М(Л-С6Н5) (CH.) (СО). + Nal
М=Мо: 268,1 141,9 260,1 149,9
M=W: 356,0 348,0
Исходя из 13,2 г (50 ммоль) Мо(СО)6 Гили соответственно из 17,6 г (50 ммоль) W(CO)6] и 6,2 г (70 ммоль) NaCsHs, по способу получения М(л-С5Н5)С1(СО)3 (М=Мо, W), описанному ниже в этой главе, приготавливают растворы анионных комплексов Na[M(T]-C5H5) (СО)з) в диметилформ-амиде.
б) Раствор NaTMfrj-CsHs) (СО)3] (М=Мо, W) в диметилформамиде упаривают в высоком вакууме при 50—80 °C. Образовавшийся маслянистый коричневый остаток растворяют в 50 мл тетрагидрофурана и при 0°С и перемешивании прибавляют 15 мл (избыток) СН31. Тотчас образуется M^-CsHs) (СН3)-(СО)3, что заметно по посветлению раствора и выпадению Nal. Для завершения реакции нагревают 3 ч при ~40°С. Летучие компоненты отгоняют в вакууме водоструйного насоса при 30—40 °C; остаток высушивают в высоком вакууме при комнатной температуре. Выделение и очистку металлоорганических соединений проводят высоковакуумной возгонкой при следующих температурах: 50—55°C для Mo^-CsHs) (СН3) (СО)3, 60—65°C для W(t]-С5Н5)(СН3)(СО)3. Выход: 10,5—11,2 г (81—86%) Мо(л-С5Н5) (СН3) (СО)3 ил» 13,7—14,6 г (79-84%) W(t)-C5H5) (СН3) (СО)3.
Свойства. Желтые, не очень устойчивые кристаллические соединения е камфарным запахом. Растворяются во всех обычных органических растворителях. Гпл 124°С (с разл.) (М=Мо), 144,5—145°C (M=W). Спектральные данные см. табл. 47.
Таблица 47. Спектральные данные метильных комплексов М(Л-С5Н5) (СН3) (СО)3
м И К (CS2) v(CO), см-1 ЯМР-'Н (СС14, ТМС) ЯМР-,3С (CDC13. ТМС)
Й(С5Н5) б(СН3) 6(СО) 6(C5HS) 6(СН3)
Мо 2023 (оч. с.), 1934 (оч. с.), 1904 (пл.) 5,27 0,34 — — —
W 2019 (оч. с.), 1924 (оч. с.) 5,38 0,40 239,2 (цис) 217,8 (транс) 92,4 —28,9
ЛИТЕРАТУРА
1. Piper Т. S., Wilkinson G., J. Inorg. Nucl. Chem., 3, 104 (1950).
□ □ Трикарбонил(т]5-инденил)метилмолибден и -вольфрам М(т]5-С9Н7)(СН3)(СО)3 (М = Мо, W)
Получение г[5-инденильных комплексов, как и соответствующих трцикло-пентадиенильных, проводят в несколько стадий без выделения промежуточных. Сначала готовят индениллитий, который при взаимодействии с карбонилам»
Алкильные и ацетильные комплексы 2027
металлов М(СО)6 (М=Мо, W) образует карбонилат-анионы [М(г]5-С9Н7) (СО)з]~. Последние метилируют избытком СН31.
а) С9Н8 + LiCHs --> LiC9H7 + СН4
116,2 22,0 122,1 22,4л
б) C9H7Li + М(СО)6 ----> Li[M(n5-C9H7) (CO)sl + ЗСО
122,1 264,0(М=Мо) 302,1 (М=Мо) 3-22,4л
351,9(M=W) 390,0(M=W)
в) Li[M(^-C9H7)(COs)] + CH3I -> M(n5-C9H7)(CH3)(CO)S + Lil
302,1 (М=Мо) 141,9 310,2(М=Мо)
390,0 (M=W) 398,1 (M=W)
В 200 мл тетрагидрофурана растворяют 5,81 г (50 ммоль) свежеперегнан-ного индена и при перемешивании добавляют по каплям раствор L1CH3 в эфире (50 ммоль активного основания). Раствор окрашивается в оранжевый цвет, интенсивно выделяется метан. Образовавшийся раствор индениллития упаривают в вакууме досуха, растворяют остаток в 50 мл абсолютного N.N-диметилформамида и кипятят с обратным холодильником с 13,2 г (50 ммоль) Мо(СО)6 [или соответственно 17,6 г W(CO)6] до тех пор, пока не закончится отщепление СО (—1 ч). После этого реакционную смесь упаривают в высоком вакууме при —70 °C до маслообразной консистенции. Красный маслянистый остаток растворяют в 200 мл тетрагидрофураиа, добавляют 8,51 г (3,70 мл, 60 ммоль) СН31 и перемешивают 3 ч при комнатной температуре. Образовавшуюся желтую суспензию полностью упаривают в вакууме. Остаток суспендируют в —20 мл бензола и фильтруют через слой кизельге-ля 60 (Merck 7734; 3X10 см). Бензолом промывают до тех пор, пока он не станет практически бесцветным. Элюат снова упаривают и желтый остаток кристаллизуют из пентана при температуре сухого льда. Выход 80—85%.
Свойства. Мо(т]5-С9Н7) (СНз) (СО)3: желто-оранжевое, очень устойчивое на воздухе кристаллическое соединение. ta„ 91—93°С (с разл.). ИК (нуйол): 1937 (с.), 1867 (с.), 1855 (с.) [v(CO)J см-1.
Производное вольфрама обладает похожими физико-химическими характеристиками, однако до сих пор не получено аналитически чистым. Во всех обычных растворителях растворяется хорошо или очень хорошо.
ЛИТЕРАТУРА
1. King R. В., Bisnette М. В., Inorg. Chem., 4, 475 (1965).
2. Alt Н. G„ Z. Naturforsch., 32b, 1139 (1977).
□ □ Гексаметилвольфрам W(CH3)6
Первоначально W(CH3)6 синтезировали из WC16 и трех эквивалентов ме-тиллития [2]. Этот метод из-за трудности в контролировании и воспроизводимости условий реакции оказался все же неудовлетворительным.
Напротив, ниже приводимый способ позволяет получать W(CH3)6 в количестве 10 г и с хорошим выходом [1]:
WC16 + 6А1(СН3)3 —> W(CH3)9 + 6А1(СН3)2С1
396,6 6-72,1 274,1 6-92,5
А1(СН3)2С1 + N(CH3)3---> (CH8)2A1C1-N(CH8)3
92,5 59,1 151,6
2028 Глава 33. Металлоорганические комплексы
Осторожно! Триметилалюминий воспламеняется на воздухе и чрезвычайно энергично реагирует с водой. О работах с пирофорными жидкими ме-таллалкилами см. [3].
W(СН3)6 совзрывом реагирует с кислородом воздуха и детонирует без всякой видимой причины в вакууме и в атмосфере инертного газа. Следует соблюдать особые предосторожности (экран из непробиваемого стекла, защитная маска, асбестовые перчатки) при работах и особенно при возгонке твердых образцов.
Трехгорлую колбу на 500 мл, снабженную краном для ввода N2, магнитной мешалкой и резиновым колпачком (Septum, см. также т. г, ч. I данного руководства), хорошо продувают сухим N2. Затем помещают 20 г (50 ммоль) свежевозогнаниого WC16 и 200 мл сухого, свободного от. О2 2-метилбутана и охлаждают колбу до —70 °C смесью сухой лед—ацетон. Далее через резиновый колпачок с помощью шприца с металлической иглой прикапывают 29 мл (0,3 моль) триметилалюминия. Перемешивание продолжают до тех пор, пока баня и содержимое колбы постепенно не нагреются до комнатной температуры. Перемешивают еще ~15 мин при этой температуре, затем снова охлаждают темно-оранжевую смесь до —70 °C и из трубки Шленка медленно при интенсивном перемешивании приливают 35 мл (0,5 моль) жидкого триметиламина. Продажный триметиламин предварительно пропускают через колонку (30X5 см), заполненную молекулярными ситами (4 А), и затем переконден-сируют в трубку Шленка.
Пока заканчивается экзотермическая реакция, двугорлую колбу на 250 мл с краном и большую воронку для фильтрования с охлаждающей рубашкой (ср. рис. 469) хорошо продувают N2. Затем колбу и фильтровальную воронку охлаждают сухим льдом с ацетоном до —70 °C и холодную реакционную смесь с помощью металлической иглы переносят на фильтр и фильтруют. Остающийся после фильтрования аддукт (CH3)3N-A1(CH3)2C1 разлагают смесью гексанол — петролейный эфир, а красно-оранжевый фильтрат при —10 °C упаривают примерно до половины исходного объема. Повторным охлаждением до —70°C и фильтрованием удаляют большую часть аддукта (см. выше). Раствор, содержащий W(CH3)6, упаривают еще и так хранят. Определение W* указывает на выход 60—70%.
Чтобы получить кристаллический W(CH3)3, раствор в петролейном эфире-осторожно еще упаривают и охлаждают примерно до —100 °C на бане, приготовленной из ацетона и жидкого N2. При этой температуре избыток растворителя отсасывают с помощью стеклянной трубки, на конце которой вставлен стеклянный пористый фильтр. Ввиду хорошей растворимости W(CH3)3 этот способ не самый удачный. Небольшие количества твердого продукта получают отгонкой растворителя при —30 °C в высоком вакууме и последующей возгонкой остатка при —20 °C на охлаждаемую до —70 °C внутреннюю поверхность сублиматора.
Свойства. Темно-красное, очень летучее кристаллическое вещество, при температуре —40 °C начинает медленно разлагаться, ниже этой температуры хранится в течение длительного времени. Растворы при —25 °C в течение нескольких дней не разрушаются и даже выдерживают кратковременное нагревание до комнатной температуры. При действии LiCH3 образует Li2[W(CH3)8]. ЯМР-'Н (CC13F, —20 °C): 6 1,8 (синглет), V(183W—Н)=3 Гц. ЯМР-13С (то-луол-<18, —50°С, ТМС, {Н}): 6 83,6, >/(:83W—13С) =43,2 Гц.
* Чтобы определить количество W, пробу обрабатывают водным раствором NH3 и некоторое время нагревают на водяной бане. Далее W можно определить фотометрически [4, 5].
Алкильные и ацетильные комплексы 2029'
ЛИТЕРАТУРА
1. Galyer A. L., Mertis К.., Wilkinson G., J. Organometal. Chem., 85, С37 (1975); 97, С65 (1975); J. Chem. Soc. Dalton, 1976, 2235.
2. Shortland A. Wilkinson G., J. Chem. Soc. Dalton, 1973, 872.
3. Harney D. W„ Meisters A., Mole T„ Aust. J. Chem., 27, 1639 (1974).
4. Snell F. D., Snell С. T„ Colorimetric Methods of Analysis, Van Nostrand, Princeton, 1959, v. 11A, p. 361.
5. Freund H., Wright M. L., Brookshier R. K., Anal. Chem., 23, 781 (1951).
□ □ Пентакарбонил(трифторацетил)марганец Mn(COCF3)(CO)s
Получают двухстадийной реакцией через пентакарбонилманганат(—I)-анион:
Mn2(CO)10 + 2Na ---> 2Na[Mn(CO)5]
390,0 2-23,0 2-218,0
Na[Mn(CO)5] + (CF3CO)2O ---> Mn(COCF3) (CO)5 -f- Na[CF3CO2]
218,0 210,0 292,0 136,0
В колбе на 250 мл готовят жидкую амальгаму из 70 г Hg и 0,7 г (0,030 моль) Na. Для этого наливают ртуть и при интенсивном перемешивании добавляют Na(~20 мин, порциями по ~0,1 г) (сильно экзотермическая реакция!).
Амальгаму заливают 60 мл тетрагидрофурана и растворяют затем 5,85 г (15 ммоль) Мп2(СО)ю. Ввиду легкой окисляемости образующегося карбонил-металлата Na[Mn(CO)s] необходимо тщательно предохранять от попадания кислорода воздуха. Перемешивают еще 1 ч и декантируют зеленовато-серый-раствор с амальгамы в другую колбу на 250 мл. Затем при перемешивании из пипетки добавляют 4,2 мл (6,3 г, 30 ммоль) трифторуксусного ангидрида, перемешивают еще 1 ч и упаривают в вакууме водоструйного насоса. Из порошкообразного остатка высоковакуумной возгонкой при 50—54 °C получают 6,5—6,8 г (74—78%) конечного продукта в виде белых иголочек.
Другие способы. Взаимодействие Li[Mn(CO)5] с трифторацетилфтори-дом. Выход 70% [2].
Свойства. Устойчивые на воздухе белые кристаллы, 1ПЛ 55—56 °C. Соединение растворяется во всех обычных органических растворителях. При нагревании выше температуры плавления переходит в Mn(CF3) (СО)5. ИК. (циклогексан): 2150 (с.), 2040 (с.), 1625 (ср. шир.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Beck W„ Hieber W., Tengler H„ Вег., 94, 862 (1961).
2. McClellan W. R„ J. Amer. Chem. Soc., 83, 1598 (1961).
□ Пентакарбонил(трифторметил)марганец Mn(CF3)(CO)5
Mn(CF3)(CO)s образуется при термическом декарбонилированиш Mn(COCF3) (СО)5:
дт
Mn(COCF3) (СО)5 ---> Mn(CF3) (СО)5 + СО
292,0 264,0 22,4 л
В колбе на 50 мл, снабженной ртутным затвором, нагревают в атмосфере-азота 2,92 г (10 ммоль) возогнанного Mn(COCF3) (СО)5 в течение 90 мин при
12030 Глава 33. Металлоорганические комплексы
температуре 110 °C. Сначала сильное выделение СО затем через —30 мии прекращается. Жидкий продукт при охлаждении до комнатной температуры застывает в виде длинных игл или листочков. Продукт достаточно чистый, его возгоняют при 35—40 °C в высоком вакууме. Выход 2,35—2,46 г (89— 93%).
Свойства. Белое, устойчивое на воздухе кристаллическое соедииеиие, 82—83 °C. Умеренно растворяется в бензоле, хорошо — в тетрагидрофуране; вообще растворяется хуже, чем исходное соединение. ИК (циклогексан): 2155 (с.), 2050 (с.), 2015 (с.), [v(CO)] см~>.
ЛИТЕРАТУРА
1. Beck W., Hieber W., Tengler H., Вег., 94, 862 (1961).
2. McClellan W. R., J. Amer. Chem. Soc., 83, 1598 (1961).
□ □ Дикарбонил(т]-циклопентадиенил)метилжелезо
Fe(T]-C5H5)(CH3)(CO)2
Связь металл—металл в [Fe^-CsHs) (СО)2]2 может восстановительно рас-.щепляться под действием Na. Взаимодействием получившегося при этом [FefrpCsHs) (СО)2]_-иона с алкилиодидами получают железоорганические комплексы типа Fe^-CsHs) (алкил) (СО)2. Нижеприведенная методика может быть применена и для высших алкилзамещенных производных.
[Fe(r]-C5H5) (СО)2[2 + 2Na -> 2Na[Fe(n-C5H5) (СО)2]
353,9 2-23,0 2-200,0
Na[Fe(-r]-C5H5) (CO)J + СН31 ->- Fe(n-CSH5) (СН3) (СО)2 + Nal
200,0 141,9 192,0 149,9
В трехгорлую колбу на 500 мл, снабженную обратным холодильником, помещают 12,0 мл (163 г) Hg. Затем порциями примерно по 0,20 г прибавляют 1,20 г (52,5 ммоль) Na. После некоторого индукционного периода бурно начинает образовываться амальгама Na. Охлаждают до комнатной температуры и добавляют 200 мл тетрагидрофурана и 7,08 г (20 ммоль) [Fe^-CsHs) (СО)2]2. Реакционную смесь 1 ч перемешивают при комнатной температуре, цвет раствора постепенно изменяется от красно-коричневого до оранжево-коричневого. Раствор с амальгамы сливают в другую колбу на 250 мл, снабженную обратным холодильником и капельной воронкой, и прикапывают при перемешивании 3,0 мл (6,92 г, 49 ммоль) метилиодида. Через 8 ч перемешивания при комнатной температуре растворитель упаривают в вакууме. Твердый остаток возгоняют в высоком вакууме при 60 °C. Желто-коричневый сублимат плавится в интервале 78—82 °C. Выход 4,76 г (62%). Повторной возгонкой получают аналитически чистый препарат.
Свойства. Желто-коричневое, воскообразное, обладающее камфорным запахом, заметно разлагающееся на воздухе соединение; 1пл 83—84 °C. Хорошо растворяется во всех органических растворителях. В растворе очень легко окисляется. ИК (CS2): 2010 (оч. с.), 1955 (оч. с.) [v(CO)] см-1. ЯМР-Ч4 (CS2, ТМС): б 4,65 [синглет, С5Н5], 0,15 {синглет, СН3]. ЯМР-13С (СНС13, ТМС): б 85,3 [С5Н5]; б 218,4 [СО]; б —23,5 [СН3].
ЛИТЕРАТУРА
4. Piper Т. S., Wilkinson G.t J. Inorg. Nucl. Chem., 3, 104 (1956).
Алкильные и ацетильные комплексы 2031
□ Тетракарбонил (перфтор-я-пропил)иодожелезо
FeI(M-C3F7)(CO)4
Fe/CO)5 + «-C3F7I --> FeI(H-C3F,) (СО)4 + СО
195,9 295,9 463,8 22,4 л
Смесь 20,5 мл (29,5 г, 0,15 моль) Fe(CO)s и 33,4 мл (44,4 г, 0,15 моль) перфтор-н-пропилиодида перемешивают в закрытом автоклаве емкостью 300 мл в течение 20 ч при 65—75 °C (ввиду высокой летучести обоих реагентов автоклав перед закрыванием охлаждают до температуры сухого льда). Образовавшийся продукт извлекают 400 мл метиленхлорида (в четыре порции) и фильтруют через стеклянный пористый фильтр (G4). Темно-красный фильтрат упаривают в вакууме масляного насоса при температуре не выше 30 °C. Остаток в виде темно-красного очень мелкого порошка очищают повторной высоковакуумной возгонкой при 35±5°С. Выход 50,8—64,7 г (73— 93%).
Свойства. Темно-красные не очень устойчивые иа воздухе кристаллы, хорошо растворимые в большинстве органических растворителей (например, СС14, С6Н6). inn 68—69 °C. При длительном нагревании до 70 °C переходит в ,[Ре1(я-СзР7)(СО)з]2. ИК (С2С14): 2145 (ср.), 2111 (оч. сл.), 2088 (оч. с.), 2054 (ср.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. King R. В., Stafford S. L., Treichel Р. М., Stone F. G. A., J. Amer. Chem. Soc., 83, 3604 (1961).
□ □ трГалогенциклопентадиенильные комплексы железа Ре(н-С3Н7)(т]-С5Х41)(СО)2 (Х=Н, С1, Вг)
Синтез проводят, исходя из Fel (и-С3р7) (СО)4, введением циклопентади-еиильного лиганда, получаемого из соответствующего замещенного диазоцик-лопеитадиена:
FeI(H-CsF7) (СО)4 + CSX4N2 -->
463,8 Х = Н: 92,1
X = С1: 229,9
Х=Вг: 407,7
---> Fe(H-CsF7) (i]-CsX4I) (СО)2 + 2СО + N2
Х=Н: 471,9 2-22,4л 22,4л
Х=С1: 609,7
Х=Вг: 787,5
Дикарбонил(т]-иодциклопеитадиенил)(перфтор-н-цропил)железо
Fe(H-C3F7)(n-C5H4I)(CO)2
В 100 мл бензола растворяют 2,27 г (4,9 ммоль) FeI(H-C3F7) (СО)4 и добавляют 600 мг (6,5 ммоль) диазоциклопентадиена CsH4N2 (получение см. в этой главе). Уже при добавлении диазосоединения наблюдается слабое выделение газа. Для ускорения реакции раствор постепенно доводят до кипения и продолжают нагревание еще в течение 2 ч. После удаления растворителя и' избытка диазоциклопентадиена остается коричневое масло, которое очищают перегонкой в высоком вакууме при 120—135 °C. Выход 1,76 г (76%).
Свойства. Темно-желтое, не очень устойчивое на воздухе масло.
.2032
Глава 33. Металлоорганические комплексы
Дикарбоиил(перфтор-н-пропил)(т]-тетрахлориодциклопентадиенил)-
железо Fe(«-C3F7)(-q-C5Cl4I)(CO)2
В 100 мл бензола растворяют 1,16 г (2,5 ммоль) FeI(«-C3F7) (СО)4 и 575 мг (2,5 ммоль) тетрахлордиазоциклопентадиена C5CI4N2 (получение см. в этой главе) и кипятят смесь в течение 2 ч. (Выделение газа начинается при нагреве смеси до 45 °C.) Желто-коричневое масло, остающееся после отгонки .растворителя в вакууме масляного насоса, закристаллизовывают путем многократного растирания в 5 мл пентана и отгонки его в атмосфере N2, а затем высоковакуумной возгонкой при 50—53°C. Выход 1,45 г (95%).
Свойства. Желто-оранжевые, устойчивые на воздухе кристаллы, tn„ 61 °C (без разл.).
Дикарбонил (перфтор-«-пропил)(т]-тетрабромиодциклопентадиенил)-
железо Fe(«-C3F7)(-r]-C5Br4l)(CO)2
В 100 мл бензола растворяют и кипятят в течение 2 ч с обратным холодильником смесь 2,32 г (5 ммоль) FeI(«-C3F7) (СО)4 и 2,04 г (5 ммоль) тетра-бромдиазоциклопентадиена CsBr4N2 (получение см. в этой главе). Темно-коричневый раствор после фильтрования (для отделения нерастворимых продуктов разложения черного цвета) упаривают в вакууме водоструйного насоса. Перекристаллизацией желто-коричневого, как правило, мелкокристаллического или, реже, порошкообразного остатка из петролейного эфира при (—35) — (—78) °C получают 3,47 г (88%) желто-оранжевого Fe(«-C3F7) (л-’ С5Вг41) (СО)2. Его можно также возогнать при 95—100°C в высоком вакууме, но с большими потерями.
Свойства. Оранжево-желтое, кристаллическое, устойчивое на воздухе соединение, tnjI 101—102°C (без разл.). Спектральные данные подробно приве-.дены в цитируемой литературе.
ЛИТЕРАТУРА
1. Herrmann W. A., Huber М„ J. Organometal. Chem., 136, СИ (1977).
2. Herrmann W. A., Huber М„ Вег., Ill, 3124 (1978).
□ Тетракис[ц3-иодо(триметил)платина] [Pt(СН3)з1]4
4K2[PtCl6] + 12MgICH3 ---> [Pt(CH3)3I]4 + 4MgI2 + 8MgCl2 + 8KC1
4-486,0 12-166,2 1468,4 4-278,1 8-95,2 8-74,6
Подготовка реагентов. Диэтиловый эфир и бензол непосредственно перед использованием перегоняют над LiAlH4 в атмосфере Аг. Метилиодид перего-.няют над безводным CaSO4 и хранят в атмосфере N2. Порошкообразный KatPtCle] несколько часов сушат в вакууме (0,2 мм рт. ст.) при 110 °C.
В атмосфере N2 прикапывают 5,0 мл (11,4 г, 80 ммоль) СН31 в 20 мл эфира к 1,1 г (45,2 ммоль) магниевой стружки с такой скоростью, чтобы эфир слабо кипел. Черную суспензию перемешивают до тех пор, пока последние частички Mg не растворятся, и фильтруют через стеклянный фильтр непосредственно в капельную воронку.
Реактив Гриньяра в течение 10 мин прикапывают к охлаждаемой льдом суспензии 2,0 г (4,12 ммоль) тонкоизмельченного Kz[PtCl6] в 10 мл эфира и 40 мл бензола. Затем постепенно доводят температуру смеси до комнатной и перемешивают далее в атмосфере N2. За 4 ч первоначально желтый раствор -становится белым. Оставляют перемешивать на ночь и дают осадку осесть. Раствор фильтруют через стеклянный пористый фильтр G3. Прозрачный, почти -бесцветный фильтрат охлаждают до 0°С и при перемешивании добавляют к
Алкильные и ацетильные комплексы 2033
нему 5 мл также охлажденного ацетона. При этом наблюдается бурная реакция ацетона с избытком реактива Гриньяра, и смесь становится сначала желтой, а затем оранжевой. По окончании реакции образуются две фазы — верхняя желтая и нижняя оранжевая. С этого момента работают уже иа воздухе. К перемешиваемой смеси добавляют 25 мл ледяной воды, при этом образуются желтое твердое вещество и оранжевый раствор. Подкисляют далее 30 мл 10%-ной НС1, твердое желтое соединение снова растворяется, и образуется желто-оранжевый органический слой над красно-оранжевой водной фазой. Небольшие количества твердых частиц оранжевого цвета, которые собираются иа границе раздела фаз, отфильтровывают, органический слой отделяют, водный экстрагируют бензолом (3x30 мл). Экстракты и органический слой объединяют. Фильтруют для высушивания через слой безводного CaSO< и упаривают в роторном испарителе. Твердый желтый остаток извлекают 30 мл СНС13, упаривают растворитель на плитке до объема ~15 мл, добавляют 25 мл ацетона и охлаждают на ледяной бане. Образовавшиеся желтые кристаллы отсасывают и сушат на воздухе. Выход 1,2 г (80%).
Свойства. Желтые, устойчивые на воздухе кристаллы, taiI 195°C (с разл.). Вещество растворяется в неполярных растворителях, не растворяется в воде и ацетоне. ИК (нуйол): 2963 (с.), 2890 (с.), 2790 (ср.) [v(CH)], 1416 (с.), 1326 (оч. сл., шир.), 1260 (оч. с.), 1223 (оч. с.), 850 (сл., шир.), 830 (оч. сл., шир.) [б(СН)3], 558 (сл.), 548 (сл., шир.) [v(PtC)] см-1. ЯМР-'Н (бензол, ТМС): б 1,84 (7=78,4 Гц) [2].
ЛИТЕРАТУРА
1. Baldwin J. С., Kaska W. С., Inorg. Chem., 14, 2020 (1975).
2. Homen J. M., Kawamoto J. M., Morgan G. L., Inorg. Chem., 9, 2533 (1970).
□ □ Метил(трифени1лфосфин)золото Au(CH3)[Р(С6Н5)з]
AuBr[P(CeH5)3] + LiCH3 --> Au(CHg) [P(C6H5)3] + LiBr
539,2 22,0 474,3 86,8
В трехгорлой колбе в атмосфере Аг суспендируют 3,24 г (6 ммоль) AuBr[P(C6H5)3] (получение см. ниже в этой главе) в 25 мл абсолютного эфира. Охлаждают до 0 °C и с помощью шприца добавляют 6,0 ммоль метиллития в эфире. Смесь перемешивают 1,5 ч, доводя температуру за это время до комнатной. Чтобы гидролизовать остатки LiCH3, добавляют небольшое количество разбавленной НС1 и фильтруют. Остаток на фильтре экстрагируют бензолом и объединяют с эфирно-водной смесью. Органический слой отделяют, промывают водой и упаривают в вакууме до половины объема и добавляют при —15 °C пентан. Получившийся продукт (неустойчивый иа свету) собирают на фильтре, промывают холодным пентаном и сушат в вакууме над Р4Ою. Выход 1,9 г (67%).
Свойства. Бесцветные кристаллы (при перекристаллизации из смеси бензол— петролейный эфир), t„„ 173—175 °C. ЯМР-'Н (CDC13, ТМС, 35 °C): б 0,90 [дублет, СН3], 37(НСАиР) =8,6 Гц.
ЛИТЕРАТУРА
1. Coates G. Е., Parkin С., J. Chem. Soc. (London), 1962, 3220.
2. Tobias R. S„ частное сообщение.
12—1361
2034 Глава 33. Металлоорганические комплексы
□ □ Триметил(трифенилфосфин)золото Аи(СН3)з[Р(С6Н5)з]
[KAuBrJ + Р(С6Н5)3 + ЗЫСН3 ---> Au(CH3)3[P(C6H5)3] + 3LiBr + КВг
555,7 262,3 3-22,0 504,4 3-86,8 119,0
В атмосфере Аг в трубке Шленка суспендируют 8,8 г (15,8 ммоль) K[AuBr<]* * и 4,2 г (16 ммоль) Р(С6Н5)3 в 30 мл свежеперегнанного эфира. Смесь охлаждают сухим льдом с ацетоном и при перемешивании по каплям добавляют 51,2 ммоль LiCH3 в эфире. Образовавшуюся серую мутную суспензию нагревают до комнатной температуры и добавляют 20 мл воды. Слои разделяют, водную фазу многократно экстрагируют эфиром, экстракты объединяют, фильтруют через стеклянный пористый фильтр и упаривают досуха. Белый осадок перекристаллизовывают растворением в теплом гексане, фильтрованием и охлаждением до 0 °C. Остаток, нерастворимый в гексане, представляет собой Аи(СН3)3[Р(С6Н5)3]. Еще одну порцию продукта получают из гексанового раствора упариванием его. Полученный продукт сушат в вакууме над P4Oio.
Другие способы. Вышеприведенная методика является вариантом метода ГЗ], по которому эфирно-водный раствор фильтруют, хроматографируют иа А12О3. После перекристаллизации из гексана получают в данном случае бесцветные кристаллы с выходом 41 %.
Свойства. Бесцветное соединение, /пл 119—120°C (с разл.), растворяется в обычных растворителях. ЯМР-'Н (диоксан-б8, ТМС): б 0,87 [СН3, транс к Р(С6Н5)3], 37(НСАиР) =9,3 Гц; б —0,23 м. д. [СН3, цис к Р(С6Н5)3], 7(НСАиР)=7,2 Гц.
литература
1. Tobias R. S., частное сообщение.
2. Block В. Р„ Inorg. Synth., 4, 16 (1953).
3. Coates G. E., Parkin C., J. Chem. Soc. (London), 1963, 421.
Карбиновые и карбеновые комплексы
□ □ Пентакарбонил[метокси(органил)карбен]хром
и пентакарбонил[метокси(органил)карбен]вольфрам M(CO)5[C(OCH3)R] (M=Cr, W; R = CH3, С6Н5)
Ниже описываемый способ является типичным примером классического синтеза карбеновых комплексов переходных металлов по Фишеру:
а) М(СО)6 + LiR -----------> Li[M{C(O)R} (CO)S)
M=Cr: 220,1 R = CH3: 22,0
M=W: 351,9 R = C6H5: 84,0
6) Li[M{C(O)R} (CO)8J + [(CH3)3O] [BF4] ->
147,9
----> M(CO)S[C(OCH3)R] + (CH3)2O + LiBF4 I. Cr(CO)5[C(OCH3)CH3): 250,1 46,1 93,7
II. Cr(CO)5[C(OCH3)C6H5]: 312,2 III. W(CO)5[C(OCH3)CH3]: 382,0
IV. W(CO)5[C(OCH3)C8HS]: 444,1
* Синтез K[AuBr4] см., например, [2].
Карбиновые и карбеновые комплексы 2035
В колбе на 1 л с капельной воронкой и ртутным затвором суспендируют 20 ммоль (4,40 г) Сг(СО)6 [или соответственно 7,04 г W(CO)s] в 500 мл эфира. К перемешиваемой суспензии прикапывают за 30 мин эквимолярное количество ~0,5 М эфирного раствора ЫСНз [имеющийся в продаже раствор, например, фирмы Merck-Schuchard) (I, II; см. реакцию б), соответственно LiC6H5 (получение см. ниже) (III, IV; см. реакцию б)]. Реакционная смесь окрашивается в интенсивно-желтый цвет, а карбонил металла полностью растворяется. Перемешивают еще 1 ч, упаривают в вакууме водоструйного насоса, растворяют желтый осадок в ~ 100 мл Н2О, добавляют 200 мл пентана, а затем присыпают к раствору при перемешивании маленькими порциями 3,11 г (21 ммоль) тетрафторобората триметилоксония. При этом тотчас образуется карбеновый комплекс, который растворяется в пентане. Окончание процесса определяют по кислой реакции водной фазы. До тех пор пока этого нет, продолжают добавлять соль оксония. После отделения пентанового раствора водный слой экстрагируют пентаном (2x100 мл). Объединенный фильтрат пропускают через слой безводного Na2SO4 и упаривают примерно до 0,1 прежнего объема. При охлаждении до —35 °C кристаллизуются длинные от желтого до оранжевого цвета иглы. После упаривания маточника и высоковакуумной возгонки остатка при 40—60 °C получают дополнительно некоторое количество комплекса. Общий выход 85—92%.
□ □ Фениллитий ЫС6Н5
2L1 + СвН6Вг ----> С6Н5Ы + LiBr
2-6,9 157,0 84,0 86,8
В трехгорлую колбу, снабженную капельной воронкой и обратным холодильником с ртутным затвором, помещают 8,0 г (1,16 моль) Li в виде мелких кусочков обезжиренной литиевой проволоки в 250 мл эфира. В течение 60 мин прикапывают раствор 80 г (0,51 моль) бромбензола в 125 мл эфира. По окончании реакции добавляют еще 100 мл эфира и кипятят еще 10 мни. Затем фильтруют через стеклянный пористый фильтр, покрытый кусочками фильтровальной бумаги. На холоду в приемнике кристаллизуется LiCeHs-LiBr в виде бесцветных или белых кристаллов. Выход практически количественный [4].
Свойства. Очень устойчивые игольчатые кристаллы от желтого до ораи-жево-красного цвета. Соединения хорошо растворяются во всех органических растворителях.
M=Cr, R=CH3: 34°C; ПК (нуйол): 2070 (с.), 1992 (с.), 1953 (оч. с.) [v(CO)] см-1; ЯМР-'Н (C6D6, ТМС): б 3,72 [синглет, ОСН3], б 2,33 [синглет, СН3].
М=Сг, R=C6H5: tn„ 46°С; ИК (нуйол): 2066 (с.), 1992 (с.), 1953 (оч. с.) [v(CO)] см-1; ЯМР-'Н (C6D6, ТМС): б 6,95 [мультиплет, CeHs], б 3,78 [синглет, ОСН3].
M=W, R=CH3: U 52°C; ИК (нуйол): 2083 (с.), 1988 (с.), 1946 (оч. с.) Гу(СО)] см-1; ЯМР-'Н (C6D6, ТМС): б 3,83 [синглет, ОСН3], б 2,31 [синглет, СН3].
M=W, R=C6H5: 59°C; ИК (нуйол): 2083 (с..), 1988 (с.), 1946 (оч. с.),
[v(CO)] см-1; ЯМР-'Н (CeDe, ТМС): б 7,04 [мультиплет, С6Н5], б 3,96 [синглет, ОСН3].
Кристаллическая структура: см. [5] и цитируемую там литературу.
ЛИТЕРАТУРА
1. Fischer Е. О., Maasbol А., Вег., 100, 2445 (1967).
2. Aumann R., Fischer Е. О., Вег., 101, 954 (1968).
12*
036 Глава 33. Металлоорганические комплексы
3. Fischer Е. О., Schubert U., Kleine W., Fischer H., Inorg. Synth., 19, 165 (1979).
4. Wittig G., Angew. Chem., 53, 243 (1940).
5. Fischer E. 0., Angew. Chem., 86, 651 (1974).
□ □ Пентакарбонил (дифенил циклопропенил идеи) хром
и пентакарбонил (дифенилццклопропенилидеи) молибден M(CO)5[C3(C6H5)2] (М = Сг, Мо)
Naa[Ma(CO)10] + C12CS(C6H5)2 ►
M=Cr: 430,1 261,1
М=Мо:518,0
---> М(СО)6[Сз(С6Н5)2] + Na[MCl(CO)e] + NaCl M = Cr: 382,3 M = Cr: 250,5 58,4
M = Mo: 426,2 M = Mo: 294,5
В колбе на 100 мл с капельной воронкой и ртутным затвором растворяют 5,0 ммоль Na2[Cr2(CO)i0] или соответственно Na2[Mo2(CO)io] (2,15 г, соответственно 2,59 г) в 60 мл тетрагидрофурана. Затем в течение 1 ч при перемешивании и охлаждении до —10 °C прибавляют раствор 1,25 г (4,8 ммоль) 3,3'ди-хлор-1,2-дифенилциклопропена* в 15 мл тетрагидрофурава. Желто-коричиевую (Cr-производное) (или соответственно темно-коричневую в случае W-произ-водного) реакционную смесь перемешивают еще 1 ч при комнатной температуре. Растворитель затем отгоняют в вакууме.
а) М=Сг: светло-коричневый, частично кристаллический остаток основательно (3x50 мл) экстрагируют бензолом, экстракт фильтруют через стеклянный пористый фильтр (G4) и упаривают в вакууме досуха. На маленьком стеклянном пористом фильтре (G3) сырой продукт промывают метанолом (3x5 мл), бензолом (5x5 мл) и высушивают в вакууме. Раствор после промывания упаривают до объема 5 мл и хроматографируют (колонка 70x2 см) на дезактивированном SiO2 (5% Н2О). Элюируют последовательно гексаном (50 мл), смесью эфир — гексан (1:2, 200 мл), бензолом (150 мл); карбеновый комплекс идет желтой зоной после коричневой полосы побочного продукта. Общий выход —70% (1,25—1,35 г).
б) М=Мо: реакционную смесь после удаления растворителя промывают на пористом фильтре (G3) последовательно бензолом (2x20 мл), водой (2Х Х20 мл), метанолом (20 мл) и высушивают в вакууме. Коричнево-желтый кристаллический продукт извлекают 20 мл тетрагидрофурана, перемешивают с 10 г SiO2, высушивают и помещают иа колонку с дезактивированным SiO2 (5% Н2О). Элюируют последовательно смесью эфир — гексан (1:2, 400 мл), эфиром (50 мл), бензолом (150 мл) и смесью бензол — тетрагидрофуран (300 мл, 1:1). Карбеновый комплекс идет желтой зоной вслед за бесцветным и коричневым исходными продуктами. Продукт, полученный после отгонки растворителя, промывают на маленьком фильтре при —25 °C небольшим количеством эфира для удаления от загрязнений (коричневого цвета) и высушивают в вакууме. Выход —51% (1,0—1,1 г).
Другие способы получения. При действии тетрафторобората 3-этокси-1,2-дифенилциклопропенильного карбкатиона вместо 3,3-дихлор-1,2-дифенилцикло-пропена. Обработка Na^[Cr(CO)5] вместо Na2[Cr2(CO)i0] приводит к существенно меньшим выходам.
* Получают из 1,2-дифенилциклопропеноиа и оксалилхлорида [3].
Карбиновые и карбеновые комплексы 2037
Свойства. Сг-производное: темио-желтые, устойчивые иа воздухе кристаллы, 1ПЛ 199—200°C (с разл.). Хорошо растворяется в тетрагидрофуране и бензоле, умеренно в эфире, плохо в гексане. Возгоняется в высоком вакууме при 90 °C. Кристаллическая структура: см. [4].
Мо-производное: светло-желтые, устойчивые на воздухе кристаллы, tnx 180°C (с разл.). Растворимость значительно хуже по сравнению с производными Сг.
ЛИТЕРАТУРА
1. Ofele К., Angew. Chem., 80, 1032 (1968).
2. Rees С. W., von Angerer E„ J. Chem. Soc. Chem. Commun., 1972, 420.
3. Fohlisch B., Burgle P., Liebigs Ann. Chem., 701, 67 (1967).
4. Huttner G., Schelle S„ Mills O. S., Angew. Chem., 81, 536 (1969).
□ □ Пентакарбонил (^М'-дифенилдиаминокарбен)хром
Cr(CO)5[C(NHC6H5)2]
Получение проводят в две стадии из карбонилметаллата, карбодиимида и кислоты, не выделяя промежуточно образующийся иа первой стадии анионный аддукт.
a) Na2[Cr(CO)5] + (C6H5N)2C-> Na2[Cr(CO)5.C(NC6Hs)2]
238,0 194,2 432,3
б) Na2[Cr(CO)6-C(NCeH5)2] + 2НС1 -> Cr(CO)5[C(NHC6H5)2] + 2NaCl
432,3 2-36,5 388,3 2-58,4
Охлажденный до —78°C раствор 2 г (8,4 ммоль) Na2[Cr(CO)5] в 100 мл тетрагидрофурана и 1,63 г (8,4 ммоль) дифенилкарбодиимида в 20 мл тетрагидрофурана помещают в предварительно прокаленную трубку Шленка и перемешивают 10 мин. Прикапывают пипеткой —18 мл —1 М раствора НС1 в эфире (18 ммоль) и дают смеси нагреться до комнатной температуры. Раствор упаривают в вакууме досуха, растворяют в 50 мл тетрагидрофурана и фильтруют через слой ~ 1 см целлюлозы. После удаления растворителя кристаллический продукт перекристаллизовывают из нагретого до 30 °C этанола (охлаждая до —30°C). Выход 2,6 г (80%).
Свойства. Длинные, зеленовато-желтые, устойчивые на воздухе игольчатые кристаллы. Вещество плохо растворяется в пентане, хорошо — в СН2С12; плавится при 139—140 °C без разложения. При попытках возогиать продукт в высоком вакууме количественно образуется изонитрильный комплекс Cr(CO)5CNC6H5. ПК (КВг): 3428, 3287 (сл.) [v(NH)l, 1504 (ср.) [v(NCN); (СН2С12): 2051 (сл.), 1929 (оч. с., шир.) [v(CO)J см-*.
ЛИТЕРАТУРА
1. Fehlhammer W. Р„ Mayr A., Ritter М., Angew. Chem., 89, 660 (1977).
□ □ Пентакарбонил (дифенилкарбен) вольфрам
W(CO)5[C(C6H5)2]
Способ 1 [1—3]
a) W(CO)6[C(OCH3)C6H5] + LiC6H5 --> Li[W(CO)6{C(OCHs) (С6Н5)2}]
444,0 84,0 528,1
2038
Глава 33. Металлоорганические комплексы
б) Li[W(CO)5{C(OCHs)(C6H5)2}]+HCl ----->
528,1 36,5
----> W(CO)5[C(C6H6)2] + LiCl + СН3ОН 490,1 42,4 32,0
В колбу на 500 мл помещают раствор 3,30 г (7,43 ммоль) W(CO)s-[С(ОСН3)С6Н6] в 250 мл эфира, охлаждают до —78 °C и при перемешивании прикапывают за 90 мин ~2 М эфирный раствор фениллития, всего 8,5 ммоль (получение см. выше в этой главе). Реакционная смесь из оранжево-красной становится красно-коричневой. Затем медленно добавляют эфирный раствор сухого НС! (10,5 ммоль НС1 в 10—15 мл эфира), при этом тотчас раствор становится темно-красным. Через 10 мин потемневший раствор доводят до комнатной температуры, добавляют 10 г кизельгеля и упаривают получившуюся суспензию в вакууме водоструйного насоса. Реакционную смесь затем хроматографируют на охлаждаемой водой колонке (60X1,2 см) иа кизельгеле, используя в качестве элюеита петролейный эфир (fKHn 40—60°C). Из темнокрасной зоны после отгонки растворителя и перекристаллизации остатка из 30 мл пентана при —25 °C получают аналитически чистый W(CO)5[C(CeH5)2]. Выход 2,14 г (59%).
Способ 2 [4]
Синтез W(CO)5[C(C6H5)2] проводят также и по следующей методике: 1,78 г (4,0 ммоль) W(CO)5[C(OCH3)C6H5] растворяют в 100 мл эфира и охлаждают до —78 °C смесью метанола с сухим льдом. Затем при интенсивном перемешивании медленно прикапывают раствор 4,4 ммоль LiC0Hs в 20 мл эфира. Раствор становится светло-коричневым. Далее его переносят на хроматографическую колонку (35x2,5 см), заполненную кизельгелем (в пентане) и охлажденную до —40 °C (кизельгель 60, Merck 7734, активность II—III степени). Тотчас появляется темво-красная зона, которая быстро перемещается. Побочные продукты не элюируются и тем самым отделяются от продукта реакции. Их смывают с колонки эфиром в охлаждаемый до —40 °C приемник. После отгонки растворителя в высоком вакууме продукт реакции повторно хроматографируют на колонке (25x2,5 см) с кизельгелем пеитаиом при —40 °C. Темно-красная зона теперь уже движется медленно. Раствор комплекса собирают в охлаждаемый приемник, отгоняют растворитель и дважды перекристаллизовывают из пентаиа. Выход 1,44 г (73%).
Свойства. Устойчивые иа воздухе кристаллы, черного цвета, /пл 65—66 °C. Растворим во всех органических растворителях, растворы на воздухе быстро окисляются (образование бензофенона). Дипольный момент р=3,48 дебай (С6Н6, 20°С). ИК (гептан): 2070 (ср.), 1971 (с.), 1963 (с.) [v(CO)l см-1. ЯМР-!Н (CS2, ТМС): б 7,2 [мультиплет, СбН5].
ЛИТЕРАТУРА
1. Casey С. Р„ Burkhardt Т. J., J. Amer. Chem. Soc., 95, 5833 (1973).
2. Casey C. P„ Burkhardt T. J., Bunnell Ch. A., Calabrese J. C., J. Amer. Chem. Soc., 99, 2127 (1977).
3. Casey С. P., Burkhardt T. J., Neumann S. M., Scheck D. M„ Tuinstra H. E., Inorg. Synth., 19, 180 (1979).
4. Fischer E. O., Held W„ Kreissl F. R., Frank A., Huttner G„ Ber., 110, 656 (1977).
Карбиновые и карбеновые комплексы 2039
□ □ трамс-Бромо(тетракарбонил) (фенилкарбин)хром и трамс-бромо(тетракарбонил)(фенилкарбин)вольфрам MBr(CO)4(CC6H5) (M=Cr, W)
M(CO)s[C(OCH3)CeH5] + ВВг3 ---> МВг(СО)4(СС6Н5) + ВВг2(ОСН3) + со
М = Сг:312,2 250,5 М = Сг: 333,1 201,7 22,4л
M = W: 444,1 M=W: 464,9
В колбе Шлейка на 250 мл растворяют 10,0 ммоль М(СО)б[С(ОСНз)С6Н5] (М=Сг: 3,12 г; M=W: 4,44 г) в 200 мл пентана. При —78 °C добавляют 3,00 г (12,0 ммоль) ВВгз и медленно нагревают раствор до —20 °C. Начинается выделение газа (СО), и постепенно выпадает желтый осадок. Через ~3 ч декантируют, при —78 °C промывают осадок пентаном (5x20 мл) и сушат в высоком вакууме при —20 °C.
Для очистки карбиновый комплекс хроматографируют при —30 °C на ки-зельгеле, который предварительно в течение 15 ч высушивают в высоком вакууме при комнатной температуре. Смесью пентан — СН2С12 (5:1) смывают сначала не вошедший в реакцию карбеновый комплекс. Затем с помощью пипетки удаляют коричневый остаток с верхней части колонки и элюируют карбиновый комплекс дихлорметаном. Раствор при —20 °C упаривают в высоком вакууме до нескольких миллилитров, добавляют 20 мл пентана и оставляют на ночь. Выпавшие из этого раствора светло-желтые игольчатые кристаллы промывают при —78 °C пентаном (3X20 мл) и сушат 24 ч при —20 °C в высоком вакууме. Выход: производное Сг — 2,7 г (81%); производное W — 4,0 г (86%).
Свойства. Желтые игольчатые кристаллы, медленно разлагаются при комнатной температуре. Хорошо растворяются в бензоле, эфире и СН2С12; умеренно растворяются в пентане. Растворы ниже —20 °C устойчивы. Спектральные и структурные данные см. [1—3].
Аналоги. Соответствующий молибденовый комплекс получают аналогично, однако он термически менее устойчив. Взаимодействием карбеновых комплексов с ВС13 и В13 получают аналогичные транс-хлоро- и транс-иодокарбиновые комплексы. Исходя из карбеновых комплексов с другими лигандами —C(OR)R, легко варьируют заместитель R у карбинового атома углерода (например, Р=алкил, —N(C2H5)2, —С=С—С6Н5).
ЛИТЕРАТУРА
1. Fischer Е. О., Kreis G„ Kreiter С. G., Miiller J., Huttner G„ Lorenz H., Angew. Chem., 85, 618 (1973).
2. Fischer E. 0., Kreis G„ Ber., 109, 1673 (1976).
3. Fischer E. O., Angew. Chem., 86, 651 (1974).
□ □ Дикарбонил (т]-циклопентадиенил) (дифенилкарбен)-марганец Mn(irC5H5) (СО)2[С(С6Н5)2]
Получение проводят диазометодом, т. е. путем переноса карбена из соответствующего диазопроизводного, дифенилдиазометана, на более реакционноспособный сольватный комплекс марганца MnfrpCsHs) (СО)2(ТГФ), получаемый в свою очередь фотолизом Mn^-CsHs) (СО)3 в тетрагидрофуране.
hv
а) Мп(т]-С5Н5) (СО)3 - Мп(т1-С6Н5)(СО)2[ОС4Н8] + СО
204,1 248,1 22,4 л
2040 Глава 33. Металлоорганические комплексы
б) Мп(л-С8Н5)(СО)2[ОС4Н3] + М2=С(СвН6)2 ----->
248,1 194,2
---> Мп(П-С5Н5) (СО)2[С(С6Н5)2] + N2 + ОС4Н8 (ОС4Н8—ТГФ) 342,3 22,4 л 72,1
В охлаждаемый водой фотореактор NORMAG с принудительным обменом жидкости (реактор типа Falling Film А 9356 по Демейеру, рис. 463) помещают раствор 9,00 г (44,1 ммоль) Мп(г]-С5Н5) (СО)3 в 1,25 л тетрагидрофурана и облучают 4—5 ч ртутной лампой (0,7 кВт) высокого давления (TQ 718)* (дюрановый фильтр, температура реакции 21—24°C). Темно-красный раствор сольватного комплекса марганца Mn^-CsHs) (СО)2(ТГФ) переносят в колбу на 2 л, добавляют 7,50 г (38,6 ммоль) кристаллического дифенилдиазометана ((Пл 28 °C, получение см. [4]) и перемешивают 40 ч при комнатной температуре. Образовавшийся темно-коричневый раствор упаривают до объема —50 мл в роторном испарителе при —40°C (температура бани), добавляют при перемешивании (водяная баня, 15 °C) 60 г кизельгеля (Merck 7734; активность I степени) и высушивают смесь в высоком вакууме в течение 3 ч. Предварительную очистку проводят хроматографированием на колонке (30x3,5 см) с кизельгелем (Merck 7734; активность I степени), элюент — бензол; смывают до тех пор, пока раствор не станет светло-желтым. Последующую очистку проводят также хроматографированием на охлаждаемой водой колонке (20x3,2 см) с кизельгелем, элюент — петролейный эфир ((кип 40—60°C). Петролейный эфир сначала элюирует 4,60 г (22,5 ммоль, 51%) ие вошедшего в реакцию Мп(грС5Н5) (СО)3 в виде желтой зоны. Смесью петролейный эфир — бензол (5:1) смывают темно-коричневую зону карбенового комплекса Мп(т]-С5Н5) (СО)2[С(С6Н5)2]. После упаривания растворителя в вакууме водоструйного насоса и перекристаллизации остатка из смеси н-пентаи — эфир (—2 : 1, —35/—80 °C) получают 5,80 г продукта [78% в пересчете на вступивший в реакцию Мп(т]-С5Н5) (СО)3], который после высушивания в высоком вакууме является аналитически чистым.
Небольшие загрузки [до 2 г Mn^-CjHs) (СО)3] проводят в обычной аппаратуре с погружаемой лампой (ср. рис. 462) емкостью 230 л. Вышеприведенную пропись корректируют с учетом загрузки. Аналогичные диарилкарбеновые комплексы получают точно по такой же методике.
Свойства. Темно-коричневые, не очень устойчивые иа воздухе ромбические кристаллы с интенсивным зеленым блеском с поверхности, (пл 91—92 °C. Вещество растворяется во всех органических растворителях, растворы неустойчивы на воздухе. ИК (гексан): 1977 (оч. с.), 1919 (оч. с.) [v(CO)] см-1. ЯМР-'Н (ацетон-de, ТМС): 6 4,88 [синглет, С5Н5]; 7,3—6,8 [мультиплет, С6Н5]. ЯМР-'ЗС (CDC13, ТМС): б(С5Н5) 90,74, б (СО) 231,45, б (С, карбеновый) 352,83, б(СеН5) 162,54, 127,17, 122,83. Кристаллическая структура: см. [5].
В атмосфере СО при повышенном давлении образует т]2-кетеновый комплекс Мп(т]-С5Н5) (СО)2[(С6Н5)2С = С=О]. Аминоацетилены реагируют, внедряясь по связи Мп — карбен [7].
ЛИТЕРАТУРА
1. Herrmann А., Вег., 108, 486 (1975).
2. Herrmann W. A., Atwood J. L., Plank J., неопубликованные результаты 1978—1980 гг.
3. Herrmann W. A., Plank L, Вег., 112, 392 (1979).
4. Smith L. 1., Howard К- L., Org. Synth, Coll. Vol. Ill, 351 (1955).
5. Haymore B. L., Hillhouse G. L., Herrmann IF. A., Inorg. Chem., в печати. 6. Herrmann W. A., Plank J., Angew. Chem., 90, 555 (1978).
7. Dotz K- H„ Pruskil I., J. Organometal. Chem., 132, 115 (1977).
* Изготовитель — фирма Original Quarzlampen-Gesellschaft mbH (Hanau). Указания по пользованию фотореактором см. [3].
Карбиновые и карбеновые комплексы 2041
□ □ Карбамоил (дикарбонил) (трциклопентадиенил )-рутений Ru(t]-C5Hs)(CONH2)(CO)2
[Ru(t]-CsH5) (СО)3] [PFJ + 2NH3 -> Ru(r]-C6H5) (CONH2) (CO)a + N^PF,
395,2 2-17,0 266,2 163,0
В трубку Шлейка, охлажденную смесью метанола с сухим льдом примерно до —60°С, помещают 400 мг (1,01 ммоль) [Ru^-CsHs) (СО)3] [PF6] (получение см. выше в этой главе) и конденсируют в нее ~30 мл сухого NH3 (сушат над Na путем конденсации в промежуточную трубку Шленка). Затем дают медленно нагреться с такой скоростью, чтобы NH3 испарился примерно за 1 ч (fKHn —33 °C). Остатки NH3 удаляют с помощью водоструйного иасоса и затем сушат в высоком вакууме. Сухой остаток перемешивают в 25—30 мл бензола (или СН2С12), NH4PFs остается при этом нераствореиным. Экстракт фильтруют, упаривают до объема 5 мл и высаживают продукт добавлением 30 мл петролейного эфира. Для большинства реакций полученный таким образом продукт является достаточно чистым. Более тонкую очистку проводят высаживанием петролейным эфиром (30 мл) из бензола (25 мл) или перекристаллизацией из бензола (25°С/8°С). Возможна также очистка возгонкой, однако она сопряжена с потерями ввиду частичного разрушения при нагревании. Выход 260 мг (96%).
Соответствующие комплексы железа и осмия, Fe(CONH2)(r]-C5H5)(CO)2 и Os(CONH2)(t]-CsH5)(CO)2 получают аналогичным образом [2].
Свойства. Белое, мелкокристаллическое соединение, 128 °C, в течение
короткого времени устойчиво иа воздухе, в растворах, однако, весьма чувствительно к окислению. Хорошо растворимо в ацетоне, СН2С12 и бензоле, очень плохо — в петролейиом эфире. ИК (СН2С12): 2036 (оч. с.), 1971 (оч. с.) (v(CO)], 1608 (с.), 1568 (ср.), 1560 (пл.) [v,5(CONH2)] см-1. ЯМР-‘Н (CD2C12, ТМС, 0 °C): 6 5,44 [синглет, С5Н5], 5,92 [мультиплет, NH2].
ЛИТЕРАТУРА
1. Behrens Н„ Jungbauer A., Z. Naturforsch., 34b, 1477 (1979).
2. Ellermann J., Behrens H., Krohberger H., J. Organometal. Chem., 46, 116 (1972).
□ □ цз-Хлорметилидин-цикло-трис(трикарбонилкобальт) (3Co—Co) p3-CCl[Co(CO)3]3
9Co2(CO)8 + 4ССЦ ---> 4p3CCl[Co(CO)3]3 %- 36CO + 6CoCl2
9-342,0 4-153,8 4-476,4 36-22,4л 6-129,8
В колбе на 500 мл, снабженной обратным холодильником, перемешивают раствор 10,3 г (30 ммоль) Со2(СО)8 в 200 мл СС1< (избыток) в течение 1 ч при 57±3°С. Охлажденный до комнатной температуры раствор встряхивают с ~ 150 мл воды, органический слой отделяют, сушат Na2SO4 и упаривают в вакууме водоструйного насоса. Сухой остаток переносят в аппаратуру для возгонки. При 40 °C в высоком вакууме получают аналитически чистые кристаллы пурпурного цвета. Выход 4,3—4,6 г (68—72%). Иначе продукт можно очищать путем экстракции сухого остатка в аппарате Сокслета гексаном.
Свойства. Пурпурные, ие очень устойчивые на воздухе кристаллы, 1ПЛ 131—133°C (с разл.). Соединение растворяется во всех органических растворителях. ИК (гексан): 2109 (сл.), 2062 (оч. с.), 2046 (с.), 2030 (сл.) [v(CO)] см-1. Кристаллическая структура р3-СНзС[Со3(СО)9]: см. [6].
2042 Глава 33. Металлоорганические комплексы
ЛИТЕРАТУРА
1. Bent W. Т., Duncanson L. A., Guy R. G., Reed Н. W. Р., Shaw В. L„ Ргос. Chem. Soc., 1961, 169.
2. Bor G., Marco L., Marco B., Chem. Ber., 95, 333 (1962).
3. Ercoli R., Santambrogio G., Tettamanti Casagrande G., Chim. Ind. (Milano), 44, 1344 (1962).
4. Ring R. B„ Organometallic Syntheses, v. 1, p. 153, Academic Press, New York, London, 1965.
5. Penjold B. R., Robinson В. H. Acc. Chem. Res., 6, 73 (1973).
6. Sutton P. W., Dahl L. F., J. Amer. Chem. Soc., 89, 261 (1967).
□ □ ц-Метиленбис[карбонил(т]-циклопентадиенил)родий] (Rh—Rh) p-CH2[Rh(n-C5H5)(CO)]2
Получение p-метиленового комплекса из И-метил-И-нитрозомочевины основано на переносе метила на металл и последующем отщеплении водорода. Приводимая ниже пропись рассчитана на получение 1,6—1,8 г u,-CH2[Rh(r]-С5Н5) (СО)]2.
Rh2(t]-C5H5)2(CO)S + (СН3) (NO)N-C(=O)NH2 -->
420,0 103,1
----> p-CH2[Rh(T]-C5Hs) (СО)]2 + СО + побочные продукты 406,0 22,4 л
К раствору 3,0 г (7,14 ммоль) РЬ2(т]-С5Н5)2(СО)з в 100 мл бензола и 20 мл тетрагидрофурана, помещенному в 500-мл колбу, добавляют 5,16 г (50,0 ммоль) перекристаллизованной И-метил-И-нитрозомочевины* (/пл 124 °C, с разл.) и кипятят в течение ~25 ч с обратным холодильником. Охлаждают до комнатной температуры, добавляют еще 5,16 г (50,0 ммоль) И-метил-И-нитрозомочевины и снова кипятят 25 ч. Эту операцию повторяют еще раз. После охлаждения темно-коричневой реакционной смеси летучие продукты отгоняют в вакууме водоструйного насоса при 30—35 °C. Маслообразный красно-коричневый остаток хроматографируют на охлаждаемой водой колонке (60x2,5 см) с SiO2 (кизельгель Merck 7734, 0,063—0,200 мм, активность II— III степени). Смесь пентан — бензол (5:1) элюирует вначале яркую желтую полосу Rh(T]-C5H5) (СО)2. Метиленовый комплекс p-CH^fRhCq-CsHs) (СО)]2 идет в виде оранжево-красной зоны в смеси пеитан — бензол (3: 1). Чистым бензолом элюируют —40 мг (0,1 ммоль) непрореагировавшего Rh2(T|-C5H5)2-(СО)з в виде быстроидущей карминово-красной полосы. После упаривания раствора желаемого комплекса и кристаллизации остатка из небольшого количества смеси пентан — эфир (при —35 °C/—78 °C) получают 1,86 г p-CT^Rh^-С5Н5)(СО)]2 [65%, считая на использованный Rh2(л-СзН^г(СО)3]. Этилиде-новый комплекс получают аналогично (выход 70%).
Свойства. Красные, преломляющие свет, устойчивые на воздухе ромбические или игольчатые кристаллы, tn„ 94 °C. Вещество растворяется во всех органических растворителях, особенно хорошо в бензоле и метилендихлориде; растворы не очень устойчивы на воздухе. ИК (пентан): 1984 (оч. с.); (КВг): 1959 (оч. с.) [v(CO)] см-1. ЯМР-'Н (ацетон-б6, ТМС): б 5,48 [триплет, С5Н5], 7,07 [мультиплет, СН2], ЯМР-13С (толуол-ds, ТМС): б(С6Н5) 89,69, б (СО) 195,38, 191,87, б(СН2) 102,25. Соединение практически изоструктурно p-CO[Rh(r)-C5H5) (СО)]2; d(Rh-Rh) =2,665 A, <£Rh—СН2—Rh=81,7°.
* Получение см. [4].
Карбиновые и карбеновые комплексы 2043
ЛИТЕРАТУРА
1. Herrmann W. A., Kruger С., Goddard R., Bernal I., Angew. Chem., 89, 342 (1977).
2. Herrmann W. A., Kriiger C„ Goddard R., Bernal I., J. Organometal. Chem., 140, 73 (1977).
3. Herrmann W. A., Plank J., Reiter В., неопубликованные результаты 1978— 1979 гг.
4. Organikum, 11, Auflage, S. 548, VEB Deutscher Verlag der Wissenschaften, Berlin, 1973.
□ □ Тетрафтороборат [р-дикарбонил-ц3-метилидин-^«кло-трис-
{(т]-циклопентадиенил)родия} ] (3Rh—Rh) [№з(тС5Н5)3(Из-СН) (|x-CO)2] [BF4]
3p-CH2[Rh(r)-C5H5) (CO)]2 4-2HBF4 ->
3-406,0 2-87,8
---> 2[Rh8(n-C6Hs)g(p3-CH)(n-CO)2][BF4] + CH4 + H2 + 2CO 2-659,9 22,4л 22,4л 2-22,4л
К раствору 912 мг (2,0 ммоль) p.-CF^RlR'n-CsHs) (СО)]2 в 30 мл тетрагидрофурана добавляют 0,8 мл (4,0 ммоль) 48%-ной водной HBF4, хорошо перемешивают в течение короткого времени и оставляют стоять реакционную смесь 2—3 дня при 10—25 °C без перемешивания. Уже через 15 мии начинают выпадать игольчатые кристаллы метилидинового комплекса, одновременно происходит углубление окраски раствора от темно-красной до черно-коричневой. По окончании реакции на дне сосуда образуются сантиметровой длины игольчатые кристаллы; их декантируют, промывают тетрагидрофураном (4Х Х5 мл), эфиром (2x5 мл) и сушат в высоком вакууме. Выход 832 мг (96%).
Свойства. Черные с металлическим блеском кристаллы, устойчивые на воздухе. При нагревании в запаянном капилляре до 280 °C не происходит никакого видимого изменения. Вещество хорошо растворяется в ацетонитриле, ацетоне, N.N-диметилформамиде и метаноле, очень плохо — в тетрагидрофуране, не растворяется в метилендихлориде, эфире и алифатических углеводородах. Кристаллы, выпадающие из ацетона, моноклинные, пространственная группа симметрии С2/с, d(Rh—Rh)=2,678—2,714 A, d(Rh—Сметилидин) = = 1,930—1,986 А, карбонильные лиганды являются несимметричными мостиковыми, ответственными за флуктуирующую геометрию комплексного катиона в растворе. ИК (КВт): 1929 (с.), 1884 (ср. с.) [v(СО)] см-'; (CH3CN): 1925 (с.), 1882 (ср. с.) i[v(CO)] см-1. ЯМР-'Н (90 МГц, диметилформамид^?, ТМС, 25 °C): б(С5Н5) 6,00 [«синглет», 15 Н], с5н6<0,4 Гц; б(СН) 16,28 [квад-
руплет, 1 Н]. ЯМР-13С (22,63 МГц, диметилформамид^т,'ТМС, 33°C): iRCsHs) 98,4, б(СН) 303,6, б (СО) 209,2. Удельная электропроводность: А=96 см2-Ом-'Хмоль-1 (CH3NO2, 21,6°С, 2-10-4 моль/л).
ЛИТЕРАТУРА
1. Herrmann W. A., Plank Ziegler М. L., Guggolz Е., Angew. Chem., 92, 660 (1980).
Бис[р-хлоро-хлоро(дифенилциклопропенилиден)палладий] [PdCl2{C3(C6H5)2}]2
Для синтеза (б) необходим абсолютно не содержащий Н2О и Os порошок Pd (палладиевая чернь). Его получают по модифицированному способу Ви
2044 Глава 33. Металлоорганические комплексы
ланда [3] (а).
a) PdCl2 + НСО2Н 4- 2NaOH ------> Pd + СО2 + 2Н2О + 2NaClj
177,3 46,0 2-40,0 106,4 22,4 л 2-18,0 2-58,4
□ □ б) 2Pd + 2С12Са(СвН5)2--------> [РбС12{Сз(С6Н6)2)]2
2-106,4 2-261,1 735,0
а) Раствор 5,0 г (28,2 ммоль) PdCl2 в 1 л Н2О нагревают до 80 °C и нейтрализуют 20%-ным раствором NaOH до тех пор, пока не перестанет выпадать Pd(OH)2. При интенсивном перемешивании добавляют 0,55 мл (14,6 ммоль) муравьиной кислоты, после начала процесса восстановления добавляют еще
Рис. 473. Фильтр-экстрактор.
I — нормальный шлиф 24; 2 — нормальный шлиф 14,5; 3 — стеклянный пористый
фильтр G3 (диаметр 30 мм).
10 мл 20%-ного NaOH и затем снова 1,1 мл (29,2 ммоль) НСООН. В течение 0,5 ч нагревания бесцветного прозрачного раствора на кипящей водяной баие дают полностью осесть черному осадку Pd, собирают его на стеклянном пористом фильтре (G4) и промывают кипящей водой (10x20 мл). Если проба препарата еще показывает щелочную реакцию, то проводят экстракцию в течение 15 ч кипящей водой. Порошок Pd сушат в высоком вакууме при 150 °C. Выход количественный. По этой методике получают черный пушистый порошок, который при доступе воздуха абсорбирует О2 с разогреванием.
б) В колбе на 10 мл с коротким обратным холодильником кипятят в течение 24 ч 0,60 г (5,64 ммоль) Pd-порошка, полученного по способу (а), 1,40 г (5,36 ммоль) 3,3'-дихлор-1,2-дифенилциклопропена и 6 мл бензола. Реакционную смесь фильтруют (стеклянный пористый фильтр G4), желтовато-серый продукт промывают СН2С12 (2x10 мл). После кратковременного высушивания продукт переносят на фильтровальную воронку-экстрактор (рис. 473) и до тех пор экстрагируют 30 мл кипящего СН2С12, пока на фильтре не останется лишь черный Pd (~ 100 ч). Экстракт фильтруют (стеклянный пористый фильтр G4), после отгонки растворителя светло-желтый кристаллический продукт промывают 5 мл СН2С12 и сушат в высоком вакууме 1 ч при 50 °C. Выход 1,5 г (72%).
Свойства. Светло-желтые, устойчивые иа воздухе кристаллы, <Пл 215 °C (с разл.). Вещество умеренно растворяется в СН2С12 и нитробензоле, хуже — в нитрометане, почти не растворяется в бензоле и эфире. С тетрагидрофураном, ацетонитрилом, пиридином и фосфинами образуются 1 : 1-аддукты состава PdCl2[C3(C6H5)2]L.
Диалоги. При взаимодействии Вг2С3(С6Н5)2 с Pd образуется соответствующий бромокомплекс. 3,3'-Дихлорциклопропены с другими заместителями в положениях 1, 2 также образуют аналогичные палладиевые комплексы.
Карбиновые и карбеновые комплексы 2045
ЛИТЕРАТУРА
1. Ofele К-, J. Organometal. Chem., 22, С9 (1970).
2. Weiss R„ Priesner C., Angew. Chem., 90, 491 (1978).
3. Wieland H„ Ber., 45, 489 (1912).
Иодид тетракис ( оксазол идин-2-и л идеи) палл адия (II) [Pd(CNHCH2CH2O)4] 12
COCh, N(C2H5)3
a) OCHNHCHaCH2OH-----------------> CNCHaCHaOH
89,1 71,1
б) Pdla + 4CNCH2CHaOH------> [Pd(CNHCH2CHa6)4]Ia
360,2 4-71,1 644,5
a) fi-Изоиианоэтанол. Исходный [3-формиламииоэтаиол получают из 6-амииоэтаиола и этилформиата (1 : 1,2) [2, 3].
В кипящий раствор 89,1 г (1 моль) OCHNHCH2CH2OH в 500 мл CH2CI2 и 500 мл триэтиламииа в течение 1 ч пропускают интенсивный ток фосгена (—100 г, 1,01 моль)*. Затем охлаждают до комнатной температуры и пропускают ток NH3 (—55 г). Образовавшуюся аммониевую соль отделяют, растворитель отгоняют в вакууме водоструйного насоса. Оставшееся масло перегоняют в высоком вакууме, собирая при 48—50 °C (10-3 мм рт. ст.) CNCH2CH2OH. Выход 50 г (70%). Свойства: ИК (пленка): 2153 (оч. с.), 2170 (пл.) [v(CN)] см-1.
б) Иодид тетракис(оксазолидин-2-илиден)палладия(П). Растворяют в 30 мл СН2С12 3,6 г (10 ммоль) Pdl2 и при интенсивном перемешивании по каплям добавляют 3,0 г (42 ммоль) 0-изоцианоэтанола в 15 мл СН2С12. Вскоре Из желтого раствора выпадает мелкий белый осадок, его отфильтровывают, промывают ацетоном и эфиром и высушивают. Выход 6,4 г (количественный).
Упариванием водного раствора или разбавлением метанольного раствора ацетоном получают крупные кристаллы соединения.
Свойства. Белый мелкокристаллический порошок или бесцветные кристаллы, /пл 156°C (с разл.). Растворяется в воде и спиртах, не растворяется в ацетоне, эфире, бензоле и хлорированных углеводородах. ИК (КВг): 3175 (с.), 3087 (с.) [v(NH)], 2973 (сл.), 2950 (сл.), 2905 (сл.) [v(CH)], 1559 (оч. с.) [Vac.B(NCO)], 1171 (оч. с.), 1152 (оч. с.) [vChm(NCO)] см-1.
Кристаллическая и молекулярная структура аналогичного хлорида моноклинная, пространственная группа симметрии Р2]/с (п=8,518 А, 6=11,714 А, с=9,185 А, 0=97,79°). Плоскость пятичленного гетероцикла приблизительно перпендикулярна квадратно-плоскостной координационной плоскости палладия [5].
ЛИТЕРАТУРА
1. Bartel К-, Fehlhammer W. A., Angew. Chem., 86, 588 (1974).
2. Mofat J., Newton M. V., Papenmeier G. J., J. Org. Chem., 27, 4058 (1962).
3. Matteson D. S., Bailey R. A., J. Amer. Chem. Soc., 90, 3761 (1968).
4. Ugi I., Fetzer U„ Eholzer U„ Knupfer H„ Offermann K„ Angew. Chem., 77, 492 (1965).
5. Fehlhammer W. P„ Liu А., неопубликованные результаты.
* С избытком фосгена образуется бис (2-изоциаиоэтил)карбонат [4].
2046 Глава 33. Металлоорганические комплексы
□ г$ис-Дихлоро[этокси(фениламино)карбен] (триэтилфосфин)-платина PtCl2[C(OC2H5)NHC6H5] [Р(С2Н5)3]
В простом синтезе карбеновых комплексов исходят из координированных изонитрилов. Ричардс с сотр. смогли впервые в 1968 г. показать, что координированный фенилизонитрил достаточно активен, чтобы присоединить такой слабый нуклеофил, как этанол, и образовать этокси (амино) карбеновый комплекс.
a) K2[PtCl4] + 2P(C2H6)s ---> PtCl2[P(C2HB)s]2 + 2КС1
415,1 2-118,2 502,3 2-74,6
б) Р1а2[Р(С2Н5)з]2 + Р1С12----> [PtCl2{P(C2H5)3}]2 .
502,3 266,0 768,3
в) [PtO2{P(C2H5)8}]24-2CNCeHe ----> 2PtCl2(CNCeH5) [Р(С2Н6)3]
768,3 2-103,1 2-487,3
г) PtCl2(CNCeH5)[P(C2He)s]4-C2H6OH ----->
487,3 46,1
----> PtCl2[C(OC2HB)NHCeHB] [Р(С2НВ)3]
533,3
а) Дихлоробис(триэтилфосфин)платииа Р1С12[Р(С2Н5)3]2 [1]
В колбу Шленка на 100 мл помещают раствор 8,3 г (20 ммоль) Ka[PtCl4] в 50 мл НгО, в атмосфере N2 добавляют 4,75 г (5,9 мл, 40 ммоль) триэтилфосфина и перемешивают 1 ч. При этом из раствора выпадает белое, иногда слегка красноватое твердое вещество. Содержимое колбы нагревают 15 мин до кипения, отфильтровывают осадок, промывают его водой и высушивают в вакууме. Выход 9,0 г (90%).
Свойства. Почти белый продукт состоит в основном из цис-Р1С12[Р(С2Нв)3]2, загрязненного небольшим количеством транс-изомера. Для большинства превращений можно использовать эту смесь изомеров без дальнейшей очистки. Обработкой диэтиловым или петролейным эфиром можно извлечь транс-изомер (<пл 140—142 °C) или изомеризовать его в более труднорастворимый цис-Р1С12[Р(С2Н6)3]2 ((пл 195—196°C). Последний комплекс слабо растворяется лишь в бензоле, этаноле и ацетоне. Нагреванием цис-изомера выше 180 °C можно провести обратную изомеризацию в тране-изомер.
б) Ди-р.-хлоро-а,[-дихлоробис(триэтилфосфии)диплатииа
[PtC12{P(C2H5>3}]2 [2-4]
Готовят тесную смесь 9,0 г (18 ммоль) PtC12[P(C2H5)3]2 и 5,0 г (19 ммоль) PtCl2 и заливают этот мелкий порошок в колбе 70 мл ксилола. Затем нагревают на масляной бане до начала закипания, хорошо перемешивают суспензию стеклянной палочкой и декантируют горячий желтый раствор с черного осадка. После охлаждения из раствора выкристаллизовывается желаемый продукт. Выход 9,7 г (70%).
Свойства. Желтое кристаллическое соединение, (пл 180—182 °C. Хорошо растворяется в бензоле и СНС13, умеренно — в холодном ацетоне, не растворяется в эфире и гексане. Соединение лучше всего кристаллизуется из ацетона.
в) цис-Дихлоро(фенилизоцианид) (триэтилфосфин)платина
PtCl2(CNCeH6)[P(C2HB)3] [5]
К раствору или суспензии 9,5 г (12,4 ммоль) i[PtCl2{P (С2Н5) з}]2 в 100 мл абсолютного бензола добавляют 2,7 мл (2,7 г, 26 ммоль) фенилизоциаиида н
Карбиновые и карбеновые комплексы 2047
перемешивают 1 ч при комнатной температуре. Выпавший продукт отфильтровывают, упаривают маточник до 1/3 первоначального объема и оставляют в холодильном шкафу для дополнительной кристаллизации. Выход 8,5 г (70%). Препарат можно перекристаллизовывать из бензола.
Отгонкой растворителя и перекристаллизацией остатка из спирта получают дополнительно третью фракцию продукта. Общий выход тем самым можно повысить до 85%.
Свойства. Белые мелкие кристаллы, £пл 161 °C, легко растворяется в CH2CI2 и СНС1з, не растворяется в эфире, пентане и ССЦ. ИК (иуйол): 2190 (оч. с.) [v(CN)], 337 (оч. с.), 277 (оч. с.) [v(Pt—Cl)] см-1.
г) цис-Дихлоро[этокси (фениламино) карбен] (триэтилфосфии) платина
Р1С12[С(ОС2Н5)ПШСвН5][Р(С2Н5)з] [6]
В течение нескольких часов кипятят с обратным холодильником 5,2 г (10,7 ммоль) PtCl2(CNCeH6){P(C2H6)3] в 150 мл абсолютного спирта. Упариванием раствора до половины исходного объема получают белый мелкокристаллический осадок, который перекристаллизовывают из спирта или смеси ацетон — эфир. Выход 4,3 г (75%).
Упариванием маточника получают вторую фракцию продукта, общий выход составляет почти 90%.
Свойства. Белые кристаллы, (пл 209—211 °C, в обычных органических растворителях растворяются очень плохо. ИК (нуйол): 3150 (ср.), 3110 (ср.-с.) [v(NH)]; 306 (оч. с.), 284 (оч. с.) [v(PtCl)]; (КВг): 1545 (с.) [vacaM(NCO)] см-1. Кристаллическая и молекулярная структура: см. [6].
ЛИТЕРАТУРА
1. Jensen К- А., 2. Anorg. Allgem. Chem., 229, 225 (1936).
2. Chatt J., J. Chem. Soc. (London), 1950, 2301.
3. Goodfellow R. J., Venanzi L. M„ J. Chem. Soc. (London), 1965, 7533.
4. Smithies A. C„ Schmidt P., Orchin M., Inorg. Synth., 12, 240 (1970).
5. Badley E. M., Chatt J., Richards R. L., J. Chem. Soc. (London), A1971, 21; Inorg. Synth, 19, 174 (1979).
6. Badley E. M., Chatt J., Richards R. L., Sim G. A., Chem. Commun., 1969, 1322.
тра«с-[Хлоро{(п-толилимино)метил)бис(триэтилфосфин)-платина] Pt(a)(CH = NC6H4-n-CH3)[P(C2H5)3]2
n2h4
a) 4«c-PtCl2[P(C2H5)3]2 ->- транс-Pt(Cl)H[P(C2H5)3]2
502,3 467,9
6) Pt(Cl)H[P(C2H5)3]2 4-CNC6H4-ra-CH3 ->
467,9 117,2
—> Pt(Cl) (CH=NCeH4-n-CHs) [P(C2H6)3]2
585,0
а) транс-Хлорогидридобис(триэтилфосфин)платина
РЦС1)Н[Р(С2Н5)з]2
На кипящей водяной бане в атмосфере N2 в течение 1 ч нагревают в колбе Шленка на 100 мл смесь 5 г (10 ммоль) чистого цис-Р1С12[Р(С2Н5)з]2, 2,5 г
2048 Глава 33. Металлоорганические комплексы
(50 ммоль) гидразингидрата и 50 мл воды. Комплекс переходит в раствор, одновременно выделяется масло. При охлаждении масло образует кристаллическую массу, которую отделяют. Из фильтрата нейтрализацией соляной кислотой выделяют дополнительно некоторое количество кристаллического вещества. Обе фракции перекристаллизовывают из небольшой порции метанола, сушат и, если необходимо, еще раз перекристаллизовывают из гексаиа. Выход 3,4 г (73%).
Свойства. Белые игольчатые кристаллы, 1ПЛ 82—83,5 °C, устойчивы на воздухе как в растворе, так и в твердом состоянии. Вещество очень хорошо растворяется в большинстве органических растворителей. ИК: 2183 (ср.) [v(PtH)] см-'. ЯМР-'Н (С6Н6, ТМС): 6=—16,9 [1:4:1-триплет, PtH]; J(PtH) = = 1276 Гц.
б) транс-[Хлоро(п-толилимино)метил)бис(триэтилфосфин)платина] Р1(С1)(СН = МСвН4-п-СНз)[Р(С2Н6)з]2
К интенсивно перемешиваемому раствору 2,0 г (4,3 ммоль) транс-Р1(С1)Н[Р(С2Н6)з]2 в 60 мл эфира в течение 0,5 ч прикапывают раствор 0,5 г (4,3 ммоль) n-толилизоцианида в 20 мл сухого эфира. Образовавшуюся суспензию откачивают досуха и остаток несколько часов перемешивают в 60 мл смеси СН2С12 и пентана (1:3). Затем отфильтровывают и фильтрат максимально откачивают. Образуется вязкое масло, которое при растирании в пентане, охлаждаемом льдом, закристаллизовывают. Выход сырого продукта практически количественный.
Для очистки растворяют в ацетонитриле и упаривают раствор до объема 5 мл. Бесцветный, устойчивый на воздухе осадок отфильтровывают и высушивают.
Свойства. Бесцветное кристаллическое соединение, /пл 100 °C, месяцами хранится, не разлагаясь. Растворяется в ароматических и хлорированных углеводородах, ацетонитриле, диметилсульфоксиде и СНзОН. В растворе существует в двух формах, син- и анти-, по отношению к связи C=N. Соединение является слабым основанием, обратимо протоиируетси (метилируется). ИК (иуйол): 1560 (с., шир.) ;[v(C = N)] см-1.
ЛИТЕРАТУРА
1. Parshall G. W„ Inorg. Synth., 12, 28 (1970).
2. Christian D. F., Clark H. C., Stepaniak R. F., J. Organometal. Chem., 112, 209 (1976).
3. Christian D. F., Clark H. C., Stepaniak R. F., J. Organometal. Chem., 112, 227 (1976).
4. Bonati F„ Clark H. С., в печати.
Трис[1-циклогекси1лимино(метокси)метилзолото] [AuC(OCH3) = NC6H1i]3
3AuCl[P(CeH5)3] + 3CNCeHu + ЗСН3ОН + 3KOH ----->
3-494,7 3-109,2 3-32,0 3-56,1
---> 3P(CeHs)3 + [AuC(OCH3)=NCeHu]3 + 3KC1 + 3H2O 3-262,3 1011,5 3-74,6 3-18,0
К суспензии 2,8 г (5,7 ммоль) АиС1[Р(СвН6)3] в 10 мл метанола при перемешивании добавляют раствор 0,33 г (5,8 ммоль) КОН в 30 мл СН3ОН н далее 0,63 г (5,8 ммоль) циклогексилизонитрила. Через 30 мин упаривают
Карбиновые и карбеновые комплексы 2049
наполовину и отфильтровывают осадок. Выход 0,6 г (31%). Продукт можно перекристаллизовать из смеси хлороформ — метанол.
Свойства. Бесцветное соединение, растворяется в ароматических и хлорированных углеводородах, хранится неограниченно долго, /пл>160°С (с разл.). ИК (нуйол): 1535 [v(C=N)] см~>.
ЛИТЕРАТУРА
1. Minghetti G., Bonati F„ Angew. Chem., 84, 482 (1972).
2. Minghetti G., Bonati F., Inorg. Chem., 13, 1600 (1974).
3. Bonati F., частное сообщение.
T етр акис( 1 -изопропилтетр азол-5-ато) аур ат( I И) тетрафениларсония [А8(СбН5)4] [Au(CN4-u3O-C3H7)4]
a) K[AuC14] + 4NaN3 + [As(CeH5)4]Cl -->
377,9 4-65,0 418,8
---> [As(CeH5)4] [Au(N3)4] 4- KC1 + 4NaCl
748,4 74,6 4-58,4
6) [As(CeH5)4] [Au(N3)4] -|- 4CN-u30-CsH7 ->
748,4 4-69,1
--> [As(C6H5)4] [Au(CN4-«3o-C3H7)4]
1024,8
а) Тетраазидоаурат(Ш) тетрафениларсония [1, 2]
К интенсивно перемешиваемому водному (~100 мл) раствору 6,5 г (0,1 моль) NaN3 медленно прикапывают раствор 3,8 г (0,01 ммоль) К[АиС14] в 50 мл воды.
Осторожно: твердый Na [Au (Na) 4] чрезвычайно взрывчат! Поэтому необходимо разбрызгивающийся по стенкам колбы (300-мл широкогорлая колба Эрленмейера) реакционный раствор постоянно смывать. По той же причине никоим образом нельзя работать с концентрированными растворами. Нельзя также добавлять твердый К[АиС14] в раствор NaN3.
Образовавшийся кирпично-красный раствор фильтруют через слой в 1 см бумажной массы и по каплям при перемешивании добавляют к нему раствор 4,2 г (0,01 ммоль) [As(CeH3)4]Cl в 20 мл Н7О. Перемешивают 30 мин, выпавший светло-оранжевый осадок отфильтровывают (стеклянный пористый фильтр G3) и тщательно сушат в высоком вакууме. Защищая от света, перекристаллизовывают из смеси CH2CI2 — петролейный эфир и получают устойчивое кристаллическое вещество красного цвета с почти количественным выходом (9— Юг).
Свойства. <пл 145°C (с разл.), соединение не чувствительно к ударам, в пламени вспыхивает без детонации. В твердом состоянии слегка, а в растворах в ацетоне или СН2С12 в течение 10 ч разрушается на свету. Особенно легко (за 15 мин) обесцвечиваются растворы в тетрагидрофуране (->[As(C3H6)4]-[Au(N3)2]+3N2) [2]. ИК (CH2CI2): 2034 (оч. с.) [vac,M(N3)] см-1.
б) Тетракис( 1-изопропилтетразол-5-ато) аурат( 111) тетрафениларсония [3, 4]
Растворяют 3,75 г (5 ммоль) [As(C6H6)4] [AufNsh] в 40 мл СН2С12 и, защищая от света, добавляют полуторакратный избыток изопропилизоциаии-13—1361
2050 Глава 33. Металлоорганические комплексы
да (2,1 г, 0,03 моль). Через ~15 мин первоначально красный раствор становится бледно-желтым, почти бесцветным. Его слегка упаривают и постепенно добавляют диэтиловый или петролейный эфир до начала помутнения. Полученный бесцветный осадок отмывают водой с эфиром от изоцианида, перекристаллизовывают из смеси CH2CI2 — эфир и сушат в высоком вакууме*. Осторожным добавлением эфира на поверхность раствора в CH2CI2 можно получить большие, хорошо выраженные кристаллы. Выход практически количественный (4,5—5,0 г).
Свойства. Бесцветные кристаллы, при быстром нагревании взрываются, /пл 177 °C (с разл.). Вещество растворяется в метилендихлориде, хлороформе и ацетоне, хуже в тетрагидрофуране. Практически не растворяется в эфире, петролейном эфире и воде. Кристаллическая и молекулярная структура моноклинная (пространственная группа симметрии Р21(Сг2): а= 11,07 А, 6=18,66 А, с= 11,361 А, 0=104,5°; четыре металл-С-о-связанных кольца тетразола расположены приблизительно перпендикулярно плоскости квадратно-плоскостной координации АиС4) [4].
Аналогичные комплексы образуются и с другими алкилизоцианидами RNC (R = CH3, С6Нц, СН2С6Н6). Взаимодействие с арилизоциаиидами ArNC (Аг=С6Н5, СвН4ОСН3-п) требует более длительного времени.
ЛИТЕРАТУРА
1. Beck W., Fehlhammer W. Р., Pollmann Р., Schuierer Е., Feldl К., Chem. Вег., 100, 2335 (1967).
2. Beck W., Fehlhammer W. P„ Angew. Chem., 79, 146 (1967).
3. Beck W„ Burger K„ Fehlhammer W. P„ Ber., 104, 1816 (1971).
4. Fehlhammer W. P., Dahl L. F„ J. Amer. Chem. Soc., 94, 3370 (1972).
Комплексы с галогеновыми лигандами
□ □ Дихлоробис(г]-циклопентадиенил)титан Tift-CjHs^Ch
T1CI4 + 2NaCsH5 --> Ti(T]-CsHs)2Cl2 + 2NaCl
Получение ведут, как это описано в гл. 22 (т. 4 данного руководства). Вместо высокоэффективной мешалки можно использовать обычную магнитную мешалку. Далее, вместо приведенного там способа получения NaC6H6 в тетрагидрофуране можно воспользоваться подробными указаниями, приведенными в методике синтеза NaCsHs (в начале гл. 33).
Описанный метод пригоден и для получения V^-CsHt^Clj.
Свойства. См. т. 4, гл. 22. Возгоняется в высоком вакууме при 160—180 °C. Кристаллическая структура: см. [4] d(Ti—Cl) =2,24 A; d(Ti—С) =2,38 А.
ЛИТЕРАТУРА
1. Wilkinson G., Birmingham J. М., J. Amer. Chem. Soc., 76, 4281 (1954).
2. Summers L., Uloth R. H„ Holmes A., J. Amer. Chem. Soc., 77, 3604 (1955).
3. Summers L., Uloth R. H„ J. Amer. Chem. Soc., 76, 2278 (1954).
4. Алексеев H. В., Ронова И. A. — Ж. структ. хим., 1966, № 7, с. 91.
* Так как кристаллы содержат значительное количество растворителя, рекомендуется сушить их в высоком вакууме при 40—60 °C, периодически растирая. Из смеси ацетон — НгО получают кристаллы, не содержащие растворителя.
Комплексы с галогеновыми лигандами 2051
□ □ Трихлоро(г|-циклопентадиенил)титан Т1(д-С5Н5)С1з
Т1(т]-С6Н6)С1з получают либо реакцией диспропорционирования лигандов (способ 1), либо циклопентадиенилированием TiC!« при действии Mg(C5H5)2 (способ 2).
Способ 1 [1]
TiCl2(n-C5H5)2 + Т1С14 -> 2Т1(т]-С5Н5)С13
249,0 189,7 2-219,4
В 100 мл ксилола в течение 2,5 ч кипятят с обратным холодильником 12,5 г (50 ммоль) Т1С12(я-С6Н6)2 и 28,5 г (0,15 моль) TiCl4. После охлаждения до комнатной температуры выпадают желтые кристаллы. Их отфильтровывают, промывают петролейным эфиром и сушат в вакууме масляного насоса. Для очистки продукт перекристаллизовывают из бензола. Для этого кипятят в минимальном количестве бензола, добавляют порошок активированного угля и фильтруют горячим. Фильтрат упаривают до начала образования кристаллов. Затем оставляют раствор медленно охлаждаться до комнатной температуры и завершают кристаллизацию охлаждением в ледяной бане. Фильтруют и сушат в высоком вакууме. Выход аналитически чистого продукта 3,4 г (15%).
Способ 2 [2]
Mg(C5H5)2 + 2TiCl4 --> 2Ti(n-C5Hs)Cls + MgCl2
154,5 2-189,7 2-219,4 95,2
К 15,5 г (0,10 моль) Mg(C6H6)2 (получение см. выше в этой главе) в 500 мл ксилола добавляют раствор 37,9 г (0,20 моль) TiCl4 в 30 мл ксилола и кипятят смесь с обратным холодильником в течение 5 ч. После охлаждения до комнатной температуры из раствора выпадают оранжевые кристаллы, которые декантацией отделяют от мутного ксилольного раствора. Последний фильтруют, получая прозрачный оранжевый раствор (на фильтре остается осадок черного цвета), и после охлаждения получают дополнительно порцию желаемого соединения. Общий выход высушенного в высоком вакууме и достаточно чистого для большинства синтезов продукта составляет 32,4 г (74%, считая на TiCl4). TiCh^-CsHs) можно перекристаллизовать из горячего бензола.
Свойства. Устойчивые на воздухе, но чрезвычайно легко гидролизуемые желтые кристаллы, /пл 216—217,5°C. Кристаллическая структура: см. [3].
ЛИТЕРАТУРА
1. Gorsich. R. D„ J. Amer. Chem. Soc., 82, 4211 (1960).
2. Sloan C. L., Barber W. A., J. Amer. Chem. Soc., 81, 1364 (1959).
3. Ganis P., Allegra P., Atti Acad. Naz. Lincei, Sci. Fis. Mat. Nat., 33, 303 (1962).
□ □ Дихлоробис(г]-циклопентади1енил)ниобий Nb(r)-C5H5)2C12 (Ниобоцендихлорид)
Синтез проводят из NbCl6 и NaC5H5 в две стадии без выделения промежуточных, через образование ц-оксокомплекса [{NbCl(T)-C5H6)2}2O]C12, восстанавливая его в той же колбе при действии SnCl2 в конечный продукт Nb(T]-C5H5)2C12. Данный способ, хорошо отработанный Лукасом, особенно пригоден для получения значительных количеств этого производного ниобия, являющегося важным исходным соединением для синтезов других комплексов ниобия.
13*
2052 Глава 33. Металлоорганические комплексы
Успешное проведение эксперимента очень зависит от подготовки исходных реагентов: NbCls предварительно возгоняют (/ВМг 120—140 °C при 10-3мм рт. ст.) и применяют в реакции с NaCsH5 в тоикоизмельчениом состоянии. Используемый NaCjHj тщательнейшим образом освобождают от Н2О и О2 и вводят в реакцию также в виде абсолютно сухого порошка. Требуемый по данной методике NaCsHs (1,1 моль) получают лишь при 50-часовом высушивании в высоком вакууме при 120—150 °C. Только тогда удается избавиться от малейших следов тетрагидрофурана, используемого при получении NaC6H6- Наибольших выходов №(г]-С5Н5)2С12 достигают, если NaCsH5 получают в виде
Рис. 474. Прибор для получения NbCl2(i]-C6H5)2.
1 — высокоэффективная мешалка с охлаждением водой; 2 — муфта, нормальный шлиф 14,5/23; 3 — кран (4 мм) для подвода азота; 4 — отогнутая трубка Шленка; 5 — кран (6 мм) для подвода азота; 6 — трехгорлая реакционная колба на 2 л, нормальный шлиф 29/32; 7 — пропеллер мешалки, выполненный нз тефлона; 8 — муфта для присоединения обратного холодильника.
снежно-белого порошка. Приготовление исходных (NbCls, NaCsH6, бензол), а также взаимодействие NbCls с NaCsH5 ведут только в^ предварительно прокаленной посуде (сушильный шкаф, 200 °C, 25 ч). Сырой же продукт, напротив, переводят в оксокомплекс (стадия б) при доступе воздуха; последнюю стадию (в) опять проводят в атмосфере N2.
a) NbCls + 6NaC5H5 -----> Nb(r]-C5H6)4 -J- 5NaCl + побочные продукты
270,2 6-88,1 353,3 5-58,4
Реакцию проводят (рис. 474) в атмосфере N2 при тщательной защите от следов О2 и Н2О. Двухлитровую колбу 6 с краном 5 для ввода N2 монтируют следующим образом; используют высокоэффективную мешалку 1, желательно, с тефлоновой пластинкой; обратный холодильник снабжают ртутным затвором и вставляют в горло 8; трубку Шленка вставляют в косо поставленное боковое горло колбы, чтобы обеспечить добавление галогенида металла порциями. Рекомендуется колбу поместить в алюминиевую или пластиковую кастрюлю, чтобы тотчас охладить льдом реакционную смесь, если реакция пойдет очень бурно.
В приготовленном таким образом приборе суспендируют в 400 мл бензола 100,0 г (1,14 моль) сухого белого (!) NaCsHs. В наклонно присоединенную трубку Шленка помещают 57,0 г (0,12 моль) мелко измельченного NbCl6. При интенсивном перемешивании суспензии порциями по 5—15 г прибавляют галогенид металла. Уже при первом добавлении NbCls смесь приобретает коричневую окраску, которая постепенно изменяется до фиолетовой. Через 30—45 мин необходимо прибавить весь NbCls. Остатки смывают бензолом в колбу. Как правило, взаимодействие протекает тем лучше, чем быстрее прибавляют NbCl6. Поскольку реакция сильно экзотермическая, необходимо охлаждение льдом, если смесь начинает закипать. Для завершения реакции перемешивают еще 15 ч при комнатной температуре.
Вг2, Г>2
б) 2Nb(r)-CsHs)4 + 4НС1 + [О] ----[{NbCl(T]-C5H5)2}2O]Cl2 + 2С6Нв
2-353,3 4-36,5 604,0 2-66,1
Комплексы с галогеновыми лигандами 2053
В вытяжном шкафу при интенсивном перемешивании и доступе воздуха вливают темио-фиолетовую суспензию в 1 л конц. НС1 (стакаи иа 5 л, магнитная мешалка с крупным якорем, например 52 мм). Реакционная смесь умеренно разогревается (~50°С) и становится серо-зеленой.
При нагревании бензол полностью отгоняют, налипающие при этом на стенках стакана твердые частицы от зеленого до черно-коричневого цвета с помощью стеклянной палочки счищают в общую массу.
Нагревая до кипения, прибавляют порциями через капельную воронку в общей сложности 60 мл Вг2. Следует точно так же поступать с налипающим на стенках стакана осадком. После того как избыток Вг2 будет полностью удален, т. е. когда перестанут выделяться пары красного цвета, порошкообразный мелкокристаллический осадок (от коричневого до черного цвета) отделяют на широком (12 см) стеклянном пористом фильтре (G3) от темно-красного раствора*.
Остаток на фильтре снова переносят в стакан (5 л) и заливают таким количеством конц. НС1, чтобы она только покрывала осадок. (Избыток НС1 уменьшает выход!) Прибавляют ~10 мл Вг2, доводят реакционную смесь до кипения и выпаривают избыток Вг2. Суспензия сильно вспенивается, поэтому постоянно хорошо перемешивают содержимое стакана (магнитной мешалкой или стеклянной палочкой). Целесообразно еще 1—2 раза добавить небольшое количество Вг2 (по 5 мл) и повторить экстракцию перед повторным фильтрованием, так как желательно общее число экстракций свести к минимальному. Экстракции проводят до тех пор, пока солянокислые вытяжки не перестанут окрашиваться в красный цвет (5—10 экстракций, общий объем фильтрата 1600—2500 мл).
B)’[{NbCl(irC5Hs)2}2O]Cl2 + [SnClsJ- + 2Н+ + ЗС1~ ->
604,0 225,0
----> 2NbCl2(n-C6H6)2 4- [SnClJ2- + HSO 2-294,0 331,4 18,0
Дальнейшие операции проводят снова в защитной атмосфере (N2). Объединенный фильтрат в зависимости от объема переносят в одну или две круглодонные колбы на 2 л, снабженные обратным холодильником и краном для ввода N2.
Для насыщения фильтрата азотом его несколько раз последовательно вакуумируют в вакууме водоструйного насоса до вспенивания и пропускают N2. Далее солянокислые вытяжки при интенсивном перемешивании нагревают на масляной бане до 100—110°С (температура бани). Одновременно в круглодонной колбе на 500 мл с обратным холодильником растворяют 45,0 г (0,20 моль) SnCl2-2H2O в 200 мл. конц. НС1 и с помощью электроплитки доводят до кипения.
Горячий раствор SnCl2 через обратный холодильник вливают в горячий раствор соединения ниобия, находящегося в двухлитровой колбе (колбах). Тотчас происходит изменение окраски до коричневой, и часть образовавшегося МЬС12(г)-СбНб)2 выпадает в виде мелкого осадка. Для полноты высаживания смесь оставляют на ночь при комнатной температуре и затем отделяют осадок на стеклянном пористом фильтре (G3) в атмосфере N2. Промывают Н2О до тех пор, пока полностью не освободятся от SnCl2, и промывают осадок окончательно 50 мл ацетона.
* Если используют недостаточно сухой и белый NaC6H5, солянокислые вытяжки окрашиваются вместо красного в желтый или буро-красный цвет. Основным продуктом является в таком случае Nb2Os, выпадающий в виде белого порошка.
2054 Глава 33. Металлоорганические комплексы
Темно-коричневый продукт сушат в вакууме масляного насоса и затем возгоняют в специально приспособленной для этой цели аппаратуре подходящего размера (рис. 475) (высокий вакуум, температура печи 290—380°C). Выход 35—44 г (57—71%, считая на NbCls)-
Хотя получившийся продукт для большинства целей достаточно чист, масс-спектроскопически можно доказать наличие 5% примеси NbBr2(T]-C5H6)2, от которого уже больше не удается избавиться. Следовательно, для спектроскопических целей приготовленный по вышеприведенной прописи препарат не при-
Рис. 475. Прибор для возгонки NbCl2(T]-C6H5)2.
/ — сырой продукт; 2 — стеклянная вата; 3 — место присоединения трубки Шленка, в которую собирают сублимат (нормальный шлиф 29/32); 4 — рабочее отверстие сублиматора (нормальный шлиф 45/50); 5 — вакуумный кран.
годен. Да и в случае дальнейших химических превращений следует учитывать наличие этой примеси. Спектрально чистый КЬС12(я-С5Н5)2 можно получить по методике [4].
Свойства. Коричневое, в течение непродолжительного времеин устойчивое иа воздухе, парамагнитное, кристаллическое соединение. Растворимость МЬС12(т]-С5Н6)2 во всех органических растворителях очень мала, перекристаллизация по этой причине невозможна. В отсутствие О2 и Н2О соединение устойчиво до 400 °C. С восстановителями, например Na, образует чрезвычайно неустойчивое на воздухе соединение Nb(n-C5H5)2C1, легко присоедиииющее обычные двухэлектронные лиганды (СО, алкены, ацетилены и т. д.). При действии СО (давление 400 бар, 135 °C, бензол) образует Nb(T]-C5H5)(CO)4, выход 52% [5]. Взаимодействием с NaBH« получают NbBH4(ri-C6H5)2 [1].
ИК (нуйол); 3090 (ср.), 1011 (ср.), 822 (с.), 725 (ср.), 308 (ср.), 290 (с.), 268 (с.) см-1. Кристаллическая структура: см. [6].
ЛИТЕРАТУРА
1. Lucas С. R„ Inorg. Synth., 16, 107 (1976).
2. Lucas С. R., Green M. L. H„ Chem. Commun., 1972, 1005.
3. Laboratoriumsvorschrift des Anorg. Chem. Inst, der Univers. Regensburg; Biersack H., частное сообщение.
4. Siegert F. W„ De Lief de Meijer H. J., J. Organometal. Chem., 23, 177 (1970).
5. Herrmann W. A., Biersack H., Ber., 112, 3942 (1979).
6. Prout K., Cameron T. S„ Forder R. A., Gritchley S. R., Denton B., Rees G. V„ Acta Crystallogr., B, 30, 2290 (1974).
Комплексы с галогеновыми лигандами 2055
□ □ Тетрахлоро(г]-циклопентадиенил)ниобий Nb(r]-C5H5)C14
□ □ Тетрахлоро(г]-циклопентадиенил)тантал Та(т]-С5Н5)С14
MCI5 + S^h-C^W-CbHs) -----------> М(т]-С6Н6)С14 + Sn(.4-C4H9)sCl
M=Nb: 270,2 355,2 M=Nb: 299,8 325,5
M= Та: 358,2 М= Та: 387,9
Все работы проводят при абсолютном исключении кислорода и, в особенности, влаги воздуха (предварительно прокаленная стеклянная аппаратура!).
В колбу на 4 л помещают 0,20 моль порошка галогенида соответствующего металла* (54,0 г NbCls или 71,6 г TaCls) и заливают ~3,3 л бензола. Получившуюся суспензию интенсивно перемешивают в течение 5—10 ч, используя крупный (52 мм) якорь для магнитной мешалки. Большая часть галогенида металла растворяется (NbCl6: темно-красный раствор; TaCls: интенсивно-желтый раствор). Смесь нагревают еще немного до 50—70°C на электронагревателе до образования гомогенного раствора, который затем быстро декантируют с осадка (обычно незначительного) (в противотоке N2I) в колбу на 6 л, снабженную капельной воронкой емкостью 500 мл. Как правило, часть галогенида металла снова выкристаллизовывается на стенках шестилитровой колбы. Тогда подогревают еще раз, чтобы получить гомогенный раствор.
К теплому раствору при постоянном перемешивании прикапывают в течение ~15 мин раствор 78,1 г (0,22 моль) Sn(H-C4H9)3(r]I-C5H5) (получение см. выше в этой главе) в 200 мл бензола. При этом тотчас выпадает в виде мелких кристаллов трудно растворимый в бензоле М(т]-С6Н6)С14. После того как суспензия охладится до комнатной температуры, большую часть раствора сливают с осадка (~2,9 л) и фильтруют остаток под давлением N2 на большом стеклянном пористом фильтре (G3). Осадок промывают несколько раз бензолом (примерно по 40 мл), затем 100 мл пентана и высушивают в вакууме. Выходы: 56,4—58,8 г (94—98%) Nb^-CsHeJCh; 67,5—71,4 г (87—92%) Ta(T]-CsH5)C14. Изменение величины загрузки практически не влияет на выход конечного продукта.
Предложенный Чаттом с сотр. [3] метод получения М(т]-С5Н5)С14 взаимодействием MCls и Mg(C5H6)2 применять нецелесообразно, поскольку и экспериментальные сложности при выделении конечного продукта значительны и выходы не воспроизводятся, да и не так просто получать сам циклопента-диенилирующий агент.
Свойства. Ржаво-красный (M=Nb) или яично-желтый (М=Та) легко гидролизуемый порошок либо соответственно кристаллы (при кристаллизации из СН2С12). Не растворяется в большинстве органических растворителей, плохо растворяется в тетрагидрофуране. При 230 °C возгоняется в высоком вакууме (М=Та). ИК (нуйол, поглощение CsHs-группы): 1420 (с.), 1260 (сл.), ИЗО (сл.), 1075 (сл.), 1020 (ср.), 900 (сл.), 870 (оч. с.), 800 (сл.) (M=Nb); 1432 (с.), 1128 (сл.), 1018 (ср.), 878 (оч. с.) (М=Та) см-1.
ЛИТЕРАТУРА
1. Bunker М. J., DeCian A., Green М. L. Н„ J. Chem. Soc, Chem. Commun.. 1977, 59.
2. Green M. L. H., частное сообщение.
3. Burt R. J., Chatt J., Leigh G. J., Teuben J. H., Westerhof A., J. Organometal. Chem., 129, C33 (1977).
* Пентагалогеииды высокой чистоты (>99%, например, фирмы Fluka или Ventron) можно использовать без предварительной возгонки.
2056 Глава 33. Металлоорганические комплексы
□ □ Бромо(пентакарбонил)хромат(0) тетраэтил аммония [N(C2H5)4] [Сг(СО)5Вг]
□ □ Хлоро(пентакарбонил)вольфрамат(0) тетраэтиламмония [N(C2H5)4] [W(CO)5C1]
Галогенопентакарбоиилметаллаты Сг и W в виде тетраалкиламмониевых солей являются очень удобными исходными соединениями для получения мо-нозамещенных производных карбонилов металлов, а именно M(CO)6L. Синтез их из М(СО)6 и X- осуществляют как термически (для предотвращения образования полизамещенных продуктов галогенид аммония берут в недостатке), так и фотохимически. Ниже приводятся оба способа.
[N(C2H6)4][Cr(CO)5Br]
АТ
Cr(CO)6 4- [N(C2H5)4]Br-> [N(C2Hb)4] [Cr(CO)5Br] + CO
220,1 210,2 402,2 22,4л
В трехгорлую колбу иа 1 л, снабженную краном для ввода азота и обратным холодильником, помещают 20,0 г (91 ммоль) Сг(СО)в и 12,6 г (60 ммоль) [N(C2H5)4]Br, предварительно высушенного в высоком вакууме при 80 °C. В атмосфере N2 добавляют 300 мл моноглима (диметилового эфира этиленгликоля) и нагревают на масляной бане до 130 °C. Как только раствор слегка пожелтеет, начинают собирать выделяющийся СО в газометр. После отщепления ~ 1,4 л СО реакцию прекращают и раствор фильтруют еще горячим через стеклянный пористый фильтр (G3), покрытый бумажной массой (можно через вату). К прозрачному оранжево-красному фильтрату добавляют 200 мл петролейного эфира и высаживают тем самым основную массу комплекса. Затем охлаждают до комнатной температуры и оставляют на ночь в холодильной камере. Кристаллы собирают на стеклянном пористом фильтре (G3), промывают петролейным эфиром (4x50 мл) и сушат в высоком вакууме (20 °C, 10-3 мм рт. ст.). Выход 22,6 г (94%, считая на [N(C2H5)4]Br).
Свойства. Оранжевые кристаллы, /пл 165 °C, при этой температуре начинается разложение и изменение окраски на зеленую. Растворяются в воде, спиртах, ацетоне и CHClg, не растворяются в эфире, петролейном эфире и бензоле. ИК (КВг): 2058 (сл.), 1906 (оч. с.) [v(CO)] см-1.
[N(C2H5)4][W(CO)SC1]
ftv
W(CO)e+[N(C2H5)4]Cl ----> [N(C2H6)4][W(CO)5C1] + CO
351,9 165,7 489,6 22,4 л
В аппаратуру для фотохимических синтезов (см., например, рис. 462) помещают эквимолярную смесь W(CO)e (3,17 г, 9 ммоль) и [N(C2H6)4]C1 (1,5 г, 9 ммоль)* в 600 мл тетрагидрофурана и облучают до прекращения выделения СО. Желтый раствор упаривают примерно до объема 100 мл и высаживают [N(C2H5)4] [W(CO)5C1] добавлением около 100 мл петролейного эфира. Большую часть избытка W(CO)6 удаляют промыванием соли холодным петролейным эфиром. Остатки сублимируют в высоком вакууме при высушивании соли (40°C). Выход 4,0 г (91%).
* Для облегчения очистки получаемого продукта в данном случае используют количество вещества немного меньше стехиометрического.
Комплексы с галогеновыми лигандами
2057
Свойства. Желтые кристаллы, растворяются в СН2С12, ацетоне, тетрагидрофуране и метаноле, не растворяются в бензоле и петролейном эфире. ИК (СН2С12): 2065 (сл.), 1967 (пл.), 1917 (с.), 1850 (ср.) [v(CO>] см-1.
ЛИТЕРАТУРА
1. Abel Е. W., Butler I. S., Reid J. G., J. Chem. Soc. (London), 1963, 2068.
2. Arbeitsvorschrift aus d. Inst. f. Anorgan. Chem., Univers. Erlangen-Ntirnberg (Prof. Dr. H. Behrens).
□ □ Хлоро(т]-циклопентадиенил)трикарбонилмолибден Mo(CO)3(r]-C5H5)Cl
□ □ Хлоро(т]-циклопентадиенил)трикарбонилвольфрам W(CO)3(r]-C5H5)Cl
Синтез осуществляют в три стадии, исходя из М(СО)в:
a) M(CO)e + NaC6Hs --------> Иа[М(СО)3(т1-С5Н5)] + ЗСО
М=Мо: 264,0 88,1 268,1 3-22,4 л
M=W: 351,9 356,0
б) Иа[М(СО)3(т]-С5Н5)]4-СН3СООН -> М(СО)3(т1-С5Н5)Н + Na[CH3CO2]
М = Мо: 268,1 60,1 246,1 82,0
M = W: 356,0 334,0
в) М(СО)3(т]-С5Н5)Н + СС14-> М(СО)3(Л-С5Н5)С1+СНС13
М=Мо: 246,1 153,8 280,5 119,4
M=W: 334,0 368,4
а) В одногорлую колбу на 500 мл с обратным воздушным холодильником помещают эквимолярные количества (50 ммоль) соответствующего гексакарбонила [13,2 г Мо(СО)6 или 17,6 г W(CO)3] и 6,2 г (70 ммоль) NaC5H5 в 100 мл N.N-диметилформамида (ДМФ). Смесь нагревают до 130 °C и выдерживают при этой температуре 2 ч. При ~75°С в случае Мо(СО)3 или при ~110°С в случае W(CO)6 начинается быстрое отщепление СО. В процессе реакции смесь приобретает светло-коричневую окраску; легко возгоняющийся гексакарбонил смывают в колбу взбалтыванием раствора. Образующаяся натриевая соль чрезвычайно неустойчива на воздухе.
б—в) Раствор натриевой соли в ДМФ охлаждают на ледяной бане и добавляют к нему 50 мл Н2О, 120 мл эфира и 18 мл ледяной уксусной кислоты. Тотчас образующийся гидридный комплекс переходит в эфирную фазу. Перемешивают 10 мин (защита от света!) и добавляют 20 мл СС14. Мгновенно образуется М(СО)з(т]-С5Н6)С1, что заметно по красному окрашиванию эфирной фазы. Перемешивают 30 мин, декантируют органический слой, оставшуюся фазу (Н2О+ДМФ) экстрагируют эфиром (5x50 мл) при встряхивании. Объединенные эфирные вытяжки упаривают в роторном испарителе. Красный или красно-коричневый остаток извлекают 80 мл ацетона, добавляют к нему мелко растертого активированного угля и безводного Na2SO4, перемешивают 30 мин и фильтруют на стеклянном пористом фильтре (G4), покрытом кусочками фильтровальной бумаги. Темно-красный фильтрат упаривают в вакууме водоструйного насоса до половины объема и оставляют кристаллизоваться в холодной камере (—35°C). Оставшийся в маточнике после его упаривания комплекс высаживают добавлением ~200 мл Н2О. Перемешивают некоторое время на магнитной мешалке, выпавшие кристаллы отделяют, промывают 20 мл Н2О, многократно петролейным эфиром (по 30 мл) и несколько часов сушат в вы-
2058 Глава 33. Металлоорганические комплексы
соком вакууме. Общий выход: 9,8—12,3 г (70—88%), когда М=Мо, и 12,5— 14,4 г (68—78%), когда M=W.
Поскольку Мо(СО)6 реагирует с NaC6H3 уже при 60°C, то при получении Мо(СО)3(т]-С5Н5)С1 можно с самого начала использовать в качестве растворителя тетрагидрофуран (см. ниже в этой главе методику синтеза Мо(т]-С6Н6) (CO)2NO). Время реакции в этом случае 15 ч. Выходы по сравнению с методикой, использующей диметилформамид, не изменяются, однако в процессе экстракции эфиром границу раздела фаз видно хуже. Это ведет к потерям и получению менее чистых препаратов.
Свойства. Мо(СО)3(т]-С6Н5)С1: блестящее красное или темно-красное кристаллическое соединение, медленно разлагающееся на воздухе, особенно в мелкокристаллической форме, ^разл 145 °C. Плохо растворяется в алифатических углеводородах, хорошо — в бензоле, CH2CI2, ацетоне. Кристаллическая структура: см. [3]. ИК (КВг): 2043, 1972, 1935 [v(CO)]; (СНС13): 2058, 1983, 1935 (пл.) [v(CO)] см-1. ЯМР-'Н (CDC13, ТМС): 6 5,29 [синглет, С5Н5].
W(CO)3(r]-C5H5)Cl: темно-красные кристаллы. /пл 157—159 °C. В твердом состоянии и растворах соединение разрушается значительно быстрее, чем Мо(СО)3(т]-С5Н5)С1. Растворимость, как и у Мо(СО)3(т]-С5Н3)С1. ИК (нуйол): 2038, 1961, 1915 ,[v(CO)] см-1.
ЛИТЕРАТУРА
1. Piper Т. S„ Wilkinson G., J. Inorg. Nucl. Chem., 3, 104 (1956).
2. Coffey С. E., J. Inorg. Nucl. Chem., 25, 179 (1963).
3. Chaiwasie S., Fenn R. H„ Acta Cryst., B, 24, 525 (1968).
□ □ Трихлоро(т]-циклопентадиенил)дикарбонилмолибден
Mo(CO)2(r]-C5H5)Cl3
Мо(СО)8(т]-С5Н5)С1-|-С12 -> Мо(СО)2(т]-С6Н6)С13 + CO
280,5 22,4 л 323,4 22,4 л
Через охлаждаемый на ледяной бане и интенсивно перемешиваемый раствор 2,81 г (10 ммоль) Мо(СО)3(т]-С5Н6)С1 в 50 мл СНС13 пропускают с помощью капилляра слабый ток С12 до тех пор, пока первоначально красный раствор не станет желто-коричневым (~2—3 мин). Затем избыточный С12 тотчас удаляют многократным вакуумированием системы водоструйным насосом и продуванием N2. Образовавшееся, практически нерастворимое в СНС13 соединение как можно быстрее отделяют на стеклянном пористом фильтре G3, промывают небольшим количеством СНС13 и высушивают в высоком вакууме. Выход 2,50 г (77%).
По точно такой же методике получают W(CO)2(t]-C5H5)C13 (3,68 г [10 ммоль W(CO)3(t]-C5H5)C1], выход 3,0 г [73%]).
Свойства. Неустойчивый на воздухе, легко гидролизуемый порошок, цвета охры или коричневый, кристаллизуется из смеси эфира с SO2. 1ПЛ 116’С (с разл.). Практически не растворяется в большинстве обычных органических растворителей. В ацетоновом растворе очень быстро разлагается прн комнатной температуре. ИК (нуйол): 2104 (с.), 2065 (с.) Tv (СО)] см-1. ЯМР-'Н (ацетон-de, ТМС): 6 6,11 [синглет, С6Н6].
ЛИТЕРАТУРА
1. Green М. L. Н., Lindsell W. Е., J. Chem. Soc. (London), А 1967, 686.
Комплексы с галогеновыми лигандами 2059
Иододикарбонил (г|-циклогептатриенил)молибден Мо(т]-С7Н7)(СО)21
[Мо(я-С7Н7) (CO)3]BF4 4- Nal -> Mofa-CrH,) (СО)21 + СО + NaBF4
357,9 149,9 370,0 22,4 л 109,8
В 700 мл ацетона растворяют 30,4 г (85 ммоль) [Мо(г]-С7Н7) (СО)з] [BF4] и добавляют при перемешивании ~75 г (0,5 моль) Nal (можно брать также Lil или KI). Раствор мгновенно окрашивается в зеленый цвет, выделяется газ. После отгонки растворителя остается темный твердый остаток. Его извлекают, интенсивно перемешивая или взбалтывая с 600 мл СН2С12 (в пять порций). Объединенный темно-зеленый экстракт упаривают в вакууме водоструйного насоса. Кристаллический остаток многократно промывают (по 50 мл) петролейным эфиром (/кип 40—60 °C) и высушивают в высоком вакууме. Выход 23,0—24,8 г (73—79%).
Свойства. Черно-зеленые, устойчивые на воздухе кристаллы, <пл 179— 184°C (с разл.). Растворимы во всех полярных органических растворителях (например, в СН2С12, ацетоне); растворы зеленого или черного цвета; в растворах постепенно разлагается. ИК (КВг): 1980, 1930 [v(CO)] см-1. ЯМР-’Н (ацетон-de, ТМС): 6=5,83 {синглет, С7Н7].
ЛИТЕРАТУРА
I. King R. В., Organometallic Syntheses, Academic Press, New York, London, 1965, v. 1, p. 141.
Хлоро (пентакар бонил )м арганец Мп (СО ) 5 С1
Мп2(СО)10 + С12 ---> 2Мп(СО)6С1
390,0 22,4 л 2-230,4
К охлаждаемой льдом суспензии 3,90 г (10 ммоль) Мп2(СО)ю в 50 мл СС14 прикапывают 25 мл СС14, насыщенного на холоду хлором. Через 5 ч перемешивания при комнатной температуре полностью заканчивается образование Мп(СО)5С1, нерастворимого в СС14. Желтоватый осадок отделяют, многократно (по 10 мл) промывают СС14 и сушат в вакууме масляного насоса. Выход сырого продукта составляет 3,9 г (85%). Для очистки препарата от нелетучих продуктов разложения, МпС12 (в основном) и [Мп(СО)4С1]2, проводят многодневную высоковакуумную возгонку при 45—50 °C. Выход 2,9— 3,2 г (63—70%).
Свойства. Устойчивые на воздухе, светло-желтые кристаллы, переходящие при нагревании свыше 60 °C в димерный [Мп(СО)4С1]2. Практически не растворяется в петролейном эфире и СС14, хорошо растворяется в бензоле, СНС13 и ацетоне. ИК (СС14): 2140 (сл.), 2054 (с.), 2021 (оч. сл.), 1998 (ср.) v[(CO)] см-1.
ЛИТЕРАТУРА
1. Abel W. Е., Wilkinson G., J. Chem. Soc. (London), 1959, 1501.
2. Reimer К. J., Shaver A., Inorg. Synth., 19, 159 (1979).
2060 Глава 33. Металлоорганические комплексы
Бромо(пентакарбонил)марганец Мп(СО)бВг
Мп2(СО)10 + Вг2 -> 2Мп(СО)6Вг
390,0 159,8 2-274,9
К раствору 7,80 г (20 ммоль) Мп2(СО)ю в 150 мл ССЦ при комнатной температуре в течение 30 мин при перемешивании добавляют раствор 1,26 мл (3,7 г; 23,1 ммоль) Вг2 в 25 мл ССЦ. Смесь перемешивают 1 ч, удаляют растворитель в вакууме водоструйного насоса и возгоняют в высоком вакууме при 55±5°С, используя охлаждаемый водой «палец» сублиматора. Выход 6,4—7,4 г (58—67%).
Свойства. Устойчивое на воздухе кристаллическое соединение оранжевого цвета. Плохо растворяется в петролейном эфире, умеренно — в СС14, хорошо —• в бензоле, эфире и ацетоне. При длительном нагревании свыше 65 °C превращается в [Мп(СО)4Вг]2. ИК (ССЦ): 2134 (сл.), 2051 (с.), 2020 (оч. сл.), 2000 (ср.) [\(СО)1 см-1.
ЛИТЕРАТУРА
1. Abel Е. W., Wilkinson G„ J. Chem. Soc. (London), 1959, 1501.
Иодо(пентакарбонил)марганец Mn(CO)sI
Mn2(CO)10 + I2 ---> 2Mn(CO)5I
390,0 253,8 2-321,9
В пробирке на 100 мл с краном медленно нагревают до 110±3°С смесь 3,90 г (10 ммоль) возогнанного, мелко измельченного Мп2(СО)ю и 2,70 г (10,7 ммоль) порошка 12. Выдерживают смесь при этой температуре 15 ч, красно-коричневый продукт охлаждают и в вакууме масляного насоса при комнатной температуре удаляют непрореагировавший 12. Рубиново-красный Мп(СО)61 возгоняют в высоком вакууме при 45 °C. Возгонка идет быстро с образованием прозрачных кристаллов. Выход 5,35 г (83%).
В методике, приведенной в работе [4], условия реакции не оптимизированы; для получения высоких выходов требуется обязательно применять порошок Мп2(СО)ю.
Другие способы. Взаимодействие Na[Mn(CO)s] с 12. Выход 75—82% [5].
Свойства. Красные, преломляющие свет кристаллы, при доступе воздуха очень медленно разлагаются с отщеплением иода. Растворяется во всех обычных органических растворителях. При нагревании выше 118 °C темно-красное окрашивание с частичной возгонкой, при более высокой температуре идет быстрое разложение. ИК (СС14): 2127 (сл.), 2045 (с.), 2016 (оч. сл., пл.), 2005 (ср.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Brimm Е. О., Lynch. М. A., Jr., Sesny W. J., J. Amer. Chem. Soc., 76, 3831 (1954).
2. Herrmann W. A., Huber M„ J. Organometal. Chem., 140, 55 (1977).
3. Abel E. W„ частное сообщение.
4. Inorg. Synth., 19, 162 (1979).
5. Quick M. H„ Angelici R. J., Inorg. Synth., 19, 161 (1979).
Комплексы с галогеновыми лигандами 2061
Хлоро(пентакарбонил)реним Re(СО)5С1
Re2(CO)10 + С12 --> 2Re(CO)sCl
652,5 22,4 л 2-361,7
В охлаждаемую льдом суспензию 3,26 г (5 ммоль) Re2(CO)io (методику синтеза см. выше в этой главе) в 80 мл СС14 при перемешивании по каплям добавляют 35 мл насыщеииого раствора С12 в СС14. Перемешивают при комнатной температуре 15 ч, отделяют мелкокристаллический осадок и многократно промывают его на фильтре СС14, охлажденном льдом (порциями по 10 мл). Выход 3,18 г (88%).
Другие способы. Карбонилирование ReCl3, ReCis или Кз[КеС1б] под давлением в присутствии порошка Си (выход >90%) [5, 6].
Свойства. Белые, устойчивые на воздухе кристаллы. Растворимость как у Re(CO)sBr. ИК (СС14): 2156 (сл.), 2045 (с.), 2016 (сл.), 1982 (ср.) [v(СО)] см-1.
ЛИТЕРАТУРА
1. Arbeitsvorschrift d. Chem. Inst. Univers. Regensburg.
2. Abel E. W., Wilkinson G., J. Chem. Soc. (London), 1959, 1501.
3. Brimm E. O., Lynch M. A., jr„ Sesny W. J., J. Amer. Chem. Soc., 76, 3831 (1954).
4. Анисимов К. H., Барышников Л. И. — Изв. АН СССР, Серия хим., 1962, с. 1241.
5. Hieber W., Schuh R„ Fuchs H., Z. Anorg. Allgem. Chem., 248, 243 (1941).
6. Schulten H., Z. Anorg. Allgem. Chem., 243, 164 (1949).
Бромо(пентакарбонил)рений Re(CO)sBr
Re2(CO)10 + Br2 --> 2Re(CO)6Br
652,5 159,8 2-406,2
К суспензии 6,53 г (10 ммоль) Re2(CO)io в 50 мл СС14 при 0°С и при перемешивании добавляют по каплям раствор 0,6 мл (1,75 г, 11 ммоль) брома в 50 мл СС14. Перемешивают 3 ч при комнатной температуре; выпавший желтоватый мелкокристаллический осадок отделяют, дважды по 10 мл промывают холодным СС14 и 1 ч сушат в высоком вакууме. Продукт аналитически чистый. Выход 8,04 г (99%).
Другие способы. Из гексаброморената (IV) и СО (200 бар) [3].
Карбонилирование гексаброморената(IV) в присутствии НСООН+НВг. Выход количественный [4].
Свойства. Белые, устойчивые на воздухе кристаллы, возгоняются при 60 °C в высоком вакууме. Плохо растворяется в петролейном эфире, хорошо — в полярных растворителях (СН2С12, тетрагидрофуране). ИК (СС14): 2150 (сл.), 2045 (с.), 2016 (сл.), 1984 (ср.) ,[v(CO)] см-'.
ЛИТЕРАТУРА
1. Vitali D., Calderazzo F., Gazz. Chim. Ital., 102, 587 (1972).
2. Brimm E. O., Lynch M. A., Jr., Sesny W. J., J. Amer. Chem. Soc., 76, 3831 (1954).
3. Hieber W„ Schulten H„ Z. Anorg. Allgem. Chem., 243, 164 (1949).
4. Colton R., Knapp J. E., Aust. J. Chem., 25, 9 (1972).
2062 Глава 33. Металлоорганические комплексы
Иодо(пентакарбонил)рений Re(CO)5I
В противоположность взаимодействию Re2(CO)i0 с С1г или Вг2 реакция с 12 не ведет к образованию исключительно моноядерного производного Re(CO)6I. Образуется смесь моно- и биядерного комплексов Re(CO)sI и [Re(CO)4I]2 соответственно. Последний может быть количественно переведен в Re(CO)6I карбонилированием при высоком давлении.
Re2(CO)10 4- 12 -> 2(1 — х) Re(CO)6I + x[Re(CO)4I]2 + 2хСО
652,5 253,8 2(1 — х)-453,2 х-850,3 2х-22,4л
2(1 —х) Re(CO)6I +x[Re(CO)4I]2 + 2хСО --> 2Re(CO)6I
2(1—х).453,2 х-850,3 2х-22,4л 2-453,2
В ампулу на 100 мл помещают 3,92 г (6 ммоль) Re2(CO)i0, хорошо размельченного на шаровой мельнице, 1,52 г (6 ммоль) порошка 12 и запаивают. Смесь нагревают 15 ч при 100±5°С. Беловато-серый сырой продукт освобождают от следов 12 в вакууме масляного насоса, а затем карбонилируют во вращающемся автоклаве при 150 °C и 300 бар СО. Через 12 ч смесь Re(CO)sI и [Re(CO)4I]2 превращается в чистый Re(CO)6I. Его извлекают 100 мл бензола, раствор фильтруют и после отгонки растворителя в вакууме водоструйного насоса (при <35 °C) получают продукт в виде белых кристаллов. Следы Re2(CO)i0 удаляют в высоком вакууме при 35 °C. Выход 4,79 г (88%).
Другие способы. Карбонилирование K2[ReIe] при высоком давлении в присутствии порошка Си (выход >90%) [4, 5].
Свойства. Белые, устойчивые на воздухе кристаллы. Возгоняются в высоком вакууме при <60 °C, при более высокой температуре превращаются В [Re(CO)4I]2. ИК (СС14): 2145 (сл.), 2042 (с.), 2013 (сл.), 1987 (ср.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Brimm Е. О., Lynch М. A., Jr., Sesny W. I., J. Amer. Chem. Soc., 76, 3831 (1954).
2. Abel E. W., Hargreaves G. B., Wilkinson G., J. Chem. Soc. (London), 1958, 3149.
3. Herrmann №. A., Ber., Ill, 2458 (1978).
4. Hieber W., Schuh R., Fuchs H., Z. Anorg. Allgem. Chem., 248, 243 (1941). 5. Schulten H., Z. Anorg. Allgem. Chem., 243, 164 (1949).
Бис [ ц-хлоро (тетр акар бонил)рений] [Re(СО)4С1]2
Re2(CO)10 + Cl2 -> [Re(CO)4Cl]2 + 2СО
652,5 22,4 л 667,4 2-22,4 л
В 250 мл СС14 растворяют 6,53 г (10 ммоль) Re2(CO)i0 и добавляют такой же объем СС14, насыщенного С12. Перемешивают 1 ч при комнатной температуре и получают суспензию белого цвета, состоящую из Re(CO)6Cl и [Re(CO)4Cl]2. В вакууме водоструйного насоса досуха отгоняют растворитель, остаток в течение 12—14 ч кипятят в 750 мл гексана, при этом Re(CO)sCl количественно превращается в [Re(CO)4Cl]2. После охлаждения до комнатной температуры фильтруют на воронке Бюхнера, осадок для очистки от загрязнений желтого цвета промывают небольшим количеством абсолютного С2Н6ОН и, наконец, отсасывают осадок досуха, промывая его несколько раз пентаном. Выход 6,27 г (94%).
Комплексы с галогеновыми лигандами 2063
Свойства. Устойчивое на воздухе кристаллическое соединение белого цвета. Не растворяется в алифатических углеводородах, плохо растворяется в горячем СС14 и большинстве других органических растворителей. ИК (СС14): 2114 (сл.), 2032 (с.), 2000 (ср.), 1959 (ср.) [v(CO)] см-1. В атмосфере СО под давлением переходит в RefCOJsCl.
ЛИТЕРАТУРА
1. Hileman J. С., Huggins D. К-, Kaesz Н. D., Inorg. Chem., 1, 933 (1962). 2. Dolcetti G„ Norton J. R., Inorg. Synth., 16, 35 (1976).
Галогено(т]-циклопентадиенил)дикарбонилжелезо Ре(СО)2(л-С5Н5)Х (X=C1, Br, I)
Галогенокомплексы типа Fe(CO)2(r)-C5H5)X (X=C1, Br, I) получают с хорошими выходами из [Fe(CO)2(T]-C5H5)]2 путем окислительного расщепления связи металл — металл, т. е. универсальным методом, характерным вообще для металлоорганической химии. Бромо- и иодопроизводные Fe(CO2) (т]-С5Н5)Вг и Fe(CO2) (т]-С6Н5)1 получают прямым окислением биядерного карбонильного комплекса [Fe(CO)2(r]-C5H5)]2 при действии Вгг и 12. Хлорокомплекс Fe(CO)2(r]-C5H5)Cl предпочтительно синтезируют из [Ре(СО)2(т]-С6Н5)]2 окислением кислородом воздуха в растворе НС1.
[Fe(CO)2(T]-C6H5)]2 + 2НС1 + 1/2О2 -> 2Fe(CO)2(T]-C.5H6)Cl 4- Н2О
353,9 2-36,5 11,2л 2-212,4 18,0
-|- Вг2 ---> 2Ре(СО)2(т]-С5Н5)Вг
159,8 2-256,9
+ 12 ----> 2Fe(CO)2(T]-C6H6)I
253,8 2-303,8
Хлоро(т]-циклопентадиенил)дикарбоннлжелезо Ре(СО)2(т]-С5Н5)С1
Через смесь из 7,1 г (20 ммоль) [Ре(СО)2(т]-С5Н5)]2, 300 мл С2Н5ОН, 60 мл СНС1з и 15 мл конц. НС1 в течение 3 ч продувают интенсивный ток воздуха. Упаривают затем досуха в вакууме масляного насоса (используют охлаждаемую ловушку) и остаток экстрагируют 600 мл Н2О. Профильтрованный красный раствор три раза встряхивают с СНС13 (по 100 мл), упаривают высушенную над Na2SO4 органическую фазу в роторном испарителе и мелкокристаллический красно-коричневый остаток перекристаллизовывают из смеси СНС1з — петролейный эфир (1 : 1). Выход 5,9—6,4 (69—75%).
Б|ромо(т]-циклопентадиенил)дикьрбонилжелезо Fe(CO)2(T]-C6H5)Br
К раствору 10,8 г (31 ммоль) [Ре(СО)2(т]-С5Н5)]2 в 150 мл СНС13 при перемешивании и охлаждении (смесь льда и соли) прибавляют по каплям раствор 1,63 мл (5,11 г, 32 ммоль) Вгг в 30 мл СНС13. Перемешивают 30 мин при комнатной температуре. Затем для удаления избытка Вг2 встряхивают в делительной воронке (3X150 мл) с 10%-ным Na2S2O3 и промывают 100 мл воды. Далее высушивают Na2SO4, фильтруют и удаляют растворитель в вакууме водоструйного насоса. Красно-коричневый мелкокристаллический осадок промывают на фильтре многократно петролейным эфиром, отсасывают и некоторое время сушат в высоком вакууме. После перекристаллизации из смеси СНС13 —петролейный эфир (1:5) получают аналитически чистый препарат. Выход 9,9— 10,5 г (63—67%).
2064 Глава 33. Металлоорганические комплексы
Иодо(т]-циклопентадиенил)дикарбонилжелезо Fe(CO)2(r]-C5H5)I
В 200 мл СНС13 в течение 30 мин кипятят с обратным холодильником 10,8 г (31 ммоль) [Ре(СО)2(т]-С6Н6)]2 и 8,9 г (35 ммоль) 12. Охлаждают до комнатной температуры, в делительной воронке отмывают от избытка 12 встряхиванием с 200 мл 15%-ного Na2S2O3 (в три приема), затем промывают 100 мл воды, органический слой сушат Na2SO4, упаривают растворитель на водоструйном насосе и черный кристаллический остаток многократно промывают на фильтре петролейным эфиром. Высушенный в вакууме продукт достаточно чистый для препаративных целей. Если потребуется, его возгоняют в высоком вакууме при 90—100 °C, однако со значительными потерями. Выход 14,2—14,8 г (77—80%).
Другие способы. Fe(CO)2(r]-C5He)Cl получают при действии С12 на раствор [Fe(CO)2(n-C5H5)]2 в СС14—СН2С12 с выходом 37% [5]. Ре(СО)2(т]-С6Н5)1 образуется при кипячении Ре(СО)2(т]-С6Н3)С1 с KI или Nal в ацетоне. Выход 65% [6, 7].
Свойства. Fe(CO)2(r]-C5H5)Cl: ие очень устойчивые кристаллы, кирпичио-красного цвета, разлагаются при 87—89°C. ИК (СС14): 2049 (оч. с.), 2008 (оч. с.) Гу (СО)] см-1.
Ре(СО)2(п-С6Н5)Вг: устойчивые на воздухе, красно-коричиевые кристаллы, <пл 105—107°C. ИК (СС14): 2044 (оч. с.), 2004 (оч. с.) [v(CO)] см-1.
Ре(СО)2(т]-С6Н5) I: устойчивые на воздухе, черные кристаллы с металлическим блеском, <пл Н9—120°C. ИК (СС14): 2035 (оч. с.), 2000 (оч. с.) (v(CO)] см-1.
Соединения ие растворяются в воде, практически не растворяются в пет-ролейном эфире, умеренно растворяются в СС14 и С3Н6, хорошо —в полярных органических растворителях, таких, как хлороформ или ацетон. При длительном воздействии кислорода воздуха из растворов выпадает осадок оксида железа (И).
ЛИТЕРАТУРА
1. Piper Т. S., Cotton F. A., Wilkinson G., J. Inorg. Nucl. Chem., 1, 165 (1955).
2. Piper T. S., Wilkinson G„ J. Inorg. Nucl. Chem., 2, 38 (1956).
3. King Д. B., Bisnette M. B., J. Amer. Chem. Soc., 86, 1267 (1964).
4. Coffey C. E„ J. Inorg. Nucl. Chem., 25, 179 (1963).
5. C. A. 53, 4298 (1959).
6. Hallam B. F., Mills O. S., Pauson P. L., J. Inorg. Nucl. Chem., 1, 313 (1955).
7. Hallam B. F., Pauson P. L„ J. Chem. Soc. (London), 1956. 3030.
Иодо(г]-иодци1клопентадиенил)дикарбонилжелезо Fe(CO)2(i)-C5H4I)I
Синтез осуществляют циклопентадиенилированием диазоциклопентадиеном:
а) С^ + N3SO2CeH4CH3 ------> C5H4N2 + H2NSO2C6H4CHs
66,1 197,2 92,1 171,2
6) Fe(CO)4I2 + C6H4N2 ---> Fe(CO)2(T]-C6H4I)I + 2CO + N^
421,7 92,1 429,8 2-22,4 л 22,4 л
а) Диазоциклопентадиен
Внимание! Обращать особое внимание на отсутствие даже следов кислоты в реакционной системе! Весьма устойчивый термически (1КИп 52—53 °C/
Комплексы с галогеновыми лигандами 2065
/50 мм рт. ст.) диаэоциклопентадиен может взорваться от каталитических количеств кислоты [2]. Перегонку его по этой причине не проводят. В растворе диазоциклопентадиен опасности не представляет [3].
Смесь 13,2 г (200 ммоль) свежеперегнанного циклопентадиена, 100 мл абсолютного ацетонитрила и 14,6 г диэтиламина охлаждают льдом и к ней же при перемешивании магнитной мешалкой по каплям прибавляют 34,4 г (205 ммоль) тозилазида с такой скоростью, чтобы температура смеси не поднималась выше +50°C. Смесь оставляют на ночь в холодильнике (+8°C), затем добавляют 400 мл Н2О и экстрагируют встряхиванием с эфиром (Зх Х200 мл). Органический слой отделяют, промывают раствором 11,2 г КОН в 200 мл Н2О и затем водой, сушат безводным Na2SO4 и упаривают в роторном испарителе при 25—30 °C. Остающееся красное масло хроматографируют на кизельгеле бензолом (колонка 2,5X80 см, водяная рубашка). Оранжевокрасный элюат содержит ~ 100 ммоль диазоциклопентадиена и может быть использован для циклопентадиенилирования без дальнейшей очистки.
б) □ Иодо(т]-иодциклопентадиеиил)дикарбонилжелезо
В колбе на 1 л к бензольному раствору (500 мл) 33,7 г (80 ммоль) Fe(CO)4I2 добавляют 100 ммоль диазоциклопентадиена в ~100 мл бензола. Смесь постепенно нагревают до кипения при перемешивании примерно за 1 ч. При 40 °C начинается интенсивное выделение газа. После двухчасового кипячения черную реакционную смесь охлаждают до комнатной температуры, добавляют 50 г кизельгеля, фильтруют через складчатый фильтр и многократно промывают смешанный с кизельгелем осадок бензолом порциями по 50 мл. Фильтрат упаривают в роторном испарителе, при этом выделяется черный маслянистый сырой продукт, который при растирании в петролейном эфире застывает в виде микрокристаллической массы. После перекристаллизации при —78 °C в эфире /пл 69—70 °C. Еще одна кристаллизация дает аналитически чистый образец, плавящийся точно при 72°C. Выход 19,9 г (58%).
При мелких загрузках (до 10 ммоль) препарат можно выделить и хроматографически (кизельгель, колонка 1,8x50 см). Продукт элюируют бензолом в виде зоны черного цвета. Продукты разложения остаются в верхней части колонки. Кристаллические иглы длиной до 6 см получают кристаллизацией из смеси эфир — СН2С12 (10:1, —35/—78°C). Потери при этом весьма значительные.
Свойства. Черные, с металлическим блеском, устойчивые на воздухе иглы или осколки. tnjI 72 °C. Растворяются без разложения в бензоле, хлороформе и других полярных растворителях. ИК (СС14): 2041 (оч. с.), 2002 (оч. с.) [v(CO)] см-1. ЯМР-’Н (C6D6, ТМС): 6(На) 4,45; 6(Hg) 4,00 [псевдотриплеты равной интенсивности]. ЯМР-13С (CDC13, 30 °C, ТМС): 6(С+ 46,3; 6(CH<a)) 83,4; б(Сн(₽>) 92,9; 6 (СО) 212,6.
ЛИТЕРАТУРА
1. Herrmann W. A., Huber М.., Вег., 111, 3124 (1978).
2. Doering W. v. Е„ DePuy С. Н., J. Amer. Chem. Soc., 75, 5955 (1953).
3. Herrmann W. A., Huber M., J. Organometal. Chem., 140, 55 (1977).
□ Дигалогено(тетракарбонил)железо Fe(CO)4X2 (X=C1, Br, I)
Соединения могут быть получены с хорошими выходами при действии С1а, Вг2 и 12 на Fe(CO)5 в петролейном или диэтиловом эфире:
Fe(CO)6 + Cl2 --> Fe(CO)4Cl2 + СО
195,9 22,4 л 248,8 22,4 л
14—1361
2066 Глава 33. Металлоорганические комплексы
+ Вг2-----> Fe(CO)4Br2 СО
158,9 327,7 22,4 л
+ 12 -----> Fe(CO)4I2 + CO
253,8 421,7 22,4 л
Д ихлоротетракарбоннлжелезо Fe (СО) 4С12
Через раствор 13,4 мл (0,10 моль) Fe(CO)5 в 150 мл петролейного эфира при охлаждении смесью льда и соли пропускают в течение 1 ч слабый ток С12. Продукт выпадает в виде желтого порошка. Его быстро отсасывают (в атмосфере N2I), промывают небольшим количеством петролейного эфира и непродолжительное время сушат в вакууме масляного насоса при —25 °C. Выход 13,3—14,6 г (53—59%).
Осторожно! Неразбавленный петролейный эфиром Fe(CO)s ни в коем случае не должен соприкасаться с С12; в противном случае моментально идет окисление с воспламенением!
Дибромотетракарбоннлжелезо Fe(CO) 4Вг2
К 13,4 мл (0,10 моль) Fe(CO)s в 50 мл петролейного эфира при перемешивании и охлаждении льдом прикапывают раствор 16,0 г (0,10 моль) Вг2 в 50 мл гексана. Тотчас выпадает красно-коричневый порошок, и наблюдается выделение СО. Осадок быстро отделяют и сушат в высоком вакууме. Выход 32,8 г (100%).
Динодотетракарбонилжелезо Fe(CO)4l2
К раствору 13,4 мл (19,6 г, 0,10 моль) Fe(CO)s в 60 мл эфира при комнатной температуре медленно прикапывают раствор 24,1 г (95 ммоль) 1г в 200 мл эфира. После окончания выделения газа растворитель и избыток Fe(CO)s удаляют в вакууме масляного насоса. Соединение при этом выпадает в виде крупных кристаллов. Выход 39,3 г (93%). Возможна очистка возгонкой в высоком вакууме прн 70 °C, однако при этом велики потери и времени и вещества.
Другие способы. Возможно получение Fe(CO)4I2 из безводного Fel2 в автоклаве уже при давлении СО >6,3 бар [4].
Свойства. Карбонилгалогениды железа светочувствительны. Термическая устойчивость изменяется в последовательности 1>Вг>С1. Хлоропроизводиое уже при комнатной температуре выделяет СО и становится серым. Соединения в атмосфере инертного газа при —35 °C хранятся месяцами без заметного разложения. Хлорид и бромид почти моментально, а иодид при нагревании реагируют с водой, образуя соответствующие соли Fe(II). Цвет: Fe(CO)4CI2 желтый, Fe(CO)4Br2 красно-коричневый, Fe(CO)4I2 фиолетово-коричневый.
Данные ИК-спектров Fe(CO4X2, Х = С1 (в С2С14): 2164, 2124, 2108, 2084; Х=Вг (в С6Н14): 2150, 2108, 2098,5, 2074; Х=1 (в С6Н14): 2131, 2086, 2062, 2047 [v(CO)] см-4. Соединения выделяются в tfuc-форме, t{nc-Fe(СО)412 фотохимически изомеризуется в транс-Ре(СО)412 [5].
ЛИТЕРАТУРА
1. Hieber W„ Bader G„ Вег., 61, 1717 (1928).
2. Hieber W., Lagally H„ Z. Anorg. Aligem. Chem., 245, 295 (1940).
3. Hieber IF., Wirsching A., Z. Anorg. Allgem. Chem., 245, 35 (1940).
4. Hieber W., Lagally H., Z. Anorg. Allgem. Chem., 245, 305 (1940).
5. Pankowski M., Bigorgne M., C. R. Acad. Sci. Paris, 263, 239 (1966).
Комплексы с галогеновыми лигандами 2067
Дииодотетракарбонилрутений Ru (СО)412
В то время как карбонилирование галогенидов рутения(III) при нормальном давлении и температуре 210—290 °C приводит к образованию полимерных карбоиилгалогенидов [Ru(CO)2X2]„ [3], Rul3 при повышенном давлении гладко образует моноядерный, хорошо изученный Ru(CO)4I2:
4-Cu
2RuIs + 8СО 2Ru(CO)4I2
2-481,8 8-22,4 л 2-466,9
10,0 г (20,8 ммоль) тщательно высушенного Rul3 (получение см. в т. 5) хорошо смешивают с ~15 г (избыток) тонкого порошка Си, не содержащего следов кислоты*, и загружают в стеклянный вкладыш 250-мл качающегося автоклава. Для лучшего перемешивания порошкообразной реакционной массы загружают также некоторое количество стеклянных шариков (диаметр 0,5 см). После того как автоклав дважды продувают СО при давлении 100 бар, начинают реакцию карбонилирования, создав рабочее давление 240 бар. Температура реакции 170 °C. Через 25 ч систему охлаждают до комнатной температуры н осторожно сжигают избыток СО. Черную порошкообразную реакционную смесь подвергают высоковакуумной возгонке при 100°C, Ru(CO)4I2 выделяется при этом в виде желтых игольчатых кристаллов. Выход 5,0—7,3 г (52— 75%).
Свойства. Золотисто-желтое, неустойчивое на воздухе кристаллическое вещество, ZpaM>140oC. ИК (СС14): 2068 (с.), 2097 (с.), 2106 (оч. с.), 2119 (сл.), 2161 (ср.) [v(CO)] см-1. Соединение выделяют в цис-форме.
ЛИТЕРАТУРА
1. Corey Е. R., Evans М. V., Dahl L. F., J. Inorg. Chem., 24, 926 (1962).
2. Dahl L. F., Wampler D. L., Acta Cryst., 15, 946 (1962).
3. Manchot W., Konig L, Ber., 57, 2130 (1924).
Бис[ц-хлоро(хлоро)трикарбонилрутений] [Ru(CO)3CI2]2
Лучшим до сих пор способом синтеза [Ru(CO)3Cl2]2 является, по-видимому, не вполне еще понятное взаимодействие Ru3(CO)i2 с СНС13 при небольшом избыточном давлении N2 при температуре около 110 °C:
СНС18
2Rus(CO)12 --->- 3[Ru(CO)8Cl2]2 4- побочные продукты
2-639,3 3-512,0
Суспензию 2,50 г (3,91 ммоль) Ru3(CO)i2 в смеси 100 мл СНС13 и 2 мл абсолютного С2Н5ОН помещают во вращающийся автоклав на 250 мл со стеклянным вкладышем, создают начальное давление азота ~5 бар и нагревают затем 13 ч при НО °C. После охлаждения и снятия давления в системе образовавшуюся суспензию белого цвета отфильтровывают на воронке Бюхнера (или Хирша)**, промывают небольшим количеством СНС13 и высушивают в вакууме. Полученный продукт (1,6—1,9 г, 53—63%) аналитически чистый.
* Чистый порошок Си перемешивают 3 ч с разбавленной НС1, отфильтровывают, промывают водой и ацетоном и сушат в сушильном шкафу прн 130 С.
** Коническая фарфоровая воронка с пористым дном в суженной части.
14'
2068 Глава 33. Металлоорганические комплексы
Бледно-желтый маточник объединяют со смывным раствором, упаривают в вакууме водоструйного насоса до объема ~25 мл и охлаждают до —10 °C. Желтоватый осадок отделяют и перекристаллизовывают из горячей смеси 1,2-дихлорэтана н и-гексана. Получают таким образом еще 250—500 мг (8— 17%) [Ru(CO)3C12]2.
Другие способы. Можно получать из RuC13-3H2O и муравьиной кислоты в присутствии НС1 [2]. Прямое хлорирование Ru3(CO)i2 приводит к трудно разделяемой смеси продуктов [3].
Свойства. Белый пушистый порошок, устойчив на воздухе. Выше 215 °C окрашивается в оранжево-коричневый цвет, разлагается выше 315 °C. Плохо растворяется в СНС13 и 1,2-дихлорэтане, хорошо — в СН3ОН и тетрагидрофуране. С последним образует сольватный моноядерный комплекс Ри(ТГФ)-(СО)3С12 [4]. ПК (СС14): 2140 (с.), 2081 (с.), 2076 (с.) [v(СО)] см-1. Кристаллическая структура: см. [5] (симметрия С2Л, два мостиковых атома С1).
ЛИТЕРАТУРА
1. Mantovani A., Cenini S., Inorg. Synth., 16, 51 (1976).
2. Colton R., Farthing R. H., Aust. J. Chem., 24, 903 (1971).
3. Johnson B. F. G„ Jonston R. D., Lewis J., J. Chem. Soc. (London), Al969, 792.
4. Bruce M. I., Stone F. G. A., J. Chem. Soc. (London), A1967, 1238.
5. Merlino S., Montagnoli G„ Acta Cryst., B, 24, 424 (1968).
Хлоро (т]-циклопентадиенил)дикарбонилрутений Ru(CO)2(t]-C5H5)C1
[Ru(CO)2(t]-C5H5)]2 + 2HC1 + l/2O2 -> 2Ru(CO)2(r]-C5H5)Cl + H2O
444,4 2-36,5 11,2л 2-257,6 18,0
К раствору 3,0 г (6,8 ммоль) [Ru(CO)2(t]-C5H5)]2 в 50 мл СНС13 добавляют 20 мл этанола, 20 мл ~2 М НС1 и, наконец, еще 3 мл конц. НС1. Через реакционную смесь в течение 6—8 ч продувают воздух. Для сохранения постоянного объема раствора время от времени подливают СНС1з- По окончании реакции органический слой отделяют, водный слой дважды экстрагируют CHCI3, хлороформные растворы объединяют и упаривают досуха. Остаток промывают водой, растворяют в СНС13 и высушивают MgSO4. После упаривания до ~10 мл высаживают светло-желтый продукт петролейным эфиром. Дальнейшую очистку проводят хроматографированием СНС1з на колонке с А12О3 (активность I степени) или перекристаллизацией из смеси CHCI3 — петролей-ный эфир. Выход 2,8 г (80%).
Свойства. Светло-желтые, устойчивые на воздухе кристаллы, /пл 103 °C, хорошо растворяются в бензоле, ацетоне, СНС1з и СН2С12, плохо — в петро-лейном эфире. ПК (СН2С12): 2056 (оч. с.), 2008 (оч. с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Blackmore Т„ Cotton J. D., Bruce М. I., Stone F. G. A., 3. Chem. Soc. (London), A 1968, 2931.
Карбонилхлориды осмия Os(CO)xCl2 (x = 3, 4)
Прямое карбонилирование OsCl3 в зависимости от температуры и давления приводит к карбонилхлоридам осмия с различным содержанием СО-групп:
Комплексы с галогеновыми лигандами 2069
Дихлоротрикарбонилосмий Os(CO)3Cl2 [1]
1 бар
20sCl3 + 7СО ------> 2Os(CO)3C12 + СОС12
2-296,6 7-22,4 л 2-345,1 98,9
В горизонтально закрепленной трубке из тугоплавкого стекла (диаметр 3 см, длина 50 см) нагревают в слабом токе СО в течение 15 ч при 260— 270 °C 2,0 г (6,7 ммоль) ОзС13. В данном случае целесообразно применять раскрывающуюся трубчатую печь. В процессе получения белый Os(CO)3Cl3 сублимируется на холодных частях трубки; поблизости от зоны нагрева продукт приобретает коричневатую окраску. Чистота получаемого таким образом препарата, как правило, достаточна для синтетических целей. Возможна дополнительная очистка кипячением в СС14. Продукт в данном случае становится снежно-белым. Выход количественный.
Свойства. Бесцветные, устойчивые на воздухе кристаллы, /пл 269—273 °C. Выше ~300°С идет разложение до металлического Os.
Дихлоротетракарбонилосмий Os(CO)4Cl2 [2]
>220 бар
20sCl3 + 9СО----------->- 2Os(CO)4C12 + СОС12
2-296,6 9-22,4л 2-373,2 98,9
В сухом качающемся автоклаве емкостью 100 мл карбонилируют 5,0 г (16,9 ммоль) OsCl3 при 125 °C (температура рубашки) и начальном давлении СО 220 бар. Для лучшего перемешивания реакционной массы применяют стеклянные шарики или кольца Рашига. Как обычно при карбонилировании под давлением, для удаления посторонних газов систему дважды продувают СО при 50—100 бар. В процессе реакции максимальное давление составляет 285 бар. Через 20 ч охлаждают до комнатной температуры, сжигают избыток СО, кристаллическое (белого цвета) содержимое автоклава экстрагируют многократно (по 50 мл) абсолютным СНС13. Желтоватую суспензию фильтруют через складчатый фильтр и до тех пор промывают горячим СНС13, пока все белые частицы не извлекутся из черно-коричневого остатка на фильтре. Фильтрат упаривают досуха при ~30°С в вакууме водоструйного насоса. Продукт получают в виде беловатого порошка, чистота которого удовлетворительна для его дальнейшего использования. Спектрально чистый препарат получают перекристаллизацией из СНС13. Выход сырого продукта 5,42—5,80 г (86— 92%).
Свойства. Бесцветные, устойчивые иа воздухе кристаллы. Свыше 250 °C разлагается с выделением металла. Растворяется в полярных органических растворителях (СНС13, ацетоне). В токе СО при 220°C быстро возгоняется. Os(CO)4Cl2 образует, по-видимому, смесь цис- и транс-изомеров.
ЛИТЕРАТУРА
1. Manchot W., Konig J., Вег., 58, 229 (1925).
2. Hieber W., Stallmann H., Ber., 75, 1472 (1942).
□ Дииодо (-q-циклопентадиенил) карбонилкобальт
Со(СО)(т]-С5Н5)12
Со(СО)2(л-С5Н5) + I2 -•> Co(CO) (t]-C5H5)I2 + CO
180,1 253,8 405,8 22,4л
2070 Глава 33. Металлоорганические комплексы
В 250-мл колбе с капельной воронкой растворяют 0,72 г (4,0 ммоль) Со(СО)2(п-С5Н5) и 40 мл эфира, охлаждают до 0 °C и по каплям прибавляют раствор 1,01 г (4,0 ммоль) 12 в 80 мл эфира. Наблюдается выделение СО и окрашивание смеси в черный цвет. После отгонки растворителя в вакууме водоструйного иасоса кристаллический остаток извлекают 15 мл ацетона, фильтруют через стеклянный пористый фильтр № 3, добавляют еще 15 мл СНС1з и оставляют раствор для кристаллизации при —35 °C. Выход 1,1 г (68%).
Свойства. Черно-фиолетовые кристаллы с металлическим блеском, /пл 185°С (с разл.). В противоположность хлор- и бромпроизводным Со(СО)(г|-C3Hs)Cl2 и Со (СО) (трС5Н5)Вг2 иодопроизводное устойчиво к действию света и кислорода воздуха. ИК (нуйол): 2068 [v(CO)] см-1.
ЛИТЕРАТУРА
1. Heck R. F., Inorg. Chem., 4, 855 (1965).
2. King R. В., Z. Naturforsch., 19b, 1160 (1964).
Бис([х-хлородикарбонилродий) [Rh(CO)2ClJ2
2RhCls-3H2O + 6CO ----> [Rb(CO)2Cl]2 + 6H2O-f-2COC12
2-263,3 6-22,4л 388,8 6-18,0 2-98,9
В длинную трубку из стекла дюран (длиной 60 см, диаметром 35 мм) помещают 10 г (38 ммоль) RhCl3-3H2O. Через аппаратуру продувают ток СО вместе с парами метанола. Чтобы концентрацию СН3ОН держать низкой, промежуточно укрепленные промывные склянки охлаждают льдом. Для контроля за уменьшением реакционной массы используют раскрывающуюся трубчатую печь. Трубку постепенно доводят до необходимой для реакции температуры 120—140 °C. Через 12 ч основная масса продукта выделяется в виде красных, сантиметровой длины кристаллических игл. Выделяющуюся в процессе реакции воду удаляют из передней части трубки осторожным подогреванием пламенем бунзеновской горелки. Соединение является аналитически чистым без дальнейшей возгонки. Выход 65%.
Особенно красивые кристаллы получают, если температуру печи доводят до 120—140 °C очень медленно, примерно за 4 ч. Скорость реакции в начальный период очень велика, однако со временем уменьшается. Количественное превращение завершается не менее чем за 15 ч. Более высокая температура реакции ведет к образованию безводного RhCl3, который при нормальном давлении с СО больше не реагирует.
Другие способы. Взаимодействие RhCl3-3H2O с СО при 100°C на стеклянном пористом фильтре. Выход 96% [2].
Автоклавный синтез из безводного RhCl3 в присутствии тонкого порошка Си [3].
Свойства. Оранжево-красные, устойчивые на воздухе, в присутствии влаги слегка гидролизуемые кристаллы, /пл 124—125 °C. Хорошо растворяется в большинстве органических растворителей (бензоле, СН2С12), однако незначительно в петролейном эфире. Растворы заметно неустойчивы на воздухе. ИК (гексан): 2105 (ср.), 2089 (с.), —2080 (оч. сл.), 2035 (с.), 2003 (сл.) [v(CO)] см-1. Кристаллическая структура: см. [4].
ЛИТЕРАТУРА
1. Hieber W„ Lagally Н„ Z. Anorg. Allgem. Chem., 251, 96 (1943).
2. McCleverty J. A., Wilkinson G., Inorg. Synth., 8, 211 (1966).
Комплексы с галогеновыми лигандами 2071
3. Manchot W., Konig J., Вег., 58, 2173 (1925).
4. Dahl L. F., Martell C„ Wampler D. L„ J. Amer. Chem. Soc., 83, 1761 (1961).
□ Ди (ц-хлоро) бис [хлоро (rj-пентаметил циклопентадиенил) -родий] [КЬ{г]-С5(СНз)5}С12]2
2С6(СН3)6 + 2RhCl3-3H2O 4-4СН3ОН ---->
2-162,3 2-263,3 4-32,0
---> [Rh{T]-C6(CH3)6JCl2]2 + 2СН3СН(ОСН3)2 + 2НС1 + 6Н2О 618,1 2-90,1 2-36,5 6-18,0
В круглодонной колбе с обратным холодильником в течение 15 ч под N2 перемешивают при 65 °C раствор 10,0 г (38,0 ммоль) RhCl3-3H2O н 15,0 г (92,4 ммоль) гексаметилбензола Дьюара* в 250 мл метанола. Затем охлаждают до комнатной температуры и уменьшают объем растворителя до ~75 мл в роторном испарителе. Выпавший продукт отделяют на пористом стеклянном фильтре, хорошо промывают эфиром (4x50 мл) для удаления избытка С6(СНз)6. Метанольный маточник досуха испаряют в роторном испарителе и остаток извлекают ~20 мл ацетона. После добавления 25 мл эфира выкристаллизовывают вторую порцию продукта. Его также отфильтровывают н дважды (по 25 мл) промывают эфиром. Объединенные фракции можно перекристаллизовывать из смеси СНС13 — бензол. Выход 11,0 (94%).
Свойства. Темно-красные, устойчивые иа воздухе кристаллы, tnjI >230 °C (с разл.). Хорошо растворяется в полярных растворителях типа СН2С12, СНС13 и Н2О. Является катализатором гомогенного гидрирования. ИК (КВг): 276 (оч. с.), 238 (с.), 206 (пл.), 184 (ср.-с.) [v(RhCl)] см-1. ЯМР-'Н (CDC13): б 1,60 [синглет, СНз]. Кристаллическая структура: моноклинная, пространственная группа P2i/c (а=8,375 А, &=9,228 А, с= 15,651 А, Р= 106,7°); центро-симметричная молекула с плоским мостиком Rh(p.-Cl)2Rh [4].
ЛИТЕРАТУРА
1. Kang J. W., Moseley К-, Maitlis Р. М., J, Amer. Chem. Soc., 91, 5970 (1969).
2. Booth В. L., Haszeldine R. N., Hill M„ J. Chem. Soc. (London), A1969, 1299.
3. Yates A., Maitlis P. M„ частное сообщение.
4. Churchill M. R., Julis S. A., Rotella F. J., Inorg. Chem., 16, 1137 (1977).
5. Schafer W., Hellmann H., Angew. Chem., 79, 566 (1967).
6. Chemische Werke Huis A. G., British Patent 1133893 (27. Mai 1976).
7. Schafer W., British Patent 1152776 (4. Jan. 1967).
Хлоротрикарбонилиридий Ir(CO)3Cl
Получение полимерного карбоиилгалогенида иридия проводят путем карбонилирования при нормальном давлении 1гС13, суспендированного в кизельге-ле в качестве носителя:
со
1гС13-ЗН2О -----> 1г(СО)3С1
352,6 311,7
* Гексаметилбензол Дьюара [гексаметилбицикло [2.2.O.] гексадиен С6(СН3)6] легко получают по реакции бутина-2 с безводным А1С13 в сухом бензоле при 33—35 °C [5—7].
2072 Глава 33. Металлоорганические комплексы
18 г кизельгеля 60 (активность II—III степени, 0,063—0,200 мм; Merck 7734) взбалтывают в растворе 3,40 г (9,64 ммоль) 1гС13-ЗН2О (56% 1г; фирма Ventron; «1гС13 водорастворимый») в 200 мл воды. Растворитель затем удаляют в роторном испарителе (60 °C) и получившийся остаток черного цвета высушивают —12 ч при 120 °C в высоком вакууме (пистолет для высушивания) . Приготовленную таким образом суспензию IrCl3/SiO2 помещают в горизонтально закрепленную стеклянную (дюран) трубку для сожжения (диаметром 2,5 см) и с обеих сторон уплотняют стеклянной ватой (слоем толщиной — 5 см; рис. 476). Реакционную зону трубки сожжения в течение 2,5 ч
Гис. 476. Прибор для получения 1гС1(СО)з.
1 — уплотнения из стекловолокна; 2 — IrCI2/SiO2; 3 — стеклянная трубка для термопары; 4 — муфта НШ 29/32 ; 5 — тефлоновый вкладыш; 6 — термоэлемент; 7 — вывод газа к счетчику пузырьков, заполненному конц. H2SO4; 8 — двухходовой кран (2,5 мм); 9 — ввод азота; 10 — ввод СО или С12 (оба газа предварительно пропускают для высушивания последовательно через H2SO4 и Р4Ою).
нагревают до 150 °C в слабом токе С12 с помощью навитой на трубку электрической спирали, заходящей примерно на 5 см за уплотнение из стеклянной ваты.
После охлаждения до комнатной температуры трубку 15 мин продувают азотом и затем проводят карбонилирование током СО при 180 °C. Скорость потока регулируют таким образом, чтобы образующийся 1г(СО)3С1 быстро выводился из зоны реакции и в то же время не улетал из аппаратуры. Примерно через 12 ч первоначально черная смесь становится почти бесцветной; одновременно на холодной части трубки оседает сублимат медного цвета. Непосредственно за суспензией SiO2/IrCI3 оседает незначительное количество желтого Ir4(CO)i2.
Очистку 1г(СО)3С1 проводят в той же аппаратуре повторной возгонкой при 180 °C в токе СО. Для выделения продукта термоэлемент вынимают и часть трубки, содержащую продукт, отрезают. Выход 0,5—1,3 г (17—43% в расчете на 1гС13-ЗН2О).
Свойства. Волокнистые, похожие на вату, тесно переплетенные между собой дихроичные иголки медно-корнчневого цвета, в течение короткого времени устойчивые на воздухе. Не растворяется практически во всех обычных органических растворителях. ИК (нуйол): 2135 (пл.), 2080 (с.), 2050 (пл.), 2020 (пл.) [v(CO)]; 320 (ср.) [v(IrCl)] см-1. Соединение имеет колончатую структуру с квадратно-плоскостной координацией у атома иридия; J(Ir—1г) = =2,844(1) А [3].
ЛИТЕРАТУРА 1 2 3
1. Fischer Е. О., Brenner К- S., Z. Naturforsch., 17b, 774 (1962).
2. Ginsberg А. Р., K.oepke J. W., Sprinkle C. R., Inorg. Synth., 19, 18 (1979).
3. Reis A. H. Jr., Hagley V. S., Peterson S. W., J. Amer. Chem. Soc., 99, 4184 (1977).
Комплексы с галогеновыми лигандами 2073
□ Ди([х-хлоро) бис [хлоро (трпентаметилциклопентадиенил )-иридий] [1г{тГС5(СН3)5}С12]2
В противоположность аналогичному производному родия образование [1г{т]-С5(СНз)5}С12]2 при непосредственном взаимодействии 1гС13-ЗН2О с гексаметилбензолом Дьюара* идет с очень низким выходом. Выход, однако, можно существенно увеличить, если синтезировать сначала промежуточный |3-1-(1-хлороэтнл)-1,2,3,4,5-пентаметилциклопентадиен C12H19CI [4, 5] [стадия а], и затем ввести его в реакцию с солью металла [стадия б].
а) С6(СН3)6 НС1 -----> С12Н19С1
162,3 36,5 198,7
-н2о
б) 2С12Н19С1 + 21гС13-ЗН2О + 4СН3ОН ----->
2-198,7 2-352,6 4-32,0
----> [ 1г{г)-С5(СНз)6)С12]2 + 2СН3СН(ОСН3)2 + 4НС1 796,7 2-90,1 4-36,5
Через раствор 3,0 г (18,5 ммоль) дьюаровского изомера гексаметилбензо-ла* в 25 мл CH2CI2, находящийся в 50-мл круглодонной колбе, продувают в течение 1 ч медленный ток сухого газообразного НС1 (~0,5 мл/с). Образовавшийся красно-фиолетовый раствор перемешивают 18 ч при 20 °C. Растворитель удаляют на роторном испарителе и получают в остатке масло, представляющее собой в основном C12H19CI. К нему добавляют раствор 1,8 (5,1 ммоль) 1гС13-ЗН2О** в 50 мл СНзОН, ставят обратный холодильник и кипятят 24 ч в атмосфере N2 при 75—80°C (температура бани). Охлаждают до комнатной температуры, упаривают в роторном испарителе до объема ~ 15 мл и добавляют 25 мл сухого эфира. Оранжево-красный осадок отфильтровывают на стеклянном фильтре, основательно (4x25 мл) промывают эфиром, перекристаллизовывают из смеси бензола с хлороформом и сушат в высоком вакууме. Выход 1,2 г (60%, считая на 1гС1з-ЗН2О).
Свойства. Устойчивые на воздухе, ораижево-красные кристаллы, /пл >230°С (с разл.), растворяются в полярных растворителях (кроме воды). ИК: 295 (оч. с.), 256 (оч. с.) [v(IrCl)]cM->. ЯМР-'Н (CDC13): 6 1,59 [синглет, СНз]. Кристаллическая структура: моноклинная, пространственная группа симметрии P2t/c (а=8,384 А, 6=9,278 А, с= 15,741 А, |3= 106,52°), цеитросим-метричная молекула с плоским мостиком 1г(р.-С1)21г [6].
ЛИТЕРАТУРА
1. Kang J. W., Moseley К, Maitlis Р. М„ J. Amer. Chem. Soc., 91, 5970 (1969).
2. Booth В. L., Haszeldine R. N„ Hill M., J. Organometal. Chem., 16, 491 (1969).
3. Yates A., Maitlis P. M„ частное сообщение.
4. Criegee R„ Gruner H„ Angew. Chem., 80, 447 (1968).
5. Paquette L. A., Krow G. R., Tetrahedron Lett., 1968, 2139.
6. Churchill M. R„ Julius S. A., Inorg. Chem., 16, 1488 (1977).
* См. предыдущее примечание.
** Черно-фиолетовое соединение, состав которого приблизительно соответствует формуле 1гС1з-ЗН2О, поставляет фирма Ventron.
2074 Глава 33. Металлоорганические комплексы
□ □ rfuc-Дихлородикарбонилплатина Pt(CO)2C12
цис-Pt (СО) 2С1г образуется из PtCla н СО в отсутствие влаги, при этом всегда идет также и образование высшего карбонилгалогенида (способ 1). Аналитически чистый препарат получают карбонилированием Н2[Р1С1б] при нормальном давлении в сульфинилхлориде (способ 2).
Способ 1 [1—3]
PtCl2 + 2СО -----> Pt(CO)2Cl2
266,0 2-22,4 л 322,0
В горизонтально расположенной тугоплавкой стеклянной трубке (диаметр 3 см, длина 50 см) нагревают в слабом токе СО 2,00 г (7,5 ммоль) безводного PtCl2 при 220 °C. Для нагрева используют раскрывающуюся трубчатую печь. В течение 3 ч на холодной части аппаратуры образуются белые иглы или белый порошок. Для предотвращения улетучивания мелкого порошка карбонилгалогенида трубку с концов уплотняют стеклянной ватой. В качестве побочного продукта, образующегося всегда и трудно отделимого от Pt(CO)2Cl2, на холодной части оседает и высший карбонилгалогенид (от желтого до красного цвета). Его наличие учитывают при проведении последующих превращений. Выход сырого продукта —2,15 г ( — 89%).
Способ 2 [4, 5]
H2[PtCl6] + ЗСО ----> Pt(CO)2Cl2 + СОС12 + 2НС1
409,8 3-22,4л 322,0 98,9 2-36,5
6,60 г (13,5 ммоль) гексагидрата гексахлороплатината (IV) водорода (40% Pt, Degussa) кипятят с обратным холодильником в отсутствие влаги воздуха — 50 ч в 100 мл сульфинилхлорида. После охлаждения до комнатной температуры коричневую суспензию насыщают сухим СО и перемешивают 15—20 дней в атмосфере СО. Небольшое избыточное давление СО создают с помощью газовой бюретки, присоединенной к реакционной колбе. Образовавшуюся смесь фильтруют, фильтрат упаривают в вакууме масляного насоса до начала образования осадка. Затем добавляют 100 мл гексана, охлаждают суспензию сухим льдом, примерно через 1 ч отфильтровывают белый осадок и сушат —10 мин в высоком вакууме. Выход 3,04 г (70%).
Иногда в процессе выделения продукта идет образование небольших количеств красного [Pt(CO)Cl2]2. Последний можно снова перевести в Pt(CO)2Cl2 обработкой СО в сульфинилхлориде (см. выше).
Свойства. Белые кристаллы, очень чувствительные к действию влаги и О2 воздуха, 1ПП 103 °C. Прн доступе влаги соединение очень быстро разрушается, приобретая окраску от коричневого до черного цвета. ИК (СН2С1г): 2180, 2140 [v(CO)] см-1.
ЛИТЕРАТУРА
1. Schutzenberger Р., Bull. Soc. Chim. France, 14, 97 (1870).
2. Schutzenberger P., C. R. Acad. Sci. Paris, 70, 1134 (1870).
3. Schutzenberger P., J. Prakt. Chem., 112, 159 (1871).
4. Belli Dell’Amico D., Calderazzo F„ Dell’Amico G., Gazz. Chim. Ital., 107, 101 П977),
5. Cclderazzo F., частное сообщение.
Комплексы с галогеновыми лигандами 2075
□ □ Хлоро(карбонил)золото Аи(СО)С1
Аи(СО)С1 лучше всего получать из Аи2С16 карбонилированием при нормальном давлении. В свою очередь Аи2С16 легко получается из НАиС14-хН2О при действии на него SOC12 (см. т. 4, гл. 18).
а) 2НАпС14 • ЗН2О + 6SOC12 -> Au2C16 + 6SO2 + 14НС1
2-393,8 6-119,0 606,7 6-22,4л 14-36,5
б) Au2Cl6 + 4СО -----> 2Au(CO)Cl + 2СОС12
606,7 4-22,4 л 2-260,4 2-98,9
При комнатной температуре, в атмосфере N2, в двугорлой колбе на 250 мл перемешивают в 60 мл сульфинилхлорида суспензию 10,0 г (25,4 ммоль) крупного порошка НАиС14-ЗН2О (50% Au, Degussa). Постепенно красно-оранжевая суспензия НАиС14-ЗН2О превращается в желтую или желто-красную суспензию Аи2С16. Через 15 ч реакционную смесь с помощью газовводиой трубки насыщают СО и перемешивают еще 15 ч. Для карбонилирования Аи2С16 достаточно примерно через каждый час в течение нескольких минут пропускать через суспензию слабый ток СО. В процессе реакции суспензия Аи2С16 растворяется и начинает выпадать белый блестящий осадок продукта карбонилирования. По окончании добавляют ~100 мл сухого петролейного эфира, выпавший осадок отфильтровывают в атмосфере СО (стеклянный пористый фильтр G3), хорошо промывают петролейным эфиром и сушат ~15 мин в вакууме масляного насоса. Выход 5,2 г (79%).
Свойства. Бесцветное, не очень устойчивое на воздухе и крайне легко гидролизующееся кристаллическое вещество. Хранят его в атмосфере сухого СО. В присутствии пиридина происходит быстрое разложение с выделением СО. В тетрагидрофураие также постепенно разлагается. В SOC12, СС14 и бензоле соединение устойчиво. ИК (СС14): 2152 [v(CO)] см-1.
ЛИТЕРАТУРА
1. Belli Dell’Amico D„ Calderazzo F., Gazz. Chim. I tai., 103, 1099 (1973).
2. Manchot W., Gall H„ Ber., 58, 2175 (1925).
3. Kharasch M. S„ Isbell H. S„ J. Amer. Chem. Soc., 52, 2919 (1930).
Гидридные комплексы
□ □ Гидридогексакис(трифторфосфин)ванадий V(PF3)6H
Путем фотоиндуцированного обмена СО на PF3 в [Na(C6H,4O3)2] [V(CO)6] образуется [Ка(СбН14О3)2}[У(РР3)б], из которого при действии кислот получают гидридный комплекс V(PF3)6H.
а) 2РС13 + 3ZnF2 -----> 2PF3 + 3ZnCl2
2-137,3 3-103,4 2-22,4л 2-136,3
hv
б) [Na(C6H14O3)2] [V(CO)g] + 6PF3 --> [Na(C6H14O3)2l [V(PF3)e] + 6CO
510,3 6-22,4л 870,1 6-22,4л
в) [Na(C6H14O3)2] [V(PF3)6] + H3PO4 -> V(PF3)6H + NaH2PO4 + 2C6H14O8
870,1 98,0 579,8 120,0 2-134,2
а) В двухлитровый нагреваемый автоклав из легированной стали загружают 1,5 кг (14,5 моль) ZnF2 (частицы меньше чем 0,063 мм), предварнтель-
2076 Глава 33. Металлоорганические комплексы
но основательно высушенного при 200 °C. Автоклав вакуумируют, затем засасывают в него при комнатной температуре 450 мл (5,2 моль) РС13. После кратковременного подогрева начинается сильно экзотермическая реакция. Смесь Выдерживают 4 ч при 130 °C. После охлаждения автоклава образовавшийся PF3 переконденсируют в трубку Шленка, охлаждаемую жидким азотом. Для очистки газ многократно пропускают через большое количество воды, чтобы гидролизовать непрореагнровавший РС13, а также не полностью фторированные продукты. PF3 затем для очистки от следов HF продувают через три-н-бутиламин. Окончательную осушку проводят над конц. H2SO4, P4Oi0 и СаН2 и собирают в промежуточный автоклав. Выход —200 г (44%, считая на РС1з).
б) Обмен СО на PF3 в [Na(C6Hi4O3)2]i[V(CO)6] проводят в аппаратуре (из стекла Solidex) для низкотемпературного фотолиза (рис. 477), в которой возможна внешнее облучение раствора при низких температурах.
Рис. 477. Аппаратура для проведения фотохимических реакций при низкой температуре.
Z — НШ 45; 2 — НШ 14,5; 3 — УФ-лампа; 4 — магнит; 5 — реакционный раствор.
В аппаратуре для фотолиза растворяют 7,0 г (13,7 ммоль) [Na (С3Н14О3)2]-rVlCOlnl в 150 мл диглима [бис(2-метоксиэтиловыи) эфир C6Hi4O3J, охлаждают до —45 °C и насыщают PF3. При интенсивном перемешивании в течение 24 ч облучают ртутной лампой высокого давления (Q 600, Hanau). В течение этого времени несколько раз реакционный раствор дополнительно насыщают PF3. Растворитель отгоняют в высоком вакууме при комнатной температуре, а остаток дважды промывают пентаном. Полученный таким путем немного окрашенный в серый цвет [Na (C6HI4O3) 2] [V(PF3h]| очишатй£°7Лрн^п« творением в диглиме и высаживанием пентаном. ИК (КВг). 867 (с.), вво (оч. с) 778 (с.) [v(PF3)] см-1. ЯМР-19Р (пентан; CFC13 — внешн. эталон): б 10,22, /(PF) = 1245 Гц.
Гидридные комплексы 2077
в) Для получения гидридного комплекса 5,0 г (5,70 ммоль) [Па(СбН14О3)2]-JV(PF3)6] вместе с 50 г (510 ммоль) кристаллической Н3РО4 помещают в колбу, соединенную через шлнф с охлаждаемой ловушкой, и нагревают до 60 °C. Образующийся в этих условиях гидриднын комплекс перекондеисируют при 10 3 мм рт. ст. в охлаждаемую до —196 °C ловушку, а затем для осушки еще раз возгоняют над Р4Ою при 50°С (10~2 мм рт. ст.). Выход 2,26 г (68%).
Свойства. Светло-желтое кристаллическое, даже при комнатной температуре заметно летучее вещество, /пл 135 °C (с разл.). Обладает типичным удушающим запахом, характерным для легколетучих РР3-производных. В атмосфере N2 при —20 °C хранится неограниченно долго, при комнатной температуре, однако, неустойчив, а иа свету разлагается в течение нескольких дней. Без каких-либо химических превращений растворяется только в насыщенных углеводородах. В кислород- или азотсодержащих донорных растворителях происходит образование ониевых солей. Это указывает на сильный кислотный характер V(PF3)eH. ИК (газ): 911 (оч. с.), 846 (с.) [v(PF3)] см"1.
ЛИТЕРАТУРА
1. Kruck Т„ Hempel H.-U., Angew. Chem., 86, 233 (1974).
2. Kruck T., частное сообщение.
□□ Тригидридобис(т|-циклопентадиенил)ниобий Nb(T]-C5H5)2H3
Получение целесообразно проводить восстановлением Nb(r]-C5H5)2C12 при действии ЫА1Н4. Сначала образуется алюминатный комплекс предположительно состава Nb(AlH4) (т]-С5Н5)2, который затем гидролизуется, давая тригид-ридный комплекс.
a) LiAlHd
б) Н2О
Nb(T]-C5H5)2Cl2 -----> Nb(T]-C5H6)2H8
294,0 226,1
В колбе на 1 л суспендируют 10,0 г (34,0 ммоль) Nb(f]-C5H5)2C12 в — 200 мл тетрагидрофурана. При охлаждении льдом и интенсивном перемешивании через воронку со шлифом прибавляют 4—5 г ЫА1Н4 (избыток), предварительно насыщенного N2- Наблюдается вспенивание и образование интенсивно окрашенного оранжевого раствора. Через 4—6 ч перемешивания при комнатной температуре образуется серый или серо-коричневый осадок, смесь Nb(T]-C5HB)2H3 и «Nb(AlH4) (т]-С5Н5)2». При 25—30 °C (температура баии) в вакууме масляного насоса полностью отгоняют растворитель, а остаток в несколько приемов извлекают 300—400 мл бензола. Объединенный экстракт фильтруют через вату на стеклянном пористом фильтре (G3). Вату предварительно тоже выдерживают в атмосфере N2, иначе произойдет синее окрашивание фильтрата из-за следов О2.
Темно-оранжевый или красный фильтрат осторожно гидролизуют 7— 10 мл воды, абсолютно исключая даже следы О2. Раствор светлеет, выделяется белый осадок, а образовавшийся Nb(r]-C5HS)2H3 полностью растворяется в бензоле. После высушивания безводным Na2SO4 бензольный раствор фильтруют через слой Na2SO4 на фильтре (—15 г). После удаления растворителя в вакууме масляного насоса белый осадок сушат в высоком вакууме. Полученный таким образом препарат является аналитически чистым, и его тотчас используют для дальнейших превращений. Выход 50—80%.
В качестве восстановителей вместо LiAlH4 можно применять другие алю-могидриды или борогидриды [1].
2078 Глава 33. Металлоорганические комплексы
Свойства. Белый, иногда слабо окрашенный в фиолетовый цвет порошок» неустойчивый на свету и на воздухе. Рекомендуется хранить при —35 °C в отсутствие О2 и Н2О. ИК (КВг): 1710 [vNbH)] см->. flMP-'H (CeD6, ТМС): 6 —2,6, —3,5 {шир., NbH]; 4,83 [С6Н8]. В кипящем бензоле в атмосфере СО (1 бар) легко образует Nb(CO) (т]-С8Н5)2Н.
ЛИТЕРАТУРА
1. Tebbe F. N., Parshall G. W., J. Amer. Chem. Soc., 93, 3793 (1971).
2. Labinger J. A., Advan. Chem. Ser. (Amer. Chem. Soc.), 167, 149 (1978).
3. Labinger J. A„ Wong K- S„ J. Organometal. Chem., 170, 373 (1979).
4. Herrmann W. A., Biersack H., неопубликованные результаты 1979 г.
□□ Гидридо(^-циклопентадиенил)три1карбонилхром,
-молибден и-вольфрам М(СО)з(т]-С5Н5)Н (M=Cr, Mo, W)
Способ 1. Двухстадийный синтез из гексакарбонилов применим для получения всех соединений этого типа:
а) М(СО)6 + NaC3H3 -------> №[М(СО)3(т]-С6Н8)] + ЗСО
М = Сг: 220,1 88,1 224,1 3-22,4л
М = Мо: 264,0 268,1
M=W: 351,9 356,0
б) NalMCCO^-CjHgyi + CHsCOOH----> М(СО)3('П-С5Н8)Н + NaICH3CO2)
М=Сг: 224,1 60,1 202,1 82,0
М=Мо: 268,1 246,1
M=W: 356,0 334,0
а) 11,0 г Сг(СО)6, 13,2 г Мо(СО)6 или 17,6 г W(CO)3 (50 ммоль) обрабатывают в 100 мл ЬГ,Я-диметилформамида при 130 °C 6,2 г (70 ммоль> NaC8H5 (см. выше методику синтеза М(СО)3(т]-С8Н8)2С1, где М=Мо, W). При нагревании первоначально желтого раствора до температуры реакции [~75°С для Мо(СО)6 или ПО °C для Сг(СО)6 и для W(CO)3] быстро начинается выделение СО. В процессе реакции выделяется примерно 3,1 л СО (0,14 моль при нормальных условиях); раствор одновременно окрашивается в светло- или темно-корнчневый цвет. Образовавшиеся комплексные анионы весьма неустойчивы на воздухе.
б) Через 2 ч растворитель отгоняют в высоком вакууме при 50—80 °C. Темио-коричневый маслянистый осадок охлаждают на ледяной бане, растворяют его в 50 мл дистиллированной воды и осторожно подкисляют 40 мл 50%-ной СНзСООН. Тотчас выпадающий интенсивно-желтый осадок отделяют иа покрытом кусочками фильтровальной бумаги стеклянном пористом фильтре G4 и промывают дистиллированной Н2О (2X30 мл). Сырой продукт сушат не менее 10 ч в вакууме масляного насоса при комнатной температуре (защищать от света!) и очищают высоковакуумной возгонкой. Для уменьшения разложения строго придерживаются следующих температур возгонки: 50— 55 °C для Сг(СО)3(т]-С8Н8)Н; 45—48 °C для Мо(СО)3(т]-С8Н8)Н; 60—67 °C для W(CO)3(t)-C3H5)H. Остатки после возгонки обычно пирофорны, и поэтому их осторожно уничтожают в атмосфере N2 обработкой водой. Выходы- 5 0—6 1 г (49-60%) Сг(СО)3(т]-С8Н8)Н; 9,6-10,1 г (78-82%) Мо(СО)3(п’с8Н5)Н; 14,4—15,0 г (86—90%) W(CO)3(ti-C5H5)H. Комплекс хрома чаще всего с поверхности слегка окрашен в сине-зеленый цвет; непосредственно перед употреб-лением его снова возгоняют.
Гидридные комплексы. 2079
Способ 2. Альтернативным способом получения Сг(СО)3(т]-С5Н5)Н является его синтез из Сг(т]-С5Н5)2 под давлением:
Сг(т]-С6Н&)2+ЗСО + 1/2Н2------> Сг(СО)3(П-С5Н5)Н+[С5Н5-]
182,2 3-22,4л 11,2л 202,1
Тщательно оберегая от попадания воздуха, в 250-мл вращающийся автоклав помещают 18,2 г (0,1 ммоль) Сг(т]-СбН5)2 и закрывают. Систему продувают дважды СО (50 бар), доводят давление необходимого для реакции Нг до 60 бар и затем наполняют СО до 180 бар. Через 20 ч реакции при 70±5°С избыток газа сжигают. Продукт реакции быстро извлекают 100 мл 3 н. NaOH, фильтруют через стеклянный пористый фильтр G3, покрытый ватой, и, охлаждая ледяной водой, высаживают из желтого фильтрата при добавлении 10 мл ледяной СНзСООН. Желтый объемистый осадок отделяют (стеклянный пористый фильтр G3, покрыт ватой), промывают 10—20 мл дистиллированной Н2О для удаления следов СНзСООН, сушат в течение ночи в вакууме масляного насоса и, наконец, возгоняют в высоком вакууме при 50—55 °C. Выход 7,3—9,1 г (35—45%).
Свойства. Светло- илн темно-желтые кристаллические соединения; в атмосфере N2 при —35 °C хранятся неограниченно долго. Светочувствительность комплексных гидридов увеличивается в ряду Cr<Mo<W, так что целесообразно, в особенности для производного W, проводить возгонку, защищая продукт от света; чувствительность же к кислороду воздуха изменяется в обратной последовательности; W(GO)3(r]-C5H5)H в течение некоторого времени устойчив на воздухе, а Сг(СО3) (т]-С5Н5)Н при контакте с О2 тотчас окрашивается в сине-зеленый цвет, иногда наблюдается самовозгорание. /разл: Сг(СО)3(ц-<5Н5)Н 57—58 °C; Мо(СО)з(г]-СвН5)Н 50—52 °C; Ш(СО)3(т]-С5Н5)Н /пл 67— 68 °C. Спектральные данные см. табл. 48. 1 2 3 4 5
Таблица 48. Данные ИК- и ЯМР-'Н-спектров гидридных комплексов М(СО)3(т]-С5Н5)Н
и к ЯМР-'Н (CeD,,; ТМС)
м v(CO), cm-1 -v(MH), см—1 «С5НБ вмн
Cr (циклогексан) 2018 (оч. c.), 1934 (c.), 1908 (оч. c.), 1888 (cp.) 4,78 —5,46
Mo (CS2) 2027 (оч. c.), 2020 (пл.), 1940 (оч. с.), 1904 (ср.) 1720 5,30 —5,42
W (циклогексан) 2026 (оч. с.), 2016 (пл.), 1935 (оч. с.), 1900 (пл.) 1845 5,35 •—7 33 [J(WH)=36 Гц]
ЛИТЕРАТУРА
1. Fischer Е. О., Hafner W., Stahl Н. О., Z. Anorg. Allgem. Chem., 282, 47 (1955).
2. Piper T. S„ Wilkinson G„ J. Inorg. Nucl. Chem., 3, 104 (1956).
3. Fischer E. O., Inorg. Synth., 7, 136 (1963).
4. Fischer E. O., Hafner W., Z. Naturforsch., 10b, 140 (1955).
5. Fischer E. O., Him Ber., 94, 2413 (1961).
2080 Глава 33. Металлоорганические комплексы
□ □ Дигидридобис(т]-циклопентадиенил)молибден Мо(т]-С5Н5)2Н2 □ □Дигидридобис(т]-циклопентадиенил) вольфрам Wfrj-CsHs^Hh
Мо(т]-С5Н5)2Н2 и W(t]-C5H5)2H2 образуются при взаимодействии соответствующих галогенидов металлов с NaC5H5, причем добавление NaBH4 существенно увеличивает выход продуктов. Уравнения реакций очень наглядны и убедительны, хотя детальное их изучение не проводилось:
MoClg -J- 3NaCsH6 -|- 2NaBH^ ->
273,5 3-88,1 2-37,8
---> Mofo-CelWig + 5NaCl + B2He + [CSHS-) 228,1 5-58,4 27,6
WC16 + 4NaC6H6 + 2NaBHt ------>
396,6 4-88,1 2-37,8
—> W(t]-C6H5)2H2 -f- 6NaCl 4- B2He + 21CSHS-} 316,1 6-58,4 27,6
Синтез осуществляют при соблюдении всех мер предосторожности для защиты от доступа влаги и воздуха в трех-горлой колбе емкостью 500 мл, снабженной обратным холодильником и эффективной мешалкой. К раствору 22,0 г (0,25 моль) NaC6Hs в 200 мл чистейшего тетрагидрофурана добавляют 4,20 г (0,11 моль) свежего NaBH+ и охлаждают затем смесью льда и соли. При интенсивном перемешивании прибавляют 11,7 г (43 ммоль) МоС15 (получение см. в т. 5), соответственно. 17,1 г (43 ммоль} WC16 (получение см. в т. 5) в 2—3 порции за время 5 мин. Реакционную смесь кипятят 5 ч с обратным холодильником и затем осторожно при 25 °C отгоняют растворитель в вакууме масляного насоса. Остающиеся масла отгоняют в высоком вакууме. Черно-коричневый сырой продукт очищают высоковакуумной возгонкой при 80—120 °C. Сублимат обычно загрязнен, поэтому необходима повторная возгонка при тех же условиях. Чистый продукт возгоняется в виде лимонно-желтых кристаллов, оседающих пучками на «пальце» сублиматора. Выход: 2,0—3,4 г (20—35%) Мо(т]-С5Н5)2Н2 или соответственно 2,4—4,9 г (18-36%) W(r]-C5H5)2H2.
Приведенная здесь методика имеет то преимущество, что дигидриды можно получать в несложной аппаратуре, однако выходы очень колеблются. Если необходимо получить большое количество продукта, то применяют методику Грина и Ноулса [3], принципиально не отличающуюся от вышеприведенной, однако требующую сложного аппаратурного оформления и больше времени (4 дня). Выходы постоянны н составляют 45% (30—45 г).
Другие способы. Соконденсация СвНв с парами металлических Мо илн W при IO-4—10-s мм рт. ст. (выход 30—60%) [4, 5]. Этот метод является учебным примером проведения синтезов с использованием паров металлов и при наличии соответствующей аппаратуры очень прост, изящен и позволяет получать оба соединения в количестве 2—3 г.
Свойства. Желтые кристаллические соединения. Комплекс Мо менее устойчив на воздухе по сравнению с W-комплексом. Тем не менее оба вещества, если они достаточно чистые, в течение короткого времени сохраняются на воздухе. В присутствии маслянистых загрязнений (т. е. препараты после первой возгонки) разрушение на воздухе идет значительно быстрее, /разл: 174 °C для Мо(т]-С6Н5)2Н2 и 194—196 °C для W(n-C5H5)2H2. Растворяются в бензоле, тетрагидрофуране и подобных растворителях. Оба комплекса в растворе кислот образуют ионы [М(т]-С5Н5)2Н3] + :
Гидридные комплексы 2081
+н+
М(л-СвНб)аН2 [М(т]-С6Нб)2Нз]+
Эту обратимую реакцию используют для очистки дигидридов.
Мо(п-С5Н5)2Н2. ИК (нуйол): 1847 [v(MoH)] см-1. ЯМР-'Н (C6D6)c б 4,55 [триплет, CsHs], J(C5H5, МоН)=0,86 Гц; б —9,12 [МоН]. Кристаллическая структура: см. [6].
№(т]-С5Н5)2Н2. ИК (нуйол): 1912 [v(WH)] см-1. ЯМР-'Н (C6D6): б 4,3» [триплет, С5Н5], /(С5Н5, WH)=0,70 Гц; 6—12,4 [WH],
ЛИТЕРАТУРА
1. Fischer Е. О., Hristidu У., Z. Naturforsch., 15b, 135 (1960).
2. Green М. L. Н„ McCleverty J. A., Pratt L., Wilkinson G., J. Chem. Soc. (London), 1961, 4854.
3. Green M. L. H., Knowles P. J., J. Chem. Soc. (London), A1973, 989.
4. VanDam M. E., Brent W. N., Silvon M. P., Skell P. S., J. Amer. Chem. Soc.,. 97, 465 (1975).
5. D’Aniello M. J. Jr., Barefield E. K, J. Organometall. Chem., 76, C50 (1974).. 6. Abrahams S. C., Ginsberg A. P„ Inorg. Chem., 5, 500 (1966).
□ □ Гидридопентакарбонилмарганец Mn(CO)5H
(Na-Hg)
Mn2(CO)10 + 2Na --------> 2Na[Mn(CO)5]
390,0 2-23,0 2-218,0
Na[Mn(CO)s] + H3PO4 ----> Mn(CO)5H + NaH2PO4
218,0 98,0 196,0 120,0
В 500-мл двугорлой колбе, снабженной капельной воронкой и обратным холодильником, готовят амальгаму натрия из 20,0 мл (272,0 г, 1,36 моль) Hg и 2,5 г (0,11 моль) Na (мелкими кусочками по 100 мг). После того как окончится экзотермическая стадия реакции, прикапывают раствор 13,0 г (33,3 ммоль) Мп2(СО)10 в 250 мл тетрагидрофурана. При этом первоначальножелтый цвет от растворенного Мп2(СО)ю переходит в серо-зеленый, вызываемый диспергированной ртутью. Сам комплексный анион [Мп(СО)5]_ бесцветен.
Через 3 ч перемешивания раствор осторожно сливают с амальгамы и упаривают в вакууме водоструйного насоса. Вязкий остаток сушат некоторое время в вакууме масляного насоса, вставляют капельную воронку (без выравнивания давления) и присоединяют к колбе систему для конденсации в высоком вакууме, состоящую из четырех охлаждаемых до —196 °C (жидкий N2) ловушек. Через капельную воронку медленно прикапывают 120 мл 85 %-ной НзРО4 (избыток). При этом образующийся Mn(CO)sH в высоком вакууме конденсируется в первой ловушке, содержащей Granusic*. Когда-кислоту полностью прикапают, колбу нагревают 30 мин до ~50°С, чтобы собрать в ловушку остатки Мп(СО)вН. Затем кран между колбой и первой-ловушкой перекрывают и переконденсируют летучий продукт последовательно в остальные ловушки, нагревая при этом их теплым воздухом из фена для полного вытеснения продукта. В результате последней переконденсации получают 10,8 г Мп(СО)5Н [83%, считая на Мп2(СО)ю]. Для предотвращения
* Granusic (фирма Baker Chemicals) представляет собой Р4О10, нанесенный на SiO2 или А12О3 для повышения эффективности осушки.
15—1361
2082 Глава 33. Металлоорганические комплексы
потерь вследствие частичного разложения продукт используют по возможности быстрее.
Свойства. Бесцветная, прозрачная, чрезвычайно неустойчивая на воздухе, легко подвижная жидкость. /пл —24,6 °C, /Кип 111 °C при 760 мм рт. ст. (экстраполировано). Из-за сильной летучести вещество очень ядовито. Обладает неприятным резким запахом. При доступе О2 тотчас превращается в Мп2(СО)ю+Н2О. С водой не реагирует. Растворяется во всех органических растворителях. В замороженном состоянии при абсолютном исключении света и О2 месяцами хранится без разложения. Кк=0,8-10~7 (Н2О). ИК (газовая фаза): 2118 (оч. сл.), 2016 (оч. с.), 2007 (с.), 1981 (сл.), 1967 (оч. сл.) [v(CO)]; (CS2): 1783 [v(MnH)] см-1. ЯМР-’Н (чистое-соединение): 6 —7,5 [синглет, МпН]. Кристаллическая структура: см. [3], С40-симметрия, d(Mn—Н) = 1,60 А.
ЛИТЕРАТУРА
1. Hieber W., Wagner G., Z. Naturforsch., 12b, 478 (1957).
2. Hieber W., Wagner G., Z. Naturforsch., 13b, 339 (1958).
3. LaPlaca S. J., Hamilton W. C., Ibers J. A., Davison A., Inorg. Chem., 8, 1928 (1969).
□ □ Трис[([х-гидридо)тетракарбонилмарганец]
(3Mn—Mn) Mn3(CO)12H3
Для получения трехъядерного карбонилгидрида декакарбонилдимарганец действием КОН переводят в зеленый, пока еще не полностью охарактеризованный, вероятно, трехъядерный карбонилманганат, из которого гидридный кластер выделяют подкислением:
а) КОН
б) Н3РО4
ЗМп2(СО)10 ------>• 2[Мп(СО)4Н]3 + побочные продукты
3-390 2-504,0
2,34 г (6,0 ммоль) Мп2(СО)ю заливают в колбе на 250 мл раствором 30 г (0,55 моль) КОН в 40 мл дистиллированной НгО и тотчас откачивают до начала испарения воды. Затем колбу закрывают и при интенсивном встряхивании выдерживают 3 ч при 60±3°С. Раствор становится изумрудио-зеленым, после охлаждения до комнатной температуры выпадают зеленые кристаллы. Их собирают на стеклянном пористом фильтре (G3) и быстро растворяют в 90 мл водного раствора 2 н. КОН. Раствор фильтруют, охлаждают льдом и при перемешивании подкисляют 15 мл 85%-ной Н3РО4. Окраска раствора постепенно через пурпурный цвет переходит в оранжево-красный. Пушистый порошкообразный осадок отфильтровывают, промывают водой до pH 7 и сушат в вакууме масляного насоса. Очистку проводят возгонкой в высоком вакууме сначала при 30 °C от небольших количеств непрореагировавшего Мп2(СО)ю, а затем при 60—70 °C. Выход 0,8—1,2 г (40—60%).
Свойства. Оранжево-красные, устойчивые на воздухе кристаллы. /разл 123—125 °C. Очень хорошо растворяется в таких полярных органических растворителях, как хлороформ и ацетон, хорошо — в бензоле, умеренно — в алифатических углеводородах. ИК (циклогексан): 2080 (ср.), 2034 (с.), 2008 (с.), 1986 (с.) [v(CO)] см-1. Кристаллическая структура: см. [5[.
Прежде рассматриваемый карбонилгидрид «Мп2(СО)9Н2» идентичен Мп3(СО)12Нз [4].
Гидридные комплексы 2083
ЛИТЕРАТУРА
1. Johnson В. F. G„ Johnston R. D„ Lewis J., Robinson В. H„ Chem. Commun., 1966, 851; J. Organometal. Chem., 10, 105 (1967).
2. Fischer E. O., Aumann R., J. Organometal. Chem., 8, P 1 (1967).
3. Smith J. Mehner K„ Kaesz H. D., J. Amer. Chem. Soc., 89, 1759 (1967).
4. Hieber W„ Beck W., Zeitler G„ Angew. Chem., 73, 364 (1961).
5. Kirtley S. W., Olsen J. P„ Bau R., J. Amer. Chem. Soc., 95, 4532 (1973).
□ □ Гидридобис(т)*циклопентадиенил)рений Ре(т]-С5Н5)2Н
2ReCl6 + 8NaCBHs + 2NaBHt---->
2-363,5 8-88,1 2-37,8
---> 2Re(r]-C5H5)2H + lONaCl + B2H6 + 4[C5H5'J
2-317,4 10-58,4 27,6
Раствор 17,6 г (0,2 моль) NaC5Hs в 150 мл тетрагидрофурана (двугорлая колба на 500 мл, магнитная мешалка и обратный холодильник) охлаждают на ледяной бане и добавляют 10,9 г (30 ммоль) ReCis. Смесь перемешивают 5 ч при 50±5°С и добавляют порциями 3,0 г (80 ммоль) NaBH4. Реакция заканчивается через 5 ч кипячения с обратным холодильником. Растворитель упаривают, остаток дважды возгоняют в высоком вакууме при 80—100 °C. Выходы сильно колеблются от 10 до 50%.
Та(т]-С5Н5)2Нз получают аналогично, выходы 5—20%.
Свойства. Желтое, неустойчивое на воздухе кристаллическое вещество, /пл 161—162 °C. Не растворяется в воде, хорошо растворяется во всех обычных органических растворителях. В водных растворах минеральных кислот образует катион [Re(r[-C5H5)2H2]+; при нейтрализации же этих растворов разбавленным NaOH снова получается Re(T]-C5HB)2H. Этот способ можно использовать для его очистки вместо вакуумной возгонки. ИК (CS2): 2030 [v(ReH)] см-1.
ЛИТЕРАТУРА
1. Green М. L. Н., Pratt L„ Wilkinson G., J. Chem. Soc. (London), 1958, 3916.
2. Green M. L. H., McCleverty J. A., Pratt L„ Wilkinson G., J. Chem. Soc. (London), 1961, 4854.
Хлоро (гидридо)трис(трифенил фосфин) карбонилрутений RuCI(H)(CO)[P(C6H5)3]3
нсно
RuCl3.3H2O + 3P(C6H5)3 -->- RuCl(H) (СО) [Р(С6Н5)3]3
261,5 3-262,3 952,4
К интенсивно перемешиваемому кипящему раствору 1,58 г (6 ммоль) Р(С6Н5)3 в 60 мл 2-метокснэтанола быстро прибавляют один за другим раствор 0,26 г (1 ммоль) RuCl3-2НгО в 20 мл 2-метоксиэтанола и 20 мл 40%-ного раствора формальдегида, кипятят 10 мин с обратным холодильником и охлаж-15*
084 Глава 33. Металлоорганические комплексы
дают. Выпавший продукт отфильтровывают, промывают 5 мл этанола, 5 мл Н2О, 5 мл этанола, гексаном (2x5 мл) и сушат в вакууме.
Свойства. Устойчивое на воздухе, мелкокристаллическое соединение кремового цвета. 209—210 °C на воздухе н 235—237 °C в запаянном под N2 капилляре. Плохо растворяется в бензоле и очень плохо — в СНС13 н СН2С12. ИК (нуйол): 2020 (ср.) [v(RuH)], 1922 (с.), 1903 (пл.) [v(CO)] cm~L
ЛИТЕРАТУРА
1. Chatt J., Shaw В. L„ Chem. Ind. (London), 1961, 290.
2. Vaska L., DiLuzio, J. Amer. Chem. Soc., 83, 1262 (1961).
3. Levison J. J., Robinson S. D., J. Chem. Soc. (London), A1970, 2947.
Хлоро(гидридо)трис(трифенилфосфин)карбонилосмий
Os(CO) [Р(С6Н5)з]3(Н)С1
HCHO
Na2[OsCl6]-6H2O + 3P(C6H5)3 -> Os(CO) [P(C6H5)3]3(H)C1
557,0 3-262,3 1041,6
Соединение получают по методике, аналогичной для его гомолога Ru(CO) [Р(С6Н5)з]з(Н)С1 (см. методику синтеза предыдущего препарата) из 0,67 г (1,2 ммоль) Na2[OsCl6]-6Н2О, 1,97 г (7,5 ммоль) Р(С6Н3)з и 15 мл 40%-ного формалина в 2-метоксиэтаноле. Выход 95%.
Свойства. Белые мелкие кристаллы. /пл 179—183 °C на воздухе илн соответственно 289—290 °C в капилляре под N2. Растворяется в бензоле, СНС13 и СН2С12. ИК (нуйол): 2099 (ср.) [v(OsH)], 1906 (оч. с.), 1891 (с.)
[v(CO)] см-1. ЯМР-'Н (бензол): 6 —6,27 [два триплета, OsH], J(РтрмсН) = = 87,0, 7(РЧ„СН)=24,5 Гц.
ЛИТЕРАТУРА
1. Vaska L„ J. Amer. Chem. Soc., 88, 4100 (1966).
2. Ahmad N., Robinson S. D., Uttley M. F., J. Chem. Soc. Dalton, 1972, 843.
Тетрагидридотрис(трифенилфосфин)осмий О8[Р(С6Н5)з]зН4
NaBH4
Na2[OsCle]-6H2O + 3P(CeHB)3 -> Os[P(C6H5)3]3H4
557,0 3-262,4 981,1
0,57 г (1 ммоль) Na2[OsCl6]-6H2O экстрагируют 20 мл холодного этанола. Профильтрованный экстракт и раствор 0,2 г NaBH4 в 20 мл этанола быстро один за другим вливают в интенсивно перемешиваемый кипящий раствор 1,57 г (6 ммоль) трифенилфосфина в 80 мл этанола. Наблюдается интенсивное вспенивание. Реакционный раствор, окрашенный в розовый цвет, кипятят 15 мин с обратным холодильником; при этом выпадает белый осадок. Его отфильтровывают, промывают этанолом (5 мл), водой (5 мл), этанолом (5 мл) и гексаном (2X5 мл) и сушат в вакууме. Выход 0,95 г (97%).
Свойства. Микрокристаллы кремового цвета. /пл 172—175 °C иа воздухе или 219—221 °C в запаянном под N2 капилляре. Плохо растворяется в бензоле, хлороформе и СН2С12. ИК (нуйол): 2086 (сл..), 2025 (ср.), 1951 (ср.), 1891 (с.) [v(OsH)] см-1. ЯМР-‘Н (бензол): 6 —7,85 [1:3:3:1-квартет, OsH], 2J(PH) = =9 Гц.
Гидридные комплексы 2085
ЛИТЕРАТУРА
1. Leigh G. Levison J. J., Robinson S. D„ J. Chem. Soc. Chem. Commun., 1969, 705; J. Chem. Soc. (London), A1970, 2947.
Гидридотетракис(трифени1лфосфит)кобальт Co[P(OC6H5)3]4H
NaBH4
Co(NO3)2-6H2O+ 4P(OCeH5)3 ----> Co[P(OC6H5)3]4H
291,0 4-310,3 1301,1
В атмосфере N2 в раствор 2,91 г (0,01 моль) Со(ПОз)2ф6Н2О в 60 мл этанола добавляют 15,5 г (0,05 моль) трифенилфосфита. К этой смеси при интенсивном перемешивании прибавляют в течение ~10 мин по каплям раствор NaBH4. Последний готовят растворением 1,0 г NaBH4 в 25 мл теплого этанола и последующим быстрым охлаждением до комнатной температуры. Фиолетовый цвет Со(II)-иона заметно бледнеет, выделяется бледно-желтое твердое вещество. Перемешивают еще 10 мин, отфильтровывают в атмосфере N2, промывают этанолом (3X10 мл) и сушат в вакууме. Выход 11,0 г (85%). Для очистки продукт растворяют в минимальном количестве бензола и высаживают добавлением по каплям метанола.
Свойства. Кристаллизуется в виде бледно-желтых гексагональных листочков, которые на воздухе постепенно изменяются. Л,л 165—168 °C (в запаянном под N2 капилляре). Растворяется в бензоле, СНС13 и СН2С12; практически не растворяется в алифатических углеводородах. ИК: нет поглощения v(CoH)! ЯМР-:Н (бензол, ТМС): 6 —13,58 [уширенный 1:4:6:4:1-квинтет, СоН], 27(РН) = 17 Гц; ЯМР-31Р (бензол, Р4О6): 6 —28,2.
ЛИТЕРАТУРА
1. Levison J. J., Robinson S. D., J. Chem. Soc. Chem. Commun., 1968, 1405; J. Chem. Soc. (London), Al970, 96.
□ □ Гексафторофосфат гидридо(т]-циклопентадиенил)-бис(триметилфосфин)кобальта(1П) '
[Co{P(CH3)3}2(n-C5H5)(H)][PF6]
Со[Р(СН3)3]2(т]-С5Н5) + NH4[PF6] ->
276,2 163,0
----* [Со{Р(СН3)3}2(т]-С5Н5) (H)] [PFJ + NH3 422,2 22,4 л
В трубке Шленка при перемешивании магнитной мешалкой растворяют в нескольких каплях ацетона 1,04 г (3,8 ммоль) Со[Р(СН3)з]2(т]-С5Н5). Затем при охлаждении на ледяной бане н перемешивании прибавляют раствор 1,0 г (6,1 ммоль) NH4[PF6] в небольшом количестве метанола. При этом из первоначально коричневого раствора выпадает желтый осадок. Для завершения высаживания добавляют 50 мл Н2О. Осадок отделяют, многократно промывают водой и высушивают. Затем растворяют его в минимальном количестве нитрометана, желто-коричневый раствор фильтруют и до тех пор добавляют к фильтрату эфир (50—100 мл), пока полностью не высадят желтый продукт реакции. Раствор остается практически бесцветным. После декантации, промывания эфиром и высушивания получают аналитически чистый продукт. Выход 1,5 г (93%).
2086 Глава 33. Металлоорганические комплексы
Свойства. Желтое, выдерживающее кратковременную экспозицию на воз>-духе соединение, хорошо растворяется в диметилсульфоксиде и нитрометане», умеренно — в спиртах и ацетоне. В воде, алифатических и ароматических углеводородах и эфире не растворяется. ИК (ацетон): 1930 [v(CoH)] см-1. ЯМР-'Н (диметил су льфоксид-Ф6, ТМС): S 1,50 [«триплет», СН3]: 5,13 [триплет». C6HS], 2Z (PH) =0,8 Гц; 6 —16,20 [триплет, СоН], 27(РН)=83;0 Гц.
ЛИТЕРАТУРА
1. Werner Н., Hofmann W., Вег., 110, 3481 (1977).
Гидридотрис(трифенилфосфин)карбонилродий
Rh(CO) [Р(С6Н5)3]зН
кон—нсно
RhCl3-3H2O + ЗР(СвН6)3 -------->• Rh(CO) [P(CeHs)3]3H
263,3 3-262,3 918,8
Раствор 0,26 г (1 ммоль) RhCl3-3H2O в 20 мл этанола в атмосфере N» приливают к интенсивно перемешиваемому кипящему раствору 2,64 г (10 ммоль) трифенилфосфина в 100 мл этанола. Примерно через 15 с быстро добавляют 10 мл 40%-ного водного раствора формальдегида, а также раствор 0,8 г КОН в 20 мл горячего этанола. Смесь кипятят 10 мин с обратным холодильником и охлаждают. Выпавшие кристаллы интенсивно-желтого цвета отделяют, промывают этанолом, водой и снова этанолом (по 5 мл), затем гексаном (2x5 мл) и сушат в вакууме. Выход 0,85 г (93%).
Реакция хорошо протекает и с большими загрузками. Если исключают из реакции КОН, то выпадает желтый Rh(CO) [Р(С6Н5)з]2С1. Действием избытка этанольного раствора NaBH4 его переводят в Rh(CO) [Р(СзН5)з]зН. О завершении этой реакции судят по исчезновению ИК-поглощения при 1960 см-', характерного для v(CO) в Rh(CO)[P(C6HS)3]2C1. В противном случае еще добавляют раствор NaBH4. Выход до 97% [3, 4].
Свойства. Устойчивое на воздухе, мелкокристаллическое соединение желтого цвета. <ял 120—122 °C на воздухе или 172—174 °C в запаянном под N2 капилляре. Растворяется в бензоле, СНС13 и СН2С12. ИК (нуйол): 2041 (с.) [v(RhH)], 1918 (оч. с.) [v(CO)] см-'. Молекулярная структура: моноклинная, пространственная группа симметрии 2Р]/п (а= 10,11 А, 6=33,31 А, с= 13,33 А, р=90°), тригонально-бипирамидальная молекула с Н и СО в аксиальном положении [5].
ЛИТЕРАТУРА
1. Bath S. S., Vaska L., J. Amer. Chem. Soc., 85, 3500 (1963).
2. Ahmad N.. Robinson S. D„ Uttley M. F„ J. Chem. Soc. Dalton, 1972, 843.
3. Strohmeier W., Hitzel E., Michel M., Stasch H.-Р., Weigelt L., частное сообщение.
4. Hallman P. S., Evans D., Osborn J. A., Yagupsky G., Wilkinson G„ J. Chem. Soc. Chem. Commun., 1967, 305; J. Chem. Soc. (London), A1968, 2660.
5. LaPlaca S. J., Ibers J. A., Acta Cryst., 18, 511 (1965).
Нитрозильные комплексы 2087
транс-Хлоро (гидриде) бис (трифенилфосфин) платина Pt[P(C6H5)3]2(H)Cl
2Pt[P(CeH6)3]2Cl2 + N2H4-H2O ->
2-790,6 50,1
----> 2Pt[P(C6H8)3]2(H)Cl + N2 + 2HC1 + H2O 2-756,1 22,4 л 2-36,5 18,0
В двугорлой колбе с обратным холодильником растворяют 2,0 г (2,53 ммоль) Pt [Р (С6Н5) 3] 2CI2 (получение см. ниже в этой главе) в смеси 50 мл этанола и 1,26 г 85%-ного гидразингидрата. Нагревают 5 мин на водяной бане до кипения и добавляют последовательно 1,2 г ледяной СН3СООН, 20 мл Н2О и 10 мл этанола. Снова нагревают до кипения и охлаждают до комнатной температуры. Бесцветные кристаллы отделяют, промывают СН3ОН и сушат в вакууме. Продукт является 1:1-аддуктом с этанолом. Выход: 1,3 г (68%).
Растиранием кристаллов и высушиванием в высоком вакууме (1 ч, 80 °C) получают комплекс, не содержащий этанола.
Другой способ. Окислительное присоединение НС1 к Р1[Р(СбН8)з]4 или Pt[P(C6H8)3]3 [3].
Свойства. Бесцветные микрокристаллы. Л,л 210—215°C (с разл.). Растворяется в бензоле, ацетоне, СНС1з, не растворяется в этаноле. ИК (нуйол): 2220 [v(PtH)], 274 [v(PtCl)] см-1; КР (тв.): 545±5 (с.) см~‘ [транс [4]]. ЯМР-31Р (СНС1з, 85% Н3РО4): 6 —28,3, V(PtP)=3001 Гц.
ЛИТЕРАТУРА
1. Chatt J., Shaw В. L., J. Chem. Soc. (London), 1962, 5075.
2. Bailar J. C., Jr., Itaiani H., Inorg. Chem., 4, 1618 (1965).
3. Cariati F., Ugo R., Bonati F., Inorg. Chem., 5, 1128 (1966).
4. MastinS. H„ Inorg. Chem., 13, 1003 (1974).
Нитрозильные комплексы
□□ Тетранитрозилхром Cr(NO)4
Cr(CO)6 4- 4NO ---> Cr(NO)4 + 6CO
220,1 4-22,4л 172,0 6-22,4л
В трубке Шленка емкостью 500 мл, снабженной трубкой для ввода газа, растворяют при перемешивании 440 мг (2 ммоль) Сг(СО)8 в 300 мл пентана и, пропуская NO, до тех пор облучают ртутной лампой высокого давления (Напо-via L-450W), пока в ИК-спектре не исчезнут полосы поглощения Сг(СО)8 (~2 ч). Растворитель пентан тщательно высушивают (лучше всего над сплавом K-Na); NO для очистки и отделения от NO2 пропускают через КОН и Р4О10.
Интенсивно окрашенный в красио-коричневый цвет реакционный раствор фильтруют в атмосфере N2, большую часть растворителя отгоняют сначала при обычном давлении. Из раствора, сконцентрированного до объема примерно 30 мл, остатки растворителя удаляют в высоком вакууме при —78 °C. Очень летучий Cr(NO)4 выделяют в виде черно-коричневого твердого соединения и очищают возгонкой при низкой температуре. Выход 175 мг (51%).
2088 Глава 33. Металлоорганические комплексы
Свойства. Черно-коричневое, очень летучее, неустойчивое на воздухе вещество. Л™ 38—39 °C (в запаянном под N2 капилляре). В некоординирующих-ся растворителях растворяется без разложения. ИК (пентан): 1721 [v(NO)] см-1.
ЛИТЕРАТУРА
1. Herberhold М., Razavi A., Angew. Chem., 84, 1150 (1972).
2. Swanson В. I., Satija S. К., J. Chem. Soc. Chem. Commun., 1973, 40; Inorg. Synth., 16, 1 (1976).
□ □ Гексафторофосфат нитрозилпентакис(ацетонитрил)-хрома(П) [Cr(NCCH3)5(NO)][PF6]2
Cr(CO)6 -f- 2NO[PF6] + 5CH3CN ->
220,1 2-175,0 5-41,1
---> [Cr(NCCH3)6(NO)] [PF6]2 + 6CO + NO
577,2 6-22,4 л 22,4 л
К перемешиваемому раствору или суспензии 550 мг (2,5 ммоль) Сг(СО)» в 20 мл ацетонитрила постепенно прибавляют 1,83 г (10,5 ммоль) твердого NO[PFe], Интенсивно перемешивают еще 5 мин, затем фильтруют и к фильтрату при перемешивании медленно добавляют 50 мл эфира. Выпавший оранжевый осадок дважды перекристаллизовывают из смеси ацетонитрил — эфир. Выход 1,28 г (89%).
Свойства. Золотисто-желтое твердое вещество, очень медленно разлагающееся на воздухе, растворы же, наоборот, разлагаются очень быстро. Растворимо в ацетоне н ацетонитриле. ИК (нуйол): 2328, 2300 (ср.) Iv(CN)l, 1797 (с.) [v(NO)J см-1.
ЛИТЕРАТУРА
1. Connelly N. Q., Louttit Т. S., частное сообщение.
□ □ (т]-Циклопентадиенил)нитрози1Лкарбонил(т]2-циклооктен)-хром Сг(т]2-С8Н14)(СО) (NO)(т]-С5Н5)
Фотолиз CrfCOJsNO^-CsHj) в растворе циклооктена ведет к образованию л-олефинового комплекса Cr(t]2-C8Hi4) (СО) (NO) (г]-С5Н5), в котором циклоолефин легко замещается другими монодентатными лигандами (например, такими, как моноолефин, алкин, фосфин). Поэтому указанное соединение используется как легкодоступный исходный комплекс для замещения СО на другие лиганды, в особенности когда их прямое введение в Сг(СО)^О(грС5Н5), принципиально возможное лишь фотохимически, не удается.
hv
Cr(CO)2NO(n-CsHs) + С8Н14 ---> Сг(п2-С8Н14) (СО) (NO) (П-С6Н6) + СО
203,1 110,2 285,3 22,4л
В трубку Шленка, изготовленную из стекла дюран, помещают 2,03 г (10 ммоль) Cr(CO)2NO(T|-C5H5) в 400 мл циклооктена, предварительно насыщенного азотом, и при интенсивном перемешивании в течение 10 ч облучают ртутной лампой высокого давления (Hanovia S, 200 Вт). Затем циклооктен
Нитрозильные комплексы
2089
удаляют в высоком вакууме при комнатной температуре. Оставшееся коричневое масло извлекают бензолом и пропускают через слой кизельгеля (10 см) на стеклянном пористом фильтре G3. Бензолом элюируют „желто-коричневый раствор, растворитель отгоняют досуха, твердый коричневый остаток извлекают пентаном ( — 100 мл), фильтруют его через стеклянный пористый фильтр G3, покрытый кусочками фильтровальной бумаги, растворитель упаривают до объема ~30 мл и охлаждают до —20 °C. Выпадают красно-коричневые кристаллы комплекса Сг(т]2-С8Н14) (СО) (NO) (т|-С5Н5). Выход 2,28 г (80%).
Из пентанового раствора возвращают около 10% непрореагировавшего <r(CO)2NO(r]-C5H5). С увеличением концентрации облучаемого раствора выход продукта снижается.
Свойства. Красно-коричневые, не очень устойчивые на воздухе кристаллы, /пл 75 °C. Хорошо растворяется в бензоле, метилендихлориде, ацетоне. ИК (СН2С12): 1957 [v(CO)], 1646 [v(NO)] см-1. ЯМР-'Н (ацетон-а!в, ТМС): 6 5,07 [синглет, С5Н5].
При пропускании этилена в бензольный раствор в течение 3 ч с выходом 75% образуется продукт замещения Сг(С2Н4) (СО) (р-С5Н5). Аналогичные реакции известны и для С2Н2 и его гомологов.
ЛИТЕРАТУРА
1. Herberhold М., Alt Н., J. Organometal. Chem., 42, 407 (1972).
2. Herberhold М., Alt Н., Justus Liebigs Ann. Chem., 1976, 292.
3. Herberhold M., Alt H., Kreiter C. G., Justus Liebigs Ann. Chem., 1976, 300.
□□ (т!-Циклопентадиенил)(тионитрозил)дикарбонилхром
Cr(CO)2NS(n-C5H5)
Комплекс Cr(CO)2NS(p-C5H5) образуется в результате обмена лиганда СО на NS при действии S3N3CI3 на Na;[Cr(СО)3(П-С5Н5)]
3Na[Cr(CO)3(n-C5H5)] + S3N3C13 -> 3Cr(CO)2NS(p-C5H5) + ЗСО + 3NaCl
3-224,1 244,6 3-219,2 3-22,4л 3-58,4
Аддукт Ма[Сг(СО)з(т]-С5Н5)] получают in situ из Cr(CO)e и NaC5H5 (получение см. выше в этой главе) и используют сразу после получения. Выход его при тщательном соблюдении необходимых условий синтеза практически количественный. В 300 мл тетрагидрофурана растворяют 13,5 г (60 ммоль) комплексной соли. К охлажденному до —78 °C раствору при интенсивном перемешивании прикапывают раствор 14,2 г (58 ммоль) S3N3CI3 (получение см. в [3]) в 120 мл тетрагидрофурана. Происходит выделение газа, и одновременно выпадает осадок, цвет раствора постепенно углубляется от темножелтого до красно-коричневого. По окончании прибавления тионитрозирую-щего реагента смесь перемешивают еще 1 ч при —78 °C, а затем медленно нагревают до комнатной температуры. Остаток, образующийся после отгонки растворителя в вакууме водоструйного насоса, в несколько порций экстрагируют — 600 мл петролейного эфира (/кип 40—60°C). Объединенный экстракт фильтруют (стеклянный пористый фильтр G3, покрытый листом фильтровальной бумаги) и растворитель отгоняют досуха в вакууме водоструйного насоса. Очистку полученного продукта проводят высоковакуумной возгонкой при 35—40 °C. Выход —2,6 г ( — 20%). Методом колоночной хроматографии из сырого продукта дополнительно извлекают p-S[Cr(CO)2(p-C5H5)]2 с выходом 34%.
Свойства. Хорошо кристаллизующееся вещество интенсивного краснофиолетового цвета. /м 68—69 °C. В кристаллическом состоянии слабо чувстви-
2090 Глава 33. Металлоорганические комплексы
тельно к действию кислорода воздуха. Растворы в органических растворителях (петролейном эфире, эфире, бензоле, СН2С12, ацетоне) при доступе воздуха разлагаются за несколько часов. ИК (гексан): 2033 (с.), 1962 (с.) [v(CO)]; 1180 [v(NS)] см-1. ЯМР-'Н (CDC13, ТМС): б(С8Н6) 5,08. ЯМР-^С (CDC13, ТМС): б(С8Н5) 92,75, 6(СО) 239,43.
ЛИТЕРАТУРА
1. Kolthammer В. W. S., Legzdins Р.. J. Amer. Chem. Soc., 100, 2247 (1978).
2. Greenhough T. J., Kolthammer В. W. S., Legzdins P., Trotter J., Inorg. Chem., 18, 3543 (1979).
3. Jolly W. L., Maguire K. D., Inorg. Synth., 9, 102 (1967).
□ □ (т|-Циклопентадиенил) (нитрозил)дикарбонилмолибден
Mo(CO)2NO(t]-C5H5)
Синтез осуществляют последовательно в три стадии без выделения промежуточных соединений.
a) Mo(CO)e + NaC8H8----> Ма[Мо(СО)3(я-С8Н8)] + ЗСО
264,0 88,1 268,1 3-22,4 л
б) Na[Mo(CO)3(n-C6Hs)] + CHjCOOH -->
268,1 60,1
----> Мо(СО)3(11-С5Н5)Н + Na[CH3COJ 246,1 82,0
в) Мо(СО)3(п-С6Н6)Н + CH3(NO)N-SO2-C^-n-CHj ----->
246,1 214,2
----> Mo(COi2NO(r|-C3H8) + СО + CH3NH—SO2—С6Н4-п-СНэ 247,0 22,4л 185,2
В колбу Шленка на 1 л, снабженную обратным холодильником с ртутным затвором, помещают 23,8 г (90 ммоль) Мо(СО)ви добавляют 11,0г (125 ммоль) NaC5H8 в —150 мл тетрагидрофурана. Реакционную смесь нагревают до кипения. Начало выделения СО, а также его протекание легко наблюдать по ртутному затвору. В процессе реакции смесь окрашивается в светло-коричневый цвет. Образующийся Na-комплекс чрезвычайно неустойчив на воздухе. Через 15 ч раствор охлаждают до комнатной температуры и при интенсивном перемешивании прикапывают к нему 15 мл ледяной уксусной кислоты. Перемешивают 15 мин и затем порциями при комнатной температуре прибавляют такое количество М-метил-Ы-нитрозо-п-толуолсульфамида*, чтобы через ртутный затвор прекратилось выделение газа. Обычно для этого требуется около 30 г ( — 0,14 моль) нитрозирующего агента. Затем в вакууме водоструйного насоса досуха отгоняют растворитель, а остаток экстрагируют 2 л петролейного эфира (/KIIn 40—60 °C) (порциями —10 раз). Раствор, в котором находится нитрозильный комплекс Мо(СО)2(т]-С5Н5), упаривают до объема 20 мл и хроматографируют затем бензолом на колонке с кизельгелем. Сухой оранжевокрасный продукт возгоняют в высоком вакууме при —65 °C. Соединение получают в виде оранжевых кристаллов, выход —13,3 г (60%).
* Нитрозирующее средство проявляет канцерогенные свойства! Работать в вытяжном шкафу! Перчатки!
Нитрозильные комплексы 2091
Аналогично синтезируют Cr(CO)2NO(r]-C5Hs). Соответствующее производное вольфрама W(CO)2NO(t)-C5H5) получают из W(CO)3(r|-C5H5)H и N-метил-N-нитрозотолуолсульфамида лишь при кипячении реагентов с обратным холодильником, однако можно использовать и вышеприведенную методику. Ввиду большей устойчивости Сг(СО)е и W(CO)3 к действию NaCjHs их комплексные соли следует готовить в диметилформамиде (см. вышеприведенную методику •синтеза М(СО)з(п-С5Н5)С1, где М=Мо, W).
Другие способы. Действием NO на На[Мо(СО)з(т|-С5Н5)] в водном растворе. Выход 60% [3].
Свойства. Оранжево-красные, не очень устойчивые на воздухе кристаллы, /пл 85 °C. Растворяется во всех обычных органических растворителях. ИК (гексан): 2021 (с.), 1948 (оч. с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Piper Т. S., Wilkinson О., J. Inorg. Nucl. Chem., 3, 104 (1956).
2. King R. В., Bisnette M. B., Inorg. Chem., 6, 469 (1967).
3. Fischer E. 0., Beckert 0., Hafner W., Stahl H. 0., Z. Naturforsch., 10b, 598 (1955).
Нитрозил(тетракарбонил)марганец Mn(CO)4NO
Mn(CO)sH + CH3(NO)N—SO2—C6H4-n- CH3 ------->
196,0 214,2
---> Mn(CO)4NO + CO + CH3NHSO2—C6H4-n-CHg 197,0 22,4л 185,2
В сухую колбу на 1 л, заполненную азотом, помещают 4,3 г (20 ммоль) Ы-нитрозо-М-метил-п-толуолсульфамида и 20 мл абсолютного эфира. Систему •охлаждают жидким азотом (—196 °C) и откачивают до высокого вакуума. Затем из присоединенного к колбе промежуточного сосуда перекоидеисируют IB вакууме 1,6 г (8,2 ммоль) Мп(СО)5Н. Вакуумированную колбу нагревают до комнатной температуры и оставляют на 16 ч. В процессе реакции первоначально желтый раствор становится интенсивно-красным. Получившийся продукт перегоняют в вакууме в охлаждаемую до —35 °C* ловушку-приемник и для очистки несколько раз переконденсируют. Растворитель же и СО собирают в соседнюю ловушку, охлаждаемую до —196°C (жидким азотом). Выход ~9,7 г ( — 60%). Ввиду неустойчивости продукт используют для дальнейших превращений по возможности быстрее.
Свойства. Темно-красная, очень легко окисляющаяся ядовитая жидкость, по внешнему виду похожая на бром, 1пл — 1 °C. На свету образует •смесь Мп2(СО)ю, Mn(CO)(NO)3 и Mn2(CO)7(NO)2. Очень легко возгоняется (Рпар —8 мм рт. ст./25°С). Неограниченно смешивается практически со всеми органическими растворителями. ИК (С2С14): 2095 (ср.), 2019 (с.), 1972 (с.) [v(CO)]; 1759 (с.) [v(NO)] см-1.
ЛИТЕРАТУРА
1. Treichel Р. М., Pitcher Е., King R. В., Stone F. G. A., J. Amer. Chem. Soc., 83, 2593 (1961).
* Эту температуру удобно поддерживать с помощью частично замерзшего 1,2-дихлорэтана, охлаждаемого жидким N2.
2092 Глава 33. Металлоорганические комплексы
□ □ Тетрафтороборат (т]-циклопентадиенил)(нитрозил)-дикарбонилмарганца(П) [MnfCO^NOfTi-CsHs)] [BF4]
Mn(CO)3(T]-CsH5) 4- NO2BF4 ---> [Mn(CO)2NO(T]-C5H6)] [BF4]
204,1 132,8 292,8
К раствору 10,0 г (49 ммоль) Мп(СО)3(т]-С5Н5) в 50 мл нитрометана прибавляют при комнатной температуре в течение 10 мин 7,0 г (53 ммоль) NO2BF4 и перемешивают смесь 1 ч при 40 °C; цвет раствора изменяется от желтого до темно-красного. После отгонки растворителя в вакууме водоструйного насоса остаток промывают эфиром, освобождаясь тем самым от непрореагировавшего Мп(СО)з(т|-С5Н5). После 30 мин перемешивания продукта, суспендированного в эфире, фильтрования и высушивания в вакууме препарат для большинства превращений является достаточно чистым. Выход 12,4 г (86%).
Свойства. Желтый, не очень устойчивый на воздухе порошок. Растворы в воде постепенно разлагаются. Растворяется в метаноле и ацетоне, ие растворяется в эфире, бензоле и петролейном эфире. ИК (КВг): 2125 (оч. с.), 2080 (оч. с.) [v(CO)]; 1840 (оч. с.) [v(NO)] см-'.
ЛИТЕРАТУРА
1. Несмеянов А. Н., Анисимов К- Н., Колобова Н. Е., Краснослободская Л. Л. — Изв. АН СССР, Сер. хим., 1970, с. 860.
□ □ ц-1,2-Бис(дифени1лфосфино)этанбис(тринитрозилмарганец) [Mn(NO)3]2[(C6H5)2PCH2CH2P(C6H5)2]
hv
а) Мп2(СО)10 + 6NO + 2С4Н8О----> 2[Мп(ОС4Н8) (NO)3j + ЮСО
390,0 6-22,4л 2-72,1 2-217,1 10-22,4л
б) 2[Мп(СС4Н8) (NO)sJ + (С6Н6)2РСН2СН2Р(СвН5)2 ->
2-217,1 398,4
---> [Mn(NO)3]2[(C6H6)2PCH2CH2P(C6H6)2] + 2С4Н3О
688,3 2-72,1
В трубку Шленка, снабженную трубкой для ввода газа, помещают раствор 0,78 г (2 ммоль) Мп2(СО)ю примерно в 200 мл тетрагидрофурана и при интенсивном перемешивании (магнитная мешалка) облучают ртутной лампой высокого давления (например, Hanovia S, 200 Вт), одновременно продувая через раствор слабый ток NO. Примерно через 2 ч, когда в спектрах ИК исчезнет полоса поглощения карбонильной группы, прозрачный желто-зеленый раствор* фильтруют через стеклянный фильтр, покрытый фильтровальной бумагой, добавляют 0,4 г (1 ммоль) 1,2-бис (дифенилфосфино) этана и оставляют на ночь перемешиваться. После удаления растворителя остаток зеленого цвета растворяют в бензоле и хроматографируют на кизельгеле бензолом. Элюат упаривают и продукт высаживают добавлением гексана. Выход 1,26 г (92%).
Свойства. Темно-зеленые кристаллы, при нагревании под N2 выше ~ 135 °C изменяют цвет и при ~ 240 °C плавятся с разложением. ИК (тетрагидрофуран): 1776, 1685/1679 [v(NO)] см-'.
* При действии различных двухэлектронных лигандов L на этот раствор можно получить комплексы типа Mn(NO)3L [2, 3].
Нитрозильные комплексы 2093
ЛИТЕРАТУРА
1. Herberhold М., Razavi A., J. Organometal. Chem., 67, 81 (1974).
2. Hleber W., Tengler H., Z. Anorg. Allgem. Chem., 318, 136 (1962).
3. Beck W., Lottes K„ Ber., 98, 2657 (1965) .
□ □ Бис[(ц-нитрозил)(т]-циклопентадиенил)железо]
(Fe—Fe) [Fe(NO)(n-C5H5)]2
[Fe(CO)2(ryC5H5)]2 + 2NO -> [Fe(NO) (n-CsHs)]2 + 4CO
353,9 2-22,4 л 301,9 4-22,4л
Этот синтез проводят в трубке Шленка (керн НШ 29/32, диаметр 35 мм^. длина 35 см), которую для уменьшения больших потерь растворителя обдувают холодным воздухом выше границы раствора с помощью фена. Растворяют 7,1 г (20 ммоль) [Fe(CO)2(T]-CsH5)]2 в 100 мл н-октана. При 120°С и постоянном перемешивании через красно-коричневый раствор в течение 2,5—3 ч-продувают слабый ток сухого NO. Чтобы избежать забивания конца трубки' для ввода газа, применяют широкую трубку диаметром не менее 1 см и погружают ее в раствор примерно на 2 см. В процессе реакции выпадают черные' кристаллы, а раствор постепенно окрашивается в темио-коричневый цвет. Реакционную смесь охлаждают до комнатной температуры, фильтруют (стек-лянный пористый фильтр G4), осадок промывают петролейным эфиром (2Х ХЗО мл). Для дальнейшей очистки проводят экстракцию в аппарате Сокслета 200 мл бензола. Экстракт упаривают в вакууме водоструйного насоса до-объема около 15 мл и разбавляют 50 мл петролейного эфира. Осадок отделяют, тщательно промывают петролейным эфиром и, наконец, для удаления непрореагировавшего i[Fe (СО) 2 (т]-С5Н5) ] 2 промывают небольшим количеством бензола. Выход 3,1 г (51°/о).
Свойства. Черные кристаллы, разрушаются при воздействии влаги и кислорода воздуха. В высоком вакууме при 120—140 °C возгоняется (с частичным' разложением). В большинстве органических растворителей растворяется плохо; Растворы окрашены в темно-зеленый цвет, в проходящем свете цвет раствора красный (в присутствии следов воздуха образуются хлопья коричневого цвета) . При нагревании образуется железное зеркало, выделяются также следы' Fe(r|-C5H5)2. Дипольный момент ц (25 °C) = 1,87 дебай (СвНв). ИК (КВг): 1514 (пл.), 1506 (оч. с.), 1498 (с.), 1490 (оч. с.) [v(NO)] см-1. Кристаллическая структура: см. [2].
ЛИТЕРАТУРА
1. Brunner Н„ J. Organometal. Chem., 14, 173 (1968)'.
2. Calderon J. L., Fontana S„ Frauendorf er E„ Day V. W., Iske D. A., J. Organometal. Chem., 64, C16 (1974).
□ □ Динитрозилдикарбонилжелезо Fe(CO)2(NO)2
Fe(CO)s + 2NaNO2 + 3NaOH + 2CH3COOH----->
195,5 2-69,0 3-40,0 2-60,0
---> Fe(CO)2(NO)2 + 2CO + Na2CO3 + 2Na[CH3CO2] -f- 2H2O
171,9 2.22,4 л 106,0 2-82,0 2-18,0'
Внимание! Fe(CO)5 и Fe(CO)2(NO)2 вследствие их высокой летучести чрезвычайно ядовиты! Все работы проводить только в хорошо дейст
2094 Глава 33. Металлоорганические комплексы
вующем вытяжном шкафу! Для всех насосов предусмотреть ловушки с жидким азотом! Выхлоп отводить прямо в вытяжной шкаф!
В колбе емкостью 500 мл, снабженной вводом для азота, обратным холодильником и капельной воронкой, при постоянном перемешивании кипятят в течение 3 ч смесь 7,5 мл (11,0 г, 56 ммоль) Fe(CO)5, 9,0 г (0,13 моль) NaNO2, 15,2 г (0,38 моль) NaOH и 120 мл воды. Затем охлаждают до 30—40 °C и пропускают через систему интенсивный ток N2; к системе присоединяют ловушку-приемник, заполненную 30 г безводного СаС12 и охлаждаемую до —78 °C (ацетон — сухой лед). В реакционную смесь прикапывают 50%-ную СН3СООН до тех пор, пока не начнут образовываться коричневые пары Fe(CO)2(NO)2. Тогда прибавление кислоты прекращают, током N2 вытесняют образовавшийся Fe(CO)2(NO)2 в охлажденную ловушку. Добавление СН3СООН и вытеснение Fe(CO)2(NO)2 повторяют до тех пор, пока не перестанет образовываться нитрозопроизводное. Общее время реакции ~ 1 ч. По окончании ловушку-приемник постепенно нагревают до расплавления Fe(CO)2(NO)2, т. е. до +18°C.
Получившуюся красную жидкость конденсируют затем в высоком вакууме в другую ловушку-приемник (—78°C), в которую помещают 30 г Р4Ою (например, марки Granusic А фирмы Baker-Chemicals) для окончательного высушивания продукта. Для тонкой очистки проводят снова вакуумную перегонку. Ввиду высокой чувствительности к О2 и перегреву синтезированный Fe(CO)2(NO)2 хранят исключительно в атмосфере N2 и при температуре не выше —20 °C. Выход до 60%.
Свойства. Красное, легколетучее, легко окисляющееся и очень ядовитое масло. /пл 18°С, /кип 110°С/760 мм рт. ст. (экстраполировано). ИК (циклогексан): 2083 (с.), 2034 (с.) [v(CO)]; 1810 (с.), 1756 (с.) [v(NO)] см-1.
ЛИТЕРАТУРА
1. Hieber W., Beutner Н., Z. Anorg. Allgem. Chem., 320, 101 (1963).
□ □ Динитрозилбис(трифенилфосфин)рутений
Ru[P(CeH5)3]2(NO)2
CH8(NO)NSO2CeH4-n-CHs
RuCl3-3H2O + 2P(C6H5)3------------------- Ru[P(CeH6)3]2(NO)2
261,5 2-262,3 685,7
К интенсивно перемешиваемому раствору 1,56 г (6 ммоль) Р(С6Н6)з в 60 мл кипящего этанола в атмосфере N2 добавляют 0,26 г (1 ммоль) RuCl3-ЗН2О в 20 мл этанола и прикапывают ~4 мл триэтиламина до тех пор, пока раствор не станет темно-красным, почти фиолетовым. Затем быстро прибавляют раствор 0,4 г И-метил-М-нитрозо-п-толуолсульфамида* в 20 мл этанола. Далее к кипящей реакционной смеси добавляют еще 6 мл триэтиламина, кипятят 5 мин и нагревание прекращают. Выпавшие после охлаждения кристаллы отфильтровывают, промывают 5 мл этанола, 5 мл воды, снова 5 мл этанола, затем гексаном (2x5 мл) и высушивают в вакууме. Выход 0,55 (80%).
Другие способы. Из Ru(CO)2[P(CeH5)3]2Cl2 через образование Ru(CO)2-[P(CeH5)3]2(NO2) с последующим внутримолекулярным переносом кислорода нитрогруппы на карбонильный лиганд; выход 95% [2]. Можно также исходить из Ru[P(C6H5)3]3H2 и NO [3].
* Осторожно! Канцероген! Работать в вытяжном шкафу в защитных перчатках!
Нитрозильные комплексы 2095
Свойства. Не очень устойчивые на воздухе кристаллические иголочки от оранжевого до темно-красного цвета. /пл 144—145°C (на воздухе), 185—186°C (под N2). Умеренно растворяется в бензоле, хлороформе и метилендихлориде. Растворы в хлорированных углеводородах нельзя хранить в течение длительного времени. На воздухе раствор комплекса поглощает О2 и превращается в-Ru[P(C6Hs)3]2(NO)(O2)(NO3). ИК (СН2С12): 1655, 1619 (с.) [v(NO)] см->. Кристаллическая и молекулярная структура ([Ru{P(CeH6)3)2(NO)2]''/гСбНе): моноклинная, пространственная группа симметрии Р2]/п (а—17,031 А; & = = 18,792 А; с= 10,800 А; 0=97,03°); молекула —в виде искаженного тетраэдра с практически линейной группой Ru—N—О [4].
ЛИТЕРАТУРА.
1. Levison J. J., Robinson S. D., Chem. Ind. (London), 1969, 1514; J. Chem.. Soc., Al 970, 2947.
2. Grundy K. R„ Laing K- R-, Roper W. R„ J. Chem. Soc. Chem. Commun.^ 1970, 1500.
3. Eliades T. I., Harris R. O., Zia M. C., J. Chem. Soc. Chem. Commun., 1970; 1709.
4. Gaughan A. P., Corden B. J., Eisenberg R., Ibers J. A., Inorg. Chem., 13; 786 (1974).
□ □ Нитрозилтрикарбонилкобальт Со(СО)зМО
Способ 1. Один из возможных способов получения, исходя из [Co(H2O)6J-(NO3)2, представляет собой двухстадийный синтез через трикарбонилкобаль-тат-анион с последующим нитрозированием:
(NH3)
а) 2[Со(Н2О)6] (NO3)2 + 8СО 4- 3Na2S2O4 ->
2-291,0 8-22,4 л 3-174,1
---->- 2Na[Co(CO)4] 4. 6SO2 4- 4NaNO3 4- 12H2O 2-194,0 6-22,4л 4-85,0 12-18,0
б) Na[Co(CO)4] 4- NaNO2 4- 2СН3СООН --->
194,0 69,0 2-60,0
----> Co(CO)3NO 4- СО 4- Н2О 4- 2Na[CH3CO.J 173,0 22,4л 18,0 2-82,0
Осторожно! Ввиду чрезвычайной ядовитости Co(CO)3NO все работы проводят в хорошо действующем вытяжном шкафу!
В четырехгорлую колбу на 2 л с эффективным холодильником, капельной воронкой, мешалкой и трубкой для ввода газа помещают 160 мл концентрированного аммиака и 240 мл Н2О. После добавления раствора 17,5 г (60 ммоль) [Со(Н2О)в] (NO3)2 в 40 мл воды воздух из реакционной колбы вытесняют через газоотводную трубку СО и охлаждают колбу до ~10°С. Продолжая перемешивание, охлаждение и пропускание слабого тока СО, медленно прикапывают раствор 20,0 г (0,11 ммоль) 80%-ного Na2S2O4 в смеси из 95 мл концентрированного аммиака и 135 мл воды( ~1 капля за 10—15 с). Ток СО прекращают, добавляют в реакционную смесь раствор 4,2 г (61 ммоль) NaNO2 в 15 мл Н2О и охлаждают смесью льда и соли примерно до —10 °C. Обратный холодильник убирают и с помощью оливы колбу соединяют с ловушкой, охлажденной до —78 °C. Затем, прикапывая по каплям 120 мл 50%-ной СН3СООН, током СО вытесняют образующиеся коричневые пары Co(CO)3NO в ловушку-приемник. Полученный продукт оседает в виде оранжевых кри
2096 Глава 33. Металлоорганические комплексы
сталлов. Ток СО продувают до тех пор, пока не прекратится образование -коричневых паров. Для удаления незначительного количества Н2О полученный -Co(CO)3NO переконденсируют в вакууме. Выход —5,2 г (—50%).
Способ 2
Со2(СО)3 + 2NO ----> 2Co(CO)3NO + 2СО
342,0 2-22,4Л 2-173,0 2-22,4л
В трубку Шленка с 4,50 г (13,2 ммоль) Со2(СО)з, помещенную в масляную 'баню, нагретую до 40 °C, продувают ток сухого чистого NO. Его получают в аппарате Киппа из NaNO2 и H2SO4 средней крепости и пропускают через две ловушки, охлаждаемые до —50 °C. Тем самым достигается очистка от побочно получающегося NO2, высших оксидов азота и воды.
Красно-коричневые кристаллы быстро превращаются в темно-красную жидкость, которую после окончания реакции осторожным нагреванием не выше 60 °C переконденсируют в охлаждаемую жидким азотом ловушку. После повторного переконденсирования (в атмосфере NO) в отпаиваемую ампулу получают 2,5 г (55%) Co(CO)3NO.
Свойства. Темно-красная, легколетучая, неустойчивая на воздухе, чрезвычайно ядовитая жидкость. /пл —1 °C, /кип 78—79“С/760 мм рт. ст. (экстраполировано). Неограниченно смешивается со всеми органическими растворителями. ИК (газ): 2109 (с.), 2047 (с.) [v(CO)]; 1825 (с.) [v(NO)] см-1. Кристаллическая структура: см. [6], </(Со—N) = l,76 A, d(N—О) = 1,10 А.
ЛИТЕРАТУРА
1. Hieber W., Fischer Е. О., Bockly Е., Z. Anorg. Allgem. Chem., 269, 308 (1952): [Na{Co(CO)4}J.
2. Seel F„ Z. Anorg. Allgem. Chem., 269, 40 (1952): [Co(CO)3NO],
3. King R. B., Organometallic. Syntheses, Academic Press, New York, London, 1965, v. 1, p. 168 f.
4. Mond R. L„ Wallis A. E., J. Chem. Soc. (London), 121, 34 (1922).
5. Reiff R„ Z. Anorg. Allgem. Chem., 202, 375 (1931).
6. Brockway L. O., Anderson J. S., Trans. Faraday Soc., 33, 1233 (1937).
IO □ Бис [ (ц-нитрозил ) (т]-циклопентадиенил ) кобальт]
(Со—Со) [CoNO(n-C5H5)]2
2Со(СО)2(т|-С5Н5) + 2NO ---> [CoNO(n-C6HB)]2 + 4СО
2-180,1 2-22,4л 308,1 4-22,4л
Реакцию проводят в двугорлой колбе емкостью 250 мл, снабженной эффективным холодильником для избежания больших потерь растворителей. В колбу помещают 18,0 г (0,1 моль) Со(СО)2(т]-С5Н5) в 150 мл гексана и продувают через раствор слабый ток NO. Последний получают в аппарате Киппа из NaNO2 и H2SO4 (не очень крепкой), пропуская его через U-образную трубку, охлажденную до —40 °C для удаления NO2, а затем высушивают пропусканием через трубку с КОН.
Через 2 ч цвет реакционной смеси меняется от красного до темно-коричневого, одновременно выпадает черный твердый осадок. Суспензию переносят на хроматографическую колонку, охлаждаемую водой и заполненную кизельгелем (30X3 см), и до тех пор промывают петролейным эфиром, пока полностью не элюируют не вошедший в реакцию красный исходный комплекс Со(СО2) (т]-С5Н5). Нитрозильный комплекс элюируют в виде темно-коричневой полосы метилендихлоридом, который затем упаривают в вакууме водоструйно
Нитрозильные комплексы 2097
го насоса до начала кристаллизации. Выпавший после добавления петролейного эфира продукт отфильтровывают на стеклянном пористом фильтре G3, промывают несколько раз небольшими порциями петролейного эфира и сушат осадок (черного цвета) в высоком вакууме. Выход 9,9—10,6 г (64—69%).
Свойства. Устойчивые на воздухе кристаллы глубокого черного цвета. Растворы в бензоле или СН2С12 неустойчивы к действию кислорода воздуха. В высоком вакууме сублимируются при 90—100 °C с частичным разложением. ИК (КВг): 1585 (с.), 1525 (оч. с., шир.) [v(NO)J см-'. ЯМР-'Н (CeDe, ТМС): 6 4,77 [синглет, С5Н5]. Кристаллическая структура: см. [2].
ЛИТЕРАТУРА
1. Brunner И., J. Organometal. Chem., 12, 517 (1968).
2. Bernal I., Korp J.-D., Reisner M. G., Herrmann W. A., J. Organometal. Chem., 139, 321 (1977).
□ Нитрозилтрис(трифенилфосфин)родий Ш1[Р(С6Н5)з]з(МО)
RhCl3-3H2O 4- 3P(CeH5)8
CH3(NO)NSO2 (п-толил)
KOH
Rh[P(C6H6)3]3(NO)
263,3
3-262,3
919,8
Этанольные растворы (примерно по 20 мл) 0,26 г (1 ммоль) RhCl3-3H2O, 0,4 г (1,9 ммоль) М-метил-Ы-нитрозо-л-толуолсульфамида* и 0,4 г (7 ммоль) КОН быстро один за другим в атмосфере N2 приливают к хорошо перемешиваемому раствору 2,62 г (10 ммоль) трифеиилфосфина в кипящем этаноле (80 мл). Нагревают 10 мин с обратным холодильником, охлаждают до 10°C и фильтруют. Красный осадок промывают последовательно этанолом, водой и снова этанолом (по 5мл), гексаном (2x5 мл) и сушат в вакууме. Выход 0,8 г (87%).
Другие способы. Пропускают NO через кипящий тетрагидрофурановый раствор RhC!3-3H2O в присутствии избытка Р(С6Н5)3 и Zn-гранул. Выход 98% [3].
Свойства. Блестящие карминово-красиые кристаллы, сравнительно устойчивые на воздухе. 1пл ~ 160°C (с разл.); в запаянном, заполиеином азотом капилляре 1ПЛ 205—206 °C. Плохо растворяется в полярных растворителях и алифатических углеводородах, умеренно растворяется в бензоле. Растворы иа воздухе очень неустойчивы. Соединение взаимодействует с хлороформом и метилендихлоридом. ИК (КВг): 1610 [v(NO)] см-1. Молекулярная структура: скошенный тетраэдр с нелинейным расположением фрагмента Rh—N—О (<RhNO= 158,9°) [4].
ЛИТЕРАТУРА
1. Hieber W., Heinicke К., Z. Anorg. Allgem. Chem., 316, 321 (1962).
2. Levison J. J., Robinson S. D„ J. Chem. Soc. (London), A1970, 2947.
3. Collman J. P., Hoffman N. W., Morris D. E., J. Amer. Chem. Soc., 91, 5659 (1969).
4. Ibers J. A., Kaduk J. A., Israel J. Chem., 15, 143 (1976).
* Осторожно! Канцероген! Работать в перчатках и вытяжном шкафу! j
16—13G1
2098 Глава 33. Металлоорганические комплексы
Дихлоронитрозилбис(трифенилфосфин)иридий Ir[P(C6H5)3]2(NO)Cl2
CH3(NO)NSO2 (п-толил)
Naa[IrCl6]-6H2O+ 2P(C6H6)j------------------ Ir[P(C6H6)3]2(NO)CIa
559,0 2-262,3 817,7
К хорошо перемешиваемому кипящему раствору 3,95 г (15 ммоль) Р(СвН6)3 в 100 мл 2-метоксиэтанола быстро добавляют один за другим растворы 1,40 г (2,5 ммоль) Na2[IrCl6] -6Н2О в 50 мл того же растворителя и 1,05 г Н-метил-И-нитрозо-п-толуолсульфамида* также в 50 мл 2-метоксиэтанола. Кипятят 5 мин с обратным холодильником и охлаждают до комнатной температуры. Выпавший осадок отфильтровывают, промывают последовательно (по 5 мл) этанолом, водой и этанолом, затем гексаном (2x5 мл) и высушивают в вакууме. Выход 1,05 г (52%).
Другие способы. Синтез из 1г(циклооктадиен-1,5) (NO)C12 и Р(С6Н5)3 [4J.
Свойства. Устойчивые на воздухе, оранжево-желтые кристаллы. !пл 247— 252 °C на воздухе и 308—309 °C под N2. Умеренно растворяется в бензоле, СНС1з и СН2С12.
ИК (нуйол): 1560 [v(NO)] см-1. Кристаллическая и молекулярная структура: пространственная группа симметрии Р2/а (а= 15,851 А; 6=9,624 А; с= = 22,050 А; (5=104,6°); молекула квадратно-пирамидальная с нелинейной координацией NO в вершине [5].
ЛИТЕРАТУРА
1. Malatesta L., Angoletta М., Caglio G„ Angew. Chem., 75, 1103 (1963).
2. Reed C. A., Roper W. R., J. Chem. Soc. Chem. Commun., 1969, 155.
3. Levison J. J., Robinson S. D., J. Chem. Soc. (London), Al970, 2947.
4, Crooks G. R., Johnson B. F. G., J. Chem. Soc. (London), A1970, 1662.
5. Mingos D. M. P., Ibers J. A., Inorg. Chem., 10, 1035 (1971).
□ □ (т]-Циклопентадиенил)нитрозилникель NiNO(r)-C5H5)
NiNOfri-CsHs) образуется при парциальном нитрозировании №(т]-СзН8)а при действии NO:
№(г]-С6Н6)2NO ------> NiNO(T)-CsH6) + lC6H6-]
188,9 22,4л 153,8
Используют двугорлую колбу на 250 мл, снабженную магнитной мешалкой и эффективным холодильником, чтобы избежать потерь растворителя в процессе реакции. В колбу помещают раствор 18,9 г (0,1 моль) Nii^-CsHs)^ в 150 мл пентана и при перемешивании в течение 2 ч пропускают слабый ток сухого NO. Газ получают в аппарате Киппа из NaNO2 и H2SO4 средней крепости и очищают его от NO2 пропусканием через охлажденную до —50 °C U-образную трубку, а затем высушивают, пропуская через трубку с КОН.
В процессе реакции цвет раствора меняется от темно-зеленого до краснокоричневого. Растворитель отгоняют при нормальном давлении. Сырой продукт очищают вакуумной перегонкой (fKHn 47—48°С/17 мм рт. ст.; охлаждаемый льдом приемник). Выход 12,3—13,4 г (80—87%).
Другие способы. Действие NO на Ni(CO)4-|-C5H6-|-(C2H5)2NH. Выход в зависимости от условий реакции до 92% [3].
* Осторожно! Канцероген! Работать в перчатках и вытяжном шкафу!
Дитиокарбонильные и тиокарбонильные комплексы 2099
Свойства. Чрезвычайно ядовитая, исключительно устойчивая на воздухе темно-красная жидкость с неприятным удушливым запахом; 1ПЛ —41 °C, /кип 144—145°С/715 мм рт. ст., 59°С/27 мм рт. ст., 47—48°С/17 мм рт. ст. Хорошо растворяется во всех органических растворителях. ИК (циклогексан): 1833 (v(NO)] см-1. Кристаллическая структура: см. [4], d(N—О) = 1,10 А (электронографически).
ЛИТЕРАТУРА
1. Fischer Е. О., Beckert О., Hafner W., Stahl Н. С., Z. Naturforsch., 10b, 598 (1955).
2. Piper Т. S., Cotton F. A., Wilkinson G„ J. Inorg. Nucl. Chem., 1, 165 (1955).
3. Feltham R. D., Carriel J. T., Inorg. Chem., 3, 121 (1964).
4. Cox A. P., Thomas L. F., Sheridan I., Nature, 181, 1157 (1958).
Дитиокарбонильные и тиокарбонильные комплексы
□ □ Дикарбонил (т]2-сероуглерод) (трмезитилен) хром
Cr[n-C6H3(CH3)3](T]2-CS2)(CO)2
а) Сг[т|-С6Н3(СН3)3] (СО)3 -J- C8Hj4 • >
256,2 110,2
---> Сг[т]-СвНз(СН3)3] (г|2-С8Н14) (СО)2 + СО
338,3 22,4 л
б) Cr[T1-CeH3(CH3)3](Ti2-C3H14)(CO)2 + CS2 ->
338,4 76,1
---> Сг[т)-С6Н3(СН3)3] (t)«CS2) (СО)2 4-СзНм
304,4 110,2
В 100 мл пентана растворяют 0,38 г (1,5 ммоль) трикарбоиил(т)-мезити-леи) хрома и 2 мл циклооктена и облучают в течение 2 ч. Образующийся цик-лооктеновый комплекс Сг[ц-СвН3(СН3)3] (i]2-C8Hu) (СО)2 (21 выделять не требуется: после добавления 2 мл CS2 он превращается в Сг[т)-С«Н3(СН3)2] (т)2-CS2) (СО) 2, выпадающий во время реакции в течение 0,5 ч из пентанового раствора. Осадок промывают пентаном и при —78 °C перекристаллизовывают из смеси тетрагидрофуран — гексан. Выход 0,23 г (50%).
Свойства. Темно-коричневые кристаллы. <пл 101 °C (с разл.). Устойчив в атмосфере N2. ИК (тетрагидрофураи): 1956, 1901 (с.) [v(CO)J см-1.
ЛИТЕРАТУРА
1. Herberhold М„ Suss-Fink М., Вег., 111, 2273 (1978).
2. Jaouen G„ Dabard R„ J. Organometal. Chem., 72, 377 (1974).
□ □ (т]-Циклопентадиенил) (нитрозил)карбонил(тиокарбонил)-хром Cr(CS)(CO)(NO)(n-C5H5)
a) Cr(CO)2NO(T1-C6He) + C8HM ---> Сг(т]а-С8Н14) (CO) (NO) (T]-CjHe) + CO
203,1 110,2 285,3 22,4 л
6) Cr(n2-CeHI4)(CO)(NO)(n-C6H5)+CS2+P(C6H6)3 ------>
285,3 76,1 262,3
----> Cr(CS) (CO) (NO) (n-C5H6) + C8H14 + (C8H5)3PS 219,2 110,2 294,3
16*
2100 Глава 33. Металлоорганические комплексы
Раствор ~1 г (4,9 ммоль) CrfCO^NO^-CjHs) в 250 мл циклооктена подвергают в течение 2—4 ч фотолизу ртутной лампой высокого давления (Напо-via S, 200 Вт) и затем отгоняют досуха. Сухой циклооктеновый комплекс Сг(т]2-С8Н14) (СО) (NO) (г|-С5Н5) в течение 2 ч при 35 °C сушат в высоком вакууме, а затем обрабатывают ~ 1,3 г (5 ммоль) трифенилфосфина и 30 мл CS2. После того как ИК-спектр зеленого раствора (примерно через 60 ч) укажет на полное превращение олефинового комплекса, CS2 отгоняют в вакууме. Для очистки раствор получившегося комплекса Cr(CS) (СО) (NO) (r|-CsHs) сначала в смеси бензол — пентан (1:5), а затем еще раз только в пентане пропускают через слой кизельгеля на стеклянном пористом фильтре (G3). Комплекс перекристаллизовывают при —78 °C из пентана. Выход 0,43 г (~40%).
Свойства. Зеленые кристаллы. /пл 42°C (под Аг). Хорошо растворяется в органических растворителях. ИК (нитрометан): 2010 [v(CO)], 1705 [v(NO)J; (КВг): 1253 [v(CS)] см-1.
ЛИТЕРАТУРА
1. Herberhold М„ Smith Р. D„ Angew. Chem., 91, 662 (1979).
□ □ (т]-Циклопентадиенил)дикарбонил(тиокарбонил)марганец Mn(CS)(CO)2(n-C5H5)
Мп(п2-С8Н14) (СО)2(п-С6Н6) + CS2 + Р(С6Н5)8->
, 286,3 76,1 262,3
---> Mn(CS) (СО)2(п-С6Н6) + (CeH5hPS + С8Н14 220,1 294,4 110,2
В круглодониой колбе на 250 мл, снабженной обратным холодильником, магнитной мешалкой и ртутным затвором, растворяют 1,15 г (4,0 ммоль) возог-ванного (трциклопентадиеиил) дикарбонил (т12-циклооктен) марганца и 1,31 г (5,0 ммоль) трифенилфосфина в 100 мл сухого, насыщенного азотом CS2* И кипятят с обратным холодильником ~36 ч. Протекание реакции контролируют по ИК-спектру**. По окончании реакции растворитель отгоняют (25°С/0,1 мм рт. ст.), а желто-коричневый кристаллический осадок возгоняют в высоком вакууме (30°C). Для более тонкой очистки проводят повторную возгонку, перекристаллизацию из пеитана или хроматографию на AI2O3/CH2CI2. Выход 0,71 г (81%).
Свойства. Золотисто-желтые кристаллы. /пл 50—52 °C. Устойчив на воздухе в течение длительного времени, однако на свету идет постепенно разложение (окраска темнеет). В холодильном шкафу можно хранить неограниченно долго. Хорошо растворяется в обычных органических растворителях. ИК (CS2): 2006, 1954 (оч. с.) [v(CO)]; 1266 (оч. с.) [v(CS)] см-'. ЯМР-'Н (CSa, ТМС): 6=4,80 [С5Н5]. В присутствии таких нейтральных лигандов L, как трифеиилфосфин или циклооктеи, легко идет фотозамещение СО и образование Mn(CS) (CO)L(t]-C5H5) или Mn(CS)L2(T]-CeH5).
ЛИТЕРАТУРА
1. Fenster А. Е., Butler I. S., Inorg. Chem., 13, 915 (1974).
2. Butler I. S., Coville N. J., Cozak D., J. Organometal. Chem., 133, 59 (1977). 3. Butler I. S., Coville N. J., Fenster A. E„ Inorg. Synth., 16, 53 (1976).
* CS2 марки Uvasol (Merck) перегоняют над P4O10 в атмосфере N2.
** Mn(Tf-C8HI4) (CO)2(n-CsH5). ИК (CS2): 1955, 1895 (оч. c.) fv(CO)] см-».
Дитиокарбонильные и тиокарбонильные комплексы 2101
□ □ (т|-Циклопентадиенил) (дикарбонил)(дитиометоксикарбо-нил)железо Ре(С88СНз)(СО)2(т]-С5Н5)
a) Na[Fe(CO)2(r]-C6H6)] + CS2 -> NalFefCOkfo-CjHJCSJ
200,0 76,1 276,1
б) Na[Fe(CO)2(n-C5H5)-CS2] + CH3I -> Fe(CSSCH3) (СО)2(П-С5Н5) + Nal
276,1 141,9 268,1 149,9
К охлажденному до —30°C раствору 20 ммоль NafFefCOWTl-CsHs)] в 200 мл тетрагидрофурана, полученному из 3,54 г [Ре(СО)2(т]-С5Н5)]2 и 6,0 мл амальгамы Na [см. методику синтеза Fe^-CsHj) (СН3) (СО)2], прибавляют сначала 20 ммоль (1,52 г, 1,21 мл) CS2 и затем 20 ммоль (2,82 г, 1,24 мл) СН31, разбавив их предварительно тетрагидрофураном в соотношении 1:1. После нагревания до комнатной температуры выпавший Nal отделяют, а растворитель упаривают в вакууме водоструйного насоса. Остаток экстрагируют пентаном (4X100 мл), пентановый раствор фильтруют и упаривают до объема ~ 150 мл. Для выделения продукта в течение длительного времени охлаждают при —40 °C, осадок отфильтровывают и высушивают в высоком вакууме. Выход 4,6 г (86%).
Свойства. Коричневый порошок или мелкие кристаллы. /пл 72 °C. Растворяется во всех обычных органических растворителях. ИК (СН2С12): 2028, .1979 с. [v(CO)J СМ-1. ЯМР-’Н (CDC13): б 4,79 [синглет, С6Н6]. 2,94 [синглет, СН3]. j
ЛИТЕРАТУРА
1. Mayr A., Dissertation, Universit. Miinchen, 1978.
2. Dombek В. D., Angelici R. J., Inorg. Synth., 17, 100 (1977).
□ Гексафторофосфат (т]-циклопентадиенил)дикарбонил-(тиокарбонил)железа(П) [Ре(С8)(СО)2(т]-С5Н5)] [PF6]
a) Fe(CSSCH3)(CO)2(T]-C6H6) + HCI ---> [FeCS(CO)2(T]-C5H6)]Cl + CH3SH
268,1 36,5 256,5 48,1
6) [FeCS(CO)2(T1-C5H6)]Cl + NH4PFe -->
256,5 163,0
----> [FeCS(CO)2(n-C5H5)] [PFJ + NH4CI
366,0 53,5
В хорошо перемешиваемый раствор 1,8 г (6,7 ммоль) Fe(CSSCH3) (СО)2(т]« С5Н5) в 150 мл пеитана в течение 10 мии продувают слабый ток НС1, затем упаривают в вакууме водоструйного насоса до объема —100 мл и снова 10 мин продувают током НС1. (Для уменьшения сильного неприятного запаха между колбой и водоструйным насосом ставят ловушку.) Дают осесть образовавшемуся осадку, осторожно декантируют и растворяют остаток в возможно меньшем количестве ацетона. Добавляют концентрированный раствор NH^PFs] (1,1 г, 6,75 ммоль) в ацетоне, фильтруют и упаривают до объема — 10 мл. Затем прикапывают эфир до тех пор, пока полностью не выпадет продукт. Выход 1,8 г (73%).
Аналитически чистый образец получают повторным высаживанием эфиром из ацетона.
2102 Глава 33. Металлоорганические комплексы
Свойства. Светло-желтый порошок или кристаллы. Очень чувствителен к гидролизу. При температуре —200°C разлагается. ИК (ацетон): 2102, 2072 (оч. с.) [v(CO)J, 1350 (с.) [v(CS)] см-1.
ЛИТЕРАТУРА
1. Busetto L., Beliuco U., Angelici R. J., J. Organometal. Chem., 18, 213 (1969).
2. Dombek B. D., Angelici R. J., Inorg. Synth., 17, 100 (1977).
3. Mayr A., Fehlhammer W. P„ неопубликованные результаты.
□ □ Бис(трифенилфосфин) (дикарбонил) (т)2-сероуглерод)железо Fe(n2-CS2)(CO)2[P(C6H5)3]2
Первый синтез этого комплекса [2] осуществлен из Fe2(CO)s. Существенно больший выход (90%) был достигнут позднее, исходя из бензилиденаце-тон (трикарбонил) железа [3].
В варианте, приводимом ниже, используют продажный Fe(CO)5 и для облегчения замещения СО добавляют триметиламииоксид.
Fe(CO)5 + 2P(C6H6)S + CS2 + 2(CH3)3NO --->
195,9 2-262,3 76,1 2-75,1
---> Fe(T]2-CS2) (CO)2[P(CeH6)s]2-f-2(CH3)3N 4-2СО3 + CO 712,6 2-59,1 2-22,4л 22,4 л
В двугорлую колбу на 500 мл с обратным холодильником (с ртутным затвором) и капельной воронкой в атмосфере N2 помещают последовательно раствор 20 г (76 ммоль) трифенилфосфина в 80 мл 96%-ного этанола, 30 мл CS2 и затем 5 мл (37 ммоль) Fe(CO)5. К хорошо перемешиваемой смеси в течение 20 мин прикапывают 8,2 г (74 ммоль) триметиламиноксида-дигидрата в 80 мл 96%-ного этанола, при этом тотчас начинается выделение газа и выпадение ржаво-красного осадка. По окончании прибавления нагревают на водяной бане до тех пор, пока газ не перестанет выделяться. Осадок отделяют на фильтре, промывают последовательно 100 мл эфира, 100 мл этанола, 100 мл эфира и высушивают в высоком вакууме. Выход 21—22 г (84—80%).
Свойства. Красный мелкокристаллический порошок. /пл 148—150 °C (с разл.). Умеренно растворяется в СН2С12, CS2 и бензоле, не растворяется в эфире и спиртах. Трифенилфосфиновые лиганды легко могут быть замещены на другие третичные фосфины или фосфиты. ИК (СН2С12): 1997, 1936 (оч. с.) [v(CO)]; (нуйол): 1135 (с.) [v(C = S)] см-‘.
ЛИТЕРАТУРА
1. LeBozec Н., Dixneuf Р., частное сообщение.
2. Baird М. С., Hartwell G. Jr., Wilkinson G., J. Chem. Soc. (London), A1967, 2037.
3. LeBozec H„ Dixneuf P„ Taylor N. J., Carty A. J., J. Organometal. Chem., 135, C29 (1977).
Цианокомплексы 2103
□ □ Бис(трифенилфосфин) (т]2-сероуглерод) платина
Pt(T]2-CS2) [Р(С«Н5)3]2
Pt[P(CeH6)3]s + CS2 -> Pt(n2-CS2)[P(C6H5)3]2+P(C6H6)3
982,0 76,1 795,8 262,3
В колбе на 100 мл, снабженной обратным холодильником с ртутным затвором, суспендируют в 50 мл эфира при перемешивании 1,47 г (1,5 ммоль) трис (трифенилфосфин) платины и нагревают до кипения. После прибавления через холодильник 3 мл CS2 желтый цвет реакционной смеси моментально изменяется на розовый. Перемешивают еще — 15 мин при нагревании, отфильтровывают осадок, дважды промывают 25 мл эфира и сушат 3 ч в высоком вакууме. Выход 1,1 г (94%). Соединение может быть перекристаллизовано из смеси CH2CI2 — эфир в присутствии небольших количеств CS2.
Pd(’n2-CS2) [Р(СвН6)3]2 получают совершенно аналогично из Pd[P(C6H5)3]3 и CS2.
Свойства. Мелкокристаллический порошок розового цвета или оранжевокрасные иглы. Соединение устойчиво на воздухе. /пл 158—165°C (с разл.). Хорошо растворяется в СН2С12, СНС13, тетрагидрофуране и бензоле, причем переходит в раствор лишь постепенно. Не растворяется в углеводородах, эфире, ацетоне, ацетонитриле и спиртах. В растворах, в особенности при нагревании, соединение медленно разлагается на воздухе. ИК (КВг): 1142 (оч. с.) [v(C=S)], 651 (ср.) [v(C-S)] см-1. ЯМР-31Р (CD2CI2, 85%-ная Н3РО4 — внешн. эталон, 25°C): б —20,9, '/(Pt—Р) =2820 Гц, б —32,2, '/(Pt—Р) = =4840 Гц.
ЛИТЕРАТУРА
1. Baird М. С., Wilkinson G., J. Chem. Soc. (London), Al967, 865. (
Цианокомплексы
□ □ Гексакис (о-толилизоцианид) хром* Сг(СМС6Н4-о-СНз)6 v
ЗСг2(Н2О)2(СН3СО2)4+ 12CNC6H4-0-CH3 -->
3-376,2 12-117,2
----> 2Cr(CNC8H4-o-CH3)6 -f- 4Cr(CH3CO2)3 4- 6H2O 2-754,9 4-229,1 6-18,0
В трубке Шленка емкостью 200 мл суспендируют 6,0 г (16 ммоль) ацетата хрома (II) в 100 мл метанола. Прикапывают 18,0 г (0,154 моль) о-толилизо-цианида*, перемешивают 1,5 ч при комнатной температуре, 30 мии кипятят с обратным холодильником. После охлаждения выпавший красный осадок отфильтровывают на стеклянном пористом фильтре G3. Продукт переосаждают из бензола гексаном, промывают холодным гексаном и сушат в вакууме. Выход 4,8—6,0 г (60—75%).
* Согласно номенклатурным правилам ИЮПАК, изложенным в книге Лидина Р. и др. «Основы номенклатуры неорганических веществ» (под ред. Б. Д. Степина, М., Химия, 1983), для цианид-иона CN- и цианат-иона OCN“ написание NC- и NCO- и названия «изоцианид» и «изоцианат» считаются устаревшими. — Прим, перев.
2104 Глава 33. Металлоорганические комплексы
Свойства. Сверкающие красные, в падающем свете отливающие зеленым цветом призмы. ;пл 149 °C. Очень хорошо растворяется в тетрагидрофуране, хорошо — в CH2CI2, плохо — в ацетоне и эфире. В пентане, метаноле и изопропиловом спирте не растворяется. ИК (СН2С12): 1950 (оч. с., шир.) (v(CN)] см-1.
ЛИТЕРАТУРА
1. Malatesta L., Sacco A., Ghielmi S., Gazz. Chim. Ital., 82, 516 (1952).
2. Essenmacher G. J., Treichel P. M., Inorg. Chem., 16, 800 (1977).
□ □ (Пентакарбонил)триметилсилилизоцианидхром Cr[CNSi(CH3)3] (CO)5
ftv
a) Cr(CO)e + C4H8O----> Cr(C4H8O) (CO)8 + CO
220,1 72,1 264,2 22,4 л
6) Cr(C4H3O) (CO)8 4- (CHgJjSiCN-> Cr[CNSi(CH3)3](CO)6 + C4H8O
264,2 99,8 291,2 72,1
В фотореактор емкостью 500 мл в атмосфере аргона помещают 1,10 г (5 ммоль) Сг(СО)8 в 450 мл тетрагидрофурана, свободного от О3, и облучают УФ-светом в течение ~3 ч. Лучшие выходы достигают при использовании тетрагидрофурана, насыщенного аргоном.
Контроль за ходом реакции осуществляют газометрически по количеству выделившегося СО. Когда СО больше не выделяется, облучение прекращают и к реакционной смеси добавляют 0,50 г (5 ммоль) триметилсилилцианида. Через 2 ч раствор становится бесцветным, тогда растворитель удаляют в вакууме водоструйного насоса при комнатной температуре. Желто-белый остаток переносят в сублиматор и в высоком вакууме возгоняют не вошедший в реакцию Сг(СО)6 при комнатной температуре. Затем температуру повышают до ~50°С и возгоняют чистый Cr[CNSi(CH3)3] (СО)3. Выход 1,05 г (72%).
По аналогичной методике получают Мо[С1Ч51(СН3)з](СО)6 и W[CNSi(CH3)31-(CO)s- t.
Другие методы. Силилирование Na[Cr(CN) (СО)5] триметилхлор-силаном [2]. Нагревание в течение 7 дней смеси Сг(СО)8 в (CH3)3SiCN с выходом 50% [3].
Свойства. Белые кристаллы, умеренно устойчивые на воздухе, одиако лабильные при нагревании. 1ПЛ 107—108 °C. Хорошо растворяется в полярных органических растворителях. ИК (КВг): 2116 (с.) [v(CN)], 2047 (с.), 1940 (оч. с.) [v(CO)], 1255 (с.), 850 (оч. с.), 765 (с.) [v{Si(CH3)3}J см-'.
ЛИТЕРАТУРА
1. Fehlhammer W. Р., Beck F„ Moll М., неопубликованные данные.
2. King R. В., Inorg. Chem., 6, 25 (1967).
3. Treichel Р. М., Shaw D. В., J. Organometal. Chem., 139, 21 (1977).
□ □ Пентакарбонил[(цианид-N) водорода]хром Cr(CNH)(CO)s
Cr[CNSi(CH3)3] (CO)6 4- CH8OH -> Cr(CNH) (CO)6 + (CH3)3SiOCH3
291,3 32,0 219,1 104,2
В маленькую трубку Шленка помещают в атмосфере азота 200 мг (0,7 ммоль) Cr[CNSi(CH3)3] (СО)8 в 10 мл метанола и перемешивают 12 ч.
Цианокомплексы 2105
Затем растворитель удаляют в вакууме водоструйного насоса, остаток возгоняют в вакууме при 40°C (10-3 мм рт. ст.). Выход ~50 мг (—33%).
Другие способы. Протонирование пентакарбонил (циано) хромата (0) с выходом 41% [2].
Свойства. Кристаллы белого или бледно-желтого цвета. 1„л 102 °C (с разл.). Медленно разлагается на воздухе. Растворяется в полярных органических растворителях. ИК (CH2CI2): 3485 (ср.) [v(NH)], 2105 (оч. сл.) [v(CN)J, 2025 (ср.), 1955 (оч. с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Fehlhammer W. Р„ Beck F., Moll М., неопубликованные данные.
2. King Р. В., Inorg. Chem., 6, 25 (1967).
□ □ Пентакарбонил(фенилизоцианид)хром Cr(CNC6H5)(CO)5
Метод, приведенный здесь, исключает получение и использование свободного фенилизоцианида. Изоцианидный* лиганд образуется в самом комплексе при расщеплении аддукта металлкарбонилата с карбодиимидом при действии фосгена.
a) Na2[Cr(CO)s] + C(CeH6N)2 --> Na2[Cr(CO)5-C(NC6H6)2]
238,0 194,2 432,3
б) Na2[Cr(CO)5 • C(NC6H5)2]-f-СОС12 ->
432,3 98,9
---> Cr(CNCeH5) (CO)6 4- C6H6NCO 4- 2NaCl 295,2 119,1 2-58,4
Работа с карбонилатами металлов требует абсолютного исключения Ог и влаги. Поэтому всю используемую посуду предварительно хорошо прогревают.
Сливают вместе насыщенные азотом растворы 2,5 г (10,5 ммоль) Иаг[Сг(СО)5] в 150 мл тетрагидрофурана и 2,04 г (10,5 ммоль) дифенилкарбо-диимида в 20 мл тетрагидрофурана при температуре —78 °C и перемешивают 10 мин. Затем из пипетки вливают —11 мл —1 М раствора фосгена в тетрагидрофуране (11 ммоль) и дают нагреться реакционной смеси до комнатной температуры. Остаток после отгоики растворителя в вакууме водоструйного насоса высушивают в высоком вакууме и экстрагируют пентаном (5x20 мл). Объединенный экстракт фильтруют через целлюлозу и в высоком вакууме отгоняют растворитель досуха. Для дальнейшей очистки проводят возгонку в высоком вакууме при 50°С. Выход 1,8 г (58%).
Другие способы. Из пентакарбонильных комплексов хрома типа Сг(ТГФ)-(СО)в или [N(C2Hs)4] [Cr(CO)sQ] и свободного фенилизоцианида [2].
Свойства. Бесцветные кристаллы. /пл 70°C (без разл.). Хорошо растворяется в пеитане. ИК (циклогексан): 2135 (с.) [v(CN)], 2052 (с.), 1965 (оч. с.), 1933 (пл.) [v(CO)J см-1.
ЛИТЕРАТУРА
1. Fehlhammer W. Р., Mayr A., Hitter М., Angew. Chem., 89, 660 (1977).
2. Connor В. J. A., Jones Е. М., McEwen G. К., Lloyd М. К.., McCleverty J. А., J. Chem. Soc. Dalton, 1972, 1246.
* См. предыдущее примечание. — Прим, перев.
2106 Глава 33. Металлоорганические комплексы
□ □ Циано(пентакарбонил)вольфрамат(0) натрия
и тетраэтиламмония M[W(CO)5(CN)] (M = Na, [М(С2Н5)4])
a) HN[Si(CH3)3]2 + NaNH2 --> Na[N{Si(CH3)3}2] + NH3
161,4 39,0 183,4 17,0
6) W(CO)6 + Na[N{Si(CH3)3}2] -> Na[W(CO)s(CN)] + O[Si(CH3)3]2
351,9 183,4 372,9 162,4
в) Na[W(CN) (CO)5] + [N(C2H5)4]Br --> [N(C2H5)4] JW(CO)5(CN)] + NaBr
372,9 210,2 480,2 102,9
а) Бис(триметилсилил)амид натрия Na[N{Si(CH3)3}2] [1, 2]
В трехгорлую колбу, снабженную обратным холодильником с ртутным затвором, помещают 27 г 30%-ной суспензии NaNH2 в толуоле (0,2 моль) и 32,3 г (0,2 моль) гексаметилдисилазана (например, фирмы Merck-Schuchardt) в 200 мл абсолютного бензола и при постоянном перемешивании нагревают так, чтобы смесь слабо кипела. Через — 2 дня выделение аммиака заканчивается. Реакционную смесь фильтруют еще теплой через широкий (диаметр 8 см) стеклянный пористый фильтр G3 и упаривают фильтрат до начала кристаллизации. При добавлении 100 мл абсолютного петролейного эфира выпадает Na[N{Si(CH3)3}2] в виде белого порошка. Его отфильтровывают в атмосфере инертного газа, промывают (3x25 мл) петролейным эфиром и сушат в высоком вакууме. Выход 24,8—29,3 г (68—80%).
Упариванием маточника и повторным высаживанием петролейным эфиром получают дополнительную порцию желаемого продукта, однако менее чистого.
Свойства. Бесцветное, хорошо кристаллизующееся вещество. /пл 165— 167 °C. Растворяется в ароматических углеводородах, плохо — в алифатических.
б) Циано(пентакарбонил)вольфрамат(0) натрия Na[W(CO)5(CN)J [3, 4]
В трубке Шленка емкостью 300 мл суспендируют 11,3 г (32,1 ммоль) W(CO)6 в 100 мл абсолютного бензола и по каплям прибавляют предварительно профильтрованный раствор 5,8 г (31,6 ммоль) бис(триметилсилил) амида натрия в 100 мл бензола. Смесь перемешивают 4—5 дней, дают осесть и затем фильтруют очень мелкий осадок на широком стеклянном пористом фильтре G3 при небольшом избыточном давлении азота. Процесс фильтрования и промывания осадка может продлиться почти целый день. Ни в коем случае нельзя фильтровать с отсасыванием, иначе выкристаллизовывающийся силазид натрия забьет поры фильтра. Многократно промывают бензолом и петролейным эфиром (до 100 мл в сумме каждого) и высушивают в высоком вакууме. Не вошедший в реакцию гексакарбонил возгоняют в высоком вакууме при 40 °C. Выход 9,9 г (83%).
Свойства. Бесцветное, иногда сероватое вещество, имеет консистенцию муки, /„л >210°C (с разл.). Хорошо растворяется в воде. ИК (поли(хлортрифторэтиленовое) масло]: 2118 (сл.) (v(CN)], 2064 (сл.), 1995 (пл.), 1970 (оч. с.), 1922 (оч. с.), 1812 (с.) [v(CO)] см-1.
в) Циано(пентакарбонил)вольфрамат(0) тетраэтиламмония [N(C2H5)4][W(CO)5(CN)] ;[4]
Фильтрование осадка Na[W(CO)3(CN)] (9,9 г, 26 ммоль), получаемого способом (б), в данном случае проводят через фильтр, предварительно покрытый ~ 1 см слоем песка. Время фильтрации, а также последующего промы
Цианокомплексы. 2107
вания значительно сокращается. Осадок на фильтре вместе с песком высушивают непродолжительное время в вакууме. Затем добавляют прямо на фильтр 500 мл воды, предварительно насыщенной азотом, и прикапывают фильтрат непосредственно в раствор 6,3 г (30 ммоль) [ЩСгЬМ^Вг в 20 мл воды. Образуется осадок цвета слоновой кости. Его отделяют, промывают водой до тех пор, пока фильтрат не станет бесцветным, отфильтровывают и сушат в высоком вакууме. Дальнейшую очистку проводят переосаждением из бензола петролейным эфиром. Выход 11,7 г [91,7%, что составляет 77% в расчете на W(CO)e].
Аналогично могут быть получены следующие комплексы хрома:
Na[Cr(CO)5(CN)]: выход 88%, /пл >247°C (с разл.). ИК (КВг): 2114 (сл.) [v(CN)J, 2079 (сл.), 1933 (с.), 1925 (оч. с.), 1824 (с.) [v(CO)] см-1.
[N(C2H5)4][Cr(CO)5(CN)]: выход 93%, <пл 98°C. ИК (СН2С12): 2098 (сл.) [v(CN)J, 2052 (сл.-ср.), 1967 (пл.), 1930 (оч. с.), 1890 (пл.) [v(CO)] см-1.
Свойства. Устойчивые на воздухе, чешуйчатые кристаллы цвета слоновой кости. /пл 123 °C. Хорошо растворяется в СН2С12, СНС13, ацетоне и тетрагидро-фураие. ИК (СН2С12): 2095 (оч. сл.) [v(CN)], 2050 (сл.), 1966 (пл.), 1927 (оч. с.), 1885 (пл.) [v(CO)J см-1.
ЛИТЕРАТУРА
1. Wannagat U., Niederprum Н., Вег., 94, 1540 (1961).
2. Kriiger С. R., Niederprum Н., Inorg. Synth., 8, 15 (1966).
3. Ring R. В., Inorg. Chem., 6, 25 (1967).
4. Arbetsvorschrift des Anorgan. — chem. Inst. Univers. Erlangen—Niirnberg.
□ Циано(т]-циклопентадиенил) (нитрозил)карбонилмарганец
Mn(CO)(NO)(T]-C5H5)CN
[Mn(CO)3NO(r]-C5H5)] [BF4] + KCN ->
292,9 65,1
---> Mn(CO) (NO) (t]-C5H5)CN + KBF4 + CO
204,1 125,9 22,4л
580 мг (2 ммоль) [Мп(СО)2НО(г|-С5Н5)] [BF4] (см. методику синтеза) в течение 2 ч перемешивают при небольшом нагревании с 130 мг (2 ммоль) KCN в 20 мл метанола или водного метанола (1:1). Выделяется газ, а раствор становится красным. После удаления растворителя остаток растворяют в СН2С12, фильтруют и хроматографируют на А120з (III степени) метилендихлоридом. Коричневая фракция, идущая вначале, содержит [Mn(CO)NO(r|-CsH5)]2. Следующую затем красную зону собирают, максимально возможное количество растворителя удаляют и добавляют петролейный эфир до начала помутнения. Затем фильтруют и раствор при —20 °C оставляют на сутки. Продукт выпадает в виде кристаллов красного цвета. Выход 140 мг (34%).
Другие способы. Из Na[Mn(CO)2(T]-C5H5)CN] и NOC1 с выходом 25% [1]. Соединение образуется в качестве побочного (5%) в реакции [Mn(CS)(СО)2(п-С5Н5)]+ cNCX- (Х=О, S) [2].
Свойства. Красные кристаллы. /пл 94°C (разлагается с выделением газа). Растворяется в СН2С12, СНС13, ацетоне, метаноле и воде. Не растворяется в бензоле, петролейном эфире и ССЦ. ИК (СН2С12): 2125 (с.) [v(CN)l, 2050 (оч. с.) [v(CO)J, 1798 (оч. с.) [v(NO)l см-1. ЯМР-'Н (CDC13, ТМС): б 5,36 [С5Н5].
«108 Глава 33. Металлоорганические комплексы
JWWKTHVb
1. Landgraf G., Behrens H., неопубликованные данные.
2. Efraty A., Arneri R„ Ruda W7. A., Inorg. Chem., 16, 3124 (1977).
□ □ Циано(т]-циклопентадиенил)дикарбонилманганат(1) натрия NatMnCCO^^-CsHsXCN)]
Mn(CO)3(n-CsH5) + Na[N(Si(CH8)8}2] ->
204,1 183,4
---> Na[Mn(CO)2(n-C5H5)CN] + OfSiXCHs^, 225,1 ’ 162,4
К раствору 1,50 г (7,3 ммоль) Мп(СО)з(т1-С8Н5) в 50 мл бензола приливают предварительно профильтрованный раствор 1,50 г (8,1 ммоль) бис(три-метилсилил) амида натрия в 150 мл бензола и нагревают смесь 14 ч в запаянной ампуле при 120 °C. По окончании реакции выпавшие светло-желтые кристаллы многократно промывают бензолом. Лучше всего для этого перемешать суспензию в бензоле, дать осесть и декантировать. Эту процедуру повторяют еще 2—3 раза, используя порции бензола по 30 мл. Выход 1,5 г (92%).
Другие способы. Из Мп(СО)з(г|-С6Н5) и NaCN с выходом 47% [2].
Свойства. Светло-желтые кристаллы. Соединение весьма неустойчиво иа воздухе. Растворяется в воде, спирте и ацетоне. С алкилирующими агентами типа [ОРз] [ВР4] образует изоцианидные* комплексы Mn(CNR) (СО)2(г|-С5Нб). ИК (КВг): 2059 (с.) [v(CN)J, 1907 (оч. с.), 1825 (оч. с.) [v(CO)l см-1. ЯМР-'Н (ацетон-<1в, ТМС): 5 4,32 {С5Н5].
ЛИТЕРАТУРА
1. Moll М., Dissertation, Univers. Erlangen—NUrnberg, 1974.
2. Fischer E. 0., Schneider R. J. J., J. Organometal. Chem., 12, P27 (1968).
□ Тетрафтороборат гексакис(4-изоциано-4-метилпеитанон-2)-железа(П) [Fe{CNC(CH3)2CH2C(O)CH3}6] [BF4]2
у-Оксоизоциаиидиый лиганд* образуется при «алкилировании» цианидного лиганда а,Р-ненасыщенными соединениями в протонной среде:
K4[Fe(CN)e] ЗН2О + 6(СН8)2С=СНСОСН8 + 6HBF4 -->
422,4 6-98,1 6-87,8
---> [Fe{CNC(CH8)2CH2C(O)CH3]6] [BF4]2 + 4KBF4 + ЗН2О 980,5 4-125,9 3-18,0
В смесь красно-коричневого цвета, состоящую из 42,8 г (0,44 моль) мези-тилоксида и 100 мл 35%-ной HBF4 (0,49 моль), порциями добавляют 21,1 г (0,05 моль) K4[Fe(CN)6]-ЗН2О. Суспензию интенсивно перемешивают 12 ч. Образуется кашицеобразная масса от темно-синего до фиолетового оттенков, в которой желаемый продукт загрязнен бесцветным KBF4 и не вошедшим в реакцию исходным комплексом. Смесь разбавляют водой до тех пор, пока не
* См. предыдущее примечание. — Прим, перед.
Цианокомплексы 2109
будут отчетливо видны две фазы. KBF4 н продукт реакции медленно оседают на дно реакционного сосуда. Осадок отделяют н тщательно промывают водой. Высушенный досуха осадок промывают на фильтре CH2CI2 порциями по 30 мл до тех пор, пока при разбавлении фильтрата петролейным эфиром (/кип 40— 60 °C) не прекратится помутнение раствора. Объединенные вытяжки СН2С1г немного упаривают, сушат СаС12, фильтруют через короткую колонку, заполненную А120з, и разбавляют смесью эфир — петролейный эфир. Выпавший осадок перекристаллизовывают из смеси CH2CI2 — петролейный эфир. Выход 40,7 (83%).
Другие способы. Указанное соединение можно получить также из K4[Fe(CN)e] и смеси ацетона с [(С2Нз)зО]Вр4, либо с BFs-эфир, либо с HBF<. В качестве «алкилирующего» средства может быть использован и диацетоновый спирт вместе с HBF4. Далее, можно исходить не из желтой кровяной соли, а из красной кровяной соли.
Свойства. Бесцветные, сравнительно устойчивые на воздухе кристаллы. /пл 149 °C. Растворяется в СНС1з и СН2С12, не растворяется в диэтиловом и петролейном эфирах. Растворы на воздухе медленно разлагаются, окрашиваясь в голубой цвет. ИК (КВг): 2215 (с.) [v(CN)], 1725 (с.) [v(CO)J, 1058 (с., шир.) fv(BF4)l см-’. ЯМР-’Н (СН2С12, ТМС): 6 1,62 [синглет, 6Н, С(СН3)2]; 2,18 [синглет, ЗН, COCHS]; 3,03 [синглет, 2Н, СН2].
ЛИТЕРАТУРА
(. Schaal М„ Dissertation, Univers. Mflnchen, 1976.
2. Schaal M., Beck W., Angew. Chem., 84, 584 (1972).
О Гексафторофосфат (т]-циклопентадиенил)(дикарбонил)-(фенилизоцианид)железа(П)* [FefCNCeHsHCOM^-CsHs)] [PF6]
a) Na[Fe(CO)2(T]-C6H6)] + C6HsNCS---> Na[Fe(CO)2(T)-C5Hs)-C(S)NCeHs]
200,0 135,1 335,1
6) Nape^^-CsHsi-QSJNCeHJ + COCh ----------->
335,1 98,9
----> (Fe(CNC6H5) (CO)2(n-C5H5)]Cl + COS + Nad 315,5 60,1 58,4
в) [Fe(CNC6H5) (CO)2(n-C5H5)]Cl + NHJPFJ -->
315,5 163,0
----> [Fe(CNC6H5) (CO)2(t1-CbH5)I [PFJ + NH4C1 425,1 53,5
К насыщенному N2 и охлажденному до —70 °C раствору 12 ммоль Na[Fe(CO)2(n-C5H5)] в 150 мл тетрагидрофурана, полученному восстановлением 2,12 г (6 ммоль) [Ре(СО)2(п-С5Н5)]2 сплавом Na/K в 150 мл ТГФ, добавляют 1,62 г (12 ммоль) фенилизотиоцианата в 20 мл ТГФ, перемешивают 10 мин и прикапывают далее избыток ~1 М раствора COCI2 в ТГФ. После отгонки растворителя в вакууме водоструйного насоса остаток высушивают в высоком вакууме, растворяют в 150 мл воды и фильтруют. С этого момента можно работать иа воздухе.
* См. предыдущее примечание. — Прим, перев.
2110 Глава 33. Металлоорганические комплексы
К фильтрату добавляют раствор 1,95 г (12 ммоль) NH4[PFe] в 10 мл воды, выпавший осадок отфильтровывают, сушат в высоком вакууме и перекристаллизовывают из смеси ацетон — эфир. Выход 3,65 г (71%).
Свойства. Оранжевые, устойчивые на воздухе кристаллы. 1ПЛ 154—155 °C (с разл.). Очень хорошо растворяется в ацетоне н ацетонитриле, несколько хуже—в СН2С12 и СНС1з и ие растворяется в эфире. ИК (СН2С12): 2192 (с.) [v(CN)J, 2083, 2046 (с.) [v(CO)J см-1.
ЛИТЕРАТУРА
1. Fehlhammer W. Р., Mayr A., J. Organometal. Chem., 191, 153 (1980).
□ □ Гексафторофосфат (трциклопентадиенил)(карбонил)-бис(фенилизоцианид)железа( II)* [Fe(CNC6H5)2(CO)(T1-C5H5)][PF6]
a) Na[Fe(CO)2(T]-C5H6)l + C(NCeH5)2 -> Na[Fe(CO)3(n-CsH5)-C(NC6H5)2}
200,0 194,2 394,2
б) Na[Fe(CO)2(ri-C5H5)-C(NCeH5)2l + COCl2 ->
394,2 98,9
----> [Fe(CNCeH5)2(CO) (n-C5H5)]Cl + СО2 + NaCl 390,7 22,4 л 58,4
в) [FefCNCgHs^CO) (т)-СвН5)]С1№Ц[РРв] -----»
390,7 163,0
---> (Fe(CNC6H5)2(CO) (П-С5Н5)1 [PFe] + NH4C1
500,2 53,5
Готовят насыщенный азотом раствор 20 ммоль Na[Fe(CO)2(r)-C5H5)} в 200 мл тетрагидрофурана из 3,54 г (10 ммоль) >[Ре(СО)2(т]-С5Н5)]2 и 6,0 мл амальгамы натрия [см. методику синтеза Fe(T)-C5H5) (CH3) (СО)2], а также насыщенный азотом раствор 3,9 г (20 ммоль) дифеиилкарбодиимида в небольшом количестве ТГФ. Эти растворы при комнатной температуре сливают и через 5 мин перемешивания охлаждают до —80 °C. Затем прикапывают раствор 20 ммоль СОС12 в ТГФ (1 М; титрование NaOH после гидролиза) или просто пропускают ток СОС12 через интенсивно перемешиваемый раствор. Дальнейшие операции можно проводить на воздухе.
Растворитель отгоняют при комнатной температуре, растворяют остаток, предварительно высушенный в высоком вакууме, в ~ 100 мл воды и добавляют водный раствор 3,3 г (20 ммоль) NH4[PF6], Выпавший осадок отфильтровывают, высушивают в высоком вакууме и перекристаллизовывают из кипящего этанола. Выход 6,1 г (63%).
Свойства. Кристаллизуется в виде иголочек медово-желтого цвета. /,,л 138—139 °C. Растворяется в ацетоне и метилендихлориде, в эфире и углеводородах ие растворяется. ИК (СН2С12): 2183, 2156 (с.) [v(CN)J, 2034 (с.) [v(CO)] см-1.
ЛИТЕРАТУРА
1. Fehlhammer W. Р., Mayr A., Ritter М., Angew. Chem., 89, 660 (1977).
* См. предыдущее примечание. — Прим, перев.
Цианокомплексы 2111
□ □ Хлорид (т)-циклопентадиенил)трис(фенилизоцианид)-железа(П) [Fe(CNC6H5)3 (п-С5Н5)]С1
Fe(CO)2(n-C5H5)Cl + 3CNC6H5 -> [Fe(CNC6H5)3(n-C5H5)]Cl + 2CO
212,4 3-103,1 465,8 2-22,4Л
В двугорлую колбу емкостью 500 мл с внутренним термометром и эффективным холодильником помещают 4,24 г (20 ммоль) Fe(CO)2(r]'C5H5)Cl и 6,18 г (60 ммоль) фенилизоцианида в 50 мл абсолютного бензола и осторожно нагревают на плитке. При ~60°С начинается сильное пенообразование. Чтобы образующая пена не заполнила весь прибор, плитку на короткое время отставляют. После прекращения выделения СО нагревают 1,5 ч с обратным холодильником, охлаждают до комнатной температуры и отфильтровывают выпавшие оранжевые кристаллы. Промывают ацетоном (5 мл) и сушат в вакууме. Выход 7,4 г (80%).
Для очистки сырой продукт растворяют в минимальном количестве СН2С12, фильтруют через слой (~1 см) целлюлозы, к фильтрату добавляют несколько капель эфира и еще раз фильтруют. Затем добавляют эфир до начала помут» нения, оставляют стоять несколько часов в холодильнике и выпавшие кристаллы отфильтровывают. Выход 3,4 г (37%).
Свойства. Кристаллы оранжевого цвета, устойчивы на воздухе. /пл 174 °C. Растворяется в СНС13, СН2С12; не растворяется в эфире и пеитане. ИК (КС1): 2190,2123 (с.) [v(CN)J см-1.
ЛИТЕРАТУРА
1. Joshi К. К., Pauson Р. L., Siubbs W. Н., J. Organometal. Chem., 1, 51 (1963).
□□ Гексацианодиникколат(1) калия (Ni—Ni) K4[Ni2(CN)6]
□ □ Тетрацианоникколат(О) калия K4[Ni(CN)4]
Восстановление легкодоступного K4[Ni(CN)4] лучше всего следует проводить в жидком аммиаке. При недостатке металлического К получают красную соль Веллуччи K4[Ni2(CN)3]. Избыток К приводит к восстановлению до K4[Ni(CN)4].
K4[Ni(CN)4]
2K2[Ni(CN)4] + 2К---> K4[Ni2(CN)6] + 2KCN
2-241,0 2-39,1 429,9 2-65,1
В охлажденный до —78 °C (смесью ацетона с сухим льдом) реакционный сосуд с впаянным стеклянным фильтром (общей емкостью 250 мл) конденсируют 100 мл аммиака, предварительно осушенного пропусканием через промывную склянку 1, заполненную КОН, и ловушку 3 с кусочками Na (рис. 478). В эту среду вносят 3,0 г (12 ммоль) твердого K2[Ni(CN)4], маленькими кусочками добавляют К (0,25 г, 6,5 ммоль, т. е. недостаток) и перемешивают на магнитной мешалке, используя запаянный в стекло сердечник магнита 4. Постепенно выпадает светло-красный осадок. Перемешивают еще 1 ч и отсасывают образующийся светло-желтый раствор через впаянный стеклянный фильтр 5, предварительно охладив до —78 °C, в другой приемник 6. Красное твердое вещество несколько раз промывают на фильтре жидким аммиаком (по 50 мл), снова конденсируя NH3 для этого в реакционный сосуд. Наконец, доводят температуру до комнатной, так что большая часть NH3 улетучивается
2112 Глава 33. Металлоорганические комплексы
Рис. 478. Аппаратура для получения K4[Ni2(CN)e] и K«[Ni(CN)4].
/ — промывная склянка, заполненная КОН (тв,); 2 — охлаждающая баня (ацетон+сухой лед); 3 — кусочки Na; 4 —’Магнит; 5 — стеклянный пористый фильтр; б —ловушка.
через ртутный затвор, а остатки растворителя удаляют в вакууме. Выход 915 мг (67%, в пересчете на К).
Свойства. Красное твердое аморфное вещество, при контакте с воздухом обесцвечивается в течение 1 ч. /пл 150°C (с разл.). Растворяется в воде (раствор темно-красного цвета), при доступе воздуха наблюдается постепенное пожелтение и выпадение Ni(OH)2. Обесцвечивание происходит и в отсутствие воздуха и других окислителей, но с выделением Н2.
K4Ni(CN)4]
K2[Ni(CN)4] + 2К-----> K4[Ni(CN)4]
241,0 2-39,1 319,2
В охлажденном до —70 °C реакционном сосуде емкостью 250 мл с впаянным фильтром (рис. 478) конденсируют —100 мл жидкого аммиака, предварительно высушенного пропусканием через КОН и над кусочками натрия. Растворяют в нем —2 г (8 ммоль) Ki[Ni(CN)4], добавляют 0,73 г (18 ммоль) К маленькими кусочками и перемешивают на магнитной мешалке, запаяв предварительно сердечник магнита в стеклянную трубку. Сначала К покрывается красной корочкой, а затем постепенно переходит в раствор, окрашивая его в голубой цвет. Одновременно образуется объемистый серый осадок. Вскоре после этого цвет реакционной смеси становится темно-синим, что указывает иа наличие избытка щелочного металла в растворе, постепенно реагирующего с растворителем с выделением Н2. Перемешивают до тех пор, пока голубой цвет не исчезнет; растворитель через фильтр удаляют и промывают желтосерый осадок на фильтре жидким аммиаком не менее 20раз порциями по 30— 50 мл для удаления непрореагировавшего Ki[Ni(CN)4]. Получившийся продукт сушат в высоком вакууме и хранят в атмосфере сухого чистого N2, свободного от Ог. Выход 2,4 г (94%).
Свойства. Аморфный порошок медного цвета, на воздухе тотчас чернеет. Растворяется в воде, однако реагирует с ней, образуя красный K4[Ni2(CN)e) и выделяя Н2. ,
ЛИТЕРАТУРА ?
1. Eastes J. W., Burgess W. M., J. Amer. Chem. Soc., 64, 1187 (1942).
Фосфиновые комплексы 211®
□ □ транс-Дифульминатобис(трифенилфосфин)платина Pt[P(C6H5)3]2(CNO)2
Для получения фульминатных комплексов, как правило, необходимы фульминаты щелочных металлов, которые в свою очередь могут быть получены» лишь из фульмината ртути. Ниже приводится простой и абсолютно безопасный способ, позволяющий получать комплексы с высокой термической устойчивостью, особенно пригодные для изучения химии координированного фуль-минатного лиганда.
Pt[P(C6H5)3]3 4- 2CHsNO2 ->
982,0 2-61,0
---> Pt[P(C6H5)3]2(CNO)2 + Р(С6Н5)3 + Н2 + 2Н2О
803,7 262,3 22,4 л 2-18,0
1,0 г (1 ммоль) Pt[P(C6H5)3]3 (или 1,25 г (1 ммоль) Р1[Р(СбН5)3]4) нагревают с обратным холодильником в смеси, состоящей из 10 мл этилового спирта, 10 мл бензола, 8 мл воды и 30 мл нитрометана, защищая от прямого-света и доступа воздуха. Выше 60 °C растворитель образует единую фазу, в которой комплекс платины (0) медленно растворяется. Приблизительно через 50 ч даже из теплого раствора начинает выпадать в осадок РЦР(СбН3)з]2-(CNO)2. Охлаждают до комнатной температуры, отфильтровывают выпавший' почти бесцветный продукт и перекристаллизовывают его из смеси СНС13 — петролейный эфир. Полученное кристаллическое соединение содержит одну молекулу СНС13 на формульную единицу. Выход 660 мг (82%).
Другие способы. Из смеси Ki[PtCl4], Р(С6Н5)3, КОН, нитрометана и точно-такой же смеси растворителей, как указано выше. Выход 70%.
Свойства. Бесцветные кристаллы. /пл 299—300 °C. Растворяется в СНС13. При температуре выше 200 °C происходит постепенное разложение и образование диизоцианатного* комплекса 4uc-Pt[P(C8H5)3]2(NCO)2. В присутствии органических карбонилсодержащих соединений эта изомеризация протекает в существенно более мягких условиях. Протонные кислоты с некоординирующимся аииоиом также вызывают указанное превращение [2]. ИК (КВг): 2325-[2Vc»m(CNO)], 2190 [VacBM(CNO)], 1153 [vs(CNO)J см-1. ЯМР-31Р (Н3РО4 — внешн. стандарт): б —13,4.
ЛИТЕРАТУРА
1. Schorpp К-, Beck W., Вег., 107, 1371 (1974).
2. Beck W„ Schorpp К., Oetker С., Вег., 107, 1380 (1974).
Фосфиновые комплексы
□ □ 1{ш>Бис(фенилфосфин)тетракарбонилмолибден
цис-Мо(СО)4(РН2С6Н5)2
Мо(п4-С7Н8) (СО)4 + 2РН2С6Н5 -> 4uc-Mo(CO)4(PH2C6H5)2 + С7Н81
300,1 2-110,1 428,2 92,1
К суспензии 45,0 г (0,15 моль) Мо(н4-С7Н8) (СО)4 (см. методику синтеза) в 200 мл гексана добавляют при охлаждении льдом в течение 1 ч 38,5 г (0,35 моль) фенилфосфина. Образующийся продукт выпадает в виде желтоватых кристаллов. Для очистки его перекристаллизовывают из петролейного» эфира (/кип 40—60°C). Выход 60 г (93%).
* См. предыдущее примечание. — Прим, перев.
17—1361
С2114 Глава 33. Металлоорганические комплексы
Свойства. Бледно-желтые кристаллы. Растворяется в эфире, тетрагндро-фуране. ИК (гексан): 2031, 1943, 1924 [v(CO)J; (нуйол): 2310 [v(PH)] см-1. -.ЯМР-'Н (CDC13, ТМС): 6 5,30 [PH], -Т.б^СвНз].
ЛИТЕРАТУРА
1. Johannsen G., Stelzer О., Вег., ПО, 3438 (1977).
Пентакис(трифторофосфин)железо Fe(PF3)5
Fe(PF3)5 получают восстановительным фторофосфииированием Fel2. Безводный активированный Fel2 получают двухчасовым нагреванием твердого .ре(СО)412 (см. методику синтеза) в вакууме при 90 °C.
Fe(CO)4I2 --> Fel2 + 4СО
421,7 309,7 4-22,4 л
Fel2 + Zn + 5PF3 —Fe(PF3)5 4-Znl2
309,7 65,4 5-88,0 495,7 319,2
20,0 г Fel2 и 16,0 г Zn-порошка, предварительно прогретого в вакууме, помещают в атмосфере сухого N2 во вращающийся автоклав на 250 мл с медными внутренними стенками. Для лучшего промешивания дополнительно кладут также латунные палочки. Затем в автоклав конденсируют сухой BF3 (см. методику синтеза), не содержащий свободной кислоты, при комнатной температуре создают рабочее давление 650 бар и нагревают автоклав в течение 48 ч при 130°C. После охлаждения до 80—100°C избыток PF3 и Fe(PF3)3 перекоиденсируют в трубку Шленка. При пониженном давлении постепенно отгоняют PF3, а оставшийся Fe(PF3)3 перегоняют над Р4О10. Выход 15,7 г (50%, в пересчете иа Fel2).
Свойства. Желтое, очень легколетучее твердое вещество с удушающим запахом. В воде не растворяется, однако хорошо растворяется в органических .растворителях. Возгоняется при 20°С (10-6 мм рт. ст.). ИК (газ): 915, 901 (оч. с.), 873, 864 (оч. сл.), 851 (оч. с.) [v(PF3)] см-’. ЯМР-19Р (пентан, CFC13— внешн. стандарт): 6 —1,9, J(PF) = 1275 Гц. ЯМР-31Р (пентан, 85%-ная Н3РО4— внешн. стандарт): 6 —163,5, J(PF) = 1270 Гц.
ЛИТЕРАТУРА
1. Kruck Th., Prasch A., Z. Anorg. Allgem. Chem., 356, 118 (1968). w
Гексафторофосфат (т]-циклопентадиенил){1,2-бис(дифенил-<фосфино)этан}карбонилжелеза(П)
;[Ре(СО){(С6Н5)2РС2Н4Р(СбН5)2}(г]-С5Н5)] [PF6]
a) Fe(CO)2(r1-C5H3)Cl + (С6Н6)2РС2Н4Р(С(.Н5)2
212,4 398,4
---> [Fe(CO) {(С6Н5)2РС2Н4Р(С6Н5)2) (П-С8Н6)]С1 + СО
582,8 22,4 л
б) [Fe(CO) {(С6Н5)2РС2Н4Р(С6Н5)2} (П-С5Н5)]С1 + NHJPFJ ->
528,8 163,0
----> [Fe(CO) {(С6Н5)2РС2Н4Р(С6Н5)2} (t]-C6H5)J [PF3] + NH4C1
692,3 53,5
В открытую колбу помещают 2,1 г (10 ммоль) Fe(CO)2(T]-C5H5)Cl и 4,2 г 4Ю,5 ммоль) (С6Н5)2РС2Н4Р(С6Н5)2 и хорошо перемешивают. Затем при лег
Фосфиновые комплексы 2115»
ком встряхивании прикапывают из пипетки —10 мл тетрагидрофураиа. В течение —30 с происходит сильное выделение газа, и образуется желтая вязкая кашица. Если реакция сразу после прикапывания ТГФ не начинается или протекает не до конца, вспенившуюся массу кратковременно нагревают при — 50 °C, чтобы в течение нескольких секунд завершить реакцию.
Тетрагидрофуран отгоняют на водоструйном насосе, остаток растворяют в 50 мл СН3ОН и добавляют раствор 1,8 г (11,0 ммоль) NH4[PF3] в 5 мл метанола. Выпавший желтый осадок отфильтровывают на стеклянном пористом фильтре G4 и перекристаллизовывают из СН2С12 (+20°C/—78°C). Выход 5,5 г' (80%).
Свойства. Желтые кристаллы. Растворяется в ацетоне, хуже — в тетрагидрофуране. ИК (КВг): 1970 [v(CO)J см-1. ЯМР-’Н (ацетон-й6, ТМС): 6 2,06, 2,76 [мультиплет, С2Н4], 5,08 [триплет, С5Н5], 7,51 [мультиплет, СбН5]._
ЛИТЕРАТУРА
1. Seltmann D., Kleinschmidt Е., J. Organometal. Chem., 140, 211 (1977).
Дихлоробис(трифенилфосфин)дикарбонилрутений Ru(CO)2[P(C6H5)3]2C12
со+нг
RuC13-3H2O + P(C6H5)3 --> Ru(CO)2[P(C6H5)3]2C12
261,5 262,3 752,6
В двугорлую колбу иа 100 мл, снабженную трубкой для ввода газа и обратным холодильником с ртутным затвором, помещают 0,26 г (1 ммоль) 37,8%-ного RuC13-3H2O, 0,7 г (2,7 ммоль) Р(С6Н5)3, 50 мл этанола и при перемешивании пропускают ток СО+Н2 (1 : 1). Когда воздух полностью будет вытеснен, смесь нагревают до кипения в течение —30 мин. Раствор из темнокрасного становится зеленым, затем почти бесцветным, и выпадают кристаллы серого цвета. Через 4 ч ток газа прекращают и оставляют смесь на ночь в атмосфере СО+Н2. Выпавшие кристаллы отделяют, промывают пентаном и высушивают в вакууме. Выход 0,63 г (84%). Для перекристаллизации продукт растворяют в атмосфере N2 при нагревании в смеси 2-метоксиэтанола с толуолом (10: 1) и ставят на ночь в холодильник.
Свойства. Белые игольчатые кристаллы. ИК (СН2С12): 2058, 1993 [v(СО)]; (CsI): 300, 380 [v(RuCl)] см-1.
ЛИТЕРАТУРА
1. Strohmeier W., Hitzel Е., Michel М„ Stasch H.-Р., Weigelt L., частное сообщение; Fahey D. R., Catalysis in Organic Synthesis, Rylander P. N., Greenfield H. Academic Press, London, 1976, p. 287.
□ Бис(трифенилфосфин)трикарбонилрутений Ru(CO)3[P(C6H5)3]2
koh+hcho
RuC13-3H2O + 2P(C6H5)3 ------->- Ru(CO)3[P(C6H5)3]2
261,5 2-262,3 709,7
В двугорлой колбе на 250 мл, снабженной обратным холодильником и краном для ввода азота, при интенсивном перемешивании нагревают раствор 2,37 г 17*
2116 Глава 33. Металлоорганические комплексы
(9 ммоль) трифенилфосфина в 90 мл 2-метоксиэтаиола. Затем максимально быстро один за одним добавляют в колбу 0,39 г (1,5 ммоль) КиСЬ-ЗНзО в 30 мл 2-метоксиэтанола, 30 мл теплого 40%-иого водного раствора формальдегида и 0,6 г (11 ммоль) КОН в 30 мл горячего 2-метоксиэтанола. Должна образоваться гомогенная смесь, которую кипятят еще 1 ч и охлаждают затем до комнатной температуры. Выпавший осадок отделяют, промывают последовательно этанолом (5 мл), водой (5 мл), этанолом (5 мл), гексаном (2x5 мл) и сушат в вакууме. Выход 0,95 г (89%).
Другие способы. Взаимодействие трифенилфосфина с Ru(CO)5 в автоклаве [5].
Свойства. Бледно-желтые, устойчивые на воздухе кристаллы. 170—
173°C (на воздухе) и 262—266°C (в запаянном под а’зотом капилляре). Растворяется хорошо в СНС1з и CH2CI2, умеренно — в бензоле; в спиртах и али--фатических углеводородах не растворяется. ИК (тетрагидрофуран): 1900; (иуйол): 1903, 1895 (с.) [v (СО) ] см-1.
ЛИТЕРАТУРА
1. Collman J. Р., Roper W. R-, J. Amer. Chem. Soc., 87, 4008 (1965).
2. Placentl F., Bianchi M., Benedetii E., Sbrana G„ J. Inorg. Nucl. Chem., 29, 1389 (1967).
-3. Levison I. J., Robinson S. D., J. Chem. Soc. (London), A 1970, 2947.
4. Ahmad N., Robinson S. D-, Uttley M. F„ J. Chem. Soc. Dalton, 1972, 843. 5. L’Eplattenier F., Calderazzo F., Inorg. Chem., 7, 1290 (1968).
DD Тетракис (триметил фосфин) кобальт Со[Р(СН3)з]4
Na-Hg
СоС12 4- 4P(CHs)g --->- Со[Р(СН3)а]4
129,8 4-76,1 363,2
В трубку Шленка емкостью 250 мл помещают 200 г ртути и при перемешивании магнитной мешалкой кусочками прибавляют 1,50 г (65,2 ммоль) Na. Растворение идет бурно, с шипением и сильным разогревом. После охлаждения к амальгаме добавляют ~ 100 мл эфира и затем 14,0 мл (147,4 ммоль) триметилфосфина. Далее прибавляют 4,0 г (30,8 ммоль) безводного C0CI2 и перемешивают сначала синюю, а позднее коричневую реакционную смесь 24 ч при комнатной температуре. Раствор декантируют, отфильтровывают от NaCl, используя кусочки бумажного фильтра. Твердый коричневый остаток, получившийся после отгонки растворителя в вакууме, возгоняют (0,1 мм ;рт. ст., вакуум масляного насоса) при ~80°С (температура масляной бани). Выход 8,1 (73%).
Свойства. Темно-коричневые кристаллы (из пентана*, —50°C). /пл>180°С (с разл.). ИК (нуйол): 1287 (ср.), 1272 (с.), 925 (оч. с., шир.), 846 (сл.), 834 (сл.), 693 (с.), 650 (с.), 376 (с.) см-1.
ЛИТЕРАТУРА
1. Klein H.-F., Karsch И. Н., Chem. Вег., 108, 944 (1975).
2. Klein H.-F., Hammer R., Angew. Chem., 88, 61 (1976).
* CH2C12 использовать нельзя, так как в нем идет реакция образования «Со[СН2=Р(СНз)з]гС12 и Со [Р (СН3) з] зС1 [2].
Фосфиновые комплексы 2117
□ □ Хлоротрис(триметилфосфин)кобальт Со[Р(СНз)з]3С1
Со[Р(СН2)3]4 + СоС12 + 2Р(СН3)3 -> 2Со[Р(СН3)3]3С1
363,2 129,8 2-76,1 2-322,6
8,1 г (22,3 ммоль) СоГР(СН3)з]4 растворяют в ~50 мл эфира и добавляют последовательно 5,3 мл (56 ммоль) триметилфосфина и 2,9 г (22,3 ммоль) СоС13. После 4 ч перемешивания выпадают темно-синие кристаллы. Для полноты высаживания в реакционную смесь добавляют гексан и затем упаривают. Кристаллы отделяют и промывают гексаном. Выход 13,65 г (95%).
Свойства. Темно-сииие кристаллы, хранятся в атмосфере N3. /пл> >90 °C (с разл.). Растворяется в бензоле, тетрагидрофураие и метаноле; плохо растворяется в пентане и эфире. ИК (нуйол): 2814 (ср.), 1298 (ср.), 1282 (с.), 974 (ср.), 943 (оч. с.), 847 (сл.), 715 (с.), 654 (с.), 343 (с.) [vP(CH3)3]; 306 (ср.) [v(CoCl)]; 278 (сл.) [v(CoP)] см-1.
ЛИТЕРАТУРА
1. Klein H.-F., Korsch Н. Н., Вег., 108, 944 (1975); Inorg. Chem., 14, 473 (1975).
□ □ (т]-Циклопентадиенил)бис(триметилфосфин) кобальт Со[Р(СНз)з]2(п-С5Н5)
Со[Р(СН3)3]3С1 + Т1С5Н5 -> Со[Р(СН3)3]2(п-С5Н5) 4- Т1С1 + Р(СН2)3
322,6 269,5 276,2 239,8 76,1
13,65 г (42,3 ммоль) Со[Р(СН3)3]3С1 растворяют в трубке Шленка в ~70 мл тетрагидрофурана и добавляют 11,4 г (42,3 ммоль) Т1С3Н5. Цвет раствора быстро изменяется от синего до коричневого. После перемешивания в течение 3 ч реакционную смесь фильтруют (Т1С1) и фильтрат упаривают в вакууме досуха. Вязкий коричневый остаток экстрагируют —80 мл пентана в снова фильтруют. Фильтрат упаривают до половины объема и охлаждают до —78 °C; выпавшие кристаллы отделяют. Выход 10,5 г (90%).
Свойства. Черные блестящие кристаллы, при растирании на воздухе разлагаются с выделением дыма. /пл 55—57 °C. В растворах при доступе воздуха тотчас разлагается. Растворяется в алифатических и ароматических углеводородах, эфире и ацетоне. Быстрое разложение происходит при растворении в нитрометане и хлорированных углеводородах. В протонных растворителях идет протонирование комплекса. В противоположность Со[Р(СН3)3]2(т]-СБНв) комплекс [Со{Р(СН3)3}2(11-С5Н5)Н] [PF6], получаемый протонированием, весьма устойчив и обработкой NaH может быть депротонирован с образованием снова нейтрального комплекса. ЯМР-'Н (C6D6, ТМС): 6 1,07 [«триплет», СН3], 4,51 [триплет, С3Н5], J(PH) = 1,4 Гц.
ЛИТЕРАТУРА
1. Werner Н., Hofmann W., Chem. Вег., ПО, 3481 (1977).
□ □ Хлоротрис(трифенилфосфин)родий RI1 |Р(С(;Н5),з] ;С1
RhCl3-3H2O + ЗР(СеН5)3 --> Rh[P(C6H5)3]3Cl
263,3 3-262,3 925,2
Трифенилфосфин предварительно перекристаллизовывают в атмосфере N2 из смеси метанол — хлороформ (4 : 1 по объему; —15 мл/г).
2118 Глава 33. Металлоорганические комплексы
К раствору 3,0 г (11,4 ммоль) Р(С6Н5)3 в 80 мл слабо кипящего этанола прикапывают интенсивио-красный раствор 0,5 г RhCl3-3H2O (38% Rh, т. е. 1,8 ммоль) в 20 мл горячего этанола и кипятят еще 30 мин. При более длительном нагревании образуется Rh(CO){P(C6H5)3]2Cl. Суспензию фильтруют еще теплой, красные кристаллы промывают 25 мл эфира и высушивают в вакууме. Выход 1,35 г (80%).
Свойства. Соединение на воздухе довольно устойчиво. /пл 155—157 °C (с разл.). Растворяется в бензоле, CH2CI2, плохо растворяется в ацетоне, спиртах, не растворяется в циклогексане. В растворе комплекс поглощает Ог- Превосходный катализатор гидрирования.
ЛИТЕРАТУРА
1. Osborn I. A., Jardine F. Н., Young J. F., Wilkinson G., J. Chem. Soc. (Loudon), A 1966, 17111.
2. Osborn J. A., Wilkinson G„ Inorg. Synth., 10, 67 (1967).
□ □ Хлоробис(триорганилфосфин)карбонилиридий
□ □ Хлоробис(трифенилфосфит)карбонилиридий Ir(CO)L2(Cl) [L=P(C6H5)3, Р(изо-С3Н7)з, Р(С6Нц)3, P(OC6H5)3]
«Комплексы Васка» образуются из 1гС13 или из [IrCle]3- и Р(СбН5)3 и СО в довольно жестких условиях. Примечательно, что источником СО при получении такого типа комплексов может служить целый ряд органических соединений (этиленгликоль, диэтиленгликоль, диметилформамид, формальдегид). Приводимый ниже вариант Бека (Beck W., Мюнхенский университет) сходен с методом Кольмана и Канга [1].
1г(СО)[Р(С6Н5)з]2(С1), «комплекс Васка»
(CH3)2NCHO
К3[1гС1„] + 2Р(СеН5)3 -------> 1г(СО) ]Р(С6Н5)3]2(С1)
522,2 2-262,3 780,3
Смесь 5,22 г (10 ммоль) К3{1гС16], 13,1 г (50 ммоль) Р(СбН5)3 и 150 мл диметилформамида помещают в круглодоиную колбу емкостью 250 мл и в течение 12 ч кипятят с обратным холодильником. Желтый раствор фильтруют еще горячим и добавляют к нему 500 мл метанола. Для полноты выпадения кристаллов колбу ставят в холодильник. Желтые кристаллы отделяют, промывают 50 мл холодного метанола и высушивают в высоком вакууме при 100—140 °C. Выход 6,0—6,5 г (77—83%).
Другие способы. Из (МН4)3[1гС1б], Р(С6Н5)3 и диэтиленгликоля с выходом 70—80% [3, 4], а также из Na^flrCle], Р(СвН3)з, формальдегида и К(С2Н5)з с выходом 58% [5].
Свойства. Желтые, устойчивые на воздухе кристаллы. Растворяется в бензоле и СНС13, не растворяется в спиртах. В растворах комплекс реагирует с кислородом, давая 1г(СО) [Р(С6Н5)3]2(т]-О2)С1 (см. также методику синтеза). ИК (нуйол): 1950 (оч. с.) [v(CO)] см-1.
Ir(CO)L2(Cl) [L=a)P(OC6H5)3, б) Р(«зо-С3Н7)3, в) Р(С6НП)3]
Исходный раствор А: в 40 мл свежеперегнанного диэтиленгликоля суспендируют при комнатной температуре 1,5 г (3 ммоль) Na3[IrCl6], Коричнево
Фосфиновые комплексы 2119
зеленую суспензию обезгаживают, в течение 10 мин продувают ток СО при комнатной температуре, затем, продолжая пропускать ток СО, медленно в течение 2 ч нагревают смесь до 130 °C и, наконец, продувают СО при 130 °C еще 30 мин. Раствор становится желтым и прозрачным.
а) Раствор А охлаждают до 50 °C и добавляют к нему при перемешивании 1,86 г (6 ммоль) Р(ОС6Н5)3, перегнанного в вакууме масляного насоса непосредственно перед употреблением. Происходит выделение СО и выпадение желтого осадка. Смесь перемешивают 15 мин, добавляют 20 мл метанола и оставляют на 2 ч при 0°С. Осадок отделяют, промывают метанолом и сушат в вакууме. Для очистки растворяют его в смеси толуол — хлороформ (3:2), раствор фильтруют горячим, добавляют 20 мл метанола и оставляют на 2 ч при 0 °C. Выпавшие кристаллы отделяют, промывают холодным метанолом и высушивают в вакууме. Выход 1,25 г (45%).
б) К раствору А добавляют 1,92 г (12 ммоль) Р(изо-СзН7)з и оставляют на 2 дня при 0 °C. Переосаждают из 10 мл горячего раствора в смеси толуол — хлороформ добавлением 15 мл метанола. Выход 650 мг (36%).
в) К раствору А, охлажденному до 70 °C, добавляют 1,68 г (6 ммоль) Р(СбНц)3, смесь перемешивают 45 мин и оставляют суспензию на 2 ч при О °C. Выпавший желтый осадок отделяют, промывают метанолом и сушат в вакууме. Затем растворяют в 150 мл смеси толуол — хлороформ (3 : 2), фильтруют раствор горячим и к фильтрату добавляют 75 мл метанола. Далее обрабатывают, как в п. «а». Выход 1,05 г (41%).
Свойства. Желтые кристаллы, довольно устойчивые на воздухе в твердом состоянии. В растворе (бензол, толуол, хлороформ) — очень эффективные катализаторы гидрирования. ИК (CS3): а)2002 (с.), б) 1936 (с.), в) 1933 (с.) (v(CO)] см-1.
ЛИТЕРАТУРА
1. Collman J. Р„ Kang J. W., J. Amer. Chem. Soc., 89, 844 (1967).
2. Collman J. P., Sears С. T., Jr., Kubota M., Inorg. Synth., 11, 101 (1968).
3. Vaska L., DiLuzio J. W., J. Amer. Chem. Soc., 83, 2784 (1961).
4. Vrieze K„ Inorg. Synth., 11, 101 (1968).
5. Levison J. J., Robinson S. D„ J. Chem. Soc. (London), A 1970, 2947.
6. Strohmeier W., Weigelt L., J. Organometal. Chem., 125, C40 (1977).
□ □ Тетракис(трифторофосфин)никель Ni(PF3)4
Ni(PF3)4 получают из металлического Ni и PF3 при высоком давлении в присутствии каталитических количеств 1г.
Ni + 4PF3 ------> Ni(PF3)4
58,7 4-88,0 410,7
Смесь 5 г (85 ммоль) порошка Ni и 0,5 г 12 помещают в специальный автоклав емкостью 100 мл с медными внутренними стенками. Для лучшего перемешивания в процессе вращения и нагревания автоклава используют латунную палочку. Автоклав вакуумируют, а затем охлаждают жидким N2. После переконденсирования в автоклав сухого PF3 (см. методику синтеза) создают при комнатной температуре рабочее давление 400 бар. Время реакции 40— 45 ч при 150°C. Далее избыток PF3 и полученный Ni(PF3)4 конденсируют в трубку Шленка. Затем при атмосферном давлении при —80 °C отгоняют PF3, а при ’-70°С — Ni(PF3)4. Выход 31,5—35 г (90—100%, в пересчете -на Ni).
Свойства. Легколетучая бесцветная жидкость. tK„n 70,5°C (760 мм рт. ст.). Пары очень ядовиты. ПК (пленка): 904, 864 (оч. с.) [v(PF)3] см-1.
2120 Глава 33. Металлоорганические комплексы
flMP-19F (пентан, CFC13 — внешн. стандарт): 6 17,5. ЯМР-31Р (пентав, 85%-ная Н3РО4— внешн. стандарт): 6 137,5, /(PF) = 1320 Гц.
ЛИТЕРАТУРА
1. Kruck Th., Baur К-, Lang W., Ber., 101, 138 (1968).
2. Kruck Th., Baur K-, Ber., 98, 3070 (1965).
□ □ Тетракис(триметилфосфит)никель Ni[P(OCH3)3]4
NiCl2-6H2O + 5P(OCH3)3 + 2(C2H5)2NH----->
237,7 5-124,1 2-73,1
----> Ni[P(OCH3)3]4 + (CH3O)3PO + 2[(C2H5)2NH2]C1 + 5H2O 555,0 140,1 2-109,6 5-18,0
В круглодонную колбу емкостью 100 мл, снабженную капельной воронкой, помещают 5,0 г (21 ммоль) NiCl2-6H2O в 50 мл метанола. К интенсивно перемешиваемому и охлаждаемому на ледяной бане зеленому раствору прикапывают в течение 1 мин 12,1 мл (13 г, 0,105 моль) свежеперегнанного Р(ОСН3)з-Цвет раствора становится темно-красным. Затем через другую капельную воронку медленно при охлаждении и интенсивном перемешивании прикапывают 5 мл (C2H5)2NH. После того как прибавят —4 мл, последующее прибавление диэтиламина проводят очень медленно (ждать ие менее нескольких минут после прибавления каждой капли!) до тех пор, пока раствор не приобретет светло-розовую окраску. Уже прн избытке в 1 каплю цвет раствора меняется на желтый и вплоть до зеленого, что свидетельствует о загрязнении продукта соединениями двухвалентного никеля. В конце прибавления диэтиламина выпадают белые кристаллы Ni[P(OCH3)3]4. Растворитель затем упаривают в вакууме водоструйного насоса до половины объема и после охлаждения метанолом с сухим льдом выделяют основное количество кристаллического продукта. Кристаллы отделяют на стеклянном пористом фильтре G3, промывают охлажденным до —70 °C метанолом до тех пор, пока кристаллы не станут чисто белого цвета, и высушивают 5 ч в вакууме масляного насоса. Выход 8,2 г (70%).
Другие способы. Замещение СО-групп в Ni(CO)4 при действии триметил-фосфита [4]. Замещение галогена на алкоголятную группу в тетракис (тригалогенофосфин) никеле [5, 6].
Свойства. Мелкие белые кристаллы. <пл 132 °C. При температуре >220 °C разлагается. Не растворяется в воде, плохо растворяется в метаноле, растворяется в большинстве неполярных органических растворителей. Выдерживает кратковременную экспозицию на воздухе, через несколько часов кристаллы становятся зелеными (Ni(II)]. ЯМР-’Н (CeDe): 6 3,7—3,9. С протонными кислотами, например CF3COOH, образует [Ni{P(OCH3)3}4H]+. ЯМР-’Н (CF3, СООН, ТМС): 5 14,5 [квинтет, N1H], У (PH) =29 Гц.
ЛИТЕРАТУРА
1. Vinal R. S., Reynolds L. Т., Inorg. Chem., 3, 1062 (1964).
2. Meier M., Basolo F., Inorg. Synth., 13, 112 (1972).
3. Werner H., Hemberger J., частное сообщение.
4. Leto J. R., Leto M. F., J. Amer. Chem. Soc., 83, 2944 (1961).
5. Irvine J. W. Jr., Wilkinson G., Science, 113, 742 (1951); J. Amer. Chem. Soc., 73, 5501 (1951).
6. Kruck Th., Hofler M., Jung H., Ber., 107, 2133 (1974).
Фосфиновые комплексы 2121
□ Тетракис(трифенилфосфит)никель М1[Р(ОС6Н5)з]4
№ВЩ
Ni(NO3)3-6H2O + 4Р(ОСвНв)3 --► Ni[P(OC6H5)3]4
290,8 4-310,3 1299,9
В атмосфере N2 к раствору 2,91 г (0,01 моль) №(ЬЮз)2-6Н2О в 60 мл этанола добавляют 15,5 г (0,05 моль) трифеиилфосфита. Затем растворяют 1,0 г NaBH4 в 25 мл теплого этанола, быстро охлаждают до комнатной температуры и прикапывают этот раствор в течение ~10 мин к интенсивно перемешиваемому раствору соли никеля. Зеленый цвет раствора Ni(II) вскоре исчезает, и начинает выпадать бледно-желтый осадок. Перемешивают еще 10 мин, фильтруют, промывают осадок (3X10 мл) этанолом н сушат в вакууме. Выход 11,0г (85%).
Другие способы. Из никелоцена (см. методику синтеза) и трифенилфос-фита с выходом 96% [3].
Свойства. Белый микрокристаллический порошок. На воздухе медленно разлагается. t„x 151—152 °C (под N2). Растворяется в бензоле, СНС13 и СН2С12, не растворяется в алифатических углеводородах.
ЛИТЕРАТУРА
1. Levison J. J., Robinson S. D., J. Chem. Soc. Chem. Commun., 1968, 1405;
J. Chem. Soc. (London), A 1970, 96.
2. Meriwether L. S., Leto J. R., J. Amer. Chem. Soc., 83, 3192 (1961).
3. Olechowski J. R., McAlister C. G., Clark R. F., Inorg. Synth., 9, 181 (1967).
□ □ Тетракис(трифенилфосфин)палладий Pd[P(C6H5)3]4
NaBH4
K2[PdCl4] + 4P(C6H5)3 ->- Pd[P(C6H5)3]4
326,4 4-262,3 1155,6
В двугорлой колбе на 500 мл, снабженной краном для ввода N2, обратным холодильником с ртутным затвором, растворяют при нагревании и перемешивании 11,0 г (42 ммоль) трифенилфосфина в 250 мл 96%-ного этанола. Затем прибавляют раствор 3,26 г (10 ммоль) K2[PdCl4] в 50 мл воды, при этом тотчас образуется желтая суспензия. Нагревание прекращают, к полученной смеси порциями прибавляют 1,7 г (45 ммоль) NaBH4 в 30 мл воды н перемешивают теплую суспензию желто-оранжевого цвета еще 1 ч. Осадок отделяют на широком стеклянном пористом фильтре G3, промывают теплым этанолом (50 мл), водой (50 мл), холодным этанолом (50 мл) и сушат 2 ч в вакууме масляного насоса. После кристаллизации из смеси толуол — пентан (толуольный раствор предварительно фильтруют через целлюлозу для освобождения от следов Pd) желтый палладиевый комплекс промывают пентаном и 4 ч сушат в высоком вакууме. Выход 9,2—10,4 г (80—90%).
Другие способы. Восстановление смеси PdCl2 и Р(СвН3)3 в диметилсульфоксиде при действии N2H4-H2O. Продукт получается ие вполне чистым и загрязнен следами азотсодержащих комплексов [3].
Свойства. Желтые ребристые листочки или микрокристаллический порошок, хранится в атмосфере инертного газа. /пл 105—110°C (с разл.). При облучении светом УФ-лампы (350 им) сильная флуоресценция. Умеренно растворяется в бензоле и СН2С12; плохо растворяется в тетрагидрофуране, аце-
2122 Глава 33. Металлоорганические комплексы
тоне, ацетонитриле, эфире; не растворяется в углеводородах и холодном этаноле. В растворе комплекс диссоциирует; весьма неустойчив на воздухе. В присутствии примесей щелочного характера быстро разлагается до металлического палладия.
ЛИТЕРАТУРА
1. Rosevear D. Т„ Stone F. G. A., J. Chem. Soc. (London), А 1968, 164.
2. Ring R. B„ Kapoor P. N., Inorg. Chem., 11, 1524 (1972).
3. Coulson D. R., Inorg. Synth., 13, 121 (1972).
Дихлоробис(трифенилфосфин)палладий Pd[P(C6H5)3]2C12
Na2[PdCl4] + 2P(CgH5)3 -> Pd[P(C6H6)3]2Cl2 + 2NaCl
294,2 2-262,3 701,9 2-58,4
Суспензию 5,0 г (17 ммоль) NaafPdCL] и 9,18 г (35 ммоль) P(C3Hj)3 в 500 мл 96%-ного этанола встряхивают в течение 12 ч в закрытой колбе. Первоначально красный цвет постепенно меняется на желтый. Выпавший осадок отделяют, промывают водой, спиртом и небольшим количеством эфира. Выход 11,35 г (95%).
Для очистки переосаждают из хлороформа петролейным эфиром.
Свойства. Желтый, микрокристаллический, устойчивый на воздухе поро-ШОК. /пл 267—270°C (с разл.). Растворяется в СНС13, CH2CI2 и бензоле, не растворяется в эфире, петролейном эфире и пентане. ИК (нуйол): 360 (с.) [v(PdCl)] 191 (ср.-с.) [v(PdP)] см-1.
ЛИТЕРАТУРА
1. Mann F. G„ Purdie D., J. Chem. Soc. (London), 1935, 1549.
Ди-ц-хлоро-а/-дихлоробис(три-н-бутилфосфин)дипалладий [Pd{P(H-C4H9)3}Cl2]2
a) Na2[PdCl4] 4- 2P(h-C4H9)3 -> Pd[P(H-C4H9)3]2Cl2 + 2NaCl
294,2 2-202,3 582 2-58,4
6) Pd[P(H-C4H9)3]2Cl2 + Na2[PdCl4] -> [Pd(P(H-C4H9)3)Cl2]2 + 2NaCi
582 294,2 759,3 2-58,4
а) транс-Р4[Р(н-С4Н9)з]2С12
К профильтрованному темно-коричиевому раствору 8,0 г (27,2 ммоль) Na^PdCh] в 25 мл воды добавляют 20 мл этанола и при перемешивании 13,5 мл (54 ммоль) три-«-бутилфосфина. Через 2—3 ч выпавший желтый осадок отфильтровывают, несколько раз промывают водой и сушат в эксикаторе над Р4О10. Выход сырого продукта —15 г (95%). Сырой продукт можно перекристаллизовать из пентаиа, однако для последующего превращения это не требуется.
Свойства. Желтые мелкие кристаллы. /пл 66—67 °C. Растворяется в полярных органических растворителях, не растворяется в воде. ЯМР-31Р (CD2CI2» 85%-ная Н3РО4 — внешн. стандарт): 6 =—10,0.
Фосфиновые комплексы 2123
б) [Pd{P(H-C4H9)3}Cl2h
Смесь 13,8 г (23,8 ммоль) транс-Р6[Р(н-С4Н9)3]2С12 и 7,0 г (23,8 ммоль) Na2[PdCl4] в 500 мл этанола кипятят с обратным холодильником 2 ч. Раствор фильтруют горячим, фильтрат упаривают досуха, красно-оранжевый остаток многократно промывают водой и перекристаллизовывают из ацетона. Выход 13,05 г. Упариванием маточника получают еще 1,5 г биядерного комплекса Pd. Общий выход 14,55 г (80%).
Свойства. Красно-оранжевые кристаллы (из этанола). 145—147°C.
Растворяется в СНС13 и бензоле, умеренно растворяется в ацетоне, плохо — в спиртах и алифатических углеводородах. ЯМР-31Р (CD2CI2, 85%-иая НзРО4 — виешн. стандарт): 6 —40,1.
ЛИТЕРАТУРА
1. Mann F. G„ Purdie D-, J. Chem. Soc. (London), 1935, 1549.
2. Mann F. G., Purdie D., J. Chem. Soc. (London), 1936, 873.
3. Laborvorschrift Inst. f. Anorgan. Chemie, LTnivers. Miinchen.
□ Тетрафторобораты ди-ц-хлоротетракис(трифенилфосфин)-диплатины(П) и -палладия(П)
[М(и-С1){P(C6H5)3}2]2[BF4]2 (M=Pt, Pd)
[Pt(g-Cl){P(C6H5)3}2b[BF4]2
2Pt[P(C6H5)3]2Cl2 + 2[O(C2H5)3] [BF4] ->
2-790,6 2-190,0
----> [{(C6H5)3P)2PtCl2Pt{P(C6H5)3}2] [BF4]2 + 2(C2H5)2O 4-2C2H5C1
1683,9 2-74,1 2-64,5
9,25 г (11,7 ммоль) Р([Р(С6Н5)3]2С12 (получение см. ниже) растворяют в 300—400 мл абсолютного CH2CI2 и при интенсивном перемешивании по каплям прибавляют 3,33 г (17,5 ммоль) тетрафторобората триэтилоксония в 60 мл абсолютного CH2CI2. Перемешивают 4 ч, фильтруют, фильтрат упаривают примерно до половины объема (соответственно до начала помутнения) в разбавляют равным объемом петролейного эфира или пентана. Образовавшуюся суспензию несколько часов охлаждают при —20 °C, осадок отфильтровывают, промывают 20 мл метанола для удаления избытка оксониевой соли н сушат в высоком вакууме. Выход 9,9 г (100%). Полученное таким образом соединение является достаточно чистым. Для более тонкой очистки переосаж-дают из CH2CI2 эфиром.
Другие способы. Из РЬ[Р(СбН5)з]2С12 и тетрафторобората триэтилоксония (1 :2) при повышенной температуре [3]. Из Pt[P(C6H5)3]2 и AgBF4 [4].
Свойства. Бесцветные кристаллы. /пл 240—250°C (с разл.). Растворяется в CH2CI2, ацетонитриле и ацетоне, не растворяется в эфире и алифатических углеводородах. Биядерный комплекс с мостиковым атомом хлора при действии донорных лигандов образует комплексы типа [Pt(L) {Р (С6Н5) з}2С1] [BF4].
ИК (нуйол): 1055 (оч. с., шир.) [v(BF4)], 310 (сл., шир.) i[v(PtCl)J см-1.
[Pd(n-C1){P(C6H5)3}2MBF4]2
Этот комплекс получают аналогично приведенному выше из Pd [Р (С6Н5) 3] 2-С1г (1,52 г, 2 ммоль; см. методику синтеза) и [О(СгН5)3] [BF4] (0,76 г, 4 ммоль) в 200 мл СН2С12. Выход 1,25—1,35 г (74—80%).
2124 Глава 33. Металлоорганические комплексы
При необходимости переосаждают из CH2CI2 или метанола эфиром.
Другие способы. Из Pd[P(C6H5)3]2Cl2 и BF3 в запаянной трубке [5].
Свойства. Микрокристаллический порошок желтого цвета. 1ПЛ 245—250 °C (с разл.). Растворяется в спиртах, CH3CN, CH2CI2 и СНС1з. ИК (нуйол): 1055 (оч. с., шир.) [v(BF4)], 300 (ср., шир.) [v(PdCl)] см-1.
ЛИТЕРАТУРА
1. Laborvorschriften Institut f. Anorgan. Chemie, Univers. Miinchen.
2. Eaborn C., Farrell N., Murphy J. L., Pidcock A., J. Chem. Soc. Dalton, 1976, 58.
3. Treichel P. M., Warren К- P-, Knebel W. J., Inorg. Chim. Acta, 6, 674 (1972).
4. Dixon K. R„ Hawke D. J., Can. J. Chem., 49, 3252 (1952).
5. Clark H. C., Dixon K. R., J. Amer. Chem. Soc., 91, 596 (1969).
{{ис-Дихлоробис(трифенилфосфин)платина Pt[P(C6H5)3]2C12
K2[PtCl4] + 2P(C6H3)3 -> Pt[P(CeH5)3]2Cl2 + 2KC1
415,1 2-262,3 790,6 2-74,6
К интенсивно перемешиваемой суспензии 6,5 г (24,8 ммоль) трифенилфосфина в 150 мл 96%-ного этанола медленно (за 1—2 ч) прикапывают раствор 5 г (12 ммоль) K2[PtCl4] в 40 мл воды и оставляют перемешиваться на ночь. Белый кристаллический осадок отделяют, промывают Н2О (2x30 мл), этанолом (30 мл), эфиром (2x30 мл) и сушат в высоком вакууме. Выход 9,24 г (97%).
Свойства. Белое микрокристаллическое вещество. Устойчив иа воздухе. Кристаллизуется из смеси СНС13 — гексан. /пл 310—312°C (с разл.). Растворяется в СН2С12. Растворы устойчивы на воздухе. ИК (КВг): 545 ±5 (с.) см-1 (цис-конфигурация см. [4]); (нуйол): 317, 293 [v(PtCl)] см-1.
ЛИТЕРАТУРА
1. Jensen К- A., Z. Anorg. Allgem. Chem., 229, 225 (1936).
2. Malatesta L„ Cariello C„ J. Chem. Soc. (London), 1958, 2323.
3. Bailar J. C. Jr., Itatani H., Inorg. Chem., 4, 1618 (1965).
4. Mastin S. H„ Inorg. Chem., 13, 1003 (1974).
□ Тегракис(грифенилфосфин)плагина Pt[P(C6H5)3]4
K2[PtCl4] + 2KOH + 4P(C6H3)3 + C2H3OH---->
415,1 2-56,1 4-262,3 46,1
----> Pt[P(C3H5»s]4 4- 4KC1 + 2H2O + CH3CHO 1244,3 4-74,6 2-18,0 44,1
В двугорлую колбу на 500 мл, снабженную краном для ввода азота, капельной воронкой и обратным холодильником, помещают 15,4 г (58,7 ммоль) Р(С6Н5)3 и 200 мл 96%-ного этанола. Смесь нагревают при перемешивании до — 65 °C. Когда образуется прозрачный раствор, добавляют 1,4 г КОН в 32 мл этанола и 8 мл Н2О. При интенсивном перемешивании в течение 1___2 ч при-
капывают раствор 5,24 (12,6 ммоль) Kz[PtCl4] в 50 мл воды, следя за тем-, чтобы не происходило разбрызгивания по стенкам колбы (и соответственно легкого восстановления до Pt). Тотчас образующийся светло-желтый осадок
Фосфиновые комплексы
2125»
перемешивают еще около 1 ч, отделяют на фильтре еще теплым (используют* фильтр с нагревательной рубашкой), промывают теплым этанолом (2X75 мл),., водой (50 мл), холодным этанолом (50 мл) и сушат 6 ч в вакууме масляного насоса. Выход 12,6—13,4 г (80—85%).
Выделенный продукт достаточно чистый для его последующего использо-вания. Дальнейшую очистку проводят перекристаллизацией из толуола или бензола в присутствии небольших количеств Р(СбН5)3.
Свойства. Светло-желтый, микрокристаллический порошок. В атмосфере-N2 хранится неограниченно долго. 1ПЛ 118—120 °C (с разл., на воздухе, расплав-красного цвета); 159—160°C (с разл., в запаянном капилляре, расплав желтого цвета). Хорошо растворяется в бензоле (диссоциация полная) и CHCfo, хуже — в эфире, не растворяется в гексане н этаноле. В СС14 идет образование ЧИС-Р{С1г[Р(СбН5)з]2. В растворе комплекс Pt[P(C6Hs)3]4 очень чувствителен» к действию воздуха [идет образование белого комплекса — карбоиатобис (трифенилфосфин) платины].
ЛИТЕРАТУРА
1. Malatesta L„ Cariello С., J. Chem. Soc. (London), 1958, 2323.
2. Ugo R., Cariati F., La Monica G., Inorg. Synth., 11, 105 (1968).
3. Hartley F. R., Organometal. Chem. Rev., A, 6, 122 (1970).
□ □ Трис(трифенилфосфин)платина Pt[P(CeH5)3]3
Pt[P(CeH5)3]4 -> Pt[P(C6H5)s]3 + P(C6H5)3
1244,3 982,0 262,3
В круглодонной колбе на 1 л, снабженной краном для ввода азота, сус-пендируют 12,44 г (10 ммоль) тетракис (трнфенилфосфин) платины (см. предыдущую методику) в 500 мл абсолютного этанола, предварительно насыщенного N2. Затем присоединяют к колбе обратный холодильник с ртутным-затвором и при перемешивании кипятят ~5 ч; смесь постепенно становится желто-оранжевой. Образовавшийся продукт отделяют еще горячим (стеклянный пористый фильтр G3 с рубашкой для нагрева); промывают теплым абсолютным этанолом (2x50 мл) и сушат в вакууме масляного насоса. Выход 5,9—6,9 г (60—70%).
Чистота полученного таким образом продукта достаточна для его дальнейшего использования в синтезах. При необходимости проводят очистку перекристаллизацией из абсолютного ацетона в атмосфере инертного газа.
Свойства. Микрокристаллический порошок от желтого до желто-ораиже-вого цвета, хранится в атмосфере инертного газа. Соединение плавится на» воздухе при 125—130 °C с разложением, образуя расплав красного цвета. Образец, запаянный в капилляре при давлении 1 мм рт. ст., плавится при 205— 206 °C. Растворимость как для Pt[P(C6H5)3]4.
ЛИТЕРАТУРА
1. Ugo R., Cariati F., La Monica G., Inorg. Synth., 11, 105 (1968).
2. Hartley F. R., Organometal. Chem. Rev., A, 6, 122 (1970).
Чис-Дихлоробис(трифенилфосфит) платина
Pt[P(OC6H5)3]2Cl2
Na2[PtCl4].4H2O + 2P(OC6H5)3 --> Pt[P(OC6H5)3J2Cl2 + 2NaCl + 4H2O
454,9 2-310,3 886,6 2-58,4 4-18,0
Смешивают растворы 0,62 г (2 ммоль) трифенилфосфита в 4 мл этанола» И 0,45 г (1 ммоль) Na2[PtCl4]-4Н2О в 8 мл этанола, осторожно подогревают».
2126 Глава 33. Металлоорганические комплексы
10 мин встряхивают при комнатной температуре и охлаждают до О °C. Через 30 мин выпавший осадок отделяют, промывают этанолом (3x3 мл) и гексаном (3x3 мл) и сушат в вакууме. Выход 0,71 г (80%).
Продукт можно перекристаллизовать из смеси CH2CI+CH3OH.
Аналогичным образом получают комплексы Pt[P(OAr)3]2C12, где Аг — о-то-. лил (выход 80%, /Пл 195—196°С), л-толил (выход 75%, /пл 151—152°С и n-толил (выход 75%, /пл 182—184°C). Исходя из Ыаг[Р6С14] или PdX2(циклооктадиен-1,5), по аналогичной методике могут быть получены соответствующие •комплексы палладия, например Р6[Р(ОСбН5)3]2С12 (выход 85%, /пл 175— 180 °C).
Свойства. Белое, кристаллическое, устойчивое на воздухе соединение, /пл 187—189°C (под N2). Растворяется в бензоле, СНС13, CH2CI2, плохо — в углеводородах и других неполярных растворителях. ИК (нуйол): 310, 335 [v(PtCl)] см-1. ЯМР-31Р (CDCI3, 85%-ная Н3РО4— внешн. стандарт): б = = —59,3, ’/(Pt—Р)=5793 Гц.
• ЛИТЕРАТУРА
1. Ahmad N., Ainscough Е. W., James Т. A., Robinson S. D., J. Chem. Soc. Dalton, 1973, 1148.
ГХлоро[трифенилфосфито(С, Р)1 (трифенилфосфит)платина [Pt{(C6H4O)P(OC6H5)2} {Р (ОС6Н5)3} ( Cl)]
При нагревании М[Р(ОАг)3]гС12 (M=Pd, Pt; Аг=Арил) в кипящем декалине происходит элимииироваиие НС1 и образование о-металлироваиного три-- арилфосфитного производного.
Pt[P(OCeH6)s]2Cl2 -> [Pt{(CeH4O)P(OCeHs)2} {P(OC6HS)3}C1J + НС1
886,6 850,1 36,5
Навеску 0,3 г Pti[P (ОС6Нз)3] 2С1г в 4—6 мл декалина кипятят с обратным холодильником в течение 2 ч и оставляют на ночь. Выпавший осадок отделяют, промывают последовательно гексаном (2x3 мл), 3 мл этанола, гексаном (2x3 мл) и сушат в вакууме. Выход 0,22 г (76%).
Свойства. Мелкие белые кристаллы, устойчивые на воздухе. /пл 160 °C. Растворяется в бензоле, СНС13 и CH2CI2. При действии НС1 в CH2CI2 постепенно превращается в исходное соединение. ИК (нуйол): 1105 (ср.), 900 (ср.), 800 (ср.) [полосы поглощения, характерные для о-металлированных трифеиил-фосфитных лигандов], 330 (ср.) [v(PtCl)] см-1.
Аналогичным образом из Pd[P(OC6H5)3]2Cl2 получают о-палладированное I I
производное [Pd{(C6H4O)P(OC6H5)2}{P(OC6H5)3}Cl] (/пл 143 °C) с выходом •50%.
- ЛИТЕРАТУРА
1. Ahmad N., Ainscough Е. W., James Т. A., Robinson S. D., J. С. S. Dalton, 1973, 1151.
Фосфиновые комплексы 2127"
Хлоро(трифенилфосфин)золото Au[P(C6H5)3]CI
a) 2Au + 11HC14-3HNO3 -------> 2HAuC14 + 3N0C1 + 6Н2О
2-197,0 11-36,5 3-63,0 2-339,8 3-65,5 6-18,0
б) НАиСЦ + 2Р(СвН6)3 ---> Аи[Р(С6Нв)3]С1 + С12Р(С6Н6)3 + НС1
339,8 2-262,3 494,7 333,2 36,5
14,6 г (74,1 ммоль) золота растворяют в «царской водке» (3 части НС1-|— + 1 часть HNO3). Затем на песчаной бане выпаривают досуха; для удаления-следов HNO3 добавляют конц. НС1 до тех пор, пока при выпаривании ие пере* станут выделяться бурые пары оксидов азота. Еще раз выпаривают почти до* суха, кристаллическую массу растворяют в 200 мл этанола и добавляют рас* твор 39,3 г (0,15 моль) Р(СбН3)3 в теплом этаноле (~800 мл). Почти мгновенно образуется белый осадок, а раствор над осадком становится светло-желтым. Перемешивают еще 0,5 ч, кристаллы отделяют, промывают этанолом и эфиром и сушат в вакууме над СаС12. Выход 33,6 г (92%).
Свойства. Бесцветное, микрокристаллическое, неустойчивое на свету соединение. /пл сырого продукта 230—240 °C, после перекристаллизации из смеси-бензол — петролейный эфир /пл 248—249°C. ИК (нуйол): 331 (с.), 323 (ср.-с.) [v(AuCl)] см-1.
ЛИТЕРАТУРА
1. Gregory В. J., Ingold С. К., J. Chem. Soc. (London), В 1969, 276.
2. Breitinger D., Kress W., частное сообщение.
Бромо(трифенилфосфин)золото Au[P(C6H5)3] Br
so2
K[AuBr4] 4- P(C6H6)3 --> Au[P(C6H5)3]Br
555,7 262,3 539,2
Через раствор 3,97 г (7,15 ммоль) KfAuBn]* в 50 мл 95%-ного этанола-пропускают SO2 до тех пор, пока изменение цвета раствора не будет свидетельствовать об окончании восстановления. Затем прибавляют 1,90 г (7,3 ммоль) Р(СеН5)3 в 10 мл тетрагидрофурана и моментально выпавший осадок отделяют иа стеклянном фильтре. Полученный продукт переосаждают из СНС13 эфиром и сушат в вакууме над Р4О10 в течение 10 ч. Выход 3,2 г (83%).
Другие способы. Из НАиС14 через образование Аи[Р(СвНб)з]С1 реакцией -обмена С1 на Вг при действии ,[N(C2H6)4]Br с выходом 64% [3].
Свойства. Белые кристаллы. /пл 245—251 °C. На свету комплекс неустойчив. Растворяется в СНС13 и бензоле, не растворяется в алифатических углеводородах. При действии Вг2 идет окислительное присоединение и образова- -иие Аи[Р(СвН5)з]Вг3. ИК (нуйол): 234 (с.) (v(AuBr)] см-1. КР (тв.): 173 (с.) [v(AuP)] см-1.
ЛИТЕРАТУРА
1. Tobias R. S., частное сообщение.
2. Block В. Р., Inorg. Synth., 4, 16 (1953).
3. Gregory В. J., Ingold С. K-, J. Chem. Soc. (London), В 1969, 276.
* О синтезе K[AuBr4] см., например, [2].
2128 Глава 33. Металлоорганические комплексы
Комплексы с кислород-, азот- и серусодержащими лигандами
□ □ гран-Трикарбонилтрис(ацетонитрил)хром, -молибден
и -вольфрам M(NCCH3)3(CO)3 (M=Cr, Мо, W)
Простейшим способом получения этих соединений, кипячением гексакар-«бонилов металлов в ацетонитриле, получают образцы, загрязненные М(МССНз)2(СО)4. Поэтому здесь приведен и другой способ, позволяющий получать более чистые препараты.
Способ 1 [1, 2]
М(СО)6 + 3NCCH3--------> M(NCCH8)3(CO)s + ЗСО
М = Сг: 220,1 3-41,1 М = Сг: 259,2 3-22,4л
М = Мо: 264,0 М = Мо: 303,1
M = W: 351,9 M = W: 391,0
В аппаратуре, приведенной на рис. 479, кипятят при перемешивании 32 ммоль гексакарбонила металла [7,0 г Сг(СО)б, 8,5 г Мо(СО)в или 11,3 с 5У(СО)6] в 50 мл (0,955 моль) ацетонитрила до тех пор, пока не прекратится
Рис. 479. Прибор для получения М(ЫССНз)з(СО)з [сгруша» для проведения синтезов с карбонилами металлов по Офеле (Ofele)].
1 — механическая мешалка; 2 — НШ 24; 3 — спираль из медной проволоки (1,5 мм);
4 — колба емкостью 100 мл.
выделение газа [для Мо(СО)6 Ю ч, для Сг(СО)з и W(CO)e 40 ч]. Выделяю-ацийся газ для контроля за протеканием реакции собирают в газометре. С помощью медленно вращающейся спирали 3 в обратном холодильнике возгоняющийся карбонил металла возвращают в реакционную колбу 4. Охлажден-
Комплексы с 0-, N- и S-содержащими лигандами 2129
ный желтый раствор фильтруют через стеклянный пористый фильтр G4, покрытый на 1 см слоем фильтровальной бумажной массы, фильтрат упаривают досуха в вакууме при комнатной температуре, а остаток 2 ч сушат в высоком вакууме. Выход 85—90%.
Свойства. Желтые, очень неустойчивые на воздухе кристаллы. Без разложения растворяются только в ацетонитриле. При растворении в ацетоне, тетрагидрофуране, диоксане и диметиловом эфире диэтиленгликоля идет более или менее быстрый обмен лигандов или разложение. Даже в высоком вакууме при температуре выше 30—40 °C идет разложение.
ИК: v(CO), см-1 М=Сг М=Мо M=W
(CH3CN) 1920 (сл.) 1794 (сл.) 1913 (сл.) 1790 (сл.)
(Нуйол) 1910 (сл.) 1915 (сл.) 1885 (сл.)
1782 (сл.) 1783 (сл.) 1778 (сл.)
Способ 2 [3, 4, 6, 7]
с2н5он
а) 2Сг(СО)6 + 10KOH-f-H2O ---------»-
2-220,1 10-56,1 18,0
---> К2Н[Сг2(СО)6(ОН)3] + 2К2СО3 + 4НСО2К + 2Н2 402,3 2-138,2 4-84,1 2-22,4л
В колбе на 500 мл, сиабжеиной обратным холодильником Либиха и ртутным затвором, в течение 24 ч нагревают при кипении 11,0 г (0,05 моль) Сг(СО)б и 22,5 г (0,40 моль) КОН в 200 мл этанола. В первые 2 ч внутри холодильника возгоняется Сг(СО)3, который время от времени очищают шпателем в реакционную колбу. По окончании реакции выпавший светло-красный осадок отделяют на стеклянном пористом фильтре G4 от оранжевого раствора, отмывают от щелочи этанолом и сушат в высоком вакууме 15 ч при комнатной температуре. Выход ~17 г. Полученный таким образом продукт хотя И содержит до 50% К2СО3 и НСО2К, однако вполне пригоден для проведения реакции обмена лигандов.
Для получения чистого К2Н[Сг2(СО)6(ОН)3] полученный комплекс экстрагируют жидким аммиаком из получившейся смеси солей. Образец, ие содержащий карбоната, получают растворением сырого продукта в небольшом количестве воды (10 г в ~40 мл), упариванием получившегося раствора в вакууме до 1/3 первоначального объема и фильтрованием при 0°С. После промывания небольшим количеством ледяной воды и этанолом получают ~4 г образца, свободного от карбоната.
Свойства. Кристаллы оранжевого цвета, очень неустойчивые на воздухе. Хорошо растворимы в воде, хуже — в этаноле и метаноле.
Аналогичные соединения: по точно такой же методике получают желтые Комплексы К3[М2(СО)3(ОН)3] (М=Мо, W). Они обладают похожими свойствами, как и описанный выше комплекс хрома [5].
б) К2Н[Сг2(СО)6(ОН)3] + 6NCCH3 + 2НС1 --->
402,3 6-41,1 2-36,5
---> 2Cr(NCCH3)3(CO)3 + 2КС1 + З^О 2-259,2 2-74,6 3-18,0
В двугорлой колбе на 500 мл, снабженной капельной воронкой и ртутным затвором, растворяют 11,3 г (0,028 моль) К2Н[Сг2(СО)3(ОН)3] (или соответст
. 18—1361
2130 Глава S3. Металлоорганические комплексы
вующее количество смешанных солей) в 50 мл Н2О и добавляют 25 мл (0,478 моль) ацетонитрила. При интенсивном перемешивании магнитной мешалкой медленно прикапывают столько 1 н. НС1, чтобы раствор имел pH 6,5; следует избегать локального избытка кислоты. Выпавший пушистый осадок желтого цвета отделяют на стеклянном пористом фильтре G3, отмывают водой от кислоты и сушат в высоком вакууме. Для очистки смывают с фильтра ацетонитрилом и растворитель отгоняют в вакууме при комнатной температуре. Выход 9,5 г (65%, считая на К2Н[Сг2(СО)8(ОН)з]). Аналогичным образом из трис-р,-гидроксогексакарбонилметаллатов Сг, Мо и W получают другие замещенные карбонильные комплексы типа М.(СО)3Ьз; в случае основных лигандов, таких, как производные пиридина, NH3, амины и т. п., подкисление не требуется.
ЛИТЕРАТУРА
1. Tate D. Р„ Knipple W. R., Augl J. М., Inorg. Chem., 1, 433 (1962).
2. Ross В. L„ Grasselli J. G., Ritchey W. M„ Raesz H. D., Inorg. Chem., 2, 1023 (1963).
3. Hieber W„ Rieger K„ Z. Anorg. Allgem. Chem., 300, 288 (1959).
4. Hieber W., Englert K.., Rieger K.., Z. Anorg. Allgem. Chem., 300, 295, 304 (1959).
5. Albano V. G., Ciani G., Manassero M., J. Organometal. Chem., 25, C55 (1970).
6. Werner H„ Prinz R., Deckelmann K., Ber., 102, 95 (1969).
7. Werner H., Deckelmann K., Schonenberger V., Helv. Chim. Acta, 53, 2002 (1970).
□ □ !1-Гидразин(1Ч,М') бис(пентакарбонилхром) p-N2H4[Cr(CO)5]2
a) Cr(CO)6 4- C4H8O ---> Cr(C4H8O) (CO)5 4- CO
220,1 72,1 264,2 22,4 л
6) 2Cr(C4H8O) (CO)6 4- N2H4 -> p.-N2H4[Cr(CO)6]2 4-2C4H8O
2-264,2 32,0 416,1 2-72,1
При 10 °C и непрерывном продувании Na облучают 7,0 г (31,8 ммоль) Сг(СО)б в 400 мл тетрагидрофурана до тех пор, пока полностью не завершится образование Сг(С4Н8О) (СО)5. В образовавшийся темно-оранжевый раствор медленно при перемешивании прикапывают 0,48 мл (15,1 ммоль) 96%-иого гидразина в 30 мл тетрагидрофурана; цвет раствора меняется иа желтый. Затем раствор упаривают при 20 °C до объема 70—80 мл и охлаждают до —30 °C, при этом выпадает основная масса кристаллического p.-N2H4[Cr(CO)s]2. Растворитель декантируют, а осадок промывают 3—5 мл тетрагидрофурана для удаления непрореагировавшего Сг(С4Н8О) (СО)5. Оставшиеся кристаллы сушат при 10 °C в высоком вакууме; сокристаллизовавшийся Сг(СО)8 при этом оседает на охлаждаемом до —30 °C «пальце» сублиматора. Остаток растворяют при 10 °C в 60 мл тетрагидрофурана и охлаждают раствор до —78 °C. Выпавший в виде желтых прозрачных кристаллов ji-N2H4 [Сг (СО)5] 2 в процессе сушки в высоком вакууме при 10 °C превращается в желтый порошок. Выход 4,9 г (74%).
Свойства. Желтые кристаллы. /пл 125—135°C (с разл.). На воздухе непродолжительное время устойчив, однако длительное время хранить рекомендуется при —30 °C в атмосфере инертного газа. Хорошо растворяется в тетрагидрофуране, метаноле, диоксане и нитрометане. Практически не растворяется в бензоле и гексане. В растворе при доступе О2 быстро разлагается. ИК (КВг):
Комплексы с 0-, N- и S-содержащими лигандами 2131
3355 (ср.), 3300 (ср.) [v(NH)], 1570 (ср.), 1180 (ср.) [S(NH)] см-1; (тетрагидрофуран): 2062 (сл.), 1927 (оч. с.), 1890 (с.) [v(CO)] см-'. ЯМР-'Н (ацетон-de, ТМС): 6 5,45.
Взаимодействие Сг(С4Н8О) (СО)5 с избытком гидразина (молярное соотношение 1 : 4) при тех же условиях и способе выделения ведет к образованию моиоядерного комплекса Cr(CO)eN2H4 с выходом 72,5% [желтые призмы, /пл 120—128 °C (с разл.); растворяется в бензоле, диоксане, СНзОН, плохо растворяется в гексане].
ЛИТЕРАТУРА
I. Sellmann D., Brandl A., Endell R., J. Organometal. Chem., Ill, 303 (1976).
□ □ ц-Диимин(М,М') бис(пентакарбонилхром) p.-N2H2[Cr(CO)5]2
;.N2H4[Cr(CO)5/2 + Pb(O2CCH3)4 -->
416,1 443,4
----> p-N2H2[Cr(CO)5]2 + Pb(O2CCH3)2 + 2CH3COOH 414,1 325,3 2-60,0
5,3 г (12,7 ммоль) ji-N2H4Cr(CO)B]2 в 50 мл тетрагидрофурана при —50 °C и интенсивном перемешивании обрабатывают порциями 8,5 г (19,2 ммоль) тетраацетата свинца. Наблюдается слабое красное окрашивание. Температуру быстро доводят до —20 °C, затем в течение 3 ч поднимают до О °C и для окончания реакции нагревают еще 15 мин при 20 °C. Раствор становится черно-красным, выделяется 200—400 мл газа. Далее в течение 5 мин через раствор пропускают ток H2S, чтобы восстановить избыток РЬ(ОгССН3)4 и осадить образовавшийся РЬ(П)-ацетат. Растворитель упаривают досуха и при —10°C экстрагируют остаток тетрагидрофураном (3X50 мл). Темнокрасный экстракт фильтруют через стеклянный пористый фильтр G3, покрытый кусочками фильтровальной бумаги и слоем Na2SO4 (10 г), упаривают до объема 60—70 мл и охлаждают до —78 °C. Выпавшие кристаллы дважды перекристаллизовывают из —70 мл тетрагидрофурана. Выход 870 мг (16,5%).
Другие способы. Из Cr(CO)3N2H4 окислением Н2О2 в присутствии Сц2+ с выходом 3%.
Свойства. Кристаллизуется при —30 °C из тетрагидрофурана в виде прозрачных красных кристаллов состава [Cr(CO)6]2N2H2-2C4H8O. При высушивании легко отдает С4Н3О и переходит в рентгеноаморфную форму черно-красного цвета с металлическим отливом. В кристаллическом состоянии в течение нескольких дней устойчив на воздухе. Растворяется в полярных растворителях, таких, как тетрагидрофуран, метанол и высшие спирты, образуя растворы темно-красного цвета; в неполярных растворителях практически не растворяется. ИК (тетрагидрофураи): 2055 (ср.), 1950 (оч. с.), 1923 (с.) [v(CO)]; (КВг): 3480 (сл.), 3250 (ср.) [v(NH)], 1415 (сл.) [v(NN)], 1352 (ср.), 1105 (ср.) [6(NH)] см-1. ЯМР-‘Н (ацетои-бб, ТМС): 6 16,20 [NH], Кристаллическая и молекулярная структура: триклинная, пространственная группа симметрии Р1 (а= 19,03 А, 6=10,27 А, с=6,37 А; а=97,3°, ₽=97,9°, у=95,9°); диимии существует в тряяс-форме [2].
ЛИТЕРАТУРА
1. Sellmann D., Brandl A., Endell R., J. Organometal. Chem., Ill, 303 (1976). 2. Huttner G., Gartzke W., Alllnger K-, Angew. Chem., 86, 860 (1974).
18*
2132 Глава 33. Металлоорганические комплексы.
□□ Моносульфан(пентакарбонил)вольфрам W(CO)5SH2
W(CO)e + H2S ----> W(CO)5SH2 + CO
351,9 22,4 л 358,0 22,4 л
В трубке Шленка емкостью 200 мл, снабженной трубкой для ввода газа и обратным холодильником, подвергают фотолизу раствор 352 мг (1 ммоль) W(CO)e в 150 мл пентана. Ртутную лампу высокого давления (Hanovia S, 200 Вт) для охлаждения помещают в охлаждаемый водой специально приспособленный патрубок. Во время облучения через раствор пропускают сухой H2S, который на выходе из колбы Шленка поглощают концентрированным раствором КОН. Через 4 ч облучение и продувание H2S прекращают, а раствор фильтруют на фильтре, покрытом кусочками фильтровальной бумаги. Зеленый фильтрат упаривают в высоком вакууме при —30 °C до объема ~50 мл и оставляют на 2 ч при —30 °C; при этом W(CO)5SH2 выкристаллизовывается почти полностью. Зеленые кристаллы для очистки от W(CO)e и побочно образующегося W2(CO)8(SH2) многократно промывают пентаном и сушат в высоком вакууме. Выход ПО мг (31%).
Свойства. Кристаллы болотно-зеленого цвета. /пл 90 °C (с разл.) (под N2). Растворяется во всех координирующихся растворителях, в растворе постепенно разлагается (запах H2S). ИК (бензол): 2076 (сл.), 1971 (ср.), 1935 (оч. с.), 1916 (пл.) [v(CO)J см-1; (КВг): 2560 [v(SH)J см-1.
ЛИТЕРАТУРА
1. Herberhold М., Siiss G., Angew. Chem., 88, 375 (1976).
2. Herberhold M., Siiss G., J. Chem. Res. (S), 1977, 246. J. Chem. Res. (Mh 1977, 2720.
□ □ (г]-Циклопентадиенил)дикарбонил(метандиазо)молибден Mo(N2CH3)(CO)2(n-C5H5)
□ □ (т]-Циклопентадиенил)дикарбонил (метандиазо) вольфрам W(N2CH3)(CO)2(TC5H5)
M(CO)3(r1-C5H6)H + CH2N2-> M(N2CH3) (CO^fo-CsHe) + CO
M = Mo; 246,1 42,0 M = Mo:26O,l 22,4л
M = W: 334,0 M = W: 348,0
К раствору 2,46 г (10 ммоль) Мо(СО)з(к]-СзН5)Н или соответственно 3,34 г (10 ммоль) W(CO)3(r|-C5H5)H (см. методику синтеза) в 150 мл тетрагидрофурана при —85 °C и перемешивании магнитной мешалкой прикапывают предварительно охлажденный до —35 °C раствор диазометана в эфире (~0,25 М раствор, не содержащий этанола). Смесь перемешивают 1 ч при —85 °C, затем нагревают в течение 5 ч до комнатной температуры и завершают реакцию, перемешивая смесь еще 10 ч при комнатной температуре. Полученный продукт хроматографируют на кизельгеле (Merck; 0,063—0,200 мм; активность II—III степени; 85x2 см; температура 10—15°C). Бензолом элюируют быстро идущую желтую полосу, растворитель отгоняют, а остаток возгоняют в высоком вакууме при 50 [или соответственно при 55 °C] и получают 70 мг Мо(СО)зСН3(т]-С5Н5) [или соответственно 55 мг W(CO)3CH3(t)-CsHs)]. Вторая, красная полоса хорошо отделяется от первой; растворитель (бензол) поч
Комплексы с 0-, К- и S-содержащими лигандами 2133
ти весь отгоняют в вакууме водоструйного насоса, а остаток повторно хроматографируют при тех же условиях. Выпадающие чаще всего в виде масел, красного цвета Mo(N2CH3) (CO)2(t]-CsHs) или соответственно W(N2CH3) (СО);г (ц-СзНз) переосаждеиием из эфира пентаном при —10 °C или соответственно при 0 °C выделяют в виде мелких кристаллов, а после кристаллизации из смеси эфир — пентан (1:2) при —80 °C и многочасового высушивания в высоком вакууме при —10 °C или соответственно при 0°С получают в аналитически чистой форме. Выходы 1,6 г (62%) или соответственно 2,3 г (66%).
Пятикратное увеличение загрузки не изменяет выхода конечных продуктов.
Свойства. Mo(N2CH3) (COJsCrpCsHs): темно-красная, чрезвычайно неустойчивая на воздухе жидкость, /пл 17—18°С. W(N2CH3) (СО)2(т1-С5Н5): темнокрасные, неустойчивые на воздухе кристаллы, А™ 36—39°C (с разл.).
Оба соединения хорошо растворяются во всех органических растворителях. При доступе воздуха растворы быстро разлагаются. Кристаллическая структура: см. [4].
Спектральные данные:
м ЯМР-1Н(СОС13, ТМС, ЯМР-13С(ацетон-г!п, ТМС, ИК, СМ-1 +33 °C) +32 °C) •v(CO), н-гексан V(N-N), КВг 6С5Н5 6СН3 6СО бС5Н5 6СН8
Мо 1987 (оч. с.) 1647 (оч. с.) 5,70 3,42 236,92 94,78 47,43 1913 (оч. с ) (синглет) (синглет)
W 1981 (оч. с.) 1627 (с., шир.) 5,83 3,63 227,08 94,32 43,13 1905 (оч. с.) (синглет) (синглет)
ЛИТЕРАТУРА
1. Herrmann W. A., Angew. Chem., 87, 358 (1975).
2. Herrmann W. A., Biersack H., Ber., HO, 896 (1977).
3. Herrmann W. A., Bistram S. A., Ber., 113, 2648 (1980).
4. Hillhouse G. L., Haymore B. L., Herrmann IF. A., Inorg. Chem., 18, 2423 (1979).
□ □ Бис[р-бромо(тетрагидрофуран)трикарбонилрений] Re2(C4H8O)2(CO)eBr2
2Re(CO)5Br + 2C4H8O--> Re2(C4H8O)2(CO)6Br2 + 4CO
2-406,2 2-72,1 844,5 4-22,4л
В колбу Шленка на 1 л, снабженную магнитной мешалкой, обратным холодильником и ртутным затвором, помещают 36,2 г (89,1 ммоль) ReBr(CO)s (см. методику синтеза) в 600 мл свежеперегнанного над LiAlH4 тетрагидрофурана. На масляной бане, нагретой до ~80°С, при интенсивном перемешивании
2134 Глава 33. Металлоорганические комплексы
содержимое колбы доводят до кипения. Для удаления СО периодически продувают азотом. После кипячения ~27 ч охлаждают до комнатной температуры и упаривают растворитель до ~200 мл. Добавлением 200 мл изооктана или гептана* высаживают большую часть продукта. Для полноты кристаллизации суспензию оставляют на несколько часов в холодильнике, затем осадок отделяют и сушат в вакууме (—5-10-2 мм рт. ст.) при комнатной температуре. Выход 30,6 г (81 %).
Свойства. Бесцветное, в твердом состоянии в течение непродолжительного времени устойчивое на воздухе соединение. Хорошо растворяется в СНС1з, гептане, CS2 и бензоле. ИК (гептан): 2036 (ср.), 1928 (с.) [v(CO)] см-1. В растворе тетрагидро фур ан а образуется моноядерный комплекс Re(C4H8O)2-(СО)зВг [ИК (ТГФ): 2030 (ср.), 1914 (с.), 1891 (ср.-с.) [v(CO)] см~1]. Кристаллическая и молекулярная структура: моноклинная, пространственная группа симметрии P2i/n (а=8,661 А, 6=11,251 А, с= 11,424 А; 0=110,36°); атомы Re, связанные двумя мостиковыми атомами Вг, гексакоординированы (2 Вг, 1 ТГФ, 3 СО [гран]) [2].
ЛИТЕРАТУРА
1. Vitali D., Calderazzo F., Gazz. Chim. Ital., 102, 587 (1972).
2. Calderazzo F„ Mavani I. P., Vitali D., Bernal I., Korp J. D., Atwood J. L., J. Organometal. Chem., 160, 207 (1978).
□ □ Тетракис(у,3-гидроксотрикарбонилрений) [Re(CO)3(OH)]4
2Reg(CO)i0 + 4H2O----> [Re(CO)s(OH)]4 + 8CO + 2Hg
2-652,5 4-18,0 1149,0 8-22.4Л 2-22,4л
Суспензию 1,35 г (2 ммоль) Re2(CO)io (см. методику синтеза) в 100 мл воды нагревают в лабораторном автоклаве 15 ч при 200 °C. После охлаждения реакционную смесь экстрагируют эфиром (5x20 мл) и растворитель удаляют досуха при —50 °C в высоком вакууме. Остаток снова растворяют в 10 мл эфира, пропускают на стеклянном фильтре через слой (1 см) кислого оксида алюминия (активность I степени, фирма Woelm) и смывают затем с носителя 100 мл эфира. Фильтрат упаривают до объема 10 мл, добавляют 20 мл пентана и охлаждают до —30 °C. Выпавшие бесцветные кристаллы отделяют, промывают пентаном и 5 ч сушат в высоком вакууме. Выход 1,1 г (96%).
Свойства. Бесцветные, устойчивые на воздухе кристаллы, темнеют прн нагревании до 160 °C, при 360 °C чернеют и разлагаются. Соединение растворяется только в координирующихся растворителях, таких, как тетрагидрофуран, эфир или ацетон. ИК (тетрагидрофуран): 2021 (с.), 1919 (оч. с.) [v(CO)] см~‘.
ЛИТЕРАТУРА
1. Herberhold М., Suss G., Ellermann J., Gabelein H., Вег., Ill, 2931 (1978). 2. Herberhold M., SUss G„ Angew. Chem., 87, 710 (1975).
* При использовании гептана для высаживания постоянно получают завышенные значения анализов на С и Н. Вероятно, указанный растворитель частично входит в кристаллическую решетку соединения.
Комплексы с 0-, N- и S-содержащими лигандами 2135
□ □ Гексафторофосфат (т]-циклопентадиенил)(дикарбонил)-
(ацетонитрил)железа(П)
[Fe(NCCH3)(CO)2(i1-C5H5)] [₽F6]
а);ре(СО)2(г]-С5Н5)С1 + А1С13 + CH3CN ->-
212,4 133,3 41,1
----> (Fe(NCCH3) (СО)2(т)-С6Н5)] [А1С14] 386,8
б) [Fe(NCCH3) (CO)2(n-C5Hs)] [А1СЦ] + NH4[PF6] + 6Н2О ->
386,8 163,0 6-18,0
----> [Fe(NCCH3) (CO)2(r]-C5H5)] [PFJ + NH4C1 + [А1(Н2О)6]С13 363,0 53,5 241,4
В колбе на 100 мл, снабженной краном для ввода азота и обратным холодильником с ртутным затвором, растворяют 2,12 г (10 ммоль) Fe(CO)2(ri-С6Н6)С1 (см. методику синтеза) в 50 мл абсолютного бензола, добавляют 4,0 г (30 ммоль) А1С13 и 5 мл абсолютного ацетонитрила и кипятят эту смесь при перемешивании ~1 ч. Последующие операции проводят уже на воздухе, ио быстро. После охлаждения летучие продукты отгоняют в роторном испарителе (0,1 мм рт. ст., 25°C). Остаток осторожно обрабатывают водой (50 мл) и получившийся оранжевый раствор фильтруют непосредственно в избыток концентрированного раствора гексафторофосфата аммония. Выпавший желтый осадок отделяют на стеклянном пористом фильтре G3, высушивают и перекристаллизовывают из смеси ацетон — эфир. Выход 1,3—1,5 г (35—40%).
Свойства. Мелкие желтые кристаллы в виде иголочек. /пл 148—151 °C (с разл.). В течение длительного времени соединение устойчиво на воздухе. Растворяется в ацетоне, ацетонитриле, тетрагидрофуране и CH2CI2, не растворяется в бензоле, эфире и углеводородах. Растворы на воздухе разлагаются. ИК (CH2CI2): 2080, 2035 (оч. с.) [v(CO)J, 845 (с.) [v(PF6)] см~‘. ЯМР-'Н (ацетон-de, ТМС, 25°C): б 5,66 [С5Н6], б 2,47 [СН3].
ЛИТЕРАТУРА
1. Treichel Р. М., Shubkin R. L., Barnett К. Reichard D., Inorg. Chem., 5, 1177 (1966).
□ □ Гексафторофосфат р-диазот (Н,М')бис[(т]-циклопента-диенил)-{1,2-бис(дифенилфосфино)этан}железа(П)] [{Fe[(C6H5)2PC2H4P(C6H5)2] (r)-C5H5)}2N2] [PF6]2
2[Fe(CO) {(C6H6)2PC2H4P(CeH6)2} (т]-СвН8)] [PFJ + N2 ->
2-692,3 22,4 л
----> [{Fe[(C6H6)2PC2H4P(C6H6)2] (t]-C5H6)}2N2] [PF6]2 + 2CO 1356,7 2-22,4 л
1,5 г (2,2 ммоль) [Fe(CO){(CeH5)2PC2H4P(C6Hs)2}(ri-C5H5)] [PF6] (см. методику синтеза) облучают в 80 мл ацетона при —30 °C [ртутная лампа высокого давления (150 Вт) фирмы Original Quarzlampen GmbH, Hanau]. Одновременно через раствор продувают сильный ток N3 для удаления выделяющегося СО. Первоначально желтый раствор через несколько минут становится черно-коричневым, однако отщепление СО заканчивается лишь через ~6 ч.
2136 Глава 33. Металлоорганические комплексы
Ацетон отгоняют при комнатной температуре, остаток растворяют в 20 мл тетрагидрофурана и продувают через раствор ток Na. Раствор приобретает оранжевую окраску, одновременно выпадает осадок от оранжевого до коричневого оттенков. После удаления растворителя оранжево-коричневый остаток дважды промывают смесью тетрагидрофураи — эфир (1 : 1), растворитель при этом окрашивается в темно-коричневый цвет; затем продукт высушивают в высоком вакууме при комнатной температуре. Выход 1,1 г (75%).
Свойства. Порошок оранжевого цвета. /пл 185—195 °C (с разл.). В твердом состоянии довольно устойчив на воздухе. Плохо растворяется в тетрагидрофуране, хорошо — в ацетоне при температуре около 0°С. При этом происходит вытеснение N2 и образование ацетонового комплекса [Ре(ацетон){(СбНб)2-PC2H4P(CeH5)2}(Ti-C5H5)][PFe]. КР: 2040 (оч. с.) [v(N2)J см~‘. ЯМР-‘Н (ацетон-de, ТМС); б 2,32, 2,53 [С2Н4]» 4,50 [триплет, С5Н5], 7,70 [мультиплет, СеНе].
ЛИТЕРАТУРА
1. Sellmann D., Kleinschmidt Е., J. Organometal. Chem., 140, 211 (1977).
□ □ (Метилсульфинато-S) (г]-циклопентадиенил)-дикарбонилжелезо 1-е(СО)2(г]-С5Н5) (SO2CH3)
Образование данного соединения идет за счет внедрения SO2 по а-связи М-С в Fe(CO)2(CH3) (т)-С8Н5):
Fe(CO)2(CH3)(T]-C6HB) + SO2 --> Fe(CO)2(r]-C5H6) (SO2CH3)
192,0 22,4 л 256,1
Через раствор 1,92 г (10 ммоль) возогнаиного Fe(CO)2(CHe) (трСеНе) (см. методику синтеза) в 100 мл петролейного эфира (1Кип 60—90°C), помещенный в колбу на 250 мл с эффективным холодильником, при перемешивании в течение 15 ч пропускают слабый ток SO2, предварительно тщательно высушенный пропусканием через промывную склянку с конц. H2SO4. Реакцию проводят при абсолютном исключении следов О2.
Темно-желтую суспензию высушивают досуха (удаляют растворитель) в вакууме водоструйного насоса. Остаток извлекают 20 мл СНС13 и пропускают через стеклянный пористый фильтр G3, заполненный нейтральным А12О3 (15x2 см). Затем вымывают хлороформом до тех пор, пока растворитель не перестанет окрашиваться. После упаривания фильтрата в вакууме водоструйного насоса до ~15 мл и добавления 150 мл петролейного эфира выпавшие кристаллы отделяют и сушат в высоком вакууме. Выход 2,1—2,3 г (82—90%).
Свойства. Желтые, устойчивые на воздухе кристаллы. /пл 134—135 °C. Растворяется в хлороформе, образуя желтый неустойчивый на воздухе раствор, а также в бензоле или ацетоне. ИК (СНС13): 2063 (оч. с.), 2053 (оч. с.), 2015 (оч. с.), 2009 (оч. с.) [v(CO)]; (нуйол): 1194 (с.), 1181 (пл.), 1061 (пл.), 1052 (с.) [v(SO2)] см-1. ЯМР-'Н (CDC13, ТМС): б 5,25 [синглет, С5Н6], б 3,15 [синглет, СН3].
ЛИТЕРАТУРА
1. Bibler J. Р., Wojcicki A., J. Amer. Chem. Soc., 86, 5051 (1964).
2. Bibler J. P., Wojcicki A., J. Amer. Chem. Soc., 88, 4862 (1966).
Комплексы с 0-, N- и S-содержащими лигандами 2137
□ □ Гидридо(?]1-диазот)трис(трифенилфосфин)кобальт Co[P(C6H5)3]3(N2)H
а) 2СоСО3 + 6С5Н8О2 + Н2О2 --> 2Со(С6Н7О2)3 + 4Н2О + 2СО2
2-118,9 6 -100,1 34,0 2-356,3 4-18,0 2-22,4л
б) Со(С6Н7О2)3 4- А1(изо-С4Н9)3 4- Ы2 4- ЗР(С6Н6)3
356,3 198,3 22,4л 3-262,3
---> Co[P(C6H5)3]3(N2)H А1(С5Н,О2)3 4~ 2 изо-С4Н8 4- ЩЮ-С4Н18
874,8 324,3 2-56,1 58,1
а) Со(С5Н7О2)з* [1]
В колбе Эрленмейера нагревают 5 г (42 ммоль) СоСО3 и 40 мл (0,4 моль) ацетилацетона С5Н8О2 до 90—100 °C. При интенсивном перемешивании добавляют по каплям за 45 мии 60 мл 10%-ного Н2О2. В начальной фазе реакции происходит сильное разогревание и выделение газа. В конце прибавления выпадает зеленый осадок, а раствор окрашивается в интенсивно-зеленый цвет. Смесь охлаждают льдом с солью, осадок отделяют и сушат при ПО °C. Для очистки кристаллы растворяют в 50 мл кипящего бензола, добавляют 300 мл гептана и для полноты кристаллизации охлаждают льдом с солью. Выпавшие кристаллы отделяют и сушат на воздухе. Выход 10—12 г (67—80%).
Свойства. Темио-зеленые кристаллы. /пл 213 °C. Растворяется в большинстве органических растворителей, не растворяется в воде.
б) Co[P(C6H5)3]3(N2)(H) [2, 3]
В 300 мл абсолютного эфира при —50 °C суспендируют 1 г (2,8 ммоль) Со(С5Н7О2)з и 2,65 г (10 ммоль) Р(С6Н3)3. К полученной смеси в атмосфере азота добавляют 3,0 мл (12 ммоль) триизобутилалюминия (осторожно!). Затем через реакционную смесь продувают ток N2 и медленно доводят температуру до 10 °C. Первоначально зеленая суспензия становится постепенно оранжевой, начинается также кристаллизация продукта реакции. Смесь перемешивают еще 3 ч при комнатной температуре, осадок отделяют от ставшего темно-красным раствора, промывают петролейным эфиром (2x20 мл) и сушат в высоком вакууме. Продукт перекристаллизовывают при низкой температуре из абсолютного толуола или переосаждают из тетрагидрофурана петролейным эфиром. Выход 1,1—1,7 г (45—69%).
Другие способы. Взаимодействие тригидридного комплекса кобальта Со[Р(СвН5)3]3Н3 (из СоС12, Р(С6Н5)з и NaBH4) с молекулярным Ь12 [5].
Свойства. Кристаллы оранжевого цвета. (Пл 80“С (с разл.). На воздухе быстро разлагается, в течение некоторого времени устойчив в атмосфере N2. Хорошо растворяется в таких неполярных растворителях, как тетрагидрофуран, бензол, толуол или эфир; не растворяется в воде. ИК (бензол): 2088 [v(N2)] см-1, отсутствует поглощение v(CoH). Кристаллическая и молекулярная структура: моноклинная, пространственная группа симметрии Р2]/с (а= = 37,159 А, 6=11,355 А, с=21,779 А, р= 101,3°); тригонально-бипирамидальное окружение атома Со, связанного с N2 и Н в аксиальных положениях [4].
* Трис (ацети л ацетон ато) кобальт Со(С5Н7О2)з поставляет, например, фирма Alfa-Ventron.
2138 Глава 33. Металлоорганические комплексы
ЛИТЕРАТУРА
I. Bryant В. Е., Fernelius W. С., Inorg. Synth., 5, 188 (1957).
2. Yamamoto A., Kitazume S„ Pu L. S., Ikeda S., Chem. Commun., 1967, 79;
J. Amer. Chem. Soc., 93, 371 (1971).
3. Misono A., Inorg. Synth., 12, 12 (1970).
4. Davis B. R., Payne N. C„ Ibers J. A., Inorg. Chem., 8, 2719 (1969).
5. Sacco A., Rossi M„ Inorg. Synth., 12, 18 (1970).
□ □ Тетракис(тетрагидрофуран)магнийбис[у,-диазоттрис-
( триметил фосфин) кобальт]
Mg(C4H8O)4[Co{P(CH3)3}3N2]2
С4Н8О
2СоС12 + 3Mg + 6P(CHS)3 + 2N2 -------►
2-129,8 3-24,3 6-76,1 2-22,4л
---> Mg(C4H8O)4[Co(N2) {P(CH3)3}s]2 + 2MgCl2
943,1 2-95,2
Реакцию проводят в приборе, изображенном на рис. 480. В левое колено 1
Рис. 480. Двухколенчатый прибор для получения
Mg (С4Н8О) 4 [CoN2{P (СНз) з)з] з •
1—2— двухколенчатая изогнутая трубка; 3 — колба на 100 мл; 4 — подвод к вакуумной системе или для ввода инертного газа, НШ 14,5; 5 — НШ 14,5; 6 — стеклянный пористый фильтр G4.
помещают 1,81 г (13,9 ммоль) безводного СоС12 и 1,00 г (0,041 моль) Mg-стружки. Затем при —78 °C конденсируют 50 мл тетрагидрофурана и 5,30 мл (55,8 ммоль) триметилфосфина*. Смесь перемешивают при 25 °C и давлении Ar 1 бар до тех пор, пока цвет раствора не изменится на оранжево-коричневый. После охлаждения до —78 °C и откачивания реакционную смесь снова интенсивно перемешивают 5 ч при давлении Na 1 бар и температуре 20°C. Ораижево-красный раствор упаривают досуха (20°C, 1 мм рт. ст.), продукт растворяют в 75 мл пентана, фильтруют в правое колено 2 и кристаллизуют при —78 °C в колбочке 3. Растворитель из 3 декантируют снова в 2 и после кратковременного высушивания в вакууме (15 мин при 20°С/1 мм рт. ст.) получают 4,48 г чистого продукта (68%, считая на СоС12).
Свойства. В замкнутом сосуде в атмосфере N2 устойчив до 82 °C; в токе N2 уже при 20 °C начинает отщеплять тетрагидрофуран. В пентане, эфире и бензоле растворяется без разложения лишь в присутствии добавок 1—2% тетрагидрофурана. На воздухе самопроизвольно воспламеняется.
ЛИТЕРАТУРА
1. Hammer R., Klein H.-F., Schubert U., Frank A., Huttner G., Angew. Chem., 88, 648 (1976).
2. Wolfsberger W„ Schmidbaur H„ Synth. React. Inorg. Metal-Org. Chem., 4, 149 (1974).
* P(CH3)3 лучше всего получать из Р(ОСвН5)3 и CH3MgI [2].
Комплексы с 0-, К- и S-содержащими лигандами 2139
Хлоро(г]-дикислород)бис(трифенилфосфин)карбонилиридий 1г(С0) [Р(С6Н5)3]2(т1-О2)С1
1г(С0) [Р(С6Н5)3]2С1 + 02 -> 1г(С0) [Р(С6Н5)3]8(т]-О2)С1
780,3 22,4 л 812,3
В затемненном реакционном сосуде растворяют 1,0 г (1,28 ммоль) 1г (СО) [Р(СбНб)з]2С1 (см. методику синтеза) в 80 мл бензола. В получившийся желтый раствор продувают слабый ток Ог до тех пор, пока в УФ-спектре реакционной смеси не исчезнут полосы 382 и 434 им (~20 ч). Светло-коричневый раствор упаривают в вакууме до объема ~10 мл, насыщают его О2 и ставят на несколько часов в холодильник. Образовавшийся оранжево-красный осадок отделяют и высушивают. Выход 850 мг (80%).
Свойства. Светло-оранжевые, устойчивые иа воздухе кристаллы, со временем, однако, темнеющие. ИК (КВг); 2010 fv(CO)], 858 [v(O2)] см-1. Кри-сталлическая структура: триклинная, пространственная группа симметрии Р1 (а= 19,02 А, 6=9,83 А, с=9,93 А; а=94,0°, 0=64,9°, у=93,2°). Молекулярная структура приблизительно тригонально-бипирамидальная с Ог, СО и С1 в экваториальном положении, d(O—О) = 1,30 А [3].
ЛИТЕРАТУРА
1. Vaska L., Science, 140, 809 (1963).
2. Geoffrey G. L., Hammond G. S„ Gray H. B., J. Amer. Chem. Soc., 97, 3933 (1975).
3. La Placa S. L, Ibers J. A., J. Amer. Chem. Soc., 87, 2581 (1965).
□ □ у.,т]2-Диазотникельлитиевый комплекс [{(LiC6H5)3NiO(C2H5)2}2N2]2
Wt(t]e-mpaHC,транс,транс-l ,5,9-Ci2Hi8) + 12LiC6H6 + 2N2 ---->-
4-221,0 12-84,0 2-22,4л
---->' [{(LiCeH6)3NiO(C2H6)2}2NT2]2 + Стране, транс, транс-1,5,9,C12Hla 1595,9 4-162,3
(транс,транс,транс-1,5,9-Ci2H18 =
= транс, транс, транс-циклододекатриен-1,5,9, CDT)
В двугорлую колбу емкостью 250 мл помещают при —78 °C 6,27 г (~28 ммоль) Ni(CDT) (см. методику синтеза) и 7,31 г (~87 ммоль) твердого LiCeH6 и добавляют 120 мл предварительно охлажденного до —78 °C эфира. При этом Ni(CDT) и LiCeH6 не переходят полностью в образующийся светло-желтый раствор. Реакционную колбу при перемешивании вакуумируют и затем присоединяют к газовой бюретке, заполненной чистым N2. Продолжая перемешивание, реакционную смесь нагревают до 0°С. Ni(CDT) и иС6Н5 полностью растворяются, раствор окрашивается в темно-красный цвет. Присоединение N2 начинается при 0 °C, раствор постепенно светлеет. В течение 1—2 ч (в зависимости от скорости перемешивания и размеров колбы) при 0 °C происходит связывание азота в соотношении IN2/2NL
По окончании присоединения N2 выпадают, как правило, 2—3 г (25%) азотсодержащего комплекса в виде желтого порошка. После промывания небольшим количеством эфира и пентана сушат в вакууме масляного насоса при
2140 Глава 33. Металлоорганические комплексы
О °C. Если раствор после присоединения N2 остается прозрачным, азотсодержащий комплекс никеля кристаллизуется при длительном стоянии в холодильнике.
Свойства. Темно-красные кристаллы (из эфира), устойчивые при комнатной температуре. Кристаллическая и молекулярная структура: моноклинная, пространственная группа симметрии P2i/a (а= 14,591 А, 6=25,503 А, с= = 14,126 А, 0=113,32°). Ядром сложной кластерной структуры являются две молекулы N2 со значительно увеличенным расстоянием d(N—N) = l,35 А [2].
ЛИТЕРАТУРА
1. Jonas К-, Angew. Chem., 85, 1050 (1973).
2. Kruger С., У i-Hung Tsay, Angew. Chem., 85, 1051 (1973).
тра«с-Дихлоробис( бензонитрил) палладий
Pd(NCC6H5)2Cl2
PdCl2 + 2CeH6CN ----> Pd(NCC6H6)2Cl2
177,3 2-103,1 383,6
Суспензию 2,0 г (11,3 ммоль) безводного PdCl2 в 50 мл бензонитрила нагревают при 100 °C на масляной бане. Красный раствор через 20 мин фильтруют еще горячим и фильтрат разбавляют 300 мл петролейного эфира. Выпавший желтый осадок отделяют, промывают петролейным эфиром и высушивают. Выход 4,0 г (92%). Для очистки перекристаллизовывают из бензола.
Свойства. Порошок желтого цвета, непродолжительное время устойчив на воздухе, постепенно отдает бензонитрил. Большинство других лигандов легко вытесняют его из координационной сферы металла. Растворяется в бензоле и СНС1з, хуже — в ацетоне, не растворяется в спиртах, эфире и алифатических углеводородах. ИК (нуйол): 2287 [v(CN)]; (CsBr): 366 [v(PdCl)] см-1.
ЛИТЕРАТУРА
1. Doyle J. R., Slade P. E., Jonassen H. B., Inorg. Synth., 6, 218 (1960).
(г)-Дикислород)бис(трифенилфосфин)платина Pt[P(C6H5)3]2(O2)
Pt[P(C6H6)3]4 4- 2O2 + (Д-----> Pt(O2) [P(C6H5)3]2 • C6H6 + 2(C6H6)SPO
1244,3 2-22,4л 78,1 829,8 2-278,3
Реакцию проводят в двугорлой колбе на 250 мл, снабженной краном для ввода азота, магнитной мешалкой и трубкой для ввода газа. В атмосфере N2 растворяют 6,22 г (5 ммоль) Pt[P(CeH5)3]4 в 150 мл бензола. Попадания воздуха избегают, так как содержащийся в нем СО2 легко образует с Pt[P (СвН5)з] 4 устойчивый карбонатный комплекс.
При перемешивании в течение 20 мни пропускают через желтый раствор интенсивный ток О2. Уже через несколько минут образуется осадок. Его отделяют, промывают бензолом (10 мл), затем несколько раз петролейным эфиром и сушат в вакууме водоструйного насоса. Выход 3,2—3,4 г (77—82%).
Свойства. Микрокристаллический порошок, буро-красного цвета. 129— 131 °C (с разл.). Растворяется в этаноле и ацетоне, не растворяется в воде, бензоле и алифатических углеводородах. ИК (КВг): 825 (с.) [v(O2)] см-1.
Комплексы с 0-, N- и S-содержащими лигандами 2141
При действии СО, NO или SO2 образуются соответственно карбонатные, нитратные и сульфатные комплексы. Кристаллическая и молекулярная структура Pt(Og) [Р(СвН5)3]2- 1,5С6Н6: моноклинная, пространственная группа симметрии Р2]/с (а= 18,331 А, 6=22,983 А, с=9,311 А; 0=92,14°), тригонально-планарио координированная Pt, d(Pt—О) =2,01 А и d(O—О) = 1,45 А [2].
ЛИТЕРАТУРА
1. Cook С. D., Jauhal G. S., Inorg. Nucl. Chem. Lett., 3, 31 (1967); J. Amer. Chem. Soc., 90, 1464 (1968).
2. Kashiwagi T., Yasuoka N., Kasai N„ Kakudo M., J. Chem. Soc. Chem. Commun., 1969, 743.
Глава 34 СПЛАВЫ И ИНТЕРМЕТАЛЛИЧЕСКИЕ СОЕДИНЕНИЯ
Г. Брауэр* (G. Brauer)
Перевод канд. хим. наук С. И. Троянова
В лабораторных условиях металлические сплавы наиболее’ часто получают прямым сплавлением компонентов; при этом заданный состав сплава достигается дозировкой исходных компонентов. При наличии сведений о фазовой диаграмме металлической системы таким путем в большинстве случаев можно синтезировать определенные интерметаллические соединения.
Иногда процесс гомогенизации конечного материала осложняется из-за потерь веществ в результате окисления и испарения компонентов или взаимодействия с материалом сосуда. Метод сплавления трудно использовать и в тех случаях, когда очень высока температура плавления и главным образом если получаемая фаза образуется по перитектической реакции и если она устойчива лишь при низких температурах. Гомогенность такого вещества достигается путем значительного увеличения продолжительности термической обработки при определенной температуре.
В ряде случаев с успехом применяют способ нагревания спрессованных смесей тонких порошков компонентов при температуре ниже температуры плавления. При этом сплав образуется благодаря диффузии атомов, что, вполне естественно, приводит к увеличению продолжительности процесса.
При больших различиях в плотности между отдельными компонентами или между исходными веществами и конечным продуктом может произойти ухудшение гомогенности вследствие разделения по плотности до или после реакции взаимодействия. По сравнению с методом сплавления остальные способы получения сплавов имеют в лаборатории меньшее значение. Однако в отдельных случаях может оказаться целесообразным получать сплав путем восстановления (химического или электрохимического) соединений металлов. Некоторые интерметаллические соединения могут быть выделены в виде остатков от растворения соответствующих сплавов. Важное значение для успеха, синтеза имеет знание фазовой диаграммы данной металлической системы. При наличии этих сведений в литературе (см. ниже) их следует привлекать для решения синтетической задачи.
Специальные способы получения или обработки интерметаллических соединений используются для выращивания монокристаллов. Монокристаллы необходимы при рентгенографическом определении структуры соединения или для измерения его физических свойств (особенно анизотропных).
Из-за чрезвычайно большого разнообразия известных к настоящему времени сплавов и интерметаллических соединений не может быть дано описание всех методов получения. Еще менее приемлемо было бы приводить отдельные-описания синтезов важнейших из этих веществ. Поэтому в данной главе рассмотрены типичные лабораторные методы, которые, как мы надеемся, могут
* Я весьма признателен проф. Т. Хойманну (университет г. Мюнстера), проф. X. Шеферу (университет г. Дармштадта) и проф. X. И. Шустеру (университет г. Кёльна) за предоставленные ими описания некоторых лабораторных процессов, а также за их согласие на публикацию этих, сведений в данной главе.
Синтезы при высоких температурах
2143
послужить основой для выбора способа синтеза в других случаях. Даны также описания методик синтеза некоторых отдельных препаратов, представляющих специальней интерес.
Следует также указать на то, что способы получения металлических соединений имеют много общего со способами синтеза полуметаллических и неметаллических соединений. Поэтому многие из описанных в настоящем разделе методов тесно связаны с препаративными методами, рассмотренными в других разделах этой книги (например, выращивание монокристаллов, зонная плавка, синтезы с подводом реагента через газовую фазу и т. д. см. в т. 1), а некоторые, наоборот, могли бы быть с успехом перенесены из области металлов на другие вещества.
ЛИТЕРАТУРА
Обзоры и справочники по диаграммам состояния и фазам металлических систем: ‘
1. Хансен М., Андерко К. Структуры двойных сплавов. Т. 1—2: Пер. с англ.— М.: Металлургиздат, 1962; 1-е доп.: Эллиот Р. П., т. 1—2, М., Металлургия, 1970; 2-е доп.: Шанк Ф. А., М., Металлургия, 1973.
2. Moffat W. G., Binary Phase Diagrams Handbook, General Electric Co., 1976.
3. Landolt—Bdrnstein, Zahlenwerte u. Funktionen, 6. Aufl., II Bd., 3. Teil, Springer, Berlin usw., 1956.
4. Lyman T., (Amer. Soc. Metals), Metal Handbook, Cleveland, 1948.
5. Гольдшмидт X. Дж. Сплавы внедрения. Т. 1—2. — М.: Мир, 1971.
6. Гишейднер К.. А. Сплавы редкоземельных металлов. — М.: Мир, 1965.
7. Pearson W. В., Handbook of Lattice Spacings and Structures of Metals and Alloys, Pergamon, London..., 1956.
8. Пирсон У. Кристаллохимия и физика металлов и сплавов. Ч. 1—2. — М.: Мир, 1977.
Синтезы при высоких температурах
Чистота исходного материала
Ожидать вообще какого-либо повышения чистоты препаратов при соединении компонентов не приходится, если не считать частных случаев, в которых незначительный эффект очистки может явиться следствием испарения при весьма высоких температурах. Поэтому следует стараться с самого начала применять металлы в возможно более чистом состоянии, т. е. использовать такие сорта или марки металлов, которые, будучи получены в промышленности или в лаборатории, содержали бы незначительное количество растворенных в них примесей (внутренние загрязнения). Внешние загрязнения, например пленки оксидов, можно удалить напильником и полировкой или химическим путем — травлением их соответствующими кислотами. На металлах, подвергаемых механическому измельчению в заводских условиях (порошок, стружка), часто остаются приставшие к ним остатки протирочных, понижающих треиие « смазочных веществ, которые приходится удалять при помощи органических растворителей, чтобы они не препятствовали образованию сплавов или не способствовали образованию карбидов. Воду и все растворители следует удалять •из материалов путем их тщательного высушивания.
В том случае, если металл образует устойчивый гидрид, последний можно с успехом использовать при синтезе сплава вместо чистого металла. Устойчивы гидриды следующих металлов: щелочные, щелочноземельные, редкоземельные, актиноиды; титан, цирконий, гафний, ванадий, ниобий, тантал, палладий. Гид
2144 Глава 34. Сплавы и интерметаллические соединения
риды легко измельчаются в порошок, и поэтому, применяя тонкий порошок гидрида, можно обеспечить хороший контакт е другим компонентом сплава. Гидриды легко разлагаются термически, так что в сравнении с процессом получения из чистых металлов образование сплава не только ие затрудняется, но благодаря высокой степени измельчения даже облегчается. Кроме того, выделяющийся при разложении водород может за счет своего восстановительного действия способствовать удалению из материала примеси оксидов. Некоторым препятствием может оказаться тот факт, что продажные препараты гидридов в большинстве случаев имеют более низкую степень чистоты, чем поступающие в продажу соответствующие металлы.
Форма исходного материала
Обычно применяют исходные материалы в виде кусков, небольших блоков, стружек или порошка. Более крупные куски в наименьшей степени подвержены внешним загрязнениям; при их плавлении обычно остается наименьшее количество материалов на стенках сосудов. Зато при использовании таких кусков иногда трудно получить однородный расплав, в особенности в тех случаях, когда компоненты сплава очень сильно отличаются друг от друга по плотности или по температурам плавления. Порошки же, несмотря на возможность их хорошего перемешивания уже в твердом состоянии, имеют тот недостаток, что оксидные пленки на их зернах затрудняют даже при достаточном нагревании соединение компонентов друг с другом, а также обусловливают нежелательное прилипание порошка к стенкам сосуда. В этом случае вместо порошков металлов лучше применять порошкообразные гидриды, о чем уже говорилось выше. Образование сплавов из порошков, мелких стружек или тонких обрезков проволоки можно очень сильно облегчить за счет того, что перемешанный материал перед нагреванием спрессовывают в небольшие таблетки. Соответствующие пресс-формы, применяемые для этой цели, описаны и изображены в т. 1. Их изготовляют из специальной стали (закаливаемой погружением в масло), например марки Rochling RCC, причем закалку их производят лишь после обработки и тщательной подгонки пуансона и матрицы.
В случае металлов, которые легко покрываются пленкой оксида, можно высверлить на токарном станке в куске металла углубление (конической формы), в которое затем вковать другой компонент, и таким образом уже с самого начала обеспечить тесный контакт между обоими металлами.
При соединении двух компонентов с сильно различающимися температурами плавления желательно создать такие условия, чтобы более легкоплавкий металл стекал по более тугоплавкому.
Навеска
Сплав заданного состава стараются получить путем простого отвешивания теоретически вычисленных количеств компонентов. Однако по различным причинам это удается сделать не всегда.
Если к составу предъявляются строгие требования, то после получения первой пробной смеси для составления сплава часто приходится проводить еще несколько дальнейших опытов с использованием результатов, полученных при первых опытах.
Важнейшие источники ошибок заключаются при этом в потере металла вследствие испарения и окисления или в результате побочных реакций с материалом сосуда, в котором производится плавление. В таких случаях предполагаемые потери компенсируют добавкой соответствующего компонента при взятии навески. При этом ориентировочно можно руководствоваться тем фактом, что при надлежащем проведении опыта в закрытых плавильных тиглях (см. ниже) даже при работе с такими химически активными металлами, как щелочные, потери не превышают 5%.
Синтезы при высоких температурах 2145»
Для дозировки щелочных металлов в особо чистом виде используют-небольшие стеклянные ампулы, наполняемые расплавленным металлом. Прш необходимости щелочной металл извлекается из ампулы путем выплавления-(см. т. 3, гл. 17 «Щелочные металлы»).
Свободные щелочные и щелочноземельные металлы можно отделить от образованных ими интерметаллических соединений, применяя растворители (например, аммиак, органические амины) или жидкости, реагирующие с этими металлами и растворяющие их (например, спирт, амиды кислот), тогда как. интерметаллиды оказываются сравнительно устойчивыми к действию этих, жидкостей. В таких случаях рекомендуется работать не со стехиометрическими количествами компонентов, а брать 2—3-кратиый избыток химически более-активного металла, который при расплавлении смеси служит «металлическим»-растворителем или средой. Это способствует образованию кристаллов интерметаллического соединения. После остывания избыток активного (щелочного-или щелочноземельного) металла удаляют с применением указанных выше-жидкостей (метод экстракции, получение остатков после растворения, см. ниже).
Тигельный и ампульный методы
Отвешенные по указанным выше основным правилам навески чистых компонентов соединяют друг с другом путем их сплавления в тиглях или в ампулах. При этом почти всегда приходится тем или иным путем заботиться о том,, чтобы потери от окисления или испарения оставались незначительными. Этого-достигают в простейших случаях при работе в открытых сосудах путем покрытия расплавленного металла защитным слоем также расплавленной соли или смеси солей или путем пропускания над металлом индифферентного газа.
При использовании тиглей их закрывают крышками, а ампулы запаивают в суженных перетяжках. Если сосуд для плавления оказывается закрытым-герметично, в нем можно создать индифферентную нли восстановительную-атмосферу либо его можно вакуумировать.
Некоторые данные о солях и смесях солей с низкой точкой плавления для использования в лабораторных условиях приведены в табл. 49—51. Однако и для промышленных целей в продаже имеется много готовых защитных средств для сплавов, которые также могут быть с успехом использованы.
Иногда наблюдаются неблагоприятные побочные явления, если гигроскопичные смеси солей, содержащие влагу, в какой-то степени реагируют со спла-
Таблица 49. Температуры плавления индивидуальных солей для солевых защитных покрытий
Соль <ПЛ' °C Соль «пл- 'C
LiNO3 255 LiF 870
NaNOs 307 Na2SOi 884
KNO3 334 kbo2- 947
LiCl 613 BaCl2 962
MgCk 708 NaBO2 966
NaoBtOy 741 K2SiO3 976
СаС12 772 NaF 988
KCl 776 Na2lSiO3 1088
NaCl KF 801 880 CaF2 1360
19—1361
2146 Глава 34. Сплавы и интерметаллические соединения
Таблица 50. Температуры плавления некоторых бинарных смесей
•солей (эвтектик)
Масс. % Соль I Масс. % Соль II и § •*» Mace. % Соль I 1 Масс. % Соль II I u
73 KNO3 27 LiNO3 132 32,8 KCl 67,2 ВаС12 645
55,3 NaNO3 44,7 LiNO3 208 51 LiBO2 49 NaBO2 648
46 LiCl 54 KCl 352 77,8 NaCl 22,2 ВаС12 654
57 LiCl 43 KCl 380 45 NaCl 55 KCl 660
61,4 KC1 38,6 MgCl2 426 35,8 LiF 64,2 MgF2 669
44 NaCl 56 MgCl2 430 72 NaCl 28 NaF 675
12 LiF 88 LiCl 485 51 KCl 49 K2SO4 690
32,8 NaCl 67,2 CaCl2 500 50 Na2COj 50 K2CO3 690
33,7 NaCl 66,3 LiCl 552 32,4 NaF 67,6 KF 700
45,8 KF 54,2 A1F3 565 35,4 LiF 64,6 A1F3 710
73,5 KC1 26,5 CaCla 600 63,7 LiF 36,3 A1F3 715
44,2 NaaSOi 55,8 Li2SO4 601 52,8 NaF 47,2 CaF2 810
-63 KCl 37 KF 605 61 NaF 39 MgF2 815
35 NaCl 65 Na2COa 620 21,5 NaF 78,5 MgF2 985
46,6 NaCl 53,4 NaiPgO? 620 90,4 KF 9,6 AIF3 835
32,8 NaCl 67,2 Na2SO4 623 87,8 BaF2 12,2 MgFa 890
26 KCl 74 CaCl2 640 78,8 BaF2 21,2 MgF2 930
85 CaCl2 15 CaFa 644
Таблица 51. Температура плавления некоторых смесей из трех
•солей (эвтектик)
Масс. % Соль I Mace. % Соль II Mace. % Соль III ^ПЛ’ °C
16,4 NaCl 24,6 KCl 59,0 540
76,4 BaCl2 14,0 KCl 9,6 N а2СОз 542
24 NaCl 37 KCl 39 Na2CO3 580
5 NaCl 9 KCl 86 Na2B4O? 640
53,5 A1F3 13,2 CaFa 33,2 NaF 705
10,1 AIF3 34,4 CaF2 55,5 NaF 780
15,9 AIF3 26,7 CaFa 57,4 NaF 825
20,5 AIF3 51,7 CaF2 27,8 NaF 1095
вом с выделением водорода. Этот эффект ослабляется при добавлении КОН. В общем расплавленными солями, широко распространенными в технике, в практике научной лаборатории следует пользоваться с известной осторожностью. Так, металлы часто способны растворяться в расплавах солей, и пренебрежение этим эффектом может в отдельных случаях привести к неудачам и ошибкам при синтезе сплава. Следует также учитывать электрохимические потенциалы компонентов в расплавах, с тем чтобы предотвратить нежелательные реакции металла с расплавленной солью. Это особенно относится к ак
Синтезы при высоких температурах 2147"
тивным металлам (например, барий в расплаве LiCl—КС! реагирует, давая: ВаС12 и металлический литий). Таблицу соответствующих электрохимических, потенциалов можно найти в руководстве [8].
Газ для создания защитной атмосферы выбирают в зависимости от металлов, входящих в состав сплава. Часто применяют водород, однако не в тех. случаях, когда присутствуют значительные количества щелочных, щелочноземельных и редкоземельных металлов, легко образующих гидриды. Применяют для этой цели и азот, за исключением тех случаев, когда среди металлов-присутствуют такие, которые образуют нитриды, как, например, литий, бериллий, магний, кальций, стронций, барий, редкоземельные металлы, актиноиды, титан, цирконий, гафний, ванадий, ниобий и тантал. Если нет основания опасаться образования карбидов, то можно с успехом использовать и моноксид углерода, тогда как СО2 и SO2 при высоких температурах могут иногда оказывать на металлы окислительное действие. Инертные газы, преимущественно аргон, являются наилучшими, хотя и наиболее дорогими защитными газами. Защитный газ при высоких требованиях к его защитному действию должен быть хорошо очищен, в особенности нежелательно присутствие в нем кислорода, даже в виде следов. Указания о способах очистки различных газов можно найти в соответствующих разделах настоящей книги [водород (гл. 1), азот (гл. 7), инертные газы]. Водород, азот и аргон высокой степени чистоты имеются в продаже или могут быть поставлены некоторыми заводами по желанию заказчика.
Желательно, чтобы плавильные тигли были узкими и высокими. В качестве-материала для изготовления таких тиглей можно использовать а) металлы, б) керамические материалы, а в качестве материала для нзготовлення ампул — в) стекло различных сортов.
а) Металлы. Естественно, что для изготовления плавильных тиглей пригодны лишь тугоплавкие металлы, в незначительной степени склонные к образованию сплавов. Часто для этой цели могут найти применение железо-и различные сорта стали, а также молибден и тантал; молибден весьма пригоден для этой цели, хотя значительно дороже железа и не так легко поддаете^ обработке. Эти металлы служат для изготовления тиглей, применяемых преимущественно при сплавлении металлов Б-подгрупп, а также химически очень активных металлов (щелочных, щелочноземельных) в соответствии с классификацией металлов, приведенной в табл. 52.
При использовании железа следует соблюдать осторожность, поскольку оно в определенных условиях способно образовывать сплавы при плавлении цинка, кадмия, алюминия, кремния, германия и олова.
Необходимо учитывать и возможность того, что в сплаве могут оказаться-в качестве компонентов такие Б-элементы, как фосфор, мышьяк, сурьма и висмут, которые иногда способны давать соединения с вышеназванными металлами для изготовления тиглей (особенно легко с танталом). Кроме того, также для тантала, известен неблагоприятный случай, когда нагревание отдельных элементов (лития, галлия илн германия) до довольно высоких температур в танталовом тигле происходило без всяких осложнений, но сплав, состоящий из этих элементов (LiGaGe), быстро разрушал танталовый тигель уже при сравнительно низких температурах (500°C).
Такие металлы, как платина, серебро, никель, которые используются, например, в химическом анализе в качестве устойчивых материалов для изготовления посуды, не могут быть применены для изготовления тиглей с целью-получения в них сплавов, так как они сами слишком легко образуют сплавы. Их можно применять, лишь если необходимо получить сплавы самих этих металлов и в то же время исключить возможность загрязнения посторонними примесями (сплавы серебра — в серебряных тиглях, сплавы меди — в медных).
При необходимости получения сплавов из двух переходных металлов применяются почти исключительно сосуды из керамических материалов (см. ниже).
19*
2148 Глава 34. Сплавы и интерметаллические соединения
Таблица 52. Классификация элементов, в частности металлов, в металловедении
Герметизация железных, молибденовых или танталовых тиглей может быть «осуществлена различными способами. Можно, например, изготовить крышку (колпачок или пробку) из того же материала так, чтобы она сильно прижимала кольцевую прокладку к тиглю н тем самым герметизировала его. Механическое усилие создается за счет завинчивания резьбы, нарезанной на самой пробке нли колпачке или на дополнительной детали, надавливающей на пробку. Уплотняемые кольцевые поверхности имеют коническую форму. Особенно надежное уплотнение создается, если угол конуса у крышки примерно на Г меньше, чем соответствующий угол у тигля (например, 59° и 60°). В такой тигель можно вставить при необходимости другой из того же или другого материала (можно и керамический). Способы уплотнения тиглей показаны на рис. 481 и 482, а также на рнс. 313 и 364 (т. 3 и 4). Если даже тигель заполняют защитным газом, следует стараться, чтобы газовое пространство, находящееся над поверхностью сплава, имело возможно меньший объем. Поэтому в случае устройства, показанного на рис. 481, внутренний цилиндр, •служащий крышкой, должен быть после внесения металлов в тигель сначала как можно глубже вбит молотком во внешний тигель, затем на высоте верхнего конца тигля он должен быть отпилен и соединен с внешним тиглем путем сваривания верхнего шва. Оформление пробки в виде полого, а не массивного цилиндра делает более удобным выполнение сварки, осуществляемой на верхнем крае. Полую пробку отбивают, придавая ей на верхнем конце коническую форму, или же, после того как пробка забита, несколько отгибают ее верхний край в виде ранта, для того чтобы шов закрылся как можно лучше н при сварке в тигель не могли проникнуть какие-либо сварочные газы. Если при •соединении металлов можно ожидать выделения значительного количества теплоты и если преждевременное начало реакции в результате нагревания при сварке до полной герметизации тигля нежелательно, то во время сваривания нижнюю часть тигля охлаждают путем его погружения в воду.
Наполнение тигля защитным газом, предшествующее привариванию к нему крышки, может быть осуществлено путем кратковременного пропускания этого газа при помощи тонкой резиновой трубки. Еще лучше тигель с содержимым и с неплотно вставленной в него пробкой поместить в достаточно широкую стеклянную пробирку с пришлифованной пробкой и со стеклянным краном соответственно рис. 482. Пробирку с тиглем сначала вакуумируют, а затем наполняют защитным газом. Таким путем достигается наиболее полное
Синтезы при высоких температурах 2149
удаление воздуха. Операцию заполнения тиглей удобно также выполнять в «сухой камере», заполненной защитным газом.
Сваривание металлических трубчатых тиглей с крышками также в виде коротких закрытых с одной стороны трубок осуществляют с помощью «авто-
Рис. 481. Стальной трубчатый плавильный тигель.
Для сравнительно небольших количеств сплавов, необходимых, например, при исследовании кристаллических структур, рекомендуются следующие размеры тигля: внешний диаметр 20—25 мм, толщина стенок 1—2 мм, высота 70—90 мм.
Рис. 482. Устройство для откачки газов из закрывающегося плавильного тигля и для введения в него защит
ного газа.
гена» (С2Н2+О2), электросваркой или электронно-лучевой сваркой. Последние два из названных способов требуют применения специальной аппаратуры из-за необходимости создания во время сварки атмосферы инертного газа (Аг) или высокого вакуума.
Для небольших количеств реагентов н прн не слишком большом перепаде давлений между внутренним объемом и наружной средой для изготовления самого тигля или сосуда для помещения в него реакционного тигля используют тонкостенные металлические трубки. Кусок такой трубки (из железа, легированной стали, титана нли тантала) нужной длины сначала сильно сжимают с одного конца, например, большими клещами так, что получающийся плоский конец оказывается закрытым. Его герметизируют путем электрической сварки в аргоне, пользуясь устройством, показанным на рис. 483, и получают таким образом нижний конец будущего тигля. После загрузки трубчатого тигля в сухой камере, наполненной аргоном, тем же способом герметично заваривают и верхний конец, охлаждая при этом нижнюю часть тигля с реагентами. Сходное устройство применительно к изготовлению тиглей и ампул из тантала с их последующим завариванием путем электросварки описано в работе [9].
Для того чтобы вскрыть толстостенный железный тигель, его крышку и дио после сплавления металлов н последующего охлаждения отпиливают и королек сплава удаляют из открытой трубки. По другому методу тигель зажимают со стороны пробки в токарный станок и стачивают его боковую стенку,
2150 Г лава 34. Сплавы и интерметаллические соединения
пока она не станет очень тонкой — толщиной 0,1—0,3 мм. Эту утонченную тонкостенную трубку теперь легко отвернуть от королька, находящегося внутри, зацепив ее в одном месте щипцами таким же путем, как открывают короб/-ку рыбных консервов.
Рис. 483. Аппарат для заваривания тиглей (по Шустеру).
1 — токовводы; 2 — вакуумиоплотиые соединения иа замазке; 3 — латунная трубка диаметром 6 мм; 4 — керн цилиндрического шлифа KPG; 5 — устройство для закрепления кериа; 6 — муфта цилиндрического шлифа; 7 — источник постоянного тока (~20 В); 8 —шаровой шлиф 40/25; 9— виит, закрепляющий электрод в металлической обойме; 10 — вольфрамовый электрод; 11 — трубка из стекла дюран диаметром 30 мм; 12 — металлическая ампула; 13 — тигель с веществом; 14—защитная кварцевая трубка диаметром 23 мм; 15 — вииты для поддерживания кварцевой трубки; 16 — латунная трубка диаметром 19 мм вверху и 8 мм внизу; 17 — нормальный шлиф 29/32; 18 — трубка из стекла дюран диаметром 23 мм; 19— трубка из стекла дюран диаметром 12 мм; 20 — нормальный шлиф 10/19.
Ввиду того что этот процесс можно осуществить весьма быстро, таким же путем можно отделять также и корольки чувствительных к действию воздуха сплавов. Еще до того, как эти корольки подвергаются сильной коррозии, нх помещают в сосуд для хранения (трубку Шленка), наполненный защитным газом. Этот прием оказывается весьма полезным и в том случае, когда королек сплава прочно приваривается к стенке тигля.
Для извлечения из тиглей веществ, отличающихся высокой чувствительностью по отношению к воздуху, как, например, сплавов, содержащих значительное количество щелочных и щелочноземельных металлов, необходимо применять специальные методы. В приспособлении (рис. 484), описанном в работе [10], распиливание железных трубчатых тиглей проводится в атмосфере инертного газа.
Железная трубка 1 соединена при помощи шлифа 2 с установкой для получения чистого азота, а также с приборами для дальнейшей обработки сплава. Тигель 3, предварительно обточенный на токарном станке до незначительной толщины стенок, зажимают в трубке 1 винтами 4 таким образом, чтобы его дно можно было отпилить через щель 5 в атмосфере азота. Щель .5 прорезана приблизительно лишь на 2/3 окружности трубки 1. Затем тигель при помощи прочной проволоки, введенной через отверстие в середине пластин-
Синтезы при высоких температурах 2151
Рис. 484. Приспособление для вскрытия железных трубчатых тиглей без доступа воздуха.
/ — железная трубка; 2 —шлиф; 3 — плавильный тигель; 4 — вииты для закрепления тигля в приборе (всего имеется 9 таких винтов, разбитых иа три группы по три винта в каждой группе; винты расположены под углом 120° по отношению друг к другу, как это показано на добавочной части рисунка); 5 — щель для введения пнлы для резки металлов; б— пластинка, закрывающая торец трубки; 7 — хомутик, закрывающий прорезь.
ки 6, сдвигают в правую сторону, где его снова прочно зажимают. После этого •отпиливают через щель 5 также и головку тигля, т. е. приваренную к нему пробку, и удаляют эту пробку, временно удалив пластинку 6 из прибора. Сплав после этого оказывается в открытой с обеих сторон тонкостенной железной гильзе. После удаления из трубки постукиванием по ней железных опилок можно вытолкнуть сплав из трубки при помощи введенного через отверстие пластинки 6 стального стержня. Однако в некоторых особых случаях дальнейшей обработке, например экстрагированию, может подвергаться и гильза с находящимся в ней сплавом. Необходимо, чтобы защитный газ выходил всегда только нз одного отверстия. Щель 5 для отпиливания может закрываться хомутиком 7, а отверстие пластинки — резиновой пробкой. Это приспособление было впервые применено при работе со сплавами Na—Pb и Na—Sn.
При применении прибора, предложенного Клеммой и Динкелакером и подробно описанного ниже (рис. 489), приходится отказаться от полного извлечения продукта; при его помощи можно лишь получить необходимое для дальнейшего изучения количество высверленных стружек образовавшегося сплава.
б)Керамические материалы. Применяемые тигли могут быть изготовлены из самых различных керамических материалов. По этому вопросу можно сослаться на данные и таблицы, приведенные в т. 1, ч. 1, в особенности на табл. 7.
Дополнительно следует указать на возможность изготовления тиглей из нитрида бора, которые можно применять для разнообразных сочетаний элементов вплоть до 1200 °C. Нитрид бора поставляется в виде круглых прутков*, которые легко обрабатываются механически. Например, из них можно высверлить трубчатый тигель. Для плавления многих металлов (но не платины) можно с успехом использовать тигли из сульфидов церия и тория до температуры 1800 °C [11].
В большинстве случаев в металловедении применяют конические тигли (так называемые высокие формы) и длинные цилиндрические (трубчатые тигли Таммана) с полукруглым, реже— с плоским дном. Керамические тигли могут быть снабжены плотно (но не герметично) закрывающими крышками из того же материала.
Только для тиглей из спеченного глинозема (AI2O3) было установлено, что самые малые из них с внешним диаметром около 15 мм могут быть сплавлены с подходящими к ним по размерам пробками в пламени ацетиленово-кислородной сварочной горелки [12]. Тигель, имеющий форму, изображенную на рис. 485, может быть наполнен металлами приблизительно на 1/4 своей высо
* Производятся, например, фирмой Elektroschmelzwerk Kempten.
2152 Глава 34. Сплавы и интерметаллические соединения
ты, затем неплотно закрыт подобранной пробкой и после вакуумирования: в устройстве, изображенном на рис. 483, наполнен соответствующим индифферентным газом. Тигель помещают на половину его высоты в сырой песок, и осторожно нагревают его верхнюю часть вместе с пробкой сначала паяльной горелкой с воздушным дутьем, а затем ацетнленово-кислородной горелкой. Наконец, боковой край пробки сплавляют с краем тигля (/пл А12О3 2050°C). После этого его очень осторожно и равномерно охлаждают. Для того чтобы: избежать образования трещин во время сварки и в особенности во время охлаждения, необходим, конечно, некоторый навык.
Рис. 485. Трубчатый тигель из спеченного глинозема (например, диаметром 15 мм и длиной 65 мм).
Также н трубчатый тигель, изготовленный из высокоглиноземистой массы (массы Пифагора), можно путем вытягивания и запаивания его верхней части закрывать наподобие ампул при помощи паяльных горелок с водородно-кислородной или с ацетиленово-кислородной газовой смесью (о методах запаивания в ампулы см. ниже).
Еще один прием заключается в том, что внутреннюю поверхность тигля покрывают другими веществами. В этом случае можно применять при получении сплавов также и такие химически устойчивые материалы, которые сами по себе непригодны для формования керамическими методами. Для сплавов кальция и вообще для работ с металлическим кальцием, который отличается особенной агрессивностью при высокой температуре, можно применять, например, футеровку из оксида кальция. Довольно трудоемкий способ футеровки железных трубчатых тиглей вместе с крышкой подробно описан в работе Ян-дера [13] (см. примечание на следующей странице).
Если работа производится с очень небольшими количествами веществ, можно воспользоваться готовыми тиглями, изготовленными из монокристаллических плавленых соединений MgO или CaF2. Эти тигли выпускаются, однако, только в виде «низких» форм и довольно дороги.
Для сплавов лития и для работы с металлическим литием при температурах до 800 °C весьма пригодной оказалась футеровка из LiF, которая относительно хорошо пристает к стенкам, например в тиглях из спеченного диоксида циркония. Подробности относительно изготовления подобной футеровки име
г
Синтезы при высоких температурах 2153
ются во втором немецком издании данного «Руководства» (т. 2, 1962, с. 1543)*.
Футеровку металлических тиглей оксидом илн фторидом кальция при получении сплавов нли соединений очень агрессивных металлов, например лития или кальция с металлическими или полуметаллическими элементами подгруппы Б, можно и не производить. При этом поступают следующим образом: берут «незащищенный» металлический тигель, но температуру повышают очень медленно, а иногда его еще выдерживают при постоянной не очень высокой температуре для прохождения первого этапа взаимодействия реагентов. Эту температуру выбирают таким образом, чтобы воздействие на материал тигля -было еще неизмеримо мало, но компоненты уже в заметной степени реагировали друг с другом. При этом высокая агрессивность активного металла сильно понижается, прежде чем наступит заметная коррозия материала тигля. В заключение можно уже нагреть до высокой температуры, при необходимости сменив тигель на новый.
Керамические тигли могут, кроме того, быть помещены в закрытые с одного конца трубки из стекла, кварца или керамических масс и находиться в них >в течение последующего процесса нагревания в вакууме или в атмосфере защитного газа. Подобная установка изображена на рис. 311 (т. 3).
Керамические тигли часто удобно помещать в железные тигли несколько большего размера, которые затем закрывают железной пробкой и герметически заваривают. Таким путем удается удачно сочетать химическую устойчивость керамического материала, в частности оксидов ВеО, MgO, AI2O3, ZrO2, ThOs, с возможностью герметизации металлических тиглей.
Наконец, все названные сорта тиглей могут быть также помещены в ампулы из стекла нли кварца, чтобы в них возможно было поддерживать во время процесса плавления вакуум или определенную газовую атмосферу.
в) Ампулы. Из всех сортов стекла, в особенности же из тугоплавких сортов (см. ч. I в т. 1), в том числе и из кварцевого стекла, а также из керамических трубок и из высокоглиноземистой массы (массы Пифагора), могут быть изготовлены ампулы для сплавления металлов. При выборе сорта стекла необ-I ходимо учитывать максимальную температуру, которая должна быть достигнута. Сосуды из йенского стекла, дюрана нли пирекса можно нагревать приблизительно до 560 °C, сосуды из стекла супремакс — до 730 °C, из кварцевого I стекла — до 1150 °C, а сосуды из высокоглнноземистой массы — до 1400 °C, не подвергая эти сосуды опасности деформации. Металлические компоненты помещают в продолговатую закрытую с одной стороны трубку из соответству-I ющего стекла или из высокоглиноземистой массы. Трубка должна иметь диаметр ~ 10—20 мм и не слишком тонкие стенки (~ 1,5—2 мм). Дно ее должно иметь полукруглую форму равномерной толщины. Эта трубка должна быть тщательно очищена и высушена. Затем ее суживают немного выше уровня загруженного в нее металла, однако не настолько близко к металлу, чтобы он при этом сильно нагревался. Суженное место трубки должно иметь несколько утолщенные стенки, подобные стейкам капилляров. Затем трубку вакуумируют и заплавляют в суженном месте так, чтобы образовавшаяся нз нижней части трубки ампула содержала металлы в вакууме. Сужение и запаивание осуществляют при помощи соответствующих паяльных горелок (воздушной, водо-, родно-кислородной и т. д.) в зависимости от температуры размягчения мате-) риала, из которого изготовлена ампула. Разумеется, можно предусмотреть и возможность наполнения ампул защитным газом. Однако в этом случае необ-I ходнмо учитывать тепловое расширение газа, например при комнатной темпе-
( ратуре нельзя наполнять ампулу газом до атмосферного давления.
* См. также перевод 1-го издания: Руководство по препаративной неорганической химии/Под ред. Р. Брауэра. —М.: ИЛ, 1956, с. 847. — Прим, перев.
2154 Глава 34. Сплавы и интерметаллические соединения
После окончания процесса плавления и затвердевания ампулу разбивают и королек металла отделяют. По данным взвешивания всех частей до и после процесса плавления можно получить отправные точки для установления состава полученного сплава. Однако надежные данные могут быть получены лишь на основании результатов тщательного химического анализа.
Нагревание и охлаждение
При определении необходимой температуры реакции или плавления следует руководствоваться данными термических диаграмм состояния. Эта температура должна превышать по меньшей мере на 30—50 °C точку ликвидуса для получаемого сплава. Еще лучше, если она будет превышать на указанное число градусов точку плавления всех компонентов сплава. Но надежнее всего, если она будет превышать на 30—50 °C наивысшую точку ликвидуса, имеющуюся в системе данных компонентов. В качестве источников нагрева служат печи различных типов (см. т. 1, ч. I).
Если можно не опасаться сильной экзотермической реакциии между компонентами или взаимодействия еще несвязанного (при образовании сплава) агрессивного реагента с материалом тигля (см. предыдущий разд.), то нагревание обычно следует вести довольно быстро. Это уменьшает степень взаимодействия между содержимым тигля и его стенками (растворимость материала тигля в расплаве редко равна нулю). Продолжительность нагревания должна поэтому не превышать того времени, которое совершенно необходимо для обеспечения однородности сплава. С учетом этого сосуды с загруженными в них компонентами следует вводить в электрические печи уже нагретые до температуры, приблизительно соответствующей ожидаемой температуре плавления.
Введение в печь, разумеется, должно проводиться не настолько быстро, чтобы в материале сосуда возникали недопустимые напряжения, которые могли бы обусловить разрыв сосуда. Стеклянные ампулы и керамические материалы с незначительной теплопроводностью подвержены в этом отношении наибольшей опасности. Во всяком случае, при выполнении этой операции необходимо всегда надевать прочные защитные очки.
Доведя нагревание до намеченной температуры плавления, однородности сплава достигают механическим перемешиванием прежде всего для того, чтобы избежать расслаивания. При открытых тиглях жидкую смесь перемешивают стержнем из подходящего материала; плотно закрытые плавильные сосуды (тигли с завинчивающимися крышками или сваренные тигли, ампулы) вынимают из печи и встряхивают или несколько раз переворачивают. Если тигли открыты сверху, но помещены для защиты в другие сосуды и вследствие опасности разбрызгивания расплавленного металла их нельзя энергично взбалтывать или наклонять, можно постараться по меньшей мере хотя бы путем постукивания или создания вибрации снаружи перемешивать находящийся в них сплав. После этого необходимо снова на короткое время нагреть сплав до высокой температуры, контролируя это нагревание.
Если образование сплава сопровождается значительным выделением теплоты, лучше брать исходные реагенты в виде не сильно размельченных кусочков и сперва производить нагревание только до температуры плавления более легкоплавкого компонента. В большинстве случаев реакции начинаются уже при этой температуре, и теплота реакции расходуется на плавление компонента, еще находящегося в твердом состоянии. При этом не очень велика и поверхность реагирующих веществ.
Если же эти предосторожности оказываются недостаточными, то можно воспользоваться следующим способом. Более легкоплавкий реагент нагревают до плавления и при этой температуре в него небольшими порциями прибавляют другой компонент. Соответствующий прибор показан на рис. 486. Гомо-
Синтезы при высоких температурах
2155
генизацию расплава производят путем механического перемешивания. Для этой цели температурный датчик вводят в прибор (рис. 486) через мягкую резиновую манжету и перемешивание осуществляют с помощью этого датчика.
Рис. 486. Прибор для получения сплавов, образование которых сопровождается значительным выделением теплоты (по Шеферу).
термопара; 2 —резиновая уплотняющая манжета; 3 — поливинилхлоридная уплотняющая манжета; 4 — стальная труб-ка; 5 — охлаждающий змеевик; 6 — электрическая печь; 7 — вещество; 8 — тигель с полукруглым дном; 9— трубка из спеченного корунда; 10 — колба для загрузки одного из реагентов (может поворачиваться «округ оси шлифа).
При охлаждении необходимо опять-таки руководствоваться данными термической диаграммы состояния, а также назначением данного сплава. Если не приходится ожидать выделения твердых растворов переменного состава или перитектических превращений или если необходимо сохранить как раз состояние, имеющееся при высокой температуре, то охлаждение производят -быстро на воздухе, начиная от максимальной температуры. Сплавы, находящиеся в металлических нли кварцевых плавильных сосудах, могут подвергаться также закалке в воде или в масле.
Наоборот, в том случае, когда при низкой температуре реакция должна •еще идти илн когда должны быть получены монокристаллы для исследования структуры (например, в случае первичной кристаллизации), необходимо проводить регулируемое медленное понижение температуры. В этом отношении надо руководствоваться особыми требованиями для каждого отдельного случая. Для образования из сплавов крупных монокристаллов необходимо обеспечить отсутствие сотрясения при медленно протекающем охлаждении. Однако в некоторых случаях благоприятным является движение расплавленной жидкости во время выделения из нее кристаллов.
О способе плавления с электродуговым или электронно-лучевым подводом энергии см. ниже. При получении монокристаллов интерметаллических соединений, плавящихся конгруэнтно, в принципе применимы такие же методы, которые используются при выращивании кристаллов чистых металлов, включая метод зонной плавки (т. 1, ч. I). В связи с возрастающим значением физических " измерений, проводимых на монокристаллах интерметаллических соединений, в виду их широкого технического использования имеется довольно обширная литература как по общим вопросам, так и для отдельных групп соединений.
2156 Глава 34. Сплавы и интерметаллические соединения
ЛИТЕРАТУРА
1. Lawson W. D., Nielsen S., Preparation of Single Crystals, London, 1958.
2. Gilman J. J. (ed.), The Art and Science of Growing Crystals, Wiley, New York, 1963.
3, Brown A., Westbrook J. H. In: Intermetallic Compounds, Westbrook J. H. (ed.), Wiley, New York, 1967, p. 303.
4. Wilke К. T., Kristallzuchtung, Berlin VEB Deutscher Verl. Wissenschaften, 1973.
5. Pfann W. G„ Zone Melting, Wiley, New York, 1958.
6. Schildknecht H., Zonenschmelzen, Verl. Chemie, Weinheim, 1964.
7. Jurisch M. In: Intermetallische Phasen, Hrg. Ringpfeil H., VEB Deutscher Verl. Grundstoffindustrie, Leipzig, 1976, S. 65.
8. Milazzo G., Elektrochemie, Springer, Berlin usw., 1952, S. 120.
9. Corbett J. D., Inorg. Synth., 22 (1980).
10. Zintl E., Harder A., Z. Physik., Chem., (B) 34, 238 (1936).
11. Eastman E. D., Brewer L., Bromley L. A., Gilles P. W., Lofgren N. L., J. Amer. Ceram. Soc., 34, 128 (1950).
12. Zintl E., Harder A., Z. Elektrochem., 41, 767 (1935).
13. Jander W., Z. Anorg. Allgem. Chem., 138, 321 (1924).
Синтезы при высоких температурах и давлениях
Особые методы необходимы в том случае, когда приходится сплавлять два металла, из которых один имеет очень низкую точку кипения (цинк, кадмий, ртуть, см. также ниже, разд. «Метод перегонки»), а другой — высокую точку плавления (металлы платиновой группы, а также другие переходные металлы). В этом случае при атмосферном давлении первый металл испаряется прежде, чем второй перейдет в жидкое состояние. Для таких случаев Новотный н сотр. [1] сконструировали особую печь, в которой металлы могут быть подвергнуты нагреванию под высоким давлением в атмосфере защитного газа.
Внутри закрытого крышками железного герметичного реактора помещена нагреваемая электрическим током угольная трубка. На рис. 487 эта установка изображена в разрезе. Во внутренней части угольной трубки 1 помещают узкий высокий трубчатый тигель для загрузки смеси. Печь присоединяют к мощному трансформатору низдого напряжения, который при напряжении во вторичной обмотке, приблизительно равном 12 В, создает в печн ток в 600—900 А в зависимости от той температуры, которую желательно получить. В качестве начального давления газа, наполняющего трубку (азот или аргон), следует выбрать давление, например, от 60 до 70 бар. Затем после включения печи давление быстро повышается до 150—200 бар н снова немного понижается на протяжении десятиминутиого плавления, например до 70—100 бар. Измерить непосредственно температуру не удается; можно только оценить ее по силе тока при прочих равных параметрах процесса при последующей градуировке (по точкам). В течение всего процесса бомбу охлаждают в проточной воде, для того чтобы находящиеся под давлением стальные детали были холодными. В этом типе печей трудно обеспечить полную герметичность.
При сплавлении цинка, кадмия с платиной или палладием вполне пригодным материалом тигля оказывается электрографитированный уголь, между тем как А12О3 при повторных плавках может служить лишь в течение незначительного времени. Потеря летучего металла из сплава может достигать 25%. Угольная трубка и приспособления для ее крепления могут выдерживать 40— 70 плавок по 10 мин каждая. Ясно, что металлы, образующие устойчивые карбиды, в этой установке нагревать нельзя.
Другая конструкция печи для работы при не очень высоких давлениях (10—20 бар) предложена в работе [2], а ее усовершенствование описано в
Синтезы при высоких температурах 2157"
работе [3]. Эта печь позволяет при пропускании тока 600 А за 90 с произвести-нагревание до —1800 °C при избыточном давлении аргона 10—20 бар. В этих условиях, например, подавляется испарение стронция при получении сплавов в системе Sr—Au.
Рис. 487. Угольная трубчатая печь для проведения синтеза при высоком давлении.
/ — угольная трубка; 2, 3 — угольные электроды; 4, 5 — латунные гильзы;
6 — стальной трубчатый кожух; 7 — фланец; 8 — прижимная крышка; 9 — завинчивающаяся крышка; 10 — уплотняющие кольца из свинца; И, 12 — кольца и гильзы из фибролита.
ЛИТЕРАТУРА
1. Nowotny Н„ Bauer Е., Stempfl A., «Alfons Leon—Gedenkschrift» der Allge-mein, Bau-Zeitung, Wien, 1951, S. 63.
2. Busch G., Hulliger F., Winkler W., Helv. Physic. Acta., 27, 74 (1954).
3. Feller—Kniepmeter M., Heumann Th., Z. Metallkunde, 51, 404 (1960).
Бестигельное плавление
В особых условиях можно осуществлять плавление небольших количеств-металлов, сплавов и других металлоподобных соединений таким образом, что-оин во время плавления вовсе или почти не соприкасаются со стенками сосуда.
Один из способов состоит в плавлении пробы, находящейся в небольшом углублении сильноохлаждаемой медной плиты, с помощью электрической дуги-
5158 Глава 34. Сплавы и интерметаллические соединения
или сфокусированного электронного луча [1, 2]. Расплавленный образец под действием сил поверхностного натяжения принимает форму несколько сплющенного шарика. Поэтому он лишь слегка соприкасается с медиой подложкой, так что за короткое время плавления загрязнения вещества не происходит. После застывания образца его переворачивают и в этом положении еще раз проплавляют. Об источниках иагрева см. также в ч. I т. 1.
Другая возможность — плавление в «подвешенном» состоянии [3—7]. Образец поддерживается на весу в индукционной катушке в условиях вакуума или в инертной атмосфере. Одновременно производится его индукционное нагревание. Этот перспективный метод требует в каждом отдельном случае подбора подходящей индукционной катушки и еще не нашел широкого применения.
'ЛИТЕРАТУРА
1. Gruber И., Z. Metallkunde, 52, 291 (1961).
2. Sperner F., Erben E., Z. Metallkunde, 55, 674 (1964).
3. Ocress E. C., Wroughton D. M., Comnetz G„ Brace P. H., Kelly J. C. R., J. Appl. Physics, 23, 545 (1952).
4. Fischer W. A., Jahnke D., Stahlschmidt K.., Arch. Eisenhiittenwes., 45, 757 (1974).
5. Fischer W. A., Schumacher J. F., Arch. Eisenhiittenwes., 49, 431 (1978). •6. Fromm E„ Jehn H„ Z. Metallkunde, 56, 599 (1965), 58, 366 (1967).
7. Pfann W. G., Hagelbarger D. W., J. Appl. Physics, 27, 12 (1956).
Измельчение материала без доступа воздуха
Во многих случаях при измельчении полученного компактного королька сплава с целью проведения химического анализа, определения плотности, съемки порошковой рентгенограммы или же для поиска и отделения монокристалла (или монокристаллического обломка) оказывается вполне достаточным поместить королек в хорошо высушенный и малогигроскопичиый органический растворитель— вазелиновое масло, лигроин, петролейный эфир и т. д. Даже в открытой чашке органическая жидкость защищает кусочки сплава от доступа воздуха (возможно взаимодействие с кислородом и влагой). Кроме того, при этом предотвращается возможность локального нагрева за счет трения, производимого измельчающим инструментом. В дополнение можно над слоем жидкости пропускать защитный газ. Труднолетучую органическую жидкость, если она в дальнейшем мешает, под конец вымывают нз материала петролейным эфиром, остатки которого удаляют в вакууме или в токе защитного газа.
При исследовании очень чувствительных по отношению к действию воздуха гигроскопичных или окисляющихся сплавов должны быть приняты особые, меры, чтобы, например, при определении плотности или при съемке порошковых рентгенограмм иметь измельченный сплав (опилки от сверления или другой материал), совершенно не содержащий продуктов разложения. Такие приспособления были описаны Цинтлем и сотр. [1], Клеимом и Динкелакером [2], а также Баронецким [3].
Способ 1 [1]. Прибор, изображенный на рис. 488, состоит из различных стеклянных частей, которые присоединяются друг к другу при помощи шлифов 1—4 и могут быть через краны Шиффа присоединены к высоковакуумиой установке, а также к прибору для получения тока очищенного и тщательно высушенного индифферентного газа. Основными частями являются две несколько расширенные камеры 5 и 6 из не слишком тонкостенной стеклянной трубки с внутренней ребристой поверхностью (из 3—4 ребер, которые получают при нажатии угольным стерженьком на размягченное стекло; они предотвращают
Измельчение без доступа воздуха 215Ф-
выскальзывание куска сплава, внесенного в камеру для обработки). К шли» фу 2 могут быть в случае необходимости присоединены приспособления, позволяющие проводить отжиг измельченного материала, заполнение стеклянных, капилляров, применяемых в рентгеновском анализе, или взятие проб для химического анализа (шарики).
Рис. 488. Приспособление для измельчения сплавов, чувствительных к действию воздуха.
1—4— шлифы; 5» 6 — камеры для помещения куска сплава; 7, 8—вакуумные краны;. 9 — вращающаяся фреза; 10 — трубка для отжига; 11 — электрическая печь; 12— капиллярная трубка; 13— защитная трубка; 14 — шарик для запанвання проб.
Перед началом эксперимента во всем приборе в .течение нескольких часов« создают высокий вакуум, осторожно нагревают, чтобы лучше высушить, и после этого присоединяют его при помощи крана 7 к источнику защитного газа, который может выходить через кран 8. Затем через шлиф 4 в камеру 5 вводят небольшой компактный кусочек сплава. Во время всех дальнейших операций заботятся о том, чтобы благодаря энергичному току защитного газа, который -проходит через шлиф 4 и выходит через неплотно надетый небольшой резиновый колпачок, воздух никоим образом не мог проникнуть в прибор. Поверхность куска сплава очищают в камере 5 при помощи небольшой вращающейся фрезы 9 (диаметром около 5 мм), которую удлиняют приблизительно на 12 см при помощи дополнительного стержня и вставляют через шлиф 4. Стержень, фрезы вставлен в обойму гибкого жгута зубоврачебной бормашины н приводится во вращение этой машиной. Если кусок сплава оказывается после такой обработки со всех сторон блестящим, то его передвигают при помощи тонкой стеклянной палочкн или толстой проволоки в камеру 6, стараясь, чтобы из камеры 5 в камеру 6 не попал порошок из отходов. Камеру 5 после этого отсоединяют от прибора.
Затем в камере 6 получают необходимое количество чистых стружек из куска сплава, причем необходимо использовать новую фрезу. Наконец шлиф 3 закрывают шлифом 4 крана 8. Путем поворачивания и постукивания полученный порошок чистого сплава переводят в трубку 10, которая до этого была хорошо прогрета и освобождена в высоком вакууме от присутствия в ней газов. В этой трубке он может быть подвергнут в течение некоторого времени при помощи небольшой электрической печи 11 отжигу для снятия напряжений-в материале. Это во многих случаях бывает необходимо для получения хороших рентгенограмм с узкими линиями.
2160 Глава 34. Сплавы и интерметаллические соединения
При работе со сплавами, отличающимися высокой чувствительностью к окислению, температура при предварительном удалении газов из трубки 10 должна быть выше, чем при последующем отжиге образца. После этого соответствующее количество порошка переносят в капилляр 12 и запаивают его, используя затем для съемки рентгенограммы. Капилляр 12 прикрепляется к шлифу на замазке. Небольшое боковое отверстие служит для выравнивания давления с давлением в защитной трубке 13.
Вместо прибора для наполнения капилляров через шлиф 2 может быть присоединен также шарик 14 для запаивания аналитических проб. Взвешивая •сначала всю насадку в незаполненном состоянии и затем, после заполнения порошком вещества и запаивания шарика, взвешивая отдельные части этой •насадки, можно определить массу порошка.
Способ 2. В установке Клемма и Динкелакера [2] очистка поверхности сплава и получение стружек для дальнейшего исследования осуществляют также прн помощи небольшой вращающейся фрезы. При этом методе, однако, •сплав не удаляют из тигля, в котором он был получен, а обрабатывают его в самом тигле. Для этого применяют изображенный на рис. 489 прибор. В тол-
Рис. 489. Приспособление для измельчения сплавов без доступа воздуха.
7 — латунная камера; 2— вращающийся цилиндр, в который вставляется тигель; 3 — ручное колесо; 4 — резиновый очиститель; 5 — насадка; 6, 7 — винты; 3 — шлнф для со--едннения с вакуумной линией; 9 — трубка для соединения с сосудом дальнейшей обработки; 10— шлнф под закрывающий колпачок.
стостенной латунной камере 1 находится вращающийся цилиндр 2. В этот цилиндр через направленную кверху в косом положении пришлифованную насадку 5 вводят тигель со сплавом, вскрытый незадолго до того путем отпиливания его головки. При помощи небольшого находящегося сбоку виита 6 тигель зажимают в цилиндре 2. Последний может быть при помощи ручного колеса 3 повернут по желанию в любое положение и закреплен в этом положении винтом 7. Через металлический шлиф 8 может быть проведено вакуумирование; к трубе 9 присоединены сосуды для дальнейшей обработки сплава {например, для анализа, получения рентгенограммы, определения плотности и т. д.). Прибор можно сделать совершенно герметичным. Вакуумировать его можно после снятия с его осн ручного колеса 3 и после закрытия шлифа 10 яришлифованным колпачком.
Сначала при помощи быстро вращающейся небольшой стальной фрезы (круглая головка диаметром приблизительно 5 мм), которая может быть приведена в действие зубоврачебной бормашиной с гибким жгутом и введена в прибор на наконечнике через отверстие в шлифе 5 (см. также описание способа 1), удаляют с поверхности сплава все загрязнения. Затем путем фрезеровки очищенного куска получают необходимое количество стружек. Путем
Перегонка 2161
поворачивания цилиндра 2 отходы и чистые стружки удаляют отдельно друг от друга через трубку 9. При этом сменный резиновый очиститель 4 удаляет с поверхности внутри прибора остатки материала, которые иначе могли бы накапливаться в приборе и в дальнейшем служить помехой.
Способ 3. В приборе Баронецкого [3] снятие с образца стружки производится вращающейся фрезой в вакууме. Прибор полностью изготовлен из тугоплавкого стекла и поэтому перед использованием может быть прогрет В вакууме для полного удаления влаги. Вращение фрезы осуществляется снаружи с помощью магнита.
ЛИТЕРАТУРА
1. Zintl Е., Harder A., Neumayr S„ Z. physik. Chem., (А) 154, 92 (1931).
2. Klemm W., Dinkelacker F., Z. Anorg. Chem., 255, 2 (1947).
3. Baronetzky E., J. Sci. Instruments, 35, 427 (1958).
Метод перегонки
Если один из компонентов бинарного сплава легколетучий, а другой труднолетучий и давление разложения продукта реакции не слишком велико, то синтез может быть осуществлен таким образом, что первый компонент добавляют ко второму путем перегонки или возгонки.
Если не принимать во внимание особых случаев, то этот метод распространяется по вполне понятным техническим причинам лишь на сплавы с легколетучими металлами и полуметаллами. При этом почти невозможно работать с металлами, имеющими температуру кипения выше 1000—1100 °C (760 мм рт. ст.), хотя сама по себе перегонка небольших количеств и несколько менее летучих металлов в высоком вакууме практически осуществима. Этот метод применяется в основном для получения сплавов со следующими летучими элементами (/пл, °C): Na (880), К (760), Rb (700), Cs (670), Mg (1107), Zn (907), Cd (767), Hg (357), P (280, 400), As (615), Se (688). Возможно использование метода перегонки и для получения сплавов щелочноземельных металлов Са, Sr, Ва и редкоземельных элементов.
Особое преимущество этого метода заключается в том, что летучий компонент непосредственно перед реакцией еще раз подвергается высокой степени очистки путем перегонки, а это для химически очень активных металлов является весьма важным. Кроме того, взаимодействие пара одного компонента с твердым порошком другого протекает не слишком энергично, а избыточное количество летучего компонента может быть после реакции просто отогнано.
Каждый компонент вносят в отдельные лодочки н обе лодочки помещают последовательно в горизонтально расположенную трубку. Прн выборе материалов для лодочек и трубки следует руководствоваться соображениями, приведенными при обсуждении материалов для изготовления тиглей (см. выше разд. «Тигельный и ампульный методы» в этой главе), и определяемыми термической и химической устойчивостью этих материалов.
Трубка должна быть совершенно непроницаемой для газов. С одной стороны она закрыта, а другим концом присоединяется иа шлифе к вакуумной системе и к источнику индифферентного газа. В другом варианте трубка иа обоих концах имеет перекрываемое кранами подсоединение к внешней системе. Два прибора подобного типа показаны иа рис. 62 и 63 (см. т. 1). Путем соответствующего подбора температур различных зон в трубке можно обеспечить поступление паров летучего компонента к партнеру по реакции и затем отгонку его избытка.
Аналогичным путем н промежуточный продукт, полученный путем сплавления обоих компонентов по тигельному или ампульному методу, может быть
20—1361
2162 Глава 34. Сплавы и интерметаллические соединения
затем освобожден от присутствия избытка летучего компонента путем его отгонки. Предпосылка для такого отделения заключается, разумеется, в том, чтобы давление пара летучего компонента в свободном состоянии было значительно выше, чем давление пара остающейся интерметаллической фазы.
К методам, в которых один из компонентов переводится в пар, принадлежит и описанный в т. 1 (ч. I, конец разд. 13) способ (так называемый «метод Фарадея»), в котором процесс проводится в запаянной ампуле. В этом случае процесс испарения регулируется только за счет создания перепада температуры. Если материал ампулы оказывается достаточно устойчивым к воздействию реагентов, то этот метод обеспечивает особенно благоприятные условия для получения чистых веществ.
Метод получения остатков после растворения
Если интерметаллическая фаза находится на термической фазовой диаграмме по соседству с чистым компонентом (a-фазой) и если между обеими фазами имеются существенные различия в отношении растворимости и соответственно реакционной способности (в смысле большей химической активности чистого компонента), то для получения желаемого препарата можно, работая по одному из методов высокотемпературного синтеза, использовать избыток соответствующего компонента и затем отделить интерметаллическое соединение от избытка при помощи подходящего растворителя. При медленном охлаждении расплава особенно хорошо образованные кристаллы интерметаллической фазы могут оказаться включенными в чистый компонент. В таких случаях говорят вообще о методе выделения фазы в качестве остатка после растворения. Растворение фаз, легче поддающихся химическому воздействию, проводят в зависимости от обстоятельств путем электрохимического окисления или при помощи растворения в водных растворах кислот, щелочей или жидком аммиаке.
Способ 1. Путем электрохимического растворения из многофазных образцов выделяют преимущественно полуметаллические соединения. Например, можно выделить Fe3C из высокоуглеродистой стали путем электрохимического окисления. Равным образом и другие карбиды, например (Fe, Сг)3С, (Сг, Fe)rC3, (Fe, Mn)3C, (V, Fe)4C3, Fe3Mo3C, были получены подобным же путем. Этот метод и специальная аппаратура, пригодная для его проведения, были разработаны главным образом Клингером н Кохом [1—4] для исследования сталей на содержание в них неметаллических составных частей. Однако этот метод может найти в металловедении и более широкое применение.
Способ 2. Соединения алюминия, например AlgTi, Al3Zr, Al3Th, A13V, Al3Nb, Al3Ta, Al4Ce, Al4La, A1B2 и др., могут быть выделены из продуктов, содержащих значительное количество алюминия и полученных при помощи соответствующих алюминотермических реакций, при действии разбавленных щелочей нли кислот. Аналогичным путем могут быть получены также в виде остатка от растворения при действии щелочей или кислот некоторые силициды, например ZrSia, ThSi2, VSi2, NbSi2, MoSi2, WSi2, USi2, из сплавов алюминия, в которых расплавленный алюминий служил «растворителем» (см. ниже).
Способ 3 [5—8]. Особый случай представляет собой выделение интерметаллических соединений щелочных н щелочноземельных металлов (за исключением бериллия и магния), а также европия и иттербия путем экстрагирования нх жидким аммиаком. Щелочной или щелочноземельный металл в большом избытке сплавляют с другими составными частями сплава, затем довольно медленно охлаждают для образования крупных кристаллов и переводят затвердевший сплав без доступа воздуха (для этой цели весьма пригоден прибор, изображенный на рнс. 484) в экстракционную аппаратуру, показанную на рис. 490.
Использование металла-растворителя
2163
Этот прибор сконструирован по образцу прибора, описанного Бильцем и, Раль-фсом (см. т. 1, рис. 79). На плотном фильтрующем слое 5 стеклянной ваты находится введенный через шлиф 7 материал сплава, подлежащего экстрагированию. Оба краиа 6 н 7 присоединены к одной и той же установке, которая служит по желанию для вакуумирования до высокого вакуума, для пропускания аммиака и для соединения трубки 2 с трубкой 3. В трубке 2 конденсируют некоторое количество аммиака и образовавшийся синий раствор щелочного металла переводят по тонкой трубочке 4 в трубку 3. В этой трубке аммиак
Рис. 490. Прибор для экстрагирования щелочных металлов жидким аммиаком.
1—7 — см. текст.
испаряют, так что свободный щелочный металл -остается в ней, газ снова пропускают в трубку 2 н конденсируют. При многократном повторении этих операций происходит полное экстрагирование из сплава активного металла и остается соединение, на которое аммиак совсем не оказывает растворяющего действия или оказывает такое действие лишь в незначительной степени.
С тем же успехом для процесса экстрагирования сплавов жидким аммиаком можно использовать аппаратуру, показанную на рис. 79 (см. т. 1).
ЛИТЕРАТУРА
1, Klinger Р„ Koch In: Handb. f d. Eisenhtittenlaboratorium, Bd. II, Diis-seldorf, 1941, S. 441.
2. Houdremont E. P., Klinger P., Blaschczyk G„ Arch. Eisenhflttenwes., 15, 257 (1941—1942).
3. Koch W., Stahl u. Eisen, 69, 1 (1949).
4. Klinger P„ Koch W., Beitrage zur metallkundlichen Analyse, Dusseldorf, 1950, S. 49.
5. Zintl E„ Kaiser H., Z. Anorg. Allgem. Chem., 211, 113 (1933).
6. Zintl E., Harder A., Z. Elektrochemie, 41, 767 (1935).
7. Haucke W., Z. Elektrochemie, 43, 712 (1937).
8. Zintl E., Haucke W., Z. Elektrochemie, 44, 104 (1938).
Метод использования металла-растворителя
По аналогии с реакциями, в которых реагенты взаимодействуют в растворе и дают неметаллическое соединение, для синтеза из простых веществ интерметаллических соединений во многих случаях используют «третий» металл, 20*
2164 Глава 34. Сплавы и интерметаллические соединения
служащий растворителем. В связи с тем что взаимная растворимость металлов (и полуметаллов) в жидком состоянии часто бывает ограниченной и поскольку после завершения взаимодействия основных компонентов металл-растворитель должен быть удален из образовавшегося соединения, применение этого метода ограничено соединениями, практически индифферентными по отношению к тому растворителю, который, применяется для удаления «третьего» металла. Таким растворителем служит преимущественно разб. HNO3 (например^ 10%-ная), а для алюминия также разб. NaOH. В большинстве случаев при растворении не удается избежать некоторых потерь выделяемого соединения. Этот метод применяется в основном прн получении силицидов, боридов, фосфидов н полуметаллов, например GaSe, GaTe, особенно в виде монокристаллов. В качестве металла-растворителя впервые было использована олово [1]. Другими успешно применяемыми металлами-растворителями являются свинец, алюминий, магний, медь и серебро. Благодаря иногда весьма значительным различиям в растворимости металлов-реагентов в металле-рас-творителе часто удается, варьируя количества реагентов, получать в расплаве металла-растворителя вполне определенное бинарное соединение из нескольких возможных в данной системе. Как металл-растворнтель можно рассматривать и ртуть при образовании амальгам (см. ниже разд. «Амальгамы»).
ЛИТЕРАТУРА
1. Jolisbois, С. R. Acad. Sci. Paris, 150, 106 (1910).
2. Blitz W., Heimbrecht M„ Z. Anorg. Allgem. Chem., 237, 132 (1938).
3. Schneider A., Stendel I., "L. Anorg. Allgem. Chem., 303, 227 (1960).
4. Petersen D. R„ Rinn H. W., Acta Cryst., 14, 328 (1961).
5. Jangg G., Kieffer R., Kogler H„ Z. Metallkunde, 59, 546 (1968).
6. Wadsten T., Chem. Commun. Univ. Stockholm, Nr. 10 (1977).
7. Andersson Y„ Rundqvist S„ Beckman O., Lundgren L., Nordblad P., Phys. Stat. Solid., (a) 49, K153 (1978).
8. Braun D.. L, Jeitschko W„ Z. Anorg. Allgem. Chem., 445, 157 (1978).
Специальные методы
Кроме описанных выше методов известны еще и другие возможности получения интерметаллических и полуметаллических соединений, которые, однако, до сего времени использовались лишь в специальных случаях, поскольку возможность их осуществления связана с довольно редким сочетанием условий.
Способ .1, Сплавы могут образовываться, когда их металлические компоненты выделяются из соответствующих неметаллических соединений в результате одновременно протекающих реакций химического восстановления. Так, например, при восстановлении водородом смеси оксида ниобия и никеля образуется сплав никеля с ниобием [1].
Аналогичное явление наблюдается, если оксид алюминия, находящийся в соприкосновении с металлической платиной, нагревают в атмосфере водорода. Образование сплава алюминия и платины в этих условиях сначала явилось нежелательной помехой, но в дальнейшем послужило основой для разработки метода получения многочисленных сплавов платины из оксидов другого металла и платины путем нагревания смесей до температуры 900—1200 °C [2]. В качестве восстановителя вместо водорода можно также применять аммиак [3]. Очень чистые силициды титана были получены при взаимодействии между TiCl4, S1C14 и водородом при температуре выше 800 °C [4]. Сплавы можно синтезировать и из смеси иодидов металлов, если последние раство- | ряются в жидком аммиаке. Эти растворы восстанавливают металлическим нат- | рием. тоже растворенным в жидком аммиаке [5]. |
’ й
'и
Специальные методы 2165
Восстановительное термическое разложение изоморфных смесей формиатов или оксалатов железа, кобальта и иикеля приводит к образованию мелкокристаллических порошков сплавов, которые заслуживают внимания в том отношении, что оии соответствуют фазовым равновесиям при относительно низких температурах [6].
Галогенид металла можно непосредственно восстановить другим металлом, взятым в избытке, и получить сплав. Таким путем, например, удалось из газообразного МоС15 и жидкого цинка (при 400—500 °C) синтезировать интерметаллические соединения состава MoZn22 и MoZn?, которые иначе трудно было бы получить из-за большой разницы температур плавления и кипения обоих металлов [7]. Единственный способ синтеза некоторых сплавов состоит в термическом разложении смеси летучих карбонилов металлов [8].
Таким путем можно реализовать фазовые равновесия при очень низких температурах, при которых еще не наступает плавление и не идет диффузия.
Способ 2. Некоторые сплавы могут быть получены путем электролитического выделения из водных растворов. Будут ли оба металлических компонента осаждаться совместно и в нужном соотношении, это зависит от состава электролита, от условий выделения н от присутствия особых способствующих выделению добавок. Гальванически осажденные сплавы, как и затвердевшие сплавы, могут представлять собой гетерогенное образование, твердый раствор или промежуточную фазу. Оии могут отличаться от соответствующих сплавов, полученных термическим путем как в отношении фазовых границ, так и по многим физико-технологическим свойствам ,[9]. Таким путем были исследованы сплавы в следующих бинарных системах: Си—Zn, Си—Sb, Си—Bi, Си—РЬ, Ag—Zn, Ag—Cd, Ag—Au, Ag—Bi, Ag—Pb, Au—Си, Au—Ni, Ni—Zn, Ni—Cd, Ni—Fe, Zn—Cd, Pb—Sn, W—Ni, W—Co, W—Fe.
Путем электролиза при высокой температуре также удается получить из расплавов некоторых соединений металлов интерметаллические или полуметаллические соединения. При этом иногда играют существенную роль вторичные реакции. Разработанный главным образом Андрие й Додеро [8] метод до сих пор был применен к получению боридов, силицидов (см. ниже), фосфидов, арсенидов и карбидов.
ЛИТЕРАТУРА
1. Grube G., Kubaschewski О., Zwiauer К-, Z. Elektrochemie, 45, 881 (1939).
2. Klemm W., Bronger W., Z. Anorg. Allgem. Chem., 319, 58 (1962).
3. Bronger W., J. Less-Common Metals, 12, 63 (1967).
4. Nickl J. J., Preparation of Intermetallic Compounds by Chemical Vapor ' Deposition. In.: 2. Intern. Conf. Chem. Vapor Deposition, ed. Blocher J. M.,
Electrochem. Soc., 1970, New York, S. 297.
5. Craven W. E„ Ostertag W. In: Proc. 7-th Rare Earth Res. Conf., Oct. 1968, Coronado Calif., S. 727.
‘ 6. Lihl F„ Metall, 5, 183 (1951); Hund F„ Z. Elektrochem., 56, 609 (1952);
Lihl F„ Wagner H., Zemsch P., Z. Elektrochem., 56, 619 (1952).
7. Heumann Th., Z. Metallkunde, 60, 438 (1969).
8. Heumann Th., Arch. Eisenhiittenwes., 34, 781 (1963).
9. Raub E., Metalloberflache, 7, (1953); Raub E., Feinwerkstechnik, 53, 205 (1949); 54, 288 (1950).
I 10. Andrieux L.. Ann. Chimie, (10) 12, 423 (1929); Chim. et Ind., 27, Sondernr.,
J 3, 411 (1932); Rev. Metallurgie, 32, 487 (1935); Congr. Chim. Ind. Nancy,
f 18, I, 124; Andrieux L., Weiss G., Bull. Soc. chim. France. Mem., (5) 15,
Г 598 (1948); Andrieux L., Dodero M., C. R. Acad. Sci. Paris, 198, 753
| (1934); Dodero M., Bull. Soc. chim. France, Мёт., (5) 17, 545 (1950).
2166 Глава 34. Сплавы и интерметаллические соединения
Бориды и силициды
Несмотря иа то что элементы бор и кремний не относятся к металлам, их соединения с металлами, в особенности с переходными металлами (Т-метал-лами в табл. 52), имеют металлический или полуметаллический характер и поэтому рассматриваются в этой главе. Указания относительно методов получения боридов и силицидов содержатся в табл. 53.
Таблица 53. Способы получения боридов и силицидов
Тип реакции Схема взаимодействия
I. Синтез из простых веществ а) путем сплавления б) путем спекания II. Алюминотермия, магние-термия III. Восстановление оксидов под действием С, В4С, Si IV. Электролиз расплавов V. Выделение из газовой фазы М+В—»МВ, МН+В^МВ+Н2 M+Si^M, MH+Si-^MSi+H2 МО+В2Оз-|-А1 (Mg) —»-МВ-|-Al (Mg) -оксид MO-j-SiCb+Al (Mg) —>MSi+Al (Mg) -оксид MF2+Al-Si—»-MSi+AlF3 MO-j-Al-Si—>MSi (в A1)+A12Os M+Al-Si—^MSi (в Al) MO+Al-Si+NaF—>-MSi (в Al)+Na3AlF6—A12O3 MO+B2O3+C—+MB+CO мо+в4с+с^мв+со MO+SiO2+C—>-MSi+CO MO+Si—^MSi+SiO (r.) МО+борат щелочного (щел.-зем.) металла+фторид щелочного (щел.-зем.) металла—>МВ+фтороборат ще-. лочного (щел.-зем.) металла M+K2SiF6^MSi+KF М (или М-галогенид)+галогенид бора+Н2—»-МВ-]-га-логеноводород M+SiCl4+H2^MSi+HCl MCL+SiCl4+H2—>MS 1+НС1
Способ 1 [1—11]. Бор и кремний поступают в продажу в виде препаратов самой различной степени чистоты. Выбор материала той или иной степени чистоты в каждом отдельном случае определяется часто лишь финансовыми соображениями. При этом, однако, следует иметь в виду, что использование компактных реагентов при синтезе боридов и силицидов металлов часто сопряжено с определенными экспериментальными трудностями, связанными с возможностью протекания слишком бурной экзотермической реакции. При обычном быстром нагревании реакционной смеси может произойти настолько сильное саморазогревание, что из-за взаимодействия со стенками сосуда или вследствие потерь при испарении получение чистого интерметаллического соединения оказывается уже невозможным. Поэтому вместо металлов в качестве реагентов используют их гидриды, которые реагируют обычно менее бурно.
Бориды и силициды 2167
Лучше, однако, вести синтез по способу 16 (табл. 53). Бориды и силициды, особенно Т-металлов (d2—d5), получают путем спекания смесей порошкообразных простых веществ при медленном повышении температуры до максимальной (1200—1500°C). Предварительное уплотнение образца при прессовании смеси порошков в таблетки облегчает диффузию компонентов. В качестве материалов для изготовления сосудов применяют оксид алюминия, графит, нитрид бора, металлические молибден или вольфрам.
Согласно Киферу и Цервенка <[9], гомогенность конечного препарата можно улучшить, если механическое давление на образец производить во время прокаливания. Однако из-за того, что при этом применяют графитовые пресс-формы, возникает опасность загрязнения сплава углеродом.
Во всех случаях нагревание следует осуществлять в вакууме или в атмосфере защитного газа — аргона.
Способ 2 [1—3, 12—14]. Методы восстановления действием алюминия или магния позволяют избежать необходимости приобретения или получения чистого бора, так как при этом можно исходить из оксидов. Неудобство этого метода состоит в том, что образовавшийся борид или силицид приходится отделять от побочного продукта реакции — оксидов алюминия или магния. Если синтез ведут, используя большой избыток металла-восстановителя, то может оказаться, что борид или силицид металла, часто в виде хорошо образованных кристаллов, оказывается внутри металла-восстановителя, тогда как побочные продукты (оксиды) всплывают наверх и могут быть в дальнейшем более легко отделены от компактного металлического королька. При применении шлакообразующих добавок, например CaF2, СаС12, Na3AlF6, CaO, Na2O, оксиды легче переводятся иа поверхность расплавленного металла-восстановителя.
После нагревания до температуры, лежащей несколько выше температуры плавления металла-восстановителя, и после последующего охлаждения образец извлекают из тигля (например, из завинченного или заваренного железного тигля) и механическим или химическим путем (нацример, соляной кислотой) удаляют шлак, а затем химически растворяют избыток алюминия (магния), в результате чего остается борид или силицид. Для растворения металлов используют растворы кислот различной концентрации, а алюминий хорошо растворяется в разбавленных растворах едких щелочей (с образованием растворов алюминатов).
В некоторых случаях вместо оксида металла для реакции берут его фторид или часто сам металл и проводят взаимодействие с раствором бора или кремния в алюминии или магнии. В целом это соответствует использованию вспомогательного металла-растворителя (см. выше разд. «Метод использования металла-растворителя» в этой главе), причем в качестве таковых можно применять и другие металлы (например, медь для силицидов).
Способ 3 [1—3, 15—17]. Этот способ заключается в восстановлении оксида металла карбидом бора (синтез боридов) либо углеродом или кремнием (синтез силицидов) при одновременном улетучивании образующихся СО или SiO при нагревании в вакууме. Для обеспечения полноты взаимодействия нагревание необходимо вести до довольно высоких температур. Реакцию, при которой SiO удаляется из реакционной смеси сублимацией, можно использовать лишь в том случае, если и металл, и восстанавливаемый оксид малолетучи.
Смеси веществ в соответствии со схемами реакций табл. 53, но взятых с учетом стехиометрических коэффициентов, сначала прессуют в таблетки, а затем нагревают в высоком вакууме. Для синтеза боридов Т-металлов (переходных металлов) за приемлемое время требуется проводить нагревание при особенно высоких температурах (1400—1900 °C).
другие способы получения. Способ 4 [1—3, 18—23]. Для получения небольших количеств боридов металлов применяют метод электролиза расплавов. Из расплавов боратов щелочных и щелочноземельных металлов на катоде
2168 Глава 34. Сплавы и интерметаллические соединения
осаждается бор, который взаимодействует с одновременно выделяющимся металлом.
При синтезе силицидов расплавами служат смеси силикатов или фторосиликатов с фторидами металлов (испытано на силицидах титана, циркония, хрома и редкоземельных элементов).
Способ 5 [1—3, 24, 25]. В результате пропускания смесей из галогенида металла, ВВг3 и водорода над раскаленными металлами, играющими роль носителей, на поверхности этих металлов образуются только слои боридов, но не компактные однородные препараты. То же справедливо для процесса сили-цидирования путем выдерживания раскаленного металла в смеси газообразных SiCl4 и водорода. Напротив, по реакции, приведенной в табл. 53 последней, могут быть получены силициды определенного состава в гомогенном виде и в существенно больших количествах.
ЛИТЕРАТУРА
1. Kiefer R„ Benesovsky F., Hartstoffe, Springer, Wien, 1963.
2. Kiefer R„ Benesovsky F., Metall, 6, 171, 243 (1952).
3. Aronsson B., Lundstrom T., Rundquist S., Borides, Silicides and Phoaphides, Methuen, London — Wiley, New York, 1965.
4. Wallbaum H. J., Z. Metallkunde, 33, 378 (1941).
5. Ehrlich P„ Z. Anorg. Allgem. Chem., 259, 1 (1949).
6. Kiessling R., Acta Chem. Scand., 4, 209 (1950).
7. Brewer L., Searcy A. W., Templeton D. H., Dauben С. H., J. Amer. Ceram. Soc., 33, 291 (1950).
8. Brewer L., Sawyer D. L., Templeton D. H„ Dauben С. H., J. Amer. Ceram. Soc., 34, 173 (1951).
9. Kieffer R., Cerwenka E„ Z. Metallkunde, 43, 101 (1952).
10. Nowothny H„ Benesovsky F„ Kieffer R., Z. Metallkunde, 50, 258, 417 (1959).
11. Самсонов Г. В., Ковальченко М. С., Верхоглядова Т. С. — Журн. неорг. хим., 1959, т. 4, с. 2759.
12. Norton J. Т., Blumenthal И., Sindeband S. J„ Trans. AIME, 185, 749 (1949).
13. Sindeband S. I., Trans. AIME, 185, 198 (1949).
14. Brauer G., HaagH., Z. Anorg. Allgem. Chem., 267, 198 (1952).
15. McKenna P. M., Ind. Ingn. Chem., 28, 767 (1936).
16. Brauer G., Mitlus A., Z. Anorg. Allgem. Chem., 249, 325 (1942).
17. Brauer G„ Haag H., Z. Anorg. Allgec. Chem., 259, 197 (1949).
18. Kieffer R„ Benesovsky F., Honak E. R., Z. Anorg. Allgem. Chem., 268, 191 (1952).
19. Меерсон Г. А., Самсонов Г. В. — Жури, прикл. хим., 1954, т. 27, с. 1115.
20. Baroch С. Т., Evans Т. Е., J. Metals, 7, 908 (1955).
21. Chene М., Ann. Chim. Paris, 15, 187 (1941).
22. Andrieux L., Rev. Metallurg., 45, 49 (1949).
23. Weiss G., Ann. Chim. Paris, 1, 446 (1946).
24. Champbell J. E„ Powell C. F., Nowicki D., Gonser B. W., J. Elektrochem. Soc., 96, 318 (1949).
25. Nickl J. J., Preparation of Intermetallic Compounds by Chemical Vapor Deposition. In: 2. Intern. Conf. Chem. Deposition, ed. Blocher J. M., Electro-chem. Soc., 1970, New York, p. 297.
Амальгамы
Получение сплавов ртути легко удается для большинства иизкоплавких металлов (tM <1000 °C) в тех случаях, когда они вообще способны образовывать амальгамы. Взаимодействие компонентов осуществляют в закрытых же-
Амальгамы 2169
лезиых тиглях и в стеклянных ампулах, в которые иногда еще помещают вставные тигли (см. общие сведения о получении интерметаллических соединений, разд. «Тигельный и ампульный методы» в этой главе). При этом имеется возможность выбирать любой состав амальгамы, образованной из основного металла и ртути, в границах, определяемых фазовыми соотношениями.
Еще один общий метод получения амальгам состоит в проведении электролиза металлической соли на ртутном катоде. Концентрация выделившегося металла в образованной таким путем амальгаме может быть повышена в значительной степени так, что при этом будут выделяться твердые фазы. Этот метод удается применить даже к металлам с исчезающе малой растворимостью в ртути (например, к железу). В этих случаях получают суспензии в ртути, обнаруживающие свойства, весьма сходные со свойствами «истинных» амальгам (см. табл. 54). С помощью амальгам истинных и псевдоамальгам удается
Таблица 54. Растворимость металлов в ртути при температуре около 20 °C [1]
Металл Растворимость, масс. % Металл Растворимость, масс. %
Ag 0,03 Na 0,62
Al 2,3 -10"3 Ni 2-Ю"’
As 0,24 Pb 1,5
Au 0,13 Pd 0,06
Ba 0,33 Pt 2-10-8
Be MO'3 Rb 1,4
Bi 1,4 Rh 0,16
Ca 0,3 Ru 0,35
Cd ~5,0 Sb 2,9 IO'3
Co <11 O'6 Si LIO'3
Cr <4-10-’ Sn 0,87
Cu 2-10'3 Sr 1,0
Fe <5-10-’ Ta 0,01
Ge 1•10-3 Th 0,016
In 57 Ti 1 • IO'6
К 0,38 U 1-Ю'5
La 0,013 V l-10-з
Li 0,036 w LIO'6
Mg 0,31 Zn 2,0
Mn 1,7-10-3 Zr 3-10'3
Mo <2-IO"6
осуществить синтез многочисленных интерметаллических соединений. При этом ртуть и амальгамы выполняют роль металла-растворителя (см. выше разд. «Метод использования металла-растворителя»). Особые преимущества применения амальгам связаны с тем обстоятельством, что синтез проводят прц относительно низких температурах, а ртуть, служащая вспомогательным ме--таллом, легко может быть удалена путем отгонки.
ЛИТЕРАТУРА
1. Jangg G., Bach Н„ Quecksilber- und Amalgammetallurgie. In: Handb. d. Techn. Elektrochemie, 2. Aufl., Bd. I, Akadem. Verl. Ges., Leipzig, 1961.
2170 Глава 34. Сплавы и интерметаллические соединения
2. Kirchmayr Н. R., Z. Metallkunde, 56, 767 (1965); Monatsh. Chem., 95, 1479 (1964); U. S. Dept. Comm. Clearing House Sci. Tech. Inform., AD627, 225, 77 (1965); C. A. 65, 431 f.
3. Lihl F„ Kjrnbauer H., Z. Metallkunde, 48, 9 (1957).
Для определенных целей, в особенности же для применения в качестве восстановителей, в лаборатории желательно располагать простыми методами получения жидких или полутвердых амальгам неблагородных металлов. Для таких случаев могут быть применены описанные ниже следующие специальные методы.
Жидкая амальгама натрия (~1% Na)
Способ 1. Очищенный металлический натрий в количестве, например, 11,5 г разрезают на кубнки с длиной ребра, приблизительно равной 5 мм, и эти кубики растворяют в 1150 г (85 мл) чистой ртути, находящейся в широ-когорлой полулитровой конической колбе. Кубики натрия быстро погружают один за другим при помощи заостренной стеклянной палочки в предварительно нагретую до 30—40 °C ртуть. Лист картона, через который вводится стеклянная палочка, закрывает отверстие конической колбы и предохраняет продукт реакции от разбрызгивания, происходящего вследствие сравнительно бурной реакции образования амальгамы.
Способ 2. 3,5 г натрия расплавляют на электрической нагревательной плитке в конической колбе емкостью 250 мл под защитным слоем 10—15 мл толуола и добавляют при помешивании или при встряхивании по каплям 340 г (25 мл) ртути. Прибавление первых миллилитров ртутн сопровождается энергичной реакцией. Затем образование амальгамы протекает менее бурно. Толуол при этом находится в состоянии непрерывного кипения; под конец его декантируют или заменяют другими жидкостями.
Можно также использовать приведенные ниже методы получения твердой амальгамы с соответственно измененными количествами реагентов.
Твердая амальгама натрия (~2—3% Na)
Способ 1. Особо чистый препарат в небольшом количестве можно получать по Физеру [5]: аккуратно разрезанный на куски натрий в количестве 6,9 г (для амальгамы с содержанием 2% натрия) нли 10 г (для амальгамы с содержанием 3% натрия) помещают в круглодонную трехгорлую колбу емкостью 250 мл. Через оба боковых горла проходят трубки для ввода и выпуска азота. В среднее горло вставлена капельная воронка с 340 г (25 мл) ртути. После тщательного заполнения колбы азотом к натрию добавляют 10 мл ртути и колбу осторожно нагревают открытым пламенем до тех пор, пока не начнется реакция. Затем медленно приливают остальное количество ртути, причем достаточно лишь слегка подогревать колбу. Когда все количество ртути будет таким образом прибавлено в колбу, еще горячую расплавленную амальгаму выливают на чистую плиту и разбивают ее на куски, пока она еще теплая и хрупкая.
Способ 2. Другой метод, по которому могут быть получены и значительные количества амальгамы, осуществляют таким образом, что сначала нагревают до плавления, например, 51 г чистого свежеразрезанного натрия, помещенного в эмалированный сосуд (диаметром около 18 см) и находящегося под слоем вазелинового масла. Толщина защитного слоя масла должна быть около 1 см. После этого к расплавленному натрию медленно и при непрерывном помешивании добавляют из капельной воронки 1650 г (122 мл) ртути. Процесс заканчивается в течение 3—4 мин. Главную массу вазелинового масла сливают. По охлаждении амальгама затвердевает при температуре около 250 °C, причем
Амальгамы 2171
в процессе охлаждения ее необходимо растолочь прочным пестиком на мелкие зериа. Можно также еще горячую жидкую амальгаму вместе с остатками масла вылить в фарфоровую чашку, дать ей затвердеть в виде плоской лепешки и растолочь твердый продукт в ступке до желаемой величины зерен.
После полного охлаждения куски амальгамы отмывают от масла петролейным эфиром или бензолом, снова высушивают для удаления промывных жидкостей и сохраняют без доступа воздуха.
Свойства. Амальгамы, содержащие до 3% натрия, не являются слишком чувствительными к действию воздуха. Однако при хранении необходимо позаботиться об их тщательной изоляции от воздуха. Температуры полного перехода в жидкость (температуры ликвидуса) составляют: при 0,5% натрия 0°С; при 1,0% — 50°С; при 1,5% —100°С; при 2% — 130°C; при 2,5% — 156°C; при 3,0% — 250°C; при 4,0% — 320°C.
Способ получения амальгам калия описан в работе [6].
ЛИТЕРАТУРА
1. Синтезы органических препаратов, сб. 1. — М.: ИЛ, 1949, с. 235.
2. Ренфроу В., Хаузер Ч. Синтезы органических препаратов, сб. 2. — М.: ИЛ, 1949, с. 482.
3. Делофе В., Герреро Т. Синтезы органических препаратов, сб. 3. — М.: ИЛ, 1952 с 299.
4. Babcock 8. И., Inorg. Synth., 1, 10 (1939).
5. Fieser L. F., Experiments in Organic Chem., New York, 1941, p. 419.
6. Boeder A., Morawietz W., Z. Elektrochemie, 60, 431 (1956).
Амальгама кальция
Отверстие небольшого стального реактора 1, изображенного на рис. 491, снабжено плоской, хорошо сделанной нарезкой, на которую герметически на-
Рис. 491. Аппарат для получения амальгамы кальция.
/—•стальной реактор длиной 19 см, внешним диаметром 5,5 см и толщиной стенок 9 мм, объем до нарезки около 45 мл; 2— головка реактора; 3 — редукционный вентиль; 4 — манометр; 5 — медный трубопровод высокого давления; 6 — подвижной поршень.
винчена выточенная из того же материала головка 2. Герметичность обеспечивается при помощи конуса. Головка просверлена и к ней сверху присоеди
2172 Глава 34. Сплавы и интерметаллические соединения
нен редукционный вентиль Россиньоля 3, затем — манометр 4 и трубопровод высокого давления 5, соединенный с азотным баллоном. В реакторе находится подвижный поршень 6, причем зазор между ним и стенками баллона весьма невелик.
Сначала в реактор вносят около 35 мл чистой ртути, затем около 7—10 г продажного очищеного рашпилем кальция, после чего вставляют поршень. Затем реактор плотно закрывают головкой и в него впускают азот до достижения давления 60—70 бар. Через несколько минут можно установить образование амальгамы по заметному разогреванию нижней части реактора. Через 10—15 мин реакция полностью заканчивается. Давление в реакторе снижают, его открывают, быстро переводят амальгаму в сухую, хорошо закрывающуюся баночку объемом — 60 мл, споласкивают стальной реактор небольшим количеством чистой ртути и наполняют ртутью баночку почти до самой пробки. При разбавлении ртутью амальгама сильно разогревается. Ее хорошо встряхивают и дают охладиться.
Свойства. Полученная таким путем амальгама содержит около 1% кальция. Она иногда затвердевает на холоду; в хорошо закрытых склянках устойчива в течение неограниченного времени.
По данным справочника i[2], кривая ликвидуса в системе Са—Hg характеризуется при содержании 2% кальция температурой около 260 °C, при содержании 1% кальция — температурой —140 °C, при 0,3% кальция — температурой —25 °C. Основная часть таких амальгам затвердевает при температуре перитектики, равной —39 °C.
ЛИТЕРАТУРА
1. Brukl A., Angew. Chem., 52, 151 (1939).
2. Хансен М., Андерко К- Структуры двойных сплавов. Т. 2: Пер. с аигл. — М.: Металлургиздат, 1962.
Амальгама стронция
Получение амальгам стронция и бария осуществляют путем электролиза растворов соответствующих хлоридов на ртутном катоде. Каждая из описанных ниже двух установок для получения определенной амальгамы может быть применена и для получения амальгамы другого металла.
Рис. 492. Прибор для получения амальгамы стронция.
1 — сосуд для электролиза; 2 — ртутный катод; 3 — токоввод (платиновая проволока); 4 — глиняная диафрагма; 5 —графитовый стержень; 6, 7 — внутренний и внешний змеевики, охлаждаемые водой.
Используют установку для электролиза, показанную на рис. 492. В сосуде для электролиза 1 содержится около 300 мл слабо подкисленного соляной кислотой насыщенного раствора очень чистого SrC12, который при необходи
Амальгамы. 2173
мости должен быть предварительно еще очищен путем перекристаллизации от следов присутствующих в нем солей натрия и железа. В качестве катода 2 служат 600 г (45 мл) чистой перегнанной ртути, к которой при помощи толстой платиновой проволоки 3 подводят электрический ток. Анодное пространство отделено при помощи подвешенной глиняной диафрагмы 4. Через нее проходит графитовый стержень толщиной приблизительно 10 мм, который является анодом. Электролиз ведут током 6,5 А, что при поверхности катода в 24 см2 соответствует плотности тока 0,27 А/см2. Вследствие сильного нагревания при прохождении тока температуру электролита понижают до 38—40 °C при помощи внутреннего и внешнего охлаждающих змеевиков 6 и 7. Эта температура является наилучшей для электролиза. Через каждые четверть часа жидкость в анодном пространстве сменяют для того, чтобы предотвратить накопление в нем хлора и избегнуть опасности перехода хлора в катодное пространство. Приблизительно через полтора часа содержание стронция в ртути повышается до 1,3%, и амальгама начинает приобретать благодаря достижению точки ликвидуса кашицеобразную консистенцию. Электролиз прерывают и выливают электролит. Затем удаляют амальгаму, промывают ее несколько раз водой и высушивают фильтровальной бумагой.
Свойства. При комнатной температуре амальгама является твердым телом и имеет серебристый блеск. Сохранять ее необходимо без доступа воздуха.
литература
1. Holleck L„ Noddack W., Angew. Chem., 50, 819 (1937).
Амальгама бария (см. также «Амальгама стронция»)
В стакан емкостью 250 мл помещают в качестве катода 250 г (18 мл) чистой ртутн, 100 мл насыщенного раствора ВаСЬ и горизонтально расположенную платиновую пластинку размером 5—10 см2 в качестве анода. Подводку тока к ртути осуществляют при помощи платиновой проволоки, которую для изоляции от электролита впаивают в стеклянную трубку.
Устанавливают напряжение 6—7 В и подвергают раствор электролизу в течение 2—2,5 ч при силе тока 1,75—2,5 А. При электролизе, продолжающемся слишком долго, наступают сильное повышение напряжения, выделение газа на катоде и разложение амальгамы. Нежелательное выделение кристаллических частичек амальгамы на поверхности ртути устраняют при помощи медленно вращающейся автоматической мешалки или путем удаления этих кристаллов палочкой, грибовидно сплющенной на одном конце.
По окончании электролиза раствор сливают, амальгаму хорошо промывают дистиллированной водой, затем спиртом и эфиром и сохраняют без доступа воздуха. Она содержит около 3% бария.
ЛИТЕРАТУРА
1. McPhail Smith G., Bennett A. C., J. Amer. Chem. Soc., 31, 804 (1909).
2. Marklein. В. C., 'West D. H., Audrieth L. F., Inorg. Synth., 1, 11 (1939).
Амальгамы цинка, кадмия, олова, свинца и висмута (жидкие) [1,2]
Для получения жидкой амальгамы циика с содержанием 2—3% цинка 4 г чистого цинка в зернах, стружках или, еще лучше, в виде фольги обезжиривают эфиром и хорошо промывают разбавленной серной кислотой. Затем цинк нагревают на водяной бане с 200 г (14,8 мл) ртути и с 2 мл 2 н. серной
2174 Глава 34. Сплавы и интерметаллические соединения
кислоты в колбе на 100 мл. Приблизительно через 20 мии происходит полное растворение цинка. Жидкую амальгаму несколько раз промывают сильно разбавленной серной кислотой и после охлаждения отделяют в делительной воронке от выделяющихся иногда твердых частей (0- или у-фаз состава, приблизительно соответствующего формуле HgZn2), которые затем можно использовать для повышения концентрации цинка в отработанной амальгаме с сильно уменьшившимся содержанием цинка.
Получение жидких амальгам кадмия и висмута с содержанием около 3% этих металлов осуществляют аналогичным путем. При получении амальгамы висмута вместо серной кислоты применяют соляную.
Для получения амальгамы олова с содержанием до 8% олова гранулированное олово нагревают с ртутью под слоем соляной кислоты.
Жидкую амальгаму свинца с содержанием около 3% этого металла получают из чистого свинца, который предварительно очищают при помощи соляной кислоты от слоя оксидов, после чего высушивают и нагревают с рассчитанным количеством ртути. Полученную амальгаму промывают водой.
Свойства. Описанные жидкие амальгамы очень устойчивы и могут сохраняться в течение долгого времени под слоем слабо подкисленной воды, так как они лишь крайне медленно реагируют с ней. Они применяются в объемном анализе в качестве восстановителей.
Амальгамы редкоземельных металлов [3]. Амальгамы редкоземельных металлов с содержанием приблизительно до 3% редкоземельного металла могут быть получены путем электролиза спиртовых растворов соответствующих безводных хлоридов с ртутным катодом и графитовым анодом. После их получения можно осуществить и дальнейшее концентрирование таких амальгам путем отгонки из них ртути.
Амальгама алюминия [4]. В большинстве случаев алюминий переводят в амальгаму не полностью, а лишь поверхностно для того, чтобы при проведении некоторых реакций произвести активацию алюминия.
ЛИТЕРАТУРА
1. Brennecke Е., Fliissige Amalgame als Reduktionsmittel in der Massanalyse, in: Brennecke, Faians, Furmann, Lang, Stamm, Neuere Massanalytische Methoden, Enke, Stuttgart, 1951.
2. Winterstein C., Z. analyt. Chem., 117, 81 (1939). .
3. Jukkola E. E., Audrieth L. F., Hopkins B. S., Inorg. Synth., 1, 15 (1939).
4. Adkins H„ J. Amer. Chem. Soc., 44, 2175 (1922).
Сплавы калия с натрием (жидкие)
На термической диаграмме состояния К—Na кривая ликвидуса проходит через минимум, соответствующий температуре —12,5 °C при 77,3 масс.% калия. Сплавы с составом, близким к этой точке, при комнатной температуре1 являются жидкостями (при +20 °C все сплавы, содержащие от 45 до 90% калия, еще являются жидкими). Реакционная способность этих сплавов повышена по сравнению с обоими щелочными металлами, взятыми в отдельности.
Эти смеси получают, осторожно нагревая, например, 3 г аккуратно нарезанного калия с 1 г так же тщательно подготовленного натрия или другие соответствующие количества обоих металлов под слоем безводных ксилола или толуола. При этом металлы сдавливают и перемешивают стеклянной палочкой с плоским концом.
По Лехеру [1], соединение обоих металлов осуществляют при комнатной температуре, растворяя в защитной жидкости незначительное количество спирта и тем самым активируя с поверхности куски металлов. Для этой цели при
Амальгамы 2175
меняют, например, сосуд Шленка, имеющий грушевидную иижнюю часть, направленное вверх узкое горло и второе боковое горло. В этот сосуд вносят 0,35 г натрия и 1,6 г калия (для органических реакций), а также лигроин, который содержит в небольшом количестве спирт, и пропускают в сосуд через боковое горло в качестве защитного газа азот. При спрессовывании металлов посредством пропущенной через верхнее горло стеклянной палочки с плоским концом оба металла соединяются с образованием жидкого сплава. Затем при непрерывном пропускании азота вымывают лигроин (содержащий спирт) вместе с суспендированными примесями чистым высушенным лигроином.
Разумеется, для получения этого сплава могут быть использованы кроме сосуда Шленка и другие приборы, обеспечивающие работу без доступа воздуха.
Жидкие сплавы натрия с калием самовоспламеняются на воздухе. Их необходимо защищать от соприкосновения с воздухом при помощи защитной жидкости или защитного газа (N2, СОг).
Данные о диаграмме состояния и кристаллических структурах в системе К—Na, а также по другим бинарным системам, образованным щелочными металлами друг с другом, содержатся в работе [2].
ЛИТЕРАТУРА
1. Lecher Н., Вег., 48, 524 (1915).
2. Helms A., Klemm W., Z. Anorg. Allgem. Chem., 242, 201 (1939).
Таблица 55. Состав и температура плавления некоторых легкоплавких сплавов
Обозначение сплава <пл> °C Состав, масс. %
Эвтектика Bi—Cd—Pb—Sn (сплав Вуда, Липовица) 71 49,5 Bi—10,1 Cd—27,3 Pb— 13,1 Sn
Эвтектика Bi—Cd—Pb 91,5 51,7 Bi—8,1 Cd—40,2 Pb
Эвтектика Bi—Pb—Sn (сплав Ньютона, Розе) 96 50 Bi—31,2 Pb—18,8 Sn
Эвтектика Bi—Cd—Sn 103 54 Bi—20 Cd—26 Sn
Эвтектика Bi—Pb 125 56,5 Bi—43,5 Pb
Эвтектика Bi—Sn 139 58 Bi—42 Sn
Эвтектика Cd—Pb—Sn 145 18,2 Cd—32 Pb—49,8 Sn
Эвтектика Pb—Sn 183 37,7 Pb—62,3 Sn
Эвтектика Sn—Zn 198,6 91,1 Sn—8,9 Zn
Эвтектика Cd—Zn 266 82,6 Cd—17,4 Zn
Эвтектика Ag—Pb 304 2,5 Ag—97,5 Pb
Эвтектика Ga—In—Sn—Zn 3 61 Ga—25 In—13 Sn—1 Zn
Эвтектика Ga—In—Sn 5 62 Ga—25 In—13 Sn
Эвтектика Ga—In—Zn 13 67 Ga—29 In—4 Zn
Эвтектика Ga—In 16 76 Ga—24 In
Эвтектика Ga—Sn 20 92 Ga—8 Sn
Эвтектика Ga—Zn 25 95 Ga—5 Zn
L 46 46,5 40,6 Bi—8,2 Cd—18 In- 22,4 Pb—10,8 Sn
L 58 58 49 Bi—21 In—18 Pb—12 Sn
Эвтектика In—Sn 117 52 In—48 Sn
2176 Глава 34. Сплавы и интерметаллические соединения /
Легкоплавкие сплавы
Для специальных целей, например для заполнения нагревательных бань, манометров, для изготовления плавких предохранителей или в качестве замазки, пользуются сплавами, имеющими низкие температуры плавления. В табл. 55 сопоставлены свойства некоторых подобных сплавов, устойчивых по отношению к действию воздуха при температуре их плавления и при несколько более высоких температурах.
При применении редких металлов галлия и индия можно получить сплавы, обладающие особыми свойствами.
Подробную сводку данных о таких сплавах с температурами плавления между —39 и +419 °C можно найти в работах [1]. Если легкоплавкие сплавы используют в качестве замазок и припоев для неметаллических материалов, то особое внимание следует обращать на соответствие коэффициентов термического расширения. Например, если для заполнения стеклянных нагревательных бань используют расплавленный сплав Вуда, то при охлаждении и затвердевании металла происходит растрескивание сосуда.
ЛИТЕРАТУРА
1. Spengler Н„ Metall, 9, 682 (1955); Z. Metallkunde, 46, 464 (1955).
2. D’Ans Г, Lax E„ Taschenbuch f. Chemiker u. Physiker, Springer, Berlin—Heidelberg, 3. Aufl., Bd. 1, S. 723 (1967).
3. Хансен M., Андерко К. Структуры двойных сплавов. Т. 1—2: Пер. с англ., 1962, l-е доп. Эллиот Р. И., 1970, 2-е доп. Шанк Ф. А., 1973, М„ Металлургия.
4. Smithells С. J. S., Metals Reference Book, London, 1949.
5. Ludwick M. T., Indium..., The Indium Corp, of Amer., New York, 1950,
6. Kroll W., Metallwirtschaft, 11, 435 (1932),
Формульный указатель
А
(Р^12^зб) 'лНзО 1908
AuBr[P(C6Hs)3] 2127
Аи(СН3)[Р(С6Н5)3] 2033
Au(CH3)3[P(CeHs)3] 2034 [AuC(OCH3)=NC6H!i]3 2048 [Au(CN4-z73O-C3H7)4] [As(C6H5)4] 2049
AuCl(CO) 2075
AuCl[P(C6H3)3] 2127 [Au(N3)4] [As(C6H5)4] 2049
В
(BWi2O40)-nH2O 1911
Ba[Cd(OH)5] 1881
Ba2[Cu(OH)e] 1880
Ba3[Fe(OH)6]2 1887
Ba2[Fe(OH)7]-0,5H2O
Ba-Hg 2173
Ba^[Mg(OH)6] 1875 Ва3(РМо1204о)иН20 1899
Ba2[Pb(OH)s[ 1890
Ba[Sn2O(OH)4] 1876 [Be(OH)4]Na2 1873 [Be (OH) 4] Sr 1873 Bi-Hg 2173
C
(C5H5)2Mg 1926
C6H5Na 1924
C5H4CH3Na 1924
C5H5TI 1925
C5H5 (C4H9) 3Sn 1927
Ca-Hg2171
Cd-Hg2173
Со(л-С3Н5)(СО)3 2007
Co(n-C5H5)2 1961 [Co(71-CsHe)2])[PF6] 1962 Co(ri-C5H5)(CO)2 1987 Co(t]-C5H3)I2(CO) 2069 [Co(t)-C3H3)NO]2 2096 Co(T)-C3H5) [P(CH3)3]2 2117 [Cofo-C5H5) (H){P(CH3)3}2] [PFe] 2085
Co(C5H7O2)3 2137
Co2(C6H5C=CC6H5) (CO)e 2007
Co2(CO)8 1946
Co4(CO)12 1948 [Co(CO)3]3(p.3-CCl) 2041 Co(CO)3NO 2095 [Co(CO)4]Tl 1948 [Co(CO)4]2[Mg(C5H5N)4] 1949 CoCl[P(CH3)3]3 2117
21—1361
Co(H)(N2) [P(CeH5)3]3 2137
CoH[P(OC6H5)3]4 2085
[CoN2{P (CH3)3}3] 2Mg (C4H8O) 4 213& [Co(OH)4]Na2 1877
Co[P(CH3)3]4 2116 Cr[r|-B3N3(CH3)6] (CO)3 1975 CrBr(CC6Hs) (CO)4 2039 [CrBr(CO)6J[N(C2H5)4] 2056 Cr(T)-C3H5)3 1995
Cr(n-C4H5N)(CO)3 1974 Cr(r]-C4H4S) (CO)3 1974 СгСп-С4Н4Те)(СО)з 1974 Cr(r)-C5H5)2 1953
Cr(n-C5H5) (q2-C3H4) (CO) (NO) 2088; Cr(n-C5H5)(CO)2(NS) 2089 Cr(r)-C5H5) (CO) (CS) (NO) 2099 Cr(r)-CSH5)H(CO)3 2078 Сг(т]-С6Н6)2 1954 Cr(r)-C6H5C1) (СО)з 1973 Cr[n-C6H3(CH3)3] (CO)2(CS)2 2099. Cr(ri4-C7H8)(CO)4 1996 Cr(CNC6H4-o-CH3)6 2103 Cr(CO)3(NCCH3)3 2128 [Cr(CO)5Br] [N(C2H5)4[ 2056.
Cr(CO)5[C3(C6H5)2] 2036 Cr(CO)5CNH 2104
[Cr (CO) 5 (CN) ] [N (C2H5) 4] 2107 [Cr(CO)5(CN)]Na 2107 Cr(CO)5(CNC6H5) 2105 Cr(CO)5[C(NHC6H5)2] 2037 [Cr(CO)5]2-p-N2H2 2131 [Cr(CO)5|2-n-N2H4 2130 Cr(CO)5C(OCH3)R 2034 Cr(CO)s[CNSi(CH3)3] 2104 [Cr(CO)5]Na2 1932 [Cr(CO)5J2N2H2 2131 [Cr(CO)5]2N2H4 2130
Cr(CO)6 1930
Cr2(CO)10Na2 1934
Cr(NO)4 2087
[Cr(NCCH3)s(NO)] [PF6]2 2088;
Cr(OH)6lNa3 1884 Cr(OH)6]2Sr3 1885
Cr3OioK2 1891 Cr40i3K2 1891 [Cu(OH)6]Ba2 1880 [Cu(OH)4]Na2 1878, 1879
F
FeBr2(CO)4 2065 [Fe(n-C3H5) (CO)3]2 2005 Fe(n-C3H3)Br(CO)3 2004 Fe(r|-C3H5)C1(CO)3 2003 Fe(n-C3H5)I(CO)3 2004 Fe(r|-C4H4) (CO)s 2001
2178 Формульный указатель
Fe(n-C4H6) (С0)3 2002 Fe('n-C4H6)2CO 2002 Fe(n-C5H5)2 1956 [Fe(ri-C5H5)2] [PF6] 1958 Fe(r)-C5H5) (CH3) (CO)2 2030 Fe(n-C5H5) (т]-С8Н4СНО) 1958 [Fefn-CsHs) (n-C5H4COCH3)] [BFJ 1958
Fe(T]-C5H5)CO]4 1984 Fe(r)-C5H5) (CO)2]2 1983 [Fe(r)-C5H5) (CO)2(NCCH3)] [PF6] 2135
[Fe(r1-CSH5)(CO)3]I[PF61 1985 Fe(Ti-C5H5)Br(CO)2 2063 Ре(я-С5Н5)С1(СО)2 2063 Fe(r)-CSH5)I(CO)2 2063 [Fe(n-C5H5) (CO)2(CNCeH5)}[PFeJ 2109
[Fe(r]-C5H5) (CO) (CNC6H5)2] [PF6] 2110
(Fe(r]-C5H5) (CNC6H5)3]C1 2111 Feh-CsHs) (CONH2) (CO)2 2041 [Fe(n-CSH5){(C6H5)2PC2H4-
P(C6H5)2}2N2IPF6]2 2135 Fe(1l-C5H5)CSSCH3(CO)2 2101 [Fe(n-C5H5) (CO){(C6H5)2PC2H4-
P(C6HS)2}] [PF6] 2114 [Fe(n-C5H5)NO]2 2093 [Fe(r]-C5H5) (CO)2CS].[PF6] 2101 Fe(r)-C5H5) (CO)2(SO2CH3) 2136 Fe(i]-C5H4COCH3)2 1957 Fe(11-C5H4I)I(CO)2 2064 [Fe(r]-CeH6)(r]-C5H5)][PF6] 1960 Fe(r)4-C7H8) (CO)3 2003 Fe(r)5-C7H9) (CO)2I 2006 [Fe(i]5-C7H9) (CO)3] [BF4] 2006 Fe(«-C3F7) (n-C5Br4I) (CO)2 2031 Fe(«-C3F7) (T]-C5C14I) (CO)2 2031 Fe(«-C3F7) (ii-C5H4I) (CO)2 2031 Fe(«-C3F7) (CO)4I 2031 [Fe{CNC(CH3)2CH2C(O)CH3}6] [BF4] 2108
Fe2(CO)9 1940 Fe3(CO)12 1941 Fe(CO)4Cl2 2065 Fe(CO)4I2 2065 [Fe(CO)4]Na2 1942 [Fe2(CO)s] Na2 1943 Fe(CO)2(NO)2 2093 Fe(CO)2(n2-CS2) [P(C6H5)3]2 2102 [Fe(OH)4]Na2 1877 Fe(PF3)5 2114
H
H6(As2Mo,8O62)-25H2O 1905
H5(BWi2O40)-nH2O 1910
H8(CeMoi2O42)-nH2O 1900 H3(CrMosO24H6)-nH2O 1901 H5(IMo6O24)-nH2O 1903 H6 (MnMo9O32) • nH2O 1906 H3(PMoi2O40)-nH2O 1898 H6(PMo9O40V3)-nH2O 1907 H4(PMO11O40V)-nH2O 1907 H5(PMo10O40V2)-nH2O 1907 H6(P2Mo18O62)-llH2O 1905 H3(PW1204o)-nH20 1910 H4(SiMoi2O40) -nH2O 1899 H4(SiWi204o)-7H2O 1909 H6(Wi2O40H2)-nH2O 1896 Hf(n-C5H5)2[C4(C6H6)4] 2023 Hf(T]5-C9H7)2(CH3)2 2023 Hg-Na 2170 Hg-Pb 2173 Hg-Sn 2173 Hg-Sr 2172 Hg-Zn 2173
I
1г(п-С5Н5)(СО)2 1993 1гС1(СО)з 2071 IrCl(CO) (PR3)2 2118 IrCl(CO) (r)-O2) [P(C6H5)3]2 2139 Г1гС12{т1-С5(СН3)5}72 2073 IrCl2(NO) [P(C6H5)3]2 2098
К
K2[A12O(OH)6] 1883
K3(AsMoi2O40)-nH2O 1901
К2Сг30ю 1891
K2Cr40i3 1891
K2 (Mn2V10O28) 12H2O 1896
K7(MnVi3O38) • I8H2O 1908
К3(Мо2(ОН)3(СО)в] 2129
K-Na 2174
K6Nb6O18-nH2O 1893
K8Nb60i9 nH2O 1892
K[PtCl3(T]-C2H4)l-H2O 2017
K4(SiWi2O40)-nH2O 1909
KsTaeOi9 • 16H2O 1893
KV3O8 1892
K3[W2(CO)6(OH)3] 2129
K2Zn2Vi0O28-16H2O 1897
L
LiC6Hs 2035 [{Li(C6H5)3NiO(C2H5)2}2N2]2 2139
M
Mg(C5H5)2 1926
Mg(r)1-C3H5)C1 1995
Формульный указатель 2179'
Mg(C4H80)4[Co{P(CH3)3}2N2]2 2138 [Mg(C5H5N)4]![Co(CO)4]2 1949 [Mg(OH)4]Na2 1874, 1878 Mg(OH)6Ba2 1875
Мп(т)-С3Н5) (C0)4 1998 MnfTf-CaHsHCOh 1997 Mn(C5H5)2 1955 Mn(r)-C5H5) (C0)3 1978 Мп(г)-С5Н5)1[С(СбН5)2] (CO)2 2039 Mn(r)-C5Br5) (CO)3 1980 Mn(n-C5H5) (CO)3 1978
Мп(т]-С5Н5) (CO) (CN) (NO) 2107 [Mn(n-C5H5) (CO)2(CN)]Na 2108 [Mn(n-C5H5) (CO)2NO] |BF4] 2092 Mn(T]-C4H4N)(CO)3 1981 Mn(n-C5H5) (r|6-C7H8) 1998 Mn(r)-C5H5) (t]2-C8H14) (CO)2 1999 Мп(я-С5Н5) (CO)2(CS) 2100 [Mn(NCCH3)3(CO)3][PF6] 1939 [Mn (NO3)3]2[ (C6H5)2PCH2CH2-
P(C6H5)2] 2092
[Mn(CO)6] [A1C14] 1938
Mn2(CO)w 1937 Mn(CO)5Br 2060 Mn(CO)s(CF3) 2029 Mn(CO)s(COCF3) 2029 Mn(CO)5Cl 2059 Mn(CO)5H 2081 Mn(CO)5I 2060 Mn3(CO)12H3 2082 Mn(CO)4NO 2091 [Mn(OH)4] Na2 1876 (Mo(T]-C5H5) (CO)2]2 1977 (MoCn-CsHs) (CO)3]2 1976 Mo.(n-C5H5) (CH3) (CO)3 2025 Mo(r)-C5H5)C1(CO)3 2057 Мо(п-С5Н5)С13(СО)2 2058 Mo(ti-C5H5)H(CO)3 2078 Мо(т]-С5Н5)Н2 2080
Mo(r)-C5H5) (CO)2(N2CH3) 2132 Mofn-CsHs) (CO)2NO 2090 Мо(т)-СбН5СНз)(СО)з 1973 [Mo(n-C7H7) (CO)3] [BF4] 1978 Mofn-C7H7)I (CO) 2 2059 Mo(i]6-C7H8) (СО)з 1997 Mo(r]4-C7H8) (CO)4 1996 Mo(r|5-C9H7) (СНз) (CO)3 2026 Мо[С3(СбНз)2] (CO)5 2036
Mo(CO)e 1935 [Mo2(CO) io] Na2 1934 Mo(CO)5[CNSi(CH3)3] 2104 Mo(CO)3(NCCH3)3 2128 Mo(CO)4(PH2C6H5)2 2113 (MoeO24AlH6)Na3-11H2O 1903 (Мо8О24СоНб)Ка3-8Н2О 1904 (MoeO24CrH6) -8H2O 1902 (Мо8О24СгН6)Нз'пН2О 1901
21*
(Mo8O24CrH6) (ЫН4)3-/гН2О 1901 (Mo6O24FeH6)(NH4)3-nH2O 1903 (MosO24I)H5-nH2O 1903 (Mo6O24I)Na5-nH2O 1903 (Mo6O24NiH6)(NH4)4-5H2O 1902 (Mo6O24RhH6)(NH4)3-7H2O 1902 Mo8O26(NH4)4-nH2O 1894 (Mo8O27) n (NH4) Qn • 4nH2O 1894 (Mo9O32Mn)He-nH2O 1906 (МозО32Мп) (NH4)6-6H2O 1905 (Mo9O32Ni)(NH4)6-6,5H2O 1906 (Моз04оРУ3)Нб-пН20 1907 (Mo10O30Co2) (NH4) 6 10H2O 1905 (Moio04qPV2) H5 • mH2O 1907 (Moi1O40PV)H4-nH2O 1907 (Moi2O40As)K3-nH2O 1901 (Mo12O42Ce)H8-nH2O 1900 (Mo,2O42Ce) (NH4)6H2- 10H2O 1901 (Moi2O42Ce) (NH4)8-8H2O 1900 (Mo12O40P)2Ba3-nH2O 1899 (Mo12O49P)H3-nH2O 1898 (Mo,2O40P)(NH4)3-nH2O 1899 (Moi2O40Si)H4-nH2O 1899 (Moi8O62As2) H6 • 25H2O 1905 (Mloi8O82As2) Na8 23H2O 1905 (Mo18O62P2)H6-llH2O 1905 (Mo18O62P2)(NH4)6-14H2O 1904
N
(NH4)6(As2W18O62) • 14H2O 1912 (NH4)8(CeMo12O42)-8H2O 1900 (NH4)e(CeMoi2O42)H2-10H2O 190C (NH4)e(Co2Mo10O30) 10H2O 1906 (NH4)3(CrMo6O24H6)-nH2O 1901 (NH4)3(FeMo6O24H6)-7H2O 1903 (NH4)6(MnMo9O32)-6H2O 1905 (NH4)4Mo3026-nH20 1894 (NH4)6„(Mo8O27)„-4nH2O 1894 (NH4)4(NiMo6O24H6)-5H2O 1902 (NH4)6(N1Mo9O32)-6,5H2O 1906 (NH4)3(PMoi2O40) -nH2O 1899 (NH4)6(P2Moi8O62)-14H2O 1904 (NH4)7(PV,2O36)-nH2O 1908 (NH4)s(P2W18O82) • 14H2O 1912 (NH4)3(RhMo5O24H6)-7H2O 1902 [N{Si(CH3)3)2]Na 2106
Na 1923
Na3(AlMo6O24H6) • 11H2O 1903 Na4(Al(OH)7] -3H2O 1882 Na4'[Al6O4(OH) iel 1883 Na4[Al4O3(OH)i0] 1883 Na6(As2Moi8O62)-23H2O 1905 Na5(BWi2O40)-nH2O 1911 Na2Be(OH)4 1873 NaC5H5 1924
2180 Формульный указатель
[Na(C6H14O3] [V(CO)6] 1929
Na2[Co(OH)4] 1877
Na3(CoMo6O24H6)-8H2O 1904
Na2[Cu(OH)4] 1878, 1879
Na2[Cr(CO)5] 1932
Na2[Cr2(CO)10] 1934
Na[Cr(CN)(CO)B] 2107
Na3(CrMo6O24H6)-8H2O 1902
•’ Cr(OH)6] 1884
Fe(CO)4] 1942
Fe2(CO)8] 1943
Fe(OH)4’-----
Fe(OH)7‘
Fe(OH)8
Fe(OH)3’
Na3 Na2 N a2 Na2 Na4 NaB NaB Na5(IMoeO24)'nH2O 1903
1877
2H2O 1885
1886
•5H2O 1886
Na2[Mg(OH)4] 1874, 1878 Na[Mn(CN) (iq-CBHB) (CO)2] 2108
Na3[Mn(OH)4] 1876, 1878
Na12[Mn(Nb6O19)2]-50H2O 1896
Na^[Mo2(CO)i0] 1934
Na8Nb6Oi9-nH2O 1892
Na2[Ni(OH)4] 1878
Na3(PW12O40)-nH2O 1910
Na2[Pb(OH)6] 1888
Na[Sn(OH)3] 1875
Na2[Sn(OH)6] 1887
NasTaeOig-nFUO 1893
Na4V2O7-15H2O 1892
Na2[W2(CO)I9] 1934
Na[W(CN)(CO)6] 2106
Na6(Wi2O40H2) -21H2O 1895
Nai0(Wi2O42H2) -27H2O 1894
Na[Zn(OH)3] 1880
Na2[Zn(OH)4] 1878, 1881
Nb(n-C5H5)2 2054
Nb(n-C5H5)(CO)4 1969, 1971, 2054
Nb3(r)-C5H6)3(CO)7 1971
Nb(Ti-C5H5)2Cl2 2051, 2054
Nb(Ti-C5H5)Cl4 2055
Nb(r)-C5H5)2H3 2077
NbgOjsKe * wH2O 1893 Nb6O19K8-nH2O 1892 Nb6Oi9Na8-nH2O 1892 (Nb6O|9Mn)Na12-50H2O 1896
Ni(r)-C3H5)2 2012 [Ni(r)-C3H5)Br]2 2011 fNi(n-C3H5)CH3]2 2012 1№{п3-С3Н3(СНз)2}(СН3)]2 2013
Ni(n-C3H5)(CH3)[P(CeH5)3] 2012
Ni(r)-C5H5)2 1962
[Ni2(n-CSH5)3][BF41 1963 [Ni(Ti-C5H5)CO]2 1994
Ni(n-C5HS)NO 2098
Ni(n4-C8H12)2 2009
Ni(n6-Ci2HI8) 2014
JNi(CN)4]K4 2111
[Ni2(CN)e]K4 2111 [Ni(OHj4]Na2 1878 [Ni(OH)6]Sr2 1879
Ni(PF3)4 2119 Ni[P(OCH3)3]4 2120 Ni[P(OC6H5)3]4 2121
О
Os(n-C5HS)(CO)2(CONH2) 2041
Os3(CO),2 1945
Os(CO)3Cl2 2068
Os(CO)4Cl2 2068
Os (CO) Cl (H) [P(C6H5)3]3 2084
OsH4[P(C6HB)3]3 2084
P
PF3 2075
(P2W13O62)(NH4)6 1912
Pd(Ti4-C3Hi2)2 2016
PdCl2Cn4-C8H12) 2015
[PdCl2{C3(C6H5)2}]2 2043
[PdCl2{P(H-C4H9)3}2] 2122
fPd(р-Cl){P (C6H5)3j2]2[BF4]2 2121
PdCl2(NCC6H5)2 2140
PdCl2[P(C6H5)3]2 2122
PdCl2[P(OC6H5)3]2 2126
fPd(CNHCH2CH2O)4]I2 2045
Pd[P(C„H5)3]4 2121 [Pt(CH3)3I]4 2032 Pt(t]-C2H4) [P(C6H5)3]2 2018 Pt(r]4-C8H12)2 2020 Pt(C6H5C^CC6H5) [P(C6H5)3]2 2019 Pt(CNO)2[P(C6H5)3]2 2113 Pt(ri2-CS2) (P(C6H5)3]2 2103 rPt(M.-Cl){P(C6H5)3}2[BF4]2 2123 Pt(Cl) (CH = NC6H4-n-CH3)-
ГР(С2Н5)3] 2047, 2048 PtCl(H)[P(C6H6)3]2 2047, 2087 PtCl2(r)4-C8H12) 2019 PtCl2(CNC6H5)[P(C2H5)3] 2046 PtCl2(CO)2 2074
PtCl2rC(OC2H8)NHC6H5] [P(C2HB)31 2046, 2047
PtCl2[P(C2HB)3]2 2046 PtCl2[P(C6HB)3]2 2124 [PtCl2{P(OCeHB)3)]2 2125 [PtCl3(r]-C2H4)]K H2O 2017 Pt(O2)[P(C6Hs)3]22140 Pt[P(C6HB)3]3 2125
Pt[P£C6HB)3]4 2124
r Pt{ (C6H4O) P (OC6H5) 2) (Cl) {P-(OC6H5)3}] 2126
Формульный указатель 2181
R
ReBr(CO)6 2061
Re2Br2(CO)6(C4H3O)2 2133
Re(T]-C5H5)2H 2083
Re(r)-C5H6) (СО)з 1981
Re(r)-C5H4Br) (СО)з 1982
Re(r)-C5H4C1) (СО)з 1982
Re(n-C5H4I) (СО)з 1983
Re2(CO)10 1939
[Re(CO)6] [A1CU] 1939
Re(CO)5Cl 2061
[Re(CO)4Cl]2 2062 [Re(CO)3(OH)]4 2134 ReI(CO)5 2062
Rh(n-C5H5)(CO)2 1989
Rh2(r)-C5H5)2(CO)3 1989
[Rh(n-C5H5)(CO)]2(|x-CH2) 2042
Rh2(n-C5H5)2(CO)2(lx-SO2) 1993 [Rh3(n-C5H5)3(li3-CH) (h-CO)2] [BF4] 2043
Rh[r)-C5(CH3)5] (CO)2 1990
Rh2[r)-C5(CH3)5]2(lx-CO) (CO)2 1990,
1991
Rh2[n-C6(CH3)5]2(n-CO)2 1990, 1992 [Rh{n-C5(CH3)5}C12]2 2071
Rh(q4-C8H12)C1]2 2008
Rh(r)2-C8H14)2C1]2 2008
Rh(CO)i2 1950
Rh6(CO)16 1951 [Rh(CO)2Cl]2 2070 Rh(CO)H[P(C6H5)3]3 2086
RhCl [P(CeH5)3]3 2117
RhNO[P(C6H5)3]3 2097
Ru(n-C6H5)2 1961
(Ru(n-C5H5)(CO)2]2 1986
[Ru(i]-CsHs)(CO)3][PFe] 1987
Ru(n-C5H5) (CO)2C1 2068
Ru(r)-C5H5) (CONH2) (CO)2 2041
Ru3(CO)12 1944
Ru(CO) (Н)С1[Р(С6Н6)з]з 2083
Ru(CO)2C12[P(C6H5)3]2 2115
[Ru(CO)3C12]2 2067
Ru(CO)3[P(C6H5)3]2 2115
Ru(CO)4I2 2067
Ru(NO)2[P(C6H5)3]2 2094
s
Sn(n1-CsH5)(H-C4He)3 1927
[Sn(OH)3]Na 1875 [Sn2O(OH)4]Ba 1876
Sr[Be(OH)4] 1873 8гз[Сг(ОН)6]2 1885 Sr2[Ni(OH)6] 1879
T
Ta(CH3)5 2024
Та(т)-С6Н5)С14 2055
Та6О19Кз- 16H2O 1893
Ta60i9Na8 • мН2О 1893
Ti(r)-C5H5)2C12 2050
Т1(т)-С5Н5)С1з 2051
Ti(n-C5H5)2[C4(C6H5)4] 2023
Т1(т]-С5Н5)2(СНз)2 2022
Ti(r)-C5H5)(CO)2 1964
Tl[Co(CO)4] 1948
V
V(r|-C5H5)2 1952
V(t)-C5H5)2C12 2050
VCq-CsHs) (П-С7Н7) 1952
V(r)-C5H5) (CO)4 1965
V2(r)-C5H5)2(CO)5 1967
УСп-С5Н4СОСНз) (CO)4 1966
У(п-С7Н7)(СО)з 1968
V(CO)6 1928
ГУ(СО)б] [Na(C6H14O3)2l 1929
У(СО)4{т1-СвНз(СНз)з}[РР6] 1969
VH(PF3)6 2075
V2O7Na4-15H2O 1892
V3O8K 1892
Vi0O28Mn2K2- 12H2O 1896
Vio02sZn2K2* 16H2O 1897 (Vi2O36P)Ag7-nH2O 1908 (У120збР) (NH4)7-nH2O 1908 (У1зОз8Мп)К7- 18H2O 1908
W
(W18As2O62)(NH4)6-14H2O 1912 W(CH3)6 2027 W(r)-C5H5)2H2 2080 W(r)-C5H5) (CH3) (СО)з 2025 W(r)-C5H5) (CO)3C1 2057 W(r)-C5H5) (СО)зН 2078 W(r)-C5H5) (СО)2(М2СН3) 2132 W(r)5-C9H7) (CH3) (СО)з 2026 [W(CN)(CO)5]N(C2H6)4 2106 (W(CN) (CO)5] Na 2106 W(CO)6 1936 [W2(CO)10]Na2 1934 W(CO)5[C(C6H5)2] 2037 W(CO)5C(OCH3)R 2034 [W(CO)sC1] [N(C2H5)4] 2056 W(CO)5[CNSi(CH3)3] 2104 [W2(CO)6(OH)3]K3 2129 W(CO)5SH2 2132 W(CO)4(CC6H5)Br 2039 W(CO)3(NCCH3)3 2128
2182 Формульный указатель
(W12O40B)H5-nH2O 1910 (W12O40B)Na5-nH2O 1911 (Wi2O40H2)H6-nH2O 1896 (W12O40H2)Na6-21H2O 1895 (WI2O42H2)Na10-27H2O 1894 (W12O40P)H3-nH2O 1910 (Wi8O62P2) (NH4)6-14H2O 1912 (Wi2O40P)Na3-/iH2O 1910 (Wi2O40Si)H4-7H2O 1909 (W12O40Si)K4-nH2O 1909 z [Zn(OH)3]Na 1880 [Zn(OH)4]Na2 1878, 1881 Zr(n-C5H5).[C4(C6H5)4] 2023 Zr(n5-C9H7)2(CH3)2 2023
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ (к 6-ти томам)
Т Г с 1—320; т. 2: с. 321—664; т. 3: с. 665—1056; т. 4: с. 1057—1504; т. 5: с. 1505—1864; т. 6: 1865—2176.
Азот
Азидоводород 495
Азотистоводородная кислота см. Азидоводород
Азотистый ангидрид см. Триоксид диазота
Азотная кислота 518
Азотноватистая кислота 519
Азотный ангидрид см. Пентоксид диазота
Аммиак NH3 484—486
Аммиака водный раствор 485
Ангидрид азотной кислоты см. Пентоксид диазота
Анджелли соль см. Триоксодинит-рат(П) динатрия
Бромофосфазены см. Фосфонитрил-бромид
Бромоциан 691
Боразол (боразин) 863
Гексаметилборазин 1975
Гексахлороантимонат(У) нитрозила
634
Гидразин 491
Гидразина сульфат 494
Гидроксиламин 503
— солянокислый см. Хлорид гидрок-силаммония
Гидроксиламмония оксалат 507
— хлорид 504
Гипонитрит натрия 520
Дейтероаммиак 173
Диазоциклопентадиен 2064
Диазоимидобнссульфат калия 540
Диоксид азота 511
Диоксид-динитрат хрома(VI) 1626
Дифторамнн 222
Дифтордиазин (дифторимин, дифто-
рид диазота) 223
Дициан 688
Закись азота см. Оксид диазота
Имидобиссульфат калия 528
— триаммония 529
Иминфторнд см. Дифторамин
Иодоциан 693
Моноамидофосфорная кислота 599
Нитрамид см. Нитроила амид
Нитрат хромила 1626
Нитридотриссульфат калия 530
Нитрозила борофторид см. Нитрозила тетрафтороборат
— бромид 516
— гексахлороантимонат(V) 634
— гидросульфат 440
— тетрафторид-хлорид 190
— тетрафтороборат 262
— фторид см. Оксофторид азота аддукт с фтороводородом 225 — хлорид 514
Нитрозобиссульфат калия 543
N-нитрозогидрокснламино-М-сульфат калия 545 <
Нитроила амид 523
— фторид см. Фторид-диоксид
— хлорид 517
Нитрокснлфторид см. Фторид-триок-сид
Оксиды 508, 510, 513
Оксогипонитрит натрия см. Триоксодинитрат (II) динатрия
Оксофторид 224
Пентоксид диазота 513
Родановодород 695
Селенид 465
Синильная кислота см. Циановодород
Сульфамид 530
Сульфоперамидная кислота 541
Тетрасульфимида соли 532
Тетратиотетранитрндимидоксида соли 526
Тетрафторид диазота (тетрафторогидразин) 222
Тиазилфторид см. Тиофторид азота Тиотритиазила хлорид 445
2184 Предметный указатель
Тиофторид 228
Тиоциановодород см. Родановодород
Триоксид диазота 512
Триоксодинитрат(П) динатрия 522
Трисерадиазотдиоксид 446
Трисерадиазотпентоксид 447
Трисульфимида соли 531
2,4,6-Трифторо-1,3,5-триазин см. Цн-анурфторид
Трихлорид 501
В-Трихлороборазии 863
В-Трихлор-М-триметилборазин 1975
Фосфонитрилбромид 598
Фосфонитрилфториды 237
Фосфонитрилхлорид 596
Фосфортиотриамид 606
Фторид 220
Фторид-диоксид 226
Фторид-триоксид 227
Хлоркарбонилизоцианат 698
Хлороазид 502
Хлороамии 498
Хлорофосфазены см. Фосфонитрилхлорид
Циановая кислота 694
Циановодород 688
Цианурфторид 252
Элементный 481
Актиний
Бромид актиния(III) 1217
Металлический 1207—1214
Оксид актиния(Ш) 1217
Сульфид актииия(Ш) 1218
Фторид актиния (III) 1214
Фторид-оксид актииия(Ш) 1215
Хлорид актиния(Ш) 1216
Хлорид-оксид актиния(Ш) 1216
Алюминий
Аланат лития см. Алюмогидрид лития
Алюминат кобальта 1772
Алюмогидрид лития 888
Амальгама алюминия 2174
Арсенид 909
Ацетат 912
Ацетилацетонат 912
Байерит 900
Бёмит 901
Бориды 855
Бромид 894
— диэтилалюминия 891
— комплекс с сероводородом 899
Гексагидроалюминат лития 889
Гексафтороалюмииат аммония 264
Гептагидроксоалюминат тетранатрия 1882
Гидраргиллит 900
Гидрид, аддукт с триметиламином 891
— полимер 890
Гидроксид 900
Диметиламид алюминия 906
Динитридоалюминат лития 905
Дифосфидоалюминат лития 907
Диэтилалюминийгидрид 893
Иодид 895
— гексааммиакат 899
Карбид 909
Криолит аммония см. Гексафтороалюминат аммония
Метилат 910
6-Молибдоалюминат аммония 1903
— натрия 1903
Нитрид 904
Оксид 901
Оксогидроксоалюминат калия 1883
— натрия 1882, 1883
Селенид 902
Сульфид 901
Теллурид 903
Тетрагидроалюмииат лития см. Алю-могидрнд лития
Тетратиофосфат 908
Тетрафтороалюминат аммония 264
Тетрахлороалюминат гексакарбоиил-марганца(1) 1938
— натрия 896
Тетрахлороалюминиевой кислоты ди-
эфират 897
эфират 897
Тетрацианоалюминат лития 910
Триэтаноламина А1-алкоголят 911
Триэтилалюминий 892
Фосфат 908
Фосфид 907
Фторид 263
Хлорид 893
— аммиакат 898
— гексагидрат 896
— комплекс с РС13 898
— соединения с графитом 680
Этилат 911
Барий
Азид 1003
Амальгама 2173
Бромат 361
Галогениды 995
Гексагидроксоферрат(Ш) 1887
Гексагидроксокадмиат 1881
Гексагидроксокупрат(П) 1880
Предметный указатель 2185
Гексагидроксомагнезиат 1875
Гексаоксоперренат(УП) 1739
Гептагидроксоферрат(Ш) 1887
Гидрид 994
Гипофосфит 583
Дигидрогипофосфат 586
Дитионат 433
Дитиофосфат 593
Иодид 996
Кислый фосфорноватистокислый барий см. Дигидрогипофосфат
Манганат 1687
Металлический 993 12-Молибдофосфат 1899 Нитрид 1002
Оксид 998
Оксогидроксостаинат(П) 1876
Октагидроксоплюмбат(1У) 1890
Ортоиодат 371
Пентаоксореиат(УП) 1738
Перренат(УП) 1737
Селенид 1001
Силициды 1008
Смешанные кристаллы BaSO< и КМпО4 1688
Сульфид 1000
Тетранитрид трибария 1002
Тетрацианоплатинат(П) 1822
Фторид 269
Хлорат 360
Хроматы 1630, 1631
Бериллий
Азид 968
Ацетаты 970, 971
Борат лития-бериллия 879
Бромид 963
Гидрид 961
Гидроксид 965
Иодид 964
Карбиды 969
Нитраты 968
Оксобериллаты щелочных металлов 965
Селенид 967
Стекло Линдемана см. Борат лития-бериллия
Сульфид 965
Теллурид 967
Тетрагидроксобериллат натрия 1873 — стронция 1873
Тетрафторобериллат аммония 268
Фторид 268
Хлорид 961
Бор
Боразии 863
Боразол см. Боразин
Бораны 857, 859
Борат висмута (III) 648
— лития-бериллия 879
Бориды 2166. См. также соответствующий металл
Борогидрид лития 860
— натрия 861
Борофторид калия см. Тетрафтороборат калия
Борофторид натрия см. Тетрафтороборат натрия
12-Вольфрамоборат натрия 1911
Гексаметилборазин 1975
Гидроксотрифтороборат калия 262
Дииодотритиоборалаи 880
Диоксоборат натрия 877
Дихлорид этилбора 883
Додекаборододекагидрид триметил-аммоиия 862
Кислота борная, метиловый эфир 880
— борофтороводородная см. фторо-борная
— н-бутилборная 885
— 12-вольфрамоборная 1910
— дигидроксодифтороборная 869
— диоксоборная 876
— метаборная см. диоксоборная
— фтороборная 261
Метаборат натрия см. Диоксоборат натрия
Нитрид 873
Нитрозилборофторид см. Тетрафтороборат нитрозила
Пентабораты щелочных металлов 878
Пероксоборат * натрия, гексагидрат 878
Свободный бор 854
Стекло Линдемана см. Борат лития-бериллия
Тетраборат натрия, декагидрат 877
Тетраборогидрид тория(IV) 1253
Тетра-н-бутиламмония триборооктагидрид 861
Тетрагидрид бора-натрия см. Борогидрид натрия
Тетрагидроксоборат натрия 875
Тетракис (диметиламино) диборан (4) 881
Тетрафенилборат натрия 886
Тетрафтороборат (т] -ацетилциклопентадиенил) (г) -циклопентадиеиил) -железа(Ш) 1958
— [|х-дикар боиил - цз -метилидин -цик-ло-трис (т) -циклопентадиеиил) родия} 2043 )
1286 Предметный указатель
— ди-р-хлоротетракис (трифенилфосфин) диплатины (II) 2123
— ди-р-хлоротетракис (трифенилфосфин) палладия (II) 2123
— калия 262
— натрия 261
— нитрозила 262
— серебра 276
— трикарбонил (т]-циклогептатриенил) молибдена (0) 1978
— трис (т]-циклопентадиеиил) диникеля (II) 1963
— (т] -циклопентадиеиил) (нитрозил)-дикарбонилмарганца (II) 2092
Тетрахлорид диборана 865
Трибромид 867
Трииодид 868
Трикарбонил (гексаметилборазин) -
хром 1975
Триметиламиноборан 862
Триметилбор 882
Триметилбороксин 884
Триоксид дибора 871
Триоксоборат лития 874
Трис (диметиламино) боран 881
Трисульфид дибора 872
Трифенилбор 886
Трифторид 259
— аммиакат 870
— дигидрат 869
— эфир ат 870
Трихлорид 864
В-Трихлороборазин 863
В-Трихлоро-М-триметилборазин 1975
Триэтилбор 883
Фенилбор, дииодид 887
— дихлорид 887
Фосфат 879
Фосфид 874
Бром
Бромид нитрозила 516
Бромилнитрат 380
Бромная кислота 366
Бромноватая кислота 361
Бромоводород 332
Дибромомоноксид 354
трет-Бутилгипобромит 357
Дибромодисульфан 422
Диоксид 353
Гидрат 328
Гипобромит калия 357
— натрия 356
Нитрат брома(I) 375
— брома(Ш) 376
Оксиды 353
Пербромат аммония 369
— калия 368
Фосфонитрилбромид 598
Фторид брома(III) 190
— брома (V) 192
Фторосульфат брома (I) 374
— брома(III) 376
Элементный 327
Ванадий
(т] -Ацетилциклопентадиенил) тетра-карбонилванадий 1966
Бис (г] -циклопент а диенил) в ан адий 1952
Бромиды 1515—1516
Ванадаты 1527, 1892, 1897
10-Ванадат дикалия-димарганца 1896 13-Ванадоманганат(1У) калия 1908 12-Ванадофосфат аммония 1908 — серебра 1908
Ванодоцен 1952
Гексакарбонилванадат(—I) [бис{ди-(2-метоксиэтиловый эфир}]натрия
Гексакарбонилванадий 1928 Гексатиоцианатованадат(Ш) калия
Г идридогексакис (трифторфосфин) -ванадий 2075
Гидроксид 1526
Ди-р-карбонилтрикарбонилбис-
[ (г]-циклопент а диенил) ванадий]
1967
Дисульфатованадиеваи (III) кислота 1534
Йодиды 1517
Карбиды 1540
Квасцы 1536
Металлический 1509
9-Молибдо-З-ванадофосфорная кислота 1907
10-Молибдо-2-ванадофосфорная кислота 1907
11 -Молибдо-1 -ванадофосфорная кислота 1907
Нитриды 1538
Оксид-дибромид 1521
Оксид-хлориды 1518—1520
Оксиды 1521—1525
Оксид-сульфаты 1537—1538
Селениды 1529
Сульфаты 1531, 1535
Сульфиды 1528, 1530
Теллуриды 1529
Тетракарбонил (т] -мезитилен) ванадия гексафторофосфат 1969
Тетратиованадат (V) аммония 1530
Предметный указатель 2187
Трикарбонил (ч -циклогептатриенил) -ванадий 1968
Фосфиды 1539
Фториды 287—288
Хлориды 1510—1514
(Ч-Циклогептатриенил) (ч -циклопентадиенил) ванадий 1952
(Ч-Циклопентадиенил) тетракарбо-иилванадий 1965
Висмут
Амальгама 2173
Борат 648
Гидрид 642
Нитрат висмута(Ш) 647
Оксид висмута(Ш) 646
Оксобромид висмута(III) 644
Оксоиодид висмута (III) 645
Оксонитрит 647
Оксохлорид 643
Тетраоксовисмутат (V) тринатрия 649
Трибромид 643
Трииодид 645
Триоксовисмутат(V) калия 650
— натрия 649
Трифенилвисмут 646
Трихлорид 642
Фосфат висмута(III) 648
Фториды 243, 244
Элементный 641
Водород
Азид см. Азидоводород
Азидоводород 495
Азотистоводородная кислота см. Азидоводород
Аммиак 484—486
Арсаиы 611, 614
Бораиы 857, 859
Бромоводород 332
Вода для измерения электропроводности 154
— наивысшей чистоты 152
Газообразный 147
Германы 780
Гидразин 491
Гидриды металлов см. соответствующий металл
Дейтероводород HD 163
Иодоводород 335
Пероксид 177
Полисиланы 721
Селеноводород 450
Сероводород 394
Силаны 715, 717, 718
Станнан 817
Стнбан 629
Сульфаны 396, 399, 402—404 Теллуроводород 467
Фосфины 556
Фтороводород 185
Хлороводород 331
Вольфрам
Бромиды 1661—1665
транс-Бромо (тетракарбонил) (фенил-карбин)вольфрам 2039
12-Вольфрамат натрия 1894 12-ВольфраМоборат натрия 1911 12-Вольфрамоборная кислота 1910 12-Вольфрамовая кислота 1896 Вольфрамовая синь 1668 18-Вольфрамодиарсенат(У) аммония
1912
18-Вольфрамодифосфат аммония 1912 12-Вольфрамокремневая кислота 1909 12-Вольфрамосиликат калия 1909 12-Вольфрамофосфат натрия 1910 12-Вольфрамофосфорная кислота
1910
Гексакарбонилвольфрам 1936 Гексаметилвольфрам 2027 Гексафенолят 1676
Г ексахлоротрипиридинвольфрам (III) 1679
Г идридо(ч-циклопентадиеиил)три-карбонилвольфрам 2078
Декакарбонилвольфрамат(—I) нат-
рия 1934
Дигидридобис(ч-циклопентадиеиил) -вольфрам 2080
Желтая вольфрамовая кислота 1668 Йодиды 1665—1666
Мета-12-вольфрамат натрия 1895 Метавольфрамовая кислота 1896 Металлический 1656
Моносульфан (пентакарбонил) вольфрам 2132
Оксид-бромиды 1672—1674 Оксид-иодиды 1674—1676 Оксид-хлориды 1669—1672 Оксиды 1666—1668 Октациановольфраматы калия 1679, 1680
Паравольфрамат натрия В 1894 Пентакарбонил [метокси (органил) -карбен] вольфрам 2034
Пентакарбонил(дифенилкарбен) -вольфрам 2037
Сульфид 1676
Трикарбонил (ч5-ииденил) метил-вольфрам 2026
граи-Трикарбонилтрис (ацетонитрил) -вольфрам 2128
2188 Предметный указатель
Трикарбонил (трциклопентадиенил) -метилвольфрам 2025
Фторид 295
Хлоро(пентакарбонил)вольфрамат (0) тетраэтиламмония 2056
Хлоро (т]-циклопентадиеиил) трикар-бонилвольфрам 2057
Хлориды 1657—1661
Циано (пентакарбонил) вольфра-мат(0) натрия 2106
— тетраэтиламмония 2106
(т] -Циклопентадиеиил) дикарбонил-(метандиазо) вольфрам 2132
Эннеахлородивольфрамат(Ш) трикалия 1677
Галлий
Антимонид 933
Арсенид 933
Бромиды 923
Галлогидрид (галланат) лития 920
Гексафторогаллат аммония 265
Гидроксиды 925
Диметилгаллий, фторид 919
— хлорид 917
Дихлорогаллан 919
Йодиды 924, 925
Квасцы галлий-аммонийные см. Сульфат галлия (Ш)-аммония
Металлический 913
Метилгаллий, дихлорид 918
Нитрат галлия (!!!) 932
Нитрид 932
Оксиды 926, 927
Перхлорат галлия (III), гексагидрат
915
Селениды 931
Сульфат галлия (!!!)-аммония, додекагидрат 930
Сульфиды 927, 928, 929
Теллуриды 931
Триметилгаллий 916
Триэтилгаллий 917
Фосфид 933
Фторид галлия (!!!) 265
Хлориды 920, 922
Гафиий
Бис(т]-инденил)диметилгафний 2023
1,1 -Бис (т] -циклопентадиеиил) -
2,3,4,5-тетрафеиилгафний 2023
Бромиды 1446, 1449, 1452
Гидрид 1425
Ди-л-циклопентадиенилгафния (IV) бромид 1491
— дихлорид 1490
— иодид 1491
Йодиды 1446, 1449, 1453
Карбид 1482
Комплексные соединения 1485
Металлический 1419
Нитрид 1474
Оксид гафнияЦУ) 1465
Отделение от циркония 1420
Хлориды 1446, 1451
Германий
Бромиды 788, 789
Гексабромодигерман 790 Гексафенилдигерман 810 Гексафторогерманат калия 257 Германат натрия 806 Германаты 806, 807 Германиды 989, 1007, 1046 Гермасиланы высшие 725 Германы 780 Гермилсилан 725
Гидриды см. Германы Дибромогерман 794 Дигермилсульфид 798 Дииодогерман 797 Диметилдибромогерман 813 Диметилтрихлорогерман 811 Йодиды 791, 792 Металлгермилы 784 Метилтрибромогерман 813 Метилтрихлорогерман 811 Монобромогерман 794 Моноиодогерман 796 Моноидодигерман 797 Монохлорогерман 793 Нитрид 803
Оксиды 799, 801
Регенерация 780
Селенид 803
Сульфиды 802
Тетраацетат 805
Тетраизоцианат 805
Тетраметилгерман 808 Тетрафенилгерман 809 Тетраэтоксигерман 804 Тиогерманаты натрия 806 Триметилбромогерман 812 Триметилхлорогерман 811 Трифенилгерман 815
Трифторогерманат(Н) цезия 807 Трихлорогерман 794
Трихлорогерманат(П) цезия 807
Фториды 255, 256
Хлориды 785, 786
Дейтерий
Аммиак тяжелый см. Дейтероаммиак Бромид 167
Предметный указатель 2189
Дейтерид урана 1287
Дейтероаммиак 173
Дейтероводород 163
Дейтеросерная кислота 172
Дейтерофосфорная кислота 174
Иодид 169
Сульфат см. Дейтеросерная кислота
Сульфид 171
Серная кислота тяжелая см. Дейтеросерная кислота
Фосфат см. Дейтерофосфорная кислота
Фосфорная кислота тяжелая см. Дейтерофосфорная кислота
Фторид 164
Элементный 156, 158
Европий см. также Редкоземельные элементы
Амальгама 1162
Гидроксид европия(П), моногидрат 1186
Карбонат европия(II) 1187
Оксалат европия (II), моногидрат 1188
Оксиды 1182, 1183
Селенид EuSe 1190
Сульфат европия(П) 1187
Сульфид европия (II) 1190
Фосфат европия (II) 1204
Фторид европия (II) 281
Хлорид европия (II), дигидрат 1186
Железо
г]-Аллил (тетракарбонил) броможелезо 2004
т]-Аллил (трикарбонил) иодожелезо 2004
Т]-Аллил (трикарбонил) хлорожелезо 2003
Ацетат основной 1759
(т]-Ацетилциклопентадиенил) (г)-циклопентадиеиил) железа (III) тетрафтороборат 1958
Бис(т]-аллил) (трикарбонил)железо 2005
Бис (т) -бутадиен) карбонилжелезо 2002
Бис [дикарбонил (т] -циклопентадиенил) железо] 1983
Бис(ц-нитрозил) (т]-циклопентадие-нил) железо 2093
Бис (трифенилфосфин) (дикарбо-иил) (т]2-сероуглерод) железо 2102
Бис (г]-циклопентадиеиил) железо 1956
Бис (т] -циклопентадиеиил) железа (III) гексафторофосфат 1958
Бромиды 1746—1747,
Бромо (т] -циклопентадиеиил) дикарбо-нилжелезо 2063
т] - Бутадиен (трикарбонил) железо 2002 Гексаацетатодигидроксотрижеле-
за(Ш) моноацетат 1759
Гексагидроксоферрат(Ш) бария 1887
Г ексакис (4-изоциано-4-метилпента-нон-2)железа(Н) тетрафтороборат 2108
Гексатиоцианатоферрат(Ш) натрия 1761
Гексацианожелезные кислоты 1759, 1760
Гексацианоферрат (II) аммония 1760 Гептагидроксоферрат(Ш) бария 1887 — натрия 1885 — стронция 1887 Гидроксид 1750
ц-Диазот (N.N') -бис [т] -циклопента-диенил) {1,2-бис (дифенилфосфино) -этан} железа (II) гексафторофосфат 2135
1,1'-Диацитилферроцен 1957
Дибромо(тетракарбоиил)железо 2065 Дииодо (тетракарбонил) железо 2065 Дикарбонил (i^-иодциклопентадиенил)-(перфтор-н-пропил) железо 2031
Дикарбонил (т] -тетрабромиодцикло-пентадиенил) (перфтор-н-пропил)-железо 2032
Дикар бонил (т] -тетр ах лориодцикло -пентадиенил) (перфтор-н-пропил)-железо 2032
Дикарбонил (т]-циклопентадиеиил) -метилжелезо 2030
Динитрозилдикарбонилжелезо 2093 Дитибферрат(Ш) калия 1758
Дихлоро (тетракарбонил) железо 2065 Додекакар бонилтрижелезо 1941 Иодид 1748
Иодо (т] -иодциклопентадиенил) ди-карбонилжелезо 2064—2065
Иодо (т] -циклопента диенил) дикарбо-нилжелезо 2063—2064
Карбид 1755 (Метилсульфинато-S) (т]-циклопентадиенил) дикарбонилжелезо 2136
Нитриды 1753
Металлическое 1745 б-Молибдоферрат(Ш) аммония 1903; Оксид-гидроксид 1751 Оксид-хлорид 1752
Оксиды 1749, 1751
Октагидроксоферрат(Ш) натрия 1886-Октакарбонилдиферрат (—I) натрия
1943
2190 Предметный указатель
Лентакис (трифторофосфин) железо 2114
Пентацианоамминферраты натрия 1761—1762
Сульфат основной 1758
Сульфид 1753
— калия-железа (Ш) 1758
Тетракарбонил (перфторо-н-пропил) -иодожелезо 2031
Тетракарбонилферрат(—II) натрия 1942
Тетр акис [к а р бонил (т) -цик л опент а -диенил) железо] 1984
Тетраоксодиферрат цинка ИЗО
Трикарбонил (г]-циклобутадиен) железо 2001
Трикар бонил (т]5 -циклогептадиенил) -железа тетрафтороборат 2006
Трикарбонил (т]4-циклогептатриен) -железо 2003
Трикарбонил (т]-циклопентадиенил)-железа (III) гексафторофосфат 1985
Феррат калия 1756
— лития 1756
Ферроцен 1956
Формилферроцен 1958
Фосфиды 1754
Фториды 301—302
Хлорида железа (III) соединения с графитом 680
.Хлоро (г] -циклопентадиенил) дикарбо-нилжелезо 2063
Хлориды 1745
Цементит 1755
(т]-Циклопентадиенил) (ц-бензол) -железа(П) гексафторофосфат 1960
(т] -Циклопентадиенил) {1,2-бис (фе-нилфосфино) этан} карбонилжелеза гексафторофосфат 2114
(трЦиклопентадиенил) (дикарбонил) -(ацетонитрил) железа (II) гекса-
фторофосфат 2135
>(т]-Циклопентадиенил) (дикарбонил) -(дитиометоксикарбонил) железо 2101
(т]-Циклопента диенил) дикарбонил-(тиокарбонил) железа (II) гексафторофосфат 2101
'(г]-Циклопентадиенил) (дикарбоиил) -(фенилизоцианид)железа (II) гексафторофосфат 2109
(т]-Циклопентадиенил) (карбонил)-бис (фенилизоцианид) железа (II) гексафторофосфат 2110
Эниеакарбонилдижелезо 1940
Золото
Ацетиленид см. Карбид
Бромо (трифенилфосфин) золото 2127 Гидроксид диметилзолота(Ш) НИ ДицианоауратЦ) калия 1110
Из остатков 1103
Иодид золота (I) 1106
Карбид золота (I) 1109
Коллоидное 1102
Метил (трифенилфосфин) золото 2033
Оксид золота (III) 1107
Особочистое 1101
Сульфиды 1108,' 1109
Тетраазидоаурат(Ш) тетрафенилар-сония 2049
Тетракис (1 -изопропилтетразол-5-ато)аурат(Ш) тетрафениларсония 2049
Тетрафтороаурат(Ш) 277
Тетрахлорозолотоводородная кислота, тетрагидрат 1106
Триметил (трифенилфосфин) золото 2034
Трис (пентафторфенил) золото (III), комплекс с трнфенилфосфином 1111
Трис[1 -циклогексилимино (метокси) -метилзолото] 2048
Трифенилфосфинзолота тетракарбо-нилкобальтат 1113
ФенилэтинилзолотоЦ) 1112
Фторид золота (III) 276
Хлорид-оксид золота(III) 1108
Хлориды 1104, 1105
Хлоро(карбонил)золото 2075
Хлоро (трифенилфосфин) золото 2127
Цианид золота (I) 1110
Иидий
Антимонид 944
Арсенид 944
Бромиды 938—940
Гексафтороиндат аммония 266
Гидроксид индия (III) 941
Диметилиндий, хлорид 936
Диэтилиндий, хлорид 937
Йодиды 939, 940
Металлический 934
Нитрид 944
Оксид индия(Щ) 941
Селениды 943
Сульфиды 942 ’
Теллуриды 943
Триметилиндий 934
Триэтилиндий 936
Фосфид 944
Фторид индия (III) 266
Хлориды 937, 940
Предметный указатель 2191
Иод
Бромид иода(1) 340
Г ексагидроксоиод (VII) гидросульфат 381
Дииодознлсульфат 379
Иодат иода(Ш) 377
Йодноватая кислота 361
Иодоводород 335
Иодозилиодат(У) 380
Иодоциан 693
Метаиодат калия 371
— натрия 369
Нитрат иода(III) 377
Ортоиодат натрия 369
Ортоиодная кислота 369
Ортопериодат никеля-натрия 1791
Пентоксид иода 354
Перхлорат дипиридиниода(1) 375
— иода(Ш) 378
Сульфат иода(Ш) 378
Тетрахлороиодат (III) тетраэтиламмо-ния 345
Тетрахлороиодная(Ш) кислота 346
Фторида иода(Ш) аддукт с пиридином 195
Фториды 193, 194
Фторосульфат иода (III) 379
Хинолиниод(1) бензоат 375
Хлорид иода(1) 339
— иода (III) 341
Элементный 328
Иридий
Васка комплекс 2118
Гексахлороиридат(1У) водорода 1837
Гексахлороиридаты 1837—1838
Дикарбонил (т]-циклопентадиенил)-иридий 1993
Ди (ц-хлоро) бис [хлоро (т] -пентаметилциклопентадиенил) иридий] 2073
Дихлоронитрозилбис (трифенилфосфин) иридий 2098
Иридийхлористоводородная кислота 1837
Металлический 1835
Оксиды 1835—1836
Фториды 308—309
Хлор (т] -дикислород) бис (трифенилфосфин) карбонилиридий 2139
Хлоробис (триорганилфосфин) карбонилиридий 2118
Хлоробис (трифенилфосфит) карбонилиридий 2118
Хлоротрикарбонилиридий 2071
Хлорид 1836
Иттербий см. также Редкоземельные элементы
Амальгама 1162
Фторид иттербия (II) 281
Кадмий
Амальгама 2173
Амид 1137
Арсениды 1140
Ацетат 1142
Бромид 1134
Бромид-триарсенид дикадмия 1140
Гексагидроксокадмиат бария 1881
Гидроксид 1135
Гидроксохлорид 1133
Дихлорид-диоксид трикадмия 1133-
Диэтилкадмий 1141
Иодид 1134
Карбонат 1141
Металлический, в иглах 1131
Нитрид Cd3N2 1137
Ортосиликат 1143
Роданид см. Тиоцианат
Сульфид 1136
Тетрацианокадмат калия 1143
Тиоцианат 1143
Трихлорид-фосфид трикадмия 1139’
Фосфиды 1138
Фторид 278
Хлорид 1132
Цианид 1142
Калий см. также Щелочные металлы
Азид 497
Антимонид 1040
Арсенид 1039
Ацетилиды (карбиды) 1043
Бромид-гексафторид 271
Бромид-тетрафторид 270
Висмутид 1041
Гексасульфид 414
Гексафторобромат (V) 271
Германид 1046
Гидрид 1024
ДибромоиодатЦ) 344
Дикалийгексасульфан см. Гексасульфид
Дикалийдисульфан см. Дисульфид
Дикалийпентасульфан см. Пентасульфид
Дикалийтетр асульфан см. Тстрасуль-фид
Дикалийтрисульфан см. Трисульфид.
Диоксид (надперокснд) 1032
Дисульфид 410
Дихлороиодат (I) 343
Иодид 338
$192 Предметный указатель
Предметный указатель 2193 15
Калийгермил 784
Металлический 1009—1019
Оксид 1028 .Пентасульфид 414 Пероксид 1031 Плюмбид 1047 ’Силилкалий 723 Силицид 1046 ‘Станнид 1047 Селенид 452 Сульфид 408 Тетрасульфид 413 Тетрафторобромат(III) 270 'Тетрафтороиодат(Ш) 271 Т’етрафторохлорат(1П) 270 Тетрахлороиодат (III) 345 Трииодид 342 Трисульфид 411
Фосфид 1036
Фторид 270
Фторосульфонат 217
Хлорид-тетр а фторид 270
Кальций
Амальгама 2171
Галогениды 995
Германиды 1007
Гидрид 994
Гидроксид 999
Гидроксилапатит 572
Карбид 1004
Металлический 990
Нитрид 1002
Оксид 996
Октакальций-фосфат 573
Ортоборат см. Триоксоборат
Ортоборат см. Триок'соборат
Селенид 1001
Силициды 1006
Сульфид 1000
Тетраоксоплюмбат(1У) 844
Триоксоборат 875
— натрия-кальция 875
Фосфид 1003
Фторид 269
Цианамид 1006
Кислород
Озон 385
Фториды 198, 200
Элементный 382
Кобальт
Аквапентамминкобальта (III) оксалат
(т]-Аллил)трикарбонилкобальт 2007
Алюминат 1772
Амид 1773
Бнс[(ц-нитрозил) (трциклопентадие-нил) кобальт] 2096
Бис(тетракарбонилкобальтат) тетракис (пиридин) магния 1949
Бис (г] -циклопентадиеиил) кобальт 1961
Бис (т] -циклопентадиеиил) кобальта (III) гексафторофосфат 1962
Бромиды 1765—1766
Виолеокобальтхлорид 1783
Гексаамминкобальта(П) иодид 1779 — хлорид 1765
Гексаамминкобальта (II) иодид 1779 — иодид 1779 — хлорид 1778
Гексакарбонил (р,т]-дифенилацети-лен) дикобальт 2007
Гексацианид калия-кобальта (111) 1786
Гексацианокобальтовая кислота 1787
Гексацианокобальтат(Ш) калия 1786 — натрия 1786
Гидр идо (тр-диазот) трис (трифенилфосфин) кобальт 2137
Г идридотетракис (трифенилфосфит) -кобальт 2085
Гидриде (г] -циклопентадиеиил) бис-(триметилфосфин) кобальта (III) гексафторофосфат 2085
Гидроксиды 1768, 1769
Декааммин-ц-пероксокобальт (III)-кобальта(1У) сульфат 1785
Диакватетрамминкобальта(Ш) сульфат 1783
Дииодо (тз-циклопентадиеннл)карбо-нилкобальт 2069
Дикарбонил (т]-циклопентадиеиил) кобальт 1987
Дикобальткарбид 1777
Дикобальтнитрид 1774
Дихлоротетрамминкобальта(Ш) хлорид 1782
Додекакарбонилтетракобальт 1948
Изоксантокобальтхлорид 1782
Йодиды 1766—1767
Карбонатотетрамминкобальта (III)-сульфат 1782
Кобальтоцен 1961
Кобальтоцения гексафторофосфат 1962
Ксантокобальтхлорид 1781
Лутеокобальтхлорид 1778
Металлический 1763
10-Молибдодикобальтат(Ш) аммония 1906
б-Молибдокобальтат(Ш) натрия 1904
Нитраты 1775
Нитрид 1775
Нитритопентамминкобальта (III) хлорид 1782
Нитрозилтрикарбонилкобальт 2095
Нитропентамминкобальта (III) хлорид 1781
Оксиды 1768
Октакарбонилдикобальт 1946
Празеокобальтхлорид 1783
Розеокобальтоксалат 1780
Сульфат 1772
Сульфоксилат кобальта 430
Сульфиды 1770—1771
Тенарова синь 1772
Тетрагидроксокобальтат(П) натрия
1877
Тетракарбонилкобальтат (—I) таллия 1948
— трифенилфосфинзолота 1113
Т етр акис (тетр агидрофуран) м агний-бис [р-диазоттристриметилфосфин) -кобальт] 2138
Тетракис (триметилфосфин) кобальт 2116
Трис(этилендиамиикобальта) (III) бромид 1784
Фосфиды 1777
Фториды 303—304
Хлориды 1764
Рз-Хлорметилидин-^икло-трис (трикарбонилкобальт) 2041
Хлоропентаммиикобальта (III) хлорид 1780
Хлоротрис (триметилфосфин) кобальт 2117
Хлорпурпуреокобальтхлорид 1780 (t] -Циклопентадиеиил) бис (триметилфосфин) кобальт 2117
Кремний
Бис (триметилсилил) амид лития 776 — натрия 776, 2106
Бис (триметилсилил) сульфан 777 Бромид кремния(II) 735 Бромоиодосиланы 743 12-Вольфрамосиликат калия 1909 Гексабромодисилаи 733 Гексаиододисилан 739 Гексаметилдисилазаи 775 Гексаметилдисилан 770 Гексафтородисилан 726 Гермилсилан 725 Декабромоциклопентасилан 735 Дииодосилан 749 Диметилдихлоросилан 770
Диметилхлорометилхлоросилан 771 Дисилан 717 Дисилилметан 756 Дисилилсульфид 753 Дисилоксан 753
Дисилтиан см. Дисилилсульфид Кислота 12-вольфрамокремневая 1909 — гексафторокремневая 255 — дикремневая полимерная 759 — кремневая 758 ----- гель 758 ----золь 758
— кремнеплавиковая см. гексафторокремневая
— кремнефтороводородиая см. гексафторокремневая
— 12-молибдокремневая 1899 — фторокремневая см. гексафторокремневая
Монобромосилан 745 Моноиододисилан 750 Моноиодосилан 748 Моносилан 715 Оксид кремния (II) 761 Органосиланы 752 (Пентакарбонил) триметилсилилизо-цианидхром 2104
Пербромосиланы 737 Перхлоросиланы 728 Полисиланы 721 Свободный кремний 714 Силаны высшие 718 Силикаты лития 760 — натрия 759, 760 — цинка 1129
Силикофтороформ см. Трифторосилан
Силикохлороформ см. Трихлоросилан Силилкалий 723 Силилфосфин 755
Силициды 2166. См. также соответствующий металл
Сульфиды 762, 763
Тетраацетат 766
Тетрабромосилан 732 Тетрагалогеносиланы 740 Тетраизоцианат 767 Тетраизотиоцианат 768 Тетраиодосилан 738 Тетраметилсилан 769 Тетраметоксисилан 765 Тетраэтилмеркаптосилан 769 Т етраэтоксисилан Трибромосилан 746 Трииодосилан 750 Триметилсилилазид 773 Триметилсилилизоциаиат 774
о-
22—1361
2194 Предметный указатель
Триметилхлорометилсилан 773
Трисилан 718
Трисилиламин 754
Трис (триметилсилил) амин 777
Трифторосилан 254
Трихлоросилан 744
Фториды 253, 727
Фторобромосиланы 741
Фтороиодосиланы 741
Фторосиликат цинка ИЗО
Фторохлоробромоиодосилан 744
Фторохлородибромосилан 743
Фторохлоросиланы 740
Хлорид кремния (II) 731
Хлоробромосиланы 742
Хлороиодосиланы 742
Хлоросилоксаны 757
Лантан см. также Редкоземельные элементы
Получение методом ионного обмена 1160
Литий см. также Щелочные металлы Азид 496
Алюмогидрид(аланат) 888
Амид 487
Антимонид 1040
Арсениды 1037, 1038
Ацетилид (карбид) 1042
Бис(триметилсилил) амид 776
Борат лития-бериллия 879
Борогидрид 860
Гексагидроалюминат 889
Гексафеиилхромат(Ш) трнлития 1618
Гидрид 1023
Гидроксид 1033
р,г]2-Диазотникельлитиевый комплекс 2139
Динитридоалюминат 905
Дифосфидоалюминат(Ш) 907
Имид 491
Литийгермил 784
Металлический 1009—1010, 1014, 1017
Метасиликат 760
Ниобаты 1573
Нитрид 1035
Оксид 1025
Оксоплутонаты 1406, 1407
Пероксид 1030
Силицид 1046
Стекло Линдемана см. Борат лития-бериллия
Танталаты 1573
Тетрагидрид бора-лития см. Борогидрид
Тетрагидроалюминат см. Алюмогид-рид
Тетрацианоалюминат 910
Триоксоборат 874
Трис (2,2'-бипиридил) титанат (—I)
1489
Трис (2,2'-бипиридил) хромат (—I) лития— тетрагидрофуран (1/4) 1609
Уранаты 1343, 1344
Фениллитий 1045, 2035
Феррат(Ш) 1756
Фосфид 1037
Фтороуранаты 1345
Магний
Азид 984
Арсениды 985
Бис(тетракарбонилкобальтат)тетра-кис(пиридин)магния 1949
Бромид 976
Германид 989
Гидрид 973
Гидроксид 979
Дициклопентадиенилмагний 1926
Иодид 978
Карбиды 987
Карналлит аммонийный 976
Металлический 972
Нитрат 984
Нитрид 983
Оксид 978
Пероксид 979
Селенид 982
Силицид 988
Сульфид 981
Теллурид 982
Тетрагидроксомагнезиат натрия 1874
Тетракис (тетрагидрофуран) магний бис[р-диазоттристриметилфосфин)-кобальт] 2138
Фосфид 985
Фторид 269
Хлориды 974, 976
Марганец
Азацимантрен 1981
(т|1-Аллил)пентакарбонилмарганец 1997
(rj-Аллил) тетракарбонилмарганец 1998
Ацетат 1693
р-1,2-Бис (дифеиилфосфино) этанбис-(тринитрозилмарганец) 2092
Бис(циклопентадиенил)марганец
1955
Предметный указатель 2195
Бромо (пентакарбонил)марганец 2060
- 10-Ванадат дикалия-димарганца
1896
1 13-Ванадоманганат(1У) калия 1908
Гексакарбонилмарганца(1) тетрахло-роалюминат 1938
? Гексацианоманганаты калия 1696—
J 1697
: Гидридопентакарбонилмарганец 2081
; Гидроксид 1682
z Декакарбонилдимарганец 1937
1 Дикарбонил(т]2-циклооктеи) (т]-цик-
лопентадиенил)марганец 1999 Дикарбонил (т) -циклопентадиенил) -
: (дифенилкарбен)марганец 2039
Диоксалатодигидроксоманганат(1\7) калия 1695
Дихлоро (дипиридин) марганец 1979
Иодо (пентакарбоиил)марганец 2060
1 Квасцы цезий-марганцовые 1691
Манганаты 1686—1688
I Металлический 1681
9-Молибдоманганат(1У)аммония 1905 9-Молибдомарганцовая (IV) кислота 1906
12-Ниобоманганат(1У) натрия 1896 Нитроз ил (тетракарбоиил) марганец 2091
Оксиды 1682—1685
Пентакарбонил (трифтороацетил) -марганец 2029
Пентакарбонил (трифторметил) марганец 2029
Пербромцимантреи 1980
Перманганат, смешанные кристаллы с BaSO4 1688
. Перренат марганца 1736
1 Сульфаты 1691
Сульфид 1689
Тетрагидроксомаиганат(П) натрия 1876
Т рикарбонил (Г] -пентабромциклопентадиенил) марганец 1980
Трикарбонил (ij-пирролил) марганец 1981
Трикарбоиил (т]-циклопентадиенил) -марганец 1978
Триоксалатоманганат(Ш) калия 1694 Триоксид-фторид 297
Трис(ц-гидридо)тетракарбонилмар-ганец 2082
Фосфиды 1692
Фтороманганаты калия 297—298
Фториды 295—296
Хлорид калия-маргаица(Ш) 1689 Хлоро (пентакарбонил) марганец 2059 Циано (г)-циклопентадиенил) дикар-
22*
бонилмаиганат(1) натрия 2108
Циано(т]-циклопентадиенил) (нитро-зил) карбонилмарганец 2107
г]6 -Циклогептатриен (т] -циклопента -диенил)марганец 1998
(т] -Циклопентадиенил) дикарбонил-(тиокарбонил) марганец 2100
(т]-Циклопентадиенил) (нитрозил)-дикарбонилмарганца (II) тетрафторобор ат 2092
Цимантрен 1978
Медь
Азид 1076
Бис (ди-2-пиридилимидо) меди (II) хлорид 1079
Бис (этилендиамии) меди (II) дииодокупрат (I) 1081
Бромиды 1064, 1068
Гексагидроксокупрат(П) бария 1880
Гидрид меди(1) 1062
Диоксалатокупрат (II) калия, дигидрат 1082
Дихлоро (ц-1,4-бутадиеи) димедь 1082
Дихлоро (ди-2-пиридилимидо) медь 1079
Иодид меди(1) 1065
Карбид 1077
Коллоидная 1061
Купрат(Ш) калия 1070
Нитрид 1075
Оксиды 1069, 1070
Пентафторфенилмедь 1079
Селенид меди(1) 1074
Сульфат меди(1) 1075
Сульфиды 1072, 1073
Теллурид меди(1) 1074
Тетраамминмеди(П) дихлорокуп-рат(1), моногидрат 1080
Тетрагидроксокупрат(1) натрия 1879 Тетраиодомеркурат(П) меди(1) 1145 Тетр ах лороку пр ат (I I) тетраэтил аммония 1083
Трифторметилтиолат 1078
Фенилмедь 1078
Фторид меди(II) 273
Хлориды 1063, 1066
Циклопентадиенилтриэтилфосфин-медь 1083
Чистая медь 1061
Молибден
Ацетат 1656
Бис (ацетилацетонато} диоксомолибден (VI) 1654
Бис [дикарбонил (г)-циклопентадиенил) молибден] 1977 ,
2196 Предметный указатель
Бис [трикарбонил (т]-циклопентадиенил) молибден] 1976
Бис [о-фениленбис(диметиларсин) ] -молибдена (II) дихлорид 1655
цис-Бис(фенилфосфин)тетракарбо-нилмолибден 2113
(т]4-Бицикло [2.2.1] гептадиен-2,5) -тетракарбонилмолибден 1996
Бромиды 1638—1640
Гексакарбонилмолибден 1935 Гексатиоцианота (N) молибдат (III)-калия 1655
Гексахлоромолибдат (III) калия 1642 — аммония 1642
Гидриде (т]-циклопентадиенил) три-карбонилмолибден 2078
Гидроксиды 1645
Декакарбонилмолибдат(—I) натрия 1934
Дигидридобис (т] -циклопентадиенил) -молибден 2080
Дипероксомономолибдат (VI) калия-водорода 1649
Йодиды 1641
Иододикарбонил (г)-циклогептатрие-нил) молибден 2059
Металлический 1632
Молибденовая кислота 1646
Молибденовая синь 1645 6-Молибдоалюминат аммония 1903 — натрия 1903 12-Молибдоарсенат(У) калия 1901 9-Молибдо-З-ванадофосфорная кислота 1907
10-Молибдо-2-ванадофосфорная кислота 1907
11 -Молибдо-1 -ванадофосфорная кислота 1907
18-Молибдодиарсенат(У) натрия 1905
Ю-Молибдодикобальтат(Ш) аммония 1906
18-Молибдодимышьяковая (V) кислота 1905
18-Молибдодифосфат аммония 1904 18-Молибдодифосфорная кислота 1905
6-Молибдоиодат натрия 1903 6-Молибдоиодная кислота 1903 б-Молибдокобальтат(Ш) натрия 1904 12-Молибдокремневая кислота 1899 9-Молибдоманганат(1У) аммония 1905
9-Молибдомарганцовая (IV) кислота 1906
б-Молибдоникколат(П) аммония 1902
9-Молибдоникколат(1\') аммония 1906
б-Молибдородат(Ш) аммония 1902 б-Молибдоферрат(Ш) аммония 1903 12-Молибдофосфат аммония 1899 — бария 1899
12-Молибдофосфорная кислота 1898
Молибдохромат(Ш) аммония 1901 б-Молибдохромат(Ш) натрия 1902 6-Молибдохромовая кислота 1901 12-Молибдоцерат(1У) аммония 1900 12-Молибдоцерат(1У) аммония-водо-рода 1900
12-Молибдоцериевая(1\7) кислота 1900
Нитрид-трихлорид 1650
— аддукт с оксид-трихлоридом фосфора 1652
Оксид-хлориды 1646—1648
Оксиды 1644—1646
Оксопентахлоромолибдат (V) аммония 1649
Оксамолибдат аммония 1894
Октахлородимолибдат (I) тетракалия 1656
Октацианомолибдат(IV) калия 1653
Пентакарбонил (дифенилци клопропе-ннлиден) молибден 2036
Пентахлороаквамолибдат(Ш) аммония 1643
Полиоктамолибдат аммония 1894
Сульфид 1652
Тетрапероксомолибдат (VI) тетрам-минцинка 1650
Тетратиомолибдат (VI) аммония 1653 Трибромотрипиридинмолибден(Ш) 1639
Трикарбонил (т]5-инденил) метилмо-либден 2026
Трикарбонил (т] -толуол) молибден 1973
гран-Трикарбонилтрис(ацетонитрил) молибден 2118
Трикарбонил (трциклогептатриенпл) -молибдена (0) тетрафторобораг 1978
Трикарбонил (т]6-циклогептатриен) -молибден 1997
Трикарбонил (т]-циклопентадиенил)-метилмолибден 2025
Трихлоротрис(мочевина)молибден (III) 1644
Трихлоро (трциклопентадиенил) ди-карбонилмолибден 2058
Фториды 294
Хлориды 1632—1638
Хлоро (т] -циклопентадиенил) трикар-
Предметный указатель 2197
бонилмолибден 2057 (т]-Циклопентадиенил) дикарбонил-(метандиазо) молибден 2132
(т]-Циклоп ентадиенил) (нитрозил)-дикарбоиилмолибден 2090
Мышьяк
Арсаи (арсин) 611
Арсенат (V) аммония 625
Арсенат гидроксиламмония 507
18-Вольфрамодиарсенат(У) аммония 1912
Гексахлороарсенат (V) триэтиламмо-ния 616
Дпарсан (диарсин) 614
Дигидроарсеиат(У) натрия 624
Дигидроарсенид натрия 614
Дитиоарсенат(V) натрия 628
Метилдииодоарсан 620 12-Молибдоарсенат(У) калия 1901 18-Молибдодиарсенат(У) натрия 1905 18-Молибдодимышьяковая (V) кислота 1905
Монотиоарсенат(V) натрия 627
Мышьяковая кислота 624
Оксид мышьяка(III) 623
— мышьяка (V)-сурьмы (V) 639
Пентафторида мышьяка и тетрафторид сульфинила аддукт 211
Пентасульфид димышьяка 625
Тетраиодид димышьяка 618
Тетрасульфид тетрамышьяка 625
Тетратиоарсенат(V) аммония 626 — натрия 626
Тетрахлороарсенат (III) тетраметил-
аммония 616
Трибромид 617
Трииодид 617
Трифеииларсан 621
Трифениларсандихлорид 622
Трихлорид 615
Фториды 239—241
Элементный 609
— желтый 609
Натрий см. также Щелочные металлы
Азид 496
Амальгама 2170
Амид 488
Антимонид 1040
Арсенид 1038
Ацетилид (карбид) 1042
— однозамещенный 1043
Бис (триметилсилил) амид 776
Германид 1046
Гидрид 1024
Гидроселенид 451
Гидросульфид 406
Динатрийдисульфан см. Дисульфид
Динатрийпентасульфан см. Пентасульфид
Диоксид (надпероксид 1031
Диселенид 453
Дисульфид 409
Металлический 1008—1022
Натрийгермил 784
Оксид 1027
Пентасульфид 413
Пероксид 1030
Порошок 1923
Селенид 452
Силицид 1046
Сульфид 406
Тетрасульфид 412
Фосфид 1037
Циклопентадиенилнатрий 1924
Неодим см. также Редкоземельные элементы
Получение методом ионного обмена 1160
Нептуний
Арсениды нептунила, двойные 1370
Бромиды 1358
Гидриды 1351
Дисилицид 1366
Дихлорид-оксид нептуния (IV) 1356
Иодид нептуния (III) 1359
Карбиды 1366
Меры предосторожности 1348
Металлический 1350
Мононитрид 1365
Монофосфид 1365
Нитраты нептунила 1369
— нептуния (IV), дигидрат 1368
— оксоиептуния(V), тригидрат 1369
Оксалаты 1370
Оксиды 1360, 1361, 1362
Оксонептунаты щелочных и щелочноземельных металлов 1367
Селениды 1364
Сульфид-оксиды 1364
Сульфиды 1363
Триацетатодиоксонептунат (VI) натрия 1371
Фосфаты нептунила, двойные 1370
Фториды 1352, 1354, 1355
Фторонептунаты щелочных и щелочноземельных металлов 1368
Хлориды 1355, 1357
Чистые соединения 1348
Никель
т]-Аллил (метил) (трифенилфосфин) -никель 2012
2198 Предметный указатель
Амид 1797
Бис (т]-аллил)никель 2012
Бис [р-бромо (л-аллил) никель] 2011
Бис [т]3 -1,3-диметилаллил (метил) никель] 2013
Бис [ц-карбонил (л -циклопентадиенил) никель] 1994
Бис Гц-метил (граллил) никель] 2012
Бис[1,2 : 5,6-л-циклооктадиен-1,5)]-никель 2009
Бис(т]-циклопентадиенил) никель 1962
Бромид 1790
Гексаммииникеля(П) хлорид 1789
Гексагидроксоникколат(П) стронция 1879
Гексафтороникколат(1У) 305
Гексацианодиникколат(1) калия 1802, 2111
Гидроксиды 1792—1794
р,Л2-Диазотннкельлитиевый комплекс 2139
Диникель (IV) -ц-дисульфотетратно-бензоат 1804
Карбид 1798
Иодид 1790
Карбонат 1800
Металлический 1788
б-Молибдоинкколат(П) аммония
1902
9-Молибдоникколат(1У) аммония
1906
Никелоцен 1962
Нитриды 1797—1798
Оксид 1791
Ортопериодат никеля-натрия 1791
Роданид 1804
Сульфиды 1795—1797
Тетракис (триметилфосфит) никель 2120
Тетракис (трифенилфосфит) никель 2121
Тетракис (трифторофосфии) никель 2119
Трис (л -циклопентадиеиил) диникеля (II) тетрафтороборат 1963
Тетрацианоникколат(Н) калия 1802
Тетрацианоникколат(О) калия 2111
Тиоцианид 1804
Трифтороникколат(П) калия 305
Фосфиды 304, 1799
Хлорид 1788
Цианид 1800
(1,2 : 5,6 : 9,10-т]-транс, транс, транс-
Циклододекатриен) инкель 2014
(Л -Циклопентадиеиил)нитрозилии-кель 2098
Ниобий
Бромиды 1551
Гексакарбоиил-Цз (т)3Ст)О) -карбонил-трис(л-циклопентадиеиил)ниобий 1971
Гептафторониобат калия 289
Гидриды 1543
Дисульфид-дихлорид 1565
Дихлоробис (л -циклопентадиеиил) ниобий 2051
Йодиды 1552—1554
Карбиды 1579
Металлический 1541
Ниобаты 1572—1573, 1892—1893
12-Ниобоманганат(1У) натрия 1896
Нитриды 1575
Оксид-галогениды 1561—1565
Оксиды 1566—1570
Пероксоииобат калия 1572
Пероксониобиевая кислота 1571
Селениды 1575
Сульфиды 1574
Теллуриды 1575
Тетрахлоро(л-циклопентадиеиил) -ниобий 2055
Тетракарбонил(л-циклопентадиенил) ниобий 1969
Тригидридобис (л-циклопентадие-нил)ниобий 2077
Фосфиды 1578
Фторид 288
Хлориды 1544—1547
Олово ‘
Амальгама 2173
Ацетат олова (IV) 836
Бромиды 823
Гексагидроксостаннат(ГУ) натрия
Гексахлорооловянная(^) кислота,
гексагидрат 821
— соли 822
Гидрид олова(IV) см. Станнан
Йодиды 824, 825
Кислоты оловянные 827
— соли 828
Металлические 816—817
Оксид олова (II) 826
Оксогидроксостаинат(П) бария 1876
Ортостаннат натрия см. Тетраоксо-станнат натрия
Станиан SnH« 817
Станниды щелочных металлов 1047
Сульфат олова (IV), дигидрат 833
Сульфиды 829, 830
Тетраметилолово 834
Тетраоксостаинат(ГУ) натрия 828
Предметный указатель -2199
Тетраэтилстаннан см. Тетраэтилолово
Тетратиостаннат(1У) натрия, октадекагидрат 833
Тетраэтилолово 835
Тригидроксостаннат(П) натрия 1875
Тритиостаннат(1У) натрия, октагидрат 832
Фториды 257
Хлориды 819, 820
т] '-Циклопентадиеиил (три-н-бутил) -олово 1927
Осмий
Гексахлороосматы 1844—1846
Дихлоро (тетракарбонил) осмий 2069
Дихлоро (трикарбонил) осмий 2069
Додекакарбонилтриосмий 1945
Металлический 1844
Нитридоосмат(УШ) калия 1847
Оксиды 1846—1847
Осмат (VI) калия 1847
Осмиамат калия 1847
Осмийхлороводородная кислота 1844
Тетрагидридобис (трифеиилфосфин) -осмий 2084
Фториды 311—312
Хлоро (гидриде) трис (трифенилфосфин) карбонилосмий 2084
Палладий
Асбест палладированный 1829
Бис [р-хлоро-хлоро (дифенилцикло-пропенилидеи)палладий] 2043
Бис[1,2 : 5,6-т)-(циклооктадиен-1,5)]-палладий 2016
Бромид 1831
транс- Дихлоробис (бензонитрил) палладий 2140
Дихлоробис (трифенилфосфин) и алла-дий 2122
траис-Дихлородиамминпалладий(П) 1834
Ди-р-хлоро-af-дихлоробис (три-н-бу-тилфосфин)дипалладий 2122
Дн-р-хлоротетракис (трифенилфосфин) палладия (II) тетрафтороборат 2123
Дихлоро(1,2 : 5,6-т]-циклооктадиен)-палладий 2015
Иодид 1832
Коллоидный 1829
Металлический 1828
Нитрат 1832
Оксид 1829
Палладиевохлористоводородная кислота 1830
Сульфат 1833
Тетракис (оксазолидин-2-илидеи) палладия (II) иодид 2045
Тетр акис (трифенил фосфин) п алл а дий 2121
Тетрахлоропалладат(П) водорода 1830
Тетрацианопалладат(П) калия 1833
Фторид 307
Хлорид 1830
Хлоропалладаты 1830—1831, 1834
Чернь палладиевая 1829
Платина
Асбест платинированный 1808
Бис(трифеиилфосфин) (^-сероуглерод) платина 2103
Бис[1,2 : 5.6-т]-(циклооктадиен-1,5)] платина 2020
Бромиды 1815
Гексагидроксоплатиновая(ГУ) кислота 1820
Гексафтороплатинат (IV) калия 307
Гексахлороплатинаты (IV) 1812—1814
(т) - Дикнслород) бис (трифенилфос-фин)платииа 2140
цис-Динитродиамминплатина (II) 1826
г] -Дифенилацетиленбис (трифенилфосфин) платина 2019
транс- Дифульминатобис (трифенилфосфин) платина 2113
цис-Дихлоробис (трифеиилфосфин) -платина 2124
цис-Дихлоробис (трифенилфосфит) -платина 2125
цис-Дихлоробис (триэтилфосфин) платина 2046
транс- Дихлородиамминплатина (II) 1826
цис-Дихлородиамминплатина (II) 1825 цис-Дихлородикарбонилплатина 2074 Ди-р-хлоро-а,f-дихлоробис (триэтил-
фосфии) диплатина 2046
Д и- р -хлоротетр акис (трифенил фосфин) диплатины (II) тетрафтороборат 2123
цис-Дихлоро (фенилизоцианид) (три-этилфосфии) платина 2046
Дихлоро [1,2 : 5,6-т1-(циклооктадиен-1,5) платина 2019
цис-Дихлоро [этокси (феииламино) -карбен] (триэтилфосфин) платина 2046
Жерара соль 1827
Йодиды 1816—1817
Клеве соль 1827
Металлическая 1805
2200 Предметный указатель
Нашатырь платиновый 1813
Оксиды 1818—1819
Пейроне соль 1825
Платинирование гальваническое 1808 — стекла и фарфора 1810 — электродов 1809
Платинохлористоводородная кислота 1812
Правила работы с платиновой посудой 1806
Регенерация платиновых остатков 1806
Рейзе основания первого хлорид 1824 — — второго хлорид 1826
Тетраамминплатины (II) хлорид 1824
Тетракис [р3-иодо (триметил) платина] 2032
Тетракис (трифенилфосфин) платина 2124
Тетранитроплатинат(II) калия 1822
Тетрахлородиамминплатина (IV) 1827 Тетрахлороплатинат (II) водорода
— тетраамминплатины(П) 1824
Тетрахлороплатиновая(П) кислота
1812
Тетрацианоплатииаты(П) 1822
Трис (трифенилфосфин) платина 2125
Трихлоро(т)-этилен)платината (II) калия моногидрат 2017
Сульфид 1821
Фториды 305—306
Хлориды 1810—1812
транс-Хлоро (гидриде) бис (трифенилфосфин) платина 2087
траяс-Хлорогидридобис(триэтилфос-фин)платина 2047
транс- [Хлоро {(п-толилимино) метил} -бис (триэтилфосфин) платина 2047
Хлоро [трифенилфосфито (С, Р)] (три-фенилфосфит) платина 2126
Пейзе соль 2017
Чернь платиновая 1807
трЭтиленбис (трифенилфосфин) платина 2018
Плутоний
Бромид плутония (III) 1393
Иодид плутония(III) 1395
Карбиды 1405
Меры предосторожности 1374
Металлический 1372, 1380
Моноарсенид 1404
Моионитрид 1402
Монофосфид 1403
Нитрат плутонила 1410
— плутония (IV) 1409
Оксиды 1396, 1397
Оксоплутонаты щелочных и щелочноземельных металлов 1406, 1407
Селениды 1400
Сульфаты 1411, 1412
Сульфиды 1398
Теллуриды 1401
Тетрасульфатоплутонат(1У) калия, дигидрат 1413
Фосфаты 1410
Фториды 1384, 1388, 1389
Фтороплутонаты щелочных и щелочноземельных металлов 1408
Хлорид плутония (III) 1390
Чистые соединения 1377
Празеодим см. также Редкоземельые элементы
Оксид празеодима (IV) 1178
Получение методом ионного обмена 1160
Протактиний
Бромид 1269
Гидрид 1260
Дибромид-оксид 1270
Дииодид-оксид 1271
Дихлорид-оксид 1268
Йодиды 1270, 1271
Металлический 1257
Нитраты 1273, 1274
Оксиды 1272, 1273
Протактинаты(У) щелочных и щелочноземельных металлов 1275
Селенат 1273
Сульфат 1273
Фториды 1261, 1262, 1264
Фторокомплексы 1274
Хлориды 1264, 1267
Чистые соединения 1254
Редкоземельные элементы см. также Европий, Иттербий, Лантан, Неодим, Празеодим, Самарий, Скандий, Тербий, Церий
Бромид-тетраоксиды 1178
Бромиды 1166, 1169
Г алогенид-окси ды 1175
Галогениды 1172
Гидриды 1164
Гидроксиды 1184
Йодиды 1166, 1170
Карбиды 1204
Карбонаты 1205
Нитраты 1199
Нитриды 1195
Оксид-фториды 283
Особо чистые 1163
Предметный указатель 2201
Полисульфиды 1191
Селениды 1188
Сульфид-диоксиды 1193
Сульфиды 1188
Теллуриды 1192
Фосфаты РЗЭ(Ш) 1203
Фосфиды 1201
Фториды 281
Хлориды 1166
Рений
Бис [u-бромо (тетрагидрофуран) три-карбонилрений] 2133
Бнс(ц-хлоротетракарбонил) рений] 2062
транс-Бис(этилендиамин)диоксоре-hhh(V) хлорид 1739
Бромиды 1714—1715
Бромо (пентакарбонил)рений 2061 Гексаброморенат(1У) калия 1729 — серебра 1729
Гексаиодоренат(IV) калия 1730
Гексаоксоперренат(УП) бария 1739
Гексафтороренат(IV) калия 301
Гексафторорениевая кислота 301 Гексахлороренат(1У) калия 1728 Г идридобис (т] -циклопентадиенил) рений 2083
Декакарбоиилдирений 1939
Диаквагептаоксид дирения 1736
Диоксотетрацианоренат (V) калия 1734
Додекахлоротриренат (III) трицезня 1733
Йодиды 1716
Иодо(пеитакарбонил)рений 2062 Металлический 1709
Нитридоренат(УП) калия 1738
Ноиагидридоренат (VII) бис(тетра-этиламмоиия) 1726
— дикалия 1727
— динатрия 1724
Оксиды 1718—1721
Оксид-тетрахлорид 1723
Оксодихлороэтоксибис (трифенилфосфин) рений (V) 1740
Оксопентахлороренат (V) цезия 1730 транс-Оксотрихлоробис (трифенил-фосфии)реиий(^) 1740
Октабромодиренат (III) тетра-н-бу-тиламмония 1732
Октабромодиренат(III) цезия 1733 Октахлородиренат(Ш) калия 1732 — тетра-н-бутиламмония 1731 Октацианоренат (V) калия 1734 Пентаоксоренат(VII) бария 1738 Перренат(УП) бария 1737
— марганца 1736
— серебра 1737
— тетраметиламмония 1737
Перрениевая кислота, раствор 1735
твердая 1736
Регенерация из остатков 1742
Сульфиды 1721—1722
Тетраацетатодибромодирений(Ш)
Т етр а ацетатодихлородирений (III)
Тетракис(|гз-гндроксотрикарбонилре-ний) 2134
Трикарбонил (г] -бромциклопентадиенил) рений 1982
Трикарбонил (триодциклопентадие-нил)рений 1983
Трикарбонил (т]-хлорциклопентадие-нил)рений 1982
Трикарбонил(т)-циклопентадиенил) -рений 1981
Триоксид-фторид 300
Триоксид-хлорид 1722
Фториды 299
Хлориды 1710—1712
Хлоро(пентакарбонил)реиий 2061
Родий
Бис [ (ц-карбонил) (т] -пентаметилцик-лопентадиенил)родий] 1992
Бис(ц-хлородикарбонилродий) 2070
Бис[ц-хлоро-{1,2 : 5,б-ц-(циклоокта-диен-1,5)} родий] 2008
Бромид 1841
Гексадекакарбонилгексародий 1951
Гексанитрородат(Ш) калия 1843
Гексахлорородаты 1840
Г идридотрис (трифенилфосфин) кар-бонилродий 2086
[ц-Дикарбоиил-цз-метилиден-чикло-трис- {(т] -циклопентадиенил) родия} тетрафтороборат 2043
Дикарбонил (т] -пентаметилциклопентадиенил) родий 1990
Дикарбонил (т] -циклопентадиенил) подий 1989
(ц-Диоксид серы) бис [карбонил (тр циклопентадиенил)родий] 1993
Ди (ц-хлоро) бис [хлоро (грпеитаме-тнлциклопентадиенил)родий] 2071
ц -Дихлоротетракис (т]2-циклооктен)-диродий 2008
Додекакарбонилтетрародий 1950
Иодид 1841
(ц-Карбонил) бис [карбонил (трпеита-метилциклопентадиенил)родий] 1991
2202 Предметный указатель
ц-Карбонилбис[карбонил(т]-Цикло-пентадиенил)родий] 1989
Металлический 1838
ц-Метилеибис [карбонил (г]-циклопентадиенил) родий] 2042
б-Молибдородат(Ш) аммония 1902
Нитрат 1843
Нитрозилтрис (трифенилфосфин) родий 2097
Оксид 1841
Сульфат 1842
Фториды 308
Хлорид 1839
Хлороамминатов родия(III) хлориды 1843
Хлоротрис (трифенилфосфин) родий 2117
Ртуть
Амидохлорид ртути(П) 1150
Ацетат ртути(I) 1155
Бис (трифторометилсульфинил) ртуть 708
Бромид ртути(1) 1144
Диамминртути(П) хлорид 1150
Димеркуроаммония бромид 1152
— гидроксид 1151
Ди (тетрахлороалюминат) триртути 1157
Ди (тионитрид) диртути (I) 1152
— ртути (II) 1152
Дихлорид-диоксид триртути 1146
Дихлорид-дисульфид триртути 1148
Дихлорид-тетраоксид пеитартути 1146
Дихлорид-фосфид диртути 1153
Диэтилртуть 1154
Имидодибромид ртути(II) 1151
Милоново основание 1151
— бромид 1152
Оксид ртути(II) 1145
Селенид 1149
Сульфид ртути (II) 1147
Тетраиодомеркурат(П) медн(1) 1145
Тетратиоцианатомеркурат (I I) калия 1156
Тионитрозилат см. Ди (тионитрид)
Тиоцианаты 1156
Трииодид-диантимонид тетрартути 1154
Трииодомеркурат(П) калия, моногидрат 1144
Фосфаты 1153
Фториды 278, 280
Рубидий см. также Щелочные металлы
Азид 497
Антимонид 1040
Арсенид 1039
Ацетилиды (карбиды) 1043
Висмутид 1041
Гексабромоталлат(Ш) 953
Гексахлоротеллурат 472
Германид 1046
Гидрид 1025
Гидроксид 1034
Диоксид (надпероксид) 1032
Дихромат(У1) 1627
Квасцы рубидий-ванадиевые 1536
Металлический 1009—1014, 1016—•
1019
Оксид 1028
Оксоплутон ат (VII) 1407
Пероксид 1031
Плюмбид 1047
Рубидийгермил 784
Силицид 1046
Станнид 1047
Тетрафтороиодат(Ш) 272
Фторид 272
Фтороплутонат(У) 1408
Фтороуранаты (IV) 1345
Хромат 1627
Рутений
Бис [дикарбоиил (т)-циклопеитадие-нил) рутений] 1986
Бис (трифенилфосфин) трикарбонил-рутений 2115
Бис [ц-хлоро(хлоро)трикарбонилру-тений] 2067
Бис (г] -циклопентадиенил) рутений 1961
Бромид 1850
Гексахлорорутенаты(1У) 1850
Дииодо(тетракарбонил)рутений 2067
Динитрозилбис (трифенилфосфин) рутений 2094
Дихлоробис (трифенилфосфин) дикар-бонилрутений 2115
Додекакарбонилтрирутений 1944
Иодид 1851
Карбамоил (дикарбонил) (т]-циклопентадиенил) рутений 2041
Металлический 1848
Оксиды 1851
Рутениевая красная 1852
Рутеноцен 1961
Трикарбонил (т] -циклопентадиеиил) -рутения (II) гексафторофосфат 1987
Фториды 310
Хлориды 1848—1850
Хлоро (гидриде) трис (трифенилфосфин) карбонилрутений 2083
Предметный указатель 2203
Хлоро (т] -циклопентадиенил) дикарбо-нилрутений 2068
Самарий см. также Редкоземельные элементы
Амальгама 1162
Фторид самария (И) 281
Свинец
Азид 847
Амальгама 2173
Ацетат свинца (IV) 851
Гексагидроксоплюмбат(1У) натрия
1888
Гексахлороплюмбат(1У) аммония
838
— калия 839
Карбонаты 851
Металлический 837
Метасиликат 761
Оксиды 841, 842
Октагидроксоплюмбат (IV) бария 1890
Ортоплюмбат натрия см. Тетраоксо-
плюмбат(1У) натрия
Плюмбид калия 1047
— рубидия 1047
— цезия 1047
Роданид см. Тиоцианат
Селенид 846
Сульфат 845
Теллурид 847
Тетраметилплюмбан см. Тетраметил-свинец
Тетраметилсвинец 848
Тетраоксоплюмбат(1У) кальция 844
— натрия 843
Тетраэтилсвинец 849
Тиоцианат 853
Трииодоплюмбат(Н) калия, дигидрат
• 840
Триоксоплюмбат(1У) натрия 843
Фториды 258
Хлорид калия-свинца (IV) см. Гекса-хлороплюмбат калия
— свинца (IV) 837
Селен
Дибромид диселена 456
Диоксид 457
Дихлорид диселена 453
Гексахлороселенаты(ТУ) 455
Монобромид селена см. Дибромид диселена
Монохлорид селена см. Дихлорид диселена
Нитрид тетраселеиа 465
Оксохлорид 460
Селенат натрия 464
Селенид азота см. Нитрид тетраселена
Селенид водорода см. Селеноводород
Селенила дифторид 218
— дихлорид см. Оксохлорид
Селенистая кислота 462
Селенит натрия 462
Селеновая кислота 463
Селеноводород 450
Селеноксодифторид см. Селеннла дн-фторид
Селенопентатионат натрия 464
Тетрабромид 457
Тетрахлорид 454
Триоксид 459
Фторид селена (IV) 218
— селена (VI) 218
Фтороселенит калия 219
Хлорюр селена см. Дихлорид диселе-иа
Элементный 448
Сера
Амидосерная кислота 527
Амидосерной кислоты фтороаигидрид
534
--- хлороангидрид 538
Амидосульфат калия 528
Амидосульфоновая кислота 527
Бис (трифторометилсульфиновой) кис-
лоты имид 710
Вакенродера жидкость 440
Гексасульфан 402
Гексатионат калия 438
Гептасульфан 402
Гидразидо-М.М'-биссерной кислоты соли 540
Г идроксиламинобиссульфат (-дисульфонат) калия 542
Гидроксиламиноизомоносульфоновая кислота 541
Гидроксиламино-О-серная кислота
541
Гидроксиламинотриссульфат калия
543
Гидросульфид аммония 405
Дейтеросерная кислота 172
Декафторид дисеры 206
Дибромодисульфан 422
Диимидобиссульфат калия 540
Динитрид дисеры 442
— тетрасеры 442
Динитрозосульфит калия 545
Дисульфан 399
Дисульфурила бисизоцианат 533
— дифторид 212
2204 Предметный указатель
— оксодифторид 213
— хлорид 425
Дитиодекафторид 206
Дитионат натрия 432
Дитионит натрия 431
Дитиохлорид см. Дихлородисульфан
Дифторид дисеры 204
Дифтородисульфан 203
Дифторосульфиминфторид см. Три-фторид-нитрид серы
Дихлорид см. Дихлоромоносульфан
Дихлорид дисеры см. Днхлородисуль-фан
Дихлорогексасульфан 417
Дихлорогептасульфан 417 Дихлородисульфан 417 Дихлоромоносульфан 416 Дихлорооктасульфан 417 Дихлоропентасульфан 417 Дихлоротетрасульфан 417 Дихлоротрисульфан 417 Имид гаптасеры 444 Имидобиссерной кислоты дифтороангидрид 535
----- дихлороангидрид 538
Имидобиссульфат калия 528 — триаммония 529
К.аро кислота см. Пероксомоиосерная кислота
Коллоидный раствор серы 391 Надсерная кислота см. Пероксомоно-серная кислота
Нитридотриссульфат калия 530
Нитрбзобиссульфат калия 543 N-нитрозогидроксиламино-М-сульфат калия 545
Нитрозодисульфонат калия 543
Нитрозосульфонилфторид 226
Оксиды низшие 424
Октасульфан 402
Пеитасульфан 402—404
Пентасульфид аммония 415
Пентатионат калия 436
Пероксиламиносульфонат калия см.
Нитрозобиссульфат калия
Пероксодисерная кислота 429
Пероксодисульфат калия 429
Пероксодисульфурилдифторид см. Дисульфурила оксодифторид
Пероксомоиосерная кислота 428 Персерная кислота см. Пероксодисерная кислота
Персульфат калия см. Пероксодисульфат калия
Перфторобутилсульфоннлизоцианат 701
Пиросульфурилфторид см. Дисульфурила дифторид
Пластическая 391
Полипероксид 424
Политионовые кислоты 440
Селенопентатионат натрия 464
Серной кислоты диамид см. Сульфамид
Сероводород 394
Сульфамид 530
Сульфаминовая кислота см. Амидо-серная кислота
Сульфан сырой 396
Сульфандисульфоновая кислота 440
Сульфанмоносульфоновая кислота 440
а-Сульфанура хлорид 446
Сульфинила бромид 427
— тетрафторид 210
— фторид 209
— хлорид 424
— хлорид-фторид 214
Сульфинилимид 524
Сульфоксилат кобальта 430
Сульфомоноперкислота см. Пероксо-моносерная кислота
Сульфонила амидофторид 534
— амидохлорид 538
— бисизоциаиат 533
— бромид-фторид 215
— дифторид 211
— фторид-изоцианат 534
— хлорид 424
— хлорид-изоцианат 536
— хлорид-фторид 214
Сульфоперамидная кислота 541
Сульфуриламид см. Сульфамид
Сульфурилбромидфторид см. Сульфонила бромид-фторид
Сульфурилдифторид см. Сульфонила дифторид
Сульфурилхлорид см. Хлорид сульфонила
Сульфурилхлоридфторид см. Сульфонила хлорид-фторид
Теллуропентатионат натрия 480
Тетраимид тетрасеры 444
Тетранитрид тетрасеры 441
Тетр асульф ан 401—403
Тетрахлорид 421
Тетрасульфимида соли 532
Тетрасульфурила хлорид 427
Тетратионат калия 435
Тиомонохлоридпентафторид см. Хлоросульфинила пентафторид
Тионилбромид см. Сульфинила бромид
Предметный указатель 2205
Тионилфторид см. Сульфинила фторид
Тионилфторидхлорид см. Сульфинила хлорид-фторид
Тионилхлорид см. Сульфинила хлорид
Тиооксотетрафторид см. Сульфинила тетрафторид
Тиотритиазила хлорид 445
Тиофосфорила триамид 606
Трисерадиазотдиоксид 446
Трисерадиазотпентоксид 447
Трисульфимида соли 531
Трисульфурила хлорид 426
Трис (трифторометилсульфиновой) кислоты нитрид 711
Трисульфан 399
Тритионат калия 434
Трифторид-нитрид 229
Трифторометилсульфинилизоцианат 707
Трифторометилсульфинилкарбонил-фторид 705
Трифторометилсульфинилхлорид 706
Трифторометилсульфиновой кислоты амид 710
Фосфорсульфидтриамид (Фосфортиотриамид) 606
Фреми соль см. Нитрозобиссульфат калия
Фторид серы (IV) 205
— серы (VI) 207
Фторосерная кислота 216
Фторосульфонилизоцнанат 534
Хлороамидобиссульфат калия 539
Хлорокарбонилсульфинилхлорид 704
Хлоросерная кислота 425
Хлоросульфинила пеитафторид 208
Хлоросульфонилизоцианат 536
Хлорсульфоновая кислота см. Хлоросерная кислота
Циклогексасера 392
Циклогептасера 393
Циклодекасера 393
Циклододекасера 392
Циклооктасера 390
Элементная 390
•Серебро
Азид 1094
Амид 1092
Ацетиленид см. Карбид
Бис (а,а'-бипиридил) серебра (II) нитрат 1097
— пероксодисульфат 1097 12-Ванадофосфат 1908 Гексаброморенат (IV) 1729
Гипонитрит 521
Иодид 1086
Карбид 1095
Кетен 1099
Коллоидное 1086
Манганат(УП) 1688
Металлическое 1083
Нитрид 1094
Оксид 1089
Пентафторфенилсеребро 1098
Пероксид 1090
Пероксодисульфат ди(о-фенантро-лин) серебра (II) 1096
Перренат(УП) 1737
Перфтор-1 -метилпропенилсеребро
1099
Перхлорат 1088
Регенерация из остатков 1084 — из фотографических ванн 1085
Селенид 1091
Сульфид 1090
Теллурид 1092
Тетрафтороаргентат(Ш) калия 276
Тетракис (трифеиилфосфин) серебра перхлорат 1100
Тетра (пиридин) серебра (II) пероксодисульфат 1101
Тетрафтороборат 276
Трис (а,а'-бипиридил) серебра (II) нит-
рат 1097
— перхлорат 1097
Трифтороацетат 1100
Трифторметилтиолат 1098
Фториды 273—275
Фтороуранаты(1У) 1345
Хлорат 1088
Цианамид 1095
Этилеибис (бигуанидин) серебра (III) сульфат 1100
Скандий см. также Редкоземельные элементы
Фторид скандия(III) 280
Чистые соединения 1158
Стронций
Амальгама 2172
Галогениды 995
Гексагидроксоникелат(П) 1879
Гексагидроксоферрат(Ш) 1887
Гексагидроксохромат(Ш) 1885
Гептагидроксоферрат(Ш) 1887
Гидрид 994
Гидроксид, октагидрат 999
Иодид 995 >
Металлический 993
Нитрид 1002
2206 Предметный указатель
Оксид 997
Селенид 1001
Силициды 1008
Сульфид 1000
Тетрагидроксобериллат 1873
Фторид 269
Сурьма
Гексабромоантимонат(1У) аммония 636
Гексахлороантимонат (V) нитрозила
634
Гексахлоросурьмяная(У) кислота 634
Гидрид сурьмы см. Стибан
Дихлорид-трифторид 243
Оксид сурьмы(Ш) 638
— сурьмы (IV) 639
— сурьмы (V)-мышьяка (V) 639
Оксида сурьмы (V) гидрат 638
Оксосульфат 640
Оксохлорид 633
Пентаоксид-хлорид тетрасурьмы 633
Пентафенилсурьма 637
Пентахлорид 632
Стибан (стибин) 629
Сульфат сурьмы(Ш) 640
Тетратиоантимонат (V) натрия 640
Трибромид 635
Трииодид 636
Трифенилсурьма 637
Трифторид 241
Трихлорид 631
Элементная 628
Таллий
Бромиды 947, 952
Гексабромоталлат(Ш) рубидия, гидрат 953
— таллия(1) 952
Гексахлороталлат(Ш) калия, дигидрат 951
— таллия (I) 950
Гексахлоротеллурат таллия 472
Гидроксид таллия(I) 954
Диметилталлия иодид 946
— хлорид 946
Дисульфатоталлиевая(Ш) кислота, тетрагидрат 958
Жидкость Клеричи 959—960
Иодид таллия (I) 947
Карбонат таллия (I) 959
Малонат таллия(I) 960
Нитрид 958
Оксиды 954, 955
Пентахлоромоноакваталлат (III) ка-
лия, моногидрат 951
Селениды 957
Сульфиды 956, 957
Сульфаты 957, 958
Тетрабромоталлат(Ш), таллия(1) 952
Тетракарбонилкобальтат (—I) таллия 1948
Тетрахлороталлат(Ш) таллия(1) 950 — тетрафениларсония 950
Тетрахлороталлиевая кислота, тригидрат 949
Трииодид таллия (I) 953
Триметилталлий 945
Формиат таллия(I) 959
Фториды 267
Фтороуранат (IV) 1345
Хлориды 947, 948
Циклопентадиенилталлий 1925
Чистый таллий 944
Эннеахлородиталлат(Ш) цезия 951
Тантал
Бромиды 1557—1559
Гексатанталат калия 1893
— натрия 1893
Гептафторотанталат калия 290!
Гидриды 1543
Йодиды 1559—1560
Карбиды 1579
Металлический 1541
Нитриды 1575
Оксид 1570
Оксид-иодиды тантала 1563
Пентаметилтантал 2024
Пероксотанталат калия 1573
Пероксотанталовая кислота 1571
Селениды 1575
Сульфиды 1574
Танталаты 1573
Теллуриды 1575
Тетр ахлоро (трциклопентадиенил) тантал 2055
Фосфиды 1578
Фторид 289
Хлориды 1554—1557
Теллур
Гексахлоротеллурат аммония 472
— калия 472
Диоксид 475
Дителлурид натрия 470
Ортотеллурат натрия 479
Ортотеллуровая кислота см. Теллуровая кислота
Теллурат натрия 479
Теллурид калия 470
— натрия 470
Теллуристая кислота 476
Теллурит натрия 476
Предметный указатель 2207
Теллуровая кислота 477
Теллуроводород 467
Теллуропентатионат натрия 480
Тетрабромид 473
Тетраиодид 474
Тетрахлорид 471
Триоксид 477
Фторид теллура(VI) 220
Элементный 466
Тербий см. также Редкоземельные элемент ы
Оксид тербия (IV) 1180
Технеций
Гексафторотехнетаты калия 1706
Гексафторофосфат дибензолтехне-
ция(1) 1707
Гексахлоротехнетат(1\7) калия 1705
Дитехнецийдекакарбонил 1704
Металлический 1700
Общие указания 1699
Оксиды 1700—1701
Октахлородитехнетат(Ш) тетра-н-бу-тиламмония 1708
Сульфиды 1702
Технетат(У1) тетраметиламмония 1704
Фторид 1703
Хлорид 1702
Эннеагидридотехнетат(УП) калия
1707
Титан
Ацетат 1485
Бис(т)-инденил)диметилтитан 2023
Бис (т) -циклопентадиенил) дикарбо-нилтитан 1964
Бис (трциклопентадиенил) диметилти-таи 2022
1,1 -Бис (г] -циклопентадиенил) -2,3,4,5-тетрафенилтитан 2023
Бромиды 1434, 1436, 1440
Галогенид-оксиды 1454
Галогениды 1426, 1429
ГексатиоцианатотитанатНУ) калия
1488
Гексахлоротитанат (IV) аммония 1439
Гидрид TiH 1425
Диацетилацетонатотитан(IV), дихлорид 1485
Д ихлоробис (трциклопентадиенил) титан 2050
Ди-л-циклопентадиенилтитан(1У, бромид 1491
— дихлорид 1490
— иодид 1491
— пентасульфид 1492
Йодиды 1435, 1443
Карбид 1480
Комплексные соединения 1485
Металлический 1414
Нитрат титана(IV) 1474
Нитрат-оксид титана(IV) 1476
Нитрид 1472
Оксиды 1458, 1460, 1462
Пероксид титана(IV) 1463
Силициды 1483
Сульфат титана (III) 1468
Сульфат-оксид титана (IV) (сульфат титанила) 1469
Сульфиды 1465
Тетракис (диэтиламид) титана (IV) 1487
Трис (2,2'-бипиридил) титан (0) 1489
Трис (2,2'-бипиридил)титанат (—I) лития 1489
Трихлоро (т|-циклопентадиенил) титан
Фосфат 1479
Фосфид 1476
Фториды 283, 285
Хлориды 1429, 1435, 1437, 1438
Торий
Арсениды 1248
Бориды 1250
Бромид тория (IV) 1229
Гидриды 1223
Дибромид-оксид tophh(IV) 1231
Дииодид-оксид тория (IV) 1235
Дифторид-оксид тория(IV) 1226
Дихлорид-оксид тория (IV) 1229
Иодат tophh(IV) 1251
Йодиды 1231, 1232, 1234
Карбиды 1248
Металлический 1218—1223
Нитрат тория(IV), гидраты 1253
Нитриды 1244
Оксид тория (IV) 1236
Оксид-динитрид дитория 1246
Перхлорат тория (IV), тетрагидрат 1251
Селениды 1243
Силициды 1249
Сульфат тория (IV) 1251
Сульфиды 1238
Теллуриды 1243
Тетраборогидрид тория(IV) 1253
Тигли из сульфидов тория 1242
— из ThO2 1237
Фосфиды 1246, 1247
Фторид тория(IV) 1225
Хлориды 1227, 1228
2208 Предметный указатель
Углерод
Блестящий 670
Бис (трифторометил) фосфан 712
Бромид-фторид карбонила 250
Бромоциан 691
Бромфторфосген см. Бромид-фторид карбонила
Гексафторотноацетон 709
Графит 673
— бромид 679
— гидроксид оксиграфита см. оксид графита
— гидросульфат 678
— кислота графитовая см. оксид графита
— нитрат 679
— оксид 675
— перхлорат 679
— пленки из графита 671
— фторид см. Монофторид углерода
— соединения с амминами щелочных металлов 674
---с хлоридом алюминия 680
---с хлоридом железа(III) 680
---с щелочными металлами 673
Диазоциклопентадиен 2064
Диоксид 682
— триуглерода 682
Диселенид 686
Дифторид карбонила 248
Дифторотиокарбонил 701
Дихлорид-оксид 684
Дициан 688
Изоцианат сульфонилфторида 534
Иодид-фторид карбонила 251
Иодоциан 693
Иодфторфосген см. Иодид-фторид
карбонила
Карбонилдиизотиоциаиат 699
Карбонилдиизоцианат 697
Кислота бис(трифторометилсульфи-
новая), имид 710
— синильная см. Циановодород
— трис (трифторометилсульфиновая), нитрид 711
— трифторометилсульфиновая, амид 710
— циановая 694
Монофторид 677
Нитрил муравьиной кислоты см. Циановодород
Оксид углерода(II) 681
Оксид-дифторид 248
Перфторобутилсульфонилизоцианат
701
Родановодород 695
Сажа 670
Селенид-оксид 685
Соединения поверхностные с кислородом 672
Сульфид-оксид 684
Тетрафторид 245
Тиокарбонилфторид см. Дифторотпо-карбонил
Тиоциановодород см. Родановодород
Трифториодметан 247
Трифторметан 246
Трифторометилгипофторид 712
Трифторометилсульфинилизоцианат 707
Трифторометнлсульфинилкарбонил-фторид 705
Трифторометилсульфинилхлорид 706
2,4,6-Трифторо-1,3,5-триазин см. Циа-нурфторид
Уголь активированный 671
Фторид тетрауглерода 678
Фторокарбонилизотиоцианат 699
Фторокарбонилизоцианат 700
Фторокарбонилсульфинилхлорид 705
Фторосульфонилизоцианат см. Изоцианат сульфонилфторида
Фторотиокарбоннлизотиоцианат 704
Фтороформ см. Трифторметан
Фтороциан 252
Фторфосген см. Оксид-дифторид
Хлорид-фторид карбонила 249
— тиокарбонила 703
Хлорокарбонилизоцианат 698
Хлорокарбонилсульфинилхлорид 704
Хлороциан 689
Хлорфторфосген см. Хлорид-фторид карбонила
Циановодород 688
Цианурфторид 252
Элементный углерод 669
Уран
Арсениды 1330
Бориды 1334
Бромид уранила см. Дибромид-диок-сид урана(VI)
Бромид-диоксид урана(V) 1313
Бромиды 1305—1307
Гептаоксид триурана 1319
Гидрид 1287
Гидрофосфат уранила, тетрагидрат 1340
Дейтерид 1287
Дибромид-диоксид ypana(VI) 1312
Дибромид-оксид урана (IV) 1314
Дифосфид 1330
Дифторид-диоксид ypana(VI) 1310
Дихлорид-диоксид урана (VI) 1311
Предметный указатель 2209'
Дихлорид-оксид урана (IV) 1313
Йодиды 1309
Меры предосторожности 1275—1280
Металлический 1282
Карбиды 1331
Нитрат уранила 1339
Нитриды 1326
Оксалаты 1341, 1342
Оксиды 1315, 1316, 1318, 1320
Пероксид урана(VI), гидраты 1315
Перхлорат уранила, гексагидрат 1335
Селениды 1323
Силициды 1334
Сульфаты 1336, 1337
Сульфиды 1321
Тетрагидроборат урана(IV) 1346
Тетрафосфид триурана 1328
Трихлорид-оксид ypana(V) 1313
Уранаты щелочных металлов 1343,
1344
Фосфид 1330
Фториды 1289, 1294, 1295, 1298
Фтороуранаты 1344, 1345
Хлориды 1299—1302
Чистые соединения 1280
Фосфор
12-Ванадофосфат аммония 1908 18-Вольфрамодифосфат аммония 1912 12-Вольфрамофосфат натрия 1910 12-Вольфрамофосфорная кислота
1910
Гексафторофосфат аммония 238
— калия 239
Гидроксилапатит 572
Гипофосфат тетранатрия 586
Гипофосфористая (фосфиновая) кислота 581
Гипофосфорная кислота 583
Гитторфа фосфор 548
Грэма соль 577
Дейтерофосфорная кислота 174
Диамидофосфорная кислота 602
Дибромид-фторид 236
Дигидрогипофосфат динатрия 585
Дигидрофосфид натрия 559
Диоксодифторофосфат аммония 238
Дитиофосфат натрия 593
Дифосфан 556
Дифосфат аммония 574
Дифосфин см. Дифосфан
Дифосфорная(1У) (гипофосфорная)
кислота 583
Дифосфорная кислота 573
Дифосфортриоксидтетрахлорид см.
Пирофосфорилхлорид
Дихлорид-трифторид фосфора (V) 234
23—1361
Дихлорид-фторид фосфора(III) 232 Дихлорофосфорная кислота 566 Дихлорофосфорной кислоты ангидрид.
см. Пирофосфорилхлорид Имидодифосфат тетранатрия 607 Курроля полифосфат натрия 578 Мадрелла соль см. Полифосфат натрия
9-Молибдо-З-ванадофосфорная кислота 1907
10-Молибдо-2-ванадофосфорная кислота 1907
11-Молибдо-1-ванадофосфорная кислота 1907
18-Молибдодифосфат аммония 1904 18-Молибдодифосфорная кислота 1905 12-Молибдофосфат аммония 1899 — бария 1899
12-Молибдофосфорная кислота 1898 Моноамидофосфат динатрия 601
Моноамидофосфорная кислота 599
Монотиофосфат натрия 591
Монотиофосфорная кислота 590 Монофосфорная кислота см. Фосфор-
ная кислота
Оксид-трихлорид фосфора — нитридтрихлорид молибдена 1652
Оксиды 568, 570
Оксобромид фосфора (V) см. Трибромид фосфорила
Ортофосфорная кислота см. Фосфорная кислота
Пероксодифосфат калия 587
Пирофосфат аммония см. Дифосфат аммония
Пирофосфорилхлорид 564
Пирофосфорная кислота см. Дифосфорная кислота
Пирофосфорной кислоты хлороаигид-рид см. Пирофосфорилхлорид
Полифосфат натрия 577
---- Курроля 578
Тетрааминофосфония иодид 606
— хлорид 607
Тетрабромид-фторид 236
Тетраиодид дифосфора 567
Тетраметафосфат натрия 579
Тетратиофосфат натрия 594 Тетрафосфат гексагуанидиния 576-— гексанатрия 575
Тетрафосфоргептасульфид 589 Тетрафосфорнонасульфид 590 Трифосфорпентанитрид 595 Тетрафосфорпентасульфид 589 Тетрафосфортриселенид 595
Тетрафосфортрисульфид 587 Тетрахлорид-фторид 234
12210 Предметный указатель
Тетрахлорофосфония гексафторофосфат 235
Тиобромид фосфора (V) см. Трибро-мнд тиофосфорила
Тиофосфорила триамид (фосфорок-сидтриамид) 604
— трибромид 563
— трихлорид (тиохлорид фосфора) 561
Трииодид 567
Триметафосфат натрия 578
Три (поли) фосфат натрия 574
Тритиофосфат натрия 593
Трифосфат(пента) натрия 574
Трифторид-оксид 235
Трихлорофосфазофосфора (V) оксид-хлорид 599
Трихлорофосфонитридофосфорила дихлорид 599
(Трихлорофосфоранилиден)амидо-
фомфорила дихлорид 599
Фосфат гидроксиламмония 506
Фосфиновая кислота 581
Фосфонитрилгалогениды 237, 596,
598
Фосфония иодид 559
Фосфоновая кислота 580
Фосфорила триамид 604
— трибромид 562
— трифторид 235
Фосфористая кислота см. Фосфоновая кислота
Фосфористый ангидрид 568 Фосфорная кислота 571 Фосфорноватая кислота см. Гипофос-форная кислота
Фосфорноватистая кислота см. Фосфиновая кислота
Фосфорноватокислый кислый натрий двузамещенный см. Дигидрогипо-фосфат динатрия
— натрий см. Гипофосфат тетранатрия
Фосфорный ангидрид 570 Фосфороксидтриамид 604 Фосфорсульфидтриамид 606 Фосфорсульфидтрихлорид см. Три-
хлорид тиофосфорила Фосфортиотриамид 606 Фториды 230, 232 Хлорид-тетрафторид 234 Элементный 547—551
Фтор
Аддукт тетрафторида сульфинила н пентафторида мышьяка 211
— фторида иода(Ш) и пиридина 195
— фторида нитрозила и фтороводорода 225
Амидофторид сульфонила 534
Бис (трифторометил) фосфан 712
Бромид-фторид карбонила 250 — сульфонила 215
Бромфторфосген см. Бромид-фторид карбонила
Гексафтородисилан 726
Гексафторотиоацетон 709
Гексафторофосфат аммония 238 — калия 239
— тетрахлорофосфония 235
Декафторид дисеры 206
Дибромид-фторид фосфора 236
Диоксид-фторид хлора 201
Диоксодифторид см. Дифторид дикислорода
Диоксодифторофосфат аммония 238 Дитиодекафторид см. Декафторид дисеры
Дифторамин 222
Дифтордиазин см. Дифторид диазота
Дифторид диазота 223
— дикислорода 198
— дисеры 204
— дисульфурила 212
— карбонила 248
— сульфонила 211
— трикислорода 198
Дифторимин см. Дифторид диазота
Дифтороангидрид имидобиссерной кислоты 535
Дифтородисульфан 203
Дифторосульфиминфторид см. Три-фторид-нитрид серы
Дифторотиокарбонил 701
Дихлорид-трифторид фосфора (V) 234
Дихлорид-фторид фосфора (III) 232
Изоцианат сульфонилфторида 534 Иминфторид см. Дифторамин Иодид-фторид карбонила 251 Иодфторфосген см. Иодид-фторид карбонила
Кислота бис (трифторометилсульфино-вая), имид 710
— трис (трифторометилсульфиновая), нитрид 711
—• трифторометилсульфиновая, амид 710
— фторосерная 216
Монофторид углерода 677
Нитрозосульфонилфторид 226
Нитроксилфторид см. Фторид-триок-сид азота
Предметный указатель 2211
Оксид-дифторид углерода 248
Оксодифторид дисульфурила 213
Оксофторид азота 224
Пентафторид хлоросульфинила 208
Пероксодисульфурилдифторид см. Оксодифторид дисульфурила
Перфторобутилсульфонилизоцианат 701
Пиросульфурилфторид см. Дифторид дисульфурила
Сульфурилбромидфторид см. Бромид-фторид сульфонила
Сульфурилдифторид см. Дифторид сульфонила
Сульфурилхлоридфторид см. Хлорид-фторид сульфонила
Тетрабромид-фторид фосфора 236
Тетрафторид диазота 222
— сульфинила 210
— углерода 245
Тетрафторид-хлорид нитрозила 190
Тетрафторогидразин см. Тетрафторид диазота
Тетрахлорид-фторид фосфора (V) 234 Тиазилфторид см. Тиофторид азота Тиокарбонилфторид см. Дифторотиокарбонил
Тиомонохлоридпентафторид см. Пентафторид хлоросульфинила
Тионилфторид см. Фторид сульфинила
Тионилфторидхлорид см. Хлорид-фто-рид сульфинила
Тиооксотетрафторид см. Тетрафторид сульфинила
Тиофторид азота 228
Триоксид-фторид хлора 202
Триоксодифторид см. Дифторид трикислорода
Трифторид фосфорила см. Трифторид-оксид фосфора
Трифторид-нитрид серы 229
Трифторид-оксид фосфора 235
Трифториодметан 247
Трифторметан 246
Трифторометилгипофторид 712
Трифторометилсульфинилизоцианат 707
Трифторометилсульфинилкарбонил-фторид 705
Трифторометилсульфинилхлорид 706
2,4,6-Трифторо-1,3,5-триазин см. Ци-анурфторид
Фосфонитрилфториды 237
Фтороангидрид амидосерной кислоты см. Амидофторид сульфонила
Фторид азота 220
— брома 190, 192
— графита см. Монофторид углерода
— дейтерия 164
— иода 193, 194
— кислорода 200
— нитрозила см. Оксофторид азота
— нитроила см. Фторид-диоксид азота
— озона см. Дифторид трикислорода
— перхлорила см. Триоксид-фторид хлора
— серы 205, 207
— сульфинила 209
— тетрауглерода 678
— фосфора 230, 232
— хлора 187, 189
— хлорила см. Диоксид-фторид хлора
— хлоротетраоксида 202
Фторид-диоксид азота 226
Фторид-триоксид азота 227
Фтороводород 185
Фторокарбонилизотиоцианат 699
Фторокарбонилизоцианат 700
Фторокарбонилсульфинилхлорид 705
Фторосульфат брома 374
— брома(Ш) 376
— иода(Ш) 379
— хлора (I) 373
Фторосульфонилизоцианат см. Изоцианат сульфонилфторида
Фторотиокарбонилизотиоциаиат 704
Фтороформ см. Трифторметан
Фтороциан 252
Фторфосген см. Оксид-дифторид углерода
Хлорид-тетрафторид фосфора (V) 234
Хлорид-фторид карбонила 249
— сульфинила 214
— сульфонила 214
— тиокарбонила 703
Хлорфторфосген см. Хлорид-фторид карбонила
Элементный 180, 183
Хлор
Гидрат хлора 326
Гипохлористая кислота см. Хлорноватистая кислота
Гипохлорит натрия 355
Диоксид 347
Диоксид-диперхлорат хрома (VI) 1626
Диоксид-фторид 201
Дихлорогексоксид 350
Дихлорогептоксид 352
-2212 Предметный указатель
Дихлоромоноксид 346
•Нитрат хлора (I) 372
Перхлорат нитрозила 365
— нитрония 366
— хлора(1) 373
— хромила 1626
Перхлорила фторид см. Триоксид-фторид
Тетрахлороиодная (Ш) кислота 346
Триоксид-фторид 202
Трихлорид азота 501
В-Трихлороборазин 863
В-Трихлор-М-триметилборазин 1975
Фторид хлора(I) 187
— хлора(Ш) 189
Фторосульфат хлора (I) 373
Хлорат аммония 359
Хлорид брома (I) 338
— нитрозила 514
— нитроила 517
Хлорид-тетрафторид калия 270
Хлорила фторид см. Диоксид-фторид хлора
Хлорит натрия 358
Хлоркарбонилизоцианат 698
Хлорная кислота 363
Хлорноватая кислота 359
Хлорноватистая кислота 355
Хлороазид 502
Хлороамин 498
Хлороводород 331
Хлоротетраоксида фторид 202
Хлорофосфазены 596
Хлороциан 689
Элементный 325
Хром
Аквапентаамминхрома(Ш) бромид 1601
— нитрат 1600
Ацетат 1614
Ацетилацетонат 1622
Бис(г]-циклопентадиенил)хром 1953
(т)4-Бисцикло [2.2.1] гептадиен-2,5) -тетракарбонилхром 1996
Бромиды 1591
Бромопентааммиихрома(Ш) бромид 1602
Бромо (пеитакарбонил) хромат(0) тетраэтиламмония 2056
транс-Бромо (тетракарбонил) (фенил-карбин)хром 2039
Бромотрис (диметиламииоэтил) ами-нохрома(П) бромид 1615
Г идридо (г] -циклопентадиеиил) три -карбонилхром 2078
Г идроксо дипероксооксохромат (VI) аммония 1630
Гексааквахрома(Ш) ацетат 1616
Гексаамминнитрат 1598 Гексаамминхлорид 1598 Гексагидроксохромат(Ш) натрия
1884
— стронция 1885
Гексакарбонилхром 1930
Г ексакис (о-толилизоцианид) хром 2103
Гекса(мочевина)хрома(Ш) хлорид 1606
Гексароданохромат(Ш) калия 1617
Гексатиоцианатохромат(Ш) калия 1617
Гексафенилизонитрилхром(О) 1610 Гексацианохромат(Ш) калия 1617 Гексафенилхромат(Ш) трилития 1618 р-Гидразин (N,N') -бис (пентакарбо-нилхром) 2130
Гидроксид 1595
Глицинат 1622
Декакарбонилхромат(—I) натрия
1934
Дибензолхром 1954
Дигидроксогексаацетатотрихро-ма(Ш) ацетат 1616
— хлорид 1616
ц-Диимин (N,N') -бис (пентакарбонил-хром) 2131
Дикарбонил (т)2-сероуглерод) (т)-ме-зитилен)хром 2099
Диоксид-динитрат 1626
Диоксид-диперхлорат 1626 Диоксодифторид 292
Дипероксотриамминхром(1У) 1630 транс-Дироданобисэтилендиаминхро-
ма(Ш) роданид 1605
Дитиохромат(III) натрия 1631
Дихлороакватриамминхрома(Ш) хлорид 1606
Дихлоро-бис[о-фенилен-бис(диметил-арсин)]хрома(Ш) перхлорат 1611
транс- Дихлоробисэтилендиаминхро-ма(Ш) хлорид 1605
цис-Дихлоробисэтилендиаминхро-ма(Ш) хлорид 1604
Дихромат (VI) рубидия 1627 — цезия 1628
Йодиды 1592—1595
Ксантогенат 1622
Лютеохромнитрат 1598
Лютеохромхлорид 1598
Предметный указатель 2213
Нитрид 1597
Металлический 1581
Молибдохромат(Ш) аммония 1901 — натрия 1902 б-Молибдохромовая(Ш) кислота 1901 Нитритопентаамминхрома(Ш) нитрат 1602
Нитрозилпентакис (ацетонитрил) хрома (II) гексафторофосфат 2088
Нитропеитаамминхрома(Ш) нитрат 1601
Оксалат 1615
Оксид-бромид 1596
Оксид-хлориды 1596, 1624
Оксид, аддукт с пиридином 1625
Пентакарбонил (N,N'-дифенилдиами-нокарбен)хром 2037
Пентакарбонил (дифенилциклопро-пенилиден)хром 2036
Пентакарбонил[метокси (органил) -карбен] хром 2034
(Пентакарбоксил)триметилсилили-зоцианидхром 2104
Пентакарбонилхромат(—II) натрия 1932
Лентакарбонил[(цианид-N)водорода] хром 2104
Пурпуреохромхлорид 1599
Рейнеке кислота 1620 — соль 1619
Роданиловой кислоты аммонийная соль 1620
Родохромхлорид 1607
Соли хрома(П), растворы 1612
Сульфат 1612
Сульфиды 1597
Тетранитрозилхром 2087
Тетрапероксохромат(V) калия 1629
Тетрароданодиамминхромат(Ш) аммония 1619
Тетратиоцианатодиамминхромат(Ш) аммония 1619
Тетрароданопиридинхромат(Ш) калия 1621
Тетрароданодиамминхромовая (III) кислота 1620
Тетратиоцианатодиамминхромовая (III) кислота 1620
Тетратиоцианатодианилинхро-мат(Ш) аммония 1620
Тетратиоцианатодипиридинхро-мат(Ш) калия 1621
Тетрахромат калия 1891
Триамминхромтетроксид 1630
Трикарбонил (гексаметилборазин) -хром 1975
Трикарбонил(т)-тиофен)хром 1974 грин-Трикарбонилтрис(ацетонит -рил)-хром 2128
Трнкарбонил (трхлорбензол) хром 1973 1р:1оксалатохромат(Ш) калия 1617
Трис(траллил)хром 1995
1 рис (2,2'-бипиридил) хром (0) 1609
Грис (2,2'-бипиридил) хрома (II) пер-
хлораты 1607—1608
Трис (2,2'-бипиридил) хромат (—I) лития, аддукт с тетрагидрофураном 1609
Трис (2,2'-бипиридил) хромат (—III) тринатрия, аддукт с тетрагидрофураном 1610
Трис (о.о'-диэтилдитиофосфат) хро-
ма(Ш) 1623
Трис (этилендиамин) хрома (111) соединения 1603
Трихромат калия 1891
Фосфат 1612
Фториды 290—292
Фторохромат(VI) калия 1628
Хлориды 1582—1590
Хлоропентаамминхрома (III) хлорид 1599
Хлорохромат (VI) калия 1629
Хромат (IV) дибария 1630
Хромат(У1) рубидия 1627
— цезия 1628
Хромат(V) трибария 1631
Хромила нитрат 1626
— перхлорат 1626
— хлорид 1624
— фторид 292
Хромоксалат калия 1617
Хромроданид калия 1617
(т]-Циклопентадиеиил) (нитрозил)-карбонил(тиокарбонил)хром 2099
(•>)-Циклопентадиеиил) иитрозилкар-бонил(т)2-циклооктен)хром 2088
(т]-Циклопентадиеиил) (тионитро-зил) дикарбонилхром 2089
Эритрохромхлорид 1607
Цезий см. также Щелочные металлы
Азид 497
Антимонид 1040
Арсенид 1039
Ацетилиды (карбиды) 1043
Висмутид 1041
Гексахлоротеллурат 472
Германид 1046
Гидрид 1025
Гидроксид 1034
Дибромоиодат(I) 344
2214 Предметный указатель
Диоксид (надпероксид) 1032
Дихлоробромат (I) 342
Дихлороиодат (I) 343
Дихромат 1628
Додекахлоротриренат (III) трицезия
1733
Иодид-тетрафторид 271
Квасцы цезий-ванадиевые 1536
— цезий-марганцовые 1691
Металлический 1009—1014, 1016—
1019
Оксид 1028
Оксопентахлороренат (V) 1730
Оксоплутонат(УН) 1407
Октабромодиренат(Ш) 1733
Пероксид 1031
Плюмбид 1047
Силицид 1046
Сульфат цезия-марганца см. Квасцы цезий-марганцовые
Станнид 1047
Тетрафтороиодат(III) 271
Трифторогерманат(И) 807
Трихлорогерманат(Н) 807
Фторид 272
ФтороплутонатСУ) 1408
Фтороуранаты(1У) 1345
Хромат 1628
Цезийгермил 784
Эннеахлородиталлат(Ш) 951
Церий см. также Редкоземельные
элементы
12-Молибдоцерат(1У) аммония 1900
— аммония-водорода 1900
12-Молибдоцериевая(1У) кислота
1900
Оксид церия(III) 1180
Фторид церия (IV) 282
Чистые соединения 1161
Цинк
Амальгама 2173
Амид 1122
Арсениды 1125
Ацетат-оксид 1128
Бромид 1117
10-Ванадат дикалия-дицинка 1897
Гидрид 1115
Гидроксид 1118
Гидроксофосфат 1125
Гидроксохлорид 1116
Гидрофосфат 1125
Дитионит 431
Диэтилцинк 1126
Иодид 1117
Карбонат 1127
Металлический 1114
Нитрид 1123
Ортосиликат 1129
Ортофосфат, тетрагидрат 1124
Ринманова зелень 1131
Селенид 1121
Сульфид 1119
Теллурид 1122
Тетрагидроксоцинкат натрия 1881
Тетраоксо диферрат ИЗО
Тетрапероксомолибдат (VI) тетрам-минцинка 1650
Тригидроксоцинкат натрия 1880 Феррит см. Тетраоксодиферрат Формальдегидсульфоксилат 1120 Фосфиды 1123
Фторид 277
Фторосиликат, гексагидрат ИЗО
Хлорид 1114
Цианид 1128
Цирконий
Ацетилацетонат 1487
Бис (т] -инденил) диметилцирконий 2023
1,1-Бис (т]-циклопентадиенил) -2,3,4,5-тетрафенилцирконий 2023
Бромиды 1446, 1449, 1452
Гексатиоцианоцирконат(IV) калия
1488
Гидрид 1425
Дихлорид-оксид 1456
Ди-л-циклопентадиенилцирко-ния(1У) бромид 1491
— дихлорид 1490
— иодид 1491
Йодиды 1446, 1449, 1453
Карбид 1482
Комплексные соединения 1485
Металлический 1419
Нитрат 1474
Нитрат-оксид 1476
Нитрид 1474
Оксид 1464
Отделение от гафния 1420
Очистка солей 1471
Силициды 1483
Сульфаты 1470
Трнацетилацетонатоциркония (IV) хлорид 1486
Фосфат 1479
Фосфиды 1476, 1478
Фториды 286
Хлориды 1446, 1450
Предметный указатель 2215
Щелочные металлы см. также Колий, Литий, Натрий, Рубидий, Цезий
Аммины, соединения с графитом
674
Германаты 806, 807
Ниобаты 1573
Оксобериллаты 965
Очистка 1014—1017
Селениды 1035
Соединения с графитом 673
Сульфиды 1035
Танталаты 1573
Теллуриды 1035
Фтороуранаты 1345
Хранение и обработка 1017—1019
Чистые 1009—1022
СОДЕРЖАНИЕ
ЧАСТЬ III. ОСОБЫЕ ГРУППЫ СОЕДИНЕНИЙ
Глава 31. Гидроксосоли. Р. Шольдер (переработано X. Шварцем) (1869).
Введение (1869). Применение концентрированных щелочей (1871). Тетрагидроксобериллат натрия (1873). Тетрагидроксобериллат стронция (1873). Тетрагидроксомагнезиат натрия (1874). Гексагидроксомагнезиат бария (1875). Тригидроксо-станнат(П) натрия (1875), Оксогидроксостаннат(П) бария (1876). Тетрагидроксоманганат(П) натрия (1876). Тетрагидроксоферрат(П) натрия (1877). Тетрагидро-ксокобальтат(П) натрия (1877). Гексагидроксоникколат(П) стронция (1879). Тетрагидроксокупрат (II) натрия (1879). Гексагидроксокупрат(П) бария (1880). Гидроксоцинкаты натрия (1880). Гексагидроксокадмиат бария (1881). Алюминаты щелочных металлов (1882). Гексагидроксохромат(Ш> натрия (1884). Гексагидроксохромат(III) стронция (1885). Гидроксоферраты(III) натрия (1885), ГидроксОферраты(Ш) бария (1887). Гексагидроксостаннат(1У) натрия (1887). Гек-сагидроксоплюмбат(1У) натрия (1888). Октагидроксоплюм-бат(1У) бария (1890).
Глава 32. Изо- и гетерополисоединения. Э. Шилль (1891).
Изополисоединения (1891).
Трихромат калия (1891). Тетрахромат калия (1891). Диванадат натрия (1892). Триванадат калия (1892). Гексаниобаг калия (1892). Метаниобат калия (1893). Гексатанталаг калия (1893). Гексатанталат натрия (1893). Октамолибдаг аммония (1894). Полиоктамолибдат аммония (1894). 12-Воль-фрамат натрия (1894). Мета-12-вольфрамат натрия (1895). 12-Вольфрамовая кислота (1896). 12-Ниобоманганат(1У) натрия (1896). 10-Ванадат дикалия-димарганца (1896). 10-Ванадат дикалия-дицинка (1897).
Гетерополисоединения (1897).
Общие методы получения (1897). 12-Молибдофосфорная кислота (1898). 12-Молибдофосфат аммония (1899). 12-Молиб-дофосфат бария (1899). 12-Молибдокремневая кислота (1899). 12-Молибдоцерат(1У) аммония (1900). 12-Молибдоцерат(1У). аммония-водорода (1900). 12-Молибдоцериевая (IV) кислота (1900). 12-Молибдоарсенат(У) калия (1901 ). 6-Молибдохро-мат(Ш) аммония (1901). б-Молибдохромовая(Ш) кислота
Содержание 2217
(1901). б-Молибдохромат(Ш) натрия (1902). 6-Молибдоро-дат(Ш) аммония (1902). б-Молибдоникколат(И) аммония (1902). 6-Молибдоиодат натрия (1903). 6-Молибдоиодная кислота (1903). 6-Молибдоалюминат натрия (1903). 6-Молибдо-феррат(Ш) аммония (1903). б-Молибдокобальтат(Ш) натрия (1904). 18-Молибдодифосфат аммония (1904). 18-Молибдоди-фосфорная кислота (1905). 18-Молибдодиарсенат(V) натрия (1905). 18-Молибдодимышьяковая(У) кислота (1905). 9-Молиб-доманганатЦУ) аммония (1905). 9-Молибдомарганцевая(1У) кислота (1906). 9-Молибдоникколат(1У) аммония (1906). Ю-Молибдодикобальтат(Ш) аммония (1906). 11-Молибдо-1-ванадофосфорная кислота (1907). 10-Молибдо-2-ванадофосфор-ная кислота (1907). 9-Молибдо-З-ванадофосфорная кислота (1907). 12-Ванадофосфат аммония (1908). 12-Ванадофосфат серебра (1908) . 13-Ванадоманганат(1У) калия (1908). 12-Вольфрамокремневая кислота (1909). 12-Вольфрамосиликат калия (1909). 12-Вольфрамофосфорная кислота (1910). 12-Вольфрамофосфат натрия (1910). 12-Вольфрамоборная кислота (1910). 12-Вольфрамоборат натрия (1911). 18-Вольфра-модифосфат аммония (1912). 18-Вольфрамодиарсенат(У) аммония (1912).
Глава 33. Металлоорганические комплексные соединения. В. П. Фельхаммер, В. А. Херманн, К- Офеле (1913).
Предварительные замечания (1913).
Общая техника работы (1914).
Исходные соединения (1923).
Натриевый порошок (1923). Циклопентадиенилнатрий (1924). Циклопентадиенилталлий (1925). Дициклопентадиенилмагний (1926). т)1-Циклопентадиенил(три-н-бутил)олово (1927).
Карбонилы металлов (1928).
Гексакарбонилванадий (1928). Гексакарбонилванадат(—I) [бис{ди(2-метоксиэтиловый эфир}]натрия (1929). Гексакарбо-нилхром (1930). Пентакарбонилхромат(П) натрия (1932). Декакарбонилхромат(—I), -молибдат(—I) и -вольфрамат(—I) натрия (1934). Гексакарбонилмолибден (1935). Гексакарбонил-вольфрам (1936). Декакарбонилдимарганец (1937). Тетрахлороалюминат гексакарбонилмарганца(1) (1938). Декакарбонилдирений (1939). Эннеакарбонилдижелезо (1940). Додекакарбонилтрижелезо (1941). Тетракарбонилферрат(—II) натрия (1942). Октакарбонилдиферрат(—I) натрия (1943). Додекакарбонилтрирутений (1944). Додекакарбонилтриосмий (1945). Октакарбонилдикобальт (1946). Додекакарбонилтетракобальт (1948). Тетракарбонилкобальтат(—I) таллия (1948). Бис (тетракарбонилкобальтат) тетракис (пиридин) магния
(1949). Додекакарбонилтетрародий (1950). Гексадекакарбонилгексародий (1951).
Ароматические сандвичевые комплексы (1952).
Бис (т]-циклопентадиенил) ванадий (1952). (т]-Циклогептатриенил) (т]-циклопентадиенил)ванадий (1952). Бис(т)-циклопен-тадиенил)хром (1953). Дибензолхром (1954). Бис (циклопентадиенил) марганец (1955). Бис (т]-циклоп ентадиенил) железо (1956). Ферроцен как представитель ароматических углеводородов (1956). 1,1'-Диацетилферроцен (1957). Формилферроцен
2218
Содержание
(1958). Гексафторофосфат бис (трциклопентадиенил) желе-за(Ш), тетрафтороборат (трацетилциклопентадиенил) (трциклопентадиенил) железа (III) (1958). Гексафторофосфат (трцик-лопентадиенил) (трбеизол) железа (00) (1960). Бис(т]-циклопен-тадиенил) рутений (1961). Бис (трциклопентадиенил) кобальт (1961). Гексафторофосфат бис (трциклопентадиенил) кобальта^!!) (1962). Бис (т]-циклопентадиеиил) никель (1962). Тетрафтороборат трис(т]-циклопентадиенил)диникеля(П) (1963).
Ароматические карбонилы металлов (1964)
Бис (г]-циклопентадиеиил) дикарбонилтитан (1964). (трЦикло-пентадиенил)тетракарбонилванадий (1965). (т) - Ацетилцикло-
пентадиенил) тетракарбонилванадий (1966). Д-ц-карбонилтри-карбонилбис[ (трциклопентадиенил)ванадий] (1967). Трикарбонил (трциклогептатриенил)ванадий (1968). Гексафторофосфат тетракарбонил (трмезитилен) ванадия (1969). Тетракарбонил (трциклопентадиенил) ниобий (1969). Гексакарбо-нил-ц3т]3Ст)2О) -карбонилтрис[ (трциклопентадиенил) ниобий] (1971).
Комплексы типа М(трароматический лиганд) (1972).
Трикарбонил (тр хлорбензол) хром (1973). Трикарбонил (трто-луол) молибден (1973). Трикарбонил (тртиофен) хром (1974). Трикарбонил (гексаметилборазин) хром (1975). Бис[трикарбо-нил (трциклопентадиенил) молибден] (1976). Бис[дикарбонил(тр циклопентадиенил) молибден] (1977). Тетрафтороборат трикарбонил (трциклогептатриенил) молибдена (0) (1978). Трикарбонил (трциклопентадиенил) марганец (1978). Трикарбоиил(трпен-табромциклопентадиенил) марганец (1980). Трикарбонил(трпир-ролил) марганец (1981). Трикарбонил (трциклопентадиенил) рений (1981). Трикарбонил (тр галогенциклопента диенил) реиий (1982). Бис[дикарбонил (трциклопентадиенил) железо] (1983). Тетракис[карбонил (трциклопентадиенил) железо] (1984). Гексафторофосфат трикарбонил (трциклопентадиенил) железа (II) (1985). Бис[дикарбонил (трциклопентадиенил) рутений] (1986). Г ексафторофосфат трикарбонил (трциклопентадиенил) руте-
ния (II) (1987). Дикарбонил (трциклопентадиенил) кобальт (1987). Дикарбонил (трциклопентадиенил) родий (1989). ц-Кар-бонилбис[карбонил(трциклопентадиенил)родий] (1989). Дикарбонил (трпентаметилциклопентадиенил) родий (1990). (р-Карбонил) бис[карбоиил (трпентаметилциклопентадиенил)родий] (1991). Бис[(р,-карбонил) (трпентаметилциклопентадиенил) родий] (1992). [у,-Диоксид серы(8)]бис[карбонил(трциклопента-диенил)родий] (1993). Дикарбонил (трциклопентадиенил)ири-^(1994) (1993)’ Бис[(х-карбонил(трциклопентадиенил)никель)
Олефиновые, ацетиленовые и аллильные комплексы (1995).
Трис (т)1аллил) хром (1995). (т]4-Бицикло[2.2.1]гептадиен-2,5)тет-ракарбонилхром, (т]4-бицикло[2.2.1]гептадие-2,5) тетракарбо-нилмолибден (1996). Трикарбонил (т]6-циклогептатриен) молибден (1997). (т]‘-Аллил)пентакарбонилмарганец (1997). (трАл-лил)тетракарбонилмаргаиец (1998). т)6-Циклогептатриен(трциклопентадиенил) марганец (1998). Дикарбонил (т]2-циклоок-тен) (трциклопентадиенил)марганец (1999). Трикарбонил(тр циклобутадиен)железо (2001). трБутадиен(трикарбоиил)желе-
Содержание 2219
зо (2002). Бис(трбутадиен)карбонилжелезо (2002). Трикарбонил (т)4-циклогептатриеи) железо (2003). т]-Аллил (трикарбонил) хлорожелезо (2003). трАллил (трикарбонил) броможелезо (2004). трАллил (трикарбонил) иодожелезо (2004). Бис[т]-ал-лил (трикарбонил) железо] (2005). Тетрафтороборат трикарбонил (т]б-циклогептадиенил) железа (2006). Дикарбонил (т]®-цик-логептадиенил) иодожелезо (2006). (т]-Аллил)трикарбонилко-бальт (2007). Гексакарбонил(ц, трдифенилацетилен) дикобальт (2007). р-Дихлоротетракис (т]2-циклооктен) диродий (2008). Бис[ц-хлоро-{ 1,2:5,6-тр (циклооктадиен-1,5) }родий] (2008).
Бис[1,2:5,6-т]-(циклооктадиен-1,5) ]никель (2009). Бис[ц-бро-мо(т]-аллил)никель] (2011). Бис[ц-метил(т]-аллил) никель], бис-(траллил) никель, траллил (метил) (трифеиилфосфин) никель
(2012). (1,2:5,6:9,10-т\-транс, транс, транс-циклододекатриен)-
никель (2014). Дихлоро (1,2:5,6-трциклооктадиен) палладий (2015). Бис[1,2:5,6-тр(циклооктадиен-1,5)]палладий (2016). Моногидрат трихлоро(трэтилен)платинзта(П) калия (2017). г)-Этиленбис (трифеиилфосфин) платина (,2018). трДифенилаце-тиленбис(трифеиилфосфин)платина (2019). Дихлоро [1,2:5,6-тр (циклооктадиен-1,5)]платина(2019). Бис[1,2:5,6-тр (циклооктадиен-1, 5)]платина (2020).
Алкильные и ацетильные комплексы (2022).
Бис (трциклопентадиенил) диметилтитан (2022). Бис(т]®-инде-нил)диметилтитан, -цирконий, -гафиий (2023). 1,1-Бис(трциклопентадиенил) -2,3,4,5-тетрафенилметаллы (2023). Пентаметил-тантал (2024). Трикарбоиил (трциклопентадиенил) метилмолиб-ден и -вольфрам (2025). Трикарбонил (т]®-ииденил)метилмолиб-ден и -вольфрам (2026). Гексаметилвольфрам (2027). Пентакарбонил (трифторацетил) марганец (2029). Пентакарбонил (трифторметил) марганец (2029). Дикарбонил(трциклопен-тадиеиил) метилжелезо (2030). Тетракарбонил(перфтор-н-про-пил) иодожелезо (2031). трГалогенциклопентадиенильные комплексы железа (2031). Тетракис[р.3-иодо(триметил)платина] (2032). Метил (трифеиилфосфин) золото (2033). Триметил (трифенилфосфин) золото (2034).
Карбиновые и карбеновые комплексы (2034).
Пентакарбонил[метокси(органил)карбен]хром и пентакарбо-нил[метокси(органил)карбен]вольфрам (2034). Пентакарбонил (дифенилциклопропенилиден) хром и пентакарбонил (дифенилциклопропенилиден) молибден (2036). Пентакарбонил-(КМ'-дифенилдиаминокарбен) хром (2037). Пентакарбонил-(дифенилкарбен) вольфрам (2037). трснс-Бромо(тетракарбо-нил) (фенилкарбин)хром и трскс-бромо(тетракарбонил) (фенил-карбин) вольфрам (2039). Дикарбонил (трциклопентадиенил) -(дифенилкарбен) марганец (2039). Карбамоил(дикарбонил)-(т]-циклопентадиеиил) рутений (2041). р,3-Хлорметилидин-
т{икло-триг(трикарбонилкобальт) (2041). р,-Метиленбис[кар-бонил (трциклопентадиенил) родий] (2042). Тетрафтороборат [ц-дикарбонил-ц3-метилидин-ч«кло-трис{ (трциклопентадиенил) родия} (2043). Бис[ц-хлоро-хлоро (дифенилциклопропенили-
ден) палладий] (2043). Иодид тетракис(оксазолидин-2-илиден)-палладия (II) (2045). чис-Дихлоро[этокси (фениламино) карбен]-(триэтилфосфин) платина (2046). трснс[Хлоро{(я-толилимино)-метил}бис(тригтилфосфин) платина] (2047). Трис[1-циклогекси-
2220
Содержание
лимино (метокси) метилзолото] (2048), Тетракис (1-изопропил-тетразол-5-ато)аурат(Ш) тетрафениларсония (2049).
Комплексы с галогеновыми лигандами (2050).
Дихлоробис(т]-циклопентадиенил)титан (2050). Трихлоро(т]-циклопентадиенил)титан (2051). Дихлоробис(т)-циклопентадие-нил)ниобий (2051). Тетрахлоро (трциклопентадиенил) ниобий, тетрахлоро (q-циклопентадиенил) тантал (2055). Бромо (пентакарбонил) хромат (0) тетраэтиламмония, хлоро (пентакарбонил) вольфрамат (0) тетраэтиламмония (2056). Хлоро (т]-циклопентадиенил) трикарбонилмолибден, хлоро (трциклопентадие-нил)трикарбонилвольфрам (2057). . Трихлоро (трциклопента-диенил)дикарбонилмолибден (2058). Иододикарбонил (трцикло-гептатриенил) молибден (2059). Хлоро(пентакарбонил)марганец (2059). Бромо (пентакарбонил) марганец (2060). Иодо (пентакарбонил) марганец (2060). Хлоро (пентакарбонил) рений (2061). Бромо (пентакарбонил) рений (2061). Иодо (пентакарбонил) рений (2062). Бис[р-хлоро(тетракарбонил)рений] (2062). Галоге-но(т]-циклопентадиенил)дикарбонилжелезо (2063). Иодо-(т)-иодцнклопентадиенил)дикарбонилжелезо (2064). Дигалоге-но (тетракарбонил) железо (2065). Дииодотетракарбонилруте-ний (2067). Бис[ц-хлоро (хлоро) трикарбонилрутений] (2067). Хлоро(т]-циклопентадиенил)дикарбонилрутений (2068). Карбо-нилхлориды осмия (2068). Дииодо (т]-циклопентадиенил) карбо-нилкобальт (2069). Бис(ц-хлородикарбонилродий) (2070). Ди(р, - хлоро)бис[хлоро(т)-пентаметилциклопентадиенил)родий] (2071). Хлоротрикарбонилиридий (2071). Ди(р-хлоро)бис[хло-ро(т)-пентаметилциклопентадиенил)иридий] (2073). чис-Дихло-родикарбонилплатина (2074). Хлоро(карбонил)золото (2075).
Гидридные комплексы (2075).
Гидридогексакис (трифторфосфин) ванадий (2075). Тригидри-добис(т)-циклопентадиенил) ниобий (2077). Гидридо(т]-циклопентадиенил) трикарбонилхром, -молибден и -вольфрам (2078). Дигидридобис (т] -циклопентадиенил) молибден, дигидридобис-(т]-циклопентадиенил) вольфрам (2080). Гидридопентакарбонил-марганец (2081). Трис[(ц-гидридо)тетракарбонилмарганец) (2082). Гидридобис(т]-циклопентадиенил)рений (2083). Хлоро-(гидридо) трис (трифенилфосфин) карбонилрутений (2083). Хлоро (гидридо) трис (трифенилфосфин) карбонилосмий (2084). Тетрагидридотрис (трифенилфосфин) осмий (2084). Гидридотетракис (трифенилфосфит) кобальт (2085). Гексафторофосфат гидридо (я-ииклопентадиенил) бис (триметилфосфин) коб а л ь-та(Ш) (2085). Гидридотрис(трифенилфосфин)карбонилродий (2087)" гРанс'Хлоро (гидридо) бис (трифенилфосфин) платина
Нитрозильные комплексы (2087).
Тетранитрознлхром (2087). Гексафторофосфат нитрозилпента-кис(ацетонитрил)хрома(П) (2088). (трЦиклопентадиенил)-нитрозилкарбонил(т]2-циклооктен)хром (2088). (т)-Циклопента-диенил) (тионитрозил)дикарбонилхром (2089). (т)-Циклопента-диенил) (нитрозил)дикарбонилмолибден (2090). Нитрозил(тет-ракарбонил) марганец (2091). Тетрафтороборат (ц-циклопента-диенил) (нитрозил)дикарбонилмарганца(П) (2092). ц-1,2-Бис-(дифенилфосфино) этанбис (триннтрозилмарганец) (2092).
Содержание 222Ь
Бис[ (ц-нитрозил) (т]-циклопентадиенил)железо] (2093). Диии-трозилдикарбонилжелезо (2093). ДинитрозилбшЦтрифенилфос-фин) рутений (2094). Нитрозилтрикарбонилкобальт (2095). Бис[(ц-иитрозил) (трциклопентадиеиил) кобальт] (2096). Нит-розилтрис (трифенилфосфин) родий (2097). Дихлоронитрозил-бис (трифенилфосфин) иридий (2098). (т]-Циклопентадиенил)-нитрозилникель (2098).
Дитиокарбонильные и тиокарбонильные комплексы (2099).
Дикарбонил (т]2-сероуглерод) (т]-мезитилен)хром (2099).
(т]-Циклопентадиенил) (нитрозил) карбонил (тиокарбонил) хром (2099). (т]-Циклопентадиенил) дикарбонил (тиокарбонил) марганец (2100). (трЦиклопентадиенил) (дикарбонил) (дитиометоксикарбонил) железо (2101). Гексафторофосфат (т]-цикло-пентадиенил)дикарбонил(тиокарбонил)железа(П) (2101). Бис-(трифенилфосфин) (дикарбонил) (т]2-сероуглерод)железо (2102). Бис (трифенилфосфин) (т]2-сероуглерод) платина (2103).
Цианокомплексы (2003).
Гексакис (о-толилизоцианид) хром (2103). (Пентакарбонил)-триметилсилилизоцианидхром (2104). Пентакарбонил(цианид-N. водорода)хром (2104). Пентакарбонил (фенилизоцианид) хром (2105). Циано (пентакарбоиил) вольфрамат (0) натрия и -тетраэтиламмония (2106). Циано (т]-циклопентадиенил) (нитрозил) -карбонилмарганец (2107). Циано(т]-циклопентадиенил)дикар-бонилманганатЦ) натрия (2108). Тетрафтороборат гексакис-(4-изоциано-4-метилпентанон-2)железа (II) (2108). Гексафторофосфат (т]-циклопентадиенил) (дикарбонил) (фенилизоцианид) -железа(II) (2109). Гексафторофосфат (т]-циклопентадиенил)-(карбонил) бис (фенилизоцианид) железа (II) (2110). Хлорид (трциклопентадиенил) трис (фенилизоцианид) железа (II) (2111). ГексацианодиникколатЦ) калия, тетрацианоникколат(О) калия (2111). транс-Дифульминатобис (трифенилфосфин) платина-(2113).
Фосфиновые комплексы (2113).
цис-Бнс(фенилфосфин)тетракарбонилмолибден (2113). Пеита-кис (трифторофосфин) железо (2114). Гексафторофосфат (т]-циклопент адиенил) {1,2-бис (дифенилфосфино)этан}карбоиил-железа(П) (2114). Дихлоробис (трифенилфосфин) дикарбоиил-рутений (2115). Бис (трифенилфосфин) трикарбонилрутеиий-(2115). Тетракис (триметилфосфин) кобальт (2116). Хлоротрис-(триметилфосфин) кобальт (2117). (т]-Циклопентадиенил)бис-(триметилфосфин) кобальт (2117). Хлоротрис(трифенилфос-фин)родий (2117). Хлоробис(триорганилфосфин)карбонили-ридий, хлоробис(трифенилфосфит)карбонилиридий (2118). Тетракис (трифторофосфин) никель (2119). Тетракис (триметил-фосфит)никель (2120). Тетракис (трифенилфосфит) никель (2121). Тетракис (трифенилфосфин) палладий (2121). Дихлоробис (трифенилфосфин) палладий (2122). Ди-ц-хлоро-cf-дихлоробис (три-н-бутилфосфин)дипалладий (2122). Тетрафторобораты, ди-ц-хлоротетракис(трифенилфосфин)диплатины (II) и -дипал-ладия(П) (2123). ^пс-Дихлоробис (трифенилфосфин) платина (2124). Тетракис (трифенилфосфин) платина (2124). Трис(три-фенилфосфин) платина (2125). чпс-Дихлоробис (трифенилфосфит) платина (2125). Хлоро[трифенилфосфито(С, Р)]трифенил-
2222
Содержание
фосфит) платина (2126). Хлоро (трйфенилфосфин) золото
(2127). Бромо (трифенилфосфин) золото (2127).
Комплексы с кислород-, азот- и серусодержащими лигандами (2128). грон-Трикарбонилтрис(ацетонитрил)хром, -молибден и -вольфрам (2128). ц-Гидразин(М,М')-бис(пентакарбонилхром) (2130). ц-Диимин(М, М')бис(пентакарбонилхром) (2131). Мо-носульфан (пентакарбонил) вольфрам (2132). (трЦиклопента-диенил) дикарбонил (метандиазо) молибден, (т]-циклопентадие-нил) дикарбо нил (метандиазо) вольфрам (2132). Бис[ц-бромо-тетрагидрофуран)трикарбонилрений] (2133). Тетракис(цз-гид-роксотрикарбонилреиий) (2134). Гексафторофосфат (т]-цикло-пентадиенил) (дикарбонил) (ацетонитрил) железа (II) (2135). Гексафторофосфат ц-диазот (N,NZ) бис[ (q-циклопентадиенил) -{1,2-бис(дифенилфосфиио)этан}железа(П)] (2135). (Метилсуль-финато-S) (г]-циклопентадиенил) дикарбонилжелезо (2136). Гидридо(т)1-диазот)трис(трифенилфосфин)кобальт (2137). Тетракис (тетрагидрофуран) магнийбис[у.-диазоттрис (триметилфос-фин)кобальт] (2138). Хлоро (т]-дикислород) бис (трифенилфосфин) карбонилиридий (2139). у., т)2-Диазотникельлитиевый комплекс (2139). транс-Дихлоробис (бензонитрил) палладий (2140). (г]-Дикислород) бис (трифенилфосфин) платина (2140).
Глава 34. Сплавы и интерметаллические соединения. Г. Брауэр (2142).
Синтезы при высоких температурах (2143).
Чистота исходного материала (2143). Форма исходного материала (2144). Навеска (2144). Тигельный и ампульный методы (2145). Нагревание и охлаждение (2154). Синтезы при высоких температурах и давлениях (2156). Бестигельное плавление (2157).
Измельчение материала без доступа воздуха (2158) Метод перегонки (2161).
Метод получения остатков после растворения (2162).
Метод использования металла-растворителя (2163).
Специальные методы (2164).
Бориды и силициды (2166).
Амальгамы (2168).
Жидкая амальгама натрия (2170). Твердая амальгама натрия (2170). Амальгама стронция (2172). Амальгама бария (2173). Амальгамы цинка, кадмия, олова, свинца и висмута (жидкие) (2173). Сплавы калия с натрием (жидкие) (2174). Легкоплавкие сплавы (2176).
•Формульный указатель (2177). Предметный указатель (2183).
содержание других томов
Том 1
Часть I. Методы препаративной химии
Часть II. Элементы и соединения
Глава 1. Водород, дейтерий, вода
Глава 2. Пероксид водорода
Глава 3. Фтор
Том 2
Глава 4. Хлор, бром, иод
Глава 5. Кислород, озои
Глава 6. Сера, селен, теллур
Глава 7. Азот
Глава 8. Фосфор
Глава 9. Мышьяк, сурьма, висмут
Том 3
Глава 10. Углерод
Глава 11. Кремний, германий
Глава 12, Олово, свинец
Глава 13, Бор
Глава 14. Алюминий
Глава 15, Галлий, индий, таллий
Глава 16. Щелочноземельные металлы»
Глава 17, Щелочные металлы
Том 4
Глава 18. Медь, серебро, золото
Глава 19. Цинк, кадмий, ртуть
Глава 20. Редкоземельные металлы
Глава 21. Актиноиды
Глава 22. Титан, цирконий, гафний
Том 5
Глава 23. Ванадий, ниобий, тантал
Глава 24. Хром, молибден, вольфраи>
Глава 25. Марганец
Глава 26. Технеций
Глава 27. Рений
Глава 28. Железо
Глава 29. Кобальт, никель
Глава 30. Платиновые металлы