/



































































































































































Text
РУКОВОДСТВО по
НЕОРГАНИЧЕСКОМУ
СИНТЕЗУ
Редактор Г. Брауэр
В шести томах
Москва«Мир» 1985
РУКОВОДСТВО по
НЕОРГАНИЧЕСКОМУ
СИНТЕЗУ
Редактор Г. Брауэр
Том 1
Перевод с немецкого
канд. хнм. наук Т. И. Почкаевой
канд. хим. наук. С. И. Троянона
Москва «Мир» 19 85
ББК 24.1
Р85
УДК 542
Handbuch der Praparativen
Anorganischen Chemie
in drei Banden
Herausgegeben von Georg Brauer
Erster Band
Unter Mitarbeit von
M. Baudler, G. Brauer, F. Feher,
F. Huber, R. Klement,
W. Kwasnik, P. W. Schenk, M. Schmeis-
ser, R. Steudel
R. Steudel
Dritte, umgearbeitete Auflage
Ferdinand Enke Verlag Stuttgart 1975
M. Баудлер., Г. Брауэр, Ф. Губер, В. Квасник, П. В. Шенк,
М. Шмайсер, Р. Штойдель
Р 85 Руководство по неорганическому синтезу: В 6-ти томах.
Т. 1. Пер. с нем./Под ред. Г. Брауэра. — М.: Мир, 1985.—
320 с., ил.
В книге коллектива авторов из ФРГ, представляющей, по существу, энцик-
лопедию неорганического синтеза, приведены методики получения более 3000 пре-
паратов. Книга выходит в 6-ти томах. 1-й том содержит описание общих при-
емов и методов работы, а также синтезов соединений водорода, дейтерия и
фтора, и представляет собой перевод первой части и гл. 1—3 1-го тома ориги-
нального издания.
Предназначена для специалистов в самых различных областях науки и
техники, а также для преподавателей и студентов химических вузов.
1802000000-090
Р---------------св. пл. подписных изданий 1985 г.
041(01)-85
ББК 24.1
540
кция литературы по химии
© 1954, 3. Auflage 1975, Ferdinand Enke
Verlag.
© Перевод на русский язык, «Мир», 1985
ПРЕДИСЛОВИЕ К ПЕРЕВОДУ
За время, прошедшее после выхода 1-го издания «Руководства по препа-
ративной неорганической химви» (1954 г.) и его перевода на русский язык
(1956 г.), в синтетической неорганической химии были достигнуты значи-
тельные успехи. Это касается как развития техники экспериментальной ра-
боты, так и расширения «ассортимента» веществ, получаемых в лаборатор-
ных условиях. К сожалению, эти достижения не нашли должного отраже-
ния в справочной литературе на русском языке. Справочник Ю. В. Карякина
и И. И. Ангелова* содержит описание методов синтеза в основном легко-
доступных препаратов, выпускаемых нашей промышленностью. Пособие
Н. Г. Ключникова «Неорганический синтез»**, весьма ценное с методической
точки зрения, из-за ограниченности объема дает лишь основы для освоения
методов синтеза прн весьма скудном фактическом материале (описан син-
тез примерно 300 препаратов).
Тем больший интерес для советского читателя должен представить пе-
ревод на русский язык 3-го издания «Руководства», содержащего описание
синтеза соединений большинства элементов периодической системы (всего
более 3000 препаратов). Третье издание вышло на немецком языке в трех
томах в 1975, 1978 н 1981 гг. с охватом литературы по 1974, 1977 и 1980 г.
соответственно. Таким образом, успехи препаративной неорганической химии
за 30 лет нашли в этом издании если не исчерпывающее, то, во всяком
случае, весьма полное отражение. Так, в вводной части, касающейся общих
приемов и методов работы, значительное внимание уделено разнообразным,
в том числе новым, материалам, используемым в современной лаборатории.
Описана техника работы с различными газами, вакуумом, высокими и низ-
кими температурами, включая способы их поддержания и измерения. Зна-
чительное место отведено подробному изложению приемов работы с лег-
когидролнзующимися и окисляющимися веществами.
Увеличение объема «Руководства» в 3-м издании произошло в основ-
ном за счет включения синтезов новых препаратов. Это, с одной стороны,
соединения таких элементов, химия которых интенсивно развивалась в пе-
риод между выходом 1-го и 3 го изданий: лантаноидов (а также иттрия и
* Карякин Ю. В., Ангелов И. И. Чистые химические вещества.—М.: Хи-
мия, 1974.
** Ключников Н. Г. Неорганический синтез. — М.: Просвещение, 1971.
6
Предисловие к переводу
скандия), технеция, актиноидов и др. Способы синтеза многих бинарных
соединений из простых веществ получили распространение в связи с тем,
что сами простые вещества в чистом виде (например, редкие, рассеянные и
радиоактивные элементы) стали гораздо доступнее благодаря прогрессу
химической технологии и металлургии. По этой же причине в 3-м издании
существенно сокращено, а иногда и опущено описание методов вскрытия
руд, разделения близких по свойствам элементов, способов получения ме-
таллов.
С другой стороны, в химии элементов, получение которых в чистом
виде и ранее не представляло трудностей, за истекший период были сде-
ланы открытия, приведшие к синтезу соединений,- относящихся к новым
классам, например галогенидных и карбонильных кластеров, соединений
инертных газов н т. д. Кроме того, многие соединения стали сравнительно
легкодоступны именно благодаря развитию техники экспериментальной ра-
боты. Так, синтез многих безводных солей, например галогенидов и метал-
лоорганических соединений, требует применения высокого вакуума, тщатель-
ного соблюдения мер предосторожности, исключающих доступ воздуха и
влаги.
Несомненным достоинством настоящего издания «Руководства» является
более точная количественная характеристика препаратов по их физическим
свойствам. Для большинства соединений приведены температуры плавления
н кипения, плотность, а для кристаллических веществ также рентгенографи-
ческие данные: параметры решетки, пространственная группа симметрии. Это
дает возможность проводить надежный контроль индивидуальности и чи-
стоты полученных препаратов.
В связи с тем, что «Руководство» было написано большим коллективом
авторов, качество переработки материала отдельных разделов при подготов-
ке 3-го издания оказалось различным. В ряде случаев выбор метода син-
теза препаратов был сделан на основании устаревших литературных данных.
В методиках некоторых синтезов имеются указания, противоречащие дейст-
вующим в СССР правилам техники безопасности (например, использование
вакуумных эксикаторов, наполненных концентрированной серной кислотой,
нагревание горелкой больших вакуумированных объемов и др.). Подобные
места снабжены соответствующими примечаниями. По возможности устра-
нены также некоторые опечатки и неточности в цифровом материале.
«Руководство по неорганическому синтезу» может оказаться полезным
весьма широкому кругу лиц, работающих экспериментально в различных
областях науки и техники. Необходимость пользования таким справочником
возникает прежде всего в любой химической лаборатории (заводской или
научно-исследовательской), в которой синтез неорганических соединений мо-
жет осуществляться с самыми разнообразными целями — для исследования
свойств, получения промежуточных соединений для других синтезов, для
проведения химического анализа и т. д. Далее, потребность в веществе, ко-
торое либо не выпускается промышленностью, либо выпускается, но недо-
статочной степени чистоты, может появиться при проведении эксперимен-
Предисловие к переводу. •
тальной работы и специалистами в других областях науки и практики — фи-
зиками, геологами, ювелирами и др. В этом случае «Руководство» может
оказаться незаменимым как раз в связи с тем, что описание синтеза обыч-
но приводится настолько обстоятельно, что в значительной степени освобож-
дает от необходимости знакомства с оригинальной литературой. Наконец, не
в последнюю очередь это — важное пособие для обучения химиков навыкам
экспериментальной работы. Преподаватель, имея богатый выбор методик,
всегда сможет подобрать для студента синтез, оптимально отвечающий по-
ставленной цели: изучению химии отдельных элементов, отработке опреде-
ленного приема работы и т. д.
Перевод 3-го издания «Руководства по неорганическому синтезу» вы-
полнен коллективом переводчиков, в основном сотрудников химического фа-
культета Московского государственного университета им. М. В. Ломоносова.
С. Троянов
ПРЕДИСЛОВИЕ РЕДАКТОРА 3-го ИЗДАНИЯ
Предыдущие два издания «Руководства по препаративной неорганиче-
ской химии» получили признание во всем мире. Многие химики из разных
стран в той или иной форме выразили свою поддержку принципам, поло-
женным в основу настоящего труда. Это руководство должно, по нашему
мнению, помочь при проведении в условиях химической лаборатории синте-
за многих неорганических соединений, будь то для научно-исследовательских
целей или при обучении студентов. Описание подготовки и проведения синте-
за отдельных препаратов, за некоторыми исключениями, приведено в до-
статочно подробном виде, так что не требуется обращаться к оригинальной
литературе. Значительная часть прописей проверена на воспроизводимость.
При отборе препаратов, подлежащих включению в руководство, мы ставил»
своей целью, отнюдь не претендуя на исчерпывающую полноту, охватить
значительное число соединений, интересных как с научной точки зрения,
так и в методическом плане. Напротив, исключены из рассмотрения веще-
ства, имеющиеся в продаже в достаточно чистом виде, или такие, получе-
ние которых можно осуществить с применением простейших лабораторных
приемов, а также вещества, представляющие узко специальный интерес.
Исходя из этих принципов, в настоящем издании по сравнению с пре-
дыдущим круг объектов довольно существенно изменен в основном с уче-
том последних достижений неорганической химии.
Редактор весьма благодарен многим коллегам, которые, не будучи ав-
торами труда, внесли в него существенный вклад, прислав свои критиче-
ские замечания, советы и пожелания, а иногда и готовые прописи, основан-
ные на нх собственном опыте.
Значительную помощь при редакционной переработке некоторых разделов
оказала доктор Б. Даммайер (Тюбинген). Мы также благодарны Б. Вила-
редт (Тенинген) за тщательное изготовление иллюстраций.
Г. Брауэр
Принятые обозначения
ДЯ’обр — энтальпия образования; d — относительная плотность; р — давление; ркрнт —
критическое давление; М — молекулярная масса; /возг — температура возгонки; /В€п —
температура вспышки; <затв — температура затвердевания; /кип — темпера! ура кипения;
Афит — критическая температура; <пл — температура плавления: /разл — температура
разложения; tCT — температура стеклования.
Часть I. Методы препаративной химии
П. В. Шенк, Р. Штойдель, Г. Брауэр
(Р. W. Schenk, R. Steudel, G. Brauer)
Перевод канд. хим. наук С. И. Троянова
В этой части рассмотрены некоторые общие принципы и методы синтеза
неорганических веществ. Мы не ставим перед собой цель дать исчерпыва-
ющий перечень всех возможных приемов, используемых в препаративной
работе. Для этого понадобилось бы значительно увеличить объем данной
главы. Кроме того, эти вопросы рассматриваются и в других книгах [1—10],
в которых можно найти описание большого числа методов, нх аппаратурного
оформления, а также обширную библиографию. Подробное описание техни-
ки лабораторных работ содержится в трехтомном руководстве «Методы ор-
ганической химии» [И]. Описание новых разработок и материалов, ис-
пользуемых в лабораторной технике, приводится в периодической литерату-
ре (например, в журналах Angewandte Chemie, Journal of Chemical Educa-
tion), в том числе в журналах, содержащих подробную и большей частью
объективную рекламу продукции различных фирм (например, LABO, Кеп-
nziffer-Zeitschrift fur Labortechnik, Darmstadt). Благодаря хорошо постав-
ленной информационной службе можно достаточно быстро наладить кон-
такты с фирмой-производителем.
' В настоящей главе рассмотрены лишь наиболее важные приемы и ме-
тоды работы, используемые химиками-синтетиками. При отборе материала
авторы руководствовались следующими основными положениями. С ростом
требований к чистоте веществ и в связи с необходимостью проведения син-
тезов в особых условиях в последние десятилетия значительно возросли тре-
бования к технике эксперимента. Так, при получении неустойчивых или
чувствительных к действию воздуха веществ обычные фарфоровые чашки,
стеклянные стаканы, перегонные колбы приходится заменять другой более
или менее сложной аппаратурой. Нередко на основании требований, необхо-
димых в отдельных, частных случаях, разрабатывались методы работы, при-
годные во многих аналогичных ситуациях и поэтому нашедшие более ши-
рокое применение. В этой главе сделана попытка объединить подобные
стандартные методики работы, выбранные из последующих глав этой книги.
В тех случаях, когда из-за недостатка места приходилось опускать подроб-
ности и ограничиваться лишь общей схемой, даны ссылки на соответствую-
щую оригинальную литературу. Кроме того, авторы попытались наряду с
известными, наиболее часто используемыми приборами описать ряд экспе- -
риментальных приемов, отражающих опыт, который с течением времени
накапливается в каждой крупной лаборатории, но не всегда находит от-
ражение в литературе.
ЛИТЕРАТУРА
1. Anger er Е., Technische Kunstgriffe
bei physikalischen Untersuchun-
gen. Friedr. Vieweg & Sohn,
Braunschweig, 1966. [Есть рус-
ский перевод 12-го нем. изд.: Ан-
герер Э. Техника физического экс-
2-143
Ю Методы препаративной химии
перимента. Пер. с нем./Под ред.
К. П. Яковлева. — М.: Физмат-
гиз, 1962.]
2. Dodd R. Е., Robinson Р. L., Ex-
perimental inorganic chemistry.
Elsevier, Amsterdam — London —
New York, 1957.
3. Grubitsch H„ Anorganisch-Prapa-
rative Chemie. Springer, Wien,
1950.
4. Jolly IT. L„ The synthesis and
characterisation of inorganic com-
pounds. Prentice Hall Englewood
Cliffs, New York, 1970.
5. Klemenc A., Behandlung und Rein-
darstellung von Gasen. Springer,
Wien, 1948.
6. Kohlrausch F.. Praktische Physik.
B. G. Teubner, Stuttgart, 1968.
7. Lux H. Anorganisch-chemische Ex-
perimentierkunst. J. A. Barth, Lei-
pzig, 1970; Лукс Г. Эксперимен-
тальные методы в неорганической
химии. Пер. с нем. — М.: Мир,
1965.
8. Pinkava J., Laboratoriumstechnik
kontinuierlicher chemischer Pro-
zesse. M. Deutsch, Frankfurt/Main,
1962; Пинкава fl. Лабораторная
техника непрерывных химических
процессов. Пер. с чеш./Под ред.
Н А. Клейменова. — М.: ИЛ,
1961.
9. Shriver D. F., The manipulation of
air-sensitive compounds, McGraw
Hill, New York, 1969.
10. Telle W... Chemische Laborato-
riumsgerate. Deutscher Verlag,
Leipzig, 1969.
11. Houben—Weyl, Methoden der or-
ganischen Chemie. Georg Thieme,
Stuttgart. Bd. I: Allg. Laborato-
riumspraxis (Teile 1, 2), 1958/59;
Bd. II: Analytische Methoden,
1953; Bd. Ill (Teile 1, 2): Physi-
kalische Methoden, 1955.
1. Сборка лабораторных установок
В настоящее время в большинстве случаев монтаж аппаратуры произ-
водится на «классическом» штативе Бунзена. Некоторые новые модели пред-
отвращают возможность смещения лапок при сильном завинчивании. При-
менение деталей из нержавеющих материалов (сталь 18/8 для штанг, спе-
циальные сплавы для лапок и муфт), хотя и дорого, но при длительном
пользовании значительно облегчает возможность поддержания аппаратуры
в чистоте.
Часто используемые установки (вакуумные, аппаратура для получения,
счистки и осушки различных газов) целесообразно монтировать на отдель-
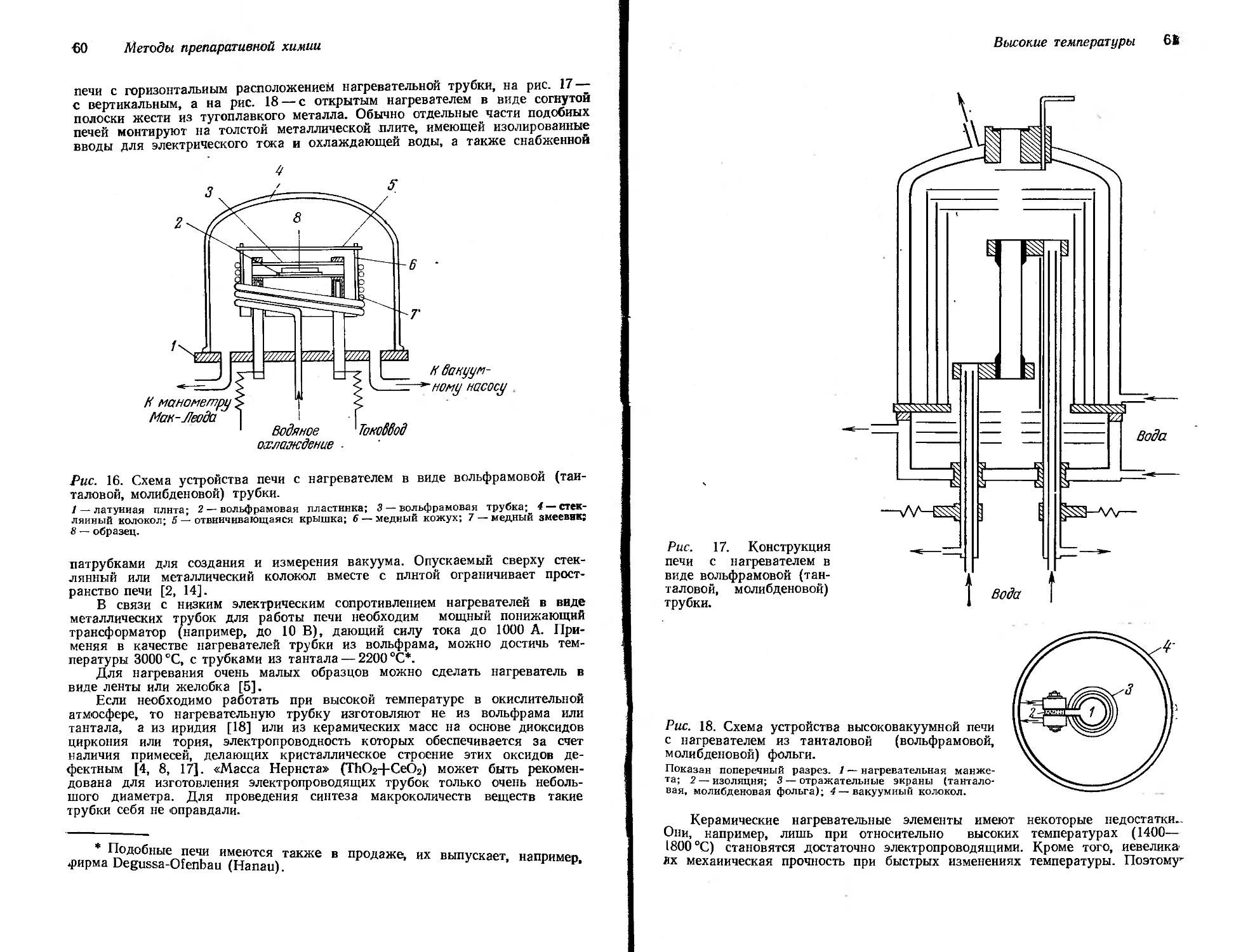
Рис. 1. Каркас для монтажа отдель-
но стоящей аппаратуры.
ных штативах, чтобы весь прибор в собранном виде можно было легко
переносить с одного места на другое или, если он не нужен, оставлять в
сторону. Более сложные установки монтируют на нескольких штативах, со-
единяя их между собой круглыми стержнями диаметром 13 мм. Для до-
стижения необходимой устойчивости установку прикрепляют к стене. При
Стекло И
помощи стержней, вмонтированных в стену, всю установку можно закрепить
только на стене, совершенно не используя в качестве опоры стол. При та-
ком способе крепления поверхность стола остается свободной и ее значи-
тельно проще содержать в чистоте. Штативы различных размеров, предназна-
ченные для монтажа на стене, имеются в продаже.
Для монтажа аппаратуры, будь то на лабораторном столе, на полу или
на передвижном основании, весьма удобны стальные перфорированные угол-
ковые профили, производимые многими фирмами и поступающие в прода-
жу под различными названиями. Из таких профилей собираются на болтах
очень устойчивые каркасы, назначение которых может быть весьма разно-
образным (рис. 1). В этих каркасах укрепляются стержни круглого сече-
ния с резьбой на обоих концах, а на стержнях с помощью муфт можно
крепить другие стержни, лапки и т. д. Легкие, но достаточно устойчивые
каркасы можно собрать из комплекта рам, сделанных из труб двух разме-
ров с соотношением длин 1 : 1,4 (как катет и гипотенуза равнобедренного
прямоугольного треугольника). Трубы с внешней резьбой на концах соеди-
няются с помощью специальных узловых элементов, имеющих внутреннюю
резьбу, в трехмерный каркас, так что отдельные трубы располагаются под
углами 45, 90, 135° друг к другу. Аппаратура укрепляется на каркасе при
помощи муфт, труб и лапок*.
При монтаже установок на каркасе необходимо также обращать вни-
мание на правильное распределение места и выбор соответствующих лапок.
Следует иметь в виду, что чрезмерно сильное завинчивание последних мо-
жет привести к поломке стеклянных приборов вследствие возникновения
напряжения, а поэтому оно так же нежелательно, как и слабое завинчива-
ние.
ЛИТЕРАТУРА
1. Monch G. Ch.. Hochvakuumtechnik. 2. Houben—Weyl. Methoden der orga-
R. A. Lang, Possneck, 1950. nischen Chemie. Band I, 1. Georg
Thieme, Stuttgart, 1958.
2. Стекло
Важнейшие сорта стекла, используемого в лаборатории, в зависимости
от их «твердости» подразделяют на группы (табл. 1). С точки зрения хи-
мика, стекла различаются по составу и, как следствие, по их химическим и
физическим свойствам.
1. Обычное или оптическое кварцевое стекло (SiO2) используют в тех
случаях, когда требуется устойчивый к действию высоких температур и к
резким колебаниям температуры материал.
Таблица 1. Типы стекол, используемых в химической лаборатории
Группа стекол Коэффициент термического расширения, град-1
Мягкие стекла Твердые стекла Специальные стекла Кварц (кварцевое стекло) 80—100-10-7 (20—100 °C) 30-50-10-7 (20—100 °C) Различные значения 6—7-10-7 (20—100 °C)
Фирма-поставщик MERO (Wiirzburg).
2*
12 Методы препаративной химии
Стекло викор (96% SiO2) получают из боросиликатного стекла, из ко-
торого после изготовления соответствующего изделия горячей кислотой вы-
щелачивают борат щелочного металла. Остающийся тонкопористый материал
в дальнейшем подвергают спеканию, после чего он частично приобретает
свойства кварцевого стекла.
2. Боросиликатные стекла (стекло для химической посуды 20, разотерм,
дюран 50, пирекс) содержат в своей основе помимо SiO2 (75—80%) еще
В2О3 (7—16%).
3. Алюмосиликатные стекла, содержащие кроме SiO2 до 20—22% А12О3
(супремакс), можно применять при высоких температурах.
4. Стекла на основе силикатов щелочных и щелочноземельных металлов
с небольшими добавками А12О3 (3—4%) известны под названием «тюринг-
ских стекол» (мягкие стекла, например марок AR, GW, или тюрингское ап-
паратурное стекло).
5. Свинцово-силикатное стекло содержит в своем составе до 50 масс. %
РЬО.
Обычным материалом для изготовления стеклянных лабораторных при-
боров и для их соединения служат стеклянные трубки круглого сечения.
В зависимости от толщины стенок различают «сгибающиеся» (толстостен-
ные) и «цилиндрические» (тонкостенные) трубки. Для обозначения сорта
стекла указывают название фирмы, а также торговое название или специ-
альный номер. Некоторые трубки имеют вдоль оси цветные полосы, ука-
зывающие на сорт стекла. Так, например, стекло фирмы Jenaer Glaswerk
Schott u. Gen. (Mainz) имеет одну цветную полосу; стекла других фирм мо-
гут маркироваться полосами двух или более цветов. В качестве дополни-
тельного отличия служит цвет самого стекла, который хорошо виден при
рассматривании свежего излома трубки при боковом освещении. Однако
цвет стекла сильно зависит от качества использованного сырья и от режи-
ма плавки, и поэтому его лишь с оговорками можно применять для рас-
познавания сортов стекла (табл. 2). В настоящее время наиболее распро-
страненным в лабораторной практике сортом стекла является дюран 50.
Кварцевое стекло (частично также викор) в отличие от стекол других
сортов прозрачно не только в видимой части спектра, но и в ультрафиоле-
Таблица 2. Наиболее распространенные сорта стекла
Сорт стекла Фирма-изготовитель Маркировка Спаивается с сортами
AR GW Стекло для хими- ческой посуды 20 Glaswerk Ruhr (Essen) Glaswerk Wertheim Glaswerk Schott u. Gen. (Mainz) Без полос Одна синяя лоса ПО- 16 III; тюрингские стекла 16 III; AR
Разотерм Glaswerk Schott u. Gen. (Jena) Одна красная лоса по- Аналогично G20
«Нормальное» стекло 16 III Дюран 50 Glaswerk Schott u. Gen. (Mainz) то же Одна темно-крас- ная полоса Без полос AR; GW Пирекс (супре- макс)
Супремакс Пиоекс » Corning Glass Works (Corning, USA) Желто-зеленая лоса Без полос по- (Дюран, пирекс) Дюран (супре- макс)
Стекло 13
Таблица За. Гидролитическая стойкость стекла8
Категория Характеристика стекла Степень выщелачивания, мг NasO
1 Очень стойкое к действию воды 0—0,03
2 Стойкое стекло 0,03—0,06
3 Тугоплавкое для приборов 0,06-0,26
4 Легкоплавкое для приборов 0,26—0,62
5 Свыше 0,62
а По стандарту DIN 12Н1 1 г стекла, измельченного до крупинок размером 0,5—
0,3 мм, выдерживают в течение 1 ч в воде, нагретой до 100 °C.
Таблица 36. Стойкость стекла к кислотам"
Категория Характеристика стекла Количество стекла, пере- шедшее в раствор, мг/1000 см2
1 Стойкое к действию кислот 0—0,7
2 Слаборастворимое в кислотах 0,7-1,5
3 Умеренно (или сильно) растворимое в кислотах Больше 1,5
а По стандарту DIN 12116 образец стекла с оплавленной поверхностью площадью
200 см2 кипятят в течение 3 ч в 20% НС1.
Таблица Зв. Стойкость стекла к щелочам"
Категория Характеристика стекла Количество стекла, пере- шедшее в раствор, мг/100 см2
1 Слаборастворимое в щелочах 0—75
2 Умеренно растворимое в щелочах 75—150
3 Сильнорастворимое в щелочах Больше 150
а По стандарту DIN 52322 оплавленные кусочки стекла кипятят в течение 3 ч в
0,5 н. NaOH+0,5 и. Na2CO3 (общая концентрация щелочи 1 н.).
товой (приблизительно до 200 нм) и ближней инфракрасной (~до
2300 см-1)- Поэтому его часто используют для проведения спектроскопи-
ческих и фотохимических исследований (изготовление окон кювет, колб для
источников излучения).
Химическая стойкость стекла к действию водных растворов характери-
зуется нормативными показателями: гидролитической стойкостью (табл. За),
стойкостью к кислотам (табл. 36), стойкостью к щелочам (табл. Зв).
Соединение отдельных частей стеклянной установки осуществляют преж-
де всего спаиванием стеклянных трубок ручной паяльной горелкой, а также
при помощи шлифов, резиновых трубок, пробок и замазок. Работа с го-
релкой несложна, ее может быстро освоить любой экспериментатор, даже не-
14
Методы препаративной химии
знакомый со стеклодувными работами. Две трубки часто легче спаять, чем
тщательно соединить их отрезком резиновой трубки. Технику стеклодувного
дела целесообразно осваивать в процессе работы под руководством спе-
циалиста, поэтому приемы работы со стеклом здесь не описываются. Одна-
ко все же приведем некоторые замечания.
1. Стеклянные трубки необходимо защищать от пыли и хранить в ле-
жачем положении. Если же из-за недостатка места трубки приходится ста-
вить вертикально, то их отверстия следует закрывать.
2. Перед употреблением стеклянные трубки следует очищать, проталки-
вая или продувая через них влажные ватные тампоны. Через трубки боль-
шого диаметра необходимо несколько раз протягивать при помощи шнура
влажную тряпку. Можно рекомендовать также промывку очень разбав-
ленной плавиковой кислотой (1%). Никогда не следует вводить в стеклян-
ные трубки железную или стальную проволоку, а также другие стеклянные
трубки. Несоблюдение этого правила может оказаться причиной внезап-
ного растрескивания трубки при нагревании.
3. Края спаиваемых трубок должны быть свежеобрезанными; их не сле-
дует касаться пальцами. Если укорачивать трубку с целью получения све-
жего среза уже невозможно, то необходимо нагреть спаиваемое место руч-
ной паяльной горелкой и снять с края стеклянной палочкой немного стекла.
Можно также запаять трубку, выдуть на ее конце тонкостенный пузырь и
обломать его.
4. При обработке боросиликатных стекол необходимо добавлять к вду-
ваемому воздуху некоторое количество кислорода. В продаже имеются со-
ответствующие ручные паяльные горелки с дополнительным вентилем для
подачи кислорода (особенно при паяльных работах со стеклом супремакс).
Однако именно в последнем случае следует остерегаться добавления слиш-
ком большого количества кислорода, так как при этом стекло «перегорает»,
т. е. становится в соответствующих местах беловатым, пузыристым и мут-
ным. Если не считать трудностей, связанных с необходимостью работы при
более высоких температурах, обращение с тугоплавкими сортами стекла,
пожалуй, даже несколько легче, чем с обыкновенным лабораторным тюринг-
ским стеклом, ввиду значительно меныпей опасности растрескивания этих
стекол при неравномерном нагревании и охлаждении. Существует непосред-
ственная связь между устойчивостью стекла к колебаниям температуры и
его коэффициентом термического расширения. Работу со стеклом ведут при
температурах, на 300—450 °C превышающих температуру его размягчения
(табл. 4).
Прозрачное кварцевое стекло получают в промышленности путем плавки
чистого кварца, а непрозрачное кварцевое стекло (ротозил)— при сплав-
лении тонкодисперсного материала. Обработка кварцевого и обычного стек-
ла производится сходным образом, так что в продажу поступает много
разнообразных изделий из кварцевого стекла. К их достоинствам относятся
прежде всего исключительная устойчивость к колебаниям температуры, про-
зрачность и относительно высокая, хотя и весьма селективная, химическая
стойкость. Используя водородно-кислородное пламя (или смесь водород+
+воздух с добавкой кислорода), изделия из кварцевого стекла (трубки,
шлифы и т. п.) можно спаивать в лабораторных условиях. Обработка квар-
цевого стекла, несмотря на высокую температуру его размягчения (около
1500°C), не намного сложнее, чем обыкновенного стекла. При этом реко-
мендуется соблюдать следующие правила.
1. При спаивании отверстия часто полностью не заплавляются; обычно
остается небольшое капиллярное отверстие. Такие места следует многократ-
но проплавлять или же «затягивать» при помощи тонкой кварцевой палочки.
Такое же затруднение возникает и при работе со стеклом супремакс.
2. Вследствие испарения SiO и SiO2 кварцевое стекло вокруг спая ста-
новится мутным. Для устранения этого недостатка следует после окончания
Стекло 15
Таблица 4. Физические и химические свойства технических стекол
Марка стекла Линейный коэффици- ент термического рас- ширения, град—1 * 3 4 * * а Температура, °C Категория стойкости, гидролити- ческая/к кислотам/к щелочам
рекрис- таллиза- ции размяг- чения, 106—105 Па-С
Рургляс (AR) 90-10-’ (20—200) 519 564 3/1/2
Стекло для химиче- 96-10-7 (20—400) 87-10-7 * * * * (20—100) 525 580 3/1—2/1—2
ской посуды «Нормальное» стекло 95-Ю-7 (20—400) (82—GO)-IO-’ (20—300) 535 708 3/1/2
16 III Стекло для химиче- (48—49)-10-7 (20—300) 565 790 1/1/2
ской посуды 20 (G20) Дюран 50 (8330) 32-Ю-7 (20—350) 530 815 1/1/2
Пирекс (солидекс) 32-Ю-7 (20—300) 519 820 1/1/3
Супремакс (8409) (37—41)-IO-’(20—300) 740 950 1/3/3
а В скобках указан интервал температур (°C).
основной работы подвергнуть всю эту область действию большого, но не
очень горячего кислородно-водородного пламени, а в случае необходимости
ополоснуть затем трубку разбавленной плавиковой кислотой.
3. В связи с быстрым увеличением вязкости кварца при его остывании
дутье необходимо проводить очень быстро и лучше всего при помощи рези-
новой трубки.
4. При охлаждении или длительном выдерживании изделия при высокой
температуре возникает опасность расстекловывания, т. е. перехода из ме-
тастабильного стеклообразного состояния в кристобалит. Этот процесс, од-
нажды начавшись, приводит к быстрому механическому разрушению. Рас-
стекловывание начинается, как правило, в местах, имевших внешние за-
грязнения, и протекает с заметной скоростью лишь при температурах выше
1000сС. Поэтому те части кварцевых приборов, которые будут нагреваться,
не следует после их тщательной очистки (водными растворами или орга-
ническими жидкостями — спиртом, ацетоном) трогать руками, поскольку да-
же следы пота (NaCl) могут вызвать расстекловывание.
В отсутствие значительных перепадов давления кварцевое стекло мож-
но применять до 1250 °C, однако вакуумированные кварцевые приборы пре-
терпевают постепенную деформацию уже при ~ 1150 °C. Оба явления, рас-
стекловывание и появление пластичности, делают кварцевое стекло непри-
годным для проведения опытов в течение длительного времени при темпе-
ратурах выше 1000 °C. Другие стекла можно использовать (с учетом тол-
щины стенок и продолжительности механических нагрузок) приблизительно
до температуры рекристаллизации. Последняя соответствует той минималь-
ной температуре, при которой в стекле постепенно снимаются механические
напряжения, что соответствует величине вязкости стекла 1012—1013 Па-с.
Приборы из неотожженного, т. е. слишком быстро и неравномерно охлаж-
денного, стекла могут неожиданно растрескиваться без видимой причины.
Часто это происходит вследствие наличия на поверхности приборов незна-
чительных повреждений, возникающих, например когда их очищают, при-
меняя слишком твердые и острые предметы, или когда прибор кладут яа
поверхность каменной настольной плиты.
16 Методы препаративной химии
Для соединения непосредственно не спаивающихся сортов стекла при-:'
меняют - переходные трубки диаметром 5—25 мм. Они состоят из ряда
очень коротких отрезков с постепенно изменяющимся коэффициентом тер-
мического- расширения. Например, при помощи 13 таких отрезков можно
соединить даже обыкновенное стекло с кварцем. Для соединения дюрана 50
и стекла для химической посуды 20 нужна лишь одна переходная трубка
(№ 3891 фирмы Schott). Стекла марок дюран 50 и пирекс схожи по мно-
гим показателям,., и поэтому допустимо их непосредственное спаивание.
Переходные трубки очень чувствительны к изменению температуры, и их не
следует подвергать нагреванию.
Для очистки стеклянные приборы обычно заполняют хромовой смесью,
оставляют стоять в течение некоторого времени и зат.ем промывают водой..
(Приготовление хромовой смеси: растворяют при помешивании порошкооб-
разный технический Na2Cr2O7 в конц. H2SO4*.) Однако необходимо иметь
в виду, что, согласно данным Лауга [4], стекло прн обработке хромовой
смесью поглощает хромовый ангидрид; последний не легко удалить полно-
стью даже при кипячении с водой. Лауг показал, что 1 г стекла поглощает
около 5 мг хромового ангидрида, нз которых 0,2 мг не удается удалить
даже при многократном кипячении с водой. Поэтому в некоторых случаях
стекло лучше промывать концентрированной азотной кислотой. Очень эффек-
тивна очистка стекла щелочным раствором перманганата калия с последую-
щим промыванием сначала водой, затем концентрированной соляной кисло-
той и, наконец, снова водой. Можно также оставить прибор на ночь в ме-
танольном растворе гидроксида калия (КОН в СН3ОН), а затем промыть
разбавленной НС1 и водой. В редких случаях приборы (за исключением
мерной посуды) быстро промывают 1—4%-ным раствором плавиковой кис-
лоты.
Стеклянные трубки и части приборов больших размеров, не помещаю-
щиеся в сушильном шкафу, ради предосторожности не следует очищать от
оставшихся на внутренних стенках следов воды органическими жидкостями
(спиртом, эфиром, ацетоном), так как последние нередко содержат нелету-
чие примеси, загрязняющие стеклянные стенки и являющиеся помехой при
получении активных веществ или при работе в высоком вакууме. В таких
случаях лучше при помощи водоструйного иасоса просасывать через труб-
ку воздух, оставляя при этом открытым для доступа воздуха только одно
отверстие, прикрыв его от попадания пыли кусочком ваты или листком
мягкой фильтровальной бумаги.
Соединения при помощи шлифов. Если отдельные части прибора при-
ходится разъединять, то места их соединений снабжают притертыми шли-
фами. Для этого применяют нормальные шлифы конусообразной формы,
шаровые или плоские, а также прозрачные прецизионные конические шли-
фы с оплавленной поверхностью. Соединение при помощи конического шли-
фа выгодно отличается определенной жесткостью, которую можно сочетать
с подвижностью шарового шлифа, например встраивая в слишком громозд-
кую аппаратуру шарнир на шаровом шлифе. <
Ко всем видам шлифов в продаже имеются соответствующие держатели, |
плотно прижимающие шлифы друг к другу. Конические шлифы рекоменду- I
ется снабжать припаянными стеклянными крючками, на которые надеваются' ;
спиральные пружины или резиновые кольца, обеспечивающие плотное со- ;
единение частей шлифа. Такне' шлифы с крючками также 'имеются в прода-'
же. Цены на шлифы отдельных- видов сильно различаются, но более деше-
вы конические шлифы. 1 :
В обозначении шаровых шлифов указываются примерный диаметр ша-
ра и внутренний диаметр трубки в миллиметрах. В связи с тем, что стан- '
* МожиО брать также К2Сг20г. Обычно растворяют ~90 г соли в 1 л
96%-ной H2SO4.— Прим, перев.
Стекло 17
дарт на шаровые шлифы DIN 12244 основан на неметрической единице
1 дюйм, размер шлифов не выражается целым числом миллиметров, но ни-
же эти значения округлены. Среди указанных размеров следует по воз-
можности использовать те, которые выделены полужирным шрифтом: KS 13/2»
KS 13/5, KS 19/9; KS 29/15, KS 35/20, KS 40/25; KS 41/25, KS 51/30, KS 64/40.
В продаже имеются шаровые шлифы и других размеров, в том числе
шлифы с размерами в метрической шкале, например:
7/1 12/6 28/(18-19) 50/(34—35)
12/1 18/7 35/20 55/(39—40)
12/2 18/(9—11) 35/25 65/45
12/3 28/12 40/25 75/50
12/5 28/15 50/30 102/75
Для плоских шлифов указывается внутренний (реже внешний) диаметр
фланца в миллиметрах. Они выпускаются следующих размеров (размеры
внутреннего фланца): 15, 25, 40, 50, 65, 75, 100. Плоские шлифы больших
размеров используются, в частности, при работе с широкогорлыми флан-
цевыми колбами.
Таблица 5. Обозначение и размеры нормальных шлифов1
Главная серия (1) Серия 0 Серия 2 Серия 3
5/13 5/20 5/9
7,5/16 7,5/25 7,5/11 —
10/19 10/30 10/13 10/10
12,5/21 12,5/32 12,5/14 12,5/12
14,5/23 14,5/35 14,5/15 14,5/12
19/26 19/38 19/17 19/12
24/29 24/40 24/20 24/12
29/32 29/42 29/22 29/12
34,5/35 34,5/45 34,5/24 34,5/12
45/40 45/50 45/27 45/12
60/46 60/55 60/31 60/12
70/50 70/60 70/33 70/12
85/55 85/70 85/37 85/12
100/60 100/80 100/40
® Конические шлифы по стандарту DIN 12242; конусность 1 : 10. Первое число ука*
зывает наибольший диаметр шлифованного конуса, второе — длину шлифованного участ~
ка. С целью упрощения работы рекомендуется по возможности использовать типы шли*
фов, выделенные полужирным шрифтом.
Обозначения конических шлифов многократно изменялись. В табл. 5
приведены принятые в настоящее время размеры для различных серий.
Кроме шлифов стандартных размеров с конусностью 1 : 10 (разность
между наибольшим и наименьшим диаметрами шлифа, деленная на длину
шлифа, равна 1 : 10) изготовляются также широкие шлифы с конусностью
1 : 5. Их используют при работах с вакуумом, поскольку в случае широких
шлифов с конусностью 1 : 10 внешнее давление воздуха на шлиф может
привести к его заклиниванию, так что разъединение деталей без их полом-:
18 Методы препаративной химии
«и становится затруднительным. При решении вопроса о том, какая часть
шлифа — муфта или керн — должна припаиваться к соответствующей части
прибора, играют роль различные факторы. Основным критерием должно
быть предотвращение загрязнения вещества материалом смазки. Если шлиф
смазывается, то при вертикальном положении муфта должна быть наверху,
•а керн — внизу. Для предотвращения попадания смазки в прибор пользуют-
ся так называемыми двухзонными шлифами, керн которых имеет кольцеоб-
разную канавку посередине шлифованного конуса. Взвешиваемые части при-
бора должны оканчиваться керном, так как его легче очищать. Вместо слоя
смазки роль уплотнителя может играть также манжета, изготовленная из
тонкой политетрафторэтиленовой (тефлоновой) пленки (см. разд. 5). Тефлон
в отличие от материалов смазок нерастворим в органических растворителях.
В табл. 5 первая цифра в обозначении шлифа указывает наибольший
диаметр шлифованной зоны (в миллиметрах), вторая — длину шлифован-
ного участка. Для выпускаемой в массовом масштабе лабораторной посуды
подходят большей частью нормальные шлифы первой серии (NS 14,5, NS 19,
NS 29 и NS 45). Если на то нет особых причин, надо стараться работать
с возможно меньшим числом различных видов шлифов. Шлифы серии О
(удлиненные шлифы) наиболее употребительны при работе с высоким ва-
куумом, шлифы серии 2 и 3 находят применение при изготовлении мерных
колб, склянок, бюксов и т. д. Шлифы с конусностью 1 : 5 выпускаются, со-
гласно стандарту DIN 12243, трех размеров: NS 60, NS 75 и NS 90.
Размеры и обозначения шлифов с оплавленной поверхностью («про-
зрачных шлифов») такие же, как конических. Соединения на таких шлифах
из-за гладкой поверхности достаточно плотны даже в отсутствие смазки;
эти шлифы легче очищать, и они не так сильно заклинивают при работе
с вакуумом или вследствие загрязнения. Прозрачные шлифы размером 14,
19, 24 и 29 изготавливаются из стекла марки дюран 50. Можно рекомендо-
вать использовать их в тех случаях, когда желательно работать без смазки,
так как она растворяется или подвергается химическому воздействию, или
когда работают с чрезвычайно небольшими количествами вещества.
При соединении на шлифах частей, изготовленных из различных сортов
стекла, муфта всегда должна быть из стекла с более высоким коэффици-
ентом расширения. В особенности это относится к соединению на шлифах
стекла и кварца. Если керн шлифа изготовлен из стекла, а муфта — из квар-
ца, растрескивание муфты в этом случае происходит уже при погружении
в кипяток. Более значительные перепады температуры могут выдерживать
лишь такие соединения на шлифах, части которых изготовлены из одного и
того же сорта стекла. Конические и шаровые шлифы из кварца и викора
производятся многими фирмами*.
Соединения на замазках. Относительно смазки кранов и шлифов, а
также о пригодных для этого смазочных материалах и замазках будет ска-
зано ниже. Во многих случаях можно с успехом вместо шлифов использовать
уплотнения при помощи замазок. Это особенно удобно для трубок очень
большого диаметра, так как соединенные таким способом части прибора
яри правильном осуществлении уплотнения можно легко разъединять без
смещения трубок. Если соединяются трубки одного диаметра, можно достичь
большей прочности, надвигая на место соединения стеклянную трубку не-
сколько большего сечения.
Для прочности соединений, осуществляемых при помощи замазок всех
сортов, важно, чтобы при подогревании соединительной трубки замазка
растекалась. Это имеет особенно большое значение для металлических ча-
стей, так как именно в этом случае весьма трудно обнаружить неплотности.
* Например, фирмами Thermal Quarzschmelze GmbH (Wiesbaden-Bieb-
rich); Westdeutsche Quarzschmelze GmbH (Geesthacht).
Стекло 1 ?
Соединение металла со стеклом. При соблюдении определенных усло-
вий металлы легко и надежно спаиваются со стеклом. Так, медную трубку
можно спаять с обыкновенным тюрингским стеклом. Для этого ее растачива-
ют на конце до толщины 0,1 мм, накаливают в горячей части пламени до
светло-красного каления, наплавляют на нее стеклянную нить, а на послед-
нюю насаживают затем стеклянную трубку (рис. 2). Впаивание в стекло
металлических деталей, особенно проволоки, можно осуществить достаточно
надежно лишь при условии, что коэффициенты термического расширения
металла и стекла в интервале температур от комнатной до температуры ре-
кристаллизации отличаются не более чем на 10%. Это имеет место, например
/Л///
Стекло
Медь
Рис. 2. Спаивание медной и стеклян-
ной трубок.
в случае платины и простого тюрингского стекла, но все же при впаивании
толстой платиновой проволоки лучше использовать специальное переходное
свинцовое стекло. Сначала по возможности в окислительном пламени его
наплавляют на проволоку в виде шарика. Затем уже вплавляют этот ша-
рик в отверстие стеклянной трубки, куда должна входить платиновая про-
волока.
__ Легкоплавкие стекла можно также спаивать со сплавами на основе ни-
кел'я, железа, хрома и марганца, например с ваковитом*. Для впаивания в
тугоплавкие стекла применяются молибден, вольфрам и сплавы железо —•
никель — кобальт, например вакон*. Все эти сплавы в виде проволоки, па-
лочек, трубок, пластин, лент, профилей и готовых изделий можно приобре-
сти через торговую сеть. Для очень тугоплавких стекол (пирекс, дюран 50,
стекло для химической посуды 20) сплав для впаивания подобрать гораздо
труднее. Обычно в этом случае используют молибден или вольфрам либо
осуществляют впаивание через промежуточную вставку из другого стекла
(например, помещают стекло № 8243 фирмы Schott между сплавом вакон 10
и стеклом для химической посуды 20). Для впаивания в кварцевое стекло
подходит лишь молибден.
Выполнение спаев стекло — металл лучше всего поручить опытному
стеклодуву. Для достижения прочного и надежного в работе соединения
стекло должно хорошо смачивать металл и удерживаться на нем. Это тре-
бует тщательной предварительной подготовки поверхности металла. Хорошо
выполненное соединение вакона или ваковита со стеклом должно быть
серого цвета и иметь матовую поверхность.
Рецепты для химического серебрения стекла можно найти в книге Ан-
герера [1]. Образующийся тонкий отражающий слой серебра не очень
прочно держится на стекле, и его нельзя использовать для припаивания к
другим изделиям из металла. Для получения прочного соединения металла
со стеклом на поверхность последнего при высокой температуре наносят
слой специального припоя (серебро, золото, платина). Такие припои посту-
пают в продажу в виде эмульсий или паст**.
* Выпускаются фирмой Vakuumschmelze AG (Hanau).
•* Фирма-изготовитель: Deutsche Gold- und Silberscheideanstalt DEGUSSA
(Frankfurt/Main).
20
Методы препаративной химии
Соединение стекла и керамики. Трубки из твердого фарфора и высо-
коглиноземистой («пифагоровой») массы можно спаять с дюрановым стек-
лом. Для этого фарфоровую трубку медленно нагревают в пламени паяль-
ной горелки, затем, добавляя к вдуваемому воздуху кислород, доводят ее
до ярко-красного каления и наплавляют немного стекла на конец трубки,
равномерно оборачивая ее стеклянной нитью (рис. 2 и 3). После этого конец
трубки соединяют в пламени с предварительно нагретой трубкой из дю-
ранового стекла. После непродолжительного пропаивания и выдувания
Стекло-дюран
Высокоглино-
земистая
масса
чммптшил
W/N/ltf/f/HIL
Рис. 3. Спаивание трубки из стекла
дюран с трубкой из высокоглиноземи-
стой массы.
стекла место соединения медленно охлаждают в пламени. Места спая в
таких случаях устойчивы даже по отношению к сильным термическим воз-
действиям. Следует отметить, что имеются также и керамические трубки с
нормальными шлифами*.
Соединение с помощью резиновых трубок. Когда пользуются для со-
единения резиновыми трубками, обычно часто не соблюдают правило, чтобы
края стеклянных трубок были кругом оплавлены. В результате оторвавшие-
Рис. 4. Уплотнительное винтовое со-
единение.
ся кусочки резины застревают между стенками резиновой и стеклянной тру-
бок и обусловливают потерю герметичности. Если вместо глицерина или
вазелинового масла для смазывания применяют силиконовое масло, то не
происходит прилипания шлангов к стеклу с течением времени. Прилипшие
резиновые шланги срезают. Потеря небольшого кусочка трубки ни в какой
степени не сравнима с теми неприятностями, которые может нанести по-
ломка стеклянной трубки. Если из пробки очень большого диаметра необ-
ходимо извлечь запекшиеся в ней термометр, трубку и т. п., то между
стеклянной трубкой и пробкой вводят хорошо смоченное глицерином про-
бочное сверло и многократно извлекают его в процессе сверления, каждый
раз добавляя свежий глицерин. Для осуществления герметичного ввода в
стеклянные аппараты термометров, капилляров, трубок и т. д. вместо про-
бок лучше использовать специальные винтовые соединения (рис. 4). Стек-
лянная трубка с резьбой из дюрана 50 либо имеет на другом конце шлиф,
либо непосредственно припаивается к прибору. Уплотнительное кольцо из-
готавливается из тефлона*. Применение таких соединений весьма многооб-
* Фирма-изготовитель: Haldenwanger (Berlin).
* Выпускается фирмами Jenaer Glaswerk Schott u. Gen. (Mainz); Quick-
fit Laborglas GmbH (Wiesbaden-Schierstein). i
Керамические материалы
21
разно благодаря тому, что они выпускаются со шлифами различных раз-
меров и с различным диаметром уплотнительных колец и прижимных го-
ловок.
При просверливании резиновой пробки никогда не следует вращать
сверло в одном направлении. После каждого полуоборота необходимо ме-
нять направление вращения; при этом нужно многократно извлекать сверло
и добавлять свежий глицерин. В противном случае отверстие по мере
углубления суживается, так как высверленный цилиндрический кусок пробки
вращается вместе со сверлом и отверстие образуется не вследствие вырезы-
вания резины острой кромкой сверла, а в результате ее отрыва.
ЛИТЕРАТУРА
1. Angerer Е., Technische Kunstgriffe
bei physikalischen Untersuchungen.
F. Vieweg & Sohn, Braunschweig,
1966. [Есть русский перевод 12-го
нем. изд.: Ангерер Э. Техника фи-
зического эксперимента. Пер. с
нем./Под ред. К. П. Яковлева. —
М.: Физматгнз, 1962.]
2. Deeg Е., Richter Н„ Gias im Labo-
ratorium. Aulendorf (Wiirtt.), 1965.
3. Espe IE., Werkstoffkunde der Hoch-
vakuumtechnik. Band II (Silikat-
werkstoffe). Deutscher Verlag d.
Wissenschaften, Berlin, 1960.
4. Laug E. P., Ind. Eng. Chem. Anal.,
6, 111 (1934); Chem. Fabrik, 7, 241
(1934).
5. Monch G. Ch., Hochvakuumtechnik.
R. A. Lang, Possnek, 1950.
6. Wilhelm H., Glas-Instrumenten-
Techn., 1, 11 (1957).
7. Druckschriften der Glashiitten und
-werke sowie der Vakuumschmelze
AG, Hanau. (Каталоги стекольных
заводов и вакуумных плавилен.)
3. Керамические материалы
Огнеупорные керамические материалы, используемые в лабораторной
практике, можно в зависимости от их свойств и состава разделить на
5 групп (табл. 6). Окончательное придание формы керамическому изде-
лию в отличие от стекла происходит либо перед высокотемпературной об-
работкой (керамический обжиг), либо во время ее (горячее прессование).
Дальнейшая обработка возможна лишь механическим путем (шлифование,
резка). Вследствие усадки при обжиге возможности получения изделий с
заданными размерами и формой ограничены определенными рамками. По-
этому для работы в лаборатории производится сравнительно небольшой на-
бор изделий из керамических материалов: прямые трубки, палочки, тигли,
чашки, лодочки и т. д. Некоторые свойства керамических материалов ’ при-
ведены в табл. 7.
Группа 1. Материалы этой группы, в основном состоящие из оксида
алюминия. и кремнезема, менее вакуумноплотны, чем чистое кварцевое стек-
ло. Проницаемость для газов сильно увеличивается при возрастании рабочей
температуры и времени эксплуатации изделия. Помимо обычного фарфора,
для лабораторной посуды различными предприятиями разработаны составы,
-обладающие более высокой химической стойкостью. Максимально допусти-
мая для применения ряда этих материалов температура возрастает по ме-
ре повышения содержания в них оксида алюминия. Глазури применяются
только для фарфора. Устойчивость к изменениям температуры у этих ма-
териалов значительно ниже устойчивости чистого кремнезема.
В химическом отношении эти материалы при высоких температурах наи-
менее стойки к щелочам, а также к веществам, обладающим сильными вос-
становительными свойствами (например, к электрохимически активным ме-
22
Методы препаративной химии
Таблица 6. Группы керамических материалов
Труп- па Состав Наименование материала Фирма-изго- а товнтель
1а<5 Алюмосиликат с силикат- ным связующим Лабораторные сорта твер- дого фарфора (20—30% А12О3) 1, 2, 3
1бб То же, что 1а, но со специ- альными огнеупорными до- бавками, например корун- дом, силлиманитом и др. Масса Пифагора, К-масса, силлиманит, димулит и др. Специальные сорта фарфо- ра 1, 2, 3, 4
2« Спеченные тугоплавкие ок- Спеченный глинозем, дегус- 1, 2, 3, 4,
Зв сиды; системы, практически не содержащие SiO2; А12О3, MgO, ВеО, ZrO2, ThO2, а также MgAl2O4 Как 1, но меньше связую- щего, частично чистые окси- ды (А12О3, MgO) сит, пирокс, рубалит, про- корунд, альсинт, спеченные оксиды магния, бериллия, циркония, тория, шпинель Шамотные массы, муллито- вые массы, SKX, силлиман- тин, корунд (связующее — каолин), альпор, пормулит, спеченный оксид магния 5, 6
4 Карбиды, нитриды, бориды, силициды, сульфиды (на- пример, SiC, ZrC, В4С, BN, Si3N4, TiB2, MoSi2, CeS) Название химического со- единения: S3N4 (5), TiB2 и ДР- (И) 5, 11
5а Углерод, более или меиее пористый, вплоть до почти газонепроницаемого Электродный уголь, элект- рографит, диабон, дюрабон 7, 9 ю
56 Углерод, газонепроницае- мый Стеклоуглерод, сиградур 7, 8
а Фирмы названы в качестве примера; указанные материалы выпускаются и Дру-
гими фирмами. 1 — Porzellanmanufaktur Haldenwanger (Berlin); 2 — Staatliche Porzellan-
manufaktur (Berlin); 3 — Rosental Technische Werke (Marktredwitz); 4— Heinrich Kop-
pers (Dusseldorf-Heerdt); 5 — Degussa, Abt. Degussit (Frankfurt/Main); 6 — Morganite-
Thermic (Dusseldorf); 7 —Sigri (Meitingen); 8 — Deutsche Carbone AG (Frankfurt/Main);
9— Schunk u. Ebe GmbH (Giessen); 10— Ringsdorf-Werke GmbH (Bad Godesberg);
11— Micropure (Dreibergen, Holland).
б Черепок плотный.
в Черепок пористый.
таллам). Химическая устойчивость возрастает по мере повышения содер-
жания глинозема.
Для особых целей (при высоких требованиях к химической стойкости)
посуду и приборы, изготовленные из материалов группы 1, снабжают еще
внутренней футеровкой из других материалов (например, из MgO, СаО»
LiF), которые сами по себе непригодны для переработки методами спекания.
Так, по данным Гёренса [7], на внутреннюю поверхность можно нанести
тесто из смеси, полуденной перемешиванием тонкоразмолотой, слабообож-
женной магнезии и крупно измельченной, сильнообожжеииой магнезии с на-
сыщенным раствором хлорида магния. Просушиванием и постепенным нагре-
ванием нанесенную массу можно превратить в хорошо приставшую защит-
ную футеровку из оксида магния. Для получения слоя оксида кальция (ко-
торый, между прочим, пригоден также и для облицовки железных сосу-
нов) применяется тесто из смеси оксида кальция и нитрата кальция или.
Керамические материалы 23
по данным Яидера [8], паста из оксида кальция и воды, нанесенная слоем’
толщиной 0,3—0,4 см. Высушивание и последующее иагреваиие следует начи-
нать от температуры 40 °C, а затем чрезвычайно медленно доводить массу
до температуры каления.
Группа 2. Для проведения синтезов при очень высоких температурах
наибольшее применение получили сосуды из оксидокерамических масс, ко-
торые изготовляются путем спекания возможно более чистых оксидов, обла-
дающих очень высокой температурой плавления. Такие массы отличаются
значительной огнеупорностью и заметной стойкостью к действию самых раз-
личных веществ при высоких температурах. Почти для каждого расплава
можно указать наиболее подходящий материал из числа оксидокерамических
масс. Наличие высокой термической и химической стойкости окупает недо-
статок, связанный с ограниченными возможностями придания приборам из
керамических материалов желаемой формы. В табл. 8—11 сведены имею-
щиеся в настоящее время практические данные, которые могут послужить
отправной точкой для выбора тех или других материалов.
В табл. 12 сведены опытные данные о нежелательных побочных явле-
ниях, происходящих при взаимном контакте поверхностей (например, реак-
ционной трубки и лодочки) для некоторых высокоогнеупорных материалов,,
а также металлических молибдена и вольфрама. Табл. 13 дает сведения о
совместимости при температурах выше 1000 °C материала тигля или лодочки
с нагреваемым в них веществом, если последнее имеет заметное давление
пара.
Группа 3. Важным дополнением к материалам из групп 1 и 2 могут
служить материалы, дающие на изломе пористый черепок и часто обладаю-
щие большей устойчивостью к колебаниям температуры. В ряде случаев их
можно применять и при более высоких температурах. Некоторые из мате-
риалов выпускаются в виде порошков, предназначенных для замешивания:
с водой и приготовления керамических масс. Часть из них состоит из пори-
стых частичек материалов группы 3 и предназначена в основном для со-
единения отдельных деталей между собой или для целей термической или
электрической изоляции (например, при монтаже нагревательных элемен-
тов). Имеются также порошкообразные массы, соответствующие по составу
чистым оксидам (А120з, ZrO2, ThO2, оксиды группы 2), которые после за-
мешивания с водой, формования, сушки и обжига при повышенной тем-
пературе дают достаточно прочные изделия.
Группа 4. В особых случаях при получении соединений или сплавов-
особо агрессивных металлов в качестве материала тиглей могут применяться
карбиды (SiC, В<С), силициды (MoSi2), бориды (TiB2), нитриды (BN, Si3N4),
сульфиды (CeS). Правда, изделия из этих материалов выпускаются лишь
периодически и в небольшом ассортименте, так как их изготовление до-
вольно трудоемко. В лабораторную практику приборы из этих материалов
еще широко не вошли.
Группа 5. Сюда относится углерод с температурой плавления около
4000°C. В продажу поступают изделия и полуфабрикаты (палочки, трубки,,
блоки, пластины) либо из чистого графитизированного угля, либо с при-
месью глинозема в качестве связующего (в основном тигли). В последнем
случае изделия несколько теряют в огнеупорности. Небольшие изделия из
чистого углерода можно изготовить путем механической обработки не очень-
твердых полуфабрикатов. Получающиеся таким образом материалы все же
в той или иной степени пористы. Их или можно рассматривать как «газо-
непроницаемые», или не считать таковыми в зависимости от конкретных
требований, предъявляемых к материалу изделия.
Стеклоуглерод получается путем проведения особого процесса — коксо-
вания высокомолекулярных углеродсодержащих соединений. Поступающие
Таблица 7. Свойства и области применения некоторых
Материал Температурный пре- дел применимости, °C Температу- ра. °C Газонепроницае- мость Стойкость к коле- баниям темпера- туры Средний коэффици- ент термического расширения, Ю-'/К Плотность, г/см3
размягчения при давлении® 2 кг/см2 К я я О) И Св *5 Я
Твердый фар- фор глазуро- ванный неглазурован- ный 1200 1400 1400 1400 ~1700 ~1700 Очень высокая То же Высокая То же 3—4 2,5
Шамот (раз- личных марок) 1500— 1700 — ~1700 Порис- тый Довольно высокая 6-9 2,2— 2,4
Силлимантнн (различные сорта) 1650 1650 1825 Порис- тый Очень высокая 5—6 2,4
К-масса, фар- фор с высокой огнеупорностью 1600 — 1800 Очень высокая Доволь- но высокая 3—4 2,5
Силлиманит ~1700 Выше 1800 Слабо- пористый или плотный Удовлет- вори- тельная 5—6 2,3— 2,85
Высокоглино- земистая масса (масса Пифа- гора) 1600 ~1700 ~1800 Очень высокая Доволь- но высокая 5 2,75
Коруид (спе- ченный, связан- ный каолином, А120з+5% SiO2) 1800 ~1700 Ниже 2000 Порис- тый Высокая 6-8 2,7— 3,6
важных керамических материалов'
Теплопро- водность, 6 ккал/(м-ч* •град) Химические свойства Область применения
*- 1 23 (при 20 °C) Хорошая химическая стой- кость, особенно к большинству кислых расплавов, за исключе- нием HF и НзРО4; частично разрушается при действии сильнощелочных расплавов Плавка металлов, сплавов и солей до 1250 °C
Высокая Стойкость к воздействию «кис- лых» и «щелочных» расплавов (в зависимости от марки) Плавка металлов (сталей)
Высокая Стойкость к летучей золе и печным газам: в восстанови- тельной атмосфере до 1600 °C, в окислительной — до 1300 °C Изготовление предохранитель- ных трубок, применяемых при- измерении температур, а так- же трубок для печей
1,72 ' (при 20 °C) Значительно большая стой- кость к химическим воздейст- виям и более сильные основ- ные свойства, чем у фарфора Плавка металлов, сплавов», стекла
2,0—2,4 Хорошая устойчивость к воз- действию расплавов, особенно кислых Плавка металлов и сплавов> (тигель Таммана). Изготовле- ние предохранительных трубок, для пирометров
1,2 Стойкость к воздействию ле- тучей пыли и компонентов золы Изготовление внешних трубою для пирометров и несущих тру- бок для нагревательных спи- ралей электропечей
Высокая Стойкость к различным хими- ческим воздействиям выше, чем у фарфора; до 1600 °C устойчив к действию большин- ства кислых расплавов Плавка металлов, сплавов, сом- лей и стекла
Материал Температурный пре- дал применимости. Температу- ра. °C Газонепроницае- мость 1 Стойкость к коле- баниям темпера- туры Средний коэффици- ент термического расширения, 10-’/К Плотность, г/см8
размягчения при давлении6 2 кг/см2 К я я о «5 я и с
Спеченный гли- «нозем (А12Оз) 1850— 1950 ~1800 2030 Высокая Средняя 6—8 3,8
Спеченная шпи- нель (MgAl2O4) 1950 2100 Высокая Средняя 8 3,3
Спеченный ок- сид бериллия (ВеО) 2200 2150 2550 Высокая Очень высокая 2,9
«Спеченный ок- сид магния (MgO) 2300 ~2000 2600 Порис- тый Средняя 13 2,5— 2,8
Спеченный ок- сид циркония (ZrO2) 2300 ~2000 2620 Высокая Низкая 10,9 5,0— 5,4
Продолжение
Теплопро- водность,б ккал/(м-ч> •1рад) Химические свойства Область применения
Высокая; 5,0 (при 1000 °C) Стойкость к щелочам, щелоч- ным и другим металлам, стек- лянным и шлаковым флюсам, а также к действию хлора, углерода, моноксида углерода, водорода, углеводородов и т. п. при самых высоких температу- рах. Почти не разрушается под действием сильных мине- ральных кислот, например HF И H2SO4 Плавка тугоплавких металлов и сплавов. Прокаливание твер- дых веществ. Изготовление трубок, тиглей, лодочек, пало- чек, пластин для самых раз- нообразных целей
Высокая; 4,7 (при 1000 °C) Стойкость к веществам щелоч- ного характера выше, чем у AI2O3 То же, что для А12О3
Очень высокая Стойкость к веществам щелоч- ного характера, а также к вос- становительному действию рас- плавленных металлов, углеро- да, моноксида углерода, водо- рода. Выше 1800 °C теряет хи- мическую стойкость; несовме- стим с диоксидом кремния Плавка тугоплавких металлов и сплавов, а также прокалива- ние твердых веществ
Высокая; 2,5 (при 1000 °C) Стойкость даже при очень вы- соких температурах к вещест- вам основного характера; мало- устойчив к сильному восстано- вительному действию углерода при высоких температурах Плавка тугоплавких металлов и сплавов; нагревание твердых веществ
Низкая 1,8 Стойкость к различным веще- ствам кислого и основного ха- рактера при очень высоких температурах. Образует кар- бид с углеродом при высоких температурах То же
Материал Температурный пре- дел применимости, °C Температу- ра, °C Газонепроницае- мость Стойкость к коле- ' банням темпера- туры Средний коэффици- ент термического расширения, 10-8/К I Плотность, г/см3
размягчения при давлении® 2 кг/см2 К S к си *5 й св Е
Спеченный ок- сид тория (ThO2) 2700 1950 3000 Высокая Низкая Высокий 9,2
Графит, сме- шанный с гли- ной —1700 1700— 1800 При гла- зуровке почти полная Очень высокая — 1,6
Уголь Выше 3000 Нет 4000 Порис- тый Очень высокая 1 ~1,5
Электрографит Выше 3000 Нет 4000 Порис- тый Очень высокая 1 1,5— 1,7
Стеклоуглерод Выше 3000 Нет 4000 Высокая Очень высокая 3,5 1,5
Карбид крем- ния (SiC) 1500 Выше 1500°С час- тично разла- гается Мало- пористый Очень высокая 5 2,2
Продолжение
Теплопро- водность, ккал/(м-ч- •град) Химические свойства Область применения
Низкая Стойкость к различным веще- ствам кислого и основного ха- рактера при очень высоких температурах. Образует кар- бид с‘углеродом при высоких температурах То же
Очень высокая Взаимодействует с основными флюсами. Во избежание науг- лероживания расплавленных малоуглеродистых железа и никеля следует применять вну- треннюю футеровку Тигли для плавки всех метал- лов и сплавов, кроме элект- рона
3,5—8 Стойкость к основным и кис- лым флюсам, если они не име- ют окислительных свойств. При плавлении металлов воз- можно незначительное загряз- нение их поверхности Плавка силикатов, процессы спекания, получение твердых сплавов, восстановление окси- дов металлов
100 (при 20 °C) Высокая стойкость к метал- лам, не образующим карбидов^ а также ко многим другим ве- ществам, за исключением окис- лителей, например воздуха при температурах выше 550 °C, водяного пара и диоксида уг-' лерода выше 900 °C. Тигли из электрографита более пригод- ны для плавления, чем уголь- ные, так как в меньшей степе- ни вызывают науглероживание металлов То же ( 4--'
- Средняя; 3-6 Аналогично графиту. Стойкость к действию фторидов Нагревание твердых веществ и расплавов, возможно и в при- сутствии агрессивных газов
8,5—4,8 Стойкость к летучей пыли и компонентам золы Изготовление внешних трубок пирометров (хорошая тепло- проводность)
Материал Температурный пре- дел применимости, °C размягчения при давлении6 п 2 кг/см2 g § О? плавления *< Газонепроницае- мость Стойкость к коле- баниям темпера- туры 1 Средний коэффици- ент термического расширения, 10~в/К Плотность, т/см3
Нитрид крем- ния (Si3N4) 1600 Воз- гоня- ется выше 1900°С Порис- тый Очень высокая 2,8 2,2— 2,5
а Подбор материалов н данные этой таблицы приведены лишь для общей ориенгн
сматривать как особую рекомендацию в отношении тех или иных материалов.
б Пересчет в единицы СИ: 1 ккалДчас-град) «4,2 кДж/(час-К)«1,2 Вт/К; 1 кг/см2«
Таблица 8. Стойкость различных оксидокерамических масс к действию
металлических расплавов®
Металл Тем перату- pa, °C Материал на основе
А12О3 ZrO2 1 MgO ВеО
Li(H2)e 700 — - ++б — — -
Na(H2) 700 +++ ++- - +++
К(Н2) 800 Н—F + +- +++
Си (окисл) 1200 +++ ++- +
Ве(Н2) 1500 —.—_ -]—1—J-
Mg(H2) 800 ++ -}—1— — 1 1 ’ —1 1— —
Са(Н2) 1000 +++ — + —
А1(Н2) 1000 +++
Si(H2) 1600 ' 1 ' }— ++- . — ——•
Ti(H2) 1800 + + —
Zr(H2) 1700 —’ ++ +++ -i—i-
Sb 800 Н- Н- ++- -
Bi 600 —।—।—}~ ++- -
Cr(окисл) 1900 —— —
Cr(H2) 1900 -}—j—|- ++ ++
Мп (окисл) 1600 — — — —
Mn(H2) 1600 ++ ++ +
Fe (окисл) 1600 — — —
Fe(H2) 1700 _|—1—1_ ++- - +--+в
Ni 1600 +++ ~ 1 I 1 —
Co 1600 +++ ++- - - —|_в
Pb 600 _|— +н—
Pt 1700 +++ ++Н - -~++в
Au 1100 +++ ++- н +++в
Обозначения: +++ очень высокая; ++ почти не разрушается под действием ре-
агента; 4- незначительно разрушается; — сильно разрушается;-----очень сильно разру-
шается; --------------------------------------------------------совершенно не стоек.
6 При условии предварительного покрытия тнгля расплавленным фторидом лнтня.
в Материал, изготовленный из неочищенных оксидов, менее устойчив.
Керамические материалы 31
Продолжение-
Теплопро- водность, ккал/(м-ч- -град) Химические свойства Область применения
Высокая; 9 Корозионная устойчивость Плавка металлов и сплавов
рсвкн; поэтому они не являются обязательными, н нх нн в коей мере не следует рас-
98100 Па (Н/м2).
Таблица 9. Стойкость различных оксидокерамических масс
к действию жидких реагентов8
Реагент Температу- ра, °C Материал иа основе
A12O3 ZrO2 ВеО
H2SO, конц. 338 ++ + —*—
НС1 конц. ПО + ++ —
HNO3 коиц. 122 + ++ ' +
HF конц. 120 ++ +++ ' —
Н3РО4 коиц. ++ ++
NaOH, 20%.-ный раствор 103 ++ ++ ’ ++
® Обозначения см. в табл. 8.
-------------------------------------------------------------i------------
продажу изделия иЗ стеклоуглерода имеют блестящую поверхность, они не
пористы с внешней стороны и газонепроницаемы. Из-за особенностей техно-
логического процесса их производства существуют определенные ограниче-
ния размеров (особенно толщины стенок) приборов из этого материала.
В кислородсодержащей атмосфере все виды материалов иа основе уг-
лерода при температурах выше 400 °C в той или иной степени подвержены
окислению.
Таблица 10. Стойкость различных оксидокерамических масс к действию
оксидов, гидроксидов и карбонатов3
Реагент Температу- Материал на основе
pa, “С А1й03 ZrO2 MgO ВеО
FIa2O2
NaOH
КОН
L12CO3
Ыа2СОз
К2СО3
<Си2О
В2О3
SiO2
РЬО
Sb2O3
Сг2О3
МоОз
WO3
Мп2О3
Мл3О4
FeO
Fe2O3
Р2О5 ’
500
500
500
1000
1000
1000
1300
1250
1780
1900
900
850
1900
800
1600
1600
1700
1500
1600
600
Все виды
оксидокерамических масс разрушаются
++ ’ I
а Обозначения см. в табл. 8.
Таблица 11. Стойкость различных оксидокерамических масс к действию
расплавленных солейа
Соль Темпера- тура, °C Материал на основе Соль Температу- ра, ”6 Материал , на основе.
Alio3J ZrO2 А12Оз ~ ZrO2
LiCI 800 4-4-4- j -4-4- KoSO, 1 9ЛЛ 1 1 1 1 I 1
Li2SiO3 T300 +++d 1 г H-+ CuS 1300 -Н—Т. ++ “Г-ГГГ
NaCl 900 +++- -++ Cu2SiO4 1400
NaCN 700 44+ -++ MgSiO» 1750 р -н-в
.NaF 1200 —t- H—F CaCl2 900 +++ +++
Na2MoO4 800 +++J -++ CaF2 1500 +ч-г
.NaNO3 600 4+f—ь - H-+ Ca3(PO4)2 1800 +++
-NaNOa 400 44+ - CaSiO3 1700
.NaPO» 800 _L_L_L _ -4-4- SrCb 1ЛЛЛ
-Na^PaO? 1200 1 1 1 1 r Sr(NO3)2 800 1—1—г +4- 14-+
-Na^SiOs 1300 +++- -++ SrSO4 1750 . + +4-
-Na2SO4 1150 [-4-4 1100 Л | ! 1 1 I
Na^SiFe -NasE^O? 1200 1000 4*4--L ++6 k4-4- BaSO4 ZnCh 1650 КПП И 1 г ++ 1 | L
-Na2WO4 700 TtT-j- 1 г 1 L 1 ККЛ П ГТ~ 1 |_ -t—f—t-
KHSO4 J 500 +++ 1 Г ++ PbB2O4 1300 J 1 4 |— 4-
KU 1000 PbSiOs 1300 1 L . ।i.i।। j1
KCN - 800 1' 1. 1 + PbSO4 1300 1 1 Г ++' 1*1
Д.Г 1000 — ++ PbS 1300 ++ —।——j—
1200 1.1 I □ L FeS 1350 □ L
П Г
K4P3O7 1200 +++
а Обозначения см. в табл. 8. ®MgO +++. вВеО —---------. rMgO ------; ВеО ++.
Керамические материалы 33
Таблица 12. Граница стойкости некоторых высокотемпературных материалов
при их взаимном контакте’
Материал w Mo А12Оз ThO2 ZrOg MgO ВеО
c 15006 15006 1400B 20006, в 16006, в 1800й 2300в
Шпинель ? ? 1900 1950 1950 1950 Неустой- чив
ВеО MgO ZrO2 ThO2 А120з Mo 1800B 2000» 1600® 2200® 1900 2000» 1800® 1600® 2200» 1900r>» 1900 1800r 1800r 17004 1800r 2100Д 2200е 2200» 19004, ж 20004 18004
а Приведенные температуры (°C) соответствуют нагреванию в вакууме при 10-*—
10~Е мм рт. ст. и времени контакта от 2 до 8 мин. Выше этой температуры наблюдаются
следующие побочные явления: 6 образование карбида; в восстановление: г взаимодей-
ствие; Л плавление; е улетучивание; ж приклеивание; ? — поведение неизвестно.
Таблица 13. Стойкость наиболее употребительных материалов для изготов-
ления тиглей к различным веществам, летучим при температурах выше
1000 °C
Л Обозначения: + -J- вполне устойчив;
б Испаряется в виде Се2Оз.
+ практически устойчив;
неустойчив.
ЛИТЕРАТУРА
1. Campbell I. Е„ High temperature
technology. J. Wiley, New York;
Chapman & Hall, London, 1956,
p.29.
2. Navias L., The technology of high-
temperature ceramics. General
Electric Res. Lab. Rep. 60-RL-
2375M, 1960.
3-143
34
Методы препаративной химии
3. Ullmanns Enzyclopadie der Tech-
nischen Chemie. Miinchen — Ber-
lin—Wien, 1966. Bd. 17, S. 569.
4. Johnson P. D., J. Amer. Ceram.
Soc., 33, 168 (1950); Ber. Dtsch.
Keram. Ges., 31, 81 (1954).
5. Eastman E. D., Brewer L., et al.,
J. Amer. Chem. Soc., 72, 2248
(1950).
6. Spedding F. H„ Daane A. H„ The
rare earths: Nonmetallic uses.
R. E. Krieger, Huntington N. Y.,
1971, p. 517.
7. Goehrens P., Einfiihrung in die
Metallographic. W. Knapp, Halle,
1948.
8. Jander W., Z. Anorg. Allgem.
Chem., 138, 321 (1924).
9. Auwarter M., Warmfeste und kor-
rosionsbestandige Sinter-Werkstof-
fe, Plansee-Seminar, Reutte, 1955,
S. 216.
10. Ryschkewitsch E„ Oxydkeramik
der Einstoffsysteme, Springer, Ber-
lin—Gottingen—Heidelberg, 1948.
11. Winzer R., Angew. Chem., 45, 429
(1932).
12. Kieffer R., Benesovsky F., Plan-
seeberichte, 5, 56 (1957).
13. Livey D. T„ Murray P.. In: Bock-
ris J. О. M., White J. L„ Macken-
zie J. D., Physiko-chemical measu-
rements at high temperatures. But-
terworths, London, 1959, p. 87.
4. Металлы
Кроме основных материалов — различных видов стекла и керамики — в
ряде случаев для изготовления приборов применяются металлы и сплавы.
Они отличаются прежде всего высокой тепло- и электропроводностью, осо-
быми механическими свойствами и высокой стойкостью к колебаниям тем-
пературы. К их достоинствам относится также хорошая устойчивость к
некоторым химическим реагентам, что используется, например, при работах
со фтором или со щелочами, щелочноземельными и земельными* металлами.
Незаменима металлическая аппаратура и при работах под высоким давле-
нием.
Медь, сплавы меди и благородные металлы
Хорошо известно применение меди в качестве проводника электричества;
кроме того, она используется как материал приборов для работ с фтором
(см. часть II, гл. 3). Из меди изготавливаются также теплообменники (на-
пример, змеевики). В продаже имеются медные трубки самых различных
размеров. Если их предварительно отжечь, то гораздо легче придать им
необходимую форму. Поскольку при изгибании твердость трубок снова воз-
растает, операцию отжига полезно повторить. Гибкие коммуникации, на-
пример между стальным баллоном с газом и аппаратурой, изготовляются из
тонких медных трубок, которые могут быть припаяны к резьбовому соедине-
нию или спаяны со стеклянными трубками (см. рис. 2). При прокалива-
нии в атмосфере водорода медь становится хрупкой.
Более гибки, чем медные, гофрированные шланги, обладающие высо-
кой подвижностью за счет прессованных волнообразных расширений. Они
изготавливаются из томпака или из высококачественной стали, причем по-
следние значительно дороже, но механически и химически гораздо устойчи-
вее, чем шланги из томпака. К другим частям аппаратуры гофрированные
шланги из томпака присоединяются либо путем тщательной пайки мягким
припоем (эти места нельзя сильно нагревать), либо при помощи специаль-
ных соединений на резьбе, а шланги из стали — пайкой твердым припоем.
Смазка, попавшая в такие шланги в процессе их изготовления, может быть
удалена путем промывания эфиром с последующей продувкой воздухом при
легком нагревании.
В тех случаях, когда важно обеспечить незначительную теплопроводность
аппаратуры, используют нейзильбер или аналогичные ему сплавы [1].
* К земельным металлам принято относить Al, Sc, Y, La. — Прим, перев.
Металлы 35
Применение серебра, также как и меди, связано с высокими значениями
тепло- и электропроводности, а также благодаря его устойчивости к
действию расплавленных щелочей. При температуре красного каления чи-
стое серебро рекристаллизуется и становится ломким. Этого не происходит
со сплавом серебра, содержащим 0,1% никеля.
Золото, как и серебро, не подвергается воздействию расплавленных ще-
лочей. Поскольку чистое золото слишком мягко, его применяют в виде
сплава с платиной. Тонким слоем золота можно покрывать стенки стальных
автоклавов, предназначенных для проведения гидротермальных синтезов. _
Из группы платиновых металлов находят применение платина, родий,
иридий и .палладий. Меры предосторожности, необходимые при работе с пла-
тиной, общеизвестны; о них можно справиться в изданиях фирм, производя-
щих благородные металлы (см. часть II, гл. 29). Родий применяется боль-
шей частью в виде сплавов (например, в термоэлементах, нагревательных
элементах). При условии принятия особых мер защиты от окисления кис-
лородом воздуха он используется и в чистом виде как материал тиглей
для работы при особо высоких температурах. Иридий имеет значительно
более высокую температуру плавления и более низкое давление пара, чем
платина. Однако в кислородсодержащей атмосфере оба металла улетучива-
ются значительно с большей скоростью, чем это соответствует их собственно-
му давлению пара, причем при сравнимых условиях потери иридия значи-
тельно больше, чем платины. Все же в особых случаях иридий применяют
как материал сосудов для нагревания сильноосновных оксидов, таких, как
ВаО, в кислородсодержащей атмосфере. К примеру, из иридия изготовля-
лись сосуды в виде желоба, нагреваемого непосредственным пропусканием
электрического тока [2]. Платино-иридиевые сплавы при достаточном со-
держании иридия устойчивы к действию хлора. Палладий дешевле плати-
ны, он применяется в основном как составная часть сплавов. Высокую про-
ницаемость палладия для водорода при температуре красного каления ис-
пользуют при получении особо чистого водорода (см. часть II, гл. 1).
Ниобий, тантал, молибден, вольфрам
Эти тугоплавкие металлы с незначительным давлением пара и высокой
механической прочностью при небольшом коэффициенте термического рас-
ширения находят разнообразное применение в качестве материалов для
изготовления сосудов и нагревательных элементов. Из этих металлов про-
изводятся готовые изделия, а также фольга, трубки, проволока и т. д. На-
гревание этих материалов до температуры выше 500 °C может производиться
лишь в атмосфере защитного газа или в вакууме. Для молибдена и воль-
фрама защитным газом, кроме инертных, может быть водород или смесь
водорода и азота (газ для синтеза аммиака), а для ниобия и тантала—•
только инертные газы. Тантал весьма устойчив к действию хлороводорода,
а молибден даже при нагревании не разрушается в контакте с щелочными,
щелочноземельными и земельными металлами. Для механической обработки
очень твердого вольфрама необходим специальный инструмент.
Железо и никель
Использование этих металлов в лабораторной практике в качестве кон-
струкционных материалов достаточно хорошо известно. Особо чистые желе-
зо (например, карбонильное железо) и никель, а также легированная сталь
служат материалом для изготовления тиглей и лодочек. Они устойчивы к
действию жидких и газообразных щелочных и щелочноземельных металлов
при повышенных температурах и в меньшей степени — к действию расплав-
ленных гидроксидов щелочных металлов. При этом следует по возможно-
сти исключить контакт с кислородом воздуха. Если требуется из тиглей
и лодочек, изготовленных из обыкновенной жести, удалить углерод, то их
начительное время прокаливают в потоке влажного водорода.
3*
36 Методы препаративной химии
ЛИТЕРАТУРА
1. D’Ans—Lax, Taschenbuch fur Che-
miker und Physiker. 3. Aufl., Bd. I.
Springer, Berlin — Heidelberg —
New York, 1967, S. 711.
2. Wagner G., Binder H„ Z. Anorg.
Allgem. Chem., 297, 328 (1959).
Ртуть
В лабораторной практике ртуть находит разнообразное применение:
в качестве рабочего тела (пары) в диффузионных насосах, для заполнения
манометров, как запирающая жидкость в вентилях особой конструкции, для
осуществления контакта в электрических реле. Важной операцией является
очистка ртути. От механических примесей ртуть очищают фильтрованием
Рис. 5. Установка для очистки ртути.
через кожу, тигель с пористым стеклянным или фарфоровым дном, а также
через бумажный фильтр, конец которого во многих местах проколот иглой.
Растворенные в ртути металлы можно удалить, либо встряхивая ртуть с
различными окислителями или кислотами, либо продувая через нее воздух;
лучше, однако, комбинировать эти методы, как показано на рис. 5. Очистку
ртути рекомендуется также производить путем последовательного встряхи-
вания ее с 5%-ным раствором нитрата ртути(1), с 15%-ной азотной кис-
лотой, с очень разбавленным раствором азотной кислоты и, наконец, с во-
дой. Весьма эффективна обработка ртути насыщенным на холоду раствором
КМпОч. Для этого ртуть многократно встряхивают со свежими порциями
раствора перманганата калия до тех пор, пока цвет раствора не будет
оставаться неизменным в течение полминуты. Затем ртуть промывают во-
дой, дают efi осесть и подкисляют небольшим количеством HNO3, при этом
диспергированные частички соединяются в общую массу жидкой ртути. По-
лученную ртуть снова промывают, высушивают, нагревая в вакууме, и, на-
конец, перегоняют.
В продаже имеется большое число приборов для перегонки ртути
(рис. 6). Приборы из стекла дюран, которые легко могут быть изготов-
лены и в лаборатории, изображены на рнс. 7 и 8. Их укрепляют на под-
ходящем штативе. После однократного отсасывания газа через кран при-
бора в последнем сохраняется вакуум в течение продолжительного времени
(даже в том случае, если в него вместе с ртутью проникли следы газа, так
как трубка 1 диаметром, не превышающим 2 мм, действует как спускная
трубка насоса Шпренгеля). Ввиду того что эти приборы должны работать
значительное время без присмотра, целесообразно включать параллельно
виткам нагревателя контрольную лампочку.
Металлы 37
Чистая ртуть ие должна при переливании оставлять налета. Для соби-
рания разбрызганной ртути (ядовита!) служат ртутные щипцы с наконечни-
ками, имеющими полусферическую форму, или ртутная пипетка (рис. 9). По-
следняя приводится в действие присоединенным к пей вакуумным иасосом.
При помощи ртутной пипетки разбрызганную ртуть можно удалить даже из
щелей.
Рис. 7. Схема установки для автоматиче-
ской очистки ртути путем перегонки.
Длина днстнлляцнонного сосуда —180 мм, диа-
метр —35 мм; изоляция толстым слоем асбеста.
Рис. 6. Схема установки для
автоматической очистки ртути
путем перегонки.
1 — трубка для слива ртути; 2 —
охлаждающие манжеты с прорезя-
ми и отгибами (изготовлены нз
алюминиевой или медной фольги).
Для сбора мелких капель ртути можно воспользоваться так называемым
«ртутным магнитом». Он представляет собой палочку металлического цин-
ка, которую предварительно опускают иа короткое^ время в разбавленную
H2SO<. Хранят ее в плотно закрытой пробирке. Действие «ртутного магни-
та» основано на образовании амальгамы.
Сварка и пайка металлов
Соединение металлических деталей путем сварки следует предпочесть
другим методам. Платиновые металлы сваривают либо непосредственно в
кислородио-водородном пламени, либо путем нагревания деталей до светло-
красного каления с последующим резким ударом по ним молотком. Свари-
38 Методы препаративной химии
ванне других металлов проводят в специализированной мастерской. Дуговая
сварка тантала или молибдена в атмосфере аргона требует наличия спе-
циальной аппаратуры.
Пайка твердым припоем дает меньшую термическую стойкость. В этом
способе применяют так называемые «твердые» (серебряные) припои (т. пл.
— 700 °C) или чистое серебро (т. пл. 960°C). Зачищенные места пайки
обильно смазывают или посыпают бурой и после достижения необходимой
температуры (с помощью паяльной или сварочной горелки) наносят при-
200
Рис. 8. Схема установки для автоматиче- Рис. 9. Пипетка для сбора
ской очистки ртути путем перегонки. ртути.
Все размеры указаны в миллиметрах.
пой в виде порошка или проволоки. Молибден и вольфрам можно паять чи-
стым серебром в присутствии большого количества буры.
Способ пайки мягким свинцово-оловянным припоем общеизвестен. Для
снятия с поверхности мест пайки оксидной пленки служит «паяльная жид-
кость» (раствор ZnCIs и NH4C1 в разб. НС1) или «паяльная смазка»; необхо-
дима также предварительная тщательная механическая зачистка мест пайки.
При пайке мягким припоем необходимо лишь незначительное нагревание
изделий; правда, и механическая прочность в этом случае наименьшая.
Алюминий и его сплавы (легкие металлы) не спаиваются обычным мяг-
ким припоем. Для этого имеются специальные припои, но при пользовании
ими требуются определенные навыки.
5. Синтетические материалы
В настоящее время в лабораторной практике используются многие син-
тетические вещества либо в готовых приборах, либо как сырье для изготовле-
ния собственными силами аппаратов, соответствующих конкретно поставлен-
Синтетические материалы 39
Hoft цели. Кроме того, синтетические материалы служат для смазывания,
уплотнения, склеивания и покрытия поверхностей других материалов. Они
обладают также весьма ценным для химиков свойством — химической стой-
костью по отношению главным образом к неорганическим веществам. При-
менение этих материалов ограничено, однако, из-за сравнительно низкой тер-
мической устойчивости и способности набухать во многих органических рас-
творителях, что снижает их твердость и химическую стойкость. Далее, сле-
дует иметь в виду, что проницаемость синтетических материалов для газов,
таких, как Н2, N2, О2, СО2, и прежде всего для паров воды значительно
выше, чем стекла и металлов. Количество газа, диффундирующего за опре-
деленное время в сосуд, например через шланг, при .прочих равных усло-
виях обратно пропорционально толщине стенки. Сравнительно высокую про-
ницаемость по отношению к кислороду имеют силиконовый и вулканизиро-
ванный натуральный каучуки. Проницаемость для паров воды уменьшается в
следующем ряду:
Ацетат целлюлозы» резина, бутадиен-натриевый каучук марок Буна S,
Буна Л/>найлои>полистирол, полиметакрилат>поливинилхлорид ^поли-
этилен > тефлон
Термопластичные синтетические материалы можно сваривать при нагрева-
нии, для чего специально изготавливают «прутки для сварки» из того же
материала. Нагревание производят при помощи простого устройства для
подачи горячего воздуха с регулируемой температурой или высокочастот-
ными нагревательными приборами.
Политетрафторэтилен (ПТФЭ, товарные наименования: тефлон, хоста-
флон, флуон)
ПТФЭ (тефлон) не термопластичен, может применяться при температу-
рах от —200 до +275 °C. Даже при повышенных температурах он устойчив
к Действию всех агрессивных газов и жидкостей, за исключением расплавлен-
ных щелочных металлов, жидкого аммиака и свободного фтора. Тефлон не
подвержен набуханию, он не хрупок и не горюч, но выше ~ 400 °C пре-
терпевает деполимеризацию. В продажу он поступает в виде полуфабрикатов
(пластины, трубки, шланги, прутки, фольга) или эмульсии, наносимой пу-
тем разбрызгивания, а также (с добавками) в виде затвердевающей пасты.
Тефлон легко поддается токарной обработке, его можно резать и сверлить.
Перед склеиванием тефлона поверхности деталей необходимо предваритель-
но придать шероховатость.
Тефлон марки ФЭП (фтор — этилен — пропилеи) в основном аналогичен
обычному тефлону, ио может применяться при температурах от —270 до
+250 С.
Политрифторхлорэтилен (ПТФХЭ, хостафлон, флуоротен, кель-f)
Этот материал инертен почти так же, как тефлон, ио легче поддается
обработке благодаря своей! термопластичности. При нагревании до —190 °C
материал становится пластичным, при 270—300 °C наступает разложение,
а при дальнейшем нагревании — плавление. На политрифторхлорэтилен не
действуют плавиковая кислота, газообразный хлор и кипящая концентриро-
ванная азотная кислота. Напротив, при кипячении в некоторых органических
растворителях, например CCli, трихлорэтилене, диэтиловом эфире и толуо-
ле, происходит его набухание (см. разд. 7).
Полиолефины
I. Полиэтилен низкого давления (хостален, луполен, марлекс, вестолен)
тт Полиэтилен высокого давления (луполен, алатон, алкатен)
III. Полипропилен (хостален ПП, моплеи, профакс)
IV. Полиметилпентилен (ТРХ)
40 Методы препаративной химии
Термостойкость, °C
Температура, при кото-
рой появляется хруп-
кость, °C
I
120
<—120
II
90
.<—55
ш
140
<—18
IV
180
Полиолефины относятся к материалам, обладающим повышенной хими-
ческой стойкостью к действию кислот, щелочей, солей, их растворов, воды.
Для всех групп полиолефинов максимальное количество абсорбированной
воды составляет менее 0,01%. Особенно устойчивы полиолефины при ком-
натной температуре к действию водных растворов аммиака, бромоводород-
ной, плавиковой (до 100%), соляной, хромовой, хлорной (10%), фосфор-
ной, азотной (до 50%), серной (до 95%) кислот, пероксида водорода (до
100%) и трихлорида фосфора. Ароматические, алифатические и хлорирован-
ные углеводороды при комнатной температуре вызывают незначительное на-
бухание. Органические кислоты, эфирные масла и галогены абсорбируются
полиолефинами или диффундируют сквозь них. При повышенных темпера-
турах возникает опасность окисления под действием воздуха или других
окисляющих веществ, особенно при одновременном освещении солнечным
светом.
Поливинилхлорид (ПВХ, хосталит, виннол, весталит, лютофан, трови-
дур, миполам)
Поливинилхлорид термопластичен; он начинает размягчаться выше 70 °C.
Абсорбция воды составляет 0,1%.
В отсутствие пластификаторов поливинилхлорид очень тверд, не имеет
запаха и чрезвычайно устойчив к действию химических веществ. Используе-
мые в лаборатории поливинилхлоридные полупрозрачные трубки достаточно
эластичны, что достигается добавкой пластификаторов, причем это приво-
дит к понижению химической стойкости. Вообще для всех синтетических ве-
ществ существует опасность снижения (иногда значительного) их коррози-
онной стойкости при абсорбции даже небольших количеств органических рас-
творителей.
Полиамиды (найлон, ультрамид, дуретан)
Полиамиды — очень вязкие и устойчивые к истиранию вещества. Они
выдерживают кратковременное нагревание до 160 °C, однако уже выше
100 °C на воздухе может наблюдаться выцветание вследствие окисления.
Полиамиды обратимо поглощают из воздуха пары воды до равновесной
концентрации. Таким же образом абсорбируются жидкая вода и алифати-
ческие спирты с низкой молекулярной массой. Абсорбированная вода при
содержании ~3% сообщает материалу повышенную эластичность. Полиами-
ды устойчивы к действию большинства органических растворителей (за ис-
ключением фенола) и щелочных водных растворов. Кислые растворы, в том
числе концентрированные муравьиная и уксусная кислоты, так же как ве-
щества-окислители, более или менее быстро взаимодействуют с полиамидами.
Поликарбонаты (например, макролон)
Макролон можно применять примерно до 140 °C, так как выше этой
температуры он размягчается. При 225 °C он плавится, а при 320 °C разла-
гается. При низких температурах механические свойства не ухудшаются:
материал не становится хрупким в жидком воздухе. Макролон устойчив в
растворах солей и кислот даже при значительных концентрациях последних,
а также к действию окислителей и восстановителей. Напротив, он разруша-
ется растворами аммиака, аминов и щелочей. Макролон легко растворим в
метиленхлориде, циклогексаноне, диметилформамиде и крезоле.
Чистые и обезвоженные растворители 41
Полистирол (луран, полистирол, вестирон, тролитул)
Полистирол — твердый, хрупкий, прозрачный как стекло или мутнова-
тый материал, устойчивый до 70°C (некоторые сорта —до 100°C). Он инер-
тен по отношению к кислотам, щелочам, спиртам, маслам и жирам, но раз-
рушается конц. HNO3, бензином, бензолом, простыми и сложными эфирами,
кетонами и хлорированными углеводородами. Сополимеры стирола с бутадие-
ном или с акрилонитрилом менее хрупки.
Полиметакрилат (плексиглас, плексигум, диакои, перспекс, резарит)
Плексиглас прозрачен, вязок, легко поддается механической обработке.
Он устойчив до 90 °C; вода абсорбируется до содержания 0,3%. Плексиглас
устойчив в разбавленных растворах кислот и щелочей, а также в растворах
солей. Многие органические растворители разрушают плексиглас, а в хло-
роформе он растворяется.
Силиконовый каучук
Силиконовый каучук и без добавок пластификаторов или других ве-
ществ сохраняет свою эластичность по крайней мере до —60 СС. Его свой-
ства незначительно зависят от температуры. Он выдерживает длительное
нагревание до 180 °C, а на короткое время может быть нагрет даже до
300 °C. Силиконовый каучук химически весьма инертен; в нем не содержит-
ся каких-либо летучих или экстрагируемых составных частей.
Силиконовый каучук выпускается также и В виде подвижной массы, за-
стывающей при комнатной температуре, и в виде ленты, способной! свари-
ваться при комнатной температуре.
Другие эластомеры
Это, например, сополимеры: пербунан (из бутадиена и акрилонитрила),
бутилкаучук (из изобутилена и изопрена) и неопрен (поли-2-хлорбутадиен).
Для очистки приборов из синтетических веществ ни в коем случае нель-
зя пользоваться продажными моющими веществами. Прибор промывают на-
гретым до кипения мыльным щелоком с небольшой добавкой поверхностно-
активных веществ (средство для ополаскивания посуды в домашнем хозяй-
стве). Оставшиеся загрязнения можно удалить разбавленной азотной кисло-
той либо кратковременным ополаскиванием спиртом.
6. Чистые и обезвоженные растворители
При проведении синтезов неорганических препаратов кроме воды и сжи-
женных газов используются в качестве растворителей также органические
жидкости, например спирты, диэтиловый эфир, бензол, тетрахлорид углерода,
пиридин, сероуглерод, ацетон и др. (см. с. 43—45 и табл. 15). Очень часто
наличие воды в органических растворителях мешает работе. Ниже будут
описаны прежде всего способы осушки растворителей, приводящие иногда к
частичному удалению и других примесей.
В продаже имеются растворители высокой степени чистоты •— для спект-
роскопических целей. Они, однако, могут быть легко получены из менее чи-
стых продуктов по методикам, описанным в литературе. Чистоту растворите-
ля определяют спектроскопически (УФ-спектр снимают при толщине слоя
1.0 мм, ПК-спектр — при толщине 0,1—I мм), сравнивая полученные резуль-
таты с данными для продажных препаратов высокой степени чистоты.
Вода особой чистоты
Подробно см. часть II, гл. 1. Для получения воды особой чистоты в
лабораторном масштабе в продаже имеются различные аппараты. . Хорошо
зарекомендовала себя, например, установка, работающая автоматически в
непрерывном режиме, в которой сначала происходит деминерализация воды
42
Методы препаративной химии
путем ионного обмена, а затем вода дважды перегоняется в кварцевом со-
суде. Воду, полученную таким образом, следует сохранять в кварцевой по-
суде. Ее чистота контролируется по значению электропроводности.
Обезвоженные растворители
Органические растворители теперь обычно обезвоживают не химическим
путем, а путем адсорбции воды на подходящем осушителе. Преимущества
этого метода заключаются в его простоте, быстроте, в более глубокой сте-
пени осушки, приводящей часто к удалению других примесей. Адсорбцион-
ный метод осушки можно применять ко всем растворителям. Многие из ад-
сорбентов легко регенерируются.
Осушка на активном оксиде алюминия. Растворитель медленно пропус-
кают через колонку с основным или нейтральным оксидом алюминия. Для
этой цели используют имеющийся в продаже А12О3 с наибольшей степенью
активности (I) и колонку высотой 30—40 см и диаметром 20—40 мм. При
наполнении сухой А12О3 несколько уплотняют путем легкого постукивания.
Растворитель подают в верхнюю колбу и по каплям со скоростью 200 мл/ч
пропускают в приемник. Степень осушивания различных растворителей на
активном А12О3 приведена в табл. 14, причем начальная фракция 50—200 мл
часто менее чистая, и поэтому ее собирают отдельно. В таблице указано
остаточное содержание воды в основной фракции. Если исходное содержание
воды в растворителе, подвергаемом осушке, меньше, чем это указано в
табл. 14, то может быть получен соответственно больший объем обезвожен-
ной фракции.
Таблица 14. Осушка растворителей с помощью оксида алюминия
Растворитель А12Оз а Диаметр колонны, мм Объем осушен- ной фракции, мл Содержание воды, %
исход- ное оста- точное
Диэтиловый эфир 100 г, нейтральный 22 50- 250 0,27 0,08
100 г, основной 22 200- 600 1,28 0,01
Бензол, насыщенный во- дой 25 г, основной 15 100- 2500 0,07 0,004
Хлороформ, насыщенный водой 25 г, основной 15 50-800 0,09 0,005
Этилацетат, насыщенный водой 250 г, нейтральный 37 150—350 3,25 0,01
а Данные приведены для А12Оз марки Akt.I, выпускаемого фирмой Woelm.
При осушке на А12О3 диэтилового эфира, а также эфиров с большой мо-
лекулярной массой и углеводородов одновременно происходит очистка от
возможной примеси пероксидов. При осушке хлороформа на основном А12О3
(марки Akt. I) из него удаляется добавленный для стабилизации этанол (ус-
ловия очистки: диаметр колонны 37 мм, масса А12О3 250 г, скорость пропус-
кания 0,1—1 л/ч, объем очищенной от спирта фракции 150—550 мл). Полу-
ченный таким образом хлороформ в охлажденном состоянии может хра-
ниться лишь несколько дней. Производить регенерацию А12О3 не рекоменду-
ется.
Чистые и обезвоженные растворители 43
Осушка на молекулярных ситах. Молекулярные сита — это синтетические
цеолиты, в кристаллической решетке которых имеются многочисленные пу-
стоты, соединенные между собой каналами определенного размера, например
диаметром ~ 0,3; 0,4 или 0,5 нм. Если воду, содержащуюся в пустотах и
Рис. 10. Схема аппарата для высу-
шивания органических растворителей
на активном оксиде алюминия или
на молекулярных ситах.
каналах, удалить путем нагревания, получается чрезвычайно активный ад-
сорбент. Благодаря строго определенному диаметру каналов в пустотах
цеолита происходит адсорбция лишь таких веществ, молекулы которых име-
ют меньшие размеры.
Для осушки органических растворителей используют молекулярные сита
с диаметром каналов 3 Л или 4 А. Лучше всего проводить осушку в ди-
намическом режиме: растворитель пропускают через колонну, заполненную
молекулярным ситом (рис. 10). В колонну высотой 60 см и диаметром 25 мм
загружают 250 г молекулярного сита в .виде шариков или палочек. Вслед-
ствие активного поглощения осушителем воды, содержащейся в воздухе,
операцию загрузки надо производить очень быстро. После заполнения ко-
лонны осушаемой жидкостью устанавливают скорость пропускания 2—3 л/ч.
Первые 250 мл собирают отдельно, так как в них содержатся еще следы
воды и частички пыли; следующие порции элюата выходят уже чистыми.
Степень очистки различных растворителей, достигаемая таким способом,
приведена в табл. 15.
При осушке описанным методом имеющихся в продаже более сухих
углеводородов и хлорированных углеводородов могут быть получены гораз-
до большие количества растворителей. Весьма глубокой осушки можно до-
стичь, применяя комбинированное. фильтрование: сначала через активный
оксид алюминия и, затем через молекулярное сито. Осушенные растворители
хранятси над регенерированным, . очищецнцм от ,пыли молекулярным ситом
(10 г/лк ' - ь \ . . ..
.44 Методы препаративной химии
Таблица 15. Осушка растворителей с помощью молекулярных сит
Растворитель Содержание воды, % Тип молеку- лярного сита Объем рас- творителя, осушаемый 250 г моле- кулярного сита (без регенера- ции), л
исходное остаточное
Диэтиловый эфира 0,12 0,001 4 А 10
Диэтиловый эфир6 1,17 0,004 4 А 3
Диизопропиловый эфир® 0,03 0,003 4 А 10
Диизопропиловый эфир6 0,53 0,002 4 А 8
Тетрагидрофуран3 0,04-0,2 0,001—0,003 4 А 7—10
Диоксана 0,08—0,28 0,002 4 А 3—10
Бензол® 0,07 0,003 4 А 10
Толуол6 0,05 0,003 4 А 10
Ксилол6 0,045 0,002 4 А 10
Циклогексан6 0,009 0,002 4 А 10
Днхлорметан6 0,17 0,002 4 А 10
Хлороформ6 0,09 0,002 4 А 10
Тетрахлорид углерода6 0,01 0,002 4 А 10
Пиридин® 0,03—0,3 0,004 4 А 2-10
Ацетонитрил® 0,05-0,2 0,002—0,004 3 А 3—4
Этилацетат® 0,015—0,21 0,003-0,006 4 А 8—10
Диметилформ амида 0,06-0,3 0,006—0,007 4 А 4—5
Метанол" 0,04 0,005 ЗА 10
Этанол" 0,04 0,003 3 А 10
Изопропанол" 0,07 0,006 3 А 7
а Продажный. Насыщенный водой. в Колонна диаметром 50 мм и длиной 200 см.
Молекулярные сита могут быть многократно регенерированы без суще-
ственной потери ими адсорбционной емкости. Для этого препарат с целью
вытеснения из него адсорбированного растворителя высыпают в большое
количество воды. Полное удаление органических примесей достигается до-
полнительной промывкой метанолом. После основательной промывки водой
проводят предварительную сушку при 200—250 °C в сушильном шкафу. Для
ряда работ бывает достаточной последующая регенерация в сушильном
шкафу при 300 °C. Обычно же более полное удаление воды осуществляют
при 300—350 °C с одновременной откачкой масляным вакуумным насосом.
При этом пары воды следует вымораживать путем конденсации в ловушке,
установленной перед насосом.
Молекулярные сита, имеющиеся в продаже, содержат 1—2% воды, так
что перед их использованием необходимо произвести регенерацию, особенно
если предстоит работа с полярными растворителями, трудно поддающимися
осушке.
Сероуглерод почти так же легко разлагается, как чистый хлороформ.
Поэтому сероуглерод очищают лишь химическим путем. Спектроскопически
чистый сероуглерод можно получить, выполняя следующие операции: 12-ча-
совое встряхивание 1 л исходного продукта с 5 г HgCl2 и небольшим ко-
личеством ртути; декантацию и промывку водой; фракционную перегонку над
СаО, после, которой сероуглерод следует оставить стоять в течение 12 ч с
конц. HNO3; промывку водой; фракционную перегонку' над Р4О10.
Замазки и смазочные вещества
45
Отметим, что особенно химически стойки фтор- н хлорпроизводные уг-
леводородов, а также полностью фторированные амины и эфиры, иногда ис-
пользуемые в качестве растворителей. В отдельных случаях применяют не-
сколько менее распространенные, но хорошо себя зарекомендовавшие рас-
творители, такие, как диметилсульфоксид (ДМСО), тетраметилепсульфон
(сульфолан, ТМС), трис (диметиламино) фосфиноксид, нитрометан, нитро-
бензол, ацетонитрил, бензонитрил, тетраметилмочевина (ТММ), диметилформ-
амид (ДМФА), тетрагидрофуран (ТГФ), диоксан, а также различные эфи-
ры этиленгликоля и диэтиленгликоля (в продажу поступают под названием
карбитол или дованол; метиловый эфир этиленгликоля — моноглим; метило-
вый эфир диэтиленгликоля — диглим).
Следует помнить, что хлорированные углеводороды и сероуглерод во
избежание взрыва нельзя осушать при помощи натрия или других щелочных
металлов. Натрий применяют для осушки углеводородов, аминов и эфиров.
Гораздо легче в обращении, чем чистый натрий, сплав, содержащий 10%
натрия и 90% свинца и представляющий собой сухие гранулы (имеется в
продаже* под названием «Dri-Na»). Сплав практически устойчив на возду-
хе, и его можно просто брать из скляики и вносить в растворитель, подле-
жащий осушке. Защищать поверхность сплава от корки оксида и гидрокси-
да, как это делают в случае использования чистого натрия, нет необходи-
мости.
ЛИТЕРАТУРА
7. Wohlleben G., Angew. Chem., 67,
741 (1955); 68, 752 (1956).
8. Каталоги фирм E. Merck (Darm-
stadt); M. Woelm (Eschwege).
9. Stephen H., Stephen T„ Solubilities
of inorganic and organic compo-
unds. Pergamon, Oxford, London,
New York, Paris, 1963—64.
10. Linke W. F., Seidell A., Solubili-
ties of inorganic and metal-orga-
nic compounds. Amer. Chem. Soc.
and Van Nostrand, Washington
and Princeton. Vol. I, 1958; Vol.
II, 1965.
1. Houben-Weyl, Methoden der orga-
nischen Chemie. Band 1/2. Georg
Thieme, Stuttgart, 1959, S. 769.
2. Hersh Ch. K., Molecular sieves.
Reinhold, New York, 1961.
3. Gruber 0., lirti P., Ralek M., Mo-
lekularsiebe. VEB Deutscher Ver-
lag der Wissenschaften, Berlin,
1968.
4. Hesse G., Schildknecht H„ Angew.
’ Chem., 67, 737 (1955).
5. Pestemer M.. Angew. Chem., 63,
118 (1951); 67, 740 (1955).
6. Stoper R., Pritze B., Z. Chem., 13,
92 (1973).
7. Замазки и смазочные вещества
Жвроподобные смазки
Их используют в основном для смазки шлифов и кранов. Имеется боль-
шое число смазочных материалов, важнейшие из которых перечислены в
табл. 16.
Смазка Рамзея особенно недорога. Ею смазывают краны и шлифы на
форвакуумных линиях. Она состоит из специальных сортов вазелина, ко-
торый за счет добавки каучука приобретает необходимую консистенцию.
Существует два сорта этой смазки: вязкая — для смазывания шлифов и
кранов и мягкая — для кранов больших размеров, эксикаторов и т. д.
Высоковакуумная смазка типа Р‘ (фирма-изготовитель Leybold) имеет
меньшее давление пара, чем смазка Рамзея, но несколько большее, чем смаз-
ка типа R. Смазка типа Р содержит высокомолекулярные, твердые или по-
выпускается фирмой Baker Chemikalien (GroB-Genau).
46
Методы препаративной химии
Таблица 16. Виды смазок, используемых в лабораторной практике
Смазка а Область приме- нения Давление пара при 26 °C, мм рт. ст. Темпера- тура разжиже- ния, сС Макси- мальная рабочая темпера- тура, °C
Смазка Рамзея, вязкая Шлифы 10-* 56 25
Смазка Рамзея, мягкая Большие шлифы, краны, эксикаторы 10-“ Неизме- 56 25
Высоковакуумная смазка типа Р Высоковакуумная смазка типа R Шлифы, краны римо мало То же 65 65 25 30
Лителен Силиконовая смазка (Шлифы и краны при высоких и низких температу- рах « 210 >200 150 150
Кель-F 90, VOLTALEF (по- литрифторхлорэтилен) Шлифы, краны <10-з 205 175
Смазка для уплотнений ти- па DD Вентили Неизме- римо мало 58 120
» Выпускаются фирмами Leybold-Heraeus GmbH u. Со. (Koln); Lehmann u. Voss
u. Co. (Hamburg) (кель-F, VOLTALEF).
лужидкие при комнатной температуре углеводороды, очищенные путем вы-
соковакуумной обработки от примесей, имеющих более высокое давление
пара.
Высоковакуумная смазка типа R* более вязка, чем смазка типа Р. Она
в еще большей степени очищена от летучих примесей. Ее используют в тех
случаях, когда требования к конечному вакууму особенно высоки или если
важно отсутствие в паре углеводородов.
Лителен отличается незначительной зависимостью своей консистенции от
температуры. Поэтому его наряду с силиконовой смазкой можно применять
как при низких, так и при высоких температурах. Лителен содержит в сво-
ем составе литиевые мыла и неустойчив к действию жидких реагентов и аг-
рессивных газов.
Высоковакуумная силиконовая смазка обладает повышенной термической
устойчивостью,- Ее смазочные свойства, однако, не остаются в той же сте-
пени неизменными при изменении температуры, как это имеет место у лите-
лена. При температурах выше 200 °C силиконовая смазка полимеризуется с
одновременным выделением газов.
Обычно силиконовую смазку используют при работах с агрессивными
веществами. Если она все же оказывается неустойчивой, применяют смазки
на основе фторированных углеводородов и фторированных хлорпроизводных
углеводородов («фторная смазка»). Хорошо себя зарекомендовал политри-
фторхлорэтилен (ПТФХЭ, кель-F 90, VOLTALEF), стойкий к действию озо-
* Эти смазки ранее назывались апиезоновыми. — Прим, перев.
Замазки и смазочные вещества 47
на, SbCl3, СгО2С12, Р0С13, дымящей HNO3, H2SO«, HSO3C1, галогенов и др.
С другой стороны, его смазочные свойства несколько хуже, чем силиконовой
смазки. Применение этого типа смазки противопоказано лишь при контакте
с сильно электроположительными металлами (например Al, Mg) при одно-
временном наличии механических нагрузок (стирание мелких частичек ме-
талла), а также при температурах выше 250 °C. Эта смазка выпускается,
кроме того, в виде масла или с консистенцией воска.
Смазка для уплотнений типа DD фирмы Leybold состоит из смеси жид-
ких и твердых углеводородов.
Все перечисленные виды смазок, выпускающихся также в виде смазочных
карандашей или в склянках с разбрызгивателем, легко могут быть удалены
при помощи ваты, смоченной в бензоле или эфире.
Смазки, не содержащие жиров
Когда шлифы или краны приходят в соприкосновение с органическими
растворителями, описанные выше смазочные вещества неприменимы. В этом
случае вместо обычных шлифов используют прецизионные прозрачные шли-
фы с оплавленной поверхностью. Такого же типа краны* применяют без
смазки, заменяя ее специальными тефлоновыми прокладками. Такой кран не
пропускает жидкости, но не является вакуумно-плотным. В особых случаях
(например, для работы при высоких температурах) соединения на шлифах
уплотняют при помощи пленок из полиэтилена или кель-Е. При этом пленку
вставляют между предварительно нагретыми частями шлифа. Если шлифы
были нагреты достаточно сильно, пленка при сжатии плавится и растекается,
давая прозрачное соединение, которое при охлаждении затвердевает, а при
нагревании снова разжижается (т. пл. пленки из полиэтилена 90—120 °C,
пленки из кель-Е— 200—230 °C). Следует указать, что при слишком силь-
ном охлаждении пленка из кель-Е становится настолько твердой, что не
обеспечивает надежного уплотнения; тогда используют пасту на основе
кель-Е. Особенно устойчивы к действию органических растворителей, соеди-
нений фтора, а также к изменению температуры манжеты, изготовленные из
тефлона, которые, однако, не дают вакуумно-плотного соединения. Тефлон
выпускается и в виде эмульсии, твердеющей после ее нанесения. В проти-
воположность кель-Е тефлон не термопластичен, но благодаря своему низко-
му коэффициенту трения обладает самосмазывающими свойствами**. Пленки
из кель-Е при 25 °C инертны по отношению к кислотам, щелочам, окислите-
лям, этанолу и в различной степени также к другим растворителям. Тефлон
устойчив при температурах от —200 до +260 °C к спиртам, высшим эфирам,
кетонам, анилину, бензолу, галогенам, трифториду бора, галогеноводородам,
щелочам, кислотам, хлориду сульфинила и др.
Иногда для уплотнений, стойких в среде эфира, можно пользоваться
смесью расплавленного сахара с глицерином. Рекомендуют также растирать
в фарфоровой чашке 25—35 г декстрина, постепенно прибавляя к нему
35 мл глицерина, а затем, помешивая, подогревать все вместе на открытом
огне до получения медообразной мази. Последняя должна быть дважды до-
ведена нагреванием до вспенивания и, наконец, профильтрована через вату.
Эта смазка отличается гигроскопичностью и немного большей вязкостью, чем
вазелин. Сохранять смазку следует в склянке с притертой пробкой. Иногда
пользуются также смазкой в виде пасты, приготовленной из очень мелкого
бентонита и глицерина.
* Например, выпускаемые фирмой R. Brand, Glashiitte (Wertheim/Main).
** Фирмы, выпускающие материалы: на основе кель-Е — Lehmann и.
Voss (Hamburg), иа основе тефлона — С. Huth u. Sohne (Bietigheim);
R. Brand, Glashiitte (Wertheim/Main).
48
Методы препаративной химии
Обратимые и необратимые замазки
Замазки должны иметь низкое давление пара, не быть слишком хруп-
кими и обладать достаточной химической стойкостью.
Пицеин (черный)—классическая вакуумная замазка. Он приготавлива-
ется из битумных материалов, термопластичен, хорошо пристает к тща-
тельно очищенной и предварительно нагретой поверхности металла или
стекла. Соединения на пицеине можно снова разъединить при нагревании.
Пицеин устойчив к действию слабых оснований, кислот, других водных рас-
творов, но неустойчив в органических растворителях (удаление эфиром или
бензолом).
Давление пара при 25 °C составляет ~10-4 мм рт. ст., температура раз-
жижжения 90 °C, максимальная рабочая температура 50 °C.
Хлорид серебра (т. пл. 455 °C) служит главным образом для вакуумно-
плотного приклеивания плоских окошек из стекла или кварца к стеклянным
приборам. Его можно применять до ~300 °C; при этом давление пара
—10~7 мм рт. ст. Хлорид серебра готовят из раствора нитрата серебра осаж-
дением разбавленной соляной кислотой, промывают, сушат при 100 °C и при
необходимости сплавляют в кварцевом или фарфоровом тигле. Он может
сравнительно легко деформироваться даже при комнатной температуре.
Соединяемые части нагревают до 500 °C, места контакта опускают в порошок
хлорида серебра или посыпают им либо покрывают пленкой из хлорида се-
ребра. Части соединяют и хлорид серебра равномерно проплавляют в пла-
мени горелки, после чего место соединения медленно охлаждают, не допу-
ская образования трещин. Для удаления замазки из хлорида серебра можно
воспользоваться раствором тиосульфата натрия.
Замазка из глицерина и свинцового глета выдерживает нагревание почти
до 300 °C. Глицерин по возможности тщательно обезвоживают, нагревая его
до высокой температуры; обезвоживают также и свинцовый глет, который
предварительно нагревают до температуры 200—400 °C. После охлаждения
20 г свинцового глета смешивают с 5 мл глицерина. Поверхности, подлежа-
щие соединению при помощи замазки, предварительно протирают глицери-
ном. Затвердение продолжается около получаса. Замазка водостойка; она
может быть удалена с помощью крепкого раствора гидроксида натрия.
Замазка из жидкого стекла водостойка и выдерживает нагревание до
высоких температур. Жидкое стекло смешивают с полевым шпатом или
тальком до образования густой кашицеобразной массы. Соединенные части
просушивают сначала на воздухе, а затем в сушильном шкафу.
Аральдитовые клеи (фирма CIBA, Basel) представляют собой эпоксидные
смолы, смешанные с отвердителем; они затвердевают при температурах от
20 до 200°С и дают вакуумно-плотные и очень прочные (несколько подвиж-
ные) соединения. На этих клеях можно соединять части из следующих ма-
териалов: металл, стекло, фарфор, графит, дерево, политетрафторэтилен,
полиэфиры. Например, вязкая смола марки AY 101 в смеси с отвердителем
HY 951 (3 масс. %) затвердевает при 20°C за 24—48 ч, а при 100°C за
30 мин, обеспечивая прочное соединение деталей, которое выдерживает ме-
ханические нагрузки в интервале температур от —60 до +60 °C. Другой тип
клея на основе смол — аральдит АТ 1 — твердое вещество, уже содержащее
в своем составе отвердитель. Эта смола сначала плавится при нагревании,
а затем затвердевает при 130—200 °C. После этого ее можно охладить до
—190 °C, не опасаясь появления трещин. Соединяемые части необходимо
предварительно обезжирить, а их поверхностям придать шероховатость.
Давление пара затвердевших аральдитовых клеев исчезающе мало. Соедине-
ние на таких клеях можно разъединить после их набухания в нитробензоле
в течение нескольких суток. Наконец можно указать на значительное число
так называемых цокольных замазок для труб [2]. Следует упомянуть также
возможность соединения стеклянных частей с помощью припоев для стекла
или металлических сплавов (например сплав Вуда).
Методы измерения температуры
49
При использовании смазок и замазок необходимо помнить, что раство-
ренный в них воздух часто может быть полностью удален лишь после мно-
гочасового обезгаживания в вакууме. Вышеуказанные значения для давле-
ния паров достигаются лишь после обезгаживания. Неблагоприятным фак-
тором может являться и то, что смазка (или замазка) слишком часто со-
прикасается с воздухом или хорошо в ней растворимыми парами. Рас-
творенные в смазке пары часто можно определить по запаху сразу после
разъединения частей шлифа. По этой причине при тщательной работе
следует стараться употреблять для смазывания как можно меньшее коли-
чество смазки.
ЛИТЕРАТУРА
1. Angerer Е„ Technische Kunstgriffe
bei physikalischen Untersuchungen.
Friedr. Vieweg & Sohn. Braunsch-
weig, 1966. [Есть русский перевод
12-го нем. изд.: Ангерер Э. Техни-
ка физического эксперимента. Пер.
с нем./Под ред. К. П. Яковлева.—
М.: Физматгиз, 1962.]
2. Espe W., Werkstoffkunde der Hoch-
vakuumtechnik. Band II, III. Deu-
tscher Verlag der Wissenschaften,
Berlin, 1960/61.
3. Jaeckel R., Kleinste Drucke, ihre
Messung und Erzeugung. Berlin,
1950.
4. Каталоги фирм Leybold-Heraeus
(Koln); CIBA (Basel); MMM-Com-
pany (Lehmann und Voss und Co.,
Hamburg).
8. Методы измерения температуры
Жидкостные термометры
Жидкостные термометры применяются для измерения температуры в
интервале от —200 до +600 °C, но их точность невысока. Чистая ртуть за-
мерзает при —38,9 °C. Ниже этой температуры используют термометры со
сплавами ртути (до —59 °C) или с органическими жидкостями (спирт, толу-
ол, пентан). Показания ртутных термометров сначала несколько изменяются,
поэтому хорошие термометры должны быть подвергнуты искусственному ста-
рению. Несмотря на это, градуировку следует время от времени проверять
по крайней мере в двух постоянных точках: точке таяния льда и точке ки-
пения воды, насыщенной воздухом. При этом необходимо учитывать, что
температура кипения tp зависит от давления воздуха р (мм рт. ст.) следую-
щим образом:
tp (°C) = 100 + 0,0367 (р — 760) — 0,000023 (р — 760)2
Для достижения точного значения 0°С тонко раздробленный лед смеши-
вают с деминерализованной водой и во время измерения интенсивно пере-
мешивают.
При заводской градуировке весь термометр, как правило, нагревают до
измеряемой температуры. Лишь у так называемых «встроенных» термомет-
ров во время градуировки столбик ртути находится при другой температуре.
В этом случае на обратной стороне шкалы указываются глубина погруже-
ния термометра и температура выступающего столбика. Обычно в резуль-
таты измерений вносят поправку иа выступающий столбик. Если длина вы-
ступающего столбика равна п (см), а его средняя температура составляет
/ср (°C), то истинная температура может быть вычислена по формуле
^ист = /иэм + Уп (4.3М-/ср)
Значения коэффициента у, представляющего собой разность коэффициен-
тов термического расширения термометрической жидкости и стекла, можно
4—143
50
Методы препаративной химии
найти в виде таблиц в справочнике [2]. Некоторая неточность такой коррек-
тировки связана с неоднозначностью в определении средней температуры
выступающего столбика. Величина поправки может составлять несколько гра-
дусов.
Тензотермометры
Принцип действия этих весьма точных термометров основан на изме-
рении давления пара жидкости. Такие термометры применяются главным
образом для измерения низких температур. Вследствие громоздкой конст-
рукции опасность их поломки довольно высока. Кроме того, интервал из-
мерения температуры таким термометром, заполненным определенной жид-
костью, невелик. Описание отдельных вариантов их устройства можно най-
ти во втором издании настоящей книги*, а также в работах [3] и [5].
Термометры сопротивления
Действие этих термометров, применяемых в температурном интервале
от —250 °C до +850 °C, основано на использовании высокого температур-
ного коэффициента электрического сопротивления платины, никеля или ме-
ди. Так, изменение сопротивления платины составляет в среднем 0,38%
ла 1 °C. Термометры сопротивления принадлежат к наиболее точным при-
Подстроечное
сопротивление
Измерительный
. инструмент
\сопротивления
Рис. 11. Схема подключения термо-
метра сопротивления в измеритель-
ную цепь (мостовая схема).
<5орам для измерения температуры. Они выпускаются различных размеров
в чехлах из слюды, стекла или керамики, с простой или двойной намоткой.
Никель можно использовать для измерения температур в интервале от
—60 до +150 °C, платину — от —250 до +850 °C и медь от +20 до +100 °C
(в калориметрах). Сопротивление такой проволочной намотки измеряется
омметром со шкалой в градусах Цельсия (мостовая измеряющая схема,
подсоединение показано на рис. 11). Для измерения используют источник
постоянного тока с напряжением в несколько вольт. Результат измерения не
зависит от небольших колебаний напряжения источника и от изменения тем-
ператур подводящих проводов вследствие взаимной компенсации. Возможны
и иные, чем показанный на рис. 11, способы подсоединения термометров
сопротивления. Выпускаются также уже градуированные термометры сопро-
тивления, например «Pt 100 DIN» с платиновым сопротивлением точно
100 Ом при 0 °C.
Термопары
Для измерения температур до +1600 °C (а в особом исполнении до
2300 °C) используют термопары. Перечень комбинаций материалов прово-
лок, обычно применяемых для изготовления термопар, приведен в табл. 17.
Числа, помещенные в скобки, соответствуют предельным температурам, до-
пустимым лишь иа короткий промежуток времени. Остальные величины со-
• Handbuch der praparativen anorganischen Chemie. Hrsg. G. Brauer. 2.
umgearb. Aufl. Bd. 1, 2. Enke, Stuttgart, 1960/62.
Методы измерения температуры 51
Таблица 17. Наиболее часто применяемые термопары
Материал термопар (по- ложительиое/отрнцатель- иое плечо) Температурные границы области применения, °C Термоэлект- родвнжущая сила, мкВ/°С , в интервале 0—100 °C Допустимая атмо- сфера при эксплуа- тации®
Платина/платина+10% 0—1700 6,4 Ba, Н, О
родия Платина/платина+13% 0—1700 6,5 Ba, Н, О
родия Нихром/никель 0—900 (1200) 41 Ba, Н, Во
Вольфрам/молибден Хромель/алюмель 0—2600 —200—1370 41 Ba, Н, Во Ba, Н, О
Железо/константан —200—700 (900) 53,7 Ba Н, Во
Медь/константан —200—400 (600) 42,8 Ba, Н, Во
® Ва — вакуум; Н — нейтральная; О — окислительная; Во — восстановительная.
ответствуют значениям температур, допустимым при длительной работе. Таб-
лицы зависимости термоэлектродвижущих сил от температуры можно най-
ти в справочниках, например [2]. Для измерения низких и средних темпе-
ратур подходят три последние термопары в табл. 17; платина — платиноро-
диевой термопарой измеряют более высокие температуры. Необходимо иметь-
в виду, что платиновые термопары не следует нагревать выше 900 °C в вос-
становительной атмосфере (Н2, СО и др.) при одновременном присутствии
материала, содержащего кремний (в том числе кварца и керамических ма-
териалов), так как в результате загрязнения механическая прочность тер-
мопар чрезвычайно сильно понижается, а также изменяются их показания.
Термоэлектродвижущую силу обычно измеряют стрелочным вольтмет-
ром хорошего класса точности, по возможности с очень высоким внутрен-
ним сопротивлением. Благодаря высокому внутреннему сопротивлению сила
тока будет весьма мала и падение напряжения в термопаре и подводящих
проводах пренебрежимо мало. При очень точных работах измерения прово-
дят компенсационным методом.
Оба плеча термопары изготавливают обычно из проволоки диаметром
0,2—0,5 мм. В продаже, однако, имеются и миниатюрные термопары, диа-
метр которых вместе с защитным чехлом составляет всего 1—2 мм*. Прово-
локи термопары можно легко сварить самостоятельно. Для сваривания, на-
пример, проволок из железа и константана их концы нагревают, окунают в
порошок буры, нагревают до плавления, соединяя в тонком язычке пламени
смеси светильного газа с кислородом. При этом образуется небольшой ша-
рик, который обрабатывают горячей водой для удаления боратов. При сва-
ривании платиновой термопары действуют аналогичным образом, но поль-
зуются водородно-кислородной горелкой и не применяют флюса. Глаза сле-
дует защищать, работая в темных очках.
Одну из проволок термопары изолируют от другой при помощи тонкой
керамической трубочки, а всю термопару помещают в защитную трубку из
кварца, фарфора, альсинта, силлимантина или металла**. Максимальная ра-
бочая температура термопары в значительной степени зависит от толщины
проволоки, а также от наличия защитного кожуха.
• Например, выпускаемые фирмой Philips Industrie Elektronik (Hamburg).
** Часто для изоляции проволоки в термопарах используют специальные
двухканальные керамические трубки. — Прим, перев.
4 *
52
Методы препаративной химии
Термопарные проволоки соединяют в так называемом месте «холодных
•спаев» с медными проводниками, ведущими к измерительному прибору.
Поскольку измерение температуры при помощи термопар по существу пред-
ставляет собой измерение разности температур «горячего» и «холодного»
«паев, температура последнего должна быть постоянной и точно известной
в момент измерения. Этого можно достичь, помещая, например, холодные
спаи термопары (или соединение на винтовых зажимах) в небольшие про-
бирки, которые для термостатирования при О °C опущены в сосуд Дьюара
Рис. 12. Схема подключения термо-
пары.
В области I (обведена рамкой) темпера-
тура любая, в области II — постоянная
температура, значение которой известно.
со смесью льда и воды. Если по каким-либо причинам провода термопары
коротки и не достигают места «холодных спаев», их удлиняют при помощи
компенсационных проводов (поставляются фирмами, производящими тер-
мопары). Компенсационные провода предотвращают возникновение допол-
нительных токов, мешающих измерениям электродвижущих сил. Они из-
готавливаются из специальных сплавов неблагородных и поэтому дешевых
металлов, выбор которых зависит от вида используемой термопары. Обе
жилы компенсационных проводов маркированы и должны подсоединяться к
соответствующим концам термопары.
На рис. 12 показана обычная схема присоединения термопары. Оба
места промежуточного соединения термопары с компенсационным проводом
должны находиться при одинаковой или почти одинаковой температуре, ко-
торая, однако, может быть произвольной.
Термопары без защитных чехлов имеют то преимущество, что они мало-
инерционны и измеряют температуру в хорошо локализованном месте, почти
в точке.
Для градуировки термометров и термопар существуют термометрические
фиксированные точки, соответствующие температурам полиморфных перехо-
дов, тройных точек, точек плавления и кипения чистых веществ (см. [2,4]).
Пирометры излучения
Для измерения температур выше 1600 °C удобнее всего пирометры из-
лучения, хотя применять их можно, уже начиная с 600 °C. Принцип рабо-
ты одного из типов пирометров основан па сравнении излучения раскален-
ного тела с излучением соответствующей лампочки накаливания в Области
длин волн от 650 нм до границ видимого спектра. При постепенном ослаб-
лении яркости светящейся нити, расположенной в поле зрения рассматривае-
Высокие температуры 53
мого раскаленного тела, она перестает быть видимой. В этот момент можно
непосредственно сделать отсчет температуры по регистрирующему прибо-
ру (пирометры частичного излучения). Действие другого типа прибора ос-
новано на том, что все лучи от светящегося тела, собранные кварцевой лин-
зой, направляют на зачерненную термопару, напряжение которой можно от-
считать по показаниям стрелки прибора (пирометры полного излучения или
радиационные пирометры). Оба типа приборов имеются в продаже в испол-
нении, очень удобном для работы с ними.
При работе с оптическими пирометрами необходимо учитывать, что из-
лучательная способность зависит от природы нагретого тела. Пирометры,
одиако, градуированы по излучению идеального «черного» тела. В связи с
тем что излучательная способность реальных тел меньше, чем «черного» те-
ла, при измерении температуры нужно вносить соответствующие поправки.
ЛИТЕРАТУРА
1. Каталоги фирм Hartmann und
Braun AG (Frankfurt), Degussa,
Zweigniederlassung (Hanau); Phi-
lips Industrie Elektronik (Ham-
burg).
2. D’Ans—Lax, Taschenbuch file Che-
miker und Physiker. Bd. 1. Sprin-
ger, Berlin — Heidelberg — New
York, 1967.
3. Лукс Г. Экспериментальные мето-
ды в неорганической химии. Пер.
с нем. — М.: Мир, 1965.
4. О’М Bockris J., White J. L., Mac-
kenzie J. D., Physico-Chemical
measurements at High Temperatu-
res. Butterworths, London, 1959,
p. 6—46, 372—374.
5. Shriver D. F„ The manipulation of
air-sensitive compounds. McGraw-
Hill, New York, 1969.
9. Высокие температуры
Нагревание за счет процесса горения
В лаборатории, за немногими исключениями, обычно применяются го-
релки, в которых происходит сгорание газа. В продаже имеются горелки
самых разнообразных типов. Они дают возможность нагревать небольшой
тигель до 700—800 °C, а при применении дополнительной насадки, концент-
рирующей газ, до 900 °C. Для получения еще более высоких температур
служат общеизвестные паяльные горелки. Очень горячее пламя можно по-
лучить, добавляя к поступающему воздуху кислород через Т-образную
трубку. В продаже имеются также горелки с точной дозировкой добавляемо-
го к воздуху кислорода. Для нагревания на открытом воздухе небольших
объектов можно воспользоваться обычной ацетиленовой сварочной горелкой.
Нагревание электрическим током
Электрические нагревательные приборы применяются в лаборатории
очень широко, во многих случаях вытесняя горелку Бунзена. Различные
плитки, колбонагреватели и т. д. для лабораторных целей выпускаются
многими фирмами. Особое преимущество электронагревательных приборов
перед горелкой Бунзена заключается в равномерном нагревании, на кото-
рое не влияет случайное движение воздуха. Выпускаются также небольшие
и относительно дешевые регуляторы, позволяющие поддерживать заданную
температуру. Так, применяют «регуляторы подводимой энергии», регулиро-
вочные трансформаторы или регулировочные сопротивления (применение
последних, одиако, сопряжено с потерями энергии). Следует упомянуть о раз-
нообразных погружаемых в жидкость кипятильниках, в том числе лалвар-
цевого стекла, которые в последнее время используют и в химических ла-
бораториях.
54
Методы препаративной химии
Для быстрого испарения жидкости без потерь путем облучения ее по-
верхности служат кварцевые радиационные • нагреватели, так называемые
поверхностные облучатели. Электрическим обогревом снабжены различные
водяные и воздушные нагреватели. Колбонагреватели, у которых нагрева-
тельные элементы не раскаляются докрасна, называемые также инфракрас-
ными лампами, особенно удобны при работе с легковоспламеняющимися
жидкостями.
Из стеклянной ткани со встроенными в нее нагревателями изготовляют-
ся подушки или ленты, служащие для равномерного обогрева колб или
стеклянных трубопроводов. Нагреватели могут также быть вмонтированы
в рубашку из силиконового каучука.
Хорошо известны электрические печи различных типов (тигельные, му-
фельные) для нагревания тиглей или чашек. Их конструкции очень хорошо
отработаны, так что изготовление их самим экспериментатором нецелесо-
образно. Напротив, электрические трубчатые печи часто приходится изготов-
лять в лаборатории, так как они должны соответствовать различным спе-
циальным целям, для которых не всегда пригодны печи, имеющиеся в про-
даже. При правильном выборе размеров таких печей можно избежать, по-
терь энергии и лишнего расхода материалов. Особенное внимание следует
обращать па хорошую теплоизоляцию печи, что предотвратит излишний рас-
ход энергии и чрезмерное нагревание помещения.
В зависимости от материала нагревательных элементов и от способа
подвода энергии различают следующие виды печей:
1) с проволочным элементом сопротивления;
2) с силитовыми стержнями, силитовыми трубками или стержнями из
силицида молибдена;
3) с нагревателями в виде угольных трубок;
4) с нагревателями в виде трубок из вольфрама, тантала, иридия или
диоксида циркония;
5) индукционные (высокочастотные);
6) катодно-лучевые, дуговые, плазменные;
7) отражательные.
Печи с проволочным нагревателем
Нагревательным элементом таких печей служит проволока или лента из
специального сплава (кантала, нихрома, хромеля, мегапура и т. п.), из пла-
тины или из молибдена (вольфрама, тантала). Большей частью проводник
наматывают на трубки винтообразно, но иногда нагревательные спирали
располагают вдоль оси трубки. Трубчатые печи с нагревателями из всех вы-
шеуказанных материалов имеются в продаже. Самостоятельно изготавли-
вают обычно печи с нагревателями из специальных сплавов*, которые про-
сты в эксплуатации и дешевы, но, с другой стороны, не позволяют произво-
дить нагревание выше ~ 1350 °C. Изготовление печей с нагревателями из
платины требуется только для специальных целей, когда, например, необходи-
мо достичь высокой температуры в печн небольшого размера. В таких случаях
изготовляют печи с внутренней спиралью. Способ их изготовления, так же
как печей с нагревателем из молибденовой проволоки, описывается ниже.
Печи с нагревателями из специальных сплавов изготавливаются следу-
ющим образом. После того как в соответствии с назначением печи выбраны
ее размеры, подбирают необходимую трубку, на которую наматывают про-
водник-нагреватель. Металлические трубки гарантируют особенно разномер-
ное распределение тепла в печи. При температурах ниже 500 °C применяют
алюминиевые трубки; до 700 °C пользуются стальными трубками, лучше все-
* Изготавливаются, например, фирмами А. В. Kanthal (Schweden), Va-
kuumschmelze AG (Hanau). . ,
Высокие температуры 55
го из специальной нержавеющей стали. При более высоких температурах
можно использовать только керамические трубки, для изготовления которых
применяют неглазурованный фарфор, высоглиноземистую массу (массу Пи-
фагора) и другие специальные алюмосиликатные массы, а для особо высо-
ких температур — оксид алюминия*. Применение керамических трубок име-
ет то преимущество, что, являясь изоляторами, они уменьшают опасность
короткого замыкания между витками обмотки. Особенно просто произве-
сти намотку печи, если пользоваться трубкой с винтовой нарезкой, в кото-
рую укладывают проволоку. Прозрачные печи для не очень высоких тем-
ператур изготовляют из отрезка стеклянной трубки, непосредственно на ко-
торую наматывают с некоторым натяжением нагревательную проволоку.
Для определения длины и поперечного сечения нагревательного провод-,
ника сначала измеряют поверхность трубки, затем при наличии теплоизоля-
ции среднего качества вычисляют энергию, необходимую для достижения за-
данной температуры, пользуясь описанным ниже эмпирическим правилом:
для нагревания до 300 °C требуется мощность 0,2 Вт/см2 затем в интервале
300—700 °C на каждые 100 °C— еще по 0,2 Вт/см2. Между 700 и 1000 °C
на каждые 100°C требуется по 0,3 Вт/см2; от 1100 до 1300°C—-по
0,4 Вт/см2; от 1300 до 1600 °C — по 0,5 Вт/см2 и свыше 1600 °C — по
0,6 Вт/см2 на каждые 100° (следовательно, для достижения 2000 °C требу-
ется мощность 7 Вт/см2).
Зная напряжение в сети, можно определить необходимую силу тока,
причем для расчета следует принять напряжение на 10% ниже, чем напря-
жение в сети. После этого вычисляют омическое сопротивление проволоки,
а также приблизительную ее длину. Для небольших печей расстояние между
витками равно — 1 мм; таким образом, можно считать, что на каждые 2 мм
длины трубки укладывается приблизительно один виток. Следовательно, дли-
на рроволоки определяется числом витков и длиной окружности трубки. По-
скольку сопротивление большинства нагревательных проводников длиной 1 м
и поперечным сечением 1 мм2 приблизительно равно 1 Ом, то таким обра-
зом устанавливают и сечение самого провода**. Такой ориентировочный ра-
счет вполне достаточен для лабораторных целей. При этом следует еще
учесть, что максимальная нагрузка нагревательной проволоки, а следователь-
но, и продолжительность ее службы, сильно зависит от диаметра. При ра-
счёте максимальной силы тока для обычно применяемых проволок принима-
ют значение тилы тока 0,6 А на каждые 0,1 мм диаметра проволоки (на-
пример, нагревательная проволока диаметром 1 мм может в течение дли-
тельного времени выдерживать ток в 6 А). Еще лучше определять макси-
мальную нагрузку по значению мощности (в ваттах), приходящейся на
1 см2 проводника. Для не очень высоких температур (например 900 °C) ее
значение не должно превышать 3,5 Вт/см2, а для высоких температур (на-
пример, 1350 °C) — 1,5 Вт/см2.
Ниже приводится расчет небольшой лабораторной трубчатой печи с
обмоткой из канталовой проволоки. Печь с трубкой диаметром 2 см и дли-
ной 30 см при напряжении 220 В должна давать температуру 900 °C. Как
указано выше, для надежности и возможности регулирования в основу ра-
счета следует положить напряжение, которое на 10% ниже напряжения
сети, т. е. 200 В. Поверхность нагрева составляет 200 см2. Согласно указан-
ному эмпирическому правилу, мощность для достижения температуры
900 °C должна быть 320 Вт. Следовательно, при напряжении 200 В сила
тока равна 1,6 А. Чтобы при напряжении 200 В получить ток 1,6 А, про-
* Например, алундовые трубки. — Прим, перев.
** Удельное сопротивление проволоки для намотки печей часто указыва-
ется изготовителями непосредственно на катушках. Таблицы значений сопро-
тивления наиболее употребительных проволок для нагревательных элемен-
тов можно иайти в справочниках, например в [7].
56
Методы препаративной химии
водник должен иметь сопротивление 200/1,6= 125 Ом. Если расстояние меж-
ду витками 2 мм и длина трубки 30 см, то по расчету необходимо 150 вит-
ков, т. е. при длине окружности трубки 6 см потребуется 9 м проводника.
Согласно обоим вышеприведенным эмпирическим оценкам максимальной на-
грузки, диаметр проволоки должен быть около 0,3 мм. Проволока такого
сечения длиной 9 м будет обладать с хорошей точностью необходимым об-
щим сопротивлением 125 Ом. Таким образом, для намотки печи необходи-
ма проволока диаметром 0,3 мм и длиной 9 м. Ее следует наматывать иа
трубку, выдерживая расстояние между витками 2 мм.
Если необходимо изготовить печь с обмоткой из платиновой проволоки,
то следует принимать во внимание ее высокий температурный коэффициент
сопротивления. Сопротивление платины при 1000 °C в 3—4 раза больше, чем
при комнатной температуре. Поэтому платиновые печи всегда следует на-
гревать медленно, постепенно выводя включенное в цепь дополнительное
сопротивление. В противном случае имеется опасность, что печь перегорит
вследствие неравномерной теплопередачи от проводника к стенке печи.
Установив длину нагревательного проводника, можно начинать намотку.
Для печей, предназначенных для работы при температуре ниже 1000 °C,
трубку рекомендуется предварительно обернуть слоем влажной асбестовой
бумаги, высушить ее и затем уже вести намотку. В печах, работающих при
более высокой температуре, проволоку непосредственно наматывают на
трубку. Начальную часть проволоки, для прочности скрученную из несколь-
ких концов, закрепляют либо при помощи петли, либо, что лучше, при по-
мощи хомутика, укрепленного на трубке. Так же закрепляется и конец об-
мотки. Для печей, предназначенных для работы при температуре до 1000 °C,
для обмазки готовится кашицеобразная масса из смеси талька с жидким стек-
лом, а для более высоких температур — из готовой изоляционной массы
(например, марки 250 фирмы Haldenwanger) или из смеси равных частей
магнезии (не содержащей карбоната), глинозема (не содержащего SiO2) и
воды; такой массой покрывают спираль слоем толщиной I мм. Изоляцион-
ные массы, содержащие свободную кремневую кислоту, при высокой тем-
пературе довольно быстро разрушают проводник. После высушивания иа
воздухе спираль помещают в сушильный шкаф и, наконец, нагревают то-
ком. После этого нагревательную трубку вкладывают в другую, более ши-
рокую трубку или в жестяной кожух. Промежуток заполняют магнезией или
инфузорной землей. Выпускаются готовые изолирующие кожухи, изготов-
ленные из оксида магния с добавками.
Для изоляции очень практичны также так называемый диатомитовый
камень и огнеупорный «легкий камень», которые можно достать на пред-
приятиях, изготовляющих огнеупорные изделия. Эти камни можно очень
легко разрезать для получения кусков нужного размера или приобрести в
готовом виде в различных формах. Хорошую изоляцию можно изготовить
из асбестового картона, проклеенного жидким стеклом. Пространство между
стенками заполняют шлаковатой, асбестовой крошкой, оксидом магния или
слоями: внутри — оксид магния, снаружи — шлаковата. Для более низких
температур достаточной изоляцией служит обмотка из нескольких слоев ас-
бестового шнура или листового асбеста, смоченного водой для придания
эластичности. Концы проволоки выводятся через керамические изоляцион-
ные бусы (приобрести можно на электротехнических предприятиях). Вместо
проволочной обмотки целесообразно применять ленточную. Для выравнива-
ния распределения температуры в печи (концевые части печи, естественно,
всегда имеют более низкую температуру, чем средняя часть) обмотку на
концах трубки рекомендуется делать чаще, чем в середине. Поскольку уста-
новить точно расстояние между витками трудно, на концах печи делают
еще дополнительную вспомогательную обмотку с отдельной регулировкой.
Печи с очень равномерным распределением температуры по всей длине
изготавливают из цилиндрического - алюминиевого или бронзового блока с
Высокие температуры 57
толщиной стенки 15—20 мм, имеющего продольный высверленный канал для
термопары. Обмотку блока ведут проволокой с надетыми на нее изоляцион-
ными бусами. В 5—10 см от обоих концов обмотки делают петлеобразные
отводы. В ограниченной таким образом средней части обмотки сила тока
будет несколько ниже, чем на концах обмотки, если параллельно ей под-
соединить шунт с регулируемым сопротивлением. Необходим также хороший
внешний кожух, более длинный, чем нагревательная трубка.
Для монтажа электрических печей целесообразно использовать подстав-
ку, показанную на рис. 13, которая позволяет устанавливать печь наклонно
и закреплять ее па различной высоте. При горизонтальном расположении
Рис. 13. Подставка для крепления
электрической печи.
трубчатой печи можно облегчить кратковременное наблюдение образца или
его быстрое закаливание, если сечь снабдить роликами, движущимися по
горизонтальным направляющим шинам. Выпускаются также печи, состоящие
из двух половинок в виде полуцилиндров с независимыми обмотками. Такая
раскрывающаяся печь позволяет класть или вынимать реакционную трубку
или вести наблюдение в процессе постепенного охлаждения, не сдвигая ре-
акционной трубки.
Печи с внутренней обмоткой
Применяя внутреннюю обмотку, можно за счет несколько более услож-
ненной конструкции повысить максимальную рабочую температуру печей с
проволочным сопротивлением (с учетом материала проволоки). Особенно эф-
фективно использование этого принципа для печей с платиновым проволоч-
ным нагревателем. Такие печи можно в течение продолжительного времени
вполне надежно применять для создания температур до 1500 °C, тогда как
платиновые печи с внешней обмоткой не выдерживают в течение долгого
времени температуру выше 1250 °C.
Если в качестве нагревательного элемента применяют платиновую про-
волоку, то в основу расчета ее длины следует принять значение сопротивле-
ния при максимальной температуре. Для изготовления такой печи вытачи-
вают круглый деревянный стержень приблизительно требующегося диамет-
ра (тоньше на 1—2 мм) и разрезают его вдоль двумя разрезами, как по-
казано на рис. 14. Потом разрезанные части стержня складывают вместе,
обертывают их слоем папиросной бумаги и обматывают виток к витку тон-
ким шпагатом. На этот слой накладывают еще несколько слоев папиросной
бумаги и, наконец, пропитав слегка маслом прокладку из папиросной бу-
маги, наматывают на нее платиновую проволоку. Производят обмазку изо-
ляционной массой и высушивают. После высушивания повторяют обмазку и
58
Методы препаративной химии
во влажном состоянии вкладывают стержень в подходящую керамическую
трубку, причем все промежутки по возможности заполняют изоляционном
массой. Все это высушивают сначала на воздухе, потом окончательно в су-
шильном шкафу. Затем осторожно вытягивают шпагат и, вытащив среднюю
клинообразную часть деревянного стержня, удаляют его. Потом еще раз
хорошо высушивают и осторожно и очень медленно нагревают до тех пор,
пока не выгорит папиросная бумага. Наконец, печь снова охлаждают, об-
мазывают ее внутри изоляционной массой и после полного высушивания
еще раз осторожно нагревают. Аналогично тому, как описано выше, можно
вмонтировать нагревательную спираль в любую теплоизоляцию.
Рис. 14. Деревянный стержень для изготов-
ления печей с внутренней обмоткой.
Печи с нагревателями из молибденовой (вольфрамовой, танталовой)
проволоки
Готовые печи с обмоткой из молибденовой проволоки или ленты могут
работать до температуры 1500 °C. Однако для предотвращения окисления
проволоки через печь следует постоянно пропускать слабый поток защит-
ного газа (водород, водяной газ СО + Н2, газ для синтеза аммиака N2+H2).
Температуру таких печей легко регулировать. Собственными силами такие
печи редко изготовляют.
Молибден, а также вольфрам и тантал благодаря их очень низкому
давлению пара используют в виде проволоки или ленты для изготовления
небольших печей, работающих в условиях высокого вакуума. Нагреватель-
ным элементом таких печей служит спираль, закрепленная в вертикальном
или горизонтальном положении иа керамической опоре. Последняя необхо-
дима, поскольку при высоких температурах спираль размягчается и может
деформироваться. Объем пространства для нагревания пробы относительно
невелик, но зато достигаются очень высокие температуры (выше 1500°C).
Чтобы уменьшить слишком большую потерю энергии за счет излучения вовне,
нагреватель окружают отражающими экранами, а пространство за ними
принудительно охлаждают. Все части подобных печей монтируют на пло-
ской плите, имеющей вакуумно-плотные вводы для электрического тока,
охлаждающей воды, а также патрубок для откачки. Плиту с печью накры-
вают навинчивающимся колоколом из стекла или металла [1] (аналогичная
конструкция показана на рис. 17).
Силитовые печи
Силитовые печи отличаются несравненно большей прочностью, хотя и
хуже поддаются регулированию и не столь экономичны, так как в них не
удается добиться достаточно хорошей теплоизоляции. Нагревательные эле-
менты изготавливают из карбида кремния (силит, глобар) или дисилицида
молибдена (мосилит). Максимальная рабочая температура силитовых печей
составляет 1400°C (непродолжительное время могут работать при 1500°C),
а мосилитовых— 1600 °C. Обычно используют нагревательные стержни,
окружающие нагреваемое пространство, причем стержни из карбида крем-
ния можно располагать горизонтально или вертикально, а стержни из ди-
силицида молибдена — только вертикально. Подобные печи имеются в про-
даже, но для специальных целей их можно изготовить самостоятельно, имея
отдельные стержни и приобретя готовые подходящие крепления, трубки и
внешние кожухи на предприятиях, выпускающих технический фарфор
(рис. 15). При этом важно обеспечить хороший контакт между нагреватель-
Высокие температуры 59
ными стержнями и подводящими проводами, для чего большей частью поль-
зуются специальными жестяными хомутиками.
Силитовые нагреватели выпускаются также в виде трубок. Хотя их
применение вместо стержней обеспечивает равномерное распределение тем-
пературы в горячей зоне, увеличиваются и трудности регулирования темпе-
ратуры. Это связано с высокой проводимостью и отрицательным температур-
ным коэффициентом сопротивления. Поэтому такие печи следует нагревать
медленно, используя регулировочное сопротивление или регулирующий
трансформатор и по мере повышения температуры снижая напряжение,
с тем чтобы сила тока не превысила максимально допустимую величину.
Рис. 15. Конструкция силитовой печи.
1 — внешний кожух из жести; 2 — изоляционный слой (шамотовая крошка, оксид магния,
инфузорная земля); 2— защитная трубка (шамот); 4 — силитовые стержни; 5— внутрен-
няя трубка (высокоглиноземнстая масса, силлиманит); 6 — крышки с отверстиями для
внутренней трубки и снлитовых стержней.
Печи с нагревателем в виде угольной трубки
В этих печах нагревательным элементом является угольная трубка.
Впервые конструкция таких печей была предложена Нерпстом и Тамманом;
часто их просто называют печами Таммана. Крупные модели таких печей
применяются в технике, а поэтому они имеются в продаже. Изготовлять та-
кие печи самому экспериментатору нецелесообразно, так как наиболее до-
рогой их частью является мощный трансформатор, дающий ток от несколь-
ких сот до 1000 А при напряжении около 10 В. Особая конструкция печи
позволяет быстро осуществлять замену угольных трубок, продолжитель-
ность работы которых при высоких температурах невелика. При помощи
таких печей можно легко осуществить нагревание выше 2000 °C. Внутри
трубки всегда существует восстановительная атмосфера. В тех случаях, когда
это нежелательно, следует применять вставную керамическую трубку. В за-
висимости от рабочей температуры можно использовать трубки из А12О3,
ВеО или ThO2. Керамическую трубку следует закреплять таким образом,
чтобы в средней горячей зоне она не касалась стенок угольной трубки.
Графит более устойчив к окислению, чем уголь. При изготовлении на-
гревательных трубок из графита для увеличения электрического сопротив-
ления в них делают продольные прорези [15]. Этот тип нагревателей мож-
но применять также при работе в вакууме и в атмосфере инертного газа.
Печи с нагревателями в виде трубок из тугоплавких металлов
Применение в печах в качестве нагревателей трубок или свернутых
полосок из вольфрама, тантала или иридия позволяет достичь очень высоких
температур. Вследствие возможности взаимодействия вольфрама с кислоро-
дом, а тантала с кислородом и азотом, а также для обеспечения лучшей
теплоизоляции такие печи применяют в условиях вакуума (реже в атмос-
фере инертного газа — гелия, аргона). На рис. 16 показана конструкция
€0
Методы препаративной химии
печи с горизонтальным расположением нагревательной трубки, на рис. 17 —
с вертикальным, а на рис. 18 —с открытым нагревателем в виде согнутой
полоски жести из тугоплавкого металла. Обычно отдельные части подобных
печей монтируют на толстой металлической плите, имеющей изолированные
вводы для электрического тока и охлаждающей воды, а также снабженной
4
охлаждение -
Рис. 16. Схема устройства печи с нагревателем в виде вольфрамовой (тан-
таловой, молибденовой) трубки.
1— латунная плита; 2— вольфрамовая пластинка; 3— вольфрамовая трубка; 4— стек-
лянный колокол; 5 — отвинчивающаяся крышка; 6 — медный кожух; 7 — медный змеевик;
8 — образец.
патрубками для создания и измерения вакуума. Опускаемый сверху стек-
лянный или металлический колокол вместе с плитой ограничивает прост-
ранство печи [2, 14].
В связи с низким электрическим сопротивлением нагревателей в виде
металлических трубок для работы печи необходим мощный понижающий
трансформатор (например, до 10 В), дающий силу тока до 1000 А. При-
меняя в качестве нагревателей трубки из вольфрама, можно достичь тем-
пературы 3000 °C, с трубками из тантала — 2200 °C*.
Для нагревания очень малых образцов можно сделать нагреватель в
виде ленты или желобка [5].
Если необходимо работать при высокой температуре в окислительной
атмосфере, то нагревательную трубку изготовляют не из вольфрама или
тантала, а из иридия [18] или из керамических масс на основе диоксидов
циркония или тория, электропроводность которых обеспечивается за счет
наличия примесей, делающих кристаллическое строение этих оксидов де-
фектным [4, 8, 17]. «Масса Нернста» (ThO2-|-CeO2) может быть рекомен-
дована для изготовления электропроводящих трубок только очень неболь-
шого диаметра. Для проведения синтеза макроколичеств веществ такие
трубки себя не оправдали.
прши“6 “ например.
Высокие температуры 66
Рис. 17. Конструкция
печи с нагревателем в
виде вольфрамовой (тан-
таловой, молибденовой)
трубки.
Рис. 18. Схема устройства высоковакуумной печи
с нагревателем из танталовой (вольфрамовой,
молибденовой) фольги.
Показан поперечный разрез. 1 — нагревательная манже-
та; 2 — изоляция; 3 — отражательные экраны (тантало-
вая, молибденовая фольга); 4— вакуумный колокол.
Керамические нагревательные элементы имеют некоторые недостатки..
Они, например, лишь при относительно высоких температурах (1400—
1800 °C) становятся достаточно электропроводящими. Кроме того, невелика
лх механическая прочность при быстрых изменениях температуры. Поэтому^
‘62 Методы препаративной химии
при их применении нужны вспомогательные нагреватели как для проведе-
ния начальной стадии нагрева, так и для обеспечения нужной скорости
• охлаждения. Ввиду наличия окислительной атмосферы эти вспомогательные
нагреватели должны изготовляться из благородных металлов — платины, пла-
•тинородия и т. д.
Индукционные (высокочастотные) печи
Энергию переменного тока высокой частоты (например, 1 МГц) можно
/при помощи катушки передать находящемуся в ней проводнику, например
тиглю из металла или графита, и тем самым нагреть его. Лабораторные
индукционные печи позволяют проводить работу в очень «чистых услови-
ях», поскольку можно поместить нагреваемый тигель в охлаждаемую квар-
цевую трубку. Последнюю либо откачивают до высокого вакуума, либо за-
полняют инертным газом. При этом следует помнить, что в определенном
интервале давлений (от 10-2 до 10 мм рт. ст.) работать нельзя вследствие
возникновения тлеющего разряда. В индукционных печах можно за не-
сколько секунд произвести нагревание до 3000 °C. К недостаткам таких
печей относится необходимость приобретения большого количества специ-
ального электрооборудования и соответственно их высокая стоимость. В про-
даже имеются генераторы индукционного тока*, работающие большей ча-
стью с большими передающими трубками. Собственно печь лучше всего
.изготовить самостоятельно в соответствии с конкретной экспериментальной
задачей. Индуктивно нагреваемый тигель делают обычно цилиндрическим и
окружают защитными экранами для уменьшения тепловых потерь за счет
излучения. Для того чтобы сами экраны не воспринимали индукционной
энергии, их делают разрезными. Для улучшения условий передачи энергии
от индукционной катушки к тиглю между ними помещают кольцеобразный
.проводник, служащий «концентратором энергии».
В особых случаях в качестве индукционно нагреваемого тела можно
/применять и керамические трубки, материал которых вследствие дефект-
ности его кристаллической структуры проводит ток при высоких темпе-
ратурах [3].
Дуговые, катодио-лучевые и плазменные печи
Дуговые печи применяют для получения сплавов и малолетучих туго-
плавких соединений, таких, как карбиды, бориды, низшие оксиды и т. п.
В условиях вакуума или при пониженном давлении подходящего газа не-
«большой компактный образец плавится в электрической дуге, горящей меж-
ду охлаждаемыми электродами ’ в виде вольфрамового прутка (сверху) и
медной пластины (снизу). В медной пластине имеются конусовидные углуб-
ления для приема расплавленных образцов. Подобные лабораторные печи
можно приобрести в готовом виде**, но их несложно изготовить и самостоя-
тельно [13, 15].
Печи, в которых энергия для нагревания производится за счет элект-
ронной бомбардировки (катодные лучи), могут работать, конечно, только в
условиях высокого вакуума и применяться лишь для нагревания нелетучих
веществ. Конструкции лабораторных печей этого типа подробно описаны в
•статье [12].
Оба типа печей в более крупном масштабе имеют важное промышлен-
ное значение для получения плавленных компактных блоков тугоплавких
металлов (Ti, Zr, iNb, Мо) из тонкодисперсного материала.
* Выпускаются, например, фирмами F. Hiittinger (Freiburg), Degussa
Hndustrieofenbau (Hanau), Elphiag-HWG (Reichenbach-Fils).
** Выпускаются, например, фирмами Leybold-Heraeus (Stuttgart), Degussa
ZIndustrieofenbau (Hanau).
Высокие температуры
63
Высокие температуры могут создаваться также за счет энергии высо-
коионизироваиного газа (плазмы). Обычные технические плазменные горел-
ки работают в открытом пространстве, т. е. при атмосферном давлении.
Для препаративных целей в лаборатории лучше использовать плазменную
горелку закрытой конструкции, работающую при пониженном давлении [16].
Она позволяет производить хорошо локализованное нагревание твердых
веществ до температуры выше 2000 °C.
Зеркальные печи
Эти печи применяются в особых случаях, например для нагревания в;
атмосфере чистого кислорода до очень высоких температур, которые недо-
стижимы в печах с проволочным сопротивлением. В этих печах при помо-
щи большого параболического или эллиптического зеркала фокусируется
широкий пучок лучей солнца или другого сильного источника света (уголь-
ная дуга, газовый разряд). В «горячей точке» такой установки, имеющей,,
разумеется, конечные размеры, за короткое время достижимы очень высо-
кие температуры. Нагреваемый объект можно заключить в охлаждаемый
сосуд, в котором поддерживается вакуум или определенная газовая ат-
мосфера [11].
ЛИТЕРАТУРА
1. Alber man К. В., J. Sci. Instr., 27,
280 (1950).
2. Buckle (Z. Metallk.),
1, 53 (1946).
3. Davenport H.. Kistler S. S. et al.,
J. Amer. Ceram. Soc., 33, 333
((950).
4. Faucher M„ Dembinski K-, Antho-
ny A.-M., Rev. Int. Hautes Temp.
Refract., 6, 17 (1969).
5. Foex M.. Traverse J.-P., Bull. Soc.
Fr. Mineral. Cristallogr., 89, 184
(1966).
6. Fricke R., Meyer F. R., Chemiker-
Ztg., 66, 53 (1942).
7. D’Ans—Lax Taschenbuch ffir Che-
miker und Physiker. 3. Aufl. Ber-
lin—Heidelberg— New York, 1967,
1—75. Справочник фирмы A. В.
Kanthal fiber Walter Kaempfert
KG (Frankfurt/Main).
8. Geller R. F., In: I. E. Campbell.
High temperature technology. J.
Wiley, New York and Chapman &
Hall, London, 1956, p. 263.
9. Glaser P. E., J. Electrochem. Soc.,
107, 226 (1960); Trombe F„ Bull.
Soc. Chim. France, 353 (1953);
Null M. R., Lozier IF. W., Rev.
Sci. Instr., 29, 163 (1958); Foex
M„ Bull Soc. Chim. France, 137
(1962); Butler С. P„ Image Fur-
nace Res. (Proc. Intern. Sympos.
High Temp. Technol. Asilomar),
I960; J. Solar Energy, 1 (1957).
10. Laszlo T. S., Image furnace tech-
niques. In: Technique of inorga-
nic chemistry (eds. H. B. Jonas-
sen, A. Weissberger). Vol. V. In-
terscience, New York — London —
Sidney, 1965.
11. Glaser P. E., Walker R. F., Ther-
mal imaging techniques. Plenunn
Press, New York, 1964.
12. Gruber H., Sperner F.. Erben E.,
Z. Metallk., 52, 291, 674 (1961).
13. Hagg G„ Kiessling G„ IVA
(Schweden), 26, 105 (1955).
14. Kieffer R., Benesovsky F., Plan-
seeberichte, 5, 56 (1957).
15. Kroll W. J., Z. Metallk., 43, 259
(1952).
16. Milller-Buschbaum H. K. Z. Anorg..
Allgem. Chem., 355, 30 (1967).
17. Rothwell E., J. Sci. Instr., 38, 191
(i961).
18. Wartenberg H„ Z. Elektrochemie,
15, 708 (1909).
19. Sherwood E. M„ Means of achie-
ving high temperatures and some
of their limitations. In: Camp-
bell I. E., High temperature tech-
nology. J. Wiley, New York and
Chapman & Hall, London, 1956.
20. Motzfeldt K, Means of attaining-
and controlling temperature. In:
Bockris J. O’M., White J. L., Mac-
kenzie J. D., Physico-chemikal?
measurements at high temperatu-
res. Butterworths, London, 1959.
<54 Методы препаративной химии
10. Низкие температуры
Для получения температур ниже 0°С применяются охлаждающие смеси
со льдом, сухим льдом (твердым диоксидом углерода) и жидким азотом
(или жидким воздухом). Кроме того, можно использовать криостаты, описан-
ные в следующем разделе, которые обеспечивают получение низких темпе-
ратур без применения внешнего охлаждающего агента.
Во многих охлаждающих смесях используется лед. Существенное значе-
ние имеет его тщательное измельчение, которое можно произодить либо в
•специальной «ледяной» мельнице, либо, что проще, деревянным молотком на
плоской бетонной плите размером 40X40 см, обшитой деревянными бортами
высотой 10 см. Даже при наличии «ледяной» мельницы лед рекомендуется
предварительно раскалывать деревянным молотком на подобной плите. Сов-
ременные производители льда поставляют его в виде больших блоков.
Составы охлаждающих смесей со льдом (в весовых частях):
3 ч. льда+1 ч. поваренной соли (—21 °C)
3 ч. льда+2 ч. MgCl2-6H2O (от —27 до —30 °C)
Г2 ч. льдаД-З ч. СаС12-6Н2О (—40 °C)
.2 ч. льда-|-1 ч. конц. HNO3 (—56 °C)
При применении последних двух смесей указанных температур можно
.достичь только в том случае, если СаС12-6Н2О или конц. НЬЮз предвари-
тельно охладить в холодильнике. Во всех случаях необходимо, чтобы лед
и соль были хорошо измельчены и перемешаны.
Еще более низкие температуры можно получить при помощи твердого
диоксида углерода (сухого льда), поставляемого в виде блоков, которые пе-
ред употреблением нужно измельчить вручную или в мельнице. Твердый ди-
оксид углерода в кусках сохраняют в латунном сосуде или тканевом мешке,
помещаемом в большой сосуд Дьюара. Ввиду того что большие дьюаровские
-сосуды очень хрупки, мешок или сосуд с кусками диоксида углерода нужно
вынимать и опускать обратно в сосуд с большой осторожностью.
Твердый диоксид углерода ввиду его плохой теплопроводности приме-
няют в смеси с подходящими жидкостями. Эфир в данном случае неприме-
ним вследствие его легкой воспламеняемости. Рекомендуется пользоваться
метанолом, а еще лучше трихлорэтиленом, в котором сухой лед плавает,
что предотвращает вспенивание смеси. Температура охлаждающей смеси не-
сколько зависит от выбранного растворителя, как это видно из табл. 18.
Таблица 18. Температура охлаждающих смесей (сухой лед + растворитель)
Растворитель Минимальная темпера- тура, °C
Этиленгликоль —15
.Этанол —75
Хлороформ —77
Ацетон —86
Этанол (при пониженном давлении) —100
Для температур до —50 °C можно применять раствор 48 г СаС12
в 100 г Н2О, доведенный при помощи конц. НС1 до pH 3,0. Такой раствор
-охлаждают, бросая в него кусочки сухого льда.
Температура сублимации сухого льда —78,5 °C достигается лишь в ат-
элосфере чистого СО2 при давлении 760 мм рт. ст. На воздухе свежеистолчен-
Низкие температуры 65
ный сухой лед охлаждается на восемь градусов ниже вследствие низкого
парциального давления диоксида углерода.
Жидкий воздух стал сейчас доступен повсеместно. В продаже имеются
небольшие лабораторные ожижители воздуха (газоохлаждающие машины),
которые могут быть дополнительно снабжены разделительной колонной для
получения жидкого азота. Для перевозки и хранения жидкого воздуха име-
ются специальные цистерны различных размеров «для сжиженных газов».
Из них жидкий воздух разливают, либо наклоняя сосуд (опрокидываемые
сосуды), либо при помощи небольшого, изображенного на рис. 19 сифона.
Рис. 19. Схема приспособления для
переливания жидкого воздуха из
транспортных цистерн.
Рис. 20. Схема приспособления для
охлаждения бань жидким воздухом.
который может быть изготовлен из стекла или металла. Необходимое дав-
ление для перекачивания малых количеств жидкого воздуха создается прн
помощи резиновой груши. Для отбора из больших цистерн лучше пользовать-
ся сжатым воздухом или азотом из баллона.
Свежеполученный жидкий воздух имеет температуру кипения —194,4 °C.
Поскольку, однако, при кипении преимущественно испаряется азот, темпе-
ратура кипения постепенно повышается (т. кип. О2 —183,0 °C). Жидкий азот
кипит при температуре —195,8 °C, но, если его испарять при пониженном
давлении (вакуумный насос), спустя короткое время получают «азотный
снег» (температура тройной точки —210,0°С). Охлаждающая способность
жидкого азота несколько хуже, чем жидкого кислорода, так как его теплота
испарения и плотность меньше. Несмотря на это, всегда, когда можно, сле-
дует использовать жидкий азот. Контакт жидкого кислорода с горючими
веществами или даже только пропитывание их жидким кислородом может
привести к разрушительным взрывам. Если все же необходимо охлаждать
горючие вещества жидким кислородом, следует изолировать охлаждаемый
стеклянный сосуд от жидкого кислорода непроницаемым защитным кожухом
из листовой меди. Это нужно делать, в частности, при охлаждении сосудов
с активированным углем, если его нельзя заменить силикагелем или молеку-
лярными ситами.
Сосуды Дьюара, изотовленные из йенского стекла, выпускаются стандарт-
ных размеров объемом от 0,2 до 50 л. Они довольно устойчивы к термическим
нагрузкам. Однако следует избегать лишних механических напряжений, по-
скольку они могут послужить причиной растрескивания сосудов (защитные
очки\). Обычно сосуды Дьюара посеребрены, но выпускаются и непосеребрен-
ные — для наблюдения хода реакций — или с узкой непосеребренной верти-
кальной полосой для контроля высоты уровня сжиженного газа. Имеются
также сосуды Дьюара, изготовленные полностью из металла. Для перелива-
5—143
66 Методы препаративной химии
ния сжиженных газов в узкогорлые сосуды используют воронки из жести
или пластмассы.
Охлаждающие бани с ацетоном, метиленхлоридом, петролейным эфиром
и пентаном охлаждают, как показано иа рис. 20, при помощи медного
змеевика, к которому припаян сосуд из листовой меди. Жидкий воздух
переливают в сосуд через сифон; испаряясь, он охлаждает жидкость, в ко-
торую погружен змеевик. Бани меньшего размера охлаждают, погружая в
них закрытую снизу медную трубку, в которую жидкий воздух просто на-
ливают или передавливают, пользуясь приспособлением, показанным на
рис. 20. Чрезвычайно опасно охлаждать горючие жидкости, непосредственно
вливая в них жидкий воздух. Никогда не следует закрывать сосуды с жид-
ким воздухом обыкновенной ватой или бумагой. Если от пламени горелки,
которой нагревают какой-либо прибор, такой там’пон из ваты воспламеняет-
ся, то ои сгорает мгновенно с образованием большого пламени; сосуд Дьюа-
ра при этом обычно лопается. В тех случаях, когда сосуд наполнен довер-
ху, ватный тампон, соприкасаясь с жидким воздухом и пропитавшись им,
может вызвать сильнейший взрыв. Во избежание таких случаев следует на-
крывать сосуд Дьюара только асбестовой ватой или асбестовым картоном.
Сосуды с жидким азотом можно закрывать соответствующим образом вы-
резанной крышкой из стиропора.
Пожаробезопасные охлаждающие бани для работы при очень низких тем-
пературах можно получить, смешивая галогенированные углеводороды
(фторсодержащие углеводороды выпускаются под названиями фригенов,
кальтронов и фреонов).
Составы охлаждающих смесей (в частях по объему):
До - 130 °C: 10 СНС13, 19 СН2С12, 22 CFC13, 7 C2F4C12, 12 С2НС13
До —155 °C и ниже: 10 СНС13, 19 СН2С12, 23 С2Н5Вг, 10 CFC13, 11 С2НС13
Таблица 19. Низкоплавкие вещества для охлаждающих бань с равновесием
твердой и жидкой фаз
Вещество Т. пл.. °C
Изопентан (при застывании вязок) —159,9
Этилхлорид —136,4
Пентан — 129,7
Метилциклогексан —126,6
Этилбромид — 118,6
Диэтиловый эфир —116,2
Сероуглерод —111,5
Трихлорфторметан —111,0
Ацетон —95,4
Дихлорметан —95,1
Толуол —95,0
Этилацетат —83,6
Хлороформ (без примеси спирта) -63,5
Хлораль (трихлорацетальдегид) —57,5
Хлорбензол —45,6
Анизол —37,4
Бромбензол —30,8
Тетрахлорид углерода —23,0
Бензиловый спирт —15,2
Низкие температуры
67
Хранить эти смеси при комнатной температуре следует над сухим К2СО3.
При наличии жидкого азота можно работать с охлаждающими банями
При постоянных температурах в интервале от —20 до —190 °C. Для этого
органическое соединение с подходящей температурой плавления охлаждают
путем медленного приливания жидкого азота при одновременном интенсив-
ном перемешивании. Охлаждение ведут до тех пор, пока ие произойдет
частичного замерзания жидкости с образованием густой массы из смеси
твердой и жидкой фаз. Если ие позаботиться о предварительном охлажде-
нии низкокипяших жидкостей, то при приливании жидкого азота значитель-
2
Рис. 22. Схема устройства охлажда-
ющего блока из алюминия.
1 — ввод жидкого азота; 2 — термометр;
3 — вывод азота; 4 — образец; 5 — волокни-
стый асбест.
Рис. 21. Разрез охлаждающего бло-
ка из алюминия.
ная часть органического соединения может испариться. Поэтому эту опера-
цию следует проводить под тягой. При достаточно большом количестве по-
лученной массы постоянная температура может поддерживаться значительное
время. В табл. 19 приведены некоторые подходящие для этих целей орга-
нические жидкости. Указанные в таблице температуры получают при ис-
пользовании чистых соединений. Если нужно работать при точно заданной
температуре, следует предотвратить попадание загрязнений в охлаждающую
смесь из недостаточно чистого сосуда Дьюара или при конденсации влаги
из воздуха. Для предотвращения снижения охлаждающей способности та-
ких смесей рекомендуется часть аппаратуры, подлежащей охлаждению
(например, ловушку), предварительно охлаждать жидким азотом почти до
температуры бани.
5*
68 Методы препаративной химии
Во многих случаях применяют также охлаждающие блоки из алюминия.
При изготовлении в блоках высверливаются отверстия, в которые можно
вставлять охлаждаемый сосуд и термометр (рис. 21). Блоки подвешивают
на крепком шнуре или металлической проволоке таким образом, чтобы снизу
можно было подставить сосуд Дьюара с жидким воздухом. Погружая в
сссуд Дьюара или поднимая из него алюминиевый блок, можно установить
в последнем необходимую температуру. Для лучшей теплопередачи пустое про-
странство в блоке заполняют одной из жидкостей, перечисленных в табл. 19.
В другом варианте (рис. 22) алюминиевый блок стоит в сосуде Дьюара на
подушке из волокнистого асбеста. Охлаждение производится путем ввода
жидкого азота через боковой широкий канал в испарителе на дне блока.
Газообразный азот выходит через несколько узких боковых каналов, коль-
цеобразно расположенных вокруг центральной цилиндрической полости, в
которой помещается охлаждаемый сосуд.
11. Постоянные температуры
Постоянные высокие температуры
Для поддержания в электрических печах постоянной температуры вы-
ше ~350 °C обычно пользуются регуляторами, описанными в разд. 12. При
этом не обязательно, но тем не менее желательно поддерживать постоян-
ными напряжение, питающее печь, и комнатную температуру. В определен-
ных пределах колебания напряжения сети можно уменьшить при помощи
стабилизаторов напряжения. Они либо работают на принципе дросселя пе-
ременного тока с магнитным насыщением, либо имеют электронную регули-
ровку. Чем больше колебания напряжения, тем с меньшей точностью проис-
ходит их выравнивание при стабилизации.
Термостаты
Для поддержания постоянной температуры от комнатной до макси-
мально 350 °C лучше всего пользоваться известными ультратермостатами,
обеспечивающими высокую точность регулирования, по крайней мере
±0,02 °C. При этом термостатная жидкость в основном сосуде подогревается
и перемешивается. Сосуды, подлежащие термостатированию, помещают не-
посредственно в жидкость или в другой сосуд (воздушную баню). Кроме
того, рабочая жидкость из термостата может циркулировать через внешний
сосуд, подсоединяемый при помощи шлангов. Таким образом удается тер-
мостатировать даже большие (до 300 л) открытые емкости. Температура в
термостатах поддерживается постоянной при помощи электрических регуля-
торов. Температурным датчиком служит либо ртутный контактный термо-
метр (для работы в различных интервалах температур от —59 до ±360°C)*,
либо платиновый термометр сопротивления.
Рабочие жидкости для термостатов выбирают в зависимости от интерва-
ла рабочих температур. Наряду с водой используют различные спирты, ми-
неральные и силиконовые масла, а также другие специальные жидкости.
Рабочая жидкость должна иметь невысокую вязкость при незначительном
давлении пара, высокую температуру воспламеняемости и не должна оказы-
вать вредного физиологического воздействия. Соединительные шланги в за-
висимости от рабочей жидкости изготавливают из пербунана, силикона, бу-
тилкаучука или из металла (сталь марки V2A, томпак).
Можно самостоятельно изготовить жидкостный термостат, используя
кипятильник небольшой мощности, контактный термометр с реле и емкость
возможно большего объема. Большое значение во всех случаях имеет хоро-
шее перемешивание рабочей жидкости. Для нагревания или охлаждения раз-
Выпускается, например, фирмой К. Mittlacher KG (Mainz-Gonzenheim).
Постоянные температуры
69
личных сосудов, трубопроводов, воронок, холодильников, хроматографичес-
ких колонн с помощью циркулирующей рабочей жидкости применяют эла-
стичные шланги с высокой теплопроводностью, поступающие в продажу под
названием калорекс («Calorex»*). Такие шланги марки «rot» могут работать
при температуре от —90 до +280 °C. Выпускаются также шланги, имеющие
в сечении форму полукруга, с шириной плоской части 8, 15 и 30 мм. Таким
шлангом можно просто обмотать сосуд, зафиксировав его положение клей-
кой лентой.
Криостаты
Для поддержания температур до —60 °C можно пользоваться описан-
ными выше ультратермостатами, снабженными охлаждающим приспособле-
нием. В криостатах с испарителем для жидкого азота достигаются темпера-
туры до —190 °C. Если, однако, необходимо поддерживать при постоянной
температуре сравнительно большие количества жидкости в течение значи-
тельного времени, используют криостаты с компрессором с минимальной ра-
бочей температурой —120 °C.
Ультратермостаты можно применять для поддержания температуры не-
много ниже комнатной. Для этого через специальный встроенный змеевик из
меди пропускают охлаждающую воду, а регулирование ведут путем нагре-
вания. Температуры в интервале между + 15 и +5 °C достигаются путем
прокачивания рабочей жидкости через теплообменник, наполненный льдом
и представляющий собой медный сосуд с двойными стенками; через его «ру-
башку» пропускают рабочую жидкость, а внутрь помещают охлаждающее
вещество. Можно также в большой сосуд Дьюара с крышкой из стиропора**
опустить медный змеевик, через который течет рабочая жидкость из тер-
мостата. При заполнении охлаждающей емкости смесью трех частей льда с
одной частью хлорида натрия достигается температура около •—3 °C, причем
в качестве рабочей жидкости можно воспользоваться, например, 20%-ным
раствором хлорида натрия. Температуру до —60 °C получают, используя
для охлаждения смесь сухой лед+метанол, а в качестве термостатной жид-
кости — метанол. В охлаждающий контур можно включить охлаждаемый
агрегат. Но все же удобнее в температурном интервале от 0 до —40 °C
пользоваться небольшими компрессорными криостатами, описанными ниже.
Криостаты с испарителем позволяют вести работу в области температур
от комнатной до температуры кипения охлаждающего агента. При этом в
течение длительного времени обеспечивается постоянство температуры с точ-
ностью не хуже, чем ±0,2 °C, что более чем достаточно для препаративных
целей. В криостатах такого типа имеется змеевик, куда поступает хладагент.
При достижении заданной температуры срабатывает электрический регу-
лирующий вентиль и поступление хладагента прекращается. На рис. 23 по-
казан надежный в работе вариант лабораторного криостата с электрической
регулировкой. Его легко можно собрать из отдельных частей. В сосуде
Дьюара вместимостью 5—10 л находится жидкий азот. Положение уровня
жидкого азота определяют по указателю в виде стеклянной палочки, ук-
репленной на поплавке 2 из пробки или пенопласта. Через патрубок 3 доли-
вают жидкий азот. Клапан избыточного давления 1, заполняемый водой, од-
новременно служит маностатом, причем давление газа, создаваемое в кол-
бе 4, передавливает жидкий азот по сифону 5 в испаритель 8. Сифон, изго-
товленный из приборного стекла, снабжен вакуумированной рубашкой, по-
серебренной также изнутри (готовые сифоны имеются в продаже). Принцип
конструкции медного или латунного испарителя соответствует рис. 20.
Выбранная в данном случае плоская форма позволяет поместить испаритель
даже в небольшой сосуд Дьюара. В другом варианте испарительная камера
* Выпускается, например, фирмой Dr. Pogacar & Со. Nachf. (Heidelberg).
** Материал, сходный с пенопластом. — Прим, перев.
70 Методы препаративной химии
располагается на дне сосуда Дьюара, а отводящая трубка для газообразно-
го азота идет широкой спиралью вдоль стенки. Внутренняя трубка сифона
должна быть несколько уже, чем входная трубка испарителя, для того что-
бы хладагент можно было подавать по каплям. Концы трубок испарителя
присоединяются к сифону и отводящей трубке короткими кусочками поли-
хлорвинилового шланга ниже уровня жидкости в сосуде 13, так как высту-
пающие над поверхностью металлические части привели бы к большому
Рис. 23. Схема устройства криостата с испарителем и с электрической регу-
лировкой.
1 — маностат; 2— поплавок; 3 — патрубок для наполнения; 4 — сосуд Дьюара с хладаген-
том; 5 — сифон; 6 — шланг; 7— крышка из пенопласта; 8 — испаритель; 9— мешалка;
10 — регулятор; // — термопара; /2 — охлаждающая баня; 13 — сосуд Дьюара; 14 — маг-
нитный вентиль; 15 — зажим.
притоку тепла. Испарившийся хладагент удаляется через длинный отрезок
шланга 6 и вентиль с электромагнитным управлением 14. При открытом
вентиле с помощью зажима 15 скорость потока газа, а следовательно, и по-
ступление хладагента регулируют до нужного уровня. Температуру охлаж-
даемой бани измеряют датчиком 11 (термопара, платиновое сопротивление),
который при отклонении температуры от заданной подает сигнал на уси-
литель 10, управляющий положением магнитного вентиля 14. Последний
при отсутствии тока должен быть закрыт. При использовании регулятора
с обратной связью точность поддержания температуры в интервале от —70
до —140 °C составляет +0,1—0,2 °C. Температуру бани измеряют, например,
платиновым термометром сопротивления, не связанным с регулятором.
Автоматические криостаты с компрессором используют в лабораторной
практике в том случае, если необходимо охлаждать большие объемы или
если надо быстро произвести охлаждение и поддерживать заданную темпе-
ратуру длительное время. Последнее необходимо, например, при работе
Регуляторы температуры
71
с сжиженными газами в качестве растворителей, причем при одной и той
же постоянной температуре должны находиться все части аппаратуры:
сосуды со сжиженным газом, реакционные сосуды, холодильники и т. п.
В качестве термостатной жидкости используют, как правило, метанол (до
—90 °C) или метилциклогексан (до —130 °C), а в качестве хладагентов в
компрессорах применяют галогенопроизводные углеводородов. Выпускаются
два конструктивно различающихся типа автоматических криостатов. В крио-
статах первого типа сжиженный в компрессоре хладагент дросселируется в теп-
лообменник и испаряется в нем. Через теплообменник с регулируемой ско-
ростью прокачивают насосом жидкость из охлаждающей бани. Точность
поддержания температуры составляет несколько сотых долей градуса.
Приборы, подлежащие охлаждению, подсоединяют к охлаждающей бане
при помощи шлангов, изолированных пористой резиной. Минимальная ра-
бочая температура несколько различается для конкретных моделей (одно-
или многоступенчатых) и достигает —128 °C. В криостатах второго типа,
предназначенных для отводов больших количеств тепла, точность регулиро-
вания температуры составляет ± 1 °C. В этом случае дросселирование хлад-
агента происходит в испаритель, находящийся непосредственно в емкости с
постоянной температурой (охлаждение путем циркуляции рассола). Термо-
статированная жидкость может также подаваться с помощью насоса в дру-
гие емкости. Лабораторные криостаты, работающие по этому принципу, вы-
пускаются также в виде погружаемых охладителей, обеспечивающих охлаж-
дение до —40 °C. Жидкий хладагент по шлангам поступает в металличес-
кий испаритель, конструктивно сходный с показанным на рис. 20, а внешне
напоминающий обычный кипятильник. В испарителе, опущенном в термо-
статируемую жидкость (в сосуде Дьюара), происходит дросселирование
хладагента и его испарение. Температура, регулируемая электрически дат-
чиком в виде, например, контактного термометра, поддерживается с точно-
стью ' 0,1—1,0 °C для различных моделей. Мощность теплоотвода этого и
других криостатов с испарителем всегда тем меньше, чем ниже рабочая
температура. В заключение следует указать на важность применения
термостатов и криостатов с кипящей жидкостью, а также на возможность
использования для охлаждения эффекта Пельтье*. Соответствующие лабора-
торные приборы имеются в продаже.
ЛИТЕРАТУРА
1. Каталоги фирм Gebruder Haake
KG (Berlin), Colora MeBtechnik
GmbH (Lorch, Wiirtt.), Mefigerate-
werk Lauda (Lauda/Tauber).
2. Engelhard U., Fischer J., lander J.,
Chemie-Ingenieur-Technik, 37, 528
(1965).
12. Регуляторы температуры
Для поддержания постоянной температуры печи, охлаждающей бани
и т. п. независимо от колебаний комнатной температуры, сетевого напряже-
ния и т. д. применяют электрический регулятор температуры. На рис. 24
показана функциональная схема регулятора. Температура измеряется ка-
ким-либо датчиком в «месте измерения», и соответствующий сигнал — «изме-
ренное значение» — подается на регулятор. В регуляторе происходит срав-
нение «измеренного значения» с предварительно установленным «заданным
значением». При определенном превышении минимальной разности этих зна-
чений регулятор воздействует иа исполнительный механизм, благодаря чему
* Выделение или поглощение тепла в месте контакта двух различных про-
водников в зависимости от направления электрического тока. — Прим, перев.
72 Методы препаративной химии
и происходит регулирование. Например в криостате, схематически показан-
ном на рис. 23, процесс регулирования состоит в открывании и закрывании
магнитного вентиля. Исполнительный механизм непосредственно воздей-
ствует на прибор, в котором поддерживается постоянная температура, так
что в определенном его месте, «месте регулирования» (охлаждающий шланг,
электрическая обмотка), происходят соответствующие изменения. Обычно
«место измерения» и «место регулирования» не совпадают между собой, а
Рис. 24. Схема процесса регулирова-
ния температуры.
/ — «заданное значение»; 2—вспомога-
тельный ток; 3 — регулятор; 4 — темпера-
турный датчик; 5 — исполнительный меха-
низм; 6 — «место измерения»; 7 — путь ре-
гулирования; 8 — «место регулирования»;
9 — прибор, в котором регулируется темпе-
ратура. Стрелками показано направление
передачи электрической или механической
энергии или информационного потока.
Рис. 25. Схема включения реле при
регулировании температуры.
1 — контакт (например, контактный термо-
метр) ; 2 — реле; 3 — печь; 4 — основное
сопротивление в цепи печи; 5 — дополни-
тельное сопротивление в цепи печи; 6 —
высокоомное сопротивление.
находятся на некотором расстоянии (путь регулирования). Вследствие этого
возникает определенная инерционность процесса регулирования.
Регуляторы температуры довольно часто двухпозиционны, т. е. их воздей-
ствие на исполнительный механизм соответствует двум крайним возможнос-
тям: «включено» или «выключено». Из-за наличия термической инерции на
пути регулирования измеренное значение периодически колеблется вблизи
заданного значения. Например, регулятор отключает ток нагревателя печи
по достижении равенства измеренного и заданного значений. Поскольку
электрическая обмотка (место регулирования) вследствие ее тепловой ем-
кости отдает тепло печи также и после отключения тока, температурный
датчик (в месте измерения) регистрирует некоторое превышение измеренно-
го значения над заданным. В связи с этим весьма целесообразно вести ре-
гулирование не способом «включено — выключено», а по принципу «боль-
ше—меньше». При этом большая часть обогревателя все время включена
(«основная нагрузка») и только другая часть («регулируемая нагрузка»)
включается или выключается при регулировании. Для достижения еще
лучшего постоянства температуры колебания измеренного значения устраня-
ют за счет применения электронных регуляторов с обратной связью. Для це-
лей регулирования температуры используют в основном не мгновенно сра-
батывающую «электронную обратную связь», а «термическую обратную
Регуляторы температуры
73
связь». Последняя отрегулирована таким образом, что срабатывание регу-
лирующего устройства происходит несколько раньше достижения соответст-
вия между измеренным и заданным значениями. Важно при этом, что влия-
ние обратной связи с течением времени затухает, так что регулятор не бу-
дет отключать нагреватель, если заданное значение не было достигнуто.
В термостатах и криостатах очень хорошо зарекомендовали себя извест-
ные ртутные контактные термометры, схема подключения которых к реле по-
казана на рис. 25. Нельзя допускать, чтобы через ртутные контакты проте-
кал сильный ток, так как в этом случае поверхность ртути загрязняется
вследствие проскакивания искры при выключении.
Сеть
Печь Термо- Регуля-
П!2Ра тор
Рис. 26. Схема подключения регуля-
тора с падающей дужкой при регу-
лировании по принципу «больше —
меньше>.
Сеть
Печь Термо- Регуля-
пара тор
Биметаллические или палочковые регуляторы являются регуляторами под-
водимой энергии. Они не позволяют производить непосредственную настройку
на требуемую температуру, однако для многих целей их вполне достаточно;
кроме того, они весьма дешевы. В биметаллических регуляторах часть тока
проходит через биметаллическую пластинку и нагревает ее. При достижении
определенной степени нагрева, которая может плавно регулироваться, пла-
стинка регулятора размыкает цепь, охлаждается, а затем снова включает-
ся. Токи через печь и регулятор при колебаниях напряжения в сети изменя-
ются пропорционально, так что регулятор сглаживает эти колебания. Анало-
гично работают палочковые регуляторы, используемые в обычных лаборатор-
ных сушильных шкафах. В металлической трубке (например, из алюминия)
находится кварцевая палочка с намотанной на ней нагревательной обмоткой,
которая включена параллельно с нагревателем печи. Различие в удлинении
алюминия и кварца при нагревании используется для включения реле, ко-
торое в свою очередь включает или выключает ток в печн посредством ртут-
ного выключателя.
В широко применяемом регуляторе с падающей дужкой (контактном
гальванометре) напряжение термопарного датчика подается на милливольт-
метр. При помощи небольшого механизма с прижимной дужкой положение
стрелки милливольтметра фиксируется через небольшие промежутки време-
ни (примерно каждые 15 с). В остальное время стрелка может свободно
перемещаться. Если в процессе работы стрелка отклоняется от заданного
положения, срабатывает выключатель, соответственно изменяющий ток в пе-
чи (рис. 26). Точность регулирования составляет примерно ±Г при 800 °C.
Действие других типов регуляторов основано не на механическом, а на
индуктивном, емкостном или фотоэлектрическом методах контроля положе-
74
Методы препаративной химии
ния стрелки измеряющего прибора, который позволяет одновременно произ-
водить прямое считывание показаний.
Наибольшую точность регулировки обеспечивают электронные регуляторы,
собранные на транзисторах и не имеющие измеряющего механизма. Регули-
ровка может вестись в температурном интервале от —250 до +1800 °C, при-
чем датчиками служат термопары или термометры сопротивления. Приборы
для осуществления электронной или термической обратной связи либо
встроены в регулятор, либо могут быть дополнительно подключены. Способ
установки заданного значения может быть аналоговым пли цифровым. Со-
ответствующий программный задатчик позволяет вести регулирование темпе-
ратуры по определенной программе. Синхронный мотор поворачивает с оп-
ределенной скоростью задающий потенциометр, что обеспечивает постоянную
•скорость изменения температуры. В другом варианте соответствующим обра-
зом вырезанная шайба вращается вокруг своей оси, в то время как положе-
ние ее края контролируется механическим или фотоэлектрическим способом.
Снятый сигнал передается на задатчик регулятора. Такой способ позволяет
производить изменения температуры по произвольной программе. Электрон-
ные регуляторы необходимо дополнять исполнительным механизмом в виде
выключающего реле. Кроме того, они не являются показывающими прибо-
рами. Для считывания показания должен подключаться независимый изме-
рительный прибор.
ЛИТЕРАТУРА
1. Pinkava J., Laboratoriumstechnik
kontinuierlicher chemischer Prozes-
se. M. Deutsch, Frankfurt/Main,
1962. Пинкава Я- Лабораторная
техника непрерывных химических
процессов. Пер. с чеш./Под ред.
Н. А. Клейменова. — М.: ИЛ, 1961.
2. Каталоги фирм Hartmann и. Braun
AG (Frankfurt/Main), Gebr. Ruhs-
trat. (Lenglern).
3. Engelhardt U., Fischer J., Jander J.,
Chemie-Ingenier-Technik, 37, 528
(1965).
13. Высокий вакуум и изоляция от воздуха
Под вакуумной техникой подразумевают специальные методы работы,
при которых используются низкие абсолютные величины давления. Высоким
вакуумом обозначают давления, меньшие чем 10~3 мм рт. ст.
Согласно принятому в настоящее время закону о единицах измерения (1969 г.) и о
порядке их применения (1970 г.), за единицу давления принят паскаль (Па). 1 Па =
= 1 Н/м2. Производными единицами давления являются 1 бар=105 Па и 1 мбар=102 Па.
В свою очередь 1 бар=750 мм рт. ст., а 1 мм рт. ст. = 1,333 мбар. Применявшаяся ранее
единица давления 1 атм эквивалентна 760 мм рт. ст. или 1,013 бар.
Для получения вакуума химики-экспериментаторы могут использовать
насосы различных типов, представленные в табл. 20. Для промышленных
целей выпускаются насосы со значительно большей скоростью откачки.
При выборе насоса или комбинации насосов следует учитывать следую-
щие факторы:
1. Давление пара рабочего тела. Масла и другие рабочие тела, исполь-
зуемые в насосах, обладают при комнатной температуре определенным соб-
ственным давлением насыщенного пара, которое ограничивает величину ко-
нечного вакуума. Несколько более высокий вакуум можно получить за счет
вымораживания паров в ловушке или их удаления при помощи специаль-
ной отсекающей пластины. Обычно применяемые охлаждаемые пластины
выполняют обе функции.
2. Предельный вакуум. При минимальном достижимом давлении насоса
скорость откачки, т. е. его производительность, равна нулю. Если в аппарату-
Высокий вакуум и изоляция от воздуха 75
Таблица 20. Типы лабораторных насосов
Тип иасоса Предельный вакуум, мм рт. ст. Скорость откачки
Водоструйный Ротационный масляный Диффузионный масляный Диффузионный ртутный Ионный а В особом исполнении нас вакуум до 10-6 мм рт. ст. 15—20 20—10-3а 10-6 (с отсекающей пласти- ной) 10-6 (с ловушкой) 10-9 (сверхвысокий ваку- ум) оса и прн полной конденсации п 0,2—0,4 м3/ч 1—10 м3/ч 25—100 л/с 3—50 л/с аров масла достигается
ре нужно создать определенное низкое давление, то всегда следует исполь-
зовать такой насос, конечное давление которого по крайней мере на поря-
док ниже.
3. Скорость откачки насоса, зависящую от давления. Приводимые значе-
ния для ротационных масляных насосов соответствуют откачке воздуха при
атмосферном давлении. Скорость откачки одноступенчатыми ротационными
насосами уменьшается, начиная с давления 10 мм рт. ст., а двухступенча-
тыми — с 1 мм рт. ст., и становится равной нулю при достижении конечного
вакуума. Скорость откачки диффузионных насосов в л/с (1 л/с=3,6 м3/ч)
указывается для давления ~1O-1 мм рт. ст., т. е. для среднего рабочего
давления насосов этого типа.
4. Газовый балласт. В том случае, если масляный насос применяют для
откачки паров, существует опасность, что при сжатии в насосе эти пары
сконденсируются. В результате могут произойти загрязнение масла, коррозия
насоса и в конечном счете ухудшение конечного вакуума. Для предотвра-
щения этих явлений выпускаются специальные насосы с вентилем газового
балласта, через который во время работы насоса в компрессионное простран-
ство впускается определенное количество воздуха. За счет промывки возду-
хом явление конденсации паров, как правило, предотвращается. Насос при
этом работает более устойчиво; однако конечный вакуум ухудшается по
сравнению с вакуумом, достигаемым при закрытом вентиле газового баллас-
та. Само собой разумеется, что пары агрессивных веществ всегда следует
вымораживать, ставя перед насосом по крайней мере одну охлаждаемую ло-
вушку.
Обычно для проведения препаративных работ вполне достаточно скоро-
сти откачки масляными насосами I—2 м3/ч, а диффузионными насосами —
Ю—20 л/с. Диффузионные насосы изготавливают из тугоплавких сортов
стекла, кварца или стали, причем первые два типа насосов особенно хорошо
поддаются очистке. Нагревание насосов ведут электрическим током. При-
менение в диффузионных насосах ртути в качестве рабочего тела имеет не-
которые преимущества по сравнению с использованием специальных масел.
Так, значение необходимого форвакуума составляет для ртутных насосов
Ю—15 мм рт. ст., тогда как для масляных — 0,5—0,1 мм рт. ст. Ртуть ме-
нее чувствительна к попаданию воздуха в нагретый насос; очистку насоса
легко проводить промыванием его азотной кислотой и водой. С другой сто-
роны, недостатки ртути состоят в ее ядовитости и в том, что при комнатной
температуре давление ее насыщенных паров недостаточно мало (2-10~а мм
рт. ст.).
Из предосторожности под стеклянный ртутиый насос подставляют боль-
шой поддон из жести. При работе масляных диффузионных насосов можно
76
Методы препаративной химии
иногда ограничиться воздушным охлаждением, применяя мощный вентиля-
тор, тогда как ртутные насосы следует всегда охлаждать проточной водой.
Весьма целесообразно систему охлаждения насоса снабжать готовым или
самодельным реле, отключающим нагреватель при отсутствии водяного ох-
лаждения. Для предотвращения толчков при кипении ртути рекомендуется
добавлять в нее немного свинцовой или цинковой пыли.
При определении размеров трубопроводов и охлаждающих ловушек,
присоединенных к насосу, необходимо учитывать сильное влияние их сопро-
тивления W на эффективную скорость откачки 5Эф вакуумной установки:
S 5
гэф — J _|_ SU7
где S—скорость откачки самого насоса.
При этом в зависимости от давления различают область ламинарного
течения (давление порядка миллиметра рт. ст.) и область молекулярного
или диффузионного течения (тысячные доли мм рт. ст.). Для прямых тру-
бок сопротивление может быть рассчитано по формуле
I I
IF (ламинарное течение) ss ; W (молекулярное течение) = -jg-
где d — диаметр трубки; I — ее длина; р — давление газа.
Соответствующие уравнения могут быть выведены и для расчета со-
противлений всевозможных частей аппаратуры, имеющих другую, иногда
неправильную форму, — отверстий, загибов, ловушек и т. д. Общее сопро-
тивление всей установки вычисляется по аддитивной схеме (см. [1]).
Из вышеприведенных формул видно, как важно применять в вакуум-
ной технике широкие трубопроводы. Сечение трубопроводов коротких вакуум-
ных линий стараются выбирать не уже, чем выходное отверстие насоса (в
зависимости от размера откачиваемых объектов), однако в любом случае
диаметр трубопроводов должен быть не менее 20 мм, а отверстий кранов —
не менее 10 мм.
Наконец, следует указать на необходимость регулярной замены масла
в ротационных и диффузионных насосах. В противном случае нельзя ожи
дать от насоса ни номинальных параметров вакуума, ни продолжительной
бесперебойной работы.
Измерение давления
Очень часто для этой цели применяют обычные U-образные манометры.
Для устранения влияния поверхностного натяжения оба плеча манометра
должны иметь одинаковое сечение (но не менее 10 мм, а для прецизионных
измерений 15—20 мм). Лучше всего деления наносить непосредственно на
трубку путем травления, а считывание показаний производить, используя
зеркало, установленное позади манометрической трубки для устранения па-
раллакса. В этом случае деления можно нанести на зеркало, а не на труб-
ку. При измерении таким способом можно вести отсчет с точностью до
0,1 мм рт. ст.
Изготовление хорошего ртутного манометра требует определенной тща-
тельности в работе. После основательной очистки трубки хромовой смесью
и промывки дистиллированной водой манометр высушивают по методике,
описанной в разд. 2 («Стекло»), и припаивают его к приспособлению для
заполнения (рис. 27). В колбу вносят необходимое количество тщательно
очищенной и высушенной ртути и запаивают трубку в точке 41. После это-
го из прибора откачивают воздух диффузионным насосом и весь прибор
основательно прогревают пламенем горелки при непрерывной работе насоса.
Под конец ртуть нагревают до тех пор, пока она не начнет кипеть в вакуу-
ме. Трубку заплавляют в точке 2 и дают прибору охладиться. Следует об-
Высокий вакуум и изоляция от воздуха
77
ращать внимание на то, чтобы при нагревании не произошло перегонки не-
которого количества ртути в манометр, что может привести к преждевре-
менному перекрыванию U-образной трубки. Затем манометр наклоняют
таким образом, чтобы ртуть перелилась в трубку, и осторожно наносят цара-
пину на месте запайки 2. Никоим образом не следует обламывать трубку в
запаянном месте. В этом случае быстро ворвавшийся воздух с такой силой
выбросит ртуть, что она разобьет манометр. Сужение поперечного сечения
манометра в точке 1, предназначенное для снижения скорости ртути, все же
Рис. 28. Схемы ртутных манометров с клапа-
нами избыточного давления (размеры указаны
в миллиметрах).
Рис. 27. Схема ртутного
манометра с приспособлени-
ем для его заполнения.
не предотвратит поломку манометра. В дальнейшем колбу удаляют и в точ-
ке 3 производят припайку манометра к аппаратуре. С помощью сжатого
воздуха ртуть передавливают в левое колено так, чтобы заполнился верхний
U-образный капилляр. Тем самым остаточный газ попадает в небольшой
верхний шарик, и при опускании столба ртути в левом колене получают
идеальную торичеллиеву пустоту. Последнюю операцию можно время от
времени повторять.
На установках, предназначенных для работы с газами при переменном
давлении (от высокого вакуума до давления несколько выше атмосферного),
применяют манометры типа изображенных на рис. 28. Эти манометры могут
выполнять роль предохранительных клапанов, если в аппаратуре возникает
слишком большое давление. Очень удобно рядом с манометрической труб-
кой иметь барометрическую трубку того же диаметра (см., например,
Ьис. 46), положение ртути в которой принимают за нулевую отметку, что
сильно упрощает считывание показаний.
78 Методы препаративной химии
При работе с агрессивными газами, взаимодействующими об ртутью, мож-
но использовать так называемые нуль-манометры (рис. 29), заполняемые
конц. H2SO4, силиконовым маслом, бромнафталином и т/п. Оба колена
нуль-манометра соединены краном. Кроме того, для удобства заполнения
одно из колен можно снабдить патрубком, который в/дальнейшем отпаи-
вается. Весьма полезной оказалась также конструкция/ртутного манометра
(рис. 30), предложенная в работе [2]; в этом приборе можно производить
замену загрязненной ртутными солями трубки 3 без/предварительного уда-
ления ртути из манометра. Сначала с помощью Воронин заполняют такой
Рис. 29. Конструкция нуль-манометра.
Рис. 30. Устройство ртутного мано-
метра для работы с агрессивными
газами (1—5 см. текст)
манометр ртутью примерно до половины через шлиф 1 (при удаленной час-
ти 4). После подсоединения части 4 при помощи сжатого воздуха вытесня-
ют ртуть в другое колено так, чтобы она попала в сосуд 5. Этот сосуд
снабжен притертой стеклянной палочкой, которая в нижнем положении пе-
рекрывает отверстие. После закрывания пришлифованной палочкой сосуда 5
столб ртути опускается лишь частично в соответствии с величиной атмосфер-
ного давления. Количество ртути должно быть выбрано таким образом, что-
бы в опущенном состоянии ртуть не доходила до шлифа 2. Теперь можно
легко вынуть трубку 3 для ее очистки. Одновременно можно при необходи-
мости удалить и заменить часть загрязненной ртути в поверхностном слое
у шлифа 2.
Особенно рекомендуем нуль-манометры с кварцевой спиралью по Боден-
штейну (рис. 31), потому что в них газы приходят в соприкосновение толь-
ко с кварцем. Эти манометры выпускаются в различном исполнении* с от-
* Фирмы-изготовители: W. С. Heraeus (Hanau), Texas-Instruments (USA)
через посредство фирмы Techmation (Dusseldorf).
Высокий вакуум и изоляция от воздуха
79
счетом с помощью микроскопа или зеркала. При перевозке их обычно на-
полняют глицерином, так как в противном случае весьма чувствительная
к сотрясениям сцираль ломается при транспортировке. Манометры с не-
сколько пониженной чувствительностью могут выдерживать перепад давле-
ний в 760 мм рт. сх. между пространством внутри и снаружи спирали; у бо-
лее чувствительных Хприборов перепад давлений не должен превышать
150 мм рт. ст. Манометры Боденштейна нужно устанавливать так, чтобы
предотвратить возмогйность их сотрясения. Они надежно выдерживают на-
гревание до 500 °C без изменения положения нулевой точки. При нагревании
спирали до более высоких температур может сместиться нулевая точка, осо-
Рис. 32. Схема присоединения нуль-
манометра (манометра Боденштей-
на).
Рис' 31. Устройство манометра с
кварцевой спиралью по Боденштей-
ну.
бенно если при этом возникают значительные перепады давлений. Если та-
ких больших перепадов давлений удается избежать, изменения нулевой точ-
ки не происходит до температуры 700 °C. Измерения давления можно прово-
дить в интервале от 760 до 10~s мм рт. ст. Способ подсоединения маномет-
ра Боденштейна показан на рис. 32.
Для измерения малых разностей давлений можно пользоваться маномет-
ром с наклонной трубкой (манометры Гюйгенса), наполненной бромнафтали-
ном или апиезоновым маслом. В манометрах с наклонной трубкой, запол-
ненных ртутью, создается слишком большое трение, потому что для изготов-
ления этих приборов приходится пользоваться трубками очень малого диа-
метра (см. [3]).
Давления индифферентных газов и паров в интервале 1—100 мм рт. ст.
можно измерять также при помощи мембранных манометров*.
Измерение малых давлений
При измерении малых давлений необходимо учитывать, что в вакуум-
ной аппаратуре обычно присутствует смесь газов и паров, сумма парциаль-
ных давлений которых равна общему давлению. Измерение парциальных
давлений хотя и возможно, но трудно осуществимо (необходим масс-спект-
рометр или масс-фильтр). Манометры для работы в интервале 10~2—10~6 мм
рт. ст., действие которых основано на зависимости теплопроводности, трения
* При помощи мембранных манометров, подсоединенных по схеме
рис. 32, можно измерять давления от 10~2 до 760 мм рт. ст. — Прим, перев.
80 Методы препаративной химии
или ионизации газов от давления, регистрируют общее давле/ие. Если же
измерение проводится компрессионным методом, то измеряется давление не-
конденсируемых газов. /
Для измерения давления в интервале 10~'—10~s мм pt. ст. издавна слу-
жит манометр Мак-Леода, работающий по принципу компрессии и кратко
называемый «маклеодом». Применяемая ртуть должна выть тщательно очи-
щена и высушена нагреванием в вакууме. Хотя маклеоды, а также вариант
исполнения по Каммереру (80—10-'1 мм рт. ст.) редко находят применение
для препаративных работ, так как они занимают сравнительно много места
и подвержены влиянию помех, их можно с успехом использовать для градуи-
Рис. 33. Конструкции укороченных манометров Мак-Леода.
а. б — для измерения давлений 1—10~3 мм рт. ст.; в — для давлений 50—10-2 мм рт. ст.
ровки других приборов для измерения давления в области ~ 10~3 мм рт. ст.
Если нужно проводить лишь ориентировочные измерения давления, применя-
ют небольшие по размерам манометры компрессионного типа по Мозеру
(рис. 33, а, б) или «вакускопы» по Гэде (рис. 33, в). Эти типы приборов
имеются в продаже. При измерении такими манометрами уже во время ком-
прессии происходит конденсация паров смазки, воды, ртути, а также эфира,
тетрахлорида углерода и т. п. За счет конденсации, а также вследствие ад-
сорбции на стеклянных стенках вклад давления этих веществ в общее дав-
ление соответствует значениям давления их насыщенных паров.
Для измерения давления инертных паров лучше всего пользоваться элек-
трическими вакуумметрами непрерывного действия (табл. 21), в которых ис-
пользуется зависимость теплопроводности, трения или ионизации газов от
давления. Следует, однако, учитывать, что выпускаемые приборы програ-
дуированы по сухому воздуху, и градуировка для других газов и паров
проводится обычно эмпирически, а не путем пересчета. При систематической
работе с неиндифферентными газами необходимо регулярно проводить про-
верку градуировки прибора. Следует защищать измерительные приборы от
попадания в них агрессивных паров путем установки охлаждающих лову-
шек. Необходимо также помнить, что ионизационный манометр действует как
миниатюрный насос. Это происходит потому, что ионы, сильно ускоренные
в поле высокого напряжения, при столкновении с электродами остаются на
поверхности металла. Поэтому измерительные приборы должны соединяться
с остальной вакуумной системой посредством коротких и широких трубок,
так как в противном случае измеренное давление может оказаться занижен-
ным.
Для быстрой ориентировки относительно примерной величины вакуума
в аппаратуре пользуются высокочастотным течеискателем. При приближении
Высокий вакуум и изоляция от воздуха
81
Таблица 21. Пределы измерения давления электрическими вакуумметрами
Принцип работы вакуумметра Пределы измерения давления, мм рт. ст.
Трение в газе Теплопроводность газа Ионизация газа с холодным катодом пеннинг магнетрон с раскаленным катодом с а-излучателем 800—Ю-3 800—10-3 10-2— ю-6 Ю-4— ю-и Ю-s— ю-w юоо—ю-4
высокочастотного электрода к вакуумированной стеклянной аппаратуре внут-
ри нее возникает свечение, характер которого зависит от величины давле-
ния. В области 150—10 мм рт. ст. внутри аппаратуры, особенно на металли-
ческих частях, проскакивают искры, переходящие при более низком давле-
нии в нитевидный разряд. При давлении 10—10-1 мм рт. ст. в газовом про-
странстве возникает красное или фиолетовое свечение, вызываемое наличием
водорода, азота и углеводородов, которое при понижении давления все бо-
лее заполняет сечение трубки. Давление ниже 10-1 мм рт. ст. характеризует-
ся появлением зеленовато-синей флуоресценции на стеклянных стенках, про-
тивоположных высокочастотному электроду. При дальнейшем снижении
давления свечение газового пространства постепенно ослабевает, так что при
10-s'mm рт. ст. заметна лишь зеленая флуоресценция стенок. При еще бо-
лее низких давлениях никакого свечения больше не наблюдается.
Обнаружение неплотностей
Поиски неплотностей в вакуумной установке требуют иногда чрезвычайно
много времени. В большинстве случаев они являются результатом недостаточ-
но тщательного замазывания щелей, плохой смазки кранов и шлифов или
плохой пропайки стеклянных спаев. Если речь идет о стеклянной аппаратуре,
при поиске таких неплотностей незаменим высокочастотный течеискатель.
Аппаратуру вакуумируют до давления 0,1—1 мм рт. ст. и затем проводят по
ней высокочастотным электродом. В аппаратуре возникает при этом слабое
свечение. В местах неплотностей через них проходит более сильный разряд,
так что такие места сразу видны по интенсивно светящимся разрядным
нитям. Однако следует избегать касаться тонких мест иа спаях, так как
через эти места при высоком напряжении может произойти прямой пробой
искры. Поэтому лучше ставить регулятор напряжения на течеискателе в сред-
нее положение. Обнаруженные неплотности в стекле необходимо заново про-
паять или закрыть их небольшой каплей пицеина, которую наносят на очи-
щенную и подогретую поверхность стекла. Вместо пицеина можно использо-
вать вакуумно-плотную замазку, например «аральдит», или специальный
состав на силиконовой основе для устранения неплотностей, наносимый при
помощи распылителя.
Труднее обнаружить неплотности в кранах. В этом случае приходится
заново смазать все вызывающие подозрение краны. Если возможно, пытают-
ся локализовать неплотность путем последовательного отключения части
кранов. Можно также рекомендовать при включенном высокочастотном тече-
искателе омывать аппаратуру углекислым газом, подаваемым из шланга. По
изменению цвета высокочастотного разряда из красноватого в белый мож-
но обнаружить местонахождение неплотности.
6—143
«2
Методы препаративной химии
Изменение цвета разряда происходит также при касании Места неплот-
ности ватным тампоном, смоченным спиртом или эфиром, что Связано с про-
никновением через неплотность паров спирта или эфира в вакуумную аппа-
ратуру. При поиске неплотности этим методом необходимо'' следить за тем,
чтобы проникшие в установку пары успевали пройти в разрядное простран-
ство. Важно также несколько выждать, прежде чем проверять следующее
подозрительное место.
Еще лучше искать неплотности при наличии в установке электрического
вакуумметра. В этом случае между аппаратурой и вакуумметром помеща-
ют охлаждаемую ловушку. При работающем насосе проводят по стенкам
аппаратуры ватным тампоном, пропитанным эфиром. Если при этом попа-
дают на неплотность, то вместо воздуха в аппаратуру преимущественно про-
никают пары эфира, которые, однако, вымораживаются в ловушке, вследст-
вие чего вакуумметр на короткое время показывает улучшение вакуума. Пре-
восходными приборами, правда дорогостоящими, являются галогенные тече-
искатели. В них вызывающее подозрение место обдувается газом, содержа-
щим атомы галогенов (например, фреоном, хлороформом), или смачивается
•соответствующей жидкостью при помощи тампона из ваты. Проникший
в аппаратуру газ благодаря каталитическому действию вызывает появление
тока в подсоединенной ионизационной трубке с нагреваемым платиновым
анодом. Этот ток после усиления преобразуется в оптический или звуковой
сигнал. Работа гелиевого течеискателя основана на том, что проникновение
через неплотности газообразного гелия регистрируется небольшим масс-спект-
рометром (см. [4]).
I
Краны 1
При выборе кранов следует обращать внимание на то, чтобы соприка-
сающиеся поверхности шлифов были достаточно большими. Наиболее упот-
ребительные формы кранов изображены на рис. 34 (а—ж). По возможности
следует применять краны Шиффа (рис. 34, в, г); они надежнее обеспечивают
герметичность, так как в них между припаянными трубками не может возник-
Рис. 34. Различные типы стеклянных кранов.
нуть соединительных каналов. Краны с закрытой с одной стороны втулкой
(рис. 34, ж) более вакуумно-плотные, чем простые проходные краны. Треххо-
довые краны (рис. 34, е) в вакуумных установках являются источниками по-
стоянных неприятностей. Их лучше заменять тремя двухходовыми кранами.
Трехходовые краны типа изображенных на рис. 34, д следует предпочесть ти-
пу рис. 34, е.
Высокий вакуум и изоляция от воздуха
8S
Краны требуют тщательного смазывания. Перед смазыванием кранов их
части тщательно очищают и нагревают до температуры 30—40 °C. Смазку
тонкими кольцами наносят деревянной, стеклянной или металлической палоч-
кой по окружности середин верхней и нижней половин пробки. Удобно так-
же дозировать смазку, выдавливая ее из отверстия пластмассового шприца.
Оба кольца смазки соединяют тонкой полоской смазывающего вещества в
месте, наиболее удаленном от отверстия канала (рис. 35). Затем пробку
крана вставляют в слегка нагретую втулку крана таким образом, чтобы по-
следний оказался в открытом положении, затем с нажимом слегка повора-
чивают пробку то в одну, то в другую сторону. При этом никогда не следу-
ет поворачивать пробку так, чтобы кран оказался в закрытом положении.
Рис. 35. Способ нанесения смазки на
кран.
Лишь после того, как смазка крана распределится совершенно равномерно
и между пришлифованными поверхностями не будет находиться ни одного
пузырька воздуха, пробку можно повертывать кругом. Только таким образом
можно добиться сплошной смазки крана. Вакуумные краиы следует пово-
рачивать мягко и не слишком быстро, чтобы не была превышена скорость
течения смазывающей жидкости и пленка смазки не разорвалась. Неизбеж-
ным последствием нарушения этого правила являются образование желоб-
ков и каналов и нарушение плотности. Предстоящий переход через границу
допустимой скорости улавливается — при наличии некоторого опыта — по осо-
бому нарастанию сопротивления пробки вращению. Если разрывы образова-
лись, то перед новым смазыванием крапы должны быть полностью очищены.
Для удаления смазки на каучуко-парафиновой основе можно использовать
алифатические углеводороды (петролейный эфир, т. кип. ~ 100 °C) или гало-
генированные углеводороды (тетрахлорид углерода, трихлорэтилен). Силико-
новую смазку, кель-Е, хостафлон лучше всего удалять ватой, смоченной в
эфире.
Для очистки каналов малого диаметра в крапах с успехом можно при-
менять «ерши для курительных трубок» — тонкие щеточки длиной около
10 см, имеющиеся в продаже в каждом табачном магазине. В других слу-
чаях пользуются тонкими деревянными палочками, обернутыми небольшим
кусочком ваты. Если газы или пары взаимодействуют со смазкой, то краны
редко сохраняют плотность достаточно долго. В таких случаях используют
вентили, не требующие смазки. Недавно стали выпускать так называемые
«плоские краны», причем трехходовой кран этого типа более плотен, чем
кран, изображенный на рис. 34.
Размер стеклянного крана определяется диаметром (в миллиметрах) про-
ходного отверстия в пробке. Обычные одноходовые краны, согласно стан-
дарту DIN 12551, выпускаются размерами 1,5; 2,5; 4; 6; 8; 10; 15; 20 мм.
Весьма целесообразно пользоваться кранами, имеющими приспособление,
предотвращающее ослабление или выпадение пробки крана.
Тефлоновые вентили
В последнее время были разработаны и оказались весьма надежными
в работе лабораторные стеклянные вентили с уплотнением в виде конуса
6*
«4
Методы препаративной химии
из тефлона*. Эти вентили, используемые при температурах от —25 до
+200 °C, могут быть проходными или угловыми (рис. 36), они хорошо «дер-
жат» избыточное давление и позволяют при полном отсутствии смазки про-
изводить точную регулировку потока газа или жидкости. Для придания
необходимой жесткости конус вентиля усилен стекловолокном. Одной своей
частью конус уплотняет проходное отверстие вентиля, а другой, при помощи
двух кольцевых уплотнительных колец из витона или мембраны, — резьбо-
вое соединение. Подобные вентили являются вакуумно-плотными вплоть до
давления 10~6 мм рт. ст. Химическая стойкость всей конструкции определи-
мые. 36. Типы стеклянных вен-
тилей с уплотняющим конусом
из тефлона.
«тся в основном материалом уплотнительных колец, которые могут быть лег-
ко заменены, хотя вообще обладают хорошей химической стойкостью. [
Вентили с уплотнением в виде тефлонового сильфона несколько менее
вакуумио-плотны. Они больше подходят для работы под избыточным дав-
лением. Их преимущество состоит в том, что поток вещества приходит в со-
прикосновение только с тефлоном.
Ртутные клапаны Штока
Из различных конструкций ртутных клапанов, предложенных Што-
ком [5], ниже рассмотрены лишь клапаны с поплавками и пористыми стек-
лянными пластинками. Поплавковые клапаны позволяют быстро пропускать
газы, однако они могут безупречно работать лишь в том случае, если строго
выдержаны указанные Штоком размеры. Поплавки должны быть массивны-
ми, изготовленными из стекла и должны иметь лишь весьма узкую, но зато
очень тщательно обработанную кольцевидную шлифованную зону (рис. 37, а).
Когда открывают крап /, в трубку 2 поступает воздух и поднимающийся
уровень ртути двигает поплавки 3 вверх. При вакуумировании трубки 2
опускающаяся ртуть освобождает поплавки 3, которые падают вниз и про-
пускают газ (если они остаются в висячем положении, нужно слегка посту-
чать по стеклянной стенке). Следует позаботиться о том, чтобы при откры-
вании клапана на обоих концах по возможности не возникало значительной
разности давлений. Когда нужно открыть какой-либо клапан, к нему мож-
но присоединить постоянно лежащий на рабочем столе гибкий шланг от
водоструйного насоса. Когда клапан срабатывает, кран 1 снова закрывают.
Клапаны с пористой пластинкой можно открывать также и тогда, когда
по обе стороны имеется значительная разность давлений. Всплывающие по-
плавки заменены в данном случае непроницаемыми для ртути пористыми
стеклянными пластинками (рис. 37,6). Недостаток подобных клапанов за-
ключается в довольно значительном торможении газовой струи. Разновид-
* Фирмы-изготовители: Jenaer Glaswerk Schott und Gen. (Mainz), R. Kuhn
KG (Hagen), Quickfit-Laborglas GmbH (Wiesbaden-Schierstein).
Высокий вакуум и изоляция от воздуха 85
ностью клапанов с пористыми стеклянными пластинками является клапан,
предложенный Вибергом (рис. 37, в). Пористая стеклянная пластина, ча-
стично или полностью погруженная в ртуть, используется в конструкции не-
большого маностата* (рис. 38). Сначала при закрытых кранах маностат че-
рез трубку 5 заполняют ртутью так, чтобы пористая пластинка была погру-
жена примерно на 3/4. Затем трубку 5 подсоединяют к аппаратуре и ваку-
умируют через трубку 4, причем краны маностата открывают в последова-
тельности 3—2—1. По достижении в аппаратуре нужного давления крапы
закрывают в порядке 1—2—3 и снова открывают кран 1. Газ, запертый во
Рис. 37. Типы ртутных клапанов Штока (а—в см. текст).
/ — кран; 2— трубка; 3— поплавки; 4 — пористые стеклянные пластинки; 5 —кран (что-
бы не загрязнять ртуть смазкой, эти краны можно исключить); 6 — припаянная стек-
лянная палочка, касающаяся пористой пластинки, служащая для облегчения стекания
ртути. Размеры указаны в миллиметрах.
внешнем объеме маностата краном 3, воспринимает колебания давления
в аппаратуре, что сказывается на положении уровня ртути во внутреннем
объеме и тем самым на поступлении газа через пористую пластинку. Путем
кратковременного открывания крана 3 маностат может быть отрегулирован
иа более низкое давление.
Клапаны Боденштейна
Эти клапаны изготовляют из стекла или кварца: их можно подвергать
нагреванию. Они представляют собой наиболее последовательное решение
проблемы конструирования клапанов без смазки, так как здесь газы прихо-
дят в соприкосновение только со стеклом (рис. 39). Клапан состоит из ка-
пилляра, к отверстию которого прижимается тщательно отшлифованный и от-
полированный шарик, укрепленный на конце стеклянной палочки. Стеклян-
ный запаянный корпус клапана отличается достаточной эластичностью, что-
бы допустить незначительные движения обеих частей относительно друг дру-
га. Обе части сжимаются пружиной, благодаря чему отверстие клапана
оказывается закрытым. При помощи винта обе части клапана могут оттяги-
ваться друг от друга, причем наличие ограничительных штифтов предотвра-
Выпускаются фирмой Normag (Hofheim/Taunus).
86
Методы препаративной химии
щает возможное раздавливание корпуса при слишком сильном закручива-
нии.
Этот клапан можно применять и для впуска жидкостей в вакуумное
пространство. В любом случае в жидкости не должно быть взвешенных
твердых частичек. Даже микроскопически малые частички, попавшие на
поверхность шлифа, очень трудно потом удалить. В дальнейшем при силь-
ном сдавливании они могут повредить шлифованную поверхность и нару-
шить герметичность клапана. В таких случаях не помогает даже длительное
промывание хромовой смесью и водой, и поэтому приходится прибегать к
замене стеклянных частей клапана. При поломке стеклянного корпуса его
легко заменить новым. Для этого сначала расплавляют влитый для фикса-
ции положения клапана сплав Вуда, вынимают осколки старого клапана из
оправы и вставляют новый. При этом стеклянную палочку с припаянным
к ней маленьким стеклянным шариком вставляют в отверстие винта, а ка-
Рис. 38. Конструкция ртутного
маностата с пористой стек-
лянной перегородкой (см.
текст).
Рис. 39. Устройство клапана Боденштейна.
пилляр с утолщением—в прорезь алюминиевого держателя. Обе части фик-
сируют в этом положении двумя кусочками асбестовой бумаги. После этого
клапан ставят вертикально на винт и в нагретую оправу винта наливают
свежерасплавлеиный сплав Вуда. Когда металл полностью затвердеет, кла-
пан (винт которого должен быть полуотвинчен) поворачивают в горизон-
тальное положение и отверстие, в центре которого находится утолщенная
часть подводящего капилляра, также заливают нагретым сплавом Вуда.
Если клапан должен работать в нагретой водяной бане, то, разумеется,
можно пользоваться вместо сплава Вуда замазкой из глицерина и свинцово
го глета.
Чтобы избежать трудностей, связанных с пришлифовкой запорных ча-
стей в подобных клапанах, было предложено [6] использовать стандартные
шаровые шлифы. В другом варианте клапанов Боденштейна, весьма надеж-
ном в эксплуатации, уплотнение осуществляется не за счет контакта стек-
ло— стекло, а благодаря сдавливанию деталей из стекла и хлорида сереб-
ра. Кроме того, выступающие гофры, сообщающие корпусу клапана некото-
рую эластичность, заменены на втянутые [7].
Разбиваемые клапаны
Для препаративных целей вместо применения довольно дорогих клана-
Высокий вакуум и изоляция от воздуха 87
нов Боденштейна часто пользуются комбинацией процессов запаивания (для
закрывания) и разбивания специальных клапанов (для открывания). Конеч-
но, это возможно только в тех случаях, когда необходимо лишь один раз
открыть или закрыть сообщение между отдельными частями прибора. По-
скольку лишь в редких случаях удается безукоризненно запаять более или
меиее широкие стеклянные трубки непосредственно в вакууме, те места,
которые впоследствии придется запаивать, предварительно сужают. При этом
их одновременно утолщают путем легкого сжатия и последующего вытягива-
ния. В дальнейшем отверстие можно запаять в вакууме, равномерно нагре-
вая трубку и вытягивая ее по оси или, когда это невозможно, оттягивая
расплавленное стекло в сторону стеклянной палочкой.
Рис. 40. Типы разбиваемых
клапанов (М — магнитный
боек).
Сообщения между отдельными частями прибора можно достигнуть пу-
тем применения разбиваемого клапана (рис. 40). Клапан состоит из широ-
кой стеклянной трубки, в которую впаяна тонкая трубочка, сначала вытя-
нутая в острие, а затем отогнутая в сторону или выдутая на конце в тонко-
стенный шарик. До встраивания клапана в установку в более широкую
трубку вкладывают боек — кусок железной проволоки, впаянный в стеклян-
ную трубку, или железную палочку в тефлоновой изоляции (от магнитной
мешалки). Осторожно при помощи сильного магнита боек в установленном
вертикально клапане спускают вниз на шарик или острый кончик. Клапаны
спаивают с трубками, соединяющими отдельные части установки. При этом
следует помнить, что тефлон нельзя нагревать выше 300 °C. Для открывания
клапана поднимают боек при помощи магнита и сбрасывают его на острый
кончик или тонкостенный шарик, тем самым разбивая их и открывая про-
ход для газа. Разбиваемый клапан можно укреплять также в горизонталь-
ном положении; в этом случае разрушение тонкого кончика происходит при
продвижении бойка по трубке при помощи магнита. На рис. 41 и 42 показа-
ны другие конструкции вакуумных «разбивалок», основанные на отламыва-
нии кончика в виде тонкого капилляра.
Клапан можно заменить U-образиой трубкой, изготовленной из капил-
лярной трубки. Охлаждая последнюю жидким воздухом, капилляр закупо-
ривают небольшой пробкой из замороженного газа. При работе с низко-
88 Методы препаративной химии
кипящими веществами, не затвердевающими даже при температуре жидкого
воздуха, в U-образный капилляр вводят капельку сплава Вуда, причем по
обе стороны изогнутой части выдувают по небольшому расширению. Клапан
оказывается открытым или закрытым в зависимости от того, дают ли метал-
лу затвердеть в расширении или в самом капилляре.
Для точного дозирования небольших количеств газа в условиях вакуума
используют описанные ранее тефлоновые вентили или клапаны Боденштей-
Рис. 41. Устройство вакуумного раз-
биваемого клапана по Штоку.
на. В некоторых случаях можно пользоваться металлическими игольчатыми
вентилями, выпускаемыми в самом разнообразном исполнении. В целом,
однако, они не позволяют проводить препаративную работу в столь же «чи-
стых» условиях, как при применении стеклянных вентилей. Уплотнение по-
Рис. 42. Устройство вакуумного раз-
биваемого клапана по Шенку.
1 — выступ иа стейке трубки; 2 тонкий
капилляр, укрепленный эксцентрично. При
вращении вставки капилляр 1 обламы-
вается.
7
движной рабочей части металлических вентилей с целью ее изоляции от ат-
мосферы производят установкой сальников, уплотнительных колец, мембран
или шлифов. Более подробное описание отдельных конструкций можно найти
во 2-м издании этой книги, с. 67. В некоторых случаях достаточно сделать
напильником в пробке стеклянного крана тонкие желобки, отходящие от его
канала в соответствующих местах. В этих случаях следует по возможности
пользоваться кранами с косыми каналами. В продаже имеются также при-
способления для напуска газа*, в которых обе части аппаратуры разделе-
ны перегородкой из пористого стекла. Изменением степени покрытия этой
перегородки ртутью регулируется скорость поступления газа (см. аналогич-
ную конструкпию маностата, рис. 38).
Вакуумная установка
Вакуумная установка состоит из насосного агрегата, приспособлений
для измерения вакуума и из специальной реакционной части, которая кон-
струируется по-разному в зависимости от проблемы, которую предстоит раз-
Фирма-изготовитель Jenaer Glaswerk Schott u. Gen. (Mainz).
Высокий вакуум и изоляция от воздуха 89
решить. Целесообразно собирать насосный агрегат в соответствии со схемой
на рис. 43. Его можно смонтировать на переносном каркасе. Однако сам
Рис. 43. Схема вакуумной установки.
1 — форвакуумный насос; 2 — диффузион-
ный насос; 3 — форвакуумная колба (3—
5 л); 4 — осушающая емкость с Р2О5; 5 —
манометр на форвакуумной линии; 6 — ма-
нометр Мозера или Мак-Леода; 7 — охлаж-
даемая ловушка для улавливания паров
ртути; 8 — двухходовой кран; 9 — кран для
напуска воздуха или для подсоединения
к насосу Тёплера.
масляный насос не следует ставить на каркас с тем, чтобы не подвергать
вибрации аппаратуру и измерительные приборы. Лучше всего поставить на-
сос на пол, подложив под него четыре большие резиновые пробки. Отсасы-
вающий штуцер насоса соединяют с аппаратурой при помощи гибких при-
способлений (резиновый шланг, тефлоновый сильфон, гофрированные метал-
лические трубки). В тех случаях, когда работа ведется с трудноконденси-
руемыми газами, которые необходимо откачивать, собирать, измерять их
количество, а в дальнейшем, возможно, и исследовать, к трубке 9 (рис. 43)
подсоединяют насос Тёплера. Последний предназначен для сбора газов и пе-
ревода их в измерительную бюретку, что достигается за счет поперемеииого
поднимания и опускания ртутного столба, а также открывания и закрыва-
ния кранов.
На рис. 44 показано одно из возможных конструкционных решений —
схема функционирующего автоматически насоса Тёплера. Насос изготовляет-
ся из стекла марки дюран 50 и снабжен впаянными в трех местах электри-
ческими контактами 2, 3, 11 из вольфрамовой проволоки. При помощи этих
контактов производится управление движением ртути в насосе. Сначала
ртуть, с помощью которой происходит перемещение газов в насосе, нахо-
дится в сборной емкости 1, как это показано на рис. 44. Здесь оиа удержи-
вается либо путем закрывания крана 5, либо специальным вспомогательным
насосом, подсоединенным через 4. В таком положении через краны 14 и 15
производят вакуумирование всех соединительных трубок, пустого шарооб-
разного сборника 7 (рабочего объема), газовой бюретки 12 и манометра 10.
Если теперь в реакционной аппаратуре выделяются газы, они, проходя через
высоковакуумный насос, заполняют и объем 7. После закрывания кранов 15
и 16 открывают кран 5 и выключают вспомогательный насос. Вследствие
напуска воздуха из атмосферы через капилляр 6 ртуть поступает из / в 7
далее в бюретку 12. Клапаны 8 и 9 установлены для того, чтобы ртуть не
могла попасть в вакуумную установку, а также для запора газа, переведен-
ного из сосуда 7 в бюретку 12. При замыкании столбом ртути контакта 11
включается вспомогательный насос, и ртуть опускается в исходное положе-
ние (/) до тех пор, пока не замкнется контакт 3, благодаря чему вспомога-
тельный насос снова отключается. Цикл этих процессов многократно повто-
ряется, пока все количество выделившегося в реакционной аппаратуре газа
не соберется в бюретке 12. При этом верхний уровень запорного столба рту-
ти следует зафиксировать в той области газовой бюретки, где имеются деле-
ния. Включение и выключение иасоса осуществляется при помощи импульс-
ного реле (пускателя, имеется в продаже), питаемого напряжением 8 В.
Схема подключения реле показана на рис. 45. Давление собранного таким
90
Методы препаративной химии
образом газа измеряют манометром 10, снабженным дополнительным объе-
мом для регулирования уровня ртути. Сечение трубки манометра и частота
делений такие же, как у газовой бюретки. Определив заранее путем калиб-
ровки величину объема, заключенного между нулевыми отметками бюретки
и левого колена манометра, можно измерить объем собранного газа. В бю-
ретке газ может быть сжат до максимального давления ~300 мм рт. ст.
Рис. 44. Схема наноса Тёплера, работающего автоматически.
1 — нижний сосуд для ртути (—300 мл); 2, 3 — впаянные тоководы; 4—подсоединение к
вспомогательному насосу; 5 — край; 6 — капилляр; 7 — рабочий объем (—200 мл); 8, 9—
стеклянные клапаны; 10 — манометр; 11 — впаянный токовод; 12 — газовая бюретка дли-
ной ~35 см; 13— тарированный сосуд ( — 100 мл); 14, 15, 16 — краны.
Если происходит переполнение бюретки, в качестве дополнительного объема
используют сосуд 13, предварительно тарированный путем взвешивания с во-
дой. Взвешивая сосуд с пробой газа, можно определить его молекулярную
массу. Этот же сосуд можно использовать для отбора газа с целью его ана-
Высокий вакуум и изоляция от воздуха 91
лиза (методами масс-спектроскопии, газовой хроматографии, ИК-спектроско-
пии). Если необходимо все количество собранного газа перевести в другой
сборник (например, для анализа газа), то в месте, обозначенном на рисунке
буквой А, устанавливают трехходовой кран с уравнительным сосудом с
ртутью, а в месте В'—трехходовой кран с капиллярной трубкой, подсоеди-
ненной к сборнику (аналогично устройству нитрометра по Лунге) [8].
насос
Рис. 45. Схема включения насоса Тёплера с электрическим управлением.
/. 2, 3 — подсоединение к соответствующим контактам насоса Тёплера. Выключатель h
при работе в автоматическом режиме ставится в положение, показанное пунктиром.
Подобные насосы Тёплера имеются в продаже*. Специальная конструк-
ция насоса, в котором ртуть ие соприкасается ни со смазкой кранов, ни с
резиновыми шлангами, описана в работе [9], а упрощенная конструкция
миниатюрного насоса Тёплера —в работе [10].
Специальные вакуумные установки
Вакуумные установки применяются в первую очередь для работы с лег-
колетучими, а также чувствительными к действию воздуха и влаги вещества-
ми. Прообразом их является установка, сконструированная Штоком для ис-
следования гидридов бора и кремния, в которой для перекрывания вакуум-
ной линии применялись только ртутные клапаны. Назначение такой установки
и ее модификаций может быть самым разнообразным. Если нельзя полно-
стью избежать использования смазываемых кранов, то путем замены в уста-
новке ртутных клапанов на краны ее можно значительно упростить. Недо-
статком кранов является то, что через слой смазки происходит медленная
диффузия воздуха и влаги; с другой стороны, ртутные клапаны Штока из-за
их значительного сопротивления потоку газа не подходят для перегонки не-
больших количеств веществ.
В случае необходимости установку для работы с химически действующи-
ми иа ртуть веществами можно снабдить клапанами Боденштейна нли
разбиваемыми клапанами, а также сужениями для запаивания. На рис. 46
показана схема части универсальной вакуумной установки, разработанной
Штоком. При работе со смесями легколетучих веществ сначала полностью
конденсируют смесь в ловушке, охлаждаемой жидким азотом. Различные
конструкции ловушек представлены на рис. 47. Рекомендуется работать с ло-
вушками типа промывных склянок (рис. 47, а, в, г), причем их важно правиль-
но подсоединять к аппаратуре, а именно: впуск газа не должен осуществлять-
ся через узкую трубку, так как в противном случае ловушка легко может
забиться.
Фирма-изготовитель Strohlein u. Со. (Dusseldorf).
92
Методы препаративной химии
После конденсации вещества в ловушке 7 (рис. 46) вход газа в установ-
ку перекрывают, производят откачку и затем часть установки между кла-
панами 6 и 9 полностью изолируют. После этого ловушку 7 медленно нагре-
вают на бане до тех пор, пока в манометре 8 не установится давление пара
30—50 мм рт. ст. Отмечают давление пара и температуру, открывают кла-
Рис. 46. Участок универсаль-
ной вакуумной аппаратуры
Штока (/—9 см. текст).
паны 6 и 4, закрывают клапан 3 и охлаждают трубку 5 жидким воздухом.
Здесь некоторой части вещества дают возможность сконденсироваться, за-
крывают клапан 4 и при той же температуре бани выжидают, пока снова
Рис. 47. Типы вымораживающих ловушек (а—е см. текст).
не установится соответствующее давление пара в манометре 8. Если давле-
ние будет иметь то же самое значение, как и прежде, снова отгоняют неко-
торую часть вещества и продолжают перегонку до тех пор, пока давление
пара в ловушке 7 не будет оставаться неизменным. Если давление понизит-
ся, то перегонку производят в направлении клапанов 1 и 2. Таким путем
можно в соответствии с величинами давления паров разделить смесь на раз-
личные фракции без какой-либо потери вещества. Если в результате ошибок
или неудачных приемов возникнут затруднения, то все вещество опять-таки
Высокий вакуум и изоляция от воздуха
93
без потерь можно сконденсировать в исходную ловушку 7j и начать весь-
процесс сначала. Можно проводить также дальнейшее разделение получен-
ных отгонов, соединять соответствующие фракции между собой и т. д. Кри-
терием чистоты может служить постоянство давления пара при постоянной,
температуре от начала до конца перегонки. Чтобы определить упругость-
пара, само собой разумеется, необходимо приостановить перегонку и выж-
дать время, достаточное для полного выравнивания температуры. Кроме
того, не следует перегонку проводить слишком быстро.
На рис. 48 изображен сосуд для сбора веществ, не конденсирующихся
при комнатной температуре. Применение таких сборников газа позволяет-
Рис. 48. Конструкция сборника
для газа.
избежать необходимости продолжительное время держать его в конденси-
рованном состоянии путем охлаждения жидким азотом. Остроумно сконструи-
рованный отборный клапан дает возможность переносить эти сборники и
подсоединять их в различных местах установки. Клапан состоит из двух
стеклянных пористых пластинок, погруженных в ртуть. Прохождение газа,
возможно только в том случае, если эти пластинки плотно прижаты своими,
плоскостями одна к другой.
Ловушки U-образной формы (рис. 48) можно подсоединить к аппара-
туре и при помощи шаровых шлифов. Такие ловушки следует использовать
при работе с пирофорными или высокотоксичными веществами, так как все
вещество конденсируется в отсоединяемой части ловушки. Напротив, при
конденсации в ловушке в форме промывной склянки (рис. 47, г) некоторое
количество вещества всегда оседает во внутренней трубке и при отделении
иижией части ловушки попадает в атмосферу.
Для фракционной перегонки смесей низкокипящих веществ предложены
специальные колонки, которые позволяют существенно ускорить работу. Во-
многих случаях, когда необходимо произвести фракционирование смеси ве-
ществ с очень близкими температурами кипения, такие колонки вообще не-
заменимы. Одна из возможных конструкций этих колонок изображена на
рис. 49 [11]; другие варианты можно найти в книге [12] и оригинальной;
литературе [13].
Весьма эффективного разделения небольших количеств веществ можно»
добиться, используя метод содистилляции, сходный с низкотемпературной:
94 Методы препаративной химии
газовой хроматографией. Перегонку проводят в потоке газа-носителя, прохо-
дящего через глубоко охлажденную колонку. В этой колонке с насадкой
осуществляется распределение температуры. При постепенном нагревании
колонки идет разделение смеси на чистые вещества за счет процессов испа-
рения и адсорбции [14].
Для более точной характеристики полученных фракций кроме значений
давления паров могут служить точки плавления и молекулярные массы, а в
некоторых случаях также инфракрасные, ЯМР- и масс-спектры. Для опреде-
Микроколонка
Рис. 49. Аппаратура для фракционной перегонки при низких температурах.
1 — сосуд для исходного газа; 2, 4, 8, 9, 14 — вакуумные краны; 3 —ловушка для про-
межуточной конденсации газа; 5, 10, 12 — манометры; 6 — микроколонка вместимостью
6—10 мл с нагревательной спиралью 7 из платино-иридиевой проволоки, с вакуумируемой
оболочкой и с ограничителем уровня в охлаждающей бане. Микроколонка изображена
несколько увеличенной по сравнению с остальной аппаратурой. Объем заключенный
между кранами 8 и 9, точно известен и служит для регулировки скорости отбора. Газ
после ректификации замораживается в 11, далее путем глубокого охлаждения перево-
дится в 15, а затем в сборник 13.
ления точки плавления Шток предложил весьма простой прибор (рис. 50). При
его применении сначала поднимают электромагнитом тоненькую стеклянную
палочку с железным сердечником и затем конденсируют в нижнем шари-
ке 1 такое количество вещества, которое может заполнить половину шари-
ка. Веществу дают расплавиться и затем его замораживают. Лишь после
этого, выключив электромагнит, опускают конец 2 стеклянной палочки на
поверхность вещества и, медленно нагревая шарик 1 в бане, наблюдают
момент, когда конец палочки начнет погружаться в вещество, что особенно
заметно по перемещению имеющейся на конце палочки капельке стекла 3.
В этот момент делают отсчет температуры, которую и можно считать точ-
кой плавления.
Молекулярную массу определяют следующим образом. Предварительно
взвешенную в эвакуированном состоянии колбочку с краном и шлифом,
объем которой заранее измерен, наполняют в установке газом под опреде-
ленным давлением. После отделения от установки и удаления смазки со
шлифа колбочку снова взвешивают. На основании данных о давлении, объе-
ме и весе легко можно вычислить молекулярную массу. Гораздо быстрее
можно достигнуть цели при помощи весов Штока с коромыслом. Принцип
устройства этого прибора основан иа измерении подъемной силы кварцево-
го шара, находящегося в исследуемом газе, давление которого можно одно-
Высокий вакуум и изоляция от воздуха
95
временно измерить. Подъемная сила кварцевого шара компенсируется при
помощи электромагнита [15].
Для съемки масс-спектра небольшую колбу, снабженную краном и шли-
фом, сначала основательно эвакуируют, затем наполняют исследуемым газом,
после чего ее непосредственно присоединяют к масс-спектрометру.
Инфракрасный спектр газообразного вещества измеряют в так называемой
газовой кювете. Она представляет собой стеклянную трубку диаметром 3—
6 см и длиной 10 см, на отшлифованных концах которой вакуумио-плотно
приклеены окна из материала, прозрачного в инфракрасной области спектра.
Список подобных материалов приведен в табл. 26 (разд. 18). Кювету, кото-
Рис. 50. Схема прибора для
определения температуры плав-
ления (1—3 см. текст). ё
рая для соединения с высоковакуумной аппаратурой должна иметь на бо-
ковой стороне отвод с краном и шлифом, сначала основательно откачивают,
а затем наполняют газом. При этом давление газа не должно быть слишком
мало (100—760 мм рт. ст).
ЛИТЕРАТУРА
1. Dushman S., Scientific foundation
of vacuum technique. 2nd ed. Wi-
ley, New York, 1962.
2. Schmidt M., Eichelsdorfer, Z.
Anorg. Allgem. Chem., 330, 116
(1964).
3. Lux H., Anorganisch-chemische
Experimentierkunst. 3. Aufl. J. A.
Barth, Leipzig, 1970, S. 451. [Есть
русский перевод более раннего
издания: Лукс Г. Эксперимен-
тальные методы в неорганической
химии. Пер. с нем. — М.: Мир.
1965.]
4. Diels К-, Jaeckel R., Leybold Va-
kuum Taschenbuch. Springer, Ber-
lin—Gottingen—Heidelberg, 1962.
5. Stock A., Z. Elektrochemie, 39,
256 (1933); Stock A.. Hydrides of
boron and silicon. Cornell Univ.,.
Ithaka, New York and Cambrid-
ge Univ., London, 1933.
6. Weigel F., Kolkenbeck K., J. Chem
Educ., 48, 448 (1971).
7. Vaughan W. E„ Rev. Sci. Instr.,
16, 254 (1945).
8. Engelhardt U., частное сообщение..
9. Zintl E., Harder A., Z. Physik-
Chem. (B), 14, 265 (1931).
D6 Методы препаративной химии
10. Seel F., Chem.-Ing.-Tech., 27, 542
(1955).
11. Clusius К-. Wolf G., Z. Natur-
forsch., 2A, 495 (1947); Clusius K.,
Schanzer W., Z. Physik. Chem.
(A), 192, 273 (1943).
12. Shriver D. F., The manipulation of
air-sensitive compounds. McGraw
Hill, New York, 1969.
13. Spielman J. R., Burg A. B., Inorg.
Chem., 2, 1139 (1963); D. J. Le
Roy, Canad. J. Chem., 28, 492
(1950); Comstock A. A., Rollef-
son G. K-, J. Chem. Phys., 19,
441 (1951).
14. Cady G. H., Siegwarth D. P„
Anal. Chem., 31, 618 (1959);
Myers H. W., Putnam R. F., Anal.
Chem., 34, 664 (1962).
15. Lehrer E„ Russ E., Z. Physik.
Chem., 163, 73 (1933).
Изоляция от кислорода и влаги воздуха
Техника экспериментальной работы с жидкими и твердыми веществами
® отсутствие воздуха может различаться в зависимости от количества веще-
ства и от его чувствительности к влаге и кислороду. Наиболее
полное удаление воздуха и влаги достигается при работе в вакууме, но при
этом однократно можно провести работу лишь с относительно небольшими
•количествами веществ. В так называемых трубках Шленка (см. ниже) и ана-
логично устроенных сосудах можно работать с количеством вещества до
100 г и более в условиях полного или почти полного отсутствия воздуха.
Рис. 51. Мешок для работы с
гигроскопичными веществами
(сухой мешок). '
В «сухих» камерах можно перерабатывать уже килограммовые количества
веществ. В зависимости от конструкции сухой камеры примесь воздуха в ее
.атмосфере мала или весьма мала, но достаточно полно исключить присутст-
вие влаги удается не всегда.
«Сухие» мешки и камеры. При относительно невысоких требованиях к от-
сутствию кислорода и воды довольно удобно проводить некоторые операции в
в «сухом» мешке (называемом также «азотным мешком»). Он представляет
собой мешок из тонкого прозрачного и эластичного полиэтилена, открытый
<с одной стороны. С другой, закрытой стороны мешок просверлен, и в отвер-
стие вклеена стеклянная трубка с краном, укрепленная дополнительно двумя
•надвинутыми резиновыми пробками (рис. 51). Через открытую часть мешка
внутрь помещают вещество и все необходимое для работы: бюксы, шпатели
;и т. д. Затем мешок герметизируют путем многократного оборачивания уз-
кой полосы краями мешка и ее последующего зажима между двумя дощеч-
ками. После этого мешок откачивают через стеклянную трубку, заполняют
его инертным газом, снова откачивают и окончательно заполняют инертным
газом. Операции проводят, беря нужные приборы руками через эластичную
стенку мешка. Можно также смонтировать мешок на круглой подставке,
сквозь которую пропущены трубки для создания вакуума и подачи инерт-
ного газа. В продаже имеются также мешки с полиэтиленовыми перчатка-
ми [1, 2].
Для работы с более чувствительными веществами и, следовательно, при
Высокий вакуум и изоляция от воздуха 97
более жестких требованиях к инертной атмосфере используют «сухие» каме-
ры. Такая камера представляет собой кожух из металла или плексигласа,
лучше всего цилиндрической формы, дно которого закрыто й к которому
сверху привернута плексигласовая крышка. Подобная конструкция доста-
точно устойчива к действию внешнего давления, и поэтому ее можно эва-
куировать при помощи масляного насоса, а затем наполнить сухим инерт-
ным газом. Камеру целесообразно закрепить на передвижном каркасе. Круг-
лые отверстия, закрываемые во время откачки камеры соответствующими по
форме заглушками, служат для внесения в камеру необходимого лаборатор-
ного оборудования, после чего па них закрепляют неопреновые перчатки для
проведения манипуляций в сухой атмосфере камеры. Более крупные прибо-
ры можно поместить в камеру, отвинтив плексигласовую крышку. Если не-
обходимо что-то дополнительно внести в уже осушенную атмосферу камеры,
это делают, используя шлюзы, которые также могут быть эвакуированы.
Неэвакунруемые камеры, большей частью в форме ящика, гораздо хуже,
поскольку в них за счет простого промывания инертным газом за ограничен-
ное время нельзя достичь необходимой степени удаления кислорода и влаги.
Убыль концентрации какого-либо компонента А (О2, СОг, Н2О и т. и.) при
простом промывании инертным газом происходит в соответствии со степен-
ной функцией от е, а именно: концентрация компонента А при промывании
n-кратным объемом инертного газа снижается до ае~п при условии полно-
го перемешивания инертного газа с атмосферной камеры. В отсутствие вен-
тилятора это условие обычно не выполняется. В камере объемом 100 л,
первоначально наполненной воздухом, после промывания ее азотом (100 л)
содержится еще по меньшей мере 7,7 об. % О2, и лишь после промывания
1000 л (!) азота содержание кислорода падает до 10~3%.
Внутри камеры и в шлюзе помещают большие плоские чашки с Р2О5
в качестве осушителя. Для создания циркуляции газа полезно иметь в ка-
мере небольшой и не очень сильный вентилятор, Токовводы располагаются
обычно в корпусе камеры. Можно, однако, приспособить вентилятор таким
образом, чтобы он постоянно продувал или просасывал газ через сосуд с гра-
нулированным осушителем. Если такой агрегат расположить вне камеры,
можно, не открывая ее, производить замену осушителя. Такие и подобные
им приспособления имеются в продаже. Их можно также применять при
работах с радиоактивными или сильнотоксичными веществами.
При проведении работ в «сухих» мешках или камерах необходимо учи-
тывать, что резина и синтетические вещества обладают определенной про-
ницаемостью для газов и прежде всего для паров воды. Из-за перепада
парциального давления воды она постепенно диффундирует в мешок или ка-
меру. Количество диффундирующего газа прямо пропорционально перепаду
парциальных давлений, времени, степени проницаемости данного материала
и его поверхности и обратно пропорционально толщине стенки. Проницае-
мость для паров воды обычно гораздо выше, чем для кислорода или диок-
сида углерода. Поэтому в камеру надо обязательно помещать осушитель,
если только работы в ней не проводятся в течение очень непродолжитель-
ного времени. Особенно легко проницаемы для паров воды целлюлоза и ее
производные; средней проницаемостью обладают резина, бутадиен-натрие-
вый каучук, найлон, полистирол, неопрен и акриловое стекло; плохопрони-
цаемы для паров воды полиэтилен и особенно политрифторхлорэтилен. Как
показывает примерный расчет, в мешок из полиэтилена с толщиной стенок
0,05 мм и поверхностью 1 м2 в час диффундируют примерно Г5 мл кисло-
рода и 20—50 мл водяного пара при 50% -ной влажности воздуха. Наряду
с постепенным ухудшением атмосферы камеры при работе в ией могут
встретиться еще дополнительные осложнения в виде накопления в сухой
атмосфере электростатического заряда на всех предметах, находящихся в
камере. Это может сильно затруднить работу с веществом, особенно в по-
рошкообразном состоянии.
7—143
98 Методы препаративной химии
ЛИТЕРАТУРА
1. Pickhardt W. Р., Safranski L. F.,
Mitchell J., Anal. Chem., 30, 1298
(1958), Ehrlich P., Hein H. L,
Kilhnl H., Chemiker-Ztg., 81, 329
(1957).
2. Glove boxes and shielded cells for
radioactive materials (ed. G. N. Wal-
ton). Sympos. Harwell, 1957; Aca-
demic Press London, 1958.
3. Barton Ch. I., Glove box techniques.
In: Technique of inorganic che-
mistry (eds. H. B. Jonassen, A.
Weissberger). Vol. Ill, New York,
1963.
4. Shriver D. F., The manipulation of
air-sensitive compounds. McGraw
Hill, New York, 1969, p. 164, 230.
Стеклянная аппаратура
Для выполнения операций с чувствительными к действию воздуха и вла-
ги жидкостями была разработана [1] техника работы, которую можно вся-
чески рекомендовать, так как она менее сложна, чем работа с универсаль-
ной аппаратурой Штока. В качестве подсобных средств при этом исполь-
зуются следующие приспособления:
1. Инъекционные шприцы, лучше всего найлоновые, поскольку только!
у этого типа шприцев уплотнительное кольцо так сильно прижимается к стен-
ке шприца, что достигается его полное опорожнение.
2. Наконечники из нержавеющей стали или других сплавов с внутренним
диаметром канала 0,5—0,7 мм и иглой длиной 5—10 см*.
3. Ампулы различных конструкций (рис. 52) с развернутым краем для
насаживания прокалываемых крышек. Небольшое углубление на дне облег-
чает полное удаление жидкости из ампулы при помощи шприца. Разновид-
ность ампул с длинным горлом служит для сохранения больших количеств
веществ при одновременном охлаждении. Удлиненное горло позволяет по-
гружать ампулу в охлаждающую баню достаточно глубоко. Отбор вещест-
ва из ампулы проводят, предварительно перелив жидкость в боковые от-
ростки.
4. Прокалываемые крышки (рис. 53, а), одеваемые на концы ампул.
Выпуклая поверхность крышек делает возможным проведение работ в высо-
ком вакууме**. При откачке ампулы выпуклая крышка сжимается и пробно,
герметизирует прокол. Напротив, при наличии в ампуле избыточного давле-
ния крышка действует как предохранительный клапан. Даже неоднократно,
(до 20 раз) проколотые крышки все еще прекрасно держат вакуум.
5. Предохранительные трубочки — короткие отрезки стеклянной трубки,
закрытые с обеих сторон прокалываемыми крышками (рис. 53,6).
6. Переходники (рис. 53, в). Их изготавливают из стеклянных шприцев,
к открытому концу которых припаивают муфту. Пользуясь такими переход-
никами, можно подсоединять наконечники в различных местах аппаратуры,
снабженной шлифами.
Способ работы с такой аппаратурой следует из рассмотрения рис. 54
и 55. Если, например, жидкое вещество из прокалываемой ампулы необхо-
димо с помощью инъекционного шприца перенести в другой реакционный
сосуд, то для этого сначала надо наполнить шприц инертным газом. Шприц
и защитную трубочку подсоединяют к вакуумной аппаратуре, откачивают из
них воздух и заполняют инертным газом, как показано на рис. 54. Затем за-
щитную трубку, находящуюся на конце шприца, плотно прижимают к крыш-
ке ампулы и прокалывают (рис. 55). После наполнения шприца жидкостью
его наконечник осторожно вынимают из ампулы так, чтобы он снова оказал-
ся в защитной трубке. В таком защищенном от доступа воздуха виде веще-
* Фирмы-изготовнтелн: Heinz Dolderer (Tuttlingen), Schoellerwerk AG
(Hellenthal).
** Например, «Крышки для инсулина» фирмы Biotest (Frankfurt/Main).
Высокий вакуум и изоляция от воздуха 99
Рис. 52. Типы ампул.
к
Рис. 53. Детали аппаратуры для рабо-
ты методом «прокалывания».
а — прокалываемая крышка; б — предохрани-
тельная трубочка; в — переходник.
ство можно перенести к реакционному сосуду и ввести его туда аналогичным
образом. Если желательно избежать создания в ампуле разрежения во вре-
мя отбора из нее жидкости, то с помощью наконечника через другую крыш-
ку в ампулу впускают сухой азот. При необходимости осуществлять посто-
янную промывку ампулы азотом через крышку вводят еще один наконечник
для выхода газа. Если же защитные трубочки ие использовать вообще, то
7*
100 Методы препаративной химии
отпадает необходимость иметь ампулы с двумя крышками. Описанную выше
технику работы можно использовать при самых разнообразных исследова-
ниях, более подробные сведения о которых содержатся в оригинальной ли-
тературе. Само собой разумеется, что материал шприцев, наконечников и
крышек должен быть в каждом отдельном случае соответственно подобран.
Рис. 54. Способ заполнения инъекционного шприца и предохранительной
трубочки азотом.
Техника работы с чувствительными к действию воздуха твердыми ве-
ществами в гораздо большей степени зависит от конкретно поставленной це-
ли. Поэтому ниже будут лишь выборочно рассмотрены некоторые приемы
работы.
На рис. 56 и 59 показаны имеющие широкое применение трубки Шленка*,
служащие для хранения и расфасовывания твердых веществ. Общий прин-
Называемые также Шленк-сосудами. — Прим, перев.
Высокий вакуум и изоляция от воздуха 101;
цип устройства этих сосудов состоит в том, что их можно эвакуировать;
после наполнения инертным газом попадание воздуха внутрь при их откры-
вании предотвращается за счет струи выходящего инертного газа. Путем
Пластмассовый
шприц
Рис. 55. Способ отбора из ампулы
жидкости, чувствительной к действию
воздуха.
Инъекционная
игла
Защитная
трубочка
Ампула
опрокидывания сосуда вещество можно переносить из одного сосуда в дру-
гой, присоединенный к нему. Инертный газ, очищаемый, например, по мето-
дике, описанной в разделе «Азот» (часть II, гл. 7), подается по полиэтиле-
новому шлангу. Необходимо, однако, учитывать, особенно при работе с не-
большими количествами высокочувствительных к действию воздуха
Рис. 56. Типы приборов для сохранения и фасовки твердых веществ, чувстви-
тельных к действию воздуха (трубки Шленка).
Отросток 1, показанный пунктиром может отсутствовать.
102 Методы препаративной химии
-веществ, что даже следы кислорода и воды, содержащиеся в очищенном
инертном газе, постепенно поглощаются веществом, и, таким образом, прн
длительных опытах и соответственно больших объемах газа значительная
часть вещества может разложиться. Поэтому нужно стараться держать ве-
щество в потоке газа не дольше, чем это необходимо.
Рис. 57. Устройство прибора для рас-
творения и перекристаллизации ве-
ществ в отсутствие воздуха.
1—4 см. текст. Трубки 2 н 4 могут нахо-
диться также в средней части прибора.
Рис. 58. Схема прибора для осажде-
ния и фильтрования в отсутствие
воздуха (см. текст).
Для перекристаллизации веществ, чувствительных к действию состав-
ных частей воздуха, а часто вообще для получения очень чистых препара-
тов используют аппаратуру, показанную на рис. 57 [2]. Прибор эвакуируют
через трубки 2 и 4, наполняют инертным газом и при необходимости созда-
ют поток газа. В колбу 1 вносят вещество так же, как в трубку Шленка,
исходный продукт растворяют, после чего поворачивают прибор на 180°, так
что колба 3 оказывается внизу. Фильтрование раствора в колбу 3 можно
проводить при охлаждении нлн прн нагревании, для чего среднюю часть
прибора с пористым стеклянным фильтром подогревают при помощи нагре-
вательной манжеты или охлаждают путем оборачивания охлаждающим
шлангом в виде трубки из свинца или синтетического материала с хорошей
Высокий вакуум и изоляция от воздуха 103
теплопроводностью («калорекс», см. разд. 11). Еще лучше применить охлаж-
дающую рубашку, которую по аналогии с холодильником Либиха припаи-
вают таким образом, чтобы она охватывала трубку с фильтром (см. рис. 58).
Осуществляя циркуляцию через рубашку жидкости из термостата или крио-
стата, можно прн фильтровании раствора, полученного в колбе 2 прн нагре-
вании или охлаждении, предотвратить как преждевременную кристаллизацию
вещества, так и его разложение при нагревании.
Кристаллизацию вещества из фильтрата в колбу 3 (рис. 57) проводят
путем упаривания нлн охлаждения раствора, после чего кристаллы и ма-
Рис. 59. Типы сосудов для
работы с растворами и
осадками в отсутствие воз-
духа.
1—3 — конические шлифы; 4 —
стеклянная пористая пластинка,
точник разделяют, снова переворачивая прибор н фильтруя через пористую
пластинку. Используя трубки 2 и 4, полученные кристаллы легко можно про-
мывать, отсасывать и сушить в вакууме (с одновременным охлаждением или
нагреванием). Выгрузку препарата осуществляют в потоке индифферентно-
го газа.
Аппаратура для осаждения, очистки и выделения легкоокисляющнхся ве-
ществ из водных растворов прн полной изоляции от воздушной атмосферы,
разработанная для получения Fe(OH)2, подробно описана во 2-ом издании
этой книги, ч. 1, с. 74—76. Однако в большинстве случаев используют при-
бор, показанный на рис. 58. В приборе, который предварительно несколько
раз попеременно откачивают и наполняют инертным газом, можно прово-
дить взаимодействие между двумя растворами с последующим отделением и
очисткой продукта реакции. Растворы, прн необходимости прокипяченные в
инертной атмосфере и насыщенные очищенным азотом и аргоном, помешают
в сосуды 1 и 2. Реакцию осаждения проводят в колбе 2 путем добавления
раствора из колбы 1 прн одновременном перемешивании и подаче инертно-
го газа. Осадок отделяют путем фильтрования на фильтре 3. Промывать
осадок можно прн охлаждении нлн нагревании; сушат осадок в вакууме.
Фильтрат из приемника 4 можно слить по трубке сифона до того, как туда
попадет промывной раствор.
Весьма похожая методика проведения работ с растворами, которая
пригодна также для осаждения, перекристаллизации, сублимации, экстрак-
ции, отбора проб, титрования нт. д. в условиях, исключающих доступ возду-
ха, была разработана рядом исследователей [3]. Аппаратуру для каждого
конкретного случая собирают из стандартных простых элементов, сходных
по принципу действия с трубками Шленка (рис. 59). С этой целью в аппа-
ратуре используют шлифы только одного размера; направление движения
инертного газа и реакционной жидкости может меняться на противополож-
ное. Сосуды соединяют либо непосредственно, либо через переходник Y- или
Х-образной формы со шлифами на концах. В цитированных работах приве-
ден целый ряд конструкционных решений для самых разных случаев.
Для проведения без доступа воздуха реакций между твердыми и газо-
образными веществами или с веществами, испаряющимися при повышенных
104 Методы препаративной химии
температурах, были предложены разнообразные приборы, приспособленные
для конкретно поставленной целя. Многие из синтезов подобного рода мож-
но провести в аппаратуре, основным элементом которой является лодочка,
помещенная в трубку, причем последнюю можно откачивать и наполнять
инертным газом. При проведении всех операций, не ограниченных воздействи-
ем извне, как, например, нагревание или действие магнитного поля, а тре-
бующих непосредственного проникновения в саму трубку (например, внесе-
ние или извлечение вещества, введение приспособлений для измельчения,
Рис. 60. Муфта шлифа, к
срезу которой приклеено
смотровое окно (!) для на-
блюдения за ходом реакции.
Рис. 61. Устройство вакуумной шаровой мель-
ницы (1, 2— шлифы).
разделения или перемещения образца), всегда открывают только одно от-
верстие для выхода подаваемого инертного газа. Можно, не опасаясь про-
никновения воздуха, проводить операции внутри трубки, если поддерживать
скорость потока выходящего газа достаточно высокой.
Что касается многочисленных конструкций, выполненных в соответствии
с этими основными положениями, то здесь можно ограничиться лишь приве-
дением некоторых литературных ссылок [6—27]. Следует упомянуть о том,
что наблюдение за процессами, протекающими внутри трубки, в большин-
стве случаев весьма затруднительное из-за оптического искажения в изогну-
тых стеклянных стенках, легко может быть осуществлено через плоскопарал-
лельные стеклянные окна. Такое окно изготавливается путем приклеивания
пластинки к коротко обрезанной трубке шлифа (рис. 60), который после
этого можно подсоединить к колбе или трубке.
Проблема измельчения чувствительного к действию воздуха веществ?
в инертной атмосфере или в вакууме решается при использовании для этой
цели вакуумной мельницы (рис. 61). Мельница состоит из полого шара
диаметром 10 см со стенками толщиной 4 мм, в котором помещаются шари-
ки диаметром 10—15 мм. Вблизи шлифа 2 трубка несколько сужена для то-
го, чтобы при высыпании перемолотого вещества она не оказалась закупо-
ренной. Вещество и шарики вводят через шлиф 1 при одновременном про-
пускании инертного газа. Затем мельницу откачивают и с помощью подхо-
дящего приспособления приводят во вращательное движение. При работе
в вакууме исключается возможность разуплотнения шлифов, кроме того,
предотвращается вынос пыли за пределы полого шара. Размол рекомендует-
ся вести в течение 1-—2 ч при скорости вращения 70—80 об/мин.
Измельчение более крупных компактных кусков твердых веществ или
же твердых веществ, прочно приставших к лодочке или тиглю, можно осу-
ществлять в атмосфере инертного газа небольшой зуботехнической фрезой.
В ч. III (гл. «Сплавы и интерметаллические соединения») в двух случаях
подробно описано применение подобной техники при работе со сплавами,
чувствительными к действию воздуха. Эти приемы можно легко модифици-
ровать и использовать для обработки неметаллических твердых веществ.
Высокий вакуум и изоляция от воздуха 105
Для проведения реакции без доступа воздуха между легколетучим и
труднолетучим веществами с образованием также труднолетучего продукт^
используют прибор, изображенный на рис. 62. Летучий компонент, взятый
в избытке, испаряется нли сублимирует при нагревании в вакууме (печью 2)
из лодочки 4. Вещество испаряется и переносится в температурном градиен-
те к другому, нелетучему компоненту, находящемуся в лодочке 5. Благодаря
трубке 1, закрытой с одного конца, происходит движение пара в нужном
направлении. Исходное вещество в лодочке 5 должно быть измельчено как
можно тоньше. Его нагревание до температуры активации ведут при помощи
Рис. 62. Способ получения
соединений путем возгонки
одного из реагентов.
I — направляющая трубка; 2,'
3 — электрические печи; 4, 5 —
лодочки; 6 — внутренняя защит-
ная трубка; 7 — внешняя реак-
ционная трубка.
печи 3. После окончания реакции температуру печи 3 несколько повышают
для того, чтобы удалить избыток летучего компонента из продукта взаимо-
действия.
В том случае, если летучий компонент представляет собой пары агрес-
сивных металлов (щелочной, щелочноземельный, редкоземельный металл),
применяют закрытую двойную ячейку из металлического молибдена [4]
(рис. 63). Испарительная камера 1 для летучего компонента и реакционная
Рис. 63. Разрез двойной ячей-
ки из молибдена для проведе-
ния реакций веществ с пара-
ми металлов.
• j/ — камера для испарения металла;
•2 — камера для твердого вещества;
—— разделительная перегородка для
'.торможения потока паров металла;
{4 — разъемное соединение; 5 — эф-
фузионное отверстие. Размеры:
длина 42 мм, внешний диаметр
20 мм.
камера 2 для нелетучего компонента и продукта реакции плотно соединяют-
ся между собой в 4 и в таком виде помещаются в нагреваемую горизон-
тальную трубку. Избыток летучего компонента по окончании реакции мож-
но удалить испарением через небольшое эффузионное отверстие 5.
При проведении синтезов без доступа воздуха с участием испаряющегося
компонента реакции и малоактивного металла последний приходится нагре-
вать до весьма высоких температур. При работе по способу, описанному в
[5], предотвращается возможность растрескивания используемого сосуда
из-за повышения давления летучего компонента при нагревании. Речь идет
при этом об ампуле из стекла супремакс или кварца, в которой под вакуу-
мом запаяны необходимые количества металла и другого компонента реак-
ции (например, серы или фосфора), причем реагирующие вещества находят-
ся в различных концах ампулы. Тот конец ампулы, в котором находится
металл, можно затем нагреть до 700—1000 °C (в зависимости от материала,
из которого сделана ампула), а другой конец (в зависимости от давления
пара второго реагента), например, до 400—500°C. Нагревание продолжают
до тех пор, пока металл не прореагирует в желаемой степени. Путем после-
106
Методы препаративной химии
дующего выдерживания при более низкой температуре избыток летучих
компонентов может быть отделен от продукта реакции с металлом. Вскрыв
ампулу (если это необходимо, в атмосфере инертного газа), продукт реак-
ции выделяют из смеси. Степень протекания реакции можно определить
путем последующего взвешивания продукта реакции.
Сходный метод работы в ампулах, помещаемых в определенный темпе-
ратурный градиент, оправдал себя при проведении реакций синтеза и раз-
ложения галогенидов металлов.
ЛИТЕРАТУРА
1. Stock A., Z. Anorg. Allgem. Chem.,
303, 294 (1960).
2. Ulich H.. Chem. Fabrik, 4, 278
(1931); Frierichs F., Chem. Fab-
rik, 4, 318 (1931).
3. Herzog S.. Dehnert J., 7. Chem., 4,
1 (1964); Herzog S.. Dehnert J.,
Liibdes K, In: Technique of inor-
ganic chemistry (eds. H. B. Jonas-
sen, A. Weissberber), Vol. VIII,
Interscience, New York—London—
Sidney, 1968, p. 119; Thomas G.,
Chemiker-Ztg., 85, 567 (1961).
4. Petzel T., Greis O. Z. Anorg.
Allgem. Chem., 396, 95 (1973).
5. Strotzer E., Blitz F„ Z. Anorg.
Allgem. Chem., 238, 69 (1938).
6. Shriver D. F„ The manipulation
of air-sensitive compounds.
McGraw-Hill, New York, 1969.
7. Metzger H., Muller E„ In: Hou-
iben-Weyl, Methoden der organi-
schen Chemie. Bd. II, 2. Georg.
Thieme, Stuttgart, 1959, S. 321.
8. Yarwood J., High vacuum techni-
que. Chapman & Hall, London,
1967.
9. Zintl E„ Baumbach H. H„ Z.
Anorg. Allgem. Chem., 198, 88
(1931).
10. Zintl E., Harder A., Meumayr S.
Z. Physik. Chem. (A), 154, 92
(1931).
11. Zintl E., Harder A., Z. Physik.
Chem. (A), 154, 47 (1931).
12. Zintl E., Harder A, Z. Physik.
Chem. (B), 14, 265 (1931).
13. Zintl E., Kaiser H„ Z. Anorg.
All gem. Chem., 211, 113 (1933).
14. Zintl E., Husemann E„ Z. Physik.
Chem. (B), 21, 138 (1933).
15. Zintl E., Harder A., Z. Elektro-
chemie, 41, 33 (1935).
16. Zintl E., Woltersdorf G„ Z. Elek-
trochemie, 41, 876 (1935).
17. Zintl E„ Harder A., Z. Physik.
Chem. (B), 34, 238 (1936).
18. Zintl E., Morawietz W., Z. Anorg.
Allgem. Chem., 236, 372 (1938).
19. Helms A., Klemm Z. Anorg.
Allgem. Chem., 241, 97 (1939).
20. Klemm W., Sodomann H., Z.
Anorg. Allgem. Chem., 225, 273
(1935).
21. Klemm W., Sodomann H., Lang-
messer P., Z. Anorg. Allgem.
Chem., 241, 281 (1939).
22. Helms A.. Klemm W., Z. Anorg.
Allgem. Chem., 242, 33 (1939).
23. Helms A., Klemm W., Z. Anorg.
Allgem. Chem., 242, 201 (1939).
24. Bohm E„ Klemm W., Z. Anorg.
Allgem. Chem., 243, 69 (1939).
25. Teichert W.. Klemm W„ Z. Anorg.
Allgem. Chem., 243, 86, 138 (1939).
26. Klemm W., Mika G., Z. Anorg.
Allgem. Chem., 248, 155 (1941).
27. Brauer G.. Z. Anorg. Allgem.
Chem., 255, 101 (1947).
28. Rihl S„ Fricke R., Z. Anorg.
Allgem. Chem., 251, 405 (1943). 7
29. Schafer H., Bayer L., Z. Anorg.
Allgem. Chem., 271, 338 (1953).
30. Schafer H„ Grau L., Z. Anorg.
Allgem. Chem., 275, 198 (1954).
31. Schafer H., Marcher B., Z. Anorg.
Allgem. Chem., 290, 279 (1957).
32. Schafer H., Etzel K-. Z. Anorg.
Allgem. Chem., 291, 294 (1957).
33. Schafer H.. Dohmann К-D., Z.
Anorg. Allgem. Chem., 300, 1
(1959).
34. Schafer H„ Sibbing E., Gerken R„
Z. Anorg. Allgem. Chem., 307, 163
(1960).
35. Schafer H„ Scholz H.. Gerken R„
Z. Anorg. Allgem. Chem., 332,
154 (1964).
36. Schafer H„ Odenbach H., Z. Anorg.
Allgem, Chem., 346, 127 (1966).
Газы 107
14. Газы [1,2]
Как видно нз изложенного выше, при работах, связанных с переходов
от вакуума к атмосферному давлению, часто приходится пользоваться ин-
дифферентными газами. Правда, их получение и очистка описаны в специаль-
ной части этой книги (ч. II), однако некоторые общие вопросы следует под-
вергнуть обсуждению уже здесь. Почти всегда применяемые для работы
азот, диоксид углерода, инертные газы, водород, кислород, хлор, диоксид се-
ры и аммиак получают в лаборатории из стальных баллонов.
Сероводород высокой чистоты также выпускается в стальных балло-
нах. Вид резьбы на вентиле стального баллона зависит от типа газа, для
которого он предназначен; левая резьба—для горючих газов, правая — для
всех остальных. Однако у баллонов с правой резьбой подсоедннительиая
резьба может быть различного размера, так что для разных газов прихо-
дится пользоваться также н различными вентилями. Кроме того, стальные
баллоны в зависимости от содержащегося в иих газа имеют опознавательные
цвета (табл. 22). Высокая степень чистоты газа в баллоне обозначается на-
рисованным на нем кольцом или буквой «/?». Благодаря такой маркировке
ошибки прн использовании газов в значительной степени исключаются.
Необходимо предотвращать возможность падения баллонов; транспор-
тировать нх следует с привинченным защитным колпачком. Для отбора га-
зов к баллонным вентилям присоединяют регулирующие вентили. Последние
могут быть вентилями точной регулировки (игольчатые вентили, впервые
сконструированы ле Россиньолем) или редукционными (понижающими
давление) вентилями, позволяющими при помощи специального механизма
Таблица 22. Маркировка стальных баллонов со сжатыми газами
Газ Окраска баллона Окраска баллона, принятая в СССРа Подсоеднннтельная резьба
Водород Н2 Красная Темно- зеленая Левая
Пропан С3На > Красная >
Ацетилен С2Н2 Желтая Белая Специальный разъем без резьбы
Кислород О2 Голубая Голубая Правая
Азот N2 Зеленая Черная Правая, как у баллонов с СО-.
Оксид углерода СО Красная — Левая
Диоксид углерода СО2 Серая Черная Правая
Инертные газы Светло- серая Серая (Аг) Коричневая (Не) Правая, как у баллонов" с СО2
Фтор F2 Серая — Правая, как у баллонов с С12
Хлор С12 3> Защитная Правая крупная
Трнфторид бора BFs —* Правая, как у баллонов с С12
Оксид диазота Ы2О » — Специальный разъем с внутренней резьбой
Сероводород H2S Красная Белая Левая крупная
а Данные для СССР составлены переводчиком.
108 Методы препаративной химии
устанавливать на выходе определенное давление газа, в значительной степе-
ни независимое от его давления внутри баллона.
Вентили ле Россиньоля в большинстве случаев портятся от чересчур
сильного завинчивания, в результате чего повреждается тонкая вентильная
игла. Обычно достаточным бывает завинчивать такой вентиль только в той
степени, в какой это удается сделать двумя пальцами без особого напряже-
ния. Если после долгого применения вентиль все-таки становится не совсем
плотным, то иглу вынимают и тщательно протирают тряпкой и лишь в слу-
чае крайней необходимости обрабатывают самой тонкой наждачной бумагой.
Если же и это не помогает, то лучше совсем закрыть газ, завинтив основной
вентиль баллона, и отдать вентиль в починку. Редукционные вентили по
возможностям дозирования и поддержания постоянного потока газа гораз-
до лучше, чем игольчатые. Однако и они не всегда могут обеспечить часто
необходимое в лабораторных условиях постоянство потока при небольших
скоростях подачи газа. Поэтому при проведении продолжительных опытов
здесь также необходим периодический контроль. Регулировку потока газа
при помощи таких вентилей ведут, открыв сначала полностью вентиль на
баллоне и установив затем регулятором давление на выходе около 0,5 бар.
Кроме газов, указанных в табл. 22, в баллонах выпускаются также такие газы, как
SO2, HF, НС1, СОС12, CCIF3, CC12F2, CH3CI, CH3NH2, HCN, NH3 и CH4. Небольшие ко-
личества (150—400 г) чистого диоксида серы и различных низкокипящих органических
жидкостей можно приобрести в маленьких баллончиках с игольчатым вентилем.
Приборы для получения газов
Наиболее распространенным из таких приборов все еще остается аппарат
Киппа. Для получения газов, не содержащих воздуха, например диоксида уг-
Рис. 64. Конструкция аппарата Кип-
па с приспособлением для изоляции
от атмосферы, служащего для полу-
чения газов без примеси воздуха.
На пористых стеклянных пластинках на-
ходится слой ртути (Hg).
I
/
лерода, применяемого для переведения других газов в азотометр, можно
снабдить этот аппарат дополнительным приспособлением, которое обеспечи-
вает присутствие выделяющегося газа и в верхнем шаре, предотвращая до-
Газы 109
ступ воздуха в кислоту, служащую для разложения. Работа этого приспо-
собления пояснена на рис. 64 и 65. Прибор, показанный на рис. 64, имеет
тот недостаток, что в нем нельзя провести достаточно полное обезгаживанне
исходных веществ. Для получения чистых газов лучше пользоваться аппара-
том, показанным на рис. 65. В нем можно получать газы как из жидкостей
и твердых веществ, так н их двух жидкостей, причем в верхний шар всегда
наливают жидкость. Прежде чем открыть кран 1, во всем приборе можно
создать вакуум. Кран 1 можно заменить шлифованной пробкой (рис. 66),
передвигаемой при помощи стеклянной палочки. Если вдоль этой пробки,
а также вдоль соприкасающейся с ней пришлифованной поверхности сделать
Рис. 65. Конструкция вакуумируемо-
го аппарата Киппа с изоляцией от
атмосферы.
Рис. 66. Конструкция вакуумируемо-
го прибора для получения Газов с
капельной воронкой (по Боденштей-
ну).
по небольшому желобку, доходящему до середины их длины, то жидкость
можно вводить также, не вынимая пробки, простым поворачиванием ее
вокруг оси. При желании удалить газы из жидкости, находящейся в верх-
нем шаре, путем его вакуумирования необходимо, разумеется, уплотнить
стеклянный стержень резиновой пробкой, предотвращающей доступ воздуха.
Аппарат Киппа и его видоизменения не позволяют получать газы, со-
вершенно не содержащие воздуха. Это было бы возможно, если бы, с одной
стороны, твердый реагент, например мрамор или гранулы цинка, не содер-
жал воздуха в виде включений, а, с другой стороны, воздух из жидкого
компонента также был полностью удален. Простым эвакуированием аппара-
та Киппа растворенный воздух полностью не удаляется. Для этого необхо-
димо производить кипячение жидкости или несколько раз попеременно ее
замораживать и эвакуировать в вакууме. Чаще можно поступить проще,
если очищать уже выделившийся газ путем его конденсации в вакууме.
Многочисленные варианты соответствующих приборов, а также аппаратов
для получения газов при взаимодействии двух жидкостей и двух твердых
веществ будут подробно описаны в ч. II.
Очистка газов
Поглотительная способность обыкновенных промывных склянок (напри-
мер, по Дрекселю, рис. 67, а) невелика. Вообще для достижения достаточно
полного взаимодействия между жидкостью, находящейся в промывной склян-
ке, и пропускаемым газом необходимо, чтобы скорость течения газов не превы-
ПО Методы препаративной химии
шала 10 л/ч. Скорость течения газов ориентировочно может быть оценена
по частоте выделения пузырьков. При диаметре трубки 5—6 мм появление
одного пузырька в секунду соответствует в обыкновенных промывных
склянках скорости течения газа 1 л/ч. Выделение 10 пузырьков в секунду,
т. е. такая скорость, при которой еще можно заметить глазом отдельные
Рис. 67. Типы промывных склянок.
а —по Дрекселю; б — по Мюнке; в» г, д — со зиеевнком; е, ж, з — с пористым фильтрам.
пузырьки, представляет собой верхний предел скорости пропускания. Еще
одним недостатком промывной склянки по Дрекселю можно считать опас-
ность засасывания промывной жидкости в соединительный шлаиг. Более
надежны в этом отношении промывные склянки по Мюнке (рис. 67,6). Го-
раздо эффективнее оказываются промывные склянки со змеевиком (рис.
67, в—д) илн с пористой стеклянной пластинкой (рнс. 67, е—з), применение
которых возможно вплоть до скоростей потока газа 60 л/ч и 150 л/ч соот-
ветственно.
В продаже имеются склянки со змеевиками двух типов. Первый тип (по
Фридрихсу, рнс. 67, в) характеризуется наличием винта (изготовляемого пу-
тем формования), прилегающего к стенкам сосуда. Преимущество склянки
этого типа заключается в том, что вследствие значительного внутреннего
пространства, занимаемого винтом в трубке, затруднено обратное движение
промывной жидкости. Недостатком ее является наличие сравнительно не-
Газы 111
большого количества промывной жидкости, а также возможность потери
жидкостью промывной способности в результате ее недостаточной циркуля-
ции как раз в наиболее ответственном месте, т. е. между стенками сосуда
и винтом. Оба этих недостатка устранены в склянке сходного типа, но дру-
гой формы, в которой спираль не прилегает к стенке, а заключена в стек-
лянный цилиндр (рис. 67, г). На рис. 67, <9 показан другой тип промывной
склянки, устроенной по типу насоса «маммут». Выходящие через небольшое
отверстие внизу газовые пузырьки увлекают за собой в змеевик промывную
жидкость и наверху снова отбрасывают ее. В результате происходит актив-
ная циркуляция сравнительно большого количества промывной жидкости.
Недостатки этой конструкции заключаются в том, что в склянке может про-
изойти обратный переброс. Чтобы с уверенностью исключить опасность
обратного переброса, приходится присоединять к промывной склянке навер-
ху еще один широкий сосуд. Кроме того, недопустимо превышать определен-
ную максимальную скорость пропускания газа, так как иначе газ будет вы-
ходить большими пузырями внизу нз отверстия, всасывающего жидкость, и
тогда эффективность промывной склянки в значительной степени понизится.
Однако при умеренных скоростях течения газа промывную склянку этого
типа вследствие очень продолжительного времени контакта газа и жидкости
можно отнести к наиболее эффективным. Кроме создания хорошего кон-
такта между газом и жидкостью необходимым условием эффективности
очистки газа является также достаточное количество правильно подобран-
ной промывной жидкости, быстро реагирующей с примесями.
Путем разделения газа иа мелкие пузырьки эффективность промывной
скляики может быть существенно повышена. Это осуществляется в обще-
известных промывных склянках с пористой стеклянной пластинкой, через ко-
торую газ поступает в промывную жидкость (рис. 67, е, ж). Пористые пла-
стинки имеют обозначения крупности спеченных зерен от GO до G3 (прибор-
ное стекло) или от DO до D3 (стекло дюран). Средний размер пор стеклян-
ных пластинок составляет:
Номер фильтра 0 12 3
Размер пор, мкм 200—150 150—90 90—40 40—15
К недостаткам подобных промывных склянок можно отнести, во-первых,
необходимость создания значительного избыточного давления, в особенности
при продавливании газа через мелкопористые фильтры. Во-вторых, в промыв-
ных склянках этого типа наблюдается особенно сильная по сравнению со
склянками других типов склонность к образованию тумана из мелких ка-
пелек промывной жидкости. В связи с тем, что улавливание тумана пред-
ставляет большие экспериментальные трудности, лучше использовать промыв-
ные склянки со змеевиком.
В продаже имеются также промывные склянки с пористой пластинкой,
в которой полностью предотвращается возможность обратного переброса
жидкости (рис. 67, з).
Для проведения продолжительных опытов, при которых промывная жид-
кость может оказаться полностью израсходованной, используют промывную
колонку, заполненную стеклянными шариками. Вверху колонки вставляют
капельную воронку, а внизу помещают сливиой кран. В этом случае оказы-
вается возможным добавлять свежую промывную жидкость и выпускать со-
ответствующее количество отработанной жидкости, не прерывая опыта.
Высушивание газов
Газы высушивают либо концентрированной серной кислотой, помещен-
ной в промывные склянки, либо пропусканием над осушающими веществами
в колонках, либо, наконец, путем глубокого охлаждения. Степень высушива-
ния различными осушителями, естественно, имеет предел. Она сильно зависит
112 Методы препаративной химии
Таблица 23. Осушители для газов
Осушитель Содержание влаги, мг/л, или прибли- зительно парциаль- ное давле- ние воды прн 25 °C, мм рт. ст. Точка ро- сы, °C Примечание
СаС12 0,2 —33 Не применяется для осушки аммиака,' аминов, спирта или фтороводорода. Хорошая влагоемкость
CaSO4 0,005 —63 Нейтральный. В США выпуска- ется под маркой «Drierite». Ре- генерация при 250 °C
СаО кон 0,003 0,002 —67 1 —70 J Неприменимы для газов, взаи- модействующих со щелочами или содержащих СО2. Умерен- ная влагоемкость
H2SO4, конц. 0,003 —65 Неприменим для H2S, NH3, HBr, С2Н2, HCN. Обладает окислительными свойствами
Силикагель 0,002 —70 Регенерация при 300 °C
А12О3 (активиро- ванный) 0,0008 —75 Регенерация при 500 °C в ва- кууме или при 700 °C на воз- духе
Молекулярные си- та 0,0008 —75 Регенерация при 350 °C в ва- кууме
Mg(C104)2 0,0005 —78 Регенерация при 250 °C в ва- кууме. Хорошая осушительная емкость. Принимать меры пре- досторожности при осушке окисляющихся веществ!
Р2О5 (Р4О10) 0,00002 —96 Не регенерируется, неприменим для NH3, галогеноводородов
СаН2 <0,00001 Реагирует с выделением водо- рода, не регенерируется х \
от условий проведения опыта. Состояние равновесия, т. е. достижение при
высушивании парциального давления воды над данным осушителем, практи-
чески никогда не достигается. Именно поэтому указания различных авторов
о степени высушивания, достигаемой при применении определенного осуши-
теля, могут весьма сильно различаться. Значения, приведенные в табл. 23,
дают ориентировочное представление об эффективности наиболее распростра-
ненных осушителей. В дополнение к табл. 23 необходимо сделать следующие
замечания.
Хлорид кальция. Значение, приведенное в табл. 23, относится только к све-
жеприготовленной безводной соли. Осушающее действие может сильно сни-
Газы 113-
жаться, особенно при повышенной температуре в помещении и после дли-
тельного применения.
Осушители, указанные в начале табл. 23 (вплоть до силикагеля), нельзя
применять в условиях высокого вакуума. Если же давление в системе должно
длительное время поддерживаться ниже 10-4 мм рт. ст., то вообще непригоден
ни один из осушителей.
Силикагель выпускается в виде зерен, иногда с цветным индикатором
(голубой гель). «К. С. — гранулированный осушитель»* представляет собой
шарики из геля диаметром около 3 мм. Преимущества применения этого,
осушителя связаны с шарообразной формой гранул и полным отсутствием
мелких пылевидных частичек. В начале использования степень высушивания
силикагелем соответствует значению точки росы ниже —55 °C. Если суще-
ствует опасность проникновения воды в виде капель или тумана, то приме-
няют силикагель в виде «К. С. — гранулированного осушителя—IF». Регене-
рацию проводят при температуре 200—250 °C. Силикагель с индикатором, ко-
торый в конце работы осушителя (при относительной влажности 10%) изме-
няет свой цвет из голубого в светло-розовый, следует регенерировать при
температуре не выше 180 °C. «К. С. — гранулированный осушитель» приме-
няется при высушивании водорода, кислорода, азота, инертных газов, диок-
сида углерода, диоксида серы, углеводородов и их галогенпроизводных. Для
осушки хлора и хлороводорода используют осушитель марки «IF». Силика-
гель, а в еще большей степени оксид алюминия, способен поглощать поми-
мо воды также другие пары, что в ряде случаев может явиться причиной
понижения выхода продукта.
При помощи молекулярных сит типа 3 А** осушают кислород, азот, ди-
оксид углерода и инертные газы, причем скорость адсорбции и адсорбционная
емкость относительно высокие. В случае необходимости можно рекомендо-
вать производить предварительное высушивание силикагелем. В противопо-
ложность многим другим осушающим средствам осушительная емкость мо-
лекулярных сит и при температуре 100 °C все еще достаточно высока. Сле-
дует, однако, учитывать, что кроме воды могут адсорбироваться и другие
вещества. Так, молекулярное сито 5 А удаляет из воздуха как воду (до
точки росы —75°C), так и диоксид углерода (до содержания 1 млн-1). Тот
же тип молекулярных епт способен поглощать из газовых смесей аммиак,
сероводород, меркаптаны, следы хлороводорода или диоксида серы. Относи-
тельно способов регенерации молекулярных сит см. разд. 6 настоящей главы.
Пентаоксид фосфора Р4Ою содержит (за исключением реактива марки
«чистый для анализа») следовые количества низших оксидов фосфора. При-
сутствие последних нежелательно, так как они при взаимодействии с водой
могут выделять фосфин. Испытание на содержание низших оксидов можно
провести путем нагревания раствора пентаокенда фосфора с AgNO3 или
HgCl2. В присутствии низших оксидов происходит восстановление до свобод-
ных металлов. При особо высоких требованиях к фосфорному ангидриду его
сублимируют в потоке кислорода непосредственно внутрь осушительной ко-
лонки (рис. 68). Для того чтобы произошло полное окисление всех примесей,
используют платиновый катализатор. Осушительную колонку спаивают с не-
сколько более широкой трубкой такой же длины при помощи узкой трубочки
диаметром 5 мм и длиной 6 см. Через осушительную колонку, заполненную
небольшим количеством стеклянной ваты, пропускают с целью осушки поток
сухого воздуха при одновременном нагревании. В пустую трубку помещают
платиновую фольгу и затем вносят такое количество пентаоксида фосфора,.,
чтобы над ним по всей длине трубки оставался свободный канал для про-
хода газа. После этого пропускают слабый поток кислорода и слегка подо-
* Фирма-изготовитель Kali-Chemie (Hannover).
** Выпускается фирмам Е. Merck AG (Darmstadt), «Sephadex» der Fa;.
Pharmacia (Uppsala, Schweden), Deutsche Pharmazia GmbH (Frankfurt/Main)..
8—143
fl 14 Методы препаративной химии
«гревают сначала колонку со стеклянной ватой, затем то место, где нахо-
дится платиновый катализатор, почти до размягчения стекла. Путем нагре-
вания приблизительно до 200—300 °C медленно перегоняют Р4О10, начав
•нагревание от платиновой фольги и распространяя его дальше н дальше до
жонца трубки. При соответствующем иагреваини колонки со стеклянной ва-
той Р4О10 распределяется в ней равномерно. Эту операцию можно выпол-
Стеклянная Вата
Рис. 68. Способ очистки пентаоксида фосфора путем сублимации.
iiimiiiiiiiiiniA
Pt- фольга
PM
ог
чяять также и в нагреваемой газом печи для сожжения. Для заполнения
•осушительных трубок меньшего размера вместо стеклянной ваты использу-
•ют стеклянные бусы. Заполнение обрезками стеклянных трубок не рекомен-
дуется, поскольку заключенный в них воздух долгое время не вымывается
из колонки и загрязняет осушаемый газ. Хорошо оправдавший себя тип лег-
жовзаимозаменяемых (вследствие вертикального положения шлифов) осу-
шительных трубок изображен на рис. 69. Перхлорат магния, а также сили-
«жагель можно вводить в осушительную трубку н без носителей, поскольку
Рис. 69. Трубка с пентаоксидом фос-
фора.
эти вещества ие так легко закупоривают трубку, как Р4О10. Последний
•выпускается также в гранулированном виде («Гранусик») или на крупно-
зернистом минеральном носителе («Сикапент»).
Наиболее полное и тщательное высушивание осуществляют путем глу-
бокого охлаждения, при котором газ пропускают через змеевик возможнб
••большей длины, опущенный в жидкий воздух (см. рис. 47, е). В обычной ло\
,-вушке полной конденсации воды не происходит.
Следует обратить внимание на то, что при глубоком охлаждении не
•всегда все количество воды осаждается на стенках ловушки. Часть ее мо-
жет в виде мельчайших кристаллов выноситься газовым потоком. Поэтому
•рекомендуется соединять последовательно несколько спиральных ловушек
так, чтобы, проходя между ловушками, газ нагревался по крайней мере
до 0 °C. При этом происходит переход воды в газовую фазу за счет испа-
рения. После столь тщательного высушивания газ следует пропускать толь-
ко через стеклянные трубки илн короткие толстостенные шланги. Давление
насыщенного пара воды при температуре —196 °C составляет около
НО-23 мм рт. ст.., а при —'80 °C — уже 4-10~4 мм рт. ст.
Газы 115”
Чистые газы для создания защитной атмосферы и газы-носители
В продаже имеются очень чистые газы, применяемые для создания за-
щитной атмосферы или в качестве газов-носителей. Гелий, аргон, водород,
н азот выпускаются чистотой по крайней мере 99,99%. В большинстве слу-
чаев затраты на очистку в лабораторных условиях инертных газов и водо-
рода себя не оправдывают. Азот, напротив, часто применяют в больших ко-
личествах для создания защитной атмосферы. Если при этом важно понн-
Рис. 70. Конструкция прибора для
получения газообразного азота из
жидкого азота.
1, 2, 3 — стеклянные трубки; 4 — сквозные
отверстия в этих трубках; 5, 6 — электри-
ческие нагреватели; 7 — сосуд Дьюара с
защитным кожухом 8; 9 — медный змеевик.
зить до минимума содержание в нем кислорода, то баллонный азот очищают
по способу, описанному в ч. П, гл. 7. В том случае, если наиболее сущест-
венное требование — минимальное содержание влаги, а кислород может
присутствовать в концентрации до 0,5%, то лучше всего получать азот ис-
парением жидкого азота из закрытого сосуда. Получаемый таким образом,
азот должен теоретически иметь точку росы —196 ”С н несколько понижен-
ное по сравнению с жидким азотом содержание кислорода (обычно <0,5%),.
так как при испарении происходит обогащение кислородом жидкой фазы.
Эт*т процесс удобно вести на установке, описанной в работе [3] и показан-
ной на рнс. 70. Сосуд Дьюара 7 вместимостью 5 л заключен в защитный*
кожух 8. Через плотно прилегающую резиновую пробку проходят три стек-
лянные трубки. Трубка 1 служит для заполнения сосуда жидким азотом, ие
содержащим взвешенных частичек льда. В трубке 2 находится поплавок,,
показывающий уровень жидкости. Трубка 3 внизу запаяна; выше пробки она
присоединяется к медному змеевику 9. Вблизи дна находится закрепленная1
8*
316 Методы препаративной химии
на трубках нагревательная спираль 6 (64 Ом), включаемая последовательно
<с другим нагревателем 5 (73 Ом), находящимся в горле сосуда Дьюара. Со-
лротивления нагревателей подобраны таким образом, чтобы испаряющийся
в 6, но еще холодный азот нагревался нагревателем 5 приблизительно до
комнатной температуры. Окончательное прогревание газа до комнатной тем-
пературы происходит при его прохождении через медный змеевик 9. Макси-
мальная производительность установки, достигаемая при ее включении на
.220 В, составляет 45 л/мнн. Регулировкой через трансформатор можно за-
давать любую меньшую скорость подачи газа.
Если из этого аппарата удалить нагревательную спираль и медный змее-
вик, то его используют для получения постоянного потока очень холодного
азота, с помощью которого можно охлаждать, например, низкотемператур-
ные колонны или другую аппаратуру небольшого размера.
Для работы с легкогидролизующимися и окисляющимися веществами
кроме сосудов Шленка н вакуумной аппаратуры необходимо иметь установ-
ку, из которой в любое время можно отбирать инертный газ. Установка
состоит из источника газа (баллона нли прибора для получения газа), кла-
пана, ограничивающего внутреннее избыточное давление значением 200 мм
рт. ст., аппаратуры для очистки и осушки газа, счетчика пузырьков, в кото-
ром исключена возможность обратного переброса, и распределительного
устройства, при помощи которого газ через краны и шланги подается в от-
дельные части реакционной аппаратуры. Если бывает нужно за короткое вре-
мя израсходовать сравнительно большое количество газа, например, для то-
го, чтобы заполнить эвакуированный сосуд, то между счетчиком пузырьков
и распределительным устройством помещают большую буферную колбу
(5 л). Находящийся в ней газ предотвращает слишком сильное понижение
давления в установке при выполнении подобных операций.
Измерение скорости подачи газа
Приблизительную оценку скорости пропускания можно произвести при
помощи счетчика пузырьков (см. разд. «Очистка газов»). Измерения прово-
дят ротаметрами или дифференциальными манометрами. Действие ротамет-
ра основано на подъеме в потоке газа вращающегося шарообразного по-
плавка, находящегося в стоящей вертикально, слегка конической градуиро-
ванной трубке. В продажу поступают уже градуированные ротаметры*.
Дифференциальный манометр представляет собой U-образную трубку, оба
колена которой соединены капилляром для прохождения газа (рис. 71, а, б).
Удобство модели, показанной на рис. 71,6, заключается в легкости замены
капилляра и возможности варьирования длины капилляра путем его укоро-
чения. Разность давлений до и после капилляра пропорциональна скорости
потока газа. Однако градуировочная кривая будет прямой линией лишь в том
случае, если использованы короткие и узкие капилляры. При заданной раз-
ности давлений скорость потока (мл/с) обратно пропорциональна вязкости
газа т], что делает возможным пересчет градуировочной кривой для дру-
гих газов.
При точных измерениях необходимо поддерживать постоянство темпера-
туры капилляра. Полезно параллельно капилляру включить стеклянный
кран. При опасном поднятии уровня жидкости в манометре перебрасывание
жидкости всегда можно предотвратить простым открыванием крана. Попыт-
ка же избежать этой неприятности путем регулировки подачи газа, как по-
казывает опыт, в большинстве случаев оказывается запоздалой. Наличие
у манометра дополнительного патрубка позволяет производить его заполне-
ние жидкостью после монтажа и высушивания манометра и затем патрубок
-отпаять.
.Фирма-изготовитель Rota (Oeflingen, Koln).
Газы 117
В качестве жидкостей для заполнения манометров применяют в зависи-
мости от конкретной задачи концентрированную серную кислоту, вазелино-
вое, апиезоновое или силиконовое масло, бромнафталин, ртуть, а также под-
крашенную воду. Необходимо учитывать, что большинство жидкостей не
только имеют заметное давление пара, но и способны в существенной степе-
ни растворять газы и пары. В манометрах с наклонной трубкой могут быть
точно измерены очень малые скорости потока лишь с небольшим торможением
потока в капилляре. (Разность давлений в 0,01 мм рт. ст. все еще поддается
измерению.) В продаже имеются удобные в работе приборы этого типа, но их
можно легко изготовить и самостоятельно.
Рис. 71. Типы дифференциальных ма-
нометров (а. б см. текст).
1 — капилляр; 2 — патрубок для запол-
нения.
Измерение количества газа
Эти измерения осуществляются при помощи градуированных газометров
или газовых бюреток. Газовая бюретка с постоянным давлением описана
Шенком [4]. Кроме того, для этой же цели применяют газовые счетчики,
имеющиеся в продаже также и для очень точных измерений. Их удобно
использовать для градуировки ротаметров.
Хранение газов
Газы обычно сохраняют в газометрах. Главный недостаток обыкновенных
газометров заключается в том, что они, за исключением газометров с коло-
колом, не допускают выпуска газа при постоянном давлении. В некоторых
газометрах этот недостаток устранен тем, что трубка из верхнего сосуда не
доходит до дна резервуара, но погружена в переливной сосуд. Такие газо-
метры легко можно изготовить из больших реактивных склянок или из бал-
лонов вместимостью 50 л. Расположение соединительных трубок показано
на рис. 72. Доходящая до дна склянки трубка 4 служит для отвода запи-
рающей жидкости снова в склянку 2, что может быть достигнуто путем
повышения давления в нижнем сосуде или понижения давления в верхнем
сосуде. Погруженная в верхний сосуд трубка 1 превращает его в сосуд Ма-
риотта. Давление газа соответствует разности высот h между уровнями
жидкости в верхнем сосуде и переливном сосуде 3. Такие газометры рабо-
тают весьма надежно. Раз установленная скорость истечения газа остается
практически постоянной в течение многих часов. Недостатком такого газо-
метра является необходимость использования значительных количеств запи-
рающей жидкости. Применение прокипяченных и насыщенных растворов
поваренной соли уменьшает опасность загрязнения газов примесями. Еще
лучше пользоваться для этой цели раствором 200 г безводного Na2’SO4 и
118 Методы препаративной химии
40 мл. конц. H2SO4 в 800 мл воды, прокипяченным с обратным холодиль-
ником. Давление пара воды над таким раствором равно ~15 мм рт. ст.
прн 20 °C, а растворимость газов в нем составляет лишь 20—30% от их рас-
творимости в чистой воде (при 25°C). Для некоторых целей применяют ва-
зелиновое масло, из которого газы удалены нагреванием в вакууме.
Газометры с колоколом, в которых колокол погружен в узкий кольцевой,
наполненный ртутью зазор, еще лучше предохраняют газ от загрязнения
примесями. Такие газометры ограничены в своих размерах вследствие все
же значительного количества ртути, требующегося для нх заполнения, не-
смотря на их особую конструкцию. Для направления движения колокола
Рис. 72. Устройство газометра с по-
стоянным давлением выходящего га-
за (1—4 см. текст).
Рис. 73. Устройство газометра с ко-
локолом (1, 2 см. текст).
(рис. 73) применяется деревянный корпус. Конечно, в тех случаях, когда
требуется неподвижное присоединение газометра к установке, для притока
и выпуска газа вместо гибкого шланга, присоединенного к крану 1, можно
воспользоваться припаянной стеклянной трубкой 2 (иа рис. 73 она изображе-
на пунктиром). Часто для хранения запасов газа проще заполнять газами
под давлением эвакуированные стальные баллоны. Для этой цели можно
воспользоваться небольшим рассчитанным на высокое давление сосудом, ох-
лаждаемым жидким воздухом. В этом случае газ конденсируют в сосуде
до жидкого состояния и затем по медной капиллярной трубке переводят еп>
в стальной баллон (рнс. 74).
Для хранения агрессивных газов пользуются предварительно эвакуирован-
ной круглой колбой, закрываемой вентилем, лучше всего тефлоновым. Есл»
при отборе газ должен находиться под большим давлением, чем в сосуде
для хранения, то его предварительно конденсируют в ловушке, температуру
которой далее поддерживают на таком уровне, чтобы создавалось необхо-
димое давление газа.
Газы 119
При желании получить смесь газов в стальных баллонах (например,
когда вводят в один и тот же баллон сначала азот под давлением 30 бар,
а затем из другого баллона водород до достижения общего давления
120 бар, чтобы получить готовую смесь для синтеза аммиака) необходимо
учитывать, что газы при столь высоких давлениях обладают значительной
вязкостью, затрудняющей полное перемешивание их друг с другом даже в
течение многих суток. Поэтому необходимо позаботиться о перемешивании
газа за счет конвекции путем нагревания части баллона, например нижней
части баллона, поставленного наклонно горлом вниз, настольной 60-ваттной
Рис. 74. Схема установки для пере-
вода конденсируемых газов в сталь-
ные баллоны.
лампой в течение 24 ч. Весьма целесообразно в этих случаях периодически
производить анализ газовой смеси до достижения постоянных результатов.
Для проведения прн повышенных температурах реакции газов н паров
с расплавленными солями Зундермейером с сотрудниками был сконструиро-
ван специальный реактор. Его можно также использовать для проведения
некоторых реакций между жидкостями и газами при менее высоких тем-
пературах.
Как показано на рис. 75, реактор состоит из толстостенного стеклянного
стакана 1 и закрывающей его плоской пришлифованной крышки 2 с четырь-
мя коническими шлифами*. Через центральный шлиф на крышке вставляет-
ся специальная мешалка 5, в которой предусмотрено охлаждение и имеется
отверстие для подвода газа**. В данном случае это отверстие используется
лишь для смазывания мешалки силиконовым маслом илн штирролом. Ме-
шалка 5 вращается со скоростью 1000—2000 об/мнн. Оиа снабжена специ-
альным «газовым перем сшивателем», в котором происходит всасывание нахо-
дящегося над жидкостью газа через боковое отверстие в верхней части и вы-
талкивание его через боковые отростки трубки, находящиеся в ннжней части
мешалки. Таким образом достигается многократное прохождение газа через
жидкость. Через другой, боковой шлиф крышки проходит трубка для газов
или паров, вступающих в реакцию. Для дозировки жидкости, пары кото-
рой вводятся в реактор, на трубку насаживается капельная воронка, а при
необходимости подводится также газ для промывки. Через другой боковой
шлиф (на рнс. 75 не показан) вставляется закрытая с ннжнего конца труб-
ка, в которую помещается термопара или термометр, измеряющие темпера-
туру расплава. Обе эти трубки, для подвода газа и для термопары, не-
* Можно использовать стакан с пришлифованной крышкой типа NW 120
из стекла дюран 50 с крышкой фирмы Schott u. Gen. (Mainz).
** Например, мешалка с одновременным вводом газа с нормальным шли-
фом NS 29 фирмы Nolrnag (Hofheim).
120 Методы препаративной химии
сколько сплющены, причем их плоскости располагаются радиально с тем,
чтобы рассекать поток движущегося расплава. Через последний шлиф крыш-
ки 3 выводятся газообразные продукты реакции, которые в случае необхо-
димости пропускают через обратный холодильник.
Рис. 75. Конструкция реактора для прове-
дения реакций газов с расплавами.
1 — стакан с плоским шлифом; 2 — крышка с пло-
ским шлифом, с одним центральным и тремя бо-
ковыми отверстиями; 3 — трубка для отвода газо-
образных продуктов реакции: 4 — ввод мешалки
через охлаждаемый цилиндрический шлиф; 5 —
мешалка, через которую проходит газ; 6 — трубка
для ввода газа или пара.
ЛИТЕРАТУРА
1. Klemenc A., Behandlung und Rein-
darstellung von Gasen. Springer,
Wien, 1948.
2. Мюллер Г., Г наук Г. Газы высо-
кой чистоты. Пер. с нем. — М.: Мир,
1968.
3. Riickert Р., частное сообщение.
4. Schenk Р. W„ Z. Anorg. Allgem.
Chem., 233, 393 (1937).
15. Работа со сжиженными газами в качестве
растворителей
Для работы со сжиженными газами, такими, как NH3, SO2, HF и др.,
разработано множество приборов, в которых все операции — осаждение,
фильтрование, промывание, сушка и др., включая титрование, можно прово-
дить при полном отсутствии воздуха и влаги. Отдельные операции выпол-
няют в вакууме, другие — под давлением паров соответствующего сжижен-
ного газа или в атмосфере инертного газа. Рассматриваемые ниже отдель-
ные конструкции приборов ни в коем случае не исчерпывают всего их мно-
гообразия, однако в приводимой литературе можно найти решение многих
экспериментальных проблем. Общие принципы работы будут объяснены иа
Работа со сжиженными газами 121
примере реакций аммонолиза. В наиболее часто встречающемся случае воз-
никает необходимость провести аммонолиз твердого вещества или раствора
твердого, жидкого или газообразного вещества в каком-либо индифферент-
ном органическом растворителе. Для аммонолиза используют либо жидкий
аммиак, либо растворы в нем амидов щелочных металлов. На рис. 76 пока-
зана установка для проведения простых реакций аммонолиза. Аппаратуру
эвакуируют при помощи вакуумного насоса, что служит и для целей осушки.
Для заполнения аппаратуры используют высушенный азот, предварительно
Рис. 76. Схема аппаратуры для проведения простых реакций аммонолиза.
/ — сосуд для конденсации; 2 — стеклянный фильтр; 3— клапан избыточного давления;
4 — манометр; 5 — счетчик пузырьков; 6 — осушительная колонка; 7 — охлаждаемая ло-
вушка.
освобожденный от примеси кислорода (см. ч. II, гл. 7). Аммиак из баллона
после соответствующей очистки конденсируют при температуре —78 °C в ло-
вушке 1 над небольшим количеством натрия. Отбор газообразного аммиака
ведут, давая жидкому аммиаку выкипать и пропуская газ через пористый
стеклянный фильтр 2 (G3) для задерживания пылевидных частичек натрия.
В случае повышения давления срабатывает ртутный клапан избыточного
давления 3.
Реакционный сосуд с шарнирным соединением показан на рис. 77.
В колбочку А помещают навеску исходного вещества и затем туда конден-
сируют аммиак. К переходнику В с пористой стеклянной пластинкой под-
соединяют маленькую колбочку С. Такая конструкция позволяет путем вра-
щения всего реакционного сосуда вокруг оси шлифа 1, производимого после
быстрого удаления охлаждающей бани, проводить декантацию или фильтро-
вание раствора из Л в В. При этом раствор не соприкасается с нагретыми
частями аппаратуры. Растворитель (NH3) может быть перегнан обратно
из С в А и затем снова использован для промывания продукта в А. Нали-
чие пористой перегородки предотвращает попадание брызг раствора из С в
122 Методы препаративной химии
Работа со сжиженными газами 123
А. Путем вращения шлифа 2 колбочки С и А удается удалить друг от дру-
га настолько, что их можно поместить в различные охлаждающие бани.
В колбочке А вещество и растворитель перемешиваются магнитной мешал-
кой. По окончании операции дают выкипеть избытку аммиака из реакцион-
ного или конденсационного сосуда, выпуская газ через осушвтельную колон-
Рис. 77. Устройство при-
бора с шарнирным со-
единением по Яндеру и
Энгельгардту в разных
проекциях (а, б).
1,2 — шлифы. В проекции б
муфта шлифа 1 направлена
перпендикулярно плоскости
рисунка. При выполнении
операции декантации прибор
вращают вокруг оси шли-
фа 1. А. В, С см. текст.
Ку 6 (рис. 76) и счетчик пузырьков, а остаток откачивают через вымора-
живающую ловушку 7. Манометр 4 позволяет производить измерение дав-
ления разложения продукта в колбе А (ср. конструкцию тензоэвдиометра на
рис. 95).
Для проведения аммонолиза легкогидролизующихся веществ использу-
ют аппаратуру, показанную на рис. 78. Исходное вещество вносят в реак-
Рис. 78. Участок аппаратуры для
проведения реакций гигроскопичных
веществ с аммиаком или амидами
щелочных металлов (1—4 см. текст).
ционный сосуд 2 в виде иавески (для твердых веществ), путем дистилля-
ции или при помощи пипетки (для жидких веществ и растворов). Можно
также сначала приготовить раствор в сосуде 1, а затем вытеснить его в 2
инертным газом. После этого в сосуде 2 конденсируют аммиак. Если нужно
провести реакцию с амидом калия, то сначала в 1 вносят калий (без приме-
си оксида) и немного Fe2O3 (катализатор), туда же конденсируют аммиак
и вытесняют раствор в 2. Именно для этого случая в 1 помещена пористая
перегородка, задерживающая частички катализатора. Находящийся в 2 про-
дукт реакции, нерастворимый в аммиаке, несколько раз промывают, конден-
сируя туда аммиак и продавливая раствор через пористую перегородку в 4.
Оттуда аммиак можно перегнать обратно в 2. Для проведения качественной
пробы на присутствие вещества в промывном растворе часть его отбирают
в сосуд 3.
Иногда для разделения смеси твердых веществ, обладающих различной
растворимостью в жидком аммиаке (или диоксиде серы), проводят их экст-
ракцию этими жидкостями в приборе, показанном на рис. 79. Исходный
-7<?Г
ЕШПШВИПП
Рис. 79. Устройство аппарата
для проведения экстракции
жидким аммиаком (1—6 см.
текст).
твердый материал помещают в колонку 1 на стеклянный пористый фильтр 2.
Через трубку 5 вводят аммиак и конденсируют его в 1. После растворения
в жидком аммиаке части вещества проводят вытеснение раствора из сосу-
да 1 в 3, для чего при соответствующем положении кранов охлаждающую
баню переносят от колонки 1 к колонке 3. Нерастворившаяся часть веще-
ства остается в колонке 1 на перегородке 2. Туда опять конденсируют ам-
миак, снова охлаждая 1 вместо 3 при открытом соединительном кране. Пу-
тем повторения этих операций можно провести многократную промывку
нерастворимого остатка за счет циркулирования одной порпии жидкого ам-
миака. Газовое пространство в подобной аппаратуре должно быть свободно
от посторонних газов. В противном случае повторная конденсация аммиака
(диоксида серы) будет сильно затруднена. Поэтому аппаратуру время от
времени эвакуируют, одновременно сильно охлаждая растворитель для пред-
отвращения его испарения.
Более простая аппаратура для проведения реакций твердых веществ с
растворами щелочных металлов в жидком аммиаке показана на рис. 80.
Щелочной металл предварительно помещают в небольшие заплавленные ам-
пулы с крючками на конце. После вскрытия ампул их на тонкой проволоке,
прикрепленной к крючкам, вводят в отросток В реакционного сосуда, где
металл выплавляют из ампул и затем туда же конденсируют аммиак. Твер-
дый реагент предварительно помещают в отросток А. Наконец, оба реагента
объединяют, наклоняя прибор. После испарения избытка аммиака продукт
реакции можно поместить в колбочки для анализа, капилляры или сосуд
для хранения (рис. 56). Аналогичную насадку для фасовки гигроскопичных
124 Методы препаративной химии
продуктов реакции можно также подсоединять к шлифу 6 прибора, изобра-
женного на рис. 79.
Если необходимо работать со сжиженными газами под избыточным
давлением, то до 15 бар пользуются небольшими стеклянными автоклавами,
до давлений около 60 бар — ампулами. Работу при более высоких давлени-
ях следует вести в металлических автоклавах (см., например, [7]). Стек-
лянные автоклавы до давлений 15 бар (рис. 81) имеются в продаже. Для
конденсации аммиака в отсутствие влаги следует крышку автоклава 3 из-
Рис. 80. Устройство простого прибо-
ра для проведения реакций в жид-
ком аммиаке.
Рис. 81. Разрез автоклава для дав-
лений до 15 бар.
1 — реакционный сосуд из стекла; 2 —
прижимное устройство с кольцевой тефло-
новой прокладкой; 3 - крышка из стали;
4 ~ игольчатый вентиль.
готовить из стали. Уплотнение осуществляется за счет сдавливания болта-
ми 2 кольца из тефлона, помещенного между стальной крышкой и плоским
фланцем реакционного сосуда 1. В крышке на резьбе, уплотняемой тефло-
ном, укреплен игольчатый вентиль 4, через который газообразный или жид-
кий аммиак напускается в сосуд 1 (о переливании жидкого аммиака c^t.
ч. II, гл. 7). Содержимое сосуда 1 можно перемешивать при помощи маг-
нитной мешалки.
Ампулы (запаиваемые трубки) изготавливают из приборного стекла 20,
из дюрана 50 и из особо ударопрочного стекла типа дюробакс (Durobax),
которое можно спаивать с приборным стеклом 20. В особых случаях для
изготовления ампул используют кварцевое стекло. Прочность ампул зависит
от их диаметра, толщины стенок и от температуры. При толщине стенок
трубок 1—3 мм и при температурах по крайней мере на 50 °C ниже темпе-
Работа со сжиженными газами 125
ратуры рекристаллизации для данного сорта стекла максимальное давление
в ампуле (в барах) можно приближенно рассчитать по формуле
р(ти2—''в2)
гн2 + гв2
где гя и гв — наружный и внутренний диаметр трубки, см; о — постоянная
для данного материала, равная для кварца максимально 500, для стекла
дюран —400, для других стекол — 300. Поскольку прочность ампулы при:
прочих равных условиях сильно зависит от качества выполнения стеклодув-
Рис. 82. Конструкция ампулы с теф-
лоновым вентилем.
ных работ, то для того, чтобы надежно предотвратить разрушение ампулы,
следует при расчете постоянную с умножить на коэффициент, который
колеблется от 0,5 до 0,2. Так, в книге [8] рекомендуется брать для расче-
тов значение о, равное 68.
Для конденсации газов трубку соединяют на шлифах или через шланг
с вакуумной аппаратурой. Для проведения реакций при комнатной темпера-
туре применяют удобные в работе ампулы (рис. 82) с припаянным тефлоно-
вым вентилем (рис. 36). Тем самым сильно облегчаются дальнейшие мани-
пуляции с продуктом реакции, проводимые в инертной атмосфере. Вентиль
выдерживает давление до 10 бар. Если ожидается, что в ампуле возникнет
более высокое давление, то следует заранее произвести отпайку вентиля.
В работе [1] приведен содержательный обзор «Техника эксперимента с
применением низкокипящих растворителей», а в статье [5] описана совре-
менная аппаратура для проведения кондуктометрического титрования в жид-
ком аммиаке при помощи растворов амида калия известной концентрации.
ЛИТЕРАТУРА
1. Nassler J., The chemistry of non-
aqueous solvents (ed. J. J. Lagow-
ski). Vol. I. Academic Press, New
York — London, 1966, p. 214.
2. Popov I., Techniques with non-
aqueous solvents. In: Technique of
inorganic chemistry (eds. H. B. Jo-
nassen, A. Weissberger). Vol. I. In-
terscience, New York — London,
1963.
3. Non-aqueous solvent systems (ed.
T. C. Waddington). Academic Press,
London, 1965.
4. Chemie in nichtwafirigen ionisiererr-
den Losungsmitteln (Hrsg. G. Jan-
der, H. Spandau, С. C. Addison).
Bd. 1/1: Jander J., Anorganische-
und allgemeine Chemie in fliissigem:
Ammoniak. Friedr. Vieweg, Braun<-
schweig, 1966.
5. Schenk P. IV, Chem.-Ing.-Tech., 38,.
576 (1966).
6. Lux H., Anorganisch-Chemische Ex-
perimentierkunst. J. A. Barth, Lei-
pzig, 1970. [Есть перевод болев'
326 Методы препаративной химии
раннего издания: Лукс Г. Экспери-
ментальные методы в неорганиче-
ской химии. Пер. с нем. — М.: Мир,
1965.]
'7. Juza R., Jacobs Н., Gerke Н., Вег.
Bunsenges. Phys. Chem., 70, 1103
(1966).
8. Shriver D. F., Manipulation of air-
sensitive compounds. McGraw Hill,
New York, 1969.
46. Проведение процессов в электрических разрядах
Различают так называемые «тихие разряды» при атмосферном давлении
я «тлеющие разряды» при пониженном давлении. Другие виды разрядов, на-
пример дуговые, можно здесь не принимать во внимание, потому что в этом
случае специфическое действие разряда перекрывается Термическими эффек-
тами.
Тихие разряды получают в озонаторе Сименса — системе, состоящей из
двух концентрических трубок, между которыми пропускают газ, подлежащий
обработке (см. ч. II, гл. 5, препарат «Озон»),
Тлеющие разряды получают при воздействии высокого постоянного или
'Переменного напряжения на газ, находящийся под пониженным давлением
(менее 10 мм рт. ст.). Ранее при получении разрядов при постоянном токе
или низкочастотных разрядов применяли разрядные трубки с впаянными в
них электродами (алюминий, железо), соединенными с мощным источником
высокого напряжения (около 6 кВ, 100—200 мА). Однако вследствие того,
•что электроды могут химически или каталитически взаимодействовать с ве-
ществами, образующимися при разряде, а места впаивания металлических
электродов, кроме того, чувствительны к воздействию термических и меха-
нических нагрузок, в настоящее время работают с «безэлектродными» раз-
рядами. При этом используемый в качестве источника энергии высокочастот-
ный генератор либо подключают к колебательному контуру, работающему в
области радиочастот (РЧ, например, 27 МГц), либо при помощи магнетро-
на или клистрона ои продуцирует излучение в микроволновой области (МВ,
например, 2,5 ГГц). В соответствии с этим различают РЧ- и МВ-разряды.
Соответствующие генераторы, применяемые в промышленности и в медици-
не, имеются в продаже.
Для получения РЧ-разряда (собственно высокочастотного разряда) ци-
линдрическую разрядную трубку подсоединяют к генератору в качестве ин-
дуктивной или емкостной нагрузки (индукционный и емкостный разряд, см.
рис. 83). Лучше всего изготовлять разрядные трубки из кварцевого стекла,
так как кварц в наименьшей степени поглощает высокочастотную энергию
и поэтому лишь незначительно нагревается. Впуск газа осуществляют через
капилляр, что ограничивает зону распространения разряда, а продукты
•обычно собирают непосредственно за разрядной зоной путем их конденсации
жидким азотом. Разрядную трубку диаметром 10—50 мм либо оборачивают
несколькими витками толстой медной проволоки (рис. 83, с), либо снабжают
двумя медными манжетами шириной 10 мм (рис. 83,6). Эти «электроды»
• соединяют с генератором при помощи коаксиального кабеля, одну из жил
которого обычно заземляют. Подстройка разрядной системы к генератору
производится при помощи переменных конденсаторов или индуктивностей
при их включении в соответствии со схемами на рис. 84. При изменении со-
става газа или его давления (при прочих равных условиях) следует произ-
вести дополнительную подстройку. В обычно используемых система?? с
РЧ-генераторами отдаваемая мощность составляет 350 Вт и более при на-
пряжении 1—3 кВ. Применяя РЧ-заряды при меньших напряжениях, можно
осуществлять более мягкое возбуждение газов.
При использовании MB-разрядов обычно отпадает проблема подстройки.
Для передачи энергии излучения кварцевой трубке применяют полый резо-
натор, в котором продуцируются стоячие волны. Разрядная трубка про-
Проведение процессов в электрических разрядах 127
ходит через резонатор в месте с максимальной напряженностью электричес-
кого поля (при Х/4 или ЗХ/4), причем ось трубки направлена перпендикуляр-
но к направлению распространения электрических волн, т. е. параллель-
но вектору электрического поля. Таким путем удается в сравнительно не-
большом объеме достичь подвода энергии до 400 Вт (включая диэлектричес-
кие потери).
Химические реакции в разряде начинаются с возбуждения и диссоциа-
ции молекул исходного сооединения (или соединений) посредством электрон-
ного удара (плазмолиз). Так, из молекулы Н2, О2, N2 или С12 образуются;
соответствующие атомы, иногда в высокой концентрации (степень диссоциа-
ции до 90%), а из молекул простых соединений, например Н2О, SO2, CS2„
СС14 — соответствующие осколки (ОН, SO, CS, СС13 и СС12). Внутри разряда
Рис. 84. Способы включения для под-
стройки разрядной трубки к РЧ-генера-
тору.
а — индукционный разряд: б — емкостный раз-
ряд. Разрядная трубка подсоединяется в Е.
И И
Рис. 83. Схемы получения высоко-
частотного (РЧ) разряда.
а — «индукционный» способ присоеди-
нения разрядной трубки к генератору;
б —* «емкостный» способ подключения,
М — медные манжеты.
и непосредственно за ним протекают реакции и процессы рекомбинации ато--
мов, ионов и радикалов между собой.
При низких давлениях газа между находящимися в разряде молекула-
ми, ионами и электронами термическое равновесие не устанавливается. Сред-
няя кинетическая энергия молекул и ионов соответствует температуре 20—
200 °C. Энергия же электронов вследствие их незначительной массы намно-
го больше и соответствует 104—10s °C.
Путем передачи энергии от ускоренных н электрическом поле электро-
нов сравнительно холодному газу уже при комнатной температуре идут
реакции, протекание которых иначе было бы возможно лишь при пиролизе
при очень высоких температурах. Наряду с техническими преимуществами
работы с почти холодными снаружи реакционными трубками определенным
недостатком метода являются необходимость создания пониженного давле-
ния и, как следствие, малые количества вводимых в реакцию веществ.
С другой стороны, низкие давления обусловливают большие скорости пото-
ка газов, что наряду с низкой температурой стенок облегчает изоляцию;
неустойчивых продуктов реакции (в случае необходимости разрядную труб-
ку погружают в охлаждающую баию).
Для препаративных целей плазмохимические методы используют тогда,,
когда должны быть получены неустойчивые или энергоемкие продукты или;
если применение этих экспериментально непростых методов оправдано от-
сутствием других сравнимых по эффективности способов получения. Из срав-
нительно устойчивых, изолируемых в чистом виде соединений можно назвать,
некоторые, получаемые преимущественно с использованием электрических
разрядов: О3 (из О2), В2С14 (из ВС13), C3S2 (из CS2 через CS), SCSe (из.
CS2 и Se), SCTe (из CS2 и Те), полипероксид серы (из SO2 и О2), фторш-
ды инертных газов (из инертного газа и фтора).
128 Методы препаративной химии
ЛИТЕРАТУРА
1. McTaggart F. К., Plasma chemistry
in electrical discharges. Elsevier,
Amsterdam, 1967.
2. Jolly W. L., The use of electrical
discharges in chemical syntheses.
In: Technique of inorganic chemist-
ry (eds. H. B. Jonassen, A. Weiss-
berger). Vol. I. Interscience, New
York — London, 1963.
3. Hollahan J. R., J. Chem. Educ., 43,
A497, A401 (1966).
4. Rummel Th., Hochspannungsentla-
dungschemie und ihre industrielle
Anwendung. Oldenbourg. Mflnchen,
1951.
17. Очистка веществ
Очистку веществ осуществляют чаще всего путем их перегонки, субли-
мации или перекристаллизации (зонную плавку можно рассматривать как
-особый вид кристаллизации). В некоторых случаях для разделения порошко-
образных смесей прибегают и к механическим приемам—-отмучиванию и
разделению по плотности. Небольшие количества летучих или растворимых
веществ могут быть разделены методами препаративной газовой, тонкослой-
ной или колоночной хроматографии.
Эффективность операций по очистке можно контролировать аналитичес-
кими методами, путем определения физических констант или спектроскопи-
чески (см. разд. 6 этой части). Высушивание веществ как особенно важный
метод их очистки рассматривается в первую очередь.
Высушивание
Методы высушивания газов и органических растворителей обсужда-
лись ранее (разд. 14 и 6 соответственно). Летучие твердые или жидкие
вещества можно высушивать сходными методами путем непосредственного
контакта с осушителем с последующей декантацией, перегонкой или субли-
мацией или за счет изотермической перегонки воды от вещества к осушителю
в эксикаторе, а при необходимости в вакууме. В случае непосредственного
соприкосновения осушитель, часто обладающий кислыми или основными свой-
ствами, выбирают таким образом, чтобы исключить возможность протекания
химического взаимодействия между ним и осушаемым веществом. При изо-
термической перегонке вещество и осушитель рассыпают по возможности
тонкими слоями для увеличения их поверхности. Понижение общего давле-
ния повышает скорбеть изотермической перегонки, зависящей от скорости
диффузии паров воды. Для веществ, устойчивых к нагреванию, можно вос-
пользоваться обогреваемым эксикатором или «сушильным пистолетом», что
особенно рекомендуется для удаления адсорбционно связанной воды. Неле-
тучие вещества весьма эффективно осушают в вакуумных сушильных шка-
фах или путем их откачки при помощи ротационных масляных насосов. Если
для эвакуирования эксикаторов или вакуумных сушильных шкафов приме-
няют водоструйный насос, то следует избегать слишком долгой откачки, так
как это ведет к обратной диффузии паров воды из насоса, что ухудшает
степень осушки.
При проведении высушивания в высоком вакууме, особенно необходимо-
го в случае термически малоустойчивых веществ, важно обеспечить хоро-
шую теплопередачу от среды к образцу (тонкий слой образца). Вследствие
затраты теплоты испарения (сублимации) воды образец сильно охлаждается,
что ведет к замораживанию в нем воды (вымораживающая сушка), к рез-
кому понижению ее давления пара и замедлению процесса высушивания.
В любом случае необходимо перед масляным насосом помещать вымора-
живающую ловушку. Таким путем можно в мягких условиях удалять из ве-
щества и органические растворители. Ниже перечислены часто употребляе-
мые осушающие вещества:
Очистка веществ 129
Кислотные
Нейтральные
Основные
Р4Ою, H2SO4
Na2SO4, MgSO4, CaSO4-0,5H2O, CuSO4, CaCl2, Mg(ClO4)2,
Ba(C104)2, Y-AI2O3, силикагель
BaO, CaO, MgO, KOH, K2CO3, CaH2, CaC2, Na, Ca, сплав
Na — К, сплав Na — Pb, амальгамы натрия, кальция, алю-
миния
Перегонка
Простой перегонкой можно разделить лишь смеси, имеющие в своем
составе только один летучий компонент, который и необходимо выделить.
С некоторым трудом разделяется смесь нескольких летучих веществ с сильно
различающимися температурами кипения. В общем случае смесь нескольких
летучих веществ (чаще всего двух) разделяют многократной фракционной
перегонкой, приводящей, однако, к значительной потере вещества, но лучше
ректификацией с применением колони. Выбор подходящей ректификацион-
ной колонны зависит от требуемой степени разделения (число «теоретичес-
ких тарелок»), количества вещества (производительность колонны и объем
удерживаемой в ней жидкости) и свойств веществ (число компонентов и их
температуры кипения, возможность разложения, коррозии, катализа). Обыч-
но сначала пытаются провести разделение на простейшей в эксплуатации ко-
лонне из имеющихся в распоряжении. Число «теоретических тарелок» колон-
ны зависит от ее нагрузки (суммарный объем флегмы и дистиллата) и при
возрастании производительности (см3/ч) обычно падает. Это связано с тем,
что при увеличении скорости пара ухудшаются условия установления рав-
новесного распределения концентрации между паром и конденсатом. С дру-
гой стороны, проведение перегонки при малой производительности колонны
требует ее очень хорошей изоляции.
Простая полая трубка длиной /ми диаметром 30—60 мм (или лучше
связка трубок диаметром 5—10 мм) имеет небольшой объем удерживаемой
жидкости и лишь при незначительной производительности может обеспечить
достаточное число «теоретических тарелок». Эффективность перегонки через
полую трубку можно повысить за счет «наколок» (колонна Вигре). Хорошая
колонна Вигре для ее теплоизоляции должна иметь эвакуированную и по-
серебренную рубашку. Такие колонны используют для проведения ректифи-
кации при пониженном давлении (рис. 85).
Часто пользуются спиральной колонной Видмера из-за ее небольших
размеров, простоты в обращении и легкости очистки, хотя эффективность
такой колонны невелика. Распределение температур в ней противоречит
принципу адиабатического противотока, согласно которому температура па-
ра и конденсата вследствие адиабатического расширения в колонне должна
понижаться снизу вверх.
Более эффективно, чем описанные выше, работают колонны с насадкой,
представляющей собой по возможности мелкозернистый материал, свободно
насыпанный в колонну. Насадка может быть изготовлена из стекла, фарфо-
ра или металла (бронза, медь, сталь). Стеклянные кольца Рашига не так
эффективны, как кольца из проволочной сетки. Применение последних ведет,
однако, из-за их большой поверхности, по которой растекается пленка жид-
кости, к сильному увеличению объема удерживаемой жидкости в колонне.
Кроме того, широкое применение в качестве насадки нашли фарфоровые
бусинки седловидной формы, разнообразные металлические спирали и др.
Очень важно обеспечить идеальное смачивание поверхности колонны и на-
садки, которое иногда нарушается за счет проникновения в колонну силико-
новой смазки из шлифов. При необходимости колонну ополаскивают сма-
чивающим составом или сильно разбавленным раствором плавиковой кисло-
ты, а шлифы смазывают порошком сульфида молибдена.
В лаборатории обычно работают с колоннами диаметром 10—30 мм и
высотой 50—100 см. Путем увеличения флегмового числа (отношение объема
9—143
130 Методы препаративной химии
флегмы к объему дистиллата) можно увеличить число «теоретических таре-
лок». Обычно на 3—4 капли флегмы приходится одна капля дистиллата, но
в особых случаях это соотношение может достигать 10—50, и тогда лучше
использовать колонну с большим числом «теоретических тарелок». Для ре-
гулирования флегмового числа служат разнообразные головки, имеющиеся
Рис. 85. Колонна Вигре.
Рис. 86. Устройство прибора для
сублимации в вакууме.
в продаже. Критерием однородности отбираемой фракции служит постоян-
ство температуры в верхней части колонны.
Очень небольшим объемом удерживаемой жидкости отличаются колон-
ны с вращающейся лентой, которые выпускаются длиной от 40 до 100 см
и предназначены для работы с количеством веществ от 2 до 100 мл. У этих
колонн, применяемых также для вакуумной перегонки число «теоретичес-
ких тарелок» несколько возрастает при увеличении скорости вращения лен-
ты (из стали или тефлона). Их разделяющая способность сопоставима с
соответствующими характеристиками насадочных колонн той же длины. '
Наконец следует указать на метод испарения в тонком слое и на рас-
ширение возможностей препаративной работы при перегонке с водяным
паром. Перегонка сконденсированных газов при низких температурах была
вкратце упомянута в разд. «Специальная вакуумная аппаратура»; соответ-
ствующая аппаратура имеется в продаже.
Очистка веществ 131
Перегонка в вакууме
Обычные приспособления для перегонки с водоструйным насосом общеиз-
вестны. Неприятные явления, связанные с перегреванием жидкости, устраня-
ют применением кипятильников, капилляров или путем перемешивания со-
держимого колбы. Для поддержания постоянства давления можно восполь-
зоваться простыми ртутными маностатами (см. рис. 38), имеющимися в про-
даже, которые, однако, быстро загрязняются и становятся ненадежными.
Применяют и более сложные и дорогие приспособления для регулирования
давления.
Перегонка при давлении от 760 до 1 мм рт. ст. обычно сопровождается
кипением вещества; напротив, в более высоком вакууме происходит испаре-
ние с поверхности тонкого слоя жидкости или твердого вещества. При этом
между паром и жидкостью не устанавливается термического равновесия,
т. е. давление в системе не равно давлению насыщенного пара. Тем самым
становится возможным перегонять в мягких условиях особенно малоустой-
чивые соединения. Молекулярная перегонка с небольшим расстоянием
между поверхностями испарения и конденсации производится при очень
низких давлениях и при относительно низких температурах. При этом важ-
но добиться того, чтобы молекулы вещества, перешедшие в пар, достигали
поверхности конденсации и там осаждались, не испытывая соударений в га-
зовом пространстве. Для этого средний свободный пробег молекулы должен
быть больше, чем расстояние между испарителем и конденсатором. При всех
видах вакуумной перегонки необходимо предварительно удалить из жид-
кости растворенные в ней газы, так как в противном случае может произой-
ти интенсивное вспенивание жидкости. Аппаратура для перегонки в тонком
слое и для молекулярной перегонки также имеется в продаже.
Сублимация
Кроме общеизвестных приспособлений для сублимации, из которых
простейшим является стакан со вставленной в него колбой, наполненной во-
дой, существуют приборы для проведения сублимации в вакууме (рис. 86);
их легко изготовить и самостоятельно. В таком приборе сублимацию можно
проводить при любой, в том числе низкой температуре. Для этого охлаждае-
мый палец заполняют охлаждающей жидкостью, например жидким азотом
или смесью сухого льда с метанолом, а сосуд с веществом погружают в ох-
лаждающую или нагревающую баню.
Перед проведением сублимации вещество необходимо хорошо измель-
чить, поскольку скорость испарения пропорциональна площади поверхности,
и, кроме того, должна быть обеспечена достаточная теплопередача от нагре-
тых стенок прибора к веществу. Сублимацию можно проводить при атмо-
сферном давлении в неподвижном инертном газе или в потоке газа-носите-
ля. Вообще наличие инертного газа препятствует диффузии молекул пара и
поэтому часто работы проводят в вакууме. При этом, однако, давление пара
вещества не должно быть настолько большим, чтобы небольшие кристаллы
могли увлекаться в конденсационное пространство.
Очень малые количества вещества можно сублимировать с поверхности
предметного стекла на покровное стекло, несколько приподнятое над образ-
цом за счет прокладки в виде стеклянного кольца.
Вакуумную сублимацию часто применяют и для очистки металлов. Необ-
ходимые для проведения этого процесса части прибора, за исключением от-
сасывающего насоса высокой производительности (так как большинство ме-
таллов выделяет при нагревании значительные количества газов), сравни-
тельно несложны. Для него необходима кварцевая, керамическая или ме-
таллическая (например, стальная) трубка большого диаметра, в которую
возгоняемый металл помещают либо непосредственно, либо в тигле. Осажде-
ние металла происходит на вставном охлаждаемом пальце, через который
циркулирует вода. По окончании сублимации вся конструкция должна легко
9*
132 Методы препаративной химии
разниматься (рис. 87). Необходимая для этого установка сходна с описанной
выше печью с вольфрамовой трубкой (рис. 16), в которой, однако, трубча-
тый нагреватель заменен на открытую лодочку, выгнутую из тонкой вольфра-
мовой фольги.
Охлаждаю-
щая Вода
К Вакууму
Рис. 87. Схема прибора для субли-
мации при высокой температуре.
Хроматографическое разделение [1—8]
Из различных хроматографических процессов, которые могут быть рас-
смотрены здесь лишь кратко, наиболее доступна давно известная колоночная
хроматография. Область ее применения, так же как и препаративного вариан-
та тонкослойной хроматографии, распространяется лишь на растворимые ве-
щества. Принцип обоих методов состоит в том, что раствор смеси веществ
пропускают через адсорбент, помещенный в колонку (рис. 10) или распре-
деленный в виде тонкого слоя на стеклянной пластинке. При этом смесь
веществ разделяется на зоны, которые далее могут быть изолированы после
проявления или элюированы при помощи другого растворителя (или смесн
растворителей). В качестве адсорбентов применяют вещества, перечисленные
в табл. 24. Элюентами служат обычные органические растворители и вода,
а при тонкослойной хроматографии — преимущественно смесн растворителей.
Большим достоинством этих методов является весьма эффективное (иногда
уже после проведения одного цикла) разделение веществ в количестве от
0,1 до 100 г, не сопровождающееся их потерей. О подробностях этого ме-
тода можно справиться в приводимой ниже литературе. Следует лишь ука-
зать на то, что элюирующая способность растворителя, как правило, возра-
стает при увеличении его полярности в следующем ряду: петролейный эфир,
циклогексан, СС14, СН2С12, СНС13, диэтиловый эфир, этанол, метанол, вода,
пиридин. Выбор растворителя всего лучше производить путем предвари-
тельных опытов с небольшими количествами веществ.
Таблица 24. Сорбенты для колоночной и тонкослойной хроматографии
Силикагель
Оксид алюминия
Животный уголь
Карбонат кальция
Инфузорная земля
Ионообменники
Целлюлоза и ее производные
Силикат кальция
Гипс (CaSO4-2H2O)
Фосфат кальция
Фторид кальция
Оксид кальция
Карбонат калия
Карбонат натрия
Карбонат магния
Оксид магния
Стеклянный порошок
Оксид хрома (III)
Тальк
Сахароза
Крахмал
Маннит
Полиамиды
Очистка веществ 133
При помощи препаративной газовой хроматографии на соответствующих
приборах можно провести разделение приблизительно 2 мл летучих веществ
за одну операцию. Компоненты газовой смеси в потоке газа-носителя раз-
дельно вымораживаются в спиральной глубокоохлаждаемой ловушке, причем
за счет туманообразования и неполной конденсации могут происходить не-
которые (иногда значительные) потери вещества. Перспективность разделе-
ния этим методом, а также времена задержки предварительно исследуют на
каком-либо простом хроматографе.
ЛИТЕРАТУРА
1. Diinnschichtchromatographie (Hrsg.
Е. Stahl). Springer, Berlin, 1967.
2. Randerath K„ Diinnschichtchroma-
tographie. Verlag Chemie, Wein-
heim/Bergstr., 1962.
3. Halpaap H., Chemiker-Ztg., 89, 835
(1965).
4. Balogh B., Anal. Chem., 36, 2498
(1964).
5. Visser R„ Anal. Chim. Acta, 38,
157 (1967).
6. Kaiser R„ Chromatographic in der
Gasphase. Bd. I—IV, Bibliographi-
sches Institut Mannheim, 1960/65,
S. 1973.
7. Houben—Weyl, Methoden der orga-
nischen Chemie. Bd. II, Thieme,
Stuttgart, 1953.
8. Brandt W. W„ Gas chromatography.
In: Technique of inorganic chemist-
ry (eds. H. B. Jonassen, A. Weiss-
berger). Vol. III. Interscience, New
York —London, 1963.
Перекристаллизация
Процесс кристаллизации в существенной степени зависит от двух фак-
торов: от скорости кристаллизации и от числа зародышей кристаллизации,
причем оба фактора сложным образом зависят от температуры при переох-
лаждении (т. е. от пересыщения). В сильно пересыщенных растворах число
зародышей кристаллизации велико, вследствие чего происходит образование
множества мелких кристаллов. В тех случаях, когда раствор пересыщен лишь
в незначительной степени, процесс определяется скоростью кристаллизации.
В этих условиях образуется лишь незначительное число кристаллов, которые
зато отличаются крупными размерами. Мнение, что особенно красиво обра-
зованные кристаллы отличаются и высокой степенью чистоты, неверно, так
как их загрязнение может быть обусловлено включениями маточного раство-
ра. На чистоту кристалла также оказывают влияние образование смешанных
кристаллов, адсорбция примесей на гранях и ребрах и на границах зерен.
Вкратце остановимся на получении более крупных кристаллов, имеющем
важное значение для кристаллографии.
Выращивание кристаллов
Крупные монокристаллы можно получить из газовой фазы, из раство-
ра, из расплава или путем рекристаллизации.
Выращивание кристаллов из газовой фазы (возгонка) осуществляется
путем запаивания вещества в продолговатые стеклянные или кварцевые ам-
пулы и поддержания в течение определенного времени по длине этих ампул
некоторого градиента температуры. Осаждение кристаллов происходит за
счет явления транспорта. Применение данного метода весьма многообразно,
если сюда включить реакции обратимого разложения с участием газовой
фазы*, а также синтез из газообразных компонентов. Таким путем в основ-
ном в хорошо закристаллизованном виде получают различные металлы, гало-
гениды и халькогениды металлов и др. Часто, однако, этот метод применим
для получения лишь небольших количеств веществ (например, до 1 г). Под-
робности можно найти в цитируемых ниже монографиях.
Химические транспортные реакции. — Прим, персе.
134 Методы препаративной химии
Выращивание кристаллов из растворов производят путем кристаллиза-
ции на заранее полученных зародышах или осколках кристаллов. Кристалл
при этом растет медленнее и будет тем совершеннее, чем меньше величина
пересыщения. Линейная скорость роста не должна превышать 1—2 мм за
сутки (это соответствует построению около 100 плоскостей в секунду). Пред-
почтительно работать при повышенных температурах, так как при этом
меньше вязкость раствора и выше скорость диффузии. При кристаллизации
из неподвижного раствора следует позаботиться о создании условий для
чрезвычайно медленного охлаждения пересыщенного раствора (хорошая теп-
лоизоляция) или о поддержании постоянной температуры при одновремен-
ном медленном испарении растворителя. Процесс можно проводить в сосуде
Рис. 88. Схема установки для выра-
щивания монокристалла из раствора
(с движущимся кристаллом).
Рис. 89. Схема установки для выра-
щивания монокристалла из расплава.
Дьюара, в термостате с медленно понижающейся температурой или при по-
стоянной температуре в эксикаторе с концентрированной серной кислотой,
поглощающей пары воды.
В сосудах большого объема крупные кристаллы получить легче, чем в уз-
ких сосудах. Зародыши кристаллов на дне сосуда являются помехой для
беспрепятственного роста выращиваемых кристаллов; их образование сле-
дует по возможности предотвращать, например проводя процесс в сосуде
с отполированным дном. После каждой кристаллизации следует отбирать
наиболее совершенно образованные кристаллы, которые можно было бы при-
менить для дальнейшего выращивания. Эти кристаллы потом снова погру-
жают в насыщенный при нагревании раствор, причем в момент их погруже-
ния степень насыщения раствора не должна быть полной. Перекладывриие
кристаллов при каждой новой кристаллизации необходимо для их равномер-
ного роста. Отдельный хорошо образованный кристалл можно также подве-
шивать в растворе на ниточке или на проволочке. Не следует допускать,
чтобы небольшие кристаллы падали с поверхности жидкости на выращивае-
Очистка веществ 135
мый кристалл. Поэтому последний защищают от такого попадания на него
кристаллов крышкой.
Можно также приклеивать зародыш кристалла к спице, приводимой во
вращение мешалкой. Постоянное движение кристалла или раствора весьма
способствует равномерному росту, так как при этом устраняется понижение
концентрации раствора вблизи кристалла. Таким образом, для получения хо-
рошо образованных крупных кристаллов вовсе не обязательно считавшееся
ранее важным условие сохранения полной неподвижности при охлаждении.
Кристалл либо поворачивают в неподвижном растворе, либо дают возмож-
ность жидкости протекать мимо неподвижного кристалла. Для первого слу-
чая предложен простой прибор, изображенный на рис. 88. Поток воздуха
регулирует скорость испарения растворителя. Над выращиваемым кристал-
лом расположена защитная крышка. Для второго случая с подвижной
жидкостью весьма пригодным оказался простой прибор в виде помещенной
в термостат с постепенно понижающейся температурой круглодоиной колбы,
находящейся в наклонном положении и приводимой в медленное вращение
вокруг ее оси.
Кристаллы малорастворимых соединений можно выращивать методом
гидротермального синтеза (например, кварц) или методом диффузии (напри-
мер, BaSO< из Ва(0Н)2 и Na2SO4), при котором ионы компонентов из двух
сосудов, наполненных соответствующими насыщенными растворами, диффун-
дируют в реакционную среду (в данном случае в воду).
При выращивании кристаллов из расплава следует избегать одновремен-
ного роста кристаллов вокруг значительного числа центров кристаллизации.
Следует позаботиться о том, чтобы выращиваемый кристалл вследствие со-
прикосновения с проводником тепла (с охлаждаемой трубкой или стержнем,
отличающимся хорошей теплопроводностью) всегда являлся самым холодным
местом в системе. Метод, особенно пригодный для выращивания кристаллов
галогенидов щелочных металлов, предложен Киропоулосом [9]. Расположе-
ние при этом методе частей прибора показано на рис. 89. Расплав в тигле,
нагреваемом электрическим током, сначала следует довести до температуры
выше точки плавления приблизительно на 150 °C, а затем путем погружения
в него охлаждающего стержня охладить до температуры выше точки плав-
ления приблизительно на 70 °C. Лишь после этого начинают интенсивно ох-
лаждать передатчик тепла. И когда на его кончике образуется кристалл,
обыкновенно имеющий полусферическую форму, этот кристалл на передаю-
щей тепло трубке осторожно поднимают при помощи микрометрического
винта на такую высоту, чтобы он едва лишь касался поверхности расплава.
Тогда, начиная от этой точки, образуется более крупный, очень правильно
образованный округленной формы кристалл (охлаждение передающей тепло
трубки необходимо при этом усилить). Наконец, этот кристалл следует так-
же поднять из расплава и очень осторожно охладить.
Выращивание металлических монокристаллов может быть проведено мно-
гими различными методами. Для медленного затвердевания соответствующих
расплавов Тамман и Бриджмен [12, 13] предложили весьма целесообразно
сконструированные приборы. Наполненную расплавом трубку равномерно и
медленно, например при помощи часового механизма, опускают через постав-
ленную вертикально электрическую трубчатую печь. Чтобы добиться начала
процесса кристаллизации в определенном месте и только от одного центра,
нижний конец трубки изготовляют в виде капилляра.
В процессе вытягивания из жидкого расплава при равномерном мед-
ленном движении (например, 0,2 мм/с) образуется монокристаллическая
металлическая нить, которая в потоке индифферентного газа (азота, диокси-
да углерода) охлаждается и предохраняется от окисления. Плавающий на
поверхности расплава продырявленный посередине листочек слюды определя-
ет диаметром своего отверстия толщину монокристаллической нити [16—18].
Расплавы могут также служить растворителями для других веществ. При
136 Методы препаративной химии
остывании такого «раствора в расплаве» идет кристаллизация вещества до
того, как расплав застынет (например, получение BaTiOs из раствора в рас-
плавленном фториде калия).
К специальным методам можно отнести метод рекристаллизации с по-
переменным чередованием механической деформации и отжига (до сих пор
этот метод применялся для некоторых металлов, полупроводников и окси-
дов), а также метод выращивания, по которому летучее соединение метал-
ла разлагают на сильно нагретой проволоке, что ведет к осаждению соответ-
ствующего металла (или неметалла). Этот метод, называемый также про-
цессом ван Аркеля и де Бура [20, 21], служит для получения некоторых
металлов, которые другим путем в столь чистом состоянии получить нельзя
(титан, цирконий, гафний, ниобий, тантал и др., см. также выше: реакции в
парах).
Небольшие кристаллы, особенно хорошо подходящие для проведения
рентгеноструктурных исследований, часто можно случайно обнаружить в
усадочных раковинах застывших расплавов. Такие же явления удается в
отдельных случаях вызвать и искусственно тем, что в расплавы металлов и
сплавов помещают клубочки согнутых из жести полосок. Просветы и ячей-
ки в этих клубочках образуют как бы усадочные раковины. При этом необ-
ходимо в продолжение медленно протекающего затвердевания расплава
двигать сосуд взад и вперед.
ЛИТЕРАТУРА
1. Smakula A., Einkristalle. Springer,
Berlin, 1962.
2. Laudise R. A.. Hydrothermal syn-
thesis of single crystals. In: Pro-
gress in inorganic chemistry (ed.
F. A. Cotton). Vol. III. Interscien-
ce, New York — London, 1962.
3. White E. A. D., The growth of oxi-
de single crystals from fluxed
melt. In: Technique of inorganic
chemistry (eds. В. H. Jonassen, A.
Weissberger). Vol. IV. Interscien-
ce, New York — London — Sidney,
1965.
4. Шефер Г. Химические транспорт-
ные реакции. Пер. с нем. —М.:
Мир, 1964.
5. Nassau К... Crystal growth techni-
que. In: Technique of inorganic
chemistry (eds. H. B. Jonassen,
A. Weissberger). Vol. VII. Inter-
science, New York — London —
Sidney, 1968.
6. Buckley И. E., Crystal growth.
New York — London, 1951.
7. Neuhaus A., Chem.-Ing.-Tech., 28,
155, 350 (1956).
8. Short M. A., The industrial che-
mist, 33, 3 (1957).
Отдельные процессы
9. Nyropoulos S., Z. Anorg. Allgem.
Chem., 154, 310 (1926).
10. Forth K., Z. Phys., 84, 677 (1933).
11. Schoeneck H„ Verleger H., Metall-
wirtsch., 26, 576 (1939).
12. Tammann G.. Lehrb. d. Metallo-
graphie. Leipzig, 1921, S. 13.
13. Bridgman P. W., Proc. Amer.
Acad. Sci., 60, 306 (1925).
14. Obremou I., Schubnikow L., Z.
Phys., 25, 31 (1924).
15. Straumanis M., Z. Physik. Chem.
(A), 147, 163 (1930).
16. Czochralsky J., Z. Physik. Chem.,
92, 219 (1918).
17. Gomperz E., Z. Phys., 8, 184
(1922).
18. Mark H., Polany M., Schmid E„
Z. Phys., 12, 60 (1923).
19. Koref F., Hoffmann H„ Fischvoigt
H., Z. Elektrochemie, 28, 511
(1922).
20. Van Arkel A. E., Physica, 3, 76
(1923).
21. Van Arkel A. E., de Boer J. H„ Z.
Anorg. Allgem. Chem., 148, 345
(1925).
22. Agte C., Moers R., 2. Anorg. All-
gem. Chem., 198, 233 (1931).
23. Van Arkel A. E., Metallwirtsch.,
13,511 (1934). .
24. Burgers W. G., Basart J. С.) M.,
Z. Anorg. Allgem. Chem., 216, 209
(1934).
25. Van Arkel A. E„ Reine Metalle.
Springer, Berlin, 1939.
Очистка веществ 137
Зонная плавка [1—3]
Этот способ очистки впервые был предложен Пфанном [1] для метал-
лов и полупроводников, но вообще он применим для всех веществ, плавя-
щихся без разложения. Температуры плавления веществ могут быть от ми-
нусовых до очень высоких.
Принцип метода состоит в продвижении образца вытянутой формы че-
рез короткую кольцеобразную зону нагрева, позади которой вещество сразу
же охлаждается. Образующаяся тонкая зона расплава постепенно переме-
щается через весь образец. Примеси, лучше растворимые в жидкой фазе,
чем в твердой, оказываются сконцентрированными в расплаве и вместе с ним
Рис. 90. Устройство прибора для
зонной плавки веществ, жидких при
комнатной температуре (по Рёку).
1 — отверстие; 2 — нагреватель; 3 — вещест-
во; 4— хладагент; S — жидкость, передаю-
щая тепло.
Рис. 91. Простое приспособление
для проведения «нормального за-
твердевания» жидкостей.
/ — жидкая фаза; 2 —твердая фаза; 3 —
охлаждающая баня; 4 — охлаждающий
рассол.
перемещаются к одному из концов образца. И наоборот, компоненты, лучше
растворимые в твердой фазе, концентрируются (после проведения нескольких
циклов) в противоположном конце образца. На эффект разделения основное
влияние оказывает коэффициент распределения данной примеси. Обычно
располагают друг за другом от 5 до 15 горячих зон, разделенных холодны-
ми зонами. Таким образом обеспечивается одновременное медленное про-
движение через образец 5—15 расплавленных зон, что позволяет достичь
высокой степени очистки основного вещества практически без его потерь.
Этот метод дает хорошие результаты при очистке металлов, полупроводни-
ков (Si, Ge), солей (KNO3, AgBr, Na2SO4), ковалентных соединений (нафта-
лин, сера) и даже тугоплавких оксидов (ТЮ2, шпинели).
Обычно образец помещают в лодочку или трубку из стекла, кварца,
оксида алюминия или другого индифферентного материала. Компактный об-
разец можно пропускать в вертикальном положении через нагреваемую зону
и без какой-либо поддерживающей внешней оболочки. Сложность экспери-
мента в основном зависит от температуры плавления вещества. В качестве
нагревателей используют термостаты, печи сопротивления, индукционные пе-
чи, а также источники теплового излучения или ускоренных электронов.
138 Методы препаративной химии
Для зонной плавки жидких при комнатной температуре веществ используют
приспособление, показанное на рис. 90. Погруженное в охлаждающую баню
вещество замораживают, в то время как через центральную трубку переме-
щают нагреватель, что приводит к медленному продвижению расплавленной
зоны через образец (эту операцию в случае необходимости повторяют не-
сколько раз).
Хороший метод очистки, применяемый также для концентрирования раз-
бавленных растворов в мягких условиях, состоит в направленной кристал-
лизации [3] расплава, осуществляемой с небольшой скоростью («нормальное
затвердевание»). Например, пробирку с бензольным или водным раствором
постепенно (2—5 см/ч) опускают в охлаждающую баню, в то время как
жидкую фазу интенсивно перемешивают. Используя часовой механизм, на
оси малой стрелки которого укреплен ролик со шнуром с подвешенной на
нем пробиркой, можно легко сконструировать необходимое для проведения
процесса приспособление (рис. 91). Дают застыть примерно 90% жидкости,
после чего ее остаток, содержащий примеси, сливают. В случае необходимо-
сти эту операцию повторяют несколько раз. Само собой разумеется, что
большие количества вещества также можно очищать таким способом, на-
пример, в колбе объемом 1 л. Для этого погружают сразу всю колбу в ох-
лаждающую баню и по мере протекания кристаллизации выдвигают лишь
мешалку.
Для проведения «нормального затвердевания» твердых веществ их пол-
ностью расплавляют в вертикальной трубчатой печи. Трубку с образцом мед-
ленно опускают из печи,’ заботясь о создании резкого температурного пере-
пада на границе печи. Этого можно добиться использованием перегородки из
асбеста, но еще лучше опускать трубку из печи непосредственно в соответ-
ствующего размера медную трубку, помещенную в вертикальном положении
в охлаждающую баню. При проведении этой операции также необходимо
перемешивать расплав непосредственно вблизи фазовой границы, что спо-
собствует лучшему установлению равновесия.
ЛИТЕРАТУРА
1. Pfann W. G., Zone melting. John
Wiley, New York, 1958.
2. Schildknecht H., Zonenschmelzen.
Verlag Chemie, Weinheim/Bergstr.,
1964.
3. Stofier R., Pritze B„ Z. Chem., 13,
92 (1973).
Разделение по плотности [1—10]
Среди механических методов разделения, которыми можно воспользо-
ваться в тех случаях, когда химическое отделение продукта реакции невоз-
можно или нецелесообразно, следует особо отметить разделение по плотно-
сти. Две фракции, подлежащие отделению друг от друга и имеющие раз-
личные плотности, можно разделять путем погружения в жидкость, имею-
щую промежуточное значение плотности. Необходимым условием при работе
по этому методу является предварительное растирание вещества в порошок
до такой степени измельчения, чтобы каждое его зернышко по возможности
было однородным и не содержало каких-либо включений, но чтобы при этом
не образовывалось слишком тонкого порошка. В качестве жидкой среды
для разделения, в которой более легкая фракция всплывала бы, а более тя-
желая опускалась на дно, были предложены различные так называемые
«тяжелые жидкости» и «тяжелые растворы». Если при этом допустимо про-
водить процесс разделения при несколько более повышенных температурах,
то в группу жидкостей можно включить также и твердые вещества с не-
высокой точкой плавления и при помощи таких «тяжелых расплавов» еще
несколько расширить набор веществ, применяемых для разделения по этому
Таблица 25. Жидкости, используемые для разделения по плотности (тяжелые
растворы и тяжелые расплавы)
Раствор ^макс’ г/см3 Свойства
Бромоформ 2,9 Очень подвижен, прозрачен, не дей- ствует на руды, несколько чувствите- лен к действию света; дешев
Раствор йодофор- ма в бромоформе 2,9—4,0а Подобен бромоформу, но интенсивно окрашен вплоть до полной непрозрач- ности. При высоком содержании СН1з (т. пл. 119 °C) плавится прн температуре выше комнатной
Тетрабромэтан (жидкость Мутма- на) 2,96 Подобен бромоформу, но устойчивее к действию света. Довольно быстро испаряется
Раствор иодомер- курата калия в во- де (раствор Туле) 3,2 Почти бесцветен, прозрачен, легко получается. (Ядовит! Разъедает ко- жу!) Гигроскопичен; сравнительно вязок. При действии железа и неко- торых оксидов и сульфидов разлага- ется с выделением ртути. Вследствие адсорбции с трудом отделяется от кристаллического порошка
Метилениодид 3,32 В чистом состоянии почти бесцветен; очень подвижен, легко отмывается бензолом; в чистом состоянии прак- тически устойчив. Чувствителен к солнечному свету, нагреванию и дей- ствию менее инертных металлов (алюминия). Сравнительно дорог
Тетрабромметан 3,42 Т. пл. 48 °C
Насыщенный рас- твор боровольфра- мата кадмия (рас- твор Клейна) 3,36 Не ядовит и безвреден; трудно полу- чается, довольно вязок. Разлагается действием железа, цинка, свинца и карбонатов
Насыщенный рас- твор иодомеркура- та бария (раствор Рорбаха) 3,59 Метод получения прост. Не разлага- ется карбонатами; разбавлять следу- ет не водой, а только раствором KI
Насыщенный рас- твор формиата тал- лия Насыщенный рас- твор формиата — малоната таллия 1 : 1 (раствор Кле- ричи) 3,17(12°С) 4,76(90 °C) 4,07(12оС) 4,65(50 °C) 5(95 °C) Наиболее тяжелые из водных раство- ров; легко подвижны, разбавляются водой. Свежеприготовленные раство- ры окрашены в слабожелтый цвет, при более долгом стоянии буреют. Окрашивание может быть устранено путем кипячения разбавленного рас- твора с активированным углем. Син- тез довольно сложен. Плотность сильно изменяется в зависимости от температуры и за счет испарения
140 Методы препаративной химии
Продолжение
Расплав t . °C ПЛ’ й, г/см3 Свойства
Нитрат серебра 198 4,1 Применяется во многих случаях. Возможно разбавление смешиванием с KNO3 или NaNOs
Нитрат ртути(1) 70 4,3 При долгом плавлении происходит разложение, поэтому рекомендуется прибавлять несколько капель HNO3. В присутствии свободных металлов возможно разложение
Нитрат таллия 206 5,3
Нитрат таллия -Ь + нитрат серебра (по Ретгерсу) 70 4,5—4,9а Получают путем растворения соот- ветствующих количеств таллия и се- ребра в HNO3. Возможно разбавле- ние водой, сопровождающееся силь- ным понижением точки плавления. Бесцветный, легко подвижный, проз- рачный расплав. При сильном дейст- вии света образуется пленка серебра, которую снова можно растворить в небольшом количестве HNOS. Суль- фиды разлагают расплав. Силикаты при действии расплава частично раз- лагаются
Нитрат таллия + + нитрат ртути(1) (по Ретгерсу) 76 5,3 Лучший тяжелый расплав; легкопо- движен; прозрачен при разбавлении водой в любом соотношении. При- годен также для сульфидов и сили- катов
8 Зависит от соотношения компонентов в смеси.
методу. В табл. 25 сопоставлены наиболее употребительные вещества, при-
годные для разделения компонентов по их плотности.
Для практического проведения разделения веществ по плотности можно
применять следующие методы.
Тяжелые жидкости и растворы. В простейшем случае применяют высокие
стаканы или обыкновенные лабораторные цилиндры. Вещество перемешива-
ют с жидкостью стеклянной палочкой, после чего смеси дают возможность
разделиться на составные части. После отстаивания более легкие части
можно слить или, еще лучше, вычерпать никелевой проволочной сеткой.
Удобнее же воспользоваться обыкновенной делительной воронкой с краном,
припаянным на расстоянии нескольких сантиметров от начала расширения.
Диаметр отверстия в пробке крана должен быть равен диаметру трубки во-
ронки. Для более тщательного разделения можно воспользоваться прибо-
ром, изображенным на рис. 92, а. Подлежащую разделению смесь помещают
в трубку большого диаметра и через воронку наливают в прибор такое ко1
личество жидкости, чтобы ее уровень достиг верхнего бокового отвода. Пос-
ле многократного перемешивания стеклянной палочкой легкие составные час-
ти всплывают кверху. Затем среднее широкое отверстие прибора закрывают
Очистка веществ 141
пробкой и через воронку наливают дополнительное количество жидкости, что
сопровождается удалением из прибора легкой фракции через сливную труб-
ку. Эту операцию рекомендуется повторить несколько раз. Другие конструк-
ции приборов можно найти в рекомендуемой литературе.
Тяжелые расплавы. Разделение можно осуществлять в пробирке. Эту про-
бирку после затвердевания расплава разбивают и сплав разделяют на части,
содержащие более легкую и более тяжелую фракции. Для более тщательно-
го разделения работу ведут в приборе, показанном на рис. 92, б. В пробир-
Рис. 92. Типы приборов для разде-
ления по плотности (1—4 см. текст).
а — для тяжелых жидкостей и растворов;
б — для тяжелых расплавов.
ке 3 из тугоплавкого стекла находится маленький сосуд 1, присоединенный
на шлифе к насадке 2. В трубку, составленную из частей 1 и 2, вводят
смесь, состоящую из порошка, подлежащего разделению, и вещества, служа-
щего расплавом. Затем части 1, 2 и 3 нагревают (например, на водяной ба-
не), причем расплав должен заполнить трубку 2. После того как произой-
дет разделение, вводят снабженную пришлифованным концом размыкающую
трубку 4, которую плотно вставляют в шлиф в положение, изображенное на
рис. 92, б. Таким путем пространственное разделение погружающегося вниз
и всплывающего вещества улучшается в такой степени, что после затверде-
вания расплава удается путем кратковременного нагревания без труда отде-
лить сосуд 1 от трубок 2 и 4. При разделении, производимом при помощи
тяжелых расплавов, всегда необходимо следить за постоянством темпера-
туры.
ЛИТЕРАТУРА
Общие работы
1. Kaiser Е., Mineralogisch-geologi-
sche Untersuchungsmethoden. In:
Keilhack, Praktische Geologie. Bd.
II. Stuttgart, 1922.
Жидкости,
используемые для разделения
2. Thoulet J., Bull. Soc. Min. France,
2, 17 (1878); Hofle J., Vervuert G„
Zentralbl. Min., 554 (1909).
3. Klein D., C. R. Acad. Sci. (Paris),
93,318 (1881).
4. Rohrbach C., Wied. Ann. Chem,,
20, 169 (1883).
5. Clerici E., Rend R. Accad. naz.
Lincei, 16, 187 (1907).
Расплавы,
используемые для разделения
6. Retgers J. W., Z. Physik. Chem.,
5, 451 (1890); Neues Jahrb., 2,
185 (1889).
142 Методы препаративной химии
Приборы
для разделения по плотности
7. Thoulet J., Bull. Soc. Min. France,
2, 17 (1879).
8. Brogger C. №.. Nenes Jahrb., I,
395 (1885).
9. Laspeyres H., Z. Kristallogr., 27,
44 (1896).
10. Penfield S. L., 2. Kristallogr., 26,
134 (1896).
18. Испытание на чистоту
При проверке чистоты вещества помимо элементного анализа пользуются
определением физических постоянных, если соответствующие величины, а воз-
можно, и их зависимость от температуры точно известны. Наибольшее рас-
пространение в лабораторной практике имеют определения температуры плав-
ления, плотности, показателя преломления и давления пара. Если эти методы
неприменимы, то можно в качестве испытания на однородность подвергнуть
вещество операциям разделения. Для этой цели применяют прежде всего не
требующие значительных затрат времени методы: газовую, тонкослойную хро-
матографию нлн хроматографию на бумаге. Высокой чувствительностью по
отношению к примесям обладают спектроскопические методы. При этом для
характеристики жидкостей (например, растворителей, см. разд. 6) и раство-
ренных веществ наиболее важны электронные спектры. Полезно иметь так-
же инфракрасный и масс-спектр, которые в соответствующем аппаратурном
оформлении могут быть сняты для образцов в твердом, жидком н газооб-
разном состоянии. Оба метода дают возможность проводить качественное и
полуколнчественное определение примесей, что очень облегчает принятие ре-
шения о целесообразности дальнейшей очистки. Например, содержание воды
в твердом препарате легко определяется по широким полосам поглощения
при 1630 н ~3400 см-1 в ИК-спектре. Разумеется, в этом случае следует
иметь в виду, что галогениды щелочных металлов, используемые при приго-
товлении таблеток для ИК-спектроскопии, гигроскопичны. Их применение
для съемки гигроскопичных объектов или для определения воды возможно
только после нх тщательной осушки и лишь прн полном отсутствии воздуха
(отмеривание, растирание с веществом, наполнение пресс-формы проводятся
в сухой камере). Другой возможностью является съемка суспензии вещест-
ва в сухом нуйоле или в другой подходящей жидкости. Подобные жидкости
должны обладать достаточно высокой вязкостью и по возможности малым
собственным поглощением в соответствующей области спектра. В качестве
материала для изготовления окон кювет для съемки ИК-спектров газов и
жидкостей применяют вещества, перечисленные в табл. 26. Если нет необхо-
димости вести съемку в области ниже 600 см-1, то следует пользоваться
сравнительно дешевыми монокристаллами хлорида натрия. Конечно, вещество
не должно реагировать с материалом окон (при необходимости предваритель-
Таблица 26. Материалы, применяемые для изготовления окон кювет
в ИК-спектроскопии
Материал Область спектра, см-1 Материал Область спект- ра, см—1
CaF2 >1000 CsI >200
BaF: >850 AgCl >400
NaCl >600 Иртран 2 (ZnS) >750
KBr >350 KRS-5(TlBr+TH) >260
CsBr >250 Полиэтилен 600—30 \
Испытание на чистоту 143
но проводят проверку в пробирке). Для съемки водных растворов применя-
ют кюветы с окнами из BaF2 и CaF2; материал иртран-2 устойчив даже к
растворам кислот средней концентрации. Для съемки спектров агрессивных
веществ окна изготовляют из стекла (область спектра >4000 см-1), кварца
(>2500 см-1), а также полиэтилена (может применяться и в виде изолиру-
ющей пленки). При использовании AgCl, который так же, как галогениды
щелочных металлов, служит для приготовления таблеток, необходимо учиты-
вать его чувствительность к свету. Нагреваемые кюветы можно (после озпа-
Рис. 94. Конструкция изотеиископа.
Рис. 93. Дифференциальная схема
подключения термопар.
1 — тигель с исследуемым веществом; 2 —
тигель с эталонным веществом; 3 — печь.
комления по литературе с лучшими их конструкциями) изготовить самостоя-
тельно, хотя они н есть в продаже.
Определение температуры плавления особенно важно вследствие просто-
ты его проведения. Правда, в некоторых случаях для веществ с высокими
температурами плавления нельзя использовать обычный метод с капилляра-
ми, погружаемыми в жидкостные бани, или воспользоваться микроскопом
с нагреваемым столиком. Для более высоких температур применяют медный
блок, нагревая образец на воздушной бане. В измеренное термометром зна-
чение температуры следует внести поправку (см. разд. 8).
Температуру плавления в области средних и высоких значений опреде-
ляют также методом дифференциального термического анализа (ДТА). Ско-
рость охлаждения расплава наблюдают при помощи погруженной в него
термопары. Процесс измерения сильно облегчается, если в расплав поместить
еще одну термопару (рис. 93), соединенную* с третьей термопарой, находя-
щейся во втором тигле с эталонным веществом (например, с мелким песком).
Оба тигля (с расплавом и эталоном) находятся в непосредственной близо-
сти друг от друга. Пока расплав и эталон имеют одинаковую температуру,
* По дифференциальной схеме. — Прим, перев.
144 Методы препаративной химии
напряжение обеих термопар взаимно компенсируется. Как только при кри-
сталлизации расплава в результате выделения теплоты кристаллизации воз-
никает разность температур, стрелка милливольтметра отклоняется, и тогда
считывается температура по основной термопаре, которая и соответствует на-
чалу кристаллизации. Этот метод помимо определения температур плавления
используют также для исследования превращений и реакций, сопровождаю-
щихся термическими эффектами. Установки ДТА имеются в продаже.
Показатель преломления жидких веществ измеряют обычным путем на
рефрактометре Аббе. Для твердых веществ проводят сравнение путем наблю-
дения в поляризационном микроскопе со стеклянным порошком или с жидко-
стями, показатель преломления которых известен (имеются в продаже).
Метод измерения давления паров низкокипящих веществ уже рассматри-
вался в разд. 13.' Для веществ, кипящих при температурах выше комнатной,
следует применят^ нагреваемые манометры. В этих случаях в качестве мано-
метрической жидкости можно воспользоваться, например, расплавленным оло-
вом. Сосуд с веществом вместе с небольшим манометром погружают в на-
гревательную баню, причем второе колено манометра присоединяют к ртут-
ному манометру и буферному объему. Давление компенсируют до тех пор,
пока уровень олова в обоих коленах не установится на одной высоте, после
чего производят отсчет по ртутному манометру.
Другим очень остроумным устройством является изотенископ. В этом
приборе само вещество играет роль манометрической жидкости. Вещество
вводят в колбочку (рис. 94), в приборе создают вакуум, жидкости дают не-
много испариться и затем наклоняют прибор таким образом, чтобы часть ве-
щества перелилась в колено манометра. Затем весь прибор доводят до же-
лаемой температуры н регулируют давление так, чтобы жидкость в обоих ко-
ленах установилась на одном уровне. После этого производят отсчет давле-
ния по ртутному манометру. Для работы прн высоких температурах лучше
всего пользоваться манометром с кварцевой спиралью (см. разд. 13), кото-
рую можно нагревать до 500°C (а в особых случаях н выше). Нулевую точ-
ку прибора проверяют после каждой серии измерений. Манометр н подводя-
щие капиллярные трубки снабжают нагревательными обмотками. Подводя-
щие трубки н спираль манометра вовсе не обязательно нагревать до той же
температуры, что н вещество. Они должны быть нагреты лишь настолько,
чтобы предотвратить конденсацию вещества (особенно в спирали)*. Отсчет
давления пара проводят по ртутному манометру после того, как проведена
компенсация давления цо нулевой точке (рис. 32).
Определение температуры кипения в тех случаях, когда нельзя проводить
работу с обычными сосудами н термометром, производится путем экстрапо-
ляции кривой давления пара.
Тензиэвдиометр был первоначально сконструирован для изучения разло-
жения аммиакатов или гидратов. Однако он может быть применен также и
для препаративных целей, например в тех случаях, когда должна быть до-
стигнута определенная ступень разложения с образованием вещества, которое
необходимо выделить. На рис. 95 изображена конструкция этого прибора.
В колбочку 1 возможно меньшего размера помешают вешество, из которого
необходимо удалить определенное количество летучего компонента. Правое
колено • манометра 8 проградуировано в миллиметрах вниз от нулевой точки
(при одинаковых уровнях). Вместимости колбочки 1, различных частей труб-
ки между кранами 5, 6 и 7, а также вспомогательных сосудов 2, 3 и 4 изме-
ряют непосредственно путем взвешивания с водой или косвенным путем с ис-
пользованием газовых законов. Откачивание летучих компонентов из колбоч-
* Это замечание верно для измерения давления ненасыщенного пара. Ес-
ли же измеряют давление насыщенного пара, то температура подводящих
трубок н спнралн должна быть равна или выше температуры вещества.— j
Прим, перев
Испытание на чистоту 145.
кн 1 производится не непрерывно, и, таким образом, количество их можно
измерить прн помощи вспомогательных сосудов 2 и 3. Благодаря наличию,
этих сосудов можно и при малых, и при больших давлениях удалять при-,
близнтельно одно и то же количество (в молях) газообразного компонента.
При низких давлениях используют все три сосуда 4, 3 н 2, при средних—.
4 и 3 или 2 и, наконец, при более высоких давлениях — только 4. После уста-,
новления равновесия кран 5 закрывают и газ откачивают через кран 7 (изо»
Рис. 95. Конструкция тензи-
эвдиометра (1—8 см. текст).
термическое разложение). Потерю в массе вещества измеряют путем взвешщ
вания закрытой краном 5 колбочки 1. Чтобы спять колбочку 1 вместе с кра-.
ном 5, через кран 6 можно впустить сухой воздух.
ЛИТЕРАТУРА
1. Wendtlandt W„ Differential thermal (eds. H. В. Jonassen, A. Weissbert
analysis; Cooper R., Stranks D. R„ ger). Vol. I, VI. Interscience, Nexy.
Vapour pressure measurements. In: York—London—Sidney, 1963, 1966,
Technique of inorganic chemistry
19. Реакции между веществами в порошкообразном
состоянии
Для проведения реакций между двумя твердыми веществами необходим
ма известная подвижность их структурных элементов, а также возможно,
большая поверхность их соприкосновения. Подвижность частиц и реакцион-
ную способность вещества (скорость диффузии) можно увеличить путем по-
вышения температуры или увеличения энергоемкости в результате нарушения
тонкой структуры. Согласно высказанному впервые Тамманом правилу (спра-
ведливому, правда, лишь в грубом приближении), хорошего выхода реакции
при нормальной продолжительности опытов (порядка 10—100% при продол-
жительности реакции от 0,1 до 100 ч) можно достичь лишь в тех случаях,
когда по крайней мере один из твердых компонентов реакции нагрет до тем-
пературы, составляющей 2/3 абсолютной температуры его плавления или вы-
ше. Так, например, быстрого протекания обменной реакции AI2O3 (/пл
2320 К) можно ожидать лишь при температуре не ниже 1550 К (~ 1300°C).
Благоприятную для диффузии большую общую поверхность компонентов
можно обеспечить путем применения тонкоизмельченных порошков, их хоро-
шего перемешивания и механического уплотнения смесей в таблетки. Простое
приспособление для изготовления таких таблеток изображено на рис. 96, а.
Такое приспособление можно легко изготовить самостоятельно из круглого
стального стержня; в продаже имеются также готовые приборы (например,
для формования образцов твердых горючих веществ, применяемых для гра-
10-143
146 Методы, препаративной химии
дуировки калориметров или изготовления таблеток из бромида калия для
спектроскопических измерений). В простейших случаях, чтобы создать необ-
ходимое давление прессования, достаточно винтового пресса соответствующе-
го размера; для получения таблеток большего поперечного сечения под вы-
сокими давлениями прессования пользуются гидравлическими прессами.
Очень часто не удается предотвратить попадания на поверхность образца
следов железа со стенок матрицы. Если это является большой помехой, то
поверхность таблетки можно поскоблить или подвергнуть шлифовке. Можно
также уплотнение таблетки вести при помощи стеклянной палочкн внутри
Рис. 96. Пресс формы для изготовления таблеток (а, б см. текст).
стеклянной трубки, плотно вставленной в прочную металлическую матрицу
(например, латунную). В таком приспособлении со стеклянной трубкой, име-
ющей внутренний диаметр 5—6 мм и внешний 7—8 мм, можно создать дав-
ление до 100 бар, все еще сохраняя целостность трубки.
На рис. 96, б изображена описанная в работе [1] разъемная прессформа,
собираемая из отшлифованных стальных частей и служащая для изготовле-
ния продолговатых прямоугольных таблеток в виде брусков.
ЛИТЕРАТУРА
1. Grube G., Schlecht Н., Z. Electrochemie, 44, 367 (1938).
Часть II. Элементы и соединения
Глава 1.
ВОДОРОД, ДЕЙТЕРИЙ, ВОДА
М. Баудлер (М. Baudler)
Перевод канд. хим. наук С. И. Троянова
Водород Н2 [1—4]
Выпускающийся в стальных баллонах водород получают электролизом или?
из водяного газа.
Электролитический водород представляет собой газ 99,7—99,8%-ной чи-
стоты, который в виде примеси содержит только воздух — и, в частности,
кислород — в количестве, меньшем 0,1%. Этот газ пропускают при 400°C че-
рез трубку, наполненную восстановленным оксидом меди (в виде проволо-
ки), нли, по методу Мейера и Ронге, через описанную в разделе «Азот» (ч. II,.
гл. 7) башню с «активной медью», или прн 30—50 °C через колонку с восста-
новленным катализатором BTS [2] фирмы BASF. Далее водород высушива-
ют хлоридом кальция и пентаоксидом фосфора. Такой водород можно при-
менять для большинства лабораторных целей, так как присутствие незначи-
тельного количества азота (около 0,2%) редко служит помехой. Если по ка-
ким-либо причинам лаборатория не располагает электролитическим водородом
в стальных баллонах, то его можно получить прн помощи прибора, описанного
в разделе «Кислород» (ч. II, гл. 5). Для этого следует, однако, произвести пе-
реключение полюсов в приборе, предназначенном для получения кислорода.
Водород, полученный из водяного газа, содержит заметные количества
примесей оксида углерода, диоксида углерода, кислорода и азота, а иногда-
также AsH3 и Fe(CO)s- Для поглощения диоксида углерода применяют гид-
роксид калия или натронную известь; AsH3 поглощают насыщенным раство-
ром перманганата калия в присутствии избытка твердого КМпО«. Для уда-
ления кислорода газ пропускают, как это описано выше, над нагретой медью-
или раскаленным докрасна платинированным асбестом (способ получения по-
следнего описан в разделе «Платиновые металлы», ч. II, гл. 29), причем од-
новременно происходит термическое разложение Fe(CO)g. Оксид углерода
удаляется при пропускании газа через восстановленный BZS-катализатор
(см. выше), а также путем вымораживания жидким азотом. Вообще для по-
лучения очень чистого водорода следует по возможности исходить из элект-
ролитического водорода.
Дальнейшую очистку водорода проводят, адсорбируя примесн на актив-
ном угле, силикагеле или молекулярных снтах [4]. Для получения наиболее
чистого, совершенно не содержащего воздуха, водорода используют следую-
щие методы:
Способ 1. Нагревание палладия, насыщенного водородом [5]
Губку палладия, полученную путем восстановления днхлорида палладия
в растворе (см. ч. II, гл. 29, «Платиновые металлы»), тщательно промывают
горячей водой и после высушивания сильно прокаливают в пламени паяль-
ной горелки. Еще в горячем состоянии губку помещают в нагретую трубку с
присоединенным к ней манометром и медленно охлаждают в вакууме. Затем
при комнатной температуре через трубку пропускают поток тщательно очи-
щенного и высушенного водорода. Поглощение палладием водорода сопро-
вождается слабым раскаливанием. Путем последующего нагревания до-
10*
Я 48 Глава 1. Водород, дейтерий, вода
200 °C поглощенный водород можно снова выделить, причем прн слабом от-
сасывании его насосом получается равномерный поток очень чистого газа.
Количество выделившегося водорода составляет при нормальных условиях
около 100 мл/г палладия.
Способ 2. Диффузия через палладий или сплав палладия с серебром
£6—13]
Метод днффузнн прн повышенной температуре через металлический пал-
ладий или его сплавы с серебром в основном используется в технике для не-
кие. 97. Разрез электролизера для
получения водорода высокой чистоты
путем электролиза воды с последую-
щей диффузией через сплав палла-
дий —серебро.
1 — стеклянный цилиндр (диаметр 10 см);
2 — концентрические платиновые аноды;
3— диффузионные трубки из сплава пал-
ладий-серебро (100 штук), нижние концы
которых закреплены путем припайки золо-
том к платиновой пластине 4; 5 — тефло-
новое уплотнение; 6 — основание в виде
стальной плиты: 7 — трубка для отвода
водорода высокой чистоты.
прерывного получения больших количеств очень чистого водорода. Получение
водорода в лабораторных условиях удобно осуществлять в приборе для
электролиза н диффузии, устройство которого показано на рнс. 97. Прибор
заполняется разбавленной серной кислотой н работает прн температуре 70 °C
и плотности тока 2 А/см2. Между двумя анодами из платины находятся
100 трубок из сплава палладия с серебром (25% Ag), служащие катодами.
Наружный диаметр трубок равен 1,6 мм прн толщине стенок 7,6-Ю~2 мм.
Прибор работает на постоянном токе (выпрямитель) и дает 2 л водорода в
минуту при содержании примесей менее 1 млн1. Прибор прост в эксплуата-
ции, поскольку не требует дополнительного подогрева, и для его заправки
необходима только дистиллированная вода.
Способ получения непрерывного потока водорода (100 мл/ч), основанный
на диффузии через подогреваемую током палладиевую трубку, описан в [13].
Способ 3. Диффузия через никель [14—16]
Очистку поступающего в продажу водорода проводят в приборе, схема
которого показана на рис. 98. Прибор позволяет получить поток очень чисто-
Водород 149
го водорода при атмосферном давлении. Пять цельнотянутых никелевых
грубок диаметром 2 мм, с толщиной стенок 0,1 мм и длиной 5 м, изогнутых
в виде спиралей, одним концом впаиваются в латунную головку, оканчиваю-
щуюся нормальным шлифом. Для того чтобы трубки было легче вставлять
в головку, нх в течение 2 ч нагревают до 1000 °C в атмосфере водорода, по-
сле чего их можно легко гнуть руками. Другие концы трубок запаивают на-
глухо. Никелевые спирали помещают в кварцевую трубку диаметром 35 мм
;и длиной 1 м. Латунные головки приклеивают на пиценне. В головке с нор-
мальным шлифом имеется игольчатый вентиль, служащий для тонкой регули-
Рис. 98. Конструкция прибора для очистки водорода путем его диффузии
через никель.
ровки избыточного давления н выжигания примесей, накапливающихся в
кварцевой трубке. В головке на другом конце кварцевой трубки подсоединен
ртутный манометр. Нагревание кварцевой трубки производят только в ее
центре так, чтобы места припаивания никелевых трубок не нагревались.
В зависимости от режима работы прибора его производительность (мл/мин)
изменяется следующим образом:
Избыточное давление, мм рт. ст.
Температура, °C 15 20 25 30
750 20 27 34 41
815 27 36 43 52
860 34 45 55 68
900 41 54 68 84
Скорость подачи газа пропорциональна величине избыточного давления,
тогда как зависимость от температуры не является линейной. Путем изме-
нения давления скорость подачи может быть практически моментально от-
регулирована до нужного уровня. При наличии хорошего вентиля на балло-
не с водородом аппаратура работает без сбоев. Продолжительность непре-
рывной работы составляет не менее 250 ч. Перед каждым новым опытом сле-
дует убедиться в герметичности аппаратуры.
Способ 4. Разложение UH3 [17]
2UH3 ——> 2U 4- ЗН2
482,11 476,06 6,05
Этот метод, в котором используется синтезированный ранее UH3, позво-
ляет получить очень чистый, без примеси инертных газов, водород в нуж-
ных количествах.
150 Глава 1. Водород, дейтерий, вода
Получение UH3 ведут в аппаратуре, схематически показанной на рис. 99.
Электролитический водород из стального баллона предварительно очищают
пропусканием через нагретые до 650—700 °C медные стружки (7) н через
Mg(ClC>4)2 (-2) Для высушивания. Дальнейшая очистка ведется в трубке, на-
полненной порошком урана, нагретым до 700—750 °C. Взаимодействие очи-
щенного таким образом водорода с урановой стружкой проводят в двугор-
лой колбе 8. Урановую стружку, заполняющую колбу 8 наполовину, предва-
рительно освобождают от оксидной пленки путем обработки разбавленной
азотной кислотой, промывки н сушки. Колбу 8 подогревают до 250 °C либо
на солевой бане с ннтрнт-нитратной смесью 9, либо в электрической печн со-
Рис. 99. Схема установки для
очистки водорода и синтеза
гидрида урана.
/ — трубка, заполненная медными
стружками; 2 —трубка с Mg(ClO4)2;
3 — трубка, содержащая порошок
урана, помещенный между двумя
тампонами из стеклянной ваты 4
и 5; 6, 7 — соединения на шлифах;
8 — колба с урановой стружкой; 9 —
баня с солевым расплавом.
противления. К колбе подсоединяют две промывные склянки — пустую и с
КОНЦ. H2SO4.
Прежде чем производить нагревание в 1, 3 или 8 и тем самым начинать
реакцию урана с водородом, приводящую к образованию UH3, аппаратуру
тщательно промывают водородом. Реакцию можно считать законченной, если
при прекращении поступления водорода не происходит засасывания серной
кислоты нз последней промывной склянки.
Полученный таким образом UH3 представляет собой темно-коричневый
самовозгорающийся порошок. Для получения очень чистого водорода гидрид
урана нагревают, в случае необходимости создавая одновременно понижен-
ное давление в системе. Если разложение ведется при 400 °C нли ниже, то
образующийся при этом порошок урана энергично реагирует с водородом
уже при комнатной температуре; при —80 °C реакция идет довольно быстро,
и лишь при —200 °C взаимодействия не наблюдается.
Способ 5. Разложение гидрида титана [18]
Для получения значительных количеств очень чистого водорода исполь-
зуют гидрид титана вследствие сравнительно низкой температуры его разло-
жения (400—900 °C), в связи с довольно высоким содержанием водорода и
благодаря его легкой регенерируемости. Важно также, что при температурах,
необходимых для разложения гидрида, оксиды и нитрид титана совершенно
устойчивы. Поскольку реакция разложения является эндотермической, при
уменьшении подвода тепла выделение газа замедляется, так что можно осу- ( |
ществлять процесс непрерывной, хорошо регулируемой дегазации. Удобно ра-
боту вести в аппаратуре, изображенной на рис. 100, в которой (в части 7)
можно одновременно проводить реакции гидрирования полученным чистым
водородом.
На кварцевую трубку 1 (наружный диаметр 34 мм, внутренний диаметр
30 мм, общая длина 1500 мм) с лодочкой из молибдена 2 намотана нагрева-
Водород 151
тельная спираль 3 (на участке длиной —650 мм), закрепленная на трубке
>при помощи массы из кварца и растворимого стекла. Отражательный экран 4
предотвращает значительную потерю тепла путем излучения наружу. Ток в
нагревательной обмотке регулируется при помощи контактного манометра 8
в зависимости от давления выделившегося водорода.
Предварительно гидрид титана получают из титановой губки обычной чи-
стоты и со средним размером зерен (например, фирмы Fluka-Chemie, Neu-
Ulm). Поверхностно адсорбированную влагу из титановой губки можно пред-
варительно удалить нагреванием ее в потоке водорода до температуры не
Рис. 100. Схема установки для получения водорода высокой чистоты из гид-
рида титана.
1 — кварцевая трубка; 2— молибденовая лодочка с титаном; 3 — нагревательная обмот-
ка; 4 — отражательный экран; 5 — отражатель для защиты пробки от нагрева; б —стек-
лянная вата; 7 — электрическая трубчатая печь, в которую для проведения реакций с
водородом высокой чистоты помещают лодочку с веществом, подлежащим гидрированию
(эта часть установки может отсутствовать, если необходимо получать чистый водород,
используемый в другой аппаратуре); 8— контактный манометр; 9— реле в цепи нагре-
вателя.
выше 400 °C. Нагревание до ~ 700 °C производят в молибденовой лодочке в
атмосфере технического водорода в течение —30 мин; охлаждение ведут в
потоке водорода. Аппаратуру тщательно откачивают, а затем полученный
гидрид титана снова нагревают. При достижении избыточного давления газа
Ю,1 бар происходит автоматическое отключение тока контактным маномет-
ром 8. Температура быстро падает, и за счет термического сжатия газа и
вследствие расходования газа на реакцию гидрирования давление понижа-
ется. При снижении давления до установленного значения печь снова вклю-
чается. Несмотря на простоту регулирующего устройства, колебания давле-
ния сравнительно малы и составляют примерно 0,1—0,2 бар.
После завершения процесса выделения водорода полностью или частично
израсходованный гидрид титана регенерируют путем его повторного нагре-
вания и последующего охлаждения в атмосфере технического водорода. При
загрузке 500 г титановой губки за один цикл может быть получено более
100 л очень чистого водорода.
Способ 6. Электролиз без доступа воздуха [19, 20]
Описание прибора, применение которого дает возможность получать пу-
тем электролиза водород или кислород (в зависимости от того, как он под-
152 Глава 1. Водород, дейтерий, вода
ключей к полюсам) в условиях полной изоляции от атмосферного воздуха,
содержится в ч. II, гл. 5 («Кислород»), Полученный газ содержит менее
4-10-6% воздуха.
Свойства. М 2,016. Бесцветный газ без запаха и вкуса; обладает, в осо-
бенности при повышенных температурах, сильным восстановительным дейст-
вием. Нагретый водород не следует пропускать через концентрированную сер-
ную кислоту, так как при этом легко происходит загрязнение газа примесью
SOg.
Температура тройной точки —259,35 °C, /кип —252,77 °C; /крит —239,91 °C;
Ркрит 12,80 бар (прн —260,353°C*); dKP„T 0,0301, йж=0,07081 (пара-водо-
род, —252,6 °C); масса 1 л при н. у. 0,08987 г. В одной объемной ча-
сти воды растворяется объемных частей водорода: прн 0°С — 0,021, при
20°C — 0,018, прн 100°C — 0,016 (давление 760 мм рт. ст.). В других жидко-
стях газ растворим тоже очень мало.
ЛИТЕРАТУРА
1. Klemenc A., Die Bchandlung und
Reindarstellung von Gasen. Sprin-
ger, Wien, 1948, S. 133.
2. Schutze M., kngew. Chem., 70,
697 (1958).
3. Muller G., Gnauck G., Reinste Ga-
se. VEB Deutscher Verlag der Wis-
senschaften, Berlin, 1965, S. 172.
4. Smolkova E., Feltl L., Gruber O.,
Z. Anal. Chem., 197, 321 (1962);
Baker C. R., Paul R. S„ Chem.
Eng. Progr., 59, 61 (1963).
5. Moser L„ Die Reindarstellung von
Gasen. Stuttgart, 1920, S. 37.
6. Darling A. S„ Chem. Ing. Techn.,
37, 18 (1965); Platinum Metals
Rev., 7, 126 (1963); Ullmanns En-
zyklopadie der techn. Chem., Bd.
18. Mtinchen, Berlin, -Wien, 1967,
S 527
7. Prescott O., Wise H. L., J. Gas
Chromatog., 4, 80 (1966).
8. Bosse К. O., Kohlmeyer F., Intern.
Z. Gaswarme, 14, 156 (1965).
9. McBride R. B., McKinley D. L.,
Chem. Eng. Progr., 61, 81 (1965).
Вода наивысшей чистоты [1—4]
10. Родина А. А., Дорониче-
ва Н. И. — Хим. пром., 1965,
т. 41, с. 902.
11. Rubin L. R., Engelgardt Ind. Inc.
Techn. Bull., 2, 8 (1961).
12. Lawson W. D., Nielsen S., Prepa-
ration of single crystals. London,
1958, p. 106.
13. Anger er E., Ebert H., Technische
Kunstgriffe bei physikalischen Un-
tersuchungen. Vieweg, Braun-
schweig, 1966, S. 28.
14. Schafer R„ Klemm W„ J. Prakt.
Chem., (4) 5, 233 (1958).
15. Snoek J. L., Haes E. J., Appl. Sci.
Res. Sect. A, 2, 326 (1950).
16. Harrison E. R., Hobbis L. C., Rev.
Sci. Instr., 26, 305 (1955).
17. Spedding F. H., Newton A. S.,
et al. Nucleonics, 4, 4 (1949).
18. Lux B., Planseeber., Pulvermetai-
lurgie, 4, 7 (1956).
19. Paneth F., Peters K. Z. Physik.
Chem., 134, 364 (1928).
20. Brauer G., Z. Anorg. Allgem.
Chem., 255, 105 (1947).
Применяемая в лабораториях дистиллированная вода обыкновенно содер-
жит еще заметные количества растворенного диоксида углерода, а также сле-
ды аммиака, органических оснований и других органических веществ. Полу-
чение очень чистой воды осуществляют в несколько этапов. Сначала в воду
на каждый 1 л добавляют 3 г NaOH (ч. д. а.) н 0,5 г КМпО4 и производят
перегонку в аппаратуре на шлифах, изготовленной из стекла типа дюран 50
или солидекс, причем собирают только среднюю фракцию. Таким путем уда-
По-видимому, данные ошибочны. — Прим, перев.
Вода 153
ляется растворенный диоксид углерода и происходит окисление органических
веществ. Удаление аммиака достигается прн проведении второй и третьей пе-
регонки с добавлением 3 г KHSO« или 5 мл 20%-ной Н3РО4, причем эти ре-
агенты предварительно нагревают с небольшим количеством КМ11О4. Чтобы
предотвратить «выползание» добавленного электролита в конденсат, при про-
ведении третьей перегонки создают «сухой участок», для чего отрезок труб-
ки между насадкой на колбу и холодильником нагревают до 150 °C. Послед-
нюю перегонку, служащую для освобождения от следов электролитов, про-
водят нз кварцевой колбы с холодильником из кварца. Верхнюю трубку хо-
лодильника, согнутую под прямым углом, вставляют без всякого уплотняю-
щего материала непосредственно в сужение колбы (рис. 101). Во избежание
попадания брызг воды целесообразно на пути пара поместить брызгоулавли-
Рис. 101. Способы присоединения
колбы к холодильнику при перегон-
ке воды особой чистоты.
а — простое (дешевое) исполнение; б — с
брызгоулавливателем.
ватель. В качестве приемника служат колбы из кварца, платины, стекла типа
дюран 50 или солидекс, которые предварительно обрабатывают водяным па-
ром. Полученная таким способом вода является «чистой по значению pH»*.
Описание другой аппаратуры для перегонки с целью получения воды осо-
бой чистоты (в том числе для специальных целей) можно найти в литерату-
ре [5—8].
Чистоту воды определяют путем измерения ее электропроводности, кото-
рая непосредственно после перегонки воды должна составлять менее
10-6 Ом-1-см-1. Испытание на содержание в воде диоксида углерода про-
изводят при помощи баритовой воды, а пробу на содержание аммиака — ре-
активом Несслера. Очень чистую воду хранят в кварцевых или платиновых
сосудах. Можно использовать для этого также и колбы из стекла дюран 50
или солндекс, предварительно обработанные паром в течение долгого време-
ни и предназначенные исключительно для этой цели. Такие сосуды лучше все-
го закрывать пришлифованными колпачками.
ЛИТЕРАТУРА
1. Lux Н„ Anorganisch-Chemische Ех-
perimentierkunst. Barth, Leipzig,
1959, S. 59.
2. Honigschtnid О., Sachtleben R., Z.
Anorg. Allgem. Chem., 221, 65
(1934).
3. Ostwald—Luther, Hand- und Hilfs-
buch zur Ausfuhrung Physiko-chemi-
scher Messungen, 5. Aufl., Leipzig,
1931, S. 633.
4. Angerer E., Ebert H„ Technische
Kunstgriffe bei physikalischen Un-
Т. e. co значением pH, равным 7,00. — Прим, перев
154 Глава 1. Водород, дейтерий, вода
tersuchungen. 14. Aufl., Braun-
schweig, 1966, S. 27. [Есть рус-
ский перевод 12-го нем. изд.: Ан-
герер Э. Техника физического экс-
перимента. Пер. с нем ./Под ред.
К. П. Яковлева. — М.: Фнзматгиз,
1962.]
5. Taylor J. Е., J. Chem. Educ., 37,.
204 (1960).
6. Holt J. M., Lux W„ Valberg L. S„
Can. J. Biochem., 41, 2029 (1963).
7. Powers R. W., Electrochem. Tech-
nol., 2, 163 (1964).
8. Ettel V., Veprek Siska J., Chem.
Listy, 60, 340 (1966).
Вода, предназначенная для измерения электропроводности
Способ 1. Получение путем перегонки [1—2]
Необходимую для проведения измерений электропроводности воду наи-
высшей степени чистоты получают путем особенно тщательной перегонки уже
предварительно очень хорошо очищенной воды. Последняя должна при 25 °C
обладать электропроводностью (х), равной 1-Ю-6—2-10-6 Ом-1-см-1. Ее
получают указанным выше методом или же путем двукратной перегонки:
а) со смесью перманганата калия и серной кислоты и б) с гидроксидом ба-
рия. Для перегонки пользуются колбой из стекла типа дюран 50 или соли-
декс с присоединенным к ней медным илн кварцевым холодильником.
КВарц
Рис. 102. Конструкция прибора для перегонки воды, предназначенной для
измерения электропроводности.
нагревательная обмотка (60 Ом); 2— колбоиагреватель (130 Ом); 3 — переходник на
Все части прибора для одноступенчатой перегонки по методу Кортюма
(рис. 102) изготовлены из стекла типа дюран 50 или солидекс, за исключе-
нием короткого кварцевого холодильника, присоединенного к перегонному
прибору на нормальном шлифе. Ведущую к холодильнику согнутую часть
нагревают при помощи нагревательного элемента (60 Ом) до температуры,''
превышающей 100 °C, во избежание увлечения жидкой воды в холодильник.
Расположенный ниже обратный холодильник высотой 60 см снабжен спи-
Вода 155
ралью Видмера. К запасной склянке холодильник присоединяется переходны-
ми шлифами. Чтобы дистиллат сохранил малую электропроводность в тече-
ние долгого времени, переходные шлифы н запасную склянку предваритель-
но необходимо в течение нескольких суток обработать горячей разбавленной
кислотой. Воду высокой чистоты (х=(1—2)-10~6 Ом-1-см-1) перегоняют,
пропуская через прибор медленный поток сжатого воздуха нз стального бал-
лона со скоростью приблизительно 1 пузырек в секунду. Воздух предвари-
тельно очищают, пропуская его через семь промывных склянок, нз которых
одна наполнена концентрированной серной кислотой, три содержат 50%-иый
раствор гидроксида калия и три — «воду для измерения электропроводности»
(последние три промывалки должны быть снабжены пористыми стеклянны-
ми пластинками). Полученную воду отбирают из запасной склянки путем вы-
теснения ее очищенным, как указано выше, сжатым воздухом. Нагревание во-
ды в колбе производят прн помощи колбонагревателя мощностью 300 Вт.
Колбу можно легко наполнить водой или опорожнить при помощи располо-
женной в середине ее вертикальной трубки. Заполнение колбы проще всего
осуществить, прекратив пропускание воздуха и выключив колбоиагреватель.
К трехходовому крану в конце холодильника присоединяют сосуд, в ко-
тором проводят измерение электропроводности перегнанной воды до тех
пор, пока не будет достигнуто желаемое значение к. После этого воду путем
переключения крана направляют в запасной сборник.
Таким путем за 1 ч можно получить 100 мл воды, для которой при 25 °C
эс=2-10“7 Ом-1-см-1. Если перегонку вести очень медленно, то электропро-
водность полученной воды может достигать значения 10-8 Ом-1-см-1.
Способ 2. Получение путем ионного обмена [3]
В больших количествах «воду для измерения электропроводности» (х от
7-Ю-8 до 1,5-10-7 Ом-1-см-1 можно получить путем ионного обмена в ап-
паратуре, схематически показанной на рис. 103.
Колонку из стекла пирекс (длиной 75 см и диаметром 7,5 см) с пористой
стеклянной пластинкой на дне наполняют смесью (750 г), состоящей из од-
яой части амберлита IR 120 (16—50 меш) и двух частей амберлита IRA 400
(20—50 меш). Смолу в колонне накрывают перфорированным полиэтилено-
вым кружком, плавающим в растворе и служащим для предотвращения
взмучивания смолы потоком воды. Через колонну пропускают обычную дис-
тиллированную воду. Как только электропроводность воды, измеряемая в
ячейке 3, достигнет достаточно низкого значения, сначала промывают, а за-
тем наполняют ею сосуд 4. Попадание в воду диоксида углерода нз воздуха
предотвращают прн помощи двух вставленных в колонну и в приемник
хлоркальциевых трубок 5, заполненных гранулированным «карбосорбом» с
индикатором.
Предварительную обработку смолы и ее регенерацию производят следу-
ющим образом. Катионообменник IR 120 несколько раз промывают дистил-
лированной водой, удаляя мелкие частицы декантацией. Затем на стеклян-
ном пористом фильтре смолу дважды обрабатывают попеременно 1 н. NaOH
и 2 н. НС1, промывая после каждой обработки дистиллированной водой до
нейтральной реакции. Аннонообмениик IRA 400 сначала также промывают
дистиллированной водой. После декантации смолу на стеклянном пористом
фильтре обрабатывают 2 н. NaOH, не содержащим карбонатов (воду для
приготовления раствора освобождают от диоксида углерода перегонкой). Об-
работку ведут до тех пор, пока концентрация ионов хлора в элюате не по-
низится до минимума. После этого смолу промывают дистиллированной во-
дой до достижения нейтральной реакции в промывных водах.
Перед регенерацией смолы смесь разделяют. В стакан вносят смолу, су-
спендируют ее в этаноле и добавляют хлороформ, причем анионообменник
собирается в верхнем слое. Смесь разделяют на составные части и проводят
раздельную регенерацию.
156 Глава 1. Водород, дейтерий, вода
При пропускании через аппаратуру обычной дистиллированной водьи
можно без регенерации получить со скоростью 1 л/мин 7000 л «воды для из-
мерения электропроводности» с х=5,52-10-8 Ом_1-см-1 при 25 °C.
ЛИТЕРАТУРА
Рис. 103. Конструкция установки для:
получения воды особой чистоты пу-
тем ионного обмена.
1 — ионообменная колонка; 2 — пористый
стеклянный фильтр; 3 — ячейка для изме-
рения электропроводности; 4 — сборник;
5 — трубка для поглощения диоксида уг-
лерода.
1. Kortum G., Chem. Fabrik, 13, 143
(1940).
2. Thiessen P. A., Herrmann K, Chem.
Fabrik, 10, 18 (1937); Z. Elektro-
chem., 43, 66 (1937).
3. Jacobs S., Chem. & Ind., 944 (1955).
Дейтерий и соединения дейтерия [1—3]
Дейтерий, а также его простые неорганические соединения имеются в
продаже*. Однако в лаборатории дейтерий часто получают из наиболее до-
ступного его соединения — D2O.
* D2 производится фирмой Linde AG, Werkgruppe Miinchen fur Gase
(Lohhof bei Miinchen); фирма, выпускающая D2O (99,75% D2O),— Merck AG
(Darmstadt).
Дейтерий 157"
Тяжелую воду (D2O) в промышленном масштабе производят в концент-
рациях 5—99,95%. В продажу она поступает в стеклянных бутылях с при-
шлифованными пробками и в запаянных стеклянных ампулах. Поскольку*
чистая тяжелая вода весьма гигроскопична, т. е. легко поглощает влагу из
воздуха, в результате чего концентрация оксида дейтерия понижается, при
вскрытии бутылей и ампул, а также при переливании из них тяжелой воды-
необходимо соблюдать некоторые меры предосторожности.
Если для проведения реакции предполагают использовать только часть-
содержимого бутыли, то отбор производят в «сухой» камере в атмосфере
Рис. 104. Способ вскрытия ампулы с
тяжелой водой.
тщательно высушенного газа. Можно также в «сухой» камере заменить проб-
ку в бутыли на присоединяемую на шлифе насадку с прокалываемым рези-
новым колпачком или перенести содержимое бутыли (или ампулы) в специ-
альную удлиненной формы колбу с такой же насадкой. После этого отбор-
порций тяжелой воды можно вести вне «сухой» камеры при помощи инъек-
ционного шприца с иглой или используя сходную технику работы согласно-
fl] (см. также ч. I, разд. 13, «Высокий вакуум н изоляция от воздуха»).
Сходным образом производят перевод содержимого сосуда с тяжелой водой
в реакционный сосуд с прокалываемым колпачком. Присоединение .реакцион-
ного сосуда к остальной аппаратуре осуществляют в потоке тщательно вы-
сушенного инертного газа.
Для отбора части содержимого ампулы с тяжелой водой можно также-
кончик ампулы оттянуть пинцетом на небольшом остром пламени в капил-
ляр. Последний обламывают (в «сухой» камере), после чего желаемое коли-
чество тяжелой воды вытесняют из ампулы путем ее легкого нагревания (на-
пример, теплом руки). Затем сосуд, служащий приемником, закрывают, а ам-
пулу снова запаивают на остром язычке пламени. Из предосторожности ам-
пулу следует хранить в эксикаторе [2].
Если при проведении опыта, при котором требуется получить продукты
реакции с очень высоким содержанием дейтерия, должно быть использовано»
все содержимое ампулы, то лучше всего вскрытие ее производить непосред-
ственно в реакционной аппаратуре. После внесения закрытой ампулы аппа-
ратуру герметизируют путем запаивания и в высоком вакууме легким нагре-
ванием пламени горелки удаляют абсорбированную на стенках «легкую» во-
ду. После этого, быстро погрузив в жидкий воздух сосуд, имеющий сходную»
с ампулой форму (рис. 104), замораживают оксид дейтерия, вследствие чего
стенки ампулы лопаются. В данном случае охлаждение смесью, содержащей
сухой лед, недостаточно эффективно, поскольку оксид дейтерия в этих усло-
виях затвердевает медленнее и его кристаллы заполняют преимущественно»
верхнюю пустую часть ампулы [3]. Можно также предварительно надлежа-
Я 58 Глава 1. Водород, дейтерий, вода
щим образом установить ампулу в приборе и затем разбить ее толстостен-
ным стеклянным бойком с железным сердечником, передвигаемым при помо-
щи электромагнита.
Из всех веществ, которые должны быть приведены во взаимодействие с
оксидом дейтерия, следует сначала тщательно удалить следы воды. Гигро-
•скопичные вещества, которые нельзя перенести в прибор без поглощения ими
хотя бы незначительных количеств воды, необходимо непосредственно перед
проведением реакции снова подвергнуть высушиванию нагреванием, перегон-
кой или сублимацией в условиях высокого вакуума в самом реакционном со-
суде.
Прн получении под вакуумом в чистом виде можно также запаивать эти
вещества в ампулы. Последние вносят затем в реакционный сосуд и разби-
вают магнитным бойком (см. выше). Гигроскопичные жидкости и растворы
можно, кроме того, вслед за операциями их очистки перенести в реакцион-
ный сосуд при помощи инъекционного шприца нли с использованием специ-
альной техники работы, описанной выше (ч. I, разд. 13). Гигроскопичные
твердые вещества в ряде случаев целесообразно вносить в тонкостенных
разбиваемых ампулах. Отдельные части установки следует по возможности
спаивать друг с другом, а число кранов в ней должно быть небольшим. Ес-
ли это окажется невыполнимым, то уплотнение мест соединения должно вы-
полняться с особой тщательностью. В местах соединения аппаратуры с ва-
куумной системой или с атмосферой помещают трубки с осушающими ве-
ществами нли, еще лучше, вымораживающие ловушки, охлаждаемые жид-
ким азотом, что предотвращает попадание в аппаратуру влаги из воздуха.
Поскольку большинство неорганических соединений дейтерия способно так
же, как и тяжелая вода, обменивать в присутствии обычной воды часть дей-
терия на водород, указанные выше меры предосторожности необходимо учи-
тывать при проведении всех описываемых ниже реакций.
Приобретение значительных количеств соединений дейтерия обходится
довольно дорого. Поэтому перед проведением каждой реакции целесообразно
предварительно поупражняться, проводя «модельные» опыты с исходными ве-
ществами, содержащими обыкновенный водород.
ЛИТЕРАТУРА
1. Feher F., Kuhlborsch G„ Luhleich
H., Z. Anorg. Allgem. Chem., 303,
294 (1960).
2. Prospekt der Norsk-Hydro-Elektrisk
Kvaelstofaktieselskab.
3. Knowlton J. W., Rossini F. D., J.
Res. nat. Bur. Stand., 19, 605
(1937).
Дейтерий D2
(Тяжелый водород)
Способ 1
2D2O + 2Na ----->• D24-2NaOD
40,06 45,98 4,03 82,01
Этот метод [1] особенно пригоден для получения небольших количеств
дейтерия (до 1 л).
В сосуде 1 стеклянного прибора, схематически изображенного на рис. 105,
находится алюминиевый тигель, в котором помещен металлический натрий,
взятый в избытке, а в сосуде 2 — оксид дейтерия, предназначенный для про-
ведения реакции. Его помещают в сосуд с предосторожностями, исключаю-
Дейтерий 159
щими возможность соприкосновения с влагой воздуха, как это описано вы-
ше. После охлаждения жидким азотом в сосуде 2 создают высокий вакуум,
открыв краны 3 и 4. Затем кран 4 закрывают и оксид дейтерия медленно пе-
регоняют в сосуд 1 с металлическим натрием, охлаждая его жидким азотом.
После перегонки для полного завершения реакции сосуд 1 нагревают в те-
чение нескольких часов при 350 °C. Потом открывают кран 4 и образовав-
шийся дейтерий перекачивают для очистки в приемник с древесным углем,
не содержащим газов. Здесь дейтерий оставляют на некоторое время при
температуре —196 °C. Дейтерий, полученный при реакции со свежей порцией
Рис. 105. Схема установки для
получения дейтерия путем
взаимодействия тяжелой воды
с натрием (1—4 см. текст).
натрия, еще содержит несколько процентов водорода, находившегося в ме-
талле в растворенном состоянии, а также в виде гидроксида натрия. Чистый
дейтерий, содержащий водород и другие посторонние газы в количестве,
меньшем 0,2%, может быть получен лишь прн повторной обработке того же
куска натрия.
Испытание чистоты полученного препарата производят измерением его>
теплопроводности или давления пара. Дейтерий получается с количественным,
выходом, соответствующим уравнению реакции.
Способ 2 [2]
D2O + Mg --------> D2 MgO
20,03 24,31 4,03 40,31
В продолговатой колбе установки, изготовленной из тугоплавкого стекла,
(пирекс, супремакс), которую предварительно вакуумируют до 10~4 мм рт..
ст., медленно испаряют 20 г оксида дейтерия. Пары поступают в вертикаль-
но присоединенную к сосуду реакционную трубку внутренним диаметром
2,4 см н длиной 55 см. В этой трубке содержится 130 г магния, причем в
ннжней части трубки магний находится в виде крупных стружек, которые по-
степенно становятся все более мелкими, а в верхней части трубки — уже в
виде рыхлого порошка. Магний снизу поддерживается перфорированной пла-
тиновой пластинкой, опирающейся на расположенные внутри трубки стек-
лянные крючки. При помощи трубчатой печи магний нагревают до 480 °C.
При продолжительной работе на стенках нагретой стеклянной трубки об-
разуется небольшое количество силицида магния. Чтобы избежать образова-
ния силицида, рекомендуется помещать магний в трубку из неглазу-
J 60 Глава 1. Водород, дейтерий, вода
фованного твердого фарфора, заключенную в целях лучшей газонепроницае-
мости в другую трубку нз тугоплавкого стекла. Обе трубки должны быть
^спаяны на одном конце кольцеобразным спаем. При работе с такой трубкой
можно нагреть магний до еще более высокой температуры и тем самым по-
высить его реакционную способность.
Полученный дейтерий как можно скорее удаляют из прибора, в кото-
ром он был получен, конденсируя его при помощи жидкого водорода или
переводя в подсоединенный к установке сборник. С целью очистки образо-
вавшийся дейтерий1 пропускают через наполненную стеклянной ватой и
охлажденную до —196 °C ловушку. Контроль за выделением газа ведут при
помощи измерителя скорости потока и манометра. Скорость выделения дей-
терия, регулируемая изменением температуры колбы с тяжелой водой, мо-
жет быть доведена до получения 1/2 моль дейтерия в час. Поскольку пер-
вые порции газа содержат прнмесь водорода, попадающего из магния и со
стеклянных стенок установки, очень полезно сначала промыть прибор неко-
торым количеством дейтерия. В последующих порциях дейтерий очень чист.
Выход количественный.
Преимуществом описанного метода является возможность быстрого по-
лучения значительных количеств дейтерия, а также его полное выделение из
^тяжелой воды.
Способ 3 [3—4]
a) 2D2O + U -------> UO2 + 2D2
40,06 238,03 270,03 8,06
б) 3D2 -J- 2U -----> 2UD3
12,08 476,06 488,14
Этот способ обладает теми преимуществами, что помимо получения дей-
терия (уравнение а) происходит его связывание в виде UDs (уравнение б).
Из последнего дейтерий высокой степени чистоты можно в любой момент вы-
делить путем термического разложения (за счет протекания реакции б в об-
ратном направлении). Прибор для медленного и вполне безопасного проведе-
ния сильно экзотермического взаимодействия паров оксида дейтерия с ура-
ном схематически показан на рис. 106. Колба 1 вместимостью 50 мл через
кран 7 соединяется с манометром 2 и с кварцевой трубкой 4. Последняя на-
гревается в печи сопротивления до 600—700 °C. К трубке присоединяется
охлаждаемая жидким азотом ловушка 5, связанная с вакуумным насосом и
с нагреваемой до 250 °C колбой 6.
Колбу 1 примерно наполовину заполняют тяжелой водой (D2O), а труб-
ку 4 и колбу 6 — урановой стружкой, которую с целью удаления поверхност-
ного слоя оксида предварительно обрабатывают разбавленной азотной кисло-
той, промывают и сушат. На концах трубки (3,3') имеются тампоны из стек-
лянной ваты. Тяжелую воду в 1 замораживают при помощи охлаждающей
смеси сухого льда с метанолом, причем охлаждение необходимо вести мед-
ленно для предотвращения растрескивания колбы. Затем весь прибор ваку-
умируют, одновременно нагревая 4 и 6. После этого тяжелую воду осторож-
но размораживают, так что начинает медленно протекать взаимодействие
паров D2O с ураном, находящимся в кварцевой трубке. Первыми порциями
образующегося дейтерия производят промывку аппаратуры, причем кран 10
ставят в закрытое положение. Затем закрывают кран 9 и открывают кран
10. В течение опыта температура колбы 1 должна быть около 30 °C. Обра-
зующийся дейтерий, проходя через ловушку 5, освобождается от случайной
примеси «проскочивших» паров D2O и затем поглощается стружкой урана
в колбе 6. Если все количество урана в колбе 6 перейдет в UD3, то избыток
газообразного дейтерия вызовет повышение давления в приборе. Это в свою
-очередь сильно замедлит испарение D2O и тем самым предотвратит дальней-
Дейтерий 161
шее выделение дейтерия. Таким образом, работающая аппаратура функцио-
нирует почти автоматически и не требует особого присмотра. В этой установ-
ке можно перевести несколько граммов DaO в UDa.
Дейтерид урана — темно-коричневый самовоспламеняющийся > порошок.
Для получения очень чистого дейтерия UD3 нагревают, создавая при необхо-
димости пониженное давление в системе (см. также препарат <Водород»,
способ получения, 4). Полученный разложением при 400 °C и ниже порошко*
Рис. 106. Конструкция прибора
для получения и связывания дей-
терия.
1 — колба для тяжелой воды; 2 — мано-
метр; 3, З7 — соединения на шлифах;
4 — кварцевая трубка с урановой струж-
кой; 5 — охлаждаемая ловушка; 6 —
реакционная колба с урановой струж-
кой; 7—10 — краны.
образный уран энергично взаимодействует с водородом или дейтерием уже
при комнатной температуре; прн —80 °C реакция идет все еще достаточно
быстро, и лишь при —200 °C взаимодействия не наблюдается.
Способ 4. Электролиз оксида дейтерия (тяжелой воды) [5]
Схема электролизера вместимостью 60 мл, части которого соединяются
посредством нормального шлифа, показана на рис. 107. Керн шлифа пере-
ходит в цилиндрическую охлаждаемую водой рубашку, внутри которой на-
ходится катод. Платиновые электроды также имеют цилиндрическую форму
и изготовлены путем сварки платиновой проволоки с платиновой фольгой.
Электролитом служит оксид дейтерия, подкисленный 25%-ным раствором
дейтеросерной кислоты (если в распоряжении экспериментатора нет дейтеро-
серной кислоты, то можно добавить тщательно обезвоженный сульфат калия
или карбонат натрия). После эвакуирования электролизера через трубки 1 и
2 начинают электролиз, причем во избежание происходящего при низких
давлениях вспенивания жидкости ток должен быть небольшим. Однако по ис-
течении короткого времени ток можно повысить до 5 А. Охлаждение следует
отрегулировать таким образом, чтобы температура электролита ие повыша-
лась. Если образующийся дейтерий должен преодолевать некоторое сопро-
тивление при прохождении через присоединенные к электролизеру части уста-
новки, служащие для очистки газа, или для проведения реакции (трубки
малого диаметра, столбы жидкостей и т. и.), то следует позаботиться о вы-
11—143
162 Глава 1. Водород, дейтерий, вода
равннванин давления путем включения на пути отводимого кислорода соот-
ветствующего клапана избыточного давления. Получаемый таким способом
дейтерий еще содержит небольшие количества кислорода и паров оксида дей-
терия. Очень чистый газ получают пропусканием дейтерия через нагретый
платинированный асбест с последующим высушиванием его жидким возду-
хом. При токе 5 А можно получить 2 л дейтерия в час.
Небольшие количества дейтерия сохраняют в запаянных стеклянных кол-
вах илн в сосудах с ртутным затвором. В качестве жидкости для аатвора
Рис. 107. Устройство прибора
для электролиза тяжелой воды.
может служить также н дистнллированвая вода. Более значительные количе-
ства дейтерия конденсируют при помощи жидкого водорода в металлической
колбе и непосредственно вслед за этим переводят его под давлением по ме-
таллическим трубкам в небольшие стальные баллоны.
Конструкции других приборов для электролиза D2O, в том числе и мень-
шей производительности, описаны в литературе [8].
Другие методы получения [6—7]
Дейтерий может быть получен путем восстановления оксида дейтерия
при высоких температурах железом или вольфрамом.
Свойства. Бесцветный газ без запаха. Химические свойства аналогичны
свойствам водорода, однако дейтерий отличается несколько меньшей активно-
стью. Смеси дейтерия с водородом в отсутствие катализаторов устойчивы до
Дейтерий 163
~500 °C. При комнатной температуре обменного взаимодействия между
дейтерием и водой также не происходит.
Температура тройной точки —254,44 °C; /КИп —249,49 °C; /крИт —234,85 °C;
Ркрит 16,282 бар; d„pHT 0,0669; d1K 0,17 (—253,1 °C). В воде н других жид-
костях растворим очень мало.
ЛИТЕРАТУРА
1. Lewis G. N., Hanson W. Т., J.
Amer. Chem. Soc., 56, 1687 (1934).
2. Knowlton J. W., Rossini F. D., J.
Res. Nat. Bur. Stand., 19, 605
(1937).
3. Spedding F. H., Newton A. S„
Warf J. C., Johnson O., Nottorf R.
W., Johns I. B„ Daane A. H„ Nuc-
leonics, 4, 4 (1949).
4. Newton A. S., U. S. At. Energy
Comm. TID-5290, Book 1, 74
(1953).
5. Wilson C. L., Wylie A. W., J. Chem.
Soc. (London), 596 (1941).
6. Zintl E., Harder A., Z. Physik.
Chem. В 28, 480 (1935).
7. Farkas A., Farkas L., Proc. Roy.
Soc. London, 144, 469 (1934).
8. NorUng F„ Phys. Z„ 36, 711 (1935);
Slack С. M., Ehrke L. F„ Rev. Sci.
Instr. (N. S.), 8, 39 (1937); Sie-
verts A., Danz W., Z. Physik Chem.,
B38, 46 (1937); Winn M. M.. J.
Sci. Instr., 28, 152 (1951); Lloyd
J. T., J. Sci. Instr., 29, 164 (1952);
Waniek R. W„ Rev. Sci. Instr., 21,
262 (1950); Reznik A., Faforge-
rie G„ Dupre G., Compt. Rend., 244,
760 (1957); Dutt P. K-, J. Sci.
Instr., 37, 352 (1960).
Дейтероводород HD [1—3]
(Гидрид дейтерия)
LiAlH4 + 4D2O ---> LiOD + A1(OD)S + 4HD
37,95 80,11 24,95 81,02 12,09
Взаимодействие проводят в двугорлой колбе вместимостью 250 мл с под*
соединенным к ней обратным холодильником. Другое горло колбы закрыва-
ют резиновым колпачком. Обратный холодильник через охлаждаемую ловуш-
ку (для конденсации увлеченного паром растворителя) подсоединяют к диф-
фузионному насосу и колбе, служащей приемником выделяющегося газа.
Наличие кранов позволяет любую часть установки эвакуировать или напол-
нить воздухом или азотом. В реакционную колбу, в которую помещена маг-
нитная мешалка, перегоняют примерно 150 мл высушенного над натрием
н-бутнлового эфира и затем в атмосфере азота добавляют 5,75 г LiAlH<
(40%-ный избыток). Содержимое колбы замораживают жидким азотом, а
аппаратуру эвакуируют. Затем путем осторожного нагревания доводят жид-
кость в колбе до кипения и кипятят 1,5 ч, после чего колбу опять замора-
живают жидким азотом и снова эвакуируют. К замороженной реакционной
смеси из инъекционного шприца прибавляют путем прокалывания резиново-
го колпачка 5 мл 99,75%-ного D2O (см. стр. 157). Газовыделение начинается
при оттаивании содержимого колбы и при одновременном перемешивании
магнитной мешалкой. Благодаря низкой температуре колба покрывается сна-
ружи льдом. Путем периодического погружения колбы в жидкий азот под-
держивают температуру на таком уровне, чтобы слой льда на наружных
стенках колбы не плавился. По мере замедления реакции добавляют еще две
порции D2O по 6,5 мл каждая (всего 18 мл, 150%-иый избыток). В итоге по-
лучают 10 л дейтероводорода чистотой 97—99%.
Свойства. Бесцветный газ без запаха. <пл —256,5 °C; <Ккп —251,02 °C
(—256,55°C при давлении 93 мм рт. ст.); /кРкт —237,2°C; рк₽ит 14,6 бар;
^крит 0,0482; 0,146 (—268,95°C).
11*
164 Глава Г.Водород, дейтерий, вода
ЛИТЕРАТУРА
1. Fookson A., Pomerantz Ph., Pich
Е. Н„ J. Res. Nat. Bur. Stand., 47,
31 (1951); Science (New York),
112,748 (1950).
2. Wender J., Friedel R. A., Orchin M.,
J. Amer. Chem. Soc., 71, 1140
(1949).
3. Scott R. B„ Brickwedde F. G.,
Phys. Rev. (2) 48, 483 (1935); 55,
672 (1939).
Фторид дейтерия DF
(Тяжелый фтороводород)
Способ 1 [1]
2CeH5COF + D2O ----> (C6H5CO)2O + 2DF
248,23 20,03 226,23 42,03
Взаимодействие осуществляют при полной изоляции от доступа воздуха
в аппаратуре, показанной на рис. 108, которую промывают сухим азотом.
К 186 г (1,5 моль) бензоилфторида, помещенного в серебряную колбу t,
охлаждаемую смесью сухой лед+ацетон, добавляют в атмосфере азота сра-
Рис. 108. Схема прибора для получения фторида дейтерия.
/ — серебряная колба; 2 — углубление для термометра; 3 — стеклянная охлаждающая ру-
башка; 4—парафинированная пробка; 5 — хлоркальциевая трубка; 6 — кварцевый при-
емник.
ву 5 г (0,25 моль) 99,75 %-ного D2O. После этого колбу присоединяют к ап-
паратуре для перегонки, также изготовленной из серебра. Через холодиль-
ник 3 циркулирует рассол при —15 °C, а кварцевый приемник охлаждают
смесью лед+ацетон до —78 °C. Затем колбу 1 перестают охлаждать и мед-
ленно нагревают до комнатной температуры, после чего производят перегон-
ку фторида дейтерия, нагревая колбу на водяной бане до 80—90 °C. Для до-
стижения высокой степени чистоты перегонку повторяют еще два раза, осво-
бождаясь таким образом от попавшего в приемник бензоилфторида. Выход
составляет 9,7 г DF (92% от теоретического).
Дейтерий. 16Й.
Способ 2 [2]
FSO3H + D2O ------> hdso4 + df
100,07 20,03 99,09 21,01
В аппаратуре из платины, никеля или монель-металла* в отсутствие вла-
ги к 1000 г фторосерной кислоты прибавляют по каплям 100 г DjO. Реакци-
онную смесь постоянно перемешивают при помощи магнитной мешалки (с
тефлоновой оболочкой). Устанавливают такую скорость добавления D2O, что-
бы при наружном охлаждении колбы температура внутри ее была в преде-
лах 50—70 °C. Образующийся фторид дейтерия непрерывно отгоняют и соби-
рают в приемнике из полиэтилена, охлаждаемом смесью сухой лед+ацетон.
По окончании реакции через колбу пропускают поток сухого азота, а тем-
пературу поднимают до 100 °C, что приводит к полному удалению растворен-
ного фторида дейтерия. С целью очистки продукт еще раз перегоняют. Вы-
ход составляет 94,5 г (90% от теоретического).
Если взаимодействие проводят в кварцевой аппаратуре, то образующий-
ся DF загрязняется тетрафторидом кремния и должен быть в дальнейшем
очищен.
Фторид дейтерия хранят в сосудах из платины, серебра или меди.
Другие способы получения:
3. Небольшие количества фторида дейтерия получают действием газооб-
разного дейтерия на фторид серебра, помещенный в серебряную колбу [3].
4. Получение больших количеств DF можно проводить синтезом из про-
стых веществ по методу, предложенному для получения фтороводорода [4].
Этот метод требует использования сложной аппаратуры.
5. Водные растворы тяжелой фтороводородной кислоты можно получать
поглощением фторида дейтерия тяжелой водой или путем взаимодействия
очень чистого CaFj с D2SO4 (см. также получение чистой фтороводородной
кислоты в соответствующем разделе).
Свойства. Бесцветная, похожая на воду жидкость с резким запахом. Ды-
мит в атмосфере влажного воздуха. Пары при вдыхании сильно ядовиты. По
химическим свойствам аналогичен фтороводороду. В присутствии ионов Н+
происходит обмен дейтерия на водород. /Пл —83,6 °C; /кип +18,36 °C. Очень
легко растворим в воде.
ЛИТЕРАТУРА
1. Olah G., Kuhn S., Z. Anorg. Allgem.
Chem., 287, 282 (1956).
2. Olah G., Kuhn S„ J. Inorg. Nucl.
Chem., 10, 164 (1959).
3. Clausen W. H„ Hildebrand J. H„
J. Amer. Chem. Soc., 56, 1820
(1934).
4. Wartenberg H., Fitzner 0., Z.
Anorg. Allgem. Chem., 151, 313
(1926).
Хлорид дейтерия DC1
(Тяжелый хлороводород)
Способ 1 [1]
f 2CeH5COCl + D2O -----► (С6Н5СО)2О + 2DC1
281,14 20,03 226,23 74,94
Взаимодействие проводят в аппаратуре, схематически показанной на
рис. 109, причем ее размеры можно изменить с целью получения больших
♦ Сплав 30% меди и 70% никеля. — Прим, перев.
166 Глава 1. Водород, дейтерий, вода
количеств хлорида дейтерия. Подсоединяемая к капельной воронке 5 длин-
ная капиллярная трубка проходит через холодильник 2 в реакционную колбу
и служит для предотвращения неравномерности прикапывания D2O к бензо-
илхлориду при незначительных колебаниях давления во время реакции. Для
задерживания следовых количеств бензоилхлорида, увлекаемых потоком га-
за через холодильник, ловушку 3 погружают в баню со льдом. Открытый с
одного конца манометр 4 служит клапаном избыточного давления. С его по-
мощью можно также следить за ходом реакции: если иа короткое время за-
крыть отводную трубку, то по манометру видно, идет ли выделение газа.
Рис. 109. Часть установки для получе-
ния хлорида дейтерия из бензоилхлори-
да и тяжелой воды.
1 — реакционная колба; 2 — обратный холо-
дильник; 3 — охлаждаемая ловушка; 4 — манен
метр, открытый с одного конца; 5 — капель-
ная воронка с капиллярной трубкой.
Для получения хлорида дейтерия берут, например, 5 мл 99,75%-ного ок-
сида дейтерия и 210 г бензоилхлорида (2—3-кратный избыток), и в колбу
помещают несколько пористых кипятильников. Сначала прибавляют по кап-
лям небольшое количество D2O и осторожно подогревают колбу до тех пор,
пока не установится умеренный поток газа из аппаратуры. Поддерживая в
колбе ту же температуру, прибавляют по каплям остальное количество тя-
желой воды. Скорость газовыделения легко регулировать изменением темпе-
ратуры. При значительном замедлении скорости выделения газа температуру
постепенно повышают до 197 °C (температура кипения бензоилхлорида) н
дожидаются, пока газ не перестанет выделяться. По окончании реакции, не
прекращая нагревания с обратным холодильником, в аппаратуру пропуска-
ют через капельную воронку небольшой поток сухого воздуха. Это способст-
вует полному вытеснению из прибора хлорида дейтерия. Продукт реакции
имеет высокую степень чистоты; выход реакции почти количественный.
Способ 2 [2]
SiCl4 + 2DSO --->- 4DCl+SiO2
169,90 40,06 149,88 60,08
Дейтерий 167
Взаимодействие проводят в колбе вместимостью 5 л со стеклянной проб-
кой, снабженной припаянным краном. В эвакуированной колбе разбивают
путем встряхивания две тонкостенные ампулы, содержащие 18 г тщательно
очищенного SiCl4 и 1,8 г DsO. Через 24 ч колбу припаивают к высоковакуум-
ной установке, после чего производят конденсацию образовавшегося газа в
ловушке, охлаждаемой жидким воздухом. Последующую очистку осущест-
вляют в низкотемпературной дистилляционной колонне (подробности см. в
оригинальной литературе).
Хлорид дейтерия хранят либо в конденсированном состоянии при низких
температурах, либо в виде газа в запаянных стеклянных колбах или в сосу-
дах с ртутным затвором.
Другие способы получения
3. Взаимодействие безводного MgCU с D2O прн 600 °C [3]:
MgCl2 + Р2О -----> 2DC1 + MgO
После перегонки получают очень чистый хлорид дейтерия.
4. Взаимодействие хлорида натрия особой чистоты с D2SO4 [4].
5. Взаимодействие PCI5 с D2O [5].
Свойства. М 37,47. Химические свойства аналогичны свойствам хлорово-
дорода. В смеси DC1 с хлороводородом в газовой фазе в отсутствие влаги и
катализаторов обмена не происходит. Однако при растворении DC1 в рас-
творителях, содержащих ионы Н+, происходит мгновенный ионогепный об-
мен, /Пл —114,7°С; /Кпп —84,75°C; /к₽пт +50,3 °C.
ЛИТЕРАТУРА
1. Brown Н. С., Groot С., J. Amer.
Chem. Soc., 64, 2223 (1942).
2. Clusius K„ Wolf G., Z. Naturforsch.,
A2, 495 (1947).
3. Lewis G. N., Macdonald R. T„
Schutz P. W., J. Amer. Chem. Soc.,
56, 494 (1934).
4. Smits A., Muller G. J., Kroger F. A.,
Z. Physik. Chem. В 38, 177 (1937);
Frioold О. E., Hassel O., Rustad S.,
Phys. Z., 38, 191 (1937).
5. Davies J. B., Hallam H. E., Trans.
Faraday Soc., 67, 3176 (1971).
Бромид дейтерия DBr
(Тяжелый бромоводород)
Способ 1 [1—2]
D2 + Br2 ----* 2DBr
4,03 159,82 163,85
Для синтеза применяют схематически изображенную иа рис. ПО стек-
лянную установку. Установку предварительно в течение долгого времени
эвакуируют через трубку 9. Из капельной воронки 4, не имеющей крана, пу-
тем передвижения штока клапана в колбу 3 вводят тщательно очищенный
бром (см. препарат «Бром»), а затем нагревают колбу 3 до 48°C. По труб-
ке 1 в колбу пропускают сухой дейтерий (см. разд. «Дейтерий») со скоро-
стью около 2 л/ч. Ои проходит через смазанный смесью фосфорной кислоты
с графитом кран 2 с ртутным затвором и насыщается в колбе 3 парообраз-
ным бромом. Бром по мере его расходования в ходе реакции добавляют из
капельной воронки. Смесь дейтерия с бромом поступает в трубку 5 из стек-
ла супремакс, наполненную кусочками фарфора и снабженную нагреватель-
ными обмотками. Температура первого отсека трубки должна быть 80 °C, а
168 Глава 1. Водород, дейтерий, вода
второго — 700 °C. Связывание дейтерия в DBr происходит иа 99%'. Избыточ-
ное количество брома задерживается в охлаждаемой до —40 °C ловушке 6
и в присоединенной к ней колонне 7, наполненной чистыми медиыми струж-
ками. Бромид дейтерия, который конденсируется в ловушке 8, охлаждаемой
жидким воздухом, можно очистить путем многократной фракционной пере-
гонки в высоком вакууме (см. ниже метод очистки иодида дейтерия, а так-
же способ 2 получения DBr). Выход почти количественный.
Рис. ПО. Конструкция установки для получения бромида дейтерия.
1 — трубка; 2 —кран, смазываемый смесью графита с фосфорной кислотой н дополни-
тельно уплотняемый ртутью; 3 — колба, в которой идет насыщение дейтерия бромом;
4— сосуд с бромом; 5 — трубка из стекла супремакс, наполненная кусочками фарфора;
6 и 8 — сосуды для конденсации; 7 — колонна с меднымн стружками; 9 — трубка для
эвакуирования.
Способ 2 [3]
РВг3 4- 3D2O -------> 3DBr + D3PO3
270,70 60,08 245,77 85,01
В эвакуированную колбу вместимостью 5 л, закрывающуюся пришлифо-
ванной пробкой с краном, помещают в заплавленных в вакууме ампулах ис-
ходные вещества и, сильно встряхивая колбу, разбивают ампулы н приводят
вещества во взаимодействие. Для завершения дейтеролиза смесь оставляют
стоять в темноте (время от времени встряхивая ее) в течение 2 сут. Реак-
цию не следует ускорять нагреванием, так как в этом случае будет проте-
кать реакция диспропорционирования:
4D3PO3 -----> 3DsPO4 + PD3
и DBr окажется загрязненным примесью PDs. Затем колбу припаивают к вы-
соковакуумной установке. Неочищенный газ конденсируют в охлаждаемом
жидким воздухом приемнике и затем очищают путем фракционной перегонки
с применением низкотемпературной дистилляционной колонны (см. ч. I,
разд. «Специальные вакуумные установки» и «Перегонка», а также ори-
гинальную литературу).
Недостатком 2-го способа по сравнению с 1-м является то, что использу-
ется лишь половина содержащегося в исходном продукте дейтерия.
Дейтерий 169
Бромид дейтерия сохраняют в конденсированном состоянии при низких
температурах или в газообразном состоянии в заплавленных стеклянных кол-
бах. В реакцию с ртутью чистый DBr вступает лишь при очень продолжи-
тельном контакте.
Способ 3. Водные растворы тяжелой бромоводородной кислоты получают
путем поглощения бромида дейтерия тяжелой водой. Для получения боль-
ших количеств концентрированной тяжелой бромоводородной кислоты про-
водят взаимодействие смеси брома и серы с тяжелой водой [4].
Свойства. М 81,92. Химические свойства аналогичны свойствам бромово-
дорода. В присутствии ионов Н+ происходит обмен дейтерия на водород.
/пл —87,63 °C; /кип —66,85 °C; /крит +88,8 °C.
ЛИТЕРАТУРА
1. Wilson С. L„ Wylie A. W„ J. Chem.
Soc. (London), 593 (1941).
2. Бойцова В. Ф., Русинова К. Д.,
Комере Г. И., Григорьева Л. П.—
Труды Гос. инет, прикл. хим., 1964,
т. 52, с. 98.
3. Clusius К., Wolf G„ Z. Naturforsch.,
A 2, 495 (1947).
4. Leitch L. C., J. Label, Compounds.,
6, 203 (1970).
Иодид дейтерия DI
(Тяжелый иодоводород)
Способ 1 [1—2]
D2 + Is -----* 2DI
4,03 253,81 257,84
Синтез проводят в стеклянной аппаратуре, схематически показанной на
рис. 111, все части которой спаяны между собой. Колбу 1 вместимостью 5 л,
в которой находится небольшое количество губчатой платины или платини-
рованного асбеста (о получении последних см. «Платиновые металлы», ч. И,
гл. 29), предварительно прокаливают в высоком вакууме до 450 °C в течение
нескольких часов (эвакуирование газов производят через патрубок 4)*. За-
тем в колбу впускают сухой воздух, не содержащий водорода (во избежа-
ние адсорбции легкого водорода на платине), и вносят в нее через патру-
бок 4 35 г тщательно очищенного нода (см. разд. «Иод»). Затем в приборе
снова создают вакуум до тех пор, пока весь воздух не окажется полностью
вытесненным из колбы парами иода. После этого при помощи насоса Тёпле-
ра в колбу вводят чистый дейтерий (см. разд. «Дейтерий»), продолжая эту
операцию до тех пор, пока давление не достигнет 120 мм рт. ст.; затем па-
трубок 4 запаивают. После этого колбу нагревают на воздушной бане при
370 °C в течение 6 ч; при этом свыше 90% дейтерия вступает в реакцию с
образованием DI. Из неочищенного газа удаляют избыток исходных веществ
путем фракционной перегонки. Для этой цели правую часть прибора, отде-
ленную от колбы 1 запаянным отростком 2, эвакуируют при открытых кра-
нах 8 и 5. Затем кран 5 закрывают и отросток 2 разбивают бойком 3 с же-
лезным сердечником, передвигаемым электромагнитом. После охлаждения
приемника 6 жидким воздухом можно, закрыв кран 8, открыть кран 5 и пе-
регнать содержимое колбы в приемник 6. Соединительную трубку между кра-
ном 5 и приемником 6 запаивают в точке Ai и, открыв кран 8, в течение ко-
* Этот прием вызывает возражения в отношении техники безопасности.—
Прим, перев.
1.70 Глава 1. Водород, дейтерий, вода
роткого времени создают в приборе вакуум, после чего вещество вторично
перегоняют из приемника 6 в приемник 7. При этом последний охлаждают
жидким воздухом, а приемник 6 нагревают охлаждающей смесью с сухнм
льдом до —78 °C. Наконец, соединительную трубку между приемниками 6
•и 7 запаивают в точке Д2. Иодид дейтерия, содержащийся в приемнике 7,
имеет чисто белый цвет, т. е. совершенно не содержит свободного иода.
Рис. 111. Конструкция установки для получения иодида дейтерия.
J — колба вместимостью 5 л с катализатором; 2— разбиваемый клапан; 3—боек; 4 —
«патрубок; 5 — кран; 6 н 7 — сосуды для конденсации; 8 — кран; Tlj и Ла—места отпайки.
Иодид дейтерия можно сохранять только в конденсированном виде прн
иизкой температуре.
Другие способы получения
2. 2P + 5I2 + 8D2O -----> 10DI + 2D3PO4 [3]
В качестве побочных продуктов образуются PD3 и PD<I, загрязняющие
иодид дейтерия. Применяя этот метод, удается использовать примерно лишь
половину взятого для реакции дейтерия.
3. Растворы тяжелой иодоводородной кислоты получают в результате
взаимодействия сульфида дейтерия с иодом в присутствии оксида дейтерия
14]: D2S+I2—»-2DI+S. Процесс проводят в циркуляционной стеклянной ап-
паратуре, части которой спаяны между собой. В суспензию иода в оксиде
дейтерия пропускают при охлаждении льдом и при встряхивании сульфид
дейтерия. Не вступивший в реакцию D2S снова вводят в реакционную смесь.
«Образовавшуюся тяжелую иодоводородную кислоту отделяют от выделив-
шейся серы фильтрованием без доступа воздуха, после чего путем создания
вакуума в течение длительного времени освобождают от растворенного в ней
сульфида дейтерия.
Свойства. М 128,92. Химические свойства аналогичны свойствам иодово-
дорода. В присутствии ионов Н+ происходит обмен < дейтерия на водород,
/пл —51,82 °C; /кип —36,2 °C; /крит +148,6 °C.
ЛИТЕРАТУРА
1. Rittenberg D., Urey Н. С., J. Amer.
Chem. Soc., 56, 1885 (1934).
2. Bates J. R., Halford J. 0., Ander-
son L. C., J. Chem. Phys., 3, 415
«(1935).
3. Clusius K„ Wolf G., Z. Naturforsch.,
A 2, 495 (1947).
4. Erlenmeyer H., Gartner H., Helv.
Chim. Acta, 19, 146 (1936).
Дейтерий 171
Сульфид дейтерия D2S
(Тяжелый сероводород)
Способ 1 [1—5]
AljSs + 6D2O -----> 3D2S-|-2Al(OD)g
150,15 120,17 108,28 162,04
Для получения сульфида алюминия [1] (см. 1 также ч. II, разд. «Алюми-
ний») глиняный гессенский тигель, выложенный серой, заполняют смесью по-
рошка алюминия и серы (препараты наивысшей , чистоты), взятых в стехио-
метрических количествах. Продажный порошок алюминия предварительно
многократно промывают для удаления приставшего к нему масла чистым , (не
оставляющим прн перегонке остатка) бензолом, а затем нагревают в течение
долгого времени,в высоком вакууме при 150°C. Реакционную смесь поджи-
гают при помощи ленты магния (осторожно — очень бурная реакция!), после
чего тигель закрывают крышкой. Образовавшийся сульфид измельчают еще
в горячем состоянии,,а затем помещают в ампулы и удаляют из него газы
путем многочасового нагревания в высоком вакууме при'150—180 °C. После
этого ампулы запаивают в вакууме. Предназначенный для проведения реак-
ции оксид дейтерия помещают в небольшие ампулы, которые после тщатель-
ного удаления из них в вакууме воздуха запаивают.
Для получения D2S около 20 г A12S3 и 7 г D2O (благодаря 'избытку
Al2Sg достигается хорошее высушивание образующегося газа) помешают в>
двух < запаянных ампулах в сосуд вместимостью 5 л с пришлифованной проб-
кой, снабженной краном. После эвакуирования до давления ~ 10-4 мм рт.
ст. кран закрывают и трубку, присоединяющую сосуд к вакуумной установ-
ке, • запаивают. После этого путем встряхивания сосуда ампулы разбивают и
таким образом приводят во взаимодействие содержащиеся в них вещества.
Пары оксида дейтерия, конденсирующиеся на верхних стенках склянки, вво-
дят в реакцию'либо путем нагревания, либо засыпая соответствующие участ-
ки не вступившим в реакцию сульфидом алюминия. После этого смесь остав-
ляют стоять в темноте (время от времени, встряхивая ее) в течение недели.
Затем сосуд припаивают к вакуумной установке с несколькими ловушками,
предназначенной для проведения фракционной конденсации < (см. ч. I, рис. 46).
Газ сначала освобождают путем вымораживания при помощи жидкого воз-
духа от небольших количеств дейтерия, а затем < фракционируют путем мно-
гократной медленной перегонки (бани с охлаждающей смесью иа основе су-
хого льда и с жидким воздухом). После этого сульфид дейтерия настолько'
чист, что он уже не оказывает действия на металлическую ртуть даже при
соприкосновении с ней в течение 'Недели. Выход несколько ниже теоретиче-
ского.
Сульфид дейтерия можно сохранять в коиденсироваииом виде при низ-
кой температуре или в газообразном виде иад сухим вазелиновым маслом.
Способ 2. Разложение CaS оксидом дейтерия в присутствии MgCl2 [6J.
Свойства. М 36,09. Химические свойства аналогичны свойствам H2S. При
растворении в содержащих ноны Н+ растворителях дейтерий обменивается
на водород. tnn —86,01 °C;' 1кржт +99,1 °C.
ЛИТЕРАТУРА
1. Fonzes-Diacon М., С. R. Acad. Sci.,
130, 1314 (1900).
2. Kruis A., Clusius К.., Z. Physik.
Chem., В 38, 156 (1937).
3. Erlenmeyer H., Gartner H„ Helv.
Chim. Acta., 19, 146 (1936).
4. Frivold О. E., Hassel O. et al., Phys.
Z„ 39, 224 (1938); 38, 191 (1937).
5. Clarke E. C. W., Glew D. N.. Ca-
na d. J. Chem., 48, 764 (1970).
6. Larsen T„ Z. Physik., Ill, 391
(1938).
172 Глава 1. Водород, дейтерий, вода
Дейтеросерная кислота (безводная) D2SO4
(Тяжелая серная кислота, сульфат дейтерия)
Способ 1 [1—4]
D2O ~Т SO3 * D2SO4
20,03 80,06 100,09
100%-ную дейтеросериую кислоту получают взаимодействием серного ан-
гидрида со стехиометрическим количеством оксида дейтерия. Для получения
необходимого • количества SO3 (около 10 г) используют прибор, схема кото-
Рис. 112. Конструкция Рис. 113. Конструкция прибора для получения дей-
прибора для получе- теросерной кислоты (2—8, А3—As см. текст).
«ия SO3.
рого показана на рис. 112. В колбу 1 помещают высокопроцентный олеум.
Колбу охлаждают, прибор эвакуируют и запаивают в точке Др-Затем пре-
кращают охлаждать колбу 1, а ловушку 2 помещают в сосуд Дьюара-с жид-
ким воздухом. После перегонки требующегося количества триоксида серы пе-
репаивают-в точке Л2. Дальнейшую очистку SO3 путем его сублимации в ва-
кууме производить, как правило, необходимости иет.
Верхний конец ловушки 2 (свободный от SO3) процарапывают стеклоре-
зом и взвешивают с точностью-0,1 мг. Затем охлаждают ловушку 2 до
—78 °C, отламывают верхний кончик и быстро переносят в открытую трубку
8 аппаратуры для проведения реакции - (рис. 113), которую сразу же запаи-
вают в точке Д3. Ловушку 4 охлаждают до —78 °C и производят очень мед-
ленную откачку аппаратуры, так как в противном случае часть SO3 может
«проскочить» через ловушку 4 и попасть в насос. После полной конденсации
серного ангидрида в ловушке 4 производят отпайку. в точке Д4. Количество
введенного в аппаратуру триоксида серы вычисляют после взвешивания обе-
их частей ловушки 2, причем следует учитывать массу воздуха (около
1,2 мг/мл внутреннего объема). Рассчитанное количество тяжелой воды
^2,5018 г на 10,0000 г SO3) вводят в-ловушку 6. Можно и здесь избежать
введения поправки на массу воздуха, если сначала взвесить эвакуированную
ловушку 6, потом заполнить ее сухим воздухом, а тяжелую воду в неболь-
шом избытке ввести через воронку с капиллярной трубкой, проходящей через
Дейтерий 173
отверстие краиа (см. рис. ИЗ). Постепенно откачивая в вакууме (при за-
крытом кране 8) избыток тяжелой воды, доводят ее количество в ловушке
до требуемого по расчету. После этого ловушку 5 охлаждают, открывают
кран и, полностью перегнав тяжелую воду в ловушку 5, отпаивают в точке
Ла. После оттаивания содержимого ловушек 4 и 5 осторожно проводят про-
цесс взаимодействия.
Полученная таким образом дейтеросерная кислота плавится при темпе-
ратуре около 11 °C. Ее можно очистить путем фракционной кристаллизации.
После пятикратного повторения кристаллизации температура плавления по-
вышается до 14,10 °C.
D2SO4 сохраняют в стеклянных сосудах.
Способ 2. Растворы D2SO4 в D2O можно получить путем электролиза
раствора безводного CuSO4 в D2O или при дейтеролизе SO2C12 [5].
Дейтеросерную кислоту (96—98%) выпускает фирма Merck (Darmstadt).
Свойства. Бесцветная жидкость с консистенцией масла. По химическим
.свойствам аналогична H2SO4. Между дейтерием и легким водородом проис-
ходит ноногенный обмен. Это следует учитывать при растворении в раство-
рителях, содержащих ионы Н+. С водой смешивается во всех отношениях,
/пл 14,35 °C; df* 1,8572.
ЛИТЕРАТУРА
1. Greenwood N. N., Thompson А., 4. Smith F.f Diehl H., Anal. Chem.,
Inorg. Synth., 6, 121 (1960). Proc. Int. Sympos., Birmingham
2. Herber R. H., Inorg. Synth., 7, 155 Univ. (U. K..), 1962, 394 (1963).
(1963) . 5. Freeman J. H., Richards С. E. C.,
3. Feher F„ Ber., 72, 1789 (1939). Atomic Energy Res. Establ. AERE
GP/R 2479 (1958).
Дейтероаммиак ND3
(Тяжелый аммиак)
Mg8N2 + 6D2O -----> 2NDS-J-3Mg(OD)2
100,95 120,17 40,10 181,02
Для проведения синтеза используют аппаратуру из стекла пирекс, схема
которой показана на рис. 114. В колбочку 3 вносят нитрид магния, а в ка-
пельную воронку 2 — тяжелую воду. Путем вращения колбы 3 нитрид маг-
ния перемещают в колбу i 1 и при интенсивном перемешивании магнитной ме-
шалкой 7 по каплям добавляют тяжелую воду. Образовавшийся в результате
реакции ND3 проходит через обратный холодильник 4,. через который про-
пускают жидкость при температуре —40 °C, причем в нем конденсируется
большая часть увлеченной с ND3. тяжелой воды. После прохождения через
две наполненные нитридом магния колонны 5 и б ND3 конденсируется в ох-
лаждаемой до —78°C ловушке.8. После этого 90% ND3 перегоняется из
ловушки 8 в ловушку 9, в которую для окончательного удаления примесей
D2O помещают немного металлического натрия. Наконец, ND3 перегоняют
в ловушку 10, а оттуда он может быть по мере необходимости переведен в
отпаиваемые ампулы 11. Ртутные манометры 12 служат также клапанами
избыточного давления.
Если желательно повысить выход, то • после проведения дейтеролиза ре-
акционный сосуд 1, содержащий гель состава Mg(OD)2-nD2O, нагревают до
300 °C и закрывают кран перед ловушкой 9. При этом в 8 конденсируется
раствор ND3 в D2O, который используют при проведении следующего опыта.
174 Глава 1. Водород, дейтерий, вода
В этом случае общий выход составляет около 90%. В описанной выше ап-
паратуре за день можно получить до 100 мл ND3.
ND3 сохраняют в конденсированном состояний при низких температурах
яли в газообразном виде в сосудах с ртутным затвором.
Рис. 114. Схема установки для получения дейтероаммиака (1—12 см. текст}.
Водные растворы дейтероаммиака получают путем конденсации ND3 в
сосудах с D2O в высоком вакууме. ND3 (в виде 20%-ного раствора в D2O>
выпускается фирмой Merck (Darmstadt).
Свойства. М 20,05. По химическим свойствам аналогичен NH;j. В присут-
ствии растворителей, диссоциирующих с образованием ионов водорода, дей-
терий обменивается на водород. 1пл —74,36 °C; —31,04 °C; <крИт 132,3 °C.
ЛИТЕРАТУРА
1. Bouclier Р., Portier Bull. Soc.
Chim. France, 738 (1967).
2. Smits A., Muller G. J., Kroger F. A.,
Z. Physik. Chem., (B), 38, 177
(1937).
3. Hart A. B.. Partington 1. R„ J.
Chem. Soc. (London), 104 (1943).
4. Frivold О. E., Hassel 0., RuStad S.,
Phys. Z„ 38, 191 (1937).
5. De Bruyne J. M. A., Smyth С. P.,
J. Amer. Chem. Soc., 57, 1203
(1935).
6. Chevallier M., Jullien J., Bull. Soc.
Chim. France, 4937 (1968).
Дейтерофосфорная кислота (безводная] D3PO4
(Тяжелая фосфорная кислота, фосфат дейтерия)
Р4О10 + 6D2O --> 4D2PO4
283,89 120,17 404,06
Безводную дейтерофосфорную кислоту получают взаимодействием в ва-
кууме возогнанного оксида фосфора (V) со стехиометрическим количеством
тяжелой воды. Сублимацию Р4Ою проводят в аппаратуре, показанной на
рис. 115, которую предварительно прогревают небольшим пламенем горелки
Дейтерий 175
в вакууме. В колено 1 помещают около 15 г Р40ю (марки ч. д. а) и затем
запаивают в точке Ai (вблизи места запаивания на стенках не должно быть
оксида фосфора во избежание расстекловывания при сильном нагревании).
При подсоединенном высоковакуумном насосе производят возгонку оксида
фосфора в колено 2 U-образной трубки, после чего отпаивают в точках Л»
и Л3. Далее проводят сублимацию в колено 3 и затем отпаивают в точке At.
Необходимую для сублимации температуру создают при помощи нагрева-
Рис. 115. Конструкция при-
бора для сублимации
Р4О10 (7—3, Ai—А4 см.
текст).
тельной спирали (500 Вт), намотанной па соответствующее место ампулы
(с прокладкой из асбестовой бумаги) н включаемой через регулирующий
трансформатор. Перетяжки в точках Л2 и А« нельзя делать очень узкими, так
как в противном случае оии могут легко забиться оксидом фосфора. Пере-
тяжки с целью их освобождения от оксида фосфора следует время от вре-
мени прогревать горелкой.
Перенос вещества в реакционную аппаратуру и его взаимодействие с тя-
желой водой осуществляют 'в основном так же, как при получении D2SO4
(см. выше). Различие состоит лишь в том, что благодаря незначительной ле-
тучести Р4О10 не нужно охлаждать соответствующую ловушку/Ампулу 3 с
оксидом фосфора взвешивают и перегоняют оксид в конец ампулы.
Свободный кончик ампулы отламывают и быстро вводят ампулу в от-
крытую трубку 4 реакционной аппаратуры (рис. 116), после чего трубку сра-
зу же запаивают. Аппаратуру эвакуируют и Р4Ою количественно переводят
сублимацией в нижнюю часть ловушки 5. Нагреванием горелкой освобожда-
ют перетяжку Д5 от последних следов оксида фосфора и затем отпаивают.
Введенное количество оксида фосфора устанавливают, взвешивая обе части
ампулы 3 с учетом массы воздуха (см. разд. D2SO4). Вычисленное количест-
во тяжелой воды (из соотношения 4,2328 г на 10,0000 г Р40,о) помещают в
ловушку 6 таким же образом, как это описано в способе 1 получении D2SO4.
Части 5 н 6 соединяют через шлиф 7, что приводит к протеканию сильно
экзотермического поглощения тяжелой воды оксидом фосфора. Через не-
сколько часов (или на следующий день) последние следы тяжелой воды пе-
регоняют из 6 в 5 путем охлаждения части 5 до —78 °C. После этого отпаи-
вают в точке Ае н для завершения дейтеролиза выдерживают ловушку 5 в
течение трех дней при температуре 95—100 °C. Полученный таким образом
продукт взаимодействия (/пл 42,6 °C) имеет стехиометрический состав D3PO4,
но содержит небольшое количество конденсированных фосфорных кислот, на-
пример ПзО+ПзРгО?-. Если от последних желательно избавиться, вещество
переносят в колено U-образной трубки и подвергают многократной медлен-
176 Глава 1. Водород, дейтерий, вода
ной фракционной кристаллизации. При этом остающуюся жидкую фракцию
всегда сливают в другое колено трубки путем декантации. Затем твердое ве-
щество расплавляют, нагревая его до 48—50 °C и оставляя лишь один крис-
талл в качестве зародыша. После повторения этой операции несколько, раз
температура плавления кислоты достигает 45,95 °C.
Рис. 116. Конструкция прибора для получения безводной дейтерофосфорной
кислоты (4—7, As, Ав см. текст).
Свойства. М 101,02. В твердом состоянии сохраняется неограниченное
время, тогда как при плавлении всегда образуется незначительное количест-
во конденсированных фосфорных кислот. По химическим свойствам аналогич-
на ортофосфорной кислоте. 49,95 °C; d425 1,9082; по20 1,4430.
ЛИТЕРАТУРА
1. Greenwood N. N., Thompson А., 2. Simon A., Schulze G., Z. Anorg.
Inorg. Synth., 6, 81 (1960). Allgem. Chem., 242, 326 (1939).
Глава 2. ПЕРОКСИД ВОДОРОДА Н2О2
М. Шмайсер, Ф. Губер
(М. Schmeieser, F. Huber)
Перевод канд. хим. наук С. И. Троянова
Правила техники безопасности
При всех работах с концентрированным пероксидом водорода, особенно»
в присутствии органических веществ или катализаторов разложения, следует
на случай возможного разложения со взрывом принимать соответствующие
меры предосторожности. К ним относится использование защитных очков и
масок, ограждение аппаратуры защитными экранами из органического илю
безосколочного стекла.
Стеклянные приборы, в которых получают пероксид водорода или рабо-
тают с ним, должны быть изготовлены из стекла пирекс или йенского стекла.
Приборы перед употреблением обрабатывают концентрированной серной кис-
лотой или олеумом (но не хромовой смесью!), основательно промывают ди-
стиллированной водой и затем высушивают, не допуская попадания пыли.
Удаление воды из Н2О2
Способ 1. По методу, впервые описанному Штеделем [1] и позднее не-
однократно применявшемуся другими авторами [2—8], значительную часть,
воды из 30%-ного Н2О2 удаляют перегонкой; полученный Н2О2 кристаллизу-
ют при охлаждении и кристаллы отделяют от маточного раствора.
Кери нормального шлифа дистилляционной колбы вместимостью 500 мда
закрывают муфтой с кипятильным капилляром. Колбу соединяют на шли-
фах со змеевиковым холодильником и далее с приемником вместимостью»
200 мл. В колбу наливают 180 мл продажного 30%-ного пероксида водорода
(ч. д. а.) и помещают иа водяную баню с температурой 45—50 °C. Перегонку
проводят при давлении 16—22 мм рт. ст. в течение ~3,5 ч. При этом отго-
няется 150—160 мл воды и немного Н2Ог, а остаток представляет собой 95—
98%-ный пероксид водорода. На колбе приемника предварительно наносят
метку в соответствии с объемом воды, который необходимо отогнать. Если>
температура водяной бани поднимается выше 52 °C, высококонцентрирован-
ный Н2О2 приобретает желтую окраску и становится непригодным для даль-
нейшей работы. Концентрированный пероксид водорода можно вылить нз-
колбы, не опасаясь разложения. (Если вместо керна горло колбы имело бы
шлифованную муфту, то при соприкосновении с шероховатой поверхностью
могло бы произойти разложение значительной части Н2О2.)
Дальнейшую переработку для получения 100%-ного Н2Ог ведут следую-
щим образом. Короткую широкую пробирку, покрытую внутри парафином?
или изготовленную из полиэтилена, хостафлона или тефлона, вместимостью
25—30 мл наполняют до половины высококонцентрированным пероксидом во-
дорода, закрывают полиэтиленовой пробкой и ставят на 0,5 ч в охлаждаю-
щую баню с температурой —35 °C. Между тем приготовляют затравочные
кристаллы, замораживая 1 мл того же Н2О2 жидком воздухом (диаграмму
плавкости системы Н2О2—Н2О см. в работе [9]). После внесения затравки
тотчас же начинают расти игольчатые бесцветные кристаллы. Выждав около-
1 мин, кристаллы быстро переносят в предварительно охлажденный прибли-
зительно до —30 °C сосуд для центрифугирования (рис. 117). После кратко-
12—143
078 Глава-2. Пероксид водорода
«временного центрифугирования (простым вращением рукой или при помощи
ручной центрифуги) кристаллы переносят в другую пробирку большого диа-
метра и снова доводят их до плавления. Для ускорения плавления пробирку
«с Н2О2 помещают в стакан с водой, подогретой до 30 °C. После полного рас-
плавления пероксид охлаждают опять до —35 °C. После выдерживания в тече-
ние 10 мин в охлаждающей бане обычно самопроизвольно начинают выпадать
(игольчатые бесцветные кристаллы, которые сразу же отделяют от маточного
раствора во втором таком же сосуде для центрифугирования. Если же кристал-
лизация не начинается самопроизвольно, вносят затравочные кристаллы.
Рис. 117. Сосуд для центрифугирования
кристаллов чистого пероксида водорода.
Полученные кристаллы при комнатной температуре очень легко разлага-
чотся с выделением кислорода. Поэтому их необходимо сохранять иа холоду
в закрытых полиэтиленовых или стеклянных парафинированных сосудах. Не-
продолжительное хранение можно осуществлять и в непарафииироваиных
стеклянных сосудах. Полученные при центрифугировании водные растворы
пероксида водорода можно снова сконцентрировать путем перегонки.
При проведении однократной кристаллизации 98%-ного раствора в про-
бирке с последующим сливанием маточного раствора можно получить не бо-
лее чем 99%-ный Н2О2.
Способ 2 [10]. 80—90%-ный продукт получают, смешав 30%-ный Н2О2
с двойным количеством пара,-цимола и отогнав из этой смеси в вакууме во-
доструйного насоса при температуре ~50°С большую часть воды и пара-
цимола. Остаток, представляющий собой двухфазную смесь пара-цимола и
шерокснда водорода, разделяют механически. Дальнейшую переработку ведут,
как это было описано выше.
Способ 3. Небольшие количества Н2О2 можно получить простым способом
[11] путем экстракции эфиром. Для этого смешивают 10 мл 90—98%-ного
•пероксида водорода и 100 мл диэтилового эфира. Из образовавшейся двух-
фазной смеси отделяют эфирный слой, который затем высушивают взбалты-
ванием с СаС12 и два или три раза с Р«Ою. Если для реакции нельзя непо-
средственно использовать полученный эфирный раствор, то безводный пер-
оксид водорода получают (после удаления эфира в вакууме водоструйного
насоса) путем фракционной перегонки в высоком вакууме. Однако, согласно
[12], полученный таким путем Н2О2 может быть загрязнен продуктами окис-
ления эфира.
Другие способы получения. Описан метод получения 99—99,7%-ного пер-
оксида водорода путем многократной фракционной перегонки 60—90 %-него
„раствора, производимой в колонне в высоком вакууме [13, 14].
Удаление воды из Н2О2 17Ф
Получение Н2О2 спектральной чистоты можно осуществить по способу,,
предложенному для синтеза D2O2 [15]. Этот способ, опирающийся на работу'
[16], основан на взаимодействии персульфата с водяным паром.
Метод получения монокристаллов пероксида водорода описан в работе.-
[17].
Свойства. М 34,015. /пл —0,43 °C; /кип (экстраполировано) 157,8 °C; d№
1,47 (0°С), 1,45 (20°C); dTB 1,64. Строение кристаллов Н2Ог описывается7
в пр. гр. P4i2i [18, 19].
ЛИТЕРАТУРА
1. Staedel W., Z. Angew. Chem., 15,
642 (1902).
2. Ahrle H„ Dissertat. Darmstadt,
1908; J. Prakt. Chem., 79, 139
(1909).
3. D’Ans J„ Friederich W„ Z. Anorg.
Allgem. Chem., 73, 326 (1912).
4. Maas O., Hatcher IV. H., J. Amer.
Chem. Soc., 42, 2548 (1920).
5. Haschke E., Diplomarbeit, Konigs-
berg, 1943.
6. Feher F., частное сообщение.
7. Foley W. T„ Gigudre P. A., Ca-
nad. J. Chem., 29, 123 (1951).
8. Giguere P. A., Bull. Soc. Chim.
France, 720 (1954).
9. Maas 0., Herzberg O. W.. J.
Amer. Chem. Soc., 42, 2569 (1920).
10. Hurd C. D., Puterbaugh M. P., J.
Amer. Chem. Soc., 52, 950 (1930).
11. Schmidt M., Bornmann P„ Z.
Anorg. Allgem. Chem., 330, 309»
(1964).
12. Dove J. E., Riddick J., Canad. J.
Chem., 46, 330 (1968).
13. Giguere P. A., Liu J. D„ Dugda-
le J. S„ Morrison J. A., Canad. J.
Chem., 32, 117 (1954).
14. Dirscherl U7., Moersler B„ Liebigs
Ann. Chem., 677, 177 (1964).
15. Feher F., Ber., 72, 1789 (1939).
16. Pietzsch A., Adolph G„ пат. ФРГ
241702, 243366, 256148, 293087.
17. Feher F., Klotzer F., Z. Elektro-
chemie, 43, 822 (1937).
18. Abrahams S. C., Collin R. L., Lips-
comb IV. N., Acta Cryst., 4, 15
(1951).
19. Linke К. H., Klaeren A., Acta>
Cryst., Sect. B, 24, 1619 (1968).
12*
(Глава 3.
ФТОР
В. Квасник (W. Kwasnik)
Перевод канд. хим. наук Т. И. Почкаевой
Общая характеристика фтора и его соединений
'Среди галогенов фтор занимает в периодической таблице совершенно исклю-
чительное положение. Поэтому методы препаративного получения соединений
«фтора отличаются от методов получения соединений других галогенов на-
столько, что должны рассматриваться особо.
Неорганические соединения фтора обычно получают с помощью следую-
щих методов.
1. Взаимодействие оксидов, гидроксидов и карбонатов с плавиковой кис-
лотой (водный раствор фтороводорода). Таким способом можно получить
'большинство фторидов, не подвергающихся гидролизу (например, фториды
щелочных и щелочноземельных металлов, бифторнды щелочных металлов,
A1F3, SbF3, ZnF2, PbF2, Hg2F2, AgF).
2. Взаимодействие соответствующих безводных хлоридов с сухим фторо-
водородом (например, TiF4, ZrF4, NbF5, TaF5, VF4, SnF4, SbF5, POF3, SOF2).
Этот метод получил наибольшее распространение из всех до сих пор пор опи-
санных в литературе. После того как безводный фтороводород стал постав-
ляться через торговые организации, значение этого метода еще более повы-
силось.
3. Взаимодействие элементов, оксидов и галогенидов со свободным фто-
фом. Таким способом получают фториды элементов в наивысшей степени
окисления (например, IF7, ReF7, UF6, SF6, BiFs, CF4, C0F3, AgF2). К разно-
видностям этого же препаративного метода относятся синтезы, выполняемые
ш кипящем слое (BiFs, PbF4, MnF4, ReF7), под давлением в автоклавах
‘(CrF5, XeFs), а также при низких температурах в неводных растворителях
(IF3). Галогениды щелочных металлов образуют с F2 комплексные соли (на-
шример, KC1F4, KBrF6). Вместо свободного фтора часто можно использовать
галогенофториды, для получения которых, однако, также необходим свобод-
«ный фтор. Неудобство применения галогенофторидов как фторирующих аген-
тов состоит в трудности отделения продукта реакции от образующегося в
результате фторирования свободного галогена. В то же время в лаборатории
« галогенофторидами легче работать (дозировать, хранить), чем со свобод-
шым фтором. Если нужно присоединить по ненасыщенной связи наряду с
'фтором еще и второй галоген (например, COC1F, COBrF, COIF, SO2BrF), то
безусловно следует предпочитать галогенофториды (если они имеются). Га-
логенофториды как фторирующие агенты обладают различной активностью,
например: C1F3 сравнительно активен, IFs — мягкий фторирующий агент.
4. Некоторые фториды получают обработкой NaF при сплавлении или в
неводных (индиферентных) растворителях (например, SOF2, SO2F2, COF2,
CSF2).
5. Взаимодействие галогенангидридов кислот с высокодисперсным «акти-
вированным» KF (фторосульфонатом калия). Так получают фториды, кото-
рые иначе можно получить только с помощью безводного фтороводорода
<SOF2, PF3, POF3, AsF3).
6. Взаимодействие оксидов с HSO3F используется для получения немно-
гих фторидов (AsF3, SiF4).
Общая характеристика 181
7. Оксиды можно переводить во фториды с помощью бензоилфторида
(например, BF3).
8. Электрохимическое фторирование оказалось весьма полезным методом
для получения некоторых фторидов (например, NF3).
9, Путем присоединения галогенов к фторидам элементов в низших сте-
пенях окисления получают смешанные фторсодержащие галогениды (PC12FS,
PC14F, PC13F2, SbCl2F3).
10. Низшие фториды получают при взаимодействии фторидов элементов
в более высокой степени окисления с соответствующим элементом (GeF2,
MoF3).
11. Оксофториды иногда получают при взаимодействии фторидов хлора
с хлорнитратом (SbO2F, SbOF3, SnOF2).
12. Широко применяется как фторирующий агент SF4 (Na2SnF4, CrO2F2).
13. Специальные реакции. Методик на основе таких реакций очень мно-
го, и их трудно систематизировать. Например, OF2 получают при взаимодей-
ствии F2 с 2%-ным гидроксидом натрия; O2F2 образуется из F2+O2 в газо-
разрядной трубке в условиях тлеющего заряда при охлаждении жидким
воздухом.
Некоторые соединения фтора можно получить с помощью разных ме-
тодов, поэтому методику синтеза выбирают, обращая внимание на имеющие-
ся в распоряжении исходные вещества и приборы.
Органические фторсодержащие соединения также получают с помощью
совершенно других методов, чем обычные органические галогенсодержащие
соединения. Поскольку CF4 и HF характеризуются очень большими теплотами
образования (967-103 и 268-103 Дж соответственно), действие фтора на ор-
ганические молекулы вызывает главным образом обугливание и осмоление,
а также образование CF4 и HF; при этом нормальный продукт замещения не
образуется даже в очень малых количествах. Обработка фтором использу-
ется в промышленности для получения высокофторированных углеводородов;
однако такая методика совершенно не подходит для лабораторных синтезов.
Для получения органических фторсодержащих соединений с удовлетвори-
тельными выходами используют в основном следующие методы.
1. Присоединение HF к олефинам (например, получение этилфторида).
2. Взаимодействие хлорсодержащих соединений с безводным фтороводо-
родом (получение, например, бензотрифторида). Этим методом можно полу-
чить только трифторированные соединения. Таким же образом можно полу-
чить фторангидриды из хлорангидрида.
3. Взаимодействие хлорсодержащих соединений с трифторидом сурь-
мы (III) (по Шварцу). Этот метод наиболее подходит для получения соеди-
нений, содержащих менее трех атомов фтора (2,2-дифторпропан).
4. Взаимодействие хлорсодержащих соединений с безводным фтороводо-
родом в присутствии сурьмы и ее соединений как катализаторов (дихлорди-
фторметан). Этот метод занимает как бы промежуточное положение между
вторым и третьим и очень часто используется.
5. Взаимодействие галогенсодержащих соединений с фторидами метал-
лов, такими, как AgF, HgF, HgF2 (получение фтороформа).
6. Диазотирование нитритов в растворах плавиковой кислоты. Этот ме-
тод подходит для получения ароматических фторсодержащих соединений
(фторбензол).
7. Термическое разложение диазоборфторидов (по Вальцу и Шиману).
Этот метод применяется главным образом для получения ароматических фто-
рированных производных, особенно подходит для лабораторных синтезов
(п-фтортолуол).
В лабораториях при выполнении работ с фтором применяют непохожие
на обычные приборы и посуду, так как приходится отказаться от традицион-
ного материала — стекла. Приборы изготовляют из никеля, железа, меди,
свинца, серебра, платины, плавикового шпата и спеченного глинозема. Ес-
182 Глава 3. Фтор
ли нельзя обойтись без прозрачных сосудов, то их изготовляют из кварца.
Все чаще применяют трубки и ловушки для газов из полиэтилена.
Для того чтобы каждый синтез фторидов не требовал длительной подго-
товки, рекомендуется иметь в лаборатории следующие часто используемые
приборы и посуду:
нескольколодочек из никеля, плавикового шпата или спеченного глино-
зема;
реакционную трубку из никеля или монеля (длина 30 см, диаметр
2,5 см), имеющую шлифы с обоих концов;
реакционную трубку из железа;
несколько цилиндров из железа;
3—5 газовых ловушек из кварца с подходящими шлифами на концах;
3 газовые ловушки из йенского стекла или полиэтилена с подходящими
шлифами на концах;
несколько осушительных («хлоркальциевых») трубок из кварца;
несколько U-образных трубок из кварца;
2 холодильника из железа;
1 газовую ловушку из железа;
несколько стальных сосудов объемом 0,5—5 л (для хранения газообраз-
ных фторидов).
Для выделения и очистки низкокипящнх фторидов преимущественно ис-
пользуется вакуумная установка со спиральным кварцевым манометром, из-
готовленная частично из кварца, частично из стекла.
Отдельные части прибора соединяют с помощью нормальных шлифов,
смазанных фторированной смазкой (см. ч. 1); герметичность шлифов можно-
обеспечить также с помощью специальных плотно закрепленных на кернах
тоиквх тефлоновых манжет или с помощью пицеина. При соединении метал-
лических шлифов с кварцевыми также используют пиценн; предварительно-
металлическую часть соединения надо разогреть, чтобы пицеин прочно дер-
жался. На краны наносят очень мало смазки, лучше всего густой фтороугле-
родной, или в очень трудных случаях используют мембранные вентили из
меди. Металлические сосуды и части прибора привинчивают друг к другу, а
также соединяют с помощью фланцев и шлифов. Кольцевые прокладки изго-
товляют из свинца с асбестовыми вкладышами или из мягких сплавов желе-
за, а также из меди и тефлона. Стальные баллоны и автоклавы должны хо-
рошо закрываться винтовыми вентилями, в которых шпиндель изготовлен нз
стали, клапаны — из латуни, а прокладки — из свинца и асбеста.
Изготовление сосудов (посуды) из плавикового шпата. Осажденный CaF2
размешивают, добавляя воду до образования густой кашицы. К пластичной
массе добавляют соляную кислоту до кислотности суспензии (шликера)
0,02 н. Для формования шликера его выливают в гипсовую форму. Форм»
для изготовления лодочек состоит из двух частей, а для трубок — из трех.
Заготовку вынимают из формы и, если необходимо, исправляют с помощью
шпателя; сушат на воздухе несколько дней. Поскольку после этого изделие
имеет незначительную твердость, все операции по дальнейшей его обработке
нужно осуществлять очень осторожно. Если нельзя использовать обычные
фабричные печи с фаянсовой керамикой, дающие 1250 °C, то обжиг изделия
проводят в печах с силитовыми стержнями. Изделие на подставке из ZrO2
помещают в фарфоровую трубку и медленно нагревают печь до 1250 °C. Од-
новременно через трубку пропускают поток сухого N2, чтобы удалить пары
воды и СО2. Полученное таким образом изделие имеет плотную и гладкую
поверхность. Изделие можно обрабатывать на увлажненном наждачном кру-
ге. Однако изделие хрупко, поэтому при его использовании надо быть осто-
рожным. Сопротивление разрыву (300 бар) совершенно недостаточно. Устой-
чивость к повышенным температурам оставляет желать много лучшего. При
высушивании и обжиге изделие усыхает на 1/3, что надо учитывать при вы*
Фтор 183
«боре .размера заготовки. Изготовление лодочек не вызывает затруднений,
в то время как трубки длиной 15 см и внутренним диаметром 1,5 см (толщи-
ща стенок 2 мм) иногда деформируются при вытаскивании из печи для об-
жига [1—3].
Если применяют кварцевую, а тем более стеклянную, посуду и приборы,
то надо учитывать, что препарат загрязняется фторсиликатами и H2SiFe- По-
лученные в таких условиях газообразные фториды часто содержат SiF<.
Если газообразные фториды конденсируются при глубоком охлаждении,
то они растворяют наряду с любыми другими низкокипящими веществами в
незначительном количестве компоненты воздуха. Следует обратить внимание
иа то, что воздух удаляется только при многократной перегонке продукта в
вакууме. Присутствие воздуха может сильно понизить температуру плавления
вещества (даже на 20 °C).
Лаборатории для работы с фтором и его соединениями должны быть
оснащены большими, хорошо работающими вытяжными шкафами; в лабора-
тории должны быть резиновые перчатки, защитные очки, противогазы, а так-
же водородно-кислородная горелка для работы с кварцевой аппаратурой.
«Сухой лед и жидкий О2 (или фракция жидкого воздуха, полученная при его
стоянии) особенно необходимы для работы с низкокипящими фторидами. Ис-
пользование жидкого N2 (/„мп —195,8 °C) нежелательно при работе с газо-
образным F2 (/квп —188°C). Возможность проводить работу с жидким N2
при более высоких, чем —190 °C, температурах показана на рис. 4. При не-
счастных случаях надо, чтобы в лаборатории обязательно имелся заранее
приготовленный раствор карбоната аммония (10%-ный); этим раствором еще
до оказания квалифицированной медицинской помощи как можно быстрее
.надо обрабатывать в течение получаса пострадавшее от HF или фторида
место. Можно также положить иа это место компресс или смазать это ме-
сто пастой из оксида магния и глицерина.
ЛИТЕРАТУРА
'1. Ruff О., Riebeth А., 7.. Anorg. Allgem. Chem., 179, 166 (1929).
Allgem. Chem., 173, 373 (1928). 3. Ruff 0., Kwasnik W., Z. Anorg.
2. Ruff 0., Fischer J.. Z. Anorg. Allgem. Chem., 209, 113 (1932).
Фтор F2
электролиз
H2F2 *" H2 F2
40 2 38
В лабораториях фтор получают электролизом при 70—100 °C расплава
смеси KF с 2—3 молями безводного HF. Конструкция прибора зависит от
цели, с которой получают фтор (демонстрация химических свойств фтора,
синтез в препаративных количествах, различные измерения).
Способ 1. Получение фтора для демонстрации его химических свойств
II].
Для этих целей используют простой прибор (рис. 118). В тигельную печь
«вставляют сварной железный сосуд, который с помощью шин соединяют с
•источником тока (отрицательный полюс). В этот сосуд наливают электролит.
Крышку сосуда также изготовляют из железа. Через центр крышки прохо-
дит широкая трубка, в которую вставляют анод; крышка снабжена, кроме
того, трубкой для отвода F2 и Н2. Вертикальная широкая трубка одновре-
менно разделяет объемы, где выделяются газы F2 и Н2; нижняя часть этой
трубки перфорирована, и вместо дна напаяна пластинка. В крышке имеется
штуцер для термометра. В качестве уплотнителя между корпусом сосуда и
184 Глава 3. Фтор
крышкой служат кружки из мягкой резины. На конец штуцера для термо-
метра в целях изоляции от агрессивной среды натягивают кусочек резинового
шланга. В широкую трубку крышки вставляют резиновую пробку с отвер-
стиями, в которые пропускают никелевый или угольный стержень (например^
от дуговой лампы). К нему вверху прикрепляют проводник, который соеди-
няют с положительным полюсом источника постоянного тока. Если пробки,
смазанные хостафлонолом, при длительном использовании уже не просто на-
греваются, а загораются, их надо поменять на новые. Через такой простей-
ший электролизер можно пропускать ток до 10 А.
Рис. 118. Устройство простой
ячейки для получения фтора в це-
лях демонстрации.
Рис. 119. Устройство прибора для полу-
чения фтора.
Электролит готовят следующим образом. Через сухой гидродпфторид ка-
лия (лучше всего высушенный в потоке фтора) длительно пропускают сухой
фтороводород до тех пор, пока с помощью анализа не будет установлено,
что смесь отвечает необходимому составу KF-(2—3)BF. При этом смесь на-
гревается и становится жидкой.
Появление первых порций фтора удобно обнаруживать с помощью смо-
ченного в масле тампона, который воспламеняется, если его поднести к вы-
ходу от анода (тампон надо укрепить на проволоке).
Для регенерации электролита через расплавленную массу пропускают
с помощью железного или медного капилляра сухой поток нагретого до 35 °C
фтороводорода из стального баллона. Пропускание фтороводорода продол-
жают до тех пор, пока объем смеси в сосуде не станет равным первоначаль-
ному.
Способ 2. Если получаемый фтор сразу используют в синтезе или при-
меняют для непосредственного фторирования, то электролиз проводят в фаб-
ричном электролизере (рис. 119) [2]. Кожух электролизера сделан из нике-
Фтороводород 185
ля, монеля или стали. Газоотводные трубки не должны быть узкими, так как
даже при слабом закупоривании их происходит захват и переброс электро-
лита. Для того чтобы не допустить попадания пузырьков Н2 в анодное про-
странство, на дно электролизера кладут резиновую пластину.
Электролизер устанавливают на обогреватель (плитку), и, таким обра-
зом, еше до включения тока можно поддерживать температуру 70—НО °C.
Во время электролиза дополнительный подогрев обычно не нужен.
При наполнении электролизера свежим электролитом через электролит
пропускают некоторое время ток, чтобы удалить следы воды. Для периоди-
ческого определения содержания фтора в анодном газе последний отбирают
с помощью продолговатой отградуированной кварцевой колбы, которая снаб-
жена двумя кранами. По наполнении колбы анодным газом ее отсоединяют
и встряхивают до тех пор, пока поверхность находящейся в ней ртути не
будет оставаться блестящей. Зная количество непрореагировавшего газа,
можно найти содержание F2 в анодном газе.
При длительном непрерывном получении фтора никелевый анод постепен-
но растворяется. Поэтому для крупных электролизеров (нагрузка 500—
1000 А) нельзя изготавливать анод из никеля. В этих случаях применяют
графит.
Выходящий из электролизера фтор пропускают через металлическую
трубку с гранулированным NaF или KF, чтобы очистить от сопутствующего
фтороводорода.
Если фтор улавливают при охлаждении с помощью ловушки, то обяза-
тельно надо предварительно пропустить его при 350 °C через никелевую труб-
ку; при этом разлагаются взрывоопасные примеси (состав которых до сих
пор неизвестен), присутствующие иногда в анодном газе. Затем ставят ло-
вушку, охлаждаемую жидким О2 (или фракцией жидкого воздуха, получен-
ной при его стоянии); в этой ловушке вымораживаются следы примесей. Для
охлаждения нельзя использовать жидкий N2, так как при температуре кипе-
ния азота (—195,8°C) конденсируется фтор (£Кип —188,3°C).
В конструкции электролизера надо избегать фланцевых соединений.
В случае крайней необходимости используют в качестве прокладок уплотня-
ющие материалы из мягких металлов (А1, отожженой Си, Ni или полифтор-
этилена).
При токовой нагрузке 6 А образуется 40 мл F2 в 1 мин. Максимальный
выход по току равен 95%. Рабочее напряжение 8—13 В. Максимальная сте-
пень чистоты получаемого F2 98%.
Свойства. Слегка зеленоватый газ с характерным запахом, немного на-
поминающим запах озона и хлора. /пл —220 °C; Аип —183,3 °C; дж 1,108
(—188°C), dr 1.31 (для воздуха d 1); /крит —130 °C; ркрит 66,2 бар. Само-
произвольно вступает в реакцию со многими веществами при комнатной тем-
пературе.
ЛИТЕРАТУРА
i. Kwasnik W., Chemie-Arbeit, 67, 172 2. Zuliani G„ Favero P. G„ Ann.
(1944). Chim., 55, 22 (1965).
Фтороводород HF
Водная фтороводородная (плавиковая) кислота [1]
Обычно в лаборатории плавиковую кислоту получать невыгодно, так как
это продажный реактив, имеющий достаточную степень чистоты. В целях до-
полнительной очистки ее можно перегонять в платиновом приборе, добавляя
в дистилляционную колбу NaF и немного PbCOs.
186 Глава 3. Фтор
Хранят фтороводородную кислоту в бутылях из полиэтилена. Для хра-
нения технической плавиковой кислоты, содержащей до 60% HF, использу-
ют покрытые резиной или оцинкованные железные емкости; плавиковую кис-
лоту с концентрацией <60% можно хранить в железных сосудах.
Свойства. Фтороводород растворяется в воде в любых соотношениях.
При перегонке образует азеотропную смесь, содержащую 38,26% HF и пере-
гоняющуюся при 112°C (750,2 мм рт. ст.). Очень агрессивная бесцветная
жидкость (беречь кожу и глаза\). Температура плавления и плотность зави-
сят от содержания HF:
Содержание HF, % 30 -40 50 60
/пл, °C —69 —42 —35 -41,6
й2о 1,149 1,154
Плотность сильно меняется в присутствии загрязнений.
Безводный фтороводород [2]
KHF2 ----> KF + HF
78,11 58,11 20
KHF2, который предварительно надо высушить (см. препарат KHF2) при
15G °C в атмосфере фтора пли в крайнем случае на воздухе, разлагается при
нагревании до 500 °C. Используют прибор для перегонки, изготовленный из
Рис. 120. Установка для перегонки безводного фтороводорода.
/ — перегонная колба (из меди); 2— холодильник (внутренняя трубка из меди); 3—ло-
вушка-приемннк из меди (—10 °C); 4 — вторая ловушка (—10 °C); 5 н 6 — конические со-
единения.
меди (рис. 120). Сосуд 1 (реторта), в котором происходит разложение, име-
ет широкое горло, которое закрывают конической насадкой, и (по возмож-
ности) хорошо впаянный и как можно ниже расположенный штуцер для тер-
мометра. Для смазки шлифов используют смесь парафина с графитом или
полихлортрифторэгилен. Насадку соединяют с нисходящим холодильником 2
(длина 1 м и диаметр 2,5 см), имеющим стеклянную рубашку. Перед нача-
лом реакции разложения медную трубку холодильника внутри обрабатыва-
ют НС1/Вг2 до тех пор, пока она не станет блестящей, потому что иначе пер-
вые порции фтороводорода окажутся окрашенными в коричневый цвет. При-
емник 3 соединяют с холодильником с помощью конического винтового со-
единения 5. Приемник 3 охлаждают в смеси льда с поваренной солью. Вто-
рой приемник 4 присоединяют к первому также с помощью конического со-
единения 6", этот приемник служит запасной емкостью. Перед соединением 5
впаивают вертикальную узкую трубку для отбора проб; открытый конец
трубки можно опустить в ванночку со ртутью.
Содержимое реторты нагревают 45 мин при 400 °C. Как только в плати-
новой чашке через трубку для отбора проб собирается ~ 10 мл, а полоска
Соединения с галогенами 187
фильтровальной бумаги под действием капель продукта становится желати-
нообразной, трубку для отбора проб опускают в Hg и усиливают нагревание
реторты, повышая температуру до 500 °C. В реторте образуется KF и бурно
выделяется фтороводород.
Через —3 ч выделение HF ослабляется. Нагревание прекращают, отсо-
единяют приемник 3 и закрывают выходы медными колпачками. После ох-
лаждения остаток в реторте удобно растворить в кипящей воде.
Для получения совершенно чистого фтороводорода продукт надо не-
сколько раз перегнать в приборе из платины или меди. Абсолютно безводный
HF получают путем пропускания F2 из стального баллона в охлажденный
препарат (в'течение 30 мин) или прибавления по каплям рассчитанного по
содержанию Н2О количества SOC12.
Из 1,2 кг KHF2 получают 250 г HF.
Свойства. Прозрачная как вода, очень легко подвижная жидкость, силь-
но дымящая на воздухе. tnn —83 °C; tHBn 19,5 °C; dK 0,987; <к₽нт 188 °C;
ркрит 66,2 бар; йкрит 0,29; электропроводность Ы0-5 Ом-1-см-1. Бурно реа-
гирует с водой и льдом. Быстро и глубоко разъедает кожу (беречь кожу и
глаза}).
Очищенный безводный фтороводород хранят в сосудах из платины или
меди. Технический фтороводород перевозят в железных баллонах, емкостях
и цистернах. В качестве материала для сосудов, в которых проводят иссле-
дования и измерения, подходят Pt, Си и нержавеющая сталь (V2A).
ЛИТЕРАТУРА
1. Ruff О., Die Chemie des Fluors. 3. Fredenhagen К., Cadenbach G.. Z.
Berlin, 1920. Anorg. Allgem. Chem., 178, 289
2. Wiechert R.. Chemie, 56, 333 (1943). (1929).
Фторид хлора|1) C1F
Способ 1 [1, 2]
Cl2 + F2 —-> 2C1F
70,92 38 108,92
Реакцию проводят в вертикальном цилиндре из никеля или монеля
(рис. 121). Через напоминающую сопло трубку подают С12, который предва-
рительно проходит через жидкостный счетчик. Сбоку поступает F2. Удлине-
ние реакционной трубки улучшает отделение твердых фторидов (NiF2,
FeFa), которые образуются при взаимодействии с материалом стенок реак-
ционного сосуда; это уменьшает опасность закупоривания. Газы из реакцион-
ного сосуда свободно поступают в горизонтально укрепленный железный хо-
лодильник (с водяным охлаждением), затем в железную, охлаждаемую су-
хим льдом (без ацетона}) ловушку (для конденсации С12 и C1FS) и, наконец,
во вторую, охлаждаемую до —183 °C жидким О2 (или каким-либо другим
способом) ловушку, в которой конденсируется C1F, в то время как F2 выхо-
дит в вытяжной шкаф.
Печь нагревают до 400 °C и через прибор пропускают F2 до тех пор, по-
ка на выходе из прибора не будет обнаружен F2 (по воспламенению смо-
ченного маслом тампона). Затем подают С12. При токовой нагрузке электро-
лизера для получения фтора 70 А С12 пропускают со скоростью 9 л/ч.
В- ловушке, охлаждаемой до —183 °C, собирается C1F, который содержит
заметные количества C1FS и С12.
По окончании фторирования C1F перегоняют в стальной сосуд (соответ-
188 Глава 3. Фтор
ствующая аппаратура схематически изображена на рис. 122). Сосуд Дьюара,
в котором находится приемник с C1F, опускают и дают продукту закипеть.
Незначительные количества примесей,'содержащих в основном F2, отсасыва-
ются водоструйным насосом. Затем открывают вентиль стального баллона и
Рис. 121. Установка для получения фторида хлора(1).
так быстро конденсируют C1F, что давление в приборе становится ~760 мм
рт. ст. Как только в кварцевой ловушке соберутся значительные количества
конденсата (Cl2, C1F3), перегонку заканчивают. Хвостовой погон'также от-
Соединения о галогенами 18!>
сасывают с помощью водоструйного насоса. Выход составляет в лучшем слу-
чае 90% в расчете на хлор.
Способ 2 [Э. 4]
Если в распоряжении имеется CIF3 (продажный реактив), то можно по-
лучить CIF по реакции
CIF3 + C12 ---*• 3C1F
92,5 70,9 163,4
Реакцию рекомендуется проводить при повышенном давлении. Стенки авто*
клава сделаны из специальной стали; автоклав с вентильными затворам»1 2
снабжен манометром из того же материала; автоклав предварительно пасси-
вируют при комнатной температуре небольшим количеством C1F3. Затем в
охлажденный до —196 °C автоклав конденсируют определенные (измерен-
ные) количества C1F3 и С12. Таким образом загруженный, а затем закрытый'
при комнатной температуре автоклав нагревают до 180 °C с помощью элект-
рической плитки или в сушильном шкафу. Согласно требованиям техники
безопасности, нагревание надо проводить в специально оборудованном месте--
в лаборатории. Через 5—6 ч реакция заканчивается, и продукты реакции из
охлажденного реакционного сосуда выводят по фракциям: в ловушку
собирают при —142 °C (расплавленный метилциклопентан) Cl2, CIF3 и FC1O2,.
в то время как C1F конденсируется при —196 °C во второй ловушке.
Свойства. М 54,46. Бесцветный газ; в жидком состоянии, как правило,
слабо окрашен в желтый цвет, в твердом состоянии — белый. tnn
—155,6°C; /кип —100,1 °C; /Крит —14°C; dK 1,67 (—108°C). Очень реакцион-
носпособное соединение, моментально разъедает стекло, так же действует в
присутствии следов влаги и на кварц. Большинство органических соединений-
воспламеняется при взаимодействии с CIF. С водой реагирует чрезвычайно •
бурно. Сильно поражает бронхи.
ЛИТЕРАТУРА
1. Ruff О., Ascher Е., Laas F., Z.
Anorg. Allgem. Chem., 176, 256
(1928).
2. Kwasnik IF., неопубликованные
данные.
3. Schmitz H., Schumacher H. J., Z.
Naturforsch., A 2, 359 (1947).
4. Schack C. J., Wilson R. D., Synth.-.
Inorg. Metal.-Org. Chem., 3 (4),.
393 (1973).
Фторид хлора(Ш) CIF3
Cl2 + 3F2 -----> 2C1F3
70,92 114,0 184,92
CIF3 получают в том же приборе, что и C1F (рис. 121). Надо только вне-
сти следующие изменения: 1) убрать вторую железную, охлаждаемую до
—183 °C ловушку или просто снять с нее охлаждение; 2) печь нагревать до
280 °C (а не до 400 °С1); 3) пропускать более слабый поток хлора. При то-
ковой нагрузке электролизера для получения фтора 70 А хлор пропускают со-
скоростью 6,2 л/ч. При недостатке F2 (относительно С12) образуется преиму-
щественно C1F, а также C1F3, который содержит много С12.
По окончании фторирования переливают жидкий C1FS из охлаждаемой
сухим льдом ловушки в охлаждаемый сухим льдом (без ацетонах) стальной
баллон (хорошая тяга, защитные очки, резиновые перчатки!) и тотчас же за-
крывают вентиль. Если баллон находится при комнатной температуре, ега>
надо снабдить железным манометром; выпускают газ из баллона при дав-
лении 2 2 бар, при этом удаляются C1F, С12 и F2.
fl 90 Глава 3. Фтор
В зависимости от соотношения С12: F2 выход продукта составляет 60—
»В0%; остальное всегда приходится на C1F.
При работе с C1FS нужно обращать внимание на то, чтобы на вентилях
не было даже следов масла или другого жира. В качестве уплотнителя для
квентиля подходит свинцовый асбест. Прокладки изготовляют из меди. Если
жидкий CIFs случайно пролили, то надо на это место положить кусочки су-
хого льда, который сильно поглощает C1FS.
Свойства C1F3. М 92,46. Бесцветный газ с удушливым запахом, сильно
^поражает бронхи. /пл —83°C; 11,3°C; dJK 2,026 (—78°C). Чрезвычайно
реакционноспособное соединение, особенно в сжиженном состоянии. Немед-
ленно разрушает стекло, так же действует в присутствии следов влаги на
кварц. Большинство органических соединений реагирует- с ним с воспламене-
нием. Реакция обмена с водой протекает со взрывом.
ЛИТЕРАТУРА
1. Ruff О., Krug Н„ Z. Anorg. Allgem. 2. Kwasnik W., Naturforschung u. Me-
Chem., 190, 270 (1930). dizin in Deutschland 1939—1946
(FIAT-Review), 23, 168.
Тетрафторид-хлорид нитрозила NOC1F4
NOF-f-ClFs ---> NOC1F4
49,01 92,46 141,47
В 0,5-литровый реакционный сосуд из полиэтилена конденсируют при
—78 °C приблизительно равные по объему количества газообразных NOF и
*C1F3 (первого — небольшой избыток). По окончании реакции удаляют с по-
мощью вакуумного насоса избыток NOF, при этом реакционный сосуд дол-
жен оставаться при температуре —25 °C. Твердое вещество в реакционном
«сосуде и есть NOC1F<.
Свойства. Белое кристаллическое вещество, может храниться в неизмен-
ном состоянии только при низких температурах. Давление диссоциации зави-
сит от температуры:
Температура, ° С —60 —25 0
Давление диссоциации, атм 0,026 0,250 1,37
Сильный окислитель, бурно реагирует с водой, воспламеняет бумагу, со спир-
том и эфиром реагирует со взрывом.
ЛИТЕРАТУРА
1. Whitney Е. D., McLaren R. О., Hur-
ley Т. J., Folge С. Е., J. Amer.
Chem. Soc., 86, 4340 (1964).
Оторид 6рома(П1] BrF3
Способ 1 [1]
Вг2 -|- 3F2 ► 2BrFg
159,82 114 273,82
Соединения с галогенами 19Ь
Получение BrF3 основано на фторировании брома при +80 °C. При этой
температуре образуются значительные количества BrFg, который далее быст-
ро и количественно реагирует с бромом:
3BrF5 + Bra ----> 5BrFs
Реакцию проводят в наклонно укрепленном железном холодильник©
(рис. 123). В холодильник впаяна железная трубка, в которую вставляют
капельную воронку. Нижний конец реакционной трубки (холодильника) при-
соединяют к разветвлению, которое изготовляют из монеля. На верхний от-
росток насаживают железный обратный холодильник. Нижний отросток име-
ет шлиф, с помощью которого можно присоединить приемник (из кварца).
Рис. 123. Установка для получения фторида брома (III).
Через прибор пропускают поток F2; в рубашку наклонного холодильникам
подают теплую воду (4-80°C); рубашку вертикального холодильника охлаж-
дают солевым раствором (около —18°C). Вг« добавляют по каплям с такой-
скооостью, чтобы в приемнике конденсировался также по каплям почти бес-
цветный BrFs. Обратный холодильник исключает потери (захват с потоком.”
F2) BrF3 или BrFs.
По окоичаиии фторирования приемник нагревают непродолжительно©-
время до 100 °C для того, чтобы отогнать из продукта растворенный в нем
BrFg. По охлаждении BrF3 переносят в железный сосуд. Выход в расчете на-
бром количественный, в расчете на фтор 90%. Степень чистоты 98% (при-
сутствует BrFg).
Способ 2 [2]
BrF3 образуется количественно и в очень чистой форме при непосредст-
венном фторировании брома, растворенного в CC13F, при —40 °C; продукт-
можно использовать в препаративных целях без дополнительной очистки. По-
лучение BrFg проводят в приборе, описанном в методике получения фторида,
иода (III).
Бром суспендируют в ~600 мл CC13F; предварительное охлаждение газа,
осуществляют с помощью охладительной бани с сухим льдом; в такие же ба-
ни помещают и предохранительные ловушки. Реакционный сосуд охлаждают
при —40 °C, опуская в прозрачный сосуд Дьюара с СН3ОН/СО2. Суспензию-
интенсивно перемешивают с помощью высокооборотной механической мешал-
ки и пропускают предварительно охлажденную смесь F2 с N2, приготовлен—
•Il 92 {Г-лава 3. Фтор
кую в соотношении 1:1. Реакция закончена, когда раствор становится свет-
ло-оранжевым, а осадок почти неокрашенным. Избыток F2, растворенный в
€C13F, вытесняют, пропуская N2, и продукт извлекают путем низкотемпера-
турного фильтрования при —78 °C, а затем высушивают при —78 °C и
10-2 мм рт. ст. BrF3 получается очень чистым, в виде неокрашенного твердо-
го вещества.
Свойства. М 136,91. Бесцветная жидкость. Кристаллизуется в виде длин-
ных призм. /Пл +8,8QC; /ИИп + 127°С; 2,84. Очень реакционноспособное
вещество, дымит на воздухе, сильно разъедает кожу.
ЛИТЕРАТУРА
’1. Kwasnik W., Naturforschung u. Me- 2. Lehmann E., Naumann D., Schmeis-
dizin in Deutschland 1939—1946 ser M., Z. Anorg. Chem., 388, 1
(FIAT-Review), 23, 168. (1972).
«Фторид 6poMa(V) BrFs
Br2 + 5F2 ----*- 2BrF6
159,82 190 349,82
В тигельную печь, позволяющую вести нагревание до 200 °C, помещают
реакционный сосуд из железа (рис. 124). Реакционный сосуд снабжен труб-
кой для подвода F2, газоотводной трубкой для BrFs, штуцером для термомет-
Рис. 124. Установка для получения фторида брома (V).
;ра и широкой трубкой (по центру), в которую вставлена капельная воронка
1И которая имеет сбоку отводную трубку для подачи Ns (поток Ns предотвра-
щает соприкосновение F2 с капельной воронкой). Газы, вступающие в реак-
цию, конденсируются в железном холодильнике. Сжиженный BrFs собирается
в железной ловушке, которую охлаждают смесью льда и поваренной соли.
Как только прибор заполняется фтором, начинают прибавлять по кап-
лям бром. При токовой нагрузке электролизера для получения фтора 150 А
скорость подачи Br2 1 капля/с. Следят за тем, чтобы F2 всегда был в из-
бытке. Сырой продукт содержит 95% BrFs и 5% BrFs. По окончании фтори-
рования продукт перегоняют в приборе, изготовленном из железа, в потоке
Соединения с галогенами 193
F2. Холодильник и приемник (при перегонке) охлаждают смесью льда с пова-
ренной солью (температура около —18 °C). Выход'87% в расчете иа бром.
Свойства. М 174,91. Бесцветная, сильно дымящая на воздухе жидкость.
До 460 °C не разлагается. /пл —61,3 °C; /кип 4-40,5 °C, dm 2,57 (0°С).
Очень реакционноспособное вещество, реагирует почти со всеми элемен-
тами с воспламенением. С водой происходит реакция, ’ часто сопровождающа-
яся взрывом. В отсутствие влаги при обычной температуре медленно разъеда-
ет стекло; практически ие действует на кварц. Ртуть покрывается коричнева-
тым налетом.
ЛИТЕРАТУРА
1. Ruff О., Menzel W., Z. Anorg. 2. Rwasnlk W., неопубликованные
Chem., 202, 49 (1931). данные.
Фторид иода(1] IF
if3 4- k------► 3IF
183,91 253,84 437,75
Получение иодомонофторида основано на восстановлении иодотрифтори-
да свободным иодом, суспендированным в CC1SF, в присутствии каталитиче-
Рис. 125. Устройство прибора для
получения фторида иода(1).
1 — охлаждающая баня (—78 °C); 2— ре-
акционный сосуд; 3 — механическая мешал-
ка; 4 — затвор, предохраняющий от вла-
ги воздуха.
ских количеств органического N-содержащего основания (пиридин, ацетонит-
рил) при —40 °C. Реакцию проводят в приборе, схематически изображенном
на рис. 125.
В реакционный сосуд из обычного стекла помещают CCI3F, охлаждают до-
—78 °C и вносят взвешенное количество IFS. Добавляют рассчитанное коли-
чество тонкодисперсного иода и 0,5 мл пиридина. При интенсивном пере--
13—143
194 Глава 3. Фтор
мешивании с помощью механической мешалки повышают температуру реак-
ционной смеси до —40 °C и оставляют при этой температуре в течение несколь-
ких дней.
Реакция окончена, как только раствор теряет обусловленную присутст-
вием иода красио-коричиевую окраску. Продукт отделяют путем фильтрова-
ния при —40 °C с отсасыванием, несколько раз промывают холодным CC13F
и сушат в вакууме (10-2 мм рт. ст.) при —40 °C. Выход в расчете на IFS
количественный.
Свойства. М 145,91. Светло-серое, почти неокрашенное вещество; твер-
дое при —40 °C, очень гигроскопичное. Плавится с разложением до 12 и IF®
при —14 °C.
ЛИТЕРАТУРА
1. Schmeisser М., Scharf Е., Angew.
Chem., 72, 324 (1960).
2. Schmeisser М., Sartori Р., Nau-
mann D., Chem. B'er., 103, 590
(1970).
Фторид иода(Ш) IF3
I2 + 3F2 ----> 2IF3
253,84 114 367,84
IFS количественно образуется при действии предварительно охлажденно-
го, разбавленного азотом F2 на суспензию 12 в CC13F при —45 °C. Прибор
для получения IFS изображен на рис. 126. Реакционный сосуд и предохрани-
тельные ловушки можно изготавливать из обычного стекла.
Рис. 126. Установка для получения фторида иода(Ш).
Прежде всего необходимо охладить предохранительные ловушки до
—78 °C, опустив их в сосуд с сухим льдом, и пропустить через собранный
пиибор сухой N2. Затем в реакционный сосуд добавляют суспензию тонкодис-
Соединения с галогенами 195
персного иода в CC1SF и температуру в реакционном сосуде повышают до
—45°C, опустив его в баню с СН3ОН/СО2 (прозрачный сосуд Дьюара), не-
смотря на то, что газы предварительно охлаждены до —78 °C. Суспензию 12
в CC13F интенсивно перемешивают с помощью механической мешалки.
Через суспензию начинают пропускать предварительно охлажденную
смесь газов F2 и N2 (в приблизительном соотношении 1:2). Фторирование
заканчивают, когда совершенно исчезнет окраска раствора, обусловленная
присутствием 12, т. е. раствор CC13F станет бесцветным. По окончании фтори-
рования F2, растворенный в CC13F, вытесняют азотом, продукт отделяют пу-
тем фильтрования с отсасыванием при —40 °C, промывают несколько раз
охлажденным CC1SF и сушат в вакууме (10-2 мм рт. ст.) при —40 °C. Выход
в расчете на иод количественный.
Аналогично (но при —78 °C) можно получить перфторалкилиоддифтори-
ды, например трифторметилиоддифторид CFSIF2:
CFSI + F2 ----> CF3IF2
Свойствам 183,91. Вещество желтого цвета, твердое при —40°C, очень
гигроскопичное. Плавится с разложением (первично образуются IF и IF8)
при —28 °C.
ЛИТЕРАТУРА
1. Schmeisser М., Ludovici W., Nau-
mann D., Sartori P., Scharf E.,
Chem. Ben, 101, 4214 (1968).
2. Schmeisser M., Scharf E., Angew.
Chem., 71, 524 (1959).
3. Baumanns J., Deneken L., Nau-
mann D„ Schmeisser M., J. Fluori-
ne Chem., 3, 323 (1973).
Аддукт фторида иода[1П) с пиридином IF3«C5H5N
IF3 4- C6H6N --------> IF3-C6H6N
183,91 79,10 263,01
В ловушке, снабженной осушительной трубкой и капельной воронкой,
охлаждают до —78 °C CC13F и добавляют взвешенное количество IFS. Затем
медленно добавляют по каплям пиридин в количестве, немного большем сте-
хиометрического. Суспензию перемешивают с помощью магнитной мешалки
и медленно поднимают температуру до комнатной. Образуется белое соеди-
нение— аддукт пиридина с фторидом иода (III), которое отделяют из рас-
твора путем фильтрования; выделенное соединение промывают CC13F, содер-
жащим пиридин, и высушивают в вакууме. Выход в расчете на введенный
в реакцию IF3 количественный.
Свойства. Белое, твердое, очень быстро гидролизующееся вещество. Рас-
творимо в CHSCN, пиридине и др. Выше +Ю0°С плавится и спекается; при
-J-166°C разлагается, выделяя иод.
Аналогично могут быть получены другие аддукты фторида иода (III) [2].
ЛИТЕРАТУРА
1. Schmeisser М., Ludovici W., Nau-
mann D„ Sartori P., Scharf E.,
Chem. Ben, 101, 4214 (1968).
2. Schmeisser M., Ludovici W., Nau-
mann D., Sartori P., Scharf E.r
Chem. Ben, 103, 590 (1970).
13*
194
Глава 3. Фтор
Фторид иода(¥) IFg
It + 5Ft ------>- 2IFS
253,84 190 443,84
В качестве реакционного сосуда служит железный барабан (рис. 127) о
охладительной рубашкой. К барабану присоединяют нисходящий железный
холодильник, а затем две газовые ловушки из кварца. Реакционный сосуд
наполняют иодом и пропускают F2, который по' возможности должен быть
очищен от HF. Реакционный сосуд охлаждают для отвода тепла и следят за
тем, чтобы накапливающийся в реакционном сосуде IFb не превратился в IF7.
Как только на выходе из прибора появится F2, реакция закончена. С реак-
Рис. 127. Установка для получения фторида иода(У).
циоииого сосуда снимают охлаждение и нагревают его снаружи пламенем
газовой горелки, при этом охладительная рубашка действует как водяная ба-
ня. IF5 перегоняют в потоке F2. При токовой нагрузке электролизера для по-
лучения F2 80 А для превращения 250 г иода достаточно 10 ч, включая пе-
регонку продукта. Выход 90% в расчете на I.
Хранят IF5 в сосудах из железа. Этот препарат удобно применять для
фторирования органических соединений.
Свойства М 221,92. Бесцветная жидкость, дымит на воздухе. /Пл
+9,6°C; /кип +98°C, d,K 3,231 (15°C); dTB 3,75 (0°С). Реагирует чрезвычай-
но бурио с водой.
ЛИТЕРАТУРА
1. Moissan F„ Bull. Soc. Chim. Fran- 2. Kwasnik W„ неопубликованные
ce [3], 29, 6 (1930). данные.
Фторид иода(УП) IF7
12 + 7F2 ------> 2IF7
253,84 266 519,84
В качестве реакционного сосуда служит цилиндр из железа, снабженный
охладительной рубашкой (рис. 128). Загрузка осуществляется через сито для
приема иода. На выходном отверстии сосуда имеется насадка высотой
-'-30 см, на которую навинчивается железный холодильник. От холодильни-
Соединения с галогенами 197
ка к ловушкам для конденсации продукта ведет железная трубка. Ловушки
изготовлены из кварца, имеют U-образную форму и сверху закрыты при*
шлифованными кварцевыми пробками, которые, однако, вставлены не очень
плотно. В то время как первая U-образная трубка служит для сбора IF?,
вторая предохраняет от попадания влаги из воздуха. Оба кварцевых сосуда
охлаждаются жидким О2 или каким-либо другим способом до температуры
*—183 С.
В реакционный сосуд через сито загружают иод и через прибор пропу-
скают F2. Предварительно F2 должен быть освобожден от HF, для чего газ
лучше всего пропустить через охлаждаемую сухим льдом железную спираль
Железо
Рис. 128. Установка для получения фторида иода (VII).
или через свежеобезвоженный KF. В реакционном сосуде иа первой стадии
иод сгорает до IFs. Реакция проводится при интенсивном пропускании воды
через охладительную рубашку реакционного сосуда. Как только на выходе
прибора появляется F2, водяное охлаждение отключают и нагревают водяную
рубашку пламенем газовой горелки, так что рубашка работает, как водяная
баня. Насадку на выходе из реакционного сосуда нагревают до 300 °C с по-
мощью намотанной спирали, подключив последнюю к электрической сети.
В потоке фтора IFS переходит в IF?; последний удаляется через железный
холодильник, собираясь в кварцевой ловушке. Для того чтобы непрореагиро-
вавший IF5 не переходил в приемник, холодильник должен хорошо работать.
И на первой, и на второй стадии реакции требуется присутствие избытка
F2. Твердый продукт, сконденсированный в коленах первой U.-образной труб-
ки, время от времени сверху вниз нагревают до плавления или лучше стря-
хивают с помощью железной проволоки. Для этой цели открывают на не-
сколько секунд пришлифованные пробки, которыми закрыты колена U-образ-
ной трубки.
Для очистки IF? перегоняют из кварцевой ловушки при атмосферном дав-
лении и 4-40 °C и собирают в другую кварцевую ловушку при —183 °C. Не-
значительный остаток IF5 вновь возвращают в реакционный сосуд. Затем
IF? перегоняют в стальной баллон, который после наполнения приводят к
комнатной температуре, после чего осторожно открывают вентиль и дают га-
зу испаряться (при этом' удаляется SiF4) до тех пор, пока ватный тампон,
смоченный спиртом и поднесенный к газовой струе, не станет вспыхивать,
что указывает на испарение IF?. Выход 83% в расчете на иод.
198 Глава 3. Фтор
_ Соединения на шлифах очень легко уплотняются (заклиниваются) под
действием фторидов иода, поэтому их надо время от времени поворачивать.
Шлифы нельзя ничем смазывать; их нельзя также уплотнять с помощью пн-
цеина; они должны быть просто хорошо вставлены друг в друга.
Свойства. М 259,84. Бесцветное соединение с затхлым кислым запахом;
в твердом состоянии — рыхлый порошок или крупные кристаллы.
tnn 6,4°C; при 4,5°C сублимируется. dIK 2,8 (6 °C).
Очень реакционноспособное вещество. По свойствам напоминает C1FS, но
в большинстве случаев более инертно. Вода растворяет газообразный IF?
(без вспышек). Гидроксид натрия абсорбирует его с сильным выделением
тепла. Серная кислота вспенивается при пропускании IF?. Разъедает стекло
н кварц.
ЛИТЕРАТУРА
1. Ruff О., Keim R„ Z. Anorg. Allgem. 2. Kwasnik W., неопубликованные
Chem., 193, 176 (1930). данные.
Дифторид трикислорода O3F2
(Триоксодифторид, фторид озона)
3O2 + 2F2 —2OsF2
96 76 172
Через смесь О2 и F? (3:2 по объему) при температуре от —196 до
—183 °C и давлении 12 мм рт. ст. пропускают электрический разряд (2100—
2400 В и 25—30 мА). Реакционный сосуд изготовляют из стекла пирекс, элек-
троды — из меди, для уплотнения используют тефлоновые пробки. Расстоя-
ние между электродами 10 см. Прибор аналогичен тому, который описан в
синтезе O2F2.
Выход O3F2 3—4 г/ч. Продукт можно перегонять почти без разложения
ври 0,1—1,5 мм рт. ст. и температуре от —177 до —160 °C. Это значительно
менее взрывоопасное вещество, чем жидкий озои.
Свойства. М 86. Темно-красная жидкость. Замерзает при —190 °C, склон-
на к переохлаждению, разлагается выше —157 °C на O2+O2F2. 4Ж 1,800
(190°C). Сильный окислитель, почти такой же, как O2F2; более реакционно-
способен, чем F2 и OF2.
ЛИТЕРАТУРА
1. Kirschenbaum A. D., v. Grosse А.,
J. Amer. Chem. Soc., 81, 1277
(1959).
Дифторид дикислорода O2F2
(Диоксодифторид)
О2 -|- F2 O2F2
32,0 38,0 70,0
Взаимодействие газов в газоразрядной трубке, охлаждаемой жидким О2,
инициируют с помощью электрического разряда; образующийся при этом не-
устойчивый O2F2 вымораживают.
Дополнительное количество F2 хранят в кварцевой ловушке, охлаждае-
мой жидким N2; его всасывают в прибор через мембранный вентиль из ме-
Соединения с кислородом 199
ди. Если располагают баллоном с Fa, то можно присоединить баллон непо-
средственно к прибору. Кислород подводят из стального баллона, который
присоединяют к прибору через железный или медный капилляр. Реакцию
проводят в стеклянном сосуде (рис. 129), который охлаждают до —183 °C,,
погружая в сосуд Дьюара с жидким О2 (или каким-либо другим способом),,
по окончании всей подготовительной работы включают разрядное устройст-
во. Электродами служат медные проволоки, которые вставлены в тонкие (ди-
аметр ~10 см) стеклянные трубки и залиты пицеином так, чтобы сохраня-
лась газонепроницаемость. Для получения газового разряда применяют мощ-
ный индуктор с прерывателем или трансформатором (50 Гц), со вторичной
обмоткой на 5000 В и 0,05 А. К разрядной трубке присоединена U-образная
трубка из кварца. После окончания синтеза она служит в качестве приемника
Рис. 129. Установка для получения O2F2.
при перегонке или в качестве сосуда для хранения продукта. К ней присо-
единяют охлаждаемую до —183 °C кварцевую ловушку, которая должна
предотвращать проникновение влаги воздуха. Для получения вакуума лучше
всего применять металлический водоструйный насос. Манометр для контроля
вакуумного разряжения не нужен, поскольку достаточно надежно можно оп-
ределять степень вакуумирования по форме газового разряда. Для работы
лучше всего подходит давление 10—20 мм рт. ст. При этих условиях разряд
имеет вид пучка.
Как только сосуды Дьюара установлены под соответствующие части при-
бора и запущен водоструйный насос, включают искровой разряд. Затем
осторожно открывают вентиль, чтобы F2 начал поступать в прибор. И лишь
после этого впускают О2, который всегда добавляют в избытке, так как ина-
че в сосуде, где происходит разряд, вымораживается озон (фиолетовое до
голубого окрашивание). O2F2 выделяется в виде красно-коричневого твердого
вещества, осаждаясь на стенках газоразрядной трубки. Время от времени
отключают на несколько минут искровой разрядник и опускают сосуд Дьюа-
ра. При этом O2F2 плавится и стекает вниз в отросток, имеющий форму ам-
пулы. Если предполагают, что образовался озон, то при плавлении необходи-
ма осторожность, так как иногда может произойти взвыв.
Как только в нижнем отростке газоразрядной трубки собирается доста-
точное количество O2F2, отключают искровой разрядник, а также прекраща-
ют подачу О2 и F2; П-образный запасной сосуд охлаждают до —183 °C и
убирают сосуд Дьюара от нижней части газоразрядной трубки. O2F2 при
15 мм рт. ст. перегоняется с частичным разложением. Следует заботиться а
том, чтобы при перегонке O2F2 температура только на очень короткое время
поднималась выше —60 °C, что позволит избежать дальнейшего разложения-
200 Глава 3. Фтор
ТВ целях очистки O2F2 можно перегонять таким же образом несколько раз.
Головной погон состоит в основном из озона н SiF«. Поскольку перегонка
всегда сопровождается образованием О2 и F2, ее обязательно производят
при постоянно включенном водоструйном насосе. Затем O2F2 перегоняют в
ампулы.
По окончании перегонки U-образный запасной сосуд отпаивают. O2F2
можно хранить только при температуре —183 °C, в крайнем случае — в су-
«ом льде.
, Свойства. Газообразный O2F2 окрашен в коричневый цвет; в виде жидко-
сти имеет вишнево-красную окраску; в твердом состоянии — оранжевую.
—163°C; /кип —57°C; dK 1,45 (—57°C); dTB 1,912 (—165°C).
ЛИТЕРАТУРА
1. Ruff О., Menzel W., Z. Anorg. 2. Ruff 0., Menzel W„ Z. Anorg.
Allgem. Chem., 211, 204 (1933). Allgem. Chem., 217, 85 (1934).
Фторид кислорода OF2
(Оксофторид)
2F, 4- 2NaOH -> OFa + 2NaF 4- H2O
76 80,0 54 83,99 18,01
С помощью платиновой трубки (внутренний диаметр ~2 мм) через 2%'-
ный раствор NaOH в реакционном сосуде из стекла барботируют F2, пропу-
ская его со скоростью 1—3 л/ч. Щелочь поступает в реакционный сосуд из
Рис. 130. Установка для получения фторида кислорода.
укрепленной несколько выше последнего склянки со скоростью 1 л/ч. Плати-
новую трубку опускают в щелочь на глубину ~2 см (рис. 130). Далее газо-
вая смесь поступает через спиральную промывную склянку, которая наполне-
на водой и должна абсорбировать F2. Наконец, OF2 конденсируют в две
стеклянные газовые ловушки, охлаждаемые жидким О2 (—183°C).
По окончании синтеза сырой продукт, сконденсированный в газовых ло-
вушках, отсасывают при —183 °C с помощью водоструйного иасоса (до
20 мм рт. ст.), при этом удаляются основные количества кислорода, раство-
ренного в жидком OF2. Для того чтобы не допустить выхода OF2 в атмосфе-
ру, перед водоструйным насосом ставят промывалку с раствором КД. При
фракционной перегонке головным погоном отгоняется О2. Уже при однократ-
Соединения с кислородом 201
дой перегонке получают продукт, содержащий 98% основного вещества. Вы»
ход 45% в расчете на фтор. •
Свойства. Бесцветный газ. В жидком состоянии окрашен в желтый
цвет с коричневым оттенком. /пл —223,8 °C; /Кип —144,8 °C; /Крит —58,0 °C;
ркрит 48,0 бар; VT 97,6 мл/мл; d„t 1,90 (—223,8°C); 1,521 (—145,3°C);
д//о6р —46 кДж. При О °C в 100 мл воды растворяется 6,8 мл газообразного
OF2.
OF2 имеет своеобразный запах. После вдыхания он вызывает сильную
одышку, которая часто может проявиться лишь через несколько часов н со-
храняться очень длительное время (несколько часов). Не разъедает стекло.
С холодной водой реагирует едва заметно. Действует иа Hg. 1
Устойчив на свету, при нагревании и при проскакивании электрической
искры. По сравнению с С12О характеризуется необычной химической инерт-
ностью. Как все низкокипящие фториды, в жидком состоянии растворяет
значительные количества воздуха.
Хранят OFj в стеклянных колбах или стальных сосудах.
ЛИТЕРАТУРА
1. Lebeau Р., Damiens А., С. R. Acad.
Sci., Paris, 188, 1253 (1928).
Фторид хлорила C1O2F
(Диоксид-фторид хлора)
2C1O24-F2 ---> 2C1O2F
134,92 38 172,92
2. Ruff О., Menzel W., Z. Anorg.
Allgem. Chem,, 190, 257 (1930). ,
Собирают прибор (рис. 131), который должен быть изготовлен полностью
из кварца. Через первую ловушку, где находится при температуре —504-
—55 °C несколько миллилитров жидкого С1О2, пропускают со скоростью
Рис. 131. Устройство прибора для
получения фторида хлорила.
500 мл/ч фтор. Трубку, по которой подводят фтор, погружают иа несколько
миллиметров в жидкий С1О2. Реакция проходит спокойно и с хорошей скоро-
стью, причем большая часть образовавшегося C1O2F остается в реакторе и
только незначительная часть попадает во вторую ловушку. Если жидкость в
первой ловушке слабо окрашена, то с нижней части реактора убирают охлаж-
дение и перегоняют C1O2F при постепенном повышении температуры в пото-
ке F2 во вторую ловушку, где продукт выпадает как неокрашенное чистое
вещество, не нуждающееся ни в какой дальнейшей очистке.
202 Г лава 3. Фтор
Согласно Шмайсер [3], методику целесообразно видоизменить следую-
щим образом. С1О2 растворяют при —78 °C в CCI3F и фторируют. После рас-
слаивания жидкости тяжелый нижний слой (образовавшийся C1O2F) отде-
ляют. Затем этот слой охлаждают и быстро отсасывают более легкую жид-
кую фазу с помощью капилляра. Синтез можно проводить в сосудах из йен-
ского стекла. Если требуется получить совершенно чистый C1O2F, то надо
(работать в кварцевых сосудах и не допускать избытка С1О2. В заключение
(продукт подвергают многократной ректификационной перегонке в кварцевой
.посуде.
Свойства М 86,46. Бесцветное вещество, очень гигроскопично, во влаж-
*ном воздухе тотчас образует туман. tHJI —115 °C; /кип —6 °C.
Термически соединение немного устойчивее, чем С1О2.
ЛИТЕРАТУРА
1. Schmitz Н., Schumacher И. J., Z.
Anorg. Allgem. Chem., 249, 242
(1942).
2. Sicre J. E., Schumacher H. I., Z.
Anorg. Allgem. Chem., 286, 232
(1956).
3. Schmeisser M., Ebenhoch F., L., An-
gew. Chem., 66, 230 (1954).
Фторид перхлорила CIO3F
(Триоксид-фторид хлора)
KC1O44-HSO3F ----> cio3f+khso4
138,56 100,07 102,46 136,17
В стеклянной круглодонной колбе, снабженной мешалкой и обратным хо-
лодильником, растворяют 10 г КСЮ4 в 100 г HSO3F. Реакция начинается при
•50 °C и заканчивается прн 85 °C. Выделяющиеся в результате реакции газы
поглощают, дважды пропуская через 10%-ный раствор гидроксида натрия
(NaOH содержит 5% Na2S2O3). Газ сушат, пропуская над твердым КОН и
конденсируют в газовой ловушке при —183 °C. Во время реакции через реак-
ционную смесь пропускают поток сухого N2. Продукт содержит 1% воздуха
и 0,4% СО2. Выход 67%- По этой методике можно получить C1O3F в кило-
граммовых количествах.
Свойства. Бесцветный газ с характерным, напоминающим OF2 запахом,
/пл —152,2 °C; /Кип —48,1 °C. Термически довольно устойчив: можно нагре-
вать в стеклянном сосуде почти до размягчения стекла. Немного растворим
в воде. Медленно реагирует с разбавленными водными щелочами.
ЛИТЕРАТУРА
1. Barth-Wehrenalp G., J. Inorg. Nucl.
Chem., 2, 266 (1956).
Фторид хлоротетраоксида C1O4F
HC1O44-Fs --->• C1O4F+HF
100,46 38,0 118,46 20,0
Прибор (рис. 132) целесообразно изготовить из кварца. Он состоит из
цилиндрической реакционной трубки (высота ~50 см), которая наполнена
сделанными из кварца кольцами Рашига. Трубка снабжена рубашкой и
охлаждается водопроводной водой. Фтор вводят через кварцевую трубку, ко-
Соединения с серой, селеном, теллуром 203
торая доходит почти до конца реакционной трубки. Через капельную ворон-
ку добавляют 70%-ную НС1О4. Жидкость стекает через енфон, а газообраз-
ные продукты реакции отводятся в верхнюю часть реакционной трубки, от-
куда они попадают в охлаждаемую жидким Ог (—183 °C) кварцевую ловуш-
ку. За ловушкой следует осушительная трубка, наполненная обезвожен-
ным KF.
НС1О4 добавляют по каплям со скоростью 1 капля/с до тех пор, пока че-
рез сифон не станет поступать жидкость. Затем начинают пропускать F2 с»
скоростью 2,5 л/ч. В охлаждаемой жидким О2 ловушке конденсируются OF2,
С12 и SiF4, а на стенках в виде твердого вещества — C1O4F. Из-за высокой
взрывоопасности за один раз можно получать не больше 4 г C1O4F. Продукт
Рис. 132. Установка для
получения фторида хло-
ротетраоксида.
очищают путем фракционной перегонки в вакууме. Хранят C1O4F в кварце-
вых ампулах охлажденным до —183 °C. При плавлении и замерзании СЮ4Е
очень легко взрывается. Выход ~60%.
Очень высокие выходы (>90%) достигаются, если реакционный сосуд
изготовить из платины. Только в крайнем случае работают в стеклянном при-
боре, поскольку при этом получают при незначительных выходах очень за-
грязненный продукт. Ни одну деталь или узел прибора нельзя изготавливать
из угля, так как он является катализатором разложения C1O4F.
Свойства. Бесцветный газ. /пл —167,3 °C; /Кип —15,9 °C.
Очень взрывоопасен; взрывается часто уже при плавлении и конденса-
ции; газообразный C1O4F взрывается, подобно NO3F, прн соприкосновении С
пылью, жиром, резиной и 2 н. раствором KI. В открытой пробирке газооб-
разный C1O4F взрывается при поднесении пламени или под действием искро-
вого разряда. Имеет резкий кислый запах, раздражает верхние дыхательные
пути и легкие, вызывает длительное удушье.
ЛИТЕРАТУРА
1. Rohrbach G. Н., Cady G. Н., J.
Amer. Chem. Soc., 69, 677 (1948).
Дифторо дисульфан FSSF
2AgF + 3S -----> FSSF + Ag2S
253,76 96 102 247,76
204 Глава 3. Фтор
В цилиндрический, закрытый с одной стороны стеклянный сосуд (диа-
метр 6 см, высота 25 см), на который сверху с помощью поворачивающегося
шлифового соединения насажена грушевидная колба (для AgF) и к которо-
му присоединены трн U-образные трубки с кранами (рис. 133), помещают
тщательно высушенный порошок серы (60 г), а в колбу — 30 г AgF. AgF
предварительно обезвоживают нагреванием прн 250 °C в сухой атмосфере.
С помощью масляной банн (125 °C) серу доводят до плавления и порциями,
поворачивая колбу, вносят в реакционный сосуд AgF. Вакуумным насосом
откачивают газообразные продукты реакции, которые конденсируются в пер-
вой охлаждаемой жидким воздухом U-образной трубке.
Процесс заканчивается через 2,5 ч. Тогда вторую U-образиую трубку
охлаждают жидким воздухом и перегоняют в нее прн возможно более ииз-
Рис. 133. Установка для получения дифтородисульфана.
ком давлении продукт реакции из первой U-образной трубки. Затем жидким
воздухом охлаждают третью U-образную трубку, а во второй создают тем-
пературу —120 °C, помещая ее в смесь метанола с жидким азотом. В этих ус-
ловиях из второй U-образной трубки удаляются SiF«, SF« и SOF2. Как толь-
ко давление в приборе понизится до 1 мм рт. ст., очистка окончена. Во вто-
рой U-образной трубке остается 1,5 мл чистого FSSF. Продукт можно хра-
нить в стеклянной посуде в замерзшем состоянии в течение нескольких дней,
не опасаясь разложения.
Свойства, /ил —133 °C; /кип 15 °C. При более высоких температурах и
давлениях превращается в SSF2 и далее в SF«+S.
ЛИТЕРАТУРА
1. Seel F., Budenz R„ Werner D.,
Chem. Ber., 97, 1369 (1964).
Дифторид дисеры SSF2
(Тиотионил фторид)
Способ 1 [1]
S2Cls + 2KF ---> SSFs + 2KC1
135 116 102 149
В этой методике в качестве фторирующего агента применяется «активи-
рованный» KF.
Соединения с серой, селеном, теллуром 205
Реактором служит вертикально установленная тефлоновая трубка (дли-
на 50 см, диаметр 2 см), которую можно нагревать до 140—145°C с по-
мощью электрической обмотки. Внизу трубка имеет сетчатое дно, и к ией на
шлифе присоединена 100-миллиметровая стеклянная колба с газоотводной
трубкой (для азота). На верхний конец реактора иавиичена охлаждаемая
трубка из политрифторхлорэтилена. От верхней части реактора отходит эла-
стичный шланг из тефлона, который соединяет реактор с двумя последова-
тельными ловушками из полипропилена, охлаждаемыми сухим льдом. После
ловушек присоединяют еще предохранительную склянку с конц. H2SO4.
В реакционную трубку помещают слой тефлонового волокна (2 см), а
поверх его — «активированный» KF, который получают путем термического
разложения в вакууме фторосульфоната калия; в стеклянную колбу помеща-
ют 35 г S2CI2. Включают электрическую обмотку на реакционной трубке и
через 0,5 ч с помощью масляной бани (155—160 °C) доводят S2CI2 до кипе-
ния. Одновременно через прибор пропускают N2. По истечении 2,5 ч дихлорид
дисеры переходит в верхнюю часть реакционной трубки. Синтез закончен.
KF расходуется на ~65%. Затем продукт реакции медленно перегоняют для
отделения от сопутствующих S2CI2 и SO2.
Способ 2 [2]
NF8-|-3S ----> S2F24-NSF
71 96 102 1 65
Выполнение синтеза см. в разд. «Тиазилфторид».
Свойства. /пл —164,6°С; /кип —10,6°С. Бесцветная жидкость. Более
устойчивое соединение, чем изомер FSSF.
ЛИТЕРАТУРА
1. Seel F., Golitz Н. D., Z. Anorg.
Allgem. Chem., 327, 32 (1964).
2. Glemser О., Biermann B„ Knaak J..
Haas A., Chem. Ber.. 98, 446
(1965).
Фторид cepbi(lV) SF4
Способ 1 [1]
S + 2F2 ----> SF4
32 76 108
SF4 образуется с очень высокими выходами при действии предварительно
охлажденного разбавленного азотом фтора иа суспензию серы в CCI3F при
—78 °C. Синтез проводят в стеклянном приборе, описанном для получения
фторида иода (III).
В реакционном сосуде суспендируют очищенную возгонкой и высушенную
в глубоком вакууме серу в 300 мл сухого CCI3F. Через суспензию пропуска-
ют сухой N2. Предохранительные ловушки, газ перед подачей в реактор и
сам реакционный сосуд медленно охлаждают до —78 °C (ловушки опускают
в бани, для предварительного охлаждения газа используют смесь Tri/СОг, а
для реакционного сосуда готовят смесь СН3ОН+СО2 в прозрачном сосуде
Дьюара). Суспензию интенсивно перемешивают с помощью высокоскоростной
механической мешалки и фторируют, пропуская через нее охлажденную газо-
вую смесь (F2: N2 от 1: 3 до 1:1). Реакция закончена, как только первона-
чально белая суспензия превращается в прозрачный, бесцветный раствор.
Для того чтобы предотвратить дальнейшее фторирование, избыточный рас-
творенный в CChF фтор вытесняют из раствора азотом.
206 Глава 3. Фтор
Полученный таким образом раствор SF4 может использоваться для по-
следующих синтезов; прн низких температурах он сохраняется без разложе-
ния в течение длительного времени. Для выделения SF4 раствор надо мед-
ленно нагреть до комнатной температуры н в потоке N2 пропустить SF4 че-
рез Hg, а затем через NaF. После фракционной перегонки получают чистый
SF«.
Способ 2 [2]
3SC12 + 4NaF -----> SF4 + S2C12 + 4NaCl
308,8 168 108 135 233,8
Реакцию проводят в 12-литровой четырехгорлой колбе (нз пнрекса).
В колбу вставляют 1-литровую капельную воронку, -мешалку, термометр и
охлаждаемый льдом обратный холодильник, который соединяют с нисходя-
щим холодильником, охлаждаемым смесью сухого льда с ацетоном (—60°C).
Подсоединение охлаждаемых такими же смесями приемников должно быть
газонепроницаемо. В реакционный сосуд помещают 2 кг тонкоизмельченного
NaF (0,8—0,4 мкм) и 6,5 л ацетонитрила и после перемешивания нагревают
до 68—70 °C. Прн этой температуре вливают 3,1 кг SC12 со скоростью
600 мл/ч. После этого реакционную смесь оставляют еще на 1,5 ч при 70—
72 °C. В приемнике собирается 1,2 кг конденсата, окрашенного в цвет от жел-
то-оранжевого до красного. Прн перегонке продукт реакции отгоняется пер-
вым. Получают 990 г 90%-ного продукта, который при вторичной ректифика-
ции дает 533 г совершенно чистого SF4. Для отделения SF4 от SOF2 доста-
точно получить аддукт BF3-SF4, из которого чистый продукт можно выде-
лить добавлением NaF (SOF2 с BF3 при комнатной температуре аддукта не
образует).
Работать с SF4 можно в сухнх стеклянных сосудах. Для хранения ис-
пользуются стальные баллоны. SF4 — очень часто применяемый фторирующий
агент (используется для получения, например, SOF4, SF5C1, Na2SnF6, CrO2F2).
Свойства. Неокрашенное вещество. /Пл —121 °C; ДИп —38 °C; /1!ГИТ 90,1 °C;
dtK 1,9191 (—73 °C). Очень гигроскопичное вещество, по токсичности похо-
жее на фосген.
ЛИТЕРАТУРА
1. Naumann D., Padma D. К., Z.
Anorg. Allgem. Chem., 401, 53
(1973).
2. Tullock C. W., Fawcett F. S.,
Smith W. C., Coffman D. D., J.
Amer. Chem. Soc., 82, 539 (1960).
Декафторид дисеры S2Fi0
(Дитиодекафторид)
2SC1F5 + H2 --> S2F10 4- 2HC1
325,0 2 254 73
Фотохимический процесс проводят в реакторе из кварца, в который вмон-
тирована трубка с излучателем — ртутной лампой. Реактор можно охлаж-
дать. С одной стороны реактора через осушительную колонку (с CaSO4) при-
соединена 20-литровая запасная емкость,, в которой находится дополнитель-
ное количество смесн SC1Fs+H2 (2:1). С другой стороны реактора при-
соединена промывная колонка, где газы движутся противотоком' с водой, и
благодаря этому удаляется НС1. Газы из промывной колонки проходят газо-
вую ловушку (—20 °C). Здесь конденсируется S2FI0. Остальная газовая смесь
с помощью диафрагменного насоса (20 л/ч) возвращается опять в запасную
емкость. Продукт реакции, собранный в ловушке, промывают водой, обраба-
Соединения с серой, селеном, теллуром 207
тывают 5 н. КОН, сушат над CaSO4, а затем над P4OJ0 и перегоняют. Выход
чистого продукта 40%.
Свойства. /пл —60 °C; /кип 29 °C; d0 2,08. Бесцветное, очень ядовитое ве-
щество.
ЛИТЕРАТУРА
1. Roberts Н. L., J. Chem. Soc. (Lon-
don), 1962, 3183.
Фторид серы(¥1) SF6
Способ 1 [1]
S + 3F2 --------> SF.
32,06 114,0 146,06
Реакцию проводят в никелевой трубке (длина ~300 мм, диаметр 25 мм),
в которую помещают никелевую лодочку с серой (рис. 134). Шлифы реакци-
онной трубки и присоединенной к ней кварцевой ловушки лучше всего вооб-
ще не смазывать (даже пнцеином), а просто плотно вставить друг в друга.
Рис. 134. Устройство прибора для получения фторида серы (VI).
В конце прибора присоединяют железную осушительную трубку со свеже-
обезвоженным KF, которая предохраняет от попадания влаги воздуха в при-
бор. Кварцевая ловушка охлаждается жидким О2 (или другим каким-нибудь
способом) до —183 °C.
В таком приборе проводят фторирование многих веществ, если необхо-
димо на твердое исходное вещество действовать газообразным фторидом (син-
тез SeFg, TeFs, AsFg, CF4, GeF4, MoF6, WFe). Для получения SFe прибор мож-
но изготовить полностью из стекла.
В потоке F2 сера сгорает синеватым пламенем. Собранный в ловушке
продукт затем пропускают через промывалку с горячим 10%-ным КОН (но
не NaOH) для очистки от примесей (HF, SF2, SF4, SOF2, S2F|o). В заключе-
ние продукт сушат в трубке над Р40ю, а для очистки от S2F(o пропускают
прн комнатной температуре над активированным углем. Выход 87%-
Способ 2 [2]
SO2 + 3F2 ----»- SFe+Os
64 114 146 32
В приборе, схематически изображенном на рис. 135, в избытке Fs диок-
сид серы сгорает до SF6. Температура должна быть возможно более высо-
208 Глава 3. Фтор
кой (~ 650 °C). Сырой SF6, собранный в ловушке, содержит как прнмесь в
основном SO2F2. Сырой продукт пропускают через несколько промывалок с
водой н с горячим 10|%-ным КОН, затем сушат над Р4Ою. Выход 70% в рас-
чете на SO2.
SF6 можно хранить в газометре под водой, в стеклянной колбе с краном
и под давлением в стальном баллоне.
Рис. 135. Установка для получения SF6.
Реакционный сосуд с ввинчивающейся горелкой изготовлен из меди; смотровые трубки
снабжены кварцевыми стеклами; холодная вода должна омывать весь реакционный
сосуд.
Свойства. Неокрашенное вещество, термически и химически очень устой-
чивое, без запаха. /пл —50,8°C (при пониженном давлении); при —63,8°C
сублимируется. Крит +45,55 °C; ркРИТ 38,33 бар; d№ 1,88 (—50,8 °C). В воде
растворимо очень слабо, немного растворимо в спирте.
ЛИТЕРАТУРА
1. Klemm W„ Henkel Р„ Z. Anorg. 2. Герм. пат. 1.72173 IVb/12 i, 1942.
Allgem. Chem., 207, 73 (1932). 3. Kwasnik W7., неопубликованные
данные.
Пентафторид хлоросульфинила SCIFs
(Тиомопохлоридпентафторид)
SF4 + C12 + CsF --* SClF6 + CsCl
108 71 151,9 162,5 168,4
172 г тонкорастертого фторида цезия, 108 г тетрафторида серы и 71 г
хлора помещают в 0,5-лнтровый автоклав (который снабжен устройством
для перемешивания), изготовленный нз нержавеющей стали, и выдерживают
1 ч при 100 °C, 1 ч при 150 °C и, наконец. 2 ч при 175 °C. Улавливаемый с по-
мощью охлаждаемой ловушки продукт (160 г) перегоняют на низкотемпера-
турной колонке (—60°C). Неокрашенная фракция (125 г), перегоняющаяся
между —23 и —21 °C, представляет собой 95%-ный SC1F6. Выход 70—80%.
Используется для получения S2Fio.
Свойства. /пл —64 °C; Кип —19°C; Крит 117,7°C; d№ 1,541 (—60°C). Не-
окрашенное вещество.
ЛИТЕРАТУРА
1. Tullock С. W., Coffman D. D., Muetterties E. L„ J. Amer. Chem.
Soc., 86, 357 (1964).
Соединения с серой, селеном, теллуром 209>
Фторид сульфинила SOF2
(Тионилфторид)
Способ 1 [1—4]
SOC12 + 2HF ----> SOF2 + 2HC1
118,97 40 86,06 72,92
Реакционным сосудом служит железная бутыль (рис. 136), к которой'
присоединена газоотводная трубка, а вслед за ней такая же бутыль, в кото-
рой должен задерживаться непрореагировавший HF. Затем следует стеклян-
ная газовая ловушка, которую опускают в жидкий О2 нли в смесь ацетона
с сухим льдом. От попадания влаги воздуха предохраняет осушительная
трубка с KF.
Рис. 136. Установка для получения фторида сульфинила.
В реакционный сосуд помещают 500 г SOC12 н 50 г SbCl5 (катализатор)»
и через газоотводную трубку начинают подавать безводный газообразный
HF. Фтороводород хорошо абсорбируется. В газовую фазу выделяется смесь
SOF2 и НС1, которые улавливаются ловушкой. Реакция сопровождается силь-
ным охлаждением, так что снаружи реакционный сосуд постепенно покрыва-
ется льдом. После полного выгорания SOC12 можно его ввести вновь, уже
не добавляя SbCl5.
Очистить SOF2 от НС1 можно либо перегонкой, либо пропусканием газо-
вой смесн через ледяную воду (НС1 полностью поглощается, в то время как:
SOF2 проходит почти не разлагаясь). В заключение газ сушат над конц-
H2SO4 илн Р4О10.
Способ 2 [5, 6]
SOCl24-2NaF -----> SOF2 + 2NaCl
118,97 83,98 86,06 116,88
Этот особенно удобный синтез проводят в очищенном перегонкой СН3СЬГ
(реакционная среда).
Реакцию проводят в колбе (из пнрекса), снабженной капельной ворон-
кой, термометром, приспособлением для перемешивания и обратным холо-
дильником с присоединенной охлаждаемой ловушкой (—78 °C). К интенсив-
но перемешиваемой суспензии, приготовленной нз 168 г высушенного тонко-
растертого NaF и 100 г CH3CN, прибавляют по каплям 60 г SOC12, при это»
реакционная смесь должна кипеть. После прибавления всего SOC12 нагрева-
ют еще 2 ч прн 80 СС, продолжая перемешивание. При этом SOF2 улетучи-
вается нз реакционной смесн и конденсируется в охлаждаемой ловушке. Для
14—143
210 Глава 3. Фтор
очистки можно использовать низкотемпературную ректификацию. Выход 77%
в расчете на введенный SOC12.
Хранят SOF2 в стальных баллонах под давлением.
Свойства. Бесцветный газ. Термически устойчив вплоть до температуры
красного каления. /пл —110,5 °C; /кип —43,7 °C; ^Рит +88 °C; 1,780
(—100°C); 2,095 (—183°C),. Не действует1 на Fe, Ni, Со, Hg, Si, Мп, В,
Mg, Al, Zn прн 125 °C. He разъедает стекло. Имеет удушливый запах. Мед-
ленно гидролизуется холодной водой.
ЛИТЕРАТУРА
1. Герм. пат. 1.53743 IVb/121.
2. Soil J., Kwasnik W., Naturforschung
u. Medizin in Deutschland 1939—
1946 (FIAT-Review), 23, 192.
3. Booth H. S., Merciola F. C., J.
Amer. Chem. Soc., 62, 640 (1940).
4. Wannagat U., Mennicken G., Z.
Anorg. Allgem. Chem, 278, 310
(1955).
5. Tulloch C. W., Coffman D. D., J.
Organ. Chem, 25, 2016 (1960).
6. Sundermeyer W., Univers. Heidel-
berg, 1973, частное сообщение.
Тетрафторид сульфинила SOF4
.(Тиооксотетрафторид)
Способ 1 [1, 2]
5SOF2 + 2BrF5 -----> 5SOF4 + Br2
-430,3 349,8 620,3 159,8
'64,5 г (0,75 моль) фторида сульфинила и 52,5 г (0,3 моль) фторида бро-
ма (V) нагревают в течение 10 ч при 280 °C в автоклаве вместимостью
300 мл, изготовленном из специальной стали. Первоначально установленное
давление в 134 бар медленно понижают до 123,6 бар. После выпуска газов
из автоклава прн комнатной температуре SOF4 получают в виде слегка жел-
товатой жидкости, которую можно подвергнуть низкотемпературной ректи-
фикации. Выход 85% в расчете на введенный SOF2.
Способ 2 [3]
NO2
sf4 + 1/2o2 —>- SOF4
108,1 16,0 124,1
В автоклав вместимостью 300 мл, изготовленный нз специальной стали,
конденсируют 51 г (0,47 моль) SF4 и 8 г (0,5 моль) Оа, а также 5 г ЬЮ2
(катализатор); затем автоклав нагревают 8 ч при 200 °C, при этом меняется
давление от 75 (первоначальное) до 60 бар. После выпуска газа и низкотем-
пературной перегонки получают 44 г чистого SOF4 (т. кип. —48°C). Выход
70% в расчете на введенный SF4.
Способ 1 более предпочтителен по сравнению со способом 2, в котором
при выполнении синтеза происходит сильная коррозия стального автоклава;
кроме того, SF4 — очень дорогой препарат.
Для получения очень чистого SOF4 сырой продукт переводят в аддукт
SOF4-AsFB (ср. следующий препарат); очищенный аддукт нагревают с KF.
Хранят SOF4 под давлением в стальных баллонах или стеклянных ампу-
лах при охлаждении сухим льдом или жидким воздухом.
Соединения с серой, селеном, теллуром 211
Свойства. Бесцветный газ. Реагирует с водой с сильным выделением теп-
ла и образованием SO2F2. Количественно абсорбируется гидроксидом натрия.
Чистый SOF4 не разъедает стекло. Имеет резкий запах. /Пл —99,6 °C; /кип.
—48,5°C; diK 1,946 (—82°C); dTS 2,55 (—183°C).
ЛИТЕРАТУРА
1. Seppelt К., Z. Anorg. Allgem. 3. Smith W. C., Engelhardt V. A., J..
Chem., 386, 289 (1971). Amer. Chem. Soc., 82, 3838 (I960).
2. Sundermeyer W., Universitat Hei-
delberg, 1973, частное сообщение.
Аддукт тетрафторида сульфинила и пентафторида мышьяка
SOF4-AsF5, [SOF3][AsF6]
SOF4 + AsF5 ----> SOF4 AsF5
124 169,9 293,9
В кварцевую ловушку конденсируют при температуре жидкого воздуха1
SOF4 н AsFs. Осторожно поднимают температуру до температуры плавления
SOF). Происходит бурная реакция, которая сопровождается частичным испа-
рением исходных веществ. Аддукт представляет собой белое твердое вещест-
во, собирающееся в ловушке. Продукт очищают путем сублимации в квар-
цевом приборе. Прн 80 °C процесс сублимации происходит со скоростью»
6 г/ч.
При взаимодействии SOF4-AsF5 с KF можно получить очень чистый SOF4.
Этот продукт присоединения также удобен для хранения SOF4 в больших ко-
личествах.
Свойства. Бесцветное вещество. Давление пара при 20 °C составляет
7 мм рт. ст.
ЛИТЕРАТУРА
1. Seel F„ Detmar О., Z. Anorg.
Allgem. Chem., 301, 113 (1959).
Дифторид сульфонила SO2F2
(Сульфурилднфторид)
SO2C12 + 2NaF --> SO2F2 -J- 2NaCl
135,0 84,0 102,1 116,9
Реакция происходит в очищенном перегонкой тетраметнлеульфоне (ТМС).
135 г SO2C12 прибавляют по каплям к интенсивно перемешиваемой суспен-
зии, приготовленной из 168 г сухого тонкодисперсного NaF в 300 г ТМС.
Реакцию проводят в 1-литровом автоклаве, который имеет приспособления
для введения жидкости по каплям без нарушения герметичности, измерения
температуры и перемешивания. Температура реакционной смеси не должна
подниматься выше 100 °C. После прибавления всего количества продолжают
перемешивание реакционной смесн, выдерживая ее последовательно при 60,
80, 100 и 125 °C; причем каждый температурный режим сохраняют в течение
1 ч; затем температуру повышают до 150 °C и оставляют на 2 ч. В течение
всей реакции автоклав под давлением компонентов реакции остается закры-
тым. Затем его открывают, газообразный SOF2 выпускают и конденсируют
14*
212 Глава 3. Фтор
да охлаждаемой (—78°C) стеклянной ловушке. Выход 85% в расчете иа вве-
денный SO2CI2.
Хранят SO2F2 в газометрах над конц. H2SO4 или под давлением в сталь-
ных баллонах.
Подобным же образом можно получить фторид селенила SeOF2:
SeOCl8 + 2NaF ----*- SeOF2-[-2NaCl
При этом нет необходимости в автоклаве. Реакцию можно проводить в ТМС
при обычном давлении и повышенной температуре (60—115 °C). Выход 28%.
Свойства. Газ без цвета и запаха. До 400 °C термически устойчив, хими-
чески очень инертен, водой гидролитически не расщепляется. Довольно быст-
рое растворение в щелочах сопровождается полным гидролизом. tna
121,4 °C; iKBn —49,7 °C; ~1,7.
Ниже приведены данные по растворимости при 16,5 °C в различных рас-
творителях:
Растворитель
Вода
Спирт
Толуол
СС14
Растворимость
SO2F2 (газ), мл/100 мл
4—5
24—27
210—220
136—137
ЛИТЕРАТУРА
1. Tullock С. \W., Coffman D. D., J.
Organ. Chem., 25, 2016 (1960).
2. Sundermeyer W„ Universitat Hei-
delberg, 1973, частное сообщение.
Дифторид дисульфурила S2O5F2
/Пиросульфурил фторид)
CaF2 + SO3 -*• FSO2OSO2F + CaSO4
78 240 182 136
В автоклав вместимостью 400 л, изготовленный из нержавеющей стали,
помещают 0,5 моль СаРг и 4,5 моль SO3. Автоклав охлаждают до —78 °C и
эвакуируют. Затем реакционную смесь при перемешивании нагревают в тече-
ние 24 ч при 200 °C. Автоклав охлаждают (до температуры 04—20 °C) и
впускают в него 500 г 92%-ной серной кислоты (для растворения непрореа-
тировавшего SO3). Обработка серной кислотой существенна для хорошего
выхода. В результате реакции получают густую кашицу. После расслоения
верхний слой, представляющий собой жидкий S2O6F2, декантируют и препарат
очищают перегонкой при атмосферном давлении. Выход 72% в расчете на
•СаРг.
Свойства. Бесцветная, весьма подвижная жидкость. «Пл —48 °C; «кип
50,8 °C; dio 1,75. До 200 °C термически устойчива. Незначительно дымит на
воздухе. Отрицательно влияет на качество табачного дыма. Подходящий рас-
творитель при реакциях фторирования. Вызывает сильное удушье, напоми-
ная по своему действию фосген.
ЛИТЕРАТУРА
4. Muetterties Е. L., Coffman D. D.,
J. Amer. Chem. Soc., 80, 5914
(1958).
Соединения с серой, селеном, теллуром 213
Оксодифторид дисульфурила S2O6F2
(Пероксодисульфурилдифторид)
2SO3 + F2 -----> S2OeFa
160,12 38,00 198,12
Синтез S2O6F2 проводят в медной трубке (длина 90 см, диаметр
~7—8 см) в присутствии AgF2 как катализатора. Катализатор получают сле-
дующим образом. Медную стружку или медную проволоку (в зависимости
•от диаметра трубки и чистоты материала берут ~2—5 кг) покрывают се-
ребром (~50—100 г), обрабатывая ее для этого раствором [Ag(CN)2]~,
промывают, высушивают и полученным материалом по возможности плотно
наполняют трубку, которая перед этим должна быть с одной стороны за-
крыта наглухо припаянной (серебряный припой) медной пластинкой; после
•наполнения другой конец таким же образом закрывают (запаивают). Через
.припаянные пластннкн на обоих концах трубки вставляют медные трубки
(диаметр ~6 мм) для подвода и отвода реакционных газов. Реакционная
трубка снабжена нагревательной обмоткой. Через наполненную трубку про-
пускают при 200 °C фтор вплоть до насыщения и получают AgF2, используе-
мый как катализатор. Триоксид серы (~ 500 г) помещают в колбу (вмести-
мостью 1 л) со шлифами, в которую опущена почти до диа трубка для под-
вода Nz. От колбы отходят газоотводные трубки, соединяющие ее с реакци-
онным сосудом. Газоподводящая и газоотводная трубки имеют краны для
того, чтобы их можно было перекрыть прн попадании жидкости. Поток су-
хого азота (8 л/ч) подают в нагретую до 30 °C колбу с SO3 и незадолго до
поступления газов в реакционную трубку добавляют в газовый поток F2,
также разбавленный азотом. Скорость подачи обоих газов и их количество
контролируют: фтор должен быть всегда в небольшом избытке. В реакци-
онной трубке устанавливают температуру 150 °C; время пребывания в ней
тазов 15 мин.
После реакционной трубки газы поступают в две стеклянные ловушки.
В первой, охлаждаемой до —78 °C, конденсируются основные количества
S2OeF2, во второй, охлаждаемой до —'183 °C, — летучие побочные продукты и
лишь немного S2OeF2. После того как оба газовых потока отрегулированы,
так что в газе, выходящем из второй ловушкн, можно обнаружить с по-
мощью KI присутствие небольшого избытка F2, содержимое первой ловушки
испаряют и конденсируют вновь при —78 °C в другой (пустой) ловушке.
Теперь уже конденсируется не содержащий SO3 продукт, и в указанных
условиях можно ожидать получения ~6 г/ч S2OcF2. Для очистки от легко-
летучнх примесей содержимое первой ловушки при —78 °C откачивают до
тех пор, пока молекулярная масса, рассчитанная из экспериментально най-
денной для взятой пробы плотности, не приблизится к 198—200. Если этого
не удается достичь, значит, не всегда пропускаемый через реакционную труб-
ку газ содержал избыток F2 и не весь SO3 прореагировал. В таком случае
продукт подвергают более совершенным методам очистки путем перегонки из
колбы в колбу в стеклянном приборе или на колонне с вращающейся метал-
лической лентой (платиновая лента, тефлоново-платиновые вентили). Краиы
должны иметь тефлоновые манжеты или смазываться фторированной смаз-
кой. Хранить S2O6F2 надо при —78 °C. Замечено, что среди побочных продук-
тов может находиться взрывоопасный FSO3F.
Свойства. Бесцветная, очень неприятно пахнущая гигроскопичная жид-
кость. Тотчас воспламеняет органические материалы. <пл —55,4 °C; /кив
67.1 °C. Давление пара при 25,8°С 146,4 мм рт. ст.; d 1,645 (35,5°C).
214 Глава 3. Фтор
Соединения с серой, селеном, теллуром 215
тп№мурк
1. Dudley F. В., Cady G. Н„ J. Amer. Amer. Chem. Soc., 83, 4521 (1961);
Chem. Soc., 79, 513 (1957). Inorg. Synth., 7, 124 (1963).
2. Shreeve J. M., Cady G. H„ J. 3. Nutkowitz P. M., Vincow G., J.
Amer. Chem. Soc., 91, 5956 (1969).
Хлорид-фторид сульфинила SOC1F
(Тионилхлоридфторид)
4SOC12 + IF5 ---> 3SOC1F + SOF2 + IC1S + Cl2
475,9 221,92 307,5 86,08 233,29 70,92
В колбу, снабженную обратным холодильником н капельной воронкой
(все из кварца), к 60 г SOC12 прибавляют медленно по каплям 45 г IFs.
Реакционная смесь разогревается в результате происходящей реакции и ста-
новится темной. Газы, проходящие через обратный холодильник, конденси-
руются в кварцевой ловушке, которую погружают в жидкий воздух. На вы-
ходе из прибора присоединяют осушительную трубку с обезвоженным KF-
Сконденсированный в ловушке продукт, окрашенный в зеленовато-жел-
тый цвет от увлеченного вместе с ним 1С13, перегоняют, пропуская через по-
рошок Sb или Hg до их обесцвечивания. В заключение продукт фракциони-
руют, причем первые погоны содержат SiF4, НС1 и SOF2, а последний —
SOC12. Фракция SOC1F отгоняется в интервале 10—18°C (760 мм от ст)
Выход 42% в расчете на SOC12. н '
Хранить SOC1F лучше всего в стеклянных ампулах при —183 °C; пр»
непродолжительном хранении в крайнем случае можно использовать стек-
лянные колбы или стальные баллоны.
Свойства. Бесцветный газ с удушливым запахом, напоминающим фтори-
ды серы. При комнатной температуре разлагается на SOC12+SOF2, а также
с Си н Hg, что обусловлено его диспропорционированием; пр»
/О С взаимодействует с Fe. Вода и гидроксид натрия вызывают гидролиз.
Со стеклом не взаимодействует.
<oJ?o^HMeeT четкой температуры плавления; плавится в интервале —110 и
—139 С (смесь двух изомеров); /кип 12,3°C; dK 1,576 (0°С).
ЛИТЕРАТУРА
1. Puff О., Jonas И., Naturforschung
u. Medizin in Deutschland 1939—
1946 (FIAT-Review), 23, 192.
2. Герм. пат. R 100 449.
3. Jonas H., Z. Anorg. Allgem. Chem_
265, 273 (1951).
Хлорид-фторид сульфонила SO2C1F
(Сульфурилхлоридфторид)
Способ 1 [1]
3SO2C12 + SbF3 ---- 3SO2C1F + SbCls
404,91 178,76 355,54 228,13
Реакцию проводят в автоклаве или стальном баллоне вместимостью 1 л
на который навинчен питаемый от водопровода железный нисходящий хо-
лодильник и который снабжен пружинным манометром и выпускным венти-
лем. Аппаратура должна выдерживать давление 11 бар.
Выпускной вентиль соединен с двумя кварцевыми ловушками, которые
погружены в жидкий воздух. На выходе установки находится осудительная
трубка с обезвоженным KF для предохранения от воздействия влаги воз-
ДУМВ реакционный сосуд загружают 220 мл (365 г) SO2C12, 187 г тонкоднс-
персного SbF3 и 40 мл SbCl5 (катализатор). Постепенно нагревают до
300 °C устанавливая давление 8 бар. Открывают вентиль и медленно выпу-
скают'реакционные газы в газовую ловушку до тех пор, пока в Реакцион-
ном сосуде не установится давление 7,3 бар. Через 2 ч собирают в ловушке
80 мл конденсата.
В заключение продукт перегоняют, причем первые погоны содержат ны
и SO2, а последний — непрореагировавший SO2C12. Получают 50 мл чистого
SO2C1F.
Способ 2 [2, 3]
С удовлетворительным выходом без применения повышенного давления
можно получить SO2C1F в CH3CN (растворитель):
SO2Cl2 + NaF ----» SO2ClF + NaCl
134,9 42,0 118,5 58,4
Синтез проводят в приборе (см. рис. 75), состоящем из колбы (из стекла
пирекс), капельной воронки, термометра, приспособления для перемешивания
и обратного холодильника с присоединенной к нему ловушкой ( /о с;.
В колбу к суспензии, приготовленной из 144 г (2,48 моль) сухого тонкодис-
персного NaF н 133 г (3,24 моль) перегнанного CH3CN, прибавляют медлен-
но по каплям 68 г (0,5 моль) SO2C12. После прибавления всего количества
SO2C12 реакционную смесь нагревают еще 3,5 ч при 80 С. При этом г>и2удг
улетучивается и конденсируется в ловушке. Выход 64% в расчете на введен-
ный в реакцию SO2C12.
Хранят SO2C1F в стальных баллонах или стеклянных колбах.
Свойства. Бесцветный газ с резким запахом, напоминающим SO2C12;
не дымит на воздухе, быстро реагирует с водой и едкими щелочами; дейст-
вует иа Hg и латунь. В чистом состоянии не действует иа стекло. Гпл
—124,7°С; /кип 7,1 °C; dx 1,623 (0°С).
ЛИТЕРАТУРА
1 Booth Н. S., Hermann V., J. Amer. 3. Sundermeyer W., Universitat Hei-
‘ Chem. Soc., 58, 63 (1936). delberg, 1973, частное сообщение.
2. Tullock C. W.. Coffman D. D„ J.
Organ. Chem., 25, 2016 (1960).
Бромид-фторид сульфонила SO2BrF
(Сульфурилбромидфторид)
Br2 + BrF3 ----* 3BrF
159,82 136,91 296,73
3BrF + 3SO2 ------► 3SO2BrF
296,73 192,18 488,91
Процесс образования продукта реакции происходит во времени. Скорость
реакции определяется обратимой реакцией Br2+BrF3^*3BrF.
В железный автоклав (при 4-12 °C) помещают смесь 20 мл Вг2 и
21 2 мл BrF3 и постепенно перегоняют туда 120 г SO2. Автоклав оставляют
на несколько дней, ежедневно встряхивая его содержимое; продукт из реак-
ционного сосуда перегоняют под собственным давлением и собирают в квар-
цевой ловушке при —183 °C. Для очистки SO2BrF пропускают через промы-
216 Глава 3. Фтор
валку с Hg (для удаления следов Вг2 и BrF3), затем через NaF (для удале-
ния HF) и, наконец, сушат над Р40ю. В заключение осуществляют фракци-
онную перегонку, причем первый погон отбрасывают, а последний вообще не
перегоняют, оставляя в перегонной колбе. Выход 88% в расчете на BrF3.
Хранят SO2BrF в запаянных кварцевых ампулах.
Свойства. М 162,98. Прн стоянии на влажном воздухе приобретает слег-
ка красноватую окраску вследствие выделения брома. Имеет удушливый за-
пах, напоминающий SO2CI2. До 300 °C термически устойчиво, при комнатной
температуре медленно реагирует со стеклом, не действует на кварц. С водой
вступает в бурную реакцию, сопровождаемую гидролизом.
<пл —86 °C; <кип 40°C; dK 2,17 (0°С); 3,16 (—183°C).
ЛИТЕРАТУРА
1. Ruff О., Jonas Н. (совместно с 2. Jonas Н., Z. Anorg. Allgem. Chem.»
Kwasnik W.), Naturforschung u. 265, 273 (1951).
Medizin in Deutschland 1939—1946
(FIAT-Review), 23, 193.
Фторосерная кислота HSO3F
Способ 1 [1]
2KHF2 + 4SO3 4- H2SO4 ---*- 4HSO3FK2SO4
156,20 320,24 98,08 400,25 174,27
К 40 мл дымящей серной кислоты, содержащей 60% SO3, в платиновой
или алюминиевой чашке, хорошо охлаждаемой смесью льда с поваренной
солью, прибавляют небольшими порциями при помешивании 20 г высушен-
ного н измельченного KHF2. Получают вязкую, немного дымящую на возду-
хе массу. Медленно нагревают ее до 100 °C для удаления непрореагировав-
ших SO3 и HF.
Затем HSO3F отгоняют (посуда из йенского стекла со шлифовымн сое-
динениями), медленно повышая температуру до 250 °C. После двухкратной
перегонки получают совершенно чистую кислоту. Выход 85%.
Способ 2 [2]
SOs4-HF -----> HSO3F
80,06 20 100,06
К 800 г SO3 в алюминиевом сосуде, температура которого поддержи-
вается в интервале 35—45 °C, с помощью капилляра из специальной стал»
(V2A) вводят 200 г HF (кончик капилляра надо погрузить в жидкость).
Поглощение HF происходит быстро, но не сопровождается взрывоподобны»*
вспениванием. Затем реакционную смесь нагревают при 100 °C для испаре-
ния избытка SO3 н HF. Продукт дважды перегоняют в приборе из алюми-
ния.
Способ 3 [3]
HSO3C1 4- HF ----> HSO3F 4- НС1
116,53 20 100,06 36,47
Серебряную колбу для перегонки, снабженную капельной воронкой из
серебра, помещают в баню со льдом и поваренной солью. В колбу через
штуцер перегоняют 50 г безводного HF. После этого штуцер соединяют с
осушительной медной трубкой, содержащей KF, для поглощения увлеченно-
го HF. Через капельную воронку в колбу вводят HSO3C1. Тотчас происхо-
дит реакция, сопровождаемая выделением и улетучиванием НС1. По оконча-
нии синтеза удаляют избыток HF и остаточные количества НС1 путем про-
Соединения с серой, селеном, теллуром 217
дувания сухого воздуха при постепенном нагревании содержимого колбы до
НО °C. Остаток в перегонной колбе представляет собой не содержащую хло-
ра HSO3F.
Совершенно чистую HSO3F хранят в запаянных стеклянных ампулах и
даже в алюминиевой посуде.
Свойства. Бесцветная жидкость. Термически устойчива до 900 °C.
/пл —87°C; /квп 163°C; dK 1,740 (18°С).
Прн комнатной температуре не действует на S, С, Se, Те, Pb, Ag, Сц,
Zn, Fe, Сг, Мп. Напротив, реагирует с Sn с незначительным выделением
газа. Также неактивно взаимодействует с Hg. Резину, пробку н сургуч быст-
ро разрушает. При нагревании довольно агрессивно действует на S, РЬ,
Sn и Hg. Дает с ацетоном (прн сильном выделении тепла) темно-красное до
коричневого окрашивание (цветная реакция на фторосульфокислоты). Взаи-
модействует с бензолом н хлороформом с отщеплением HF. Эфир вызывает
сильное разогревание и вспучивание, причем образуется этиловый эфир фторо-
серной кислоты. Чистая HSO3F не действует на стекло. Реакция с водой про-
исходит настолько бурно, что близка к взрыву. Кислота дымнт на воздухе.
ЛИТЕРАТУРА
1. Meyer J., Schramm G., Z. Anorg. 2. Герм. пат. I. 52953 IVb/12i, 1935.
Allgem. Chem., 206, 25 (1932). 3. Wlechert H„ Z. Anorg. Allgem.
Chem., 261, 310 (1950).
Получение фторосульфатов C1OSO2F, BrOSO2F, Br(OSO2F)3 и I(OSO2F)s
описано в части 11, гл. 3. Получение производных фторосерной кислоты
HiNSO2F, FSO2NCO и HN(SO2F)s описано в части II, гл. 7.
Фторосульфонат калия FSO2K
KF + SO2 -------> FSO2K
58,10 64,06 122,16
1 кг безводного тонкодисперсного KF и 2 кг сжиженного SO2 в желез-
ном автоклаве вместимостью 4 л (снабженном приспособлением для пере-
мешивания) медленно перемешивают при комнатной температуре в течение
5 сут. Затем выпускают избыточный SO2 и получают 2 кг KSO2F (продукт
содержит 95% (мол.) KSO2F). Синтез можно осуществлять также следую-
щим образом. В автоклав загружают KF, присоединяют через капилляр
стальной баллон с сжиженным SO2, вытесняют воздух из автоклава, про-
пуская SO2, и затем закрывают вентиль автоклава. После включения ме-
шалки начинается бурное поглощение SO2, которое прекращается после то-
го, как KF поглотит 1 кг SO2.
Фторосульфонат калня можно использовать в качестве «активированно-
го фторида калия». Он взаимодействует со многими неорганическими и орга-
ническими галогенсодержащими кислотами с образованием соответствующих
фторидов н поэтому часто применяется как фторирующий агент в реакциях,
где иначе был бы необходим безводный HF (получение, например, SOF2,
PF3, POF3, AsF3, C6H6COF).
Свойства. Бесцветное твердое вещество. Разлагается прн 170—180 °C.
Растворимость в жидком SO2 (при 0°С) 3,85 мг/100 г. Растворение в воде
сопровождается гидролизом. С С12, Вг2 и F2 образует фторид сульфонила.
ЛИТЕРАТУРА
I. Seel F., Riehl L., Z. Anorg. Allgem.
Chem., 282, 293 (1955).
218 Глава 3. Фтор
Фторид селена(У1) SeF6
Se + 3FS ------*- SeFe
78,96 114 192,96
Селен фторируют в приборе, описанном в методике получения SFe.
Можно использовать прибор, изготовленный из стекла. В потоке F2 селен-
сгорает (причем реакция ие требует нагревания). Время от времени реак-
ционную трубку надо охлаждать. Собранный в ловушке при —,183 °C про-
дукт затем пропускают через промывалку с 10%-ным гидроксидом калия и;
сушат над Р40ш. После фракционирования получают совершенно чистый
SeFe.
SeFe можно хранить в стеклянных колбах и в газометрах под водой.
Свойства. Бесцветный газ. Термически очень устойчив. Инднферентеи к
стеклу, однако немного действует на Hg. После вдыхания вызывает одышку
и щемление сердца. /пл —34,8 °C > ^возг —46,6 °C; /крит 72 °C; d№ 2,108-
(—10°C); dTB 3,478 (—195°C).
ЛИТЕРАТУРА
1. Klemm W., Henkel P„ Z. Anorg.
Allgem. Chem., 207, 74 (1932).
Фторид селена[ IV) SeF4
Se -|- 2F2 ----SeF4
78,96 76 154,96
Селен, высушенный при 200 °C, помещают в широкий реакционный со-
суд нз пирекса или кварца, который охлаждают льдом. На дне сосуда дол-
жен находиться тонкий слой селена. Пропускают со скоростью 1 л/ч фтор,
разбавленный N2 в отношении 1:1. Важно обеспечить хорошее охлаждение
и фторирование проводить осторожно, так как иначе образуется SeF6. В за-
ключение жидкий продукт реакции перегоняют в вакууме.
Свойства. Бесцветная жидкость: смешивается с H2SO4, спиртом, эфиром:
и IF6. /пл —9,5 °C; /кип 106 °C; d 2,72 (25 °C)
NaF, KF, RbF, CsF и T1F растворяются в SeF4 с образованием комплек-
сов типа MSeF5. Вода бурно разлагает SeF4. При нагревании с Hg в колбе
с обратным холодильником в течение нескольких часов образуется HgSeF4-.
Медленно действует на стекло пирекс.
ЛИТЕРАТУРА
1. Aynsley Е. Е., Peacock R. D., Ко- 2. Peacock R. D., J. Chem. Soc. (Lon-
binson P. L„ J. Chem. Soc. (Lon- don), 1953, 3617.
don), 1952, 1231.
Дифторид селенила SeOF2
Селенилдифторид, селеноксодифторид)
SeOCl2 + 2KF ----> SeOF24-2KCl
165,87 116,2 132,96 149,11
Прежде всего в стеклянной колбе на 100 мл обезвоживают 1 моль KF,
нагревая его в течение 8 ч прн 200—300°C в вакууме (масляный насос).
Соединения с серой, селеном, теллуром 219
После охлаждения на колбу надевают насадку Аншютца, в которую встав-
лены капельная воронка н трубка с СаС12. К фториду калия начинают при-
бавлять по каплям SeOCl2 с такой скоростью, чтобы температура реакцион-
ной смесн не поднималась выше 60—70 °C. Затем на колбу насаживают
обратный холодильник и реакционную смесь нагревают 8 ч в вакууме при
100 °C. Как только из обратного холодильника перестает возвращаться кон-
денсат, реакцию считают законченной. Теперь к колбе присоединяют необ-
ходимые для вакуумной перегонки устройства, в том числе водоструйный
насос, н отгоняют SeOF2, нагревая содержимое перегонной колбы пламенем
горелки Бунзена; в приемнике собирают желтоватую жидкость.
Для того чтобы получить чистый, прозрачный как вода продукт, сырой
продукт добавляют по каплям в стеклянную колбу, в которой находится KF.
При этом образуется KSeOFs (реакция сопровождается сильным разогрева-
нием). Продукт присоединения разлагается при нагревании в вакууме (кол-
-ба из тугоплавкого стекла) и отгоняется чистый SeOF2. Выход 79%.
Свойства. Бесцветное вещество. /пл 15,5°C; /кип 34,5°C (9 мм рт. ст.).
ЛИТЕРАТУРА
1. Paetzold R., Aurich К., Z. Anorg.
Allgem. Chem., 314, 72 (1962).
Фтороселенит калия FSeO2K
Способ 1
KF-|-SeO2 -----> FSeO2K
58 111 169
В платиновом тигле сплавляют при 250 °C KF (предварительно обезво-
женный путем 6-часового нагревания при 300 °C и 10 мм рт. ст.) и SeO2
(в мольном отношении 1:1). Для предотвращения окрашивания продукта в
розовый цвет, что обусловлено присутствием следов селена, в расплав в те-
чение 10 мин пропускают поток сухого содержащего i\'O2 кислорода. При
охлаждении расплава FSeO2K кристаллизуется в виде хорошо образованных
игл. С помощью платиновой проволоки, которую погружают в еще расплав-
ленную массу, плав вытаскивают из тигля н размельчают в ступке в атмо-
сфере сухого азота.
Способ 2
K2SeO3 + SeOF2 ---> 2FSeO2K
205 133 338
В тигель, имеющий приспособление для защиты от попадания влаги, по-
мещают хорошо высушенный К25еОз и прибавляют к нему по каплям рас-
считанное количество SeOF2. Реакция протекает очень бурно. Продукт реак-
ции представляет собой жидкость. Дальнейшую обработку проводят, как
описано в первом способе.
Свойства. Белое, кристаллическое, очень гигроскопичное вещество.
/пл 250 °C; df° 2,98.
ЛИТЕРАТУРА
1. Paetzold R-, Aurich К„ Z. Anorg.
Allgem. Chem., 335, 281 (1965).
220 Глава 3. Фтор
Фторид Tennypa(VI) TeF6
Те + 3F2 ------>• TeFe
127,61 114 241,61
Теллур фторируют в приборе, описанном в синтезе SFe, который в этом
случае можно изготовить полностью нз стекла. Происходит экзотермическая
реакция, однако, как правило, пламени не появляется, если снаружи произ-
водить охлаждение.
Собранный в ловушке при —183 °C продукт фракционируют. Газы нель-
зя пропускать для промывания через гидроксид калия, так как TeFe в этих
условиях гидролизуется.
TeFe можно хранить в стеклянных колбах.
Свойства. Бесцветный газ с неприятным запахом; химически не такой
индиферентный, как SeFe и SFe. Водой медленно, но полностью гидролизует-
ся; действует на Hg.
TeFe вызывает затруднение дыхания и щемление в области сердца. Пос-
ле вдыхания ощущается отвратительный запах теллура, 1ПЛ —37,6 °C;
*суОл —38,9°С; Zkpht 83°С; dx 2,499 (—10°С); dTB 4,006 (—191 °C).
ЛИТЕРАТУРА
1. Klemm W„ Henkel P., Z. Anorg.
Allgem. Chem., 207, 74 (1932).
2. Yost D. М., Clausen W. Н., J. Amer.
Chem. Soc., 55, 885 (1933).
Фторид азота(Ш) NF3
(Фтор азот)
Способ 1 [1—6]
4NH3 + 3F2 -->• NF3 + 3NH4F
68,12 114,0 71,0 111,12
Фторирование NH3 происходит под действием фтора в момент его вы-
деления из расплава прн электролизе NH4HF2.
В электролизере, описанном в методике получения F2, проводят электро-
лиз по возможности сухого NH4HF2, не содержащего хлора. Состав распла-
ва должен соответствовать интервалу МН4Р-1,1 HF—NH4F-1,8 HF. Электро-
лиз проводят при температуре 130—140 °C. В качестве анода используют
никель или графит. Процесс осуществляют при 10 А н 5,6—5,9 В. Плотность
тока на аноде 0,05—0,1 А/см2. (Плотность тока не оказывает существенного
влияния на выход.)
Поступающие из электролизера газы для очистки от HF и Н2О пропу-
скают через железную трубку со свежеобезвоженным KF. Далее следуют две
стеклянные газовые ловушки, охлаждаемые до —183 °C с помощью жидкого
О2 или каким-нибудь другим способом. На выходе прибора присоединена
железная осушительная трубка с KF.
Как только начинается электролиз, в газовых ловушках конденсируются
прежде всего только твердые, частично окрашенные (в цвет от лилового до
голубого) продукты (N2O, N2O3, О3). Это может происходить уже в первые
часы нли через несколько дней, что зависит от содержания в электролите
влаги. Начиная электролиз, надо иметь в виду возможность взрыва, кото-
рый вызывается озоном. По мере того как из расплава с помощью электро-
лиза удаляется влага, уменьшаются количества твердых продуктов в кон-
Соединения с азотом 221
денсате и все больше №3 конденсируется в виде жидкости. Выход по току,
однако, не удалось получить более чем 42% (от теоретического).
Заметим, что состав продуктов электролиза сильно зависит от материа-
ла анода. Если при использовании определенных сортов никеля и угля для
изготовления анода были получены плохие результаты, то надо выбрать
другие марки.
Сконденсированную в ловушках (частично в виде жидкости, частично в
виде твердых веществ) смесь промывают прежде всего гидроксидом калия
для удаления компонентов кислотного характера. Затем продукт фракцио-
нируют для того, чтобы удалить основные количества N2O. При этом NF3.
появляется уже как бесцветная жидкость, покрытая белым слоем твердого
N2O. Для окончательного их разделения NF3 многократно, очень осторожно1 2 3 4
и тщательно фракционируют. Удобнее последние следы N2O отделить путем1
низкотемпературного фильтрования при —183 °C. Уже после одного фильтро-
вания получают совершенно чистый NF3. В заключение из жидкого iNF3
удаляют растворенный воздух, для чего продукт в газовой ловушке, охлаж-
даемой при —'183 °C, откачивают в течение нескольких часов с помощыа
масляного насоса. Чистоту NF3 лучше всего контролировать по молекуляр-
ной массе, рассчитанной из измерений плотности для отобранной пробы.
Способ 2 [7]
Таким же образом, как описано в первом способе, проводят электролиз1
гидрофторида гуаниднния. Преимущества метода состоят в том, что полу-
чают высокий выход по току и можно работать при более низких темпера-
турах. Недостаток метода связан с тем, что при этом одновременно обра-
зуется CF4, который очень трудно отделить от NF3.
Прежде всего получают исходный гидрофторид гуаниднния действием1
на карбонат гуаниднния 40 %-ной HF. Упаривают раствор в вакууме и осаж-
дают ацетоном чистый кристаллический продукт (<Пл 198°C).
Электролиз 40%-ного раствора гуанидина в безводном фтороводороде-
проводят при температуре 15 °C и напряжении 15 В. На никелевом аноде
выделяется смесь газов: NF3, CF4 и N2. Выход NF3 47%, суммарный выход
всех фторированных продуктов 66%.
Хранят NF3 в стеклянных колбах или в газометрах под водой; можно<
хранить под давлением в стальных баллонах. Препарат применяют как жид-
кий наполнитель для термометров, действие которых основано иа изменении
давления [7].
Свойства. Бесцветное вещество со своеобразным гнилостно-тленным за-
пахом. При комнатной температуре термически довольно устойчиво и ие ре-
агирует с водой и гидроксидом калия, но может реагировать прн прохожде-
нии искры. Со стеклом и Hg также не реагирует. Работы с чистым NF3, еслт
только не вдыхать его в больших количествах, безопасны. Неочищенный NF3,
однако, из-за присутствия примесей гораздо более неприятное вещество; вы-
зывает головную боль, рвоту и расстройство желудка. /Пл —208,5 °C; /кип.
—129 °C; ДНобр 109 кДж; dx 1,885 (—129 °C).
ЛИТЕРАТУРА
1. Ruff О., Luft F., Fischer J., Z.
Anorg. Allgcm. Chem., 172, 417
(1928).
2. Ruff O., Z. Anorg. Allgem. Chem.,
197, 273 (1931).
3. Ruff O., Staub L„ Z. Anorg.
Allgem. Chem., 198, 32 (19?1).
4. Kwasnik W., Naturforschung u. Me-
dizin in Deutschland 1939—1946
(FIAT-Review), 23, 204.
5. Glemser 0., Schroeder J., Knaak A,
Chem. Ber., 99, 371 (1966).
6. Engelbrecht A.. Nachbaur E„ Mo-
natsh. Chem., 90, 371 (1959).
7. Menzel W.. Mohry F„ Z. Anorg,
Allgem. Chem., 210, 257 (1933).
'222 Глава 3. Фтор
.Дифторамин NHF2
(Иминфторид)
N2F4 + 2CeH5SH --2NHF2 + C6H5SSCeH5
104 220 106 218
В литровую круглодонную колбу, снабженную магнитной мешалкой и
присоединенную к вакуумной установке, наливают 12 мл тиофенола. После
обезгаживаиия тиофенола колбу охлаждают жидким азотом и конденсируют
«в нее 483 мл газообразного N2F4. Затем смесь приводят к комнатной темпе-
ратуре и помещают реакционную колбу в масляную- баню. При перемешива-
нии смесь выдерживают 4 ч при 50 °C. Убирают масляную баню и охлаж-
дают реакционную смесь.
Содержимое реакционной колбы отгоняют в вакууме, конденсируя фрак-
ции при —80, —128 и —196 °C. Продукт, собранный во второй ловушке, за-
тем вновь перегоняют и конденсируют в приемник при —196 °C. Этот кон-
денсат содержит 99% NHF2. Получают 587 мл газа, что соответствует вы-
воду 75%. В ловушке, охлаждаемой при —196°C, собирается не вступив-
ший в реакцию N2F4.
Дифторамип в виде газа можно хранить в сосудах из стекла пирекс при
.комнатной температуре. В не очень чистых металлических сосудах он посте-
пенно разлагается.
Свойства. М 53. /пл —117°C; <кип —23,6°C; d 1,587 (—80,5°C).
Внимание! Дифторамин иногда имеет склонность к чрезвычайно сильным
-взрывам. Это может касаться также ловушек в вакуумной системе, если оии
отогреваются или в них попал воздух.
-ЛИТЕРАТУРА
<1. Freeman J. Р., Kennedy A., Col-
burn С. В., J. Amer. Chem. Soc.,
82, 5304 (1960).
'Тетрафторогидразин N2F4
<(Тетрафторид диазота)
Способ 1 [1]
2NF3-|-2Hg ---> N2F4 + Hg2F2
142 401 104 439
В вертикальной закрытой с одной стороны трубке из никеля (длина
-50 см, диаметр 2,5 см), которую можно нагревать и охлаждать, через слой
фтути пропускают при 320—330 °C поток NFs 1(8 г/ч). Необходимы очень
чистые исходные соединения. Газообразные продукты реакции улавливают с
помощью ловушек при температуре жидкого воздуха. Из 50 г NF3 около
14 г остается иепревращениым. Остальное количество NF3 вступает в ре-
акцию с 616 г Hg; получается 16 г N2F4 (соответствует выходу 60%) и не-
умного qnc-N2F2. Продукт реакции фракционируют на низкотемпературной
колонке.
Другие способы получения. Реакция NF3 с металлической медью. Через
.нагретую до 375 °C медную трубку, наполненную медными стружками, про-
шускают NF3. Если время пребывания газа в трубке составляет 13 мин, пре-
Соединения с азотом 22?
вращение идет на 42—62%. Полученный N2F4 очищают путем фракциониро-
вания при —131 °C [2].
Свойства, /кип —74,1 °C; Арит 36°С.
ЛИТЕРАТУРА
1. Dresdener R. D„ Flumac F. N.,
Young J. A., J. Inorg. Nucl. Chem.,
14, 299 (1960).
2. Colburn С. B., Kennedy A., J. Amer.
Chem. Soc., 80, 5004 (1958).
Дифторид диазота N2F2
(Дифтордиазин, дифторимии)
2NaN34-2F2 ---> N2F2 + 2N2 + 2NaF
130 76 66 56 84
Получение больших количеств N2F2 (3 г rpaHC-N2F2 в 1 ч) осуществля-
ют при комнатной температуре во вращающемся трубчатом реакторе из ни-
келя (рис. 137) путем непрерывного фторирования технического азида нат-
рия.
Установка состоит из наклонного вращающегося трубчатого реактора иа
никеля, который закрыт с обоих концов крышками, служащими одновремен-
Рис. 137. Установка для получения N2F2.
но подшипниками для вращающейся трубки. Трубка помещена в кожух»,
который можно нагревать и охлаждать. Подобно вращающейся трубчатой
печи, реактор приводится в движение электромотором через зубчатую пере-
дачу. В реактор вмонтирована спираль, с помощью которой можно переме-
шивать и продвигать твердые продукты. Газы перемещаются по трубке сни-
зу вверх. Верхний конец трубки соединен со змеевиком из никеля, погружен-
ным в масляную баню, в котором разлагается промежуточно образующийся.
FN3. Далее следуют две газовые ловушки.
Во вращающуюся трубку загружают влажный NaNs, нагревают никеле-
вый змеевик до 60 °C, охлаждают газовые ловушки жидким воздухом, уста-
навливают необходимую скорость вращения (2 об./мин) и пропускают F2 со
скоростью 90 пузырьков в 1 мин (~12 л/ч). Азид натрия должен быть взят
в большом избытке. Добавление к фтору небольшого количества HF повы-
шает выход.
В ловушках собираются N2F2 и N2O. Сырой продукт перегоняют в высо-
ком вакууме. Максимальный выход 40%.
224 Глава 3. Фтор
Полученный таким образом продукт представляет собой транс-NzFa.
Если никелевый змеевик нагреть до 90 °C, то наряду с транс-изомером об-
фазуется час-аналог. Изомеры можно разделить фракционной перегонкой.
Свойства. транс-Изомер: /пл —187 °C; 4ип —111,4 °C.
Час-Изомер: tnn <—195 °C; ^кип —105,7 °C.
Бесцветный газ, устойчивый при комнатной температуре, по запаху по-
хож на NO2. Не обнаруживает склонности к взрывам. Реагирует с F2 с об-
разованием NFs. Разлагается при 300 °C на К2 и F2. Совершенно не подвер-
гается кислому и щелочному гидролизу, однако разлагается подкисленным
фаствором KI.
ЛИТЕРАТУРА
1. Roesky Н. R„ Glemser О., Bor-
mann D., Angew. Chem., 76, 713
«Фторид нитрозила NOF
.(Оксофторид азота)
Способ 1 [1]
NOBF44-NaF -----> NOF-|-NaBF4
116,83 42 49,01 109,82
(1964); Chem. Вег., 99, 1589
(1966).
Реакцию проводят в закрытой с одной стороны никелевой трубке
Чрис. 138). На выступающем нз печи конце трубки имеется свинцовый змее-
вик, через который протекает холодная вода. Никелевая трубка соединена
<с двумя кварцевыми ловушками, охлаждаемыми жидким воздухом. Кроме
НСарцевый
манометр,
'ртутный ’
насос,Впуск
воздуха
-183° С -183 °С.
Рис. 138. Установка для получения нитрозилфторида.
того, в установку должны входить кварцевый спиральный манометр и труб-
ка с Р4О10 на выходе в атмосферу. Установку присоединяют к ртутному
диффузионному насосу (для смазки кранов используют смесь вазелина и
графита). Перед насосом включают охлаждаемую жидким воздухом газо-
вую ловушку для того, чтобы предотвратить засасывание в иасос кислых
паров.
Никелевую трубку наполняют по возможности наиболее чистым
NOBF< и избытком сухого NaF. Все операции надо производить в атмосфе-
ре сухого азота. Затем присоединяют кварцевые ловушки. Шлифы нельзя
•смазывать обычной смазкой, содержащей жиры, для этой цели можно нс-
Соединения с азотом 225
пользовать только пицеин. Наконец, включают ртутный насос и откачивают
до давления 0,01 мм рт. ст. Печь постепенно нагревают до 300 °C. Реакция
начинается уже при ~ 100 °C, при 200 °C превращение протекает с заметной
скоростью, а при 300 °C — очень бурно. 3 первой и частично во второй ло-
вушках конденсируется голубой NOF. Из 30 г NOBF4 получают 10 мл сы-
рого NOF. По окончании. реакции NOF фракционируют в кварцевый сосуд.
Редко удается получить совершенно неокрашенное вещество. Чаще всего оно
имеет слегка голубоватый оттенок.
Преимущество методики состоит в том, что в синтезе ие используется
свободный фтор.
Способ 2 [2]
2NO-|-F2 ---*• 2NOF
Смесь газов NO и Fz реагирует без дополнительного нагревания, экзо-
термически. Реакцию проводят в никелевой трубке. Образующийся NOF
улавливают в охлаждаемой ловушке и для очистки подвергают фракционной
перегонке. Методика синтеза описана во 2-м издании данной книги (1960 г.);
о подготовке и сборке прибора см. там же.
Хранить NOF лучше всего в кварцевых ампулах при охлаждении жид-
ким воздухом.
Свойства. Чистый NOF — бесцветное соединение, в жидком состоянии
часто имеет голубоватый оттеиок, обусловленный присутствием загрязнений.
При растворении в воде сначала появляется голубое окрашивание, ио затем
NOF очень быстро превращается в NO и HNO3. Бурио реагирует со стеклом,
ноне взаимодействует с кварцем. /пл —132,5°C; /кип —59,9°C; dx 1,326
(—59°C); </,„ 1,719.
ЛИТЕРАТУРА
1. Balz G., Mailander Е., Z. Anorg. 2. Ruff О., Menzel W., Neumann
Allgem. Chem., 217, 166 (1934). Z. Anorg. Allgem. Chem., 208, 293
(1932).
Аддукт фторида нитрозила с фтороводородом NOF*3HF
NOC14-4HF -----> NOF-3HF+ НС1
65,5 80,0 109,0 36,5
Для проведения реакции необходима бутыль из тефлона или металли-
ческий сосуд, соответствующим образом облицованный внутри тефлоном (ре-
акционный сосуд должен иметь винтовой запор), а также две тефлоновые
трубки для ввода и отвода газов. В широкую в виде буквы «Т» газоотвод-
ную трубку надо плотно вставить (так чтобы не нарушить газонепроницае-
мости) короткую закрытую снизу трубку из никеля или специальной стали,
служащую «карманом» для термометра. Перед началом опыта присоединяют
в качестве холодильника никелевую трубку (длина ~30 см, диаметр
'v 1 см) и осушительную трубку из полиэтилена. Бутыль охлаждают снаружи
кусочками сухого льда.
В литровую бутыль конденсируют прежде всего 500 г (25 моль, или
500 мл) HF и 330 г (5 моль) 'NOC1. При этом следят за тем, чтобы HF по
возможности был безводным, a NOC1 содержал как можно меньше NzO3.
Приготовленную смесь медленно нагревают. При —40 °C начинается выделе-
ние газообразного НС1, который, улетучиваясь, захватывает немного NOC1
и HF. При комнатной температуре жидкость, остающаяся в тефлоновой бу-
тыли, уже почти не содержит НС1. Теперь бутыль можно опустить в масля-
15—143
226 Глава Л. Ф’юр
ную баню и перегонять ее содержимое в приемник — вторую бутыль из
тефлона. Прежде всего отгоняется легкокипящая обогащенная HF фракция.
Азеотроп, отвечающий составу NOF-3HF, перегоняется при 735 мм рт. ст. н
94 °C (выход ~75%). Если исходные вещества содержали большие коли-
чества воды или N2O3, то дополнительно получают кипящий при 68 °C и
735 мм рт. ст. азеотроп состава NOF-5,5 HF-0,5 Н2О или N2O3-13 HF.
Свойства. Бесцветная, тяжелая, похожая на коиц. H2SO4 жидкость.
Сильно разъедает кожу. Плавится в интервале 0,6—5,6 °C. /кип 94 °C;
1,5.
NOF-3 HF представляет собой раствор HF в плавящемся при 5.6 °C
соединении NOF-2.94HF, которое при охлаждении выделяется в виде длин-
ных толстых призм.
Аддукт — хороший фторирующий агент, например при хлор-фторзаме-
гцепиях, в реакциях с оксидами и -свободными элементами. Используется
при получении HgF2.
ЛИТЕРАТУРА
1. Seel F., Angew. Chem., 77, 689 (1965).
Нитрозосульфонилфторид FSO2NO
nof + so2 —*- fso2no
49,01 64 113,01
Вслед за NOF конденсируют очень большое количество SO2, так чтобы
после оттаивания получилась суспензия FSO2NO в жидком SO2. Путем фрак-
ционного выпаривания и конденсации получают чистый нитрозосульфонил-
фторид.
В препаративном отношении использование FSO2NO имеет преимущест-
во, поскольку реагент действует как «стабилизированный NOF». FSO2NO лег-
ко получить чистым и можно хранить в стеклянных сосудах в низкотемпера-
турном холодильнике. Для проведения реакций с FSO2NO подходит посуда
из хостафлона (политрифторхлорэтилеиа).
Свойства. Бесцветные, блестящие кристаллы в форме листочков, легко
сублимируются. /пл 8°C (ниже 31,00 мм рт. ст.). При 19°C вещество более
чем иа 70% разлагается на компоненты.
ЛИТЕРАТУРА
1. Seel F., Massat Н., Z. Anorg.
Allgem. Chem., 280, 186 (1955).
Фторид нитроила NO2F
Способ 1 [1]
N2O5 + BF3 + HF ----* NO2[BF4) + HNO3
108,0 67,82 20 132,82 63
NO2[BF4] + NaF ---> NO2F4-NaBF4
132,82 42 65,0 109,82
Синтез осуществляют без применения фтора. В раствор N2O5 в нитроме-
тане вводят эквивалентные количества безводного HF и BF3. Из раствора в
Соединения с азотом 227
нитрометане выпадает фтороборат нптроила. Кристаллы отфильтровывают,
не допуская попадания влаги, и затем нагревают с NaF в платиновом или ни-
келевом сосуде при 240 °C (аналогично получению NOF по первому спо-
собу) .
Поскольку N2O5 легко отщепляется, удобно сначала получить из N2Os
и BFS устойчивое без доступа влаги и при комнатной температуре соедине-
ние N2O5-BF3, которое затем растворяют в нитрометане и вводят в реакцию
с безводным фтороводородом.
Способ 2 [2]
2NO2 + F2 ----> 2NO2F
Реакция между NO2 и F2 происходит с небольшим выделением тепла,
поэтому не требуется дополнительного нагревания. Реакцию осуществляют в
никелевой трубке. NO2F улавливают в охлаждаемой ловушке (—120 °C) и
очищают путем фракционной перегонки. Описание методики и прибора см.
во 2-м издании данной книги (1960 г.).
Хранить NO2F лучше всего в кварцевых ампулах при температуре жид-
кого воздуха.
Свойства. Газообразный и жидкий NO2F бесцветен, в твердом состоя-
нии — белое вещество. Гидролизуется; полностью абсорбируется ртутью.
Реагирует с большинством металлов и неметаллов, кроме того, бурно взаи-
модействует со спиртом, эфиром, бензолом и хлороформом. NO2F имеет
резкий запах и сильно поражает слизистые оболочки. /пл —166,0 °C; /кип
—72,4°C; dK 1,796, f—72°C); dIB 1,924.
ЛИТЕРАТУРА
1. Schmeisser М„ Elischer S., Z. Na- 2. Ruff O., Menzel W., Neumann W..
turforsch., В 7, 583 (1952). Z. Anorg. Allgem. Chem., 208, 298
(1932).
Триоксид-фторид азота NO3F
( H итроксил фторид)
HNO34-F2 -----> NO3F + HF
63,01 38 81,01 20
Фтор, сохраняемый в кварцевой ловушке при охлаждении жидким азо-
том и жидким воздухом, всасывают через медный мембранный вентиль в
прибор (рис. 139). Можно использовать F2 из стального баллона. В кварце-
вый реактор помещают 100%-ную HNO3, полученную путем перегонки сме-
си дымящей азотной кислоты и конц. H2SO4 при давлении 20 мм рт. ст. и
комнатной температуре. (При хорошем охлаждении она не должна разла-
гаться.) К реактору присоединяют кварцевую осушительную трубку со све-
жеобезвоженным KF для того, чтобы удалить HF. Далее следуют три квар-
цевые ловушки, охлаждаемые жидким О2 или каким-нибудь другим спосо-
бом до —183 °C. Две первые служат приемниками, последняя только предо-
храняет от попадания влаги из атмосферы. Затем следуют манометр для
контроля вакуума и стеклянный кран. Для получения вакуума служит водо-
струйный насос, изготовленный из металла.
Скорость подачи F2 регулируют с помощью мембранного вентиля так,
чтобы через реактор при 20 мм рт. ст. проходило 2—3 пузырька в секунду.
При этой скорости потока незаметно разогревание реактора. Для того чтобы
не забивались ловушки, надо каждые 15 мин на непродолжительное время
15*
228 Глава 3. Фтор
убирать с иих охлаждение, при этом сконденсированный в них твердый
NOSF плавится и собирается иа дие ловушки.
При проведении синтеза надо обращать внимание на то, чтобы все ча-
сти и соединения прибора были обезжирены, так как иначе возможен взрыв.
Полученный NO3F очищают путем фракционной перегонки при 100 мм
рт. ст. иа кварцевой колонке. Фракция, отгоняющаяся при —79 °C (99 мм
рт. ст.), и есть чистый NO3F. Первые погоны состоят из S1F4, последние —
главным образом 113 H2SiF6 и HF.
Мембран-
ный Вентиль
из меди
Жидкий. 0а
Рис. 139. Установка для получения NO3F.
NO3F можно хранить расплавленным в кварцевых или стеклянных ам-
пулах в вакууме (<0,1 мм рт. ст.), однако при этом необходима осторож-
ность, так как иногда происходят взрывы. Наиболее надежны в хранении
ампулы, охлаждаемые жидким воздухом.
Свойства. Бесцветный газ с удушливым, отвратительным запахом вызы-
вает режущую боль в носу, головную боль и перебои в дыхании, которые
сохраняются в течение нескольких дней. При сильных сотрясениях жидкий
NO3F может взрываться. В воде гидролизуется до OF2, Ог, HF и HNO3.
Хорошо растворим в ацетоне. Со спиртом, эфиром и анилином тотчас про-
исходит взрыв. /пл —175°С; /кип —45,0°С; d« 1,507 (45,0°С); dTB 1,951
(—193,2 °C).
ЛИТЕРАТУРА
1. Ruff О., Kwasnik W., Angew. Chem.,
48, 238 (1935).
Тиофторид азота NSF
(Тиазил фторид)
NFs4-3S ---> NSF4-S2F2
71 96 65 102
Эту методику можно использовать для препаративного получения двух
продуктов — NSF и SSFz.
Соединения с азотом 229
В нагреваемой снаружи никелевой ловушке (длина 40 см, диаметр
3 см; см. рис. 140) через расплавленную серу (высота слоя 4 см) при 400 °C
пропускают NFs (время пребывания в реакции 12 мин).
Опущенная в ловушку трубка, через которую подается газ, внизу рас-
ширена в виде воронки и имеет отверстия для лучшего распределения пото-
ка газа. Крышка с тефлоновым кольцом плотно завинчена. Из прибора газы
попадают в ловушку из тефлона (—78°C), а затем проходят через две ох-
лаждаемые жидким воздухом кварцевые ловушки. В тефлоновой ловушке
коиденснруется желтоватая легко подвижная жидкость. Первая кварцевая
ловушка служит для сбора непрореагировавшего NF31 а вторая — для предо-
хранения от попадания влаги.
NF3
Рис. 140. Установка для получения NSF.
Продукты реакции, попавшие в тефлоновую ловушку, разделяют только
путем фракционной перегонки в высоком вакууме. Сначала отгоняются
SiF4, SFe и SOF2, а при —80 °C — чистый SSF2. Высококипящая фракция
представляет собой тиофторид азота. Получают 60% SSF2 и 30% NSF.
Свойства. /пл —89 °C; /кип 0,4 °C. Газ с резким запахом, термически ие
устойчив, очень реакционноспособен, разлагается водой.
ЛИТЕРАТУРА
1. Glemser О., Biermann U., Knaak J.,
Haas A., Chem. Ber., 98, 446
(1965).
Трифторид-нитрид серы SNF3
(Дифторсульфиминфторид)
S4N4+12AgF2 ----> 4SNF3+12AgF
184 1751 412 1523
Газовую смесь SNF+SN2F2, образующуюся при взаимодействии S4N4 с
AgF2 в СС14, пропускают при 20 °C над AgF2.
В 250 мл СС14 вносят 2,5 г S4N4 и при перемешивании 1,5 г свежепри-
готовленного AgF2 (рис. 141). Медленно нагревают до температуры кипения
И кипятят смесь с обратным холодильником в течение 2 ч. Образующаяся
бесцветная газовая смесь (SNF+SN2F2) конденсируется в газовой ловушке
при —78 °C.
По окончании реакции смесь, находящуюся в газовой ловушке, медлен-
но испаряют и газы пропускают при нормальном давлении через кварцевую
230 Глава 3. Фтор
трубку (длина 30 см), которая по всей длине загружена свежеприготовлен-
ным дифторидом серебра (слой 3—5 мм в высоту). Снаружи трубки с по-
мощью охладительной рубашки поддерживают температуру около 20 °C.
К реакционной трубке присоединяют две ловушки, охлаждаемые смесью су-
хого льда с ацетоном; в первой из них конденсируется SNF3. Продукт очи-
щают путем фракционной перегонки. Выход 90%.
Рис. 141. Установка для по-
лучения SNF3.
Свойства. М 103. Бесцветное, термически устойчивое соединение.
<пл —72,6 °C; /КИп —27,1 °C; dt~№ 1,92. Медленно гидролизуется водой и бы-
стро— гидроксидом натрия до NH4+, F~ и SO32-.
ЛИТЕРАТУРА
1. Glemser О., Schroder Н., Z. Anorg. 2. Glemser О., Schroder Н.. Haase-
Allgem. Chem., 284, 97 (1956). ler H., Z. Anorg. Allgem. Chem.,
279, 28 (1955).
Фторид фосфора(Ш) PF3
Способ 1 [1]
PC13 + 3HF ----> PF3-|-3HC1
137,35 60 87,98 109,38
Реактором служит закрытая с одного конца кварцевая или железная
трубка (длина ~70 см, диаметр 4 см; см. рис. 142). В нее вводят через ре-
зиновую пробку железный капилляр, доходящий почти до дна сосуда. На ре-
актор насаживают обратный холодильник из кварца или железа. За ним
следуют конденсаторные ловушки (из кварца или стекла), охлаждаемые
жидким воздухом.
Реакционный сосуд наполняют РС13 и вводят в него газообразный HF.
При 50—60 °C процесс происходит гладко. По окончании синтеза собранную
в ловушках смесь PF3 и НС1 быстро пропускают через промывалки с ледя-
ной водой для поглощения НС1, высушивают над Р4О10 н перегоняют. Вы-
ход >90% в расчете па РС13.
Для проведения реакции в более крупных масштабах используют уста-
новку, схематически изображенную на рис. 136.
Способ 2 [2]
2РС13 + 3ZnF2 ---> 2PF34-3ZnCl2
274,7 310,1 176,0 408,8
Соединения с фосфором и мышьяком 231
Порошок фторида цинка сушат в течение ночи при 140—150 °C. 50—60 г
еще теплого фторида помещают в перегонную колбу на 300 мл, снабженную
капельной воронкой и двумя присоединенными последовательно ловушками
для конденсации (сухой лед). На выходе из прибора устанавливают пузырь-
ковый газовый счетчик (конц. H2SO4) и ловушку, охлаждаемую жидким
воздухом. Из капельной воронки к ZnF2 по каплям прибавляют 45 мл РС13,
вначале очень осторожно (в течение 2 мин со скоростью ~1 капля/с). После
индукционного периода начинается реакция, сопровождаемая сильным выде-
лением тепла. Если реакция протекает очень бурно, реакционный сосуд ох-
Рис. 142. Установка для получения фторида фосфора (III).
лаждают водой. К концу реакции, напротив, необходимо нагревание с по-
мощью теплой водяной бани. Реакция заканчивается через 1,5—2 ч. Продукт,
собранный в охлаждаемых при —194 °C ловушках, пропускают над Р4Ою и
перегоняют, разделяя на фракции.
Хранят PF3 в стальных баллонах или стеклянных колбах. Для этой цели
также можно использовать газометры с Hg.
Свойства. Бесцветный газ, ие дымящий на воздухе, почти без запаха,
ядовит (вызывает удушье, головную боль и рвоту). /пл —151,5°C; 1Яп
—101,8 °C; /Крит —2 °C; ркрит 44 бар. Медленно гидролизуется водой; не дей-
ствует на стекло.
ЛИТЕРАТУРА
1. Kwasnik W., Naturlorschung u. Me- 2. Chatt J., Williams A., J. Chem. Soc.,
dizin in Deutschland 1939—1946 1951, 3061.
(FIAT-Review), 23, 213.
232 Глава 3. Фтор
Фторид фосфора( V) PFs
3PC15+5AsF3 ----► 3PFe4-5AsClg
624,8 659,6 377,9 906,8
К PCls в стеклянной колбе добавляют по каплям из капельной воронки
AsFa (рис. 143). К газоотводной трубке с помощью шлифов присоединяют
газовую ловушку, охлаждаемую жидким воздухом; оиа служит приемником
PF5. Далее следует осушительная трубка с обезвоженным KF для предохра-
нения от попадания влаги.
Реакция начинается без подвода тепла. В газовой ловушке собирается
в виде твердого белого вещества PF's, загрязненный- AsFa. По окончании ре-
акции сырой PFs перегоняют, разделяя иа фракции.
Рис. 143. Прибор для получения фторида
фосфора (V).
PFs можно хранить под давлением в стальных баллонах или в стеклян-
ных колбах.
Свойства. М 125,98. Бесцветный газ, сильно дымит на воздухе, разъедает
кожу и поражает легкие.
Лтя —83 °C; /Кип —75 °C. PFs быстро гидролизуется водой. При комнат-
ной температуре не действует на сухое стекло.
ЛИТЕРАТУРА
1. Ruff О., Die Chemie des Fluors.
Berlin, 1920, S. 29.
Дихлорид-фторид фосфора( 111) PC12F
(Дихлормонофторфосфин)
PC13 + SbF3 ---> PC12F + SbClF2
137,35 178,76 120,90 195,21
Реакцию проводят в двухлитровой трехгорлой круглодонной колбе
(рис. 144). Через среднее горло вводят мешалку с вакуумным затвором, вто-
рое горло служит для внесения SbF3 (это осуществляют с помощью пово-
рачивающегося шнека пли просто с помощью прикрепленной на резиновом
шланге круглодонной колбочки). В третье горло вставляют метровую стек-
Соединения с фосфором и мышьяком 233
лянную колонку, верхняя часть которой служит дефлегматором. Далее реак-
ционные газы поступают в ловушку, опущенную в жидкий воздух. На выхо-
де присоединена осушительная трубка со свежеобезвожейным KF и стеклян-
ный кран, с помощью которого отсоединяют прибор от манометра и водо-
струйного насоса.
В реакционную колбу помещают 130 г РС13 и 2 г РС15 (катализатор).
Затем прибор эвакуируют до давления 250 мм рт. ст.; это давление сохра-
няют в течение всего времени синтеза. Дефлегматор охлаждают, подключив
Рис. 144. Установка для получения дихлорид-фторида фосфора(Ш).
к водопроводной сети/Теперь в реакционный сосуд постепеиио в течение 3 ч
добавляют 175 г высушенного порошка SbF3. Надо позаботиться о том, что-'
бы можно было охлаждать и нагревать реакционный сосуд снаружи (тем-
пература должна сохраняться приблизительно постоянной, ~49°С).
Сырой PClsF собирается в газовой ловушке. По окончании синтеза про-
дукт перегоняют, разделяя на фракции..Выход 60%.
Лучше всего хранить PC12F в запаянных стеклянных ампулах при
—78 °C, в крайнем случае можно непродолжительное время хранить его в
стальных баллонах при комнатной температуре.
Свойства. Бесцветный газ, при комнатной температуре термически
не устойчив. На воздухе PC12F не дымит, гидролизуется водой, без остатка
с выделением тепла поглощается гидроксидом натрия. /пл —144 °C; /кип
13,85 °C; d 1,507.
ЛИТЕРАТУРА
1. Booth Н. S., Bozart А. Р„ J. Amer.
Chem. Soc., 61, 2927 (1939).
234 Глава 3. Фтор
Дихлорид-трифторид фосфора(У) PC12F3
pf3 + ci2 —> рад
88,02 70,92 158,94
Одинаковые объемы PF3 и С12 (поток каждого газа проходит через
соответствующую аппаратуру для' контроля количества газа) подают в квар-
цевую трубку (длина ~1 м), где и происходит реакция — экзотермическое
присоединение С12 к PF3. К кварцевой трубке с помощью шлифа присоединя-
ют кварцевую ловушку, которую погружают в жидкий воздух. Для предо-
хранения от попадания влаги далее присоединяют осушительную трубку
с KF. .
Продукт реакции, собранный в ловушке, затем перегоняют, разделяя иа
фракции.
PF3CI2 можно хранить в стеклянных колбах.
Свойства. Бесцветный газ с очень резким запахом. /пл —125-9—‘130 °C:
/кип 7,1° С. Поражает органы дыхания. На воздухе образует плотный белый
туман. Разлагается при нагревании до 200 °C по реакции диспропорциониро-
вания. Вода (в избытке) поглощает PCI2F3 без остатка с образованием
Н3РО4, HF и НС1. С небольшим количеством воды идет с увеличением объ-
ема образование POF3 и НС1. При поглощении спиртом происходит алкого-
лиз. PC^Fs-, медленно превращается в солеподобное соединение PC14-PF5.
ЛИТЕРАТУРА
1. Poulenc С., С. R. Acad. Sci., Paris, 2. Schomaker V., Hatscher J. B., J.
113, 75 (1891). Amer. Chem. Soc., 60, 1837 (1938).
Тетрахлорид-фторид фосфора PCI4F
PC12F4-C12 ---> PCI4F
120,9 71 191,9
В стеклянную ампулу c PCI2F при —196 °C медленно пропускают чистый
С12, в то ,}ке время температура поднимается приблизительно до —78 °C;
Пропусканйе С12 продолжают до тех пор, пока реакционная смесь в ампуле
ие начнет сохранять желтую окраску, что свидетельствует об избытке С12.
Для того чтобы превращение прошло достаточно полно, реакционную смесь
оставляют еще на полчаса при —78 °C. Затем избыточный С12 отгоняют в
вакууме. При температуре бани —35 °C в остатке находится РОД7.
Свойства. /Пл —59 °C; /кип 67 °C. При комнатной температуре медленно
переходит в солеподобиое соединение фосфония. Очень быстро реагирует
при 50 °C с PFg с образованием PCU-PFe-
ЛИТЕРАТУРА
1. Holmes R. R., Gallagher W. Р„
Inorg. Chem., 2, 433 (1963).
Хлорид-тетрафторид фосфора PCIF4
PCl2F34-SbF3 ----> PClF4 + SbClF2
158,9 178,7 142,4 195,3
Синтез проводят в приборе, подобном описанному в методике получения
PC12F. В колбу на 100 мл помещают PCI2F3 и оставляют при —95 °C. Три
Соединения с фосфором и мышьяком 235
ловушки'для конденсации охлаждают до —186 °C. Надо установить в при-
боре давление 25 мм рт. ст.
Реакция начинается при введении небольшого количества SbF3. Затем
SbF3 добавляют с такой скоростью, чтобы поддерживать температуру реак-
ционной смеси в интервале —40ч—50 °C. Хотя добавление SbF3 продол-
жается только ~ 1 ч, реакция заканчивается лишь через ~6 ч. Продукт ре-
акции перегоняют, разделяя на фракции. Выход чистого продукта 20%.
Свойства. 1ПЛ —'132 °C; Аип —43,4 °C. При комнатной температуре про-
дукт постепенно переходит в полярную форму, поэтому хранить его целе-
сообразнее при охлаждении.
ЛИТЕРАТУРА
1. Carter R. Р; Holmes R. R.. Inorg.
Chem., 4, 738 (1965).
Трифторид фосфорила POF3
(Трифторпд-оксид фосфора)
POCl3-f-3HF ----*- POF34-3HC1
153,35 60 104,0 109,38
Реакцию проводят в приборе, описанном в методике получения PF3. Га-
зообразный HF вводят при +65 °C в РОС13. Добавляют 5% SbCls (как ка-
тализатор). Продукты реакции (POF3+3 НО), собранные в ловушке, раз-
деляют путем фракционной перегонки. Выход >90% в расчете на РОС13.
Свойства. Бесцветный газ с резким запахом, слабо дымит на воздухе.
1ПЛ —39,4 °C; /субл —39,8 °C; /крит 7i3,3°C; ркрит 43 бар.
Хранят POF3 в стеклянных колбах или в стальных баллонах.
ЛИТЕРАТУРА
1. Kwasnik W„ Naturforschung u. Me- 1
dizin in Deutschland 1939—1946
(FIAT-Review), 23, 213.
Гексафторофосфат тетрахлорофосфония PCI4-PF6
P2C11O 4- 2AsF3 -+ PCl4-PFe + 2AsCl3
416,53 263,82 317,89 362,46
46 г PC15 растворяют в 300 мл AsCl3 и при перемешивании и легком
охлаждении смешивают с 29,8 г AsF3, который добавляют по каплям. Выпа-
дает белый мелкокристаллический осадок PCL.PF;;. Конец реакции можно
определить по выделению PF5 (появление белого густого тумана). Осадок
отфильтровывают на пористом фильтре G4 с крышкой и приспособлением
для защиты от влаги воздуха; продукт промывают AsCl3 и удаляют остав-
шийся иа кристаллах AsCl3, продувая сухой воздух. Выход составляют 35 г,
т. е. равен теоретическому.
Свойства. Белая кристаллическая соль. Сублимируется прн 135 °C с час-
тичным разложением. /пл 161 °C (при уменьшенном давлении). Плохо раство-
рима в AsCl3.
236 Глава 3. Фтор
PCU-PFe — удобный исходный продукт для получении гексафторофосфа-
тов (гидролиз соответствующими гидроксидами; см. синтез KPFe) и PF»
(термическое разложение при 80°C).
ЛИТЕРАТУРА
1. Kolditz L., Z. Anorg. Allgem. Chem.,
284, 144 (1956).
Дибромид-фторид фосфора PBr2F
PBr3-|-SbF3 ----> PBr2F + SbBrF2
270,7 178,8 209,8 239,7
В трехгорлой колбе, снабженной поворачивающимся шнеком для дози-
рованного введения SbF3, мешалкой и вертикальным воздушным холодиль-
ником, нагревают до 85 °C 50 мл РВг3, который смешан с небольшим коли-
чеством РВг5 (катализатор фторирования), и порциями прн постоянном пере-
мешивании добавляют 914 г SbF3. Для того чтобы не допустить протекания
вторичных реакций, надо как можно быстрее выводить из зоны реакции об-
разующийся PBr2F. Для этой цели во время синтеза в приборе поддержива-
ют давление 200 мм рт. ст. Воздушный холодильник возвращает в реакци-
онную смесь РВг3.
В двух ловушках, присоединенных к обратному холодильнику и охлаж-
даемых жидким воздухом, конденсируется PBr2F. Затем продукт перегоня-
ют при комнатной температуре и 60 мм рт. ст., разделяя на фракции. В за-
ключение очищенный таким образом продукт еще раз подвергают фракци-
онной перегонке при нормальном давлении. Выход составляет 21,8 г, т. е.
11 % теоретического. Используют для получения PBr4F.
Свойства. ^КИП 78,5 °C.
ЛИТЕРАТУРА
1. Kolditz L., Bauer К., Z. Anorg.
Allgem. Chem., 302, 241 (1959).
Тетрабромид-фторид фосфора PBr4F
(Фторид тетрабромофосфора)
PBr2F + Br2----»-PBr4F
209,82 159,84 369,66
12,1 г свежеперегнанного PBr2F в колбе грушевидной формы охлаждают
до температуры сухого льда (рис. 145). Затем через жидкость пропускают в
потоке азота пары брома ,(9,2 г). Выделяющиеся на стейках колбы кристал-
лы брома постепенно растворяются в жидкости. По окончании реакции сосуд
оставляют на ночь в охлаждающей бане, температура которой повышается
приблизительно до —30 °C. За это время большая часть жидкости затверде-
вает. Тогда реакционный сосуд переворачивают, осадок отсасывают на пори-
стом фильтре (G4) и сушат 30 мин в .вакууме. Выход составляет 17,4 г
PBr4F, т. е. 82% теоретического.
При бромировании в охлаждающей бане образуется прежде всего го
мополярное соединение PBr4F, которое при —30 °C постепенно превращается
Соединения с фосфором и мышьяком 23Т
в твердое гетерополяриое соединение, устойчивое в отличие от гомополяриой
формы при комнатной температуре.
Свойства. Желтоватое, очень гигроскопичное вещество. Плавится при
87 °C с частичным разложением.
Рис. 145. Прибор для получения тет-
рабромид-фторида фосфора.
ЛИТЕРАТУРА
I. Kolditz L., Bauer К.. Z. Anorg.
Allgem. Chem., 302, 241 (1959).
Фосфонитрилфториды (PNF2)3 и (PNF2)4
(PNC12)3 + 6KSO2F --*- (PNF2)3 + 6KC1 + 6SO2
347,7 732,96 248,94 447,36 384,36
(PNC12)4 + 8KSO2F --> (PNF2)4 + 8KC1 + 8SO2
463,6 977,28 331,92 596,48 512,48
Тример или тетрамер фосфоиитрилхлорида (порошок) взаимодействует
при 120—125 °C с фторосульфоиатом калия. В результате реакции ие меняет-
ся степень полимеризации.
Свойства. Оба фосфонитрилфторида при комнатной температуре пред-
ставляют собой твердые, неокрашенные, хорошо кристаллизующиеся, легко
летучие вещества, термически устойчивые до 300 °C. Тример кипит при 51,8 °C
и кристаллизуется по моноклинно-призматическому типу. Тройная точка при
27,1 °C. В результате 15-часового нагревания при 350 °C ои полимеризуется
в фосфоиитрилфторидный каучук. Тетрамер кипит при 89,7 °C и кристалли-
зуется по триклиино-пинакоидальному типу. Тройная точка при 30,4 °C.
ЛИТЕРАТУРА
I. Seel F., Langer J., Angew. Chem.,
68, 461 (1956).
238 'Глава 3. Фтор
Гексафторофосфат аммония NH4|PFf,]
(фосфоргексафторид аммония)
Способ 1 [1]
РС15 + 6NH4F ----► NU4[PFb] + 5NH4C1
208,31 222,24 163,06 267,45
В пробирке, встряхивая, смешивают 9,4 г РС15 и 11,6 г высушенного
NI'UF. Пробирку, пе закрывая ее, укрепляют почти горизонтально в лапках
штатива и нагревают смесь небольшим пламенем ближе к открытому концу
пробирки до тех пор, пока пе начнется реакция (тяга, защитные очки!). Пре-
вращение самопроизвольно распространяется до дна’пробирки и происходит
с выделением тяжелых дымяших на воздухе паров. По охлаждении спек-
шееся вещество в пробирке растворяют в 2 л воды. В раствор медленно
приливают при перемешивании 100 мл уксуснокислого раствора, содержаще-
го 9 г нитрона. При этом выпадает гексафторофосфат нитрона.
После двухчасового стояния иа льду соль отфильтровывают, несколько
раз промывают ледяной водой и еще мокрую встряхивают в делительной
воронке с хлороформом и 25%-ным раствором аммиака. Таким образом
удается вытеснить нитрон. Водный раствор переносят в платиновую чашку
и выпаривают на водяной бане досуха. Получают 4 г NH4[PF6].
Для очистки соль растворяют в небольшом количестве воды, фильтруют,
выпаривают вновь в платиновой чашке; так повторяют до тех пор, пока не
получат густую кашицу кристаллов, которую размазывают на фарфоровой
пластинке и сушат на воздухе.
Способ 2 [2]
(NPCl2)n + 6/г HF --> п NH4[PF6] + 2л НС1
116,06 120 163,06 73
Фосфоиитрилхлорид приливают к плавиковой кислоте в Платоновой чаш-
ке, при этом наблюдается сильное разогревание. Содержимое чашки выпари-
вают досуха на водяной бане. Выход количественный. Данная методика
имеет преимущество перед первой в том, что сразу получается чистый про-
дукт, поэтому не требуется очистка через гексафторофосфат нитрона.
Свойства. Неокрашенная соль. Как правило, кристаллизуется в форме
квадратных, реже прямоугольных листочков или толстых табличек. Структу-
ра кубическая, б^10 2,180. Легко растворима в воде: при 20 °C в 100мл во-
ды растворяется 74,8 г. Кроме того, растворима в ацетоне, этиловом н мети-
ловом спирте. Разлагается при сильном нагревании еще до достижения
плавления. При комнатной температуре не действует на стекло. Медленно
гидролизуется при кипячении с сильными кислотами
Препарат применяют для получения многих солей гексафторофосфорной
кислоты.
ЛИТЕРАТУРА
1. Lange W., Muller Е., Вег., 63, 1063 3. Bode И., Clausen Н., Z. Anorg.
(1930). Allgem. Chem., 265, 229 (1951).
2. Lange W„ v. Krueger G., Ber., 65,
1265 (1932).
Диоксодифторофосфат аммония NH4[PO2F2]
P4Olo+6NH4F ----->- 2NH4[PO2F2]-1-2(NH4)2[PO3F]
284,08 222,24 238,12 268,2
Соединения с фосфором и мышьяком 2 39
В никелевом или медном тигле вместимостью 300 мл нагревают 23,5 г
Р4Ою и 18,5 г NH4F до тех пор, пока не начнется реакция. Процесс проис-
ходит далее самопроизвольно, надо только реакционную смесь хорошо пере-
мешивать. По охлаждении содержимое тигля измельчают в порошок и ки-
пятят в стеклянной колбе с 600 мл абсолютного спирта. Фильтруют горячим
через складчатый фильтр, фильтрат тотчас охлаждают и нейтрализуют ам-
миачным раствором спирта. При этом выделяется 8,3 г монофторофосфата
аммония, который отделяют при отсасывании. Фильтрат выпаривают в пла-
тиновой чашке на водяной бане досуха.’Получают 11,6 г (70% теоретиче-
ского) сырой соли. Еще неочищенный продукт iNH4[PO2F2] уже можно ис-
пользовать для многих целей.
Для очистки продукт перекристаллизовывают из 6 мл горячей воды и
высушивают над P4Oi0. Получают 3,2 г (20% теоретического) соли квали-
фикации «ч. д. а.»,, которую можно хранить без доступа воздуха в стеклян-
ных сосудах.
Свойства. Л1 134,1. Неокрашенная соль. tnj, 213 °C (плавится без разло-
жения). Структура ромбическая. Первоначально реагирует с водой, давал
нейтральную реакцию, Но через некоторое время гидролизуется. Легкораство-
рима в воде, этиловом, й метиловом спирте и ацетоне.
I 1 I
ЛИТЕРАТУРА— - ---- -1
I. Lange IF., Вег., 62~ 790 (1929).
Гексафторофосфат калия K[PF6]
PCl4-PFe + 7КОН ----> K[PFe] + К2(НРО4] + 4КС1 + ЗН2О
317,89 392,51 183,98 174,18 298,24 54
0,78 PC14-PF6 .гидролизуют путем обработки 20 мл гидроксида калия.
Объем раствора уменьшают до 3 мл путем испарения в вакууме. Кристал-
лический осадок, появляющийся при этом, отфильтровывают, промывают
спиртом и сушат.
Свойства. Кристаллизуется в виде квадратных и прямоугольных толстых
табличек с кубической гранецентрированной решеткой. Плавится с частич-
ным разложением при температуре красного каления. При нагревании с
твердым NaOH при 400 °C происходит бурное превращение с образованием
фторида и фосфата.
ЛИТЕРАТУРА
1. Kolditz L., Z. Anorg. Allgem. Chem.,
284, 144 (1956).
Фторид мышьяка[1П) AsF3
Способ 1 [1]
As2O3 + 6HF -----*- 2AsF3-f-3H2O
197,80 120,06 263,82 54,04
Реакцию проводят в приборе для перегонки, изготовленном целиком из
железа (рис. 146). Через As2O3 при температуре бани 140 °C пропускают без-
водный HF. При этом стальной сосуд с HF ставят в водяную баню при
+35 °C. Отгоняющийся AsF3 конденсируют в холодильнике, в котором с по-
240 Глава 3. Фтор
мощью охладительной смеси (солевая смесь) поддерживается температура
—18 °C. HF пропускают с такой скоростью, чтобы AsF3 из холодильника
вытекал спокойной струей.
После отключения потока HF отсоединяют реакционный сосуд; к сыро-
му AsF3 приливают H2SO4 (1 : 10), присоединяют приемник и перегоняют.
Между 50 и 85 °C собирается основной погон — AsF3. Выход 80% в расчете
на AS2O3. За день можно получить 6 кг AsF3.
Рис. 146. Устанэвка для получения фторида мышьяка(III).
Способ 2 [2]
2As2Os + 8HSO3F -----> 2AsF3 + 2SO3 + 3H2SO4 4- Asj(SO4)3 + 2HF
395,64 800,56 263,82 160 294,24 437,84 40,0
В стеклянной круглодоииой колбе со шлифом, на которую насажен ши-
рокий обратный холодильник и к которой присоединены нисходящий холо-
дильник и приемник, охлаждаемый льдом, смешивают 144 г As2O3 и 247 г
HSO3F (40%-ный избыток). При этом происходит сильное разогревание.
С помощью обратного холодильника HSOjF удерживается в реакции, при-
чем колбу нагревают на открытом пламени. В течение 1,5 ч при 58—-62 °C
перегоняется 60 г AsF3. Выход 78% в расчете на введенный As2O3.
Свойства. М 131,9. Бесцветная, легкоподвижная, очень гигроскопичная
жидкость. —8,5°C; /КИп +63°C; dx 2,73 (15°C). Дымит.на воздухе,
разъедает стекло. Разлагается водой, как только прибавлены эквивалентные
количества последней. Растворим в спирте, эфире и бензоле.
Хранят AsF3 в железных сосудах. С этой целью можно перевести его в
легкоразлагаемое соединение с SOs (см. ниже).
ЛИТЕРАТУРА
1. Kwasnik W., неопубликованные 2. Engelbrecht A., Aignesberger А.,
данные. Hayek Е., Monatsh. Chem., 86, 47
(1955).
Аддукт 2AsF3«3SO3
2AsFs + 3SOs :—2AsFs*3SOg
263,8 - -240,2 504,0
Соединения с фосфороА и мышьяком 241
Реакцию проводят в стеклянной колбе со шлифами, в которую вставле-
ны обратный холодильник и капельная вороика. К 123 г свежеперегнаиного
SO3 (20%-иый избыток) по каплям медленно прибавляют 84 г AsF3. Реакция
происходит очень бурно. По окончании реакции сырой продукт перегоняют.
Аддукт может служить для безопасного хранения AsF3. Для того чтобы
освободить последний, достаточно прибавить NaF. Эта реакция даже при
охлаждении иа льду протекает очень бурио. После последующего прибав-
ления аддукта к NaF реакционную смесь нагревают в течение 1 ч с обрат-
ным холодильником и отгоняют затем при 62 °C чистый AsF3, ие содержа-
щий SO3.
Свойства. При —70 °C застывает, образуя твердую стеклоподобпую мас-
су. /кип 138 °C (709 мм рт. ст.), d2o 2,415. Перегоняется без разложения, неог-
раниченно растворим в SO2CI2, S2O5CI2 и РОС13.
ЛИТЕРАТУРА
1. Engelbrecht A., Aignesberger А., и.
Hayek Е„ Monatsh. Chem., 86, 470
(1955).
Фторид мышьяка(У) AsFs
2As + 5F2 -----* 2AsFe
149,82 190,0 339,82
Для фторирования используют прибор, описанный в методике получе-
ния SF6. Мышьяк помещают в лодочку из Ni или А12О3. Продукт, собранный
в газовых ловушках, несколько раз перегоняют, разделяя иа фракции, в
кварцевом приборе.
Свойства. М 169,91. Бесцветный газ. На влажном воздухе образует бе-
лое облако, /пл —79,8 °C; /КВп —52,9 °C; 2,33 (—52,8 °C). Водой сразу же
гидролизуется. Растворим в спирте, эфире и бензоле. ,
Хранят AsF3 в стальных баллонах. ,
ЛИТЕРАТУРА I
1. Ruff О., Menzel W., Plaut Н„ Z.
Anorg. Allgem. Chem., 206, 61
(1931).
Фторид сурьмы(Ш) SbF3
Sb2O3 + 6HF ---> 2SbFa-|-3H2O
291,52 120 357,52 54,03
Способ 1 i[l], Sb2O3 растворяют в избытке водной плавиковой’кислоты
и раствор быстро выпаривают досуха иа электроплитке. Продукт отгоняют
в медном приборе. Для этого используют конический вверху перегоииый со-
суд, иа который надета широкая короткая иасадка. Во время перегонки не-
обходимо следить, чтобы иасадка оставалась достаточно теплой, иначе про-
изойдет закупорка.
Способ 2 [2]. В конический, изготовленный из магниевой жести сосуд,
закрытый крышкой нз магния, помещают Sb2O3 и пропускают через серебря-
ный капилляр газообразный HF. Сосуд слабо нагревают. газовым пламенем
Если HF больше не поглощается, нагревание усиливают для того, чтобы
16 —143
242 Глава 3. Фтор
удалить образовавшуюся воду. Пропускание HF и удаление Н2О повторяют
до тех пор, пока больше не образуется водной плавиковой кислоты. Затем
образовавшуюся массу плавят, расплав выливают на пластинку нз Mg, раз-
бивают на куски и складывают в плотно закрывающийся бюкс из жести.
SbF3 можно, кроме того, перегнать, как описано выше.
Свойства. М 178,76. Бесцветные расплывающиеся кристаллы. /пл 292 °C;'
7кип 376°C; йтв 4,379 (20°C). Структура ромбическая. Легко растворяется в'
воде (с частичным гидролизом): при 20°C 443 г/100 мл, прн 30°C
562-r/lOO мл.
ЛИТЕРАТУРА
I. Ruff О., Die Chemie des Fluors.
1920, S. 39.
2. Soil J., Naturforschung u. Medizin
in Deutschland (FIAT-Review), 23,
276.
Фторид сурьмы(У) SbF5
SbF3 + F2 -----> SbF5
178,76 38 216,76
На рис. 147 приведена схеца прибора из кварца для синтеза. SbFg.
Фтор вводят в реакцию с газообразным SbF3. Нагревание производят горел-
кой Буизена так, чтобы SbF3 слабо кипел. Через алюминиевую трубку вво-
для получения
дят F2 со скоростью 10 г/ч. SbF3 при этом реагирует, время от времени вспы-
хивая, и SbF6 отгоняется. Продукт для разделения на фракции можно пе-
регонять нз запасного кварцевого сосуда.
Свойства. Бесцветная, густая жидкость. /пл 6 °C; /КИп 150 °C; </ж 2,993
(22°C). Очень реакционноспособна; шипит при вливании в воду; разъедает
кожу. Слабо взаимодействует со стеклом, Си и РЬ, не действует на кварц.
Pt и А1. . -
Соединения с сурьмой й висмутом 243
Хранят SbFs в закрывающихся алюминиевых флягах нли в крайнем
случае в кварцевых сосудах. Подходящими являются и небольшие емкости
из платины. ,
ЛИТЕРАТУРА
1. Soil J., Naturforschung u. Medizin
in Deutschland 1939—1946 (FIAT-
Review), 23, 276.
Дихлорид-трифторид сурьмы SbCl2F3
SbF3 + Cl2 -----> SbCl2F3
178,76 70,91 249,67
В стальной цилиндр, снабженный манометром и винтовым вентилем,
вносят иавеску SbF3. Емкость эвакуируют, закрывают вентиль и емкость,
взвешивают. Затем присоединяют через стальной капилляр баллон с хло-
ром, открывают вентиль и впускают в реакционный сосуд С12, который быст-
ро поглощается SbF3 с выделением тепла. Время от времени реакционный
сосуд отсоединяют от баллона с хлором и встряхивают. После этого вновь
пропускают С12. Синтез заканчивают после поглощения рассчитанного ко-
личества С12.
Свойству. Вязкая жидкость.
Хранят SbCl2F3 в железных сосудах.
Применяют как катализатор прн получении многих органических соеди-
нений.
ЛИТЕРАТУРА
1. Неппе A. L., Organ. Reactions, II, S. 61.
Фторид висмута) 111) BiF3
BiOCl + 3HF -г-—> BiF3 + H2O + HC1
260,43 60,02 J'265,98 18,01 36,46
Около 1 г свежеосажденного и высушенного при 400 °C BiOCl взвеши-
вают в лодочке из стеклографита, которую помещают в реакционную трубку
из такого же материала, находящуюся в трубчатой печи. После тщательного
вытеснения из реакционной трубки воздуха потоком N2 высокой степени чи-
стоты поднимают температуру до 300 °C, пропуская в это время поток сухо-
го HF. После однодневного нагревания при 300 °C реакционную трубку ох-
лаждают, продолжая пропускать HF, а затем отключают подачу HF и тща-
тельно вытесняют последний азотом.
Надо предостеречь от часто рекомендуемого в литературе синтеза путем
обработки Bi2O3 или Bi(OH)3 водной плавиковой кислотой с последующим
удалением летучих продуктов. При этом получается не чистый BiF3, а обра-
зуются оксид-фториды. Принимая последние за BiF3, сведения о кристалличе-
ской решетке (кубическая решетка), относящиеся к оксид-фторидам, ошибоч-
но адресуют BiF3.
Свойства. Белый кристаллический порошок. Кристаллическая структур:»
орторомбическая (тип YF3). /пл 727 °C; d 7,92.
Может храниться па воздухе в стеклянной посуде.
16*
244 Глава 3. Фтор
ЛИТЕРАТУРА
1. Greis О., Univers. Freiburg, частное
сообщение (1974).
2. Zachariasen Н. W„ U. S. Atomic
Energy Commission, Argonne Labo-
ratory Report ANL-4400 (Jan. 1950).
3. Zalkin A., Templeton D. H„ J.
Amer. Chem. Soc., 75, 2453 (1953).
4. Aurivillius B„ Acta Chem. Scand.,
9, 1206 (1955).
Фторид висмута(У) BiF5
Способ 1 [1]
BiF3 + F2 ---> BiFs
266 38 304 ’
Лодочку из спеченного глинозема илн плавикового шпата задвигают с
помощью никелевой проволоки в трубку из спеченного глинозема (рнс. 148).
Оба конца трубки закрывают медными колпачками, которые охлаждаются
Колпа-
чок из меди
Никеледая про до- Г ZJ
пока ___________
Стеклянная
насадка
Колпачок
из меди
ВЦ
4
Место Ампула
отпаива-
ния ам-
пулы
Рис. 148. Участки установки для получения фторида висмута (V).
подои. Отверстия между трубкой и колпачками заделывают пнцеином. Це-
лесообразно устанавливать прибор так, чтобы можно было поворачивать
печь в вертикальной плоскости почти на 90°. Для подвода Fz используют
очень гибкий 5-метровый медный капилляр. Пропускают F? со скоростью
20 мл/мин и нагревают печь до 550 °C. Прн 460 °C нз лодочки начинает
сублимироваться BiFs, который осаждается на конце реакционной трубки,
образуя тонкие, белые иглы длиной ~3 мм. Лучше всего сублимация про-
исходит при 500 °C. При более высокой температуре этот процесс настолько
ускоряется, что BiFs продвигается против потока Fz н оседает в начале
трубки.
По окончании фторирования фтор заменяют потоком N2, очищенного от
СО2 и Oz. Тем временем вытаскивают с помощью никелевой проволоки ло-
дочку, вытягивая ее из реакционной трубки навстречу потоку газа и как
следует укрепляют медный колпачок. Другой медный колпачок заменяют на
стеклянную насадку (рис. 148). Теперь трубку поворачивают на 90°, так что
выход из нее опускается вниз, а вход (ввод газов) поднимается вверх. Раз-
рыхляют с помощью длинной никелевой проволоки слипшиеся нглы BiFs.
Через стеклянную насадку иглы падают в ампулу, которую затем отпаивают.
Способ 2 [2]
2Bi + 5F2 ----► 2BiFs
418 190 608
Соединения с углеродом 245
Для взаимодействия висмута с фтором используют реактор с кипящим
слоем. Заимствованная из техники методика синтеза в кипящем слое имеет
следующие преимущества: быстрое установление теплового равновесия в ре-
акционной смеси, отсутствие спекания твердых продуктов реакции, хороший
тепловой обмен со стенками трубки, большая поверхность твердых исход-
ных веществ и поэтому быстрое превращение.
Лабораторное оборудование состоит из вертикальной никелевой трубки
(длина 60 см, диаметр 1,5 см, толщина стенок 2 мм), на нижнем конце ко-
торой находится мелкоперфорнрованное никелевое сито для приема порошка
металла. Фтор поступает снизу через сито. Выше сита на расстоянии 30 см
друг от друга на трубке реактора с кнпящнм слоем привинчены клеммы.
Клеммы присоединены к вторичной обмотке трансформатора печи Таммана.
Таким образом, никелевая трубка нагревается. К верхнему концу трубки
реактора присоединяют две газовые ловушки из кварца, которые охлажда-
ются жидким воздухом.
После вытеснения фтором воздуха нз прибора в трубку реактора загру-
жают 500—1000 мг висмута с размером зерен 0,062—0,075 мм. Прибор эва-
куируют, а затем устанавливают такой поток фтора, чтобы протяженность
кипящего слоя составила 10 см длины трубки. Одновременно трубку нагре-
вают до 600—700 °C. Реакция заканчивается за несколько минут. BiFs соби-
рается в ловушках. В конце избыток F2 отсасывают из газовой ловушки в
высоком вакууме прн —60 °C.
Анализ лучше всего проводить путем восстановления навески препарата
водородом, сильно разбавленным СО2, прн 80—'150 °C (1 ч) в платиновой
трубке с последующим взвешиванием образующегося BiF3.
Свойства. Белые, очень чувствительные к влаге кристаллы. /возг~550 °C.
На влажном воздухе BiFs тотчас окрашивается в цвет от желтого до ко-
ричневого. С водой реагирует иногда со вспышками, причем образуются
озон н BiF3. Выше 50 °C реагирует с парафином.
ЛИТЕРАТУРА
1. V. Wartenberg Н„ Z. Anorg. Allgem. 2. Roesky Н. W., Glemser О., Hell-
Chem., 244, 344 (1940). berg К.-H., Chem. Ber., 98, 2046
(1965).
Тетрафторид углерода CF4
Способ 1 [1, 2]
С + 2F# ----»- CF4
12 76 88
В приборе, описанном в методике получения SF6, сжигают в никелевой
лодочке в токе F2 обезгаженную сажу. Чтобы реакция протекала с умерен-
ной скоростью, применяют внешнее охлаждение. В кварцевой ловушке, ох-
лаждаемой до —183 °C жидким Оз или другим каким-нибудь способом, соби-
рается в виде жидкости сырой CF4. По окончании фторирования продукт
откачивают в течение ~1 ч на водоструйном насосе, поддерживая в ловушке
температуру —183 °C. Таким образом удаляют большую часть растворенных
газов. Затем продукт пропускают через несколько промывалок с пористыми
стеклянными пластинками, заполненных 20%-ным КОН (не NaOH), при этом
вымывают F2CO, SiF4 и HF. Наконец, CF4 пропускают над Р4О10 и затем
вновь конденсируют при —183 °C. Теперь продукт очень тщательно фрак-
ционируют, чтобы отделить последующие гомологи CF4 (C2F6, C3F3). В за-
ключение продукт в течение нескольких часов откачивают на масляном на-
сосе от растворенного воздуха, держа ловушку в жидком воздухе (жидком
азоте).
246 Глава 3. Фтор
Вся посуда, используемая уже после окончания фторирования, может
быть изготовлена из стекла.
Способ 2 [3, 4]
2CO-4-4F, ----->- 2CF. + О2
56 152 176 32
Способ получения CF4 из СО н F2 имеет то преимущество перед описан-
ным выше, где фторируют углерод, что продукты реакции совершенно не со-
держат других членов гомологического ряда CF4. Синтез проводят в приборе,
собранном согласно рис. 135 [подробно см. 2-е издание этой книги (1960 г.)].
Для получения хороших выходов CF4 надо позаботиться о том, чтобы СО по-
возможности был предварительно нагретым (до ~ 400 °C). Прн нагрузке по-
току электролизера для получения фтора 1000 А выход составляет 80—85%
в расчете на СО. При значительно меньшей скорости потока, например если
нагрузка по току электролизера была только 10 А, максимальный выход.
CF4—15% (побочный продукт в основном F2CO).
Обработка сырого CF4 проводилась так же, как описано в первом спо-
собе. Степень чистоты препарата можно легко контролировать путем опре-
деления точки плавления, так как в присутствии растворенных воздуха и
C2Fc она сильно понижается.
Свойства. Бесцветный, не имеющий запаха газ. /пл —183,6 °C; /кИП
—127,8°С; dTB 1,98 (—195°С); 1,89 (—183°С). Термически очень устой-
чив, а прн комнатной температуре и химически индиферентен.
Хранят CF4 в стеклянных колбах илн стальных баллонах. Применяется
как наполнитель термометров, принцип действия которых основан на изме-
нении давления пара [5].
ЛИТЕРАТУРА
1. Ruff О., Keim R., Z., Anorg. Allgem.
Chem., 192, 249 (1930),
2. Ruff О., Reim R., Z. Anorg. Allgem.
Chem., 201, 255 (1931).
3. Kwasnik W„ Naturforschung u. Me-
dizin in Deutschland 1939—1946
(FIAT-Review), 23, 168.
4. Golibeau J.. Bues IF., Kampmann It7.,
Z. Anorg. Allgem. Chem., 283, 123
(1956).
5. Menzel W., Mohry F., Z. Anorg.
Allgem,-Chem,, 210, 256 (1933).
Трифторметан CHF3
(Фтороформ)
Способ 1 [1]
CHI3 + 3HgF ----> CHF34-3HgI
393,76 658,83 70,0 982,59
Смесь CHI3, HgF и порошка плавикового шпата (в качестве разбавите-
ля) в отношении 20 : 33,4 : 40 растирают и помещают в стеклянную колбу на
100 мл. К выходному штуцеру присоединяют газовую ловушку, погруженную
в жидкий воздух. В конце присоединяют осушительную трубку с Р4Ою.
Колбу нагревают на сернокислотной’бане. При ~80 °C начинается ре-
акция; далее температура повышается до ~ 150 °C благодаря выделяемому
во время реакции теплу. В ловушке собирается сырой, окрашенный кодом
Соединения с углеродом 247
По окончании реакции продукт перегоняют, разделяя на фракции. По-
гон, переходящий прн температуре бани в интервале —404—30 °C, и есть
практически чистый CHFs. Его промывают 2 н. NaOH и сушат над Р4О10.
Выход 45%.
Способ 2 [2]
CHC134-3HF ------> CHF3 + 3HC1
119,39 60 70,0 109,41
В автоклав из специальной стали, имеющий приспособление для пере-
мешивания и железный обратный холодильник, помещают 360 г CHCI3 и
600 г SbCl5 (катализатоор). Затем создают давление путем введения 200 г
безводного HF н нагревают автоклав в течение 1,5 ч прн 130 °C. Давление
при этом повышается до 77 бар. Постепенно снижают давление с помощью
вентиля вверху обратного холодильника. Выходящие газы пропускают через
ледяную воду и разбавленный NaOH, высушивают над Р4О10 и перегоняют,
разделяя на фракции.
С тем же самым количеством катализатора можно много раз повторять
синтез, возобновляя только исходные вещества, так что в автоклав каждый
раз загружают 360 г СНС13 и вводят 60 г HF. Выход 95%. ,
Такой способ фторирования, когда промежуточно образующееся соеди-
нение SbF3C12-2HF действует как катализатор, нашел разнообразные при-
менения, например, при получении CCIF3, CCI2F2, C2CI3F3, C2CI2F4 и
C2HC13F2.
Свойства. Бесцветный газ. /Ш[ —160°C; /кип —84,4°C. 1,52 (—100°C);
4в 1,935. Термически устойчив вплоть до 1150 °C. В химическом отношении
проявляет удивительную индифферентность.
CHF3 можно хранить в стеклянных колбах и газометрах под водой.
ЛИТЕРАТУРА
1. Ruff О., Вег., 69, 299 (1936). 2. Whallay В., J. Soc. Chem. Ind., 66,
429 (1947).
Трифториодметан CF3I
Способ 1 [1—3]
CF3COONa + I2 ---->- CF3I + CO2 + Nal
136,01 253,84 195,91 44 149,84
Смесь 27,2 г (200 ммоль) сухого CFsCOONa н 38 г (300 ммоль) 12
в круглодонной колбе на 500 мл, снабженной обратным холодильником,
взмучивают в 80 мл безводного диметилформамида и нагревают. К водяно-
му холодильнику присоединяют две ловушки, охлаждаемые жидким возду-
хом. Трубки, подводящие к ловушкам продукт реакции, должны иметь по
возможности большой диаметр. Иод испаряется, пары газов, слабо окрашен-
ные в фиолетовый цвет, проходят через холодильник и конденсируются сов-
местно с небольшими количествами иода н СО2 в обеих ловушках. CF3I
очищают путем низкотемпературной перегонки или промыванием разбавлен-
ным NaOH. Выход 22,4 г (70%).
Способ 2 [4]
CF3COOAg+ 12 ----->- CF3I + Agl +СО2
220,89 253,84 195,92 234,80 44,01
248 Глава 3. Фтор
Прежде всего получают трифторацетат серебра, для чего Ag2O вносят в
50%-ную трифторуксусную кислоту н выпаривают реакционную смесь в ва-
кууме досуха.
100 г растертого в порошок трифторацетата серебра смешивают с ПО—
300 г растертого в порошок иода и наполняют этой смесью закрытую с од-
ного конца трубку из стекла пирекс. Трубку закрепляют в горизонтальном
положении, присоединяют к открытому концу ловушку, охлаждаемую ледя-
ной водой, затем еще две ловушки, охлаждаемые сухим льдом, и наконец
счетчик пузырьков, заполненный водой. Реакционную смесь постепенно на-
гревают пламенем газовой горелки выше 100 °C, причем интенсивность на-
гревания устанавливают, ориентируясь па скорость выделения газа, опре-
деляемую с помощью счетчика пузырьков. В первой ловушке собирается
нод, в последней — CIFs. Затем CIF3 промывают разбавленным NaOH н очи-
щают путем фракционной перегонки. Выход 80—95%.
Свойства. Бесцветный, светочувствительный газ. (кип —22,5 °C. Давление
пара (в мм рт. ст.): 1g р= 7,5665—1174,29 t. Теплота испарения 5357 кал.
ИК-спектр (см->): 19Ю (сл), 4814 (ел), 1714 (сл), 1611 (сл), 1441 (сл),
1334 (сл), 1274 (оч. сл),1187 (оч. сл), 1080 (оч. сл), 1030 (оч. сл), 924 (сл),
811 (ср), 777 (ср), 747 (оч.сл), 737 (оч. сл), 559 (ср), 537 (ср).
ДМР 19F: бегз 5 м. д. (внутр, стандарт CFCla).
Хранят CIF3 в стеклянных ампулах.
C1F3 образует прн нагревании н облучении УФ-светом свободный радикал
CF3 н поэтому очень подходит для синтезов различных соединений типа
СРз(СРг)п-Х, металлоорганических соединений, а также органических произ-
водных неметаллов.
ЛИТЕРАТУРА
1. Paskovich D., Gaspar Р., Ham-
mond G. S„ J. Org. Chem., 32, 833
(1967).
2. Haszeldine R. N„ J. Chem. Soc.
(London), 1951, 584.
3. McGee P. R., Cleveland F. F„ Mil-
ler S. I., J. Chem. Phys., 20, 1044.
4. Henne A. L„ Finnegan IF. G., J.-
Amer. Chem. Soc., 72, 3806 (1950).
Оксид-дифторид углерода F2CO
(Дифторид карбонила, фторфосген)
С12СО + 2NaF ---* F2CO + 2NaCl
98,9 84,0 66,0 116,9
Синтез проводят в реакторе, сконструированном для проведения реак-
ций с расплавами солей н снабженном специальным устройством, позволяю-
щим использовать пропускание газа для перемешивания' реакционной смеси
(рис. 75). В реактор помещают 252 г сухого фторида натрия н 1 л очищен-
ного перегонкой ацетонитрила и суспендируют. В течение ~6 ч вводят при
интенсивном перемешивании 300 г С12СО. Синтез проводят прн 82 °C. Мощ-
ный обратный холодильник, укрепленный на крышке реактора; охлаждается
с помощью криостата до —30 °C, благодаря чему растворитель н непроре-
агнровавший С12СО возвращается в реакционную смесь. Реакция протекает
вяло.
F2CO отводится из обратного холодильника, проходит для вторичной-
очистки через ловушку, охлаждаемую до —78 °C, и конденсируется прн ох-
лаждении жидким воздухом. В результате низкотемпературной перегонки
получают 104 г исходного СОС12 и 128 г F2CO.
Выход 96% в расчете на вступивший в реакцию СОС12.
Соединения с углеродом 24?
Другие способы.
FjCO получают в значительных количествах наряду с BrFCO прн взаи-
модействии BrFs с СО, а также прн синтезе CF4 по второму способу.
Свойства. Бесцветный газ с отвратительным запахом. /пл —'114,0 °C;
/кип —83,Г°С; dTB 1,388 (— 190°С); dx 1,139 (—114°С). Очень гигроскопи-
чен, сразу гидролизуется водой.
ЛИТЕРАТУРА
1. Tullock С. W., Coffman D. D., J.
Organ. Chem., 25, 2016 (1960).
2. Sundermeyer IF., Univers. Heidel-
berg, частное сообщение (1974).
Хлорид-фторид карбонила C1FCO
(Хлорфторфосген)
Способ 1 [1]
CFC13 + SO3 ---> C1FCO + SO2C12
137,4 80,0 82,5 134,9
Синтез проводят в литровой трехгорлой колбе, которая снабжена термо-
метром, капельной воронкой с длинным, погружающимся в реакционную
смесь «хвостиком», магнитной мешалкой и двумя установленными друг за
другом обратными холодильниками, имеющими внутренний холодильный
змеевнк (холодильник Димрота). В колбу помещают 470 г (5,87 моль)
65%-ного олеума и медленно прибавляют по каплям 227 г (1,66 моль)
CFC13. Предварительно к олеуму добавляют 1 г Hg2SO4 и 1 г HgSO4 (ка-
тализаторы). Нижний обратный холодильник охлаждается жидкостью с
температурой +10 °C, верхний — жидкостью с температурой —30 °C. Темпе-
ратура реакционной смеси достигает —60 °C. Газ, выделяющийся нз реакцион-
ной смеси, проходит выше холодильников и после промывалкн с H2SO4 кон-
денсируется в ловушке, охлаждаемой прн —78 °C. Конденсат (142 г) содер-
жит CiFCO (выход ~ 60%), CFC13 и СО2. Путем низкотемпературного фрак-
ционирования получают 114 г чистого COFC1.
Способ 2 [2]
С12СО + SbF3 ----> C1FCO 4- SbClF2
99 178 82 195
В облицованном медью автоклаве нагревают в течение 1 ч при 135 °C
1 моль SbF3, высушенного при 120 °C и растертого в порошок, 4 моль фосге-
на и 10 мл SbFg. Через вставленный в автоклав обратный холодильник цир-
кулирует вода. После охлаждения реакционные газы выпускают из автокла-
ва и конденсируют в стеклянных газовых ловушках, охлаждаемых сухим
льдом. Содержимое ловушек несколько раз фракционируют при низких тем-
пературах. Выход составляет 2,1 моль C1FCO.
Свойства. Бесцветный газ, по запаху трудно отличимый от фосгена.
/пл — 148°С; /кип —47,2°С; dx 1,506 (—78°С), 1,323 (0°С), 1,277 (+18°С).
Давление пара 13 бар (+19 °C) - £нрит +85 °C.
C1FCO и в газообразном, и в жидком состоянии устойчив прн комнатной
температуре. Водой гидролизуется в течение 0,5 ч. Гидроксид натрия тотчас
полностью поглощает его с выделением тепла. Стекло в течение недели
остается практически устойчивым, только становится мутным. Кварц ока-
зывается более прочным, чем стекло, но также медленно мутнеет. Hg разъ-
едается под действием C1FCO; резина становится почти твердой после не-
250 Глава 3. Фтор
дельного воздействия. Хорошую устойчивость против действия C1FCO прояв-
ляют сталь специальных марок, латунь и алюминий, среднюю устойчивость—
никель, монель, олово, цинк н сплав электрон, еще менее устойчивы железо,
медь, свинец н серебро.
Лучше всего хранить C1FCO в кварцевых ампулах при температуре
жидкого воздуха, можно также использовать сосуды высокого давления из
специальной стали.
ЛИТЕРАТУРА
1. Siegemund G., Angew. Chem., 85, 2. Emeleus H. J., Wood J. F., Chem.
982 (1973). Soc., 1948, 2183.
Бромид-фторид карбонила BrFCO
(Бромфторфосген)
Способ 1 [1]
BrF3 + 2CO ----> BrFCO-|-F2CO
136,9 56,0 126,9 66
Синтез проводят в трехгорлой кварцевой колбе, снабженной термомет-
ром (в никелевом кожухе), трубкой для подвода газа и обратным холо-
дильником. В колбу помешают 68,5 г BrF3 н пропускают при 10—30 °C мо-
ноксид углерода (для очистки СО пропускают через раствор пирогаллола,
конц. H2SO4 и Р4Ою). Газообразные продукты проходят через обратный хо-
лодильник и конденсируются в двух ловушках: основные количества
BrFCO — в ловушке, охлаждаемой смесью сухого льда с ацетоном, a F2CO
и небольшие количества BrFCO — в другой, охлаждаемой жидким возду-
хом. Для очистки BrFCO от Вг2 его пропускают над порошком сурьмы; за-
тем фракционируют в кварцевом приборе. Прн этом чистый COF2 отгоняет-
ся при —854—60 °C. В интервале —304—15 °C улавливается BrFCO. В ре-
зультате повторной фракционной перегонки можно получить совершенно
чистый продукт. Получают 53,3 г BrFCO. Из конденсата второй ловушки
можно выделить путем фракционной перегонки 20 г COF2.
Способ 2 [2, 3]
CFBr34-SO3 ----> BrFCO 4- SO2Br2
270,7 80,0 126,9 223,8
В приборе, описанном в способе 1 получения C1FCO, к 200 г (2,5 моль)
SO3 при 37—43 °C в течение 2 ч прибавляют 135 г (0,5 моль) CFBr3. В ниж-
нем обратном холодильнике поддерживается температура +10 °C, в верх-
нем— 0 °C. Газ, выделяющийся из реакционной смеси, перед конденсацией
пропускают через промывалку с трихлорэтиленом, освещаемую УФ-светом;
при этом связывается бром. Путем фракционирования содержимого ловушек
(80 г) получают 60 г продукта (в интервале —17,54—10 °C), который наря-
ду с BrFCO содержит незначительные количества SO2 и СО2.
Свойства. Бесцветный газ. По запаху похож на фосген, однако если
иметь некоторый опыт, то запах BrFCO можно хорошо отличать от запаха
СОС12. /пл —120°С; /кпп —20,6°С. Давление пара 4,56 бар (+18°С); /крит
+ 124°C; ркр„т -62 бар; dx 1,944 (0°С).
Газообразный BrFCO термически устойчив вплоть до +125 °C. Сжижен-
ный BrFCO не может длительное время сохраняться при комнатной темпе-
ратуре. Вода вызывает гидролиз до СО2, НВг и HF. Эта реакция происходит
Соединения с углеродом 251
количественно за ~30 мин. Гидроксид натрия мгновенно поглощает COBrF.
Стекло длительное время устойчиво к действию COBrF. Резина под действи-
ем BrFCO чернеет и становится хрупкой, a Fe н Hg разрушаются.
Лучше всего BrFCO хранить в кварцевых ампулах прн температуре
жидкого воздуха. Для этих целей можно также использовать кварцевые
сосуды или баллоны высокого давления из специальной стали, однако при
таком хранении через некоторое время BrFCO окрашивается в желто-корич-
невый цвет и нуждается в дополнительной перегонке.
ЛИТЕРАТУРА
1. Kwasnik W., Naturforschung u. Ме-
dizin in Deutschland 1939—1946
(FIAT-Review), 23, 242.
2. Siegemund G.. Angew. Chem., 85,
982 (1973).
3. Olah G. A.. Kuhn S. I., J. Org.
Chem., 21, 1318 (1956).
Иодид-фторид карбонила IFCO
(Иодфторфосген)
IF6 + 3CO -----> IFCO4-2F„CO
221,93 84,0 173,93 132,0
Во вращающийся автоклав вместимостью I л помещают 50 г IFs. Затем
из бомбы под давлением 122 бар впускают СО и поворачивают автоклав в
наклонном положении в течение недели. Приводят содержимое автоклава к
атмосферному давлению, выпуская избыточный газ; прн этом улетучиваются
F2CO и избыточный СО. Наконец, к вентилю автоклава присоединяют квар-
цевую лову'шку, охлаждаемую жидким воздухом, в которой находится поро-
шок сурьмы. Установку эвакуируют в течение 1 ч до 200 мм рт. ст. При
этом IFCO отгоняется и конденсируется в газовой ловушке. Автоклав мож-
но, не загружая вновь IFs, опять заполнить СО.
Собранный в ловушке конденсат перегоняют над Sb, которая уже ранее
находилась там. Небольшие количества F2CO перегоняются до —15 °C. Затем
из-за разложения IFCO перегонку производят при уменьшенном давлении
(~300 мм рт. ст.). В интервале —15++20°С перегоняется IFCO. После
этого еще раз перегоняют над Sb при уменьшенном давлении. Выход 12%
в расчете на IFs. Основными продуктами этой реакции являются 12 и F2CO.
IFCO можно хранить в кварцевых ампулах прн температуре сухого льда
или лучше жидкого воздуха.
Свойства. Чистый IFCO бесцветен. По удушливому запаху похож на
BrFCO, но хорошо отличим от С12СО.
Плавится при —904—91 °C; /кип +23,4°C (с разложением); dx 2,618
(0°С); 2,470 (+16°C).
При хранении выше —20 °C разлагается с заметным выделением иода.
Газообразный IFCO разлагается также при комнатной температуре. Подоб-
но BrFCO, медленно гидролизуется водой. Полностью поглощается гидрокси-
дом натрия. Кварц н стекло при контакте с жидким IFCO при комнатной
температуре покрываются желтым налетом.
ЛИТЕРАТУРА
1. Kwasnik IF., Naturforschung u. Me-
dizin in Deuschland 1939—1946
(FIAT-Review), 23, 242.
252 Глава 3. Фтор
Цианурфторид (ICN)3
(2,4,6-Трифторо-1,3,5-триазин)
Способ 1 [1]
(C1CN)3 + 3HF ---*• (FCN)3 + 3HC1
184 60 135 109
В цианурхлорнд, находящийся в железном сосуде, пропускают при 40 °C
газообразный безводный фтороводород. Затем продукт перегоняют в прибо-
ре, изготовленном из железа, причем сначала отгоняется HF. Основные ко-
личества цианида фтора переходят в интервале 55-^75 °C. В остатке нахо-
дится непрореагнровавший (C1CN)3. Для очистки сырой (FCN)3 перегоняют
над KF.
Способ 2 [2]
(C!CN)3 4- 3NaF ---* (FCN)3 + 3NaCl
184 126 135 175
В автоклав загружают суспензию фторида натрия в ацетонитриле и
цианурхлорида и нагревают 0,2 ч при 134 °C. По охлаждении (FCN)3 отго-
няют при атмосферном давлении.
Свойства. /пл —38 °C; tKKn 74 °C. Устойчив к воде. Гидролизуется раз-
бавленной плавиковой кислотой до циануровой кислоты. Цианурфторид бо-
лее реакционноспособен, чем цианурхлорнд.
ЛИТЕРАТУРА
1. Kwasnik W., Klemm W., Naturfor- 2. Tullock C. W., Coffman D. D„ J.
schung u. Medizin in Deutschland, Org. Chem., 25, 2016 (1960).
23 (Teil I), 243 (1949).
Фтороциан FCN
(FCN)3 ---► 3FCN
135 135
Через промывную склянку с цианурфторидом пропускают при 60 "С и
50 мм рт. ст. поток азота, который, захватывая цианурфторид, проходит при
1300 °C со скоростью 50 г/ч через вертикальную угольную трубку, наполнен-
ную угольной крошкой. Реакционная трубка индукционно нагревается. Про-
дукт, улавливаемый в охлаждаемую жидким азотом ловушку, перегоняют
затем на колонке из стекла при низких температурах. Сначала отгоняются
CF4, C2F6 и CF3CN. Степень превращения >50%.
Свойства. А1 45,02. Неокрашенное соединение. /пл —82 °C; /кип —46 °C.
При комнатной температуре полимеризуется, давая цианурфторид. В при-
сутствии BF3 или HF эта реакция проходит очень бурно.
Фтороцнан можно хранить при —78 °C в сосудах из нержавеющей стали.
ЛИТЕРАТУРА
1. Fawcett F. S„ Lipscomb R. D„ J.
Amer. Chem. Soc., 82, 1509 (1960);
86, 2576 (1964).
Соединения с кремнием 253
Получение других фторпроизводных углерода, а именно: F2C6, FC(S)NCS,
FC(O)SC1, F3CSCOF, F3CSC1, F3CSNO, Hg(SCF3)2, FaCC(S)CF3>
F3CSNH2, (F3CS)2NH, (F3CS)3N, F3COF, (F3C)2PH, описано в разд. «Кова-
лентные соединения углерода».
Фторид кремния SiF4
Способ 1 [1,2]
2CaF2 + 2H2SO4 ----> 2CaSO4 + 4HF
156,14 196,15 272,25 80,04
4HF4-SiO2 ------> SiF4 4- 2H2O
80,04 60,05 104,06 36,03
Измельченный в порошок плавиковый шпат обрабатывают в платиновой
чашке плавиковой кислотой для удаления карбоната.
Синтез проводят в приборе, изготовленном полностью из стекла
(рис. 149). В реакционную колбу помещают смесь эквивалентных количеств
Рис. 149. Прибор для получения фторида
кремния (способ 1).
Рис. 150. Прибор для по-
лучения фторида кремния,
(способ 2).
порошка плавикового шпата, очень чистого (99,9%) кварцевого песка (берут
в небольшом избытке) н конц. H2SO4 и колбу слабо подогревают на песча-
ной бане. Выделяющийся SiF4 проходит через нисходящий холодильник, за-
тем для отделения возможных примесей (HF) — через ловушку, охлаждае-
мую смесью ацетона с сухим льдом, н конденсируется в ловушке, охлаждае-
мой жидким воздухом. Для предотвращения попадания влаги воздуха при-
соединяют трубку с Р4О10. Для очистки продукт сублимируют в закрытых
стеклянных сосудах или перегоняют при небольшом повышенном давлении,
причем первую н последнюю фракции отбрасывают.
Способ 2 [3]
H2[SiFe] (+конц. H2SO4) ----> SiF4 + 2HF
144,06 104,06 40
254 Глава 3. Фтор
Вместо стеклянной колбы, как описано в первом способе, реакцию про-
водят в сосуде из кованого железа (рис. 150). К 1 л 60%-ной H2SiF6 добав-
ляют по каплям из капельной воронки, которая с помощью резиновой проб-;
кн вставлена в железную воронку с удлиненным «хвостиком», 2 л конц.
H2SO4. Воронка с удлиненной трубкой вставлена в запаянную железную
трубку: H2SO4 поступает в эту железную трубку, через открытый верхний
конец которой она вытекает и попадает в реакционный сосуд. Образующий-
ся в реакции HF весь удерживается конц. H2SO4.
Способ 3 [4]
SiO2 4- 4HSO3F + 2Н2О ----► SiF4 + 4H2SO4
60 400 36 104 392
Синтез проводят в стеклянной колбе на 250 мл, снабженной мешалкой,
капельной воронкой, газоотводной трубкой и обратным холодильником (не-
плотно набитым стеклянной ватой). К 24 г мелкогранулированного силикаге-
ля (37,5% Н2О) прибавляют по каплям в течение 45 мин 100 г фторосерной
кислоты. Реакционную смесь выдерживают 2 ч при 100 °C. К газоотводной
трубке присоединены последовательно три ловушки. В первой, охлаждаемой
.до —78 °C, конденсируется непрореагнровавшая фторосерная кислота, во вто-
рой и третьей (при —183 °C)—SiF4. Получают 21,8 SiF4, что соответствует
84 %-ному выходу.
Свойства. Бесцветный газ, очень гигроскопичный, образует на влажном
воздухе густой туман. /СУбл —95,7 °C; /пл —90,2 °C (при уменьшенном дав-
лении); dK 1,590 (—78°C); /КрИТ —15 °C; рцр„т 52 бар. Быстро разлагается
водой, почти не взаимодействует со смазкой кранов.
Хранят SiF4 в стеклянных колбах с краном, в газометрах над Hg или
конц. H2SO4 нли в стальных баллонах.
ЛИТЕРАТУРА
1. Lebouche L., Fischer IF., Biltz IF.,
Z. Anorg. Allgem. Chem., 207, 64
(1932).
-2. Ruff O., Ascher E., Z. Anorg.
Allgem. Chem., 196, 413 (1913).
3. Soil J., Naturforschung u. Medizin
in Deutschland 1939—1946 (FIAT-
Review), 23, 257.
4. Belf L., J. Chem. & Ind., 1955,
1296.
Трифторосилан S1HF3
(Силикохлороформ)
4SiHCl3 + 3TiF4 -► 4SiHF3 + 3TiCl4
541,76 371,7 344,28 569,19
SiHCl3 или TiF4 нагревают в автоклаве на масляной бане при 100—
200 °C в течение 8 ч. В крайнем случае реакцию проводят в запаянной труб-
ке. После охлаждения автоклава газы медленно выпускают и улавливают в
кварцевых или стеклянных ловушках, погруженных в жидкий воздух. За-
тем продукты фракционируют путем перегонки. В автоклаве остаются TiF<
и TiCU
Свойства. М 86,07. Бесцветный, горючий газ, образует с воздухом взры-
воопасную смесь. /Пл —ПО °C; /кип —80 °C. При комнатной температуре
медленно, а при нагревании до 400 °C быстро разлагается па Н2, Si и SiF4.
Водой гидролизуется. Разлагает спирт и эфир; восстанавливает конц. HNO3.
Соединения с кремнием 255
Рекомендуется хранить SiHF3 при температуре сухого льда или жидко-
го воздуха.
ЛИТЕРАТУРА
1. Ruff О., Albert С.. Вег., 38, 56 (1905).
Гексафторокремневая кислота H2[SiF6]
(Фторокремневая кислота, кремнефтороводородная кислота,
кремнеплавиковая кислота) .
6HF + SiO2 ----> H2[SiF6] + 2Н2О
120 60,06 144,03 36,03
Способ 1 [1]
В железный сосуд наливают 70—95%-ную плавиковую кислоту, к кото-
рой предварительно добавляют немного HztSiFe]. Порциями вносят квар-
цевую муку (99,9%) до тех пор, пока она не перестанет растворяться. При
необходимости реакционную смесь охлаждают льдом. Предварительное до-
бавление H2[SiF6] необходимо для спокойного начала реакции. По оконча-
нии превращения избыток кварцевой муки отделяют и декантируют 60—
70%-ную HatSiFe].
Другие способы: пропускание SiF< в воду [2]; взаимодействие конц.
H2SO4 с Ba[SiFe] [3].
Свойства. Бесцветная жидкость, в безводном состоянии при комнатной-
температуре почти на 50% распадается на SiF4 и HF. Перегонять без разло-
жения можно только 13%-ный водный раствор. Водные растворы при 17,5 °C
имеют следующую плотность: 1,049 (6%-ный); 1,173 (20%-ный); 1,314-
(34%-ный). Водный раствор H2SiF6 не действует на стекло.
Для хранения оказались подходящими железные кувшины. Высокопро-
центная гексафторокремневая кислота замерзает прн —19 °C, при этом вы-
деляется кристаллический тетрагидрат; перед выливанием нз кувшина она-
должна быть расплавлена при небольшом нагревании.
Применяется для получения фторосиликатов н SiF4.
ЛИТЕРАТУРА
1. Soil J., Naturforschung u. Medizin 2. Hempel W., Ber., 18, 1438 (1885).
in Deutschland 1939—1946 (FIAT- 3. Baur E., Glaessner A., Ber., 36, 4215-
Review), 23, 257. (1903).
Фторид германия(П) GeF2
GeF4 + Ge ----> 2GeF2
148,6 72,6 221,2
В качестве исходного вещества применяют преимущественно GeF4, кото-
рый получают из Ge и F2, разбавленного N2 (см. способ 2 получения GeF4).
По такой методике получают более чистый и сухой препарат GeF4, чем полу-
чаемый по другим методикам.
В горизонтально установленную кварцевую трубку, охлаждаемую с обе-
их концов и имеющую также с обеих концов ловушки для конденсации, по-
мешают мелкогранулированный Ge (4—5 мм в диаметре). Трубку нагревают
256 Глава 3. Фтор
до 150—300 °C и пропускают через нее несколько раз то в одном, то в 'дру-
гом направлении пары GeF4. Образующийся GeF2 конденсируется в реакци-
онной трубке на охлаждаемых концах. По окончании синтеза и удалении
сублимацией непрореагировавшего GeF« полученный GeF2 перегоняют в ва-
кууме н собирают в ампулу.
Свойства. М 110,6. Белое твердое вещество. /пл ПО °C. Может перего-
няться в вакууме прн 130 °C. Выше 160 °C разлагается с окрашиванием. Рас-
творяется в 48%-ной плавиковой кислоте. Обладает сильными восстанови-
тельными свойствами.
ЛИТЕРАТУРА
J. Bartlett N.. Yu К. С., Сап. J. Chem.,
39, 80 (1961).
Фторид германия(1У) GeF4
Способ 1 [1—3]
Ba[GeFe] ---* GeF4 -j- BaFa
- 323,96 148,60 175,36
В платиновой чашке растворяют GeO? в плавиковой кислоте и добавля-
ют BaCh; в осадок выпадает комплексная соль BafGeFe]. Зернистый осадок
промывают, сушат и нагревают в кварцевой трубке в потоке сухого азота.
Выделение GeF4 начинается при 500 °C и более интенсивно происходит прн
700 °C. Постепенно температуру увеличивают до 1000 °C. Выделяющиеся га-
зы поступают в охлаждаемую жидким воздухом кварцевую ловушку, где
‘GeF4 выделяется в виде твердого вещества. Продукт затем фракционируют в
кварцевом приборе. Прн этом первый погон представляет собой SiF4. Выход
•€7%.
Способ 2 [4]
Ge-}-2F2 ---*- GeF4
72,6 76 148,6
Синтез проводят в приборе, описанном в методике синтеза SF6. Герма-
ний обрабатывают фтором, разбавленным азотом. После воспламенения гер-
мания, которое начинается благодаря слабому нагреванию снаружи на не-
большом пламени газовой горелки, германий сгорает с сине-зеленым окраши-
ванием. Продукт собирается в газовой ловушке, охлаждаемой до —183 °C
жидким кислородом (илн каким-нибудь другим способом). Затем температу-
ру в ловушке поднимают до —90 °C, прн этом удаляются легколетучие при-
меси (например, SiF4). В заключение продукт фракционируют.
Свойства. Бесцветный газ. Вплоть до 1000 °C термически устойчив; силь-
но дымит на воздухе, имеет резкий чесночный запах, поражает дыхательные
пути н вызывает хрипоту. /пл —15°С; /Субл —36,5°С; dx 2,162 (0°С); dTB
3,148 (—195°C).
Хранят GeF4 в стеклянных колбах илн лучше под давлением в сжижен-
ном виде в кварцевых ампулах.
ЛИТЕРАТУРА
1. Dennis L. М., Laubengayer A. W.,
Z. Physik. Chem., 130, 520 (1927).
2. Dennis L. M„ Z. Anorg. Allgem.
Chem., 174, 119 (1928).
3. Le Boucher L„ Fischer W., Biltz W„
Z. Anorg. Allgem. Chem., 207, 65
(1932).
4. Bartlett N., Yu К. C„ Can. J.
Chem., 39,80 (1961).
Соединения с германием, оловом, свинцом 257
Гексафторогерманат калия KsfGeFe]
GeO2 + 6HF + 2КС1 ----* KjGeFJ + 2НС1 + 2Н2О
104,6 120,0 149,1 264,8 72,9 36,0
В платиновой чашке растворяют 2 части GeO2 в 12 частях 20%-ной HF
и добавляют концентрированный раствор 3 частей КС1. Жидкий раствор
превращается в студень, который прн перемешивании вновь разжижается, и
из него выпадают мельчайшие кристаллы, образуя густую кашицу. Кристал-
лы фильтруют, промывают сначала небольшим количеством воды, а в заклю-
чение спиртом и сушат. Вместо КС1 можно добавлять раствор КгСО3.
Свойства. Белый, очень гигроскопичный кристаллический порошок. При
перекристаллизации из воды образует пластинки. Решетка гексагональная.
/пл ~ 730 °C; /кип ~ 835 °C. При 18 °C в 184,4 г Н2О растворяется 1 г
Ka[GeF6], при 100 °C в 34,0 г Н2О — 1 г препарата.
ЛИТЕРАТУРА
1. Winkler Cl., J. Prakt. Chem., [2], 3. Krilss G„ Nilson O., Ber., 20, 1697
36, 199 (1887). (1887).
2. Muller J. H„ J. Amer. Chem. Soc.,
48, 1089 (1921).
Фторид олова(Н) SnF2
SnO + 2HF -----> SnF2 + H2O
143,70 40 156,70 18,01
В платиновой чашке растворяют SnO в 40%-ной плавиковой кислоте,
раствор выпаривают досуха без доступа воздуха.
SnF2 получают в виде хорошо образованных кристаллов следующим об-
разом. В полиэтиленовый стакан на 200 мл наливают <15—20 мл не содер-
жащей газов воды и всыпают 67,4 г (0,5 моль) SnO. Затем стакан, нагрева-
ют на паровой бане до 60 °C в атмосфере азота, свободного от О2, и мед-
ленно, по каплям, добавляют 46 г 48%-ной плавиковой кислоты (1,1 моль);
при этом содержимое стакана все время перемешивают, покачивая стакан.
С началом реакции происходит разогревание. Когда все растворится, стакан
ставят в эксикатор и охлаждают; выпадают кристаллы. Через 2 ч маточный
раствор декантируют во второй стакан. Оба стакана ставят в эксикатор над
СаС12 и КОН (1 :1). Через 2 сут их ставят в эксикатор над Mg(ClO4)2. Еще
через 4 сут маточный раствор еще раз декантируют и получают вторую пор-
цию кристаллов, которая высушена так же, как первая. Выход 86%.
Свойства. Бесцветные призмы; растворяются в воде, давая прозрачный
раствор. Решетка моноклинная. /пл 2104-215 °C.
ЛИТЕРАТУРА
1. Gay-Lussac I. L., Thenard L. J.,
Mem. Phys. Chim., 2, 317 (1809).
Фторид олова(1У) SnF4
SnCl4 + 4HF ----> SnF4 + 4HC1
260,53 80 194,7 145,84
2. Nebergdll H., Muhler J. C„ Day
H. G., J. Amer. Chem. Soc., 74,
1604 (1952).
17—143
258 Глава 3. Фтор
Также, как описано в методике получения TiF4, к двукратному по срав-
нению с расчетным количеству безводного фтороводорода добавляют по кап*
лям SnCl4. Прн этом образуется соединение SnCl4-SnF4. Реакционный сосуд
снабжают медным обратным холодильником (в качестве охладителя — ох-
лаждающая смесь) н нагревают с избытком HF, пока не прекратится выде-
ление НС1. С нисходящим холодильником отгоняют HF, затем остаток нагре-
вают до 130—220 °C, при это-м соединение SnCl4-SnF4 разлагается и SnCl4
отгоняется. Реакционный сосуд снабжают специальной насадкой, н SnF«
сублимируется при температуре красного каления. При этом нисходящую
часть насадки целесообразно покрыть влажной асбестовой бумагой. SnF4
тотчас переносят в закрытый сосуд из железа илн меди.
Свойства. Снежно-белая искристая кристаллическая масса; чрезвычайно
гигроскопичная. /ВОЗг 705 °C; d 4,78 (19 °C).
ЛИТЕРАТУРА
1. Ruff О., Plato W.. Вег., 37, 673 (1904).
Фторид свинца(П) PbF2
PbCO3-|-2HF -----> PbF2 + Н2О + СО2
267,21 40 245,21 18,01 44,0
В плавиковую кислоту, которая находится в свинцовой или платиновой
чашке, вносят порциями РЬСО3 (не содержащий нитратов и сульфатов). При
этом плавиковая кислота должна присутствовать в избытке. Затем реакци-
онную смесь нагревают в течение приблизительно одного дня до тех пор,
пока прекратится выделение СО2. Раствор, содержащий избыток кислоты,
сливают; остаток нагревают на электроплитке досуха. Для разложения кис-
лых фторидов свинца продукт быстро плавят, для чего платиновую чашку
с продуктом вносят на несколько минут в электрическую печь, предваритель-
но нагретую до красного каления.
Из-за низкой степени чистоты РЬО не подходит как исходное вещество
для синтеза PbF2, однако можно использовать РЬ(ОН)2.
Свойства. Белый кристаллический порошок. /пл 824 °C; /кип 1293 °C;
d 8,24. Растворимость в 100 г воды; 0,507 г (0°С), 0,065 г (20°C). В при-
сутствии HNO3 и нитратов растворимость увеличивается. PbF2 может суще-
ствовать в двух кристаллических модификациях: ромбической a-PbF2 (тип
РЬС12), которая выше 316 °C переходит в кубическую 0-PbF2 (тип плавико-
вого шпата).
ЛИТЕРАТУРА
1. Ruff О., Die Chemie des Fluors.
Springer, Berlin, 1920, S. 33.
Фторид свинца( IV) PbF4
Способ 1 [1]
PbF2 + F2 ----♦- Pl)F4
245,21 38 283,21
В приборе, описанном в методике получения BiFa, при 300°C фторируют
PbF2 в лодочке из спеченного глинозема. До подачн в прибор F2 разбавляют
Соединения с бором 259
диоксидом углерода или азотом и затем, медленно поднимая температуру до
550 °C, постепенно повышают содержание фтора в газовой смеси. Прн этом
pbF4 в виде белых длинных (.1—2 мм) кристаллов остается в лодочке н
только незначительное количество улетучивается.
По окончании фторирования PbF4 вместе с лодочкой вытаскивают на
подставленную стеклянную крышку и выскребают содержимое лодочки с по-
мощью никелевой проволоки. Стеклянную ампулу с кристаллами тотчас за-
паивают.
Способ 2 [2]
Pb + 2F2 ----► PbF4
207 76 283
Таким же образом, как описано в способе 2 получения BiFs, свинец из-
мельчают до опилок, просеивают и фторируют в реакторе в кипящем слое.
По окончании синтеза препарат освобождают от F2 н HF в вакууме.
Свойства. Белое кристаллическое вещество, очень чувствительное к вла-
ге, тотчас окрашивается на влажном воздухе в коричневый цвет (РЬО2).
Плавится при —600 °C. d 6,7. Кристаллизуется в тетрагональной сингонии.
ЛИТЕРАТУРА
1. V. Wartenberg Н., Z. Anorg. Allgem. 2. Roesky Н. W., Glemser 0., Hell-
Chem., 244, 339 (1940).
berg К.-H., Chem. Вег., 98, 2046
(1965).
Фторид бора BF3
Способ 1 [1]
KBF4 + 2BaO3 ---> BF3 + KF-B4Oe
125,92 139,28 67,82 197,38
В запаянной с одного конца, наклонно установленной железной трубке
(длина 40 см, диаметр 3 см) медленно нагревают до ~600 °C смесь нз 80 г
высушенного или лучше плавленного KBF4 и 30 г В2Оз. На железную трубку
надет фланец .(в качестве прокладки — медное кольцо), в отверстие которого
вставлена железная трубочка (шириной —10 мм); к трубочке присоединена
•осушительная трубка, наполненная стеклянной ватой для задержки пыли.
Далее поставлена газовая ловушка (из стекла или кварца), охлаждаемая
жидким воздухом. На выходе присоединена осушительная трубка со свеже-
обезвоженным KF. Получают 17 г BF3. Продукт для очистки несколько раз
перегоняют, разделяя на фракции.
Способ 2 [2, 3]
6NaBF4 + В2О3 + 6H2SO4
8BF3 -f- 6NaHSO4 4- ЗН2О
658,92 69,44 588,40 542,56 720,36 54,04
В литровой стеклянной колбе со шлифом (рис. 151) смешивают 300 г
N'aBF4, 50 г В20з и 300 мл конц. HaSO4 и реакционную смесь осторожно на-
гревают до начала выделения газов. Затем нагревание можно усилить. Вы-
деляющийся газ проходит через холодильник, абсорбционную трубку с В2Оз,
рыхло наполненную стеклянной ватой, н, наконец, конденсируется прн
—183 °C в газовой ловушке. Трубка с KF на выходе нз прибора служит для
защиты от влаги воздуха.
37*
260 Глава 3. Фтор
Преимущество данного способа состоит в том, что в колбе остается
растворимый в воде остаток, что облегчает очистку реакционного сосуда.
Более высокие выходы BF3 (80%) получают [3], если применяют
200%-ный избыток 105,9%-ной серной кислоты (олеум) и реакционную смесь
нагревают до 180 °C.
Рис. 151. Прибор для получе-
ния фторида бора.
Способ 3 [4]
Н3ВО3 + 3HSOsF ------> BF3 + ЗН^Од
61,84 300,22 67,82 294,24
Синтез проводят в железном сосуде, имеющем вверху два штуцера для
подачи жидкости и один для выпуска газа, а также один закрывающийся
штуцер внизу — для слива жидкости. В реакционный сосуд помещают конц.
H2SO4 и вводят при 85—435 °C 20—25%-ный раствор борной кислоты в конц.
H2SO4 и HSO3F. С умеренной скоростью выделяется BF3. Образовавшуюся
H2SO4 время от времени сливают и добавляют новые порции борной кис-
лоты.
Другие способы: термическое разложение фтороборатов диазония [5];
метод, в котором 40 г KBF4 с 8 г В2О3 и 120 мл конц. H2SO4 нагревают до
270 °C на песчаной бане [6]; метод [7], основанный на реакции
В2О3 + 6CeH6COF ----> 2BF3 + 3(С6Н5СО2)2О
Эта методика особенно подходит для получения BF3 в небольших количест-
вах; используют вакуумную аппаратуру. Исходные вещества удобны тем,
что нз них в любое время можно получить необходимое точно измеренное
количество BF3.
Старая методика синтеза BF3, исходя нз CaF2, считается теперь невы-
годной, так как дает низкие выходы и препарат сильно загрязняется SiF4.
Свойства. Бесцветный газ с удушливым запахом, дымит на влажном
воздухе, термически очень устойчив. /пл —128 °C; /кип —101 °C; /крнт
—12,25°C; р„рпт 50,7 бар; 1,769 (—128°C); rfTB 1,87 (—150°C). Гидроли-
зуется водой до Н3ВО3 и HBF4. В виде газа разъедает резину, поэтому при
Соединения с бором 2б1
проведении синтеза надо по возможности избегать резиновых пробок и шлан-
гов.
Хранят BF3 в стеклянных сосудах над ртутью или в стальных баллонах.
Получение препаратов BF3-2H2O, H[BF2(OH)2]r BF3-NH3 и BF3-O(C2H5)2
описано в гл. 13, посвященной соединениям бора.
ЛИТЕРАТУРА
1. Hellriegel W., Вег., 70, 689 (1937).
2. Booth Н. S„ Willson К. S.. J. Amer.
Chem, Soc., 57, 2273 (1935).
3. Рысс Г., Полякова Ю. М. — Ж.
общ. хим., 1949, т. 19 (81), с. 1596.
4. Пат. США, 2 416 133.
Фтороборная кислота H[BF4]
(Борофтороводородная кислота)
Н3ВО3 + 4HF ----> H[BF4] + ЗН2О
61,82 80,04 87,82 54,04
5. Balz G., Schiemann G., Ber., 60,
1186 (1927).
6.. Baumgarten P., Henning H., Ber.,
72, 1747 (1931).
7. Seel F„ Budenz R., Gruner K„ Lieb.
Ann. Chem., 684, 1 (1965).
В железный сосуд помещают Н3ВО3 (немного больше рассчитанного ко-
личества) и прн охлаждении льдом добавляют порциями 70—90%-ную пла-
виковую кислоту. Реакция сопровождается сильным выделением тепла. По
окончании превращения дают отстояться избыточной Н3ВО3 н путем декан-
тации получают чистую H[BFJ.
Свойства. Бесцветная жидкость. Прн комнатной температуре не дейст-
вует на стекло. При нагревании разлагается водой с образованием оксофто-
роборных кислот. Ядовита; даже при сильном разбавлении тормозит процес-
сы брожения. Хранят H[BF4] в стеклянных сосудах.
ЛИТЕРАТУРА
1. Mathers F. С., Stewart С. О., Нои- 2. Fichter F„ Thiele К., Z. Anorg.
setnann Н. V., Lee I. E., J. Amer. Allgem. Chem., 67, 302 (1910).
Chem. Soc., 37, 1516 (1915).
Тетрафтороборат натрия Na[BF4]
(Борофторид натрия)
2HSBOS + 8HF + Na2CO3 -----> 2Na[BF4] + 7H2O + CO2
123,64 160,08 105,99 219,63 126,1 44,0
В платиновой чашке при охлаждении льдом к 25 г 40 %-ной плавиковой
кислоты добавляют 6,2 г Н3ВО3. Реакционную смесь оставляют на 6 ч прн
комнатной температуре, затем вновь охлаждают льдом и добавляют 5,3 г
безводного Na2CO3. Раствор концентрируют до начала кристаллизации. Соль
можно перекристаллизовать затем из воды, при этом можно получить пре-
красные крупные кристаллы. Na[BF<] сушат в вакууме.
Свойства. М 109,815. Неокрашенная соль; кристаллизуется в безводном
состоянии в виде прозрачных прямоугольных призм, затупленных с концов.
Ромбические кристаллы, изоморфные Na[ClO4], Не разъедает стекло, если
оно абсолютно сухое. Легко растворяется в воде.
ЛИТЕРАТУРА
1. Balz G., Wilke-Dorfurt Е., Z. Anorg.
Allgem. Chem., 159, 197 (1927).
262 Глава 3. Фтор
Тетрафтороборат калия K[BF4]
(Борофторид калия)
Н3ВО3 + 4HF + КОН --* K[BF4J + 4H2O
61,82 80,04 56,11 125,92 72,05
К 25 г 40 %-ной плавиковой кислоты, находящейся в охлаждаемой льдом
платиновой чашке, добавляют 6,2 г Н3ВО3. Реакционную смесь оставляют па
6 ч при комнатной температуре, затем вновь охлаждают во льду и при по-
мешивании добавляют 5 и. КОН до перехода окраски метилоранжа. При
этом кристаллизуется K[BF<]. Маточный раствор декантируют, кристаллы
промывают водой и высушивают в вакууме. Выход 90%.
Свойства. Белая кристаллическая соль; не гигроскопична. tBn 530 °C;
d42C 2,505. Растворимость в 100 мл воды: 0,45 г (20°С), 6,3 г (100°С). Обра-
зует две кристаллические модификации: ромбическую бипирамндальную и ку-
бическую (точка перехода 276—280°C).
ЛИТЕРАТУРА
1. Vorlander D„ Hollatz J., Fischer J.,
Ber., 65, 535 (1932).
Гидроксотрифтороборат калия K[BF3OH]
2КНЕ, 4- H3BO3 --*- K[BF3OH] + KF 4- 2H,0
156,22 61,84 123,96 58,1 36
В полиэтиленовом стакане растворяют в 250 мл воды 100 г KHF2. Через
несколько часов отделяют фильтрованием KafSiFe] и нерастворявшийся
KHF2; прозрачный раствор охлаждают в ледяной воде и прибавляют при пе-
ремешивании 40 г порошка борной кислоты, которая быстро растворяется.
Через час выпадают мелкие кристаллы. Их отсасывают через стеклянный
фильтр, промывают небольшим количеством ледяной воды, затем 95%-ным
метаиолом и ацетоном. Соль высушивают при 120 °C.
Свойства. Плавится без разложения. Растворяется в воде хуже, чем
K[BF<]. Не осаждается ацетатом нитрония; легче гидролизуется раствором
КОН, чем K[BF<]. Не удается перекристаллизовать из воды без разложения.
ЛИТЕРАТУРА
1. Wamser С. A., J. Amer. Chem. Soc.,
60, 1209 (1948).
Тетрафтороборат нитрозила NO[BF4]
(Нитрозилборофторид)
2H[BF4] + NSO3 --> 2NO[BF4] + H2O
175,65 76,01 233,65 18,01
В высококонцентрированный раствор фтороборной кислоты (см. синтез
H[BF«]) в платиновой чашке пропускают поток сухого N2O3 (получают из
концентрированной азотной кислоты и As2O3) до тех пор, пока содержимое
платиновой чашки полностью' не застынет в густую кашицу, a N2O3 больше
Соединения с алюминием ,галлием, индием, таллием 263
не поглощается. Выделившиеся кристаллики отделяют при отсасывании через
платиновый тигель с фильтрующим диом и отжимают. Маточный раствор в
платиновой чашке концентрируют до появления густого белого дыма; в полу-
ченный раствор пропускают еще раз N2O3. Таким способом получают допол-
нительные количества продукта.
Отделенные кристаллы NO [BF<] • Н2О сушат в течение 2 сут над Р4Ою
в вакууме. Затем переносят в склянку для работы под давлением и обраба-
тывают при —15 °C жидким N2O3. Через несколько суток склянку открыва-
ют при этом избыток N2O3 улетучивается. Наконец; препарат оставляют на
несколько суток в вакууме над Р4Ою и СаО.
Для получения высокочистого NO[BF4] продукт сублимируют в вакууме
при 0,01 мм рт. ст. Сублимацию удобно проводить в закрытой с одного кон-
ца трубке из йенского стекла, в которую введен водяной холодильник. Сбоку
имеется штуцер, ведущий к ртутному насосу. Нагревание до 200—250 °C осу-
ществляют с помощью парафиновой бани. На холодильнике осаждается
NO[BF4] в виде неокрашенной твердой кристаллической корки, которую со-
скабливают.
Свойства М 116,83. Бесцветные, двоякопреломляющие, гигроскопичные
листочки. d425 2,184. Кристаллизуется в ромбической сингонии. Водой разла-
гаются с выделением оксидов азота. Если соединение ие содержит влаги, то
оно не разъедает стекла. Можно хранить в газовых баллонах.
ЛИТЕРАТУРА
1. Wilke-Dorfurt Е„ Balz G., Z. Anorg.
Allgem. Chem., 159, 219 (1927).
Фторид алюминия AIF3
(NH4)s[AlFeJ ---* A1F3 + 3NH4F
195,09 83,97 111,12
2. Balz G„ Mailander E„ Z. Anorg.
Allgem. Chem., 217, 162 (1934).
(NH4)3[A1F6] в платиновой лодочке нагревают в потоке N2 при темпера-
туре красного каления до постоянной массы.
Обезвоживанием A1F3-3H2O можно получить фторид алюминия, загряз-
ненный оксид-фторидами.
Свойства. Белый порошок. Плавится выше 1260 °C; /воаг 1260 °C; d 2,882.
Кристаллизуется в гексагональной сингонии. Слаборастворим в воде (0,559 г
в 100 мл воды при 25 СС), кислотах и щелочах. Устойчив даже при обработ-
ке конц. H2SO4. Гидролизуется водяным паром при 300—400 °C.
ЛИТЕРАТУРА
1. Blitz W., Rahlfs Е., Z. Anorg.
Allgem. Chem., 166, 370 (1927).
Тригидрат фторида алюминия A1F3«3H2O
Al + 3HF-f-3H2O -----> AlF3-3H2O + 3/2H2
27,0 60,0 54,0 138,0 3,0
Кусочки алюминиевой фольги добавляют постепенно к 15%-иой плавико-
вой кислоте в платиновой чашке. Путем кратковременного многократного по-
гружения платиновой чашки в ледяную воду поддерживают температуру ре-
акционной смеси <25 °C. Через некоторое время идущая вначале очень бур-
264 Глава 3. Фтор
но реакция становится при дальнейшем прибавления алюминия практически
незаметной. Реакционную смесь фильтруют через пластмассовую воронку в
пластмассовую чашку, к фильтрату добавляют еще один кусочек алюминие-
вой фольги и ставят для кристаллизации в лед. Через 24 ч кристаллизация
закончена. Кристаллы промывают небольшим количеством воды и сушат при
комнатной температуре на глиняной пластинке.
Если при О °C не происходит кристаллизации A1F3-3H2O, то раствор упа-
ривают на водяной бане до ноявления первых кристаллов; при этом получа-
ют вторую модификацию A1Fs-3H2O, которая отличается от выше. описанной
растворимостью в воде и рентгенограммами.
Свойства. Белое кристаллическое вещество. Высушивание происходит
медленно. При .нагревании на водяной бане может .отщепить две молекулы
гидратной воды и перейти в моногидрат.
ЛИТЕРАТУРА
1. Ehret W. F., Frere F. J., J. Amer.
Chem. Soc., 67, 64 (1945).
2. Fischer W„ Bock E., Z. Anorg.
Allgem. Chem., 262, 54 (1950).
Гексафтороалюминат аммония [NH4)3[A1F6]
(Криолит аммония)
6NH4F+A1(OH)3 ---*• (NH4)3[A1FcJ + 3NH4OH
222,24 77,97 195,06 105,15
В горячий достаточно концентрированный раствор NH4F добавляют пор-
циями свежеосажденный А1(ОН)3. Выпадает желатинообразный осадок, ко-
торый при нагревании легко отделяется от раствора. Осадок декантируют или
фильтруют с отсасыванием, промывают водно-спиртовой смесью и высушива-
ют при 105 °C.
Свойства. Белый мелкокристаллический порошок. Термически устойчив
при температурах >100 °C. Растворимость в 1 л воды: 4 г (0°С), 7,7 г
(25°C). Не разъедает стекла.
ЛИТЕРАТУРА
I. v. Helmolt И., Z. Anorg. Allgem. 2. Petersen Е., J. Prakt. Chem., (2),
Chem., 3, 127 (1893). 40, 55 (1889).
Тетрафтороалюминат аммония NH4[A1F4]
Способ 1
(Nll4)3[AlFeJ -> NH4[A1F4] + 2NH4F
195,1 121,0 74,1
Термическое разложение (NH4)3[AlFe] до A1F3 и NH4F при определенных
условиях проходит через стадию образования NH4[A1F<].
Исходный препарат в никелевой или медной лодочке помещают в труб-
ку из кварца или стекла пирекс, пропускают . сухой N2 и нагревают в печи
до 300 °C. Возгоняющийся NH4F собирается в холодной части реакционной
трубки и в присоединенном к. ней приемнике. В лодочке остается чистый
NH4[A1F4], При повышении температуры >350 °C происходит дальнейшее
разложение до A1F3. При этом надо совершенно исключить попадание влаги.
Соединения с алюминием, галлием, индием, таллием 265
Способ 2. NH4[A1F4] можно получить также «мокрым» методом: путём
осаждения из концентрированного раствора A1F3, подкисленного HF, при
пропускании аммиака.
Свойства. Кристаллизуется в тетрагональной сингонии.
ЛИТЕРАТУРА
1. Thilo Е„ Naturwiss., 26, 529 (1938).
Фторид галлия(Ш) GaF3
Способ 1 [1]
(NH4)3[GaF6] ---> GaF34-3NH4F
237,84 126,72 111,12
2. Brosset С., Z. Anorg. Allgem.
Chem., 239, 301 (1938).
(NH4)3[GaFe] в лодочке из спеченного глинозема нагревают несколько
часов при 400 °C в никелевой трубке, пропуская F2.
Путем обезвоживания GaF3-3H2O в вакууме в потоке F2 также можно
получить GaFs, не содержащий оксофторидов.
Способ 2 [2]
Ga + 3HF --------► GaF3 + »/2H2
69,7 60,0 126,7 3
Никелевую лодочку с металлическим галлием помещают в никелевую
трубку и нагревают в потоке HF до 550 °C. HF можно разбавить азотом.
Через несколько часов галлий покрывается коркой из GaF3; в лодочке также
находится немного кристаллического GaF3.
Свойства. Бесцветное вещество, /пл >1000 °C; /КИп 950 °C. Не разлагается
в холодной и горячей воде. В воде GaF3 (в отличие от GaF3-3H2O) раство-
рим очень слабо. В 100 мл горячей соляной кислоты растворяется 0,0028 г
препарата. При нагревании в потоке N2 выше 800 °C сублимируется не разла-
гаясь.
ЛИТЕРАТУРА
1. Hannebohn О., Klemm W., Z. Anorg. 2. Brewer F. M.. Garton G., Goodga-
Allgem. Chem., 229, 342 (1936). me D. M. L„ J. Inorg. Nucl. Chem.,
9, 56 (1959).
Гексафторогаллат аммония (NH4]3[GaF6]
Г ~ Ga(OH)3 -f- 3HF 4- 3NH4F -> (NH4)3[GaF6] + 3H2O
[120,74 ; 60,03 111,12 237,84 54,05
В платиновой чашке растворяют 2 г Ga(OH)3 в 40 %-ной плавиковой кис-
лоте и выпаривают почти досуха. Добавляют по возможности насыщенный
на холоду раствор, содержащий 6 г NH4F. (NH4)3GaF6 тотчас выделяется
в виде хорошо образованных кристаллов.
Свойства. Неокрашенная, кристаллизирующаяся в виде октаэдров соль.
При нагревании на воздухе переходит в Ga2O3; при нагревании в вакууме
при 220°C образуется GaN (в несколько стадий).
ЛИТЕРАТУРА J
1. Hannebohn О., Klenlm W., Z. Anorg.
Allgem. Chem., 229, 341 (1936).-
266 Глава 3. Фтор
Фторид индия(111] InF3
Способ 1
(NH4)3[lnFJ -----> lnF3-|-3NH4F
282,88 171,76 111,12
(NFUJalnFe в корундовой лодочке помещают в пикечевую трубку и на-
гревают в потоке F2 до постоянной массы.
Способ 2
21n„O34-6F2 ----->- 4InF34-3O,
555,04 228 687,04 96
1п2О3 в лодочке из спеченного глинозема фторируют в кварцевой трубке
(прибор такой же, как в методике получения T1F3). После небольшого подо-
гревания реакция не требует в дальнейшем подвода тепла (иногда даже про-
исходит раскаливание). Далее желтый оксид обесцвечивается и продукт за-
полняет весь объем. Для получения препарата, не содержащего оксофторидов,
продукт выдерживают несколько часов в никелевой трубке в потоке F2 прн
температуре по меньшей мере 500 °C.
Способ 3
InCl3 + 3HF -----> ЗНС1 + InF3
221,2 69,0 109,4 171,8
Не содержащий воды 1пС13 в платиновой лодочке нагревают в устойчивой
к HF металлической трубке в потоке HF при 100 °C. По истечении несколь-
ких часов 1пС13 превращается в lnF3. Выход количественный.
Свойства. Неокрашенное вещество. /пл 1170 °C; 4ип>1200°С; d 4,39.
Не разлагается холодной и горячей водой. В отличие от InF3-3H2O очень
слабо растворимо в воде (0.040 г/100 мл при комнатной температуре). Лег-
ко растворимо в разбавленных кислотах. В очень медленном потоке Н2 при
300 °C восстанавливается до (почти чистого) InF2; при интенсивном потоке
Н2 восстановление идет до металла.
ЛИТЕРАТУРА
1. Hannebohn О., Klemm W., Z. Anorg. 2. Roberts J. E., Laubengayer A. W.,
Allgem. Chem., 229, 342 (1936). J. Amer. Chem. Soc., 79, 5895
(1957).
Гексафтороин дат аммония |NH4]3[InF6]
In(OH)3 + 3HF + 3NH4F -----> (NH4)3[InFe] 4-3H2O
165,78 60,63 111,12 282,88 54,03
В платиновой чашке растворяют 2 г 1п(ОН)3 в 40%-ной плавиковой кис-
лоте и выпаривают почти досуха. Добавляют по возможности небольшое ко-
личество воды, приливают насыщенный на холоду раствор 6 г NH4F и кон-
центрируют раствор до начала кристаллизации.
Свойства. Неокрашенное, кристаллизирующееся в виде октаэдров соеди-
нение; разлагается при нагревании в вакууме с образованием InN.
Соединения с алюминием, галлием, индием, таллием 267
ЛИТЕРАТУРА
1. Hannebohn О.,
Anorg. Allgem.
(1936).
Klemm W„ Z.
Chem., 229, 342
Фторид таллия[1] T1F
T12CO3 + 2HF -----> 2T1F + CO2 + H20
468,79 40 446,78 44,0 18,01
В платиновой чашке растворяют Т12СО3 в избытке 40%-ной плавиковой
кислоты и дважды выпаривают досуха. Затем продукт в платиновом тигле
доводят до плавления.
Свойства. М 223,39. Твердые блестящие белые кристаллы. Структура ром-
бическая (деформированная решетка каменной соли). /пл 327 °C; /кип 655 °C;
dtQ0 8,36. В расплаве имеет желтоватую окраску. Негигроскопично, однако
расплывается, если на него подышать, но сразу вновь застывает. Раствори-
мость в воде при 20 °C: 78,8 г на 21,2 г Н2О. Концентрированный водный
раствор имеет сильнощелочную реакцию. Немного растворим в спирте.
Применяется при получении фторсодержащих эфиров.
ЛИТЕРАТУРА
1. Ketelaar I. A. A., Z. Kristallogr., 2. Hayeck Е., Z. Anorg. Allgem.
92, 30 (1935). Chem., 225, 47 (1935).
Фторид таллия(Ш) T1F3
2TI2O34-6F2 -----> 4T1F3 + 3O2
913,56 228 1045,56 96
В кварцевую трубку (рис. 152)* помещают кварцевую лодочку с Т12О3 н
фторируют. F2 вводят через трехметровый медный капилляр, который до-
Рис. 152. Установка для получения фторида таллия(Ш).
пускает поворот реакционной трубки на 90°. Реакция происходит уже при
комнатной температуре. При этом шоколадно-коричневый Т12О3 меняет свой
цвет на черный, а затем на коричнево-красный. Наконец, препарат становится
* Этот прибор подходит для любых синтезов, где необходимо фториро-
вание свободным фтором, если продукты представляют собой нелетучие фто-
риды (CuF2, AgF2 CeF<, CoF3, GaF3, InF3).
268 Глава 3. Фтор
совершенно белым. Фторирование надо проводить очень медленно, так как
иначе продукт оплавляется, образуя желтоватую массу, и фторирование не
проходит до конца. В конце синтеза температуру повышают до 300 °C.
Как только реакция закончена, удаляют осушительную трубку и при-
соединяют Т-образное приспособление из кварца с ампулой (см. рис. 152).
Реакционную трубку наклоняют на 90° и стряхивают препарат в потоке F2
в кварцевую ампулу. В запаянной кварцевой ампуле TlFj можно хранить без
разложения достаточно длительное время.
Свойства. М 261,39. Белое вещество. /Пл 550 °C; /к™>550°С; d425 8,36.
«Очень чувствительно к влаге, тотчас реагирует с водой с образованием чер-
ного осадка. При нагревании на воздухе претерпевает разложение; однако
может плавиться в атмосфере фтора.
ЛИТЕРАТУРА
1. Hannebohn О., Klemm. W., Z. Anorg.
Allgem. Chem.,' 229, 343 (1936).
Фторид бериллия[11| Bel 2
Способ 1 [1]
(NH4)2[BeF4J ---> BeF2 + 2NH4F
121,10 47,02 74,08
(NH4)2[BeF4] нагревают в платиновой лодочке при температуре красного
каления, по возможности предохраняя от влаги воздуха; при этом возгоня-
ется NH4F. BeF2 остается в виде стеклообразной полупрозрачной массы.
Способ 2 [2]
BeO + 2HF ------> BeF2 + H2O
25 40 47 18
Кальцинированный ВеО в приборе из никеля обрабатывают при 220 °C в
течение 1 ч безводным HF (скорость подачи HF 15 мл/мин). Степень превра-
щения 98,4%.
Свойства. Неокрашенное, очень гигроскопичное вещество, /пл 800 °C
(в зависимости от сорта стекла плавится при средних температурах его раз-
мягчения). d425 1,986. Кристаллизуется в тетрагональной сингонии. Растворя-
ется в воде в любых соотношениях; нерастворимо в безводном HF; немного
растворимо в абсолютном спирте; достаточно хорошо растворимо в смеси
спирта и эфира. BeF2 заметно летуч при 800 °C.
ЛИТЕРАТУРА
1. Lebeau Р.. С. R. Acad. Sci., Paris, 2. Hyde К. R-. O’Connor D. J., Wal-
126, 1418 (1898). te E.. J. Inorg. Nucl. Chem., 6, 14
(1958).
Тетрафторобериллат аммония [NH4)2[BeF4]
4NH4F-|-Be(OH)2 ---> (NH4)2[BeF4] + 2NH4OH
148,06 43,14 121,10 70,10
В горячий раствор NH4F порциями вносят Ве(ОН)2. При выпаривании и
охлаждении близкого к насыщенному прозрачного раствора очень быстро
Соединения с бериллием и щелочноземельными металлами 269
выпадают мелкие бесцветные кристаллы в форме игл или призм. Их отфиль-
тровывают с отсасыванием и промывают небольшим количеством спирта; су-
шат при 105 °C.
Свойства. Бесцветные кристаллы. При нагревании растрескиваются, за-
тем плавятся и выделяют NH4F. Кристаллизуются в ромбической бипирами-
дальиой сингонии.
ЛИТЕРАТУРА
1. V. Helmolt И., Z. Anorg. Allgem.
Chem., 3. 129 (1893).
Фториды щелочноземельных металлов MgF2, CaF2, SrF2, BaF2
MgCO3 + 2HF - 84,3 40 > MgF2 4" CO2 + H2O 62,3 44 18
CaCO3 + 2HF - 100,1' 40 —> CaF2 + CO2 + H2O 78,1 44 1 8
SrCO34-2HF — 147,6 40 —SrF2 + CO2 + H2O 125,6 44 18
BaCO3 + 2HF - 197,3 40 —* BaF2 + CO2 + HaO 175,3 44 18
В платиновой чашке растворяют 3 г соответствующего карбоната щелоч-
ноземельного металла в горячей разбавленной уксусной кислоте и добавляют
затем 40%-ную плавиковую кислоту. (Растворение именно в уксусной кисло-
те, а не в соляной, имеет весьма существенное значение для получения чисто-
го фторида.) Раствор трижды упаривают и каждый раз вновь добавляют пла-
виковую кислоту. Затем еще раз добавляют 40%-ную плавиковую кислоту,
а также 3—5 г NH4F или NH4F-HF. Раствор конденсируют до появления бе-
лых паров NH4F и охлаждают. Твердый осадок разбивают и переносят в
платиновую лодочку, которую помещают в реакционную трубку, нагреваемую
в трубчатой печи (см. разд. «Трифториды редкоземельных элементов» прибор
для получения трифторидов редкоземельных элементов). Для удаления лету-
чих HF, Н2О, NH4F продукт медленно нагревают до 400 °C в потоке N2 вы-
сокой чистоты. Для получения хорошо образованных кристаллов нагревают
еще 2—3 сут при 700 °C в потоке инертного газа.
Свойства. Белые соли. Характеристики приведены ниже: Растворимост ь в воде при 18 °C, г/л J
СОЛЬ 1 ип кристалличе- ской решетки d
MgF2 Рутил CaF2 Флюорит SrF2 Флюорит BaF2 Флюорит 1290 1480 1400 1320 3,15 3,18 4,34 4,89 0,087 0,015 0,117 1,6
Эти соли можно хранить в стеклянной посуде на воздухе.
ЛИТЕРАТУРА
1. Grets О., Chem. Laborator. Univers.
Freiburg, 1972, неопубликованные
данные.
270 Глава 3. Фтор
Фторид калия KF *
KF достаточной степени чистоты имеется в продаже. Тонкодисперсный1
так называемый активированный фторид калия, чрезвычайно подходящий для
фторирования многих соединений, получают путем термического разложения
фторосульфоната калия KSO2F при 150 °C в вакууме. «Активированный KF»
имеет насыпной вес 170 мл/моль и средний размер частицы 4 мкм (см. пре-
парат KSO2F). I
ЛИТЕРАТУРА
1. Seel F„ Gorlitz Н. D., Z. Anorg.
Allgem. Chem., 327, 32 (1964).
Тетрафторохлорат[Н1) калия K[C1F4]
(Хлорид-тетрафторид калия)
KF + C1F3 ---*- K[C1F4]
58 92,5 150,5
KF помешают в никелевый автоклав, присоединенный к вакуумной уста-
новке (из стекла пирекс или кварца), и вводят путем перегонки избыток
CIF3. Реактор отсоединяют от вакуумной установки и нагревают в течение
1 ч при ~ 100 °C при периодическом покачивании автоклава. По охлаждении
реактор вновь присоединяют к вакуумной установке и отгоняют при комнат-
ной температуре в вакууме избыток CIF3. Если автоклав сохраняет постоян-
ную массу, то это значит, что удалены все летучие компоненты. Автоклав
можно заполнить сухим азотом, открыть крышку и вынуть препарат.
Свойства. Белое, твердое вещество; d 2,58. Бурно реагирует с водой,
взрывоподобно с органическими растворителями.
Хранят K[C1F4] в атмосфере сухого азота.
ЛИТЕРАТУРА
1. Whitney Е. D„ McLaren R. О., Fol-
ge С. F„ Hurley Т. J., J. Amer.
Chem. Soc., 86, 2583 (1964).
Тетрафторобромат[Н1] калия K[BrF4]
(Бромид-тетрафторид калия)
3KCl+4BrF3 ----> 3K[BrF4l + 1/2Br2+3/2С12
223,68 547,64 585,0 79,91 106,41
К ~0,5 г КС1 в кварцевом сосуде медленно по каплям добавляют боль-
шой избыток BrF3. Нагревают около 2 мин до 120 °C и вновь охлаждают.
Затем кварцевый сосуд соединяют с кварцевой ловушкой, охлаждаемой
жидким воздухом, и с вакуумным насосом и отгоняют избыток BrF3 в ваку-
уме в кварцевую ловушку.
Свойства. Белый, кристаллический порошок. Разлагается при нагревании
с отщеплением BrF3. Быстро реагирует с водой с разложением, одиако менее
Соединения со щелочными металлами 271
бурио, чем BrF3. Не взаимодействует с СС14 ацетоном и диоксаном. При на-
гревании действует на платину.
ЛИТЕРАТУРА
1, Sharpe A. G.. Emeleus Н. 1., J.
Chem. Soc. (London), 1948, 2135.
Гексафторобромат(У) калия K[BrF6j
(Бромид-гексафторид калия)
KF4-BrFs -----> K[BrFe]
58 175 233
Синтез проводится ио той же методике, что и получение K[C1F4], только
исходным веществом является KF. Дальнейшую переработку продукта про-
изводят аналогичным образом.
Свойства. Белое вещество, бурно реагирует с водой и взрывоподобпо е
органическими растворителями.
ЛИТЕРАТУРА
1. Whitney Е. D„ McLaren R. О., Fol-
ge С. F., Hurley Т. J., J. Amer.
Chem. Soc., 86, 2583 (1964).
Тетрафтороиодат(Ш) цезия Cs[1F4]
(Иодид-тетрафторид цезия)
CsF -J- IF3 --* Cs[IF4]
151,91 183,91 335,82
Реакцию проводят при —40 °C в ловушке, соединенной с осушительной
трубкой. В ловушку помещают взвешенные количества сухого ацетонитрила
и IF3. К раствору добавляют сухой CsF в количестве ~90% от стехиометри-
ческого. Суспензию при —40 °C почти целый день хорошо перемешивают с
помощью магнитной мешалки, а затем температуру медленно поднимают до
комнатной (это осуществляют с помощью бани с холодной водой). Образу-
ется белый CsIF<, который отделяют фильтрованием на специальной воронке
(без доступа воздуха), промывают ацетонитрилом и сушат в вакууме. Выход
количественный в расчете на введенный CsF.
Свойства. Белое вещество, очень чувствительное к гидролизу. Разлагает-
ся выше 120 °C.
ЛИТЕРАТУРА
1. Schmeisser М., Ludovici W„ Nau-
mann D., Sartori P„ Scharf E.,
Chem. Ber., 101, 4214 (1968).
Соли калия и рубидия получают аналогично Cs[1F4],
Тетрафтороиодат(Ш) калия K[1F4]. М 242,0. Белое вещество, очень чув-
ствительное к гидролизу. Термически разлагается выше 20 °C.
272 Глава 3. Фтор
Тетрафтороиодат(Ш) рубидия Rb[IF<]. М 288.4. Белое вещество, очецВ
чувствительное к гидролизу. Разлагается выше 70 °C. ' <
Гексафтороиодат(У) калия K[.IF6] 1
(Иодид-гексафторид калия) .. i
KF + IFS -------> K[IF6]
58,11 174,91 233,02
В кипящем фториде иода(У), находящемся в кварцевом сосуде, раство-
ряют фторид калия. В 100 г IFs растворяется 1 г KF. При охлаждении вы-
деляется K[IF6] в виде белых кристаллов. Избыточный фторид иода(У) уда-
ляют путем выпаривания при 15—20 °C и 2—5 мм рт. ст.
Свойства. Белые кристаллы. 1ПЛ —200 °C. Немного растворяется в холод-
ном, лучше в горячем фториде иода(У). При нагревании до 200 °C разлагает- ,
ся. Гидролизуется водой (экзотермическая реакция); не взаимодействует
с ССЦ. 1
I
3
ЛИТЕРАТУРА
j
1. Emeleus Н. J., Sharpe A. G., J.
Chem. Soc. (London), 1949, 2206. I
Фториды рубидия и цезия RbF, CsF
RbaCO3-|-2HF -----> 2RbF 4-СО2 4-H2O
230,9 40 208,9 44 18 j
Cs2CO3 + 2HF ----->• 2CsF + CO2H2O |
325,8 40 303,8 44 18 |
В платиновой чашке в небольшом количестве горячей воды растворяют 5
~3 г соответствующего карбоната щелочного металла и осторожно прибав- ]
ляют 40%-ную плавиковую кислоту. Раствор трижды упаривают, каждый -
раз добавляя плавиковую кислоту. Затем еще раз добавляют 40%-ную пла-
виковую кислоту и 3—5 г NH4F или NH4F-HF. Расплавленный продукт ре-
акции переносят в платиновую лодочку, которую помещают в трубку, нагре-
ваемую в трубчатой печи (оборудование и приборы см. в разд. «Трифториды
редкоземельных элементов»). Летучие компоненты (HF, Н2О, NH4F) удаляют
нагреванием до 400 °C в потоке азота высокой чистоты. Через 48 ч нагрева-
ние прекращают и прибор охлаждают. Для хранения кристаллический, но
очень гигроскопичный фторид щелочного металла переводят в потоке инерт-
ного газа в трубку Шленка.
Свойства. Белые соли. Кристаллическая решетка Типа NaCl. Характери-
стики приведены ниже:
Соль AI 'пл- °c 'кип’ 'c d
RbF 104,47 775 1410 3,56
CsF 151,9 682 1251 4,12
ЛИТЕРАТУРА
1. Greis О., Chem. Labor. Univers.
Freiburg, 1972, неопубликованные
Данные'
2. Ruff О., Schmidt G., Mugdan S„
Z. Anorg. Allgem. Chem., 123, 84
(1922).
3. Lannung A., Z. Phys. Chem. (A),
161,260 (1932).
Соединения с медью, серебром, золотом 27&
Фторид меди(Н] CuF2
Способ 1
CuCl2 F2 ------*- CuF2 + C1?
134,48 38 101,57 70,$2
, На обезвоженный СиС12 в медной лодочке (в приборе, описанном в ме-
тодике получения T1F3, рис. 152) действуют F2 или C1F3 при 400 °C.
Способ 2
CuO-f-2HF -----* CuF2 + Н2О
i‘ 79,57 40 101,57 18,01
В платиновой чашке растворяют СиО в избытке 40%-ной плавиковой»
кислоты, так что образуется CuF2-5H2O-5HF. Затем эту соль переносят в пла-
5 типовую лодочку, которую помещают в медную или никелевую трубку; обез-
воживание проводят в потоке совершенно сухого HF при 400 °C (см. рис..
I 152). Избыток HF вытесняют потоком N2 и затем прибор охлаждают.
: Если получен ие совершенно безводный CuF2, то прн нагревании он слег-
4 ка окрашивается в серый цвет.
Свойства. Белый, кристаллический, неустойчивый на воздухе порошок-
7Пл 950 °C; d<25 4,23. A/Ass —542 кДж/моль. Немного растворяется в холод-
ной воде; гидролитически разлагается горячей водой. При 20 °C в 100 мл во-
ды растворяется 4,7 г.
ЛИТЕРАТУРА
1. Henkel Р., Klemm W„ Z. Anorg. 2. о. Wartenberg W., Z. Anorg;.
Allgem. Chem., 222, 74 (1935). Allgem. Chem., 241, 381 (1939).
Фторид дисеребра Ag2F
Способ 1 [1]
Ag 4- AgF ------> Ag2F
107,87 126,87 234,74
В раствор Ag2CO3 в 47 %-ной плавиковой кислоте, взятой в количестве,-,
строго необходимом для растворения, добавляют свежеосажденный AgF, по-
лученный путем восстановления раствора AgNO3 в метаформальдегиде. Ре-
акционную смесь осторожно концентрируют на водяной бане до тех пор, пока-
над осадком еще остается жидкость. Затем добавляют новую порцию раство-
ра AgF и также осторожно упаривают. После декантации осадок несколько-
раз промывают абсолютным спиртом и сушат в вакууме при комнатной тем-
пературе. Выход 225 г (96%).
Другие способы получения. Электрохимический метод [2, 3]. Катодное-
восстановление при 60 °C конц. водного раствора AgF при низких плотностях
тока (при более высокой плотности тока образуется Ag).
Свойства. Медно-желтые кристаллы с зеленоватым оттенком; при месяч-
ном (или более) облучении светом их поверхность становится серо-черной.
Решетка гексагональная, d 8,64. A№2SB —210,9 кДж/моль. Водой сразу раз-
лагается на AgF и Ag. Нерастворим в органических растворителях. При на-
гревании в потоке N2 или СО2 до 90—115 °C разлагается, образуя серую мас-
су, состоящую из AgF и Ag.
18—143
274 Глава 3. Фтор
ЛИТЕРАТУРА
1. Payer L., Fischer М., Harrison Н.,
Bryant В. Е., Inorg. Synth., 5, 18
(1957).
2. Hettich A., Z. Anorg. Allgem. Chem.,
167, 67 (1927).
3. Scholder R.. Traulsen K., Z. Anorg.
Allgem. Chem., 197, 56 (1931).
Фторид серебра(1] AgF
Способ 1 [11
Ag2CO3 + 2HF ------> 2AgF + CO2 + H2O
275,77 40 253,76 44,0 18,01
Для получения AgF в виде чистого безводного препарата растворяют
Ag2CO3 (в избытке) в 40%-ной плавиковой кислоте; раствор фильтруют и
упаривают при помешивании до кристаллизации. Оставшуюся воду удаляют,
.добавляя три раза абсолютный метанол с последующей его декантацией.
В заключение таким же образом продукт промывают абсолютным эфиром.
Продукт, окрашенный в цвет от желтого до светло-коричневого, сушат при
‘60—70 °C. Из-за чувствительности AgF к свету надо избегать прямого осве-
.щения.
Способ 2 [2J
2Ag + 2HF4-H2O2 ------> 2AgF + 2H2O
216 40 34 254 36
Очень чистый AgF получают при обработке порошка Ag (полученного
при быстром смешивании при 60 °C конц. раствора AgNO3 и раствора форми-
ата аммония) 25,3%-ной водной плавиковой кислотой (в одно- или полуто-
ракратном избытке). В течение 0,5 ч при интенсивном перемешивании добав-
ляют по каплям 6%-ный Н2О2 (в одно- или полуторакратном избытке), при
этом в большинстве случаев образуется буроватый осадок. Реакционную
•смесь нагревают в течение 1 ч на водяной бане до тех пор, пока прекратит-
ся выделение газа. Затем бурый осадок отфильтровывают и концентрируют
«фильтрат досуха.
Другие способы
3. Для получения чистого продукта достаточно полученный, согласно
описанной выше методике, сырой продукт перекристаллизовать из воды, для
дегидратации добавить избыток бензола и затем медленно перегнать; при
этом вода удаляется с азеотропной смесью. Коричневый порошок сушат в ва-
кууме при 140 °C [3].
4. Термическое разложение Ag[BrF<] при 200 °C [4].
5. Сухим путем: над Ag2CO3 в платиновой трубке пропускают безводный
HF, температуру повышают до 310 °C [5].
6. Чистый препарат можно получить также термическим разложением
Ag[BrF«] [6].
Свойства. М 126,88. Белая, слоистая кристаллическая масса, по эластич-
ности напоминающая изделия из рога, лишь с трудом поддается растиранию
в порошок, /пл 433-4-435 °C; d 5,852 (15,5°C). Решетка кубическая (тип
NaCl). Д//°298 —203,8 кДж/моль. Очень гигроскопичное и светочувствительное
соединение. При 16 “С в 100 мл Н2О растворяется 177,52 г AgF; растворяется
в HF, СНзСООН и CH3CN; нерастворимо в диэтиловом эфире. Хранят AgF
в темных парафинированных склянках.
Соединения с медью, серебром, золотом 275>
ЛИТЕРАТУРА
1. Anderson F. A., Bak В., Hillebert А.,
Acta Chem. Scand., 7, 236 (1953).
2. Choi Q. W„ J. Amer. Chem. Soc.,
82, 2686 (1960).
3. Buckler F. J., Pattison F. L. M.,
Saunders B. C., J. Chem. Soc., 1949,
1471.
4. Sharpe A. G„ J. Chem. Soc., 1952,
4538.
5. Ruff 0., Die Chemie des Fluors.
Berlin, 1920, S. 37.
6. Hoppe R„ 20 Jahre Fonds Chem.
Ind. Beitr. Wiss. Veranstaltung Ba-
den-Baden, 1970, S. 15.
Фторид серебра(Н) AgF2
Способ 1
2AgCl+2F2 ----* 2AgF2 + Cl2
286,67 76 291,76 70,91
Над AgCl в никелевой лодочке, находящейся в никелевой трубке (см:,
прибор в методике получения TIFs, рис. 152), пропускают F2. Вначале трубку
необходимо охлаждать снаружи так, чтобы температура не поднималась вы-
ше 80 °C, потому что иначе образуется тройная смесь, состоящая из AgCl,.
AgF и AgF2, которая плавится, и дальнейшее взаимодействие с F2 будет за-
труднено. Постепенно в потоке F2 температуру понижают и вытесняют F2 из.
прибора потоком сухого N2. Выход 90% в расчете на AgCl.
Способ 2
Ag + F2 -------► AgF2
107,88 38 145,88
Реакцию проводят в приборе, описанном в первом способе. Фторируют-
очень тонкодисперсное, так называемое «молекулярное» серебро (см. гл. 18,.
«Медь, серебро, золото»). Реакция начинается уже при комнатной темпера-
туре и сопровождается разогреванием и образованием продукта, окрашенно-
го в желто-коричневый цвет. Надо следить за тем, чтобы температура не под-
нималась выше 60°C (применять наружное охлаждение). При затихании-
бурной реакции постепенно повышают температуру до 250 °C. В заключение-
продукт охлаждают в потоке F2 и вытесняют F2 из прибора потоком сухого-
азота.
Другие способы. Взаимодействие C1F3 с Ag в автоклаве при 120 °C [4].
Пропускание 5—8%-ной смеси F2 и N2 при 25—30°C над смесью AgOCNl
и KF (1:1 по массе) [5].
Свойства. Чистый кристаллический AgF2 имеет синеватый цвет (цвет ста-
ли). Желтый продукт, полученный при фторировании Ag, рассматривается
как бедная фтором форма AgF2. /пл 690 °C; d 6,1. —347 кДж/моль.
AgF2 термически стабилен вплоть до 700 °C. Химически очень реакционноспо-
собен. Чрезвычайно чувствителен к гидролизу.
Хранят AgF2 в запаянных кварцевых ампулах или сосудах из железа.
Применяют при фторировании органических соединений, а также при полу-
чении COF2.
ЛИТЕРАТУРА
1. Struve W. S., et al. Ind. Eng.
Chem., 39, 353 (1947).
2. Ruff 0., Giese M., Z. Anorg.
Allgem. Chem., 219, 143 (1934).
3. v. Wartenberg H„ Z. Anorg.
Allgem. Chem., 242, 406 (1939).
4. Hiickel IF., Nachr. Ges. Wiss, Got-
tingen Math. Phys. KI. 36, 1946;
C. A., 1949, 6793.
5. Якубович А. FL, Энглин M. А., Ма-
каров С. П. — Ж. общ. хим., 1960,
т. 30, с. 2374.
18*
276 Глава 3. Фтор
.Тетрафтороборат серебра Ag[BF4]
Способ 1 [1]
AgF + BF3 ------> Ag[BF4]
126,87 67,81 194,68 ;
В суспензию фторида серебра в бензоле пропускают трифторид бора до
-тех пор, пока почти все твердое вещество не растворится. Нерастворенный
•осадок фторида серебра отфильтровывают, раствор концентрируют путем ис-
парения в вакууме; при этом выпадают кристаллы состава Ag[BF4] • (С6Н6)2.
Отгоняют бензол при давлении 1 мм рт. ст. и 50 °C; остается чистый
Ag[BF4J.
Другие способы
2. Ту же реакцию можно проводить в нитрометане [2].
3. Менее рекомендуемый метод основан на взаимодействии Ag2O и BF3
13]:
2Ag2O + 4BF3 ---> 3Ag[BF4] + AgBO2
так как сообщается, что при реакции наблюдалась сильная детонация [4].
Свойства. Белый, кристаллический порошок, незначительно светочувстви-
телен; очень гигроскопичен. Хранят Ag[BF4] под пентаном.
ЛИТЕРАТУРА
1. Неу ns К.., Paulsen Н., Angew. 3. Meerwein И., Wunderlich К., Ап-
Chem., 72, 349 (1960). gew. Chem., 69, 481 (1957).
2. Olah G. A., Quinn H. W., J. Inorg. 4. Lemal D. M., Flu A. J., Tetrahed-
Nucl. Chem., 14, 295 (1960). ron Letters, 1961, 775. ;
Тетрафтороаргентат[Н1] калия K[AgFJ
Свободный фтор пропускают при температуре 200—400 °C над смесью
KCI-|-AgNO3 или KNO3-|-AgNO3 в сосуде из спеченного корунда.
Свойства. М 222,97. Желтое вещество чрезвычайно чувствительно к воде.
Обладает диамагнитными свойствами.
ЛИТЕРАТУРА
1. Hoppe R„ Z. Anorg. Allgem. Chem.,
292, 28 (1957).
Фторид золота(Ш) AuF3
Способ 1
Au + 2BrF3 ------*• Au[BrF6] + 1/2Br2
197 274 391 80
Au[BrFe] --->• AuF3 + BrF3
391 254 137
1—2 г порошка золота (полученного восстановлением из водного раство-
ра) нагревают в кварцевом сосуде с 15—20 г BrF3 до тех пор, пока не пре-
кратится образование брома. Бром сливают. При растворении золота рас-
твор быстро желтеет. Избыточный BrF3 отгоняют в вакууме и конденсируют
Соединения с медью, серебром, золотом 277
в кварцевой ловушке, охлаждаемой жидким воздухом. Остающийся в ампуле
желтый Au[BrF6] разлагается при нагревании до 300 °C в вакууме. Получают
практически чистый AuF3.
Другие способы
АнС13-хН2О медленно переходит в AuF3 при взаимодействии с F2 при
200 °C.
Порошок золота при пропускании F2 при 250 °C образует AuF3 в смеси
с Ан.
Свойства. Оранжевый порошок. Термически устойчив до 500 °C; выше
500 °C разлагается на Ан и F2. Бурно реагирует с водой, гидролизуется 40% -
ной плавиковой кислотой. Сильный фторирующий агент; с органическими
растворителями реагирует с воспламенением.
ЛИТЕРАТУРА
1. Sharpe A. G., J. Chem. Soc., 1949, 2. Asprey L. В., Kruse F. H., Jack K-
2901. H-, Maitland R.. Inorg. Chem., 3,
602 (1964).
Тетрафтороаурат[П1) калия K[AuF4]
K[AuC14]+2F2 ----*• K[AuF4] + 2C12
378 76 312 142
K[AuC14]-0,5H2O или К [AuC14] • H2O обезвоживают при 105 °C. Затем пре-
парат в корундовой лодочке фторируют свободным фтором при 200—300 °C.
Свойства. Светло-желтое очень гигроскопичное вещество.
ЛИТЕРАТУРА
1. Hoppe R., Klemm W„ Z. Anorg.
Allgem. Chem., 268, 364 (1952).
Фторид цинка(П) ZnF2
ZnCO3-}-2HF -----> ZnF24-CO2 + H2O
125,39 40 103,38 44 18,01
ZnCO3 вносят в горячую плавиковую кислоту, взятую в избытке. Вна-
чале раствор остается прозрачным. При дальнейшем добавлении ZnCO3 вы-
падает ZnF2 в виде белых непрозрачных кристаллов. Реакционную смесь вы-
паривают на электроплитке досуха.
Если ZnF2 используют как фторирующий агент, то в дело пускают имен-
но этот препарат, представляющий собой не совсем безводную форму ZnF2.
Абсолютно безводный ZnF2 намного инертнее в химическом отношении и по-
этому менее подходящ как фторирующий агент.
Для получения безводного ZnF2 продукт надо нагревать без доступа вла-
ги воздуха при 800 °C. В присутствии NH4F это приводит к получению более
крупных кристаллов.
Свойства. Прозрачные иглы. Решетка типа рутила. tn„ 872 °C; /Кнп
1500 °C; d 4,84. Трудно растворяется в воде (5-10~5 моль/л), немного рас-
творяется в разбавленной плавиковой кислоте; растворяется в соляной и
азотной кислотах и в аммиаке.
278 Глава 3. Фтор
ЛИТЕРАТУРА
1. Ruff О., Die Chemie des Fluors. 2. Частное сообщение из Anorg.
Berlin, 1920, S. 36. Chem. Institut d. Univ. Munster.
Фторид кадмия(Н) CdF2
CdCO34-2HF -------> CdFg + COj + HjO •
172,52 40 150,51 44 18,01 ?
В 40%-ную плавиковую кислоту, взятую в избытке, в платиновой чашке '
вносят CdCOs. Раствор упаривают на электроплитке досуха и продукт обез- i
воживают в вакууме прн 150 °C. I
Свойства. Неокрашенное соединение. Решетка кубическая (типа флюори- 1
та), /пл 1049°C; /КИп 1748°C; d 6,33. При 25°C в 100 мл воды растворяется |
4,3 г CdF2. Растворяется в плавиковой и др. минеральных кислотах; не рас- *
творяется в спирте и жидком аммиаке.
ЛИТЕРАТУРА j
1. Klemm W., Tilk W7., о. Mullen- ?
helm S., Z. Anorg. Allgem. Chem.,
176, 13 (1928). 1
Фторид ртути(1] Hg2F2 *
Hg2CO3 + 2HF -----* Hg2F2 + CO2 + H2O j
461 40 439 44 18
К ~450 мл воды добавляют ~60 мл разбавленной азотной кислоты и |
растворяют 150 г Hg2(NO3)2‘, полученный раствор тонкой струей при силь- ]
ном встряхивании вливают в раствор, содержащий 50 г КНСО3 в 1 л воды. |
После нескольких декантанций водой, насыщенной СОг (в воду бросают ку- *
сочки сухого льда), осадок быстро отделяют с отсасыванием на стеклянном j
фильтре. Не высушивая, Hg2CO3 вносят небольшими порциями при постоян-
ном перемешивании в платиновую чашку с 40 %-ной плавиковой кислотой. i
Hg2F2 выпадает в осадок в виде желтого порошка. Hg2COs добавляют до тех >
пор, пока еще происходит выделение СО2; затем отстоявшуюся сильно раз-
бавленную плавиковую кислоту сливают, добавляют новую порцию 40%-ной
плавиковой кислоты н выпаривают на водяной бане досуха. Продукт раз- ;
мельчают и сушат в сушильном шкафу при 120—150 °C. Затем продукт сразу
перекладывают в плотно закрывающийся сосуд из меди. Синтез Hg2F2 надо' ’
проводить в темноте или по крайней мере на рассеянном свету.
Свойства. Желтоватый, на свету быстро темнеющий кристаллический по- 1
рошок. Решетка тетрагональная, /пл 570 °C; d15 8,73. В воде растворим луч- j
ше, чем Hg2Cl2; при растворении гидролизуется. ;
ЛИТЕРАТУРА ?
1. Ruff О.. Die Chemie des Fluors. 2. Неппе A. L., Renoll М. W„ J. ’
Berlin, 1920, S. 34. Amer. Chem. Soc., 60, 1060 (1938). 1
Фторид ртути(11] HgF2
HgCl2 + F2 -----> HgF2 + Cl2
271,52 38 238,61 70,92
Соединения с цинком, кадмием, ртутью 279
Реакцию проводят в горизонтально расположенном медном цилиндре
(рис. 153), который наподобие вращающегося барабана может поворачивать-
ся вокруг своей оси (со скоростью 20 об/мин). Вдоль оси цилиндр ймеет две
трубки: в одну вводится F2, в другую выводятся реакционные газы.
Медный барабан наполняют сухим порошком HgClj и помещают несколь-
ко кусочков меди (для предотвращения образования корки). Как только
впускают F2, начинается экзотермическая реакция. Протекание реакции кон-
тролируют путем отбора проб. Пробы растворяют азотной кислотой и опре-
Рис. 153. Прибор для получения фторида ртути(П).
деляют С1_. Если С1~ больше не обнаруживается, реакцию заканчивают. Пре-
парат переносят в закрывающийся медный сосуд. Выход 75% в расчете на
HgCl2.
Способ 2 [2]
H3O + 2HF ------* HgF2 + Н2О
216,62 40 238,61 18,01
Реакцию проводят в приборе, описанном в методике получения CoFj
(см. рис. 157). HgO в никелевой лодочке в течение 4,5 ч при 380—450°C фто-
рируют смесью безводного HF с О2 (HgO : HF : Ог=22 : 60 : 4 по массе)
Способ 3 [3]
Hg2F2 + 2NOF-3HF ------> 2HgF2 + 2NO + 6HF
439,2 218,0 477,2 60 120
Реакцию проводят в автоклаве из специальной стали, имеющем свинцо-
вую или тефлоновую прокладку и вводную трубку из тефлона с винтовым
запором. При загрузке автоклава надо обращать внимание на свободный объ-
ем автоклава, так чтобы максимальное давление газов NO и HF не превыша-
ло допустимого значения для автоклава. Прежде всего в автоклаве при не-
туго закрытом запоре тефлоновой трубки нагревают в течение 6 ч при 200 °C
смесь Hg2F2 и NOF-3HF, взятых в молярном отношении 1 :3. Затем авто-
клав охлаждают и выпускают NO. Такую операцию по нагреванию, охлажде-
нию и выпуску газа проводят дважды. Затем к автоклаву присоединяют U-
образную трубку из никеля, с которой можно работать под вакуумом, отка-
чивают избыточный NOF-3HF в вакууме и U-образную трубку опускают в
жидкий воздух. В автоклав осторожно впускают сухой воздух и •Открывают
крышку.
Другие способы
Если имеют в распоряжении баллон с Fa, то лучше всего для получения
HgF2 фторировать HgBr2 газообразным фтором {4]. При этом одновремен-
но получают BrFs.
280 Глава 3. Фтор
Небольшие количества HgF2 можно также получить в приборе, описанг
ном в методике получения T1F3 (ем. рис. 152). '
Свойства. Белый, очень чувствительный к. влаге порошок. tn„ 645°C;
Б<ип>650°С; d 8,95 (15°C). Решетка типа флюорита. Водой сразу гидроли-
зуется, давая желтое окрашивание. Применяют как фторирующий агент прн
получении соответствующих соединений.
Полученный HgF2 лучше всего хранить в трубке из тефлона, которая
должна быть вставлена в точно подогнанную трубку из полиэтилена (или по-
липропилена) с толщиной стенок 1 см. В тонкостенных сосудах из этих ма-
териалов из-за их пористости HgF2 сохраняется без разложения, только если
эти сосуды помещают в эксикатор. HgF2 можно хранить также в цилиндри-
ческих емкостях, изготовленных из никеля или нержавеющей стали с винто-
вым запором и прокладками из тефлона; однако эти сосуды открывают толь-
ко в сухой камере в атмосфере абсолютно сухого воздуха.
ЛИТЕРАТУРА
1. Неппе A. L., Midgley Th., J. Amer. 3. Seel F., Angew. Chem., 77, 689
Chem. Soc., 58, 886 (1936). (1965).
2. Пат. США 2757070. 4. Seel F., частное сообщение, 1973.
Дигидрат фторида ртути(Н) HgF2»2H2O
HgO + 2HF + Н2О -----> HgF2-2H2O
21,7 тонкодисперсного желтого HgO в полиэтиленовой бутылке интенсив-
но механически встряхивают с 20 мл 40 %-ной плавиковой кислоты. Встряхи-
вание продолжают до тех пор, пока образуется белая паста, однако макси-
мальное время встряхивания не должно превышать 1 ч. Затем добавляют
5 мл эфира и продолжают встряхивание еще 15 мин. HgF2-2H2O отделяют от
избытка плавиковой кислоты центрифугированием. Влажный препарат целе-
сообразно непосредственно применять для фторирования. Выход 97%.
Свойства. Белое, немного светочувствительное вещество. Не растворяет-
ся в безводном фтороводороде, растворяется в плавиковой кислоте. При на-
гревании >60 °C происходит гидролитическое расщепление. Хранят HgF2-
•2Н2О в полиэтиленовых сосудах в темноте.
ЛИТЕРАТУРА
1. Doetzer R„ Meuwsen A., Z. Anorg.
Allgem. Chem., 308, 79 (1961).
Фторид скандия[1П) ScF3 |
Sc(OH)3 + 3HF ----> ScF3 + 3H2O . |
96,13 60 102,10 54,03
Насыщенный раствор Sc(OH)3 или Sc2O3 в 40 %-ной плавиковой кислоте ;
выпаривают в платиновой чашке, отфильтровывают образовавшийся осадок ’s
и сушат его в вакууме при 150—180 °C.
Свойства. Белый порошок. Решетка гексагональная. Очень мало раство-
ряется в воде, но достаточно хорошо — в растворах карбонатов щелочных
металлов и аммония. При сплавлении со щелочами ScF3 полностью разлага-
ется.
ЛИТЕРАТУРА
1. Gmelin—Kraut, VI, 2, S. 681.
Соединения со скандием и редкоземельными элементами 281
Трифториды редкоземельных элементов LnF3
(Ln = La—Lu, Y)
Ln2O3 + 6HF ----> 2LnF3 + 3H2O
В стеклянном стакане растворяют 3 г Ln2O3 [соответственно РгвОц или
Се(1\Ю3)з] в конц. НС1; в некоторых случаях при затруднениях в растворе-
нии добавляют немного конц. HNO3. Чтобы не образовались нитраты, реак-
ционную смесь выпаривают, удаляя летучие компоненты, и добавляют раз-
бавленную НС1. Раствор LnCl3 переливают в платиновую чашку и добавляют
40%-ную HF, раствор трижды выпаривают с 40%-ной HF, добавляют еще
раз плавиковую кислоту и 3—5 г NH4F или NH4F-HF. Раствор концентриру-
ют до тех пор, пока не образуется белый туман (NH4F), и охлаждают. Твер-
дую массу дробят и переносят в большую платиновую лодочку, которую по-
мещают в реакционную трубку, нагреваемую трубчатой печью. Реакционная
трубка может быть изготовлена из высокоглниоземистой массы (массы Пи-
фагора), но в этом случае необходимо защищать ее внутреннюю поверхность
в горячей зоне от воздействия NH4F и HF (путем футеровки платиновой
жестью нли с помощью трубки из А12О3). Реакционная трубка имеет с обоих
открытых концов шлифы; с одной стороны она присоединена к установке для
очистки Ns, а с другой стороны — к промывной склянке (конц. H2SO4) для
защиты от влаги воздуха. Нагревание в средней зоне осуществляется с по-
мощью передвигающейся трубчатой печи. • '
Для удаления летучих компонентов через трубку пропускают высокочи-
стый N2 и медленно нагревают до 400 °C. Для получения хорошо образован-
ных кристаллов нагревание продолжают еще 2—3 сут при 700 °C, а затем
медленно в течение 3 сут охлаждают.
Свойства. Фториды редкоземельных элементов при комнатной температу-
ре могут образовывать кристаллические решетки двух типов. При получении,
согласно описанной здесь методике, трифторнды ряда La — Nd имели гекса-
гональную решетку, а трифториды ряда Sm — Lu и YFs — решетку ортором-
бического типа [1]. Температуры плавления трифторидов редкоземельных
элементов приведены в работе [2]. Трифториды редкоземельных элементов
можно хранить на воздухе в стеклянной посуде.
ЛИТЕРАТУРА
1. Greis О., Petzel Т„ Z. Anorg. 2. Thoma R. Е„ Brunton G. D., Inorg.
Allgem. Chem., 403, 1 (1974). Chem., 5, 1937 (1966).
Дифториды редкоземельных элементов SmF2, EuF2 и YbF2
Способ 1. Восстановление редкоземельными элементами [1—3]
2SmF3 4* Sm — —>- 3SmF2
414,69 150,35 565,04
2EuF3 + Eu — —*• 3EuF2
417,91 151,96 569,87
2YbF3 4- Yb — —> 3YbF2
460,07 173,04 633,11
Дифториды редкоземельных элементов SmF2, EuF2 и YbF2 можно полу-
чить из трифторидов путем их восстановления соответствующим металлом.
Для получения особо чистого продукта рекомендуется взаимодействие с па-
рами металла. Восстановление проводят в двухкамерной ячейке из модпбде-
282 Глава 3. Фтор
на (см. рис. 63); в камере 1 находится редкоземельный элемент в трехкрат-
ном избытке, а в камере 2 — трифторид. Эти камеры сообщаются Друг с
другом, и в реакционную трубку помещают молибденовую лодочку. Затем
нагревают в высоком вакууме. При получении YbF2 поддерживается темпе-
ратура 700 °C, при получении EuF2 и SmF2— 950 °C; температурный режим
сохраняют в течение 12—24 ч, что зависит от размера эффузионного отвер-
стия 5 в реакционной ячейке. Наконец, продукт охлаждают.
Способ 2. Восстановление водородом [4—7]. Дифторид европия (но не
SmF2 и YbF2) можно получить восстановлением водородом.
2EuF3-|-H2 -----► 2EuF2 + 2HF
417,9 2 379,9 40
Молибденовую лодочку с навеской EuF3 вдвигают в реакционную труб-
ку из А12Оз, облипованную внутри молибденовой фольгой. С помощью пере-
двигающейся трубчатой печи в течение 2—4 ч нагревают до 1100 °C, пропу-
ская высокочистый N2. Затем заменяют N2 на высокочистый Н2; при тех же
температурных условиях водород пропускают 24 ч н охлаждают также при
пропускании Нз- Ранее рекомендованную методику получения EuF2 в плати-
новой лодочке нельзя применять, так как в указанных условиях эксперимен-
та платиновая фольга разлагает EuF2 и легируется им.
Свойства. Характеристики солей приведены ниже:
а (решетка
Соль м Цвет О флюорита). А 'пл- °с
SmF2 188,35 Черный 5,8710 Разлагается
EuF2 189,96 Светло-желтый 5,8423 1403
YbF2 211,04 Серый 5,5991 1407
Дифториды редкоземельных элементов занимают достаточно широкую фазо-
вую область в направлении состава, богатого фтором; приведенные значения
параметра решетки а являются максимальными до состава LnF2,oo- EuF2 и
YbF2 можно хранить на воздухе в стеклянной посуде. Напротив, черный
SmF2 очень неустойчив и должен храниться только в атмосфере аргона.
ЛИТЕРАТУРА
1. Petzel Т., Greis О., Z. Anorg.
Allgem. Chem., 396, 95 (1973).
2. Catalano E., Bedford R. G., Silvei-
ra V. G., Hickman H. H., J. Phys.
Chem. Solids, 30, 1613 (1969).
3. Stezowski J. J., Eik H. A., Inorg.
Chem., 9, 1102 (1970).
4. Petzel T., Greis O., Z. Anorg.
Allgem. Chem., 388, 137 (1972).
Фторид церия(1У) CeF4
2CeF3 + F2 ----> 2CeF4
394,26 38 432,26
CeO2 + 2F2 -----* CeF4 + O2
172 76 216 32
5. Beck G., Nowacki W., Naturwiss.,
26, 495 (1938).
6. Klemm W., Doll W„ Z. Anorg.
Allgem. Chem., 241, 233 (1939).
7. Bedford R. G., Catalano E„ Proc.
Sth Rare Earth Res. Conf., April
1970, Reno, Nevada, Vol. I, S. 388.
В приборе, описанном в методике получения T1F3 (рис. 152), фторируют
прн 500 °C CeF3 или СеО2 в лодочке из спеченного глинозема.
Соединения со скандием и редкоземельными элементами 283
Свойства. Белая кристаллическая соль. /ПЛ>65О°С; d 4,77. Нераствори-
ма в воде; очень медленно гидролизуется холодной водой. Восстанавливает-
ся при 300 °C водородом до CeF3.
ЛИТЕРАТУРА
1. v. Wartenberg Н.. Z. Anorg. Allgem. 3. Asber A. J., Wylie A. W., Austr.
Chem., 244, 343 (1940). Journ. Chem., 18, 959 (1965).
2. Klemm W., Henkel P„ Z. Anorg.
Allgem. Chem., 220, 181 (1934).
Оксид-фториды редкоземельных элементов LnOF
|Ln = La—Lu, Y)
Способ 1
LnF34-Ln2O3 ----> 3LnOF
Навески LnF3 и Ln2O3 в молярном отношений 1 : 1 тщательно перетира-
ют и вносят в платиновую трубочку, которую по возможности тщательно
(газонепроницаемо) закрывают, отодвигая смесь от края и сплющивая тру-
бочку. Для установления состояния равновесия смесь в трубочке нагревают
в атмосфере инертного газа или условиях высокого вакуума при 1100 °C в
течение нескольких часов. Для получения хорошо образованного кристалли-
ческого продукта температуру до комнатной надо понижать очень медленно
в течение нескольких суток.
Другие способы
Сомнительно, что рекомендуемое нагревание смеси указанного выше со-
става в открытой лодочке в условиях высокого вакуума или в атмосфере
инертного газа приводит к успеху, так как LnF3 может теряться при испаре-
нии (давление пара LaF3 при 1000°C равно 10~8 мм рт. ст.).
Гидролиз LnF3 на воздухе при высокой температуре (600—800 °C) также
при случае можно использовать для препаративного получения оксид-фтори-
дов редкоземельных элементов. Однако при этом нельзя дать никаких гаран-
тий, что образуется единственный однородный конечный продукт, так как
известны оксид-фториды и других составов, отличных от LnOF.
Свойства. Образуются решетки двух типов (ромбическая и кубическая).
Физические свойства см. в работе [5, 6].
Получение фторидов актиноидов описано в гл. 21.
ЛИТЕРАТУРА
1. Klemm W., Klein Н., Z. Anorg.
Allgem. Chem., 248, 167 (1941).
2. Zachariasen W. H., Acta Cryst., 4,
231 (1951).
3. Bevan D. J. M., Acta Cryst., 26,
2129 (1970).
4. Erfahrungen im Chem. Laborator.
Univers. Freiburg, Lehrstuhl Anorg.
Chem.
5. Gibson J. A., Havey G. S., Proper-
ties of the Rare Earth Metals and
Compounds, Batelle Inst., Techn.
Rep., AFML-TR-65/430 (1966).
6. Niihara K., Yajima S., Bull. Chem.
Soc. Japan, 44, 643 (1971).
Фторид титана(Ш) TiF3
Ti (в виде гидрида) ф 3HF ----*- TiF3 + 3/2H2
47,9 60 104,9 3
284 Глава 3. Фтор
Металлический титан гидрируют при 600—700 °C (см. гл. 22 «Титан>)
и фторируют газообразным HF в никелевой лодочке 3, помещенной в гори-
зонтально установленную с одной стороны закрытую никелевую трубку ]
Нг
Рис. 154. Установка для получения фторида титана (III).
/ — трубка из никеля; 2 —никелевая крышка; 3— никелевая лодочка.
(рис. 154). Ближе к открытому концу на трубке имеется охладительная ру-
башка; трубка закрывается медной крышкой 2, укрепленной с помощью пи-
цеина. В крышку прочно впаяны две медиые трубки: одна для ввода, дру-
Рис. 155. Прибор для сублимации
фторида титана(Ш).
I — кварцевая трубка; 2 — пальчиковый хо-
лодильник; 3 — никелевый тигель; 4 —
трубчатая печь.
гая для вывода Н2; кроме того, концентрически впаяна серебряная трубка
для ввода HF. Выходящие газы проходят пустую склянку из полиэтилена,
счетчнк пузырьков с вазелиновым маслом и газовую ловушку из полиэтилена
для вымораживания избыточного HF. Закрытый конец реакционной трубки
вводят в трубчатую печь. Температуру контролируют снаружи трубки.
Для вытеснения других газов через прибор пропускают Н2, затем фто-
рируют газовой смесью (H2:HF=1: 4) в течение 4—5 ч при 70°. Подачу
HF начинают при температуре >200 °C. Водяная рубашка на конце реакци-
Соединения с титаном и цирконием 285»
онной трубки должна создавать температурный режим >20 °C, чтобы не бы-
ло конденсации HF. По окончании фторирования прибор охлаждают в по-
токе Н2. Выход >90%.'
В заключение T1F3 очищают путем возгонки в вакууме (рис. 155). В на-
клонно установленную трубчатую печь вводят закрытую с одной стороны
кварцевую трубку; на дно трубки помещают никелевый тигель с возгоняемым-
веществом. Открытый конец кварцевой трубки закрыт крышкой со штуцером»
ведущим к вакуумному насосу. В крышку плотно (с помощью пицеина)
вставлен пальчиковый холодильник из меди, охлаждаемый водой. На нижнем-
конце пальчикового холодильника имеется медный стержень, который способ-
ствует росту кристаллов TiFa. Продукт начинает возгоняться при 10-2—
IO-3 мм рт. ст. Через 4 ч при 1000 °C возгоняется ~80% продукта, осаждае-
мого на пальчиковом холодильнике в виде сверкающих синих кристаллов.
В тигле остается серо-черный остаток. Возогнанный TiFa настолько чист»,
что может применяться для измерения магнитных свойств.
Свойства. Синие ромбические кристаллы. <пл >1100 °C; d^s 2,98. TiFg-
устойчив на воздухе, необычно стоек к действию кислот и оснований, начи-
нает возгоняться при давлении <0,1 мм рт. ст. при ~930 °C. Не растворяет-
ся в воде и спирте. При 950 °C начинает диспропорционировать до TiF4 и Th.
ЛИТЕРАТУРА
1. Ehrlich Р., Pietzka G., Z. Anorg.
Allgem. Chem., 275, 121 (1954).
Фторид титана(1У) TiF4
TiCl4 + 4HF ----> TiF« + 4HC1
189,74 80 123,9 145,84
Реакцию проводят в колбе Эрленмейера, изготовленной из меди или пла-
тины, кроме того, необходимы насадка (шлем), нисходящий медный холо-
дильник, медная осушительная трубка (СаС12).
В колбу Эрленмейера, охлаждаемую смесью льда с поваренной солью,
наливают взвешенное количество охлаждаемого во льду безводного фторово-
дорода (тяге!). Затем в пробирке взвешивают половину рассчитанного по
вышеприведенному уравнению количества TiCl4 и добавляют его по каплям
к HF. Каждая капля вызывает буриую реакцию с выделением НС1. Затем
смесь (на колбу надевают насадку (шлем), соединенную с осушительной'
трубкой) оставляют на несколько часов до тех пор, пока лед не растает.
Колбу Эрленмейера помещают в масляную баню, присоединяют между на-
садкой и осушительной трубкой медный холодильник, ставят свинцовую чаш-
ку в качестве приемника и постепенно нагревают реакционную смесь до-
200 °C; при этом отгоняется плавиковая кислота, содержащая НС1. Убирают
масляную баню, отсоединяют холодильник и T.iF4 возгоняют, нагревая пла-
менем бунзеновской горелки; продукт поступает через насадку и конденси-
руется в медном закрывающемся приемнике, в котором TiF4 можно и хра-
нить. Приемник вдвигают в горло реторты и с помощью свинцового шланга-
охлаждают водой. При возгонке надо обратить внимание на то, чтобы на-
садка всегда оставалась теплой во избежание закупоривания. Выход 90% в
расчете на TiCl4.
Свойства. Бесцветный, рыхлый, очень гигроскопичный порошок. /Пл>
>400°C (при уменьшенном давлении); /субл 284°C; d 2,798 (20°C). С ши-
пением реагирует с водой; растворяется в спирте с разогреванием; нераство-
рим в эфире.
286 Глава 3. Фюр
ЛИТЕРАТУРА
1. Ruff О., Die Chemie des Fluors. 3. Ruff О.. Plato W'., Ber., 37, 673
Berlin, 1920, S. 48. (1904).
2. Ruff O., Ipsen R„ Ber., 36, 1777
(1903).
Фторид циркония(Ш) ZrF3
Zr (в виде гидрида) -f- 3HF -> ZrF3 + 3/2Ha
91 60 148 3
Таким же способом, как описано в методике получения TiF3, гидрируют
>при 800 °C стружки циркония. Количество присоединенного водорода соответ-
ствует формуле ZrHi.ss. Гидрированный препарат прежде всего нагревают
при 50—100 °C в потоке Н2 а затем уже пропускают смесь HF+H2 (2:1),
причем температуру повышают до 750 °C и оставляют при этой температуре
на 6 ч. Реакция почти количественная. Надо точно поддерживать указанный
температурный режим, так как иначе образуется ZrF4 или реакция пройдет
яе полностью.
Свойства. Голубовато-серое вещество. Решетка типа ReO3. d425 4,26. Тер-
мически устойчиво до 300 °C. Трудно растворяется в горячей воде, легко —
в горячих кислотах; не растворяется в растворах щелочей и аммиака.
ЛИТЕРАТУРА
1. Ehrlich Р., Ploger F„ Koch Е„ Z.
Anorg. Allgem. Chem., 333, 209
(1964).
Фторид циркония(1У) ZrF4
Способ 1 [1, 2]
ZtCl44-4HF -----* ZrF4-|-4HCl
233,06 80 167,22 145,84
Способом, описанным в методике получения TiF4, в 120—150 г безвод-
ного HF вносят порциями 50 г ZrCl4. Дальнейшая работа здесь, однако, уп-
рощается, поскольку ZrF4 не нужно возгонять. После отгонки HF колбу Эр-
ленмейера нагревают до начала красного каления диа. ZrF4 получается в чи-
стой форме, и после охлаждения его можно раскладывать в закрывающиеся
-сосуды из меди.
Способ 2 [3]
ZrO2 + 4HF (aq) ----> ZrF4 + 2H2O
123 80 167 36
ZrO2 растворяют в водной плавиковой кислоте; раствор концентрируют
до выделения ZrF4-3H2O. Гидрат обезвоживают в условиях высокого ваку-
ума. Затем обезвоженный продукт возгоняют при 625 °C в условиях высокого
вакуума (см. синтез TiF3); ои осаждается на охлаждаемом пальчиковом хо-
лодильнике. Получают очень чистую соль.
Другие способы: термическое разложение (NH4)2ZrFe [4].
Соединения с ванадием, ниобием, танталом 287
Свойства. Белая, сильно блестящая, прозрачная масса. /сувл >600 °C’,
d 4,6 (16°С). В 100 мл воды растворяется 1,32 г. При 50°С гидролизуется:
водой.
ЛИТЕРАТУРА
1. Ruff О., Die Chemie des Fluors.
Berlin, 1920, S. 49.
2. Wolter L., Chemiker-Ztg., 51, 607
(1908).
3. Ehrlich P., Pio ger F„ Koch E., Z.
Anorg. Allgem. Chem., 333, 209"
(1964).
4. v. Hevesy G., Dullenkopf W., Z-
Anorg. Allgem. Chem., 221, 16Г.
(1935).
Фторид ванадия[Н1) VF3
Способ 1 [1]
VC13 + 3HF -----*• VF3 + 3HC1
157,32 60 107,95 109,37
В приборе, описанном в методике получения CoF2 (см. рнс. 157), 4 г
VC13 в лодочке из никеля, спеченного глинозема или платины обрабатывают*
безводным HF. Прежде всего прибор заполняют сухим N2, чтобы удалить-
кислород воздуха. Во время фторирования трубку медленно нагревают до*
200 °C. Лишь через 1,5 ч температуру повышают до красного каления. Реак-
цию считают законченной, когда выходящие газы больше не содержат HCL
Охлаждают до 100 °C в потоке HF, вытесняют избыток HF азотом и остав-
ляют охлаждаться. Выход 95% в расчете на VC13.
Способ 2 [2]
V4-3HF -------> VFs + s/2H2
51 60 108 3
Реакцию проводят в стальном автоклаве вместимостью 200 мл. Автоклав--
внутри футерован золотой фольгой (для того, чтобы продукт не был загряз-
нен компонентами материала автоклава). Перед началом синтеза автоклав-
эвакуируют при 200 °C и по охлаждении заполняют сухим N2. Затем вносят
5 г порошка ванадия и вновь эвакуируют. Теперь в охлаждаемый при
—60°C автоклав перегоняют взвешенное количество безводного HF (30 г)..
Автоклав нагревают в электропечи 12 ч при 500 °C. Затем при 300—400 °C
выпускают газообразные продукты и автоклав эвакуируют.
Свойства. Желто-зеленый порошок. /п л >800 °C; сублимируется при тем-
пературе красного каления; d 3,63. Почти не растворяется в воде, спирте, аце-
тоне, уксусноэтиловом эфире, уксусном ангидриде, ледяной уксусной кислоте,
толуоле, CCI4, хлороформе и CS2. При действии гидроксида натрия VF3 чер-
неет.
ЛИТЕРАТУРА
1. Ruff О., Lickfett Н., Вег., 44, 2539
(1911).
2. Roesky Н. W., Glemser О., Hell-
berg К.-H., Chem. Ber., 99, 459>
(1966).
Фторид ванадия[1У) VF4
УСЦ + 4HF ------> VF4 + 4НС1
192,79 80 126,95 145,84
288 Глава 3. Фтор
Реакцию проводят в полиэтиленовом сосуде (на 250 мл), снабженном
•нисходящим холодильником также из полиэтилена. Перемешивание реакцион-
ной смеси осуществляют с помощью магнитной мешалки, сердечник которой
изолирован тефлоновым кожухом. В реакционный сосуд с 20 г CC13F (рас-
творитель) через специальный штуцер добавляют 10 г VC14; реакционный со-
суд охлаждают до —78 °C. В то же время, чтобы не допустить попадания
«влаги, в сосуд пропускают сильный поток сухого N2 и при интенсивном пере-
мешивании вводят 25—30 мл безводного HF. В течение 3 ч реакционную
•смесь доводят до комнатной температуры. Путем нагревания до 30—35 °C за-
тем отгоняют избыток HF н растворитель. В реакционном сосуде остается чи-
нный ,VF4. При проведении реакции без растворителя и без перемешивания
полученный продукт содержит 3,5% примесей.
Свойства. Зеленый рыхлый порошок; d 2,975 (23°C). Очень гигроскопи-
чен, расплывается на воздухе, образуя синюю жидкость. Легко растворяется
•в воде с синим окрашиванием. Растворяется в ацетоне и ледяной уксусной
кислоте с темно-зеленым и сине-зеленым окрашиванием соответственно.
Труднорастворим в SO2C12, спирте и хлороформе. VF4 не летуч, однако вы-
•ше 100 °C разлагается по реакции диспропорционирования, давая VF3 и
'VFs. Хранят VF4 в закрытых сосудах из железа и меди.
ЛИТЕРАТУРА
1. Cavell К. G„ Clark Н. С., J. Chem.
Soc., 1962, 2692.
Фторид ванадия(У) VF5
2V ф- 5F2 (под давлением) --> 2VFS
102 190 292
Порошок ванадия вступает в реакцию со свободным фтором, если ее про-
водить в автоклаве из никеля при давлении 200 бар и температуре 300 °C.
После снятия давления при комнатной температуре продукт отгоняют из ав-
токлава и фракционируют в кварцевом приборе.
Свойства. М 146. Бесцветное вещество, /пл 19,0 °C; /кип 47,9 °C. Очень
гигроскопично, растворяется в воде с красно-желтым окрашиванием. Легко
растворяется в спирте, хлороформе, ацетоне и лигроине; не растворяется в
«сероуглероде. Вызывает разложение толуола н эфира. Хранят VFS в сосудах
из никеля, железа или платины.
ЛИТЕРАТУРА
.4. Trevorrow L. Е., Fischer J., Steu-
nenberg R. К., J. Amer. Chem. Soc.,
79, 5167 (1951).
Фторид нйобия(У) NbF5
Способ 1 [1, 2]
NbCls + 5HF -----> NbF5 + 5HCl
270,20 100 187,91 182,30
Методика работы такая же, как при получении TiF4. NbCls вносят в без-
водный HF, взятый в двукратном по сравнению со стехиометрическим коли-
Соединения с ванадием, ниобием, танталом 289
честве. Используют обратный холодильник из меди или железа, охлаждае-
мый с помощью охладительной смеси. Реакционную смесь с избытком HF
кипятят несколько часов до тех пор, пока не улетучится весь HCI. Затем
с нисходящим холодильником отгоняют HF. Наконец, заменяют холодильник
на специальную насадку, надевая ее на реакционный сосуд, и отгоняют NbFs.
Способ 2 [3]
2Nb + 5F2 ------*• 2NbF5
185,82 190 375,82
В приборе, описанном в методике получения SF& металлический ниобий
при 300 °C взаимодействует со свободным фтором.
Свойства. Бесцветные, сильно светопреломляющие кристаллы, очень гиг-
роскопичны, расплываются ‘иа воздухе. /пл 78,9°C; Л«п 233,3°C; d 3,293.
Растворение NbFs в воде и спирте сопровождается сольволизом. Немного
растворим в CS2 и хлороформе. Гидролизуется щелочами. NbFg растворяет-
ся в‘конц. H2SO< немного легче, чем TaF5.
ЛИТЕРАТУРА
1. Ruff О., Schiller Е., Z. Anorg.
Allgem. Chem., 72, 329 (1911).
2. Ruff О., Zedner J., Ber., 42, 492
(1909).
3. Junkins J. H„ Farrar jr. R. A., Bar-
ber E. J., Bernhardt H. A., J. Amer.
Chem. Soc., 74, 3464 (1952).
Гептафторониобат калия К2[МЬР7]
Nb2O5 + 6HF -f- 4KHF2 ---> 2K2[NbF7] + 5H2O
265,82 120 312,44 608,18 90,05
Nb2O5 в платиновой чашке растворяют в 40%-ной плавиковой кислоте и
добавляют раствор KHF2 до тех пор, пока начнет образовываться ‘неисчеза-
ющий осадок. Реакционную смесь охлаждают. K2[NbF7] перекристаллизовы-
вают из разбавленной плавиковой кислоты; «кристаллы отжимают между ли-
стами фильтровальной бумаги и сушат в вакууме.
Свойства. М 304,09. Небольшие очень блестящие иглы; можно перекри-
сталлизовывать • из плавиковой кислоты. При 18 °C в 100 мл воды растворя-
ется 8 г KatNbF?]. Ион [NbF?]’- легко гидролизуется до [NbOFs]2-.
ЛИТЕРАТУРА
1. Kriiss G„ Nilson L. F., Ber., 20,
1688 (1887).
Фторид тантала|У) TaF5
ТаС15 + 5HF ----*• TaFs + 5НС1
358,17 100 275,83 182,30
Реакцию проводят в приборе, описанном в методике получения TiFi.
К‘50—60 г безводного HF в реакционном сосуде добавляют 30 г ТаС1в. При-
соединяют медный обратный холодильник, охлаждаемый охладительной
смесью, и кипятят реакционную смесь в течение нескольких часов до тех пор,
пока весь НС11 не улетучится. Избыток HF отгоняют, заменяя обратный хо-
лодильник на нисходящий. Затем вместо холодильника присоединяют насад-
19—143
290 Глава 3. Фтор
ку и отгоняют TaF5 из реакционного сосуда в платиновый пальчиковый'ти-
гель. Выход 65% в расчете на ТаС15.
Свойства. Бесцветные, сильно светопреломляющие призмы, расплывающие-
ся на воздухе. tnn 96,8 °C; /кип 229,5 °C; d20 4,74. Растворяется в воде с ши-
пением. Дымящая и • концентрированная азотная кислота растворяет TaFe ие
так активно, как вода. Конц. H2SO4 на холоду растворяет лишь незначи-
тельные количества TaF6, едкие щелочи инициируют бурную 1 реакцию. CS2 I
ч СС14 при нагревании растворяют небольшие количества TaF6. С эфиром >
происходит бурная реакция. При комнатной температуре очень медленно *
разъедает сухое стекло, но при более высокой — быстро.
Хранят TaFs в закрытых сосудах из меди или железа.
ЛИТЕРАТУРА
1. Ruff О., Schiller Е., Z. Anorg. 2. Ruff О., Zedner J., Вег., 42, 492
Allgem. Chem., 72, 329 (1911). (1909).
Гептафторотанталат калия Кг[ТаГ7]
Та2О5 + 6HF + 4KHF2 ---* 2K2[TaF,] + 5Н2О
441,76 120 312,44 784,14 90,05
В платиновой-чашке на водной бане растворяют Ta2Os в 40%-ной пла-
виковой кислоте и добавляют раствор KHF2 до выпадения осадка. Осадок
KjfTaF?] можно перекристаллизовать из плавиковой кислоты. Его отжима-
ют и'сушат при 120 °C.
Свойства. М 392,07. Блестящие, тонкие, короткие иглы; можно перекри-
сталлизовывать из плавиковой кислоты. Решетка моноклинная (псевдоромби-
ческая). При 15°С в'100 мл воды растворяется 0,5 г TaFs.
ЛИТЕРАТУРА
1. Berzelius J. J., Pogg. Ann., 4, 6 (1825).
Фторид хрома(Н) CrF2
2CrF3 + Cr ---*• 3CrF2
218 52 270
В закрывающийся никелевый тигель помещают эквимолярные количества
безводного CrF3 и порошка Сг, хорошо перемешивают и нагревают до
1000 °C.‘ Препарат содержит 0,2% Ni, переходящего из стенок тигля.
Свойства. М 90. Темно-зеленая кристаллическая масса с перламутровым
блеском, /пл 894°С; /кип>1200оС; d 4,11. Решетка моноклинная; немного
растворяется в воде; не - растворяется в спирте; растворяется в кипящей со-
ляной кислоте. Серная и разбавленная азотная кислоты ие взаимодействуют
с CrF2. При нагревании на воздухе переходит в‘Сг2О3.
ЛИТЕРАТУРА
1. Sturm В. J., Anorg. Chem., 1, 665 (1962).
Соединения с хромом, молибденом, вольфрамом 291
Фторид хрома(Ш) CrF3
СгС13 + 3HF ----*• CrF3 + ЗНС1
158,38 60 109,01 109,38
Синтез проводят так же, как при получении CoF2. CrCI3 нагревают в по-
токе HF-до тех пор, пока не перестанет выделяться НС1. Температура долж-
на доходить до 550 °C. Затем избыток HF вытесняют потоком сухого N2;
в потоке сухого азота и охлаждают прибор. При‘1200 °C в потоке HF в пла-
тиновой трубке CrF3 может плавиться и при этом частично отгоняться. Та-
ким образом‘получают кристаллический продукт.
При взаимодействии Сг(ОН)3 с плавиковой кислотой получают тригид-
рат CrFs-3H2O, ио не безводный фторид.
Свойства. Зеленоватые иглы, нерастворимые в воде'и спирте. tBJI 1404°C;
/кип>1Ю0°С; d 3,8.
ЛИТЕРАТУРА
1. Poulenc С., С. R. Sci., Paris, 116,
254 (1893).
2. Poulenc С., Ann. Chim. Phys., (7),
2, 62 (1894).
Фторид хрома(1У) CrF4
2CrCl3 + 4F2 ---> 2CrF4-f-3Cl2 или Cr-f-2F2 -------> CrF4
316,75 152 256,02 212,73 52,01 76 128,01
Синтез проводят в приборе, описанном в методике получения SFe- По-
рошок электролитического хрома'или СгС13 в лодочке из плавикового шпата
или спеченного глинозема фторируют при 350—500 °C. В приемнике собира-
ются и CrFs, и CrF4. Основные количества CrF4 находятся • в реакционной
трубке позади лодочки в виде похожего иа лак блестящего коричневого
утолщения. По окончании фторирования прибор промывают потоком N2
или СО2 и сразу же переносят CrF4 в стеклянную ампулу, которую запаи-
вают.
Свойства. М 128,01. Коричневый аморфный, гигроскопичный продукт;'па-
ры имеют иитенсивио синий цвет. 1ПЯ ~ 200 °C; ZKl,n~400°C; d 2,89. Раство-
рениев воде‘сопровождается гидролизом.
ЛИТЕРАТУРА
1. V. Wartenberg Н„ Z. Anorg. Allgem.
Chem., 247, 136 (1941).
Фторид хрома(У) CrF5
2Cr-f-5Fa ---> 2CrF6
104 190 294
Порошок хрома взаимодействует co свободным фтором в никелевом ав-
токлаве под давлением 200 бар при температуре 400 °C в течение 1 ч. По
окончании реакции газы из автоклава выпускают в газовую ловушку, охлаж-
даемую жидким -воздухом. Конденсируется преимущественно красный CrFs
и немного желтого CrF6. После откачивания избытка F2 создают высокий ва-
куум и охлаждают до —80 °C; при этом CrF6 разлагается на CrFs и F2.
19*
292 Глава 3. Фтор |
Свойства. М 147. Вещество огненно-красного цвета, /пл 30 °C. Хранят
CrFB в кварцевых ампулах.
ЛИТЕРАТУРА
1. Glemser О., Roesky Н„ Hell-
berg К.-H., Angew. Chem., 75, 346
(1963).
Фторид хрома(У1] CrF6
Cr+3F, ------> CrF6
52 114 166 ' '
Методика работы подобна описанной в синтезе CrFs. Реакцию проводят ~
в никелевом автоклаве при 350 бар и 400 °C в течение 1 ч. На порошок хро-
ма, содержащий в качестве примеси порошок марганца, действуют в этих
условиях свободным фтором. Наряду с образованием 200 мг легколетучего
CrF6 реакционные газы содержат очень много CrFB. Реакционные газы из ав-
токлава выпускают через кварцевую ловушку, охлаждаемую жидким возду-
хом. После отсасывания избытка F2 в высоком вакууме остается CrFB.
Свойства. Лимонно-желтое неустойчивое вещество; быстро разлагается
уже при —100 °C до CrFB и F2. Хранят CrFB в ампулах, охлаждаемых жид- j
ким воздухом. ?
ЛИТЕРАТУРА f
1. Glemser О., Roesky Н., Hell- j
berg К-H., Angew. Chem., 75, 346 i
(1963). |
i
i
Фторид хромила CrO2F2
(Диоксодифторид хрома)
Способ 1 [1]
CrO2C 12 + F2 --*- CrO2F2 + Cl2
154,92 38 122,01 70,91
Через кварцевую ловушку с СгО2С12, температура которого при помощи
глицериновой бани поддерживается не выше 100 °C, пропускают N2 со ско-
ростью ~50 мл/мин (рис. 156). Азот захватывает СгО2С12 и поступает далее {
в Т-образную железную трубку, в которую подается фтор со скоростью 1
~60—70 мл/мин. Газовая смесь проходит реакционную трубку из никеля ;
(диаметр ~3 см), которая нагревается электрической печью до 200°C.
Вместо никелевой трубки можно использовать трубку из спеченного глинозе-
ма с пробками-крышками из меди (см. синтез BiFB). Затем реакционная
смесь достигает U-образной кварцевой трубки, колена которой должны иметь
внутренний диаметр не менее 15 мм. Здесь продукты реакции конденсируют-
ся (охлаждение смесью сухого льда с ацетоном). Для предотвращения вса-
сывания влаги воздуха на выходе из прибора присоединяют железную осу-
шительную трубку, наполненную свежеобезвоженным KF.
Когда в U-образной трубке конденсируется достаточное количество про-
дукта (коричневая, похожая на войлок масса), глицериновую баню заменя-
ют на ледяную и в прибор вместо F2 подают N2 до тех пор, пока F2 не будет
полностью вытеснен. Теперь в потоке N2 снимают охлаждение с'U-образной
трубки. Коричневая масса обесцвечивается, и образуется грязновато-белая, s
Соединения с хромом, молибденом, вольфрамом 293
похожая на войлок пробка. После отключения • потока N2 оба колена U-об-
разной трубки отпаивают (на высоте утончений колен) и сохраняют CrO2F2
в виде белой устойчивой модификации.
Рис. 156. Установка для получения фторида хромнла.
Способ 2 [2]
CrO34-SF4 -----> CrO2F2 + SOF2
100 108 122 86
Вертикально укрепленную трубку длиной 20 см из термостойкого стекла
дюран (или пирекс), имеющего рубашку для охлаждения, неплотно загружа-
ют крупнокристаллическим безводным СгО3. При 5 °C через трубку с оксидом
медленно пропускают SF41 разбавленный сухим N2, при этом тотчас начина-
ется реакция с образованием коричневого газообразного CrO2F2. Отходящие
газообразные продукты реакции конденсируются в нескольких соединенных
последовательно газовых ловушках при —78 °C, причем CrO2F2 осаждается
в менее охлажденных частях этой системы в виде длинных красно-коричне-
вых игл, a SOF2 и непрореагировавший SF4 конденсируются лишь при более
низких температурах. CrO2F2 затем сублимируют в потоке N2.
Свойства. Газообразный CrO2F2 окрашен в красно-коричневый цвет. Твер-
дый CrO2F2 существует в виде двух модификаций. Одна из них неустойчива;
вещество окрашено в цвета от красно-коричневого до черно-красного. Давле-
ние пара 24 мм рт. ст. (0°С). /субл+30 °C; <Пл 31,6 °C. Устойчивость этой мо-
дификации особенно понижается на свету, при освещении УФ-светом или при
нагревании. Эту модификацию удается сохранить только для свежеконденси-
рованного продукта при температуре-жидкого воздуха (около —190 °C) в от-
сутствие освещения.
Вторая модификация — полимерное соединение. Это грязно-белое устой-
чивое вещество, которое начинает улетучиваться только при >200°C, причем
образуются красно-коричневые'пары CrO2F2.
ЛИТЕРАТУРА
1. V. Wartenberg Н„ Z. Anorg. Allgem. 2. Kraus Н. L„ Schwarzbach F„ Chem.
Chem., 247, 140 (1941). Ber., 94, 1205 (1961).
294 Глава 3. Фтор
Фторид молибдена[П1) MoF3
3MoFs + 2Mo ---*• 5MoFs
573 192 765
В сухой инертной атмосфере смешивают MoFs (в 50 %-ном избытке) с
порошком молибдена. Этой смесью наполняют никелевый реактор, который
эвакуируют. После этого реактор нагревают в течение 2 ч при 180°C и 4 ч
при 400 °C. Затем избыток MoFs отгоняют в глубоком вакууме. Остается
MoFs.
Свойства. М 153. Светло-зеленое вещество; при нагревании до 800 °C чер-
неет, при 900 °C становится темно-красным. Кристаллизуется по типу VF3;
d 4,64. До'500 °C термически устойчиво.
ЛИТЕРАТУРА
1. LaValle D. Е., Steele R. М„ Wilkin-
son М. К-, Yakel И. L„ J. Amer.
Chem. Soc., 82, 2433 (1960).
Фторид молибдена(У) MoF5
2Мо(СО)6 + 5F2 -----* 2MoF5 ф- 12СО
528 190 382 336
1 г (не больше!) карбонила молибдена обрабатывают фтором при —78 °C.
Образуются оливково-зеленое твердое вещество и СО. Так как при этом ча-
сто происходят необъяснимые взрывы, на одну загрузку можно использовать
не более 1 г‘карбонила молибдена.
По окончании превращения твердый продукт постепенно нагревают в ва-
кууме до 170 °C. Отгоняется желтый MoF6. В остатке остается немного свет-
ло-зеленого MoF4.
Если фторирование проводят при 50 °C, то получают MoF6tCOF2 вме-
сто желаемых продуктов MoFs+CO.
Свойства. М 191. Твердое вещество желтого цвета, /пл 64 °C. Дымит на
воздухе; легко гидролизуется.
ЛИТЕРАТУРА
1. Peacock R. D., Proc. Chem. Soc.,
1957, 59.
Фторид молибдена(У1) MoF6
Mo + 3F2 -------> MoF6
96,0 114,0 210,0
Молибден фторируют в приборе, описанном в методике получения SF6
(реакционная трубка из никеля, ловушки для конденсации из кварца). В ре-
акционную трубку помещают в лодочке из Л12О3 или платины порошок мо-
либдена. Конденсаторные ловушки охлаждают жидким кислородом (—183 °C)
или в крайнем случае смесью сухого льда с ацетоном. • После того как при-
бор заполнится фтором, осторожно нагревают никелевую трубку до тех пор,
пока’ не начнется реакция. Во время фторирования реакционную трубку на-
до иногда охлаждать (наиболее простой способ — прикладывание влажной
Соединения с хромом, молибденом, вольфрамом 295
тканн). В кварцевой ловушке конденсируется белый MoF6, а также немного
оксид-фторидов (MoOF4 н MoO2F2), которые образуются, если F2 содержит
О2. По окончании фторирования MoF6 для очистки от побочных продуктов
несколько раз перегоняют в кварцевом приборе.
Для контроля чистоты продукта подходит определение температуры
плавления.
Свойства. Белое кристаллическое, очень гигроскопичное и реакционноспо-
собное вещество. /пл 17,5°C; /кип 35,0°С; d 2,543 (19°С). С водой реагирует
с шипением, на влажном воздухе образует голубовато-белые иглы.
MoF6 хранят в запаянных кварцевых ампулах.
ЛИТЕРАТУРА
1. Ruff О., Escher A., Z. Anorg.
Allgem. Chem., 196, 418 (1931).
Фторид вольфрама(У1] WF6
W + 3F2 ------> WF6
184,0 114,0 298,0
Порошок вольфрама в лодочке из А12О3 сжигают в потоке F2 в приборе,
описанном в методике получения SFe- Для очистки несколько раз перегоняют.
Для контроля степени > чистоты наряду с определением температуры плав-
ления можно использовать определение молекулярной массы путем измере-
ния плотности газов в кварцевой колбе.
Свойства. Вещество в виде газа t бесцветно, в виде жидкости слабо окра-
шено в желтоватый цвет, в виде твердого вещества белое. Очень гигроско-
пично. /пл +2,3°С; /кип +17,5°С; dH! 3,441 (+15°С). Кристаллизуется в
ромбической сингонии.
Хранят WFc в стеклянных или кварцевых ампулах.
Фторид вольфрама (V) WF5 можно получить исходя из WF6 и W. Пары
WF6 восстанавливаются над вольфрамовой проволокой при 600—700 °C; твер-
дый WF5 осаждается иа стенках сосуда. Таким образом, из 3 мл жидкого
WF6 получают за 8 ч 1,5—2 г WF5. Описание кварцевого прибора для син-
теза см. в работе [3].
ЛИТЕРАТУРА
1. Ruff О., Escher A., Z. Anorg. 3. Schroder J., Grewe F. J., Chem.
Allgem. Chem., 196, 413 (1931). Ber., 103, 1546 (1970).
2. Henkel P., Klemm W„ Z. Anorg.
Allgem. Chem., 222, 68 (1935).
Фторид марганца(И) MnF2
MnCO3-f-2HF ----> MnF2 + CO2 + H2O
114,94 40 92,93 44 18
В 40%-иую плавиковую кислоту, взятую в избытке, в платиновой или
свинцовой чашке вносят МпСО3. Затем с бледно-красного MnF2 сливают рас-
твор и сушат продукт при 110 “С.
Свойства. Кристаллизуется в виде розовых квадратных призм. /Пл
856 °C; d 3,98. Структура типа рутила. В 100 мл воды растворяется 1,06 г
296 Глава 3. Фтор
MnF2. Растворяется в разбавленной плавиковой кислоте; легко растворяется
о конц. НС1 и HNO3.
ЛИТЕРАТУРА
1. Moissan Н., Venturi, С. R. Acad.
Sci., Paris, 130b, 1158 (1900).
Фторид марганца(Ш) MnF3
2MnI2-)-13F2 ----> 2MnF3 + 4IF5
617,54 494 223,86 887,68
Так же как описано в методике получения MoF6, действуют фтором на
свежеплавленный измельченный в порошок Мп12 в лодочке из платины или
А12О3. Происходит саморазогревание, и IFS улетучивается. Реакционную труб-
ку нагревают до 250 °C, затем охлаждают в потоке фтора, вытесняют фтор
сухим азотом и препарат тотчас переносят в ампулы.
Таким же путем MnF2 можно перевести при 250 °C в MnF3.
Согласно Хоппе [3], для получения MnF3 можно исходить из
(NH4)2MnFs, который вводят в реакцию с F2. При этом соблюдают все пред-
осторожности, необходимые при работе с гигроскопичными веществами, ка-
ковыми являются исходные соединения. Кроме того, фторирование легче идет
до продуктов полного превращения, так как суммарный объем газообразных
исходных веществ больше, чем объем газообразных продуктов реакции.
Свойства. М 111,93. Соль винно-красного цвета; d 3,54. Термически устой-
чива до 600 °C, гидролизуется водой.
ЛИТЕРАТУРА
1. Moissan И., С. R. hebd. Seances
Acad. Sci., Paris, 130c, 622 (1900).
2. v. Wartenberg H„ Z. Anorg. Allgem.
Chem., 244, 346 (1940).
3. Hoppe R., частное сообщение.
Фторид марганца[1У) MnF4
Mn+2F2 ------> MnF4
55 76 131
В приборе, описанном в методике получения BiFs, действуют фтором в
кипящем слое на порошок металлического марганца (0,5—1 г). Прежде всего
прибор эвакуируют, затем впускают поток фтора так, чтобы в трубке обра-
зовался кипящий слой высотой 10 см. Одновременно никелевую трубку вы-
ше кипящего слоя нагревают до 600 °C. Фторирование продолжается лишь
несколько минут. MnF4 собирают в присоединенную к реакционной трубке ло-
вушку, охлаждаемую жидким воздухом. Непрореагировавший фтор отгоня-
ют из конденсата при —60 °C в высоком вакууме.
Свойства. После сублимации вещество окрашено в светло-голубой цвет;
очень реакционноспособно, сильно гигроскопично. С керосином реагирует со
взрывом. При 0 °C в высоком вакууме медленно отщепляет F2, переходя в
MnF3.
ЛИТЕРАТУРА
1. Roesky Н. W., Glemser О., Hell-
berg К.-H., Angew. Chem., 75, 920
(1963); Chem. Вег., 98, 2046
(1965).
Соединения с марганцем и рением 297
Триокси д-фторид марганца MnO3F
KMnO4 + 2HSOsF ------> MnO3F + KSO3F + H2SO4
158 200 122 138 98
Реакцию проводят в сосуде из меди, который можно присоединить к ло-
вушке из политрифторхлорэтилена. В реакционный сосуд помещают 250 г
HSO3F, сосуд охлаждают до —78 °C и очень медленно прибавляют 100 г тон-
корастертого КМпО4. Когда КМпО4 введен полностью, реакционный сосуд
через охлаждаемую ловушку присоединяют к вакуумной системе. Систему
откачивают и затем медленно повышают температуру до 20 °C. В охлаждае-
мой в сухом льду ловушке конденсируется сырой MnO3F. Путем отгонки в
вакууме отделяют основное количество легколетучего HF. За одну загрузку
получают 40 г MnO3F. Для дальнейшей очистки от захваченного HF продукт
перегоняют над сухим KF или КМпО4 (в первом случае образуется KF-HF,
во втором — MnO3F).
Свойства. Темно-зеленая жидкость. /пл —38 *С; /Кип +60 °C. Термически
устойчива только до 0°С. Выше 0°С разлагается (иногда со взрывом) на
MnF2, МпО2 и О2. Сильно гигроскопичное вещество. Очень бурно реагирует
с органическими соединениями.
Продукт хранят при —78 °C в сосудах из политрифторхлорэтилена или
меди.
ЛИТЕРАТУРА
1. Engelbrecht A., v. Grosse A., J.
Amer. Chem. Soc., 76, 2042 (1954).
Трифтороманганат(Н) калия K[MnF3]
3KF + MnCla -----* K[MnF3J + 2KCl
174 126 151 149
Водный 10%-иый раствор KF медленно при комнатной температуре сме-
шивают с концентрированным раствором МпС12 (К:Мп=5:1). Выпадает
тонкодисперсный осадок, который отфильтровывают и сушат в вакууме. .
Используется для получения K[MnF6],
Свойства. Нежно-розовый мелкокристаллический осадок, незначительно
растворим в растворе KF. На воздухе при комнатной температуре устойчив,
окрашивается при более высокой температуре. "• .-
Аналогичным способом можно получить следующие соединения:
Na[MnF3] (насыщенный раствор NaF, Na : Mn=4-?-5 : 1, реакция при темпе-
ратуре кипения); Rb[MnF3] (40%-ный раствор RbF, Rb:Mn=4:l, комнат-
ная температура); Cs[MnF3] (раствор CsF, концентрация которого в два ра-
за меньше, чем насыщенного, комнатная температура); NH4[MnF3] (раствор
NH4F, концентрация которого в два раза меньше, чем насыщенного, комнат-
ная температура). Все эти препараты имеют нежно-розовый цвет.
ЛИТЕРАТУРА
1. Hoppe R.. Liebe W., Dahne W., Z.
Anorg. Allgem. Chem., 307, 276
(1961).
298 Глава 3. Фтор
Соединения с марганцем и рением 299
Тетрафтороманганат(Ш) калия K[MnF4]
2K[MnFs] + Н2 --> 2K[MnF4] + 2HF
378 2 340 40
Реакцию проводят в трубке из йенского стекла, нагреваемой снаружи с
помощью печи с алюминиевым блоком. В трубку в лодочке из А12О3 поме-
щают K[MnFs] и пропускают тщательно высушенный Н2, при этом темпера-
туру повышают до 250 °C. Реакция заканчивается за ~2ч.
Свойства. М 170. Твердое вещество, окрашенное в цвета от коричнево-
фиолетового до темно-коричневого.
Аналогичным способом можно получить RbfMnF4] (температура реакции
275 °C).
ЛИТЕРАТУРА
1. Hoppe R„ Liebe W., Dahne W., Z.
Anorg. Allgem. Chem., 307, 276
(1961).
Пентафтороманганат(1У) калия K[MnF5]
K[MnF3] + F2 ----> K[MnF5]
151 38 189
Ha K[MnF3] действуют фтором в приборе (из никеля), описанном в ме-
тодике получения T1F3. Сначала поддерживают комнатную температуру, за-
тем в течение 2 ч температуру медленно повышают до 450 °C и продолжа-
ют фторирование при этой температуре. При более высокой температуре
(500 °C) K[MnFs] начинает спекаться, что затрудняет дальнейшее фториро-
вание. Продукт охлаждают в потоке F2.
Свойства. Вещество кирпично-красного цвета. На воздухе медленно раз-
лагается.
Препарат используют для получения K[MnF4].
Аналогичным способом можно получить Rb[MnFs] (500 °C, продолжи-
тельность синтеза 4,5 ч) и Cs[MnF3] (500 °C, продолжительность синтеза
3 ч).
ЛИТЕРАТУРА
1. Hoppe R„ Liebe W., Dahne W., Z.
Anorg. Allgem. Chem., 307, 276
(1961).
Гексафтороманганат[1¥] калия IQMnFe]
МпС12 + 2KCI + 3F2 ---►- K2[MnFe]-f-2C12
125,84 149,12 114 247,16 141,8
Синтез проводят в приборе, описанном в методике получения T1F3. Смесь
2 моль КС1 и 1 моль МпС12 нагревают в потоке F2 при 375—400 °C. По ох-
лаждении избыток F2 вытесняют сухим N2.
Свойства. Золотисто-желтые прозрачные пластинки. Имеет гексагональ-
ную структуру. При нагревании KafMnFe] становится красно-коричневым, а
при охлаждении принимает свой первоначальный цвет. Водой разлагается с
выделением МпО2.
Аналогичным способом из смесей соответствующих хлоридов или соот-
ветствующих оксоманганатов можно получить гексафтороманганаты щелоч-
ноземельных элементов: Ba [MnF6] (из ВаМпО4 при 400 °C); Sr [MnF6] (из
SrMnO4 при 300—400°C); Ca[MnF6] (из СаС12+МпС12 при 500—550°C);
Mg[MnF6] (из MgCl2+MnCl2 при 500—550°C). Все эти соединения либо
имеют светло-желтый цвет, либо отсвечивают желтизной; Mg[MnF6] имеет
слегка оранжевый оттенок [2].
ЛИТЕРАТУРА
1. Huss Е., Klemm W„ Z. Anorg. 2. Hoppe R., Blinne К, Z. Anorg.
Allgem. Chem., 262, 25 (1950). Allgem. Chem., 291, 269 (1957).
Фторид рения(У1) ReF6
Re + 3F2 ----->- ReFe
186,3 114 300,3
Порошок рения нагревают в автоклаве вместимостью 250 мл в атмосфе-
ре фтора. В автоклав помещают рений; автоклав эвакуируют, погружают в
жидкий азот и конденсируют в автоклаве фтор. Автоклав закрывают и на-
гревают в течение 1 ч при 125 °C. Необходимо поддерживать заданную тем-
пературу, чтобы фторирование не привело к получению каких-либо других
продуктов. При этом давление газов в автоклаве должно быть ~22 бар.
Автоклав оставляют охлаждаться, выпускают избыток F2; ReFe перегоняют
и конденсируют в ловушке; продукт очищают сублимацией в вакууме. Для
того чтобы получить ReFe, не содержащий ReF?, сырой фторнд нагревают при
400 °C или помещают в сосуд из никеля, добавив порошок металлического
рения, и оставляют на несколько часов при 250—400 °C. Выход ReFe достав-
ляет 95%.
Свойства. Желтое вещество. /пл 18,6°С; /кип 33,7°С; d1K 3,58 (22°С).
Очень гигроскопично.
ReFe можно хранить при комнатной температуре в ампулах из стёкла
пирекс.
ЛИТЕРАТУРА
1. Ruff О., Kwasnik W., Z. Anorg.
AUgem. Chem., 219, 65 (1934).
2. Slivnik J., Smale A., Zemljic A.,
Kurzreferat Int. Fluorsymposium,
1965, 184.
3. Malm J. G„ Selig H„ J. Inorg.
Nucl. Chem., 20, 189 (1961).
Фторид рения(УИ) ReF7
2Re + 7F2 ----> 2ReF,
372 266 638
Порошок рения взаимодействует с фтором в кипящем слое при 600—
700 °C (методику см. в синтезе BiFs). Отходящие газообразные продукты
реакции улавливают ловушками для конденсации; для очистки продукты
перегоняют.
300 Глава 3. Фтор
Можно также проводить реакцию получения ReFy в автоклаве, фторируя
порошок рения; при этом необходимы давление 200 бар и температура
400 °C.
Свойства. М 319. Вещество желтого цвета; tnn 48,3 °C; /кип 73,7 °C; dm
3,65 (52°C).
ReF? можно хранить при комнатной температуре в ампулах из стекла
пирекс.
ЛИТЕРАТУРА
1. Roesky Н. W., Glemser О., Hell-
berg К.-H., Chem. Ber., 98, 2046
(1965).
Триокси д-фторид рения ReO3F
Способ 1 [1]
ReO3Cl + HF ------> ReO3F + НС1
269,8 20 253,3 36,5
В охлажденную в жидком азоте ловушку из политрифторхлорэтилена пе-
регоняют безводный HF в трехкратном избытке, а затем ReO3CL Присоеди-
няют обратный холодильник из того же материала и медленно нагревают ло-
вушку до температуры плавления фтороводорода. Начинается выделение га-
зообразного НС1. Для того чтобы избежать бурного протекания реакции, ло-
вушку надо время от времени охлаждать. Затем ловушку с реакционной
смесью несколько часов нагревают с присоединенным обратным холодильни-
ком. Наконец, избыток HF отгоняют при 80 °C в вакууме. Не допуская по-
падания влаги воздуха, переносят сырой окрашенный в голубоватый цвет
ReO3F в платиновую трубку, которая плотно (так, чтобы мог сохраняться ва-
куум) соединена с трубкой из политрифторхлорэтилена. Препарат сублими-
руют в вакууме. В пластмассовой трубке осаждается чистый ReO3F. Это
желтое стеклообразное вещество. Выход 70% теоретического.
Способ 2 [2]
KReO4 + IF5 -----> ReO3F + IOF3 + KF
289,4 221,9 253,3 199,9 58,1
В никелевую колбочку, которую можно соединять с обратным холодиль-
ником и вакуумной системой, помещают 2 г KReO4 и конденсируют 9 г IFs-
При комнатной температуре начинается очень слабое взаимодействие, причем
реакционная смесь окрашивается в желтый цвет. Присоединяют обратный
холодильник н нагревают реакционную смесь до 97 °C, так что содержимое
колбы полностью растворяется, становится прозрачным и может застыть.
При этом образуется шлам, окрашенный в желтый цвет. Избыток IFg отго-
няют в вакууме и нагревают (также в вакууме) желтый остаток на парафи-
новой бане до 140 °C. Отгоняются остаточные количества IFS, и в колбе оста-
ется ReO3F в виде стеклообразного желтого вещества.
Свойства. Желтый мелкокристаллический порошок. /Пл 147 °C; Дип
164 °C. Гидролизуется водой до HReO4 и HF.
ЛИТЕРАТУРА
1. Engelbrecht A., v. Grosse A., J. 2. Aynsley Е. F., Hair М. L„ J. Chem.
Amer. Chem. Soc., 76, 2042 (1954). Soc., 1958,3747.
Соединения с железом и никелем 301
Гексафтороренат[1У) калия KajReFg]
K2[ReBr6] + 3KHF2 -----> K2[ReF6] + 3K.Br -f- ЗНВг
744,3 234 378,3 357 243
1 г K2[ReBr6] и 10 г KHF2 хорошо перемешивают и нагревают в плати-
новом тигле на небольшом пламени до тех пор, пока не получится совершен-
но прозрачный расплав. Плав дробят и выщелачивают холодной водой. Эта
операция занимает почти целый день: КВг и KF переходят в раствор, а
K2[ReF6] остается в остатке. Последний отфильтровывают, растворяют в ки-
пящей воде и еще раз фильтруют. Бледно-розовый фильтрат упаривают в
платиновой чашке. При охлаждении из него кристаллизуется KztReFs]. Его
можно перекристаллизовать из разбавленной плавиковой кислоты. Кристаллы
отфильтровывают, промывают спиртом и эфиром и сушат при 80 °C.
Выход 85—95% теоретического. Потери при перекристаллизации состав-
ляют ~30%.
Свойства. Белые кристаллы (тригональная сингония). Незначительно рас-
творяется в холодной, лучше в горячей воде. При комнатной температуре со-
вершенно не гидролизуется.
ЛИТЕРАТУРА
1. Weise Е., Z. Anorg. Allgem. Chem., 2. Brauer G., Allardt H.-D., Z. Anorg.
283, 377 (1956). Allgem. Chem., 316, 134 (1962).
Гексафторорениевая кислота H2[ReF6]
иоиообменник
K2|ReF6] ---------> HJReFJ
378,3 302,3
Реакцию проводят в вертикальной трубке (шланг) из полиэтилена (дли-
на 40 см, внутренний диаметр 1 см), внизу трубку закрывают зажимом. Над
зажимом находится сито из хосталена. Трубку заполняют катионообменни-
ком (например, амберлитом IR 120) в Н+-форме. Раствор Ka[ReF6] в подо-
гретой, очень разбавленной плавиковой кислоте пропускают через колонку.
Затем промывают водой. Элюат содержит разбавленную свободную кислоту
HafReFs]. Элюат концентрируют при комнатной температуре в вакуум-экси-
каторе над Р4О10 или H2SO4 до тех пор, пока не выделятся бледио-розовые
кристаллы кристаллогидрата H2[ReF6] -Н2О.
Свойства. Концентрированный водный раствор слабо окрашен в розовый
цвет. Из этого раствора кристаллизуются бледно-розовые кристаллы кри-
сталлогидрата.
ЛИТЕРАТУРА
1. Brauer G„ Allardt H.-D., Z. Anorg.
Allgem. Chem., 316, 134 (1962).
Фторид жепеза(И) FeF2
FeCl2 + 2HF -----> FeF2 + 2HCl
126,76 40 93,84 72,92
В приборе, описанном в методике получения CoF2, обрабатывают FeCl2
безводным фтороводородом. Реакция происходит уже при комнатной темпе-
302 Глава 3. Фтор
ратуре. Полученный таким способом продукт, аморфен. Для получения в
кристаллической форме продукт надо нагреть до 1000 °C.
Чистый белый FeF2 получают только при работе с совершенно безвод-
ными веществами. Обычно после перекристаллизации препарат окрашен в
серо-коричневые тона.
Свойства. Белый порошок. Структура типа рутила. £Пл>1100°С; субли-
мируется при 1100 °C; d 4,09. В воде растворяется незначительно; не раство-
ряется в спирте, эфире и бензоле.
ЛИТЕРАТУРА
1. Poulenc С., С. R. Acad. Sci., Paris, 2. Poulenc C„ Ann. Chim. Phys., (7),
115,942 (1892). 2,53 (1894).
Фторид железа[Н, III] Fe2F5
(Пентафторид дижелеза)
FeF2-aq4-FeFs-aq ----> Fe2Fr,-aq
94 113 207
Для получения гептагидрата Fe2F5 вносят порошок железа в кипящую.
48%-ную плавиковую кислоту до тех пор, пока не прекратится растворение.
Почти бесцветный раствор фильтруют и оставляют в открытом стакане на
воздухе приблизительно на 2 недели. За это время постепенно через (проме-
жуточный) зеленый FeF2-4FI2O образуется желтый Fe2F5-7H2O. Кристаллы
отделяют от маточного раствора, промывают водой, спиртом, эфиром и су-
шат в эксикаторе над Р4Ою-
Если хотят приготовить тригидрат Fe2F5, то в разбавленную (1:1) пла-
виковую кислоту на водяной бане вносят небольшими порциями порошок же-
леза до тех пор, пока прекратится растворение, добавляют равный объем
раствора FeF3 (полученный растворением Fe(OH)3 в плавиковой кислоте) и
раствор фильтруют. Фильтрат концентрируют в закрытом сосуде при 100 °C,
пропуская N2 (сосуд снабжен трубками для отвода и подвода газа). В те-
чение 40—60 ч образуются темно-красные кристаллы Fe2F5-3H2O.
При нагревании в слабом потоке N2 до 180 °C гепта- и тригпдраты обез-
воживаются за 2 сут. Получают безводную соль, которая представляет собой
небольшие голубовато-серые чешуйки со стальным отливом.
Свойства. Fe2F5-7H2O — желтые кристаллы, малорастворимые в воде;
d 2,20.
Fe2F5-3H2O — темно-красные кристаллы; d 2,43.
Fe2F5 — темные голубовато-серые чешуйки со стальным отливом; d 3,34.
Очень гигроскопичное вещество. На влажном воздухе быстро переходит в
красный тригидрат. С избытком воды дает желтый гептагидрат.
ЛИТЕРАТУРА
1. Brauer G., Eichner М., 7. Anorg.
Allgem. Chem., 296, 13 (1958).
Фторид железа(Ш) FeF3
FeCl3 + 3HF ----*• FeF3 + 3HCI
162,22 60 112,84 109,38
В приборе, описанном в методике получения CoF2 (см. рис. 157), прово-
дят реакцию между безводным FeCl3 и безводным HF. Реакцию прекращают.
Соединения с железом и никелем 303
когда НО больше не выделяется. При комнатной температуре образуется
аморфный FeFs- Для получения кристаллической формы продукт надо нагре-
вать при 1000СС.
Свойства. Зеленоватый порошок, d 3,87. Структура гексагональная. Суб-
лимируется выше 1000 °C. Мало растворяется в воде (0,091 г/100 мл при
25 °C); легко растворяется в разбавленной плавиковой кислоте; не растворя-
ется в спирте, эфире и бензоле.
ЛИТЕРАТУРА
1. Poulenc С., С. R. Acad. Sci., Paris,
115, 944 (1892).
2. Poulenc C., Ann. Chim. Phys., (7),
2, 57 (1894).
Фторид кобальта(П) CoF2
CoCl2 + 2HF ------> CoF24-2HC1
129,89 40,02 96,97 72,94
Кристаллический хлорид кобальта СоС12-2Н2О обезвоживают в стеклян-
ной трубке в потоке НС1 (200°C). При обезвоживании окраска препарата
меняется от розовой до голубой.
Рис. 157. Установка для получения фторида кобальта(П).
Лодочку из плавикового шпата (рис. 157) с СоС12 помещают в железную
трубку, через которую пропускают при 300 °C безводный HF до тех пор, по-
ка не прекратится выделение НС1*. В заключение пропускают поток сухого
N2 для удаления избытка HF.
Свойства. Розовато-красный порошок. /КПп 11004-1200 °C, d 4,43. Струк-
тура типа рутила. Незначительно растворяется в воде; при нагревании рас-
творяется в минеральных кислотах.
ЛИТЕРАТУРА
1. Ruff О., Ascher Е„ Z. Anorg.
Allgem. Chem., 183, 193 (1929).
2. Burford W. В., Ind. Eng. Chem.,
39,321 (1947).
* Установка, собранная по этой схеме, подходит для всех фторирований
безводным HF; в ней удобно получать нелетучие фториды (CuF2, AgF, HgF2,
CrF2, CrF$, VF3, FeF2, FeF3).
304 Глава 3. Фтор
Фторид кобальта[Н1) C0F3
Способ 1 [1]
2CoF2 J- F2 -----► 2CoF3
193,88 38,0 231,88
В приборе, описанном в методике получения T1F3, обрабатывают CoF2
фтором. Реакция вначале идет очень медленно, но при подогревании реак-
ционной трубки до 75 °C ускоряется. Благодаря выделению тепла во время
реакции температура повышается до 200 °C. По окончании реакции прибор
охлаждают в потоке F2, а затем вытесняют фтор сухим азотом. Выход 91%
в расчете на CoF2.
Способ 2 [2, 3]
2CoC124-3F2 -----> 2CoF3H-2Cl2
259,71 114 231,88 141,8
Co2Os + 3F2 -----> 2CoF3 + 3/2O2
165,88 114 231,88 48,0
В приборе, описанном в методике получения T1F3, действуют фтором на
безводный СоС12 или Со2О3. Реакционную трубку постепенно нагревают до
300 °C и строго поддерживают эту температуру до тех пор, пока на выходе
из прибора не появится F2. Затем из прибора вытесняют F2 сухим азотом.
Свойства. М 115,94. Светло-коричневый порошок, на влажном воздухе
становится сразу темно-коричневым, d 3,88. Структура гексагональная. Летуч
в потоке F2 при 600—700 °C, при этом разлагается на CoF24-F2. С водой реа-
гирует с выделением О2.
Хранят CoF3 в закрытых воздухонепроницаемых сосудах из стекла, квар-
ца или металла. Применяют как фторирующий агент в реакциях с органиче-
скими соединениями.
ЛИТЕРАТУРА
1. Belmore Е. A., Ewalt W. М., Woj-
cik В. Н„ Ind. Eng. Chem., 39, 341
(1947).
2. Ruff О., Ascher E„ Z. Anorg.
Allgem. Chem., 183, 193 (1929).
3. McBee E. T., ei., Ind. Eng. Chem.,
39,310 (1947).
Фторид никеля(П) NiF2
NiCl2+F2 -----> NiF2 + Cl2
129,6 38 96,69 70,92
В приборе, описанном в методике получения T1F3, фторируют при 150 °C
обезвоженный NiCl2 (в никелевой лодочке). Затем находящийся в лодочке
продукт (имеющий состав, близкий NiF2,5) нагревают в потоке N2 или СО2
при 400—500 °C, причем продукт с более высоким содержанием фтора от-
щепляет фтор и переходит в NiF2.
Свойства. Желтовато-зеленый порошок, d 4,63. Структура типа рутила.
Сублимируется в потоке HF при температуре выше 1000 °C. Очень мало рас-
творяется в воде; не растворяется в спирте и эфире.
ЛИТЕРАТУРА
1. Henkel Р„ Rlemm W., Z. Anorg.
Allgem. Chem., 222, 74 (1935).
Соединения с железом и никелем 305
Трифтороникелат[П) калия K[NiF3]
NiF2 + KHF2 ----> K[NiFs] + HF
97 78 155 20
Стехиометрические количества безводных NiF2 и KHF2 нагревают при
800 °C в потоке N2 или в вакууме, не допуская попадания влаги воздуха,
Из-за категорического требования об отсутствии воды применяют вместо гиг-,
роскопичного KF гидрофторид калия.
Свойства. Светло-зеленое вещество; кубическая решетка. Обладает анти,
ферромагнитными свойствами. Растворяется в кислотах.
Аналогичным способом получают из NiF2-p2KHF2 тетрафтороиикелат(П)
калия K2[NiF4] (вещество светло-зеленого цвета; тетрагональная структура;
антиферромагнитные свойства).
ЛИТЕРАТУРА
1. Rildorff W., Randler J., Babel D..
Z. Anorg. Allgem. Chem., 317, 261
(1962).
Гексафтороникелат[1У) калия K^NiFe]
2KC1 + NiCl2 + 3F2 ---> KjNiF.1 + 2CL
149,12 129,6 114 250,88 141,8
В приборе, описанном в методике получения T1F3, смесь 2 моль КС1-к
+ 1 моль NiCl2 нагревают в течение 3 ч в потоке F2 при 275 °C. По охлажден
нии прибора фтор вытесняют сухим азотом. Эта методика синтеза неоднократно
использовалась для получения других соединений, например K2[MnF6], K2[CrF6l.
K2[FeF6], Кз[СоР7], MVFJ и K3[CuF6].
Свойства. Блестящая соль красного цвета, d 3,03. Кристаллизуется по ти-_
пу KifPtCle]. Реагирует с водой с выделением газа (OF2 ?) и образованием,
черного осадка; восстанавливается водородом при 200 °C.
Хранят K2[NiF6] в запаянных вакуумированных стеклянных ампулах или,
стеклянных бутылях под азотом.
ЛИТЕРАТУРА
1. Klemm W„ Huss Е., Z. Anorg.
Allgem. Chem., 258, 221 (1949).
Фторид платины(У) PtF5
2PtCl2 + 5F2 ---> 2PtFb-4-2Cl2
532 190 580 142
Через нагретую до 350 °C кварцевую трубку, в которой находится в плд-
тиновой лодочке PtCl2, пропускают смесь газов F2 н N2 (1:3). В более хо^
лодиых местах трубки осаждается PtF6 в виде красного сублимата. По окон-
чании реакции препарат плавят и переносят без доступа воздуха в ампулы.
Свойства. М 290. Темно-красное вещество, tnn 80 °C. При плавлении об-
разует красную вязкую жидкость. Начиная с 130 °C, диспропорционирует на
PtF6 и P1F«. Реагирует с Н2О. Растворяется в BrF3.
ЛИТЕРАТУРА
1 Bartlett N., Lohmann D. Н., J. Chem.
Soc., 1964, 619.
20—143
306 Глава 3. Фтор
Фторид платины[У1) PtF6
Pt + 3F2 ----* PtF6
195 114 309
Платиновую проволоку фторируют в приборе (рис. 158 и 159), изготов-
ленном из стекла или латуни.
При пропускании через платиновую проволоку тока она разогревается и
загорается. На расстоянии всего нескольких миллиметров от зоны реакции
•Лсосуду со фтором
К Вакуумной системе
I
Рис. 159. Прибор для получения фтори-
да платины (VI), изготовленный из ла-
туни.
Рис. 158. Прибор для получения
фторида платины (VI), изготов-
ленный из стекла.
находится сосуд, охлаждаемый жидким азотом, на его стенках и осаждается
PtF6. Затем PtF6 несколько раз перегоняют под вакуумом в приборе, состоя-
щем из U-образной трубки, изготовленной из никеля или монеля (охлажде-
ние до —78 °C).
Выход 8,8% при работе в стеклянном приборе и 71%—в приборе из
латуни.
Свойства. В твердом и жидком состоянии окрашен в темно-красный цвет,
в виде газа — коричнево-красный. ;пл 61,3°C; /1:ип 69,1 °C; d1K 3,826 (64,3°C).
До 200 °C термически устойчив. Давление пара 29 мм рт. ст. (0°С), 96 мм
рт. ст. (21 °C). Сильный фторирующий агент. С его помощью, например, уже
при комнатной температуре можно получить из низших фторидов BrFs,
NpF6 и PuF6.
Хранят PtF6 в сосудах из никеля. В кварцевых сосудах он постепенно
разлагается.
Соединения с платиновыми металлами 307
ЛИТЕРАТУРА
1. Weinstock В., Claassen Н. И., Malm
J. G., J. Amer. Chem. Soc., 79, 5832
(1957).
2. Weinstock B„ Malm J. G., Wea-
ver E. F., J. Amer. Chem. Soc., 83,
4310 (1961).
ГексафтороплатинатрУ) калия K2[PtF6]
3KOH + 2H2O 4- PbO2 --> K2[Pb(OH)6]-KOH
168,3 36,0 239,2 443,5
Ka[Pb(OH)6]-KOH + 8HF ---> 3KF-HF-PbF4 + 7H2O
443,5 160,0 477,5 126,1
Pt 4- 2(3KF-HF PbF4) -> K2[PtF6] + 2PbF22KHF2 + 2KF
195 955 387 490 156 116
В золотой чашке готовят плав из 42 г КОН и 9 мл Н2О. При температур
ре 100—150 °C в чашку с плавом вносят 60 г РЬО2. Когда РЬО2 растворится,,
удаляют избыток воды, длительно нагревая чашку с реагентами при 100 °C,
В чашке остается гидроксоплюмбат калия, состав которого приблизительна
выражается формулой К2[РЬ(ОН)6]-КОН.
Этот «плюмбат» осторожно, небольшими порциями, вносят в 48%-ную
плавиковую кислоту. Прозрачный раствор сливают с небольшого количества
осевшей на дно чашки соли. Фильтрат концентрируют до тех пор, пока из
него не выпадут моноклинные игольчатые кристаллы соли 3KF-HF-PbF4.
В платиновой чашке смешивают тонкоизмельченную платину с рассчитан-
ным количеством 3KF-HF-PbF< и смесь нагревают. Масса плавится и пере-
ходит в красную жидкость. При охлаждении жидкость застывает в красно-
коричневый «пирог». Последний многократно обрабатывают 48%-ной HF до
тех пор, пока сливаемые вытяжки не будут бесцветными. Раствор фильтру-
ют. Свинец осаждают несколькими каплями конц. H2SO4 и вновь фильтруют.
Фильтрат упаривают на водяной бане до начала кристаллизации. Затем
охлаждают до 0°С; при этом выпадают кристаллы K2[PtF6], которые можно
перекристаллизовать из горячей воды.
Свойства. Бледно-желтое кристаллическое вещество. Разлагается при тем-
пературе красного каления. В 100 г Н2О при 25 °C растворяется 0,0023 г пре-
парата.
ЛИТЕРАТУРА
1. Schlesinger И. J., Tapley М. W..
J. Amer. Chem. Soc., 46, 278 (1924).
2. Schlesinger H. J., Palmateer R. E.,
J. Amer. Chem. Soc., 52, 4319
(1930).
3. Clark G. L., J. Amer. Chem. Soc.,
41, 1477 (1919).
Фторид палладия(Ш) PdF3
2Pd4-3F2 ------*- 2PdF3
214 114 328
В приборе, описанном в методике получения IrF6 (реакцию можно про-
водить в кварцевой трубке, а не в трубке, изготовленной из плавикового
шпата), хлорируют порошок палладия при температуре темно-красного кале-
ния. Продукт хлорирования измельчают в порошок и еще раз хлорируют,
чтобы не осталось даже следов металлического палладия. Затем хлор вытес-
20*
308 Глава 3. Фтор
няют из прибора фтором, а температуру поднимают до 200—250 °C. По
охлаждении фтор вытесняют азотом, лодочку с PdF3 помещают в сухую тру-
бочку, в которой и хранят продукт в эксикаторе.
Свойства. М 164. Вещество черного цвета, решетка ромбическая; d 5,06.
Водой гидролизуется. Растворяется в кислотах и щелочах. Восстановление
водородом при комнатной температуре до металла может сопровождаться
вспышками.
ЛИТЕРАТУРА
1. Ruff О., Ascher Е„ Z. Anorg.
Allgem. Chem., 183, 1 (1929).
Фторид родия(П1) RhF3
2Rh + 3F2 ---> 2RhFs
206 114 320
Порошок родия в лодочке из плавикового шпата помещают в трубку из
'плавикового шпата (см. синтез IrF6). Прежде всего переводят родий под дей-
ствием хлора при температуре темно-красного каления в коричнево-красный
RhCl3. Полученный хлорид хорошо измельчают и фторируют при 600 °C в том
же самом приборе, не допуская попадания влаги. По охлаждении избыток
F2 из прибора вытесняют азотом.
Свойства. М 160. Негигроскопичное вещество красного цвета с ромбиче-
ской решеткой; d 5,38. Не растворяется в воде, разбавленных кислотах и ще-
лочах.
ЛИТЕРАТУРА
1. Ruff О., Ascher Е., Z. Anorg.
Allgem. Chem., 183, 1 (1929).
Фторид родия[¥1] RhF6
Rh + 3F2 ----> RhF6
103 114 217
Таким же способом, как описано в методике получения RuF6, на поро-
шок родия действуют фтором.
Свойства. Соединение черного цвета, в газовой фазе — темно-красио-ко-
ричневого; кубическая кристаллическая решетка. ~80°С. При комнат-
ной температуре термически неустойчиво. Давление пара 15 мм рт. ст. (0°С),
49,6 мм рт. ст. (20°C). Действует на стекло.
ЛИТЕРАТУРА
1. Cher nick С. L„ Claassen Н. Н.,
Weinstock В., J. Amer. Chem. Soc.,
83, 3165 (1961).
Фторид иридия(У) 1г15
2Ir + 5F2 ---► 2IrFb
386 190 576
Соединения с платиновыми металлами 309
В сосуде из монеля нагревают при 350—380 °C порошок иридия с рассчи-
танным количеством фтора. Крышку, закрывающую сосуд, охлаждают воз-
духом. С нижней стороны на крышке осаждаются желтые кристаллы субли-
мирующегося IrFs.
Свойства. М 288. Желтые моноклинные кристаллы. tn,, 104,5 °C.
ЛИТЕРАТУРА
1. Bartlett N., Rao Р. R., Chem.
Comm., 1965, 252.
Фторид иридия( VI) IrFp
Способ 1
Ir + 3F2 ------> IrFe
192,2 114 306,2
В трубку из плавикового шпата, снабженную электрическим обогревате-
лем, помещают лодочку из плавикового шпата с порошком иридия (рис. 160).
Иридий фторируют при 240 °C. Реакцию можно проводить также в никеле-
Рис. 160. Установка для получения фторида иридия(У1).
вой или платиновой трубке. Сначала F2 поступает в кварцевую ловушку 1,
охлаждаемую до —1704—183 °C; здесь вымораживается HF. Затем газ по-
ступает в зону реакции (печь). Газы нз зоны реакции проходят две U-образ-
ные трубки или ловушки (2 и 3) из кварца. Ловушка 2 охлаждается до
—78 °C, а ловушка 3— до —183 °C. Затвором от внешней среды служит ло-
вушка 4 (—183°C), которая не допускает проникновения влаги воздуха.
Трубка из плавикового шпата соединяется с кварцевыми частями уста-
новки посредством шлифов, которые уплотняют, смазывая снаружи смесью
асбеста с жидким стеклом.
Прежде всего установку промывают сухим азотом, а ее кварцевые части
прогревают для удаления остатков влаги. Затем к установке подключают по-
310 Глава 3. Фтор
ток F2, а газовые ловушки охлаждают. Как только F2 достигает лодочки с
иридием, из реакционной трубки появляются желтые пары IrF6, которые кон-
денсируются в ловушках. По окончании фторирования установку вновь про-
мывают азотом для вытеснения избыточного F2. Наконец, IrF6 очищают пу-
тем фракционной перегонки в вакууме (используют кварцевую установку без
кранов). Продукт собирают в кварцевые ампулы, которые быстро отпаивают.
Другие способы. IrFs можно получать также путем фторирования при
повышенном давлении (51 бар) в никелевом автоклаве при 240 °C (2 ч). Ме-
тодика работы аналогична описанной в синтезе ReF6 [2].
Свойства. Светло-желтые блестящие чешуйки и . иглы, которые выше
—15 °C окрашиваются в золотисто-желтый цвет и становятся похожими на
стеклянные. /11Л 44,4°C; /!ШП 53°C; dTB ~6 (—190°C). Кристаллизуется в
тетрагональной сингонии. IrF6 — очень гигроскопичное вещество; разъедает
стекло. Выше 400 °C разъедает платину. Восстанавливается галогенами при
комнатной температуре до IrF«.
Хранят IrF6 в кварцевых ампулах.
ЛИТЕРАТУРА
1. Ruff 0., Fischer J., Z. Anorg.
Allgem. Chem., 179, 166 (1929).
2. Slivnik J., Smale A., Zemljic А.,
Kurzreferat Intern. Fluor-Sympo-
sium, Miinchen, 1965, 184.
Фторид рутения(У) RuF5
2Ru -|- 5F2 --> 2RuFs
203,4 190 293,4
Через никелевую трубку, в которой в никелевой лодочке находится по-
рошок рутения, пропускают со скоростью 6 л/ч при 300 °C фтор, разбавлен-
ный азотом. На холодных местах трубки осаждается зеленый RuF5. Для
очистки препарат перегоняют в вакууме.
Свойства. М 196,7. Вещество изумрудно-зеленого цвета. tn л 86,5 °C; Ашп
227 °C. Кристаллизуется в моноклинной сингонии.
RuFs можно хранить при комнатной температуре в стеклянных сосудах,
совершенно исключив доступ влаги воздуха.
ЛИТЕРАТУРА
1. Holloway J. Н., Peacock R. D., J.
Chem. Soc., 1963, 527.
Фторид рутения(VI) RuF6
Ru-|-3F2 -—-> RuF6
102 114 216
В цилиндрический кварцевый сосуд (например, 3 см в диаметре), кото-
рый соединен с вакуумной установкой, помешают неглубокую никелевую чаш-
ку с порошком рутения. На 3 мм выше чашки с рутением находится пальчи-
ковый холодильник, изготовленный из золота или в крайнем случае из квар-
ца. Стенки сосуда, в котором проводится реакция, и пальчикового холодиль-
ника охлаждаются жидким азотом.
Перед началом синтеза и перед внесением порошка рутения сначала че-
рез установку пропускают водород, а затем нагревают; после откачивания во-
Соединения с платиновыми металлами 311
дорода установку заполняют фтором и нагревают, так как обычно RuF6 реа-
гирует с никелем. Для проведения реакции установку заполняют фтором, соз-
давая давление 300 мм рт. ст. Порошок рутения нагревают с помощью ин-
дукционной печи до тех пор, пока он не сгорит в атмосфере фтора при
~ 750 °C. Затем реакционный сосуд тотчас же охлаждают жидким азотом,
•благодаря чему RuF6 осаждается на холодных поверхностях в виде твердого
коричневого продукта. По окончании фторирования продукт перегоняют в
вакууме. Выход 49% теоретического.
Свойства. Темно-коричневые октаэдрические кристаллы. /,1Л 54 °C,
,р 23 мм рт. ст. (0°С), 67 мм рт. ст. (17,6°C). Термически устойчив до 200°C.
RuF6 очень сильно разъедает стекло, посуду, слабо действует на кварце-
вое стекло и может храниться длительное время в никелевых сосудах.
ЛИТЕРАТУРА
1. Claassen Н. Н., Selig Н„ Malm I. Amer. Chem. Soc., 83, 2390 (1961);
G., Chernick C. L„ Weinstock B„ J. Chem. Eng. News, 39, 56 (1961).
•Фторид осмия(У) OsFs
10OsF6 + I2 ----> 10OsFs + 2IF5
3040 254 2850 444
В сосуд из боросиликатного стекла к известному отвешенному количест-
ву 12 вводят путем конденсации избыток OsF6 и 5-кратное количество IF5 в
качестве растворителя. Систему приводят к комнатной температуре и далее
подогревают до 50 °C. Затем отгоняют OsFg и IF5. Зеленое вещество в остат-
ке перегоняют в вакууме; OsF5 отгоняется при 120 °C. Остаток содержит еще
немного OsF«, который перегоняется в вакууме в интервале 280—300 °C и в
приемнике при 200 °C застывает в желтую массу.
Свойства. Af 285. Вещество голубовато-зеленого цвета. Кристаллизуется
в моноклинной сингонии. 1Пл 70 °C; /кип 233 °C.
ЛИТЕРАТУРА
1. Hargreaves G. В., Peacock R. D.,
J. Chem. Soc., 1960, 2618.
Фторид осмия[У1) OsF6
Os-}-3F2 ---->- OsF6
190 114 304
Лодочку из пирекса с порошком осмия помещают в реакционную трубку
из пирекса. В потоке F2 осмий сгорает прн 250 °C (методика аналогична опи-
санной в синтезе SF6). К реакционной трубке присоединена ловушка, кото-
рую охлаждают до —100°C (—78°C недостаточно). В ловушке количествен-
но осаждается OsF6. Следом за этой ловушкой-приемником присоединена еще
одна ловушка, охлаждаемая жидким азотом, для защиты от влаги воздуха.
По окончании фторирования OsF6 фракционируют в вакууме. Выход количе-
ственный.
Свойства. Вещество желтого цвета. tna 32,1 °C; £KIln 45,9 °C.
ЛИТЕРАТУРА
1. Weinstock В., Malm J. G., J. Amer.
Chem. Soc., 80, 4466 (1958).
312 Глава 3. Фтор
Фторид осмия(УН) OsF7
2Os-f-7F2 ---> 2OsF,
380 266 646
Реакцию проводят в автоклаве из никеля (длина 230 мм, внешний диа-
метр 80 мм, внутренний диаметр 20 мм). Автоклав закрывается крышкой,
имеющей коническую прокладку, которая с помощью перекидной гайки из
стали плотно прижимается к автоклаву. Коническая прокладка просверлена
по центру (диаметр отверстия 4 мм) и вверху имеет внешнюю винтовую резь-
бу, на которую навинчивается короткий водяной холодильник и вентиль (из
специальной стали) с мембранным уплотнением. Перед синтезом автоклав,
заполненный фтором, нагревают при 500 °C; после охлаждения автоклав эва-
куируют и заполняют азотом или аргоном. Вносят 0,3—1 г порошка осмия
(или OsF6); автоклав вновь эвакуируют, охлаждают до —196 °C и конденси-
руют в него F2. После этого автоклав постепенно нагревают до 500—600 °C,
причем устанавливают в нем давление 350—400 бар. По достижении необхо-
димой температуры содержимое горячего автоклава выпускают через никеле-
вую ловушку, охлаждаемую жидким азотом, а затем отсасывают избыточный
F2. В ловушке остается голубовато-желтый OsF7.
Свойства. М 323. Вещество голубовато-желтого цвета, чрезвычайно гиг-
роскопичное. Разлагается уже ниже —100 °C на OsF6 и F2.
Препарат можно хранить в сосудах из никеля, охлаждаемых жидким
азотом или жидким воздухом.
ЛИТЕРАТУРА
1. Glemser О., Roesky Н. W., Hell-
berg Н.-Н., Werther H.-U., Chem.
Ber., 99, 2652 (1966).
Фторид криптона(Н) KrF2
Кг + Fa ------> KrF2
83,7 38 121,7
Через установку, включающую газоразрядную трубку из пирекса, цирку-
лирует со скоростью 1300 мл/мин смесь Кг и F2 (1:2) с помощью магнитно-
го насоса. Установка состоит из газоразрядной трубки, двух последовательно
соединенных U-образных трубок (в которых имеются места для отпаивания
и вентили для прерывания потока) и магнитного насоса. Вентили из моиеля
служат для введения исходных веществ и эвакуирования установки.
В газоразрядной трубке (диаметр 60 мм, высота 200 мм) находятся в
качестве электродов две медные пластинки (диаметр 20 мм, толщина 5 мм),
впаянные на расстоянии 75 мм. Газоразрядную трубку можно погружать в
сосуд Дьюара с жидким кислородом (—183°C).
Перед синтезом в установке создают высокий вакуум и прогревают, за-
полняют фтором и оставляют на 12 ч, затем вновь эвакуируют. Газоразряд-
ную трубку охлаждают в жидком кислороде и строго поддерживают в уста-
новке давление 12—50 мм рт. ст. Включают магнитный насос, а затем и га-
зоразрядную трубку (3000—4000 В, 15 мА). Образующийся KrF2 осаждается
на стенках газоразрядной трубки.
По окончании синтеза откачивают насосом избыток газов, отключают
Магнитный насос и создают в газоразрядной трубке температуру —78 °C
(смесью сухого льда с трихлорэтиленом), оставляя включенный вакуумный
насос еще на 1 ч. Далее охлаждают одну из U-образных трубок в сухом
Соединения с инертными газами 313
льду и постепенно поднимают температуру газоразрядной трубки до комнат-
ной; вакуумный насос все это время должен работать. Таким образом, KrF2
сублимируется и переводится в U-образную трубку. Сублимацию можно по-
вторить несколько раз. Части установки, в которых больше не нуждаются,
отпаивают.
Таким образом, за 1 ч получают 0,25 г KrF2. Выход составляет 75% в
расчете на прореагировавший криптон.
Свойства. Белое вещество. Кристаллизуется в тетрагональной сингонии.
Самопроизвольно разлагается при комнатной температуре. Гидролизуется во-
дой с образованием Кг, HF и О2. Окисляет C1F3 до ClFg, Хе до XeF6. С SbFs
образует при —20 °C аддукт KrF2-2SbF5.
KrF2 можно хранить в сосудах из пирекса или полиэтилена при —78 °C.
ЛИТЕРАТУРА
1. Schreiner F., Malm J. G., Hind-
man J. C„ J. Amer. Chem. Soc., 87,
25 (1965).
Фторид ксенона(П) XeF2
Xe -|- F2 > XeF2
131 38 169
В приборе для облучения (рис. 161), изготовленном из никеля и снабжен-
ном окошками из синтетического сапфира толщиной 3 мм (вакуумное уплот-
нение), облучают смесь Хе и F2 (1 :2) при комнатной температуре.
Рис. 161. Прибор для получения
фторида ксенона (II).
В качестве источника облучения служит ртутная лампа (например, мощ-
ностью 1000 Вт, General Electric АН-6), которая дает свет с длинами волн в
области 230—350 нм. Перед окошком помещают в качестве фильтра кварце-
вую кювету с водным раствором CoSO4 и NiSO4 (45 г CoSO4-7H2O и 500 г
NiSO4-6H2O на 1 л раствора). Для быстрого удаления образовавшегося
XeF2 из зоны реакции используют циркуляцию потока, которая осуществля-
ется благодаря тому, что одна часть вертикальной циркуляционной трубки
находится при 100°C, а другая—при —78°C. Во время облучения давление
в трубке понижается, и на стенках конденсируется бесцветный кристалличе-
ский налет. После облучения препарат сублимирует в вакууме, причем обра-
зуются кристаллы в несколько -миллиметров длиной. За 1—2 сут можно по-
лучить 2 г XeF2.
314 Глава 3. Фтор
Свойства. Бесцветные тетрагональные кристаллы. /пл 140 °C; р 3,8 мм
рт. ст. (25°C), 318 мм рт. ст. (100°C); d 4,32. При комнатной температуре-
устойчив. Имеет удушливый запах, вызывает тошноту.
ЛИТЕРАТУРА
1. Weeks J. L., Chernick С. L., Mathe- 2. Hoppe R., Mattauch H., Rodder R.
son M. S. J., J. Amer. Chem. Soc., M., Angew. Chem., 74, 903 (1962),
84, 4612 (1912).
Фторид ксенона(1У) XeF4
Xe-j-2F2 ----> XeF4
131 76 207
В автоклав из никеля (вместимостью 130 мл) конденсируют при темпе-
ратуре жидкого азота 0,36 г Хе и 0,52 F2; автоклав нагревают в течение 4 ч
при 400 °C. При этом давление повышается максимально до 10 бар. Затем ав-
токлав быстро охлаждают до —78 °C и откачивают избыток F2 через ловуш-
ку (—195 °C); при этом в ловушке конденсируется также непрореагировав-
ший ксенон, a XeF4 остается в реакторе. Вся аппаратура предварительно про-
гревается в потоке F2. Необходимо обращать внимание на то, чтобы в реак-
тор не попала влага, так как это может быть причиной сильных взрывов из-
за образования ХеО3. XeF4 можно легко сублимировать в вакууме и при
этом получить крупные бесцветные кристаллы.
Если не располагают никелевым автоклавом, то можно пропустить через
никелевую трубку при 300 °C газовую смесь Xe+F2, причем время пребыва-
ния газов в зоне реакции должно составлять 1 мин. При этом XeF4 осажда-
ется в кристаллической форме на холодных участках установки. Выход поч-
ти 100%.
Свойства. Бесцветные кристаллы с моноклинной структурой. tna 114 °C,
р 3 мм рт. ст. (20°C); d 4,10. Умеренный по активности фторирующий агент.
Реагирует с Н2 при 70—130 °C. Дает с Хе при 400 °C XeF2, а с F2 при
300 °C — XeF6. С SF4 дает SF6+Xe.
XeF4 можно хранить (практически неограниченно) в емкостях из никеля
или монеля при комнатной температуре, если исключить доступ влаги воз-
духа.
ЛИТЕРАТУРА
1. Claassen Н. Н., Selig Н., Malm J. 2. Templeton D. Н„ Zalkin A., Forres-
G., J. Amer. Chem. Soc., 84, 3593 ter J. D., Williamson S. M., J.
(1962). Amer. Chem. Soc., 85, 242 (1963).
Фторид ксенона( VI) XeF6
Xe-}-3F, ----> XeF6
131 114 245
В никелевом автоклаве под давлением 60 бар при 300 °C проводят реак-
цию (в течение 16 ч) между Хе и F2, взятыми в молярном отношении 1 :20
(например, 0,68 г Хе+4,18 г F2). Предпочтительно, однако, работать при дру-
гом режиме: 700 °C и 200 бар. Продукты реакции охлаждают до —78 °C и
откачивают избыток F2. Остаток сублимируют в вакууме. Надо не допускать
попадания влаги, потому что это приводит к образованию ХеОз, который
сильно взрывает.
Формульный указатель 315
Свойства. Ниже 42 °C — это бесцветное вещество, выше 42 °C окрашено
в желтый цвет, так же как и в газовой фазе. £Пл 46 °C; р пара 3 мм рт. ст.
(О°C), 20 мм рт. ст. (20°C). Реагирует с водой и кварцем с образованием
XeOF4 и ХеО3. С водородом бурно реагирует с образованием Хе и HF.
С BF3 и AsFs дает аддукты XeF6-BF3 и XeF6-AsF6. Взаимодействует с Hg.
XeF6 можно хранить в емкостях из никеля неограниченно долго при ком-
натной температуре.
ЛИТЕРАТУРА
1. Malm J. G., Sheft L, Chernick C. L„
J. Amer. Chem. Soc., 85, 110
(1963).
Формульный указатель
A C0F3 304 G
Ag[BF4] 276 CrF2 290 rrF„ 9Q1 GaF3 265
AgF 274 Ag2F 273 AgF2 275 A1FS, A1F3 • 3H2O 263 AsF3 239 2AsF3 • 3SO3 240 AsF5 241 A11F3 276 CrF4 291 CrFs 291 CrF6 292 CrO2F2 292 CsF 272 Cs[IF<] 271 CuF2 273 GeF2 255 GeF4 256 H H2 147 H[BFJ 261 HD 163 HF 185
D H2O 152, 154
В H2O2 177
BF3 259 D2 156, 158 H2[ReF6] 301
BaF2 269 DBr 167 HSO3F 216
BeF2 268 DC1 165 H2SiF6 255
BiF3 243 DF 164 HgF2 278
BiFs 244 DI 169 Hg2F2 278
BrF3 190 D3PO4 174 HgF2 2H2O 280
BrFs 192 D2S 171
BrFCO 250 D2SO4 172 1
BrFSO2 215 IF 193
E IF з 194
C EuF2 281 IF з C5H5N 195
CF4 245 IF s 196
CHF3 246 F IF 7 196
CIF3 247 IFCO 251
COBrF 250 F2 183 InF3 266
COC1F 249 FCN 252 IrFs 308
CaF2 269 (FCN)3 252 IrF6 309
CdF2 278 F2CO 248 К
CeF4 282 F2SO 209
C1F 187 F2SO2 211 К [AgF4] 276
CIF3 189 FSO2K 217 к AuF4] 277
C1FCO 249 FSO2NO 226 к BFJ 262
C1FSO2 214 FSSF 203 к BF3OH] 262
C1O2F 201 FSeO2K 219 к BrF4] 270
CIO3F 202 FeF2 301 к BrF6] 271
CIO4F 202 FeF3 302 к C1FJ 270
C0F2 303 Fe2F5 302 KF 270
316 Формульный указатель
K2[GeF6] 257 NO3F 227 SOF2 209
К IF4] 271 NOSO2F 226 SOF4 210
К IF6] 272 NSF 228 SO2F2 211
к MnF3 297 Na[BF4] 261 S2O5F2212
к MnF4 298 NbFs 288 S2O6F2 213
к MnF5' 298 NiF2 304 SOF4-AsF5 21F
К2 MnFe 298 [SOF3][AsF6] 211
KlNbbj] 289 0 SSF2 204
K[NiF3] 305 OF2 200 SbCl2F3 243
K2[NiF6] 305 O2F2 198 O3F2 198 OsFs 311 SbF3 241
К [PFeJ 239 SbFs 242
K2[PtF6] 307 . ScF3 280
K2[ReF6] 301 vv 01 Oil OsF6 311 SeF4 218
KSO2F 217 OsF7 312 SeF6 218
KSeO2F 219 p SeOF2 218
K2[TaF7] 290 KrF2 312 PBr2F 236 PBr4F 236 SiF4 253 SiHF3 254
PCIF4 234 SmF2 281
L PC12F 232 SnF2 257
LaF3 281 PC12F3 234 SnF4 257
LaOF 283 POUF 234 SrF2 269
LnF3 281 PC14 • PF6 235 T
LnOF 283 PF3 230 1
PF5 232 TaF3 289
M (PNF2)3 237 TeF6 220
MgF2 269 MnF2 295 MnF3 296 MnF4 296 MnO3F 297 MoF3 294 (PNF2)4 237 POF3 235 PbF2 258 PbF4 258 PdF3 307 PtF5 305 TiF3 283 TiE> 285 T1F 267 T1F3 267 V
MoFs 294 PtF6 306 VF3 287
MoF6 294 n VF4 287
К VF5 288
N RbF 272
ND3 173 Rb[lF4] 272 W
NF3 220 N2F2 223 ReF6 299 ReF7 299 WF6 295
N,F4 222 ReO3F 300 X
(NH4)[A1F4] 264 (NH4)3[A1F6J 264 (NH4)2[BeF4J 268 NHF2 222 RhF3 308 RhF6 308 RuFs 310 RuF6 310 XeF2 313 XeF4 314 XeF6 314
(NH4)3[GaF6] 265 c v
(NH4)3[InF6] 266 0 I
NH4[PF6] 238 SC1F6 208 YbF2 281
NH4[PO2F2] 238 SF4 205
NO[BF4] 262 SF6 207
NOC1F4 190 S2Fio 206 Z
NOF 224 SNF3 229 ZnF2 277
NOF 3HF 225 SO2BrF 215 ZrF3 286
NO2F 226 SOC1F 214 ZrF4 286
SO2C1F 214
СОДЕРЖАНИЕ
Предисловие к переводу (5)
Предисловие редактора 3-го издания (8)
Часть I. Методы препаративной химии./7. В. Шенк.Р. Штой-
дель, Г. Брауэр (9)
1. Сборка лабораторных установок (10). 2. Стекло (11).
3. Керамические материалы (21). 4. Металлы (34). 5. Синте-
тические материалы (38). 6. Чистые и обезвоженные раство-
рители (41). 7. Замазки и смазочные вещества (45). 8. Ме-
тоды измерения температуры (49). 9. Высокие температуры
(53). 10. Низкие температуры (64). 11. Постоянные темпера-
туры (68). 12. Регуляторы температуры (71). 13. Высокий
вакуум и изоляция от воздуха (74). 14. Газы (107). 15. Ра-
бота со сжиженными газами в качестве растворителей (120).
16. Проведение процессов в электрических разрядах (126).
17. Очистка веществ (128). 18. Испытание на чистоту (142).
19. Реакции между веществами в порошкообразном состоя-
нии (145).
Часть II. Элементы и соединения (147)
Глава 1. Водород, дейтерий, вода. М. Баудлер (147).
Водород (147). Вода наивысшей чистоты (152). Вода, пред-
назначенная для измерения электропроводности (154). Дей-
терий и соединения дейтерия (156). Дейтерий (158). Дейтеро-
водород (163). Фторид дейтерия (164). Хлорид дейтерия
(165). Бромид дейтерия (167). Иодид дейтерия (169). Сульфид
дейтерия (171). Дейтеросерная кислота (безводная) (172).
Дейтероаммиак (173). Дейтерофосфорная кислота (безвод-
ная) (174).
Глава 2. Пероксид водорода. М. Шмайсер, Ф. Губер (177).
Правила техники безопасности (177). Удаление воды из Н2О2
(177).
Глава 3. Фтор. В. Квасник (180)
Общая характеристика фтора и его соединений (180). Фтор
(183). Фтороводород (185). Фторид хлора(1) (187). Фторид
хлора(Ш) (189). Тетрафторид-хлорид нитрозпла (190). Фто-
рид брома(Ш) (190). Фторид брома(У) (192). Фторид ио-
да(1) (193). Фторид иода(Ш) (194). Аддукт фторида
иода(Ш) с пиридином (195). Фторид иода(У) (196). Фторид
иода(VII) (196). Дифторид трикислорода (198). Дифторид
-318 Содержание
днкислорода (198). Фторид кислорода (200). Фторид хлори-
ла (201). Фторид перхлорила . (202). Фторид хлоротетраокси-
да (202). Дифтородисульфан (203). Дифторид дисеры (204).
Фторид серы (IV) (205). Декафторид дисеры (206). Фторид
серы (VI) (207). Пентафторид хлоросульфинила (208). Фто-
рид сульфинила (209). Тетрафторид сульфинила (210). Ад-
дукт тетрафторида сульфинила и пентафторида мышьяка
(211). Дифторид сульфонила (211). Дифторид дисульфурила
(212). Оксодифторид дисульфурила (213). Хлорид-фторид
сульфинила (214). Хлорид-фторид сульфонила (214). Бромид-
фторид сульфонила (215). Фторосерная кислота (216). Фто-
росульфонат калия (217). Фторид селена(У1) (218). Фторид
селена(1\7) (218). Дифторид селениЛа (218). Фтороселенит
калия (219). Фторид теллура^!) (220). Фторид азота(III)
(220). Дифторамин (222). Тетрафторогидразин (222). Ди-
фторид диазота (223). Фторид нитрозила (224). Аддукт фто-
рида нитрозила с фтороводородом (225). Нитрозосульфонил-
фторид (226). Фторид нитроила (226). Триоксид-фторид азота
(227). Тиофторид азота (228). Трифторид-нитрид серы
(229). Фторид фосфора(III) (230). Фторид фосфора (V)
(232). Дихлорид-фторид фосфора (III) (232). Дихлорид-три-
фторид фосфора (V) (234). Тетрахлорид-фторид фосфора
(234). Хлорид-тетрафторид фосфора (234). Трифторид фосфо-
рила (235). Гексафторофосфат тетрахлорофосфония (235).
Дибромид-фторид фосфора (236). Тетрабромид-фторид фос-
фора (236). Фосфонитрилфториды (237). Гексафторофосфат
аммония (238). Диоксодифторофосфат аммония (238). Гекса-
фторофосфат калия (239). Фторид мышьяка(Ш) (239). Ад-
дукт 2 AsF3-3SO3 (240). Фторид мышьяка (V) (241). Фторид
сурьмы (III) (241). Фторид сурьмы (V) (242). Дихлорид-три-
фторид сурьмы (243). Фторид висмута(Ш) (243). Фторид
висмута (V) (244). Тетрафторид углерода (245). Трифторме-
таи (246). Трифториодметан (247). Оксид-дифторид углерода
(248). Хлорид-фторид карбонила (249). Бромид-фторид кар-
бонила (250). Иодид-фторид карбонила (251). Цианурфторид
(252). Фтороциан (252). Фторид кремния (253). Трифторо-
силан (254). Гексафторокремневая кислота (255). Фторид
герМания(П) (255). Фторид repMaHHH(IV) (256). Гексафто-
рогермаиат калия (257). Фторид олова(П) (257). Фторид
олова(IV) (257). Фторид свинца(II) (258). Фторид свин-
na(IV) (258). Фторид бора (259). Фтороборная кислота
(261). Тетрафтороборат натрия (261). Тетрафтороборат ка-
лия (262). Гидроксотрифтороборат калия (262). Тетрафто-
роборат нитрозила (262). Фторид алюминия (263). Тригидрат
фторида алюминия (263). Гексафтороалюминат аммония
(264). Тетрафтороалюминат аммония (264). Фторид гал-
лия(Ш) (265). Гексафторогаллат аммония (265). Фторид
индия (III) (266). Гексафтороиндат аммония (266). Фторид
таллия(I) (267). Фторид таллия(III) (267). Фторид берил-
лия (II) (268). Тетрафторобериллат аммония (268). Фторнды
щелочноземельных металлов (269). Фторид калия (270). Тет-
рафторохлорат(Ш) калия (270). Тетрафторобромат(Ш) ка-
лия (270). Гексафторобромат(V) калия (271). Тетрафторо-
иодат(Ш) цезия (271). Гексафтороиодат(V) калия (272).
Фториды рубидия и цезия (272). Фторид меди (II) (273).
Фторид дисеребра (273). Фторид серебра (I) (274). Фторид
серебра(П) (275). Тетрафтороборат серебра (276). Тетрафто-
Содержание 319
роаргентат(Ш) калия (276). Фторид золота(Ш) (276). Тет-
рафтороаурат(Ш) калия (277). Фторид цинка(П) 277). Фто-
рид кадмия(П) (278). Фторид ртути(1) (278). Фторид рту-
ти (II) (278). Дпгидрат фторида ртути(II) (280). Фторид
скандия (III) (280). Трифториды редкоземельных элементов
(281). Дифториды редкоземельных элементов (281). Фторид
церия (IV) (282). Оксид-фториды редкоземельных элементов
(283). Фторид титана(III) (283). Фторид титана(IV) (285).
Фторид циркония(III) (286). Фторид циркония(IV) (286).
Фторид ванадия (III) (287). Фторид ванадия (IV) (287). Фто-
рид ванадия (V) (288). Фторид ниобия (V) (288). Гептафто-
рониобат калия (289). Фторид тантала(У) (289). Гептафто-
ротанталат калия (290). Фторид хрома(II) (290). Фторид
хрома(Ш) (291). Фторид xpoMa(IV) (291). Фторид хро-
Ma(V) (291). Фторид xpoMa(VI) (292). Фторид хромила
(292). Фторид молибдена(Ш) (294). Фторид молибдена (V)
(294). Фторид молибдена(У1) (294). Фторид вольфрама(VI>
(295). Фторид марганца(П) (295). Фторид маргапца(Ш)
(296). Фторид марганца (IV) (296). Триоксид-фторид марганца
(297). Трифтороманганат(П) калия (297). Тетрафтороманга-
нат (П1) калия (298). Пентафтороманганат(IV) калия (298).
Гексафтороманганат(IV) калия (298). Фторид рения(У1)
(299). Фторид рения(УП) (299). Триоксид-фторид рения (300).
Гексафтороренат (IV) калия (301). Гексафторорениевая кис-
лота (301). Фторид железа(П) (301). Фторид желе-
за (II, III) (302). Фторид железа(III) (302). Фторид ко-
бальта(П) (303). Фторид кобальта(Ш) (304). Фторид нике-
ля(П) (304). Трифтороникелат(П) калия (305). Гексафторо-
никелат(ГУ) калия (305). Фторид платины (V) (305). Фторид
платины (VI) (306). Гексафтороплатинат(IV) калия (307).
Фторид палладия (III) (307). Фторид родия (III) (308). Фто-
рид родия (VI) (308). Фторид иридия (V) (308). Фторид ири-
дия (VI) (309). Фторид рутения (V) (310). Фторид руте-
hhh(VI) (310). Фторид осмия(У) (311). Фторид осмия(У1)
(311). Фторид осмия(УП) (312). Фторид криптона(П) (312).
Фторид ксенона(П) (313). Фторид ксенона(1У) (314). Фто-
рид ксенона(VI) (314).
Формульный указатель (315).
СОДЕРЖАНИЕ ДРУГИХ ТОМОВ
Том 2
Глава 4. Хлор, бром, иод
Тлава 5. Кислород, озон
Глава 6. Сера, селен, теллур
Глава 7. Азот
Глава 8. Фосфор
Тлава 9. Мышьяк, сурьма, висмут
Том 3
Глава 10. Углерод
Тлава 11. Кремний, германий
Глава 12. Олово, свинец
Глава 13. Бор
Глава 14. Алюминий
Глава 15. Галлий, индий, таллий
Тлава 16. Щелочноземельные метал-
лы
Глава 17. Щелочные металлы
Том 4
Глава 18. Медь, серебро, золото
Глава 19. Цинк, кадмий, ртуть
Глава 20. Редкоземельные металлы
Глава 21. Актиноиды
Глава 22. Титан, цирконий, гафний
Том 5
Глава 23. Ванадий, ниобий, тантал
Глава 24. Хром, молибден, вольфрам
Глава 25. Марганец
Глава 26. Технеций
Глава. 27. Рений
Глава 28. Железо
Глава 29. Кобальт, никель
Глава 30. Платиновые металлы
Том 6 ' ”
Часть III. Особые группы со-
единений
Глава 31. Гидроксосоли
Глава 32. Изо- и гетерополисоедине-
ния
Глава 33. Металлоорганические комп-
лексные соединения
Глава 34. Сплавы и интерметалли-
ческие соединения
Предметный указатель
М. Баудлер, Г. Брауэр, Ф. Губер и др.
РУКОВОДСТВО ПО НЕОРГАНИЧЕСКОМУ СИНТЕЗУ
Редактор Георг Брауэр
Том 1
Научный редактор Н. А. Васина
Мл. научный редактор И. А. Колчин
Художник И. Б. Кравцов
Художественный редактор М. Н. Кузьмина
Технический редактор Т. А. Максимова
Корректор В. И. Киселева
ИБ № 3943
Сдано в набор 22.05.84. Подписано к печати 23.10.84. Формат бОХЭО’Лв.
Объем 10 бум. л. Бумага типографская № 1. Гарнитура литературная.
Печать высокая. Усл. печ. л. 20. Усл. кр.-отт. 21. Уч.-изд. л. 24,65.
Изд. № 3/3092. Тираж 8.000 экз. Зак. 143. Цена 1 р. 80 к.
ИЗДАТЕЛЬСТВО «МИР».
129820, Москва, И-110, ГСП, 1-й Рижский пер., д. 2.
Московская типография № II Союз полиграф пром а при Государственном
комитете СССР по делам издательств, полиграфии и книжной торговли. .
Москва, 113105, Нагатинская ул., д. 1.