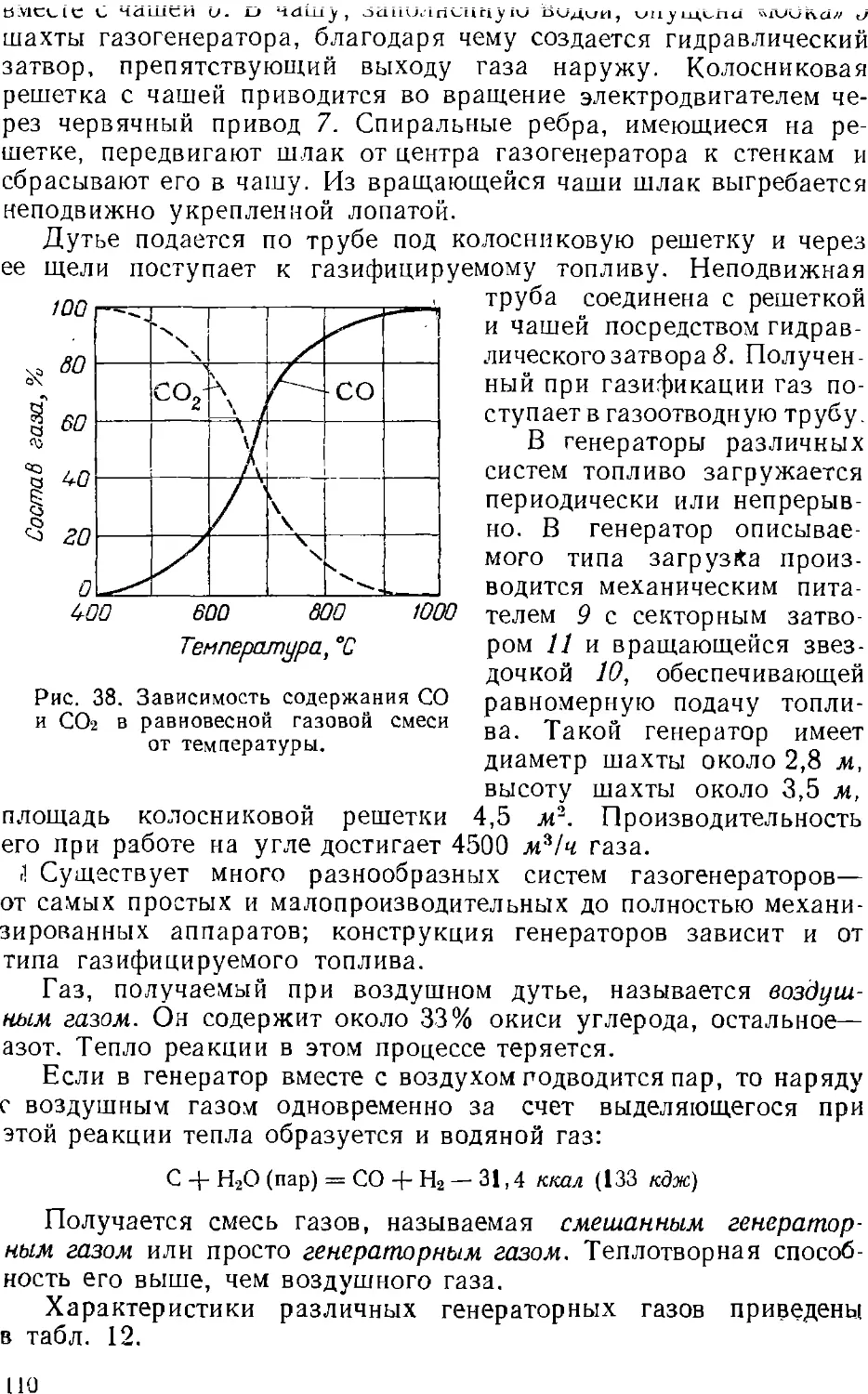
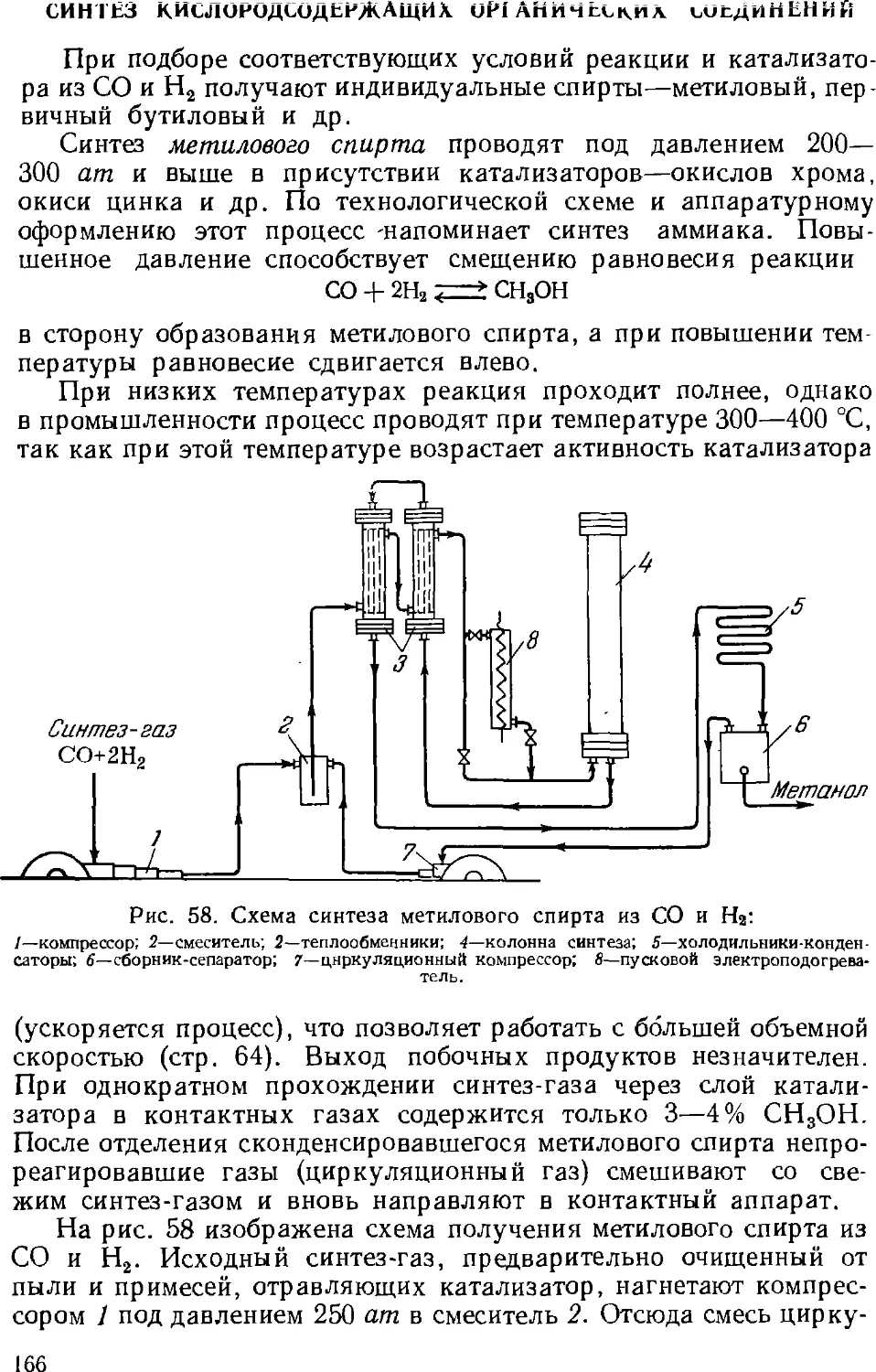
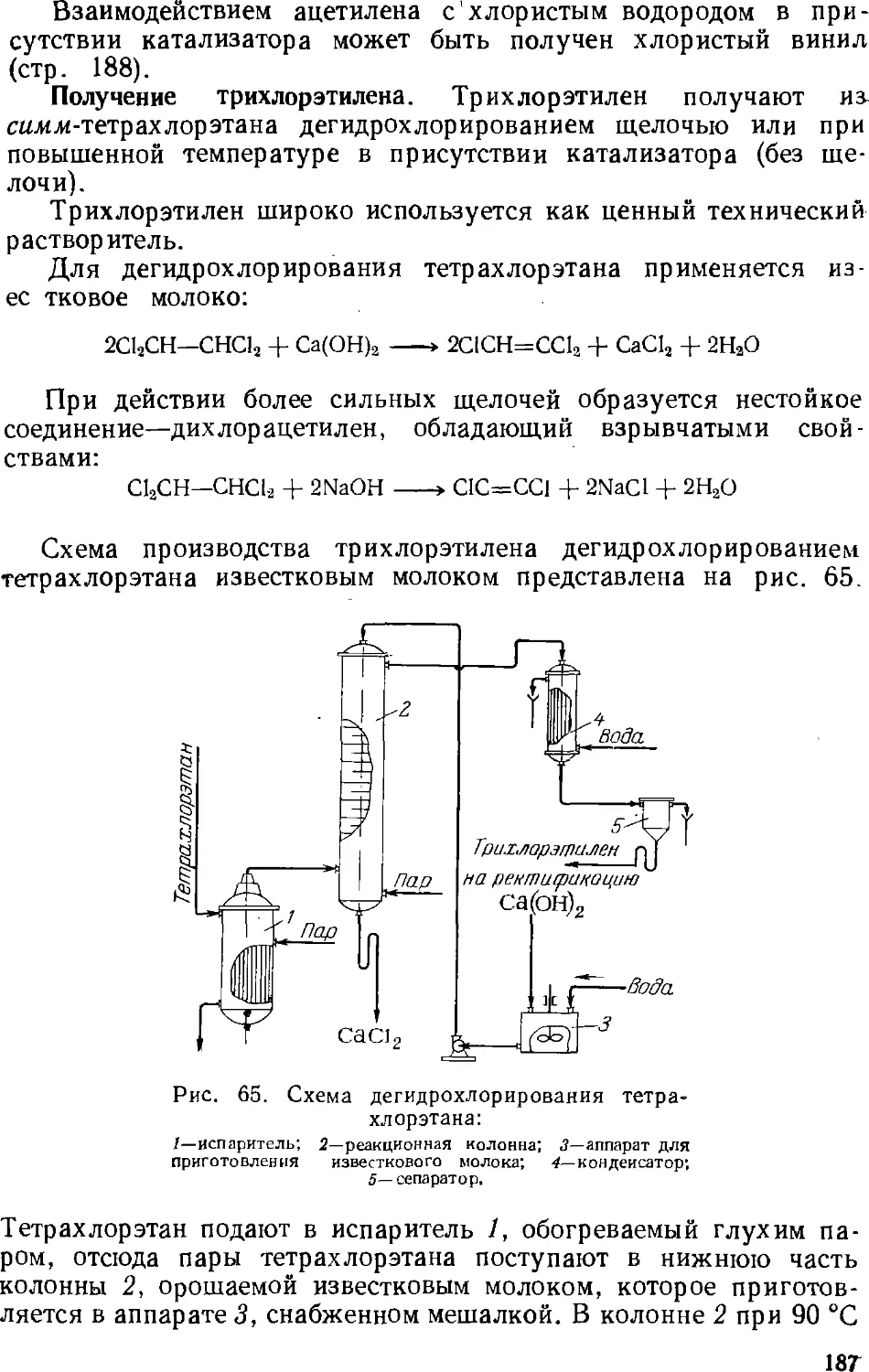
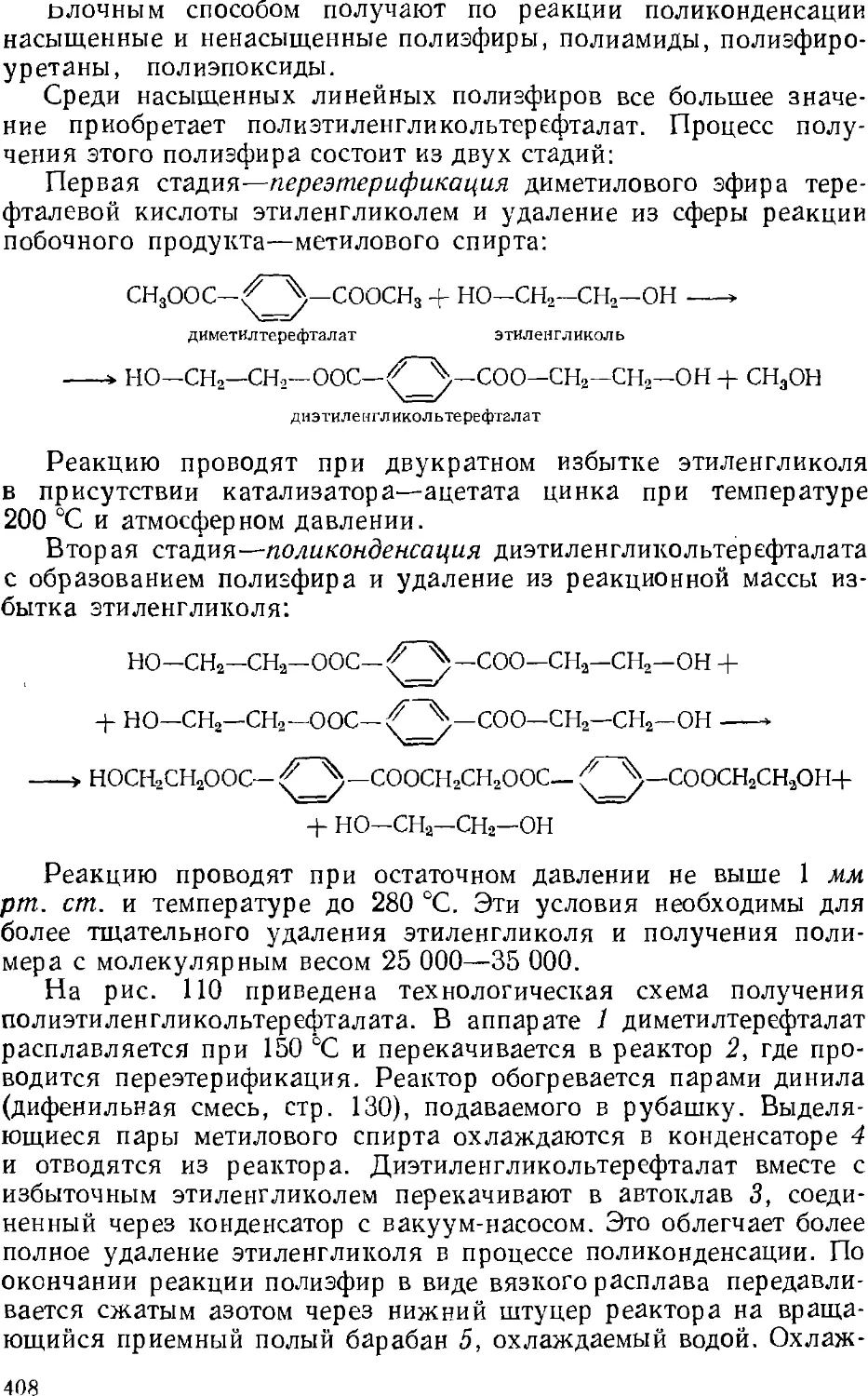
/


































































































































































































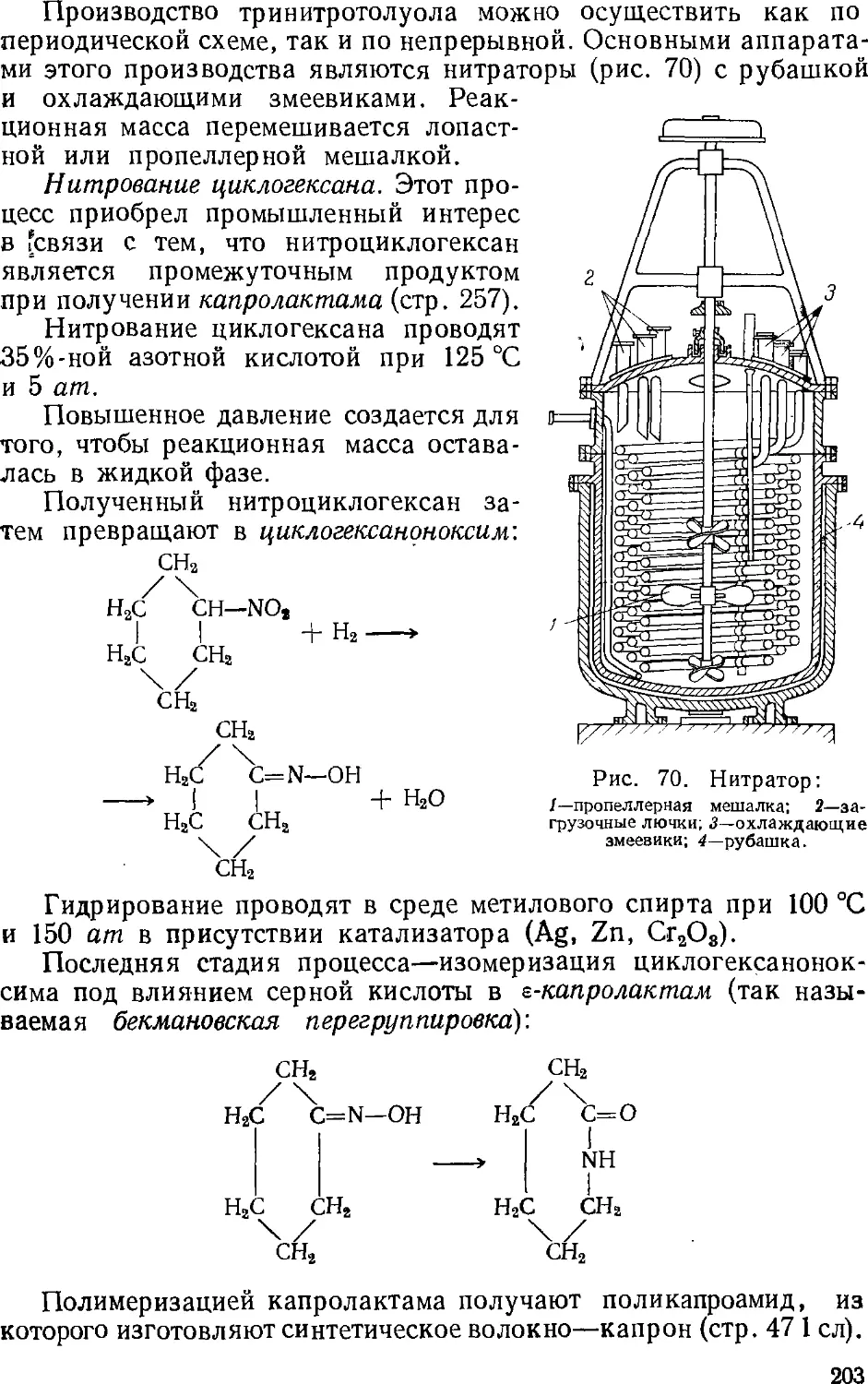




















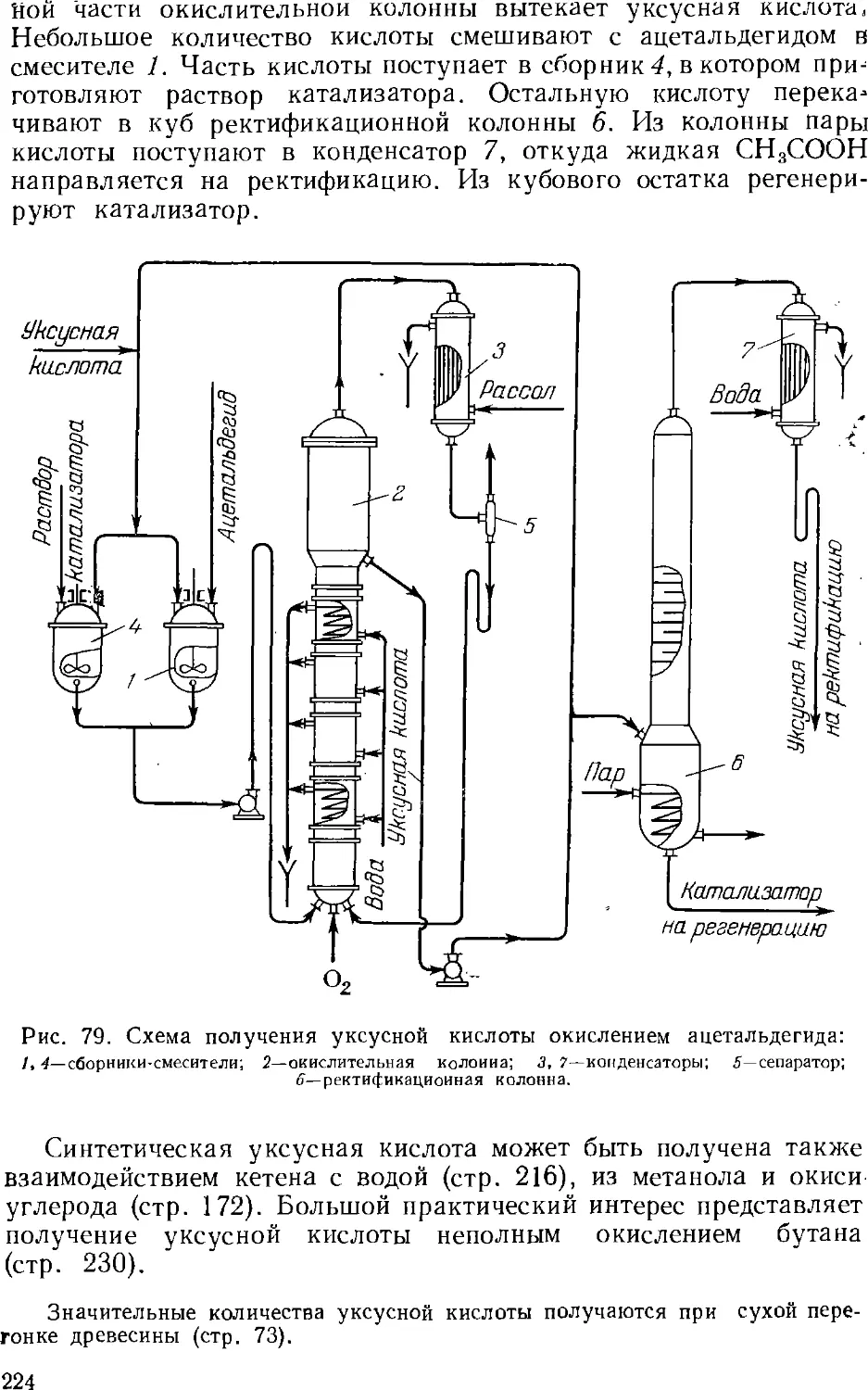
























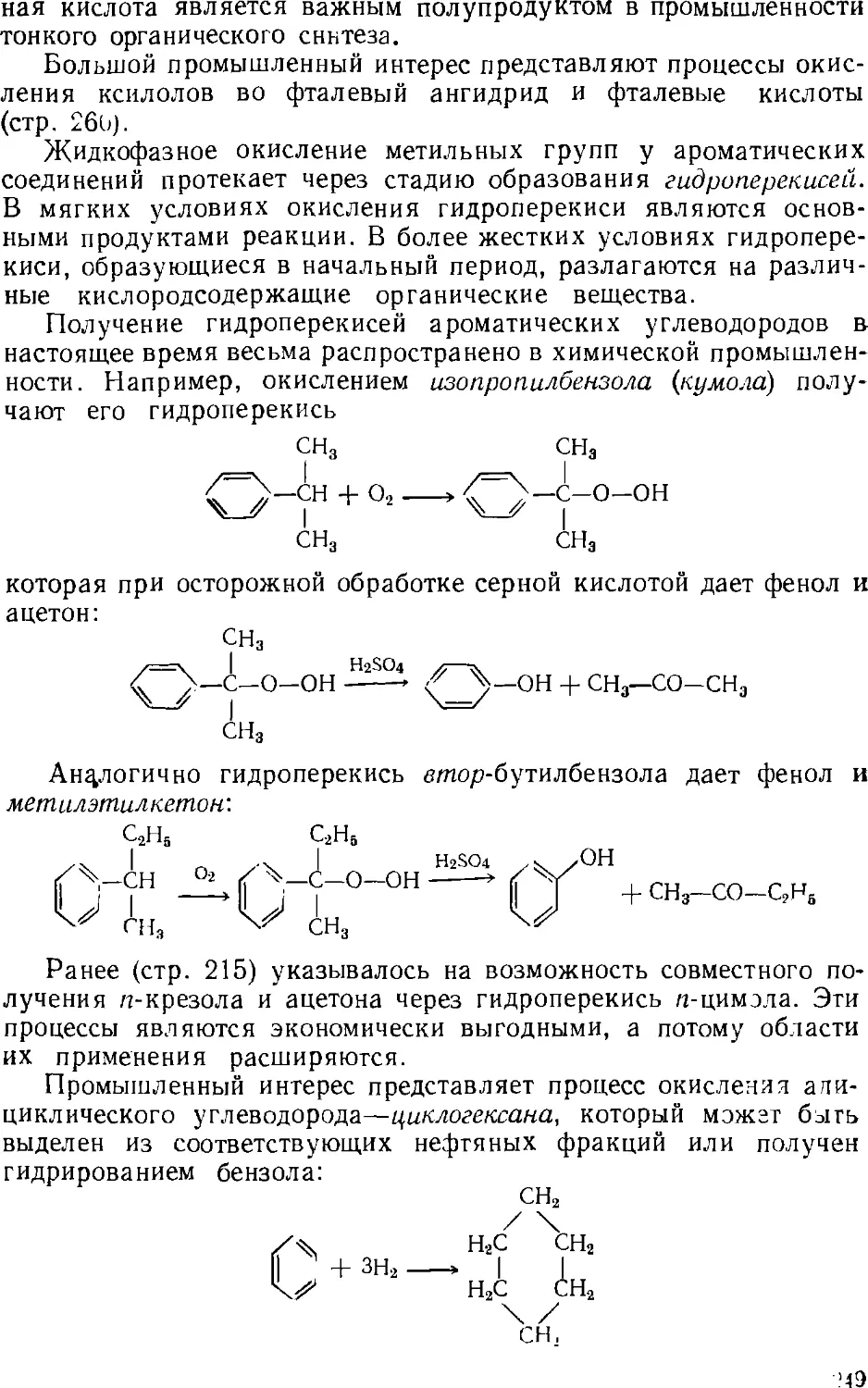

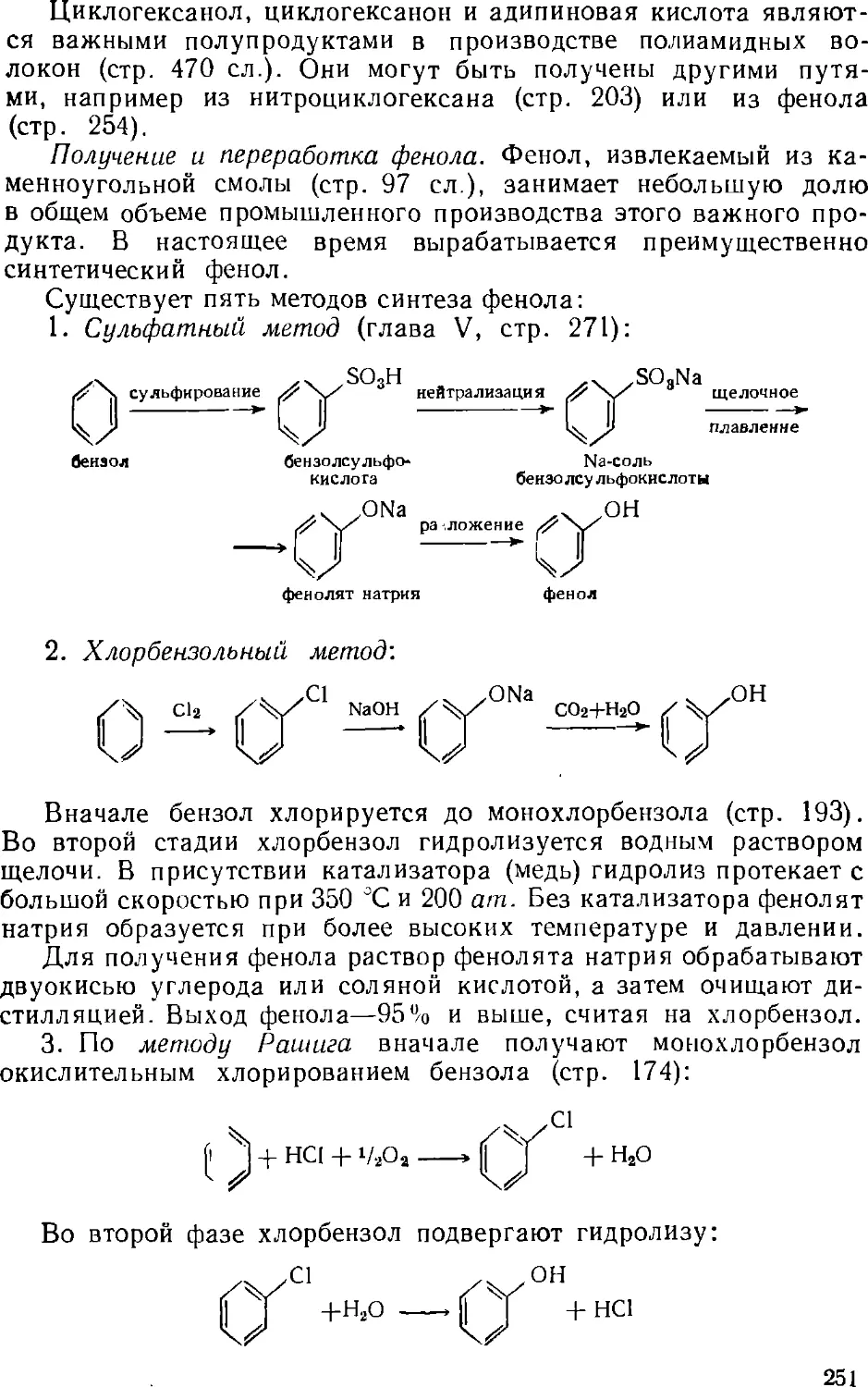




























































































































































































































































































































































Author: Зыков Д.Д. Деревицкая В.А.
Tags: химическая технология химическая промышленность химия органическая химия
Year: 1966




Text
Д. Д. ЗЫКОВ, В. А. ДЕРЕВИЦКАЯ,
Е. Б. ТРОСТЯ НСКАЯ, М. А. ЧЕК А ЛИН,
И. И. ЮКЕЛЬСОН, Ф. О. ЯШУНСКАЯ
ОБЩАЯ
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
¦ ИЗДАНИЕ ВТОРОЕ, ПЕРЕРАБОТАННОЕ
Под редакцией доктора технических наук
профессора Д. Д. ЗЫКОВА
Допущено Министерством
высшего и среднего специального образования СССР
в качестве учебного пособия
для химических техникумов
ИЗДАТЕЛЬСТВО «ХИ/ЧИЯ»
Москва 1966
УДК 660@75 3)
3 30
В книге описаны важнейшие процессы и способы химической
переработки топлив {природного газа, нефти, древесины,
торфа, углей и сланцев), производства продуктов основного
органического синтеза {кислородсодержащих органических веществ,
хлор- и ф пор производных углеводородов, нитросоединений и
других продуктов) и тонкого органического синтеза
{промежуточных продуктов, синтетических красителей, средств
химической защиты растений, поверхностно-активных веществ и
других химикатов). Значительная часть книги посвящена
технологии высокомолекулярных соединений {синтез полимеров
и переработка их в химические волокна и пластические пассы,
технология каучука и резины).
Книга предназначается в качестве учебного пособия для
учащихся химических техникумов; она может быть также
полезна для широкого круга инженерно-технических
работников, желающих получить краткие сведения по технологии
основных органических п одуктов.
СОДЕРЖАНИЕ
Предисловие
Часть первая
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА ТОПЛИВА
Введение. Горючие вещества органического происхождения .
Общие сведения . . . .
Классификация топлив
Запасы и потребление топлив
Состав и свойства топлив
Глава I. Технология природного газа и иефти
1. Общие сведения
Происхождение нефти и газа
2. Добыча нефти и газа
3. Переработка газов
Состав газов
Осушка и очистка газов
Улавливание газового бензина
Газовая сажа
4. Переработка нефти
Состав и свойства нефти
Подготовка нефти к переработке
Перегонка нефти
Важнейшие нефтепродукты
Очистка нефтепродуктов
Крекинг и риформинг нефтепродуктов
Г лава II. Технология твердого топлива и продуктов его переработки
1. Общие сведения
2. Переработка древесины
Состав и свойства древесины
Сухая перегонка древесины
Получение целлюлозы
Гидролиз древесины
3. Добыча и переработка торфа
Происхождение и свойства торфа
Добыча торфа гэ
Химическая переработка торфа 79
4. Термическая переработка углей и сланцев 80
Происхождение и состав углей 81
Классификация углей 83
Добыча и обогащение углей 84
Коксование 86
Улавливание летучих продуктов коксования 92
Переработка продуктов коксования 97
Полукоксование 101
Использование и переработка продуктов полукоксования 105
Глава III. Газификация и гидрогенизация топлив 106
1. Газификация топлив 107
Получение водяного газа Ill
Другие способы газификации 113
2. Гидрогенизация топлив 114
Литература к части первой 118
Часть вторая
ПРОМЫШЛЕННЫЙ ОРГАНИЧЕСКИЙ СИНТЕЗ
Введение 119
Глава IV. Технология основного органического синтеза 124
1. Продукты промышленности основного органического синтеза 124
2. Исходные вещества для промышленных синтезов 131
3. Катализаторы 133
4. Термические превращения углеводородов 134
Получение ацетилена 135
Дегидрирование алифатических углеводородов 141
Дегидроциклизация алифатических углеводородов .... 146
Дегидрирование жирно-ароматических углеводородов . . 147
5. Изомеризация и алкилирование 148
6. Разделение смесей углеводородов 155
Разделение газовых смесей 155
Разделение жидких смесей 159
7. Синтезы на основе окиси углерода 162
Синтез кислородсодержащих органических соединений . . 166
Оксосинтез 168
Получение карбоновых кислот 172
8. Галоидирование углеводородов 174
Хлорирование парафинов 175
Хлорирование и гидрохлорирование олефииов 181
Хлорирование пропилена и получение синтетического
глицерина 184
Галоидирование и гидрогалоидирование ацетиленовых
углеводородов 186
Хлорирование бензольных углеводородов 192
Фторирование углеводородов 194
9. Нитрование углеводородов , 196
Нитрование парафиновых углеводородов 196
Нитрование ароматических и алициклических
углеводородов 200
10. Гидратация непредельных углеводородов , . . . 204
Гидратация олефинов 204
Гидратация ацетилена 217
Дегидратация спиртов 228
11. Окисление углеводородов 229
Окисление парафиновых углеводородов 229
Окисление олефинов 241
Окисление ароматических и алициклических
углеводородов 248
Литература 262
Глава]/. Технология тонкого органического синтеза 263
1. Промежуточные продукты 264
Сульфирование 265
Замена сульфогруппы на гидроксильную методом
щелочного плавления 270
Восстановление цитросоединений 272
Ацилирование аминосоединений 276
Алкилирование и арилирование ароматических амино- и
оксисоединений 279
Реакции конденсации 282
Реакции перегруппировки 283
2. Технология синтетических красителей 286
Краткие сведения о цвете и строении органических
соединений 287
Классификация красителей 290
Азокрасители 291
Антрахиноновые красители 311
Индиго и индигоидные красители 314
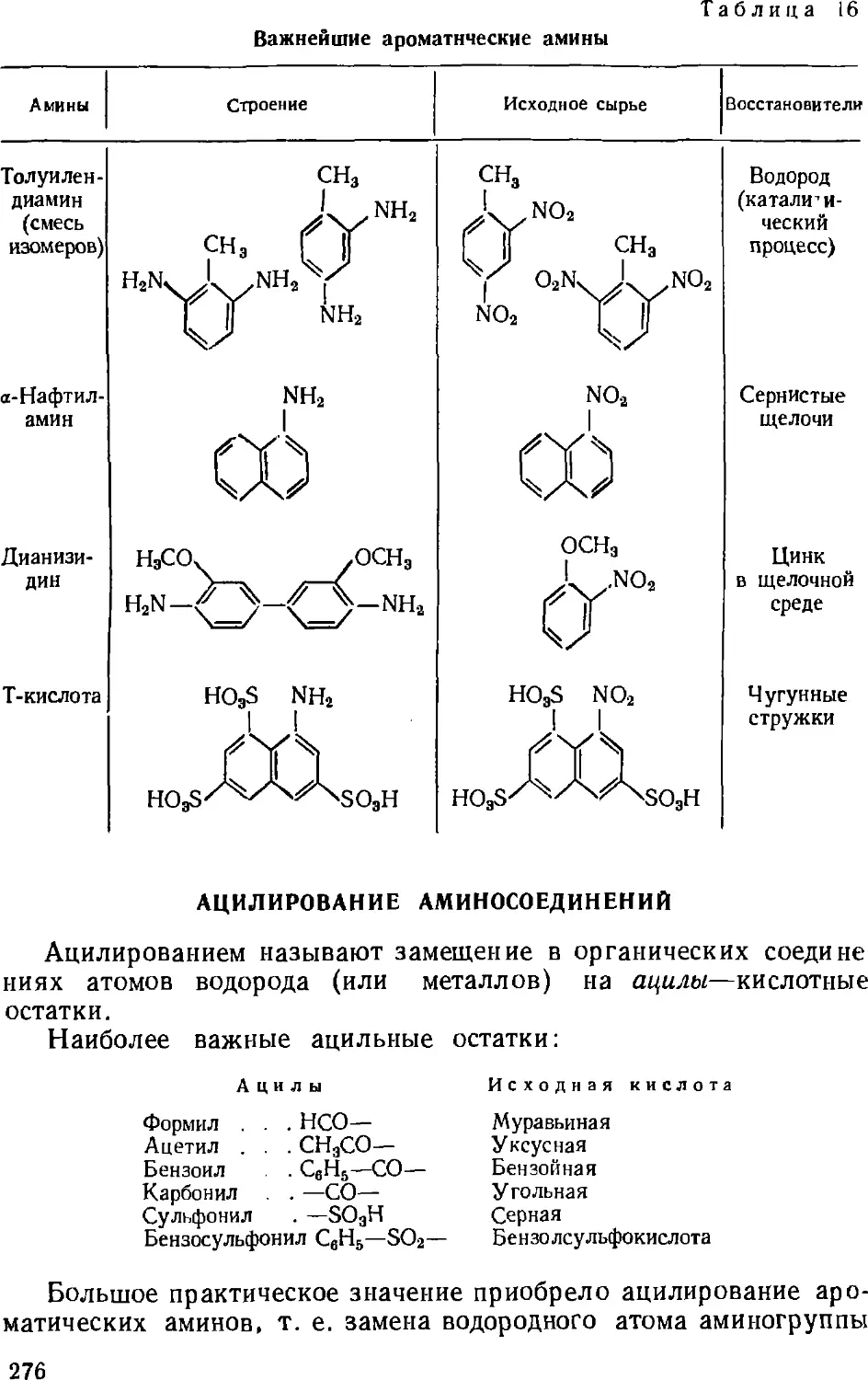
Кубовые полициклические красители 317
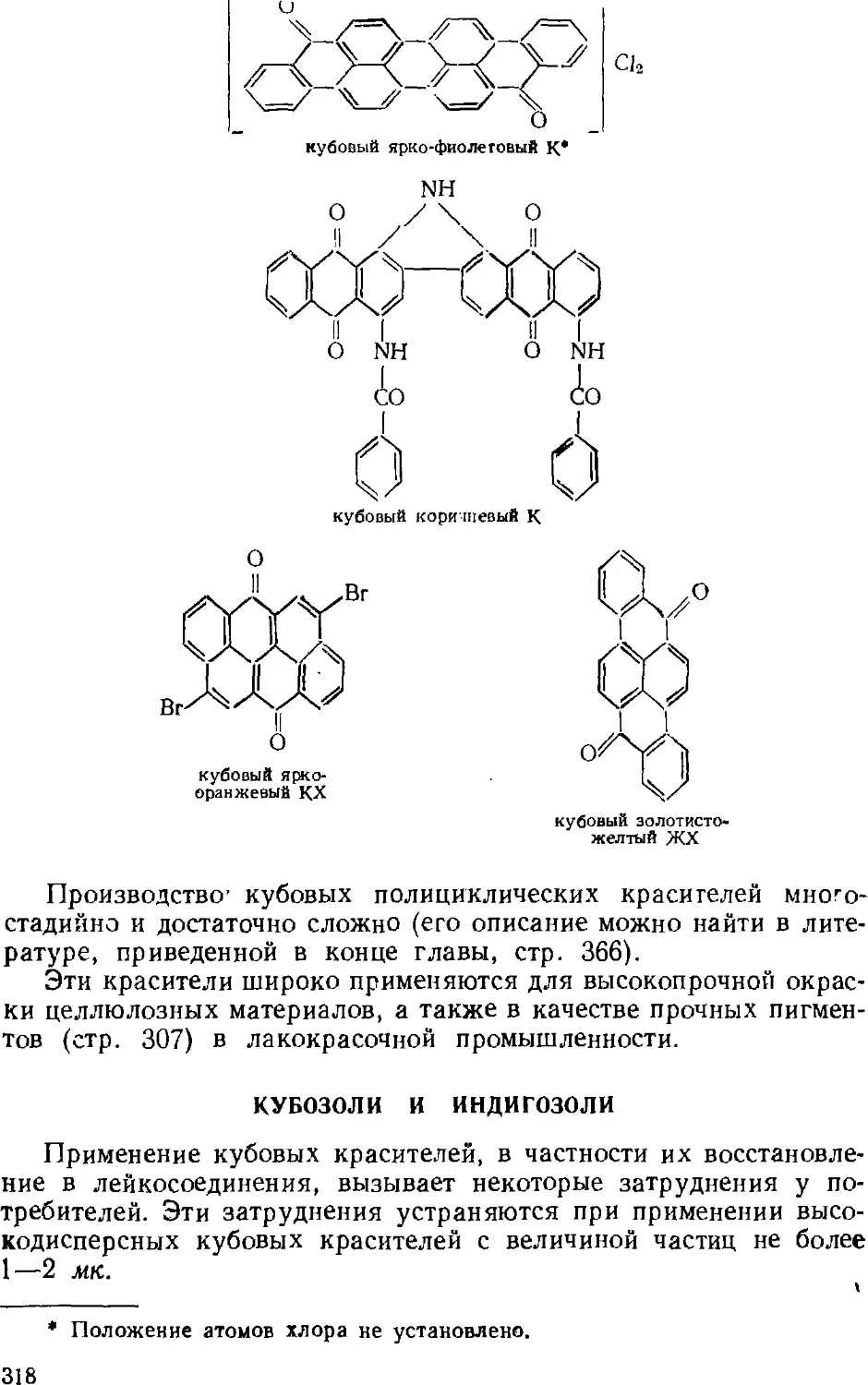
Кубозоли и нидигозоли 318
Сернистые красители 319
Фталоцианиновые красители 321
Арилметановые красители 322
Азнновые, тиазиновые и оксазиновые красители (хинонн-
миновые красители) 324
Оптически отбеливающие и осветляющие вещества .... 326
3. Поверхностно-активные вещества 328
4. Органические пестициды 335
Гербициды 338
Дефолианты и деснкаиты 350
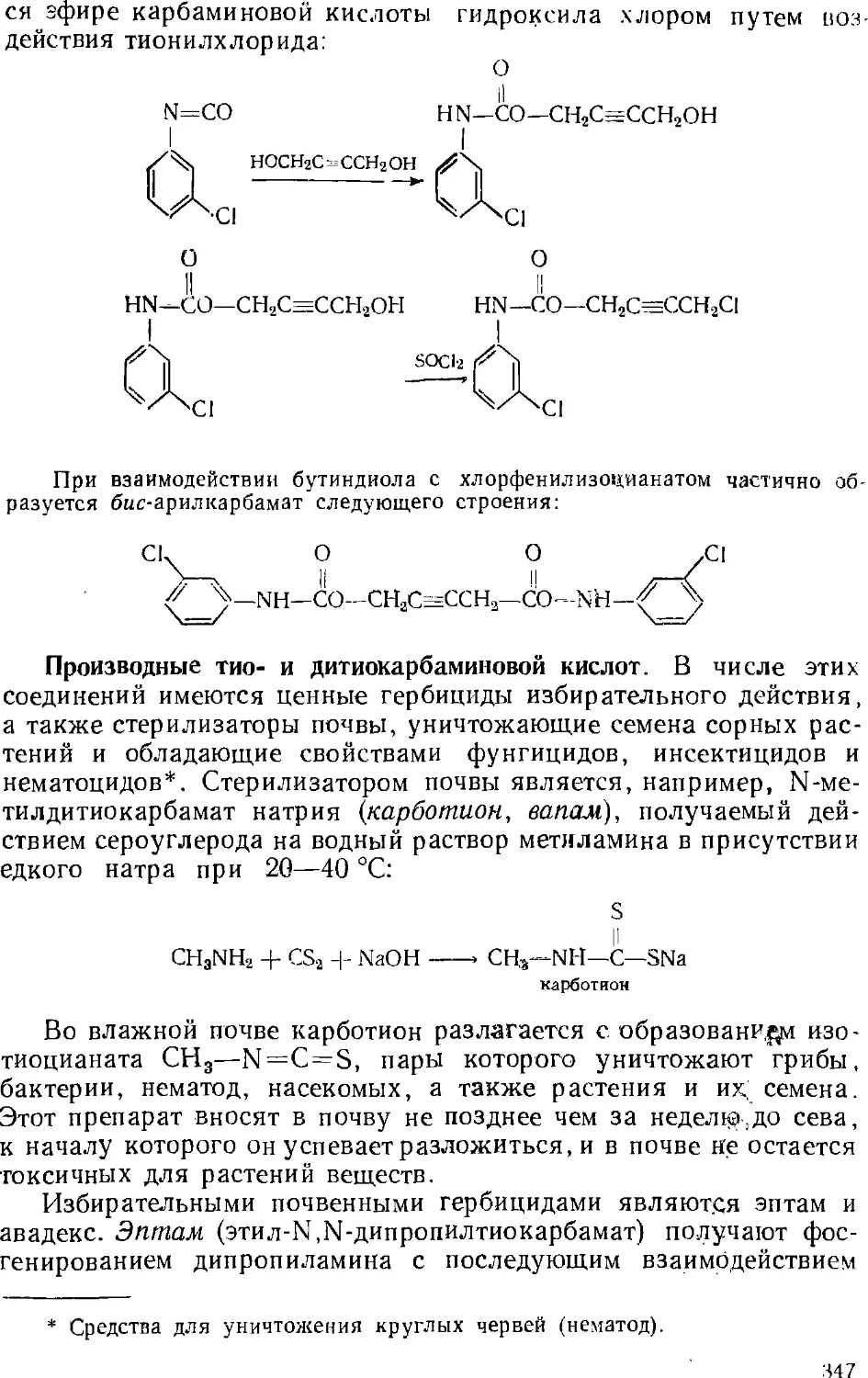
Инсектициды 350
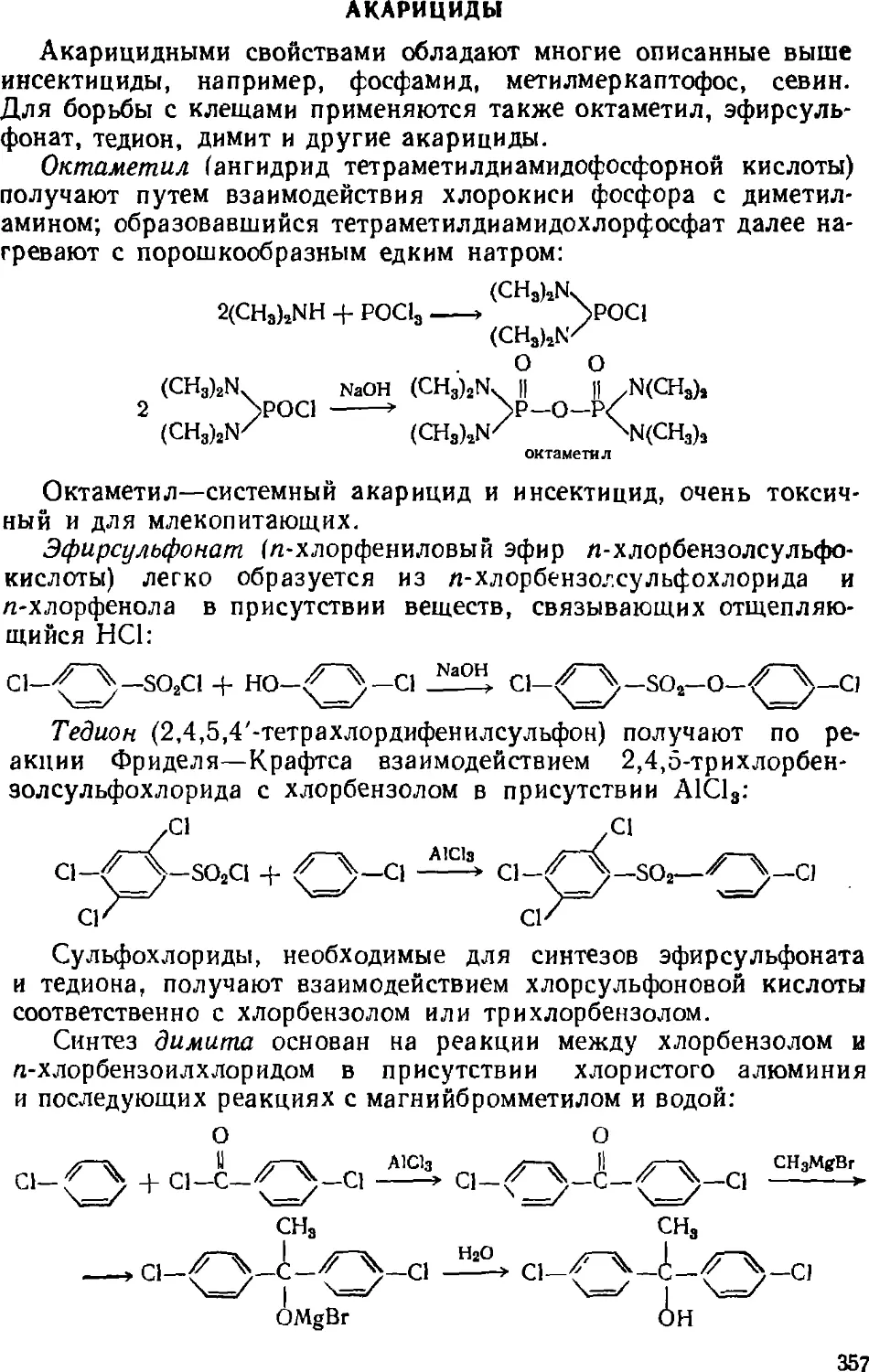
Акарициды 357
Фунгициды 358
Зооциды 360
5. Препараты для резиновой промышленности 361
Литература 366
Часть третья
ТЕХНОЛОГИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
Введение 367
Классификация н номенклатура высокомолекулярных
соединений 377
5
Глава VI. Технология изготовления полимеров 381
1. Общие закономерности синтеза полимеров 385
Поликонденсация 385
Полимеризация 394
2. Способы изготовления полимеров 402
Периодические способы 405
Непрерывные способы 425
3. Модифицирование свойств полимеров 430
Полимераналогичные превращения 431
Блоксополимеризация и привитая полимеризация .... 435
Превращение линейных полимеров в пространственные . 437
Совмещение полимеров 438
Г лава VII. Технология химических волокон 439
1. Общие сведения 439
2. Искусственные волокна на основе целлюлозы 447
Производство вискозного волокна 447
Производство медноаммиачного волокна 459
Производство ацетатного волокна 462
3. Синтетические волокна 464
Производство карбоцепных волокон 464
Гетероцепные волокна 471
Глава VIII. Технология каучука и резины 476
1. Общие сведения 476
2. Сырье для резиновой промышленности 479
Натуральный каучук 481
Синтетические каучуки 482
Синтетические латексы 495
Синтетические смолы 496
Регенерат 497
Ингредиенты резиновых смесей 497
Армирующие материалы 501
Вспомогательные материалы 503
3. Общие технологические процессы резинового производства 504
Рецептура резиновых смесей 505
Подготовительные процессы 507
Заготовительно-сборочные процессы .• 513
Вулканизация 519
Отделка и разбраковка резиновых изделий 523
Глава IX. Пластические массы 526
1. Состав пластических масс 526
2. Термопластичные пластические массы 531
Методы переработки термопластов 531
Свойства термопластов 538
Термопластичные стекла 545
Термопластичные пенопласты 548
3. Термореактилные пластические массы и изделия из них . . . 550
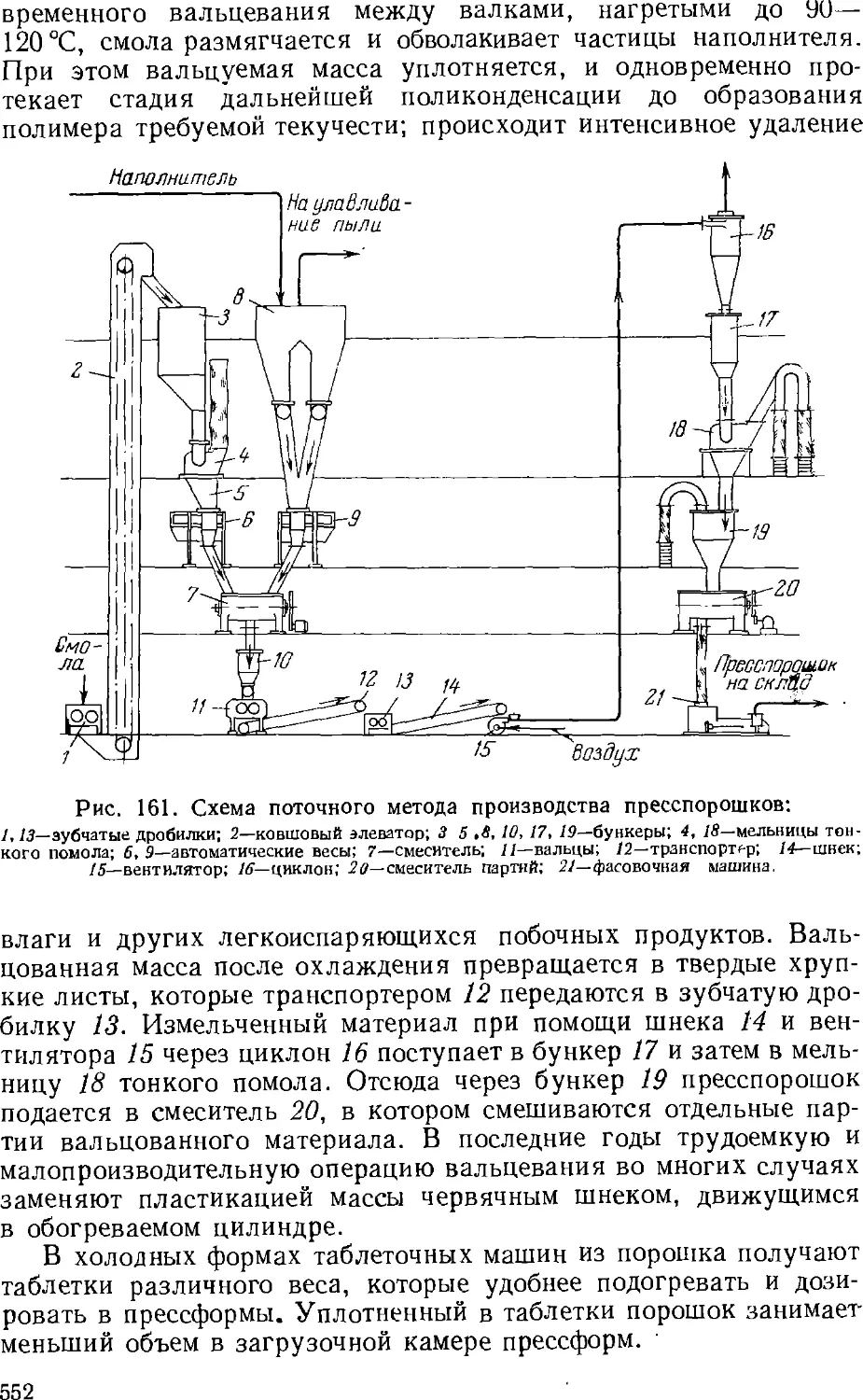
Термореактивные пресспорошки 551
Волокниты 556
Слоистые пластики 564
Термореактивные пенопласты 576
Литература к части третьей 578
Предметный указатель ¦ 579
ПРЕДИСЛО В ИЕ
При подготовке нового издания учебного пособия по
технологии органических веществ авторы стремились отразить в нем
направления развития этой отрасли промышленности,
соответствующие решениям XXII съезда КПСС, майского A958 г.) и
декабрьского A963 г.) пленумов ЦК КПСС о необходимости
дальнейшей ускоренной химизации народного хозяйства. В связи с
этим учебный материал, излагаемый в данной книге, весьма
существенно обновлен, особенно разделы, посвященные технологии
высокомолекулярных соединений.
Круг рассматриваемых в книге вопросов ограничен, как и
в предыдущем издании, изложением технологии синтетических
органических веществ. Общая структура книги сохранена. Она
состоит из трех частей, однако несколько изменена
последовательность изложения, введены новые разделы и исключены
некоторые разделы, входившие в предыдущее издание.
В первой части книги, посвященной химической переработке
топлива, последовательно описаны газообразное, жидкое и
твердое топливо как сырье для промышленности органического
синтеза. Такой порядок изложения соответствует вниманию,
уделяемому отдельным видам топлива в качестве источников
химического сырья, и позволяет создать определенную
последовательность рассмотрения технологических процессов и приемов
переработки топлива. Разделы, относящиеся к облагораживанию
ископаемых топлив, сокращены.
Вторая часть книги, в которой рассматриваются важнейшие
промышленные производства органического синтеза, состоит из
двух разделов. В разделе основного органического синтеза
описаны процессы производства многотоннажных органических
продуктов жирного и ароматического ряда. В разделе, посвященном
тонкому органическому синтезу, изложены принципы технологии
промежуточных продуктов и синтетических красителей,
приведены методы получения поверхностно-активных и вспомога-
тельных веществ для текстильной, резиновой и других отраслей
промышленности и даны сведения об органических пестицидах.
Исключены главы о технологии лекарственных и душистых
веществ, так как они не входят в программу курса общей
химической технологии.
Третья часть книги, составляющая около 40% ее объема,
отведена технологии высокомолекулярных соединений. В нее
включена новая глава, в которой рассмотрены методы синтеза и
свойства важнейших полимеров. Последующие процессы их
переработки в изделия и полимерные материалы излагаются в порядке
постепенного возрастания сложности этих технологических
процессов (вначале описаны химические волокна, затем каучуки и
резина и, наконец, пластические массы).
За годы, прошедшие со времени выпуска в 1955 г. первого
издания книги, скончались три его автора (проф. Б. М.
Богословский, проф. А. А. Стрепихеев и П. Н. Змий). Поэтому авторский
коллектив пополнен новыми участниками—специалистами
соответствующих отраслей технологии органических веществ.
Предисловие и первая часть книги написаны Д. Д. Зыковым,
которым осуществлена также общая редакция всей книги; им же
совместно с И. И. Юкельсоном написано введение к ее второй
части. Глава IV написана И. И. Юкельсоном, глава V—М. А. Че-
калиным. Введение к третьей части и главу VII написала В. А. Де-
ревицкая, автором VI и IX глав является Е. Б. Тростянская,
глава VIII написана Ф. О. Яшунской.
Д. Д. Зыков
Часть первая
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА ТОПЛИВА
ВВЕДЕНИЕ
ГОРЮЧИЕ ВЕЩЕСТВА ОРГАНИЧЕСКОГО ПРОИСХОЖДЕНИЯ
ОБЩИЕ СВЕДЕНИЯ
Горючими веществами называются вещества, способные
окисляться с интенсивным выделением тепла. Существует много
горючих веществ, однако для получения тепла и для химической
переработки используются только широко распространенные
горючие вещества органического происхождения. Главной
составной частью этих веществ является углерод.
Широкое использование горючих веществ органического
происхождения объясняется тем, что запасы их достаточно велики и
доступны для добычи, органические горючие вещества обладают
большой реакционной способностью, при горении выделяют много
тепла, продукты их сгорания не действуют губительно на животных
и растения. Основная часть органических горючих веществ
сжигается для получения тепла (в свое время сжигание было
единственным способом их использования), поэтому они получили
название топлив. Наиболее распространены в природе твердые
топлива—уголь, сланцы, торф и древесина. Жидкие и
газообразные топлива—нефть и природные газы—распространены
значительно меньше. Однако добыча и использование жидких и
особенно газообразных топлив значительно легче, чем твердых, в том
числе и основного твердого топлива—угля.
Значительная часть топлив подвергается химической
переработке. Отрасль производства, осуществляющая такую
переработку, называется промышленностью химической переработки
топлива, а наука о методах этой переработки—химической
технологией топлива.
Топливо (как источник тепла и химическое сырье) и пищевые
продукты играют главную роль среди веществ, потребляемых
человеком (рис. 1), и имеют огромное значение для всего народ-
ного хозяйства. Широкое использование человеком этих веществ,
являющихся соединениями углерода, не должно казаться
удивительным, хотя углерод является одиннадцатым по
распространенности среди элементов земной коры и содержание его в коре
составляет только 0,35%. Огромные масштабы потребления угле-
родсодержащих соединений объясняются их чрезвычайным
многообразием, связанным со способностью углерода соединяться с
многими другими элементами и образовывать соединения
различного строения, содержащие большое число углеродных атомов,
достигающее сотен и даже тысяч. Благодаря этому из
органического сырья (угля, нефти,.древесины и др.) можно получать
самые различные полезные продукты.
Углерод является основой всей
живой природы. Из органических
углеродных соединений состоят
ткани человеческого тела.
Естественно, что для поддержания своей
жизни человек берет от природы
в основном органические
вещества—пищу, топливо, волокнистые
материалы (лен, хлопок, шерсть)
для тканей, кожу и др. До
недавнего времени человек потреблял
только имеющиеся в природе
естественные продукты. Теперь все
возрастающее количество веществ
и материалов, потребляемых и
применяемых человечеством,
изготовляется синтетическими
методами. Причем многие синтетические
вещества обладают ценными
специфическими свойствами,
отсутствующими у природных веществ.
Источником получения
разнообразных синтетических продуктов являются естественные
запасы реакционноспособного углерода, содержащиеся в
горючих веществах органического происхождения. Основная их
масса накопилась в земной коре за прошедшие геологические
эпохи в результате жизнедеятельности существовавших тогда
растений и организмов, базировавшейся на процессах фотосинтеза
органических соединений под воздействием солнечных лучей.
В настоящее время запасы горючих веществ являются для
человечества кладовой углерода и солнечной энергии, и эти богатства
следует расходовать умно и бережливо.
Для переработки доступен сейчас далеко не весь углерод,
имеющийся на земле. Большая его часть, превышающая почти
в 2000 раз запасы каменных углей, скована в виде углекислых
10
Рис. 1. Диаграмма потребления
важнейших видов сырья.
солей в горных породах, а также рассеяна в виде двуокиси
углерода в атмосфере и в воде океанов, морей, рек, озер.
Запасы «органического» углерода, которые могут быть
использованы человеком в виде топлива и органического сырья,
составляют ориентировочно 16 триллионов тонн A6 000 000 000 000 т).
Почти весь этот углерод содержится в угле; на остальные виды
органического сырья приходится незначительная его доля:
Нефть и газ 1,65
Древесина 0,07
Пищевые продукты (годовое
потребление) 0,005
Примечание. Углерод, содержащийся в
¦именном угле, принят за 100.
В настоящее время потребляется лишь небольшая часть
доступных запасов углерода, но и она преимущественно сжигается в
топочных устройствах и тепловых двигателях, где в лучшем
случае используется лишь 40% потенциальной тепловой энергии
топлива. В последние десятилетия доля топлив, подвергаемых
химической переработке, непрерывно увеличивается и все
уменьшается доля расточительно сжигаемого топлива. Возможности же
развития и совершенствования процессов химической переработки
топлив (стр. 14) безграничны.
КЛАССИФИКАЦИЯ ТОПЛИВ
Органические горючие вещества пока используются главным
образом как топливо, поэтому классификация их основывается
в первую очередь на оценке топливных свойств и условий добычи.
Классификация топлив с точки зрения возможностей их
химического использования изменяется по мере развития отдельных
отраслей промышленности органического синтеза.
Как известно, в качестве топлива применяются горючие газы,
нефть, каменный и бурый уголь, сланцы, торф, дрова. В некоторых
случаях как топливо используют также отходы сельского
хозяйства и лесной промышленности и другие неудобные для перевозки
материалы, сжигание которых целесообразно только на месте их
накопления. Эти виды горючих веществ получили название
местных топлив.
Все ископаемые и растительные топлива в необработанном
виде носят название естественных топлив. В некоторых случаях
естественные топлива не удовлетворяют требованиям, которые
предъявляют к ним потребители. Поэтому естественные топлива
подвергают переработке для получения новых видов топлива—
искусственных топлив. Так, уголь перерабатывают в кокс для
доменных печей, размалывают, чтобы получить пылевидное топ-
П
ливо для котлов электростанций, газификацией угля получают
газ, используемый для нужд промышленности и в быту.
Классификация основных видов топлива по их физическому
состоянию и происхождению показана в табл. 1.
Таблица I
Классификация топлив
Физическое
состояние
Естественные топлива
Искусственные топлива
Газообразное
Жидкое
Твердое
Естественный (природный) газ
газовых месторождений и
попутный, добываемый
одновременно с нефтью
Нефть
Ископаемые—торф, бурый и
каменный уголь, горючие сланцы
Газы нефтепереработки,
коксовый, светильный, водяной,
генераторный , доменный,
смешанный, карбюрированный газ и
др-
Бензин, лигроин, керосин,
соляровое масло, мазут
(получаются из нефти, могут быть
получены также из угля и
сланца), этиловый спирт
Кокс, полукокс, древесный уголь,
брикеты, пылевидное топливо
Все перечисленные виды топлив, в том числе и местные, во
все большей степени используются как химическое сырье. Более
подробные классификации топлив, учитывающие все
направления их использования, приводятся при описании способов
переработки твердых, жидких и газообразных топлив.
ЗАПАСЫ И ПОТРЕБЛЕНИЕ ТОПЛИВ
Советский Союз обладает неисчерпаемыми запасами сырья.
По запасам угля, превышающим, по современным оценкам,
8 000 млрд. т, наша страна занимает первое место в мире. Быстро
увеличиваются разведанные запасы нефти и особенно природного
газа. Так, за период 1955—1960 гг. разведанные запасы нефти в
СССР увеличились в 1,5 раза, газа—почти в 4 раза.
Потенциальные ресурсы природного газа в нашей стране исчислялись (на
начало 1960 г.) в 60 000 млрд. м3, что составляет 38% мировых
ресурсов газа. Запасы торфа в Советском Союзе составляют около
60% мировых запасов, древесины—примерно 35%.
Распределение запасов топлив по территории СССР показано
на рис. 2.
По возможностям добычи и дальнейшего использования
жидкие и особенно газообразные топлива более удобны и экономичны,
чем твердые, в связи с чем доля нефти и газов в общем топливном
балансе СССР увеличивается. Это облегчает также задачу дальней-
12
,'у'/УХ-у',- Зона, богатая торфом
Лесная зо на
Рис. 2. Распределение основных вапасов топлив по территории СССР.
шего быстрого развития народного хозяйства. При этом запасы
нефти и газа расходуются быстрее запасов угля, но это
компенсируется увеличением разведанных запасов в новых нефтяных и
газовых месторождениях, значительно опережающим рост
потребления жидкого топлива и газа. Древесина, являющаяся ценным
поделочным материалом и химическим сырьем, в качестве топлива
будет расходоваться во все меньшем количестве. **•¦ ¦
Табл. 2 характеризует относительные запасы и'добычу
топлива в СССР.
Таблица 2
Запасы и добыча топлива в СССР
(в пересчете на условное топливо)
Топливо
Доля
в общих
запасах
%
95,8
0,7
0,1
3,4
—
Доля в добыче, %
1940 г.
59,1
18,7
1,9
5,1
14,9
0,3
1950 г.
66,1
17,4
2,3
4,8
9,0
0,4
I960 г.
53.9
30,5
7,9
2,9
4,1
0,7
1963 г.
45,9
34,8
12,4
2,5
3,6
0,8
Уголь . .
Нефть . .
Газ ...
Торф . .
Древесина
Сланцы .
Данные о мировом потреблении топлива и энергии в 1963 г.
приведены в табл. 3.
Таблица 3
Мировое потребление топлива и энергии в 1963 г.
Топливо и энергия
Уголь
Нефть
Газ
Гидроэнергия
Древесина
Торф
Потребление
млн. т
условного
топлива
2175
1700
810
300
205
25
%
41,7
32,6
15,5
5,8
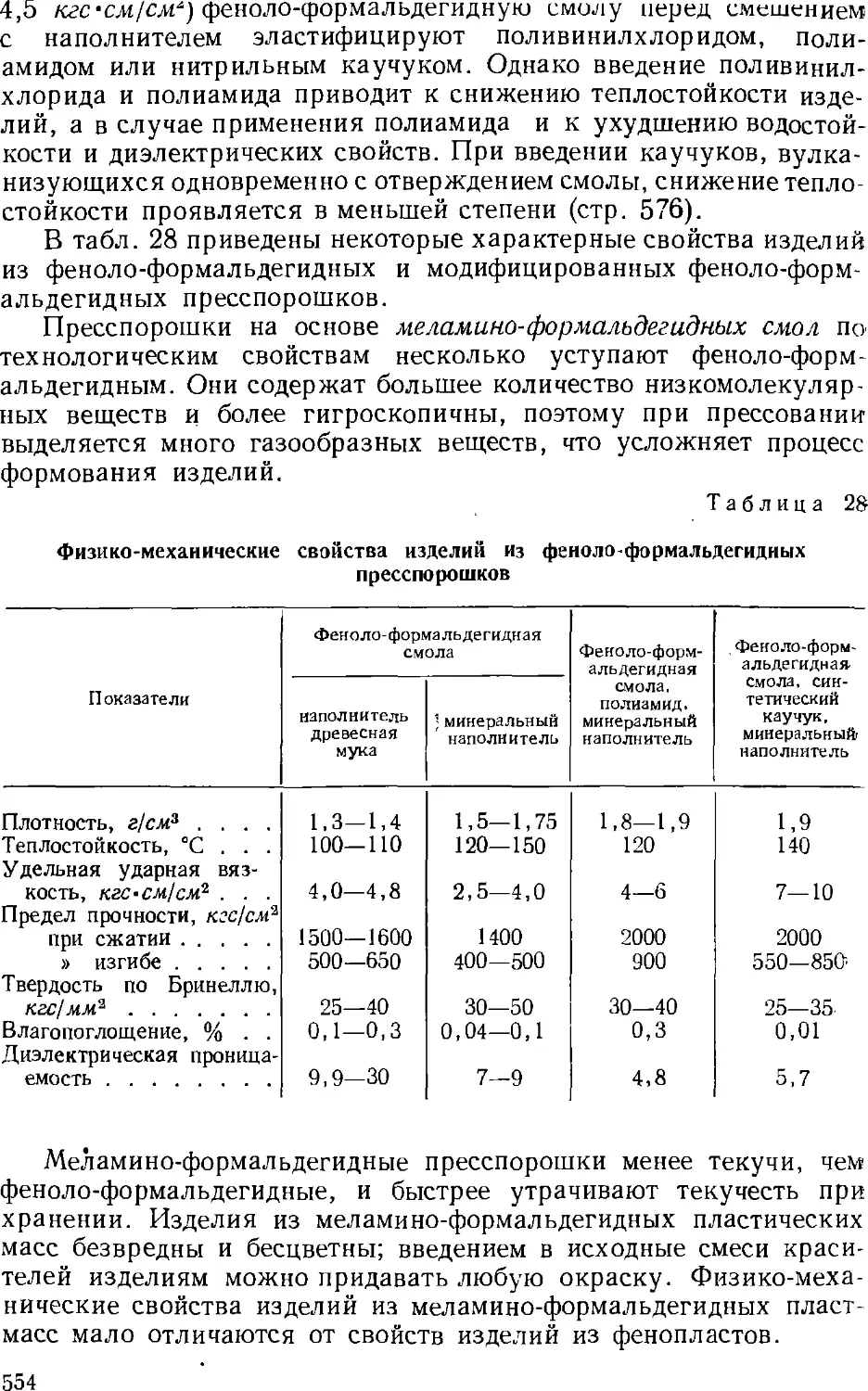
3,9
0,5
В настоящее время естественные топлива даже для сжигания
используются все в меньшей степени. Обычно их подвергают
переработке для получения облагороженных искусственных топ-
лив, более пригодных в тех или иных условиях их сжигания—
в доменной г?чи, в двигателях внутреннего сгорания и т. д. или
в процессах получения исходных веществ для промышленности
органического синтеза. В большинстве случаев эти оба вида про-
дуктов переработки топлив параллельно получаются на топливо-
перераба тывающих предприятиях. Введены в эксплуатацию
также специальные заводы по переработке органических горючих
веществ только в химическое сырье или готовые синтетические
продукты; эта отрасль переработки топлив все время
расширяется. В настоящее время химической переработке подвергается до
25% всего добываемого угля, нефть практически вся проходит
предварительную переработку. Таким образом, промышленность
химической переработки топлива не только перерабатывает
топливное сырье в облагороженное искусственное топливо, но и в
большом объеме производит разнообразные органические
вещества, используемые далее во многих процессах органического
синтеза.
СОСТАВ И СВОЙСТВА ТОПЛИВ
Качество топлив оценивают в зависимости от
предполагаемых способов их использования. Например, при использовании
топлива как горючего вещества важно знать количество тепла,
которое способен выделить 1 кг данного топлива при его
сжигании, т. е. теплотворную способность (по
интернациональной системе единиц «СИ»—удельную теплоту сгорания).
Теплотворная способность и ряд других свойств топлива
определяются его химическим элементарным составом. При химической
переработке топлива зачастую необходимо знать характер
веществ, входящих в его состав, их химическое строение; в этих
случаях топливо следует подвергать более глубоким
химическим исследованиям, различным при разнообразных способах его
использования.
Химическим анализом прежде всего определяют в топливе
содержание его органической горючей части и сопутствующей ей
негорючей минеральной части—золы. Вредным балластом в
топливе является также содержащаяся в нем влага. Состав топлива
может быть выражен следующим образом:
рабочее топливо = сухое топливо + влага
сухое топливо = горючая масса + зола
Негорючим балластом газообразного топлива являются
двуокись углерода, азот и другие инертные газы. Влага содержится
в газе в виде пара. Газы и отдельные виды жидкого топлива иногда
лишены балласта, твердые топлива, наоборот, могут содержать
много золы и влаги. Так, зольность некоторых углей и сланцев
достигает 50% и более.
Твердые топлива и тяжелые высококипящие жидкие топлива
обладают свойством, широко используемым при их химической
переработке. При сильном нагревании без доступа воздуха они
разлагаются, образуя сухой обуглившийся твердый остаток—
15
/соке и газоооразные летучие вещества. Такой процесс разложения
топлива называется сухой перегонкой. Выделяющиеся летучие
вещества (в них не включается одновременно выделяющаяся влага)
после охлаждения разделяются на жидкие продукты перегонки и
газ. Таким образом, состав топлив, способных подвергаться сухой
перегонке, может быть выражен еще одной схемой:
рабочее топливо = твердый остаток -f- летучие вещества + влага
(кокс)
Характер горючей массы топлива зависит от ее элементарного
состава, т. е. от соотношения составляющих ее химических
элементов. Основными элементами, входящими в состав горючей
массы топлив, являются: углерод, водород, кислород, азот, сера;
в топливе содержатся также незначительные количества фосфора
и некоторых других элементов.
В СССР состав топлива принято обозначать следующим образом: V—ле-
гучая часть, А—зола, W—влага.
Содержащиеся в топливе различные элементы обозначаются их
химическими символами (С, Н, О, N, S, Р и др.)- При химическом символе ставят индекс—
букву, показывающую, к какому составу топлива относится найденное
анализом процентное содержание данного элемента. Так, буква «р» обозначает, что
проценты вычислены от состава рабочего топлива; буква «л»—проценты
вычислены от массы лабораторного, т. е. частично подсушенного, топлива;
буква «с»—для анализа брали сухое топливо, не содержащее влаги; буква «г»—
проценты вычислены в расчете на горючую массу топлива.
Например 1/р=21,5% означает, что рабочая масса данного топлива
дает 21,5% летучих веществ; Сг=79,0%—горючая масса топлива
содержит 79,0% углерода. *
Исследование состава топлива может быть проведено с
большей или меньшей полнотой. При кратком техническом анализе
определяют только содержание в топливе влаги, золы, летучих
веществ (если требуется) и теплотворную способность. Полный
элементарный анализ включает количественное определение
химических элементов, входящих в состав топлива. При оценке
топлива как химического сырья проводится еще более глубокое
изучение топлива.
Содержание отдельных составных частей и элементов в
топливе определяют путем его химического анализа*. Влага
определяется путем высушивания топлива, зола—выжиганием всей
горючей массы. Горючая масса определяется по разности между
общей массой топлива и содержанием в нем золы и влаги.
Теплотворная способность топлив обозначается буквой Q и выражается
в ккал/кг (в системе СИ удельная теплота сгорания Q—в кдж/кг).
Для любого топлива она может быть точно определена в
специальном приборе, называемом калориметром.
* Анализ топлив и определение их теплотворной способности описаны
в книге А. П. Грошева, Технический анализ, Госхимиздат, 1958.
16
Из элементов, составляющих горючую массу топлива, при
сгорании углерода выделяется 8137 ккал/кг или 34 068 кдж/кг,
при сгорании водорода—34 180 ккал/кг или 143 112 кдж/кг.
Таким образом, сгорание этих элементов обусловливает
выделение основного количества тепла при сжигании топлива.
Горючим веществом в топливе является также сера,
содержащаяся в виде органических соединений (Sopr<), а в твердых топли-
вах—в виде сульфида железа, или пирита (Sni)pj. Сера
содержится в топливе также в виде негорючих соединений,
преимущественно в виде сульфатов (Scyj1bAJ. Общее содержание серы в топливе
(So6iu.) составляет:
. = Sopr. T SnHp_ -|- асульф.
Присутствие серы в топливе нежелательно (хотя при ее
сгорании выделяется 2200 ккал/кг тепла (9211 кдж/кг), так как при ее
горении образуется сернистый ангидрид SO2, вредно действующий
на людей, животных, растительность и вызывающий коррозию
аппаратуры, с которой соприкасаются продукты горения.
Некоторые виды топлив содержат значительные количества
кислорода и азота, которые понижают теплотворную способность
топлива, потому что они не горят, а кислород, кроме того,
связывает часть углерода и водорода, уменьшая количество тепла,
выделяемого ими при горении.
При сгорании в промышленных устройствах (топки, печи и т. д.)
килограмм топлива выделяет меньше тепла, чем определено в
калориметре. Это происходит потому, что в производственных
условиях влага (содержащаяся в топливе и образовавшаяся при
сгорании водорода) уходит в дымовую трубу вместе с продуктами
сгорания (топочными газами) в виде водяного пара, унося с собой
часть тепла F00 ккал или 2510 кдж на каждый килограмм влаги).
При сжигании же топлива в бомбе калориметра вся эта влага
конденсируется, отдавая теплоту конденсации прибору. Поэтому
различают две величины теплотворной способности: QB—высшая
теплотворная способность, включающая теплоту конденсации
влаги, и Q„—низшая теплотворная способность, которая меньше
QB на величину теплоты конденсации влаги, содержащейся в
продуктах сгорания топлива.
Практически качество топлива оценивают по величине
низшей теплотворной способности рабочего топлива, обозначаемой Qhp.
Зная элементарный состав топлива, можно приближенно
определить его теплотворную способность (в ккал/кг). Ниже
приведена формула, предложенная Д. И. Менделеевым для
определения QB:
QB = 81С + 300Н — 26 (О—S)
где С, Н, О и S—соответственно содержание в топливе
углерода, водорода, кислорода и горючей серы, %.
2—805 17
Для сравнительной оценки разнообразных видов топлива и
учета общих его запасов введена единая мера—единица условного
топлива. Условным топливом принято считать топливо с
теплотворной способностью 7000 ккал/кг (или ~29 000 кдж/кг).
Таблица 4
Свойства основных видов топлива
(средние характеристики)
Топливо
Теплотворная
способность
«5
ккал/кг
Количество,
соответствующее I m
условного
топлива
Состав горючей части
%
Сг
Ог
Содержание
в рабочем
топливе
влага
Wp
зола
(округленно)
АР
Твердое
дрова
торф
бурый уголь . . . .
каменный уголь . .
каменный уголь кок
совый
антрацит
сланец
Жидкое
сырая нефть . . . .
бензин
керосин
спирт
Газообразное
природный газ . .
коксовый газ . .
генераторный газ
2 950
2 900
3 300
6 500
7 100
6 700
2 700
10 300
10 700
10 650
6510
8 600*
4 300*
1 500*
2,4
2,4
2,1
1,1
0,99
1,05
2,6
0,67
0,65
0,66
1,1
5,2
4,0
3,0
1,35
1,1
1,05
2,9
0,79
0,84
0,79
1,75
810
1 630
4 650
50,0
58,0
70,0
81,0
87,0
94,0
75,0
86,0
84,0
84,5
55,0
6,0
5,8
5,0
5,4
5,0
2,0
9,0
13,0
16,0
15,5
13,0
43,0
33,0
20,0
8,6
4,0
1,5
13,5
1,0
35,0
30
36
30
5,5
3,5
5,5
15,0
1,0
0,7
5,5
15,0
10,0
10,0
10,0
50,0
Содержание, объемн. %
СО
6,0
26,0
На
57,
14,
0
0
СН4
94
26
,0
,0
* Теплотворная способность—в ккал/м& (считая на объем газа, приведенного к нормаль-
«ьш услопчям).
В табл. 4 приведены свойства различных видов топлива.
Теплотворная способность их колеблется в пределах от 3000 до
10 000 ккал/кг и более.
При сгорании жидких и газообразных топлив совсем не
образуется золы. После сгорания твердых топлив остается большое
количество золы, для удаления которой требуются специальные
устройства. Если зола легкоплавка,.ее удаление особенно
осложняется.
18
Газ коксовый
ІВЗОм3
Дрова 5,2мЗ
Торф 4-м5
Бурый уголь Зм3
'{амепный
/уголь 1,1 м3
.Бензил 06м3
Одни виды топлива легко загораются и горят, другие трудно
разжечь и для поддержания их горения необходимо дутье.
Условия хранения и доставки топлива к месту сжигания, а
также процесс сжигания зависят в значительной степени от
физического состояния топлива. Так, жидкие и газообразные
топлива хранят в специальных емкостях (цистернах или
газохранилищах), подают к месту сжигания по трубам, сжигают в
специальных горелках. Для промышленного использования твердых топ-
лив требуются значительно более сложные устройства.
Очень важной характеристикой
топлива является количество тепла,
которое может быть выделено при
полном сгорании единицы его
объема,—энергоемкость топлива. Для
сравнения энергоемкости различных
топлив на рис. 3 сопоставлены
объемы топлив, необходимые для
получения одного и того же количества
тепла. Наиболее ценными видами
топлив, удобными для использования,
являются высококалорийные газы и
жидкие нефтяные топлива.
Все эти оценки топлива в
известной мере условны и с развитием
техники в них вносятся поправки и
уточнения. В результате
совершенствования методов переработки
топлив создаются широкие возможности
получения более ценных видов
топлива из менее ценных (например,
легких жидких топлив из тяжелых
или даже из твердых—угля и сланца, газообразного топлива из
твердого и жидкого и т. д.), замены одних топлив другими и т. д.
Наряду с облагораживанием топлива для
развивающейся промышленности органического синтеза требуются все большие
количества топлив в качестве сырья. При переработке их для этой
цели производится как извлечение из топлива содержащихся в
нем ценных природных веществ, так и разрушение сложных
соединений, имеющихся в топливе, на более простые активные
вещества, используемые как химическое сырье. Запасы природных
ценных веществ в топливе и его химическая активность, т. е.
способность давать при переработке большие выходы продуктов,
необходимых для последующих синтезов, являются теми свойствами,
по которым топливо оценивается как химическое сырье.
Топлива, являющиеся универсальным источником углеродсо-
держащих веществ, все более широко используют как химическое
сырье для производства самых разнообразных ценных продуктов.
Рис. 3. Объемы топлив,
соответствующие 1 т
условного топлива.
Глава І
ТЕХНОЛОГИЯ ПРИРОДНОГО ГАЗА И НЕФТИ
/. ОБЩИЕ СВЕДЕНИЯ
Промышленность по добыче и переработке природного газа
и нефти является одной из ведущих, сложных отраслей народного
хозяйства. Она играет исключительную роль в экономике страны,
обеспечивая ее снабжение удобным эффективным газовым
топливом, моторными топливами для двигателей внутреннего сгорания,
смазочными материалами, сырьем для химической
промышленности, котельным топливом, такими материалами, как битумы,
парафин и церезин, сажа и многие другие.
С развитием этой промышленности связано обеспечение
обороноспособности страны.
Развитие добычи и промышленного использования нефти
началось с середины XIX в. До этого нефть использовали в
ограниченных масштабах—для отопления, освещения, лечебных и других
целей. Начало развития переработки нефти и резкое увеличение ее
потребления связаны с открытием способов получения из нефти
керосина для освещения. С конца 70-х годов XIX в., когда из
нефти стали получать кроме керосина также смазочные масла для
машин, начался керосиново-масляный период нефтепереработки.
Особенно быстрый рост потребления нефти вызвало внедрение в
практику легких двигателей внутреннего сгорания, для которых
потребовались большие количества бензина,—наступил
бензиновый период использования нефти. В последнее время начался
новый этап переработки нефти—использование ее также в
качестве химического сырья.
Потребление нефти непрерывно растет. Особенно увеличился
спрос на нефтяные продукты во время второй мировой войны, в
которой, по подсчетам, участвовало до 40 млн. автомашин и
тягачей, 150 тыс. танков и 200 тыс. самолетов. Мировая добыча нефти
20
в настоящее время достигает 3 млн. т в сутки. Особенно быстро
растет потребление нефтепродуктов и добыча нефти в СССР и
странах социалистического лагеря.
В 1960 г. в СССР было добыто 148 млн. т нефти, в 1964 г.—
224 млн. т.
По запасам нефти наша страна, как известно, занимает одно из ведущих
уест в мире. Главнейшими нефтеносными районами в Советском Союзе являются
(см. рис. 2, стр. 13): Апшеронский полуостров (Баку), район г. Грозного, Ку-
бано-Черноморский район (Майкоп), «Второе Баку» (Волго-Уральский район),
Эмба (северное побережье Каспийского моря), среднеазиатские месторождения,
Ухта, Западная Украина, Сахалин, Камчатка и др. За последние годы открыты
новые перспективные месторождения нефти, в частности в Сибири;
разведанные запасы нефти все увеличиваются.
Добыча и переработка нефти—ценнейшего источника моторных топлив
в химического сырья—также непрерывно возрастают.
Промышленное использование природного газа началось позже,
чем нефти. Вначале газ считался нежелательным спутником
нефти, создававшим пожароопасные условия на нефтяных промыслах.
Затем газ стали использовать как топливо на месте его добычи
(для обогрева аппаратуры нефтеперерабатывающих заводов).
Когда была разработана техника передачи газа по трубопроводам
на дальние расстояния, его стали широко применять как очень
удобное, высококалорийное и дешевое топливо для бытовых
целей и в промышленности. Широкое использование газа как
топлива в США началось в 30-х годах. В СССР природный газ стал
потребляться в больших масштабах в последние годы войны,
в 1960 г. потребление газа приблизилось к 50 млрд. м3 в год и
в ближайшие годы будет увеличиваться быстрее потребления
других видов топлива. В 1964 г. в СССР добыча природного газа
составила 110 млрд. м3. Особое значение приобретает все
развивающееся использование природного газа в качестве химического
сырья в промышленности органического синтеза и для синтеза
аммиака.
Природный газ, жидкая нефть и природные битумы
представляют собой группу веществ, основной составной частью которых
являются углеводороды. Природные смеси легких углеводородов,
газообразных в обычных условиях (метан, этан, пропан, бутан),
получили название природного газа. Природная смесь жидких
углеводородов (от пентана до очень тяжелых, высококипящих),
содержащая примеси органических соединений кислорода, серы,
азота, называется нефтью. Смеси высококипящих
(воскообразных или твердых) углеводородов и некоторых других
родственных им веществ получили название природных битумов. Все
перечисленные углеводородные смеси, особенно нефть и газ,
связаны общностью происхождения, условий залегания, методов их
добычи и в определенной мере—методов переработки.
21
ПРОИСХОЖДЕНИЕ НЕФТИ И ГАЗА
Вопрос о происхождении нефти и газа имеет большое
теоретическое и практическое значение. Зная, как образуются залежи
того или иного полезного ископаемого, можно правильно
организовать и вести его поиски, добычу и переработку. В настоящее
время достаточно хорошо известно, как и в каких геологических
условиях скапливаются нефть и природный газ. Вопрос же о
происхождении нефти и газа до сих пор окончательно еще не решен.
Существуют два основных направления в объяснении происхождения нефти
и~ природного газа—неорганическое и органическое.
Согласно первому из них углеводородные смеси, встречающиеся в природе,
образовались при взаимодействии воды или водных растворов солей натрия,
магния и других металлов с карбидами металлов. Карбидную теорию высказал
в 1877 г. Д. И. Менделеев, предположивший, что в земной оболочке, особенно
в ее нижних слоях, содержатся карбиды, к которым по трещинам в земной коре
проникает влага, вступающая во взаимодействие с карбидами. В результате
образуются различные углеводороды, например этан, по реакции:
2FeC + ЗН2О » Fe2O3 + С2Нв
Поднимаясь из области, где протекала реакция, к поверхности земли, эти
углеводороды претерпевали химические превращения и скапливались в
пустотах и пористых породах земной коры. По мнению Менделеева, такой процесс
происходил не только в давние геологические эпохи, но протекает и теперь.
Карбидная теория, имеющая своих сторонников и сейчас, оставила не-
объясненным ряд вопросов, связанных со свойствами нефтей и образованием
их залежей, и потому не разделяется большинством исследователей нефти.
Органические теории образования нефти и газа исходят из того, что эти
горючие вещества являются продуктами разложения отмиравшей живой
материи. Разница между отдельными органическими теориями заключается в том,
какой вид живой материи принимается за материнский при образовании
залежей. Сторонники теории животного происхождения нефти считают, что она
образовалась из остатков погибших животных (рыб, земноводных и др.),
сторонники теории растительного происхождения принимают за материнское
вещество остатки растений. Наиболее вероятная из них—теория смешанного
происхождения—принимает за исходное вещество остатки мелких
простейших животных и водорослей. Отмирая, они образуют слои гниющего ила—
сапропеля, при гниении которого образуются метан (болотный газ) и другие
углеводороды. Накопление сапропеля в водоемах активно происходит и сейчас,
в предшествующие геологические эпохи оно протекало значительно более бурио
и могло служить источником образования больших запасов органического
вещества (порядка сотен миллионов тонн).
Каким бы путем не образовывались нефть и газ, во всех
случаях, они, будучи текучими, способны перемещаться
(мигрировать) в пористых, проницаемых слоях земной коры. Как легкие
вещества, нефть и газ стремятся переместиться к поверхности
земли. Если на их пути встречается куполообразно сложившаяся
непроницаемая горная порода или выпуклая кверху складка
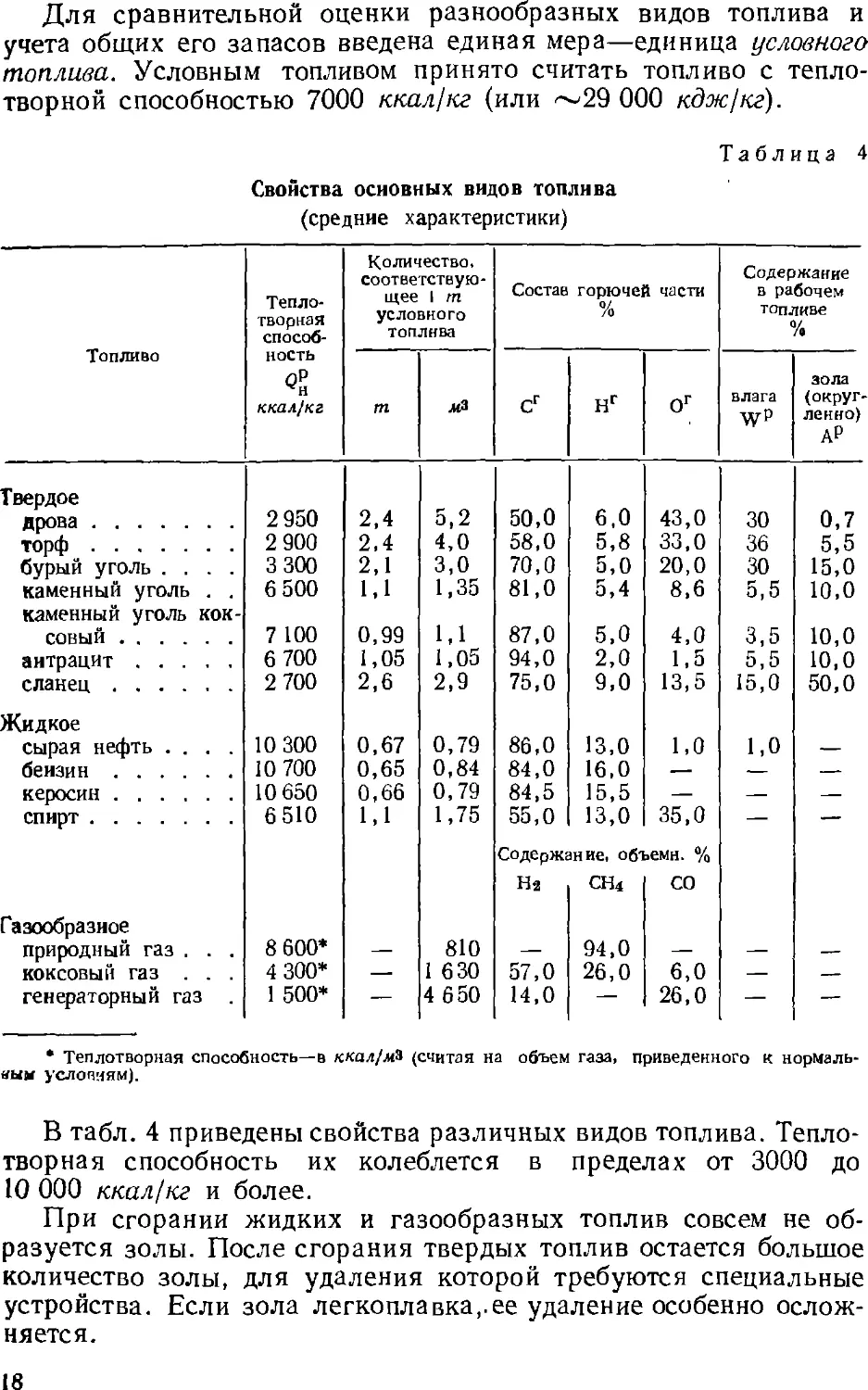
земной коры (антиклиналь), нефть (или газ) скапливается в таком
куполе или складке, где зачастую находится под очень высоким
давлением. Такое скопление называется соответственно нефтяным
или газовым месторождением.
22
2. ДОБЫЧА НЕФТИ И ГАЗА
Добыча нефти и газа заключается в нахождении мест их
накопления, проникании (иногда на очень большую глубину)
через толщу горных пород, образующих и покрывающих купол,
к нефтяному или газовому пласту и извлечении топлива из
пласта. Поэтому работа на нефтепромысле, который заранее найден
и изучен геологами, подразделяется на два этапа:
1) бурение скважин;
2) добыча нефти или газа—эксплуатация скважин.
Бурение скважин. Скважиной (нефтяной или газовой)
называют колодец круглого сечения, пробуриваемый в земле. Верх
скважины называется ее устьем, а дно, в котором буровой
инструмент разрушает породу,—забоем. Совершенствование техники
бурения позволяет проникать все глубже в толщу земной коры и
вскрывать все новые запасы нефти и газа. Глубина скважин иногда
достигает 4000—6000 м и более.
Раньше при прохождении скважины породу долбили
специальным долотом, закрепленным на длинной штанге. Такой метод
назывался ударным бурением. Затем перешли к методу
вращательного бурения, по которому разрушение породы производится
бурильным инструментом, прикрепленным к вращающейся,
периодически наращиваемой, бурильной трубе. Непрерывный поток
воды со взмученной в ней глиной (глинистый раствор) промывает
скважину, вынося из нее раздробленную породу, охлаждая
инструмент, закрепляя стенки скважины и создавая в скважине
гидравлический затвор, препятствующий выбросам нефти и газа.
Буровая установка (рис. 4) состоит из двигателя 5,
вращающего буровой инструмент* 1 и несущие его буровые трубы 2;
буровой вышки 7 с лебедкой 4 для подъема и опускания буровых
труб; насосной установки 9 для нагнетания глинистого раствора
и из других подсобных механизмов и устройств. Высота
современной буровой вышки—более 50 м. С помощью такой буровой
установки можно пробурить в течение месяца скважину глубиной
до 5000 м.
В 1924 г. М. А. Капелюшников (СССР) предложил метод
турбинного бурения, по которому механизм, вращающий буровой
инструмент, переносится в забой. Таким механизмом явилась
специальная турбина, вращаемая подаваемым в нее глинистым
раствором. Применение турбобура исключило необходимость
вращать всю тяжелую колонну бурильных труб, что упростило
технику бурения и сделало ее более экономичной. Позже, также в
СССР, был сконструирован электробур—буровой инструмент,
вращаемый специальным электродвигателем, также опускаемым
* Буровым инструментом здесь является сложное устройство, несущее
дробящие зубчатки, но сохранившее название «долото» (см. на рис. 4 справа).
23
Рис. 4. Буровая установка (справа показано долото):
/—долото; 2—буровые трубы; 3—шланг для глинистого раствора; 4—лебедка;
5—электродвигатель; 5—вертлюг; 7—буровая вышка; 8—талевый блок; S—насооная.
в забой. При помощи турбо- и электробуров можно бурить не
только вертикальные, но и наклонные скважины. Это позволяет
пробуривать скважины под морское дно, под здания и сооружения,
бурить с одной площадки 8—12 расходящихся скважин (кустовое
бурение).
Схема расположения скважин на нефтяном месторождении
показана на рис. 5.
Эксплуатация скважин. Способ добычи нефти и газа зависит
от того, находятся ли они в пласте под давлением или нет. В
первом случае, как только скважина пройдет через непроницаемый,
Рис. 5. Примерная схема расположения скважин на нефтяном
месторождении:
/—антиклинальная складка непроницаемой горной породы; 2—скопление газа;
Я—скопление нефти; 4—пластовая (подпирающая) вода; Л—скважина
вертикальная; 6—наклонные скважины; 7—застроенный^ участок; 8—море.
сдерживавший слой, газ или нефть вырываются по ней на
поверхность. При этом нефть бьет из земли фонтаном, такой метод ее
добычи называется фонтанным. Открытый фонтан нефти опасен
в пожарном отношении, легкие углеводороды нефти и газ при
этом совсем теряются. Чтобы их уловить, над устьем скважины
ставят специальную запорную арматуру, через которую газ или
нефть отводят по трубам в специальные приемные устройства.
Под пластовым давлением нефть, и особенно газ, можно
перемещать на значительные расстояния, что повышает экономичность
данного метода добычи. По мере эксплуатации скважины давление
в пласте падает, его удается искусственно поддерживать,
например, закачивая в пласт (по его контуру) воду через исчерпанные
скважины.
Во втором случае, когда давление в пласте отсутствует, нефть
можно откачивать из пласта (газ так нельзя добывать).
25
В хранилище Газ
Нефть
'/////////Л
Рис. 6. Схема газополъемной установки:
1—газовая трубі; 2—подъемная трубі. З- скважина; 4 —компрессор;
5—газоотделитель; 6—поплавковый регулятор уровня.
Ж№тТ№Щ?ЩвЩ
Рис. 7. Схема
глубинного насоса:
/— обсядная трубі; 2—насосная
трубі. З—пигметмтельный
клапан. 4—приемный (всас'ыв^ю-
шиЙ) кл.|П:ш; 5 з ібой
скважины; *>—двигатель,
7—качающееся коромысло, в—штанга;
3—трос.
По этому методу осуществляется подъем нефти при
помощи газа (или воздуха) и откачиванием ее глубинными насосами.
По первому способу (рис. 6) сжатый газ, подаваемый в пласт
по трубе /, соприкасается с нефтью и вспенивает ее. Легкая
газонефтяная пена по подъемной трубе 2 поступает в газоотделитель,
а из него—в приемное хранилище. Такие же газоотделители
используются и для отделения нефти от газа при совместном выходе
их из пласта под давлением.
Другой более распространенный способ добычи нефти
заключается в откачивании ее глубинными насосами (рис. 7). Штанга 8
насоса поднимается и опускается специальным механизмом 7.
Вместе со штангой движется поршень, несущий шаровой
нагнетательный клапан 3. Нефть входит в рабочую полость насосной
трубы 2 через приемный клапан 4.
Из скважин нефть и газ поступают в специальные
резервуары-хранилища, откуда расходуются по назначению.
Описанными способами удается извлечь из пласта далеко не
всю находящуюся в нем нефть. В зависимости от способа добычи
отдача нефти составляет 40%, редко—до 60% ее запасов в пласте.
Увеличить отдачу нефти удается только специальными приемами.
2. ПЕРЕРАБОТКА ГАЗОВ
На химическую переработку могут поступать различные
нефтяные газы:
природный (сухой, тощий) газ чисто газовых месторождений,
состоящий преимущественно из метана СН4;
попутный {жирный) газ, получаемый из скважин попутно с
нефтью и содержащий, кроме метана, различные количества
насыщенных углеводородов (от С2 и выше);
газы нефтепереработки (нефтезаводские газы)—искусственные
газы, получаемые в процессах переработки нефти, связанных с
ее разложением. Состав нефтезаводских газов особенно
разнообразен, большей частью они содержат значительные количества
непредельных углеводородов, являющихся ценным сырьем для
промышленности органического синтеза.
Методы переработки газов зависят от ее целевого назначения
и от свойств и состава перерабатываемого газа. Основными
методами переработки газов являются:
1) очистка от вредных примесей;
2) улавливание легкокипящих углеводородов (газового
бензина), содержащихся в газе в виде паров;
3) разделение газов на индивидуальные компоненты или
группы компонентов;
4) химические превращения газового сырья;
5) получение из газа сажи.
27
Ьсли газ используется как топливо, его химическая оораоотка
ограничивается очисткой и освобождением от паров газового
бензина (если они присутствуют). Остальные процессы связаны
с более глубокой химической переработкой газов и подробнее
описываются во второй части книги (стр. 131 и ел.). В частности,
разделение газа на компоненты является подготовкой к
химической переработке индивидуальных предельных газообразных
углеводородов в непредельные (например, этана в этилен).
Углеводородные газы необходимы во все больших количествах для
разнообразных синтезов, которые, как правило, проводятся с
использованием однообразного по составу, простого химически
активного сырья. Таким сырьем являются индивидуальные
непредельные углеводороды, которые приходится получать из предельных
углеводородов, так как ресурсы непредельных в газах
нефтепереработки становятся недостаточными. Химическая переработка
газов приобретает все большее значение и является одним из
основных направлений дальнейшего развития химической
промышленности в нашей стране.
Разделение смесей газообразных и легколетучих
углеводородов используют также для выделения из них пропан-бутановой
фракции, по составу занимающей промежуточное положение
между сухим газом и газовым бензином, которые могут содержать
Газы
Природный
саратовский
ставропольский
Попутный
бугурусланский
новогрозненский
ишимбайский (сернистый) ....
Искусственные (нефтезаводские)**
термического крекинга (стр. 58)
термического пиролиза (стр. 62)
каталитического крекинга
(тяжелое сырье, стр. 64)
платформинга (стр. 66) ....
Относительная
плотность
(плотность
воздуха = [)
0,592
0,94
1,2—1,4
1,0
Состав
различных
Содержание,
н2
3
14
1,69
32
предельные
сн4
94
98
70—73
40—50
44—45
50
40
8,8
24
С2Н6
1—2
0,3
10
10—13
17—20
17
12
8,4
16
• По книге В. Е. Пархоменко, Технология переработки нефти и газа, Гостоптех-
издат, 1959, стр. 15 и 227.
** Усредненные составы газов, огвобожденных от пентана.
28
пропан и бутан, часто являющиеся нежелательной примесью этих
газовых смесей. Между тем эти углеводороды (С3Н8 и С4Н1в)
представляют собой легко ожижаемое газообразное топливо и
имеют самостоятельное применение как химическое сырье. При
использовании пропан-бутановой смеси в качестве топлива ее
подвергают ожижению (под избыточным давлением до 12 am)
и доставляют потребителю в стальных баллонах. При выпуске
из баллона жидкая смесь превращается в газ и сгорает в горелках
бытовых, коммунальных и промышленных топок. Ожиженные
газы используют также как топливо для автомобилей и для резки
металлов.
СОСТАВ ГАЗОВ
Из горючих веществ органического происхождения
газообразное топливо наиболее просто по составу. Так, число
углеводородов Q—С5 (нормального и изостроения), из которых могут состоять
нефтяные газы, ограничено (менее двух десятков). По
количественному содержанию отдельных компонентов углеводородные
газы сильно отличаются друг от друга (табл. 5). Размеры добычи
природных и попутных газов более чем в десять раз превышают
ресурсы газов нефтепереработки.
Таблица 5
нефтяных газов*
объемн. %
углеводороды
с3н8
0,5—0,7
0,1
7,5
15—24
16
10
15,1
24
С4Н10
0,5
0,02
8,3
14—22
5,5
5
1,0
30,3
4
С5 и выше
0,2
5-10
2,5
—
—
непредельные
QH4
z
—
2
17
2,52
с3ни
—
—
8
9
16,90
углеводороды
с4н8
—
—
5
5
16,29
с4нв
—
1,0
—
со2
0,3
0,3
0,8
0,1
0,3
—
—
5алласт
H2S
—
1,0
5,0
—
—
првчяе
3,0
1,0
—
—
29
C!w~UiKA И OMFiCiXA Глоиб
Перед использованием и подачей в линии дальнего
газоснабжения нефтяные газы должны быть освобождены от вредных
примесей и балласта—влаги, сероводорода и других сернистых
соединений, двуокиси углерода, азота.
Водяные пары являются не только балластом, снижающим
теплотворную способность газа, но и затрудняют его
использование. При снижении температуры газа влага конденсируется и
заливает трубы. При более низких температурах она замерзает,
создавая ледяные пробки. С углеводородами, входящими в состав
газа, влага способна образовывать твердые гидраты (например,
СН4-7Н2О, С2Н6-7Н2О и др.) в следующих условиях:
Условия Углеводороды
СН4 CjHg C3Hj C4H10
Температура, ниже которой образуются гидраты,
°С 21,5 15 5,6 0,9
Давление, выше которого образуются гидраты,
кгс/см2 300 33 5,4 1,2
Выделяющиеся гидраты могут закупоривать трубопроводы и
газовые приборы.
Присутствующая в газе влага зачастую затрудняет его
использование в качестве химического сырья, так как вредно действует
на катализаторы и другие вещества, участвующие в процессах
переработки газов.
Осушка газа производится при помощи веществ, поглощающих
влагу. Для этого используются твердые и жидкие поглотители.
В качестве твердых поглотителей применяют гранулированный
хлористый кальций, плавленый едкий натр и др. Их употребляют,
когда требуется полная осушка от остатков влаги при
повышенном давлении газа (более 50 am). Установки для осушки газа
твердыми поглотителями компактны, но эксплуатация их обходится
дороже, чем жидкостных установок.
В качестве жидкого поглотителя для осушки газа чаще всего
применяется диэтиленгликоль (НОСН2—СН2JО (d*„=l,12; темп,
кип. 244,5 °С, темп. пл. —6,5 °С), хорошо поглощающий влагу.
Можно применять также водный раствор СаС12. Осушка обычно
проводится в поглотительных колоннах—абсорберах (стр. 32),
в которых газ барботирует через несколько слоев поглотительного
раствора. После насыщения влагой абсорбент регенерируется
путем кипячения.
Сероводород и другие сернистые соединения являются крайне
нежелательной примесью в газе. Сероводород вызывает коррозию
аппаратуры. При сжигании газа присутствующие в нем
серосодержащие соединения образуют сернистый ангидрид SO2, вредно
действующий на аппаратуру и приборы, с которыми соприкаса-
30
ются продукты горения газа. Кроме того, такие продукты горения
загрязняют воздух. При химической переработке газов сернистые
соединения вредно действуют на катализаторы.
Для очистки газа от сероводорода и других сернистых
соединений, как и для осушки, можно применять твердые и жидкие
поглотители. В качестве твердых сорбентов используется
специально подготовленная гидроокись железа Fe(OHK,
реже—активированный уголь. Способы очистки газов от серы твердыми и
некоторыми жидкими поглотителями описаны в курсе химической
технологии неорганических веществ. Очистка нефтяных газов
твердыми поглотителями применяется редко.
При очистке жидкими поглотителями (мокрые методы очистки)
различают два типа процессов—с превращением сероводорода в
другие вещества и без его превращения. Первые из этих процессов
обычно связаны с окислением сероводорода до элементарной серы
переносчиками кислорода, входящими в состав поглотительной
жидкости:
HaS + і/зОа » Н2О + S
Такие методы чаще применяются для очистки искусственных
газов, получаемых из твердого топлива (коксовый газ,
генераторные газы).
Для очистки нефтяных газов наиболее широко используются
жидкостные круговые процессы, в которых циркулирующий в
поглотительной системе абсорбент образует с сероводородом
легко разрушаемые соединения. При образовании таких
соединений сероводород извлекается из газа, при их разрушении—
регенерируется абсорбент и выделяется свободный сероводород.
В качестве абсорбентов используются:
1. Соли сильных оснований и слабых кислот (карбонаты, фос-
-фаты, феноляты натрия или калия), взаимодействующие с
сероводородом по реакции:
Me2R -f HaS ;—» MeHR + MeHS
(здесь Me—щелочной металл; R—кислотный остаток). В условиях
процесса поглощения (нормальная температура, целесообразно
также повышенное давление) реакция идет слева направо, при
регенерации (повышенная температура, целесообразен вакуум)—
справа налево.
2. Слабые органические основания, из которых особенно
широко используются, например, этаноламины. Процесс
поглощения сероводорода идет по следующей реакции:
2(CH2CH2OH)NHa + H2S » [(CH2CH,OH)NH3]aS
моноэтаноламин аминосульфид
При поглощении она протекает слева направо, при
регенерации—справа налево.
31
Аппаратура и технологическая схема всех круговых процессов
сходны (рис. 8). Очищаемый газ пропускают снизу вверх через
абсорбер (скруббер) /, где
противотоком ему движет-
Очйщенныйт
4H,S
газ -
ся абсорбент — раствор,
промывающий газ. Чтобы
обеспечить хороший
контакт газа и
поглотительного раствора, абсорбер
заполняют насадкой
(деревянные решетчатые
круги из реек, укладываемые
друг на друга, спирали
из нержавеющей
металлической ленты, фигурные
тела из химически
стойкого
материала—керамики, металла), по которой
стекает раствор, или
оборудуют абсорбер
специальными полками
(тарелками). Перерабатываемая
жидкость собирается на тарелках абсорбера, через слой жидкости
барботирует (пробулькивает) очищаемый газ.
¦ Газ ,|
на очистку
Рис. 8. Схема кругового абсорбционного
процесса (улавливания HiS):
/—абгорбер; 2—десорбер; 3—теплообменник;
4—подогреватель; Л—холодильник; в—насосы.
Рис. 9. Типы насадок:
/—кольца, уложенные беспорядочно; 2—кольца с перегородками, правильне
уложенные; 3—спиральные кольца; 4—шары; 5—пропеллерная насадка;
6—седлообразная насадка; 7—хордовая насадка.
Различные типы насадок для контактирования газов с
жидкостями показаны на рис. 9.
32
Насыщенный поглотительный раствор подается в регенератор
(десорбер) 2 (см. рис. 8), где при повышенной температуре (а иногда
при разрежении) из раствора отдувается сероводород. Устройство
десорберов и абсорберов аналогично. Большей частью
применяются тарельчатые десорберы. Поскольку в процессе очистки
газа поглотительный раствор то нагревается, то охлаждается,
большое значение приобретает 6ar.ee полнее испех/ьзоваьие
физического теп„.а раствора. Горячий регенерированный раствор,
движущийся в теплообменнике 3 по одну сторону труб, отдает
тепло раствору, направляемому на регенерацию. Окончательны^
подогрев раствора до температуры десорбции и его охлаждение,
необходимое для лучшего поглощения, производится
соответственно в подогревателе 4 и холодильнике 5. Циркуляция раствора в
системе осуществляется при помощи насосов 6.
Сероводород, получаемый в результате очистки газа, может
быть использован для производства серной кислоты (сжигание
H2S).
Степень очистки газа от серы зависит от направления erofc
дальнейшего использования. Газ для технических целей (обогрев-
промышленных печей и др.) может содержать от 1—2 до 20 гім*
H2S, для бытовых нужд—до 0,02 г/м3, при использовании газ*
в каталитических процессах подчас требуется очистка до
содержания 0,2 мгім3 серы и менее. Особенно трудно удаляются из газа»,
органические соединения серы, для очистки от которых
приходится применять более сложные методы, чем описанные здесь.
УЛАВЛИВАНИЕ ГАЗОВОГО БЕНЗИНА
Улавливание из газа паров легкокипящих углеводородов
газового бензина является первичным грубым разделением
углеводородной части газа на группы компонентов: парообразные
углеводороды (С4 и выше) и газообразные углеводороды (С,—С3). При
этом получаются два продукта—газ и газовый бензин, дальнейшие
направления использования которых различны. Газ используется
как ценное газообразное топливо и химическое сырье (чаще всего
после разделения на индивидуальные компоненты). Газовый
бензин, являющийся смесью низкокипящих углеводородов,
применяется в основном в качестве компонента легкого моторного
топлива. Из газового бензина выделяют также отдельные
компоненты, например пентан, для их химической переработки. Пред
варительное разделение сырого газа облегчает его дальнейшую^
тонкую переработку.
Основными методами улавливания газового бензина являются
компрессия и охлаждение, абсорбция и адсорбция. Обычно эт%
процессы дополняются процессом стабилизации, газового бенви~
на, заключающимся в отделении от него пропана и частично
бутана. Стабилизация производится методом ректификация
3-805 33
^CT[J. Ч2.) ііиД ДаБЛСпгісМ и HiVitCi цсЛїли у гїгсппшп і с, uiuij-icvia і ci3\j-
вого бензина и испарение его при хранении, предотвратить
образование газовых пробок в топливопроводах двигателей внутреннего
сгорания.
Компрессия и охлаждение. Чаще применяется не
самостоятельно, а р комбинации с другими процессами. Для отделения
газового бензина этим методом газ сжимают (компримируют) и
охлаждают, р зависимости от примененных давления и температуры
соответствующая часть углеводородов переходит в жидкое
состояние (конденсируется) и конденсат отделяется от газа. Чем выше
давление сжатия и ниже температура охлаждения газа, тем больше
выход конденсата.
Описанным методом не достигается четкое разделение
компонентов сырого газа, так как при его сжатии и охлаждении
одновременно присутствуют все компоненты исходной паро-газовой смеси,
поэтому они же будут содержаться и в газе и в конденсате. Однако
относительное содержание их в жидкой и газовой фазах будет
различно: в конденсате (газовом бензине) будут преобладать
более тяжелые компоненты, в газе—более легкие. В конце процесса,
когда между составами газа и конденсата установится равновесие,
соотношение концентраций каждого компонента в газовой и
жидкой фазах будет определяться уравнением:
У = КХ
где X и У—молекулярные концентрации (мольные доли)*
данного компонента соответственно в жидкой и газовой
(паровой) фазах;
К—константа равновесия, определяющая
распределение вещества между газовой и жидкой фазами.
Каждый компонент смеси в данных условиях характеризуется
определенной величиной id которая увеличивается с повышением
температуры и уменьшается с ростом давления.
Как видно из приведенного выше уравнения, компоненты, для
которых в данных условиях TOI, преимущественно остаются в
газе, а те компоненты, для которых і(<1, переходят в конденсат.
Чем ниже конечная температура процесса, тем меньше может
быть давление, необходимое для ожижения заданной части
исходного газа. Четкость разделения компонентов также улучшается
со снижением температуры процесса. Обычно для выделения
газового бензина искусственный холод не применяется, и
температура охлажденного газа определяется температурой охлаждающей
технической воды; чаще всего процесс ведут при 25—30 °С.
Давление выбирается в зависимости от конкретных условий процесса
разделения, пределы применяемых давлений довольно широкие.
* Молекулярной концентрацией (мольной долей) компонента называется
количество его молей, содержащихся в 1 моль смеси, в которую входит данный
компонент.
34
Общий вид компрессионной установки для улавливания
бензина из природного газа показан на рис. 10.
Абсорбция и десорбция. Эти процессы дают возможность
более полно и четко отделить газовый бензин от газа. Процесс
проводится по схеме кругового процесса аналогично очистке газа от
H2S (см. рис. 8, стр. 32). В абсорбере происходит выделение
(вымывание) из газа газового бензина, в десорбере
(регенераторе)—отдувка острым водяным паром" поглощенного гаюзого
Рис. 10. Общий вид компрессионной установки для улавливания бензина
из природного газа.
бензина из поглотителя. Различие заключается лишь в том, что
при серосчистке псглсщгемыи компонент (H2S) реагирует с
поглотителем, а при улавливании газового бензина происходит его
растворение в поглотителе. Распределение компонентов сырого газа
между газовой фазой и жидкой фазой (поглотителем) подчиняется
той же закономерности, которая была рассмотрена при описании
процесса разделения методом компрессии, т. е. для улучшения
улавливания компонентов газа надо уменьшать величину f(,
т. е. понижать температуру и повышать давление. В процессе
* Острым называется пар, подаваемый непосредственно в
обрабатываемое вещество. Пар, подаваемый для нагрева в трубчатку, отдающий тепяо
через ее стенку и не соприкасающийся с нагреваемым веществом, называется
глухим.
3* 35
десорбции, наоборот, требуется повышение температуры и
снижение даоления.
Как указывалось, методом абсорбции (при прочих равных
условиях) удается более полно отделить бензин, чем
компрессионным методом, и лучше разделить компоненты газовой смеси на
бензин и газ. Это объясняется применением поглотителя и
контактом газа и поглотителя в противотоке на насадке или на бар-
ботажных тарелках (стр. 3 ). Таким образом, абсорбционный
рроцесс разделения протекает последовательно, в несколько
этапов, при компрессии же происходит однократное разделение.
В качестве поглотителей обычно применяют жидкие
нефтепродукты, кипящие при 100—200 °С. При разделении газа абсорбцион-
рые установки зачастую применяют после компрессионных для
дополнительного улавливания части бензина, оставшегося в газе.
Адсорбция—процесс, основанный на способности адсорогнтов
{твердых пористых тел с сильно развитыми мелкими порами)
поглощать вещества, с которыми соприкасается поверхность адсорбента.
Из смеси углеводородов адсорбент активнее извлекает вещества
с более высоким молекулярным весом. Если адсорбент, ранее
поглотивший низкомолекулярный углеводород, привести в
соприкосновение с высокомолекулярным углеводородом, то последний
вытеснит из адсорбента более легкий компонент. Этой особенностью
процессу адсорбции пользуются для избирательного
(селективного) поглощения отдельных углеводородов и разделения
углеводородных смесей на компоненты. Поэтому адсорбция чаще
примеряется не для улавливания газового бензина, а для более тонкой
ререработкй газов, которая описана далее (стр. 15 j ел.).
В качестве адсорбентов используют активированный уголь,
Кйликагель, активированные глины. Адсорбент загружают в
вертикальные полые аппараты (адсорберы), через которые пропускают
ja3. По насыщении адсорбента отключают поток газа от адсорбера
ц продувают через него острый водяной пар для отгонки бензина
wji адсорбента. После отдувки адсорбент охлаждают, сушат и
снова пропускают через адсорбер газ до насыщения адсорбента
бензином.
Применяются также адсорберы с подвижным слоем
адсорбента (стр. 156),
ГАЗОВАЯ САЖА
В ряде отраслей химической промышленности используется
Сажа—пгрошкесбразрый амсгфный углерод (размеры частиц
порядка 30—50 мк). Главным истребителем сажи является
резиновая промышленность, применяющая сажу в качестве активного
наполнителя резиновых смесей, улучшающего механические
свойства резигы (стр. 4S9). Сажа потребляется также в
лакокрасочной,' пелигргфической, электротехнической и других отраслях
промышленности.
36
Одним из лучших видов сажи является газовая сяжа. Она
получается путем неполного сжигания газа. При недостатке воздуха
газ горит сильно коптящим пламенем, образующуюся при этом
сажу собирают, просеивают, размалывают или перетирают на
вальцах и упаковывают в мешки. Для получения сажи
применяются также методы термического разложения метана.
Ввиду горючести, взрывоопасное™ и токсичности газов во
всех технологических процессах с их участием требуются
исключительная внимательность и четкость проведения операций.
Особую опасность представляет образование взрывчатых газо-воз-
душных смесей при утечке газов.
Для быстрого обнаружения утечки газ при подаче его в
газопроводы и сети одорируют—добавляют к нему небольшое
количество летучего вещества, обладающего сильным запахом (обычно
меркаптаны). При появлении характерного запаха одоранта
необходимо быстро обнаружить и ликвидировать причину утечки
таза.
Взрывы и пожары, возможные при несоблюдении правил
работы с газами, могут иметь весьма тяжелые последствия. Вопросы
техники безопасности при работе с углеводородными газами более
подробно рассмотрены на стр. 154.
4. ПЕРЕРАБОТКА НЕФТИ
Состав нефти гораздо сложнее состава природных газов. Нефть
представляет собой смесь большого числа разнообразных веществ,
в основном углеводородов, значительно более сложных, чем
углеводороды газа. Чем выше температура кипения углеводорода
нормального строения, тем больше число его возможных
изомеров:
Углеводороды
с4н10
сьн12
Температура кипе- Число
ния нормального возможных
углеводорода, °С изомеров
—0,6
36,3
69
125,7
214,5
2
3
5
18
355
Углеводороды
с,ян38
С4|Н8
Температура кипе- Чисто
ния нормального возможны*
углеводорода, °С изомэроа
318
Разлагаются до
начала
кипения при
атмосферном
давлении
Тоже
(Ю- 10'
36.8-10*
62.|0'а
Поэтому чем выше температурные пределы выкипания
нефтепродукта (стр. 39), тем сложнее его состав. Сложность состава
и разнообразие типов (клесов) нефтей обусловливают
возможность получения из них большого числа различных продуктов и
многообразие методов переработки нефти. Выбор метода
переработки определяется свойствами нефти и продуктами, которые должны
быть получены из нее.
37
Основное количество добываемой нефти перерабатывается на
моторные топлива (бензин, керосин и др.) и смазочные масла.
В меньших количествах получают трансформаторное,
медицинские масла и другие продукты. В последние десятилетия быстро
развивается использование продуктов переработки нефти в
качестве химического сырья. Нефтехимическая промышленность
становится одной из ведущих отраслей мировой экономики.
В сыром виде нефть сейчас не используется даже как топливо.
Из нее, как правило, получают различные нефтепродукты
(стр. 4с), свойства которых четко регламентируются в
соответствии с у.л..ьияг».и потреб..е..ия этих продуктов.
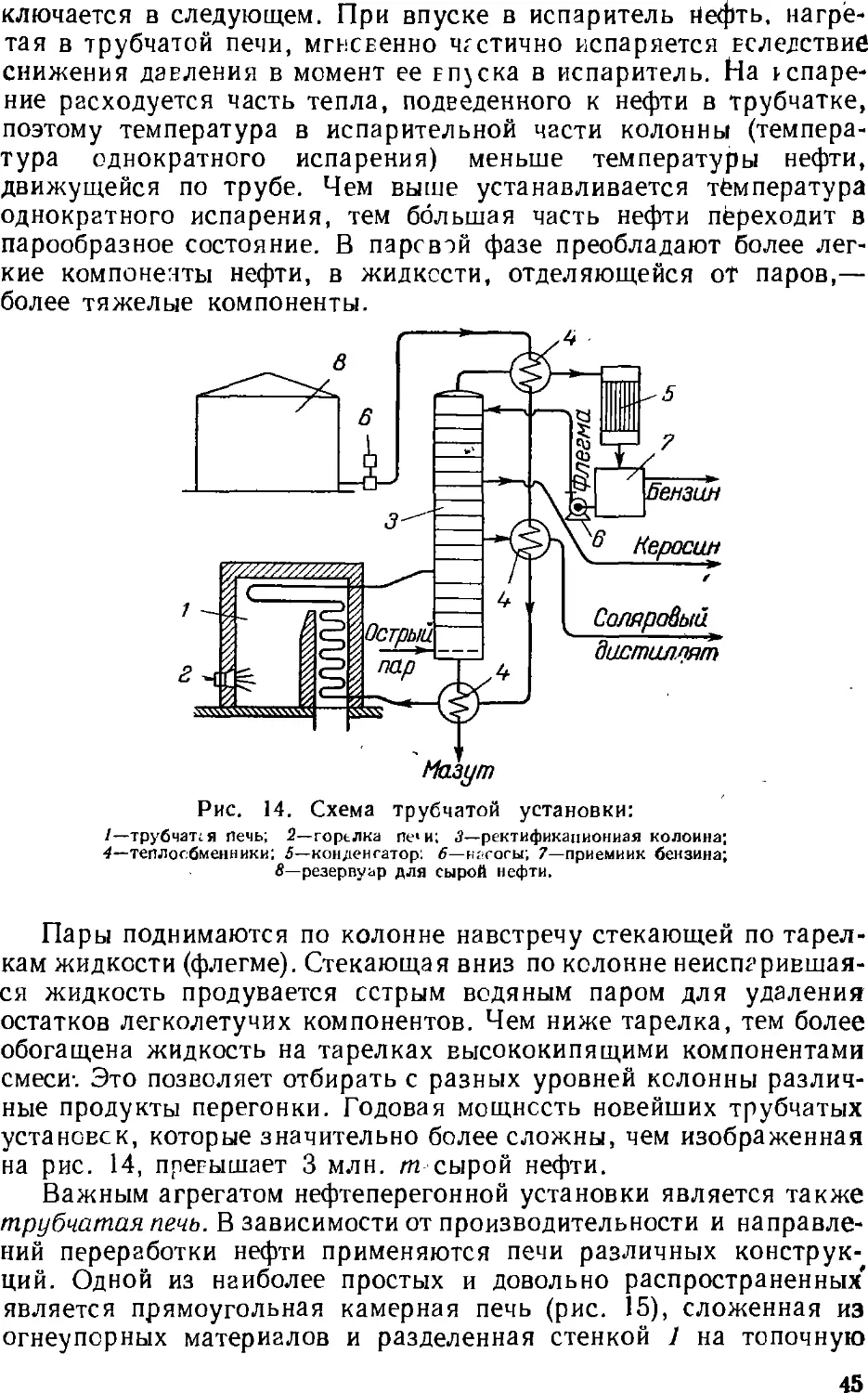
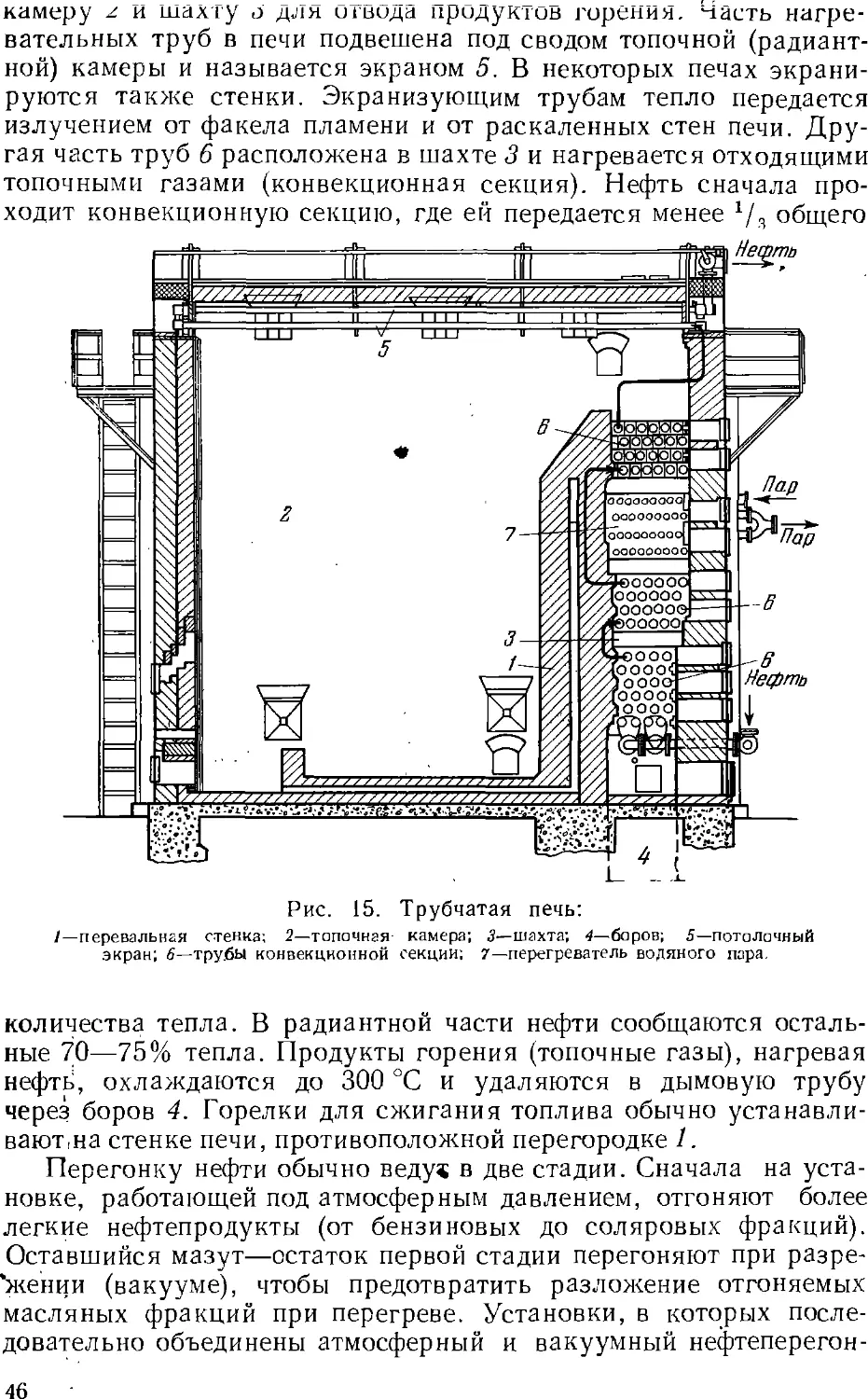
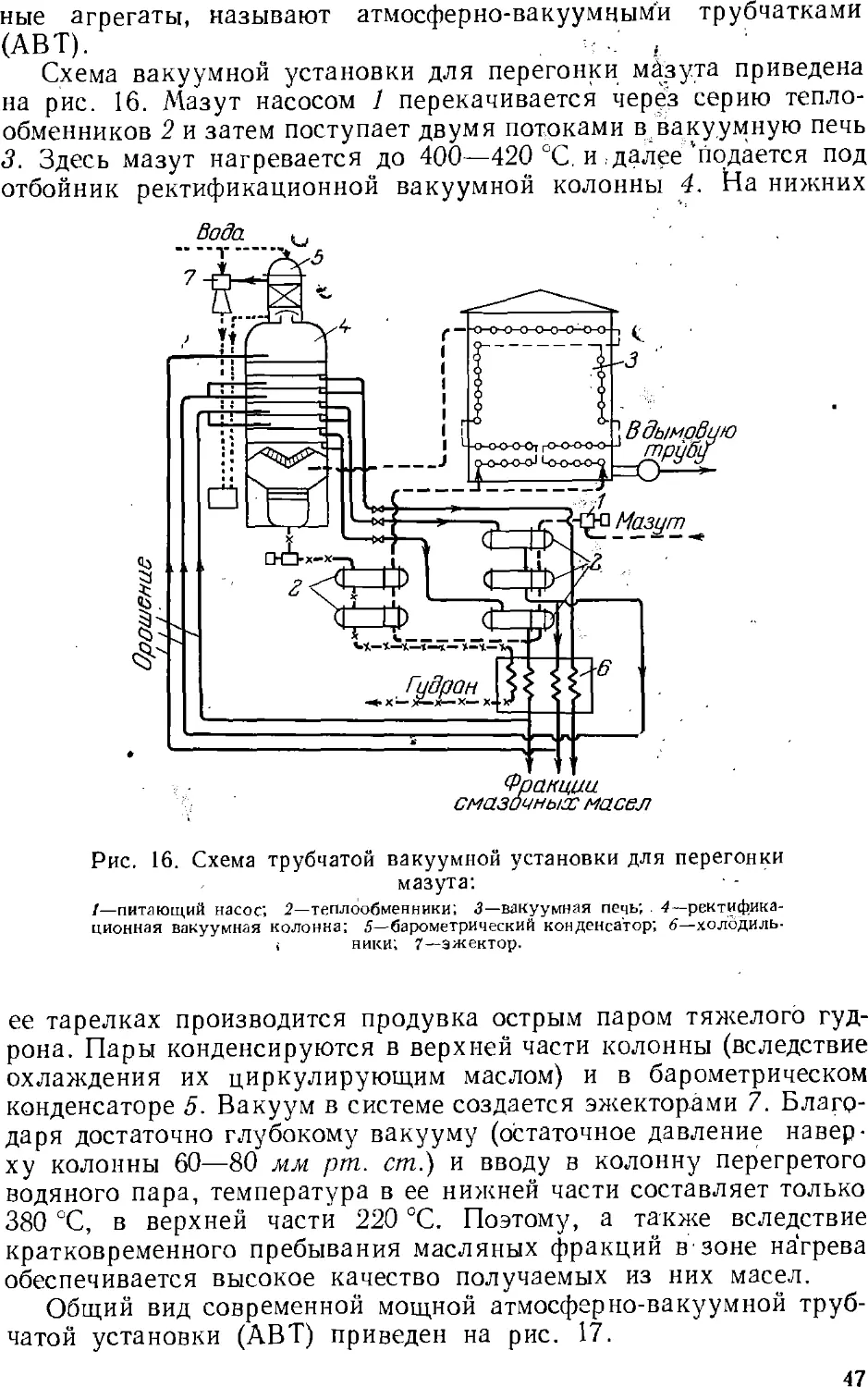
Основным процессом переработки нефти является разделение
ее перегонкой (стр. 4z) на фракции, выкипающие в определенных
температурных пределах, наиболее дефицитны обычно легкоки-
пящие (бензиновые) фракции. Для увеличения их выхода нефть
подвергают химической переработке, в результате которой менее
дефлцитные ьысококипящле компоненты нефти превращаются
в легкокипящие продукты заданного состава.
Методы химической переработки перечислены ниже.
Крекинг и пиролиз—процессы термической обработки нефти и
нефтепродуктов, в ходе которой происходит расщепление
молекул их компонентов с одновременной изомеризацией и соединением
(уплотнением) части продуктов расщепления. Такой термический
процесс, проводимый при температуре ниже 600 UC с целью
увеличения выхода бензина, называется крекингом (стр. 5с). Если
для ускорения крекинга применяют катализатор, то процесс
называется каталитическим крекингом (стр. 64). Термический
процесс, проводимый при температурах выше 700 °С для
получения ароматических соединений (бензола, толуола и др.) и
непредельных углеводородов, называется пиролизом (стр. 62).
Крекинг в присутствии водорода, гидрирующего продукты
расщепления нефтяного сырья, называется гидрогенизационным
крекингом (стр. 67). Обработка нефтяных остатков водородом,
связанная с более глубоким расщеплением молекул, для
превращения этих веществ в легкие продукты называется деструктивной
гидрогенизацией.
Для выделения из нефти и нефтепродуктов отдельных ценных
веществ (например, парафина) или вредных примесей применяются
также кристаллизация, избирательное растворение, обработка
химическими реактивами (последняя чаще при очистке
нефтепродуктов).
Получение нефтехимического сырья, его очистка и подготовка
к дальнейшему использованию обычно составляют
заключительные операции нефтепереработки. Они проводятся на
нефтеперерабатывающем заводе, а иногда на химическом заводе,
потребляющем указанное сырье. Эти процессы подробнее описываются ниже
(стр. 131 ел.).
28
СОСТАВ И СВОЙСТВА НЕФТИ
В зависимости от качества сырая нефть представляет собой
более или менее густую красно-коричневую жидкость (легкие
нефти—желтого цвета с зеленоватым оттенком, тяжелые—черного
цвета) с характерным запахом. Нефть состоит в основном из
углеводородов и содержит примеси смолистых веществ, органических
кислот, сернистых и азотистых органических соединений, а также
минеральные примеси. Сырая нефть может содержать, кроме того,
воду.
Элементарный состав углеводородной части нефти: 82—88%
углерода, 12—18% водорода. По характеру углеводородной части
нефти делятся на следующие классы*:
1) парафино-нафтеновые, наиболее богатые бензиновыми
фракциями и содержащие твердый парафин;
2) нафтено-парафиновые;
3) нафтено-ароматические;
4) парафино-нафтено-ароматические (асфальтовые)—наиболее
тяжелые из нефтей.
Чисто парафиновых, нафтеновых или ароматических нефтей,
т. е. содержащих углеводороды только одного гомологического
ряда, как правило, не бывает, ненасыщенные углеводороды редко
присутствуют в нефтях.
Для оценки качества нефти и выбора методов ее дальнейшей
переработки большое значение имеет распределение содержащихся
в ней углеводородов по температурам кипения. Особенно ценны
нефти с преобладанием легкокипящих углеводородов, плотность
таких нефтей ниже и они называются легкими. Чем больше высо-
кокипящих углеводородов содержит нефть, тем более тяжелой
она считается.
Температурные пределы выкипания нефтяных фракций:
Фракции Пределы кипения
°С
Бензиновые До 170—200
Лигроиновые 160—200
Керосиновые 180—280C00)
Газойле-соляровые . . . 300—350
Остаток после отгона перечисленных фракций называется
мазутом. Фракции нефти, из которых получаются смазочные
масла, имеют еще более высокую температуру кипения.
Фракционный состав нефти характеризуется ее кривой разгонки,
показывающей, какие количества исследуемой нефти отгоняются в
определенных температурных пределах.
* В названии класса углеводороды перечисляются в порядке их убывания
в составе нефти.
39
Кроме углеводородов, из остальных соединений, входящих
в состав нефтей, наибольшая доля приходится на кислородные
соединения—смолисто-асфальтовые вещества и органические
кислоты. При большом содержании смолистых веществ переработка
нефти сильно осложняется. Органические (как правило,
нафтеновые) кислоты нефтей образуют со щелочами мазеобразные мыла—
(мылонафт) и могут быть использованы для получения этого
продукта.
Весьма вредной примесью нефти являются сернистые
соединения. Их присутствие отрицательно сказывается в процессе
переработки нефти (они вызывают коррозию аппаратуры) и на
качестве получаемых нефтепродуктов. Сера присутствует в нефти в
виде сероводорода H,S, меркаптанов RSH, сульфидов (тиозфи-
ров) RSR', тисфена, его гидропроизводных и других соединений,
в нефтях встречается и элементарная сера. Содержание серы в
нефтях колеблется в широких пределах; так, малосернистые
бакинские и грозненские нефти содержат около 0,1% S, волжские
и башкирские—до 2,5—3,0%, нефть Чусовских Городков—5,5%.
Для переработки и использования нефти и нефтепродуктов
большое значение имеют следующие свойства:
Относи/' ельная плотность при 20 °С (отнесенная к плотности
воды при 4 °С) di" характеризует состав и качество нефти и
легкость ее отстаивания от воды. Обычно относительная плотность
нефтей находится в пределах 0,80—0,95.
Вязкость определяет расход энергии при перекачивании нефти
по трубопроводам. Вязкость разных нефтей может сильно
различаться и зависит от температуры, резко уменьшаясь при ее
повышении.
Температура застывания характеризует условия, при которых
нефть теряет текучесть; для разных нефтей колеблется от —80
до +20 °С.
Температура вспышки и воспламенения является показателем
степени огнеопасности нефтепродукта.
Взрывоопасность нефтепродуктов определяется верхним или
нижним пределами концентраций газов или паров нефтепродукта
в воздухе, при которых газо- или паро-воздушная смесь
взрывается при поднесении к ней пламени или проскоке электрической
искры (стр. 154).
Горючесть и взрывоопасность нефти и нефтепродуктов
обусловливают необходимость соблюдения целого ряда мер
предосторожности при обращении с ними и переработке их. В рабочих
помещениях должна обеспечиваться хорошая вентиляция, чтобы в них
не скапливались горючие газы и пары. Надо также применять
специальные герметизированные электроприборы и
электропроводки, чтобы исключить возможность образования искр. В
лабораториях надо соблюдать осторожность при обращении с открытым
огнем, а в производственных помещениях вообще не пользоваться
40
им. Необходимо также принимать меры к предотвращению искро-
образования при падении или ударе металлических и других
предметов. Несоблюдение правил безопасной работы с горючими
.продуктами может привести к несчастным случаям и тяжелым авариям.
Более подробные сведения о свойствах нефтей и
нефтепродуктов и методах их определения можно найти в специальной
литературе (см., например, стр. 118).
ПОДГОТОВКА НЕФТИ К ПЕРЕРАБОТКЕ
Сырая нефть не может быть непосредственно направлена на
переработку, так как содержит примеси, которые надо удалить.
От нефти отделяется попутный газ, являющийся денным
самостоятельным продуктом; к тому же, если он не отделен, то мешает
переработке нефти и вызывает увеличений ее потерь при хранении,
так как с газом будет улетучиваться часть бензиновых фракций.
/ г
и
Пар
т
\
Вода
-7 6-
-Вода
нефти
Рис. 11. Схема обезвоживания и обессоливания
электрическим методом;
/—сырая нефть; 2—насос; 3— нагреватель; 4—аппарат для
обезвоживания; 5—пластинчатые электроды; Л—водоотделитель; 7—хранилище
обезвоженной и обессоленной нефти.
Предварительное (грубое) отделение газа производится в
газоотделителях (см. рис. 6, стр. 26), окончательное (четкое)
отделение—в стабилизационных установках, где газ отгоняется от нефти
в специальных ректификационных колоннах (стр. 41).
Вредными примесями нефти являются вода, присутствующая в
ней в виде крупных капель, минеральные соли (NaCl, M^Cl,,
СаС12 и др.) и механические примеси (песок и глина). Вода и
механические примеси отделяются путем отстаивания, для
облегчения которого зачастую применяется подогрев нефти. Если вода
образует с нефтью трудно разделимые эмульсии, содержащие 20—
30 и даже до 60% воды в виде мельчайших капелек @,1—100 мк),
приходится применять деэмульгашоры—поверхностно-активные
вещества, способствующие разрушению пленок, которые окружают
водные частицы эмульсии. Особенно активно разрушаются эмуль-
41
сии при пропускании их между электродами, включенными в
цепь переменного тока высокого напряжения C0—40 тыс. б). При
этом капельки эмульсии деформируются, сливаются в крупные
капли, которые затем отделяются от нефти при ее отстаивании.
Схема установки для обезвоживания и обессоливания нефти
электрическим методом изображена на рис. 11. Такая обработка нефти
производится предварительно на промысле и окончательно на
нефтеперерабатывающем заводе. Кроме того, на заводе проводится
сортировка и смешение нефпгей для получения более равномерного
по составу сырья, поступающего на дальнейшую переработку, и
защелачивание нефти (добавление растворов щелочей или аммиака
для нейтрализации кислых и сернистых примесей, вызывающих
коррозию аппаратуры при переработке нефти).
ПЕРЕГОНКА НЕФТИ
Разделение нефти и нефтепродуктов перегонкой на фракции
основано на том, что при кипении смесей жидких веществ
образуются пары, содержащие все компоненты смеси, но с
преобладанием легколетучих низкокипящих компонентов. Содержание
каждого компонента в парах тем больше, чем выше его летучесть и
концентрация в кипящей смеси. По мере выкипания легколетучих
компонентов температура паров, отходящих из перегонного
аппарата (температура перегонки), возрастает. Отгоняемые пары
конденсируют; конденсаты, полученные в определенных
температурных пределах перегонки, образуют соответствующие фракции,
каждая последующая фракция состоит из более высококипящих
компонентов, чем ранее отогнанные фракции.
По этой схеме прежде проводили дробную перегонку нефтей,
которая, однако, не приводила к четкому разделению
компонентов на фракции. Для более четкого разделения
многокомпонентных смесей используют метод ректификации; при этом между
кипятильником, из которого отгоняются пары, и конденсатором
паров устанавливают ректификационную колонну.
Ректификационная колонна (рис. 12) представляет собой вертикальный
цилиндрический аппарат, заполненный насадкой (насадочная колонна, стр. 32)
или снабженный большим количеством горизонтальных тарелок 5,
расположенных одна над другой (тарельчатая колонна). В верхнюю часть колонны
подается орошающая жидкость (флегма), которая стекает по насадке или перетекает
с тарелки на тарелку навстречу поднимающимся из кипятильника парам.
Орошение колонны ведется легкокипящей жидкостью (обычно частью
получаемого дистиллята). На тарелках (или насадке) осуществляется тесный контакт
между парами и стекающей жидкостью, при этом более высококипящие
компоненты паров конденсируются и переходят в жидкий поток, а за счет тепла их
конденсации из жидкости испаряются более летучие компоненты. Поэтому
стекающая по тарелкам колонны жидкость (орошение) постепенно обогащается
высококипящими компонентами, а поднимающийся вверх паровой поток—легко-
кипящими. Таким образом, колонну покидают пары наиболее легких
компонентов перегоняемого сырья, достаточно четко отделенных от высококипящих
компонентов, образующих остаток.
42
Тарелка ректификационной колонны представляет собой горизонтальную
перегородку в колонне, на тарелке находится слой стекающей по колонне
жидкости (орошение), сквозь которую барботируют поднимающиеся снизу пары.
Тарелки бывают разнообразных конструкций и различаются по типу барботаж-
ных устройств. Наиболее распространены колпачковые тарелки (рис. 13), в
которых имеются паровые патрубки 2, перекрытые колпачками 3 с
зазубренными нижними краями. Пары, выходя из-под колпачка, барботируют через
слой жидкости на тарелке /, жидкость (флегма) стекает с тарелки на тарелку
по переливным патрубкам 4. Чем больше в колонне тарелок и чем интенсивнее
ее орошение, тем более четко удается разделить компоненты исходной смеси.
Ректификационные установки могут
работать по непрерывной или
периодической схеме. Кипятильник
ректификационной колонны периодического действия
представляет собой емкость (куб), куда
заливают порцию сырья, из которой
постепенно, одна за другой, отгоняются все
более тяжелые (высококипящие) фракции.
Остаток
Рис. 13. КолпаЧковая тарелка:
1—тарелка; 2—паровые патрубки; 3—колпачки; 4—переливные
патрубки (сплошными стрелками показано движение жидкости,
пунктирными—движение паров).
Рис. 12 Схема
ректификационной колонны:
/—ректификационная колонна,
2—конденсатор; 3— приемная
емкость для отгона; 4—насос
для подачн орошения в
колонну; 5—тарелкн колонны;
й—кипятильник.
Современные высокопроизводительные ректификационные
установки работают по непрерывной схеме (см. рис. 12). Подогретое
или частично испаренное сырье непрерывно подается в среднюю
часть колонны и разделяется в ней на два продукта—отгон и
остаток. Если исходную смесь надо разделить на несколько
фракций, устанавливают несколько колонн. Иногда их конструктивно
соединяют в одну колонну с промежуточным отбором фракций в
нескольких точках по высоте колонны (см. рис. 14). Каждая
колонна снабжена конденсатором, в котором конденсируются
покидающие колонну пары. Часть конденсата направляется на
орошение колонны, остальной конденсат отводится из системы как
продукт разделения.
43
Схемы промышленных ректификационных установок весьма
разнообразны в зависимости от характера перегоняемого сырья
и продуктов его разделения, которые должны быть получены.
Обогрев кипятильника может быть огневой или паровой в
зависимости от требуемых условий перегонки. Перегонку высоко-
кипящих термочувствительных продуктов для снижения
температуры ведут при разрежении или с подачей острого пара.
Ректификацию углеводородных газов и легкокипящих жидкостей
проводят под давлением, чтобы перегоняемые продукты оставались в
ожижением состоянии.
Переработка нефти методом перегонки в России возникла еще в 1745 г.
Промышленник Ф. С. Прядунов организовал тогда на Ухте производство
сбелой нефти» (керосина), которую он продавал для освещения в Петербурге,
Москве и других городах; завод просуществовал более 30 лет. В 1823 г. такое
производство в более крупном масштабе организовали на Северном Кавказе
бргтья Дубинины, выходцы нз Владимирской области, которые перенесли в
нефтепереработку опыт промышленности сухой перегонки дерева, развитой
в России. В Америке аналогичные производства возникли позже, в середине
XIX в.
Первсначрльно перегонка нефти проводилась на установках
периодического действия. Нефть загружали в цилиндрические, обычно горизонтальные,
металлические од бы, обогреваемые огнем. Пары отводились нз куба по
специальной трубе и конденсировались непосредственно в холодильнике, без
ректификации, поэтему отгоняемые фракции были нечеткими. Остаток после отбора
легких погсиов—мазіт, охлаждглся в кубе, затем мазут сливали и в куб
загружали новую порцию сырья. В технику перегонки нефтн в дальнейшем
были введены многочисленные усовершенствования. Большую роль в этом
сыграли такие крупные ученые, как Д. И. Менделеев A834—-1907 гг.), В. Г. Шу-
хон A853—1939 гг.) и др.
Были созданы, кубовые батареи непрерывного действия, в которых нефть
непрерывно перетекала из одного куба в другой, в каждом из них отбиралась
соответствующая фракция. Позже при кубах стали устанавливать сначала
примитивные, затем все более совершенные ректнфикациокныз колонны
В 1890 г. В. Г. Шухов и С. Г. Гаврилов сконструировали первую трубчатую
печь, н которой нефть нагревалась, проходя по змеевиковым трубам. Змеевик
имел хорошо развитую певерхность нагрева и прн этом вмещал малое
количество нефти в нагревательной зоне, что создавало значительно более безопас -
ные условия перегонки,, нежели в больших кубах.
В настоящее время вся перегонка нефти осуществляется на трубчатых
установках с ректификационными колоннами. Кубы используются только
в специальных процессах, обычно ограниченного масштаба, например, при
получении нефтяных битумов, нефтяного кокса.
Трубчатая установка (рис. 14) для перегонки нефти состоит
из трубчатой печи / (см. ниже), ректификационной колонны 3,
теплообменной аппаратуры 4 и 5 и другого вспомогательного
оборудования. Нефть подается на перегонку из резервуара 8 насосом
б через теплообменники 4, где для сокращения расхода топлива
нагревается теплом отходящих продуктов перегонки и затем
поступает в трубчатую печь /. Здесь нефть, проходя по трубам
змеевика, нагревается до требуемой конечной температуры и подается
в испарительную часть колонны 3, где происходит так
называемое однократное испарение нефти. Сущность этого процесса за-
44
ключается в следующем. При впуске в испаритель йефть,
нагретая в трубчатой печи, мгнсвенно чгстично испаряется вследствие
снижения давления в момент ее еп^скэ в испаритель. На рспаре-
ние расходуется часть тепла, подеєдєнного к нефти в трубчатке,
поэтому температура в испарительной части колонны
(температура однократного испарения) меньше температуры нефти,
движущейся по трубе. Чем выше устанавливается температура
однократного испарения, тем большая часть нефти переходит в
парообразное состояние. В парсвой фазе преобладают более
легкие компоненты нефти, в жидкости, отделяющейся от паров,—
более тяжелые компоненты.
в
Мазут
Рис. 14. Схема трубчатой установки:
/—трубчаті я печь; 2—roptJiKa пе1и; 3—ректификационная колонна;
4—теплообменники; 5—конденгатор: 6—насосы: 7—приемник бензина;
в—резервуар для сырой нефти.
Пары поднимаются по колонне навстречу стекающей по
тарелкам жидкости (флегме). Стекающая вниз по колонне неисп?риЕшая-
ся жидкость продувается сстрым водяным паром для удаления
остатков легколетучих компонентов. Чем ниже тарелка, тем более
обогащена жидкость на тарелках еысококипящими компонентами
смеси-. Это позволяет отбирать с разных уровней колонны
различные продукты перегонки. Годовая мощность новейших трубчатых
устаноЕск, которые значительно более сложны, чем изображенная
на рис. 14, превышает 3 млн. т сырой нефти.
Важным агрегатом нефтеперегонной установки является также
трубчатая печь. В зависимости от производительности и
направлений переработки нефти применяются печи различных
конструкций. Одной из наиболее простых и довольно распространенных'
является прямоугольная камерная печь (рис. 15), сложенная из
огнеупорных материалов и разделенная стенкой / на топочную
45
камеру ^ и шахту 6 для иіьида придукюи горения, пасть
нагревательных труб в печи подвешена под сводом топочной
(радиантнои) камеры и называется экраном 5. В некоторых печах
экранируются также стенки. Экранизующим трубам тепло передается
излучением от факела пламени и от раскаленных стен печи.
Другая часть труб 6 расположена в шахте 3 и нагревается отходящими
топочными газами (конвекционная секция). Нефть сначала
проходит конвекционную секцию, где ей передается менее х/з общего
Нефть
Рис. 15. Трубчатая печь:
/—перевальная стенка; 2—топочная камера; 3—шахта; 4—баров; 5—потолочный
экран; 6—трубы конвекционной секции; 7—перегреватель водяного пара.
количества тепла. В радиантнои части нефти сообщаются
остальные 70—75% тепла. Продукты горения (топочные газы), нагревая
нефть', охлаждаются до 300 °С и удаляются в дымовую трубу
через боров 4. Горелки для сжигания топлива обычно устанавли-
вают,на стенке печи, противоположной перегородке /.
Перегонку нефти обычно веду* в две стадии. Сначала на
установке, работающей под атмосферным давлением, отгоняют более
легкие нефтепродукты (от бензиновых до соляровых фракций).
Оставшийся мазут—остаток первой стадии перегоняют при разре-
*жении (вакууме), чтобы предотвратить разложение отгоняемых
масляных фракций при перегреве. Установки, в которых
последовательно объединены атмосферный и вакуумный нефтеперегон-
46
ные агрегаты, называют атмосферно-вакуумными трубчатками
(АВТ). ¦-,' 4.
Схема вакуумной установки для перегонки мазута приведена
на рис. 16. Мазут насосом / перекачивается через серию
теплообменников 2 и затем поступает двумя потоками в вакуумную печь
«3. Здесь мазут нагревается до 400—420 °С. и далее'подается под
отбойник ректификационной вакуумной колонны 4. На нижних
1
I
<§¦¦
а
} В дымовую
щруб
Фракции
смазочных масел
Рис. 16. Схема трубчатой вакуумной установки для перегонки
мазута:
/—питающий насос; 2—теплообменники; 3—вакуумная печь; 4—
ректификационная вакуумная колонна; 5—барометрический конденсатор; в—холодиль-
і ники; 7—эжектор.
ее тарелках производится продувка острым паром тяжелого
гудрона. Пары конденсируются в верхней части колонны (вследствие
охлаждения их циркулирующим маслом) и в барометрическом
конденсаторе 5. Вакуум в системе создается эжекторами 7.
Благодаря достаточно глубокому вакууму (остаточное давление
наверху колонны 60—80 мм рт. ст.) и вводу в колонну перегретого
водяного пара, температура в ее нижней части составляет только
380 °С, в верхней части 220 °С. Поэтому, а также вследствие
кратковременного пребывания масляных фракций в зоне нагрева
обеспечивается высокое качество получаемых из них масел.
Общий вид современной мощной атмосферно-вакуумной
трубчатой установки (АВТ) приведен на рис. 17.
47
A\*
Чг"-
|У**~'
V.
-^'«аапс^-М"»," ' "¦'¦ v - ^'" лліХ
Рис. 17. Общий вид современной мощной атмосферио-вакуумной
трубчатой установки (АВТ).
ВАЖНЕЙШИЕ НЕФТЕПРОДУКТЫ
Многочисленные и разнообразные продукты, получаемые при
переработке нефти (кроме газов нефтепереработки, стр. 29 и ел.),
молено подразделить на следующие группы:
1) светлые продукты (бензин, лигроин, керосин);
2) соляровые масла (газойль, дизельные топлива);
3) смазочные масла;
4) мази (масленочные смазки, вазелины и пр.), парафин;
5) остаточные продукты (мазут и гудрон);
6) нефтехимическое сырье—разнообразные продукты
переработки нефти (газообразные и жидкие легкие углеводороды, пара-
48
фин и др.). используемые в качестве исходных веществ в
промышленности органического синтеза (стр. 131 и ел.).
Области применения нефтепродуктов определяются их
свойствами, которые регламентируются соответствующими стандартами
и техническими условиями.
Светлые продукты. Главным продуктом переработки нефти
являются моторные бензины, используемые как горючее в
карбюраторных двигателях внутреннего сгорания. В цилиндре такого
двигателя горючая смесь бензина с воздухом сжимается,
зажигается искрой и сгорает с большой скоростью. Газообразные продукты
горения расширяются, приводя в движение поршень, а через
него и вал двигателя. Чем выше степень сжатия горючей смеси,
тем экономичнее, мощнее и компактнее двигатель. Некоторые
из бензинов при повышении температуры смеси во время ее
сжатия самовоспламеняются раньше, чем закончится сжатие, и
детонируют, т. е. сгорают со скоростью вчрыва. Такое
неправильное сгорание рабочей смеси сопровождается резкими колебаниями
давления в цилиндре и вызывает падение мощности и перегрев
двигателя.
Антидетонационные свойства, относящиеся к основным
показателям качества моторных бензинов, характеризуются так
называемым октановым числом. Оно определяется путем сравнения
антидетонационных свойств испытуемого образца бензина и
стандартных смесей изооктана и «-гептана, взятых в различных
соотношениях.
Октановое число гептана, который очень легко детонирует, принято за
нуль. Изооктан B,2,4-триметилпентан) обладает высокими
антидетонационными свойствами, его октановое число принято за 100. Смешивая в различных
отношениях эти два углеводорода, можно получить образцы моторного
топлива, которые по детонационным свойствам будут соответствовать разным сортам
бензина. Процентное содержание изооктана в смеси, детонирующей при тех
же условиях сжатия в цилиндре двигателя, что и испытуемый бензин, является
мерой антидетонационных свойств бензина и называется его октановым числом.
Обычно оно колеблется в пределах 70—87. Авиационные бензины имеют
октановое число 100—125, т. е. еще менее способны к детонации, чем изооктан
Для повышения октанового числа бензинов их смешивают с продуктами,
обладающими высоким октановым числом, или со специальными веществами,
называемыми антидетонаторами. В качестве антидетонатора обычно
применяется тетраэтнлевинец.
Испаряемость, также являющаяся важным показателем
качества бензина, характеризуется температурами, при которых
испаряется 10, 50 и 90 объемн. % бензина. Для разных сортов
бензина эти температуры различны.
Нефтепродукты, перегоняющиеся в интервале температур
180—280 °С (и даже до 315 °С), называются керосинами.
Плотность керосинов 0,788—0,835 г/см3. Промежуточное положение
между керосином и бензином занимают лигроины.
4—805 49
Керосины применяются главным образом как моторное
топливо для тракторов, но все больше используются и как сырье для
химической переработки. Известное количество керосина еще
употребляется для бытовых нужд (освещение и др.)-
Показателем безопасности применения керосина является его температура
вспышки, которая должна быть не ниже 28 °С (обычно 50—80 °С).
Цвет керосина характеризует степень его очистки.
Соляровые масла перегоняются при температуре выше 250
и примерно до 350 °С. Плотность их равна 0,87—0,88 г/см3,
температура вспышки—около 130 °С, температура застызания
—20 °С. Соляровые масла обладают значительной вязкостью.
Они широко применяются в качестве топлива для дизельных
двигателей, а в смеси с другими продуктами используются и как
сырье в процессе крекинга (стр. 58).
По аналогии с бензинами свойства дизельного топлива
характеризуются цетановым числом.
Для этого вида моторных топлив валена способность самовосплачгняться
при сжатии в цилиндре двигателя. В качестве эталонного легко
самовоспламеняющегося углеводорода выбран цетан (я-гексадекан), за углевоцород,
обладающий низкой (нулевой) способностью к самовоспламенению, принят
а-метилнафталин. Процентное содержание цетана в смеси с а-метилнафтали-
ном, самовоспламеняющейся в аналогичных условиях с испытуемым образцом
дизельного топлива, является его цетановым числом.
Смазочные масла являются вторым по важности
нефтепродуктом после бензинов. Кроме смазки всевозможных машин и
механизмов, эти масла употребляются при обработке металлов
резанием, в электротехнической промышленности (для
трансформаторов и электрических масляных выключателей), при
флотационном обогащении руд и для многих других целей. Огновныэ
показатели, по которым оценивается качество смазочных масел:
температуры вспышки, воспламенения, застывания, вязкость, цвет,
плотность, окисляемость, электропроводность, склонность к
образованию стойких эмульсий.
Смазочные масла подразделяются на следующие основные
группы:
индустриальные и трансмиссионные масла, применяемые для
смазывания станков и машин и не подвергающиеся
непосредственному воздействию пара, горячего воздуха или газов;
трансмиссионные масла особо вязкие;
масла для двигателей внутреннего сгорания, применяемые для
смазки цилиндро-поршневой группы двигателей;
масла для турбин, паровых машин и компрессоров,
работающих в особо жестких условиях, различных для разных типов
перечисленных машин;
специальные масла.
50
Мази и парафин. К ним относятся консистентные густые
смазки, вазелины, используемые в производстве медицинских и
парфюмерных препаратов, и парафин.
Парафин выделяется из нефтяных фракций путем
вымораживания. В состав парафина входят углеводороды, содержащие в
молекуле более 16 углеродных атомов. Хорошо очищенный
парафин широко употребляется в электротехнической, бумажной,
текстильной и других отраслях промышленности, для
изготовления свечей и все шире применяется как сырье в промышленности
органического синтеза. При окислении парафина на специальных
установках получают высокомолекулярные спирты и кислоты,
используемые для синтеза моющих веществ, искусственных
жиров и др.
Остаточные продукты. Среди них наибольшее значение имеют
мазуты, широко применяемые в качестве топлива и исходного
продукта для получения смазочных масел. Основными
показателями качества мазута является его вязкость (условная вязкость
примерно 30 °Е при 50 °С), а также плотность @,9—1,0 г/см3) и
температура вспышки (80—95 °С).
Остаток после отгонки масел из мазута, называемый гудроном,
используется непосредственно или после соответствующей
обработки в качестве нефтяного битума различных марок. Из гудрона
могут быть получены также смазочные масла с высокой
температурой вспышки.
Нефтяные битумы и асфальты представляют собой
высокоплавкие вещества буро-черного цвета. Они используются в
дорожном строительстве (асфальтирование), в производстве кровельных
материалов (толь, рубероид и др.), некоторых дешевых
пластических масс и др. Природные битумы и асфальты обладают лучшими
свойствами, чем нефтяные.
Коксованием остаточных продуктов нефтепереработки в
специальных кубах или печах получают нефтяной кокс. Благодаря
почти полному отсутствию золы (беззольный кокс) он
употребляется при изготовлении электродов для электрических печей,
различных изделий для электропромышленности, а также
искусственных графитовых изделий.
ОЧИСТКА НЕФТЕПРОДУКТОВ
При получении нефтепродуктов в их состав переходят вредные
примеси, содержавшиеся в сырой нефти или образовавшиеся при
ее переработке. Примеси сернистых соединений вызывают
коррозию аппаратуры, образуют при сгорании SO2, отравляют
катализаторы при химической переработке нефтепродуктов;
органические кислоты также оказывают коррозионное действие;
смолистые примеси образуют осадки и затрудняют сгорание топ-
лив; непредельные соединения снижают химическую стабильность
4* 51
нефтепродуктов. Чтобы товарный нефтепродукт был пригоден
для использования,, транспортирования и хранения, из него надо
удалить вредные примеси. Это достигается путем очистки
нефтепродуктов, методы которой весьма разнообразны, так как
выбираются в зависимости от характера перерабатываемого сырья
и требований к конечным нефтепродуктам.
Для очистки применяются как химические, так и
физико-химические методы. При химической очистке нефтепродукт
обрабатывают реагентом, взаимодействующим с удаляемой примесью,
которая при этом разрушается или уплотняется (зачастую до
полного осмолення). Реагент в таких случаях обычно теряется.
Физико-химические методы очистки основаны на том, что реагент,
не смешивающийся с очищаемым продуктом, растворяет или
сорбирует примеси, которые таким образом удаляются из
нефтепродукта. При последующей регенерации очистного реагента
поглощенная им примесь выделяется в неизменном виде или
разрушается. Если применяемый очистной агент обладает каталитическим
действием, вызывающим уплотнение или другие изменения
примесей, облегчающие их удаление, очистка называется
каталитической.
В зависимости от того, в каком состоянии продукт подвергается
очистке, различают жидкофазные и парофазные процессы очистки.
Применяемый для очистки реагент может обладать
универсальным действием и удалять многие примеси иди действовать изби:
рательно (селективная очистка). При очистке: зачастую
последовательно применяют несколько различных способов удаления
примесей из нефтепродуктов.
Щелочная очистка является наиболее распространенным
способом обработки светлых нефтепродуктов для удаления
сероводорода, меркаптанов, фенолов, нафтеновых кислот» а также для
нейтрализации, например после очистки нефтепродукта серной
кислотой (стр. 54). При небольшом содержании в нефтепродукте
других примесей, например непредельных соединений, можно
ограничиться только щелочной очисткой. Вредное действие
непредельных соединений (смолообразование) предотвращают в
этом случае ингибированием—добавлением антиокислителей
(ингибиторов), препятствующих воздействию кислорода воздуха на
непредельные соединения. Наиболее простой формой щелочной
очистки является защелачивание высокосернистых
нефтепродуктов (непосредственно при их получении) для удаления
сероводорода, который далее может окислиться до трудно удаляемой
элементарной серы.
Схема более глубокой щелочной очистки бензина приведена
на рис. 18. Бензин очищается в экстракционной колонне /
стекающим ему навстречу концентрированным раствором щелочи.
На середине высоты колонны в раствор вводится 30—40%
метанола для более полного извлечения меркаптанов, которые не мо-
гут оыть целиком удалены только щелочной обработкой
(сероводород извлекается при этом полностью). Степень извлечения
меркаптанов и H2S при щелочной очистке бензина приведена ниже:
' олекулярный вес .
Степени извлечения
щелочью, % ...
H2S C2H5SH
34 62
100 97,1
n-C3H7SH
76
n-C4H,SH
93
Ь9,2
Очистка проводится при 40 °С и давлении 5—7 am.
Захваченный бензином метанол извлекается из него щелочным
раствором в верхней части колонны /. В колонне 2 из щелочного раствора
Очищенный
Меркаптаны
Рис. 18. Схема щелочной очистки бензина в присутствии
метанола:
I—экстракционная колонна; 2—колонна для отделения меркаптанов; 3—ме-
танолыгая колонна; 4—сепаратор; 5—конденсаторы; В—насос; 7—кипятильники.
отгоняются метанол и меркаптаны. Эта смесь расслаивается в
сепараторе 4, метанол направляется на регенерацию в колонну 3
и затем возвращается в цикл очистки, меркаптаны отводятся из
системы.
Ошстха адсорбентами широко используется для удаления
примесей из светлых нефтепродуктов и масел. Наиболее
распространенными адсорбентами являются: природные отбеливаюцле
земли и глины, активированный уголь, силикагель,
алюмосиликаты и т. д. Адсорбенты обладают весьма развитой активной
поверхностью; так, например, 1 г силикагеля имеет поверхность
около 503 м2. На такой поверхности адсорбируются смэлистыз и
асфальтовые вещества, непредельные соединения и другие
примеси нефтепродукта. Ненасыщенные соединения могут пэлимери-
зоваться на поверхности адсорбента, сернистые примеси
разрушаются.
53
ОЧИСіКЛ исНЗИНОБ И ДруГИл ,/ісГКиа
в паровой фазе. Адсорбент в виде крупки загружают в
вертикальные цилиндрические башни слсем высотой до 3 м, через адсорберы
сверху вниз пропускают пары очищаемого нефтепродукта при
температуре 230—240 °С и давлении 4—5 am. На 1 т
отбеливающей глины можно очистить до 700 т нефтепродукта (до потери
активности сорбента).
Очистка высококипящих масел адсорбентами ведется в
жидкой фазе. Для этого подогретое масло смешивают в мешателях
Сжатый. |
воздух 1
Рис. 19. Схема комбинированной (кислотно-адсорбционной)
очистки масел:
1—кислотный мешатель; 2—отстойник; 3—смеситель; 4—испаритель; 5—трубчатая
печь; в— насосы; 7—холодильник; 8—фнльтрпрессы; 9—сборник; 10—сборник-
смеситель.
с порошкообразным адсорбентом, дополнительно нагревают смесь,
выдерживают ее, затем фильтрованием на фильтрпрессах отделяют
масло от отработанного сорбента. Можно производить очистку
также фильтрованием продукта через слой адсорбента (перколя-
ция).
Сернокислотная очистка—один из первых методов очистки
нефтепродуктов, получивший распространение еще в конце
XIX в. При обработке нефтепродукта серной кислотой удаляются
смолистые примеси, разрушаются сероводород и меркаптаны,
сульфиды RSR' и дисульфиды RSSR' растворяются в серной
кислоте, непредельные соединения тоже образуют растворимые
в кислоте алкилсерные кислоты RSO2OH. В качестве отхода
сернокислотной очистки получается кислый гудрон, в который пере-
ходят удаляемые примеси. После отделения кислого гудрона
нефтепродукт промывают водой и нейтрализуют щелочью.
В зависимости от условий очистки применяется кислота
различной концентрации (до 96%-ной HaSO4 и олеума). Расход
кислоты при очистке светлых нефтепродуктов составляет 0,5—5%
от их веса, при очистке обычных масел 1,5—6%, специальных
масел—до 10%, медицинских и так называемых белых масел—до
50% и более. Сернокислотную очистку проводят в специальных
мешателях периодического действия или по непрерывной схеме в
аппаратах, состоящих из смесителей и отстойников.
Из-за больших, потерь нефтепродуктов при сернокислотной
очистке, дороговизны кислоты и образования обременительных
отходов использование этого метода сокращается. Он сохраняет
значение лишь для очистки масел.
Схема комбинированной очистки масел серной кислотой с
доочисткой и нейтрализацией адсорбентами приведена на рис. 19.
Масло обрабатывается кислотой в мешателе / при перемешивании
сжатым воздухом. После отделения кислого гудрона в отстойнике
2 масло смешивается с тонкоизмельченным пылевидным
адсорбентом в смесителе .3. Затем смесь выдерживается и отпаривается в
испарителе 4.
Для поддержания необходимой температуры процесса смесь
непрерывно циркулирует через печь 5.
После охлаждения смесь через сборники-смесители 10
поступает на двухступенчатое фильтрование. Перекачивание масла в
системе производится насосами 6.
Очистка избирательными растворителями, получившая
распространение в последние годы, применяется в основном для
обработки масел и светлых нефтепродуктов. Селективные
растворители не смешиваются с нефтепродуктом, но растворяют примеси
смолистых, сернистых, непредельных соединений, нафтеновых
кислот и др., извлекая их таким образом из очищаемого
нефтепродукта. В качестве таких растворителей применяются фурфурол,
фенолы, жидкий сернистый ангидрид, нитробензол и другие
вещества.
Процесс селективной очистки может быть описан следующим
уравнением:
С.2/Сх = К
где С;—концентрация извлекаемого вещества в нефтепродукте;
С2—то же, в растворителе;
/С—коэффициент распределения вещества при данной
температуре.
Из этой формулы видно, что однократной обработкой
растворителем полностью очистить нефтепродукт от примесей нельзя.
Достаточно высокая степень их извлечения достигается лишь при
большой величине К. При ограниченном значении К для тщатель-
55
ной очистки продукта неооходима его многократная (в несколько
ступеней) обработка растворителем или создание противотока
А/еж^у счрщгемь'м сырьем и растворителем. Растворитель выби-
ргкт в зэеисрмссти от сесйств сырья, примесей, требуемой
степени счистки. Креме того, рзстЕсритель должен легко
регенерироваться и не теряться с нефтепродуктом.
Помимо перечисленных основных методов очистки, используются также
методы преврашения сернистых примесей в неактивные соединения, например
обработка нефтепродукта плюмбитом натрия
2RSH + Pb(ONaJ = R-S-Pb—S—R + 2NaOH
меркаптан меркаптнд
R—S—Pb—S—R + S = R—S—S—R + PbS
неактивный
дисульфид
или обработка гипохлоритом:
2RSH + NaOCl = R-S-S-R + Н2О + NaCl
Применяются также гидроочисткс—ограниченная гидрогенизация
нефтепродукта с превращением непредельных примесей в насыщенные соединения
и сернистых в легко удаляемый сероводород. С процессами очистки связаны
также освобождение масел от твердого парафина (депарафинизация) и от
асфальтовых веществ (деасфальтизация).
КРЕКИНГ И РИФОРМИНГ НЕФТЕПРОДУКТОВ
Требования к качеству продуктов переработки нефти
непрерывно изменяются, быстро растет и потребление этих продуктов.
Увеличивается количество машин, оборудованных двигателями
внутреннего сгорания,—автомобилей, самолетов,
сельскохозяйственных машин и др.; конструкции двигателей (бензиновых,
дизельных, реактивных) совершенствуются, для них становятся
необходимы новые виды жидких топлив и смазочных материалов
с определенными строго регламентированными свойствами и
составом. Еще более быстро возрастает потребление продуктов
нефтепереработки в химической промышленности.
Разнообразие процессов промышленного органического
синтеза определяет различие требований к химическим свойствам
разных видов нефтехимического сырья, используемого в этой
отрасли химической промышленности. Спрос на это сырье не
удается обеспечить за счет тех веществ, которые входят в состав
сырой нефти и получаются простым фракционированием и
очисткой. Отсюда возникает необходимость путем искусственного
преобразования углеводородов нефти увеличить выход наиболее
ценных легких углеводородов и придать им требуемые свойства
путем изменения их химической структуры. Методы таких
преобразований нефтепродуктов должны непрерывно
совершенствоваться в соответствии с направлениями развития нефтехимии.
Простейшим промышленным приемом преобразования тяжелых углево-
56
С-С-С С—С—С -
дородов нефти в легкие является термический крекинг*—разрыв
(расщепление) больших молекул этих углеводородов под действи-
€м тепла и образование меньших молекул более легких
углеводородов. Однако, расщепление молекул в процессе крекинга
протекает хаотически и не может быть проведено направленно с
получением углеводородов заданного строения. Частично это
достигается при ведении процесса в присутствии специально
подобранных катализаторов, т. е. при каталитическом крекинге.
Дальнейшее преобразование строения углеводородов, полученных в
результате крекинга, осуществляется в процессах риформинга—
изомеризации, алкилирования, дегидроциклизации и др.
Схемы процессов крекинга и риформинга углеводородов нефти**
приведены ниже:
1. Крекинг и дегидрирование (стр. 58 и 141)
С=С—С— • ¦ + С—С—С— ¦ (крекинг)
С—С—С С—С=С + Н2 (дегидрирование)
2. Гидрокрекинг (стр. 115)
С-С-С С-С-С + Н2 » С-С-С+С-С-С
3. Изомеризация (стр. 148)
С—С—С С—С—С > С—С—С С—С
' С
4. Дегидроциклизация (стр. 146)
С-С-С—С-С-С-С.
5. Дегидрирование шестичленных нафтенов
І ні —, М) + зн2
6. Алкилирование (стр. 149 ел.)
'Ч С=С_
7. Полимеризация
С=С—С + С=С—С » С=С—С—С—С—С
* Krack (англ.)—давать трещину, трескаться, раскалываться,
расщепляться.
** В схемах черточками обозначены связи С—С (связи углерода с
атомами, водорода не показаны); многоточия обозначают продолжение углеродной
цепи молекул; циклы с буквой Н в середине полностью насыщены.
57
На указанных выше страницах данной книги каждый из
процессов описывается более подробно, некоторые из них
рассматриваются во второй части данной книги.
Как следует из приведенных схем, в процессах крекинга
изменяется в основном количество атомов углерода в
молекулах углеводородов нефтепродукта, процессы риформинга
приводят к изменениям химического строения углеводородных
молекул.
Последний из приведенных выше процессов применяется для
получения полимеризационного бензина из непредельных
газообразных углеводородов, содержащихся в газах нефтепереработки,
и является каталитическим процессом, как бы обратным крекингу.
Таким способом эти углеводороды могут быть использованы для
получения дополнительного количества высококачественных
легких моторных топлив. Однако, по-видимому, более перспективно
непредельные углеводороды газов нефтепереработки направлять,
после соответствующей подготовки, на синтез полимеров
(полиэтилена, полипропилена и др., стр. 415 и ел.).
Термический крекинг
Расщепление углеводородов нефти под действием тепла было
известно давно. Широкое же промышленное использование этого
процесса для увеличения выходов бензина при переработке нефти
началось в 20—30-е годы. В настоящее время установки
термического крекинга входят в состав почти каждого крупного
нефтеперерабатывающего завода. Свыше 60% получаемого бензина
производится этим способом.
Первые соображения о возможности технического использования
термического разложения нефти были высказаны Д. И. Менделеевым в 1876 г. В это
же время А. А. Летний опубликовал свои данные об изучении этого процесса.
В 1891 г. В. Г. Шухову и С. Г. Гаврилову был выдан первый патент на
промышленный способ крекинга. В США такой патент был взят в 1912 г. Бартоном,
который соорудил первую примитивную промышленную крекинг-установку.
В настоящее время обычный термический крекинг как процесс
получения моторных бензинов постепенно вытесняется
каталитическим крекингом и применяется преимущественно для получения
из тяжелых нефтяных остатков широкой фракции—сырья для
каталитического крекинга.
Разновидностями процесса термического расщепления
нефтепродуктов, как указывалось (стр. 38), являются: собственно
крекинг на бензин, проводимый под высоким давлением,
коксование нефтяных остатков под низким давлением, направленное на
получение широкой фракции и нефтяного кокса, и пиролиз.
Общая закономерность поведения углеводородов в этих процессах:
чем больше атомов углерода в молекуле, тем легче углеводороды
58
подвергаются крекингу. В остальном различные классы
углеводородов при термическом расщеплении ведут себя по-разному.
Парафины подвергаются крекингу наиболее легко. Разрыв
цепи нормальных углеводородов обычно происходит в середине
с образованием насыщенного и ненасыщенного углеводородов,
параллельно может происходить дегидрирование. Бензины,
получаемые из парафинового сырья, по качеству хуже других.
Нафтены (цикланы) в рассматриваемом процессе
дегидрируются, отщепляют боковые цепочки и даже претерпевают разрыв
кольца. Бензины из циклансвого сырья лучше, чем из
парафинового.
Олефины, образующиеся в процессе крекинга, могут в
соответствующих условиях пслимеризоваться или отщеплять мелкие
молекулы более высокой непредельности
с5н10 —- с4н„ + сн4
амгглен дивинил метан
а также образовывать ароматические соединения и
конденсироваться с ними.
Ароматические углеводороды наиболее термоустойчивы, они
подвергаются уплотнению
2СвН(з = СвН5—С6Н5 + Нг
бензол дифемпл
образуют конденсированные ароматические соединения (нафталин
и др.), отщепляют боковые цепи. По мере углубления процесса
крекинга в образующихся продуктах накапливаются
ароматические соединения, конечные продукты пиролиза состоят почти
нацело из этих соединений.
Сернистые примеси под действием нагрева в процессе крекинга
разлагаются. Их разложение начинается раньше разложения
углеводородов (уже около 200 °С), при этом образуются
сероводород и элементарная сера, вызывающие коррозию оборудования.
Крекинг-газы, полученные при термическом расщеплении
сернистого сырья, богаты сероводородом, а бензин обычно содержит
значительно меньше сернистых соединений, чем исходное сырье.
Важнейшими факторами, влияющими на протекание
термического крекинга, являются температура и продолжительность
процесса. Скорость реакций крекинга, определяющая
продолжительность выдерживания крекируемого сырья в данных
условиях, необходимую для достижения требуемой глубины его
преобразования, сильно зависит от температуры. Скорость реакции
примерно удваивается при повышении температуры на 10 °С.
Существенную роль в процессе крекинга играет также давление.
Применяемое давление определяет выбор процесса обработки
сырья—парофазный или жидкофазный процесс. От этого, в свою
очередь, зависят размеры аппаратуры, в которой проводится кре-
59
кинг. Кроме того, давление сильно сказывается на реакциях
конденсации и уплотнения, протекающих в паровой фазе. При
повышенном давлении уменьшаются выход газа и содержание
нежелательных непредельных углеводородов в бензине.
Установки термического крекинга. Схемы и аппаратурное
оформление этого процесса зависят как от свойств крекируемого
сырья, так и от заданного направления его переработки, т. е.
целевых продуктов крекинга. В соответствии со свойствами
углеводородов крекинг легкого, термически стойкого сырья следует
вести в более жестких условиях—при более высоких температуре
Сырье
Газ
Рис. 20. Упрошенная схема установки термического
крекинга (насосы не показаны):
/. 2—трубчатые печи, 3—режтор; 4— испаритель: 5
6—ректификационные колонны; 7—редукционные нентили, 8— холодильник;
5—конденсатор; 10—сепаратор.
и давлении, тяжелое углеводородное сырье целесообразно
подвергать крекингу в более мягких условиях. Однако в последнем
случае схемы крекинг-установок сложнее, так как процесс
приходится осуществлять в два потока. В одном потоке крекируется
исходное тяжелое сырье—мазут, в другом—его более легкие
фракции и средний циркулирующий в системе продукт, полученный
в результате крекинга исходного мазута.
Упоощенная ,схема установки термического крекинга мазута
приведена на рис. 20. Исходное сырье, нагретое в
теплообменниках за счет тепла остатка (на схеме не показано), вводится в
среднюю часть колонны 5, где разделяется на легкую и тяжелую
фракции пои нагревании парами, поступающими из испарителя 4.
Тяжелая фракция—остаток из низа колонны 5 подается в
трубчатую печь 1 на легкий коекинг (температура около 470 °С. давление
40—45 am). Отгоняемая из колонны 5 фракция поступает в
колонну б, где разделяется на легкие продукты—газ и бензин и более
тяжелую керосино-газойлевую фракцию, направляемую далее
60
в печь 2 на глубокий крекинг (температура 500—510 °С, давление
около 50 am). Из обеих печей продукты крекинга передаются в
выносной реактор 3, где этот процесс завершается. Из реактора
продукты поступают через редукционный вентиль 7 в испаритель 4
и разделяются здесь на тяжелый остаток, удаляемый из системы
после охлаждения, и пары, идущие на ректификацию в колонны 5
и 6, которые работают под давлением порядка 10 am. Покидающие
колонну 6 пары охлаждаются и частично конденсируются в
холодильнике 9 и затем в сепараторе 10 разделяются на конечный
(целевой) продукт—бензин—и газы крекинга. Часть бензина
возвращается на орошение колонны 6.
Баланс процесса крекинга в установках описанного типа
характеризуется следующими округленными данными (в %)'¦
Бензин ... ¦... 13
Газ 5
Остаток 80
Потери 2
Выход бензина при термическом крекинге мазута составляет
7—15%, при крекинге керосино-соляровой фракции 15—25%,
при крекинге лигроина достигает 70%.
Коксование нефтяных остатков используется для более
глубокого термического разложения тяжелого сырья, крекинг которого
под давлением дает небольшие выходы бензина. В процессе
коксования не получается жидкого тяжелого остатка, разложение сырья
идет до образования кокса. При этом получается в среднем 20%
кокса, 10% газа, содержащего много непредельных соединений,
15% бензина, 55% широкой фракции, которая поступает на
каталитический крекинг для получения дополнительных количеств
бензина. В результате проведения этих процессов суммарный выход
бензина из тяжелого сырья достигает 40%. Получаемый при
коксовании остатков беззольный нефтяной кокс самостоятельно
используется как сырье для производства угольных электродов.
Процесс коксования может быть проведен в кубах
периодического действия, полунепрерывным способом в печах с камерами
из огнеупорного кирпича или на установках непрерывного
действия, где коксуемое сырье приводится в соприкосновение с
мелким нефтяным коксом, полученным в этом же процессе и
перегретым выше температуры коксования остатков. Сырье обволакивает
частицы кокса и коксуется на них в тонком слое. Существует два
типа процессов коксования остатков, отличающихся по размерам
частиц применяемого кокса E—15 мм или 0,02—0,3 мм). Особый
интерес представляет процесс коксования в кипящем (псевдоожи-
женном) слое более мелкого порошкообразного кокса.
Порсшкссбразный материал, лежащий на решетке или пористой
перегородке, при продувании через него с определенной скоростью воздуха, г'за
или пара переходит в состояние кипящего слоя (рис. 21). При малой скорости
* 01
дутья слои твердых частиц остается в неподвижном состоянии, и газ проходит
(фильтруется) через него. Если скорость подачп дутья превышает ее
определенное критическое значение, твердые частицы приходят в интенсивног движение
и слой приобретает текучесть и внешнее сходство с кипящей жидкостью. При"
еще более высокой скорости дутья поток газа захватывает и уносит с собой
частицы порошка.
Технологические процессы, проводимые в кипящем слое, характеризуются
высокой интенсивностью тепло- и массопередачи, обусловленной активным
перемешиванием частиц порош'<а и газа в кипящем слое и непреоызны.1
обновлением поверхности контакта ме-к^у ними. Благодаря этим достоинствам
процессы в кипящем слое получают все большее распространение в химической
и других отраслях промышленности.
В отя.ачяэл в лиг гпэдэггг кэхгэзания порошкообразные
4acrrj.ii цчг<улаэу.ог мэ-кту дзумя аппаратами: реактором и
кок :э «грез іге лам. В реакторе на пэроиэк подается сырье,
происходит его коксование и охлаждение
(температура 510—550 °С, избыточное
давление 0,7—0,8 am). В коксонагреватель
подводится воздух и кокс нагревается за
счет тепла сгорания некоторой его части
(температура 570—620 °С, избыточное
давление 0,8—1,0 am). Избыток
образующегося кокса отводится из нагревателя. Газы
и пары удаляются из реактора в
ректификационную колонну через циклон, где
отделяется коксовая пыль.
Пиролиз нефтяного сырья является
наиболее жесткой формой термического
крекинга. Этот процесс проводится при
Дутое
Рис. 21. Схема
кипящего слоя:
/—распределителен, і я решетка;
2-зона подвод, д/-ья; 3-кипя- 650 — 750 °С И аТМОСферНОМ ДаВЛЄНИИ.
Целью процесса пиролиза является
получение ароматических углеводородов и газа с высоким
содержанием непредельных соединений C0—40%). В связи с
внедрением каталитических методов ароматизации нефтепродуктов
значение процесса пиролиза как источника получения
ароматических соединений уменьшается, но возрастает его значение как
способа получения непредельных газообразных углеводородов.
Последовательность превращений углеводородов в процессе
пиролиза может быть представлена следующей схемой:
Парафины
Олефины
Диолефины
Нафтены
I Ароматические
| углеводороды
Раньше для получения ароматических углеводородов методом
пиролиза ориентировались на относительно легкое нефтяное
сырье—керосин, в настоящее время в качестве сырья в этом
процессе используют более тяжелые нефтяные продукты.
Распространенными установками пиролиза являются пироген-
ные трубчатые печи непрерывного действия. Схема такой установки
показана на рис. 22.
Исходное сырье, подаваемое на пиролиз, подогревается в
теплообменнике / и через конвекционную секцию печи 2
поступает в испаритель 3, где происходит разделение паров (более
легких фракций) и жидкости (более тяжелой части сырья). В
сепараторе 4 пары отделяются от капель жидкости и направляются
сначала в радиантную секцию печи 2, где нагреваются до температуры
реакции, затем переходят в реакционную камеру 5. Здесь при
Меисларбнное
сырье
Тяжелое масло
пиролиза
/идравличная
смола
Рис. 22. Схема трубчатой установки для пиролиза нефтепродуктов:
/—теплоебменшк; 2—трубчатая печь; 3—исгаритель; 4—сепаратор; 5—реакционная камера;
S—гидравлика; 7—грязевик для отстаиваьия <ажи; 8—емкость для гидр*пличной смолы;
9—холодильник; 10—ректификационная колонна; //—конденсатор; 1'2—газосепаратор.
высокой температуре происходит образование ароматических
углеводородов. Из камеры 5 парообразные продукты пиролиза
поступают в аппарат 6, называемый гидравликой и состоящий из двух
башен, орошаемых смолой. Нижняя часть первой (по ходу паров)
башни погружена в смолу и образует гидравлический затвор.
В гидравлике из продуктов пиролиза удаляется сажа и тяжелая
(гидравличная) смола, далее пары направляются в
ректификационную колонну 10. Из ее верхней части отбираются газ и легкое
масло, которые разделяются затем в газосепараторе 12, из нижней
части отводится жидкий остаток—тяжелые масла с большим
содержанием ароматических углеводородов.
Из жидких продуктов пиролиза на ректификационных
установках получают бензол, толуол, ксилол, зеленое масло
(применяется в производстве сажи), нафталиновое масло, из которого
выделяется нафталин, и пек—ценное сырье для получения
беззольного кокса.
63
Каталитический крекинг
Увеличение выхода и улучшение качества крекинг-бензина
могут быть достигнуты при крекинге нефтяного сырья в
присутствии катализаторов. Бензины каталитического крекинга
содержат меньше непредельных соединений и потому более стабильны.
Октановые числа таких бензинов выше, так как они богаче
углеводородами разветвленного строения и ароматическими
соединениями.
В табл. 6 приведены составы бензинов различного
происхождения.
Таблица 6
Состав различных бензинов
Бензины
Прямой гонки
из парафинового сырья ....
из наф-1 енового сырья
Термического крекинга
из парафинового сырья ....
из нафтенового сырья ....
Каталитического крекинга
из парафинового сырья ....
из наф'енового сырья
Октановое
число
20—60
20-60
—
65—80
—
74—82
Содержание углеводородов, %
парафины
65
35
37
30
46
33
дельные
—
—
39
30
16
13
маф
тены
30
55
16
25
23
32
тические
5
10
8
15
15
22
Описанные свойства бензина каталитического крекинга
связаны с тем, что формирование составляющих его углеводородов
происходит в присутствии активного алюмосиликатного
катализатора, вызывающего изомеризацию углеводородов и подавляющего
образование непредельных соединений и газов крекинга. Большую
роль в разработке методов каталитического крекинга сыграли
советские ученые академики Н. Д. Зелинский, С. В. Лебедев,
С. С. Наметкин.
Процесс каталитического крекинга проводится при
температуре около 480 °С и небольшом давлении. Сырье подается на
катализатор в испаренном виде. Глубина крекинга зависит от
температуры и длительности контакта сырья с катализатором. Время
контакта определяется объемной скоростью*. Например, при
* Объемной скоростью называют количество кубических метров пара или
газа (приведенного х нормальным условиям), проходящего через 1 м3
катализатор ной массы за 1 ч.
64
Газ а бензин
подаче 1 объема жидкого сырья на 1 объем катализатора в час
выход бензина составляет 45%, а при подаче 2 объемов сырья,
т. е. при сокращении вдвое времени контакта, выход бензина в
тех же условиях понижается до 37%.
Сырье в процессе крекинга распадается на газ, бензин, остаток
(газойль) и кокс. Пары бензина и газойля вместе с газом отводятся
на ректификацию, кокс отлагается на катализаторе, вследствие
чего его активность постепенно снижается. Для регенерации
катализатора к нему подводят воздух и выжигают кокс. Во
избежание порчи катализатора
необходимо строго соблю-
дать температурный
режим регенерации. Чтобы
неподвижный слой
катализатора можно было попере- ш
менно использовать в
процессе крекинга и
регенерировать, необходимо
систематически переключать
аппарат, в котором
находится катализатор, то на
рабочий цикл, то на
регенерацию. Такой способ
ведения процесса неудобен
и взрывоопасен, поэтому
Воздух', ¦--,
Рис. 23.
Остаток
Схема каталитического крекинга
в кипящем слое:
;—реактор; ^/—разделительная ректификационная
колонна; Ш— регенератор; '. 7—циклоны;
2—холодильники; 3—линия возврата регенерированного
катализатора; 4—отпарная секция реактора; 5—линия
подачи катализатора на регенерацию; S—воздушный
компрессор; 8—дымовая труба (по ней отходящие
газы могут направляться в котел-утилизатор).
сейчас от него отказались
и осуществляют' крекинг
в аппарате с движущимся
слоем катализатора,
который перемещается из
реактора в регенератор,
аналогично тому, как
описано для непрерывного процесса коксования (стр. 61). Крекинг
можно также проводить с кусковым и мелко измельченным
катализатором, в последнем случае в реакторе образуется кипящий
слой.
Схема установки каталитического крекинга в кипящем слое
показана на рис. 23. Она состоит из трех основных аппаратов:
реактора /, разделительной колонны // и регенератора ///.
Испаренное сырье вводится в поток регенерированного
катализатора, движущийся по трубе 5, и поступает вместе с ним в
реактор /. Парообразные продукты крекинга отделяются от катали-
заторной пыли в циклонах 1 и поступают в колонну //, где
частично конденсируются (при охлаждении в холодильниках 2) и
разделяются на газ и бензин, жидкие крекинг-фракции и остаток.
Часть остатка циркулирует в нижней части колонны, охлаждая
пары, поступающие из реактора, и отмывая их от остатков ката-
5—805
65
лизаторнои пыли. Одновременно приислидт ^индсис^и.«;-; па^^
остатка, который после отстаивания частично возвращается на
крекинг. Отработанный катализатор через отпарную секцию 4
поступает по трубе 5 в регенератор ///. В эту трубу и нижнюю
часть регенератора подается сжатый воздух, инжектирующий
катализатор и способствующий выжиганию кокса с его
поверхности. Газы, образующиеся при регенерации катализатора,
отводятся через циклоны 7 в дымовую трубу 8 и далее могут быть
использованы в котлах-утилизаторах для получения пара.
Способы повышения качества бензинов
Улучшение свойств бензина как моторного топлива в
основном сводится к уменьшению содержания в нем предельных
углеводородов нормального строения, обладающих крайне низким
октановым числом, и снижению количества непредельных
соединений, присутствие которых при хранении и окислении бензина
является причиной образования смол, нагаров в двигателе и
возникновения других трудностей использования топлива. Однако
улучшение свойств бензина надо вести без его потерь (в виде газа
или тяжелого остатка), иначе сокращение расхода бензина
вследствие улучшения его качества сведется на нет из-за общего
уменьшения количества бензина. Поэтому для повышения качества
бензина используются спрсобы прямого преобразования (риформинга)
строения молекул его компонентов без существенного изменения
их молекулярного веса (стр. 57): изомеризация, дегидроцикли-
зация, дегидрирование нафтенов, гидрирование непредельных
углеводородов без разрыва молекул совместно с изомеризацией
и др.
Частично эти процессы протекают и при каталитическом
крекинге, что способствует улучшению качества получаемого
бензина. В промышленности они осуществляются и как
самостоятельные процессы. Наиболее распространены из них процессы
гидроформинга, при которых одновременно протекают все
перечисленные реакции. На практике они проводятся двояким
способом: в кипящем слое молибденового катализатора с циркуляцией
водорода или на неподвижном слое платинового катализатора
(платформинг), также с циркуляцией водорода. Температура
в обоих способах порядка 450—530 °С, давление 14—56 am. Для
гидрирования непредельных углеводородов используется водород,
выделяющийся в реакциях дегидроциклизации парафинов и
дегидрирования нафтенов (стр. 57).
Особенностью метода гидроформинга в кипящем слое является
повышенное коксообразование и необходимость регенерации
катализатора, но зато молибденовый катализатор нечувствителен
к сере, присутствующей в сырье, и к фракционному составу сырья.
Следовательно, на таком катализаторе можно подвергать рифор-
66
мингу различное сырье с более широкими пределами температур
кипения.
Платиновый катализатор более экономичен, отличается
длительным сроком службы (кокс практически не образуется), но
для платформинга требуется сырье лучшего качества и содержащее
не более 0,1% серы.
Обработка сырья водородом, ограничивающаяся гидрированием
непредельных соединений, называется гидростабилизацией, или
гидроочисткой. Если проводится относительное мягкое
гидрирование нефтяных дистиллятов, сопровождающееся разрывом
молекул компонентов сырья и присоединением водорода по месту
разрыва (стр. 57), процесс носит название гидрокрекинга. В том
случае, когда при гидрировании с разрывом молекул
осуществляется более глубокая переработка нефтяных остатков в бензин,
процесс называется деструктивной гидрогенизацией (стр. 114).
Все описанные процессы преобразования углеводородов,
входящих в состав нефтепродуктов, осуществляются на
нефтеперерабатывающих предприятиях применительно к многокомпонентным
смесям углеводородов различных классов. При этом
одновременно протекают разнообразные реакции и конечный результат
процесса определяется выбранными условиями и целевым
направлением переработки нефтепродукта. Б химической же
промышленности отдельные реакции преобразования проводятся с
индивидуальными углеводородами и протекают более четко, в определенном
направлении. Подробнее они описаны во второй части книги.
Глава II
ТЕХНОЛОГИЯ ТВЕРДОГО ТОПЛИВА И ПРОДУКТОВ
ЕГО ПЕРЕРАБОТКИ
/. ОБЩИЕ СВЕДЕНИЯ
Использование твердых топлив. Основную массу запасов
топлива на земле составляет твердое топливо—угли, а также сланцы,
торф и древесина. Большая часть добываемых твердых топлив
сжигается для получения тепла. Около четверти добываемых
твердых топлив подвергается химической переработке. Из них
получают кокс для металлургии и другие виды облагороженного
топлива, .сырье для производства продуктов основного
органического синтеза, красителей, синтетических полимеров, взрывчатых
и лекарственных веществ и многих других продуктов,
потребляемых человеком. Промышленность химической переработки
твердых топлив по масштабам, количеству перерабатываемого сырья
и разнообразию применяемых методов занимает одно из первых
мест среди химических производств.
В народном хозяйстве нашей страны твердые топлива играют
огромную роль. Поэтому добыча и переработка твердых топлив в
СССР непрерывно возрастает: в 1960 г. добыча угля достигла
513 млн. т, в 1964 г. 554 млн. т. В 1965 г. и в последующие
годы добыча угля будет увеличиваться. Возрастет также добыча
торфа и сланцев, заготовка и переработка древесины,
главным образом как поделочного материала и химического сырья.
Правильное использование богатейших топливных запасов невозможно
без глубокого знания свойств органических горючих веществ—топлив и
способов их переработки. Эти знания накапливались в течение тысячелетий. Еще
первобытный человек умел сжигать древесину и другие растительные топлива
для приготовления пищи и согревания. Позже люди научились обжигать
древесину и пользоваться получаемым древесным углем для выплавки металлов
из руд. Деготь, образующийся одновременно с древесным углем, стали
употреблять для осмолки лодок, судов, канатов, сетей и для других целей.
68
С развитием промышлениости потребность в топливе стала быстро
возрастать. Особенно остро недостаток топлива стал ощущаться в конце XVIII—
начале XIX в., когда сильно развилась металлургия и в качестве двигателя
широкое распространение приобрела паровая машина. Леса быстро
уничтожались и возникла иеобходимость замены древесииы другими видами топлива.
Таким заменителем оказался каменный уголь.
В России первые систематические поиски угля были начаты при Петре I.
В 20-х годах XVIII в. уголь был найден иа берегу р. Северного Донца (Донбасс)
и в районе Верхнетомского острога (Кузбасс). Однако донецкие угли начали
разрабатывать только в середине XIX в., а широкое промышленное
использование углей Кузбасса началось лишь при советской власти.
В середине XVIII в. были открыты способы обжига каменного угля и
производства из него кокса для доменных печей. При обжиге каменного угля
получалась смола, отличавшаяся, однако, от древесного дегтя и долго не
находившая применения. Только после открытия Н. Н. Зининым A842 г.) способа
промышленного получения анилина из бензола, выделенного из каменноугольной
смолы, оиа превратилась в ценное сырье, сначала для производства красителей,
а затем лекарственных препаратов, взрывчатых веществ, пластических масс и
многих других продуктов. В настоящее время многочисленные коксохимические
заводы ежегодно перерабатывают миллионы тонн каменноугольной смолы.
В начале XIX в. были разработаны способы получения горючих газов из
твердых топлив, и вскоре возникла новая отрасль химического производства—
газовая промышленность. Наконец, в первой четверти XX в. уголь стали
применять для получения искусственных моторных жидких топлив.
В открытии и усовершенствовании способов химической переработки
топлив большую роль сыграли русские специалисты и ученые. Особенно
широкую перспективу использования топлив наметил в своих трудах великий
русский химик Д. И. Менделеев.
Виды и состав твердых топлив. Все твердые топлива являются
продуктами растительного происхождения. Растения,
произраставшие в давно прошедшие геологические периоды, скапливались
в виде огромных залежей, составляющих современные запасы
горючих ископаемых. Эти залежи образовывались в разные сроки
и в различной степени испытывали давление покрывавших их
горных пород, нагревание и другие воздействия. Поэтому
растительный материал в таких залежах подвергался изменениям в
неодинаковой степени.
В настоящее время человек располагает различными
твердыми топливами: древесиной, полностью сохраняющей состав живой
растительной ткани, торфом, в котором растительная ткань уже
значительно изменилась, бурыми и каменными углями, в которых
состав растительной ткани почти полностью изменился—она
обогатилась углеродом и уплотнилась. В антраците—наиболее
древнем горючем ископаемом—растительная ткань подверглась еще
большим изменениям и превратилась в почти чистый углерод.
Состав твердых топлив и их теплотворная способность
характеризуются такими же показателями, как и другие виды горючих
(стр. 15).
Основные способы переработки твердых топлив. Вещество
твердых топлив способно изменяться под воздействием тепла и
химических реагентов. Механической обработкой можно менять
69
только форму и размеры кусков этих тоилив, сортировать топливо
ио размеру кусков или зольности. Ниже рассмотрены основные
процессы переработки твердых тоилив.
Механическая обработка. Она заключается в
дроблении топлива, просеивании, обогащении (отделение от
топлива пустой породы) и брикетировании (прессование трудно
сжигаемой топливной мелочи со связующим в брикеты, более удобные
для перевозки и сжигания). Механической обработке подвергают
угли, сланцы и торф.
Экстрагирован и е—применяется к древесине, торфу
и бурым углям. Из топлива при этом извлекаются ценные
вещества, например канифоль—из
древесины, воск—из торфа, горный воск—
из углей и др.
Термическая обработ-
к а. Такой обработке подвергают
почти все виды твердого топлива, за
исключением тощих углей и
антрацитов. Такой процесс проводится в
закрытых камерах без доступа
воздуха к перегоняемому сырью (сухая
перегонка). По характеру он
соответствует термическому крекингу нефти
и является простейшим методом
переработки вещества твердых топлив
(см. схему на стр. 16). Строение их
органической части много сложнее,
чем у нефти, поэтому в процессе
термического разложения твердых
тоилив образуется больше твердого
остатка (кокса) и соответственно
меньше летучих веществ (газа и
жидких продуктов).
На рис. 24 показан состав
газового угля (см. также табл. 9, стр.
83) и схема его расщепления при
сухой перегонке.
Поскольку перегонка топлива производится в аппаратах,
обогреваемых огнем, этот процесс называют пирогенным*.
Получаемый при этом твердый остаток представляет собой обугленный
материал (древесный уголь—из древесины, разные сорта кокса—
из углей и торфа), летучие продукты являются смесью многих
химических соединений разнообразного состава. После
охлаждения эта смесь разделяется на газообразные продукты, водный
конденсат, содержащий растворимые в воде вещества, и деготь (или
Рис. 24. Состав газового угля
и схема его расщепления при
сухой перегонке. (Влага и зола
даны в расчете на рабочее
топливо, элементарный состав—на
горючую массу.)
* От греческих слов пирос—огонь и генезис—происхождение.
70
смолу)—масляный конденсат, не смешивающийся с водным.
Состав и свойства получаемых продуктов в значительной степени
зависят от исходного сырья и условий его перегонки.
Принципиальная же схема процесса во всех случаях одинакова. Его
можно разделить на следующие стадии:
1. До 100 °С—сушка (удаление содержащейся в топливе влаги).
2. От 100 до 300—350 °С—разложение менее стойких,
преимущественно кислородсодержащих, органических соединений и
отгонка легколетучих веществ.
3. До 500—550 °С—образование твердого остатка, дегтя
(смолы) и газа—так называемое полукоксование.
4. До 1000—1100 °С—окончательное прокаливание твердого
остатка—коксование с выделением из кокса труднолетучих
веществ. Летучие вещества, соприкасаясь с раскаленным коксом,
превращаются из соединений преимущественно жирного ряда в
циклические соединения—происходит ароматизация дегтя (смолы).
В зависимости от вида топлива, т. е. от его состава, основное
значение могут иметь различные стадии перегонки. Кроме того,
не во всех случаях процесс доводят до последней стадии. Чем
моложе топливо, тем ниже температура окончания процесса
перегонки. Она зависит также и от того, какие конечные
продукты требуется получить в результате перегонки.
Более глубокими методами переработки твердых топлив
являются газификация и ожижение.
Газификацию осуществляют в тех случаях, когда
отсутствует природный газ и требуются большие количества
газообразного топлива или газа для химических синтезов.
Газифицировать можно все виды твердых топлив. Этот процесс заключается
в частичном сжигании, т. е. окислении кислородом углерода
топлива с превращением его в горючий газ—окись углерода.
2С + О2 = 2СО + 52,8 ккал
Воздействием на раскаленный уголь водяным паром получают
так называемый водяной газ—смесь окиси углерода и водорода:
С + Н2О (пар) = СО 4- Н3 — 31,4 ккал
Ожижение угля (гидрогенизация)
заключается в расщеплении сложных органических веществ, входящих
в состав угля, и присоединении водорода к продуктам распада с
образованием жидких углеводородов. Процесс идет под давлением
водорода до 700 am и при температуре порядка 500 °С.
Дальнейшим шагом в области химической переработки топлив
является синтез разнообразных органических продуктов из
водяного газа, полученного, в свою очередь, из твердых топлив (см.
главу IV, стр. 162 и ел.).
71
2. ПЕРЕАБРОТКА ДРЕВЕСИНЫ
Древесина употребляется как строительный и поделочный
материал, а также в качестве топлива и химического сырья. На
переработку она обычно поступает в виде дров. Используются
также древесные отходы (сучья, опилки, пни и др.), составляющие
более 40% от общего количества заготовляемой древесины.
Методами химической переработки древесины являются сухая
перегонка и гидролиз. Обработкой древесины соответствующими
растворителями получают целлюлозу. Из древесной смолы (дегтя)
извлекают канифоль и скипидар. Древесина используется также
для получения горючего генераторного газа, в частности в
транспортных генераторах, питающих газом (вместо бензина)
автомобили и тракторы._
СОСТАВ И СВОЙСТВА ДРЕВЕСИНЫ
Действительная плотность древесины довольно постоянна и
для всех пород дерева равна 1,51 —1,56 тім}. Кажущаяся
плотность* зависит от сорта древесины, ее возраста и влажности. Так,
для березы она равна 0,65, для ели—0,45 тім}. Свежая
древесина содержит до 60% влаги; ее теплотворная способность в
абсолютно сухом состоянии примерно 5000 ккал/кг B1 Мдж/кг).
Химический состав древесины очень сложен и для разных
пород дерева неодинаков. Поэтому при переработке древесины
разнообразных хвойных и лиственных пород получают различные
продукты. Элементарный же состав древесины почти одинаков и
округленно может быть принят следующим (в %):
Углерод 50
Водород 6
Кислород .... 43
Азот Около 0,1
Основными веществами, составляющими растительную ткань,
являются полисахариды—целлюлоза (клетчатка) и гемицеллюло-
зы, а также лигнин. В сухой древесине содержится 30—50%
целлюлозы. Минеральные вещества, образующие золу, распределены в
дереве неравномерно: собственно в древесине их содержится
0,3—0,7%, в коре—до 3—3,5%.
Целлюлоза имеет волокнистое строение, ее эмпирическая
формула (СвН10О5)„ или [С6Н7О3(ОНK1„. Целлюлоза нерастворима
в воде и органических растворителях. Под действием
минеральных кислот целлюлоза гидролизуется в сахара. Наличие в каждом
глюкозном остатке С6Н10О5 трех гидроксильных групп позволяет
получать сложные эфиры целлюлозы, используемые для произ-
* Действительная плотность—плотность сплошной массы чистого
вещества, кажущаяся плотность—масса единицы объема данного вещества,
включая поры, присутствующую в веществе влагу и различные примеси.
72
водства искусственного волокна, лаков, взрывчатых и других
веществ.
Гемицеллюлозы состоят из пентозанов (С5Н8О„)Л,
преобладающих в лиственных породах древесины, и гексозанов (С6Н10О5)„,
преобладающих в хвойных породах. Содержание гемицеллюлоз
в сухой древесине достигает 24—30%; они менее химически стойки,
чем целлюлоза, и легче гидролизуются кислотами.
Лигнин является важнейшей, после целлюлозы, составной
частью древесины (содержание 25—30%). Строение лигнина
окончательно еще не установлено. Известно, что он не является
индивидуальным веществом, имеет ароматический характер и
содержит метоксильные (СН3О), гидроксильные (ОН), ацетильные
(СН3СО) и другие группы.
Кроме перечисленных трех основных компонентов, на долю
которых приходится около 96% состава сухой древесины, она
содержит также смолы (из них выделяют канифоль и скипидар),
дубильные и некоторые другие вещества.
СУХАЯ ПЕРЕГОНКА ДРЕВЕСИНЫ
Старейшим и наиболее распространенным способом химиче-
-ской переработки древесины является ее сухая перегонка. Она
производится путем нагревания древесины в замкнутом
пространстве, без доступа воздуха. При этом древесина разлагается на
древесный уголь, остающийся в камере перегонки, и отгоняемые
летучие продукты, состоящие из газов и паров, конденсирующихся
при охлаждении. Образующийся конденсат разделяется на два
слоя: водный (надсмольную или подсмольную воду), представляющий
собой разбавленный раствор уксусной кислоты, метилового спирта,
ацетона и других ценных продуктов, и деготь (осадочную смолу).
Качество древесного угля, а также состав и выход летучих
продуктов зависят от породы перерабатываемой древесины (табл. 7)
я условий сухой перегонки.
Таблица 7
Основные продукты сухой перегонки хвойной и лиственной. древесины
(в %)
Порода древесины
Сосна
Береза
Древесный
уголь
38
32
Газ
15
14
Деготь
12
8
Уксусная
кислота
3,5
7,0
Метиловый
спирт
0,9
1,6
Водный
конденсат*
26
37
Примечание. Количество, недостающее до 100 %. приходится на долю растворимых
мол (около 8%) и других органических продуктов второстепенного значення.
До выделения СНзСООН и СНЭОН.
73
Если целью сухой перегонки древесины является только
получение древесного угля, процесс называется углежжением.
Собственно сухая перегонка древесины производится с улавливанием
и использованием также летучих продуктов. Если же цель
перегонки—только получение смолы и скипидара, процесс называют
смолокурением.
Печи для сухой перегонки древесины. Обычно применяются
камерные печи периодического или непрерывного действия. Одной
из более новых конструкций является канальная печь с
циркуляционным обогревом, в которой древесина обогревается
циркулирующими в печи горячими газами. В печах такого типа
перерабатывается до 100 жя дров в сутки.
Движение вагвье/по/г
Движение газов
Ввод теплоносителя
Рис. 25. Печь для сухой перегонки древесины:
/—камера сушки; 2—камера переугливания; 3—камера охлаждения; 4—тамбуры;
5—подогреватель (рекуператор); 6—топка рекуператора; 7—установка улавливания;
8—газоход в камеру сушкн; 9—дымовая труба.
Печь, показанная на рис. 25, состоит из длинного коридора,
разделенного на камеру сушки /, камеру переугливания
(перегонки) 2 и камеру охлаждения 3. Камера сушки расположена
отдельно, сама печь разделена на части тремя переходными
тамбурами 4. Камеры сушки и переугливания—кирпичные, камера
охлаждения на 2/3 ее длины выполнена из листовой стали. Дрова
подаются в печь в вагонетках, передвигаемых в печи по рельсам
специальным толкателем. Передвижку вагонеток производят через каждые
2,5—3 ч. Сначала из камеры охлаждения выводят одну вагонетку,
все остальные вагонетки сдвигают и на освободившееся место
подают через тамбур последнюю вагонетку из камеры
переугливания, а взамен из камеры сушки подают вагонетку с чурками.
В качестве греющего агента (теплоносителя), циркулирующего
в печи, используют газ, образующийся при сухой перегонке
древесины. Этот газ из камеры переугливания направляется в
установку улавливания 7 (в смолоотделители и промыватели), где
выделяются увлеченные газом деготь и падсмольная вода. Затем
74
основная часть газа поступает для подогрева в рекуператор 5
и возвращается в камеру переугливания. Небольшая часть газа
сжигается в топке 6 рекуператора, откуда топочные газы
направляются по газоходу 8 в камеру сушки и в дымовую трубу 9.
При нагревании до 100 °С из древесины удаляется влага. С
повышением температуры от 150 до 270 °С постепенно усиливается
выделение водяных паров и начинается образование двуокиси
и окиси углерода и паров уксусной кислоты. Процесс протекает
с поглощением тепла. Начиная от температуры 270—280 °С, сухая
перегонка становится экзотермическим процессом. Вследствие
выделения тепла реакций разложения древесины температура
повышается до 380—400 °С, при этом начинают отгоняться деготь,
метиловый спирт, уксусная кислота и газы, содержащие около 40%
углеводородов, 5% водорода и до 35% СО2.
Имеются также печи для сухой перегонки щепы и других
отходов переработки древесины.
Продукты сухой перегонки древесины. Состав древесного угля:
до 80% углерода, 4% водорода, до 16% кислорода и азота, 3—
5% влаги, 1% золы. Древесный уголь применяется при выплавке
некоторых сортов чугуна, в кузницах и литейных. Малая зольность,
незначительное содержание фосфора и отсутствие серы в
древесном угле позволяют выплавлять на нем металл особо высокого
качества. Путем специальной обработки (например, паром) может
быть получен пористый древесный уголь, так называемый
активированный уголь, обладающий хорошей поглотительной
способностью и применяемый для наполнения коробок противогазов и
улавливания (адсорбции) паров летучих веществ в химических
производствах.
Летучие продукты перегонки, как указывалось, разделяются
при охлаждении на деготь (осадочная смола) и водный конденсат.
Из дегтя лиственных пород древесины получают медицинский
препарат—креозот, масла, применяемые для флотационного
обогащения руд, вещества для пропитки древесины и др. Из смолы
древесины хвойных пород получают также скипидар.
Водный конденсат содержит 4—9% уксусной кислоты и 2—
3% метилового спирта. Их выделяют из конденсата порошковым
и экстракционным способами.
По первому способу надсмольная вода перегоняется из кубов.
При этом в кубе остается «кубовая» смола, а пары, содержащие
уксусную кислоту и метиловый спирт, пропускают через
известковое молоко, связывающее уксусную кислоту по реакции:
2СН3СООН + Са(ОНJ = (СН3СООJСа + 2Н2О
Далее конденсируются пары, содержащие только спирт,
получаемый при этом спирт-сырец содержит 10% СН3ОН.
Концентрированный метиловый спирт получают перегонкой спирта-сырца.
75
ную соль сушат. Образуется так называемый древесный порошок,
путем обработки которого серной кислотой получают уксусную
кислоту:
(СН3СООJСа -I- H.2SO4 = CaSO4 + 2СН3СООН
Сухой перегонкой древесного порошка при 450 °С можно
получить ацетон:
(СН3СООJСа = СаСО3 + СН3СОСН3
Более совершенный и экономичный экстракционный способ
извлечения уксусной кислоты заключается в обработке водного
конденсата (после отгонки метилового спирта) серным эфиром или
крезолами.
Уксусная кислота, ацетон и метиловый спирт находят
широкое применение в промышленности органического синтеза
(стр. 168, 214 ел., 225 ел.).
Для получения из хвойной древесины канифоли и скипидара
используют смолу живого дерева (живицу) или смолу старых пней
хвойных деревьев, в которых накапливается до 30% смолистых
веществ (так называемый пневой осмол). Для получения живицы
на стволе дерева делают надрез (подсочка) и собирают
вытекающую смолу. Смола из пневого осмола извлекается путем
перегонки (смолокурение) или экстракцией. После очистки смолу
разделяют перегонкой на легкокипящий скипидар и остающуюся в
кубе канифоль, которую применяют в целлюлозной,
лакокрасочной, мыловаренной, электротехнической и других отраслях
промышленности.
Скипидар употребляется как растворитель и для получения
искусственной камфоры, используемой в производстве целлулоида
и кинопленки.
ПОЛУЧЕНИЕ ЦЕЛЛЮЛОЗЫ
Целлюлозно-бумажная промышленность вырабатывает
целлюлозу для производства бумаги, искусственного волокна,
взрывчатых и других веществ. Целлюлозу выделяют из древесины путем
ее обработки реагентами, растворяющими все составные части-
древесины, кроме целлюлозы. В зависимости от реагента,
применяемого для такой обработки, различают два основных способа
получения целлюлозы:
сульфитный—обработка древесины при 130—150 СС
разбавленным раствором бисульфита кальция Ca(HSO3J, содержащим
свободный SO2;
сульфатный—обработка древесины при 150—180 °С
раствором едкого натра и сульфида натрия Na2S. Предварительно
древесина обычно подвергается предгидролизу—обрабатывается
очень слабым раствором серной кислоты (до 0,5% H2SO4) или
водой при повышенной температуре (до 180 СС).
76
Наиболее распространен сульфитный способ, по которому
вырабатывается до 3/4 мирового производства целлюлозы.
Процесс получения целлюлозы из древесины по любому
способу состоит из стадий подготовки древесины (очистка,
сортировка, измельчение), варки (обработка соответствующими
реагентами), промывки целлюлозы, отбелки (обработка хлором или
хлорной водой и гипохлоритом натрия) с последующей промывкой.
Если полученная целлюлоза является товарным продуктом и не
перерабатывается на месте, а отправляется на другие предприятия
(заводы искусственного волокна и т. д.), то ее высушивают при
100—ПО °С.
ГИДРОЛИЗ ДРЕВЕСИНЫ
Гидролизная промышленность является сравнительно молодой,
бурно развивающейся отраслью лесохимии. Ее развитию
придается большое значение.
Гидролиз древесины производится для превращения
содержащихся в ней полисахаридов (гемицеллюлоз и целлюлозы) в_моно-
сахариды—сахара (осахари-
вание). В дальнейшем сахара
можно сбраживать для
получения этанола,
восстановлением превращать их в
многоатомные спирты,
дегидратацией—в фурфурол,
окислением—в моно- и дикарбо-
новые оксикислота.
Используя моносахариды как
питательную среду, получают
кормовые белково-витамин-
ные дрожжи и антибиотики.
Лигнин, не
разрушающийся при гидролизе, после
сушки используется главным
Вода -... • Пар
Пар Д СВ/рье
Раствор
caxapo?
Шлам
Гидролизный
лигнин
Рис. 26. Схема аппарата для
гидролиза древесины:
/—гидролизер; 2—циклон для отделения
лигнина; 3—испаритель гидролизата; 4—нейтрзли-
затор; 5—отстойник.
образом как топливо, а
также применяется в качестве
наполнителя в производстве
пластических масс, для
изготовления перевязочных
материалов и лигниновой
бумаги (для салфеток).
Схема установки для гидролиза древесины показана на рис. 26.
Основной аппарат—гидролизер 1 представляет собой
вертикальный автоклав емкостью 18—50 м3, изнутри футерованный
кислотоупорным материалом. Измельченная древесина загружается в
автоклав сверху, после чего в него непрерывно подается 0,5—
77
1%-ная серная кислота и пар при температуре ibU—1»и С и
давлении до 12 am.
Полисахариды гидролизуются по реакции:
НоО
(C0Hi0O5)„ > лС6Н12О0
целлюлоза
Образовавшиеся сахара переходят в раствор и непрерывно
выводятся через фильтр снизу автоклава. По окончании процесса
гидролиза остающийся в автоклаве лигнин выдувается из него.
В новейших установках весь процесс гидролиза идет непрерывно
при противоточном движении реагентов.
Раствор сахаров после нейтрализации, отстаивания и
фильтрования поступает на сбраживание для получения этилового спирта:
СвН1ЭО<, * 2СО2 + 2С,Н5ОН
Из продуктов брожения отгоняют этиловый спирт. В
результате такой переработки из 1 т сухой древесины удается получить
150—180 л 95%-ного этилового спирта, до 120 кг жидкой
двуокиси углерода, 250—300 кг лигнина, до 40 кг белковых дрожжей и
другие продукты.
Сбраживанию можно подвергать и сульфитные
щелока—отходы производства целлюлозы.
3. ДОБЫЧА И ПЕРЕРАБОТКА ТОРФА
ПРОИСХОЖДЕНИЕ И СВОЙСТВА ТОРФА]
Торф является одним из распространенных и простых по
способам добычи видов топлива (его добывают без подземных
разработок). Недостатками торфа являются его малая плотность,
высокая влажность и относительно низкая теплотворная
способность. Поэтому перевозить торф нерационально и его обычно
потребляют вблизи от места добычи (местное топливо). Запасы торфа
велики, и он имеет существенное значение для народного
хозяйства. С 1961 по 1964 гг. добыча торфа возросла в 1,5 раза и
достигает 84 млн. т в год.
По составу торф занимает промежуточное положение между
древесиной, содержащей живую растительную ткань, и углем—
полностью минерализованным горючим ископаемым. Местами
залегания торфа обычно являются торфяные болота, где остатки
растений скапливаются под слоем воды, препятствующим полному
истлеванию растительной ткани. Болота, в которых накопление
торфа продолжается, называются живыми; торфяники, где этот
процесс прекратился, являются мертвыми. Торфяные болота
делятся на низинные, верховые и переходные. Низинные
торфяные болота образуются в низких местах, залитых водой; в них
78
встречаются остатки деревьев, кустарников и крупных
травянистых растений (осока, тростник, хвощ). В низины водами
заносятся минеральные вещества, поэтому низинные торфы отличаются
большой зольностью. Верховые торфяные болота образуются на
возвышенных местах; в них преобладают мелкие растения (мхи).
Зольность верховых торфов меньше и качество их лучше.
Переходные болота занимают промежуточное положение.
Горючие продукты неполного разложения отмерших
растительных остатков, из которых состоит торф, постепенно
обогащаются углеродом и приобретают бурый или черный цвет; такие
продукты носят название перегноя, или гумуса.
Влажность сырого торфа достигает 80—90% и может быть
снижена путем подсушки только до 40—30%. В зависимости от
типа торфа зольность его колеблется от 0,5 до 25%. Содержание
серы в торфах не превышает 0,2—0,5%.
В настоящее время торф используется главным образом как
топливо. Его сжигают в специальных топках на крупных
электростанциях, построенных вблизи торфяных залежей (например,
Шатурская электростанция и др.), в промышленных топочных
устройствах и в обычных печах.
ДОБЫЧА ТОРФА
Торф добывают открытой разработкой. Массовая добыча
торфа у нас механизирована и производится специальными машинами.
Торф используют в виде мелочи (мелкие куски) или размалывают
и формуют из полученной массы кирпичи, которые далее сушат.
Одним из наиболее длительных процессов при заготовке торфа
является его сушка. Сушка торфа на воздухе длится месяцами,
искусственными приемами продолжительность его обезвоживания
удается резко сократить.
Эффективным способом добычи торфа является размывание
его залежей сильной струей воды. Образующуюся пульпу
перекачивают из торфяника насосами и разливают на поверхности
земли слоем высотой 250—300 мм. После стекания воды и
подсушки затвердевший слой разрезают на кирпичи, которые затем сушат.
Такой торф получил название гидроторфа.
ХИМИЧЕСКАЯ ПЕРЕРАБОТКА ТОРФА
Большинство перечисленных на стр. 70 способов химической
переработки твердых топлив может быть применено и к торфу.
Основным из них является газификация торфа, позволяющая
получать из него высококалорийный горючий газ (стр. 107).
Экстрагированием из торфа удается извлечь некоторые ценные
вещества; сухой перегонкой (полукоксование) могут быть
получены торфяной кокс и летучие продукты; нерациональна только
79
гидрогенизация торфа. Однако химическая переработка торфа еще
слабо развита.
В настоящее время освоены экстракционный метод переработки
торфа (обработка органическими растворителями) и сухая
перегонка. Экстракционным методом из торфа в специальных
аппаратах—экстракторах извлекают битумы. Из торфяных битумов
затем выделяют воска, широко применяемые в промышленности.
Сухая перегонка торфа может производиться в замкнутых
аппаратах, обогреваемых снаружи (печи с внешним обогревом),
и в аппаратах, в которых через массу торфа продувают струю
перегретого газа (печи с внутренним обогревом). Техника ведения
этих процессов будет рассмотрена ниже (стр. 101 и ел.). При
сухой перегонке торфа, с выходом около 40%, получается
малозольный (но и малопрочный) бессернистый высокоактивный кокс,
который используется в некоторых процессах выплавки особо
чистых металлов, а также при электротермической возгонке
фосфора из природных фосфатов и др. Теплотворная способность
такого кокса около 6500 ккал/кг (^27 Мдж/кг).
Летучие продукты сухой перегонки торфа состоят из дегтя,
газа и водных погонов, содержащих аммиак и органические
кислоты, преимущественно уксусную. Деготь, выход которого
составляет в среднем 10%, представляет собой ценное химическое
сырье, состоящее в основном из углеводородов (в том числе 3—
8% твердых парафинов), а также восков D—8%), фенолов A5—
20%) и др. Содержание N2 в дегте (характеризует содержание в
нем соединений азота) составляет 4%; карбоновых кислот 1,5—
2%. Асфальтены (вещества, нерастворимые в петролейном эфире)
в дегте верховых торфов присутствуют в количестве 8—15%,
в дегте низинных торфов—в количестве 17—40%. Фракция
углеводородов, выкипающая в пределах 180—280 °С (бензино-кероси-
новая фракция), получается при перегонке дегтя в количестве
14—20%. Выход газа составляет около 20%, или примерно 200 м3
на 1 т торфа (теплотворная способность газа 1600 ккал/м3 или
6,7 Мдж/м3). Газ содержит метан, водород, окись углерода и
относительно много балласта (СО2 и N2).
Генераторный газ, получаемый при газификации торфа,
имеет низшую теплотворную способность примерно 1500 ккал/м3
F,3 Мдж/м3).
4. ТЕРМИЧЕСКАЯ ПЕРЕРАБОТКА УГЛЕЙ И СЛАНЦЕВ
Из твердых топлив наибольшее значение в народном хозяйстве
СССР имеют угли. Доля их в общих запасах ископаемого топлива
у нас превышает 95% (стр. 14). В. И. Ленин говорил: «Уголь—
это настоящий хлеб промышленности». В угле содержатся
основные запасы энергии и органического химического сырья нашей
страны, в процессе добычи он извлекается из недр практически
80
нацело. В единице массы уголь содержит больше горючего
вещества, чем любое другое твердое топливо. Однако техника
угледобычи более сложна и трудоемка, чем добыча нефти и газа, что
заставляет сейчас отдавать предпочтение развитию нефтяной и
газовой промышленности. Одна из наиболее важных задач для
угольной промышленности—максимальная механизация
угледобычи и ее удешевление. Следует отметить, что особенно интенсивно
в ближайшее время будет развиваться добыча угля в восточных
районах страны.
Более 70% добываемого угля непосредственно расходуется
в качестве топлива. Потребителями угля являются крупные
электростанции, промышленность, железнодорожный и водный
транспорт; большое количество угля идет также на отопление жилищ
и для других бытовых нужд.
Угли, содержащие много золы и влаги (большей частью
бурые угли), целесообразнее использовать на месте их добычи—
сжигать на электростанциях или превращать в горючий газ.
Полученную электроэнергию или горючий газ направляют к
месту их потребления (даже на дальние расстояния) по линиям
электропередачи или газопроводом.
Основная часть углей, поступающих на химическую
переработку (более 20% от общей их добычи), употребляется на
коксование—для получения металлургического кокса, коксового газа
и различных химических продуктов. Коксование и
полукоксование являются методами
термической
переработки углей и сланцев.
Более сложные
методы термохимической
переработки углей—их
ожижение и
газификация.
ПРОИСХОЖДЕНИЕ
И СОСТАВ УГЛЕЙ
Уголь представляет
собой минерализованные
скопления растительных
остатков. Эти остатки после
торфяной стадии превращения
переходят в уголь.
Растительное
происхождение угля доказано бесспорно; существовавшее до середины XIX в.
мнение о его минеральном происхождении теперь полностью отвергнуто.
Одним из первых о растительном происхождении угля писал М. В.
Ломоносов в своем сочинении «О слоях земпых», утверждая, что каменные
угли произошли из первобытных торфяников под действием подземного
¦огня. В дальнейшем растительное происхождение угля было подтверждено
изучением его под микроскопом (рис. 27). Кроме того, в угольных залежах
найдены отпечатки различных частей растений.
Рис. 27. Вид угля под микроскопом:
/—остатки древесной ткани (фюзен),
2—бесструктурная масса (внтрен).
6-805
81
Таблица
Промышленная классификация углей
Методы использования
Химико-технологические
Химико-металлургические
Энергохими ческие
Энергетические
Промышленный класс
Экстракционные
Перегонные
Коксовые*
Доменные**
Газогенераторные
Непосредственно
сжигаемые
Брикетируемые
Виды углей
Некоторые бурые угли,
богхеды
Сапропелиты и каменные
угли, богатые
смолистыми веществами
Каменные угли марок Г,
пж, к, пс
Некоторые тощие
каменные угли
Все виды углей
* Донецкие, кузнецкие, карагандинские н др.
** Некоторые кузнецкие угли и др.
Отдельную группу твердых топлив составляют горючие сланцы,
органические вещества которых образовались не из наземных растений, а из остатков
низших организмов, ранее населявших водоемы. В результате химической
переработки сланцев получают ценные жидкие погоны, заменяющие некоторые
нефтепродукты.
Сланцам родственны по происхождению менее распространенные богхеды
и сапропелиты, содержащие повышенное количество летучих веществ.
Классификация углей
Марки
д
г
пж
к
ПС
т
Типы
Длиннопламенные
Газовые
Паровичные жирные
Коксовые . . .
Паровичные спекающиеся . . .
Тощие
Выход
летучих*
%
Более 42
44-35
35—26
26—18
18—12
Менее 17
Теплотворная
способность*
ккал/кг
7650—8100
7900—8300
8300—8700
8400—8750
8450—8720
8300—8700
Элементарный
С
76—86
78—89
84—90
87—92
89—94
90—95
*В расчете на горючую массу топлива.
82
Ископаемые угли по степени их обуглероживания—так
называемому условному возрасту—делятся на бурые угли
(«молодые»), каменные угли и самые обуглероженные антрациты
(«древние угли»). В табл. 4 (стр. 18) было показано изменение состава
твердых горючих веществ в зависимости от их возраста. По мере
перехода от древесины к антрациту уменьшается содержание в
топливе кислорода и водорода и увеличивается содержание углерода.
Основные виды углей—бурые и каменные—подразделяются
на ряд сортов (марок). Состав углей разных марок различен.
Отличительной особенностью бурых углей является наличие
в них гуминовых кислот, образовавшихся еще на торфяной
стадии превращения. В каменных углях гуминовые кислоты
превратились в глубоко минерализованные гуминовые вещества. Бурые
угли более гигроскопичны, чем каменные (рабочая влажность
бурых углей достигает 30%, каменных—3,5%), и не спекаются.
Состав продуктов сухой перегонки бурых и каменных углей
различен. Самыми «молодыми» из бурых углей являются так
называемые лигниты, еще сохранившие структуру древесины.
КЛАССИФИКАЦИЯ УГЛЕЙ
Для характеристики разных типов каменных углей
предложены различные классификации их. Классификация углей,
характеризующая возможности их промышленного использования,
приведена в табл. 8.
Ни одна из существующих классификаций (даже самая
распространенная классификация Грюнера) не охватывает всего
многообразия разновидностей ископаемых углей. Поэтому для отдель-
Таблица 9
Донецкого бассейна
состав органической массы, %
Н
5—6
4,5—5,5
4—5,4
4—5,2
3,8-4,9
3,4—4,4
N
(в среднем)
1,8
1,7
1,7
1,5
1,5
1,2
О
10—17,5
6,8—16
5—10,5
3-8
2—5
1,6-4,5
Характеристика кокса
Неспекшийся, порошкообразный или
слипшийся
Спекшийся, сплавленный, иногда
вспученный (рыхлый)
Спекшийся, сплавленный или умеренна
плотный
То же
Спекшийся или сплавленный, от
плотного до умеренно плотного
Неспекшийся, порошкообразный или
слипшийся
S3
ных угольных оассеинив составлены свои уточненные
классификации. Представление о самых распространенных типах
донецких углей дает табл. 9. Угли Кузнецкого бассейна, содержащие
меньше серы и золы и обладающие более высокой теплотворной
способностью, чем донецкие, не укладываются в эту
классификацию.
ДОБЫЧА И ОБОГАЩЕНИЕ УГЛЕЙ
Уголь распределен довольно равномерно на территории
нашей страны. Основными угольными месторождениями СССР (см.
рис. 2 на стр. 13) являются: Донецкий, Кузнецкий и
Карагандинский каменноугольные бассейны. Большое значение имеют также
Кизеловский бассейн на Урале, Печорский бассейн, угли Средней
Азии, Дальнего Востока (Сучан и др.), Восточной Сибири (Черем-
ховский бассейн) и Кавказа (Тквибули, Ткварчели).
Богатые залежи бурых углей имеются на правобережной
Украине, в Подмосковном бассейне, на Восточном Урале, в
Средней Азии и в Восточной Сибири. Огромные угленосные площади
на севере азиатской части СССР еще недостаточно разведаны.
Как правило, залежи угля погребены под пластами горных
пород, и добыча угля связана с горными работами. При
неглубоком залегании угля (чаще всего бурого) достаточно снять слой
почвы, покрывающей уголь, после чего его можно добывать
открытыми разработками при помощи экскаваторов. В последнее
время успешно развивается метод гидродобычи, по которому и
добыча угля из пласта и его транспортирование производятся
при помощи воды. Если уголь залегает глубоко под землей, пла~
стами, добыча его производится в шахтах.
Из схемы шахты (рис. 28) видно, что для проникания к
угольному пласту приходится сооружать глубокий ствол шахты. От
него к разрабатываемым угольным пластам проходит серия
горизонтальных ходов, так называемых квершлагов. В стороны от
квершлагов в толще угольного пласта (по его простиранию)
пробивают ходы—штреки. Между двумя соседними штреками
прокладывают наклонный ход, (уклон), в котором и производится
разработка угольного пласта. Вырубаемый уголь спускают в нижний
штрек, транспортируют к стволу шахты и далее на поверхность.
Процесс добычи угля в СССР механизирован полнее, чем в
какой-либо другой стране мира,—от зарубки угля в шахте до
погрузки его в вагоны.
Балласт, входящий в состав бурых и каменных углей (зола
и влага), затрудняет использование их в качестве
энергетического топлива и сырья для химической переработки. Топливо
необходимо, по возможности, освобождать от балласта. Избыточную
влагу удаляют путем сушки угля. Значительно сложнее отделить
от угля «золу». Для этой цели сооружают специальные
углеобогатительные фабрики.
84
Уголь содержит «золу» двух типов: внутреннюю и внешнюю,
Внутренняя зола отлагалась одновременно с накоплением
углистого материала и вошла в его состав; отделение ее очень
затруднено, а подчас и невозможно. Внешняя зола представляет собой
куски породы, находившейся в угле в виде прослоек или
составляющей почву или кровлю угольного пласта. Внешняя зола
может быть отделена от угля механическими способами—путем его
мокрого или сухого обогащения.
Рис. 28. Схема угольной шахты.
При сухом обогащении предварительно просеянный уголь
направляют на специальные столы с рифленой поверхностью,
где, благодаря разной степени сцепления с поверхностью стола,
куски гладкого, скользкого угля отделяются от шероховатой
породы, уголь и порода ссыпаются в разные бункеры.
Более широко распространено мокрое обогащение, основанное
на отделении угля от породы вследствие различия их плотности
(плотность угля 1,2—1,3 г/см3, углистого сланца и породы—
1,4 г/см3 и более). В воде или в водных суспензиях минеральных
порошков—барита, магнетита (плотность их суспензий может
достигать 1,4 г/см3 и выше) минеральные частицы осаждаются, а
уголь всплывает. Для обогащения углей применяется также
флотация. Методы обогащения сырья описаны в курсе
химической технологии неорганических веществ*.
* См., например, А. П. Егоров, А. И. Ш е р е ш е в с к и й,
И. В. Шманенков, Общая химическая технология неорганических
веществ, нзд. «Химия», 1964, 1965.
85
очень затруднено, особенно в небольших топках. Угольную
мелочь превращают в кусковое топливо путем брикетирования—
прессования в формах под давлением с добавлением к углю в
случае необходимости связующего вещества.
КОКСОВАНИЕ
Основным процессом переработки каменных углей является
коксование. В процессе коксования из угля получают кокс для
доменных печей, коксовый газ, обладающий высокой теплотворной
способностью и разнообразные ценные химические продукты
(аммиак, бензол, толуол, нафталин и др.). Переработкой угля путем
коксования с одновременным получением химических продуктов
занимается коксохимическая промышленность.
В развитых промышленных странах мира коксованию
подвергают до 20% всего добываемого угля. По уровню развития
коксохимической промышленности, от которой зависят металлургия и
в значительной степени промышленность органического синтеза,
можно судить об общем уровне промышленного развития страны.
Отечественная коксохимическая промышленность непрерывно
развивается и совершенствуется. В настоящее время по объему
продукции она сравнялась с коксохимической промышленностью
США и занимает первое место в мире. С 1913 по 1963 гг.
производство кокса в нашей стране увеличилось примерно в 15 раз.
В 1964 г. в СССР было произведено 66,3 млн. т кокса.
Процесс коксования заключается в сухой перегонке
каменного угля при высоких температурах. Каменный уголь загружают
в специальные закрытые камеры—коксовые печи и нагревают до
температуры выше 1000 °С. При этом образуются летучие
вещества (газо- и парообразные продукты, пары воды и аммиак) и
твердый нелетучий остаток—кокс. Процесс коксования протекает в
несколько стадий. При температуре порядка 100 °С уголь
подсушивается; далее—до 600 °С органическая масса угля начинает
постепенно разлагаться на летучие продукты и твердый остаток—
полукокс, еще содержащий значительное количество летучих
веществ. Процесс, заканчивающийся на этой стадии, называется
полукоксованием. При дальнейшем повышении температуры из
полукокса выделяются остатки летучих веществ, и он
превращается в кокс. В процессе коксования уголь подвергается наиболее
глубоким изменениям. Летучие продукты проходят при этом зону
печи, нагретую до 1000 °С, и органическая часть их (пары смолы
и более легких углеводородов) претерпевает глубокое
разложение, происходит так называемая ароматизация летучих.
Главной целью процесса коксования является получение
металлургического кокса, который должен обладать высокой прочностью, чтобы не
разрушаться под огромным давлением слоя материалов, загружаемых в доменную
печь.
-86
Для получения кокса могут быть применены только угли, способные
хорошо спекаться и образовывать кокс требуемого качества,—так называемые
коксовые угли. Такие угли при 350—500 °С переходят в пластическое состояние,
размягчаются, образуя угольный «расплав». При дальнейшем повышении
температуры пластичная масса выделяет летучие продукты, твердеет и
превращается в кокс.
Пригодные для коксования угли составляют лишь около 20% общих
запасов угля. Практика показала, что путем смешения углей разных сортов-
можно составить такую смесь—шихту, из которой будет получаться кокс
требуемого качества. Кроме коксовых углей, в состав такой шихты можно
вводить близкие им по свойствам угли марок ПС, ПЖ, Г (стр. 82).
Современный коксохимический завод средней мощности
потребляет в сутки около 15 тыс. т углей. Для составления шихты
из этих углей их специально обрабатывают в углеподготовитель-
ном отделении: усредняют их состав на складе, дробят,
составляют из них нужную смесь, подвергают ее тонкому дроблению
с перемешиванием и только после этого подают шихту к печам.
Печи для коксования углей. Современная коксовая печь
представляет собой обогреваемую камеру из огнеупорных кирпичей
(высота камеры около 4 м, длина 14 м, ширина 0,4 м),
вмещающую 15 т угля и более. Новейшие печи в СССР имеют высоту
5 м, их единовременная загрузка превышает 20 т. Разрез
коксовой печи с обслуживающими механизмами показан на рис. 29.
Основным огнеупорным материалом для сооружения
современных коксовых печей служит динасовый кирпич, который хорошо
проводит тепло и выдерживает высокие температуры (до 1350—
1400 °С), не подвергаясь разрушению.
Для превращения угля в кокс проводятся следующие
последовательные операции, выполняемые специальными машинами.
Заготовленная шихта из угольной башни / ссыпается
определенными порциями в специальный загрузочный вагон 2, отвозится
к загружаемой печи и ссыпается в камеру 5 через загрузочные
люки, плотно закрываемые после загрузки. Уголь выдерживают
в печи 14—17 ч, т. е. до тех пор, пока он не спечется в сплошную
массу, так называемый коксовый пирог, в котором в концу
коксования образуются трещины.
Летучие продукты коксования покидают камеру через стояк 4
и по газосборнику 3 и газопроводу отводятся на
коксохимический завод. |
Выделение летучих почти прекращается при 800 °С, и при
дальнейшем нагревании до 900—1000 °С происходит
окончательный выжиг кокса. Когда кокс готов, с помощью особых
механизмов снимают двери камеры, и кокс выгружается из печи
коксовыталкивателем 8. Коксовыталкиватель, загрузочный вагон и
другие механизмы, обслуживающие печь, передвигаются вдоль
батареи к той печи, в которой кокс готов к выдаче.
Выданный из печи раскаленный коксовый пирог падает в
тушильный вагон 12 и отвозится в тушильную башню //, где кокс
87'
Нонсовый газ
'N.Y
Рис. 29. Коксовая печь:
1 угольная башня- 2-загрузочный вагон; S-газосборник; 4-стояк (газоотвод); J-камера коксовой печн; 5-штанга
коксовыталкивателя- 7-башмак коксовыталкивателя; «-коксовыталкиватель; 9-регенераторы; JO-борова; Л-тушильная
коксовикі^ 1С ». б J2—тушильный вагон; JJ-площадка для кокса; М-транспортер.
охлаждают водой (гашение кокса). Охлажденный кокс ссыпается
на наклонную площадку 13, откуда транспортером 14 подается на
сортировку по величине кусков. Наиболее крупный кокс (куски
размером более 25 мм) используется в доменных печах. Такой
кокс называется металлургическим. Внешний вид куска кокса
показан на рис. 30. Чтобы использовать тепло, выделяемое
раскаленным коксом, его гашение можно производить в закрытой
камере инертными газами, которые при этом нагреваются и
поступают в котел-утилизатор, где используются для получения пара.
Такой метод охлаждения кокса называется сухим тушением.
Существующие в настоящее
время многочисленные типы
коксовых печей различаются
главным образом системой
обогрева (рис. 31), которая состоит
из так называемых горелочных
каналов3, расположенных вдоль
стенок камер / печи. В каналах
сжигается коксовый газ
(полученный в процессе коксования)
или другой (бедный) газ с
более низкой теплотворной
способностью (например, доменный
или генераторный).
Чтобы в камерах печи
поддерживалась температура
порядка 1100—1200 °С,
продукты горения газа в горелочных
каналах (дымовые газы) должны даже на выходе иметь еще более
высокую температуру. Для использования тепла дымовых газов их
пропускают через регенераторы 2, находящиеся под камерами
коксовых печей. Регенераторы заполнены насадкой из мелких
кирпичей, установленных в виде решетки. Горячие дымовые газы
нагревают решетку и уходят в дымовую трубу. Подачу горячих газов в
регенераторы периодически прекращают и начинают пропускать
через него холодный воздух или бедный газ, которые нагреваются,
соприкасаясь с накаленной кирпичной насадкой регенераторов.
Таким образом, в горелочные каналы поступает горячий воздух
(или газ), благодаря чему температура пламени в каналах
повышается и расход топлива на обогрев печи сокращается. Для
равномерного и непрерывного обогрева камеры под каждой печью
устанавливают две группы регенераторов. Пока одна из них
нагревается отходящими дымовыми газами, в другой нагревается
воздух или (в раздельных частях регенератора) воздух и
низкокалорийный газ, поступающие на отопление печи. Через каждые
20—30 мин регенераторы переключаются, что позволяет вести
равномерный обогрев печи.
89
Рис. 30. Вид куска кокса.
образующих батарею коксовых печей, в которой чередуются
разделенные обогревательными простенками горелочные каналы и
камеры. Одна батарея включает до 70 камер и более. Под камерами
батареи располагают регенераторы и борова для отвода дымовых
газов в общую для всей батареи дымовую трубу. Периодическое
Рис. 31. Обогревательная система коксовой печи (схематический
разрез):
/—камеры печи;1 2—регенераторы; J—горелочные каналы (вертикалы); 4—косые
ходы, соединяющие вертикалы с регенераторами; 5—колосники,
поддерживающие насадку регенераторов (заполнение насадкой показано пунктиром);
6—подовый (распределительный) канал.
аереключение отопительных" устройств и регенераторов, так
называемая кантовка, производится специальной кантовочной
лебедкой.
Количество печей в батарее обычно таково, что при
последовательной их разгрузке перерывы в выдаче кокса составляют
всего 15—20 мин. Таким образом, процесс коксования угля
проводится как периодический в каждой отдельной камере, но во всей
коксовой батарее в целом приближается к непрерывному.
Общий вид коксовой батареи представлен на рис. 32.
Качество металлургического кокса регламентируется в СССР
общесоюзными стандартами (ГОСТ 513—54 и 2014—53).
Содержание в коксе золы не должно превышать 9—10%, серы—1,7%,
влаги—5%.
По опытным данным, каждый процент золы в коксе повышает
на 2,5% его расход на 1 т чугуна; каждая десятая доля процента
Рис. 32. Общий вид батареи коксовых печей.
серы в коксе снижает производительность доменной печи на 1—2%.
Присутствие фосфора в коксе увеличивает хрупкость
выплавляемого на нем металла.
Тепловой и материальный баланс коксования. На коксование
1 кг угольной шихты расходуется в среднем около 600 ккал тепла
(~2,5 Мдж), в том числе:
%
На нагревание коксового пирога 45
На нагревание летучих продуктов 18
На испарение влаги из угля 12
С дымовыми газами 14
Потери в окружающую сред1.' 11
9Г
Из 1 т шихты с влажностью 6% при коксовании получается
¦примерно следующее количество продуктов (округленно):
кг
Кокс сухой ... 730
Газ сухой 140 C00 м3)
Смола каменноугольная 30
Бензольные углеводороды 10
Аммиак 3
Водный конденсат* 80
Итого.. 993
Потери при коксовании 7
* Состоит из влаги, содержавшейся в угле и образовавшейся
при его разложении.
Получение светильного Ггаза. Процесс получения светильного газа из
-каменного угля аналогичен процессу коксования. Отличие заключается лишь
в том, что сырьем служат газовые угли (марки Г), дающие при перегонке
больше газа (до 400 ма на 1 т. угля), но меньше кокса (и худшего качества). Период
коксования при получении светильного газа сокращенный.
Мощность газовых заводов меньше, чем коксохимических. Типы газовых
печей и их конструкции отличаются от коксовых печей.
УЛАВЛИВАНИЕ ЛЕТУЧИХ ПРОДУКТОВ КОКСОВАНИЯ
Полное улавливание и переработка летучих продуктов
коксования углей входят в число основных процессов
коксохимического и газового производств. Летучие продукты покидают камеру
коксования при 700—800 °С в виде смеси паров и газов.
Дальнейшая переработка этой паро-газовой смеси заключается в ее
охлаждении до 25—30 °С, выделении продуктов, конденсирующихся
при этой температуре (смола и водный конденсат), и улавливании
из газа оставшихся в нем аммиака, бензола, а часто и
сероводорода. Очищенный таким образом коксовый газ называется
обратным газом.
Состав летучих продуктов коксования зависит от качества
исходного угля и режима коксования. Примерный состав этой
паро-газовой смеси:
Коксовый газ*, м3 300—350 (на 1 т угля)
Пары смолы (дегтя), г 85—130 1
Пары сырого бензола, г 20—40
Аммиак, г 10 (на 1 м? газа)
Пары воды, г 200—300
Сероводород, г S—30
• Считая на объем сухого газа, приведенного к нормальным условиям
@ "С, 760 мм рт-ст.).
В летучих продуктах содержатся также другие примеси (пыль,
цианистый водород, пары фенолов, пиридиновых оснований
и др.). Наибольшую ценность представляют газ, смола, сырой
бензол и аммиак.
92
Коксовый газ, очищенный от всех сопутствовавших ему
продуктов, представляет собой смесь газов примерно следующего
среднего состава (в объемн. %):
Hg
сн,
со ¦ . .
Непредельные
углеводороды . . .
57
26
6
1,7
СО,
02
N.
2,0
. . 0,3
7,0
Плотность коксового газа 0,47—0,5 кг/м3, теплотворная
способность 3800—5000 ккал/м3. Коксовый газ является ценным
газообразным топливом и химическим сырьем.
Сырой бензол—легкоподвижная, резко пахнущая жидкость
от светло-желтого до бурого цвета (плотность 0,88—0,89 г/см3),
содержащая преимущественно бензол, толуол, ксилолы и другие
вещества, кипящие при температурах до 180 °С.
Каменноугольная смола—темно-бурая или черная густая
жидкость (плотность 1,18—1,23 г/см3), содержит в основном также
ароматические вещества, но кипящие при более высоких
температурах, чем компоненты сырого бензола.
Смола и сырой бензол служат сырьем для многих органических
синтезов в производстве полимеров, красителей, лекарственных
препаратов, взрывчатых веществ и др. Из смолы получают также
технические масла, например шпалопропиточное (консервант
древесины), и пек, широко используемый в производстве
кровельных материалов (например, толя), дорожных битумов,
беззольного кокса, употребляемого в производстве электродов для
электрических печей, и др.
Аммиак коксового газа был единственным техническим
источником получения NH3 до создания промышленного способа
синтеза аммиака. На коксохимических заводах аммиак улавливают
преимущественно в виде сульфата аммония (NH4JSO4 или
концентрированной аммиачной воды. Оба эти продукта используются
в качестве азотных удобрений.
На некоторых коксохимических и на всех газовых заводах
коксовый газ очищают также от сероводорода. Сероводород,
полученный при очистке газа, перерабатывают в элементарную
(газовую) серу или в серную кислоту.
Первой ступенью в процессе улавливания летучих продуктов
коксования (рис. 33) является охлаждение газа и конденсация
паров с выделением смолы и надсмольной воды. Чтобы сразу
быстро охладить газ, в газосборник 2 и в стояки, по которым в него
поступает газ из камер коксовой печи, вбрызгивают надсмольную
воду. Благодаря этому температура газа понижается от 700 "С
(температура на выходе газа из камер) до 80—85 °С. В
газосборнике конденсируется большая часть смолы и удаляются
угольная и коксовая пыль, сажа и другие механические примеси (так
называемые фусы).
jj^jia икинчагельниги охлаждении до zo—du и газа по
газопроводу 5 направляется в трубчатые холодильники 5 и 6, по
трубкам которых протекает холодная вода, а в межтрубном простран-»
/аз
\ і
\ і
\ і
в
І-—.—
/7
—и
Смола
Рис. 33. Схема улавливания летучих продуктов коксования:
\—коксовая печь", 2—газосборник; 3—газопровод; 4—отстойник-фусоотделителъ; 5. 6—трубчатые
холодильники; 7 — отстойник конденсата; в—сборник смолы; 9—сборник аммиачной воды;
10—турбогазодувка; 11—электрофильтр; 12, 13—аммиачные колонны; 14—фенольная установка;
IS— центрифуга; /«—сатуратор; 17—каплеотбойник; 18—конечный холодильник; 19.
10—скрубберы; 21—бензольная колонна; 22—подогреватель масла; 13—дефлегматор; 24—колонна для
разделения бензола на легкий и тяжелый; 25—конденсатор; 16— сепаратор; 27— холодильник;
28—холодильник масла; 19— отстойник; 30—регенератор масла.
стве движется охлаждаемый газ. Жидкость, образовавшаяся
вследствие конденсации паров при охлаждении газа, отделяется
в фусоотделителе 4, откуда направляется в отстойники 7 (отстой-
94
ные ямы). Здесь жидкость разделяется на Два слоя: более тяжелая
смола (плотность 1,18—1,23 г/см3) образует нижний слой на дне
хранилища, в верхнем водном слое растворена часть аммиака
коксового газа. Этот верхний слой называется надсмольной,
или аммиачной водой. Аммиачная вода поглощает из газа также
двуокись углерода и сероводород с образованием углекислых и
сернистых солей. Смолу и аммиачную воду после отстаивания
раздельно направляют в хранилища 8 и 9 и затем—на переработку.
Для дальнейшего использования и переработки охлажденный
коксовый газ тщательно очищают от взвешенных в нем капелек
конденсата (тумана) в электрофильтрах 11.
Удаление газа из печей и перемещение его через
конденсационную и улавливающую аппаратуру производятся турбогазо-
дувкой 10, создающей напор порядка 1,5—2,5 м вод. ст. Каждый
эксгаустер нагнетает до 70 тыс. м31ч газа. Конденсат,
выделяющийся из газа в эксгаустере и электрофильтрах, также поступает
в отстойник 7.
Улавливание аммиака. Часть аммиака, образовавшегося при
коксовании, остается и после охлаждения в газе, частично аммиак
растворяется в водном конденсате, выделяющимся из газа при его
охлаждении. Образовавшаяся при этом аммиачная вода содержит
аммиак в виде NH4OH, а также в виде растворенных летучих солей
(карбонатов и сульфидов) и нелетучих соединений (хлоридов и
других солей).
Для последующей переработки аммиак (газообразный и
растворенный) соединяют вместе, для этого в колоннах 12 и 13
отгоняют NH3 из аммиачной воды. Колонны устроены по типу ранее
описанных ректификационных аппаратов (стр. 42). Обычно они
выполняются из чугуна, который более устойчив к коррозии,
вызываемой кислыми соединениями аммиака. В колонне 12
отгоняется весь «летучий» аммиак (см. выше), затем аммиачная вода
направляется на установку 14 для извлечения растворенных
фенолов. Далее вода возвращается в нижнюю часть колонны 12,
где смешивается с известковым молоком, и перетекает в колонну 13.
Здесь отгоняется аммиак, выделяющийся из нелетучих солей
аммония при разложении их гидроокисью кальция Са(ОНK.
Отгоняемый аммиак направляется в газопровод перед сатуратором 16.
В настоящее время аммиак коксового газа используют для
получения сульфата аммония. Цех производства этого удобрения
оборудован сатураторами 16, в которых NH3 улавливается серной
кислотой, центрифугами 15, сушилками и другими аппаратами.
Производство сульфата аммония описано в курсе химической
технологии неорганических веществ*.
* См., например, А. П. Егоров, А. И. Ш е р е ш е в с к и й,
И. В. Шманенков, Общая химическая технология неорганических
веществ, изд. «Химия», 1964, 1965, стр. 588.
95
щенной концентрированной аммиачной воды*. Для этого
оставшийся в коксовом газе NH3 поглощают в скрубберах водой и
перерабатывают скрубберную аммиачную воду в колоннах,
аналогичных описанным выше, однако без добавления известкового
молока, так как в скрубберной воде отсутствуют нелетучие соли
аммония.
Для связывания аммиака начали применять также
фосфорную кислоту, при этом получается концентрированное азотно-
фосфорное удобрение—аммофос*.
Улавливание сырого бензола. Очищенный от аммиака газ
направляется на улавливание сырого бензола. Наиболее
распространено улавливание бензола из газа соляровым маслом (пределы
кипения 260—350 °С) или каменноугольным маслом (пределы
кипения 250—300 °С). Этот процесс аналогичен улавливанию
газового бензина (стр. 33).
Коксовый газ и поглотительное масло противотоком подаются
в скрубберы 19 и 20 (см. рис. 33). Соприкасаясь с газом, масло
извлекает из него бензол. Насыщенное бензолом поглотительное
масло нагревается глухим паром в подогревателях 22 до 120—
130 °С и направляется в бензольную колонну 21. В нижнюю часть
колонны подается острый водяной пар, который отгоняет из масла
поглощенный сырой бензол. Далее в аппаратах 23—27 он
разделяется на легкий бензол (темп. кип. до 150 °С) и тяжелый
бензол (температура кипения выше 150 °С). Масло охлаждается после
отгонки бензола, в холодильнике 28, отделяется в отстойниках 29
от шлама и водной эмульсии и снова направляется на орошение
скрубберов 19 и 20.
Перед подачей в эти скрубберы газ дополнительно охлаждается
в холодильнике 18, при этом из газа выделяется нафталин.
Охлажденный газ из бензольных скрубберов направляется на
дальнейшее использование.
Очистка газа от серы (в основном от сероводорода H2S)
зачастую необходима перед использованием газа в качестве топлива
или химического сырья. Содержание H2S в газе составляет обычно
12—18 г/м3, а иногда и более. Газ, используемый в металлургии
при выплавке специальных сталей, должен содержать не более
2—3 г/м3 H2S, бытовой газ—не более 2 г H2S на 100 м3. Из
газа, предназначенного для органических синтезов, сера должна
быть удалена полностью, так как она является ядом для
катализаторов.
Методы очистки газа от серы были кратко рассмотрены на
стр. 31 и ел. Более подробно они описаны в курсе химической
технологии,неорганических веществ*.
* См. сноску на стр. 95.
?6
ПЕРЕРАБОТКА ПРОДУКТОВ КОКСОВАНИЯ
Сырой бензол и каменноугольная смола представляют собой
сложные смеси разнообразных индивидуальных химических
соединений, многие из которых являются ценными химическими
продуктами только в чистом виде. Поэтому для получения из сырого
бензола и каменноугольной смолы товарных продуктов их
подвергают дополнительной переработке.
Переработка сырого бензола. Вещества, входящие в состав
сырого бензола (табл. 10), широко используются в производстве
полимеров, красителей, лекарственных препаратов, взрывчатых
веществ и многих других продуктов.
Состав сырого бензола
Таблица 10
Компоненты
Легкокипящие сернистые и
непредельные соединения (в основном
сероуглерод CS2 и циклопентадиен С6Н6) .
Бензол С6Н„
Толуол С6Н6СН3
Ксилолы С6Н4(СН3Ь
Сольвенты (неразделяемая смесь
углеводородов)
Фенол QHjOH
Содержание
%
До 3
55—65
10—16
5—7
5—8
0,6
Температура
кипения
°С
До 80
80,1
110,6
138,5—144,4
150—180
182,1
Примечание. Сырой беизол содержит также небольшие количества тиофеяа
C4H4S, пиридина C5H5N и других соединений.
Как видно из табл. 10, компоненты сырого бензола имеют
различные температуры кипения, и потому отдельные чистые
вещества могут быть выделены из него путем ректификации (стр. 42).
При разгонке сырого бензола отбираются следующие фракции;
Фракции
Сероуглеродная ...
Бензольная
Толуольная
Ксилольная
Тяжелый бензол (сольвенты)
Пределы
кипения, °С
До 80
До 100
100—125
125—150
150—180
Примечание. В современных схемах переработки
отбирается обідав фракция БТК (бензоло-толуоло ксклоль-
иая), которую после химической очнсткн разделяют
ректификацией на чистые продукты.
В этих фракциях наряду с основными веществами
присутствуют примеси; некоторые из них являются вредными и
препятствуют или затрудняют дальнейшее использование основных про-
7—805
97
nVKTOB. ІЛЛЯ получения ЧМСЇЬіХ ПрС/ДуКїиь фракции CDipui u uctidujia
подвергают химической очистке (обычно серной кислотой и
щелочью) и повторной (окончательной) ректификации.
В результате переработки фракций сырого бензола получают
чистые бензол, толуол, ксилолы или смеси чистых продуктов—
так называемые моторные бензолы, служащие добавками к
топливу для авиационных и автомобильных двигателей.
Переработка каменноугольной смолы. Каменноугольная смола,
как и сырой бензол, представляет собой смесь преимущественно
ароматических углеводородов. В состав каменноугольной смолы
входят главным образом вещества, кипящие выше 180 °С.
Соединения, температура кипения которых близка к 180 °С, содержатся
и в сыром бензоле.
Состав каменноугольной смолы изучен далеко не полно, так
как многие компоненты содержатся в ней в незначительных
количествах и представляют собой сложные соединения, кипящие при
температурах выше 400 °С.
Смолу разделяют перегонкой на ряд кипящих в определенных
пределах фракций, называемых маслами, каждое из них имеет
свою специфическую область применения. Характеристика этих
масел приведена в табл. 11.
Таблица II
Фракционный состав
Фракции
Легкое масло
Фенольное масло ....
Нафталиновое масло . . .
Тяжелое (поглотительное)
масло ¦ •
Антраценовое (шпалопро-
питочное) масло ....
Пек
Выход
% от смолы
0,5—1,0
1,5-2,0
8,0
10—13
18-25
55—60
каменноугольной смолы
Пределы
кипения
°С
До 180
180—210
210—230
230—270
270—360
Выше 360
Плотность
г/смЗ
0,915—0,930
0,95—1,00
1,01—1,02
1,04—1,05
1,10—1,13
Продукт,
ВЫД1 ЛЯ-'МЫЙ
из фракции
Фенолы
Нафталин
Антрацен
Примечание. Три первые масла (фракции) иногда отбирают вмгсге, получая так на
зываемое легкосреднее масло (темп. кип. до 230 °С)-
Разгонка каменноугольной смолы производится на трубчатых
установках. Наиболее четкое разделение смолы на фракции с
высоким содержанием основных продуктов достигается на
высокопроизводительных смолоразгонных установках непрерывного
действия.
Схема такой установки, перерабатывающей в сутки более
200 т смолы, представлена на рис. 34. Для освобождения от
примесей и влаги смола отстаивается, после чего подогревается в
теплообменнике 1 и подается в первую секцию 2 нагревательной
трубчатой печи 5 (стр. 46). Из секции 2 печи смола, нагретая до
98
150—170 °С, поступает в отпарную колонну 8, где отделяются пары
воды и легкое масло, направляемые в конденсатор 9 и далее в
сборники.
Отделенная от воды и легкого масла смола из колонны 8
подается во вторую секцию 4 трубчатой печи 5, где нагревается до
360—380 °С; все масла при этом испаряются, а в жидком
состоянии остается только пек. Паро-жидкостная смесь поступает в пе-
ковую колонну //, в нижнюю часть которой для отделения масел
от пека подают из перегревателя 3 перегретый водяной пар.
Смола
Рис. 34. Схема смолоразгонной установки:
/—теплообменник; 2—первая секция трубчатой печи; 3—пароперегреватель; 4—вторая
секция печи; 5—трубчатая печь; «—горелка; 7—боров; #—отпарная колонна;
9—конденсаторы; 10—водоотделитель легкой фракции; 11—пековая колонна; /2,
13—ректификационные колонны; Н—холодильники.
Степенью подогрева смолы и количеством подаваемого пара
регулируют количество и качество отгоняемых масел. Пек стекает из
колонны 11 в теплообменник /, где его тепло используется для
нагревания свежей смолы.
Смесь паров каменноугольных масел из колонны //
направляется в первую ректификационную колонну 12, орошаемую
тяжелым маслом. При его испарении конденсируются антраценовые
масла, раздельно отводимые из колонны: из нижней
части—тяжелое масло, из средней части—более легкое антраценовое масло.
Аналогично работает колонна 13 для разделения тяжелого,
нафталинового и фенольного масел.
7* 99
Из масел, полученных при разгонке каменноугольной смолы,
выделяют индивидуальные вещества, после чего масла поступают
на дальнейшее использование. Например, тяжелое масло
применяется на коксохимических заводах для улавливания бензола,
антраценовое масло используется для пропитки древесины с
целью предохранения ее от гниения и т. п.
Из соединений, входящих в состав каменноугольной смолы,
наиболее широко используются в промышленности фенолы
(смесь фенола, о-, м- и /г-крезолов и некоторых высших фенолов),
нафталин и антрацен.
Фенолы извлекаются из масел путем их промывки раствором
едкого натра:
CeHaOH + NaOH > CeH5ONa + Н2О
фено фенолят
натрия
Растворимые в воде феноляты разлагаются кислотами или
двуокисью углерода:
CeH5ONa + СО, + Н2О * NaHCO3 + СвН6ОН
Для регенерации едкого натра образовавшийся бикарбонат
натрия обрабатывают известковым молоком и затем упаривают:
NaHCO3 + Са(ОНJ > NaOH + СаСОэ + Н-2О;
Фенолы применяют главным образом в производстве
пластмасс (стр. 392, 551 ел.) и в фармацевтической промышленности
Нафталин С10Н8 (темп. кип. 218 °С, темп. пл. 80,3 °С)
получается из нафталинового масла путем кристаллизации с
последующей очисткой кристаллического нафталина. Среднее
содержание нафталина в смоле—7%. Он используется для производства
фталевого ангидрида (стр. 260), применяемого в производстве
пластмасс, красителей, взрывчатых веществ, употребляется как сред
сгво для уничтожения моли и для других целей.
Антрацен С14Н10 (темп. кип. 342 °С, темп. пл. 216 °С)
получается из антраценового масла. Среднее содержание антрацена
в смоле—1%. Антрацен используется для производства
красителей (стр. 282), синтетических дубителей и др. Антраценовое
масло для выделения содержащихся в нем веществ поступает в
кристаллизаторы. В результате кристаллизации из масла выпадает
илистая масса зеленого цвета, состоящая из смеси кристаллов
антрацена, карбазола и фенантрена:
(А А,
NH
антрацен карбазол
фенаятрен
100
Антрацен и карбазол применяются в промышленности
тонкого органического синтеза. Фенантрен получается в большем
количестве, чем антрацен и карбазол, но используется пока
в незначительной степени.
Каменноугольный пек представляет собой блестящий черный
материал (темп. пл. от 70 до 120—150 °С), широко используемый
в дорожном строительстве, производстве кровельных материалов,
беззольного (электродного) кокса и др. Области применения пека
в настоящее время все более расширяются.
полукоксование
В отличие от процесса коксования, полукоксование
проводится при температурах, не превышающих 600 °С. Полукоксованию
подвергают разнообразные виды топлив, содержащих много
летучих,—молодые каменные и бурые угли, богхеды, горючие
сланцы. В результате этого процесса получается полукокс, газ
полукоксования, органические летучие продукты {первичный деготь
н'газовый бензин) и водный дистиллят.
Продукты полукоксования носят название первичных
продуктов, так как получаются при более слабом тепловом воздействии,
в результате первичного разложения топлива. Все продукты
полукоксования отличаются от продуктов коксования меньшей
степенью разложения органической массы топлива. Так, полукокс
содержит больше летучих (около 10%), менее прочен и более реак-
ционноспособен (лучше загорается), чем кокс. Первичный
деготь, при перегонке которого получаются продукты, сходные с
бензином и керосином, отличается от смолы, получаемой при
коксовании, меньшей плотностью (около 1 гкмъ и ниже) и почти не
содержит ароматических углеводородов, особенно высших.
Зачастую первичный деготь содержит также повышенное
количество фенолов (до 20% и более), преимущественно бысококипящих.
Газ полукоксования содержит меньше водорода B0% Н2), чем
коксовый газ (~57% Н2), но больше углеводородов и непредельных
соединений, и отличается повышенной теплотворной способностью
(8000—9000 ккалім3, коксовый газ—5000 ккал/м3). Удельный
выход газа полукоксования (на 1 т угля) в три раза меньше, чем
коксового газа.
Водный конденсат является отходом процесса полукоксования.
Он почти не содержит аммиака и имеет кислую реакцию
вследствие того, что в нем растворены фенолы.
Полукоксование использовалось еще в XVIII в., т. е. раньше
процессов производства светильного газа и коксования.
Полукоксование проводилось для получения осветительных масел и
бездымного топлива для домашних очагов. В настоящее время
полукоксование сохраняет значение в странах, богатых углями с
большим содержанием летучих, но бедных нефтью (например,
101
в Англии, где полукокс используют в каминах).
Полукоксование применяется и как способ первичной подготовки угля к
дальнейшему использованию, например для отделения от угля
ценных летучих веществ перед его сжиганием.
Как и перегонку торфа, полукоксование углей можно вести
в печах с внутренним и внешним обогревом.
Печь полукоксования с внешним обогревом (рис. 35)
представляет собой реторту /, обмурованную снаружи огнеупорной
кладкой с горелочными каналами. По ним циркулируют горячие газы,
образующиеся при сжигании топлива (например, газа
полукоксования) в топке 6. Газы нагревают стенки реторты и отсасываются
Уголь
Отходя'щие
газы
Масло
I Газ пол и*
X сования
' "і —>-
Генератор -
ныа газ
Рис. 35. Печь полукоксования с внешним обогревом:
/—реторта; 2—холодильник; 3—смолоотделитель; 4—эксгаустер; 5—скруббер;
6—топка.
вентилятором в трубу. Уголь загружают в печь сверху, готовый
полукокс выгружается снизу. Продолжительность периода
полукоксования зависит от толщины слоя загружаемого угля и
колеблется от нескольких часов до 15—20 мин. Летучие продукты
полукоксования удаляются эксгаустером 4 через холодильнику 2
в смолоотделитель 3. Смесь легких углеводородов, не
конденсирующихся вместе со смолой, поступает далее в скруббер 5 для
улавливания газового бензина. Часть газа полукоксования
расходуется на обогрев реторты, для этого может быть использован
также генераторный газ, полученный газификацией (стр. 107)
части полукокса или другого твердого топлива. Для
полукоксования мелких кусков неспекающихся бурых углей получили
распространение печи, в которых этот процесс происходит в тонком
C0 мм) слое топлива.
Печь полукоксования с внутренним обогревом представляет
собой вертикальную шахту, в которой перерабатываемое топливо
102
движется сверху вниз. Продолжительность пребывания топлива
в такой печи 3—8 ч. Производительность печи 200—300 кг/ч угля
на 1 м3 емкости. Противотоком движению топлива в печь
поступают циркуляционные газы, нагретые до 600 °С и выше. Для
обогрева печи используют газ полукоксования, генераторный или
топочный газ, водяной пар. Эти газы непосредственно
соприкасаются с топливом и нагревают его. Смесь циркуляционных газов
и летучих продуктов перегонки поступает в холодильники, смоло-
отделитель и другие конденсационные и улавливающие аппараты.
Размеры этих аппаратов при одинаковой производительности печи
по углю больше, чем в установке печи с внешним обогревом, так
как объем циркулирующего газа при внутреннем обогреве больше.
После очистки и охлаждения газы разделяются на несколько
потоков. Постоянное количество газа (циркуляционная часть) снова
направляется на обогрев печи, причем некоторая его доля подается
в нижнюю часть печи для охлаждения выгружаемого полукокса.
Большая часть циркуляционного газа до поступления в печь
нагревается в специальном подогревателе до температуры,
необходимой при сухой перегонке топлива. Избыток газа,
соответствующий количеству летучих продуктов полукоксования,
выводится из цикла.
Печи обоих типов имеют свои достоинства и недостатки. В
печах с внешним обогревом можно получать кусковой полукокс
из мелких спекающихся углей и подвергать полукоксованию как
кусковое топливо, так и угольную мелочь. Содержание паров
смолы и бензина в газах этих печей значительно выше, чем в
газах, получаемых из печей с внутренним обогревом.
Печи с внутренним обогревом проще по конструкции,
производительнее (до 300 т/сутки и выше), но для них требуется более
громоздкая газовая аппаратура, так как постоянный объем
циркуляционных газов в этих печах 1000 м9 и более на 1 т угля.
В печах с внутренним обогревом можно перерабатывать только
кусковое топливо.
При перегонке сланцев получаются высокие выходы ценных
жидких фракций, заменяющих нефтепродукты, и относительно
высокозольный малоценный полукокс. Для этой цели применяются
так называемые туннельные печи (рис. 36). В этих печах (с
внутренним обогревом) сланец медленно продвигается в вагонетках 2
по туннелю 1 цилиндрического сечения. Туннель с обеих сторон
герметически закрыт дверями 3 и разделен внутренними
перегородками на три зоны: сушки, полукоксования и охлаждения.
Между зонами имеются шлюзовые перепускные камеры 4.
Через туннельную печь продувают подогретые газы и пары
летучих продуктов полукоксования (см. разрез А). При помощи
вентиляторовб газы непрерывно циркулируют через сланец в
туннеле, а затем через трубчатые подогреватели б (калориферы),
обогреваемые газами, образующимися при сжигании сланца в спе-
103
циальных топках, іа^ьі, выделяилдйсо; «и ї,ш,.цп u o„Si„ .*-_..-
коксования, отводятся на установку для конденсации и улавлиня-
ния летучих продуктов.
Вагонетки передвигают в печи через каждые 8—20 мин с таким
расчетом, чтобы время пребывания сланца в зоне
полукоксования составляло около 2 ч. Диаметр туннеля печи 2 м (в свету),
длина 53 м. В туннеле помещается 19 вагонеток, из них 12—в зоне
полукоксования. Каждая вагонетка вмещает 2,2 т сланца. В
туннельной печи можно перерабатывать до 400 т сланца в сутки.
Рис. 36. Туннельная печь (А—разрез
по зоне полукоксования):
/—туннель; 2—аагонеткн; 3—даерн туннеля;
4—шлюзовые перепускные камеры; 5—вентиляторы;
й-калориферы зоны полукоксования; 7—калориферы зоны
сушки.
В связи с начинающимся развитием энергохимического
использования твердых топлив разрабатываются новые приемы их
полукоксования. В энергохимических установках
полукоксование является одной из стадий процесса переработки угля. Перед
сжиганием из топлива отгоняются все содержащиеся в нем ценные
химические продукты. Обычно в стадии полукоксования
производится внутренний обогрев топлива газообразным или твердым
теплоносителем и процесс осуществляется скоростными методам!-;
в аппаратуре большой производительности.
104
ИСПОЛЬЗОВАНИЕ И ПЕРЕРАБОТКА
ПРОДУКТОВ ПОЛУКОКСОВАНИЯ
Состав и выход продуктов полукоксования зависят в
основном от качества исходного сырья.
Получаемый в этом процессе кусковой полукокс является
товарным продуктом. Мелкий полукокс обычно сжигают или
подвергают газификации на месте его получения. Жидкие продукты
(деготь и газовый бензин) перерабатывают на моторные топлива
и другие продукты. Газ большей частью используется на месте
как высококалорийное топливо.
Из водного конденсата обычно извлекают только
растворенные в нем фенолы, после чего конденсат сливают в промышленную
канализацию, предварительно очистив сточные воды от вредных
примесей.
Первичный деготь перерабатывают преимущественно для
получения погонов, по составу примерно соответствующих
нефтепродуктам—бензину, керосину, дизельному топливу. Выход
моторных топлив из первичных дегтей обычно невелик, кроме того,
эти топлива отличаются высоким содержанием непредельных
соединений и потому мало стабильны. Для увеличения выхода
ценных продуктов первичные дегти подвергают крекингу (стр. 56)
или гидрогенизации (стр. 114). Летучие продукты полукоксования
содержат значительное количество фенолов и могут служить
существенным источником их получения.
Глава III
ГАЗИФИКАЦИЯ И ГИДРОГЕНИЗАЦИЯ ТОПЛИВ
При промышленном использовании горючих в качестве топ-
лив и особенно в качестве химического сырья для синтезов
зачастую возникает необходимость в более глубокой химической
переработке топлива. Такая глубокая переработка обычно
предпринимается для полного превращения всей горючей массы
исходного топливного сырья в новый, существенно отличающийся
от этого сырья продукт с заданными качествами. Применение этого
вида переработки особенно важно в странах, богатых, например,
твердым топливом, но не имеющих собственных запасов нефти или
природного газа.
Направление такой глубокой химической переработки при
промышленном облагораживании топлив можно представить
следующей схемой:
Твердые топлива » ЖиДкие топлива » Горючке газы
При использовании природных горючих в качестве химического
сырья целью их переработки является превращение сложных
веществ природного' горючего в простые химические соединзния,
которые затем служат исходными веществами в процессах синтеза
или реагентами (например, СО, Н2) для обработки других веществ.
Очевидно, указанные превращения всей органической горючей
массы более обуглероженного топлива в простые менее обуглеро-
женные соединения не могут быть осуществлены с участием
только тех веществ, которые входят в состав перерабатываемого
исходного топлива. Для таких превращений необходимо
взаимодействие топлива с каким-либо реагентом. В качестве подобных
реагентов при глубокой переработке топлив обычно используют
кислород и водород.
Неполное окисление топлива применяется в процессах
газификации, приводящих к получению из углерода твердых и тяже-
106
лых жидких топлив горючего газа—окиси углерода, которая
служит газообразным топливом и ценным химическим сырьем.
Присоединение водорода к топливам (обычно твердым или
тяжелым жидким) происходит в процессах деструктивной
гидрогенизации топлива и связано с его одновременным разрушением
и образованием гораздо более простых соединений. В результате
таких превращений обычно получаются смеси легкокипящих
жидких углеводородов, используемых далее как легкое моторное
топливо или химическое сырье.
Разнообразные химические превращения индивидуальных
углеводородов, подобные описанным выше, широко используются в
промышленности органического синтеза и рассматриваются в
главе IV. Эти процессы глубокого химического воздействия на сырье
при переработке топлив протекают очень сложно и, как правило,
не могут быть описаны одной элементарной реакцией; возможно
показать лишь общее направление химических изменений
перерабатываемого сырья. Поскольку химическая сущность этих
процессов однотипна для всех видов топлива, их можно
рассматривать в обобщенном виде для различного сырья. Аппаратурное
же оформление весьма разнообразно, так как конструкция
применяемых аппаратов зависит от агрегатного состояния
перерабатываемого сырья, его состава, зольности и других свойств.
/. ГАЗИФИКАЦИЯ ТОПЛИВ
Как указывалось, газификация топлива заключается в
неполном его окислении. Процесс проводится при температуре выше
700 °С и протекает с выделением тепла:
2С + О2 = 2СО + 52,8 ккал (-220 кдж)
Наряду с этим процессом происходит тепловое разложение
органического вещества топлива и взаимодействие кислорода и
водяных паров с продуктами разложения; в результате
образуются СО2, СО и Н2. В зависимости от свойств и состава топлива,
подвергаемого газификации, от характера окислительной среды
(воздух, обогащенный кислородом; смесь воздуха с водяным паром;
водяной пар или просто воздушное дутье) и от того, все ли
органические «осколки» топлива подверглись воздействию кислорода,
состав и, следовательно, свойства получаемых газов могут быть
весьма различны.
Частичное окисление углеводородных газов (природного и др.)
протекает по реакции: J
2CH4:+P;,W4H24-2GO+ 16>06 ккал (~67 кдж)
Цель процесса—получение водорода или газа для синтезов
на основе СО и Н2 (стр. 162). Этот и другие процессы конверсии
107
углеводородных газов подроинее рассмотрены в курсе
химической технологии неорганических веществ*.
Газификацию жидких топлив ведут, когда это требуется, так,
чтобы оставалась незатронутой большая доля углеводородных
газов, образовавшихся при расщеплении исходного тяжелого
нефтяного сырья. Таким способом удается получить из этого
сырья бытовой газ с теплотворной способностью 4000—4200 ккал1мг
(~17 Мдж/м3) .
Наиболее сложно аппаратурное оформление газификации
твердых топлив.
Газогенераторы. Газификацию твердых топлив производят в
аппаратах, называемых газогенераторами. Газогенератор
распространенного типа (рис. 37) представляет собой вертикальную
шахту, в которую сверху загружают топливо, а снизу подводят дутье—
воздух или другой газифицирующий агент (паро-воздушная смесь,
пар, паро-кислородная смесь). В шахте генератора топливо
постепенно опускается вниз, проходя зоны /—IV подсушки, перегонки,
газификации (восстановления) и горения. Снизу из шахты
выгружают шлак—остаток после полной газификации топлива. Весь
процесс протекает непрерывно.
Кислород, поступающий в генератор с дутьем, в зоне горения
полностью сжигает топливо с образованием двуокиси углерода:
С + О2 = СО2 + 94 ккал C94 кдж)
Двуокись углерода поднимается в зону восстановления и там
восстанавливается углем до окиси углерода:
— 41,2 ккал (-173 кдж)
Если газификация протекает при пониженных температурах
в получаемой газовой смеси преобладает двуокись углерода:
повышение температуры ведет к образованию больших количеств
окиси углерода.
На рис. 38 приведены кривые, характеризующие изменение
состава газа в зависимости от температуры процесса газификации.
Шахта газогенератора (см. рис. 37) обычно цилиндрическая и
перекрыта сводом с отверстием для загрузки угля. Свод и стенки
шахты сложены из огнеупорного кирпича (футеровка 2) и
заключены в металлический кожух /. Для использования тепла,
выделяющегося при газификации, нижняя часть шахты может быть
снабжена водяной рубашкой 3, служащей парообразователем.
Днище газогенератора—колосниковая решетка 4—служи і
опорой для топлива и шлака; через нее же подается и
распределяется дутье и удаляется шлак. Современные газогенераторы
имеют колосниковые решетки, медленно вращающиеся @,1 — 1 обіч)
* См. сноску на стр. 95.
108
Топливо
Рис. 37. Газогенератор с вращающейся колосниковой решеткой:
/—зона подсушки: //—зона перегонки; ///—зона газификации; IV—зона горения; /—кожух:
5—футеровка; 3- водяная руб.ішка (котел); 4—колосников/ я решетка; 5—«юбка» генератора;
в—чаша; 7—червячный привод; 8—гидравлический затвор: 9- механический питатель;
10—вращающаяся звездочка; //—секторный затвор; 12— привод питания; 13— шу ровочные отверстия.
too
во
60
20
О
ш
4
\
co2
У
X
/¦
\
\
4
CO
600 800
Температура, °С
1000
шахты газогенератора, благодаря чему создается гидравлический
затвор, препятствующий выходу газа наружу. Колосниковая
решетка с чашей приводится во вращение электродвигателем
через червячный привод 7. Спиральные ребра, имеющиеся на
решетке, передвигают шлак от центра газогенератора к стенкам и
сбрасывают его в чашу. Из вращающейся чаши шлак выгребается
неподвижно укрепленной лопатой.
Дутье подается по трубе под колосниковую решетку и через
ее щели поступает к газифицируемому топливу. Неподвижная
труба соединена с решеткой
и чашей посредством
гидравлического затвора 8.
Полученный при газификации газ
поступает в газоотводную трубу.
В генераторы различных
систем топливо загружается
периодически или
непрерывно. В генератор
описываемого типа загрузка
производится механическим
питателем 9 с секторным
затвором // и вращающейся
звездочкой 10, обеспечивающей
равномерную подачу
топлива. Такой генератор имеет
диаметр шахты около 2,8 м,
высоту шахты около 3,5 м,
площадь колосниковой решетки 4,5 м2. Производительность
его при работе на угле достигает 4500 ма/ч газа.
г1 Существует много разнообразных систем газогенераторов—
от самых простых и малопроизводительных до полностью
механизированных аппаратов; конструкция генераторов зависит и от
типа газифицируемого топлива.
Газ, получаемый при воздушном дутье, называется
воздушным газом. Он содержит около 33% окиси углерода, остальное—
азот. Тепло реакции в этом процессе теряется.
Если в генератор вместе с воздухом подводится пар, то наряду
с воздушным газом одновременно за счет выделяющегося при
этой реакции тепла образуется и водяной газ:
С + Н2О (пар) = СО + Н2 — 31,4 ккал A33 кдж)
Получается смесь газов, называемая смешанным
генераторным газом или просто генераторным газом. Теплотворная
способность его выше, чем воздушного газа.
Характеристики различных генераторных газов приведены
в табл. 12.
Рис. 38. Зависимость содержания СО
и СОг в равновесной газовой смеси
от температуры.
110
Таблица 12
Свойства генераторных газов
Газы
Воздушный . .
Смешанный
генераторный .
Водяной ....
Паро-кислород-
ный
Паро-кислородный,
полученный под
давлением . . .
Газ подземной
газификации
Газифицирующий
агент
(дутье)
Воздух
Воздух+пар
Пар
Кислород 4-
-1-пар'
Кислород+
+пар
Воздух
Преобладающая
реакция*
1
1 и 2
2
1 и 2
1, 2,3
1
Содержаріие основных
компонентов газа, объемн. %
СО
33
26
40
30
20
15
н2
14
50
40
55
15
СН4
4
18
балласт
65
55
25
4
70
Низшая
теплотворная
способность
газа
ккал '.и3
900—1100
1200—1500
2400—2700
2300—2500
3600—3800
800—1100
Реакции обозначены следующими цифрами:
1. 2С + О2 . > 2СО + 52,8 ккал
2. С + №О » СО +Н2-ЗЫ ккал
3. С + 2Н2 > CH4 + 18 ккал
ПОЛУЧЕНИЕ ВОДЯНОГО ГАЗА
Для получения чистого водяного газа, используемого,
например, для синтезов (стр. 162), в генератор надо подавать
водяной пар без воздуха. В этом случае тепло, необходимое для
разложения пара, подводят в зону газификации путем внешнего
обогрева шахты газогенератора, или сильно перегревают подаваемый
в газогенератор водяной пар, или же накаливают газифицируемое
топливо, продувая его воздухом. Наиболее распространен
последний способ, по которому процесс газификации осуществляется
как периодический, циклами. Сначала в слой топлива вдувают
воздух (период горячего или воздушного дутья)—происходит
образование воздушного газа с выделением тепла, слой топлива при
этом накаляется. Когда топливо достаточно накалится,
прекращают воздушное дутье и начинают подачу водяного пара (период
холодного дутья или газования). Реакция образования водяного
газа протекает с поглощением тепла, вследствие чего слой топлива
охлаждается. Когда топливо охладится до определенного предела,
цикл начинается снова.
Периодические переключения дутья, изменения его направле-,
ния для равномерного нагревания и охлаждения слоя топлива в
m
генераторе, а также необходимость раздельного отвода воздушного
и водяного газов усложняют систему трубопроводов на установ
ках получения водяного газа. Схема такой установки представлена
на рис. 39.
В генератор /, снабженный водяной рубашкой 2 с паросбор
ником 3, подаются попеременно воздух и пар. Образующиеся в
генераторе горячие газы последовательно проходят регенератор 6
с кирпичной насадкой, пароперегреватель 7 и котел-утилизатор 8
затем поступают в стояк 9. Далее газ в период газования через
водяной
газ
11 ^
Рис. 39. Схемы установки для получения водяного газа:
/—генератор; 2—водяная руб.ішка; 3—паросборник; 4—воздуходувка; 5—топка регенератора;
5—регенератор; 7— л^рилеригревателн; 8—котел-утнлнзатор; 9—стояк; (й-гидравлический
затвор; 11—холодильник; 12—клапан; 13—дымовая труба; а—д—задвижки.
гидравлический затвор 10 и холодильник 11 направляется в линию
водяного газа, а в период горячего дутья отводится через клапан 12
а дымовую трубу 13. Пар из котла-утилизатора, имеющий
давление 17—19 am, подается потребителям, а пар из паросборника 3
используется в качестве парового дутья. Полный цикл работы
такой установки длится примерно 4 мин и состоит из шести
стадий: горячее дутье; продувка генератора паром для удаления
воздушного газа; газование с подачей пара снизу; газование с
подачей пара сверху; заполнение нижней части генератора паром,
(чтобы избежать взрыва в начале подачи воздуха);
предварительная продувка воздухом. На каждой стадии, путем соответствующих
переключений задвижек, газовые потоки в установке получают
определенное направление.
112
Свойства водяного и других генераторных газов зависят
не только от условий ведения процесса, но и от состава
топлива, подвергавшегося газификации. Так, например, большое
количество балластных газов (CO2+H.2S) в паро-кислородном газе
(см. табл. 12) обусловлено тем, что он получается из
низкосортных топлив.
Коэффициент полезного действия процесса газификации,
измеряемый процентным отношением теплотворной способности
полученного газа к теплотворной способности исходного топлива,
колеблется в зависимости от свойств перерабатываемого топлияа
и метода газификации. Примерные к. п. д. газификации кокса
и антрацита при получении различных газов:
Газ ы к. п. д.
%
Водяной 6 0
Воздушный 7 3
Смешанный .... 80
ДРУГИЕ СПОСОБЫ ГАЗИФИКАЦИИ
Процессы газификации непрерывно совершенствуются. Для
получения смешанного газа в газогенераторах стали применять
паро-кислородное дутье вместо паро-воздушного. Это позволило
увеличить подачу пара в генератор (и, следовательно, повысить
долю водяного газа в получаемом смешанном газе) и исключить
из состава получаемого газа азот—балластную примесь,
неизбежную при паро-воздушном дутье. Переход на паро-кислородное
дутье дал также возможность резко повысить теплотворную
способность генераторного газа (см. табл. 12), увеличить на 5—8%
к. п. д. газогенераторной установки и проводить газификацию как
непрерывный процесс благодаря одновременному протеканию
эндотермических реакций, требующих подвода тепла, и
экзотермических реакций, компенсирующих его расход.
Дальнейшее повьшение теплотворной способности газа
возможно путем применения в процессе газификации паро-кислород-
ного дутья, подаваемого под давлением до 20 am. В этих условиях
в генераторе образуется метан по суммарной реакции:
С + 2Н2 = СН4 + 18 ккал G5,5 кдж)
В действительности процесс протекает гораздо сложнее, по
нескольким реакциям—через окисление углерода топлива до
СО и СО2 и последующую гидрогенизацию их до метана, например:
СО + ЗН2 = СН4 + Н2О (пар) + 49 ккал (-206 кдж)
Давление способствует образованию метана, так как обе
реакции протекают со значительным уменьшением объема газа. В
таком же направлении на процесс влияет и понижение температуры,
поскольку он идет с выделением тепла.
8-805 ИЗ
В последние годы большое внимание уделяется разработке
конструкций генераторов, работающих на пылевидном и мелком
топливе, которое газифицируется во взвешенном состоянии в
струе направленного снизу вверх дутья. Газификация жидких
топлив проводится в присутствии катализаторов.
Особое место среди процессов газификации занимает способ
превращения угля в газ под землей, на месте его залегания. При
таком способе газификации шахтная добыча угля может быть
заменена непосредственным превращением его в газ. Подземная
газификация была задумана и впервые предложена Д. И.
Менделеевым в 1888 г. Идея подземной газификации угля получила
высокую оценку В. И. Ленина. Этот способ может значительно
облегчить использование угольных богатств и дать
возможность извлекать из-под земли только горючую часть топлива,
оставляя там балласт. Подземную газификацию начали применять
и как способ извлечения из пластов остающейся в них нефти.
2. ГИДРОГЕНИЗАЦИЯ ТОПЛИВ
Обогащая тяжелое жидкое или твердое топливо водородом,
удается превратить его в смесь легкокипящих углеводородов
со значительно более низким средним молекулярным весом, чем
На 100кг С в топливе содержится:
О, N, S, кг
Н,кг
Бурый уголь
Газовый уголь
Коксовый уголь
Коксовая смола
Первичный деаоть
Мазут
Сырая нефть
Легкая ароматика
Керосин
Бензин
Рис. 40. Состав органической массы различных горючих
(в кг на 100 кг углерода).
для исходного сырья. О количестве водорода, которое должно
быть добавлено к сырью для таких превращений, можно судить
по диаграмме, представленной на рис. 40. На этой диаграмме пока-
114
зано содержание водорода и других элементов (в кг) на каждые
100 кг углерода в различных горючих. Стрелками обозначены
направления промышленной переработки различных тяжелых
топлив путем деструктивной гидрогенизации в более легкие
продукты.
Процессы деструктивной гидрогенизации подразделяются на
два типа*.
По одному из них процесс гидрогенизации ведут до
образования из сырья легких насыщенных углеводородов (типа бензина);
цель этого процесса—получение из тяжелого углеводородного
сырья моторных топлив (бензинов, дизельного топлива и др.)
и химического сырья.
В процессе второго типа из углей получают важнейшие виды
химического сырья—легкие ароматические углеводороды,
пиридиновые основания и фенолы**. В основу решения этой задачи
положено то обстоятельство, что вещество угля представляет
собой скопление высокомолекулярных образований (мицелл),
имеющих углеродный решетчатый скелет в виде
шестичленных колец (структура, подобная графиту). При разрушении
(деструкции) такой решетки и гидрогенизации по месту
освобождающихся связей из углеродных шестичленных колец образуются
ароматические соединения, из колец, содержащих азот,
кислород или серу,—гетероциклические соединения. Из колец, имеющих
присоединенные гидроксильные группы, образуются фенолы.
Расщепление молекул углеводорода при деструктивной
гидрогенизации всегда сопровождается присоединением атомов
водорода по месту разрыва цепи:
R'-CHa-CH2-R" + Н2 » R'-СНз + R"-CH3
где R' и R" —углеводородные радикалы.
Если в гидрируемом соединении имеются двойные связи, то
в результате гидрогенизации сначала образуется насыщенное
соединение.
Гидрогенизация соединений ряда нафтенов начинается с
разрыва кольца.
При гидрогенизации ароматических циклов сначала
присоединяется водород, т. е. происходит превращение ароматических
соединений в нафтены, а затем уже разрываются кольца. В
многоядерных соединениях кольца гидрируются, разрываются и отде-
* Менее глубокие процессы гидроочистки и гидроформинга (стр. 66)
здесь не рассматриваются.
** В этом процессе в ароматическое сырье превращается почти вся
органическая масса угля, при коксовании-^примерно 1,5% ее.
8* 115
генизации нафталина:
СН3
+Но.
ч/сн2
сн2
+н2
сн3—сн2—сн2—сн3
При гидрогенизации гетероциклов, содержащих азот, серу,
кислород, водород присоединяется по месту двойных связей,
после чего происходит разрыв кольца и образование
соответственно аммиака, сероводорода и воды. Эти же соединения
образуются из перечисленных элементов на более ранних стадиях
гидрирования, если азот, сера и кислород не входят в гетеро-
цикл, а присутствуют в боковой цепи.
Все процессы гидрогенизации протекают со значительным
уменьшением обьема реакционной среды и потому
интенсифицируются при повышении давления. В промышленности они
осуществляются под давлением водорода до 700 am и выше. Кроме того,
давление способствует лучшему растворению водорода в
жидком гидрируемом сырье и улучшает проникание Н2 к
гидрируемым соединениям. Легко гидрируемое сырье можно обрабатывать
и при меньших давлениях. Применяемая температура должна
быть такой, чтобы обеспечивалось расщепление гидрируемых
веществ на активные осколки (остатки молекул). В зависимости
от условий процесса гидрогенизацию обычно ведут при
температуре 350—500 °С. Ускорение процесса достигается в присутствии
катализаторов.
Основная доля расходов в процессе гидрогенизации относится
на стоимость водорода, затраты на создание требуемого
давления, а также на стоимость катализатора и сложного
оборудования. Особенно сложно оформление процесса гидрогенизации
углей, поскольку в этом случае необходим ввод в аппаратуру,
работающую под давлением, твердого вещества—молотого угля
и отвод из аппаратов шлама (золы), с которым обычно теряется
и катализатор. Все эти трудности приходится учитывать при
выборе сырья, пригодного для гидрогенизации. На стадии терми
ческой деструкции оно должно легко образовывать реакционно-
способные продукты расщепления; кроме того, в сырье должно
содержаться минимум золы и возможно меньше кислорода, серь:
и азота, на связывание которых в процессе гидрогенизации будет
непроичводительно расходоваться водород; необходимо также
как можно более высокое соотношение концентраций водорода
и углерода в сырье A00Н : С должно быть не менее 6,5). В этом
116
отношении лучшим сырьем для гидрогенизации являются тяжелые
нефтяные остатки (остатки крекинга, мазут), молодые угли и угли
сапропелевого происхождения. Бурые угли, хотя и содержат
довольно много водорода (см. рис. 40), но вследствие высокого
содержания кислорода и других балластных элементов зачастую
непригодны для гидрогенизации.
В промышленности процесс деструктивной гидрогенизации
проводится в несколько стадий. Первой стадией гидрогенизации
угля является его подготовка—сушка, размол, замешивание
с циркуляционным (затирочным) маслом до получения пасты,
легко перекачиваемой специальными насосами, и смешение с
катализатором. Далее для угольной пасты (и для тяжелых
нефтяных остатков) следует первая ступень гидрогенизации в жидкой
Нг+еазы
__ - Сырье +
'катализатор
1 ОстатН
Рис. 41. Упрощенная схема гидрогенизации нефтяных
остатков:
/—реактор первой ступени: 2—горячие сепараторы; 3—реактор второй
ступени: 4—ректификационная колонна.
фазе в присутствии суспендированного катализатора. В этой
стадии процесса горючая масса топлива превращается
преимущественно в легко испаряемые вещества, что создает возможность их
последующей переработки в паровой фазе. В первой ступени
получаются, кроме того, небольшие количества бензина и горючего
газа, являющегося в данном случае малоценным, продуктом
(выход его стремятся сократить). В качестве отхода образуется
тяжелый остаток, вместе с которым из цикла отводятся зола,
катализатор и др.
Содержащееся в остатке масло может быть отделено от него
и возвращено на гидрогенизацию. При использовании углей в ка-
117
Катализатором первой ступени процесса гидрогенизации обычно
являются соединения железа; они недороги, но активность их
невысока.
Жидкофазный гидрогенизат поступает на окончательную
гидрогенизацию во вторую парофазную ступень. Процесс
осуществляется в стационарном слое активного катализатора,
размещенного на полках реактора. Катализаторами второй ступени
обычно являются сульфиды вольфрама или молибдена на носителе—
окиси алюминия. Парофазная гидрогенизация зачастую
подразделяется на две стадии—предварительное гидрирование и бензи-
нирование.
Упрощенная схема двухступенчатой гидрогенизации
нефтяных остатков с получением бензина приведена на рис. 41.
Схемы процессов гидрогенизации и их аппаратурное
оформление могут быть весьма различны в зависимости от характера
гидрируемого сырья и конечных продуктов его переработки. Вся
аппаратура, используемая в процессе гидрогенизации, работает
под высоким давлением и сходна с аппаратами, применяемыми
в промышленном синтезе аммиака.
ЛИТЕРАТУРА К ЧАСТИ ПЕРВОЙ
ГГ^а рхоменко В. Е., Технология переработки нефти и газа, Гостоптех-
издат, 1959.
Раковский В. Е., Общая химическая технология торфа, Госэнергоиздат,
1949.
Караваев Н. М., Пильский И. Я., Шепелев И. Г., Машины
и аппараты коксохимического производства, Металлургиздат, 1955.
Коляндр Л. Я., Улавливание и переработка химических продуктов
коксования, Металлургиздат, 1962.
Гойхрах И. М., Пинягин Н. Б., Химия и технология
искусственного жидкого топлива, Гостоптехиздат, 1954.
Грош ев А. П., Технический анализ, Госхимиздат, 1958.
Часть вторая
ПРОМЫШЛЕННЫЙ ОРГАНИЧЕСКИЙ СИНТЕЗ
ВВЕДЕНИЕ
До середины XIX в. практика переработки органических
веществ не выходила за пределы извлечения из растительного и
животного сырья содержащихся в нем ценных продуктов
(например,- красителей, Сахаров, дубителей и др.). Для выделения их
использовались простейшие механические и тепловые процессы
обработки сырья: дробление, растворение, фильтрование, отжим,
выпаривание, перегонка и т. д. При получении спирта, уксусной
кислоты и некоторых других органических веществ
использовались биохимические процессы (в частности, брожение). Некоторые
органические продукты были выделены при термическом
разложении природного сырья. Так, при сухой перегонке древесины
наряду с древесным углем получали уксусную кислоту,
древесный спирт, деготь.
В начале XIX в. термическое разложение угля стали
использовать для производства светильного газа.
Условия для развития промышленного производства
синтетических органических веществ появились в середине XIX в.,
когда бурное развитие текстильной и других отраслей
промышленности вызвало увеличение спроса на ряд продуктов, ранее
получавшихся из растительного и животного сырья. Благодаря
развитию металлургии и связанного с ним увеличения производства
кокса одновременно была создана сырьевая база (смола, сырой
бензол), необходимая для синтеза органических продуктов.
Замечательные научные открытия Ф. Велера, Н. Н. Зинина,
А. М. Бутлерова, Ф. Кекуле, М. Вертело и других ученых
позволили решить практические задачи, связанные с организацией
первых производств органического синтеза. К этому времени в
ранее возникших отраслях химической промышленности,
производивших соду, серную кислоту и другие минеральные вещества,
уже был накоплен большой опыт конструирования различной
химической аппаратуры и проведения разнообразных химических
процессов.
119
Промышленность химической переработки минеральных
веществ была в состоянии обеспечить новые органические
производства необходимыми неорганическими реагентами, а в отраслях
промышленности, перерабатывающих природное сырье, были
хорошо освоены приемы работы с органическими продуктами.
Особое значение для развития промышленного органического
синтеза имела теория химического строения, созданная
А. М. Бутлеровым в 1861 г. и явившаяся основой современной
органической химии. Эта теория позволила выяснить строение
сложнейших органических веществ, а затем и синтезировать их из
более простых соединений.
На примере развития промышленного органического синтеза
хорошо видно, что каждая новая отрасль производства возникает
лишь тогда, когда для этого создаются необходимые
материальные предпосылки. Поэтому не случайно первыми органическими
продуктами, синтезированными в промышленном масштабе, были
красители, пользовавшиеся большим спросом в интенсивно
развивавшейся текстильной промышленности. Этим объясняется и
то, что из двух первых синтезов красителей, осуществленных в
1856 г. независимо друг от друга польским ученым Я. Натаисо-
ном и англичанином В. Перкиным, был реализован только
синтез Перкина, так как в промышленной Англии для этого имелись
необходимые условия. Вслед за производством красителей
возникают и другие производства органических продуктов
ароматического ряда—лекарственных, душистых, взрывчатых и других
веществ.
Второе десятилетие XX в. ознаменовалось дальнейшим
совершенствованием методов химической технологии—внедрением
процессов, проводимых с применением катализаторов, высоких
давлений и температур, глубокого холода, а также разработкой
непрерывных процессов.
В результате освоения этих новых методов и процессов
появилась возможность организовать промышленный синтез
метанола из окиси углерода и водорода под давлением, осуществить
производство искусственного жидкого топлива. В связи с
возрастающим использованием двигателей внутреннего сгорания стали
развиваться и процессы переработки нефти в моторные топлива
(крекинг) и связанные с ними производства, в которых продукты
переработки нефти используются как химическое сырье. На этой
основе возник и начал бурно развиваться промышленный
органический синтез соединений алифатического ряда.
Промышленное производство синтетических органических
продуктов (промышленный органический синтез), возникшее во
второй половине прошлого века, к настоящему времени достигло
уровня крупнейших отраслей мировой химической
промышленности. Промышленный органический синтез охватывает
производство сотен продуктов—от многотоннажных относительно про-
120
стых по строению веществ до очень сложных синтетических
органических препаратов, выпускаемых в сравнительно небольших
количествах.
К первой группе относятся такие продукты, как исходные
вещества (мономеры) для получения полимеров, из которых
изготовляются химические волокна, пластические массы, и мономеры
для синтеза разнообразных каучуков; растворители;
многотоннажные промежуточные продукты для различных процессов
органического синтеза; некоторые химические препараты,
используемые в сельском хозяйстве; антидетонаторы и многие другие
вещества.
Производство этих веществ условно объединяется в отрасль
химической промышленности, называемую промышленностью
основного органического синтеза. Важнейшие технологические
процессы этой отрасли промышленности описаны в главе IV.
К второй группе производств, также условно объединяемой
под названием промышленности тонкого органического синтеза,
относится производство синтетических красителей и некоторых
малотоннажных органических полупродуктов, производство
поверхностно-активных и вспомогательных веществ, химических
средств защиты растений; препаратов для резиновой, текстильной
и других отраслей промышленности; получение лекарственных
и душистых веществ и многих других. Технологические процессы,
используемые в этой отрасли промышленного органического
синтеза, рассматриваются в главе V. Однако четкое разграничение
промышленности органического синтеза по указанным
признакам затруднительно и в известной мере условно. Многие
продукты, производство которых ранее было начато в небольших
масштабах и могло быть отнесено к промышленности тонкого
органического синтеза, становятся теперь массовыми продуктами,
вырабатываемыми в огромных количествах. Различие обеих групп
промышленности органического синтеза в гораздо большей степени'
проявляется при рассмотрении применяемых в этих отраслях
методов производства синтетических продуктов.
В многотоннажных производствах основного органического,
синтеза применяются, как правило, непрерывные процессы с
ограниченным числом стадий. Производства тонкого органического
синтеза обычно меньше по масштабу, в них применяются
многостадийные процессы, осуществляемые чаще всего в аппаратуре
периодического действия. Особенностями промышленности
тонкого органического синтеза является также необходимость
применения исходных индивидуальных веществ высокой чистоты и
возможность получения разнообразных конечных продуктов
сложного состава из сравнительно немногих продуктов более простой
химической переработки—так называемых полупродуктов.
Поэтому структура ряда отраслей тонкого органического синтеза
(например, производства красителей, многих лекарственных,
121
-- S--^- ¦- ---!-.=- --r .
дующем виде:
Производство углеводородного сырья из топлив
I
Производство полупродуктов
1
Производство конечных продуктов
В возникшей позднее промышленности основного
органического синтеза (производство алифатических соединений) такого
резкого разграничения производственных стадий провести нельзя.
С развитием промышленного органического синтеза постепенно
стираются грани и между процессами производства соединений
алифатического и ароматического рядов. Такие важные продукты,
как, например, стирол, гексахлоран, ДДТ, изопропилбензол,
являются жирно-ароматическими соединениями.
На первых ступенях переработки углеводородного сырья,
т. е. главным образом в процессах основного органического
синтеза, используется сравнительно ограниченное число химических
реакций, основными из которых являются термическое
разложение, гидрирование и дегидрирование, гидратация, окисление,
галоидирование и гидрогалоидирование, нитрование и др. Для
получения конечных веществ из продуктов первичной переработки
сырья требуется их дополнительная обработка, в процессе
которой кроме перечисленных выше реакций используются реакции
этерификации, конденсации и т. д. Широко применяются также
методы взаимного превращения углеводородов как в пределах
одного гомологического ряда, так и с переходом их из одного
гомологического ряда в другой. Для таких превращений
используются реакции изомеризации, алкилирования, полимеризации.
Комбинируя применяемые методы и реакции и используемое в них
сырье, осуществляют промышленные синтезы самых
разнообразных продуктов.
Проводя различные химические процессы, можно получить
один и тот же продукт из разных видов органического сырья
или различные продукты одинаковыми методами.
Число органических продуктов, получаемых синтетическими
методами, непрерывно увеличивается. Промышленность
органического синтеза сейчас уже далеко переросла рамки производства
заменителей природных веществ. Современный уровень развития
химической технологии позволяет получать синтетические
материалы с заранее заданными ценными свойствами, иногда такими,
каких не имеют природные вещества.
В настоящее время особенно интенсивно развиваются новые
отрасли химической промышленности, например производство
122
полимерных материалов—синтетических каучуков, пластических
масс, химических волокон, пленок и др. Полимерные материалы
и изделия из них благодаря ценным специфическим свойствам
приобрели огромное значение в современной технике.
Методы получения и переработки полимеров в готовые
изделия также имеют свою специфику, в связи с чем производство
полимеров выделилось в самостоятельную отрасль технологии
органических веществ. В данном учебном пособии технология
высокомолекулярных соединений рассматривается в отдельной
третьей части книги.
Исключительно быстрое развитие промышленности
органического синтеза можно видеть из следующих примеров. За
последние тридцать лет вся химическая промышленность США выросла
примерно в три раза, а производство пластических масс за это
же время—более чем в 80 раз; к середине 50-х годов объем
производства пластических масс составлял 70% от общего количества
вырабатываемых цветных металлов (алюминия, меди, цинка,
свинца, олова).
Еще более ускоренными темпами развивается отечественная
химическая промышленность, особенно производство продуктов
органического синтеза и полимеров. Решения Партии и
Правительства о резком увеличении выпуска химической продукции
успешно выполняются. За последние годы в нашей стране весьма
значительно увеличился и продолжает возрастать объем
производства минеральных удобрений, пестицидов, пластических масс,
химических волокон, автомобильных шин, лакокрасочных и
пленочных материалов, синтетического спирта, синтетических
моющих средств и других химических продуктов.
Глава IV
ТЕХНОЛОГИЯ ОСНОВНОГО ОРГАНИЧЕСКОГО СИНТЕЗА
/. ПРОДУКТЫ ПРОМЫШЛЕННОСТИ ОСНОВНОГО
ОРГАНИЧЕСКОГО СИНТЕЗА
Промышленность основного органического синтеза в больших
масштабах производит разнообразные химические продукты,
причем число их и объем выработки возрастают с каждым годом. Эти
продукты находят широкое применение в различных отраслях
народного хозяйства.
Так, например, фенол используется в качестве исходного
продукта в производстве феноло-формальдегидных смол (является
основой некоторых видов пластических масс), капролактама
(сырье для получения синтетического волокна—капрона),
фармацевтических препаратов (аспирин, салол, салициловая кислота),
душистых веществ, синтетических дубителей и др.
Этиловый спирт служит полупродуктом для производства
синтетического каучука, распространенного инсектицида ДДТ (ди-
хлордифенилтрихлорметилметан), является хорошим
растворителем и т. д.
Уксусную кислоту перерабатывают в уксусный ангидрид
(используемый для получения ацетатного волокна), в 2,4-дихлор-
феноксиуксусную кислоту (гербицид 2,4-Д), уксусная кислота
также служит полупродуктом в производстве пигментов.
Ацетон является промежуточным продуктом в производстве
органического стекла (полиметилметакрилат) и широко
применяется в качестве растворителя.
Ниже приводятся основные группы продуктов основного оргя
нического синтеза.
Мономеры. Мономеры—простые органические вещества, из
которых путем полимеризации или пол и конденсации получают
синтетические смолы, каучуки и другие высокомолекулярные
124
продукты. Эти синтетические полимеры применяются для
производства резиновых изделий, пластических масс, синтетических
текстильных волокон, лакокрасочных изделий, пленочных
материалов (стр. 384 ел.).
В качестве мономеров используются органические соединения
многих классов—углеводороды, их хлор- и фторпроизводные.
альдегиды, органические кислоты, нитрилы, лактамы, лактоны
и др.
Важнейшие мономеры, перерабатываемое в
высокомолекулярные соединения методами полимеризации и сополимеризации:
Этилен . .
Пропилен
Изобутилен
. . .СНо=СН2
. СН2=СН-СН3
. СН2=СНС1
Хлористый винил .
Винилиденчлорид СН2=СС1а
Тетрафторэтилен CFi=CF2
Бутадиен-1,3 . . СН2=СН-СН=СН2
(дивинил)
2-Метилбутадиен-1,3 |
(изопрен) СН2=С—СН=СН2
С1
2-Хлорбутадиен-1,3 |
(хлоропрен) .... СН2—С—СН=СН2
Винилбензол (стирол) . СвН6—СН=СН2
СНа
а-Метилстирол СвН6—С=СН2
Акриловая кислота . СН2=СН—СООН
Метиловый эфир
метакриловой
кислоты
(метилметакрилат) . .
Акрилонитрил .
Винилацетат . .
а-Винилпиридин .
Капролактам .
СНа
. СН2=С—COOCHj
. . .CH2=CH-CN
. СН2=СНООССН3
ЯП
1>с(СН"СН'1Н'
ЧЮ-NH-CH,,
Ниже приведены мономеры, перерабатываемые в
высокомолекулярные соединения методом поликонденсации:
Первый мономер
Адипиновая кислота
НООС—(СНіL—СООН
Диизоцианат
O=CN-R—NC=O
Диметиловый эфир терефта-
й
левой кислоты
СНзООС-
-СООСНз
Дихлордиэтиловый эфир
¦С1СН2-СН2 —О—С На- СН2С1
Второй мономер
Гексаметилендиамин
H2N—СН2—(СН2L—СН2—NH2
Этиленгликоль
НОСНі—СН..ОН
Этиленгликоль
НОСН2-СН2ОН
Полисульфид натрия
Na2S4
Полимер
Найлон
(синтетическое
волокно)
Полиуретановый
каучук
Лавсан
(синтетическое волокно'
Тиокол
(полисульфидный
каучук)
125
Глицерин
НОСН2—СНОН—СН2ОН
Фенол
C,HSOH
Карбамид
/NH2
с=о
Фталевый ангидрид
СО
свн
Формальдегид
неон
Формальдегид
неон
со
Глиф' алевые
смолы (для
производства лаков)
Феноло-формаль-
дегидные смолы
(для
производства пластмасс)
Карбамидо-форм-
альдегидные
смолы (для
производства
пластмасс)
Растворители. Органические растворители применяются в
качестве реакционной среды во многих технологических
процессах, как экстрагенты, а также для изготовления лаков и т. д.
В качестве растворителей используются углеводороды, спирты,
кетоны, кислоты, простые и сложные эфиры, галоидорганические
соединения.
Наиболее распространенными органическими растворителями
являются:
Алкилацетаты . . .
Ацетон
Бензин
Диметилформамид .
Диоксан
Дихлорэтан ....
Метиловый спирт
. . .СН3—COOR
. .CIV-CO-CHa
. . . HCON/
nch3
О
/\
Н2С СН2
I
Н2С СН2
\/
О
. . ан2с—снгсі
сн,он
Метиленхлорид .
Метилэтилкетон .
Трихлорэтилен . .
Уксусная кисло-іа
Хлорбензол . . .
Циклогексанол
Четырех хлористый
Этиловый спирт .
Зтнленгликолі. - -
¦ . - СН2С12
. СН3—СО—С2Н3
.... СНС1=СС12
. . сн3соон
. . .С6Н5С1
. .с,нпон
углерод . . . СС14
СоН5ОН
. HOCH,—СН.2ОН
В экстракционной технике органические растворители служат
для извлечения различных компонентов из смесей твердых или
жидких веществ. Из подсолнечных, льняных, хлопковых и других
126
растительных семян органические растворители эффективно
извлекают масла, из бурых углей—весьма ценный воск (монтан-
воск) и т. д.
В нефтяной и нефтехимической промышленности при помощи
растворителей из нефтепродуктов извлекают ароматические и
непредельные соединения (стр. 160). Из легких масляных фракций
выделяют твердый парафин—смесь высших парафиновых
углеводородов нормального строения; из тяжелых мазутов—церезин
и вазелин, являющиеся смесями твердых парафинов, главным
образом разветвленных.
В ряде случаев действие растворителей основано на
избирательном извлечении отдельных компонентов смеси, поэтому
такие растворители называют селективными (или избирательными).
Легколетучие органические растворители используются в
лакокрасочной промышленности для растворения
пленкообразующих веществ. Они быстро удаляются в процессе образования
пленки, их можно улавливать и снова использовать (рекуперация
летучих растворителей).
Нелетучие растворители, применяемые для лакокрасочных
материалов, сохраняются в пленке после ее образования и
сообщают ей пластичность; они называются пластификаторами.
Общие требования, предъявляемые к растворителям: высокая
растворяющая способность, большая селективность, значительное
различие температур кипения растворителя и обрабатываемого
вещества, достаточная термическая устойчивость и стабильность
к окислению, отсутствие коррозионного действия на аппараты и
трубопроводы, минимальная токсичность. Кроме того, при выборе
растворителя учитываются степень его горючести и взрывоопас-
ности, доступность и стоимость.
Исключительно большое значение для повышения
продуктивности сельского хозяйства и сокращения затрат труда на уход
за посевами имеют органические пестициды—химические средства
защиты растений, гербициды, стимуляторы роста (стр. 335). Их
ассортимент и масштабы производства непрерывно увеличиваются.
Среди органических препаратов, применяемых для повышения
урожайности, большое место занимают хлор- и фторпроизводные
ароматических, алициклических и алифатических углеводородов,
фосфорорганические соединения, различные эфиры и пр.
Синтетические моющие средства. Большое развитие приобрело
производство разнообразных синтетических моющих средств
(детергентов). Эти препараты, получаемые из продуктов нефте- и
газоперерабатывающей промышленности, во многих отношениях
превосходят обычные жировые мыла, получаемые из
растительных и животных жиров. Синтетические моющие средства,
используемые в разнообразных отраслях промышленности и в быту,
проявляют моющее действие в нейтральной, кислой и в жесткой
воде. Они не гидролизуются, их водные растворы в ряде случаев
127
v-ip имічпт hip unuunu пряКГІИИ ЧТП Плягпппиятнп гкачипяртса яя
ности обрабатываемых шелковых и шерстяных тканей.
Технология синтетических моющих средств рассматривается
в главе V (стр. 328 ел.).
Антидетонаторы и высокооктановые добавки. Благодаря при
менению антидетонаторов и высокооктановых добавок к
моторному топливу возрастает экономичность работы двигателей
внутреннего сгорания (стр. 49).
Наиболее распространенным антидетонатором моторного
топлива для автомобилей является тетраэтилсвинец РЬ(С2Н5)+
(стр. 181), вводимый в бензин в виде так называемой этиловой
жидкости. В ее состав входит 50—60% тетраэтилсвинца, а также
галоидные соединения (дибромэтан, дихлорэтан, хлорнафталин).
Последние при сгорании смеси в цилиндрах двигателя образуют
галоидоводородные кислоты, взаимодействующие с окисью свинца
(продукт сгорания тетраэтилсвинца). При этом выделяются
летучие бромид или хлорид свинца, удаляемые с выхлопными газами.
Таким образом предотвращается осаждение окиси свинца на
стенках цилиндров двигателя и уменьшается их износ. Чтобы отличить
бензин с добавкой тетраэтилсвинца*, в состав этиловой жидкости
вводят растворимый в бензине краситель судан.
Добавление 2—4 мл этиловой жидкости к 1 кг бензина резко
улучшает его антидетонационные свойства (октановые числа
повышаются на 20—22 единицы). Это особенно важно для бензинов,
состоящих преимущественно из парафиновых углеводородов с
прямой цепью.
В качестве высокооктановых добавок используются изооктан,
изопентан, неогексан и некоторые другие разветвленные
парафиновые углеводороды, этилбензол, кумол и др. Синтетические
добавки вводятся в бензины в количествах, достигающих 30—40%,
и потому они являются компонентами моторного топлива.
Увеличение октановых чисел бензинов позволяет повышать степень
сжатия е горючей смеси в цилиндрах двигателя, что в свою очередь значительно
повышает его экономичность:
?=5,25 6=8,0 е=1О.З
Сравнительная длина пробега на одном и том же
количестве бензина при скорости 32 км/ч . . . 100 158 179
Некоторые продукты основного органического синтеза
используются в качестве смазочных материалов для двигателей и
механизмов, работающих как при очень низких (—60 °С), так и при
высоких температурах C00 °С и выше). Для этих целей
применяют некоторые сложные эфиры многоатомных спиртов и
двухосновных карбоновых кислот, низкомолекулярные продукты
полимеризации этилена и других ненасыщенных соединений, кремний-
* Тетраэтилсвинец является вредным ядовитым веществом. Он может
попадать в организм человека через кожу.
128
органические соединения, фтор- и хлорфторпроизводные
углеводородов.
Фреоны. Хлорфторуглеводороды, известные под названием
фреонов, в больших количествах используются в качестве хладо-
агентов в компрессионных и абсорбционных холодильных
установках. Как хладоагенты они обладают большими достоинствами.
В отличие от наиболее распространенного
хладоагента—аммиака фреоны практически не токсичны, не воспламеняются и не
образуют с воздухом взрывоопасных смесей. Наиболее безвредным
хладоагентом является дихлордифторметан CF2C12 (фреон-12),
который опасен лишь при концентрации в воздухе около 30%.
Фреоны используются также для получения фторкаучуков
(стр. 493) и других фторорганических соединений.
Антифризы. Некоторые органические жидкости,
замерзающие при низких температурах, так называемые антифризы, нашли
применение для охлаждения автомобильных и авиационных
двигателей и в качестве гидравлической жидкости в машинах,
работающих на холоду (для гидравлических приводов механизмов
экскаваторов, дорожных машин и т. д.). В качестве антифризов могут
применяться водные растворы метилового и этилового спиртов,
этиленгликоля, глицерина.
Смеси антифриза с водой (растворы) должны иметь температуру
кипения, не превышающую значительно температуру кипения
воды. Кроме того, к антифризам предъявляются следующие
требования: низкая температура замерзания, отсутствие запаха,
негорючесть, отсутствие коррозионного действия (не должен портить
отделку автомобиля). Водные растворы антифризов должны
обладать высокой теплоемкостью и низкой вязкостью.
Наиболее соответствует этим требованиям этиленгликоль
НОСН2—СН2ОН. Как можно видеть, температура замерзания
водных растворов этиленгликоля понижается пропорционально
его концентрации (до 66 объемн. %):
Содержание
этиленгликоля, объемн. % .... 25 30 35 40 55 66
Температура замерзания,
°С —9,8 —13 —16 —21 —40 —75
При температуре замерзания в жидкости могут появляться
кристаллики льда, и она становится несколько более вязкой.
Однако такой замерзающий раствор способен свободно
циркулировать по охлаждающим трубкам.
Высококипящие органические теплоносители. Если химический
процесс протекает при температурах выше 150—180 °С,
применение насыщенного водяного пара становится затруднительным,
так как его давление сильно растет (при 200 °С—около 16 am,
при 250 °С—40,5 am). Использование водяного пара таких
параметров усложняет конструкцию реакционных аппаратов из-за
9—805 129
НеООХОДИМОСТИ ЗНаЧИТеЛЬНОГО увеличения ИЛ ЛриЧНпсдИ. ііргіЛіс-
нение в качестве теплоносителя топочных газов не всегда
рационально, поскольку коэффициент теплоотдачи топочных газов
очень мал, возможны местные перегревы, затруднено точное
регулирование температур. Такой метод обогрева огнеопасен и
требует громоздкой аппаратуры. Нагревание электрическим током
также не всегда экономически выгодно, особенно в районах, где
отсутствуют дешевые источники электроэнергии.
В связи с этим большой интерес представляютвысококипящие
органические теплоносители (ВОТ), которые могут быть
использованы для подогрева до высоких температур. В качестве таких
теплоносителей применяют дифенил и его производные, дифе-
ниловый эфир, дитолилметан, глицерин, фтор- и кремнийоргани-
ческие соединения и т. д.
Наиболее распространенными высокотемпературными
теплоносителями являются эвтектическая и азеотропная смесь дифенила
и дифенилоксида (дифенильная смесь) и дитолилметан,
обладающие высокой температурой кипения и низким давлением
насыщенных паров:
_ Давление насыщенного пара, am
Температура
кипения дифенильная дитолилметан вода
С смесь
250 0,86 0,55 40,55
Q00 2,38 1,03 87,60
,50 5,31 2,25 168,6
Коэффициент теплоотдачи от конденсирующихся паров дифе-
нильной смеси в среднем составляет 1400—1500 ккал/ (м2-ч-град),
т. е. в несколько десятков раз больше коэффициента
теплоотдачи от топочных газов.
Недостатками дифенильной смеси как теплоносителя
являются ее горючесть и нестабильность при температурах 370—
380 °С. В настоящее время изыскивают новые органические
теплоносители, сохраняющие термическую стойкость при
температуре выше 400 °С.
Полупродукты. Промышленность основного органического
синтеза вырабатывает большое количество полупродуктов для других
отраслей химической промышленности—анилинокрасочной,
лакокрасочной, фармацевтической, витаминной, шинной,
резинотехнической, производства душистых веществ и т. д. К таким
полупродуктам относятся: анилин, хлорбензол, одноатомные
спирты, гликоли, глицерин, фенолы, ацетон и другие кетоны, карбо-
новые кислоты и их ангидриды, эфиры, нитросоединения,
нитрилы, аминосоединения и т. п.
В большинстве случаев к полупродуктам предъявляются
определенные требования в отношении чистоты, которая является
необходимым условием успешного проведения синтезов конечных
продуктов.
130
2. ИСХОДНЫЕ ВЕЩЕСТВА ДЛЯ ПРОМЫШЛЕННЫХ СИНТЕЗОВ
Главными исходными веществами для производства
многочисленных продуктов основного органического синтеза являются
различные углеводороды—парафины, олефины, диолефины,
ацетиленовые и ароматические углеводороды, нафтоны, а также окись
углерода и водород.
Неисчерпаемым источником углеводородов служат нефть и
природные горючие газы, продукты их переработки и
переработки каменных углей.
Наиболее распространенный в природе углеводород—метан,
ресурсы его огромны, он является главным, а иногда единственным
компонентом природных горючих газов (стр. 28). Ресурсы
природного метана во много раз превышают ресурсы этана,
пропана и бутанов, вместе взятых. Метан содержится в отходящих
газах нефтеперерабатывающих заводов (см. табл. 5), в коксовом газе
содержание метана составляет около 27 объемн. %. В состав
природных газов и газов нефтепереработки также входят
ближайшие гомологи метана (С2—С5). Большие количества этих
углеводородов получают при стабилизации сырой нефти (стр. 41), бу-
таны и частично пентаны выделяют, крометого, при стабилизации
газового бензина (стр. 33).
Источниками получения этилена, пропилена, бутиленов и
амиленов являются газы, образующиеся в результате
высокотемпературной деструктивной переработки нефтяного сырья —
термического крекинга и пиролиза (стр. 28, табл. 5). Меньшие
количества этих углеводородов находятся в газах
каталитического крекинга (стр. 64).
Олефины, как ненасыщенные соединения отличаются
высокой реакционной способностью и поэтому представляют
исключительную ценность как исходные соединения для синтезов. В
связи с быстрым развитием промышленности органического синтеза
количество олефиновых углеводородов (и в первую очередь
этилена), получаемых как побочные продукты при обычных
процессах деструктивной переработки нефтяного сырья на моторное
топливо, является недостаточным. Для увеличения ресурсов
этилена некоторые нефтяные дистилляты (бензин, лигроин, газойль
и др.) подвергают специальному пиролизу. При повышении
температуры этого процесса, снижении парциальных давлений
паров образующихся продуктов и сокращении времени
пребывания их в реакционной зоне выход этилена достигает 20% и более
от количества перерабатываемого нефтяного сырья.
Большие количества этилена и пропилена получают пиролизом
этана и пропана:
СН3—СН3 » СН3=СН, + Н2
СНз-СНз-СНз * СН,=СН2 + СН4
СНз—СН2—СН3 » СН2=СН—СН3 + Н2
9* 131
деструктивной переработки нефтяного сырья образуются вГ
небольших количествах. Промышленное получение таких важных ди-
олефинов, как бутадиен-1,3 (дивинил) и 2-метилбутадиен-1,3
(изопрен), основано на каталитическом разложении парафинов и оле-
финовС4 и С5. Так, дивинил получают каталитическим разложением
«-бутана или бутилена (стр. 142), изопрен—разложением изопен-
тана или амиленов (стр. 145). Дивинил получается также
каталитическим разложением этилового спирта по методу С. В.
Лебедева (стр. 211). На основе этого метода в СССР в начале 30-х
годов было создано первое в мире производство
синтетического каучука.
Разработаны методы получения диеновых углеводородов
совместно с олефинами путем деструктивной переработки жидких
нефтепродуктов.
Ацетилен в течение многих лет получали только разложением
карбида кальция водой (стр. 139):
СаС2 + 2Н2О > Са(ОНJ + СН=СН
Сырьем для производства ацетилена по карбидному методу
являются каменные угли, так как СаС2 образуется при
сплавлении извести СаО с коксом и антрацитом. В последние
десятилетия разработаны и внедрены способы получения
ацетилена из метана и его гомологов (стр. 135). Для получения
ацетилена может быть использовано также жидкое нефтяное сырье.
Бензольные углеводороды получают выделением из продуктов
коксования каменных углей, из фракций бензинов прямой
перегонки нефти и крекинг-бензинов, а также из продуктов пиролиза
керосина и некоторых других нефтяных дистиллятов.
Ароматические углеводороды экстрагируются из этих фракций
селективными растворителями.
Выход бензола и толуола из 1 т нефтяных дистиллятов
(продуктов пиролиза) составляет (в кг):
Бензол Толуол
Лигроин : S6.5 56,0
Керосин 64,0 52,0
Газойль 55,0 50,0
Мазут 52,0 36,0
Для увеличения ресурсов бензольных углеводородов
применяются процессы ароматизации нефтяного сырья, основанные на
дегидрировании нафтеновых углеводородов и дегидроциклизации
парафинов (стр. 146 и ел.), а
Используемые в промышленности окись углерода и водород
получают газификацией твердого топлива (кокс, антрацит и др.,
стр. 107) или конверсией природного газа. Эти процессы подробно
рассмотрены в курсе химической технологии неорганических
веществ.
132
3. КАТАЛИЗАТОРЫ
Большинство реакций, используемых в технологии основного
органического синтеза, протекает в присутствии катализаторов.
Применяемые катализаторы как по составу, так и по способам их
приготовления весьма разнообразны. В качестве катализаторов
могут употребляться газы (окислы азота, хлористый водород,
аммиак и др.), жидкости (минеральные кислоты, органические
основания и др.) и твердые вещества (металлы, их окислы, соли и др.).
Реакции, протекающие в присутствии катализаторов, принято
разделять на реакции гомогенного и гетерогенного катализа.
К реакциям гомогенного катализа относятся такие, в которых
катализатор и реагенты находятся в газовой или жидкой фазе. В этом
случае границы раздела между катализатором и реагирующими
веществами отсутствуют. При гетерогенном катализе катализатор
и реагенты находятся в разных фазах и взаимодействие протекает
на поверхности раздела фаз. В процессах гетерогенного катализа
особенно широко применяются твердые катализаторы.
Катализаторы должны удовлетворять следующим требованиям:
обладать высокой активностью (значительно ускорять
основные химические реакции или подавлять побочные), иметь
большую удельную поверхность, быть достаточно прочными и
стойкими к действию высоких температур и т. д.
С увеличением удельной поверхности катализатора повышается
его производительность. Под производительностью
катализатора понимается количество целевых продуктов, образующихся
за единицу времени на единицу массы (или объема) катализатора.
Обычно удельная поверхность промышленных катализаторов
очень велика: 100—300 м2 и более на 1 г катализатора. Для
получения катализаторов с такой развитой поверхностью применяются
специальные методы их приготовления. Окисные катализаторы
чаще всего изготовляют одним из следующих методов: сухим
методом (обжиг), влажным методом (осаждение) или сплавлением.
При обжиге неустойчивые соединения металлов, например
нитраты или соли органических кислот, в результате сильного
нагревания разлагаются с образованием окислов. Методом обжига
получают такие катализаторы, как окислы цинка, железа, меди
и др.
При получении катализаторов влажным методом металлы
осаждаются в виде гидроокисей или карбонатов из водных растворов
солей этих металлов, например нитратов. Для осаждения
применяют аммиачную воду, соду, двуокись углерода. Выделяемые
осадки подвергают сушке и легкому прокаливанию. В случае
необходимости окислы могут быть восстановлены до металлов
водородом при повышенной температуре.
Для увеличения активности катализатора путем развития его
поверхности катализатор часто осаждают на инертный материал,
133
гель, уголь, пемзу, асбест и т. д.
Плавленые катализаторы получают сплавлением смесей окислов
металлов. Такой сплав измельчают до образования гранул
требуемой величины и затем восстанавливают водородом. Плавленые
катализаторы отличаются большой механической прочностью.
Большой интерес представляют так называемые скелетные
катализаторы, получаемые сплавлением никеля или другого
металла с алюминием. Такой сплав выщелачивают—обрабатывают
раствором щелочи. Получаемый в результате выщелачивания
пористый металл («скелет») обладает высокой каталитической
активностью. Впервые скелетный катализатор был получен Ре-
неем A924 г.). Необходимо отметить, что восстановленные
катализаторы и катализаторы Ренея обладают пирофорными
свойствами, т. е. способны разогреваться и даже раскаляться на воздухе,
что может быть причиной возникновения пожара. Во избежание
этого катализаторы необходимо хранить в атмосфере водорода
или инертного газа (N2, CO2).
4. ТЕРМИЧЕСКИЕ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ
При нагревании углеводородов до определенной температуры
происходит их разложение. Разложение углеводорода может
произойти как с размыканием углерод-углеродной связи
{расщепление), так и с размыканием углерод-водородной связи
(дегидрирование). В зависимости от условий—температуры, давления,
наличия катализатора, продолжительности пребывания
реагентов в зоне высоких температур—образуются различные новые
углеводороды с меньшим молекулярным весом. Например,
разложение н-бутана может протекать по следующим схемам:
СНз—СН3 + СНо=СН2
этан этилен
сн,=сн-сн3 + сн4
пропилен метан
сн2=сн—сн2—сн3 + н2
я-бутилен
У-л\ъ — ^^3 1 ^-'^4 і ^
2СН2:^СН2 ~\~ Н2
ацетилен
С)
B)
C)
D)
E)
F)
Реакция A) протекает главным образом при низких
температурах (~^400 =С), реакция B)—в широком диапазоне температур
(до 400 °С и выше). Преимущественное дегидрирование бутана по
реакции C) происходит в присутствии катализатора (стр. 142).
При температурах выше 1000 °С происходит пиролиз бутана по
134
реакциям E) и F) с образованием этилена B5—28%) и ацетилена
B0—25%).
Для углеводородов С5 и более характерно еще большее
разнообразие продуктов разложения. При создании соответствующих
условий процесс разложения можно направить таким образом,
чтобы получались требуемые углеводороды.
[ПОЛУЧЕНИЕ АЦЕТИЛЕНА
При обычных процессах деструктивной переработки
нефтепродуктов ацетилен образуется в ничтожных количествах. Для
промышленного получения ацетилена подвергают специальной
переработке метановые углеводороды. Сырьем могут служить
индивидуальные углеводороды (например, метан) или их смеси:
2Г:н4 * СН=СН + ЗН2
С2На » СН=СН + 2М2
2С3Н8 » 3CHsCH + 5Н2 и т, д.
Для этой цели используются также жидкие нефтяные фракции.
Методы получения ацетилена из метановых углеводородов
основаны на их термическом разложении и различаются только
способом достижения высоких температур. Основными
промышленными методами производства ацетилена из углеводородного сырья
являются:
1) разложение углеводородов при высокой температуре,
развивающейся при сжигании некоторой части исходного сырья—
так называемый окислительный пиролиз;
2) разложение углеводородов в регенеративных печах;
3) электрокрекинг—разложение их в электрической дуге.
Общим условием достижения максимальных выходов
ацетилена является применение высоких температур при
кратковременном пребывании углеводорода в реакционной зоне, а также
быстрое охлаждение продуктов реакции. Это предотвращает
протекание побочных реакций и разложение ацетилена на элементы.
Окислительный пиролиз. На рис. 42 схематически показано
устройство печи для крекинга метана. Природный газ с высоким
содержанием метана и кислород, предварительно нагретые до
550—600 °С, вводят в верхнюю часть печи, где они смешиваются.
Содержание кислорода в смеси составляет 37—38%. У нижнего
края футерованной части печи установлена плита, выполненная
из высокоогнеупорного материала и имеющая большое
количество отверстий диаметром 8 мм. Горящая ниже плиты смесь
метана и кислорода, проходя через отверстия плиты, образует
отдельные факелы. Скорость движения метано-кислородной смеси
значительно превышает скорость распространения фронта
пламени в газе. Это обеспечивает безопасность ведения процесса.
135
Процесс, протекающий в печи, можно выразить следующей
суммарной реакцией:
11СН4
2СН=СН + 6СО + 14Нг + СО2 + 6Н2О
Метан
Вода
Тепла, выделяющегося при протекании этой реакции,
достаточно для поддержания требуемой температуры процесса
разложения (~1500Т).
В продуктах реакции содержится
8% ацетилена, 26% окиси углерода и
54% водорода, остальное—азот,
двуокись углерода, этилен и неразложи-
вшийся метан, количество которого
составляет около 4%. Степень
превращения метана в ацетилен достигает
30—32%.
Из реакционных газов извлекают
концентрированный ацетилен. Смесь
окиси углерода и водорода может быть
использована как синтез-газ (стр. 163).
Крекинг углеводородов в
регенеративных печах. В процессе термическо
го крекинга углеводородов поэтому
способу тепло подводится к реагентам от
нагретой стенки печи. В печах
имеется огнеупорная насадка в виде
параллельно расположенных горизонтальных
пластин, образующих цилиндрические
ходы диаметром около 6 мм. по всей
Г$одааи Длине печного канала (рис. 43). Рабо-
: чиє циклы процесса, отличающиеся
направлением газовых потоков, состоят
Рис. 42. Схема реакцион- каждый из четырех периодов. В первом
ной печи для получения ПЄрИОдЄ насадка / или 3 (поочередно)
ацетилена из метана в ^ , ,лп іслл о/-
присутствии кислорода, нагревается до 1400-1600 °С
сжигаемым в топке 2 газом; продукты
сгорания отводятся в дымовую трубу. По
достижении требуемой температуры клапаны автоматически
переключаются, и пламя гаснет. Во втором периоде насадку
продувают водяным паром. В третьем периоде на раскаленной насадке
происходит разложение (крекинг) углеводородов, и насадка
охлаждается.
При снижении температуры до определенного уровня клапаны
снова автоматически переключаются и насадка продувается паром
(четвертый период).
Далее начинается новый цикл с обратным направлением
газовых потоков.
136
Продукты реакции содержат 7% ацетилена при крекинге
метана и 10% С,Н2 при крекинге пропана.
1 ' ' I ' ' ' ' ' ' ' ' ' ' ' Л
Рис. 43. Регенеративная печь:
/, 3—регенеративная насадка; 2—топочная
камера; а—деталь кладки регенератора;
4—фасонные огнеупорные плитки; 5—каналы
я диаметром 6 мм.
Электрокрекинг. По этому способу разложение метановых
углеводородов проводят в электродуговом реакторе (рис. 44).
Электрическая дуга создается в нем между электродами.
Исходные углеводороды (чаще всего природный газ с высоким
содержанием метана) под абсолютным давлением 1,5 am поступает в
цилиндрическую реакционную камеру через патрубок по
касательной к стальной стенке камеры. Газ совершает в камере
вращательное движение и входит в вертикальную реакционную трубу.
Под действием электрической дуги углеводородный газ
разогревается до 1600 °С (метан) или до 1300—1400 °С (гомологи
метана). Скорость прохождения газа в реакционной камере
достигает 100 м/сек, и более, а в реакционной трубе—600 м/сек,.
Продукты реакции вместе с неразложившимися углеводородами быстро
охлаждаются по выходе из реакционной трубы впрыскиванием
в нее воды.
Мощность, потребляемая реактором, достигает 7000 квт,
производительность составляет 4000 т ацетилена в год. При
электрокрекинге природного газа, содержащего 92—93% метана,
в продуктах реакции содержится 13—14% ацетилена и его
гомологов, 1% этилена, 50—55% водорода и 30—35% непрореагиро-
вавшего СН4. Степень превращения метана—около 50%.
Удельный расход электроэнергии на 1 кг ацетилена 9—10 кьт-ч.
При электрокрекинге образуется большое количество сажи
{50 кг на 1 т ацетилена). Она является товарным продуктом и
используется в качестве активного наполнителя резиновых смесей
(стр. 499). Водород, образующийся при разложении углеводородов,
можно использовать в различных промышленных синтезах.
127
В случае электрокрекинга углеводородов, более тяжелых, чем
метан, концентрация ацетилена в продуктах реакции повышается
до 16% и снижается удельный расход электроэнергии.
Продукты
реакции
Рис. 44. Электродуговая реакционная печь для
получения ацетилена из углеводородов:
/—шина; 2—электрод; 3—фарфоропый изолятор; 4—реакционная
камера; 5—заземленный электрод; 6—реакционная труба; 7—компен
саторы; 8—пусковой электрод.
Концентрация ацетилена в продуктах термического
разложения углеводородов невелика (8—16%). После охлаждения
реакционную газовую смесь направляют на выделение из нее ацетиле-
138
на методом абсорбции водой под давлением A8—20 am) или
селективными растворителями — диметилформамидом, N-ме-
тилпирролпдоном и др.
Получение ацетилена из карбида кальция. Сущность карбидного метода
.заключается во взаимодействии карбида кальция с водой (стр. 132).
Выход ацетилена из технического карбида кальция зависит от чистоты
исходного продукта и полноты его разложения. Литраж технического
карбида кальция (количество литров ацетилена, полученного при полном
разложении водой 1 кг карбида кальция) составляет от 230 до 320 л/кг.
Количество тепла, выделяющегося при образовании ацетилена, соответственно
колеблется в пределах 400—500 ккал на 1 кг технического карбида кальция.
Карбид кальция получают путем сплавления в электрических печах
измельченной смеси окиси кальция с углеродистым материалом (коксом,
антрацитом) :
СаО + ЗС » СаС, + СО
У концов угольных электродов электрической печи развивается
температура до 3500 °С. Образующийся карбид кальция выпускают в расплавленном
виде в изложницы, где он застывает, или непрерывно отводят из печи через
барабанный охладитель специальной конструкции, орошаемый снаружи водой.
Затвердевшие куски карбида далее дробят в мельницах. Для безопасности
работы процесс проводят в атмосфере азота.
Карбид кальция очень быстро разлагается влагой воздуха, поэтому его
необходимо хранить в герметически закрытой таре.
Разложение карбида кальция водой для получения ацетилена проводят
р. аппаратах, называемых ацетиленовыми генераторами. По принципу
действия эти аппараты могут быть разделены на генераторы «вода на карбид»
(г подачей воды на карбид) и генераторы «карбид в воду» (с загрузкой
карбида в воду).
Генераторы системы «вода на карбид» более компактны; расход воды в
таких генераторах значительно меньше, чем в генераторах системы «карбид
в воду». Они очень удобны для получения ацетилена непосредственно на
месте выполнения сварочных работ (автогенная сварка). На химических
заводах обычно применяются ацетиленовые генераторы системы «карбид в
воду» производительностью до 500 м3/ч ацетилена и так называемые «сухие*
или бесшламовые генераторы производительностью 2000 м3/ч ацетилена.
На рис. 45 показана одна нз конструкций ацетиленовых генераторов
системы «карбид в воду.». Карбид непрерывно подается секторным
питателем 2 в шахту 3 генератора, распределяется по сечению генератора коп\-
сом 4 и поступает на решетки 5. Побочно образующаяся гидроокись
кальция удаляется в виде известкового молока по трубе 7. Во избежание
«заиливания» карбида (осаждения извести на его поверхности) над решетками 5
установлены гребки 6, непрерывно перемешивающие куски карбида.
Ацетилен отводится в газгольдеры.
Подача карбида в генератор автоматически регулируется в зависимости
от запаса ацетилена в газгольдере, из которого газ поступает потребителям.
Если ацетилена потребляется больше, чем поступает из генератора, давление
в газгольдере уменьшается, и колокол газгольдера, опускаясь, автоматически
включает электродвигатель, вращаюший барабан питателя ацетиленового
генератора.
Достоинства генераторов системы «карбид в воду»: высокий коэффициент
полезного действия* (96—98%); возможность получения более чистого ацетп-
* Коэффициентом полезного действия ацетиленовых генераторов
называют отношение количества фактически полученного ацетилена к количеству
ацетилена, которое может быть получено при полном разложении исходного
карбида.
139
лена, чем в генераторах системы «вода на карбид»;гббльшая безопасность
работы. К недостаткам генераторов этого типа надо отнести сложность
конструкции, большие габариты, повышенный расход воды и значительные количества
отходов—шлама.
Рис. 45. Ацетиленовый генератор системы
«карбид в воду»:
/—бункеры для карбида; 2—секторный питатель;
3—шахта; 4—конус; 5—решетки; б—гребки; Г—труба
для удаления известкового молока; В—
предохранительный гидравлический затвор; 9—шлюзовой затвор
для вывода твердых примесей; 10—труба для
продувки генератора азотом.
Более рациональны так называемые «сухие» бесшламовые ацетиленовые
генераторы. Разложение карбида кальция в них производится при небольшом
избытке воды по сравнению с теоретическим. Благодаря выделению
значительного количества тепла реакции избыточная вода , полностью испаряется, и
образуется гашеная известь в виде сухого порошка (пушонки), а температура
ацетилена обычно не превышает 100—ПО °С.
При разложении карбида кальция водой на поверхности его кусков обра -
зуется слой извести, препятствующий дальнейшему контакту реагентов. Для
удаления извести с поверхности кусков карбида в сухнх генераторах имеется
перемешивающее устройство.
На рис. 46 изображена схема бесшламового ацетиленового генератора.
Генератор состоит из цилиндрического металлического кожуха / (диаметром
3,5 м), неподвижно укрепленных тарелок 2 и вертикального вала 3 с гребками4,
Взаимодействие между карбидом и водой происходит на тарелках с отверстия-
140
ми. На нечетных тарелках (считая сверху) отверстия расположены в
центральной части, на четных—по периферии. Сухая известь собирается в нижней
части генератора и удаляется шнеком 5. Благодаря большому количеству
тарелок достигается почти полное разложение карбида.
Рис. 46. Схема бесшламового
ацетиленового генератора:
/—кожух; 2—тарелки; 3—вертикальней
вал; 4—гребки; 5—шнек.
ДЕГИДРИРОВАНИЕ АЛИФАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Дегидрированием алифатических соединений получают
этиленовые и диеновые углеводороды, обладающие значительной
реакционной способностью и нашедшие широкое применение
в химической промышленности. Например/дегидрированием этана
можно получить этилен:
СНз—СНз * СН2=СН2 + Н2
Дегидрированием н-бутилена—бутадиен-1,3 (дивинил):
СН2=СН-СН2-СН3 » СН2=СН-СН=СН2 + Н2
Дегидрированием изопентана—изопрен B-метилбутадиен-1,3):
СН3 снз
СНз-СН—СН2-СН3 > СН2=С-СН=СН2 + 2Н3
Этан и пропан дегидрируют в трубчатых печах (стр. 46)
чисто термически. При дегидрировании бутана и высших
парафинов применяют катализаторы—окислы некоторых металлов (окись
хрома и др.).
В последние годы производство олефинов и диолефинов
приобрело большое народнохозяйственное значение. Например,
получение бутадиена дегидрированием бутана позволяет
значительно расширить сырьевую базу для производства синтетических
141
каучуков, которые обходятся дешевле, чем при производстве
каучука на основе бутадиена, получаемого из этилового спирта
(стр. 211). Дегидрирование изопентана в изопрен создает широкие
возможности для производства некоторых ценных видов
синтетического каучука (стр. 486 и 494).
Получение бутадиена дегидрированием бутана
Этим способом бутадиен (дивинил) может быть получен в две
либо в одну стадию. В первом случае сначала получают бутилен,
который далее дегидрируют до бутадиена. Во втором случае
бутан дегидрируют непосредственно в бутадиен.
Дегидрирование и-бутана. На первой стадии н-бутан
подвергают дегидрированию до н-бутилена:
СН3-СН2-СН2-СН3 7=^ СН2=СН-СН2—СН3 + Н2
Реакция дегидрирования бутана обратима и протекает с
увеличением объема. Согласно принципу Ле-Шателье, понижение
давления благоприятно влияет на процесс дегидрирования. На
практике для упрощения его проводят под атмосферным давлением
при 550—575 °С в присутствии алюмохромового катализатора.
Активность катализатора понижается вследствие отложения на
его поверхности углеродсодержащих соединений, образующихся
в результате протекания побочных процессов глубокой
деструкции углеводорода. Для восстановления активности катализатора
его часто подвергают регенерации путем продувки воздухом,
предварительно смешанным с топочными газами.
Алюмохромовый катализатор необратимо дезактивируется
водяными парами, поэтому все газы, поступающие в контактные
реакторы, должны быть предварительно тщательно осушены.
Степень конверсии (превращения) н-бутана за один проход
газа через катализатор составляет 40—60%; при более высоких
степенях конверсии значительно увеличивается протекание
побочных реакций, главным образом расщепления углеводородов.
Выход н-бутиленов составляет 75—80% от количества
конвертированного бутана и 35—40% от количества сырья (н-бутана),
пропущенного через катализатор.
При дегидрировании н-бутана в стационарном (неподвижном)
слое катализатора контактные аппараты работают
полунепрерывно, так как после непродолжительного рабочего цикла
необходима регенерация катализатора (выжигание углеродсодержащих
отложений). На восстановленном катализаторе затем вновь
начинается процесс дегидрирования. Такое чередование циклов
дегидрирования и регенерации делает процесс неустойчивым и
обусловливает низкую производительность контактных
аппаратов этого типа.
Более эффективен непрерывный процесс в движущемся слое
катализатора.
142
На рис. 47 показана схема агрегата для дегидрирования н-бу-
тана в кипящем слое пылевидного катализатора. Агрегат состоит
из двух аппаратов—реактора / и регенератора 2.
Предварительно нагретый бутан поступает под распределительную решетку 3
со скоростью, при которой катализатор поддерживается в псев-
доожиженном состоянии. Газообразные
продукты реакции удаляются через
циклоны, где газы отделяются от
увлекаемых ими частиц катализатора и затем
направляются на разделение. Непроре-
агировавший н-бутан возвращают в
производственный цикл.
Пылевидный катализатор увлекается
из нижней части реактора топочными
газами и по стояку 4 непрерывно
перемещается в регенератор. Под
распределительную решетку 7 подают воздух,
который поддерживает катализатор в кипящем
состоянии. В регенераторе при 600—650 °С
углеродистые отложения выжигаются с
поверхности катализатора.
Регенерированный катализатор по стояку 6
непрерывно возвращается в реактор.
При дегидрировании бутана в
движущемся слое катализатора обеспечивается
непрерывность и устойчивость процесса,
что улучшает эксплуатационные
показатели и увеличивает производительность
реакторов.
Дегидрирование «-бутиленов. н-Бути-
лены, выделенные из продуктов реакции
дегидрирования н-бутана*, подвергают
дальнейшему дегидрированию (вторая
стадия):
СН2=СН-СН2-СН3 -
Рис, 47. Агрегат для
дегидрирования бутана в
«кипящем» слое
катализатора:
/—реактор; 2—регенератор;
3. 7—распределительные
решетки; 4, 6—трубы для
перемещения катализатора; 5—циклоны.
СН,—СН=Ш—СН, —
сн,=сн—сн=сн, + н3
В качестве катализаторов применяют смеси окислов металлов,
например MgO, Fe2O3, CuO, K3O. Процесс проводят при 600—
650 °С. Олефины предварительно смешивают с перегретым водяным
паром (соотношение исходного сырья и водяного пара от 1 : 10
до 1 : 20). При этом понижается парциальное давление
реагирующих газов, что способствует увеличению выхода бутадиена;
кроме того, водяной пар нагревает углеводороды до температуры реак-
* Для получения бутадиена используют также н-бутилены, выделяемые
из иефтезаводских газов.
143
дни. ми смешения с yi леводиридами пар перегревают в трубчатых
печах до 720—750 СС.
Дегидрирование я-бутиленов чаще всего проводится в так
называемых адиабатических контактных реакторах. Реактор
представляет собой цилиндрический шахтный аппарат (рис. 48), в
Пары
]н-оутшгенов
\ Водяной
"пар
Рис. 48. Реактор (адиабатический конвертор) для дегидрирования
к-бутиленов:
/—устройство для смешения паров воды и н-бутилеиов; 2—люк для загрузки
катализатора; 3—съемная корзина с насадочными кольцами; 4— слой катализатора; 5—люк
для выгрузки катализатора; 6—слой насадочных колец; 7—термопары.
144
котором на решетке укладывают слои колец и поверх него слои
катализатора. Эндотермическая реакция дегидрирования
протекает за счет подвода тепла с перегретым водяным паром.
Периодически реактор переключают на регенерацию катализатора,
производимую горячим воздухом. Обычно устанавливают несколько
реакторов, поэтому процесс дегидрирования в целом
осуществляется непрерывно.
Степень конверсии бутиленов составляет 22—28%, выход
бутадиена 80—85% на прореагировавший газ и 18—23% на
введений в процесс.
Технологическая схема дегидрирования бутиленов в
адиабатических реакторах аналогична схеме дегидрирования этил-
бензола в стирол (стр. 148, рис. 49).
Получение изопрена
Дегидрирование изопентана, как и бутана, можно проводить
в одну или две стадии. При двухстадийном процессе в результате
каталитического дегидрирования изопентана (первая стадия)
образуются три изомерных амилена:
СН3
сн.2=с-сн.г—сн3
і ' 2-метилбутен-1
сн3—сн—сн2—сн3 *¦ сн3—с=сн—сн3
СН3
-На
сн.
13
2-метилбутен-2
сн.г=сн—сн—сн„
сня
із
З-метилбутен-1
Этот процесс протекает в интервале 520—600 °С. В качестве
катализаторов предложены смеси различных окислов—хрома,
молибдена, вольфрама, нанесенных на окись алюминия. Выход
изоамиленов составляет 78—87% на прореагировавший изопентан
и 33—40% на изопентан, пропущенный через катализатор.
Вторая стадия—дегидрирование изоамиленов в изопрен (ме-
тилбутадиен-1,3)
СН2=С-СН2-СН3 - ^Нз
¦ СН,=С—СН=СН2
СН3—С(СН3)=СН—СН3—
-На
СН2=СН-СН-СН3 — ¦
СНз
10—805 145
проводится в условиях, аналогичных условиям дегидрирования
н-бутиленов в бутадиен (в присутствии водяного пара, на таких
же катализаторах и примерно при тех же температурах).
Другой промышленный метод получения изопрена основан
на реакции взаимодействия формальдегида с изобутиленом с
последующим разложением образующегося диметилдиоксана (стр.
235). Промышленное значение приобрел и трехстадийный процесс
получения изопрена из ацетона и ацетилена, разработанный
впервые Е. А. Фаворским.
ДЕГИДРОЦИКЛИЗАЦИЯ АЛИФАТИЧЕСКИХ УГЛЕВОДОРОДОВ
Советские ученые Б. А. Казанский и др. показали, что
алифатические углеводороды с шестью и более атомами углерода в цепи
в определенных условиях превращаются в ароматические
соединения, содержащие такое же число углеродных атомов, например:
СНа-(СН2L-СН3 * ( ] + 4Н2
з—\упгН—^п3 » | у -f чпг
н-гептан
СН,-(СБа).,-СНя » І II + 4Н
СН3-(СН2)в -СН3 > || І + 4Н2
н-октан
Этот процесс получил название дегидроциклизации
{дегидроароматизации). Дегидроароматизации можно подвергать не
только парафины, но и олефины, которые превращаются в
ароматические соединения с большей скоростью, чем парафины. В качестве
катализаторов процесса дегидроциклизации применяют окислы
хрома, алюминия, титана и других металлов, осажденные на
носителях. Например, толуол можно получить из н-гептана с
90%-ным выходом, проводя дегидроциклизацию при температуре
около 500 °С и применяя в качестве катализатора окись хрома,
нанесенную на окись алюминия. Активатором в данном случае
служит небольшое количество двуокиси церия.
Н. Д. Зелинским была открыта реакция дегидрирования цикло-
парафинов. Пропуская циклогексан при 250—300 °С и
атмосферном давлении над катализатором (платина, палладий, никель),
Зелинский получил бензол:
СН2
Н2С СН2
\ /
сн2
146
Аналогично из метилциклогексана оэразуется толуол:
СН2
н2с сн-сн3 ^\/СНз
I I —» [ If + зн2
в,с сн2 Ч/
сн2
Процессам дегидроциклизации и дегидрирования подвергают
«е только индивидуальные углеводороды, ной сложные смеси их—
нефтяные фракции. Сочетание этих процессов на практике
применяется в различном технологическом оформлении (платфор-
минг, гидроформинг и др., см. стр. 66).
ДЕГИДРИРОВАНИЕ ЖИРНО-АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
При дегидрировании жирно-ароматических углеводородов—¦
этил- и изопропилбензола образуются соответственно стирол и
¦а-метилстирол:
¦v 2~ 3 /v ^ 2
-г н2
СНо CHq
3 | 3
—С—Н <ґ\—C=CH
от -а
н2
сн3
Стирол (винилбензол или фенилэтилен) и а-метилстирол в
больших количествах применяют для производства синтетических
каучуков (стр. 487) и пластических масс (стр. 426 ел.).
На рис. 49 показана технологическая схема производства
стирола дегидрированием этилбензола.
Дегидрирование проводят при 560—600 °С в адиабатических
контактных реакторах (стр. 144). В качестве катализаторов
применяют обычно окислы металлов (железа, магния, цинка, меди
и др.).
Процесс образования стирола протекает с увеличением объема,
поэтому течению реакции благоприятствует пониженное
давление. Пары этилбензола смешивают с водяным паром (на 1 кг
этилбензола 2,6 кг водяного пара), благодаря чему парциальное
давление паров этилбензола снижается примерно до 0,1 am.
Смесь этилбензола и водяного пара направляют в испаритель 3,
обогреваемый контактными газами, и затем в перегреватель 2.
Отсюда при температуре 530 °С смесь подают в адиабатический
реактор; тепло, необходимое для дегидрирования, подводится
с перегретым водяным паром. Тепло контактных газов
используется в испарителе, перегревателе и котле-утилизаторе для по-
10* 147
лучения водяного пара. По выходе из котла-утилизатора
контактные газы последовательно поступают в конденсаторы 5 и 6,
охлаждаемые водой и рассолом. Конденсат собирают в
отстойнике 7, где углеводороды отделяются от воды. Несконденсиро-
вавшиеся газы направляют на использование в качестве топлива.
Воздух
на регенерацию
Опходя-
газы
вода
Рис. 49. Схема производства стирола дегидрированием этилбензола:
/—контактный аппарат; 2—теплообменник-перегреватель; 3—испаритель; 4—котел-утилизатор;
5, 6—конденсаторы; 7—отстойник.
Углеводороды (печное масло), содержащие стирол, отводятся
из верхней части отстойника. Для предотвращения
преждевременной полимеризации в печное масло добавляют гидрохинон или
элементарную серу и направляют на ректификацию.
5. ИЗОМЕРИЗАЦИЯ И АЛКИЛИРОВАНИЕ
Процесс изомеризации углеводородов заключается в
изменении строения молекулы углеводорода без изменения его
молекулярного веса и состава. Например, изомеризация н-пентана в
изопентан:
СНзч
СН3—(СН2)з—СН3 > ^>СН—СНг—СН3
Процесс изомеризации имеет большое практическое значение.
Присутствие углеводородов с разветвленной цепью в моторном
топливе улучшает его антидетонационные свойства (стр. 49).
Так, октановое число н-пентана составляет 88,7, изопентана—
148
105,7. Еще большая разница октановых чисел я-гексана F5,3)
и изогексанов (93,1 —108,9).
Не меньший интерес представляет процесс изомеризации
н-бутана в изобутан—ценное исходное соединение для
производства изобутилена, получившего широкое применение для синтеза
некоторых типов каучуков, в качестве добавки к смазочным
маслам, для изготовления моющих средств, пластмасс (стр. 418).
Возросшая потребность в изобутилене не могла быть
удовлетворена тем количеством его, которое выделяют из газов крекинга.
Это вызвало необходимость разработки промышленного процесса
изомеризации н-бутана. В промышленной установке процесс:
осуществляется при температуре 175—225 °С и давлении 12—15 am
в присутствии катализатора—безводного хлорида алюминия. За
один проход газа через катализатор степень превращения
н-бутана составляет 40—50%; при повторном пропускании газа
выход изобутана достигает 90%.
Изомеризацию н-бутиленов в изобутилен впервые
осуществили советские химики, применившие в качестве катализатора
фосфорную кислоту, нанесенную на шамот. При 265—420 °С и
однократном прохождении газов через катализатор выход
изобутилена составляет 50%.
Большой интерес представляет изомеризация
жирно-ароматических углеводородов, в частности превращение о- и л*-ксило-
лов в га-ксилол:
СН3
с
Это значительно увеличивает ресурсы га-ксилола—исходного
углеводорода для получения синтетического волокна—лавсан
(стр. 474).
Изомеризацию смеси ксилолов проводят при 400—500 °С и
давлении 10—25 am в присутствии катализатора—платины на
носителе. Продукты реакции, содержащие 19% га-ксилола,
конденсируются и подвергаются кристаллизации (стр. 162), в
результате выделяется более половины пара-изомера. Маточный раствор
ксилолов (содержащих около 8% «-ксилола) вновь направляют
на изомеризацию и т. д., практически до полного превращения
изомеров в м-ксилол.
Алкилированием называют введение алкильной группы в
молекулу органического вещества.
149>
Процесе алкилирования углеводородов жирного ряда
заключается во взаимодействии олефинов с изопарафином, содержащим
третичный атом углерода. Например, при взаимодействии
«-бутилена с изобутаном образуется октан с сильно разветвленной
цепью—2,2,3- тримепшлпентан
СНд СНд
снзч I f
сн2=сн—сн2—сн3 + >сн—сн3 —»сн3—с—сн—сн2—сн3
сн/ ¦ I
сн,
который частично изомеризуется в 2,2,4-тримгтилпентан:
СН3 СН3 СН3 СН3
II II
СН3—С СН—СН2—СН3 » СНз—С—СН2—СН—СН3
I I
СН3 СН3
Процесс алкилирования можно осуществить как в
присутствии катализаторов, так и без них. Каталитический процесс
протекает при обычной температуре (или даже при охлаждении) и
давлении, близком к атмосферному. В качестве катализаторов
применяются 96%-ная серная кислота, жидкий фтористый водород
и др. В отсутствие катализаторов алкилирование предельных
углеводородов проводят под высоким давлением C00—350 am)
при температуре около 500 °С.
Алкилирование углеводородов жирного ряда используется
для получения компонентов моторного топлива с высокими
антидетонационными свойствами (стр. 128).
В промышленности органического синтеза применяется
алкилирование ароматических углеводородов, в первую очередь
взаимодействие бензола с этиленом или пропиленом с получением
соответственно этилбензола
. .х ^СН,—СН3
! + сн2=сн, Г
" Ч.
или изопропилбензола:
/СН3
1 + СН2=СН-СН3 > (| РС.Н
К/ ЧСН3
Этилбензол и изопропилбензол являются важными
промышленными продуктами. Этилбензол используется для получения
стирола (стр. 147), изопропилбензол перерабатывают в а-метилсти-
рол и в фенол совместно с ацетоном (стр. 252).
При алкилировании бензола олефинами применяют
различные катализаторы, главным образом кислотного типа (А1С13,
150
Н3РО4, А1Вг3, BF3, HF, H2SO4), чаще всего безводный
хлористый алюминий. Процесс проводят в жидкой фазе. Синтез алкил-
бензолов алкилированием бензола олефинами осуществил в 1879 г.
Бальсон, продолжавший работы Фриделя, Крафтса и Густавсона,
которые впервые открыли реакции алкилирования
ароматических углеводородов галоидалкилами.
При алкилировании бензола олефинами наряду с моноалкил-
бензолом образуются полиалкилбензолы (ди-, три-, тетраалкил-,
а иногда пента- и гексаалкилбензолы). Под воздействием
катализатора (хлористого алюминия) частично протекает обратная
реакция—деалкилирование полиалкилбензолов, например:
где R — алкильная группа.
Реакция алкилирования, осуществленная в промышленных
условиях, протекает в присутствии катализатора
(порошкообразный безводный хлористый алюминий или катализаторный
комплекс А1С13 с диэтилбензолом и хлористым водородом).
На рис. 50 показана технологическая схема получения этил-
бензола алкилированием бензола этиленом в присутствии ката-
лизаторного комплекса. Жидкий катализаторный комплекс
приготовляют в аппарате /, откуда он подается в реакционный
аппарат 2 (алкилатор). Одновременно в нижнюю часть алкилатора
непрерывно подают бензол и этилен. Процесс проводят при
температуре около 100 °С. Реакционное тепло отводится главным
образом за счет испарения бензола. Пары бензола, выходящие
из верхней части алкилатора, конденсируются в обратном
холодильнике 3 и возвращаются в процесс.
Продукты реакции (алкилшп) отделяют в отстойнике 5 от
механически унесенного катализаторного комплекса и
обрабатывают водой в аппарате 6. После отстаивания от воды в аппарате 7
и нейтрализации (на схеме не показана) алкилат направляют на
ректификацию.
Алкилатор (рис. 51) представляет собой колонну высотой 10 м,
футерованную изнутри графитовыми плитками. Аппарат состоит
151
из четырех царг диаметром 1,4 м, снабженных рубашками, в
которые подают пар для подогрева реакционной массы при пуске
аппарата. При установившемся процессе для отвода части
реакционного тепла в рубашки подают воду.
Большой практический интерес представляет процесс алкили-
рования бензола тетрамером пропилена—додеценом:
Н
2
с12н25
Ч
Образующийся при этом додецилбензол используется для
производства моющих средств (стр. 331). Технический додецилбензол
Y
бензол
Эгтищн-
Рис. 50. Схема получения этилбензола алкилированием
бензола:
/—аппарат для приготовления катализаторного комплекса;
2—реакционный аппарат; 3—холодильник; 4—сепаратор; 5, 7—отстойники;
6—промыватель.
представляет собой жидкость плотностью 0,870—0,875 г/см3;
пределы кипения жидкости 275—330 °С.
Додецен получают при 180—200 °С и 40—60 am по реакции:
4СН2=СН—СН3 > С12Н24
Катализатором служит ортофосфорная кислота. Реакцию
проводят в полочном или трубчатом аппарате высотой 5 лі и
диаметром 180 мм с электрообогревом и охлаждающими змеевиками.
152
Паро-газобая
смесь
--L—ft
Натализатор-
ный комплекс
Рис. 51. Алкилатор:
/—термопары; 2—предохранительный клапан; 3—графитовые
плитки.
іехника безопасности при раооге и
При получении, переработке, транспортировании и хранении
углеводородов обслуживающий персонал должен строго
соблюдать правила и нормы техники безопасности. Это обусловлено
горючестью и взрывоопасностью углеводородо-воздушных смесей
и токсичностью углеводородов.
Одной из важнейших характеристик газо- или паро-воздуш-
ных горючих смесей являются нижний и верхний пределы взрывае-
мости.
Нижним (или верхним) пределом взрываемости называется
наименьшее (или наибольшее) содержание горючего газа или пара
в смеси с воздухом, при котором может произойти взрыв. При
наличии источника воспламенения (открытый огонь, искра и т. д.)
газовая смесь может взрываться при концентрациях горючего
газа или пара между верхним и нижним пределами.
При концентрации углеводородов (газа или пара) в воздухе
меньше нижнего или больше верхнего пределов взрываемости не
происходит взрыва.
К наиболее взрывоопасным углеводородам относится ацетилен.
Он является эндотермическим соединением, легко разлагающимся
с выделением тепла:
С2Н2 * 2С + Н2 + 54 ккал
В определенных условиях даже в отсутствие кислорода
ацетилен склонен к взрывному распаду. Ацетилено-воздушная смесь
взрывоопасна практически при любых соотношениях
компонентов смеси. Разложение чистого ацетилена, находящегося под
давлением выше 2 ат, может произойти с сильным взрывом. Поэтому
особые меры предосторожности необходимы при компримировании
ацетилена. Не допускается хранение и транспортировка
ацетилена в обычных баллонах. Баллоны для ацетилена должны быть
заполнены активированным углем, пропитанным растворителем
(ацетон), который абсорбирует значительное количество газа,
нагнетаемого под абсолютным давлением 15—20 am.
Углеводороды очень токсичны. Многие из них являются
нервными ядами, обладающими наркотическим действием разной
степени. Хроническое отравление ароматическими углеводородами
вызывает тяжелые заболевания крови и кроветворных органов и
изменения в сосудистой системе. Для успешного предотвращения
пожаров, взрывов и отравлений необходимо учитывать
перечисленные свойства углеводородов. Технологический процесс необходимо
проводить в строгом соответствии с установленным регламентом.
Все установки должны быть полностью герметизированы, чтобы
исключить возможность утечки газов, паров или жидкости из
аппаратов и трубопроводов или проникания в них атмосферного
154
воздуха. Нельзя допускать возникновения источников огня в
производственных помещениях и складах, где хранится или
перерабатывается углеводородное сырье. Они должны быть
оборудованы эффективной искусственной вентиляцией с достаточной
кратностью воздухообмена. Обслуживающий персонал
допускается к работе только после тщательного инструктирования и
ознакомления с правилами техники безопасности.
7. РАЗДЕЛЕНИЕ СМЕСЕЙ УГЛЕВОДОРОДОВ
РАЗДЕЛЕНИЕ ГАЗОВЫХ СМЕСЕЙ
Ацетилен, получаемый разложением карбида кальция, и
«сухой» природный газ, содержащий в основном метан, могут быть
непосредственно использованы для дальнейшей переработки.
Углеводородные газы крекинга и пиролиза нефтяных
дистиллятов, коксовый газ, а также «жирные» природные газы являются
сложными смесями веществ различного состава. Они могут
использоваться в качестве химического сырья только после
предварительного разделения на компоненты. В зависимости от
требований, предъявляемых к сырью при дальнейшей переработке, газы
разделяют на индивидуальные углеводороды (четкое разделение)
или на группы (фракции) углеводородов с близкими свойствами
(грубое разделение).
Разделение газовых смесей в промышленности осуществляется
тремя методами: абсорбцией, адсорбцией и охлаждением компри-
мированной (сжатой) газовой смеси.
. Абсорбция обычно применяется для грубого разделения смесей
газообразных углеводородов. Она основана на поглощении
газообразных веществ селективным растворителем (селективным
абсорбентом). Разделяемую газовую смесь пропускают через
абсорбционную колонну, орошаемую растворителем. Содержащиеся
в газе низшие углеводороды (Сх—С,) не поглощаются
растворителем и отводятся из верхней части колонны. Более тяжелые
углеводороды абсорбируются орошающей жидкостью, из нижней части
абсорбционной колонны непрерывно вытекает раствор
углеводородов в абсорбенте. В отгонной колонне (десорбере) газы
выделяются из раствора. Растворитель охлаждают и возвращают
на абсорбцию, выделенные углеводороды разделяют
ректификацией. Для более полного извлечения углеводородов из газов
абсорбцию обычно проводят при повышенном давлении A2—20 am)
и охлаждении исходного газа и растворителя.
Адсорбция основана на поглощении газов твердыми
мелкопористыми телами (активный уголь, силикагель и др.) с
последующей отгонкой и ректификацией поглощенных углеводородов.
Через адсорберы с неподвижным слоем активного угля
пропускают разделяемую смесь газов до определенной степени насыще-
155
газы
Газовая
смесь
if
її
Десорбиро-
8анны'п?аз
ш
на
переработку
чают и отгоняют углеводороды из адсорбента острым водяным паром.
Отогнанные углеводороды конденсируют, отделяют от воды и
при необходимости подвергают повторному разделению. После
отгонки углеводородов адсорбент сушат, охлаждают, продувают
инертным газом и вновь включают адсорбер в систему поглощения.
Таким образом, адсорберы с непо-
отходящас движным слоем поглотителя
работают периодически. Ввиду малой
производительности таких
аппаратов и значительного расхода пара
описываемый процесс разделения
углеводородных газов адсорбцией
получил сравнительно
ограниченное распространение.
В промышленности все больше
находит применение гиперсорбция—
непрерывное разделение газовых
смесей избирательным поглощением
отдельных компонентов газа
медленно движущимся слоем активного
угля.
Гиперсорбер (рис. 52)
представляет собой вертикальный
цилиндрический аппарат (высота 30 м и
более), в котором непрерывно
движется сверху вниз зернистый активный
уголь. В верхней части аппарата
уголь, проходя вертикальные
трубки, сушится и охлаждается (зона
охлаждения /). Ниже находится
зона адсорбции //. Поступающая в нее
газовая смесь движется
противотоком углю, который адсорбирует
более тяжелые компоненты смеси.
Непоглощенные легкие
углеводороды отводятся из верхней части
аппарата. В нижней трубчатой части—в зоне десорбции III уголь
продувается паром, при этом из адсорбента выделяются
поглощенные углеводороды. Чтобы продукты десорбции не
смешивались с поступающим в гиперсорбер исходным газом, разделяемую
газовую смесь вводят на 5—6 м выше точки вывода десорбирован-
ного газа.
При таком размещении входного и выходного
патрубков гидравлическое сопротивление слоя взвешенного угля
достаточно, чтобы исключалась возможность проникания десорбиро-
ванного газа в зону адсорбции.
Рис. 52. Схема работы гипер-
сорбера:
I— зонз охлаждения; //—зонз
адсорбции; /II—зона десорбции.
J56
Регенерированный адсорбент удаляется из нижней части
аппарата с помощью специального регулирующего механизма и
пневматически подается в верхнюю часть гиперсорбера.
Компримирование с одновременным охлаждением применяется
для грубого разделения паро-газовых смесей углеводородов.
Разделяемую смесь сжимают до давления 15—20 am и затем
охлаждают водой (реже рассолом); парообразные углеводороды при
этом конденсируются и отделяются от газов, не
конденсирующихся в этих условиях*. Такой метод используется для выделения
газового бензина из «жирных» природных газов (стр. 27).
Для более четкого разделения газовых смесей применяют
конденсационно-ректификационный метод. Смесь сжимают и
охлаждают до очень низкой температуры (порядка —100 °С).
При этом углеводороды С2—С5 сжижаются, а метан и водород
остаются в газовой фазе. Сжиженные углеводороды затем
подвергают ректификации под давлением при низких температурах.
Для достижения низких температур используют испарение
сжиженных газов (аммиак, этилен и т. д.), а также способ
дросселирования, основанный на свойстве газов сильно охлаждаться при
быстром понижении давления.
На рис. 53 изображена схема разделения газов пиролиза
керосина конденсационно-ректификационным методом. Исходный газ,
очищенный от сероводорода и двуокиси углерода, поступает в
трехступенчатый компрессор 1, где сжимается в первых двух
ступенях до давления 15 am. Из второй ступени компрессора газ
через теплообменник 2 поступает в конденсационно-отпарную
колонну 3, верхняя часть которой охлаждается испаряющимся в
вакууме жидким аммиаком. При температуре до —40 °С
конденсируются углеводороды С4—С5, а также вода и бензол, выпадающие
в виде кристаллов. Растворяющиеся в конденсате этилен, этан и
пропилен отпариваются в кубе колонны глухим паром. Жидкие
углеводороды С4—С5 из нижней части колонны 3 направляются
на ректификацию. Колонну периодически очищают от
кристаллов льда и бензола.
Газообразные углеводороды, выходящие из верхней части
колонны 3, нагреваются в теплообменнике 2 и поступают в третью
ступень компрессора /, где сжимаются до 30 am. Сжатые газы
охлаждаются в комплексном теплообменнике 4 возвратными
продуктами разделения газовой смеси и испаряющимся аммиаком и
направляются на конденсацию в конденсационно-отпарную
колонну 5. Эта колонна в верхней части имеет несколько трубчатых
секций, охлаждаемых различными хладоагентами. Нижняя
трубчатая секция / колонны 5 охлаждается испаряющимся этаном
* Если температура, до которой проведено охлаждение, выше
критической для данного углеводорода,—он является газом, если температура ниже
критической,—углеводород находится в состоянии насыщенного или
ненасыщенного пара.
157
, .- ,.-,, ,IWV.._. ,!..,..-, ., -, - 1 .-. - ^
лонны 7. Секция // охлаждается испаряющимся этиленом (темп.
кип. —104,0 °С), поступающим из ректификационной колонны 6.
В секцию /// со специальной компрессорной установки (на схеме
не показана) подается под давлением 20—25 am жидкий этилен,
который дросселируется и испаряется в этой секции,
дополнительно охлаждая разделяемую смесь.
Испарившийся этилен из секции /// возвращается на
компрессорную установку для сжижения и вновь поступает в эту же
секцию аппарата на испарение, совершая замкнутый цикл.
Рис. 53. Схема установки разделения газов конденсащюнно-ректификацион-
ным методом:
/—компрессор; 2—теплообменники: 3. 5—конденсационно-отпарные колонны; 4— комплексный
теплообменник; 6, 7—ректификационные колонны; 5—дроссельные вентили.
Самая верхняя секция IV колонны 5 предназначена для
отделения газа, дросселированного с помощью вентиля 8 с 30 до
1 am и не сжижающегося в колонне (метан и водород). Этот газ
по выходе из колонны имеет температуру —100 °С.
В колонне 5 конденсируются этилен, этан, пропилен и
пропан; в отпарной части аппарата отгоняется от смеси метан, а
сжиженные углеводороды С2—С3 направляются в
ректификационную колонну 6. В колонне, обогреваемой в нижней части паром,
а в верхней—охлаждаемой кипящим аммиаком, под давлением
20—25 am производится ректификация поступившей сжиженной
смеси. Этилен из верхней части колонны 6 направляется на
испарение в секцию // колонны 5, кубовая жидкость, содержащая этан,
158
пропилен и пропан, поступает на ректификацию в колонну /.
Получаемый здесь дистиллят—этан—направляется на испарение
в секцию / аппарата 5, а пропан-пропиленовая фракция из куба
колонны 7 поступает в комплексный теплообменник 4.
Описанный способ разделения газов на компоненты может
быть осуществлен в различных вариантах. Выбор той или иной
схемы с использованием различных сочетаний температур и
давлений и применением соответствующей аппаратуры определяется
составом исходной газовой смеси, целевым назначением
продуктов разделения, заданным числом фракций и требуемой чистотой
индивидуальных углеводородов.
В табл. 13 приведен примерный состав фракций, получаемых
при разделении газа пиролиза керосина конденсационно-ректифи-
кационным методом.
Таблица 13
Примерный состав фракций, получаемых при разделении газа
пиролиза коиденсационно-ректификационным методом
Компоненты
н2
сн4
с2н6
с3н8
С4Н10
С2Н4
сэн6
С4Н8
Дивинил
Амилены и пентаны
Высшие углеводороды
Содержание компонентов во фракциях,
объемн. %
метан
о-водородная
35
62
3
этиленовая
1—4
3—6
90—95
1—3
эта новая
90
6
4
пропи-
леновая
2
9
88
1
бутиле-
новая
.8
2
65
22
3
аннленовая
1
7
80
12
РАЗДЕЛЕНИЕ ЖИДКИХ СМЕСЕЙ
Наиболее распространенным методом разделения жидких
углеводородных смесей на индивидуальные компоненты является
ректификация—разделение путем многократного повторения
протекающих одновременно процессов частичного испарения и
конденсации компонентов смеси (стр. 42).
Ректификацию проводят под атмосферным давлением или под
давлением больше или меньше атмосферного. Ректификация под
избыточным давлением применяется в тех случаях, когда
компоненты разделяемой смеси в обычных условиях находятся в га-
зэобразном состоянии. Вакуум-ректификация используется при
необходимости понижения температуры в процессах разделения.
159
¦Расринат
Ч-Удіісінч U jj Л/і^ ^чіу ЧііСи jjfiO^J^iCiilld ЛиіДЛПЛ J 1 іЛСОидиридПОІЛ
смесей на индивидуальные компоненты при помощи обычной
ректификации затруднительно или вообще невозможно. Например,
обычной ректификацией нельзя разделить смесь компонентов,
образующих азеотропные смеси*. Затруднительно также
разделение компонентов с
близкими температурами
кипения. Для разделения
таких смесей применяют
экстракцию, азеотропную
или экстрактивную
перегонку, фракционную
кристаллизацию и т. д.
Экстракция.
Разделение жидких смесей
методом экстракции основано
на применении
селективных растворителей (стр.
127). Такие вещества, как
жидкий сернистый
ангидрид, фенол, диэтиленгли-
коль, хорошо растворяют
бензол и его гомологи и
В в меньшей
степени—углеводороды с прямой цепью.
Для повышения
растворяющей способности и
селективности часто
используют смеси
различных растворителей.
По технологическому
оформлению процесс
экстракции сходен с абсорбцией. На рис. 54 показана схема
экстракционной установки, используемой в тех случаях, когда
растворитель имеет большую плотность, чем разделяемые жидкости, и
очень мало растворим в легких компонентах смеси. Исходная
разделяемая смесь вводится в среднюю, растворитель—в
верхнюю часть экстракционной колонны. При контакте с разделяемой
смесью растворитель экстрагирует часть компонентов,
образующийся раствор (экстракт) стекает по насадке и отводится из
колонны снизу. Экстракт направляют на ректификацию, в
процессе которой выделяются растворенные компоненты исходной
смеси. Регенерированный растворитель возвращают на орошение
экстракционной колонны. При дополнительной ректификации
* Азеотропными или постояннокипящими смесями называют жидкие смеси,
состав которых одинаков с составом равновесных с ними паров, например смесь
этиловый спирт (95,57%)—вода D,43%), темп. кип. смеси 78,13 СС.
Рис. 54. Схема установки для экстракции
растворителями:
/—экстракционная колонна; 2—холодильник;
Л—дефлегматор; 4—ректификационная колонна; і—насос;
Ь—кипятильник; 7— теплообменник.
160
Азеотропний Низкокипящий
агент компонент
выделенных компонентов могут быть получены индивидуальные
вещества. Из верхней части экстракционной колонны вытекают
компоненты исходной смеси (рафинат), не растворяющиеся в
зкстрагенте.
Азеотропная ректификация применяется для выделения
компонентов из нераздельно кипящих (азеотропных) смесей. Этот
способ ректификации основан на том, что в исходную разделяемую
смесь вводят новый компонент,
называемый азеотропним, или
разделяющим агентом. Он образует с
выделяемым компонентом азеотропную
смесь с минимальной температурой
кипения*.
На рис. 55 показана схема
ректификации с применением
азеотропного агента, который образует с
компонентом разделяемой смеси гетеро-
азеотропную** систему. Исходную
смесь и азеотропний агент подают
в ректификационную колонну /, из
которой отбирают в качестве
дистиллята азеотропные смеси
разделяющего агента с отгоняемыми
компонентами. Из верхней части пары,
близкие по составу к гетероазеотропу,
направляются в дефлегматор 2, а
затем в сепаратор 3, где
конденсат расслаивается. Слой, богатый Рис- 55- Сх?ма установки для
/ „ азеотропнои ректификации:
азеотропним агентом (в данной схе- , і ,
г , /—ректификационная колонна; 2—де-
МЄ бОЛЄЄ ЛеГКИИ), В ВИДЄ фЛЄГМЬІ флегматор; 3—сепаратор: 4—кнпятнль-
возвращают на орошение в колон- ннк'
ну /.Из нижней части сепаратора отводят выделенный из
исходной смеси низкокипящий компонент.
Кубовую жидкость, которая также является продуктом
разделения, содержащим незначительное количество азеотропного
агента, подвергают обычной ректификации.
Экстрактивная ректификация. Для разделения жидких
смесей методом экстрактивной ректификации в исходную смесь
вводится новый компонент. В отличие от азеотропнои при
экстрактивной ректификации новый компонент представляет собой
селективный растворитель с более высокой температурой кипения, чем
компонент
* Температура кипения азеотропнои смеси может быть ниже температур
кипения входящих в нее компонентов (смеси с минимальной температурой
кипения) или выше температур кипения компонентов (смеси с максимальной
температурой кипения).
** Гетероазеотропной (расслаивающейся) системой называется азеотроп-
пая смесь двух компоиептов с ограниченной взаимной растворимостью.
11—805
161
разделяемая смесь углеводородов. Раствор тяжелых компонентов
в растворителе отводится из нижней части ректификационной
колонны.
Выделение индивидуальных углеводородов из продуктов
переработки нефтяного сырья и природных горючих газов является
одной из важнейших областей применения методов азеотропной
и экстрактивной ректификации. Для выделения ароматических
углеводородов с помощью экстрактивной ректификации в качестве
разделяющих агентов используют полярные растворители—одно-
или двухатомные спирты, фенол, анилин и др. При азеотропной
ректификации для выделения ароматических углеводородов
применяют ацетонитрил, ацетон, метилэтилкетон, уксусную
кислоту и др.
Фракционная кристаллизация. Этот метод, основанный на
выделении кристаллов из растворов или расплавов при их
охлаждении, позволяет достигать сравнительно четкого разделения
компонентов. Примером применения этого метода в заводских
масштабах может служить разделение смеси п- и лі-ксилолов. Смесь
осушают и охлаждают в кристаллизаторе до —57 °С, при этом
выпадают крупные кристаллы п-ксилола, которые отделяют на
фильтрующей центрифуге. Отфугованный продукт, содержащий 75—
80% п-ксилола, направляется на повторную кристаллизацию.
После перекристаллизации и последующего центрифугирования
получают 85—98%-ный п-ксилол, являющийся целевым продуктом.
Для выделения ж-ксилола фильтрат первой кристаллизации
смешивают с равным объемом четырех хлор истого углерода и
охлаждают до —79 °С. Четырех хлор истый углерод образует с
л-ксилолом кристаллический комплекс л-СвН4(СН3J-ССІ4
(темп. пл. —3,9 °С), который отфильтровывают.
7. СИНТЕЗЫ НА ОСНОВЕ ОКИСИ УГЛЕРОДА
В начале XX в. было доказано, что окись углерода,
считавшаяся ранее малоактивной, в определенных условиях способна
взаимодействовать с водородом, образуя углеводороды.
Пропуская смесь окиси углерода и водорода в объемном
соотношении 1 : 3 над мелко раздробленным никелем, французский
ученый Сабатье A902 г.) впервые получил простейший
углеводород—метан:
со + з.н, —»сн4 + н2о
В 1908 г. русский ученый Е. И. Орлов установил, что при
каталитическом восстановлении окиси углерода водородом
образуется связь С—С, т. е. могут быть получены не только метан, но
и высшие углеводороды. При нагревании до 100 °С смеси окиси
углерода и водорода A:2) в присутствии никель-палладиевого
катализатора Орлов получил этилен:
2СО + 4Н2 » СН2=СН2 + 2Н2О
162
В результате обширных дальнейших исследований в области
синтезов на основе окиси углерода и водорода были разработаны
способы получения углеводородов с любой длиной углеродной
цепи (вплоть до твердых парафинов), а также кислородсодержащих
органических соединений—спиртов, альдегидов, кетонов, кислот.
Промышленные синтезы на основе окиси углерода и водорода
имеют большое народнохозяйственное значение, так как для
получения ценных органических продуктов могут быть использованы
любые виды твердого, жидкого и газообразного топлива. В
зависимости от применяемого катализатора, температуры, давления
и соотношения СО : Н2 в исходной газовой смеси (табл. 14) могут
быть получены продукты различного состава.
Смесь окиси углерода и водорода, употребляемая для
синтезов, носит название синтез-газ. В обычном водяном газе (стр. 71)
соотношение СО : Н2 составляет примерно 1 : 1, но во многих
случаях требуется синтез-газ с большим содержанием водорода.
Для обогащения водородом водяной газ или часть его
обрабатывают водяным паром при 500—600 °С в присутствии
катализаторов (смесь окислов железа и магния, активированная калием,
алюминием и хромом):
СО + Н2О . > СО2 + Н2
Такой процесс носит название конверсии окиси углерода.
Большое развитие в настоящее время получили процессы
получения синтез-газа конверсией метана
СН4 + Н2О > СО + ЗН2
СН4 + СО2 > 2СО + 2Н2
СН4 + V2O2 > СО + 2Н2
и других углеводородов.
На рис. 56 показана технологическая схема получения синтез-
газа конверсией метана водяным паром. Метан нагревают в
теплообменнике 3 до 400 °С контактными газами, отходящими из
трубчатой печи (конвертора /) и направляется в смеситель 2.
Одновременно в смеситель 2 из теплообменника 4 подается водяной пар
при температуре 400 °С. Полученную паро-газовую смесь
направляют в конвертор /. В трубы конвертора, выполненные из
жароупорной стали и обогреваемые снаружи, помещают никелевый
катализатор (высота слоя 7 м).
При конверсии метана водяным паром получают смесь окиси
углерода и водорода в соотношении 1 : 3. Если требуется другое
соотношение СО : Н2 в газе (например 1:2), часть водяного пара
заменяют двуокисью углерода. На рис. 57 показана зависимость
отношения Н2 : СО в продуктах конверсии от соотношения
СО2 : СН4 в исходной смеси при различных температурах и
равном соотношении водяного пара и метана в конвертируемой
газовой смеси (Н2О:СН4=1).
11* 163
Таблица 14
Синтезы на основе окиси углерода и водорода
Синтезируемые вещества
Метан
Высшие алифатические
углеводороды
Ароматические
углеводороды
Метанол
Высшие спирты
Синтол
Катализаторы и активаторы
Ni, ThO2, MgO
Fe, Co, Ni, ThO2, MgO, A12O3,
K2O
ThO2> ZnO, Al2Oa, K2O
Ru
Cr2O3, ThO2
ZnO, Cu, Cr2O3, MnO
ZnO, Cu, Cr2O3, MnO, K2O
Fe, K2O
Температура
°С
250—500
150—350
400—500
150-250
475—500
200—400
300—450
400—450
Давление
am
1
1—30
100—1000
100—1000
30
100—1000
100—1000
100-150
Объемное
соотношение
CO : Н2
1 : 3
1 : 1
или 1 : 2
1 : 1
1 : 1
или 1 : 2
1 : 1
1 : 2
1 : 2
от 1 : П/2
до 1 : 2
Состав продуктов реакции
Преимущественно метан
Парафины (от метана до твердого
парафина) и олефины прямого
строения
Газообразные и жидкие парафины
и олефины разветвленного
строения
Высшие парафины
Преимущественно ароматические
углеводороды
Метанол
Метанол и высшие спирты
Смесь алифатических спиртов,
альдегидов, кетонов, эфиров,
органических кислот и т. д.
со+н2
Метан
топливо
Рис. 56. Схема получения синтез-газа конверсией метана
водяным паром:
/—конвертор; 2—смеситель: 3, 4—теплообменники.
¦ц
\
\
\
\500t
\
\
-CL.
-о-
Iі
I
О
о
ots і 1,5 г г,5
СО2:СН^ В исходной смеси
Рис. 57. Зависимость отношения Нг : СО от
условий процесса конверсии метана водяным паром и
двуокисью углерода
(сплошная линия—при Н2О:СН4=1,
пунктирная линия—при Н2О : CH.j = 0,5).
СИНТЕЗ КИСЛОРОДСОДЕРЖАЩИХ ОРіАНИЧЬ.Січііл ілісДИпЕНИЙ
При подборе соответствующих условий реакции и
катализатора из СО и Н2 получают индивидуальные спирты—метиловый,
первичный бутиловый и др.
Синтез метилового спирта проводят под давлением 200—
300 am и выше в присутствии катализаторов—окислов хрома,
окиси цинка и др. По технологической схеме и аппаратурному
оформлению этот процесс -напоминает синтез аммиака.
Повышенное давление способствует смещению равновесия реакции
СО + 2Н2 <—» СН3ОН
в сторону образования метилового спирта, а при повышении
температуры равновесие сдвигается влево.
При низких температурах реакция проходит полнее, однако
в промышленности процесс проводят при температуре 300—400 °С,
так как при этой температуре возрастает активность катализатора
Синтез-газ
СО+2Н2
Рис. 58. Схема синтеза метилового спирта из СО и J-fe:
/—компрессор; 2—смеситель; 2—теплообменники; 4—колонна синтеза;
5—холодильники-конденсаторы; б—сборник-сепаратор; 7—циркуляционный компрессор; 8—пусковой
электроподогреватель.
(ускоряется процесс), что позволяет работать с большей объемной
скоростью (стр. 64). Выход побочных продуктов незначителен.
При однократном прохождении синтез-газа через слой
катализатора в контактных газах содержится только 3—4% СН3ОН.
После отделения сконденсировавшегося метилового спирта непро-
реагировавшие газы (циркуляционный газ) смешивают со
свежим синтез-газом и вновь направляют в контактный аппарат.
На рис. 58 изображена схема получения метилового спирта из
СО и Н2. Исходный синтез-газ, предварительно очищенный от
пыли и примесей, отравляющих катализатор, нагнетают
компрессором / под давлением 250 am в смеситель 2. Отсюда смесь цирку-
166
ляционного и синтез-газа направляют в теплообменники 3 и далее
в колонну синтеза 4. Контактные газы выходят из колонны,
охлаждаются в теплообменниках 3 и в холодильнике 5. Пары метилового
спирта конденсируются и отделяются в сборнике-сепараторе 6
от непрореагировавших газов. Циркуляционные газы
компрессором 7 нагнетаются в смеситель 2. При накоплении в
циркуляционном газе примесей его выводят
из системы, которую затем
заполняют свежей газовой смесью.
Колонна синтеза выполняется из
качественной стали и футеруется
изнутри медью. Это предотвращает
взаимодействие окиси углерода с
железом и образование пентакарбонила
железа Fe(COM, являющегося
вредной примесью.
Нарис. 59 схематически показана
колонна синтеза с полочной
насадкой. Диаметр колонны 800 мм,
высота 12 м, толщина стенок корпуса
90 мм. В верхней части колонны
размещается катализаторная
коробка с полками для катализатора и
электроподогревателем, в нижней
части—теплообменник.
Поток синтез-газа вводится
сверху и проходит вниз по кольцевому
пространству между корпусами
колонны и катализатор ной коробки.
Далее газ поступает в межтрубное
пространство теплообменника и
подогревателя и нагревается за счет
тепла продуктов реакции,
движущихся по трубам. В межтрубном
пространстве имеются перегородки,
направляющие часть газового
потока поперек труб. Из теплообменника
нагретый газ через центральную
трубу 2 поступает в катализаторное
пространство, где образуется
метиловый спирт. Продукты реакции
проходят по трубам
теплообменника, охлаждаясь поступающим
свежим газом, и через боковой отвод в нижней крышке выводятся
из колонны синтеза.
Для предотвращения перегрева катализаторной массы
предусмотрена подача холодного байпасного газа. Для этого на каж-
Рис. 59. Колонна синтеза
метилового спирта:
/—корпус катализаторной коробки;
2—труба для
электроподогревателя; 3—полки для катализатора;
4—теплообменник; 5—ввод
холодного газа.
167
дую полку аппарата подведены трубки с мелкими отверстиями,
через которые холодный газ вводится в контактную массу.
Количество поступающего холодного газа регулируется. В пусковой
период колонна синтеза обогревается электронагревательными
элементами, установленными в верхней части аппарата (в трубе 2).
Важнейшей областью применения метанола является
производство формальдегида путем неполного окисления СН3ОН
(стр. 232).
Кроме того, метанол используется как растворитель и как
полупродукт в синтезе диметилсульфата, диметиланилина и т. д.
Следует иметь в виду, что метиловый спирт—сильный,
преимущественно нервный и сосудистый яд с резко выраженным
кумулятивным действием. При попадании внутрь СН3ОН вызывает
тяжелое поражение зрения и смерть.
Из окиси углерода и водорода (СО : На= 1 : 2) при 400—450 °С и 100—
150 am можно получить смесь кислородсодержащих органических соединений.
Процесс проводят в присутствии катализатора—железных стружек,
обработанных поташом. Эта смесь, так называемый синтол, содержит 25% альдегидов,
29% спиртов, 10% органических кислот, 5% кетонов и 4% сложных эфиров.
Содержание углеводородов в синтоле составляет только 2—2,5%.
ОКСОСИНТЕЗ
При взаимодействии окиси углерода и водорода с олефинами
(гидроформилирование) образуются альдегиды:
лО
* R—CH2-CH2-C<f
R—СН=СН, + СО + Но —
R—СН—СН3
Гидроформилирование протекает при 100—200 °С и давлении
100—250 am в присутствии катализатора (нафтената или олеата
кобальта).
Образующиеся альдегиды содержат на один атом углерода
больше, чем исходный олефин, например:
СН2=СН2 -г СО + Н2 » СН3-СН2-С
этилен пропионовый альдегид
Альдегиды затем восстанавливают водородом до одноатомных
спиртов. 1 акой двухстадийный процесс носит название оксо-
синтеза.
Методом оксосинтеза мой но получить широко используемые
первичные спирты (пропиловый, бутиловый, амиловый и высшие),
в то время как при гидратации олефинов (стр. 204) получаются
только вторичные и третичные спирты (за исключением этилового).
168
При взаимодействии альдегидов с водой (во второй стадии
оксосинтеза) образуются спирты и кислоты:
.о
<f
..О .
2R-C<f + Н2О » R-CH2OH + R-C<f
Далее они могут реагировать между собой с образованием
сложных эфиров.
Окислением альдегидов можно получать соответствующие
кислоты, по реакции В. Е. Тищенко—сложные эфиры, альдольной
конденсацией—альдегидсспирты и т. д. Процесс оксосинтеза имеет
большое значение для производства спиртов и жирных кислот из
олефиновых углеводородов С8—С12. Получаемые этим способом
спирты и кислоты используются в производстве синтетических
моющих средств (стр. 331 ел.), пластификаторов и т. д.
Технологическое оформление промышленных установок
оксосинтеза определяется главным образом типом применяемого
кобальтового катализатора.
Все известные схемы процесса оксосинтеза можно разделить
на пять групп:
1) схемы, в которых в качестве катализатора используются
соединения кобальта, нанесенные на кизельгур и суспедирован-
ные в углеводороде в количестве 3—5% от веса сырья. После
окончания реакции катализатор отфильтровывают;
2) схемы, где катализатором является раствор карбонила
кобальта в инертном растворителе (если исходный олефин—газ)
или в исходном жидком олефине. Разложение карбонила
кобальта (декобальтизация) производится в отдельном аппарате;
3) периодические схемы со стационарным слоем катализатора.
Реакционный аппарат заполняют пемзой с осажденным на ней
кобальтом. При пропускании через насадку окиси углерода,
водорода и олефина образуется катализатор—карбонил кобальта
и одновременно протекает реакция гидроформилирования.
Карбонил кобальта вымывается жидкими продуктами реакции и
выводится с ними из реактора;
4) схемы, в которых используют растворы солей кобальта
в исходных олефиновых углеводородах или в инертных
органических растворителях. В этом случае удобнее всего применять
хорошо растворимый нафтенат кобальта, так как при гидроформи-
лировании нафтенат сравнительно легко превращается в
карбонил—активный катализатор процесса. Декобальтизация
образующихся продуктов производится кислотной обработкой с
последующим превращением солей кобальта в нафтенат;
5) схемы с применением водорастворимых солей кобальта
(например, ацетата кобальта). В условиях процесса
гидроформилирования водные растворы солей также образуют активный
карбонил кобальта, являющийся катализатором реакции. Декобаль-
169
ІИіации ииралуіиЩ,ИЛС.Я ІІрОДуКТОЬ іїрииаьидиіі,« раоойБЛспгіии
уксусной кислотой. При этом получают хорошо растворимый в
воде и не растворимый в углеводороде ацетат кобальта.
На рис. 60 показана упрощенная технологическая схема
процесса гидроформилирования олефинов с применением в качестве
катализатора нафтената кобальта. Сырье смешивается в аппарате
/ с катализатором—нафтенатом кобальта (концентрация кобальта
NaOH
или
Органический
рост
RCOONa I I I CoSO4
гидрирование
Рис. 60. Схема процесса гидроформилирования в присутствии
нафтената кобальта:
/, //—смесители; 2—подогреватель; 3—колонна синтеза; 4—холодильник;
5—сепаратор; б—скруббер; 7—декобальтизатор; 8—промынной аппарат;
9, 10, 12— отстойники.
составляет около 0,2%). Смесь подогревают до 160 °С и направляют
в реактор 3 (колонна синтеза), заполненный насадкой.
Одновременно в реактор подают смесь окиси углерода и водорода.
Процесс проводят под давлением 210—245 ат и при температуре
170—180 °С.
Продукты реакции, содержащие растворенные карбонилы и
непрореагировавшие газы, охлаждают и направляют в
сепаратор 5, где газы отделяются от жидкости.
Далее газы промываются в скруббере 6 и затем возвращаются
в реактор 3.
Жидкие продукты из сепаратора 5 частично возвращают в
реактор для отвода реакционного тепла, остальное количество
направляют в декобальтизатор 7. Туда же подают 5—10%-ную
170
серную кислоту и перекись водорода. Процесс декобальтизации
проводят при 60—70 °С. В отстойнике 10 жидкость разделяется;
нижний слой—водный раствор сульфата кобальта—направляют
в смеситель // для получения нафтената кобальта. Верхний слой
передают в аппарат 8 на промывку водой или водным раствором
щелочи. Далее продукты реакции, содержащие альдегиды,
направляют на гидрирование для получения одноатомных первичных
спиртов.
Синтез спиртов в одну стадию. Одностадийный синтез
первичных спиртов осуществляют исходя из олефина, окиси углерода и
воды:
R'-CH=CH-R" + ЗСО + 2Н2О
R'—CH2—CH—CH2OH
СН2ОН
R'—CH-CH2—R"
2СО2
где R' и R"—алкильные радикалы.
Окись углерода присоединяется к обоим атомам углерода при
двойной связи, что приводит к образованию двух изомерных
спиртов.
Этот процесс отличается от оксосинтеза тем, что его проводят
при сравнительно низких давлениях и температурах. Для
низших олефинов оптимальными условиями реакции являются:
давление 10—15 am и температура 90—110вС. При использовании
вышекипящих олефинов требуются более жесткие условия—
более высокие температуры и давления. Олефины, имеющие
разветвление у атома углерода, связанного двойной связью, в эту
реакцию вступают с трудом. В качестве катализатора применяют
производные гидрокарбонила железа, получаемые из пентакар-
бонила железа, амина и воды. Установлено, что для получения
катализатора могут применяться лишь немногие амины.
Особенно легко взаимодействуют с карбонилами железа N-алкилпирро-
лидины, алкильный остаток которых должен содержать такое
же число атомов углерода, что и синтезируемый спирт.
Например, для синтеза бутилового спирта целесообразно применять
N-бутилпирролидин:
I
СН2—
2\
>N-C4H9
Амин загружают вместе с пентакарбонилом железа, водой и
бутиловым спиртом (растворитель). При нагревании до 100 "С
в течение 4 ч образуется раствор катализатора. При пропускании
через этот раствор окиси углерода и пропилена под давлением
15 am получают бутиловый спирт.
171
КАРьиНОиЫл
Важным промышленным синтезом является получение
органических кислот при непосредственном взаимодействии окиси
углерода с олефином и водой:
R—СН=СН2 + СО + М2О R—СН—СН3
СООН
Например, исходя из этилена, можно получить пропионовую
кислоту:
СН2=СН2 + СО + Н2О » СН3—СН2—СООН
Эти процессы проводят под высоким давлением (до 1000 am)
при 300—400 °С в присутствии катализаторов (фосфорная или
соляная кислоты, фтористый бор BF3 и др.).
Из ацетилена этим способом получают непредельную
акриловую кислоту, которую затем можно использовать для синтеза
ряда производных (эфиры, тиоэфиры, амиды и др.), имеющих
большое значение в промышленности пластических масс (стр. 546).
В качестве источника окиси углерода используют карбони-
лы металлов, например Ni(COL. Реакция
4CHsCH + Ni(COL + 2НС1 + 4Н2О » 4СН2=СН—СООН + NiCl„ + Н2
акриловая кислота
протекает под атмосферным давлением при 40—50 °С в
присутствии кислот или галоидов (для удаления никеля из сферы реакции).
Органические кислоты можно получить также при
взаимодействии СО с одноатомным спиртом, например, уксусную кислоту
из метилового спирта и окиси углерода:
СН3ОН + СО —-* СН3—СООН
Синтезы органических кислот на основе окиси углерода весьма
перспективны, так как дают возможность получать в
промышленных масштабах как низшие, так и высшие индивидуальные
кислоты.
Техника безопасности при работе с окисью углерода
Окись углерода—бесцветный газ, без вкуса и практически без
запаха, что особенно затрудняет его органолептическое
определение. Окись углерода является сильным ядом кумулятивного
действия, т. е. обладает способностью постепенно накапливаться в
организме. При взаимодействии СО с гемоглобином крови
образуется карбоксигемоглобин—стабильное соединение, не
поглощающее кислород воздуха и тем самым мешающее крови быть
переносчиком О2. Результат длительного воздействия окиси
углерода в малых концентрациях (хроническое отравление)
проявляется у человека через 2—3 месяца. При этом наблюдаются головные
боли, головокружение, потеря зрения и чувствительности кожи.
172
Острое отравление вызывает расстройство центральной
нервной системы, координации движений, обморочное состояние,
судороги и смерть.
Предельно допустимое содержание СО в воздухе
производственных помещений 20 мг/м3.
Концентрация окиси углерода в воздухе производственных
помещений выше 500 мг/м3 опасна, при концентрации 4500—
5000 мг/м3 смерть наступает через несколько минут.
Для обеспечения безопасных условий труда при получении и
переработке окиси углерода обслуживающий персонал, кроме
общих правил техники безопасности, должен строго соблюдать
специальные меры предосторожности.
В цехах устанавливаются чувствительные автоматические
газоанализаторы, регистрирующие содержание СО в пределах от
0,002 до 0,1% с точностью до 0,002%. При неблагоприятных
результатах анализа немедленно подается автоматический сигнал
(звонок, сирена), оповещающий о том, что каждый рабочий должен
сразу же принять меры личной безопасности согласно инструкции.
При проектировании цехов необходимо учитывать, что окись
углерода легче воздуха (относительная плотность по воздуху
0,97), при попадании в помещение она накапливается главным
образом в верхних слоях воздуха. Обслуживающий персонал,
находящийся на площадках или верхних этажах, подвергается при
этом наибольшей опасности. Поэтому производственные
помещения необходимо обеспечить вентиляционными устройствами
достаточной мощности и предусмотреть возможность быстрой
эвакуации работающих из помещения.
Следует особенно тщательно следить за герметичностью
оборудования, трубопроводов, запорной аппаратуры. Проникание
окиси углерода в производственное помещение создает опасность
отравления этим газом, ^опадание же воздуха в аппараты
или трубопроводы с окисью углерода может привести к взрыву
или пожару (пределы взрываемости газо-воздушной смеси 12,4—
75,0 объемн. % СО).
При работе с СО необходимо пользоваться гопкалитовыми
противогазами марки СО (белая окраска коробки), так как окись
углерода не адсорбируется активированным углем обычных
фильтрующих противогазов.
Действие гопкалитовых противогазов основано на
каталитическом окислении СО в СО2 кислородом воздуха
2СО + О2 * 2Со2
под влиянием гопкалита*. Образующаяся при этом двуокись
углерода связывается химическим поглотителем.
* Гопкалит—зернистый катализатор, содержащий активную двуокись
марганца МпСЬ н другие окислы.
173
вилах техники безопасности и о мерах оказания первой помощи
пострадавшим (удаление из отравленной атмосферы, вдыхание
кислорода, искусственное дыхание и т. д.).
8. ГАЛОИДИРОВАНИЕ УГЛЕВОДОРОДОВ
Процессы галоидирования широко применяются при
переработке как алифатических, так и ароматических углеводородов.
В ряде случаев введение одного или нескольких атомов галоида
придает этой молекуле повышенную реакционную способность.
Однако замена водородных атомов молекул углеводорода атомами
галоида может привести и к образованию весьма инертных
соединений, мало склонных к химическим превращениям. Свойства
галоидпроизводных отличаются от свойств исходных
углеводородов: обычно галоидпроизводные мало горючи или совсем
негорючи, термически более стойки, чем углеводороды, менее летучи
и т. д. Замещая галоид другими атомами или группами, можно
получить разнообразные вещества, синтез которых
непосредственно из углеводородов затруднителен или вообще невозможен.
Наиболее широко используются и вырабатываются в наибольших
количествах хлорпроизводные углеводородов. Их ценные свойства
и доступность элементарного хлора, необходимого для их
получения, в значительной степени способствовали развитию
промышленного производства хлорорганических соединений.
Взаимодействие углеводородов с хлором протекает с
присоединением хлора по месту ненасыщенных связей между углеродными
атомами
R—сн=сн2 -ь ci2 —»R—сна—сн2а
R—С=СН + 2С12 > R—СС12—СНС12
или с замещением атомов водорода атомами хлора:
->- R—СНС1— СН3 + НС]
R-CH2—Ch3 + а.2 -
-»- R—сн2—сн2а + на
Для хлорирования углеводородов можно применять не только
элементарный хлор, но и хлорсодержащие соединения, такие,
как хлористый сульфурил SO2C12, фосген СОС12, четыреххлори-
стый углерод СС14 и др.
Большой практический интерес представляет получение хлор-
производных взаимодействием углеводородов с хлористым
водородом. Этот процесс может быть проведен либо путем
окислительного хлорирования ¦
С1
Ч/ Ч-
174
либо путем гидрохлорирования ненасыщенных углеводородов:
СНа=СНа + НС1 , СН3—СН2С1
СН=СН + HCI СНа=СНС1
Некоторые хлориды углеводородов (метиленхлорид СН2С12,
четыреххлористый углерод СС14, трихлорэтилен СНС1=СС12,
дихлорэтан СН2С1—СН2С1) используются непосредственно как
растворители.
При дальнейшей переработке многих хлорпроизводных
получают спирты (амиловые, аллиловые), каучукоподобные материалы
(тиокол, полихлоропрен), синтетические полимеры (поливинил-
хлорид, энант), антидетонаторы (тетраэтилсвинец) и др.
В последние десятилетия стали производить в промышленных
масштабах хлорфтор- и фторпроизводные углеводородов.
Хлорфторпроизводные используются как хладоагенты в
холодильных машинах (фреоны, стр. 129), для наполнения
огнетушителей, в качестве высокотемпературных теплоносителей,
смазочных и трансформаторных масел.
Большой интерес представляют
перфторуглероды—производные углеводородов, у которых все атомы водорода замещены
фтором: C„F2n, C„F2„+2 и т. д. Перфторуглероды—химически очень
устойчивы. Они могут применяться в качестве инертных
растворителей, неокисляющихся высокотемпературных смазок,
теплоносителей, диэлектриков для токов высокой и сверхвысокой частоты
и исходных веществ в синтезах химически и термически стойких
полимеров (фторопластов, стр. 543).
ХЛОРИРОВАНИЕ ПАРАФИНОВ
При хлорировании парафинов атомы водорода в них
замещаются атомами хлора с отщеплением хлористого водорода. При этом
получают моно-, ди-, три- и полихлориды. При непосредственном
хлорировании парафиновых углеводородов нельзя выделить
индивидуальное хлорзамещенное в чистом виде—практически
образуется смесь хлоридов:
+ С12 » CnH2n+1Cl -\- НС]
СЛН2Л+1С1 + С1, * СлН2/!С1а + HCI
С„Н2„С12 + С1„ » СяН^СІз + НС1 и т. д.
При определенных условиях процесса хлорирования можно
получить преимущественно одно из хлорзамещенных.
Хлорирование парафиновых углеводородов проводят в паровой
и в жидкой фазах различными способами: нагреванием
реакционной смеси (термическое хлорирование), в присутствии различных
катализаторов (каталитическое хлорирование), при специальном
освещении компонентов реакции (фотохимическое хлорирование).
175
і-салдйіи ллиупуиЬаппН МоЖгіО ЦриьиДИіь в среде ИНерТНОГО
растворителя или в расплавленных средах.
Термическое хлорирование применяется чаще всего для
получения хлоридов низших углеводородов. Наиболее трудно
вступает во взаимодействие с хлором метан. Его хлорируют при
температуре 400—460 °С; повышение температуры хлорирования
сверх 500 °С может привести к взрывной реакции:
СН4 + 2С[2 » С + 4НС1 + 69,7 ккал
Гомологи метана вступают в реакцию с хлором легче, чем
СН4; этан хлорируется при 300—375 °С, н-пентан—при 250—
300 °С. При низких температурах атомы водорода у третичных и
вторичных углеродных атомов в молекуле углеводорода
замещаются с большей скоростью, чем у первичных; с повышением
температуры реакции это различие в скоростях уменьшается, и при
600 °С замещение проходит с одинаковой скоростью.
Каталитическое хлорирование углеводородов проводят чаще
всего в жидкой фазе, причем газообразные углеводороды
предварительно растворяют в хлорорганических растворителях.
Каталитическое хлорирование парафинов протекает при более низких
температурах, чем термическое хлорирование. Например, в
присутствии катализатора четыреххлористый углерод может быть
получен при 250—300 °С, без катализатора—при 460 °С.
В качестве катализаторов применяют как хлориды металлов
(Си, Sb, Sn, Si) и металлоидов (J, S), так и высокопористые
материалы (активный уголь, пемза, силикагель). Наиболее
активные из них—хлористая медь СиС12, нанесенная на
высокопористый материал, а также органические соединения—перекись бен-
зоила и некоторые нитрилы.
Хлорирование метана. При хлорировании метана образуются
все четыре его хлорпроизводных:
СН4 + С12 • СН3С1 + НС1
СН3С1 + С12 » СН2С12 + НС1
СН2С12 + С12 » СНСіз + НСІ
+ С12 , СС14 + НС[
Из приведенных уравнений видно, что образование
хлоридов происходит путем последовательного замещения водородных
атомов в углеводороде на атомы хлора. Эта реакция была
открыта французским ученым Дюма и получила название металепсии.
Соотношение количеств образующихся хлорзамещенных
метана зависит в значительной мере от соотношения объемов хлора
и метана в исходной смеси (рис. 61).
Хлористый метил СН3С1 преимущественно образуется при
большом избытке метана. При соотношении СН4 : С12 = 5 : 1
содержание хлористого метила в смеси образующихся хлоридов со-
176
ставляет 80%. Для преимущественного получения четыреххлори-
стого углерода требуется стехиометрическое соотношение метана
и хлора.
0,8 I? г ft 3,2
Молярное отношение хлора к метану
Рис. 61. Зависимость состава продуктов хлорирования
метана и теплового эффекта реакции от соотношений хлора
и метана.
Рис. 62. Схема хлорирования метана:
/—смеситель; 2—хлоратор; 3—холодильник; 4—колонные аппараты; 5—сепараторы;
в—компрессор; 7—конденсатор.
На рис. 62 показана схема хлорирования метана. Природный
газ с высоким содержанием метана или метано-водородную смесь
с установок разделения газов пиролиза (стр. 157) хлорируют
в стальном вертикальном цилиндрическом аппарате—хлораторе 2,
12—805 177
шу і cuUoctiiri'jm кипу l piri Шсїі?ЇО7 ііііїі«і Кііріііі пиш . и u,\_n luv. uiniuuuiu
имеется цилиндр из шамотного кирпича, установленный на
кирпичную решетку, через которую свободно проходят реакционные
газы. Процесс хлорирования протекает в кольцевом пространстве
между внутренней стенкой аппарата и цилиндром.
Выходящие из хлоратора продукты реакции охлаждаются и
последовательно проходят три колонны 4. В первой колонне
хлористый водород поглощается водой. Во второй колонне хлорме-
таны нейтрализуются водным раствором NaOH и в третьей
колонне осушаются серной кислотой. Далее паро-газовая смесь компри-
мируется до 10 am, затем охлаждается в аппарате 7 до —40 *С.
Сконденсировавшиеся хлорметаны направляются на
ректификацию для выделения индивидуальных продуктов, газы, содержащие
непрореагировавший метан и некоторые количества хлористого
метила, возвращают на хлорирование. Выход продуктов—85%.
В промышленности органического синтеза хлористый метил
(темп. кип. —23,7 °С, темп. пл. —97,6 °С, плотность при
температуре кипения 0,992 г/см3) используется для введения метильной
группы в органические соединения:
АІСІз
R-H + СН3С1 > R-СНз + НС1
Хлористый метил является исходным продуктом в синтезе
кремнийорганических соединений. В результате взаимодействия
хлористого метила со сплавом меди и кремния при 300 °С
образуется дихлордиметилсилан
2СН3С1 + Si > (CH3JSiCl2
используемый для получения силоксановых каучуков (стр. 491).
Хлористый метилен СН2С12 (темп. кип. 40,1 °С, темп. пл.
—96,8 °С, плотность при температуре кипения 1,335 г/см3)
хорошо растворяет эфиры целлюлозы, жиры и масла, с трудом гидро-
лизуется, не горюч. Пары хлористого метилена образуют с
воздухом взрывчатые смеси с узкими пределами взрываемое™ (8,8—
11,1 объемн. %).
Перечисленные выше ценные свойства хлористого метилена
определили его применение в качестве хорошего растворителя.
Он применяется в производстве негорючей кинопленки, для
очистки материалов от красок и смазочных масел от парафина.
Хлористый метилен обладает слабым наркотическим действием и менее
ядовит, чем хлороформ и четыреххлористый углерод.
Хлороформ (трихлорметан) СНС13 (темп. кип. 61,2 °С, темп,
пл. —63,5 °С, плотность при температуре кипения 1,489 г/см3)
хорошо растворяется в спирте и эфире. В присутствии кислорода
(в особенности на свету) хлороформ разлагается с образованием
очень ядовитого фосгена:
СНСІз + VaO2 * СОС12 + НС1
ITS
Хлороформ обладает сильным наркотическим действием и
ранее применялся в медицине для наркоза. Хлороформ растворяет
некоторые эфиры целлюлозы, гуттаперчу и многие синтетические
смолы. Однако наркотическое действие хлороформа препятствует
широкому использованию его в качестве растворителя.
Хлороформ применяют также для получения хлорфторугле-
водородов (стр. 129), для синтеза красителей и фармацевтических
препаратов.
Ранее хлороформ получали взаимодействием гипохлорнта кальция с
втиловым спиртом или с ацетоном при температуре 85 °С:
2C,HSOH + 4Са(ОС1J » 2СНС13 + (НСООJСа + 2Са(ОНJ + СаС[2 + 2Н2О
2(СН3)гСО + ЗСа(ОС[J » 2СНС13 + (СН3СООJСа + 2Са(ОНJ
Образующийся хлороформ вместе с парами воды выводили из реактора и
после конденсации отделяли от воды.
В настоящее время хлороформ совместно с четыреххлористым
углеродом получают фотохимическим хлорированием метилен-
хлорида (рис. 63). Процесс хлорирования проводят при 50 °С в
эмалированном или освинцованном хлораторе колонного типа.
Сі
Рис. 63. Схема фотохимического хлорирования хлористого метилеиа:
/—хлоратор; 2—холодильник; Л—сепаратор; 4—сборник; 5—ректификационные
колонны; 6—конденсаторы.
В аппарате имеются лючки для установки ртутных светильников.
Для отвода реакционного тепла хлоратор снабжают рубашкой и
теплообменными элементами, устанавливаемыми внутри.
12*
179
ной жидкостью, непрерывно подают метиленхлорид и хлор.
Образующийся побочно хлористый водород вместе с парами хлор-
метанов проходит холодильник-конденсатор 2, где
конденсируются хлорметаны, возвращаемые обратно в хлоратор, газообразный
НС1 направляют на абсорбцию для получения товарной соляной
кислоты.
Жидкие продукты реакции из хлоратора отводят в сборник 4,
затем подают в ректификационные колонны, где последовательно
выделяют непрореагировавшие метиленхлорид, хлороформ и че-
тыреххлористый углерод. Метиленхлорид возвращают на
хлорирование.
Четыреххлористый углерод (тетрахлорметан) СС14 (темп,
кип. 76 °С, темп. пл. —23 °С, плотность при температуре кипения
1,594 г/см3) хороший, практически негорючий растворитель, слабо
гидролизующийся водой. В присутствии металлов скорость
гидролиза возрастает. Четыреххлористый углерод используется в
качестве огнетушащей добавки к горючим фумигантам
(сероуглерод, дихлорэтан, стр. 350).
Старым способом получения четыреххлористого углерода является
хлорирование сероуглерода:
CS2 + ЗС12 » СС14 + S2CI2
Реакцию проводили при 50—70 °С в присутствии
катализатора—порошкообразной металлической сурьмы в количестве 0,2% от веса CS2. Из
реакционной массы отгоняли четыреххлористый углерод, а образовавшуюся
монохлористую серу использовали для получения дополнительного количества
четыреххлористого углерода. Для этого к S2CI2 прибавляли свежие порции
сероуглерода и катализатора (железные опилки). При 60 °С и перемешивании
происходило образование четыреххлористого углерода:
CSs + 2S2C12 > CCI4 + 6S
Выпавшую элементарную серу отфильтровывали, отмывали от
монохлористой серы и направляли на производство сероуглерода.
Сероуглерод получают в стальных ретортах при температуре красного
каления взаимодействием паров серы с древесным углем:
С + 2S * CS2
Пары сероуглерода затем конденсируют и очищают.
Хлорирование этана. Хлористый этил С2Н5С1 (темп. кип.
12,4 °С, темп. пл. —138,7 °С, плотность при температуре кипения
0,924 г/см3)—первый продукт замещения при хлорировании этана:
СН3—СН3 + С12 » СН3—СН2—CI + НС1
Хлористый этил получают с хорошим выходом при
значительном избытке этана в реакционной смеси (8 объемов этана на 1 объем
хлора). Хлорирование проводят при 300—375 °С. Газообразные
продукты реакции охлаждают, направляют в скрубберы, орошае-
180
мые водой, для поглощения хлористого водорода, затем сжимают
компрессором до 8—10 am и охлаждают до —25 °С. При этом
хлористый этил конденсируется, далее его направляют на очистку
и ректификацию. Непрореагировавший этан возвращают в
производственный цикл.
Этан вступает в реакцию с хлором значительно легче, чем
метан, что позволяет использовать для хлорирования не только
чистый углеводород, но и природный газ, содержащий 90% СН4
и 10% С2Нв. В указанных выше условиях этан хлорируется почти
полностью, хлорпроизводные метана практически не образуются.
В настоящее время наибольшее значение имеет способ
получения хлористого этила гидрохлорированием этилена:
СН2=СН2 + HCI
Реакцию проводят при —15 °С в присутствии катализатора—
порошкообразного безвод'ного хлористого алюминия.
Предварительно исходные хлористый водород и этилен тщательно осушают.
Процесс проводится в вертикальном стальном реакторе,
заполненном хлористым этилом с взвешенным в нем катализатором. Через
жидкость пропускают смесь хлористого водорода и этилена. Для
равномерного суспендирования катализатора в жидкости и более
полного соприкосновения газа с реакционной средой реакцию
проводят при интенсивном механическом перемешивании. Тепло
реакции отводится рассолом, циркулирующим в рубашке
аппарата. В результате образования хлористого этила объем реакционной
смеси увеличивается, избыток ее непрерывно удаляют.
Вытекающий из реактора жидкий хлористый этил содержит взвешенные
частицы катализатора и растворенный хлористый водород. Для
очистки хлористого этила его испаряют, промывают в скруббере
10%-ным раствором щелочи, осушают серной кислотой и
конденсируют.
Взаимодействием хлористого этила со сплавом свинца и
металлического натрия получают тетраэтилсвинец
4PbNa + 4GjH5Cl > Pb(C2H5L + 4NaCl + ЗРЬ
применяемый как антидетонатор моторного топлива (стр. 49).
ХЛОРИРОВАНИЕ И ГИДРОХЛОРИРОВАНИЕ ОЛЕФИНОВ
При хлорировании предельных углеводородов атомы водорода
замещаются на атомы хлора. Для непредельных углеводородов
характерна не только реакция замещения, но и присоединение
атомов галоида с образованием более насыщенных соединений.
Этиленовые углеводороды присоединяют хлор с образованием
дихлорпроизводных парафинового ряда, например:
СН2=СН2 + С12 * С1СН2—СНаС1
этилен 1,2-днхлорэтам
181
Скорость присоединения галоидов возрастает с увеличением
молекулярного веса олефина; наиболее медленно реагирует с
галоидами этилен.
Монохлорпроизводные парафиновых углеводородов можно
получить путем гидрохлорирования олефинов:
СНЗЧ
СН2=СН—СН3 + НС1 * >СНС1
сн3/
пропилен хлористый
изопропил
Согласно правилу В. В. Марковникова, атом галоида
присоединяется к наименее гидрогенизироваиному атому углерода.
Поэтому при гидрогалоидировании непредельных углеводородов
образуются только вторичные и третичные галоидпроизводные.
Одновременное присоединение галоидов и замещение водородных
атомов галоидами приводит к образованию полигалоидных
соединений. Повышенная температура и избыток галоида по отношению
к олефину способствуют протеканию реакции замещения. При
низких .температурах и избытке олефина продукты замещения
образуются в ничтожных количествах. В процессах
взаимодействия олефинов с хлором при высоких температурах вместо
присоединения атомов хлора по месту двойной связи происходит
замещение хлором атомов водорода метильной группы. Например,
в то время как при хлорировании этилена при низкой температуре
•образуется дихлорэтан
СН2=СН2 + С12 > С1СН2—СН2С1
¦при температуре выше 500 °С —хлористый винил
СНа=СН2 + С1а » CHCl=CHj + НС1
Аналогично из пропилена при низкой температуре получают
дихлорпропан
СН2=СН—СН3 + С12 » СН2С1—СНС1—СН3
а при 500 °С—хлористый аллил
СН2=СН—СН3 + С12 » СН2=СН—СН2С1 + НС1
В молекулах третичных олефинов (изоолефинов) замещение в
метильной группе происходит и при низкой температуре.
Реакция между хлором и изобутиленом даже при —50 °С приводит
в основном к получению метилхлоропрена (хлористого метал-
лила):
СНЗЧ СН2С1
>С=СН2 + С12 » ^С=СН2 + НС1
av СНз'
ш
Получение дихлорэтана. В промышленности дихлорэтан .
получают путем присоединения хлора к этилену:
СН2=СН2 + С12 » С1СН2—СН2С1
Сухие газообразные хлор и этилен при обыкновенной
температуре реагируют чрезвычайно медленно. Скорость реакции
значительно увеличивается при проведении ее в растворе дихлорэтана.
Кроме того, газообразный хлор образует с этиленом
взрывоопасные смеси, а хлорирование в растворе делает этот процесс
безопасным. В жидкой фазе улучшаются также условия теплопередачи от
Вода
На
ректификацию
Рис. 64. Схема производства дихлорэтана из хлора и этилена:
/—хлоратор; 2—сборник дихлорэтана-сырца; Л—колонный конденсатор
смешения; 4—холодильник; 5—промывной скруббер; 6— нейтрализатор;
7—сепаратор; 8—сборник щелочи; 9—сборник нейтрализованного дихлорэтана.
реакционной массы к охлаждающей воде, благодаря чему
значительно облегчается отвод реакционного тепла и устраняется
опасность местных перегревов.
При хлорировании этилена может протекать и реакция
замещения, которая приводит к образованию небольшого количества
полигалоидных соединений, главным образом трихлорэтана:
СН2=СН2 + 2С12
• CICHj-CHClj + HCI
Процессы замещения ускоряются с повышением температуры.
Схема получения дихлорэтана показана на рис. 64. Исходные
хлор и этилен предварительно тщательно осушаются, так как
хлор в присутствии влаги частично гидролизуется и разрушает
стальную аппаратуру. Этилен (этиленовую фракцию) осушают
твердым хлористым кальцием или вымораживанием влаги с
помощью охлажденного рассола. Не рекомендуется применять для
осушки этиленовой фракции серную кислоту, чтобы избежать
осмолення и сульфирования непредельных углеводородов.
Хлор и этилен раздельно подают в хлоратор
/—цилиндрический стальной аппарат, заполненный дихлорэтаном. Оба газа
барботируют через раствор дихлорэтана при 30 °С. Выделяющееся
тепло отводят холодной водой, циркулирующей в рубашке и
змеевиках, установленных внутри аппарата. Отходящие из реактора
газы (непрореагировавший этилен, хлористый водород и пары
дихлорэтана) поступают в колонну 3, орошаемую охлажденным
дихлорэтаном. Сильное охлаждение (до —15 °С) приводит к
понижению парциального давления паров дихлорэтана.
Сконденсировавшийся дихлорэтан смешивают в сборнике 2 с дихлорэтаном,
вытекающим из реактора. Газы, отходящие из колонны 3,
проходят скруббер 5, где их отмывают водой от хлористого водорода, и
удаляются из системы. Из сборника 2 дихлорэтан перекачивают в
аппарат б для промывки водой и нейтрализации 5%-ным раствором
щелочи, затем отделяют от воды в сепараторе 7 и подвергают
ректификации.
Дихлорэтан приобрел большое значение в качестве исходного
продукта для получения хлористого винила (стр. 188), из
которого синтезируют поливинилхлорид (стр. 542).
Применение дихлорэтана в качестве промышленного
растворителя ограничено из-за его токсичности.
Дихлорэтан иногда применяется в сельском хозяйстве для
фумигации (обеззараживания помещения или зерна парами или
газами, ядовитыми для насекомых и возбудителей грибковых
заболеваний).
ХЛОРИРОВАНИЕ ПРОПИЛЕНА И ПОЛУЧЕНИЕ
СИНТЕТИЧЕСКОГО ГЛИЦЕРИНА
1,2-Дихлорпропан получают при взаимодействии пропилена
с хлором
СН-2=СН-СН3 + С12 » СНаС1—СНСІ-СНз
в условиях, аналогичных условиям синтеза дихлорэтана.
При высоких температурах E00 °С) пропилен реагирует с
хлором с образованием хлористого аллила:
СНа=СН—СН3 + С1а * СНа=СН—СН2С1 + НС1
пропилен хлористый аллнл
Этот процесс положен в основу синтетического способа
получения глицерина СН2ОН—СНОН—СН2ОН.
184
Хлористый аллил гидролизуют раствором едкого натра или
соды при 150—160 °С и давлении 14 am:
СН2=СН—СН2С1 + NaOH » СН2=СН— СН2ОН + NaCl
хлористый аллил аллиловый спирт
Затем в результате присоединения к аллиловому спирту
хлорноватистой кислоты получают хлоргидрины глицерина
— НОСН2—СНС1—СН2ОН
СН2=СН—СН2ОН + НОСІ —
— С1СН2—СНОН—СН2ОН
которые в свою очередь гидролизуют едким натром или водой с
образованием глицерина без применения хлора:
НОСН2—СНС1—СН2ОН + NaOH , НОСН2—СНОН—СН2ОН + NaCl
глицерин
Образовавшийся глицерин отделяют от поваренной соли и
других примесей и подвергают тщательной перегонке в вакууме.
Полученный продукт содержит 99% глицерина.
Большой интерес представляет получение глицерина
синтетическим путем из пропилена без применения хлора:
1/202 /О Н2О2
СНо=СН-СН3 * СНа=СН—C<f
пропилен акролеин
/О н2
* СН2ОН—СНОН—О? > СН2ОН—СНОН—СНаОН
хн
глицериновый альдегид глицерин
. При нормальном давлении глицерин кипит при 290 °С с
частичным разложением, поэтому перегонку его обычно проводят
в вакууме. Глицерин очень гигроскопичен и на воздухе поглощает
до 50% воды.
Большую часть производимого сейчас глицерина получают как побочный
продукт мыловарения—при омылении растительных и животных жиров и масел
(глицериды высших жирных кислот):
СН2О-ОС—С17Н,5
СНО—ОС—С17Н35 4
1
СН2О-ОС-С17Н35
глицерид стеариновой
кислоты
СН2ОН
NaOH |
- ЗН2О »¦ СНОН 4
СН2ОН
глицерин
- ЗС17Н35СООН
стеариновая
кислота
Ранее глицерин получали при спиртовом брожении Сахаров в присутствии
бикарбоната натрия NaHCO3 и сульфита натрия Na2SO3 с выходом до 14%
от исходного сахара.
Глицерин нашел широкое применение в лакокрасочной
промышленности. В больших количествах он используется для изгсь
185
товления полиэфирных (алкидных) смол путем поликонденсации
•его с фталевым ангидридом или алифатическими многоосновными
кислотами.
Применение глицерина весьма многообразно. Он используется
в парфюмерной и в ликерно-водочной промышленности, в
полиграфии—для предохранения краски от высыхания, в качестве
смазочного материала, как теплоноситель и как составная часть
незамерзающих жидкостей для охлаждения двигателей и т. д.
При нитровании глицерина азотной кислотой в присутствии
•серной кислоты (водоотнимающее средство) получается полный
-сложный эфир—глицеринтринитрат (называемый также
нитроглицерином):
+3HNO3
НОСН2—СНОН—СН2ОН * OoNOCH2—CHONO2—CH2ONO2 + 3Н2О
H2SO4
Нитроглицерин является сильным взрывчатым веществом,
применяемым для изготовления некоторых видов бездымных по-
рохов.
ГАЛОИДИРОВАНИЕ И ГИДРОГАЛОИДИРОВАНИЕ
АЦЕТИЛЕНОВЫХ УГЛЕВОДОРОДОВ
Прямое взаимодействие ацетилена и хлора сопровождается
сильным взрывом:
СН=СН + С12 » 2С + 2НС1 + 98,8 ккал
Безопасный способ хлорирования ацетилена был разработан
акад. Е. А. Фаворским с сотрудниками. По этому способу
хлорирование ацетилена проводят в жидкой фазе (в среде тетрахлорэта-
на) в присутствии катализатора—железа, вводимого в зону
реакции в виде насадки из стальных колец.
В результате присоединения хлора к ацетилену образуется
насыщенное соединение—смлш-тетрахлорэтан, обладающий
некоторой токсичностью и легко гидролизующийся. Он почти не
применяется как растворитель, но является исходным продуктом для
получения ряда других хлорпроизводных:
+2СІ2
сн=сн > сі2сн-снсі2
ацетилен силл-тетрахлорэтан
Са(ОНJ
С12СН-СС13 <+С1а С1СН=СС12
пентахлорэтан трнхлорэтилен
Са(ОНJ +СІ2
¦ С13С=СС12 — ¦-» СС13-СС13
тетрахлорэтилен гексахлорэтан
186
Взаимодействием ацетилена с хлористым водородом в
присутствии катализатора может быть получен хлористый винил
(стр. 188).
Получение трихлорэтилена. Трихлорэтилен получают иі
силш-тетрахлорэтана дегидрохлорированием щелочью или при
повышенной температуре в присутствии катализатора (без
щелочи).
Трихлорэтилен широко используется как ценный технический
растворитель.
Для дегидрохлорирования тетрахлорэтана применяется из-
ес тковое молоко:
2С1аСН—СНС1а + Са(ОНJ
2С1СН=СС12 + СаС12 + 2Н2О
При действии более сильных щелочей образуется нестойкое
соединение—дихлорацетилен, обладающий взрывчатыми
свойствами:
С12СН—СНСІ2 + 2NaOH > С1С=СС1 + 2NaCl + 2Н2О
Схема производства трихлорэтилена дегидрохлорированием
тетрахлорэтана известковым молоком представлена на рис. 65.
Трихлорэтилен
на ректшрикоцит
са(онJ
Рис. 65. Схема дегидрохлорирования
тетрахлорэтана:
/—испаритель; 2—реакционная колонна; 3—аппарат для
приготовления известкового молока; 4—конденсатор;
5— сепаратор.
Тетрахлорэтан подают в испаритель /, обогреваемый глухим
паром, отсюда пары тетрахлорэтана поступают в нижнюю часть
колонны 2, орошаемой известковым молоком, которое
приготовляется в аппарате 3, снабженном мешалкой. В колонне 2 при 90 °С
18Г
образуется трихлорэтилен. Для поддержания требуемой
температуры реакции в нижнюю часть колонны подается острый пар. Из
верхней части колонны пары трихлорэтилена, непрореагировав-
шего тетрахлорэтана и воды поступают в конденсатор 4.
Конденсат стекает в сепаратор 5, где жидкость расслаивается. Верхний
слой (вода) сливается в канализацию, нижний слой,
представляющий собой смесь трихлорэтилена и тетрахлорэтана,
направляется на ректификацию для отделения СНС1=СС12,
передаваемого далее на склад. Тетрахлорэтан возвращается в испаритель /
и вновь поступает на омыление.
Получение трихлорэтилена пиролизом тетрахлорэтана
заключается в пропускании паров последнего при температуре около
500 °С над катализатором (активный уголь, силикагель, пемза
и др.):
С12СН—СНС|2 » С1СН=СС12 + НС|
При этом способе отпадает необходимость расхода щелочи,
а отщепляемый хлористый водород может быть использован для
получения соляной кислоты.
Возможно также получение трихлорэтилена из этилена по
следующей схеме:
+СІ2 +СІ2 +СІ2
СН2=СН2 * С1СН2—СН2С1 * С1СН2—СНС|2 *
дихлорэтан трнхлорэган
> С1СН2—СС13 > С1СН=СС13
—неї
несимм-
тетрахлорэтан трихлорэтнлен
Получение хлористого винила. Хлористый винил СН2=СНС1
(как и другие моногалоидные производные олефинов, содержащие
галоид при углеродном атоме, имеющем двойную связь) может
быть получен двумя путями: гидрохлорированием ацетилена
CHsCH + HCI » СН2=СНСІ
или отщеплением хлористого водорода (дегидрохлорирование)
от дигалоидного производного, в данном случае от дихлорэтана:
С1СН2—СН2С1 > СН2=СНС1 + НС1
В промышленности хлористый винил получают
гидрохлорированием ацетилена в жидкой и в газовой фазе. Второй способ более
совершенный, так как требует аппаратуры меньшего объема
и может быть легко автоматизирован.
На рис. 66 показана схема гидрохлорирования ацетилена в
газовой фазе. Синтез проводят при 170 °С в трубчатом контактном
аппарате 2, трубки которого заполнены катализатором —
хлорной ртутью, нанесенной на активированный уголь A0%
HgCl2 от веса угля). Тепло реакции отводят циркулирующим в
межтрубном пространстве холодным минеральным маслом. По
188
мере снижения активности катализатора температуру в
контактном аппарате повышают до 200 °С, для чего в некоторых случаях
масло предварительно подогревается. В контактный аппарат
непрерывно подают очищенный ацетилен и концентрированный
хлористый водород. Контактные газы, содержащие 93%
хлористого винила и 5% НО, поступают в скрубберы 3, где хлористый
Раствор
на
ректадзикацию
HCJ
Рис. 66. Схема получения хлористого винила гидрохлорированием
ацетилена в газовой фазе:
/—смеситель; 2—контактный аппарат; 3—скрубберы; 4—осушитель; 5—конденсатор.
винил последовательно отмывается водой, щелочью и далее
подвергается осушке в аппарате 4. Осушенный винилхлорид
конденсируется в конденсаторе 5 и направляется на ректификацию для
отделения от примесей (ацетальдегид, 1,1-дихлорэтан). После
ректификации из продукта отгоняют ацетилен, очищенный
жидкий хлористый винил передается в сборник.
В промышленности для получения хлористого винила из
дихлорэтана применяются два способа дегидрохлорирования
щелочью: в жидкой фазе
—СН2С1 + NaOH > С1СН=СН2 + NaCl + Н2О
в газовой фазе
С1СН2-СН2С1
• СН,=СНС1 + НС1
Жидкофазное дегидрохлорирование дихлорэтана проводят в
вертикальных цилиндрических реакторах, снабженных рубашкой
189
и пропеллерной мешалкой. В аппарат загружают 42%-ный
водный раствор NaOH и метиловый (или этиловый) спирт, затем
постепенно приливают дихлорэтан. Процесс протекает при 60—
70 °С в среде спирта, растворяющего как дихлорэтан, так и щелочь.
Чтобы предотвратить возможность образования ацетилена по
реакции
CJCH2—СН2С1 + 2NaOH > СН=СН + 2NaCl + 2Н2О
избыток щелочи не должен превышать установленного
регламентом.
При получении хлористого винила путем термического
пиролиза в газовой фазе процесс проводят при 480—500 °С в реакторе,
состоящем из двух вставленных друг в друга труб (диаметром
100 мм и 70 мм). Для обогрева аппарата в горелках,
установленных в нижней части внутренней трубы, сжигается газ. Пары
дихлорэтана поступают в кольцевое пространство между внутренней
и наружной трубами. Продукты реакции через смолоотделитель
и холодильник подаются в абсорбер. Хлористый винил
извлекается дихлорэтаном. Затем хлористый винил отгоняется из раствора
и подвергается очистке и конденсации.
Пиролиз дихлорэтана имеет ряд преимуществ перед
щелочным способом получения хлористого винила—не требуются
затраты щелочи и спирта, возможна организация непрерывного
процесса и его полная автоматизация.
Хлористый винил—один из важнейших мономеров.
Полимеризацией и сополимеризацией хлористого винила получают ценные
полимеры для производства пластических масс (стр. 542) и
синтетического волокна (стр. 466).
Хлорированием поливинилхлорида получают перхлорвини-
ловую смолу, содержащую 64—65% хлора (поливинилхлорид
содержит 55—56% хлора). Хлорирование поливинилхлорида
проводят в среде хлорорганического растворителя (хлорбензол,
дихлорэтан и др.) при ПО—115 °С. При применении низкокипя-
щего растворителя процесс проводится под давлением. Перхлор-
виниловая смола нашла применение в производстве лаков, эмалей
и синтетического волокна (стр. 467).
При взаимодействии хлористого винила с хлором образуется
1,1,2-трихлорэтан
СН2=СНС1 + С12 > СН2С1—СНС12
полупродукт для получения винилиденхлорида. Реакция
протекает с высоким выходом при пропускании смеси хлора и паров
хлористого винила через колонну с насадкой из железных колец.
Хлор образует с железом FeCl3, являющееся катализатором этого
процесса. Получаемый таким методом трихлорэтан достаточно
чист и может быть непосредственно использован для синтеза
винилиденхлорида без предварительной ректификации.
190
Трихлорэтан получают также хлорированием дихлорэтана:
С!СН2—СН2С1 + С12 • СН2С1—CHCU + НС1
В этом случае в качестве побочных продуктов образуются
высшие полихлориды.
Полимеры и сополимеры винилиденхлорида используются в
производстве пластических масс и синтетического волокна.
Винилиденхлорид получают дегидрохлорированием трихлор-
этана в жидкой фазе известковым молоком при температуре около
100 °С
+Са(ОНJ
СН2С1—СНС12 *• СН2=СС12
или в газовой фазе при 500 °С:
СН2С1-СНС12 » СН2=СС12 + НС1
Гидрохлорирование винилацетилена (получение хлоропрена).
Ацетилен в присутствии водного раствора хлористой меди Си2С12
и хлористого аммония NH4C1 образует димер, тример и тетрамер:
2СН=СН » СН=С—СН=СН2
ЗСН=СН . СН2=СН—С=С—СН=СН2
4СН=СН » СН2=СН—СН=СН—С=С—СН=СН2
Димер ацетилена—винилацетилен СН=С—СН=СН2 (темп,
кип. 5,5 °С)—промежуточный продукт в производстве полихлоро-
прена—синтетического каучука (стр. 488).
Винилацетилен получают в горизонтальных аппаратах с
мешалками или в аппаратах колонного типа. Ацетилен барботирует
в реакторах через раствор катализатора, процесс проводят при
80—100 °С.
В газообразных продуктах реакции, кроме винилацетилена,
содержатся дивинилацетилен, непрореагировавший ацетилен,
водяные пары и др. Паро-газовую смесь разделяют ступенчатым
охлаждением: водяные пары и дивинилацетилен конденсируются
при более высоких температурах, для выделения винилацетилена
газовую смесь охлаждают до —70 °С. Несконденсировавшийся
газ (главным образом ацетилен) возвращают в производственный
цикл. Кроме того, винилацетилен можно выделить из газов
адсорбцией его растворителями (например, толуолом).
Хлоропрен получают взаимодействием винилацетилена с
хлористым водородом. Первая стадия этого процесса—образование
4-хлорбутадиена-1,2
СН2=СН—CsCH + НС1 » СН2С1—СН=С=СН2
который изомеризуется в 2-хлорбутадиен-1,3 (хлоропрен):
СН2С1—СН=С=СН2 > СН2=СС1—СН=СН2
191
СС ОСуЩССІЬЛИЮІ При HrUcHunbnUlvl HCjjCMCmriDuhria, щои-
образные винилацетилен и хлористый водород барботируют через
раствор катализатора—Си2С12 в соляной кислоте. Выходящая из
реактора паро-газовая смесь хлоропрена, непрореагировав-
шего винилацетилена и некоторых побочных продуктов
реакции поступает далее на установку выделения и очистки
хлоропрена.
ХЛОРИРОВАНИЕ БЕНЗОЛЬНЫХ УГЛЕВОДОРОДОВ
В зависимости от условий взаимодействие бензольных
углеводородов с хлором может протекать с присоединением атомов хлора
или с замещением атомов водорода атомами хлора.
Реакция присоединения протекает при фотохимическом
хлорировании, например:
СНС1
)С1НС СНС1
+ ЗС12 » | |
С1НС СНС1
СНС1
сн,
4- ЗС1,
гексахлорциклогексан
н3с
<
/
С1НС
1
-* 1
CIHC
С1
/
\
СНС1
1
СНС1
СНС1
гексахлорметилциклогексан
Получаемый этим методом гексахлорциклогексан нашел
широкое применение в качестве инсектицида, известного под названием
гексахлоран (стр. 352).
Замещающее хлорирование бензола проводят в присутствии
катализаторов (железо или его соли). При этом сначала образуется
монохлорбензол
Fe /Ч/С1
г сі, —ЛТ + неї
затем дихлорбензол и продукты более глубокого хлорирования—
вплоть до гексахлорбензола.
При хлорировании толуола получают большее число хлор-
производных, чем при хлорировании бензола, так как в этом
случае кроме замещения атомов водорода в бензольном кольце воз-
192
можно также замещение водорода в метильной группе.
Продуктами замещения водородных атомов в ядре являются:
сня
сня
сн.
сн.
С1
С1
о-хлортолуол
СІ
я-хлор-
толуол
(У
С1
2,4-дихлор-
толуол
С1
С1
С1
2,4.6-трихлор-
толуол
бензилхлорид
бензотрихлорид
бензол
При замещении водородных атомов боковой цепи получают;
СН2С1 СНС12 СС13
I I I
о
бензальхлориД
Хлорирование боковой цепи в отсутствие железа обычно
протекает при температуре, близкой к температуре кипения толуола.
Получение хлорбензола. Хлорбензол С6Н5С1 нашел широкое
промышленное применение. Он
используется как полупродукт
в производстве синтетических
красителей (стр. 275), в
производстве фенола (стр. 251),
инсектицида ДДТ (стр. 351),
является хорошим растворителем
некоторых зфиров целлюлозы
и многих других полимеров.
На рис. 67 показана
технологическая схема
получения хлорбензола.
Хлорирование проводят в колонном
хлораторе 2, футерованном изнутри
кислотоупорной плиткой и
заполненном насадкой из
железных колец. Бензол и осушенный
хлор непрерывно подают в
нижнюю часть хлоратора. Реакция
протекает с большим
выделением тепла, реакционная масса
разогревается до температуры
кипения G6—83 °С). Тепло
реакции отводится за счет испарения избытка бензола,
вводимого в хлоратор. При большом избытке бензола получается
главным образом монохлорпроизводное, в то время как высшие
полихлориды образуются в небольших количествах.
Рис. 67, Схема получения
зола:
/ — напорный бак лля бензола:
хлорбен-
3—холодильник; 4—сепаратор.
ор,
13-805
1ГЗ
Выделяющийся при хлорировании хлористый водород вместе
с парами бензола, хлорбензола, остатками влаги и газообразными
примесями, содержащимися в хлоре, отводят из верхней части
хлоратора и охлаждают в графитовом холодильнике 3 до —2 °С.
¦При этом пары бензола конденсируются. Из расширенной части
хлоратора отбирают жидкие продукты реакции—хлорбензол,
полихлориды и непрореагировавший бензол и разделяют их в
двухколонном ректификационном агрегате. Дистиллят первой
колонны—бензол возвращается в производственный цикл.
Дистиллят второй колонны, работающей при
разрежении,—товарный хлорбензол.
ФТОРИРОВАНИЕ УГЛЕВОДОРОДОВ
При непосредственном взаимодействии фтора с углеводородами
реакция протекает с воспламенением и даже со взрывом.
Происходит полное разрушение молекулы углеводорода с образованием
углерода, фтористого водорода и четырех фтор истого углерода.
Поэтому для получения перфторуглеродов используют
следующие два основных метода:
1. Пары фторируемого вещества разбавляют азотом и
пропускают через слой трехфтор истого кобальта CoF3 при температуре
250—350 °С, например:
CH3-(CH2)a—СН3 + 32CoF3 » CF3-(CF3M-CF3 + 32CoF2 + 16HF
Степень замещения зависит от продолжительности пребывания
исходного соединения в реакционной зоне, температуры процесса
и свойств углеводорода. Образующийся двухфтористый кобальт
CoF2 регенерируют до трехфтористого кобальта CoF3 обработкой
элементарным фтором при 200—250 °С.
2. Пары фторируемого вещества и фтор раздельно разбавляют
азотом и смешивают при 200—325 °С в катализаторной массе,
состоящей из медных стружек, покрытых фтористым серебром
AgF. При этом протекают следующие реакции:
2AgF + F2 * 2AgF2
32AgF2 + С7Н1в * C7F16 + 16HF + 32AgF
2AgF + F2 * 2AgF2 и т. д.
Перфторуглероды могут быть также получены из хлорфтор-
производных углеводородов. Так, например, тетрафторэтилен
CF2=CF2, являющийся важным мономером для синтеза фторо-
нласта-4 (тефлон, стр. 543), получают пиролизом монохлордифтор-
метана при 650 °С (без участия катализатора):
2CHF2C1 > CF2=CF2 + 2НС1
Выход тетрафторэтилена—90% и выше.
Другой метод получения тетрафторэтилена заключается во
взаимодействии тетрафтордихлорэтана с цинковой пылью в среде
194
растворителя (метиловый или этиловый спирт, ацетон, диоксан)
при 110—113 СС и давлении до 40 am:
CFiCl-CFaCl + Zn > CFa=CFa + ZnCl2
ХлорфторпроизЕОдные углеводородов преимущественно с
одним и двумя атомами углерода (фреоны, стр. 129) получают
обычно є результате взаимодействия хлорпроизводных с фтористым
Еодородом. Так, важнейший фреон—дихлордифторметан (фреон-12)
получают действием фтористого водорода на четыреххлористый
углерод в присутствии небольших количеств пятихлористой
сурьмы:
SbCls
СС!4 + 2HF » CC!2Fa + 2НС1
Аналогично, исходя из гексахлорэтана СС13^СС13< можно
получить силш-дифтортетрахлорэтан CCljF—CC^F (фреон-112)
и силш-дихлортетрафторэтан CCl^—CCIjFjj (фреон-113).
При взаимодействии хлороформа с фтористым водородом
образуется дифторхлорметан:
СНС13 + 2HF > CHF2C1 + 2НС1
Техника безопасности при работе с хлором
и галоидорганическими соединениями
Хлор—ядоеитый газ со своеобразным запахом. Он сильно
раздражает слизистые оболочки дыхательных путей, разрушает
стенки альвеол и ЕЫзыЕает отек легких, который может привести
к смерти.
Предельно допустимая концентрация хлора в єоздухе произ-
водственных помещений—1 мг/м3. При концентрации хлора
500 мг/м3 смерть наступает после 15 мин пребывания в зараженной
атмосфере.
Для обеспечения безопасной работы с хлором кроме общих
правил по технике безопасности необходимо соблюдать праєила,
учитывающие специфические свойства хлора.
Плохо осушенный хлор оказывает сильное коррозионное
действие на стальную аппаратуру и трубопроводы, в результате чего
хлор может проникнуть є производственное помещение.
Необходимо тщательно контролировать степень осушки хлора.
Трубопроводы и аппараты для транспортирования и переработки
влажного хлора следует выполнять из коррозионноустойчиєьіх
материалов.
Активированный уголь хорошо адсорбирует хлор, поэтому
промышленные противогазы (марка В, цеєт коробки—зеленый)
13*
195
могут служить хорошим средством индивидуальной защиты
работающих при сравнительно небольших концентрациях хлора. При
очень высоких концентрациях С12 возможно «пробивание»
фильтрующего противогаза, поэтому в таких случаях рекомендуется
пользоваться кислородно-изолирующими противогазами.
Хлорпроизводные углеводороды в той или иной степени
вредно действуют на человеческий организм, причем введение атомов
хлора усиливает не только наркотическое действие вещества, но
и в целом его токсичность. Наркотическое действие хлоруглево-
дородов (хлороформ СНС13, хлористый этил С2Н5С1) позволяет
применять их для общей и местной анестезии, однако в настоящее
время для наркоза применяют менее вредные вещества.
Некоторые галоидуглеводороды оказывают значительное токсическое
действие на обмен веществ и внутренние органы (хлороформ),
сильное раздражающее действие на слизистые оболочки и глаза
(галоидбензолы), вызывают нервные заболевания (трихлорэтилен,
хлоропрен).
Фторорганические соединения обладают определенным
физиологическим действием на человеческий организм. Среди
соединений этой группы довольно токсичны 1-фторпарафины
(особенно с четным числом углеродных атомов), а также фтор ацетаты
(CFraH,_nCOOR), которые оказывают вредное действие как при
вдыхании, так и при попадании на кожу.
Хлорфторуглероды (фреоны) практически нетоксичны. О і а
проявляют себя лишь при значительных концентрациях в воздухе
производственных помещений.
9. НИТРОВАНИЕ УГЛЕВОДОРОДОВ
НИТРОВАНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
Попытки ввести нитрогруппу в углеводороды жирного ряда
прямым воздействием азотной кислоты в течение многих лет были
безуспешны. Предполагалось, что азотная кислота более склонна
к окислению, чем к нитрованию углеводородов. Для синтеза нитро-
парафинов изыскивались сложные обходные пути, например
действие солей азотистой кислоты на галоидные алкилы.
В 1888—-1893 гг. М. И. Коновалов, изучая углеводороды
нефти, доказал возможность непосредственного получения нитропара-
финов действием азотной кислоты на предельные углеводороды
в паровой фазе. Это открытие, представляющее большой
теоретический интерес, в течение нескольких десятков лет не находило
применения. Лишь в последние десятилетия нитрование
парафинов стало промышленным процессом. Многочисленные
теоретические исследования, в частности работы С. С. Наметкина,
А. В. Топчиева и многих других советских и зарубежных ученых,
196
позволили в настоящее время осуществить ряд технологических
процессов получения нитропарафинов и их производных.
Нитрование парафиновых углеводородов, как и хлорирование
их, представляет большой интерес как процесс производства
многих химических продуктов. Введение в молекулу углеводорода
нитрогруппы позволяет превратить стабильные молекулы низших
парафиновых углеводородов в реакционноспособные соединения.
Нитропроизводные парафинов находят широкое применение
в промышленности. Так, нитрометан CH3NO2, нитроэтан C2H5NO2,
нитропропан C3H7NO2 являются хорошими растворителями
некоторых высокомолекулярных соединений (эфиров целлюлозы, ви-
нильных полимеров, лаков, красок). Нитропарафины используются
в качестве добавок, снижающих температуру самовоспламенения
дизельных топлив, как промежуточные продукты в синтезах ряда
ценных органических соединений. Восстановлением
нитропарафинов в кислой среде можно получить амины (аналогично реакции
Н. Н. Зинина), восстановлением динитропарафинов в других
условиях—альдегиды и кетоны, действием кислот—жирные
кислоты, конденсацией с альдегидами—нитроспирты и далее аминоспир-
ты и т. д.
При нитровании парафинов атомы водорода замещаются
нитрогруппами:
С„Н2/1+2 + HNO3 » C„H2fl+1NO2 + Н2О
C„H2/mNO2 + HNO3 —¦* C„H2n(NO2J + Н2О и т. д.
Таким образом, в зависимости от условий введения нитро-
групп в молекулу углеводорода, получают моно-, ди- или поли-
нитросоединения.
Нитрование можно проводить в жидкой или в паровой фазе,
используя азотную кислоту различной концентрации, жидкую или
в виде паров, а также окислы азота.
Скорость реакции и выход нитросоединений зависят от
температуры, давления, продолжительности процесса, а также от
концентрации применяемой азотной кислоты. С повышением
концентрации HNO3 реакция нитрования ускоряется. Если
взаимодействие парафиновых углеводородов с 47,5%-ной азотной кислотой
протекает достаточно быстро при атмосферном давлении, то при
нитровании разбавленной азотной кислотой A0—13,5%-ной) для
ускорения реакции повышают температуру и давление.
Наибольший выход при минимальном содержании побочных продуктов
достигается в случае нитрования 13,5%-ной кислотой при
температуре НО—140 °С под давлением, равным парциальному
давлению насыщения паров реакционной смеси при этих
температурах.
При взаимодействии парафинов с разбавленной азотной
кислотой наиболее легко замещается атом водорода при третичном
197
углеродном атоме (в группе >СН—) и образуется устойчивое
третичное нитросоединение:
Я Я
I I
R'_CH + HONOa * R'—С—NO2 + Н2О
I I
Я" Я"
При отсутствии третичного атома в молекуле углеводорода
нитрогруппа легче присоединяется к вторичному углеродному
атому. G повышением концентрации кислоты и температуры
реакционной смеси увеличивается количество образующихся вторичных и
первичных нитросоединений.
Нитрование сопровождается частичным окислением первичных
углеводородов в альдегиды, а вторичных—в кетоны и далее в
соответствующие карбоновые кислоты; при этом азотная кислота
восстанавливается до элементарного азота. Как установил
С. С. Наметкин, при продолжительном кипячении парафинов с
большим избытком азотной кислоты в продуктах реакции
преобладают кислородсодержащие соединения; с уменьшением избытка
азотной кислоты и продолжительности нагревания повышается
выход нитросоединений.
При нитровании углеводородов в жидкой фазе образуются
большие количества полинитросоединений. Это можно объяснить
тем, что исходные углеводороды и азотная кислота взаимно
нерастворимы, а образующееся нитросоединение лучше растворяется
в азотной кислоте и подвергается дальнейшему нитрованию. Паро-
фазное нитрование позволяет смешивать реагенты в любых
соотношениях и проводить процесс при высоких температурах D00—
450 °С). При этом увеличивается скорость реакции и значительно
уменьшается выход полинитросоединений.
Высокая температура процесса парофазного нитрования
способствует образованию первичных нитропроизводных за счет
вторичных и третичных. Одновременно с нитрованием в паровой фазе
протекают процессы окисления углеводородов. Азотная
кислота в этом случае восстанавливается до окиси азота,
которую можно вновь превратить в HNO3- При парофазном
нитровании азотная кислота используется полнее, чем при жидкефазном.
Особенностью процесса парофазного нитрования парафиновых
углеводородов является образование наряду с нитропроизводным
исходного углеводорода нитросоединений с меньшим числом
углеродных атомов в молекуле, чем в исходном углеводороде.
Например, при нитровании этана в паровой фазе в нитропродуктах
содержится не только нитроэтан C2H5NO2, но и нитрометан CH3NO2;
при нитровании в паровой фазе пропана наряду с нитропропа-
нами (CH3CHNO2CH3, CH3CH2CH2NOS) образуются нитрометан
CH3NO2 и нитроэтан CaH5NOa. Это позволяет получать нитроме-
198
тан из его гомологов, что важно, так как метан нитруется труднее
других парафинов. В качестве исходного углеводорода в
промышленных условиях чаще всего используется пропан. Пропан и
азотная кислота взаимодействуют в паровой фазе под давлением около
10 am и при 400—425 ЬС, причем в продуктах реакции содержится
25% 1-нитропропана, 40% 2-нитропропана, 25% нитрометана и
10% нитроэтана. Общий выход нитропарафинов достигает 96%
от теоретического.
Азотная Углеводород
кислота
*
| Испарение |
Пары азотной кислоты
| Смешение |
Смесь парод
| НигпроВание |
Продукты нитродания
[ Охлаждение и кондвисация\-
Воздрат
В цикл
Сода-
Т
Жидкие продукты
1
Нейтрализация и отделение
карбоноВых кислот
Окислы азота
и уалеВодороды
_Г
Т
Кйрбонобые
кислоты
Смесь нитропарафинов
I Разгонка I
Углеводороды
Разделение
Индивидуальные
нитропарафины
Окислы азота
Регенерация
азотной, кислоты
Рис. 68. Принципиальная схема парофазного процесса нитрования
парафиновых углеводородов.
Принципиальная схема процесса нитрования в паровой фазе
показана на рис. 68. Смесь паров азотной кислоты (или окислов
азота) и газообразного или испаренного углеводорода подается
а нитратор, представляющий собой трубчатый аппарат. Чтобы
избежать образования взрывчатых смесей, углеводород подают с
большим молярным избытком.
199
того, чтобы исключить возможность местных перегревов,
вызывающих протекание нежелательных побочных реакций, заданную
температуру нитрования в промышленных условиях поддерживают
с точностью ± 1 °С.
По выходе из нитратора продукты нитрования охлаждаются и
конденсируются. После удаления окислов азота и непрореагиро-
вавших газообразных углеводородов жидкие продукты реакции
нейтрализуют раствором соды и отделяют кислородсодержащие
соединения (в основном карбоновые кислоты). Нитропарафины
разделяют путем фракционированной перегонки на индивидуальные
нитросоединения (нитромеТан, нитроэтан и др.). Смесь
газообразных окислов азота и углеводородов также разделяют:
углеводороды возвращают на нитрование, а окислы азота поступают на
установку для получения азотной кислоты.
НИТРОВАНИЕ АРОМАТИЧЕСКИХ И АЛИЦИКЛИЧЕСКИХ
УГЛЕВОДОРОДОВ
Реакция нитрования бензольных углеводородов в общем виде
может быть представлена следующим уравнением*:
H2SO4
ArH + HONOa » ArNO2 + Н2О
При этом возможно введение в молекулу углеводорода
нескольких нитрогрупп.
В процессе нитрования выделяется эквимолекулярное
количество воды, вследствие чего концентрация азотной кислоты
постепенно уменьшается. Это вызывает нежелательную побочную
реакцию окисления, так как разбавленная азотная кислота обладает
более окислительным, чем нитрующим действием. Чтобы избежать
этого, нитрование проводят смесью азотной и серной кислот
(нитрующая смесь); H2SO4 добавляют для связывания выделяющейся
воды (обычно на 1 моль 50—60%-ной HNO3 3—4 моль H2SO4).
Соотношения серной и азотной кислот и воды зависят от свойств
получаемого нитросоединения. Вместо азотной кислоты часто
применяют так называемый меланж-—смесь концентрированной
азотной кислоты и 7—9% серной кислоты. Меланж не оказывает
коррозионного действия на обычную углеродистую сталь, тогда как
азотная кислота без примеси серной разрушает черные металлы.
Поэтому меланж можно хранить и транспортировать в стальных
цистернах.
При получении мононитросоединений азотную кислоту вводят
примерно в стехиометрическом количестве A—1,5% избытка или
5% недостатка против расчетного количества). При получении по-
линитросоединений применяют 10—20%-ный избыток азотной
* В действительности процесс нитрования более сложен.
200
кислоты. Количество серной кислоты рассчитывают по
концентрации отработанной кислоты. Так, при получении мононитросоеди-
нения концентрация H2SO4 в отработанной кислоте составляет
обычно 68—72%.
Большое значение имеет температура процесса нитрования.
Незначительное повышение температуры сверх заданной
значительно ускоряет реакцию окисления, сопровождающуюся
образованием газообразных окислов азота. В некоторых случаях это
может привести к взрыву. При нитровании выделяется большое
количество реакционного тепла, поэтому реакторы (нитраторы)
Окислы азота
кислота
Нитробензол -
Рис. 69. Схема получения нитробензола:
/—нитратор; 2—«дозреватель»; З—холодильник; 4—сепаратор: 5~ сборник
отработанной кислоты; 6—промывная колонна; 7—нейтралнзационная колона.
снабжаются теплообменными элементами (рубашки, змеевики
и пр.), в которых циркулирует холодная вода или охлаждающий
рассол. Вместо азотной кислоты для нитрования иногда
применяют окислы азота, например двуокись азота.
Получение нитробензола. Для получения нитробензола
обычно применяют нитрующую смесь, содержащую 32% HNO3 и 60%
H2SO4- Процесс проводят при температуре около 40 °С. В
заводских масштабах нитробензол получают в реакторах как
периодического, так и непрерывного действия.
На рис. 69 схематически показан процесс получения
нитробензола по непрерывному методу. В реактор (нитратор) 1
непрерывно подают нитрующую смесь и бензол. Реактор, выполненный
из нержавеющей хромоникелевой стали, снабжен змеевиками и
пропеллерной мешалкой, установленной в диффузоре, представ-
201
ляюшем собой цилиндрический стакан. Наличие диффузора
значительно улучшает циркуляцию жидкости во всем объеме
аппарата. Из верхней части нитратора избыток реакционной массы
непрерывно перетекает в «дозреватель» 2, в котором заканчивается
реакция нитрования. Продукты реакции охлаждаются в водяном
холодильнике 3 и поступают в отстойник-сепаратор 4. Здесь смесь
расслаивается: нижний кислотный слой, содержащий до 72%
серной кислоты, поступает в сборник 5, откуда его направляют на
денитрацию (для извлечения из отработанной кислоты
растворенного нитробензола, азотной кислоты и окислов азота) и последующее
концентрирование. Сырой нитробензол промывают в колонне б
кодой и в колонне 7 слабым раствором щелочи.
Нитробензол является важным промежуточным продуктом в
анилинокрасочной и фармацевтической промышленности. Исходя
из нитробензола, получают анилин, бензидин (стр. 274), п-амино-
салициловую кислоту (ПАСК) и др.
Получение тринитротолуола. Тринитротолуол (тротил, тол)
C6H2(NO2KCH3 является одним из высокобризантных взрывчатых
веществ. В промышленных масштабах его получают нитрованием
толуола:
СвН5СН3 + 3HNO3 * CeH2(NO2KCH3 + ЗН2О
Этот процесс в производственных условиях осуществляется в
несколько стадий (фаз)—в бензольное ядро толуола вводят
последовательно первую, вторую и третью нитрогруппы.
В первой фазе нитрование проводят при 40 °С нитрующей смесью,
содержащей около 70% H2SO4, 12% HNO3 и 20—25% воды.
При этом образуется смесь изомерных мононитротолуолов:
о-нитротолуол (~59%), п-нитротолуол C6%) и лі-нитротолуол
E%).
Во второй фазе применяют нитросмесь, содержащую около
74% H2SO4 и 8% HNO3; температуру процесса доводят почти до
75 °С. В результате получают в основном 2,4- и 2,6-динитротолуол.
Остальные четыре изомера составляют около 5%.
В третьей фазе применяют нитрующую смесь, состоящую из
83% H2SO4 и 17% HNO3 при 85 °С. Процесс заканчивается после
нагревания реакционной массы до 110 °С. В продуктах реакции
содержится до 95% 2,4,6-тринитротолуола.
Трехстадийное нитрование толуола для получения
тринитротолуола экономически целесообразно, так как позволяет вначале
применять более слабые нитрующие смеси.
При этом во второй фазе используются отработанные
кислотные смеси третьей фазы, а в первой фазе—смеси второй фазы с
добавлением в них некоторого количества азотной кислоты. Трех-
стадийный процесс менее взрывоопасен, так как температура
повышается постепенно от стадии к стадии.
202
Производство тринитротолуола можно осуществить как по
периодической схеме, так и по непрерывной. Основными
аппаратами этого производства являются нитраторы (рис. 70) с рубашкой
и охлаждающими змеевиками.
Реакционная масса перемешивается
лопастной или пропеллерной мешалкой.
Нитрование циклогексани. Этот
процесс приобрел промышленный интерес
в 'связи с тем, что нитроциклогексан
является промежуточным продуктом
при получении капролактама (стр. 257).
Нитрование циклогексана проводят
35%-ной азотной кислотой при 125 °С
и 5 am.
Повышенное давление создается для
того, чтобы реакционная масса
оставалась в жидкой фазе.
Полученный нитроциклогексан
затем превращают в циклогексаноноксим:
сн2
/\
Н2С СН—N0»
I I + н2 —>
н2с сн2
\/
сн2
сн2
Н2С C=N—ОН
—> II + н2о
Н2С СН2
сн2
Гидрирование проводят в среде метилового спирта при 100 °С
и 150 am в присутствии катализатора (Ag, Zn, Cr2O8).
Последняя стадия процесса—изомеризация циклогексанонок-
сима под влиянием серной кислоты в ^.-капролактам (так
называемая бекмановская перегруппировка):
Рис. 70. Нитратор:
/—пропеллерная мешалка;
2—загрузочные лючки; 3—охлаждающие
змеевики; 4—рубашка.
СНг
Н2С C=N—ОН
Н2С СНг
оС
сн2
С
c=o
NH
CH2
Полимеризацией капролактама получают поликапроамид, из
которого изготовляют синтетическое волокно—капрон (стр. 47 1 ел).
203
in гигтрлтлпыа ирттпр /тр гтііііг у уг>
ГИДРАТАЦИЯ ОЛЕФИНОВ
Гидратацией называется процесс присоединения элементов
воды к молекуле непредельного углеводорода. При гидратации
олефинов образуются одноатомные спирты:
слн2л + н2о —»с„н2п+1он
Согласно правилу В. В. Марковникова, атом водорода
присоединяется к наиболее гидрогенизированному атому углерода, а
гидроксил—к наименее гидрогенизированному. Поэтому все оле-
фины, кроме этилена, при гидратации образуют вторичные или
третичные спирты:
ОН
I
R—СН=СН2 + Н2О * R—СН—СН3
R\ R\ .ОН
>С=СН2 + Н2О » >С<
R"/ R"' NCH3
Исключение составляет этилен, образующий при гидратации
первичный (этиловый) спирт:
СН2=СН2 + Н2О » СН3—СН2ОН
Присоединение воды к олефинам может быть осуществлено
методами прямой и непрямой гидратации.
Прямая гидратация заключается в непосредственном
взаимодействии олефина с водой или водяным паром в присутствии
катализаторов (фосфорная кислота и ее соли, серная кислота, окись
алюминия, окись вольфрама, некоторые органические соединения
и др.). Этот метод получения одноатомных спиртов впервые
открыл А. М. Бутлеров в 1876 г. Гидратацией изобутилена и гепти-
лена в запаянных трубках в присутствии небольших количеств
серной кислоты он получил изобутиловый и гептиловыи спирты.
Процесс гидратации газообразных олефинов происходит с
уменьшением объема, поэтому, согласно принципу Ле-Шателье,
повышенное давление способствует протеканию реакции. Скорость
реакции гидратации зависит от строения олефина и длины его
углеродной цепи. С наибольшей скоростью протекает гидратация
олефинов с разветвленной углеродной цепью; чём короче углеродная
цепь олефина, тем труднее происходит его гидратация.
При получении спиртов методом непрямой гидратации (так
называемый сернокислотный метод) сначала к олефину
присоединяется серная кислота с образованием алкилсерной кислоты
R— СН=СН2 + НО—SO2—ОН » R—СН—О—SO2—ОН
сн3
204
и частично диалкилсульфата:
СН3 СН3
2R—СН=СН2 + НО—SO2—ОН > R—СН—О—SO2—О—СН—R
При гидролизе полученных соединений выделяется спирт и
регенерируется кислота:
R—СН—О—SO2—ОН + Н2О > R—СНОН -f- H2SO4
I !
СН3 СН3
R—СН—O-SO2-O-CH-R + 2НоО , 2R—СНОН + H2SO4
! I I
СН3 СНз СНз
Зависимость условий гидратации от длины углеродной цепи
олефина видна на следующих примерах. Для получения этилсер-
ной кислоты из зтилена требуется, концентрированная серная
кислота (примерно 95—98%-ная—так называемое купоросное
масло), парциальное давление зтилена над серкой кислотой должно
составлять 10 am, температура 70—80 СС. При получении изопро-
пилсерной кислоты можно применять 70—80%-ную серную
кислоту при 5 am и температуре 25—35 СС, а для н-бутиленов 60—
75%-ную серную кислоту, причем реакция протекает при
атмосферном давлении и охлаждении до —10 °С.
Большинство алкилсерных кислот, образованных высшими
олефинами, гидролизуется холодной водой, однако для получения
этилового и изопропилового спиртов гидролиз проводят при
100 СС и более высокой температуре. При гидратации олефинов
наряду с образсванием спиртов протекают побочные процессы,
приводящие к образованию простых зфиров и продуктов
полимеризации олефиков. Кроме того, алкилсерные кислоты в условиях
гидролиза частично разлагаются на олефины и серную кислоту,
а выделившийся олефин полимеризуется.
При проведении сернокислотной гидратации необходимо
строго соблюдать установленные оптимальные условия процесса.
Уменьшение концентрации кислоты и понижение температуры
приводят к замедлению и даже прекращению реакции, увеличение
концентрации кислоты и повышение температуры способствуют
образованию продуктов полимеризации олефиков (смол).
Производство спиртов методом гидратации олефинов—одна
из наиболее важных отраслей промышленности органического
синтеза.
Производство этилового спирта
Этиловый спирт или этанол С2Н5ОН (темп. кип. 78,3 СС, темп,
пл. —110,5 СС, плотность при температуре кипения 0,7893 гісм3)—
важный органический продукт, вырабатываемый в очень больших
количествах. Он применяется как растворитель и как исходное
205
фармацевтической, парфюмерной, пищевой промышленности, в
производстве синтетического волокна, взрывчатых веществ, по-
рохов, в медицине, в лабораторной практике, для бытовых нужд,
в качестве горючего и т. д. В СССР большие количества этилового
спирта расходуются на производство синтетического каучука по
методу академика С. В. Лебедева (стр. 211).
Этиловый спирт применяется как исходный продукт для
производства хлороформа, хлораля, диэтилового эфира, диэтилсуль-
фата, этилацетата и многих других продуктов органического
синтеза.
Раньше этиловый спирт получали в промышленном масштабе
исключительно нз пищевого сырья.
Этот наиболее старый промышленный способ получения этилового спирта
заключается в сбраживании осахаренного крахмала, картофеля или зерна
Осахаривание производится с помощью солода (измельченные проросшие
зерна ячменя). Под влиянием находящихся в солоде сложных биологических
катализаторов (энзимы) происходит расщепление крахмала с образованием
дисахарида—мальтозы C12H22OU. Под действием других энзимов, вводимых в
осахаренный раствор с дрожжами, мальтоза расщепляется с присоединением
воды на две молекулы глюкозы С6Н12О6.
В результате брожения глюкозы образуется этиловый спирт и двуокись
углерода:
СвН12Ов ». 2QiHaOH + 2СО2
При получении этилового спирта этим способом расходуется большое
количество ценного пищевого сырья (9—9,5 т картофеля, или 3—3,5 т ржи,
или 18—19 т сахарной свеклы на 1 т этилового спирта). В связи с этим важное
значение приобрели способы получения спирта из непищевого сырья.
В настоящее время большие количества этилового спирта получают
сбраживанием растительного сырья, содержащего крахмалистые вещества,—оса-
харенной древесины (стр. 77) и сульфитных щелоков (отходы целлюлозного
производства). Большие перспективы имеет использование для этой цели
сульфитных щелоков, содержащих 1 —1,5% Сахаров. В процессе получения
этилового спирта щелока нейтрализуют и охлаждают, отфильтровывают от
взвешенных частиц и заливают в деревянные или'железобетонные бродильные
чаны, куда добавляют дрожжи. Образовавшийся в результате брожения
Сахаров этиловый спирт затем подвергают очистке и ректификации.
Синтетический этиловый спирт получают методами
сернокислотной или прямой гидратации этилена.
Прямая гидратация этилена проводится в паровой и жидкой
фазе:
СН2=СН2 + Н2О > СН3-СН2ОН
Катализатором парофазного процесса является фосфорная
кислота на носителе, в качестве которого используют силикагель
или синтетический алюмосиликат. Фосфорнокислотный
катализатор содержит 38—40% активной кислоты, вводимой в виде
свободной фэсфэрной кислоты или гидролизующихся фосфатов.
Синтез этилового спирта прямой гидратацией этилена
сопровождается уменьшением объема, поэтому, согласно правилу Ле-
206
Шателье, увеличению выхода спирта за один проход паро-газовой
смеси через катализатор (сдвиг равновесия реакции, вправо)
способствуют низкие температуры и высокие давления."
Однако применяемые в настоящее время промышленные
катализаторы активны лишь при температурах выше 250 СС. При 260 СС
и 70 am (молярное отношение Н2О : С2Н4=0,65 : 1) конверсия
этилена составляет 3%; при повышении температуры до 280 СС
конверсия увеличивается до 5%. Величина давления в данном
процессе ограничивается точкой росы водяных паров, т. е.
условиями начала их конденсации.
Рис. 71. Схема получения этилового спирта прямой гидратацией
этилена:
/—коь.прсг ор; 2—смеситель; а— н«ос высокого давления; 4, 5—теплообменники;
в—трубчатая печь; 7—колонна синтеза (гидрататор); 8—тройник; 9, 11,
/2—сепараторы; 10—дроссельный вентиль.
На рис. 71 показана упрошенная технологическая схема
получения этилового спирта методом прямой гидратации. Свежий и
оборотный этилен компрессором 1 нагнетается в систему под
давлением 70 am и в смесителе 2 смешивается с водой (в соотношении
Н2О : С2Н4=0,65 : 1), подаваемой насосом высокого давления 3.
В теплообменниках 4 и 5 смесь нагревается до 200 СС за счет
физического тепла продуктов реакции. В змеевиках трубчатсй печи 6
с огневым нагревом паро-газовая смесь перегревается до 290 °С
и проходит сверху вниз через слой находящегося в гидрататоре 7
фосфорнокислотного катализатора. Продукты реакции,
содержащие этанол, из нижней части гидрататора поступают в тройник 8,
где нейтрализуются щелочью и отделяются от образующихся солей
207
5 продукты реакции охлаждаются и в сепараторе // отделяются
от непрореагировавшего этилена, который после промывки водой
(на схеме не показана) возвращается
в производственный цикл. Из
сепаратора // жидкость через дроссельный
вентиль 10 поступает в сепаратор 12
низкого давления. Выделяющиеся газы
отводят из системы, а слабый водный
раствор спирта A5—16%-ный)
направляют на очистку и ректификацию.
Гидрататор (рис. 72) представляет
собой цилиндрическую полую колонну
диаметром 1,5 м и высотой 10 м,
выполненную из углеродистой стали. Стенки
и днища ее для защиты от
коррозионного действия фосфорной кислоты
обкладывают изнутри листами красной
меди.
Катализатор загружают в аппарат
слоем высотой 8,5 м. Для
удлинения срока службы катализатора его
дополнительно насыщают, вводя в
гидрататор вместе с паро-газовой смесью
распыленную фосфорную кислоту. При
объемной скорости циркуляции
парогазовой смеси, равной 1800—2000 м3
газа на 1 м3 катализатора в час,
производительность гидрататора
составляет 180—200 кг/ч спирта с 1 м3
катализатора.
Сернокислотная гидратация
этилена. Процесс сернокислотной гидратации
(см. реакцию на стр. 204) проходит
последовательно через стадии образования промежуточных
продуктов— этилсерной кислоты C2H,SO,H и диэтилсульфата
(C2H5OJSO2 и гидролиза алкилсульфатов водой до этилового
спирта. Последний затем поступает на очистку и ректификацию.
При взаимодействии этилена с серной кислотой (первичная
реакция) сначала образуется этилсерная кислота, а затем диэтил-
сульфат (вторичная реакция).
Распределение серной кислоты в продуктах реакции в
зависимости от молярного соотношения этилена и кислоты в реакционной
массе приведено на рис. 73.
Скорость взаимодействия между этиленом и серной кислотой
зависит от концентрации кислоты, температуры процесса,
парциального давления этилена и от поверхности контакта между
Рис. 72. Гидрататор:
1—предохранительный клашн;
2—корпус аппарата; 3—медная
футеровка; 4—приспособление для
выгрузки катализатора.
208
реагентами. Оптимальная концентрация кислоты 97—98%.
Температуру процесса обычно поддерживают в пределах 70—80 °С.
При более низких температурах и концентрациях кислоты
реакция протекает медленно. Повышение концентрации и температуры
сверх оптимальной ускоряет побочные процессы, в первую очередь
полимеризацию этилена. С повышением парциального давления
этилена увеличиваются скорость взаимодействия и степень
адсорбции этилена серной
кислотой, оптимальное
парциальное давление этилена
составляет 15 am.
Схема процесса
получения этилового спирта
показана на рис. 74.
Этан-этиленовая фракция, очищенная от
высших олефинов и
ацетилена и содержащая 60—70%
С2Н4, поступает под
давлением 20—25 am в
реакционный аппарат 1 колонного
типа, орошаемый
концентрированной серной кислотой.
Колонна имеет две глухие
тарелки и переточные трубы
для жидкости. Тепло
экзотермической реакции
гидратации отводится в выносных
кожухотрубных
холодильниках 7, через которые
непрерывно циркулируют жидкие
продукты реакции,
возвращаемые затем в аппарат /.
Из нижней части
реакционной колонны непрерывно
вытекает смесь этилсульфатов и непрореагировавшей серной
кислоты. При дросселировании из жидкой смеси выделяются этилен,
этан и другие газы и пары диэтилового эфира (побочный
продукт взаимодействия этилена с серной кислотой). Гидролиз
алкилсульфатов проводится под давлением около 5 am и при 90—
100 °С. Паро-газовую смесь отводят из верхней части гидролизера
в скрубберы 3 и 4 на промывку и нейтрализацию следов серной
кислоты, увлеченной газами.
Промытые и нейтрализованные газы вместе с газами,
отходящими из реакционной колонны /, направляют в цех пиролиза
этана (стр. 141).
Из нижней части гидролизера 2 жидкость, содержащая
этиловый спирт, воду, серн/ю кислоту, диэтиловый эфир, а также не-
14—805 . 209
° О,Ь D? 1,2 ,
Молярное отношение С2Н4'- HjSO^
Рис. 73. Распределение 98%-ной
серной кислоты в продуктах реакции r
зависимости от молярного
соотношения этилена и серной кислоты:
/—свободная серная кислота; 2—серная
кислота, связанная в этилсер.чую кислоту;
Л—серная кислота, связанная в диэтилсульфат.
і идрилизиванные з'іил<_ульіраш, подается на верхние іарелки
отгонной колоннв! 5, в которую снизу вводят острый водяной
пар для завершения гидролиза этилсульфатов и отгонки спирта
и эфира. Из кубовой части отпарной-'колонны отводят на
концентрирование 45—47%-ную серную кислоту. Паро-газовую смесь,
отбираемую из верхней части отпарной колонны, нейтрализуют
Щелочь
Вода
Этилен
45%-ная
H2SO4
Рис. 74. Схема получения этилового спирта сернокислотной
гидратацией этилена:
/—реакционная колонна (абсорбер); 2—гидролиаер; 3. 4—скрубберы;
5—отгонная колонна; 6—конденсатор; 7—выносные холодильники; 8—дроссельный
вентиль.
(на схеме не показано) и конденсируют. Сконденсировавшаяся
жидкость, содержащая 25—35% этилового спирта, подвергается
ректификации. Каждый из двух описанных выше способов
гидратации этилена обладает своими достоинствами и недостатками.
Основным недостатком сернокислотного способа является
необходимость применения серной кислоты и сложная схема ее
регенерации. При использовании разбавленной серной кислоты (отход спир-
210
тового производства) для получения суперфосфата или сульфата
аммония процесс сернокислотной гидратации в значительной мере
упрощается и удешевляется.
При прямой гидратации серная кислота не применяется,
благодаря этому отпадает сложная система кислотного хозяйства,
уменьшается коррозия аппаратуры и трубопроводов. Однако для
прямой гидратации требуется высококонцентрированный этилен,
в то время как при сернокислотном процессе можно использовать
этан-этиленовую фракцию с содержанием этилена 60—70% (в
некоторых случаях и ниже).
Синтезы на основе этилового спирта
Получение 1,3-бутадиена по методу акад. С. В. Лебедева. При
нагревании этилового спирта выше 500 °С происходит его
разложение на окись углерода, метан и водород. В присутствии
катализаторов разложение спирта протекает при более низких
температурах. В зависимости от применяемого катализатора и
температуры образуются различные продукты реакции.
Дегидратирующие катализаторы (Al2O3j ThO2, W2Ob) направляют реакцию в
сторону отщепления воды с образованием этилена или диэтило-
вого эфира:
ж400°С
СН3-СН2ОН * СН2=СН2 + Н2О
;=250°С
22Н3—СН2ОН * СН3—СНа—О—СНа—СНа + Н2О
В присутствии дегидрирующих катализаторов (Си, Zn, Mg
и др.) происходит отщепление водорода и образуется ацетальдегид:
»400°С
сн3—сн2он —-»• сн3—сно + н2
В 1910 г. С. В. Лебедев показал, что смешанные
дегидратирующие и дегидрирующие катализаторы, например А12О3+2п,
направляют каталитическое разложение этилового спирта в сторону
одновременного отщепления воды и водорода и образования
1,3-бутадиена (дивинила):
2СН3—СН2ОН > СН2=СН—СН=СН2 4- 2Н2О + Н2
Этот способ получения бутадиена был осуществлен в СССР в
промышленном масштабе. Полимеры и сополимеры бутадиена
представляют собой синтетические каучуки высокого качества
(стр. 483 ел.).
Контактное разложение этилового спирта производится в
ретортной контактной печи, схема которой представлена на рис. 75.
Печь состоит из муфеля (верхняя часть печи) и спиртоперегрева-
тельной нижней части печи. Цилиндрический муфель, выложен-
14* 211
вое пространство между стенками (высота около 3 м) является
топкой печи 1, в которой сжигается жидкое или газообразное
топливо, подаваемое форсунками 2. Топочные газы проходят
через каналы 3 (в нижней части топки) в спиртоперегревательную
часть печи и во внутреннее пространство муфеля—газовую
камеру 4. В газовой камере по окружности, вблизи внутренней стенки
муфеля, на равном расстоянии друг от друга расположены
реторты 5. Они представляют собой
стальные прямоугольного се-
13 чения трубы высотой 4—5 м,
заполненные катализатором.
Реторты обогреваются за
7 счет тепла раскаленной
внутренней стенки топки.
Дополнительное тепло, в случае
необходимости, вводится с
топочными газами,
количество которых регулируется
шибером 6. В нижней части
печи, состоящей из ряда
отдельных камер (число камер
равно числу реторт), находятся
змеевики 8, в которых спирт
нагревается топочными
газами. Топочные газы далее
поступают в боров
печи 9.
Пары спирта, выходящие
из змеевиков 8 при
температуре контактирования,
поступают в реторты 5, где
проходят через слой катализатора. В ретортах спирт разлагается
и образующиеся продукты реакции выводятся из верхней части
реторт в коллектор 13.
При разложении спирта наряду с образованием бутадиена
протекает ряд побочных реакций. Поэтому в контактных газах
содержится, кроме водорода и паров неразложившегося этанола,
более тридцати различных соединений—углеводороды, спирты,
альдегиды, эфиры, окислы углерода и др.
Схема производства бутадиена из этилового спирта показана
на рис. 76.
Выходящие из контактной печи 3 продукты реакции
охлаждаются в водяных и рассольных теплообменниках 6 и 7 до
температуры —7 °С. При этом конденсируются пары воды и этилового
спирта и частично углеводороды, эфиры, альдегиды и высшие
спирты. Конденсат после отделения углеводородов подвергают ректи-
212
Рис. 75. Ретортная контактная печь для
получения бутадиена из этилового
спирта:
(—топка; 1—форсунка; 3— канал; 4—газовая
камера; 5— реторта; б—шибер; 7—тяга; S— змеепиковый
подогреватель; 9— боров; 10, И—площадки для
обслуживания печи; 12—коллектор паров спирта;
13— коллектор контактных газов.
фикации. Выделенный этиловый спирт возвращают на
разложение. Несконденсировавшиеся газы, содержащие бутадиен,
поступают в абсорберы, орошаемые этиловым спиртом. Из раствора
бутадиена в спирте в колонне 11 отгоняют «сырой» бутадиен,
после отмывки водой от альдегидов его направляют на
ректификацию.
Бутадиен-ректификат поступает далее на полимеризацию
(стр. 429). Побочные продукты, образующиеся при синтезе
бутадиена, с успехом используются в химической промышленности,
что значительно снижает его себестоимость.
Сырой
бутадиен
на
отмывку и
ректификацию
Пар
Спирт
со оклада
Возвратный
спирт
8 Конденсат на вы дел е-
ниє спирта
Рис. 76. Схема производства бутадиена из этилового спирта:
/—сборник спирта; 2— испаритель спирта; 3— контактная ретортная печь; 4—перегреватель;
5—реторта; б, 7—теплообменники; в—сборник конденсата; 9— абсорбер; 10—
теплообменник; II—отгонная колонна; 12— испаритель; 13—дефлегматор; 14—холодильник.
Получение хлораля (трихлоруксусного альдегида).
Современные промышленные методы получения хлораля основаны на
реакции хлорирования этилового спирта, открытой Либихом в 1832 г.:
,0
СН3—СН2ОН + С12 »СС13—C<f + НС1
Хлорирование спирта осуществляется периодическим или
непрерывным способом. При периодическом процессе в
эмалированный реактор с мешалкой и обратным холодильником заливают
94%-ный спирт и пропускают через него хлор. В начале
хлорирования поддерживают температуру 20—25 "С. По мере
протекания реакции температуру повышают до 60 °С, а затем до 85 °С.
Процесс хлорирования длится 2—3 суток. По окончании
хлорирования полученное «хлорированное масло» смешивают с серной
213
кислотой (соотношение 1,0 : і) и подвергают перегонке. Нервую
¦фракцию, содержащую продукты неполного хлорирования
спирта, возвращают на хлорирование. Чистый хлораль отбирают в
интервале температур 90—100 °С.
Непрерывный процесс хлорирования проводится в
футерованных кислотоупорной плиткой аппаратах колонного типа с
насадкой. Реакционное тепло отводится водой, циркулирующей в
змеевиках (свинцовых или из кислотоупорной стали). Этиловый
спирт орошает сверху насадку, снизу в колонну поступает хлор.
Для ускорения процесса иногда применяют
катализатор—активированный уголь, который служит также насадкой колонны.
Хлораль—бесцветная маслянистая жидкость с резким
специфическим запахом (темп. кип. 98 °С, относительная плотность
1,512 г/см3 при 20 °С). Он используется в больших количествах
для производства эффективного инсектицида ДДТ (стр. 351).
Производство изопропилового спирта и синтезы на его основе
В промышленности сернокислотная гидратация пропилена
освоена раньше гидратации этилена. Пропилен гораздо легче
реагирует с серной кислотой, чем этилен, в связи с чем в этом процессе
устанавливают более мягкий режим: давление газа (при высоком
содержании пропилена) 4—5 am, температура 25—30 "С,
концентрация серной кислоть; 75—-80%.
После ректификации образующегося разбавленного
спиртового раствора получают 88%-ный изопропиловый спирт
(азеотропная смесь, кипящая при 80,4 °С). Технологическая схема синтеза
изопропилового спирта из пропилена примерно такая же, что и
этилового спирта из этилена.
Изопропиловый спирт применяется как растворитель, во
многих случаях заменяющий этиловый спирт, например для
масел и смол, и используется также для получения ацетона.
Получение ацетона. Ацетон может быть получен
дегидрированием или неполным окислением изопропилового спирта:
(СН3JСНОН » СН3СОСН3 + Н2
1/2OZ
!(СНаJСНОН -» СНз—СО—СН3 + Н2О
Процесс производства ацетона дегидрированием
изопропилового спирта протекает в паровой фазе при 380—400 "С над
катализатором (окись цинка, нанесенная на пемзу). По
технологическому оформлению этот процесс аналогичен дегидрированию цикло-
гексанола в циклогексанон (стр. 256).
При синтезе ацетона неполным окислением изопропилового
спирта смесь паров спирта и воздуха пропускают над металличе
ским катализатором при температуре 500—600 °С. В качестве
катализатора используют серебряную сетку или серебро, осаж-
214
денное на пемзе, при этом ацетон получают с высоким выходом.
Технологическая схема получения ацетона этим методом
аналогична схеме получения формалина неполным окислением
метилового спирта (стр. 232).
Неполное окисление изопрспилового спирта можно проводить
и в жидкой фазе под давлением при 90—140 °С. Образующаяся
гидроперекись изопропила распадается далее на ацетон и
перекись водорода. Реакция протекает по цепнсму механизму:
СН3—СН-СН3 + О » СН3—СН—СН3
I I
ОН О—ОН
СН3—СН—CH3_2i-^CH3—СО—СН3+ Н2О2+О и т. д.
о-он
• На 1,0 кг перекиси водорода получается 1,7 кг ацетона.
Продукты реакции—ацетон, перекись водорода и непрореаги-
ровавший изопропиловый спирт разбавляют водой, вводят
вещества, стабилизирующие перекись водорода, и разделяют смесь
фракционированной перегонкой.
Этот метод представляет большой практический интерес, так
как открывает возможность одновременного получения дешевой
перекиси водорода. До сих пор высокая стоимость перекиси
водорода, получаемой электрохимическим методом, ограничивала
ее применение в промышленных органических синтезах. В
крупных промышленных масштабах ацетон получают совместно с
фенолом разложением гидроперекиси изопропилбензола (стр. 252),
а совместно с п-крезолом—разложением гидроперекиси метилизо-
пропилбензола (л-цимола):
СН3
СНз-f ^-СН » СНз-
I
сн3
СН3—СО—СН3 + СНз—<f^/—он
Новый способ получения ацетона заключается в прямом
окислении пропилена в присутствии хлористого палладия и хлорной
меди:
PdCb
CH2=CH-CH3 + i/2O2 -ZTTT СН3СОСН3
С11СІ2
Он аналогичен способу получения ацетальдегида прямым
окислением этилена (стр. 220).
Ацетон—один из наиболее распространенных растворителей.
Он применяется в огромных количествах в лакокрасочной
промышленности, в производстве искусственного шелка, киноплен-
215
ки, во всевозможных экстракционных процессах и широко
используется во многих органических синтезах. В смеси с
ароматическими углеводородами ацетон применяют при отделении парафина
от смазочных масел. Ацетон используют при наполнении
баллонов ацетиленом (стр. 154).
При действии синильной кислоты на ацетон образуется окси-
нитрил—ацетонциангидрин
снзч сн3
>СО + HCN > \С-СЫ
сн/ сн/ і
он
который при взаимодействии с метиловым спиртом и серной
кислотой дает метиловый эфир метакриловой кислоты (стр. 239),
получаемый в промышленности в сравнительно больших
количествах .
Акриловая и метакриловая кислоты, их нитрилы, и особенно
эфиры, используются для получения полиакрилатов—ценных
пластических масс (стр. 414, 546).
Действием на ацетон гидроокиси бария или других щелочей получается
диацетоновый спирт
Ва(ОНJ СН3х
2СН3—СО—СН3 > >С(ОН)—СН2—СО—СН3
сн/
хороший растворитель нитратов целлюлозы и ацетилцеллюлозы, применяемый
для изготовления лаков.
Ацетон вступает в реакцию с непредельными соединениями с образованием
алкилированных кетонов. Так, этилен в присутствии катализаторов под
давлением выше 50 am при 450—500 °С дает метилпропилкетон
СН3—СО—СН8 + СН2=СН2 » СН3—СО—С3Н7
а пропилен—метилбутилкетон:
СН3—СО—СН3 + СН2=СН—СН3 ». СН3—СО—С4Н9
Катализаторы этих реакций—NiO, ZnO, Cr2O3, V2O6.
Разработан промышленный метод получения кетена на основе
ацетона. Кетен образуется при пропускании паров ацетона над
пористыми телами (глинозем и др.) при 500 °С:
СН3—СО—СН3 »• СН2=С=О + СН4
Он может служить исходным соединением в синтезе уксусной
кислоты
СН2=С=О + Н2О ». СН3—СООН
и уксусного ангидрида:
СН2=С=О + СН3— СООН » (СН3СОJО
216
Кетен может быть использован для введения ацетильной
группы в различные органические соединения.
ГИДРАТАЦИЯ АЦЕТИЛЕНА
Получение ацетальдегида и синтезы на его основе
Непосредственная гидратация ацетилена приводит к
образованию уксусного альдегида:
СН=СН + Н2О > СН3—C<f
хн
Эта реакция была открыта М. Г. Кучеровым в 1881 г.
Ацетилен пропускают через сернокислотный раствор катализатора—окиси
ртути HgO. Образующийся ацетальдегид, будучи сильным
восстановителем, восстанавливает двухвалентную ртуть до
металлической:
HgO * Hg2O * 2Hg
Это уменьшает количество окисной ртути в растворе и сильно
замедляет реакцию гидратации. Для поддержания постоянной
концентрации окиси ртути в реактор вводят окисное железо,
которое в присутствии серной кислоты вначале окисляет
металлическую ртуть до закисной, а затем последнюю в окисную.
Кроме того, ацетальдегид в кислой среде при повышенной
температуре вступает в реакцию конденсации, образуя кротоновыи
альдегид и смолообразные продукты:
2СН3-с/ —-> СН3—СН=СН—с/ + Н2О
\Н ХН
Поэтому ацетальдегид необходимо удалять из сферы
реакции.
Технологическая схема производства ацетальдегида
гидратацией ацетилена показана на рис. 77. Ацетилен, тщательно
очищенный от вредных примесей (РН3, NH3, H2S, AsH3 и др.),
подают) в газовый смеситель /. Сюда же псступает циркуляционный
газ из скруббера 8, содержащий главным образом непрореагиро-
вавший ацетилен. Газ из смесителя компрессором 3 нагнетается
под давлением 0,5 am в гидрататор 2.
Гидрататор представляет собой вертикальный аппарат
колонного типа из нержавеющей или углеродистой стали (в последнем
случае его изнутри гуммируют). В гидрататор загружают 20%-ную
H2SO4, окись ртути A% HgO от веса раствора серной кислоты)
и окислы железа D0 г/л). Ацетилен барботирует через раствор и
при 80 °С гидратируется с образованием ацетальдегида.
217-
І моль
v_»L»-
ккал или
разовании І моль ацетальдегида выделяется 33,8 ккал или
141,6 кдж тепла). Однако это не компенсирует расход тепла на
испарение влаги, поэтому для поддержания требуемой
температуры реакции (80 °С) в гидрататор вводят острый пар.
При прохождении через раствор катализатора гидратируется
около 30% всего количества ацетилена. Остальной ацетилен вместе
с парами воды и ацетальдегида выходит из гидрататора, унося с
собой некоторое количество ртути, улавливаемой в ловушке 5.
Оборотный ацетилен
ицетальдегиё
Пар
Рис. 77. Схема получения ацетальдегида гидратацией ацетилена:
/—смеситель; 2—гидрататор; 3—компрессор; 4, 7—сборники; 5—ловушки;
6—холодильник; A—скруббер; 9—ректификационная колонна; 10—дефлегматор;
II—конденсатор; 12—кипятильник.
Паро-газовая смесь проходит через холодильник 6, где
конденсируются пары воды, и поступает в скруббер 8, орошаемый водой
для поглощения несконденсировавшегося ацетальдегида.
Конденсат из холодильника 6 и раствор ацетальдегида из скруббера
поступают в сборник 7. Из верхней части скруббера выходит отмытый
от ацетальдегида газ, содержащий в основном ацетилен, который
снова возвращается в смеситель /. В циркулирующем ацетилене
постепенно накапливаются инертные газы и, что особенно опасно,
кислород. Поэтому аппаратуру периодически продувают, а
циркуляционный газ выпускают в атмосферу.
Из сборника 7 разбавленный водный раствор ацетальдегида
непрерывно перекачивают в ректификационную колонну 9 с вы-
218
носным кипятильником 12. В колонне ацетальдегид отделяется^
воды и в виде паров поступает в дефлегматор 10 и далее в
конденсатор // (охлаждаемый рассолом), из которого вытекает
жидкий ацетальдегид.
Жидкость из гидрататора сливают в сборник 4 и далее
направляют на регенерацию.
Процесс гидратации ацетилена можно проводить и в газовой
фазе, пропуская смесь ацетилена и водяных паров при
температуре около 400 °С над катализаторами (окислы некоторых
металлов, ванадат цинка и др.). По этому способу в ацетальдегнд может
быть превращено 75—80% ацетилена, содержащегося в паро-га-
зовой смеси.
А. Е. Фаворским и М. Ф. Шостаковским разработан интересный метод
синтеза уксусного альдегида. Первая стадия процесса—конденсация ацетилена
со спиртом в присутствии КОН с образованием винилового эфира:
CHsCH + R-OH * CH2=CH-O-R
Далее виниловый эфир гидролизуется разбавленной кислотой с
образованием ацетальдегида и спирта:
.О
СН2=СН—O-R + Н2О » CHg-C/ -f R—ОН
ЧН
Спирт вновь поступает в процесс.
Ацетальдегид получают также неполным окислением или де*
гидрированием этилового спирта:
/Р
СН3—СН2ОН 4- VaOj => СН3—Cf + Н2О
ХН
Неполное окисление этилового спирта в ацетальдегид
осуществляют при 450—550 *С над катализатором (медь, серебро,
осажденное на высокопористый материал, медно-серебряные
сплавы).
Для уменьшения теплового эффекта процесса неполного
окисления вводят меньшее количество воздуха, чем это требуется по
стехиометрическому расчету. Например, спирт и воздух
смешивают в отношении 1 : 1,1, что соответствует 44% кислорода от
теоретического количества. В результате в продуктах реакции
содержится около 10% несвязанного водорода.
При дегидрировании этилового спирта без участия кислорода
применяют медные или медно-цинковые катализаторы. Хорошим
катализатором является медь с добавкой 5% окиси кобальта и
2% окиси хрома, нанесенная на асбест. В этом случае процесс
проводят при сравнительно низких температурах B75—3Q0 *С).
219
Большой интерес для промышленности представляет новый
способ получения ацетальдегида путем окисления этилена:
.О
СН2=СН2 + i/2O2 > СН3-С/
хн
В этом процессе в качестве катализатора применяют водный
раствор смеси СиС12 и небольшого количества PdCl2. Действие
катализатора можно выразить следующими реакциями:
СН2=СН2 + PdCl2 + Н2О » СН3—СЄ + 2НС1 + Pd A)
NH
Pd + 2CuCl2 * PdCl2 + 2CuCl B)
2CuCl + 2HC1 + V2O2 > 2CuCl2 + H2O C)
Для окисления можно использовать технический кислород либо
воздух.
При окислении этилена кислородом процесс проводят в одну
стадию (рис. 78). Реактор /, стенки и днище которого футерованы
титаном, заполняют катализаторным раствором. Через раствор
пропускают этилен и оборотный газ, содержащий в основном не-
прореагировавшие этилен и кислород.
Реакцию проводят при нагревании катализаторного раствора
до 110—115 °С под давлением 3—7 am. Тепло экзотермической
реакции окисления E2 ккал/моль или 217,88 кдж/моль) отводится
за счет испарения воды и продуктов реакции. Для поддержания
определенной концентрации катализатора в реактор непрерывно
подают воду в количестве, равном испаряющейся части жидкости.
Образующийся ацетальдегид вместе с парами воды и непро-
реагировавшими газообразными реагентами (С2Н4 и О2) по выходе
из реактора охлаждают и пропускают через скруббер 3. Водный
раствор ацетальдегида из скруббера направляют затем на
ректификацию в колонны 4 и 5 я последующую стабилизацию
(освобождение от растворенных газов). Из верхней части скруббера 3 не-
прореагировавшие этилен и кислород возвращаются в
производственный цикл.
В процессе ректификации продуктов реакции, проводимом
в двухколонном ректификационном агрегате 4 и 5, в первой
колонне 4 от воды отделяют ацетальдегид и газообразные продукты.
Далее эти продукты поступают в колонну 5, где ацетальдегид
является кубовым продуктом. Выход ацетальдегида составляет
95% и более (в пересчете на этилен).
В отличие от описанного выше одностадийного процесса,
в котором окисление этилена и регенерация катализатора
происходят в одном реакционном аппарате, при окислении этилена
воздухом процесс проводится в две стадии. В этом случае окисление
этилена и регенерацию катализатора производят раздельно в двух
аппаратах.
220
Ацетальдегид в присутствии небольшого количества серной
кислоты легко полимеризуется, образуя паральдегид
О
Нч
| |
1°
Н3С Н
жидкость с темп. кип. 124 °С и темп. пл. 12 °С. Наряду с паральде-
гидом образуется небольшое количество кристаллического мет-
альдегида (С2Н4ОL.
Вода \
Дцетальдвгид
Рис. 78. Схема получения ацетальдегида окислением этилена:
/—реакционная колонна; 2—холодильники; 3—скруббер; 4. 5—ректификационные
колонны; 6—кипятильники.
Мономерный ацетальдегид перевозят в специальных
железнодорожных цистернах. В некоторых случаях ацетальдегид
превращают в паральдегид, который транспортируют и на месте
потребления деполимеризуют при нагревании.
221
Ацетальдегид является сырьем для производства многих
химических продуктов.
Окислением ацетальдегида получают уксусную кислоту
(стр. 223).
Конденсация ацетальдегида в присутствии щелочи приводит
к образованию альдоля:
ОН
2СНа—С<^ » СНа—СН—С^
Гидрированием альдоля с последующей дегидратацией
образующегося 1,3-бутиленгликоля можно получить бутадиен:
ОН
СНа—СН—СН2-с/ -^ СНа—СН—СН2—СН2ОН -
I ХН
он
» СН2=СН—СН=СН2
Этот способ в годы первой мировой войны применялся в
Германии для получения синтетического каучука.
Из ацетальдегида через альдоль получают кротоновый
альдегид СН3—СН = СН—СНО, при гидрировании которого образуется
н-бутиловый спирт. Из ацетальдегида получают также ацеталь
СН3—СН(ОС2Н5J, используемый в качестве растворителя и в
производстве некоторых мономеров (исходный продукт в синтезе
винилэтилового эфира); альдегидаммиак, нашедший применение
в производстве пластических масс; этилацетат; молочную кислоту
и многие другие продукты.
Паральдегид, взаимодействуя в жидкой фазе с аммиаком,
образует 2-метил-5-этилпиридин
Л/Сн°
4(СНаСНОK + 3NH3 > Г J
Н3С-Н2С
который при дегидрировании в присутствии катализатора дает
2-метил-5-винилпиридин
А/сн° „ Л/™*
—Н2
ценный мономер для получения сополимерных синтетических
латексов (стр. 495 ел.).
222
Получение уксусной кислоты из ацетальдегида. В настоящее
время большую часть уксусной кислоты получают окислением
ацетал ьдегида. ,
Процесс окисления ацетальдегида протекает в две фазы.
Сначала выделяется надуксусная кислота
,0 ,0
CH3-C<f + О2 » CH3-C<f
Н ХО— ОН
которая далее снова реагирует с ацетальдегидом, образуя
уксусную кислоту:
/Р />° /Р
CH3-C<f + СНз-С/ ». 2CH3-C<f
хо-он хн. \ш
Надуксусная кислота—очень нестойкое соединение и
разлагается с выделением кислорода:
сн3—с/ —> сн3—соон + о
хо—он
Распад ее сопровождается выделением большого количества
тепла, поэтому накопление надуксусной кислоты в продуктах
реакции может привести к взрыву. Выделяющийся кислород
энергично окисляет ацетальдегид с образованием двуокиси уг/.
рода и воды.
Чтобы избежать накопления надуксусной кислоты, стремятся
создать такие условия процесса, в которых обе фазы реакции
протекают с одинаковой скоростью, что достигается подбором
соответствующего катализатора. Некоторые катализаторы (соли
железа, меди, кобальта) ускоряют окисление альдегида в надуксус-
ную кислоту, но недостаточно повышают скорость второй фазы
реакции, что вызывает накопление надуксусной кислоты.
Применение марганцевого катализатора (в виде ацетата
марганца) способствует восстановлению надуксусной кислоты в
уксусную.
Схема получения уксусной кислоты каталитическим
окислением ацетальдегида в жидкой фазе показана на рис. 79. Смесь
ацетальдегида и уксусной кислоты из смесителя 1 подают вместе
с раствором катализатора (ацетат марганца) в уксусной кислоте
в окислительную колонну 2. Прямотоком окисляемой жидкости
в нижнюю часть колонны 2 вводят воздух (или кислород). Для
отвода тепла экзотермического процесса окисления внутри
колонны 2 установлены охлаждающие водяные змеевики.
Газы, выходящие из окислительной колонны, содержат пары
ацетальдегида и уксусной кислоты. Паро-газовая смесь
поступает в конденсатор 3, охлаждаемый рассолом. Конденсат стекает
обратно в колонну, а газы отводятся в атмосферу. Из расширен-
223
Нои "части окислительной колонны вытекает уксусная кислота,
Небольшое количество кислоты смешивают с ацетальдегидом в
смесителе /. Часть кислоты поступает в сборник 4, в котором
приготовляют раствор катализатора. Остальную кислоту
перекачивают в куб ректификационной колонны 6. Из колонны пары
кислоты поступают в конденсатор 7, откуда жидкая СН3СООН
направляется на ректификацию. Из кубового остатка
регенерируют катализатор.
Уксусная
на регенерацию
Рис. 79. Схема получения уксусной кислоты окислением ацетальдегида:
/, 4— сборники-смесители; 2—окислительная колоииа; 3, 7—конденсаторы; Л—сепаратор;
Є—ректификационная колонна.
Синтетическая уксусная кислота может быть получена также
взаимодействием кетена с водой (стр. 216), из метанола и окиси
углерода (стр. 172). Большой практический интерес представляет
получение уксусной кислоты неполным окислением бутана
(стр. 230).
Значительные количества уксусной кислоты получаются при сухой
перегонке древесины (стр. 73).
224
Пищевую уксусную кислоту получают биохимическим окислением
этилового спирта под влиянием бактерий (уксусный грибок). Бактерии размножаются
на древесных стружках, помещенных в деревянные чаны большой емкости.
Чаны заполняют спиртом и пропускают через него воздух. Процесс окисления
спирта в уксусную кислоту идет сравнительно быстро.
Вырабатываемую по этому способу уксусную кислоту выпускают в виде
80%-ной уксусной эссенции или в виде уксуса C%-иого водного раствора
уксусной кислоты).
Уксусная кислота смешивается во всех отношениях с водой,
спиртом, эфиром и многими другими растворителями. Она хорошо
растворяет серу, фосфор, галоидоводороды и другие
неорганические продукты. Ледяная уксусная кислота является хорошим
растворителем многих органических веществ. Уксусная кислота
широко используется в текстильной и пищевой промышленности,
в производстве искусственного волокна, пластических масс и т. д.
Значительную часть уксусной кислоты перерабатывают в
уксусный ангидрид (стр. 226). Большое применение нашли также
соли уксусной кислоты (ацетаты). Ацетат свинца используется
в медицине, для получения пигментов: свинцовых белил,
свинцового крона и пр.; ацетат меди—для производства средств борьбы
с болезнями растений; ацетаты алюминия—в качестве протрав
при крашении тканей; ацетат натрия является промежуточным
продуктом для получения уксусного ангидрида и хлористого
ацетила.
Важное промышленное значение в настоящее время приобрела
надуксусная кислота, используемая в производстве эпоксидных
соединений. Эпоксисоединения могут быть получены в
результате взаимодействия надуксуснои кислоты с олефинами или с
другими ненасыщенными соединениями
\с=с/ + сна-с<( —. >с—с/ + сна-с/
/ \ ХО-ОН / \ / \ ХОН
О
Иадуксусная кислота для этих целей получается из уксусной
кислоты и перекиси водорода:
/Р уР
СНа—C<f + Н2О2 -, СНа—C<f + Н2О
хон хо-он
или окислением ацетальдегида в присутствии небольших
количеств озона как катализатора:
у О оа / О
CHa-C<f + Оа . СНЭ— С/
ХН ХО—ОН
Хлоруксусная кислота С1СН2—СООН, получаемая
хлорированием уксусной кислоты в присутствии катализатора, нашла
15—80S 225
Применение ДЛЯ получения ИНДИГО V-ф- о'Ю). П насіинщсс ьрсмп
она является важным полупродуктом для производства одного
из самых эффективных гербицидов—2,4-дихлорфеноксиуксусной
кислоты, известного под названием 2,4-Д (стр. 341). Этот препарат
получают из натриевой соли хлоруксусной кислоты и 2,4-дихлор-
фенолята натрия в водном растворе или в среде органического
растворителя:
.СІ
С1—f x)—ONa + С1СН2—COONa >
>— О—СН2—COONa + NaCl
)—СН2—COONa + HCI >
СІ—fj>—О—СН2—СООН + NaCl
Сложные эфирьг уксусной кислоты, получаемые ее этерифика-
цией спиртами в присутствии H2SO4, являются хорошими
растворителями эфиров целлюлозы и применяются в производстве
лаков, кинопленки и др.
Уксусная кислота при взаимодействии с ацетиленом в
присутствии катализатора образует сложный виниловый эфир—
винилацетат:
СНзСООН + СН=СН » СНаСО—О—СН=СН2
Процесс проводят в жидкой или в паровой фазе. При паро-
фазном процессе смесь паров уксусной кислоты и ацетилена
пропускают при температуре около 200 "С над ацетатом цинка,
нанесенным на высокопористый материал (активированный уголь,
силикагель).
Полимер винилацетата—поливинилацетат используется в
производстве лаков, клеев, пластических масс (стр. 428).
Получение уксусного ангидрида. С развитием
промышленности химических волокон и, в частности, производства волокна
из ацетилцеллюлозы (стр. 462) возросло потребление уксусного
ангидрида, широко применяемого также как ацетилирующее
средство в производстве красителей, лекарственных и душистых
веществ.
Уксусный ангидрид в промышленных масштабах получают
взаимодействием кетена с уксусной кислотой:
СН3СОЧ
СНзСООН + СН2=С=О »• р>0 + Н2О
226
Кетен в свою очередь получают пиролизом ацетона (стр. 216)
или уксусной кислоты:
СН3—СООН ї=Г± CH2=G=O + Н2О
На рис. 80 изображена схема процесса получения уксусного
ангидрида по этому методу. Пиролиз проводят при 700—720 "С
в трубчатом реакторе с газовым или электрическим обогревом
Вода
Уксусная
кислота
Уксусная
кислота'
Рис. 80. Схема получения уксусного ангидрида из уксусной
кислоты:
/—трубчатый реактор; 2—холодильник; Д—колонные абсорберы.
В качестве катализатора применяют пары триэтилфосфата
РО(ОС2Н5K, вводимые в реактор вместе с парами уксусной
кислоты. Для замедления обратной реакции
СН2=С=О + Н2О * СН3—СООН
к продуктам, выходящим из реактора и содержащим кетен,
добавляют небольшое количество аммиака. После охлаждения
продукты реакции поступают в колонные абсорберы 3, где происходит
взаимодействие кетена с уксусной кислотой. Выход уксусного
ангидрида—90% (считая на уксусную кислоту).
Известен метод получения уксусного ангидрида
взаимодействием ацетилена и уксусной кислоты в присутствии катализатора
15*
227
(сульфат двухваленінси pry in) при їй—ou ^. hjjm 3i
образуется этилидендиацетат
CH=CH + 2CH3—COOH » СН3—СН(ОСОСН3J
который при разложении дает уксусный ангидрид:
СН3—СН(ОСОСН3J » (СН3СОJО + СН3—
ДЕГИДРАТАЦИЯ СПИРТОВ
Для некоторых процессов переработки олефинов
существенное значение имеет чистота [сходного углеводорода. Например,
в производстве полимеров и сополимеров на основе изобутилена
Изоіїутилеп на
\рвнтифинацию
Иэооутиловый
спирт
Т
¦Водный
слой
Рис. 81. Схема получения изобутилена дегидратацией изобутилового
спирта:
/—испаритель спирта; 2—теплг-обм<нрик; 3- контактный аппарат; 4—кої'денсатср для
ртути; 5—печь с испарителем ртути; S—конденсатор; 7—сепаратор.
требуется высокая чистота исходного углеводорода, так как
примеси з?херживают прсцесс его полимеризации.
Олефины высокой чистоты можно получать при дегидратации
спиртов:
С„Н2П+1ОН » С„Н.2П + Н,0
Из этилового спирта таким способом получают чистый этилен
(стр. 211), дегидратацией иэобутилового спирта (СН3JСН—СН.,ОН
или третичнсго бутилового спирта (СН3KСОН—чистый изобути-
лен и т. д.
На рис. 81 изображена технологическая схема получения
изобутилена дегидратацией изобутилового спирта. Пары спирта
228
из аппарата / под давлением 4 am при 150 С поступают в
теплообменник 2. где оми перегреваются до 300—310 °С за счет тепла
газов, выходящих из контактного аппарата 3. В контактном
аппарате пары спирта разлагаются при 360—370°С с образованием
изобутилена:
СН3ч СНЗЧ
>СН— СН2ОН > >С=СН3 + НіО
СИ/ СН„у
Контактный аппарат 3 представляет собой трубчатый
теплообменник, трубки которого заполнены катализатором.
Контактные газы, содержащие 65% изсбутилена, 24% воды, 2% неразло-
жившегося из бутилового спирта и 9% других органических
продуктов, выходят из аппарата 3, охлаждаются в
теплообменнике 2 до 180—210 °С и поступают в конденсатор 6. Отсюда
конденсат, содержащий изсбутилен и воду, перетекает в сепаратор 7,
где изсбутилен отделяется и далее направляется на ректификацию.
Контактный аппарат обогревается парами ртути,
образующимися в испарителе печи 5. Они поступают в нижнюю часть
межтрубного пространства контактного аппарата 3 и, конденсируясь
на трубках, нагревают катализатор. Конденсирующаяся ртуть
стекает обратно в испаритель печи 5, туда же направляют ртуть
из конденсатора 4-
Преимуществами ртутного обогрева являются низкое
давление паров ртути при высокой температуре (при 400 аС давление
паров ртути 2 am) и равномернее нагревание катализатора по
есєй высоте слоя. К недостаткам такого способа обогрева следует
отнести токсичность паров ртути и ее высокую стоимость.
По аналогичной схеме с применением такого же оборудования
возможно получение этилена дегидратацией этилового спирта.
Этот способ получения олефинов осуществляется на установках
небольшей производительности при отсутствии других
источников олефинов.
//. ОКИСЛЕНИЕ УГЛЕВОДОРОДОВ
Переработка углеводородов путем их неполного окисления
занимает в современной промышленности органического синтеза
ви^нэе место ввиду дешевизны этого способа и возможности
получения с его помощью разнообразных кислородсодержащих
органических соединений. Неполным окислением различных
углеводородов могут быть получены спирты, фенолы, альдегиды, ке-
тоны, органические кислоты, их ангидриды, эпоксисоединения.
ОКИСЛЕНИЕ ПАРАФИНОВЫХ УГЛЕВОДОРОДОВ
Неполное окисление парафиновых углеводородов проводится
как в жидкой, так и в газовой фазе. Окисление
низкомолекулярных углеводородов (Сі—С6) в спирты, альдегиды и другие соеди-
292
и температуре порядка 500 СС в присутствии различных
катализаторов: металлов, их окислов и солей. В ряде случаев в процессе
окисления применяют повышенное давление и более низкие
температуры. Окисление высших парафинов, а также нафтенов
целесообразно проводить в жидкой фазе, так как при высоких
температурах, требуемых для испарения этих углеводородов, они
расщепляются.
В качестве окислителя чаще всего применяется воздух, в
некоторых случаях—технический кислород, озон, водяной пар,
двуокись и другие окислы азота. Иногда окислы азота могут
служить катализаторами.
Наиболее трудно окисляется метан, при последовательном
переходе от метана к бутану протекание реакций окисления
облегчается. Особенностью реакции окисления гомологов метана
является образование соединений с меньшим числом
углеводородных атомов, чем в молекуле исходного углеводорода. Так, при
неполном окислении этана С2Нв образуются метиловый СН3ОН
и этиловый С2Н5ОН спирты, уксусный альдегид СН3СНО и
уксусная кислота СН3СООН, формальдегид НСНО и муравьиная
кислота НСООН. При окислении пропана С3Н8 получается ацетон,
СН3СОСН3, уксусный альдегид, формальдегид и т. д. Это
доказывает, что окисление гомологов метана при высокой
температуре происходит с расщеплением (деструкцией) молекулы по
связи С—С.
Проводя процесс окисления парафиновых углеводородов при
температуре около 200 °С в присутствии бромистого водорода
(катализатор), можно значительно затормозить процессы разрыва
углеродных цепей и повысить выход целевых продуктов. Так, из
этана при 220 °С в присутствии НВг получается уксусная
кислота с выходом 63% (молярных); в результате окисления пропана
при 180 °С образуется до 75% ацетона и 8% пропионовой кислоты.
Исключительный интерес для промышленности представляет
процесс неполного окисления бутана, позволяющий получить
большие количества уксусного альдегида и уксусной кислоты.
Окисление метана. При неполном окислении метана вначале
образуются метиловый спирт и формальдегид:
ШІ 1 / ҐЛ Ґ*ХД ҐЛТ-Т
4 ~| /2^2 > Ч_«Г1д^_>Г1
СН3ОН + ЇДА у HC<f + Н2О
Однако эти соединения подвергаются дальнейшему окислению
значительно легче, чем исходный углеводород, т. е. процесс
окисления продолжается до образования муравьиной кислоты, а за-
?30
тем двуокиси углерода (продукты полного окисления метана):
,0
,0
не/ +
не/
4
HC<f
.О
он
С02 + Н20
Мегпан
5
Воздух
Вода
Окисление метана в присутствии платины и палладия в
качестве катализаторов до образования муравьиной кислоты и
продуктов полного окисления (С02 и Н20) происходит так быстро,
что выделить
промежуточные продукты окисления
(формальдегид, метанол)
не удается.
Для
преимущественного получения
формальдегида окислением метана
при атмосферном
давлении в качестве
катализаторов применяют
соединения меди и а^>р^па, а
также окислы' т'а , ч -ер-
живающие'^сс ^
ления на си' '0~ _
ния пром-^-'ль їх
дуктов. , г ..уиокое
окисление ы'ожет быть
частично предотвращено при
быстром удалении
продуктов реакции из зоны
высоких температур. Для этого
газовую смесь (метан и
кислород) пропускают
через реакционную зону с
большой скоростью.
Окисление метана при
атмосферном давлении
приводит главным образом к образованию формальдегида. При
высоком давлении и большом избытке метана в газовой смеси
основным продуктом является метиловый спирт.
Получение формальдегида. В промышленности применяются
два метода получения формальдегида—окисление метана и его
гомологов и окисление метилового спирта.
На рис. 82 показана схема процесса получения
формальдегида неполным окислением метана кислородом воздуха при
объемном отношении 98%-ного метана к воздуху, равном 3,7 : 1.
Раствор
формальдегида
Рис. 82. Схема получения формальдегида
окислением метянл:
/—трубчятый реактор; 1 -теплообменник;
3—холодильник. 4—скруббер; 5 —імяодувка.
231
Нагретая в теплообменнике 2 до 400 С смесь свежего и
оборотного газа (метан и воздух) поступает в трубчатый реактор I (на
1 объем свежей газовой смеси 9 объемов рециркулирующей
смеси). Трубчатый реактор состоит из трубок длиной 3 м, выполненных
из окалиностойкой стали. Вследствие выделения тепла реакции
температура в реакторе повышается до 500 °С и выше. В
начальный (пусковой) период для подогрева реактора сжигают часть
метана.
На входе в реактор в газовую смесь вводят пары азотной
кислоты @,08 объемн. %), которая под действием высокой
температуры распадается на окислы (катализаторы процесса).
Soda
пар
Метиловый
спирт
Формалин
Рис. 83. Схема получения формальдегида окислением метилового
спирта:
/— испаоиттль; 2 — подогреватель; Л контакт ый аппарат; 4—холодильник. 5-б.ірбо-
тажпый хол* дильник; tf— колої і ы& :бсіб-р. 7 —ресивер; 8— а^ку>м кс N.nptjcrop;
9 — водоо гд ели тель.
Горячие продукты по выходе из реактора охлаждаются в
теплообменнике 2 и холодильнике 3. В скруббере 4 газы промываются
водой, поглсщающей формальдегид. Последний в виде 5—10%-но-
го водного раствора вытекает из нижней части скруббера и
направляется лалее на ректификацию.
Основным промышленным способом получения формальдегида
в настоящее время является неполное окисление синтетического
метилового спирта:
/Р
СН3ОН + і/2О2 » НС< + Н2О
ХН
Технологический процесс, схема которого представлена на
рис. 83, был изучен и разработан Е. И. Орловым. Очищенный от
232
пыли воздух при 45 С барботирует через метиловый спирт,
находящийся в испарителе /. Образующаяся здесь спирто-воздуш-
ная смесь, содержащая 0,5—1,0 г паров спирта в 1 л воздуха,
через подогреватель 2 поступает в контактный аппарат 3.
Большое значение для нормального ведения процесса имеет
постоянство состава паро-воздушной смеси, так как изменение ее
состава может понизить выход формальдегида, а уменьшение
концентрации спирта в смеси создает опасность взрыва. Это
постоянство достигается автоматическим регулированием уровня
спирта в испарителе, температуры воздуха и спирта и давления
в системе.
В контактном аппарате на серебряном катализаторе при
температуре около 600 °С происходит неполное окисление метанола
в формальдегид.
Этот процесс можно описать двумя основными реакциями:
дегидрирование метанола
О
СН3ОН » HC<f + Н2
Н
и окисление образующегося водорода кислородом воздуха
Н2 + V2O2 » Н2О
Чтобы затормозить дальнейшие превращения формальдегида,
продукты реакции из зоны катализатора сразу же отводят в
холодильник 4, смонтированный как одно целое с контактным
аппаратом. Из холодильника 4 контактные газы, охлажденные до
125 °С, поступают в холодильник 5, а далее в абсорбер 6
колонного типа, орошаемый холодной водой, которая поглощает
формальдегид. Из контактных газов тепло абсорбции отводится в
выносных холодильниках.
Из нижней части абсорбера вытекает формалин—водный
раствор формальдегида, содержащий 37,6% НСНО и около 10%
СН3ОН. Метиловый спирт стабилизирует формальдегид,
предотвращая его полимеризацию. Формалин направляют на склад как
товарную продукцию. Для получения концентрированного
формальдегида формалин подвергают ректификации. Освобожденные
от формальдегида газы, отходящие из абсорбера (смесь N2, Н2,
СН4, СО2, О2), через рессивер 7 удаляются ротационным вакуум-
компрессором 8 в атмосферу.
Выход формальдегида—85% и более от количества метилового
спирта. Чистый сухой газообразный формальдегид довольно
стоек, но в присутствии следов влаги начинается его
полимеризация с образованием полиоксиметиленов (СН2О)„Н.2О (п=8—100).
При упаривании водных растворов можно получить твердый
продукт—параформальдегид (СН2О)П. Нагревание таких полимерных
продуктов в присутствии небольших количеств серной кислоты
вызывает их распад с выделением формальдегида.
233
симетилен (триоксан)
О СН2
4 СН2—
впервые полученный А. М. Бутлеровым. Триоксан плавится при
64 °С и кипит при 115 °С; при нагревании с H2SO4 деполимеризует-
ея в формальдегид, как и другие его полимеры.
Формальдегид (в виде формалина или полимеров) находит
большое применение во многих органических промышленных
синтезах, а также употребляется как средство для дезинфекции
и дезинсекции. Большие количества формальдегида
используются в производстве феноло-формальдегидных полимеров, амино-
пластов, полиформальдегида (полимер НСОН) и других
высокомолекулярных продуктов.
В промышленности пластических масс, а также в
производстве взрывчатых веществ используется продукт взаимодействия
формальдегида с аммиаком—гексаметилентетрамин или
уротропин [CH2I6N4, впервые синтезированный А. М. Бутлеровым:
6HC<f
\снГ\
I
о / I \
¦ 4NH3 »Н2С N-CH2—N
СН,
Уротропин является также лекарственным веществом.
В промышленности уротропин получают двумя способами:
в жидкой фазе и в газовой фазе.
При проведении жидкофазного процесса формалин и 25%-ная
аммиачная вода тщательно перемешиваются при температуре не
выше 50 °С. Образующийся слабощелочной раствор уротропина
обрабатывают активированным углем, фильтруют и выпаривают
в вакууме до кашицеобразного состояния. После охлаждения его
центрифугируют, выделяют и сушат влажный продукт при 30—
35 °С.
При газофазном способе контактные газы, получаемые при
окислении метилового спирта (см. рис. 83, стр. 232), направляют в
реактор, заполненный насыщенным раствором уротропина. Туда
же направляют газообразный аммиак. Реакция взаимодействия
протекает при 70 °С и абсолютном давлении 0,5 am. По мере
образования уротропин кристаллизуется из насыщенного раствора,
кристаллы затем отделяют в центрифугах; после сушки
уротропин отправляют на склад как готовый продукт.
Весьма интересен в практическом отношении двухстадийный
процесс получения изопрена из формальдегида. В первой стадии
234
формальдегид, взаимодействуя с изобутиленом, дает 4,4-диметил-
1,3-диоксан
О СН2
/О СНЗЧ / \
2НС/ + (СН3JС=СН3 ——¦ >С О
СН2—СН2
2—СН2
который при 220—240 °С в присутствии производных фосфорной
кислоты расщепляется на изопрен, формальдегид и воду (вторая
стадия):
О—СН2 СН3
OV / \ | ,0
\С О » СНа=С—СН=СН, + HC<f + Н2О
сн/ \ / хн
сн2—сн2
При взаимодействии водного раствора формальдегида с ацет-
альдегидом в присутствии гидроокиси кальция образуется
четырехатомный спирт—пентаэритрит:
X) Са(ОНJ
CH.^Cf » С(СН2ОНL + НСООН
ХН
Пентаэритрит—твердое вещество (темп. пл. 141 °С),
применяется в производстве синтетических смол, пластификаторов и
взрывчатых веществ.
Получение и применение синильной кислоты. Окислением
метана в смеси с аммиаком получают синильную кислоту:
СН4 + NH3 + 1V2O2 * HCN + 3H2O
Процесс протекает при 1000 °С, такая высокая температура
поддерживается за счет тепла этой сильно экзотермической
реакции. К исходной смеси, содержащей примерно 12% метана, 11%
аммиака и 77% воздуха, добавляют азот (чтобы избежать
образования взрывоопасных концентраций) и направляют газовую смесь
в контактный аппарат, который напоминает конвертор для
окисления аммиака в производстве азотной кислоты.
Катализатором являются сетки из платиново-родиевого сплава,
расположенные друг над другом.
В продуктах реакции содержится около 7 мол. % синильной
кислоты и 23% водяного пара, образующегося в результате
основной и побочных реакций. Остальное—N2, СО, СО2, NHa, CH4,
Н2, О2. Чтобы синильная кислота не разлагалась при высокой
температуре, газы, выходящие из контактного аппарата, быстро
охлаждают в трубчатом котле-утилизаторе, присоединенном
непосредственно к нижней части контактного аппарата. Водяной
пар, образующийся при охлаждении контактных газов в котле-
утилизаторе, используют для технологических целей. Далее из
контактных газов синильную кислоту абсорбируют водой и из
235
водного раствора путем десорбции выделяют 99%-ную синильную
кислоту.
Чистота синильной кислоты имеет очень большое значение,
так как присутствие примесей вызывает ее полимеризацию с
образованием темной аморфней массы (HCN)„ и выделением
газообразных продуктов AМН3, СО). Полимеризация проходит с счень
большей скоростью и носит взрывной характер, в результате
возможен разрыв стальных оболочек аппаратов и баллоь.ов, в
которых находится HCN. К веществам, вызывающим полимеризацию
синильней кислоты, относятся: аммиак, щелочи (NaOH, КОН),
цианиды щелочных металлов, органические амины и даже вода
(в количестве более 3—4%). Поэтому в синильную кислоту обычно
вводят стабилизаторы, предотвращающие ее полимеризацию
(серная кислота в количестве 0,005—0,01% от веса HCN, эфиры хлор-
угольней кислоты, SnCl2 и др.). Роль стабилизаторов заключается
в связывании веществ, ускоряющих полимеризацию.
Синильная кислота—бесцветная, подвижная и летучая
жидкость с запахом горького миндаля (темп. кип. 25,7 °С, темп. пл.
—14,9 СС); смешивается во всех отношениях с водой, спиртом,
бензолом, эфиром; HCN—очень слабая кислота, даже такие
кислоты, как угольная или сероводородная, вытесняют
синильную кислоту из ее солей. Поэтому цианиды (соли синильной
кислоты) легко разлагаются на воздухе под действием двуокиси
углерода-
KCN + СО2 + Н2О HCN + КНСО3
Синильная кислота—наиболее сильный и опасный яд. Она
оказывает отравляющее действие как при попадании в
желудочно-кишечный тракт или в кровь, так и при вдыхании воздуха,
содержащего пары HCN. При большом содержании в воздухе
синильная кислота проникает в организм через кожные покровы.
Концентрация синильной кислоты в воздухе, равная 0,06 е/м3,
уже смертельна; при концентрации около 1 г/м3 смерть наступает
моментально.
Синильная кислота плохо адсорбируется активированным
углем, поэтому при работе с нею в качестве средств
индивидуальной защиты должны применяться противогазы со специальными
химическими поглотителями или кислородные изолирующие
противогазы.
Несмотря на исключительную токсичность, в настоящее время
синильная кислота широко применяется в ряде важных
технических синтезов.
С окисью этилена HCN образует этиленциангидрин (нитрил
гид ракр иловой кислоты)
СН2-ОН
Н2С СН2 + HCN . |
СИ,-1
\ / CH2-CN
О
236
представляющий собой легкоподвижную ядовитую жидкость с
темп. кип. 220 °С.
Процесс протекает при 55 °С под давлением около 2,5 am в
присутствии катализаторов—алифатических или ароматических
аминсв. В продуктах реакции содержится около 90% этиленциан-
гидрина. Для получения чистого вещества продукты реакции
разгоняют в вакууме.
Акриловая кислота образуется при нагревании этиленциан-
гидрина с разбавленной серной кислотой. При этом этиленциан-
гидрин сначала взаимодействует с двумя молекулами воды и дает
гидра<риловую кислоту, которая затем дегидратируется в
акриловую:
СН2ОН 2н2о СН2ОН СН2
СН2—CN СН2—СООН ~На0 СН—СООН
В результате реакции этиленциангидрина с одноатомными
спиртами в присутствии кислоты получают эфиры акриловой
кислоты:
сн,он сн2
| +ROH > I!
CH2-CN 'л-^4 CH2COOR
Акриловая кислота и ее э|)яры (особенно метиловый)
применяются для получения полимерных материалов.
Дегидратацией этиленциангидрина получают нитрил
акриловой кислоты:
СН2ОН
| -—- CH2=CH-CN
CH2-CN -Ha0
Реакцию проводят в паровой или в жидкой фазе. Жицкофаз-
ную /егилратацию осуществляют при 160—210 °С в аппарате
колонного типа, обогреваемом высококипящим органическим
теплоносителем (ВОТ, стр. 119). Пары акрилонитрила и воды,
содержащие небольшое количество этиленциангидрина, отводят из
верхней части реакционной колонны и направляют в конденсатор.
Верхний слой конденсата—акрилонитрил—отделяют, прометают
серной кислотой и после нейтрализации подвергают
ректификации.
Другой способ получения акрилонитрила—прямой синтез из
синильной кислоты и ацетилена в присутствии катализа гора-
хлор истей меди:
СН=СН + HCN . СН2=СН—CN
Реакцию проводят в стальном цилиндрическом аппарате ]
(рис. 84) высотой 16 м и диаметром 1,6 м. Верхняя расширенная
часть реактора имеет диаметр 3 м. Изнутри реактор гуммирован
и футерован кислотоупорными плитками. Реактор заполнен на
237
Вода
'/в катализатирным раствором, лцеіилен смешиьают с азотом,
нагревают смесь до 90 СС в подогревателе 2 и пропускают снизу
через катализаторную жидкость в аппарате /. Газы захватывают
часть жидкости и увлекают ее в верхнюю расширенную часть
реакционного аппарата. Здесь благодаря значительному
уменьшению скорости газо-жидкостного потока жидкость отделяется от
іцетилена и отводится в нижнюю часть аппарата через боковую
циркуляционную трубу. В
эту же трубу подают
синильную кислоту. Такое
устройство обеспечивает
интенсивную циркуляцию
и хороший контакт
реагентов.
Процесс проводят при
80 СС, образующийся
акрилонитрил (темп. кип.
77,3 °С) в виде паров
удаляется вместе с непроре-
агировавшим ацетиленом
из верхней части аппарата.
Продукты реакции
охлаждаются в водяном
холодильнике 3 и поступают
в абсорбер 4, где
акрилонитрил извлекается из
Растворакрилонит- газов водой. Непоглощен-
ные газы (ацетилен, азот)
очищаются в адсорбере 5,
заполненном
активированным углем, от винилаце-
тилена (побочный продукт
реакции) и возвращаются
в реакционный аппарат / .
Из нижней части абсорбера 4 вытекает слабый раствор
акрилонитрила, направляемый далее на выделение и очистку.
Новые перспективные промышленные способы синтеза нитрила
ікриловой кислоты:
1. Взаимодействие акролеина с аммиаком и кислородом:
Ацетилен
рила на дыоЁлвнае
Рис. 84. Схема прямого синтеза нитрила
акриловой кислоты из синильной кислоты
и ацетилена:
/—реакционный аппарат; 2—подогреватель; 3—
холодильник; 4—абсорбер; 5—адсорбер.
СН2=СН—C<f
Р
NH3
СН2=СН—CN + 2Н2О
Превращение акролеина в акрилонитрил проводится в паровой
фазе при 550—600 СС в присутствии металлических
катализаторов, например окиси молибдена, нанесенной на окись алюминия.
Выход превышает 90% (считая на акролеин). Наиболее перспек-
,238
тивныи метод получения акролеина—прямое окисление
пропилена в присутствии катализатора (закись меди на алунде):
/Р
СН2=СН—СН8 + О2 * СНа=СН—C<f + Н2О
ХН
Этот процесс проводят при 320—350 °С под давлением 8—10 am.
2. Прямое превращение пропилена в нитрил акриловой
кислоты:
СН3=СН-СН3 + NH3 + U/A * СН2=СН—CN + ЗНаО
Катализаторами этого процесса могут служить окислы
молибдена и кобальта, фосфоромолибдат висмута на носителе (силика-
гель); оптимальные соотношения реагентов—кислород : пропилен
1 : 1 или 2:1, аммиак : пропилен 1 : 1 или 2,5 : 1. Степень
превращения пропилена в нитрил акриловой кислоты—около 53%.
3. Прямой синтез из пропилена и аммиака:
СН2=СН-СН3 + NH3 > CH2=CH-CN + ЗН2
Эта реакция была известна давно, однако из-за низких выходов
акрилонитрила она не имела промышленного значения. При
атмосферном давлении наряду с акрилонитрилом образуется
главным образом ацетонитрил CH3CN. При 200 am и 350—400 °С в
присутствии никелевого или кобальтового катализатора выход
нитрила акриловой кислоты значительно повышается,
благодаря чему этот способ представляет для промышленности существен
ный интерес.
4. Взаимодействие пропилена с окисью азота:
СН2=СН—СН3 + 6NO > 4СН2=СН—CN + Na + 6Н2О
При температуре процесса 460—500 °С катализаторами могут
служить соединения серебра. За один проход реакционных газов
через катализатор выход акрилонитрила достигает примерно 12%.
Нитрил акриловой кислоты имеет большое значение как
мономер для производства синтетического каучука (стр. 489),
химического волокна нитрон (стр. 465), а также применяется в синтезе
пластификаторов.
Продукт взаимодействия HCN с ацетоном—ацетонциангидрив
(стр. 2І6) в основном перерабатывается в метиловый эфир мет-
акриловой кислоты—метилметакрилат:
сн
СНЗЧ hcn СНз\ /0Н снзон I * JO
\С=О > С ——* СН3=І-С/
сн/ ch/\cn H2S°4 >СНз
К ацетонциангидрину при 75 °С и при интенсивном
перемешивании приливают сначала серную кислоту, а затем метиловый
239
спирт, і емпературу повышают до oU—1UU (^ и прибавляют
безводный сульфат натрия (дегидратирующий агент). Реакционную
смесь разделяют путем фракционированной перегонки.
Метакриловая кислота получаемая из ацетонциангидрина
ОН ОН СН3
СН3ч | +2Н2О СНЗЧ | |
\C-CN -> >С-СООН ——^ СН2=С—СООН
СН3Х СН3/ -Н2°
ацетон- оксиизомасляная метакриловая
циангидрин кислота кислота
й ее эфиры также находят широкое применение (стр. 414, 546).
Окисление высших гомологов метана
Окислением высших гомологов метана получают карбоновые
кислоты, близкие по свойствам к кислотам, образующимся при
расщеплении жиров. Карбоювые кислоты С10—С2,, вполне
пригодны для изготовления хозяйственного и туалетного мыла,
технических моющих средств, смазочных масел, пленкообразующих
веществ и т. д. Сырьем для получения этих кислот служит
твердый парафин (стр. 127). Акад. С. С. Наметкин, исследовавший
состав парафина, установил, что он состоит в основном из
предельных углеводородов нормального строения и лишь на ]/4—из
углеводородов изостроения, нафтенов и др. Поэтому при окислении
твердого парафина возможен высокий выход насыщенных карбо-
новых кислот с неразветвленной углеродной цепью.
Перед окислением парафин очищают от механических
примесей и воды и обрабатывают 96%-ной серной кислотой. При этом
олефины, изопарафины, нафтены и ароматические углеводороды,
содержащиеся в парафине, растворяются в кислоте, образуя суль-
фокислоты, которые удаляются при промывке водным раствором
щелсчи. Очищенный (рафинированный) парафин разгоняют в ди-
стилляиионных аппаратах, для окисления отбирают фракцию,
кипящую в пределах 320—450 СС.
Парафин окисляют кислородом воздуха при 100—120 °С в
присутствии катализаторов (смеси перманганата калия К.МпО4
с солями щелочных металлов и др.).
На рис. 85 изображена схема окисления парафина. Смесь
расплавленного парафина с катализатором поступает из смесителя 1
в окислительную колонну 2, куда воздуходувкой нагнетается
очищені ый от пыли воздух, барботирукщии через реакционную
массу. Для подсгрева смеси до ПО—115 °С в змеевики окислительной
колонны в начале прсиесса г охают есдянсй пар. Реакция скисле-
ния преходит с выделением большого количества тепла, поэтому
в дальнейшем его отводят, пропуская по змеевикам холодную воду.
240
Продукты окисления из колонны 2 после отделения катали-
заторного шлама в отстойнике 4 промываются в колонне 5 водой.
В сепараторе 6 отделяются растворимые в воде низшие жирные
Шсжигание
Лара/рш катализатор
Воздух
Водшй раствор
низших Нардо-
ttoffb/x />'і/слот
Рис. 85. Схема окисления парафина:
I—смеситель; 2—окислительная колонн^; 3—воздушный фильтр; 4—отстойник
катализаторного н;Лома; 5—промывная колонна; а—сепаратор
кислоты (муравьиная, уксусная, пропионовая, масляная,
образующие нижний слей). Окисленный парафин из верхней части
сепаратора поступает на выделение высших карбоновых кислот.
ОКИСЛЕНИЕ ОЛЕФИНОВ
Окисление олефинов может протекать по следующим схемам:
1. Окисление метиленовей группы, расположенной рядом с
углеродом, образующим двойную связь, до гидроперекиси:
R-CH,—СН=СН,
Оа
R—СНООН—СН=СН2
При разложении гидроперекиси могут образоваться спирт
или кетон
-02
• R—СНОН—СН=СН2
R—СНООН—СН=СН2 —
R—СО—СН=СН,
-н3о
окисляющиеся далее до кислот.
16—815
24!
бонильную, а затем в карбоксильную группу:
R—СН=СНа * R— СК2—СНО » R—СН2—СООН
3. Размыкание двойной связи с образованием циклической
перекиси, далее превращающейся в соответствующий кетон или
альдегид:
о2 R.
С=СН2 * >С О » R—СО—R + НСНО
R R I I
СН2-О
о2
R—СН=СН2 » R—СН—О » R—СОН + НСОН
СН,—
О
4. Образование окисей олефинов (эпоксидов):
1/aOu
R—СН2—СН=СН2 * R—СН2—СН—СН2
5. Прямое каталитическое окисление олефинов в присутствии
смеси водных растворов PdCl2 и СиС12 до альдегидов и кетонов
(стр. 215, 220):
,0
СН2=СН2 + V2O2 > СН3-С/
ХН
СН2=СН-СН3 + V2O2 » СНз-СО—СН3
СН2=СН-СН2-СН3 + !/2О2 » СНз-СО—СН2-СН3
За последние годы этот процесс приобрел большое значение.
Из перечисленных реакций наибольший практический интерес
представляет четвертая реакция. Окиси олефинов—весьма реак-
ционноспособные соединения, они легко взаимодействуют с га-
лоидоводородами, спиртами, органическими кислотами и их
хлорангидридами, под действием катализаторов изомеризуются
в альдегиды и кетоны, а также могут вступать в реакции
полимеризации, приводящие к образованию соединений с большим
молекулярным весом.
Получение окиси этилена. В промышленных масштабах окись
этилена
Н2С СН2
\/
о
может быть получена окислением этилена или дегидрохлориро-
ванием этиленхлоргидрина.
242
Для получения окиси этилена по первому способу смесь
воздуха и этилена пропускают при 220—280 °С и давлении 5—25 am
над серебряным катализатором. Для предотвращения
образования взрывоопасной газовой смеси содержание в ней этилена не
должно превышать 3 объемн. % (нижний предел взрываемости
этилена в этилено-воздушной смеси составляет 3,4%).
При окислении одновременно протекают две реакции:
СН2=СН2 + i/2O2 . Н2С СН2
V
СН2=СН2 + ЗО2 » 2СО2 + 2Н2О
Процесс проводят в трубчатых аппаратах с большим
количеством трубок диаметром 20—25 мм. Процесс окисления проходит
с выделением большого количества тепла (особенно по второй
реакции). Чтобы предотвратить перегрев катализатора,
необходим интенсивный отвод реакционного тепла. Поэтому в межтрубном
пространстве контактного аппарата циркулирует охлаждающая
жидкость (дифенильная смесь или вода под давлением).
Выходящие из контактного аппарата продукты реакции
охлаждаются и проходят через абсорбер для извлечения окиси этилена
под давлением. Далее окись этилена выделяют из водного
раствора ректификацией. Газы, содержащие непрореагировавший
этилен, выходят из абсорбера, смешиваются со свежим С2Н4 и
возвращаются в контактный аппарат. Выход окиси этилена—около
60%.
По второму способу окись этилена получают взаимодействием
этиленхлоргидрина со щелочью (обычно с концентрированным
известковым молоком):
2С1СН2—СН2ОН + Са(ОНJ » 2Н2С СН2 + СаС12 + 2Н2О
Этиленхлоргидрин образуется при взаимодействии этилена с
хлорноватистой кислотой в водной среде. При пропускании в
воду этилена и хлора протекают следующие реакции:
С12 + НОН » НОСІ + НС1
НОСІ + СН2=СН2 » НОСН2—СН2С1
При небольшом содержании в воде хлорноватистой кислоты
этилен, пропускаемый в воду, реагирует с хлорноватистой
кислотой с образованием этиленхлоргидрина. При накоплении в
растворе соляной кислоты взаимодействие хлора с водой замедляется
и начинает сказываться наличие молекулярного хлора,
образующего с этиленом дихлорэтан:
СН2=СН2 + С12 > С1СН2—СН2С1
16* 243
Окись этилена
Этиленхлор- *
гидрини из-
дестковое
молоко
Поэтому па практике, во избежание образования Оольших
количеств дихлорэтана, процесс ведут таким образом, чтобы
содержание этиленхлоргидрина в воде не превышало 7—8 "и.
Гипохлорирование этилена
производится в реакционной
колонне высотой около 15 м.
Воду, хлор и этилен подают в
нижнюю часть колонны. Этилен
вводят несколько выше, чем
хлор, благодаря чему
отсутствует непосредственный
контакт между этими газами,
уменьшается образование
дихлорэтана, а этилен реагирует
с образовавшейся
хлорноватистой кислотой.
Температуру процесса
поддерживают в пределах 48—
52 °С. Продукты реакции
отводят из верхней части аппарата
и отделяют от газов.
Полученный водный раствор
этиленхлоргидрина обрабатывают
щелочью C0%-ное известковое
молоко) для получения окиси
этилена:
2НОСН2—СН2С1 + Са(ОНJ >
» 2Н2С СН2 + СаСЬ + 2Н2О
О
На рис. 86 изображен
реакционный
аппарат—вертикальный стальной цилиндр, внутри
которого расположена стальная
спиральная перегородка5
высотой около 2,5 м. Смесь
этиленхлоргидрина с известковым
молоком поступает в
центральную часть аппарата и
движется по спирали к его перифе-
рии. Реакционная масса подо-
"^ гревается острым водяным па-
Рис. 86. Реакционный аппарат для Р°м- Образующаяся окись эти-
получения окиси этилена: лена вместе с парами воды и
/-корпус апшр.тл; 2-паровые трубы; 3-галь- ПОбоЧНЫХ ПРОДУКТОВ, ПРОХОДЯ
ник; 4—СЛНВНІ.Я труба; 5—спиральная перего- J „ г д
' сквозь каплеотбоиную насадку,
родка; 6—каплеотбойная насадка.
244
удаляется через горловину аппарата. Далее пары
конденсируются, конденсат подвергается ректификации.
При сравнении этих способов получения окиси этилена мэжно
считать, что прямое окисление этилена более экономично, так как
по этому способу не требуется затрат хлора и щелочи, которые
при хлоргидринном способе фактически теряются.
Синтезы на основе окиси этилена. Благодаря чрезвычайно
высоксй реакционной способности окись этилена является одним
из важнейших промежуточных продуктов для многочисленных
промышленных синтезов. На основе окиси этилена получают
растворители, пластические массы, взрывчатые вещества,
антифризы, синтетические душистые вещества и т. д.
Окись этилена способна к полимеризации и изомеризации.
Под влиянием третичных аминов, хлорного олова и некоторых
других катализаторов окись этилена полимеризуется, образуя
твердую белую массу, представляющую смесь полимеров состава
(С2Н4О)„. Процесс полимеризации протекает с выделением
большого количества тепла и может привести к взрыву.
При нагревании до 200—250 °С в присутствии А13О3 окись
этилена перегруппировывается в ацетальдегид:
Н. ,н | u
У—С/ Н-С-С'
У
О Н
Гидратацией окиси этилена при 180—195 °С и давлении 17-
18 am получают двухатомный спирт—этиленгликоль:
Н2С СН, + I |2О » HOCH,—СН2ОН
\ /
О
Водные растворы этиленгликоля имеют низкие температуры
замерзания, благодаря чему этиленгликоль широко применяется
для приготовления антифризов (стр. ІІ9). Этиленгликоль очень
гигроскопичен и хорошо растворяет сложные эфиры, смоты,
красители и некоторые вещества растительного происхождения.
Сочетание этих свойств обусловило применение этиленгликоля при
крашении текстильных изделий, в ситцепечатании, для
приготовления штемпельных красок, косметических препаратов, для
увлажнения табака и т. п.
При нитровании этиленгликоля смесью азотной и серной
кислот легко образуется динитрат гликоля (называемый иногда ди-
нитроэтиленгликоль) О2КОСН2—CH2ONO2, применяемый для
изготовления взрывчатых веществ. В водной среде окись этилена
¦24S
с этиленгликолем образует ди-, три- и полиэтиленгликоли:
—СН2ОН > НОСН2—СН2—О—СН2—СН2ОН
H2C СН2 4- НОСН2-СН2-О-СН2-СН2ОН *
* НОСН2—СН2—О—СН2—СН2—О—СН2—СН2ОН и т. д.
Диэтиленгликоль используется как растворитель, а также как
жидкость для тормозных гидравлических приспособлений и при
отделке и крашении тканей в текстильной промышленности.
Триэтиленгликоль и тетраэтиленгликоль—растворители и
пластификаторы. Полиэтиленгликоли применяются в качестве
пластификаторов, смазочных материалов и т. д.
Путем присоединения к окиси этилена одноатомных спиртов
получают простые моноэфиры гликоля
Н2С СН2 + ROH > ROCH2-CH2OH
используемые в качестве растворителей эфиров целлюлозы (цел-
лозольвы).
Моноэфиры гликоля могут быть получены этим способом при
атмосферном давлении в присутствии катализаторов (серная
кислота) или при повышенном давлении и высокой температуре без
катализатора. Для увеличения выхода моноэфиров и подавления
реакций образования эфиров высших гликолей процесс следует
вести при большом избытке спирта (в несколько раз больше, чем
окиси этилена).
Пропуская окись этилена над сульфатом и бисульфатом натрия
(Na2SO4, NaHSO4) при 150 °С, получают димер окиси этилена—
диоксан
^Г12—v^rta
О
сн2
- -сн2
применяемый в качестве растворителя эфиров целлюлозы, масел,
жиров и смол.
Диоксан был впервые получен А. Е. Фаворским в 1906 г.
перегонкой этиленгликоля с 5%-ной серной кислотой:
СН2—СН2
2НОСН2—СН2ОН » О О
\ /
снг—сн2
'246
При взаимодействии с аммиаком окись этилена образует
этаноламины:
Н2С СН2 + NH3 » Н2—N—СН2—СН2ОН
О
.СН2—СН2ОН
2Н2С СН2 + NH3 » Н—N<Q
О
.сн2—сн2он
ЗН2С СН2 + NH3 » N—СН2—СН2ОН
\^ \сн2—сн2он
Аналогично с окисью этилена вступают в реакцию первичные
и вторичные амины, давая соответствующие алкиламино- и арил-
аминоспирты:
RNH2
» RNH—СН2—СН2ОН
Н2С СН2 — r,nh
\ / > R2N-CH2-CH2OH
О
С двуокисью углерода, сероводородом и синильной кислотой
этаноламины образуют на холоду комплексные соединения,
разрушающиеся при нагревании. Это свойство этаноламинов
используют при очистке нефтяного, коксового, генераторного и других
газов от примесей СО2, H2S и HCN. Кроме того, этаноламины
применяются как добавки к моющим средствам, для приготовления
косметических препаратов, а также пластификаторов,
потребляемых в кожевенной и резиновой промышленности. При нитровании
этаноламинов образуются взрывчатые соединения.
При дегидратации диэтаноламина получается циклическое
соеди нен ие—морфолин:
СН2—СН2
HN
сн.
-сн2
Морфолин обладает большой растворяющей способностью и
превосходит в этом отношении диоксан, пиридин, бензол.
Триэтаноламин при взаимодействии с тионилхлоридом SOCla
или пятихлористым фосфором РС15 образует трихлортриэтиламин
N(CH2CH2CIK—тяжелую маслянистую жидкость, обладающую
сильным кожнонарывным действием.
Окись этилена с сероводородом образует тиодигликоль:
.СН2—СН2ОН
2Н2С СН2 + H2S > S<
\ / х:н2—сн2он
о
247
ii|jn дспсівии на іиидпкіплило ічигіцспі уприааппип си.їимии
кислоты получают Р,Р'-дихлордиэтилсульфид S(CH2—СН2С1J,
применявшийся в первую мировую войну как отравляющее
вещество (иприт). Его можно также получить взаимодействием
этилена с монохлористой или дихлористой серой (S3CI2, SC13).
ОКИСЛЕНИЕ АРОМАТИЧЕСКИХ И АЛИЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ
Бензол является одним из углеводородов, наиболее стойких
к окислительному действию кислорода. К самым интересным
направлениям процесса прямого окисления бензола можно было бы
отнести синтез фенола по схеме:
С6Н6+ V2O2 , С„Н5ОН
Однако, несмотря на многочисленные исследования, процесс,
пригодный к реализации в промышленных масштабах, до сих пор
¦еще не разработан, так как получаются низкие выходы целевого
продукта—фенола. В настоящее время фенол получают из
производных бензола (стр. 251). Прямое окисление бензола приводит
к его расщеплению до малеинового ангидрида
СН—СО
:н—со
применяемого в значительных количествах для получения
полиэфирных смол для лаков, пластических масс (стеклопластиков,
¦стр. 409, 558) и моющих средств (стр. 328 ел.). Реакцию окисления
проводят в паровой фазе, пропуская смесь паров бензола с
воздухом над катализатором—пятиокисью ванадия.
Окислением толуола получают с хорошим выходом бензальде-
гид:
,0
+ Н2О
Толуол нагревают вместе с водой и кислородом в автоклаве
при 60 am и 235—240 °С в присутствии катализатора (окись
железа, скись хрома, окись меди). Бензальдегид применяют в
производстве многих красителей арилметанового ряда (стр. 322) и
в парфюмерной промышленности.
Толуол можно окислить также до бензойной кислоты:
нз v
J +г/2о2—>!| [ + н2о
Непрерывный процесс окисления протекает при 149 °С и 2,5—
3,0 am в присутствии нафтената кобальта (катализатор). Бензой-
248
ная кислота является важным полупродуктом в промышленности
тонкого органического синтеза.
Большой промышленный интерес представляют процессы
окисления ксилолов во фталевый ангидрид и фталевые кислоты
(стр. 260).
Жидкофазное окисление метальных групп у ароматических
соединений протекает через стадию образования гидроперекисей.
В мягких условиях окисления гидроперекиси являются
основными продуктами реакции. В более жестких условиях
гидроперекиси, образующиеся в начальный период, разлагаются на
различные кислородсодержащие органические вещества.
Получение гидроперекисей ароматических углеводородов в
настоящее время весьма распространено в химической
промышленности. Например, окислением изопропилбензола (кумола)
получают его гидроперекись
СН3 СН.,
которая при осторожной обработке серной кислотой дает фенол и
ацетон:
сн3
—ОН + СН3—СО—СНЭ
Аналогично гидроперекись emop-бутилбензола дает фенол и
метилэтил кетон:
С*Н.
ОН
Vch
і , »у j | И | +СН3—СО—С?Н5
* сня \г СНз
Ранее (стр. 215) указывалось на возможность совместного
получения и-крезола и ацетона через гидроперекись /г-цимола. Эти
процессы являются экономически выгодными, а потому области
их применения расширяются.
Промышленный интерес представляет процесс окисления
эпициклического углеводорода—циклогексани, который мэжгт быть
выделен из соответствующих нефтяных фрзкций или получен
гидрированием бензола:
СН2
НаС СНя
|| + зн2 —> | |
KS н,с сн2
с
l2v-
сн,
¦?4Э
С.СЛИ окисление циклогексана воздухом проводить в
уксуснокислом растворе в присутствии хлористого кобальта, то
образуется адипиновая кислота:
сн2
н2с сна
НаС СНа
сна
. НООС—(СН2L—СООН + Н2О
Она может быть получена из циклогексана без применения
уксусной кислоты. Этот процесс протекает в две стадии. В первой
стадии циклогексан окисляют воздухом при 140 °С и 15—20 am
в присутствии катализатора—нафтената кобальта. При этом
получается смесь продуктов, основные из которых—циклогексанол и
циклогексанон—образуются в результате распада гидроперекиси
циклогексана:
СН2
НаС СНОН
сн2 сна „ нас сна
\ /\ /н
СН2 оа Н2С С—О—С
нас сна *-1 -'
ОН
сн
н
\
V
сн2
нас с=о
НчС СНч
На второй стадии смесь продуктов подвергают дальнейшему
окислению 60%-ной азотной кислотой
(катализаторы—соединения меди и ванадия). В результате окисления образуется
адипиновая кислота:
СНа
/\
HSC СНОН
н,с сна
СНа HNO3
нас
Н2С
сна
\=0
LHa
¦ НООС-(СНаL-СООН
250
Циклогексанол, циклогексанон и адипиновая кислота
являются важными полупродуктами в производстве полиамидных
волокон (стр. 470 ел.). Они могут быть получены другими
путями, например из нитроциклогексана (стр. 203) или из фенола
(стр. 254).
Получение и переработка фенола. Фенол, извлекаемый из
каменноугольной смолы (стр. 97 ел), занимает небольшую долю
в общем объеме промышленного производства этого важного
продукта. В настоящее время вырабатывается преимущественно
синтетический фенол.
Существует пять методов синтеза фенола:
1. Сульфатный метод (глава V, стр. 271):
,х ,SO3H
/у \ сульфирование х? \/ нейтрализаиля
плавление
бензол бензолсульфо Na-соль
кислота бензолсульфокислоты
ONa ^х ОН
разложение # >у/
—ч/
фенолят натрия фенол
2. Хлорбензольный метод:
NaOH /%У а СО2+Н2О
Вначале бензол хлорируется до монохлорбензола (стр. 193).
Во второй стадии хлорбензол гидролизуется водным раствором
щелочи. В присутствии катализатора (медь) гидролиз протекает с
большой скоростью при 350 °С и 200 am. Без катализатора фенолят
натрия образуется при более высоких температуре и давлении.
Для получения фенола раствор фенолята натрия обрабатывают
двуокисью углерода или соляной кислотой, а затем очищают
дистилляцией. Выход фенола—95% и выше, считая на хлорбензол.
3. По методу Рашига вначале получают монохлорбензол
окислительным хлорированием бензола (стр. 174):
^а
+ HCI + V20a , (| j + Н20
Во второй фазе хлорбензол подвергают гидролизу:
+н2о —. Т + неї
251
вание. Теоретически в этом процессе должны расходоваться
только бензол и кислород воздуха, на практике, однако, необходимо
компенсировать частичные потери хлористого водорода.
Окислительное хлорирование бензола проводят в паро-газо-
вой фазе при 200—230 °С над хлоридами железа и меди.
Хлорбензол гидролизуется водяным паром при 600 °С над катализаторами
Са3(РО4J или MgCl2, последний применяется на силикагеле или
SiO2 в качестве носителей. Выход фенола составляет 75—85%
от теоретического.
4. Кумольный метод проводится по схеме:
—CO—
Изопропилбензол (кумол) получают алкилированием бензола
пропиленом (стр. 150). Окисление кумола и разложение
образующейся гидроперекиси на фенол и ацетон проводят по способу,
разработанному советскими учеными П. Г. Сергеевым, Б. Д. Кру-
жаловым, Р. Ю. Удрисом и Б. С. Немцовым.
Упрощенная технологическая схема этого способа приведена
на рис. 87. Окисление изопропилбензола проводят при 3 am
и I Ю "С в барботажнсй колонне / из нержавеющей стали. Воздух,
уносящий из колонны пары изопропилбензола, проходит
конденсатор-холодильник 2, где изопропилбензол конденсируется, и
возвращается на окисление. Жидкие продукты, содержащие 27—
30% гидроперекиси изопропилбензола, направляют на выделение
изопропилбензола в насадочную колонну 3, работающую при
остаточном давлении 30 мм рт. ст. Здесь отгоняется основное
количество неирореагировавшего изопропилбензола. Остаток,
содержащий до 70—75% гидроперекиси, направляется в вакуум-дистил-
ляционную колонну 5, в которой при остаточном давлении менее
10 мм рт. ст. отгоняется остаток изопропилбензола.
Затем 90—92%-ную гидроперекись разлагают при 40—60 °С
серной кислотой в реакторе 10, снабженном мешалкой и
охлаждающей рубашкой. Продукты разложения, содержащие главным
образом фенол и ацетон, далее нейтрализуют и подвергают
ректификации для выделения технических продуктов.
5. Получение фенола из толуола. Толуол окисляется
кислородом воздуха в присутствии катализатора (нафтенат кобальта)
252
до бензойной кислоты (стр. 248). Кислота отделяется от примесей
(непрореагировавший толуол, бензальдегид) в отпарной колонне
и затем экстрагируется из кубовой жидкости горячей водой. После
охлаждения водного слоя бензойная кислота выпадает в осадок.
Изопропил-
бензол
Рис 87. Схема производства фенола кумольным способом:
/—налипни для ски( лгния; 9 кон.чгн атогы хпл< дили ики. J-птіонж.я ниг; /ь чм. я кплс»нма;
4— кичятилы ик. 5 иакуум ди тилляци* 'И;- я кс л« їм а. *•- ко*и*иг;ітор; ^-регишр. Я
вакуум к ммр'ччор. 9 іборники; 10- р. і.ктор для р;ізлпж»'Ния гидропірркиги; // - подоотде-
ЛИГіЛЬ.
Ее отфильтровывают, расплавляют и в смеси с катализатором
направляют в реактор, гге в грисутствии воздуха и водяного пара
бензсйная кислота декарбсксилируется в фенол:
соон
он
+ СО
Еыхсд бензсйнсй кислсты за один проход через реактор
достигает 75%. Продукты ^екарбсксилиргвания подвергаются
ректификации для Еы^елекия чисте го фенола. Непрореагировавшая
¦бензсйная киелста экстрагируется из кубевей жидкости горячей
водей и возвращается в процесс декарбсксилирования.
253
рех способов получения фенола позволяет считать кумольный
метод наиболее перспективным.
Получение фенола из толуола тоже следует отнести к
перспективным методам, так как в связи с развитием промышленных
процессов гидроформинга и платформинга (стр. 66) могут быть
получены очень большие количества толуола.
Рис. 88. Схема производства циклогексанола
гидрированием фенола:
/—сборник фенола; 2— насос для перекачивания фенола; 3—напорный
бак; 4—испаритель; 5—контактный аппарат; в—конденсатор;
7—теплообменник; в—разделитель; 9—компрессор; 10— сборник.
Фенол является одним из наиболее многотоннажных
продуктов промышленности основного органического синтеза и находит
разнообразное применение в химической, нефтяной,
фармацевтической, парфюмерной и других отраслях промышленности. В
химической промышленности фенол служит полупродуктом в
производстве феноло-формальдегидных полимеров, полиэпоксидов и
полиамидов (стр. 392, 390, 396), некоторых красителей,
применяется также для получения салициловой кислоты (стр. 284),
аспирина и других лекарственных соединений, моющих средств
(стр. 334). В нефтяной промышленности фенол используют для
селективной очистки масел и в качестве азеотропного агента для
выделения толуола из бензина ректификацией.
Гидрирование фенола в циклогексанол—исходное вещество в
синтезе капролактама—проводят обычно в паро-газовой фазе
над металлическим никелем, осажденным на носителе—окиси
254
алюминия. Процесс осуществляют при давлении около 18 am и
температуре 130—150 "С.
На рис. 88 показана схема производства циклогексанола.
Трубчатый контактный аппарат 5 смонтирован вместе с
испарителем 4, снабженным паровыми змеевиками. В испарителе фенол,
через который барботирует водород, поддерживается на
определенном уровне. Насыщенный парами фенола, водород проходит
через трубки контактного аппарата, заполненные таблетирован-
ным катализатором. Тепло экзотермической реакции гидрирования
фенола отводится за счет испарения воды в межтрубном
пространстве аппарата 5. Пары конденсируются по выходе из аппарата 5
в конденсаторе 6 и вновь возвращаются в межтрубное
пространство контактного аппарата.
За один проход паро-газовой смеси через катализатор в
реакцию вступает 10% водорода. Непрореагировавший водород вместе
с парами циклогексанола выходит из верхней части контактного
аппарата и охлаждается до 90 °С в теплообменнике 7, подогревая
до 110 °С водород, поступающий на гидрирование фенола.
Паро-газовую смесь из теплообменника (испарителя) 4
направляют в конденсатор (на схеме не показан) и разделитель 8.
Конденсат далее поступает на ректификацию для выделения
циклогексанола, водород возвращают на гидрирование.
Получение капролактамі. Циклогексанол перерабатывают в
диклогексанон неполным окислением
или дегидрированием:
СНа CHj
нгс снон нх с=о
II —• I I +н2
НаС СНг Н^С СНа
СНд СНд
На рис. 89 показана технологическая схема получения цикло-
гексанона дегидрированием циклогексанола. Концентрированный
(99,9%-ный) циклогексанол-ректификат подается через фильтр 3
в подогреватель 4, где он нагревается водяным паром до 100—
ПО'С, и далее направляется в испарительно-перегревательную
-систему, состоящую из трех последовательно соединенных труб-
255
Чсі 1 ыл сишаиа іuo и, u, / . i-i ппл L\ni\jiui i-ix^anvA иі_ііирлсіел л при
температуре 430—450 °С поступает в трубы контактного
аппарата #, заполненные катализатором (оцинкованное железо в виде
Z-образных пластинок толщиной 0,2—0,4 мм). Выходя из
контактного аппарата, продукты реакции поступают в конденсатор 10
и далее в сепаратор 12, где конденсат (циклогексанон-сырец)
отделяется от водорода. Циклогексанон-сырец направляют на
ректификацию, водород после очистки используют для гидрирования
фенола в циклогексанол.
Вода
ТоплиВный
Циклогексанан -
сырец на ректи- і
фикацию
Рис. 89. Схема дегидрирования циклогексанола:
1 — насос; 2—циркуляционная газодувка; З—фильтр; 4, 5—подогреватели, б—испаритель;
7—перегреватель паров; 8- контактный аппарат; 9— тспка; 10—конденсатор; // — гидравлический затвор;
12—сепаратор; 13—воздуходувка; Л.Д.—дроссельный вентиль.
Для обогрева контактного аппарата и подогревательно-испа-
рительной системы в нижней части топки 9 сжигают газообразное
топливо, при горении которого развивается высокая температура
(^— 1000— 1100 °С). Во избежание перегрева катализатора и
разложения продуктов реакции топочные газы в верхней части топ-
.ки 9 смешивают с газами, возвращаемыми с помощью
циркуляционной газодувки 2 из борова печи. Температура топочных газов,
поступающих в межтрубное пространство контактного аппарата, не
превышает 500—550 °С.
Циклогексанон-ректификат подвергают далее оксимирова-
нию действием на циклогексанон водного раствора сернокислой
соли гидроксиламина, и при интенсивном перемешивании и
охлаждении получают сернокислый циклогексаноноксим
CHS
н/\ о
2 L 1 + (NH2OHJ-H2SO4:=i
H,C CH2
CH~
H~C
H~C
C=N—OH
CH2
_2
который далее нейтрализуют щелочью или аммиаком с
образованием оксима:
- сна
/ \
HjC C=N-OH
1 ^
•H2SO4-bNHa »
г
CH2
H2C
H~C
\
C=N-
CH2
он
+ (NH4JSO4
В зависимости от условий проведения реакции оксим
получают в кристаллическом либо в жидком виде. В процессе обработки
20—36%-ным олеумом циклогексаноноксим изомеризуется
(перегруппировка Бекмана) в лактам г-аминокапроновой кислоты
(капролактам), представляющий собой бесцветное вещество,
маслянистое на ощупь (темп. кип. 262,5 °С).
Изомеризация циклогексаноноксима протекает с большой
скоростью и носит взрывной характер, поэтому олеум
предварительно смешивают с готовым капролактамом (на 95%
капролактама 5% олеума). К этой смеси при интенсивном охлаждении и
перемешивании добавляют расплавленный оксим и олеум. Продукты
реакции нейтрализуют щелочью, отделяют лактам и подвергают
его вакуум-дистилляции.
Полимеризацию капролактама проводят непосредственно на
фабриках, изготовляющих капроновую нить или пленку (стр. 410).
Интересным способом получения капролактама является
гидрирование анилина до циклогексиламина с одновременным
гидролизом последнего в циклогексанол при 200—250 °С на никелевом
катализаторе:
Ш2
гидрирование
бензол
циклогекснламин
цичлогексанол
17—805
257
,н
дегидрирование
У\ /Ч бекмановская
і С=0 оксимирование C=N ОН перегруппировка
іщклогексаНон цнклогексаноноксим
сн2
Н2С СН3
N—Н
Н.2С С=О
сн2
капролактам
На стр. 250 рассматривалось получение адипиновой кислоты
НООС—(СН2L—СООН окислением циклогексана кислородом и
азотной кислотой. Этот способ, по-видимому, является наиболее
перспективным. Наряду с этим в промышленности адипиновую
кислоту получают также окислением циклогексанола 60%-ной
азотной кислотой при 70 °С:
СН2
ОН
Н„С СМ
Н„С СН„
4HNO3 » НООС—(CHaL-COOH + 2NaO3 + ЗН3О
На рис. 90 показана схема этого процесса. В реакторе 3 цик-
логексанол постепенно приливают к азотной кислоте. Аппарат,
выполненный из хромоникелевой стали, имеет змеевики и
рубашку для отвода тепла. Продукты окисления передают в
кристаллизатор 5, где адипиновую кислоту дважды перекристаллизовывают
для удаления побочных продуктов (дикарбоновые кислоты).
Кристаллы адипиновой кислоты отделяют на нутч-фильтре б и сушат
воздухом при 80 *С в сушилке 7.
Из адипиновой кислоты получают гексаметилендиамин.
Процесс протекает в две стадии. В первой стадии получают адиподи-
нитрил:
НООС—(СН2L—СООН + 2NH3 > NC-(CH2L—CN+4H2O
Реакцию проводят двумя путями: пропуская аммиак при
200 *С через расплавленную адипимовую кислоту в присутствии
катализатора (соли фосфорной кислоты) или пропуская смесь
паров адипиновой кислоты и аммиака при 350 °С над
катализатором (фосфорная кислота на сил и ка геле).
258
Выделенный адиподинитрил гидрируют при 50—80 am и
100 °С (вторая стадия), в результате образуется гексаметилен-
диамин:
NC-(CHSL-CN + 4Н2 » H2N-(CH2)e-NHs
В процессе гидрирования применяют скелетный (стр. 134)
кобальтовый катализатор. Процесс проводится в горизонтальном
автоклаве с мешалкой и рубашкой, гексаметилендиамин-сыреи
перегоняется в вакууме. Выход диамина достигает 90%.
Цинлогексанал
Пар
HNO3
ДдипиноВа*
кислота
Рис. 90. Схема окисления циклогексанола:
/, 2—мерники; 3—реактор; 4—холодильник;
5—кристаллизатор; 6—нутч-фильтр; 7—сушильная камера;
в—калорифер:
Для точной дозировки и стабилизации"этих веществ в
процессе хранения адипиновую кислоту и гексаметилендиамин
растворяют в метаноле, перед использованием метанол отгоняют.
Образующиеся кристаллы смеси обоих компонентов отжимают
от маточного раствора на центрифугах. Такая смесь, известная
17*
259
пгш названием соли АГ, используется для получения полиамидного
волокна анид (стр. 4/ij.
Получение фталевого ангидрида. Фталеаый ангидрид
О
о
применяется для производства глифталевых смол, пластифика.
торов (стр. 500, 529) и красителей (стр. 321).
Промышленным продуктом стали и фталевые кислоты С6Н4(СООНJ;
о-фталевая, л*-фталевая (изофталевая) и л-фталевая (тере-
фталевая).
Фталевый ангидрид получают каталитическим окислением
нафталина или о-ксилола:
О
j
I I + 4V2O2.
j
С\-\
О + 2H2O + 2СО2
I
О
о
II
II
о
Катализатором этих процессов является пятиокись ванадия
VSO5 на носителе (окись алюминия, окись кремния).
Нафталин предварительно плавят, испаряют и смешивают с
большим избытком воздуха (на 1 кг нафталина подают более 30 м3
воздуха). Паро-газовую смесь направляют в контактный аппарат
с сильно развитой поверхностью теплообменных элементов. По
конструкции он напоминает контактный аппарат для синтеза
углеводородов из СО и Н2 под атмосферным давлением.
Температура процесса 350—450 °С.
Продукты реакции по выходе из контактного аппарата
охлаждают, при этом кристаллизуется фталевый ангидрид. Его
выделяют и направляют для удаления влаги в плавильный котел, за-
260
полненный инертным газом. Окончательная очистка фталевого
ангидрида достигается двухступенчатой возгонкой.
Сублимированный продукт поступает на охлаждаемые вальцы, где
срезается ножом в виде чешуек.
Описанный выше процесс окисления о-ксилола во фталевый
ангидрид осуществляют примерно в тех же условиях, что и
окисление нафталина.
На многих предприятиях процесс окисления нафталина во
фталевый ангидрид проводят в «кипящем» слое пылевидного
катализатора.
Такой способ позволяет в значительной степени
интенсифицировать работу контактных аппаратов и облегчает отвод
реакционного тепла, выделяющегося в очень большом количестве в
процессе окисления.
Фталевый ангидрид при взаимодействии с одноатомными
алифатическими спиртами образует фталатЗ. (алкильные эфиры
о-фталевой кислоты)
О
COOR
О + 2ROH
4 ҐУ
L II
•<S ^^^COOR
II
О
применяемые в качестве пластификаторов (стр. 500).
Наиболее распространенным из них является дибутилфталат
С6Н4(СООС4НвJ.
При нагревании фталевого ангидрида с водой образуется
о-фталевая кислота:
О
V
о
Большой интерес представляет реакция неполного окисления
изомерных ксилолов. Например, при окислении я-ксилола в
жидкой фазе молекулярным кислородом в присутствии нафтенатов
кобальта или марганца, при 120 °С образуется я-толуиловая
кислота:
^ч/СНз „ч .СООН
Hfi/^' HaC'
261
П- 1 ОЛуИЛОВаЯ KHCJLUTd за і см
метиловый эфир
я-толуиловой кислоты
.СООСНз
монометиловый эфир диметиловый эфир
терефталевой кислоты терефталевой кислоты
Диметилтерефталат является важным полупродуктом в
производстве полимеров, из которых вырабатывают синтетическое
волокно—лавсан. Такие полимеры получают путем
поликонденсации диметилтерефталата с этиленгликолем (стр. 408).
ЛИТЕРАТУРА
Вольфкович С. И., Роговин 3. А., Руденко Ю. П.,
Шманенков И. В., Общая химическая технология, под ред. акад.
Вольфковича С. И., т. II, Госхимиздат, 1959.
Юкельсон И. И., Технология основного органического синтеза,
Госхимиздат, 1959.
Основы технологии нефтехимического синтеза, под ред. Динцеса А. И. и По-
толовского Л. А., Гостоптехиздат, 1960.
Д а л и н М. А., Нефтехимические синтезы, Госхимиздат, 1961.
Волков А. Е., Л а п и д у с А. С, Техника безопасности в производстве
ацетилена из природного газа, Изд. «Химия», 1964.
Глава V
ТЕХНОЛОГИЯ ТОНКОГО ОРГАНИЧЕСКОГО СИНТЕЗА
Отрасли химической промышленности, относящиеся к
тонкому органическому синтезу, характеризуются главным образом
очень широким ассортиментом продукции при относительно
небольших масштабах производства отдельных веществ.
Деление промышленного органического синтеза на основной
(см. главу IV) и тонкий—условно, так как с развитием техники
появляются новые области применения тех или иных продуктов
и благодаря этому быстро растет производство этих продуктов.
Так, анилин и фталевый ангидрид, которые ранее являлись
только полупродуктами анилинокрасочной промышленности, теперь
стали применяться в производстве полимерных материалов и
вырабатываются в количестве десятков и сотен тысяч тонн.
Таким образом, производства анилина и фталевого ангидрида уже
относятся к области основного органического синтеза.
Производства тонкого органического синтеза обычно
отличаются большим количеством технологических операций,
проводимых с участием разнообразных реагентов; получаемые вещества,
как правило, более сложны по составу, чем продукты основного
органического синтеза. Масштабы и многостадийность многих
производств тонкого органического синтеза в известной мере
ограничивают внедрение в них прогрессивных непрерывных
методов, однако в Советском Союзе в этой области в последние годы
все же достигнуты определенные успехи.
Основные виды продукции, производимые промышленностью
тонкого органического синтеза:
промежуточные продукты (полупродукты), применяемые для
•синтеза красителей и других веществ;
красители и оптически отбеливающие (осветляющие) вещества;
поверхностно-активные вещества;
препараты для сельского хозяйства—пестициды (ядохимикаты);
препараты для резиновой промышленности.
263
органическому синтезу относятся также производства
лекарственных и душистых веществ, препаратов для
кино-фотопромышленности, химически чистых реактивов и ряд других, которые
рассматриваются в специальных курсах.
/. ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ
Термин «промежуточные продукты» или «полупродукты»
возник в промышленности синтетических красителей (анилинокра-
сочная промышленность). Он относится к получаемым в этой
промышленности, преимущественно ароматическим, соединениям,
содержащим одну или несколько следующих групп и атомов: С1,
SO3H, NO2, NH2, ОН и др. Полупродукты применяются для
синтеза красителей, а также в производстве душистых,
лекарственных, взрывчатых и других веществ. В последние годы большие
количества полупродуктов используются в промышленности
полимерных материалов.
Основным сырьем для получения полупродуктов являются
ароматические углеводороды угля и нефти, главным образом
бензол, толуол, ксилолы, нафталин и антрацен.
Для синтеза полупродуктов применяются следующие
основные реакции:
Замещение водородных атомов исходных углеводородов
другими атомами или группами, например: сульфирование (замещение
группой SO8H); нитрование (замещение группой NO2);
хлорирование (замещение атомами хлора) и др.
Превращение или замена имеющихся заместителей новыми,
например замена сульфргруппы гидроксилом методом щелочного
плавления, восстановление нитросоединений до аминов и другие
реакции.
Реакции конденсации, приводящие к образованию новых
связей между углеродными атомами (в ряде случаев эти реакции
приводят к образованию гетероциклических соединений), а также
внутримолекулярные перегруппировки.
Ниже рассматриваются важнейшие из перечисленных здесь
реакций.
Реакции замещения водородных атомов в ароматических
соединениях проводятся как с незамещенными ароматическими
углеводородами*, так и с веществами, содержащими различные
заместители.
В последнем случае реакции замещения более сложны и
относятся к области тонкого органического синтеза.
• Хлсрнрованре и нитрование ароматических углеводородов рассмотрены
в главе IV (стр. 174 и ел.).
264
При проведении реакций замещения наблюдаются следующие
закономерности:
1) группы NO2) SO3H, COOH и некоторые другие (так
называемые заместители первого рода) направляют вновь вступающие
в молекулу атомы и группы преимущественно в мета-положение к
имеющимся заместителям. Реакция протекает труднее, чем с
незамещенными углеводородами;
2) группы и атомы ОН, NH2, СІ, СН3 и др. (заместители
второго рода) направляют новые заместители в пара- и орто-положения
к этим группам (часто образуются смеси изомеров). Реакция
проходит легче, чем с незамещенными соединениями (исключение
составляет хлор, затрудняющий вступление нового заместителя).
СУЛЬФИРОВАНИЕ
Введение группы SO3H чаще всего производится путем
сульфирования концентрированной серной кислотой или олеумом, реже—
серным ангидридом, бисульфатами, бисульфитами и хлорсульфо-
новой кислотой SO2(OH)C1.
Сульфирование—реакция обратимая; в присутствии воды
образующаяся сульфокислота гидролизуется, поэтому схематически
реакция сульфирования бензола изображается следующим
образом:
SO3H
f J + H2SO4 ї=ї f J) + H2O
бензолсуль-
фокислота
Скорость и направление реакции сульфирования зависят от
строения сульфируемого вещества, концентрации серной кислоты,
температуры и наличия катализаторов. Например, нафталин
сульфируется легче, чем бензол; заместители первого рода затрудняют
сульфирование, а заместители второго рода облегчают его. Для
легко сульфируемых веществ в качестве сульфирующего агента
применяют серную кислоту, для трудно сульфируемых—олеум.
При использовании серной кислоты недостаточно высокой
концентрации реакция сульфирования резко замедляется и часто
практически прекращается.
Влияние температуры на результаты сульфирования можно
наблюдать на примере нафталина, который при температуре 35—
60 СС образует главным образом а-сульфокислоту, а при 160 *С—
преимущественно ?-сульфокислоту:
SO3H
\ +H2SO4 ^\/% +H2SO4
35-60 "С 1^ IX J 160 °С
265
Большой интерес представляет открытое выдающимся русским
ученым М. А. Ильинским влияние солей ртути на сульфирование
антрахинона олеумом. В то время как в отсутствие катализатора
образуется ?-сульфокислота антрахинона, в присутствии даже
малых количеств солей ртути получают а-сульфокислоту:
О
О О
Р-»нтрахинонсульфо- антрахшюи а-антрахннонсульфо-
кислота кислота
В ряде случаев для сульфирования применяют смесь серной
кислоты или олеума с сульфатом натрия, что позволяет получать
сульфокислоти с лучшими выходами, например:
SO3H SO3H
SO3H
л-бензолдвсульфо-
кнслота
Сульфирование чаще всего проводится в чугунных или стальных
котлах, так называемых сульфураторах, оборудованных
мощными мешалками для перемешивания густых масс. Реакционная
смесь нагревается паром, поступающим в рубашку аппарата,
иногда используют электрообогрев. Реакция сульфирования
ведется обычно при повышенной температуре и большом избытке
серной кислоты или олеума. Это объясняется тем, что
образующаяся при сульфировании вода снижает концентрацию серной
кислоты, скорость реакции при этом резко снижается.
При большом избытке серной кислоты понижение ее
концентрации в процессе сульфирования незначительно.
В 1928—1931 гг. в СССР под руководством проф. Р. К. Эйхма-
на было разработано и освоено сульфирование бензола «в парах».
Образующаяся при сульфировании вода вместе с парами бензола
непрерывно отгоняется из реакционной массы. Это позволяет
существенно снизить расход серной кислоты.
На рис. 91 схематически показана установка для получения
бензолсульфокислоты. В испарителе 1 бензол, проходя по трубам,
обогреваемым снаружи паром, перегревается до 150—160 °С.
Далее перегретые пары бензола поступают в сульфуратор
2—чугунный котел с рубашкой (рис. 92), где барботируют через серную
кислоту при температуре реакции 150—160 °С. Образующаяся
при сульфировании вода испаряется и вместе с парами непрореа-
266
гировавшего бензола конденсируется в холодильнике 3. В
отстойнике 4 конденсат расслаивается и бензол отделяется от воды. После
того как в реакцию вступит около 95% взятой серной кислоты,
подачу бензола прекращают. Сульфомассу, которая содержит
около 4% бензола, передают в котел 5, откуда она перекачивается в
колонну б, обогреваемую паром. В колонне, работающей при
разрежении, отгоняется почти все количество бензола. Большая часть
бензола конденсируется в холодильнике 7, остальной бензол
поглощается в адсорбере 8 активированным углем.
Рис. 91. Схема сульфирования бензола «в парах»:
/—испаритель бензола; 2—сульфуратор; 3. 7—холодильники; 4—отстойник; 5, 9—котлы для
сульфошссы; б—колонна для отгонки бензола; 8—адсорбер паров бензола; 10—аппарат для
непрерывной нейтрализации сульфомассы.
После отгонки бензола сульфомассу передавливают в котел 9,
из которого ее одновременно с раствором сульфита натрия
непрерывно подают в аппарат 10. Образующийся раствор натриевой
соли бензолсульфокислоты поступает далее на производство
фенола. Выделяющийся при нейтрализации сернистый ангидрид
Na2SO3
— SO3Na+SO2+H2O
также используется в производстве фенола.
Сульфирование аминов в ряде случаев проводят методом
«запекания». Этот метод заключается в том, что кислую сернокислую
соль амина, образующуюся при взаимодействии эквимолекуляр-
267
ных количеств амина и серной кислоты, нагревают без
растворителя до температуры 200 °С («запекание»).
Схема реакции:
NHa
I
0^\
NH2-H2SO4
NH,
запекание
SO3H
сульфаниловая
кислота
В последние годы вместо «запекания» сернокислые соли
аминов нагревают в высококипящих органических растворителях
(стр. 129), например в техническом о-ди-
хлорбензоле. В этих условиях при 175 °С
сернокислая соль амина
перегруппировывается в соответствующую
сульфокислоту, например:
NH,-HjSO4
NH2
I
SO3H
нафтноновая
кислота
Рис. 92. Сульфуратор
для сульфирования
бензола «в парах».
В табл. 15 приведены некоторые
ароматические сульфокислоты и условия их
получения.
Сульфокислоты применяются в
больших количествах для получения
фенолов и нафтолов методом щелочного
плавления (стр. 270). Ряд сульфокислот
используется в качестве моющих и других поверхностно-активных
веществ (стр. 331). Сульфокислоты, содержащие группы NH2,
ОН и др., являются промежуточными продуктами для синтеза
красителей (стр. 291 и ел.), так как присутствие сульфогруппы
придает красителям ряд ценных свойств: хорошую растворимость
в воде, способность окрашивать белковые волокна, улучшение
цвета, прочности окраски и т. д.
268
Таблица 15
Ароматические сульфокислоти
Сульфокислота
Строение
Исходное
сырье
Сульфирующий
Температура
сульфирования
°С
Бензолмоно-
сульфокислота
л-Бензолди-
сульфокислота
Сульфаниловая
кислота
1 -Нафталин-
сульфокислота
2-Нафталин-
сульфокислота
1,3,6-Нафталин-
трисульфо-
кислота
1-Антрахинон-
сульфокислота
2-Антрахинон-
сульфокислота
SO,H
SO3H
I
SO3H
NH2
0
о
^
SO.H
о
Бензол
Бензолмоно-
сульфо-
кислота
Анилин
Нафталин
То же
2-Нафталин-
сульфо-
кислота
Антрахинон
То же
Концентрированная
HaSO4
Олеум
65%-ный+
+NaaSO4
Серная
кислота
96%-ная
Концентрированная
HaSO4
Серная
кислота
96%-ная
Олеум
20%-ный
Олеум
20о/0-иый
(HgSO4)
Олеум
30%-ный
150—160
(сульфирование «в парах»)
200
(«запекание»)
ЗАМЕНА СУЛЬФОГРУППЫ НА ГИДРОКСИЛЬНУЮ
МЕТОДОМ ЩЕЛОЧНОГО ПЛАВЛЕНИЯ
Ароматические сульфокислоты превращаются в оксисоеди-
нения—фенолы, нафтолы и др.—при нагревании солей
сульфокислот с щелочами, чаще всего с едким натром:
ArSO3Na + 2NaOH > ArOH + Na,SO3 + HaO
Фенолы широко используются в производстве пластических
масс (стр. 550), фенолы и нафтолы—в синтезе красителей
(стр. 294), лекарственных препаратов и др.
Щелочное плавление сульфокислот осуществляют двумя
способами:
1) твердый едкий натр нагревают с 5—20% воды до 275—325 °С
в открытом сосуде и к образовавшемуся жидкому плаву
постепенно добавляют натриевую соль сульфокислоты в виде порошка или
водного раствора. Затем реакционную массу перемешивают в
течение часа при 300—350 °С до окончания реакции. Образующаяся
вода в этих условиях испаряется;
2) щелочное плавление под давлением ведется в закрытом
аппарате—автоклаве—в водном растворе щелочи при температуре
не выше 250 °С. Давление в автоклаве может достигнуть 30 am и
более.
Таким образом, при использовании метода щелочного
плавления под давлением создаются более мягкие условия (ниже
температура, менее концентрированная щелочь). Этот метод часто
применяется для замены одной из нескольких сульфогрупп гидр-
оксилом, например:
H2N SO,Na H2N ОН
II І
•SO3Na
Na-соль Аш-кислоты
Необходимо отметить, что в нафталиновом ядре сульфогруп-
пы, находящиеся в а-положении, замещаются легче, чем в ?-поло-
жении. В антрахиноновом ряду сульфогруппа замещается на
гидроксил при обработке сульфокислоты гидроокисью кальция
под давлением (М. А. Ильинский). При нагревании же с едким
натром и NaNO3 образуется 1,2-диоксиантрахинон (ализарин,
стр. 311):
NaO 0 0 О
SO3Na
Са(ОНJ
1,2-диоксиантрахинон
(динатриевая соль)
О
Na-соль антрахинон-
2-сульфокислоты
2-оксиантрахннон
270
Важнейшими ароматическими оксисоединениями являются
фенол, резорцин и ?-нафтол.
Схема производственного получения фенола из бензолсульфо-
кислоты показана на рис. 93.
NaOH НатриеВая соль
\ ^ \бендалсульфакисіютьі
. Кубовый
Раствор J остаток
Na2SO3
Рис. 93. Схема получения фенола из бензолсульфокислоты:
/—бак для раствора едкого натра: 2—плавильный котел; 3—печь: 4—мерник
раствора натриевой соли бензолсульфокиелоты; 5—котел; б—центрифуга;
7—аппарат для выделения фенола; В—отстойник; в—колонка;
/О—дефлегматор.
Из бака / в плавильный котел 2, оборудованный мешалкой,
наливают 70—75%-ный раствор едкого натра и нагревают его до
300 °С топочными газами, получаемыми в печи 3, при этом в
результате испарения части воды концентрация щелочи возрастает
до 80—85%.
При температуре около 325 °С к раствору щелочи из
мерника 4 постепенно приливают раствор натриевой соли
бензолсульфокислоты (стр. 267).
По окончании реакции плав сливают в котел 5, заполненный
водой. Выпавший сульфит натрия отделяют на центрифуге б и
промывают горячей водой. Промывные воды могут
использоваться при обработке плава в котле 5. Сульфит натрия
применяется в производстве химических волокон и
целлюлозно-бумажной промышленности.
271
иаствор, содержащий фенолят натрия и часть сульфита натрия,
затем обрабатывают сернистым газом, при этом фенолят
превращается в фенол:
О№ ОН
I \
2 f jj + SO2 + Н2О > 2 Ґ Jj + Na2SO3
Эту реакцию проводят непрерывным методом в аппарате 7.
Образующийся фенол отделяют от раствора сульфита натрия в
отстойнике 8, а затем очищают путем вакуум-перегонки в
колоннах 9.
На стр. 251 и ел. описаны другие методы получения фенола.
Резорцин получают при нагревании динатриевой соли .м-бен-
золдисульфокислоти с твердым едким натром при 300—340 "С".
SO3Na О Na
J
NaOH
Резорцин находит широкое применение для изготовления
лекарственных веществ (л-аминосалициловая кислота—ПАСК—
средство для лечения туберкулеза), пластических масс,
красителей.
?-Нафтол образуется при взаимодействии натриевой соли
?-нафталинсульфокислоты с 87—88%-ным раствором едкого натра
при 300—325 °С:
?-нафтол
?-Нафтол применяется главным образом для производства
неозона Д—препарата резиновой промышленности (противоста-
рителя, стр. 365), а также различных полупродуктов
нафталинового ряда и синтетических красителей.
ВОССТАНОВЛЕНИЕ НИТРОСОЕДИНЕНИЙ
Восстановлением нитросоединений получают амины—важные
промежуточные продукты промышленности органического синтеза.
Ниже описаны наиболее распространенные способы
восстановления нитросоединений.
Восстановление чугунными стружками в присутствии
электролитов—чаще всего растворов хлористого железа или
хлористого аммония. Это один из старых способов, в настоящее время
272
по этому способу получают о-толуидин и другие ароматические
амины:
4 f |[
о-нитротолуол
+ 9Fe + 4HaO
о-толуиднн
+ 3Fe3O4
На рис. 94 показана схема процесса восстановления о-нитро-
толуола чугунными стружками. В редуктор / наливают воду,
о- Толуидин
Рис. 94. Схема получения о-толуидина из о-нитротолуола:
г—редуктор: 2—мерник соляной кислоты; л—мерник нитротолуола, 4—обратный холодильник:
5—отстойник; б—фильтр; 7—куб; 8—конденсатор.
вносят чугунную стружку и небольшое количество соляной
кислоты. Реакционную смесь нагревают до кипения при
перемешивании. В этих условиях железо взаимодействует с соляной
кислотой (травление) с образованием FeCl2. Затем к полученной смеси
постепенно из мерника приливают о-нитротолуол, периодически
добавляя чугунную стружку. Процесс восстановления ведут при
температуре кипения реакционной массы, поддерживаемой за
счет тепла реакции. Пары воды конденсируются в обратном
холодильнике 4. После окончания загрузки о-нитротолуола
реакционную массу кипятят до окончания реакции восстановления
(около 1 ч), соли железа осаждают гашеной известью и о-толуидин
отгоняют с водяным паром. о-Толуидин и вода, охлаждаясь в
холодильнике 4, поступают в отстойник 5. После отстаивания
о-толуидин пропускают через фильтр 6 и перегоняют в вакууме из
куба 7.
Разбавленные водные растворы о-толуидина используют
вместо воды в процессе восстановления,
18—805 273
Основным недостатком метода восстановления железом
является большое количество отходов—окислов железа (прокаливанием
они могут быть превращены в железоокисные пигменты,
применяемые в лакокрасочной промышленности).
Каталитическое восстановление нитросоединений водородом—
новейший, более экономичный, непрерывный способ получения
ароматических аминов, применяемый, например, при
производстве анилина. Нитробензол по этому способу восстанавливают
в паровой фазе при температуре от 170 до 350 °С в присутствии
медного катализатора:
NO2 NH2
I I
О катализатор ^\
+ ЗН, -Г j+2H2O
Восстановление можно вести в жидкой фазе под давлением до
100 am в присутствии никелевого катализатора.
Восстановление нитросоединений сернистыми щелочами. Этот
распространенный способ получения аминов применяется чаще
всего для частичного восстановления нитросоединений, например,
по реакции:
NO2 NH2
I I
4 I ll + 6NaaS + 7Ha° * 4 I 1 + 3Na2S2O3 + 6NaOH
jk-дин итробензол л-нитроанилин
Кроме сернистого натрия в этой реакции могут применяться
Na2S2, Na2S3, NaSH и др.
Один из недостатков способа восстановления сернистыми
щелочами—образование побочного продукта тиосульфата натрия,
загрязняющего сточные воды.
Восстановление нитросоединений металлами в щелочной среде.
Способ нашел наибольшее применение для получения
ароматических диаминов, в частности бензидина. В качестве
восстановителя применяют цинк. Процесс протекает в несколько стадий:
NHOH
феинлгидроксил-
амнн
274
азобензол
О
азоксибензол*
гидразобензол
Гидразобензол при нагревании с соляной (или серной)
кислотой перегруппировывается в диамин—бензидин:
бензидин
Ароматические амины получают не только восстановлением
нитросоединений, но и другими методами. Большое
промышленное значение имеет реакция замещения хлора аминогруппой:
—С1 + 2NH3 * /~\—NH2 + NH4C1
Реакцию проводят при 200—210 °С и 70 am.
Амины находят широкое применение для получения
полимерных материалов (в частности, полиамидных волокон),
препаратов для резиновой промышленности, сельского хозяйства, кино-
фотопромышленности, взрывчатых, лекарственных веществ,
красителей, текстильных вспомогательных веществ, в качестве
компонентов ракетного топлива и др. В табл. 16 приведены
наиболее важные ароматические амины, кроме о-толуидина, анилина и
бензидина, описанных выше.
Ароматические амины (кроме содержащих сульфогруппы)
являются высокотоксичными веществами. Острое отравление
ароматическими аминами (анилин, о-толуидин и др.) приводит к
нарушению деятельности нервной и сердечно-сосудистой системы.
Кроме того, попадание в организм некоторых ароматических
аминов, например ?-нафтиламина, 3,3'-дихлорбензидина,
бензидина, служит причиной возникновения злокачественных
опухолей.
В Советском Союзе прекращено производство токсичных
?-нафтиламина и 3,3'-дихлорбензидиыа. Производство бензидина
осуществляется непрерывным методом и полностью
автоматизировано.
В рабочих помещениях концентрация аминов в воздухе должна
быть не более Ьмгім'. Чтобы избежать попадания аминов на одежду
и тело работающих, транспортирование и загрузка этих
веществ механизированы.
* Стрелка обозначает координационную связь кислорода с азотом.
18* 275
Таблица 16
Важнейшие ароматические амины
Амины
Строение
Исходное сырье
Восстановители
СН,
СНЭ
NH,
VV*T
NH2
OCH3
—NHa
HO3S NH2
CH,
NO2
СНЯ
NO2
NO,
OCH3
..NO,
HO3S NO2
HO3S
NO2
¦SO3H
Водород
(каталитический
процесс)
Сернистые
щелочи
Цинк
в щелочной
среде
Чугунные
стружки
АЦИЛИРОВАНИЕ АМИНОСОЕДИНЕНИЙ
Ацилированием называют замещение в органических соедине
ниях атомов водорода (или металлов) на ацилы—кислотные
остатки.
Наиболее важные ацильные остатки:
Ацилы
Формил
Ацетил
Бензоил
Карбонил
Сульфонил
.НСО—
. СН3СО-
. С6Н5-СО-
.—СО—
. -SO3H
Бензосульфонил CeH5—SO2—
Исходная кислота
Муравьиная
Уксусная
Бензойная
Угольная
Серная
Бензолсульфокислота
Большое практическое значение приобрело ацилирование
ароматических аминов, т. е. замена водородного атома аминогруппы
на ацильную группу. Реже производится ацилирование оксисоеди-
нений—в этом случае ацил замещает водород оксигруппы.
Ацилирование проводится кислотами, их ангидридами и хлор-
ангидридами; наиболее энергично вступают в реакцию хлорангид-
риды, всего слабее—кислоты. Анилин ацилируют муравьиной
кислотой при нагревании:
NH2 HNCOH
O
О
4- HCOOH
форманилид
Аш-кислоту ацетилируют уксусным ангидридом:
НО NHa HO NHCOCHa
4- (СН3СОJО
Н(
N-ацетил-Аш-кислота
Карбонильную группу СО вводят в органическое соединение
при обработке его хлорангидридом угольной кислоты (фосген):
он
f
он он
<алая> кислота
Ацильная группа придает органическим соединениям новые
свойства: особую яркость, прочность и хорошую
растворимость—красителям, специальные качества—препаратам для
резиновой промышленности, лекарственным,
текстильно-вспомогательным веществам и т. д.
Кроме того, ацилирование используется в некоторых синтезах
для «временной защиты» высоко реакционноспособных групп
NH2, ОН, SH путем превращения содержащих эти группы веществ
в менее активные ацильные производные. После проведения
стадий синтеза, на которых необходима такая защита, ацильные
производные гидролизуют, снова превращая их в соответствующие
амино-, окси- и другие соединения. Так, при получении п-нитро-
277
анилина нитруют не анилин (который при нитровании окисляется
и дает побочные продукты), а форманилид:
HNCOH HNCOH
HNCOH
HNCOH
OHNOa
H2SO4 l^
л-нитроформанилид
Полученный л-нитроформанилид затем подвергают гидролизу
нагреванием с разбавленной серной кислотой:
NHCOH NH,
NO2
NOa
л-нитроаиилин
На рис. 95 показана схема получения л-нитроанилина. В
алюминиевый котел /, снабженный дефлегматором 2 и алюминиевым
Отгон
H2SO4
ю
Рис. 95. Схема получения л-нитроанилина:
/—аппарат для ацилировання; 2—дефлегматор; 3—холодильник; 4—мерник анилина; 5—мерник
муравьиной кислоты; 5—мерник форманилнда; 7—интратор; S, 13— меринки серной кислоты:
9~мерник азотной кислоты; 10—чан; //, /4—фильтры; 12—аппарат для гидролиза.
278
змеевиковым холодильником 3, наливают из мерника 4 анилин,
а из мерника 5 муравьиную кислоту и кипятят около 1 ч. После
этого из реакционной смеси отгоняют сначала воду и непрореаги-
ровавшую муравьиную кислоту, а затем—остаточный анилин.
Расплавленный форманилид из котла / через мерник 6 поступает
в нитратор 7, в который из мерника 8 предварительно наливают
100%-ную серную кислоту. Полученный раствор охлаждают
рассолом, циркулирующим через рубашку, и постепенно при
охлаждении (ниже О °С) из мерника 9 приливают азотную кислоту.
По окончании нитрования реакционную массу постепенно
передавливают в чан 10, заполненный холодной водой. Выпадающий
при этом осадок л-нитроформанилида отделяют на фильтре // и
промывают водой. В котле 12 л-нитроформанилид гидролизуют
разбавленным водным раствором серной кислоты, прибавляемым
из мерника 13. Процесс ведут при нагревании.
Когда гидролиз закончится, реакционную массу охлаждают,
полученный /г-нитроанилин отфильтровывают на фильтре 14
и промывают водой.
В настоящее время я-нитроапилин чаще получают при
взаимодействии /г-нитрохлорбензола с аммиаком. /г-Нитроанилин
используют главным образом в синтезе красителей (стр. 292).
АЛКИЛИРОВАНИЕ И АРИЛИРОВАНИЕ АРОМАТИЧЕСКИХ
АМИНО- И ОКСИСОЕДИНЕНИЙ
В главе IV было рассмотрено алкилирование углеводородов.
Эта реакция часто применяется также и для замещения алкилами
водородных атомов в амино- или оксигруппах. Такое же
замещение ароматическими остатками—арилами называют арилирова-
нием. В результате алкилирования и арилирования ароматических
амино- и оксисоединений получают ряд важных веществ,
используемых в качестве лекарственных препаратов, вспомогательных
веществ для текстильной промышленности, полупродуктов для
синтеза красителей, гербицидов (препаратов для борьбы с
сорняками, стр. 338) и др.
Алкилирование аминов в ряде случаев осуществляется при
нагревании их в автоклаве со спиртами под давлением в
присутствии катализаторов кислого характера. Например, анилин эти-
лируют большим избытком этилового спирта в присутствии
соляной кислоты при 200 °С и 30 am. При этом образуется смесь моно-
этиланилина, диэтиланилина и соли четвертичного основания:
HNCjH6 I^QHsh N(CaH5KCr
моноэтиланилнн диэтнланилин соль
четвертичного основания
2 79
Полученную смесь разделяют фракционной перегонкой или
другими способами.
Диметиланилин получают при нагревании смеси анилина и
метанола до 205—215 °С в присутствии серной кислоты:
NHS N(CH3J
H2SO4
диметиланилин
Соль четвертичного аммониевого основания, полученную в
качестве побочного продукта, разлагают при нагревании с едким
натром:
N(CH3K-HSO4 N(CH3J
NaOH ^\
* I \\ + СН3ОН + Na2SO4
В последние годы все большее значение приобретают методы
непрерывного каталитического парофазного алкилирования
аминов.
Процесс заключается в пропускании смеси паров амина и
спирта над катализатором (например, активная окись алюминия)
при 300—400 °С.
В некоторых случаях вместо спирта при каталитическом
алкилировании применяют эфир, например:
NH2 N(CH3J
+ C)*» М) + Н2О
Для введения в аминогруппу бензильного остатка \^Ъ—СНа
используют хлористый бензил:
/QH
HNC2HS CH2C1 N<
I I ГСН2
о +о-<
ыоноэтил- хлористый N-зтилбензиланилин
анилин бензил
280
Оксиэтильную группу —СН2СНаОН вводят с помощью окиси
этилена (стр. 242) по реакции:
СН
С.Н,
N—СНаСН2ОН
о
М-этил-М-оксиэтиланилин
Алкилирование ароматических оксисоединений протекает
труднее, чем алкилирование аминов. В этом случае применяют более
активные реагенты—хлористые алкилы (СН3С1, СаН5С1), зфиры
серной кислоты—диметилсульфат (CH3)aSO4 и др.
Для замещения водородного атома оксигрупп на карбокси-
метильную группу —СН2СООН используют хлоруксусную
кислоту:
ОН OCHjCOOH
I
+NaOH
феноксиуксусная
кислота
Арилирование аминосоединений (или араминирование) произ^
водится путем взаимодействия между собой одинаковых или
различных ароматических аминов при повышенной температуре в.
присутствии катализаторов (например, треххлористого фосфора.
РС13).
Таким способом получают дифениламин:
NH,
О
В последние годы дифениламин получают из анилина чаще
всего непрерывным методом в трубчатых контактных аппаратах
в присутствии катализаторов А12О3 или TiO2.
Дифениламин применяют в производстве синтетических
красителей, препаратов для резиновой промышленности и некоторых
других продуктов.
Вторичные ароматические амины (диариламины) образуются
также при взаимодействии ароматических аминов с оксисоедине-
ниями.
Так, неозон Д получается из ?-нафтола, анилина и
небольшого количества хлоргидрата анилина.
Этот процесс более подробно описан на стр. 365.
281
PF лізини і<г>нЛЕНСДЦИм
Реакциями конденсации называют реакции уплотнения,
приводящие к образованию новых углерод-углеродных связей или к
образованию гетероциклов. Реакции конденсации часто
сопровождаются отщеплением от реагентов некоторых атомов или групп,
которые образуют молекулы водорода, воды, НС1 и других,
большей частью несложных, веществ.
Реакции конденсации широко используются в производстве
полупродуктов, красителей и других синтетических органических
веществ. Реакции описанного типа используются также при
синтезе ряда важных полимеров методом поликонденсации (стр. 385).
Рассмотрим некоторые реакции конденсации, используемые в
промышленности органического синтеза. Так, получение
инсектицида ДДТ (стр. 351) осуществляют путем конденсации хлораля
и хлорбензола, сопровождающейся отщеплением воды:
СС13—СНО + 2 <f\— СІ . Cl—<f^—CH—/Л— СІ + Н2О
Важный промежуточный продукт—антрахинон получают не
только окислением антрацена воздухом в присутствии
катализатора
О
О
антрахннон
но и конденсацией фталевого ангидрида с бензолом:
О
о-бенэоилбензойная
кислота
На рис. 96 изображена схема получения антрахинона. В котел /
из мерника 2 наливают безводный бензол, вносят хлористый
алюминий, нагревают и прибавляют из котла 3 бензольный
раствор фталевого ангидрида. При температуре сколо 70 СС
образуется комплекс о-бензоилбензойной кислоты с хлористым алюми-
282
ниєм, который под давлением азота передают в котел 4,
наполненный водой для разложения комплекса. Сюда же добавляют
серную кислоту (для полноты разложения) и затем отгоняют бензол
с водяным паром. К остатку после удаления бензола приливают
холодную воду, в результате о-бензоилбензойная кислота выпадает
в осадок. Ее отфильтровывают на фильтре 5 и сушат в сушилке 6.
Бензол
ФталеВый
Рис. 96. Схема получения антрахинона методом конденсации:
/—котел для получения о-бенаоилбемзойной кислоты; 2—мерннк бензола;
3—котел для растворения фталевого ангидрида; 4—котел для осаждения
•-бензоилбензоймой кислоты; 5. в—фильтры; б—сушилка; 7—котел для
получения антрахинона.
В котел 7 наливают 100%-ную серную кислоту и прибавляют
сухую о-бензоилбензойную кислоту. Образующийся антрахинон
осаждают, добавляя к реакционной массе холодную воду. Антра-
хинон отфильтровывают на фильтре 8 и промывают горячей водой.
РЕАКЦИИ ПЕРЕГРУППИРОВКИ
Реакции перегруппировки, имеющие большое значение в
синтезах полупродуктов, приводят к изменению положения
отдельных атомов или групп в молекуле органических соединений с
сохранением, как правило, их элементарного состава.
281
КрНЧТ-ТТТЫНППЯ а ПРПРГт'ППНПППКЯ ПТЛГГа т^пі*ґ*ЧГі'ГПґ\їїп р пнтигппп
бензидина из гидразобензола (стр. 275).
Здесь будут приведены синтезы некоторых оксикарбоновых
кислот, в ходе которых, по-видимому, протекают
внутримолекулярные перегруппировки.
Салициловая кислота получается при взаимодействии СО2
и фенолята натрия. Предполагается, что вначале образуется
Na-соль кислого фенилового эфира угольной кислоты, которая
Салициловая]
-* - >
кислота
Рис. 97. Схема получения салициловой кислоты:
/—котел для получения фенола; 2—мерник фенола; 3—автоклав;
4—холодильник; 5—приемник фенола; 6—чан для осаждения салициловой кисло-
ты; 7—центрифуга; 8— сушилка.
под действием избытка двуокиси углерода, при нагревании и
повышенном давлении перегруппировывается в натриевую соль
салициловой кислоты*:
ONa
СО2-
OCOONa
С
Na-соль кислого
фекнлового эфира
угольной кислоты
.COONa
Na-соль
салициловой кислоты
* Существуют и другие представления о механизме образования
салициловой кислоты.
2S4
Аналогично из ?-нафтолята натрия получают 2-окси-З-нафтой-
ную кислоту—полупродукт в синтезе красителей:
ONa
2-окси-З-нафтойная
кислота
На рис. 97 приведена схема получения салициловой кислоты.
В котел 1 наливают водный раствор едкого натра и приливают
из мерника 2 предварительно расплавленный фенол. Смесь
нагревают до полного
растворения фенола и
передают в автоклав 3,
оборудованный мощной
мешалкой и
специальными стальными
ножами, прикрепленными к
стенкам автоклава и
входящими в
промежуток между лопастями
мешалки. Раствор
упаривают досуха при
размешивании, при
этом масса
измельчается между лопастями
мешалки и ножами. После
этого в автоклав при
температуре не выше
200 °С, давлении 8 am
и перемешивании
пропускают сухую
двуокись углерода. В этих
условиях в автоклаве
Рис. 98. Автоклав для получения
салициловой кислоты:
1—лопасти мешалкн; 2—змеевик для обогрева;
3—теплоизоляция автоклава.
одновременно
протекают реакция
присоединения СО2 и
перегруппировка. Не вошедший
в реакцию фенол
отгоняют в вакууме через холодильник 4 в приемник 5, оставшийся
в автоклаве салицилат натрия растворяют в горячей воде.
Полученный раствор передают в чан 6, где салициловая кислота
осаждается при добавлении разбавленной серной кислоты. На
центрифуге 7 салициловую кислоту отделяют от маточного раствора,
промывают холодной водой и высушивают в сушилке 8.
На рис. 98 схематично показан автоклав для получения
салициловой кислоты.
285
о трунл nrirua ruf-i-rp-rifupruuv isn л гит& гтрр
Красителями называются органические вещества,
обладающие цветом и способностью окрашивать различные материалы
и изделия—текстильные волокна, кожу, меха, пластические
массы, резину и т. д.
Нерастворимые в воде красители называют пигментами.
Смеси красителей или пигментов с некоторыми другими веществами,
например связующими (олифа, клей, синтетические смолы),
растворителями, наполнителями (белила и другие инертные вещества),
называют красками.
До 1857 г. применялись только естественные
красители/Например, синее индиго получали из листьев растений индигоноска
красильная и вайда красильная, ализарин, применявшийся
преимущественно для окраски в красный цвет,—из корней марены.
Красный кармин получали из некоторых высушенных насекомых,
знаменитый античный пурпур—из моллюсков Средиземного моря
и т. п. Применялось всего лишь несколько десятков природных
красителей, дорогих, большей частью непрочных и недостаточно
ярких.
Открытию синтетических красителей предшествовало бурное
развитие текстильной промышленности и большие
успехи^органической химии. Из наиболее крупных 'достижений того
времени, сыгравших значительную роль в развитии химии
синтетических красителей, следует отметить получение Э. Митчерли-
хом в 1834 г. нитробензола и, особенно, открытие Н. Н. Зини-
ным в 1842 г. способа получения анилина и других
ароматических аминов.
В 1855 г. польский химик Я- Натансон на основе анилина и
дихлорэтана получил первый синтетический краситель—красного
цвета, позднее названный парарозанилином. В 1856 г.
английский химик В. Перкин в результате окисления смеси анилина
и толуидинов синтезировал красновато-фиолетовый краситель
мовеин. Уже в следующем году Перкин сумел организовать
промышленное производство мовеина. Вслед за этим был открыт ряд
других очень ярких, но мало прочных синтетических красителей
различных цветов.
Первоначально исследования в области синтеза красителей
носили в основном случайный характер. Гениальная теория
строения органических соединений А. М. Бутлерова, а также
выдающиеся работы Ф. Кекуле, объяснившие строение бензола и других
органических соединений, создали предпосылки для синтеза
новых красителей на научной основе. В 1869 г. К. Гребе и К. Либер-
^ан установили состав одного из прочных натуральных
красителей—ализарина и получили его синтетическим путем; в 1883 г.
А. Байеру удалось установить строение и синтезировать второй
прочный естественный краситель—синее индиго.
286
К концу XIX в. синтетические красители, более дешевые и
яркие, практически полностью вытеснили природные.
Дальнейшие исследования в области синтетических красителей
были направлены на расширение их ассортимента, удешевление и
улучшение прочности окрасок.
В настоящее время мировой ассортимент синтетических
красителей, имеющих промышленное значение, насчитывает
несколько тысяч наименований и продолжает пополняться новыми
яркими, прочными и удобными в применении красителями.
Интенсивно проводятся дальнейшие работы по созданию
специальных красителей для химических волокон и других
полимерных материалов. Мировое производство синтетических
красителей в настоящее время превышает 400 тыс. т в год, причем
Советский Союз и США вырабатывают по 80—90 тыс. т, далее
следуют ФРГ, Англия и другие страны.
КРАТКИЕ СВЕДЕНИЯ О ЦВЕТЕ И СТРОЕНИИ ОРГАНИЧЕСКИХ
СОЕДИНЕНИЙ
Предметы, воспринимаемые нами как окрашенные, отражают (точнее,
рассеивают) или излучают световые лучи определенной длины волн в пределах
от 390—до 800 нм*. Наряду с этими видимыми лучами, солнце и другие
источники света испускают невидимые лучи—ультрафиолетовые (длина волн от 2
до 390 нм) и инфракрасные (длина волн от 800 до 5-Ю5 нм).
Белы» солнечный свет при прохождении через стеклянную призму может
быть разложен на составляющие его части, отличающиеся длиной волны и
цветом (так называемый спектр). Обычно спектр условно разделяют на
следующие главные части:
Цвет
Фиолетовый .
Синий . . .
Голубой . .
Сине-зеленый
Зеленый . .
Длина
волны
1-М
. 390—450
. 450—480
. 480—490
. 490—510
. 510—550
Цвет
Желто-зеленый
Желтый . . .
Оранжевый . .
Оранжево-красный ....
Красный . . .
Длина
волны
нм
550—575
575—585
585—605
605—625
625—800
Изменение цветов спектра от фиолетового к красному происходит
непрерывно, через промежуточные цвета и оттенки (глаз различает в спектре свыше
100 цветов и оттенков). Так, среди желтых цветов спектра глаз замечает
зеленовато-желтые (или лимонные) и оранжевато-желтые (золотисто-желтые)
оттенки и т. д.
Белый цвет можно вновь получить смешением всех лучей спектра, а также
смешением лучей двух определенных цветов. Два цвета, дающие при смешении
их лучей белый цвет, называются дополнительными. К дополнительным
относятся такие пары цветов, как красный и сине-зеленый, оранжевый и голубой.
Цвет тел зависит главным образом от поглощения ими света. Белые тела
рассеивают почти весь падающий на них свет, не поглощая его; черные поглощают
• 1 нанометр (или 1 миллимикрон)=10~9л=10 ' мм. 1 ммк.= \ юи=10А.
287
степени все лучи спектра.
Цветные тела поглощают цвет избирательно, т. е. один лучи сильнее, а
другие слабее. Тело, сильно поглощающее лучи одного какого-либо цвета,
окрашено в дополнительный цвет; например, при поглощении сине-зеленых
лучей цвет тела красный, при поглощении оранжевых лучей цвет голубой и т. д.
Если тело сильно поглощает лучи нескольких цветов, например все, кроме
красных и фиолетовых, оно будет иметь цвет последних, т. е. красно-фиолетовый.
В химии красителей часто употребляются термины углубление и повышение
цвета. Под этим понимают не усиление или потемнение оттенка, а изменение
цвета; например, при углублении—переход от желто-зеленого через желтый,
красный, синий к зеленому или при повышении—наоборот, от зеленого к
желто-зеленому:
Желто -зеленый
желтый
оранжевый
красный
фнолетовы й
синий
голубой
сине-зеленый
зеленый
Углубление и повышение цвета связано обычно с введением в молекулу
красителя определенных атомов или групп атомов.
Щ Цвет и строение органических соединений. Зависимость между
строением и цветом органических веществ давно привлекала
внимание ученых. В конце XIX в. наибольшее влияние имела теория
О. Витта A876 г.), согласно которой неокрашенные органические
соединения становятся окрашенными при введении в их молекулы
особых групп атомов—хромофоров (от греч. слов: хромое—цвет,
форео—ношу). Например, дифенил не имеет окраски, тогда как
азобензол, содержащий азогруппу —N = N—, обладает желтым
цветом:
дифенил
(бесцветный)
азобензол
(светло-желтый)
Кроме азогруппы к хромофорам относятся нитрогруппа NO2.
нитрозогруппа N0, карбонильная группа СО и некоторые другие.
Окрашенные вещества, содержащие хромофоры, были
названы хромогенами. Кроме хромофоров, Витт отметил значение
других групп, усиливающих и углубляющих (см. выше) цвет,
например гидроксильной ОН и аминогруппы NH2. Эти группы
названы ауксохромами (ауксо—увеличиваю). Влияние ауксохро-
мов иллюстрировалось на примере аминоазобензола
который окрашен в более глубокий и интенсивный желтый цвет,
чем азобензол.
288
Последующие исследования показали недостаточность теории
Витта, было получено большое число органических соединений,
не содержащих хромофоров, но обладающих цветом, например
нафтаден
(оранжевый цвет)
В настоящее время теории цветности органических
соединений основываются на новейших данных физики и органической,
химии. Согласно этим данным, в поглощении ультрафиолетовых
и видимых лучей света участвуют главным образом электроны
молекул. При этом энергия фотонов (квантов света) затрачивается
на возбуждение электронов, т. е. перевод их на более высокий,
энергетический уровень.
Энергия фотона выражается формулой:
с
? = *• —
где h—постоянная Планка F,62 10~27 эрг-сек*);
с—скорость света C-Ю10 смісек);
X—длина волны света, см.
Из этого уравнения следует: чем больше длина волны света,
тем меньше его энергия. Поэтому энергия ультрафиолетовых
лучей больше энергии видимых лучей.
Если обозначить Ео—энергию молекулы до поглощения ею
света, а Ь1—энергию возбужденной светом молекулы, то
Е1 — Еа=Е =h--j-
Отсюда видно, что возбуждению каждой молекулы
соответствует поглощение фотона с определенной длиной волны.
Неокрашенные органические соединения поглощают
ультрафиолетовые лучи, обладающие большой энергией, но энергия
видимых лучей недостаточна для возбуждения электронов в
молекулах таких соединений. В окрашенных органических соединениях
возбудимость электронов настолько велика, что для их
перемещения на высший энергетический уровень достаточно
относительно небольшой энергии видимых лучей.
Повышенная возбудимость электронов в молекулах красителей
объясняется в основном следующим:
1. Все красители принадлежат к ненасыщенным соединениям,
содержащим двойные связи, которые отличаются от простых
(ординарных) связей тем, что двойные связи образуются не только с-элек-
тронами, но и тс-электронами**, которые гораздо более возбудимы.
* По СИ единицей энергии является 1 дж=10" эрг.
** Точнее, электронами в а- и ^-состояниях.
19-805 ctg
2. В КраСИТеЛЯХ, ОТНОСЯЩИХСЯ К Лримйшча.лш« ї
соединениям с многочисленными сопряженными двойными
связями*, z-электроны значительно более возбудимы, чем в
веществах с одиночными двойными связями.
3. Наличие электронодонорных групп (отдающих электроны),
например ОН, NH2, и электроноакцепторных групп
(притягивающих электроны), например NO2 и —N=N—, особенно в системе
сопряженных двойных связей, увеличивает возбудимость
u-электронов и вызывает углубление цвета.
Таким образом, окраска органических соединений объясняется
настолько легкой возбудимостью их тс-электронов, что они могут
поглощать даже фотоны видимого света, обладающие небольшой
энергией. Причины повышенной возбудимости
тс-электронов—взаимодействие их в системе многих сопряженных двойных связей,
которое усиливается группами, являющимися акцепторами и
донорами электронов.
Красители должны не только обладать цветом, но и прочно
удерживаться на окрашиваемых материалах. Эта способность,
как и цвет, зависит от строения красителя. Такая зависимость
далее будет рассмотрена для каждой группы красителей.
КЛАССИФИКАЦИЯ КРАСИТЕЛЕЙ
Наиболее распространены две классификаций красителей.
Первая—химическая исходит из особенностей химического
строения и обычно употребляется в учебной и научной литературе.
Важнейшими группами этой классификации являются:
1) азокрасители, 5) сернистые,
2) антрахиноновые, 6) фталоцианиновые,
3) индигоидные, 7) арилметановые,
4) полициклические] 8) хияониминовые краси-
кубовые, тели.
Другая классификация—техническая исходит из технических
свойств красителей, способов и областей их применения.
Техническая классификация используется потребителями красителей
при планировании и учете производства красителей и т. д.
Согласно этой^классификации выделяют следующие важнейшие
группы красителей:
1) кислотные, 8) красители, образуе-
2) протравные, мые на волокне,
3) прямые, 9) «активные»,
4) основные, 10) для химических во-
5) кубовые, локон,
6) сернистые, 11) растворимые в органи-
7) пигменты и лаки, ческих растворителях.
* Сопряженными двойными связями называют чередующиеся двойные и
ординарные связи.
290
Свойства и особенности красителей различных групп этой
классификации рассматриваются далее.
Номенклатура красителей. Иностранные фирмы обычно
присваивают красителям случайные названия, например, сириус
голубой (Сириус—звезда), индантрен (от слов индиго и антрацен)
и др.
В СССР названия красителей построены рационально. Первое
слово названия характеризует принадлежность красителя к
группе технической классификации, а второе—его цвет. Цифры и
буквы, следующие за названием, обозначают оттенок цвета.
Например, прямой черный 3, кислотный синий 2К, кубовый
ярко-зеленый Ж и т. д. Буквы К, 3, Ж, С означают соответственно
красноватый, зеленоватый, желтоватый, синеватый оттенок цвета,
буква О—основной (главный, наиболее важный) цвет. Цифры
перед буквами указывают на интенсивность оттенка BК.—более
красноватый оттенок, чем К, и т. д.). К названиям иногда
добавляются некоторые специальные слова и буквы, значение которых
будет расшифровано при рассмотрении отдельных групп краси*
телей.
АЗОКРАСИТЕЛИ
Азокрасители—самая многочисленная группа красителей; их
ассортимент гораздо шире, чем всех остальных вместе взятых, а
доля в общем объеме производства красителей достигает
35—40%.
Азокрасители входят почти во все группы технической
классификации (стр. 290); они обладают самыми разнообразными
свойствами и широко применяются во многих отраслях народного
хозяйства.
Строение и методы получения азокрасителей. Азокрасители
содержат одну или несколько азогрупп —N = N—, связанных с
ароматическими, реже с гетероциклическими или жирноаромати-
ческими радикалами. Кроме групп —N = N— азокрасители
содержат группы ОН или NH2, причем водород этих групп может
быть замещен на алифатические или ароматические радикалы.
Красители, растворимые в воде, почти всегда содержат сульфо-
группы SO3H. Азокрасители часто содержат такие заместители,
как NO2, CHg, Cl, COOH и др., оказывающие существенное
влияние на свойства красителей (цвет, прочность и т. д.).
Одним из простейших азокрасителей является кислотный
оранжевый
NaO,Sf У-N=N—,
19*
Кислотный оранжевый принадлежит к моноазокрасителям,
содержащим одну азогруппу. Красители с двумя азогруппами
называются дисазокрасителями, с тремя азогруппами—трисазо-
красителями; для красителей, содержащих более двух азогрупп,
принят также термин полиазокрасители.
Важнейший способ получения азокрасителей основан на
последовательном проведении реакций диазотирования и азосоче-
тания (или просто сочетания).
Реакция диазотирования заключается во взаимодействии
ароматических или некоторых гетероциклических аминов с
азотистой кислотой в присутствии соляной или серной кислоты.
Поскольку азотистая кислота неустойчива, ее получают в
процессе диазотирования действием разбавленной соляной или
серной кислоты на нитрит натрия.
Азотистая кислота диазотирует амины (например, анилин)
по схеме:
NH2 N=NCr
о
HNO2 + НС1 » II +2Н2О
хлористый
бензолдиазоиий
Диазосоединения, образующиеся в результате этой реакции
в кислой среде, находятся в виде солей, например из анилина
образуется хлористый бензолдиазоний.
Диазосоединения—неустойчивые вещества, в сухом виде сильно взрывчаты, поэтому их
получают обычно при пониженных температурах (от 0 до 10—15 °С)
и перерабатывают в виде растворов непосредственно после
получения.
Амины, вступающие в реакцию диазотирования при получении
азокрасителей, называются диазосоставляющими.
Ниже приведено строение некоторых важнейших диазосостав-
ляющих (анилин упомянут ранее):
Диазосоставляющие
.и-Ксилидин НзС—'/ у-
л-Нитроанилин O2N— vZ3~NHa
Метаниловая кислота <; ,—NHS
Сульфаниловая кислота HO3S— " J—NH2
292
Диаэос оставляющие
Строение
а-Нафтшіамин ? Г Ті І
NH2
І
і-Нафтиламин-5-сульфокислота .... [ II І
І
HO3S
HO NH2
Аш-кислота {ill
HO3S^/^-^SO3H
Бензидин H2N— \~\— \~/~ NH2
H3COv /ОСНз
Дианизидин H2N—4? \—'f \—NH2
Диамины, например бе'нзидин и дианизидин, образуют при
диазотировании бисдиазосоединения:
HaN~~C=/~\=3~NH2 + 2HN°2 + 2HC1 —*
, Cl~N=lNi—-f^S—/~~Ч—N=NC1~ + 4H2O
хлористый дифенил-бис-диазоний
Диазосоединения легко вступают в различные химические
реакции, одна из важнейших—реакция азосочетания, в
результате которой образуются азокрасители.
В реакцию азосочетания с диазосоединениями могут вступать
так называемые азосоставляющие, к которым принадлежат многие
ароматические амино- и оксисоединения специфического
строения, а также некоторые гетероциклические и
жирно-ароматические соединения.
Так, .и-фенилендиамин реагирует с хлористым бензолдиазо-
нием с образованием азокрасителя—хризоидина по схеме:
+ _
NH2 хризоидин
Некоторые азосоставляющие могут вступать в реакцию
азосочетания не один раз, а дважды и трижды. Например, Аш-кислота
293
сочетается дважды: первый рал и ^
ной, образуя дисазокрасители,—по схеме:
H2N ОН
кислая
щелочная среда
H2N OH
дисазокраситель кислотный сиие-черный
Ниже приведено строение важнейших азосоставляющих.
Звездочками отмечены атомы углерода, к которым при азосочетании
преимущественно присоединяются диазосоединения (обычно
образуются также примеси изомеров). Место сочетания в кислой
среде отмечено буквой К, в щелочной^-Щ.
Азосоставляющие Строение
*
Фенол *<^ \—ОН
NHa
л-Фенилендиамин *<^ \—NHa
•СООН
Салициловая кислота »'f ^—ОН
?-Нафтол
Р-соль
о-Нафтиламин *\ /—NHj
294
Азосоставляющие Строение
NH2
Набтионовая кислота
К леве-кислота-1,7 -^
SO3H
l2
Ч03Н
И-кислота
ОН
НО
I К
Гамма-кислота
H2C C—CH3
|-Фенил-3-метил-5-пиразолон-4'-сульфо- | ||
кислота С N
Ацетилацетанилид CH3COCH2CONH—
Ароматические азосоставляющие сочетаются в
пара-положение к группам —NH2 и —ОН. Если это положение занято или
соседние группы (например, SO3H) препятствуют сочетанию в
пара-положение, оно происходит в орто-положение к группам NH2
и ОН, но не в мета-положение.
Бисдиазосоединения могут сочетаться с двумя молекулами
одинаковых или разных азосоставляющих, образуя дисазокра-
сители, например:
OONa
295
хлористый днфенил-бис-диазоний Na-соль
салициловой кислоты
NH2
щелочная
COONa
дисазокраситель прямой коричневый КХ
Азокрасители, содержащие аминогруппу, можно диазоти-
ровать повторно*, а полученные диазосоединения сочетать с
различными азосоставляющими. Таким образом получают дис-
и трисазокрасители, например кислотный черный С:
-NH2 hno2
кислотный черный С
Реакция азосочетания в производстве обычно проводится в
водной среде при пониженной температуре @—10 °С).
Образующиеся азокрасители осаждаются из растворов при добавлении
поваренной соли в количестве 10—20% от объема раствора
(так называемое высаливание). Осадки красителей
отфильтровывают, высушивают, размалывают в порошок и стандартизируют.
Чаще всего красители образуют аморфные, медленно
отфильтровываемые осадки, поэтому для этой операции применяют
фильтрпрессы с большой фильтрующей поверхностью и
механическим зажимом таблиц и рам (рис. 99). Относительно редко
используются барабанные вращающиеся вакуум-фильтры
непрерывного действия (рис. 100) с механическим съемом осадка
специальным ножом. Такие фильтры обычно применяются для отделе-
* Исключение составляют азокрасители нафталинового ряда, содержащие
аминогруппы в орто-положении к азогруппе.
296
ния кристаллических, хорошо отделяющихся осадков. В
последнее время в СССР разработаны конструкции автоматических
фильтрпрессов (ФПАК.) с большой фильтрующей поверхностью.
Рис. 99. Рамный фильтрпресс с гидравлическим затвором.
Такие фильтры, при обслуживании которых не требуется затрат
ручного труда, начинают применяться и в производстве
красителей.
Рис. 100. Барабанный вакуум-фильтр:
/—фильтрующий барабан; 2—корыто: 3—скребок (нож); 4—вал; 5—шкив.
Влажные осадки красителей (пасты) высушивают при
температуре 50—150 °С, в зависимости от термической устойчивости
красителя. Для этой цели применяются сушилки различных
типов. Наибольшее распространение получили двухвальцовые не-
297
прерывнодействующие сушилки (рис. 1U1), в которых сушка
красителя происходит на вращающихся вальцах, обогреваемых
изнутри паром. Сырой краситель поступает на вальцы по
трубопроводу, высушенный—механически снимается с вальцов
специальным ножом.
Рис. 101. Двухвальцовая сушилка:
/—вальцы; 3—пастомеситель; 3—нож.
Сухие красители размалывают в мелкий порошок чаще[ всего
в центробежных мельницах—дезинтеграторах (рис. 102) с двумя
дисками, вращающимися в противоположных направлениях со
скоростью около 2500 обімин. На дисках укреплены стальные
етержни, размалывающие краситель при вращении.
Рис. 102. Дезинтегратор (кожухснят, диски раздвинуты).
Полученные после размола порошки неоднородны по
концентрации красящего вещества. Поскольку одним из основных
требований, предъявляемых к красителям, является их
однородность и постоянство концентрации, порошкообразные красители
предварительно стандартизируют. Эту операцию в производстве
298
называют установкой на тип (тип—типовой эталон стандартного
красителя). Установка на тип заключается в тщательном
перемешивании порошкообразных красителей, к которым, как правило,
добавляют небольшое количество A0—20%) наполнителей с
таким расчетом, чтобы концентрация полученного красителя
равнялась концентрации эталонного. В качестве наполнителей
используют дешевые вещества, не только не ухудшающие, а иногда
и улучшающие качество красителей—поваренную соль, сульфат
натрия, декстрин и некоторые другие.
Установка на тип производится во вращающихся смесовых
барабанах, внутри которых установлены перегородки для
лучшего смешивания порошков. Барабан попеременно вращается в
противоположных направлениях. Загрузка и выгрузка красителя
происходят автоматически с помощью шнека.
Эти операции (фильтрование, сушка, размол и
стандартизация) являются общими для всех видов красителей.
Кислотные азокрасители содержат группы кислотного
характера, чаще всего SO3H. Их натриевые, аммонийные или калийные
соли достаточно хорошо растворимы в воде.
Кислотные красители окрашивают белковые вoлqкнa и
материалы, в первую очередь шерсть и кожу, реже они применяются
для окраски натурального шелка и синтетических полиамидных
волокон. Многие марки этих красителей, обычно яркого цвета
и часто недорогие, используются для окраски бумаги, древесины,
для изготовления чернил, подкраски мыла и т. д.
При крашении белковых и синтетических полиамидных
волокон кислотными красителями группы основного характера,
имеющиеся в макромолекулах волокон, вступают в ионную связь с
кислотными группами красителя, например:
НОч
ш—NH2 + HO3S—<f ^_N=N—,
X
где со—NH2—условное обозначение молекулы белкового
вещества шерсти (кератина).
Ионная связь, как известно, легко разрывается в водных
средах, поэтому окраски, получаемые при помощи обычных
кислотных азокрасителей, мало прочны к стирке. Значительно
более прочно окрашивают шерсть кислотные азокрасители,
состоящие из молекул больших размеров, например кислотный
299
черный С (см. выше). Повышенная устойчивость окрасок
объясняется тем, что такие красители удерживаются на волокне не
только ионными связями, но и силами адсорбции. Чем больше
молекула красителя, тем значительнее силы адсорбции, в
которой в большей или меньшей мере участвуют все атомы
молекулы красителя.
Кислотные азокрасители обычно являются моно- или дисазо-
красителями. Это — одна из наиболее многочисленных групп
азокрасителей, включающая красители всех цветов. Строение
отдельных кислотных азокрасителей (кислотный оранжевый,
кислотный сине-черный, кислотный черный С) и схемы их
синтеза приводились выше.
Протравные и металлсодержащие кислотные азокрасители.
Протравными называют красители, образующие с солями
некоторых металлов — протравами — при крашении прочные,
большей частью комплексные, соединения. Различают протравные
красители, применяемые для окраски целлюлозных волокон и
протравные красители для шерсти.
Наибольшее значение имеют протравные красители для
шерсти, образующие очень прочные окраски. Протравой для
них служат соли хрома. Большая часть этих красителей
является азокрасителями соответствующего строения, например:
ОН НО HNCOCH,
I NH2
NO2 SO3Na
хромовый коричневый К
,COONa
NaO3S— *¦ J—N=N—^ ^>—OH
хромовый желтый К
Как видно из этих примеров, протравные азокрасители
должны содержать две группы ОН в соседних ядрах в орто-положении
к азосвязи —N=N—; или по одной группе ОН и NH2 в том же
положении; или же группы СООН и ОН в орто-положении. Эти
группы служат для образования связей с трехвалентным хромом
в комплексном соединении.
300
Хромовые протравные красители прочно удерживаются на
шерсти благодаря образованию комплекса, в котором атом хрома
связан и с красителем, и с группами NH2 или ОН
макромолекул шерсти. Методы получения хромовых азокрасителей мало
отличаются от применяемых в синтезе кислотных азокрасителей.
Промышленность вырабатывает хромовые протравные азокра-
сители всех цветов, оттенки их обычно неяркие.
Более ярки и удобны для применения кислотные
металлсодержащие азокрасители. Они отличаются от протравных тем,
что образуют комплекс с металлом не в процессе крушения, а
при изготовлении красителей. Чаще всего это тоже хромовые
комплексы, реже кобальтовые. К этой группе красителей
относятся комплексы, содержащие на один атом металла одну
молекулу азокрасителя (комплексы 1 : 1), а также открытые в последние
десятилетия комплексы 1 : 2, содержащие на один атом металла
две молекулы азокрасителя. Комплексы 1 : 2 не содержат сульфо-
групп, растворимость их в воде обусловлена присутствием
сульфамидных групп SO2NH2 и некоторых других. Комплексы 1 : 2
особенно удобны, так как окрашивают шерсть из слабокислых
и нейтральных растворов и пригодны для окраски смесей шерсти
с волокнами, неустойчивыми в кислой среде. Ниже приведены
примеры металлсодержащих азокрасителей—комплексов 1 : 1 и 1 : 2:
О Cr—NH
O,N
X
кислотный зеленый ЖМ
(комплекс 1 : 1)
H2NO2S
N=N—С С—СН3
і А
/V
¦ Nah
ЩС-С С—N=N—/~^4-осуш?
кислотный алый прочный НЖМ (комплекс 1 : 2)
Буква М указывает на присутствие в молекуле кислотного
красителя комплексносвязанного металла, а Н означает, что краситель
может окрашивать шерсть в нейтральной среде.
301
Прямые азокрасители—красители, спосооные
непосредственно (прямо) без протрав или каких-либо специальных обработок
окрашивать целлюлозные волокна (хлопковое, вискозное). Почти
все многочисленные прямые красители являются азокрасителями.
Анилин
Раствор Раствор | H2SO4
'аствор
НС1
16
Бензидин
яг
, Краситель
прямой черный 3
Рис. 103. Схема производства красителя прямого черного 3:
/—монтежю (хранилище бенэидина); 2—чан для диазотировання бензидина; 3, 4, S, 8,
10, 12—мерники; 5—ча^ для растворения Аш-кислоты; 7—чан для первого сочетания; 9—чаи
для дназотирования анилина; 13—чан для второго и третьего сочетаний; /¦)—чан для
растворения л-фенилен диамина; /5—насос; IS— горизонтальної монтежю для фильтрации;
17—фильтрпресс; 18—монтежю для влажного красителя; 19—вальцовая сушилка;
20—дезинтегратор; 2/—смесовый барабан.
Прямые красители удерживаются на волокне силами
адсорбции и водородными связями*.
Почти все красители этой группы принадлежат к дис- и трис-
азокрасителям. Формула одного из прямых дисазокрасителей—
прямого коричневого КХ изображена на стр. 296. Важным
представителем прямых трисазокрасителей является прямой черный
* Водородные связи образуются между атомами или молекулами с участием
водорода; пример—димер муравьиной кислоты (водородная связь показана
тремя точками):
302
3. Схема его производства приведена на рис. 103 и является
типичной для красителей этой группы.
Процесс начинают с диазотирования бензидина (см. стр. 293).
Из цеха полупродуктов бензидин в виде 10—15%-ной водной
суспензии поступает в монтежю /. Рассчитанное количество
бензидина передавливают сжатым воздухом из монтежю / в чан 2
и наливают из мерника 3 соляную кислоту. Смесь охлаждают до
5 °С дробленым льдом или рассолом, циркулирующим через
змеевики, затем из мерника 4 при включенной мешалке
постепенно в чан 2 приливают 30%-ный раствор нитрита натрия.
Вследствие выделения тепла реакции температура повышается до 15 °С.
После введения нитрита натрия реакционную массу
перемешивают в течение 1—2 ч.
В ходе диазотирования в реакционной массе периодически
контролируют содержание НС1 (индикатор—азокраситель конго
красный) и HNO2 (индикатор—йодистый калий и крахмал).
Во время проведения реакции диазотирования бензидина в чане
5 готовят водный раствор Na-соли Аш-кислоты, добавляя из
мерника 6 кАш-кислоте раствор соды до нейтральной реакции
раствора.
По окончании диазотирования из чана 2 переливают
(самотеком) раствор диазосоединения в чан 7 и охлаждают до 10—12 °С.
При этой температуре и энергичном перемешивании из чана 5
приливают в течение нескольких часов раствор Аш-кислоты; для
нейтрализации выделяющейся соляной кислоты из мерника 8
прибавляют раствор соды. Реакция азосочетания протекает в
кислой среде по следующей схеме:
H2N ОН
C1N = N—<fr~^>—/~~\ —N = NCP +
хлористый дифеннл-бис-диазоннй Аш-кнслота
(динатриевая соль)
ОН
+ НС1
моноазосоединение
В ходе реакции сочетания контролируют рК среды и наличие
в реакционной массе Аш-кислоты и хлористого дифенил-бие-
диазония (качественные пробы); процесс заканчивают, когда все
диазосоединение вступит в реакцию.
Параллельно с процессом получения моноазоссединения диазо-
тируют анилин. В чан 9 наливают воду, из^мерника 10 добавляют
зоз
необходимое количество анилина, загружают лед, а затем из
мерника //—серную кислоту. При 0—2 °С из мерника 12 приливают
раствор Нитрита натрия. Диазотирование длится 1 ч. В
результате образуется сернокислый бензолдиазоний:
NH2 N=NHSO7
!
I I
Q) + NaNO2 + H2SO4 * f J)
По окончании процесса первого сочетания реакционную массу
из чана 7 сливают в чан 13, охлаждают до О °С, прибавляют
раствор сернокислого бензолдиазония из чана 9 и очень быстро
загружают такое количество кальцинированной соды, чтобы был
нейтрализован избыток кислоты, а в реакционной массе
образовался 1%-ный раствор соды. Реакция второго сочетания в
щелочной среде заканчивается в течение 30—40 мин:
H2N ОН
дисаэосоединение
Дисазосоединение содержит диазогруппу, способную к
дальнейшему азосочетанию. В щелочной среде эта группа существует
в форме диазотата R—N=N—ONa в равновесии с диазокатионом
RN^N, который участвует в азосочетании.
К полученному дисазосоединению прибавляют поваренную
соль (около 10% от объема реакционной массы), приливают из
чана 14 раствор лі-фенилендиамина и перемешивают 2 я. Затем
подогревают раствор до 25 °С и размешивают еще 1 ч. В результате
реакции третьего сочетания образуется трисазокраситель прямой
черный 3:
H2N ОН NH2
NaO-N=N—<f~\-f~^S—N=N. I \ '
304
NaO3S -
прямой черный З
Далее реакционную массу нагревают до 80—85 °С, прибавляют
NaCl и отфильтровывают выпавший в осадок краситель на фильтр-
прессах 17. Осадок выгружают из фильтрпрессов в монтежю 18,
откуда влажный краситель поступает на сушку в непрерывную
вальцовую сушилку 19. Сухой краситель размалывают в порошок
дезинтегратором 20, порошок тщательно перемешивают с
поваренной солью (около 10% от количества красителя) в барабане 21 и
выгружают в тару.
Прямой черный 3 и другие прямые азокрасители самых
различных цветов благодаря удобным методам крашения
применяются в текстильной, трикотажной и кожевенной
промышленности .
Однако прочности окрасок многими прямыми красителями к
свету и стирке невысоки, что следует учитывать при выборе
областей их применения.
Повышенной светопрочностью обладают медные
комплексы прямы азокрасителей, например краситель прямой синий
светопрочный ^ 3:
NaO,S О
Строение ряда азокрасителей, способных образовывать
комплексы с металлами, рассмотрено на стр. 300.
Медные комплексы прямых красителей получаются как в
процессе синтеза красителей, так и при крашении,
непосредственно на волокне, при обработке ткани медным купоросом или другими
соединениями меди.
Способность прямых азокрасителей прочно удерживаться
целлюлозными волокнами (за счет сил адсорбции или водородных
связей) связана с особенностями их строения. Это вытянутые,
подобно макромолекулам целлюлозы, молекулы, содержащие не
менее восьми сопряженных двойных связей. В молекулах прямых
красителей атомы находятся в одной плоскости (копланарны).
Группы NH2, ОН и —N=N—. по-видимому, участвуют в
образовании водородных связей с целлюлозой.
Активные азокрасители появились только в последнее
десятилетие. Основной их особенностью является способность в
процессе крашения вступать в прочную химическую (ковалентную*)
связь с окрашиваемым материалом. Активные красители содер-
* Сущность ковалентной (гомеополярной) связи рассматривается в курсе
органической химий.
20-805 305
жат подвижные атомы или группы, олагодаря чему ини b^ja^i
в реакцию замещения или присоединения с реакционноспособными
группами макромолекул волокна (ОН, NH2).
В настоящее время наибольшее значение имеют активные
красители для крашения целлюлозных волокон.
В качестве примера можно привести схему получения
активного ярко-красного красителя 5СХ (X означает, что крашение
холодное, ведется без нагревания) и принцип его взаимодействия
с целлюлозным волокном.
Исходными продуктами являются моноазокраситель (I) и
цианурхлорид:
ОН NH
моноаэокраситель (I)
I
СІ
цианурхлорнд
С\
А'— (
HO HN С
) 1 \ч=<
актннный ярко-красный 5СХ
HO HN С
Z-OH
крашение
/O-Z
^N
SO3Na
іде Z—OH—условное обозначение макромолекул
целлюлозы.
Цианурхлорид получают при полимеризации хлорциана CNC1
и применяют для синтеза ряда активных красителей, а также
для получения оптически отбеливающих веществ (стр. 326) и
гербицидов (стр. 345).
Активные красители очень ярки; благодаря образованию
прочных химических связей с волокном получаемая окраска
особенно устойчива к стирке, трению и другим обработкам. Про-
306
•изводство этого нового вида красителей в настоящее время
интенсивно развивается.
Азопигменты и лаки. Как уже упоминалось, пигментами
называют нерастворимые в воде красители. В промышленности
синтетических красителей понятие «лак» отличается от принятого
в лакокрасочной промышленности; в химии красителей лаки —
нерастворимые в воде, чаще всего бариевые или кальциевые соли
растворимых красителей.
Большая часть пигментов принадлежит к азокрасителям. Это
обычно моноазокрасители, реже дисазокрасители. Пигменты
нерастворимы в воде, так как они не содержат групп SO3H, COOH
и NH2, образующих соли.
Одним из наиболее важных широко применяемых пигментов
является пигмент желтый светопрочный, схема синтеза которого
приведена ниже:
NH2 NsN.Cr v
О N І О N I CH3COCHiCONH-^=/)
2 Ч^\ NaNO2+HCl 2 *\^ ^ ацетилацетаиилид
Y
сн.
і
Н
а
3-нитро- диазосоединенне
4-аминотолуол
Н8С—{%- N=N—CH—СОСН
пигмент желтый светопрочный
Яркие и прочные пигменты различных цветов широко
применяются для окраски пластических масс, резины, в лакокрасочной
промышленности. С развитием производства полимеров пигменты
стали широко применяться для окраски текстильных материалов.
Так, при изготовлении химических волокон, в частности
вискозных, в прядильную массу (стр. 443) вводят измельченные
тонкодисперсные пигменты (величина частиц 1—2 мк), после
пропускания прядильной массы через фильеры получают прочно
окрашенные волокна.
Другой метод окрашивания—пигментная
печать—заключается в том, что мельчайшие частицы пигментов приклеиваются к
ткани тонкой и прочной пленкой полимера, образующейся при
нагревании непосредственно на ткани.
20* 307
Примером лака может служить лак оранжевый, получаемый
действием хлористого бария на краситель кислотный оранжевый
ОН
2NaO3S—f~\—N=N—.
кислотный оранжевый
BaCI,
OH
Ва++
лак оранжевый
Лак содержит примеси неокрашенных наполнителей—
А1(ОН)Я и BaSO4, вводимых в процессе синтеза.
Обычно лаки менее устойчивы, чем пигменты. Главные области
применения лаков—полиграфия, окраска резины, изготовление
обоев, цветных карандашей и др.
Азокрасители, образуемые на волокне («холодные» азокраси-
тели). Принцип так называемого холодного крашения волокон
(преимущественно целлюлозных) заключается в том, что реакция
азосочетания проводится на волокне, пропитанном растворами
азосоставляющей и диазосоединения.
Необходимые для холодного крашения неустойчивые
диазосоединения (стр. 292) ранее получали диазотированием аминов
непосредственно на текстильных фабриках, эти амины получили
название азоаминов.
Однако процессы диазотирования более рационально проводить
на красочных заводах. Поэтому разработаны устойчивые формы
диазосоединений—диазоли, в отличие от обычных диазосоеди-
нений безопасные при хранении и транспортировании. В качестве
диазолей чаще всего применяются двойные соли диазосоединений
с хлористым цинком.
Диазоли, как правило, содержат до 50% инертных
наполнителей—Na2SO4, A12(SO4K идр., повышающих стабильность
диазосоединений. Примером может служить диазоль алый К,
получаемый из азоамина алого К".
NH,
O,N'
ОСНЧ
" ОСН;
(У
N=N-HSO4
• ZnCl2
азоамин алый К
NO2
диазоль алый К
308
Диазоль алый К содержит в качестве наполнителя Na,SO4.
Азосоставляющие для холодного крашения называются азо-
толами, например азотол А:
Азоамины и азотолы отличаются от прочих диазо- и азосостав-
ляющих тем, что в них отсутствуют группы SO3H и СООН,
придающие веществам способность растворяться в воде. Благодаря
этому получаемые окраски устойчивы к стирке.
Азокрасители для синтетических и ацетатных волокон.
Синтетические и ацетатные волокна обладают некоторыми свойствами,
которые затрудняют их окраску красителями, применяемыми для
натуральных волокон.
При смачивании водой поры натуральных текстильных волокон
расширяются до размеров, допускающих проникание в них
довольно больших молекул красителей. Синтетические и
искусственные волокна гидрофобны,т.е. плохо смачиваются водой,
размеры их пор невелики и доступны только для красителей с
относительно небольшими размерами молекул.
В связи с этим для окраски искусственных и синтетических
волокон были разработаны специальные красители. К ним,
властности, принадлежат так называемые дисперсные красители,
хорошо окрашивающие ацетатное, полиэфирное, полиамидное и
другие химические волокна. Дисперсные красители нерастворимы
в воде и находятся в ней в виде очень мелких дисперсных частиц.
Молекулы таких красителей имеют небольшие размеры и легко
проникают в поры волокна.
Большое значение имеют дисперсные азокрасители,
например дисперсный алый Ж:
В последние годы для окраски полиамидных волокон стали
применяться активные дисперсные красители, содержащие атомы
и группы, которые в процессе крашения способны реагировать
с имеющимися на концах макромолекул полиамида аминогруппами
с образованием прочной химической связи.
Примером активных дисперсных красителей может служить
активный оранжевый П (П означает, что краситель предназначен
для окраски полиамидного волокна). Взаимодействие этого кра-
309
сителя с полиамидным волокном поясняется следующей схемой:
(HOH«C2JN —\~\—N=N—/~^y— SO2NHCH2CH2C1 + H2N—П >
-n + НС1
где H2N—П—условное обозначение макромолекул полиамида,
образующего волокна.
Окраски активными дисперсными красителями по устойчивости
к стирке, трению и ряду других обработок превосходят окраски,
получаемые при применении неактивных дисперсных красителей.
Волокно нитрон содержит группы кислотного характера,
поэтому в водной среде оно обладает свойствами аниона. Катион-
ные красители представляют собой четвертичные аммониевые соли,
диссоциирующие в водном растворе с образованием окрашенных
катионов, которые легко адсорбируются и прочно удерживаются
волокном нитрон.
Катионные азокрасители относятся к группе моноазокраси-
телей и содержат гетероциклические остатки. Примером может
служить катионный синий К- Для получения катионного синего К
диазотируют 2-амино-6-метоксибензтиазол, образующееся диазосо-
единение сочетают с N-метилдифениламином и моноазокраситель I
метилируют диметилсульфатом:
N N
<?\/ \, NaNO2+H2SO4
HSCO^V\/ НзСО^4
2-амино-6-метокси-
бенатиазол
СНз
N-метнлдифен ил-
амии
моноазокраситель I
катионный синий К
310
АНТРАХИНОНОВЫЕ КРАСИТЕЛИ
Эта важная группа красящих веществ объединяет протравные
кислотные, дисперсные активные и катионные красители,
являющиеся производными антрахинона (стр. 282). Как указывалось
выше (стр. 270), один из важнейших природных
красителей—ализарин является 1,2-диоксиантрахиноном.
Наиболее распространенный способ получения ализарина
заключается в сульфировании антрахинона олеумом при 130—
140 °С до 2-антрахинонсульфокислоты, которую далее
обрабатывают едким натром в присутствии окислителя (NaNO3) при
температуре около 200 °С и давлении (процесс щелочного плавления):.
О
динатриевая соль
ализарина
О
ализарин
Ализарин принадлежит к протравным красителям. Он
образует с солями алюминия на целлюлозных волокнах чрезвычайно'
прочные и яркие красные окраски. Несмотря на исключительную
прочность, в настоящее время ализарин находит очень
ограниченное применение и успешно заменяется холодными азокрасителями
(стр. 308), значительно более удобными. Другие протравные антра-
хиноновые красители находят еще меньшее применение.
Кислотные антрахиноновые красители очень прочны, ярки
и широко применяются для окраски шерсти. Типичным их
представителем является кислотный зеленый антрахиноновый.
Кислотные антрахиноновые красители обычно получают
замещением атомов СГ или Вг или же групп ОН" или N0^ в а-про-
изводных антрахинона на остатки ароматических аминов. Эти
реакции проводят при нагревании и в присутствии катализаторов. В
реакционную смесь часто вносят также восстановители,
превращающие производные антрахинона в белее активные производные
антрацена. Образующиеся промежуточные продукты—«основания»
кислотных антрахиноновых красителей во многих случаях
нерастворимы в воде, но хорошо растворяются в органических жидкостях
311
{жидкие горючие, масла и др.) и часто применяются для их
подкраски. Сульфируя такие «основания», получают растворимые в
воде кислотные антрахиноновые красители.
На рис. 104 показана схема производства этого красителя.
В стальном эмалированном котле / расплавляют толуидин,
прибавляют из мерника 2 соляную кислоту, а затем борную кислоту
и 1,4-диоксиантрахинон (хинизарин). Смесь нагревают до 80 °С,
прибавляют цинковую пыль, затем перемешивают 4 ч при 100 °С.
п-Тол у и дин, Zn
жнцзарин, Н3ВО3
/6
Рис. 104. Схема производства красителя кислотного зеленого антрахинонового:
/—котел для получения 1,4-дитолиламииоантрахинона; 2. 5. //. /2—мерники; 3— котел для
перекристаллизации 1,4-дитолиламиноаитрахинона; 4—холодильник; 5—пароструйный насос; 7—нутч-
фальтр; 8— монтежю для маточного раствора; S— сушилка; to—сульфураторг 13— чан для
осаждения красителя; 14—монтежю для растворения поваренной соли; /5—центробежный насос;
16—фильтрпресс.
При этом протекает реакция замещения гидроксильных групп в
1,4-диоксиантрахиноне на остатки л-толуидина:
-г- 2H2N-
НСІ+НзВОз-t-Zn
»
нагревание
О HN
-сн3
М-диголиламиноантрахшюн
312
Цинк и соляная кислота в этом процессе необходимы для
восстановления 1,4-диоксиантрахинона в более реакционноспособ-
й 1,4,9,10-тетраоксиантрацен
становления 1,4диоксиантрахи
ный 1,4,9,10-тетраоксиантрацен
ОН ОН
ОН ОН
1,4,9! Ш-тетраоксиан грацен
Борная кислота является катализатором реакции замещения.
Полученный 1,4-дитолиламиноантрахинон, содержащий
примесь л-толуидина, перекристаллизовывают из метилового спирта
в котле 3, снабженном холодильником 4, и отфильтровывают на
нутч-фильтре 7. Фильтрат из сборника 8 поступает на
регенерацию метилового спирта и содержащегося в растворе л-толуидина.
Осадок 1,4-дитолиламиноантрахинона высушивают в сушилке 9
и сульфируют в котле 10 3%-ным олеумом при 35 °С в течение
10 ч:
NaO3Sv
О HN—\[3~СНз ° HN—ч ~^—
О HN-
NaO3S'
кислотный зеленый антрахиноновый
Сульфирующую смесь получают при смешивании 20%-ного
олеума и концентрированной серной кислоты, поступающих
из мерников 11 и 12.
Реакционную массу из котла 10 передают в чан 13, где
краситель осаждают добавлением поваренной соли. Суспензию
красителя фильтруют через фильтрпресс 16, осадок промывают
раствором NaCl, приготовленным в монтежю 14. Далее краситель
поступает на сушку и размол.
Анилинокрасочная промышленность вырабатывает несколько
десятков различных кислотных антрахиноновых красителей,
преимущественно фиолетового, синего, голубого и черного цветов. По
строению и методу получения они сходны с кислотным зеленым
антрахиноновым. Эти красители содержат сульфогруппы и,
как правило, замещенные или незамещенные аминогруппы.
Активные антрахиноновые красители отличаются от кислотных
антрахиноновых тем, что содержат активные атомы или группы
313
(стр. 305). Наибольшее значение имеют красители
цвета, например:
О NH2
,/SO3Na
—SO3Na
N
—С С—'
¦С]
N
N
активный синий КХ
Дисперсные антрахиноновые красители, как и дисперсные
азокрасители (стр. 309), имеют большое значение для окраски
ацетатного и синтетических волокон, дополняя цветовую гамму
более глубокими—фиолетовыми, синими и зелеными оттенками.
Типичным представителем дисперсных антрахиноновых
красителей является дисперсный синий К, получаемый по следующей
схеме:
О SO3H О HNCH3
І
V +h2nch3 f^Y\(f\ +Br2
1-анграхинон-
сульфокислота
О HNCH,
1-метнламиио-
антрахинои
О HNCH3
I
+н2мсн2сн2он
О
1-метиламино-
4-бромантрахинон
О HNCH2CH2OH
дисперсный синий К
ИНДИГО И ИНДИГОИДНЫЕ КРАСИТЕЛИ
Индиго добывалось с древних времен из диких, а позднее из
культурных растений. Это—один из прочных природных
красителей, окрашивающий животные и растительные волокна в синий
314
цвет. Строение индиго было установлено в 1878 г. С 1896 г.
началось производство синтетического индиго, которое вскоре
вытеснило более дорогой природный краситель.
Индиго получают, исходя из хлоруксусной кислоты С1СН2СООН
и анилина, в результате следующих процессов:
FeSO4 + 2NaOH -
Fe(OHJ + 2CICH2COOH.
NH2
¦ Fe(OHJ + Na„SO«
¦r, (ClCH2COOJFe + 2H2O
NHCH2COCT
О нагревание
+ (ClCH2COOJFe *¦
NHCH,COO"
С
железная соль
фени лг ліщина
NHCBjCOOK
І
Fe
Fe
2KOH
калиевая соль
фенилглицина
2Na + 2NH3 > Н2
амид
натрия
NHCH2COOK
NH
+ NaNH2
сплавление Ix
Ak
калиевая
индокси
NH
\ o2
CH э
О
II
NH С
NH
A,
О
индиго
Индиго принадлежит к кубовым красителям, нерастворимым
в воде. При нагревании индиго в щелочной среде е восстановнте-
315
ЛЯМИ (ГИДрОСуЛЬфИТ натрия; образуются
косоединения*, например:
о
II
NH С
:=с
. / \ /Ч
С NH
+Na2S2O4+Na0H
восстановление
+О2
(окисление на волокне)
II
О
индиго
NH
\
ONa
с-с
Ч
NH
А
Na
лейкоиндиго
Лейкосоединения кубовых красителей хорошо адсорбируются
текстильными волокнами, на которых в результате последующего
окисления воздухом образуются исходные, нерастворимые в воде
кубовые красители.
Окраски кубовыми красителями устойчивы к стирке.
После того как был синтезирован индиго, получили ряд его
замещенных и аналогов, из которых, например, бромпроизводное
представляет собой более яркий и прочный краситель, чем
исходный индиго:
О
II Вг
Вг С NH I
||
о
Вг
броминдиго (синего цвета)
В настоящее время большое значение приобрели аналоги
индиго—так называемые тиоиндигоидные красители, содержащие
вместо группы NH атом серы.
* От греч. слова лейкоз—белый. Лейкосоединения ранее известных
кубовых красителей были почти не окрашены, позднее лейкосоединениями стали
называть и глубоко окрашенные продукты восстановления красителей.
316
К таким красителям относится, например, тиоиндиго красный С:
О
II
S С
сх>..
С S
II
о
Тиоиндигоидные красители самых различных цветов широко
применяются для печати по тканям из целлюлозных волокон,
однако большая часть таких красителей обладает умеренной свето-
прочиостью.
КУБОВЫЕ ПОЛИЦИКЛИЧЕСКИЕ КРАСИТЕЛИ
Первый представитель кубовых полициклических
(многоядерных) красителей—индантрон* (кубовый синий О) был
получен в 1901 г. Метод синтеза сохранил значение до настоящего
времени и заключается в нагревании выше 200 °С ?-аминоантрахино-
на со смесью! едких щелочей в присутствии натриевой селитры:
О О
U-f у
мн
NaOH+KOH+NaNO3
кубовый синий О
В последующие годы было открыто большое число кубовых
полициклических красителей различных цветов, отличающихся
чрезвычайной прочностью. Ниже приведено строение нескольких
наиболее важных красителей:
Н3СО ОСН3
'кубовый ярко-зеленый С
* В технике применяется также название индантрен.
317
а2
кубовый ярко-фиолеговый К*
о
о
кубовый ярко-
оранжевый КХ
кубовый золотисто-
желтый жх
Производство- кубовых полициклических красителей
многостадийно и достаточно сложно (его описание можно найти в
литературе, приведенной в конце главы, стр. 366).
Эти красители широко применяются для высокопрочной
окраски целлюлозных материалов, а также в качестве прочных
пигментов (стр. 307) в лакокрасочной промышленности.
КУБОЗОЛИ И ИНДИГОЗОЛИ
Применение кубовых красителей, в частности их
восстановление в лейкосоединения, вызывает некоторые затруднения у
потребителей. Эти затруднения устраняются при применении
высокодисперсных кубовых красителей с величиной частиц не более
1—2 мк.
* Положение атомов хлора не установлено.
318
Большое применение находят кубовые красители в виде
растворимых в воде солей сернокислых эфиров лейкосоединений, так
называемых индигозолей и кубозолей. Индигозоли и кубозоли
получают при обработке соответственно индигоидных
(индигозоли) или кубовых полициклических красителей (кубозоли)
смесью пиридина, хлорсульфоновой кислоты и
порошкообразного железа.
Железо восстанавливает кубовые красители до
лейкосоединений, которые образуют с хлорсульфоновой кислотой и пиридином
пиридиниевые соли кислых сернокислых эфиров. При
последующей обработке раствором соды получают натриевые соли.
Например, из кубового золотисто-желтого ЖХ получают кубо-
золь золотисто-желтый Ж:
кубовый золотисто- кубоэоль
желтый ЖХ золотисто-желтый Ж
Индигозоли и кубозоли хорошо растворимы в воде. Этими
растворами пропитывают ткани, в порах которых при окислении
азотистой кислотой (или другими окислителями) осаждается
исходный кубовый краситель.
СЕРНИСТЫЕ КРАСИТЕЛИ
Сернистые красители были открыты в 1861 г. Раньше их
получали нагреванием различных органических веществ (опилки,
обрезки кожи, меха и т. д.) с серой и полисульфидами натрия
Na2S„ (где п=2—9), причем красители обладали малой
красящей способностью.
В настоящее время сернистые красители получают из
индивидуальных химических веществ, преимущественно ароматических
нитро- и аминосоединений и их производных двумя методами:
«запеканием», которое заключается в нагревании сухих
органических соединений с серой или полисульфидами натрия, и
«варкой», при которой сера вводится в молекулу красителя с помощью
полисульфида натрия в водном (реже спиртовом) растворе.
Например, краситель сернистый черный получают по схеме,
приведенной на рис. 105.
В монтежю / расплавляют 2,4-динитрохлорбензол. Расплав
отстаивается от примеси воды в аппарате 2 и затем поступает
через мерник 4 в котел 3. Здесь динитрохлорбензол нагревают
319
С раСТВОрОМ еДКОГО Натра, ПрИЛИВаеМЫМ ИЗ мерника и, а
тате образуется 2,4-динитрофенолят натрия:
С1
ONa
NO,
+NaOH
нагревание
NO2
2,4-диннтро-
хлорбенэол
NO2
2,4-дииитро-
феиолят натоня
В аппарате б при нагревании серы с раствором сернистого
натрия (из мерника 7) получают полисульфид NaaS4.
Образующийся сероводород поглощается в скруббере 8. В котле 9,
снабженном холодильником 10, к раствору полисульфида натрия
постепенно при 85—90 °С приливают раствор динитрофенолята натрия и
H2S
Рис. 105. Схема производства красителя сернистого черного:
1, 12—монтежю; 2—отстойник; 3—котел для смьрления дннитрохлорбензола; 4, 5, 7, 14, 16—
мерники; 6— котел для получения полисульфида натрия; 8, 11—скрубберы для поглощения
сероводорода; 9—котел для получения сернистого черного; 10, If.— холодильники; 13—котел.
нагревают реакционную массу в течение суток, постепенно
упаривая ее. Выделяющийся сероводород улавливают в скруббере 11.
По окончании «варки» реакционную массу передают в котел 13,
где ее упаривают еще около суток, прибавляют Na2S и
полученную массу применяют непосредственно для крашения или
осаждают из нее краситель, продувая воздух. Осадок сернистого
черного отфильтровывают и высушивают.
Строение сернистого черного и других сернистых красителей
сложно и точно еще не установлено.
320
Аналогично сернистому черному получают красители других
цветов, из которых наиболее важны синие, зеленые и коричневые
красители.
Например, синий сернистый краситель образуется при осер-
нении индотолуидина, полученного конденсацией о-толуидина и
1-нитрозофенола по схеме:
п-в итрозофен ол
—oh-hso;
индотолуидин
Коричневые сернистые красители получают осернением 2,4-ди-
нитротолуола или смеси 1,5- и 1,8-нитронафталинов.
Сернистые красители по свойствам напоминают кубовые:
они нерастворимы в воде, но при нагревании с восстановителем
(Na2S) образуют растворимые в воде лейкосоединения, которые
хорошо адсорбируются целлюлозными волокнами и после
окисления на волокне дают нерастворимые красители.
Сернистые красители неярки и обладают умеренной прочностью,
однако, благодаря их дешевизне и удобным методам применения,
широко используются для окраски целлюлозных волокон в
тех случаях, когда к яркости и прочности окрасок не
предъявляются повышенные требования.
ФТАЛОЦИАНИНОВЫЕ КРАСИТЕЛИ
Фталоцианиновые красители были открыты только в 1927 г.
Важнейшим представителем фталоцианиновых красителей
является пигмент голубой фталоцианиновый. Один из методов его
получения заключается в нагревании карбамида с фталевым ангидридом
и однохлористой медью:
О
II
С NH4
,=О -і- CiiaClj »
Д карбамид
фталевыВ
ангидрид
пигмент голубой фталоцизнниовый
21—805 321
Пигмент голубой фталоцианиновый обладает ярким цветом,
нерастворим в воде, очень устойчив и благодаря этим ценным
свойствам находит широкое применение в полиграфической и других
отраслях промышленности.
Хлорированием голубого пигмента получают зеленый пигмент,
а сульфированием—растворимый в воде краситель прямой
бирюзовый светопрочный.
Известны фталоцианиновые красители во временно
растворимой форме—так называемые алцианы, превращающиеся в
процессе крашения в нерастворимые пигменты, а также активные
(стр. 305) фталоцианиновые красители.
Интересны фталогены—фталоцианиновые пигменты,
получаемые непосредственно на волокне при нагревании промежуточных
продуктов, например 1-амино-З-иминоизоиндоленина
NH
ОС >
NH2
с солью меди или никеля.
АРИЛМЕТАНОВЫЕ КРАСИТЕЛИ
Эта одна из старейших групп синтетических красителей
(открыты в 1855 г.), в настоящее время в значительной степени
утерявшая свое значение, особенно для текстильной промышленности,
в которой применяются более прочные красители других групп
(кубовые, антрахиноновые, фталоцианиновые, азокрасители).
Арилметановые красители—производные метана, в котором
два или три атома водорода замещены арильными остатками, чаще
всего фенильными группами, поэтому такие красители часто
называют ди- и трифенилметановыми. Незамещенные арилметаны
неокрашены, цвет появляется при введении в арильные остатки
ауксохромных окси- или аминогрупп и окислении полученных
лейкосоединений в кислой среде, например:
лейкофуксин (иеокрашен)
322
NH2
фуксин*
(сииевэти-красного цвета)
•сг
Для получения фуксина смесь анилина, о-толуидина и я-толуи-
дина окисляют нитробензолом в присутствии хлористого железа
и хлористого цинка при 200 °С.
Чаще арилметановые красители получают конденсацией
альдегидов с ароматическими аминами в присутствии водоотнимаю-
щих веществ (хлористый цинк, серная кислота и др.)- Так,
основной зеленый получают при длительном нагревании бензальдегида
и диметиланилина в присутствии хлористого цинка и
последующем окислении образовавшегося лейкосоединения:
ZnCls
N(CH3J
лейкосоедннение
основного зеленого
N(CH3J
основной зеленый
Основной зеленый, фуксин и ряд других арилметановых
красителей принадлежат к основным красителям, окрашивающим
белковые волокна без протрав, а целлюлозные с протравой танни-
ном**. Окраски очень ярки, но непрочны, поэтому основные
арилметановые красители почти не используются в текстильной
промышленности, однако их применяют в тех случаях, когда не
требуется высокая прочность окраски, например для окраски бумаги,
изготовления чернил, карандашей и т. д.
* Приведена упрощенная формула строения фуксина. Строение его—
см. И. М. Коган, Химия красителей, Госхимиздат, 1956.
** Таннин—продукт растительного происхождения, сложная смесь
продуктов взаимодействия глюкозы и галловой, лі-дигалловой и, возможно, тригал-
ловой кислот.
21*
323
Значительно более прочны основные лаки—соли красителей
с фосфорновольфрамомолибденовыми кислотами, например
Н7 [P(W2O7L(Mo2O7Ji; они ярки и успешно применяются в
полиграфии и для других целей.
Кислотные арилметановые красители отличаются от
основных наличием в молекуле красителя сульфогрупп. В качестве
примера приведем кислотный фиолетовый С:
Эти красители ярки, но большей частью мало прочны, они
применяются обычно для окраски кожи, бумаги, чернил и т. д.
АЗИНОВЫЕ, ТИАЗИНОВЫЕ И ОКСАЗИНОВЫЕ КРАСИТЕЛИ
(хиноннминовые красители)
Красители этой группы были открыты вскоре после арилмета-
новых A856 г.) и имеют много общего с ними. Они также очень
ярки, но большей частью непрочны, поэтому применяются
ограниченно и вытесняются более прочными красителями.
Азиновые, тиазиновые и оксазиновые красители содержат
гетероциклические группировки диазина (пиразина), тиазина и
оксазина:
N
нс Чсн
II 1
НС СН
\ //
гшпазин
NH
НС СН
II II
НС СН
V
тиазнн
NH
«С СН
II II
НС СН
V
оксазни
Наиболее многочисленны из них азиновые красители. К этой
группе относятся основные и кислотные красители и пигменты.
Представителем основных азиновых красителей является ярко-
красный сафранин, получаемый при окислении хромпиком смеси
анилина, о-толуидина и 2,5-диаминотолуола:
NH2 CH3 СН3
324
0
Сґ
Сафранин дает яркие, но непрочные окраски. Значительно
более прочны кислотные азиновые красители, например
кислотный темно-голубой
Очень прочен пигмент глубоко-черный, получаемый окислением
анилина. Окисление проводят также непосредственно на волокне,
тогда краситель называют анилиновый черный. Точное строение
пигмента глубоко-черного не установлено.
К этой же группе красящих веществ принадлежат красители
для меха. Это различные ароматические амино- и оксисоединения.
Их нанссят на мех и окисляют обычно перекисью водорода в
присутствии солей хрома, железа и других металлов, получая
окраски, имитирующие цвет натуральных мехов.
Тиазиновые красители применяются крайне ограниченно.
Наиболее известен метиленовый голубой
N
(СН3ЫЧ
очень яркий, но непрочный краситель.
Оксазиновые красители более важны, особенно ценны прямые
оксазиновые красители, например прямой ярко-голубой свето-
* К сафранину и другим хинониминовым красителям относится ранее
сказанное о строении арилметановых красителей (см. сноску на стр. 323).
325
ПрОЧНЫИ, Пил у чаем шм ГфН і-;ОНД'_и:_ЛШг>-
кислотой:
•SO.h
СІ
прямой ярко-голубой светопрочный
ОПТИЧЕСКИ ОТБЕЛИВАЮЩИЕ И ОСВЕТЛЯЮЩИЕ ВЕЩЕСТВА
Обычно белые тела имеют желтоватый оттенок, вызываемый
поглощением дополнительных голубых или синих лучей (стр. 287).
Применение оптически отбеливающих и осветляющих веществ
основано на их свойстве флуоресцировать, т. е. превращать
ультрафиолетовые лучи в видимые—голубые или синие, компенсируя
недостаток этих лучей в отражаемом телом свете; поэтому
желтизна оттенка исчезает, и тело кажется совершенно белым.
Известный прием «подсиньки» не дает такого же эффекта, подсиненный
материал имеет сероватый оттенок.
Оптические отбеливатели, открытые в 1929—1934 гг., нашли
в настоящее время широкое-применение. В текстильной
промышленности они используются не только для отбелки
натуральных и синтетических волокон, но и для повышения яркости
окрашенных тканей. Ими также отбеливают пластические массы,
резину, бумагу, мыло, кинопленку, но главным образом они
применяются в качестве добавок к моющим средствам.
Оптически отбеливающие вещества являются ароматическими
или гетероциклическими соединениями. Один из довольно старых
препаратов—прямой белый применяется для отбелки целлюлоз-
326
ных волокон. Его получают из диаминостильбендисульфокисло-
ты и фенилизоцианата
SO3H SO3H
4,4'-диаминостильбеп-2,2'-дисульфокислота фенилизоцианат
о о
SO3H HO3S
прямой белый
Более ценными отбеливающими свойствами и повышенной
светопрочнсстью обладает бланксфор Б, образующийся по схеме:
N
H2N—<{\—СН=СН— (f/—NH2+2C1—С С—СІ
С >
SO3H HO,S
Cl
цианурхлорид
N N
7 с—ын-'!;ГЛ-сн=сн— f~\—n\vL c-ci
NN \n „ m ( NN
С С
СІ СІ
о с
HN N Н Н N NH
+2CeH5NH2. н2о С XC-N-<f~^,-CH=CH-^^-N-C/ С
и і \ / і її
NN Чсо и нп / NN
х у ьи3н ми3ь х /
С . С
ОН ОН
бланк офор Б
327
Для отбелки шерсти применяется кислотный белый, получаемым
из бензоина и карбамида:
Н NH2
Известно большое число других отбеливающих веществ,
применяемых, в частности, для отбелки новых полимерных
материалов.
Продукция современной анилинокрасочной промышленности
по многообразию, яркости и прочности получаемых красящих
веществ далеко обогнала лабораторию природы; однако эта
отрасль химической промышленности продолжает бурно
развиваться. Изобретаются все более яркие, прочные и удобные в
применении красители. В связи с развитием производства полимеров
особое внимание уделяется синтезу красителей для прочной и
красивой окраски новых синтетических материалов.
3. ПОВЕРХНОСТНО-АКТИВНЫЕ ВЕЩЕСТВА
К поверхностно-активным относятся вещества, способные
адсорбироваться на границе раздела фаз гетерогенной системы
(жидкость—газ или пар, жидкость—жидкость,
жидкость—твердое тело) в виде тонкого слоя толщиной в одну или несколько
молекул.
Свойства поверхностно-активных веществ (ПАВ) находятся в
тесной зависимости от их специфического строения. Линейные
молекулы ПАВ, имеющие большую длину, состоят из двух
частей—гидрофильной, придающей ПАВ способность растворяться
в воде, и гидрофобной (отталкивающейся от воды).
Гидрофобной частью молекулы ПАВ являются остатки
алифатических или жирноароматических углеводородов с длинной
углеродной цепочкой, как известно, нерастворимых в воде. Гидро-
фильность обусловливается присутствием групп COO", SO*".
SOr и т. д.
328
Ионы, а также некоторые неионизированные группы и атомы,
в частности атомы кислорода, образуют с молекулами воды
водородные связи (см. сноску на стр. 302), в результате чего
поверхностно-активные вещества растворяются в воде.
Общеизвестными ПАВ являются мыла на основе природных
жиров (соли высших жирных кислот), например соли стеариновой
кислоты:
СН3—(СН2I4—СН2—COONa
гидрофобная часть
гидрофильная часть
Важнейшим свойством ПАВ является способность снижать
поверхностное натяжение на границе раздела фаз. В водном
растворе молекулы ПАВ располагаются на поверхности раздела
фаз гидрофильной частью
а сторону воды (рис. 106).
На этом рисунке
изображено строение молекул
ПАВ в виде
вертикальных линий с кружками
на конце (линия
обозначает гидрофобную часть,
кружок — гидрофильную).
Благодаря
ориентированному расположению
слоя ПАВ неводная фаза
(на рис. 106 воздух)
соприкасается уже не с водой, а
с гидрофобной частью
молекул. Поверхностное
натяжение гидрофобных
веществ (например,
углеводородов) значительно ниже, чем воды, поэтому добавление ПАВ
уменьшает поверхностное натяжение водного раствора.
Эта специфичность строения ПАВ и способность
адсорбироваться на поверхности раздела фаз обусловили их многие ценные
свойства: они являются хорошими моющими веществами (применяются
в быту и технике), способствуют смачиванию, диспергированию,
образованию эмульсий, предотвращая их расслаивание, и т. д.
Поверхностно-активные вещества широко применяются в
самых разнообразных областях народного хозяйства. Так, в тек-
гтильной промышленности ПАВ используются на всех стадиях
обработки волокон, пряжи и тканей в качестве моющих веществ,
для получения равномерной, яркой и прочной окраски изделий,
для придания тканям мягкости, водоотталкивающих свойств,
уменьшения сминаемости, усадки тканей и загрязняемости; при
прядении—для предотвращения возможности образования заря-
Г
ы
Тер
ас
Xh
db
ос
гт
ь
Уг
л
/
Группа
ж
дс
ч
W
13
%
ОС
Чп
X.
<л
4L
СООН
Вода
Рис. 106. Распределение молекул поверх
ностно-активного вещества на поверхности
раздела фаз вода—воздух.
329
ботке синтетических волокон. В кожевенной и меховой
промышленности ПАВ также используются как моющие, смачивающие,
диспергирующие средства.
В нефтяной промышленности поверхностно-активные вещества
добавляются в глинистый раствор при бурении скважин (стр. 23),
что облегчает удаление из них породы и способствует укреплению
стенок забоя. ПАВ находят также применение в качестве деэмуль-
гаторов при обезвоживании и обессоливании нефти (стр. 41),
в процессах очистки нефтепродуктов в качестве присадок.
Добавление ПАВ значительно улучшает свойства многих
материалов: красителей, лаков, пестицидов, фармацевтических
препаратов и т. д.
С помощью ПАВ можно ускорить многие химические реакции,
протекающие в гетерогенной среде. ПАВ нашли применение
также при эмульсионной полимеризации, флотационном обогащении
руд, предотвращении пылеобразования в шахтах, во многих
других процессах.
Области применения ПАВ непрерывно расширяются и
потребность в них увеличивается. В связи с этим возрастает объем
выработки ПАВ, например в 1956 г. в США было выработано
314 тыс. т синтетических поверхностно-активных веществ, а в
1963 г.—891 тыс. т.
В зависимости от строения поверхностно-активные вещества
делят на три основные группы:
1. Анионоактивные вещества, гидрофильная часть которых
образует в растворе анионы, чаще всего [СОО] М+ или
[SO3]~M+ (где М—Na, реже К и др.). К числу анионоактивных
веществ относятся мыла, смачиватель НБ (стр. 331).
2. Катионоактивные вещества, содержащие гидрофильные
группы—катионы, например препарат 101 (стр. 333).
3. Неионогенные вещества, гидрофильная часть которых не
образует ионов. Например, препарат ОП-10, являющийся смесью
нескольких соединений следующей общей формулы:
Л'
R-<f>-O(CH2CH2O)„H
гидрофобная гидрофильная
часть часть
где R—алкильный остаток линейного строения, содержащий
8—10 атомов углерода;
R'—алкильная группа или атом водорода;
п—в среднем 10.
Необходимо отметить, что в технике ПАВ обычно применяются
в виде смесей различных веществ, чаще гомологов или изомеров.
330
І акие смеси значительно дешевле индивидуальных веществ и
во многих случаях обладают более эффективным действием.
Лнионоактивные вещества. Старейшие анионоактивные
вещества—мыла—в настоящее время заменяются и дополняются
разнообразными синтетическими препаратами, обладающими
многими ценными свойствами.
К основным недостаткам мыл относятся: чувствительность к
жесткости воды; щелочная реакция раствора, что вызывает
разрушение некоторых волокон (шерсть, полиамид); невозможность
использования в кислых средах.
Кроме того, для получения мыл расходуется пищевое сырье
(растительные масла, жиры).
Синтетические анионоактивные вещества обычно не
чувствительны к жесткой воде и не дают щелочной реакции. Они обладают
моющими, диспергирующими, смачивающими и другими
специфическими свойствами и успешно применяются для различных
целей, обычно в щелочной среде.
Среди многочисленных анионоактивных препаратов
наибольшее значение в качестве моющего вещества имеет додецилбензол-
сульфонат (сульфонол).
Его получают сульфированием додецилбензола (стр. 152),
сульфокислоту нейтрализуют едким натром и высушивают. В
настоящее время применяется непрерывное сульфирование
додецилбензола, проводимое в двух последовательно соединенных сульфура-
торах из нержавеющей стали. В первый сульфуратор снизу
поступает додецилбензол и 5%-ный олеум. Поскольку в процессе
сульфирования выделяется большое количество тепла,
реакционная масса из первого сульфуратора охлаждается путем
циркуляции через холодильник.
Реакция сульфирования заканчивается во втором сульфура-
торе, откуда реакционная масса передается в аппарат для
разбавления водой, затем отстаивается, отделяется от разбавленной
серной кислоты и нейтрализуется 15%-ным раствором NaOH до
рН=7,5—8.
Технический додецилбензолсульфонат
содержит до 40% сульфата натрия в качестве наполнителя.
Додецилбензолсульфонаты (сульфонолы) благодаря невысокой
стоимости и доступности исходного сырья принадлежат к
наиболее распространенным синтетическим моющим веществам,
выпускаемым в СССР.
К группе алкиларилсульфонатов относится также смачиватель
НБ, получаемый путем нагревания смеси нафталина, бутилового
спирта и серной кислоты. При этом одновременно протекают реак-
331
ции алкили рования нафталина бутиловым спиртом и
сульфирования:
-I- С4Н9ОН
H2SO4
-4П9
бутилнафталин-
сульфокислота
Бутилнафталинсульфокислоту обработкой едким натром
превращают в натриевую соль.
В промышленных условиях получение смачивателя НБ
проводится в котле, оборудованном обратным холодильником,
рубашкой для нагревания и механической мешалкой. Смесь нафталина и
бутилового спирта нагревают до 80—85 вС и постепенно
прибавляют концентрированную серную кислоту. Реакционную смесь
перемешивают в течение 2 ч до тех пор, пока проба реакционной
массы не будет полностью растворяться в воде. По окончании
реакции бутилнафталинсульфокислоту отделяют от отработанной
серной кислоты отстаиванием, нейтрализуют продукт раствором
едкого натра, затем прибавляют гипохлорит натрия для
осветления раствора, отфильтровывают от механических загрязнений и
высушивают (в некоторых случаях смачиватель применяют в виде
влажного продукта).
Анионоактивный диспергатор НФ получают сульфированием
нафталина и последующей конденсацией смеси изомерных моно-
сульфокислот (в основном ?-изомер) с формальдегидом.
Образующийся продукт является смесью веществ, содержащих
от двух до девяти остатков нафталинсульфокислоты, соединенных
в а-положениях группой СН2. Димер имеет строение:
SO3Na
диспергатор НФ
Нафталин сульфируют концентрированной серной кислотой в
течение 8 ч при 130—165 *С, конденсацию с формальдегидом
проводят при 80—100 *С, продолжительность реакции 15—20 ч.
Избыток серной кислоты нейтрализуют гашеной известью,
отфильтровывают гипс, раствор сульфокислоты нейтрализуют едким
натром, упаривают и высушивают продукт,
332
Катионоактивные вещества. Эта группа
поверхностно-активных веществ обладает интересными специфическими свойствами,
которые обусловили их широкое применение в текстильной
промышленности. Катионоактивные вещества способствуют вырав
ниванию и упрочнению окраски, придают тканям несминаемость.
гидрофобность (водоотталкивающие свойства), облегчают удаление
с поверхности ткани статического электричества и т. д.
Примером катионоактивногоПАВ может служить препарат 101.
Его получают путем взаимодействия стеариновой кислоты с
метиламином:
С17Н35СООН + H2NCH3 > CI7H35CONHCH3
Образующийся N-метиламид стеариновой кислоты далее
реагирует с формальдегидом и солянокислым пиридином по
схеме:
Ci7H35CONHCH3 + НСНО
СН3
* Ci7H35CO—N—СН2—
препарат 101
Неионогенные ПАВ. Большую часть неионогенных
поверхностно-активных веществ получают взаимодействием окиси этилена
с различными гидрофобными соединениями—высшими спиртами,
алкилфенолами, высшими карбоновыми кислотами и некоторыми
другими.
При взаимодействии таких соединений с окисью этилена
образуется смесь продуктов, отличающихся числом присоединенных
остатков окиси этилена. Например, препарат леониль ОХ,
синтезируемый из октадецилового спирта и окиси этилена, содержит
в среднем 15 таких остатков:
С18Н37ОН + 15Н2С СН2 » С18Н37О—(CH2CH2O)i4—СН2СН2ОН
О леониль ОХ
При синтезе леониля ОХ 800 кг октадецилового спирта и 80 г
@,01%) едкого натра нагревают до 160—180 *С, создают в котле
вакуум и затем заполняют аппарат азотом (инертная среда
необходима, так как смесь окиси этилена с воздухом взрывоопасна).
Постепенно, в течение 15 ч, через слой жидкости пропускают
газообразную окись этилена. Реакция сопровождается выделением
тепла. Процесс оксиэтилирования заканчивают, когда
полученный продукт будет полностью растворяться в воде, а при
нагревании раствора до 40 °С—мутнеть (растворимость неионоген-
333
ных ПАВ в воде при повышении температуры обычно
уменьшается). Леониль ОХ применяют в виде 30%-ного водного
раствора.
В Советском Союзе выпускают аналогичный препарат ОС-20,
представляющий собой продукт взаимодействия смеси высших
жирных спиртов С;6—С,8 с 20 моль окиси этилена.
Большую группу неионогенных ПАВ составляют препараты
ОП-4, ОП-7, ОП-10 и ОП-20. Синтез препаратов ОП включает
две основные стадии. Сначала фенол алкилируют полимердистил-
лятом—смесью олефинрв, получаемых полимеризацией бутиленов
и амиленов из газов крекинга нефти (стр. 28). В результате алки-
лирования образуется смесь моно- и диалкилфенолов:
ОН ОН ОН
J
0
J Ч
<U„„ ін,„,
где n=8 и 10.
Затем смесь алкилфенолов взаимодействует с окисью этилена,
причем при синтезе ОП-4 на 1 моль алкилфенола берут 4 моль
окиси этилена, для ОП-7, ОП-10 и ОП-20—соответственно 7, 10
и 20 моль:
О(СН2СН2О)тН
где т—соответственно 4, 7, 10 и 20.
Получение ОП-10. Первую стадию—алкилирование фенола
проводят следующим образом. В эмалированный котел,
снабженный греющей рубашкой, загружают фенол и бензолсульфокисло-
ту (катализатор). Смесь, нагревают до 80 °С, вытесняют воздух
азотом и приливают в течение примерно 5 ч полимердистиллят.
Затем реакционную массу нагревают до 110 °С и перемешивают
еще 2 ч. По окончании реакции из смеси экстрагируют горячей
водой бензолсульфокислоту.
После отстаивания смеси в делительной воронке сливают
нижний водный слой. Затем для удаления не вступивших в
реакцию олефинов к смеси добавляют небольшое количество
раствора едкого натра и отгоняют в вакууме воду, олефины и другие
примеси при температуре до 120 °С.
Вторую стадию—оксиэтилирование—проводят при
температуре около 160 °С, аналогично описанному выше процессу
получения леониля ОХ, при молярном соотношении алкилфенола и окиси
334
этилена, равном 1 : 10. После завершения реакции аппарат
продувают азотом для удаления токсичной окиси этилена,
реакционную массу охлаждают, подкисляют, если это необходимо, уксусной
кислотой до рН = 7—7,5 и сливают в тару.
Неионогенные ПАВ нечувствительны к жесткости воды, они
могут применяться как в кислой, так и в щелочной среде в
качестве смачивающих, моющих, эмульгирующих веществ, для
снятия статического электричества (в текстильной
промышленности), как выравниватели цвета при крашении и для других
целей.
Недостатком препаратов ОП и некоторых других
поверхностно-активных веществ является их устойчивость к
микроорганизмам, что, в связи с широким применением синтетических моющих
средств в быту и промышленности, приводит к загрязнению
водоемов. Поэтому в настоящее время во всех странах проводятся
изыскания ПАВ, окисляемых микроорганизмами, для замены так
называемых биологически неразлагаемых препаратов типа ОП.
4. ОРГАНИЧЕСКИЕ ПЕСТИЦИДЫ
Пестицидами называются вещества, применяемые для борьбы
с сорняками, болезнями и вредителями растений и животных, а
также используемые в здравоохранении и в целях защиты
продуктов питания и неметаллических материалов от вредных
насекомых и грызунов. В зависимости от назначения пестициды*
разделяются на следующие основные группы:
1) гербициды, с помощью которых уничтожают сорняки или
подавляют их развитие. К гербицидам близки по свойствам
дефолианты, удаляющие листву, и десиканты, вызывающие
подсыхание растений, а также регуляторы роста и плодоношения
растений;
2) инсектициды—препараты для уничтожения насекомых. По
назначению к ним близки репелленты, отпугивающие насекомых;
3) акарициди—средства для уничтожения клещей;
4) фунгициды, применяемые для борьбы с грибными и другими
заболеваниями растений;
5) зооциды, употребляемые для уничтожения грызунов.
Многие пестициды используются для различных целей,
например для борьбы с вредными насекомыми, клещами и грибными
заболеваниями. Применяются также смешанные пестициды; так,
для протравливания семян часто употребляются смеси
фунгицидов и инсектицидов.
Применение пестицидов позволяет резко снизить ущерб,
наносимый вредителями растений, и избежать огромных потерь
* Другие названия пестицидов—сельскохозяйственные ядохимикаты,
химические средства защиты растений.
335
сельскохозяйственной продукции. Ранее наблюдавшееся массовое
распространение вредителей растений, например саранчи, клопа -
черепашки, в ряде случаев приводило к полной гибели растений на
больших площадях. Однако и в настоящее время мировые поте
ри урожая от вредителей и болезней растений оцениваются
суммой в 30 млрд. долл. в год; велик также ущерб, причиняемый
распространением сорняков.
Использование гербицидов, дефолиантов и десикантов дает
возможность механизировать прополку урожая, а также
уничтожить сорную растительность вдоль дорог, на строительных
площадках, в каналах и т. п. Уничтожение вредных насекомых,
грызунов и клещей химическими средствами позволяет предотвратить
распространение тифа, малярии, дифтерии и других опасных
болезней.
Первоначально в качестве пестицидов применялись преиму
щественно неорганические вещества—соединение мышьяка, ртути,
меди и др. С 30-х годов текущего столетия начали проводиться
интенсивные исследования в области синтеза и испытаний
органических пестицидов, которые в последние десятилетия
приобрели наибольшее значение. Органические пестициды более
эффективны и экономичны, так как расходуются в небольших
количествах (часто 1—10 кг и менее на 1 га посевов), многие органические
пестициды обладают избирательным действием, например
поражают сорняки, но практически безвредны для культурных
растений. Многообразие растительного и животного мира (только
насекомых существует свыше 750 000 видов), различия климата,
почв и других условий вызывают необходимость применения
широкого ассортимента пестицидов, который к настоящему времени
насчитывает несколько сотен препаратов и быстро пополняется
новыми все более эффективными веществами. Большое внимание
уделяется получению пестицидов избирательного действия,
препаратов, мало токсичных для человека и домашних животных, а
также пестицидов, не накапливающихся в почве.
Пестициды применяются в виде эмульсий, суспензий,
растворов, тонкоизмельченных порошков (дусты), в виде гранул, газов
я паров (фумиганты, стр. 350), аэрозолей. Все они должны
хорошо распределяться на обрабатываемой поверхности и
удерживаться на ней. Растворами, эмульсиями и суспензиями
пестицидов опрыскивают растения и почву, порошки служат для опыли-
вания, гранулы вносят в почву. Водорастворимые пестициды
обычно выпускаются в виде порошков, однако большая часть
пестицидов плохо растворима в воде, поэтому часто их выпускают
в виде так называемых смачивающих порошков, которые при
размешивании с водой легко образуют устойчивые суспензии.
В состав таких суспензий входят высокодисперсные пестициды
(величина частиц 1—5 мк), поверхностно-активные вещества
(стр. 328), придающие суспензии устойчивость и хорошую смачи-
336
наемость, а также вещества, улучшающие прилипание
пестицидов к листьям растений (карбоксиметилцеллюлоза, клей и т. д.),
и наполнители (каолин, тальк, силикагель и др.)-
Порошкообразные инсектициды—дусты тоже содержат тонко молотые
наполнители, что облегчает внесение необходимой минимальной дозы
пестицида, способствует равномерному распределению на
обрабатываемой поверхности и улучшают адгезию (прилипание)
препарата.
Пестициды, применяемые в виде эмульсий, растворяют сначала
н органических растворителях (например, в нефтепродуктах) и
смешивают с эмульгаторами (препарат ОП-10 и др., стр. 334).
Перед употреблением такой раствор разбавляют водой, при этом
образуется водная эмульсия пестицида.
В последние годы все большее применение находят
гранулированные пестициды (размеры гранул обычно от 0,1 до 1,5 мм).
Не пылящие при разбрасывании гранулы в ряде случаев более
эффективны и обладают более избирательным действием, чем дусты.
Гранулированные пестициды содержат дешевые наполнители
(каолин и др.) и связующие вещества, необходимые для
образования гранул.
Аэрозольный способ применения пестицидов заключается в
превращении их в дымы или туманы. Дымы получаются при
нагревании пестицидов, изготовленных, например, в виде дымовых
шашек, которые содержат, кроме пестицида, горючее, окислитель
(чаще всего хлорат калия) и дымообразующие вещества
(например, тиомочевину). Туманы образуются в результате механического
рассеивания растворов пестицидов специальными форсунками.
В ряде случаев к пестицидам добавляют вещества,
усиливающие их действие,—так называемые синергисты.
В связи с широким распространением химических средств
защиты растений необходимо учитывать их технические свойства,
чтобы избежать вредных последствий для здоровья людей и для
полезных флоры и фауны. При производстве и использовании
пестицидов следует строго выполнять правила обращения с ними
и точно соблюдать регламентированные условия и дозы
применения ядохимикатов для борьбы с вредителями
сельскохозяйственных культур.
Важнейшими мероприятиями, обеспечивающими нормальные
и безопасные условия работы в производстве ядохимикатов
являются: ведение процессов в строгом соответствии с технологическим
регламентом, тщательная герметизация аппаратуры и
коммуникаций, устройство мощной приточно-вытяжной вентиляции,
снабжение работающих спецодеждой, спецобувью, индивидуальными
средствами зашиты (очки, респираторы, противогазы и др.),
молоком и жирами (противоядия от многих вредных веществ), а
также постоянный врачебный надзор за состоянием здоровья
работающих и санитарными условиями производства,
•22—805 337
і іНЬііцИДЬі
Наибольший ущерб сельскому хозяйству причиняет
распространение сорняков; так, в США ежегодные потери урожая от
сорняков оцениваются суммой 4 млрд. долл. Применение
гербицидов для уничтожения сорной растительности во много раз
производительнее ручной прополки, например один самолет
обрабатывает гербицидами за 1 ч более 100 га. Борьба с сорняками при
помощи гербицидов является одним из важнейших элементов
современного высокопроизводительного земледелия.
Различают гербициды сплошного действия, уничтожающие
всю растительность, и избирательные гербициды, поражающие
одни растения, но безопасные при правильной дозировке для
других растений. Гербициды сплошного действия применяются для
уничтожения растительности на железнодорожных путях,
обочинах шоссейных дорог, строительных площадках и т. д., а также
для борьбы с водяными сорняками в каналах и ирригационных
сооружениях. В сельском хозяйстве гербициды сплошного
действия используются преимущественно для уничтожения сорняков
на полях под паром с тем, чтобы ко времени посева культурных
растений гербицид отсутствовал в почве. Метод «направленного»
применения гербицидов сплошного действия используют,
например, для уничтожения сорняков в междурядьях на посевах
хлопчатника. В последние годы все больше вводится в практику
«химическая пахота»—уничтожение дерна гербицидами, не
нарушающее структуру почвы.
Избирательные гербициды находят наибольшее применение в
сельском хозяйстве. Особенно ценны избирательные гербициды
широкого действия, уничтожающие многие виды сорняков, но
безопасные для культурных растений. При увеличении норм
расхода почти все избирательные гербициды становятся гербицидами
сплошного действия.
По характеру действия различают контактные гербициды,
поражающие растения только в местах непосредственного
соприкосновения с ними, и системные гербициды, перемещающиеся по
сосудистой системе растений. Известны также почвенные
гербициды корневого действия, действующие на семена, проростки и
корни растений.
Первоначально в качестве гербицидов применялись хлораты
натрия и магния, арсенат и арсенит натрия и другие
неорганические вещества, а также нефтяные масла. Эти гербициды обладают
преимущественно контактным действием и мало избирательны.
Нормы их расхода достигают нескольких сот килограммов на
1 га обрабатываемой площади.
Многочисленные органические гербициды, синтезированные в
течение последних 20 лет, обладают избирательным действием и
более экономичны (нормы расхода 0,25—10 кг/га), что сделало
338
возможным их широкое применение в сельском хозяйстве.
Органические гербициды большей частью токсичны для теплокровных
и в ряде случаев могут переходить из растений в пищевые
продукты. Это следует учитывать при выборе сроков внесения и других
условий применения гербицидов. Большой интерес представляют
гербициды, относительно быстро разлагающиеся в почве, которые,
в отличие ст более устойчивых, не мешают росту растений в
последующие годы, при смене культур. Для применения вне
сельского хозяйства (например, для уничтожения растительности на
дорогах) иногда необходимы устойчивые гербициды длительного
действия.
Важнейшие гербициды принадлежат к следующим группам
органических соединений:
1) хлорированные алифатические кислоты;
2) амиды карбоновых кислот;
3) нитро- и хлорфенолы;
4) арилоксикарбоновые кислоты;
•5) производные карбамида;
¦6) симметричные триазины;
7) эфиры карба.мииовой кислоты;
8) производные тио- и дитиокарбаминовой кислоты.
Хлорированные алифатические кислоты. Среди гербицидов
этой группы наибольшее значение приобрели натриевые соли
трихлоруксусной кислоты CCl3COONa (тр их лор ацетат натрия
ТХА) и а,а-дихлорпропионовой кислоты CH3CCl2COONa
(далапон). Они хорошо растворимы в воде и уничтожают
преимущественно злаковые сорняки; далапон является системным гербицидом,
ТХА обладает контактным действием. В больших дозах эти
препараты используются как гербициды сплошного действия.
Трихлоруксусную и дихлорпропионовую кислоты получают
хлорированием соответственно уксусной и пропионовой кислот
при температуре 170—175 °С под давлением, в присутствии
катализаторов (РС13, иод и др.). В результате хлорирования
образуются хлорангидриды трихлоруксусной и дихлорпропионовой
кислот, которые при действии разбавленной щелочи превращаются
в соли, например:
NaOH
ССІ3СОСІ > CCl3COONa
— N3.CI
Процессы хлорирования проводят как периодическими, так и
непрерывными способами.
Амиды карбоновых кислот. В этой группе гербицидов большое
значение имеет Ы,Ы-диаллилхлорацетамид (рэндокс), получаемый
действием хлорацетилхлорида на диаллиламин;
ClCH.COCl + Ш(СН2—СН=СНаJ —н> C1CH?CON(CH2—СН=СНа)8 + HCJ
22* 339
В ПРИСУТСТВИИ ВОДЫ ХЛОраЦеТЙЛХЛОрИД ОЫСТри
поэтому реакцию проводят в среде органического растворителя,
например дихлорэтана, при температуре —10 °С. В охлажденный
раствор диаллиламина в дихлорэтане при энергичном
перемешивании вливают хлорацетилхлорид, реакция заканчивается очень
быстро. Затем добавляют небольшое количество воды, отделяют
раствор рэндокса в дихлорэтане от водного слоя, содержащего
НС1, промывают раствором NaCl и отгоняют в вакууме сначала
дихлорэтан, потом рэндокс.
Рэндокс—почвенный гербицид, поражающий семена многих
сорняков, особенно злаковых. Он ценен тем, что быстро разла
гается в почве, благодаря чему в ней не остается токсичных
веществ.
Нитро- и хлорфенолы. Гербициды этой группы стали приме
няться ранее других синтетических гербицидов (в середине 30-*
годов). Они обладают контактным действием и токсичны главным
образом для широколистных растений. Наибольшее значение
имеют 2-метил-4,6-динитрофенол (динитро-о-крезол, ДНОК, ди
нок), 2-втор-бутил-4,6-динитрофенол (ДНБФ, диносеб) и пента
хлорфенол. Обычно эти гербициды применяются в виде
водорастворимых натриевых, аммонийных и других солей.
Препарат ДНОК получают нитрованием о-крезола азотной
кислотой в присутствии серной кислоты:
ДНОК применяется не только как гербицид, но и в качестве
инсектицида (для опрыскивания садовых культур).
Диносеб получается в результате нитрования o-emop-бутилфе-
нола:
HNO3 2
Haso4
По гербицидному действию диносеб превосходит ДНОК
примерно в два раза.
340
Наиболее рациональным методом синтеза пентахлорфеноло/
является гидролиз гексахлорбензола в присутствии едкого натра:
С1 ONa
NaOH
Cl Cl
пентахлорфеноля і
натрия
Процесс проводится под давлением при температуре около
250 °С. Исходный гексахлорбензол целесообразно получать из
отходов производства гексахлорана—нетоксичных изомеров гекса-
хлорциклогексана (стр. 192), образующих при нагревании в
присутствии FeCl3 трихлорбензол, который далее хлорируют до гек
сахлорбензола.
Пентахлорфенол используется как гербицид, фунгицид и анти -
септик.
Нитро- и хлорфенолы токсичны для человека и животных „
ДНОК и диносеб также взрыво- и пожароопасны, поэтому их часто
применяют в смеси с инертными наполнителями, например
сульфатом натрия.
Арилоксиалкилкарбоновые кислоты. Гербицидным действием
обладают арилоксиалкилкарбоновые кислоты, содержащие в арома
тическом ядре атомы хлора. Это рчень активные системные гер
бициды, даже в малых дозах @,5—1,5 кгіга) поражающие двудоль
ные сорняки в посевах зерновых и других культур.
Наибольшее применение находит 2,А-дихлорфеноксиуксусна^
кислота B,4-Д). В промышленности ее получают двумя способами
По одному из них сначала проводят реакцию между хлоруксуснок
кислотой и фенолятом натрия в концентрированном водном
растворе в присутствии едкого натра и при нагревании:
ONa • OCHjCOONa
NaOH >N>
-f CICHjCOOH -»¦ Г I - NaQ
Затем образовавшуюся натриевую соль феноксиуксусной
кислоты хлорируют, пропуская в раствор хлор при 30—40 °С и pH
раствора около 8 (для поддержания этой величины pH в. раствор,
добавляют едкий натр):
OCH2COONa OCH2COONa
4/ ч
4-Д
341
По второму способу сначала хлорируют фенол до 2,4-дихлор-
фенола, который затем взаимодействует с хлоруксусной кислотой:
ОН ОН OCH2COONa
СІ І СІ
+CICHaCOOH+NaOH ' '
Ч
сі
Реакцию 2,4-дихлорфенола с хлоруксусной кислотой проводят
в концентрированном водном растворе при 60—80 °С и pH
реакционной массы около 10 (для чего добавляют раствор едкого натра).
В реакцию вводят избыток дихлорфенола для более полного
использования ценной хлоруксусной кислоты. По окончании
процесса в раствор добавляют соляную кислоту, доводя pH раствора
до 5. В этих условиях натриевая соль дихлорфенола превращается
в дихлорфенол (кислоту), который отгоняют с водяным паром и
снова возвращают в процесс. Очищенный от дихлорфенола раствор
натриевой соли 2,4-Д подкисляют соляной кислотой до рН=1,
при этом соль выпадает в осадок, который затем
центрифугированием отделяют от жидкости. Этот способ синтеза 2,4-Д проще,
но получается менее чистый гербицид, небольшая примесь
остающегося в нем 2,4-дихлорфенола придает препарату неприятный
запах.
Ценными гербицидами этой группы являются: 2-метил-4-хлор-
феноксиуксусная кислота BМ-4Х, метоксон), более пригодная,
чем 2,4-Д, для обработки посевов риса и льна, а также 2,4,5-mpu-
хлорфеноксиуксусная кислота B,4,5-Т), рекомендуемая, в
частности, для уничтожения кустарников на лугах, и некоторые
другие препараты.
Гербицид 2М-4Х получают аналогично препарату 2,4-Д,
заменяя фенол о-крезолом, который взаимодействует с
хлоруксусной кислотой и далее с хлором по схеме:
ОН OCH2COONa OCH2COOH
j + С1СН2СООН ^ V
При синтезе гербицида 2,4,5-Т проводят реакцию между
1,2,4,5-тетрахлорбензолом и едким натром.
¦U42
Образующийся 2,4,5-трихлорфенолят натрия ацилируют
хлоруксусной кислотой:
С1 ONa OCH2COONa
СІ ' СІ СІ
NaOH <^\/ СІСН2СООН, NaOH ^v '
СІ
Гербициды типа 2,4-Д плохо растворимы в воде. Обычно они
применяются в виде водорастворимых солей или зфиров, почти
нерастворимых в воде, но хорошо растворимых в органических
растворителях. Более активны соли, образованные
алифатическими аминами—триэтиламином N(C2Ha)g, триэтаноламином
N(C2H4OHK и др. В качестве примера приведем строение соли,
образуемой триэтаноламином и 2М-4Х:
OCHjCOO" HN+(C2H4OHK
Менее активны, но дешевле натриевые соли. Эфиры в 2—3 раза
активнее солей и отличаются более широким гербицидным
действием. Недостатком зфиров низших спиртов (этилового, изопропи-
лового, бутилового) является их летучесть, которая при
повышенной температуре может привести к поражению некоторых культур,
произрастающих вблизи от обрабатываемых гербицидом участков.
Менее летучи и более удобны при хранении зфиры высших
спиртов (изооктилового, бутоксиэтилового и др.).
Бутоксиэтиловый зфир 2,4-Д имеет следующее строение:
ОСН2СООС2Н4ОС4Н9
.С1
Эфиры получают нагреванием арилоксиуксусных кислот с
соответствующими спиртами в присутствии катализаторов,
например, ионитов, содержащих сульфогруппы. Выделяющаяся
в ходе реакции вода отгоняется вместе с избыточным
спиртом или другим органическим растворителем (дихлорэтаном и др.).
343
Препарат 2,4-Д и другие хлорарилоксиуксусные кислоты а
небольших дозах стимулируют рост растений и применяются,
например, для ускорения созревания томатов.
Производные карбамида. Среди производных карбамида
найдены ценные гербициды, токсичные для многих сорняков.
Наибольшее значение имеют несимметричные М-алкил-ІМ'-арилпроизводньїе
карбамида—фенурон. монурон, диурон, небурон:
_МН_ СО—N(CH3k
Фенурон
-NH- CO—WCH-,),
монурон
СІ— (' ^—NH—CO—ї>
небурон
Гербициды, являющиеся производными карбамида, вносятся
u почву и проникают в растения через корни. Эти препараты
довольно длительно A—2 года) сохраняются в почве. В малых доза*
они уничтожают многие однодольные и двудольные сорняки, Е
том числе многолетние.
В больших дозах поражают всю растительность и успешно
применяются для уничтожения растений с глубокими корнями.
я также растительности на дорогах, строительных площадках
и т. д.
Синтез гербицидов этой группы проводят путем
взаимодействия ароматических изоцианатов с алифатическими аминами.
Например, для получения монурона растворяют л-хлоранилин в
безводном хлорбензоле, насыщают раствор сухим НС1 при 75—
90 °С и пропускают сухой фосген.
По окончании фосгенирования растйор образовавшегося изо
цианата продувают азотом для удаления непрореагировавшего
фосгена, охлаждают до 10 °С, прибавляют раствор диметил-
амина в хлорбензоле, нагревают до 55 °С и перемешивают в
течение часа. В результате образуется монурон [Ы-диметил-Ы'-(л-хлор-
бензол)-карбамид]:
СІ—
—NH—CO—N(CH,b
CI—<^^>-N=CO -і- HN(CH3)a
і-хлорфенилнзоцютат
Затем отгоняют большую часть хлорбензола, остаток
охлаждают, при этом монурон выпадает в осадок, который отфильтро
еывают.
Симметричные триазины. Производные симметричного триази-
на уже свыше 30 лет применяются в качестве красителей.
Ценные гербицидные свойства этих веществ были открыты только
в 1952—1955 гг. Наибольшее значение в качестве гербицидов
получили монохлоралкилзамещенные силш-триазина—симазин.
344
атразин и пропазин:
СІ СІ.
А \
Н N і/ N
І II І II
H6Cj—HN—С С—NH—СгН, HsCj—HN—С С—NH—GH(CHi,h-
V V
Симазив «травив
сі
І
с
f
—HN—C
<СН3JНС—HN—C С—NH—СШСНоЬ
V
пролазив
Эти гербициды эффективно применяются для уничтожения раэ
личных сорняков в посевах кукурузы. Атразин, несколько лучше
растворимый в воде, рекомендуется для засушливых районов и
уничтожения горняков с глубокими корнями. Плохо растворимые
симазин и пропазин дольше удерживаются во влажной почве.
Гербициды этой группы получают взаимодействием цианур-
хлорида с соответствующими алифатическими аминами, при
мольном соотношении реагентов 1 : 2. Реакцию проводят в водной
среде, причем вначале при температуре 0—5 °С вводят в реакцию
1 моль амина, затем при 20—30 °С еще 1 моль. Выделяющуюся
соляную кислоту нейтрализуют содой или едким натром.
В качестве примера приведена схема синтеза атразина:
С1 С1
А А
N N H2N-CH(CH3J N N HsN-c2h6
С С =™ ~* С С -HCI
цванурхлорвд
С1
і
1 я
HsQi—HN—С С—NH—CH(CH3J
V
атразии
346
Цианурхлорид очень плохо растворим в воде, поэтому для
введения в реакцию его предварительно тонко измельчают и
энергично перемешивают реакционную массу.
Эфиры арилкарбаминовой кислоты. Наибольшее применение
из гербицидов этой группы находят изопропил-1Ч-фенилкарбамат
(ИФК, изопропиловый зфир фенилкарбаминовой кислоты), изо-
пропил-М-C'-хлорфенил)-карбамат (хлорИФК) и 4-хлор-2-бути-
нил-1М-C'-хлорфенил)-карбамат (карбин, барбан, хлоринат):
О С1Ч О
) —NH-CO—CH(CH3J <f>-NH-CO-CH(CH3J
ИФК хлорИФК
С1Ч О
-CH2CsCCH2Cl
карбин
Препараты ИФК и хлорИФК—почвенные гербициды,
уничтожающие преимущественно однодольные сорняки. В почве ИФК
довольно быстро разлагается, особенно при повышенных
влажности и температуре. ХлорИФК дольше сохраняется в почве, а
по гербицидному действию превосходит ИФК в несколько раз и
потому находит большее применение. Карбин обладает узко
избирательным действием, поражая устойчивый к другим
гербицидам овсюг в посевах пшеницы, ячменя и других культур.
Для получения ИФК проводят реакцию между фосгеном
и изопропиловым спиртом; образующийся при этом изопропи-
ловый зфир хлоругольной кислоты далее взаимодействует с
анилином:
(СН3JСН—ОН + СОС12 ¦¦ (СН3JСН—О—СОС1 + H2N-
--НС1
о
» <f~^-NH-CO-CH(CH3J '
Заменяя анилин 3-хлоранилином, получают хлорИФК.
другой способ получения ;фиров карбаминовой кислоты осно-
вчц па взаимодействии арилизоцианатов (стр. 344) со спиртами,
например:
С1 С1 О
N=CO + (СН3JСНОН »
Преимущество этого способа синтеза—отсутствие токсичных
сточных вод.
Карбин (хлоринат) получается действием 3-хлорфенилизоциа-
ната на бутин-2-диол с последующим замещением в оСразовавшем-
346
ся зфире карбаминовой кислоты гидроксила хлором путем поз-
действия тионилхлорида:
О
її
N=CO HN—CO—CH2CsCCH2OH
I I
HOCH2C = CCH2OH
О О
II II
HN—CO—CH2C=CCHaOH HN—CO—СН2С==ССНаС|
•СІ
При взаимодействии бутиндиола с хлорфенилизоідианатом частично
образуется бис-арилкарбамат следующего строения:
О О „СІ
ІІ !!
—NH—СО—СНаС=ССН2—CO--NH—і
Производные тио- и дитиокарбаминовой кислот. В числе этих
соединений имеются ценные гербициды избирательного действия,
а также стерилизаторы почвы, уничтожающие семена сорных
растений и обладающие свойствами фунгицидов, инсектицидов и
нематоцидов*. Стерилизатором почвы является, например, N-ме-
тилдитиокарбамат натрия (карботион, вапам), получаемый
действием сероуглерода на водный раствор метиламина в присутствии
едкого натра при 20—40 °С:
S
II
CH3NH2 + CSa Н- NaOH > СН,—NH—С—SNa
карботиои
Во влажной почве карботион разлагается с образованием изо-
тиоцианата СН3—N=C=S, пары которого уничтожают грибы,
бактерии, нематод, насекомых, а также растения и их, семена.
Этот препарат вносят в почву не позднее чем за недели?-, до сева,
к началу которого он успевает разложиться, и в почве не остается
токсичных для растений веществ.
Избирательными почвенными гербицидами являются эптам и
авадекс. Эптам (этил-Ы,Ы-дипропилтиокарбамат) получают фос-
генированием дипропиламина с последующим взаимодействием
* Средства для уничтожения круглых червей (нематод).
447
Образовавшегося ДИПрО1Ш.Лкариаминии,дл.дс/}7ида и
тана:
О
СОСІ2 +HSC2H6 II
(C3H7JNH * (C3H,)aN—COCl - (C,H7)aN—С—S—С„Н,
эптам
Авадекс E-2,3-дихлораллил-Ы,Ы-диизопропилтиокарбамат)
синтезируют по аналогичной схеме из диизопропилкарбаминоил-
хлорида и 2,3-дихлораллилмеркаптана-1:
О
(СН3JСН ||
}N—CC1 + HSCH2CC1=CHQ *
(CHgJCH
О
(СН,JСНЧ II
\n—С—SCHaCCl=CHCl
авадекс
Некоторые свойства и области применения описанных
гербицидов приведены в табл. 17.
Таблица 17
Свойства и области применения некоторых гербицидов
Гербициды
ТХА
Далапон
Рэндокс
днок
Диносеб
Физическое
состояние
при 20 °С
Твердое
То же
Жидкое
Твердое
То же
Токсичность
для человека
и животных
Действует на
кожу
Мало токсичен
Токсичен
Сильно токсичен
(взрыва- и
пожароопасен)
То же
Действие
на растения
Избирательное
на однолетние
травы и
пырей;
сплошное—в
больших дозах
Избирательное
на злаковые
сорняки,
сплошное—в
больших дозах
Избирательное
на травы
Избирательное
на двудольные
сорняки
То же
Защищаемые культуры
Сахарная свекла, хлоп
чатник, картофель
Сахарная свекла, хлоп
чатник, леи
Кукуруза, ячмень, лен
сахарная свекла,
хлопчатник, овош-
. ные культуры
Пшеница, картофель
Садовые культуры
(инсектицид)
Пшеница, соя, фасоль
кукуруза
Хлопчатник
(дефолиант)
Картофель (десикант)
Продолжение табл. 17
Гербициды
Лентахлорфенол
¦2,4-Д
2М-4Х
2,4,5-Т
Фенурон
Монурон
Диурон
Небурон
Симазин
Атразин
Пропазин
Кароин (хло-
ринат)
ИФК
ХлорИФК
Карботион
Эптам
Авадекс
Физическое
состояние
при 20 °С
Твердое
То же
,
і
і
і
t
і
•
Жидкое
То же
Токсичность
для человека
и животных
Токсичен
То же
1
>
Слабо токсичен
То же
і
>
Токсичен
Токсичен
ъ
Мало токсичен
То же
Токсичен
То же
Мало токсичен
Токсичен
Действие
иа растения
Спло шное
Избирательное
на двудольные
сорняки
То же
То же и
уничтожение кус-
mrt ГЧІ і ЖЖ ІУ Ґ\ҐІ
TdpHHKUD
Избирательное;
сплошное—в
больших дозах
То же
•
>
Избирательное;
сплошное—в
больших дозах
Избирательное
Избирательное
на овсюг
Избирательное
на
однодольные сорняки
То же
Сплошное
Избирательное
Избирательное
на овсюг и др
Защищаемые культури
Многолетние травы, соя
(инсектицид,
фунгицид, дефолиант)
Пшеница, ячмень, овес,
просо, кукуруза, рис
То же и лен
Травы (на лугах,
пастбищах)
Хлопчатник, кукуруза,
пшеница, соя
Хлопчатник, картофель,
виноград
То же
Горох, соя, томаты,
виноград
Кукуруза, томаты,
виноград
Кукуруза
К\'куруза, просо
Овес, пшеница, горох,
ячмень, лен,
сахарная свекла, картофель
Овощные культуры
Хлопчатник,
подсолнечник, овощные
культуры, горох
Стерилизация почвы в
парниках (фунгицид,
инсектицид, немато-
ЦИД)
Сахарная свекла,
хлопчатник,
подсолнечник, люцерна,
картофель
Сахарная свекла,
ячмень, хлопчатник,
подсолнечник,
пшеница, овес, горох
349
діі<4>иЛИАНі Ьі и ДЬСігіічАіі і і»
Обработка растений дефолиантами вызывает опадение листьев,
благодаря чему ускоряется созревание растений и облегчгется их
уборка. Наибольшее значение имеет дефолиация для возде/ывания
хлопчатника, позднее созревание которого приводит к
затягиванию уборки и в конечном итоге—к снижению урожая. Особенно
необходима дефолиация при механизированной уборке хлопка,
поскольку наличие листьев затрудняет работу хлопкоуборочных
машин.
Десиканты применяются для высушивания растений на корню.
Благодаря этому ускоряется, например, раскрытие недозрелых
коробочек хлопчатника. Дефолиация и десикация применяются
также для облегчения механизированной уборки и ускорения
созревания семенных посевов сахарной свеклы, клевера, люпина,
люцерны, особенно в северных районах и зонах избыточного
увлажнения. В последнее время десиканты начинают применяться при
обработке посевов риса, ячменя, пшеницы, кукурузы.
В качестве дефолиантов и десикантов используются хлорат
магния Mg(ClO3J, цианамид кальция CaCN2, описанные выше
гербициды пентахлорфенол и диносеб, а также трибутилтисфос-
фат (бутифос), бис-этилксантогентрисульфид (БЭКТ) и некоторые
другие вещества.
Бутифос получают взаимодействием бутилмеркаптана с хлор-
окисью фосфора в присутствии пиридина:
NC5H5
3C4H9SH + POCI3 * (QH9SKPO
БЭКТ синтезируют, проводя реакцию между хлористой серок
и этилксантогенатом натрия:
S s
II II
2С2Н5—ОС—SNa + S2CI, » (CaHB—ОС—SJb
ИНСЕКТИЦИДЫ
По характеру действия на насекомых инсектициды разделяются
на кишечные яды, фумиганты, контактные и системные. Свойствами
кишечных ядов обладают неорганические
инсектициды—парижская зелень Си(СН3СОО).2-ЗСі^АбО^з, арсениты и арсенаты натрия
и кальция, ВаС12, кремнефторид натрия и др. В качестве
фумигантов для борьбы с вредителями в зернохранилищах, на складах
и т. п. применяются синильная кислота HCN, бромистый метил
СН3Вг, дихлорэтан (стр. 183), хлорпикрин CC13NO2 и другие лег-
колетучие яды.
350
Наибольшее значение приобрели контактные инсектициды,
поражающие насекомых при соприкосновении с ними и
проникании в их организм, и системные инсектициды, которые
поглощаются корнями или листьями растений и оказывают
токсическое действие на насекомых, питающихся соком растения. Часто
системные инсектициды одновременно обладают контактным
действием. Те и другие инсектициды обычно являются также
кишечными ядами, а в ряде случаев—фумигантами.
Контактные и системные инсектициды высокотоксичны для
насекомых и вместе с тем во многих случаях относительно
безопасны для теплокровных. Эти препараты представляют собой более
или менее сложные органические соединения, которые можно
подразделить на три основные группы: хлорированные углеводороды,
фосфорорганические соединения и зфиры карбаминовой кислоты
(карбаматы).
Применение инсектицидов привело к появлению насекомых,
устойчивых к некоторым из этих ядохимикатов. Вследствие этого
и огромного разнообразия видов насекомых возникла
необходимость в большом ассортименте инсектицидов и непрерывном
расширении его.
Хлорированные углеводороды. Наиболее распространенным
инсектицидом является 4,4'-дихлордкфенилтрихлорметилме-
тан—так называемый ДДТ, применение которого началось в
40-е годы.
ДДТ получают конденсацией хлораля с хлорбензолом в
присутствии олеума (стр. 282).
Наряду с 4,4'-изомером ДДТ содержит примеси 2,4'- и 2,2'-изо-
меров.
Хлорбензол не только реагирует с хлоралем, но и частично
сульфируется олеумом, образуя /г-хлорбензолсульфокислоту,
которая используется в синтезе полупродуктов для красителей.
С учетом этого при синтезе ДДТ на 1 моль хлораля
обычно берут 3,5 моль хлорбензола и 4,5 моль 5—7%-кого олеума.
Конденсацию проводят в течение примерно 7 ч при 20—25 GC и
энергичном перемешивании реакционной массы. По завершении
процесса реакционная масса отстаивается, затем сливают нижний
слой, содержащий серную кислоту и гс-хлорбензолсульфокислоту.
Остаток в реакторе промывают ведой, отгоняют избыток
хлорбензола острым паром и передают расплавленный ДДТ в
барабанный кристаллизатор, откуда продукт в виде чешуек поступает на
упаковку.
ДДТ—сильный инсектицид контактного действия и
кишечный яд, поражающий многие виды насекомых. Недостатком
ДДТ является его токсичность для человека и домашних
животных.
Синтезирован ряд инсектицидов, близких по химическому
строению к ДДТ, но менее токсичных для теплокровных и обла-
351
ДаЮЩИХ НеСКОЛЬКО ИНЬШ ДеИСТВИем, например
ДДД:
СС13
метоксихлор
НСС12
ДДД
Метоксихлор получают аналогично ДДТ—при замене хлор
бензола анизолом СвН5ОСН3. Для получения ДДД хлорируют
при 25—30 °С этиловый спирт, образовавшийся дихлорацеталь
дегид СНС!2СНО конденсируют с хлорбензолом.
Во время второй мировой войны начал применяться и в
дальнейшем получил большое значение в качестве инсектицида гек-
сахлорциклогексан (гексахлоран):
CHCI
CIHC сна
анс снсі
сна
Из восьми пространственных изомеров гексахлорциклогек
сана, отличающихся различным расположением атомов хлора
относительно углеродных атомов, наибольшим инсектицидным
действием обладает у-изомер.
Гексахлоран получается в результате присоединения трех
молекул хлора к бензолу в присутствии специфических
катализаторов (например, перекисей) или при воздействии света:
С.Н«, + ЗС12 > CeHeCle
Производство гексахлорана осуществляется непрерывным
методом (рис. 107).
В колонну-хлоратор 3 сверху поступает бензол, в нижнюю
часть (примерно на 1/i высоты колонны)—хлор. Хлоратор пред
варительно заполняется бензолом, в котором растворяется
поступающий хлор, а затем образующийся гексахлоран.
Реакционная смесь освещается ртутно-кварцевыми лампами,
установленными внутри колонны. Требуемая температура процесса C0 —
50 °С) поддерживается путем пропускания холодной воды в
рубашку и трубчатку, которыми снабжен хлоратор.
Раствор, непрерывно вытекающий из нижней части колонны
в сборник 4, содержит около 30% гексахлорана, бензол и
небольшие количества хлора и хлористого водорода (часть побочно вы-
352
деляющегося хлористого водорода поглощается в скруббере 9
водой). Бензол отгоняют острым паром в кубе 5, конденсируют в
холодильнике 10, отделяют от воды в сосуде 7 и вновь направляют
на хлорирование.
После отгонки бензола гексахлоран 'поступает в
кристаллизатор 6, где смешивается с водой и застывает в виде гранул,
которые отфильтровывают, промывают водой, а иногда высушивают.
бензол
>
— 1
Вканала-
у зацию
Рис. 107. Схема получения гексахлорана:
/—напорный бак; 2—фильтр; 3—хлоратор; 4—сборник реакцивн-
ного раствора; 5—куб; S—кристаллизатор; 7—разделительный
сосуд; в—сборник обратного бензола; 9—скруббер для поглощения
паров бензола; 10—холодильник-конденсатор.
Технический гексахлоран содержит около 12% активного
-^-изомера. Перекристаллизацией из метанола и другими
способами получают гексахлоран, обогащенный до содержания 20 —
99% у-изомера.
Гексахлоран—эффективный контактный инсектицид и
кишечный яд, при повышении температуры—фумигант, действует
быстрее, чем ДДТ. Недостатком гексахлорана является его неприят-
23-806
353
ный запах. ^-Изомер не имеет неприятного запала и р
ние более целесообразно, чем смеси изомеров. Другие изомеры
гексахлорана, практически не являющиеся инсектицидами,
токсичны для теплокровных и, накапливаясь в почве, могут попадать
в продукты питания. Однако эти мало токсичные для насекомых
изомеры после отделения от ^-изомера могут быть переработаны
в некоторые полупродукты, например в трихлорбензол.
Ценные инсектициды найдены среди хлорпроизводных
терпенов. Так, хлорированием камфена, извлекаемого из скипидара,
можно получить полихлоркамфен (строение его точно не
установлено).
По реакции Дильса—Альдера (диеновый синтез) получают
гептахлор, альдрин и другие инсектициды близкого к ним
строения. Например, гептахлор образуется при взаимодействии цик-
лопентана с гексахлорциклопентадиеном и последующем
хлорировании:
у.СС1
О '
,СС\
1С | НС_
Инсектициды, относящиеся к хлорированным углеводородам,
представляют собой твердые вещества, более или менее
токсичные для человека, устойчивые и практически не растворимые в
воде, но растворимые в жирах, маслах и других органических
растворителях. При систематическом применении они
накапливаются в почве и через растения могут попадать в организм
теплокровных, а затем в продукты животноводства. Поэтому хлорорга-
нические инсектициды чередуют с менее стабильными
препаратами, а иногда ограничивают их применение в сельском
хозяйстве.
Фосфорорганические инсектициды обладают рядом
преимуществ перед инсектицидами типа хлорированных
углеводородов. Они менее устойчивы и при их внесении в почве и
растениях не остается токсичных веществ. Среди фосфорорганических
инсектицидов имеются мало токсичные для теплокровных
препараты, а также системные яды, поражающие паразитирующих на
растениях вредителей, но безопасные для пчел и других полезных
насекомых. Фосфорорганические инсектициды принадлежат
преимущественно к различным эфирам тиофосфорной кислоты.
Одним из наиболее распространенных фосфорорганических
инсектицидов является метилмеркаптофос, содержащий два изо-
354
мера—тионовый и тиоловый. Синтез метилмеркаптофоса состоит
из следующих стадий:
1) взаимодействие тиотреххлористого фосфора с метиловым
спиртом в присутствии едкого натра при пониженной температуре
с образованием диметилхлортиофосфата
S
II
PSC13 + 2СН3ОН > (CHjOfoPCl
2) реакция между окисью этилена (стр. 242) и этилмеркаптаном,
приводящая к образованию 2-оксидиэтилсульфида
Н2С СН2 + HSQHs » НОСНїСН2—S—С,Н,
О
3) получение из диметилхлортиофосфата и
2-оксидиэтилсульфида тионового изомера метилмеркаптофоса
S S
В II
(CH3O)jPCl -і- НОСИцСН*—S—C2Hb * (СН3ОJ—PO—СН2СН2—S—СЬЦ
4) частичная перегруппировка тионового изомера при
нагревании в более токсичный для вредителей тиоловый изомер
S О
о а
(СНЭОJРО— СН2СН2— S—С2Н5 * (СН3ОJР—S—СН2СН2—S—QH,
Метилмеркаптофос—токсичная жидкость, обладает
эффективным контактным и системным инсектицидным действием.
Другим важным инсектицидом является хлорофос,
получаемый конденсацией хлора ля с диметилфосфитом:
О
II
СС)з—СОН + (СН3ОJРОН • (СН3ОJР—СН(ОН)-СС1,
Хлорофос менее токсичен для теплокровных, чем
метилмеркаптофос, и широко применяется в сельском хозяйстве и
здравоохранении.
В табл. 18 приведены некоторые другие фосфорорганические
инсектициды.
Карбаматы. В этой группе инсектицидов большое значение
имеет севин—эфир а-нафтола и N-метилкарбаминовой кислоты*.
* Карбаминовая кислота H^NCOOH известна только в виде солей, хлор-
ангидрида и эфиров.
23* 355
Некоторые фосфорорганические инсектициды
Инсектициды
Метафос
Тиофос
Метнлиитрофос
Трихлормета-
фос-3
Фосфамид
Метилацетофос
Карбофос
Строение
S
(CHSOJPO-Q)-NO2
S
С ^>т т
сн3с\ 5 ^с1
QHeO/ ~)=/'~
S О
(СН3ОJР—S-CH2—C-NH—CHS
S
(CHSOJP—S—CH2COOC2He
s
II
(CHsO^P-S-CHCOOQHj
Характер действия,
токсичность для
теплокровных
Контактный, токсичен
Контактный, более
токсичен, чем метафос
Контактный, мало
токсичен
То же
Контактный и
системный, относительно
мало токсичен
Контактный, мало
токсичен
Контактный,
относительно мало токсичен
Севин получают фосгенированием а-нафтолята натрия и
последующим воздействием метиламина на образовавшийся а-нафтило-
вый эфир хлоругольной кислоты:
ONa
Севин—эффективный и мало токсичный для теплокровных
инсектицид, практически не оставляющий вредных веществ в
почве и продуктах питания; он находит большое применение в
земледелии и животноводстве.
356
АКАРИЦИДЫ
Акарицидными свойствами обладают многие описанные выше
инсектициды, например, фосфамид, метилмеркаптофос, севин.
Для борьбы с клещами применяются также октаметил, эфирсуль-
фонат, тедион, димит и другие акарициды.
Октаметил (ангидрид тетраметилдиамидофосфорной кислоты)
получают путем взаимодействия хлорокиси фосфора с диметил-
амином; образовавшийся тетраметилдиамидохлорфосфат далее
нагревают с порошкообразным едким натром:
(CH3JN4
2(CH3JNH + POCI3 > >РОС1
(CH)N/
О О
(CH3JN4 NaOH (CH3JN4 И И ,N@*3),
2 \POC1 * >Р-О-Р<
(СН3JЫ/ (CH3JNK XN(CH3K
октаметил
Октаметил—системный акарицид и инсектицид, очень
токсичный и для млекопитающих.
Эфирсульфонат (л-хлорфениловый эфир я-хлорбензолсульфо-
кислоты) легко образуется из я-хлорбенз&г.сульфохлорида и
/z-хлорфенола в присутствии веществ, связывающих
отщепляющийся НО:
Тедион B,4,5,4'-тетрахлордифенилсульфон) получают по
реакции Фриделя—Крафтса взаимодействием 2,4,5-трихлорбен-
эолсульфохлорида с хлорбензолом в присутствии А1С13:
Сульфохлориды, необходимые для синтезов эфирсульфоната
и тедиона, получают взаимодействием хлорсульфоновой кислоты
соответственно с хлорбензолом или три хлорбензолом.
Синтез димита основан на реакции между хлорбензолом и
/і-хлорбензоилхлоридом в присутствии хлористого алюминия
и последующих реакциях с магнийбромметилом и водой:
О
/Г^ь. U /Г-Ъ. АІСІз ув—ь II /Г~\ СНзМгВг
OMgBr
357
ФУНГИЦИДЫ
Фунгициды широко применяются для протравливания семян
защиты плодовых, ягодных, овощных и других культур и для со
хранения продуктов питания—зерна, плодов и
материалов—древесины, тканей, бумаги, красок, клеев и др. Фунгициды часто не
только предотвращают грибные заболевания растений, но и
защищают их от бактерий, клещей, нематод и насекомых.
В качестве фунгицидов все большее применение находят
органические препараты, широко используются также элементарная
сера, полисульфиды кальция и бария, смесь раствора CuSO4 с
Са(ОНJ (бордосская жидкость), хлорокись меди ЗСиО СиС12 -4Н2О
и другие неорганические вещества. Для фумигации хранилищ,
амбаров и складов применяют SO2, обычно получаемый сжиганием
серы.
Для протравливания семян используются ртутьорганические
соединения, например этилмеркурхлорид C2H5HgCl (гранозан),
получаемый действием ртути на хлористый этил, и фенилмеркур-
ещетат C6HsHgOOCCH3, образующийся в результате
взаимодействия ртути с раствором бензола в ледяной уксусной
кислоте.
Ртутьорганические соединения, особенно легколетучий
этилмеркурхлорид, очень токсичны. Менее токсичны органические
соединения меди, из которых для протравливания семян
применяются трихлорфенолят меди и 8-оксихинолят меди.
Трихлорфенолят меди получается нагреванием 1,2,4,5-тетра-
хлорбензола с NaOH и последующим взаимодействием с CuSO4:
ONa
І Сі С1
NaOH <f\/ CuS0*
—* JL —*
I xi cv
Синтез 8-оксихинолята меди производят нагреванием о-амино-
фенола, глицерина и концентрированной серной кислоты и
последующим взаимодействием с CuSO4:
+ 2СН2(ОН)СН(ОН)СН2ОН
С" -О
358
8-Оксихинолят меди применяется в сельском хозяйстве и для
защиты тканей от гниения.
Важную группу фунгицидов составляют производные дитио-
карбаминовой кислоты, мало токсичные для теплокровных. Так,
тиурам (стр. 363) как фунгицид, обозначаемый также ТМТД,
широко используется для протравливания семян, для опиливания
и внесения в почву. В качестве фунгицидов применяются также
диметилдитиокарбамат цинка (цирам) и железа (фербам):
S S S S
II У II
(CH3JNC—S-Zn—S—CN(CHa)j (CHaJNC—S—Fe—S—CN(CHaJ
цирам фербам
Взаимодействием CS2 и NaOH с этилендиамином получают
этилен-бис-дитиокарбамат натрия—фунгицид набам:
S S
II II
NaS—С—NH—СН2—СН2- N Н—С—SNa
Манеб—марганцевая соль этого соединения—один из наиболее
важных фунгицидов, применяемых для обмывания листьев
растений; широкое применение находит также цинковая
соль—фунгицид цинеб.
Для обработки плодовых и овощных культур и
протравливания семян применяется 2,3-дихлор-1,4-нафтохинон—дихлон,
получаемый хлорированием 1,4-нафтохинона:
О О
II г\
о о
Ценным фунгицидом является каптан (N-трихлорметилтиоимид
тетрагидрофталевой кислоты), превосходящий по эффективности
бордосскую жидкость и вместе с тем мало токсичный для
теплокровных. Для получения каптана конденсируют малеиновый
ангидрид с бутадиеном, затем действием аммиака превращают
ангидрид тетрагидрофталевой кислоты в амид:
СН2
JI НС-CO СО СО
СН " ЧО II I О
CHa
359
Далее амид реагирует с перхлорметилмеркаптаном:
СО СО
\ ciscci3 /~^У \
NH * [| [ N—S-CC1,
СО СО
каптан
Каптан применяется для протравливания семян хлопчатника
и овощных культур, защиты листьев плодовых и других растений
и для предохранения фруктов от порчи.
ЗООЦИДЫ
Грызунов травят в складах, а также в норах ядовитыми
газами, например HCN, образующимся при действии влаги на циан-
плав, содержащий цианиды натрия и кальция, или уничтожают
при помощи хлорпикрина CC13NO2. В приманках для
уничтожения грызунов применяют фосфид цинка 2п3Р2 и некоторые другие
яды. Особенно сильным действием обладают органические зоо-
циды—ратиндан и зоокумарин.
Ратиндан получают конденсацией диэтилфталата с 1,1-ди-
фенилацетоном:
Ч-СНз—СО-СН(СвН6)а-
COOQHs
СО
\
СН=-СО—СН(С„Н6),
Синтез зоокумарина проводится по следующей схеме:
1) конденсация бензальдегида с ацетоном с образованием бен-
зальацетона
NaOH
С„Н6СНО + СНзСОСНз » СвН5СН=СНСОСН3
2) получение метилового эфира ацетилсалициловой кислоты
ОСОСНз
•СООСН,
360
3) получение 4-оксикумарина действием металлического
натрия на метилацетилсалицилат
ОН
ОСОСНз
соосн,
4) конденсация 4-оксикумарина с бензальацетоном
он
-A/ Vh
' ' ¦ сн=сн—со—снэ
Z0" "
V:—сн—сн,—со-сн
зоокумарин
5. ПРЕПАРАТЫ ДЛЯ РЕЗИНОВОЙ ПРОМЫШЛЕННОСТИ
В резиновой промышленности, кроме основного
сырья—каучука, используются различные добавки—ингредиенты резиновых
смесей (стр. 497). Эти добавки придают смесям необходимые
технологические свойства и улучшают качество резиновых изделий.
Применяются как неорганические ингредиенты (сера,
кремнекислота, некоторые минералы, соли, окислы металлов и др.). так
и вещества органического происхождения (сажа, продукты
переработки нефти и каменного угля, разнообразные синтетические
препараты). Важнейшими синтетическими препаратами,
потребляемыми резиновой промышленностью, являются:
ускорители вулканизации каучука, улучшающие также физико-
химические свойства получаемых резиновых изделий, некоторые
ускорители предохраняют резиновые изделия от старения;
противостарители (антиоксиданты), замедляющие процесс
окисления каучука и повышающие таким образом устойчивость
резиновых изделий к различным воздействиям;
ускорители пластикации, облегчающие подготовку жестких
каучуков к переработке их в изделия.
361
ІЗ ООЛЬШИНСТВе Случаев ЗГИ [фСИіфаіьі иредсіавлмпл Lui/un o^
щества сложного строения. Сущность их действия и условия
применения рассматриваются в главе VI11 (стр. 476 и ел.).
Ниже в качестве примеров приводятся схемы синтеза некоторых
наиболее распространенных препаратов.
Ускорители вулканизации. Одним из наиболее
часто применяемых ускорителей высокой активности является
каптакс B-меркаптобензотиазол), получаемый нагреванием
анилина с серой и сероуглеродом:
А
s
каптакс
Для получения каптакса в автоклав загружают серу и анилин,
содержащий около 15% нитробензола, вытесняют воздух азотом
и добавляют сероуглерод (CS2 ядовит, с воздухом образует
взрывчатую смесь). Смесь сначала нагревают до 150 °С (электрообогрев),
далее температура повышается до 200—250 °С вследствие
выделения тепла реакции, при этом давление может превысить 50 am.
Процесс синтеза каптакса длится несколько часов. По окончании
реакции из автоклава удаляют выделившийся сероводород,
который поглощается в ловушке раствором едкого натра, и непро-
реагировавший сероуглерод, поглощаемый анилином и
используемый затем в производстве тиокарбанилида.
Реакционную массу из автоклава передавливают паром в чан
с раствором едкого натра, в котором каптакс растворяется с
образованием натриевой соли. Раствор отфильтровывают от смолистых
примесей и осаждают каптакс разбавленной серной кислотой.
Продукт центрифугируют, промывают водой и высушивают.
К ускорителям группы тиазолов относится также альтакс
(дибензотиазолдисульфид), получаемый окислением натриевой
соли каптакса азотистой кислотой:
N NaNO2-t-H2SO4 f \ N N-
II II
С—S—S—Сч
S S
Синтез альтакса несложен. Каптакс растворяют в водном рас
творе едкого натра, вводят нитрит натрия и постепенно приливают
эту смесь к разбавленной серной кислоте. После перемешивания
реакционную массу нейтрализуют аммиаком и центрифугируют.
Полученный альтакс промывают водой и высушивают.
362
В качестве ускорителей вулканизации используются
соединения группы гуанидина, наиболее употребительным из них
является дшренилгуанидин.
Для получения дифенилгуанидина вначале пропускают хлор
через охлажденный водный раствор цианистого натрия.
Образующийся при этом раствор хлорциана прибавляют при 30—40 °С
к смеси воды, анилина и солянокислого анилина, нагревают до
95 °С и выдерживают при этой температуре 1 ч. Схема синтеза
NH2 Н—N—CN
0+оп-0 ^
дифеннлгуаиидин
Реакционную массу нейтрализуют едким натром и отгоняют
с водяным паром не вошедший в реакцию анилин. Раствор
дифенилгуанидина очищают активированным углем, осаждают продукт
добавлением едкого натра и отфильтровывают. Осадок
дифенилгуанидина промывают водой и высушивают.
К наиболее распространенным ускорителям вулканизации
группы тиурамов относится тетраметилтиурамдисульфид,
обычно называемый просто тиурамом.
Тиурам синтезируют в две стадии. Сначала при
взаимодействии сероуглерода с диметиламином в растворе едкого натра при
20—30 °С получают диметилдитиокарбамат натрия
(CH3JNH 4- CS2 + NaOH ¦ (CH3JN—С—SNa
I!
S
который затем окисляют при 20 °С азотистой кислотой,
образующейся при взаимодействии серной кислоты и нитрита натрия:
2(CH3)aN—С—SNa -f 2NaNO2 -f 2H2SO4 »
II
S
> (CHgbN—C—S—S—C—N(CH„),
II II
• s s
гвурам
В настоящее время для окисления диметилдитиокарбамата с
успехом применяется также перекись водорода. Этот способ более
экономичен, так как позволяет значительно упростить очистку
сточных вод от вредных примесей.
По окончании реакции тиурам отделяют от маточного раствора
на центрифуге и высушивают.
363
Диметилдитиокарбамат натрия при оораоотке /,тла оиразуеі
цинковую соль—цимат, очень активный ускоритель
вулканизации:
2(CHaJN—С—SNa + ZnCla » [(СНзJ1М—С—S—]2Zn + 2NaCl
циыат
Тиурам и цимат (называемый также цирам) применяются^
качестве фунгицидов в сельском хозяйстве (стр. 359).
При вулканизации многих синтетических каучуков широко
применяются сульфенамиды. В качестве представителей
ускорителей этого типа можно назвать сульфенамид Ц (М-циклогексан-2-
бензотиазолсульфенамид), получаемый из каптакса (см. выше).
Сначала каптакс путем обработки едким натром превращают
в натриевую соль
/\ к р N N
|| + NaOH
ч /C-SH
S S
которая затем реагирует с циклогексиламином:
"",—СН2
NH.
C-SNa
g2
-N / \
a—CH СНа
\ /
СН2—СН2
циклогексиламин
N СНа—СН»
U їм v-na—^-па
|| +-^-- / \
v .С—S-HgN—CH СН
S \ /
СН2—СНа
Далее образовавшуюся циклогексиламиновую соль каптакса
окисляют в сульфенамид Ц:
NaOCl
СН2 »
СНа—СН2
/\ N СНа—СНа
* L J с—S—NH—СН СН2
сульфенамид Ц
364
Противостарители. В качестве противостарителеи
в большинстве случаев применяются ароматические амины,
продукты их конденсации с альдегидами, фенолы. К этой группе
ингредиентов резиновых смесей относятся и противоутомители,
замедляющие старение резиновых изделий, которые
подвергаются различным многократным деформациям в процессе
эксплуатации.
Один из наиболее распространенных противостарителеи
неозон Д (фенил-2-нафтиламин) получают при нагревании 2-нафтола
с анилином в присутствии небольшого количества солянокислого
анилина:
NH2
Г
неозон Д
Процесс проводят в котлах с мешалками при температуре около
250 °С. Смесь реагентов нагревают в течение нескольких часов.
Образующаяся вода отгоняется из реакционной массы. По
окончании реакции в котел загружают едкий натр (для превращения
солянокислого анилина в свободное основание) и отгоняют
оставшийся анилин с водяным паром. Затем отгоняют воду и
направляют расплавленную массу в кристаллизатор. Неозон Д
кристаллизуется на стенках вращающегося барабана, охлаждаемого изнутри
водой. Затвердевший продукт размалывают в мелкий порошок
в токе двуокиси углерода, чтобы предотвратить образование
взрывоопасной смеси пыли неозона с воздухом.
Эффективным протнвостарителем и противоутомителем, широко
применяемым в настоящее время, является дифенил-п-фенилен-
диамин, получаемый нагреванием гидрохинона с анилином:
—ОН + 2
Н Н
дифенил-л-фенилендиамин
У с к о р~и тели пластикации каучука.
Активным ускорителем пластикации является пептон 22 (ди-
о-бензамидофенилдисульфид), получаемый в две стадии. На
первой стадии при нагревании анилина с избытком серы образуется
2,2'-диаминодифенилдисульфид:
NH2
С
365
На агорой стадии его бензоилируют:
NH2 NH2 (rOa
бензоилхлорид
HNCO—
пептон 22
При повышенных температурах более активно ускоряет
пластикацию каучука ренацит П (трихлортиофенол). Его получают
при действии хлористой серы на 1,2,4-трихлорбензол и
последующем восстановлении образующегося дисульфида (частично
содержащего моносульфид):
С1 С1
трихлортиофенол
Для удобства дальнейшего использования ускорители
пластикации и другие препараты резиновой промышленности обычно
гранулируют.
ЛИТЕРАТУРА
ворожцов Н. Н., Основы синтеза промежуточных продуктов и краси
телей, Госхнмиздат, 1955.
воронцов И. И., Полупродукты аиилинокрасочиой промышленности
Госхимиздат, 1955.
Коган И. М., Химия красителей, Госхимиздат, 1956.
Чекали и М. А., Еремии Ф. Ф., Химия азокрасителей, Госхимиз
дат, 1952.
Кархаров А. А., Калонтаров И. Я-, Активные красители и
их применение в текстильной промышленности, Изд. научно-техн. лит
РСФСР, 1961.
Ф р от ш е р Г., Химия и физическая химия текстильных вспомогательных
веществ, Гизлегпром, 1958.
Шварц А., Перри Дж., Бер q Дж., Поверхностно-активные
вещества и моющие средства (перев. с англ.), Издатинлит, 1960.
Мельников Н. Н., Баскаков Ю. А., Химия гербицидов и
регуляторов роста растений, Госхимиздат, 1962.
Б е л о з е р о в Н. В., Технология резины, Изд. «Химия», 1964.
Журн. ВХО им. Менделеева, № 6 A963); № 3 A960).
Часть третья
ТЕХНОЛОГИЯ ВЫСОКОМОЛЕКУЛЯРНЫХ
СОЕДИНЕНИЙ
ВВЕДЕНИЕ
В химической технологии органических вешеств все большее
значение приобретают процессы производства и переработки
высокомолекулярных соединений. Сюда относятся: производство
пластических масс, каучуков и резины, химических волокон,
пленок, целлюлозы и бумаги, лаков и красок, кинопленки и др.
Все в большей степени от произволства высокомолекулярных
соединений зависит технический прогресс машиностроения,
приборостроения, электротехнической промышленности,
ракетной техники, производства строительных материалов, предметов
широкого потребления и і. д.
Еще сравнительно недавно технология высокомолекулярны*
соединений сводилась исключительно к переработке природных
вешеств растительною или животного происхождения—волокна,
кожи, древесины и и \ В последние десятилетия быстрыми темпами
развивается производство синтетических высокомолекулярных
соединений, из которых получают не только полноценные
заменители природных продуктов, но и вещества, обладающие
свойствами, отсутствующими у природных материалов. В связи с этим
высокомолекулярные соединения находят большое применение в
самых различных областях современной техники.
Интенсивный рост потребления пластических масс и других
материалов на основе высокомолекулярных соединений
обусловлен их ценными свойствами. Так, детали из пластмасс при
одинаковой прочности с металлическими деталями много легче
последних (например, на 1000 пог. м водопроводных труб расходуется
пластмассы 250 кг, а металла 2 т) и отличаются большей
химической стойкостью. Это позволяет облегчить конструкции машин и
изделий, в которых применяются пластмассовые детали,
сократить затраты на их ремонт, повысить срок службы и надежность.
Кроме того, для производства деталей из пластмасс затрачивается
в 7,5—10 раз меньше труда, чем на изготовление их из металлов.
Трудозатраты на производство 1 т. химических волокон в 2—
4 раза меньше, чем на производство такого же количества волокна
367
на ириридныл материалив. і іи шиисшам лимичеекие виликна не
уступают природным, а в ряде случаев превосходят их по
прочности, сопротивлению истиранию, несминаемости, химической
стойкости. Капиталовложения на производство 1 т продукции из
синтетических высокомолекулярных соединений обычно в 2, а
иногда в 5 раз меньше капиталовложений на 1 т заменяемого
природного материала.
Необходимость дальнейшего значительного расширения
производства полимеров отмечалась в решениях XXII съезда КПСС,
майского A958 г.) и декабрьского A963 г.) Пленумов ЦК КПСС.
Эти решения успешно реализуются отечественной химической
промышленностью.
Так, за 1960—1964 гг. производство пластических масс
возросло в 2,4 раза, объем производства химических волокон за этот
период увеличился в 1,8 раза. Бурно развивается і роизводство
полимеров и в зарубежных странах.
Несмотря на значительное разнообразие свойств различных
видов высокомолекулярных соединений, они обладают и рядом
общих свойств, в связи с чем их выделяют в особый класс
химических соединений.
Для всех высокомолекулярных соединений характерен очень
большой молекулярный вес, который колеблется в пределах от
10 000 до нескольких миллионов и редко снижается до несколь
ких тысяч.
Вследствие этого молекулы таких соединений обычно
называются макромолекулами*. Понятие о молекулярном весе
высокомолекулярных соединений имеет ряд особенностей по сравнению
с понятием о молекулярном весе низкомолекулярных соединений.
Характерным отличием высокомолекулярных соединений
является то, что они представляют собой смесь макромолекул различной
величины, т. е. высокомолекулярные соединения полидисперсны
(неоднородны) по молекулярному весу. Поэтому их
молекулярный вес является средней величиной для смеси составляющих
его макромолекул, молекулярный вес которых колеблется около
этого среднего значения.
Макромолекулы большинства высокомолекулярных
соединений построены из одинаковых, многократно повторяющихся групп
атомов—элементарных звеньев А, например:
А—А-А-А—А—А—А-А—•
К таким высокомолекулярным соединениям относятся
полиэтилен (—СН2—СН2—)„, полиакриловая кислота (—СН2—СН—)„,
:оон
полиизопрен (—СН2—СН(СН9)=СН—СН2—)„ и другие. Поэтому
* Макро (греч.)—большой.
368
высокомолекулярные соединения называют также высокополи-
мерами или просто полимерами, в отличие от мономеров—этилена,
изопрена и других низкомолекулярных соединений, используемых
для синтеза полимеров (стр. 125, 126).
Величина макромолекулы полимера может быть
охарактеризована также числом элементарных звеньев, входящих в ее
состав. Это число носит название степени полимеризации*. Степень
полимеризации Р связана с молекулярным весом полимера М
уравнением:
М
Р = — или М = Рт
где т—молекулярный вес элементарного звена.
г Любой полимер, как говорилось выше, является смесью
макромолекул с различным числом элементарных звеньев в них. Эти
макромолекулы образуют так называемый полимергомологический
ряд с гомологической разницей между его соседними членами,
равной молекулярному весу одного элементарного звена.
При достаточно больших значениях степени полимеризации,
свойственных полимерам, разница молекулярных весов (и,
следовательно, свойств) двух соседних гомологов столь мала, что смеси
полимергомологов не могут быть разделены на индивидуальные
соединения методами, которые имеются в настоящее время
в распоряжении науки и техники. Таким образом, полимеры, в
отличие от химически чистых низкомолекулярных соединений, не
являются молекулярно однородными, а представляют собой смеси
различных членов данного полимергомологического ряда.
Не все высокомолекулярные соединения построены из
повторяющихся звеньев одинакового химического состава.
Макромолекулы некоторых полимеров построены из различных по составу
элементарных звеньев, как правило, нерегулярно расположенных
в молекулярной цепи, например:
А—А—В— А—В—В—В—А- В—А—А—А—В—А
или
А—В—В—С—А—С—С—В—А—В
где А, В и С—элементарные звенья, различные по химическому
составу.
* При написании суммарной формулы полимера степень полимеризации
обычно обозначают буквой и, например /—СНа—СН—\ полипропилен.
/—СНа—СН—\
\ СН3 /„
24—805 369
Такие высокомолекулярные соединения называются шпили-
мерами. В качестве примера приведем сополимер акрилонитрила
СН2=СН и винилпиридина СН2=СН—\__yN
CN
СН2—СН—СНа—СН—СНа—СН—СН2—СН—СН2—СН
CN ^Ч CN CN
N
N
Макромолекулы некоторых сополимеров построены путем
чередования не отдельных звеньев, а целых участков молекулярной
цепи (блоков), различающихся по химическому составу и
строению, например:
(-А)я (-Б-)т (-А-)„, (-Б-)«.
Такие сополимеры называются блпксополимерами. К этому
типу полимеров относятся, по-видимому, многие белки. Блок-
сополимеры получают синтетически. Например, блоксополимер
полиэтиленоксида и полиэфира:
Сна—СНг—О—СНа—СНа—О— С—R—С—O-R'
II II
О О
Синтез различных сополимеров значительно расширяет
разнообразие свойств и областей применения полимерных
материалов.
Высокополимерные соединения в зависимости от
геометрической формы макромолекул разделяют на линейные, разветвленные
и пространственные (сетчатые).
Рис. 108. Схематическое изображение строения
: полимеров:
а—линейных: б— разветвленных; а—пространственных.
На рис. 108 схематически изображено строение линейного,
разветвленного и пространственного полимеров.
Макромолекулы линейных полимеров представляют собой
длинные цепи, причем длина макромолекулы во много раз A00—1000 и
370
оолее) превышает ее поперечный размер (высокая степень
асимметрии). Поэтому макромолекулы линейных полимеров часто
называют молекулярными цепями.
К линейным полимерам относятся некоторые природные
полимеры, например, целлюлоза, натуральный каучук, некоторые
белки (в частности, казеин) и очень большое число синтетических
полимеров (полиамиды, полиэфиры, полиолефины и др.).
Полимеры, макромолекулы которых имеют ответвления от
основной линейной цепи, носят название разветвленных полимеров.
К разветвленным полимерам относится амилопектин—составная
часть крахмала. Разветвленные полимеры могут быть получены
и синтетически. Часто основная цепь полимера отличается
по химическому составу от боковых цепочек, которые как бы при-
питы к основной цепи:
(_А-)ч,-А-(-А-)„„-А-(-А-)„„,
(В)т. (В)т„
Такие разветвленные полимеры называют привитыми
сополимерами. В последние годы синтез привитых сополимеров (стр. 436)
получил широкое развитие.
Пространственные полимеры, (рис. 108, в) построены из
линейных цепей, соединенных друг с другом поперечными химическими
связями.
К природным полимерам этой группы относится шерсть,
известно также большое количество синтетических полимеров,
обладающих пространственной структурой (феноло-формальдегид-
ные полимеры, вулканизованные каучуки).
Геометрическая форма макромолекулы, наряду с другими
особенностями химического строения, сильно влияет на свойства
высокомолекулярных соединений.
¦ Все линейные полимеры могут быть переведены в раствор. Их
растворы при относительно небольших концентрациях обладают
значительной вязкостью вследствие высокой степени асимметрии
макромолекул. Вязкость растворов полимеров в десятки и сотни
раз превышает вязкость соответствующих растворов
низкомолекулярных соединений.
Многие линейные полимеры могут плавиться без разложения,
причем их расплавы также обладают очень высокой вязкостью.
Макромолекулы линейных полимеров в зависимости от
химического строения и условий (температура, концентрация раствора,
растворитель и др.) могут быть более или менее гибкими,
закручиваться различным образом, приобретать форму спирали или даже
клубка (глобулы). Но во всех случаях под влиянием внешних
условий (механическая нагрузка, изменение температуры) они могут
приобретать более вытянутую форму и определенным образом
ориентироваться по отношению друг к другу.
24* 3
В результате ориентации макромолекул создается плотная
структура полимера, в которой отдельные участки макромолекул
почти параллельны друг другу. Расстояния между
макромолекулами малы, что способствует возникновению многочисленных
межмолекулярных связей (Ван-дер-Ваальса, водородных связей)
с большой суммарной энергией. Благодаря этим особенностям
линейные полимеры отличаются высокими физико-механическими
показателями (высокая прочность и эластичность).
Разветвленные полимеры также могут быть переведены в
раствор, однако прочность разветвленных полимеров и вязкость их
растворов обычно ниже, чем соответствующих линейных
полимеров.
Пространственные полимеры резко отличаются по свойствам
от линейных и разветвленных полимеров. Они не могут быть
переведены в раствор или расплавлены при нагревании. Это
объясняется тем, что молекулярные цепи пространственных полимеров
соединены поперечными химическими связями, разрыв которых
приводит к разрушению полимера.
При одинаковой геометрической форме макромолекул
полимеры могут очень сильно различаться по свойствам в зависимости
от их химического строения. Так, полиамиды, содержащие амид-
ные группировки —CONH— (например, поликапроамид или
капрон)
...—NH—(СН2M—СО—NH—(СН2M—СО—NH—(СН2M—СО
обладают высокой разрывной прочностью, сопротивлением
истиранию, эластичностью и высокой температурой плавления B00—
300 °С), что позволяет использовать их в различных отраслях
производства. Полиуретаны типа
NH—(СН2L—NHCOO—(СН2L—COONH—(CH2)e—NHCOO—...
отличающиеся от полиамидов только тем, что к амидной
группировке присоединен еще один атом кислорода —NHCOO—,
имеют значительно более низкую температуру плавления, A50—
180 °С) и находят более ограниченные области применения.
Полимеры акриловой кислоты
/—СНа—СН —\
\ соон!
очень хрупки, растворимы в очень небольшом числе
растворителей и также находят ограниченное применение. Полимер
нитрила акриловой кислоты
Г- СНа-СН -1
*- CN -L
372
обладает высокой эластичностью, термо- и светостойкостью и с
успехом используется в производстве синтетических волокон -
Полимер метилового эфира метакриловой кислоты
СН,
3
-С-
:ООСН3_
отличается высокой светопрозрачностью, ударопрочностью и
используется для производства органических стекол.
Свойства полимеров, обладающих одинаковым химическим
составом, зависят также от пространственного расположения
атомов и групп атомов макромолекулы. Особенно отчетливо эта
зависимость проявляется для полиолефинов. Например, полипропилен,
в котором метильные группы расположены беспорядочно по цепи
сн3 сн3
СН2—СН—СНа—СН—СН2—СН—СН2—СН—СН2—СН
СН3 СН3 СН3
имеет очень низкие физико-механические показатели и
поэтому не может быть использован для переработки в волокна »
другие изделия. Полипропилен, полученный в присутствии
специальных стереоспецифических катализаторов (стр. 419 ел.),
имеет строго определенное расположение метильных групп по
цепи, обладает хорошими физико-механическими свойствами и
успешно используется в промышленности (стр. 469, 492, 540).
Полимеры, имеющие строго определенное расположение
боковых групп в пространстве, называют стереорегулярными.
Строение стереорегулярных полипропиленов, изображенное в
плоскости, схематически может быть представлено следующим образом:
СН2—СН—СНа—СН—СНа—СН—СН2—СН—
СН L L ГН,
1_.Н3 <-П3 *-jH3 ЬНз
изотактический полипропилен
ИЛИ
СН, СНз
CHj—СН—СН2—СН—СН2—СН—CHj—СН
сн3 сн3
сииднотактическиВ полипропилен
В действительности строение стереорегулярных полимеров
сложнее, так как их молекулы закручены в спирали.
373
ррур ПОЛИМерЫ ПОДраЗДеЛЯЮТ на UJUihu^nlu^
и синдиотактические в отличие от атактических полимеров
с беспорядочным расположением боковых групп. Изотактиче-
скими называются полимеры, у которых боковые группы
расположены по одну сторону молекулярной цепи, синдиотактическими
полимерами—полимеры с чередующимся расположением
боковых групп по обе стороны молекулярной цепи.
Стереорегулярные полимеры по свойствам значительно
отличаются от атактических полимеров. Они обладают более высокой
температурой плавления и меньшей растворимостью.
Все рассмотренные особенности строения высокомолекуляр
ных соединений определяют их физико-механические
свойства и возможность изготовления из них изделий самого
разнообразного назначения. Эти свойства полимеров, как и других
веществ, обусловливаются характером и величиной деформации
(изменений формы до разрушения) под влиянием внешних
усилий. Деформации, возникающие под влиянием нагрузки, могут
быть обратимыми (полностью исчезать после снятия нагрузки^
и необратимыми (сохраняться после снятия нагрузки).
Деформация полимеров может происходить за счет изменения
межатомных расстояний, способности молекул изменять свою
форму (вытягиваться или изгибаться), а также за счет перемещения
молекул полимера друг относительно друга. Каждое из этих
явлений может происходить независимо одно от другого или они
могут совмещаться и происходить одновременно. В последнем
случае преобладающее явление определяет характер
деформации полимера и, следовательно, его поведение в условиях
эксплуатации.
Вследствие большой величины молекулярного веса
подвижность макромолекул полимеров очень мала и резко падает с
понижением температуры. Поэтому характер деформации полимера,
возникающей под влиянием внешних условий, в большой степени
зависит от температуры.
При низких температурах подвижность макромолекул
ничтожно мала, а энергия межмолекулярного взаимодействия велика
•и значительно превышает энергию химической связи.
Единственно возможным видом деформации полимера в этих
¦условиях является упругая деформация, возникающая в
результате изменения межатомных расстояний под влиянием нагрузки.
Так как межатомные расстояния могут быть изменены до
разрушения полимера на очень небольшую величину, то и деформация,
возникающая даже при относительно больших усилиях, очень
невелика. После снятия нагрузки деформация быстро исчезает
Механические свойства полимеров при низких температурах
близки к механическим свойствам кристалла.
Є повышением температуры тепловое движение макромолекул
усиливается. Энергия этого движения еше недостаточна для того.
374
чтобы молекулы имели возможность взаимного перемещения, но
они уже способны под влиянием нагрузки изменять свою форму
(от изогнутой, спиральной к более вытянутой). Вследствие этого-
возникает так называемая высокоэластическая деформация. Она
тоже обратима, т. е. исчезает после снятия нагрузки, и
макромолекулы принимают первоначальную форму. Однако, в отличие от
упругой деформации, исчезающей почти мгновенно,
высокоэластическая деформация возникает и исчезает в течение некоторого
времени, которое зависит от гибкости цепи полимера и
температуры. Продолжительность этого времени называется периодом
релаксации.
Высокоэластическая деформация, связанная с гибкостью
длинных цепных молекул или участков этих молекул,
свойственна только высокомолекулярным соединениям.
С дальнейшим повышением температуры энергия теплового
движения макромолекул становится выше энергии
межмолекулярного взаимодействия, при этом возможно передвижение
макромолекул друг относительно друга и появляется пластическая
деформация полимера, называемая также вязким течением. В
отличие от упругой и высокоэластической деформации, пластическая
деформация необратима—она сохраняется и после снятия
нагрузки.
Особенностью физической структуры полимеров является то.
что они представляют собой полностью аморфные вещества
(аморфные полимеры) или содержат одновременно кристаллические
и аморфные области (кристаллические полимеры).
Изменения, происходящие в аморфных областях
кристаллического полимера ниже температуры его плавления, в значительной
степени определяют характер деформации всего полимера.
С точки зрения их использования все полимеры условно
подразделяются в первую очередь по физико-механическим
свойствам, т. е. по видам деформаций, которые возникают в полимере
под влиянием внешних усилий при комнатной температуре.
Вследствие этого полимеры можно подразделить на три группы:
1. Твердые полимеры, для которых характерна только упруга»
деформация. Они обладают достаточно высокой твердостью и
хрупкостью и по свойствам близки к силикатным стеклам.
Поэтому состояние, в котором находятся твердые аморфные полиме
ры, называют стеклообразным.
2. Эластичные полимеры, для которых кроме упругой
характерна также высокоэластическая деформация. Под влиянием
сравнительно небольших усилий эластичные полимеры способны к
большим обратимым деформациям.
3. Текучие полимеры, которые по свойствам очень близки к
обычным жидкостям. Они могут относительно легко необратимо
менять свою форму. Однако вследствие большой вязкости
полимеров в текучем состоянии для деформации их требуются боль-
375
шиє усилия. К этой группе принадлежат полимеры с
низким молекулярным весом, занимающие, таким образом,
промежуточное положение между низкомолекулярными и
высокомолекулярными соединениями и называемые олигомерами.
С изменением температуры полимеры способны переходить
из одного состояния в другое. При нагревании твердого полимера
он последовательно переходит в эластическое и вязкотекучее
состояние. Температура перехода полимера из твердого в
эластическое состояние (или, наоборот, из эластического в твердое)
называется температурой стеклования и обозначается Тс. Температура,
при которой полимер переходит в вязкотекучее состояние,
называется температурой текучести полимера и обозначается Гт.
Эти переходы осуществляются постепенно в более или менее
широком интервале температур и без химических изменений
полимера.
Химические изменения в полимере вызывают обычно
резкие изменения его физических свойств.
Твердые полимеры чаще всего используются для изготовления
пластмассовых изделий. Эластичными полимерами, с очень
большими деформациями при малых усилиях, являются каучук и
некоторые резины.
Полимеры с умеренной величиной деформации при
относительно больших усилиях используются для переработки в волокна
« пленки.
Процесс производства изделий из высокомолекулярных
соединений состоит из двух основных стадий: производства (синтеза)
полимера или выделения его из природных материалов и
переработки полимера в изделия.
Первая стадия рассматривается в главе VI, вторая, в
зависимости от направления использования,—в главах VII, VIII и IX.
На стадии переработки полимеров получают изделия заданной
-конфигурации, при этом полимер приобретает определенную
молекулярную структуру. Такие процессы осуществляются при
формовании резиновых изделий путем прессования, каландрования,
литья под давлением с последующей или одновременной
вулканизацией (стр. 519 ел.), изготовления изделий из пластических
масс методом литья, прессования и др. (стр. 531), при отливке
пленок из раствора полимера, при изготовлении химических
волокон (формование, вытяжка, стр. 443).
Процесс переработки высокомолекулярных веществ в
подавляющем большинстве случаев включает стадию взаимной
перегруппировки макромолекул, в результате которой изделие
приобретает требуемую форму, и стадию фиксации (закрепления)
достигнутого расположения макромолекул в полимере.
Взаимное перемещение макромолекул возможно при переходе
ролимера в вязкотекучее состояние в условиях повышенной тем
376
пературы, при его растворении или набухании под действием
растворителей. Из растворов или расплавов формуют изделия, форма
которых и молекулярная структура полимера фиксируются затем
при охлаждении или удалении растворителя. Во многих случаях,
например при вулканизации каучука, отверждении феноло-аль-
дегидных смол, «высыхании» масляных пленок, фиксация
макромолекул происходит в результате специально проводимых
химических процессов превращения линейных полимеров в
пространственные. В результате этих процессов исключается
возможность последующего плавления или растворения
полимера.
Так как пространственные полимеры непригодны для
формования изделий (пленок, нитей, пластмассовых и резиновых изделий),
то в процессе синтеза высокомолекулярных соединений
получают линейные полимеры, которые сохраняют в готовых
изделиях линейное строение, либо приобретают пространственную
структуру в процессе переработки. Лишь в немногих случаях
пространственные полимеры являются первичным продуктом
синтеза (например, резольные смолы, стр. 421). При этом в
начальных стадиях технологического процесса получают олигомеры с
молекулярным весом около 1000, еще сохраняющие способность
плавиться или растворяться и, следовательно, поддающиеся
формованию. В последующих стадиях переработки такие
олигомеры превращаются в пространственные полимеры, теряя
растворимость и способность плавиться (процесс отверждения резоль-
ных смол. стр. 553).
КЛАССИФИКАЦИЯ И НОМЕНКЛАТУРА
ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
Все высокомолекулярные соединения по химическому
строению основной цепи делятся на два больших класса.
1. Высокомолекулярные соединения, основные цепи которых
построены из одинаковых атомов. Например, если они построеньк
из атомов углерода, то полимеры называют карбоцепными:
или
Атомы углерода основной цепи могут быть связаны с
водородом или с какими-либо другими атомами или группами.
Из природных органических полимеров к карбоцепным
относится натуральный каучук, из неорганических—различные
модификации элементарного углерода (графит, алмаз).
377
К синтетическим карбоцепным полимерам относятся
высокомолекулярные углеводороды (предельные, непредельные и
ароматические) и их производные.
2. Высокомолекулярные соединения, основная цепь которых
построена из двух (или большего числа) разных элементов,
например из углерода и кислорода, углерода и азота, углерода и
серы, кремния и кислорода. Такие высокомолекулярные
соединения образуют класс гетероцепных полимеров.
Основная цепь таких полимеров может быть представлена
следующим образом:
или
О—С—
К гетероцепным органическим полимерам относятся
важнейшие природные высокомолекулярные соединения: белки, нуклеи-
но' ые кислоты, полисахариды, лигнин и др. К синтетическим
геироцепным полимерам относятся: полиамиды, сложные и
простые полиэфиры, полиуретаны, полиалкиленсульфиды и др.
Карбоцепные полимеры. Название карбоцепных полимеров
складывается обычно из названия исходного мономера, входящего
в качестве элементарного звена в состав макромолекулы
полимера, и приставки поли. Так, полимер, полученный из винилхло-
рида СН2=СНС1, называется поливинилхлоридом; полимер,
полученный из этилена СН2=СН2,—полиэтиленом; полимер хлоро-
прена СН2=СС1—СН=СН2—полихлоропреном и т. д.
Полимеры, полученные из монозамещенных производных
этилена, содержащих винильный радикал СН2=СН—,
объединяют под общим названием ванильных полимеров. К этой группе
относятся, например:
1— СНа—CHF—]„ Г—СН2—СН— ] Г— СН2—СН— ]
L
-СН— "І Г—СН2—СН-
ОСОСНз L | ОН
поливйнилфторид поливинилацетат поливиниловый
спирт
Полимеры, полученные из 1,1-дизамещенных этилена,
содержащих винилиденовый радикал СН2=С<, называют винилидено-
выми полимерами. К ним относятся:
CN"
[-СНа-СС12-]л [_CH2-CF2-]n
-CHj-C-
_ п
поливниилнден- поливинилиден- поливннилидеи-
хлорид фторид цианид
378
В соответствии с классификацией и номенклатурой органиче
ской химии все карбоцепные полимеры можно подразделить на
определенные классы соединений.
Ниже приведена классификация наиболее важных карбоцеп-
ных высокомолекулярных соединений.
П р е д е л ь н'ые угле вод oj) оды и их производные
Название Формула
Предельные углеводороды
Полиэтилен .
Полипропилен
Полиизобуі плен
Полининилбензол (полистиролГ
[-СНа-СНа-]„
Г—СН,—СН—
L СНз
—СН,—С—
сна_
-_сн,-сн--|
С)
Галоадопроиэводные предельных углеводородов
Поливинилхлорид . .
Поливинилиденхлорид
Политетрафторэтилен ....
[-CH2-CHCI-Jn
f-GH1-CCl,-]„
f-CFo-CF2-l„
Спирты, их простые и сложные эфиры
Поливиниловый спирт . . . . Г—СН-2—СН—
Простые Эфиры ПОЛІ1ШІНИЛОНОГО
спирта
Подивинилацетат
—СНа—СН—¦
Ar
—СН2—СН-
ососн.
* В основу принятой классификации высокомолекулярных соединений
положено строение их основных цепей. Поэтому полистирол рассматривается в ряду
предельных углеводородов, хотя его свойства в значительной степени
определяются наличием боковых фенильных групп.
379
Название
Формул«
Ацетили
Поливинилформаль . .
Поливинилбутираль
Альдегиды
Полиакроленн
Г—СНа—СН—СНа—СН—І
L О-СНа—O ]„
"—СНа—СН—СН,—СН—"
О—СН—О
сан,
г_сн2—СН—1
Кислоты и их производные
Полиакриловая кислота
Поли-а-метакриловая кислота
Полиметнлакрнлат
Полиметил-а-метакрилат . .
—СНа—СН— "І
соон!
СН3
—СН,—С-
і
ООН
—СНа—СН-
COOCHj
СН3 -
—СН,—С—
СООСНз.
Полнакриламид
Полиакрилонитрнл (поливинилцианид)
Поливинилиденцианнд .
Г—СНа—СН— І
I CONH.L
Г-CHj-CH-l
L CN J„
CN"
_сна-с-
CN
380
Непредельные углеводороды и их производные
Название Формула
Непредельные углеводороды
Полибутадиен [—СН2—СН=СН—СН2—]п
Полиизопрен (натуральный и син- I —CHS—С=СН—CHS— I
тетический каучуки) |
L СН3 J„
Галоидпроизводныс непредельных углеводородов
Полихлоропрен
. . Г—СН2—С=СН-СН2—"
А.
Ароматические углеводороды
Полиметиленоксифенилены (феноло-
формальдегидные полимеры) См. стр. 392
Гетероцепные полимеры. Гетероцепные высокомолекулярные
соединения в зависимости от гетероатома, входящего в состав
основной цепи, подразделяются на кислородсодержащие,
азотсодержащие, серосодержащие и элементорганические полимеры.
Эти большие группы полимеров подразделяются на подгруппы
в соответствии с принятой в органической химии классификацией.
. Название гетероцепных полимеров складывается из названия
класса соединений, к которому относится данный полимер, и
приставки поли, например полиэфиры, полиамиды, полиуретаны
и т. д.
Наиболее технически важные гетероцепные
высокомолекулярные соединения приведены ниже.
Кислородсодержащие полимеры
Название Формула
Простые полиэфиры (полиоксисоединения)
Полиоксиэтилен и его производные
Пэгсчо ксипропилен и его произво-
ные
1\
—СНо-С-О—
R'
-CHs-CHj-C-O-
і. .
381
Названии -*м^-и
Полиацепшли
І Іолиоксиформаль (полиоксиметилен) (—СН2—О—1„
Целлюлоза См1 стр 432
Сложные полиэфиры
Полиэтиленгликольмалеинат, поли- Н—[—О—R—ООС—R'—СО—f„.
этиленгликольтерефталат и др. . где к_радикал ГЛИКОля;
R'—радикал непредельной или аром&
тической двухосновной кислоты
Азотсодержащие полимеры
Полиамиды
Н—JNH—(СНа),—СО—J„—ОН
г е х > 1
или
Н—[NH—R—NHCO—R'—СО—J„—ОН
где R и R'—группы (СН2)Ж или бензольны»-
кольца
Полиуретаны
Полимочевины
Г—С—NH—(CH^—NHCO—(CH2}„—О— 1
I II II
„о о . \„
Г_с—NH—(СНа)л—NHCNH—(CHa)„-NH-
Г II .11
О О
Карбамидо-формальдегидные полиме-
ры
Меламино-формальдегидные полиме-
ры
См. стр. 392, 393
То же
Серосодержащие полимеры
Простые политиоэфиры
{полисульфиды)
Полисульфоны
О
О
Кремнийорганические полимеры .
Элементортанические полимеры
R
—Si—О—
і
38?
Многие из приведенных выше полимеров находят весьма
разнообразное применение. Так, полиэтилен, полипропилен,
полиамиды, полиуретаны, полиэфиры применяются в производстве
пластических масс, пленок и химических волокон. Полиакрилаты
и полиметакрилаты перерабатываются главным образом в
пластические массы, а полиакрилонитрил используется для получения
химического волокна нитрон. Полибутадиен и его производные
(полиизопрен, полихлоропрен) являются синтетическими
каучуками, некоторые полиуретаны и кремнииорганические
полимеры также используются в качестве синтетических каучуков,
обладающих ценными свойствами.
Глава VI
ТЕХНОЛОГИЯ ИЗГОТОВЛЕНИЯ ПОЛИМЕРОВ
Подавляющее большинство полимеров, применяемых в
производстве химических волокон, резиновых изделий, пленок,
пластических масс, лаков, получают, как указывалось, синтетическим
путем из низкомолекулярных соединений (мономеров).
Соединение молекул низкомолекулярных веществ между собой с
образованием макромолекул полимера может происходить в результате
различных реакций, в зависимости от строения исходных
мономеров. Если в молекулах мономеров имеются функциональные
группы, вступающие в реакцию между собой, и процесс
присоединения молекул друг к другу сопровождается выделением
побочных низкомолекулярных продуктов, то процесс синтеза полимера
носит название реакции поликонденсации. В случае, когда синтез
полимера является следствием перегруппировок внутри
функциональных групп без изменения элементарного состава, такой
процесс называют ступенчатой полимеризацией. Если же молекулы
мономера содержат кратные связи или представляют собой
циклические соединения и образование макромолекул происходит в
результате раскрытия двойных связей или разрушения циклов
и не сопровождается выделением побочных продуктов, то процесс
получения полимера называется реакцией цепной
полимеризации. Поликонденсация и цепная полимеризация являются
наиболее распространенными способами получения полимеров.
Свойства полимеров, как синтетических, так и природных,
можно существенно изменить путем их химических превращений.
В результате подбора реагентов и определенных условий их
взаимодействия изменяются химический состав элементарных звеньев
или строение макромолекул {отверждение, вулканизация).
Все большее значение приобретают различные способы
модифицирования полимеров: совмещение их в виде сплавов или
химическое соединение макромолекул разных полимеров (синтез
блоксополимеров и привитых сополимеров, стр. 435).
384
/. ОБЩИЕ ЗАКОНОМЕРНОСТИ СИНТЕЗА ПОЛИМЕРОВ
Выбор аппаратуры для изготовления полимеров, подбор
режимов и способов проведения процесса определяются теми
закономерностями, которые характерны для реакций их образования.
ПОЛИКОНДЕНСАЦИЯ
К числу общеизвестных конденсационных процессов относится,
например, реакция этерификации:
RCOOH + R'OH т~^ Н2О + RCOOR'
и
или реакция получения амидов:
RCOOH + R'NH2 " Н2О + RCONHR'
Для реакции конденсации характерна обратимость процесса.
На начальных стадиях скорость прямой реакции vx во много раз
превышает скорость обратного процесса и2. Однако, по мере того
как в реакторе возрастает количество продуктов реакции,
увеличивается и скорость обратного процесса. С наступлением
равновесия, когда при выбранных режимах v2 становится равной vv
система приобретает стабильность. Для подавления обратных
реакций (в данных примерах образования эфира или амида) реакцию
необходимо вести при избытке одного из исходных компонентов и
по мере протекания реакции извлекать из реакционной массы
целевой или побочный продукт.
Чтобы реакция конденсации не заканчивалась только одним
актом присоединения молекул двух веществ с образованием
эфира, амида или других низкомолекулярных веществ, а многократно
повторялась и образующиеся продукты вовлекались бы в новые
реакции конденсации, необходимо наличие в молекулах исходных
веществ нескольких функциональных групп, участвующих в этой
реакции. При строгом соблюдении эквимолекулярности исходных
веществ удается получить высокомолекулярные соединения. Эфир
или амид, полученные из эквимолекулярных количеств
двухосновной кислоты и двухатомного спирта или, соответственно,
диамина, сохраняют функциональные группы, присущие исходным
реагентам:
НООС—R—СООН + НО—R'—ОН ; » НООС—R—COO—R'—ОН + Н2О
НООС—R—СООН + NH2—R'—NH2 ; * НООС—R—CONH—R'—NH2 + H2O
Поэтому полученные соединения могут вторично вступать в
реакцию конденсации. Во втором акте процесса конденсации вновь
образуется эфир или амид, сохраняющие прежние функциональ-
25—805 385
НЫе ГруППЫ, НО С более ВЫСОКИМ Молекулярным tJCUJM, -icm npvj^j.v
ты предыдущей реакции. Следовательно, полифункциональные
вещества могут вступать в одну и ту же реакцию конденсации
я-ное число раз, причем на каждой стадии новое вещество
становится более высокомолекулярным, сохраняя прежние
функциональные группы:
НООС—\~\—СООСН2—СН2—ОН +
НООС—\%— СООСН2—СН2—ОН (—»
—COOCHa-CH.OH + Н„о
НООС-(СН2L—CONH-(CH2)e—NH2 -f
4- НООС—(СНаL—CONH—(CHa)e—NHa -—*
ZZZl HOOC-(CH2L-CONH-(CH2)eNHOC(CH!!L—CONH—(CH2)e—NH24- HaO
Многократно повторяющийся процесс конденсации
полифункциональных веществ назван поликонденсацией.
Поликонденсация, проводимая в растворе или в расплаве,
так же как и одноактная реакция конденсации, подчиняется
закономерностям обратимых процессов. При подавлении обратной
реакции повторение одного и того же процесса конденсации даже
между веществами, молекулы которых имеют только по две
функциональные группы, должно происходить до тех пор, пока все
молекулы, находящиеся в реакторе, не соединятся в единую
макромолекулу. Однако нарушение строгой эквимолекулярности
исходных веществ приводит к прекращению процесса
поликонденсации на более ранних стадиях, вследствие того, что на обоих
концах макромолекул появляются одинаковые функциональные
группы. Например:
2НООС—R—СООН 4- ЗНО—R'— ОН -—*
7—* НО— R'-OOC-R—COO-R'—ООС—R-COO—R'—ОН 4- Н2О
ИЛИ
ЗНООС—R— СООН 4- 4NH2—R'—NHa •;—>
•t—» NH2—R'NHOC—R— CONHR'NHOC—R— CONHR'NHOC—R-CONHR'-NH2
Необходимость сохранения эквимолекулярности
бифункциональных веществ часто вызывает большие затруднения, так как
поликонденсация обычно протекает при температурах выше
100 °С. В этих условиях компонент с более низкой температурой
кипения (большей летучестью) не успевает полностью вступить
в реакцию и частично удаляется из реакционной среды вместе с
побочными продуктами.
386
Таким образом, равновесная реакция поликонденсации будеї
смещаться в сторону получения высокомолекулярного полимера
при обычных способах ее проведения только в том случае, если
будут создаваться благоприятные условия для своевременного
и полного удаления непрерывно образующихся побочных
продуктов. Однако это также встречает известные затруднения,
поскольку быстро возрастающая вязкость реакционной среды с
повышением молекулярного веса полимера препятствует
удалении! из сферы реакции даже низкокипящих побочных продуктов
реакции. Повышение температуры процесса поликонденсации,
облегчая удаление побочных продуктов, может в то же время
вызвать термическую деструкцию образующегося полимера.
Поэтому реакция полнконденсации бифункциональных веществ
прекращается на сравнительно ранних этапах (п^ЮО—150).
Форма макромолекул определяется количеством
функциональных групп в исходных продуктах, которые при выбранных
условиях участвуют в реакции поликонденсации. Если каждый из
реагентов содержит по две функциональные группы, то их
количество остается неизменным на всем протяжении процесса и
образуется полимер линейного строения. При большем количестве
функциональных групп в молекулах исходных мономеров
развитие цепи макромолекулы полимера происходит одновременно по
месту нескольких таких групп. При этом каждый новый акт
взаимодействия приводит к увеличению числа функциональных групп
в макромолекуле.
Примером может служить реакция полиэтерификации дикар-
боновой кислоты с пентаэритритом:
НОН2С ,СН2ОН
С + НООС—(СН2),—СООН z=z±
НОНаС/ \:Н2ОН себацииовая кислота
пеитааритрит
НОН2С CHjOOC—(СН2)„—СООСНа СН2ОН
^Н2О+ V V
СООНїС CHjOOC НОНаС СНаООС—(СНа)8—СООН
(СН2), (СН2)8
СООН СООСНа СНаОН
С СНаООС—
НООС—(СН2)8—СООН2С СНаООС—(СН2)8—СООН
Уже на третьей стадии этерификации молекула содержит
восемь функциональных групп. По мере дальнейшего присоедине-
25* 387
ния молекул друг к другу все быстрее нарастает число
функциональных групп в макромолекулах и становится все сложнее их
форма.
Возрастание полифункциональности макромолекул
способствует сохранению высокой реакционной способности полимера,
несмотря на быстро нарастающую вязкость среды и малую
подвижность макромолекул столь сложной формы. Образовавшиеся
побочные продукты, которые и в этом случае частично остаются
в реакционной массе, также смещают равновесие реакции,
вызывая гидролиз некоторых новых функциональных групп,
возникающих в макромолекулах полимера (например, сложно-
эфирных). Однако количество побочного продукта слишком мало,
чтобы порвались все связи, которые успели возникнуть между
такими полифункциональными макромолекулами, и полимер
превращается в единую пространственную макромолекулу.
Обратимость реакции в данном случае приводит лишь к появлению
разрывов (дефектов) в пространственной сетке полимера, число
которых тем больше, чем больше побочных продуктов остается в
реакционной массе. Такие разрывы в пространственной сетке
снижают твердость, прочность и теплостойкость полимера.
Поскольку реакция поликонденсации обратима на всех
стадиях, в реакционной среде всегда содержится некоторое
количество непрореагировавших исходных веществ (амины, спирты,
фенолы и др.). Эти вещества принимают участие в различных
побочных процессах, особенно в процессах разрушения
макромолекул, вступая в реакцию с возникшими в макромолекулярной цепи
новыми функциональными группами:
.. ._oc_R_co_o_R'_oloc-R-CO-0-R'-0— ¦ ¦ адкого^
Н—OR'
„ .. .—ОС—R—СО—О—R'—ОН + R'O—ОС—R—СО—О— R'— О
Вероятность таких побочных процессов возрастает с
увеличением длины макромолекул, а скорость реакции—с повышением
температуры. Взаимодействие между не вошедшими в реакцию
исходными веществами и макромолекулами полимера (аминолиз,
фенолиз, алкоголиз) приводит к резкому снижению среднего
молекулярного веса линейных полимеров. Поскольку в реакцию
вовлекаются преимущественно высокомолекулярные фракции,
размеры макромолекул линейных полимеров, получаемых методом
поликонденсации, мало отличаются друг от друга, т. е. такие
полимеры обладают сравнительно малой полидисперсностью.
Побочные реакции между полимером и не вошедшими в реакцию
низксмолекулярными веществами наблюдаются и в случае
поликонденсации полифункциональных веществ. При этом происходит
разрыв некоторого количества новых связей между макромоле-
388
кулами, что, так же как и обратимость реакции, приводит к
появлению разрывов в пространственной сетке полимера и
увеличивает число дефектов в ней.
Для процесса поликонденсации характерно то, что на всех
последующих стадиях реакция протекает по такому же механизму,
что и на первоначальных, и в тоже время каждая стадия
поликонденсации представляет собой самостоятельную реакцию, не
зависящую от предыдущих. Для осуществления каждой стадии
поликонденсации необходима затрата энергии, количество которой
определяется типом исходных веществ. Количество энергии
остается неизменным на протяжении всех актов присоединения и не
зависит от размера растущих макромолекул, поэтому скорость
реакции поликонденсации возрастает с повышением температуры.
Снижением температуры можно приостановить процесс
образования полимера на любой стадии.
На этом основана технология переработки полимеров,
получаемых из мономеров, которые содержат более чем по две
функциональные группы.
Реакцию поликонденсации таких мономеров проводят до
образования сравнительно низкомолекулярного полимера, легко
растворимого и легкоплавкого. Снижением температуры можно
настолько замедлить, поликонденсацию, что полимер, называемый
на этой стадии термореактивным, будет сохраняться в таком
состоянии в течение нескольких недель или месяцев.
*¦¦-*¦ Термореактивный полимер удобно подготовить в виде
пластической массы, клея, лака, заливочного компаунда, пенопласта,
затем сформовать изделие (или покрытие) и завершить при
нагревании реакцию поликонденсации, в ходе которой полимер
приобретает пространственную структуру и становится неплавким,
твердым, прочным и нерастворимым.
К числу полимеров с линейной структурой макромолекул
(термопластичные полимеры), получаемых методом
поликонденсации, относятся следующие:
1. Линейные полиэфиры (насыщенные и ненасыщенные),
получаемые из двухосновных кислот (соответственно насыщенных
или ненасыщенных) и гликолей:
пНООС-СН=СН-СООН + пНО-СН2-СН2-О-СН2—СН2-ОН ; >
малеиновая кислота диэтиленгликоль
-t— НО—[ОС—СН=СН—COOCRj— СН2— О—СН2—СН2—О—]„—Н +
полидиэтиленгликольмалеинат
+ Bп— 1)Н2О
Линейные полиэфиры применяются в производстве
пластических масс и химических волокон (например, лавсана).
389
2. Полиамиды—продукты поликонденсации двухосновных
насыщенных кислот и диаминов или аминокислот:
яНООС—(СН2L—СООН + /iH2N—(CHa)e—NHa ^Г^
адипиновая кислота гексаметнленднамин
Z=i НО—[— ОС—(СНаL—COHN—(СНа)„—NH—]„—Н + Bя + 1) Н2О
пол и гексаметил єна дипамкд
^-HaN-(CH3)e-COOH + ~HaN-(CH2)„-COOH ^=*
амииоэнантовая кислота
<—» Н—[—HN—(СН2)в—СО—]п—ОН + (я + 1) НаО
полнэнантоамид
Из полиамидов производят химические волокна (капрон,?най-
лон, энант), пластические массы и стойкие к истиранию покрытия
для трущихся поверхностей металлических деталей.
3. Полиэпоксиды, получаемые из двухатомных фенолов и эпи-
хлоргидрина:
СН8
пНО—<^~Ч—С—
~ СН,
—ОН + лСІСНо—СН—CHj
V
дифекилолпропан
н
СН,
-о—
СН3
эпихлоргидрин
ь—О—СН2—СН—СНа—
I
он
СН3
Bп — 1) НС1 + СНа-СН—СНа—О—
о
сня
Полиэпоксиды применяются в производстве пластических
масс, лаковых покрытий, клеев, заливочных компаундов,
герметизирующих составов.
4. Линейные полисилоксаны, образующиеся при поликонден-
еации силандиолов
СН3
СН3
-^-HO-Si—ОН + -4-НО—Si-OH
2 2 !
СН, СН,
но—
СНа
сн3 .
—Н + Н,0
димьтнлеиланднол
полиднметиленлоксан
390
используют в качестве синтетического каучука, смазок
масел.
5. 'Полисульфиды (тиоколы)—продукты поликонденсации
хлоралкилов с полисульфидом натрия:
яС|—СН2—СН„—О—СН2—СН2—С1
дихлордиэтиловый эфир
ди-
CI—Г—СН2—СН2—О—СНа—СНа—S—S—]—Na
II I!
S S
NaCl
Uo
но—г—сн2—сн2—о—сна—сн2—s—s—і—и
ss
s—і
s і
Они находят применение в качестве каучуков, герметизирующих
составов и др.
Для современной техники требуются линейные полимеры
различного молекулярного веса, например полисилоксаны с
молекулярным весом около 1000 и около 1 000 000. Первые имеют
жидкую консистенцию и применяются в качестве
трансформаторного масла, обладающего повышенной теплостойкостью и
хорошими диэлектрическими свойствами. Полисилоксаны с более
высоким молекулярным весом представляют собой густо-вязкие
продукты, используемые в качестве смазок для деталей, работающих
при низких (ниже —20 °С) и высоких B00—350 °С) температурах.
Высокомолекуля ные полисилоксаны служат сырьем для
производства теплост йких резин.
Чтобы прекратить поликонденсацию на стадии образования
полимеров сравнительно низкого молекулярного веса—олигомеров,
охлаждают реакционную смесь, нарушают эквимолекулярность
соотношения бифункциональных мономеров или вводят в
реакцию наряду с бифункциональными веществами
монофункциональные компоненты:
СНд СНу
mHO—Si—ОН + 2Н3С—Si-OH ^=1
СНо СНо
СНЯ
Н3С—Si—О—
сня
сня
_Si—О—
Шя
сн3
СН3+Н2О
Олигомеры, получаемые при несоблюдении эквимолекулярности
исходных веществ, не сохраняют на концах макромолекул
прежних функциональных групп, поэтому при последующем нагрева-
391
ний они не могут вновь вступить в реакцию поликонденсации.
Олигомеры, синтез которых приостановлен путем охлаждения
реакционной смеси на термореактивной стадии, сохраняют
функциональные группы на концах макромолекул. К таким олигомерам
относятся резольные. феноло-формальдегидные, карбамидные, крем-
нийорганические полимеры.
Феноло-формальдегидные полимеры получают
поликонденсацией смеси изомерных оксибензиловых спиртов и диметилол-
фенолов (образующихся в результате взаимодействия фенола с
формальдегидом, взятым в избытке). Молекулы каждого
компонента такой смеси содержат по три функциональные группы (гидр-
оксильные, метальные группы и незамещенные водородные атомы
в орто- и пара-положениях, отмеченные в формуле звездочками),
которые могут принять участие в реакции поликонденсации:
ОН ОН
I О |
он
он
При избытке фенола реакция заканчивается на стадии
образования олигомера, называемого новолаком.
Исходными продуктами для синтеза карбамидных полимеров
служат смеси моно- и диметилолкарбамида или ди- и триметилол-
меламина. Метилольные производные карбамида, получаемые
присоединением к карбамиду формальдегида, взятого в избытке,
392
содержат по четыре функциональные группы, участвующие в
реакции поликонденсации:
NH2 О HN—СН2ОН HN—СН2ОН
I II I I
1,5лС=О + 2лСН2 > лС=О + 0,5лС=О < >
| | | -н2о
NH2 NH2 HN-CH2OH
мокометклол- диметилол-
карбамид карбамид
N-CH2-N-CH2-N-CH2
¦^zz t=o с=о с=о
NH/ NH NH2
СН2
N—CH2—N—СН2
t==O С=О
NH2 NH2
Метилольные производные меламина, содержащие по шесть
функциональных групп, образуются в результате присоединения
формальдегида к меламину:
N
J \ О
H2N-C С—NH2 I!
2 | || +5СН2 >
N N
V
NH2
НОН2С N СН2ОН НОН2С N СН2ОН
—» I II + I II
NN NN
V V
NH2 HN—CH2OH
диметилолмеламик триметилолмеламин
3. Кремнийорганические полимеры получаются путем
поликонденсации смеси силандиолов (I) и силантриолов и (II):
СН3 СН3
НО—Si—ОН НО—Si—ОН
сн, он
'3
II
394
Реакция протекает по схеме:
СН3 СН3
| I -нао
«НО—Si—ОН+лНО—Si—ОН^ІГІ
сн, он
сн„ сна
—Si-O—Si— О—
сн3
о
СН, СН3 СН3 СН3 СНз | СНЭ
7~^ —Si—О—Si—О—Si—О—Si—О—Si—О—Si—О—Si—О—
СН„ СН3 I СН3 СНэ СН8 |
СИ, I СН3 I СНэ
_О—Si—О—Si—О—Si—О— — Si—О—Si—О—
СН3 СНз СНЭ СНЭ СН3
В "отличие^от низкомолекулярных полимеров, образующихся
в результате поликонденсации бифункциональных веществ при
нарушении эквимолекулярности их соотношения или в
присутствии монофункциональных добавок, термореактивные полимеры
при хранении или быстро при нагревании превращаются в
высокомолекулярные соединения пространственного строения.
ПОЛИМЕРИЗАЦИЯ
В зависимости от протекания этой реакции различают цеп
ную и ступенчатую полимеризацию. В обоих случаях реакция
происходит без выделения побочных продуктов и не обратима.
Особенностью цепной полимеризации является зависимость
всех последующих актов роста макромолекулы от
предшествующих, поскольку энергия, приобретенная в первом акте
(активация), сохраняется неизменной в течение многочисленных
последующих актов присоединения новых молекул мономера к растущей
цепи. Энергия активации затрачивается на превращение части
молекул мономера в радикалы (радикальная цепная полимеризация)
или в ионы (ионная цепная полимеризация), которые становятся
центрами образования макромолекулярных цепей, сохраняющих
в течение всего процесса их роста строение радикалов или ионов.
В реакциях ступенчатой полимеризации на каждый акт
присоединения молекулы мономера к макромолекуле требуются
затраты энергии для внутримолекулярных перегруппировок. В
любой промежуточной стадии роста на концах макромолекул
сохраняются функциональные группы, что обеспечивает
продолжение реакции.
394
Цепная полимеризация
Реакции цепней полимеризации характерны для
ненасыщенных или циклических веществ. К числу ненасыщенных соединений,
особенно широко применяемых в производстве полимеров,
относятся этилен и ряд винильных соединений, а также производные
акриловой кислоты и бутадиена:
—CH2—CH2—)п
полиэтилен
С1
пСН2=СН
хлористый
ВИНИЛ
nCF2=CF2
тетрафтор-
этнлен
пСН2=СН *
ОСОСНа
винилацетат
СН2=СН
1
¦ 0-
стирол
СН
пСИ -СН
3
*l-CH,-CH-J„
пропилен полипропилен
г сі і
.!_—сн2—сн—\п
поливинилхлорид
> (_CF2-CF2-)n
пол итет ра фто р-
этилен
Г—CHj-CH "I
L 0COCH3J„
пол ивн н ил ацетат
-_сн2—сн—-
1
. с
полистарол
п
сна
;=С
-СН2-С
соосн.
метилметакрилат
пСНа=СН—СН=СН2 —
бутадиен
СНа
пСН2=С—СН=СН2
взопрей
С1
пСН2=С—СН=СНа —
хлоропрен
пол »метилметакрилат
_¦ (—СН2—СН=СН—СН2—)„
полибутадиен
f Г 1
-¦ L-CH2—СН=СН— СН2—1
полиизопреи
С1
1
-. (—СН2—С=СН—СНа—)„
полихлоропрен
В реакцию цепной полимеризации легко вступают и
неустойчивые циклические соединения, содержащие в цикле атом
кислорода или атом азота, например:
пСН2—СН2 •
V
окись этилена
сн2
о/чо
л | -
СН-2 Сгі2
\/
О
триоксиметилен
СН2—СН2—СН2 ^
п
со —
сн2-сн2— nh/
капролактам
полиэтиленоксид
— (-СН2-О-)„
полиформальдегид
¦* [-NH-(CH2-M-CO-J„
поликапроамид
Для образования макромолекулы одна из молекул
ненасыщенного или циклического вещества должна быть переведена в
состояние высокой активности. Такая молекула приобретает
способность вступать в реакцию с неактивированными молекулами,
последовательно присоединяя их. Реакционная способность
растущей цепи при этом не утрачивается. Активация молекул
ненасыщенного или циклического соединения связана с разрывом
двойной связи или разрушением цикла. Если в результате
разрыва связи молекула превращается в радикал, происходит
радикальная полимеризация. Разрыв кратной связи молекулы
может привести к образованию иона, в этом случае реакция
протекает по законам ионной полимеризации. Если начальный ион
приобретает положительный заряд, происходит катионная
полимеризация, а в случае образования отрицательно заряженного
иона—анионная полимеризация.
Разрыв двойной связи ненасыщенных соединений с
превращением их в радикалы обычно осуществляется под действием очень
неустойчивых веществ—перекисей, гидроперекисей, диазосоеди-
нений, которые служат инициаторами радикально-цепной
полимеризации. С повышением температуры такие вещества легко
образуют высокоактивные радикалы:
О О
перекись бензоила радикал
Распад перекисей и гидроперекисей ускоряется при
добавлении малых количеств так называемых промоторов, в качестве ко-
396
торых применяются соли двухвалентного железа, нафтената
кобальта и другие вещества.
Если распад инициатора на радикалы происходит в среде
ненасыщенного мономера, то радикалы, по мере их возникновения,
присоединяются к молекулам мономера, образуя начальные
радикалы реакции полимеризации. Этот первый акт процесса
полимеризации назван реакцией активации:
А. + СН2=СН А-СН2-СН
I I
R R
радикал молекула начальный радикал
инициатора мономера полимеризации .
Для завершения активации молекулы мономера требуется
довольно длительное время, в течение которого происходит
накапливание энергии, достаточной для распада молекулы инициатора
на свободные радикалы и присоединения его к молекуле
мономера. С повышением температуры увеличиваются скорость
активации и количество одновременно образовавшихся начальных
радикалов реакции полимеризации.
Каждый начальный радикал присоединяет неактивированные
молекулы мономера, сохраняя приобретенную энергию
свободного радикала и превращаясь в макрорадикал, длина которого
непрерывно возрастает. Этот процесс носит название роста цепи:
R R
. +СН2=СН • +СН2=СН
A—CH.J—СН *- А—СНг—СН—СНа—СН *¦
> А—СН2—СН—СН2—СН—СН2—*СН
I I I
R R R
Рост цепи происходит с очень высокой скоростью.
Если распад молекул инициатора протекает в смеси двух
ненасыщенных веществ, то в росте макрорадикала принимают
участие молекулы обоих мономеров. Такой процесс назван
совместной полимеризацией или сополимеризацией:
R' R
I I
+СНа=СН • 4-СНа=СН
А-СНа-СН *¦ А-СН2—СН-СНг-СН *¦
R
А—СНа—СН—СНа—СН—СНа—СН
R R' R
397
Соотношение элементарных звеньев оооих мономеров в ма
молекуле сополимера и их взаимное расположение в ней
определяются условиями проведения реакции, соотношением
мономеров в исходной смеси и их активностью в процессе
полимеризации.
Рост макрорадикалов заканчивается в результате их
столкновения между собой, приводящего к соединению макрорадикалов
в макромолекулу или к перераспределению атомов в конечных
»веньях макрорадикалов (обрыв цепи). Этот процесс можно
представить в виде следующей схемы:
А—(—СНа—СН—)„,—СНа—СН + СН—CHj—(—СН—СНа—)„,—А .
і к к к
». А—(—СН2— СН)„, —СНа—СН—СН—СНа—(—СН—СНа—)да—А
і і і к
ИЛИ
-(—СНа—СН-)„,-СН,—*СН + СН—СН2—(—СН-СНа—)„„— А
к к к к
)„+ 2(
к к к к
> А—(-Ша-СН—)„,—СН=СН + СНа—СНа-(—СН-СНа—)„»—А
R R R R
Чем выше концентрация одновременно растущих макроради
калов, тем больше вероятность прекращения их роста вследствие
столкновения друг с другом. Прекращение роста одной
макромолекулы может послужить источником начала роста другой
(передача цепи). В качестве примера приведем реакцию
полимеризации в присутствии растворителя, легко вступающего в реакцию
с макрорадикалами с образованием нового радикала:
А—(—СНа—СН—)„—СНа—СН + ССІ* >
R
CC13 + A—(—СНа—СН—)„—СНа—СНС1
Если вновь образовавшийся радикал достаточно активен дли
разрыва двойной связи молекул мрномера, то начинается рост
нового макрорадикала:
С13С + СН2=СН > С13С—СН2—СН
398
Такие растворители носят название телогенов. Изменяя
концентрацию телогена в мономере, можно регулировать среднюю
длину макромолекул и получать методом радикально-цепной
полимеризации олигомеры или полимеры различного
молекулярного веса.
Мономеры, молекулы которых имеют кратные связи или
представляют собой неустойчивые циклы, могут вступать в реакцию
с ионами (катионами или анионами). Этот процесс
сопровождается изменением количества электронов в молекуле мономера, т. е
превращением ее в катион или анион:
Н++ СН2=С:Н » Н-СН2-СН
R
начальный катноп
Є
2=СН
НО" + СН2=СН » НОСН2—СН
А
R
начальный аннон
НО +¦ СНа-СН2 * НО—СН2-СН2-О
і _2Х начальный аннон
Вещества, легко диссоциирующие в среде мономера на ионы,
вызывающие реакцию полимеризации, названы катализаторами
полимеризации. Начальные ионы инициируют рост
макромолекул, который на каждой ступени сопровождается перераспреде
лением электронов и сохранением в конечном звене цепи струк
туры иона:
ОД,—СН » ОД,—СН—СНа—СН * СН3—СН-СН2-СН-СН3-СН
I,
Прекращение роста макроиона происходит в результате
столкновения его с противоионом катализатора или какими-либо
веществами, специально вводимыми в реактор (регуляторы моле
кулярного веса). Диссоциация катализатора и возникновение на
чальных ионов полимеризации требуют весьма малой энергии
активации, поэтому процесс ионно-цепной полимеризации можеі
происходить при очень низкой температуре (от —40 до —60 °С).
Рост каждого макроиона происходит за доли секунды. Реакция
обрыва цепи в процессе ионной полимеризации менее вероятна и
протекает с меньшей скоростью, чем для макрорадикалов
(поскольку одинаково заряженные макроионы не могут реагиро
вать между собой). Поэтому средний молекулярный вес полимеров,
получаемых методом ионно-цепной полимеризации, выше
молекулярного веса полимеров, образующихся в процессе
радикально-цепной полимеризации.
Рост макрорадикала и макроиона может прекратиться также
в результате столкновения их с молекулами примесей,
присутствующих в мономере. Поэтому для получения полимеров с высоким
молекулярным весом необходима тщательная очистка исходных
мономеров.
Рост цепи макромолекулы и его прекращение сопровождаются
выделением тепла, которое расходуется на активацию роста
новых макромолекул или на разнообразные побочные
процессы.
Так, под влиянием тепла часть молекул мономера или звеньев
макромолекул могут вступить в реакцию с кислородом воздуха,
в результате чего появляются новые структурные элементы,
например:
ООН
СН2—СН + О2 » СН2—С »
С
о«
-» СН2—С ЬНОі
Возникшие новые радикалы могут стать источником роста
боковых ответвлений в макромолекуле:
СН2-С
Одновременно могут происходить и побочные реакции между
функциональными группами звеньев одной и той же
макромолекулы
СН2-СН—СН2—СН > СН.2-СН-СН2-СН
¦ | -Н«О | ,
соон соон о=с—о—с=о
или между группами соседних цепей:
СН2-СН-СН2-СН— — СН2—СН-СН2-СН
соон соон с=о соон
-ЫаО |
соон ¦ с=о
сн2—сн—сн2—сн— —сн2—сн—сн2—сн
соон соон
Побочные процессы нежелательны, поскольку они приводят
к изменению формы макромолекул, их состава и, следовательно,
к изменению свойств полимера. Поэтому для многих процессов
полимеризации требуется интенсивный отвод тепла, или их
следует проводить в среде азота и при возможно более низкой
температуре.
Полимеризация, инициируемая перекисями или
гидроперекисями, распад которых происходит при температуре 60—100 °С
приводит к образованию полимера с наиболее низким средним
молекулярным весом и наименее регулярным строением
макромолекул. Введение промоторов снижает температуру распада
инициатора и дает возможность проводить радикально-цепную
полимеризацию при 5—10 °С (низкотемпературная
полимеризация), что в значительной степени способствует повышению
качества полимера.
Ионную полимеризацию можно проводить при очень низких
температурах. Это предотвращает протекание побочных
процессов, поэтому данным методом получают полимеры бо„;ее
регулярной структуры. При соответствующем подборе катализатора
ионной полимеризации возможна строгая взаимная ориентация в
пространстве боковых групп в молекулах мономера в момент их
присоединения к макроиону. Это позволяет придать строению
макромолекул высокую регулярность и получить стереорегуляр-
ные полимеры. Чем регулярнее строение макромолекул, тем выше
степень кристалличности полимера.
Ступенчатая полимеризация, также как и поликонденсация,
является процессом взаимодействия функциональных групп,
имеющихся в молекулах мономера, и протекает через стадии
образования димеров, тримеров, тетрамеров и последующих полимер-
гомологов. На каждой стадии этого процесса образуются
молекулы (но не радикалы или ионы), которые сохраняют способность
в данных условиях вступать в реакцию дальнейшего
присоединения. Однако в отличие от реакции поликонденсации
ступенчатая полимеризация не сопровождается образованием побочных
продуктов и не является обратимой реакцией, рост
макромолекулы происходит с высокой скоростью и с выделением тепла.
Реакция останавливается, когда система становится слишком
2Г>—805 401
вязкой, вследствие чего функциональные группы громоздких
макромолекул лишаются возможности приблизиться друг к другу.
Повышая температуру, можно продолжить реакцию до более
глубокой стадии превращения (полимеризации).
Методом ступенчатой полимеризации синтезируют ряд
полимеров, в том числе полиуретаны, полиэфироуретаны.
Полиуретаны получают из двухатомных спиртов и диизоциа-
натов:
НО—R'—ОН + О=С=Іі—R—N=C=O »
HO-R'-OH
* НО—R'—О-С—N—R-N=C=O
II I
о н
* HO—R'—О—С—N—R—N—С—О—R'—ОН и т. д.
ОН НО
При синтезе полиэфироуретанов вместо двухатомных спиртов
применяют низкомолекулярные полиэфиры (молекулярный вес
до 5000), на концах цепей которых находятся гидроксильные
группы:
О
II
НО—[—R—ООС—R'—СОО—]„—R—ОН C=N
4- R' *
НО—[—R—ООС—R'—COO—]„--R—ОН C=N
II
О
НО—[-R—ООС—R'— СОО—]„—R-0
HN—С=.О
HN—С=О
О—R—[OOC-R'— COO—R—]„—ОНит а
2. СПОСОБЫ ИЗГОТОВЛЕНИЯ ПОЛИМЕРОВ
Синтез полимеров методами полимеризации или
поликонденсации можно проводить в среде только исходного
вещества или в смеси исходных веществ и катализатора или инициа-
402
тора реакции. Такой процесс изготовления называют блочным
способом или синтезом полимера в среде мономера. По мере того
как мономер вступает в реакцию образования полимера,
концентрация мономера в реакторе постепенно снижается, а вязкость
среды значительно возрастает вследствие увеличения
концентрации полимера в мономере(в случае полимеризации) или
увеличения среднего молекулярного веса продуктов реакции (для
процесса поликонденсации). Блочный способ изготовления полимеров
связан с рядом технических трудностей. В значительной мере они
вызваны необходимостью строго соблюдать тепловой режим
процесса и равномерно распределять тепло в реакционной среде,
так как от этих условий зависит средний молекулярный вес
полимера и регулярность строения макромолекул. В ходе процесса
синтеза полимера вязкость реакционной массы возрастает
настолько, что равномерная передача тепла от стенок реактора или от
внутренних слоев реакционной массы даже при ее перемешивании,
взбалтывании, вспенивании и др. становится все более
затруднительна, а во многих случаях и невозможна.
Цепную полимеризацию необходимо вести при возможно более
низких температурах, чтобы получился полимер с наиболее
высоким молекулярным весом и наиболее регулярной структурой
макромолекул. Однако начальная температура радикально-цепной
полимеризации определяется температурой распада инициатора
и скоростью активации молекул мономера. В то же время рост
макрорадикалов протекает с высокой скоростью и
сопровождается выделением тепла. Накапливание тепла в реакционной массе
повышает скорость распада еще неиспользованного инициатора
и, следовательно, скорость образования новых центров роста
макромолекул. В конце концов в системе может накопиться
такое количество тепла, что произойдет термическая
деструкция образующегося полимера, а еще непрореагировавший
мономер будет вступать в нежелательные побочные реакции.
Ионно-цепная полимеризация проходит со значительно
большей скоростью, чем радикальная, поэтому своевременный отвод
тепла, выделяющегося во время реакции, становится особенно
трудным. Поскольку образование начальных ионов может
происходить и при низких температурах, мономер охлаждают до
минус 60—минус 80 °С еще до начала реакции, что предотвращает
перегрев полимера хотя бы на первых стадиях процесса
полимеризации. Однако при низких температурах вязкость раствора
полимера в мономере быстро нарастает по мере повышения
концентрации полимера, затрудняя дальнейший отвод тепла из глубинных
слоев.
Таким образом, в начале реакции радикальной
полимеризации требуется подвод тепла или применение высокоэффективных
промоторов, а на всех последующих стадиях (при любых способах
полимеризации) необходим интенсивный отвод тепла.
26* 403
Реакция поликонденсации на всех ее стадиях является
эндотермическим процессом, но для смещения равновесия этой реакции
в сторону образования полимера (особенно в конечных стадиях
процесса) требуется непрерывный отвод побочных продуктов из
сферы реакции, что становится все более трудным по мере
повышения вязкости реакционной массы.
Процессы поликонденсации полифункциональных веществ
приостанавливают на стадии образования сравнительно
низкомолекулярного промежуточного продукта (термореактивного
полимера), т. е. задолго до наступления равновесного состояния
системы. На таких ранних стадиях процесса побочные вещества
можно оставлять в реакционной массе, так как в этих условиях
скорость обратной реакции еще невелика. При поликонденсации
бифункциональных веществ (кроме синтеза олигомеров) обычно
стремятся получить полимер с наиболее высоким молекулярным
весом, т. е. сдвинуть равновесие системы в сторону реакции
поликонденсации. Для этого требуется удалять из системы побочные
продукты и непрореагировавшие компоненты, а также
увеличивать подвижность макромолекул снижением вязкости среды.
Следовательно, поликонденсацию бифункциональных веществ
целесообразно проводить при температуре кипения побочных
продуктов и постепенно повышать ее по мере того, как нарастает
вязкость среды и затрудняется удаление газообразных побочных
веществ.
Увеличение вязкости среды затрудняет равномерное
распределение в системе тепла, подводимого через стенки реактора,
что приводит к местным перегревам реакционной массы и
частичной деструкции полимера.
Отсюда можно сделать вывод, что изготовление полимеров
блочным способом возможно лиївь в следующих случаях:
1. При малом объеме реагирующих веществ и достаточно
большой поверхности теплообмена (например, поверхность формы),
когда обеспечивается равномерное распределение тепла в
реакционной массе.
2. При получении полимера, достаточно термостабильного
(хотя бы в среде азота), который при высокой температуре
сохраняет жидкотекучее состояние. Это позволяет интенсивно
перемешивать полимер в реакторе, т. е. облегчается регулирование
подвода (или отвода) тепла и удаление побочных продуктов.
3. Если реакцию можно заканчивать на ранних стадиях, т. е.
при низкой концентрации полимера в мономере (в процессе
полимеризации) или по образовании полимера со сравнительно
низким средним молекулярным весом (в процессе поликонденсации),
когда реакционная масса еще сохраняет низкую вязкость.
Блочным способом получают полимеры или заготовки простой
конфигурации, заливая мономер в формы, в которых одновременно
с образованием полимера происходит формование заготовки.
404
Блочной полимеризацией метилметакрилата изготовляют,
например, органические стекла, полимеризацией капролактама в
формах—заготовки поликапроамида. Блочный способ применяется при
синтезе полиамидов и полиэфиров поликонденсацией
бифункциональных веществ при высоких температурах (средний
молекулярный вес получаемых полимеров 10 000—12 000). Процесс блочной
полимеризации винилацетата или стирола заканчивают при
частичном превращении мономера в полимер; после отделения
полимера возвращают мономер в новый цикл полимеризации.
Большинство процессов синтеза полимеров проводят в какой-
либо жидкой среде. В зависимости от характера среды и степени
диспергирования в ней продуктов реакции различают четыре
способа проведения указанных процессов: лаковый, в среде оса-
дителя, эмульсионный, суспензионный или гранульный.
Лаковым способом называют процесс синтеза полимера,
проводимый в растворителе, который растворяет исходные вещества
и полученный полимер, но не принимает участия в реакции.
При изготовлении полимера в среде осадителя в качестве
жидкой фазы подбирается вещество, которое растворяет только
мономер и не растворяет образующийся полимер.
Весьма распространенный эмульсионный способ заключается
в том, что синтез полимера проводится в жидкой среде, не
являющейся растворителем ни для мономера, ни для полимера.
Мономеры и образующийся полимер эмульгируются в этой среде при
помощи специально вводимых эмульгаторов. Реакция начинается
в мицеллах эмульгатора, содержащих также инициатор.
Мономер постепенно проникает в мицеллы, где взаимодействует с
инициатором и превращается в полимер.
Предварительно растворив инициатор в исходном мономере,
можно путем интенсивного перемешивания диспергировать
мономер в виде мелких кгпель в нерастворяющей его жидкой среде.
При нагревании такой смеси каждая капля постепенно
превращается в полимер. После прекращения перемешивания мелкие
гранулы полимера осаждаются на дно реактора. Такой способ
синтеза полимера назван суспензионным способом.
ПЕРИОДИЧЕСКИЕ СПОСОБЫ
Независимо от того, каков механизм процесса синтеза
полимера и каким из описанных выше методом его
получают, изготовление полимера может быть осуществлено
периодическим или непрерывным способом. Периодический способ
является первой ступенью развития технологии полимеров. Переход
на непрерывные методы синтеза полимеров в ряде случаев
затрудняется некоторыми особенностями реакций. Применение
непрерывного способа иногда ограничивается малым объемом
производства отдельных видов полимеров.
405
іиііовое оборудование
Несмотря на многообразие применяемых исходных веществ
и процессов, приводящих к образованию полимеров*, а также на
различие способов и технологических режимов, получение
большого числа полимеров периодическими методами проводится в
типовом оборудовании.
Основным аппаратом установок периодического действия в
производстве полимеров служит реактор. В производстве
полимеров, получаемых поликонденсацией, реактор принято
называть варочным котлом, получаемых
полимеризацией—полимеризатором. Емкость реакторов различна. Корпус реактора / (рис. 109)
'Вакуумная линия
Катализатор
Сырье
Іобавки
Рис. 109. Типовая установка для синтеза полимеров периодическим
методом:
/—реактор; 2—люк для слива готового продукта; 3—конденсатор; 4—сборник; 5—смотровые
фонарн.
имеет цилиндрическую форму и заканчивается
сферическим днищем, в центре которого предусмотрен люк 2 для слива
готового продукта. Реактор закрывают крышкой, на которой
установлен привод якорной мешалки. В крышке имеются люки
для чистки котла и штуцеры для взятия проб, замера температуры
в реакторе, загрузки компонентов, отвода газообразных веществ.
* Полимеры, применяемые в резиновой промышленности, называют
синтетическими каучуками; полимеры, предназначенные для изготовления
изделий из пластмасс, получения лаков и клеев, иногда называют смолами.
406
Внутренняя поверхность реактора и мешалка покрыты
термоустойчивой и химически стойкой эмалью. Реактор, а также
крепление штуцеров и болты, соединяющие крышку реактора с
корпусом, рассчитаны на возможное избыточное давление внутри
реактора A0—20 am). В некоторых случаях реактор рассчитывают нз
работу при более высоком давлении, такие реакторы называются
автоклавами. Обогрев или охлаждение реакционной массы
производится через стенки реактора жидкостью или паром,
циркулирующими в рубашке (кожухе), приваренной к котлу на а/3 его
высоты. Для равномерного распределения тепла в реакционной
массе и смешения исходных веществ реактор снабжается
мешалкой якорного или пропеллерного типов, в зависимости от условий
проведения процесса. При нагревании продуктов реакции часть
из них может перейти в парообразное состояние. Пары поступают
в трубчатый конденсатор 3, конденсируются и возвращаются в
реактор. На конечных стадиях реакции, когда требуется удалить
мономер, не вступивший в реакцию, или получить полимер с
более высоким молекулярным весом и одновременно удалить легко-
испаряющиеся побочные продукты, или же освободить полимер
от растворителя, к конденсатору подключают сборник 4, куда
отводят удаляемые вещества. Для более интенсивного извлечения
.низкомолекулярных продуктов сборник в ряде случаев
подключают к вакуум-насосу.
Некоторые полимеры, даже при повышенной температуре,
могут иметь такую высокую вязкость, что затрудняется
перемешивание реакционной массы и выгрузка продукта через нижний
штуцер. В этом случае реакцию проводят без перемешивания
(мешалка в котле отсутствует), а в реактор вставляют металлический
стакан или форму, заполняемые реакционной массой. По
окончании реакции стакан или форму с полученным полимером
выгружают из реактора и заменяют другим стаканом или формой.
На установках описанного типа изготовляют полимеры,
при синтезе которых не требуются интенсивное охлаждение и
высокое давление, а также отсутствует необходимость непрерывного
получения полимера.
В типовом оборудовании периодического действия изготовляют
линейные полимеры методом поликонденсации; проводят
поликонденсацию полифункциональных веществ до термореактивной
стадии; получают некоторые синтетические каучуки; проводят
начальную стадию полимеризации стирола, метилметакрилата,
получая так называемые форполимеры—растворы полимера в
мономере. Синтез проводится блочным, лаковым, суспензионным
и эмульсионным методами.
Блочный способ. На установках периодического действия
блочным способом изготовляют полибутадиены (бутадиеновые
каучуки), форполимеры стирола, метилметакрилата, винилаце-
тата.
407
олочным способом получают по реакции поликонденсации
насыщенные и ненасыщенные полиэфиры, полиамиды, полизфиро-
уретаны, полиэпоксиды.
Среди насыщенных линейных полизфиров все большее
значение приобретает полиэтиленгликольтерефталат. Процесс
получения этого полиэфира состоит из двух стадий:
Первая стадия—переэтерификация диметилового эфира тере-
фталевой кислоты этиленгликолем и удаление из сферы реакции
побочного продукта—метилового спирта:
СН3ООС— \~\—СООСНз + НО—СН2—СН2—ОН *
диметилтерефталат этиленгликоль
* НО—СН2—СН,-ООС— (^^>-СОО-СН2-СН3-ОН + СНЭОН
диэтиленгликольтерефталат
Реакцию проводят при двукратном избытке этиленгликоля
в присутствии катализатора—ацетата цинка при температуре
200 °С и атмосферном давлении.
Вторая стадия—поликонденсация диэтиленгликольтерефталата
с образованием полиэфира и удаление из реакционной массы
избытка этиленгликоля:
HO-CH2-CH2-OOC-^J^-COO-CH2-CH2-OH +
+ НО—СН2—СН2— ООС— \~\—СОО—СН2—СН2—ОН *
—соосн2сн2оос_ <!у%—соосн2сн2он+
+ НО-СНа-СН2—ОН
Реакцию проводят при остаточном давлении не выше 1 мм
рт. ст. и температуре до 280 °С. Эти условия необходимы для
более тщательного удаления этиленгликоля и получения
полимера с молекулярным весом 25 000—35 000.
На рис. ПО приведена технологическая схема получения
полиэтиленгликольтерефталата. В аппарате / диметилтерефталат
расплавляется при 150 °С и перекачивается в реактор 2, где
проводится переэтерификация. Реактор обогревается парами динила
(дифенильная смесь, стр. 130), подаваемого в рубашку.
Выделяющиеся пары метилового спирта охлаждаются в конденсаторе 4
и отводятся из реактора. Диэтиленгликольтерефталат вместе с
избыточным этиленгликолем перекачивают в автоклав 3,
соединенный через конденсатор с вакуум-насосом. Это облегчает более
полное удаление этиленгликоля в процессе поликонденсации. По
окончании реакции полиэфир в виде вязкого расплава
передавливается сжатым азотом через нижний штуцер реактора на
вращающийся приемный полый барабан 5, охлаждаемый водой. Охлаж-
408
денная лента полимера подхватывается направляющими валками
и передается в дробилку 6 на измельчение.
Полиэтиленгликольтерефталат, известный под названием
лавсан, применяют для изготовления химических волокон и пленок,
отличающихся высокой прочностью, атмосфероустойчивостью и
повышенной (по сравнению с природными волокнами)
теплостойкостью.
К числу ненасыщенных линейных політЦшров, получаемых
блочным способом, относится полималеинат—продукт совместной
полиэтерификации малеиновой и фталевой кислот с этилен- или
диэтиленгликолем.
Диметил-
тересрталат
Зтилен- '
гликоль
/ранулы
полимера
Рис. 110. Схема получения полиэтиленгликольтерефталата:
/—аппарат для расплавления диметилтерефталата; 2— реактор; 3—автоклав
для поликонденсации; 4—конденсаторы; 5—барабан; в—дробилка.
Схема получения полималеината, типичная для производства
ненасыщенных полиэфиров, приведена на рис. 111. Синтез
полиэфира проводится в реакторе 4, снабженном конденсатором 5,
который соединен с вакуум-насосом для более полного удаления
реакционной воды. В реактор из бункера 3 загружают фталевую
кислоту, из бункера 2—малеиновую кислоту, а из сборника 1—
этилен- или диэтиленгликоль. Полиэтерификацию проводят при
170—220 °С до тех пор, пока кислотное число реакционной массы
не снизится до 40 мг КОН на 1 г смеси. Это соответствует
образованию полиэфира с молекулярным весом около 3500. Реакционную
смесь далее охлаждают до 50 °С и передавливают сжатым азотом
в аппарат 6, где полизфир растворяют в мономере, например в
стироле, подаваемом из сборника 7. Полученный раствор пили-
409
^.j-i.jy^ ^..діііууіиі icptJ фи^іьтр о и передают и шорник у, откуда
переливают в тару 70.
^ Раствор полималеината в мономере применяют в производстве
стеклопластиков (стр. 564 ел.) или используют в качестве
заливочного компаунда.
Для синтеза полиамидов обычно используют аминокислоты
(например, аминоэнантовую кислоту), капролактам или соли
диаминов и дикарбо*ювых кислот (например, АГ-соль, стр. 260).
Во всех случаях образование полимера сопровождается
выделением воды, которую необходимо непрерывно удалять из реактора.
/
(¦'
\
\
/
4~~
$4
Рис. 111. Схема производства ненасыщенных полиэфиров
(полималеината):
/. 7, 9—сборники; 2, 3—бункеры для загрузки мономеров; 4—реактор;
J—конденсатор; 6—аппарат для приготовления растворов; 8—фильтр; 1и—тара для
растворенного полиэфира.
Обычно полиамиды получают в две стадии (рис. 112). В
реакторе / мономер расплавляют в присутствии небольшого
количества воды, затем через сборник 2 передают в обогреваемый
автоклав 3, где проводится поликонденсация при 250—270 °С. Чтобы
предотвратить окисление аминогрупп, поликонденсацию
ведут в атмосфере азота. При нагревании мономера давление в
аппарате повышается до 10—15 am. По достижении указанной
выше температуры к автоклаву подключают конденсатор и,
снизив давление до атмосферного, удаляют воду из реакционной
смеси. После окончания реакции поликонденсации расплав
полимера через нижний щелевой штуцер автоклава выдавливается в
виде ленты в ванну 4 для охлаждения. Затем полимер поступает
в измельчитель 5.
Полиамиды—энант, капрон (поликапроамид), полиамид 66
(полигексаметиленадипамид или найлон)—применяют для
производства высокопрочных волокон (стр. 471); капрон и полиамид
66 находят также широкое применение как конструкционные
материалы в производстве деталей приборов и машин, которые
должны обладать длительным сопротивлением истиранию.
410
Полиэпоксиды получают поликонденсацией дифенилолпропана
с эпихлоргидрином (стр. 390). Соотношение дифенилолпропана
к эпихлоргидрина в исходной смеси определяет молекулярный
вес полиэпоксида.
Рис. 112. Схема производства поликапроамида:
I—реактор; 2—сборник расплавленного капролактама; 3—обогреваемый
автоклав; 4—ванна для охлаждения ленты полимера; 5—валки для наматывания
ленты и измельчитель; 6—бункер; 7—конденсатор.
На рис. 113 приведена схема производства полиэпоксидов.
Низковязкий полиэпоксид с молекулярным весом порядка 400—
500 получают при 60—65 °С. В реакторе / периодического дей-
Толуол
'исренилолпропан
Толуол
, —«j—
Зпихлоргидрин
Вода
PacmBop NaOH
Смола
Рис. 113. Схема производства полиэпоксидов:
/, Ч реакторы; 2, 6, 7—сборники; 4, 9—конденсаторы; 5, 10—холодильники; 8—выпарной
аппарат.
411
для обогрева и охлаждения, загружают дифенилолпропан и эпи-
хлоргидрин. Выделяющийся хлористый водород нейтрализуют,
постепенно вводя 15%-ный водный раствор NaOH таким образом,
чтобы температура в реакторе не превышала 65 °С. Реакция
длится около 6 ч. Для удаления образовавшегося хлористого натрия
смолу растворяют в толуоле и промывают теплой водой,
подаваемой в реактор. Отмытый раствор смолы, содержащий некоторое
количество вл'аги, через сборник 2 перекачивается в реактор 3
для удаления воды. Пары воды, охлаждаясь в конденсаторе 4
и холодильнике 5, собираются в виде конденсата в сборнике 6.
Высушенный раствор смолы через сборник 7 передают в выпарной
аппарат 8, обогреваемый снаружи паром, где при 60—110 °С и
остаточном давлении 50—100 мм рт. ст. из раствора смолы
удаляют толуол. Пары толуола охлаждаются в конденсаторе 9 и
холодильнике 10, а расплавленная смола спускается через нижний
штуцер аппарата 8.
Для получения более высоковязкого полиэпоксида
(молекулярный вес 1500—2000), к расплаву ранее синтезированного
низковязкого полимера добавляют некоторое количество дифенилол-
пропана, тщательно перемешивают и нагревают при 170—180 °С
в течение 4 ч. Быстро охлгдив реакционную массу, выливают еще
теплый вязкий полимер на охлаждаемый барабан.
Полиэпоксиды отличаются высокой адгезией (клейкостью)
по отношению к любым материалам. Поэтому на основе поли-
эпоксидов приготовляют универсальные клеи. Их добавляют
в защитные составы для повышения прочности сцепления
покрытий с металлической поверхностью. В сочетании со
стекловолокном полиэпоксиды дают наиболее прочные стеклопластики.
В производстве полиэфироуретанов применяют следующие
диизоцианаты:
N=C=O
O=C=N—(CH2N—N=C=O
гексаметилендиизоцианат толуилендиизоцианат
В качестве полиэфиров в этих синтезах используют продукты
этерификации адипиновой, себациновой или терефталевой кислот
гликолями (стр. 408).
Для получения полидиэтиленгликольадипината р реактор
загружают адипиновую кислоту и расплавляют при температуре
выше 175 °С. В расплав вводят диэтиленгликоль, повышают
температуру до 220 °С и нагревают при этой температуре до тех пор,
пока из реактора не выделится в конденсатор рассчитанное
количество реакционной воды. Диэтиленгликоль берут в некотором
избытке с таким расчетом, чтобы в кснце процесса на каждую
макромолекулу полиэфира приходилось по 1,5—1,7 гидроксильных
412
групп. Молекулярный вес получаемых таким способом
полиэфиров колеблется в пределах 3000—5000, а температура плавления
50—70 °С.
В расплавленный полиэфир загружают диизоцианат и при
80—90 °С непрерывно перемешивают в течение 30 мин.
Молекулярный вес образующегося при этом поли?фироуретана достигает
15 000—18 000. Если эту реакцию проводить при избытке диизо-
цианата, то на концах макромолекул полинфироуретана будут
преимущественно изоцианатные группы. Такой полигфироуре-
тан легко реагирует с водой, при этом его молекулярный вес
еще более возрастает, а в макромолекулах появляются карбамид-
ные звенья:
R__N=C=O + НоО + O=C=N-R *
> R-KH-C—NH-R і-СОї
II
О
Реакцию с водой проводят в том же аппарате, где протекал
синтез полизфироуретана. Количество добавляемой воды
составляет 1% от массы полимера. Смесь нагревают при 85 °С в
течение 30—40 мин. Полимер термопластичен и растворим, однако
после нагревания при 130—170 °С в течение 4—6 ч он утрачивает
эти свойства, приобретая сетчатую структуру.
Полизфироуретаны нашли применение в качестве исходных
веществ для изготовления жестких и мягких пенопластов,
универсальных клеев, прочных эластичных пленок и резиновых
изделий.
В типовых реакторах периодическим блочным методом
изготовляют также форполимеры—начальные продукты
полимеризации или поликонденсации. Эти продукты достаточно текучи при
нагревании и легко заполняют формы, а также могут быть
подвергнуты дальнейшим превращениям в более
высокомолекулярные вещества в аппаратах непрерывного действия. Такой способ
применяется, например, при изготовлении листов и плит из
органического стекла блочной полимеризацией метилметакрилата, а
иногда используется при поликонденсации фенола с
формальдегидом для получения литого карболита.
Процессы полимеризации и поликонденсации сопровождаются
значительными усадками реакционной массы, а в случае
поликонденсации, как известно, и выделением побочных продуктов.
Изготовление изделий, не имеющих усадочных раковин, трещин
или газообразных включений, представляет значительные
трудности, так как для этого требуется тщательное регулирование
температуры и максимально возможное снижение скорссти процесса.
Поэтому изготовление полимеров в формах применяется лишь
в исключительных случаях. Для облегчения этого процесса формы
заполняют не исходными веществами, а фсрполимерсм, так как
113
}«_<і,цп.<і ыис»_й и пилйЧоЛЬи ЬЫДеЛЯЮЩИХСЯ ПОООЧНЫХ ПрОДуКТОВ
особенно велики на начальной стадии превращения мономеров в
полимер.
форполимер, полученный в типовом реакторе периодического
действия в виде вязкой массы, переливают в формы (в случае поли-
конденсации массу следует тщательно отделить от побочных и
исходных продуктов). Так, при получении полиметилметакрилата
(рис. 114) форполимеризацию прекращают как только раствор
полимера в мономере (форполимер) приобретет сиропообразную
консистенцию. Затем форполимер выливают через нижний сливной
люк реактора 3 в заранее приготовленные формы—кюветы 4. Они
выполняются из двух листов силикатного стекла, между которыми.
Рис. 114. Схема* производства органического стекла:
/, 2—мерникн; 3—реактор для форполимериэации; 4—кювета для
заливки форполимера; 5—стойка для кювет; а—термокамера.
находятся прокладки. Торцовые стенки кюветы
обклеены''пропитанной бумагой, непроницаемой для мономера. Кюветы
установлены вертикально на легко передвигаемых тележках (стойки для
форм) 5, которые вкатывают в термокамеру 6. В камере
происходит дальнейшая полимеризация метилметакрилата до
образования прозрачной твердой стекловидной массы. Затем кюветы
помещают в горячую воду, в среде которой отклеиваются
бумажные торцовые стенки и легко отделяются листы силикатного
стекла от находящегося между ними листового полиметилметакрилата
(органического стекла).
Массивные блоки нецелесообразно изготовлять этим способом,
так как даже при умеренных температурах реакции отвод тепла
из центральных слоев вязкой массы весьма затруднителен.
Повышение температуры внутри блока способствует испарению
мономера, еще не вступившего в реакцию. Пузырьки пара, остающиеся
внутри вязкой массы, нарушают монолитность блока. К, тому же
полимеризация отдельных слоев толстостенного блока происходит
при неодинаковой температуре, поэтому молекулярный вес
полимера и его механические свойства в разных слоях различны.
414
Блочная поликонденсация полифункциональных веществ в
формах сопряжена с еще большими затруднениями, что
обусловлено не только неравномерным распределением тепла в массе
материала, но и необходимостью удалять из полимера все
побочные продукты реакции до ее окончания, т. е. до перехода полимера
в нерастворимое состояние. Поэтому форполимер, полученный на
установке периодическою действия и тщательно отделенный от
низкомолекулярных веществ, заливают в формы и устанавливают
в термокамеру, снабженную вакуумной установкой Дальнейшую
поликонденсацию проводят в течение длительного времени,
медленно повышая температуру. Постепенным прогреванием и
тщательным вакуумированием стремятся достигнуть равномерного
повышения вязкости всей массы полимера с одновременным
удалением непрерывно выделяющихся побочных продуктов.
Если исходное вещество представляет собой газ с низкой
температурой сжижения, а для превращения его в полимер требуется
нагревание до сравнительно высоких температур, реакцию
необходимо проводить в автоклаве. Блочным способом в аппаратах
периодического действия, рассчитанных на высокое избыточное
давление, получают, например, полиэтилен и полиизобутилен.
Полимеризацию этилена проводят под давлением 1500—
3000 am и при температуре 190—200 °С. Инициатором процесса
является кислород @,05—0,15% от количества этилена),
образующий в указанных условиях с отдельными молекулами этилена
неустойчивые перекисные соединения. Эти соединения
распадаются, и в результате возникают начальные радикалы
полимеризации.
Схема полимеризации этилена показана на рис. 115. Этилен
с небольшой добавкой воздуха (присутствие в этилене более
0,16% Оо может привести к взрыву при полимеризации)
воздуходувкой / из смесителя 10 подается через счетчик 2 в компрессор 3,
где смесь сжимается до давления 300 am. Пройдя
маслоотделитель 4, газ поступает во второй компрессор 5, где сжимается до
давления 2000 am и вновь проходит маслоотделитель 6. Этилен
полимеризуется в змеевиковом реакторе 7, за время прохождения
газа по змеевику степень превращения этилена в полимер
достигает 15—20%. Далее реакционная смесь поступает в испаритель 8,
где давление снижается до 200 am, затем в газосепаратор 9,
работающий при атмосферном давлении. Здесь газообразный этилен
отделяется от полимера и после очистки от побочных
низкомолекулярных продуктов снова возвращается в цикл.
Порошкообразный полимер (молекулярный вес 25 000—30 000) гранулируют
в таблетки, удобные для дальнейшей переработки.
Полиэтилен применяют для изготовления химически стойких
труб, электроизоляции проводов и кабеля, газонепроницаемых
защитных пленок, легкой и упругой тары, а также используют
для совмещения с каучуками (стр. 438, 496).
415
соба перед блочным заключается в возможности равномерного
распределения тепла в реакционной массе и сохранения ее
сравнительно низкой вязкости в течение всей реакции. Это позволяет,
интенсифицируя процесс, получать более однородный по
свойствам полимер и достигать большей глубины химических
превращений. Однако при производстве полимеров лаковым способом
необходим тщательный подбор растворителя. Поскольку
поликонденсацию бифункциональных веществ приходится проводить
при температурах 160 °С и выше (для более полного удаления
побочных продуктов), то и применяемый растворитель должен
Этилен
Рис. 115. Схема производства полиэтилена при высоком
давлении:
/—воздуходувка; 2—счетчик; 3, 5—компрессоры; 4, в—
маслоотделители; 7—змеевиковый реактор; 8—испаритель этилена; 9—газосепаратор;
/ 0—смесит ель.
иметь высокую температуру кипения. Выбор же доступных высо-
кокипящих растворителей ограничен, а для их последующего
удаления из полимеров требуется установка вакуум-сушилок и
строгое соблюдение соответствующего режима сушки.
Процессы полимеризации можно проводить при температурах
до 70—80 °С, что значительно облегчает выбор растворителя.
Однако многие растворители являются телогенами или
замедлителями реакции полимеризации и оказывают существенное
влияние на рост макромолекул, увеличивая полидисперсность
полимера. Чем меньше становится концентрация мономера в
реакционной смеси, тем в большей степени сказывается влияние
растворителя на реакцию полимеризации (возрастает
неоднородность полимера по величине молекулярного веса, сни-
416
жается средний молекулярный вес полимера, а во многих
случаях и замедляется реакция полимеризации).
Следует также отметить, что удаление даже сравнительно
легколетучего растворителя из массы высокомолекулярного
полимера является весьма трудоемкой операцией.
Лаковый способ синтеза используется, когда полимер в
дальнейшем применяют в виде лака на основе того же растворителя,
в среде которого проводится полимеризация, или когда
необходимо равномерное распределение тепла в реакционной среде,
или же, когда часть исходных компонентов является твердыми
веществами и для проведения реакции в гомогенной среде
требуется подобрать общий растворитель, и, наконец, когда катализатор
полимеризации нерастворим в мономере и его необходимо
диспергировать в реакционной среде.
По лаковому способу поликонденсации на типовой установке
периодического действия получают карбамидные полимеры, к
числу которых относятся продукты поликонденсации
формальдегида с карбамидом (иногда называемые мочевино-формальдегид-
ными полимерами) или меламином.
В начальной стадии процесса поликонденсации образуются
метилольные производные карбамида или меламина (стр. 393).
Они характеризуются высокой реакционной способностью и легко
взаимодействуют между собой с выделением побочного
продукта—воды. Поликонденсация проходит в водном растворе,
который постепенно становится все более вязким, не теряя при этом
прозрачности и оставаясь бесцветным. Только на последних
стадиях образования пространственного полимера он утрачивает
растворимость в воде и становится стекловидным и хрупким.
Скорость реакции заметно возрастает с повышением температуры
или при добавлении катализаторов (кислоты и соли сильных
кислот). Поликонденсацию заканчивают на стадии образования
водного раствора полимера требуемой вязкости. Такие растворы
можно сохранять при комнатной температуре в течение 1—3 месяцев.
Термореактивные карбамидные полимеры получают при 25—
40 °С и мольном отношении карбамида к формальдегиду 1 : 1,5
и меламина к формальдегиду 1 : 3. Для образования метилоль-
ных производных pH среды должно быть в пределах 7—8, что
достигается введением в реакционную смесь водного раствора
аммиака или гексаметилентетрамина. Смесь метилольных
производных нагревают в течение 30—45 мин, затем вводят 10%-ный
раствор щавелевой кислоты, доводя pH среды до 6,5—7,5 для
ускорения реакции поликонденсации образовавшихся производных.
Полученный раствор используют в производстве слоистых
пластиков, для пропитки хлопчатобумажных тканей или
древесины, а также в качестве клея в производстве фанеры.
Способ лаковой полимеризации используется для
производства термореактивных смол—полиакрилатов. Их получают эте-
27—805 417
рификациеи смеси метакриловой и фталевой кислот гликоля-
ми (этиленгликоль, диэтиленгликоль, триэтиленгликоль, бутан-
диол) или глицерином и последующей частичной полимеризацией
эфиров. Каждая макромолекула образовавшегося смешанного
полиэфира содержит по две и более несопряженных кратных
связей, что придает полиэфиру высокую активность в процессе
дальнейшей полимеризации. Рост макромолекулы происходит
одновременно с разрывом всех кратных связей, поэтому на
конечной стадии полимеризации образуется полимер
пространственной структуры. Процесс приостанавливают на стадии образования
форполимера в виде массы сиропообразной консистенции. Для
предотвращения дальнейшей полимеризации и увеличения (до
2—3 месяцев) длительности сохранения термореактивной смолы
в нее добавляют ингибитор (гидрохинон).
Синтез полиакрилатов проводят в среде толуола. В реактор
загружают гликоль, толуол, метакриловую кислоту,
двухосновную насыщенную кислоту (в качестве растворителя) и небольшое
количество серной кислоты—катализатора процесса этерифика-
ции. Реакцию проводят при температуре кипения растворителя,
при этом пары толуола уносят с собой пары воды, выделяющиеся
в процессе этерификации; далее смесь паров конденсируется в
холодильнике, и конденсат расслаивается в ловушке. Толуол
вновь возвращают в реактор, вода стекает в сборник. Этерифика-
ция сопровождается также частичной полимеризацией. По
окончании процесса не вступившие в реакцию кислоты нейтрализуют
и удаляют путем промывки водой. Затем реактор соединяют
через прямой конденсатор с вакуум-насосом и отделяют оставшийся
толуол от форполимера. Чтобы не происходила дальнейшая
полимеризация форполимера, при отгонке толуола -в раствор
добавляют гидрохинон.
* Форполимеры полиакрилатов, представляющие собой вязкие
жидкости светло-желтого цвета, применяются в производстве
стеклопластиков, заливочных компаундов и лаковых покрытий.
Процесс ионной полимеризации, используемый, например,
в производстве полиизобутилена, часто сопровождается весьма
интенсивным повышением температуры реакционной смеси, что,
как известно, приводит к снижению среднего молекулярного веса
полимера, увеличению его полидисперсности и разветвленности
макромолекул. Поэтому указанный процесс приходится
проводить при интенсивном охлаждении продуктов полимеризации.
Полиизобутилен получают методом анионной полимеризации
(рис. 116), охлаждая изобутилен жидким этиленом.
Катализатором реакции служит комплекс фтористого бора с эфиром. Жидкий
этилен поступает под давлением 16 am и при температуре —40 °С
в трубы теплообменника 1, где дополнительно охлаждается
газообразным этиленом, возвращающимся из испарителя 2.
Вследствие охлаждения давление жидкого этилена снижается до атмо-
418
сферного и далее он подается в аппарат 2, где, частично испаряясь,
охлаждается до -г-104 °С. Газообразный этилен возвращается в
теплообменник 1, жидкий этилен перекачивается в дозатор 3 и,
проходя по трубам, охлаждает до —85 °С изобутилен,
поступающий в межтрубное пространство. Охлажденный изобутилен поли-
меризуется на ленте полимеризатора 5. Реакция протекает в
среде жидкого этилена, подаваемого по трубопроводам из дозатора 3.
Катализатор полимеризации BF3 поступает в трубопровод из
приемника 4 и смешивается с жидким этиленом. На ленту
полимеризатора подается стабилизатор (mpem-бутилфенилсульфид) для
предотвращения деполимеризации полиизобутилена.
Полученный полимер передается в дробилку 6.
Изобутилен - рвктисрикат
Полшзобутилен
на холодильную
, установку
Рис. 116. Схема полимеризации изобутилена:
.'—теплообменник; 2—испаритель; J—дозатор: 4— приенник кгт?лизатора:
5—полимеризатор; в—дробилка.
Поли изобутилен используют в качестве синтетического
каучука, а также совмещают с другими полимерами (стр. 496) для
повышения их упругости или эластичности.
Получение полимеров в среде осадителя. Недостатков
способа лаковой полимеризации, которые связаны с
трудностью удаления растворителя из высоковязкого расплава
полимера, можно избежать, проводя реакцию в жидкой среде,
растворяющей только мономеры и инициатор или диспергирующий
катализатор. Такой прием облегчает регулирование теплового режима
реакции, предотвращает (при необходимости) увлажнение
катализатора и упрощает отделение полимера. По этому способу
проводят полимеризацию этилена при низком давлении,
полимеризацию пропилена на стереоспецифических катализаторах,
поликонденсацию фенола с формальдегидом.
27*
419
На рис. 117 приведена принципиальная схема получения сте-
реорегулярного полипропилена в среде осадителя.
Катализатор этого процесса—комплекс треххлористого титана и три-
Пропилен
и
> «-гептан
Триэтилалю-
миний и
м-гептан '
Треххлористый
титан
Очистка от
примесей и сушка
Полимеризация
Центрифугирование
«-Гептаи и
катализатор
1 1
Отмывка от
катализатора
Осушка н-гептана
Полипропилен-
сырец
Вода
\ і І
Отмывка от катализатора
и «-гептана
Метанол
t
Сушка полимера
Ректификация
спирта
і
Полипропилен
Рис. 117. Принципиальная схема производства полипропилена.
этилалюминия—теряет активность в присутствии влаги, поэтому
реакцию проводят в среде тщательно осушенных углеводородов
(чаще в н-гептане).
420
В реактор сначала загружают 4%-ный раствор триэтилалю-
миния в я-гептане и треххлористый титан, затем раствор
пропилена в к-гептане, процесс продолжается 10 ч при 60 °С, давление
в реакторе при этом достигает 3 am. По окончании реакции
отделяют полимер от «-гептана на центрифуге. Полимер многократно
промывают метиловым спиртом, затем водой для
извлечения следов катализатора и передают в термокамеры для
высушивания.
В реакторах периодического действия в среде, растворяющей
только мономеры, получают также полимеры методом
поликонденсации. К таким процессам относится приготовление так
называемых резольных смол—начальная стадия поликонденсацни
смеси моно- и диметилольных производных фенола, устойчивых
только в щелочных средах и при низкой температуре. В
слабощелочных и особенно в слабокислых средах происходит
поликонденсация метилольных производных до образования
пространственного полимера (стр. 392).
Реакцию проводят в водном растворе исходных веществ.
Резольная смола даже на начальной стадии поликонденсации (т. е.
еще в виде олигомера) нерастворима в воде и легко отделяется от
раствора. В этой стадии резольная смола представляет собой
хрупкий полимер с низкой температурой размягчения F5—75°С),
легко растворимый в спирте, ацетоне, бензоле. В производстве
лаковой смолы фенол заменяют л-бутилфенолом.
Как только средний молекулярный вес резольной смолы
достигнет 700—1000, реакцию поликонденсации замедляют
снижением температуры.
Для получения резольной смолы в реактор вводят фенол и
38%-ный водный раствор формальдегида. Реакцию проводят в
присутствии аммиака A,5—2 вес. ч. на 100 вес. ч. фенола).
Реакционную смесь нагревают водяным паром до 90—95 °С и
выдерживают при этой температуре до достижения требуемой вязкости.
Резольная смола легко отделяется от водной фазы. Остаточное
количество воды вместе с непрореагировавшими
низкомолекулярными веществами отгоняют в вакууме. Расплавленную и
осушенную смолу через нижний штуцер реактора выгружают в
металлические сборники, где она охлаждается, превращаясь в
прозрачную хрупкую массу, которую при комнатной температуре можно
сохранять в течение 4—6 месяцев.
Эмульсионный способ. Реакцию эмульсионной
полимеризации проводят в среде, не растворяющей ни мономер, ни
полимер. Чаще всего такой средой служит вода, в которой растворяют
инициатор полимеризации.
Для образования устойчивой эмульсии мономера применяют
эмульгирующие добавки. Эмульсию легко перемешивать на любой
стадии образования полимера, что позволяет достигнуть равномер-
421
ного распределения тепла в реакционной массе и, следовательно,
значительно повысить скорость полимеризации (по сравнению со
скоростью полимеризации по блочному способу). Полимер
образуется в виде латекса—мельчайших частиц, взвешенных в воде.
Вводя в латекс некоторое количество коагулянта (соли, кислоты),
полимер осаждают в виде крошки. Этот процесс обычно связан
с некоторой потерей полимера вследствие неполноты коагуляции
н загрязнением полимера эмульгаторами и коагулянтами,
которые трудно извлечь из него.
Эмульсионную полимеризацию можно проводить в
присутствии промоторов (стр. 396), что дает возможность снизить
температуру процесса до 5—6 °С (низкотемпературная эмульсионная
полимеризация) и, следовательно, повысить средний
молекулярный вес полимера и получить полимер более регулярного
строения. Такой способ полимеризации применяется в производстве
синтетических каучуков из хлоропрена, бутадиена или его
сополимеров со стиролом или акрилонитрилом и др. Резины на
основе каучуков, получаемые низкотемпературной эмульсионной
полимеризацией, обладают повышенной прочностью и
эластичностью.
Примером технологического процесса эмульсионной
полимеризации может служить процесс получения поливикилхлорида.
Исходным веществом является газообразный хлористый винил
(стр. 188), имеющий температуру кипения —13,9 °С. Под
небольшим избыточным давлением A—2 am) он сохраняется в
сжиженном состоянии при комнатной температуре.
Сжиженный хлористый винил перекачивают в реактор,
заполненный водным раствором персульфата аммония или калия
(инициатор полимеризации) и натриевых солей алифатических или
ароматических сульфокислот (эмульгаторы). При перемешивании
хлористый винил эмульгируется в растворе, далее раствор
нагревают до 60 °С, при этой температуре и происходит полимеризация.
Давление в реакторе в начальный период реакции достигает 5—
7 am. По окончании процесса полимеризации образуется
стабильная водная эмульсия поливинилхлорида, для разрушения которой
и осаждения полимера в реактор вводят электролит (соль или
кислоту). Полимер отделяется от жидкости в виде тонкого
порошка, который отфильтровывают, тщательно отмывают от
водорастворимых примесей и сушат.
Поливинилхлорид находит очень широкое и разнообразное
применение. Его используют в качестве кислото- и солестойких
облицовок гальванических ванн, для изготовления
кислотостойких труб, арматуры, аккумуляторных баков. Из
поливинилхлорида получают также эластичные материалы, из которых
изготовляют шланги, прокладки, изоляцию электрических
проводов, защитные пленки, эластичные и жесткие пенопласты,
заменители кожи.
422
Большинство исходных веществ, применяемых в процессе
поликонденсации, растворимо в воде, поэтому эмульсионным
способом этот процесс обычно не проводят. Исключение
составляет синтез тиоколов (полиалкилсульфидов), получаемых из
таких водонерастворимых веществ, как дихлоралкилы, например
?-хлорэтиловый эфир этиленгликоля. Дихлоралкилы вступают
в поликонденсацию с полисульфидами натрия (например, с тет-
расульфидом) по следующей схеме:
пС1-СН2—СНа-О-СНа-СНа—С1 + rtNa2S4 >
> [-СН2—СН2—О—СН2—СН2— S—S—]„ + 2nNaCl
II II
S S
Реакция между дихлоридами и полисульфидами экзотермична.
В водной среде, эмульгирующей исходные вещества, облегчается
регулирование температуры в реакторе. Чтобы получить полимер
в виде эмульсии, в исходную смесь добавляют диспергаторы
(например, гидроокись магния). Поскольку поликонденсация
сопровождается образованием различных побочных продуктов,
ухудшающих качество полимера, реакцию проводят в
присутствии спирта или ацетона, улучшающих растворение побочных
продуктов. Требуемая температура в реакторе поддерживается
путем регулирования скорости подачи дихлоралкила в смесь
водного раствора полисульфида, гидроокиси магния и спирта,
предварительно нагретую до 85—95 °С. По мере введения в
реакционную смесь дихлоралкила образуется эмульсия полимера. По
окончании реакции полимер осаждают, добавляя к эмульсии полимера
в реакционной среде соляную кислоту. Полиалкилсульфид
тщательно промывают водой и высушивают.
Полиалкилсульфиды находят применение в качестве бензо- и
маслостойких каучуков и герметизирующих составов.
Полимер, получаемый эмульсионным методом, всегда
содержит следы веществ, применяемых в качестве коагулянтов и
эмульгаторов; кроме того, он несколько загрязняется пылью во время
сушки и хранения. Для большего удобства транспортирования,
хранения и последующей переработки полимера в изделия
методами прессования или литья под давлением полученный порошок
подвергают гранулированию в шнек-машинах или развальцовывают
в виде листов (в производстве синтетических каучуков).
Суспензионный способ получения полимеров заключается в том,
что нерастворимый в воде мономер, содержащий растворенный
в нем инициатор или катализатор, диспергируют в водной среде
путем интенсивного перемешивания до образования мелких
капель.
Чтобы капли не слипались, повышают вязкость раствора,
вводя в него крахмал, водорастворимые синтетические полимеры
423
или тальк. В каждой капле мономера, содержащей инициатор
или катализатор, происходит процесс полимеризации.
Получаемый таким методом полимер представляет собой мелкие
гранулы. Тепло реакции отводится от капли (или подводится к ней)
водой. По окончании реакции и прекращении перемешивания
мелкие стекловидные частицы полимера (гранулы) осаждаются
на дне реактора. Размер частиц регулируется вязкостью водной
среды и скоростью перемешивания.
Суспензионная полимеризация облегчает отделение полимера
от дисперсионной среды и обеспечивает полноту его осаждения,
что невозможно при коагуляции эмульсии полимера.
Сферические гранулы полимеров, полученных суспензионным
способом, лишены указанных выше недостатков полимеров,
изготовленных эмульсионным способом, и поэтому в производстве
пластмассовых изделий методами прессования или литья под
давлением предпочитают применять полимеры, полученные
суспензионным способом. В связи с этим все большее количество полимеров
стирола, метилметакрилата, хлористого винила, сополимеров
бутадиена получают суспензионным способом. Однако
отрицательным качеством данного способа пока является трудность
оформления его в виде непрерывного процесса.
Гранульная форма полимеров весьма удобна также в случае
применения их в качестве ионообменных поглотителей (ионитов).
Наиболее распространенным исходным веществом для
получения ионитов является сетчатый сополимер стирола и ди-
винилбензола, получаемый методом суспензионной
полимеризации:
сн=сн. сн=сн2
I I
О
СН—СНа-СН—СНз-СН-СНа-СН—СН,
¦
f
СН—СН
424
Процесе проводят в 2—3%-ном водном растворе
поливинилового спирта или крахмала, присутствие которых предотвращает
слипание гранул. Затем в водный раствор вводят смесь стирола,
дивинилбензола и перекись бензоила (инициатор сополимериза-
ции). Мономеры, с растворенным в них инициатором,
диспергируют в воде при интенсивном перемешивании и нагревают при
при 70—80 °С до тех пор, пока каждая капля не заполимеризуется
в твердую стекловидную гранулу. После прекращения
перемешивания гранулы легко отделяются от воды.
Некоторые иониты получают методом поликонденсации.
Технологические различия суспензионной поликонденсации от
суспензионной полимеризации заключаются в том, что при
поликонденсации дисперсионной средой служит обычно не вода, в которой
растворимы исходные вещества и начальные продукты
поликонденсации, а минеральные масла, высококипящие органические
растворители или полненлоксановые жидкости. В этих средах
диспергируют не исходные вещества, как в случае полимеризации,
а начальные продукты их поликонденсации (олшополимеры). В
процессе гранульной поликонденсации необходимо особенно
тщательно соблюдать режим начальной стадии процесса, в
противном случае затрудняется диспергирование олигополимера и полу«
чение полимера в виде гранул одинакового размера.
НЕПРЕРЫВНЫЕ СПОСОБЫ
В связи с резким увеличением объема производства
"полимеров потребовалось коренное усовершенствование технологии их
изготовления.
Для повышения производительности оборудования и более
экономичного его использования исключительное значение
приобретает внедрение в промышленность непрерывных процессов,
связанное в свою очередь с необходимостью применения машин
и аппаратов нового типа. Ряд способов получения полимеров—
блочный, лаковый, эмульсионный—оформляют в виде
непрерывных процессов.
Установки непрерывного действия работают по принципу
аппаратов вытеснения или аппаратов смешения. В первом случае
реакция образования полимера происходит в процессе
перемещения смеси реагирующих веществ в аппарате. При этом смесь
должна тщательно перемешиваться. Из аппарата непрерывно вытекает
расплав или раствор полимера.
Чтобы исходные вещества успели вступить в реакцию во время
движения в аппарате, он должен иметь форму змеевика, длина
и диаметр которого определяются скоростью процесса и вязкостью
среды. Такие реакторы целесообразно использовать для
процессов, протекающих с большой скоростью. Змеевик обогревается
или охлаждается омывающими его газами или жидкостями. По-
425
скольку реакционная смесь на конечных стадиях процесса
должна сохранять высокую текучесть, реакцию обычно проводят
лаковым или эмульсионным способами. г
Аппараты смешения непрерывного действия применяются в тех
случаях, когда реакция образования полимеров протекает с
малыми скоростями. Реактор состоит из нескольких последовательно
соединенных секций, каждая из которых снабжена мешалкой,
рубашкой для обогрева или охлаждения и в некоторых случаях
конденсатором. Реакционная смесь перемещается из секции в
секцию до тех пор, пока не закончится реакция. Для упрощения
конструкции реактора и уменьшения количества его секций можно
снизить скорость перемещения реакционной смеси из секции ;в
секцию. На рис. 118 представлена секционная колонна
непрерывного действия. Мешалки 2 всех секций укреплены на общем
валу 3 и приводятся в движение от одного электропривода 5. Вал
движется в патрубках 4, верхние концы которых приподняты
над уровнем реакционной массы.
Непрерывным способом блочной полимеризации в реакторе
типа многосекционного аппарата смешения изготовляют
полистирол, непрерывным способом блочной поликонденсации—фе-
ноло-формальдегидные полимеры.
На рис. 119 приведена технологическая схема производства
полистирола. Полимеризацию проводят в две стадии. Первая
стадия—форполимеризация—протекает в реакторах /
периодического действия, куда из сборника поступает стирол, в котором
растворен инициатор полимеризации. Температуру в реакторе
поддерживают в пределах 60—80 °С. При этой температуре
инициатор распадается и начинается полимеризация стирола. Процесс
форполимеризации проводят при перемешивании, что
обеспечивает равномерное распределение тепла в реакционной смеси.
Вязкий раствор полимера B7—29%-ный) в мономере из реакторов /
передается в многосекционную колонну 2 непрерывного действия.
Она состоит из шести—восьми секций, в каждой из которых
поддерживается определенный температурный режим—от 80—85 °С
в первой секции, до 212 °С в последней секции. Внизу колонна
заканчивается конусом, в котором температура достигает 215 °С.
Для обеспечения непрерывной работы колонны ее соединяют с
двумя реакторами форполимеризации. Форполимер медленно
стекает по колонне в течение 25—30 ч, постепенно обогащаясь
полимером. Пары мономера поднимаются вверх по колонне и
отводятся на охлаждение и конденсацию. Конденсат возвращается
в сборник. Из конуса колонны расплавленный полистирол,
полностью освобожденный от мономера, непрерывной струей стекает
на шнек-пресс 3, который выдавливает полимер в воздушный
холодильник 4. Здесь полистирол охлаждается, образуя
прозрачную стекловидную массу, которая затем измельчается в
грануляторе и сбрасывается в приемник.
426
Рис. 118. Многосекционная
колонна:
1—секция; 2—мешалка; 3—вал;
4—патрубки; 5—электропривод.
Азот
Стирол
Рис. 119. Схема непрерывной
блочной полимеризации
стирола: .
/—реакторы; 2—полимеризационная
колонна; 3—шнек-пресс; 4—воздушный
холодильник.
Средний молекулярный вес полистирола, получаемого таким
способом, равен 150 000—200 000. Суточная производительность
установки—около 1 т полистирола.
Реакция поликонденсации выгодно отличается от реакции
полимеризации отсутствием длительного периода активации
мономеров. Это дает возможность исключить первую стадию,
осуществляемую в случае реакции полимеризации в аппаратах
периодического действия, и проводить процесс (например, изготовление
Поли.Ви.нила.цета.т
Рис.
120. Схема производства поливинилацетата
непрерывным лаковым способом:
/, 2—аппараты для приготовления исходных компонентов; 3, 4—по-
лиыеризационные блшни; 5, 6—реакторы дополнительной
полимеризации; 7—холодильники, 6—насосы.
феноло-формальдегидной смолы до термореактивной стадии)
сразу в колонне с четырьмя-пятью секциями, работающими при
одинаковом температурном режиме.
Непрерывным способом лаковой полимеризации получают по-
ливинилацетат (рис. 120). Полимеризацию проводят
последовательно в двух башнях (высота 9 м, диаметр 0,6 м) при температуре
кипения растворителя (бензол, этилацетат или метиловый спирт).
В первой башне 3 имеется мешалка, состоящая из серии лопастей,
перемешивающих раствор в каждой секции башни. В аппарате /
приготовляют раствор инициатора полимеризации, в аппарате 2—
раствор винилацетата. Оба раствора подают в верхнюю часть
башни 3. Реакционная смесь последовательно проходит все секции
башни. При этом растворитель частично испаряется, пары
поступают в холодильник 7 и конденсируются в нем. Конденсат
возвращается в реактор. Из башни 3 раствор подается в башню 4
и проходит ее снизу вверх до разгрузочного штуцера. В верхней
428
части башни в растворе остается около 5—8% мономера, не
вступившего в реакцию. Полимеризация завершается в реакторе 5
или 6, где раствор полимера дополнительно нагревают до тех пор,
пока количество свободного мономера не снизится до 1 —1,5%,
Раствор
инициатора
в мономере
Мономеры
Водный.
Раствор
промотора
вмономере
О О
раствор
эмульгатора
Промывные
воды
Рис. 121. Схема непрерывной эмульсионной сополимеризации стирола
и дивинила:
/—эмульгатор; 2—реактор; 3—отгонные колонны; 4—сборник; 5—сетка для промывки пали-
мера; 6—каляндр; 7—транспортер; В—сушильная шахта.
Часовая производительность подобной установки достигает 150 кг
поливинилацетата. Средний молекулярный вес полимера
колеблется в пределах 14 000—32 000.
Растворы поливинилацетата находят применение в
производстве клеев, лаков, пропиточных составов, а также в качестве
исходного вещества для получения поливинилового спирта (стр. 434).
В последние годы созданы установки для непрерывного
производства стереорегуляторных полимеров дивинила и изопрена в
среде растворителя.
Метод непрерывной эмульсионной полимеризации особенно
широко используют для синтеза полихлоропрена, полиизопрена,
полимеров и сополимеров дивинила. В этом процессе более
удобен многосекционный реактор, каждая секция которого является
самостоятельным аппаратом.
Сополимеризацию стирола и дивинила (рис. 121) проводят при
различных соотношениях мономеров в зависимости от назначения
429
эмульгирующего агента непрерывно подается в эмульгатор 1,
обогреваемый паром.
Мономеры эмульгируются при интенсивном перемешивании,
после чего в эмульсию вводят инициатор и промотор,
обеспечивающие инициирование полимеризации при 48—50 °С, и
переводят смесь в полимеризационную батарею. Последняя состоит из
12 последовательно соединенных реакторов 2 (на рисунке показан
один аппарат), снабженных мешалками и рубашками, в которых
циркулирует горячая вода. Эмульсия непрерывно переливается
из одного реактора в другой со скоростью, обеспечивающей
заданную степень сополимеризации в последнем реакторе. Далее
эмульсия полимера (латекс) подается в колонны 3, где водяным паром
из нее отгоняют незаполимеризовавшиеся мономеры, снова
возвращаемые в цикл. В сборнике 4 полимер коагулируют раствором
поваренной соли. Затем полимер передается на движущуюся
сетку 5 для промывки от эмульгатора и коагулятора. Масса полимера
отжимается на каландре бив виде ленты с помощью
транспортера 7 подается в шахту 8, где высушивается, проходя по
направляющим роликам.
Сополимер применяют в больших количествах в качестве
синтетического дивинил-стирольного каучука (стр. 487).
13. МОДИФИЦИРОВАНИЕ&СВОЙСТВ ПОЛИМЕРОВ\
В ряде случаев синтетическим полимерам, полученным одним
из описанных способов, или природным полимерам (например,
целлюлозе) необходимо придать новые свойства, которыми они
не обладают. Такие изменения свойств полимеров
(модифицирование) осуществляются методами химического воздействия. При
химических воздействиях функциональные группы
макромолекул полимера вступают в такие же реакции, как и аналогичные
группы низкомолекулярных соединений.
В ряде случаев модифицирование полимера химическими
методами может сопровождаться его частичной термической или
окислительной деструкцией, которая приводит к снижению
среднего молекулярного веса полимера и появлению в некоторых
звеньях макромолекул новых функциональных групп. Если
полимер находится в растворе, то процесс окислительной
деструкции проходит интенсивнее, так как облегчается доступ кислорода
к макромолекулам. Для предотвращения термо- и окислительной
деструкции процессы модифицирования полимеров стремятся
проводить при умеренной температуре.
Химическое воздействие может быть различным. Если оно
направлено только на изменение функциональных групп в звеньях
макромолекул без нарушения их структуры, такой процесс
обычно называют полимераналогичным превращением. Если нужно по-
430
лучить полимер, обладающий совокупностью свойств двух
различных полимеров, состоящих из звеньев А и Б, то соединяют
между собой макромолекулы или части макромолекул двух таких
полимеров в новые макромолекулы линейного (блоксополимера)
или разветвленного (привитого) сополимера (стр. 435 ел.).
Блоксополимеризацию часто применяют для соединения
макромолекул с целью получения полимера высокого молекулярного
веса из полимеров значительно более низкого молекулярного веса,
поскольку прямым синтезом не удается достигнуть требуемой
высокий степени полимеризации. Привитые сополимеры получают,
присоединяя к макромолекуле одного полимера длинные боковые
ответвления макромолекул другого полимера так, чтобы они
были расположены на значительном расстоянии друг от друга.
Если бифункциональное низкомолекулярное соединение первой
функциональной группой присоединяется к звену одной
макромолекулы полимера, а второй функциональной группой—к Дру
гой макромолекуле, то оно как бы сшивает эти макромолекулы,
создавая между ними мостик. При образовании множества таких
мостиков полимер приобретает пространственное (сетчатое)
строение (стр. 437). Таким приемом пользуются, например, при
вулканизации каучука серой и другими бифункциональными
веществами.
Модифицировать свойства полимера можно также совмещением
(сочетанием) его с другим полимером (стр. 438).
ПОЛИМЕРАНАЛОГИЧНЫЕ ПРЕВРАЩЕНИЯ
Методом полимераналогичных превращений можно
значительно изменить свойства полимера, заменяя большинство имеющихся
в нем функциональных групп другими функциональными
группами. Однако условия для проведения подобных превращений
необходимо подбирать так, чтобы по возможности избежать
нарушения первоначальной структуры и длины макромолекул.
Для этого при полимераналогичных превращениях полимер
переводят в раствор или вызывают его набухание. Растворитель
раздвигает макромолекулы полимера, облегчая таким образом
доступ реагентов к звеньям макромолекул. Процессы
полимераналогичных превращений стремятся вести при возможно более
низкой температуре, избегая применения сильных кислот, щелочей
и тем более окислителей.
Методом полимераналогичных превращений получают ценные
полимеры, используемые в производстве целлулоида, волокон,
лаков, пластических масс, резин. Полимераналогичные
превращения природных и синтетических полимеров позволяют
расширить области их применения. Чрезвычайно большое значение
приобрели полимеры, получаемые полимераналогичным
превращением целлюлозы и поливинилацетата.
431
Целлюлоза—основной компонент всех растительных клеток—
является наиболее распространенным природным
высокомолекулярным соединением. Она широко используется как
промышленное сырье, и процессы полимераналогичных превращений в
переработке целлюлозы играют большую роль.
Целлюлозу получают из хлопка и древесины. Хлопок
содержит до 94% целлюлозы, остальное—белки и масла, отделяемые
от целлюлозы промывкой хлопка разбавленными растворами
щелочей. В древесине содержится 45—50% целлюлозы, которую
извлекают химической обработкой древесины. При этом н раствор
переходят все компоненты древесины, кроме целлюлозы (стр. 72).
Целлюлоза принадлежит к классу высокомолекулярных
углеводов, макромолекулы которых имеют регулярное и строго
линейное строение. Она сильно набухает в растворах солей.
Это свойство целлюлозы используют в производстве бумажной
тары и фибры. В набухшем состоянии целлюлоза превращается
в вязкую массу, из которой затем можно формовать изделия.
После высушивания, а в некоторых случаях и пропитки растворами
смол, масса становится твердой, и фиксируется приданная ей форма.
В органических растворителях целлюлоза не растворяется;
при нагревании в пластическое состояние не переходит, поэтому
ее нельзя применять в производстве защитных покрытий,
волокон, изделий сложных форм, применяя существующие для этих
целей технологические приемы. Полимераналогичными
превращениями удается заменить в целлюлозе функциональные группы,
придав ей способность растворяться в органических растворителях.
В каждом звене макромолекул целлюлозы имеется потригидр-
оксильные группы:
н
СН2ОН
ОН
«о«
сн2он
он
Поэтому в химических реакциях целлюлоза ведет себя как
полиатомный спирт, образуя алкоголяты, сложные и простые
эфиры. Среди сложных эфиров целлюлозы особенно большое
значение приобрели ее ксантогенат и ацетат, применяемые в
производстве химических волокон и пленок (стр. 450, 462), а также
азотнокислые эфиры целлюлозы (нитраты), имеющие самостоятельное
применение.
Азотнокислые эфиры целлюлозы получают ее нитрованием,
обрабатывая целлюлозу смесью азотной и серной кислот (весовое
432
соотношение HNO3 : H2SO4 в нитрующей смеси от 1 : 1 до 3 : 1).
Для равномерного смачивания целлюлозы нитрующую смесь
берут с большим избытком. Степень замещения гидроксильных
групп целлюлозы нитратными группами регулируется введением
в нитрующую смесь определенного количества воды. Глубоко про-
нитрованная целлюлоза представляет собой взрывчатое вещество—
так называемый пироксилин. Для получения менее нитрованной
целлюлозы—коллоксилинов, применяемых в производстве
нитролаков, пленок, целлулоида, подбирают такие условия процесса,
чтобы происходило неполное замещение гидроксильных групи
(на 70—80%). При этом в нитрующую смесь вводят
18—20% воды, а нитрование проводят при низкой
температуре 15—20 °С в течение 30 мин. Нитрат целлюлозы,
нерастворимый в нитрующей смеси, отжимают и тщательно промывают водой.
Основное количество воды затем снова отжимают, остаток воды
вытесняют этиловым спиртом. Сушить нитрат целлюлозы при
повышенной температуре опасно.
Нитраты целлюлозы хорошо растворимы в спирте, ацетоне,
сложных эфирах и образуют вязкие растворы, используемые для
дальнейшей переработки.
Существенным недостатком сложных эфиров целлюлозы
является их сравнительно малая химическая стойкость,
способность омыляться в растворах кислот и щелочей. Для
изготовления химически стойких лаков применяют простые эфиры
целлюлозы—продукты взаимодействия целлюлозы с хлористым этилом
(этилцеллюлоза) или с хлористым бензилом (бензилцеллюлоза).
Для получения простых эфиров требуются более жесткие
условия, чем в процессе этерификации. Это приводит к значительной
деструкции полимера, снижению его среднего молекулярного
веса и, следовательно, к уменьшению прочности и твердости пленок
и лаковых покрытий на основе простых эфиров целлюлозы по
сравнению с этими показателями изделий из сложных эфиров
целлюлозы.
Полимераналогичные превращения поливинилацетата
Такие полимеры, как поливиниловый спирт и поливинилаце-
тат, получили большое промышленное значение. Однако
поливиниловый спирт невозможно получить непосредственной
полимеризацией винилового спирта, так как последний крайне
неустойчив и при попытках его получения немедленно изомеризуется в
ацетальдегид:
СН2=СН-ОН > СНз-СНО
Поливиниловый спирт приходится синтезировать методом поли-
мераналогичных превращений путем гидролиза синтетического
28—805 433
сложного эфира—поливинилацетата, который легко получается
полимеризацией винилацетата:
яСНа=СН » Г—сн2—СН— |
ОСОСНз L ОСОСНз L
Поливинилацетат гидролизуют спиртовым раствором щелочи
при температуре кипения спирта в течение 10 ч. Получаемый
поливиниловый спирт
NaOH, Н2О
Г«-СН2—СН- Л *- Г-СН2-СН-1 +CH3COONa
[ ОСОСНз 1 L ОН J„
в отличие от поливинилацетата нерастворим в спирте и
выделяется из него в виде тонкого порошка при омылении 60% сложно-
эфирных групп. Полученный полимер отделяют от жидкой фазы,
промывают спиртом и сушат при температуре 45—60 °С.
Поливиниловый спирт можно использовать в производстве
волокон, пленок, бензо- и маслостойких изделий. Он служит
также исходным сырьем для поливинилацеталей, на основе которых
изготовляют пластические массы и синтетические клеи.
Поливинилацетали получают путем дальнейшего полимер-
аналогичного превращения поливинилового спирта. Из
разнообразных поливинилацеталей наибольшее практическое значение
приобрели поливинилформаль (формвар)—продукт
взаимодействия поливинилового спирта с формальдегидом
+сн2о
СН2—СН-СН,—СН * СНа-СН—CHa—СН
ОН ОН О—СН,—6
и поливинилбутираль (бутвар), образующийся при
взаимодействии с масляным альдегидом:
+с3н7сно
..—СН2—СН-СН2-СН »- СН2-СН-СН2-СН
он
ОН O—СН—O
С3Н,
Реакцию ацеталирования поливинилового спирта проводят
в его водном растворе. Катализатором этой реакции обычно
служит фосфорная кислота. Степень ацеталирования определяется
соотношением поливинилового спирта и альдегида.
Поливинилацетали не растворимы в воде, но хорошо
растворяются в спирте. Растворы их отличаются высокой клейкостью, а
пленки, образующиеся после удаления растворителя, обладают
хорошей эластичностью и светостойкостью.
434
БЛОКСОПОЛИМЕРИЗАЦИЯ И ПРИВИТАЯ ПОЛИМЕРИЗАЦИЯ
В последнее время все большее значение приобретают
процессы блоксополимеризации, направленные на сращивание
линейных макромолекул различных полимеров между собой с
сохранением линейного строения новых макромолекул. Используя
метод блоксополимеризации, можно значительно повысить
молекулярный вес полимеров. Например, блоксополимеризацией
полиэфиров и полиуретанов получают полиэфироуретаны
(стр. 576 ел.):
O=C=N—(А)я—N=C=O + НО—(B)m—ОН »
полиуретан полнэфнр
О
„ O=C=N—(A)n—NH—С—О—(В)т—ОН
При синтезе полиэпоксидов получаются полимеры низкого
молекулярного веса (до 700—1500). Конечные звенья макромолекул
таких полимеров имеют этиленоксидные группы, которые
отличаются высокой химической активностью. Это используют для
повышения молекулярного веса полиэпоксида путем
присоединения к нему макромолекул другого также сравнительно
низкомолекулярного полимера, в конечных звеньях которого имеются
легко подвижные атомы водорода. Примером может служить блок-
сополимеризация полиэпоксида с полиамидом (молекулярный
вес 3000—5000), конечные звенья макромолекул которого
содержат аминогруппы:
СН2— СН—(А)я—СН—СН2 + H2N—(B)m— NH2 *
q' полиэпоксид q' полиамид
»CH2—СН—(А)„—CH(OH)—СН,—NH—(B)m—NH2
о
Если полимеры имеют высокий молекулярный вес и целью
блоксополимеризации является получение нового материала,
сочетающего свойства обоих полимеров, применяют совместную
пластикацию. Процессом пластикации полимеров принято
называть любое воздействие на них, направленное на снижение
среднего молекулярного веса полимера без изменения состава звеньев
макромолекул. При этом снижение молекулярного веса
происходит вследствие разрыва макромолекул по их длине. В местах
разрывов возникают свободные валентности, т. е. макромолекулы при
этом превращаются в макрорадикалы, но длина их становится
много меньше длины макромолекул. Макрорадикалы соединяются
между собой в новых сочетаниях либо присоединяют кислород
воздуха, насыщая свободную валентность и превращаясь в ста-
28* 435
бильные макромолекулы полимера с оолее низким молекулярным
весом.
Разрушения макромолекул полимера для снижения его
среднего молекулярного веса можно достигнуть механическим
воздействием, например путем истирания, измельчения (механоокисли-
тельная пластикация), действием повышенных температур
(термоокислительная пластикация) или ультразвука. Метод
пластикации широко применяется в технологии переработки полимеров,
так как при уменьшении молекулярного веса полимера снижается
температура его перехода в пластическое состояние, повышается
пластичность, одновременно с этим возрастает растворимость
полимера и снижается вязкость его растворов. Все эти изменения
свойств полимера облегчают формование изделий, волокон,
изготовление пленки, нанесение лакового покрытия.
В процессе пластикации специально подобранной смеси
разных полимеров, сводя к минимуму контакт этой смеси с
кислородом воздуха, создают условия для соединения между собой
образующихся макрорадикалов в новых сочетаниях, в том числе и для
соединения макрорадикалов различных полимеров, что приводит
к образованию блоксополимера.
Средний молекулярный вес блоксополимеров, полученных
методом совместной пластикации в отсутствие кислорода воздуха,
мало изменяется по сравнению со средним молекулярным весом
исходных полимеров, но свойства блоксополимера становятся
иными. В результате механического дробления макромолекул
(пластикации) смеси полимеров в среде азота возникают
макрорадикалы, которые соединяются в новых сочетаниях, образуя блок-
сополимеры.
Для образования привитых сополимеров необходимо создать
такие условия, при которых в некоторых звеньях макромолекул
основного полимера боковые атомы или группы отщепляются или
окисляются до гидроперекисей. Отщепление боковых атомов
(водорода, хлора, брома, фтора) может происходить под влиянием
радиоактивного или ультрафиолетового облучения или
ультразвукового воздействия. В результате такой обработки в
макромолекуле полимера возникает несколько свободных валентностей,
т. е. образуется полимакрорадикал. При окислении отдельных
звеньев макромолекул до гидроперекисей также могут
образоваться полимакрорадикалы. Особенно эффективным окислителем
является озон. Под влиянием повышенной температуры или
восстановителей гидроперекисные группы разрушаются, тоже
образуя свободные валентности.
Отщепление подвижных атомов или групп в полимере или
разрушение гидроперекисных групп проводят в присутствии какого-
либо мономера. В этом случае полимакрорадикалы основного
полимера служат начальными радикалами полимеризации
мономера и в местах возникших свободных валентностей присоединя-
436
ются боковые ответвления новых полимерных цепей. Примером
практического использования метода привитой сополимеризации
для модифицирования свойств полимера может служить прививка
полиоксиэтилена к полиамиду. Образование боковых полиокси-
этиленовых ответвлений повышает эластичность и гидрофильность
полимера.
При радиоактивном облучении поверхности изделий из
политетрафторэтилена к ним прививают полистирольные цепи,
благодаря чему эти изделия приобретают способность склеиваться;
к полиэтиленовым пленкам прививают поливинилпиридин, что
придает привитому сополимеру свойства полиэлектролита.
ПРЕВРАЩЕНИЕ ЛИНЕЙНЫХ ПОЛИМЕРОВ В ПРОСТРАНСТВЕННЫЕ
Химические реакции между функциональными группами
отдельных звеньев линейных макромолекул с низкомолекулярными
веществами, имеющими две функциональные группы или две
свободные валентности, приводят к соединению макромолекул с
образованием полимеров сетчатой структуры. Этот процесс получил
широкое распространение в технологии переработки полимеров.
К наиболее широко применяемым процессам этого типа относится
вулканизация (стр. 519). В процессе вулканизации линейные
полимеры превращаются в сетчатые с редким расположением по-
леречных связей, что придает полимеру нерастворимость, лишает
его термопластичности, но сохраняет его высокую эластичность.
При полимеризации дивинильных соединений в звеньях
макромолекул сохраняется по одной двойной связи, например:
(_СН,—СН-СН—СН,—)„
Под влиянием повышенной температуры A00—150 °С) и
активаторов по месту двойных связей полимера могут протекать
реакции с серой. Если ввести небольшое количество серы, то в реакции
примет участие только часть звеньев макромолекулы и возникнет
небольшое количество поперечных мостичных связей с соседними
макромолекулами, т. е. полимер приобретет редкосетчатую
структуру. В упрощенном виде образование таких связей можно
представить следующим образом:
-(_СН,—СН=СН-СНа—)л-СН«—СН—СН—СН,-(—СН,—СН=СН—СН„—)„-
S
—СН,—СН—СН-СН,—
Вулканизация может происходить и в результате
окислительной деструкции отдельных звеньев полимера. Такой процесс
практически применяется при переработке полисилоксанов. При нагре-
437
до 180—200 °С в присутствии перекиси некоторая часть звеньев
полимера подвергается окислительной деструкции. Этот
процесс затрагивает наиболее слабую связь, например вызывает
окисление метильных групп, что приводит к появлению свободных
валентностей и соединению макромолекул полимера
поперечными связями:
СН, СИ« СН, СН,
—Si—О—Si-O—Si—О—Si—О—
сн3
СН., СН3 СН
СН-2
2 у jj
I I I
—О—Si—О—Si—О—Si—
СН3 СН3 СН3
3
Соединение линейных макромолекул в сетчатые полимеры
с частыми поперечными связями придает полимеру
нерастворимость, твердость, хрупкость и лишает его термопластичности и
эластичности. Этот процесс называется отверждением—по
аналогии с самопроизвольным переходом термореактивных
полимеров промежуточной стадии образования в полимеры
пространственного строения. Вещества, применяемые для соединения
макромолекул линейного полимера в полимер густосетчатой
структуры, называются отвердителями. Примером таких процессов
может служить реакция между полиэпоксидом и диамином или
дикарбоновой кислотой, реакции между линейными
ненасыщенными полиэфирами и ненасыщенными мономерами.
СОВМЕЩЕНИЕ ПОЛИМЕРОВ
Совмещением полимеров можно достигнуть значительного
изменения их свойств. Обычно совмещенные полимеры получают
сплавлением или смешиванием их растворов. В большинстве
случаев совмещение приводит к образованию стабильного твердого
раствора одного полимера в другом, а иногда—к частичному
образованию блоксополимеров. Среди совмещенных полимеров
большое значение приобрели сочетания резольных феноло-формальде-
гидных смол с различными полимерами. При совмещении с поли-
винилацеталями повышается клейкость растворов этих смол;
сочетание с полиамидами или каучуками приводит к уменьшению
хрупкости отвержденной смолы; при совмещении с анилино-форм-
альдегидной смолой улучшаются диэлектрические свойства;
при сочетании с полисилоксанами повышается термостойкость
смол. Совмещенные полимеры применяют в качестве клеев
(например, клеи марок БФ, ВК-32, стр. 574), а также в производстве
пластических масс и резиновых изделий.
438
Глава VII
ТЕХНОЛОГИЯ ХИМИЧЕСКИХ ВОЛОКОН
/. ОБЩИЕ СВЕДЕНИЯ
Химические волокна получают путем химико-технологической
переработки природных или синтетических линейных полимеров.
Химические волокна можно разделить на две большие группы—
искусственные и синтетические волокна. Сырьем для
производства искусственных волокон служат природные
высокомолекулярные соединения, важнейшим из которых является целлюлоза.
Синтетические волокна изготовляются из полимеров, исходным
сырьем для которых служат низкомолекулярные соединения.
Синтетические волокна соответственно исходным полимерам
подразделяются на карбоцепные и гетероцепные волокна.
Промышленное производство искусственных волокон возникло
60—70 лет тому назад, производство синтетических волокон—
лишь 25—30 лет назад и особенно бурно развивается в последние
годы.
В настоящее время производится большое количество
искусственных и синтетических волокон различных видов (рис. 122).
Мировое производство химических волокон в настоящее время
превышает 4 млн. т в год; около 20% этого количества
приходится на долю синтетических волокон.
Химические волокна получаются в виде филаментных нитей,
штапельного волокна и моноволокна.
Филаментная нить состоит из пучка параллельно
расположенных тонких непрерывных волокон (элементарных нитей) большой
протяженности, проходящих вдоль всей длины нити. Для
повышения прочности и упругости нитей их скручивают. Число
волокон в нитях, получаемых на заводах химических волокон,
колеблется в весьма широких пределах, в зависимости от назначения
нитей.
439
Органические химические волокна
Искусственные волокна
Целлюлозные
Гидрат-
целлюлозные
Эфироцел-
люлозные
Синтетические волокна
Карбоцепные
Из полиакрило-
нитрила
На основе по-
ливинилхлорида
\
Гетероцепные
Полиамидные
Полиэфирные
Вискозные и
медноаммиачные
Ацетатные,
триацетатные
Из
полипропилена
Из фторпроиз-
водных этилена
Рис. 122. Важнейшие виды химических волокон
Для изделий и тканей широкого потребления
изготовляются текстильные нити с числом волокон от 3 до 100; в кордных
нитях, применяемых при изготовлении тканей для авиационных
и автомобильных покрышек, число волокон может превышать
2000.
Моноволокно, представляющее собой одиночное волокно
большой длины, вырабатывается в небольшом количестве и имеет
ограниченное применение (в качестве искусственного волоса,
щетины, струн).
Штапельное волокно, похожее на волокнистую массу хлопка
или шерсти, состоит из коротких волокон длиною примерно от 35
до 150 мм- Для получения нитей штапельного волокна
требуется механическое прядение, подобное прядению хлопка или
шерсти.
Качество химических волокон характеризуется комплексом
их свойств, важнейшими показателями которых являются:
прочность и удлинение (в сухом и мокром состояниях), эластичность,
устойчивость к многократным деформациям, гигроскопичность,
стойкость к действию света, высоких и низких температур и
атмосферным воздействиям, химическая стойкость, сопротивление
старению.
Важными показателями качества волокон являются также их
равномерность по толщине, прочности и удлинению, отсутствие
внешних изъянов (оборванных и склеенных волокон, узлов, пятен,
различия оттенков и т. д.). При оценке качества волокон,
применяемых для изготовления товаров народного потребления,
дополнительно определяются сминаемость, усадка, равномерность
окрашивания, легкость очистки от загрязнений, гигиенические и
некоторые другие свойства.
Толщина волокон выражается различными способами. Лишь в редких
случаях она характеризуется площадью поперечного сечения волокна (только
для элементарных нитей) или его диаметром (только для элементарных нитей
круглого сечения). Обычно толщина химических волокон как элементарных,
так и филаментных нитей, выражается номером волокна или элементарной
нити. Номер волокна (нити) означает число метров волокна (нити), масса
которых равна 1 г, и выражается формулой:
где L—длина волокна (нити), м;
g—масса волокна (нити) этой длины, г.
Чем больше толщина волокна (нити), тем меньше его номер.
Прочность волокна (предел его прочности при растяжении) выражается
в кгсЫм? или в разрывных километрах (ркм). Прочность в разрывных
километрах—условная величина, указывающая длину волокна (нити) в километрах,
разрыв которого произойдет под действием собственной тяжести (например,
при мысленном поднятии волокна за его конек над землей).
441
Соотношение между ПрОЧНОСІЬЮ, dblpdAcritiuii
ляется следующей формулой:
p
где Е—прочность, кгс/мм2;
Lp—разрывная прочность, ркм;
р—удельный вес волокна (нити), гс/см3.
Общее удлинение волокна определяют как величину приращения длины
образца в момент разрыва по отношению к первоначальной его длине:
/=
где /—удлинение, %;
Lx—длина образца в момент разрыва, м;
Lf,—первоначальная длина образца, м.
Эластичность волокна характеризуется отношением обратимого удлинения,
обусловленного упругой и эластической деформацией, к общему удлинению.
Устойчивость к действию многократных деформаций определяется числом
двойных изгибов волокна до его разрушения.
Получение химических волокон. Высокая прочность и большая
гибкость волокон в значительной мере обусловлены их
молекулярной структурой. Огромное большинство природных и химиче-
Рис. 123. Процесе ориентации линейных
макромолекул при формовании волокна.
Расположение макромолекул: Л—беспорядочное; Б—частичне
ориентированное', В—ориентированное.
ских волокон состоит из длинных линейных макромолекул,
располагающихся в большей или меньшей степени вдоль волокна, т. е.
ориентированных в направлении его оси (рис. 123).
Основным процессом в производстве любых химических
волокон является перемещение нитевидных макромолекул полимера
в пространстве, расположение их в образованном волокне более
или менее параллельно его оси с последующей фиксацией такого
взаимного положения макромолекул.
Макромолекулы исходных полимеров, применяемых в
производстве химических волокон, не ориентированы в определенном
442
направлении, поэтому для получения прочного химического
волокна исходное высокомолекулярное вещество необходимо
подвергнуть обработке, в результате которой образовалось бы
волокно, состоящее из линейных макромолекул, ориентированных
вдоль оси этого волокна. Возможность перемещения
макромолекул друг относительно друга в твердом полимере крайне
ограничена, вследствие высокой энергии межмолекулярного
взаимодействия. Для повышения подвижности макромолекул полимер
переводят в раствор или расплавляют, а из растворов и расплавов
полимера формуют химические волокна. Первой стадией процесса
получения химических волокон является приготовление
прядильной массы—расплава или раствора полимера, второй стадией—
формование из прядильной массы волокна. Формование
(прядение) производится путем вытягивания с большой скоростью
весьма тонких струек раствора или расплава полимера. В процессе
формования линейные макромолекулы в той или иной мере
ориентируются вдоль оси вытягиваемых струй, подобно бревнам,
сплавляемым по быстрой реке.
Формование волокна заканчивается отвердеванием
образовавшихся элементарных волокон с сохранением ориентации
макромолекул. Часто для повышения степени ориентации волокна
(т. е. для достижения большей параллельности расположения
макромолекул вдоль оси волокна) прибегают к последующей
вытяжке еще не совсем отвердевших волокон.
При формовании из расплава струйки расплавленного
полимера, охлаждаясь, затвердевают и превращаются в волокна. Если
формование производится из раствора полимера в сравнительно
легколетучем растворителе, волокна образуются в результате
испарения растворителя из струек прядильного раствора,
обдуваемых воздухом («высыхание» струек). Такой метод образования
волокна носит название сухого формования. Прядильные растворы
полимеров в труднолетучих растворителях перерабатывают в
химические волокна методом так называемого мокрого формования.
По этому методу волокна образуются из струек прядильного
раствора под действием веществ, содержащихся в жидкой осадитель-
ной ванне (раствор реагентов), в которую поступают струйки.
Обычно формование волокна из струек происходит в результате
разбавления растворителя, при этом полимер как бы выпадает
в осадок. В некоторых процессах мокрого формования
компоненты прядильного раствора вступают в химическое взаимодействие
с компонентами осадительной ванны, при этом состав
образующихся волокон может отличаться от состава растворенного полимера.
Последовательность операций при получении химических
волокон описанными методами показана на рис. 124.
Ко всем полимерам, применяемым для получения химических
волокон, независимо от их химического строения, предъявляются
следующие общие требования:
443
ДОЛЖНЫ
липсгї ншм
нием (асимметрической формой), это позволяет придать
макромолекулам ориентированное вдоль оси волокна расположение.
Если полимер имеет пространственное строение, то он непригоден
для производства химических волокон, так как исключается
возможность его расплавления или растворения.
Полимер
, Нагревание
Расплод
полимера
| Охлаждение^
}
Волокно
Полимер Растворитель
Воздух
РастВор полимера Осадительная
' - " Sauna
Сухое
формование
Рекуперация
растворителя
[ Мокрое
формование
Регенерация оса-
дительной оанны
Волокно
Волокно
Химический
реагент
Растборитель
Полимер
Про~изЬодное
полимера
Раствор
производного
полимера
Осадительная
Ванна¦
Регенерация
исходного
полимера
Регенерация^
. о садит ель ной
ванны
Волокно
Отработанная
осадительная -
Ванна
В
Рис. 124. Схема процессов получения химических волокон:
А—формование из расплава; Б— формование из раствора полимера (сухое и мокрое
формование); В—формование из раствора производного полимера.
2. Полимер должен иметь достаточно высокий молекулярный
вес (в пределах 15 000—100 000). Уменьшение молекулярного
веса полимера (ниже 15 000) приводит к резкому падению
прочности и гибкости волокон, а чрезмерное увеличение его
молекулярного веса осложняет формование волокна вследствие
уменьшения подвижности макромолекул и увеличения вязкости растворов.
444
3. Величина сил межмолекулярного взаимодействия должна
быть достаточной для предотвращения взаимного перемещения
макромолекул в сформованном волокне. Если величина
межмолекулярных сил мала, то в результате взаимного перемещения
макромолекул волокно будет подвергаться необратимым
пластическим деформациям или обладать каучукоподобной
эластичностью.
4. Полимер должен содержать возможно меньше посторонних
примесей, затрудняющих приготовление однородного
прядильного раствора или расплава и осложняющих формование волокна.
Прядильные машины. В зависимости от типа и назначения
химических волокон и способа их формования конструкции
прядильных машин весьма различны. Однако все они включают одни
и те же рабочие элементы, так как
основы процесса формования всех химических
волокон одинаковы.
Прядильная машина состоит из
большего или меньшего числа совершенно
одинаковых (для данной конструкции)
прядильных мест (в некоторых случаях
более двухсот). На каждом прядильном
месте происходит формование одной фи-
ламентной нити, состоящей из заданного
числа отдельных волокон.
Индивидуальные элементы прядильного места:
1) насосик, с помощью которого
раствор или расплав полимера подается
непосредственно к месту формования нити—
к фильере (от равномерности подачи
прядильной массы зависит равномерность
толщины получаемого волокна);
2) фильера (рис. 125), представляющая собой колпачок с очень
малыми отверстиями, число которых равно числу волокон
формуемой нити. Прядильная масса, проходя через отверстия
фильеры, разделяется на тонкие струйки;
3) пространство, в котором струйки прядильной массы
отвердевают по выходе из фильеры, превращаясь в волокна
(элементарные нити), т. е. происходит формование волокон. При мокромфор-
мовании струйки поступают из фильеры в осадительную ванну,
заполненную раствором реагентов. При сухом формовании это
пространство заполнено паро-воздушной смесью или воздухом,
охлаждающим струйки расплава, и носит название шахты
прядильной машины;
4) приемное устройство, при помощи которого сформованное
волокно наматывается или непрерывно передается на последующие
операции. Формование волокна всегда производится под
натяжением, которое создается системой приемных и натяжных приспо-
445
Рис.г 125. Фильера.
соблениіщбобина, диск, центрифуга, приемные ролики или
цилиндры), і
Питающие трубопроводы, по которым прядильная масса
подается в машину, а также осадительная ванна, вытяжные,
приемные устройства и механический привод прядильной машины
могут быть общими для нескольких прядильных мест.
Производительность прядильных машин зависит от числа
прядильных мест, скорости формования волокна и толщи ны
получаемой нити. Чем ниже номер волокна и больше число волокон,
входящих в нить, тем она толще и тем выше производительность^ G
прядильной машины:
nvz 60-24
0
где п—число отверстий в фильере, равное числу волокон нити;
v—скорость формования (по скорости вращения приемного
устройства), м/мин;
z—число прядильных мест на машине;
№—номер волокна.
Прядильная машина для производства штапельного волокна
снабжена фильерами с числом отверстий до 3600. Волокна,
выходящие из нескольких фильер, соединяются затем вместе в один
штапельный жгут (нить).
Отделка химических волокон. Кроме основных процессов
приготовления прядильной массы и формования, технология
химических волокон включает ряд вспомогательных операций.
После формования волокно еще не представляет собой
готовую продукцию и до поступления на текстильную переработку
должно пройти отделку, включающую удаление загрязнений,
в ряде случаев отбелку, а иногда и окраску волокна. Текстильную
и кордную нить подвергают крутке. Для облегчения текстильной
переработки (прядение штапельного волокна, ткачество, вязание
и др.) все виды волокна подвергаются специальной операции—
замасливанию. Отделка включает также сушку, перемотку
волокна, а при изготовлении штапельного волокна—и рыхление с
последующей упаковкой в кипы или рулоны. Последовательность
и количество проводимых операций отделки определяются
назначением волокна и требованиями к его качеству.
Отделка филаментных нитей производится на комбайнах
(непрерывный способ); при прерывных способах формования
отделочные растворы просасываются через толщу намотанного волокна
под действием вакуума или под небольшим давлением @,3—0,2 am),
последующая сушка волокна производится в специальных
сушилках. В производстве штапельного волокна операции
формования и отделки выполняются на одной и той же машине
(агрегате).
446
В процессе обработки непрерывной нити на комбайнах она
проходит по роликам, погруженным в отделочный раствор или
орошаемым этим раствором. Для увеличения продолжительности
обработки нить обвивает ролик многовитковой спиралью. Сушка
нити производится на роликах, обогреваемых паром или горячей
водой.
Штапельное волокно отделывается в жгуте или в резаном виде
на оборудовании непрерывного действия, объединенном с
прядильными машинами. Жгут протягивают через ряд ванн с
соответствующими отделочными растворами, режут и высушивают
в ленточных сушилках. Резаное штапельное волокно при отделке
распределяют ровным слоем на непрерывно движущейся
бесконечной сетке и орошают отделочными растворами.
2. ИСКУССТВЕННЫЕ ВОЛОКНА НА ОСНОВЕ ЦЕЛЛЮЛОЗЫ
ПРОИЗВОДСТВО ВИСКОЗНОГО ВОЛОКНА
Вискозное волокно получают химико-технологической
переработкой целлюлозы по так называемому вискозному методу.
Это волокно состоит из регенерированной целлюлозы (гидрат-
целлюлозы), которая химически идентична природной целлюлозе,
но имеет измененную физико-химическую структуру.
Основным сырьем для производства вискозного волокна
является древесная целлюлоза (стр. 76), чаще всего еловая,
поступающая на заводы искусственного волокна в виде листов картона.
Она должна содержать не менее 88% а-целлюлозы (целлюлоза,
не растворимая в 17,5%-ном растворе NaOH при комнатной
температуре). Молекулярный вес исходной целлюлозы колеблется
в пределах 100 000—150 000, что соответствует степени
полимеризации 650—900. Отдельные партии поступающей целлюлозы
могут несколько отличаться по свойствам, поэтому для придания
большей однородности исходному сырью партии сначала
смешивают, а затем составляют навески целлюлозы, соответствующие
объемам производственного оборудования.
Приготовление прядильного раствора. Первая стадия
технологического процесса получения вискозного волокна заключается
в обработке листов целлюлозного картона 17,5%-ным раствором
едкого натра для удаления из целлюлозы растворимых веществ
и придания ей большей реакционной способности. Этот процесс
называется мерсеризацией и проводится в мерсеризационных
ваннах-прессах (рис. 126).
Листы целлюлозного картона пачками устанавливают
вертикально между стальными перфорированными листами мерсери-
зационного ванны-пресса и заливают 17,5%-ным раствором щелочи.
В процессе мерсеризации а-целлюлоза набухает и образует с
447
едким натром новое химическое соединение—щелочную целлюлозу
(алкалицеллюлозу):
[СвНА(ОНK]„
целлюлоза
NaOH
С,Н7О2(ОНJО
Na
щелочная целлюлоза
Процесс мерсеризации продолжается около часа, после чего
с помощью гидравлических приспособлений отжимают избыток
щелочи и вынимают целлюлозу из ванны-пресса вместе с
перфорированными стальными листами. Для улучшения условий труда
и ускорения процесса разгрузка ванн-прессов механизирована.
Рис. 126. Мерсеризационный ванна-пресс:
/—поршень гидравлического насоса; 2—стальные листы; 3—приспособление
для выгрузки отжатой мерсеризованной целлюлозы.
Щелочь многократно используется в процессе мерсеризации.
Диализом щелочь освобождают от растворенных гемицеллюлоз
(низкомолекулярная целлюлоза и примеси полисахаридов,
растворимых в 17,5%-ной щелочи), добавляют свежий, раствор
щелочи, охлаждают и вновь направляют раствор на мерсеризацию.
После мерсеризации и отжима щелочную целлюлозу для
ускорения последующих процессов измельчают в специальных
аппаратах периодического или непрерывного действия.
Измельчитель периодического действия (рис. 127) представляет собой
аппарат с седлообразным днищем, снабженным стальными
зубьями. Щелочная целлюлоза перетирается и измельчается в зазоре
между вращающимися коленчатыми валами 2 и седловиной /
448
днища. Для лучшего измельчения и перемешивания материала
направление вращения валов через определенные промежутки
времени автоматически меняется на противоположное. Корпус
измельчителя снабжен охлаждающей рубашкой.
Продолжительность процесса измельчения—около 2 ч.
Измельчитель непрерывного действия, показанный на рис. 128,
выполнен в виде мельницы с двумя дисками (жерновами), один
из них неподвижен, другой вращается. Щелочная целлюлоза
измельчается в зазоре между дисками, степень измельчения
регулируют изменением расстояния между ними.
Измельченную щелочную целлюлозу помещают в
специальные камеры, в которых в течение 30—50 ч при 20—22 °С (или 8—
10 ч при 36—40 °С)
происходит процесс предсоз-
ревания. Этот процесс
заключается в уменьшении
степени полимеризации
целлюлозы в 2—2,5 раза
вследствие ее окисления,
сопровождающегося
разрывом макромолекул.
Снижение степени
полимеризации целлюлозы,
зависящее от
продолжительности и температуры процесса
предсозревания,
необходимо для уменьшения
вязкости получаемых
прядильных растворов и
облегчения условия формования
из них волокна.
Предсозревание можно проводить также на непрерывно
движущихся лентах, на которые помещают слой
щелочной'целлюлозы высотой 400—500 мм, или в медленно вращающихся трубах
диаметром около 3 м и длиной 30—40 м. Трубу устанавливают с
уклоном в 1° и снабжают водяной рубашкой для нагревания или
охлаждения. Вследствие вращения трубы (один оборот за 4—
5 мин) щелочная целлюлоза постепенно перемещается от одного
конца трубы к другому.
Целлюлоза относится к полимерам, растворимым в очень
ограниченном числе малодоступных растворителей, поэтому для
осуществления процесса ее дальнейшей переработки в вискозное
волокно приходится получать производное целлюлозы—ее
ксантогенат, растворимый в разбавленных растворах щелочей. После
предсозревания щелочную целлюлозу обрабатывают
сероуглеродом C3—38% от количества а-целлюлозы), при взаимодействии
с которым щелочная целлюлоза ^превращается в ксантогенат—
Рис. 127.
периодического
Измельчитель
действия:
/—седловина днища; 2—коленчатые валы.
29—805
449
лоты:
кислого эгЬира целлюлозы и дитиоугольнои
кисcs2
ГСвН7О2(ОНJО
L N
-Q,H7O2(OHJO
C=S
SNa
ксантогенат целлюлозы
В ходе этого процесса, называемого ксантогенированием,
сероуглерод реагирует не только со щелочной целлюлозой, но и
с едким натром, образуя ряд
побочных продуктов. Скорость
ксантогенирования возрастает с
повышением температуры.
Обычная продолжительность
ксантогенирования 1—2 ч при 22—30 °С
с постепенным повышением
температуры к концу процесса.
Ксантогенирование проводят
во вращающихся стальных
барабанах, называемых ксантатбара-
банами или баратами. Щелочную
целлюлозу загружают в ксантат-
барабан через люк, сероуглерод
вводят в герметически закрытый
аппарат через цапфу.
Температуру ксантогенирования регулируют
при помощи водяной
охлаждающей рубашки. Ксантатбарабан
медленно вращается, при этом
щелочная целлюлоза
перемешивается.
Ксантогенат целлюлозы
представляет собой сыпучее вещество
оранжевого цвета, растворимое
в разбавленной D—5%-ной)
щелочи с образованием вязкого прядильного раствора, называемого
вискозой. Вискоза содержит обычно 8—9% целлюлозы и 6—7%
NaOH.
Ксантогенат растворяется в специальных
аппаратах-растворителях, снабженных мешалкой и охлаждающей рубашкой. С
понижением температуры растворимость ксантогената повышается,
поэтому растворение проводят при 6—10 °С в течение 4—5 ч.
Значительные достоинства имеет последовательное ведение
процессов ксантогенирования и растворения в одном аппарате—
вакуум-ксантатсмесителе. Оно позволяет исключить операцию
Рис. 128. Измельчитель
непрерывного действия.
450
перегрузки ксантогената из аппарата в аппарат и улучшить
условия труда, так как уменьшается выделение паров сероуглерода.
Вакуум-ксантатсмеситель (рис. 129) представляет собой
аппарат емкостью до 15 м3, нижняя часть которого состоит из двух
полуцилиндрических днищ и снабжена рубашкой для
охлаждения. В полуцилиндрических днищах расположены два
горизонтальных Z-образных (коленчатых) вала, перемешивающих массу
в процессе ксантогенирования. В верхней части аппарата
установлена вертикальная мешалка, улучшающая условия
перемешивания при растворении. После загрузки щелочной целлюлозы
в аппарат при разрежении вводят сероуглерод (разрежение
способствует лучшему
распределению сероуглерода в
целлюлозе). По окончании
ксантогенирования в
вакуум-ксантатсмеситель загружают
разбавленную щелочь и растворяют
полученный ксантогенат при
постоянном вращении
горизонтальных коленчатых валов и
верхней вертикальной мешалки
и при непрерывном охлаждении
вакуум-ксантатсмесителя
рассолом, подаваемым в рубашку.
Приготовленную вискозу со
строго определенными
концентрациями целлюлозы и щелочи
и постоянной вязкостью
передают по трубам в так
называемый вискозный погреб, где
происходит созревание вискозы.
В производстве штапельного волокна и корда режим
получения вискозы обычно отличается от описанного. Все стадии этого
процесса объединяются в одном аппарате—так называемом
аппарате ВА (рис. 130). Особенностью режима получения вискозы
в аппарате ВА является отсутствие операции отжима щелочной
целлюлозы после мерсеризации и резкое уменьшение
продолжительности предсозревания щелочной целлюлозы.
Процесс получения вискозы в аппарате ВА состоит из
следующих стадий: загрузка целлюлозы, предварительно разрезанной
на довольно мелкие куски E0x50 мм), и обработка ее щелочью
при 60—62 °С в течение 1—2,5 ч. При этом одновременно
протекают процессы мерсеризации и предсозревания целлюлозы.
Поскольку в аппарате ВА щелочная целлюлоза не отжимается,
количество щелочи, расходуемой на мерсеризацию, значительно
меньше, а концентрация NaOH выше, чем в отдельном аппарате
мерсеризации. Кроме того, в аппарате В А гемицеллюлоза не вы-
Рис. 129. Вакуум-ксантатсмеситель.
29*
451
мывается из щелочной целлюлозы, поэтому очень желательно
применять облагороженное сырье с . высоким содержанием
а-целлюлозы.
Для ускорения процесса предсозревания, помимо повышения
температуры мерсеризации к щелочи добавляют часто около 1%
перекиси водорода. По окончании предсозревания аппарат В А
охлаждают до 20—24 "С и обрабатывают щелочную целлюлозу
сероуглеродом C8—42% CSa от количества а-целлюлозы) в
течение 1 ч при температуре не выше 28 °С.
Рис. 130. Аппарат ВА.
Полученный ксантогенат целлюлозы растворяют в воде при
10 °С C0—40 мин), образовавшийся вязкий раствор далее
поступает на дорастворение, которое продолжается 2 ч, а затем вискозу
передают в вискозный погреб. Общая продолжительность одного
цикла работы аппарата ВА 6—8 я.
При созревании вискозы протекают сложные химические и
физико-химические процессы. Ксантогенат целлюлозы является
нестойким химическим соединением, поэтому в процессе
созревания он частично гидролизуется с отщеплением тиокарбоновых
групп —C=S:
5
+н2о
1СН
SNa
Выделяющиеся при частичном гидролизе сероуглерод и едкий
натр, реагируя между собой, образуют различные серосодержащие
соединения, главным образом тритиокарбонат натрия:
3CSa -|- 6NaOH * 2Na2CSa -f Na2CO3 + 3H2O
452
В результате гидролиза степень этерификации ксантогената
целлюлозы постепенно понижается и содержание свободного
едкого натра в растворе уменьшается. Это приводит к повышению
зрелости и уменьшению устойчивости вискозы.
Устойчивость вискозы определяется ее способностью
коагулировать при добавлении водных растворов солей (обычно
хлорида натрия или аммония). Чем меньше раствора соли требуется
для коагуляции вискозы, тем ниже ее устойчивость, тем выше
зрелость.
В зависимости от вида вырабатываемых вискозных волокон
их формуют из вискозы различной зрелости. Наименее зрелые
(молодые) вискозы применяются в производстве корда, наиболее
зрелые (старые)—в производстве штапельного волокна.
Слишком продолжительное созревание может привести к
застудневанию вискозы и, следовательно, к невозможности
формования из нее волокна. Слишком молодые вискозы также
непригодны для формования волокон, так как выходящие из
фильеры тонкие струйки очень молодой вискозы не отвердевают в оса-
дительной ванне.
Обычно при созревании температуру поддерживают в пределах
14—16 °С. при этом созревание продолжается 25—48 ч, в
зависимости от требуемой зрелости вискозы.
После созревания несколько партий вискозы смешивают
для получения однородного по вязкости, зрелости и концентрации
раствора. Смешение производится в горизонтальных баках
большой емкости, снабженных охлаждающими рубашками и
горизонтальными мешалками. Для получения более однородной
вискозы перемешивание часто проводят в двух или трех последовательно
установленных баках.
Из смесителей вискозу передают на фильтрование. Эта
операция обычно проводится последовательно на 2—4 фильтр прессах.
Вначале вискоза подвергается грубому фильтрованию для
отделения наиболее крупных частиц механических примесей, далее
фильтрование проводится через более плотный фильтрующий
материал. Вискоза продавливается через фильтрпресс сжатым
воздухом или подается зубчатыми насосами под давлением 5—6 am.
В качестве фильтрующего материала употребляют
хлопчатобумажную ткань или ткань из волокна хлорин (стр. 467) с
прокладкой гигроскопической ваты.
Отфильтрованная вискоза отстаивается при разрежении в
стальных баках для удаления из нее пузырьков воздуха (обезвоздуши-
вание). Продолжительность этой операции 10:—20 ч.
Приготовленную описанным способом вискозу передают по
трубам на формование вискозного волокна, перед формованием
ее еще раз фрільтруют через фильтрпрессы.
Формование вискозного волокна. Вискоза подводится по общему
трубопроводу 1 к прядильной машине (рис. 131, А). Насосик 2
453
подает строго определенное количество вискозы в фильтр 3 све-
чевого типа, где происходит тщательное фильтрование прядильного
раствора. Далее вискоза по стеклянной трубке 4 поступает к
фильере 6 и продавливается тонкими струйками сквозь отверстия
фильеры в осадительную ванну, заполняющую желоб 5. Фильеры
для формования
вискозного волокна изготовляют
из сплавов платины с
золотом или иридием или
из тантала, устойчивых
к действию осадительной
ванны. Отверстия
фильеры имеют диаметр 0,05—
0,1 мм, число отверстий в
фильере—от 24 до 3200,
в зависимости от типа
изготовляемого волокна
(текстильная нить,
кордная нить, штапельное
волокно).
Осадительная ванна
для формования
вискозного волокна содержит
120—150 г! л H2SO4, 250—
300 г/л сульфата натрия и
Л—бобинный способ формования;
?—центрифугальний способ формования.
/—трубопровод для подачи вискозы; 2—насосик;
3—фильтр свечевого типа; 4—стеклянная трубка;
5—желоб; 6—фильера; 7—бобина; 5—направляющий
диск; 3—воронка; 10—центрифуга.
Рис. 131. Схема прядильной машины:
14—20 г]л сульфата
цинка, иногда в нее вводят
сульфаты аммония р!ли
магния (сульфаты уско-
ряют коагуляцию и
замедляют разложение ксантогената целлюлозы в волокне серной
кислотой). Температура осадительной ванны 45—48 °С. При
поступлении в осадительную ванну струйки вискозы коагулируют,
серная кислота нейтрализует свободный едкий натр и
разлагает ксантогенат целлюлозы:
СвН7О2(ОН)аО—C=S
SNa
rH2SO4
—CS2, Na2SO4
[С6Н7О2(ОНK]Л
Регенерированная целлюлоза (гидратцеллюлоза) образует
волокна, которые собираются в нить.
Скорость формования колеблется в пределах 45—70 м/мин,
в зависимости от конструкции прядильной машины, способа
формования и вида получаемого волокна.
Состав и температура осадительной ванны в процессе
формования постоянно изменяются вследствие протекания химических
реакций. Для сохранения постоянства температуры и состава
454
ванну непрерывно подогревают и добавляют в нее свежие
реагенты. Непрерывная циркуляция осадительной ванны между
желобами прядильной машины и аппаратами, в которых проводится
регенерация, фильтрование, нагревание и упаривание
осадительной ванны, осуществляется при помощи насосов, изготовленных
из кислотоупорных материалов. Осадительная ванна
представляет собой чрезвычайно агрессивную жидкость, поэтому всю
соприкасающуюся с ней аппаратуру во избежание коррозии
тщательно защищают кислотоупорными материалами.
Вследствие выделения сероводорода и сероуглерода в
результате разложения тритиокарбоната в процессе формования,
производство вискозного волокна является токсичным и
взрывоопасным. Для уменьшения вредности производственные цехи
оборудуются максимально герметизированными аппаратами и
коммуникациями, взрывобезопасным оборудованием и мощной приточ-
но-вытяжной вентиляцией с очисткой воздуха, удаляемого из
производственного помещения. На стадии ксантогенирования
предусматривается также наружное искусственное освещение.
Для увеличения прочности волокна свежесформованную нить
по выходе из осадительной ванны вытягивают с помощью двух
дисков, установленных на прядильной машине и вращающихся
с различной окружной скоростью. После вытяжки нить поступает
на приемное устройство. Волокно может быть вытянуто без
обрыва с увеличением первоначальной длины на 25—30%
(кратность вытяжки 1,25—1,3), что приводит к повышению его
прочности на 30—35%. Дальнейшее увеличение кратности вытяжки
и, следовательно, прочности, может быть достигнуто путем
вытягивания пластифицированного волокна.
Пластификацию* применяют для получения особо прочных
кордных нитей, используемых для изготовления тканей,
служащих каркасом для авиационных и автомобильных покрышек
(стр. 502). Кратность вытяжки пластифицированного волокна
достигает 1,7—1,8. Пластификацию проводят при 80—85 °С в
особой пластификационной ванне, содержащей 6—10 г/л серной
кислоты. Изменением условий вытяжки удается в широких
пределах изменять физико-механические свойства волокна и
получать волокно требуемого качества.
В производстве штапельного волокна получаемый жгут нитей
разрезают на куски длиной 38—40 мм (для переработки совместно
с хлопком) или длиной 60—80 мм (для переработки совместно
с шерстью).
По типу устройств для приема сформованной нити способы
формования волокна делятся на бобинный и центрифугальный.
* В общем виде под пластификацией полимеров понимают процессы их
обработки, приводящие к уменьшению суммарной энергии межмолекулярного
взаимодействия и увеличению подвижности макромолекул.
455
При бобинном способе формования (см. рис. 131, А) нить после
вытяжки (на рисунке не показана) наматывается на бобину 7.
При центрифугальном способе (см. рис. 131, Б) нить через диск 8
направляется в воронку 9, а из нее в центрифугу 10, приводимую
во вращение электродвигателем. Нить откладывается по
внутренним стенкам центрифугальної"! кружки в виде прядильного
кулича (паковки).
Бобинный и центрифугальний способы приема нитей являются
прерывными, так как волокно получают в виде отдельных мотков
и затем передают на промывку и отделку. При непрерывном
способе прядения на комбайнах сформованная нить проходит через
ряд роликов, на которых отделывается, сушится и в виде готового
волокна непрерывно принимается на крутильное веретено.
В табл. 19 приведены данные, позволяющие сравнить
производительность прядильных машин при различных процессах
получения вискозного волокна.
Прерывные способы (бобинный и центрифугальный), несмотря
на простоту аппаратурного оформления, недостаточно
совершенны. Более совершенные непрерывные способы приема нити дают
возможность получить равномерное по свойствам волокно при
меньшем числе ручных операций, повысить производительность
прядильных машин и создать лучшие условия труда. Однако
применяемые для этих способов машины более сложны, причем
сложность их возрастает с увеличением числа отделочных операций.
Чем выше производительность каждого прядильного места, тем
целесообразнее применение более сложных' машин непрерывного
действия. В производстве волокна, требующего сложных
отделочных операций, рационально применять только непрерывные
прядильно-отделочные агрегаты; в производстве кордного волокна
наряду с центрифугальними машинами прерывного действия
используются комбайны; при получении тонкого текстильного
волокна целесообразно применение только более простых бобинных
и центрифугальних машин. По мере совершенствования техники
машиностроения и автоматизации процессов машины
непрерывного действия приобретают все большие преимущества по сравнению
с бобинными и центрифугальними.
Современная центрифугальная машина схематически
изображена на рис. 132. Такая машина имеет 77—120 прядильных мест
(на рисунке показано одно прядильное место). В нижней ее части
расположены насосики, фильтры свечевого типа, желоба с осади-
тельной ванной, фильеры, а также центрифуги и воронки,
направляющие нить. В верхней части машины находятся диски для
приема и вытяжки нити. Центрифуги вращаются со скоростью
7—8 тыс. об/мин. Под действием центробежной силы нить
отбрасывается к стенкам, вращаясь вокруг своей оси, и таким образом
скручивается. Нить равномерно раскладывается по высоте
центрифуги движущейся вверх и вниз стеклянной воронкой.
456
Т а б л и ц а 19
Характеристика способов формований вйскозкых волоком
Виды
ВИСКОЗНОГО
волокна
Текстильная
нить
То же,
упрочненная
Кордная пить
[Итапелыгое
волокно
Способ формования
Центрифугальний, бобин-
иый, без вытяжки . .
Центрифугальний, бобин-
ный, с вытяжкой, без
пластификации ....
Центрифугальний,
непрерывный (комбайн), с
вытяжкой и
пластификацией
Непрерывный (штапелыю-
отделочньш агрегат),
без вытяжки или с вы-
. тяжкой в жгуте . . .
Количество
отверстий
D фиЛЬТре
24-60
24-100
30—1G00
1600—3200
Помер
волокна
м/е
4500—2250
6000—4500
6000—4500
6000—2260
І Іомер
пити
м/е
t
150—45
200—90
14,3—3,6
0,02—0,005
(жгут)
Скорость
формования
м/лшн
60—80
50—70
40-70
45—55
Протподп-
ТСЛІ.ПОСТІ,
одного
прядильного
места
кг/сутки
0,5-2,0
0,3—1,5
3,5-20
30—75
Фіпико механические
свинства волокна
прочное гь
ркм
14—16
18—22,5
26-30
14—22,5
разрывное
удлинение
%
20—24
14—18
10—14
14—24
Величина крутки, зависящая от числа оборотов центрифуги
и скорости формования волокна, выражается числом витков на
1 м длины нити. При центркфугальном прядении величина крутки
нити Л' может быть вычислена по формуле:
где М—скорость вращения центрифуги, об/мин;
v—скорость формования (приема нити в центрифугу), мімин.
Рис. 132. Центрифугальная прядильная машина:
1—трубопровод для вискозы; 2—насосик; 3—фильтр свечевого
типа; 4—фильера; 5— диск; 6—кружка (центрифуга).
Центрифугальная (прядильная) кружка изготовляется из
алюминия, поверхность которого для защиты от коррозии
покрывают бакелитовым лаком. Внутренний диаметр кружки 160—
170 мм, высота 90—152 мм. Заполненную волокном кружку
снимают с оси электроверетена, на котором она вращается, и
вытряхивают из нее волокно в виде цилиндрической полой паковки—
458
кулича. Такие куличи (по 200—600 г, считая на сухое волокно)
далее поступают на отделку.
Отделка вискозного волокна. Сформованное вискозное волокно
находится в сильно набухшем состоянии и содержит около 80%
осадительного раствора C,5—4,5% серной кислоты, 12—15%
сульфатов и около 2% прочих примесей, главным образом свободной
серы). Серная кислота и соли вымываются из волокна водой при
40—50 СС, серу удаляют обработкой 1—2,5%-ным раствором
сульфита натрия, едкого натра или сернистого натрия при 40—75 °С.
Для придания текстильным нитям чистого белого цвета
производят отбелку волокна раствором гипохлорита натрия, содержащим
1 гіл активного хлора при 20—25 °С (кордные нити и штапельное
волокно не отбеливают). После каждой обработки отделочными
растворами волокно тщательно промывают и подвергают мылов-
ке 0,1—0,5%-ным раствором олеинового мыла или других
поверхностно-активных веществ (стр. 328 ел.). Общая
продолжительность отделки волокна 3—4 ч.
На качество химических волокон большое влияние оказывает
чистота воды, применяемой в процессах отделки. Обычно
применяют умягченную воду, не содержащую соединений тяжелых
металлов (особенно железа) и органических веществ.
После отделки волокно отжимают и направляют в сушилку
(температура сушки не выше 60—70°С), сушка в куличах
продолжается 30—60 ч. Высушенные куличи перематываются на бобинно-
перемоточных машинах на конические шпули, вмещающие 500—
1500 г волокна.
Текстильные нити, сформованные бобинным способом, в
отличие от нитей, получаемых на центрифугальных машинах, не
имеют крутки, поэтому после сушки нить передают на крутильные
машины и также перематывают на конические шпули. Часто во
время заключительной операции—кручения или перемотки—
на сухое волокно при помощи вращающегося ролика наносят
1—3% замасливателя. Нижняя часть ролика погружена в корыто
с замасливателем, а верхняя* соприкасается с перематываемой
нитью. В качестве замасливателей обычно применяют безводные
смеси минеральных масел с поверхностно-активными веществами
(оксиэтилированными алкилфенолами, эфирами сульфированных
жирных кислот и т. д., стр. 330 и ел.) или водные эмульсии
масел.
Готовое волокно, намотанное на шпули, направляют на
трикотажные или ткацкие фабрики. •
ПРОИЗВОДСТВО МЕДНОАММИАЧНОГО ВОЛОКНА
Процесс производства этого вида искусственного целлюлозного
волокна основан на способности аммиачного раствора гидроокиси
меди растворять целлюлозу с образованием вязких концентриро-
459
бежными потерями меди D0—50 кг на 1 m волокна) и
применяется лишь в небольших масштабах для производства штапельного
волокна.
При взаимодействии гидроокиси меди с водным раствором,
аммиака образуется растворимый в воде медноаммиачный
комплекс:
Cu(OHJ + nNH3 * [Cu(NHg)n](OHJ
Это соединение взаимодействует с гидроксильными группами
целлюлозы, благодаря чему происходит ее растворение. Водно-
аммиачный, раствор гидроокиси меди принадлежит к числу
немногих реагентов, растворяющих целлюлозу.
Ниже приводится состав медноаммиачного прядильного
раствора (в %):
Целлюлоза 9,5—10,0
Аммиак 7,0—8,0
Медь 4,2—4,3
При разбавлении этого раствора водой происходит коагуляция
с выделением студнеобразной темно-синей массы
медноаммиачного соединения целлюлозы, состав которого приближенно может
быть выражен формулой:
[С6НА(ОН)з1л-[Си(ЫН3ЫОНJ]т
(пит приблизительно одинаковы, х—колеблется в пределах
1-2).
Серная кислота разлагает это соединение с выделением гидрат-
целлюлозы
[QH7O2(OHK]„.[Cu(NH3WOHJ]m + т. [ 1 - -J-) H2SO4 >
* [С6Н7О,(ОН)Э]Я + (т- — j (NH4JSO4 + 2mHa0
На этих двух процессах—коагуляции медноаммиачного
раствора целлюлозы водой и разложении студнеобразного
медноаммиачного соединения целлюлозы серной кислотой—основано
формование медноаммиачного волокна.
Таким образом, по химическому составу вискозное и медно-
аммиачное волокна совершенно одинаковы и представляют собой
чистую регенерированную целлюлозу (гидратцеллюлозу).
Сырьем для получения медноаммиачного волокна чаще всего
служит хлопковая целлюлоза, получаемая из линтера—отхода
очистки хлопковых семян на маслобойных заводах, непригодного
из-за малой длины волокон для выработки хлопчатобумажных
тканей.
460
Приготовление прядильного раствора. Для приготовления
прядильных растворов вначале получают основную сернокислую
соль меди Cu(OHJ-CuSO4, которая растворяется в охлажденной
до 6—10 СС 25%-ной аммиачной воде с образованием комплексной
соли:
Cu(OHJ-CuSO4 + 8NH3 » [Cu(NH3L](OHJ-[Cu(NH3L]SO4
Раствор комплексной соли заливают в аппарат—растворитель
с винтовой лопастной мешалкой (рис. 133), туда же при
непрерывном перемешивании раствора загружают целлюлозу, которая
сильно набухает в этом растворе. К набухшей массе добавляют
раствор едкого натра, разлагающего комплексную соль:
[Cu(NH3L](OHJ-[Cu(NH3L]SO4 + 2NaOH » 2[Cu(NH3L](OH)a + Na2SO4
Целлюлоза растворяется в растворе медноаммиачного
комплекса, образуя очень вязкий темно-синий прядильный раствор.
Рис. 133. Аппарат-растворитель с винтовыми лопастями.
Формование и отделка медиоаммиачного волокна. Перед
формованием растворы целлюлозы фильтруют через никелевые сетки
и удаляют из раствора пузырьки воздуха. Медноаммиачное
волокно формуют двухванным способом, по которому в первой оса-
дительной ванне коагулирует медноаммиачное соединение
целлюлозы, разлагаемое во второй ванне с выделением гидратцеллю-
лозы.
Прядильный раствор продавливается через фильеру с
отверстиями диаметром 0,5—1 мм, закрепленную в верхней части
стеклянной воронки. В воронку непрерывно поступает вода (осади-
тельная ванна), поток которой увлекает за собой струнки пря-
461
дильного раствора. При этом прядильный раствор коагулирует,
отверстия воронки. Свежесформованные нити собираются со
всей прядильной машины в жгут, непрерывно поступающий на
другую машину во вторую ванну, содержащую 1%-ный раствор
H2SO4, в котором медноаммиачное соединение целлюлозы
разлагается при температуре 40—45 °С. После обработки серной
кислотой жгут волокон, состоящих из гидратцеллюлозы, тщательно
промывают и сушат. Для уменьшения расхода реагентов медь
и аммиак регенерируют из отработанных растворов.
Медноаммиачным способом можно получать также штапельное
волокно и текстильные нити.
производство ацетатного волокна
Ацетатное волокно занимает в общем мировом производстве
химических волокон второе место после вискозного волокна и
вырабатывается главным образом в виде текстильных нитей.
Ценным сырьем для текстильной промышленности является также
ацетатное штапельное волокно. Технология производства
ацетатного шелка и ацетатного штапельного волокна одинакова.
Исходным сырьем для производства ацетатного волокна
служит ацетилцеллюлоза (уксуснокислый эфир целлюлозы),
получаемый ацетилированием целлюлозы:
h2so4
[С0Н7О2(ОНK]„ + Зя(СН3СОJО * [С6Н7О2(ОСОСН3K]Л + ЗлСН3СООН
Первичным продуктом ацетилирования является триацетил-
целлюлоза (первичный ацетат), которая частичным омылением
может быть превращена во вторичную ацетилцеллюлозу,
содержащую 54—55% связанной уксусной кислоты и примерно
соответствующую формуле
[C0H7O2(OCOCH3),,5(OH)Ui5];I
Первичный ацетат растворим в метиленхлориде, хлороформе,
уксусной кислоте. Вторичная ацетилцеллюлоза растворима во
многих органических растворителях, в том числе в смеси 85%
ацетона и 15% этилового спирта. Растворы ацетилцеллюлозы в
этой смеси служат прядильными растворами для получения
ацетатного волокна. Триацетатное волокно формуют из его
растворов в метиленхлориде.
Ацетатное волокно, в отличие от вискозного и медноаммиачного,
состоит из эфира целлюлозы. Это определяет специфические
свойства ацетатного волокна—большую эластичность (меньшая
сминаемость тканей) и меньшую термическую стойкость
(деформируется при температуре выше 140—150 °С). При крашении
изделий, в состав которых входят как целлюлозные, так и ацетат-
462
сые волокна, отличающиеся по химическому составу, получают
неодинаковую окраску. Этим пользуются для придания тканям
специальных цветовых эффектов, значительно улучшающих
внешний вид изделий.
Приготовление прядильных
растворов. Вторичную ацетилцеллюлозу
растворяют в смеси ацетона со спиртом,
триацетилцеллюлозу—в
метиленхлориде или смеси метиленхлорида со
спиртом. Растворение проводят в
герметических аппаратах—растворителях,
получаемый прядильный раствор должен
содержать 18—24% ацетилцеллюлозы
(вязкость такого раствора в 5—10 раз
выше вязкости вискозы и составляет
400—800 пз). Перед формованием
раствор подвергают трех- или
четырехкратной фильтрации на фильтрпрессах
с последующим удалением пузырьков
воздуха.
Во избежание коррозии вся
аппаратура, применяемая для работы с
прядильными растворами, выполняется из
легированных сталей или футеруется
фосфористой бронзой.
Формование ацетатного волокна.
Волокно из вторичной ацетилцеллюлозы
формуют сухим методом (рис. 134).
Прядильный раствор, нагретый до
40—50 °С, подается зубчатым насоси-
ком и продавливается через фильеру
2 в вертикальную шахту. Через
шахту продувается горячий воздух F0—
65 °С). Летучие растворители
испаряются, а струйки ацетилцеллюлозы
затвердевают, образуя волокна. Высоту
прядильной шахты рассчитывают таким
образом, чтобы в ней происходило
полное испарение растворителей. Обычно
высота шахты равна 3—4 м.
Выходящие из фильеры волокна движутся в шахте прядильной
машины сверху вниз и по выходе из шахты собираются в нить,
которая наматывается на бобину 6. В прядильной шахте в
результате испарения образуется паро-воздушная смесь, содержащая
25—40 г летучих растворителей на 1 м3 воздуха. Растворители
улавливают из паро-воздушной смеси путем адсорбции
активированным углем и после отгонки из него снова возвращают в
'463
Рис. 134. Схема машины
для формования
ацетатного волокна:
/—фильтр; 2—фильера;
3—дырчатые трубы для отсоса паров
растворителя; 4—коллектор для
отвода отсасываемых паров;
5—обогревающее
приспособление; б—бобина; 7—нитеводи-
тель.
производство. Скорость формования ацетатного волокна сухим
методом составляет 200—300 мімин.
Формование волокна из триацетилцеллюлозы производят
преимущественно мокрым методом. В качестве осадительной ванны
применяется метиловый спирт. Для уменьшения потерь
метилового спирта и метиленхлориде формование волокна проводят
при 20—30 °С.
После формования не требуется отделки ацетатного волокна,
поэтому его замасливают непосредственно на прядильной машине
перед намоткой на бобину. Далее волокно подвергают кручению,
затем перематывают, сортируют и упаковывают для отправки
лотребителям.
3. СИНТЕТИЧЕСКИЕ ВОЛОКНА
ПРОИЗВОДСТВО КАРБОЦЕПНЫХ ВОЛОКОН
Общие методы получения карбоцепных волокон. В настоящее
время исходными веществами для промышленного производства
карбоцепных синтетических волокон являются: полимеры и
сополимеры акрилонитрила, поливкяилхлорид,сополимер винилхло-
рида с акрилонитрилом, полиэтилен и полипропилен, полимеры
фторпроизводных этилена.
Для получения карбоцепных волокон используют методы
сухого и мокрого формования (из раствора), а также формование
из расплава или размягченного полимера.
Сухое формование карбоцепных волокон аналогично
формованию ацетатного волокна. При использовании мокрого метода
формования карбоцепных волокон в отличие от формования
вискозного волокна не происходит химических реакций между
компонентами прядильного раствора и осадительной ванны. Струйки
прядильного раствора по выходе из фильеры попадают в осади-
тельную ванну, разбавляющую растворитель, в результате
полимер коагулирует в форме волокон. Они собираются в нить или
жгут и поступают, в соответствующий приемный механизм. Нити
обычно наматываются на бобину, жгут штапельного волокна
непрерывно поступает в отделочный агрегат, где промывается,
отделывается и сушится.
Формование карбоцепных синтетических волокон всегда
производится с большей или меньшей вытяжкой (фильерная вытяжка),
величина которой зависит от требуемых физико-механических
показателей готового волокна. Скорость мокрого формования
карбоцепных волокон невелика и обычно не превышает 50 мімин.
Отделка карбоцепных волокон значительно проще отделки
вискозных и медноаммиачных волокон. Она заключается в
удалении из волокна компонентов осадительной ванны или остатков
неиспарившегося растворителя, нанесении на волокно замаслива-
теля, сушке, кручении нитей и их перемотке.
464 -
Специфической операцией в производстве синтетических
волокон является последующая вытяжка сформованного волокна
(на холоду или при повышенной температуре) для повышения его
прочности и снижения разрывного удлинения. При этом волокно
вытягивается в 4—10 раз. Вытянутое волокно выдерживают при
высокой температуре (термофиксация).
Производство волокна нитрон
Исходным сырьем для производства волокна нитрон служит
полиакрилонитрил (ПАН, стр. 380). Для приготовления
прядильного раствора полиакрилонитрил растворяют в диметилформ-
амиде в две стадии: сначала в аппарате-смесителе на холоду
замешивают порошкообразный полиакрилонитрил с таким
количеством диметилформамида, чтобы в последующем получился
15—16%-ный раствор полимера. Суспензию набухшего полимера
передают в аппарат-дорастворитель, где полимер полностью
растворяется при 85—90 °С и непрерывном перемешивании.
Полученный прядильный раствор два или три раза фильтруют
при температуре 50—80 °С и, для повышения скорости обезвоз-
душивания, удаляют из раствора пузырьки воздуха в вакууме
при 45—50 °С.
Прядильный раствор можно приготовить также растворением
полиакрилонитрила в диметилсульфоксиде, в азотной кислоте или
водном растворе солей, чаще всего роданистого натрия.
Волокно нитрон формуют из растворов полиакрилонитрила
в диметилформамиде по мокрому и сухому методам. Принцип
мокрого формования волокна нитрон аналогичен с формованием
триацетатного волокна. В качестве осадительной ванны используют
разбавленный водный раствор диметилформамида или смеси
многоатомных спиртов с диметилформамидом. Вода или спирты,
содержащиеся в осадительной ванне, смешиваясь с
диметилформамидом, вымывают его из поступающих в ванну струек прядильного
раствора. При этом полиакрилонитрил выделяется из раствора
в виде тонких волокон, которые далее поступают на приемные
приспособления. Диметилформамид отгоняют из осадительной
ванны и возвращают в производство.
Формование волокна нитрон сухим методом проводится так
же, как формование волокна из вторичного ацетата (стр. 463).
Однако поскольку температура кипения диметилформамида
A53 °С) гораздо выше температуры кипения ацетона E6 СС), то
в шахте прядильной машины поддерживается более высокая
температура (около 200 °С). Пары диметилформамида, выходящие из
шахты, улавливаются водой или сорбируются углем.
Для улучшения физико-химических свойств волокно
подвергают вытяжке при повышенной температуре (термофиксация) и
последующей отделке.
30—805 465
Нитрсн выпускается главным образом в виде штапельного
волскна и используется ь качестве заменителя шерсти как caivrc-
стоятельно, так и в смеси с натуральной шерстью. Поэтому обычно
перед резкой волокно подвергают гофрировке для придания изьт:-
тости, свойственной волокнам натуральной шерсти. Прочность
штапельного волокна 20—25 ркм, удлинение 25—35%.
Волокно нитрон обладает очень высокой светостойкостью и
хорошими теплоизоляционными свойствами. Его используют для
производства трикотажных изделий и искусственного меха. К
недостаткам этого волокна относится очень низкая
гигроскопичность и, как следствие этого, плохая окрашиваемость.
Для улучшения окрашиваемости и некоторых
физико-механических свойств полиакрилонитрильного волокна в качестве
исходных полимеров в производстве волокон подобного типа
применяют сополимеры акрилонитрила с другими винильными
соединениями—винилпиридином, акриламидом, метилакрилатом и др.,
количество которых, как правило, не превышает 20%.
В некоторых случаях используют сополимеры, содержащие
значительное количество винильного компонента. Из таких
сополимеров промышленное применение получили сополимеры
акрилонитрила B0—60%) с винилхлоридом (80—40%). Из них
вырабатывается шелк виньон N и штапельное волокно дайнель.
Получение и отделка этих волокон аналогичны получению волокна
нитрон, однако указанные сополимеры акрилонитрила и винил-
хлорида растворимы в ацетоне, что значительно упрощает и
удешевляет производство волокна из них. По светостойкости и
показателям физико-механических свойств виньон N и дайнель
несколько уступают нитрону. Области их применения такие же, что
и волокна нитрон.
Производство поливинилхлоридных волокон
Поливинилхлорид (стр. 422) растворяется с образованием
вязких растворов, пригодных для формования волокна, в очень
ограниченном числе растворителей: в смеси ацетона с бензолом или
сероуглеродом и в тетрагидрофуране. При формовании волокон
сухим способом поливинилхлорид растворяют в смеси ацетона и
сероуглерода, для формования используют прядильные машины,
применяемые в производстве ацетатного волокна.
Из растворов полимера в тетрагидрофуране формуют
штапельное волокно по мокрому способу, используя аппаратурный
принцип процесса формования медноаммиачного волокна.
Волокно, полученное из поливинилхлорида, имеет низкую
термостойкость (при 70—75 °С начинает деформироваться), трудно
окрашивается, но обладает высокой устойчивостью к
истиранию, негорючестью и очень высокой химической стойкостью.
Благодаря перечисленным выше ценным свойствам поливинил-
466
хлоридное волокно с успехом применяется в производстве
фильтровальных тканей и спецодежды. Кроме того, оно легко
электризуется и поэтому используется для изготовления лечебного белья
рекомендуемого к носке при заболеваниях радикулитом,
ревматизмом и т. п. Однако производство волокна из поливинилхлорида
очень ограничено в связи с необходимостью применять
малодоступный растворитель (тетрагидрофуран) или взрывоопасный
растворитель (сероуглерод).
Более широкое
распространение получило
волокно хлорин,
получаемое из хлорированного
поливинилхлорида,
содержащего 64—65% хлора
(стр. 190). Этот полимер
хорошо растворим в
ацетоне, что используется для
приготовления
прядильных растворов.Перед
формованием волокна хлорин
25—30%-ный раствор
полимера в ацетоне трижды
фильтруют и удаляют из
раствора воздух.
Волокно хлорин
формуют мокрым способом,
используя в качестве оса-
дптельной ванны
разбавленный водный раствор
ацетона. Для нормального
формования необходимо,
чтобы ВОЛОКНО ДОВОЛЬНО Рис. 135. Прядильная машина для формс-
прОДОЛЖИТелЬНОе время вания иолокна хлорин мокрым способом.
находилось в осадитель-
ной ванне; это достигается увеличением пути нитей в ванне
до 250 см. Такая особенность процесса формования волокна
хлорин обусловила необходимость применения прядильной
машины (рис. 135), конструкция которой отличается от
конструкций машин, используемых в производстве других химических
волокон.
Осадительная ванна помещается не в горизонтальном желобе,
как на других машинах, а подается в вертикальные трубки, куда
поступают также струйки прядильного раствора из фильеры.
Пройдя через трубку с осадительной ванной, струйки раствора
в результате вымывания растворителя превращаются в волокна,
которые поступают на приемные диски, расположенные в
верхней части машины. Вследствие различной скорости вращения дис-
30*
467
и далее поступает на сушку, в процессе которой происходит
терморелаксация волокна.
Ацетон регенерируют из осадительной ванны отгонкой.
Волокно хлорин выпускается в виде текстильной нити и
штапельного волокна. Свойства и области применения волокна
хлорин примерно такие же, как и поливинилхлоридного
волокна.
Производство волокна из фторсодержащих полимеров
Из фторсодержащих полимеров получают два вида волокон—
тефлон и фторлон.
Исходным сырьем для получения волокна тефлон служит тет-
рафторэтилен (стр. 194). Он не растворяется ни в каких известных
растворителях и не плавится без разложения. Поэтому из тетра-
фторэтилена не может быть получен прядильный раствор пли
расплав для формования волокна.
Волокно тефлон формуют из суспензий, получаемых при
синтезе политетрафторэтилена методами эмульсионной или
суспензионной полимеризации.
Волокно тефлон обладает совершенно исключительной
устойчивостью к действию химических реагентов, его свойства не
изменяются даже при нагревании в концентрированных серной и
азотной кислотах и в щелочах. Волокно отличается также очень
высокой светостойкостью и термостабильностью (выдерживает
нагревание до 350 °С). Кроме того, тефлон—наиболее
гидрофобное из всех химических волокон, однако прочность его
относительно невелика A5—16 ркм). Следует отметить, что, поскольку
плотность тефлона B,3 г/см3) почти в два раза выше плотности других
волокон, прочность в ркм для этого волокна мало показательна.
Сочетание весьма высокой химической и термической стойкости
делает это волокно незаменимым для некоторых технических
изделий.
Для получения волокна фторлон в качестве исходного сырья
применяется растворимый в ацетоне сополимер фтор производных
этилена с различным содержанием фтора. Из прядильного раствора
такого сополимера в ацетоне волокно фторлон можно формовать
мокрым или сухим методами. По мокрому методу в качестве
осадительной ванны используют разбавленный водный раствор
ацетона. Формование фторлона проводят на машине, подобной
прядильной машине, применяемой в производстве триацетатного
волокна.
Волокно фторлон несколько уступает по термостойкости и
химической стойкости волокну тефлон, но процесс его формования
очень прост.
468
Производство полиолефиновых волокон
Для получения волокон этого типа используют полиэтилен
высокого и низкого давления (стр. 415) и стереорегулярный
полипропилен (стр. 420). Эти полимеры растворяются в алифатических
и ароматических углеводородах при температуре 130—175 СС,
превышающей температуру плавления полимеров.
Растворы полиэтилена и полипропилена могут быть
использованы для формования волокон, но технологическое оформление
этого процесса гораздо сложнее, чем при сухом формовании.
Формование волокна из расплавов этих полимеров
производится на шнек-машине (рис. 136) специальной конструкции и
принципиально отличается от способов формования других
синтетических волокон. Это отличие обусловлено очень высокой вязкостью
Рис. 136. Схема шнековой машины для формования полипропиленового
моноволокна:
/—шнек-машина; 2—прядильная головка; 3—ванна для охлаждения волокна; 4, 6—вальцы;
S—вытяжная камера; 7—приемное устройство.
расплавленной массы полимеров, в связи с чем для формования из
нее волокон необходимо прилагать весьма большие механические
усилия.
Шнековая машина снабжена дозирующим устройством,
обеспечивающим равномерность подачи полимера. Отношение
диаметра шнека к его длине—от 1:16 до 1 : 20. Шнек имеет 3—5 зон
обогрева, температура в которых зависит от вязкости полимера.
Температурный режим формования волокна зависит также от
вида получаемого волокна. Так, при формовании текстильных
нитей из полипропилена, имеющего молекулярный вес порядка
100 000, применяется следующий температурный режим
формования:
Зоны Температура
°С
Загрузки 15—20
Предварительного обогреш .... 100—110
Расплавления 175—180
Головки и насоса 265
Фильерного комплекта 270—275
469
Дальнейшая обоаботка волокна производится различными
СПОСОбаМИ В заВИСИМОСТИ ОТ ВИДа иилучаелШЛ »илипип ^«ипи-
волокно, штапельное волокно, текстильные нити).
Моноволокно получают непрерывным способом. Сформованное
волокно охлаждается в водяной ванне, здесь же одновременно
осуществляется фильерная вытяжка. Затем волокно поступает
в вытяжную ванну, температура которой около ПО—120 °С.
После вытягивания волокно подвергается термофиксации и
поступает на приемные устройства.
Сформованное штапельное волокно и текстильные нити после
фильерной вытяжки принимаются на шпули. Жгуты
штапельного волокна далее подвергаются вытяжке в паровой камере
при 110 °С с последующей термофиксацией при 130—140 °С.
Затем волокно поступает на замасливание, гофрировку и
упаковку.
Текстильные полипропиленовые нити подвергаются
предварительному кручению, затем вытяжке в среде водяного пара или
на металлических нагретых каркасах. Далее следует
термофиксация и перемотка на конические шпули.
В таблице 20 приведены физико-механические свойства поли-
олефиновых волокон и, для сравнения,—волокна из найлона 66.
Таблица 20
Физико-механические свойства полиолефиновых волокон и иайлоиа 66
Полимеры
Полиэтилен высокого
давления ....
Полиэтилен низкого
давления . ...
Полипропилен стереорегу-
лярный
Найлон 66 (сухой) . . .
Плотность
г/смЗ
0,92
0,95
0,90
1,14
Прочность
ркм
18—20
35—45
40—70
До 70
Удлинение
%
25—35
10—20
15—25
До 30
Усадгса
за 20 мин
при 100 °С
%
40—60
5—10
10-15
5—10
Потеря
прочности
при 100 °С
по сравнению
с прочностью
при 20 °С
%
80
60
40
20
Из таблицы видно, что волокна из полиэтилена и
полипропилена обладают хорошими физико-механическими свойствами.
Кроме того, полиолефиновые волокна отличаются высокой
устойчивостью к действию щелочей и кислот, значительной
морозостойкостью и низкой плотностью. К недостаткам этих волокон
относится значительная потеря прочности при повышенных
температурах, гораздо большая, чем, например, для полиамидов.
470
ГЕТЕРОЦЕПНЫЕ ВОЛОКНА
В отличие от заводов, вырабатывающих карбоцепные волокна
и в большинстве случаев получающих исходные полимеры с
других предприятий, на заводах, где производятся гетероцеппые
волокна, имеются химические цехи для синтеза исходных
полимеров.
Производство полиамидных волокон
Наиболее распространенные полиамидные волокна получают
из поликапроамида—волокно капрон (перлон, силон, найлон 6);
из полигексаметиленадипамида—волокно анид (найлон 66); из
полиэнантоамида —волокно энант (найлон 7):
Н—[—HN—(—СН2—)a—СО—]„—ОН
поликапроамид
Н—[HN—(—СН2—)в—NHCO—(—СН2—M—СО—]ч—ОН
полигексаметиленадипамид
Н -[—HN—(—СНа—)„—СО- ]я—ОН
полиэиаитоамид
Кроме того, полиамидные волокна изготовляют из продуктов
поликонденсации гексаметилендиамина и себациновой кислоты,
а также из полиундеканамида.
При синтезе полиамида периодическим способом расплавленная
масса полимера выдавливается сжатым азотом через щелевидное
отверстие автоклава под давлением 6—8 am в ванну с холодной
водой. Затвердевший в виде ленты полимер наматывают на
мотовила, затем измельчают в крошку—на кусочки размером 4—5 мм-
Крошка полимеров, применяемых в производстве волокон анид,
и энант, поступает непосредственно на формование волокна.
В крошке поликапроамида содержится незаполимеризовавшийся
мономер, который удаляют путем экстракции горячей водой.
Перед формованием волокна капрон крошку сушат до содержания
влаги менее 0,1%. Полиамиды образуют вязкие расплавы, из
которых можно получать изделия любой формы, в том числе
формовать полиамидные волокна.
Формование полиамидных волокон. Прядильная машина для
формования волокна из расплава полиамида имеет высоту до
8,5 м и размещается в четырехэтажном здании. Партии сухой
крошки полимера периодически загружают в бункер, который
герметически закрывают и продувают очищенным азотом для
предотвращения возможности окисления полиамида. Из бункера
сыпучая крошка поступает на плавильную решетку. Решетка пред-
471
ставляет собой плоскую плотную спираль, изготовленную из
стальной трубки, внутри которой циркулирует нагретый до 270—
290 °С органический теплоноситель (обычно дифенильная смесь
стр. 130). Попадая на решетку, крошка плавится в атмосфере азота,
расплав стекает в приемный конус, из которого насосом через
фильтр подается к фильере.
Фильера представляет собой стальной диск толщиной 5 мм,
в котором по окружности просверлены отверстия диаметром
0,25 мм. Число отверстий в фильере колеблется от 3 до 90.
Все рабочие детали фильерной головки—от плавильной
решетки до фильеры—постоянно обогреваются парами
теплоносителя (температура 270—290 °С), чтобы полимер оставался в
расплавленном состоянии. Струйки расплавленного полиамида из
фильеры попадают в высокую вертикальную прядильную шахту,
где их равномерно обдувает поток воздуха. Затвердевая, струйки
расплава превращаются в волокна—элементарные нити, которые
при соприкосновении со специальными дисками замасливаются
и далее наматываются на бобину.
Скорость формования из расплава значительно выше скорости
формования другими способами и достигает 500—1500 м/мин.
Такая высокая скорость достигается потому, что остывание нити
расплавленного полимера происходит значительно быстрее, чем
протекают процессы формования волокна из растворов.
При формовании волокна капрон полиамид частично деполи-
меризуется до мономера, для удаления которого нити капрона
промывают горячей водой.
Сформованное полиамидное волокно имеет очень низкую
прочность A0—12 ркм) и большое пластическое удлинение C00—
500%), так как в процессе формования из расплава
макромолекулы полимера почти совсем не ориентируются вдоль оси волокна.
Для придания волокну требуемых физико-механических свойств
его после предварительного кручения подвергают «холодной»
вытяжке (при комнатной температуре) до 3—5-кратного
увеличения длины, при этом происходит значительное повышение
степени ориентации макромолекул, а прочность волокна возрастает
в 4—7 раз, остаточное удлинение уменьшается до 12—25%, и
волокно перестает быть пластичным.
Вытяжку волокна производят на специальных крутильно-
вытяжных машинах. При получении текстильных и кордных
нитей применяют раздельную вытяжку волокон, при выработке
штапельного волокна вытягиванию подвергают толстый жгут,
состоящий из большого числа отдельных волокон. После вытяжки
волокно замасливают, сушат и направляют на кручение и
перемотку.
При производстве полиамидных волокон непрерывным
методом расплавленный полиамид из трубы, где происходит
непрерывная полимеризация или поликонденсация, подается по обогре-
472
ваемому трубопроводу на прядильную машину. Этот метод
позволяет упростить конструкцию п уменьшить высоту прядильной
машины, так как отпадает необходимость в бункере для подачи
Растворитель -
Фильтрование
Полимеризащ
лактамО-Вав/
• клаВв
Расплавление
полимера
Лантам
Растворение
Измельчение
ленты
Лактам
Растворитель
Нагревание
УДозированис
Очистка '_
капроновой.
крошки
Поіимбризааиї
ам а
Формование
волокна
Готовая
продукция
д
Рис. 137. Схема производства волокна капрон:
А—прерывный гроцесс; Б—непрерывный процесс.
крошки полимера к плавильной решетке и, кроме того,
значительно сокращается продолжительность производственного цикла.
На рис. 137 приведены схемы производства волокна капрон
прерывным (рис. 137, А) и непрерывным (рис. 137, Б)
методами.
473
казателями физико-механических свойств. Они имеют высокую
прочность, эластичны, устойчивы к истиранию.
Капроновое волокно и волокно анид выпускаются в виде
текстильных и кордных нитей, штапельного волокна и моноволокна
(щетина). В производстве указанных видов волокон
используются полимеры соответствующего (большего или меньшего)
молекулярного веса. Число отверстий в применяемой фильере и
степень вытяжки готового волокна также различны. Наибольшей
вытяжке (в 5 раз) подвергают кордные нити, прочность их
составляет 75—80 ркм..
Основные показатели физико-механических свойств полиамид-
лых волокон капрон и анид приведены в табл. 21.
Таблица 21
Физико-механические свойства полиамидных волокон
Волокна
Капрон
обычное
высокопрочное . . .
Анид
обычное
высокопрочное . . .
Прочность
ркм
40—50
60—75
35—40
65—70
Удлинение
%
20—30
16—20
20—30
13—18
Производство волокна энант, разработанное и полученное
советскими химиками, только развивается. Это волокно
отличается светостойкостью и эластичностью и превосходит по этим
показателям другие полиамидные волокна.
Производство полиэфирного волокна лавсан
Исходным полимером для получения волокна лавсан служит
полиэтиленгликольтерефталат (стр. 408). Волокно лавсан
(терилен) формуют из расплава полимера (аналогично полиамидным
волокнам). Полиэтиленгликольтерефталат в виде крошки из
химического цеха поступает в прядильную головку, где на
плавильной решетке при 275—285 °С расплавляется и дозирующим насо-
сиком подается к фильерам. Струйки расплава, выходящие из
фильеры, проходят обдувочную камеру, затем прядильную
шахту, куда подается термостатированный воздух, и в виде
затвердевших нитей поступают на приемные приспособления.
Скорость приема нити значительно выше скорости ее выхода
из фильеры, благодаря чему в прядильной шахте нить подвер-
474
гается вытяжке и макромолекулы полимера в нити приобретают
упорядоченное расположение (ориентируются вдоль оси волокна).
На приемном устройстве нить замасливается и после прогрева
подвергается дальнейшей вытяжке и кручению. При получении
штапельного волокна вытяжке подвергается жгут волокон,
который затем поступает на гофрировку и резку.
Волокно лавсан обладает достаточно высокой прочностью
D0—65 ркм при удлинении 30—10%), хорошей упругостью и
высоким сопротивлением истиранию. Лавсан очень устойчив к
действию высоких температур, света, к воздействию кислот и
окислителей, но быстро разрушается горячими растворами щелочей.
Из волокна лавсан в смеси с шерстью часто вырабатывают
немнущиеся ткани и трикотаж.
Глава VIII
ТЕХНОЛОГИЯ КАУЧУКА И РЕЗИНЫ
/- ОБЩИЕ СВЕДЕНИЯ
Резиновая промышленность занимает важное место в народном
хозяйстве, являясь, с одной стороны, крупным потребителем
ценнейших химических материалов (синтетических каучуков,
волокон и др.), а с другой стороны—поставщиком самых
разнообразных резиновых изделий и деталей. Трудно назвать вид
транспорта, отрасль промышленности, сельского хозяйства, культуры и
быта, которые не нуждались бы в резиновых изделиях.
Резиновые шины, транспортерные ленты, приводные ремни,
рукава, резино-пневматические подвески, муфты сцепления,
губчатые сидения и матрацы, резиновые или обрезиненные валы и
валики для бумажной, текстильной, химической и других отраслей
промышленности, гуммированные цистерны, баки, трубопроводы,
разнообразные резиновые уплотнители (в том числе и для
космических кораблей), изоляция кабелей и проводов—вот лишь
краткий перечень некоторых важнейших резиновых изделий, без
которых не может обойтись современная техника. К
многочисленным резиновым изделиям медицинского и бытового назначения
относятся: резиновые трубки для переливания крови, грелки,
желудочные зонды, соски, спринцовки, хирургические перчатки,
резиновая обувь, подошвы, резиновые клеи, прорезиненные ткани,
спортивные и детские мячи и т. д.
Ассортимент резиновых изделий насчитывает многие десятки
тысяч предметов. Обычно их подразделяют на следующие три
основные группы: шины, резиновые технические изделия (включая
изделия для кабельной промышленности), резиновые изделия
народного потребления (резиновая обувь и др.). К резиновой
промышленности примыкает также промышленность асбестовых
технических изделий, потребляющая каучук и некоторые другие
материалы, используемые в производстве резиновых и резино-тех-
476
нических изделий. Резиновая промышленность выпускает как
чисто резиновые изделия, так и резино-тканевые и резино-метал-
лические изделия, однако главным носителем специфических
¦свойств этих изделий является резина.
Резина представляет собой высокоэластичный материал,
получаемый в результате вулканизации (стр. 519) смесей каучука
со специальными добавками—ингредиентами резиновых смесей.
Отличительной особенностью резины как конструкционного
материала является ее эластичность, т. е. способность к очень
большим практически полностью обратимым деформациям, например
к растяжению в 10 и более раз под действием относительно
небольших сил.
Эластические свойства резины определяются ее главным
компонентом—синтетическим или натуральным каучуком. Для любых
каучуков и резин характерен низкий модуль упругости*. Так,
модуль упругости резины находится в пределах 10—100 кгс/см2,
тогда как модули упругости текстильных материалов, кожи,
пластических масс составляют 100—100 000 кгс/см2, модуль упругости
металлов—800 000—2 000 000 кгс/см2. Эластические свойства
резин проявляются в широких температурных пределах—в
среднем от —50 до -(-150 °С для обычных резин. Морозо- и
теплостойкие резины сохраняют эластичность при гораздо более низких
или высоких температурах.
Резина великолепно растягивается и сжимается в линейном
направлении, но очень плохо или почти совсем не поддается
объемному сжатию, что также является важной особенностью резины
как конструкционного материала. Резина способна выдерживать
•без разрушения миллионы циклов многократных деформаций
растяжения, сжатия, сдвига. Однако при этом часть
механической энергии, расходуемой на деформацию резины, теряется
на внутри- и межмолекулярное трение в каучуке и на трение
между макромолекулами каучука и частицами
наполнителей (стр. 499 ел.). Энергия, затрачиваемая на трение,
преобразуется в тепло. Потери энергии на внутреннее трение
называют гистерезисными потерями (явление механического
гистерезиса).
В тонкостенных резиновых изделиях это тепло быстро
излучается в окружающую атмосферу. В многослойных толстостенных
резиновых изделиях под действием многократных деформаций
выделяется значительное количество тепла, вызывающее
нарастание температуры, что обусловлено плохой теплопроводностью
резины. Например, движущаяся шина всегда, даже зимой,
нагрета до 50—100 °С и выше.
а
* Модуль упругости Е— ——отношение напряжения а в материале к его
относительному удлинению 3 (обычно измеряется при комнатной температуре).
477
Чем выше эластичность резины, тем меньше внутреннее
теплообразование в резине при многократных деформациях и,
следовательно, выше долговечность резиновых изделий.
Эластические свойства резины сочетаются с другими важными
техническими свойствами—высокой прочностью при растяжении
и раздире (разрыв при растяжении надрезанного материала),
высоким сопротивлением истиранию, газо- и
водонепроницаемостью, химической стойкостью, ценными электрическими
свойствами, малой плотностью. Особенно следует отметить высокую
износостойкость резин, подвергающихся внешнему трению. Такие
резины применяются для изготовления бегового слоя протектора
шины, резиновой подошвы или каблука, для обкладки приводного
ремня и транспортерной ленты. По износостойкости резина
значительно превосходит металлы, кожу, древесину и многие другие
материалы.
Все резины должны обладать также стойкостью к старению,
т. е. в течение длительного времени не разрушаться под действием
кислорода п озона воздуха, света и тепла.
Как упоминалось, в состав резины, наряду с главным
компонентом—каучуком, входят и другие вещества, называемые
ингредиентами. Смесь сырого (невулканизованного) каучука с
ингредиентами называется резиновой смесью- Она должна быть
однородной и пластичной, что необходимо в процессах ее дальнейшей
обработки для получения заготовок (полуфабрикатов) резиновых
изделий или деталей.
Важнейшим завершающим процессом резинового
производства является вулканизация. В процессе вулканизации под
действием нагрева, химических добавок (например, серы и др.) или
радиационного излучения сырая резиновая смесь преобразуется
в резину или вулканизат. В процессе вулканизации происходит
сшивание макромолекул каучука с образованием
пространственной структуры, отличающей резину от сырого каучука, что
коренным образом изменяет свойства материала (стр. 519).
Вулканизующим агентом, образующим межмолекулярные
мостики, чаще всего служит сера, являющаяся ингредиентом
резиновой смеси. Мостичные связи могут быть образованы и другими
вулканизующими веществами, возможна также непосредственная
сшивка макромолекул вследствие возникновения между ними
связей С—С и др.
Сырой каучук без добавок и последующей обработки, особенно
без вулканизации, как правило, почти не имеет самостоятельного
применения. Однако именно масштабы потребления каучука, как
главного компонента резины, являются мерилом развития
резиновой промышленности. В 1964 г. в капиталистических странах
потребление каучука превысило 4,5 млн. га. В экономически
развитых странах примерно 50—60% каучука расходуется на
производство шин.
478
2. СЫРЬЕ ДЛЯ РЕЗИНОВОЙ ПРОМЫШЛЕННОСТИ
Основной компонент любой резиновой смеси—каучук
относится к высокомолекулярным соединениям. Иногда каучуки
называют эластомерами, т. е. полимерами с высокой эластичностью.
Гибкие длинные цепи макромолекул каучука состоят из
десятков тысяч и более атомов. В ненапряженном состоянии
макромолекулы каучука находятся в свернутом состоянии, а при
растяжении—в значительной мере распрямляются, при снятии
растягивающей нагрузки—вновь самопроизвольно свертываются. Этим
и объясняется способность каучуков и резин к большим обратимым
деформациям.
Различают натуральный каучук, добываемый из
каучуконосных растени_й, и синтетические каучуки, получаемые химической
переработкой определенных низкомолекулярных веществ,
преимущественно углеводородов.
Натуральный каучук представляет собой углеводородный
полимер—полиизопрен
сн3
—сн,—-:=ск—сн2—
Синтетические каучуки могут иметь такой же состав и
строение, как натуральный каучук, или значительно отличаться от
него. Возможности синтеза разнообразных по составу и свойствам
каучуков практически безграничны.
В зависимости от химического состава каучуки можно
разделить на органические, состоящие в основном из углеводородов
и их производных, элементорганические, в которых главная
молекулярная цепь состоит из неорганических элементов—атомов
кремния, олова, кислорода и др., и лишь боковые цепи содержат
углеводородные группы, и неорганические, т. е. не содержащие
атомов органических веществ. Подавляющее число товарных
каучуков—натуральный и большинство синтетических—относятся
к группе органических каучуков.
Каучуки, состоящие из звеньев одного мономера, являются
гомополимерными каучуками. Если для синтеза каучуков
используют два или более исходных мономера, то каучуки являются
сополимерами.
Каучуки поступают на заводы резиновой промышленности
в сухом виде—безводные твердые или жидкие каучуки, либо в
виде водных эмульсий—латексов, обычно содержащих около 30%
сухого каучукового вещества. К жидким каучукам относятся
олигомеры (стр. 376), имеющие тот же химический состав, что и
твердые каучуки, но более низкую степень полимеризации.
Олигомеры и другие низкомолекулярные каучуковые полимеры
занимают весьма незначительную долю в потреблении каучуков. Наи-
479
оольшам доли іфигкаА-—'""' I^^pyj,^... ^_, ..„;.; „UJ ^„„..^ .„. .
латексам.
В зависимости от назначения каучуки обычно разделяются
на две группы: каучуки общего назначения, пригодные для
производства массовых резиновых изделий—шин, ремней,
транспортерных лент, обуви и др.; и каучуки специального назначения,
используемые для производства изделий с особыми свойствами,
например, высоко химически стойких, теплостойких,
морозостойких, нефте- и маслостойкпх изделий и т. д.
Довольно обширная группа каучуков может использоваться
в изделиях как общего, так и специального назначений.
Латексы как товарный продукт образуют особую группу
каучуков и применяются в различных производствах (стр. 495).
Основными техническими характеристиками любых видов
натурального и синтетических каучуков являются их
эксплуатационные свойства, проявляющиеся в работающем резиновом изделии,
и технологические свойства, проявляющиеся в процессе
изготовления резиновых изделий.
К эксплуатационным свойствам каучуков относятся
механические свойства—предел прочности при растяжении,
сопротивление раздиру, износостойкость, комплекс эластических
характеристик (относительные и остаточные удлинения, упругий отскок,
напряжение при заданном удлинении и др.), а также физические
и химические свойства—тепло- и морозостойкость (способность
выдерживать кратковременное воздействие высоких и низких
температур при обратимых потерях механических свойств),
стойкость к тепловому старению (способность выдерживать
длительное воздействие тепла при минимальных необратимых потерях
механических свойств), свето- и озоностойкость, масло- и бензо-
стойкость, стойкость к действию химически активных веществ,
газонепроницаемость, электрические свойства и др. Важным
показателем эксплуатационных свойств каучука считается также
его плотность (чем она ниже, тем лучше каучук).
Технологическими свойствами сырых каучуков являются их
пластичность, способность к пластикации, к смешению с
ингредиентами, к дальнейшей переработке, усадка, клейкость,
способность вулканизоваться, стойкость к преждевременной
вулканизации и др.
До 30-х годов текущего столетия вся мировая резиновая промышленность
потребляла только натуральный каучук, добываемый в тропических странах
из каучуконосов.
В 1932 г. в Со'ветском Союзе были введены в эксплуатацию первые в мире
крупные заводы синтетического каучука общего назначения. Это стало
началом эры крупнопромышленного производства синтетического каучука. Наша
страна явилась не только родиной синтетического каучука, но и первых в мире
мощных резиновых заводов, на которых переработка этого каучука была
освоена в массовых промышленных масштабах.
480
Германия начала создавать собственную крупную промышленность
синтетического каучука на 5 лет позднее, а США—на 10 лет позднее, чем СССР; с
опозданием более чем на четверть века вступили на этот путь Англия, Франция,
Италия и некоторые другие страны.
Синтез метилкаучука (диметилбутадиенового каучука), начатый в
Германии в 1915 г., не имел крупнопромышлеиного значения и к концу первой
мировой войны был прекращен как нерентабельный. Синтезы тиокола,,
предпринятые в 1929 г., и хлоропренового каучука в 1931 г. в США также
осуществлялись в очень малых масштабах; эти каучуки предназначались только
для производства резиновых изделий специального назначения.
В настоящее время во всех промышленно развитых странах имеются
или строятся заводы синтетического каучука. В ближайшие годы
возрастающая потребность мировой резиновой промышленности в каучуке будет
удовлетворяться в основном путем расширения производства синтетических кау-
чуков.
НАТУРАЛЬНЫЙ КАУЧУК
Натуральный каучук (НК) получают из млечного сока
(латекса) каучуконосных растений. Основным мировым
промышленным каучуконосом практически является дерево гевея,
выращиваемое на плантациях в тропических странах. Попытки создания
в умеренных широтах каучуконосных культур
крупнопромышленного значения (каучуконосные растения кок-сагыз, гвайюла и др.)
не увенчались успехом, поскольку эти культуры оказались
недостаточно экономичными.
В настоящее время основное количество натурального
каучука добывается в Малайзии и Индонезии, меньшие количества НК
добываются в других странах юго-восточной Азии, а также в
Африке.
Обычный натуральный каучук неоднороден по скорости
вулканизации и механическим свойствам. В ограниченных
количествах начат выпуск технически классифицированного, более
стандартного НК-
Техника добычи натурального каучука заключается в
подсочке коры деревьев гевеи. С подсоченного участка коры латекс по
каплям стекает в подвешенную чашку. Сборщики латекса
периодически сливают латекс из чаш в ведра и относят их на завод.
Здесь латекс перемешивают и коагулируют, добавляя в него
разбавленную муравьиную или уксусную кислоту. Полученный коа-
гулюм отделяют от жидкой фазы, промывают, сушат, коптят или
отбеливают.
К высшим сортам натурального каучука относятся марки
смокед шитс («копченые листы») и риббед смокед шитс
(«рифленые копченые листы»). Некопченый коагулюм высокой степени
очистки, выпускаемый под названием светлый креп, представляет
собой высококачественный натуральный каучук. Менее чистые,
второсортные марки НК называются темными крепами или
бланкед-крепами.
Кроме того, выпускают специальные марки хорошо
обрабатывающегося натурального каучука (СП-раббер). Их получают при
31—805 481.
добавлении В ООЫЧНЫИ „idicK^ псишишііі nu.;u ,vv.« — ,
подвулканизованного латекса.
Несмотря на огромные успехи в области синтеза каучука и
непрерывное увеличение объема производства синтетических кау-
чуков, а также расширение их ассортимента, натуральный
каучук частично сохраняет значение как один из наиболее
высококачественных каучуков общего назначения, но доля его в
мировом потреблении каучуков резко снижается.
СИНТЕТИЧЕСКИЕ КАУЧУКИ
Процесс получения синтетического каучука (СК) на
химических заводах обычно состоит из двух стадий: синтеза мономеров
и их полимеризации.
Синтез мономеров, применяемых в производстве каучуков,
принципиально не отличается от синтеза мономеров,
используемых для получения ряда других полимерных материалов, и
описан в главе IV данной книги (стр. 124 и ел.).
Напомним, что диеновые мономеры для синтеза каучука
(бутадиен и изопрен) получаются преимущественно методом
дегидрирования парафиновых и олефиновых углеводородов (стр. 141).
Эти мономеры должны отличаться высокой степенью чистоты, что
особенно важно для процессов стереоспецифической
полимеризации (стр. 419 и ел.).
Важнейшими мономерами в современном производстве
синтетических каучуков являются бутадиен (дивинил) и изопрен.
Из бутадиена вырабатывают бутадиеновые каучуки разных типов
и сополимеры: бутадиен-стирольные, бутадиен-метилстирольные,
бутадиен-нитр ильные, бутадиен-метилвинилпиридиновые, бута-
диен-карбоксилатные каучуки и др. Из изопрена—изопреновые
каучуки и бутилкаучуки—сополимеры изобутилена с изопреном.
Для синтеза каучуков широко применяются и другие
мономеры: стирол, метилстирол, хлоропрен, изобутилен, нитрил
акриловой кислоты и др.
Вторая стадия процесса получения синтетических каучуков—
полимеризация осуществляется различными методами,
описанными в главе VI (стр. 384 ел.).
В настоящее время пока еще подавляющее большинство
синтетических каучуков и латексов—бутадиен-стирольные, бутадиен-
метилстирольные, хлоропреновые, бутадиен-нитрильные
изготовляют эмульсионным методом (стр. 421). Преимущества
эмульсионной полимеризации по сравнению со старым методом блочной
полимеризации (стр. 407) заключаются в том, что эмульсионная
полимеризация технически проще и безопаснее, ее применение
позволяет легче регулировать отвод выделяющегося тепла, вести
процесс непрерывно, синтезировать самые разнообразные
сополимеры, причем более высокого молекулярного веса и более одно-
482
родные по свойствам. Огромным достоинством процесса
эмульсионной полимеризации является также легкость получения
каучука в виде латексов, что очень важно для процессов пропитки
корда, тканей, бумаги и других материалов, для производства
губчатых и тонкостенных изделий (например, сосок или перчаток
методом макания) и др.
Наибольшее распространение приобрели процессы
эмульсионной полимеризации при сравнительно низких температурах,
например при 5 °С (вместо 50 °С), что позволяет значительно
улучшить качество получаемых каучуков. Промышленное применение
низкотемпературной эмульсионной полимеризации стало
возможным благодаря разработке специальных
окислительно-восстановительных систем инициирования этого процесса.
Другим усовершенствованием производства каучуков методом
эмульсионной полимеризации является введение в каучук
некоторых ингредиентов резиновых смесей (мягчителей, саж ^или
других наполнителей) на стадии изготовления латекса и
получение на заводах СК'так называемых наполненных каучуков
(стр. 488).
В последние годы в СССР и других странах интенсивно
ведутся работы в области промышленного синтеза высококачественных
стереорегулярных полимеров (стр. 486).
Стереорегулярные каучуки, главным образом полибутадиен
и полиизопрен, обладают высокой эластичностью и другими
ценными свойствами. Во многих отношениях они равноценны
натуральному каучуку и даже превосходят его и имеют значительные
преимущества перед другими синтетическими каучуками общего
назначения. Для получения стереорегулярных каучуков
используется метод полимеризации в растворах в присутствии стерео-
специфических катализаторов—лития и его органических
производных (например, бутиллития), комплексных катализаторов
(алкилалюминия в сочетании с галогенидами титана, кобальта,
ванадия) и др.
Рост производства синтетических каучуков в СССР ив ряде
других экономически развитых стран направлен на расширение
производства стереорегулярных каучуков при сокращении доли
производства бутадиен-стирольных каучуков.
Бутадиеновые каучуки. Все бутадиеновые каучуки являются
полимерами бутадиена и имеют общую формулу:
(—СНа—СН=СН—СН2—)„
12 3 4
Отдельные^типы бутадиеновых каучуков различаются
пространственным расположением атомов и звеньев в
макромолекулах полимера.
31* ' 483
Первый в мире бутадиеновый каучуяг был получен- русским ученым'
С. В. Лебедевым и продемонстрирован в качестве опытного образца в 1909 г.
на заседании Русского физико-химического общества в Петербурге. В 1928 г.
лебедевский бутадиеновый каучук оказался йобедителем международного
конкурса, объявленного Высшим советом народного хозяйства (ВСНХ) СССР
на лучший промышленный способ синтеза каучука. На конкурс были
представлены первые два килограмма полибутадиена, а уже в 1932 г. советские
заводы синтетического каучука выпускали значительные количества
бутадиенового каучука марки СКВ (первые две буквы обозначают «синтетический
каучук», буква Б—название опытного завода «Литер Б»т где был разработан
этот каучук).
СКС
СХИ
-75
100"
1
1
12S-\
СКД
100-
ни
СКС
СКИ
СКВ
Рис. 138. Предел прочности при растяжении и
износостойкость резин на основе различных каучуков
(показатели свойств НК приняты за 100%):
СКВ—натрийбутадиеновый каучук; СКД—стереорегулярный цае-
бутадиеноаый каучук; CKG—бутадиен-стнрольный каучук;
СИ—стереорегулярный ця^-изопреновый каучук;
НК—натуральный каучук.
Этот каучук получается полимеризацией бутадиена в присутствии натрия
(катализатора) и поэтому называется также натрийбутадиеновым. Каучук
СКВ был первым в мире синтетическим каучуком общего назначения,
вырабатывавшимся в крупном промышленном масштабе. Организация
производства синтетического каучука значительно уменьшила зависимость отечественной
промышленности от импорта натурального каучука. На базе производства
СКВ в Советском Союзе была создана мощная промышленность синтетического
каучука и заложены основы ее дальнейшего развития. В 50-х годах СКВ
начинают вытеснять другие типы синтетических каучуков, превосходящих его
по техническим качествам, особенно по износостойкости и прочности при
растяжении (рис. 138). Роль СКБ как каучука общего назначения в сырьевом
балансе отечественной резиновой промышленности непрерывно снижается, а
объем его производства характеризуется относительно небольшими
масштабами.
484
Новый бутадиеновый каучук СКД (синтетический каучук
дивиниловый) одинаков по химическому составу с каучуком
СКВ. Различие между ними заключается в большей регулярности
строения каучука СК.Д. Звенья его молекулярной цепи
расположены в строго линейном порядке:
СН2—СН=СН— СН,—СН2—СН=СН—СН3
1 2 3 4 1 2 3 4
Такое расположение звеньев полимерной цепи называют
соединением в положение 1—4.
150-L
ски
сне
СКВ
ска
нк
100
ЯТІ
СКВ
1
I
1 1«H
J I
Y25* |
СКС;
СКИ
СКД
нк
Рис. 139. Эластические свойства и внутреннее трение резин
на основе различных каучуков (показатели свойств НК
приняты за 100%, обозначения те же, что на рис. 138).
Большинство звеньев в макромолекулах каучука СКВ
присоединено в положение 1—2 (первый углеродный атом следующего
звена соединен не с четвертым, а со вторым углеродным атомом
предыдущего звена).
12 12
¦ ¦ -СНз—СН—СН3—СН
сн2
3. 4
СН
II
С Н3
3, 4
Наличие в макромолекулах СКВ большого количества боковых
винильных групи (большая разветвленность полимера) заметно
ухудшает эластические свойства этого каучука, повышая его
внутреннее трение (рис. 139) и теплообразование.
Строение СКД отличается также тем, что звенья его
макромолекул имеют преимущественно цме-конфигурацию, т. е. первый
485
и четвертый атомы углерода в каждом овспс
сторону плоскости основной цепи полимера:
н
\2
н
з/
с=с
—сна
1
1
сн
4
jue-1,4
н н
\2 3/
с=с
2—СН2 СН;
1 4
-полибутадиен
Синтезированы также полибутадиены, большинство звеньев
которых имеет /л/7<2«с-конфигурацию:
Н СН2-СН2 Н
\2 з/ \2 г/
С=С С=С
—СН2 Н Н СН,—
1 4
т ране-1,4-пол ибутадиен
Чем больше звеньев в макромолекуле полимера имеет транс-
конфигурацию, тем он жестче и тем легче кристаллизуется.
Стереорегулярный цш>1,4-полибутадиен—каучук СКД
существенно превосходит каучук СКБ по прочности, в два раза—па
величине упругого отскока и намного лучше СКБ по величине
гистерезисных потерь. По эластическим свойствам СКД
соответствует натуральному каучуку, а по износостойкости значительно
превосходит все массовые синтетические каучуки и натуральный
каучук. Эти достоинства каучуковтипаСКД позволяют отнести его
к наиболее перспективным каучукам общего назначения.
Изопреновые каучуки. Химическими способами долго не
удавалось воспроизвести натуральный цис-1,4-полиизопрен,
создаваемый природой в каучуконосном растении. Синтетические поли-
изопрены имели такой же химический состав, как натуральный
каучук, но другое строение макромолекул и иные свойства.
Полные синтетические аналоги натурального каучука,
соответствующие ему по структуре и свойствам, удалось получить
сравнительно недавно советскому ученому А. А. Короткову с сотр.
Лишь спустя несколько лет после их открытия появились
публикации о синтезе подобных полиизопренов за рубежом. В
настоящее время осуществлено промышленное получение этого каучука
(рис. 140).
Синтетические цис- 1,4-полиизопрены—стереорегулярные
изопреновые каучуки, названные в СССР каучуками СКИ (СКИ-3),
могут полностью заменить натуральный каучук в изделиях,
которые должны сочетать высокую эластичность с хорошими
показателями других свойств.
486
Рис. 140. Завод синтетического изопренового каучука (общий вид
технологических установок).
Каучуки СКИ уступают каучукам СКД по износостойкости,
но значительно превосходят последние по технологическим
свойствам, прочности при растяжении и сопротивлению раздиру, а по
комплексу свойств так же перспективны, как каучуки СКД.
Бутадиен -стирольные и бутадиен-метилстирольные каучуки.
Как следует из названия этих каучуков, они являются
сополимерами бутадиена и стирола (или метилстирола) следующего
строения:
"—СН2—СН=СН—СН2 CHj—CH—"
бутадиен-стирольный каучук
—СН,—СН=СН—СН„ СН.
сн3-
,—с—
бутадиеи-метилстирольный каучук
487
В настоящее время бутадиен-стирольные сополимеры по
объему производства занимают главное место в мире среди
синтетических каучуков общего назначения. На их основе получаются
резиновые изделия хорошего качества. Однако для производства
некоторых наиболее ответственных изделий, например
автомобильных или авиационных шин, работающих с большой
нагрузкой или при высоких скоростях, предпочтительнее использовать
более высокоэластичные каучуки, например стереорегулярные
СКД, СКИ или натуральный каучук.
В Советском Союзе выпускаются различные марки бутадиен-
стирольных каучуков, например СКС-ЗО АРКМ* и др.
Бутадиен-стирольные каучуки получают преимущественно
методом низкотемпературной эмульсионной полимеризации при
5 °С. Получаемый латекс коагулируют и коагулюм выпускают в
в виде сухого каучука; часть продукции выпускается в виде
латекса как конечного товарного продукта.
В обычных сополимерах, получаемых эмульсионной
полимеризацией, бутадиеновые и стирольные или метилстирольные звенья
чередуются нерегулярно. В настоящее время исследуется
возможность получения стереорегулярных бутадиен-стирольных
каучуков.
Значительную часть каучуков СКС или СКМС выпускают в
виде масляных каучуков, получаемых при добавлении в латекс
дешевых мягчителей (нефтяных масел). Заправленный мягчите-
лями латекс подвергают затем коагуляции. Масляные каучуки
дешевле ненаполненных обычных. Причем введение в их состав
масла-разбавителя не ухудшает эксплуатационные свойства
полимера, а некоторые из них даже улучшает, например повышает
сопротивление разрастанию трещин при многократных
деформациях. Сохранение высоких технических свойств каучуков при
наполнении их маслами в больших дозировках достигается тем,
что для производства масляных каучуков используются более
высокомолекулярные, очень жесткие исходные полимеры. Такие
каучуки обладают повышенной прочностью ц эластичностью, но
недоступны для переработки без добавления масел.
Большие перспективы имеет применение так называемых саже-
масляных бутадиен-стирольных каучуков. Их получают при
введении в латекс масел и углеродных саж, что приводит к
повышению износостойкости резин на основе таких каучуков.
Хлоропреновые каучуки. В отличие от чисто углеводородных
полимеров—натурального каучука, синтетических полибутадие-
* СКС—синтетический каучук стпрольный; цифра 30 указывает
содержание стиролышх звеньев в сополимере, буквы АРКМ обозначают:
А—низкотемпературный, Р—регулированный (по молекулярному весу), не требующий
термопластикации, К—канифолевый (полученный с эмульгатором—канифолью)v
М—маслонаполненный или масляный каучук.
СКМС—синтетический каучук метилстирольный.
488
нов, полиизопренов и бутадиен-стирольного каучука, хлоропре-
новые каучуки являются полимерами хлорпроизводных
углеводородов и содержат в макромолекулах атомы хлора:
(—СН2—СС1=СН—СН2—)„
Это сильно влияет на свойства полимера. В макромолекуле
полихлоропрена атом хлора экранирует двойную связь и
повышает полярность полимера, поэтому хлоропреновые каучуки
обладают высокой масло-, бензо- и озоностойкостью (см.
рис. 141).
Хлоропреновые каучуки в настоящее время производятся
методом эмульсионной полимеризации.
В СССР эти каучуки получают по оригинальной технологии,
разработанной проф. А. Л. Клебанским с сотр. и выпускаются под
названием наирит (происходит от древнего названия Армении,
где впервые начали выпускать этот каучук).
Наирит обладает великолепными эксплуатационными
свойствами и вместе с тем очень дешев. Однако он имеет более высокую
плотность, чем углеводородные каучуки A,25 и 0,91—0,93 гісм9
соответственно), меньшую морозостойкость и несколько более
труден в обработке. Хлоропреновые каучуки применяются
преимущественно в производстве резиновых технических изделий и в
кабельной промышленности. Кроме того, хлоропреновые каучуки
добавляют в некоторые теплостойкие смеси, используемые,
например, для изготовления варочных камер и диафрагм,
применяемых в процессе вулканизации шин.
Наряду сполихлоропреном выпускаются также некоторые
сополимеры хлоропрена (в частности, хлоропрен-стирольные каучуки
и др.).
Особый интерес представляет наирит марки НТ—заменитель
природной гуттаперчи, но имеющий лучшие свойства и более
дешевый.
Гуттаперча—изомер каучука добывается из сока некоторых
растений; представляет собой жесткий, твердый при комнатной
температуре полимер, размягчающийся при нагревании;
применяется для изоляции подводных кабелей, производства клеев
и др.
Как и другие эмульсионные каучуки, полихлоропрен и
сополимеры хлоропрена выпускаются в виде сухих каучуков (твердых
и жидких) и в виде латексов.
Бутадиен-нитрильиые каучуки представляют собой
сополимеры бутадиена и нитрила акриловой кислоты
СН2—СН=СН—СН2—СН2—СН
C=N
489
нк
в обычных сополимерах бутадиена со стиролом или метил
стиролом.
В Советском Союзе выпускаются бутадиен-нитрильные
каучуки марок СК.Н-18, СКН-26 и СКН-40 (цифры обозначают
количество нитрила акриловой кислоты в кг на 100 кг бутадиена в
исходной смеси полимеризуемых мономеров).
Нитрильная группа —C=N обладает еще большим
экранирующим действием, чем атом хлора. Поэтому бутадиен-нитрильные-
каучуки отличаются более высокой масло- и бензостойкостью,
чем хлоропреновые (рис. 141). Кроме того, бутадиен-нитрильные
каучуки имеют повышенную теплостойкость. Чем выше
содержание звеньев акрилонитрила в сополимере, тем больше его масло-
и бензостойкость, но ни-
-150 же морозостойкость.
Практически для изготовления
резиновых изделий,
которые должны сочетать мас-
лостойкость и
морозостойкость, чаще всего
применяются каучуки типа
СКН-18.
Нитрильные каучуки
используются в
производстве бензо- и масло-
стойких рукавов,
уплотнителей, прокладок и
других резиновых изделий
для нефтяной
промышленности, авиации и т. д.
Бутилкаучуки получают совместной полимеризацией двух
мономеров, из которых основным является изобутилен, имеющий
одну двойную связь:
сн3
DKH-4O
ЩО КОЛ,
5
!
скс
-100 «Г
Л
Л
Л
У50Л
Рис. 141. Набухание
различных каучуков
24 ч, при
резин на
в бензине
20 °С).
основе
(после
СН,
В качестве второго мономера обычно применяется изопрен,
вводимый в количестве 1,5—3%. Исходную смесь мономеров поли-
меризуют в среде растворителя при глубоком охлаждении.
Образующийся сополимер—бутилкаучук имеет следующее строение:
сн3 сн3 сн3
І І І
С—СНа—С—СН2 СНа—С=СН—СН3
СН,
СН,
4 90
Особенностью химического строения макромолекул бутилкау-
чука является во много раз меньшее количество двойных связей
а них, чем в каучуках, полученных полностью или
преимущественно на основе диолефинов. Двойная связь, особенно в главной
молекулярной цепи,—наиболее уязвима и больше всего
подвержена разрушающему воздействию кислорода и озона воздуха.
Очень малое количество двойных связей в молекулах бутил-
каучука обусловливает его выдающуюся озоностойкость, а также
теплостойкость. Кроме того, бутилкаучук отличается
исключительно высокой газонепроницаемостью, превосходя в 10 раз в
этом отношении натуральный, бутадиеновые и бутадиен-стироль-
ные каучуки. В связи с этим бутилкаучук является наилучшим
материалом для изготовления таких газо- или
воздухонепроницаемых изделий, как камера автомобильной шины, герметизирующий
слой бескамерной шины, детали противогазов, варочные камеры
и диафрагмы, применяемые при вулканизации шин и т. д.
Бутилкаучук медленно вулканизуется и плохо совмещается
с каучуками, макромолекулы которых содержат большое
количество двойных связей. Галоидпроизводные бутилкаучука,
например бромбутилкаучук и хлорбутилкаучук, лишены этих
недостатков.
Полисульфидные каучуки, известные также под названием
тиоколы, являются продуктами поликонденсации дигалоидпроиз-
водных углеводородов, например дихлоргидрина глицерина, и
полисульфидов щелочных металлов (например, Na.2S4). В процессе
образования полимера побочно образуется хлористый натрий.
Строение полисульфидного каучука примерно следующее:
_СН,-СН2—S—S—
II II
S S
Полисульфидные каучуки выпускаются в сухом и жидком
виде, причем жидкие тиоколы используются для изготовления
герметизирующих паст. Полисульфидные каучуки отличаются
очень высокой масло- и бензостойкостью, стойкостью к другим
органическим растворителям (см. рис. 141), и заметно
превосходят в этом отношении хлоропреновые и даже нитрильные каучуки.
Однако их механические свойства (предел прочности при
растяжении, износостойкость) и, главное, теплостойкость намного ниже,
чем других каучуков.
Полисульфидные каучуки применяются для изготовления
резиновых изделий, обладающих наибольшей стойкостью к
действию нефтяных топлив, разрушающих резины на основе
каучуков общего назначения.
Силоксаиовые (кремнийорганические) каучуки отличаются от
описанных выше тем, что главные цепи их макромолекул не со-
491
держат атомов углерода, а состоят из чередующихся атимив
кремния и кислорода:
R R
—Si—О—Si —О—
Боковые углеводородные радикалы R, соединенные с
атомами кремния, представляют собой метальные, этильные или
другие группы.
Отечественная марка силоксанового каучука—СКТ
(синтетический каучук теплостойкий). Обычные натуральный и
органические синтетические каучуки в значительной степени теряют свои
эксплуатационные свойства при 140—160 °С и разлагаются при
температурах около 200 °С, тогда как силоксановые каучуки
великолепно выдерживают длительное действие температур
порядка 250 °С, кратковременное действие температур до 350 °С и
одновременно являются морозостойкими.
Каучук СКТ применяется для изготовления различных тепло-
и морозостойких резиновых уплотнителей и прокладок, ряда
электроизоляционных деталей, а также некоторых медицинских
изделий, как физиологически безвредный.
Силоксановые каучуки, несмотря на дороговизну, незаменимы
для специальных целей.
Этилен-пропиленовые каучуки представляют собой
сополимеры этилена с пропиленом, получаемые примерно по следующей
схеме:
Н2С=СНа + СН2=СН > Г—СН2—СН2 СН2—СН— 1
сн3 L сн3 J„
этилен пропилен полимер
Хотя синтез этих каучуков проводится с применением стерео-
специфических катализаторов (стр. 483), они не имеют
регулярного строения, но отличаются очень высокой эластичностью в
сочетании с хорошими механическими свойствами.
Этилен-пропиленовые сополимеры (советская марка СКЭП)—
самые легкие из всех известных каучуков, их плотность—0,86 г/см*
(для обычных каучуков общего назначения плотность 0,90—
0,93 г/см*), что является большим достоинством этих каучуков,
так как позволяет уменьшить вес резиновых изделий на их основе
и, следовательно, машин, в которых применяются такие изделия.
В этилен-пропиленовых сополимерах отсутствуют двойные
связи, поэтому каучуки СКЭП обладают высокой озоностойкостью
и химической стойкостью, великолепно сопротивляются старению
и морозостойки. Кроме того, их получают из весьма дешевых
и доступных мономеров. Однако из-за отсутствия двойных связей
492
сополимеры этилена с пропиленом не вулканизуются серой, для
их вулканизации приходится применять перекиси, что
технологически менее удобно. При введении третьего
мономера—диенового углеводорода—в исходную смесь этилена с пропиленом с
получением тройного сополимера этот недостаток устраняется.
Хорошие эксплуатационные свойства и относительная
дешевизна этилен-пропиленовых сополимеров (особенно тройных)
позволяют отнести их к весьма перспективным видам каучуков.
В 1963 г. начат промышленный выпуск этилен-пропиленовых
сополимеров. Работы по изучению и освоению этих каучуков
продолжаются.
Уретановые каучуки получают путем ступенчатой
полимеризации. Исходными мономерами служат двухосновные карбоно-
вые кислоты и эфиры гликолей; последующее структурирование
(сшивка) образующегося полимера производится изоцианатами
(стр. 402).
Уретановый каучук можно получать из сложных и простых
эфиров.
Уретановые каучуки (советская марка СКУ) отличаются от
других полимеров исключительно высокой износостойкостью и
обладают также маслостойкостью и газонепроницаемостью.
Однако они недостаточно температуростойки и довольно дороги.
Полиуретаны нашли применение в производстве мелкопористых
губчатых пенорезин, а также некоторых масло- и бензостойких
резиновых изделий.
Фторкаучуки. Для современного самолетостроения,
ракетостроения, атомной энергетики и других специальных областей
техники требуются полимеры, обладающие особенно высокой
теплостойкостью и химической стойкостью. В этом отношении
несомненный интерес представляют фтор содержащие полимеры, в
том числе каучуки.
Для* синтеза таких каучуков могут применяться различные
фторпроизводные этилена: четырехфтористый этилен F2C=CF2
(стр. 194), образующий при полимеризации политетрафторэтилен
или трифторхлорэтилен F2C=CFC1 и винилиденфторид F2C=CH2,
совместная полимеризация которых дает один из фторкаучуков
следующего строения:
[—CF2—CFCI-CFu—СН2—]я
Отечественная промышленность выпускает фторкаучуки
марки СКФ (синтетический каучук фтор содержащий).
Фторкаучуки стойки к действиям озона, концентрированной
азотной кислоты и других агрессивных сред. Они обладают
высокой теплостойкостью, а некоторые из них и морозостойкостью.
Как и силоксановые каучуки, фторкаучуки пока дороги, но
незаменимы для производства ряда изделий специального
назначения.
493
Важнейшие технические свойства
Каучуки
Бутадиеновые
СКД
СКБ
Изопреновые
СКИ-3
натуральный . . .
Бутадиен-стирольные
СКС-30
СКС-30-АРКМ . .
Бутадиен-нитрнльные
СКН-18 .
СКН-26
СКН-40 . . .
Хлоропреновые (наирит)
Бутилкаучук
Силоксановый ....
Полисульфидный . . .
Этилен-пропиленовый
(СКЭП)
Плотность
сырого
каучука
г/смЗ
0,90—0,92
0,90—0,92
0,91—0,93
0,91
0,94
0,92-0,95
0,943
0,962
0,986
1,2-1,25
0,92
1,7-2,0
1,3—1,4
0,86
предел прочности при
растяжении, кгс/см2
прн 20 °С
175
130—160
315
325—340
240—280
250—275
250—270
280—310
300—330
200—265
160—240
35—80
38—42
230—250
при 100 °С
97
85—105
200
200
105—115
80 .
80—95
90-100
100—130
70—100
45—110
30—80
—
160
Наполненные f
нстнранне
емЗ/квт-ч
прн 20 °С
120—150
400—500
280—300
240—300
220—280
240—300
220—250
200—230
190—200
290—350
200—250
—
1000—1800
220
іулканизатьі
сопротивление
разднру
кгс/смг
50—60
50—60
100-115
100
85—95
66—70
55—65
60—70
75—85
60—90
65—95
15—20
4—5
55
Наряду с описанными выше каучуками в промышленных или
опытных масштабах изготовляются многие другие виды каучуков—
акриловые, хлорсульфоэтиленовые, бутадиен-метилвинилпириди-
новые, карбоксилатные и др.
Данные о^свойствах большинства рассмотренных каучуков и
их стандартных вулканизатов, содержащих углеродную сажу,
приведены в табл. 22. Поскольку многие резиновые изделия,
например шины, приводные ремни и др., во время работы
разогреваются, характеристики свойств резин приводятся при
температурах 20 и 100 °С.'Следует учитывать, что в зависимости от
рецептуры резиновых смесей, условий их переработки в изделия и
режимов эксплуатации резин их свойства могут сильно колебаться.
Для полной технической характеристики каучуков необходимо
знать не только механические и другие эксплуатационные
свойства вулканизатов, но и способность сырых каучуков к
переработке на оборудовании резинового завода, т. е. технологические свой-
494
Таблица 22
каучуков и вулканизатов
стандартных смесей
модуль
при 300%
удлинения
кгс/см%
60—75
65
35
40
90—ПО
92
90—110
100—120
110-120
150
30—60
30—40
—
90—120
относительное удлинение
при разрыве, %
при 20 °С
500—700
500—600
700—880
600—700
550—650
500
450—550
550—650
600—700
600—750
650—800
360
250—430
550—650
при 100 °С
400—600
330—430
750—850
750—950
380—450
380
300-350
300—400
300—400
300
450—550
200—300
—
350—500
остаточное
удлинение
при разрыве, %
при 20 °С
10—15
35—50
28
25—40
15-25
28
15—20
20—30
20—30
12
30—45
4
40—80
6—8
эластичность г
при 20 °С
52
26—28
45
55
40—42
26—29
37-40
28—30
15—20
30—40
8—12
45-50
20
53
ю огскоку, %
при 100 "С
55
41
55
65
47
42
—
68
35-50
54
15
56
ства каучуков и резиновых смесей, зависящие не только от качеств
каучука, но и от рецептуры резиновых смесей, технологии их
приготовления и переработки в изделия.
СИНТЕТИЧЕСКИЕ ЛАТЕКСЫ
Синтетические латексы нашли широкое применение не только
в производстве каучука, но и как конечный товарный продукт.
Смесь латекса с различными добавками используют для пропитки
корда и других технических тканей, формования изделий
маканием, получения пенорезин и в других процессах.
Для пропитки корда и других технических тканей
используются главным образом бутадиен-стирольные, а также
бутадиеновые карбоксилатные, бутадиен-метилвинилпиридиновые и
другие латексы.
495
высокую причиисіь chmjh резины і_ ікапьіи uuci_iit-
чивают карбоксилатные латексы типа СКД-1 (бутадиеновый или
дивиниловый латекс, содержащий в среднем по два элементарных
звена метакриловой кислоты на 100 бутадиеновых звеньев в
макромолекулах полимера) и дивинил-метилвинилпиридиновый
латекс марки ДМВП-10, получаемый совместной полимеризацией
бутадиена и метилвинилпиридина в соотношении 90 : 10.
Пропиточный состав на основе латекса ДМВП-10 по сравнению
¦с бутадиен-стирольным латексом повышает прочность связи
волокон корда с обкладочной резиной в полтора-два раза, особенно
если эта резина изготовлена на основе изопреновых или
бутадиеновых каучуков.
Хлоропреновые латексы применяются для изготовления
хирургических перчаток и других тонкостенных изделий, резиновых
нитей круглого сечения, резиновых клеев, в производстве
искусственной кожи и картона и для других целей.
Путем вспенивания синтетических латексов получают
губчатые изделия (пенорезины).
Латексы применяются в производстве бумаги, заменителей
кожи, уплотнительных прокладок для консервов, для изоляции
кабелей, промазки тканей на клеепромазочных машинах, в
производстве асбестотехнических изделий, водостойких
строительных красок и многих других жизненно важных промышленных
и бытовых изделий.
Возможности дальнейшего расширения ассортимента и
создания новых типов синтетических каучуков безграничны. Для зтого
используются новые виды исходного сырья, новые способы
технологии синтеза мономеров, новые комбинации их и новые методы
полимеризации с применением различных наполнителей и
модификаторов.
СИНТЕТИЧЕСКИЕ СМОЛЫ
Наряду с каучуками и латексами резиновая промышленность
потребляет некоторые жесткие полимеры, обладающие
значительно меньшей эластичностью, чем каучук, и применяемые главным
образом в производстве пластических масс. К таким полимерам
относятся полиизобутилены, бутадиен-стирольные сополимеры
с высоким содержанием стирола; феноло- и резорцино-феноло-
формальдегидные полимеры, поливинилхлорид и др. Они
применяются как добавки к каучуку в резиновых смесях и в
пропиточных составах. Например, современные пропиточные составы
для обработки технических тканей, особенно корда,
изготавливаются на основе латексов и термореактивных полимеров,
преимущественно резорцино-феноло-формальдегидных.
В последние годы в рецептуру многих резин массового
назначения стали вводить небольшие добавки полипропилена и других
полимеров, применяемых в производстве пластических масс и
496
химических волокон. В присутствии этих добавок повышается
износостойкость и прочность резиновых изделий, регулируется
эластичность или облегчается технологическая обработка
резиновых смесей. Некоторые из таких полимеров добавляют в латекс
(с последующей совместной коагуляцией и получением
«смоляного» каучука) либо вводят на стадии приготовления резиновых
смесей при работе с сухим каучуком.
Синтетические полимеры типа феноло-формальдегидных все
шире применяются для вулканизации резиновых смесей на
основе бутилкаучука и других каучуков. Некоторые виды
транспортерных лент и других резиновых изделий изготовляются с
применением поливинилхлорида и других жестких полимеров.
РЕГЕНЕРАТ
Регенерат представляет собой продукт переработки старых
резиновых изделий (изношенных шин и др.) и вулканизованных
отходов резинового производства. Его вводят в резиновые смеси
для частичной замены каучука. Чтобы получить регенерат,
старую резину измельчают, подвергают тепловой обработке в
присутствии мягчителей (пластификаторов) и других веществ.
Содержащийся в регенерате каучук химически связан с серой,
частично с сажей и другими примесями, так как при регенерации
происходит лишь частичная деструкция резины (девулканизация).
В результате регенерации старая резина превращается в мягкую
пластичную массу. Регенерат, содержащий около 40—50%
углеводорода каучука, является его ценным заменителем. В ряде
случаев введение регенерата облегчает обработку резиновых
смесей, увеличивает химическую стойкость резин, в том числе
их сопротивление старению.
В настоящее время объем производства регенерата составляет
около 10—15% от количества производимых каучуков.
Регенерат в 3—5 раз дешевле каучука, поэтому применение
регенерата позволяет значительно удешевить производство резиновых
изделий.
Наиболее перспективные разновидности регенерата
получаются методом непрерывной термомеханической обработки старой
резины и методом ее диспергирования. Регенерат, получаемый
этими двумя методами, отличается значительно более высокими
эксплуатационными свойствами, чем регенерат, получаемый
старыми способами.
ИНГРЕДИЕНТЫ РЕЗИНОВЫХ СМЕСЕЙ
Наряду с каучуком—носителем уникальных
высокоэластических свойств резины и регенератом, частично заменяющим
каучук, в резиновой смеси содержится ряд других ингредиентов.
32—805 497
лить на следующие важнейшие группы: химические ускорители
пластикации каучука, группа вулканизующих веществ,
усилители каучука, мягчители (пластификаторы), защитные вещества,
разбавители каучука (инертные наполнители).
Ускорители пластикации каучука. Некоторые каучуки
(например, натуральный и многие синтетические) обладают
недостаточной пластичностью и нуждаются в предварительном
размягчении перед их дальнейшей переработкой. Для размягчения
некоторых типов каучуков необходима их окислительная
деструкция.
Окислительную деструкцию каучука можно ускорить
добавлением некоторых органических веществ. К числу таких
ускорителей пластикации относятся, например, тионафтол, тиофенолы
и др. Товарные марки ускорителей пластикации — ренациты,
пептоны и т. д. (стр. 365, 366).
Группа вулканизующих веществ. Для образования полимеров
пространственного строения из линейных или разветвленных
макромолекул сырого каучука, т. е. для их соединения (сшивки)
между собой в поперечном направлении, используют процессы
вулканизации. Наиболее распространенным способом
вулканизации является нагревание каучука с серой, которая при
повышенных температурах образует мостичные химические связи
между его макромолекулами. В данном процессе сера является
вулканизующим веществом. Обычно ее вводят в резиновые смеси в
количестве 0,5—3,5% от веса каучука. При увеличении дозировки
серы до 45% от количества сырого каучука и нагревании
резиновой смеси в течение достаточно длительного времени получается
не мягкая эластичная резина, а жесткий роговидный эбонит,
сходный с термореактивными полимерами. Эбонит значительно
превосходит резину по прочности, электроизоляционным
свойствам, химической стойкости, но не обладает эластическими
свойствами.
Каучук можно вулканизовать также при помощи других
химических соединений или элементов, например органических
перекисей, некоторых синтетических смол, окисей металлов,
кислорода, селена и др. Путем ядерного облучения сырой
резиновой смеси можно непосредственно, без участия каких-либо
вулканизующих веществ, соединить макромолекулы каучука
поперечными связями, образующимися между углеродными или
другими атомами соседних молекулярных цепей.
Представляет интерес также комбинированная радиационно-
химическая вулканизация—с' участием ядерного облучения и
вулканизующих химических добавок.
Наряду с серой и другими вулканизующими веществами в
резиновые смеси вводят ускорители
вулканизации—преимущественно органические соединения.
49Я
Наиболее широко применяются ускорители группы тиазола
(каптакс, альтакс, сульфенамиды), тиурамы и дифенилгуанидины,
соли карбаминовой кислоты (стр. 362 и ел.). Эти добавки не
только ускоряют процесс вулканизации, но и улучшают
физико-механические свойства вулканизатов. Действие ускорителей
вулканизации активируется при введении в резиновую смесь окиси
цинка или окислов других металлов.
Некоторые ускорители вулканизации настолько эффективны,
что могут вызвать преждевременную вулканизацию (подвулка-
низацию) резиновых смесей в процессе их обработки. Подвулкани-
зованная смесь может необратимо потерять пластичность
прежде, чем из нее будет изготовлен полуфабрикат. Чтобы
предотвратить зто нежелательное явление, в ряде случаев применяют
фталевый ангидрид или другие замедлители вулканизации.
Усилители каучука. Подавляющее большинство
синтетических каучуков как в сыром, так и в вулканизованном виде,
обладает низкой прочностью при растяжении, невысоким
сопротивлением раздиру и малой износостойкостью. Введение в резиновую
смесь специальных усилителей, например сажи или минеральных
наполнителей, приводит к значительному улучшению
прочностных свойств резиновых изделий, а также облегчает обработку
резиновых смесей. Усилители добавляются практически ко всем
каучукам в количестве от 15 до 100 вес. ч. и более на 100 вес. ч.
каучука (в среднем около 30—50 вес. ч,), в зависимости от типа
каучука и назначения резиновых изделий. На каждый миллион
тонн перерабатываемого каучука расходуется около 0,5 млн. т
сажи.
Повышение износостойкости резины Гв результате введения
сажи особенно важно для изделий, подвергающихся внешнему
трению, например протекторов автомобильных или велосипедных
и других шин, подошвенных резин, обкладочной резины
приводных ремней, трущихся о шкив передачи, и т. д.
Сажа повышает также прочность связи резины с тканью или
с металлом.
Сажи маркируются в зависимости от способа их производства
и исходного сырья (нефтяные масла, углеводородные газы и др.),
например: печные сажи из жидкого сырья, печные газовые сажи,
канальные газовые сажи, форсуночная сажа и др. Значительная
часть саж выпускается в гранулированном виде. Это устраняет
пыление сажи, придает ей «текучесть», облегчает ее
транспортирование и автоматизацию процессов приготовления резиновых
смесей.
Все сажи принято делить на две главных группы—активные
и полуактивные. Активные сажи используются преимущественно
для повышения износостойкости, прочности резин при растяжении
и сопротивления их раздиру, а также для повышения прочности
связи резин с металлом.
32* 499
обладающие большим усиливающим действием. Полуактивные
сажи вводятся в резины, которые наряду с прочностью должны
иметь высокую эластичность. Широко применяются комбинации
различных саж в одной и той же резиновой смеси.
Резина, содержащая сажу, обычно имеет черный цвет. В
резиновых смесях, предназначенных для изготовления светлых и
цветных резиновых изделий (красиво окрашенных велосипедных шин,
медицинских грелок, светлых подошв и т. д.), углеродную сажу
заменяют минеральными усилителями, например коллоидной
кремнекислотой (так называемой белой сажей), каолином, окисью
цинка, активированным мелом. Применяются также усиливающие
органические добавки—полипропилен, полистирол и др.
Мягчители (пластификаторы). В любую резиновую смесь,
в том числе в смеси на основе масляного каучука, обычно вводят
некоторое количество мягчителей.
В качестве мягчителей могут служить нефтяные масла,
например, типа ПН-6, мазут, петролятум; каменноугольная и
сосновая смола, рубракс (окисленный битум), жирные кислоты, как
синтетические, так и получаемые из растительных масел и животных
жиров, кумароно-инденовые смолы и др. Мягчители облегчают
приготовление и обработку резиновых смесей, что особенно важно
для смесей, сильно наполненных сажей или другими
порошкообразными материалами. Кроме того, введение мягчителей позволяет
регулировать эксплуатационные свойства резин, например
придавать им большую мягкость. Некоторые виды мягчителей
повышают морозостойкость резин. К таким мягчителям относятся
органические соединения типа дибутилфталата и некоторые другие
сложные эфиры, применяемые также в качестве пластификаторов
в производстве пластических масс (стр. 529).
Защитные вещества. С течением времени каучуки и резины
стареют, т. е. свойства их необратимо ухудшаются, даже если
изделия или материалы не находятся в эксплуатации, а хранятся в
обычных условиях. Признаки старения нередко можно заметить
на глаз—поверхность прорезиненной ткани, шины и других
резиновых изделий покрывается трещинами, резины твердеют,
становятся ломкими и хрупкими, теряя свое ценнейшее свойство—
эластичность.
Старение резин происходит в результате деструкции или
структурирования (сшивки) молекул каучука под действием кислорода
воздуха, причем эти химические процессы ускоряются при
нагревании, облучении и других воздействиях. Чтобы предотвратить
или замедлить старение резин, т. е. сохранить их технические
свойства в течение длительного времени, в состав резиновых
смесей вводят так называемые противостарители. Обычно они
представляют собой ароматические амины или фенолы, например фе-
нил^-нафтиламин (неозон Д, стр. 365). Большинство противоста-
50,0
рителей является антиоксидантами, защищающими каучуки от
теплового и окислительного старения.
Противостарители вводят не только в резиновые смеси, но и
заправляют ими сырой каучук непосредственно на
заводах-изготовителях для защиты его от окисления и стабилизации свойств
каучука и получаемых из него вулканизатов.
Каучук в резине стареет не только под действием тепла, света,
кислорода, но и в результате многократных деформаций
растяжения, сжатия, сдвига, которым большинство резиновых изделий
длительно подвергается в процессе их эксплуатации. Для защиты
каучука от утомления в этих условиях в резиновые смеси вводят
специальные противоутомители—производные /г-фениленди-
амина и другие, являющиеся одновременно антиоксидантами.
Чтобы предотвратить образование в резине глубоких трещин,
появляющихся в ней под действием озона, наряду с химическими
противостарителями—антиозонантами, применяют
противостарители физического действия: особого рода воски, церезин,
парафин, петролятум. Сочетание химических антиозонантов с восками
позволяет создать наиболее эффективную защиту резин от
озонного старения.
Разбавители и другие ингредиенты. Для удешевления
резиновых изделий менее ответственного назначения (например,
резиновые коврики, шины для детских колясок и гужевого транспорта,
стельки и др.) дорогостоящий каучук в резиновых смесях
разбавляют мелом или другими доступными и дешевыми наполнителями.
Эти ингредиенты называют неактивными или инертными
наполнителями, потому что они, в отличие от активных усилителей, не
улучшают прочностных свойств резины.
В состав смесей, применяемых для изготовления белых и
цветных резиновых изделий (грелки, игрушки, отдельные виды и
детали шин), вводят органические красители или неорганические
пигменты. В производстве губчатых изделий из резины широко
применяются порофоры—порообразователи (газообразователи),
выделяющие газы при повышенной температуре, что приводит к
образованию пор в материале. В качестве таких порообразую-
щих веществ в резиновые смеси вводят, например, карбонат
аммония или диазоаминобензол.
АРМИРУЮЩИЕ МАТЕРИАЛЫ
Большинство резиновых изделий должны одновременно
обладать эластичностью, высокой прочностью и сохранять под
нагрузкой свои определенные габаритные размеры, т. е.
деформироваться лишь в установленных пределах. Для сочетания этих свойств
большинство резиновых технических изделий армируют, т. е.
вводят в них каркас—прочный, жесткий высокомодульный
материал—ткань или металл. Таким образом изготовляются пневма-
501
... i„_.»«._ шипы, іранспортерньїе ленты, приводные ремни, рукава
л др. Резина придает этим изделиям требуемую эластичность,
армирующий материал воспринимает основную механическую
нагрузку в процессе эксплуатации изделий, регулируя их
деформируемость. Например, эластичная резиновая шина, армированная
кордной тканью, не может растянуться при накачивании сверх
допустимых габаритов.
Текстильные волокна, металлическая проволока, применяемые
в качестве армирующих материалов, по модулю упругости
во много раз превосходят резину; удлинение обычного
текстильного корда при разрыве составляет 10—25%, удлинение
большинства резин—500% и более. Текстильные ткани и нити входят
в конструкцию многих резиновых изделий—автомобильных
авиационных, тракторных, сельскохозяйственных, мотоциклетных,
'велосипедных и других шин, конвейерных и транспортерных
лент, приводных ремней, рукавов и шлангов, резино-пневма-
тических рессор и муфт, резиновой обуви и многих других
изделий и деталей. Выпускаются также различные изделия из
прорезиненных тканей.
Наиболее распространенным текстильным материалом,
используемым в производстве шин и других резиновых технических
изделий, является корд. Кордом называются ткани из прочных
крученых нитей. Обычно он используется в виде кордной ткани,
а частично в виде нитей (безуточный корд) или корд-шнура. Для
резино-тканевых изделий ответственного назначения (тяжелые
автомобильные, авиационные и некоторые другие виды шин)
наиболее пригоден корд из полиамидных волокон (капроновый корд)
и частично металлокорд, изготовляемый из прочной стальной
проволоки. В шинах для легковых и грузовых автомобилей малой
и средней мощности, а также для тракторов и других
сельскохозяйственных машин успешно применяется вискозный корд.
В последние годы начато промышленное применение
полиэфирных волокон типа лавсан, а также стекловолокна для
изготовления корда и других технических тканей.
Хлопчатобумажный корд (из хлопковых волокон) уступает
корду из высокопрочных химических волокон: по прочности при
растяжении в 2—4 раза, по усталостной прочности—в 100 и более
раз, по ударопрочное™—в 10—15 раз и по теплостойкости—во
много раз. Поэтому хлопчатобумажный корд все более
вытесняется кордом из химических волокон.
При изготовлении некоторых резиновых технических изделий
пока еще широко применяются хлопчатобумажные ткани.
Например, прочная высококачественная ткань бельтинг
используется в прокладках транспортерных лент, приводных ремней,
рукавов и др. Однако и в этих изделиях преимущества химических
волокон перед хлопковыми (особенно их более высокая прочность
и теплостойкость) бесспорны, Так, транспортерные ленты, арми-
502
рованные полиамидной тканью типа капрон или анид, служат
вдвое дольше, чем ленты с прокладками из хлопчатобумажных
тканей. Переход на ткани из химических волокон позволил
увеличить срок службы некоторых резинотканевых изделий во
много раз.
Металлическая проволока является упрочняющим элементом
борта пневмлтической шины. В последние годы в шинах новых
конструкций все шире применяется м«таллокорд, частично
заменяющий корд из химических волокон. Весьма перспективны новые
конструкции шин высокой грузоподъемности с комбинированной
армировкой текстильным кордом и металлокордом.
В качестве армирующих элементов для рукавов тяжелого типа,
транспортерных лент и некоторых других изделий также
применяются стальная проволока и стальные тросы. Разнообразные
амортизаторы и прокладки и многие детали, широко
используемые в современном машиностроении, в автомобильной и
авиационной промышленности, выполняются в виде всевозможных
соединений резиновых и металлических деталей.
ВСПОМОГАТЕЛЬНЫЕ МАТЕРИАЛЫ
В производстве резиновых изделий, кроме перечисленных выше
веществ и материалов, потребляются клеи, смазки, растворители,
прокладки и другие вспомогательные материалы.
Растворители каучука (бензин, этилацетат, амилацетат,
скипидар и др.) применяются главным образом для
изготовления клеев, употребляемых в процессах сборки резиновых
изделий и для промазки резиновых тканей. Наиболее широко
применяется и наименее токсичен бензин марки «Галоша» (плотность
не более 0,73 г 1см9, температура начала перегонки не ниже 80 °С).
Бензин «Галоша» используется для изготовления клеев из изо-
преновых (синтетических и натурального), бутадиеновых, бута-
диен-стирольных и бутадиен-метилстирольных каучуков. Кроме
того, бензин применяется для увеличения клейкости
поверхностей склеиваемых или стыкуемых резиновых, резино-тканевых
и резино-металлических деталей при сборке (конфекции).
При работе с растворителями, которые являются
воспламеняющимися и взрывоопасными, а также токсичными веществами,
необходимо строго соблюдать правила техники безопасности и
оборудовать производственные помещения надлежащей
вентиляцией.
Из различных клеев в резиновой промышленности наибольшее
применение имеют резиновые клеи. Они изготовляются путем
растворения хорошо пластифицированного или измельченного
каучука в бензине или другом растворителе. Этот процесс
проводится в лопастных клеемешалках. Клеи могут содержать
большее или меньшее количество каучука (густые клеи называют
503
мазями) и различные дооавки—сажу, красители и т. д. Некоторые
клеи содержат вулканизующие вещества и способны
вулканизоваться при нагреваьии или при комнатной температуре
(самовулканизующиеся клеи, необходимые для ремонта резиновых изделий).
Резиновые клеи употребляются при сборке сложных заготовок
из многих деталей, особенно из неклейких и малоклейких
резиновых смесей. Это облегчает процесс сборки и повышает
монолитность вулканизованных изделий.
Кроме резиновых клеев на основе каучука, в резиновых
производствах применяются и другие клеи. Для крепления резин к
металлу в производстве резино-металлических изделий или
деталей на тщательно очищенную поверхность металла наносятся изо-
ционатные клеи или клеи из хлорированного или окисленного
каучука. Кроме того, резины крепятся к металлу через
эбонитовую прослойку, латунированием, оцинкованием и другими
методами.
Для покрытия изделий и внутренней поверхности прессформ
перед вулканизацией применяются смазки самого
разнообразного состава, включая мыльные растворы, суспензии слюды,
каолина, силиконовые масла и др.
Многие полуфабрикаты, например автокамеры, детали
резиновых рукавов и т. д., необходимо опудривать, чтобы избежать
слипания. В качестве опудривающих веществ чаще всего
применяется тальк, а также крахмал и др. В некоторых случаях на
поверхность заготовок или изделий наносят суспензию талька, что
устраняет его пыление, неизбежное при опудривании.
Многие полуфабрикаты, например невулканизованные
прорезиненные ткани, профилированные заготовки, подаются на
обработку или хранятся на прокладочных холстах или полотнищах.
Наиболее удобны и долговечны прокладки из тканей, покрытых
нитролаками и др.
3. ОБЩИЕ ТЕХНОЛОГИЧЕСКИЕ ПРОЦЕССЫ
РЕЗИНОВОГО ПРОИЗВОДСТВА
Процесс производства резиновых изделий, начиная с
подготовки поступивших на завод исходных сырых материалов и кончая
выпуском готовой продукции, можно условно разделить на
следующие общие технологические стадии:
1) подготовительные процессы—подготовка и развеска
каучука и ингредиентов, приготовление резиновых смесей (смешение),
2) заготовительно-сборочные процессы—шприцевание,
пропитка тканей, каландрование, раскрой деталей, сборка изделий
или их предварительное формование;
3) вулканизация и отделочные операции.
На разных заводах при изготовлении резиновых изделий по
различным технологическим схемам применяются разнообразные
504
варианты и сочетания перечисленных стадии производственного
процесса. На многих участках предприятий резиновой
промышленности производство изделий механизировано,
автоматизировано и осуществляется поточными методами.
Производственные процессы на каждом резиновом или шинном
заводе можно разделить на процессы общей технологии резины
(подготовка сырья, приготовление резиновых смесей и др.) и
процессы специальной технологии изготовления различных видов
резиновых изделий. Специфичность процессов специальной
технологии определяется особенностями конструкции
изготовляемых изделий.
В настоящем разделе описываются преимущественно
процессы и оборудование общей технологии резины.
РЕЦЕПТУРА РЕЗИНОВЫХ СМЕСЕЙ
Состав или рецепт резиновой смеси зависит не только от типа
изготовляемого из нее изделия (или детали), но и от условий
дальнейшей эксплуатации резиновых изделий. Так, одинаковые по
размерам и конструкции шины, изготовляются из смесей разной
рецептуры в зависимости от того, для каких дорожных и
климатических условий предназначены выпускаемые шины, например для
работы в умеренных широтах, на Крайнем Севере или в тропиках.
В табл. 24 приводятся примерные рецептуры некоторых
резиновых смесей.
Высокоэластичные резины изготовляют из наиболее
эластичного каучука, содержание которого в таких резинах составляет
от 60 до 70 вес. ч. на 100 вес. ч. резиновой смеси. При этом
для изготовления наиболее ответственных высокоэластичных
резиновых изделий стремятся подбирать стереорегулярные каучуки
с минимальными гистерезисными потерями (например, изопрено-
вые, бутадиеновые или натуральный каучуки).
В качестве примера одной из рецептур смесей для
высокоэластичных резин может служить приведенная в табл. 23 смесь
для наиболее ответственной детали шин—брекера, расположенного
в центре массива покрышки пневматической шины.
Износостойкие резины, которые должны обладать высокой
прочностью и эластичностью и оптимальным сопротивлением
истиранию, получают на основе цис-бутадиеновых каучуков в
комбинации с изопреновыми или бутадиен-стирольными к-'учуками
и с введением в смесь большого количества
высокоактивной сажи.
При изготовлении ответственных износостойких резиновых
изделий, подвергающихся многократным деформациям, в
рецептуру резиновой смеси вводят гораздо больше каучука, чем в
подошвенную резину, и применяют более износостойкие каучуки и
высокоактивные сажи.
Таблица 23
Примерные рецептуры резиновых смесей
(в вес. ч.)
Компоненты смеси
Каучук
Синтетические смолы ....
Вулканизующие вещества:
сера
ускорители вулканизации
замедлители ,
вулканизации
активаторы ускорителей
Противостарители
Мягчители
Усилители
Смеси
для брекера
Смеси
для резиновых
подошв
Смеси
для беговой части
шины
(протекторные)
Тип каучука
стереореіуляр.іьій
СКИ или НК
и комбинации их
сСКД
100
1—2,5
1,2—0,8
0,3—0,5
3,5—5
0,5—2
7—8
40—50
(активная сажа
или ее
комбинации с
полуактивной)
бутадиек-стироль-
кый с добавкой
синтетических
смол
50—100
50-0
2—4
0,8—2
1,5—5
1—2
3—20
100—185
(сажа, каолин)
цце-бутадиеновый,
бутадиен-стироль -
ный. их
комбинации*
100
1,5-2,5
0,7—1
0,3—0,5
0,3—5
0,5—2
8,8-14
50—60
(активная сажа]
* Для шин наиболее ответственного назначения в протекторные смеси вводят добавки
стереорегулярного СКИ-3 или НК-
Для получения^газонепроницаемых резин стремятся
использовать бутилкаучук и его модификации. Для резин,
предназначенных для работы в среде растворителей и масел, наиболее
пригодны маслостойкие каучуки, например тиоколовые, хлоропре-
новые, нитрильные (причем нитрильные наиболее теплостойки).
Резиновые прокладки, работающие при температурах 200 °С и
выше, целесообразно изготовлять на основе силоксановых кау-
чуков, фторкаучуков и др.
При составлении рецептуры резиновых смесей весьма важен
также подбор соответствующих противостарителей, противоуто-
мителей, антиозонантов и правильное соотношение дозировки
серы и ускорителей вулканизации.
Из сказанного следует, что рецептуры резиновых смесей
исключительно многообразны и для получения резиновых изделий
с требуемыми свойствами при составлении рецептуры смесея
необходимо учитывать многие факторы. К ним относятся также и
технологические свойства смеси (способность к вулканизации и др.),
506
иначе рецептуру резиновой смеси, обеспечивающая высокие
эксплуатационные показатели изделий, может оказаться негодной
в технологическом отношении.
ПОДГОТОВИТЕЛЬНЫЕ ПРОЦЕССЫ
Подготовка каучука. Каучуки поступают на резиновые
заводы в кипах, рулонах, в виде крошки, латекса и др. Некоторые типы
каучуков достаточно пластичны и для дальнейшей обработки не
нуждаются в дополнительном размягчении или пластикации.
Натуральный и некоторые синтетические каучуки,
поступающие на резиновые заводы в кипах, обычно очень жестки.
Для размягчения натурального каучука его перед дальнейшей
обработкой подвергают декристаллизации, т. е. переводят в
аморфное состояние под действием тепла. Для этого натуральный
каучук, разрезанный ка более или менее мелкие куски,
обрабатывается на специальных установках токами высокой частоты или
подвергается распарке в камерах, обогреваемых паром, при 70—90 °С.
Распарка продолжается 1—3 суток.
На многих резиновых заводах распарку натурального каучука
проводят в камерах непрерывного действия. Декристаллизация
токами высокой частоты значительно более производительна, она
длится всего 40 мин при температуре 40—50 °С.
Декристаллизованный натуральный каучук еще сохраняет
некоторую жесткость и подвергается дальнейшему размягчению
при помощи механической пластикации.
Механическая пластикация необходима для размягчения
натурального и некоторых синтетических каучуков. Этот процесс
осуществляется путем перетирания жесткого полимера между
валками вальцов (стр. 509), или в рабочей камере резиносмеси-
теля (стр. 511), или же в червячном прессе-пластикаторе (стр. 5]3).
Под действием механических усилий растяжения и сдвига
молекулярные цепи полимера рвутся и становятся более короткими.
При этом протекают и химические процессы, в том числе
окислительная деструкция каучука под влиянием кислорода
воздуха. Частично (в меньшей степени) происходит и обратный
процесс—структурирование (сшивка). В результате механо-хими-
ческого процесса пластикации молекулярный вес каучука
уменьшается.
В резиносмесителях, используемых для пластикации,
обрабатываемый каучук подвергается более эффективным механическим
воздействиям, чем при пластикации на вальцах. Кроме того, ре-
зиносмесители служат как для пластикации, так и для смешения,
что позволяет объединить в одной машине оба этих процесса.
В отличие от натурального каучука его синтетический
аналог—каучук СКИ-3 не требует специального процесса
пластикации и легко размягчается при обработке на смесительном обору-
507
^,„„t.11.,^. lulv«4v. nc путд<ііии.н в иредиарительнои пластикации
современные регулированные (мягкие) сополимеры бутадиена и
стирола (например, каучуки типа СКС-30 АРК и СКС-30 АРКМ),
хлоропреновые каучуки, бутилкаучуки, мягкие нитрильные
каучуки. Стереорегулярный бутадиеновый каучук типа СКД при
механической обработке не деструктируется, но при совместной
обработке с изопреновыми или бутадиен-стирольными каучуками
приобретает необходимые технологические свойства.
Процессы декристаллизации натурального каучука и
термоокислительной деструкции ряда жестких синтетических каучуков
(например, бутадиен-стирольных типа СКС-30 или СКС-10 и др.)
интенсифицируются в присутствии специальных
добавок—ускорителей пластикации. Термоокислительная пластикация таких
каучуков проводится в специальных котлах.
Для облегчения автоматической развески и подачи каучуков
в резиносмесительное оборудование их можно измельчать на
червячных прессах с гранулирующей головкой. Получаемые при
этом «шнуры» разрезаются ножами на куски или гранулы
размером 12—25 мм, затем на них наносится каолиновая суспензия
или другие опудривающие вещества. Таким образом каучук
превращается в сыпучий материал. Грануляция каучука принята
на некоторых шинных заводах.
Подготовка ингредиентов. Различные ингредиенты резиновой
смеси должны поступать на заводы в виде сухих
гранулированных материалов или паст, не требующих дополнительной
обработки перед смешением.
Однако пока еще большинство применяемых сыпучих
ингредиентов приходится предварительно высушивать и просеивать перед
приготовлением резиновых смесей. >Кидкие и легкоплавкие
ингредиенты (мазут, гудрон, петролятум и др.) до введения в смесь
расплавляют и очищают от механических примесей на сетчатых
фильтрах. Тугоплавкие ингредиенты, например мягчители,
очищают с поверхности, затем дробят и загружают в резиносмеситель.
Ускорители вулканизации вводят в резиновые смеси, как
правило, в очень малых дозах. Для лучшего распределения их в
каучуке и устранения потерь при пылении на заводах приготовляют
так называемые ускорительные пасты—смеси ускорителя с мяг-
чителем и красителем.
Более совершенной выпускной формой ускорителей
вулканизации и других химикатов, применяемых в малых дозах, являются
гранулы, наиболее удобные для автоматической развески и
загрузки.
Развеска каучука и ингредиентов. На многих резиновых
заводах ручная развеска каучука и ингредиентов все более
заменяется автоматической. Это особенно важно при использовании
современного высокопроизводительного оборудования для
скоростного приготовления резиновых смесей. Исходные компоненты
508
хранятся в специализированных бункерах с питателями и
указателями уровня. При быстрой загрузке смесителя
предусматриваются промежуточные емкости для заранее взвешенных порций
компонентов смеси.
Современное отделение приготовления резиновых смесей
оборудовано сложной системой транспортных устройств, питающих
резиносмесители, и разнообразными автоматическими весами.
Широкое применение получили автоматические весы с
дистанционной настройкой. Жидкие компоненты поступают в смесители через
весовые дозаторы, управляемые с пульта. При автоматической
развеске и загрузке ингредиентов резиновой смеси значительно
уменьшается возможность ошибок и неточностей при взвешивании,
приводящих к получению некачественных резиновых смесей.
Смешение. Резиновые смеси
приготовляют на различном
смесительном оборудовании: сравнительно
мелкие навески—на открытых
смесительных вальцах., крупные
навески смесей для изделий массового
производства—в мощных закрытых
резиносмесителях.
Вальцы состоят из двух горизон- Рис ш Схема приготовления
тальных полых валков, укрепленных резиновой смеси на вальцах:
В СТаНИНе (рИС. 142). ВаЛКИ Вра- /—передний, медленнее вращающийся
тпяттга HancmAUV nnvr rmvrv r пяч валок; 2—задний, быстрее вращающийся
ЩаЮТСЯ Навстречу Друг другу С pd3- ваЛок; 3-резиновая смесь на переднем
ЛИЧНОЙ СКОРОСТЬЮ. СоОТНОШЄНИЄ ЭТИХ валке; -/-запас резиновой смеси между
г , „ валкамн.
скоростей, называемое фрикцией, для
смесительных вальцов обычно
составляет 1,08, в зависимости от типа и состава смеси оно может
колебаться в пределах 1,04—1,25. Для вальцов другого типа
(подогревательных, питательных, рафинировочных) фрикции могут
быть более высокими. На вальцах, используемых для
пластикации натурального каучука, соотношение окружных скоростей
валков составляет 1,25.
Вследствие разницы окружных скоростей вращения валков в
обрабатываемом материале, проходящем через зазор между
валками, возникают деформации сдвига, что способствует более
равномерному распределению ингредиентов смеси в каучуке.
Наиболее распространены смесительные вальцы с длиной валков 2130
и 1530 мм. Изнутри валки интенсивно охлаждаются
циркулирующей в них холодной водой.
Закрытые резиносмесители имеют различные
производительность, габариты, мощность, скорость вращения и форму валков.
Один из новейших видов смесительного
оборудования—скоростной резиносмеситель высокого давления типа РСВД-140-40
изображен на рис. 143 и 144. В рабочей камере резиносмесителя
емкостью 245 л имеются два овальных валка, которые вращаются
509
H^ fJ фр на пииерхнисіи
каждого валка расположены два винтообразных гребня или
лопасти. В отличие от тихоходных смесителей со скоростью вращения
валков 20 об/мин и менее, современные скоростные резиносмеси-
тели имеют скорости вращения валков от 30 до 40 об/мин
и более. Мощность двигателя для резиносмесителя описываемого
типа составляет 700 кет, но применяются и более мощные
двигатели A000 кет и выше).
Компоненты резиновой смеси подаются в смеситель
автоматически в^загрузочную воронку через верхний затвор по заданной
программе смешения. В
верхнем затворе имеется плунжер,
создающий давление на
обрабатываемую смесь, благодаря
чему компоненты резиновой смеси
интенсивнее перемешиваются.
В современных резиносмесите-
лях давление плунжера на
смесь составляет 5—б am и
более, в старых резиносмесите-
лях 0,8—1,5 am.
Вследствие высоких
скоростей вращения валков и
большой массы перемешиваемого
материала (до 160 кг) в смеси
создаются деформации сдвига,
что также значительно
улучшает условия перемешивания.
Температура смеси,
выгружаемой из нижнего затвора 3,
может достигать 150 °С.
При высокой температуре
смешения возможно
необратимое ухудшение свойств каучука, поэтому современные резино-
смесители интенсивно охлаждаются.
При смешении на вальцах распределение ингредиентов в
каучуке происходит только в зазоре между валками. В закрытом
резиносмесителе ингредиенты распределяются в каучуке в зазоре
между валками и в пространстве между лопастями или гребнями
вращающихся валков и неподвижной стенкой рабочей камеры.
Поэтому резиносмесители намного производительнее
смесительных вальцов. Для смешения на вальцах требуется 9—10 мин, в
резиносмесителе процесс заканчивается за 2—4 мин. К.
преимуществам резиносмесителей относятся также лучшая
обрабатываемость и однородность получаемых смесей, большие возможности
механизации процессов загрузки и выгрузки, экономия рабочих
площадей, электроэнергии и др.
510
Рис. 143. Резиносмеситель
РСВД-140-40 (общий вид).
Интенсификация смешения целесообразна только при условии
автоматической развески и загрузки ингредиентов в резиносмеси-
тель.
Рис. 144. Схема устройства резиносмесителп:
/—стенки смесительной камеры; 2—овальные валки; Л—нижний скользящий
затвор; 4—откидная дверца; 5—вытяжное устройство.
Качество резиновой смеси, тщательность и равномерность
распределения ингредиентов в каучуке зависят от режима смешения—
порядка введения компонентов резиновой смеси, длительности
обработки каждой порции вводимых компонентов, общей
длительности (цикла) процесса и температурных условий. Для каждого
511
шения.
В резиновой промышленности приняты одностадийные и двух-
стадийные режимы смешения. При одностадийном режиме в рези-
носмеситель вводят каучук и затем в определенной
последовательности загружают все ингредиенты, кроме серы. Этот режим
особенно пригоден при переработке саже-масляных бутадиен-сти-
рольных каучуков, поскольку самая энергоемкая операция-—
К протекторному
агрегату
Рис. 145. Схема двухстадийного приготовления протекторных смесей:
I—бункеры для сажи; 2—емкости для мягчителен; 3—бункеры для защитных веществ
замедлителей вулканизации, окиси цинка и канифоли; 4—бункеры для каучуков; 5—автоматические
весы; 6—ленточные конвейеры; 7—резиносмеситсль (скорость вращения 40 об/мин)'. 8—шприц
машина с гранулирующей головкой; 9—вибрационный конвейер; 10— элеватор; II—охлаждающая
камера; 12—линия пневматической подачи гранулированной маточной смеси; 13—бункеры для
ускорителей вулканизации и серы; 14—бункеры для гранулированной маточной смеси; /5—рези-
носмяситель (скорость вращения 30 об/мин); 16—вальцы.
введение сажи в каучук—уже выполнена на заводе-изготовителе
СК. Описанный режим лишь условно может быть назван
одностадийным, так как дополнительной конечной стадией смешения
являегся последующее введение серы на вальцах. Обычно при
одностадийном смешении используют резиносмесители, валки
которых совершают 20—30 об/мин.
При двухстадийном смешении (рис. 145) в первой стадии
изготовляются промежуточные смеси непластицированного
каучука с большей частью ингредиентов, а затем в
промежуточную резиновую смесь вводят остальные ингредиенты, включая
512
серу и ускорители вулканизации (вторая стадия). Окончательное
смешение на второй стадии можно проводить на тихоходном рези-
носмесителе после некоторой выдержки и охлаждения смеси.
Двухстадийный цикл смешения продолжается всего 2—4 мин,
т. е. близок к непрерывному процессу смешения с частой
порционной подачей навесок.
В настоящее время в резиновой промышленности ведутся
работы по созданию резиносмесителей непрерывного действия,
типа червячных прессов.
Отбор готовой смеси из резиносмесителя можно осуществлять
разными способами. В отличие от старых агрегатов с тихоходными
резиносмесителями, из которых приготовленные порции
резиновой смеси поступают на листовальные вальцы, в новых более
совершенных агрегатах с мощными скоростными резиносмесителями
готовая смесь подается на крупногабаритные червячные прессы
или шприц-машины, где проходит дополнительную обработку,
очистку и приобретает заданную выпускную форму гранул,
листов и т. д. Описанные новые агрегаты значительно удобнее для
организации поточного производства с прямой подачей
резиновой смеси от резиносмесителей на последующие производственные
операции.
Для получения резиновой смеси в виде гранул применяют
шприц-машину со специальной гранулирующей головкой.
Диаметр червяка такой шприц-машины 380/450 мм, длина червяка
свыше 2 м. Для выпуска резиновой смеси в виде листов
используют шприц-машины с листующей головкой.
В агрегате с резиносмесителем устанавливают также шприц-
машины типа червячных фильтрпрессов для очистки смеси от
посторонних примесей.
ЗАГОТОВИТЕЛЬНО-СБОРОЧНЫЕ ПРОЦЕССЫ
Шприцевание или выдавливание при помощи шприц-машин
резиновой смеси в виде жгута, пластины, трубки или лент более
сложного фигурного сечения широко распространено как в
резиновой промышленности, так и в других отраслях переработки
полимеров*. Методом шприцевания формуют резиновую смесь,
получая из нее невулканизованные профилированные заготовки
различных изделий или деталей (камеры автомобильных шин,
рукава, шланги, трубки всевозможных видов и т. д.), а также
профилированные массивные резиновые заготовки с различным
фигурным сечением (протекторная лента для шин, шнуры4
облицовочной резины для штампования галош, уплотнительные шнуры
и ленты и т. д.).
• В переработке пластмасс машины, аналогичные шприц-машинам,
получили название экструдеров (стр. 535), а процесс шприцевания—экструзии.
33—805 513
p^UhDiiri дйші«„ ш1?,щщаШПІ,,
применяемых в резиновом производстве, являются: рабочий
цилиндр с загрузочным отверстием и червяк, вращающийся внутри
этого цилиндра от привода; головка, станина и опорная плита
На рис. 146 приведена типовая схема шприц-машины для выпуска
резиновых камер, рукавов и других трубчатых заготовок.
Форма шприцуемого полуфабриката определяется
конструкцией головки и мундштука.
Устройство шприц-машины для выпуска заготовок в
принципе аналогично устройству шприц-машин, применяемых на
стадии приготовления резиновых смесей. Шприц-машины могут быть
включены в поточную линию приготовления резиновых смесей
Загрузка
Пар
Z
Рис. 146, Схема устройства шприц-машины:
/—червяк; 2—рабочий цилиндр; 3~голрвка; 4—мундштук; 5—Дорн-
G—центрирующие болты.
и непосредственно загружаться гранулированной смесью. В
случае необходимости дополнительной очистки готовой смеси от
посторонних включений перед дальнейшей обработкой смесь
пропускают через червячный фильтрпресс (стрейнер) с
перфорированной шайбой в головке машины. Очистку резиновой смеси в таких
шприц-машинах нередко сочетают с листованием (рис. 147) или
грануляцией.
Прямую передачу смеси от резиносмесительного агрегата на
профилирующую шприц-машину можно обеспечить и при такой
системе работы, когда между этими производственными участками
размещаются подогревательные и питательные вальцы. В этом
случае резиновая смесь с питательных вальцов срезается в виде
ленты и системой транспортеров подается в загрузочную воронку
шприц-машины для выпуска заготовок.
При шприцевании, как и при смешении, применяется
охлаждение машин и строго соблюдается установленный температурный
режим.
В результате механических воздействий, которым
подвергается резиновая смесь на шприц-машине, в выходящей из нее заго-
514
товке создаются внутренние напряжения. Вследствие этого по
выходе из мундштука машины эластичная резиновая заготовка
начинает самопроизвольно деформироваться—сокращается по
длине и увеличивается в поперечном направлении. Такие
деформации получили название усадки. Подбирая рецептуру смеси
и соответствующий режим ее обработки, удается регулировать
степень усадки.
При шприцевании трубчатых заготовок (например, камер или
шлангов) их опудривают изнутри для того, чтобы предотвратить
слипание стенок. Для этой цели шприц-машины оснащаются
Рис. 147. Шприц-машина с листующей головкой (общий вид).
опудривающими устройствами. Обычно применяемый для опудри-
вания тальк подается в воронку, захватывается воздушной струей
и вдувается внутрь шприцуемой камеры или трубки. Вместо опуд-
ривания применяется также опрыскивание заготовок
каолиновыми суспензиями и др.
Размеры и вес резиновых заготовок, получаемых на шприц-
машинах, систематически контролируются.
Пропитка тканей. Большинство технических тканей,
являющихся элементами конструкции пневматических шин,
транспортерных лент и других ответственных резино-тканевых изделий,
подвергается предварительной пропитке для увеличения
прочности связи между волокнами и резиной. Пропиточные составы
изготовляются преимущественно на основе синтетических латексов
и водорастворимых синтетических смол (например, резорцино-
феноло-формальдегидных). Во многих случаях в пропиточные
составы вводят сажу и некоторые другие химические добавки.
33* 515
ПрОПИТКа ЯВЛЯеТСЯ Обязательным процессом Оираишки і
из химических волокон и рекомендуется также для
хлопчатобумажных тканей.
Пропитку тканей из химических, особенно полиамидных
волокон, совмещают с горячей вытяжкой. На ряде предприятий
пропиточные машины работают в одном агрегате с каландрами.
Каландрование. На каландрах, представляющих собою
машины валкового типа, проводятся следующие операции:
формование резиновой смеси для получения полуфабрикатов
в виде гладких или профилированных листов или лент;
обрезинивание тканей (обкладка резиной) и промазка их
резиновой смесью.
В зависимости от назначения применяются двух-, трех-,
четырех- и пятивалковые каландры с различным расположением
и размерами валков, различной скоростью их вращения, разной
мощностью и другими конструкционными различиями.
Наибольшее распространение получили трехвалковые каландры (рис. 148)
и четырехвалковые. Схема работы трехвалковых каландров для
листования резиновой смеси и обрезинивания ткани показана
на рис. 149.
Валки каландров, служащих для формования гладких или
профилированных резиновых листов или лент и для обкладки тканей,
обычно имеют одинаковую скорость вращения, однако применяются
и валки с фрикцией. Каландры для промазки тканей
(втирания резиновой смеси в ткань между переплетениями нитей)
всегда работают с фрикцией между валками. Каландры, которые
можно использовать как листовальные, обкладочные и промазочные,
т. е. устанавливать на разные соотношения скоростей их валков,
называются универсальными.
Основной конструкционной деталью каландров являются
горизонтальные валки, вмонтированные в станину. Валки двух-
и трехвалковых каландров обычно расположены друг над другом
в вертикальной плоскости. Однако в новых конструкциях трех-
и четырехвалковых каландров валки расположены в разных
вертикальных плоскостях, как показано на рис. 148.
Современный четырехвалковый
каландр—высокоэффективная машина, работающая с большой скоростью (до 90 м/мин) и
высоким классом точности. Такие каландры используются для
обработки шинного корда. Толщину полотнища, каландруемого
на этой машине, можно регулировать с точностью до 0,005 мм.
Валки каландров изготовляются из чугуна, стального литья,
хромированной стали и др. и выполняются полыми, что облегчает
их нагревание или охлаждение. Зазор между валками, куда
подается резиновая смесь или смесь совместно с тканью, следует
точно регулировать. От точности регулирования зависит
постоянство каландрованного материала по заданному калибру
(толщине) и весу.
516
Зазор между валками каландра можно регулировать при
помощи бомбировки или небольшого смещения осей валков. Под
бомбировкой понимают такую обточку рабочей поверхности вал-
Рмс. 148. Современный трехвалковый каландр (внешний вид).
ков, в результате которой в середине валка образуется некоторая
выпуклость. При прогибе валков под влиянием распорных
усилий зазор между бомбированными валками выравнивается, а в
~~ Резиновая
смесь
Ткань для
оокшдки. разимой
Резиновая
смесь
Прокладочная
ткань
Прокладочная
/ ткань
Рис. 149. Схема работы трехвалкового каландра:
а—листование резиновых смесей; б—обкладка ткани резиной; /—валки каландра (верхние
н средние—горячие, нижние—холодные); 2—закаточные устройства.
отсутствие бомбировки зазор в середине валков становится
больше, чем по краям, что может явиться причиной выпуска
некачественных полуфабрикатов.
517
ками заключается в небольшом смещении или перекрещивании
осей валков. Этот прием компенсации прогибов валков дает
возможность выравнивать калибры каландруемых полуфабрикатов
до установленных размеров.
Калибр каландрованных полотнищ постоянно контролируется.
В последние годы ручные методы измерения все более заменяются
механизированными и автоматическими методами. В качестве
примера можно привести ?-лучевой калиброметр, работающий
по принципу измерения интенсивности радиационного излучения,
проходящего через контролируемое полотнище. С помощью этого
прибора автоматически регулируют также зазор между валками.
Калибр каландрованных материалов можно контролировать
путем пропускания их через весы непрерывного действия.
Все большее распространение в резиновой промышленности
получают поточные линии, включающие резиносмесительное
оборудование, пропиточные агрегаты, каландры и участки
последующих заготовительно-сборочных операций.
Обрабатываемые на каландрах ткани (особенно ткани
тяжелого типа) должны быть хорошо высушены. Сушка производится
на сушильных барабанах, режим работы которых строго
контролируется, включая также контроль влажности корда.
При выпуске каландрованных резиновых пластин,
прорезиненных тканей (промазанных резиновой смесью) или обрезинен-
ных тканей (обложенных резиновой смесью) применяются
прокладочные холсты, опудривание пластин и тканей и другие средства,
предотвращающие слипание заготовок или прилипание их к
поверхности сборочных и других приспособлений при последующих
технологических операциях.
Промазка тканей на клеепромазочных машинах. Для проре
зинивания тонких легких тканей, например одежных, или для
нанесения на ткань очень тонких слоев резиновой смеси
используют клеепромазочные машины, обычно горизонтальные.
Ткань, подлежащая обработке, с раскаточного
приспособления подается на рабочий вал машины. Над валом подвижно
укреплен промазочный нож. Перед ножом на ткань накладывают
резиновый клей. Ткань движется непрерывно между рабочим валом и
ножом. Подвижной нож можно опускать до образования
требуемого зазора между ножом и рабочим валом и таким образом
регулировать толщину клеевого слоя, втираемого в ткань.
Промазанная клеем ткань поступает на обогреваемую плиту клеепромазоч-
ной машины, где нагревается до 70—90 °С. При этом из клея
испаряется растворитель, пары которого улавливаются на
специальной рекуперационной установке. Промазка резиновыми клеями
может быть однократной и многократной.
Раскрой деталей и сборка заготовок изделий. Сложные
резиновые изделия (шины, транспортерные ленты, ремни, рукава, рези-
518
новая обувь и др.) в процессе их производства собирают из
предварительно заготовленных, раскроенных деталей, которые
соединяют между собой путем склеивания или подпрессовки.
Полученную таким образом сырую заготовку изделия затем подвергают
вулканизации. Детали для сборки изделий заготавливают из
сырой резиновой смеси, прорезиненных или обрезиненных тканей
и других материалов, включая резино-металлические детали.
Технологическое оформление процессов раскроя и сборки весьма
разнообразно в зависимости от типа изделий. Эти операции
относятся к специальной технологии резиновых производств и
здесь не рассматриваются. Следует лишь отметить, что процесс
производства подавляющего большинства резиновых изделий
включает заготовительно-сборочные операции и формование,
которое осуществляется обычно на завершающей стадии—во
время вулканизации, но может проводиться и как самостоятельная
операция.
Довольно широкий ассортимент резиновых изделий
изготовляется без применения сборочных операций. Так, трубки, шнуры
или ленты формуются методом шприцевания, некоторые
резиновые технические детали—методом литья под давлением и т. д.
Необходимо подчеркнуть все более широкое внедрение так
называемой латексной технологии—изготовление изделий
непосредственно из латекса. В качестве примера можно привести
производство пенорезин (пористых резин), получаемых
вспениванием латекса и используемых в качестве матрацев, сидений,
обивок и т. д. Методом макания формочек в латекс, содержащий
соответствующие ингредиенты, и в результате последующих
операций—коагуляции, сушки и вулканизации—получают многие
тонкостенные резиновые изделия (перчатки, соски и т. п.).
Существуют и другие способы производства изделий непосредственно
из латекса или с частичным его применением.
ВУЛКАНИЗАЦИЯ
В этом важнейшем завершающем процессе резинового
производства происходит поперечное сшивание макромолекул каучука
под действием вулканизующих веществ. Благодаря этому
принципиально изменяются технические качества вулканизуемой
смеси: почти полностью теряются пластические свойства, повышаются
эластичность вулканизатов, их прочность, износостойкость,
стойкость к набуханию в растворителях, химическая стойкость и др.
Большинство резиновых- изделий вулканизуется под
давлением в прессформах, при этом происходит отпрессовка внутренних
деталей и профилирование наружной поверхности изделий.
Таким образом, физико-химический процесс вулканизации часто
сочетается с механическим процессом окончательного формования
изделий.
519
Шины, ремни, рукава, резиновая обувь и многие другие виды
резиновых изделий вулканизуются горячим способом—при
нагревании, так как наиболее употребительные вулканизующие
вещества (сера, окислы металлов, синтетические смолы и др.),
присутствующие в сырой резиновой смеси, вступают в химическое
взаимодействие с каучуком только при повышенных температурах.
Теплоносителями служат преимущественно горячий воздух, пар,
перегретая вода, применяются также их различные комбинации.
Источником тепла в процессе вулканизации может служить также
электроэнергия.
Некоторые резиновые изделия вулканизуются холодным
способом, т. е. при комнатной температуре. Особенно
распространена холодная вулканизация при ремонте и починке резиновых
изделий. В этих случаях часто пользуются самовулканизующимися
резиновыми смесями или клеями.
Режимы вулканизации—длительность, температура,
чередование температурных интервалов, давление, послевулканизацион-
ное охлаждение могут быть весьма различными. Они зависят от
габаритов изделия, состава резиновых смесей, характера вулка-
низационного оборудования, типа греющей среды и других
факторов.
Как правило, продолжительность вулканизации тонкостенных
изделий меньше, чем массивных толстостенных. Так,
сравнительно тонкостенная камера автомобильной шины вулканизуется
в течение 5—12 мин при 150—165 °С, тогда как длительность
вулканизации тяжелой многослойной пневматической шины при
температуре 143 °С может достигать нескольких часов.
В процессе горячей вулканизации механические свойства
резиновой смеси постепенно улучшаются до определенного предела
и затем, при продолжающемся нагревании, некоторое время
остаются на достигнутом уровне. Продолжительность периода
вулканизации, в течение которого сохраняются оптимальные для
данного вулканизата свойства, называют плато вулканизации. При
дальнейшем нагревании изделий происходит перевулканизация—
ухудшение свойств резины. Характер изменения свойств
резиновых изделий в процессе вулканизации показан на рис. 150.
Рецептуру резиновой смеси и условия вулканизации стремятся
подобрать таким образом, чтобы достигалось возможно более
широкое плато вулканизации. Это позволяет уменьшить возможность
перевулканизации как однослойных изделий при
кратковременном цикле вулканизации, так и многослойных изделий при
длительных процессах вулканизации, когда наружные и внутренние
слои резиновой смеси нагреваются с различной скоростью.
В зависимости от типа и конструкции резиновых изделий
для горячей вулканизации применяются разнообразные виды
оборудования: прессы, автоклавы, котлы, барабанные
вулканизаторы, вулканизационные камеры непрерывного действия. Много-
520
слоиные изделия тяжелого типа, нуждающиеся в дополнительном
прессовании при вулканизации (например, шины,
транспортерные ленты), а также многие формовые резиновые изделия
(амортизаторы, грелки, формовая резиновая обувь и т. д.) вулканизуют
в прессах и автоклавах под давлением, причем такие формовые
изделия, как шины, вулканизуются в прессформах.
Галоши, изготовляемые методом клейки на конвейере или
методом штампования, резиновые перчатки и другие тонкостенные
изделия, различные пластинчатые и кольцевые уплотнители,
резиновые нити, амортизационные шнуры вулканизуют в котлах
или других вулканизационных аппаратах без применения пресс-
форм.
Продолжительность Вулканизации.
Рис. 150. Зависимость предела прочности резин при
растяжении от длительности вулканизации.
Периоды процесса; /—предварительный нагрев резиновой смеси
до начала ее структурирования; 2—структурирование резиновой смеси до
достижения оптимальной совокупности свойств резины; Л—плато
вулканизации (нагрев без дальнейшего изменения качеств вулканизата);
4—перевулканизация с ухудшением технических качеств вулканизата.
Для вулканизации бесконечных приводных ремней,
транспортерных и элеваторных лент широкое применение получили
рамные челюстные (одно- и двухэтажные) прессы с плитами. Длина
таких прессов достигает 7—10 м, ширина—2,5 м. Управление
работой таких прессов в ряде случаев осуществляется дистанционно.
Агрегат для вулканизации лент включает раскаточную стойку
с рулоном невулканизованной ленты, вулканизационный пресс
с зажимным и растяжным устройствами, закаточную стойку для
приема вулканизованной ленты из пресса. Ленты вулканизуются
отдельными последовательными участками по их длине. По
завершении вулканизации одного участка ленты ее протягивают на
прессе так, чтобы последующий участок, подлежащий вулканизации,
непосредственно примыкал к вулканизованному участку. При
этом небольшие смежные зоны отдельных участков ленты могут
перевулканизоваться, что является одним из недостатков
данного периодического процесса.
521
этот недостаток устраняется при непрерывной вулканизации
ремневых пластин в вулканизаторах барабанного типа. На
обогреваемый барабан диаметром 1,0—1,5 ж накладывается
вулканизуемая ремневая или транспортерная лента, прижимаемая к
барабану стальными бесконечными лентами. Огибая нагретую
поверхность барабана, изделие вулканизуется под давлением
стальной ленты.
Однако в аппаратах подобного типа пока еще не обеспечиваются
достаточно высокие прессующие усилия, необходимые при
вулканизации обрезиненных ремней и лент.
Наиболее совершенным видом современного оборудования для
вулканизации пневматических шин является
форматор-вулканизатор. Один из таких аппаратов
изображен на рис. 151.
Форматор-вулканизатор
представляет собой сдвоенный
пресс, т. е. аппарат с двумя
прессформами. В центре
нижней половины каждой пресс-
формы прикреплена
тонкостенная складная резиновая
диафрагма, на которую сверху
автоматически подается с конвейера
заготовка сырой шины в виде
плоского браслета. В диафрагму
вводят пар. Под его давлением
при одновременном опускании
верхней половины прессформы
браслетообразная заготовка
шины сворачивается, приобретая
торообразную форму*. При этом
ее борта сближаются друг с другом, беговая зона становится
выпуклой. В результате сырая заготовка приобретает общие
очертания шины. Когда обе половины прессформы, обогреваемой паром,
сомкнутся, в диафрагму и паровую камеру подается
теплоноситель и начинается процесс вулканизации, с одновременным от-
прессовыванием соответствующего рельефного рисунка на
беговой поверхности шины. По окончании вулканизации пресс
автоматически открывается, вулканизованная покрышка снимается
с диафрагмы и автоматически сбрасывается на отборочный
транспортер.
Вулканизационные аппараты снабжаются
контрольно-измерительными и регулирующими приборами, что позволяет автомати-
Рис. 151. Форматор-вулканизатор для
пневматических шин.
* Тор—геометрическое тело, образуемое при вращении круга вокруї
прямой, лежащей с ним в одной плоскости и не пересекающей его. Форму тора
имеют, например, баранка, спасательный круг.
522
чески поддерживать заданный режим процесса (температуру,
длительность и давление).
В производстве массовых формовых изделий применяются
литьевые червячные прессы. Это позволяет осуществить комплексную
механизацию и автоматизацию производственных процессов, а в
ряде случаев совместить литье под давлением с процессом
вулканизации.
Ведутся также работы по созданию вулканизациониых
аппаратов нового типа, в которых процесс вулканизации может
происходить под действием токов высокой частоты, инфракрасного
облучения, ионизирующих излучений.
ОТДЕЛКА И РАЗБРАКОВКА РЕЗИНОВЫХ ИЗДЕЛИЙ
Вулканизованные резиновые изделия подвергают различным
отделочным операциям. Для изделий, полученных путем
вулканизации в прессах, необходимо удаление заусенцев или
небольших выпрессовок вулканизованной резины на линии разъема
прессформ. Некоторые резиновые технические изделия после
вулканизации следует обточить, отшлифовать или зачистить.
Многие изделия, например автомобильные шины, окрашивают
для защиты от старения. В качестве защитных составов
используют смеси парафина с сажей, воскообразные вещества и др.
Такие изделия, как, например, галоши, детские мячи и др., для
улучшения их внешнего вида покрывают лаком. Ряд изделий в
процессе отделки оснащают арматурой. Например, заключительными
операциями изготовления резиновых камер являются: изгиб
корпуса вентиля, вставка золотника, проверка герметичности камеры
и навинчивание колпачка на вентиль.
Все готовые резиновые изделия подвергаются внешнему
осмотру для проверки сортности. Небольшие поверхностные дефекты
в ряде случаев устраняют путем ремонта без ущерба для
эксплуатационных качеств изделий.
Контроль качества сырья и продукции
На заводы резиновых изделий синтетические и натуральные
каучуки поступают партиями. Каждая партия снабжается
паспортом, в котором отражены важнейшие показатели технологических
и физико-механических свойств каучука. Основными
технологическими свойствами каучука, подлежащими дополнительной
проверке на резиновом заводе, являются: его пластичность и
эластическая восстанавливаемость, способность к пластикации, скорость
вулканизации резиновых смесей и склонность к подвулканиза-
ции. Показатели механических свойств—эластичность по
отскоку, сопротивление разрыву и раздиру, износостойкость, относи-
тельные удлинения и др.—проверяются на образцах вулкани-
затов.
Контроль качества корда и других технических тканей
заключается в определении их разрывной прочности, относительных
удлинений, однородности нитей и тканей по этим показателям,
усталостной выносливости, теплостойкости и др.
Сажи и другие наполнители подвергаются контрольным
испытаниям для выяснения дисперсности наполнителей, их
усиливающих свойств, чистоты (отсутствие посторонних примесей),
влажности и др. Ускорители и замедлители вулканизации, противо-
старители и другие ингредиенты резиновых смесей поступают на
химические анализы для проверки соответствия их качества
нормам технических условий и ГОСТ.
Резиновые смеси, пропитанные и обрезиненные ткани,
профилированные и другие заготовки проверяются по габаритам,
калибрам, весу, однородности и т. д.
Широкая система контроля качества готовых резиновых
изделий включает следующие виды испытаний:
1) лабораторные испытания образцов резин, тканей или
конструкционных элементов готовых изделий. При этом
проверяются, например, физико-механические свойства резин, включая
ударную и усталостную прочность, морозо- и теплостойкость,
стойкость к старению и др.;
2) станочные или стендовые испытания изделий как для
определения их общей долговечности (например, усталостной
прочности в условиях станочных испытаний), так и какого-либо
специального свойства (монолитности конструкции, сопротивления
истиранию и др.);
3) ускоренные эксплуатационные испытания при
форсируемых и строго контролируемых условиях нагружения, скорости
и т. д. К таким видам контроля качества изделий относятся
ускоренные дорожные испытания автомобильных шин на специальных
экспериментальных автомобилях с балластом (заданной
постоянной нагрузкой), проводимые на автодромах или специально
выбранных дорожных участках;
4) эксплуатационные испытания в обычных условиях работы
изделия (учитываемые пробеги шин в различных автомобильных
хозяйствах, проверка работы транспортерных лент на угольных
или рудных месторождениях и на заводах, опытная носка
резиновой обуви, проверка качества буровых рукавов,
используемых в горно-геологической разведке и т. п.).
Условия испытаний должны в известной мере воспроизводить
практические условия эксплуатации изделий на практике. Обычно
испытания проводятся в более жестких условиях, чтобы ускорить
разрушение образца или изделия и оценить его
эксплуатационную годность. Однако при испытаниях не всегда удается точно
установить соотношение сроков работоспособности изделий в ла-
524
бораторных и эксплуатационных условиях. Эксплуатационная
проверка изделий также имеет ряд недостатков, поскольку в
практических условиях на работоспособности изделий могут
отрицательно сказаться случайные факторы, которые приведут к
искажению оценки качества резины. Кроме того, эксплуатационная
проверка в обычных условиях очень длительна.
Оценка качества серийной продукции резиновых заводов 'и
опытных образцов изделий проводится комплексно, путем
различных лабораторных и эксплуатационных испытаний.
Глава IX
ПЛАСТИЧЕСКИЕ МАССЫ
Пластические массы все шире используются в качестве
конструкционных и поделочных материалов в различных областях
машиностроения, в приборостроении, электротехнике,
радиотехнике и многих других отраслях промышленности. Сочетание
ряда ценных свойств обусловливает широкое применение
пластических масс в современной технике. В отличие от металлов
пластические массы являются теплоизоляционными материалами,
хорошими диэлектриками, могут быть оптически или
радиопрозрачными, высокоупругими и даже эластичными. Все это совершенно
не свойственно металлам, поэтому пластическая масса стала
неотъемлемой частью любого прибора, аппарата, машины.
Плотность пластических масс не превышает 2 г/см3, они не
подвергаются коррозии, легко формуются в изделия, могут выдерживать
высокие механические нагрузки. Благодаря этому пластмассы
во многих случаях успешно заменяют металлы, особенно цветные
(при изготовлении деталей машин, приборов, аппаратов), а также
легкие сплавы (в производстве обшивок летательных аппаратов,
автомобилей, вагонов, судов или корпусов приборов и аппаратов).
/. СОСТАВ ПЛАСТИЧЕСКИХ МАСС
В большинстве случаев пластические массы представляют
собой сложные смеси, основным и непременным компонентом
которых являются полимеры.
Для производства пластических масс применяют
термопластичные и термореактивные полимеры. Температура перехода
из стеклообразного состояния в высокоэластическое (стр. 376)
для термопластичных полимеров, предназначенных для
изготовления пластмасс, должна быть выше температуры эксплуатации
изделия (температура теплостойкости термопласта). Выбираемые
526
для изготовления пластмасс термореактивные полимеры при
нагревании должны отверждаться в твердые стекловидные
материалы, утрачивающие текучесть после того, как будет отформовано
изделие.
В зависимости от типа выбранного полимера пластические
массы делят также на термопластичные и термореактивные. К
числу термопластичных пластических масс относятся материалы,
основным компонентом которых являются полимеры с линейным
строением макромолекул, сохраняющие это строение (и
следовательно, способность вновь переходить в пластическое состояние)
при повышенной температуре и после того, как из них будет
изготовлено изделие. Основным компонентом термореактивных
пластических масс являются термореактивный самоотверждаю-
щийся полимер либо смесь полимера линейного строения и от-
вердителя, при определенных условиях вступающих между собой
в реакцию отверждения.
В процессе изготовления изделия из термореактивной
пластической массы отверждение полимера, входящего в ее состав,
происходит непосредственно после придания материалу требуемой
формы. При этом материал утрачивает способность вновь
переходить в пластическое состояние при повышении температуры,
т. е. становится термостабильным.
Кроме полимера или смеси полимера и отвердителя, в состав
пластических масс входят наполнители, пластификаторы, эласти-
фикаторы, ускорители отверждения, а иногда ингибиторы
отверждения, смазывающие вещества, красители, замутнители, газо-
образователи.
Наполнители в большинстве случаев применяют в сочетании
с полимерами, отверждающимися в процессе формования изделий.
Назначение наполнителей весьма разнообразно. Процесс
отверждения любого термореактивного полимера сопровождается его
значительной уовдкой, затрудняющей изготовление неперенапря-
женных изделий с заданными размерами. Добавление какого-
либо инертного порошкообразного вещества к термореактивному
полимеру позволяет в значительной мере уменьшить усадки при
формовании изделий, повысить их качество и точность
изготовления. Количество порошкообразного наполнителя достигает
40—50%, поэтому его присутствие в определенной степени влияет
на физико-механические свойства изделия. Однако такой
наполнитель не ухудшает текучести материала и смесь вполне
пригодна для формования из нее изделий сложной конфигурации.
К числу широко применяемых порошкообразных наполнителей
пластических масс относится древесная мука (высушенные и
дополнительно измельченные древесные опилки), асбестовый
порошок, кварцевая мука и др. Древесную муку можно пропитать
раствором или расплавом термореактивной смолы, это придает
изделиям определенную монолитность. Присутствие древесной муки
527
не снижает прочностных свойств, присущих выоранному полимеру,
но заметно уменьшает термостойкость материала и ухудшает
диэлектрические характеристики изделий. Асбестовый порошок
вводят в тех случаях, когда в готовом изделии необходимо
сохранить повышенную термостойкость, характерную для выбранного
отвержденного полимера.
При введении в состав пластмассы кварцевой муки изделия не
только сохраняют термостойкость, свойственную отвержденному
полимеру, но и улучшаются их диэлектрические характеристики.
Однако присутствие кварцевой муки не только повышает
хрупкость пластмассовых изделий, но и снижает их прочность при
статических нагрузках. К тому же кварцевая мука очень тверда
и вызывает быстрый износ прессформ, в которых производится
изготовление изделий.
При замене порошкообразного наполнителя волокнистым
прочность изделий в условиях динамических нагрузок (а при
ориентированном расположении волокон и в условиях статических
нагрузок) значительно возрастает. В этих случаях вместо
древесной муки можно использовать хлопковые очесы, а вместо
асбестового или кварцевого порошка—асбестовое, стеклянное,
кварцевое, графитированное волокно. Применение волокнистого
наполнителя (особенно асбестового или хлопкового) затрудняет
формование изделий сложной конфигурации и мелких деталей, в которых
должно быть запрессовано большое количество тонкой
металлической армировки. Этот дефект в меньшей степени проявляется
когда формуемая масса содержит стеклянное волокно.
Обладающее высокой хрупкостью стеклянное волокно частично
разрушается, когда масса под давлением заполняет формы сложной
конфигурации, и волокно вместе со смолой обтекает арматуру.
При изготовлении изделий, которые должны обладать высокой
механической прочностью при статических и динамических
нагрузках, в качестве наполнителей применяют листовые
материалы,—бумагу, хлопчатобумажную, асбестовую или стеклянную
ткани, древесный шпон. Применение таких наполнителей часто
вынуждает использовать более сложные способы производства
изделий по сравнению со способами переработки пластмасс,
содержащих волокнистые, а тем более порошкообразные
наполнители. В зависимости от применяемого наполнителя пластические
массы разделяют на пресспорошки, волокниты и слоистые
материалы (слоистые пластики).
Не меньшее значение в качестве компонента пластических
масс имеют пластификаторы и эластификаторы. Для формования
изделий из термопластичных материалов наиболее пригоден
метод литья под давлением. Однако этот метод может применяться
лишь для материалов, обладающих высокой текучестью при
повышенной температуре и выдерживающих длительное время
действие температуры формования без термической или окислительной
528
деструкции. Совокупностью этих свойств наделены лишь
немногие термопластичные полимеры. Назначением пластификатора
является повышение текучести термопластичного полимера в
размягченном состоянии и снижение температуры перехода
полимера в вязкотекучее состояние, что позволяет снизить
температуру формования, предупреждая этим его
термоокислительную деструкцию.
Пластификаторами служат высококипящие вязкие жидкости,
например сложные эфиры фталевой и себациновой кислот,
растворимые в полимере, а также легкоплавкие синтетические во-
скоподобные вещества, хорошо совмещающиеся с полимером.
В присутствии пластифицирующих добавок облегчается
скольжение макромолекул размягченного полимера друг относительно
друга, т. е. повышается текучесть материала. Пластификатор
должен оставаться и в готовых изделиях, благодаря чему
повышается их упругость, эластичность и морозостойкость, но
снижается теплостойкость и ухудшаются диэлектрические характеристики,
увеличивается коэффициент объемного термического расширения
и возрастает ползучесть (хладотекучесть) материала под
нагрузкой. Жидкие пластификторы постепенно улетучиваются из
изделий, что вызывает их коробление и изменение
физико-механических свойств (старение пластифицированных полимеров).
Поэтому в производстве пластических масс стремятся использовать
воскоподобные пластификаторы. Количество пластификатора,
вводимого в состав термопластичного полимера, можно
варьировать в широких пределах в зависимости от требований, которые
предъявляются к готовым изделиям.
Термореактивные пластические массы обладают достаточно
высокой текучестью, которую к тому же легко регулировать,
останавливая реакцию образования полимера на стадии, наиболее
удобной для его применения в последующих процессах
приготовления пластической массы и формования изделий. Поэтому
термореактивные пластические массы не нуждаются в
пластифицировании.
Однако отвержденные термореактивные полимеры
характеризуются высокой хрупкостью, которую не удается устранить в
изделиях сложной конфигурации введением порошкообразного
наполнителя. В этих случаях используют эластификатор,
назначением которого является повышение ударопрочное™ изделий.
В качестве эластификаторов для термореактивных смесей
служат термопластичные полимеры или каучуки, хорошо
совмещающиеся с термореактивным полимером.
Присутствие термопластичного полимера в составе
термостабильного материала снижает теплостойкость последнего и
несколько повышает его ползучесть под нагрузкой. В связи с этим
количество добавляемого термопласта не превышает 5—20% от
количества термореактивного полимера.
34—805 529
Введение эластификатора придает термореактивным пласі-
массам повышенную ударопрочность. В состав некоторых
термореактивных пластических масс вводят также ингибиторы
процесса отверждения полимера, что позволяет удлинить сроки
хранения приготовленной пластической массы до ее формования
в изделия. В качестве ингибиторов применяют вещества,
замедляющие процесс отверждения пластической массы при
комнатной температуре (т. е. в условиях ее хранения) и разрушающиеся
с повышением температуры в процессе формования изделий.
В медленно отверждающиеся пластические массы и в
пластические массы, рассчитанные на кратковременное хранение до
формования их в изделия, вводят ускорители процесса
отверждения. Ускорителями отверждения феноло-формальдегидных
полимеров являются минеральные и органические кислоты;
отверждение меламино- и карбамидо-формальдегидных полимеров
ускоряется в присутствии органических кислот и солей сильных
минеральных кислот; в качестве ускорителей отверждения
ненасыщенных полиэфиров применяются перекиси или гидроперекиси,
часто в сочетании с восстановителями.
Термопластичные полимеры приобретают густосетчатую
структуру при взаимодействии с некоторыми полифункциональными
соединениями, называемыми в производстве пластических масс
отвердителями. Для низкомолекулярных
феноло-формальдегидных полимеров типа новолаков отвердителем служит гексамети-
лентетрамин, для полиэпоксидов—феноло-формальдегидные
полимеры резольного типа и лишь в отдельных случаях полиамины
(полиэтиленполиамины), для полисилоксанов в зависимости от
их строения—перекиси или тетраэтоксисилан; для линейных
ненасыщенных полиэфиров (полималеинатов)—ненасыщенные
мономеры (стирол, винилацетат, диаллилфталат).
В состав порошкообразных пластических масс обычно вводят
1—2% смазывающего вещества (парафина или стеарина). При
формовании изделий в нагретых металлических формах
легкоплавкий и высокотекучий парафин или стеарин выплавляется из
материала, образуя сплошную пленку между металлической формой
и поверхностью формуемого изделия, что облегчает удаление
изделий из формы.
Если приготовляемая композиция предназначена для
изготовления пенопластичного материала, то в состав ее добавляют
газообразователь (порофор). Порофоры представляют собой
порошкообразные вещества, разлагающиеся при повышенной
температуре с образованием большого количества газов (N2 или СО2).
Для придания пластмассовым изделиям требуемых
декоративных эффектов в исходные смеси вводят красители минерального
или органического происхождения. Большинство органических
красителей растворяется в полимере, и он, окрашиваясь,
сохраняет оптическую прозрачность. Если по условиям работы изделия
530
его прозрачность нежелательна, в исходную смесь вводят
небольшое количество замутнителя—порошка, не совмещающегося с
полимером.
2. ТЕРМОПЛАСТИЧНЫЕ ПЛАСТИЧЕСКИЕ МАССЫ
К числу термопластичных полимеров, применяемых в
производстве пластических масс, относятся: изготовляемые методом
полимеризации полиэтилен, полипропилен, полистирол, поли-
винилхлорид, политетрафторэтилен и политрифторхлорэтилен,
полиформальдегид, полиметилметакрилат и некоторые их
сополимеры и блоксополимеры; полиамиды, поликарбонаты,
получаемые в процессе поликонденсации, и полимеры, получаемые поли-
мераналогичным превращением целлюлозы.
Термопластичные пластические массы применяются для
изготовления деталей приборов и машин, тары, изделий бытового
назначения, облицовочных и декоративных листовых материалов,
химически стойких труб, электроизоляции, деталей остекления
(например, самолетов), пенопластичных материалов.
МЕТОДЫ ПЕРЕРАБОТКИ ТЕРМОПЛАСТОВ
В зависимости от выбранного метода переработки пластические
массы предварительно подготовляют в виде гранул, таблеток или
заготовок, в форме листов или плит различных размеров и
толщины, в виде труб и стержней различного диаметра.
Переработка пластической массы в изделия производится методом
литья под давлением
(литьевые массы),
реже—прессованием, штамповкой, вакуум- или
пневмовытяжкой листовых ма- 4
териалов, в некоторых
случаях — механической
обработкой (резанием, сверлением,
фрезерованием листов, плит,
труб, стержней). Механическую
обработку целесообразно
применять только при изготовлении
небольшого числа изделий
данного типа.
Прессование изделий осуществляют в металлических пресс-
формах на гидравлических прессах. Прессформа (рис. 152)
состоит из двух основных частей—матрицы 2, расположенной
на нижней плите пресса, и пуансона 1, укрепленного на верхней
подвижной плите пресса. Прессуемый материал 3 загружают в
гнезда матрицы (формующие полости), очертания которых соот-
Рис. 152. Конструкция прессформы:
/—пуансон; 2—матрица; 3—формуемый
материал; 4—направляющие колонки;
5—выталкиватель.
34*
531
ветствуют конфигурации изделии, давление на материал ш
подвижной плиты пресса передается пуансоном. Для равномерной
посадки гнезд пуансона в гнезда матрицы при опускании верхней
плиты пресса пуансон снабжается направляющими колонками 4,
которые первыми входят в пазы, имеющиеся в обойме матрицы, что
предотвращает возможность перекосов при посадке пуансона
в матрицу и смятия формующих гнезд. В обоймах матрицы и
пуансона предусмотрены каналы для обогрева прессформы паром или
электрическим током и каналы для охлаждения водой.
Термопластичный материал нагревается в прессформе до
перехода в текучее состояние, под давлением, создаваемым прессом,
распределяется в формующей полости и уплотняется в ней.
Полное смыкание прессформы происходит в момент окончательного
оформления изделия. Не снижая давления, охлаждают пресс-
форму и находящийся в ней материал, затем прессформу
открывают,! поднимая верхнюю плиту пресса вместе с пуансоном. При
этом изделие остается в гнезде матрицы. Далее с помощью
выталкивателя 5 изделие поднимают над гнездом. В гнездах матрицы
и пуансона на направляющих шпильках можно укрепить
металлическую арматуру, которая обволакивается термопластичным
материалом и надежно запрессовывается в изделие.
В местах стыка пуансона и матрицы и в местах подхода
выталкивателя к гнезду на поверхности изделия остается тонкая пленка
вытекшего материала. Эту пленку (заусенец) снимают с изделия
на станках механической обработки. Гнезда матрицы и пуансона
тщательно полируют и хромируют, чтобы изделие, извлекаемое
из прессформы, имело ровную и глянцевую (словно лакированную)
поверхность.
Для сокращения цикла прессования изделий из термопласта
таблетку материала подогревают в термошкафах до пластичного
состояния, а затем, повышая давление, прессуют изделия в пресс-
формах, нагретых до температуры, несколько выше температуры
стеклования полимера. В этих условиях размягченный материал,
не успевая охладиться о стенки прессформы, заполняет полость
гнезда. В то же время на охлаждение пластической массы до
стеклообразного состояния и последующее нагревание прессформы для
нового цикла прессования требуется меньшее время. Этот метод
назван ударным прессованием термопластов.
На рис. 153 приведен общий вид гидравлического пресса,
применяемого для прессования изделий из пластических масс.
Гидравлический пресс приводится в действие давлением
жидкости (обычно воды или масла), подаваемой в рабочий цилиндр /
пресса. Под давлением жидкости верхняя подвижная плита
пресса 2 вместе с плунжером продвигается по направляющим
колоннам 3 и опускает пуансон формы на матрицу, установленную на
неподвижной нижней плите 4 пресса. При размыкании пресса
жидкость из рабочего цилиндра передается в выталкивающий ци-
532
линдр, установленный под нижней плитой, и поднимает
выталкивающий плунжер. Последний в свою очередь поднимает
выталкиватели, расположенные в матрицах формы.
Достоинством формования изделий прессованием является
возможность использования термопластичных материалов, не
обладающих высокой текучестью, или материалов,
выдерживающих лишь кратковременное действие повышенной температуры.
Однако в связи с необходимостью последующих операций
охлаждения и нагревания массивных пресс-
форм цикл формования изделий
удлиняется. Переход на способ ударного
прессования сокращает затраты
времени на изготовление изделия, но
продвижение материала, еще не
обладающего достаточной текучестью и
находящегося под давлением пуансона, может
вызвать смещение или поломку тонкой
металлической арматуры, которая
должна быть запрессована в изделие, и
плохо заполняет формы сложной
конфигурации. Все эти недостатки
существенно ограничивают применение
способа ударного прессования.
Литье под давлением. При
формовании изделий из термопластичных
материалов наиболее предпочтителен метод
литья под давлением.
На рис. 154 изображена литьевая
машина, в которой проводится этот
процесс, на рис. 155—схема ее работы.
В бункер 4 загружают гранулирован-, Рис 153. Общий вид гидрав-
ный материал. Через дозирующее
устройство 3 при движении плунжера 2
влево материал подается в отверстие
цилиндра /. При движении поршня
вправо подача прекращается и
материал уплотняется и проталкивается в
обогреваемую часть цилиндра к соплу 7. Нагретый до пластичного
состояния материал из сопла передается по каналам в формующие
полости прессформы (пуансон //и матрицу 13), которые в это
время сомкнуты. Заполняя формующие полости, разогретый
материал охлаждается при соприкосновении с холодными стенками
прессформы. При обратном движении плунжера форма
открывается и с помощью толкателя 12 изделия поднимаются над
гнездами.
Метод литья под давлением позволяет тщательно прогреть
материал до вязкотекучего состояния, пока он медленно дви-
лического пресса:
1—рабочий цилиндр пресса;
2—верхняя подвижная плита пресса;
3—направляющие колонны; 4—нижняя
неподвижная плита пресса.
5 33
Рис. 154. Общий вид горизонтальной автоматической литьевой машины.
жется в цилиндре и в узком сопле (этим создаются условия для
формования изделий сложной конфигурации и предотвращается
возможность смещения или повреждения арматуры), а затем быстро
охладить его в холодной форме.
Рис. 155. Схема работы литьевой
Цикл формования изделий в литьевой машине продолжается
всего несколько десятков секунд, температурный режим
цилиндра машины и прессформы постоянный, процесс формования
полностью автоматизирован. Однако при литье под давлением
необходимы высокая пластичность материала в нагретом состоянии,
широкий интервал между температурами перехода материала в
вязкотекучее состояние и начала его термической деструкции.
высокая упругость в охлажденном состоянии и сравнительно ма-
534
лая усадка при переходе из пластичного в твердое состояние.
Два последних требования особенно важны при формовании
крупных или толстостенных изделий. При интенсивном охлаждении
термопластов, представляющих собой малотеплопроводные
материалы, прежде всего охлаждаются наружные слои стенок изделия,
а внутри материал еще остается горячим и оказывает давление
на наружные сжавшиеся при охлаждении слои стенок, вызывая
появление мелких трещин иа поверхности изделия. Эти
недостатки литья под давлением крупных изделий усугубляются также
тем, что давление на термопласт, находящийся в форме,
передается только через материал, заполняющий цилиндр, сопло и
литниковые каналы формы, поэтому усадочные раковины,
возникающие при охлаждении изделия, не заполняются новыми
порциями материала.
Вода
Рис. 156. Схема экструдера:
/—шнек; 2—загрузочный бункер; 3—сопло; 4—оформляющая головка; 5—обогревающая
рубашка; fi—охлаждающая полость.
При изготовлении непрерывным способом профильных
деталей, трубок, стержней в конструкцию литьевой машины вводят
некоторые изменения (рис. 156). Материальный плунжер
заменяют вращающимся шнеком /, который непрерывно подает
материал от загрузочного бункера к соплу 3. Вместо формы к
соплу машины прикрепляют оформляющую головку 4, придающую
непрерывно выдавливаемому материалу требуемую форму (на
рисунке—трубы). Материал, перемещаясь по цилиндру,
нагревается теплоносителем, циркулирующим в рубашке 5, до текучего
состояния и обтекает формующую головку.
Во внутреннюю полость оформляющей головки подается
воздух, давлением которого труба, выходящая из машины,
увеличивается в диаметре и, касаясь стенок охлаждающей полости 6
машины, фиксирует приобретенную форму. Непрерывный способ
изготовления профильных изделий называют экструзионным
формованием.
Штампование. Этот метод используют для формования
изделий из листовых термопластичных материалов. Лист / (рис. 157)
нагревают до Температуры высокоэластического состояния и
укладывают на матрицу 2 штампа, установленного на неподвижной
плите пресса. На матрицу опускают пуансон 3 и выдерживают
535
¦з
штамп в сомкнутом состоянии до тех пор, пока деформированный
лист не охладится и не зафиксируется принятая им форма.
При пневматическом формовании жесткий пуансон заменяют
сжатым воздухом, под давлением которого нагретый лист
термопласта прогибается и принимает
форму, соответствующую
внутреннему контуру матрицы. Штамп
для пневматического формования
закрывается крышкой с
отверстием для подачи сжатого
воздуха.
При формовании некоторых
листовых термопластов, которые
в нагретом состоянии
приобретают повышенную липкость,
следует избегать их
соприкосновения с поверхностями матрицы и
пуансона. В этом случае
прибегают к вакуум-формованию, схема
которого приведена на рис. 158.
Лист термопласта /, нагретого
в термошкафу до температуры высокоэластического состояния,
укладывают на протяжную рамку 2 и закрепляют на ней
прижимным кольцом 3 и зажимами 4. Протяжная рамка опирается на
777
Рис. 157. Схема штамповки
листовых термопластов:
f—листовой термопласт; 2—матрица;
3—пуансон.
v}//t)/nhtii/ / г /1 /1/ /і}// п / / / / / //////t/і//і////)}/!/А Г
Рис. 158. Схема установки для вакуумного формования
листовых термопластов:
f—лист термопласта; 2—протяжная рамка; 3— прижимное кольцо; 4—зажимы,
5—цилиндр; 6—вакуум-насос; 7—указатель глубины вытяжки.
цилиндр 5, присоединенный к вакуум-насосу 6. После
закрепления нагретого листа из цилиндра 5 откачивают воздух. Под
давлением наружного воздуха нагретый лист втягивается внутрь
рамки 2 до тех пор, пока его нижняя точка не коснется
указателя 7 глубины вытяжки.
Детали, выполненные из термопластичных материалов,
собирают, применяя механическое крепление (шпильки, заклепки,
536
Рис. 159. Сварка с применением
присадочного материала:
/—свариваемые детали; 2—присадочный
материал (пруток); Л—подача горячего воздуха.
винты, болты), сваривая или склеивая эти детали. Крепежную
металлическую арматуру целесообразно заранее вводить в детали
во время их изготовления литьем под давлением или
прессованием. При затяжке болтов и
установке крепежной арматуры
необходимо учитывать
большое различие коэффициентов
теплового расширения металла
и термопластов.
Склеивание
термопластичных деталей производят в том
случае, когда полимер
(полиэтилен, полипропилен,
фторопласты) при обычных
температурах не растворяется в
общедоступных растворителях.
Однако этот способ соединения
наименее надежен и
применяется лишь в редких
случаях. Наибольшее
распространение приобрел метод сварки
деталей. Этот метод основан на том, что в определенных
условиях макромолекулы поверхностных слоев соприкасающихся
деталей, приобретая повышенную
подвижность, диффундируют в этих
слоях, способствуя сращиванию
материала в единое целое. Чтобы повысить
подвижность макромолекул
термопластичного полимера, его нагревают
выше температуры стеклования (в случае
аморфного полимера) или выше
температуры плавления (в случае
кристаллического полимера). Макромолекулы
термопластичного полимера
приобретают подвижность и при набухании
аморфного материала, что достигается
смачиванием растворителем соединяемых
поверхностей,
Сварку производят с присадочным
Давление прижима
материалом или без него в
зависимоРис. 160. Сварка без
присадочного материала с
электронагревом: сти от степени пластичности, или на-
/—свариваемые вгтык листы термо- буХанИЯ СОЄДИНЯЄМЬІХ ТерМОШШСТОВ,
а^ижимнте™пТрРи°споГ«ллени"; или от качества обработки
соединяемых поверхностей. При тепловой
сварке присадочным материалом обычно служит тот же полимер
(часто пластифицированный для ускорения его диффузии в
свариваемые поверхности), вытянутый в форме прутка (рис. 159).
537
При сварке набухающего полимера на соединяемые
поверхности наносят тонкий слой 2—2,5%-ного раствора этого
полимера в выбранном растворителе. Это ускоряет диффузию
макромолекул на этих поверхностях и облегчает прочное соединение
деталей, в которых трудно обеспечить надежный контакт по всей
плоскости соединения.
Нагревание свариваемых поверхностей при тепловой сварке
производят с помощью электронагревателя (рис. 160), токов
высокой частоты или ультразвука.
СВОЙСТВА ТЕРМОПЛАСТОВ
К числу термопластичных материалов, пригодных для
переработки методом литья под давлением и экструзионного
формования, относятся: полиэтилен, полипропилен, полистирол и его
сополимеры, пластифицированный поливинилхлорид (пластикат),
политрифторхлорэтилен (фторопласт-3), полиформальдегид,
полиамиды, поликарбонат, этролы.
Политрифторхлорэтилен, поступающий на переработку в виде
белого порошка или белых матовых гранул, не содержит каких-
либо добавок. Гранулированные полиамиды (полиамид 54,
полиамид 548, полиамид 6, полиамид 66) могут содержать
стабилизаторы, повышающие их стойкость к термоокислительной
деструкции, краситель и замутнитель. В литьевую массу на основе
поликарбоната иногда вводят краситель и замутнитель, материал
поступает на переработку в гранулированном виде.
Полиформальдегид—белый матовый порошок—окрашивают в различные
цвета.
Полиэтилен и полипропилен, выпускаемые в виде порошка или
гранул, являются бесцветными матовыми материалами и легко
окрашиваются в различные цвета.
В состав литьевой массы на основе полиэтилена или
полипропилена кроме красителя входит стабилизатор, снижающий
скорость окислительной деструкции полимеров.
Пластифицированный поливинилхлорид (пластикат)
представляет собой более сложную композицию, в которой кроме
полимера содержатся стабилизатор, снижающий скорость термической
деструкции полимера в процессе формования; пластификатор,
повышающий текучесть полимера в нагретом состоянии и
придающий ему эластичность после охлаждения изделий, а также
краситель и в некоторых случаях—замутнитель. Пластикат
изготавливают в виде порошка.
Литьевые массы, получаемые из ацетата целлюлозы и этил-
целлюлозы, называют этролами. Для придания эфирам
целлюлозы термопластичности их смешивают с жидким
пластификатором, затем в пластифицированную массу вводят краситель,
замутнитель и небольшое количество A0—15%) хлопковых очесов
538
для повышения механической прочности материала. Этролы
выпускают в гранулированном виде.
Наиболее высокой текучестью в размягченном состоянии
обладают полиэтилен, полипропилен, полиамиды; текучесть
пластиката и этролов зависит от количества содержащегося в них
пластификатора. Наименьшая текучесть характерна для
фторопласта-3.
Температура материала на выходе его из цилиндра литьевой
машины или экструдера определяется температурой перехода
термопласта в вязкотекучее состояние. Например,
полиэтиленовая литьевая масса, в состав которой входит полиэтилен с
молекулярным весом 20 000—30 000, формуется при температуре 130—
140 °С. Если для литьевой массы был использован полиэтилен с
молекулярным весом выше 100 000, температура литья
повышается до 170—280 °С. Фторопласт-3 необходимо нагревать до 320—
330 °С. Все остальные материалы формуют при температуре
150—230 °С. Температуру литьевой формы поддерживают равной
25—60 °С.
Давление на материал в цилиндре литьевой машины должно
быть в пределах 800—2000 кгс/см2 в зависимости от сложности
изделий. Исключение составляют полиэтилен и полипропилен,
формуемые под давлением 70—200 кгс/см2, и полиамиды, которые
формуют под давлением 150—800 кгс/см2. Длительность выдержки
изделия в форме под давлением составляет 10—180 сек, в
зависимости от типа материала и формы изделий.
Свойства изделий из литьевых масс приведены в табл. 24.
Наиболее низкой плотностью обладают изделия из
полипропилена и полиэтилена, наиболее высокой—изделия из фтороплас-
та-3. Высокая эластичность в сочетании с морозостойкостью
характерна для изделий из пластиката: шлангов, пленок, трубок,
электроизоляционных оболочек проводов, уплотнительных колец
и прокладок, защитных пленок, заменителей кожи. Менее
эластичен полиэтилен, из которого помимо перечисленных изделий
(за исключением заменителей кожи) изготовляют тару различных
объемов, химическую посуду, детали приборов. Высокой
упругостью отличаются изделия из полиамидов, фторопласта-3 и
особенно из поликарбоната. Наименее упруги изделия из
полистирола. Изделия из полиамидов и полиформальдегида отличаются
высокой стойкостью к истиранию и низким коэффициентом
трения (особенно по стальным поверхностям), поэтому полиамиды
и полиформальдегид рекомендуется использовать для
изготовления деталей машин, подвергающихся трению скольжения
(подшипники, вкладыши, зубчатые передачи, шестерни). Этролы
применяют для изготовления рукояток, кнопок, рулей
управления, деталей корпусов приборов. Изделия из поликарбоната и
полиформальдегида имеют наиболее высокую прочность и
наименьшую ползучесть под нагрузкой при нагревании до 90—100 °С,
539
Физико-механическне свойства
Показатели
Полимеры, составляющие
полиэтилен
полипропилен
полистирол
Плотность, г/'см3
Теплостойкость, °С
Морозостойкость, °С
Температура размягчения, °С
Удельная ударная вязкость, кгс-см/см2
Предел прочности, кгс/см2
при растяжении
» изгибе
» сжатии
Твердость по Бринеллю, кгс/мм2 . . .
Относительное удлинение при разрыве,
%
Модуль упругости при растяжении,
кгс/см*
Температура разложения, °С
Влагопоглощение за 24 ч, % ....
Горючесть
0,92—0,93
60
-60
110—135
0,90
65
-10
160—170
Не разрушаются*
100—300
120—170
4,3—5,2
16—150
900—1050
0
Горит
і
330—350
700-900
600—700
400—800
0
Горит
1,1
80
-60
12—18
350-400
800—900
1000
15-20
0,4—0,7
14000—32000
260—300
0,03
Горит
* В стандартных условиях испытания.
поэтому поликарбонат и полиформальдегид используют
преимущественно для изготовления деталей машин общего назначения.
При низкой температуре длительнее других полимеров
сохраняет свои упругие свойства фторопласт-3, не утрачивая их
даже при температуре —150 °С. Самой низкой морозостойкостью
из перечисленных термопластов обладают полипропилен и
полиамиды. Ползучесть изделий из полиэтилена становится
заметной при 60 °С, из полистирола, полиамидов, фторопласта-3—при
70—80 "С. Наибольшей теплостойкостью (способностью
сохранять форму при одновременном действии повышенной
температуры и нагрузки) обладают полиформальдегид и поликарбонат.
Термическая деструкция пластиката начинается при 145—150 °С,
остальные литьевые массы начинают разрушаться при
температуре выше 200 °С.
Полиэтилен и полипропилен растворяются в ароматических
углеводородах при температуре выше 60 °С и выпадают в осадок
при снижении температуры. Полистирол легко растворим в
бензоле, толуоле, дихлорэтане, полиамиды—в фенолах, в диметил-
формамиде, поликарбонат—в эфирах, кетонах, хлор производных
углеводородов.
Для повышения прочности к ударным нагрузкам и
сопротивления внутренним напряжениям, возникающим при формовании,
540
изделий из литьевых масс
Таблица 24
основу литьевых масс
фторопласт-3
2,14
70
—195
208—210
20—30
350—400
600—800
500—570
10—13
100—200
.
Выше 315
0
Не горит
полиамид 66
1,14
50-55
от —20
до -25
250—260
до 100
700—1000
800—1000
700—1000
5—8
50—200
Выше 280
0,12—0,6
полиформальдегид
1, 475
80
170—180
10—15
700
1000
—
13-75
_
0,40
поликарбонат
1,17—1,22
115—125
180-200
180—200
890
800—1000
9—10
до 85%
22 000
0,16
пластифицированный*!
поливиннл-
хлорид
50—60
от —15
до —50
—
100—180
100—180
—
120—280
_
Выше 145
—
Не горит
пластифицированный
ацетат
целлюлозы
1,32—1,4
40—50
15—35
400—500
700—1000
250—400
3,5-8
—
20000—25000
Выше 200
0,7—0,8
Не горит
полистирол эластифицируют бутадиен-нитрильным каучуком
СКН (стр. 490). Этот процесс осуществляют при совместном
вальцевании полистирола (80—85%) с каучуком B0—15%) при
повышенной температуре. С увеличением содержания каучука
повышается и упругость полистирола, но ухудшаются его
диэлектрические свойства.
Наиболее высокой химической стойкостью обладает
фторопласт-3, который в обычных условиях не разрушается при
действии кислот, щелочей и окислителей. Полиэтилен, полипропилен
и полистирол устойчивы к действию кислот, щелочей, но
разрушаются под влиянием окислителей—кислорода воздуха, озона,
перекисей, азотной кислоты и т. д. Под влиянием кислорода
воздуха изделия из полиэтилена и полипропилена (особенно
тонкостенные) со временем становятся более твердыми, жесткими и
хрупкими. Изделия из полистирола и полиамидов постепенно
желтеют и приобретают хрупкость. Пластикаты разрушаются в
растворах щелочей. Полиамиды нестойки к действию кислот и
кислорода воздуха при повышенной температуре. Этролы
разрушаются в растворах кислот и щелочей. Под влиянием
атмосферных воздействий из пластиката и этролов постепенно удаляется
часть пластификатора и полимеры становятся менее
эластичными.
541
Все литьевые массы (за исключением полиамидов) являются
диэлектриками и применяются не только в качестве механически
прочных, часто химически- или атмосфероустойчивых
материалов, но и используются для изготовления электроизоляционных
деталей электро- и радиоаппаратуры, эластичных
электроизоляционных пленок или лаков.
Наилучшими диэлектрическими свойствами обладают
изделия из фторопласта-3, полиэтилена и полистирола.
Полистирол прозрачен и бесцветен, его светопрозрачность
выше светопрозрачности обычных минеральных стекол. К тому
же полистирол много легче минерального стекла, легко
обрабатывается на станках, шлифуется, полируется и более упруг, чем
минеральное стекло. Поэтому полистирол находит широкое
применение в производстве небольших оптических линз для
приборостроения. Полистирольные линзы, защищенные от прямого
действия ультрафиолетовых лучей, желтеют медленно и могут
использоваться в течение длительного времени.
К термопластам, перерабатываемым в изделия
преимущественно методом прессования, относятся винипласт и фторопласт-4.
Винипласт представляет собой непластифицированный
порошкообразный поливинилхлорид с добавкой 3—6% стабилизатора.
При температуре выше 170 °С поливинилхлорид начинает
приобретать текучесть, достаточную для формования сравнительно
простых изделий методом прессования. Однако уже при этой
температуре заметно возрастает скорость его термической деструкции.
Продуктом начальной стадии деструкции зтого полимера
является хлористый водород, который, накапливаясь в материале,
становится катализатором дальнейшего процесса разрушения.
Чтобы из поливинилхлорида сформовать изделие, необходимо на 60—
90 мин замедлить деструкцию поливинилхлорида путем
непрерывного связывания выделяющегося хлористого водорода. Это
достигается введением стабилизатора (кальциевые или свинцовые
соли угольной или стеариновой кислоты).
Винипласт обычно перерабатывают в изделия методом
ударного прессования. Учитывая плохую текучесть материала и
невозможность его длительного прогрева, из порошка прессуют изделия
несложной формы и при последующей механической обработке
придают им требуемую конфигурацию. Большую часть винипласта
изготавливают в виде листов, плит разной толщины и труб
различных диаметров. Такие заготовки получают прогреванием и
сплавлением порошкообразного винипласта на нагретых вальцах,
каландрованием нагретой массы до образования тонкой пленки,
которую складывают в пакеты или наматывают на металлический
стержень. Из таких пакетов прессуют листы, плиты или трубы,
внутренний диаметр которых определяется диаметром
металлического стержня, а толщина—количеством слоев поливинилхло-
ридной пленки. Прессование проводят при 170—190 °С в много-
542
этажных гидравлических прессах, укладывая пакеты между
отполированными нагретыми плитами пресса или помещая
стержни из намотанной пленки в пазы нагретой прессформы.
Длительность процесса формования определяется продолжительностью
действия стабилизатора при указанной температуре.
Отпрессованный винипласт представляет собой твердый
матовый материал, имеющий цвет от светло-желтого до темно-бурого.
Винипласт характеризуется высокой поверхностной твердостью и
упругостью в сочетании с достаточно хорошей механической
прочностью и стойкостью к атмосферным воздействиям, к действию
кислот и растворов солей. Он легко обрабатывается резанием,
детали из винипласта надежно соединяются методом сварки.
Существенным недостатком винипласта является его заметная
ползучесть под нагрузкой, значительно снижающая прочность
этого материала при длительном нагружении. Поэтому изделия из
винипласта приходится изготовлять с двойным запасом прочности.
Ползучесть материала заметно возрастает с повышением
температуры, одновременно уменьшается механическая прочность.
Снижение прочности и увеличение ползучести становятся особенно
заметными при температуре выше 60 °С, вследствие чего
изделия из винипласта не рекомендуется использовать под нагрузкой
при температуре, превышающей 60 °С. Упругость винипласта при
20 °С достаточно высока, но резко уменьшается при температуре
ниже нуля.
Листовой винипласт обладает некоторой пластичностью при
170—190 °С, поэтому из него можно штамповать изделия простой
формы в нагретых металлических штампах.
Сварные конструкции из винипласта находят широкое
применение в качестве кислотоупорных трубопроводов,
аккумуляторных и напорных баков, вентиляторов, насосов, фильтров,
внутренней облицовки гальванических ванн, корпусов приборов, деталей
машин.
Физико-механические свойства изделий из винипласта
приведены в табл. 25.
Вторым термопластичным материалом, который не
перерабатывают методом литья под давлением, является фторопласт-4
(политетрафторэтилен). Он применяется без каких-либо добавок
и выпускается в виде тонкого белого порошка. Фторопласт-4
начинает разрушаться при температуре выше 330 °С, причем
скорость деструкции заметно возрастает с дальнейшим повышением
температуры. При нагревании до 360 °С деструкция полимера
не отражается на его физико-механических свойствах, однако
следует учитывать, что при деструкции отщепляется фтор,
оказывающий вредное действие на организм человека. Одновременно
с появлением признаков деструкции при 330—360 °С полимер
приобретает некоторую пластичность. Нагревать фторопласт-4
до более высокой температуры не рекомендуется, а формо-
543
Таблица 25
Физико-механические свойства изделий из винипласта и фторопласта-4
Показатели
Винипласт
Фторопласт-4
Плотность, г/см3
Теплостойкость, °С
Температура разрушения, °С ....
Морозостойкость, °С
Удельная ударная вязкость, кгс-см/см2
Предел прочности, кгс/см2
при растяжении
» изгибе
» сжатии
Твердость по Бринеллю, кгс/мм- . .
Относительное удлинение при разрыве,!
%'
Влагопоглощение, %
1,38
65
Выше 160
от—20 до —25
120 (при 20°С)
30 (при —20 °С)
550 (при 20 °С)*
750
800
15—16
10—15 (при 20 °С)
140 (при 80 °С)
I
2,1—2,3
60
Выше 330
—250
Более 100
До 250
До 150
3-4
200—250
0,0
* При 80 °С 200 кг
вать изделия из такого малотекучего материала трудно, поэтому
изделия из фторопласта изготовляют преимущественно методом
спекания, и лишь такие несложные тонкостенные изделия, как
прокладки, шайбы, уплотнительные кольца, формуют методом
ударного прессования.
При формовании изделий методом спекания порошкообразный
фторопласт-4 предварительно таблетируют в металлических пресс-
формах под давлением 250—300 кгс/см2 и при комнатной
температуре. Затем таблетки помещают в герметические термошкафы,
снабженные смотровыми окнами и вентиляцией, и выдерживают
при температуре 330—360 °С. В этих условиях зерна
порошкообразного полимера оплавляются и спекаются между собой, в
результате чего образуется монолитный блок полимера. Спекание
считают законченным, когда заготовка (блок) становится
полупрозрачной. Ее извлекают из термошкафа и охлаждают водой, при
этом материал становится матово-белым с воскоподобной
поверхностью. В процессе спекания и последующего охлаждения
заготовка несколько коробится. Детали получают механической
обработкой заготовок. Для получения из фторопласта пленки
цилиндрическую заготовку укрепляют на токарном станке, вращая ее,
срезают непрерывный слой требуемой толщины и полируют пленку на
вальцах, пропуская между нагретыми валками.
Изделия из фторопласта-4 отличаются наиболее высокой
химической и морозостойкостью, хорошими диэлектрическими
свойствами и наиболее низким коэффициентом трения скольжения
по металлическим поверхностям. Фторопласт-4 может работать
в интервале температур от —200 до +250 °С без заметного измене-
544
ния упругости и других физических качеств; выдерживает
длительное действие кислот, щелочей и окислителей при любых
концентрациях и даже при повышенных температурах, заметно не
изменяется под влиянием атмосферных воздействий и
ультрафиолетового облучения. Однако механическая прочность фторопласта-4
невелика (см. табл. 25), при нагрузках, намного меньших
предела прочности, в нем появляется высокая ползучесть. Фторо-
пласт-4 находит широкое применение как диэлектрик, не
меняющий своих характеристик в широком диапазоне частот
переменного тока и температур. Из фторопласта-4 изготавливают также
уплотнительные прокладки, сальники, трубопроводы, надежно
работающие в любых агрессивных средах и в широком диапазоне
температур. Для изготовления шлангов, труб, герметизирующих
профилей, нанесения электроизоляции используют
политетрафторэтилен марки фторопласт-4Д. Этот полимер имеет
сферическую форму частиц в отличие от фторопласта-4, частицы которого
имеют волокнистое строение. Фторопласт-4Д дает устойчивые
суспензии во многих средах, что позволяет изготавливать из него
пасты, содержащие небольшое количество жидкой фазы. Пасты
легко формуются в изделия литьем под давлением или экструзион-
ным способом. Изделия поступают в термокамеры для удаления
из них жидкой фазы и спекания полимера в монолитную массу.
ТЕРМОПЛАСТИЧНЫЕ СТЕКЛА
Высокой оптической прозрачностью обладают три типа
термопластичных материалов: полистирол, целлулоид и полиметил-
метакрилат. Полистирол легко перерабатывается в изделия
методом литья под давлением, поэтому, кроме других направлений
использования этого полимера, он применяется и для изготовления
небольших оптических линз. Попытки наладить производство
крупных прозрачных изделий из полистирола литьем под
давлением в большинстве случаев не увенчались успехом, поскольку
при формовании таких изделий в полимере возрастают
внутренние напряжения, что приводит к появлению в изделиях мелких
трещин.
В качестве деталей остекления, получаемых блочной
полимеризацией в формах, полистирол существенно уступает полиметил-
метакрилату по показателям прочности (особенно к ударным
нагрузкам), теплостойкости, твердости и атмосфероустойчивости.
Исходным полимером для изготовления целлулоида служит
нитрат целлюлозы (стр. 433). Для придания ему
термопластичности и монолитности полимер растворяют в этиловом спирте и
смешивают со спиртовым раствором пластификатора (камфоры).
Из образующейся однородной бесцветной высоковязкой массы
при вальцевании (температура вальцов 65—70 °С) удаляют
большую часть спирта. Твердые листы охлажденного сырого целлу-
35—805 545,
ЛОИДа ^ДО ZUU Kd) ук.'іадоШ<ііиі и и,и»;НлДрПЧ^;,..,^ ...„.ґ...^,„. Z
пресса и выдерживают в течение 10—14 ч под давлением при
температуре 60—65 °С до полного сплавления отдельных листов.
Блок целлулоида, еще содержащий около 20% этилового спирта,
вынимают из блок-пресса, разрезают на листы требуемой толщины
и развешивают в термокамере для полного удаления из
целлулоида этилового спирта. В процессе сушки листы целлулоида
сильно коробятся, поэтому их выпрямляют и одновременно
полируют между нагретыми полированными металлическими плитами
пресса.
Прозрачный целлулоид гораздо легче минерального стекла
(плотность целлулоида 1,4 г/см2) и отличается значительно
большей упругостью и светопрозрачностью. Целлулоид легко
поддается всем видам механической обработки. Отдельные
целлулоидные детали легко соединяются при обработке их поверхности
ацетоном или раствором целлулоида в ацетоне. Нагретые листы
целлулоида можно штамповать, причем штампованные изделия не
имеют заметных оптических искажений.
К недостаткам целлулоида относятся горючесть, низкая
теплостойкость (до 40 °С) и низкая атмосфероустойчивость. Последнее
проявляется в том, что целлулоид постепенно желтеет, причем
скорость пожелтения возрастает под действием прямого солнечного
освещения. Эти отрицательные качества целлулоида ограничивают
области его применения. Целлулоид используется для
изготовления прозрачных деталей приборов (шкалы, смотровые и защитные
стекла, защитные колпаки) и как декоративный материал для их
отделки (ручки, клавиши, облицовочные панели приборов). При
получении декоративного целлулоида в раствор нитрата
целлюлозы и камфоры вводят краситель, а в некоторых случаях и за-
мутнитель.
Более широкое и разнообразное применение в качестве
термопластичного стекла находит полиметилметакрилат, получаемый
методом блочной полимеризации в кюветах из минерального стекла
(стр. 414). Термопластичные стекла, получаемые на основе поли-
метнлметакрилата, называют органическими стеклами. При
изготовлении органического стекла марки СОЛ в исходную смесь
кроме мономера и инициатора процесса полимеризации вводят
6—9% пластификатора. Если полиметилметакрилат
предназначен для остекления сигнальных фонарей, в смесь добавляют
краситель, а при изготовлении органического стекла для
электроосветительной арматуры замутнитель. Органическое стекло на основе
полиметилметакрилата легче и пластичнее целлулоида. Для него
характерна также более высокая прочность при статических
нагрузках (табл. 26).
С понижением температуры до —50 °С полиметилметакрилат
утрачивает упругость и по хрупкости приближается к
минеральному стеклу.
546
Таблица 26
Физико-механнческие свойства органических стекол
Показатели
Целлулоид
1,4
40
100—120
150
450-550
600
—
6
2
61-82
[в зависимости
от толщины)
Органическое
стекло
1,18
80
220—240
600—700
1000
700-850
18
0,3
90—99
Плотность, г/см3
Теплостойкость по Мартенсу ....
Температура начала разрушения, °С .
Удельная ударная.вязкость, кгс-см/см2
Предел прочности, кгс/см2
при растяжении
» изгибе
» сжатии
Твердость по Бринеллю, кгс/мм2 . .
Влагопоглощение, %
Светопрозрачность, %
Многоосной ориентацией полиметилыетакрилата, т. е. вытя^
гиванием его одновременно в нескольких направлениях при 120—
130 °С, удается значительно упорядочить взаимное расположение
макромолекул полимера и заметно увеличить его прочность при
статических и динамических нагрузках. В результате ориентации
возрастает морозостойкость органического стекла, хрупкость его
начинает проявляться лишь при минус 60—минус 65 °С. При
температуре выше 80 СС листы органического стекла утрачивают
твердость и становятся эластичными (наибольшая эластичность
появляется в интервале 120—180 °С). Выше 180 °С начинается
разложение пластификатора, и в листе появляются газовые пузырьки.
Одновременно начинает нарастать пластичность материала, и
листы деформируются. Около 220—240 °С происходит термическая
деструкция полимера до исходного мономера.
Изделия из листового пластифицированного полиметилмет-
акрилата (детали остекления самолетов, автомобилей, вагонов,
телевизионные линзы и т. д.) формуют при температуре 120—
160 °С. В этих условиях пластифицированное органическое
стекло сохраняет высокие показатели оптических свойств. Изделия
сложной конфигурации формуют в штампах; более простые
изготовляют вакуум-вытяжкой с протяжной рамкой (см. рис. 158).
Поверхность органического стекла, нагретого до эластического
состояния, становится мягкой и липкой, поэтому во всех возможных
случаях предпочитают формование изделий вакуум-вытяжкой.
Органическое стекло легко поддается механической обработке
любым способом (резание, сверление, фрезерование, шлифовка,
полировка). Оно надежно сваривается при набухании
соединяемых поверхностей в растворе органического стекла в дихлор-
35*
547
этане или при нагреве. Светопрозрачность органического стекла
выше прозрачности целлулоида и обычного минерального стекла;
оптические искажения невелики, длительное ультрафиолетовое
облучение полиметилметакрилата не вызывает его пожелтения,
а различные атмосферные воздействия не приводят к заметному
изменению свойств материала. При резких сменах температуры,
высоких скоростях резания или тугой затяжке органического
стекла металлическими болтами или металлическим каркасом
на поверхности полимера образуются мелкие трещины вследствие
больших внутренних напряжений. Органическое стекло
растворяется в хлорпроизводных углеводородов, в концентрированной
уксусной кислоте, сложных эфирах, кетонах. Теплостойкость
изделий из органического стекла можно повысить до 110 °С,
удалив из него пластификатор, однако это ухудшает формуемость
полимера, снижает его ударопрочность и морозостойкость.
ТЕРМОПЛАСТИЧНЫЕ ПЕНОПЛАСТЫ
-и
Пенопласта отличаются своеобразной структурой. Они
представляют собой совокупность мельчайших газовых пузырьков
(ячеек) одинакового размера, разделенных тонкими стенками
полимерного материала. Пенопласта с особенно низкой объемной
массой @,1—0,2 г/сма) содержат в единице объема весьма большое
количество газовых ячеек, стенки которых очень тонки.
Естественно, что такие материалы должны обладать низкой
механической прочностью. В нагруженных конструкциях листы
пенопласта упрочняют тонкой обшивкой листами металла или
стеклопластика, которые наклеивают на пенопласт. В этом
случае пенопласт заменяет ребра жесткости, что приводит к резкому
снижению веса конструкций, способствует более равномерному
распределению нагрузки по площади обшивки и ускоряет сборку
изделий. Из пенопластов изготавливают также изделия, для
которых ранее применялся такой дорогой материал, как пробка
(поплавки, спасательные круги и пояса). Будучи
теплоизоляционным материалом, пенопласт заменяет природные
теплоизоляционные материалы (шерсть, мех), применяется для облицовки стенок
хладостатов, в строительстве сборных домов облегченной
конструкции. Изделия из пенопластов отличаются исключительно
высокими диэлектрическими свойствами, прозрачны для
радиоволн, поэтому их используют в производстве антенных
обтекателей, электроизоляционных деталей и пр.
Пенопласта сочетают низкую объемную массу с высокой
эластичностью, поэтому такие материалы применяют вместо
металлических пружин в производстве мягкой мебели или в качестве
амортизаторов деталей приборов.
Пенопласты можно изготовлять из любого термопластичного
полимера. Обычно их получают из полистирола, пластифициро-
548
ванного и непластифицированного поливинилхлорида. В состав
термопластичных материалов, образующих жесткие пенопласты,
входят два компонента—полимер и газообразователь (порофор).
При изготовлении эластичных пенопластов в смесь добавляют
также пластификатор. Полимеры, используемые для
производства пенопластов, получают эмульсионным способом. Исходный
полимер обычно представляет собой порошок, который тщательно
смешивают с небольшим количеством порофора. В качестве поро-
форов применяют органические вещества, преимущественно диазо-
соединения, разлагающиеся при нагревании с выделением
большого количества азота. Температура разложения порофора
должна быть несколько выше температуры начала текучести полимера.
Смешение порошкообразного полимера с порофором
проводится в шаровых мельницах с керамической облицовкой и
керамическими шарами. Применение керамики предотвращает попадание
металлической пудры в термопластичный материал. Соотношение
полимера и порофора в смеси определяется требуемым
количеством ячеек в единице объема пенопласта (его объемной массой)
и количеством азота, образующегося при термическом разложении
порофора. Из приготовленной смеси в герметических прессформах
прессуют плиты или диски при температуре, достаточной для
размягчения полимера и его сплавления в монолитную массу (для
полистирола и поливинилхлорида 140—150 °С). В этих условиях
порофор постепенно разлагается, а выделяющийся азот создает
в прессформе давление, которое компенсируется давлением
пресса B50—300 кгс/см2). При этом азот равномерно распределяется
в полимере.
По окончании процесса, формования заготовки, не снижая
давления, охлаждают прессформу. Охлажденный полимер
становится твердым и упругим материалом, мельчайшие ячейки газа
в нем сжимаются, внутреннее давление в материале исчезает.
После этого заготовку вынимают из формы и проводят
вспенивание. Для этого каждую заготовку помещают в легкие
ограничительные формы, внутренняя полость которых соответствует
конфигурации заготовки, но имеет значительно большие размеры.
Формы устанавливают в термокамеру и нагревают до температуры
перехода полимера в высокоэластическое состояние (для
полистирола и полихлорвинила 95—100 °С). Полимер при нагревании
становится эластичным, и каждый мельчайший пузырек газа,
равномерно расширяясь в объеме, раздвигает стягивающие его стенки
полимера. Объем изделия увеличивается до тех пор, пока
материал не коснется стенок ограничительной формы.
Быстрым охлаждением изделия материал снова переводят в
твердое стекловидное состояние, фиксируя таким образом
приданную ему пористую структуру. Она сохраняется до тех пор,
пока пенопласт не будет нагрет выше температуры
стеклования (для пенополистирола и пенополивинилхлорида до
549
60—70 С). При нагревании выше этой предельной температуры-
полимер становится эластичным, давление газов в нем начинает
превышать давление наружного воздуха и газы диффундируют
из материала. Пенопласт сплющивается и постепенно утрачивает
ячеистую структуру. В зависимости от соотношения полимера и
порофора можно изготовить пенопласты с объемной массой 0,1;
0,2; 0,3 г/см'3. Более легкие пенопласты имеют слишком низкую
прочность. При равной объемной массе пенополистиролы более
прочны, чем пенополивинилхлориды, и обладают лучшими
диэлектрическими свойствами. Однако полистиролы легко
воспламеняются, растворяются в органических растворителях, сильно
набухают в керосине, бензине и смазочных маслах.
Детали из пенопластичных материалов хорошо склеиваются
между собой; их можно приклеивать к деревянным и
металлическим поверхностям и к стеклопластикам при помощи
синтетических клеев, отверждающихся на холоду (стр. 570). В табл. 27
приведены свойства пенополистирола и пенополивинилхлорида.
Таблица 27
Физико-механические свойства термопластичных пе но пластов
Показатели
Объемная масса, г/см3
Теплостойкость, °С
Удельная ударная вязкость, кгс-см/см2
Предел прочности, кгс/см2
при сжатии
» растяжении
» изгибе
Коэффициент теплопроводности,
ккалі(м-ч-град)
Горючесть
Влагопоглощение, кг/см2
Диэлектрическая проницаемость . .
Пенополистирол
0,2
60
1,8
30
42
65
0,044
Горюч
0,06
1,28
Пенополиви-
н илхлорид
0,2
60
1,5
26
45
40
0,045
Не горюч
0,3
2,4
3. ТЕРМОРЕАКТИВНЫЕ ПЛАСТИЧЕСКИЕ МАССЫ
И ИЗДЕЛИЯ ИЗ НИХ
Для изготовления термореактивных пластических масс
применяют феноло-формальдегидные, карбамидные, меламино-форм-
альдегидные полимеры, полисилоксаны, полиэпоксиды и
ненасыщенные полиэфиры.
Отверждение полимеров сопровождается большими усадками,
нарушающими монолитность изделий и создающими внутри них
большие внутренние напряжения, что приводит к разрушению
и растрескиванию таких изделий. Усадки при формовании
термореактивных пластических масс значительно больше, чем при фор-
55 0
мовании изделий из термопластов. Кроме обычных объемных
изменений материала, связанных с переходом его из жидкотеку-
чего в стекловидное состояние, в процессе отверждения возникают
•большие объемные изменения, обусловленные превращением
сравнительно низкомолекулярных полимеров в полимеры
пространственной структуры. Чтобы снизить усадки материала при
формовании изделий, в состав термореактивных пластических масс в
качестве обязательного компонента цводят наполнитель.
Быстрая потеря текучести термореактивными композициями
при повышенных температурах затрудняет формование их методом
литья под давлением, поэтому такие композиции формуют в
изделия или заготовки методом прессования, применяя различные
способы передачи давления на материал. Применение метода
прессования термореактивных материалов связано не только с
необходимостью перевести их сначала в пластическое состояние,
придать им требуемую форму и уплотнить, но и с необходимостью
последующего отверждения материала. Следовательно, в
процессе прессования термореактивных материалов происходит не
только формование изделий, как в случае термопластов, но и
протекают сложные химические реакции. Условия и режимы
прессования должны соответствовать условиям отверждения выбранной
композиции (температура, количество выделяющихся побочных
продуктов, их свойства, скорость отверждения и т. д.).
ТЕРМОРЕАКТИВНЫЕ ПРЕССПОРОШКИ
Для производства пресспорошков применяют феноло-форм-
альдегидные, меламино-формальдегидные, карбамидо-формальде-
гидные и полисилоксановые полимеры. Полимеры, находящиеся
в термореактивной стадии, часто называют смолами, по
отношению к наполнителям смолы являются связующими.
Наибольшее распространение получили пресспорошки на
основе феноло-формальдегидных смол (фенопласты) и их различных
модификаций. Измельченную в порошок феноло-формальдегидную
смолу смешивают с древесной мукой или каким-либо
порошкообразным минеральным веществом. Количество наполнителя в смеси
достигает 45—50%. В смесь добавляют смазку и краситель, а
также гексаметилентетрамин, если в качестве связующего служит
новолачная смола (стр. 392). Смесь приготовляют (рис. 161) в
шаровых мельницах с керамической футеровкой. Крупные куски
смолы измельчают на зубчатой дробилке 1 и ковшовым
элеватором 2 через бункер 3 передают в мельницу тонкого помола 4,
из которой порошок смолы, пройдя бункер 5 и весы 6, передается
на смешение. Наполнитель из бункера 8 поступает на весы 9 и
периодически загружается в смеситель 7. Сюда же вводятся
краситель и смазка. Смесь передается в бункер 10, из которого
небольшими порциями поступает на вальцы 11. В результате кратко-
551
временного вальцевания между валками, нагретыми до 9U—
120 °С, смола размягчается и обволакивает частицы наполнителя.
При этом вальцуемая масса уплотняется, и одновременно
протекает стадия дальнейшей поликонденсации до образования
полимера требуемой текучести; происходит интенсивное удаление
Наполнитель
I?
Воздух
Рис. 161. Схема поточного метода производства пресспорошков:
/, 13— зубчатые дробилки; 2—ковшовый элеватор; 3 5 .*, 10, 17, 19—бункеры; 4, 18—мельницы
тонкого помола; б, 9—автоматические весы; 7—смеситель; //—вальцы; 12—транспортер; /4—шнек;
15— вентиляггор; 16— циклон; 2о— смеситель партий; 21— фасовочная машина.
влаги и других легкоиспаряющихся побочных продуктов.
Вальцованная масса после охлаждения превращается в твердые
хрупкие листы, которые транспортером 12 передаются в зубчатую
дробилку 13. Измельченный материал при помощи шнека 14 и
вентилятора /5 через циклон 16 поступает в бункер 17 и затем в
мельницу 18 тонкого помола. Отсюда через бункер 19 пресспорошок
подается в смеситель 20, в котором смешиваются отдельные
партии вальцованного материала. В последние годы трудоемкую и
малопроизводительную операцию вальцевания во многих случаях
заменяют пластикацией массы червячным шнеком, движущимся
в обогреваемом цилиндре.
В холодных формах таблеточных машин из порошка получают
таблетки различного веса, которые удобнее подогревать и
дозировать в прессформы. Уплотненный в таблетки порошок занимаег
меньший объем в загрузочной камере прессформ. ¦
552
Изделия из феноло-формальдегидных пресспорошков с
древесным наполнителем прессуют при температуре 145—155 °С,
пресспорошки с минеральным наполнителем—при 155—180 °С.
Давление при прессовании достигает 150—350 кгс/см2,
длительность пребывания материала в замкнутой прессформе 0,8—1,5 мин
на 1 мм толщины изготовляемого изделия (считая по наиболее
толстой стенке). Такая длительная технологическая выдержка
необходима для перевода прессматериала в пластическое
состояние, заполнения им прессформы и для последующего
отверждения материала. Для уменьшения технологической выдержки
можно предварительно подогревать таблетки до перехода полимера
в пластическое состояние. Высокое давление прессования
требуется для заполнения сложных контуров прессформы, для большего
уплотнения прессуемого материала и для создания
противодействия давлению выделяющихся газообразных побочных
продуктов. Из пресспорошков на основе феноло-формальдегидных смол
можно прессовать изделия сложных конфигураций с большим
количеством различно расположенной арматуры, отпрессовывать
знаки, отверстия, резьбу, сводя таким образом последующую
механическую обработку деталей лишь к снятию
заусенцев—тонкой смоляной пленки, образующейся в зазоре между пуансоном
и матрицей.
Изделия из пресспорошков отличаются высокой твердостью
(особенно если в качестве наполнителя используют кварцевую
муку) и имеют глянцевую поверхность. Отвержденный материал
нерастворим, поглощает ничтожное количество влаги (до 0,3%
после 2 ч пребывания в воде), устойчив к действию растворов
кислот, но постепенно разрушается под влиянием окислителей
и щелочных растворов.
Феноло-формальдегидные смолы окрашены в темный цвет,
поэтому в пресспорошки можно вводить только черный,
коричневый или темно-красный краситель. В изделиях сохраняется
некоторое количество не вступившего в реакцию фенола, поэтому
из феноло-формальдегидных смол не рекомендуется изготовлять
посуду, детские игрушки, тару для пищевых продуктов и
медикаментов.
Детали из фенопластов, наполненных древесной мукой, могут
длительное время работать под нагрузкой при температуре 100—
ПО °С; если наполнителем является минеральный порошок,
температура эксплуатации может быть повышена до 180—200 °С.
При более высокой температуре наблюдается некоторое
коробление нагруженного изделия и снижение его прочности. Заметная
термическая деструкция начинается при температуре выше 250 °С.
Фенопласты обладают удовлетворительными диэлектрическими
свойствами (особенно если наполнителем служит кварцевая
мука), удачно сочетающимися с высокой теплостойкостью. Для
снижения хрупкости изделия (удельная ударная вязкость 3,5—
553
4,5 кгссм/см'') феноло-формальдегидную смолу перед смешением
с наполнителем эластифицируют поливинилхлоридом,
полиамидом или нитрильным каучуком. Однако введение поливинил-
хлорида и полиамида приводит к снижению теплостойкости
изделий, а в случае применения полиамида и к ухудшению
водостойкости и диэлектрических свойств. При введении каучуков,
вулканизующихся одновременно с отверждением смолы, снижение
теплостойкости проявляется в меньшей степени (стр. 576).
В табл. 28 приведены некоторые характерные свойства изделий
из феноло-формальдегидных и модифицированных феноло-форм-
альдегидных пресспорошков.
Пресспорошки на основе меламино-формальдегидных смол по
технологическим свойствам несколько уступают феноло-форм-
альдегидным. Они содержат большее количество
низкомолекулярных веществ и более гигроскопичны, поэтому при прессовании
выделяется много газообразных веществ, что усложняет процесс
формования изделий.
Таблица 28
Физико-механические свойства изделий из феноло-формальдегидных
пресспорошков
Показатели
Плотность, г/см3 ....
Теплостойкость, °С ...
Удельная ударная
вязкость, кгс-см/см2 . . .
Предел прочности, кгс/см2
при сжатии
» изгибе
Твердость по Бринеллю,
кгс/мм2
Влагопоглощение, % . .
Диэлектрическая
проницаемость
Феноло- форма льдегидная
смола
наполнитель
древесная
мука
1,3 — 1,4
100—110
4,0—4,8
1500—1600
500—650
25—40
0,1—0,3
9,9—30
; минеральный
наполнитель
1,5—1,75
120—150
2,5—4,0
1400
400—500
30—50
0,04—0,1
7-9
Феноло-форм-
альдегидная
смола,
полиамид.
минеральный
наполнитель
1,8—1,9
120
4—6
2000
900
30—40
0,3
4,8
Феноло-форм-
альдегидная
смола,
синтетический
каучук.
минеральный
наполнитель
1,9
140
7-Ю
2000
550—850
25—35
0,01
5,7
Меламино-формальдегидные пресспорошки менее текучи, чем
феноло-формальдегидные, и быстрее утрачивают текучесть при
хранении. Изделия из меламино-формальдегидных пластических
масс безвредны и бесцветны; введением в исходные смеси
красителей изделиям можно придавать любую окраску.
Физико-механические свойства изделий из меламино-формальдегидных
пластмасс мало отличаются от свойств изделий из фенопластов.
554
Карбамидо-формальдегидные смолы имеют сравнительно
низкую термическую стойкость (начало деструкции при 145—150 °С),
поэтому их нецелесообразно совмещать с минеральными
наполнителями. Эти смолы безвредны и бесцветны, легко окрашиваются
в различные цвета. Основное применение пресскомпозиции на
основе карбамидо-формальдегидных смол получили в
производстве деталей приборов и бытовых изделий с яркой декоративной
или опознавательной окраской. Поэтому в качестве
наполнителей рекомендуется использовать дешевые, бесцветные
материалы, хорошо совмещающиеся с карбамидо-формальдегидной
смолой и не усложняющие процесса формования.
Карбамидо-формальдегидная смола в термореактивной стадии
получается в виде водного раствора, полное удаление воды
связано с большими затруднениями, поэтому желательно, чтобы
наполнитель был наделен повышенной гигроскопичностью.
Указанным выше требованиям удовлетворяет древесная
целлюлоза, которую пропитывают водным раствором карбамидо-
формальдегидной смолы и высушивают в вакуум-камерах.
Полученная после сушки твердая и хрупкая масса легко измельчается
в порошок.
Порошкообразная карбамидо-формальдегидная пресскомпо-
зиция (аминопласт) имеет сравнительно малую текучесть и
утрачивает ее при хранении в течение 1—3 месяцев. В процессе
прессования аминопластов выделяется большое количество летучих
веществ. Повышение температуры прессформы сверх 145 °С
вызывает пожелтение материала; технологическая выдержка амино-
пласта в прессформе длительнее, чем для фенопласта.
Изделия из аминопластов уступают изделиям из фенопластов
по тепло-и водостойкости.
Полисилоксановые смолы применяются для изготовления
деталей приборов, длительно работающих при высоких температурах,
или деталей, которые должны обладать высокими показателями
диэлектрических свойств и сохранять их в широком диапазоне
температур. В изделиях первого типа в качестве наполнителя
можно использовать асбестовую муку, при изготовлении
диэлектриков в пресскомпозицию следует вводить кварцевую муку или
аналогичные ей минеральные порошки. Отверждение полисилокса-
яов происходит при 180—200 °С в присутствии перекисей или при
взаимодействии смолы с тетраэтоксисиланом. Выдержка изделий
в прессформе составляет 2—2,5 мин на 1 мм толщины, для
окончательного отверждения материал подвергают дополнительной
термообработке в шкафах при 200 °С. Отвержденные полисилокса-
ны наиболее хрупки по сравнению с обычно применяемыми от-
верждаемыми смолами. При введении кварцевой муки в пресс-
композицию на основе полисилоксана хрупкость изделий
возрастает (удельная ударная вязкость 2,5—3,0 кгс-см/см2) . Для
снижения хрупкости полисилоксановую смолу можно совмещать
555
с термопластами, однако это приводит к резкому ухудшению
термостойкости—основного положительного качества полисилокса-
нов. Поэтому иолисилоксановую смолу совмещают только с феноло-
формальдегидной смолой, которая в наименьшей степени
уступает полисилоксанам по теплостойкости.
Изделия на основе полисилоксанов могут подвергаться
длительной эксплуатации при 200—220 °С без заметного изменения
прочностных качеств и кратковременно A00—150 ч) работать
при 300 °С. Диэлектрические свойства таких изделий мало
изменяются в широком интервале температур. Полисилоксановые
пресскомпозиции предназначены для производства электро- и
радиотехнических изделий, работающих при высоких
температурах.
волокниты
В состав волокнитов в качестве наполнителей вводят
хлопковые очесы, асбестовое, стеклянное, кварцевое или графитирован-
ное волокно. Для изготовления волокнитов с хлопчатобумажными
наполнителями применяют фе-
ноло-формальдегидную смолу
резольного типа или карб-
амидную смолу. Пропитку
хлопковых очесов раствором смолы
производят в специальных
смесителях (рис. 162). После
пропитки растворитель удаляют
при нагревании материала в
термошкафах, а пропитанные
смолой хлопковые очесы
прессуют в нагретых прессформах.
Волокниты непригодны для
изготовления мелких и сложных
изделий с тонкой
металлической арматурой. При
формовании изделий даже простых форм
требуется более высокое
давление, чем для формования пресс-
порошков. Последующая обработка изделий резанием вызывает
не меньшие затруднения, так как хлопковые очесы легко
вырываются из материала резцом или сверлом, что ухудшает
поверхность среза. Волокниты применяют для формования изделий
простой конфигурации, не имеющих тонких стенок, резьбы, острых
углов и т. д., не требующих дополнительной механической
обработки. Изделия из волокнитов должны работать в условиях
повышенных динамических нагрузок (удельная ударная вязкость 10—
15 кгс-см/см2) при отсутствии ползучести под нагрузкой,
характерной для термопластов, и обладать теплостойкостью до 110*С.
Рис. 162. Смеситель для
производства волокнита.
556
Изделия с наполнителем—природным асбестом имеют еще более
грубую внешнюю форму. Асбоволокниты используют для
формования простых крупногабаритных изделий, способных выдерживать
без деформации длительные нагрузки при повышенных
температурах A50—160 °С—для асбоволокнита на основе феноло-
формальдегидных смол, 200—220 °С—для волокнита на основе
полисилоксанов).
В последние годы найдены способы изготовления
синтетического асбестового волокна с диаметром 3—5 мк. Это волокно
значительно прочнее природного волокна и превышает по прочности
лучшие стеклянные волокна (предел прочности при разрыве
достигает 330 кгс/мм?). Асбестовое волокно плавится при
температуре 1600 °С, в то время как стеклянное волокно полностью
теряет прочность при 500—600 °С, кварцевое—при 1000—1200°С.
Синтетическое асбестовое волокно в сочетании с феноло-формаль-
дегидной или меламино-формальдегидной смолой применяют для
изделий, которые должны выдерживать кратковременные тепловые
удары в несколько тысяч градусов (например, в качестве
теплозащитного слоя на поверхности летательного аппарата) или
работать под нагрузкой при температуре 600—1000 °С в течение
нескольких минут.
В табл. 29 приведены некоторые характеристики свойств
изделий из асбоволокнитов, полученных из природного и
синтетического асбестового волокна.
Все большее значение приобретают стекловолокниты,
обладающие такими ценными свойствами, как прочность при статических
и динамических нагрузках, атмосфероустойчивость, химическая
стойкость, хорошая теплостойкость, высокие диэлектрические
показатели и в ряде случаев радиопрозрачность. Из стекловолок-
нитов можно формовать разнообразные изделия, в том числе
мелкие армированные детали радиоаппаратуры, сложные по
конфигурации, крупногабаритные детали в виде труб различного
диаметра, деталей ракет, высокопрочных деталей машин различного
назначения, кузовов автомашин, корпусов лодок, сборных
ангаров, элементов кровли и др.
Стеклянное волокно, применяемое как наполнитель, не
пропитывается связующим. Поэтому при изготовлении стекловолокнитов
приходится в качестве связующих подбирать смолы, хорошо
смачивающие стеклянные волокна и склеивающие их между собой,
и изыскивать такие технологические приемы, которые
обеспечивают нанесение равномерной пленки связующего на волокно. Эти
обстоятельства послужили причиной для разработки рецептур
разнообразных стекловолокнитов, отличающихся типом
связующего, составом стекломассы, толщиной волокна, способами
последующей обработки, а также технологией покрытия стеклянных
волокон связующим и методом изготовления изделий. В настоящее
время в качестве связующих в стекловолокнитах применяются
557
і л и л и ц a
Физико-механические свойства изделий из асбоволокнитов
(связующее—феноло-формальдегидная смола)
Показатели
Плотность, г/см3
Теплостойкость, °С
Удельная ударная вязкость, кгс-см/см2
Предел прочности, кгс/см2
при изгибе
» растяжении
» сжатии
Влагопоглощение, %
Асбоволокнит на основе
природного
волокна
1,85
200—250
20
700
650
800
0,8
синтетического
волокна
1,85
250—280
130—140
2950—4080
1410—2180
1690—2180
0,8
феноло-формальдегидные смолы, совмещенные с поливинилбутира-
лем, нитрильным каучуком, фурфуролом; полисилоксаны, мел-
амино-формальдегидные смолы, различные ненасыщенные
полиэфиры (например, раствор полималеината в стироле), полиэпок-
сиды, полиакрилаты.
По взаимному расположению стеклянных волокон в изделии
различают неориентированные и ориентированные стекловолок-
ниты. Формование изделий производят прессованием при высоком
давлении A00—350 кгс/см2) в прессформах, установленных между
плитами гидравлического пресса; при низком давлении
A—10 кгс/см2) в формах облегченного типа и давлении,
создаваемом сжатым воздухом (контактное прессование); напылением на
форму струи рубленого волокна, смоченного смолой (метод
напыления); намоткой на вращающуюся форму стекловолокна или
жгута из стеклянных волокон (метод намотки).
При одном и том же способе формования свойства изделий
из стекловолокнита в значительной степени определяются
выбором связующего (табл. 30).
Наиболее высокую теплостойкость в сочетании с хорошими
диэлектрическими характеристиками имеют стекловолокниты на
основе полисилоксанов, но формование их возможно лишь при
высоком давлении с длительной технологической выдержкой
под прессом и последующим отверждением в термошкафах.
Модифицированная феноло-формальдегидная смола придает
стекловолокниту более высокие механические свойства при
достаточной теплостойкости, но для формования изделий из такого
прессматериала требуются в большинстве случаев высокие
давления (до 300 кгс/см2). Стекловолокниты на основе полиэфиров
и полиэпоксидов можно формовать при низком давлении и
применять для изготовления изделий методом напыления и намотки,
проводя отверждение при умеренной или при комнатной
температурах.
558
Таблица 30
Физико-механические свойства изделий из стекловолокнитов
Показатели
Связующие
модифицированная
феноло-форм-
альдегидиая
смола
полиси-
л океан
полиэфир
Плотность, e/cMJ
Теплостойкость, °С
Удельная ударная вязкость, кгс-см/см2
Предел прочности, кгс/см2
при сжатии
» растяжении
» изгибе .
Влагопоглощение, %
Диэлектрическая проницаемость . .
1,7-1,8
до 200
25—30
1300
800
1000
0,8—1,1
8
1,8—2,0
до 300
30—40
1000
500 |
0,3—0,5
4,5
1,7—1,9
до 140
30
750
1000
1,3
3
Полиэфиры придают изделиям из стекловолокнита наиболее
высокие диэлектрические качества, радиопрозрачность, иногда
оптическую прозрачность. Однако такие изделия обладают более
низкой механической прочностью, чем стекловолокниты,
полученные на основе других связующих. Теплостойкость полиэфирных
стекловолокнитов лежит в пределе 80—140 °С.
Изделия на основе полиэпоксидов отличаются наибольшей
прочностью, но более низкими показателями диэлектрических
свойств и меньшей теплостойкостью, чем изделия,
изготовленные из феноло-формальдегидных смол.
Стекловолокниты с неориентированным расположением
волокон получают из мотков стеклянного волокна, из стекломатов
(стр. 560) и путем нанесения волокна на поверхность формы
методом напыления. Характерным примером стекловолокнита,
формуемого в изделия при высоком давлении и имеющего
неориентированное расположение волокон, является прессматериал АГ-4в,
изготовленный смешением разрыхленных мотков стеклянного
волокна с раствором модифицированной феноло-формальдегид-
ной смолы. Смешение проводят в аппарате с Z-образными
лопастями (см. рис. 162), в котором мотки стекловолокон дробятся, хао-.
тично располагаются относительно друг друга и смачиваются
раствором смолы. Полученную массу раскладывают на противни и
сушат в термошкафах для удаления растворителя. Прессование
проводится в прессформах, нагретых до 145—165 °С, под
давлением 300—700 кгс/см2 в течение 1—1,5 мин на 1 мм толщины
изделия.
Для изготовления простых по форме крупногабаритных
изделий, к которым не предъявляются особенно высокие требования
в отношении их прочностных и других свойств, применяется кон-
559
тактное прессование или напыление. Получаемые изделия
отличаются хаотичным расположением стеклянного волокна.
Такие стекловолокнитовые изделия можно формовать в
специальных формах, изготовленных из древесины, дуралюмина или
пластических масс и повторяющих внутренний или наружный
контур изделия.
На рис. 163 изображена схема контактного прессования стек-
ловолокнита. В форму / закладывается стекломат 2—войлок,
изготовленный из стекловолокна, толщина стекломата
подбирается соответственно толщине стенки изделия. На стекломат
распылителем наносят раствор
полиэфирной смолы в мономере,
содержащий инициатор реакции
полимеризации, или расплав полиэпок-
сида и отвердителя. Стекломаты
имеют разрыхленную структуру,
поэтому сравнительно
низковязкий раствор или расплав
полимера легко проникает в толщу
войлока, смачивая и склеивая
стеклянные волокна и заполняя
пустоты между ними. Затем
заготовку покрывают целлофановой
пленкой и резиновым мешком 3.
На форму укрепляют
контрформу 4, прикрепляя ее к основанию
формы 5 замками 6. В резиновый
мешок через штуцер подают
подогретый сжатый воздух, под давлением которого заготовка
уплотняется, а повышенная температура воздуха способствует быстрому
отверждению связующего.
Для изготовления изделий, имеющих более сложный контур
или различную толщину стенок, а также изделий с заостренными
краями или отбортовками невозможно использовать стекломаты.
В этих случаях формование проводят методом напыления при
помощи трехходового пульверизатора. В качестве связующего
обычно применяют полиэфирную смолу, в которую для повышения
скорости отверждения смолы вводят инициатор и ускоритель
инициирования. Жгут стекловолокна непрерывно втягивается
в центральное сопло распылителя и с помощью резального
устройства нарезается на отрезки заданной длины, которые тут же
подхватываются струей воздуха и, вылетая вместе с ним из сопла,
направляются к форме. Одновременно из второго сопла под
давлением сжатого воздуха подается раствор полиэфира и инициатора
в стироле. Струя раствора смачивает стекловолокно, еще
находящееся во взвешенном состоянии (в виде отдельных волоконец).
Из третьего сопла также под давлением сжатого воздуха в общую
Рис. 163. Схема контактного
формования изделий из
стеклопластиков:
/—форма; 2—стекломат, пропитанный свя-
аующим (заготовка); Л—резиновый мешок;
4— контрформа; 5—основание формы;
if—замок, скрепляющий форму и контрформу.
560
струю подается раствор полиэфира и ускорителя в стироле. Смесь
аопадает на форму. Под влиянием инициатора и ускорителя
вязкость смолы нарастает настолько быстро, что смесь удерживается
на форме. Перемещая распылитель, всю форму покрывают слоем
склеенного стекловолокна. Стекловолокно уплотняют на форме
арокаткой резиновыми роликами или контрформой, после чего
для полного отверждения смолы изделие выдерживают несколько
часов на форме при комнатной температуре, а затем при
повышенной температуре в термошкафу для еще более глубокого
отверждения.
Применение неориентированных стекловолокнитов в виде
стекломатов, склеенных быстроотверждающейся смолой, или
в виде склеенных стекловолокон, напыленных на форму,
позволило использовать пластмассы для изготовления
крупногабаритных изделий при помощи простых легкозаменяемых
приспособлений. Однако такие изделия менее прочны, чем изделия из
стекловолокнита с ориентированным расположением волокон.
Для повышения прочности стекловолокнита необходимо
придать стеклянным волокнам ориентированное взаимное
расположение и обеспечить их более тщательное и равномерное
обволакивание связующим. Для изготовления такого стекловолокнита
на стенде укрепляют несколько сот шпуль со стеклянным
волокном и непрерывно сматывают его, заставляя перемещаться по
направляющим валкам. В результате волокна располагаются в один
ряд параллельно друг другу в виде узкой ленты, которая далее
поступает в пропиточную ванну, заполненную раствором смолы.
Избыток смолы отжимается с ленты валками, укрепленными над
ванной, а растворитель испаряется при движении ленты в
сушильной камере, куда подается нагретый воздух. Из камеры выходит
хрупкая узкая лента, состоящая из склеенных между собой
стеклянных волокон, которая нарубается на куски. Далее куски
стекловолокнита загружают в прессформу, где под давлением 200—
350 кгс/см2 и при температуре 145—165 аС отпрессовывают
изделия. Таким образом могут быть получены изделия сложной
конфигурации с большим количеством металлических деталей.
В табл. 31 приведены физико-механические свойства изделий,
отпрессованных из неориентированного АГ-4в и
ориентированного стекловолокнита. Чем тщательнее склеены соседние волокна,
равномернее распределена смола в материале и чем плотнее его
структура, тем выше прочность стекловолокнитовых изделий.
Метод ориентации волокна и его равномерного склеивания
используют для изготовления из стекловолокнита высокопрочных
труб, листов, плит и панелей. Стеклянные нити (или волокна)
перематываются со шпулей на вращающийся барабан (рис 164)
или подаются на него непосредственно из электропечи, где происходит
формование волокна. Каждый слой нитей, наматываемых на
барабан, предварительно смачивается раствором смолы, поступающим из
36—805 561
Таблица Зі
Свойства изделий из стекловолокиитов, полученных различными методам»
Показатели
Плотность, г/см3
Удельная ударная вязкость.
кгс-см/см2
Предел прочности, кгсісм2
при растяжении
» сжатии
» изгибе
Прессование при 300—400 кге/сл&
неориентированный стекло-
волокннт
1,7-1,8
25
800
1300
1000
ориентированный стекло-
волок ннт
1,7-1,8
100
2000
1300
2000
СВ AM
1,74
100
4400
3500
распылителя, избыток смолы отжимается щеткой. После
наматывания на барабан требуемого количества слоев стеклянные нити
обрезают, барабан снимают с вала и насаживают на него
следующий барабан. С первого барабана снимают смоченные смолой
нити, разрезая их по образующей, выпрямляют заготовку и сушат
для удаления растворителя. Высушенный лист укладывают между
нагретыми металлическими плитами пресса, при этом происходит
уплотнение заготовки и отверждение термореактивной смолы.
При производстве труб слои стеклянных нитей наматывают на
оправку, диаметр которой соответствует внутреннему диаметру
изготовляемой трубы. Не снимая с оправки, стекловолокнитовую
заготовку спрессовывают и отверждают в преесформах.
Стекловолокниты, получаемые описанным способом, названы
СВАМ (стекловолокнистый анизотропный материал).
Анизотропность этого материала обусловлена ориентацией в нем волокон
в одном направлении. Поэтому СВАМ отличаются особенно
высокой прочностью в продольном направлении и значительно меньшей
—в поперечном.
На рис. 164 показана схема установки для производства СВАМ.
Для многих изделий (в том числе для труб), работающих при
высоких внутренних давлениях, особенно необходима равнопроч-
ность изделия во всех направлениях. Для этого заготовку
укрепляют на опорах, допускающих не только вращение заготовки,
но и изменение угла между осью вращения и направлением
движения стеклянного волокна. Благодаря этому слои волокон
накладываются на заготовку под различным углом, способствуя
этим равномерному распределению наполнителя по детали.
Применение ориентированного стекловолокнита в
производстве изделий сложной конфигурации (прессованием ленты
стекловолокнита), а также изделий, имеющих форму тел вращения или
плит (прессованием СВАМ), или заготовок, полученных методом
намотки, дало возможность изготовлять изделия и детали, проч-
562
ность которых в ряде случаев не уступает прочности стальных
конструкций того же веса и сочетается с хорошими
теплоизоляционными и диэлектрическими свойствами, присущими всем
стеклопластикам. Изделия"; из стеклопластиков могут длительное
время без заметной потери прочности работать при температуре
200—250 °С, что выгодно отличает их от таких легких сплавов,
как алюминиевые и тем более магниевые. Для изготовления
изделий, находящихся под нагрузкой в течение длительного времени
при температуре 250—350 °С или
кратковременно при 450 °С,
применяют кварцевое волокно.
Однако в таких условиях
стеклопластики быстрее теряют свои
прочностные свойства, чем асбоволок-
ниты, но превосходят последние
по диэлектрическим свойствам и
радиопрозрачности,
несвойственной изделиям из асбоволокнита.
Благодаря низкой
теплопроводности внутренние слои стенок
конструкций из стеклопластиков
сохраняют работоспособность при
тепловом ударе на наружный слой
порядка 5000—6000 °С в течение
50—70 сек.
В последние годы появился
новый вид наполнителя—графити-
рованное волокно — материал,
обладающий наиболее высокой
теплостойкостью (температура раз-
Рис. 164. Схема установки для
производства СВАМ:
МЯГЧРНИЯ ПН111Р ЧООО °О " ГпягЬи- /—электропечь; 2-стеклянные волокна;
МЯІЧЄНИЯ ВЫШе JUUU Ц|. І рафИ ,з__распылитель для нанесения связующего
ТИроВаННОЄ ВОЛОКНО ОТЛИЧаеТСЯ на волокно; 4—наматывающий барабан.
от других наполнителей тем,
что оно не утрачивает своих прочностных свойств при
температуре, близкой к температуре размягчения (предел
прочности при разрыве графитированного волокна при 20 °С равен
25 кгс/см2, при 2500 °С—22 иге/см2).*
Графитированное волокно получают пиролизом вискозного
волокна в инертной среде с постепенным повышением
температуры пиролиза до 3000 °С. В этих условиях макромолекулы
целлюлозы утрачивают водород и кислород, а атомы углерода
группируются в макромолекулы, состоящие из конденсированных
ароматических циклов. Анализ волокна показывает, что его
химический состав, взаимное расположение углеродных атомов и
* Д. Шмидт, Л. Донат, Химия и технология полимеров, вып. 11,
24 A964).
36*
563
характер кристаллической решетки соответствуют структуре
графита.
Графитированное волокно в сочетании с феноло-формальде-
гидной смолой применяют для прессования изделий—графито-
пластов, которые в отличие от стекло- и асбопластов не обладают
хорошими теплоизоляционными свойствами и не могут
использоваться в качестве диэлектриков или радиопрозрачных
материалов, однако преимуществом графитопластов над всеми известными
в настоящее время материалами является возможность с помощью
простых методов прессования переработать материал в изделия
сложных форм, обладающих низкой плотностью, большой ударо-
прочностью и способностью при минимальных потерях
выдерживать под большими нагрузками действие температур порядка
3000 °С. Графитопласты нашли широкое применение в
производстве наиболее ответственных деталей ракетных двигателей.
Показатели свойств графитопластов приведены ниже:
Плотность, г/см3 1,44
Предел прочности, кгс/см2
при разрыве 1160
» изгибе 1850
» сжатии 2800
СЛОИСТЫЕ ПЛАСТИКИ
Для изготовления слоистых пластиков, таких, как гетинаксы
(на основе бумаги), текстолиты (на основе хлопчатобумажной
ткани) и древесные слоистые пластики ДСП (на основе древесного
шпона), применяются феноло-формальдегидные смолы резольного
типа. В тех случаях, когда слоистый пластик используется в
качестве декоративного облицовочного материала, сохраняющего
текстуру и окраску листов наполнителя, в качестве связующего
употребляют карбамидо-формальдегидную смолу. Для
изготовления стеклотекстолитов (наполнитель—стеклянная ткань)
применяют смолы, обладающие адгезией к стекловолокну, а в ряде
случаев—смолы, отверждающиеся без выделения побочных
продуктов. Широко известны стеклотекстолиты, в которых
связующим являются бутваро-феноло-формальдегидные смолы (БФ).
Бутвар (поливинилбутираль, стр. 434) вводится для придания фе-
ноло-формальдегидной смоле более высокой упругости и для
повышения адгезии к стеклянным тканям. Во многих случаях в
качестве связующего применяют полиэпоксидные смолы с отвердите-
лем или полиэфирные смолы, содержащие инициатор
отверждения.
Смазанная смолой стеклянная ткань после удаления
растворителя очень жестка и хрупка, поэтому полиэпоксидная или
полиэфирная смолы обычно наносятся на ткань в процессе
изготовления изделия. Все остальные слоистые пластики изготовляют
заранее, их можно сохранять до формования в течение 5—б месяцев.
564
На рис. 165 приведена схема горизонтальной пропиточной
машины для нанесения смолы на непрерывную ленту бумаги или
ткани. Наполнитель (бумага или ткань) в виде непрерывной лен-
Рис. 165. Внешний вид и схема горизонтальной пропиточной машины:
/—рулон наполнителя; 2, S—валы для просушки; 3, 8, 10—направляющие вяликн; 4— ванна
с раствором смолы; 5—отжимные валики; 7—сушильная камера; 9—паровой калорифер;
//—приемный барабан.
ты сматывается с рулона /, подсушивается на нагретом валу 2 и,
проходя под направляющим валиком 3, опускается в ванну 4,
где находится раствор смолы. Избыток смолы отжимается от
материала валиками 5. Для удаления растворителя лента, пропи-
565
тайная смолой, проходит сушильную камеру 7, в которой
циркулирует нагретый воздух. Высушенный материал наматывается на
приемный барабан 11.
Количество смолы, остающейся в наполнителе, колеблется от
30 до 50%. Чем больше ее содержание, тем выше влагостойкость
и диэлектрические характеристики отпрессованного слоистого
пластика, но тем ниже его механическая прочность. В текстолиты
и стеклотекстолита, предназначенные для электротехнических
изделий, вводят 40—50% смолы, слоистые пластики для изделий
конструкционного назначения содержат 30—40% смолы.
Рис. 166. Детали, изготовленные из плит ДСП (зубчатые
колеса и фрикционные соединения).
Наполнителем древеснослоистых пластиков служат листы
тонкой древесной стружки—шпона (толщина 0,1—0,3 мм), который
пропитывают смолой маканием в ее раствор и высушивают для
удаления растворителя. Древеснослоистые пластики
используются в качестве конструкционных материалов. Для достижения
оптимальной прочности содержание смолы в ДСП не должно
быть выше 22—25%. Из слоистых пластиков изготовляют плиты,
листы, трубы и заготовки, форма которых близка к форме
изделия, например диски определенных толщин и диаметров для
производства шестерен (рис. 166).
В процессе отверждения феноло-формальдегидной и карбами-
до-формальдегидной смол выделяется большое количество
побочных газообразных продуктов, под давлением которых пластик
может расслаиваться с нарушением его монолитности. Поэтому,
несмотря на простые формы изделий из слоистых пластиков, их
приходится прессовать под высоким давлением (до 125 кгс/см2).
Этот процесс проводят в многоэтажном прессе, где одновременно
запрессовываются заранее подготовленные пакеты, состоящие из
многих листов ткани, бумаги или шпона, пропитанных смолой.
566
Плиты пресса нагревают до 120—160 °С и выдерживают между
ними пакеты в течение времени, достаточного для размягчения
смолы, склеивания отдельных листов и отверждения полимера.
Длительность процесса 3—7 мин на 1 мм толщины прессуемого
листа или плиты. По окончании процесса прессования в каналы
плит подают воду для охлаждения материала. Одновременно
снижается давление газов, выделившихся при отверждении и
находящихся между слоями наполнителя, что предотвращает
последующее расслаивание материала или коробление изделий.
В табл. 32 приведены свойства изделий из слоистых
пластиков, изготовляемых описанным способом. Для слоистых
пластиков характерна значительная анизотропность, связанная с тем,
что основа (продольные нити) любой ткани прочнее уточных
(поперечных) нитей, а листы шпона более прочны в направлении
расположения волокон древесины.
Таблица 32
Физико-механические свойства изделий из слоистых пластиков
Показатели
Плотность, г/см2
Теплостойкость,
°С
Удельная
ударная вязкость,
кгс-см/см'1 . .
Предел
прочности, кгс/см2
ПГМТ Г\*1 ПГГСТ
іірИ UdLTH-
жении
» сжатии
» изгибе
В лагопог
лощение, % ...
Диэлектрическая
проницаемость . . .
Гетинакс
1,3—1,4
До 150
16—18
800—1000
2400—3400
1300
1-2,5
8
Текстолит
1,3—1,4
До 125
25—36
1100
2300—2500
1600
0,8—1,5
8
ДСП
1,4
До 120
2600
1700
1600
5—18
—
Стеклотекстолиты
на смоле
БФ
1,85
До 200
60—70
2350—2700
1600—1800
2200—2800
0,8—2,0
5-6
на поли-
эпоксидиой
смоле,
отверди-
тель—фено-
ло-формаль-
дегидиая
смола
1,62—1,71
До 250
150
4000
2600
4200
0,1—0,5
5—7
на
полиэфирной
смоле
1,6—1,75
До 200
500
3900—4300
1500
2100—2700
1,3—1,65
4,5—5,0
Для изготовления плит, лишенных этого недостатка, сборку
пакетов производят так, чтобы нити основы или волокна шпона
в соседних листах были расположены взаимно перпендикулярно.
Гетинакс, прочный материал с удовлетворительными
диэлектрическими свойствами, используется в электротехнической
промышленности. Текстолит, близкий по свойствам к гетинаксу, на-
567
Пропитка
шпона смолой
ходит широкое применение как прочный диэлектрик, работающий
под нагрузкой при температуре до 110—120 °С. Благодаря
высокой прочности к истиранию и к вибрационным нагрузкам,
текстолит используется также для изготовления деталей машин (втулок,
шестерен, подшипников). Стеклотекстолит обладает наилучшими
диэлектрическими свойствами по сравнению с другими
слоистыми пластиками и большей теплостойкостью.
Широкое применение находят трубы из текстолита и
стеклотекстолита. Вследствие высокой прочности, химической
стойкости, атмосфероустойчивости,
малого веса такие трубы удобны
в процессах сборки, кроме того,
они достаточно упруги и
герметичны. Трубы изготовляют путем
намотки пропитанных смолой
тканей на металлические формы
и последующего прессования в
многогнездных прессформах
(режим прессования такой же, как
для плит).
Высокопрочные древесные слои-
^ стые пластики находят все боль-
(( шее применение в качестве раз-
v гйЗЭШШйЙй- личных деталей машин (см. рис.
166), панелей, облицовочных и
строительных плит. Прочность
таких материалов в несколько
раз больше прочности древесины,
они гораздо менее гигроскопичны,
не подвержены грибковым
заболеваниям, поражающим древесину.
На рис. 167 приведена схема
производства ДСП.
Для многих изделий из дре-
веснослоистых пластиков не
требуется такая высокая прочность,
которая достигается
прессованием ДСП под давлением 80—100 кгс/см2. В связи с этим можно
упростить технологию их изготовления, заменив гидравлические
прессы и громоздкие прессформы автоклавами. Количество смолы
в ДСП не превышает 25%, и потому расслаивание материала под
давлением газов, побочно выделяющихся при отверждении
полимера, не столь велико, как в текстолите и гетинаксе. Вследствие
этого давление при прессовании изделий может быть снижено до
20 кгс/см2 (т. е. в пять раз), хотя прочность отпрессованного
изделия снижается лишь вдвое (без расслаивания материала). При
таком пониженном давлении целесообразно формовать изделия в
568
Рис. 167. Схема производства
древеснослоистого пластика.
автоклавах, в которых давление нагретого сжатого воздуха
равномерно передается на пакет пропитанного шпона, уложенного
на легкую форму и прижатого к ней крепежными лентами. Пакет
накрывается резиновым чехлом. В автоклаве смола размягчается
и склеивает отдельные листы шпона, при этом пакет уплотняется
и обжимает форму. В процессе отверждения смолы фиксируется
приданная пакету форма, а поверхность изделия покрывается
полимерной пленкой. Таким способом изготовляют корпуса шлюпок,
спортивных лодок, отдельные отсеки спортивных самолетов.
Применение полиэфиров и полиэпоксидов в качестве
связующих в стеклопластиках позволило без заметного снижения
прочности изделий уменьшить давление при прессовании до 2—
5 кгс/см2. Благодаря этому появилась возможность изготовлять
крупногабаритные изделия простых конфигураций в еще более
несложных приспособлениях, чем при формовании ДСП в
автоклавах. Эти приспособления применяются для формования
изделий из стекломатов, пропитанных полиэфирной или
полиэпоксидной смолой. На. форму, покрытую целлофаном (что
предотвращает прилипание к ней изделия), укладывают слой раскроенной
стеклянной ткани и промазывают его раствором полиэфира и
инициатора в каком-либо мономере или смесью полиэпоксида и
отвердителя. Затем на эту поверхность натягивают новый слой
ткани и вновь промазывают ее смолой. Таким образом, сборка
пакета производится непосредственно на форме. Пакет покрывают
целлофаном, прижимают к форме резиновым чехлом и
устанавливают на пакет контрматрицу, соединяя ее с матрицей. В зазор
между контрформой и резиновым чехлом нагнетают
подогретый сжатый воздух, под давлением которого пакет
уплотняется и одновременно при повышенной температуре отверждается
смола.
Изготовление изделий из стеклотекстолитов—более
трудоемкий процесс по сравнению с изготовлением изделий из
стекломатов, однако прочность, однородность и водостойкость стеклотек-
столитовых изделий много выше. Кроме того стеклоткань легче
выкладывается на формах несколько более сложной
конфигурации. На рис. 168—175 показаны некоторые детали и изделия,
выполненные из стеклотекстолита.
При изготовлении крупногабаритных изделий из отверждаю-
щихся пластических масс особое значение приобретает вопрос
надежности соединения деталей из пластиков между собой или
их присоединения к элементам конструкции, выполненным из
металла. Во многих случаях для соединения применяют
механическое крепление (заклепки, резьбы, болты). Часто элементы
механического крепления отпрессовывают из стекловолокнитов.
Детали из гетинакса, текстолита, ДСП с достаточной надежностью
соединяются клеевым способом. Однако прочность клеевого шва
в изделиях из стеклопластиков при повышенных температурах
569
снижается более интенсивно, чем прочность самого материала.
Поэтому изделия, предназначенные для работы при повышенных
температурах, помимо склеивания дополнительно соединяют
заклепками или болтами, располагая их сравнительно редко вдоль
шва.
-*
Рис. 168. Трехслойные панели сотовой конструкции.
Преимущества клеевого соединения деталей перед
механическим креплением заключаются в том, что материал не ослабляется
отверстиями, изделие не утяжеляется, в местах соединений
равномерно распределяются напряжения, соединение получается
достаточно герметичным, поверхности соединяемых деталей не
повреждаются и остаются гладкими.
Известно, что растворы полимеров отличаются высокой
клейкостью (адгезией) к различным материалам. Полимерная
пленка может прочно удерживаться на поверхности другого материала
и после удаления растворителя. Пленки некоторых полимеров
обладают избирательной адгезией, т. е. прочно удерживаются
только на поверхности какого-либо одного материала; другие
полимерные пленки прочно прилипают к поверхностям различных
материалов, т. е. обладают универсальной адгезией. Особенно
большое практическое значение имеют клеевые материалы на основе
термореактивных полимеров и полимеров, переходящих в
термостабильное состояние при помощи отвердителей. Широко
известны клеи на основе феноло-формальдегидных и модифицированных
феноло-формальдегидных полимеров, полиуретанов и. полиэпок-
сидов.
Феноло-формальдегидная смола относится к клеям
избирательной адгезии и прочно соединяет только картон, древесину, пено-
пласты и пластические массы, связующим в которых является
также феното-формальдегидная смола.
570
Рис, 169. Детали оборудования из стеклопластиков:
/—распорные клинья; 2—кликья для крепления пазовой изоляции",
3. 4—стеклотекстолитовые цилиндры.
Рис. 170. Обтекатели радиолокационных станций
Рис. 171. Корпус прицепа и рама, изготовленные из стеклопластика
на полиэфирном связующем.
Рис. 172. Катер из стеклопластика.
572
Рис. 173. Обтекатель винта из стеклопластика.
Для склеивания различных деталей из перечисленных выше
материалов можно пользоваться водным или спиртовым раствором
резольной смолы. Часть растворителя удаляется с поверхностей,
на которые нанесен такой клей, часть диффундирует в
склеиваемые материалы. Поверхности, на которые нанесен клеевой состав,
слегка прижимают друг к другу и выдерживают в таком состоянии
до отверждения полимера.
Не рекомендуется повышать температуру склеивания
пластмасс с органическим наполнителем, деревянных изделий и изделий
из термопластичных пенопластов выше 60 °С. Однако при более
низкой температуре процесс отверждения длится несколько
месяцев, поэтому непосредственно перед склеиванием в раствор
полимера вводят катализатор процесса отверждения. В качестве
катализатора может быть использована любая кислоту, чаще
всего применяют продукты сульфирования керосиновой фракции
нефти, так называемый контакт Петрова. Смесь, содержащая
10—14% контакта, через 1—2 ч после приготовления становится
настолько вязкой, что ее трудно наносить на склеиваемые
поверхности. Для удлинения срока хранения клеевого состава до
3—4 ч в него добавляют стабилизатор (ацетон или спирт).
Раствором резольной смолы, смешанным с контактом,
изделия склеивают при обычной температуре с выдержкой в течение
24 ч под давлением 1—2 кгс/см2 (для пенопласта 0,5 кгс/см2).
Прочность клеевого соединения деревянных и пенопластовых
изделий выше прочности склеиваемого материала. Клеевая
пленка обладает влагостойкостью и устойчивостью против грибковых
заболеваний, выдерживает более высокие температуры, чем
склеиваемый материал. Однако клеевые материалы на основе феноло-
формальдегидной смолы имеют ряд недостатков. Кислота,
необходимая для отверждения смолы, может постепенно разрушать
573
Рис. 174. Легковая машина с кузовом из стеклопластика.
древесину в слоях, прилегающих к клеевой пленке, швы феноло-
формальдегидного клея имеют темную окраску, что может портить
внешний вид изделий. Кроме того, при работе с феноло-форм-
альдегидной смолой необходимо соблюдать определенные меры
предосторожности (применение спецодежды, перчаток, тщательная
вентиляция помещений и т. д.).
В связи с этим в ряде случаев (особенно при склеивании
деревянных деталей) феноло-формальдегидные клеевые материалы
заменяются карбамидо-формальдегидными составами.
Карбамидо-формальдегидная смола используется в виде
водного раствора, катализатором отверждения ее служат соли
(ZnCl2, NH4C1). Клеевой шов обладает такой же прочностью,
как феноло-формальдегидный, но уступает ему по водостойкости
(при повышенной влажности прочность карбамидо-формальдегид-
ного клеевого шва снижается на 25—30%).
Феноло-формальдегидная смола имеет малую адгезию к
стеклопластикам и к металлам, ее можно повысить, добавив в
клеевой состав поливинилбутираль (клей БФ), некоторые
синтетические каучуки (например, клей ВК-32) или эпоксидную
смолу. Для ускорения процесса отверждения феноло-формальде-
гидной смолы при склеивании металлов нельзя применять
кислоты, так как они вызывают сильную коррозию металлических
изделий. Поэтому отверждение феноло-формальдегидной смолы
в таких случаях ускоряют путем повышения температуры
склеивания.
Клеи готовят в виде спиртовых растворов, наносят их на
склеиваемые поверхности и выдерживают до полного испарения
растворителя, затем зажимают в специальных приспособлениях
и постепенно повышают температуру до 150—200 "С. Такие
клеевые соединения отличаются высокой прочностью (особенно
резольно-полиэпоксидные клеи) и длительно выдерживают
действие повышенных температур—до 180—200 °С (особенно резольно-
574
Рис. 175. Грузовая машина с кузовом из стеклопластика.
каучуковые клеи). Высокопрочное, хотя и менее теплостойкое,
клеевое соединение, пригодное для крепления деталей из любых
материалов, возможно при применении клеевых составов на основе
полиэпоксидных смол. Отвердителями таких смол обычно служат
диамины (например, полиэтиленполиамин) или ангидриды кислот
(например, малеиновый ангидрид). При 60—65 °С полиэпоксиды
становятся маловязкими жидкостями, в этом состоянии их легко
смешивать с отвердителем и наносить на склеиваемые
поверхности.
Для образования прочного клеевого соединения достаточно
избыточного давления порядка 0,5—1 кгс/см2. Если отвердителем
полиэпоксидов является полиамин, то отверждение происходит
при нормальной температуре, теплостойкость клеевого шва
достигает 80—100 °С. Для отверждения с помощью малеинового
ангидрида требуется 10—12-часовое прогревание склеиваемого
изделия при температуре до 180 °С, прочность и теплостойкость
клеевого шва при этом возрастают. Процесс отверждения можно
ускорить также введением диэтиланилина в смесь смолы и
малеинового ангидрида.
Универсальной адгезией обладает смесь полиэфира и
диизоцианата. Отверждение клея на основе этой смеси
происходит в течение нескольких минут по удалении
растворителя из клеевой пленки, однако прочность такого клеевого шва,
отвержденного на холоду, уступает прочности эпоксидных и
модифицированных резольных клеевых пленок. Если полиэфироурета-
новый клей отверждают при 70—80 °С, прочность клеевого
соединения заметно повышается.
575
-"¦—-.р..г.г- ПРЫЛПГОАГТЫ
Применение термореактивных полимеров для получения пено-
пластов позволяет значительно расширить температурные
интервалы эксплуатации этих материалов. Так, термопластичные
пенопласты можно использовать при температурах, не
превышающих 60 "С, а более теплостойкие пенопласты на основе
термореактивных полимеров выдерживают температуру до 250 *С.
Пенопласты изготовляют из феноло-формальдегидных, поли-
силоксановых, полиуретановых смол. Отвержденные феноло-
формальдегидные и особенно полисилоксановые смолы
отличаются высокой хрупкостью, которая еще более усиливается в пено-
пластах на основе этих полимеров. Для повышения упругости
пенопластов новолачную феноло-формальдегидную смолу
совмещают с бутадиен-нитрильным каучуком СКН (стр. 490). Обычно
применяют композиции, содержащие до 40% каучука СКН, с
повышением содержания каучука теплостойкость пенопласта
несколько снижается.
Новолачную смолу измельчают до порошкообразного
состояния и смешивают с газообразователем, гексаметилентетрамином
(отвердитель новолачной смолы), с серой (вулканизующее
вещество) и с ускорителями процесса вулканизации каучука. Смесь
обрабатывают вместе с синтетическим каучуком на вальцах при
температуре не выше 60 °С. Полученную однородную массу
измельчают в порошок или вытягивают в экструдерах в тонкие
стержни, измельчая их затем в гранулы. Порошок (или гранулы)
засыпают в формы-ограничители, закрывают их и подвергают
материал термообработке.
В начале разогрева (до 120 *С) происходит размягчение смеси
и разрушение газообразователя. Вследствие отверждения
полимера его вязкость нарастает и образующиеся газовые пузырьки
не могут выйти из материала. При дальнейшем повышении
температуры до 150—200 °С происходит полное отверждение полимера
с сохранением ячеистой структуры.
Феноло-формальдегидо-каучуковые пенопласты ФК-20 и
ФК-40 достаточно прочны при объемной плотности 0,1 г/сма.
ФК-20 может длительное время работать при 130 *С, ФК-40—при
температуре до 150 "С. В тех случаях, когда изделие в процессе
эксплуатации должно выдерживать более высокие температуры,
применяют пенопласты на основе полисилоксанов. Они могут
длительно работать при 250 *С и кратковременно при 350 *С, но более
хрупки, чем пенопласты ФК (табл. 33).
Большое распространение получили жесткие, и особенно
эластичные, пенополиуретаны (ПУ). Они получаются взаимодействием
низкомолекулярных полиэфиров с молекулярным весом
до 3500 (слаборазветвленные полиэфиры—для эластичных
пенопластов и сильноразветвленные—для жестких) с диизоцианатом
576
Таблица 33
Физико-механические свойства различных пенопластов
(объемная масса 0,2 г/сд8)
Показатели
Пено-
полистирол
ЛС-1
Пенополн-
виннл-
хлорид
ЛХВ-1
Сплав
ФК-20
Пено-
полиси-
локсан
Пенополиуретан
ПУ-10ІА
жесткие эластичный
Рабочая
температура, °С
Коэффициент
теплопроводности,
ккалЦм-ч-град) .
Удельная ударная
вязкость,
кгс-CMJcM2 . . .
Предел прочности,
кгс/см2
при растяжении
j> изгибе
j> сжатии . .
Влагопоглощен ие,
кг/см2 .....
Диэлектрическая
проницаемость .
До 60
0,044
1,9
42
65
30
0,06
1,28
До 60
До 130
0,045 0,052
1.5
45
40
26
0,3
2.4
0,8
18
32
20
0,28
1,2
До 250
0,043
0,16
б
8
0,1
1,31
До 170
0,049
1,6
18
42
28
1,25
До 130
5,5
0,1
1,2
(чаще всего применяют толуилендиизоцианат). Иэоцианат берут
в избытке по отношению к полиэфиру. В реакционную смесь
вводят небольшое количество воды. При избытке диизоцианата
получаются полиэфироуретаны (стр. 402), сравнительно низкого
молекулярного веса, причем на концах их макромолекул остаются изо-
цианатные группы, которые реагируют с водой. При этом
образуются полимеры пространственного строения. Процесс
сопровождается выделением двуокиси углерода.
Реакция между полиэфиром и дииэоцианатом протекает при
обычной температуре и сопровождается быстрым нарастанием
вязкости реакционной смеси, одновременно происходит
поперечное сшивание макромолекул полиэфироуретана. Выделяющаяся
СО2 не может преодолеть сопротивления вязкой массы и остается
в ней, образуя множество мелких ячеек. Путем подбора
соответствующего эмульгатора, вводимого в реакционную смесь,
достигается равномерное распределение и одинаковый объем ячеек
пенопласта. Процесс поликонденсации можно проводить при 20 "С,
но для повышения прочности и теплостойкости пенополиуретана
его рекомендуется прогревать в течение нескольких часов при
90—100 *С. При этом теплостойкость жестких ПУ возрастает до
150 *С, а эластичных—до 120 'С. В зависимости от свойств
исходного полиэфира могут быть получены пенополиуретаны
различной эластичности. Морозостойкость, устойчивость к Действию
кислорода,, бензо- и маслостойкость эластичных,
ценопластоо^значительно ВЫШе, ЧёМ ПОрИСТЫХ реЗИВОЙЫХ ИЗДеЛИА« и,- і...... ¦¦":' •'¦ 'і
37-805
577
МЯГКИе ПОЛИЭфироуретанОВЫе пенОПЛасты Иримситиі iijjri noiu-
товлении мягкой мебели, в качестве покрытий для полов, звуко-
и теплоизоляционных материалов, амортизаторов в приборах.
Жесткие пенополиуретаны используются в качестве легких
конструкционных материалов.
Исходная жидковязкая смесь, приготовленная для
вспенивания, очень удобна для заполнения пустот (полостей) в
конструкциях даже при весьма узких зазорах между стенками. Проникая
в такие полости, жидкая масса заполняет их. Значительная
усадка при отверждении полимера компенсируется в процессе
вспенивания массы, благодаря чему отвержденный пенопласт
равномерно заполняет пустоты и плотно приклеивается к внутренним
стенкам.
ЛИТЕРАТУРА К ЧАСТИ ТРЕТЬЕЙ
Лосев И. П., Тростянская Е. Б., Химия синтетических
полимеров, Изд. «Химия», 1964.
Стрепих е"е*в А. А., Деревицкая В. А., Основы химии
высокомолекулярных соединений, Госхимиздат, 1960.
Роговин 3. А., Основы химии и технологии производства химических
волокон, т. I и II, Изд. «Химия», 1964.
Ряузов А. Н., Груздев В. А., Артеменко М. А.,
Технология искусственного волокна, Изд. «Химия», 1965.
Таиров С. А., Ч а ч х и а и и А. Б., Машины и аппараты заводов
искусственных волокон, Гизлегпром, 1955.
П е Tjy х о в Б. В., Полиэфирное волокно, Госхимиздат, I960.
П а к ш в е р А. Б., Геллер Б. Э., Химия и технология производства
волокна нитрон, Госхимиздат, I960.
К о н к и и А. А., Производство шиннога корда, Изд. «Химия», 1964.
Литвин О. Б., Основы технологии синтеза каучуков, Изд. «Химия»,
1964.
Б ел оз ер ов Н. В., Технология резины. Изд. «Химия», 1964.
Кошелев Ф. Ф., Климов Н. С, Общая технология резины,
Госхимиздат, 1958.
Салтыков А. В., Основы современной технологии автомобильных шин,
Госхимиздат, 1960.
Яшуиская Ф. И., Синтетический каучук и его применение в народном
хозяйстве, Госхимиздат, 1958.
Крючков А. П., Общая технология синтетических каучуков, изд. 3-е, Изд.
«Химия», 1965.
Крючков А. П., Каучук (научно-популярный очерк), Изд. «Химия», 1965.
Б а р г Э. И., Технология синтетических пластических масс, Госхимиздат,
1954.
Николаев А. Ф., Синтетические полимеры и пластические массы иа их
основе. Изд. «Химия», 1964.
Кардашов Д. А., Синтетические клеи, Изд. «Химия», 1964.
Киселев Б. А., Стеклопластики, Госхимиздат, 1961.
Чегодаев Д. Д., Наумова 3. К., Дунаевская Ц. С,
Фторопласты, Госхимиздат, 1960.
Хувинк Р., Ставерман А., Химия и технология полимеров (перев. с
нем.), Изд. «Химия», 1965.
Новые материалы в технике, под ред. Е. Б. Тростяиской, Б. А. Колачева,
С. И. Снльверстровнча, Изд. «Химия», 1964.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбенты 30 ел.
Абсорберы 30, 32, 155 ел., 209, 210
Абсорбция
аммиака 95
ацетилена 139
бензина 35 ел.
углеводородов 155
Авадекс (8-2,3-Дихлораллил-М,М-
диизолропилтиокарбамат) 348,
349
Автоклавы 77, 285
АГ соль 260
АГ-4в, прессматериал 559, 561
Адгезия 570
Адиабатический конвертор 144 ел.,
147, 148
Адипиновая кислота 250, 251, 258,
390
Адсорбенты 30, 36, 53 ел.
Адсорберы 36
Адсорбция
бензина 36
красителей 300, 305, 316, 321
углеводородов 155
Азеотропная ректификация 161
Азеотропные смеси 160, 161
Азиновые красители 324, 325
Азоамины 308, 309
Азобензол 275, 288
Азокрасители 291 ел.
активные 305 ел., 309 ел.
дисперсные 309 ел.
катионные 310
кислотные 299 ел.
прямые 302 ел.
«холодные» 308
Азоксибензол 275
Азопигменты 307
Азосоставляющие 293 ел.
Азосочетание 293 ел., 302 ел., 310
Азотолы 309
Акарициды 335, 357
Акрилаты 237, 239
Акриловая кислота 237
полимеры 368, 372, 380
синтез 172
эфиры 237, 239
Акрилонитрил 237 ел.
полимеры 372, 373, 380, 383, 465,
466
сополимеры 466, 489, 490, 494,
495, 506, 541
Акролеин 238, 239
Активация мономеров 393, 394, 397
Активированный уголь 75, 195
Активные красители 305 ел., 309 ел.
синий КХ 314
ярко-синий 5СХ 306
Активные наполнители 137, 499, 500
Активные сажи 499, 500
«Алая» кислота 277
Ализарин 270, 286, 311
Алкалицеллюлоза 448 ел.
Алкилатор 151 ел.
Алкилирование 57, 149 ел.
ароматических соединений 150,
279 ел., 334
катализаторы 150 ел., 280, 334
Алкилсульфаты 208, 209
Алкоголиз 388
Аллил хлористый 182, 184 ел.
Аллиловый спирт 185
Алцнаны 322
Альдегиды 168, 242
ацетальдегид см. Ацетальдегид
беизальдегид 248, 323, 360
кротоновый 222
прошюновый 168
формальдегид ем. Формальдегид
Альдольная конденсация 222
Альтакс (Дибензотиазолдисульфид)
362, 499
37*
579
Амиды 339 ел., 385
полимеры см. Полиамиды
Амилены 145
Аминированне хлорбензола 275
Амииоазобеизол 288
Аминокислоты 202, 309
Аминолиз 388
Аминопласш 555
я-Аминосалициловая кислота 202
Аминоспирты 247
Амииоэнантовая кислота 390
Амины ароматические 275, 276, см.
также Анилин, Бензидин,
Нафтиламин
алкилирование 279, 280
диазотирование 292 ел.
получение 272 ел.
сульфирование 267, 268
фосгенирование 277
Аммиак коксового газа 93
улавливание 95
Аммиачная вода 95, 96
Аммофос 96
Анализ топлива 16
Анид (Найлои 66) 470 ел., 503
Аиизол 352
Анилии 69, 202, 280, 281
ацилирование 277
диазотирование 292, 304
получение 272 ел.
синтезы на его основе 362, 363,
365
Анионная полимеризация 396, 399,
418, 419
Анионоактивиые ПАВ 330 ел.
Антидетонаторы 49, 128, 175
Аитиоксиданты (Противостарители)
361, 365, 500, 501
Антифризы 129
Антрахинои
синтез 282, 283
сульфирование 266
Антрахиноновые красители 311 ел.
активные 313
дисперсные 314
кислотные 311 ел.
протравные 311
Антрахинонсульфокнслоты 269, 270
Антрацен 100, 101
окисление 282
Антраценовое масло 98, 100
Антрацит 18, 69, 83
Араминированне 281
Арилирование 279 ел.
Арилметаиовые красители 322 ел.
Арилоксиалкилкарбоновые кислоты
341 ел.
Армирующие материалы 501 ел,
SSO
Ароматизация продуктов коксования
71, 86
Ароматические амииы см. Амины
ароматические
Ароматические углеводороды
алкилирование 150
нитрование 200 ел.
окисление 248 ел., 260 сл„ 282
получение 63, 115, 132, 164
сульфирование 251, 265 ел., 332
уплотнение 59
хлорирование 192 ел., 251, 352 ел.
Асбестовые наполнители 527, 528,
555, 557
Асбоволокниты 557, 558
Асфальтены 80
Асфальтовые нефти 39
Асфальти 51
Атактические' полимеры 374
Атмосферио-вакуумная трубчатка
(АВТ) 47 ел.
Атразии 345, 349
Ауксохромы 288
Ацеталирование 434
Ацеталь 222
Ацетальдегид
окисление 223, 225
полимеризация 221
получение 217 ел., 230, 245
применение 222 ел.
Ацетатное волокно 462 ел.
Ацетаты 225, 226
целлюлозы 462 ел., 541
Ацетилаиилид 295
Ацетилен 154
абсорбция 139
гидратация 217 ел.
гидрохлорнроваиие 188 ел.
карбидный 139 ел.
получение 132, 134
синтезы на его основе 146, 237 ел.
хлорирование 186 ел.
Ацетиленовые углеводороды высшие
галоидирование 186 ел.
гидрохлорирование 191
Ацетилирование 277, 462
Ацетилцеллюлоза 462
Ацетон 76, 124, 215 ел., 466, 467
конденсация с беизальдегидом 360
пиролиз 216, 227
получение 214 ел., 239
— совместно с фенолом 248 ел.,
252
Ацетонциангидрин 216, 239, 249
Ацилирование 276 ел., 342, 343
Ацилы 276
Аш-кислота 270, 293, 294, 302, 303
ацетилирование 277
Баллоны ацетиленовые 154
Барабанные
вакуум-фильтры 296, 297
вулканизаторы 522
Барабаны смесовые 299
Бараты (ксантатбарабаны) 450
Барбан (Хлорбутннил-Ы-хлорфеиил-
карбамат) 346
Батареи
коксовых печей 90, 91
кубовые 44
Беззольный кокс 51, 61, 101
Бекмана перегруппировка 203, 258
Белая сажа 500
Бельтинг 502
Бензальацетон 360
Бензальдегид 248, 323
конденсация с ацетоном 360
Бензидин 202, 293
диазотирование 293, 302
синтез 274 ел.
Бензидиновая перегруппировка 27а
Бензин 12, 20, 38, 39, 66, 114
абсорбция 35 ел.
адсорбция 36
газовый 33 ел., 101, 105, 157
«Галоша» 503
десорбция 35
испаряемость 49
моторный 49
очистка 52 ел.
полимеризационныи 58
свойства 18, 49
состав 64
стабилизация 33 ел.
энергоемкость 19
Бензоила перекись 396, 425
Бензонлнрование 366
Бензоин 328
Бензойная кислота 248, 249, 253
Бензол 63, 132, 466
алкилированне 150 ел.
гидрирование 249
конденсация с фталевым
ангидридом 282
моторный 98
окисление 248
получение 146
сульфирование 251, 265 ел.
сырой 93, 96, 97
улавливание нз коксового газа 96
хлорирование 192 ел., 251, 352 ел.
Беизолсульфокислота 269
Бисдиазосоединении 293
Битумы
нефтяные 51
ириродные 21
торфяные 80
Бланкед-крепы 481
Блаикофор Б 327
Блоксополимеризация 431, 435 ел.
Блоксополимеры 370, 431, 435, 436
Блочная поликоиденсация 404, 405,
408 ел., 415
Блочная полимеризация 402 ел.,
407 ел., 415
непрерывная 426 ел.
Бобинное формование 454, 456, 457
Богхеды 82
Бомбировка валков 517
Брекер 505, 506
Брикетирование 70, 86
Брикеты 12
Броминдиго 316
Бурение скважин 23 ел.
Бурый уголь И, 12, 69, 82, 83
свойства 18
состав 114
энергоемкость 19
Бутадиен-1,3 (Дивинил)
полимеризации 395
полимеры 381, 383, 395, 407,
483 ел., 494, 495
синтез 132, 141 ел., 211, 222
сополимернзацни со стиролом
429, 430
сополимеры 429, 430, 487 ел., 490,
494 ел., 506, 541
Бутаднен-метилвиннлпирндиновый
латекс 496
Бутаднен-метилстнрольные каучуки
487, 488
Бутадиен-нитрнльные каучуки 489,
490, 494, 506, 541
применение 554, 558, 576
Бутадиеновые каучуки 381, 383, 395,
407, 483 ел., 494, 505
Бутадиен-стирольные каучукн 430,
487, 488, 494
Бутан
дегидрирование 141 ел.
изомеризации 149
пиролиз 134 ел.
Бутвар (Поливинилбутираль) 380,
434, 564, 574
Бутнлбензол 249
Бутнлены
дегидрирование 141, 143 ел.
изомеризация 149
получение 134, 142
Бутнлкаучукн 482, 490, 491, 494, 506
Бутиловый спирт 171
N-Бутилпирролиднн 171
Бутилфенол, нитрование 340
Бутифос (Трибутилфосфат) 350
Бутоксиэтиловый эфнр 2,4-Д 343
581
БФ клей 438, 575
БЭКТ (бис-Эти л исантогентри
сульфид) 350
ВА аппарат 451 ел.
Вазелин 127
Вакууы-ксантатсмеситель 450, 451
Вакуумное формование 536
Вакуум-перегонка 46 ел.
Вакуум-ректификация 159
Вакуум-фильтры барабанные 296,
297
Валки каландров 516, 517
Вальцевание 545
Вальцы 509
Ванна осадительная 445, 454, 455,
461, 462, 464, 465, 467, 468
Ваниа-пресс мерсеризационный 447,
448
Вапам (N-Метилдитиокарбамат
натрия) 347, 349
Варочный котел 406, 407
Взрываемости пределы 154
Взрывоопасность нефти 40
Винилацетат 226, 530
полимеризация 395, 405, 428
полимеры см. Поливинилацеіат
Винилацетилен 191
Винилбензол см. Стирол
Винилиденовые полимеры 378, 493
Винилиденфторид, сополимеры 493
Винилиденхлорид 191
Винилхлорид
полимеризация 395, 422
полимеры см. Поливинилхлорид
получение 182, 187 ел.
свойства 190
сополимеры 466
Винильные полимеры 378, 466
Винипласт 542 ел.
Виньон N 466
Вискоза 450 ел.
созревание 451, 453
устойчивость 453
Вискозное волокно 447 ел.
вытяжка 455
отделка 459
пиролиз 563
свойства 457
формование 453, 457
Витрен 81
Ватта теория цветности 288
ВК-32, клей 438, 574
Водород
получение 107
теплота сгорания 17
Водяной газ 12, 71, ПО ел., 163
Воздушное дутье 111
Воздушный газ ПО ел.
волокна
Волокнистые наполнители 528
Волокиты 528, 556 ел.
Воск 70, 80
Восстановление
катализаторы 274
нитросоединений 272 ел.
ВОТ 129, 130, 288, 472
Вращательное бурение 23
Вспенивание 549, 576 ел.
Вулканизаторы барабанные 522
Вулканнзаты 478, 494, 495
Вулканизация 384, 431, 437, 438, 478,
519 ел.
горячая 520
замедлители 499
плато 520
преждевременная 499
ускорители 361 ел., 498, 499, 503
холодная 520
Вулканизующие вещества 478, 498
Высокомолекулярные соединения
367 ел., см. также Полимеры
Высокооктановые добавки 128
Высшая теплотворная способность
топлива 17
Вытяжка химических волокон 465
вискозного 455
полиолефиновых 470
холодная 472
Вязкость
нефти 40
растворов полимеров 371, 372
Вязкотекучее состояние 375, 376
%
Газификация то пли в 12, 71, 106 ел.,
132
жидких 107, 132
коэффициент полезного действии
' 113
паро-кислородная 113
подземная 114
торфа 79
Газование 111, 112
Газовая промышленность 20
Газовая сажа 36, 37
Газовый бензин 33 ел., 101, 105, 157
Газовый уголь 70, 114
Газогенераторы 108 ел., 111, 112, 114,
139 ел.
Газойль 39, 132
Газообразное топливо 9, 12, 18, 21
Газоподъемная установка 26 ел.
Газы
водяной 12, 71, ПО сл„ 163
воздушный ПО ел.
генераторные 12, 18, 71, 72, 80,
ПО ел., 163
582
Газы
для синтезов 107, 163 ел.
доменный 12
карбюрированный 12
коксовый 12, 18, 86, 92, 93
компрессия 34, 157
крекинга 28, 29, 59
нефтяные 27 ел.
осушка 30
очистка см. Очистка газов
переработка 27 ел.
пиролиза 28, 29, 157
платформинга 28, 29
подземной газификации 111
полукоксования 101
попутные 12, 21, 27 ел.
природные см. Природные газы
разделение 27 ел., 33, 155 ел., 159
светильный 12, 92
сжижение 28, 29, 34
смешанный 12
сухой 27
топочные 17
тощий 27
углеводородные 27 ел.
Галоидированне углеводородов
174 ел., 180 ел., 186 ел., 192 ел.,
251, 252, 352 ел.
Гамма-кислота 295
Гевея 481
Гексаметилендиамин 390
Гексаметилендиизоцианат 412
Гексаметилентетрамин (Уротропин)
234, 530, 551, 576
Гексахлоран (Гексахлорциклогек-
сан) 192, 352 ел.
Гексахлорбензол 341
Гексахлорметилциклогексан 192
Гексахлорэтан 186
Гексахлорциклогексан (Гексахлоран)
192, 352 ел.
Гемицеллюлозы 72, 73, 77
Генераторные газы 12, 18, 71, 72, 80,
ПО ел., 163
Генераторы ацетиленовые 139 ел.
Гептахлор 354
Гербициды 335, 338 ел., 346 ел.
токсичность 339 ел., 348, 349
Гетероазеотропная система 161
Гетерогенный катализ 133
Гетероцепные волокна 440, 471 ел.
полиамидные 390, 410, 470 ел., 502
полиэфирные 389, 409, 440, 474,
475
Гетероцепные полимеры 378, 381 ел.
Гетероциклы, гидрогенизация 116
Гетинаксы 564, 567
Гидравлический пресс 532, 533
Гидразобензол 275
Гидракриловая кислота 237
Гидрататор 208, 217
Гидратация 204 ел.
ацетилена 217 ел.
катализаторы 204, 206, 207, 217
окиси этилена 245
олефинов 204 ел., 211, 214
Гидратцеллюлоза 454, 460
Гидрирование сы. также
Гидрогенизация
катализаторы 254, 259
углеводородов 115, 249
фенола 254 ел.
Гидрогенизационный крекинг 38, 57,
67.
Гидрогенизация 71, см. также
Гидрирование
гетероциклов 116
двухступенчатая 117 ел.
деструктивная 38, 67, 107, 115 ел.
жидкофазная 117
нафталина 116
топлив 106 ел., 114 ел.
условия 116
Гидрокрекинг 38, 57, 67
Гидролиз
алкилсульфатов 209
гексахлорбензола 341
глицеридов 185
древесины 72, 77
ксантогената целлюлозы 452, 453
поливинилацетата 434
тетрахлорбензола 342
хлорбензола 251
хлористого аллила 185
Гидролизеры 77, 209, 210
Гидроочистка 56, 67
Гидроперекиси 215, 241, 249, 250
Гидростабилизация 67
Гидроторф 79
Гидроформилирование 168 ел.
Гидроформинг 66, 147
Гидрохлорирование 175, 181 ел.,
188 ел.
Гидроэнергия, потребление 14
Гиперсорбция 156 ел.
Гипохлорирование этилена 243
Гистерезисные потери 477
Глифталевые смолы 126
Глицерин 184 ел.
Глицеринтринитрат (Нитроглицерин)
186
Глубинный насос 26 ел.
Глухой пар 35
Гомогенный катализ 133
Гомополимеры 369, 479
Гопкалит 173
Горелочные каналы 89, 90
Горный воск 70
Горючие вещества органические
9 ел., 114
583
р y
Горячее дутье 111, ]]2
Гранозан (Этнлмеркурхлорид) 358
Графитированное волокно 563, 564
Графитопласты 564
Гудрон 51, 54 ел.
Гуминовые вещества и кислоты 83
Гумус 79
Гуттаперча 381, 489
2,4-Д B,4-Дихлорфеноксиуксусная
кислота) 124, 226, 341 ел., 348
Дайнель 466
Далапон (а,а-Дихлорпропионат
натрия) 339, 348
Двухвальцовая сушилка 297, 298
ДДД 352
ДДТ (Дихлордифенилтрихлор-
метнлметан) 124, 214, 282, 351
Деалкнлирование 151
Деасфальтизация 56
Дегидратация
катализаторы 2] ]
спиртов 211, 228 ел.
этиленцнангидрина 237
Дегидрирование 28, 29, 57, 59, 132,
134, 141 ел., 147 ел.
амиленов 145
бутана 141 ел.
бути ленов 141, 143 ел.
в кипящем слое 143
изопентана 141, 145
катализаторы 141 ел., 147, 211,
214, 219, 256
нафтенов 57, 59, 132, 146
спиртов 211, 214, 219, 233
циклогексанола 255 ел.
этана 141
этилбензола 147, 148
Дегндроароматизация (Дегидроцик-
лнзация) 57, 132, 146 ел.
Дегндрохлорирование 187 ел.
Дегидроциклизация 57, 132, 146 ел.
Деготь 68, 70, 72, 73, 75, 80
первичный 101, 105, 1І4
Дезинтеграторы 298
Декобальтизация 169 ел.
Декристаллизацня каучука 507
Депарафнинзация 56
Десиканты 335, 350
Десорберы 32, 33, 155
Десорбция бензина 35
Деструктивная гидрогенизация 38,
67, 107, 115 ел.
Деструкция 388, 497
окислительная 498, 507
поливннилхлорида 542
фторопласта 543
Детергенты 127, 128, 329 ел.
Детонация 49 -, . , ']
п „*„„„„„,.„ адг; 350 СЛ.
Деформации полимерии oii, o/.j, -,,,
Деэмульгаторы 41
Диазоли 308, 309
Диазосоединення 292
Дназосоставляющие 292 ел.
Диазотнрование 292 ел., 310
анилина 292, 304
бензидина 293, 302
Диаллиламин 339, 340
Диаллилфталат 530
Г^Ш-Диаллилхлорацета.мид (Рэндокс>
339, 340, 348
Днанизидин 276, 293
Диариламины 281
Диацетоновый спирт 216
Днбензотиазолднсульфид (Альтакс)
362, 499
Дивинил см. Бутаднен-1,3
Дивини лбензол, сополимеризация-
424, 425
Диеновый синтез 354
Диизоцианаты 125, 412, 576, 577
Дильса—Альдера реакция 354
Днметнланилин 280, 323
4,4-Диметил-1,3-дноксан 235
Диметнлдитиокарбаматы 359, 363,
364
Днметилснландиол 390
Диметилтерефталат 262, 408
Диметилтнокарбамат натрия 363
Диметнлформамнд 139, 465
Димнт 357
Днннтрат этиленгликоля 245
Динитробеизол, восстановление 274
Динок (Дннитро-о-крезол) 340, 341
Днносеб B-вгор-Бутил-4,6-дннитро-
фенол) 340, 341, 348, 350
Диоксан 246
Днолефнны 132, см. также Б\тадие«
и Изопрен
Дисазокраснтели 292, 294 ел., 300,
302, 307
Диспергаторы 332, 419, 423
Дисперсные красители 309 ел.
алый Ж 309
оранжевый П 309
синий К 314
Дитиокарбаминовая кислота 347 ел.
соли 359
Днтолилметан 130
Диурон 344, 349
Дифенил 130, 288, 472
Дифениламин 281
Днфенилгуанидин 363
2,2-Дифеннлдисульфнд 365
Днфеннлолпропан (Дноксидифенил-
изопропан) 390, 412
Дифеннл-я-фенилендиачин 365
Дифенильная смесь 130, 472
Дихлон B,3-Дихлор-1,4-нафтохинон)
359
Дихлорацетилен 187
Дихлордиметилснлан 178
Дихлордифенилтрихлорметилметан
(ДДТ) 124, 214, 282, 351
Дихлорднфторметан (Фреон-12) 129
Дихлорднэтиловый эфир 391
Дихлорпропан 182, 184
Дихлорпропионовая кислота 339
2,4-Дихлорфенокснуксусная кислота
B,4-Д) 124, 226, 341 ел., 349
Дихлорэтан 181 ел., 188, 350
дегидрохлорирование 189 ел.
пиролиз 190
синтез 243
хлорирование 191
Диэлектрические свойства
пенопластов 550, 577
полненлоксаиов 556
слоистых пластиков 567
термопластов 542
фенопластов 554
фторопласта 544
Диэтиленгликоль 30, 246
Диэтилеигликольтерефталат 408
ДНБФ B-вгор-Бутнл-4,6-динитрофе-
нол) 340, 341
ДНОК (Дннитро-о-крезол) 340, 341,
348
Добыча топлив 9, 14
нефти 21, 23 ел.
природного газа 21
торфа 78, 79
угля 68, 84
Додецен 152
Додецилбензол 152, 331
Додецилбензолсульфонат (Сульфо-
нол) 331
Долото 23, 24
Доменный газ 12
Древесина 9 ел., 69
гидролиз 72, 77
запасы 12 ел.
переработка 72 ел.
свойства 18, 72
состав 72
сухая перегонка 72 ел.
энергоемкость 19
Древесная мука 527, 528, 551, 553,
554
Древесная смола см. Деготь
Древесно-слоистые пластики (ДСП)
564, 566 ел.
Древесный порошок 76
Древесный уголь 12, 73, 75
Дробная перегонка нефтей 42
Дресселнрование 157
Естественное топливо 11 ел. ]
Живица 76
Жидкие каучуки 479, 491
Жидкое топливо 9, 12
газификация 107, 132
свойства 18
Жидкофазные процессы
гидрогенизации 117
нитрования 198
очистки нефтепродуктов 52
Жирный газ 27
Жировые мыла 185, 329
Забой скважины 23
Замасливание волокон 446, 459, 464
Замасливателн 459
Замедлители вулканизации 499
Замутннтели пластических масс 531
«Запекание» 267, 268, 319
Защелачивание 42, 52
Зеленое масло 63
Зелинского реакция 146
Зола 15, 18,72, 85
Зоокумарин 360, 361
Зооциды 335, 360, 361
Избирательные (ая)
адгезия 570
гербициды 338, 346, 347
растворители 127, 139, 155, 160,
162
Измельчение
алкалицеллюлозы 448 ел.
каучуков 508
красителей 298
пресспорошков 551
Износостойкость
резин 478, 484, 494
термопластов 539
Изобутилеи
полимеризация 418, 419
получение 149, 228 ел.
сополимеры 490, 494, 495
Изобутнловый спирт, дегидратация
228 ел.
Изомеризация 57, 148 ел.
окисей олефинов 242, 245
циклогексаноиоксима 257
Изооктан 49
Изопентан
дегидрирование 141, 145
получение 149
Изопрен 369, 482
полимеризация 395
полимеры 368, 381, 383, 395, 486.
487, 494, 505
синтез 132, 141, 145 сл„ 234 235
сополимеры 490, 494, 495
585
Изопреновые каучуки 368, 3ol, doo,
395, 486, 487, 494, 505
Изопропил хлористый 182
Изоиропплбензол (Кумол) 147, 150,
249, 252
Изопропиловый спирт 214, 215, 346
Изопропил-ІМ-феннлкарбамат (ИФК)
345, 346, 349
Изопропил-N- (З-хлорфенил)-карба-
мат (ХлорИФК) 346, 349
Изотактические полимеры 373, 374
Ингибиторы
окисления 52
отверждения 530
Ингредиенты резиновых смесей
364 ел., 497 ел.
вулканизующие вещества 478, 493
замедлители вулканизации 499
контроль качества 524
мягчители (пластификаторы) 488,
500
подготовка 508 ел.
противостарители 361, 365, 500,
501
противоутомители 365, 50]
разбавители 501
усилители 489, 499, 500
ускорители вулканизации 361 ел.,
498, 499, 508
пластикации 361, 365, 366, 498, 508
Индантрон 317
Индиго 286, 314 ел.
Индигозоли 318, 319
Индигоидные красители 316, 317
Индустриальные масла 50
Инертные наполнители 501
Инициаторы полимеризации 396, 401,
422, 425
Инсектициды 335, 347, 350 ел.
фосфорорганические 354 ел.
Иониты 424, 425
Ионная полимеризация 394, 396,
399 сл„ 401, 403, 418
Иприт 248
Искусственное топливо 11 ел., 15
Искусственные волокна 439, 440,
447 ел.
ацетатное 462 ел.
вискозное см. Вискозное волокно
медноаммиачное 459 ел.
ИФК (Изопропил-]М-фенилкарбамат)
345, 346, 349
Каландрование 516 ел.
Каландры 516 ел.
Калориметр 16, 17
Каменноугольная смола 69, 93,
98 ел., 114
Каменноугольное масло 96, 98 ел.
Каменноугольный пек 93, 98, 101
392,
каменные угли 11, iz, иэ, oz, c_
коксование 81, 86 ел.
свойства 18
энергоемкость 19
Камфора 545
Канальная печь 74
Канифоль 70, 72, 73, 76, 488
Кантовка 90
Капролактам 124, 203
полимеризация 257, 396, 405
полимеры см. Капрон
синтез 254 ел.
Капрон (Поликапроамид) 203, 372,
396, 405, 410, 411, 471, 502, 503
Каптакс B-Меркаптобензотиазол)
362, 364, 499
Каптан (N-Трихлорметилтиоимид те-
трагидрофталевоц кислоты)
360
Карбазол 100, 101
Карбаматы 351, 355 ел.
Карбамид, производные 344 ел
о9о
Карбамидные полимеры 382
применение 550, 551, 556
синтез 392, 393, 417
Карбамидо-формальдегидные
полимеры 126, 382, 555
отверждение 566
применение 564, 574
Карбид кальция 132, 139 ел.
Карбидная теория происхождения
нефти 22
Карбин (Хлорбутинил-М-хлорфенил-
карбамат) 346, 347, 349
Карбоксилатные латекса 495, 496
Карбонилы
железа 167
кобальта 169 ел.
Карбоновые кислоты
адипиновая 250, 251, 258, 390
амиды 339 ел., 385
арилоксиалкилированные 341 ел.
бензойная 248, 249, 253
малеиновая 389
масляная 241
муравьиная 230, 241
пропионовая 230, 241
себациновая 387
синтез 169, 172 ел., 240 ел., 248,
250, 253, 258
стеариновая 333
уксусная см. Уксусная кислота
хлорированные 339
Карботион (N-Метилдіітнокарбамат
натрия) 347, 349
Карбофос 356
Карбоцепные волокна 440, 464 ел.
нитрон 465, 466
отделка 464
586
Карбоцепные волокна
поливинилхлоридные 466 ел.
полиолефиновые 469, 470
формование 464
фторсодержащие 468
хлорин 467, 468
Карбоцепные полимеры 377 ел.
Карбюрированный газ 12
Кармин красный 286
Катализаторы 133 ел.
алкилирования 150 ел., 279 ел.,
334
ацеталирования 434
восстановления 274
гидратации 204, 206, 207, 217
гидрирования 254, 259
гидроформилнрования 168 ел.
гидрохлорирования 188
дегидратации 211
дегидрирования 141 ел., 147, 211,
214, 219, 256
дегидрохлорирования 191
дегидроциклизации 146
изомеризации 149
носители 134
окисления 2І5, 219, 220, 223, 3^5.
230, 231, 235, 240, 242, 243, 248,
250, 252, 261
окисные 133, 143, 145, 147, 164,
231
оксосинтеза 169 ел.
отверждения 573, 575
пиролиза 227
плавленые 134
полимеризации 245, 399, 418, 420,
483
получение 133
производительность 133
пылевидный 143
регенерация 142, 143
Ренея 134
¦синтеза карбоновых кислот 172
— спиртов 171
скелетные 134, 259
стереоспецифические 420, 483
сульфирования 266
удельная поверхность 133
хлорирования 176, 187, 190, 192,
339
Каталитический крекинг 38, 57, 64 ел.
Катионная полимеризация 396, 399
Катионные азокраентели 310
Катионоактивные ПАВ 330, 333
Каучуки 479 ел.
вулканизация см. Вулканизации
гомополкмерные 479
декристаллизация 507
жидкие 479, 491
измельчение 508
.контроль качества 523, 524
Каучуки
масляные 488
наполненные 483
натуральный см. Натуральный
каучук
общего назначения 480
пластикация см. Пластикация
подготовка 507 ел.
потребление 478
развеска 508, 509
растворители 503
сажемасляные 488
свойства 480, 483, 485 ел., 490 ел.
синтетические см. Синтетические
каучуки
специального назначения 480,
489 ел.
усилители 489, 499, 500
Каучуконосы 481
Кварцевая мука 527, 528, 553, 555
Керосин 12, 20, 38, 39, 114, 132
свойства 18, 49 ел.
Кетен 216, 217, 226 ел.
Кетоны 216
ацетон см. Ацетон
метилпропилкетон 216
метилэтилкетон 249
получение 242
Кипящий слой 62
дегидрирование 143
коксование 61 ел.
крекинг 65 ел.
окисление нафталина 261
М-Кчслота 295
Кислотные красители 299 ел., 311 гл.
алый прочный НЖМ 301
арилметановые 322 ел.
белый 328
зеленый антрахнноновый 311 ел.
— ЖМ 301
оранжевый 291, 308
сине-черный 294
хромовые 300, 301
черный С 296
Кислоты карбоновые см. Карбоновм»».
кислоты
Кислый гудрон 54 ел.
Кишечные яды 350, 351, 353
Классификация
красителей 290, 291
нефтей 39
полимеров 377 ел.
топлив 11 ел.
углей 82 ел.
Клеве-кислота 295
Клеепромазочная машина 518
Клеи синтетические 412, 438, 503 504
518, 570, 573 ел.
Кокс 11, 12. 16, 70, 86
беззольный 51, 61, 101
587
металлургический 89, 91
нефтяной 51, 61
торфяной 80
Коксование 69. 71
в кипящем слое 61 ел.
материальный баланс 92
нефтепродуктов 51, 58, 61 ел.
тепловой баланс 91
углей 81, 86 ел.
Коксовая смола 114
Коксовые
печи 86 ел.
угли 67, 114
Коксовый
газ 12, 18, 19, 86, 92, 93
пирог 87
Коксохимическая промышленность 86
Коллоксилин 433
Колонны
гипохлорирования 244
дегидрохлорирования 187 ел.
отпарные 158
полимеризационная 426
ректификационные 42, 43, 158, 160,
161
синтеза метанола 167
хлорирования 214
экстракционные 160
Колпачковая тарелка 43
Компрессионная установка 35
Компрессия газов 34, 157
Конверсия
метана 132, 163 ел.
окнеи углерода 163
Конверторы 61, 144 ел., 147, 148, 165
Конденсацнонно-ректификационное
разделение газов 157 ел.
Конденсация, реакции 282, 283, 385
альдольная 222
бензальдегида с ацетоном 360
бензола с фталевым ангидридом
282
диэтилфталата ¦ с дифенилацето-
ном 360
толуидина с нитрозофенолом 321
хлораля с диметилфосфитом 355
с хлорбензолом 282, 351
хлоранила с нероловой кислотой
326
Контакт Петрова 573
Контактная печь 211, 212
Контактное прессование 559, 560
Контактные пестициды
гербициды 338, 340
инсектициды 351, 353, 356
Контактный аппарат 229
Корд 502, 524
Кордная нить 457, 474
Котел варочный 406, 407
588
полезного действия газификации
113
теплопроводности пенопластов
550, 577
Красители 286 ел.
адсорбция на волокнах 300, 305,,
316, 321
азиновые 324, 325
азокрасители см. Азокрасители
активные см. Активные красители
антрахиноновые см.'Антрахиноно-
вые красители
арилметановые 322 ел.
дисперсные см. Дисперсные краси-
•тели
для меха 325
индигоидные 316, 317
катнонные 310
кислотные см. Кислотные
красители
классификация 290, 291
кубовые 315 ел.
металлсодержащие 301, 321
номенклатура 291
оксазиновые 325, 326
основные 323
полициклические 317 ел.
протравные 300, 311
прямые см. Прямые красители
размол 298
сернистые 319 ел.
сушка 297, 298
тназиновые 325
установка на тиі 298, 299
фталоцианиновые 321 ел.
хинониминовые 324 ел.
«холодные» 308
хромовые 300, 301
цвет и строение 287 ел.
Краски 286
Крашение волокон 299 ел., 305,
307 ел., 321
Крезолы
нитрование 340
получение 215
Крекинг (а)
в кипящем слое 65 ел.
газы 28, 29, 59
гидрогрнизационный 38, 57, 6?
каталитический 38, 57, 64 ел.
мазута 60 ел.
материальный баланс 61
нефтепродуктов 56 ел.
нефти 38
термический 57 ел.
углеводородов 59, 135, 13&
условия 59 ел., 64
Крекииг-установка 60 ел. і
Кремнийоргаиические полимеры 382
каучуки 491, 492, 494, 506
применение 383
синтез 393, 394
Креозот 75
Кривая разгонки 39
Кристаллизация фракционная 162
Кристаллические полимеры 375
Кротоновый альдегид 222
Крутка нити 458
Ксантатбарабаны (Бараты) 450
Ксантогенирование 449, 450
л-Ксилидин 292
Ксилолы 63, 97
изомеризация 149
окисление 249, 260, 261
разделение 162
Кубовые красители 315 ел.
Кубозоли 318, 319
Кубы перегонные 44
Кумол (Изопропилбензол) 147, 150,
249, 252
Кустовое бурение 25
Кучерова реакция 217
Лавсан (Полиэтиленгликольтерефта-
лат) 125, 262, 408 ел., 474, 502
Лаки 307, 308, 324
Лаковая
поликонденсация 416, 417
полимеризация 405, 416 ел., 428,
429
Латексы 481
синтетические 222, 422, 479, 495,
496
Легкие нефти 39
Легкое масло 98
Лейкосоединения 316, 319, 321 ел.
Леониль ОХ 333
Лигнин 72, 73, 77, 78
Лигниты 83
Лигроин 12, 39, 49, 132
Линейные полимеры 370 ел., 389 ел.
ориентация 442, 547, 561
сшивка 377, 387. 392 ел., 401, 413,
424, 431, 437, 438, см. также
Вулканизация
Листовые термопласты 535 ел., 542,
543
Литраж 139
Литьевая машина 533, 534
Литьевые пластмассы 540, 541
Литье под давлением 523, 528,
533 ел., 539, 545
Мази 51
Мазут 39, 44, 114, 132
крекинг 60 ел.
перегонка 46 ел.
свойства 51
Макроионы 399
Макромолекулы 368, 369
ориентация 442, 547, 561
сшичка 478, 493
фиксация 376, 377
Макрораднкалы 398, 436
Малеиновая кислота 389
Малеиновый ангидрид 248
Манеб 359
Марковна/сова правило 182, 204 '
Масла
антраценовое 98, 100
зеленое 63
каменноугольное 96, 98 ел.
легкое 98
медицинские 38
нафталиновое 63, 98, 100
очистка 53 ел.
смазочные 20, 38, 50, 128, 175
соляровое 12, 50, 96
технические 93
трансмиссионные 50
трансформаторное 38
тяжелое 98
фенольное 98
Масляная кислота 241
Масляные каучуки 488
Материальный баланс
коксования 92
крекинга 61
Матрицы 531, 532, 535, 536
Медицинские масла 38
Медноаммиачное волокно 459 ел.
Меламии 393
Меламино-форм альдегидные
полимеры 417, 550, 551, 554, 558
Мелаиж 200
Менделеева
теория происхождения нефти 22
формула теплотворной
способности 17
Меркаптаны 53
2-Меркаптобензотназол (Каптакс)
362, 364, 499
Мерсеризация 447, 448, 451, 452
Местное топливо 11
Метакриловая кислота 240
Металепсия 176
Металлокорд 502, 503
Металлоорганические пестициды
357 ел.
Металлургический кокс 89, 91
Метальдегид 221
Метан 131
конверсия 132, 163 ел.
крекинг 135, 136
окисление 230 ел.
получение 112, 162, 164
хлорирование 176 ел.
589
Метаниловая кислота 292
Метанол см. Метиловый спирт
Метафос 3S6
Метил
бромистый 350
хлористый 176, 178
Метилаллил хлористый (Метилхлоро-
прен) 182
Метиламин 333
Мет.илацетофос 356
2-Метилбутадиен-1,3 см. Изопрен
Метилбутилкетон 216
2-Метил-5-винилпиридин 222
N-Метилдитиокарбамат натрия (Кар-
ботион, Вапам) 347, 349
Метиле н хлористый 178
Метнлкаучук 481
Метилмеркаптофос 354 ел.
Метилметакрилат 216, 239, 240, 373
полимеризация 395, 405, 414
а-Метилнафталин 50
Метилнитрофос 356
Метиловый спнрт 73, 75, 76
дегидрирование 233
окисление 232 ел.
получение 164, 166, 230
применение 168
синтезы на его основе 355
Метилолкарбамиды 393
Метилолмеламины 393
N-Метилпирролидон 139
Метилпропилкетон 216
а-Метилстирол 147
применение 487, 488
Метилхлоропрен 182
Метилэтилкетон 249
Метоксихлор 352
Метоксон B-Метил-4-хлорфенокси-
уксусная кислота, 2М-4Х) 342,
343, 349
Механическая пластикация 507
Механоокислительная пластикация
436
Многоэтажный пресс 566, 567
Мовенн 286
Модифицирование полимеров 384,
430, 431, 438, 541, 554, 558, 559,
564, 576, S77
Модуль упругости
резины 477
термопластов 540, 541
Мокрое обогащение 85
Мокрое формование химических
волокон 443
вискозного 453 ел.
карбоцепных 464 ел.
медноаммиачного 460
Молекулярная концентрация 34
Моноазокрасители 292, 300, 306, 307
Моноволокно 441, 470, 474
590
Мономеры 124, 125
активация 393, 394, 397
для синтеза каучука 482
Моптан-воск 127
Монурон 344, 349
Морозостойкость
каучуков 490, 492, 493
термопластов 540, 541, 544
Морфолин 247
Моторное топливо 38, 49, 98, 105, 128
Моющие средства (Детергенты) 127,
128, 329 ел.
Мочевина см. Карбамид
Мочевино-формальдегидные
полимеры см. Карбамидо-формальде-
гидиые полимеры
Муравьиная кислота 230, 241
Муравьиный альдегид см.
Формальдегид
Мыла жировые 329
Мягчители 488, 500
Набам (Этилен-бис-дитиокарбамат
натрия) 359
Надсмольная вода 73, 95
Надуксусная кислота 223, 225
Наирит 489, 494
Найлон 125, 470 ел., 503
Намотка стекловолокна 558, 561 ел.
Наполненные каучуки 483
Наполнители
активные 137, 499, 500
инертные 501
красителей 299
пестицидов 337
пластических масс 527, 528, 554,
555, 564, 566
резиновых смесей 489, 499 ел.
Напыление 558, 560
Насадкн абсорберов 32
Насос глубинный 26 ел.
Натуральный каучук 381, 480 ел.,
494, 505
декристаллизация 507
пластикация 507, 508
свойства 494
строение 479
Нафталин 63, 100
гидрогенизация 116
окисление 260 ел.
сульфирование 265, 332
Нафталиновое масло 63, 98, 100
Нафталинсульфокнслоты 269 332
Нафтацен 289
Нафтено-ароматические нефти 39
Нафтено-парафиновые нефти 39
Нафтены 115
дегидрирование 57, 59, 132, 146
окисление 230, 249, 250
а-Нафтиламин 276, 293, 294
сульфирование 268
Нафтиламинсульфокислота 293
Нафтионовая кислота 268, 295
?-Нафтол 272, 281, 294
Нафтохинон 359
Небурон 344, 349
Неионогенные ПАВ 330, 333, 334
Нематоциды 347
Неозон Д (Фенил-2-нафтнламнн) 281,
365
Неорганические волокна 528, 557, 563
Непрямая гидратация 204, 205
Нероловая кислота 326
Нефтезаводские газы 27 ел.
Нефтепродукты 48 ел.
коксование 51, 58, 61 ел.
крекинг 56 ел.
остаточные 51, 61
очистка 51 ел.
пиролиз 58, 62
риформннг 57 ел., 66
светлые 49
Нефтехимическое сырье 38, 48
Нефть 9 ел., 20 ел.
взрывоопасность 40
добыча 21, 23 ел.
запасы 12 ел., 21
защелачивание 42, 52
классификация 39
крекинг 38
месторождения 21, 22
обезвоживание и обессоливание
41 ел.
однократное испарение 44, 45
отделение от газа 4]
очистка 38
перегонка 38, 42 ел.
переработка 20, 37 ел.
пиролиз 38
происхождение 22
ректификация 42 ел.
свойства 18, 40
сортировка и смешение 42
состав 39 ел., 114
НефТЯНОЙ "'
кокс 51, 61
пек 63
Нефтяные
битумы 51
газы 27 ел.
фракции 39
Нижний предел взрываемости 154
Низкотемпературная полимеризация
483, 488
Низшая теплотворная способность
17, 111
Нитрат целлюлозы
(Нитроцеллюлоза) 433, 545
Нитраторы 199 ел.
Нитрилы
акриловой кислоты см. Акрило-
нитрил
гидракриловой кислоты 236, 237
Нитроанилин 274, 278, 279, 292
Нитробензол 201 ел.
восстановление 273 ел.
Нитрование
углеводородов 196 ел., 200 ел.
фенолов 340
целлюлозы 433 ,
этаноламинов 247
этиленгликоля 245
Нитроглицерин 186
Нитрометан 197 ел.
Нитрон 465, 466
Нитропропан 197 ел.
оНитротолуол, восстановление 273
Нитрофенолы 340
Нитроэтан 197 ел.
Нитрующая смесь 200, 433
Новолаки 392, 530, 551, 576
Номер волокна 441
Носители катализаторов 134
Обезвоживание нефти 41 ел.
Обессоливание иефти 41 ел.
Облагораживание топлив 106
Облагороженное топливо 15, 68
Обогащение 70
углей 84 ел.
Обратный газ 92
Обрыв цепи полимера 399, 400
Объемная скорость 64
Однократное испарение 44, 45
Ожижение угля 71
Окиси олефинов 242 ел., 396, см.
также Окись этилена .
Окисление
антрацена 282
ацетальдегида 223, 225
бензола 248
бутилбензола 249
в кипящем слое 261
диметнлтнокарбамата натрия 363
изопропилбензола 249
изопропилового спирта 214, 215
ингибиторы 52
катализаторы 215, 219, 220, 223,
225, 230, 231, 235, 240, 242. 243,
248, 250, 252, 261
ксилолов 249, 260, 261
метанола 232 ел.
нафталина 260 ел.
нафтенов 230, 249, 250
591
Окисление
олефинов 215, 220, 221, 241 ел.
парафинов 51, 107, 229 ел., 240 ел.
толуола 248, 252 ел.
топлива 106 ел.
циклогексанола 258, 259
этилового спирта 219
Окислители 230
Окислительная деструкция полимеров
498, 507
Окислительное хлорирование 174, 251,
252
Окислительный пиролиз 135
Окись углерода
конверсия 163
синтезы на ее основе 162 ел.
токсичность 172
Окись этилена
полимеризация 396
получение 242 ел.
синтезы на ее основе 245 ел., 333,
334, 355
Оксазиновые красители 325, 326
Оксиантрахинон 270
Оксикарбоновые кислоты 284 ел., 294
Оксикумарин 360, 361
Оксимирование 256, 257
Оксинафтойная кислота 285
8-Оксихинолят меди 358, 359
Оксиэтилирование 281, 333, 334
Оксосинтез 168 ел.
Октадециловый спирт 333
Октаметил 357
Октановое число 49, 64, 128, 148, 149
Олефины
гидратация 204 ел., 211, 214
гидроформилирование 168 ел.
гидрохлорирование 182
крекинг 59
окиси 242 ел., 396, см. также
Окись этилена
окисление 215, 220, 221, 241 ел.
полимеризация 395, 415, 416,
418 ел.
получение 131, 164, 228
хлорирование 181 ел., 188
Олигомеры 375 ел., 391, 392, 399,
404, 425, 479
Омыление жиров 185
Оптические отбеливатели и
осветлители 326 ел.
ОП препараты 330, 334, 335, 337
Опудривание 515
Опудривающие вещества 504
Органическая теория происхождения
нефти 22
Органические стекла 414, 545 ел.
Осадительная ванна 445, 454, 455,
461, 462, 464, 465, 467, 468
Осахаривание 77
Основной органический синтез
121 ел.
методы 122
продукты 124 ел.
сырье 131 ел.
Основные красители 323
Острый пар 35
Осушка газов 30
Отбелка 326, 459
Отверждение полимеров 384, 438
527, 550, 551, 560, 561, 575,
576
ингибиторы 530
катализаторы 573, 575
полисилоксанов 555
термореактивных 566, 574 ')
ускорители 530
Отделка
вискозного волокна 459
карбоцепных волокон 464
медноаммиачного волокна 462
резиновых изделий 523
филаментных нитей 446
штапельного волокна 446, 447
Очистка газов 31 ел.
коксового 92 ел.
мокрая 31
от серы 31 ел., 93, 96
Очистка
масел 53 ел. '
нефтепродуктов 51 ел.
нефти 38
Паральдегид 221, 222
Парарозанилин 286
Парафин 51, 127, 240, 530
Парафино-нафтеновые нефти 39
Парафины 21, 22, 27 ел., 37
дегидроциклизация 132, 146 ел.
крекинг 59, 135, 136
нитрование 196 ел.
окисление 51, 107, 229 ел., 240 ел.
пиролиз 131, 135
получение 164
хлорирование 175 ел., 180 ел.
Параформальдегид 233
Парофазные процессы
нитрования 198 ел.
очистки нефтепродуктов 52, 54
сульфирования 266 ел.
Пек
каменноугольный 93, 98, 101
нефтяной 63
Пенопласты
термопластичные 548 ел.
термореактивные 576 ел.
Пенополивинилхлорид 549, 550, 577
Пенополисилоксаны 577
592
Пенополистирол 549, 550, 577
Пенополиуретаны 577 ел.
Пенорезины 495, 496, 519
Пентакарбонил железа 167
Пентан, изомеризация 148
Пентахлорфенол 340, 341, 349, 350
Пентахлорэтан 186
Пентаэритрит 235, 387
Пептон 22 (Ди-о-бензамидофенилди-
сульфид) 365, 366
Перевулканизация 520
Перегонка
мазута 46 ел.
нефти 38, 42 ел.
сухая см. Сухая перегонка
Перегонные кубы 44
Перегруппировки реакции 203, 258,
275, 283 сл„ 355
Перекись бензоила 396, 425
Переатерификация 408
Период релаксации 375
Перлон 471
Перфторуглероды 175, 194, 195
Перхлорвинилоаая смола 190, 467,
468
Пестициды 127, 335 ел.
Петрова контакт 573 ;.¦, а •
Печи Si*.
канальная 74
коксовые 86 ел.
¦ контактная 211, 212
крекинга метана 135, 136
і полукоксования 102 ел.
регенеративные 136, 137
трубчатые 44 ел., 63
туннельные 103 ел.
электродуговая 137, 138
Пигменты 286, 307, 322
Пиридин 333 ;
Пирогенные процессы 70
Пироксилин 433
Пиролиз
ацетона 216, 227
вискозного волокна 563 .
газы 28, 29, 157
дихлорэтана 190
катализаторы 227
нефтепродуктов 58, 62
нефти 38
окислительный 135
парафинов 131, 134 ел.
уксусной кислоты 227
Пластикат 538, 541 '
Пластикация 498
механическая 507
механоокислительиая 436
совместная 435, 436
термоокислительная 436, 508
ускорители 361, 365, 366, 498, 508
Пластики слоистые 528, 564 ел.
38—805
Пластификаторы 127, 488, 500, 528,
529, 545
Пластификация 455, 500, 520
Пластическая деформация 375
Пластические массы 123, 367, 383,
526 ел.
замутнители 531
литьевые 540, 541
наполнители 527, 528, 554, 555,
564, 566
пластификаторы 528, 529
применение 526, 539, 542, 545
свойства 526 ел., 538 ел., 544, 545
состав 526 ел.
текучесть 529, 554
термопластичные см. Термопласты
термореактивные см.
Термореактивные пластмассы
эластификаторы 529, 530, 540,
541, 554
Плато вулканизации 520
Платформинг 66, 147
Плотность каучуков 489, 492, 494
Плотность пластмасс 526
винипласта 544
волокнитов 558 ел.
графитопластов 564
органического стекла 547 , ,.
слоистых 567
1 стекловолокнитов 562
термопластов 539 ел.
фенопластов 554
фторопласта 544
целлулоида 546, 547 - ц
Пневой осмол 76
Поверхностно-активные вещества
(ПАВ) 328 сл.; 336, 459
Повышение цвета 288 '";
Подвулканизация 499 -U
Подземная газификация 114
Подошвенная резина 506
Подсмольная вода 73 v
Ползучесть полимеров 540, 543
Полиазокрасители 292
Полиакриламид 380
Полиакрил аты 216, 380
получение 417 ел. '
применение 383, 558
Полиакриловая кислота 368, 372, 380
Полиакрилонитрил 372, 373, 380 383
465, 466
Полиакролеин 380
Полиамидные волокна 390, 410 440
470 ел., 502
Полиамиды 382, 390, 470, 541
переработка 471 ел., 538 ел.
применение 383, 410, 502, 554
свойства 539 ел.
синтез 390, 408, 410, 411
сополимеры с полиэпоксидами 435
593
Полиамины 530
Полиацетали 380, 382, 434, 564, 574
Полибутадиен 381, 383, 395, 407,
483 ел., 494, 505
По дивинил ацетали 380, 434, 564, 574
Поливинилацетат 226, 378, 379, 395
полимераналогичные превращения
433, 434
применение 429
синтез 428, 429
Поливинилбутираль (Бутвар) 380,
434, 564, 574
Поливинилиденфторид 378
Поливинилиденхлорид 378
Поливинилиденцианид 378, 380
Поливиниловый спирт 378, 379, 433,
434
Поливинилформаль (Формвар) 380,
434
Поливииилфторид 378
Поливинилхлорид 190, 378, 379, 395,
422
деструкция 542
переработка 538 ел., 542
пластифицированный 538, 541
применение 466 ел., 549, 554
свойства 541, 543
хлорированный 190, 467
Поливинилхлоридные волокна 466 ел.
Полигексаметиленадипамид 390, 471
Полидисперсность полимеров 388
Полиизобутилен 379, 418, 419
Полиизопрен 368, 381, 383, 395, 486,
487, 494, 505
Поликапроамид (Капрон) 203, 372,
396, 405, 410, 411, 471, 502, 503
Поликарбонаты 538 ел.
Поликонденсация 384 ел.
блочная 404, 405, 408 ел., 415
в среде осадителя 419, 421
диэтиленголикольтерефталата 408
лаковая 416, 417
непрерывная 426, 428
полиэфиров с диизоцианатом
576 ел.
суспензионная 425
эмульсионная 423
Полималеинаты 409, 410, 530
Полимераналогичные превращения
430 ел.
Полимергомологический ряд 369
Полимердистиллят 334
Полимеризатор 406, 4<O
Полимеризационный бензин 58
Полимеризация 57
анионная 396, 399, 418, 419
ацетальдегида 221
блоксополимеризация 431, 435 ел.
блочная 402 ел., 407 ел., 415,
426 ел.
594
Полимеризация
бутадиена 395
в среде осадителя 405, 419 ел.
винилацетата 395, 405, 428
изобутилена 418, 419
изопрена 395
инициаторы 396, 401, 422, 425
ионная 394, 396, 399 ел., 40!. 403
418
капролактама 203, 257, 396. 405
катализаторы 245, 399, 418 420
483
катионная 396, 399
лаковая 405, 416 ел., 428, 429
метилметакрилата 395, 405
« непрерывная 425 ел.
низкотемпературная 483, 488
оборудование 406 ел.
обрыв цепи 399, 400
окисей олефинов 242. 396
олефинов 395, 415, 416, 418 ел.
привитая 436, 437
радикальная 394 ел., 397, 398, 403
рост цепи 397, 400
синильной кислоты 236
совместная (Сополямеризация)
397, 398, 424, 425, 429, 430,
435, 436
степень 369
стирола 395, 405, 426 ел.
ступенчатая 384, 394, 401. 402
суспензионная 405, 423 ел.
тетрафторзтилена 395
триоксиметилена 396
формальдегида 233
хлоропрена 395
цепная 384, 394 ел., 403
эмульсионная 405. 421 ст.. 429,
430, 482, 483, 488
Полимеры 123, 367 ел.
атактические 374
гетероцепные 378. 381 ел.
деструкция см. Деструкция
деформация 374, 375, 477
для химических волокон 444 ел.
изотактические 373, 374
карбоцепные 377 ел.
классификация 377 ел.
кремнийорганические см. Крем-
нийорганические полимеры
кристаллические 375
линейные см. Линейные полимеры
модифицирование 384, 430, 431,
438, 541, 554, 558, 559, 554, 576,
577
молекулярный вес 368, 369. 401,
539
обрыв цепи 399, 400
отверждение см. Отверждение
полимеров
Полимеры
пластификация 455, 500, 520
ползучесть 540, 543
по.-шдисперсиость 388
полимераналогичные превращения
430 ел.
привитые 371, 431, 436, 437
пространственные 371, 372, 377,
392 ел., 437, 438, 577, 578
разветвленные 371, 372, 431, 436,
437
растворимость 372, 459 ел., 462,
465, 466, 540
рост цепи 397
свойства 371 ел, 483 ел., 494, 495,
538, 541
синдиотактические 373, 374
синтез 384 ел., 402 ел., см. также
Поликонденсация и
Полимеризация
совмещение 415, 419, 431, 438,
541, 554
сополимеры см. Сополимеры
стабилизация 542, 548, 573
стереорегулярные 373, 374, 401,
420, 421, 469, 482, 483, 486
твердые 375, 376
текучесть 375, 376, 529, 539, 554
температура стеклования и
течения 376
термопластичные 389 ел., 526 ел.,
531, 545 ел.
термореактивные 389, 394, 404,
417, 526 ел., 566, 576 ел.
усадка 515, 550, 551, 578
эластичность см. Эластичность
полимеров
Полиметакриловая кислота 380
Полиметилакрилат 380
Полиметилнетакрилат 124, 380, 395,
405, 414, 545 ел.
Полимочевины 382
Полиоксиметилены 233, см. также
Полиформальдегид
Полиолефиновые волокна 469, 470
Полиолефины см. Полипропилен и
Полиэтилен
Полипропилен 369, 373, 379, 395, 496,
497
переработка 538 ел.
применение 383
свойства 469, 539 ел.
стереорегулярный 373, 420, 421,
469
Полисахариды 72, 77, 78, см. также
Целлюлоза
Полисилоксаны 530, 555, 556, 577
применение 491, 492, 494, 506,
550, 551, 558, 576
синтез 390, 391
38*
Полистирол 379, 395
переработка 538 ел.
применение 542, 545, 548, 549
свойства 539 ел.
синтез 426
эластификаторы 540, 541
Полисульфидные каучуки (Тиоколы)
125, 391, 423, 491, 494, 506
Полисульфоны 382
Политетрафторэтилен 379, 395, 493
переработка 543 ел.
применение 468, 545
Политиоэфиры 382
Политрифторхлорэтилен 538 ел.
Полиуретаны 125, 372, 382, 493
применение 383, 576
синтез 402
сополимеризация 435
Полиформальдегид 396
переработка 538 ел.
свойства 539 ел.
Полвхлоропрен 378, 381, 383, 395,
488, 489, 494, 506
Полициклические красители 317 ел.
Полиэнантоамид 390, 471
Полиэпоксиды 390
применение 412, 550, 558, 559, 564,
569, 575
синтез 408, 411 ел.
сополимеризация с полиамидами
435
Полиэтерификацин 385 ел., 417, 418
Полиэтилен 368, 378, 379, 395
высокого давления 415, 416
низкого давления 419
переработка 538 ел.
применение 415, 383, 469
свойства 469, 539 ел.
Полиэтиленгликоли 246
Полиэтиленгликольмалеинат 382, 389
Полиэтиленгликольтерефталат 125,
262, 382, 408 ел., 474, 475, 502
Полиэтиленоксид 396
Полиэфирные волокна 389, 409, 440,
474, 475
Полиэфироуретаны 577, 578
применение 413, 576 ел.
синтез 402, 408, 412, 413, 435
Полиэфиры 381 ел., 385 ел., 410, 530
линейные 389
поликонденсацин с диизоциана-
том 577
применение 383, 389, 409, 440, 474,
475, 550, 558 ел., 564, 569
синтез 408
Полуактивные сажи 499, 500
Полукокс 12, 86, 101 ел., 105
Полукоксование 71, 101 ел.
торфа 79
углей 81, 86 ел.
595
Полупродукты 121, 130, 264 ел,
Попутные газы 12, 21, 27 ел.
Пористая резина 496, 519
Порообразователи (Порофоры) 501,
530, 549
Почвенные гербициды 338, 346
Пределы взрываемости 154
Предсозревание целлюлозы 449, 452
Препараты
для резиновой промышленности
361 ел.
ОП 330, 334, 335, 337
101 (ПАВ) 333
Прессование
контактное 559, 560
пенопластов 549
слоистых пластиков 566 ел.
стекловолокиитов 558, 559
термопластов 531 ел., 542
термореактивиых пластмасс 551
ударное 532, 533, 542
фенопластов 553
Пресспорошки 528, 551 ел.
Прессформы 521, 531 ел.
Прессы
гидравлический 532, 533
мерсеризационный 447, 448
многоэтажный 566, 567
рамные (челюстные) 521
Привитые сополимеры 371, 431, 436,
437
Природные газы 9, 11, 12, 18, 20 ел.,
27 ел.
добыча 21, 23 ел.
запасы 12 ел.
конверсия 132, 163 ел.
месторождения 22
происхождение 22
свойства 18
Присадочные прутки 537
Промазка ткаии 518
Промоторы 396, 397, 401, 422
Пропазин 344, 345, 349
Пропан
окисление 230
пиролиз 131
Пропан-бутановая фракция 28, 29
Пропилен 131
гидратация 214
гндрохлорирование 182
окисление 215
полимеризация 395, 419 ел.
полимеры см. Полипропилен
получение 131, 134
синтезы иа его основе 239
сополимеры 492 ел.
хлорирование 184 ел.
Пропионовая кислота 230, 241
Пропионовый альдегид 16S
Пропитка тканей 515 ел.
596
Пропиточная машина ot>t>
Пространственные полимеры 371,
372, 377, 392 ел., 437, 438, 577,
578
Протекторная резина 506, 512
Противогазы 173, 195, 236, 337
Противостарители (АнтиоксидантыУ
361, 365, 500, 501
Противоутомители 365, 501
Протравные красители 300, 311
Протравы 300
Прочность
винипласта 544
волокиитов 558, 559, 562
графитопластов 564
клеевых швов 573 ел.
пенопластов 550, 577
резин 484, 494
слоистых пластиков 567
стекловолокнитов 562
термопластов 539 ел.
фенопластов 554
фторопласта 544, 545
химических волокон 441, 442
вискозных 457
нитрона 466
полиамидных 470, 474
поливинилхлоридного 466
полиолефиновых 470
тефлона 468
Прядение см. Формование
химических ВОЛОКОН
Прядильная масса 443
Пряднльиые машины 445, 446, 454,
456, 458, 467
Прядильные растворы 447, 448, 450,
460, 461, 463, 465, 466, 468
Прямая гидратация 204, 206, 211
Прямые красители 302 ел.
белый 326, 327
коричневый КХ 296
медные комплексы 305
черный 3 303 ел.
ярко-голубой светопрочный 326
Пуансон 532, 535, 536
Пылевидные
катализаторы 143
топливо 11 ел.
Раббед смокед-шитс 481
Рабочее топливо 15
Радикальная полимеризация 394 ел.
397, 398, 403
Разбавители резиновых смесей 501
Разветвленные полимеры 371 372
431, 436, 437
Разделение углеводородных газов
27 ел., 33, 155 ел., 159
Размол красителей 298
Рамный пресс 521
Растворители 126
каучуков 503
полиакрилонитрила 465
поливинилацеталей 434
поливииилхлорида 466
полиолефинов 469, 540
полистирола 540
рекуперация 127
селективные 127, 139, 155, 160,
162
феноло-формальдегидных смол
575
целлюлозы 459
эфиров целлюлозы 433, 450, 464
Растворитель лопастной 461
Ратин дан 360
Рашига метод получения феиола 251,
252
Реакции
Дильса—Альдера 354
Зелинского 146
Кучерова 217
Фриделя—Крафтса 357
Регенерат 497
Регенераторы 89, 136, 137
Регенерация катализаторов 142, 143
Резиновая промышленность 476
Резиновые изделия 476, 477
армирование 501 ел.
вулканизация см. Вулканизация
контроль качества 524
маканые 519
отделка и разбраковка 523 ел.
пористые 496, 519
производство 504 ел., 513 ел.
раскрой 518, 519
сборка 519
Резиновые клеи 503, 504, 518, 574
Резиновые смеси 478
ингредиенты см. Ингредиенты
резиновых смесей
каландрование 516 ел.
контроль качества 524
приготовление 509 ел.
рецептуры 505 ел.
усадка 515
шприцевание 513 ел.
Резиносмесители 507, 509 ел.
Резины 476 ел.
брекерная 505, 506
крепление к металлам 504
набухание 490
подошвенная 506
пористая 496, 519
протекторная 506, 512
регенерация 497
свойства 477, 478, 484, 485, 494,
495
старение 500
Резолы 421, 564, 573, 574
Резорцин 272
Ректификация 33, 42 ел., 63, 97,
159 ел.
азеотропная 161
вакуумная 159
углеводородов 157
экстрактивная 161 ел.
Рекуперация растворителей 127
Ренацит II (Трихлортиофенол) 365
Ренея катализатор 134
Репелленты 335
Рецептуры резиновых смесей 505 ел..
Решетка плавильная 471, 472
Риформинг 57 ел., 66
Рост цепи полимера 397, 400
Р-соль 294
Рэндокс (N.N-Диаллилхлорацетамид)
339, 340, 348
Сажа 137, 499
белая 500
газовая 36, 37
Сажемасляные каучуки 488
Салициловая кислота 284 ел., 294-
Сапропелиты 82
Сапропель 22
Сафранин 324, 325
Сборка изделий
из термопластов 536, 537
резиновых 519
СВАМ 562, 563
Сварка термопластов 537, 538
Светильный газ 12, 92
Свинец 18
Связующие 551 ел.
Себациновая кислота 387
Севин 355 ел.
Селективная очистка 38, 55 ел.
Селективные растворители 127, 139,
155, 160, 162
Сера
содержание в газах 33
— в топливе 16, 17, 79
теплота сгорания 17
Сернистые красители 319 ел.
Сернокислотная гидратация 204, 205,.
208, 214
Сероводород 30 ел.
Сероуглерод 449, 450
хлорирование 180
Сжижение газов 28, 29, 34
Силоксаиовые каучуки 491, 492, 494
506
Силон 471
Симазин 345, 349
Синдиотактические полимеры 373
374
Синергисты 337
Синильная кислота 235 ел., 350
Синтез-газ 107, 163 ел.
597'
Синтетические волокна 439, 440,
464 ел.
гетероцепные см. Гетероцепные
волокна
карбоцепные см. Карбоцепные
волокна
нитрон 465, 466
полиамидные 275, 390, 410, 440,
470 ел., 502
поливинилхлоридные 466 ел.
полиолефиновые 469, 470
полиэфирные 389, 409, 440, 474,
475
фторсодержащие 468
хлорин 467, 468
Синтетические каучуки 381, 383, 406,
419, 479 ел., 482 ел.
бутадиен-метилстирольные 487,
488
бутадиен-нитрильные см. Бута-
диен-нитрильные каучуки
бутадиеновые 381, 383, 395, 407,
483 ел, 494, 505
бутадиен-стирольные 430, 487,
488, 494
бутилкаучуки 482, 490, 491, 494,
506
жидкие 479, 491
изопреновые 368, 381, 383, 395,
486, 487, 494, 505
масляные 488
наполненные 483
полисульфидные 125, 391, 423,
491, 494, 506
сажемасляные 488
силоксановые 491, 492, 494, 506
стереорегулярные 482, 483, 486
уретановые 493
фторкаучуки 493, 506
хлоропреновые 378, 381, 383, 395,
488, 489, 494, 506
этилен-пропиленовые 492 ел.
Синтетические клеи 412, 483, 503,
504, 518, 570, 573 ел.
Синтетические смолы 406, 496 497,
515, 551 ел.
глифталевые 126
карбами до-форм альдегидные см.
Карбамидо-формальдегидные
полимеры
меламино-формальдегидные 417,
550, 551, 554, 558
модифицированные 384, 430, 43!,
438, 541, 554, 558, 559, 564, 574,
576
перхлорвнниловая 190, 467, 468
полиэфирные см. Полиэфиры
силоксановые см. Полисилоксаны
совмещенные 438, 555, 556
598
Синтетические смолы
феноло-формальдегидные см. Фе-
ноло-формальдегидные
полимеры
эпоксидные см. Полиэпоксиды
Синтол 164, 168
Системные пестициды
гербициды 338, 344
инсектициды 351, 356
Скважины 23 ел.
эксплуатация 25 ел.
Скелетные катализаторы 134, 259
Скипидар 72, 73, 75, 76
Склеивание
резин 504, 519
слоистых пластиков 569, 570,
573 ел.
термопластов 537
Сланцы 9, 11, 12, 82
запасы 14
перегонка 103
Слоистые пластики 528, 564 ел.
Смазочные масла 20, 38, 50, 128, 175
Смачиватель НБ 331, 332
Смесители
волокнитов 556, 559
резиновых смесей 507, 509 ел.
Смешанный газ 12
Смокед шитс 481
Смолокурение 74, 76
Смолоразгонная установка 98 ел.
Смолы
каменноугольная 69, 93, 98 ел.,
114
синтетические см. Синтетические
смолы
Совмещение полимеров 415, 419, 431,
438, 541, 554
Созревание вискозы 451, 453
Соль АГ 260
Сольвенты 97
Соляровое масло 12, 50, 96
Сополимеризация 397, 398, 424, 425,
429, 430, 435, 436
Сополимеры 369, 370, 479
акрнлонитрила 422, 466, 489, 490,
494, 495, 506, 541
бутадиена 422, 429, 430, 487 ел.,
494 ел., 506, 541
винилиденфторида 493
винилхлорида 466
дивинилбензола 424
изобутилена и изопрена 482, 490,
494, 495
метилвинилпиридина 496
метилстирола 487, 488
полиамидов и полнэпокендов 435
нолиуретанов и полиэфиров 435
привитые 371, 431, 436, 437
Сополимеры
стирола 422, 424, 429, 430, 487 ел.,
494, 495, 506, 541
трнфторхлорэтнлена 493
хлоропрена 489
этилена и пропилена 492 ел.
Спекание 544
Спирты
аллпловый 185
бутиловый 171
высшие, получение 164
дегидратация 211, 228 ел.
дегидрирование 211, 214, 219, 233
диацетоновый 216
изобутиловый 228 ел.
изопропиловый 214, 215
метиловый см. Метиловый спирт
многоатомные 30, 184 ел., 235,
246, 387
октадециловый 333
поливиниловый 378, 379, 433, 434
свойства 18
синтез 166, 168, 169, 171
этиловый см. Этиловый спирт
СП-раббер 481, 482
Стабилизаторы
газового бензина 33 ел.
клеев 573
полимеров 542, 548, 573
синильной кислоты 236
формальдегида 233
Старение резин 500
Стеарин 530
Стеариновая кислота 333
Стекла органические 414, 545 ел.
Стекловолокниты 557 ел., 564
анизотропные 562, 563
Стеклом аты 560, 569
Стеклообразное состояние 375, 376
Стеклопластики 557 ел., 562 ел.
Стеклотёкстолиты 564, 567 ел.
Стеклянное волокно 528
намотка 558, 561 ел.
применение 557 ел.
Степень полимеризации 369
Стереорегулярные полимеры 373, 374,
401, 420, 421, 469, 482, 483,486
Стереоспецифнческие катализаторы
420, 483
Стирол 530
полимеризация 395, 405, 426 ел.
получение 147 ел.
сополимеры 424, 429, 430, 487 сл„
494, 495, 506, 541
Структурирование 507
Ступенчатая полимеризация 384, 394,
401, 402
Сульфаниловая кислота 268, 269, 292
Сульфат аммония 93, 95
Сульфатная целлюлоза 76
Сульфенамид Ц AМ-циклогексан-2-
бензтиазолсульфенамид) 364
Сульфирование
аминов 267, 268
антрахинона 266
бензола 251, 265 ел.
додецилбензола 331
катализаторы 266
нафталина 265, 332
хлорбензола 351
Сульфитная целлюлоза 76
Сульфитные щелока 78, 206
Сульфокислоты 266 ел.
щелочное плавление 270 ел.
Сульфонол (Додецилбензолсуль-
фонат) 331
Сульфураторы 266, 268
Сульфурил хлористый 174
Суспензионная полимеризация 405,.
423 ел.
Сухая перегонка 16, 70
древесины 72 ел.
сланцев 103
торфа 79, 80
углей 86 ел., 101 ел.
Сухое
обогащение 85
топливо 15
тушение кокса 89
формование волокон 443
ацетатного 463
карбоцепных 464 ел.
Сушилка двухвальцовая 297, 298
Сушка
газов (осушка) 30
красителей 297, 298
тканей 518
торфа 79
химических волокон 459
2,4,5-Т B,4,5-Трихлорфеноксиуксус-
ная кислота) 342, 349
Тарелка колпачковая 43
Твердое топливо 9, 12
добыча 68
коксование см. Коксование
переработка 68 ел.
происхождение 69
свойства 18
Тедион B,4,5,4'-Тетрахлордифеиил-
сульфон) 357
Текстильные нити 457, 470, 474
Текстолиты 564, 567, 568
Текучесть полимерных материалов
375, 529, 539, 554
Телогены 398, 399
Температура вспышки
керосина 50
мазута 51
нефти 40
599-
Температура
застывания нефти 40
стеклования 376
течения 376
Теории
происхождения нефти 22
цветности 288 ел.
Тепловой баланс коксования 91
Теплоносители высококипящие 129,
130, 288, 472
Теплостойкость
винипласта 544
волокнитов 556, 557, 559, 563'
клеевых швов 576
•органических стекол 547, 548
пенопластов 550
слоистых пластиков 567
термопластов 526, 540, 541
фенопластов 554
фторопласта 544
целлулоида 547
Теплотворная способность топлива
15 ел.
древесины 72
низшая 17, 111
торфяного кокса 80
углей 82
Терилен (Лавсан) 125, 262, 406 ел.,
474, 475, 502
Термический крекинг 57 ел.
Термическое хлорирование
парафинов 175 ел.
Термоокислительная пластикация 436,
508
Термопластичные полимеры 389 ел..
526 ел., 531, 545 ел.
Термопласты 526 ел., 531 ел.
вакуум-формование 536
листовые 535 ел., 542, 543
литье под давлением 523, 528,
533 ел., 539, 545
органические стекла 124, 380, 395,
405, 414, 545 ел.
пенопластичные 548 ел.
прессование 531 ел., 542
применение 539
сборка изделий 536, 537
сварка и склеивание 537, 538
: свойства 538 ел.
химическая стойкость 468, 493,
541, 543 ел.
штампование 535 ел.
экструзия 513, 535
Термореактивные пластмассы 530,
550 ел.
волокниты 528, 556 ел.
пенопласты 576 ел.
пресспорошки 528, 551 ел.
слоистые 528, 564 ел.
600
Термореактивные полимеры 389, 394,
404, 417, 526 ел., 566, 576 ел.
Термофиксация 465, 470
Термохимическая переработка углей
81
Терпены 354
Тетрагидрофуран 466
Тетраметилдисульфид (Тиурам) 359,
363
Тетрафторэтилен 194, 195
полимеризация 395
полимеры см.
Политетрафторэтилен
Тетрахлорбензол 342
2,4,5,4'-Тетрахлордифенилсульфон
(Тедион) 357
Тетрахлорметан (Четыреххлористын
углерод) 162, 174, 179, 180, 195
Тетрахлорэтан 186 ел.
Тетрахлорэтилен 186
Тетраэтиленгликоль 246
Тетраэтилсвинец 49, 128, 181
Тефлон см. Политетрафторэтилен и
фторлон
Техника безопасности
в нефтепереработке 40 ел.
— производстве химических
волокон 455
при работе
с аминами 275
— галоидами 195 ел.
— окисью углерода 172 ел.
— пестицидами 337
— синильной кислотой 236
— углеводородами 154 ел.
Технические масла 93
Технический анализ топлива 16
Тиазиновые красители 325
Тиодигликоль 247, 248
Тиоиндиго красный С 317
Тиокарбаминовая кислота 347 ел.
Тиоколы (Полисульфидные каучуки)
125, 391, 423, 491, 494, 506
Тионилхлорид 347
Тиофос 356
Тиурам (Тетраметилдисульфид) 359,
363
Ткани 502, 503, 564
промазка 518
пропитка 515 ел.
сушка 518
Т-кислота 276
Токсичность
гербицидов 339 ел., 348, 349
инсектицидов 351, 354 ел.
металлоорганических соединений
357, 358
окиси углерода 172
Токсичность
углеводородов 154
хлора 195
о-Толуидин 273
Толуилендиамин 276
Толуилендиизоцианат 412, 577
n-Толуиловая кислота 262
Толуол 63, 97, 132
нитрование 202 ел.
окисление 248, 252 ел.
получение 147
хлорирование 192, 193
Тонкий органический синтез 121,
263 ел.
Топливо 9
анализ 16
балласт 15
безбалластное 15
газификация см. Газификация
топлив
газообразное 9, 12, 18
гидрогенизация 106 ел., 114 ел.
горючая масса 15 ел.
добыча см. Добыча топлив
естественное 11 ел.
жидкое см. Жидкое топливо
запасы 12 ел.
зольность 15
искусственное 11 ел., 15
классификация 11 ел.
местное 11
моторное 38, 49, 98, 105, 128
облагораживание 106
облагороженное 15, 68
окисление 106 ел.
переработка 19
потребление 14
пылевидное 11 ел.
рабочее 15
свойства 15 ел., 18
сжигание 18, 19
состав 15 ел., 114
сухая перегонка см. Сухая перс-
гонка
сухое 15
твердое см. Твердое топливо
теплотворная способность см.
Теплотворная способность
топлива
термохимическая переработка 81
условное 18
химическая переработка 9, 14 ел.
хранение 19
энергоемкость 19
Топочные газы 17
Торф 9, 11, 12, 69
газификация 79
добыча 78, 79
запасы 12 ел.
Торф
переработка 79, 80
происхождение 78 ел.
свойства 18, 79
состав 78
сушка 79
энергоемкость 19
Торфяники 78, 79
Тощий газ 27
Трансмиссионные масла 50
Трансформаторное масло 38
Триазины симметричные 344 ел.
Трибутилфосфат (Бутифос) 350
Триметилпентаны 150
Тринитротолуол (Тротил) 202 ел.
Триоксан 234
Триоксиметилен, полимеризация 396
Трисазокрасители 292, 296, 302
Трифторхлорэтилен 493
Трихлорацетат натрия (ТХА) 339,
348
Трихлорметафос-3 356
Трихлортиофенол (Ренацнт II) 366
Трихлорэтиламин 247
Трихлоруксусная кислота 339
Трихлоруксусный альдегид см.
Хлораль
Трихлорфеноксиуксусная кислота
B,4,5-Т) 342, 349
Трихлорфенолят меди 358
Трихлорэтан 186, 187, 190, 191
Трихлорэтилен 186 ел., 196
Триэтиленгликоль 246
Трубчатые установки
перегонная 44 ел.
пиролиза 63
Туннельные печн 103 ел.
Турбинное бурение 23
Турбобур 23 ел.
ТХА (Трихлорацетат натрия) 339,
348
Тяжелое масло 98
Углеводороды
ароматические см. Ароматические
углеводороды
ацетиленовые см. Ацетилен н
Ацетиленовые углеводороды
газообразные 27 ел.
галоиднрование 174 ел., 180 ел.,
186 ел., 192 ел., 251, 252,
352 ел.
гидрирование 115, 249
дегидрирование 28, 29, 57, 59, 132,
134, 141 ел., 147 ел.
дегидроцнклизация 57, 132, 146 ел
диеновые 132, см. также
Бутадиен и Изопрен
изомеризация 148 ел.
60 1-
і глеводороды
крекинг 59, 135, 136
нитрование 196 ел., 200 ел.
окисление 51, 107, 215, 220, 221,
229 ел., 240 ел., 248 ел., 252 ел.,
260 ел., 282
олефиновые см. Олефины
пиролиз 131, 135
получение 164
предельные см. Парафины
разделение 27 ел., 33, 155 ел., 159
разложение 134
сорбция 155
токсичность 154
хлорированные, см. Хлорирование
Углежжение 74
Углерод 10
окись см. Окись углерода
содержание в земной коре 10, 11
теплота сгорания 17
четыреххлористый 162, 174, 179,
180, 195
Угли 9, 80 ел.
активированный 75, 195
бурые см. Бурые угли
газификация см. Газификация
топлнв
газовый 70, 114
добыча 68, 84
древесный 12, 73, 75
запасы 12 ел.
каменные см. Каменные углн
классификация 82 ел.
коксование 81, 86 ел.
коксовые 87, 114
месторождения 84
обогащение 84 ел.
ожижение 71
переработка 80 ел.
происхождение 81
свойства 18
состав 81 ел., 114
строение 115
Углубление цвета 288
Ударное прессование 532, 533, 542
Удельная ударная вязкость
волокнитов 556, 558 ел., 562
органического стекла 547
пенопластов 550, 577
слоистых пластиков 567
термопластов 540, 541
фенопластов 554
целлулоида 547
Уксусная кислота 73, 75, 76, 124
пиролиз 227
свойства и применение 225
синтез 172, 216, 223 ел., 230, 241
хлорирование 225
гид
Уксусный ангидрид 216, 226 ел.
Улавливание
аммиака 95
газового бензина 33 ел., 157
продуктов коксования 92 ел.
сероводорода 32
сырого бензола 96
Универсальная адгезия 570, 575
Уретановые каучуки 493
Уротропин (Гексаметилентетрамин)
234, 530, 551, 576
Усадка полимеров 515, 550, 551, 578
Усилители каучука 489, 499, 500
Ускорители
вулканизации 361 ел., 498, 499,
508
отверждения полимеров 530
пластикации 361, 365, 366, 498,
508
Условное топливо 18
Устье скважины 23
Фенантрен 100, 101
Фенигидроксиламин 274
л-Фенилендиамин 293, 294
Фенилмеркурацетат 358
Фенилметилпиразолонсульфо-
кислота 295
Фенилэтилен см. Стирол
Феноксиуксусная кислота 281
Фенол 97, 100, 105, 124, 294, 392
алкилирование 334
гидрирование 254 ел.
нитрование 340
применение 254, 281
синтез 215, 251, 252, 271, 272
— совместно с ацетоном 248 ел.,
252
хлорирование 342
Фенолиз 388
Феноло-формальдегидные полимеры
126, 381, 419, 421, 496, 497, 530
модифицирование 558, 559, 564,
576, 577
отверждение 566, 575, 576
применение 556 ел., 564, 570, 573,
575 ел.
синтез 392, 426
совмещение 438
Фенольное масло 98
Фенопласты 551 ел.
модифицированные 576
Фенурон 344, 349
Фербам (Диметнлдитиокарбамат
железа) 359
Фиксация макромолекул 376, 377,
465, 470
602
Филаментная нить 439, 446
Фильерная вытяжка 464, 470
Фильеры 445, 454, 461, 472
Фильтрование
вискозы 453
красителей 296, 297
Фильтрпрессы 296, 297
ФК, пенопласты 576, 577
Флегма 42, 43
Флотация 85
Формальдегид 392, 393
полимеризация 233
получение 230, 231 ел.
синтезы на его основе 234, 292,
421
стабилизация 233
Формалин 233
Форманилид 277, 278
Форматор-вулканизатор 522
Формвар (Поливинилформаль) 380,
434
Формование пластических масс
531 ел.
вакуумное 536
контактное 559, 560
органических стекол 547
спеканием 544
стекловолокнитов 558, 560
стеклопластиков 569
термореактивных 551 ел., 576
экструзионное 535, 576
Формование химических волокон 443
ацетатного 463, 464
бобинное 454, 456, 457
вискозного 453, 457
двухванное 461, 462
из расплавов 443, 444, 469, 471 ел.
карбоцепных 464
медноаммиачного 461, 462
мокрое см. Мокрое формование
волокон
нитрона 465
полиамидных 471 ел.
поливинилхлоридного 466, 467
полиолефиновых 469
полизфіірньїх 474
сухое см. Сухое формование
волокон
фторлона 468
центрифугальное 454 ел.
Формовочная машина 463
Форполимеры 407, 413, 414, 418
Фосген 174, 178
Фосгенирование
аминов 277
дипропиламина 347
нзопропилового спирта 346
а-нафтолята натрия 356
хлоранилина 344
Фосфамид 356, 357
Фосфорорганические инсектициды
354 ел.
Фотосинтез 10
Фотохимическое хлорирование 175,
179, 180, 352
Фракции нефтяные 39
Фреоны 129, 175, 195, 196
Фриделя—Крафтса реакция 357
Фрикция 509
Фталаты 261
Фталевые кислоты 249, 260, 261
Фталевый ангидрид 100, 249, 321
конденсация с бензолом 282
получение 260
применение 261
Фталогены 322
Фталоцианиновые красители 321 ел.
Фторирование 194
Фторкаучуки 493, 506
Фторлон 468
Фторопласты 129, 194, 379, 395, 468,
493, 538 ел., 543 ел.
Фуксин 322, 323
Фумиганты 350, 351, 353
Фунгициды 335, 347, 358 ел,
Фурфурол 77
Фусы 93
Фюзен 81
Химическая стойкость
каучуков 490, 491
термопластов 468, 493, 541,543 ел.
Химические волокна 367, 368, 383,
439 ел.
вытяжка см. Вытяжка
химических волокон
гетероцепные см. Гетероцепные
волокна
графитированные 563, 564
искусственные см. Искусственные
волокна
карбоцепные см. Карбоцепные
волокна
крашение 299 ел., 305, 307 ел., 321
методы получения 442, 443
неорганические 528, 557, 563
номер 441
отделка 446, 447, 459, 462, 464
синтетические см. Синтетические
волокна
стеклянное см. Стеклянное
волокно
сушка 459
формование см. Формование
химических волокон
штапельное см. Штапельное
волокно
603
Хинониминовые красители ол* oi.
>Сладоагенты 175
>Сладотекучесть 529
Хлораль (Трихлоруксуснып
альдегид) 213, 214, 282
конденсация с диметилфосфитом
355
— с хлорбензолом 351
Хлоранил 326
Хлоранилин 344
Хлораторы 177 ел., 193, 352
Хлорацетилхлорид 339, 340
Хлорбензол 192 ел.
аминирование 275
гидролиз 251
конденсация с хлоралем 282, 351
синтезы на его основе 357
сульфирование 351
Хлорин 467, 468
Хлоринат 346
Хлорирование
ароматических углеводородов
192 ел., 251, 352 ел.
ацетилена 186 ел.
дихлорэтана 191
карбоновых кислот 339
катализаторы 178, 187, 190, 192,
339
1,4-нафтохинона 359
окислительное 174, 251, 252
олефинов 181 ел., 188
парафинов 175 ел., 180 ел.
поливинилхлорида 190
сероуглерода 180
термическое 175 ел.
терпенов 354
уксусной кислоты 225
фенола 342
фотохимическое 175, 179, 180, 352
этилового спирта 213 ел.
Хлористый сульфурил 174
"ХлорИФК [Изопропил-Ы-C-хлорфе-
нил)-карбамат] 346, 349
Хлоропрен 191, 192, 196
полимеризация 395
полимеры 378, 381, 383, 395, 488,
489, 494, 506
сополимеры 489
Хлоропреновые
каучуки 378, 381, 383, 395, 488,
489, 494, 506
латексы 496
Хлороформ 178, 179, 195, 196
Хлорофос 355
Хлорпикрин 350
.Хлоруксусная кислета 225 ел 341
342
Хлорфенолы 348 ел.
лииидпап
вулканизация 520
вытяжка 472
Холодное дутье 111
«Холодные» красите-чи 308
Хризоидин 293
Хромовые красители 300, 301
Хромогены 288
Хромофоры 288
Целлозольвы 246
Целлулоид 545, 546
Целлюлоза 72, 73, 382
ацетилирование 462
ксантогенат 449, 450, 452
нитрование 433
переработка в волокно 447 ел.
полимераналогичные
превращения 432, 433
получение 76 ел.
эфиры 433, 450, 464, 538, 541, 545
Центрифугальное формование 454 ел.
Цепная полимеризация 384, 394 ел.,
403
Церезин 127
Цетановое число 50
Цианиды 236
Цианурхлорид 306, 327, 345, 346
Циклогексан 249, 250
нитрование 203
Циклогексанол 250, 251, 254, 255
окисление 258, 259
Циклогексанон 250, 251, 254 ел.
Циклогексаноноксим 203, 257
Цимат (Диметилдитиокарбамат
цинка) 364
ге-Цимол 215
Цинеб 359
Цирам (Диметилдитиокарбамат
цинка) 359, 364
Челюстные прессы 521
Четыреххлористый углерод 162, 174,
179, 180, 195
Число
октановое 49, 64, 128, 148, 149
цетановое 50
Шахта угольная 85
Шины резиновые 505
Шнхта 87
Шнек-машина 469
Шприцевание резиновых смесей
513 ел.
Шприц-машина 513 ел.
Штампование 535 ел.
Штапельное волокно 441, 457, 466,
470, 474
отделка 446, 447
604
Щелока сульфитные 78, 206
Щелочная очистка 52
Щелочное плавление сульфокислот
270 ел.
Щетина 474
Экстрактивная ректификация 161 ел.
Экстракция 70, 79, 80, 126, 127,
160 ел.
Экструзия 513, 535
Эластификаторы 529, 530, 540, 541,
554
Эластичность полимеров 375, 376,
539 ел.
резин 474, 478, 485, 494
химических волокон 442
Электробур 23 ел.
Злектродуговая печь 137, 138
Злектрокрекинг 135, 137
Эмульгаторы 422
Эмульсионная
поликонденсация 423
полимеризация 405, 421 ел., 429,
430, 482, 483, 488
Знант 471, 474
Энергоемкость топлива 19
Зпихлоргидрин 390
Зпоксиды 242 ел., 396
полимеры см. Полиэпоксиды
Зптам (Этил-М,М-дипропилтиокар-
бамат) 347 ел.
Этан
дегидрирование 141
екисление 230
пиролиз 131
хлорирование 180 ел.
Этанол см. Этиловый спирт
Этаиоламины 31, 247
Этерификация 343, 385
Этил хлористый 180 ел.
N-Этилбензиланилин 280
Этилбензол 147, 148, 150 ел. '
Этилеи
гидратация 204, 206 ел.
гидрохлорирование 181
гипохлорирование 243
окисление 220, 221, 242 ел.
окись см. Окись этилена
получение 131, 134, 141, 162
полимеризация 395, 415, 416
полимеры см. Полиэтилен
сополимеры 492 ел.
хлорирование 183 ел., 188
Этилеигликоль 129, 245, 389, 408
Этилен-пропилеиовые каучуки 492 ел.
Этилеихлоргидрин 243 ел.
Этнленциангидрин 236, 237
Этилидеидиацетат 228
Этилксантогентрисульфид 350
Этилмеркурхлорид (Гранозан) 358
Этиловая жидкость 128
Этиловый спирт 12, 77, 78, 124
дегидратация 211
дегидрирование 211, 219
окисление 219
получение 205 ел., 230
применение 205, 206
свойства 205
синтезы на его основе 211 ел.
хлорирование 213 ел.
Этролы 538 ел., 541
Эфирсульфонат 357
Эфиры
акриловой кислоты 237, 239
арилкарбаминовых кислот 346 ел.
2,4-Д 343
дихлордиэтиловый 391
карбаминовой кислоты 345 ел.,
355 ел.
целлюлозы 433, 450, 464, 538, 541,
545
Ядохимикаты см. Пестициды
Яды
катализаторные 96
кишечные 350, 351, 353 . .;
Дмитрий Дмитриевич Зыков,
Варвара Андреевна Деревицкая,
Елена Борисовна Тростяиская,
Михаил Александрович Чекалин,
Илья Исаевич Юкельсон,
Фелиция Иосифовна Яшуиская
ОБЩАЯ ХИМИЧЕСКАЯ ТЕХНОЛОГИЯ
ОРГАНИЧЕСКИХ ВЕЩЕСТВ
608 с. УДК 660@75.3)
Редакторы И. С, Абрамова, И. В. Лебедева
Технический редактор М. 3. Васина
Корректоры М. С. Хрипунова, В. И. Советникова
Т02234 Подписано к печати 25/1 1966 г.
Формат бумаги 60Х901Дй = 19 бум- л.=38печ. л. Уч.-изд. л. 37,5:
Тираж 15000 экз. Типогр. бум. № 2. Цена 1 р. 44 к- Зак. 805
Московская типография № 21 Главполнграфпрома
Комитета по печати при Совете Министров СССР.
'Москва, 88, Угрешская, 12.
ОПЕЧАТКИ
Стр
185
189
195
222
284
301
311
311
315
354
392
393
397
418
446
457
465
474
Заказ
Строка
so
10 сверху
6 и 7
сверху
14 сверху
12 сверху
подпись
к рис. 97
Формула
кислотного
зеленого
ЖМ
3 снизу
8 снизу
5, 6 снизу
14, 15 сверху
12 сверху
Нижняя
часть
формулы карба-
мидного
полимера
1 сверху
16, 17 сверху
21 сверху
В 3 графе
таблицы,
2 снизу
2 снизу
17 снизу
5.
Напечатано
образованием глицерина
без применения хлора:
дегидрохлорирования
щелочью: в жидкой фазе
CC12F2—CC12F2
первой
получения фенола
О — Cr-NH
/ | \
/? V. М М >у \i
У X
O2N \_S
антрацена
С1~ или Вг~ или же
групп ОН" или N0^
калиевая
индокси
циклопентане
метильные
1
N-CH2-N-CH2-• ¦ •
1 1
с=о с=о
NH2 NH2
нафтената
толуол, метакриловую
кислоту, двухосновную
насыщенную кислоту
(в качестве растворителя)
жгут (нить).
30—1600
температуре
(термофиксация)
разработанное и
полученное
Следует читать
образованием глицерина:
дегидрохлорирования: щелочью
в жидкой фазе
CC1F2—CC1F2
второй
получения фенолята
О — Cr-NH
/ А \
оксиантрацена
СІ или Вг или же групп
ОН или NO2
калиеваи соль
индоксила
ци клопеніадиена
метилольные
I
N—СН2—N—СН2—
] 1
с=о с=о
NH, NH2
нафтенат
метакриловую кислоту,
двухосновную насыщенную кислоту, толусл
(в качестве растворителя)
жгут.
300—1600
температуре, термофиксации
разработанного и полученного
ИЗДАТЕЛЬСТВО ,,ХИМИЯ'
готовятся
К ПЕЧАТИ
АНДРЕЕВ Ф. А., КОЗЛОВ Л. И.,
ПРИСТАВКО В. Ф.
Технология связанного азота.
35 л. Цена 1 р. 30 к. в пер.
В книге описаны методы получения газовой
(азотноводородной) смеси конверсией
углеводородных газов, газификацией твердого и жидкого
топлива, глубоким охлаждением коксового газа
и воздуха, а также рассмотрены процессы
очистки азотноводородной смеси, направляемой на
синтез аммиака.
В книге изложены также теоретические основы
и технология синтеза аммиака, получения
разбавленной и концентрированной азотной кислоты.
Значительное внимание уделено аппаратурному
оформленню рассматриваемых процессов,
приведены примеры технологических расчетов,
принципы контроля и регулирования процессов,
организации техники безопасности на предприятиях
азотной промышленности.
Книга предназначена в качестве учебного
пособия для учащихся химико-технологических
техникумов. Она может быть полезна также для
широких кругов работников химической и газовой
промышленности, желающих получить
представление о производстве таких важных продуктов,
как синтетический аммиак и азотная кислота.
ЭРИХ В. Н.
Химия нефти и газа.
15 л. Цена 60'коп. в пер.
В учебнике рассмотрены химический состав
нефти, физические и моторные свойства нефти и
нефтепродуктов, а также химические процессы
переработки нефти и газа.
Помимо химизма процессов термического
крекинга и пиролиза, термокаталитических процессов
переработки нефтяного сырья и синтеза
компонентов моторного топлива в книге рассмотрены
химические процессы, лежащие в основе
переработки углеводородных газов и нефтехимического
синтеза.
Книга предназначена в качестве учебника для
учащихся химических техникумов. Она может
служить также руководством для
инженерно-технических работников и мастеров
нефтехимической, нефтеперерабатывающей и газовой
промышленности.